ES2779152T3 - Derivados de (2-(5-isoindolin-2-il)pirimidin-4-il)-amina como inhibidores de Rho-quinasa para tratar enfermedades autoinmunes - Google Patents
Derivados de (2-(5-isoindolin-2-il)pirimidin-4-il)-amina como inhibidores de Rho-quinasa para tratar enfermedades autoinmunes Download PDFInfo
- Publication number
- ES2779152T3 ES2779152T3 ES14852268T ES14852268T ES2779152T3 ES 2779152 T3 ES2779152 T3 ES 2779152T3 ES 14852268 T ES14852268 T ES 14852268T ES 14852268 T ES14852268 T ES 14852268T ES 2779152 T3 ES2779152 T3 ES 2779152T3
- Authority
- ES
- Spain
- Prior art keywords
- alkyl
- compound
- mmol
- amine
- independently selected
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 0 C[C@@](*)(C=C)c(cc1)c(C=C)cc1*(C(C12)=*C(*(C3)CC(C=C4)=C3CC4OC)=*C1C2N)=C Chemical compound C[C@@](*)(C=C)c(cc1)c(C=C)cc1*(C(C12)=*C(*(C3)CC(C=C4)=C3CC4OC)=*C1C2N)=C 0.000 description 8
- FYZSHLZHWDBLAP-UHFFFAOYSA-N CC1=[I][IH]C=CC(NC(C2)C=CC(N)=C2C=N)=N1 Chemical compound CC1=[I][IH]C=CC(NC(C2)C=CC(N)=C2C=N)=N1 FYZSHLZHWDBLAP-UHFFFAOYSA-N 0.000 description 1
- XUAHQIMFEWIIBE-UHFFFAOYSA-N COC(C(NC(N1)S)=O)C1=O Chemical compound COC(C(NC(N1)S)=O)C1=O XUAHQIMFEWIIBE-UHFFFAOYSA-N 0.000 description 1
- RFGAFUSDCBRIFT-UHFFFAOYSA-N Fc1cccc(CBr)c1CBr Chemical compound Fc1cccc(CBr)c1CBr RFGAFUSDCBRIFT-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D495/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having sulfur atoms as the only ring hetero atoms
- C07D495/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having sulfur atoms as the only ring hetero atoms in which the condensed system contains two hetero rings
- C07D495/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/506—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/519—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/395—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum
- A61K39/39533—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum against materials from animals
- A61K39/3955—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum against materials from animals against proteinaceous materials, e.g. enzymes, hormones, lymphokines
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/04—Drugs for disorders of the alimentary tract or the digestive system for ulcers, gastritis or reflux esophagitis, e.g. antacids, inhibitors of acid secretion, mucosal protectants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/16—Drugs for disorders of the alimentary tract or the digestive system for liver or gallbladder disorders, e.g. hepatoprotective agents, cholagogues, litholytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/06—Antiasthmatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
- A61P15/10—Drugs for genital or sexual disorders; Contraceptives for impotence
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/04—Antipruritics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/06—Antipsoriatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/04—Drugs for skeletal disorders for non-specific disorders of the connective tissue
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/08—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/08—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease
- A61P19/10—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease for osteoporosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P21/00—Drugs for disorders of the muscular or neuromuscular system
- A61P21/04—Drugs for disorders of the muscular or neuromuscular system for myasthenia gravis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/18—Antipsychotics, i.e. neuroleptics; Drugs for mania or schizophrenia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
- A61P27/06—Antiglaucoma agents or miotics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/10—Antimycotics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P33/00—Antiparasitic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/08—Antiallergic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
- A61P5/48—Drugs for disorders of the endocrine system of the pancreatic hormones
- A61P5/50—Drugs for disorders of the endocrine system of the pancreatic hormones for increasing or potentiating the activity of insulin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/02—Antithrombotic agents; Anticoagulants; Platelet aggregation inhibitors
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/12—Antihypertensives
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D491/00—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00
- C07D491/02—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00 in which the condensed system contains two hetero rings
- C07D491/04—Ortho-condensed systems
- C07D491/044—Ortho-condensed systems with only one oxygen atom as ring hetero atom in the oxygen-containing ring
- C07D491/048—Ortho-condensed systems with only one oxygen atom as ring hetero atom in the oxygen-containing ring the oxygen-containing ring being five-membered
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D491/00—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00
- C07D491/02—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00 in which the condensed system contains two hetero rings
- C07D491/04—Ortho-condensed systems
- C07D491/044—Ortho-condensed systems with only one oxygen atom as ring hetero atom in the oxygen-containing ring
- C07D491/052—Ortho-condensed systems with only one oxygen atom as ring hetero atom in the oxygen-containing ring the oxygen-containing ring being six-membered
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/2863—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against receptors for growth factors, growth regulators
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/70—Immunoglobulins specific features characterized by effect upon binding to a cell or to an antigen
- C07K2317/76—Antagonist effect on antigen, e.g. neutralization or inhibition of binding
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Medicinal Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Immunology (AREA)
- Physical Education & Sports Medicine (AREA)
- Diabetes (AREA)
- Neurology (AREA)
- Biomedical Technology (AREA)
- Epidemiology (AREA)
- Rheumatology (AREA)
- Oncology (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Neurosurgery (AREA)
- Endocrinology (AREA)
- Communicable Diseases (AREA)
- Hematology (AREA)
- Pulmonology (AREA)
- Ophthalmology & Optometry (AREA)
- Heart & Thoracic Surgery (AREA)
- Cardiology (AREA)
- Urology & Nephrology (AREA)
- Psychiatry (AREA)
- Dermatology (AREA)
- Obesity (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Molecular Biology (AREA)
- Microbiology (AREA)
- Genetics & Genomics (AREA)
Abstract
Un compuesto de fórmula II: **(Ver fórmula)** R2 se selecciona del grupo que consiste en arilo, heteroarilo, C3-C7 cicloalquilo, un anillo heterocíclico de tres a doce miembros que contiene hasta 3 heteroátomos, cada uno de los cuales puede estar opcionalmente sustituido de 1 a 3 sustituyentes seleccionados independientemente de halo y C1-C6 alquilo; R4 y R5 se seleccionan independientemente del grupo que consiste en H, halo, C1-C8 alquilo, C2-C8 alquenilo, C2-C8 alquinilo, C1-C6 alcoxi, -(C1-C6 alquil)-O-(C1-C6 alquilo), -NR41R42, - (CH2)xNR41R42 y -O-(C1-C6 alquilo)-O-(C1-C6 alquilo); R41 y R42 se seleccionan independientemente del grupo que consiste en H, C1-C8 alquilo, C2-C8 alquenilo, C2-C8 alquinilo y -(C1-C6 alquil)-O-(C1-C6 alquilo); o R41 y R42 pueden tomarse juntos para formar un cicloalquilo o anillo heterocíclico de tres a doce miembros que tiene hasta 3 heteroátomos que es opcionalmente sustituido de 1 a 3 sustituyentes seleccionados independientemente de halo, C1-C6 alquilo y C1-C6 alcoxi; x se selecciona de 0 a 6; o R4 y R5 pueden tomarse juntos para formar un anillo heterocíclico o aromático de tres a doce miembros que tiene hasta 3 heteroátomos que es opcionalmente sustituido de 1 a 3 sustituyentes seleccionados independientemente de halo y C1-C6 alquilo y C1-C6 alcoxi; cada R6 se selecciona independientemente del grupo que consiste en halo, C1-C6 alquilo, C2-C6 alquenilo, C1-C6 alcoxi, -(C1-C6 alquilo)-O- (C1-C 6 alquilo), C3-C7 cicloalquilo, oxo, acilo, - S-(C1-C 6 alquilo), OH, NH2, CN y C1-C3 perfluroalquilo; y d se selecciona de 0 a 3.
Description
DESCRIPCIÓN
Derivados de (2-(5-isoindolin-2-il)pirimidin-4-il)-amina como inhibidores de Rho-quinasa para tratar enfermedades autoinmunes
CAMPO DE LA INVENCIÓN
La invención se refiere a inhibidores de ROCK1 y/o ROCK2 y su uso para inhibir ROCK1 y/o ROCK2 útiles para el tratamiento de enfermedades.
REFERENCIA A SOLICITUDES RELACIONADAS
Esta solicitud reivindica la prioridad de la Solicitud U.S. N.° 61/887.935, depositada el 7 de octubre de 2013.
ANTECEDENTES DE LA INVENCIÓN
La proteína quinasa asociada a Rho (ROCK) es un regulador intracelular clave de la dinámica del citoesqueleto y la motilidad celular. La Rho-quinasa regula varias dianas posteriores de RhoA a través de la fosforilación, que incluyen, por ejemplo, la cadena ligera de miosina, la subunidad de unión a fosfatasa de la cadena ligera de miosina y la LlM-quinasa 2. Estos sustratos regulan la organización y la contractilidad de los filamentos de actina. En las células de músculos lisos, la Rho-quinasa media la sensibilización al calcio y la contracción de los músculos lisos. La inhibición de la Rho-quinasa bloquea la contracción muscular inducida por el agonista de 5-HT y fenilefrina. Cuando se introduce en células musculares no lisas, la Rho-quinasa induce la formación de fibras de estrés y es necesaria para la transformación celular mediada por RhoA. La Rho-quinasa participa en una variedad de procedimientos celulares, que incluyen, entre otros, la adhesión celular, la motilidad y la migración celular, el control del crecimiento, la contracción celular y la citocinesis. La Rho-quinasa también está involucrada en la activación del sistema de transporte de intercambio de Na/H, la formación de fibras de estrés, la activación de aducina y procedimientos fisiológicos tales como la vasoconstricción, la constricción de músculos lisos bronquiales, la proliferación de células de músculos lisos vasculares y de células endoteliales, la agregación plaquetaria y otros.
La inhibición de la actividad de la Rho-quinasa en modelos animales ha demostrado una serie de beneficios de la inhibición de la Rho-quinasa para el tratamiento de enfermedades humanas. Estos incluyen modelos de enfermedades cardiovasculares como hipertensión, aterosclerosis, reestenosis, hipertrofia cardíaca, hipertensión ocular, isquemia cerebral, vasoespasmo cerebral, disfunción eréctil peniana, trastornos del sistema nervioso central como degeneración neuronal y lesión de la médula espinal, y en neoplasias. Se ha demostrado que la inhibición de la actividad de la Rhoquinasa inhibe el crecimiento de las células tumorales y la metástasis, la angiogénesis, los trastornos trombóticos arteriales como la agregación plaquetaria y la agregación de leucocitos, el asma, la regulación de la presión intraocular y la resorción ósea. La inhibición de la actividad de la Rho-quinasa en pacientes tiene beneficios para controlar los vasoespasmos cerebrales y la isquemia después de una hemorragia subaracnoidea, la reducción de la presión intraocular, el aumento de la secreción acuosa ocular mediante la relajación del tejido de malla trabecular, mejorando el flujo sanguíneo al nervio óptico y la protección de células ganglionares saludables.
En los mamíferos, la Rho-quinasa consta de dos isoformas, ROCK1 (ROCKp; p160-ROCK) y ROCK2 (ROCKa). ROCK1 y ROCK2 se expresan y regulan diferencialmente en tejidos específicos. Por ejemplo, ROCK1 se expresa de manera ubicua a niveles relativamente altos, mientras que ROCK2 se expresa preferentemente en el músculo cardíaco y en el cerebro y músculos esqueléticos. Las isoformas también se expresan en algunos tejidos y en una etapa del desarrollo de manera específica. ROCK1 es un sustrato para la escisión por caspasa-3 durante la apoptosis, mientras que ROCK2 no lo es. La calponina básica específica de músculos lisos es fosforilada solo por ROCK2.
Dado el alcance de los procedimientos y enfermedades celulares implicados, son deseables compuestos que inhiban selectivamente una Rho-quinasa, o que inhiban ROCK1 y ROCK2.
WO 03/059913 describe inhibidores de la Rho-quinasa potencialmente indicados para el tratamiento del crecimiento tumoral, la disfunción eréctil y muchas otras posibles indicaciones mediadas por la Rho-quinasa, por ejemplo, la enfermedad coronaria. WO 2012/048222 está dirigida a compuestos de furo[3,2-d]pirimidina como inhibidores de quinasas propuestos en el tratamiento de afecciones y enfermedades especialmente inflamatorias y trastornos y afecciones proliferativas, por ejemplo, cánceres. WO 2009/152325 describe compuestos de diamino-piridina, pirimidina y piridazina como moduladores del receptor H4 en composiciones farmacéuticas y tratamiento de estados de enfermedad, trastornos y afecciones mediadas por la actividad del receptor H4, por ejemplo, alergia, asma y enfermedades autoinmunes. WO 01/40215 incluye compuestos de 2,4-diaminopirimidina propuestos como agentes inmunosupresores, composiciones farmacéuticas que los incluyen, y procedimientos para el tratamiento de, por
ejemplo, enfermedades autoinmunes, inflamación, alergia, rechazo de trasplantes. WO 2010/065721 se refiere a compuestos para estabilizar células y procedimientos de su uso. WO 2010/019392 abarca derivados de purina como moduladores de gamma secretasa y composiciones farmacéuticas que los comprenden, propuestos para tratar enfermedades asociadas con el depósito del péptido beta-amiloide en el cerebro, como la enfermedad de Alzheimer, o para impedir o retrasar la aparición de demencia asociada con tales enfermedades. WO 2008/054599 se refiere a inhibidores de quinasa de ROCK1 y ROCK2, que pueden ser selectivos para ROCK2, y procedimientos para modular las propiedades farmacocinéticas y/o farmacodinámicas de tales compuestos. WO 2015/157556 se refiere al tratamiento de la enfermedad de injerto contra huésped (GVHD) usando compuestos que inhiben ROCK2. WO 2012/112674 describe compuestos, composiciones, kits y procedimientos para tratar afecciones relacionadas con neurodegeneración o enfermedad ocular. US 2012/094999 se refiere a pirimidina, pirrolo-pirimidina, pirrolo-piridina, piridina, purina y compuestos de triazina que se proponen para modular el receptor del factor de crecimiento epidérmico (EGFR), incluidas las HER-quinasas, utilizadas en el tratamiento de enfermedades, trastornos o afecciones. WO 2012/135697 describe materiales y procedimientos para el tratamiento potencial de enfermedades y trastornos asociados con la expresión de quinasas asociadas a Rho (ROCK), por ejemplo, trastornos oncológicos, enfermedades cardiovasculares y trastornos inflamatorios.
RESUMEN DE LA INVENCIÓN
La presente invención se refiere a compuestos que tienen la: fórmula II:

R2 se selecciona del grupo que consiste en arilo, heteroarilo, C3-C7 cicloalquilo, un anillo heterocíclico de tres a doce miembros que contiene hasta 3 heteroátomos, cada uno de los cuales puede estar opcionalmente sustituido de 1 a 3 sustituyentes seleccionados independientemente de halo y C1-C6 alquilo;
R4 y R5 se seleccionan independientemente del grupo que consiste en H, halo, C1-C8 alquilo, C2-C8 alquenilo, C2-C8 alquinilo, C1-C6 alcoxi, -(C1-C6 alquil)-O-(C1-C6 alquilo), -NR41R42,-(CH2)x NR41R42 y -O-(C1-C6 alkyl)-O-(C1-C6 alquilo);
R41 y R42 se seleccionan independientemente del grupo que consiste en H, C1-C8 alquilo, C2-C8 alquenilo, C2-C8 alquinilo, y -(C1-C6 alquil)-O-(C1-C6 alquilo);
o R41 y R42 pueden tomarse juntos para formar un cicloalquilo o anillo heterocíclico de tres a doce miembros que tiene hasta 3 heteroátomos que es opcionalmente sustituido de 1 a 3 sustituyentes seleccionados independientemente de halo, C1-C6 alquilo y C1-C6 alcoxi;
x se selecciona de 0 a 6;
o R4 y R5 pueden tomarse juntos para formar un anillo heterocíclico o aromático de tres a doce miembros que tiene hasta 3 heteroátomos que es opcionalmente sustituido de 1 a 3 sustituyentes seleccionados independientemente de halo y C1-C6 alquilo y C1-C6 alcoxi;
cada R6 se selecciona independientemente del grupo que consiste en halo, C1-C6 alquilo, C2-C6 alquenilo, C1-C6 alcoxi, -(C1-C6 alquil)-O-(C1-C6 alquilo), C3-C7 cicloalquilo, oxo, acilo,-S-(C1-C6 alquilo), OH, NH2, CN y C1-C3 perfluroalquilo; y
d se selecciona de 0 a 3.
La presente invención incluye composiciones farmacéuticas que comprenden los compuestos de la invención y un vehículo y/o diluyentes farmacéuticamente aceptables.
La presente invención incluye composiciones que comprenden un compuesto sustancialmente puro de la invención y una sal, estereoisómero o hidrato farmacéuticamente aceptable de los mismos, y un excipiente y/o diluyentes farmacéuticamente aceptables.
La invención proporciona un compuesto de Fórmula II para su uso en la inhibición de una Rho-quinasa en un mamífero. La invención proporciona un compuesto de Fórmula II para su uso en el tratamiento de un paciente que padece una enfermedad que comprende administrar al paciente que necesita dicho tratamiento una cantidad terapéuticamente efectiva de un compuesto de Fórmula II. En ciertas de tales realizaciones, el compuesto de Fórmula II inhibe su ROCK2. En ciertas de tales realizaciones, el compuesto de Fórmula II inhibe selectivamente ROCK2. Las enfermedades y afecciones no limitantes tratadas según la presente invención incluyen enfermedades cardiovasculares tales como hipertensión, aterosclerosis, reestenosis, hipertrofia cardíaca, hipertensión ocular, isquemia cerebral, vasoespasmo cerebral, disfunción eréctil peniana, trastornos del sistema nervioso central tales como degeneración neuronal y lesión de la médula espinal, trastornos trombóticos arteriales tales como agregación plaquetaria y agregación de leucocitos, asma, regulación de la presión intraocular y resorción ósea. En las neoplasias, la inhibición de la Rho-quinasa inhibe el crecimiento de las células tumorales y la metástasis, y la angiogénesis. La invención proporciona un compuesto de Fórmula II para su uso en el tratamiento de un trastorno autoinmune en un sujeto que comprende administrar al sujeto una cantidad terapéuticamente efectiva de un compuesto de Fórmula II. Los trastornos autoinmunes incluyen, entre otros, artritis reumatoide (esclerosis múltiple), lupus eritematoso sistémico (SLE; lupus), psoriasis, enfermedad de Crohn, dermatitis atópica, eczema o enfermedad de injerto contra huésped (GVHD).
La invención proporciona un compuesto de Fórmula II para su uso en el tratamiento de un trastorno cardiovascular en un sujeto que comprende administrar al sujeto una cantidad terapéuticamente efectiva de un compuesto de Fórmula II. Los trastornos cardiovasculares incluyen, entre otros, hipertensión, arteriosclerosis, reestenosis, hipertrofia cardíaca, hipertensión ocular, isquemia cerebral, vasoespasmo cerebral o disfunción eréctil.
La invención proporciona un compuesto de Fórmula II para su uso en el tratamiento de una inflamación en un sujeto que comprende administrar al sujeto una cantidad terapéuticamente efectiva de un compuesto de Fórmula II. La inflamación incluye, entre otros, asma, inflamación cardiovascular, inflamación renal o arteriosclerosis.
La invención proporciona un compuesto de Fórmula II para su uso en el tratamiento de un trastorno del sistema nervioso central en un sujeto que comprende administrar al sujeto una cantidad terapéuticamente efectiva de un compuesto de Fórmula II. Los trastornos del sistema nervioso central incluyen, entre otros, degeneración neuronal o lesión de la médula espinal, así como enfermedad de Huntington, enfermedad de Parkinson, enfermedad de Alzheimer, esclerosis lateral amiotrófica (ALS) o esclerosis múltiple.
La invención proporciona un compuesto de Fórmula II para su uso en el tratamiento de un trastorno trombótico arterial en un sujeto que comprende administrar al sujeto una cantidad terapéuticamente efectiva de un compuesto de Fórmula II. Ejemplos no limitantes de trastornos trombóticos arteriales son la agregación plaquetaria o la agregación de leucocitos.
La invención proporciona un compuesto de Fórmula II para su uso en el tratamiento de un trastorno fibrótico en un sujeto que comprende administrar al sujeto una cantidad terapéuticamente efectiva de un compuesto de Fórmula II. Ejemplos no limitantes de trastornos fibróticos son fibrosis hepática, fibrosis pulmonar o fibrosis renal.
La invención proporciona un compuesto de Fórmula II para su uso en el mantenimiento de la estabilidad epitelial que comprende administrar al sujeto una cantidad terapéuticamente efectiva de un compuesto de Fórmula II.
La invención proporciona un compuesto de Fórmula II para su uso en el tratamiento del glaucoma o la regulación de la presión intraocular en un sujeto que comprende administrar al sujeto una cantidad terapéuticamente efectiva de un compuesto de Fórmula II. Los ejemplos no limitantes de glaucoma incluyen glaucoma primario de ángulo abierto, glaucoma agudo de ángulo cerrado, glaucoma pigmentario, glaucoma neovascular, glaucoma congénito, glaucoma de tensión normal o glaucoma secundario.
La invención proporciona un compuesto de Fórmula II para su uso en el tratamiento de una enfermedad neoplásica en un sujeto que comprende administrar al sujeto una cantidad terapéuticamente efectiva de un compuesto de Fórmula II. Las enfermedades neoplásicas incluyen, entre otras, un linfoma, carcinoma, leucemia, sarcoma o blastoma, como cáncer de células escamosas, cáncer de pulmón de células pequeñas, cáncer pituitario, cáncer de esófago, astrocitoma, sarcoma de tejidos blandos, cáncer de pulmón de células no pequeñas, adenocarcinoma de pulmón, carcinoma escamoso de pulmón, cáncer del peritoneo, cáncer hepatocelular, cáncer gastrointestinal, cáncer de páncreas, glioblastoma, cáncer cervical, cáncer de ovario, cáncer de hígado, cáncer de vejiga, hepatoma, cáncer de mama, cáncer de colon, cáncer colorrectal, carcinoma endometrial o uterino, carcinoma de glándulas salivales, cáncer de riñón, cáncer de hígado, cáncer de próstata, cáncer de vulva, cáncer de tiroides, carcinoma hepático, cáncer de cerebro, cáncer de endometrio, cáncer de testículo, colangiocarcinoma, carcinoma de vesícula biliar, cáncer gástrico, melanoma o cáncer de la cabeza y cuello.
La invención también proporciona un compuesto de Fórmula II para uso en el tratamiento del síndrome metabólico, resistencia a la insulina, hiperinsulinemia, diabetes tipo 2 o intolerancia a la glucosa en un sujeto que comprende administrar al sujeto una cantidad terapéuticamente efectiva de un compuesto de Fórmula II.
Además, la invención proporciona un compuesto de Fórmula II para su uso en el tratamiento de la osteoporosis o promover la formación ósea en un sujeto que comprende administrar al sujeto una cantidad terapéuticamente efectiva de un compuesto de Fórmula II.
La invención también proporciona un compuesto de Fórmula II para uso en el tratamiento del síndrome metabólico, resistencia a la insulina, hiperinsulinemia, diabetes tipo 2 o intolerancia a la glucosa en un sujeto que comprende administrar al sujeto una cantidad terapéuticamente efectiva de un compuesto de Fórmula II.
La invención también proporciona un compuesto de Fórmula II para su uso en la regulación de TH17 y la función Treg, así como la producción de IL-17 e IL-21 en células del sistema inmune. Por consiguiente, la invención proporciona un compuesto de Fórmula II para su uso en la regulación de respuestas inmunológicas usando inhibidores de Rhoquinasa de Fórmula II.
La invención proporciona un compuesto de Fórmula II para su uso en el tratamiento de un trastorno ocular que tiene un componente angiogénico que comprende administrar al sujeto una cantidad terapéuticamente efectiva de un compuesto de Fórmula II y un inhibidor de la angiogénesis. Los ejemplos no limitantes de tales trastornos oculares incluyen la degeneración macular relacionada con la edad (AMD), la neovascularización coroidea (CNV), el edema macular diabético (DME), la neovascularización del iris, la uveítis, el glaucoma neovascular o la retinitis del prematuro (ROP).
Entre los inhibidores de la angiogénesis se encuentran los antagonistas de VEGR, incluidos los anticuerpos anti-VEGFR2. También se describe en esta invención una región variable de cadena pesada aislada que comprende una secuencia CDR-1H, CDR-2H y CDR-3H, donde:
(i) la secuencia CDR-1H es GFTFSWYX1MX2 (SEQ ID NO:185), donde X1 es V o I, X2 es G o L,
(ii) la secuencia CDR-2H es SIX1X2SGGX3TX4YADSVKG (SEQ ID NO:186), donde X1 es Y o G, X2 es P o S, X3 es A o F, X4 es N o D, y
(iii) la secuencia CDR-3H es GNYFDY (SEQ ID NO:3) o GLAAPRS (SEQ ID NO:11).
También se describe en esta invención una región variable de cadena ligera aislada que comprende un CDR-1L, CDR-2L y CDR-3L, donde
(i) la secuencia CDR-1L es X1GX2X3LX4X5X6X7X8S (SEQ ID NO:187), donde X1 es S, Q, o T, X2 es D, E, o Q, X3 es K, S, N, I, o A, X4 es G o R, X5 es D, S, H, E, o N, X6 es E, Y, Q, R, o N, X7 es Y, F, o S, y X8 es A o S, o SGSX1SNX2X3X4X5X6X7X8 (SEQ ID NO:188), donde X1 es S, o T, X2 es I o L, X3 es E o G, X4 es T, S, o N, X5 es N o Y, X6 es T. P, A, o Y, X7 es V o L, y X8 es N, I, o Y, o X1GX2SX3DX4GX5YDYVS (SEQ ID NO:189), donde X1 es A o T, X2 es S o T, X3 es H, S, o N, X4 es I o V, y X5 es S o A,
(ii) la secuencia CDR-2L es X1X2X3X4X5PS (SEQ ID NO:190), donde X1 es Q, D, T, Y, S, o A, X2 es D, N, S, T, V, o V, X3 es D, N, S, T, o Y, X4 es Q, K, N, o L, y X5 es R o L, y
(iii) donde la secuencia CDR-3L es QX1WX2X3X4X5X6X7X8 (SEQ ID NO:191), donde X1 es A o T, X2 es D o G, X3 es R o ningún aminoácido, X4 es S, F, en N, X5 es S, T, en N, X6 es S, T, o P, X7 es A, V, L, I, o Y, y X8 es V o L, o AX1WDDX2LX3X4X5X6 (SEQ ID NO:192), donde X1 es A, S, o T, X2 es N o S, X3 es N, I, o G, X4 es G o S, X5 es P, W, o V, y X6 es V o L, o MYSTITX1LL (SEQ ID NO:193), donde X1 es A o T.
También se describe en esta invención una región variable de cadena ligera aislada que comprende un CDR-1L, CDR-2L y CDR-3L, donde
(i) la secuencia CDR-1L es RASX1X2X3X4X5X6X7YX8X9 (SEQ ID NO:194), donde X1 es Q, E, o H, X2 es S, R, o N, X3 es V, I, o L, X4 es S, R, G o N, X5 es S o N, X6 es S, N, W, o D, X7 es G o ningún aminoácido, X8 es L o F, y X9 es A, G, M, o S,
(ii) la secuencia CDR-2L es GASX1RAT (SEQ ID NO:195), donde X1 es S, T, I o N, y
(iii) la secuencia CDR-3L es QQX1X2X3X4X5X6X7X8 (SEQ ID NO:196), donde X1 es F o Y, X2 es D, G, o Y, X3 es S, T, o N, X4 es S, L, o W, X5 es P o ningún aminoácido, X6 es P o T, X7 es L, I, V, P, W, o Y, y X8 es T o S.
BREVE DESCRIPCIÓN DE LAS FIGURAS
La Figura 1 muestra los compuestos de la invención.
La Figura 2 muestra que ROCK2 siRNA, y no ROCK1 siRNA, inhibe la secreción de IL-17 e IL-21. Panel A, izquierda: Anti-ROCK1 siRNA redujo la expresión de Rock1 en aproximadamente un 75%. Anti-ROCK2 siRNA redujo la expresión de Rock2 en aproximadamente un 85%. Panel A, derecha: ROCK2 siRNA, y no ROCK1 siRNA, inhibe la expresión de IL-17 e IL-21. No se observó inhibición de IFN-y. Panel B: ROCK2 siRNA, y no ROCK1 siRNA, inhibió la fosforilación de Stat3, IRF4 y RORyt. Panel C: ROCK2 siRNA, y no ROCK1 siRNA, inhibió la fosforilación de MLC. La Figura 3 muestra que los compuestos de la invención regulan la secreción de citoquinas. El panel A muestra que el compuesto del Ejemplo 1 inhibe la secreción de IL-21, pero no INF-y o IL-2. El panel B muestra que la inhibición de la secreción de IL-17 depende de la dosis. Leyenda (A y B): barras de la izquierda 1: KD025 (un inhibidor selectivo de ROCK2); barra central: compuesto del Ejemplo 1 disuelto en HCL; barra derecha; compuesto del Ejemplo 1 disuelto en DMSO.
Las Figuras 4A-C muestran secuencias de región variable de cadena pesada humana, cadena ligera lambda y cadena ligera kappa, respectivamente, de anticuerpos anti-VEGFR2 útiles en la invención. Las regiones encuadradas indican residuos de aminoácidos en regiones determinantes de complementariedad definidas por Kabat y/o Chothia.
La Figura 5 muestra un inhibidor de ROCK (el compuesto del Ejemplo 1) que reduce la parálisis asociada con encefalomielitis autoinmune experimental (EAE). Ratones C57BL/6 fueron inmunizados con péptido MOG35-55 en adyuvante completo de Freund (CFA). Los ratones se trataron a continuación con los compuestos indicados (FTY720 a 10 mg/kg y Compuesto 1 a 150 mg/kg una vez [QD] o dos veces al día [BID]) cuando individualmente tenían signos de parálisis durante tres días. Los cambios de dosis del Compuesto 1 durante el curso del tratamiento están indicados para el grupo 150 BID. FTY720 es un modulador del receptor de 1-fosfato de esfingosina.
La Figura 6A representa secuencias de aminoácidos de cadena pesada de cuatro anticuerpos maduros por afinidad derivados de Mab 138, que contiene el dominio Vh que tiene SEQ ID NO:4. La Figura 6B representa secuencias de aminoácidos de cadena ligera de los mismos cuatro anticuerpos madurados por afinidad derivados de SEQ ID NO:160. La Figura 7A representa la unión de los anticuerpos descritos en esta invención al VEGFR2 humano y murino en comparación con DC101 (un Ab monoclonal murino que se une al VEGFR2 murino) y un anticuerpo de control que se une solo al VEGFR2 humano. Mab 147 se une a VEGFR2 humano y murino. Mab 106 se une al VEGFR2 humano, pero no al VEGFR2 murino. La Figura 7B representa el bloqueo de ligando. Mab 147 bloquea la unión de VEGF humano con VEGFR2 humano y la unión de VEGF murino con VEGFR2 murino. Mab 106 bloquea la unión de VEGF humano con VEGFR2 humano, pero no la unión de VEGF murino con VEGFR2 murino.
La Figura 8A representa la unión de Mab106 y Mab 147 al VEGFR2 humano en HUVEC (células endoteliales de vena umbilical humana) y células endoteliales aórticas porcinas (PAE) porcinas que sobreexpresan KDR (KDR-PAE). La Figura 8B muestra que Mab 147, y no Mab 106, se une a VEGFR2 en células endoteliales murinas MS1. En las Figs.
8A y 8B, el control es un anticuerpo que se une a hVEGFR2, pero no a mVEGFR2.
La Figura 9 muestra la inhibición de la transducción de señal mediada por VEGFR2 por Mab 106 y Mab 147. Mab 106 y Mab 147 inhiben la fosforilación de KDR y p44/42 en células KDR-PAE (Fig. 9A) y en HUv Ec (Fig. 9B) de una manera dependiente de la dosis.
La Figura 10A representa la inhibición de la proliferación de células KDR-PAE por Mab 106, Mab 147 y un anticuerpo de control que se une a hVEGFR2. La Figura 10B muestra la inhibición de la migración celular inducida. La migración de células Kd R-PAE fue inducida con un gradiente de VEGF (50 ng/mL de VEGF (arriba), 100 ng/mL de VEGF (bajo)). El gráfico representa los recuentos de células en presencia de 0,6 |jg/mL, 3 |jg/mL o 15 |jg/mL de anticuerpo Mab 106 o Mab 147.
DESCRIPCIÓN DETALLADA
La presente invención se refiere a compuestos que tienen la: fórmula II:

R2 se selecciona del grupo que consiste en arilo, heteroarilo, C3-C7 cicloaiquiio, un anillo heterocíclico de tres a doce miembros que contiene hasta 3 heteroátomos, cada uno de los cuales puede estar opcionalmente sustituido de 1 a 3 sustituyentes seleccionados independientemente de halo y C1-C6 alquilo;
R4 y R5 se seleccionan independientemente del grupo que consiste en H, halo, C1-C8 alquilo, C2-C8 alquenilo, C2-C8 alquinilo, C1-C6 alcoxi, -(C1-C6 alquil)-O-(C1-Ca alquilo), -NR41R42,-(CH2)x NR41R42 y -O-(C1-Ca alkyl)-O-(C1-Ce alquilo);
R41 y R42 se seleccionan independientemente del grupo que consiste en H, C1-C8 alquilo, C2-C8 alquenilo, C2-C8 alquinilo, y -(C1-C6 alquil)-O-(C1-Ca alquilo);
o R41 y R42 pueden tomarse juntos para formar un cicloalquilo o anillo heterocíclico de tres a doce miembros que tiene hasta 3 heteroátomos que es opcionalmente sustituido de 1 a 3 sustituyentes seleccionados independientemente de halo, C1-C6 alquilo y C1-C6 alcoxi;
x se selecciona de 0 a 6;
o R4 y R5 pueden tomarse juntos para formar un anillo heterocíclico o aromático de tres a doce miembros que tiene hasta 3 heteroátomos que es opcionalmente sustituido de 1 a 3 sustituyentes seleccionados independientemente de halo y C1-C6 alquilo y C1-C6 alcoxi;
cada R6 se selecciona independientemente del grupo que consiste en halo, C1-C6 alquilo, C2-C6 alquenilo, C1-C6 alcoxi, -(C1-C6 alquil)-O-(C1-C6 alquilo), C3-C7 cicloalquilo, oxo, acilo,-S-(C1-C6 alquilo), OH, NH2, CN y C1-C3 perfluroalquilo; y
d se selecciona de 0 a 3.
También se describen en esta invención compuestos que tienen la fórmula I:

o una sal farmacéuticamente aceptable de esta, donde:
R1 se selecciona del grupo que consiste en H o C1-C6 alquilo;
R2 se selecciona del grupo que consiste en arilo, heteroarilo, C3-C7 cicloalquilo, un anillo heterocíclico de tres a doce miembros que contiene hasta 3 heteroátomos, cada uno de los cuales puede estar opcionalmente sustituido de 1 a 3 sustituyentes seleccionados independientemente de halo y C1-C6 alquilo;
X se selecciona de N o CR3;
Y se selecciona de N o CR3;
Z se selecciona de N o CR4;
W se selecciona de CR5;
donde X, Y y Z se seleccionan independientemente y al menos uno de los cuales es N;
donde cada R3 se selecciona independientemente del grupo que consiste en H, C1-C8 alquilo, C2-C8 alquenilo, C2-C8 alquinilo, -(C1-C6 alquil)-O-(Ci -C6 alquilo), -NR31R32, y C1-C6 alquil) -O-(Ci -C6 alquilo);
R31 y R32 se seleccionan independientemente del grupo que consiste en H, C1-C8 alquilo, C2-C8 alquenilo, C2-C8 alquinilo, y -(C1-C6 alquil)-O-(C1-C6 alquilo);
R4 y R5 se seleccionan independientemente del grupo que consiste en H, halo, C1-C8 alquilo, C2-C8 alquenilo, C2-C8 alquinilo, C1-C6 alcoxi, -(C1-C6 alquil)-O-(C1-Ca alquilo), -NR41R42,-(CH2)xNR41R42 y -O-(C1-Ca alquil)-O-(C1-Ce alquilo); R41 y R42 se seleccionan independientemente del grupo que consiste en H, C1-C8 alquilo, C2-C8 alquenilo, C2-C8 alquinilo, y -(C1-C6 alquil)-O-(C1-C6 alquilo);
o R41 y R42 pueden tomarse juntos para formar un cicloalquilo o anillo heterocíclico de tres a doce miembros que tiene hasta 3 heteroátomos que es opcionalmente sustituido de 1 a 3 sustituyentes seleccionados independientemente de halo, C1-C6 alquilo y C1-C6 alcoxi;
x se selecciona de 0 a 6;
o R4 y R5 pueden tomarse juntos para formar un anillo heterocíclico o aromático de tres a doce miembros que tiene hasta 3 heteroátomos que es opcionalmente sustituido de 1 a 3 sustituyentes seleccionados independientemente de halo y C1-C6 alquilo y C1-C6 alcoxi;
a se selecciona de 0 a 2;
b se selecciona de 0 a 2;
donde a o b son seleccionados independientemente y uno de los cuales es al menos 1
A es un anillo heterocíclico o aromático de tres a doce miembros que tiene hasta 3 heteroátomos que está opcionalmente sustituido de 1 a 3 sustituyentes seleccionados independientemente de halo, C1-C6 alquilo, C2-C6 alquenilo, C1-C6 alcoxi, -(C1-C6 alquil)-O-(C1-C6 alquilo), C3-C7 cicloalquilo, oxo, acilo, -S-(C1-C6 alquilo), OH, NH2, CN y C1-C3 perfluroalquilo.
Como se describe en esta invención, R1 es H;
En una realización de la invención, R2 se selecciona de indazol, ciclohexilpiridina, fenilpiridina, fenil-1 W-pirazol, 1H-pirrolo[2,3-b] piridina, 1H-pirazol, piridina, isoquinolina, quinolina y 1,3-tiazolil piridina;
En una realización de la invención, R2 se selecciona de:

Como se describe en esta invención,
X, Z = N; W = CR3, o
X, Y = N; Z = CR4.
En una realización de la invención, R4 y R5 son cada uno H;
En otra realización, R4 y R5 son cada uno -CH3;
En otra realización más, R4 y R5 se toman juntos para formar un anillo de cinco miembros que incluye, entre otros, dihidrofurano.
La invención proporciona compuestos de la fórmula II:

donde R2, R4 y R5 son como se definieron anteriormente;
cada R6 se selecciona independientemente del grupo que consiste en halo, C1-C6 alquilo, C2-C6 alquenilo, C1-C6 alcoxi, -(C1-C6 alquil)-O-(C1-C6 alquilo), C3-C7 cicloalquilo, oxo, acilo, -S-(C1-C6 alquilo), OH, NH2, CN y C1-C3 perfluroalquilo; y
d se selecciona de 0 a 3.
En ciertas realizaciones, la invención proporciona compuestos de la fórmula III:

donde R4 y R5 son como se definieron anteriormente;
cada R6 se selecciona independientemente del grupo que consiste en halo, C1-C6 alquilo, C2-C6 alquenilo, C1-C6 alcoxi, -(C1-C6 alquil)-O-(C1-C6 alquilo), C3-C7 cicloalquilo, oxo, acilo, -S-(C1-C6 alquilo), OH, NH2, CN y C1-C3 perfluroalquilo; y
d se selecciona de 0 a 3.
El término "heteroátomo", tal como se usa en esta invención, significa un átomo de cualquier elemento que no sea carbono o hidrógeno. Heteroátomos preferidos son nitrógeno, oxígeno y azufre.
El término "alquilo" se refiere al radical de grupos alifáticos saturados, que incluyen grupos alquilo de cadena lineal y grupos alquilo de cadena ramificada. En realizaciones preferidas de la invención, un alquilo de cadena lineal o ramificada tiene 10 átomos de carbono o menos en su cadena principal (por ejemplo, C1-C10 para cadena lineal, C3-C10 para cadena ramificada). Asimismo, cicloalquilos preferidos tienen entre3y 10 átomos de carbono en su estructura de anillo, y más preferentemente, tienen de 3 a 6 carbonos en la estructura del anillo.
A menos que el número de carbonos sea especificado de otra forma, "alquilo inferior" como se usa en esta invención significa un grupo alquilo, como se definió anteriormente, pero que tiene de uno a seis carbonos, y más preferentemente de uno a cuatro átomos de carbono. Del mismo modo, "alquenilo inferior" y "alquinilo inferior" tienen longitudes de cadena similares (C2-C6). Grupos alquilo preferidos son alquilos inferiores. En realizaciones preferidas de la invención, un sustituyente designado en esta invención como alquilo es un alquilo inferior.
El término "cicloalquilo" se refiere a grupos carbocíclicos saturados que tienen de 3 a 7 carbonos en el anillo. Grupos cicloalquilo preferidos incluyen ciclopropilo, ciclobutilo, ciclopentilo y ciclohexilo.
El término "aralquilo", tal como se utiliza en esta invención, hace referencia a un grupo alquilo sustituido con un grupo arilo (por ejemplo, un grupo aromático o heteroaromático).
Los términos "alquenilo" y "alquinilo" hacen referencia a grupos alifáticos insaturados análogos en longitud y posible sustitución a los alquilos descritos anteriormente, pero que contienen al menos un enlace doble o triple respectivamente.
El término "arilo", como se usa en esta invención, incluye grupos aromáticos de anillo simple de 5 y 6 miembros que pueden incluir de cero a cuatro heteroátomos, por ejemplo, benceno, pireno, pirrol, furano, tiofeno, imidazol, oxazol, tiazol, triazol, pirazol, piridina, pirazina, piridazina y pirimidina, y similares. Aquellos grupos arilo que tienen heteroátomos en la estructura del anillo también se pueden denominar "heterociclos arilo", "heteroaromáticos" o "heteroarilo". El anillo aromático se puede sustituir, en una o más posiciones del anillo, por sustituyentes como los descritos anteriormente, por ejemplo, halógeno, azida, alquilo, aralquilo, alquenilo, alquinilo, cicloalquilo, hidroxilo, alcoxilo, amino, nitro, sulfhidrilo, imino, amido, fosfonato, fosfinato, carbonilo, carboxilo, sililo, éter, alquiltio, sulfonilo, sulfonamido, cetona, aldehído, éster, heterociclilo, fracciones aromáticas o heteroaromáticas, -CF3, -CN o similares. El término "arilo" también incluye sistemas de anillos policíclicos con dos o más anillos cíclicos donde dos o más carbonos son comunes a dos anillos contiguos (los anillos son "anillos fusionados") donde al menos uno de los anillos es aromático, por ejemplo, los demás anillos cíclicos pueden ser cicloalquilos, cicloalquenilos, arilos y/o grupos heterocíclilos.
Los términos "heterociclilo" o "grupo heterocíclico" se refieren a estructuras de anillo de 3 a 10 miembros, más preferentemente anillos de 5 o 6 miembros, cuyas estructuras de anillo incluyen de 1 a 4 heteroátomos. Los heterociclos también pueden ser policiclos. Los grupos heterocíclicos incluyen, por ejemplo, tiofeno, tiantreno, furano, pirano, isobenzofurano, cromeno, xanteno, fenoxatiin, pirrol, imidazol, pirazol, isotiazol, isoxazol, piridina, pirazina, pirimidina, piridazina, indolizina, isoindol, indol, indazol, purina, quinolizina, isoquinolina, quinolina, ftalazina, naftiridina, quinoxalina, quinazolina, cinolina, pteridina, carbazol, carbolina, fenantridina, acridina, pirimidina, fenantrolina, fenazina, fenarsazina, fenotiazina, furazano, fenoxazina, pirrolidina, oxolano, tiolano, oxazol, piperidina, piperazina, morfolina, lactonas, lactamas tales como azetidinonas y pirrolidinonas, sultamas, sultonas y similares. El anillo heterocíclico se puede sustituir, en una o más posiciones por sustituyentes como los descritos anteriormente, por ejemplo, halógeno, alquilo, aralquilo, alquenilo, alquinilo, cicloalquilo, hidroxilo, alcoxilo, amino, nitro, sulfhidrilo, imino, amido, fosfonato, fosfinato, carbonilo, carboxilo, sililo, éter, alquiltio, sulfonilo, cetona, aldehído, éster, un heterociclilo, una fracción aromática o heteroaromática, -CF3, -CN o similares.
Los términos "policiclilo" o "grupo policíclico" se refieren a dos o más anillos (por ejemplo, cicloalquilos, cicloalquenilos, arilos y/o heterociclilos) en los que dos o más carbonos son comunes a dos anillos adyacentes, por ejemplo, los anillos son "anillos fusionados". Los anillos que se unen a través de átomos no adyacentes se denominan anillos "puenteados". Cada uno de los anillos de un grupo policíclico se puede sustituir por sustituyentes como los descritos anteriormente, por ejemplo, halógeno, alquilo, aralquilo, alquenilo, alquinilo, cicloalquilo, hidroxilo, amino, nitro, sulfhidrilo, imino, amido, fosfonato, fosfinato, carbonilo, carboxilo, sililo, éter, alquiltio, sulfonilo, cetona, aldehído, éster, un heterociclilo, una fracción aromática o heteroaromática, -CF3, -CN o similares.
Como se usa en esta invención, el término "nitro" significa -NO2. El término "halógeno" o "halo" designa -F, -Cl, -Br o -I. El término "hidroxilo" significa -OH.
Los términos "amina" y "amino" se refieren tanto a aminas no sustituidas como aminas sustituidas, por ejemplo, una fracción que puede representarse mediante la fórmula general:
R

k»
donde R, R' y R" representan cada uno independientemente H, alquilo, alquenilo, alquinilo, aralquilo, arilo y grupos heterocíclicos, y más preferentemente H o alquilo inferior.
Los términos «alcoxilo» o «alcoxi», tal como se utilizan en esta invención, hacen referencia a un grupo alquilo, tal como se definió anteriormente, que tiene un radical oxígeno unido a este. Los grupos alcoxilo representativos incluyen metoxi, etoxi, propiloxi, terc-butoxi y similares. El término alcoxi inferior se refiere a un grupo alcoxi que tiene de 1 a 6 átomos de carbono.
El término "oxo", como se usa en esta invención, se refiere a un átomo de oxígeno que tiene un doble enlace con otro átomo, particularmente carbono o azufre.
Tal como se usa en esta invención, la definición de cada expresión, por ejemplo, alquilo, m, n, R, etc., cuando se produce más de una vez en cualquier estructura, pretende ser independiente de su definición en otra parte en la misma estructura.
Se entenderá que "sustituido", "sustitución" o "sustituido con" incluye la condición implícita de que dicha sustitución está según la valencia permitida del átomo sustituido y el sustituyente, y que la sustitución da como resultado un compuesto estable, por ejemplo, que no sufre una transformación espontánea, como por reordenamiento, ciclación, eliminación, etc.
Como se usa en esta invención, se contempla que el término "sustituido" incluya todos los sustituyentes permitidos de compuestos orgánicos. En un aspecto amplio, los sustituyentes permisibles incluyen sustituyentes acíclicos y cíclicos, ramificados y no ramificados, carbocíclicos y heterocíclicos, aromáticos y no aromáticos de compuestos orgánicos. Sustituyentes ilustrativos incluyen, por ejemplo, aquellos descritos anteriormente en esta invención.
Ciertos compuestos de la presente invención pueden existir en formas geométricas o estereoisoméricas particulares. La presente invención contempla todos estos compuestos, incluidos cis- y frans-isómeros, R - y S -enantiómeros, diastereómeros, las mezclas racémicas de los mismos y otras mezclas de los mismos, dentro del alcance de la invención. Pueden estar presentes átomos de carbono asimétricos adicionales en un sustituyente tal como un grupo alquilo. Todos estos isómeros, así como sus mezclas, están incluidos en esta invención.
Ciertas realizaciones de los presentes compuestos pueden contener un grupo funcional básico, tal como amino o alquilamino, y son, por lo tanto, capaces de formar sales farmacéuticamente aceptables con ácidos farmacéuticamente aceptables. El término "sales farmacéuticamente aceptables" en este contexto, se refiere a las sales de adición de ácido inorgánico y orgánico relativamente no tóxicas de los compuestos de la presente invención. Sales representativas incluyen las sales de bromhidrato, clorhidrato, sulfato, bisulfato, fosfato, nitrato, acetato, estearato, laurato, benzoato, lactato, fosfato, tosilato, citrato, maleato, fumarato, succinato, tartrato, naftilato y mesilato y similares. (vER, Por ejemplo, Berge y col. "Pharmaceutical Salts", J. Pharm. Sci. (1977) 66:1-19).
En otros casos, los compuestos de la presente invención pueden contener uno o más grupos funcionales ácidos y, por lo tanto, son capaces de formar sales farmacéuticamente aceptables con bases farmacéuticamente aceptables. Sales representativas incluyen sales alcalinas o alcalinotérreas como las sales de litio, sodio, potasio, calcio magnesio y similares. Aminas orgánicas representativas útiles para la formación de sales de adición de bases incluyen etilamina, dietilamina, etilendiamina, etanolamina, dietanolamina, piperazina y similares. (Ver, por ejemplo, Berge y col., supra). En un aspecto, la presente invención proporciona compuestos de Fórmula II que son inhibidores de Rho-quinasa. La Rho-quinasa (ROCK), una serina/treonina quinasa, sirve como proteína diana para la pequeña proteína de unión a GTP Rho, y es un mediador importante de numerosas funciones celulares, incluidas las adhesiones focales, la motilidad, la contracción del músculo liso y la citocinesis. En el músculo liso, ROCK juega un papel importante en la sensibilización a Ca2+ y el control del tono vascular. Modula el nivel de fosforilación de la cadena ligera de miosina II de la miosina II, principalmente a través de la inhibición de la miosina fosfatasa, y contribuye a la sensibilización al Ca2+ inducido por agonistas en la contracción del músculo liso.
La Rho-quinasa se encuentra en dos formas, ROCK 1 (ROCKp; p160-ROCK) y ROCK 2 (ROCKa). En algunas realizaciones, el compuesto de Fórmula II inhibe selectivamente ROCK1. En algunas realizaciones, el compuesto de Fórmula II inhibe selectivamente ROCK2. En algunas realizaciones, el compuesto de Fórmula II no es selectivo con respecto a la inhibición de ROCK1 y ROCK2.
Los procedimientos para determinar la inhibición de la quinasa son bien conocidos en la técnica. Por ejemplo, la actividad de la quinasa de una enzima y la capacidad inhibitoria de un compuesto de prueba se pueden determinar midiendo la fosforilación específica de enzima de un sustrato. Se pueden emplear ensayos y kits comerciales. Por ejemplo, la inhibición de la quinasa se puede determinar usando un ensayo IMAP® (Molecular Devices). Este método de ensayo implica el uso de un sustrato peptídico marcado con fluorescencia. La fosforilación del péptido marcado por una quinasa de interés promueve la unión del péptido a una nanopartícula basada en metal trivalente a través de la interacción específica de alta afinidad entre el grupo fosfo y el metal trivalente. La proximidad a las nanopartículas da como resultado un aumento de la polarización de fluorescencia. La inhibición de la quinasa por un inhibidor de quinasa impide la fosforilación del sustrato y, por lo tanto, limita la unión del sustrato marcado con fluorescencia a la nanopartícula. Tal ensayo puede ser compatible con un formato de ensayo con micropocillos, lo que permite la determinación simultánea de IC50 de múltiples compuestos.
En otro aspecto de la presente invención, se proporciona un compuesto de Fórmula II para su uso en el tratamiento de un paciente que padece una enfermedad que comprende administrar a un paciente que necesita dicho tratamiento una cantidad terapéuticamente efectiva de un compuesto de la presente invención. La frase "cantidad terapéuticamente efectiva" como se usa en esta invención significa la cantidad de un compuesto, material o
composición que comprende un compuesto de la presente invención que es efectivo para producir algún efecto terapéutico deseado en al menos una subpoblación de células en un animal con una relación beneficio/riesgo razonable aplicable a cualquier tratamiento médico, por ejemplo, efectos secundarios razonables aplicables a cualquier tratamiento médico.
Los compuestos de la invención que inhiben la fosforilación mediada por Rho-quinasa y/o Rho-quinasa son útiles para el tratamiento de pacientes que padecen enfermedades cardiovasculares y no cardiovasculares que implican la función de la Rho-quinasa, tales como hipertensión, hipertensión pulmonar, aterosclerosis, reestenosis, enfermedad coronaria, hipertrofia cardíaca, hipertensión ocular, retinopatía, enfermedades isquémicas, isquemia cerebral, vasoespasmo cerebral, disfunción eréctil peniana, trastorno circulatorio periférico, enfermedad oclusiva de la arteria periférica, glaucoma (por ejemplo, regulación de la presión intraocular), pulmón fibroide, hígado fibroide, riñón fibroide, enfermedad pulmonar obstructiva crónica (COPD), síndrome de dificultad respiratoria del adulto, trastornos del sistema nervioso central como degeneración neuronal y lesión de la médula espinal. Además, los inhibidores de Rho-quinasa de la invención pueden usarse para tratar trastornos trombóticos arteriales tales como la agregación de plaquetas, la agregación de leucocitos y la resorción ósea.
En una realización de la invención, los compuestos se usan para tratar la malformación cavernosa cerebral (CCM). Las CCM son lesiones vasculares que consisten en grupos de capilares permeables y dilatados y están asociadas con trastornos del sistema nervioso central (CNS), que incluyen convulsiones y derrames cerebrales. Se cree que la pérdida de integridad vascular implica la activación de RhoA y la activación de ROCK, lo que lleva a cambios en la estabilidad del citoesqueleto y al aumento de la permeabilidad vascular. Los compuestos de la invención inhiben la activación de ROCK y restablecen la función endotelial vascular.
Como se indica, en ciertas realizaciones, se usa un compuesto de Fórmula II para tratar el glaucoma. Existen varios tipos de glaucoma que se pueden tratar, incluidos, entre otros, los siguientes. Los dos más comunes, el glaucoma primario de ángulo abierto y el glaucoma agudo de ángulo cerrado se caracterizan por una alta presión ocular. El glaucoma pigmentario y el glaucoma congénito también se caracterizan por secreción de fluido reducida y alta presión intraocular (IOP). Se cree que el glaucoma de tensión normal se debe a otro mecanismo, en particular flujo sanguíneo deficiente al nervio óptico. El glaucoma secundario puede resultar de una lesión, infección, inflamación, tumor o cataratas, y también se asocia con el uso prolongado de esteroides, hipertensión sistémica, retinopatía diabética y oclusión de la vena central de la retina.
En ciertas realizaciones, se usa un inhibidor de Rho-quinasa de la invención para tratar la inflamación, que incluye, pero no se limita a, asma, inflamación cardiovascular, inflamación renal, aterosclerosis y arteriosclerosis.
La invención proporciona un compuesto de fórmula II para uso en el tratamiento del glaucoma que comprende administrar, a un paciente que lo necesite, una cantidad efectiva de un inhibidor de Rho-quinasa. El inhibidor de Rhoquinasa es un compuesto de cualquiera de las Fórmulas I-XXV. El inhibidor de Rho-quinasa puede ser no selectivo con respecto a ROCK1 y ROCK2, o puede ser un inhibidor selectivo de ROCK1 o un inhibidor selectivo de ROCK2. Generalmente, se prefiere que el inhibidor inhiba ROCK1, es decir, inhiba tanto ROCK1 como ROCK2 o sea selectivo para ROCK1. En el contexto de esta invención, selectivo significa que el inhibidor demuestra un IC50 que es al menos 2 veces, al menos 5 veces, al menos 10 veces o al menos 25 veces menor para una Rho-quinasa en comparación con el IC50 para la otra Rho-quinasa. Como se discutió anteriormente, hay varios tipos de glaucomas, los compuestos selectivos para ROCK1 o ROCK2 pueden ser beneficiosos para tratar ciertos tipos. Además, ciertos glaucomas que tienen un componente neovascular pueden beneficiarse de la administración de un inhibidor de angiogénesis además de un inhibidor de ROCK.
Los inhibidores de Rho-quinasa de la invención inhiben el crecimiento y la metástasis de las células tumorales y la angiogénesis, y son útiles para tratar enfermedades neoplásicas. Las enfermedades neoplásicas incluyen cualquier crecimiento o tumor maligno causado por una división celular anormal o incontrolada, y pueden extenderse a otras partes del cuerpo a través del sistema linfático o el torrente sanguíneo. La enfermedad neoplásica incluye, entre otros, linfoma (una neoplasia de tejido linfático que generalmente es maligno), carcinoma (cualquier tumor maligno derivado del tejido epitelial), leucemia (neoplasia maligna de tejidos formadores de sangre; caracterizada por una proliferación anormal de leucocitos), sarcoma (un tumor generalmente maligno que surge del tejido conectivo (hueso o músculo, etc.), y blastoma (malignidad en células precursoras). Ejemplos no limitativos incluyen cáncer de células escamosas, cáncer de pulmón de células pequeñas, cáncer pituitario, cáncer de esófago, astrocitoma, sarcoma de tejidos blandos, cáncer de pulmón de células no pequeñas, adenocarcinoma de pulmón, carcinoma escamoso de pulmón, cáncer del peritoneo, cáncer hepatocelular, cáncer gastrointestinal, cáncer de páncreas, glioblastoma, cáncer cervical, cáncer de ovario, cáncer de hígado, cáncer de vejiga, hepatoma, cáncer de mama, cáncer de colon, cáncer colorrectal, carcinoma endometrial o uterino, carcinoma de glándulas salivales, cáncer de riñón, cáncer de hígado, cáncer de próstata, cáncer de vulva, cáncer de tiroides, carcinoma hepático, cáncer de cerebro, cáncer de endometrio, cáncer de testículo, colangiocarcinoma, carcinoma de vesícula biliar, cáncer gástrico, melanoma o cáncer de la cabeza y cuello.
Según la invención, inhibidores de ROCK se usan para efectuar pérdida de peso y/o limitar aumento de peso. En una realización preferida de la invención, el inhibidor de ROCK es selectivo para ROCK2. Los inhibidores de ROCK-2 promueven la pérdida de peso en sujetos normales y limitan el aumento de peso en sujetos propensos a la obesidad.
En una realización de la invención, se usa un inhibidor de ROCK para reducir o impedir la resistencia a la insulina o restaurar la sensibilidad a la insulina. Por consiguiente, en una realización, los compuestos de la invención se usan para promover o restaurar la absorción de glucosa dependiente de insulina. En otra realización de la invención, se usa un inhibidor de ROCK de la invención para promover o restaurar la tolerancia a la glucosa. En otra realización de la invención, se usa un inhibidor de ROCK de la invención para tratar síndrome metabólico. En otra realización, se usa un inhibidor de ROCK de la invención para reducir o impedir la hiperinsulinemia. En una realización de la invención, se usa un inhibidor de ROCK para tratar la diabetes (particularmente diabetes tipo 2). Los inhibidores de ROCK de la invención también se pueden usar para promover o restaurar la relajación mediada por insulina de las células del músculo liso vascular (VSMC). En realizaciones preferidas de la invención, el inhibidor de ROCK es selectivo para ROCK2.
Los compuestos de fórmula II demuestran la penetración efectiva de la barrera hematoencefálica (BBB) y la distribución de tejidos a los tejidos del sistema nervioso central. Por lo tanto, los compuestos de la invención son útiles para el tratamiento de trastornos del sistema nervioso central, así como trastornos, tales como ciertos trastornos oculares, que se benefician de la capacidad de cruzar la BBB. Dichos trastornos pueden implicar degeneración neuronal o lesiones físicas en el tejido neural, incluyendo, entre otros, la enfermedad de Huntington, la enfermedad de Parkinson, la enfermedad de Alzheimer, la esclerosis lateral amiotrófica (ALS) o la esclerosis múltiple.
Las células Th17 son un subconjunto nuevo de células CD4+ T auxiliares que secretan IL-17, IL-21 e IL-22. La actividad proinflamatoria de las células Th17 puede ser beneficiosa para el huésped durante la infección, pero la función Th17 no controlada se ha relacionado e involucrado activamente en varias patologías autoinmunes y en el desarrollo de la enfermedad de injerto contra huésped aguda y crónica (GVHD), una enfermedad caracterizada por daño epitelial selectivo a los órganos diana que es mediada por células T maduras presentes en las células madre o inóculos de médula ósea. De hecho, se detectan altos niveles de IL-17 en los sueros y biopsias de pacientes con artritis reumatoide (RA) y pacientes con lupus eritematoso sistémico (SLE) que se correlaciona con la destrucción del tejido sinovial y la actividad de la enfermedad. El papel patológico de IL-17 en las articulaciones artríticas se asocia con su estimulación de la producción de citocinas proinflamatorias y el aumento del reclutamiento de células T y células inmunes innatas. Además, el número de células Th17 aumenta significativamente en la sangre periférica de pacientes con RA, así como se observaron concentraciones elevadas de IL-17 en los sobrenadantes de sus PBMC después de la estimulación con anticuerpos anti-CD3/CD28 ex vivo. Además, en pacientes con esclerosis múltiple (MS), las células Th17 reactivas a mielina también se enriquecen y producen altas cantidades de IL-22 e IFN-y. Además, un número significativamente mayor de células IL-17+ se detectan en áreas intestinales afectadas por la enfermedad en comparación con áreas sanas de los mismos sujetos con enfermedad de Crohn (CD).
El desarrollo y la función de las células Th17 depende de la activación de rutas específicas de señalización intracelular. El receptor nuclear de tipo receptor de esteroide RORYt se expresa selectivamente en células Th17 y parece ser necesario para la producción de IL-17. Se ha observado que la inducción de RORYt está mediada por IL-6, IL-21 e IL-23 a través de un mecanismo dependiente de STAT3. STAT3 también se une directamente a los promotores de IL-17 e IL-21. Además de RORYt y STAT3, se requiere el factor regulador de interferón 4 (IRF4) para la diferenciación de las células Th17 ya que los ratones IRF4 KO no lograron montar la respuesta a Th17 y fueron resistentes al desarrollo de respuestas autoinmunes. Estudios recientes han demostrado que la fosforilación de IRF4 por Rho-quinasa 2 (ROCK2) regula la producción de IL-17 e IL-21 y el desarrollo de la autoinmunidad en ratones.
Según la invención, alcanzar las células Th17 (secretadoras de IL-17) mediante la inhibición de la roquinasa se refiere a un compuesto de fórmula II para su uso en el tratamiento de enfermedades mediadas por células Th17, que incluyen, pero no se limitan a, trastornos autoinmunes tales como RA, MS, SLE, Psoriasis y enfermedad de Crohn y GVHD en humanos. En una realización de la invención, el inhibidor de Rho-quinasa es un compuesto de Fórmula II. En algunas realizaciones, el inhibidor de la Rho-quinasa inhibe ROCK1 y ROCK2. En algunas realizaciones, el inhibidor de la Rhoquinasa inhibe selectivamente ROCK2. La inhibición selectiva de ROCK2 proporciona el tratamiento de enfermedades mediadas por células Th17 y reduce o impide las toxicidades asociadas con la inhibición completa de la actividad de ROCK.
Las células T reguladoras (Tregs) juegan un papel crítico en el mantenimiento de la tolerancia inmunológica a los autoantígenos y la inhibición de las respuestas autoinmunes, pero, al mismo tiempo, impiden una respuesta inmune efectiva contra las células tumorales. De hecho, Tregs aisladas de la sangre periférica de pacientes con enfermedades autoinmunes, como la artritis reumatoide (RA) y la esclerosis múltiple (MS), muestran un defecto en su capacidad para suprimir la función de las células T efectoras, mientras que una mayor acumulación de Tregs se correlaciona con un mal pronóstico en muchos tipos de cáncer. Por lo tanto, el nivel de la función de las Treg produce un equilibrio entre
la inmunidad efectiva y la evitación de la autorreactividad patológica.
El desarrollo y la función de Tregs dependen de la activación de rutas de transducción de señalización específicas. TGF-p e IL-2 activan la expresión de factores de transcripción Foxp3 y STAT5 que juegan un papel esencial en el control de la función supresora de Treg. Por otro lado, las citocinas proinflamatorias inhiben la expresión de Foxp3 a través de la regulación positiva de la fosforilación de STAT3. Según la invención, la inhibición farmacológica de ROCK2 (por ejemplo, con inhibidores selectivos de ROCK2 como KD025, inhibición mediada por siRNA específica de ROCK2 de ROCK2), pero no ROCK1, conduce a una baja regulación de la fosforilación de STAT3, factor regulador de interferón 4 (IRF4) y los niveles de proteína RORYt del receptor nuclear de tipo esteroide en células T humanas. Por lo tanto, los inhibidores de ROCK2 regulan la función de Treg.
La invención proporciona compuestos de fórmula II para su uso en el tratamiento de enfermedades y trastornos con un componente angiogénico. Según la invención, en ciertas realizaciones, tales enfermedades y trastornos se tratan administrando a un sujeto una cantidad efectiva de un inhibidor de Rho-quinasa. En tales realizaciones, se prefiere un inhibidor de ROCK2. En ciertas realizaciones, el inhibidor es un inhibidor selectivo de ROCK2. Según la invención, tales enfermedades y trastornos también pueden tratarse administrando una cantidad efectiva de un inhibidor de Rhoquinasa que inhibe ROCK2, y puede ser selectivo para ROCK2, y una cantidad efectiva de un inhibidor de angiogénesis. Según la invención, las enfermedades y trastornos oculares que tienen un componente angiogénico se tratan de esta manera. En una realización, la invención proporciona un compuesto de fórmula II para su uso en el tratamiento de la degeneración macular relacionada con la edad (AMD), que ocurre en formas "secas" y "húmedas". La forma "húmeda" de AMD causa pérdida de visión debido al crecimiento anormal de los vasos sanguíneos (neovascularización). La hemorragia, las fugas y las cicatrices de estos vasos sanguíneos retinianos eventualmente causan daños irreversibles a los fotorreceptores. La forma seca (no exudativa) resulta de la atrofia de la capa epitelial del pigmento de la retina, que causa pérdida de visión a través de la pérdida de fotorreceptores (bastones y conos) en la parte central del ojo. En otra realización, la invención proporciona un compuesto de fórmula II para su uso en el tratamiento de la neovascularización coroidea (CNV). La neovascularización coroidea es un procedimiento en el que crecen nuevos vasos sanguíneos en la coroides, a través de la membrana de Bruch e invaden el espacio subretiniano, y es un síntoma de, entre otras causas, degeneración macular relacionada con la edad, miopía y trauma ocular. En otra realización, la invención proporciona un compuesto de fórmula II para su uso en el tratamiento del edema macular diabético (DME). En otra realización, la invención proporciona un compuesto de fórmula II para su uso en el tratamiento del edema macular que es secundario a la oclusión de la vena de la retina ramificada (BRVO) u oclusión de la vena de la retina central (CRVO). En otras realizaciones, las enfermedades a tratar incluyen, entre otros, neovascularización de la retina, infecciosa y no infecciosa, neovascularización corneal infecciosa y no infecciosa, neovascularización del iris, uveítis, glaucoma neovascular y retinitis del prematuro (ROP). El tratamiento puede ser profiláctico, como evitar la neovascularización corneal después del trasplante corneal o modular el procedimiento de cicatrización de heridas en la cirugía de trabeculectomía. Estas enfermedades y trastornos pueden caracterizarse por tener un componente angiogénico. Según la invención, tales trastornos se tratan administrando un inhibidor de Rho-quinasa, preferentemente un inhibidor de Rho-quinasa selectivo ROCK2, y un inhibidor de angiogénesis.
Por consiguiente, en una de tales realizaciones, la enfermedad o trastorno es AMD, y a un sujeto que necesita tratamiento para AMD se le administra una cantidad de un inhibidor de ROCK2 efectivo para tratar AMD. En otra realización, al sujeto se le administra un inhibidor de ROCK2 y un inhibidor de angiogénesis en cantidades efectivas para tratar la AMD. En tales realizaciones, se prefiere un inhibidor selectivo de ROCK2. En algunas realizaciones, el inhibidor de la angiogénesis es un antagonista de VEGFR2. En ciertas de tales realizaciones, el antagonista de VEGFR2 se une a VEGF. En ciertas de tales realizaciones, el antagonista de VEGFR2 se une a VEGFR2. Dichos inhibidores de unión a VEGFR2 incluyen agentes que se unen al dominio extracelular de VEGFR2, incluidos, entre otros, anticuerpos y fragmentos de unión a VEGFR2 de los mismos, y agentes que interactúan con el dominio intracelular de VEGFR2 y bloquean la activación de la transducción de señales dependiente de VEGFR2. Los antagonistas de VEGFR2 incluyen además agentes que interactúan con otros componentes celulares para bloquear la transducción de señales dependiente de VEGFR2. En otras realizaciones de la invención, otras enfermedades y trastornos oculares que tienen un componente angiogénico, tal como se indicó anteriormente, se tratan de manera similar.
Según la invención, un inhibidor de ROCK y un inhibidor de angiogénesis se administran a un sujeto en cantidades efectivas para tratar o impedir una afección patológica caracterizada por una angiogénesis excesiva. Tales afecciones, que implican, por ejemplo, vascularización y/o inflamación, incluyen aterosclerosis, artritis reumatoide (RA), hemangiomas, angiofibromas y psoriasis. Otros ejemplos no limitantes de enfermedad angiogénica son la retinopatía del prematuro (fibroplástico retrolental), el rechazo del injerto corneal, la neovascularización corneal relacionada con las complicaciones de la cirugía refractiva, la neovascularización corneal relacionada con las complicaciones de los lentes de contacto, la neovascularización corneal relacionada con el pterigion y el pterigion recurrente, la enfermedad de la úlcera corneal y enfermedad de la superficie ocular no específica, diabetes mellitus insulinodependiente, esclerosis múltiple, miastenia gravis, enfermedad de Chron, nefritis autoinmune, cirrosis biliar primaria, pancreatitis aguda, rechazo alográfico, inflamación alérgica, dermatitis de contacto y reacciones de hipersensibilidad retardada,
enfermedad inflamatoria intestinal, shock séptico, osteoporosis, osteoartritis, defectos cognitivos inducidos por inflamación neuronal, síndrome de Osler-Weber, restinosis e infecciones fúngicas, parasitarias y virales, incluidas las infecciones por citomegalovirus.
La invención proporciona inhibidores de pan-ROCK (es decir, compuestos que inhiben ROCK1 y ROCK1), así como inhibidores de ROCK que son selectivos de isoformas. Como se discutió anteriormente, en ciertas realizaciones de la invención, se puede preferir un inhibidor selectivo de ROCK2. Por ejemplo, un estudio observó que ROCK2 con frecuencia se sobreexpresa en el cáncer hepatocelular en comparación con los hígados no timóricos, mientras que la expresión de ROCK1 no se altera. Otros cánceres que pueden beneficiarse del tratamiento con un inhibidor selectivo de ROCK2 incluyen, entre otros, cáncer de colon y vejiga. En contraste, se ha observado que los niveles de expresión de ROCK1 son más altos en los tumores mamarios. Se puede analizar cualquier cáncer para determinar si hay una sobreexpresión de ROCK1 y/o ROCK2 y tratarlo en consecuencia. En ciertas circunstancias, las isoformas ROCK 1 y ROCK2 muestran similitud en la regulación de ciertas dianas posteriores y ninguna isoforma parece ser predominante. En tales casos, se prefiere un inhibidor de pan-ROCK.
En otro aspecto, la presente invención proporciona composiciones farmacéuticamente aceptables que comprenden una cantidad terapéuticamente efectiva de uno o más de los compuestos de Fórmula II, formulados junto con uno o más vehículos (aditivos) y/o diluyentes farmacéuticamente aceptables. Tal como se describe en detalle más adelante, las composiciones farmacéuticas de la presente invención pueden formularse especialmente para la administración en forma líquida o sólida, inclusive las adaptadas para lo siguiente: (1) administración por vía oral, por ejemplo, pociones (soluciones o suspensiones acuosas o no acuosas), comprimidos (por ejemplo, las que se dirigen a la absorción bucal, sublingual y/o sistémica), bolos, polvos, gránulos, pastas para aplicación en la lengua; (2) administración parenteral, por ejemplo, mediante inyección subcutánea, intramuscular, intravenosa o epidural, por ejemplo, como una solución o suspensión estéril o una formulación de liberación sostenida; (3) aplicación tópica, por ejemplo, como una crema, un ungüento o un parche o aerosol de liberación controlada que se aplica a la piel; (4) por vía intravaginal o intrarectal, por ejemplo, como un pesario, una crema o una espuma; (5) por vía sublingual; (6) por vía ocular; (7) por vía transdérmica o (8) por vía nasal.
La frase “farmacéuticamente aceptable” se emplea en esta invención para referirse a los compuestos, materiales, composiciones y/o formas de dosificación que, según un criterio médico sensato, son adecuados para su uso en contacto con tejidos humanos y animales sin provocar una toxicidad excesiva, irritación, respuesta alérgica u otros problemas o complicaciones, con una relación beneficio/riesgo correspondiente razonable.
La frase "vehículo farmacéuticamente aceptable" según se usa en esta invención significa un material, una composición o un portador farmacéuticamente aceptables, tales como un relleno líquido o sólido, diluyente, excipiente, auxiliar de fabricación (por ejemplo, lubricante, magnesio en talco, estearato de calcio o cinc o ácido estérico) o un material para encapsulado de solvente, involucrado en el traslado o transporte del compuesto objeto de un órgano o parte del cuerpo a otro órgano o parte del cuerpo.. Cada excipiente debe ser “aceptable” en el sentido de que sean compatible con los demás ingredientes de la formulación y no perjudicial para el paciente. Algunos ejemplos de materiales que pueden servir como portadores farmacéuticamente aceptables incluyen: (1) azúcares, tales como lactosa, glucosa y sacarosa, (2) almidones, tales como almidón de maíz y almidón de papa, (3) celulosa y sus derivados, tales como carboximetilcelulosa de sodio, etilcelulosa y acetato de celulosa, (4) tragacanto en polvo, (5) malta, (6) gelatina, (7) talco, (8) excipientes, tales como manteca de cacao y ceras para supositorios, (9) aceites, tales como aceite de maní, aceite de algodón, aceite de cártamo, aceite de sésamo, aceite de oliva, aceite de maíz y aceite de soja, (10) glicoles, tales como propilenglicol, (11) polioles, tales como glicerina, sorbitol, manitol y polietilenglicol, (12) ésteres, tales como oleato de etilo y laurato de etilo, (13) ágar, (14) agentes amortiguadores, tales como hidróxido de magnesio e hidróxido de aluminio, (15) ácido algínico, (16) agua libre de pirógenos, (17) solución salina isotónica, (18) solución de Ringer, (19) alcohol etílico, (20) soluciones amortiguadoras de pH; (21) poliésteres, policarbonatos y/o polianhídridos; y (22) otras sustancias compatibles no tóxicas empleadas en formulaciones farmacéuticas.
Como se expuso anteriormente, ciertas realizaciones de los presentes compuestos pueden contener un grupo funcional básico, tal como amino o alquilamino, y son, por lo tanto, capaces de formar sales farmacéuticamente aceptables con ácidos farmacéuticamente aceptables. El término "sales farmacéuticamente aceptables" a este respecto, se refiere a las sales de adición de ácido inorgánico y orgánico relativamente no tóxicas de los compuestos de la presente invención. Estas sales pueden prepararse in situ en el vehículo de administración o en el procedimiento de fabricación de la forma de dosificación, o haciendo reaccionar por separado un compuesto purificado de la invención en su forma de base libre con un ácido orgánico o inorgánico adecuado, y aislando la sal así formada durante la purificación posterior. Las sales representativas incluyen las sales de bromhidrato, clorhidrato, sulfato, bisulfato, fosfato, nitrato, acetato, valerato, oleato, palmitato, estearato, laurato, benzoato, lactato, fosfato, tosilato, citrato, maleato, fumarato, succinato, tartrato, naftilato, mesilato, glucoheptonato, lactobionato y laurilsulfonato y similares. (Ver, por ejemplo, Berge y col. (1977) "Pharmaceutical Salts", J. Pharm. Sci. 66:1-19).
Las sales farmacéuticamente aceptables de los compuestos en cuestión incluyen las sales no tóxicas convencionales
o sales de amonio cuaternario de los compuestos, por ejemplo, de ácidos orgánicos o inorgánicos no tóxicos. Por ejemplo, tales sales no tóxicas convencionales incluyen aquellas derivadas de ácidos inorgánicos tales como clorhidrato, bromhídrico, sulfúrico, sulfámico, fosfórico, nítrico y similares; y las sales preparadas a partir de ácidos orgánicos tales como acético, propiónico, succínico, glicólico, esteárico, láctico, málico, tartárico, cítrico, ascórbico, palmítico, maleico, hidroximaleico, fenilacético, glutámico, benzoico, salicílico, sulfanílico, 2-acetoxibenzoico, fumárico, toluensulfónico, metansulfónico, etanodisulfónico, oxálico, isotiónico y similares.
En otros casos, los compuestos de la presente invención pueden contener uno o más grupos funcionales ácidos y, por lo tanto, son capaces de formar sales farmacéuticamente aceptables con bases farmacéuticamente aceptables. El término "sales farmacéuticamente aceptables" en estos aspectos se refiere a las sales de adición de bases inorgánicas y orgánicas relativamente no tóxicas de los compuestos de la presente invención. Estas sales también pueden prepararse in situ en el vehículo de administración o en el procedimiento de fabricación de la forma de dosificación, o haciendo reaccionar por separado el compuesto purificado en su forma de ácido libre con una base adecuada, como el hidróxido, el carbonato o el bicarbonato de un catión metálico farmacéuticamente aceptable, con amoníaco o con un amina primaria, secundaria o terciaria orgánica farmacéuticamente aceptable. Las sales alcalinas o alcalinotérreas representativas incluyen las sales de litio, sodio, potasio, calcio, magnesio y aluminio y similares. Aminas orgánicas representativas útiles para la formación de sales de adición de bases incluyen etilamina, dietilamina, etilendiamina, etanolamina, dietanolamina, piperazina y similares. (Ver, por ejemplo, Berge y col., supra).
En las composiciones también pueden estar presentes agentes emulsionantes y lubricantes, como laurilsulfato de sodio y estearato de magnesio, así como agentes colorantes, agentes de liberación, agentes de recubrimiento, agentes edulcorantes, saborizantes y perfumantes, conservantes y antioxidantes.
Ejemplos de antioxidantes farmacéuticamente aceptables incluyen: (1) antioxidantes solubles en agua, tales como ácido ascórbico, clorhidrato de cisteína, bisulfato de sodio, metabisulfito de sodio, sulfito de sodio y similares; (2) antioxidantes solubles en aceite, tales como palmitato de ascorbilo, hidroxianisol butilado (BHA), hidroxitolueno butilado (BHT), lecitina, propil galato, alfa-tocoferol y similares; y (3) agentes quelantes de metal, tales como ácido cítrico, ácido etilendiamina tetraacético (EDTA), sorbitol, ácido tartárico, ácido fosfórico y similares.
Las formulaciones de la presente invención incluyen las adecuadas para administración por vía oral, nasal, tópica (incluida bucal y sublingual), rectal, vaginal y/o parenteral. Las formulaciones se pueden presentar convenientemente en forma de dosificación unitaria y se pueden preparar mediante cualquier método bien conocido en la técnica de farmacia. La cantidad de ingrediente activo que se puede combinar con un material portador para producir una forma de dosificación única variará en función del hospedador que se esté tratando y el modo particular de administración. La cantidad de ingrediente activo que se puede combinar con un material portador para producir una forma de dosificación única será generalmente la cantidad del compuesto que produce un efecto terapéutico. Generalmente, del cien por ciento, esta cantidad variará de aproximadamente 0,1 por ciento a aproximadamente noventa y nueve por ciento de ingrediente activo, preferentemente de aproximadamente 5 por ciento a aproximadamente 70 por ciento, lo más preferentemente de aproximadamente 10 por ciento a aproximadamente 30 por ciento.
En ciertas realizaciones, una formulación de la presente invención comprende un excipiente seleccionado del grupo que consiste en ciclodextrinas, celulosas, liposomas, agentes formadores de micelas, por ejemplo, ácidos biliares y vehículos poliméricos, por ejemplo, poliésteres y polianhídridos; y un compuesto de la presente invención. En ciertas realizaciones, una formulación mencionada anteriormente hace que un compuesto de la presente invención sea biodisponible por vía oral.
Los procedimientos para preparar estas formulaciones o composiciones incluyen la etapa de asociar un compuesto de la presente invención con el vehículo y, opcionalmente, uno o más ingredientes accesorios. En general, las formulaciones se preparan al asociar de manera uniforme e íntima un compuesto de la presente invención con vehículos líquidos o vehículos sólidos finamente divididos, o ambos, y a continuación, si es necesario, dándole forma al producto.
Las formulaciones de la invención adecuadas para administración por vía oral pueden encontrarse en forma de cápsulas, sellos, píldoras, comprimidos, grageas (usando una base saborizada, normalmente sacarosa y acacia o tragacanto), polvos, gránulos o como una solución o una suspensión en un líquido acuoso o no acuoso, como una emulsión líquida de aceite en agua o de agua en aceite, como un elíxir o jarabe, o como pastillas (usando una base inerte, como gelatina y glicerina o sacarosa y acacia) y/o como enjuagues bucales y similares, cada uno con una cantidad predeterminada de un compuesto de la presente invención como un ingrediente activo. Una composición de la presente invención también se puede administrar como bolo, electuario o pasta.
En formas de dosificación sólida de la invención para administración oral (cápsulas, comprimidos, píldoras, grageas, polvos, gránulos, pastillas y similares), el ingrediente activo se mezcla con uno o más vehículos farmacéuticamente aceptables, tales como citrato de sodio o fosfato dicalcio y/o cualquiera de los siguientes: 1) rellenos o extensores,
como almidones, lactosa, sacarosa, glucosa, manitol y/o ácido silícico; 2) aglutinantes, tales como, por ejemplo, carboximetilcelulosa, alginatos, gelatina, polivinilpirrolidona, sacarosa y/o acacia; 3) humectantes, tales como glicerol; 4) agentes desintegrantes, tales como agar-agar, carbonato de calcio, almidón de patata o tapioca, ácido algínico, algunos silicatos y carbonato de sodio; 5) agentes retardantes de solución, tales como parafina; 6) aceleradores de la absorción, como compuestos de amonio cuaternario y tensioactivos, como poloxámero y laurilsulfato de sodio, 7) agentes humectantes, tales como, por ejemplo, alcohol cetílico, monoestearato de glicerol y tensioactivos no iónicos, 8) absorbentes, tales como arcilla de bentonita y caolín; 9) lubricantes, tales como talco, estearato de calcio, estearato de magnesio, polietilenglicoles sólidos, lauril sulfato de sodio, estearato de cinc, estearato de sodio, ácido esteárico y mezclas de estos; 10) agentes colorantes; y 11) agentes de liberación controlada, tales como crospovidona o etilcelulosa. En el caso de las cápsulas, comprimidos y pastillas, las composiciones farmacéuticas también pueden comprender agentes amortiguadores. Las composiciones sólidas de un tipo similar también se pueden emplear como rellenos en cápsulas de gelatina rellenas duras y blandas usando excipientes tales como lactosa o azúcares de la leche, así como polietilenglicoles de alto peso molecular y similares.
Se puede fabricar un comprimido mediante compresión o moldeo, opcionalmente con uno o más ingredientes accesorios. Los comprimidos se pueden preparar usando un aglutinante (por ejemplo, gelatina o hidroxipropilmetilcelulosa), lubricante, diluyente inerte, conservante, desintegrante (por ejemplo, glicolato de almidón de sodio o carboximetilcelulosa de sodio reticulada), agente tensioactivo o de dispersión. Los comprimidos moldeados se pueden fabricar moldeando en una máquina adecuada una mezcla del compuesto en polvo humedecido con un diluyente líquido inerte.
Los comprimidos y otras formas de dosificación sólidas de las composiciones farmacéuticas de la presente invención, tales como grageas, cápsulas, píldoras y gránulos, opcionalmente se pueden marcar o preparar con revestimientos y conchas, tales como revestimientos entéricos y otros revestimientos bien conocidos en la técnica de formulación farmacéutica. También pueden formularse para proporcionar una liberación lenta o controlada del ingrediente activo en el mismo utilizando, por ejemplo, hidroxipropilmetil celulosa en proporciones variables para proporcionar el perfil de liberación deseado, otras matrices de polímeros, liposomas y/o microesferas. Pueden formularse para una liberación rápida, por ejemplo, liofilizados. Pueden esterilizarse, por ejemplo, mediante filtración a través de un filtro de retención de bacterias o mediante la incorporación de agentes esterilizantes en forma de composiciones sólidas estériles que pueden disolverse en agua en condiciones estériles o algún otro medio inyectable estéril inmediatamente antes de su uso. Estas composiciones también pueden, opcionalmente, contener agentes opacificantes y pueden ser de una composición tal que libere únicamente el o los ingredientes activos o, preferentemente, en una parte determinada del tracto gastrointestinal, opcionalmente, de manera retardada. Los ejemplos de composiciones de inclusión que se pueden emplear incluyen ceras y sustancias poliméricas. Los ingredientes activos también pueden encontrarse en forma microencapsulada, si corresponde, con uno o más de los excipientes antes mencionados. Las formas de dosificación líquida para administración oral de los compuestos de la invención incluyen emulsiones, microemulsiones, soluciones, suspensiones, jarabes y elíxires farmacéuticamente aceptables. Además del ingrediente activo, las formas de dosificación líquida pueden contener diluyentes inertes que se utilizan comúnmente en la técnica, tales como, por ejemplo, agua u otros solventes, agentes solubilizantes y emulsionantes, como alcohol etílico, alcohol isopropílico, carbonato de etilo, acetato de etilo, alcohol bencílico, benzoato de bencilo, propilenglicol, 1,3-butilenglicol, aceites (en particular, aceite de semilla de algodón, cacahuate, maíz, germen, oliva, ricino y sésamo), glicerol, alcohol de tetrahidrofurilo, polietilenglicoles y ésteres de ácidos grasos de sorbitán y mezclas de estos.
Además de los diluyentes inertes, las composiciones orales también pueden incluir adyuvantes, tales como agentes humectantes, emulsionantes y de suspensión, agentes edulcorantes, saborizantes, colorantes, perfumantes y conservantes.
Las suspensiones, además de los compuestos activos, pueden contener agentes de suspensión tales como alcoholes isostearílicos etoxilados, polioxietileno sorbitol, y ésteres de sorbitán, celulosa microcristalina, metahidróxido de aluminio, bentonita, agar-agar y tragacanto y mezclas de los mismos.
Las formulaciones de las composiciones farmacéuticas para administración rectal, vaginal o uretral pueden presentarse como un supositorio, que puede prepararse al mezclar uno o más compuestos de la invención con uno o más excipientes o portadores adecuados no irritantes que comprenden, por ejemplo, manteca de cacao, polietilenglicol, una cera para supositorios o un salicilato y que es sólido a temperatura ambiente, pero líquido a temperatura corporal y, por lo tanto, se derretirá en el recto o cavidad vaginal y liberará el compuesto activo.
Las formulaciones adecuadas para administración vaginal incluyen formulaciones en pesarios, tampones, cremas, geles, pastas, espumas o aerosoles que contienen dichos portadores que se consideran adecuados en la técnica. Las formas de dosificación para administración tópica o transdérmica de un compuesto de la presente invención incluyen polvos, aerosoles, ungüentos, pastas, cremas, lociones, geles, soluciones, parches e inhaladores. El
compuesto activo se puede mezclar en condiciones estériles con un vehículo farmacéuticamente aceptable y con cualquier conservante, amortiguador o propelente que sea necesario.
Los ungüentos, pastas, cremas y geles pueden contener, además de un compuesto activo de esta invención, excipientes, tales como grasas animales y vegetales, aceites, ceras, parafinas, almidón, tragacanto, derivados de celulosa, polietilenglicoles, siliconas, bentonitas, ácido silícico, talco y óxido de cinc o mezclas de estos.
Los polvos y aerosoles pueden contener, además de un compuesto de esta invención, excipientes tales como lactosa, talco, ácido silícico, hidróxido de aluminio, silicatos de calcio y poliamida en polvo, o mezclas de estas sustancias. Los aerosoles pueden contener adicionalmente propelentes habituales, tales como clorofluorohidrocarburos e hidrocarburos volátiles no sustituidos, tal como butano y propano.
Los parches transdérmicos tienen la ventaja adicional de proporcionar al cuerpo una administración controlada de un compuesto de la presente invención. Tales formas de dosificación se pueden elaborar al disolver o dispersar el compuesto activo en el medio apropiado. Los potenciadores de absorción también se pueden usar para aumentar el flujo del compuesto a través de la piel. La velocidad de dicho flujo se puede controlar ya sea al proporcionar una membrana de control de la velocidad o al dispersar el compuesto en un gel o una matriz poliméricos.
Las formulaciones oftálmicas, ungüentos, polvos, soluciones oculares y similares también se encuentran contempladas dentro del alcance de la presente invención.
Las composiciones farmacéuticas de esta invención adecuadas para administración parenteral comprenden uno o más compuestos de la invención en combinación con una o más soluciones, dispersiones, suspensiones o emulsiones acuosas o no acuosas isotónicas estériles farmacéuticamente aceptables, o polvos estériles que pueden reconstituirse en soluciones inyectables estériles o dispersiones justo antes de su uso, que pueden contener azúcares, alcoholes, antioxidantes, tampones, bacteriostatos, solutos que hacen que la formulación sea isotónica con la sangre del receptor previsto o agentes de suspensión o espesantes.
Ejemplos de portadores acuosos y convenientes que pueden ser empleados en las composiciones farmacéuticas de la invención incluyen agua, etanol, polioles (como glicerol, propileno glicol, polietileno glicol y similares) y mezclas adecuadas de los mismos, aceites vegetales, como aceite de oliva, ésteres orgánicos inyectables, como oleato de etilo. La fluidez adecuada se puede mantener, por ejemplo, mediante el uso de materiales de revestimiento, como, por ejemplo, lecitina, mediante el mantenimiento del tamaño de partícula requerido en el caso de dispersiones y mediante el uso de tensioactivos.
Estas composiciones pueden también contener adyuvantes tales como conservantes, agentes humectantes, agentes emulsionantes y agentes dispersantes. La prevención de la acción de los microorganismos sobre los compuestos objeto se puede asegurar mediante la inclusión de diversos agentes antibacterianos y antifúngicos, por ejemplo, parabeno, clorobutanol, ácido fenólico sódico y similares. También puede ser deseable incluir en las composiciones agentes isotónicos, tales como azúcares, cloruro de sodio y similares. Asimismo, la absorción prolongada de la forma farmacéutica inyectable puede provocarse mediante la inclusión de agentes que retrasan la absorción, tales como monostearato de aluminio y gelatina.
En algunos casos, para prolongar el efecto de un fármaco, es conveniente enlentecer la absorción del fármaco de la inyección subcutánea o intramuscular. Esto se puede lograr mediante el uso de una suspensión líquida de material cristalino o amorfo que tenga baja solubilidad en agua. Entonces, la velocidad de absorción del fármaco depende de su velocidad de disolución, la que, a su vez, puede depender del tamaño del cristal y de la forma cristalina. De manera alternativa, la absorción retardada de una forma de fármaco administrado parenteralmente se logra al disolver o suspender el fármaco en un vehículo oleoso.
Las formas de depósito inyectables se preparan formando matrices de microencapsulas de los compuestos en cuestión en polímeros biodegradables tales como polilactida-poliglicólido. En función de la relación del fármaco respecto al polímero y de la naturaleza del polímero particular utilizado, se puede controlar la velocidad de liberación del fármaco. Los ejemplos de otros polímeros biodegradables incluyen poli(ortoésteres) y poli(anhídridos). Las formulaciones de depósito inyectables también se preparan atrapando el fármaco en liposomas o microemulsiones que son compatibles con los tejidos corporales.
Cuando los compuestos de la presente invención se administran como productos farmacéuticos, a humanos y animales, se pueden administrar per se o como una composición farmacéutica que contiene, por ejemplo, del 0,1 al 99% (más preferentemente, del 10 al 30%) de ingrediente activo en combinación con un vehículo farmacéuticamente aceptable.
Las preparaciones de la presente invención pueden administrarse por vía oral, parenteral, tópica o rectal. Por
supuesto, se dan en formas adecuadas para cada ruta de administración. Por ejemplo, se administran en forma de comprimido o cápsula, por inyección, inhalación, loción ocular, ungüento, supositorio, etc. administración por inyección, infusión o inhalación; tópica por loción o ungüento y rectal por supositorios. Se prefieren las administraciones orales. Las frases "administración parenteral" y "administrada por vía parenteral" como se usan en esta invención se refieren a modos de administración distintos de la administración enteral y tópica, habitualmente mediante inyección, e incluyen, sin limitación, inyección e infusión intravenosa, intramuscular, intraarterial, intratecal, intracapsular, intraorbital, intracardíaca, intradérmica, intraperitoneal, transtraqueal, subcutánea, subcuticular, intraarticular, subcapsular, subaracnoidea, intraespinal, epidural e intraesternal.
Las frases "administración sistémica", "administrado sistémicamente", "administración periférica" y "administrado periféricamente" como se usa en esta invención significan la administración de un compuesto, fármaco u otro material que no sea directamente en el sistema nervioso central, de modo que ingrese al sistema del paciente y, por lo tanto, está sujeto a metabolismo y otros procedimientos similares, por ejemplo, administración subcutánea.
Estos compuestos pueden administrarse a humanos y otros animales para terapia por cualquier vía de administración adecuada, incluyendo oral, nasal, como, por ejemplo, por aerosol, rectal, intravaginal, parenteral, intracisternal y tópica, como polvos, ungüentos o gotas, incluyendo bucal y sublingualmente.
Independientemente de la ruta de administración seleccionada, los compuestos de la presente invención, que pueden usarse en una forma hidratada adecuada y/o las composiciones farmacéuticas de la presente invención, se formulan en formas de dosificación farmacéuticamente aceptables por procedimientos convencionales conocidos por expertos en la materia.
Los niveles de dosificación reales de los ingredientes activos en las composiciones farmacéuticas de la presente invención pueden variar para obtener una cantidad de ingrediente activo efectiva para alcanzar la respuesta terapéutica deseada para un paciente, composición y modo de administración particulares, sin ser tóxica para el paciente.
El nivel de dosificación seleccionado dependerá de una diversidad de factores, incluyendo la actividad del compuesto particular de la presente invención empleado, o el éster, sal o amida del mismo, la ruta de administración, el tiempo de administración, la tasa de excreción o metabolismo del compuesto particular que se está empleando, la velocidad y el grado de absorción, la duración del tratamiento, otros fármacos, compuestos y/o materiales usados en combinación con el compuesto particular empleado, la edad, el sexo, el peso, la condición, la salud general y el historial médico anterior del paciente que se está tratando, y factores similares ya conocidos en la técnica médica.
Un médico o veterinario con experiencia en la técnica puede determinar e indicar fácilmente la cantidad efectiva de la composición farmacéutica necesaria. Por ejemplo, el médico o veterinario puede comenzar las dosis de los compuestos de la invención empleados en la composición farmacéutica en niveles inferiores a los requeridos para lograr el efecto terapéutico deseado y aumentar gradualmente la dosificación hasta alcanzar el efecto deseado. En general, una dosis diaria adecuada de una composición de la invención será la cantidad del compuesto que es la dosis efectiva más baja para producir un efecto terapéutico. Dicha dosis efectiva generalmente dependerá de los factores que se describieron anteriormente. Generalmente, las dosis orales, intravenosas, intracerebroventriculares y subcutáneas de los compuestos de esta invención para un paciente, cuando se usan para los efectos analgésicos indicados, variarán en un intervalo de aproximadamente 0,0001 a aproximadamente 100 mg por kilogramo de peso corporal por día.
En ciertas realizaciones, se administra una dosis de un compuesto o una composición a un sujeto todos los días, cada dos días, cada par de días, cada tercer día, una vez a la semana, dos veces a la semana, tres veces a la semana o una vez cada dos semanas. Si se desea, la dosis diaria efectiva del compuesto activo se puede administrar como una, dos, tres, cuatro, cinco, seis o más subdosis administradas por separado en intervalos apropiados a lo largo del día, opcionalmente, en formas de dosificación unitaria. En algunas realizaciones, se administra una o más dosis de un compuesto o una composición durante 2 días, 3 días, 5 días, 7 días, 14 días o 21 días. En ciertas realizaciones, se administra una dosis de un compuesto o una composición durante 1 mes, 1,5 meses, 2 meses, 2,5 meses, 3 meses, 4 meses, 5 meses, 6 meses o más.
Los cronogramas de administración descritos anteriormente se proporcionan solo con fines ilustrativos y no deben considerarse limitantes. Un experto en la materia comprenderá fácilmente que todas las dosis están dentro del alcance de la invención.
Si bien es posible que un compuesto de la presente invención se administre solo, es preferible administrar el compuesto como una formulación (composición) farmacéutica.
El paciente que recibe este tratamiento es cualquier animal que lo necesite, que incluye primates, en particular seres humanos y otros mamíferos tales como equinos, ganado, cerdos y ovejas y aves de corral y mascotas en general. El compuesto de la invención puede administrarse como tal o en mezclas con vehículos farmacéuticamente aceptables y también puede administrarse junto con agentes antimicrobianos tales como penicilinas, cefalosporinas, aminoglucósidos y glucopéptidos. La terapia conjuntiva, por lo tanto, incluye la administración secuencial, simultánea y separada del compuesto activo de manera que los efectos terapéuticos del primero administrado no desaparezcan por completo cuando se administra el subsiguiente.
La adición del compuesto activo de la invención al alimento para animales se realiza preferentemente preparando una premezcla de alimento apropiada que contiene el compuesto activo en una cantidad efectiva e incorporando la premezcla en la ración completa.
Alternativamente, un concentrado intermedio o suplemento alimenticio que contiene el ingrediente activo se puede mezclar en la alimentación. La forma en que tales premezclas de alimentos y raciones completas se pueden preparar y administrar se describe en los libros de referencia (como "Applied Animal Nutrition", W.H. Freedman and CO., San Francisco, U.S.A., 1969 o "Livestock Feeds and Feeding" O and B books, Corvallis, Ore., U.S.A., 1977).
La tecnología de microemulsificación puede emplearse para mejorar la biodisponibilidad de los agentes farmacéuticos lipofílicos (insolubles en agua). Los ejemplos incluyen trimetrina (Dordunoo, S. K., y col., Drug Development and Industrial Pharmacy, 17(12), 1685-1713, 1991) y REV 5901 (Sheen, P. C., y col., J Pharm Sci 80(7), 712-714,1991). Entre otras cosas, la microemulsificación proporciona una biodisponibilidad mejorada al dirigir preferentemente la absorción al sistema linfático en lugar del sistema circulatorio, que evita el hígado e impide la destrucción de los compuestos en la circulación hepatobiliar.
En un aspecto de la invención, las formulaciones contienen micelas formadas a partir de un compuesto de la presente invención y al menos un vehículo anfifílico, en el que las micelas tienen un diámetro promedio de menos de aproximadamente 100 nm. Las realizaciones más preferidas de la invención proporcionan micelas que tienen un diámetro promedio inferior a aproximadamente 50 nm, e incluso realizaciones más preferidas de la invención proporcionan micelas que tienen un diámetro promedio inferior a aproximadamente 30 nm, o incluso menos de aproximadamente 20 nm.
Si bien se contemplan todos los vehículos anfifílicos adecuados, los vehículos actualmente preferidos son generalmente aquellos que tienen el status de Reconocimiento general como seguro (GRAS), y que pueden solubilizar el compuesto de la presente invención y microemulsionarlo en una etapa posterior cuando la solución entra en contacto con una fase acuosa compleja (como la que se encuentra en el tracto gastrointestinal humano). Por lo general, los ingredientes anfifílicos que satisfacen estos requisitos tienen valores de HLB (equilibrio hidrofílico a lipofílico) de 2-20, y sus estructuras contienen radicales alifáticos de cadena lineal en el intervalo de C-6 a C-20. Ejemplos son los glicéridos grasos de polietilenglicol y los polietilenglicoles.
Los vehículos anfifílicos particularmente preferidos son los glicéridos de ácidos grasos polietilenglicolizados saturados y monoinsaturados, tales como los obtenidos de diversos aceites vegetales total o parcialmente hidrogenados. Dichos aceites pueden consistir ventajosamente en glicéridos de ácidos grasos tri, di y mono grasos y ésteres de di y mono polietilenglicol de los ácidos grasos correspondientes, con una composición de ácido graso particularmente preferida que incluye ácido cáprico 4-10, ácido cáprico 3-9, ácido láurico 40-50, ácido mirístico 14-24, ácido palmítico 4-14 y ácido esteárico 5-15%. Otra clase útil de vehículos anfifílicos incluye sorbitán y/o sorbitol parcialmente esterificados, con ácidos grasos saturados o monoinsaturados (serie SPAN) o análogos etoxilados correspondientes (serie TWEEN). Los portadores anfifílicos disponibles en el mercado se contemplan particularmente, incluidas las series Gelucire, Labrafil, Labrasol o Lauroglycol (todos fabricados y distribuidos por Gattefosse Corporation, Saint Priest, Francia), PEG-mono-oleato, PEG-di-oleato, PEG-mono- laurato y di-laurato, lecitina, polisorbato 80, etc. (producidos y distribuidos por varias empresas en EE.UU. y en todo el mundo).
Los polímeros hidrofílicos adecuados para su uso en la presente invención son aquellos que son fácilmente solubles en agua, se pueden unir covalentemente a un lípido formador de vesículas, y que se toleran in vivo sin efectos tóxicos (es decir, son biocompatibles). Los polímeros adecuados incluyen polietilenglicol (PEG), ácido poliláctico (también denominado polilactida), ácido poliglicólico (también denominado poliglicólido), un copolímero de ácido polilácticopoliglicólico y alcohol polivinílico. Los polímeros preferidos son aquellos que tienen un peso molecular de aproximadamente 100 o 120 daltons hasta aproximadamente 5.000 o 10.000 daltons, y más preferentemente de aproximadamente 300 daltons a aproximadamente 5.000 daltons. En una realización particularmente preferida de la invención, el polímero es polietilenglicol que tiene un peso molecular de aproximadamente 100 a aproximadamente 5.000 daltons, y más preferentemente tiene un peso molecular de aproximadamente 300 a aproximadamente 5.000
daltons. En una realización particularmente preferida de la invención, el polímero es polietilenglicol de 750 daltons (PEG (750)). Los polímeros usados en la presente invención tienen un peso molecular significativamente menor, aproximadamente 100 daltons, en comparación con el gran MW de 5000 daltons o mayor que se usa en técnicas de pegilación estándar. Los polímeros también pueden definirse por el número de monómeros en el mismo; una realización preferida de la presente invención utiliza polímeros de al menos aproximadamente tres monómeros, tales polímeros de PEG que consisten en tres monómeros (aproximadamente 150 daltons).
Otros polímeros hidrófilos que pueden ser adecuados para su uso en la presente invención incluyen polivinilpirrolidona, polimetoxazolina, polietiloxazolina, polihidroxipropilmetacrilamida, polimetacrilamida, polidimetilcrilamida y celulosas derivatizadas tales como hidroximetilcelulosa o hidroxietilcelulosa.
En ciertas realizaciones, una formulación de la presente invención comprende un polímero biocompatible seleccionado del grupo que consiste en poliamidas, policarbonatos, polialquilenos, polímeros de ésteres acrílicos y metacrílicos, polímeros de polivinilo, poliglicólidos, polisiloxanos, poliuretanos y copolímeros de los mismos, celulosas, polipropileno., polietilenos, poliestireno, polímeros de ácido láctico y ácido glicólico, polianhídridos, poli(orto)ésteres, poli(ácido butílico), poli(ácido valérico), poli(lactida-co-caprolactona), polisacáridos, proteínas, ácidos polihialurónicos, policianoacrilatos y mezclas, combinaciones o copolímeros de los mismos
Las características de liberación de una formulación de la presente invención dependen del material de encapsulación, la concentración del fármaco encapsulado y la presencia de modificadores de liberación. Por ejemplo, la liberación se puede manipular para que sea dependiente del pH, por ejemplo, usando un recubrimiento sensible al pH que se libera solo a un pH bajo, como en el estómago, o a un pH más alto, como en el intestino. Se puede usar un recubrimiento entérico para impedir la liberación hasta después del paso por el estómago. Se pueden usar múltiples recubrimientos o mezclas de cianamida encapsulados en diferentes materiales para obtener una liberación inicial en el estómago, seguida de una liberación posterior en el intestino. La liberación también se puede manipular mediante la inclusión de sales o agentes formadores de poros, que pueden aumentar la absorción de agua o la liberación del fármaco por difusión desde la cápsula. Los excipientes que modifican la solubilidad del fármaco también se pueden usar para controlar la velocidad de liberación. También se pueden incorporar agentes que mejoran la degradación de la matriz o la liberación de la matriz. Pueden agregarse al fármaco, agregarse como una fase separada (es decir, como partículas), o pueden codisolverse en la fase de polímero dependiendo del compuesto. En todos los casos, la cantidad debe estar entre 0,1 y treinta por ciento (p/p de polímero). Los tipos de potenciadores de degradación incluyen sales inorgánicas como sulfato de amonio y cloruro de amonio, ácidos orgánicos como ácido cítrico, ácido benzoico y ácido ascórbico, bases inorgánicas como carbonato de sodio, carbonato de potasio, carbonato de calcio, carbonato de zinc e hidróxido de zinc, y bases orgánicas como sulfato de protamina, espermina, colina, etanolamina, dietanolamina y trietanolamina y tensioactivos como Tween®. y Pluronic®. Los agentes formadores de poros que agregan microestructura a las matrices (es decir, compuestos solubles en agua como sales inorgánicas y azúcares) se agregan como partículas. El intervalo debe estar entre uno y treinta por ciento (p/p de polímero).
La absorción también se puede manipular alterando el tiempo de residencia de las partículas en el intestino. Esto se puede lograr, por ejemplo, recubriendo la partícula con, o seleccionando como material de encapsulación, un polímero adhesivo de la mucosa. Los ejemplos incluyen la mayoría de los polímeros con grupos carboxilo libres, tales como quitosano, celulosas y especialmente poliacrilatos (como se usa en esta invención, los poliacrilatos se refieren a polímeros que incluyen grupos acrilatos y grupos acrilatos modificados tales como cianoacrilatos y metacrilatos). Los cronogramas de administración descritos anteriormente se proporcionan solo con fines ilustrativos y no deben considerarse limitantes. Un experto en la materia comprenderá fácilmente que todas las dosis están dentro del alcance de la invención.
Los compuestos de la invención pueden administrarse ventajosamente con segundos agentes a pacientes que los necesitan. Cuando un inhibidor de Rho-quinasa se administra con un segundo agente, el inhibidor de Rho-quinasa y el segundo agente pueden administrarse secuencialmente o concomitantemente. Secuencialmente significa que un agente se administra durante un tiempo seguido de la administración del segundo agente, que puede ser seguido por la administración del primer agente. Cuando los agentes se administran secuencialmente, el nivel o un agente puede no mantenerse a un nivel terapéuticamente efectivo cuando se administra el segundo agente, y viceversa. Concomitantemente significa que el primer y el segundo agente se administran según un programa que mantiene a ambos agentes en un nivel sustancialmente terapéuticamente efectivo, aunque los agentes no se administran simultáneamente. Cada agente puede administrarse en dosis únicas o múltiples, y las dosis pueden administrarse en cualquier horario, incluyendo, entre otros, dos veces al día, diariamente, semanalmente, cada dos semanas y mensualmente.
La invención también incluye la administración complementaria. La administración complementaria significa que se administra un segundo agente a un paciente además de un primer agente que ya se está administrando para tratar una enfermedad o síntoma de la enfermedad. En algunas realizaciones, la administración complementaria implica
administrar un segundo agente a un paciente en el que la administración del primer agente no trató suficientemente una enfermedad o síntoma de la enfermedad. En otras realizaciones, la administración complementaria implica la administración del segundo agente a un paciente cuya enfermedad ha sido tratada eficazmente mediante la administración del primer agente, con la expectativa de que el tratamiento complementario mejore el resultado del tratamiento. En algunas realizaciones, el efecto de administrar el primer y segundo agentes es sinérgico. En algunas realizaciones, la administración del primer y segundo agente impide o alarga el tiempo hasta la recaída, en comparación con la administración de cualquiera de los agentes solos. En algunas realizaciones, la administración del primer y segundo agente permite una dosis y/o frecuencia de administración reducidas del primer y segundo agente.
En una realización de la invención, se administran un inhibidor de Rho-quinasa de la invención y un agente antineoplásico a un sujeto que lo necesite. En otra realización, se administran un inhibidor de Rho-quinasa de la invención y un inhibidor de angiogénesis a un sujeto que lo necesitan En otra realización, se administran un inhibidor de Rho-quinasa de la invención y un agente antiinflamatorio a un sujeto que lo necesita. En otra realización más, se administran un inhibidor de Rho-quinasa de la invención y un inmunosupresor. El segundo agente puede ser, entre otros, una molécula pequeña, un anticuerpo o fragmento de unión a antígeno del mismo, o radiación.
Los agentes antineoplásicos incluyen, entre otros, agentes quimioterapéuticos citotóxicos, moléculas pequeñas específicas y moléculas biológicas y radiación. Los compuestos y agentes que pueden administrarse para el tratamiento oncológico, además de un inhibidor de la Rho-quinasa de la invención, incluyen los siguientes: irinotecán, etopósido, camptotecina, 5-fluorouracilo, hidroxiurea, tamoxifeno, paclitaxel, capcitabina, carboplatino, cisplatino, bleomicina, dactomicina, gemcitabina, doxorrubicina, danorrubicina, ciclofosfamida y radioterapia, que pueden ser externas (por ejemplo, radioterapia de haz externo (EBRT)) o internas (por ejemplo, braquiterapia (BT)).
Las moléculas pequeñas específicas y las moléculas biológicas incluyen, entre otros, inhibidores de componentes de vías de transducción de señales, tales como moduladores de tirosina quinasas e inhibidores de receptores de tirosina quinasas, y agentes que se unen a antígenos específicos de tumor. Los ejemplos incluyen inhibidores del receptor del factor de crecimiento epidérmico (EGFR), incluidos gefitinib, erlotinib y cetuximab, inhibidores de HER2 (por ejemplo, trastuzumab, trastuzumab emtansina (trastuzumab-DM1; T-DM1) y pertuzumab), anticuerpos y fragmentos anti-VEGF (por ejemplo, bevacizumab), anticuerpos que inhiben CD20 (por ejemplo, rituximab, ibritumomab), anticuerpos anti-VEGFR (por ejemplo, ramucirumab (IMC-1121B), IMC-1C11 y CDP791), anticuerpos anti-PDGFR e imatinib. Los inhibidores de la quinasa de moléculas pequeñas pueden ser específicos para una tirosina quinasa particular o pueden ser inhibidores de dos o más quinasas. Por ejemplo, el compuesto N-(3,4-dicloro-2-fluorofenil)-7-({[(3aR,6aS)-2-metiloctahidro ciclopenta[c]pirrol-5-il] metil}oxi)-6-(metiloxi)quinazolin-4-amina (también conocida como XL647, EXEL-7647 y KD-019) es un inhibidor in vitro de varios receptores tirosina quinasas (RTK), incluidos EGFR, EphB4, KDR (VEGFR), Flt4 (VEGFR3) y ErbB2, y también es un inhibidor de la SRC quinasa, que está involucrada en vías que provocan la falta de respuesta de tumores a ciertos TKI. En una realización de la invención, el tratamiento de un sujeto necesitado comprende la administración de un inhibidor de Rho-quinasa de Fórmula II y la administración de KD-019.
Dasatinib (BMS-354825; Bristol-Myers Squibb, Nueva York) es otro inhibidor de Src competitivo de sitio ATP biodisponible por vía oral. Dasatanib también se dirige a Bcr-Abl (aprobado por la FDA para uso en pacientes con leucemia mielógena crónica (CML) o leucemia linfoblástica aguda (ALL) cromosómica positiva (Ph+) Filadelfia), así como c-Kit, PDGFR, c-FMS, EphA2, y quinasas de la familia Src. Otros dos inhibidores orales de la tirosina quinasa de Src y Bcr-Abl son bosutinib (SKI-606) y saracatinib (AZD0530).
Según la invención, los inhibidores de angiogénesis se pueden administrar a un sujeto junto con los compuestos de la invención. Los inhibidores de angiogénesis incluyen cualquier sustancia que inhiba el crecimiento de nuevos vasos sanguíneos. Por ejemplo, los inhibidores de angiogénesis incluyen antagonistas de los receptores VEGF, PlGF y VEGF, incluidos los anticuerpos descritos en esta invención. Por inhibidor se entiende un inhibidor de un procedimiento biológico o un inhibidor de una diana. A este respecto, un inhibidor de la angiogénesis es un agente que reduce la angiogénesis. Un inhibidor de Rho-quinasa es un agente, como un inhibidor competitivo de la unión a ATP, que inhibe una actividad intrínseca o bloquea una interacción de Rho-quinasa. Por antagonista se entiende una sustancia que reduce o inhibe una actividad o función en una célula asociada con una diana. Por ejemplo, un antagonista de VEGF reduce o bloquea una función en una célula que está asociada con VEGF. Un antagonista de VEGF puede actuar sobre VEGF, uniéndose a VEGF y bloqueando la unión a sus receptores y/o puede actuar sobre otro componente celular implicado en la transducción de señales mediada por VEGF. De manera similar, un antagonista de VEGFR2 es un agente que reduce o bloquea la transducción de señales mediada por VEGFR2 uniéndose a VEGFR2 y bloqueando la unión o interacción del ligando con un sustrato de VEGFR2, o actúa sobre otro componente celular para reducir o bloquear la transducción de señales mediada por VEGFR2. Por lo tanto, los inhibidores de angiogénesis incluyen anticuerpos anti-VEGFR2 establecidos en esta invención (Tabla 1, Tabla 2, Fig. 10) y antagonistas de, entre otros, VEGF, VEGFR1, VEGFR2, PDGF, PDGFR-p, neuropilina-1 (NRP1), y complemento.
También se describen en esta invención anticuerpos anti-VEGFR2, que incluyen ácidos nucleicos que codifican dichos anticuerpos y composiciones que comprenden dichos anticuerpos. También se describe en esta invención una región
variable de cadena pesada de anticuerpo aislada que comprende una secuencia CDR-1H, CDR-2H y CDR-3H, donde: (i) la secuencia CDR-1H es GFTFSWYX1MX2 (SEQ ID NO:185), donde Xi es V o I, X2 es G o L,
(ii) la secuencia CDR-2H es SIX1X2SGGX3TX4YADSVKG (SEQ ID NO:186), donde X1 es Y o G, X2 es P o S, X3 es A o F, X4 es N o D, y
(iii) la secuencia CDR-3H es GNYFDY (SEQ ID NO:3) o GLAAPRS (SEQ ID NO:11).
También se describe en esta invención una región variable de cadena ligera aislada que comprende un CDR-L1, CDR-L2 y CDR-L3, donde
(i) la secuencia CDR-L1 es X1GX2X3LX4X5X6X7X8S (SEQ ID NO:187), donde X1 es S, Q, o T, X2 es D, E, o Q, X3 es K, S, N, I, o A, X4 es G o R, X5 es D, S, H, E, o N, X6 es E, Y, Q, R, o N, X7 es Y, F, o S, y X8 es A o S, o SGSX1SNX2X3X4X5X6X7X8 (SEQ ID NO:188), donde X1 es S, o T, X2 es I o L, X3 es E o G, X4 es T, S, o N, X5 es N o Y, X6 es T, P, A, o Y, X7 es V o L, y X8 es N, I, o Y, o X1GX2SX3DX4GX5YDYVS (SEQ ID NO:189), donde X1 es A o T, X2 es S o T, X3 es H, S, o N, X4 es I o V, y X5 es S o A,
(ii) la secuencia CDR-L2 es X1X2X3X4X5PS (SEQ ID NO:190), donde X1 es Q, D, T, Y, S, o A, X2 es D, N, S, T, o V, X3 es D, N, S, T, o Y, X4 es Q, K, N, o L, y X5 es R o L, y
(iii) donde la secuencia CDR-L3 es QX1WX2X3X4X5X6X7X8 (SEQ ID NO:191), donde X1 es A o T, X2 es D o G, X3 es R o ningún aminoácido, X4 es S, F, o N, X5 es S, T, o N, X6 es S, T, o P, X7 es A, V, L, I, o Y, y X8 es V o L, o AX1WDDX2LX3X4X5X6 (SEQ ID NO:192, donde X1 es A, S, o T, X2 es N o S, X3 es N, I, o G, X4 es G o S, X5 es P, W, o V, y X6 es V o L, o MYSTITX1LL (SEQ ID NO:193), donde X1 es A o T.
También se describe en esta invención una región variable de cadena ligera aislada que comprende un CDR-L1, CDR-L2 y CDR-L3, donde
(i) la secuencia CDR-L1 es RASX1X2X3X4X5X6X7YX8X9 (SEQ ID NO:194), donde X1 es Q, E, o H, X2 es S, R, o N, X3 es V, I, o L, X4 es S, R, G o N, X5 es S o N, X6 es S, N, W, o D, X7 es G o ningún aminoácido, X8 es L o F, y X9 es A, G, M, o S,
(ii) la secuencia CDR-L2 es GASX1RAT (SEQ ID NO:195), donde X1 es S, T, I o N, y
(iii) la secuencia CDR-L3 es QQX1X2X3X4X5X6X7X8 (SEQ ID NO:196), donde X1 es F o Y, X2 es D, G, o Y, X3 es S, T, o N, X4 es S, L, o W, X5 es P o ningún aminoácido, X6 es P o T, X7 es L, I, V, P, W, o Y, y X8 es T o S.
También se describe en esta invención un anticuerpo que comprende un dominio variable de cadena pesada que comprende uno, dos, tres, cuatro, cinco o seis de las secuencias CDR de dominio variable de cadena ligera y dominio variable de cadena pesada establecidas anteriormente.
Se proporcionan ejemplos no limitantes de secuencias de anticuerpos de unión a VEGFR2. Como se describe en esta invención, a partir de bibliotecas de visualización de fagos Fab humanos, se identificaron dos anticuerpos neutralizantes que se unen al VEGFR2 humano, bloquean la unión del ligando VEGFA al hVEGFR2 e inhiben la fosforilación del VEGFR2 y la transducción de las señales posteriores estimuladas por VEGFA. La Tabla 1 indica las secuencias de aminoácidos de los CDR y los dominios variables de los anticuerpos. Las K¿s de Mab 101 y Mab 102 son aproximadamente 6,6 mM y 1,7 nM, respectivamente.

La cadena pesada de Mab 101 se reorganizó con genes de cadena ligera k (biblioteca k) y genes de cadena ligera A (biblioteca A). Se encontraron 20 variantes únicas de la cadena ligera A mediante el barrido de la biblioteca A contra VEGFR2 humano y VEGFR2 de ratón. Se encontraron 22 variantes únicas de cadena ligera k mediante el barrido de la biblioteca k contra VEGFR2 humano y VEGFR2 de ratón. La Tabla 2 indica las secuencias de aminoácidos de los CDR y los dominios variables de las cadenas ligeras. Los KD de las Mabs 105, 106 y 107 se incrementaron aproximadamente 10 veces (0,24 nM, 0,22 nM y 0,12 nM, respectivamente).

También se describe en esta invención un anticuerpo VEGFR2 aislado y fragmentos de unión a VEGFR2 del mismo, que comprende una, dos o tres CDR de cadena pesada y uno, dos o tres CDR de cadena ligera seleccionadas de las secuencias expuestas en la Tabla 1 y la Tabla 2. En un anticuerpo descrito en esta invención, cuando se selecciona más de un CDR de las secuencias presentadas en la Tabla 1 y la Tabla 2, los diferentes CDR no necesitan seleccionarse del mismo anticuerpo monoclonal presentado en esas tablas, pero pueden seleccionarse de dos o más dominios de anticuerpos variables presentados en las tablas. El anticuerpo VEGFR2 aislado comprende uno, dos o tres CDR de cadena pesada que tienen SEQ ID NO:1, SEQ ID NO:2 y SEQ ID NO:3. El anticuerpo comprende uno, dos o tres CDR de cadena ligera que tienen SEQ ID NO:5, SEQ ID NO:6 y SEQ ID NO:7. El anticuerpo comprende uno, dos o tres CDR de cadena ligera que tienen secuencias como se establece en la Tabla 1 o 2. Los ejemplos no limitativos incluyen una región variable de cadena ligera que comprende uno o más de SEQ ID NO:25, SEQ ID NO:26 y SEQ ID NO:27, uno o más de SEQ ID NO:29, SEQ ID NO:30, y SEQ ID NO:31, o uno o más de SEQ ID NO:33, SEQ ID NO:34 y SEQ ID NO:35. El anticuerpo VEGFR2 comprende un dominio variable de cadena pesada que comprende SEQ ID NO:4 o SEQ ID NO:12. El anticuerpo VEGFR2 comprende un dominio variable de cadena ligera que comprende SEQ ID NO:8, SEQ ID NO:16, SEQ ID NO:27, SEQ ID NO:31 o SEQ ID NO:35. Los anticuerpos comprenden uno de los dominios variables de cadena pesada mencionados anteriormente y uno de los dominios variables de cadena ligera mencionados anteriormente. Los anticuerpos VEGFR2 o sus fragmentos de unión comprenden uno o más CDR o uno o más dominios variables con una secuencia de aminoácidos al menos 85%, al menos 90%, al menos 95%, al menos 97%, al menos 98% o al menos 99%, idéntico a las secuencias de dominio variable y CDR establecidos en la Tabla 1, 2 o 3. Los anticuerpos tienen aminoácidos CDR idénticos a los descritos en esta invención y estructuras que son al menos 85%, al menos 90%, de al menos 95% idénticos.
"Identidad" se refiere al número o porcentaje de posiciones idénticas compartidas por dos secuencias de aminoácidos o de ácido nucleico, teniendo en cuenta el número de espacios, y la longitud de cada espacio, que es necesario
introducir para el alineamiento óptimo de las dos secuencias. «Sustancialmente idéntica» significa una secuencia de aminoácidos que difiere solo en sustituciones conservativas de aminoácidos, por ejemplo, la sustitución de un aminoácido por otro de la misma clase (por ejemplo, valina por glicina, arginina por lisina, etc.) o en una o más sustituciones, deleciones o inserciones no conservativas situadas en posiciones de la secuencia de aminoácidos que no destruyen la función de la proteína. Preferentemente, la secuencia de aminoácidos es al menos el 80 %, más preferentemente al menos aproximadamente el 85 %, y de la forma más preferente al menos aproximadamente el 90 % similar a otra secuencia de aminoácidos. Los procedimientos y programas informáticos para determinar la similitud de secuencia están disponibles públicamente, incluyendo, entre otros, el paquete de programa GCG (Devereux y col., Nucleic Acids Research 12: 387, 1984), BLASTP, BLASTN, FASTA (Altschul y col., J. Mol. Biol. 215:403 (1990), y el programa ALIGN (versión 2.0). El bien conocido algoritmo de Smith Waterman también se puede usar para determinar la similitud. El programa BLAST está disponible públicamente en NCBI y otras fuentes (BLAST Manual, Altschul, y col., NCBI NLM NIH, Bethesda, Md. 20894; BLAsT 2.0 en http:/www.nebi.nlm.nih.gov/blast/). Al comparar secuencias, estos procedimientos consideran diversas sustituciones, deleciones y otras modificaciones. Las sustituciones conservativas incluyen normalmente sustituciones dentro de los siguientes grupos: glicina, alanina, valina, isoleucina, leucina, ácido aspártico, ácido glutámico, asparagina, glutamina, serina, treonina, lisina, arginina y fenilalanina, tirosina.
También se describe en esta invención el uso de fragmentos de anticuerpos de unión a VEGFR2. Un Fv es el fragmento más pequeño que contiene un dominio variable completo de cadena pesada y ligera, incluidos los seis lazos hipervariables (CDR). Al carecer de dominios constantes, los dominios variables están asociados de forma no covalente. Las cadenas pesadas y ligeras pueden conectarse en una cadena de polipéptidos única (un "Fv de cadena sencilla" o "scFv") usando un conector que permite que los dominios Vh y Vl se asocien para formar un sitio de unión a antígeno. El enlazador es (Gly-Gly-Gly-Gly-Ser)3. Dado que los fragmentos scFv carecen de los dominios constantes de los anticuerpos completos, son considerablemente más pequeños que los anticuerpos completos. Los fragmentos scFv también están libres de interacciones normales de dominio constante de cadena pesada con otras moléculas biológicas que pueden ser indeseadas en ciertas realizaciones.
También se pueden usar fragmentos de un anticuerpo que contiene Vh, Vl y opcionalmente Cl, Ch1, u otros dominios constantes. Los fragmentos monovalentes de anticuerpos generados por la digestión con papaína se denominan Fab y carecen de la región de articulación de la cadena pesada. Fragmentos generados por la digestión de pepsina, denominada F(ab')2, retienen la articulación de la cadena pesada y son divalentes. Dichos fragmentos también se pueden producir de forma recombinante. Muchos otros fragmentos de anticuerpos de unión a antígeno útiles son conocidos en la técnica e incluyen, entre otros, diacuerpos, triacuerpos, anticuerpos de dominio único y otras formas monovalentes y multivalentes.
También se describen en esta invención proteínas de unión a antígeno multivalentes, que pueden estar en forma, entre otros, de anticuerpos, fragmentos de unión a antígeno de los mismos y proteínas que comprenden la totalidad o parte de porciones de anticuerpos que se unen a antígeno. Las proteínas de unión a antígeno multivalentes pueden ser monoespecíficas, biespecíficas o multiespecíficas. El término especificidad se refiere al número de diferentes tipos de determinantes antigénicos a los que puede unirse una molécula particular. Si una molécula de inmunoglobulina se une a un solo tipo de determinante antigénico, la molécula de inmunoglobulina es monoespecífica. Si la molécula de inmunoglobulina se une a diferentes tipos de determinantes antigénicos, entonces la molécula de inimunoglobulina es multiespecífica.
Por ejemplo, un anticuerpo bivalente de cadena sencilla multivalente permite el reconocimiento de dos tipos diferentes de epítopos. Ambos epítopos pueden estar en el mismo antígeno (por ejemplo, VEGFR2). Alternativamente, un epítopo puede estar en un antígeno (por ejemplo, VEGFR2), y el segundo epítopo en un antígeno diferente.
Un anticuerpo de cadena sencilla multivalente incluye un fragmento de cadena ligera variable unido a un fragmento de cadena pesada variable (similar a un scFv), que se une adicionalmente por otro conector peptídico a al menos otro dominio de unión a antígeno. Típicamente, el enlazador peptídico está compuesto por aproximadamente quince residuos de aminoácidos. El número de dominios Vl y Vh es equivalente. Por ejemplo, un anticuerpo bivalente de cadena sencilla se puede representar de la siguiente manera: Vl-L1-Vh-L2-Vl-L3-Vh o Vl-L1-Vh-L2-Vh-L3-Vl o Vh-L i-Vl-L2-Vh-L3-Vl o Vh-L1-Vl-L2-VL-L3-Vh. Los anticuerpos multivalentes de cadena sencilla que son trivalentes o mayores tienen uno o más fragmentos de anticuerpos unidos a un anticuerpo bivalente de cadena sencilla por enlaces peptídicos adicionales. Un ejemplo de un anticuerpo trivalente de cadena sencilla es: Vl-L1-Vh-L2-Vl-Li-Vh-L2-Vl-Li-Vh.
Se pueden combinar dos anticuerpos de cadena sencilla para formar un diacuerpo, también conocido como dímero bivalente. Los diacuerpos tienen dos cadenas. Cada cadena del diacuerpo incluye un dominio Vh conectado a un dominio Vl por un conector corto de aproximadamente 5-10 residuos de aminoácidos, por ejemplo (Gly-Gly-Gly-Gly-Ser), (Gly-Gly-Gly-Gly-Ser)2. Dichos enlazadores son lo suficientemente cortos para impedir el emparejamiento intracadena entre dominios en la misma cadena, lo que impulsa el emparejamiento entre cadenas entre dominios complementarios en diferentes cadenas y recrea dos sitios de unión a antígeno. La estructura del diacuerpo es rígida
y compacta, con sitios de unión a antígeno en los extremos opuestos de la molécula. Los diacuerpos pueden ser monoespecíficos o biespecíficos.
Se pueden combinar tres anticuerpos de cadena sencilla para formar un triacuerpo, también conocido como trímeros trivalentes. Los triacuerpos se construyen con el extremo carboxi de un dominio Vl o Vh directamente fusionado al extremo amino de un dominio Vh o Vl, es decir, sin ninguna secuencia enlazadora. El triacuerpo tiene tres cabezas de Fv con los polipéptidos dispuestos de forma cíclica, de la cabeza a la cola. Una posible conformación de la molécula de triacuerpo es plana con los tres sitios de unión ubicados en un plano en un ángulo de 120 grados entre sí. Los triacuerpos pueden ser monoespecíficos, biespecíficos o triespecíficos.
Se entiende que los anticuerpos anti-VEGFR2, cuando se usan en un mamífero con fines de profilaxis o tratamiento, se administrarán en forma de una composición que comprende adicionalmente un vehículo farmacéuticamente aceptable. Los vehículos farmacéuticamente aceptables adecuados incluyen, por ejemplo, uno o más de agua, solución salina, solución salina amortiguada con fosfato, dextrosa, glicerol, etanol y similares, así como combinaciones de los mismos. Los vehículos farmacéuticamente aceptables pueden comprender además cantidades menores de sustancias auxiliares tales como agentes de humectación o emulsionantes, conservantes o amortiguadores, que mejoran la vida útil o la efectividad de los anticuerpos.
Como se establece en esta invención, la invención proporciona la administración de inhibidores de Rho-quinasa con inhibidores de angiogénesis. Por consiguiente, los anticuerpos de VEGFR pueden administrarse con cualquier inhibidor de Rho-quinasa, incluidos, entre otros, los descritos en esta invención, compuestos descritos en WO 2014/055996, WO 2014/055999 y WO 2006/105051.
Los anticuerpos anti-VEGFR-2 pueden administrarse con agentes que se dirigen al VEGF. Ejemplos no limitantes de agentes de unión a VEGF incluyen anticuerpos de VEGF y trampas de VEGF (es decir, dominios de unión a ligando de receptores de VEGF). En general, una trampa de VEGF es una proteína que comprende dominios de unión a VEGF de una o más proteínas receptoras de VEGF. Las trampas de VEGF incluyen, entre otros, VEGFR-1 soluble, neuropilina 1 soluble (NRP1), VeGFR-3 soluble (que une VeGF-C y VEGF-D) y aflibercept (Zaltrap; Eyelea; VEGF Trap R1R2), compuesto por segmentos de los dominios extracelulares de receptores de factor de crecimiento endotelial vascular humano VEGFR1 y VEGFR2 fusionados a la región constante (Fc) de IgG1 humano. Conbercept (KH902) es una proteína de fusión que contiene el dominio extracelular 2 de VEGFR-1 (Flt-1) y el dominio extracelular 3, 4 de VEGFR-2 (KDR) fusionado a la porción Fc de IgG1 humano. Se describen varias trampas de VEGF que contienen dominios similares a KDR y FLT-1 Ig en diversas combinaciones en U.S. Patente 8,216,575. Las DARPins (un acrónimo de proteínas repetidas de anquirina diseñadas) son proteínas miméticas de anticuerpos modificadas genéticamente que típicamente exhiben una unión a proteínas diana altamente específicas y de alta afinidad. DARPin® MP0112 es un inhibidor del factor de crecimiento endotelial vascular (VEGF) y ha entrado en ensayos clínicos para el tratamiento de la degeneración macular húmeda y el edema macular diabético.
La expresión de VEGF puede ser alcanzada. Por ejemplo, el inhibidor de VEGF PTC299 se dirige a VEGF posttranscripcionalmente uniendo selectivamente las regiones 5'- y 3'- no trasladadas (UTR) del RNA mensajero de VEGF (mRNA), evitando así la traslación de VEGF. Pegaptanib (Macugen) es un aptámero de RNA dirigido contra VEGF-165.
El factor de crecimiento placentario (P1GF) se ha implicado en la angiogénesis patológica. P1GF está estructuralmente relacionado con VEGF y también es un ligando para VEGFR-1. En consecuencia, las trampas de VEGF que comprenden el dominio extracelular de VEGFR1 (ver arriba) son útiles para alcanzar a P1GF.
PDGF se compone de cuatro cadenas de polipéptidos que forman los homodímeros PDGF-AA, BB, CC y DD, así como el heterodímero PDGF-AB. Los receptores PDGF (PDGFR) -a y -p median las funciones PDGF. Específicamente, PDGFRa se une a PDGF-AA, -BB, -AB y -CC, mientras que PDGFRp interactúa con -BB y -DD. Los ejemplos no limitantes de agentes de unión a PDGF incluyen anticuerpos anti-PDGF y trampas de PDGF. Los agentes que se dirigen a PDGF incluyen Fovista™ (E10030, Ophthotech), un aptámero pegilado dirigido a PDGF-B y AX102 (Sennino y col., 2007, Cancer Res. 75(15):7359-67), un aptámero de oligonucleótidos de DNA que se une a PDGF-B. Los agentes que se dirigen a los receptores PDGF incluyen ramucirumab (IMC-3G3, IgG1 humano) un anticuerpo anti-PDGFRa, crenolanib (CP-868596), un inhibidor selectivo de PDGFRa (IO50 = 0,9 nM) y PDGFRp (IC50 = 1,8 nM), y nilotinib (Tasigna®), un inhibidor de PDGFRa y PDGFRp y otras tirosina quinasas.
Los inhibidores de angiogénesis incluyen agentes intracelulares que bloquean la transducción de señales mediada por, por ejemplo, VEGF, PDGF, ligandos de receptores VEGF o PDGF, o complemento. Los agentes intracelulares que inhiben los inhibidores de angiogénesis incluyen los siguientes, entre otros. Sunitinib (Sutent; SU11248) es un inhibidor específico de moléculas pequeñas de VEGFR1-VEGFR3, PDGFRa y PDGFRp, receptor del factor de células madre (cKIT), Flt-3 y receptor del factor 1 estimulante de colonias (CSF-1R). Axitinib (AG013736; Inlyta) es otro
inhibidor de tirosina quinasa de molécula pequeña que inhibe VEGFR-1-VEGFR-3, PDGFR y cKIT. Cediranib (AZD2171) es un inhibidor de VEGFR-1-VEGFR-3, PDGFRp y cKIT. Sorafenib (Nexavar) es otro inhibidor molecular pequeño de varias proteínas tirosina quinasas, incluidas las VEGFR, PDGFR y Raf quinasas. Pazopanib (Votrient; (GW786034) inhibe VEGFR-1, -2 y -3, cKIT y PDGFR. Foretinib (GSK1363089; XL880) inhibe VEGFR2 y MET. CP-547632 es como un potente inhibidor de VEGFR-2 y de las quinasas del factor básico de crecimiento de fibroblastos (FGF). E-3810 ((6-(7-((1-aminociclopropil)metoxi)-6-metoxiquinolin-4-iloxi) -N-metil-1-naftamida) inhibe VEGFR-1, -2 y -3 y FGFR- 1 y -2 quinasas en el intervalo nanomolar. Brivanib (BMS-582664) es un inhibidor de VEGFR-2 que también inhibe la señalización del receptor de FGF. CT-322 (Adnectin) es una proteína pequeña basada en un dominio de fibronectina humana y se une e inhibe la activación de VEGFR2. Vandetanib (Caprelas; Zactima; ZD6474) es un inhibidor de las tirosina quinasas VEGFR2, EGFR y RET. X-82 (Xcovery) es un inhibidor de indolinona de molécula pequeña de señalización a través de los receptores del factor de crecimiento VEGFR y PDGFR
Los antiinflamatorios e inmunosupresores incluyen fármacos esteroides como los glucocorticoides (por ejemplo, dexametasona), FK506 (tacrolimus), ciclosporina, fingolimod, interferón, como IFNp o IFNy, una proteína de unión al factor de necrosis tumoral alfa (TNF-a) como infliximab (Remicade), etanercept (Enbrel) o adalimumab (Humira) y ácido micofenólico.
Debe entenderse y esperarse que un experto en la técnica pueda realizar variaciones en los principios de invención descritos en esta invención y se pretende que tales modificaciones se incluyan dentro del alcance de la presente invención.
EJEMPLOS
Las abreviaturas utilizadas en los siguientes ejemplos y preparaciones incluyen:
Ac2O anhídrido acético
AcOH ácido acético
Bn Bencilo
Celite® tierra de diatomeas
DCM diclorometano
DIEA diisopropiletilamina
DMAP 4-dimetilamino piridina
DME 1,2-dimetoxiletano
DMF dimetil formamida
DMSO dimetil sulfóxido
EDC 1-(3-dimetilaminopropil)-3-etilcarbodimida hidrocloruro
EtOAc acetato de etilo
EtOH alcohol etílico o etanol
Et2O éter etílico
EtaN trietilamina,
g gramos
HOBt 1-hidroxibenzotriazol
HPLC cromatografía líquida de alta presión
h hora(s)
MeCN acetonitrilo
min minuto(s)
MeOH alcohol metílico o metanol
mL mililitro
mmol milimoles
MS espectrometría de masas
NMR resonancia magnética nuclear
iPrOH iso-propanol
PyBOP® benzotriazol-1-il-oxitri pirrolidinofosfonio
rt temperatura ambiente
s singlete
t triplete
THF tetrahidrofurano
La espectrometría de masas fue realizada por: SynPep Co., 6905 Ct. Dublin, CA 94568, o se registró en una LC-MS: Waters 2695 Módulo de separaciones con un detector MS cuadrapolo simple Waters ZQ2000. A menos que se indique, toda la espectrometría de masas se ejecutó en modo ESI. 1. Los espectros de NMR H se registraron en una máquina Varian a 400 MHz utilizando el software Mercury. En términos generales, la síntesis de los siguientes ejemplos de compuestos de la presente invención no se describe explícitamente en dicho ejemplo, la síntesis es como se describe
en esta invención en términos generales y el material de partida apropiado puede seleccionarse fácilmente para sintetizar el compuesto del ejemplo.
Ejemplo 1
W-(2-(5-Metox¡¡somdolm-2-yl)p¡r¡midm-4-¡l)-1H-mdazol-5-amma

W-(2-Cloropirimidin-4-il)-1H-mdazol-5-amina

A una solución de 2,4-dicloropirimidina (2,0 g, 13,4 mmol) en etanol (67 mL) se le añadió 5-amino-indazol (1,79 g, 13,4 mmol) y trietilamina (2,81 mL, 20,1 mmol). La mezcla se calentó a reflujo durante la noche. La mezcla de reacción se concentró y el residuo se recristalizó con metanol para proporcionar el compuesto del título como un sólido rosa (3,1 g, 97%).
W-(2-(5-Metoxiisoindolin-2-yl)pirimidin-4-il)-1H-indazol-5-amina

Una mezcla de /V-(2-cloropirimidin-4-il) -1H-indazol-5-amina (0,500 g, 2,04 mmol), clorhidrato de 5-metoxiisoindolina (0,404 g, 2,18 mmol) y K2C03 (0,844 g, 6,11 mmol) en DMF (4,07 mL) se agitó a 115°C durante la noche. La mezcla se diluyó con agua y se extrajo con EtOAc dos veces. Las capas orgánicas combinadas se lavaron con agua y salmuera, se secaron sobre Na2SO4, se concentraron y purificaron por cromatografía con MeOH en DCM. El producto se purificó adicionalmente por recristalización en EtOAc para proporcionar el compuesto del título como un sólido naranja claro (215 mg, 30%). 1H NMR (300 MHz, DMSO-d6) 5 13,15 (s, 1H), 9,45 (s, 1H), 8,52 (s, 1H), 8,29 (s, 1H), 8,16 (d, J = 5,8 Hz, 1H), 7,81 - 7,66 (m, 2H), 7,59 (s, 1H), 7,25 (d, J = 19,2 Hz, 1H), 7,10 (d, J = 8,4 Hz, 1H), 6,29 (d, J = 5,8 Hz, 1H), 5,01 (d, J = 16,0 Hz, 4H), 3,99 (s, 3H). MS (ES+) m/e 359 (M+H)+.
Ejemplo 2
W-(2-(Isomdolm-2-¡l)pyr¡m¡dm-4-¡l)-1H-mdazol-5-amma

Una mezcla de W-(2-cloropirimidin-4-il)-1H-indazol-5-amina (0,200 g, 1,03 mmol), isoindolina (0,140 g, 1,03 mmol) y K2CO3 (0,171 g, 1,24 mmol) en DMF (6,89 mL) se agitó a 80°C durante 4 h seguido de rt durante la noche. La mezcla se diluyó con agua y se extrajo con EtOAc. La capa orgánica se lavó con agua, salmuera y se secó sobre MgSO4 para proporcionar un aceite marrón crudo que se recristalizó en MeOH/DCM al 5% seguido de una recristalización adicional
en EtOH para proporcionar el compuesto del título como un sólido gris (36,1 mg, 11%). 1H NMR (300 MHz, DMSO-d6) 5 12,95 (s, 1H), 9,25 (s, 1H), 8,25 (s, 1H), 8,07 (s, 1H), 7,95 (d, J = 4,4 Hz, 1H), 7,31-7,51 (m, 6H), 6,08 (d, J = 4,4 Hz, 1H), 4,82 (d, J = 24,7 Hz, 4H). MS (ES+) m/e 329 (M+H)+.
Ejemplo 3
W-(5-Fluoro-2-(5-metox¡¡somdolm-2-M)pmmidm-4-¡l)-1H-mdazol-5-amma

W-(2-Cloro-5-fluoropirimidin-4-il)-1H-indazol-5-amina

A una solución de 2,4-didoro-5-fluoropirimidina (0,800 g, 4,55 mmol) en EtOH (40 mL) se le añadió Na2CO3 (6,27 g, 45,5 mmol) y compuesto 1H-indazol-5-amina (0,605 g, 4,55 mmol). La mezcla resultante se agitó durante 12 h a 100°C. Después de que LCMS mostró que la reacción se había completado, el disolvente se eliminó a presión reducida y el residuo se puso en agua (50 mL) y se extrajo con EtOAc (3 x 100 mL). La fase orgánica se secó sobre Na2SO4, se filtró y se concentró a presión reducida para dar un residuo que se purificó por cromatografía en columna sobre gel de sílice (eluido con PE:EA = 1:1) para dar el compuesto del título como un sólido (120 mg, 9,7%).
W-(5-Fluoro-2-(5-metoxiisoindolin-2-il)pirimidin-4-il)-1H-indazol-5-amina
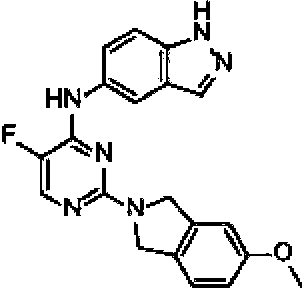
A una solución de N-(2-cloro-5-fluoropirimidin-4-il) -1H-indazol-5-amina (300 mg, 1,14 mmol) en acetonitrilo (4 mL) se añadieron 5-metoxiisoindolina (170 mg, 1,14 mmol ) y DIEA (441 mg, 3,42 mmol). La mezcla resultante se calentó a 90°C durante 12 h. Después de que LCMS mostró que la reacción se había completado, la mezcla se concentró y se disolvió en metanol y se purificó por HPLC preparativa, y se liofilizó para dar el compuesto del título como un sólido blanco (52 mg, 12,1%). 1H NMR (300 MHz, DMSO-d6) 513,0 (s, 1H), 9,26 (s, 1H), 8,32 (s, 1H), 8,09 (s, 1H), 8,03 (d, J = 4,0 Hz, 1H), 7,74 (d, J = 8,8 Hz, 1H), 7,50 (d, J = 9,2 Hz, 1H), 7,30 (b, 1H), 7,00 (b, 1H), 6,86 (dd, J = 8,4 y 2,8 Hz, 1H), 4,72 (s, 2H), 4,69 (s, 2H), 3,76 (s, 3H). MS (ES+) m/e 377 (M+H)+.
Ejemplo 4
W-(5-Cloro-2-(5-methxMsomdolm-2-¡l)p¡rim¡dm-4-M)-1H-mdazol-5-amma

W-(2,5-Dicloropirimidin-4-il)-1H-indazol-5-amina

A una solución de 2,4,5-tridoropirimidina (1,00 g, 5,49 mmol) en EtOH (20 mL) se añadieron Na2CO3 (3,03 g, 28,6 mmol) y compuesto 1H-indazol-5-amina (0,657 g, 4,94 mmol). La mezcla resultante se agitó durante 12 h a 15°C. Después de que LCMS mostró que la reacción se había completado, a continuación, se eliminó el disolvente a presión reducida y el residuo se puso en agua (50 mL) y se extrajo con EtOAc (3 x 50 mL). La fase orgánica se secó sobre Na2SO4, se filtró y se concentró a presión reducida para dar al compuesto el compuesto del título como un sólido (800 mg, 52,3%).
W-(5-Cloro-2-(5-methoxiisoindolin-2-il)pirimidin-4-il)-1H-indazol-5-amina

A una solución de compuesto N-(2,5-dicloropirimidin-4-il)-1H-indazol-5-amina (281 mg, 1,01 mmol) en acetonitrilo (5 mL) se le añadió compuesto 5-metoxilisoindolina (150 mg, 1,01 mmol) y DIEA (391 mg, 3,03 mmol). La mezcla resultante se calentó a 90°C durante 12 h. Después de que LCMS mostró que la reacción se había completado, la mezcla se concentró y se disolvió en metanol y se purificó por HPLC preparativa, y se liofilizó para dar el compuesto del título como un sólido blanco (115 mg, 29,1%). 1H NMR (300 MHz, DMSO-d6) 513,00 (s, 1H), 8,73 (s, 1H), 8,14 (s, 1H), 8,08 (s, 2H), 7,69 (d, J = 8,8 Hz, 1H), 7,50 (s, J = 8,8 Hz, 1H), 7,27 (b, 1H), 6,97 (b, 1H), 6,84 (d, J = 8,4 Hz, 1H), 4,67 (t, J = 16,4 Hz, 4H), 3,74 (s, 3H). MS (ES+) m/e 393 (M+H)+.
Ejemplo 5
W-(2-(5-MetoxMsomdolm-2-¡l)-5-met¡lpmm¡dm-4-¡l)-1H-mdazol-5-amma

W-(2-Cloro-5-metilpyrimidin-4-il)-1H-indazol-5-amina
A una solución de compuesto de 2,4-dicloro-5-metilpirimidina (1,00 g, 6,17 mmol) en EtOH (20 mL) se le añadió Na2CO3 (3,27 g, 30,9 mmol) y compuesto 1H-indazol-5-amina (0,821 g, 6,17 mmol). La mezcla resultante se agitó durante 12 h a 90°C. Después de que LCMS mostró que la reacción se había completado, a continuación, se eliminó el disolvente a presión reducida y el residuo se puso en agua (50 mL) y se extrajo con EtOAc (3 x 50 mL). La fase orgánica se secó sobre Na2SO4, se filtró y se concentró a presión reducida para dar un residuo que se purificó por HPLC preparativa para dar al compuesto el compuesto del título como un sólido (400 mg, rendimiento: 25%).
W-(2-(5-Metoxiisoindolin-2-il)-5-metilpirimidin-4-il)-1H-indazol-5-amina

A una solución del compuesto N-(2-cloro-5-metilpirimidin-4-il) -1H-indazol-5-amina (261 mg, 1,01 mmol) en DMF (5
mL) se le añadió el compuesto 5-metoxiisoindolina (150 mg, 1,01 mmol) y DIEA (391 mg, 3,03 mmol). La mezcla resultante se calentó a 110°C durante 12 h. Después de que LCMS mostró que la reacción se había completado, la mezcla se concentró y se disolvió en metanol y se purificó por HPLC preparativa, y se liofilizó para dar el compuesto del título como un sólido blanco (105 mg, 27,0%). 1H NMR (300 MHz, DMSO-da) 512,92 (s, 1H), 8,25 (s, 1H), 8,18 (s, 1H), 8,06 (s, 1H), 7,81 (s, 1H), 7,72 (d, J = 9,2 Hz, 1H), 7,48 (d, J = 8,8 Hz, 1H), 7,25 (d, J = 8,0 Hz, 1H), 6,98 (s, 1H), 6,83 (d, J = 8,0 Hz, 1H), 4,70 (s, 2H), 4,65 (s, 2H), 3,75 (s, 3H), 2,09 (s, 3H). MS (ES+) m/e 373 (M+H)+.
Ejemplo 6
N-(2-(5-Metoxiisoindolin-2-il)-6-metilpirimidin-4-il)-1H-indazol-5-amina

N-(2-Cloro-6-metilpirimidin-4-il)-1H-indazol-5-amina

A una solución del compuesto 2,4-dicloro-6-metilpirimidina (1,00 g, 6,17 mmol) en EtOH (20 mL) se le añadió Na2CO3 (3,27 g, 30,9 mmol) y compuesto 1H-indazol-5-amina (0,821 g, 6,17 mmol). La mezcla resultante se agitó durante 12 h a 90°C. Después de que LCMS mostró que la reacción se había completado, a continuación, se eliminó el disolvente a presión reducida y el residuo se puso en agua (50 mL) y se extrajo con EtOAc (3 x 50 mL). La fase orgánica se secó sobre Na2SO4, se filtró y se concentró a presión reducida para dar un residuo que se purificó por HPLC preparativa para dar el compuesto del título como un sólido (400 mg, 25%).
N-(2-(5-Metoxiisoindolin-2-il)-6-metilpirimidin-4-il)-1H-indazol-5-amina

A una solución del compuesto N-(2-cloro-6-metilpirimidin-4-il) 1H-indazol-5-amina (261 mg, 1,01 mmol) en DMF (5 mL) se le añadió el compuesto 5-metoxiisoindolina (150 mg, 1,01 mmol) y DIEA (391 mg, 3,03 mmol). La mezcla resultante se calentó a 110 °C durante 12 h. Después de que LCMS mostró que la reacción se había completado, la mezcla se concentró y se disolvió en metanol y se purificó por HPLC preparativa, y se liofilizó para dar el compuesto del título como un sólido blanco (80 mg, 21,4%). 1H NMR (300 MHz, DMSO-d6) 512,91 (s, 1H), 9,08 (s, 1H), 8,25 (s, 1H), 8,06 (s, 1H), 7,47-7,53 (m, 2H), 7,29 (b, 1H), 7,00-7,05 (m, 1H), 6,87 (d, J = 10,4 Hz, 1H), 5,93 (s, 1H), 5,76 (b, 4H), 3,77 (s, 3H), 2,17 (s, 3H). MS (ES+) m/e 373 (M+H)+.
Ejemplo 7
N-(1H-Indazol-5-M)-2-(5-metoxMsomdoMn-2-M)-5,7-dihidrofuro[3,4-d]pirimidm-4-amma
2-Cloro-N-(1H-indazol-5-il)-5,7-dihidrofuro[3,4-d]pirimidin-4-amina

A una solución del compuesto 2,4-dicloro-5,7-dihidrofuro[3,4-d] pirimidina (1,00 g, 5,30 mmol) en EtOH (30 mL) se le añadió Na2CO3 (1,70 g, 15,8 mmol) y compuesto 1H-indazol-5-amina (711 mg, 5,3 mmol). La mezcla resultante se agitó durante 12 h a 15°C. Después de que LCMS mostró que la reacción se había completado, el disolvente se eliminó a presión reducida y el residuo se disolvió en EtOAc (100 mL), se lavó con agua (2 x 50 mL). La fase orgánica se secó sobre Na2SO4, se filtró y se concentró a presión reducida para dar al compuesto el compuesto del título como un sólido (800 mg, rendimiento: 38,3%).
N-(1H-indazol-5-il)-2-(5-metoxiisoindolin-2-il)-5,7-dihidrofuro[3,4-d]pirimidin-4-amina

A una solución del compuesto 2-cloro-N-(1H-indazol-5-il)-5,7-dihidrofuro[3,4-d]pirimidin-4-amina (346 mg, 1,21 mmol) en acetonitrilo (30 mL) se añadieron el compuesto 5-metoxiisoindolina (150 mg, 1,00 mmol) y DIEA (273 mg, 2,11 mmol). La mezcla resultante se calentó a 90°C durante 20 h. Después de que LCMS mostró que la reacción se había completado, la mezcla se concentró y se disolvió en metanol y se purificó por HPLC preparativa, y se liofilizó para dar el compuesto del título (69,0 mg, 17,2%). 1H NMR (300 MHz, DMSO-d6) 5 12,90 (s, 1H), 8,83 (s, 1H), 8.27 (s, 1H), 8,09 (s, 1H), 7,64 (d, J = 8,8 Hz, 1H), 7,50 (d, J = 8,8 Hz, 1H), 7,00-7.40 (m, 2H), 6,86 (dd, J = 8,4 y 2,8 Hz, 1H), 4.73 4.89 (m, 8H), 3,77 (s, 3H). MS (ES+) m/e 401 (M+H)+.
Ejemplo 8
Ejemplo comparativo N-(4-(5-metoxMsoindolin-2-il)pirimidin-2-il)-1H-indazol-5-amina
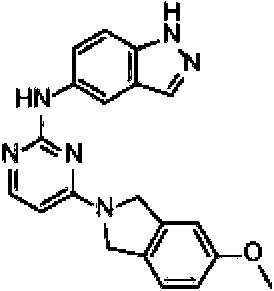
2-(2-Cloropirimidin-4-il)-5-metoxiisoindolina
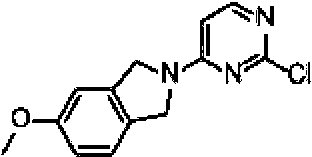
A una solución del compuesto 2,4-dicloropmmidina (298 mg, 2,01 mmol) en EtOH (10 mL) se le añadió Na2CO3 (1,11 g, 10,5 mmol) y compuesto 5-metoxiisoindolina (300 mg, 2,01 mmol). La mezcla resultante se agitó durante 12 h a 90°C. Después de que LCMS mostró que la reacción se había completado, a continuación, se eliminó el disolvente a presión reducida y el residuo se puso en agua (50 mL) y se extrajo con EtOAc (3 x 50 mL). La fase orgánica se secó sobre Na2SO4, se filtró y se concentró a presión reducida para dar un residuo que se purificó por TLC preparativa para dar el compuesto del título como un sólido (250 mg, rendimiento: 47,8%).
N-(4-(5-Metoxiisoindolin-2-il)pirimidin-2-il)-1H-indazol-5-amina

A una solución del compuesto 2-(2-cloropirimidin-4-il) -5-metoxiisoindolina (250 mg, 0,958 mmol) en DMF (20 mL) se le agregaron el compuesto 5-aminoindazol (127 mg, 0,958 mmol) y DIEA (370 mg, 2,87 mmol). La mezcla resultante se calentó a 110 °C durante 20 h. Después de que LCMS mostró que la reacción se había completado, la mezcla se concentró y se disolvió en metanol y se purificó por prep-HPLC, y se liofilizó para dar el compuesto del título (51,7 mg, 17%). 1H NMR (300 MHz, DMSO-da) 512,83 (s, 1H), 9,03 (s, 1H), 8,33 (s, 1H), 8,00 (s, 1H), 7,99 (s, 1H), 7,62 (dd, J = 9,2 y 2,0 Hz, 1H), 7,42 (d, J = 8,8 Hz, 1H), 7,30 (b, 1H), 7,07 (b, 1H), 6,90 (d, J = 8,4 Hz, 1H), 6,02 (d, J = 6,4 Hz, 1H), 4,83 (d, J = 14,4 Hz, 2H), 4,65 (d, J = 17.6 Hz, 2H).3,77 (s, 3H). MS (ES+) m/e 359 (M+H)+.
Ejemplo 9
N-(2-(5-Fluoroisoindolin-2-il)pirimidin-4-il)-1H-indazol-5-amina

A una solución de N-(2-cloropirimidin-4-il)-1H-indazol-5-amina (457 mg, 1,87 mmol) en DMF (5 mL) se le añadieron el compuesto 5-fluoroisoindol (250 mg, 1,87 mmol) y DIEA (724 mg, 5,61 mmol). La mezcla resultante se calentó a 110°C durante 20 h. Después de que LCMS mostró que la reacción se había completado, la mezcla se concentró y se disolvió en metanol y se purificó por prep-HPLC, y se liofilizó para dar el compuesto del título (200 mg, 31,0%). 1H NMR (300 MHz, DMSO-d6) 5 12,92 (s, 1H), 9,24 (s, 1H), 8,28 (s, 1H), 8,06 (s, 1H), 7,94 (d, J = 6,0 Hz, 1H), 7,11-7,53 (m, 5H), 6,08 (d, J = 6,4 Hz, 1H), 4,80 (b, 4H). MS (ES+) m/e 347 (M+H)+.
Ejemplo 10
N-(2-(5-Cloroisoindolin-2-il)pirimidin-4-il)-1H-indazol-5-amina

A una solución de N-(2-cloropirimidin-4-il)-1H-indazol-5-amina (384 mg, 1,57 mmol) en acetonitrilo (5 mL) se añadieron el compuesto 5-cloroisoindolina (240 mg, 157 mmol) y DIEA (608 mg, 4,71 mmol). La mezcla resultante se calentó a 90°C durante 17 h. Después de que LCMS mostró que la reacción se había completado, la mezcla se concentró y el crudo se lavó con MeOH para dar el compuesto del título como un sólido marrón (230 mg, rendimiento: 40,5%). 1H NMR (300 MHz, DMSO-d6) 513,00 (s, 1H), 9,65 (s, 1H), 8,31 (s, 1H), 8,10 (s, 1H), 7.93 (d, J = 6.0 Hz, 1H), 7,35-7,57 (m, 5H), 6,18 (d, J = 6,0 Hz, 1H), 4,82 (b, 4H). MS (ES+) m/e 363 (M+H)+.
Ejemplo 11
Ejemplo comparativo N-(2-(6-Metoxi-3,4-dihidroisoquinolin-2(1H)-il)pirimidin-4-il)-1H-indazol-5-amina

Una mezcla de N-(2-doropirimidin-4-il)-1H-indazol-5-amina (100 mg, 0,41 mmol), 6-metoxi-1,2,3,4-tetrahidroisoquinolina (81,3 mg, 0,41 mmol) y K2CO3 (168 mg, 1,22 mmol) en DMF (1,2 mL) se agitó a 120°C durante la noche, se enfrió a temperatura ambiente, se diluyó con agua y se extrajo con EtOAc. La capa orgánica se concentró y el residuo se purificó por cromatografía con 1-20% de MeOH/DCM para proporcionar el compuesto del título como un sólido blanco (42 mg, 28%). 1H NMR (300 MHz, DMSO-d6) 5 12,96 (s, 1H), 9,21 (s, 1H), 8,13 (d, J = 1.6 Hz, 1H), 8,04 (t, J = 1,1 Hz, 1H), 7,93 (d, J = 5,7 Hz, 1H), 7,56-7,41 (m, 2H), 7,15 (d, J = 8,2 Hz, 1H), 6,84 - 6,72 (m, 2H), 6,03 (d, J = 5,7 Hz, 1H), 4,79 (s, 2H), 3,95 (t, J = 5,8 Hz, 2H), 3,73 (s, 3H), 2,85 (t, J = 6,0 Hz, 2H). MS (ES+) m/e 373 (M+H)+.
Ejemplo 12
Ejemplo comparativo N-(2-(6,7-dihidrotieno[3,2-c]piridin-5(4H)-il)pirimidin-4-il)-1H-indazol-5-amina

Una mezcla de N-(2-cloropirimidin-4-il)-1H-indazol-5-amina (100 mg, 0,41 mmol), 4-H tieno[3,2] piridina (71,5 mg, 0,41 mmol) y K2CO3 (168 mg, 1,22 mmol) en DMF (1,2 mL) se agitó a 120°C durante la noche, se enfrió a temperatura ambiente, se diluyó con agua y se extrajo con EtOAc. La capa orgánica se concentró y el residuo se purificó por cromatografía con 1-20% de MeOH/DCM para proporcionar el compuesto del título como un sólido blanco (33 mg, 23%). 1H NMR (300 MHz, DMSO-d6) 512,97 (s, 1H), 9,23 (s, 1H), 8,12 - 8,01 (m, 2H), 7,93 (d, J = 5,7 Hz, 1H), 7,56 -7,40 (m, 2H), 7,34 (d, J = 5.1 Hz, 1H), 6,97 (d, J = 5,2 Hz, 1H), 6,04 (d, J = 5,7 Hz, 1H), 4,80 (s, 2H), 4,07 (t, J = 5,6 Hz, 2H), 2,93 - 2,82 (m, 2H). MS (ES+) m/e 349 (M+H)+.
Ejemplo 13
Ejemplo comparativo N-(6-(5-Metoxiisoindolin-2-il)pirimidin-4-il)-1H-indazol-5-amina

N-(6-Cloropirimidin-4-il)-1H-mdazol-5-amina

Una mezcla de 4,6-dicloropirimidina (300 mg, 2,01 mmol), 5-amino-1H-indazol-1-carboxilato de terc-butilo (470 mg, 2,01 mmol), diisopropiletilamina (0,74 mL, 3,03 mmol) y DMF ( 2,01 mL) se agitó a 80°C durante la noche seguido de 120°C durante 4 h. La mezcla de reacción se enfrió hasta temperatura ambiente, se diluyó con agua y se extrajo con EtOAc. La capa orgánica se concentró al vacío para proporcionar el compuesto del título que se llevó directamente para la reacción de la siguiente etapa sin purificación adicional.
N-(6-(5-Metoxiisoindolin-2-il)pirimidin-4-il)-1H-indazol-5-amina

Una mezcla de N-(6-doropirimidin-4-il)-1H-indazol-5-amina (150 mg, 0,61 mmol), cloruro de hidrógeno y 5 metoxiisoindolina (121 mg, 0,65 mmol), K2CO3 (253 mg, 1,83 mmol) en DMF (1,22 mL) se agitó a 100°C durante 2 h seguido de 120°C durante 12 h. La mezcla de reacción se enfrió hasta temperatura ambiente, se diluyó con agua y se extrajo con EtOAc. La capa orgánica se concentró y se purificó por cromatografía con 0-20% de MeOH en DCM para proporcionar el producto que se purificó adicionalmente por trituración con DCM/Hex para proporcionar el compuesto del título puro (30 mg, 14%). 1H NMR (300 MHz, DMSO-d6) 512,95 (s, 1H), 9,02 (s, 1H), 8,22 (d, J = 0,9 Hz, 1H), 8,01 (d, J = 2,1 Hz, 2H), 7,55 - 7,36 (m, 2H), 7,31 (d, J = 8,4 Hz, 1H), 7,00 (d, J = 2,3 Hz, 1H), 6,89 (dd, J = 8,4, 2,4 Hz, 1H), 5,74 (d, J = 1,1 Hz, 1H), 4,65 (s, 4H), 3,77 (s, 3H). MS (ES+) m/e 359 (M+H)+.
Ejemplo 14
Ejemplo comparativo N-(6-(5-MetoxiisomdoMn-2-N)-2-metMpirimidm-4-M)-1H-mdazol-5-amma
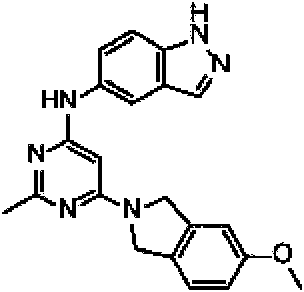
Una mezcla de N-(6-cloropirimidin-4-il)-1H-indazol-5-amina (159 mg, 0,61 mmol), cloruro de hidrógeno y 5 metoxiisoindolina (121 mg, 0,65 mmol), K2CO3 (253 mg, 1,83 mmol) en DMF (1,22 mL) se agitó a 100°C durante 2 h seguido de 120°C durante 12 h. La mezcla de reacción se enfrió hasta temperatura ambiente, se diluyó con agua y se extrajo con EtOAc. La capa orgánica se concentró y se purificó por cromatografía con 0-20% de MeOH en DCM para proporcionar el producto que se purificó adicionalmente por trituración con DCM/Hex para proporcionar el compuesto del título puro (25 mg, 11%). 1H NMR (300 MHz, DMSO-d6) 5 12,95 (s, 1H), 8,90 (s, 1H), 7,98 (d, J = 15,8 Hz, 2H), 7,54 - 7,36 (m, 2H), 7,29 (d, J = 8,5 Hz, 1H), 6,99 (d, J = 2,0 Hz, 1H), 6,88 (dd, J = 8,3, 2,4 Hz, 1H), 5,60 (s, 1H), 4,63 (s, 4H), 3,76 (s, 3H), 2,34 (s, 3H). MS (ES+) m/e 373 (M+H)+.
Ejemplo 15
Ejemplo comparativo N-(2-(1H-Pirrolo[3,4-c]piridin-2(3H)-il)pirimidin-4-il)-1H-indazol-5-amina

Una mezcla de N-(2-cloropirimidin-4-il)-1H-indazol-5-amina (100 mg, 0,41 mmol), cloruro de dihidrógeno 2,3-dihidro-1H-pirrolo[3,4-c]piridina (102 mg, 0,53 mmol), diisopropiletilamina (0,74 mL, 1,22 mmol) en DMF (0,81 mL) se agitó a 110°C durante la noche. La mezcla de reacción se enfrió hasta temperatura ambiente, se diluyó con agua y se extrajo con EtOAc. La capa orgánica se concentró y se purificó por cromatografía con MeOH al 0-20% en DCM para proporcionar el compuesto del título (13 mg, 10%) como un sólido ligeramente amarillo. 1H NMR (300 MHz, DMSO-d6) 512,95 (s, 1H), 9,29 (s, 1H), 8,69 (s, 1H), 8,52 (d, J = 5,0 Hz, 1H), 8,29 (s, 1H), 8,08 (s, 1H), 7,97 (d, J = 5,7 Hz, 1H), 7,61 - 7,46 (m, 3H), 6,11 (d, J = 5,8 Hz, 1H), 4,88 (s, 4H). MS (ES+) m/e 330 (M+H)+.
Ejemplo 16
N-(2-(5-Metoxiisoindolin-2-il)-5,6-dimetilpirimidin-4-il)-1H-indazol-5-amina

N-(2-Cloro-5,6-dimetilpirimidin-4-il)-1H-indazol-5-amina
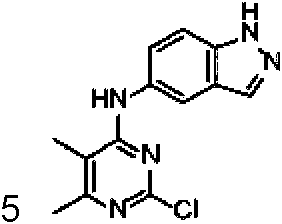
A una solución de 2,4-dicloro-5,6-dimetilpirimidina (0,800 g, 4,55 mmol) en EtOH (40 mL) se le añadió Na2CO3 (2,42 g, 22,8 mmol) y 1H-indazol-5-amina (0,605 g, 4,55 mmol). La mezcla resultante se agitó durante 12 h a 100°C. El disolvente se eliminó a presión reducida y el residuo se vertió en agua (50 mL) y se extrajo con EtOAc (3 x 100 mL). La fase orgánica se secó sobre Na2SO4, se filtró y se concentró a presión reducida para dar un residuo que se purificó por cromatografía en columna sobre gel de sílice (eluido con PE:EA = 1:1) para proporcionar el compuesto del título (120 mg, rendimiento: 9,7%) como un sólido blanco.
N-(2-(5-Metoxiisoindolin-2-il)-5,6-dimetilpirimidin-4-il)-1H-indazol-5-amina

A una solución de N-(2-cloro-5,6-dimetilpirimidin-4-il)-1H-indazol-5-amina (1,0 g, 3,66 mmol) en CH3CN (60 mL) se añadieron 5-metoxiisoindolina (545 mg, 3,66 mmol) y K2CO3 (1,01 g, 7,33 mmol). La mezcla resultante se calentó a 90°C durante 72 h. Después de que LCMS mostró que la reacción se había completado, la mezcla se concentró y el crudo se lavó con MeOH para dar el compuesto del título (423 mg, rendimiento: 30,0%) como un sólido marrón. 1H NMR (300 MHz, DMSO-d6) 512,95 (s, 1H), 8,08 (s, 1H), 8,01 (d, J = 8,8 Hz, 1H), 7,30 (d, J = 8,0 Hz, 1H), 7,00 (s, 1H), 6,90-6,83 (m, 4H), 4,81 (s, 2H), 4,77 (s, 2H), 3,75 (s, 3H), 2,44 (s, 3H), 2,77 (s, 3H). MS (ES+) m/e 387 (M+H)+. Ejemplo 17
N-(1H-Indazol-5-il)-2-(isoindolin-2-il)-5,7-dihidrofuro[3,4-d]pirimidin-4-amina
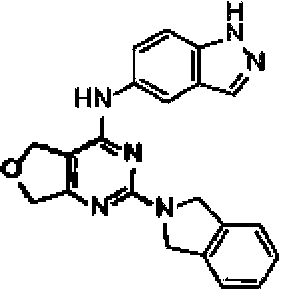
El compuesto del título se sintetizó utilizando el mismo procedimiento que el descrito para la síntesis del Ejemplo 7 (KL-00230). 1H NMR (400 MHz, DMSO-d6) 512,94 (s, 1H), 8,82 (s, 1H), 8,25 (s, 1H), 8,06 (s, 1H), 7,64-7,28 (m, 6H), 4,87-4,72 (m, 8H). MS (ES+) m/e 371 (M+H)+.
Ejemplo 18
N-(4-(1H-pirazol-4-il)fenil)-2-(5-metoxiisoindolin-2-il)pirimidin-4-amina
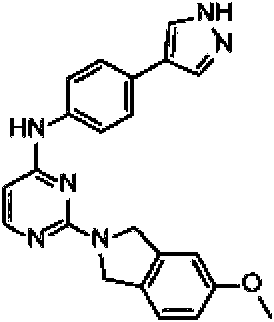
N-(4-Bromofenil)-2-doropirimidin-4-amina
hn.
N=/ ~ \ = 0 ^ Br
cl^ O
A una solución del compuesto N-(4-bromofenil)-2-doropirimidin-4-amina (2,00 g, 13,4 mmol) en EtOH (20 mL) se le añadió Na2CO3 (4,26 g, 40,2 mmol) y compuesto 4-bromoanilina (2,3 g, 13,4 mmol). La mezcla resultante se agitó durante 12 h a 15°C. Después de que LCMS mostró que la reacción se había completado, a continuación, se eliminó el disolvente a presión reducida y el residuo se puso en agua (50 mL) y se extrajo con EtOAc (3 x 50 mL). La fase orgánica se secó sobre Na2SO4, se filtró y se concentró a presión reducida para dar un residuo que se purificó por lavado con EA para dar el compuesto del título (1,20 g, rendimiento: 31,6%) como un sólido.
N-(4-Bromofenil)-2-(5-metoxiisoindolin-2-il)pirimidin-4-amina

A una solución del compuesto N-(4-bromofenil)-2-cloropirimidin-4-amina (1,00 g, 3,53 mmol) en acetonitrilo (5 mL) se le agregaron el compuesto 5-metoxiisoindolina (526 mg, 3,53 mmol) y DIEA (1,36 g, 10,6 mmol). La mezcla resultante se calentó a 90°C durante 17 h. Después de que LCMS mostró que la reacción se había completado, la mezcla se concentró y el crudo se lavó con MeOH para dar el compuesto del título (600 mg, rendimiento: 42,9%) como un sólido marrón.
2-(5-Metoxiisoindolin-2-il)-N-(4-(1-((2-(trimetilsilil)etoxi)metil)-1H-pirazol-4-il)fenil)pirimidin-4-amina

A la solución del compuesto N-(4-bromofenil)-2-(5-metoxiisoindolin-2-il) pirimidin-4-amina (300 mg, 0,758 mol), compuesto 4-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)-1-((2-(trimetilsilil)etoxi)metil)-1 H-pirazol (492 mg, 1,52 mol) y K2CO3 (209 mg, 1,52 mol) se disolvieron en dioxano/H2O (2 mL/2 mL) y se desgasificaron con nitrógeno durante 10 minutos. Se añadió Pd(dppf) Cl2 (27,7 mg, 0,0379 mmol) y la mezcla de reacción se agitó bajo nitrógeno a 85°C durante 17 h. Después de que LCMS mostró que la reacción se había completado, a continuación, la mezcla se concentró y se usó para la siguiente etapa.
N-(4-(1H-Pirazol-4-il)fenil)-2-(5-metoxiisoindolin-2-il)pirimidin-4-amina

La solución del compuesto 2-(5-metoxiisoindolin-2-il)-N-(4-(1-((2-(trimetilsilil)etoxi)metil)-1H-pirazol-4-il)fenil)pirimidin-4-amina (350 mg, 0,0195 mol) se disolvió en TFA (2 mL) y se agitó a 85°C durante 1 h. Después LCMS mostró que la reacción estaba completa. a continuación, se eliminó el TFA y el residuo se purificó mediante prep-HPLC para dar el compuesto del título (53,1 mg, rendimiento: 20,3%) como un sólido marrón. 1H NMR (400 MHz, DMSO-d6) 59,55 (s, 1H), 7,99 (s, 1H), 7,94 (d, J = 6,0, 1H), 7,76 (d, J = 8,4, 1H), 7,57 (d, J = 8,4, 1H), 7,31 (s, 1H), 7,00 (s, 1H), 6,89 (d, J
= 8,4, 1H), 6,13 (d, J = 5,6, 1H), 4,77 (m, 4H), 3,76 (s, 3H). MS (ES+) m/e 385 (M+H)+.
Ejemplo 19
2-(5-Metoxiisoindolin-2-il)-N-(piridin-4-il)pirimidin-4-amina

2-(5-Metoxiisoindolin-2-il)pirimidin-4-amina
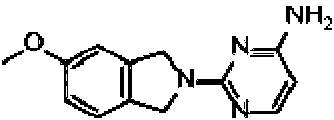
A una solución, compuesto 2-d oropirimidin-4-amina (381 mg, 2,95 mmol) y DIEA (692 mg, 5,37 mmol) en CH3CN (20 mL) compuesto 5-metoxiisoindolina (400 mg, 2,68 mmol). A continuación, la reacción se agitó a 90°C durante 16 h. Después LCMS mostró que la reacción estaba completa. El producto crudo se purificó por prep-TLC para dar el compuesto del título (230 mg, rendimiento: 35,4 %).
2-(5-Metoxiisoindolin-2-il)-N-(piridin-4-il)pirimidin-4-amina

A una solución de 2-(5-metoxiisoindolin-2-il)pirimidin-4-amina (270 mg, 1,12 mmol) y 4-bromopiridina (193 mg, 1,23 mmol) en dioxano (20 mL) se añadió CS2CO3 (1,09 g, 3,35 mmol) y se agitó bajo N2. A continuación xphos (32,2 mg, 0,0558 mmol) y Pd2(dba)3 (51,1 mg, 0,0558 mmol) se añadieron y la reacción se agitó a 85°C bajo N2 por 16 h. Después LCMS mostró que la reacción estaba completa. El producto crudo se purificó por prep-HPLC para dar el compuesto del título (96,0 mg, rendimiento: 26,9 %).1H NMR (400 MHz, DMSO-d6) 59,69 (s, 1h ), 8,38 (dd, J = 4,8 y 1,6, 2H), 8,07 (d, J = 5,2, 1H), 7,80 (dd, J = 4,8 y 1,2, 2H), 7,33-7,89 (m, 1H), 7,04-6,98 (m, 1H), 6,86 (d, J = 8,4, 1H), 6,16 (d, J = 5,2, 1H), 4,87-4,71 (m, 4H), 3,76 (s, 3H). MS (ES+) m/e 320 (M+H)+.
Ejemplo 20
2-(5-Metoxiisoindolin-2-il)-N-(4-(piridin-4-il)fenil)pirimidin-4-amina
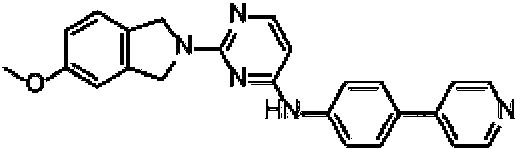
Una mezcla de compuesto N-(4-bromofenil)-2-(5-metoxiisoindolin-2-il)pirimidin-4-amina (297 mg, 0,748 mmol), ácido piridin-4-ilborónico (307 mg, 1,496 mmol), K2CO3 (206 mg, 1,496 mmol), PdCh (dppf) (30,5 mg, 0,0374 mmol) y dioxano (20 mL) y H2O (20 mL) se agitó a 110°C bajo N2 por 12 h. Después de que LCMS mostró que la reacción se había completado, la mezcla se enfrió a 28°C, se filtró y la torta del filtro se lavó con MeOH (20 mL), se secó para dar el compuesto del título (200 mg, rendimiento: 67,6%) como un sólido marrón. 1H NMR (400 MHz, DMSO-d6) 59,54 (s, 1H), 8,56 (s, 2H), 8,00-7,69 (m, 7H), 7,29-6,87 (m, 3H), 6,14 (s, 1H), 4,78-4,75 (m, 4H), 3,75 (s, 3H). MS (ES+) m/e 396 (M+H)+.
Ejemplo 21
N-(1H-Indazol-5-il)-2-(5-metoxiisoindolin-2-il)-7,8-dihidro-5H-pirano[4,3-d]pirimidin-4-amina

Etil 4-oxotetrahidro-2H-piran-3-carboxilato
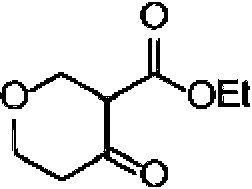
A una solución de dihidro-2H-piran-4 (3H)-ona (5,00 g, 50 mmol) en tolueno (300 mL) se le añadió LDA en hexano (27,5 mL, 55 mmol) rápidamente a 0°C. Después de agitar durante 10 minutos, se añadió carboncianidato de etilo (4,45 g, 45 mmol) a la solución en una porción y la mezcla se agitó durante 10 minutos más. La mezcla se inactivó con AcOH (30 mL) y agua (100 mL), y se extrajo con EtOAc (150 mL x 3). Las capas orgánicas combinadas se lavaron con NaHCO3 y salmuera, secada sobre Na2SO4 y se concentró para dar el compuesto del título (1,50 g, rendimiento: 19.4%), que se utilizó para la siguiente etapa directamente.
2-(Metiltio)-7,8-dihidro-3H-pirano[4,3-d]pirimidin-4(5H)-ona

A una solución de metóxido de sodio (62,8 mg, 1,16 mmol) en MeOH (5 mL) se le añadieron Etil 4-oxotetrahidro-2H-piran-3-carboxilato (100 mg, 0,581 mmol) y metil carbamimidotioato (78,5. Mg, 0,872 mmol ) La mezcla se calentó a reflujo durante 12 h. LCMS mostró que la reacción estaba completa. La solución se concentró para dar el producto crudo, que se purificó mediante prep-TLC para dar el compuesto del título (52 mg, rendimiento: 45,2%). 1H NMR (400 MHz, MeOH) 54,44 (2H, s), 3,92 (2H, t, J=5,6 Hz), 2,64 (2 H, t, J=5,6 Hz), 2,54 (3H, s).
4-Cloro-2-(metiltio)-7,8-dihidro-5H-pirano[4,3-d]pirimidina

A una solución de 2-(metiltio)-7,8-dmidro-3H-pirano[4,3-d]pmmidin-4(5H)-ona (1,00 g, 5,05 mmol) en MeCN (9,6 mL)/DMF ( 0,3 mL) se añadió POCl3 (6,4 mL, 50,5 mmol) a 0°C. La mezcla se calentó a 80°C y se agitó durante 16 horas bajo N2. Después de que LCMS mostró que la reacción se completó, la mayoría de los POCl3 se eliminó al vacío y el residuo se vertió en agua con hielo. La mezcla resultante se neutralizó a pH=7-8 con Na2CO3 ac. y se extrajo con EtOAc (100 mL x 3). Las capas orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4 y se concentraron para dar el compuesto del título (900 mg, rendimiento: 82,6%), que se utilizó para la siguiente etapa directamente.
1H NMR (CDCla, 400 MHz) 54,69 (2 H, s), 4,03 (2 H, t, J=5,6 Hz), 2,92 (2 H, t, J=5,6 Hz), 2,58 (3 H, s).
N-(1H-Indazol-5-il)-2-(metiltio)-7,8-dihidro-5H-pirano[4,3-d]pirimidin-4-amina

A una solución de 4-doro-2-metiltio)-7,8-dihidro-5H-pirano[4,3-d]pirimidina (100 mg, 0,463 mmol) en dioxano (5 mL) se le agregaron KI (76,4 mg, 0,463 mmol), HCl ac. (0,2 mL) y 1 H-indazol-5-amina (73,9 mg, 0,556 mmol) uno por uno. La mezcla resultante se calentó a 100°C durante 12 horas. Después LCMS mostró que la reacción estaba completa. La mezcla se concentró para dar el producto crudo, que se purificó mediante prep-HPLC para dar el compuesto del título (45 mg, rendimiento: 31,1%). 1H NMR (400 MHz, DMSO-cfe) 59,23 (1 H, s), 8,06 (1H, s), 7,88 (1 H, s), 7,45-7,53(2 H, m) 4,60 (2 H, s), 3,91 (2H, t, J=5,2 Hz), 2,70 (2 H, t, J=5,2Hz), 2,35 (3H, s).
N-(1H-Indazol-5-il)-2-(metilsulfonil)-7,8-dihidro-5H-pirano[4,3-d]pirimidin-4-amina

A una solución de N-(1H-indazol-5-il)-2-(metiltio)-7,8-dihidro-5H-pirano[4,3-d] pirimidin-4-amina (1,4 g, 4,47 mmol) en CH2O 2 (10 mL)/dioxano (6 mL) se añadió m-CPBA (1,54 g, 8,94 mmol) en porciones a 15°C. La mezcla se agitó a 15°C durante 2 ~3 horas. LCMS mostró que la reacción estaba completa. La mezcla de reacción se concentró bajo N2 para dar 1,5 g del compuesto del título, que se usó para la siguiente etapa directamente.
N-(1H-Indazol-5-il)-2-(5-metoxiisoindolin-2-il)-7,8-dihidro-5H-pirano[4,3-d]pirimidin-4-amina

A una solución de N-(1H-indazol-5-il)-2-(metilsulfonil)-7,8-dihidro-5H-pirano[4,3-d] pirimidin-4-amina (1,5 g, 4,34 mmol) en dioxano (20 mL) se añadieron TEA (2,19 g, 21,7 mmol) y 5-metoxiisoindolina (1,30 g, 8,69 mmol). El tubo sellado se calentó a 150°C durante 2 horas en microondas. LC-MS mostró que el material de partida fue consumido. La mezcla se concentró al vacío. El residuo se purificó por cromatografía en columna sobre gel de sílice y prep-HPLC para dar el compuesto del título (130 mg, rendimiento: 7,23%). 1H NMR (400 MHz, CD3OD) 58,10 (1 H, s), 8,02(1 H, s), 7,68 (1 H, d, J=9,2 Hz), 7,49 (1 H, d, J=8,8 Hz), 7,19 (1 H, d, J=8,4 Hz), 6,88 (1H, s), 6,82 (1 H, d, J=8,4 Hz), 4,74 (2H, s), 4,70 (2 H, s), 4,63 (2H, s), 3,99 (2H, t, J=5,2 Hz), 3,78 (3H, s), 2,72 (2H, t, J=5,2 Hz). MS (ES+) m/e 415,2 (M+H)+. Ejemplo 22 (KL-00254)
N-(1H-Indazol-5-il)-2-(5-metoxiisoindolin-2-il)-6-metil-6,7-dihidro-5H-pirrolo[3,4-d]pirimidin-4-amina

tert-Butil 4-((1H-indazol-5-il)amino)-2-cloro-5H-pirrolo[3,4-d]pirimidina-6(7H)-carboxilato

Una mezcla de tert-butil 2,4-dicloro-5H-pirrolo[3,4-d]pirimidina-6(7H)-carboxilato (1,0 g, 3,46 mmol), 1H-indazol-5-amina (0,55 g, 4,15 mmol) y Na2CO3 (1,1 g, 10,38 mmol) en DMF (20 mL) se calentaron a 100°C durante 2,5 h. La mezcla de reacción se enfrió a temperatura ambiente, se diluyó con agua y se extrajo con EtOAc (3 x 20 mL). Las capas orgánicas combinadas se concentraron al vacío. El residuo se purificó por cromatografía en columna sobre gel de sílice para dar el compuesto del título (0,9 g, rendimiento: 67,7%). 1H NMR (400 MHZ, MeOD) 5 13,05 (1 H, S), 9,57(1 H, S), 8,06 (1 H, S), 7,95 (1 H, M), 7,53-7,47 (2 H, M), 4,41-4,39 (4H, S), 1,43 (9 H, S).
tert-Butil 4 -((1H-indazol-5-il)amino)-2-(5-metoxiisoindolin-2-il)-5H-pirrolo[3,4-d] pirimidina-6 (7H)-carboxilato

Una mezcla de tert-butil 4-((1H-indazol-5-il) amino)-2-cloro-5H-pirrolo[3,4-d] pirimidina-6(7H)-carboxilato (1,50 g, 3,89 mmol), 5-metoxiisoindolina (0,87 g, 5,38 mmol) y TEA (1,18 g, 11,7 mmol) en NMP (30 mL) se calentó a 150°C durante 1 h. La mezcla de reacción se enfrió a temperatura ambiente, se filtró y se purificó mediante prep-HPLC para dar el compuesto del título (0,95 g, 48,9%). 1H NMR (400 MHZ, DMSO) 58,20-8,10 (2 H, M), 7,75-7,57 (2 H, M), 7,25-7,23 (1 H, D), 6,91-6,88 (2 H, M), 4,89 (4 H, M), 4,59-4,48 (3 H, M), 3,79 (3 H, S), 1,49 (9 H, S).
N-(1H-Indazol-5-il)-2-(5-metoxiisoindolin-2-il)-6,7-dihidro-5H-pirrolo[3,4-d]pirimidin-4-amina
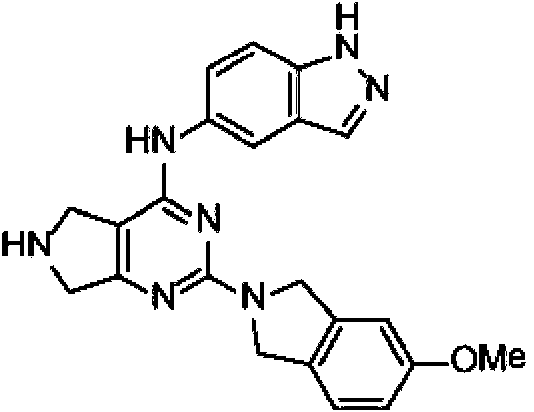
Una mezcla de tert-butil 4-((1H-indazol-5-il)amino)-2-(5-metoxiisoindolin-2-il)-5H-pirrolo[3,4-d]pirimidina-6 (7H)-carboxilato (0,95 g, 1,9 mmol) y TFA (20 mL) en Dc M (20 mL) se agitó a 25°C durante 1 h. lCm S mostró que la reacción estaba completa. La mezcla se concentró al vacío y se purificó mediante prep-HPLC para dar el compuesto
del título (380 mg). 1H NMR (400 MHZ, MeOD) 58,13-8,11 (2 H, M), 7,66-7,60 (2 H, M), 7,27-7,25 (1 H, D), 6,91-6,88 (2 H, M), 4,91-4,86 (4 H, M), 4,57-4,51 (4 H, M), 3,79 (3 H, S). MS (ES+) M/E 400,2 (M+H)+.
Ejemplo 23
N-(1H-Indazol-5-il)-2-(5-metoxiisoindolin-2-il)-6-metil-6,7-dihidro-5H-pirrolo[3,4-d]pirimidin-4-amina

Una mezcla de N-(1H-indazol-5-il)-2-(5-metoxiisoindolin-2-il)-6,7-dihidro-5H-pirrolo[3,4-d]pirimidin-4-amina (1,0 g, 2,5 mmol) y HCHO (4 mL) en MeOH (20 mL) se agitó a 25°C durante 1 h. Se añadió NaBHaCN (100 mg,3mmol) y la mezcla se agitó a 25°C durante 10 h. LCMS mostró que la reacción estaba completa. La mezcla se concentró al vacío para dar el producto crudo, que se usó directamente para la siguiente etapa.
N-(1H-Indazol-5-il)-2-(5-metoxiisoindolin-2-il)-6-metil-6,7-dihidro-5H-pirrolo[3,4-d]pirimidin-4-amina

Una mezcla de ((1H-indazol-5-il)(2-(5-metoxiisoindolin-2-il)-6-metil-6,7-dihidro-5H-pirrolo[3,4-d]pirimidin-4-il) amino)metanol (1,2 g, 2,7 mmol) y HCl (5 mL) en MeOH (20 mL) se agitó a 25°C durante 1,5 h. LCMS mostró que la reacción estaba completa. La mezcla se concentró y se purificó mediante prep-HPLC para dar el compuesto del título (100 mg, 9.0%). 1H NMR (400 MHZ, MeOD) 58,13-8,10 (2 H, M), 7,66-7,58 (2 H, M), 7,27-7,25 (1 H, D), 6,94-6,87 (2 H, M), 4,85 (4 H, M), 4,61-4,56 (4 H, M), 3,80 (3 H, S). MS (ES+) M/E 414,1 (M+H)+.
Ejemplo 24
N-(1H-Indazol-5-il)-2-(5-metoxiisoindolin-2-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-amina

tert-Butil 4-((1H-indazol-5-il) amino)-2-doro-5,6-dihidropirido[3,4-d]pirimidina-7(8H)-carboxilato

Una mezcla de tert-butil 2,4-didoro-5,6-dihidropirido[3,4-d] pirimidina-7(8H)-carboxilato (1,50 g, 4,95 mmol), 1H-indazol-5-amina (0,66 g, 4,95 mmol) y Na2CO3 (1,05 g, 9,9 mmol) en DMF (30,0 mL) se calentó a 100°C durante 3 h. LCMS mostró que la reacción estaba completa. El sólido insoluble se eliminó por filtración y los filtrados se purificaron por HPLC para dar el compuesto del título (400 mg, 20,2%). 1H NMR (400 MHz, MeOD) 58,02 (1 H, S), 7,96 (1 H, S), 7,52 (2 H, S), 4,41 (2 H, M), 3,74 (2 H, M), 2,65-2,62 (2 H, M), 1,49 (9 H, S).
tert-Butil 4-((1h-indazol-5-il)amino)-2-(5-metoxiisoindolin-2-il)-5,6-dihidropirido[3,4-d] pirimidina-7(8h)-carboxilato

Una mezcla de tert-butil 4-((1H-indazol-5-il) amino)-2-cloro-5,6-dihidropirido[3,4-d] pirimidina-7(8H)-carboxilato (360 mg, 0,9 mmol), 5-metoxiisoindolina (201 mg, 1,35 mmol) y TEA (272 mg, 2,7 mmol) en NMP (20 mL) se calentó a 150°C durante 1 h. La mezcla de reacción se enfrió a temperatura ambiente, se filtró y se purificó mediante prep-HPLC para dar el compuesto del título (110 mg, 29,6%). 1H NMR (400 MHz, MeOD) 58,08-8,02 (2 H, M), 7,64-7,56 (2 H, M), 7,17 (2 H, B), 6,84-6,83 (2 H, M), 4,70-4,53 (6 H, M), 3,84-3,82 (5 H, M), 2,68-2,57 (2 H, M), 1,53 (9 H, S).
n-(1H-Indazol-5-il)-2-(5-metoxiisoindolin-2-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-amina
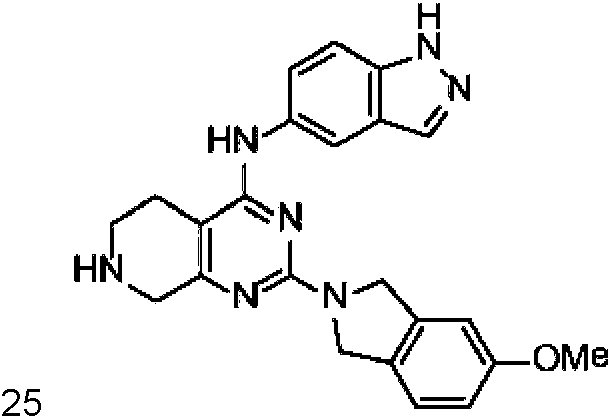
Una mezcla de terc-butil 4-((1H-indazol-5-il)amino)-2-(5-metoxiisoindolin-2-il)-5,6-dihidropirido[3,4-d]pirimidina-7 (8H)-carboxilato (110 mg, 0,21 mmol) y TFA (3 mL) en dCm (3 mL) se agitó a 25°C durante 1 h. Después de que LCMS mostró que la reacción se había completado, la mezcla se concentró para dar el compuesto del título (75 mg, 88%).
1H NMR (400 MHz, MeOD) 58,12 (1 H, s), 8,00 (1 H, s), 7,63 (2 H, m), 7,22 (2 H, brs), 6,90-6,87 (2 H, m), 4,80-4,79
(4 H, m), 4,40 (2 H, s), 3,76 (2 H, s), 3,66-3,63 (2 H, m), 2,95-2,92 (2 H, m). MS (ES+) m/e 414,1 (M+H)+. Ejemplo 25
N-(1H-Indazol-5-il)-2-(5-metoxiisoindolin-2-il)-7-metil-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-amina
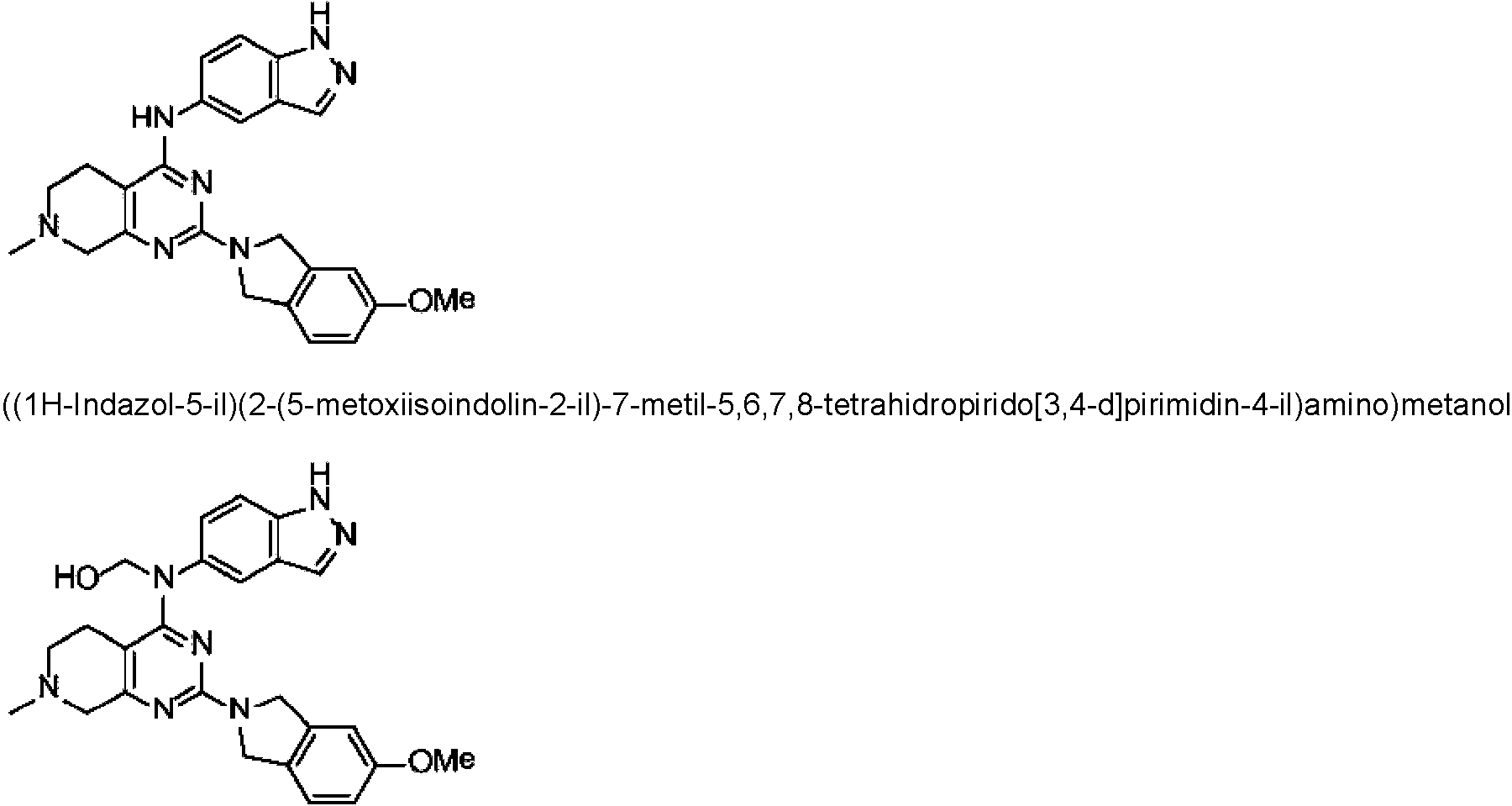
Una mezcla de N-(1H-indazol-5-il)-2-(5-metoxiisoindolin-2-il)-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-amina (300 mg, 0,73 mmol) y HCHO (1 mL) en MeOH (10 mL) se agitó a 25°C durante 1 h. A continuación, se añadió NaBHaCN (30 mg, 1 mmol) y la mezcla se agitó a 25°C durante 10 h. LCMS mostró que la reacción estaba completa. La mezcla se concentró y se usó en la siguiente etapa.
n-(1H-Indazol-5-il)-2-(5-metoxiisoindolin-2-il)-7-metil-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-amina
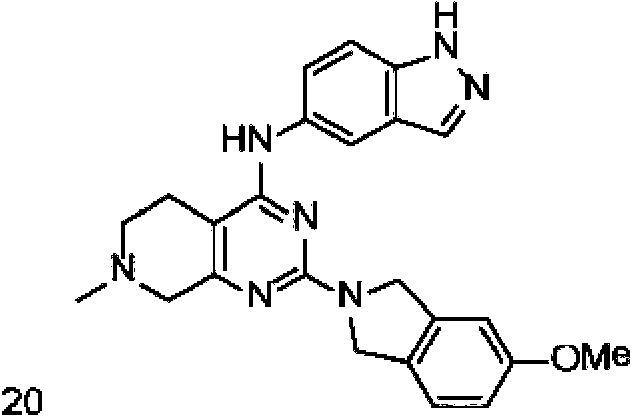
Una mezcla de ((1H-indazol-5-il)(2-(5-metoxiisoindolin-2-il)-7-metil-5,6,7,8-tetrahidropirido[3,4-d]pirimidin-4-il)amino)metanol (400 mg, 0,87 mmol) y HCl (2 mL) en MeOH (10 mL) se agitó a 25°C durante 1,5 h. Después de que LCMS mostró que la reacción se había completado, la mezcla se concentró y se purificó por HPLC para dar el compuesto del título (86 mg, 23,2%). 1H NMR (400 MHZ, MeOD) 58,12 (1 H, S), 8,01 (1 H, S), 7,63 (2 H, M), 7,23 (2
H, B), 6,90-6,88 (2 H, M), 4,80 (4 H, M), 4,46 (2 H, S), 3,77-3,70 (5 H, M), 3,15 (3 H, S), 3,03-3,00 (2 H, M). MS (ES+)
M/E 428,2 (M+H)+.
Ejemplo 26
2-(4-((1H-Indazol-5-il)amino)pirimidin-2-il)isoindolina-5-carbonitrilo

A una solución de 5-bromoisoindolina-1,3-diona (25,0 g, 0,11 mmol) en THF (1000 mL) se agregaron BH3Me2S (100 mL). La mezcla resultante se calentó a 80 °C durante 17 h. Después de que LCMS mostró que la reacción se había completado, la mezcla de reacción se enfrió con MeOH y se concentró. El residuo se purificó por cromatografía en columna sobre gel de sílice (eluido con PE: EA = 20:1) para dar el compuesto del título (3,98 g, 18,2%). 1H NMR (400 MHz, MeOD) 57,62 (1 H, s) 7,55 (1 H, d, J = 8,0 Hz) 7,35 (1 H, d, J = 8,0 Hz) 4,62 (2 H, s) 4,58 (2 H, s).
terc-Butil 5-bromoisoindolina-2-carboxilato

A una solución de 5-bromoisoindolina (3,98 g, 20,2 mmol) en DMF (20 mL) se añadió DMAP (40,0 mg) y BOC2O (8,81 g, 40,4 mmol). La mezcla se agitó a 25°C durante 10 h. Después LCMS mostró que la reacción estaba completa. La mezcla de reacción se concentró para dar el producto crudo, que se lavó con Pe para dar el compuesto del título (5,00 g, 83,4%). 1H NMR (400 MHz, CDCla) 57,39-7,43 (2 H, m) 7,09-7,18 (1 H, m) 4,61-4,68 (4 H, m) 1,53 (9 H, s). tert-Butil 5-cianoisoindolina-2-carboxilato

A una solución de terc-butil 5-bromoisoindolin-2-carboxilato (5,00 g, 16,8 mmol) en DMF (150 mL) se le añadió ZN(CN)2 (3,94 g, 33,5 mmol) y Pd(PPh3)4 (3,62 g). La mezcla se agitó a 80°C durante 3 h. Después de que LC-MS mostró que la reacción se había completado, la mezcla se concentró y el residuo se purificó por cromatografía en columna sobre gel de sílice (eluido con Pe : EA = 30:1) para dar el compuesto del título (4,00 g, 97,6%). 1H NMR (400 MHz, CDCl 3) 5 7,53-7,60 (2 H, m) 7,33-7,42 (1 H, m) 4,71-4,76 (4 H, m) 1,53 (9 H, s).
Isoindolina-5-carbonitrilo

La solución de terc-butil 5-cianoisoindolin-2-carboxilato (4,00 g, 16,4 mmol) en TFA/DCM (2M, 100 mL) se agitó a 15°C durante 120 min. Después de que LC-MS mostró que la reacción se había completado, la solución de reacción se concentró para dar el compuesto del título (2,36 g, 99,9%), que se usó para la siguiente etapa directamente.
2-(4-((1H-Indazol-5-il)amino)pirimidin-2-il)isoindolina-5-carbonitrilo

A una solución de compuesto isoindolina-5-carbonitrilo (200 mg, 1,39 mmol) en DMF (5 mL) se le añadió TEA (200 mg) y 4-metil-N,N-di(prop-2-in-1-M) bencenosulfonamida (170 mg, 0,694 mmol). Se agitó la mezcla a 80°C durante 5 h. Después de que LC-MS mostró que la reacción se había completado, la solución se concentró para dar el producto crudo, que se purificó mediante prep-hplc para dar el compuesto del título (130 mg, 53,0%). 1H NMR (400 MHz, DMSO) 5 10,81 (1 H, b), 8,29 (1 H, b) 8,17 (1 H, s), 8,01 (1 H, d, J=8,0 Hz), 7,94 (1 H, d, J=6,8 Hz) 7,85 (1 H, d, J=6,8 Hz) 7,65-7,67 (1H, m), 7,60 (3 H, Br. s.), 6,42 (1 H, Br. s.), 4,92-5,03 (4 H, m). MS (ES+) M/E 354,1 (M+H)+.
Ejemplo 27
N-(2-(5-(Metoximetil)isoindolin-2-il)pirimidin-4-il)-1H-indazol-5-amina

4-Metil-n,n-di(prop-2-in-1-il)bencenosulfonamida

A una solución de 4-metilbencenosulfonamida (10 g, 48,1 mmol), K 2CO3 (12,7 g, 120 mmol), en CH3CN (200 mL) se añadió bromuro de propargilo (10,4 g, 144 mmol). La mezcla se calentó a 60°C y se agitó durante 5 h. Después de que LCMS mostró que la reacción se había completado, la mezcla se diluyó con EtOAc y agua. Se extrajo la fase acuosa con EtOAc. La capa orgánica se lavó con salmuera y se secó sobre Na2SO4, se filtró y se concentró a presión reducida para obtener un residuo, que se recristalizó en EtOH para dar el compuesto del título (12 g, 83,1%) como un sólido amarillo.
5-(Metoximetil)-2-tosilisoindolina

A una solución de 4-metil-N,N-di(prop-2-in-1-il)bencenosulfonamida (5 g, 0,02 mol), 3-metoxiprop-1-ino (3,5 g, 0,05 mmol) en EtOH (100 mL) se añadió Rh(PPh3)4Cl (2,37 g, 2 mmol) a 0°C. A continuación la mezcla se agitó a 20°C durante 18 h. Después de que LCMS mostró que la reacción se había completado, a continuación, la reacción se concentró a presión reducida para obtener un residuo, que se purificó por cromatografía en columna sobre gel de sílice para proporcionar el compuesto del título (2 g, 31,2%). 1H NMR: (400 MHz, CD3OD) 52,34 - 2,44 (3 H, m), 3,31 (3 H, s), 4,39 (2 H, s), 4,56 (4 H, s), 7,10 - 7,24 (3 H, m), 7,38 (2 H, d, J=7,94 Hz), 7,76 (2H, d, J=7,94 Hz).
5-(Metoximetil)isoindolina

A una solución de 5-(metoximetil)-2-tosilisoindolina (2,5 g, 7,89 mmol), Mg (480 mg, 20 mmol) en MeOH (50 mL) se agregó Et3N (2,0 g, 20 mmol) y se agitó a 75°C por 24 h. La conversión es ~50%, a continuación, la reacción se
concentró a presión reducida para obtener un residuo, que se usó directamente sin purificación adicional.
N-(2-(5-(Metoximetil)isoindolin-2-il)pirimidin-4-il)-1H-indazol-5-amina

A una solución de 5-(metoximetil)isoindolina (1,5 g crudo), N-(2-cloropmmidin-4-il)-1H-indazol-5-amma (2,5 g, 0,01 mol) en MeCN (50 mL) se añadió DIEA (5,2 g, 0,04 mmol) y se agitó a 120°C en microondas durante 6 h. A continuación, la reacción se concentró a presión reducida para obtener un residuo, que se purificó por prep-HPLC para dar el compuesto final (94 mg) como un sólido blanco. 1H NMR (400 MHz, DMSO-cfe) 54,43 (2 H, s), 4,80 (4 H, d, J=18,52 Hz), 6,07 (1 H, d, J=5,73 Hz), 7,25 (1 H, d, J=7,50 Hz), 7,42 (2 H, Br. s.), 7,45 - 7,59 (2 H, m), 7,94 (1 H, d, J=5,73 Hz), 8,06 (1 H, Br. s.), 8,29 (1 H, Br. s.), 9,22 (1 H, Br. s.), 12,91 (1 H, Br. s.), MS (ES+) m/e 373 (M+H)+. Ejemplo 28
N-(2-(4-Metoxiisoindolin-2-il)pirimidin-4-il)-1H-indazol-5-amina
t etil-1 3,2-dioxaborolan-2-il)isoindolina-2-carboxilato
etil-1 3,2-dioxaborolan-2-il)isoindolina-2-carboxilato
Una mezcla de tert-butil 4-bromoisoindolina-2-carboxilato (1,00 g, 3,37 mmol), 4,4,4',4'5,5,5',5'-octametil-2,2'-bi(1,3,2-dioxaborolano) (1,03 g, 4,04 mmol), AcOK (661 mg, 6,74 mmol) y Pd(dppf)Cl2 (50,0 mg) en dioxano (20 mL) se agitó a 90°C durante 16 h en atmósfera de nitrógeno. Después de enfriar a 20°C y LCMS mostró que la reacción se había completado, la mezcla se filtró y el filtrado se concentró a presión reducida para obtener el compuesto del título (1,2 g, rendimiento 103%) como un sólido marrón, que fue directamente a la siguiente etapa sin más purificación. 1H NMR (CDCla, 400 MHz) 57,69 (d, J = 7,2 Hz, 1 H) 7,36 - 7,27 (m, 2 H) 4,87 - 4,63 (m, 4 H) 1,52 (s, 9 H) 1,31 (s, 12 H). tert-Butil 4-hidroxiisoindolina-2-carboxilato

A una mezcla de tert-butil 4-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)isoindolina-2-carboxilato (1,2 g, 3,48 mmol) en THF (20 mL) se agregó NH4Cl (188 mg, 3,48 mmol) en H2O (10 mL) y H2O2 (20 mL) gota a gota, que se agitó a 19°C durante 16 h. Después de que LCMS mostró que la reacción se había completado, la reacción se detuvo mediante solución ac.Na2SO3 y se extrajo con EtOAc (3 x 30 mL). La capa orgánica se secó sobre Na2SO4, se filtró y se concentró a presión reducida para proporcionar el compuesto del título (800 mg, 97,9%), que se usó directamente en
la siguiente etapa sin purificación adicional. 1H NMR (CDCI3, 400 MHz) 57,19 - 7,11 (m, 1 H) 6,83 - 6,68 (m, 2 H) 4,78 - 4,63 (m, 4 H) 1,24 (s, 9 H).
tert-Butil 4-metoxiisoindolina-2-carboxilato

A una mezcla de tert-butil 4-hidroxiisoindolina-2-carboxilato (750 mg, 3,19 mmol) y K2CO3 (880 mg, 6,38 mmol) en DMF (10 mL) se añadió Mel (680 mg, 4,79 mmol) gota a gota, que se agitó a 100°C durante 6 horas. Después de que LCMS mostró que la reacción se había completado, la mezcla se concentró, a continuación, se disolvió en EtOAc (20 mL) y se filtró, el filtrado se concentró para dar el compuesto del título crudo (700 mg, 88,0%) como un sólido marrón, que fue usado directamente a la siguiente etapa sin más purificación. 1H NMR (CDCb , 400 MHz) 57,18 - 7,14 (m, 1 H) 6,81 - 6,64 (m, 2 H) 4,62 - 4,52 (m, 4 H) 3,78 (s, 3 H) 1,44 (s, 9 H).
4-Metoxiisoindolina

Una mezcla de tert-butil 4-metoxiisoindolina-2-carboxilato (700 mg, 2,81 mmol) en HCl/EtOAc (20 mL, 4 M) se agitó a 18°C durante 16 h. Después de que la TLC (EtOAc) mostró que la reacción se había completado, la mezcla se concentró, a continuación, se disolvió en EtOAc (20 mL) y se filtró, la torta del filtro se lavó con EtOAc (3 x 10 mL) y se concentró para dar el compuesto del título (500 mg, 96,1%) como un sólido marrón. 1H NMR (DMSO-cfe, 400 MHz) 5 9,95 (br.s, 2 H) 7,35 - 7,31 (m, 1 H) 6,95 (d, J = 7,6 Hz, 2 H) 4,46 - 4,45 (m, 2 H) 4,37 (t, J = 4,8 Hz, 2 H) 3,81 (s, 3 H).
N-(2-(4-Metoxiisoindolin-2-il)pirimidin-4-il)-1H-indazol-5-amina

Una mezcla de 4-metoxiisoindolina (200 mg, 1,08 mmol), N-(2-cloropirimidin-4-il)-1H-indazol-5-amina (291 mg, 1,19 mmol) y DIPEA (559 mg, 4,32 mmol) en n-BuOH (10 mL) se agitó a 150°C bajo microondas durante 2 h. Después de que LCMS mostró que la reacción se había completado, la mezcla se concentró y se purificó en gel de sílice (PEEtOAc = 10:1 —5:1 ~1:1 ~0:1) para dar el producto crudo, que fue purificado por prep-HPLC básico para dar el compuesto del título (40 mg, 10,3%) como un sólido amarillo. 1H NMR (DMSO-d6, 400 MHz) 5 12,93 (Br.s, 1 H) 9,21 (s, 1 H) 8,25 (Br.s, 1 H) 7,93 (d, J = 5,6 Hz, 2 H) 7,49-7,47 (m, 2 H) 7,31-7,27 (m, 1 H) 7,03 - 6,97 (m, 1 H) 6,90 (d, J = 8,4 Hz, 1 H) 6,06 (d, J = 5,6 Hz, 1 H) 4,84 - 4,68 (m, 4 H) 3,84 (Br.s, 3 H). MS (ES+) m/e 359,2 (M+H)+.
Ejemplo 29
N-(2-(4-Fluoroisoindolin-2-il)pirimidin-4-il)-1H-indazol-5-amina

1,2-bis(Bromometil)-3-fluorobenceno

Una mezcla de 1-fluoro-2,3-dimetilbenceno (1,00 g, 3,37 mmol), NBS (2,87 g, 16,1 mmol) y BPO (19,5 mg) en CCI4 (50 mL) se agitó a 80°C durante 1 h. Después de enfriar a temperatura ambiente, y la TLC (PE) haber mostrado que la reacción se había completado, la mezcla se filtró, el filtrado se concentró a presión reducida para obtener el producto crudo, que se purificó por cromatografía en columna sobre gel de sílice (PE) para dar el compuesto del título (1,7 g, 74,9%) como un aceite incoloro. 1H NMR (CDCla, 400 MHz) 57,32 - 7,26 (m, 1 H) 7,17 (d, J = 7,6 Hz, 1 H) 7,08 - 7,01 (m, 1 H) 4,70 (s, 2 H) 4,63 (s, 2 H).
2-Bencil-4-fluoroisoindolina

Una mezcla de 1,2-bis(bromometil)-3-fluorobenceno (1,9 g, 6,74 mmol), BnNH2 (1,08 g, 10,1 mmol) y K2CO3 (1,86 g, 13,9 mmol) en tolueno (30 mL) se agitó a 100°C durante 12 h. Después de que TLC (PEEtOAc = 10:1) mostró que la reacción se había completado, la mezcla se filtró, el filtrado se concentró y se purificó por cromatografía en columna sobre gel de sílice (pE:EtOAc = 1:0— 100:1 — 50:1—30:1—20:1) y prep-TLC (PEEtOAc = 10:1, Rf = 0,6) para dar el compuesto del título (580 mg, 37,9%) como un aceite marrón. 1H NMR (CDCl3, 400 MHz) 57,42 - 7,33 (m, 4 H) 7,30 - 7,27 (m, 1 H) 7,20 - 7,11 (m, 1 H) 6,95 (d, J = 7,6 Hz, 1 H) 6,86 (t, J = 8,8 Hz, 1 H) 3,99 (s, 2 H) 3,95 (s, 2 H) 3,91 (s, 2 H).
4-Fluoroisoindolina

A una mezcla de 2-bencil-4-fluoroisoindolina (580 mg, 2,56 mmol) y Pd-C (200 mg) en MeOH (10 mL) se añadió HCl (1 mL), a continuación, la mezcla de reacción se agitó a 50°C. bajo 50 psi de H2 por 16 horas. Después de que LCMS mostró que la reacción se había completado, la mezcla se filtró, el filtrado se concentró para dar el compuesto del título (350 mg, 79,7%) como un sólido marrón claro, que se usó directamente en la siguiente etapa sin purificación adicional. 1H NMR (CDCla, 400 MHz) 57,43 - 7,37 (m, 1 H) 7,22 (d, J = 7,6 Hz, 1 H) 7,09 (t, J = 8,8 Hz, 1 H) 4,57 (br.s, 4 H).
N-(2-(4-Fluoroisoindolin-2-il)pirimidin-4-il)-1H-indazol-5-amina

Una mezcla de 4-fluoroisoindolina (350 mg, 2,02 mmol), N-(2-doropirimidin-4-il)-1H-indazol-5-amina (495 mg, 2,02 mmol) y DIPEA (521 mg, 4,04 mmol) en n-BuOH (5 mL) se agitó a 150°C durante 2 horas en microondas. Después de que LCMS mostró que la reacción se había completado, la mezcla se concentró, a continuación, se le añadió MeOH (20 mL), el precipitado se recogió por filtración, se lavó con MeOH (3 x 10 mL) y se secó al vacío para dar el compuesto del título (450 mg, 64,3%) como un sólido marrón claro. 1H NMR (DMSO-cfe, 400 MHz) 5 12,93 (s, 1 H) 9,25 (s, 1 H) 8,22 (br.s, 1 H) 7,96 - 7,94 (m, 2 H) 7,54 - 7,47 (m, 2 H) 7,38 - 7,29 (m, 2 H) 7,15 - 7,10 (m, 1 H) 6,09 (d, J = 5,6 Hz, 1 H) 4,85 (br.s, 4 H). MS (ES+) m/e 347,2 (M+H)+.
Ejemplo 30
2-(5-Fluoroisoindolin-2-il)-N-(1H-indazol-5-il)-5,7-dihidrofuro[3,4-d]pirimidin-4-amina

Una mezcla de 2-cloro-N-(1H-indazol-5-il)-5,7-dihidrofuro[3,4-d] pirimidin-4-amina (0,5 g, 1,3 mmol), TEA (0.4 g, 3.9 mmol) y clorhidrato de 5-fluoroisoindolina (0,29 g, 1,3 mmol) en NMP (10 mL) se calentó a 150°C durante 1 h. La mezcla de reacción se enfrió a temperatura ambiente, se filtró y se purificó mediante HPLC para dar el compuesto del título (54 mg, 8,05%). 1H NMR (400 MHz, METANOL-cfe) 58,18 (1 H, s) 8,04 (1 H, s), 7,65 (1 H, d, J=8,10 Hz), 7,52 (1 H, d, J=8,40 Hz), 7,34 (1 H, m), 7,10 (1 H, d, J=7,60 Hz), 7,02 (1 H, m), 4,79-4,91 (8 H, m). MS (ES+) m/e 389 (M+H)+. Ejemplo 31
2-(5-Cloroisoindolin-2-il)-N-(1H-indazol-5-il)-5,7-dihidrofuro[3,4-d]pirimidin-4-amina

Una mezcla de 2-cloro-N-(1H-indazol-5-il)-5,7-dihidrofuro[3,4-d] pirimidin-4-amina (1,0 g, 2,6 mmol), TEA (0,8 g, 7,8 mmol) y clorhidrato de 5-cloroisoindolina (0,58 g, 2,6 mmol) en NMP (20 mL) se calentó a 150°C durante 1 h. La mezcla de reacción se enfrió a temperatura ambiente, se filtró y se purificó mediante HPLC para dar el compuesto del título (110 mg, 9,12%). 1H NMR (400 MHz, DMSO-cfe) 512,95 (1 H, s), 8,83 (1 H, s), 8,26 (1 H, s), 8,08 (1 H, s), 7,62 (1 H, d, J=8,80 Hz), 7,49 (1 H, d, J=8,80 Hz), 7,34 (1 H, d, J=7,60 Hz), 4,86-4,71 (8 H, m). MS (ES+) m/e 405 (M+H)+. Ejemplo 32
N-(2-(Isoindolin-2-il)-5,6-dimetoxipyrimidin-4-il)-1H-indazol-5-amina

A MeOH (30 mL) se añadió Na (2,4 g, 104,3 mmol) a temperatura ambiente Después de que Na desapareció, se añadió la mezcla de 2-metoximalonato de dimetilo (6,8 g, 41,9 mmol) y tiourea (4,8 g, 61,3 mmol). La mezcla resultante se agitó durante 10 h a 80°C. Después de que la TLC mostró que la reacción se había completado, el disolvente fue evaporado. Se añadió agua y se lavó con EtOAc (3x 30 mL). La capa acuosa se acidificó a pH = 1 con HCl. El precipitado se recogió por filtración y se secó al vacío para dar el compuesto del título (5 g, 64,1%). 1HNMR (400 MHz, DMSO) 510,41 (s, 2 H) 3,75 (s, 3 H).
5-Metoxi-2-(metiltio)pirimidina-4,6(1H, 5H)-diona

Se añadió Mel (4,45 g, 31,3 mmol) gota a gota a la mezcla de 5-metoxi-2-(metiltio)pirimidina-4,6(1H, 5H)-diona (5 g, 28,6 mmol) y NaOH (1,36 g, 34 mmol) en agua (40 mL). Después de la adición, la mezcla resultante se agitó durante 16 h. La mezcla se filtró y el filtrado se acidificó a pH = 1 y el precipitado se recogió por filtración, se lavó con agua y se secó al vacío para dar el compuesto del título (2 g, 37%). 1HNMR (400 MHz, MeOD) 53,71 (s, 3 H) 2,52 (s, 3 H) 4,6-Dicloro-5-metoxi-2-(metiltio)pirimidina

Se calentó una mezcla de 5-metoxi-2-(metiltio)pirimidina-4,6(1H,5H)-diona (2 g, 10,6 mmol) y N,N-dietilanilina (1 mL) en oxicloruro de fósforo (15 mL) a 100°C durante 3 h. La reacción se detectó completamente por LCMS. El exceso de reactivo se eliminó al vacío y el residuo se vertió en hielo y se extrajo con MTBE. La fase orgánica se lavó con agua (2 x 10 mL), se secó sobre Na2SO4 y se concentró al vacío para dar el producto crudo. El producto crudo se purificó por cromatografía en columna sobre gel de sílice (PE: EtOAc = 10:1) para dar el compuesto del título (2,3 g, 96%) como un sólido blanco. 1HNMR (400 MHz, MeOD) 53,90 (s, 3 H) 2,53 (s, 3 H).
N-(6-Cloro-5-metoxi-2-(metiltio)pirimidin-4-il)-1H-indazol-5-amina

Se calentó una mezcla de 4,6-didoro-5-metoxi-2-(metiltio)pirimidina (2,0 g, 89 mmol) y 1H-indazol-5-amina (1,54 g, 11,6 mmol) en etanol (15 mL) mediante reflujo durante 16 h, a continuación, el disolvente se eliminó a presión reducida. El producto crudo se purificó por cromatografía en columna sobre gel de sílice (PE: EtOAc = 1:1) para dar el compuesto del título (2,3 g, 82%) como un sólido blanco. 1HNMR (400 MHz, DMSO) 59,44 (s, 1H) 8,02 (d, J = 8,0 Hz, 1 H) 7,58 (d, J = 7,6 Hz, 1 H) 7,50 (d, J = 8,8 Hz, 1 H) 3,80 (s, 1H) 2,38 (s, 1 H).
N-(5,6-Dimetoxi-2-(metiltio)pirimidin-4-il)-1H-indazol-5-amina

A MeOH (30 mL) se añadió Na (0,36 g, 15,5 mmol) a temperatura ambiente. Después de que el Na desapareció, se añadió N-(6-cloro-5-metoxi-2-(metiltio)pirimidin-4-il)-1H-indazol-5-amina (1,0 g, 3,1 mmol). La mezcla resultante se agitó durante 10 h a 80°C. Después de que la TLC mostró que la reacción se había completado, el disolvente se concentró a presión reducida, el residuo se disolvió en CH2Cl2, se lavó con agua (2 x 10 mL), se secó sobre Na2SO4 y se evaporó al vacío para dar el compuesto del título (0,9 g, 92%).
N-(5,6-Dimetoxi-2-(metilsulfonil)pirimidin-4-il)-1H-indazol-5-amina

Una mezcla de N-(5,6-dimetoxi-2-(metiltio)pirimidin-4-il)-1H-indazol-5-amina (0,9 g, 3,1 mmol) y ácido 3-cloroperoxibenzoico (2,03 g, 9,3 mmol) en diclorometano (30 mL) se agitó a 25°C durante 3 h. La TLC mostró que la reacción se había completado, la mezcla se lavó con ac. Na2SO3 (2 x 20 mL), ac.NaHCO3 (2 x 20 mL), H2O (20 mL), se secó sobre Na2SO4 y se evaporó al vacío para dar el compuesto del título (0,9 g, 90%) como un sólido blanco.
N-(2-(Isoindolin-2-il)-5,6-dimetoxipyrimidin-4-il)-1H-indazol-5-amina

Una mezcla de N-(5,6-dimetoxi-2-(metilsulfonil)pirimidin-4-il)-1H-indazol-5-amina (0,9 g, 2,5 mmol) e isoindolina (0,58 g, 3,75 mmol) en 1,4-dioxano (30 mL) se hizo reaccionar en microondas a 140°C durante 5 h. La LCMS mostró que la mayor parte del material de partida se consumió, y a continuación el disolvente se eliminó a presión reducida. El
producto crudo se purificó por prep-HPLC neutro para dar el compuesto del título (175 mg, 18%) como un sólido marrón. 1H NMR (MeOD, 400 MHz) 5 12,88 (s, 1 H) 8,50 (s, 1 H) 8,28 (s, 1 H) 8,02 (s, 1 H) 7,74 (d, J = 8,8 Hz, 1H) 7,46 - 7,41 (m, 3 H) 7,29-7,27 (m, 2H), 4,77 (s, 4 H) 3,91 (s, 3 H) 3,64 (s, 3 H). MS (ES+) m/e 389 (M+H)+.
Ejemplo 33
ROCK2 siRNA, y no ROCK1 siRNA, inhibe la expresión de IL-17 e IL-21.
Para confirmar el papel de ROCK2 en la regulación de la secreción de IL-17 e IL-21 en células T humanas, silenciamos específicamente la expresión de ROCK1 y ROCK2 por interferencia de RNA. El RNA (siRNA) interferente pequeño específico en ROCK1 y ROCK2 redujo los niveles de expresión de proteínas en un 72% y 84% respectivamente. El silenciamiento de ROCK2, pero no de ROCK1, redujo significativamente la IL-17 y la IL-21, con un efecto mínimo sobre la secreción de IFN-y en células T humanas (Fig. 2).
Ejemplo 34
Selectividad compuesta de ROCK1 y ROCK2
Las curvas de respuesta a la dosis para la inhibición de Rho-quinasa se derivaron de un kit de ensayo de quinasa Invitrogen Z'-LYTE™ (número de catálogo Invitrogen PV3793). ROCK1 y ROCK2 activos purificados se obtuvieron de Invitrogen (números de catálogo ROCK1, PV3691 y ROCK2, PV3759). Los componentes del kit incluyen un péptido marcado con cumarina y fluoresceína basado en la cadena ligera 2 de miosina (KKRPQRRYSNVF), un reactivo de desarrollo que contiene proteasa patentado y un amortiguador Stop patentado utilizado para terminar la reacción de desarrollo. Las actividades inhibidoras de los compuestos se miden según el protocolo del fabricante. Brevemente, se agregan concentraciones decrecientes de compuestos de prueba o el inhibidor de ROCK conocido Y-27963, de 10 uM a 2,56 x 10' 5 uM al amortiguador de reacción que contiene 50 mM HEPES pH 7,5, 10 mM MgCh , 5 mM EGTA y Brij-35 al 0,05% y de ROCK1 a 0,18 ug/mL o ROCK2 a 0,8 ug/mL en amortiguador de dilución de ensayo. Esta mezcla se superpone en una placa blanca de media área de 96 pocillos y la reacción se inicia con la adición de ATP 5 uM para ROCK1 o ATP 12 uM para ROCK2. El ensayo continúa a temperatura ambiente durante 1 hora seguido de la adición de reactivo de desarrollo, y una incubación adicional durante 1 hora a temperatura ambiente. A continuación, se agrega el reactivo STOP a la reacción e inmediatamente las señales de emisión de cumarina y fluoresceína se leen en un lector de placa de fluorescencia Tecan Infinite M1000 (excitación: 400 nm; emisión 445 y 520 nm, respectivamente). Al comparar las relaciones de emisión de las muestras de prueba con las muestras de control, se calculan los valores del porcentaje de fosforilación y la concentración de inhibidor que produce 1/2 inhibición de la actividad de la quinasa (IC50) se determina utilizando Prism. La Tabla 1 proporciona concentraciones IC50 para compuestos de los ejemplos anteriores. Varios de los compuestos también demostraron actividad en un ensayo preliminar que midió la inhibición de la fosforilación de la cadena ligera de miosina (pMLC). Para los compuestos marcados con ND, la actividad no fue determinable en las condiciones de prueba empleadas. Los datos que muestran la inhibición de ROCK1 y ROCK2, y la selectividad de ciertos compuestos para la inhibición de ROCK2, se presentan en la Tabla 1.
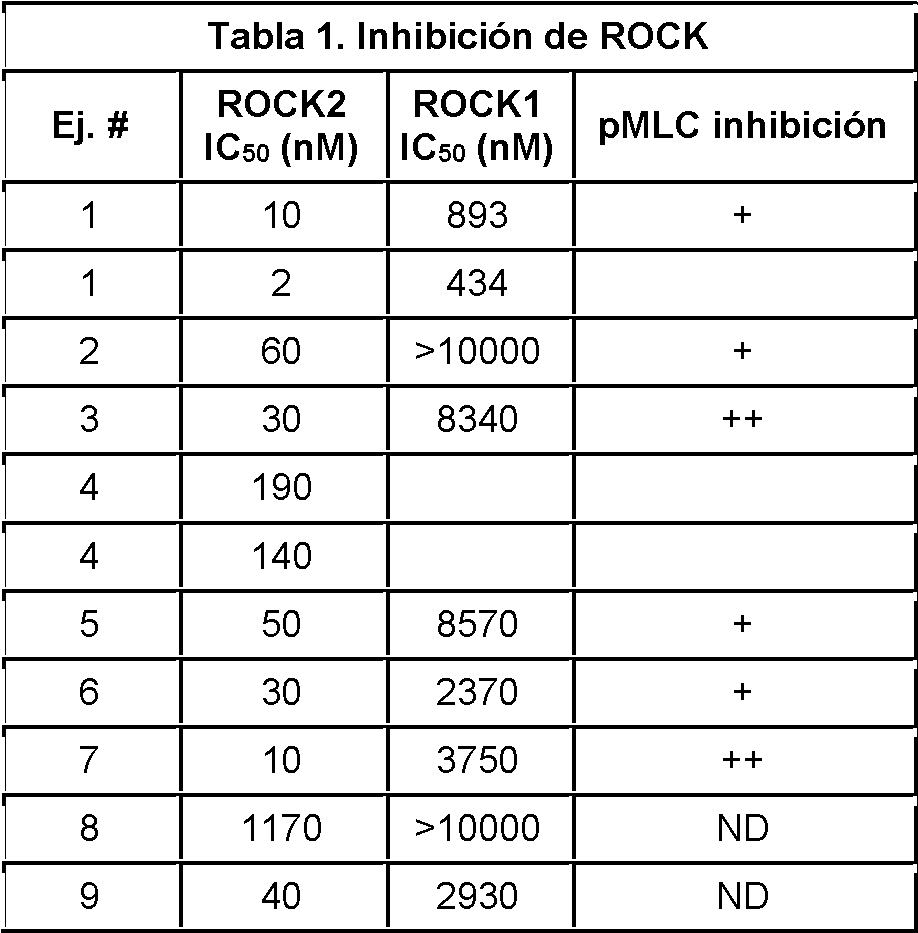

E j e m p l o 35
El i n h i b i d o r s e l e c t i v o R O C K 2 d e l E j e m p l o 1 i n h i b e la s e c r e c i ó n d e I L -17 e IL-21, p e r o n o la s e c r e c i ó n d e I NF -y o IL-2, e n c é l u l a s T C D 4 h u m a n a s in vitro.
La activación de las células T en reposo, que resulta en la secreción y proliferación de citoquinas, involucra dos señales distintas de las células presentadoras de antígeno (APC), imitadas por la coestimulación del complejo receptor de células T (TCR)/CD3 y el receptor CD28. Usando células T humanas CD4+ recién purificadas y anticuerpos estimuladores contra CD3 y CD28 para estimular la secreción de IL-17 e IL-21 en respuesta a la activación de TCR, se encontró que el tratamiento con el inhibidor selectivo ROCK2 del Ejemplo 1 inhibía significativamente la secreción de IL-17 e IL-21 de una manera dependiente de la dosis. En las mismas condiciones, no se observó la inhibición de IFN-y e IL-2 (Fig. 2).
E j e m p l o 36
El i n h i b i d o r d e R O C K 2 d e l E j e m p l o 1 r e d u c e la p a r á l i s i s a s o c i a d a c o n la e n c e f a l o m i e l i t i s a u t o i n m u n e e x p e r i m e n t a l ( EAE) .
Para la inducción de EAE, los ratones se inmunizaron subcutáneamente el día 0 con péptido MOG 35-55 de 150 mg/ratón (Molecular Biology Core Facilities, Dana-Farber Cancer Institute, Harvard University), emulsionado en CFA
(CFA suplementado con 400 mg/mL Mycobacterium tuberculosis)
Los ratones se trataron a continuación con los compuestos indicados (FTY720 a 10 mg/kg y Compuesto 1 a 150 mg/kg una vez [QD] o dos veces al día [BID]) cuando individualmente tenían signos de parálisis durante tres días. Los cambios de dosis del tratamiento con Compuesto 1 están indicados solo para el grupo de 150 BID. FTY720 es un modulador del receptor de 1-fosfato de esfingosina. FTY720 induce una disminución en el número de linfocitos de sangre periférica y ejerce actividad inmunomoduladora en varios modelos experimentales de aloinjertos y enfermedades autoinmunes. La puntuación EAE se basó en Ivanov, y col., 2006. Brevemente: 0-sin enfermedad, 1-cola flácida, 2-patas traseras débiles/parcialmente paralizadas, 3-patas traseras completamente paralizadas, 4 parálisis completa de las patas traseras y parcial de las delanteras, 5-parálisis completa/muerte. Las puntuaciones clínicas medias de los ratones tratados se representan en la Figura 5. Los sujetos tratados con el compuesto 1 demostraron una mejora sustancial en sus puntuaciones clínicas.
Ejemplo 36
Anticuerpos VEGFR2
En esta invención se describen ejemplos no limitantes de secuencias de anticuerpos de unión a VEGFR2. Como se describe en esta invención, a partir de bibliotecas de visualización de fagos Fab humanos, se identificaron dos anticuerpos neutralizantes que se unen al VEGFR2 humano, bloquean la unión del ligando VEGFA al hVEGFR2 e inhiben la fosforilación del VEGFR2 y la transducción de las señales posteriores estimuladas por VEGFA. La Tabla 1 indica las secuencias de aminoácidos de los CDR y los dominios variables de los anticuerpos. Las K¿s de Mab 101 y Mab 102 son aproximadamente 6,6 mM y 1,7 nM, respectivamente.
La cadena pesada de Mab 101 se reorganizó con genes de cadena ligera k (biblioteca k ) y genes de cadena ligera A (biblioteca A). Se encontraron 20 variantes únicas de la cadena ligera A mediante el barrido de la biblioteca A contra VEGFR2 humano y VEGFR2 de ratón. Se encontraron 22 variantes únicas de cadena ligera k mediante el barrido de la biblioteca k contra VEGFR2 humano y VEGFR2 de ratón. La Tabla 2 indica las secuencias de aminoácidos de los CDR y los dominios variables de las cadenas ligeras. Los KD de las Mabs 105, 106 y 107 se incrementaron aproximadamente 10 veces (0,24 nM, 0,22 nM y 0,12 nM, respectivamente).
Mab 138 (Tabla 2), que contiene la cadena pesada de Mab 4, se seleccionó para maduración por afinidad. Se introdujeron mutaciones en CDR3 de la cadena ligera y CDR1, CDR2 y CDR3 de la cadena pesada. La biblioteca resultante se analizó en VEGFR2 humano y murino. La Tabla 3 indica las secuencias de aminoácidos de los CDR de cadena pesada y ligera y los dominios variables de cuatro de los anticuerpos resultantes. La Figura 6 muestra una comparación de las secuencias con la cadena pesada de Mab 4 y la cadena kappa de Mab 138 (es decir, SEQ ID NO:160).

Con el análisis de Biacore se determinaron las constantes de unión de Mab 147 y Mab 149, así como la Mab 160 original para VEGFR2 humano, murino y de rata (Tabla 4).




Mab 147 fue examinada por ELISA por sus propiedades de unión al receptor y bloqueo del ligando. El bloque Mab 147 se une a hVEGFR2 y mVEGFR2 con afinidad similar (Fig. 7A). Mab 147 bloquea la unión del ligando a hVEGFR2 similar a un anticuerpo de control específico de hVEGFR y también bloquea la unión de ligandos a mVEGFR2 similar a un anticuerpo de control específico de mVEGFR2 (Fig. 7b ).
También se confirmó la unión de Mab 147 a hVEGFR2 y mVEGFR2 expresado en las membranas celulares. La Fig. 8A muestra la unión a hVEGR2 expresada por las células entoteliales de la vena umbilical humana (HUVEC), así como las células endoteliales aórticas procinas (PAE) que sobreexpresan KDR (es decir, VEGFR2 humano). Mab 147 también se unió al mVEGFR expresado por células endoteliales murinas MS1 (Fig. 8B).
Mab-147 inhibe la transducción de señal mediada por VEGFR-2 en células KDR-PAE, como lo indica la fosforilación reducida de KDR y p42/44 en células KDR-PAE (Fig. 9A) y en HUVEC (Fig. 9B). Mab-106 y Mab-147 inhiben la proliferación de células KDR-PAE (Fig. 10A), así como también inhiben la migración inducida por VEGF de células KDR-PAE (Fig. 10B).
Claims (13)
1. Un compuesto de fórmula II:
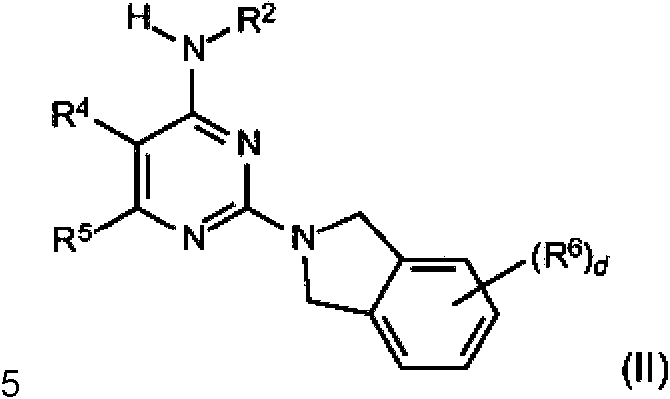
R2 se selecciona del grupo que consiste en arilo, heteroarilo, C3-C7 cicloaiquiio, un anillo heterocíclico de tres a doce miembros que contiene hasta 3 heteroátomos, cada uno de los cuales puede estar opcionalmente sustituido de 1 a 3 sustituyentes seleccionados independientemente de halo y C1-C6 alquilo;
R4 y R5 se seleccionan independientemente del grupo que consiste en H, halo, C1-C8 alquilo, C2-C8 alquenilo, C2-C8 alquinilo, C1-C6 alcoxi, -(C1-C6 alquil)-O-(C1-C6 alquilo), -NR41R42, -(CH2)xNR41R42 y -O-C 1-C6 alquilo)-O-(C1-C6 alquilo);
R41 y R42 se seleccionan independientemente del grupo que consiste en H, C1-C8 alquilo, C2-C8 alquenilo, C2-C8 alquinilo y -(C1-C6 alquil)-O-(C1-C6 alquilo);
o R41 y R42 pueden tomarse juntos para formar un cicloalquilo o anillo heterocíclico de tres a doce miembros que tiene hasta 3 heteroátomos que es opcionalmente sustituido de 1 a 3 sustituyentes seleccionados independientemente de halo, C1-C6 alquilo y C1-C6 alcoxi;
x se selecciona de 0 a 6;
o R4 y R5 pueden tomarse juntos para formar un anillo heterocíclico o aromático de tres a doce miembros que tiene hasta 3 heteroátomos que es opcionalmente sustituido de 1 a 3 sustituyentes seleccionados independientemente de halo y C1-C6 alquilo y C1-C6 alcoxi;
cada R6 se selecciona independientemente del grupo que consiste en halo, C1-C6 alquilo, C2-C6 alquenilo, C1-C6 alcoxi, -(C1-C6 alquilo)-O-(C1-C 6 alquilo), C3-C7 cicloalquilo, oxo, acilo, - S-(C1-C 6 alquilo), OH, NH2 , CN y C1-C3 perfluroalquilo; y
d se selecciona de 0 a 3.
2 El compuesto de la reivindicación 1, que tiene la fórmula

R4 y R5 se seleccionan independientemente del grupo que consiste en H, halo, C1-C8 alquilo, C2-C8 alquenilo, C2-C8 alquinilo, C1-C6 alcoxi, -(C1-C6 alquilo)-O-(C1-C6 alquilo), -NR41R42, -(CH2)x NR41R42 y -O-(C1-C6 alquilo)-O-(C1-C6 alquilo;
R41 y R42 se seleccionan independientemente del grupo que consiste en H, C1-C8 alquilo, C2-C8 alquenilo, C2-C8 alquinilo y -(C1-C6 alquil)-O-(C1-C6 alquilo);
o R41 y R42 pueden tomarse juntos para formar un cicloalquilo o anillo heterocíclico de tres a doce miembros que tiene hasta 3 heteroátomos que es opcionalmente sustituido de 1 a 3 sustituyentes seleccionados independientemente de halo, C1-C6 alquilo y C1-C6 alcoxi;
x se selecciona de 0 a 6;
o R4 y R5 pueden tomarse juntos para formar un anillo heterocíclico o aromático de tres a doce miembros que tiene hasta 3 heteroátomos que es opcionalmente sustituido de 1 a 3 sustituyentes seleccionados independientemente de halo y C1-C6 alquilo y C1-C6 alcoxi;
cada R6 se selecciona independientemente del grupo que consiste en halo, C1-C6 alquilo, C2-C6 alquenilo, C1-C6 alcoxi, -(C1-C6 alquilo)-O-(C1-C6 alquilo), C3-C7 cicloalquilo, oxo, acil, -S-(C1-C6 alquilo), OH, NH2, CN y C1-C3 perfluroalquilo; y
d se selecciona de 0 a 3.
3. El compuesto de la reivindicación 1, donde R2 se selecciona de indazol, ciclohexilpiridina, fenilpiridina, fenil-1H-pirazol, 1H-pirrolo[2,3-£)]piridina, 1H-pirazol, piridina, isoquinolina, quinolina y 1,3-tiazolil piridina.
4. El compuesto de la reivindicación 1, donde R2 se selecciona de:

5. El compuesto de las reivindicaciones 1 o 2, donde R4 y R5 se toman juntas para formar un anillo de cinco miembros.
6. El compuesto según cualquiera de las reivindicaciones 1 a 5 para su uso en el tratamiento de una afección seleccionada de: un trastorno autoinmune, un trastorno cardiovascular, inflamación, un trastorno del sistema nervioso central, un trastorno trombótico arterial, un trastorno fibrótico, glaucoma o regulador de presión intraocular, enfermedad neoplásica y osteoporosis.
7. El compuesto para su uso según la reivindicación 6, donde el trastorno autoinmune es artritis reumatoide (esclerosis múltiple), lupus eritematoso sistémico (SLE; lupus), psoriasis, enfermedad de Crohn, dermatitis atópica, eccema o enfermedad de injerto contra huésped (GVHD).
8. El compuesto para su uso según la reivindicación 6, donde el trastorno cardiovascular es hipertensión, arteriosclerosis, restenosis, hipertrofia cardíaca, hipertensión ocular, isquemia cerebral, vasoespasmo cerebral o disfunción eréctil.
9. El compuesto para su uso según la reivindicación 6, donde la inflamación es asma, inflamación cardiovascular, inflamación renal o arteriosclerosis.
10. El compuesto para su uso según la reivindicación 6, donde el trastorno fibrótico es fibrosis hepática, fibrosis pulmonar o fibrosis renal.
11. El compuesto según cualquiera de las reivindicaciones 1 a 5 para su uso en el tratamiento de una afección seleccionada de síndrome metabólico, resistencia a la insulina, hiperinsulinemia, diabetes tipo 2 o intolerancia a la glucosa, donde una cantidad terapéuticamente efectiva del compuesto debe administrarse a un sujeto.
12. El compuesto según cualquiera de las reivindicaciones 1 a 5 para su uso en el tratamiento de un trastorno ocular que tiene un componente angiogénico, donde una cantidad terapéuticamente efectiva del compuesto y un inhibidor de la angiogénesis deben administrarse a un sujeto.
13. El compuesto según cualquiera de las reivindicaciones 1 a 5 para su uso en el tratamiento de un trastorno que tiene un componente angiogénico seleccionado de aterosclerosis, artritis reumatoide (RA), hemangioma, angiofibroma, psoriasis, rechazo de injerto corneal, diabetes mellitus dependiente de insulina, esclerosis múltiple, miastenia gravis, enfermedad de Chron, nefritis autoinmune, cirrosis biliar primaria, pancreatitis aguda, rechazo de alografía, inflamación alérgica, dermatitis de contacto, hipersensibilidad retardada, enfermedad inflamatoria intestinal, shock séptico, osteoporosis, osteoartritis, inflamación neuronal, síndrome de Osler-Weber, reestenosis, infección fúngica, infección parasitaria e infección viral, donde una cantidad terapéuticamente efectiva del compuesto y un inhibidor de angiogénesis deben administrarse a un sujeto.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201361887935P | 2013-10-07 | 2013-10-07 | |
| PCT/US2014/059572 WO2015054317A1 (en) | 2013-10-07 | 2014-10-07 | Rho kinase inhibitors |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2779152T3 true ES2779152T3 (es) | 2020-08-13 |
Family
ID=52813600
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES14852268T Active ES2779152T3 (es) | 2013-10-07 | 2014-10-07 | Derivados de (2-(5-isoindolin-2-il)pirimidin-4-il)-amina como inhibidores de Rho-quinasa para tratar enfermedades autoinmunes |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US10125144B2 (es) |
| EP (1) | EP3055301B1 (es) |
| JP (1) | JP6507157B2 (es) |
| CN (1) | CN104903312B (es) |
| ES (1) | ES2779152T3 (es) |
| WO (1) | WO2015054317A1 (es) |
Families Citing this family (25)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| SI3186241T1 (sl) * | 2014-08-29 | 2021-03-31 | Chdi Foundation, Inc. | Sonde za slikanje Huntingtonovega proteina |
| CN107531785B (zh) * | 2014-10-07 | 2021-06-29 | 卡德门企业有限公司 | 人抗-vegfr-2/kdr抗体 |
| CN105002178B (zh) * | 2015-07-09 | 2017-09-12 | 广东省人民医院 | 抑制STAT3表达和防治慢性移植物抗宿主病的shRNA及应用 |
| KR102563325B1 (ko) * | 2017-06-30 | 2023-08-03 | 베이징 타이드 파마슈티컬 코퍼레이션 리미티드 | Rho-관련 단백질 키나아제 억제제, Rho-관련 단백질 키나아제 억제제를 함유하는 약학 조성물, 제조 방법 및 약학 조성물의 용도 |
| JP7311228B2 (ja) * | 2017-06-30 | 2023-07-19 | ベイジン タイド ファーマシューティカル カンパニー リミテッド | Rho-関連プロテインキナーゼ阻害剤、それを含む医薬組成物並びにその調製方法及び使用 |
| CN110582491B (zh) | 2017-06-30 | 2023-09-29 | 北京泰德制药股份有限公司 | Rho相关蛋白激酶抑制剂、包含其的药物组合物及其制备方法和用途 |
| US11926596B2 (en) | 2017-07-21 | 2024-03-12 | Kadmon Corporation, Llc | Inhibitors of Rho associated coiled-coil containing protein kinase |
| EP3675861A4 (en) * | 2017-09-01 | 2021-01-06 | Kadmon Corporation, LLC | ALPHA SUPERWINDING PROTEIN KINASE INHIBITORS ("COILED-COIL") ASSOCIATED WITH RHO |
| WO2019079369A1 (en) | 2017-10-19 | 2019-04-25 | Effector Therapeutics, Inc. | BENZIMIDAZOLE INDOLE INHIBITORS OF MNK1 AND MNK2 |
| GB201801226D0 (en) | 2018-01-25 | 2018-03-14 | Redx Pharma Plc | Modulators of Rho-associated protein kinase |
| CA3110661A1 (en) | 2018-08-29 | 2020-03-05 | University Of Massachusetts | Inhibition of protein kinases to treat friedreich ataxia |
| CN112969694B (zh) * | 2018-11-09 | 2023-06-13 | 北京泰德制药股份有限公司 | Rho相关蛋白激酶抑制剂、包含其的药物组合物及其用途 |
| CN111825675B (zh) * | 2019-04-15 | 2023-08-01 | 武汉朗来科技发展有限公司 | Rock抑制剂及其制备方法和用途 |
| CN110200972A (zh) * | 2019-05-10 | 2019-09-06 | 南京市儿童医院 | Kd025在制备防治慢性肾纤维化的药物中的用途 |
| TW202110878A (zh) * | 2019-05-16 | 2021-03-16 | 法商賽諾菲公司 | 在神經系統中表現抗原結合蛋白 |
| CN114502163A (zh) * | 2019-07-22 | 2022-05-13 | 安吉昂生物医药公司 | 作为rho相关卷曲螺旋激酶(rock)抑制剂的乙炔基杂环 |
| WO2021109737A1 (zh) * | 2019-12-02 | 2021-06-10 | 上海璎黎药业有限公司 | 一种含氧杂环化合物、其制备方法及应用 |
| US12014835B2 (en) | 2020-02-19 | 2024-06-18 | Vanderbilt University | Methods for evaluating therapeutic benefit of combination therapies |
| CN113559121B (zh) * | 2020-04-29 | 2023-09-19 | 首都医科大学附属北京同仁医院 | 一种用于角膜移植免疫排斥反应治疗的药物 |
| CN113929678B (zh) * | 2020-07-14 | 2025-09-30 | 武汉朗来科技发展有限公司 | 一种rock抑制剂及其制备方法和用途 |
| CN114573566B (zh) * | 2021-03-04 | 2023-05-30 | 杭州邦顺制药有限公司 | 选择性rock2激酶抑制剂 |
| JPWO2023085369A1 (es) | 2021-11-11 | 2023-05-19 | ||
| CN114380806B (zh) * | 2022-03-24 | 2022-06-10 | 中国药科大学 | 2-氨基-4-吲哚基嘧啶类化合物及其制备方法与应用 |
| CN120035442A (zh) * | 2022-10-13 | 2025-05-23 | 北京泰德制药股份有限公司 | 治疗尘肺病的方法 |
| WO2025189104A1 (en) * | 2024-03-07 | 2025-09-12 | Neurelis, Inc. | Treatment of cavernous angiomas with a 4-substituted piperidine derivative |
Family Cites Families (35)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR0144833B1 (ko) * | 1992-12-28 | 1998-07-15 | 김태훈 | 신규의 퀴나졸린 유도체 및 그의 제조방법 |
| JP2896532B2 (ja) * | 1994-08-13 | 1999-05-31 | ユーハン コーポレーション | 新規なピリミジン誘導体およびその製造方法 |
| IN188411B (es) * | 1997-03-27 | 2002-09-21 | Yuhan Corp | |
| MXPA02005350A (es) | 1999-11-30 | 2002-12-11 | Pfizer Prod Inc | Compuestos de 2,4-diamino pirimidina utiles como supresores de inmunidad. |
| EP1246823A1 (en) | 1999-12-28 | 2002-10-09 | Pharmacopeia, Inc. | Pyrimidine and triazine kinase inhibitors |
| WO2002022605A1 (en) * | 2000-09-15 | 2002-03-21 | Vertex Pharmaceuticals Incorporated | Pyrazole compounds useful as protein kinase inhibitors |
| CA2671776C (en) * | 2001-01-31 | 2012-12-18 | H. Lundbeck A/S | Gal3 receptor antagonists for the treatment of depression |
| US20030078271A1 (en) * | 2001-01-31 | 2003-04-24 | Blackburn Thomas P. | Use of GAL3 receptor antagonists for the treatment of depression and/or anxiety and compounds useful in such methods |
| UY27224A1 (es) | 2001-03-23 | 2002-10-31 | Bayer Corp | Inhibidores de la rho-quinasa |
| ATE407678T1 (de) * | 2001-10-17 | 2008-09-15 | Boehringer Ingelheim Pharma | Pyrimidinderivate, arzneimittel enthaltend diese verbindungen, deren verwendung und verfahren zu ihrer herstellung |
| JP4505228B2 (ja) * | 2002-01-10 | 2010-07-21 | バイエル・シェーリング・ファルマ・アクチェンゲゼルシャフト | Rho−キナーゼ阻害剤 |
| WO2004014307A2 (en) * | 2002-08-07 | 2004-02-19 | Synaptic Pharmaceutical Corporation | Gal3 antagonists for the treatment of neuropathic pain |
| WO2004014376A1 (en) * | 2002-08-07 | 2004-02-19 | Synaptic Pharmaceutical Corporation | Gal3 receptor antagonists for the treatment of affective disorders |
| US7220775B2 (en) * | 2002-08-07 | 2007-05-22 | H. Lundbeck A/S | Compound useful for the treatment of neuropathic pain |
| OA13050A (en) | 2003-04-29 | 2006-11-10 | Pfizer Ltd | 5,7-diaminopyrazolo [4,3-D] pyrimidines useful in the treatment of hypertension. |
| WO2005009980A1 (en) | 2003-07-22 | 2005-02-03 | Neurogen Corporation | Substituted pyridin-2-ylamine analogues |
| MX2008002207A (es) * | 2005-08-16 | 2008-03-27 | Memory Pharm Corp | Inhibidores de fosfodiesterasa 10. |
| CA2622372A1 (en) * | 2005-09-13 | 2007-03-22 | Palau Pharma, S.A. | 2-aminopyrimidine derivatives as modulators of the histamine h4 receptor activity |
| NL2000323C2 (nl) * | 2005-12-20 | 2007-11-20 | Pfizer Ltd | Pyrimidine-derivaten. |
| US20100035863A1 (en) * | 2006-09-12 | 2010-02-11 | Ucb Pharma, S.A. | 2 Amino-Pyrimidine Derivatives As H4 Receptor Antagonists, Processes For Preparing Them And Their Use In Pharmaceutical Compositions |
| WO2008054599A2 (en) | 2006-09-27 | 2008-05-08 | Surface Logix, Inc. | Rho kinase inhibitors |
| MX2010013726A (es) * | 2008-06-12 | 2011-01-14 | Janssen Pharmaceutica Nv | Moduladores de diamino-piridina, pirimidina, y piridazina del receptor h4 de histamina. |
| EP2312945A4 (en) | 2008-08-13 | 2012-05-09 | Merck Sharp & Dohme | PURE DERIVATIVES FOR THE TREATMENT OF MORBUS ALZHEIMER |
| EP3623374B1 (en) * | 2008-12-03 | 2021-09-08 | The Scripps Research Institute | Stem cell cultures |
| WO2010085246A1 (en) | 2009-01-21 | 2010-07-29 | Praecis Pharmaceuticals Inc | 2,4-diamino-1,3,5-triazine and 4, 6-diamino-pyrimidine derivatives and their use as aggrecanase inhibitors |
| US8263632B2 (en) * | 2009-03-24 | 2012-09-11 | Vertex Pharmaceuticals Incoporated | Aminopyridine derivatives having Aurora A selective inhibitory action |
| US9908884B2 (en) | 2009-05-05 | 2018-03-06 | Dana-Farber Cancer Institute, Inc. | EGFR inhibitors and methods of treating disorders |
| RU2013120966A (ru) * | 2010-10-08 | 2014-11-20 | Эббви Инк. | ФУРО[3,2-d]ПИРИМИДИНОВЫЕ СОЕДИНЕНИЯ |
| US9382229B2 (en) | 2011-02-15 | 2016-07-05 | The Johns Hopkins University | Compounds and methods of use thereof for treating neurodegenerative disorders |
| US9616064B2 (en) | 2011-03-30 | 2017-04-11 | H. Lee Moffitt Cancer Center And Research Institute, Inc. | Rho kinase inhibitors and methods of use |
| KR101394534B1 (ko) * | 2012-03-02 | 2014-05-13 | 한국화학연구원 | 신규한 1h-인다졸-5-일아미노-치환 헤테로사이클릭 화합물 및 이를 포함하는 약제학적 조성물 |
| WO2014081718A1 (en) * | 2012-11-20 | 2014-05-30 | Genentech, Inc. | Aminopyrimidine compounds as inhibitors of t790m containing egfr mutants |
| CN103626741A (zh) * | 2013-11-26 | 2014-03-12 | 苏州大学 | 具有腺苷受体拮抗剂活性的杂环氨基嘧啶化合物 |
| CN103664908A (zh) * | 2013-12-10 | 2014-03-26 | 苏州大学 | 一种具有腺苷受体拮抗活性的氨基嘧啶杂环化合物 |
| WO2015157556A1 (en) | 2014-04-09 | 2015-10-15 | Kadmon Corporation, Llc | Treatment of gvhd |
-
2014
- 2014-10-07 CN CN201480003391.0A patent/CN104903312B/zh active Active
- 2014-10-07 JP JP2016521259A patent/JP6507157B2/ja active Active
- 2014-10-07 WO PCT/US2014/059572 patent/WO2015054317A1/en not_active Ceased
- 2014-10-07 EP EP14852268.3A patent/EP3055301B1/en active Active
- 2014-10-07 US US15/027,975 patent/US10125144B2/en active Active
- 2014-10-07 ES ES14852268T patent/ES2779152T3/es active Active
Also Published As
| Publication number | Publication date |
|---|---|
| JP6507157B2 (ja) | 2019-04-24 |
| EP3055301A4 (en) | 2017-05-03 |
| US10125144B2 (en) | 2018-11-13 |
| EP3055301B1 (en) | 2019-11-20 |
| WO2015054317A1 (en) | 2015-04-16 |
| JP2016534038A (ja) | 2016-11-04 |
| EP3055301A1 (en) | 2016-08-17 |
| US20160237095A1 (en) | 2016-08-18 |
| HK1214813A1 (zh) | 2016-08-05 |
| CN104903312A (zh) | 2015-09-09 |
| CN104903312B (zh) | 2019-01-29 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2779152T3 (es) | Derivados de (2-(5-isoindolin-2-il)pirimidin-4-il)-amina como inhibidores de Rho-quinasa para tratar enfermedades autoinmunes | |
| US20260034127A1 (en) | Treatment of gvhd | |
| ES2924915T3 (es) | Inhibidores de RHO cinasa | |
| JP7399848B2 (ja) | Rho関連コイルドコイル含有プロテインキナーゼの阻害剤 | |
| WO2014055999A2 (en) | Treatment of ocular disorders | |
| HK1214813B (zh) | Rho激酶抑制剂 | |
| HK40062732A (en) | Rho kinase inhibitors | |
| HK40062730A (en) | Rho kinase inhibitors | |
| HK40085319A (en) | Rho kinase inhibitors | |
| HK40064586A (en) | Rho kinase inhibitors | |
| HK40062731A (en) | Rho kinase inhibitors |