ES2775213T3 - Análisis estadístico para la determinación no invasiva de aneuploidías de los cromosomas sexuales - Google Patents
Análisis estadístico para la determinación no invasiva de aneuploidías de los cromosomas sexuales Download PDFInfo
- Publication number
- ES2775213T3 ES2775213T3 ES14811485T ES14811485T ES2775213T3 ES 2775213 T3 ES2775213 T3 ES 2775213T3 ES 14811485 T ES14811485 T ES 14811485T ES 14811485 T ES14811485 T ES 14811485T ES 2775213 T3 ES2775213 T3 ES 2775213T3
- Authority
- ES
- Spain
- Prior art keywords
- chromosome
- nucleic acid
- fetal
- sample
- copies
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6876—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes
- C12Q1/6883—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes for diseases caused by alterations of genetic material
-
- G—PHYSICS
- G16—INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR SPECIFIC APPLICATION FIELDS
- G16B—BIOINFORMATICS, i.e. INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR GENETIC OR PROTEIN-RELATED DATA PROCESSING IN COMPUTATIONAL MOLECULAR BIOLOGY
- G16B20/00—ICT specially adapted for functional genomics or proteomics, e.g. genotype-phenotype associations
- G16B20/20—Allele or variant detection, e.g. single nucleotide polymorphism [SNP] detection
-
- G—PHYSICS
- G16—INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR SPECIFIC APPLICATION FIELDS
- G16B—BIOINFORMATICS, i.e. INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR GENETIC OR PROTEIN-RELATED DATA PROCESSING IN COMPUTATIONAL MOLECULAR BIOLOGY
- G16B20/00—ICT specially adapted for functional genomics or proteomics, e.g. genotype-phenotype associations
-
- G—PHYSICS
- G16—INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR SPECIFIC APPLICATION FIELDS
- G16B—BIOINFORMATICS, i.e. INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR GENETIC OR PROTEIN-RELATED DATA PROCESSING IN COMPUTATIONAL MOLECULAR BIOLOGY
- G16B20/00—ICT specially adapted for functional genomics or proteomics, e.g. genotype-phenotype associations
- G16B20/10—Ploidy or copy number detection
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q2600/00—Oligonucleotides characterized by their use
- C12Q2600/156—Polymorphic or mutational markers
Landscapes
- Life Sciences & Earth Sciences (AREA)
- Health & Medical Sciences (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Physics & Mathematics (AREA)
- Chemical & Material Sciences (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Analytical Chemistry (AREA)
- Genetics & Genomics (AREA)
- Molecular Biology (AREA)
- Biotechnology (AREA)
- Biophysics (AREA)
- General Health & Medical Sciences (AREA)
- Spectroscopy & Molecular Physics (AREA)
- Theoretical Computer Science (AREA)
- Bioinformatics & Computational Biology (AREA)
- Medical Informatics (AREA)
- Evolutionary Biology (AREA)
- Organic Chemistry (AREA)
- Wood Science & Technology (AREA)
- Zoology (AREA)
- Microbiology (AREA)
- General Engineering & Computer Science (AREA)
- Biochemistry (AREA)
- Immunology (AREA)
- Pathology (AREA)
- Measuring Or Testing Involving Enzymes Or Micro-Organisms (AREA)
Abstract
Un procedimiento para calcular un riesgo de aneuploidía cromosómica de X o Y fetal en una muestra materna, en el que las etapas de determinación, estimación y cálculo se realizan en un ordenador, comprendiendo el procedimiento las etapas de: consultar uno o más locus cromosómicos en Y in vitro; consultar uno o más locus cromosómicos en X in vitro; consultar uno o más locus polimórficos en al menos un primer autosoma in vitro; calcular una proporción de ácido nucleico fetal en la muestra materna analizando una frecuencia de polimorfismos en el primer autosoma; determinar si la proporción de ácido nucleico fetal en la muestra materna es adecuada para realizar el análisis estimar una frecuencia del cromosoma I relativa para un cromosoma Y y un cromosoma X en la muestra materna en vista de la proporción de ácido nucleico fetal; usando la proporción de ácido nucleico fetal calculada, calcular los valores de probabilidad de que el cromosoma Y fetal no esté presente en ninguna copia, esté presente en una copia o dos o más copias en la muestra materna; calcular un riesgo de aneuploidía del cromosoma Y fetal en la muestra materna comparando los valores calculados de probabilidad con un primer modelo matemático que supone ninguna copia del cromosoma Y, un segundo modelo matemático que supone una copia del cromosoma Y y un tercer modelo matemático que supone dos o más copias del cromosoma Y; usando la proporción calculada de ácido nucleico fetal, calcular los valores de probabilidad de que el cromosoma X fetal esté presente en una copia, dos copias o tres o más copias en la muestra materna; y calcular un riesgo de aneuploidía del cromosoma X fetal en la muestra materna comparando los valores calculados de probabilidad con un primer modelo matemático que supone una copia del cromosoma X, un segundo modelo matemático que supone dos copias del cromosoma X y un tercer modelo matemático que supone tres o más copias del cromosoma X.
Description
DESCRIPCIÓN
Análisis estadístico para la determinación no invasiva de aneuploidías de los cromosomas sexuales
CAMPO DE LA INVENCIÓN
La presente invención se refiere al análisis estadístico para la determinación no invasiva del sexo de un feto o de anomalías en la frecuencia cromosómica de X e Y detectando y determinando la contribución relativa de secuencias genéticas de los cromosomas X e Y en vista del porcentaje de contribución fetal en una muestra mixta materna. ANTECEDENTES DE LA INVENCIÓN
En el siguiente análisis se describirán determinados artículos y procedimientos con propósitos preliminares e introductorios. Nada contenido en el presente documento se ha de interpretar como una "admisión" de la técnica anterior. El solicitante se reserva expresamente el derecho a demostrar, cuando resulte apropiado, que los artículos y procedimientos a los que se hace referencia en el presente documento no constituyen una técnica anterior en virtud de las disposiciones estatutarias aplicables.
Las anomalías genéticas representan un amplio número de patologías, incluyendo los síndromes provocados por aneuploidía cromosómica (por ejemplo, síndrome de Down) y los provocados por mutaciones germinales que dan como resultado enfermedades o trastornos monogénicos o bien poligénicos. La detección tanto de anomalías cromosómicas evidentes, tales como trisomías, translocaciones e inserciones o deleciones grandes, como de rasgos debidos a un único gen, tales como mutaciones debidas a un único gen o polimorfismos asociados con el estado del grupo sanguíneo Rh, trastornos autosómicos dominantes o ligados al cromosoma X, o trastornos autosómicos recesivos son útiles en la detección de patologías y trastornos reales y potenciales que puedan afectar a un feto. Por ejemplo, todas las anomalías cromosómicas, tales como las trisomías 13, 18 y 21, las translocaciones robertsonianas y las deleciones más grandes, tales como las encontradas en el cromosoma 22 en el síndrome de DiGeorge, tienen un impacto significativo sobre la salud fetal.
Aunque la tecnología convencional proporciona procedimientos de detección para estas diferentes anomalías genéticas, hasta hace poco diferentes anomalías genéticas requerían diferentes técnicas para consultar diferentes clases de mutaciones. Por ejemplo, los procedimientos convencionales de pruebas de diagnóstico prenatal para determinar una aneuploidía cromosómica requerían la extracción de una muestra de células fetales directamente del útero para su análisis genético, usando una biopsia de vellosidades coriónicas (BVC) entre las 11 y 14 semanas de gestación o bien amniocentesis después de las 15 semanas. Sin embargo, dichos procedimientos invasivos conllevan un riesgo de aborto espontáneo de alrededor de un uno por ciento (véase Mujezinovic y Alfirevic, Obstet. Gynecol., 110:687-694 (2007)). Otros análisis de células fetales típicamente implican el cariotipado o hibridación in situ con fluorescencia (FISH) y no proporcionan información sobre rasgos debidos a un único gen; por tanto, se requieren pruebas adicionales para la identificación de enfermedades y trastornos debidos a un único gen.
La detección no invasiva de secuencias de ADN heredadas por vía paterna que están ausentes en el genoma materno, por ejemplo, secuencias cromosómicas de Y para la identificación del sexo fetal y el gen RHD para el genotipado del grupo sanguíneo, ha sido posible desde mediados de la década de 1990. Sin embargo, la reciente aparición de tecnologías de recuento de moléculas únicas, tales como reacción en cadena de la polimerasa digital y, en particular, secuenciación masivamente paralela, ha permitido que se use ADN fetal circulante para el diagnóstico prenatal no invasivo de aneuploidías cromosómicas fetales y enfermedades monogénicas, aunque otras anomalías fetales y/o parámetros de control de calidad para las pruebas permanecen sin abordar.
Existe una necesidad en la técnica de obtener una determinación exacta del sexo fetal, la frecuencia cromosómica de X y la frecuencia cromosómica de Y. La presente invención aborda esta necesidad.
El documento US 2013/0029852 describe un procedimiento para determinar las variaciones del número de copias (VNC) de una secuencia de interés en una muestra de prueba que comprende una mezcla de ácidos nucleicos que se sabe o se sospecha que difieren en la cantidad de una o más secuencias de interés.
El documento US 2012/0191358 describe procedimientos para determinar probabilidades de riesgo exactas para aneuploidías fetales.
SUMARIO DE LA INVENCIÓN
Se proporciona el presente sumario para introducir una selección de conceptos de forma simplificada que se describen adicionalmente a continuación en la descripción detallada. No se pretende que el presente sumario identifique rasgos característicos clave o esenciales de la materia objeto reivindicada, ni tampoco se pretende que se use para limitar el alcance de la materia objeto reivindicada. Otros rasgos característicos, detalles, utilidades y ventajas de la materia objeto reivindicada serán evidentes a partir de la siguiente descripción detallada escrita, incluyendo los aspectos ilustrados en los dibujos adjuntos y definidos en las reivindicaciones adjuntas.
En un aspecto, los procedimientos utilizan amplificación y detección multiplexadas de regiones de ácido nucleico seleccionadas en los cromosomas sexuales y uno o más autosomas (es decir, autosomas) para calcular la frecuencia de los cromosomas X e Y en relación con el porcentaje de contribución de ácido nucleico fetal en una muestra mixta materna. Las cantidades relativas de las regiones de ácido nucleico seleccionadas se determinan para las regiones genómicas de interés (por ejemplo, secuencias cromosómicas sexuales, así como secuencias de una o más secuencias de cromosomas autosómicos) usando los procedimientos analíticos como se describe en el presente documento. Dichos procedimientos se usan para determinar el sexo de un feto, posibles aneuploidías cromosómicas de X e Y y mosaicismos intersexuales, así como para evaluar la probabilidad de contaminación de la muestra mixta materna.
Estos y otros aspectos, rasgos característicos y ventajas se proporcionarán con más detalle como se describe en el presente documento.
BREVE DESCRIPCIÓN DE LAS FIGURAS
La figura 1 es un diagrama de flujo simplificado de un procedimiento de ensayo de acuerdo con la presente invención.
La figura 2 ilustra un sistema de ensayo multiplexado para la detección de dos o más regiones de ácido nucleico seleccionadas.
La figura 3 ilustra un sistema de ensayo multiplexado alternativo para la detección de dos o más regiones de ácido nucleico seleccionadas.
La figura 4 ilustra aún otro sistema de ensayo multiplexado alternativo para la detección de dos o más regiones de ácido nucleico seleccionadas.
La figura 5 ilustra aún otro sistema de ensayo multiplexado alternativo para la detección de dos o más regiones de ácido nucleico seleccionadas.
La figura 6 ilustra aún otro sistema de ensayo multiplexado alternativo para la detección de regiones de ácido nucleico seleccionadas.
La figura 7 ilustra aún otro sistema de ensayo multiplexado alternativo para la detección de regiones de ácido nucleico seleccionadas.
La figura 8 es un diagrama de flujo simplificado de un procedimiento ejemplar para realizar un análisis estadístico de acuerdo con la presente invención.
La figura 9 es aún otro diagrama de flujo simplificado de un procedimiento ejemplar para realizar un análisis estadístico de acuerdo con la presente invención.
DESCRIPCIÓN DETALLADA DE LA INVENCIÓN
Los procedimientos descritos en el presente documento pueden emplear, a menos que se indique de otro modo, técnicas y descripciones convencionales de la tecnología de biología molecular (incluyendo técnicas recombinantes), biología celular, bioquímica, y micromatrices y secuenciación, que estén dentro de la habilidad de quienes las ponen en práctica en la técnica. Dichas técnicas convencionales incluyen síntesis de matrices poliméricas, hibridación y unión de oligonucleótidos, secuenciación de oligonucleótidos y detección de hibridación usando un marcador. Se pueden tener ilustraciones específicas de técnicas adecuadas por referencia a los ejemplos en el presente documento. Sin embargo, por supuesto, también se pueden usar procedimientos convencionales equivalentes. Dichas técnicas y descripciones convencionales se pueden encontrar en manuales de laboratorio estándar, tales como Green, et al., Eds., Genome Analysis: A Laboratory Manual Series (vols. I-IV) (1999); Weiner, et al., Eds., Genetic Variation: A Laboratory Manual (2007); Dieffenbach, Dveksler, Eds., PCR Primer: A Laboratory Manual (2003); Bowtell y Sambrook, DNA Microarrays: A Molecular Cloning Manual (2003); Mount, Bioinformatics: Sequence and Genome Analysis (2004); Sambrook y Russell, Condensed Protocols from Molecular Cloning: A Laboratory Manual (2006); y Sambrook y Russell, Molecular Cloning: A Laboratory Manual (2002) (todos de Cold Spring Harbor Laboratory Press); Stryer, L., Biochemistry (4.a ed.) W.H. Freeman, Nueva York (1995); Gait, "Oligonucleotide Synthesis: A Practical Approach" IRL Press, Londres (1984); Nelson y Cox, Lehninger, Principles of Biochemistry, 3.a Ed., W. H. Freeman Pub., Nueva York (2000); y Berg et al., Biochemistry, 5.a Ed., W.H. Freeman Pub., Nueva York (2002). Antes de que se describan las presentes composiciones, herramientas de investigación y procedimientos, se ha de entender que la presente invención no se limita a los procedimientos, composiciones, objetivos y usos específicos descritos, ya que, por supuesto, como tales pueden variar. También se ha de entender que la terminología usada en el presente documento es solo para el propósito de describir aspectos particulares y no se pretende que limite el alcance de la presente invención, que solo se limitará por las reivindicaciones adjuntas.
Cabe destacar que como se usa en el presente documento y en las reivindicaciones adjuntas, las formas en singular "un", "una" y "el/la" incluyen referentes al plural a menos que el contexto lo indique claramente de otro modo. Por tanto, por ejemplo, la referencia a "una región de ácido nucleico" se refiere a una, más de una, o mezclas de dichas regiones, y la referencia a "un procedimiento" incluye la referencia a etapas y procedimientos equivalentes conocidos por los expertos en la técnica, etc.
Cuando se proporciona un intervalo de valores, se ha de entender que cada valor intermedio, entre el límite superior e inferior de ese intervalo y cualquier otro valor establecido o intermedio en ese intervalo establecido se engloba dentro de la invención. Cuando el intervalo establecido incluye los límites superior e inferior, los intervalos que excluyan cualquiera de esos límites también se incluyen en la invención.
En la siguiente descripción, se exponen numerosos detalles específicos para proporcionar un entendimiento más completo de la presente invención. Sin embargo, será evidente para un experto en la técnica que la presente invención se puede poner en práctica sin uno o más de estos detalles específicos. En otros casos, no se han descrito rasgos característicos y procedimientos bien conocidos para los expertos en la técnica para evitar eclipsar la invención.
Definiciones
Se pretende que los términos usados en el presente documento tengan el significado simple y ordinario como se entiende por los expertos en la técnica. Se pretende que las siguientes definiciones ayuden al lector en el entendimiento de la presente invención, pero no se pretende que varíen o de otro modo limiten el significado de dichos términos a menos que se indique específicamente.
El término "ácido nucleico amplificado" es cualquier molécula de ácido nucleico cuya cantidad se ha incrementado al menos dos veces por cualquier procedimiento de amplificación o replicación de ácido nucleico realizado in vitro en comparación con su cantidad de partida.
El término "anomalía cromosómica" se refiere a cualquier variante genética para todo o parte de un cromosoma. Las variantes genéticas pueden incluir, pero sin limitarse a, cualquier variante del número de copias, tal como duplicaciones o deleciones, translocaciones, inversiones y mutaciones. El término también incluye mosaicismo cromosómico en tejido fetal o materno.
El término "herramienta de diagnóstico" como se usa en el presente documento se refiere a cualquier composición o procedimiento de la divulgación usado, por ejemplo, en un sistema para llevar a cabo una prueba o ensayo de diagnóstico en una muestra del paciente.
El término "mosaicismo intersexual" o "mosaicismo de los cromosomas sexuales" o "mosaico de los cromosomas sexuales" se refiere a la presencia de dos o más poblaciones de células con diferentes genotipos de los cromosomas sexuales en un individuo. Los mosaicismos intersexuales surgen cuando algunas células en un individuo tienen, por ejemplo, dos cromosomas X (XX) y otras células en el individuo tienen un cromosoma X y un cromosoma Y (XY); cuando algunas células en un individuo tienen un cromosoma X (XO) y otras células en el individuo tienen un cromosoma X y un cromosoma Y (XY); o cuando algunas células en un individuo tienen dos cromosomas X y un cromosoma Y (XXY) y otras células en el individuo tienen un cromosoma X y un cromosoma Y (XY).
El término "hibridación" significa, en general, la reacción por la que se produce el emparejamiento de cadenas complementarias de ácido nucleico. Normalmente el ADN es bicatenario, y cuando las cadenas se separan, se rehibridarán en las condiciones apropiadas. Se pueden formar híbridos entre ADN-ADN, ADN-ARN o ARN-ARN. Se pueden formar entre una cadena corta y una cadena larga que contenga una región complementaria a la corta. También se pueden formar híbridos imperfectos, pero cuanto más imperfectos sean, menos estables serán (y será menos probable que se formen).
El término "probabilidad" se refiere a cualquier valor logrado calculando directamente la probabilidad o cualquier valor que se pueda correlacionar o de otro modo indique una probabilidad.
El término "locus" como se usa en el presente documento se refiere a una región de ácido nucleico de localización conocida en un genoma.
El término "muestra materna" como se usa en el presente documento se refiere a cualquier muestra tomada de una hembra gestante que comprenda ácidos nucleicos tanto fetales como maternos (por, ejemplo, ADN). Preferentemente, se obtienen muestras maternas para su uso en la invención a través de medios relativamente no invasivos, por ejemplo, extracción de sangre u otras técnicas estándar para extraer muestras periféricas de un sujeto.
"Micromatriz" o "matriz" se refiere a un soporte en fase sólida que tiene una superficie, preferentemente, pero no exclusivamente una superficie plana o sustancialmente plana, que tiene una matriz de sitios que contienen ácidos nucleicos, de modo que cada sitio de la matriz comprenda copias sustancialmente idénticas o idénticas de oligonucleótidos o polinucleótidos y esté espacialmente definido y no se superponga con otros sitios miembros de la matriz; es decir, los sitios sean espacialmente discretos. La matriz o micromatriz también puede comprender una estructura que se puede consultar no plana con una superficie tal como una microesfera o un pocillo. Los oligonucleótidos o polinucleótidos de la matriz pueden estar unidos de forma covalente al soporte sólido, o pueden estar unidos de forma no covalente. La tecnología de micromatrices convencional se revisa, por ejemplo, en Schena, Ed., Microarrays: A Practica! Approach, IRL Press, Oxford (2000). "Análisis con matriz", "análisis por matriz" o "análisis por micromatriz" se refiere a un análisis, tal como, por ejemplo, aislamiento de ácidos nucleicos específicos o análisis de secuencias de una o más moléculas biológicas usando una micromatriz.
Por "no polimórfica" o "independiente de polimorfismo", cuando se usa con respecto a la detección de regiones de ácido nucleico seleccionadas, se entiende la detección de una región de ácido nucleico, que puede contener uno o más polimorfismos, pero en la que la detección no es dependiente de la detección del polimorfismo específico dentro de la región. Por tanto, una región de ácido nucleico seleccionada puede contener un polimorfismo, pero la detección de la región usando los procedimientos de la invención se basa en la aparición de la región en lugar de en la presencia o ausencia de un polimorfismo particular en esa región.
El término "oligonucleótidos" como se usa en el presente documento se refiere a oligómeros lineales de monómeros de ácido nucleico naturales o modificados, incluyendo desoxirribonucleótidos, ribonucleótidos, formas anoméricas de los mismos, monómeros de ácido peptidonucleico (PNA), monómeros de ácido nucleico bloqueado (LNA), y similares, o una combinación de los mismos, que se pueden unir específicamente a un polinucleótido monocatenario por medio de un patrón regular de interacciones monómero con monómero, tal como el tipo de emparejamiento de bases de Watson-Crick, apilamiento de bases, tipos de emparejamiento de bases de Hoogsteen o Hoogsteen inverso, o similares. Normalmente, los monómeros se enlazan por enlaces fosfodiéster o análogos de los mismos para formar oligonucleótidos que varían en tamaño de unas pocas unidades monoméricas, por ejemplo, 8-12, a varias decenas de unidades monoméricas, por ejemplo, 100-200 o más.
Como se usa en el presente documento, el término "polimerasa" se refiere a una enzima que enlaza nucleótidos individuales entre sí en una cadena larga, usando otra cadena como molde. Existen dos tipos generales de polimerasa: las ADN polimerasas, que sintetizan ADN, y las ARN polimerasas, que sintetizan ARN. Dentro de estas dos clases, existen numerosos subtipos de polimerasas, dependiendo de qué tipo de ácido nucleico puede funcionar como molde y de qué tipo de ácido nucleico se forme.
Como se usa en el presente documento, "reacción en cadena de la polimerasa" o "PCR" se refiere a una técnica para replicar una parte específica de ADN diana in vitro, incluso en presencia de ADN no específico en exceso. Se añaden cebadores al ADN diana, donde los cebadores inician la copia del ADN diana usando nucleótidos y, típicamente, Taq polimerasa o similares. Realizando ciclos de la temperatura, el ADN diana se desnaturaliza y copia repetidamente. Se puede amplificar una única copia del ADN diana, incluso si se mezcla con otro ADN aleatorio, para obtener mil millones de réplicas. La reacción en cadena de la polimerasa se puede usar para detectar y medir cantidades muy pequeñas de ADN y para crear partes de ADN adaptadas. En algunos casos, se pueden usar procedimientos de amplificación lineal como alternativa a la PCR.
El término "polimorfismo" como se usa en el presente documento se refiere a cualquier cambio genético en un locus que puede ser indicativo de ese locus particular, incluyendo, pero sin limitarse a, polimorfismos mononucleotídicos (SNP), diferencias en la metilación, repeticiones cortas en tándem (STR) y similares.
En general, un "cebador" es un oligonucleótido usado, por ejemplo, para cebar la extensión, unión y/o síntesis de ADN, tal como en la etapa de síntesis de la reacción en cadena de la polimerasa o en las técnicas de extensión del cebador usadas en determinadas reacciones de secuenciación. También se puede usar un cebador en técnicas de hibridación como un medio para proporcionar complementariedad de una región de ácido nucleico a un oligonucleótido de captura para la detección de una región de ácido nucleico específica.
El término "herramienta de investigación" como se usa en el presente documento se refiere a cualquier procedimiento de la invención usado para la exploración científica, de naturaleza académica o comercial, incluyendo el desarrollo de tratamientos farmacéuticos y/o biológicos. No se pretende que las herramientas de investigación de la invención sean terapéuticas ni estén sujetas a autorización; en su lugar, se pretende que las herramientas de investigación de la invención faciliten la investigación y ayuden en dichas actividades de desarrollo, incluyendo cualquier actividad realizada con la intención de producir información para respaldar una presentación del expediente de registro.
El término "región de ácido nucleico seleccionada" como se usa en el presente documento se refiere a una región de ácido nucleico correspondiente a un cromosoma individual. Las regiones de ácido nucleico seleccionadas se pueden aislar y enriquecer directamente de la muestra para la detección, por ejemplo, en base a la hibridación y/u
otras técnicas basadas en secuencias, o se pueden amplificar usando la muestra como un molde antes de la detección de la secuencia.
Los términos "amplificación selectiva" y "amplificar de forma selectiva" y similares se refieren a un procedimiento de amplificación que depende completamente o en parte de la hibridación de un oligonucleótido a una secuencia en una región de ácido nucleico seleccionada. En determinadas amplificaciones selectivas, los cebadores usados para la amplificación son complementarios a una región de ácido nucleico seleccionada. En otras amplificaciones selectivas, los cebadores usados para la amplificación son cebadores universales, pero solo dan como resultado un producto si una región del ácido nucleico usado para la amplificación es complementaria a una región de ácido nucleico seleccionada de interés.
Los términos "secuenciación" y "determinación de secuencias" y similares, como se usa en el presente documento, se refieren, en general, a cualquiera y a todos los procedimientos bioquímicos que se pueden usar para determinar el orden de las bases nucleotídicas en un ácido nucleico.
Los términos "se une específicamente" y "unión específica" y similares como se usa en el presente documento, cuando se refieren a un compañero de unión (por ejemplo, una sonda o cebador de ácido nucleico, anticuerpo, etc.) dan como resultado la generación de una señal positiva estadísticamente significativa en las condiciones de ensayo designadas. Típicamente, la interacción posteriormente dará como resultado una señal detectable que es al menos dos veces la desviación estándar de cualquier señal generada como resultado de interacciones no deseadas (fondo). El término "universal", cuando se usa para describir un procedimiento de amplificación, se refiere al uso de un único cebador o conjunto de cebadores para una pluralidad de reacciones de amplificación. Por ejemplo, en la detección de 96 secuencias diana diferentes, todos los moldes pueden compartir secuencias de cebado universales idénticas, lo que permite la amplificación multiplexada de las 96 secuencias diferentes usando un único conjunto de cebadores. El uso de dichos cebadores simplifica en gran medida la multiplexación en tanto que solo se necesitan dos cebadores para amplificar una pluralidad de secuencias de ácido nucleico seleccionadas. El término "universal" cuando se usa para describir un sitio de cebado es un sitio al que se hibridará un cebador universal. También cabe destacar que se pueden usar "conjuntos" de secuencias de cebado/cebadores universales. Por ejemplo, en reacciones altamente multiplexadas, puede ser útil usar varios conjuntos de secuencias universales, en lugar de un único conjunto; por ejemplo, 96 ácidos nucleicos diferentes pueden tener un primer conjunto de secuencias de cebado universales, y los segundos 96 un conjunto diferente de secuencias de cebado universales, etc.
La divulgación en general
La invención se refiere a procedimientos como se define en las reivindicaciones. La presente divulgación proporciona procedimientos mejorados para identificar variantes del número de copias de los cromosomas X e Y. Los procedimientos de la divulgación son útiles para determinar el sexo de un feto, evaluar la probabilidad de una aneuploidía del cromosoma X, una aneuploidía del cromosoma Y o mosaicismo de los cromosomas sexuales en un feto, o para determinar la posible contaminación de una muestra materna. En algunos aspectos, los procedimientos de la divulgación también son útiles para detectar aneuploidías o mosaicismos del cromosoma X en la madre.
Los procedimientos de ensayo de la divulgación incluyen el enriquecimiento selectivo de regiones de ácido nucleico seleccionadas del cromosoma X y del cromosoma Y y uno o más cromosomas de referencia no sexuales (autosomas). Una clara ventaja de la divulgación es que las regiones de ácido nucleico seleccionadas se pueden analizar además usando una variedad de técnicas de detección y cuantificación, incluyendo, pero sin limitarse a, técnicas de hibridación, PCR digital y, preferentemente, técnicas de determinación de secuenciación de alto rendimiento. Se pueden diseñar cebadores frente a cualquier número de regiones de ácido nucleico seleccionadas para cualquier cromosoma además de los cromosomas X e Y. Aunque la amplificación antes de la identificación y cuantificación de las regiones de ácido nucleico seleccionadas no es obligatoria, es preferente una amplificación limitada antes de la detección.
La presente divulgación es una mejora sobre las técnicas más aleatorias, tal como la secuenciación con disparo masivamente paralela (por ejemplo, secuenciación aleatoria) o el uso de PCR digital aleatoria, que se han usado recientemente para detectar variaciones del número de copias en muestras maternas, tales como sangre materna. El enfoque mencionado anteriormente se basa en la secuenciación de todos o una población estadísticamente significativa de fragmentos de ADN en una muestra, seguido de cartografía de o de otro modo asociación o alineación de los fragmentos a sus cromosomas apropiados. A continuación, los fragmentos identificados se comparan entre sí o con alguna otra referencia (por ejemplo, una muestra con un complemento cromosómico euploide conocido) para determinar la variación del número de copias de cromosomas particulares. Los procedimientos de secuenciación aleatoria o con disparo son inherentemente ineficaces en comparación con la presente divulgación, ya que los datos generados en las regiones cromosómicas de interés solo constituyen una minoría de los datos que se generan.
Las técnicas que son dependientes de un muestreo muy amplio de ADN en una muestra proporcionan una cobertura amplia del ADN analizado, pero, de hecho, están muestreando el ADN contenido dentro de una muestra
en una base de 1x o menos (es decir, submuestreo). Por el contrario, las técnicas de amplificación y/o enriquecimiento selectivo (tal como hibridación) usadas en los presentes procedimientos proporcionan una profundidad de cobertura de solo las regiones de ácido nucleico seleccionadas; y como tales proporcionan un "supermuestreo" de las regiones de ácido nucleico seleccionadas con una cobertura de secuencia promedio de preferentemente 2x o más, más preferentemente una cobertura de secuencia de 100x o más, 200x o más, 250x o más, 500x o más, 750x o más o incluso más preferentemente una cobertura de secuencia de 1000x o más de las regiones de ácido nucleico seleccionadas.
Por tanto, en los ensayos de la presente divulgación, la mayoría sustancial de las secuencias analizadas para la cuantificación son informativas de la presencia de una o más regiones de ácido nucleico seleccionadas en el cromosoma X e Y y uno o más autosomas. Los procedimientos de la divulgación no requieren el análisis de grandes cantidades de secuencias que no sean de los cromosomas de interés y que no proporcionen información sobre la cantidad relativa de los cromosomas de interés.
Detección y cuantificación de los cromosomas sexuales
La presente divulgación proporciona procedimientos para determinar la frecuencia de secuencias de X e Y en una muestra materna. Estas frecuencias se pueden usar, por ejemplo, para determinar el sexo fetal y/o para identificar aneuploidías cromosómicas de X, aneuploidías cromosómicas de Y y/o mosaicismos de los cromosomas sexuales. Las muestras son muestras maternas que comprenden ADN tanto materno como fetal, tal como muestras de sangre materna (es decir, sangre completa, suero o plasma). Los procedimientos enriquecen y/o aíslan y amplifican una o, preferentemente, de varias a muchas regiones de ácido nucleico seleccionadas en una muestra materna que corresponden a los cromosomas X e Y y uno o más autosomas que se usan para determinar la presencia o ausencia y/o cantidad relativa o frecuencia de las secuencias cromosómicas de X e Y en vista del porcentaje de ADN fetal presente en la muestra. Como se describe en detalle supra, los procedimientos de la divulgación emplean preferentemente una o más etapas de amplificación, unión o enriquecimiento selectivo (por ejemplo, usando uno o más ácidos nucleicos que se hibridan específicamente a las regiones de ácido nucleico seleccionadas) para potenciar el contenido de las regiones de ácido nucleico seleccionadas en la muestra. Las etapas de amplificación, unión y/o enriquecimiento selectivo típicamente incluyen mecanismos para genomanipular copias de las regiones de ácido nucleico seleccionadas para su aislamiento, amplificación y análisis adicional. Este enfoque selectivo contrasta directamente con el enfoque de amplificación aleatoria usado por otras técnicas, por ejemplo, secuenciación con disparo masivamente paralela, ya que dichas técnicas implican, en general, la amplificación aleatoria de todo o una porción sustancial del genoma.
En determinados aspectos, los procedimientos de la divulgación pueden identificar aneuploidías maternas, incluyendo mosaicismo materno. Las frecuencias del material cromosómico de X e Y maternos se deben analizar en vista del ADN fetal en la muestra materna. Los perfiles de riesgo determinado de dichas aneuploidías maternas pueden variar dependiendo de la aneuploidía particular. Por ejemplo, en el caso donde una madre tenga un mosaico XX/XO, la probabilidad determinada será dependiente del nivel de mosaicismo materno, de los tejidos afectados, así como del porcentaje fetal en la muestra materna.
Los ejemplos anteriores demuestran que si se han de detectar de forma sólida ácidos nucleicos específicos presentes a un porcentaje tan bajo a través de los procedimientos descritos en el presente documento, la variación en la medición del cromosoma adicional tiene que ser significativamente menor que el porcentaje de incremento del cromosoma adicional.
La figura 1 es un diagrama de flujo simplificado de un procedimiento 100 de acuerdo con la presente divulgación. En una primera etapa, se obtiene una muestra materna 101. La muestra materna comprende ácidos nucleicos tanto maternos como fetales. Las muestras maternas pueden ser cualquier muestra tomada de una hembra gestante que comprenda ácidos nucleicos tanto fetales como maternos (por, ejemplo, ADN). Preferentemente, las muestras maternas para su uso en la divulgación están libres de células y se obtienen a través de medios relativamente no invasivos, tales como extracción de sangre u otras técnicas estándar para extraer muestras periféricas de un sujeto.
En la siguiente etapa 103, los cebadores oligonucleotídicos específicos para regiones de ácido nucleico seleccionadas en el cromosoma X y en el cromosoma Y y en al menos un autosoma (y preferentemente más de un autosoma) se hibridan a los ácidos nucleicos seleccionados en la muestra materna. Los cebadores oligonucleotídicos se usan para amplificar de forma selectiva las regiones de ácido nucleico seleccionadas en la etapa 105 para producir copias de las regiones de ácido nucleico seleccionadas. Como se describe en detalle infra, las regiones de ácido nucleico seleccionadas se someten a una etapa de amplificación selectiva, pero también se pueden someter a una etapa de amplificación universal antes de la etapa de amplificación selectiva o bien, preferentemente, después de la etapa de amplificación selectiva. Además, se pueden realizar una o más etapas de enriquecimiento como se describe infra. Además, como alternativa a la amplificación, se puede realizar una etapa de enriquecimiento, tal como por hibridación selectiva, que separa las regiones de ácido nucleico seleccionadas de los demás ácidos nucleicos en la muestra.
En la etapa 107, las regiones de ácido nucleico seleccionadas amplificadas o copiadas se secuencian y cuantifican a continuación. Los modos de realización preferentes utilizan técnicas de secuenciación de alto rendimiento o de última generación, aunque opcionalmente se pueden usar otras técnicas, como se describe infra. La secuenciación de alto rendimiento permite la paralelización masiva de la determinación de secuencias y la etapa de cuantificación.
En la etapa 109, se determina el porcentaje de ADN fetal en la muestra materna. A continuación, en la etapa 111, las frecuencias de las regiones de ácido nucleico seleccionadas del cromosoma X y del cromosoma Y se determinan en vista del porcentaje de ADN fetal determinado en la etapa 109. Como se describe en detalle en el presente documento, la frecuencia de las regiones de ácido nucleico seleccionadas del cromosoma X y del cromosoma Y en vista del porcentaje fetal permite en la etapa 113 la evaluación del riesgo de anomalías en la frecuencia cromosómica de X, tales como las que surgen de la aneuploidía cromosómica de X, mosaicismo cromosómico de X o contaminación cromosómica de X y/o anomalías en la frecuencia cromosómica de Y, tales como las que surgen de la aneuploidía cromosómica de Y, mosaicismo cromosómico de Y o contaminación cromosómica de Y de una muestra materna de una mujer embarazada de un feto de sexo femenino.
Por tanto, en general, las regiones de ácido nucleico seleccionadas que corresponden a múltiples locus en el cromosoma X se detectan y suman para determinar la frecuencia relativa de los cromosomas X en la muestra materna. Las regiones de ácido nucleico seleccionadas que corresponden a múltiples locus en uno o más autosomas se detectan y suman para determinar la frecuencia relativa del uno o más cromosomas adicionales en la muestra materna, lo que permite el cálculo del porcentaje fetal. Una vez que se determina el porcentaje fetal, se observa la frecuencia del cromosoma X en vista del porcentaje fetal para evaluar si existe una anomalía cromosómica en X. De forma similar, las regiones de ácido nucleico seleccionadas que corresponden a múltiples locus en el cromosoma Y se detectan y suman para determinar la frecuencia relativa del cromosoma Y en la muestra materna y se observa la frecuencia del cromosoma Y en vista del porcentaje fetal para evaluar si existe una anomalía cromosómica en Y.
Los procedimientos de la divulgación analizan múltiples regiones de ácido nucleico seleccionadas que representan locus seleccionados en al menos tres cromosomas: el cromosoma X, el cromosoma Y y al menos un autosoma y la frecuencia relativa de cada región de ácido nucleico seleccionada se analiza y se cuantifica independientemente para determinar una frecuencia relativa para cada región de ácido nucleico seleccionada en la muestra. Las sumas de las regiones de ácido nucleico seleccionadas en la muestra se usan para determinar el porcentaje de ADN fetal en la muestra y se comparan para determinar estadísticamente si existe una aneuploidía cromosómica u otra anomalía cromosómica en relación con las secuencias cromosómicas de X y/o Y.
En otro aspecto, se analizan subconjuntos de regiones de ácido nucleico seleccionadas en cada cromosoma para determinar si existe una anomalía cromosómica o anomalía en la frecuencia cromosómica. Las frecuencias de las regiones de ácido nucleico seleccionadas se pueden sumar para un cromosoma particular, y las sumas de las regiones de ácido nucleico seleccionadas se pueden usar para determinar anomalías. Este aspecto de la divulgación suma las frecuencias de las regiones de ácido nucleico seleccionadas individuales de cada cromosoma y, a continuación, compara la suma de las regiones de ácido nucleico seleccionadas en el cromosoma X con uno o más cromosomas no sexuales y compara la suma de las regiones de ácido nucleico seleccionadas en el cromosoma Y con uno o más autosomas. Se pueden elegir de forma aleatoria los subconjuntos de regiones de ácido nucleico seleccionadas, pero con números suficientes para proporcionar un resultado estadísticamente significativo al determinar si existe una anomalía cromosómica. Se pueden realizar múltiples análisis de diferentes subconjuntos de regiones de ácido nucleico seleccionadas en una muestra materna para proporcionar más capacidad estadística. Por ejemplo, si existen 100 regiones de ácido nucleico seleccionadas para el cromosoma Y y 100 regiones de ácido nucleico seleccionadas para el cromosoma 2, se podría realizar una serie de análisis que evaluaran menos de 100 regiones para cada uno de los cromosomas. Por ejemplo, se podría realizar una serie de análisis que evaluaran menos de 50 regiones, tal como menos de 30 regiones, menos de o igual a 16 regiones, menos de 10 regiones u 8 regiones. En otro aspecto, se pueden seleccionar regiones de ácido nucleico seleccionadas particulares en cada cromosoma que se sabe que tienen menor variación entre las muestras, o los datos usados para la determinación de la frecuencia cromosómica se pueden limitar, por ejemplo, ignorando los datos de las regiones de ácido nucleico seleccionadas con frecuencias muy altas o muy bajas dentro de una muestra.
Aún en otro aspecto, la proporción de las frecuencias de las regiones de ácido nucleico seleccionadas se compara con una proporción media de referencia que se ha determinado para una población estadísticamente significativa de sujetos genéticamente euploides, es decir, sujetos que no tienen una anomalía cromosómica en X o un anomalía cromosómica en Y.
Se debe entender por los expertos en la técnica que los procedimientos para determinar la frecuencia del cromosoma X y del Y en vista del porcentaje de ADN fetal en una muestra materna se pueden combinar con otras técnicas no invasivas de diagnóstico prenatal, tales como las técnicas que evalúan el riesgo de una aneuploidía fetal de autosomas, o técnicas que detectan secuencias polimórficas en el feto.
Procedimientos de ensayo
Se puede emplear una serie de diferentes procedimientos de ensayo en la presente divulgación, incluyendo ensayos que usan conjuntos de oligonucleótidos que consisten solo en oligonucleótidos fijos, o conjuntos de oligonucleótidos que consisten en oligonucleótidos fijos y uno o más oligonucleótidos puente. Adicionalmente, los oligonucleótidos en un conjunto se pueden hibridar a las secuencias de ácido nucleico seleccionadas inmediatamente adyacentes entre sí donde se pueden unir, o los oligonucleótidos en un conjunto pueden no hibridarse a las secuencias de ácido nucleico seleccionadas inmediatamente adyacentes entre sí, y, por tanto, se emplea una reacción de extensión del cebador usando una polimerasa y dNTP antes de la unión de los oligonucleótidos en un conjunto. Las figuras 2 a 7 ilustran algunos procedimientos de ensayo ejemplares.
La figura 2 ilustra un modo de realización del procedimiento ejemplar donde se detectan dos diferentes regiones de ácido nucleico seleccionadas en un único ensayo de reacción en tándem. Dichos modos de realización del procedimiento, sistemas de ensayo y modos de realización relacionados se describen en detalle, por ejemplo, en los documentos US 20120034603, presentado el 25 de enero de 2011; US 20120077185, presentado el 26 de septiembre de 2011; US 20120040859, presentado el 8 de agosto de 2011; US 20130004950, presentado el 10 de noviembre de 2011; US 8.771.085, presentado el 8 de agosto de 2011; US 20120034685, presentado el 8 de agosto de 2011; US 20120164646, presentado el 29 de febrero de 2012; US 20120190018, presentado el 15 de octubre de 2011; US 20120190557, presentado el 9 de diciembre de 2011, y US 20120191358, presentado el 28 de diciembre de 2011. Dos conjuntos de oligonucleótidos de secuencia fijos (201 y 203, 223 y 225) que se hibridan específicamente a dos diferentes regiones de ácido nucleico seleccionadas 215, 231 se introducen 202 en una muestra genética y se deja que se hibriden 204 a las regiones de ácido nucleico seleccionadas respectivas. Cada conjunto de oligonucleótidos de secuencia fijos comprende un oligonucleótido 201, 223 que tiene una región específica de secuencia 205, 227, una región de cebador universal 209 y una región de índice 221, 235. El otro oligonucleótido de secuencia fijo en un conjunto comprende una región específica de secuencia 207, 229 y una región de cebador universal 211. Los oligonucleótidos de secuencia fijos varían, en general, en tamaño de aproximadamente 30-200 nucleótidos de longitud, o de aproximadamente 30-150 nucleótidos de longitud, o de aproximadamente 35-120 nucleótidos de longitud, o de aproximadamente 40-70 nucleótidos de longitud. Si se emplean oligonucleótidos puente, los oligonucleótidos puente varían, en general, en tamaño de aproximadamente 4 a aproximadamente 80 nucleótidos de longitud, o de aproximadamente 4 a aproximadamente 60 nucleótidos de longitud, o de aproximadamente 5 a aproximadamente 50 nucleótidos de longitud, o de aproximadamente 7 a aproximadamente 40 nucleótidos de longitud, o de aproximadamente 10 a aproximadamente 40 nucleótidos de longitud, o de aproximadamente 12 a aproximadamente 30 nucleótidos de longitud, o de aproximadamente 15 a aproximadamente 25 nucleótidos de longitud.
Tras la hibridación, los oligonucleótidos de secuencia fijos no hibridados se separan preferentemente del resto de la muestra genética (etapa no mostrada). Los oligonucleótidos puente 213, 233 se introducen en el par hibridado de regiones de oligonucleótido de secuencia fijo/ácido nucleico y se deja que se hibriden 206 a estas regiones. Aunque se muestra en la figura 2 como dos oligonucleótidos puente diferentes, de hecho, el mismo oligonucleótido puente puede ser adecuado para ambos acontecimientos de hibridación (que suponen que las secuencias son iguales o sustancialmente similares), o pueden ser dos oligonucleótidos de una agrupación de oligonucleótidos de secuencia redundante. Los oligonucleótidos hibridados se unen 208 para crear un ácido nucleico contiguo que se extienda y sea complementario a cada región de ácido nucleico seleccionada de interés. Cabe destacar que, aunque este modo de realización particular ejemplifica un procedimiento que usa dos oligonucleótidos de secuencia fijos y un oligonucleótido puente para amplificar cada región de ácido nucleico seleccionada, se pueden emplear procedimientos que usan solo dos oligonucleótidos de secuencia fijos que se hibridan inmediatamente adyacentes entre sí, o se pueden emplear procedimientos que usan solo dos oligonucleótidos de secuencia fijos que no se hibridan inmediatamente adyacentes entre sí, pero donde se llena un "hueco" usando una polimerasa y dNTP.
Tras la unión, se introducen los cebadores universales 217, 219 para amplificar 210 los oligonucleótidos unidos para crear 212 los productos de amplificación 237, 239 que comprenden la secuencia de las regiones de ácido nucleico seleccionadas de interés. Estos productos de amplificación 237, 239 se aíslan (opcionalmente), se detectan (es decir, se secuencian) y se cuantifican para proporcionar información sobre la presencia y cantidad de las regiones de ácido nucleico seleccionadas en la muestra.
Se pueden usar numerosos procedimientos de amplificación para amplificar de forma selectiva las regiones de ácido nucleico seleccionadas que se analizan en los procedimientos de la divulgación, incrementando el número de copias de las regiones de ácido nucleico seleccionadas de una manera que permita la conservación de la cantidad relativa de las regiones de ácido nucleico seleccionadas en la muestra inicial. Aunque no todas las combinaciones de amplificación y análisis se describen en detalle en el presente documento, está dentro de la habilidad de los expertos en la técnica utilizar diferentes procedimientos de amplificación y/o análisis comparables para analizar las regiones de ácido nucleico seleccionadas consecuentes con la presente memoria descriptiva, ya que dichas variaciones deben ser evidentes para un experto en la técnica tras la lectura de la presente divulgación.
Los procedimientos de amplificación útiles en la presente invención incluyen, pero no se limitan a, reacción en cadena de la polimerasa (PCR) (pat. de EE. UU. n.os 4.683.195 y 4.683.202; y se describen en PCR Technology: Principies and Applications for DNA Amplification, ed. H. A. Erlich, Freeman Press, NY, N.Y., 1992); reacción en
cadena de la ligasa (LCR) (Wu y Wallace, Genomics 4:560, (1989); Landegren et al., Science 241:1077 (1988)); amplificación por desplazamiento de cadena (SDA) (pat. de EE. UU. n.os 5.270.184 y 5.422.252); amplificación mediada por transcripción (TMA) (pat. de EE. UU. n.° 5.399.491); amplificación lineal enlazada (LLA) (pat. de EE. UU. n.° 6.027.923), replicación de secuencia autosostenida (Guatelli et al., PNAS USA, 87:1874 (1990) y el documento WO90/06995); amplificación selectiva de secuencias de polinucleótidos diana (pat. de EE. UU. n.° 6.410.276); reacción en cadena de la polimerasa cebada con secuencias consensos (CP-PCR) (pat. de EE. UU. n.° 4.437.975); reacción en cadena de la polimerasa cebada arbitrariamente (AP-PCR) (pat. de EE. UU. n.os 5.413.909 y 5.861.245); y amplificación de secuencias basada en ácidos nucleicos (NASBA) (véanse las pat. de EE. UU. n.os 5.409.818, 5.554.517 y 6.063.603). Otros procedimientos de amplificación que se pueden usar incluyen: q-beta replicasa, descrito en la solicitud de patente PCT n.° WO8706270; procedimientos de amplificación isotérmica, tales como SDA, descritos en Walker et al., Nucleic Acids Res. 20(7):1691-6 (1992); y amplificación en círculo rodante, descrita en la pat. de EE. UU. n.° 5.648.245. Aún otros procedimientos de amplificación que se pueden usar se describen en las pat. de EE. UU. n.os 5.242.794, 5.494.810, 4.988.617 y en el documento US20050123943 y la pub. de EE. UU. n.° 20030143599. En aspectos preferentes, el ADN se amplifica por PCR multiplexada específica de locus. En algunos aspectos, el ADN se amplifica usando unión a adaptador y PCR con cebador único. Otros procedimientos de amplificación disponibles incluyen PCR equilibrada (Makrigiorgos et al., Nat. Biotechnol., 20:936-39 (2002)) y la replicación de secuencia autosostenida (Guatelli et al., PNAS USA, 87:1874 (1990)). En base a dichas metodologías, un experto en la técnica puede diseñar fácilmente cebadores en cualquier región 5' y 3' adecuada con respecto a una región de ácido nucleico seleccionada de interés. Dichos cebadores se pueden usar para amplificar ADN de cualquier longitud siempre que contenga la región de ácido nucleico seleccionada de interés en su secuencia.
La longitud de las regiones de ácido nucleico seleccionadas elegidas es lo suficientemente larga como para proporcionar suficiente información de secuencia para distinguir las regiones de ácido nucleico seleccionadas entre sí. En general, una región de ácido nucleico seleccionada tiene al menos aproximadamente 16 nucleótidos de longitud, y más típicamente, una región de ácido nucleico seleccionada tiene al menos aproximadamente 20 nucleótidos de longitud. En un aspecto preferente de la divulgación, las regiones de ácido nucleico seleccionadas tienen al menos aproximadamente 30 nucleótidos de longitud. En un aspecto más preferente de la divulgación, las regiones de ácido nucleico seleccionadas tienen al menos aproximadamente 32, 40, 45, 50 o 60 nucleótidos de longitud. En otros aspectos de la divulgación, las regiones de ácido nucleico seleccionadas pueden tener aproximadamente 100, 150 o hasta 200 de longitud.
En algunos aspectos, el procedimiento de amplificación selectiva usa una o unas pocas tandas de amplificación con pares de cebadores que comprenden ácidos nucleicos complementarios a las regiones de ácido nucleico seleccionadas (es decir, un procedimiento de amplificación específica de secuencia). En otros aspectos, la amplificación selectiva comprende una etapa de amplificación lineal inicial (también un procedimiento de amplificación específica de secuencia). Los procedimientos de amplificación lineal pueden ser útiles en particular si la cantidad de partida de ADN es limitada. La amplificación lineal incrementa la cantidad de moléculas de ADN de una forma que es representativa del contenido de ADN original, lo que ayuda a reducir el error de muestreo en casos tales como la presente divulgación donde se necesita una cuantificación exacta de las regiones de ácido nucleico seleccionadas.
Por tanto, en aspectos preferentes, se realiza un número limitado de ciclos de amplificación específica de secuencia en la muestra materna de partida que comprende ADN libre de células. El número de ciclos, en general, es menor que el usado para una amplificación por PCR típica, por ejemplo, de 5-30 ciclos o menos.
Los oligonucleótidos en los conjuntos de oligonucleótidos están diseñados para hibridarse a la muestra de una manera específica de secuencia y para amplificar las regiones de ácido nucleico seleccionadas. Los cebadores para la amplificación selectiva están diseñados preferentemente para 1) amplificar eficazmente las regiones de ácido nucleico seleccionadas del/de los cromosoma(s) de interés; 2) tener un intervalo predecible de expresión de fuentes maternas y/o fetales en diferentes muestras maternas; y 3) ser distintivos con respecto a las regiones de ácido nucleico seleccionadas, es decir, no amplificar regiones de ácido nucleico no seleccionadas. Los cebadores o sondas se pueden modificar con un marcador de extremo en el extremo 5' (por ejemplo, con biotina) o en otra parte a lo largo del cebador o sonda, de modo que los productos de amplificación se puedan purificar o acoplar a un sustrato sólido (por ejemplo, microesfera o matriz) para su aislamiento o análisis adicional. En un aspecto preferente, los cebadores se genomanipulan para que tengan, por ejemplo, temperaturas de fusión compatibles para que se usen en reacciones multiplexadas que permitan la amplificación de muchas regiones de ácido nucleico seleccionadas de modo que una única reacción proporcione múltiples copias de ADN de diferentes regiones de ácido nucleico seleccionadas y preferentemente todas las regiones de ácido nucleico seleccionadas. A continuación, los productos de amplificación de la amplificación selectiva se pueden amplificar con procedimientos de PCR estándar o con amplificación lineal.
Se puede aislar ADN libre de células, por ejemplo, de sangre completa, plasma o suero de una mujer gestante, e incubar con cebadores genomanipulados para que amplifiquen un número establecido de regiones de ácido nucleico seleccionadas que correspondan a los cromosomas de interés. Preferentemente, el número de pares de cebadores usados para la amplificación inicial de secuencias específicas del cromosoma X (y, por tanto, el número de regiones
de ácido nucleico seleccionadas en el cromosoma X) será de 8 o más, tal como de 16 o más, de 32 o más, de 48 o más o de 96 o más. De forma similar, el número de pares de cebadores usados para la amplificación inicial de secuencias específicas del cromosoma Y (y, por tanto, el número de regiones de ácido nucleico seleccionadas en el cromosoma Y) y en cada uno de los uno o más cromosomas de referencia autosómicos será de 8 o más, tal como de 16 o más, de 32 o más, de 48 o más o de 96 o más. Cada uno de los pares de cebadores corresponde a una única región de ácido nucleico seleccionada, y los pares de cebadores se marcan opcionalmente para la identificación (por ejemplo, usando índices como se describe supra) y/o aislamiento (por ejemplo, comprenden una secuencia de ácido nucleico o resto químico que se utiliza para la captura). Se realiza un número limitado de ciclos de amplificación, preferentemente de 10 o menos. Los productos de amplificación (las regiones de ácido nucleico amplificadas seleccionadas) se aíslan posteriormente opcionalmente por procedimientos conocidos en la técnica. Por ejemplo, cuando los cebadores se enlazan a una molécula de biotina, los productos de amplificación se pueden aislar por medio de unión a avidina o estreptavidina en un sustrato sólido. A continuación, los productos de amplificación se pueden someter a procedimientos bioquímicos adicionales, tales como amplificación adicional con otros cebadores (por ejemplo, cebadores universales) y/o técnicas de detección, tales como hibridación y determinación de secuencias.
Las eficacias de amplificación pueden variar entre regiones de ácido nucleico seleccionadas y entre ciclos, de modo que en determinados sistemas se puede usar la normalización (como se describe infra) para garantizar que los productos de la amplificación de las regiones de ácido nucleico seleccionadas sean representativos del contenido de ácido nucleico de la muestra. Uno que ponga en práctica los procedimientos de la divulgación puede extraer los datos con respecto a la frecuencia relativa de los productos amplificados para determinar la variación en las regiones de ácido nucleico seleccionadas, incluyendo la variación en regiones de ácido nucleico seleccionadas dentro de una muestra y/o entre regiones de ácido nucleico seleccionadas en diferentes muestras (en particular, de las mismas regiones de ácido nucleico seleccionadas en diferentes muestras) para normalizar los datos.
Como alternativa a la amplificación selectiva, se pueden enriquecer regiones de ácido nucleico seleccionadas por técnicas de hibridación (por ejemplo, hibridación de captura o hibridación a una matriz), opcionalmente seguido de una o más tandas de amplificación. Opcionalmente, las regiones de ácido nucleico seleccionadas hibridadas o capturadas se liberan (por ejemplo, por desnaturalización) antes de la amplificación y determinación de secuencias. Las regiones de ácido nucleico seleccionadas se pueden aislar de una muestra materna usando diversos procedimientos que permiten el enriquecimiento selectivo de las regiones de ácido nucleico seleccionadas usadas en el análisis. El aislamiento puede ser una retirada de ADN en la muestra materna no usado en el análisis y/o la retirada de cualquier oligonucleótido en exceso usado en la etapa de enriquecimiento o amplificación inicial. Por ejemplo, se pueden aislar las regiones de ácido nucleico seleccionadas de la muestra materna usando técnicas de hibridación (enriquecimiento), por ejemplo, capturar usando unión de las regiones de ácido nucleico seleccionadas a oligonucleótidos complementarios en un sustrato sólido, tal como una microesfera o una matriz, seguido de la retirada de los ácidos nucleicos no unidos de la muestra. En otro ejemplo, cuando se usa una técnica con sonda de tipo precircular para la amplificación selectiva (véanse, por ejemplo, Barany et al., pat. de EE. UU. n.os 6.858.412 y 7.556.924, y la FIG. 7), los productos de ácido nucleico con forma circular se pueden aislar de los ácidos nucleicos lineales, que están sujetos a degradación selectiva. Otros procedimientos de aislamiento útiles serán evidentes para un experto en la técnica tras la lectura de la presente memoria descriptiva.
Las copias amplificadas de forma selectiva de las regiones de ácido nucleico seleccionadas se pueden amplificar opcionalmente en una etapa de amplificación universal tras la amplificación selectiva (o etapa de enriquecimiento), antes o bien durante la etapa de detección (es decir, secuenciación u otra tecnología de detección). Al realizar la amplificación universal, se usan secuencias de cebador universal añadidas a la región de ácido nucleico seleccionada copiada en la etapa de amplificación selectiva para amplificar además las regiones de ácido nucleico seleccionadas en una única reacción de amplificación universal. Como se describe, se pueden añadir secuencias de cebador universal a las regiones de ácido nucleico seleccionadas copiadas durante el procedimiento de amplificación selectiva, si se realiza, usando cebadores para la etapa de amplificación selectiva que tienen secuencias de cebador universal, de modo que las copias amplificadas de las regiones de ácido nucleico seleccionadas incorporen la secuencia de cebado universal. De forma alternativa, los adaptadores que comprenden secuencias de amplificación universal se pueden unir a los extremos de las regiones de ácido nucleico seleccionadas tras la amplificación o enriquecimiento, si se realiza, y el aislamiento de las regiones de ácido nucleico seleccionadas de la muestra materna.
Se pueden introducir sesgo y variabilidad en una muestra durante la amplificación de ADN, y se sabe que esto sucede durante la reacción en cadena de la polimerasa (PCR). En los casos donde una reacción de amplificación está multiplexada, existe el potencial de que las regiones de ácido nucleico seleccionadas se amplifiquen a diferentes tasas o eficiencias, ya que cada conjunto de cebadores para una región de ácido nucleico seleccionada dada se puede comportar de forma diferente en base a la composición base del cebador y ADN molde, condiciones del tampón u otras condiciones. Una amplificación de ADN universal para un sistema de ensayo multiplexado introduce, en general, menos sesgo y variabilidad. Otra técnica para minimizar el sesgo de amplificación implica variar las concentraciones de cebador para diferentes regiones de ácido nucleico seleccionadas para limitar el número de ciclos de amplificación específicos de secuencia en la etapa de amplificación selectiva. Se pueden usar las mismas condiciones o diferentes (por ejemplo, polimerasa, tampones y similares) en las etapas de amplificación,
por ejemplo, para garantizar que no se introduce de forma inadvertida sesgo y variabilidad debido a las condiciones experimentales.
En un aspecto preferente, se realiza un pequeño número (por ejemplo, 1-10, preferentemente 3-5) de ciclos de amplificación selectiva o enriquecimiento de ácido nucleico, seguido de amplificación universal usando cebadores universales. El número de ciclos de amplificación usando cebadores universales variará, pero preferentemente será de al menos 5 ciclos, más preferentemente de al menos 10 ciclos, incluso más preferentemente de 20 ciclos o más. Al pasar a la amplificación universal tras uno o unos pocos ciclos de amplificación selectiva, se reduce el sesgo de que determinadas regiones de ácido nucleico seleccionadas se amplifiquen a tasas mayores que otras.
Opcionalmente, los procedimientos incluyen una etapa entre la amplificación selectiva y la amplificación universal para retirar cualquier ácido nucleico en exceso que no se amplifique específicamente en la amplificación selectiva. El producto completo o una alícuota del producto de la amplificación selectiva se puede usar para la amplificación universal.
Las regiones universales de los cebadores usados en los procedimientos están diseñadas para ser compatibles con procedimientos multiplexados convencionales que analizan simultáneamente grandes cantidades de ácidos nucleicos en una reacción en un recipiente. Dichos procedimientos de cebado "universal" permiten un análisis eficaz y de alto volumen de la cantidad de regiones de ácido nucleico presentes en una muestra materna, y permiten la cuantificación exhaustiva de la presencia de regiones de ácido nucleico dentro de una muestra materna de este tipo para la determinación de una aneuploidía.
Los ejemplos de procedimientos de amplificación universal incluyen, pero no se limitan a, procedimientos de multiplexación usados para amplificar y/o genotipar simultáneamente una variedad de muestras, tales como los descritos en Oliphant et al., pat. de EE. UU. n.° 7.582.420.
En determinados aspectos, el sistema de ensayo de la divulgación utiliza una de las siguientes técnicas de amplificación selectiva y universal combinadas: (1) la reacción de detección de la ligasa ("LDR") acoplada a la reacción en cadena de la polimerasa ("PCR"); (2) PCR primaria acoplada a PCR secundaria acoplada a LDR; y (3) PCR primaria acoplada a PCR secundaria. Cada una de estas combinaciones tiene una utilidad particular para una detección óptima. Sin embargo, cada una de estas combinaciones usa detección multiplex donde los cebadores oligonucleotídicos de una fase preliminar del sistema de análisis contienen secuencias que se utilizan en una fase posterior del sistema de análisis.
Barany et al., pat. de EE. UU. n.os 6.852.487, 6.797.470, 6.576.453, 6.534.293, 6.506.594, 6.312.892, 6.268.148, 6.054.564, 6.027.889, 5.830.711, 5.494.810, describen el uso del ensayo de reacción en cadena de la ligasa (LCR) para la detección de secuencias específicas de nucleótidos en una variedad de muestras de ácido nucleico. Barany et al., pat. de EE. UU. n.os 7.807.431, 7.455.965, 7.429.453, 7.364.858, 7.358.048, 7.332.285, 7.320.865, 7.312.039, 7.244.831, 7.198.894, 7.166.434, 7.097.980, 7.083.917, 7.014.994, 6.949.370, 6.852.487, 6.797.470, 6.576.453, 6.534.293, 6.506.594, 6.312.892 y 6.268.148, describen el uso de LDR acoplada con PCR para la detección de ácido nucleico. Barany et al., pat. de EE. UU. n.os 7.556.924 y 6.858.412, describen el uso de sondas precirculares (también llamadas "sondas candado" o "sondas de inversión múltiple") con LDR y PCR acopladas para la detección de ácido nucleico. Barany et al., pat. de EE. UU. n.os 7.807.431, 7.709.201 y 7.198.814, describen el uso de reacciones de escisión y unión por endonucleasas combinadas para la detección de secuencias de ácido nucleico. Willis et al., pat. de EE. UU. n.os 7.700.323 y 6.858.412, describen el uso de sondas precirculares en la amplificación, detección y genotipado multiplexados de ácido nucleico. Ronaghi et al., pat. de EE. UU. n.° 7.622.281 describe técnicas de amplificación para marcar y amplificar un ácido nucleico usando un adaptador que comprende un cebador único y un código de barras. Los procedimientos ejemplares útiles para amplificar y/o detectar regiones de ácido nucleico seleccionadas incluyen, pero no se limitan a, los procedimientos descritos en el presente documento. Además de las diversas técnicas de amplificación, numerosos procedimientos de determinación de secuencias son compatibles con los procedimientos de la divulgación. Preferentemente, dichos procedimientos incluyen procedimientos de secuenciación de "última generación". Los procedimientos ejemplares para la determinación de secuencias incluyen, pero no se limitan a, procedimientos basados en hibridación, tal como se divulga en Drmanac, pat. de EE. UU. n.os 6.864.052, 6.309.824, 6.401.267 y pub. de EE. UU. n.° 2005/0191656; secuenciación por procedimientos de síntesis, por ejemplo, Nyren et al., pat. de EE. UU. n.° 7.648.824, 7.459.311 y 6.210.891; Balasubramanian, pat. de EE. UU. n.os 7.232.656 y 6.833.246; Quake, pat. de EE. UU. n.° 6.911.345; Li et al., PNAS, 100: 414-19 (2003); secuenciación por pirofosfato, como se describe en Ronaghi et al., pat. de EE. UU. n.os 7.648.824, 7.459.311, 6.828.100 y 6.210.891; y procedimientos de determinación de secuenciación basados en unión, por ejemplo, Drmanac et al., pub. de Ee . UU. n.° 2010/0105052, y Church et al., pub. de EE. UU. n.os 2007/0207482 y 2009/0018024.
Se puede realizar la secuenciación usando cualquier aparato de secuenciación adecuado que pueda secuenciar en paralelo en altos órdenes de multiplexación, tal como MiSeq (Illumina), Ion PGM™ (Life Technologies), Ion Torrent™ (Life Technologies), HiSeq 2000 (Illumina), HiSeq 2500 (Illumina), plataforma 454 (Roche), Illumina Genome
Analyzer (Illumina), SOLiD System (Applied Biosystems), tecnología SMRT™ en tiempo real (Pacific Biosciences) y secuenciadores de nanoporos y/o nanocanales adecuados.
De forma alternativa, las regiones de ácido nucleico seleccionadas se pueden seleccionar y/o identificar usando técnicas de hibridación. Los procedimientos para llevar a cabo ensayos de hibridación de polinucleótidos para la detección se han desarrollado bien en la técnica. Los procedimientos y condiciones del ensayo de hibridación variarán dependiendo de la aplicación y se seleccionan de acuerdo con los procedimientos de unión generales conocidos, incluyendo los que se hace referencia en: Maniatis et al., Molecular Cloning: A Laboratory Manual (2.a ed. Cold Spring Harbor, N.Y., 1989); Berger y Kimmel, Methods in Enzymology, vol. 152; Guide to Molecular Cloning Techniques (Academic Press, Inc., San Diego, Calif., 1987); y Young y Davis, PNAS, 80:1194 (1983). Se han descrito procedimientos y aparatos para llevar a cabo reacciones de hibridación repetidas y controladas, por ejemplo, en las pat. de EE. UU. n.os 5.871.928, 5.874.219, 6.045.996, 6.386.749 y 6.391.623.
La presente divulgación también contempla la detección de señales de hibridación entre ligandos en determinados aspectos preferentes; véanse las pat. de EE. UU. n.os 5.143.854, 5.578.832, 5.631.734, 5.834.758, 5.936.324, 5.981.956, 6.025.601, 6.141.096, 6.185.030, 6.201.639, 6.218.803 y 6.225.625, en el documento US2004/0012676 y en la solicitud PCT PCT/US99/WO99/09997 (publicada como el documento WO99/47964).
Se divulgan procedimientos y aparatos para la detección de señales y el procesamiento de datos de intensidad, por ejemplo, en las pat. de EE. UU. n.os 5.143.854, 5.547.839, 5.578.832, 5.631.734, 5.800.992, 5.834.758, 5.856.092, 5.902.723, 5.936.324, 5.981.956, 6.025.601, 6.090.555, 6.141.096, 6.185.030, 6.201.639, 6.218.803 y 6.225.625, en el documento US2004/0012676 y en la solicitud PCT PCT/US99/06097 (publicada como el documento WO99/47964).
En la FIG. 3, se usan dos conjuntos de oligonucleótidos de secuencia fijos que comprenden sustancialmente las mismas regiones específicas de secuencia 305, 307, pero que comprenden diferentes índices, 321, 323. Las reacciones de unión se llevan a cabo con material de la misma muestra genética 300, pero en tubos separados con los diferentes conjuntos de oligonucleótidos específicos de alelo. Los oligonucleótidos puente 313, 333 correspondientes a los dos posibles SNP en la región de ácido nucleico seleccionada 313, 333 se usan para detectar la región de ácido nucleico seleccionada en cada reacción de unión. Se pueden usar dos índices de alelos 321, 323 que son indicativos de los SNP para identificar los productos de amplificación, de modo que la determinación de secuencias de la secuencia real de los ácidos nucleicos de interés y SNP no sea necesariamente necesaria, aunque estas secuencias todavía se pueden determinar para identificar y/o proporcionar la confirmación del alelo. Cada uno de los oligonucleótidos de secuencia fijos comprende una región complementaria a la región de ácido nucleico seleccionada 305, 307 y secuencias de cebador universal 309, 311 que se usan para amplificar las diferentes regiones de ácido nucleico seleccionadas tras la selección y/o aislamiento inicial. Las secuencias de cebador universal se localizan en los extremos de los oligonucleótidos de secuencia fijos 301, 303 y 323 que flanquean los índices y las regiones complementarias al ácido nucleico de interés, conservando, por tanto, las secuencias específicas de ácido nucleico y los índices de alelos en los productos de amplificación. Los oligonucleótidos de secuencia fijos 301, 303, 323 se introducen en la etapa 302 en una alícuota de la muestra genética 300 y se deja que se hibriden a regiones de ácido nucleico seleccionadas 315 o 325. Tras la hibridación, los oligonucleótidos de secuencia fijos no hibridados se separan preferentemente del resto de la muestra genética (no mostrado).
Los oligonucleótidos puente correspondientes a un SNP A/T 313 o un SNP G/C 333 se introducen en la etapa 304 y se deja que se unan en la región de la región de ácido nucleico seleccionada 315 o 325 entre las primera 305 y segunda 307 regiones complementarias al ácido nucleico de los oligonucleótidos de secuencia fijos. De forma alternativa, los oligonucleótidos puente 313, 333 se pueden introducir simultáneamente en la muestra con los oligonucleótidos de secuencia fijos. Los oligonucleótidos unidos se unen en la etapa 306 en la mezcla de reacción para crear un oligonucleótido contiguo que se extienda y sea complementario a la región de ácido nucleico de interés. Además, cabe destacar que en algunos ensayos algunos oligonucleótidos puente serán específicos de polimorfismo y otros no lo serán, ya que tanto las regiones de ácido nucleico no polimórficas (o independientes de polimorfismo) de interés como las regiones de ácido nucleico polimórficas de interés se pueden consultar en un único ensayo.
Tras la unión, las reacciones separadas se combinan preferentemente para las etapas de detección y amplificación universal. Los cebadores universales 317, 319 se introducen en la etapa 308 en las reacciones combinadas para amplificar los oligonucleótidos unidos y crean en la etapa 310 los productos 327, 329 que comprenden la secuencia de la región de ácido nucleico de interés que representa los SNP en la región de ácido nucleico seleccionada. Los productos 327, 329 se detectan y cuantifican secuenciando los productos o porciones de los productos, a través de la identificación del índice de alelos, la región del producto que contiene el SNP de la región de ácido nucleico seleccionada, o ambos. Preferentemente, los productos de los procedimientos de la FIG. 3 se detectan y cuantifican a través de la secuenciación de última generación de los índices de alelos, obviando, por tanto, la necesidad de determinar las secuencias reales de la región del producto complementaria a la región de ácido nucleico seleccionada o de todo el producto. Sin embargo, en otros aspectos, puede ser deseable determinar la secuencia de
tanto el índice como de la región del producto complementaria a la región de ácido nucleico seleccionada, por ejemplo, para proporcionar confirmación de los resultados.
En los procedimientos de la FIG. 3 (y en los procedimientos ilustrados en las demás figuras), se ha descrito un índice de alelos. Sin embargo, los índices mostrados en 321 y 323 pueden ser índices de alelos, índices de muestras, índices combinados de alelos y muestras, índices de locus o cualquier otro índice o combinación de índices descrito en el presente documento o de otro modo usado en la técnica.
Además, se pueden emplear procedimientos donde el nucleótido distintivo se localice en los oligonucleótidos de secuencia fijos en vez de en un oligonucleótido puente. Por tanto, en un sistema de ensayo ejemplar de este tipo, un índice de alelos se asocia con un oligonucleótido de secuencia fijo específico de alelo, y la detección de los alelos resulta de la secuenciación del índice de alelos. El índice de alelos se puede incluir en el primer oligonucleótido de secuencia específico de alelo o bien en el segundo oligonucleótido de secuencia fijo. En aspectos específicos, está presente un índice de alelos tanto en el primer como en el segundo oligonucleótido de secuencia fijo para detectar dos o más polimorfismos dentro de las regiones de ácido nucleico seleccionadas. El número de oligonucleótidos de secuencia fijos usados en dichos aspectos puede corresponder al número de posibles alelos que se están evaluando para determinar una región de ácido nucleico seleccionada, y la determinación de secuencias del índice de alelos puede detectar la presencia, cantidad o ausencia de un alelo específico en la muestra genética.
La FIG. 4 ilustra este aspecto de la divulgación. En la FIG. 4, se usan tres oligonucleótidos de secuencia fijos 401, 403 y 423. Dos de los oligonucleótidos de secuencia fijos 401, 423 son específicos de alelo, que comprenden una región complementaria a un alelo en una región de ácido nucleico que comprende, por ejemplo, un SNP A/T o G/C, respectivamente. Cada uno de los oligonucleótidos de secuencia fijos específicos de alelo 401, 423 también comprende un correspondiente índice de alelos 421, 431 y una secuencia de cebador universal 409. El segundo oligonucleótido de secuencia fijo 403 tiene una segunda secuencia de cebador universal 411, y estas secuencias de cebador universal se usan para amplificar las regiones de ácido nucleico seleccionadas, hibridación y unión de los conjuntos de oligonucleótidos a las regiones de ácido nucleico seleccionadas de la muestra genética. Las secuencias de cebador universal se localizan en los extremos de los oligonucleótidos de secuencia fijos 401, 403, 423 que flanquean los índices y las regiones en los oligonucleótidos de secuencia fijos complementarios a las regiones de ácido nucleico seleccionadas de interés; capturando, por tanto, las secuencias específicas de ácido nucleico y los índices en los productos de cualquier procedimiento de amplificación universal.
Los oligonucleótidos de secuencia fijos 401, 403, 423 se introducen en la etapa 402 en la muestra genética 400 y se deja que se hibriden a regiones de ácido nucleico seleccionadas 415, 425. Tras la hibridación, los oligonucleótidos de secuencia fijos no hibridados se separan preferentemente del resto de la muestra genética (no mostrado). Los oligonucleótidos puente 413 se introducen y se deja que se hibriden en 404 al ácido nucleico 415 en la región entre la región del primer oligonucleótido de secuencia fijo específico de alelo 405 y la otra región de oligonucleótido de secuencia fijo 407 o al ácido nucleico 425 complementario a la región entre la región del segundo oligonucleótido de secuencia fijo específico de alelo 435 y la otra región de oligonucleótido de secuencia fijo 407. De forma alternativa, los oligonucleótidos puente 413 se pueden introducir simultáneamente en la muestra con los conjuntos de oligonucleótidos de secuencia fijos. Como se establece previamente en relación con la figura 3, cabe destacar que en algunos ensayos algunos conjuntos de oligonucleótidos serán específicos de polimorfismo y otros no lo serán, ya que tanto las regiones de ácido nucleico no polimórficas (o independientes de polimorfismo) de interés como las regiones de ácido nucleico polimórficas de interés se pueden consultar en un único ensayo.
Los oligonucleótidos hibridados a las regiones de ácido nucleico seleccionadas se unen en la etapa 406 para crear un oligonucleótido contiguo que se extienda y sea complementario a las regiones de ácido nucleico seleccionadas de interés. La unión se produce principalmente solo cuando los extremos específicos de alelo de los oligonucleótidos de secuencia fijos específicos de alelo son complementarios al SNP en la región de ácido nucleico seleccionada. Tras la unión, se introducen los cebadores universales 417, 419 para amplificar en la etapa 408 el oligonucleótido unido para crear en la etapa 410 los productos 427, 429 que comprenden la secuencia de las regiones de ácido nucleico de interés. Estos productos 427, 429 se detectan y cuantifican a través de la determinación de secuencias de todo o una porción del producto, y, en particular, la región del producto que contiene el SNP en la región de ácido nucleico seleccionada y/o el índice de alelos. Aquí, el nucleótido específico de alelo se muestra como que está en el extremo del oligonucleótido de secuencia fijo específico de alelo, aunque el nucleótido específico de alelo no necesita estar localizado así. Sin embargo, para hacer que la unión sea específica de alelo, el nucleótido específico del alelo debe estar cerca del extremo unido. Típicamente, el nucleótido específico de alelo debe estar dentro de los 5 nucleótidos del extremo unido. En un aspecto preferente, el nucleótido específico de alelo es el penúltimo o último nucleótido (terminal).
Aún en otro ejemplo del ensayo de la presente divulgación, la detección de los alelos resulta de la hibridación de un índice de locus a una matriz. Cada alelo se detecta a través de una etapa de marcado específico de alelo, donde cada alelo se marca, por ejemplo, con un marcador fluorescente espectralmente distinto durante la amplificación universal. La FIG. 5 ilustra este aspecto. En la FIG. 5, se usan tres oligonucleótidos de secuencia fijos 501, 503 y 523. Dos de los oligonucleótidos de secuencia fijos 501, 523 son específicos de alelo, y cada uno comprende una región que coincide con un alelo diferente en la misma región de ácido nucleico seleccionada, un índice de locus
521 y secuencias de cebador universal específico de alelo 509, 539. El tercer oligonucleótido de secuencia fijo no específico de alelo 503 comprende otra secuencia de cebador universal 511. Las secuencias de cebador universal se usan para amplificar las regiones de ácido nucleico seleccionadas tras la hibridación y unión de los oligonucleótidos. Se incorporan marcadores en los productos de amplificación que distinguen cada alelo. Como en el ejemplo previo, las secuencias de cebador universal se localizan en los extremos proximales de los oligonucleótidos de secuencia fijos 501, 503, 523 y, por tanto, capturan las secuencias específicas de alelo y los índices en los productos de cualquier procedimiento de amplificación universal. Los oligonucleótidos de secuencia fijos 501, 503, 523 se introducen en la etapa 502 en la muestra genética 500 y se deja que se unan específicamente a regiones de ácido nucleico seleccionadas 515, 525. Tras la hibridación, los oligonucleótidos de secuencia fijos no hibridados se separan preferentemente del resto de la muestra genética (no mostrado). Los oligonucleótidos puente 513 se introducen y se deja que se unan en la etapa 504 a la región de las regiones de ácido nucleico seleccionadas 515, 525 entre los primer (específico de alelo) 505 y segundo (no específico de alelo) 507 oligonucleótidos de secuencia fijos y entre los primer (específico de alelo) 535 y segundo 507 (no específico de alelo) oligonucleótidos de secuencia fijos. De forma alternativa, los oligonucleótidos puente 513 se pueden introducir simultáneamente en la muestra con los oligonucleótidos de secuencia fijos.
Los oligonucleótidos unidos se unen en la etapa 506 para crear un oligonucleótido contiguo que se extienda y sea complementario a las regiones de ácido nucleico seleccionadas de interés. La unión se produce principalmente cuando los extremos específicos de alelo coinciden. Tras la unión, se introducen los cebadores universales 517, 519, 537 para amplificar en la etapa 508 el oligonucleótido unido para crear en la etapa 510 los productos 527, 529 que comprenden la secuencia de la región de ácido nucleico seleccionada de interés. Los cebadores universales 517 y 537 tienen marcadores fluorescentes espectralmente distintos, de modo que la información específica de alelo se capture y se pueda leer a través de estos marcadores fluorescentes. Los productos 527, 529 se detectan y cuantifican a través de la hibridación del índice de locus 521 a una matriz y formación de imágenes. Como se describe en relación con la FIG. 4, es importante destacar que la unión 506 es preferentemente específica de alelo; por tanto, el nucleótido distintivo se localiza al menos 5 nucleótidos desde el extremo del oligonucleótido de secuencia fijo específico de alelo y preferentemente se localiza como el penúltimo o último nucleótido. El ejemplo mostrado en la FIG. 5, donde se usa un índice de locus para la hibridación a una matriz, se puede usar en cualquiera de los diversos procedimientos descritos en el presente documento, tal como procedimientos donde los oligonucleótidos de secuencia fijos y el oligonucleótido puente no se hibridan de forma adyacente y se usan una polimerasa y dNTP para cerrar el "hueco" entre los oligonucleótidos seguido de unión. De forma similar, se pueden usar procedimientos de hibridación del índice de locus en protocolos donde solo se usan oligonucleótidos de secuencia fijos, es decir, ningún oligonucleótido puente está presente, y donde los oligonucleótidos de secuencia fijos se hibridan de forma adyacente y se enlazan por unión o donde los oligonucleótidos de secuencia fijos se hibridan con un hueco entre ellos y se enlazan usando una polimerasa y dNTP seguido de unión.
En un aspecto alternativo, está presente un índice de alelos tanto en el primer como en el segundo oligonucleótido de secuencia fijo para detectar polimorfismos en los extremos de cada oligonucleótido de secuencia fijo usando un correspondiente marcador fluorescente espectralmente distinto para cada oligonucleótido de secuencia fijo para un alelo dado. En este procedimiento, el número de oligonucleótidos de secuencia fijos corresponde al número de posibles alelos que se están evaluando para determinar una región de ácido nucleico seleccionada. En las figuras y ejemplos anteriores, los oligonucleótidos de secuencia fijos se representan como dos oligonucleótidos distintos. En otro aspecto, los oligonucleótidos de secuencia fijos pueden ser los extremos opuestos del mismo oligonucleótido (véase, por ejemplo, la FIG. 7, supra).
En los aspectos descritos anteriormente, los oligonucleótidos puente usados se hibridan a regiones del ácido nucleico de interés adyacentes a las regiones complementarias a los oligonucleótidos de secuencia fijos, de modo que cuando el/los oligonucleótido(s) de secuencia fijo(s) y puente se hibridan específicamente son directamente adyacentes entre sí para la unión. Sin embargo, en otros aspectos, el oligonucleótido puente se hibrida a una región que no es directamente adyacente a la región complementaria a uno o ambos de los oligonucleótidos de secuencia fijos, y es necesaria una etapa intermedia que requiera la extensión de uno o más de los oligonucleótidos antes de la unión. Por ejemplo, como se ilustra en la FIG. 6, cada conjunto de oligonucleótidos contiene preferentemente dos oligonucleótidos de secuencia fijos 601, 603 y uno o más oligonucleótidos puente 613. Cada uno de los oligonucleótidos de secuencia fijos comprende una región complementaria a la región de ácido nucleico seleccionada 605, 607, y preferentemente secuencias de cebador universal 609, 611; es decir, regiones de oligonucleótido complementarias a los cebadores universales. Las secuencias de cebador universal 609, 611 se localizan en o cerca de los extremos de los oligonucleótidos de secuencia fijos 601, 603, y, por tanto, capturan las secuencias específicas de ácido nucleico en los productos de cualquier procedimiento de amplificación universal.
Los oligonucleótidos de secuencia fijos 601, 603 se introducen en la etapa 602 en la muestra genética 600 y se deja que se unan específicamente a porciones complementarias de la región de ácido nucleico seleccionada de interés 615. Tras la hibridación, los oligonucleótidos de secuencia fijos no hibridados se separan preferentemente del resto de la muestra genética (no mostrado). A continuación, el oligonucleótido puente se introduce y se deja que se una en la etapa 604 a la región de la región de ácido nucleico seleccionada 615 entre los primer 601 y segundo oligonucleótidos de secuencia fijos 603. De forma alternativa, el oligonucleótido puente se puede introducir simultáneamente en la muestra con los oligonucleótidos de secuencia fijos. En el aspecto ejemplar mostrado aquí, el
oligonucleótido puente se híbrida a una región directamente adyacente a la región del primer oligonucleótido de secuencia fijo 605, pero se separa en uno o más nucleótidos de la región complementaria del segundo oligonucleótido de secuencia fijo 607. Tras la hibridación de los oligonucleótidos de secuencia fijos y puente, el oligonucleótido puente 613 se extiende en la etapa 606, por ejemplo, usando una polimerasa y dNTP, para llenar el hueco entre el oligonucleótido puente 613 y el segundo oligonucleótido de secuencia fijo 603. Tras la extensión, los oligonucleótidos hibridados se unen en la etapa 608 para crear un oligonucleótido contiguo que se extienda y sea complementario a la región de ácido nucleico seleccionada de interés 615. Después de la unión, se introducen los cebadores universales 617, 619 en la etapa 610 para amplificar el oligonucleótido unido para crear en la etapa 612 los productos 623 que comprenden la secuencia de la región de ácido nucleico de interés. Estos productos 623 se aíslan, detectan y cuantifican para proporcionar información sobre la presencia y cantidad de las regiones de ácido nucleico seleccionadas en la muestra genética. Preferentemente, los productos se detectan y cuantifican a través de secuenciación de última generación de un índice de alelos 621, o, de forma alternativa, la determinación de secuencias de la porción del producto de amplificación complementaria al ácido nucleico seleccionado de interés 615 dentro del producto de amplificación 623.
La FIG. 7 ilustra cómo los oligonucleótidos de secuencia fijos pueden ser parte de la misma molécula. En aspectos específicos, el único oligonucleótido de secuencia fijo 701 es complementario a la región de ácido nucleico seleccionada 715 en ambos extremos. Cuando este único oligonucleótido de secuencia fijo 701 se hibrida a la región de ácido nucleico seleccionada 715 forma un oligonucleótido precircular 703 donde los extremos se separan por varios nucleótidos. A continuación, el oligonucleótido puente 713 se une entre las regiones complementarias 705, 707 del oligonucleótido precircular 703 para llenar este hueco. A continuación, las regiones de oligonucleótido 705, 707 del oligonucleótido precircular 703 unido a la muestra genética 715 se unen entre sí con el oligonucleótido puente 713, formando un círculo completo. Al igual que con otros procedimientos ejemplificados en el presente documento, el uso de un oligonucleótido puente no es necesario, y en dichos modos de realización, los oligonucleótidos de secuencia fijos se pueden hibridar de forma adyacente, o se pueden usar una polimerasa y dNTP para llenar un hueco si los oligonucleótidos de secuencia fijos no se hibridan de forma adyacente. El molde circular es preferentemente el escindido y amplificado usando uno o más de los sitios de cebador universal. En aspectos específicos, se usa una única región de cebador universal para replicar el molde usando técnicas tales como replicación en círculo rodante, como se divulga en Lizardi et al., pat. de Ee . UU. n.° 6.558.928.
Como se ilustra en la FIG. 7, el oligonucleótido de secuencia fijo tiene dos sitios de cebado universales 709, 711 en el molde circular y opcionalmente uno o más índices 721 entre los extremos de la construcción que son complementarios a la región de ácido nucleico seleccionada. Aquí se muestra que un sitio de escisión 723 existe entre los dos sitios de cebado universales. La construcción 701 se introduce en la muestra genética en la etapa 702, se deja que se hibride a la región de ácido nucleico seleccionada de interés, y en la etapa 704 se introduce el oligonucleótido puente y se deja que se hibride a la región de ácido nucleico seleccionada. A continuación, la construcción se dispone en forma circular a través de unión en la etapa 706 en el oligonucleótido puente 713, y se puede usar una nucleasa para retirar todos o la mayoría de los oligonucleótidos sin forma circular. Después de la retirada de los oligonucleótidos sin forma circular, el oligonucleótido con forma circular se escinde, conservando y, en algunos aspectos, exponiendo los sitios de cebado universales 709, 711. Se añaden cebadores universales 717, 719 en la etapa 708 y se produce una amplificación universal 710 para crear 712 los productos 725 que comprenden la secuencia de la región de ácido nucleico seleccionada de interés. Los productos 725 se detectan y cuantifican a través de, por ejemplo, la secuenciación de última generación de la porción del producto complementaria a la región de ácido nucleico seleccionada o, de forma alternativa, el índice, lo que obvia la necesidad de secuenciar toda la construcción. Sin embargo, en otros aspectos, es deseable determinar el producto que comprende secuencias tanto del índice como de la región de ácido nucleico seleccionada, por ejemplo, para proporcionar una confirmación interna de los resultados o donde el índice proporcione información sobre la muestra y no sea informativo de la región de ácido nucleico seleccionada. Como se menciona anteriormente, se puede aplicar esta metodología con único oligonucleótido de secuencia fijo a cualquiera de los ejemplos en las figuras 2-7. Además, nuevamente, cabe destacar que en algunos ensayos, algunos conjuntos de oligonucleótidos serán específicos de polimorfismo y otros no lo serán, ya que tanto las regiones de ácido nucleico no polimórficas (o independientes de polimorfismo) de interés como las regiones de ácido nucleico polimórficas de interés se pueden consultar en un único ensayo.
Uso de índices en los procedimientos de la divulgación
Como se describe en relación con las figuras 2-7, anteriormente, en determinados aspectos, los oligonucleótidos de secuencia fijos en un conjunto comprenden uno o más índices que, por ejemplo, identifican las regiones de ácido nucleico seleccionadas (un índice de locus), SNP dentro de una región de ácido nucleico seleccionada (un índice de alelos) y/o una muestra particular que se está analizando (un índice de muestra). Por ejemplo, la detección de uno o más índices de locus puede servir como un sustituto para la detección de toda la región de ácido nucleico seleccionada como se describe a continuación, o la detección de un índice puede servir como confirmación de la presencia de una región de ácido nucleico seleccionada particular si se determinan tanto la secuencia del índice como la secuencia del producto oligonucleotídico complementaria a la propia región de ácido nucleico. Los índices se asocian preferentemente con las regiones de ácido nucleico seleccionadas durante la etapa de amplificación selectiva usando cebadores que comprenden tanto el índice como una región que se hibrida específicamente a la región de ácido nucleico seleccionada (es decir, secuencias específicas de región de ácido nucleico seleccionada).
Los índices son típicamente secuencias únicas no complementarias usadas dentro de un cebador de amplificación para proporcionar información pertinente para la región de ácido nucleico seleccionada que se aísla y/o amplifica usando el cebador. El orden y disposición de los índices, así como la longitud de los índices, pueden variar, y se pueden usar los índices en diversas combinaciones. De forma alternativa, los índices y/o secuencias de amplificación universal se pueden añadir a las regiones de ácido nucleico seleccionadas amplificadas de forma selectiva tras la amplificación selectiva inicial usando la unión de adaptadores que comprendan estas secuencias. La ventaja de emplear índices es que se puede obtener la presencia (y, en definitiva, la cantidad o frecuencia) de las regiones de ácido nucleico seleccionadas sin la necesidad de secuenciar toda la longitud de las regiones de ácido nucleico seleccionadas, aunque en determinados aspectos puede ser deseable hacerlo así. En general, sin embargo, la capacidad de identificar y cuantificar una región de ácido nucleico seleccionada a través de la identificación de uno o más índices disminuirá la longitud de la secuenciación requerida, en particular, si la secuencia de índice se captura en el extremo 3' o 5' de la región de ácido nucleico seleccionada aislada proximal a donde se puede localizar un cebador de secuenciación. El uso de índices como un sustituto para la identificación de regiones de ácido nucleico seleccionadas también puede reducir los errores de secuenciación, puesto que las lecturas de secuenciaciones más largas son más propensas a la introducción o error. Además, como se describe anteriormente en relación con la figura 5, se puede usar el índice de locus, junto con, por ejemplo, marcadores fluorescentes para identificar y cuantificar las regiones de ácido nucleico seleccionadas por hibridación a una matriz.
En un ejemplo de un índice, los cebadores usados para la amplificación selectiva de las regiones de ácido nucleico seleccionadas están diseñados para incluir un índice de locus entre la región complementaria a las regiones de ácido nucleico seleccionadas y el sitio de cebador de amplificación universal. Un índice de locus típicamente es único para cada región de ácido nucleico seleccionada, de modo que la cuantificación del número de veces que se produce un índice de locus particular en una muestra se puede relacionar con el número relativo de copias de la única región de ácido nucleico correspondiente y el cromosoma particular que contiene la única región de ácido nucleico. En general, el índice de locus es lo suficientemente largo como para marcar cada única región de ácido nucleico conocida de forma única. Por ejemplo, si el procedimiento usa 192 únicas regiones de ácido nucleico conocidas, existen al menos 192 únicos índices de locus, identificando cada uno de forma única una única región de ácido nucleico de un locus particular en un cromosoma. Los índices de locus usados en los procedimientos de la divulgación pueden ser indicativos de diferentes únicas regiones de ácido nucleico en un único cromosoma, así como únicas regiones de ácido nucleico conocidas presentes en diferentes cromosomas dentro de una muestra. El índice de locus puede contener nucleótidos adicionales que permiten la identificación y corrección de errores de secuenciación, incluyendo la detección de deleción, sustitución o inserción de una o más bases durante la secuenciación, así como cambios de nucleótidos que se pueden producir fuera de la secuenciación, tales como síntesis de oligonucleótidos, amplificación o cualquier otro aspecto de los procedimientos.
En otro ejemplo, se pueden diseñar los cebadores usados para la amplificación de las regiones de ácido nucleico seleccionadas para proporcionar un índice de alelos (como alternativa a un índice de locus) entre la región complementaria a la región de ácido nucleico seleccionada y el sitio de cebador de amplificación universal. Un índice de alelos es único para un alelo particular de una región de ácido nucleico seleccionada, de modo que la cuantificación del número de veces que se produce un índice de alelos particular en una muestra se puede relacionar con el número relativo de copias de ese alelo y la suma de los índices alélicos para una región de ácido nucleico seleccionada particular se puede relacionar con el número relativo de copias de esa región de ácido nucleico seleccionada en el cromosoma particular que contiene la región de ácido nucleico seleccionada. En modos de realización donde se usan tanto conjuntos de oligonucleótidos específicos de polimorfismo como conjuntos de oligonucleótidos independientes de polimorfismo o SNP en un único ensayo, se pueden emplear tanto índices de alelos como de locus.
Aún en otro ejemplo, se pueden diseñar los cebadores usados para la amplificación de las regiones de ácido nucleico seleccionadas para proporcionar un índice de identificación entre la región complementaria a una región de ácido nucleico seleccionada y el sitio de cebador de amplificación universal. En un aspecto de este tipo, está presente un número suficiente de índices de identificación para identificar de forma única cada molécula amplificada en la muestra. Las secuencias de índice de identificación tienen preferentemente 6 o más nucleótidos de longitud. En un aspecto preferente, el índice de identificación es lo suficientemente largo como para tener una probabilidad estadística de marcar cada molécula con una única región de ácido nucleico de forma única. Por ejemplo, si existen 3000 copias de una única región de ácido nucleico particular, existen sustancialmente más de 3000 índices de identificación, de modo que cada copia de una única región de ácido nucleico particular esté probablemente marcada con un único índice de identificación. Al igual que con otros índices, el índice de identificación puede contener nucleótidos adicionales que permiten la identificación y corrección de errores de secuenciación, incluyendo la detección de deleción, sustitución o inserción de una o más bases durante la secuenciación, así como cambios de nucleótidos que se pueden producir fuera de la secuenciación, tales como síntesis de oligonucleótidos, amplificación y cualquier otro aspecto del ensayo.
El índice de identificación se puede combinar con cualquier otro índice para crear un índice que proporcione información para dos propiedades. El locus de identificación también se puede usar para detectar y cuantificar el
sesgo de amplificación que se produce en dirección 3' del aislamiento inicial de las regiones de ácido nucleico seleccionadas de una muestra y estos datos se pueden usar para normalizar los datos de la muestra.
Además de los demás índices descritos en el presente documento, se puede emplear un índice de corrección. Un índice de corrección es una secuencia de nucleótidos corta que permite la corrección de la amplificación, secuenciación u otros errores experimentales, incluyendo la detección de una deleción, sustitución o inserción de una o más bases durante la secuenciación, así como cambios de nucleótidos que se pueden producir fuera de la secuenciación, tales como síntesis de oligonucleótidos, amplificación u otros aspectos del ensayo. Los índices de corrección pueden ser índices independientes que sean secuencias separadas, o se pueden incluir dentro de otros índices para ayudar en la confirmación de la exactitud de las técnicas experimentales usadas, por ejemplo, un índice de corrección puede ser un subconjunto de secuencias de un índice de locus o un índice de identificación.
En algunos aspectos, se usan índices que indican la muestra de la que se aíslan las regiones de ácido nucleico seleccionadas para identificar la fuente de las regiones de ácido nucleico seleccionadas en un sistema de ensayo multiplexado. En dichos aspectos, las regiones de ácido nucleico seleccionadas de un individuo se asignarán y asociarán con un único índice de muestra particular. Por tanto, se puede usar el índice de muestra para ayudar en la identificación de regiones de ácido nucleico para la multiplexación de diferentes muestras en un único recipiente de reacción (es decir, en el caso de la agrupación de muestras), de modo que se pueda identificar cada muestra en base a su índice de muestra. En un aspecto preferente, existe un único índice de muestra para cada muestra en un conjunto de muestras, y las muestras se agrupan durante la secuenciación. Por ejemplo, si se agrupan doce muestras en una única reacción de secuenciación, existen al menos doce únicos índices de muestra de modo que cada muestra se marque de forma única. Después de que se realiza la etapa de secuenciación, en primer lugar, los datos de secuenciación se disocian preferentemente por índice de muestra antes de determinar la frecuencia de cada región de ácido nucleico seleccionada para cada muestra y antes de determinar si existe una anomalía cromosómica para cada muestra.
Minimización de la variación dentro de las muestras
Una dificultad con la detección de anomalías cromosómicas en una muestra mixta es que a menudo el ADN del tipo de célula con la anomalía cromosómica (es decir, ADN fetal) está presente en una abundancia mucho menor que el ADN de un tipo de célula euploide (es decir, ADN materno). En el caso de una muestra materna que contiene ADN fetal y materno libre de células, el ADN fetal libre de células como un porcentaje del ADN libre de células total puede variar de menos de un uno a un cuarenta por ciento, y lo más comúnmente está presente en o por debajo de un veinte por ciento y con frecuencia en o por debajo de un diez por ciento. Por ejemplo, en la detección de una aneuploidía del cromosoma Y en el ADN fetal de dicha muestra materna mixta, el incremento relativo de las secuencias cromosómicas de Y es un múltiplo del porcentaje esperado de las secuencias de Y si el feto es un varón normal, y, por tanto, como un porcentaje del ADN total en una muestra mixta donde, como ejemplo, el ADN fetal es de un 5 % del total, el incremento de la contribución del cromosoma Y como un porcentaje del total es un 1/47 de un 5 % (0,11 % del porcentaje total de ADN en la muestra). Si se ha de detectar de forma sólida esta diferencia a través de los procedimientos descritos en el presente documento, la variación en la medición del cromosoma Y tiene que ser mucho menor que el porcentaje de incremento del cromosoma Y.
En algunos aspectos, se normaliza la cantidad medida de una o más regiones de ácido nucleico seleccionadas en un cromosoma para tener en cuenta la variación conocida de fuentes tales como el sistema de ensayo (por ejemplo, temperatura, diferencias entre los lotes de reactivos), biología subyacente de la muestra (por ejemplo, contenido de ácido nucleico), diferencias entre los operarios o cualquier otra variable. Además, los datos usados para determinar la frecuencia de las regiones de ácido nucleico seleccionadas pueden excluir datos con valores atípicos que parece que se deben a un error experimental, o que tienen niveles elevados o disminuidos en base a un sesgo genético idiopático dentro de una muestra particular. En un ejemplo, los datos usados para la suma pueden excluir regiones de ácido nucleico con una frecuencia en particular elevada en una o más muestras. En otro ejemplo, los datos usados para la suma pueden excluir regiones de ácido nucleico seleccionadas que se encuentran en una abundancia en particular baja en una o más muestras.
La variación entre las muestras y/o para regiones de ácido nucleico seleccionadas dentro de una muestra se puede minimizar usando una combinación de procedimientos analíticos. Por ejemplo, la variación se reduce usando una referencia interna en el ensayo. Un ejemplo de una referencia interna es el uso de un cromosoma presente en una abundancia "normal" (por ejemplo, disomía para un autosoma) para compararla frente a los cromosomas X e Y que pueden estar presentes en abundancia anómala, es decir, una aneuploidía u oligocontaminante, en la misma muestra. Aunque el uso de un único cromosoma "normal" de este tipo como cromosoma de referencia puede ser suficiente, es preferente usar de dos a varios autosomas como cromosomas de referencia interna para incrementar la capacidad estadística de la cuantificación.
Una utilización de una referencia interna es calcular una proporción de abundancia de la frecuencia de los cromosomas X y/o Y supuestamente anómala con respecto a la abundancia de los autosomas en una muestra, llamada proporción cromosómica. Al calcular la proporción cromosómica, la abundancia o recuentos de cada una de las regiones de ácido nucleico seleccionadas para cada cromosoma se suman para calcular los recuentos totales
para cada cromosoma. A continuación, los recuentos totales de un cromosoma se dividen por los recuentos totales de un cromosoma diferente para crear una proporción cromosómica para esos dos cromosomas.
De forma alternativa, se puede calcular una proporción cromosómica para cada cromosoma sumando en primer lugar los recuentos de cada una de las regiones de ácido nucleico seleccionadas para cada cromosoma, y, a continuación, dividiendo la suma para un cromosoma por la suma total para dos o más cromosomas. Una vez calculada, la proporción cromosómica se compara, a continuación, con la proporción cromosómica promedio de una población euploide.
El promedio puede ser la media, la mediana, la moda u otro promedio, con o sin normalización o exclusión de datos con valores atípicos. En un aspecto preferente, se usa la media. Al desarrollar el conjunto de datos para la proporción cromosómica de la población euploide, se calcula la variación normal de los cromosomas medidos. Esta variación se puede expresar de una serie de formas, lo más típicamente como el coeficiente de variación o CV. Cuando la proporción cromosómica de X de la muestra se compara con la proporción cromosómica promedio de una población euploide, si la proporción cromosómica de X para la muestra se encuentra estadísticamente fuera de la proporción cromosómica promedio para la población euploide, la muestra contiene una anomalía cromosómica en X indicativa, por ejemplo, de una aneuploidía de X y/o un mosaicismo del cromosoma X. De forma similar, cuando la proporción cromosómica de Y de la muestra se compara con la proporción cromosómica promedio de una población euploide, si la proporción cromosómica de Y para la muestra se encuentra estadísticamente fuera de la proporción cromosómica promedio para la población euploide, la muestra contiene una anomalía cromosómica en Y indicativa, por ejemplo, de una aneuploidía de Y y/o un mosaicismo del cromosoma Y.
Los criterios para establecer el umbral estadístico para declarar una aneuploidía dependen de la variación en la medición de la proporción cromosómica y las tasas de positivos falsos y negativos falsos aceptables para el procedimiento deseado. En general, este umbral puede ser un múltiplo de la variación observada en la proporción cromosómica. En un ejemplo, este umbral es tres o más veces la variación de la proporción cromosómica. En otro ejemplo, es cuatro o más veces la variación de la proporción cromosómica. En otro ejemplo, es cinco o más veces la variación de la proporción cromosómica. En otro ejemplo, es seis o más veces la variación de la proporción cromosómica. En el ejemplo anterior, la proporción cromosómica se determina sumando los recuentos de regiones de ácido nucleico seleccionadas por cromosoma. Típicamente, se usa el mismo número de regiones de ácido nucleico seleccionadas para cada cromosoma.
Un procedimiento alternativo para generar la proporción cromosómica sería calcular los recuentos promedio para las regiones de ácido nucleico seleccionadas para cada cromosoma o región cromosómica. El promedio puede ser cualquier estimación de la media, la mediana o la moda, aunque típicamente se usa un promedio. El promedio puede ser la media de todos los recuentos o alguna variación, tal como un promedio truncado o ponderado. Una vez que se hayan calculado los recuentos promedio para cada cromosoma, los recuentos promedio para cada cromosoma se pueden dividir por los otros para obtener una proporción cromosómica entre dos cromosomas, los recuentos promedio para cada cromosoma se pueden dividir por la suma de los promedios para todos los cromosomas medidos para obtener una proporción cromosómica para cada cromosoma como se describe anteriormente. Como se resalta anteriormente, la capacidad de detectar un cromosoma X, frecuencia del cromosoma X, cromosoma Y o frecuencia cromosómica de Y en una muestra materna donde el ADN fetal está en baja abundancia relativa depende en gran medida de la variación en las mediciones de diferentes regiones de ácido nucleico seleccionadas en el ensayo. Se pueden usar numerosos procedimientos analíticos que reduzcan esta variación y, por tanto, mejorar la sensibilidad de este procedimiento para detectar una aneuploidía.
Un procedimiento para reducir la variabilidad del ensayo es incrementar el número de regiones de ácido nucleico seleccionadas usadas para calcular la abundancia de los cromosomas. En general, si la variación medida de una única región de ácido nucleico seleccionada de un cromosoma es B % y se miden C diferentes regiones de ácido nucleico seleccionadas en el mismo cromosoma, la variación de la medición de la abundancia cromosómica calculada sumando o promediando la abundancia de cada región de ácido nucleico seleccionada en ese cromosoma será aproximadamente B % dividida por C1/2. Dicho de otra forma, la variación de la medición de la abundancia de los cromosomas sería aproximadamente la variación promedio de la medición de la abundancia de cada región de ácido nucleico seleccionada dividida por la raíz cuadrada del número de regiones de ácido nucleico seleccionadas.
En un aspecto preferente, el número de regiones de ácido nucleico seleccionadas medidas para cada cromosoma (el cromosoma X, el cromosoma Y y el uno o más autosomas) es al menos de 8. En otro aspecto preferente, el número de regiones de ácido nucleico seleccionadas medidas para cada cromosoma es al menos de 24. Aún en otro aspecto preferente, el número de regiones de ácido nucleico seleccionadas medidas para cada cromosoma es al menos de 32. En otro aspecto preferente, el número de regiones de ácido nucleico seleccionadas medidas para cada cromosoma es al menos de 100. En otro aspecto preferente, el número de regiones de ácido nucleico seleccionadas medidas para cada cromosoma es al menos de 200. Existe un coste incremental incrementado en la medición de cada región de ácido nucleico seleccionada y, por tanto, es importante minimizar el número de regiones de ácido nucleico seleccionadas mientras que todavía se generan datos estadísticamente sólidos. En un aspecto preferente, el número de regiones de ácido nucleico seleccionadas medidas para cada cromosoma es menor que
2000. En un aspecto preferente, el número de regiones de ácido nucleico seleccionadas medidas para cada cromosoma es menor que 1000. En un aspecto lo más preferente, el número de regiones de ácido nucleico seleccionadas medidas para cada cromosoma es al menos de 32 y menor que 1000.
En un aspecto, tras la medición de la abundancia para cada región de ácido nucleico seleccionada, se puede usar un subconjunto de las regiones de ácido nucleico seleccionadas para determinar la presencia o ausencia de una anomalía cromosómica en X o Y. Existen muchos procedimientos estándar para elegir el subconjunto de regiones de ácido nucleico seleccionadas, incluyendo la exclusión, donde las regiones de ácido nucleico seleccionadas con niveles detectados por debajo y/o por encima de un determinado percentil se descartan del análisis. En un aspecto, el percentil puede ser el 5 % más bajo y más alto como se mide por la frecuencia. En otro aspecto, el percentil que se va a descartar puede ser el 10 % más bajo y más alto como se mide por la frecuencia. En otro aspecto, el percentil que se va a descartar puede ser el 25 % más bajo y más alto como se mide por la frecuencia.
Otro procedimiento para elegir un subconjunto de regiones de ácido nucleico seleccionadas incluye la eliminación de regiones que se encuentran fuera de algún límite estadístico. Por ejemplo, las regiones que se encuentran fuera de una o más desviaciones estándar de la abundancia media se pueden retirar del análisis. Otro procedimiento para elegir el subconjunto de regiones de ácido nucleico seleccionadas puede ser comparar la abundancia relativa de una región de ácido nucleico seleccionada con la abundancia esperada de la misma región de ácido nucleico seleccionada en una población sana y descartar cualquier región de ácido nucleico seleccionada que no cumpla la prueba de expectativas. Para minimizar además la variación en el ensayo, se puede incrementar el número de veces que se mide cada región de ácido nucleico seleccionada. Como se analiza, a diferencia de los procedimientos aleatorios de detección de anomalías en la frecuencia de los cromosomas X e Y, donde se mide el genoma en promedio menos de una vez, los procedimientos de la presente invención miden de forma intencionada cada región de ácido nucleico seleccionada múltiples veces. En general, cuando se cuentan acontecimientos, la variación en el recuento se determina por la estadística de Poisson, y la variación en el recuento es típicamente igual a uno dividido por la raíz cuadrada del número de recuentos. En un aspecto preferente, las regiones de ácido nucleico seleccionadas se miden cada una en promedio al menos 5 veces. En un determinado aspecto, las regiones de ácido nucleico seleccionadas se miden cada una en promedio al menos 10, 50 o 100 veces. En un determinado aspecto, las regiones de ácido nucleico seleccionadas se miden cada una en promedio al menos 250 veces. En un determinado aspecto, las regiones de ácido nucleico seleccionadas se miden cada una en promedio al menos 500 veces. En un determinado aspecto, las regiones de ácido nucleico seleccionadas se miden cada una en promedio al menos 1000 veces o al menos 5000 o al menos 10.000 veces.
En otro aspecto, se pueden elegir de forma aleatoria subconjuntos de regiones de ácido nucleico seleccionadas usando números suficientes para proporcionar un resultado estadísticamente significativo al determinar si existe una anomalía cromosómica. Se pueden realizar múltiples análisis de diferentes subconjuntos de regiones de ácido nucleico seleccionadas dentro de una muestra materna para proporcionar más capacidad estadística. En este ejemplo, puede ser necesario o no retirar o eliminar cualquier región de ácido nucleico seleccionada antes del análisis aleatorio. Por ejemplo, si existen 100 regiones de ácido nucleico seleccionadas para el cromosoma Y y 100 regiones de ácido nucleico seleccionadas, por ejemplo, para el cromosoma 2, se podría realizar una serie de análisis que evaluaran menos de 100 regiones para cada uno de los cromosomas.
Los recuentos de secuencias también se pueden normalizar eliminando sistemáticamente los sesgos de muestra y ensayo usando pulido por la mediana en recuentos transformados por logaritmo. Se puede calcular una métrica para cada muestra como la media de recuentos para una región de ácido nucleico seleccionada dividida por la suma de la media de recuentos para regiones de ácido nucleico seleccionadas en un cromosoma particular y la media de recuentos para las regiones de ácido nucleico seleccionadas en un cromosoma diferente. Se puede usar una prueba de la Z estándar de proporciones para calcular la estadística de la Z:

donde pj es la proporción observada para un cromosoma dado de interés en una muestra dada j, p0 es la proporción esperada para el cromosoma de prueba dado calculado como la mediana de pj, y nj es el denominador de la métrica de la proporción. La estandarización de la estadística de la Z se puede realizar usando censura iterativa. En cada iteración, se retiran las muestras que se encuentran fuera de, por ejemplo, tres medianas de la desviación absoluta. Después de diez iteraciones, la media y la desviación estándar se calcularon usando solo las muestras sin censura. A continuación, todas las muestras se estandarizan frente a esta media y desviación estándar. Se pueden usar la prueba de Kolmogorov-Smirnov (véase Conover, Practica! Nonparametric Statistics, pp. 295-301 (John Wiley & Sons, Nueva York, NY, 1971)) y la prueba de Shapiro-Wilk (véase Royston, Applied Statistics, 31:115-124 (1982)) para someter a prueba la normalidad de la estadística de la Z de las muestras euploides.
Además de los procedimientos anteriores para reducir la variación en el ensayo, se pueden usar en combinación otras técnicas analíticas, muchas de las cuales se describen anteriormente en la presente solicitud. Por ejemplo, la
variación en el ensayo se puede reducir cuando todas las regiones de ácido nucleico seleccionadas para cada muestra se consultan en una única reacción en un único recipiente. De forma similar, se puede reducir la variación en el ensayo cuando se usa un sistema de amplificación universal. Además, la variación del ensayo se puede reducir cuando el número de ciclos de amplificación es limitado.
Determinación del contenido de ADN fetal en una muestra materna
La determinación del porcentaje de ADN fetal en una muestra materna incrementa la exactitud de los cálculos de la frecuencia para las regiones de ácido nucleico seleccionadas, ya que el conocimiento de la contribución fetal proporciona información importante sobre la presencia estadística esperada de las regiones de ácido nucleico seleccionadas de los cromosomas X e Y. Es importante, en particular, tener en cuenta el porcentaje fetal en circunstancias donde el nivel de ADN fetal en una muestra materna sea bajo, ya que se usa el porcentaje de contribución fetal para determinar la significación estadística cuantitativa en las secuencias cromosómicas de X e Y en la muestra. Es importante tener en cuenta el porcentaje fetal cuando se evalúa la presencia de una aneuploidía cromosómica de X, aneuploidía cromosómica de Y o mosaicismo de los cromosomas sexuales y/o se determina si existe contaminación de la muestra.
La contribución materna relativa del ADN materno en un alelo de interés se puede comparar con la contribución no materna en ese alelo para determinar la concentración de ADN fetal aproximada en la muestra. En aspectos preferentes, se usa la cantidad relativa de secuencias derivadas exclusivamente por vía paterna, por ejemplo, polimorfismos específicos por vía paterna en autosomas, para determinar la concentración relativa de ADN fetal en una muestra materna. Otro enfoque ejemplar para determinar el porcentaje de contribución fetal en una muestra materna es a través del análisis de fragmentos de ADN con diferentes patrones de metilación del ADN entre el ADN fetal y materno.
Debido a que las secuencias de los cromosomas X e Y típicamente no se usan para calcular el porcentaje fetal en los presentes procedimientos, la determinación de polimorfismos fetales requiere un análisis de SNP y/o mutaciones dirigido para identificar la presencia de ADN fetal en una muestra materna. En cada muestra derivada por vía materna, el ADN de un feto tendrá aproximadamente un 50 % de sus locus heredados de la madre y un 50 % de los locus heredados del padre. La determinación de los locus contribuidos al feto de la fuente paterna permite la estimación de ADN fetal en una muestra materna y, por tanto, proporciona información usada para calcular las diferencias estadísticamente significativas en frecuencias cromosómicas para los cromosomas de interés. En algunos aspectos, se puede realizar el uso de genotipado previo del padre y de la madre. Por ejemplo, los padres se pueden haber sometido a la determinación del genotipo para la identificación de marcadores de enfermedad, por ejemplo, la determinación del genotipo para trastornos tales como fibrosis quística, distrofia muscular, atrofia muscular espinal o incluso se puede determinar el estado del gen RhD. Si así fuera, se puede usar la diferencia en los polimorfismos, variantes del número de copias o mutaciones para determinar el porcentaje de contribución fetal en una muestra materna.
En un aspecto preferente alternativo, el porcentaje de ADN fetal libre de células en una muestra materna se puede cuantificar usando detección de SNP multiplexada sin conocimiento previo del genotipo materno o paterno. En este aspecto, se usan regiones de ácido nucleico polimórficas seleccionadas con uno o más SNP conocidos en cada región. En un aspecto preferente, las regiones de ácido nucleico polimórficas seleccionadas se localizan en cromosomas autosómicos que es improbable que sean aneuploides, por ejemplo, el cromosoma 6. Además, en un aspecto preferente, las regiones de ácido nucleico polimórficas seleccionadas se amplifican en una reacción en un recipiente. Cada alelo de las regiones de ácido nucleico polimórficas seleccionadas en la muestra materna se determina y se cuantifica usando, por ejemplo, secuenciación de alto rendimiento. Tras la determinación de secuencias, se identifican los locus donde los genotipos materno y fetal son diferentes, por ejemplo, el genotipo materno es homocigótico y el genotipo fetal es heterocigótico. Las secuencias heredadas por vía paterna se pueden identificar por polimorfismos detectados que se producen a una frecuencia baja pero estadísticamente pertinente. La identificación se consigue observando una alta frecuencia relativa de un alelo (>60 %) y una baja frecuencia relativa (<20 % y >0,15 %) del otro alelo para una región de ácido nucleico seleccionada particular. El uso de múltiples locus es ventajoso, en particular, ya que reduce la cantidad de variación en la medición de la abundancia de los alelos. Se usan todos o un subconjunto de los locus que cumplan este requisito para determinar la concentración fetal a través de análisis estadístico.
En un aspecto, la concentración fetal se determina sumando los alelos de baja frecuencia de dos o más locus entre sí, dividiendo entre la suma de los alelos de alta y baja frecuencia y multiplicando por dos. En otro aspecto, el porcentaje de ADN fetal libre de células se determina promediando los alelos de baja frecuencia de dos o más locus, dividiendo entre el promedio de los alelos de alta y baja frecuencia y multiplicando por dos.
Para muchos alelos, las secuencias maternas y fetales pueden ser homocigóticas e idénticas, y como esta información no distingue entre ADN materno y fetal, no es útil en la determinación del porcentaje de ADN fetal en una muestra materna. Los presentes procedimientos utilizan información alélica donde existe una diferencia entre el ADN fetal y materno (por ejemplo, un alelo fetal que contiene al menos un alelo que difiere del alelo materno) en los cálculos del porcentaje fetal. Los datos relativos a regiones alélicas que son iguales para el ADN materno y fetal no
se seleccionan, por tanto, para el análisis, o se retiran de los datos pertinentes antes de la determinación del porcentaje de a Dn fetal para no enturbiar los datos útiles. Se pueden encontrar procedimientos ejemplares para cuantificar ADN fetal en plasma materno, por ejemplo, en Chu et al., Prenat Diagn, 30:1226-29 (2010).
En un aspecto, las regiones de ácido nucleico seleccionadas se pueden excluir si la cantidad o frecuencia de la región parece ser un valor atípico debido a un error experimental o de sesgo genético idiopático dentro de una muestra particular. En otro aspecto, los ácidos nucleicos seleccionados se pueden someter a un ajuste estadístico o matemático, tal como normalización, estandarización, agolpamiento o transformación antes de la suma o el promedio, por ejemplo, como es conocido en la técnica o como se describe supra. En otro aspecto, los ácidos nucleicos seleccionados se pueden someter tanto a normalización como a exclusión de errores experimentales de los datos antes de la suma o el promedio. En un aspecto preferente, se usan 12 o más locus para el análisis. En otro aspecto preferente, se usan 24 o más locus para el análisis. En otro aspecto preferente, se usan 32 o más locus, 48 o más locus, 72 o más locus, 96 o más locus, 100 o más locus o 200 o más locus para el análisis.
En un aspecto preferente, se puede cuantificar el porcentaje de contribución fetal en una muestra materna usando detección de SNP en tándem en los alelos maternos y fetales. Se divulgan técnicas para identificar SNP en tándem en ADN extraído de una muestra materna en Mitchell et al., pat. de EE. UU. n.° 7.799.531 y los documentos USSN.
12/581.070; 12/581.083; 12/689.924 y 12/850.588. Estas referencias describen la diferenciación de los locus fetales y maternos a través de la detección de al menos un polimorfismo mononucleotídico (SNP) en tándem en una muestra materna que tiene un diferente haplotipo entre el genoma fetal y materno. La identificación y cuantificación de estos haplotipos se puede realizar directamente en la muestra materna, como se describe en las divulgaciones de Mitchell et al., y usar para determinar el porcentaje de contribución fetal en la muestra materna.
Aún en otra alternativa, se han identificado determinados genes como que tienen diferencias epigenéticas entre las copias génicas maternas y fetales, y dichos genes son locus candidatos para marcadores de ADN fetal en una muestra materna. Véase, por ejemplo, Chim, et al., PNAS USA, 102:14753-58 (2005). Estos locus, que pueden estar metilados en el ADN fetal, pero no metilados en el ADN materno (o viceversa), se pueden detectar fácilmente con alta especificidad por el uso de PCR específica de metilación (MSP) incluso cuando dichas moléculas de ADN fetal estaban presentes entre un exceso de ADN en plasma de fondo de origen materno. Se puede usar la comparación de los productos de amplificación metilados y no metilados en una muestra materna para cuantificar el porcentaje de contribución de ADN fetal a la muestra materna calculando la proporción alélica epigenética para una o más de dichas secuencias que se sabe que están reguladas de forma diferencial por la metilación en el ADN fetal en comparación con ADN materno.
Para determinar el estado de metilación de los ácidos nucleicos en una muestra materna, los ácidos nucleicos de la muestra se someten a conversión con bisulfito de las muestras y, a continuación, se someten a MSP, seguido de extensión del cebador específico de alelo. Los procedimientos convencionales para dicha conversión con bisulfito incluyen, pero no se limitan a, el uso de kits disponibles comercialmente, tal como el kit de modificación de ADN Methylamp™ (Epigentek, Brooklyn, NY). Las frecuencias alélicas y las proporciones se pueden calcular directamente y exportar a partir de los datos para determinar el porcentaje relativo de ADN fetal en la muestra materna.
Uso del porcentaje de ADN fetal libre de células en el análisis de la frecuencia de los cromosomas X e Y
Una vez que se ha calculado el porcentaje de ADN fetal libre de células, estos datos se combinan con procedimientos para la detección y cuantificación de las secuencias de los cromosomas X e Y para determinar la probabilidad de que un feto pueda ser de sexo femenino, de sexo masculino, aneuploide para el cromosoma X, aneuploide para el cromosoma Y, un mosaico del cromosoma X, un mosaico del cromosoma Y. También se puede usar en la determinación de aneuploidías maternas, incluyendo mosaicismo, o para identificar si la muestra materna que se está sometiendo a prueba está contaminada.
Por ejemplo, en una muestra materna que sea de un 10 % de ADN fetal, cada cromosoma contribuirá 1/46 de un 10 % (o de aproximadamente un 0,22 %) en un feto euploide. En un feto de sexo masculino euploide, el cromosoma Y contribuirá, por tanto, 1/46 de un 10 % (0,22 %), el cromosoma X contribuirá 1/46 de un 10 % (0,22 %) y los pares de autosomas contribuirán 2/46 (es decir, 1/23) de un 10 % (un 0,44 %, ya que existen dos de cada autosoma). Por tanto, al determinar si un feto es un feto de sexo masculino euploide, la frecuencia de las secuencias específicas del cromosoma Y en una muestra que sea un 10 % fetal debe ser de un 0,22 % y la frecuencia, por ejemplo, de las secuencias específicas del cromosoma 3 debe ser de un 0,44 %, puesto que un feto de sexo masculino tiene dos cromosomas 3. Al determinar si existe una aneuploidía cromosómica de Y (es decir, dos o más cromosomas Y), la frecuencia de las secuencias específicas del cromosoma Y sería de aproximadamente un 0,44 % para dos cromosomas Y y de aproximadamente un 0,66 % para tres cromosomas Y. Al determinar si un feto puede ser un mosaico del cromosoma Y, la frecuencia de las secuencias específicas del cromosoma Y debe ser menor y puede ser sustancialmente menor de un 0,22 % para un mosaico XX/XY, y lo mismo sería cierto para evaluar la probabilidad de la contaminación de la muestra de una muestra materna con ácidos nucleicos de un feto de sexo femenino contaminada por una muestra materna con ácidos nucleicos de un feto de sexo masculino. En un mosaico XY/XYY, la frecuencia de la secuencia específica del cromosoma Y debe estar entre un 0,22 % y un 0,44 %. En otro
ejemplo, en una muestra materna que sea de un 5 % de ADN fetal, cada cromosoma contribuirá 1/46 de un 5 % (o de aproximadamente un 0,11 %) en un feto euploide. En un feto de sexo masculino euploide, el cromosoma Y contribuirá, por tanto, 1/46 de un 5 % (0,11 %) y los pares de autosomas contribuirán 2/46 o 1/23 de un 5 % (un 0,22 %, ya que existen dos de cada autosoma).
En otro ejemplo, en un feto de sexo femenino euploide, el cromosoma X contribuirá 2/46 (es decir, 1/23) de un 10 % (un 0,44 %, ya que existen dos cromosomas X en un feto de sexo femenino euploide) y los pares de autosomas contribuirán 2/46 o 1/23 de un 10 % (un 0,44 %, ya que existen dos de cada autosoma). Por tanto, al determinar si un feto es un feto de sexo femenino euploide, la frecuencia de las secuencias específicas del cromosoma X en una muestra que sea un 10 % fetal debe ser de un 0,44 % y la frecuencia, por ejemplo, de las secuencias específicas del cromosoma 3 debe ser de un 0,44 %, puesto que un feto de sexo femenino tiene dos cromosomas 3. Al determinar si existe una aneuploidía cromosómica de X (es decir, uno, tres o más de tres cromosomas X), la frecuencia de las secuencias específicas del cromosoma X sería de aproximadamente un 0,22 % para un cromosoma X, 0,66 % para tres cromosomas X y de aproximadamente un 0,88 % para cuatro cromosomas Y. Al determinar si un feto puede ser un mosaico del cromosoma X, la frecuencia de las secuencias específicas del cromosoma X debe ser menor y puede ser sustancialmente menor de un 0,44 % para un mosaico XX/X0 o de entre un 0,44 % y un 0,66 % para un mosaico XX/XXX. En otro ejemplo, en una muestra materna que sea de un 5 % de ADN fetal, cada cromosoma contribuirá 1/46 de un 5 % (o de aproximadamente un 0,11 %) en un feto euploide. En un feto de sexo femenino euploide, el cromosoma X contribuirá, por tanto, 2/46 o 1/23 de un 5 % (0,22 %) y los pares de autosomas contribuirán 2/46 o 1/23 de un 5 % (0,22 %, ya que existen dos de cada autosoma).
La figura 8 es un diagrama de flujo simplificado de un procedimiento 800 ejemplar para realizar un análisis estadístico de acuerdo con la presente divulgación. En la etapa 801 del procedimiento 800, se consultan locus en los cromosomas X e Y, así como en al menos un autosoma. En la etapa 803, se estiman las frecuencias de los cromosomas para cada uno de los cromosomas consultados en la etapa 801. En la etapa 805, se calculan los valores de probabilidad de que el cromosoma Y no esté presente en ninguna copia, esté presente en una copia o dos o más copias, y en la etapa 807 se calcula un riesgo de aneuploidía de Y comparando los valores calculados de probabilidad con modelos matemáticos que suponen que el cromosoma Y está presente en 0, 1 o 2+ copias. De forma similar, en la etapa 809 se calculan los valores de probabilidad de que el cromosoma X esté presente en una copia, dos copias o tres o más copias, y en la etapa 811 se calcula un riesgo de una aneuploidía de X comparando los valores calculados de probabilidad con modelos matemáticos que suponen que el cromosoma X está presente en 1, 2 o 3+
La figura 9 es un diagrama de flujo simplificado de otro modo de realización de un procedimiento ejemplar 900 para realizar análisis de acuerdo con la presente invención. En el procedimiento 900, se determina la proporción fetal de la muestra materna y se calcula el riesgo de aneuploides de X e Y fetales. En la etapa 901, se consulta al menos un locus polimórfico en al menos un autosoma. En la mayoría de los modos de realización de la invención, se consultarán de varios a muchos locus polimórficos en al menos dos y, a menudo, más de dos autosomas. En la etapa 903, la proporción de ácido nucleico fetal (porcentaje fetal) de la muestra materna se calcula usando la información sobre locus polimórficos. En la etapa 905, se realiza una determinación en cuanto a si la proporción de ácido nucleico fetal de la muestra materna es adecuada para realizar análisis adicionales. En la etapa 907, se consultan locus (polimórficos o no polimórficos) en el cromosoma Y. En la mayoría de los modos de realización, la consulta de locus en el cromosoma Y y en el cromosoma X se realizará al mismo tiempo y preferentemente en el mismo recipiente en la consulta de locus en el al menos un autosoma. En la etapa 909, usando la proporción de ácido nucleico fetal calculada, se calculan los valores de probabilidad de que el cromosoma Y no esté presente en ninguna copia, esté presente en una copia o dos o más copias, y en la etapa 911 se calcula un riesgo de aneuploidía fetal de Y comparando los valores calculados de probabilidad con modelos matemáticos que suponen 0, 1 o 2+ copias del cromosoma Y. En la etapa 913, se consultan locus (polimórficos o no polimórficos) en el cromosoma X, y como se destaca previamente, la consulta de los locus en el cromosoma X y en el cromosoma Y se realizará al mismo tiempo y preferentemente en el mismo recipiente en la consulta de locus en el al menos un autosoma. En la etapa 915, usando la proporción de ácido nucleico fetal calculada, se calculan los valores de probabilidad de que el cromosoma X esté presente en una copia, dos copias o tres o más copias, y en la etapa 9171 se calcula un riesgo de aneuploidía fetal de X comparando los valores calculados de probabilidad con modelos matemáticos que suponen 1, 2 o 3+ copias del cromosoma X.
Como se menciona anteriormente, en un aspecto preferente, las reacciones para consultar tanto las regiones de ácido nucleico polimórficas seleccionadas para la determinación del porcentaje de ADN fetal en la muestra como las regiones de ácido nucleico seleccionadas de los cromosomas X e Y se realizan en una única reacción (es decir, en un único recipiente). La única reacción ayuda a minimizar el riesgo de contaminación o sesgo que se puede introducir durante las diversas etapas en el sistema de ensayo que de otro modo puede desviar los resultados cuando se utiliza el contenido de ADN fetal para ayudar a determinar la presencia o ausencia de una anomalía cromosómica. Por lo tanto, como se menciona cuando se describen los ensayos, algunos conjuntos de oligonucleótidos usados para consultar regiones de ácido nucleico seleccionadas serán específicos de polimorfismo para medir la fracción fetal y algunos conjuntos de oligonucleótidos usados para consultar regiones de ácido nucleico seleccionadas serán polimórficos o independientes de SNP para determinar el sexo fetal o aneuploidías de los cromosomas X e Y.
En otros aspectos, una región o regiones de ácido nucleico seleccionada(s) se puede(n) utilizar tanto para la determinación del porcentaje del contenido de ADN fetal como para la detección de anomalías cromosómicas de X e Y. Los alelos para regiones de ácido nucleico seleccionadas se pueden usar para determinar el contenido de ADN fetal y estas mismas regiones de ácido nucleico seleccionadas se pueden usar a continuación para detectar anomalías cromosómicas fetales ignorando la información alélica o específica de SNP. La utilización de las mismas regiones de ácido nucleico seleccionadas tanto para el contenido de ADN fetal como para la detección de anomalías cromosómicas ayuda además a minimizar cualquier sesgo debido a error experimental o contaminación.
En un modo de realización, se mide la contribución de la fuente fetal en una muestra materna independientemente del sexo fetal usando SNP autosómicos (véase, Sparks, et al., Am. J. Obstet & Gyn., 206:319.e1-9 (2012)). Los procedimientos utilizados no requieren un conocimiento previo del genotipo paterno, ya que los alelos no maternos se identifican durante los procedimientos independientemente del conocimiento de la herencia paterna. Se puede usar una estimación de probabilidad máxima usando la distribución binomial para calcular la contribución de ácido nucleico fetal estimada en varios locus informativos en cada muestra materna. Los procedimientos para el cálculo de la contribución de ácido nucleico fetal usados se describen, por ejemplo, en el documento US 20130024127, presentado el 19 de julio de 2012. Las regiones polimórficas usadas para la determinación de la contribución fetal pueden ser de los cromosomas 1-12, y preferentemente no se dirigen a los antígenos de grupo sanguíneo.
En determinados aspectos, la determinación del número de cromosomas Y en el ADN fetal se puede realizar independientemente de la determinación del número de cromosomas X en el ADN fetal.
En determinados aspectos, la estimación de la contribución fetal de los ensayos polimórficos se usa para determinar un valor de la frecuencia fetal del cromosoma Y (YFF). Por ejemplo, en determinados aspectos, la frecuencia fetal se puede definir como
PF Poly - PF ChrY
YFF = 1 - (— ~ ~
PF_Poly------- )
donde PF_Poly es la estimación del porcentaje de contribución fetal de los ensayos polimórficos y PF_ChrY es la fracción normalizada de los recuentos para el cromosoma Y, donde PF_ChrY se puede determinar calculando, por ejemplo, una proporción de la mediana de recuentos por ensayo para el cromosoma Y y la mediana de recuentos por cada ensayo para autosoma o cromosoma de referencia. Cuando el valor de YFF esté cerca de cero, el ADN fetal probablemente no comprenda un cromosoma Y. Cuando el valor de YFF esté cerca de uno, el ADN fetal probablemente comprenda un único cromosoma Y. Si el valor de YFF está cerca de dos, el ADN fetal probablemente comprenda dos copias del cromosoma Y, y así sucesivamente, para copias adicionales de Y. En determinados aspectos, el valor de YFF se usa para determinar la presencia de más de dos copias del cromosoma Y en ADN fetal de una muestra, tal como tres, cuatro o cinco copias.
En determinados aspectos, la estimación de PF_Poly se usa para determinar un valor de la frecuencia fetal del cromosoma X (XFF). En determinados aspectos, la XFF se puede definir usando diferentes definiciones para cada composición del cromosoma X potencial en una muestra. Por ejemplo, en determinados aspectos, la probabilidad fetal de X (p 1) para una muestra j se puede definir para ADN fetal que comprende un único cromosoma X como
_ ( 1-°,5*fj)*p0
P1 (l-0,5*fj)*p° +(1-p° )
donde p 1 es la probabilidad de que el ADN fetal comprenda un único cromosoma X, p0 es la proporción de referencia para un autosoma y f j es la fracción fetal de la muestra. Preferentemente, la fracción fetal usada es la PF_Poly. En determinados aspectos, la probabilidad fetal de X (£2) se puede definir para ADN fetal que comprende dos cromosomas X como
P 2=P°
donde p2 es la probabilidad de que el ADN fetal comprenda dos cromosomas X y p0 es la proporción de referencia para un cromosoma dado.
En determinados aspectos, la probabilidad fetal de X (£3) para una muestra j se puede definir para ADN fetal que comprende tres cromosomas X como
_ ( 1+°,5*fj )*p0
P 3 (1+0,5*fj)*p° +(1-p° )
donde p3 es la probabilidad de que el ADN fetal comprenda tres cromosomas X, p0 es la proporción de referencia para un cromosoma dado y f es la fracción fetal de la muestra.
En determinados aspectos, el número de cromosomas X en el ADN fetal se puede estimar comparando el valor de probabilidad normalizada para cada uno de p1, p2 y p3 que, en determinados modos de realización, se calcula dividiendo los valores de probabilidad calculados anteriormente por la suma total de los valores de probabilidad:

En determinados aspectos, la probabilidad normalizada más alta corresponde al número estimado de cromosomas X en el ADN fetal. Por ejemplo, si p2 es mayor que p1 y p3, entonces el ADN fetal comprende probablemente dos cromosomas X. En determinados aspectos, los cálculos del valor de probabilidad normalizada X se usan para determinar la presencia de más de tres copias del cromosoma X en el ADN fetal de una muestra, tal como cuatro, cinco o seis copias.
Los valores de XFF e YFF medidos se pueden ver afectados por las variaciones que se producen durante el análisis, tales como la variación del sistema de ensayo, diferencias entre los operarios u otras variables. En determinados aspectos, se puede excluir un intervalo particular de valores de diferencias fetales como fuera de un nivel de referencia de certeza con propósitos de informar de un resultado. En algunos aspectos, un valor de YFF por debajo de cero se considerará claramente indicativo de que el feto no tiene un cromosoma Y. En algunos aspectos, un valor de YFF de entre cero y uno no proporciona el nivel de certeza requerido para determinar la presencia o ausencia de un cromosoma Y. Por lo tanto, en determinados aspectos, los valores de YFF que se encuentran en un determinado intervalo, tal como de 0 a 1, se consideran fuera del intervalo de certeza, tal como los valores de diferencias fetales en un intervalo de 0,1 a 0,9, tal como de 0,2 a 0,8. Dichas determinaciones, sean concluyentes o no, se pueden usar para calcular una puntuación de riesgo en base al valor de la probabilidad como se demuestra anteriormente. Dichas puntuaciones de riesgo se pueden usar, por ejemplo, para realizar un asesoramiento sobre la atención clínica de la madre y/o feto.
Implementación informática de los procedimientos de la invención
Los procedimientos de la presente divulgación se pueden implementar por medio de un ordenador o sistema informático. Por ejemplo, los datos brutos de la "lectura" de los procedimientos, es decir, secuenciación de alto rendimiento de los productos de amplificación o hibridación a una matriz, se comunican a un ordenador o procesador, y el ordenador puede ejecutar un programa informático que, por ejemplo, "cuente" o "registre" la frecuencia de aparición de diversas secuencias de interés, compare las frecuencias, normalice las frecuencias, realice el control de calidad y/o el análisis estadístico, calcule la proporción o porcentaje fetal de una muestra materna, calcule la dosificación o frecuencia de regiones genómicas y/o cromosomas en vista del porcentaje de ácidos nucleicos fetales, determine las probabilidades de riesgo o realice otros cálculos para determinar las anomalías cromosómicas. En un modo de realización, el ordenador puede comprender un ordenador personal, pero el ordenador puede comprender cualquier tipo de máquina que incluya al menos un procesador y memoria.
La salida del componente informático comprende un informe, por ejemplo, con un valor de probabilidad de que una región genómica y/o un cromosoma (tal como, en este caso, un cromosoma X y/o Y) tenga una anomalía de dosificación. En algunos aspectos, este informe es un valor de la probabilidad de que una región o cromosoma tenga dos copias (por ejemplo, sea disómico) y un valor de la probabilidad de que una región o cromosoma tenga más copias (por ejemplo, sea trisómico) o menos copias (por ejemplo, sea monosómico). El informe puede ser un documento que se imprime, o electrónico, que se puede visualizar en un monitor y/o comunicar electrónicamente a los usuarios por medio de correo electrónico, FTP, mensaje de texto, publicar en un servidor, y similares. Aunque el procedimiento de normalización de la divulgación se describe como que está implementado como programa informático, también se puede implementar como una combinación de equipo físico y programa informático. Además, se puede implementar el programa informático para la normalización como múltiples componentes que funcionan en el mismo ordenador o en diferentes. Tanto un servidor, si está presente, como el ordenador pueden incluir componentes de equipo físico de dispositivos informáticos típicos (no mostrados), incluyendo un procesador, dispositivos de entrada (por ejemplo, teclado, dispositivo de señalización, micrófono para comandos de voz, botones, pantalla táctil, etc.) y dispositivos de salida (por ejemplo, un dispositivo de visualización, altavoces y similares). El servidor y el ordenador pueden incluir medios legibles por ordenador, por ejemplo, dispositivos de memoria y almacenamiento (por ejemplo, memoria flash, disco duro, unidad de disco óptico, unidad de disco magnético y similares) que contienen instrucciones informáticas que implementan la funcionalidad divulgada cuando se ejecutan por el procesador. El servidor y el ordenador pueden incluir además interfaces de comunicación en red alámbrica o inalámbrica para la comunicación.
EJEMPLOS
Se proponen los siguientes ejemplos para proporcionar a los expertos en la técnica una divulgación y descripción completas de cómo preparar y usar la presente invención, y no se pretende que limiten el alcance de lo que los inventores consideran su invención, ni tampoco se pretende que representen o impliquen que los experimentos a continuación sean todos o los únicos experimentos realizados. Por lo tanto, los presentes aspectos se han de considerar en todos los respectos como ilustrativos y no restrictivos.
Se han realizado esfuerzos para garantizar la exactitud con respecto a los números usados (por ejemplo, cantidades, temperatura, etc.) pero se deben tener en cuenta algunos errores experimentales y desviaciones. A menos que se indique de otro modo, las partes son partes en peso, el peso molecular es el peso molecular promedio en peso, la temperatura está en grados centígrados y la presión es igual o cercana a la atmosférica.
Ejemplo 1: Preparación de ADN para su uso en procedimientos de unión en tándem
Se obtuvo ADN genómico de sujetos de los depósitos de células de Coriell (Camden, Nueva Jersey) y se fragmentó por cizallamiento acústico (Covaris, Woburn, MA) hasta un tamaño de fragmento medio de aproximadamente 200 pb. El ADN se biotiniló usando procedimientos estándar. En resumen, el ADN fragmentado de Covaris se sometió a reparación final generando la siguiente reacción en un microtubo de 1,5 ml: 5 ug de ADN, 12 pl de tampón ligasa 10x T4 (Enzymatics, Beverly MA), polinucleótido cinasa 50 U de T4 (Enzymatics, Beverly MA) y H2O hasta 120 pl. Esto se incubó a 37 °C durante 30 minutos. El ADN se diluyó usando Tris 10 mM, EdTa 1 mM, pH 8,5 hasta la concentración final deseada de ~0,5 ng/pl.
Se dispusieron 5 pl de ADN en cada pocillo de una placa de 96 pocillos, y la placa se selló con un sellador de placa adhesivo y se centrifugó durante 10 segundos a 250 x g. A continuación, la placa se incubó a 95 °C durante 3 minutos, y se enfrió a 25 °C, y se centrifugó nuevamente durante 10 segundos a 250 x g. Se preparó una mezcla maestra de biotinilación en un microtubo de 1,5 ml hasta una concentración final de: 1x tampón TdT (Enzymatics, Beverly MA), 8 U TdT (Enzymatics, Beverly MA), CoCl2250 pM, 0,01 nmol/pl de biotina-16-duTp (Roche, Nutley NJ), y H2O hasta 1,5 ml. Se añadieron alícuotas de 15 pl de la mezcla maestra a cada pocillo de una placa de 96 pocillos, y la placa se selló con sellador de placa adhesivo. La placa se centrifugó durante 10 segundos a 250 x g y se incubó a 37 °C durante 60 minutos. Tras la incubación, la placa se centrifugó nuevamente durante 10 segundos a 250 x g, y se añadieron 7,5 pl de mezcla de precipitación (1 ng/pl de azul dextrano, NaOAc 3 mM) a cada pocillo.
La placa se selló con un sellador de placa adhesivo y se mezcló usando un vórtex de placas IKA durante 2 minutos a 3000 rpm. Se añadieron 27,5 pl de isopropanol a cada pocillo, la placa se selló con sellador de placa adhesivo y se agitó en vórtex durante 5 minutos a 3000 rpm. La placa se centrifugó durante 20 minutos a 3000 x g, el sobrenadante se decantó y la placa se invirtió y centrifugó a 10 x g durante 1 minuto sobre una toallita absorbente. La placa se secó al aire durante 5 minutos y el sedimento se resuspendió en 10 pl de Tris 10 mM, pH 8,0, EDTA 1 mM. Se creó una agrupación equimolar (40 nM cada uno) de conjuntos de los primer y segundo oligonucleótidos fijos específicos de locus a partir de los oligonucleótidos preparados como se expone anteriormente. Asimismo, se creó una agrupación equimolar separada (20 pM cada una) de oligonucleótidos puente para los procedimientos de ensayo en base a las secuencias de los locus genómicos seleccionados.
Se transfirieron 10 pg de microesferas de estreptavidina a los pocillos de una placa de 96 pocillos, y se retiró el sobrenadante. Se añadieron 60 pl de tampón de unión (Tris 100 mM, pH 8,0, EDTA 10 mM, NaCL 500 mM, formamida al 58 %, Tween-80 al 0,17 %), 10 pl de agrupación de oligonucleótidos de secuencia fijos 40 nM y 30 pl del ADN molde biotinilado preparado en el ejemplo 2 a las microesferas. La placa se selló con un sellador de placa adhesivo y se agitó en vórtex a 3000 rpm hasta que se resuspendieron las microesferas. Los oligonucleótidos se hibridaron al ADN molde por incubación a 70 °C durante 5 minutos, seguido de un enfriamiento lento a 30 °C.
La placa se dispuso en una placa magnética con varilla elevada durante 2 minutos para poner las microesferas magnéticas y a Dn asociado al lado de los pocillos. El sobrenadante se retiró por pipeteo y se reemplazó por 50 pl de tampón de unión al 60 % (v/v en agua). Las microesferas se resuspendieron por agitación en vórtex, se dispusieron nuevamente sobre el imán y se retiró el sobrenadante. Este procedimiento de lavado de microesferas se repitió una vez usando 50 ul de tampón de unión al 60 %, y se repitió dos veces más usando 50 pl de tampón de lavado (Tris 10 mM, pH 8,0, EDTA 1 mM, NaCl250 mM).
Las microesferas se resuspendieron en 37 pl de una mezcla de reacción de unión que consistía en 1X de tampón Taq ligasa (Enzymatics, Beverly MA), 10 U de Taq ligasa y una agrupación de oligonucleótidos puente 2 uM (dependiendo del formato de ensayo), y se incubaron a 37 °C durante una hora. Cuando resultó apropiado, y dependiendo del formato de ensayo, se incluyó una polimerasa termoestable sin corrección más cada dNTP 200 nM en esta mezcla. La placa se dispuso en una placa magnética con varilla elevada durante 2 minutos para poner las microesferas magnéticas y ADN asociado al lado de los pocillos. El sobrenadante se retiró por pipeteo y se reemplazó por 50 pl de tampón de lavado. Las microesferas se resuspendieron por agitación en vórtex, se dispusieron nuevamente sobre el imán y se retiró el sobrenadante. Se repitió una vez el procedimiento de lavado.
Para eluir los productos de las microesferas de estreptavidina, se añadieron 30 pl de Tris 10 mM, EDTA 1 mM, pH 8,0, a cada pocillo de la placa de 96 pocillos. La placa se selló y mezcló usando un vórtex IKA durante 2 minutos a 3000 rpm para resuspender las microesferas. La placa se incubó a 95 °C durante 1 minuto, y el sobrenadante se aspiró usando una pipeta de 8 canales. Se transfirieron 25 pl de sobrenadante de cada pocillo a una nueva placa de 96 pocillos para la amplificación universal.
Ejemplo 2: Amplificación universal de productos unidos
Los ácidos nucleicos polimerizados y/o unidos se amplificaron usando cebadores de PCR universales complementarios a las secuencias universales presentes en los primer y segundo oligonucleótidos de secuencia fijos hibridados a las regiones de ácido nucleico de interés. Se usaron 25 pl de cada una de las mezclas de reacción del ejemplo 3 en cada reacción de amplificación. Una reacción PCR universal de 50 pl que consistía en 25 pl de producto de unión eluido más 1x tampón Phusion (Finnzymes, Finlandia), betaína 1 M, cada dNTP 400 nM, 1 U de ADN polimerasa termoestable con corrección de errores de Phusion y pares de cebadores con marcas de muestra usadas para identificar de forma única muestras individuales antes de la agrupación y secuenciación. La PCR se llevó a cabo en condiciones rigurosas usando un termociclador BioRad Tetrad™.
Se agruparon 10 pl de producto de PCR universal de cada una de las muestras y el producto de PCR agrupado se purificó y cuantificó usando Quant-iT™ PicoGreen, (Invitrogen, Carlsbad, CA). Los productos de PCR purificados se secuenciaron en un solo carril de un portaobjetos en un Illumina HiSeq™ 2000. Las tandas de secuenciación típicamente dan lugar a ~100 M lecturas brutas, de las que ~85 M (85 %) se cartografían con respecto a las estructuras de ensayo esperadas. Esto se tradujo en un promedio de ~885 K lecturas/muestra en todo el experimento, y (en el caso de un experimento usando 96 locus) 9,2 K lecturas/réplicas/locus en 96 regiones de ácido nucleico seleccionadas.
Ejemplo 3: Análisis de locus polimórficos para evaluar el porcentaje de contribución fetal
Para evaluar la proporción de ácido nucleico fetal en las muestras maternas, se diseñaron ensayos frente a un conjunto de locus que contenían SNP en los cromosomas de 1 a 12, donde se usaron dos oligonucleótidos puente que diferían en una base para consultar cada SNP (véase, por ejemplo, la figura 3). Los SNP se optimizaron en cuanto a la frecuencia del alelo minoritario en el conjunto de datos HapMap 3. Duan, et al., Bioinformation, 3(3):139-41(2008); Epub 2008 Nov 9.
Los oligonucleótidos se sintetizaron por IDT (Coralville, lowa) y se agruparon entre sí para crear una única agrupación de ensayo multiplexado. Se generaron productos de PCR a partir de cada muestra de sujeto como se describe previamente. Los locus polimórficos informativos se definieron como locus donde los alelos fetales diferían de los alelos maternos. Debido a que el ensayo presenta especificidades de alelo superiores a un 99 %, los locus informativos se identificaron fácilmente cuando se midió que la proporción de alelos fetales de un locus estaba entre un 1 y un 20 %. Se estimó una probabilidad máxima usando una distribución binomial, tal como la descrita en la solicitud pendiente de tramitación US 20130024127, presentada el 19 de julio de 2012, para determinar la proporción fetal más probable en base a mediciones de varios locus informativos. Los resultados se correlacionaron bien (R2 > 0,99) con el enfoque de promedio ponderado presentado por Chu y sus compañeros (véase, Chu, et al., Prenat. Diagn., 30:1226-29 (2010)).
Ejemplo 4: Detección de anomalías en la frecuencia del cromosoma Y usando sitios no polimórficos en regiones genómicas específicas de cromosoma
En un primer modo de realización, se usaron ensayos dirigidos frente a regiones genómicas específicas en el cromosoma Y para identificar la presencia o ausencia de una anomalía en la frecuencia del cromosoma Y. El presente sistema de ensayo permitió la identificación de la presencia o ausencia de una anomalía de este tipo en el ADN de múltiples individuos usando un sistema altamente multiplexado.
Se prepararon múltiples consultas usando oligonucleótidos complementarios a o derivados del cromosoma Y (chrY) y los cromosomas 13, 18 y 21 (chr13, chr18 y chr21). Todos los oligonucleótidos usados en los formatos de unión en tándem se sintetizaron usando química convencional en fase sólida. Los oligonucleótidos del primer conjunto fijo y los oligonucleótidos puente se sintetizaron con restos fosfato en 5' para posibilitar la unión a los extremos hidroxilo en 3' de oligonucleótidos adyacentes. Se desarrollaron treinta y dos ensayos no polimórficos en chrY y se compararon con ensayos desarrollados para chr13, chr18 y chr21 (véase, por ejemplo, Sparks, et al., Prenat. Diagn., 32(1):3-9 (2012) y Sparks, et al., Am J. Obstet. Gynecol. (2012), doi:10.1016/j.ajog.2012.01.030). La fracción fetal se midió usando un conjunto de locus que contenían SNP en los cromosomas 1 a 12 o chr13, chr18 y chr21. Se calculó una estimación de la fracción fetal a partir de estos cromosomas usando la detección de polimorfismo y un algoritmo de probabilidad, como se describe en los documentos US 20120190557, presentado el 9 de diciembre de 2011, y US 20120191358, presentado el 28 de diciembre de 2011.
La fracción de recuentos para el cromosoma Y se detectó y se calculó para determinar la PF_ChrY. La PF_ChrY se dividió por la PF_Poly calculada. El resultado se restó de uno para proporcionar la YFF.
PF Poly - PF ChrY
YFF = 1 -(— ~ ’ „ , ~
v PF_Poly ------ )
J
La varianza de la YFF se calculó en este caso por muestreo con reposición de los recuentos por ensayo para Y, recuentos por ensayo para autosoma y la PF_Poly calculada. Usando modelos de 0 copias de Y, 1 copia de Y y 2+ copias de Y, se realizó un análisis bayesiano para estimar la probabilidad de tener 0, 1 o 2+ copias. Una implementación usó un modelo normal truncado para 0 copias, y modelos de distribución normal para 1 o 2+ copias, usando las desviaciones estándar realizadas en la etapa precedente. Cuando la diferencia esté cerca de 0, el feto es probablemente de sexo femenino, y cuando la diferencia esté cerca de 1, el feto es probablemente de sexo masculino. En el presente ejemplo, cuando los resultados para YFF estaban entre 0,25 y 0,65 o por encima de 3,5, se consideró que el resultado estaba fuera del intervalo declarable; sin embargo, se pueden usar otros umbrales derivados de las observaciones para establecer lo que es declarable.
Las distribuciones de este valor de diferencias fetales se calcularon usando un muestreo con reposición, y se calculó una razón de posibilidades con log 10 comparando la probabilidad de que la diferencia sea de una muestra que se ajusta a los modelos de 0, 1 o 2+ copias de chrY.
Los resultados obtenidos muestran que la exactitud de la prueba para el sexo fetal fue de un 100 %, con 745 mujeres identificadas correctamente y 797 varones identificados correctamente. Se logró una concordancia de un 99,8 % entre el cariotipado y el uso del análisis para chrY como se describe en el presente documento para la determinación del sexo fetal y la determinación de aneuploidías cromosómicas de Y. La tabla 1 muestra la concordancia del estado del cromosoma Y fetal para el conjunto de validación.

Ejemplo 5: Determinación de los genotipos XO, XX, XY, XXX, XXY, XYY y XXYY fetales
Para permitir el análisis de los cromosomas X fetales, se usaron conjuntos de oligonucleótidos de secuencia fijos y puente como se describe para consultar los locus del cromosoma X además de los conjuntos de oligonucleótidos para consultar el cromosoma Y y los cromosomas 13, 18 y 21. Los cálculos para calcular la probabilidad del número de cromosomas X presentes en el feto se añadieron a los cálculos que calculan la probabilidad del número de chrY, chr13, chr18 y chr21, así como la determinación de PF_Poly.
En este estudio, se procesó un conjunto de 432 muestras de plasma. Todos los sujetos fueron parte de un estudio enmascarado de casos y controles, donde todos los sujetos gestantes se habían sometido a pruebas invasivas. Los resultados obtenidos se compararon con el cariotipo obtenido de las pruebas invasivas. Todas las muestras de plasma de individuos gestantes cariotipados se procesaron de acuerdo con los protocolos detallados en los ejemplos 1-4, supra. Los criterios de aceptación de muestras usados para seleccionar las muestras fueron una edad materna igual o superior a 18 años, edad gestacional igual o superior a 10 semanas y embarazos únicos. Se permitieron embarazos con donantes de ovocitos tanto para donantes de ovocitos propios como no relacionados genéticamente.
En primer lugar, se calculó el riesgo de T21, T18 y T13 para cada muestra (véase, por ejemplo, los documentos US 20120190557, presentado el 9 de diciembre de 2011, y US 20120191358, presentado el 28 de diciembre de 2011). Las puntuaciones de riesgo se limitaron a un 99 % en el extremo superior y a un 0,01 % en el extremo inferior. Un riesgo superior a un 1 % clasificó a un sujeto como de alto riesgo para T21, T18 o T13. Se identificó la concordancia de la clasificación basada en la puntuación de riesgo para T21, T18 y T13 determinada usando los ensayos y el estado genético como se determina por el cariotipo.
El análisis del cromosoma Y se realizó como se describe en el ejemplo 4. Para el cromosoma X, se usó la proporción de recuentos para chrX frente a chr13, chr18 y chr21. Se derivaron modelos para 1, 2 o 3+ copias fetales de chrX usando la pérdida o ganancia esperada en base a PF_Poly medida en la muestra.
La probabilidad fetal de X (pi) para una muestra j se definió para el ADN fetal que comprendía un único cromosoma X como
_ ( 1-°,5*fj)*Po
P1 ( 1-° ,5*fj)*Po +(1-p° )
donde p 1 es la probabilidad de que el ADN fetal comprenda un único cromosoma X, p0 es la proporción de referencia para un cromosoma dado y f j es la fracción fetal de la muestra. La probabilidad fetal de X (P2) se puede definir para ADN fetal que comprende dos cromosomas X como
P 2=P°
donde p2 es la probabilidad de que el ADN fetal comprenda dos cromosomas X y p0 es la proporción de referencia para un cromosoma dado. La probabilidad fetal de X (p3) para una muestra j se puede definir para ADN fetal que comprende tres cromosomas X como
_ ( 1+0,5*fj )*p0
P 3 ( 1+°,5*fj )*Po +(1-p° )
donde p3 es la probabilidad de que el ADN fetal comprenda tres cromosomas X, p0 es la proporción de referencia para un cromosoma dado y f j es la fracción fetal de la muestra. A continuación, se estimó el número de cromosomas X en el ADN fetal comparando el valor de probabilidad normalizada para cada una de p1, P2 y p3 que se calculó dividiendo los valores de probabilidad calculados anteriormente por la suma total de los valores de probabilidad:

La probabilidad normalizada más alta correspondió al número estimado de cromosomas X en el ADN fetal.
Los criterios de aceptación de datos para la determinación de la probabilidad del número de cromosomas X presentes en el feto fueron esencialmente los mismos que para el cromosoma Y, excepto en que se usan tres cálculos para calcular las posibilidades con log 10 de una muestra del paciente que se ajusta a los modelos para 1, 2, o 3+ copias del cromosoma X fetal, respectivamente: En combinación con los cálculos realizados para el número de cromosomas Y fetales, la prueba que calculó la probabilidad del número de cromosomas X presentes en el feto usó modelos bayesianos para evaluar los datos de los cromosomas X e Y y comparar las hipótesis para los genotipos XO, XX, XY, XXX, XXY, XYY y XXYY. Si la probabilidad de un sexo fetal (masculino frente a femenino) fue < 99 %, se generó un "sin resultado" para el estado de los cromosomas X e Y fetales. XFF representa aproximadamente el número de cromosomas X fetales perdidos o ganados.
414 de las 432 muestras de plasma superaron las métricas de CC para una tasa de aprobación de muestra de un 95,8 %. Los resultados de t 21, T18 y T13 dan > 99 % de concordancia con el cariotipado y la tasa "sin resultado" para informar del estado de los cromosomas X e Y fetales fue < 1 %. El análisis de los cromosomas X e Y fetales dio una esPecificidad Para cada aneuPloidía de los cromosomas sexuales (XO, XX, XY, XXX, XXY, XYY y XXYY) de > 99 %; para las muestras, dio una sensibilidad para la aneuploidía de los cromosomas XO (que se asocia con el síndrome de Turner) de > 8° %; y el análisis de los cromosomas X e Y fetales dio exactitud para el sexo fetal (varón/mujer) > 99 %.
Usando una puntuación de riesgo de un 1 % como delimitador para la clasificación para T21 frente a no T21, se observó un 100 % de concordancia entre el cálculo de la probabilidad y el cariotipado; usando una puntuación de riesgo de un 1 % como delimitador para la clasificación para T18 frente a no T18, se observó un 100 % de concordancia entre el cálculo de probabilidad y el cariotipado; y usando una puntuación de riesgo de un 1 % como delimitador para la clasificación para T13 frente a no T13, se observó un 100 % de concordancia entre el cálculo de probabilidad y el cariotipado. Los resultados para la determinación del sexo fetal y la aneuploidía se resumen en la tabla 2:

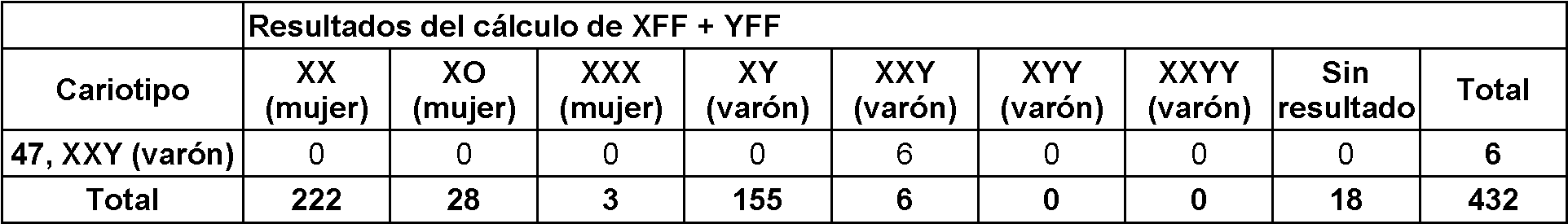
Los resultados de la prueba de XFF YFF dieron una especificidad para cada muestra con aneuploidía XO y XXX de un 99,5 % (intervalo de confianza de Wilson al 95 % 98,1-99,9) y una especificidad para cada muestra con aneuploidía XXY, XYY y XXYY de un 100 % (intervalo de confianza de Wilson al 95 % 99-100). La prueba de XFF YFF dio sensibilidad para muestras con aneuploidía XO de un 96,3 % (intervalo de confianza de Wilson al 95 % 82 -99,8), que supera los criterios de aceptación de >80 %. Los resultados de la prueba de XFF XYY dieron una exactitud para el sexo fetal de un 100 % (intervalo de confianza de Wilson al 95 % 99-100).
Aunque la presente invención se cumple por los aspectos en muchas formas diferentes, como se describe en detalle en conexión con aspectos preferentes de la invención, se entiende que la presente divulgación se ha de considerar como ejemplar de los principios de la invención y no se pretende que limite la invención a los aspectos específicos ilustrados y descritos en el presente documento. El alcance de la invención se medirá por las reivindicaciones adjuntas. El resumen y el título no se han de interpretar como limitantes del alcance de la presente invención, ya que su propósito es posibilitar que las autoridades apropiadas, así como el público en general, determinen rápidamente la naturaleza general de la invención. En las reivindicaciones que siguen, a menos que se use el término "medios", ninguno de los rasgos característicos o elementos enumerados en las mismas se debe interpretar como limitaciones de medio más función de conformidad con la sección 112.6 del 35 U.S.C.
Claims (11)
1. Un procedimiento para calcular un riesgo de aneuploidía cromosómica de X o Y fetal en una muestra materna, en el que las etapas de determinación, estimación y cálculo se realizan en un ordenador, comprendiendo el procedimiento las etapas de:
consultar uno o más locus cromosómicos en Y in vitro;
consultar uno o más locus cromosómicos en X in vitro;
consultar uno o más locus polimórficos en al menos un primer autosoma in vitro;
calcular una proporción de ácido nucleico fetal en la muestra materna analizando una frecuencia de polimorfismos en el primer autosoma;
determinar si la proporción de ácido nucleico fetal en la muestra materna es adecuada para realizar el análisis estimar una frecuencia del cromosoma I relativa para un cromosoma Y y un cromosoma X en la muestra materna en vista de la proporción de ácido nucleico fetal;
usando la proporción de ácido nucleico fetal calculada, calcular los valores de probabilidad de que el cromosoma Y fetal no esté presente en ninguna copia, esté presente en una copia o dos o más copias en la muestra materna; calcular un riesgo de aneuploidía del cromosoma Y fetal en la muestra materna comparando los valores calculados de probabilidad con un primer modelo matemático que supone ninguna copia del cromosoma Y, un segundo modelo matemático que supone una copia del cromosoma Y y un tercer modelo matemático que supone dos o más copias del cromosoma Y;
usando la proporción calculada de ácido nucleico fetal, calcular los valores de probabilidad de que el cromosoma X fetal esté presente en una copia, dos copias o tres o más copias en la muestra materna; y
calcular un riesgo de aneuploidía del cromosoma X fetal en la muestra materna comparando los valores calculados de probabilidad con un primer modelo matemático que supone una copia del cromosoma X, un segundo modelo matemático que supone dos copias del cromosoma X y un tercer modelo matemático que supone tres o más copias del cromosoma X.
2. Un procedimiento para calcular un riesgo de una aneuploidía cromosómica de X fetal en una muestra materna, en el que las etapas de estimación y cálculo se realizan en un ordenador, comprendiendo las etapas de: consultar uno o más locus cromosómicos en X in vitro;
consultar uno o más locus polimórficos en al menos un primer autosoma in vitro;
calcular una proporción de ácido nucleico fetal en la muestra materna analizando una frecuencia de locus polimórficos en el primer autosoma;
usando la proporción de ácido nucleico fetal calculada, calcular los valores de probabilidad de que el cromosoma X fetal esté presente en una copia, dos copias o más de dos copias en la muestra materna; y
calcular un riesgo de aneuploidía fetal del cromosoma X fetal en la muestra materna comparando los valores calculados de probabilidad con un primer modelo matemático que supone una copia del cromosoma X, un segundo modelo matemático que supone dos copias del cromosoma X y un tercer modelo matemático que supone tres copias del cromosoma X.
3. El procedimiento de una cualquiera de las reivindicaciones precedentes, en el que se miden al menos 96 locus polimórficos para calcular el porcentaje de proporción de ácido nucleico fetal.
4. El procedimiento de una cualquiera de la reivindicación 1 o la reivindicación 2, en el que se consultan al menos diez o más locus polimórficos.
5. El procedimiento de la reivindicación 4, en el que se calcula una proporción de ácido nucleico fetal en la muestra materna analizando la frecuencia de los polimorfismos de
(a) al menos dos autosomas; o
(b) al menos tres autosomas.
6. El procedimiento de la reivindicación 1, en el que:
(a) se consultan al menos 24 locus en cada uno del cromosoma X, el cromosoma Y y el al menos un autosoma; o (b) se consultan al menos 32 locus en cada uno del cromosoma X, el cromosoma Y y el al menos un autosoma.
7. El procedimiento de la reivindicación 6, en el que cada locus se mide al menos 100 veces.
8. El procedimiento de una cualquiera de las reivindicaciones precedentes, en el que el riesgo calculado de aneuploidía fetal se ajusta usando información extrínseca sobre el riesgo anterior.
9. El procedimiento de la reivindicación 1, en el que se calcula el riesgo de aneuploidía del cromosoma Y fetal utilizando YFF (frecuencia fetal del cromosoma Y), en el que YFF se define como
PF_Poly - PF_ChrY
YFF = 1 -PF_Poly
y en el que PF_Poly es la estimación del porcentaje de contribución fetal de los ensayos polimórficos y PF_ChrY es la fracción normalizada de los recuentos para el cromosoma Y, donde PF_ChrY se puede determinar calculando una proporción de la mediana de recuentos por ensayo para el cromosoma Y y la mediana de recuentos por cada ensayo para autosoma.
10. El procedimiento de una cualquiera de las reivindicaciones precedentes, en el que calcular los valores de probabilidad de que el cromosoma Y fetal no esté presente en ninguna copia, esté presente en una copia o dos o más copias en la muestra materna y calcular los valores de probabilidad de que el cromosoma X fetal esté presente en una copia, dos copias o tres o más copias en la muestra materna se realiza por muestreo con reposición.
11. El procedimiento de una cualquiera de las reivindicaciones precedentes, en el que el cálculo de una etapa de riesgo se realiza usando una razón de posibilidades con log 10.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US13/917,329 US11270781B2 (en) | 2011-01-25 | 2013-06-13 | Statistical analysis for non-invasive sex chromosome aneuploidy determination |
| PCT/US2014/017081 WO2014200579A1 (en) | 2013-06-13 | 2014-02-19 | Statistical analysis for non-invasive sex chromosome aneuploidy determination |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2775213T3 true ES2775213T3 (es) | 2020-07-24 |
Family
ID=52022643
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES19219999T Active ES2971972T3 (es) | 2013-06-13 | 2014-02-19 | Análisis estadístico para la determinación no invasiva de aneuploidía del cromosoma Y |
| ES14811485T Active ES2775213T3 (es) | 2013-06-13 | 2014-02-19 | Análisis estadístico para la determinación no invasiva de aneuploidías de los cromosomas sexuales |
Family Applications Before (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES19219999T Active ES2971972T3 (es) | 2013-06-13 | 2014-02-19 | Análisis estadístico para la determinación no invasiva de aneuploidía del cromosoma Y |
Country Status (7)
| Country | Link |
|---|---|
| EP (2) | EP3663414B1 (es) |
| JP (3) | JP6426162B2 (es) |
| CN (2) | CN112037860B (es) |
| AU (2) | AU2014278730B2 (es) |
| CA (1) | CA2898747C (es) |
| ES (2) | ES2971972T3 (es) |
| WO (1) | WO2014200579A1 (es) |
Families Citing this family (28)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US11270781B2 (en) | 2011-01-25 | 2022-03-08 | Ariosa Diagnostics, Inc. | Statistical analysis for non-invasive sex chromosome aneuploidy determination |
| US20130309666A1 (en) | 2013-01-25 | 2013-11-21 | Sequenom, Inc. | Methods and processes for non-invasive assessment of genetic variations |
| CN112037860B (zh) * | 2013-06-13 | 2024-02-23 | 豪夫迈·罗氏有限公司 | 用于非入侵性性染色体非整倍性确定的统计分析 |
| US10612080B2 (en) * | 2014-09-22 | 2020-04-07 | Roche Molecular Systems, Inc. | Digital PCR for non-invasive prenatal testing |
| WO2019227420A1 (zh) * | 2018-05-31 | 2019-12-05 | 深圳华大临床检验中心 | 确定男性待测样本是否存在三倍体的方法、系统和计算机可读介质 |
| JP2020146314A (ja) * | 2019-03-14 | 2020-09-17 | 株式会社三洋物産 | 遊技機 |
| JP2020146312A (ja) * | 2019-03-14 | 2020-09-17 | 株式会社三洋物産 | 遊技機 |
| JP2020146303A (ja) * | 2019-03-14 | 2020-09-17 | 株式会社三洋物産 | 遊技機 |
| JP2020146307A (ja) * | 2019-03-14 | 2020-09-17 | 株式会社三洋物産 | 遊技機 |
| JP2020146315A (ja) * | 2019-03-14 | 2020-09-17 | 株式会社三洋物産 | 遊技機 |
| JP2020146308A (ja) * | 2019-03-14 | 2020-09-17 | 株式会社三洋物産 | 遊技機 |
| JP2020146311A (ja) * | 2019-03-14 | 2020-09-17 | 株式会社三洋物産 | 遊技機 |
| JP2020146316A (ja) * | 2019-03-14 | 2020-09-17 | 株式会社三洋物産 | 遊技機 |
| JP2020146300A (ja) * | 2019-03-14 | 2020-09-17 | 株式会社三洋物産 | 遊技機 |
| JP2020146298A (ja) * | 2019-03-14 | 2020-09-17 | 株式会社三洋物産 | 遊技機 |
| JP2020146317A (ja) * | 2019-03-14 | 2020-09-17 | 株式会社三洋物産 | 遊技機 |
| JP2020146301A (ja) * | 2019-03-14 | 2020-09-17 | 株式会社三洋物産 | 遊技機 |
| JP2020146297A (ja) * | 2019-03-14 | 2020-09-17 | 株式会社三洋物産 | 遊技機 |
| JP2020146302A (ja) * | 2019-03-14 | 2020-09-17 | 株式会社三洋物産 | 遊技機 |
| JP2020146310A (ja) * | 2019-03-14 | 2020-09-17 | 株式会社三洋物産 | 遊技機 |
| JP2020146313A (ja) * | 2019-03-14 | 2020-09-17 | 株式会社三洋物産 | 遊技機 |
| JP2020146309A (ja) * | 2019-03-14 | 2020-09-17 | 株式会社三洋物産 | 遊技機 |
| JP2020146318A (ja) * | 2019-03-14 | 2020-09-17 | 株式会社三洋物産 | 遊技機 |
| JP2020146305A (ja) * | 2019-03-14 | 2020-09-17 | 株式会社三洋物産 | 遊技機 |
| JP2020146306A (ja) * | 2019-03-14 | 2020-09-17 | 株式会社三洋物産 | 遊技機 |
| JP2020146304A (ja) * | 2019-03-14 | 2020-09-17 | 株式会社三洋物産 | 遊技機 |
| CN112899358A (zh) * | 2021-01-06 | 2021-06-04 | 南京普济生物有限公司 | 一种无创产前胎儿染色体非整倍体的检测方法及其试剂盒 |
| KR102712453B1 (ko) * | 2024-03-13 | 2024-10-04 | (주)에스씨엘헬스케어 | 상염색체 및 성염색체의 이수성을 동시에 진단 가능한 비침습적 산전 검사 방법 |
Family Cites Families (90)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4437975A (en) | 1977-07-20 | 1984-03-20 | Mobil Oil Corporation | Manufacture of lube base stock oil |
| US5242794A (en) | 1984-12-13 | 1993-09-07 | Applied Biosystems, Inc. | Detection of specific sequences in nucleic acids |
| US4683202A (en) | 1985-03-28 | 1987-07-28 | Cetus Corporation | Process for amplifying nucleic acid sequences |
| US4683195A (en) | 1986-01-30 | 1987-07-28 | Cetus Corporation | Process for amplifying, detecting, and/or-cloning nucleic acid sequences |
| CA1340843C (en) | 1987-07-31 | 1999-12-07 | J. Lawrence Burg | Selective amplification of target polynucleotide sequences |
| JP2650159B2 (ja) | 1988-02-24 | 1997-09-03 | アクゾ・ノベル・エヌ・ベー | 核酸増幅方法 |
| CA1340807C (en) | 1988-02-24 | 1999-11-02 | Lawrence T. Malek | Nucleic acid amplification process |
| US4988617A (en) | 1988-03-25 | 1991-01-29 | California Institute Of Technology | Method of detecting a nucleotide change in nucleic acids |
| CA2005589C (en) | 1988-12-16 | 2001-02-06 | Akzo Nobel Nv | Self-sustained, sequence replication system |
| US5856092A (en) | 1989-02-13 | 1999-01-05 | Geneco Pty Ltd | Detection of a nucleic acid sequence or a change therein |
| US5143854A (en) | 1989-06-07 | 1992-09-01 | Affymax Technologies N.V. | Large scale photolithographic solid phase synthesis of polypeptides and receptor binding screening thereof |
| US5800992A (en) | 1989-06-07 | 1998-09-01 | Fodor; Stephen P.A. | Method of detecting nucleic acids |
| US5871928A (en) | 1989-06-07 | 1999-02-16 | Fodor; Stephen P. A. | Methods for nucleic acid analysis |
| US5547839A (en) | 1989-06-07 | 1996-08-20 | Affymax Technologies N.V. | Sequencing of surface immobilized polymers utilizing microflourescence detection |
| CA2020958C (en) | 1989-07-11 | 2005-01-11 | Daniel L. Kacian | Nucleic acid sequence amplification methods |
| US5494810A (en) | 1990-05-03 | 1996-02-27 | Cornell Research Foundation, Inc. | Thermostable ligase-mediated DNA amplifications system for the detection of genetic disease |
| DE69128545D1 (de) | 1990-08-24 | 1998-02-05 | Univ Tennessee Res Corp | Technik des genetischen fingerabdrucks mit dns-vervielfältigung |
| WO1992007095A1 (en) | 1990-10-15 | 1992-04-30 | Stratagene | Arbitrarily primed polymerase chain reaction method for fingerprinting genomes |
| US5270184A (en) | 1991-11-19 | 1993-12-14 | Becton, Dickinson And Company | Nucleic acid target generation |
| US5422252A (en) | 1993-06-04 | 1995-06-06 | Becton, Dickinson And Company | Simultaneous amplification of multiple targets |
| US6027923A (en) | 1993-07-23 | 2000-02-22 | Bio-Rad Laboratories, Inc. | Linked linear amplification of nucleic acids |
| US6401267B1 (en) | 1993-09-27 | 2002-06-11 | Radoje Drmanac | Methods and compositions for efficient nucleic acid sequencing |
| US6045996A (en) | 1993-10-26 | 2000-04-04 | Affymetrix, Inc. | Hybridization assays on oligonucleotide arrays |
| US5578832A (en) | 1994-09-02 | 1996-11-26 | Affymetrix, Inc. | Method and apparatus for imaging a sample on a device |
| US6090555A (en) | 1997-12-11 | 2000-07-18 | Affymetrix, Inc. | Scanned image alignment systems and methods |
| US5631734A (en) | 1994-02-10 | 1997-05-20 | Affymetrix, Inc. | Method and apparatus for detection of fluorescently labeled materials |
| US5648245A (en) | 1995-05-09 | 1997-07-15 | Carnegie Institution Of Washington | Method for constructing an oligonucleotide concatamer library by rolling circle replication |
| US5545531A (en) | 1995-06-07 | 1996-08-13 | Affymax Technologies N.V. | Methods for making a device for concurrently processing multiple biological chip assays |
| US6852487B1 (en) | 1996-02-09 | 2005-02-08 | Cornell Research Foundation, Inc. | Detection of nucleic acid sequence differences using the ligase detection reaction with addressable arrays |
| US6114122A (en) | 1996-03-26 | 2000-09-05 | Affymetrix, Inc. | Fluidics station with a mounting system and method of using |
| WO1997043611A1 (en) | 1996-05-16 | 1997-11-20 | Affymetrix, Inc. | Systems and methods for detection of labeled materials |
| EP1736554B1 (en) | 1996-05-29 | 2013-10-09 | Cornell Research Foundation, Inc. | Detection of nucleic acid sequence differences using coupled ligase detection and polymerase chain reactions |
| US6312892B1 (en) | 1996-07-19 | 2001-11-06 | Cornell Research Foundation, Inc. | High fidelity detection of nucleic acid differences by ligase detection reaction |
| GB9620209D0 (en) | 1996-09-27 | 1996-11-13 | Cemu Bioteknik Ab | Method of sequencing DNA |
| US6309824B1 (en) | 1997-01-16 | 2001-10-30 | Hyseq, Inc. | Methods for analyzing a target nucleic acid using immobilized heterogeneous mixtures of oligonucleotide probes |
| US6201639B1 (en) | 1998-03-20 | 2001-03-13 | James W. Overbeck | Wide field of view and high speed scanning microscopy |
| US6185030B1 (en) | 1998-03-20 | 2001-02-06 | James W. Overbeck | Wide field of view and high speed scanning microscopy |
| EP1985714B1 (en) | 1998-03-25 | 2012-02-29 | Olink AB | Method and kit for detecting a target molecule employing at least two affinity probes and rolling circle replication of padlock probes |
| US5936324A (en) | 1998-03-30 | 1999-08-10 | Genetic Microsystems Inc. | Moving magnet scanner |
| US6787308B2 (en) | 1998-07-30 | 2004-09-07 | Solexa Ltd. | Arrayed biomolecules and their use in sequencing |
| US6949370B1 (en) | 1998-10-30 | 2005-09-27 | Cornell Research Foundation, Inc. | High fidelity thermostable ligase and uses thereof |
| CA2356697C (en) | 1999-01-06 | 2010-06-22 | Cornell Research Foundation, Inc. | Accelerating identification of single nucleotide polymorphisms and alignment of clones in genomic sequencing |
| WO2000040758A2 (en) | 1999-01-06 | 2000-07-13 | Hyseq Inc. | Enhanced sequencing by hybridization using pools of probes |
| GB9901475D0 (en) | 1999-01-22 | 1999-03-17 | Pyrosequencing Ab | A method of DNA sequencing |
| US7014994B1 (en) | 1999-03-19 | 2006-03-21 | Cornell Research Foundation,Inc. | Coupled polymerase chain reaction-restriction-endonuclease digestion-ligase detection reaction process |
| US6506594B1 (en) | 1999-03-19 | 2003-01-14 | Cornell Res Foundation Inc | Detection of nucleic acid sequence differences using the ligase detection reaction with addressable arrays |
| US6218803B1 (en) | 1999-06-04 | 2001-04-17 | Genetic Microsystems, Inc. | Position sensing with variable capacitance transducers |
| US6818395B1 (en) | 1999-06-28 | 2004-11-16 | California Institute Of Technology | Methods and apparatus for analyzing polynucleotide sequences |
| WO2001023610A2 (en) | 1999-09-29 | 2001-04-05 | Solexa Ltd. | Polynucleotide sequencing |
| US7582420B2 (en) | 2001-07-12 | 2009-09-01 | Illumina, Inc. | Multiplex nucleic acid reactions |
| EP1303639A2 (en) | 2000-04-14 | 2003-04-23 | Cornell Research Foundation, Inc. | Method of designing addressable array for detection of nucleic acid sequence differences using ligase detection reaction |
| US6386749B1 (en) | 2000-06-26 | 2002-05-14 | Affymetrix, Inc. | Systems and methods for heating and mixing fluids |
| GB0021977D0 (en) | 2000-09-07 | 2000-10-25 | Pyrosequencing Ab | Method of sequencing DNA |
| DE60131903T2 (de) | 2000-10-24 | 2008-11-27 | The Board of Trustees of the Leland S. Stanford Junior University, Palo Alto | Direkte multiplex charakterisierung von genomischer dna |
| DE60137582D1 (de) | 2000-12-01 | 2009-03-19 | Cornell Res Foundation Inc | Nachweis von nukleinsäureunterschieden mit einer kombination aus endonukleasespaltungs- und ligationsreaktionen |
| US6787063B2 (en) | 2001-03-12 | 2004-09-07 | Seiko Epson Corporation | Compositions, methods for producing films, functional elements, methods for producing functional elements, methods for producing electro-optical devices and methods for producing electronic apparatus |
| JP2005535283A (ja) | 2001-11-13 | 2005-11-24 | ルビコン ゲノミクス インコーポレイテッド | ランダムフラグメント化により生成されたdna分子を用いたdna増幅および配列決定 |
| WO2005035725A2 (en) * | 2003-10-08 | 2005-04-21 | The Trustees Of Boston University | Methods for prenatal diagnosis of chromosomal abnormalities |
| WO2005044086A2 (en) * | 2003-10-30 | 2005-05-19 | Tufts-New England Medical Center | Prenatal diagnosis using cell-free fetal dna in amniotic fluid |
| CA2555704A1 (en) | 2004-02-10 | 2005-08-25 | Cornell Research Foundation, Inc. | Method for detection of promoter methylation status |
| US7622281B2 (en) | 2004-05-20 | 2009-11-24 | The Board Of Trustees Of The Leland Stanford Junior University | Methods and compositions for clonal amplification of nucleic acid |
| WO2006073504A2 (en) | 2004-08-04 | 2006-07-13 | President And Fellows Of Harvard College | Wobble sequencing |
| CN101124337B (zh) | 2004-08-24 | 2010-11-17 | 康乃尔研究基金会有限公司 | 使用内切核酸酶剪切/连接酶重新密封反应和毛细管电泳或微阵列检测核酸差异 |
| WO2007120208A2 (en) | 2005-11-14 | 2007-10-25 | President And Fellows Of Harvard College | Nanogrid rolling circle dna sequencing |
| GB0523276D0 (en) * | 2005-11-15 | 2005-12-21 | London Bridge Fertility | Chromosomal analysis by molecular karyotyping |
| EP2351858B1 (en) | 2006-02-28 | 2014-12-31 | University of Louisville Research Foundation | Detecting fetal chromosomal abnormalities using tandem single nucleotide polymorphisms |
| LT2557517T (lt) * | 2007-07-23 | 2023-01-10 | The Chinese University Of Hong Kong | Nukleino rūgščių sekos disbalanso nustatymas |
| US12180549B2 (en) * | 2007-07-23 | 2024-12-31 | The Chinese University Of Hong Kong | Diagnosing fetal chromosomal aneuploidy using genomic sequencing |
| US8518640B2 (en) | 2007-10-29 | 2013-08-27 | Complete Genomics, Inc. | Nucleic acid sequencing and process |
| EP3770255A1 (en) * | 2008-09-16 | 2021-01-27 | Sequenom, Inc. | Processes and compositions for methylation-based enrichment of fetal nucleic acid from a maternal sample useful for non-invasive prenatal diagnoses |
| US8312249B1 (en) | 2008-10-10 | 2012-11-13 | Apple Inc. | Dynamic trampoline and structured code generation in a signed code environment |
| JP5886197B2 (ja) * | 2009-08-04 | 2016-03-16 | エフ.ホフマン−ラ ロシュ アーゲーF. Hoffmann−La Roche Aktiengesellschaft | 脈管形成インヒビターに対する応答性 |
| US8563242B2 (en) * | 2009-08-11 | 2013-10-22 | The Chinese University Of Hong Kong | Method for detecting chromosomal aneuploidy |
| US20120185176A1 (en) * | 2009-09-30 | 2012-07-19 | Natera, Inc. | Methods for Non-Invasive Prenatal Ploidy Calling |
| EA034241B1 (ru) * | 2009-11-06 | 2020-01-21 | Те Чайниз Юниверсити Ов Гонконг | Способ пренатальной диагностики дисбаланса последовательности |
| US9260745B2 (en) * | 2010-01-19 | 2016-02-16 | Verinata Health, Inc. | Detecting and classifying copy number variation |
| US9323888B2 (en) * | 2010-01-19 | 2016-04-26 | Verinata Health, Inc. | Detecting and classifying copy number variation |
| US20130123120A1 (en) * | 2010-05-18 | 2013-05-16 | Natera, Inc. | Highly Multiplex PCR Methods and Compositions |
| EP2572003A4 (en) * | 2010-05-18 | 2016-01-13 | Natera Inc | METHOD FOR NONINVASIVE PRANATAL PLOIDIE ASSIGNMENT |
| US20120190557A1 (en) * | 2011-01-25 | 2012-07-26 | Aria Diagnostics, Inc. | Risk calculation for evaluation of fetal aneuploidy |
| US11031095B2 (en) * | 2010-08-06 | 2021-06-08 | Ariosa Diagnostics, Inc. | Assay systems for determination of fetal copy number variation |
| US8700338B2 (en) * | 2011-01-25 | 2014-04-15 | Ariosa Diagnosis, Inc. | Risk calculation for evaluation of fetal aneuploidy |
| CA2822439A1 (en) * | 2010-12-23 | 2012-06-28 | Sequenom, Inc. | Fetal genetic variation detection |
| AU2012209421B2 (en) * | 2011-01-25 | 2016-02-11 | F. Hoffmann-La Roche Ag | Risk calculation for evaluation of fetal aneuploidy |
| WO2012118745A1 (en) * | 2011-02-28 | 2012-09-07 | Arnold Oliphant | Assay systems for detection of aneuploidy and sex determination |
| AU2011365507A1 (en) * | 2011-04-14 | 2013-05-02 | Verinata Health, Inc. | Normalizing chromosomes for the determination and verification of common and rare chromosomal aneuploidies |
| DK2561103T3 (da) * | 2011-06-29 | 2014-10-20 | Bgi Diagnosis Co Ltd | Ikke-invasiv påvisning af føtal genetisk anomali |
| JP6161607B2 (ja) * | 2011-07-26 | 2017-07-12 | ベリナタ ヘルス インコーポレイテッド | サンプルにおける異なる異数性の有無を決定する方法 |
| CN103074416B (zh) * | 2012-06-20 | 2017-12-08 | 宁波海尔施基因科技有限公司 | 一种检测五条染色体数目异常的方法 |
| CN112037860B (zh) * | 2013-06-13 | 2024-02-23 | 豪夫迈·罗氏有限公司 | 用于非入侵性性染色体非整倍性确定的统计分析 |
-
2014
- 2014-02-19 CN CN202010934438.2A patent/CN112037860B/zh active Active
- 2014-02-19 AU AU2014278730A patent/AU2014278730B2/en not_active Ceased
- 2014-02-19 EP EP19219999.0A patent/EP3663414B1/en active Active
- 2014-02-19 WO PCT/US2014/017081 patent/WO2014200579A1/en not_active Ceased
- 2014-02-19 CA CA2898747A patent/CA2898747C/en active Active
- 2014-02-19 ES ES19219999T patent/ES2971972T3/es active Active
- 2014-02-19 JP JP2016519499A patent/JP6426162B2/ja active Active
- 2014-02-19 CN CN201480019119.1A patent/CN105074011B/zh active Active
- 2014-02-19 EP EP14811485.3A patent/EP3008215B1/en active Active
- 2014-02-19 ES ES14811485T patent/ES2775213T3/es active Active
-
2018
- 2018-10-24 JP JP2018199881A patent/JP6662983B2/ja active Active
-
2020
- 2020-02-13 JP JP2020022362A patent/JP6878631B2/ja active Active
-
2021
- 2021-01-28 AU AU2021200510A patent/AU2021200510A1/en not_active Abandoned
Also Published As
| Publication number | Publication date |
|---|---|
| AU2014278730A1 (en) | 2015-07-23 |
| JP6878631B2 (ja) | 2021-05-26 |
| EP3008215B1 (en) | 2020-01-01 |
| CN112037860B (zh) | 2024-02-23 |
| JP2020108377A (ja) | 2020-07-16 |
| WO2014200579A1 (en) | 2014-12-18 |
| CN112037860A (zh) | 2020-12-04 |
| JP6662983B2 (ja) | 2020-03-11 |
| CN105074011B (zh) | 2020-10-02 |
| CA2898747C (en) | 2021-09-21 |
| CA2898747A1 (en) | 2014-12-18 |
| JP6426162B2 (ja) | 2018-11-21 |
| JP2016526377A (ja) | 2016-09-05 |
| EP3008215A4 (en) | 2017-01-25 |
| CN105074011A (zh) | 2015-11-18 |
| EP3663414A1 (en) | 2020-06-10 |
| EP3663414B1 (en) | 2023-11-22 |
| JP2019047793A (ja) | 2019-03-28 |
| AU2021200510A1 (en) | 2021-02-25 |
| ES2971972T3 (es) | 2024-06-10 |
| EP3008215A1 (en) | 2016-04-20 |
| AU2014278730B2 (en) | 2020-12-10 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2775213T3 (es) | Análisis estadístico para la determinación no invasiva de aneuploidías de los cromosomas sexuales | |
| US20220157400A1 (en) | Statistical analysis for non-invasive sex chromosome aneuploidy determination | |
| US20210343361A1 (en) | Assay systems for determination of fetal copy number variation | |
| US10718019B2 (en) | Risk calculation for evaluation of fetal aneuploidy | |
| ES2943669T3 (es) | Cálculo de riesgo para la evaluación de aneuploidía fetal | |
| AU2019283856B2 (en) | Non-invasive fetal sex determination | |
| US20130310262A1 (en) | Noninvasive detection of robertsonian translocations |