ES2776145T3 - 3,4-Dihidro-1h-[1,8]naftiridinonas sustituidas con homopiperidinilo antibacterianas - Google Patents
3,4-Dihidro-1h-[1,8]naftiridinonas sustituidas con homopiperidinilo antibacterianas Download PDFInfo
- Publication number
- ES2776145T3 ES2776145T3 ES12744016T ES12744016T ES2776145T3 ES 2776145 T3 ES2776145 T3 ES 2776145T3 ES 12744016 T ES12744016 T ES 12744016T ES 12744016 T ES12744016 T ES 12744016T ES 2776145 T3 ES2776145 T3 ES 2776145T3
- Authority
- ES
- Spain
- Prior art keywords
- alkyl
- compound
- formula
- halo
- hydrogen
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- -1 homopiperidinyl Chemical group 0.000 title claims abstract description 32
- 230000000844 anti-bacterial effect Effects 0.000 title description 7
- KQJVDEBWHJRJBT-UHFFFAOYSA-N 3,4-dihydro-1h-1,8-naphthyridin-2-one Chemical group C1=CN=C2NC(=O)CCC2=C1 KQJVDEBWHJRJBT-UHFFFAOYSA-N 0.000 title 1
- 150000001875 compounds Chemical class 0.000 claims abstract description 156
- 239000001257 hydrogen Substances 0.000 claims abstract description 40
- 229910052739 hydrogen Inorganic materials 0.000 claims abstract description 40
- 125000003118 aryl group Chemical group 0.000 claims abstract description 24
- 125000001424 substituent group Chemical group 0.000 claims abstract description 24
- 125000001072 heteroaryl group Chemical group 0.000 claims abstract description 22
- 150000003839 salts Chemical class 0.000 claims abstract description 21
- 239000002253 acid Substances 0.000 claims abstract description 18
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims abstract description 17
- 125000001475 halogen functional group Chemical group 0.000 claims abstract description 16
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims abstract description 14
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 claims abstract description 11
- 125000004178 (C1-C4) alkyl group Chemical group 0.000 claims abstract description 9
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical group [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 claims abstract description 8
- 229910052799 carbon Inorganic materials 0.000 claims abstract description 8
- 125000004356 hydroxy functional group Chemical group O* 0.000 claims abstract description 8
- 125000000168 pyrrolyl group Chemical group 0.000 claims abstract description 7
- 125000000335 thiazolyl group Chemical group 0.000 claims abstract description 7
- 125000001544 thienyl group Chemical group 0.000 claims abstract description 7
- 125000001425 triazolyl group Chemical group 0.000 claims abstract description 7
- 125000003545 alkoxy group Chemical group 0.000 claims abstract description 6
- 125000004093 cyano group Chemical group *C#N 0.000 claims abstract description 6
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 claims abstract description 4
- 125000003226 pyrazolyl group Chemical group 0.000 claims abstract description 4
- 125000002541 furyl group Chemical group 0.000 claims abstract description 3
- 125000002883 imidazolyl group Chemical group 0.000 claims abstract description 3
- 125000001786 isothiazolyl group Chemical group 0.000 claims abstract description 3
- 125000000842 isoxazolyl group Chemical group 0.000 claims abstract description 3
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 claims abstract description 3
- 125000001715 oxadiazolyl group Chemical group 0.000 claims abstract description 3
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 claims abstract description 3
- 125000003373 pyrazinyl group Chemical group 0.000 claims abstract description 3
- 125000002098 pyridazinyl group Chemical group 0.000 claims abstract description 3
- 125000004076 pyridyl group Chemical group 0.000 claims abstract description 3
- 125000000714 pyrimidinyl group Chemical group 0.000 claims abstract description 3
- 125000002943 quinolinyl group Chemical group N1=C(C=CC2=CC=CC=C12)* 0.000 claims abstract description 3
- 125000003507 tetrahydrothiofenyl group Chemical group 0.000 claims abstract description 3
- 125000003831 tetrazolyl group Chemical group 0.000 claims abstract description 3
- 125000001113 thiadiazolyl group Chemical group 0.000 claims abstract description 3
- 102000004190 Enzymes Human genes 0.000 claims description 25
- 108090000790 Enzymes Proteins 0.000 claims description 25
- 125000000217 alkyl group Chemical group 0.000 claims description 20
- 208000035143 Bacterial infection Diseases 0.000 claims description 15
- 208000022362 bacterial infectious disease Diseases 0.000 claims description 15
- 150000002431 hydrogen Chemical group 0.000 claims description 15
- 241000894006 Bacteria Species 0.000 claims description 13
- 239000003937 drug carrier Substances 0.000 claims description 11
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 10
- 239000008194 pharmaceutical composition Substances 0.000 claims description 9
- 239000003814 drug Substances 0.000 claims description 6
- 125000006684 polyhaloalkyl group Polymers 0.000 claims description 5
- 239000003513 alkali Substances 0.000 claims description 2
- 238000004519 manufacturing process Methods 0.000 claims 2
- 239000012458 free base Substances 0.000 claims 1
- 125000001188 haloalkyl group Chemical group 0.000 claims 1
- 239000000543 intermediate Substances 0.000 description 153
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 135
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 102
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 102
- 239000000203 mixture Substances 0.000 description 76
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 66
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 64
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 58
- 239000000243 solution Substances 0.000 description 55
- 235000019439 ethyl acetate Nutrition 0.000 description 49
- 239000012044 organic layer Substances 0.000 description 48
- 239000002904 solvent Substances 0.000 description 46
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 40
- 238000002360 preparation method Methods 0.000 description 38
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 34
- 238000006243 chemical reaction Methods 0.000 description 32
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 32
- 235000019341 magnesium sulphate Nutrition 0.000 description 32
- 238000000034 method Methods 0.000 description 32
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 30
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 28
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 description 26
- 239000012071 phase Substances 0.000 description 25
- 239000011541 reaction mixture Substances 0.000 description 23
- 239000012267 brine Substances 0.000 description 20
- 239000000741 silica gel Substances 0.000 description 20
- 229910002027 silica gel Inorganic materials 0.000 description 20
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 20
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 19
- 125000005843 halogen group Chemical group 0.000 description 19
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 18
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 17
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 17
- 235000011114 ammonium hydroxide Nutrition 0.000 description 17
- 229910000027 potassium carbonate Inorganic materials 0.000 description 17
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 16
- 235000015320 potassium carbonate Nutrition 0.000 description 16
- 238000010898 silica gel chromatography Methods 0.000 description 16
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 15
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 14
- 239000003480 eluent Substances 0.000 description 14
- 239000012299 nitrogen atmosphere Substances 0.000 description 14
- 230000001580 bacterial effect Effects 0.000 description 13
- 238000010992 reflux Methods 0.000 description 13
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical compound [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 12
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 12
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 12
- 238000011282 treatment Methods 0.000 description 12
- 230000000694 effects Effects 0.000 description 11
- 239000010410 layer Substances 0.000 description 11
- 239000007787 solid Substances 0.000 description 11
- 235000019270 ammonium chloride Nutrition 0.000 description 10
- 239000007864 aqueous solution Substances 0.000 description 10
- 238000002347 injection Methods 0.000 description 10
- 239000007924 injection Substances 0.000 description 10
- 238000004262 preparative liquid chromatography Methods 0.000 description 10
- 239000000047 product Substances 0.000 description 10
- 0 C*N1CCC(C)=CCC1 Chemical compound C*N1CCC(C)=CCC1 0.000 description 9
- 239000000725 suspension Substances 0.000 description 9
- 238000012360 testing method Methods 0.000 description 9
- DTQVDTLACAAQTR-UHFFFAOYSA-N trifluoroacetic acid Substances OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 9
- MZRVEZGGRBJDDB-UHFFFAOYSA-N N-Butyllithium Chemical compound [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 description 8
- 239000007832 Na2SO4 Substances 0.000 description 8
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 8
- 238000003818 flash chromatography Methods 0.000 description 8
- 238000009472 formulation Methods 0.000 description 8
- 238000001727 in vivo Methods 0.000 description 8
- 230000002401 inhibitory effect Effects 0.000 description 8
- 229910052938 sodium sulfate Inorganic materials 0.000 description 8
- 235000011152 sodium sulphate Nutrition 0.000 description 8
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 7
- 230000008901 benefit Effects 0.000 description 7
- 239000003054 catalyst Substances 0.000 description 7
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 7
- 235000019441 ethanol Nutrition 0.000 description 7
- 239000000706 filtrate Substances 0.000 description 7
- 239000000796 flavoring agent Substances 0.000 description 7
- 235000019634 flavors Nutrition 0.000 description 7
- 208000015181 infectious disease Diseases 0.000 description 7
- 239000000843 powder Substances 0.000 description 7
- 150000003254 radicals Chemical class 0.000 description 7
- LMDZBCPBFSXMTL-UHFFFAOYSA-N 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide Chemical compound CCN=C=NCCCN(C)C LMDZBCPBFSXMTL-UHFFFAOYSA-N 0.000 description 6
- FPQQSJJWHUJYPU-UHFFFAOYSA-N 3-(dimethylamino)propyliminomethylidene-ethylazanium;chloride Chemical compound Cl.CCN=C=NCCCN(C)C FPQQSJJWHUJYPU-UHFFFAOYSA-N 0.000 description 6
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 6
- 241000699670 Mus sp. Species 0.000 description 6
- 241000191967 Staphylococcus aureus Species 0.000 description 6
- 238000003556 assay Methods 0.000 description 6
- 239000002585 base Substances 0.000 description 6
- 239000012043 crude product Substances 0.000 description 6
- 239000002552 dosage form Substances 0.000 description 6
- 235000003599 food sweetener Nutrition 0.000 description 6
- 239000003112 inhibitor Substances 0.000 description 6
- 230000005764 inhibitory process Effects 0.000 description 6
- 239000007788 liquid Substances 0.000 description 6
- 230000008018 melting Effects 0.000 description 6
- 238000002844 melting Methods 0.000 description 6
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 6
- 229910052757 nitrogen Inorganic materials 0.000 description 6
- 238000000746 purification Methods 0.000 description 6
- 239000003765 sweetening agent Substances 0.000 description 6
- 239000003826 tablet Substances 0.000 description 6
- ASOKPJOREAFHNY-UHFFFAOYSA-N 1-Hydroxybenzotriazole Chemical compound C1=CC=C2N(O)N=NC2=C1 ASOKPJOREAFHNY-UHFFFAOYSA-N 0.000 description 5
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 5
- 102100022089 Acyl-[acyl-carrier-protein] hydrolase Human genes 0.000 description 5
- 108010039731 Fatty Acid Synthases Proteins 0.000 description 5
- 239000004480 active ingredient Substances 0.000 description 5
- 230000015572 biosynthetic process Effects 0.000 description 5
- 239000003795 chemical substances by application Substances 0.000 description 5
- 201000010099 disease Diseases 0.000 description 5
- NPZTUJOABDZTLV-UHFFFAOYSA-N hydroxybenzotriazole Substances O=C1C=CC=C2NNN=C12 NPZTUJOABDZTLV-UHFFFAOYSA-N 0.000 description 5
- 239000003208 petroleum Substances 0.000 description 5
- 230000002829 reductive effect Effects 0.000 description 5
- 229920006395 saturated elastomer Polymers 0.000 description 5
- 239000003643 water by type Substances 0.000 description 5
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 4
- 238000002835 absorbance Methods 0.000 description 4
- 239000000654 additive Substances 0.000 description 4
- 239000002775 capsule Substances 0.000 description 4
- 230000008859 change Effects 0.000 description 4
- 235000014113 dietary fatty acids Nutrition 0.000 description 4
- UAOMVDZJSHZZME-UHFFFAOYSA-N diisopropylamine Substances CC(C)NC(C)C UAOMVDZJSHZZME-UHFFFAOYSA-N 0.000 description 4
- 238000010790 dilution Methods 0.000 description 4
- 239000012895 dilution Substances 0.000 description 4
- 239000000194 fatty acid Substances 0.000 description 4
- 229930195729 fatty acid Natural products 0.000 description 4
- 150000004665 fatty acids Chemical class 0.000 description 4
- 239000002054 inoculum Substances 0.000 description 4
- 230000000144 pharmacologic effect Effects 0.000 description 4
- 239000007858 starting material Substances 0.000 description 4
- PLDWAJLZAAHOGG-UHFFFAOYSA-N 1-bromo-3-methoxybenzene Chemical compound COC1=CC=CC(Br)=C1 PLDWAJLZAAHOGG-UHFFFAOYSA-N 0.000 description 3
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 3
- 101710146995 Acyl carrier protein Proteins 0.000 description 3
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 3
- 241000588724 Escherichia coli Species 0.000 description 3
- 239000007821 HATU Substances 0.000 description 3
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 3
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 3
- 238000004458 analytical method Methods 0.000 description 3
- 239000003242 anti bacterial agent Substances 0.000 description 3
- 230000037396 body weight Effects 0.000 description 3
- 239000000460 chlorine Substances 0.000 description 3
- 238000005859 coupling reaction Methods 0.000 description 3
- 231100000135 cytotoxicity Toxicity 0.000 description 3
- 230000003013 cytotoxicity Effects 0.000 description 3
- 229940079593 drug Drugs 0.000 description 3
- 125000001153 fluoro group Chemical group F* 0.000 description 3
- 238000011534 incubation Methods 0.000 description 3
- 238000001990 intravenous administration Methods 0.000 description 3
- 229910052740 iodine Inorganic materials 0.000 description 3
- 239000011630 iodine Substances 0.000 description 3
- VMGAPWLDMVPYIA-HIDZBRGKSA-N n'-amino-n-iminomethanimidamide Chemical compound N\N=C\N=N VMGAPWLDMVPYIA-HIDZBRGKSA-N 0.000 description 3
- YJVFFLUZDVXJQI-UHFFFAOYSA-L palladium(ii) acetate Chemical compound [Pd+2].CC([O-])=O.CC([O-])=O YJVFFLUZDVXJQI-UHFFFAOYSA-L 0.000 description 3
- BOLDJAUMGUJJKM-LSDHHAIUSA-N renifolin D Natural products CC(=C)[C@@H]1Cc2c(O)c(O)ccc2[C@H]1CC(=O)c3ccc(O)cc3O BOLDJAUMGUJJKM-LSDHHAIUSA-N 0.000 description 3
- 210000002966 serum Anatomy 0.000 description 3
- 239000000377 silicon dioxide Substances 0.000 description 3
- 238000001228 spectrum Methods 0.000 description 3
- 239000012086 standard solution Substances 0.000 description 3
- 239000000375 suspending agent Substances 0.000 description 3
- 208000024891 symptom Diseases 0.000 description 3
- 239000006188 syrup Substances 0.000 description 3
- 235000020357 syrup Nutrition 0.000 description 3
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 description 3
- COIOYMYWGDAQPM-UHFFFAOYSA-N tris(2-methylphenyl)phosphane Chemical compound CC1=CC=CC=C1P(C=1C(=CC=CC=1)C)C1=CC=CC=C1C COIOYMYWGDAQPM-UHFFFAOYSA-N 0.000 description 3
- IPWBFGUBXWMIPR-UHFFFAOYSA-N 1-bromo-2-fluorobenzene Chemical compound FC1=CC=CC=C1Br IPWBFGUBXWMIPR-UHFFFAOYSA-N 0.000 description 2
- FAJGKPMGLMWSKW-UHFFFAOYSA-N 1-phenyl-1,4-diazepane Chemical compound C1CCNCCN1C1=CC=CC=C1 FAJGKPMGLMWSKW-UHFFFAOYSA-N 0.000 description 2
- MIDXCONKKJTLDX-UHFFFAOYSA-N 3,5-dimethylcyclopentane-1,2-dione Chemical compound CC1CC(C)C(=O)C1=O MIDXCONKKJTLDX-UHFFFAOYSA-N 0.000 description 2
- USFZMSVCRYTOJT-UHFFFAOYSA-N Ammonium acetate Chemical compound N.CC(O)=O USFZMSVCRYTOJT-UHFFFAOYSA-N 0.000 description 2
- 239000005695 Ammonium acetate Substances 0.000 description 2
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 2
- 241000606768 Haemophilus influenzae Species 0.000 description 2
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 2
- WTKZEGDFNFYCGP-UHFFFAOYSA-N Pyrazole Chemical compound C=1C=NNC=1 WTKZEGDFNFYCGP-UHFFFAOYSA-N 0.000 description 2
- KAESVJOAVNADME-UHFFFAOYSA-N Pyrrole Chemical compound C=1C=CNC=1 KAESVJOAVNADME-UHFFFAOYSA-N 0.000 description 2
- WINXNKPZLFISPD-UHFFFAOYSA-M Saccharin sodium Chemical compound [Na+].C1=CC=C2C(=O)[N-]S(=O)(=O)C2=C1 WINXNKPZLFISPD-UHFFFAOYSA-M 0.000 description 2
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 2
- WQDUMFSSJAZKTM-UHFFFAOYSA-N Sodium methoxide Chemical compound [Na+].[O-]C WQDUMFSSJAZKTM-UHFFFAOYSA-N 0.000 description 2
- 150000007513 acids Chemical class 0.000 description 2
- 235000019257 ammonium acetate Nutrition 0.000 description 2
- 229940043376 ammonium acetate Drugs 0.000 description 2
- 230000009286 beneficial effect Effects 0.000 description 2
- 239000011230 binding agent Substances 0.000 description 2
- 230000006696 biosynthetic metabolic pathway Effects 0.000 description 2
- YNHIGQDRGKUECZ-UHFFFAOYSA-L bis(triphenylphosphine)palladium(ii) dichloride Chemical compound [Cl-].[Cl-].[Pd+2].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 YNHIGQDRGKUECZ-UHFFFAOYSA-L 0.000 description 2
- 238000002815 broth microdilution Methods 0.000 description 2
- 239000007853 buffer solution Substances 0.000 description 2
- 235000013736 caramel Nutrition 0.000 description 2
- 125000004432 carbon atom Chemical group C* 0.000 description 2
- 230000003833 cell viability Effects 0.000 description 2
- 239000003153 chemical reaction reagent Substances 0.000 description 2
- 229910052801 chlorine Inorganic materials 0.000 description 2
- 125000001309 chloro group Chemical group Cl* 0.000 description 2
- 125000004122 cyclic group Chemical group 0.000 description 2
- 229940043279 diisopropylamine Drugs 0.000 description 2
- 125000002147 dimethylamino group Chemical group [H]C([H])([H])N(*)C([H])([H])[H] 0.000 description 2
- 229910001873 dinitrogen Inorganic materials 0.000 description 2
- 208000035475 disorder Diseases 0.000 description 2
- 229940023064 escherichia coli Drugs 0.000 description 2
- XWRLQRLQUKZEEU-UHFFFAOYSA-N ethyl(hydroxy)silicon Chemical class CC[Si]O XWRLQRLQUKZEEU-UHFFFAOYSA-N 0.000 description 2
- 239000008103 glucose Substances 0.000 description 2
- 125000005842 heteroatom Chemical group 0.000 description 2
- 239000001866 hydroxypropyl methyl cellulose Substances 0.000 description 2
- 235000010979 hydroxypropyl methyl cellulose Nutrition 0.000 description 2
- 229920003088 hydroxypropyl methyl cellulose Polymers 0.000 description 2
- UFVKGYZPFZQRLF-UHFFFAOYSA-N hydroxypropyl methyl cellulose Chemical compound OC1C(O)C(OC)OC(CO)C1OC1C(O)C(O)C(OC2C(C(O)C(OC3C(C(O)C(O)C(CO)O3)O)C(CO)O2)O)C(CO)O1 UFVKGYZPFZQRLF-UHFFFAOYSA-N 0.000 description 2
- 238000001802 infusion Methods 0.000 description 2
- 239000004615 ingredient Substances 0.000 description 2
- 238000004811 liquid chromatography Methods 0.000 description 2
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 2
- 239000000314 lubricant Substances 0.000 description 2
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 2
- LRDVDFVDHDYJCO-UHFFFAOYSA-M magnesium;1-chloro-3-fluorobenzene-5-ide;bromide Chemical compound [Mg+2].[Br-].FC1=C[C-]=CC(Cl)=C1 LRDVDFVDHDYJCO-UHFFFAOYSA-M 0.000 description 2
- 238000001819 mass spectrum Methods 0.000 description 2
- 229930027945 nicotinamide-adenine dinucleotide Natural products 0.000 description 2
- BOPGDPNILDQYTO-NNYOXOHSSA-N nicotinamide-adenine dinucleotide Chemical compound C1=CCC(C(=O)N)=CN1[C@H]1[C@H](O)[C@H](O)[C@@H](COP(O)(=O)OP(O)(=O)OC[C@@H]2[C@H]([C@@H](O)[C@@H](O2)N2C3=NC=NC(N)=C3N=C2)O)O1 BOPGDPNILDQYTO-NNYOXOHSSA-N 0.000 description 2
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 2
- 239000012074 organic phase Substances 0.000 description 2
- 229910052763 palladium Inorganic materials 0.000 description 2
- NFHFRUOZVGFOOS-UHFFFAOYSA-N palladium;triphenylphosphane Chemical compound [Pd].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 NFHFRUOZVGFOOS-UHFFFAOYSA-N 0.000 description 2
- 239000002245 particle Substances 0.000 description 2
- 230000003389 potentiating effect Effects 0.000 description 2
- 239000010970 precious metal Substances 0.000 description 2
- 239000003755 preservative agent Substances 0.000 description 2
- 230000008569 process Effects 0.000 description 2
- 238000011321 prophylaxis Methods 0.000 description 2
- 102000004169 proteins and genes Human genes 0.000 description 2
- 108090000623 proteins and genes Proteins 0.000 description 2
- 230000002441 reversible effect Effects 0.000 description 2
- 235000017550 sodium carbonate Nutrition 0.000 description 2
- 229910000029 sodium carbonate Inorganic materials 0.000 description 2
- 239000012453 solvate Substances 0.000 description 2
- 239000011877 solvent mixture Substances 0.000 description 2
- 235000010356 sorbitol Nutrition 0.000 description 2
- 239000000600 sorbitol Substances 0.000 description 2
- 238000010561 standard procedure Methods 0.000 description 2
- 230000000707 stereoselective effect Effects 0.000 description 2
- 238000003756 stirring Methods 0.000 description 2
- KDYFGRWQOYBRFD-UHFFFAOYSA-N succinic acid Chemical compound OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 2
- 235000019408 sucralose Nutrition 0.000 description 2
- 230000004083 survival effect Effects 0.000 description 2
- 238000003786 synthesis reaction Methods 0.000 description 2
- 230000008719 thickening Effects 0.000 description 2
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 2
- 239000000080 wetting agent Substances 0.000 description 2
- NUFKRGBSZPCGQB-FLBSXDLDSA-N (3s)-3-amino-4-oxo-4-[[(2r)-1-oxo-1-[(2,2,4,4-tetramethylthietan-3-yl)amino]propan-2-yl]amino]butanoic acid;pentahydrate Chemical compound O.O.O.O.O.OC(=O)C[C@H](N)C(=O)N[C@H](C)C(=O)NC1C(C)(C)SC1(C)C.OC(=O)C[C@H](N)C(=O)N[C@H](C)C(=O)NC1C(C)(C)SC1(C)C NUFKRGBSZPCGQB-FLBSXDLDSA-N 0.000 description 1
- 125000006273 (C1-C3) alkyl group Chemical group 0.000 description 1
- DYLIWHYUXAJDOJ-OWOJBTEDSA-N (e)-4-(6-aminopurin-9-yl)but-2-en-1-ol Chemical compound NC1=NC=NC2=C1N=CN2C\C=C\CO DYLIWHYUXAJDOJ-OWOJBTEDSA-N 0.000 description 1
- 125000001607 1,2,3-triazol-1-yl group Chemical group [*]N1N=NC([H])=C1[H] 0.000 description 1
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 1
- OSIGJGFTADMDOB-UHFFFAOYSA-N 1-Methoxy-3-methylbenzene Chemical group COC1=CC=CC(C)=C1 OSIGJGFTADMDOB-UHFFFAOYSA-N 0.000 description 1
- TVIICJDDSIMWQT-UHFFFAOYSA-N 1-benzylazepan-4-one;hydrochloride Chemical compound Cl.C1CC(=O)CCCN1CC1=CC=CC=C1 TVIICJDDSIMWQT-UHFFFAOYSA-N 0.000 description 1
- QBELEDRHMPMKHP-UHFFFAOYSA-N 1-bromo-2-chlorobenzene Chemical compound ClC1=CC=CC=C1Br QBELEDRHMPMKHP-UHFFFAOYSA-N 0.000 description 1
- HTDQSWDEWGSAMN-UHFFFAOYSA-N 1-bromo-2-methoxybenzene Chemical compound COC1=CC=CC=C1Br HTDQSWDEWGSAMN-UHFFFAOYSA-N 0.000 description 1
- QSSXJPIWXQTSIX-UHFFFAOYSA-N 1-bromo-2-methylbenzene Chemical compound CC1=CC=CC=C1Br QSSXJPIWXQTSIX-UHFFFAOYSA-N 0.000 description 1
- JRGGUPZKKTVKOV-UHFFFAOYSA-N 1-bromo-3-chlorobenzene Chemical compound ClC1=CC=CC(Br)=C1 JRGGUPZKKTVKOV-UHFFFAOYSA-N 0.000 description 1
- NHDODQWIKUYWMW-UHFFFAOYSA-N 1-bromo-4-chlorobenzene Chemical compound ClC1=CC=C(Br)C=C1 NHDODQWIKUYWMW-UHFFFAOYSA-N 0.000 description 1
- IIZPXYDJLKNOIY-JXPKJXOSSA-N 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCC\C=C/C\C=C/C\C=C/C\C=C/CCCCC IIZPXYDJLKNOIY-JXPKJXOSSA-N 0.000 description 1
- 125000001462 1-pyrrolyl group Chemical group [*]N1C([H])=C([H])C([H])=C1[H] 0.000 description 1
- OWEGMIWEEQEYGQ-UHFFFAOYSA-N 100676-05-9 Natural products OC1C(O)C(O)C(CO)OC1OCC1C(O)C(O)C(O)C(OC2C(OC(O)C(O)C2O)CO)O1 OWEGMIWEEQEYGQ-UHFFFAOYSA-N 0.000 description 1
- QWENRTYMTSOGBR-UHFFFAOYSA-N 1H-1,2,3-Triazole Chemical compound C=1C=NNN=1 QWENRTYMTSOGBR-UHFFFAOYSA-N 0.000 description 1
- ZFRUGZMCGCYBRC-UHFFFAOYSA-N 1h-1,8-naphthyridin-2-one Chemical class C1=CC=NC2=NC(O)=CC=C21 ZFRUGZMCGCYBRC-UHFFFAOYSA-N 0.000 description 1
- MEKOFIRRDATTAG-UHFFFAOYSA-N 2,2,5,8-tetramethyl-3,4-dihydrochromen-6-ol Chemical compound C1CC(C)(C)OC2=C1C(C)=C(O)C=C2C MEKOFIRRDATTAG-UHFFFAOYSA-N 0.000 description 1
- MWVMYAWMFTVYED-UHFFFAOYSA-N 2,3,4,5-tetrahydro-1h-3-benzazepine Chemical compound C1CNCCC2=CC=CC=C21 MWVMYAWMFTVYED-UHFFFAOYSA-N 0.000 description 1
- QXISTPDUYKNPLU-UHFFFAOYSA-N 2-bromo-1,4-dimethylbenzene Chemical compound CC1=CC=C(C)C(Br)=C1 QXISTPDUYKNPLU-UHFFFAOYSA-N 0.000 description 1
- RNUBPKHSQXYYCV-UHFFFAOYSA-N 2-bromo-1-fluoro-3-methoxybenzene Chemical compound COC1=CC=CC(F)=C1Br RNUBPKHSQXYYCV-UHFFFAOYSA-N 0.000 description 1
- JIQXVIJARQLCOY-UHFFFAOYSA-N 2-bromo-4-fluoro-1-methoxybenzene Chemical compound COC1=CC=C(F)C=C1Br JIQXVIJARQLCOY-UHFFFAOYSA-N 0.000 description 1
- SFGFOJPGCOYQJK-UHFFFAOYSA-N 2-bromo-4-fluoro-1-methylbenzene Chemical compound CC1=CC=C(F)C=C1Br SFGFOJPGCOYQJK-UHFFFAOYSA-N 0.000 description 1
- TUCRZHGAIRVWTI-UHFFFAOYSA-N 2-bromothiophene Chemical compound BrC1=CC=CS1 TUCRZHGAIRVWTI-UHFFFAOYSA-N 0.000 description 1
- 125000004493 2-methylbut-1-yl group Chemical group CC(C*)CC 0.000 description 1
- 125000000175 2-thienyl group Chemical group S1C([*])=C([H])C([H])=C1[H] 0.000 description 1
- JMTMSDXUXJISAY-UHFFFAOYSA-N 2H-benzotriazol-4-ol Chemical compound OC1=CC=CC2=C1N=NN2 JMTMSDXUXJISAY-UHFFFAOYSA-N 0.000 description 1
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 description 1
- JGHLRHACKQVDPU-UHFFFAOYSA-N 4,4-difluoroazepane Chemical compound FC1(F)CCCNCC1 JGHLRHACKQVDPU-UHFFFAOYSA-N 0.000 description 1
- QJPJQTDYNZXKQF-UHFFFAOYSA-N 4-bromoanisole Chemical compound COC1=CC=C(Br)C=C1 QJPJQTDYNZXKQF-UHFFFAOYSA-N 0.000 description 1
- AHKQOPMYYACTFC-UHFFFAOYSA-N 4-phenylazepane Chemical compound C1CCNCCC1C1=CC=CC=C1 AHKQOPMYYACTFC-UHFFFAOYSA-N 0.000 description 1
- VJEOGGNIBLORIJ-UHFFFAOYSA-N 6-bromo-3,4-dihydro-1h-1,8-naphthyridin-2-one Chemical compound N1C(=O)CCC2=CC(Br)=CN=C21 VJEOGGNIBLORIJ-UHFFFAOYSA-N 0.000 description 1
- 244000215068 Acacia senegal Species 0.000 description 1
- WBZFUFAFFUEMEI-UHFFFAOYSA-M Acesulfame k Chemical compound [K+].CC1=CC(=O)[N-]S(=O)(=O)O1 WBZFUFAFFUEMEI-UHFFFAOYSA-M 0.000 description 1
- 108700037654 Acyl carrier protein (ACP) Proteins 0.000 description 1
- 102000048456 Acyl carrier protein (ACP) Human genes 0.000 description 1
- 229920001817 Agar Polymers 0.000 description 1
- 239000004377 Alitame Substances 0.000 description 1
- 235000019489 Almond oil Nutrition 0.000 description 1
- 239000005995 Aluminium silicate Substances 0.000 description 1
- 206010053555 Arthritis bacterial Diseases 0.000 description 1
- 108010011485 Aspartame Proteins 0.000 description 1
- 241000193738 Bacillus anthracis Species 0.000 description 1
- 206010066798 Bacterial tracheitis Diseases 0.000 description 1
- 241000167854 Bourreria succulenta Species 0.000 description 1
- 208000004020 Brain Abscess Diseases 0.000 description 1
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 1
- MUXOGJXSEJQIQL-UHFFFAOYSA-N C(CNCC1)C=C1c1ncc[s]1 Chemical compound C(CNCC1)C=C1c1ncc[s]1 MUXOGJXSEJQIQL-UHFFFAOYSA-N 0.000 description 1
- UDTDGZLOTIXWLG-UHFFFAOYSA-N CC(C)(C)OC(N(CCC1)CCC1(c1cc(Cl)cc(Cl)c1)O)=O Chemical compound CC(C)(C)OC(N(CCC1)CCC1(c1cc(Cl)cc(Cl)c1)O)=O UDTDGZLOTIXWLG-UHFFFAOYSA-N 0.000 description 1
- XCJLUYALDCIDHH-UHFFFAOYSA-N CC(C)(C)OC(N(CCC1)CCC1(c1cccc(C(F)(F)F)c1)O)=O Chemical compound CC(C)(C)OC(N(CCC1)CCC1(c1cccc(C(F)(F)F)c1)O)=O XCJLUYALDCIDHH-UHFFFAOYSA-N 0.000 description 1
- OVLAPXXKUAKLCV-UHFFFAOYSA-N CC(C)(C)OC(N(CCC1)CCC1[n]1nccc1)=O Chemical compound CC(C)(C)OC(N(CCC1)CCC1[n]1nccc1)=O OVLAPXXKUAKLCV-UHFFFAOYSA-N 0.000 description 1
- KIANTQVZCDHCNV-VMPITWQZSA-N CC(C)(C)[O](C(/C=C/c1cc(CCC(N2)=O)c2nc1)=O)=C Chemical compound CC(C)(C)[O](C(/C=C/c1cc(CCC(N2)=O)c2nc1)=O)=C KIANTQVZCDHCNV-VMPITWQZSA-N 0.000 description 1
- GAQKGXSLDKWFSB-UHFFFAOYSA-N CCC(CCC=C)CC1C=CCCC1 Chemical compound CCC(CCC=C)CC1C=CCCC1 GAQKGXSLDKWFSB-UHFFFAOYSA-N 0.000 description 1
- 206010007882 Cellulitis Diseases 0.000 description 1
- 206010007918 Cellulitis orbital Diseases 0.000 description 1
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 1
- 241000873310 Citrobacter sp. Species 0.000 description 1
- MROOLBNEBSIANU-UHFFFAOYSA-N ClC1=C(C=CC=C1Cl)[Mg].[Br] Chemical compound ClC1=C(C=CC=C1Cl)[Mg].[Br] MROOLBNEBSIANU-UHFFFAOYSA-N 0.000 description 1
- MIYFUKOVVNEJOI-UHFFFAOYSA-N Clc1cc(C2=CCCNCC2)cc(Cl)c1 Chemical compound Clc1cc(C2=CCCNCC2)cc(Cl)c1 MIYFUKOVVNEJOI-UHFFFAOYSA-N 0.000 description 1
- 241000694440 Colpidium aqueous Species 0.000 description 1
- 206010010741 Conjunctivitis Diseases 0.000 description 1
- 229920002261 Corn starch Polymers 0.000 description 1
- UDIPTWFVPPPURJ-UHFFFAOYSA-M Cyclamate Chemical compound [Na+].[O-]S(=O)(=O)NC1CCCCC1 UDIPTWFVPPPURJ-UHFFFAOYSA-M 0.000 description 1
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 1
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 1
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 1
- 206010011844 Dacryocystitis Diseases 0.000 description 1
- 206010012735 Diarrhoea Diseases 0.000 description 1
- 206010014568 Empyema Diseases 0.000 description 1
- 241000792859 Enema Species 0.000 description 1
- 108010060861 Enoyl-(Acyl-Carrier-Protein) Reductase (NADH) Proteins 0.000 description 1
- 206010016936 Folliculitis Diseases 0.000 description 1
- 235000016623 Fragaria vesca Nutrition 0.000 description 1
- 240000009088 Fragaria x ananassa Species 0.000 description 1
- 235000011363 Fragaria x ananassa Nutrition 0.000 description 1
- 241000589602 Francisella tularensis Species 0.000 description 1
- 239000005715 Fructose Substances 0.000 description 1
- 229930091371 Fructose Natural products 0.000 description 1
- RFSUNEUAIZKAJO-ARQDHWQXSA-N Fructose Chemical compound OC[C@H]1O[C@](O)(CO)[C@@H](O)[C@@H]1O RFSUNEUAIZKAJO-ARQDHWQXSA-N 0.000 description 1
- 230000005526 G1 to G0 transition Effects 0.000 description 1
- 244000068988 Glycine max Species 0.000 description 1
- 235000010469 Glycine max Nutrition 0.000 description 1
- 238000003747 Grignard reaction Methods 0.000 description 1
- 229920000084 Gum arabic Polymers 0.000 description 1
- 241000606790 Haemophilus Species 0.000 description 1
- 238000007341 Heck reaction Methods 0.000 description 1
- 206010021531 Impetigo Diseases 0.000 description 1
- 208000004575 Infectious Arthritis Diseases 0.000 description 1
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 1
- 229910010082 LiAlH Inorganic materials 0.000 description 1
- 241000186779 Listeria monocytogenes Species 0.000 description 1
- GUBGYTABKSRVRQ-PICCSMPSSA-N Maltose Natural products O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@@H]1O[C@@H]1[C@@H](CO)OC(O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-PICCSMPSSA-N 0.000 description 1
- 229930195725 Mannitol Natural products 0.000 description 1
- 241001465754 Metazoa Species 0.000 description 1
- 239000012359 Methanesulfonyl chloride Substances 0.000 description 1
- RJQXTJLFIWVMTO-TYNCELHUSA-N Methicillin Chemical compound COC1=CC=CC(OC)=C1C(=O)N[C@@H]1C(=O)N2[C@@H](C(O)=O)C(C)(C)S[C@@H]21 RJQXTJLFIWVMTO-TYNCELHUSA-N 0.000 description 1
- 229920000168 Microcrystalline cellulose Polymers 0.000 description 1
- 108050004114 Monellin Proteins 0.000 description 1
- 241000588655 Moraxella catarrhalis Species 0.000 description 1
- 241000699666 Mus <mouse, genus> Species 0.000 description 1
- 241000187479 Mycobacterium tuberculosis Species 0.000 description 1
- 241000588650 Neisseria meningitidis Species 0.000 description 1
- 208000000493 Orbital Cellulitis Diseases 0.000 description 1
- 206010031252 Osteomyelitis Diseases 0.000 description 1
- 206010033078 Otitis media Diseases 0.000 description 1
- 229910020667 PBr3 Inorganic materials 0.000 description 1
- 229910019142 PO4 Inorganic materials 0.000 description 1
- 241001494479 Pecora Species 0.000 description 1
- 206010034531 Perinephric abscess Diseases 0.000 description 1
- 206010057182 Periorbital cellulitis Diseases 0.000 description 1
- 241000588770 Proteus mirabilis Species 0.000 description 1
- 241000588767 Proteus vulgaris Species 0.000 description 1
- PLXBWHJQWKZRKG-UHFFFAOYSA-N Resazurin Chemical compound C1=CC(=O)C=C2OC3=CC(O)=CC=C3[N+]([O-])=C21 PLXBWHJQWKZRKG-UHFFFAOYSA-N 0.000 description 1
- 206010057190 Respiratory tract infections Diseases 0.000 description 1
- 206010038975 Retroperitoneal abscess Diseases 0.000 description 1
- 240000001890 Ribes hudsonianum Species 0.000 description 1
- 235000016954 Ribes hudsonianum Nutrition 0.000 description 1
- 235000001466 Ribes nigrum Nutrition 0.000 description 1
- 235000011034 Rubus glaucus Nutrition 0.000 description 1
- 244000235659 Rubus idaeus Species 0.000 description 1
- 235000009122 Rubus idaeus Nutrition 0.000 description 1
- 240000004808 Saccharomyces cerevisiae Species 0.000 description 1
- 241000607149 Salmonella sp. Species 0.000 description 1
- 206010040070 Septic Shock Diseases 0.000 description 1
- 241000607714 Serratia sp. Species 0.000 description 1
- 241000607758 Shigella sp. Species 0.000 description 1
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 1
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 1
- DBMJMQXJHONAFJ-UHFFFAOYSA-M Sodium laurylsulphate Chemical compound [Na+].CCCCCCCCCCCCOS([O-])(=O)=O DBMJMQXJHONAFJ-UHFFFAOYSA-M 0.000 description 1
- 241000191963 Staphylococcus epidermidis Species 0.000 description 1
- 229920002472 Starch Polymers 0.000 description 1
- 241000122973 Stenotrophomonas maltophilia Species 0.000 description 1
- UEDUENGHJMELGK-HYDKPPNVSA-N Stevioside Chemical compound O([C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@H]1O[C@]12C(=C)C[C@@]3(C1)CC[C@@H]1[C@@](C)(CCC[C@]1([C@@H]3CC2)C)C(=O)O[C@H]1[C@@H]([C@@H](O)[C@H](O)[C@@H](CO)O1)O)[C@@H]1O[C@H](CO)[C@@H](O)[C@H](O)[C@H]1O UEDUENGHJMELGK-HYDKPPNVSA-N 0.000 description 1
- 239000004376 Sucralose Substances 0.000 description 1
- 229930006000 Sucrose Natural products 0.000 description 1
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 1
- 244000299461 Theobroma cacao Species 0.000 description 1
- FZWLAAWBMGSTSO-UHFFFAOYSA-N Thiazole Chemical compound C1=CSC=N1 FZWLAAWBMGSTSO-UHFFFAOYSA-N 0.000 description 1
- 206010044248 Toxic shock syndrome Diseases 0.000 description 1
- 231100000650 Toxic shock syndrome Toxicity 0.000 description 1
- OKJPEAGHQZHRQV-UHFFFAOYSA-N Triiodomethane Natural products IC(I)I OKJPEAGHQZHRQV-UHFFFAOYSA-N 0.000 description 1
- 239000007983 Tris buffer Substances 0.000 description 1
- 241000251539 Vertebrata <Metazoa> Species 0.000 description 1
- 206010052428 Wound Diseases 0.000 description 1
- 206010048038 Wound infection Diseases 0.000 description 1
- 208000027418 Wounds and injury Diseases 0.000 description 1
- 238000002441 X-ray diffraction Methods 0.000 description 1
- TVXBFESIOXBWNM-UHFFFAOYSA-N Xylitol Natural products OCCC(O)C(O)C(O)CCO TVXBFESIOXBWNM-UHFFFAOYSA-N 0.000 description 1
- WSWOKFODOPIESP-UHFFFAOYSA-M [Br-].FC(F)(F)C1=CC=CC([Mg+])=C1 Chemical compound [Br-].FC(F)(F)C1=CC=CC([Mg+])=C1 WSWOKFODOPIESP-UHFFFAOYSA-M 0.000 description 1
- BMWRQNIRQHXSLA-UHFFFAOYSA-M [Br-].FC(F)(F)OC1=CC=CC([Mg+])=C1 Chemical compound [Br-].FC(F)(F)OC1=CC=CC([Mg+])=C1 BMWRQNIRQHXSLA-UHFFFAOYSA-M 0.000 description 1
- SISJEYQGFBUPMU-UHFFFAOYSA-M [Br-].FC(F)(F)OC1=CC=CC=C1[Mg+] Chemical compound [Br-].FC(F)(F)OC1=CC=CC=C1[Mg+] SISJEYQGFBUPMU-UHFFFAOYSA-M 0.000 description 1
- TXBIJPMTUNMCHF-UHFFFAOYSA-N [Mg]CBr Chemical compound [Mg]CBr TXBIJPMTUNMCHF-UHFFFAOYSA-N 0.000 description 1
- 210000001015 abdomen Anatomy 0.000 description 1
- 206010000269 abscess Diseases 0.000 description 1
- 238000010521 absorption reaction Methods 0.000 description 1
- 239000000205 acacia gum Substances 0.000 description 1
- 235000010489 acacia gum Nutrition 0.000 description 1
- 235000010358 acesulfame potassium Nutrition 0.000 description 1
- 229960004998 acesulfame potassium Drugs 0.000 description 1
- 239000000619 acesulfame-K Substances 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 230000009471 action Effects 0.000 description 1
- 208000013086 acute epiglottitis Diseases 0.000 description 1
- 241001148470 aerobic bacillus Species 0.000 description 1
- 239000008272 agar Substances 0.000 description 1
- 150000001298 alcohols Chemical class 0.000 description 1
- 235000019409 alitame Nutrition 0.000 description 1
- 108010009985 alitame Proteins 0.000 description 1
- 150000001336 alkenes Chemical class 0.000 description 1
- 239000008168 almond oil Substances 0.000 description 1
- 235000012211 aluminium silicate Nutrition 0.000 description 1
- VSCWAEJMTAWNJL-UHFFFAOYSA-K aluminium trichloride Chemical compound Cl[Al](Cl)Cl VSCWAEJMTAWNJL-UHFFFAOYSA-K 0.000 description 1
- 150000001412 amines Chemical class 0.000 description 1
- 125000003277 amino group Chemical group 0.000 description 1
- 239000000908 ammonium hydroxide Substances 0.000 description 1
- 230000000845 anti-microbial effect Effects 0.000 description 1
- 238000002829 antibacterial sensitivity test Methods 0.000 description 1
- 229940088710 antibiotic agent Drugs 0.000 description 1
- 238000009635 antibiotic susceptibility testing Methods 0.000 description 1
- 239000008365 aqueous carrier Substances 0.000 description 1
- 239000008346 aqueous phase Substances 0.000 description 1
- 239000008135 aqueous vehicle Substances 0.000 description 1
- 239000000605 aspartame Substances 0.000 description 1
- 235000010357 aspartame Nutrition 0.000 description 1
- IAOZJIPTCAWIRG-QWRGUYRKSA-N aspartame Chemical compound OC(=O)C[C@H](N)C(=O)N[C@H](C(=O)OC)CC1=CC=CC=C1 IAOZJIPTCAWIRG-QWRGUYRKSA-N 0.000 description 1
- 229960003438 aspartame Drugs 0.000 description 1
- 125000004429 atom Chemical group 0.000 description 1
- 229940065181 bacillus anthracis Drugs 0.000 description 1
- 208000029212 bacterial myositis Diseases 0.000 description 1
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 1
- GUBGYTABKSRVRQ-QUYVBRFLSA-N beta-maltose Chemical compound OC[C@H]1O[C@H](O[C@H]2[C@H](O)[C@@H](O)[C@H](O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@@H]1O GUBGYTABKSRVRQ-QUYVBRFLSA-N 0.000 description 1
- 238000004166 bioassay Methods 0.000 description 1
- 230000001851 biosynthetic effect Effects 0.000 description 1
- 208000010217 blepharitis Diseases 0.000 description 1
- 230000000903 blocking effect Effects 0.000 description 1
- 210000004369 blood Anatomy 0.000 description 1
- 239000008280 blood Substances 0.000 description 1
- 210000000988 bone and bone Anatomy 0.000 description 1
- 210000004556 brain Anatomy 0.000 description 1
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 1
- 229910052794 bromium Inorganic materials 0.000 description 1
- 125000001246 bromo group Chemical group Br* 0.000 description 1
- 230000005587 bubbling Effects 0.000 description 1
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 239000001506 calcium phosphate Substances 0.000 description 1
- 229910000389 calcium phosphate Inorganic materials 0.000 description 1
- 235000011010 calcium phosphates Nutrition 0.000 description 1
- GSHUZVSNIBLGMR-UHFFFAOYSA-N calcium;1,1-dioxo-1,2-benzothiazol-3-one Chemical compound [Ca].C1=CC=C2C(=O)NS(=O)(=O)C2=C1 GSHUZVSNIBLGMR-UHFFFAOYSA-N 0.000 description 1
- PFKFTWBEEFSNDU-UHFFFAOYSA-N carbonyldiimidazole Chemical compound C1=CN=CN1C(=O)N1C=CN=C1 PFKFTWBEEFSNDU-UHFFFAOYSA-N 0.000 description 1
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 1
- 230000000747 cardiac effect Effects 0.000 description 1
- 239000000969 carrier Substances 0.000 description 1
- 150000001768 cations Chemical class 0.000 description 1
- 210000003169 central nervous system Anatomy 0.000 description 1
- 238000012512 characterization method Methods 0.000 description 1
- 238000001311 chemical methods and process Methods 0.000 description 1
- 235000019693 cherries Nutrition 0.000 description 1
- 235000019219 chocolate Nutrition 0.000 description 1
- 238000004587 chromatography analysis Methods 0.000 description 1
- 235000019506 cigar Nutrition 0.000 description 1
- 229940110456 cocoa butter Drugs 0.000 description 1
- 235000019868 cocoa butter Nutrition 0.000 description 1
- 230000001332 colony forming effect Effects 0.000 description 1
- 238000004440 column chromatography Methods 0.000 description 1
- TUFGVZMNGTYAQD-UHFFFAOYSA-N comins' reagent Chemical compound FC(F)(F)S(=O)(=O)N(S(=O)(=O)C(F)(F)F)C1=CC=C(Cl)C=N1 TUFGVZMNGTYAQD-UHFFFAOYSA-N 0.000 description 1
- 239000008120 corn starch Substances 0.000 description 1
- 229940099112 cornstarch Drugs 0.000 description 1
- 230000008878 coupling Effects 0.000 description 1
- 238000010168 coupling process Methods 0.000 description 1
- KFWWCMJSYSSPSK-PAXLJYGASA-N crotonoyl-CoA Chemical compound O[C@@H]1[C@H](OP(O)(O)=O)[C@@H](COP(O)(=O)OP(O)(=O)OCC(C)(C)[C@@H](O)C(=O)NCCC(=O)NCCSC(=O)/C=C/C)O[C@H]1N1C2=NC=NC(N)=C2N=C1 KFWWCMJSYSSPSK-PAXLJYGASA-N 0.000 description 1
- 239000013078 crystal Substances 0.000 description 1
- 238000009109 curative therapy Methods 0.000 description 1
- 239000000625 cyclamic acid and its Na and Ca salt Substances 0.000 description 1
- 125000000753 cycloalkyl group Chemical group 0.000 description 1
- 238000002784 cytotoxicity assay Methods 0.000 description 1
- 231100000263 cytotoxicity test Toxicity 0.000 description 1
- 230000006378 damage Effects 0.000 description 1
- 239000008367 deionised water Substances 0.000 description 1
- 229910021641 deionized water Inorganic materials 0.000 description 1
- 230000001934 delay Effects 0.000 description 1
- 230000001419 dependent effect Effects 0.000 description 1
- 230000001627 detrimental effect Effects 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- 125000001028 difluoromethyl group Chemical group [H]C(F)(F)* 0.000 description 1
- BEPAFCGSDWSTEL-UHFFFAOYSA-N dimethyl malonate Chemical compound COC(=O)CC(=O)OC BEPAFCGSDWSTEL-UHFFFAOYSA-N 0.000 description 1
- 239000007884 disintegrant Substances 0.000 description 1
- 239000002270 dispersing agent Substances 0.000 description 1
- 239000006185 dispersion Substances 0.000 description 1
- 238000000132 electrospray ionisation Methods 0.000 description 1
- 239000003995 emulsifying agent Substances 0.000 description 1
- 239000000839 emulsion Substances 0.000 description 1
- 206010014665 endocarditis Diseases 0.000 description 1
- 206010014801 endophthalmitis Diseases 0.000 description 1
- 239000007920 enema Substances 0.000 description 1
- 229940079360 enema for constipation Drugs 0.000 description 1
- 201000010063 epididymitis Diseases 0.000 description 1
- 208000001606 epiglottitis Diseases 0.000 description 1
- 150000002148 esters Chemical class 0.000 description 1
- BEFDCLMNVWHSGT-UHFFFAOYSA-N ethenylcyclopentane Chemical compound C=CC1CCCC1 BEFDCLMNVWHSGT-UHFFFAOYSA-N 0.000 description 1
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 1
- 238000011156 evaluation Methods 0.000 description 1
- 230000005284 excitation Effects 0.000 description 1
- 239000003925 fat Substances 0.000 description 1
- 235000019197 fats Nutrition 0.000 description 1
- KTWOOEGAPBSYNW-UHFFFAOYSA-N ferrocene Chemical compound [Fe+2].C=1C=C[CH-]C=1.C=1C=C[CH-]C=1 KTWOOEGAPBSYNW-UHFFFAOYSA-N 0.000 description 1
- 239000000945 filler Substances 0.000 description 1
- 239000013020 final formulation Substances 0.000 description 1
- 229910052731 fluorine Inorganic materials 0.000 description 1
- 238000001640 fractional crystallisation Methods 0.000 description 1
- 229940118764 francisella tularensis Drugs 0.000 description 1
- 229940076988 freshmint Drugs 0.000 description 1
- 239000007789 gas Substances 0.000 description 1
- 230000002496 gastric effect Effects 0.000 description 1
- 239000007903 gelatin capsule Substances 0.000 description 1
- 150000002303 glucose derivatives Chemical class 0.000 description 1
- 125000005456 glyceride group Chemical group 0.000 description 1
- 150000002334 glycols Chemical class 0.000 description 1
- 230000009036 growth inhibition Effects 0.000 description 1
- 229940047650 haemophilus influenzae Drugs 0.000 description 1
- 230000036541 health Effects 0.000 description 1
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 238000004128 high performance liquid chromatography Methods 0.000 description 1
- 235000012907 honey Nutrition 0.000 description 1
- 238000000338 in vitro Methods 0.000 description 1
- 239000012442 inert solvent Substances 0.000 description 1
- 230000002458 infectious effect Effects 0.000 description 1
- 201000007119 infective endocarditis Diseases 0.000 description 1
- 208000014674 injury Diseases 0.000 description 1
- 238000011081 inoculation Methods 0.000 description 1
- 238000010255 intramuscular injection Methods 0.000 description 1
- 239000007927 intramuscular injection Substances 0.000 description 1
- 238000007912 intraperitoneal administration Methods 0.000 description 1
- 238000010253 intravenous injection Methods 0.000 description 1
- 125000002346 iodo group Chemical group I* 0.000 description 1
- INQOMBQAUSQDDS-UHFFFAOYSA-N iodomethane Chemical compound IC INQOMBQAUSQDDS-UHFFFAOYSA-N 0.000 description 1
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- NLYAJNPCOHFWQQ-UHFFFAOYSA-N kaolin Chemical compound O.O.O=[Al]O[Si](=O)O[Si](=O)O[Al]=O NLYAJNPCOHFWQQ-UHFFFAOYSA-N 0.000 description 1
- 206010023332 keratitis Diseases 0.000 description 1
- 239000008101 lactose Substances 0.000 description 1
- 235000010445 lecithin Nutrition 0.000 description 1
- 239000000787 lecithin Substances 0.000 description 1
- 229940067606 lecithin Drugs 0.000 description 1
- 231100001231 less toxic Toxicity 0.000 description 1
- 239000003446 ligand Substances 0.000 description 1
- 229960003907 linezolid Drugs 0.000 description 1
- TYZROVQLWOKYKF-ZDUSSCGKSA-N linezolid Chemical compound O=C1O[C@@H](CNC(=O)C)CN1C(C=C1F)=CC=C1N1CCOCC1 TYZROVQLWOKYKF-ZDUSSCGKSA-N 0.000 description 1
- 229940115931 listeria monocytogenes Drugs 0.000 description 1
- 239000007937 lozenge Substances 0.000 description 1
- 201000003453 lung abscess Diseases 0.000 description 1
- 230000002101 lytic effect Effects 0.000 description 1
- 239000011777 magnesium Substances 0.000 description 1
- 235000019359 magnesium stearate Nutrition 0.000 description 1
- IWCVDCOJSPWGRW-UHFFFAOYSA-M magnesium;benzene;chloride Chemical compound [Mg+2].[Cl-].C1=CC=[C-]C=C1 IWCVDCOJSPWGRW-UHFFFAOYSA-M 0.000 description 1
- ZMPYQGQHGLLBQI-UHFFFAOYSA-M magnesium;chlorobenzene;bromide Chemical compound [Mg+2].[Br-].ClC1=CC=C[C-]=C1 ZMPYQGQHGLLBQI-UHFFFAOYSA-M 0.000 description 1
- 230000014759 maintenance of location Effects 0.000 description 1
- 239000000594 mannitol Substances 0.000 description 1
- 235000010355 mannitol Nutrition 0.000 description 1
- 230000000873 masking effect Effects 0.000 description 1
- 238000005259 measurement Methods 0.000 description 1
- 238000002483 medication Methods 0.000 description 1
- HEBKCHPVOIAQTA-UHFFFAOYSA-N meso ribitol Natural products OCC(O)C(O)C(O)CO HEBKCHPVOIAQTA-UHFFFAOYSA-N 0.000 description 1
- 229910052751 metal Inorganic materials 0.000 description 1
- 239000002184 metal Substances 0.000 description 1
- 229940098779 methanesulfonic acid Drugs 0.000 description 1
- QARBMVPHQWIHKH-UHFFFAOYSA-N methanesulfonyl chloride Chemical compound CS(Cl)(=O)=O QARBMVPHQWIHKH-UHFFFAOYSA-N 0.000 description 1
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 1
- 229920000609 methyl cellulose Polymers 0.000 description 1
- 235000010270 methyl p-hydroxybenzoate Nutrition 0.000 description 1
- 239000001923 methylcellulose Substances 0.000 description 1
- 235000010981 methylcellulose Nutrition 0.000 description 1
- 125000000325 methylidene group Chemical group [H]C([H])=* 0.000 description 1
- 229960003085 meticillin Drugs 0.000 description 1
- 239000008108 microcrystalline cellulose Substances 0.000 description 1
- 235000019813 microcrystalline cellulose Nutrition 0.000 description 1
- 229940016286 microcrystalline cellulose Drugs 0.000 description 1
- 239000002480 mineral oil Substances 0.000 description 1
- 235000010446 mineral oil Nutrition 0.000 description 1
- 150000007522 mineralic acids Chemical class 0.000 description 1
- 125000000896 monocarboxylic acid group Chemical group 0.000 description 1
- 125000002950 monocyclic group Chemical group 0.000 description 1
- VHSIAYLBCLUAFT-UHFFFAOYSA-N n-[3-acetyl-6-(4-chlorophenyl)-7-(2,4-dichlorophenyl)-1-methyl-2-oxo-1,8-naphthyridin-4-yl]acetamide Chemical class C=1C=C(Cl)C=CC=1C=1C=C2C(NC(=O)C)=C(C(C)=O)C(=O)N(C)C2=NC=1C1=CC=C(Cl)C=C1Cl VHSIAYLBCLUAFT-UHFFFAOYSA-N 0.000 description 1
- 239000006199 nebulizer Substances 0.000 description 1
- 230000018791 negative regulation of catalytic activity Effects 0.000 description 1
- 125000004433 nitrogen atom Chemical group N* 0.000 description 1
- 235000013615 non-nutritive sweetener Nutrition 0.000 description 1
- 231100000252 nontoxic Toxicity 0.000 description 1
- 230000003000 nontoxic effect Effects 0.000 description 1
- 239000003921 oil Substances 0.000 description 1
- 235000019198 oils Nutrition 0.000 description 1
- 239000002674 ointment Substances 0.000 description 1
- 239000006186 oral dosage form Substances 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- 230000008520 organization Effects 0.000 description 1
- 125000002734 organomagnesium group Chemical group 0.000 description 1
- 238000010653 organometallic reaction Methods 0.000 description 1
- MUJIDPITZJWBSW-UHFFFAOYSA-N palladium(2+) Chemical compound [Pd+2] MUJIDPITZJWBSW-UHFFFAOYSA-N 0.000 description 1
- NXJCBFBQEVOTOW-UHFFFAOYSA-L palladium(2+);dihydroxide Chemical compound O[Pd]O NXJCBFBQEVOTOW-UHFFFAOYSA-L 0.000 description 1
- LXNAVEXFUKBNMK-UHFFFAOYSA-N palladium(II) acetate Substances [Pd].CC(O)=O.CC(O)=O LXNAVEXFUKBNMK-UHFFFAOYSA-N 0.000 description 1
- 238000002638 palliative care Methods 0.000 description 1
- 238000007911 parenteral administration Methods 0.000 description 1
- 244000052769 pathogen Species 0.000 description 1
- 230000037361 pathway Effects 0.000 description 1
- 239000003961 penetration enhancing agent Substances 0.000 description 1
- 125000001147 pentyl group Chemical group C(CCCC)* 0.000 description 1
- 239000000546 pharmaceutical excipient Substances 0.000 description 1
- ANRQGKOBLBYXFM-UHFFFAOYSA-M phenylmagnesium bromide Chemical compound Br[Mg]C1=CC=CC=C1 ANRQGKOBLBYXFM-UHFFFAOYSA-M 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 1
- 239000010452 phosphate Substances 0.000 description 1
- KGHBSRNCHAEYEG-UHFFFAOYSA-N phosphoric acid Chemical compound OP(O)(O)=O.OP(O)(O)=O.OP(O)(O)=O.OP(O)(O)=O KGHBSRNCHAEYEG-UHFFFAOYSA-N 0.000 description 1
- IPNPIHIZVLFAFP-UHFFFAOYSA-N phosphorus tribromide Chemical compound BrP(Br)Br IPNPIHIZVLFAFP-UHFFFAOYSA-N 0.000 description 1
- 239000006187 pill Substances 0.000 description 1
- 229920001184 polypeptide Polymers 0.000 description 1
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 1
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 1
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 1
- 235000011181 potassium carbonates Nutrition 0.000 description 1
- 229920001592 potato starch Polymers 0.000 description 1
- 239000002244 precipitate Substances 0.000 description 1
- 238000002953 preparative HPLC Methods 0.000 description 1
- 230000002335 preservative effect Effects 0.000 description 1
- 108090000765 processed proteins & peptides Proteins 0.000 description 1
- 102000004196 processed proteins & peptides Human genes 0.000 description 1
- 238000004393 prognosis Methods 0.000 description 1
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 235000010232 propyl p-hydroxybenzoate Nutrition 0.000 description 1
- QELSKZZBTMNZEB-UHFFFAOYSA-N propylparaben Chemical class CCCOC(=O)C1=CC=C(O)C=C1 QELSKZZBTMNZEB-UHFFFAOYSA-N 0.000 description 1
- 229940007042 proteus vulgaris Drugs 0.000 description 1
- JUJWROOIHBZHMG-UHFFFAOYSA-O pyridinium Chemical compound C1=CC=[NH+]C=C1 JUJWROOIHBZHMG-UHFFFAOYSA-O 0.000 description 1
- 238000003908 quality control method Methods 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 210000002345 respiratory system Anatomy 0.000 description 1
- 208000020029 respiratory tract infectious disease Diseases 0.000 description 1
- 230000004044 response Effects 0.000 description 1
- 239000012465 retentate Substances 0.000 description 1
- 150000004671 saturated fatty acids Chemical class 0.000 description 1
- 235000003441 saturated fatty acids Nutrition 0.000 description 1
- 229930195734 saturated hydrocarbon Natural products 0.000 description 1
- 238000009738 saturating Methods 0.000 description 1
- 201000009881 secretory diarrhea Diseases 0.000 description 1
- 238000010956 selective crystallization Methods 0.000 description 1
- 230000035945 sensitivity Effects 0.000 description 1
- 201000001223 septic arthritis Diseases 0.000 description 1
- 238000013207 serial dilution Methods 0.000 description 1
- 235000017557 sodium bicarbonate Nutrition 0.000 description 1
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 1
- 229910000033 sodium borohydride Inorganic materials 0.000 description 1
- 239000012279 sodium borohydride Substances 0.000 description 1
- 229960001462 sodium cyclamate Drugs 0.000 description 1
- 239000012312 sodium hydride Substances 0.000 description 1
- 229910000104 sodium hydride Inorganic materials 0.000 description 1
- 235000019333 sodium laurylsulphate Nutrition 0.000 description 1
- 229940079832 sodium starch glycolate Drugs 0.000 description 1
- 239000008109 sodium starch glycolate Substances 0.000 description 1
- 229920003109 sodium starch glycolate Polymers 0.000 description 1
- 239000007909 solid dosage form Substances 0.000 description 1
- 230000007928 solubilization Effects 0.000 description 1
- 238000005063 solubilization Methods 0.000 description 1
- 235000010199 sorbic acid Nutrition 0.000 description 1
- 239000004334 sorbic acid Substances 0.000 description 1
- 229940075582 sorbic acid Drugs 0.000 description 1
- 241000894007 species Species 0.000 description 1
- 201000005789 splenic abscess Diseases 0.000 description 1
- 239000003381 stabilizer Substances 0.000 description 1
- 230000000087 stabilizing effect Effects 0.000 description 1
- 235000019698 starch Nutrition 0.000 description 1
- 239000008223 sterile water Substances 0.000 description 1
- 229940013618 stevioside Drugs 0.000 description 1
- OHHNJQXIOPOJSC-UHFFFAOYSA-N stevioside Natural products CC1(CCCC2(C)C3(C)CCC4(CC3(CCC12C)CC4=C)OC5OC(CO)C(O)C(O)C5OC6OC(CO)C(O)C(O)C6O)C(=O)OC7OC(CO)C(O)C(O)C7O OHHNJQXIOPOJSC-UHFFFAOYSA-N 0.000 description 1
- 235000019202 steviosides Nutrition 0.000 description 1
- 238000010254 subcutaneous injection Methods 0.000 description 1
- 239000007929 subcutaneous injection Substances 0.000 description 1
- 239000000126 substance Substances 0.000 description 1
- 229960005137 succinic acid Drugs 0.000 description 1
- BAQAVOSOZGMPRM-UHFFFAOYSA-N sucralose Chemical compound OC1C(O)C(Cl)C(CO)OC1OC1(CCl)C(O)C(O)C(CCl)O1 BAQAVOSOZGMPRM-UHFFFAOYSA-N 0.000 description 1
- BAQAVOSOZGMPRM-QBMZZYIRSA-N sucralose Chemical compound O[C@@H]1[C@@H](O)[C@@H](Cl)[C@@H](CO)O[C@@H]1O[C@@]1(CCl)[C@@H](O)[C@H](O)[C@@H](CCl)O1 BAQAVOSOZGMPRM-QBMZZYIRSA-N 0.000 description 1
- 239000005720 sucrose Substances 0.000 description 1
- 235000000346 sugar Nutrition 0.000 description 1
- 150000008163 sugars Chemical class 0.000 description 1
- 239000000829 suppository Substances 0.000 description 1
- 239000002511 suppository base Substances 0.000 description 1
- 239000000454 talc Substances 0.000 description 1
- 229910052623 talc Inorganic materials 0.000 description 1
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 1
- 150000003536 tetrazoles Chemical class 0.000 description 1
- 230000001225 therapeutic effect Effects 0.000 description 1
- 206010043778 thyroiditis Diseases 0.000 description 1
- 230000000699 topical effect Effects 0.000 description 1
- 231100000331 toxic Toxicity 0.000 description 1
- 230000002588 toxic effect Effects 0.000 description 1
- QORWJWZARLRLPR-UHFFFAOYSA-H tricalcium bis(phosphate) Chemical compound [Ca+2].[Ca+2].[Ca+2].[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O QORWJWZARLRLPR-UHFFFAOYSA-H 0.000 description 1
- 125000004205 trifluoroethyl group Chemical group [H]C([H])(*)C(F)(F)F 0.000 description 1
- 125000000876 trifluoromethoxy group Chemical group FC(F)(F)O* 0.000 description 1
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 1
- 108010050327 trypticase-soy broth Proteins 0.000 description 1
- 210000001635 urinary tract Anatomy 0.000 description 1
- 239000003981 vehicle Substances 0.000 description 1
- 235000012431 wafers Nutrition 0.000 description 1
- 239000000811 xylitol Substances 0.000 description 1
- 235000010447 xylitol Nutrition 0.000 description 1
- HEBKCHPVOIAQTA-SCDXWVJYSA-N xylitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)CO HEBKCHPVOIAQTA-SCDXWVJYSA-N 0.000 description 1
- 229960002675 xylitol Drugs 0.000 description 1
- WRSWIWOVJBYZAW-UHFFFAOYSA-M zinc;methanidylbenzene;bromide Chemical compound Br[Zn+].[CH2-]C1=CC=CC=C1 WRSWIWOVJBYZAW-UHFFFAOYSA-M 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
- A61P31/06—Antibacterial agents for tuberculosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Public Health (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Pharmacology & Pharmacy (AREA)
- Veterinary Medicine (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Oncology (AREA)
- Communicable Diseases (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Pulmonology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Measuring Or Testing Involving Enzymes Or Micro-Organisms (AREA)
- Agricultural Chemicals And Associated Chemicals (AREA)
Abstract
Un compuesto de fórmula (I) **(Ver fórmula)** donde X representa C y uno de los dos enlaces **(Ver símbolo)** representa un doble enlace (y el otro representa un enlace sencillo); o X representa N (en cuyo caso ambos enlaces **(Ver símbolo)** representan enlaces sencillos); Z1 representa CH o N; R1 es hidrógeno, alquilo C1-4 o halo; R2 es hidrógeno, alquilo C1-4 o halo; R3 es hidrógeno, alquilo C1-6, hidroxi o halo; R4 es hidrógeno, alquilo C1-6, halo, arilo, heteroarilo, alquilo C1-6 sustituido con arilo o alquilo C1-6 sustituido con heteroarilo; y, cuando los sustituyentes R3 y R4 están situados en posiciones adyacentes, dichos R3 y R4 se pueden juntar para formar un radical de fórmula =CH-CH=CH-CH=, siempre que X represente carbono y los dos enlaces representen un enlace sencillo; arilo es fenilo; fenilo sustituido con uno, dos o tres sustituyentes seleccionados cada uno individualmente entre halo, hidroxi, alquilo C1-4, polihaloalquilo C1-4, alquiloxi C1-4, polihaloalquiloxi C1-4, ciano, nitro y amino; heteroarilo es furanilo, tiofenilo, pirrolilo, pirazolilo, imidazolilo, isoxazolilo, tiazolilo, triazolilo, tetrazolilo, isotiazolilo, tiadiazolilo, oxadiazolilo, piridinilo, piridazinilo, pirimidinilo, pirazinilo, benzo[1,3]dioxolilo, benzofuranilo, benzotiazolilo, indolilo, 2,3-dihidro-1H-indolilo, tetrahidrotiofenilo o quinolinilo; donde cada heteroarilo puede estar sustituido con uno o dos sustituyentes seleccionados cada uno independientemente entre halo, ciano, alquilo C1-4, alquiloxi C1-4, (alquil C1-4)carbonilo o fenilo; o una de sus sales de adición de ácido farmacéuticamente aceptables.
Description
DESCRIPCIÓN
3,4-dihidro-1H-[1,8]naftiridinonas sustituidas con homopiperidinilo antibacterianas
La presente invención se refiere a compuestos de fórmula (I) novedosos que inhiben la actividad de la enzima FabI, los cuales son por lo tanto útiles en el tratamiento de infecciones bacterianas. También se refiere a composiciones farmacéuticas que comprenden estos compuestos y a procesos químicos para la preparación de estos compuestos.
Los compuestos de la presente invención son compuestos antibacterianos que inhiben la proteína FabI, una enzima enoil-ACP (siglas en inglés de la proteína portadora de acilos)-reductasa dependiente de NADH en la vía biosintética de los ácidos grasos. La ácido graso-sintasa (FAS, por sus siglas en inglés) participa en la vía biosintética general de los ácidos grasos saturados en todos los organismos, pero la organización estructural de FAS varía considerablemente entre ellos. Las características distintivas de FAS de vertebrados y levaduras son que todas las actividades enzimáticas están codificadas en una o dos cadenas polipeptídicas, y que la proteína portadora de acilos (ACP) existe en forma de complejo. En cambio, en la FAS bacteriana, cada uno de los pasos sintéticos es catalizado por una enzima monofuncional diferente y la ACP es una proteína discreta. Por lo tanto, es posible inhibir la FAC bacteriana de forma selectiva mediante el bloqueo de uno de los pasos sintéticos utilizando un agente inhibidor. La enoil-ACP-reductasa dependiente de NADH (FabI) participa en el último paso de los cuatro pasos de reacción incluidos en cada ciclo de biosíntesis de ácidos grasos bacterianos. Así pues, la enzima FabI es la enzima biosintética en la vía sintética global de la biosíntesis de los ácidos grasos bacterianos.
Se ha demostrado que la enzima FabI constituye una diana esencial en patógenos importantes tales como E. coli (Heath et al. J. Biol. Chem. 1995, 270, 26538; Bergler et al. Eur. J. Biochem. 2000, 275, 4654). Por ende, los compuestos que inhiben FabI pueden ser útiles como agentes antibacterianos.
En WO-01/26652, WO-01/26654 y WO-01/27103 se han descrito compuestos con actividad inhibidora de la enzima FabI. En WO-03/088897, WO-2007/043835 y WO-2008/098374 se han descrito compuestos naftiridinónicos sustituidos con actividad inhibidora de FabI. La solicitud de patente internacional WO 2007/053131 también describe diversos compuestos naftiridónicos para su potencial uso como inhibidores de FabI. Sin embargo, ninguno de estos documentos describe un compuesto en el que haya un grupo amino cíclico unido directamente a un resto carbonilo en posición a respecto a un alqueno. La solicitud de patente internacional WO 2011/061214 también describe diversos compuestos para su potencial uso como inhibidores de FabI. Sin embargo, este documento no describe específicamente, inter alia, compuestos en los que haya un grupo cíclico de 7 miembros que contenga nitrógeno que contenga opcionalmente un doble enlace.
La presente invención se refiere a un compuesto de fórmula (I)

donde
X representa C y uno de los dos enlaces ...... representa un doble enlace (y el otro representa un enlace sencillo); o
X representa N (en cuyo caso ambos enlaces ...... representan enlaces sencillos);
Z1 representa CH o N;
1
R es hidrógeno, alquilo C o halo;
2
R es hidrógeno, alquilo C o halo;
3
R es hidrógeno, alquilo C16, hidroxi o halo;
4
R es hidrógeno, alquilo C16, halo, arilo, heteroarilo, alquilo C1-6 sustituido con arilo o alquilo C1-6 sustituido con heteroarilo;
y 3 3 4ando los sustituyentes R 4, cu y R están situados en posiciones adyacentes, dichos R y R se pueden juntar para formar un radical de fórmula =CH-CH=CH-CH=, siempre que X represente carbono y los dos enlaces representen un enlace sencillo;
arilo es fenilo; fenilo sustituido con uno, dos o tres sustituyentes seleccionados cada uno individualmente entre halo, hidroxi, alquilo C polihaloalquilo C alquiloxi C polihaloalquiloxi C14, ciano, nitro y amino;
heteroarilo es furanilo, tiofenilo, pirrolilo, pirazolilo, imidazolilo, isoxazolilo, tiazolilo, triazolilo, tetrazolilo, isotiazolilo, tiadiazolilo, oxadiazolilo, piridinilo, piridazinilo, pirimidinilo, pirazinilo, benzo[1,3]dioxolilo, benzofuranilo, benzotiazolilo, indolilo, 2,3-dihidro-1H-indolilo, tetrahidrotiofenilo o quinolinilo;
donde cada heteroarilo puede estar sustituido con uno o dos sustituyentes seleccionados cada uno independientemente entre halo, ciano, alquilo C 4, alquiloxi C 4, (alquil C 4)carbonilo o fenilo;
o una de sus sales de adición de ácido farmacéuticamente aceptables.
Tal como se utilizan en las definiciones anteriores:
- halo es un término genérico para fluoro, cloro, bromo y yodo;
- alquilo C_4 define radicales hidrocarbonados saturados de cadena lineal y ramificada que contienen de 1 a 4 átomos de carbono tales como, por ejemplo, metilo, etilo, propilo, butilo, 1 -metiletilo, 2-metilpropilo y similares;
- se pretende que alquilo C^g incluya alquilo C_4 y sus homólogos superiores que contengan 5 o 6 átomos de carbono tales como, por ejemplo, 2-metilbutilo, pentilo, hexilo y similares;
- polihaloalquilo C 4 se define como alquilo C 4 polisustituido con halo (según se ha definido anteriormente en la presente) sustituido con de 2 a 6 átomos halógenos tales como difluorometilo, trifluorometilo, trifluoroetilo y similares. Tal como se utiliza en la descripción, cuando se emplea la expresión “compuesto de fórmula (I)”, se pretende que esta incluya también las sales de adición farmacéuticamente que los compuestos de fórmula (I) sean capaces de formar y los solvatos que los compuestos de fórmula (I) o las sales de adición de ácido farmacéuticamente aceptables de los compuestos de fórmula (I) sean capaces de formar.
La definición de “compuestos de fórmula (I)” incluye de forma inherente todos los estereoisómeros del compuesto de fórmula (I) ya sea como un estereoisómero puro o como una mezcla de dos o más estereoisómeros. Los enantiómeros son estereoisómeros que son imágenes especulares no superponibles el uno del otro. Una mezcla 1:1 de un par de enantiómeros es un racemato o mezcla racémica. Los diastereómeros (o diastereoisómeros) son estereoisómeros que no son enantiómeros, es decir, que no están relacionados como imágenes especulares. Si un compuesto contiene un grupo cicloalquilo disustituido, los sustituyentes pueden estar en la configuración cis o trans. Por lo tanto, la invención incluye los enantiómeros, diastereómeros, racematos, isómeros cis, isómeros trans y mezclas de estos.
La configuración absoluta se especifica de acuerdo con el sistema de Cahn-Ingold-Prelog. La configuración en un átomo asimétrico se especifica como R o S. Los compuestos resueltos cuya configuración absoluta se desconoce se pueden designar como (+) o (-) dependiendo de la dirección en la que hagan rotar el plano de la luz polarizada. Cuando se identifica un estereoisómero específico, esto quiere decir que dicho estereoisómero está sustancialmente exento, es decir, asociado con menos de un 50%, preferentemente menos de un 20%, más preferentemente menos de un 10%, aún más preferentemente menos de un 5%, en particular menos de un 2% y aún más preferentemente menos de un 1%, de los otros isómeros. Por lo tanto, cuando se especifica que un compuesto de fórmula (I) es, por ejemplo, (R), esto quiere decir que el compuesto está sustancialmente exento del isómero (S); cuando se especifica que un compuesto de fórmula (I) es, por ejemplo, E, esto quiere decir que el compuesto está sustancialmente exento del isómero Z; cuando se especifica que un compuesto de fórmula (I) es, por ejemplo, cis, esto quiere decir que el compuesto está sustancialmente exento del isómero trans.
Las expresiones “estereoisómeros” o “formas estereoquímicamente isoméricas” se utilizan de forma indistinta anteriormente y en lo sucesivo en la presente.
La configuración estereoquímica absoluta de los compuestos de fórmula (I) y de los intermedios utilizados en su preparación puede ser determinada fácilmente por los expertos en la técnica utilizando métodos muy conocidos tales como, por ejemplo, difracción de rayos X.
Algunos de los compuestos de fórmula (I) también pueden existir en su forma tautomérica. Se pretende que tales formas, aunque no se indique de forma explícita en la fórmula anterior, queden incluidas dentro del alcance de la presente invención.
Además, algunos compuestos de fórmula (I) y algunos de los intermedios utilizados en su preparación pueden exhibir polimorfismo. Se debe sobreentender que la presente invención engloba cualesquiera formas polimórficas que posean propiedades útiles en el tratamiento de las afecciones mencionadas anteriormente en la presente. Se pretende que las sales de adición de ácido farmacéuticamente aceptables como las mencionadas anteriormente en la presente comprendan las formas salinas de adición de ácido atóxicas terapéuticamente activas que los
compuestos de fórmula (I) sean capaces de formar. Estas sales de adición de ácido farmacéuticamente aceptables se pueden obtener convenientemente tratando la forma básica con un ácido adecuado de este tipo. Los ácidos adecuados comprenden, por ejemplo, ácidos inorgánicos tales como haluros de hidrógeno, p. ej., ácido clorhídrico o bromhídrico, sulfúrico, nítrico, fosfórico y los ácidos similares; o ácidos orgánicos tales como, por ejemplo, ácido acético, propanoico, hidroxiacético, láctico, pirúvico, oxálico (es decir, etanodioico), malónico, succínico (es decir, ácido butanodioico), maleico, fumárico, málico, tartárico, cítrico, metanosulfónico, etanosulfónico, bencenosulfónico, p-toluenosulfónico, ciclámico, salicílico, p-aminosalicílico, pamoico y los ácidos similares.
Y a la inversa, dichas formas salinas se pueden convertir en la forma básica libre tratándolas con una base adecuada.
Los compuestos de fórmula (I) pueden existir tanto en formas solvatadas como no solvatadas. El término "solvato" se utiliza en la presente para describir una asociación molecular que comprende el compuesto de la invención y una o más moléculas de disolvente farmacéuticamente aceptable, por ejemplo, agua o etanol. El término "hidrato" se utiliza cuando dicho disolvente es agua.
El término "FabI" está aceptado en la técnica y se refiere a la enzima bacteriana que se cree que funciona como enoil-ACP (proteína portadora de acilos)-reductasa en el paso final de las cuatro reacciones incluidas en cada ciclo de biosíntesis de ácidos grasos bacterianos. Se cree que esta enzima está extensamente distribuida en las bacterias.
Los compuestos de fórmula (I) que se pueden mencionar incluyen aquellos en los que:
(i) Z1 representa CH y, por lo tanto, el compuesto de fórmula I representa lo siguiente:

donde
(ii) cuando R1 o R2 representan halo, entonces son preferentemente F o Cl;
(iii) R1 representa hidrógeno o alquilo C1-4; y/o
(iv) R2 representa hidrógeno o alquilo C1-4.
Los compuestos interesantes de fórmula (I) son aquellos compuestos de fórmula (I) en los que se aplican una o más de las siguientes restricciones:
2 a) R 1 y R representan hidrógeno; o
b) R representa hidrógeno; o
3
c) R representa alquilo C o halo; o
d) R representa halo, arilo, heteroarilo o alquilo C14 sustituido con arilo; o
e) R3 y R están situados en posiciones adyacentes y se juntan para formar un radical de fórmula =CH-CH=CH-CH=, siempre que X represente carbono y los dos enlaces representen un enlace sencillo; y f) heteroarilo es tiofenilo, pirrolilo, tiazolilo o triazolilo.
Un primer grupo de compuestos son los compuestos de fórmula (I)

donde
X representa C y uno de los dos enlaces ...... representa un doble enlace (y el otro representa un enlace sencillo); o
X representa N (en cuyo caso ambos enlaces ...... representan enlaces sencillos);
Z1 representa CH o N;
R1 es hidrógeno;
R2 es hidrógeno;
3
R es hidrógeno, alquilo C1-6 o halo;
4
R es halo, arilo, heteroarilo o alquilo C16 sustituido con arilo;
3 4y, cuando los sustituyentes R 3 y R 4 están situados en posiciones adyacentes, dichos R y R se pueden juntar para formar un radical de fórmula =CH-CH=CH-CH=, siempre que X represente carbono y los dos enlaces representen un enlace sencillo;
arilo es fenilo; fenilo sustituido con uno o dos sustituyentes seleccionados cada uno individualmente entre halo, alquilo C , polihaloalquilo C , alquiloxi C y polihaloalquiloxi C ;
heteroarilo es tiofenilo, pirrolilo, tiazolilo o triazolilo;
o una de sus sales de adición de ácido farmacéuticamente aceptables.
Los compuestos de fórmula (I) que se pueden mencionar incluyen aquellos que en los que X representa C, los dos enlaces representan enlaces sencillos, y R3 y R4 están presentes y situados en posiciones adyacentes y se juntan para formar un radical de fórmula =CH-CH=CH-CH=. Sin embargo, los compuestos de fórmula (I) que son particularmente preferidos incluyen aquellos en los que:
X representa C y uno de los dos enlaces representa un doble enlace (y el otro representa un enlace sencillo); o
X representa N (en cuyo caso, ambos enlaces ...... representan enlaces sencillos) y, por lo tanto, los siguientes anillos que contienen X son particularmente preferidos:
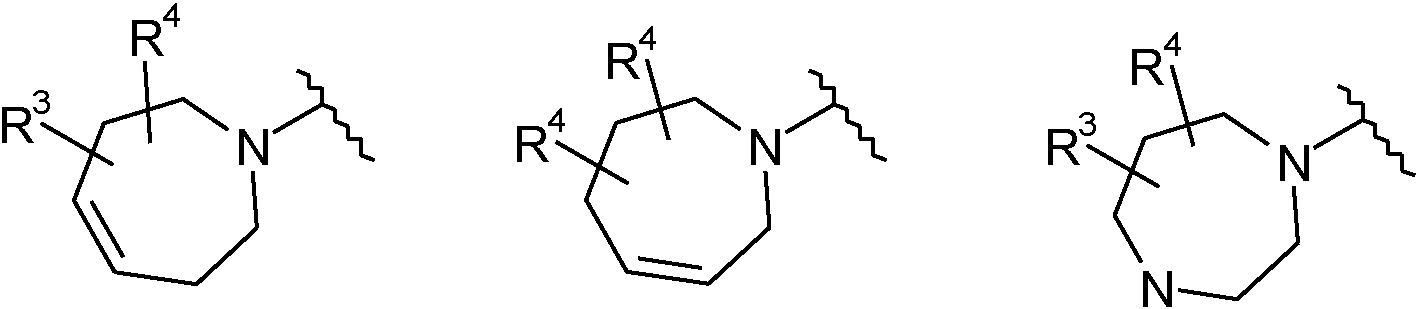
En este caso, se prefiere que los grupos R3 y R4 adyacentes no se junten para formar un radical.
En los compuestos de fórmula (I), se prefiere que:
(i) Haya al menos un sustituyente R3 o R4 presente que no represente hidrógeno;
(ii) Uno de los grupos R3 y R4 (p. ej., R3) represente hidrógeno, halo, alquilo C1-3 o hidroxi, y el otro de los grupos R3 y R4 (p. ej., R4) represente un sustituyente que no sea hidrógeno;
(iii) R3 represente hidrógeno, alquilo C1-4 (p. ej., metilo) o halo (p. ej., fluoro) y aún más preferentemente represente hidrógeno (es decir, que R3 esencialmente no esté presente);
(iv) R4 represente un sustituyente que no sea hidrógeno (es decir, que haya un sustituyente R4 que esté presente y que no represente hidrógeno);
(v) R4 represente un sustituyente que no sea hidrógeno, el cual esté unido a X,
en donde cualesquiera de los apartados anteriores se pueden considerar de forma conjunta o combinados. Por ejemplo, los apartados (iii), (iv) y/o (v) se pueden considerar combinados para proporcionar los siguientes compuestos de fórmula (I) particularmente preferidos:

en donde R4 representa un sustituyente que no sea hidrógeno. Los anillos que contienen X más preferidos en los compuestos de fórmula (I) son:

en donde R4 representa un sustituyente que no sea hidrógeno. Los sustituyentes particularmente preferidos que R4 puede representar (en este caso y en otros) incluyen:
(i) arilo (opcionalmente sustituido según se define en la presente);
(ii) heteroarilo (opcionalmente sustituido según se define en la presente);
(iii) alquilo C1-6 sustituido con arilo o heteroarilo (estos dos últimos grupos arilo y heteroarilo están a su vez opcionalmente sustituidos según se ha definido en la presente).
Se prefiere particularmente que el grupo R4 contenga un resto aromático y, por lo tanto, se prefieren particularmente los apartados (i), (ii) y (iii) anteriores.
En el caso en que R4 represente el apartado (i) anterior, entonces el grupo arilo es preferentemente fenilo, y este grupo puede no estar sustituido o estar sustituido con uno o dos (p. ej., uno) sustituyentes seleccionados entre halo (p. ej., cloro, fluoro), alquilo C1-4 (p. ej., metilo), polihaloalquilo C1-4 (p. ej., -CF3), alquiloxi C1-4 (p. ej., -OCH3), polihaloalquiloxi C1-4 (p. ej., -OCF3).
En el caso en que R4 represente el apartado (ii) anterior, entonces el grupo heteroarilo es preferentemente un anillo monocíclico de 5 o 6 miembros que contiene de uno a cuatro heteroátomos (p. ej., uno o dos heteroátomos) para formar así, p. ej., un tiazolilo (p. ej., 2-tiazolilo), tienilo (p. ej., 2-tienilo), pirazolilo (p. ej., 1- o 2-pirazolilo), triazolilo (p. ej., 1,2,3-triazol-1-ilo) o pirrolilo (p. ej., 1-pirrolilo).
En el caso en que R4 represente el apartado (iii) anterior, entonces el grupo alquilo C1-6 es preferentemente metilo, es decir, -CH3, y este resto alquilo está sustituido con arilo (p. ej., fenilo, tal como un fenilo no sustituido).
Aún más preferentemente, el grupo R4 representa el apartado (i) o (ii) anterior, es decir, arilo o heteroarilo. Incluso más preferentemente, el grupo R4 representa el apartado (i) anterior, especialmente fenilo no sustituido.
Se indica previamente en la presente que los siguientes anillos que contienen X son particularmente preferidos:

y particularmente aquellos en los que R4 es como se ha definido anteriormente. Tales compuestos que contienen un resto N(R4) o un resto C(R4) adyacente a un doble enlace pueden ser beneficiosos. Esto se debe a que la forma del átomo de nitrógeno (p. ej., que es más planar de por sí, en comparación con un resto CR4 que no sea adyacente a un doble enlace) o la presencia del doble enlace en el anillo que contiene X puede ayudar a orientar el grupo R4 (si está presente), de modo que el total del compuesto (p. ej., teniendo en cuenta la orientación del sustituyente R4) presente unas propiedades de unión a la enzima bacteriana FabI mejores/mejoradas. Por ende, estos compuestos de la invención pueden ser convenientes en el sentido de que la presencia del doble enlace puede hacer que mejore
la unión con la enzima FabI o su inhibición. Por consiguiente, los compuestos de la invención pueden ser compuestos convenientes (p. ej., en comparación con los compuestos conocidos) en virtud de estas propiedades que pueden hacer que, como consecuencia, mejore la potencia, eficacia, etc.
Los compuestos de fórmula (I), por lo general, se pueden preparar haciendo reaccionar un intermedio de fórmula (II) con un intermedio de fórmula (III), en al menos un disolvente inerte en la reacción y opcionalmente en presencia de al menos un reactivo de acoplamiento adecuado y/o una base adecuada, donde además dicho proceso comprende opcionalmente convertir un compuesto de fórmula (I) en una de sus sales de adición y/o preparar formas estereoquímicamente isoméricas de este.

Puede que sea conveniente activar el ácido carboxílico de fórmula (III) añadiendo una cantidad eficaz de un promotor de la reacción. Los ejemplos no limitantes de tales promotores de la reacción incluyen carbonildiimidazol, W,W-diciclohexilcarbodiimida o 1-(3-dimetilaminopropil)-3-etilcarbodiimida, hidroxibenzotriazol, hexafluorofosfato de benzotriazoliloxitris(dimetilamino)fosfonio, hexafluorofosfato de tetrapirrolidinofosfonio, hexafluorofosfato de bromotripirrolidinofosfonio o uno de sus derivados funcionales.
Los compuestos de fórmula (I) también se pueden preparar haciendo reaccionar un intermedio de fórmula (II) con un intermedio de fórmula (IV), donde Y representa hidroxi o halo. La reacción se puede llevar a cabo en un disolvente inerte en la reacción tal como, por ejemplo, diclorometano o dimetilformamida, y opcionalmente en presencia de una base adecuada tal como, por ejemplo, diisopropiletilamina (DIPEA).

Los materiales de partida y algunos de los intermedios son compuestos conocidos y se pueden adquirir de proveedores comerciales o se pueden preparar de acuerdo con procedimientos de reacción convencionales generalmente conocidos en la técnica.
Los compuestos de fórmula (I) también se pueden preparar haciendo reaccionar un intermedio de fórmula (V) con un intermedio de fórmula (VI),

donde Xai representa un grupo saliente adecuado tal como un grupo halo adecuado (p. ej., cloro, yodo y especialmente bromo) y los otros elementos son como se han definido anteriormente en la presente, en condiciones de reacción adecuadas para la reacción, por ejemplo, en condiciones de reacción de acoplamiento con un catalizador metálico (p. ej., condiciones de reacción de acoplamiento con metales preciosos, donde el metal precioso es, p. ej., paladio), en particular en las condiciones de reacción de Heck en las que se utiliza preferentemente un catalizador a base de paladio tal como acetato de paladio, tetrakis(trifenilfosfina)paladio (0), dicloruro de bis(trifenilfosfina)paladio (II), dicloruro de [1,1'-bis(difenilfosfino)ferroceno]paladio (II) o similar (preferentemente, el catalizador es acetato de paladio), por ejemplo, opcionalmente en presencia de un disolvente adecuado (p. ej., acetonitrilo o similar), una base (p. ej., una base amínica tal como W,W-diisopropilamina o similar) y un ligando (p. ej., trifenilfosfina, tri-o-tolilfosfina o similar). La reacción se puede llevar a cabo en un tubo sellado y/o en un microondas.
Los materiales de partida y algunos de los intermedios son compuestos conocidos y se pueden adquirir de
proveedores comerciales o se pueden preparar de acuerdo con procedimientos de reacción convencionales generalmente conocidos en la técnica.
Los intermedios de fórmula (II-a), definidos como intermedios de fórmula (II) donde X representa carbono y R4 está situado en la posición 4 del anillo homopiperidinílico, se pueden preparar de acuerdo con el siguiente esquema de reacciones general.

En el esquema de reacciones anterior, el radical PG en los intermedios (V) y (VI) es un grupo protector de nitrógeno tal como, p. ej., ferf-butiloxicarbonilo, que se puede eliminar fácilmente en condiciones ácidas. El reactivo 4
organomagnésico R -MgBr se puede obtener utilizando reacciones organometálicas conocidas en la técnica tales como la reacción de Grignard.
Para los compuestos en los que Z1 representa CH, los intermedios (IV) y (VI) se pueden preparar según se ha descrito en la presente o de acuerdo con procedimientos de reacción convencionales generalmente conocidos en la técnica. Este también puede ser el caso para los intermedios correspondientes en los que Z1 representa N. Sin embargo, tales compuestos también se pueden preparar de acuerdo con el siguiente esquema:

Condiciones:
a) NBS, ACN, reflujo, 3 h, 70% ; b) LiAlH 1 M en THF, THF, 5 °C-TA, durante la noche, 20%; c) PBr3, DCM, TA, durante la noche, 90%; f) malonato de dimetilo, NaOMe en MeOH, MeOH, TA, durante la noche, 25%; g) NaOH, MeOH, reflujo, 4 h, HCl, reflujo, durante la noche; h) DIEA, Pd(OAc)2, tri-o-tolilfosfina, ACN, DMF, microondas, 180 °C, 25 min.
Los compuestos de fórmula (I) como los preparados en los procesos descritos anteriormente en la presente se pueden sintetizar en forma de mezclas racémicas de enantiómeros que se pueden separar unos de otros mediante los procedimientos de resolución conocidos en la técnica. Aquellos compuestos de fórmula (I) que se obtienen en forma racémica se pueden convertir en las formas salinas diastereoméricas correspondientes por reacción con un ácido quiral adecuado. Dichas formas salinas diastereoméricas se separan posteriormente, por ejemplo, mediante una cristalización fraccionada o selectiva, y los enantiómeros se liberan de estas con álcali. Una manera alternativa de separar las formas enantioméricas de los compuestos de fórmula (I) implica la cromatografía líquida utilizando una fase estacionaria quiral. Dichas formas estereoquímicamente isoméricas puras también pueden obtenerse a partir de las formas estereoquímicamente isoméricas puras correspondientes de los materiales de partida adecuados, siempre que las reacciones tengan lugar de manera estereoespecífica. Preferentemente, si se desea obtener un estereoisómero específico, dicho compuesto se sintetizará mediante métodos de preparación estereoespecíficos. En estos métodos, se emplearán convenientemente materiales de partida enantioméricamente
puros.
Los compuestos descritos en la presente son inhibidores de la enzima FabI, tal como se demuestra en el Ejemplo farmacológico 1. En vista de estas propiedades inhibitorias de la enzima FabI, los compuestos descritos en la presente son útiles para tratar infecciones bacterianas. Por ejemplo, estos compuestos son útiles para tratar infecciones bacterianas, tales como, por ejemplo, infecciones de las vías respiratorias altas (p. ej., otitis media, traqueítis bacteriana, epiglotitis aguda, tiroiditis), de las vías respiratorias bajas (p. ej., empiema, absceso pulmonar), cardíacas (p. ej., endocarditis infecciosa), gastrointestinales (p. ej., diarrea secretora, absceso esplénico, absceso retroperitoneal), del SNC (p. ej., absceso cerebral), oculares (p. ej., blefaritis, conjuntivitis, queratitis, endoftalmitis, celulitis orbital y preseptal, dacriocistitis), renales y de las vías urinarias (p. ej., epididimitis, absceso intrarrenal y perinéfrico, síndrome del choque tóxico), cutáneas (p. ej., impétigo, foliculitis, abscesos cutáneos, celulitis, infección de heridas, miositis bacteriana), y óseas y articulares (p. ej., artritis séptica, osteomielitis). Además, los compuestos pueden ser útiles combinados con antibióticos conocidos.
Por lo tanto, la presente invención también se refiere a compuestos de fórmula (I) para su uso como medicina especialmente para su uso en el tratamiento de infecciones bacterianas, en particular infecciones bacterianas provocadas por una bacteria que expresa una enzima FabI. Subsecuentemente, los presentes compuestos se pueden utilizar con el fin de elaborar una medicina para tratar infecciones bacterianas, en particular infecciones bacterianas provocadas por una bacteria que expresa una enzima FabI.
Además, la presente invención proporciona un compuesto de fórmula (I) que inhibe la enzima FabI para su uso en un método para tratar infecciones bacterianas que comprende administrar dicho compuesto a un sujeto que lo necesite.
Un sujeto que necesite tratamiento padece una infección bacteriana o ha estado expuesto a una bacteria infecciosa y sus síntomas se pueden aliviar administrando una cantidad terapéuticamente eficaz de los compuestos de la presente invención. Por ejemplo, un sujeto que necesite tratamiento puede padecer una infección para la cual se pueden administrar los compuestos de fórmula (I) como tratamiento. En otro ejemplo, un sujeto que necesite tratamiento puede tener una herida abierta o una lesión por quemadura para la cual se pueden administrar los compuestos de fórmula (I) como tratamiento profiláctico. Normalmente, un sujeto se tratará para combatir una infección bacteriana existente.
Un sujeto puede padecer una infección bacteriana provocada por Bacillus anthracis, Citrobacter sp., Escherichia coli, Francisella tularensis, Haemophilus influenza, Listeria monocytogenes, Moraxella catarrhalis, Mycobacterium tuberculosis, Neisseria meningitidis, Proteus mirabilis, Proteus vulgaris, Salmonella sp., Serratia sp., Shigella sp., Stenotrophomonas maltophilia, Staphylococcus aureus o Staphylococcus epidermidis. Preferentemente, el sujeto se trata (profiláctica o terapéuticamente) para combatir una infección bacteriana provocada por una bacteria que expresa una enzima FabI.
Los términos "tratar" y "tratamiento", tal como se utilizan en la presente, se refieren al tratamiento curativo, paliativo y profiláctico que incluye revertir, aliviar, prevenir o inhibir el avance de la enfermedad, trastorno o afección a la cual se aplican dichos términos, o uno o más síntomas de tal enfermedad, trastorno o afección.
Una "cantidad terapéuticamente eficaz" de un compuesto de la presente invención es aquella cantidad que, cuando se administra a un sujeto que necesita tratamiento, mejora el pronóstico del sujeto, p. ej., retrasa la aparición de uno o más de los síntomas asociados con una infección bacteriana en el sujeto y/o reduce su intensidad. La cantidad del compuesto descrito que se ha de administrar a un sujeto dependerá de la enfermedad particular, el modo de administración y las características del sujeto, tales como su estado de salud general, edad, sexo, genotipo, peso corporal, tolerancia a los fármacos y otras enfermedades. Un experto será capaz de determinar las dosis adecuadas dependiendo de estos y otros factores.
Los compuestos se pueden evaluar en uno de los diversos ensayos biológicos para determinar la concentración de compuesto necesaria para ejercer un efecto farmacológico determinado.
Además, la presente invención proporciona composiciones farmacéuticas que comprenden al menos un portador farmacéuticamente aceptable y una cantidad terapéuticamente eficaz de un compuesto de fórmula (I).
Para preparar las composiciones farmacéuticas de esta invención, se combina una cantidad eficaz del compuesto particular, en forma de sal de adición de ácido o base, como principio activo en mezcla íntima con al menos un portador farmacéuticamente aceptable, y dicho portador puede adoptar una gran variedad de formas dependiendo de la forma del preparado deseada para la administración. Es preferible que estas composiciones farmacéuticas se presenten en una forma farmacéutica unitaria adecuada, preferentemente, para la administración oral, administración rectal, administración percutánea o inyección parenteral.
Por ejemplo, en la preparación de composiciones en una forma farmacéutica oral, se puede emplear cualquiera de los portadores farmacéuticos líquidos habituales tales como, por ejemplo, agua, glicoles, aceites, alcoholes y similares en el caso de preparados líquidos orales tales como suspensiones, jarabes, elixires y soluciones; o portadores farmacéuticos sólidos tales como almidones, azúcares, caolín, lubricantes, aglutinantes, agentes
desintegrantes y similares en el caso de polvos, pastillas, cápsulas y comprimidos. Debido a su fácil administración, los comprimidos y las cápsulas representan las formas farmacéuticas unitarias orales más convenientes, en cuyo caso se emplean obviamente portadores farmacéuticos sólidos. Para las composiciones de inyección parenteral, el portador farmacéutico comprenderá principalmente agua estéril, aunque se pueden incluir otros ingredientes para mejorar la solubilidad del principio activo. Las soluciones inyectables se pueden preparar, por ejemplo, utilizando un portador farmacéutico que comprenda una solución salina, una solución glucosada o una mezcla de ambas. También se pueden preparar suspensiones inyectables utilizando portadores líquidos, agentes de suspensión y aditivos similares que sean adecuados. En las composiciones adecuadas para la administración percutánea, el portador farmacéutico puede comprender opcionalmente un agente potenciador de la penetración y/o un agente humectante adecuado, opcionalmente combinados con proporciones minoritarias de aditivos adecuados que no provoquen ningún efecto perjudicial significativo en la piel. Dichos aditivos se pueden seleccionar de modo que faciliten la administración del principio activo sobre la piel y/o sean útiles para preparar las composiciones deseadas. Estas composiciones tópicas se pueden administrar de varias formas, p. ej., como un parche transdérmico, una unción dorsal puntual o una pomada. Las sales de adición de los compuestos de fórmula (I), debido a su mayor solubilidad en agua en comparación con la forma básica correspondiente, son obviamente más adecuadas para la preparación de composiciones acuosas.
Es especialmente conveniente formular las composiciones farmacéuticas de la invención en formas farmacéuticas unitarias debido a su fácil administración y a la uniformidad de la dosis. La expresión "forma farmacéutica unitaria", tal como se utiliza en la presente, se refiere a unidades físicamente discretas adecuadas como dosis unitarias, donde cada unidad contiene una cantidad predeterminada de principio activo, calculada para producir el efecto terapéutico deseado, asociada con el portador farmacéutico requerido. Los ejemplos de tales formas farmacéuticas unitarias son comprimidos (incluidos los comprimidos recubiertos o ranurados), cápsulas, pastillas, sobres de polvos, obleas, soluciones o suspensiones inyectables, cucharaditas, cucharadas y similares, y múltiplos segregados de estos.
Para la administración oral, las composiciones farmacéuticas de la presente invención pueden presentarse como formas farmacéuticas sólidas, por ejemplo, comprimidos (tanto deglutibles como masticables), cápsulas o cápsulas de gelatina, preparadas de forma convencional con excipientes y portadores farmacéuticamente aceptables tales como aglutinantes (p. ej., almidón de maíz pregelatinizado, polivinilpirrolidona, hidroxipropilmetilcelulosa y similares), rellenos (p. ej., lactosa, celulosa microcristalina, fosfato de calcio y similares), lubricantes (p. ej., estearato de magnesio, talco, sílice y similares), agentes desintegrantes (p. ej., almidón de papa, glicolato sódico de almidón y similares), agentes humectantes (p. ej., laurilsulfato de sodio) y similares. Tales comprimidos también se pueden recubrir mediante métodos muy conocidos en la técnica.
Los preparados líquidos para la administración oral pueden presentarse en forma de, p. ej., soluciones, jarabes o suspensiones, o se pueden formular como un producto seco que se ha de mezclar con agua y/u otro portador líquido adecuado antes de usarlo. Tales preparados líquidos se pueden preparar de forma convencional, opcionalmente con otros aditivos farmacéuticamente aceptables tales como agentes de suspensión (p. ej., jarabe de sorbitol, metilcelulosa, hidroxipropilmetilcelulosa o grasas comestibles hidrogenadas), agentes emulsionantes (p. ej., lecitina o goma arábiga), portadores no acuosos (p. ej., aceite de almendras, ésteres oleosos o alcohol etílico), edulcorantes, saborizantes, agentes enmascarantes y conservantes (p. ej., p-hidroxibenzoatos de metilo o propilo, o ácido sórbico).
Los edulcorantes farmacéuticamente aceptables útiles en las composiciones farmacéuticas de la invención comprenden preferentemente al menos un edulcorante intenso tal como aspartamo, acesulfamo potásico, ciclamato de sodio, alitamo, un edulcorante dihidrocalcónico, monelina, esteviosida, sucralosa (4,1 ',6'-tricloro-4,1 ',6'-tridesoxigalactosacarosa) o preferentemente sacarina, sacarina sódica o cálcica, y opcionalmente al menos un edulcorante espesante tal como sorbitol, manitol, fructosa, sacarosa, maltosa, isomaltitol, glucosa, jarabe de glucosa hidrogenada, xilitol, caramelo o miel. Los edulcorantes intensos se utilizan convenientemente en concentraciones bajas. Por ejemplo, en el caso de la sacarina sódica, dicha concentración puede estar comprendida entre aproximadamente un 0.04% y un 0.1% (peso/volumen) de la formulación final. El edulcorante espesante se puede utilizar de forma efectiva en concentraciones más altas comprendidas entre aproximadamente un 10% y aproximadamente un 35%, preferentemente entre aproximadamente un 10% y un 15% (peso/volumen).
Los saborizantes farmacéuticamente aceptables que pueden enmascarar los ingredientes con sabor amargo en las formulaciones de dosis baja son preferentemente saborizantes con sabor a fruta tal como sabor a cereza, frambuesa, grosella negra o fresa. Una combinación de dos saborizantes puede proporcionar muy buenos resultados. En las formulaciones de dosis alta puede que se necesiten saborizantes farmacéuticamente aceptables más potentes tales como caramelo de chocolate, menta fresca, fantasía y similares. Cada saborizante puede estar presente en la composición final en una concentración comprendida entre aproximadamente un 0.05% y un 1% (peso/volumen). Las combinaciones de tales saborizantes potentes se emplean de forma beneficiosa. Preferentemente, se utiliza un saborizante que no sufra ninguna pérdida o cambio de sabor y/o color en las condiciones de la formulación.
Los compuestos de fórmula (I) se pueden formular para la administración parenteral por inyección, convenientemente inyección intravenosa, intramuscular o subcutánea, por ejemplo, por inyección en bolo o infusión
intravenosa continua. Las formulaciones para inyección se pueden presentar en formas farmacéuticas unitarias, p. ej., en ampollas o recipientes multidosis, que incluyan un conservante añadido. Se pueden presentar en formas tales como suspensiones, soluciones o emulsiones en vehículos oleosos o acuosos, y pueden contener agentes de formulación tales como agentes isotonizantes, de suspensión, estabilizantes y/o dispersantes. Como alternativa, el principio activo se puede presentar en forma de polvo que ha de ser mezclado con un vehículo adecuado, p. ej., agua estéril exenta de pirógenos, antes de usarlo.
Los compuestos de fórmula (I) también se pueden formular en composiciones rectales tales como supositorios o enemas de retención, p. ej., que contengan bases de supositorios convencionales tales como manteca de cacao y/u otros glicéridos.
Los expertos en el tratamiento de enfermedades antibacterianas asociado con la inhibición de la enzima FabI podrán determinar fácilmente la cantidad terapéuticamente eficaz de un compuesto de fórmula (I) a partir de los resultados de las pruebas que se presentan más adelante en la presente. En general, se contempla que una dosis terapéuticamente eficaz estará comprendida entre aproximadamente 0.001 mg/kg y aproximadamente 50 mg/kg de peso corporal, más preferentemente entre aproximadamente 0.01 mg/kg y aproximadamente 10 mg/kg de peso corporal del paciente que se desee tratar. Podría resultar adecuado administrar la dosis terapéuticamente eficaz en forma de dos o más subdosis en intervalos adecuados a lo largo del día. Dichas subdosis se pueden formular como formas farmacéuticas unitarias, por ejemplo, que contengan cada una de aproximadamente 0.1 mg a aproximadamente 1000 mg, más particularmente de aproximadamente 1 a aproximadamente 500 mg del principio activo por forma farmacéutica unitaria.
La dosis exacta y la frecuencia de administración dependen del compuesto particular de fórmula (I) utilizado, la afección particular que se esté tratando, la gravedad de la afección que se esté tratando, la edad, el peso y el estado físico general del paciente particular, así como también de otra medicación que el paciente pueda estar tomando, como bien sabrán los expertos en la técnica. Además, dicha "cantidad terapéuticamente eficaz" se puede reducir o incrementar dependiendo de la respuesta del paciente tratado y/o dependiendo de la evaluación del médico que prescriba los compuestos de la presente invención. Por consiguiente, los intervalos de la cantidad diaria eficaz que se han mencionado anteriormente son solamente orientativos.
Los compuestos de fórmula (I) pueden presentar la ventaja de que pueden ser más eficaces, menos tóxicos, tener una acción más prolongada, ser más potentes, producir menos efectos secundarios, ser más fácilmente absorbidos y/o presentar un perfil farmacocinético mejor (p. ej., mayor biodisponibilidad oral y/o menor eliminación) y/o presentar otras propiedades farmacológicas, físicas o químicas útiles en comparación con compuestos conocidos en la técnica anterior, ya sea para su uso en las indicaciones mencionadas anteriormente o en otras.
Por ejemplo, los compuestos de fórmula (I) pueden presentar la ventaja de que poseen una solubilidad termodinámica buena o mejorada (p. ej., en comparación con compuestos conocidos en la técnica anterior; y, por ejemplo, según se determina mediante un método conocido y/o un método descrito en la presente). Los compuestos de fórmula (I) también pueden presentar la ventaja de que poseen un espectro de actividad amplio frente a agentes antibacterianos (p. ej., un espectro más amplio de actividad antibacteriana en comparación con compuestos conocidos en la técnica anterior; y, por ejemplo, según se determina mediante pruebas conocidas y/o pruebas descritas en la presente). Los compuestos de fórmula (I) también pueden presentar la ventaja de que poseen una biodisponibilidad oral y unas propiedades farmacocinécticas in vivo buenas o mejoradas. También pueden presentar la ventaja de que presentan una eficacia in vivo buena o mejorada. Por ejemplo, los compuestos de la invención se pueden adaptar para la administración/formulación intravenosa y, por lo tanto, pueden presentar una eficacia in vivo mejorada cuando se administran por vía intravenosa.
Los compuestos de fórmula (I) pueden presentar sorprendentemente las ventajas mencionadas anteriormente o pueden ser sorprendentemente equiparables a compuestos conocidos en la técnica anterior. En particular, puede ser sorprendente que los compuestos de fórmula (I), en virtud de la presencia del anillo que contiene X de 7 miembros relativamente grande, presenten propiedades convenientes o incluso equiparables. Además, ciertos compuestos de fórmula (I) pueden presentar otras ventajas (como las mencionadas anteriormente en la presente), por ejemplo, los compuestos en los que el anillo que contiene X contiene NR4 y en particular aquellos en los que contiene un resto CR4 (p. ej., en donde X es CR4), que es adyacente a un doble enlace. Cualquiera de estas propiedades beneficiosas adicionales se puede atribuir a la presencia de los restos NR4 o CR4 adyacentes a un doble enlace.
Parte experimental
"DMF" se define como W,W-dimetilformamida, "DCM" o "CH2Ch" se define como diclorometano, "MeOH" se define como metanol, "EtOH" se define como etanol, "MgSO4" se define como sulfato de magnesio y "THF" se define como tetrahidrofurano; HATU es hexafluorofosfato de 3-óxido de 1-[bis(dimetilamino)metileno]-1H-1,2,3-triazolo[4,5-ó]piridinio; AcOEt o EtOAc es acetato de etilo; DIPEA es diisopropiletilamina; EDCI se define como monoclorhidrato de W-(etilcarbonimidoil)-W,W-dimetil-1,3-propanodiamina; HOBT significa 1-hidroxi-1H-benzotriazol; K2CO3 significa carbonato de potasio; NH OH se define como hidróxido de amonio; NH Cl se define como cloruro de amonio; N es nitrógeno gaseoso; y TFA significa ácido trifluoroacético.
A. Síntesis de los intermedios
Ejemplo A.1
a) Preparación de  intermedio (1)
intermedio (1)
Una solución de 6-bromo-3,4-dihidro-1H-[1,8]naftiridin-2-ona (1.0 g, 4.4 mmol), acrilato de ferf-butilo (2.56 mL, 17.62 mmol) y W,W-diisopropiletilamina (1.46 mL, 8.81 mmol) en acetonitrilo (20 mL) y DMF (7 mL) se agitó y desgasificó con nitrógeno gaseoso durante 10 minutos. Se añadieron tri-o-tolilfosfina (0.27 g, 0.88 mmol) y acetato de paladio (II) (al 47% en Pd) (0.099 g, 0.44 mol), y la mezcla resultante se sometió a microondas (1600 W, 180 °C, 35 minutos). La mezcla de reacción se evaporó a sequedad, se añadió una mezcla de DCM/metanol (8/2) (50 mL), se filtró a través de un lecho corto de Celite y se lavó con DCM. La capa orgánica se lavó con agua, se secó con MgSO4, se filtró y se evaporó a sequedad. Se añadió etanol frío (10 mL) al residuo y se agitó a 5 °C durante 5 minutos, el precipitado se filtró, se lavó con etanol frío (3 mL) y se secó al vacío para obtener 950 mg del intermedio (1).
b) Preparación de  .CF3COOH interm edio (2)
.CF3COOH interm edio (2)
El intermedio (1) (4.1 g, 14.95 mmol) se disolvió en una mezcla de ácido trifluoroacético (23.2 mL) en DCM (41 mL). La reacción se agitó a temperatura ambiente durante 30 minutos. La mezcla de reacción se concentró a presión reducida. El sólido resultante se lavó con éter dietílico, se separó por filtración y se secó al vacío para obtener 3.97 g del intermedio (2).

El intermedio (2) se lavó durante la noche en una mezcla de HCl en dioxano (4 M, 48 mL), y el sólido se separó por filtración, se lavó con éter dietílico y se secó al vacío para obtener 3.7 g del intermedio (3).
Ejemplo A.2
a) Preparación de  intermedio (4)
intermedio (4)
Una mezcla de clorhidrato de W-bencilhexahidroazepin-4-ona (25.0 g, 104.3 mmol), dicarbonato de di-ferf-butilo (25.0 g, 114.7 mmol) y catalizador de Pearlman (4.46 g, 31.3 mmol) en EtOAc (550 mL) y trietilamina (17.4 mL, 125.13 mmol) se hidrogenó a temperatura ambiente durante la noche en un reactor Parr. La mezcla de reacción se filtró a través de un lecho corto de Celite®, la masa retenida se lavó con EtOAc, el filtrado se lavó con agua y a continuación con salmuera, se secó (MgSÜ4) y se evaporó a sequedad para obtener 23.4 g del intermedio (4).
b) Preparación de  intermedio (5)
intermedio (5)
Reacción en atmósfera de N2. Se añadió cloruro de fenilmagnesio (93.8 mL, 169 mmol) gota a gota a una solución del intermedio (4) (30 g, 141 mmol) en THF (300 mL) a 0 °C y a continuación la mezcla se agitó durante 3 horas a 5 °C. Se añadieron NH4Cl acuoso al 10% y EtOAc, la capa orgánica se separó, se lavó con agua y salmuera, se secó (MgSÜ4) y se evaporó a sequedad para obtener 39.2 g del intermedio (5).
c) Preparación de  intermedio (6)
intermedio (6)
y  intermedio (7)
intermedio (7)
Una solución del intermedio (5) (38.85 g, 133.3 mmol) en HCl (al 35% en agua, 200 mL) se agitó a temperatura ambiente durante 1 hora. La mezcla de reacción se vertió sobre hielo picado y se añadió K2CO3 sólido en porciones (hasta pH = 9-10), y a continuación se extrajo dos veces con DCM. Las capas orgánicas se combinaron, se lavaron con agua, se secaron (MgSO4) y se evaporaron a sequedad. El residuo se purificó mediante cromatografía de líquidos preparativa (en gel de sílice, 20-45 pm, 1000 g, fase móvil (1% de NH4OH, 93% de DCM, 7% de MeOH)). Se recogieron las fracciones puras y se evaporó el disolvente para obtener el intermedio (6) y el intermedio (7).
Ejemplo A.3
a) Preparación de  intermedio (8)
intermedio (8)
Reacción en atmósfera de N2. Se añadió n-butillitio 1.6 M en hexano (6.35 mL, 9.31 mmol) gota a gota a -20 °C a una solución de diisopropilamina (1.43 mL, 10.2 mmol) en THF (15 mL) y a continuación la mezcla se agitó a -20 °C durante 20 minutos. Posteriormente, se añadió una solución del intermedio (4) (1.9 g, 8.46 mmol) en THF (20 mL) a -78 °C y la mezcla resultante se agitó durante 30 minutos a -78 °C. Se añadió una solución de 2-[N,N-bis(trifluorometilsulfonil)amino]-5-cloropiridina (3.8 g, 9.31 mmol) en THF (10 mL) a -78 °C, y a continuación se dejó que la mezcla alcanzara la temperatura ambiente, se agitó durante la noche y se concentró. El residuo se purificó mediante cromatografía en columna de fase normal (gel de sílice, 20-45 pm, 450 g, fase móvil (80% de heptano, 20% de acetato de etilo)). Se recogieron las fracciones puras y se evaporó el disolvente para obtener 1.34 g del intermedio (8).
b) Preparación de  intermedio (9)
intermedio (9)
Reacción en atmósfera de N2. Una solución del intermedio (8) (0.24 g, 0.695 mmol) en THF (2 mL) y bromuro de bencilzinc en THF (0.5 M, 3.34 mL, 1.67 mmol) se desgasificó burbujeando nitrógeno durante 10 minutos y a continuación se añadió 1,1'-bis(difenilfosfino)ferrocenodicloropaladio (II) (0.102 g, 0.139 mmol). La mezcla se sometió a microondas durante 20 minutos, se enfrió hasta temperatura ambiente, se añadieron agua y acetato de etilo, la mezcla se filtró a través de un lecho corto de Celite, y la capa orgánica se separó, se lavó con agua seguida de salmuera, se secó (MgSO4) y se evaporó a sequedad. El residuo se purificó mediante cromatografía flash en un cartucho de gel de sílice corto con una mezcla de heptano a heptano/EtOAc (90/10). Se recogieron las fracciones puras y se evaporaron a sequedad para obtener 0.11 g del intermedio (9).
c) Preparación de  intermedio (10)
intermedio (10)
Una mezcla del intermedio (9) (0.11 g, 0.383 mmol) y TFA (0.3 mL) en DCM (2 mL) se agitó a temperatura ambiente durante 30 minutos y a continuación la mezcla de reacción se vertió sobre K2CO3 (solución acuosa al 10%) y se extrajo con DCM. La capa orgánica se separó, se lavó con agua, se secó (MgSO4) y se evaporó a sequedad para obtener 0.058 g del intermedio (10).
Ejemplo A.4
a) Preparación de  intermedio (11)
intermedio (11)
Reacción en atmósfera de N2. Se añadió bromuro de 3-clorofenilmagnesio (100 mL, 50.0 mmol) gota a gota a una
solución del intermedio (4) (8.9 g, 41.7 mmol) en THF (90 mL) a 0 °C y a continuación la mezcla se agitó durante 3 horas a 5 °C. Se añadieron NH4Cl (solución acuosa al 10%) y EtOAc, la capa orgánica se separó, se lavó con agua y salmuera, se secó (MgSO4) y se evaporó a sequedad. El residuo se purificó mediante cromatografía flash en un cartucho de gel de sílice [15-40 pm, de 80/20 de heptano/EtOAc a 60/40 de heptano/EtOAc]. Se recogieron las fracciones puras y se evaporaron a sequedad para obtener 4.4 g del intermedio (11).
b) Preparación de  intermedio (12)
intermedio (12)
y  intermedio (13)
intermedio (13)
Una solución del intermedio (11) (4.4 g, 13.5 mmol) en HCl en agua (al 35%, 22 mL) se agitó a temperatura ambiente durante 1 hora. La mezcla de reacción se vertió sobre hielo picado y se añadió K2CO3 sólido en porciones (hasta pH = 9-10), y a continuación se extrajo dos veces con DCM. Las capas orgánicas se combinaron, se lavaron con agua, se secaron (MgSO4) y se evaporaron a sequedad. La capa acuosa se evaporó, se añadió DCM y se filtró. Se combinó con el primer extracto y se evaporó a sequedad. El residuo se purificó mediante cromatografía flash en gel de sílice (15-40 pm, 90 g, de DCM a DCM/MeOH/NH4OH: 90/10/0.5). Las fracciones puras se recogieron y se evaporaron a sequedad. El residuo se purificó mediante cromatografía de líquidos preparativa [en gel de sílice, 15 40 pm, 300 g, fase móvil (0.5% de NH4OH, 90% de DCM, 10% de MeOH)]. Se recogieron las fracciones puras y se evaporó el disolvente para obtener 1 g del intermedio (12) y 0.4 g del intermedio (13).
Ejemplo A.5
a) Preparación de  intermedio (14)
intermedio (14)
Reacción en atmósfera de N2. Se añadió n-butillitio en hexano (1.6 M, 3.52 mL, 5.63 mmol) gota a gota a -78 °C a una solución de tiazol (0.366 mL, 5.16 mmol) en éter dietílico (5 mL) y la mezcla se agitó durante 30 minutos. Se añadió una solución del intermedio (4) (1.0 g, 4.69 mmol) en éter dietílico (5 mL), a continuación la mezcla se agitó y se dejó que alcanzara temperatura ambiente durante 2 horas. Se añadieron agua y EtOAc, y la capa orgánica se separó, se lavó con agua seguida de salmuera, se secó (MgSO4) y se evaporó a sequedad. El residuo se purificó mediante cromatografía de líquidos preparativa (gel de sílice, 15-40 pm, 25 g, fase móvil (70% de heptano, 30% de EtOAc)) para obtener 1.05 g del intermedio (14).
b) Preparación de  intermedio (15)
intermedio (15)

El intermedio (14) (710 mg, 2.38 mmol) y HCl concentrado (2 mL) en acetonitrilo (6 mL) se agitaron a reflujo durante 2 días. El disolvente se evaporó. Se añadieron agua y DCM. Se añadió K2CO3 en polvo para basificar la capa acuosa y se retiró la capa orgánica. La capa acuosa se extrajo de nuevo con DCM después de saturar la capa acuosa con K2CO3. Las capas orgánicas combinadas se concentraron, y el residuo se purificó y se separó mediante cromatografía en columna de gel de sílice (15-40 pm, 25 g) para obtener 137 mg del intermedio (15) y 65 mg del intermedio (16).
Ejemplo A.6
a) Preparación de  intermedio (17)
intermedio (17)
Reacción en atmósfera de N2. Se añadió bromuro de 3-(trifluorometil)fenilmagnesio (1.4 g, 5.6 mmol en 10 mL de
éter dietílico) gota a gota a una solución del intermedio (4) (1 g, 4.69 mmol) en THF (15 mL) a 0 °C y a continuación la mezcla se agitó durante 3 horas a 5 °C. Se añadieron NH4Cl (solución acuosa al 10%) y EtOAc, la capa orgánica se separó, se lavó con agua y salmuera, se secó (MgSO4) y se evaporó a sequedad. La purificación se llevó a cabo mediante cromatografía flash en gel de sílice (40 g, heptano/EtOAc partiendo de 85/15). Las fracciones puras se recogieron y se concentraron para obtener 520 mg del intermedio (17).
b) Preparación de  intermedio (18)
intermedio (18)
y  intermedio (19)
intermedio (19)
Una solución del intermedio (17) (400 mg, 1.13 mmol) en HCl (al 37% en agua, 15 mL) se agitó durante 30 minutos a reflujo y después se enfrió hasta temperatura ambiente. La mezcla de reacción se vertió sobre hielo picado y se añadió K2CO3 sólido en porciones (hasta pH = 9-10), y a continuación se extrajo dos veces con DCM. Las capas orgánicas se combinaron, se lavaron con agua, se secaron (MgSO4) y se evaporaron a sequedad. El residuo se purificó mediante cromatografía de líquidos preparativa (gel de sílice, 5 pm, 150x30.0 mm, fase móvil (gradiente de 0.2% de NH4OH, 98% de DCM y 2% de MeOH a 1.2% de NH4OH, 88% de DCM y 12% de MeOH)). Se recogieron las fracciones puras y se evaporó el disolvente para obtener 140 mg del intermedio (18) y 42 mg del intermedio (19). Ejemplo A.7

Reacción en atmósfera de N2. Se añadió bromuro de 3-cloro-5-fluorofenilmagnesio (5 M en THF) (14.1 mL, 7 mmol) gota a gota a una solución del intermedio (4) (1 g, 4.7 mmol) en THF (20 mL) a 0 °C y a continuación la mezcla se agitó durante 3 horas a 5 °C. Se añadieron NH4Cl (solución acuosa al 10%) y EtOAc, la capa orgánica se separó, se lavó con agua y salmuera, se secó (MgSO4) y se evaporó a sequedad. La purificación se llevó a cabo mediante cromatografía flash en gel de sílice (40 g, heptano/EtOAc partiendo de 85/15). Las fracciones puras se recogieron y se concentraron para obtener 900 mg del intermedio (20).
b) Preparación de  intermedio (21)
intermedio (21)

Una solución del intermedio (20) (900 mg, 2.5 mmol) en HCl (al 37% en agua, 30 mL) se agitó durante 30 minutos a reflujo y después se enfrió hasta temperatura ambiente. La mezcla de reacción se vertió sobre hielo picado y se añadió K2CO3 sólido en porciones (hasta pH = 9-10), y a continuación se extrajo dos veces con DCM. Las capas orgánicas se combinaron, se lavaron con agua, se secaron (MgSO4) y se evaporaron a sequedad. El residuo se purificó mediante cromatografía de líquidos preparativa (gel de sílice, 5 pm, 150x30.0 mm, fase móvil (gradiente de 0.2% de NH4OH, 98% de DCM y 2% de MeOH a 1% de NH4OH, 90% de DCM y 10% de MeOH)). Se recogieron dos fracciones y se evaporó el disolvente para obtener 290 mg del intermedio (21) y 80 mg del intermedio (22).
Ejemplo A.8
a) Preparación de intermedio (23)
Reacción en atmósfera de N2. Se añadió bromuro de 3-cloro-5-fluorofenilmagnesio (0.5 M en THF, 18.7 mL, 9.37 mmol) gota a gota a una solución del intermedio (4) (1 g, 4.7 mmol) en THF (20 mL) a 0 °C y a continuación la mezcla se agitó durante 3 horas a 5 °C. Se añadieron NH4Cl (solución acuosa al 10%) y EtOAc. La capa orgánica se separó, se lavó con agua y salmuera, se secó (MgSO4) y se evaporó a sequedad. La purificación se llevó a cabo mediante cromatografía flash en gel de sílice (40 g, heptano/EtOAc partiendo de 85/15). Se recogieron las fracciones puras y se evaporó el disolvente para obtener 650 mg del intermedio (23).
b) Preparación de  intermedio (24)
intermedio (24)
y  intermedio (25)
intermedio (25)
Una solución del intermedio (23) (800 mg, 2.33 mmol) en HCl (al 37% en agua, 25 mL) se agitó durante 30 minutos a reflujo y después se enfrió hasta temperatura ambiente. La mezcla de reacción se vertió sobre hielo picado y se añadió K2CO3 sólido en porciones (hasta pH = 9-10), y a continuación se extrajo dos veces con DCM. Las capas orgánicas se combinaron, se lavaron con agua, se secaron (MgSO4) y se evaporaron a sequedad. El producto crudo se purificó mediante cromatografía de líquidos preparativa (gel de sílice, 5 pm, 150x30.0 mm, fase móvil (gradiente de 0.2% de NH4OH, 98% de DCM y 2% de MeOH a 1% de NH4OH, 90% de DCM y 10% de MeOH)). Se recogieron dos fracciones y se evaporó el disolvente para obtener 325 mg del intermedio (24) y 90 mg del intermedio (25).
Ejemplo A.9
a) Preparación de  intermedio (26)
intermedio (26)
Reacción en atmósfera de N2. Se añadió n-butillitio (1.6 M en hexano, 10.55 mL, 16.88 mmol) gota a gota a -78 °C a una solución de 2-bromotiofeno (1.5 mL, 15.47 mmol) en éter dietílico (7.5 mL) y a continuación la mezcla se agitó durante 30 minutos. Se añadió una solución del intermedio (4) (3 g, 14.07 mmol) en éter dietílico (7.5 mL). La mezcla se agitó y se dejó que alcanzara la temperatura ambiente durante 2 horas. Se añadieron agua y EtOAc, y la capa orgánica se separó, se lavó con agua seguida de salmuera, se secó (MgSO4) y se evaporó a sequedad. El residuo se purificó mediante cromatografía de líquidos preparativa (en gel de sílice, 15-40 pm, 90 g, fase móvil (80% de heptano, 20% de EtOAc)). Se recogieron las fracciones puras y se evaporó el disolvente para obtener 2.65 g del intermedio (26).
b) Preparación de  intermedio (27)
intermedio (27)
El intermedio (26) (6.3 g, 21.18 mmol) y HCl concentrado (15 mL) en ácido acético (45 mL) se agitaron a reflujo durante 45 minutos. Se evaporaron los disolventes. Se añadieron agua y DCM. Se añadió K2CO3 en polvo para basificar y se retiró la fase orgánica. La fase acuosa se saturó con K2CO3 en polvo y se extrajo con una mezcla de disolventes constituida por DCM y metanol (95/5). Ambas fases orgánicas se combinaron, se evaporaron a sequedad y el residuo se purificó mediante cromatografía en columna de gel de sílice (15-40 pm, 100 g) con una mezcla de disolventes constituida por DCM/metanol/NH4OH (92/7/1) para obtener el intermedio (27).
Ejemplo A.10

Reacción en atmósfera de N2. Se añadió bromo(2,3-diclorofenil)magnesio (3.75 g, 15 mmol en 20 mL de éter dietílico) gota a gota a una solución del intermedio (4) (2.1 g, 10 mmol) en THF (20 mL) a 0 °C y a continuación la
mezcla se agitó durante 3 horas a 5 °C. Se añadieron NH4CI (solución acuosa al 10%) y EtOAc, la capa orgánica se separó, se lavó con agua y salmuera, se secó (MgSO4) y se evaporó a sequedad. El producto crudo se cristalizó en 80/20 de heptano/EtOAc y se secó al aire para obtener 700 mg del intermedio (29).
b) Preparación de  intermedio (30)
intermedio (30)
y  intermedio (31)
intermedio (31)
Una solución del intermedio (29) (700 mg, 1.694 mmol) en HCl (al 37% en agua, 20 mL) se agitó durante 30 minutos a reflujo y después se enfrió hasta temperatura ambiente. La mezcla de reacción se vertió sobre hielo picado y se añadió K2CO3 sólido en porciones (hasta pH = 9-10), y a continuación se extrajo dos veces con DCM. Las capas orgánicas se combinaron, se lavaron con agua, se secaron (MgSO4) y se evaporaron a sequedad. El producto crudo se purificó mediante cromatografía de líquidos preparativa (gel de sílice, 5 pm, 150x30.0 mm, fase móvil (gradiente de 0.2% de NH4OH, 98% de DCM y 2% de MeOH a 1.1% de NH4OH, 89% de DCM y 11% de MeOH)). Se recogieron las fracciones puras y se evaporó el disolvente para obtener el intermedio (30) y una segunda fracción. La segunda fracción se purificó mediante cromatografía de líquidos preparativa (gel de sílice, 5 pm, 150x30.0 mm, fase móvil (gradiente de 0.2% de NH4OH, 98% de DCM y 2% de MeOH a 1.1% de NH4OH, 89% de DCM y 11% de MeOH)). Se recogieron las fracciones puras y se evaporó el disolvente para obtener el intermedio (31).
Ejemplo A.11
a) Preparación de  intermedio (32)
intermedio (32)
Reacción en atmósfera de N2. Se añadió bromo[3-(trifluorometoxi)fenil]magnesio (1.1 g, 4.15 mmol en 10 mL de éter dietílico) gota a gota a una solución del intermedio (4) (0.6 g, 2.77 mmol) en THF (10 mL) a 0 °C y a continuación la mezcla se agitó durante 3 horas a 5 °C . Se añadieron NH4Cl (solución acuosa al 10%) y EtOAc, y la capa orgánica se separó, se lavó con agua y salmuera, se secó (MgSO4) y se evaporó a sequedad. La purificación se llevó a cabo mediante cromatografía flash en gel de sílice (40 g, heptano/EtOAc partiendo de 80/20). Las fracciones puras se recogieron y se concentraron para obtener 250 mg del intermedio (32).
b) Preparación de  intermedio (33)
intermedio (33)
y  intermedio (34)
intermedio (34)
Una solución del intermedio (32) (240 mg, 0.639 mmol) en HCl (al 37% en agua, 10 mL) se agitó durante 30 minutos a reflujo y después se enfrió hasta temperatura ambiente. La mezcla de reacción se vertió sobre hielo picado y se añadió K2CO3 sólido en porciones (hasta pH = 9-10), y a continuación se extrajo dos veces con DCM. Las capas orgánicas se combinaron, se lavaron con agua, se secaron (MgSO4) y se evaporaron a sequedad. El residuo (136 mg) se purificó mediante cromatografía en columna de gel de sílice (15-40 pm, 25 g) con una mezcla de disolventes constituida por DCM/metanol/acetonitrilo (92/7/1) para obtener 86 mg del intermedio (33) y 33 mg del intermedio (34).
Ejemplo A.12
n-C -o-a) Preparación de  i intermedio (35)
i intermedio (35)
1
CF3
Reacción en atmósfera de N2. Se añadió bromo[2-(trifluorometoxi)fenil]magnesio (3.63 g, 13.7 mmol en 15 mL de éter dietílico) gota a gota a una solución del intermedio (4) (1.95 g, 9.1 mmol) en THF (20 mL) a 0 °C y a continuación la mezcla se agitó durante 3 horas a 5 °C. Se añadieron NH4Cl (solución acuosa al 10%) y EtOAc, la capa orgánica se separó, se lavó con agua y salmuera, se secó (MgSO4) y se evaporó a sequedad. La purificación se llevó a cabo mediante cromatografía flash en gel de sílice (40 g, heptano/EtOAc partiendo de 80/20 ). Las fracciones puras se recogieron y se concentraron para obtener 550 mg del intermedio (35).
b) Preparación de  intermedio (36)
intermedio (36)
y  intermedio (37)
intermedio (37)
El intermedio (35) (450 mg, 1.2 mmol) y HCl concentrado (1.5 mL) en ácido acético (4.5 mL) se agitaron a reflujo durante la noche. Se evaporaron los disolventes. Se añadieron agua y DCM. Se añadió K2CO3 en polvo para basificar. Se separó la capa orgánica y se evaporó, y el producto crudo (350 mg) se purificó mediante cromatografía de líquidos preparativa (gel de sílice, 5 pm, 150x30.0 mm, fase móvil (gradiente de 0.2% de NH4OH, 98% de DCM y 2% de MeOH a 1.2% de NH4OH, 88% de DCM y 12% de MeOH)). Se recogieron dos fracciones y se evaporó el disolvente para obtener 140 mg del intermedio (36) y 63 mg del intermedio (37).
Ejemplo A.13
Preparación de  intermedio (38)
intermedio (38)
Se añadió clorhidrato de hexahidro-1-(fenilmetil)-4H-azepin-4-ona (56 g, 233 mmol) a Na2CO3 (solución acuosa saturada, 1000 mL) y EtOAc (1000 mL). La mezcla se agitó durante 30 minutos. La capa orgánica se separó y la capa acuosa se extrajo con EtOAc (1000 mL). Las capas orgánicas combinadas se secaron con Na2SO4, se filtraron y se evaporó el disolvente del filtrado. El residuo y dicarbonato de ferf-butilo (66 g, 300 mmol) en EtOAc (800 mL) se hidrogenaron a temperatura ambiente (0.4 MPa) con Pd(OH)2 (15 g) como catalizador. Después de la captación de hidrógeno (1 eq.), el catalizador se separó por filtración y el filtrado se evaporó. El residuo se purificó mediante cromatografía en columna de gel de sílice (eluyente: 3/1 de éter de petróleo/EtOAc). Se recogieron las fracciones del producto y se evaporó el disolvente para obtener 49 g del intermedio (38).
Ejemplo A.14
a) Preparación de  intermedio (39)
intermedio (39)
Se introdujo Mg (0.34 g, 14 mmol), unas pocas gotas de una solución de 1-bromo-3-metoxibenceno (1.1 mL, 9.28 mmol) en THF (5 mL) y yodo (0.01 g) en THF (30 mL) en un matraz de tres bocas anhidro dotado de un suministro de nitrógeno, un embudo y un condensador de reflujo. La mezcla se calentó suavemente hasta el comienzo de la reacción, a continuación se añadió el resto de la solución de 1-bromo-3-metoxibenceno gota a gota a una velocidad que mantuvo el reflujo. Se continuó agitando hasta que el yodo desapareció por completo (aproximadamente 1 hora). A continuación, la mezcla se enfrió hasta 0 °C. Se añadió la solución del intermedio (38) (2.0 g, 9.38 mmol) en THF (10 mL) a la mezcla. La mezcla de reacción se agitó en un baño de hielo y después se calentó hasta temperatura ambiente. La mezcla de reacción se desactivó con NH4Cl saturado (20 mL) y se agitó a temperatura ambiente durante la noche. La capa orgánica se separó y la capa acuosa se extrajo con EtOAc (3 x 50 mL). Las capas orgánicas combinadas se secaron con Na2SO4, se filtraron y se evaporó el disolvente del filtrado. El residuo se purificó mediante cromatografía en columna de gel de sílice (eluyente: 10/1 de éter de petróleo/EtOAc). Se recogieron las fracciones del producto y se evaporó el disolvente para obtener 2.3 g del intermedio (39).

A una solución del intermedio (39) (2.0 g, 6.5 mmol) en DCM (30 mL) se añadió TFA (20 mL) gota a gota a 0 °C. Tras la adición, la mezcla se agitó durante 2 horas a temperatura ambiente. La mezcla de reacción se concentró (<35 °C). La mezcla se repartió entre salmuera (20 mL), Na2CÜ3 (5 g) y EtOAc (20 mL), y la capa acuosa se extrajo con EtOAc (3 x 20 mL). Las capas orgánicas combinadas se secaron con Na2SO4, se filtraron y se evaporó el disolvente. El residuo se purificó mediante cromatografía en columna de gel de sílice (eluyente: 30/1 de DCM/MeOH). Se recogieron las fracciones puras y se evaporó el disolvente para obtener 0.2 g del intermedio (40). Los siguientes compuestos se prepararon utilizando el mismo procedimiento que en el Ejemplo A.14 en el cual el 1-metoxi-3-metilbenceno se reemplazó por 1-bromo-2-metilbenceno, 2-bromo-4-fluoro-1-metoxibenceno, 1-bromo-4-clorobenceno, 1-bromo-2-metoxibenceno, 2-bromo-4-fluoro-1-metilbenceno, 1-bromo-4-metoxibenceno, 1-bromo-3-metoxibenceno, 1-bromo-3-clorobenceno o 1-bromo-2-clorobenceno respectivamente.

Ejemplo A.15

Una solución de 1-bromo-2-fluorobenceno (1.48 g, 8.5 mmol) en THF anhidro (50 mL) se agitó en atmósfera de nitrógeno a -78 °C durante 30 minutos y después se añadió n-butillitio (2.5 M en hexano, 3.5 mL, 10.1 mmol) gota a gota a -78 °C en 5-10 minutos y la mezcla formada se agitó durante 30 minutos. A continuación, se añadió el intermedio (38) (1.5 g, 101 mmol) en THF (10 mL) a través de una jeringa. Tras la adición, se retiró el baño de hielo. La mezcla de reacción se agitó durante 1 hora, después se desactivó con HCl 1 N (200 mL) y la mezcla se extrajo con DCM (3 x 100 mL). Las capas orgánicas combinadas se separaron y se secaron con Na2SO4 anhidro, después se filtraron y se concentraron al vacío.
El residuo se purificó mediante cromatografía en columna de gel de sílice (eluyente: 10/1 de éter de petróleo/EtOAc).
Se recogieron las fracciones puras y se evaporó el disolvente para obtener 1.54 g del intermedio (53).

A una solución del intermedio (53) (1 g, 3.2 mmol) en DCM (20 mL) se añadió TFA (15 mL) gota a gota a 0 °C. Tras la adición, la mezcla se agitó durante 2 horas a temperatura ambiente. La mezcla de reacción se concentró (<35 °C). La mezcla se repartió entre salmuera (5 mL), Na2CO3 (5 g) y EtOAc (50 mL), y la capa acuosa se extrajo con EtOAc (3 x 50 mL). Las capas orgánicas combinadas se secaron con Na2SO4, se filtraron y se evaporó el disolvente. El residuo se purificó mediante cromatografía en columna de gel de sílice (eluyente: 30/1 de DCM/MeOH). Se recogieron las fracciones puras y se evaporó el disolvente para obtener 0.6 g del intermedio (54).
Los siguientes compuestos se prepararon utilizando el mismo procedimiento que en el Ejemplo A.15 en el cual el 1-bromo-2-fluorobenceno se reemplazó por 2-bromo-1-fluoro-3-metoxibenceno o 2-bromo-1,4-dimetilbenceno respectivamente.
Ejemplo A.16

A una solución del intermedio (38) (5 g, 23 mmol) en THF (100 mL) se añadió la sal lítica de /V-(1-metiletil)-2-propanamina (23 mL, 46 mmol) a -78 °C. La mezcla se agitó durante 0.5 horas a -50 °C. Se añadió yodometano (6.5 g, 46 mmol) a la mezcla y se agitó durante la noche a temperatura ambiente. La mezcla de reacción se desactivó con 100 mL de salmuera. La capa orgánica se separó y la capa acuosa se extrajo con EtOAc. Las capas orgánicas se combinaron y se concentraron. El producto crudo se purificó mediante cromatografía en columna de gel de sílice (eluyente: 9/1 de éter de petróleo/EtOAc). Se recogieron las fracciones del producto y se evaporó el disolvente para obtener 3 g del intermedio (57).

A una solución del intermedio (57) (1.7 g, 7.5 mmol) en THF (50 mL) se añadió bromofenilmagnesio (3.7 mL, 11.2 mmol) a 0 °C. La mezcla se agitó durante la noche a temperatura ambiente. La mezcla de reacción se desactivó con 50 mL de salmuera. La capa orgánica se separó y la capa acuosa se extrajo con EtOAc. Las capas orgánicas se combinaron y se concentraron. El producto crudo se purificó mediante cromatografía en columna de gel de sílice (eluyente: 1/1 de éter de petróleo/EtOAc). Se recogieron las fracciones del producto y se evaporó el disolvente para obtener 0.5 g del intermedio (58).

Una mezcla del intermedio (58) (0.5 g, 1.64 mmol) en HCl (10 mL, 6 mol/L en agua) se calentó a reflujo durante la noche. Se eliminó el disolvente a presión reducida. El residuo se disolvió en 20 mL de agua. La solución formada se basificó hasta pH 10 con K2CO3. La solución resultante se extrajo con EtOAc (4 x 50 mL). Las capas orgánicas se
combinaron y se concentraron para obtener 0.3 g del intermedio (59).
Ejemplo A.17

El intermedio (45) (4 mmol) en MeOH (40 mL) se hidrogenó a 40 °C (0.1 MPa) con PtÜ2 (0.5 g) como catalizador. Después de la captación de hidrógeno (1 eq.), el catalizador se separó por filtración y el filtrado se evaporó. El residuo se purificó mediante cromatografía en columna de gel de sílice (eluyente: 40/1 de DCM/MeOH). Se recogieron las fracciones del producto y se evaporó el disolvente para obtener 1 g del intermedio (60).
Ejemplo A.18

Se añadió borohidruro de sodio (0.35 g, 9.38 mmol) lentamente a una solución del intermedio (38) (2 g, 9.38 mmol) en MeOH (20 mL) con flujo de nitrógeno a 0 °C. La mezcla se agitó durante 2 horas a temperatura ambiente. La mezcla se vertió en agua. La capa orgánica se extrajo con EtOAc, se lavó con salmuera, se secó con MgSO4, se filtró y se concentró para obtener 1.62 g del intermedio (61).

Se añadió una solución de cloruro de metanosulfonilo (0.88 mL, 11.35 mmol) en DCM (10 mL) gota a gota a una solución del intermedio (61) (1.88 g, 8.73 mmol) y trietilamina (3.64 mL, 26.2 mmol) en dCm (10 mL). La solución se agitó a temperatura ambiente durante 2 horas. Se añadieron agua y DCM, y la capa orgánica se separó, se secó con MgSO4, se filtró y se concentró para obtener 2.53 g del intermedio (62). El producto se utilizó sin purificación adicional.
Reacción en atmósfera de N2. Se añadió hidruro sódico (dispersión al 60% en aceite mineral, 0.082 g, 2.05 mmol) en porciones a 5 °C a una solución de pirazol (0.14 g, 2.05 mmol) en DMF (10 mL) y la mezcla se agitó durante 30 minutos. Se añadió el intermedio (62) (0.506 g, 1.71 mmol) en DMF (5 mL) gota a gota, se dejó que la mezcla de reacción alcanzara la temperatura ambiente y se agitó durante la noche. Se añadieron agua y EtOAc. La capa orgánica se separó, se lavó con agua seguida de salmuera, se secó (MgSO4) y se evaporó a sequedad para obtener 446 mg del intermedio (62a). El residuo se utilizó como tal en el siguiente paso.

Se añadió TFA (1.23 mL, 15.97 mmol) a una solución del intermedio (62a) (0.446 g, 1.6 mmol) en DCM (4 mL). La mezcla de reacción se agitó a temperatura ambiente durante 3 horas, se añadieron agua y DCM, se añadió K2CO3 al 10% para basificar, y la capa orgánica se separó, se lavó con agua, se secó (MgSO4) y se evaporó a sequedad para obtener 78 mg del intermedio (63).
Los siguientes compuestos se prepararon utilizando el mismo procedimiento que en el Ejemplo A.18 en el cual el 1H-pirazol se reemplazó por 1H-pirrol o 1H-[1,2,3]triazol respectivamente.

Ejemplo A.19

A una solución del intermedio (38) (3 g, 14.1 mmol) en THF (30 mL) se añadió bromometilmagnesio (5.64 mL , 16.92 mmol) a 0 °C. Se añadió solución acuosa saturada de NH4Cl (10 mL). La mezcla formada se extrajo con DCM (2 x 20 mL). Las capas orgánicas combinadas se secaron con MgSO4, se filtraron y se evaporó el disolvente. El residuo se purificó mediante cromatografía en columna de gel de sílice (eluyente: 100/1 de DCM/MeOH). Se recogieron las fracciones deseadas y se evaporó el disolvente para obtener 1.74 g del intermedio (66).

A una solución del intermedio (66) (1.5 g , 6.55 mmol) en benceno (50 mL) se añadió tricloruro de aluminio (4.37 g, 32.75 mmol). La mezcla formada se calentó a reflujo durante la noche. La mezcla de reacción se vertió en hielo. La solución formada se basificó hasta pH 8, se extrajo con DCM (2 x 50 mL). Las capas orgánicas combinadas se secaron con MgSO4, se filtraron y se evaporó el disolvente. El residuo se purificó mediante cromatografía en columna de gel de sílice (eluyente: 10/1 de DCM/MeOH). Se recogieron las fracciones deseadas y se evaporó el disolvente para obtener 520 mg del intermedio (67).
Algunos compuestos intermedios empleados en la preparación de los compuestos finales se pueden adquirir de proveedores comerciales tales como hexahidro-4-fenil-1H-azepina, 2,3,4,5-tetrahidro-1H-3-benzazepina, hexahidro-1-fenil-1H-1,4-diazepina y 4,4-difluorohexahidro-1H-azepina.
B. Síntesis de los compuestos finales y compuestos de referencia
Ejemplo B.1

Una mezcla del intermedio (2) (0.192 g, 0.577 mmol), intermedio (7) (0.15 g, 0.866 mmol), EDCI (0.133 g, 0.693 mmol), HOBT (0.0936 g, 0.693 mmol) y trietilamina (0.193 mL, 1.39 mmol) en DCM (4 mL) y THF (4 mL) se agitó durante la noche a temperatura ambiente. Se añadieron agua y DCM, y la capa orgánica se separó, se lavó con agua, se secó (MgSO4 ) y se evaporó a sequedad. Se añadió etanol al residuo, se filtró y se secó (al vacío) para obtener el compuesto (1).
Ejemplo B.2

Una mezcla del intermedio (40) (0.98 mmol), intermedio (2) (1 mmol), trietilamina (0.5 g) y HATU (0.4 g) en DMF (10 mL) se agitó a temperatura ambiente durante una noche. El disolvente se evaporó. Se añadió DCM (20 mL) al residuo y se lavó con agua (20 mL x 2). La capa orgánica separada se secó (Na2SO4), se filtró y se evaporó el disolvente. El residuo se purificó mediante cromatografía en columna de gel de sílice (eluyente: 5/1 de DCM/MeOH). Se recogieron las fracciones del producto y se evaporó el disolvente para obtener 0.07 g del compuesto (6).
Ejemplo B.3
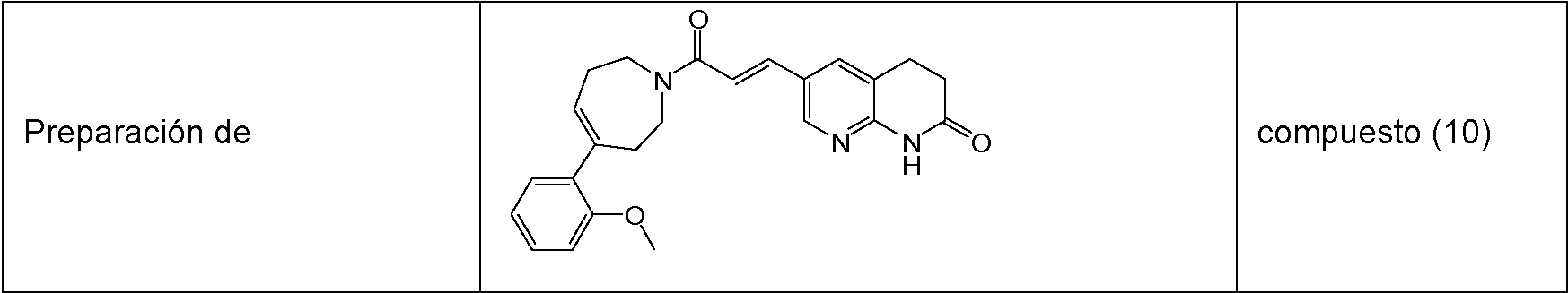
A una mezcla del intermedio (45) (2.03 g, 10 mmol), intermedio (2) (3.3 g, 10 mmol) y HATU (3.80 g, 10 mmol) en DCM (100 mL) se añadió DIPEA (8 mL, 846 mmol) gota a gota en atmósfera de nitrógeno a 0 °C. Una vez finalizada la adición, la mezcla resultante se agitó durante la noche a temperatura ambiente. La mezcla de reacción se repartió entre agua (300 mL) y EtOAc (300 mL), y la capa acuosa se extrajo con EtOAc (4 x 200 mL). Las capas orgánicas combinadas se secaron con Na2SO4, se filtraron y se evaporó el disolvente del filtrado. El residuo se purificó mediante cromatografía en columna de gel de sílice (eluyente: EtOAc). Se recogieron las fracciones del producto y se evaporó el disolvente. El residuo se cristalizó en EtOAc para obtener 1.5 g del compuesto (10).
Ejemplo B.4

Una solución de hexahidro-1-fenil-1H-1,4-diazepina (0.085 g, 0.48 mmol), intermedio (2) (0.16 g, 0.48 mmol), 1-hidroxibenzotriazol (HOBT) (0.078 g, 0.58 mmol), clorhidrato de 1-(3-dimetilaminopropil)-3-etilcarbodiimida (EDCI) (0.11 g, 0.58 mmol) y trietilamina (0.23 mL, 1.69 mmol) en DCM (4 mL) y THF (4 mL) se agitó durante la noche a temperatura ambiente. La mezcla se vertió en agua. La capa orgánica se extrajo con DCM. Las capas orgánicas combinadas se lavaron con salmuera, se secaron con MgSO4, se filtraron y se concentraron. El residuo se cristalizó en acetonitrilo, se filtró y se secó al vacío a 60 °C. El residuo se secó al vacío a 70 °C para obtener 0.079 g del compuesto (11) (pf = 156 °C).
Ejemplo B.5

Una mezcla del intermedio (42) (1.5 g, 6.8 mmol), intermedio (2) (2.7 g, 8.14 mmol), trietilamina (2.2 g, 17 mmol) y EDCI (1.55 g, 8.14 mmol) en DCM (100 mL) se agitó a temperatura ambiente durante una noche. El disolvente se evaporó. El residuo se trató con DCM (100 mL) y la mezcla resultante se lavó con agua (2 x 50 mL). La capa orgánica separada se secó (Na2SO4), se filtró y se evaporó el disolvente. El residuo se purificó mediante
cromatografía en columna de gel de sílice (eluyente: 20/1 de DCM/MeOH). Se recogieron las fracciones del producto y se evaporó el disolvente para obtener 1 g del compuesto (27).
Ejemplo de referencia B.6

Una mezcla del intermedio (67) (0.13 g, 0.688 mmol), intermedio (2) (0.251 g, 0.757 mmol), EDCI (0.145 g, 0.757 mmol), HOBT (0.102 g, 0.757 mmol) y DIPEA (0.445 g, 3.44 mmol) en DCM (50 mL) se agitó durante la noche a temperatura ambiente. Se añadió solución acuosa saturada de NH4Cl (50 mL). La mezcla formada se extrajo con DCM (2 x 20 mL). Las capas orgánicas combinadas se lavaron con solución acuosa saturada de NaHCO3 (30 mL) y salmuera (30 mL), se secaron con MgSO4, se filtraron y se evaporó el disolvente del filtrado. El residuo se purificó mediante cromatografía en columna de gel de sílice (eluyente: 20/1 de DCM/MeOH) y HPLC preparativa. Se recogieron las fracciones deseadas y se evaporó el disolvente para obtener 55 mg del compuesto (28).
La Tabla F-1 enumera los compuestos que se prepararon de acuerdo con alguno de los Ejemplos anteriores.
Tabla F-1





C. Identificación de los compuestos
C1. LCMS
Para la caracterización por LCMS de los compuestos de la presente invención, se emplearon los siguientes métodos.
Procedimiento general A
La medición por LC se llevó a cabo utilizando un sistema UPLC (cromatografía líquida de ultrarresolución) Acquity (Waters) que comprendía una bomba binaria con desgasificador, un automuestreador, un detector de haz de diodos (DAD) y una columna según se especifique en los respectivos métodos más adelante, la columna se mantiene a una temperatura de 40 °C. El flujo procedente de la columna se desvió a un espectrómetro MS. El detector MS se configuró con una fuente de ionización por electronebulización. El voltaje de la aguja capilar fue de 3 kV y la temperatura de la fuente se mantuvo a 130 °C en el Quattro (espectrómetro de masas de triple cuadrupolo de Waters). Se empleó nitrógeno como gas nebulizador. La adquisición de datos se realizó con un procesador de datos Micromass MassLynx-Openlynx de Waters.
Método 1
Además del procedimiento general A: la UPLC en fase inversa se llevó a cabo en una columna C18 Acquity BEH de Waters (partículas híbridas con puente de etilsiloxano/sílice) (1.7 pm, 2.1 x 100 mm) con una tasa de flujo de 0.35 mL/min. Se emplearon dos fases móviles (fase móvil A: 95% de acetato de amonio 7 mM/5% de acetonitrilo; fase móvil B: 100% de acetonitrilo) para un análisis con unas condiciones de gradiente de un 90% de A y un 10% de B (se mantuvo durante 0.5 minutos) a un 8% de A y un 92% de B en 3.5 minutos, se mantuvo durante 2 min y se restauraron las condiciones iniciales en 0.5 min, manteniéndolas durante 1.5 minutos. Se empleó un volumen de inyección de 2 |iL. El voltaje del cono fue de 20 V para los modos de ionización positivo y negativo. Los espectros de
masas se adquirieron por barrido entre 100 y 1000 en 0.2 segundos con tiempo mínimo de lectura entre cada barrido de 0.1 segundos.
Método 2
Además del procedimiento general A: la UPLC en fase inversa se llevó a cabo en una columna C18 Acquity BEH de Waters (partículas híbridas con puente de etilsiloxano/sílice) (1.7 pm, 2.1 x 100 mm) con una tasa de flujo de 0.343 mL/min. Se emplearon dos fases móviles (fase móvil A: 95% de acetato de amonio 7 mM/5% de acetonitrilo; fase móvil B: 100% de acetonitrilo) para un análisis con unas condiciones de gradiente de un 84.2% de A y un 15.8% de B (se mantuvo durante 0.49 minutos) a un 10.5% de A y un 89.5% de B en 2.18 minutos, se mantuvo durante 1.94 min y se restauraron las condiciones iniciales en 0.73 min, manteniéndolas durante 0.73 minutos. Se empleó un volumen de inyección de 2 pL. El voltaje del cono fue de 20V para los modos de ionización positivo y negativo. Los espectros de masas se adquirieron por barrido entre 100 y 1000 en 0.2 segundos con tiempo mínimo de lectura entre cada barrido de 0.1 segundos.
Método 3
Además del procedimiento general A: la UPLC en fase inversa se llevó a cabo en una columna C18 Halo (2.7 pm, 4.6 x 50 mm) con una tasa de flujo de 1.8 mL/min. Se emplearon dos fases móviles (fase móvil A: H O (0.05% de FA); fase móvil B: acetonitrilo (0.05% de FA) para realizar un análisis con unas condiciones de gradiente de un 95% de A y un 5% de B a un 5% de A y un 95% de B desde t = 0 hasta pasado 1 minuto, a continuación se mantuvieron durante 1 minuto, y después de nuevo un 95% de A en 1 minuto y se mantuvieron estas condiciones durante 0.5 minutos. Se empleó un volumen de inyección de 2 pL. El voltaje del cono fue de 20V para los modos de ionización positivo y negativo. Los espectros de masas se adquirieron por barrido entre 100 y 1000 en 0.2 segundos con tiempo mínimo de lectura entre cada barrido de 0.1 segundos.
Tabla C.1 : Datos de LC/MS

C2. Puntos de fusión
Para varios compuestos, se obtuvieron los puntos de fusión con una placa caliente de Kofler que consistía en una placa térmica con un gradiente de temperatura lineal, un puntero movible y una escala de temperatura en grados Celsius.
Para varios compuestos, los puntos de fusión se determinaron con un calorímetro diferencial de barrido (CDB). Los puntos de fusión se midieron con un gradiente de temperatura de 10 °C/minuto. La temperatura máxima fue de 400 °C.
El resto de los puntos de fusión se determinaron con tubos capilares abiertos.
Tabla C.2: Datos de los puntos de fusión


D. Ejemplos farmacológicos
D.1 Inhibición de la enzima FabI: Ensayo de inhibición de la enzima FabI de Staphvlococcus aureus.
Los ensayos de inhibición de la enzima FabI se realizaron en placas de microvaloración de 384 pocillos de área media. Los compuestos se evaluaron en mezclas de ensayo de 40 pL que contenían NaADA 100 mM, pH 6.5 (ADA = ácido W-[2-acetamido]-2iminodiacético), crotonoil-CoA 250 pM, NAd H 625 pM y 50 pg/mL de FabI de S. aureus ATCC 29213. Los inhibidores normalmente variaron en el intervalo de 50 a 0.39 pM. Las mezclas de reacción se incubaron durante 30 minutos a temperatura ambiente y la reacción se detuvo añadiendo tampón Tris 200 mM (pH 9.0) para crear un cambio de pH. El consumo de NADH se monitorizó midiendo el cambio en la absorbancia a 340. Comparando las lecturas de las muestras con aquellas de los controles negativos (ausencia de compuesto) y positivos (ausencia de enzima), se determinó el porcentaje de inhibición de la actividad enzimática de los compuestos. Se creó una curva de ajuste óptimo mediante un método de mínimos cuadrados. A partir de esta curva, se obtuvo el valor de CI50 (expresado en pg/mL) que provocaba el 50% de la inhibición enzimática.
Tabla D.1 : Valores de CI50 para FabI de S. aureus



* = Ref.
D.2 Método in vitro para evaluar los compuestos y determinar su actividad antibacteriana contra varias cepas bacterianas
Preparación de suspensiones bacterianas para las pruebas de susceptibilidad
Se utilizaron las siguientes bacterias: Staphylococcus aureus ATCC 29213, Staphylococcus aureus resistente a la meticilina (MRSA, por sus siglas en inglés) ATCC 700788 y Escherichia coli ATCC 35218. Las bacterias utilizadas en este estudio se cultivaron durante la noche en matraces que contenían 100 mL de caldo de Mueller-Hinton (Difco, núm. de cat. 0757-17) en agua desionizada estéril con agitación a 37 °C. Las soluciones patrón se conservaron a -70 °C hasta su uso.
Las bacterias se incubaron en una placa de agar tripticasa de soya que contenía 5% de sangre de oveja (Becton Dickinson, núm. de cat. 254053) durante 18-24 horas a 35 °C en condiciones aeróbicas (primer pase). Para el segundo pase, se inoculan 5-10 colonias en caldo de Mueller-Hinton virgen y se cultiva durante la noche a 35 °C hasta que se observa turbidez (alcanzando la fase logarítmica) en condiciones aeróbicas. A continuación, la suspensión bacteriana se ajusta hasta 0.5 de densidad de McFarland y se diluye aún más con un factor de dilución de 1:100 en medio de caldo de Mueller Hinton. Esta suspensión se utiliza como inóculo.
Los resultados (para STA (Staphylococcus aureus) ATCC 29213) se presentan más adelante en la tabla D2.
Pruebas de susceptibilidad antibacteriana: Determinación de CI 50
Los ensayos para determinar la CIM (concentración inhibitoria mínima) se realizaron mediante el método de microdilución de caldo en un formato de 96 pocillos (placas de microvaloración de fondo plano) con un volumen final de 0.1 mL de caldo de Mueller Hinton que contenía diluciones en serie con un factor de 2 de los compuestos y en el que se inocularon 5x105 CFU/mL de bacterias (tamaño de inóculo estándar de acuerdo con las directrices del CLSI). Los inhibidores normalmente varían en el intervalo de 63 a 0.49 pM. La concentración final en DMSO en el ensayo fue del 1.25% (concentración tolerable máxima en DMSO = 6%). En los ensayos en los que se evaluó el efecto del suero humano sobre la actividad de los compuestos contra S. aureus, se añadió suero humano en una concentración final del 10%. Las placas se incubaron a 35 °C durante 16-20 horas. Al final de la incubación, se cuantificó el crecimiento bacteriano fluorimétricamente. Para esto, se añadió resazurina a todos los pocillos y las placas se volvieron a incubar. El tiempo de incubación depende del tipo de bacterias. Un cambio de color de azul a rosado indicó el crecimiento de bacterias. La fluorescencia se leyó en un fluorímetro controlado por computadora (Fluoroskan Ascent FL, Labsystems) a una longitud de onda de excitación de 540 nm y una longitud de onda de emisión de 590 nm. El % de inhibición del crecimiento conseguido por los compuestos se calculó de acuerdo con métodos estándar. La CI90 (expresada en pg/mL) se definió como la concentración que produce el 90% de inhibición del crecimiento bacteriano. Un panel de compuestos de referencia se evaluó simultáneamente para superar el control de calidad.
Los resultados se presentan más adelante en la tabla D2 (STA 10% de SH (suero humano)).
Ensayos de citotoxicidad
La citotoxicidad de los compuestos se evaluó usando el ensayo de MTT. Se expusieron células HelaM humanas cultivadas en placas de 96 pocillos a diluciones en serie de los compuestos evaluados (volumen final de 0.2 mL) y se incubaron durante 72 horas a 37 °C y con un 5% de CO2. Los inhibidores normalmente varían en el intervalo de 25 a 0.8 pM. La concentración final en DMSO en el ensayo fue del 0.5%. Se añadió MTT (bromuro de 3-(4,5-d i meti lti azol-2-il)-2,5-d ife n i ltetrazol i o, un tetrazol) y se redujo a formazán púrpura únicamente en las células vivas. La solubilización de los cristales de formazán se consiguió añadiendo 100 pL de 2-propanol. La viabilidad celular se determinó midiendo la absorbancia del formazán reducido, que proporcionaba un color púrpura, a 540 nm y 690 nm. La absorbancia medida a 690 nm se restó automáticamente de la absorbancia a 540 nm para eliminar los efectos de la absorción no específica. El porcentaje de citotoxicidad conseguido por los compuestos se calculó de acuerdo con métodos estándar. La citotoxicidad se reporta como CC50, la concentración que provoca un 50% de reducción de la viabilidad celular.
Los resultados se presentan más adelante en la tabla D2 (TOX HELAM).
Tabla D2 - Datos para los ejemplos representativos


Ejemplo E
E.1 Solubilidad termodinámica/solubilidad en solución acuosa
El perfil de solubilidad en función del pH se obtuvo a temperatura ambiente durante un periodo de 4 días. Se realizó un estudio de la solubilidad en el momento de la saturación para determinar la solubilidad máxima en una solución tampón particular. El compuesto se añadió a la respectiva solución tampón hasta alcanzar el punto de saturación. A continuación, se agitó el matraz durante 4 días a temperatura ambiente. Una vez transcurridos los 4 días, las soluciones se filtraron y se inyectaron en un UPLC, y la concentración se determinó utilizando un método de HPLC genérico.
Resultados

NE = no evaluado
E.2 Espectro antimicrobiano de actividad
Las concentraciones inhibitorias mínimas (CIM) se determinaron de acuerdo con la metodología del Instituto de Estándares Clínicos y de Laboratorio (CLSI, por sus siglas en inglés) contra bacterias aerobias (CLSI M07-A8) (remítase al Instituto de Estándares Clínicos y de Laboratorio. 2009. Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically. documento M07-A8 del CLSI, Vol. 29, N.° 2) mediante el método de microdilución con caldo en medio de caldo de Mueller-Hinton con ajuste de cationes (CA-MHB) para la mayoría de los organismos, salvo para Haemophilus influenza, en cuyo caso se utilizó caldo del medio de prueba de Haemophilus (HTM). En la tabla se pueden encontrar descripciones de los organismos particulares. Cuando fue posible, se evaluaron cepas estándar ATCC.
La densidad del inóculo para las pruebas de susceptibilidad se normalizó para obtener un inóculo final de aproximadamente 5x105 CFU/mL. La CIM del caldo se determinó como la concentración más baja del fármaco que previno un crecimiento visible después de 16-24 horas (según la especie) de incubación a una temperatura comprendida entre 35 °C y 37 °C.
Tabla: Descripción de los organismos particulares evaluados

Se prepararon soluciones patrón de los compuestos en DMSO en concentraciones de 1 mg/mL. Se preparó linezolida en DMSO en una concentración de 2 mg/mL. Se diluyeron las soluciones patrón de todos los compuestos en CA-MHB para obtener un intervalo de diluciones con un factor de 2, dependiendo de la sensibilidad del organismo que se estuviera evaluando.
Resultados (cuando se dispuso de ellos)


E.3 Propiedades farmacocinéticas y biodisponibilidad oral in vivo
Las propiedades farmacocinéticas y biodisponibilidad oral in vivo del compuesto de los ejemplos se investigó/investiga en ratones Swiss macho (alimentados) después de una única administración en bolo intravenosa (i.v.) y oral (p.o.). Para las formulaciones i.v. y p.o., el compuesto se disolvió/disuelve en una solución de HP-p-CD al 20%. El pH de las formulaciones era/es de aproximadamente 4. Todas las formulaciones i.v. fueron isotónicas. Resultados

E.4 Eficacia in vivo
El concepto del estudio del efecto in vivo de un compuesto antibacteriano mediante el tratamiento de ratones infectados por vía intraperitoneal se introdujo en 1911 para la optocina contra los neumococos (Morgenroth y Levy, 1911). La popularidad del modelo se debe a su facilidad de uso con experimentos de corta duración, infecciones reproducibles y parámetros de valoración simples.
Método
Se emplea una cepa ATCC 29213 de S. aureus sensible a la meticilina para infectar ratones Swiss hembra albinos. Un cultivo bacteriano de caldo de infusión de cerebro y corazón (BHI) se inocula el día antes de la infección, se incuba a 37 °C durante la noche y se diluye en caldo BHI virgen hasta la concentración deseada. Se realiza una inyección intraperitoneal (i.p.) de ~5x109 unidades formadoras de colonias (CFU) en uno de los cuadrantes inferiores laterales del abdomen. Tras la inoculación, los ratones se guardan en sus jaulas con observación diaria para detectar el desarrollo de signos de infección o su muerte. Para el tratamiento de ratones, se puede utilizar tanto la vía p.o. como i.v., y cada ratón se trata de forma individual por una sonda nasogástrica o por inyección i.v. En este modo se evalúan tanto soluciones como suspensiones. El parámetro empleado para monitorizar el curso de la infección y el efecto del tratamiento es la muerte o la supervivencia de los animales a los 3 días posinfección. Ya que la muerte también puede deberse a efectos secundarios tóxicos, se incluye un grupo de control no infectado
constituido por 3 ratones tratados con la dosis más alta del compuesto evaluado.
Resultados
Los compuestos de la invención/ejemplos presentan unas buenas propiedades de eficacia in vivo, por ejemplo, los compuestos pueden exhibir tales propiedades según se determina por el % de supervivencia (tras la prueba anterior).
Claims (19)
1. Un compuesto de fórmula (I)

donde
X representa C y uno de los dos enlaces ;r7T;: representa un doble enlace (y el otro representa un enlace sencillo); o
X representa N (en cuyo caso ambos enlaces :;rT;: representan enlaces sencillos);
Z1 representa CH o N;
1
R es hidrógeno, alquilo C o halo;
2
R es hidrógeno, alquilo C o halo;
3
R es hidrógeno, alquilo C 6, hidroxi o halo;
4
R es hidrógeno, alquilo C halo, arilo, heteroarilo, alquilo C sustituido con arilo o alquilo C sustituido con heteroarilo;
3 4 tes, dichos R 3y, cuando los sustituyentes R y R están situados en posiciones adyacen y R 4 se pueden juntar para formar un radical de fórmula =CH-CH=CH-CH=, siempre que X represente carbono y los dos enlaces representen un enlace sencillo;
arilo es fenilo; fenilo sustituido con uno, dos o tres sustituyentes seleccionados cada uno individualmente entre halo, hidroxi, alquilo C 4, polihaloalquilo C 4, alquiloxi C 4, polihaloalquiloxi C 4, ciano, nitro y amino;
heteroarilo es furanilo, tiofenilo, pirrolilo, pirazolilo, imidazolilo, isoxazolilo, tiazolilo, triazolilo, tetrazolilo, isotiazolilo, tiadiazolilo, oxadiazolilo, piridinilo, piridazinilo, pirimidinilo, pirazinilo, benzo[1,3]dioxolilo, benzofuranilo, benzotiazolilo, indolilo, 2,3-dihidro-1H-indolilo, tetrahidrotiofenilo o quinolinilo;
donde cada heteroarilo puede estar sustituido con uno o dos sustituyentes seleccionados cada uno independientemente entre halo, ciano, alquilo C 4, alquiloxi C 4, (alquil C 4)carbonilo o fenilo;
o una de sus sales de adición de ácido farmacéuticamente aceptables.
2. El compuesto según se reivindica en la reivindicación 1, donde el anillo que contiene X representa:

3. El compuesto según se reivindica en la reivindicación 1 o reivindicación 2, donde:
Z1 representa CH;
1
R es hidrógeno o alquilo C 4¡ 2
2
R es hidrógeno o alquilo C .
4. El compuesto según se reivindica en la reivindicación 1 o reivindicación 3 (o si procede, en la reivindicación 2), donde
X representa C y uno de los dos enlaces representa un doble enlace (y el otro representa un enlace sencillo); o
X representa N (en cuyo caso ambos enlaces :;rT;: representan enlaces sencillos);
R 1 es hidrógeno;
R2 es hidrógeno;
3
R es hidrógeno, alquilo C1-6 o halo;
4
R es halo, arilo, heteroarilo o alquilo C16 sustituido con arilo;
4 3 4y, cuando los sustituyentes R 3 y R están situados en posiciones adyacentes, dichos R y R se pueden juntar para formar un radical de fórmula =CH-CH=CH-CH=, siempre que X represente carbono y los dos enlaces representen un enlace sencillo;
arilo es fenilo; fenilo sustituido con uno o dos sustituyentes seleccionados cada uno individualmente entre halo, alquilo C , polihaloalquilo C , alquiloxi C y polihaloalquiloxi C ;
heteroarilo es tiofenilo, pirrolilo, tiazolilo o triazolilo;
o una de sus sales de adición de ácido farmacéuticamente aceptables.
5. El compuesto según se 1 2 reivindica en cualquiera de las reivindicaciones 1-4, donde R es hidrógeno y R es hidrógeno.
3
6. El compuesto según se reivindica en cualquiera de las reivindicaciones 1-5, donde R es representa hidrógeno.
3
7. El compuesto según se reivindica en cualquiera de las reivindicaciones 1-5, donde R representa alquilo C1-4 o halo.
4
8. El compuesto según se reivindica en cualquiera de las reivindicaciones 1-7, donde R es arilo.
4
9. El compuesto según se reivindica en cualquiera de las reivindicaciones 1-7, donde R es heteroarilo.
4
10. El compuesto según se reivindica en cualquiera de las reivindicaciones 1-7, donde R es alquilo C1-6 sustituido con arilo.
11. El compuesto según se reivindica en cualquiera de las reivindicaciones 1 o 3-9, donde X representa carbono y los 3 4 dos enlaces ...... representan un enlace sencillo, y R y R están situados en posiciones adyacentes y se juntan para formar un radical de fórmula =CH-CH=CH-CH=.
12. Un compuesto según se reivindica en la reivindicación 1 seleccionado entre los siguientes:




o una de sus sales farmacéuticamente aceptables.
13. Un compuesto seleccionado entre los siguientes:

o una de sus sales farmacéuticamente aceptables.
14. Una composición farmacéutica que comprende un portador farmacéuticamente aceptable y una cantidad terapéuticamente activa de un compuesto según se reivindica en cualquiera de las reivindicaciones 1-13.
15. Un proceso para preparar una composición farmacéutica según se reivindica en la reivindicación 14, donde una cantidad terapéuticamente activa de un compuesto según se reivindica en cualquiera de las reivindicaciones 1-13 se combina en mezcla íntima con un portador farmacéuticamente aceptable.
16. Un compuesto de fórmula (I) según se define en cualquiera de las reivindicaciones 1-12 o un compuesto de la reivindicación 13 para su uso como medicina.
17. Un compuesto de fórmula (I) según se define en cualquiera de las reivindicaciones 1-12 un compuesto de la
reivindicación 13 para su uso en el tratamiento de infecciones bacterianas.
18. Un compuesto para su uso según se reivindica en la reivindicación 17, donde la infección bacteriana es provocada por una bacteria que expresa una enzima FabI.
19. Un proceso para preparar compuestos de fórmula (I), según se definen en la reivindicación 1:
(i) haciendo reaccionar un intermedio de fórmula (II) con un intermedio de fórmula (III),

(ii) haciendo reaccionar un intermedio de fórmula (V) con un intermedio de fórmula (VI),

donde Xa1 representa un grupo saliente adecuado y los otros elementos son como se han definido en la reivindicación 1;
o; si se desea; un compuesto de fórmula (I) se convierte en una sal de adición de ácido farmacéuticamente aceptable o, por el contrario, una sal de adición de ácido de un compuesto de fórmula (I) se convierte en una forma de base libre con álcali.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP11177115 | 2011-08-10 | ||
| PCT/EP2012/065729 WO2013021051A1 (en) | 2011-08-10 | 2012-08-10 | Antibacterial homopiperidinyl substituted 3,4 dihydro 1h [1,8]naphthyridinones |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2776145T3 true ES2776145T3 (es) | 2020-07-29 |
Family
ID=46639542
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES12744016T Active ES2776145T3 (es) | 2011-08-10 | 2012-08-10 | 3,4-Dihidro-1h-[1,8]naftiridinonas sustituidas con homopiperidinilo antibacterianas |
Country Status (13)
| Country | Link |
|---|---|
| US (1) | US9394295B2 (es) |
| EP (1) | EP2742044B1 (es) |
| JP (2) | JP6521631B2 (es) |
| KR (1) | KR101996866B1 (es) |
| CN (2) | CN108003155A (es) |
| AU (1) | AU2012293618B2 (es) |
| BR (1) | BR112014003146B1 (es) |
| CA (1) | CA2842526C (es) |
| EA (1) | EA038317B1 (es) |
| ES (1) | ES2776145T3 (es) |
| HK (2) | HK1200445A1 (es) |
| MX (1) | MX360856B (es) |
| WO (1) | WO2013021051A1 (es) |
Families Citing this family (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9394295B2 (en) * | 2011-08-10 | 2016-07-19 | Janssen Sciences Ireland Uc | Antibacterial homopiperidinyl substituted 3,4-dihydro-1H-[1,8]naphthyridinones |
| JO3611B1 (ar) | 2011-08-10 | 2020-08-27 | Janssen Sciences Ireland Uc | سايكلو بنتا (سي (بيرول 4,3 ثاني هيدرو 1 hمستبدله [8,1] نافثيريدينونات مضادة للجراثيم |
| US9062075B2 (en) | 2011-12-02 | 2015-06-23 | Aurigene Discovery Technologies Limited | Tetrahydropyridine derivatives as FabI inhibitors |
| US9062002B2 (en) | 2011-12-02 | 2015-06-23 | Aurigene Discovery Technologies Limited | Substituted pyridine derivatives as FabI inhibitors |
| WO2014072930A2 (en) * | 2012-11-09 | 2014-05-15 | Aurigene Discovery Technologies Limited | Fused pyridine derivatives as antibacterial agents |
| WO2014195844A1 (en) | 2013-06-04 | 2014-12-11 | Aurigene Discovery Technologies Limited | TETRAHYDROPYRIDINE DERIVATIVES AS FabI INHIBITORS |
| WO2015071780A1 (en) * | 2013-11-12 | 2015-05-21 | Aurigene Discovery Technologies Limited | Alkylidine substituted heterocyclyl derivatives as anti-bacterial agents |
| US12398151B2 (en) | 2019-03-19 | 2025-08-26 | Suvalent Therapeutics, Inc. | Sumo inhibitor compounds and uses thereof |
Family Cites Families (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4215123A (en) | 1979-05-07 | 1980-07-29 | American Home Products Corporation | Antisecretory 4-oxy-3-carboxy or cyano-1,2-dihydro-2-oxo-1,8-naphthyridine derivatives |
| HU230030B1 (hu) | 1999-10-08 | 2015-05-28 | Debiopharm International Sa | Fab I inhibitorok |
| WO2001026652A1 (en) | 1999-10-08 | 2001-04-19 | Smithkline Beecham Corporation | Fab i inhibitors |
| ATE294578T1 (de) | 1999-10-08 | 2005-05-15 | Affinium Pharm Inc | Fab i hemmer |
| EP1560584B1 (en) | 2001-04-06 | 2009-01-14 | Affinium Pharmaceuticals, Inc. | Fab i inhibitors |
| DK1828167T3 (da) * | 2004-06-04 | 2014-10-20 | Debiopharm Int Sa | Acrylamidderivater som antibiotiske midler |
| US7973060B2 (en) | 2005-10-13 | 2011-07-05 | Crystalgenomics, Inc. | Fab I inhibitor and process for preparing same |
| JP5468899B2 (ja) | 2006-07-20 | 2014-04-09 | アフィニウム ファーマシューティカルズ, インク. | Fabiインヒビターとしてのアクリルアミド誘導体 |
| EP2125802A4 (en) | 2007-02-16 | 2014-08-20 | Debiopharm Int Sa | SALTS, PRODRUGS AND POLYMORPHES OF FAB I INHIBITORS |
| WO2011061214A1 (en) | 2009-11-18 | 2011-05-26 | Fab Pharma Sas | Novel heterocyclic acrylamides and their use as pharmaceuticals |
| US9394295B2 (en) * | 2011-08-10 | 2016-07-19 | Janssen Sciences Ireland Uc | Antibacterial homopiperidinyl substituted 3,4-dihydro-1H-[1,8]naphthyridinones |
-
2012
- 2012-08-10 US US14/237,841 patent/US9394295B2/en active Active
- 2012-08-10 ES ES12744016T patent/ES2776145T3/es active Active
- 2012-08-10 WO PCT/EP2012/065729 patent/WO2013021051A1/en not_active Ceased
- 2012-08-10 CA CA2842526A patent/CA2842526C/en active Active
- 2012-08-10 KR KR1020147004303A patent/KR101996866B1/ko not_active Expired - Fee Related
- 2012-08-10 HK HK15100736.2A patent/HK1200445A1/xx unknown
- 2012-08-10 MX MX2014001598A patent/MX360856B/es active IP Right Grant
- 2012-08-10 CN CN201711445665.3A patent/CN108003155A/zh active Pending
- 2012-08-10 JP JP2014524408A patent/JP6521631B2/ja not_active Expired - Fee Related
- 2012-08-10 AU AU2012293618A patent/AU2012293618B2/en not_active Ceased
- 2012-08-10 EP EP12744016.2A patent/EP2742044B1/en active Active
- 2012-08-10 EA EA201490436A patent/EA038317B1/ru not_active IP Right Cessation
- 2012-08-10 BR BR112014003146-0A patent/BR112014003146B1/pt not_active IP Right Cessation
- 2012-08-10 CN CN201280038607.8A patent/CN104039782A/zh active Pending
-
2017
- 2017-06-07 JP JP2017112304A patent/JP2017222646A/ja not_active Withdrawn
-
2018
- 2018-10-22 HK HK18113482.8A patent/HK1254403A1/zh unknown
Also Published As
| Publication number | Publication date |
|---|---|
| CA2842526A1 (en) | 2013-02-14 |
| KR101996866B1 (ko) | 2019-07-05 |
| MX360856B (es) | 2018-11-20 |
| KR20140072025A (ko) | 2014-06-12 |
| WO2013021051A1 (en) | 2013-02-14 |
| HK1200445A1 (en) | 2015-08-07 |
| AU2012293618B2 (en) | 2017-04-20 |
| JP6521631B2 (ja) | 2019-05-29 |
| BR112014003146B1 (pt) | 2022-03-15 |
| US9394295B2 (en) | 2016-07-19 |
| EP2742044A1 (en) | 2014-06-18 |
| JP2017222646A (ja) | 2017-12-21 |
| EA038317B1 (ru) | 2021-08-09 |
| MX2014001598A (es) | 2014-04-25 |
| CN104039782A (zh) | 2014-09-10 |
| CN108003155A (zh) | 2018-05-08 |
| CA2842526C (en) | 2019-07-23 |
| EP2742044B1 (en) | 2019-12-04 |
| JP2014531403A (ja) | 2014-11-27 |
| HK1254403A1 (zh) | 2019-07-19 |
| EA201490436A1 (ru) | 2014-07-30 |
| BR112014003146A2 (pt) | 2021-01-26 |
| AU2012293618A1 (en) | 2014-02-06 |
| US20140171418A1 (en) | 2014-06-19 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2881681T3 (es) | 3,4-Dihidro-1H-[1,8]naftiridinonas sustituidas con piperidinilo antibacterianas | |
| ES2776145T3 (es) | 3,4-Dihidro-1h-[1,8]naftiridinonas sustituidas con homopiperidinilo antibacterianas | |
| US10526331B2 (en) | Antibacterial cyclopenta[C]pyrrole substituted 3,4-dihydro-1H-[1,8]naphthyridinones | |
| EA043636B1 (ru) | АНТИБАКТЕРИАЛЬНЫЕ 3,4-ДИГИДРО-1Н-[1,8]НАФТИРИДИНОНЫ, ЗАМЕЩЕННЫЕ ЦИКЛОПЕНТА[с]ПИРРОЛОМ |