ES2784240T3 - Composiciones farmacéuticas que comprenden DGLA y uso de las mismas - Google Patents
Composiciones farmacéuticas que comprenden DGLA y uso de las mismas Download PDFInfo
- Publication number
- ES2784240T3 ES2784240T3 ES15727395T ES15727395T ES2784240T3 ES 2784240 T3 ES2784240 T3 ES 2784240T3 ES 15727395 T ES15727395 T ES 15727395T ES 15727395 T ES15727395 T ES 15727395T ES 2784240 T3 ES2784240 T3 ES 2784240T3
- Authority
- ES
- Spain
- Prior art keywords
- dgla
- study
- approximately
- dose
- subjects
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- HOBAELRKJCKHQD-QNEBEIHSSA-N dihomo-γ-linolenic acid Chemical compound CCCCC\C=C/C\C=C/C\C=C/CCCCCCC(O)=O HOBAELRKJCKHQD-QNEBEIHSSA-N 0.000 title claims abstract description 269
- HOBAELRKJCKHQD-UHFFFAOYSA-N (8Z,11Z,14Z)-8,11,14-eicosatrienoic acid Natural products CCCCCC=CCC=CCC=CCCCCCCC(O)=O HOBAELRKJCKHQD-UHFFFAOYSA-N 0.000 title claims abstract description 227
- 239000008194 pharmaceutical composition Substances 0.000 title claims abstract description 34
- 238000011282 treatment Methods 0.000 claims abstract description 84
- 239000002775 capsule Substances 0.000 claims abstract description 74
- 206010012438 Dermatitis atopic Diseases 0.000 claims abstract description 53
- 201000008937 atopic dermatitis Diseases 0.000 claims abstract description 53
- 235000014113 dietary fatty acids Nutrition 0.000 claims abstract description 49
- 229930195729 fatty acid Natural products 0.000 claims abstract description 49
- 239000000194 fatty acid Substances 0.000 claims abstract description 49
- 150000004665 fatty acids Chemical class 0.000 claims abstract description 47
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 claims description 30
- 108010010803 Gelatin Proteins 0.000 claims description 25
- 239000008273 gelatin Substances 0.000 claims description 25
- 229920000159 gelatin Polymers 0.000 claims description 25
- 235000019322 gelatine Nutrition 0.000 claims description 25
- 235000011852 gelatine desserts Nutrition 0.000 claims description 25
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Chemical compound O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 claims description 11
- 208000037851 severe atopic dermatitis Diseases 0.000 claims description 9
- IIZPXYDJLKNOIY-JXPKJXOSSA-N 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCC\C=C/C\C=C/C\C=C/C\C=C/CCCCC IIZPXYDJLKNOIY-JXPKJXOSSA-N 0.000 claims description 5
- 239000000787 lecithin Substances 0.000 claims description 5
- 229940067606 lecithin Drugs 0.000 claims description 5
- 235000010445 lecithin Nutrition 0.000 claims description 5
- 229940057917 medium chain triglycerides Drugs 0.000 claims description 5
- 239000008213 purified water Substances 0.000 claims description 5
- 235000019738 Limestone Nutrition 0.000 claims description 3
- 239000006028 limestone Substances 0.000 claims description 3
- 239000000126 substance Substances 0.000 claims description 2
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 claims 2
- RTAQQCXQSZGOHL-UHFFFAOYSA-N Titanium Chemical compound [Ti] RTAQQCXQSZGOHL-UHFFFAOYSA-N 0.000 claims 1
- 229910002092 carbon dioxide Inorganic materials 0.000 claims 1
- 239000001569 carbon dioxide Substances 0.000 claims 1
- 229910052719 titanium Inorganic materials 0.000 claims 1
- 239000010936 titanium Substances 0.000 claims 1
- 235000021298 Dihomo-γ-linolenic acid Nutrition 0.000 description 220
- 239000003814 drug Substances 0.000 description 103
- 229940079593 drug Drugs 0.000 description 102
- 239000000902 placebo Substances 0.000 description 56
- 229940068196 placebo Drugs 0.000 description 56
- BSYNRYMUTXBXSQ-UHFFFAOYSA-N Aspirin Chemical compound CC(=O)OC1=CC=CC=C1C(O)=O BSYNRYMUTXBXSQ-UHFFFAOYSA-N 0.000 description 47
- 229960001138 acetylsalicylic acid Drugs 0.000 description 46
- 210000003491 skin Anatomy 0.000 description 45
- 238000011156 evaluation Methods 0.000 description 41
- 238000004458 analytical method Methods 0.000 description 39
- SONNWYBIRXJNDC-VIFPVBQESA-N phenylephrine Chemical compound CNC[C@H](O)C1=CC=CC(O)=C1 SONNWYBIRXJNDC-VIFPVBQESA-N 0.000 description 39
- 229960001802 phenylephrine Drugs 0.000 description 39
- 230000009467 reduction Effects 0.000 description 38
- 238000000034 method Methods 0.000 description 34
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 33
- 230000004872 arterial blood pressure Effects 0.000 description 30
- 238000012360 testing method Methods 0.000 description 30
- 238000012216 screening Methods 0.000 description 29
- 201000004624 Dermatitis Diseases 0.000 description 28
- 230000002354 daily effect Effects 0.000 description 27
- 208000010668 atopic eczema Diseases 0.000 description 26
- 239000000203 mixture Substances 0.000 description 26
- 230000008859 change Effects 0.000 description 24
- 239000012530 fluid Substances 0.000 description 24
- 208000003251 Pruritus Diseases 0.000 description 22
- 241000700159 Rattus Species 0.000 description 21
- 230000036772 blood pressure Effects 0.000 description 21
- 201000010099 disease Diseases 0.000 description 21
- 230000002411 adverse Effects 0.000 description 20
- 206010020772 Hypertension Diseases 0.000 description 18
- 241001465754 Metazoa Species 0.000 description 18
- 210000004369 blood Anatomy 0.000 description 18
- 239000008280 blood Substances 0.000 description 18
- 230000036470 plasma concentration Effects 0.000 description 18
- 230000009885 systemic effect Effects 0.000 description 17
- 239000000523 sample Substances 0.000 description 16
- 238000001990 intravenous administration Methods 0.000 description 15
- 230000036572 transepidermal water loss Effects 0.000 description 14
- 229960004195 carvedilol Drugs 0.000 description 13
- NPAKNKYSJIDKMW-UHFFFAOYSA-N carvedilol Chemical compound COC1=CC=CC=C1OCCNCC(O)COC1=CC=CC2=NC3=CC=C[CH]C3=C12 NPAKNKYSJIDKMW-UHFFFAOYSA-N 0.000 description 13
- 238000012552 review Methods 0.000 description 13
- 206010012735 Diarrhoea Diseases 0.000 description 12
- 208000035475 disorder Diseases 0.000 description 12
- 230000000694 effects Effects 0.000 description 12
- 238000005259 measurement Methods 0.000 description 12
- 230000035935 pregnancy Effects 0.000 description 12
- 238000009597 pregnancy test Methods 0.000 description 12
- 206010024438 Lichenification Diseases 0.000 description 11
- 239000003246 corticosteroid Substances 0.000 description 11
- 229960001334 corticosteroids Drugs 0.000 description 11
- 238000002483 medication Methods 0.000 description 11
- 206010015150 Erythema Diseases 0.000 description 10
- 208000035874 Excoriation Diseases 0.000 description 10
- 239000003433 contraceptive agent Substances 0.000 description 10
- 238000001647 drug administration Methods 0.000 description 10
- 231100000321 erythema Toxicity 0.000 description 10
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 9
- 238000009826 distribution Methods 0.000 description 9
- 230000003203 everyday effect Effects 0.000 description 9
- 238000003304 gavage Methods 0.000 description 9
- 238000011160 research Methods 0.000 description 9
- 230000004044 response Effects 0.000 description 9
- 230000000699 topical effect Effects 0.000 description 9
- GWEVSGVZZGPLCZ-UHFFFAOYSA-N Titan oxide Chemical compound O=[Ti]=O GWEVSGVZZGPLCZ-UHFFFAOYSA-N 0.000 description 8
- 230000008901 benefit Effects 0.000 description 8
- 230000007717 exclusion Effects 0.000 description 8
- 230000001631 hypertensive effect Effects 0.000 description 8
- 230000007803 itching Effects 0.000 description 8
- 238000009533 lab test Methods 0.000 description 8
- 230000000873 masking effect Effects 0.000 description 8
- 239000013641 positive control Substances 0.000 description 8
- 239000011550 stock solution Substances 0.000 description 8
- 210000002700 urine Anatomy 0.000 description 8
- 229940084778 1,4-sorbitan Drugs 0.000 description 7
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 7
- 230000015271 coagulation Effects 0.000 description 7
- 238000005345 coagulation Methods 0.000 description 7
- 239000003974 emollient agent Substances 0.000 description 7
- 239000000047 product Substances 0.000 description 7
- 229960002920 sorbitol Drugs 0.000 description 7
- 230000002269 spontaneous effect Effects 0.000 description 7
- 238000011699 spontaneously hypertensive rat Methods 0.000 description 7
- 238000002562 urinalysis Methods 0.000 description 7
- JNYAEWCLZODPBN-JGWLITMVSA-N (2r,3r,4s)-2-[(1r)-1,2-dihydroxyethyl]oxolane-3,4-diol Chemical compound OC[C@@H](O)[C@H]1OC[C@H](O)[C@H]1O JNYAEWCLZODPBN-JGWLITMVSA-N 0.000 description 6
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 6
- 206010030113 Oedema Diseases 0.000 description 6
- 208000033809 Suppuration Diseases 0.000 description 6
- 230000033228 biological regulation Effects 0.000 description 6
- 229940046731 calcineurin inhibitors Drugs 0.000 description 6
- 230000009246 food effect Effects 0.000 description 6
- 235000012631 food intake Nutrition 0.000 description 6
- 238000002360 preparation method Methods 0.000 description 6
- 231100000279 safety data Toxicity 0.000 description 6
- 208000024891 symptom Diseases 0.000 description 6
- 238000002560 therapeutic procedure Methods 0.000 description 6
- 230000000007 visual effect Effects 0.000 description 6
- 102000012673 Follicle Stimulating Hormone Human genes 0.000 description 5
- 108010079345 Follicle Stimulating Hormone Proteins 0.000 description 5
- 102000015696 Interleukins Human genes 0.000 description 5
- 108010063738 Interleukins Proteins 0.000 description 5
- 230000005856 abnormality Effects 0.000 description 5
- 239000002253 acid Substances 0.000 description 5
- 239000000090 biomarker Substances 0.000 description 5
- 208000002352 blister Diseases 0.000 description 5
- 230000001684 chronic effect Effects 0.000 description 5
- 229940124558 contraceptive agent Drugs 0.000 description 5
- 230000002254 contraceptive effect Effects 0.000 description 5
- 230000034994 death Effects 0.000 description 5
- 231100000517 death Toxicity 0.000 description 5
- 238000013461 design Methods 0.000 description 5
- 235000015872 dietary supplement Nutrition 0.000 description 5
- 230000008030 elimination Effects 0.000 description 5
- 238000003379 elimination reaction Methods 0.000 description 5
- 229940028334 follicle stimulating hormone Drugs 0.000 description 5
- 230000003054 hormonal effect Effects 0.000 description 5
- 238000012544 monitoring process Methods 0.000 description 5
- 210000000056 organ Anatomy 0.000 description 5
- 238000011002 quantification Methods 0.000 description 5
- 230000001105 regulatory effect Effects 0.000 description 5
- 210000002966 serum Anatomy 0.000 description 5
- 238000007619 statistical method Methods 0.000 description 5
- -1 1,4-sorbitan sugar alcohols Chemical class 0.000 description 4
- 241000725303 Human immunodeficiency virus Species 0.000 description 4
- 206010039509 Scab Diseases 0.000 description 4
- 238000010521 absorption reaction Methods 0.000 description 4
- 230000001174 ascending effect Effects 0.000 description 4
- 239000007844 bleaching agent Substances 0.000 description 4
- 239000003795 chemical substances by application Substances 0.000 description 4
- 238000011161 development Methods 0.000 description 4
- 230000018109 developmental process Effects 0.000 description 4
- 238000002474 experimental method Methods 0.000 description 4
- 235000020937 fasting conditions Nutrition 0.000 description 4
- 230000037406 food intake Effects 0.000 description 4
- 239000007903 gelatin capsule Substances 0.000 description 4
- 206010022437 insomnia Diseases 0.000 description 4
- 230000003902 lesion Effects 0.000 description 4
- 235000012054 meals Nutrition 0.000 description 4
- 239000004014 plasticizer Substances 0.000 description 4
- 238000011076 safety test Methods 0.000 description 4
- 238000003860 storage Methods 0.000 description 4
- 208000011117 substance-related disease Diseases 0.000 description 4
- 238000001356 surgical procedure Methods 0.000 description 4
- 239000004408 titanium dioxide Substances 0.000 description 4
- 239000011782 vitamin Substances 0.000 description 4
- 229940088594 vitamin Drugs 0.000 description 4
- 229930003231 vitamin Natural products 0.000 description 4
- 235000013343 vitamin Nutrition 0.000 description 4
- KIUKXJAPPMFGSW-DNGZLQJQSA-N (2S,3S,4S,5R,6R)-6-[(2S,3R,4R,5S,6R)-3-Acetamido-2-[(2S,3S,4R,5R,6R)-6-[(2R,3R,4R,5S,6R)-3-acetamido-2,5-dihydroxy-6-(hydroxymethyl)oxan-4-yl]oxy-2-carboxy-4,5-dihydroxyoxan-3-yl]oxy-5-hydroxy-6-(hydroxymethyl)oxan-4-yl]oxy-3,4,5-trihydroxyoxane-2-carboxylic acid Chemical compound CC(=O)N[C@H]1[C@H](O)O[C@H](CO)[C@@H](O)[C@@H]1O[C@H]1[C@H](O)[C@@H](O)[C@H](O[C@H]2[C@@H]([C@@H](O[C@H]3[C@@H]([C@@H](O)[C@H](O)[C@H](O3)C(O)=O)O)[C@H](O)[C@@H](CO)O2)NC(C)=O)[C@@H](C(O)=O)O1 KIUKXJAPPMFGSW-DNGZLQJQSA-N 0.000 description 3
- NVKAWKQGWWIWPM-ABEVXSGRSA-N 17-β-hydroxy-5-α-Androstan-3-one Chemical compound C1C(=O)CC[C@]2(C)[C@H]3CC[C@](C)([C@H](CC4)O)[C@@H]4[C@@H]3CC[C@H]21 NVKAWKQGWWIWPM-ABEVXSGRSA-N 0.000 description 3
- RZVAJINKPMORJF-UHFFFAOYSA-N Acetaminophen Chemical compound CC(=O)NC1=CC=C(O)C=C1 RZVAJINKPMORJF-UHFFFAOYSA-N 0.000 description 3
- 206010067484 Adverse reaction Diseases 0.000 description 3
- 206010002091 Anaesthesia Diseases 0.000 description 3
- PMATZTZNYRCHOR-CGLBZJNRSA-N Cyclosporin A Chemical compound CC[C@@H]1NC(=O)[C@H]([C@H](O)[C@H](C)C\C=C\C)N(C)C(=O)[C@H](C(C)C)N(C)C(=O)[C@H](CC(C)C)N(C)C(=O)[C@H](CC(C)C)N(C)C(=O)[C@@H](C)NC(=O)[C@H](C)NC(=O)[C@H](CC(C)C)N(C)C(=O)[C@H](C(C)C)NC(=O)[C@H](CC(C)C)N(C)C(=O)CN(C)C1=O PMATZTZNYRCHOR-CGLBZJNRSA-N 0.000 description 3
- 108010036949 Cyclosporine Proteins 0.000 description 3
- 206010013654 Drug abuse Diseases 0.000 description 3
- 208000005176 Hepatitis C Diseases 0.000 description 3
- 206010020751 Hypersensitivity Diseases 0.000 description 3
- FBOZXECLQNJBKD-ZDUSSCGKSA-N L-methotrexate Chemical compound C=1N=C2N=C(N)N=C(N)C2=NC=1CN(C)C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 FBOZXECLQNJBKD-ZDUSSCGKSA-N 0.000 description 3
- 235000008753 Papaver somniferum Nutrition 0.000 description 3
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Chemical compound NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 3
- 230000002159 abnormal effect Effects 0.000 description 3
- 208000026935 allergic disease Diseases 0.000 description 3
- 230000037005 anaesthesia Effects 0.000 description 3
- 239000012491 analyte Substances 0.000 description 3
- 238000012550 audit Methods 0.000 description 3
- LMEKQMALGUDUQG-UHFFFAOYSA-N azathioprine Chemical compound CN1C=NC([N+]([O-])=O)=C1SC1=NC=NC2=C1NC=N2 LMEKQMALGUDUQG-UHFFFAOYSA-N 0.000 description 3
- 229960002170 azathioprine Drugs 0.000 description 3
- 210000000746 body region Anatomy 0.000 description 3
- 239000004202 carbamide Substances 0.000 description 3
- 229940106189 ceramide Drugs 0.000 description 3
- 229960001265 ciclosporin Drugs 0.000 description 3
- 238000012790 confirmation Methods 0.000 description 3
- 229930182912 cyclosporin Natural products 0.000 description 3
- 230000003247 decreasing effect Effects 0.000 description 3
- 230000001419 dependent effect Effects 0.000 description 3
- 238000004090 dissolution Methods 0.000 description 3
- 235000020664 gamma-linolenic acid Nutrition 0.000 description 3
- 230000002496 gastric effect Effects 0.000 description 3
- 210000003128 head Anatomy 0.000 description 3
- 208000002672 hepatitis B Diseases 0.000 description 3
- 229920002674 hyaluronan Polymers 0.000 description 3
- 229960003160 hyaluronic acid Drugs 0.000 description 3
- 230000009610 hypersensitivity Effects 0.000 description 3
- 208000015181 infectious disease Diseases 0.000 description 3
- 239000004615 ingredient Substances 0.000 description 3
- 238000011835 investigation Methods 0.000 description 3
- 229960000485 methotrexate Drugs 0.000 description 3
- 235000020660 omega-3 fatty acid Nutrition 0.000 description 3
- 229940012843 omega-3 fatty acid Drugs 0.000 description 3
- 235000020665 omega-6 fatty acid Nutrition 0.000 description 3
- 229940033080 omega-6 fatty acid Drugs 0.000 description 3
- 238000003305 oral gavage Methods 0.000 description 3
- 229960005489 paracetamol Drugs 0.000 description 3
- 230000002085 persistent effect Effects 0.000 description 3
- 239000000546 pharmaceutical excipient Substances 0.000 description 3
- 239000013589 supplement Substances 0.000 description 3
- 230000002123 temporal effect Effects 0.000 description 3
- 208000004998 Abdominal Pain Diseases 0.000 description 2
- 208000007848 Alcoholism Diseases 0.000 description 2
- 238000000959 Cochran–Mantel–Haenszel (CMH) test Methods 0.000 description 2
- 239000011627 DL-alpha-tocopherol Substances 0.000 description 2
- 235000001815 DL-alpha-tocopherol Nutrition 0.000 description 2
- 206010013786 Dry skin Diseases 0.000 description 2
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 2
- VSNHCAURESNICA-UHFFFAOYSA-N Hydroxyurea Chemical compound NC(=O)NO VSNHCAURESNICA-UHFFFAOYSA-N 0.000 description 2
- 102000003820 Lipoxygenases Human genes 0.000 description 2
- 108090000128 Lipoxygenases Proteins 0.000 description 2
- 206010028813 Nausea Diseases 0.000 description 2
- 239000006057 Non-nutritive feed additive Substances 0.000 description 2
- 206010033733 Papule Diseases 0.000 description 2
- 102000057297 Pepsin A Human genes 0.000 description 2
- 108090000284 Pepsin A Proteins 0.000 description 2
- 208000024777 Prion disease Diseases 0.000 description 2
- 102000004005 Prostaglandin-endoperoxide synthases Human genes 0.000 description 2
- 108090000459 Prostaglandin-endoperoxide synthases Proteins 0.000 description 2
- 108010094028 Prothrombin Proteins 0.000 description 2
- 102100027378 Prothrombin Human genes 0.000 description 2
- 206010037660 Pyrexia Diseases 0.000 description 2
- QJJXYPPXXYFBGM-LFZNUXCKSA-N Tacrolimus Chemical compound C1C[C@@H](O)[C@H](OC)C[C@@H]1\C=C(/C)[C@@H]1[C@H](C)[C@@H](O)CC(=O)[C@H](CC=C)/C=C(C)/C[C@H](C)C[C@H](OC)[C@H]([C@H](C[C@H]2C)OC)O[C@@]2(O)C(=O)C(=O)N2CCCC[C@H]2C(=O)O1 QJJXYPPXXYFBGM-LFZNUXCKSA-N 0.000 description 2
- 206010048222 Xerosis Diseases 0.000 description 2
- 238000009825 accumulation Methods 0.000 description 2
- 230000001154 acute effect Effects 0.000 description 2
- 230000006838 adverse reaction Effects 0.000 description 2
- 206010001584 alcohol abuse Diseases 0.000 description 2
- 208000025746 alcohol use disease Diseases 0.000 description 2
- 125000005907 alkyl ester group Chemical group 0.000 description 2
- 229960003473 androstanolone Drugs 0.000 description 2
- 239000000427 antigen Substances 0.000 description 2
- 102000036639 antigens Human genes 0.000 description 2
- 108091007433 antigens Proteins 0.000 description 2
- 239000000739 antihistaminic agent Substances 0.000 description 2
- 229940125715 antihistaminic agent Drugs 0.000 description 2
- 206010003549 asthenia Diseases 0.000 description 2
- 230000004888 barrier function Effects 0.000 description 2
- 239000002876 beta blocker Substances 0.000 description 2
- 229940097320 beta blocking agent Drugs 0.000 description 2
- 229960000074 biopharmaceutical Drugs 0.000 description 2
- 235000021152 breakfast Nutrition 0.000 description 2
- RYYVLZVUVIJVGH-UHFFFAOYSA-N caffeine Chemical compound CN1C(=O)N(C)C(=O)C2=C1N=CN2C RYYVLZVUVIJVGH-UHFFFAOYSA-N 0.000 description 2
- 238000004364 calculation method Methods 0.000 description 2
- 150000001783 ceramides Chemical class 0.000 description 2
- 238000000546 chi-square test Methods 0.000 description 2
- HVYWMOMLDIMFJA-DPAQBDIFSA-N cholesterol Chemical compound C1C=C2C[C@@H](O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H]([C@H](C)CCCC(C)C)[C@@]1(C)CC2 HVYWMOMLDIMFJA-DPAQBDIFSA-N 0.000 description 2
- 238000011260 co-administration Methods 0.000 description 2
- 150000001875 compounds Chemical class 0.000 description 2
- GVJHHUAWPYXKBD-UHFFFAOYSA-N d-alpha-tocopherol Natural products OC1=C(C)C(C)=C2OC(CCCC(C)CCCC(C)CCCC(C)C)(C)CCC2=C1C GVJHHUAWPYXKBD-UHFFFAOYSA-N 0.000 description 2
- 230000006378 damage Effects 0.000 description 2
- 206010012601 diabetes mellitus Diseases 0.000 description 2
- 238000003745 diagnosis Methods 0.000 description 2
- 231100000673 dose–response relationship Toxicity 0.000 description 2
- 239000003651 drinking water Substances 0.000 description 2
- 235000020188 drinking water Nutrition 0.000 description 2
- 230000037336 dry skin Effects 0.000 description 2
- 235000004626 essential fatty acids Nutrition 0.000 description 2
- 230000001815 facial effect Effects 0.000 description 2
- 230000027950 fever generation Effects 0.000 description 2
- 235000021588 free fatty acids Nutrition 0.000 description 2
- 230000036541 health Effects 0.000 description 2
- 230000000004 hemodynamic effect Effects 0.000 description 2
- 239000000938 histamine H1 antagonist Substances 0.000 description 2
- 208000010544 human prion disease Diseases 0.000 description 2
- 230000001976 improved effect Effects 0.000 description 2
- 230000006698 induction Effects 0.000 description 2
- 230000002452 interceptive effect Effects 0.000 description 2
- 238000002372 labelling Methods 0.000 description 2
- 150000002632 lipids Chemical class 0.000 description 2
- 229940057995 liquid paraffin Drugs 0.000 description 2
- 230000003908 liver function Effects 0.000 description 2
- 210000003141 lower extremity Anatomy 0.000 description 2
- 230000014759 maintenance of location Effects 0.000 description 2
- 230000005906 menstruation Effects 0.000 description 2
- 238000012986 modification Methods 0.000 description 2
- 230000004048 modification Effects 0.000 description 2
- 230000008693 nausea Effects 0.000 description 2
- 230000000926 neurological effect Effects 0.000 description 2
- 229940094443 oxytocics prostaglandins Drugs 0.000 description 2
- 229940111202 pepsin Drugs 0.000 description 2
- 238000001126 phototherapy Methods 0.000 description 2
- KASDHRXLYQOAKZ-ZPSXYTITSA-N pimecrolimus Chemical compound C/C([C@H]1OC(=O)[C@@H]2CCCCN2C(=O)C(=O)[C@]2(O)O[C@@H]([C@H](C[C@H]2C)OC)[C@@H](OC)C[C@@H](C)C/C(C)=C/[C@H](C(C[C@H](O)[C@H]1C)=O)CC)=C\[C@@H]1CC[C@@H](Cl)[C@H](OC)C1 KASDHRXLYQOAKZ-ZPSXYTITSA-N 0.000 description 2
- 229960005330 pimecrolimus Drugs 0.000 description 2
- 229920000642 polymer Polymers 0.000 description 2
- 239000000955 prescription drug Substances 0.000 description 2
- 230000002265 prevention Effects 0.000 description 2
- 230000008569 process Effects 0.000 description 2
- 238000012545 processing Methods 0.000 description 2
- 150000003180 prostaglandins Chemical class 0.000 description 2
- 229940039716 prothrombin Drugs 0.000 description 2
- 230000000241 respiratory effect Effects 0.000 description 2
- 238000010206 sensitivity analysis Methods 0.000 description 2
- 230000005808 skin problem Effects 0.000 description 2
- 239000000243 solution Substances 0.000 description 2
- 239000002904 solvent Substances 0.000 description 2
- 239000000934 spermatocidal agent Substances 0.000 description 2
- 238000000528 statistical test Methods 0.000 description 2
- 210000002784 stomach Anatomy 0.000 description 2
- 230000036561 sun exposure Effects 0.000 description 2
- 230000035488 systolic blood pressure Effects 0.000 description 2
- 229960001967 tacrolimus Drugs 0.000 description 2
- QJJXYPPXXYFBGM-SHYZHZOCSA-N tacrolimus Natural products CO[C@H]1C[C@H](CC[C@@H]1O)C=C(C)[C@H]2OC(=O)[C@H]3CCCCN3C(=O)C(=O)[C@@]4(O)O[C@@H]([C@H](C[C@H]4C)OC)[C@@H](C[C@H](C)CC(=C[C@@H](CC=C)C(=O)C[C@H](O)[C@H]2C)C)OC QJJXYPPXXYFBGM-SHYZHZOCSA-N 0.000 description 2
- 239000011269 tar Substances 0.000 description 2
- 229960000984 tocofersolan Drugs 0.000 description 2
- 230000001052 transient effect Effects 0.000 description 2
- 210000001364 upper extremity Anatomy 0.000 description 2
- 229940045136 urea Drugs 0.000 description 2
- 239000003981 vehicle Substances 0.000 description 2
- 238000012795 verification Methods 0.000 description 2
- GVJHHUAWPYXKBD-IEOSBIPESA-N α-tocopherol Chemical compound OC1=C(C)C(C)=C2O[C@@](CCC[C@H](C)CCC[C@H](C)CCCC(C)C)(C)CCC2=C1C GVJHHUAWPYXKBD-IEOSBIPESA-N 0.000 description 2
- AKHONHCNYRUTLT-NXBFSHDWSA-N (8R,9S,10R,13S,14S,17S)-15,15,17-trihydroxy-10,13-dimethyl-2,6,7,8,9,11,12,14,16,17-decahydro-1H-cyclopenta[a]phenanthren-3-one Chemical compound O=C1CC[C@]2(C)[C@H]3CC[C@](C)([C@H](CC4(O)O)O)[C@@H]4[C@@H]3CCC2=C1 AKHONHCNYRUTLT-NXBFSHDWSA-N 0.000 description 1
- IUKXMNDGTWTNTP-OAHXIXLCSA-N 15(S)-HETrE Chemical compound CCCCC[C@H](O)\C=C\C=C/C\C=C/CCCCCCC(O)=O IUKXMNDGTWTNTP-OAHXIXLCSA-N 0.000 description 1
- IUKXMNDGTWTNTP-RHDCIPCHSA-N 15-HETrE Chemical compound CCCCCC(O)\C=C\C=C/C\C=C/CCCCCCC(O)=O IUKXMNDGTWTNTP-RHDCIPCHSA-N 0.000 description 1
- 206010003645 Atopy Diseases 0.000 description 1
- 208000035143 Bacterial infection Diseases 0.000 description 1
- 108010039209 Blood Coagulation Factors Proteins 0.000 description 1
- 102000015081 Blood Coagulation Factors Human genes 0.000 description 1
- 208000019838 Blood disease Diseases 0.000 description 1
- 241000283690 Bos taurus Species 0.000 description 1
- 206010006482 Bronchospasm Diseases 0.000 description 1
- YDNKGFDKKRUKPY-JHOUSYSJSA-N C16 ceramide Natural products CCCCCCCCCCCCCCCC(=O)N[C@@H](CO)[C@H](O)C=CCCCCCCCCCCCCC YDNKGFDKKRUKPY-JHOUSYSJSA-N 0.000 description 1
- 208000024172 Cardiovascular disease Diseases 0.000 description 1
- 208000002177 Cataract Diseases 0.000 description 1
- 206010007764 Cataract subcapsular Diseases 0.000 description 1
- 102000019034 Chemokines Human genes 0.000 description 1
- 108010012236 Chemokines Proteins 0.000 description 1
- 102000011022 Chorionic Gonadotropin Human genes 0.000 description 1
- 108010062540 Chorionic Gonadotropin Proteins 0.000 description 1
- 206010053567 Coagulopathies Diseases 0.000 description 1
- 208000035473 Communicable disease Diseases 0.000 description 1
- 208000032170 Congenital Abnormalities Diseases 0.000 description 1
- 206010010356 Congenital anomaly Diseases 0.000 description 1
- 206010010741 Conjunctivitis Diseases 0.000 description 1
- 206010010904 Convulsion Diseases 0.000 description 1
- 206010011224 Cough Diseases 0.000 description 1
- 102000004127 Cytokines Human genes 0.000 description 1
- 108090000695 Cytokines Proteins 0.000 description 1
- 206010062918 Dennie-Morgan fold Diseases 0.000 description 1
- 206010015719 Exsanguination Diseases 0.000 description 1
- 206010061958 Food Intolerance Diseases 0.000 description 1
- 206010017543 Fungal skin infection Diseases 0.000 description 1
- 206010018852 Haematoma Diseases 0.000 description 1
- 206010019233 Headaches Diseases 0.000 description 1
- 206010019280 Heart failures Diseases 0.000 description 1
- 208000032843 Hemorrhage Diseases 0.000 description 1
- 208000009889 Herpes Simplex Diseases 0.000 description 1
- 241000282412 Homo Species 0.000 description 1
- 206010060708 Induration Diseases 0.000 description 1
- 206010061218 Inflammation Diseases 0.000 description 1
- LPHGQDQBBGAPDZ-UHFFFAOYSA-N Isocaffeine Natural products CN1C(=O)N(C)C(=O)C2=C1N(C)C=N2 LPHGQDQBBGAPDZ-UHFFFAOYSA-N 0.000 description 1
- 201000002287 Keratoconus Diseases 0.000 description 1
- 206010066295 Keratosis pilaris Diseases 0.000 description 1
- OYHQOLUKZRVURQ-HZJYTTRNSA-N Linoleic acid Chemical compound CCCCC\C=C/C\C=C/CCCCCCCC(O)=O OYHQOLUKZRVURQ-HZJYTTRNSA-N 0.000 description 1
- 206010025323 Lymphomas Diseases 0.000 description 1
- CRJGESKKUOMBCT-VQTJNVASSA-N N-acetylsphinganine Chemical compound CCCCCCCCCCCCCCC[C@@H](O)[C@H](CO)NC(C)=O CRJGESKKUOMBCT-VQTJNVASSA-N 0.000 description 1
- 244000061176 Nicotiana tabacum Species 0.000 description 1
- 235000002637 Nicotiana tabacum Nutrition 0.000 description 1
- 206010068319 Oropharyngeal pain Diseases 0.000 description 1
- 208000001132 Osteoporosis Diseases 0.000 description 1
- 102000035195 Peptidases Human genes 0.000 description 1
- 108091005804 Peptidases Proteins 0.000 description 1
- 201000007100 Pharyngitis Diseases 0.000 description 1
- 239000004743 Polypropylene Substances 0.000 description 1
- 201000004681 Psoriasis Diseases 0.000 description 1
- 206010040799 Skin atrophy Diseases 0.000 description 1
- 206010040860 Skin haemorrhages Diseases 0.000 description 1
- 206010040925 Skin striae Diseases 0.000 description 1
- 244000061456 Solanum tuberosum Species 0.000 description 1
- 235000002595 Solanum tuberosum Nutrition 0.000 description 1
- 241000191967 Staphylococcus aureus Species 0.000 description 1
- 208000031439 Striae Distensae Diseases 0.000 description 1
- 210000001744 T-lymphocyte Anatomy 0.000 description 1
- 206010065173 Viral skin infection Diseases 0.000 description 1
- 238000003677 abuse test Methods 0.000 description 1
- 230000001944 accentuation Effects 0.000 description 1
- 230000009471 action Effects 0.000 description 1
- 230000006978 adaptation Effects 0.000 description 1
- 239000002671 adjuvant Substances 0.000 description 1
- 239000000951 adrenergic alpha-1 receptor antagonist Substances 0.000 description 1
- 150000001299 aldehydes Chemical class 0.000 description 1
- 102000030619 alpha-1 Adrenergic Receptor Human genes 0.000 description 1
- 108020004102 alpha-1 Adrenergic Receptor Proteins 0.000 description 1
- 125000003277 amino group Chemical group 0.000 description 1
- 229940069428 antacid Drugs 0.000 description 1
- 239000003159 antacid agent Substances 0.000 description 1
- 230000003466 anti-cipated effect Effects 0.000 description 1
- 230000003276 anti-hypertensive effect Effects 0.000 description 1
- 230000000845 anti-microbial effect Effects 0.000 description 1
- 239000004599 antimicrobial Substances 0.000 description 1
- 206010003246 arthritis Diseases 0.000 description 1
- 238000003556 assay Methods 0.000 description 1
- 201000002463 atopic dermatitis 2 Diseases 0.000 description 1
- 210000003050 axon Anatomy 0.000 description 1
- 235000015241 bacon Nutrition 0.000 description 1
- 230000001580 bacterial effect Effects 0.000 description 1
- 208000022362 bacterial infectious disease Diseases 0.000 description 1
- 230000002146 bilateral effect Effects 0.000 description 1
- 239000003124 biologic agent Substances 0.000 description 1
- 230000015572 biosynthetic process Effects 0.000 description 1
- 230000007698 birth defect Effects 0.000 description 1
- 230000000740 bleeding effect Effects 0.000 description 1
- 239000010836 blood and blood product Substances 0.000 description 1
- 230000017531 blood circulation Effects 0.000 description 1
- 239000003114 blood coagulation factor Substances 0.000 description 1
- 238000004820 blood count Methods 0.000 description 1
- 231100001015 blood dyscrasias Toxicity 0.000 description 1
- 229940125691 blood product Drugs 0.000 description 1
- 210000004204 blood vessel Anatomy 0.000 description 1
- 230000036760 body temperature Effects 0.000 description 1
- 206010006451 bronchitis Diseases 0.000 description 1
- 229960001948 caffeine Drugs 0.000 description 1
- VJEONQKOZGKCAK-UHFFFAOYSA-N caffeine Natural products CN1C(=O)N(C)C(=O)C2=C1C=CN2C VJEONQKOZGKCAK-UHFFFAOYSA-N 0.000 description 1
- 150000001720 carbohydrates Chemical class 0.000 description 1
- 235000014633 carbohydrates Nutrition 0.000 description 1
- 229910052799 carbon Inorganic materials 0.000 description 1
- 210000000748 cardiovascular system Anatomy 0.000 description 1
- 230000001364 causal effect Effects 0.000 description 1
- 230000001413 cellular effect Effects 0.000 description 1
- ZVEQCJWYRWKARO-UHFFFAOYSA-N ceramide Natural products CCCCCCCCCCCCCCC(O)C(=O)NC(CO)C(O)C=CCCC=C(C)CCCCCCCCC ZVEQCJWYRWKARO-UHFFFAOYSA-N 0.000 description 1
- 238000012512 characterization method Methods 0.000 description 1
- 238000006243 chemical reaction Methods 0.000 description 1
- 239000003153 chemical reaction reagent Substances 0.000 description 1
- 235000012000 cholesterol Nutrition 0.000 description 1
- 229940015047 chorionic gonadotropin Drugs 0.000 description 1
- 238000003759 clinical diagnosis Methods 0.000 description 1
- 230000035602 clotting Effects 0.000 description 1
- 239000011280 coal tar Substances 0.000 description 1
- 235000012716 cod liver oil Nutrition 0.000 description 1
- 239000003026 cod liver oil Substances 0.000 description 1
- 239000003086 colorant Substances 0.000 description 1
- 239000002131 composite material Substances 0.000 description 1
- 238000011970 concomitant therapy Methods 0.000 description 1
- 229940124301 concurrent medication Drugs 0.000 description 1
- 239000002285 corn oil Substances 0.000 description 1
- 235000005687 corn oil Nutrition 0.000 description 1
- 238000004132 cross linking Methods 0.000 description 1
- 238000013481 data capture Methods 0.000 description 1
- 238000013480 data collection Methods 0.000 description 1
- 230000006735 deficit Effects 0.000 description 1
- 210000004443 dendritic cell Anatomy 0.000 description 1
- 230000006866 deterioration Effects 0.000 description 1
- 238000010586 diagram Methods 0.000 description 1
- 230000035487 diastolic blood pressure Effects 0.000 description 1
- 230000003205 diastolic effect Effects 0.000 description 1
- 235000005911 diet Nutrition 0.000 description 1
- 230000037213 diet Effects 0.000 description 1
- 238000007922 dissolution test Methods 0.000 description 1
- 208000002173 dizziness Diseases 0.000 description 1
- 230000035622 drinking Effects 0.000 description 1
- 229940000406 drug candidate Drugs 0.000 description 1
- 206010013663 drug dependence Diseases 0.000 description 1
- 210000005069 ears Anatomy 0.000 description 1
- 230000002526 effect on cardiovascular system Effects 0.000 description 1
- 230000002500 effect on skin Effects 0.000 description 1
- 239000012636 effector Substances 0.000 description 1
- 235000013601 eggs Nutrition 0.000 description 1
- 150000002066 eicosanoids Chemical class 0.000 description 1
- 230000002996 emotional effect Effects 0.000 description 1
- 238000005538 encapsulation Methods 0.000 description 1
- 230000007613 environmental effect Effects 0.000 description 1
- 210000003979 eosinophil Anatomy 0.000 description 1
- 150000002148 esters Chemical class 0.000 description 1
- 125000004494 ethyl ester group Chemical group 0.000 description 1
- 239000003777 experimental drug Substances 0.000 description 1
- 210000001508 eye Anatomy 0.000 description 1
- 235000019197 fats Nutrition 0.000 description 1
- 210000001105 femoral artery Anatomy 0.000 description 1
- 210000003191 femoral vein Anatomy 0.000 description 1
- 230000001605 fetal effect Effects 0.000 description 1
- 229960003592 fexofenadine Drugs 0.000 description 1
- RWTNPBWLLIMQHL-UHFFFAOYSA-N fexofenadine Chemical compound C1=CC(C(C)(C(O)=O)C)=CC=C1C(O)CCCN1CCC(C(O)(C=2C=CC=CC=2)C=2C=CC=CC=2)CC1 RWTNPBWLLIMQHL-UHFFFAOYSA-N 0.000 description 1
- 239000011888 foil Substances 0.000 description 1
- 235000013305 food Nutrition 0.000 description 1
- 210000002683 foot Anatomy 0.000 description 1
- 238000009472 formulation Methods 0.000 description 1
- 230000006870 function Effects 0.000 description 1
- VZCCETWTMQHEPK-UHFFFAOYSA-N gamma-Linolensaeure Natural products CCCCCC=CCC=CCC=CCCCCC(O)=O VZCCETWTMQHEPK-UHFFFAOYSA-N 0.000 description 1
- VZCCETWTMQHEPK-QNEBEIHSSA-N gamma-linolenic acid Chemical compound CCCCC\C=C/C\C=C/C\C=C/CCCCC(O)=O VZCCETWTMQHEPK-QNEBEIHSSA-N 0.000 description 1
- 229960002733 gamolenic acid Drugs 0.000 description 1
- 239000000499 gel Substances 0.000 description 1
- 230000036449 good health Effects 0.000 description 1
- 230000005484 gravity Effects 0.000 description 1
- 231100000869 headache Toxicity 0.000 description 1
- 208000014951 hematologic disease Diseases 0.000 description 1
- 208000018706 hematopoietic system disease Diseases 0.000 description 1
- 210000000548 hind-foot Anatomy 0.000 description 1
- 210000003630 histaminocyte Anatomy 0.000 description 1
- 238000009802 hysterectomy Methods 0.000 description 1
- 206010021198 ichthyosis Diseases 0.000 description 1
- 230000028993 immune response Effects 0.000 description 1
- 230000036039 immunity Effects 0.000 description 1
- 230000001771 impaired effect Effects 0.000 description 1
- 230000006872 improvement Effects 0.000 description 1
- 238000001727 in vivo Methods 0.000 description 1
- 230000002757 inflammatory effect Effects 0.000 description 1
- 230000004054 inflammatory process Effects 0.000 description 1
- 230000028709 inflammatory response Effects 0.000 description 1
- 229940125369 inhaled corticosteroids Drugs 0.000 description 1
- 230000002401 inhibitory effect Effects 0.000 description 1
- 230000000977 initiatory effect Effects 0.000 description 1
- 238000002347 injection Methods 0.000 description 1
- 229940090044 injection Drugs 0.000 description 1
- 239000007924 injection Substances 0.000 description 1
- 238000007689 inspection Methods 0.000 description 1
- 230000003993 interaction Effects 0.000 description 1
- 229940047122 interleukins Drugs 0.000 description 1
- 238000010253 intravenous injection Methods 0.000 description 1
- 210000002510 keratinocyte Anatomy 0.000 description 1
- 210000003734 kidney Anatomy 0.000 description 1
- 230000003907 kidney function Effects 0.000 description 1
- 210000000265 leukocyte Anatomy 0.000 description 1
- 235000020778 linoleic acid Nutrition 0.000 description 1
- OYHQOLUKZRVURQ-IXWMQOLASA-N linoleic acid Natural products CCCCC\C=C/C\C=C\CCCCCCCC(O)=O OYHQOLUKZRVURQ-IXWMQOLASA-N 0.000 description 1
- 238000012006 liquid chromatography with tandem mass spectrometry Methods 0.000 description 1
- 229960003088 loratadine Drugs 0.000 description 1
- JCCNYMKQOSZNPW-UHFFFAOYSA-N loratadine Chemical compound C1CN(C(=O)OCC)CCC1=C1C2=NC=CC=C2CCC2=CC(Cl)=CC=C21 JCCNYMKQOSZNPW-UHFFFAOYSA-N 0.000 description 1
- 210000002540 macrophage Anatomy 0.000 description 1
- 238000012423 maintenance Methods 0.000 description 1
- 230000003211 malignant effect Effects 0.000 description 1
- 230000008774 maternal effect Effects 0.000 description 1
- 230000007246 mechanism Effects 0.000 description 1
- 230000010534 mechanism of action Effects 0.000 description 1
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 1
- 208000037852 mild atopic dermatitis Diseases 0.000 description 1
- 230000000116 mitigating effect Effects 0.000 description 1
- 230000000877 morphologic effect Effects 0.000 description 1
- RTGDFNSFWBGLEC-SYZQJQIISA-N mycophenolate mofetil Chemical compound COC1=C(C)C=2COC(=O)C=2C(O)=C1C\C=C(/C)CCC(=O)OCCN1CCOCC1 RTGDFNSFWBGLEC-SYZQJQIISA-N 0.000 description 1
- 229960004866 mycophenolate mofetil Drugs 0.000 description 1
- SQMWSBKSHWARHU-SDBHATRESA-N n6-cyclopentyladenosine Chemical compound O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1N1C2=NC=NC(NC3CCCC3)=C2N=C1 SQMWSBKSHWARHU-SDBHATRESA-N 0.000 description 1
- 201000009240 nasopharyngitis Diseases 0.000 description 1
- VVGIYYKRAMHVLU-UHFFFAOYSA-N newbouldiamide Natural products CCCCCCCCCCCCCCCCCCCC(O)C(O)C(O)C(CO)NC(=O)CCCCCCCCCCCCCCCCC VVGIYYKRAMHVLU-UHFFFAOYSA-N 0.000 description 1
- 210000002445 nipple Anatomy 0.000 description 1
- 239000000820 nonprescription drug Substances 0.000 description 1
- 210000001331 nose Anatomy 0.000 description 1
- 239000004006 olive oil Substances 0.000 description 1
- 235000008390 olive oil Nutrition 0.000 description 1
- 238000001543 one-way ANOVA Methods 0.000 description 1
- 238000009806 oophorectomy Methods 0.000 description 1
- 229940127234 oral contraceptive Drugs 0.000 description 1
- 239000003539 oral contraceptive agent Substances 0.000 description 1
- 230000003647 oxidation Effects 0.000 description 1
- 238000007254 oxidation reaction Methods 0.000 description 1
- 230000036961 partial effect Effects 0.000 description 1
- 239000000825 pharmaceutical preparation Substances 0.000 description 1
- 229940127557 pharmaceutical product Drugs 0.000 description 1
- 230000003285 pharmacodynamic effect Effects 0.000 description 1
- 210000003800 pharynx Anatomy 0.000 description 1
- 229940071370 phenylephrine 10 mg Drugs 0.000 description 1
- 229940036309 phenylephrine injection Drugs 0.000 description 1
- 230000037081 physical activity Effects 0.000 description 1
- 206010035111 pityriasis alba Diseases 0.000 description 1
- 229920001155 polypropylene Polymers 0.000 description 1
- 235000012015 potatoes Nutrition 0.000 description 1
- 229940124606 potential therapeutic agent Drugs 0.000 description 1
- 230000002028 premature Effects 0.000 description 1
- 230000000770 proinflammatory effect Effects 0.000 description 1
- 238000000275 quality assurance Methods 0.000 description 1
- 238000003908 quality control method Methods 0.000 description 1
- 238000011552 rat model Methods 0.000 description 1
- 230000009257 reactivity Effects 0.000 description 1
- 230000007115 recruitment Effects 0.000 description 1
- 230000000306 recurrent effect Effects 0.000 description 1
- 230000002829 reductive effect Effects 0.000 description 1
- 230000002040 relaxant effect Effects 0.000 description 1
- 230000029058 respiratory gaseous exchange Effects 0.000 description 1
- 230000036387 respiratory rate Effects 0.000 description 1
- 230000000717 retained effect Effects 0.000 description 1
- 230000002441 reversible effect Effects 0.000 description 1
- 102220047090 rs6152 Human genes 0.000 description 1
- 208000037921 secondary disease Diseases 0.000 description 1
- 230000035945 sensitivity Effects 0.000 description 1
- 230000001568 sexual effect Effects 0.000 description 1
- 208000017520 skin disease Diseases 0.000 description 1
- 206010040872 skin infection Diseases 0.000 description 1
- 238000013112 stability test Methods 0.000 description 1
- 238000010561 standard procedure Methods 0.000 description 1
- 230000001954 sterilising effect Effects 0.000 description 1
- 238000004659 sterilization and disinfection Methods 0.000 description 1
- 230000037359 steroid metabolism Effects 0.000 description 1
- 238000013517 stratification Methods 0.000 description 1
- 201000009032 substance abuse Diseases 0.000 description 1
- 230000008093 supporting effect Effects 0.000 description 1
- 230000001629 suppression Effects 0.000 description 1
- 239000000725 suspension Substances 0.000 description 1
- 230000035900 sweating Effects 0.000 description 1
- 238000009121 systemic therapy Methods 0.000 description 1
- 238000012353 t test Methods 0.000 description 1
- 229940124597 therapeutic agent Drugs 0.000 description 1
- 229960005196 titanium dioxide Drugs 0.000 description 1
- 239000011732 tocopherol Substances 0.000 description 1
- 229960001295 tocopherol Drugs 0.000 description 1
- 229940026191 topical antipsoriatics tars Drugs 0.000 description 1
- 229940025703 topical product Drugs 0.000 description 1
- 150000003626 triacylglycerols Chemical class 0.000 description 1
- 125000005457 triglyceride group Chemical group 0.000 description 1
- 238000009810 tubal ligation Methods 0.000 description 1
- 208000019206 urinary tract infection Diseases 0.000 description 1
- 238000010200 validation analysis Methods 0.000 description 1
- 230000002792 vascular Effects 0.000 description 1
- 230000002861 ventricular Effects 0.000 description 1
- 230000003612 virological effect Effects 0.000 description 1
- 238000011179 visual inspection Methods 0.000 description 1
- 238000005303 weighing Methods 0.000 description 1
- 230000036642 wellbeing Effects 0.000 description 1
- 235000008939 whole milk Nutrition 0.000 description 1
- 210000002268 wool Anatomy 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/48—Preparations in capsules, e.g. of gelatin, of chocolate
- A61K9/4816—Wall or shell material
- A61K9/4825—Proteins, e.g. gelatin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/185—Acids; Anhydrides, halides or salts thereof, e.g. sulfur acids, imidic, hydrazonic or hydroximic acids
- A61K31/19—Carboxylic acids, e.g. valproic acid
- A61K31/20—Carboxylic acids, e.g. valproic acid having a carboxyl group bound to a chain of seven or more carbon atoms, e.g. stearic, palmitic, arachidic acids
- A61K31/201—Carboxylic acids, e.g. valproic acid having a carboxyl group bound to a chain of seven or more carbon atoms, e.g. stearic, palmitic, arachidic acids having one or two double bonds, e.g. oleic, linoleic acids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/185—Acids; Anhydrides, halides or salts thereof, e.g. sulfur acids, imidic, hydrazonic or hydroximic acids
- A61K31/19—Carboxylic acids, e.g. valproic acid
- A61K31/20—Carboxylic acids, e.g. valproic acid having a carboxyl group bound to a chain of seven or more carbon atoms, e.g. stearic, palmitic, arachidic acids
- A61K31/202—Carboxylic acids, e.g. valproic acid having a carboxyl group bound to a chain of seven or more carbon atoms, e.g. stearic, palmitic, arachidic acids having three or more double bonds, e.g. linolenic
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/60—Salicylic acid; Derivatives thereof
- A61K31/612—Salicylic acid; Derivatives thereof having the hydroxy group in position 2 esterified, e.g. salicylsulfuric acid
- A61K31/616—Salicylic acid; Derivatives thereof having the hydroxy group in position 2 esterified, e.g. salicylsulfuric acid by carboxylic acids, e.g. acetylsalicylic acid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K8/00—Cosmetics or similar toiletry preparations
- A61K8/02—Cosmetics or similar toiletry preparations characterised by special physical form
- A61K8/11—Encapsulated compositions
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K8/00—Cosmetics or similar toiletry preparations
- A61K8/18—Cosmetics or similar toiletry preparations characterised by the composition
- A61K8/30—Cosmetics or similar toiletry preparations characterised by the composition containing organic compounds
- A61K8/33—Cosmetics or similar toiletry preparations characterised by the composition containing organic compounds containing oxygen
- A61K8/36—Carboxylic acids; Salts or anhydrides thereof
- A61K8/361—Carboxylic acids having more than seven carbon atoms in an unbroken chain; Salts or anhydrides thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K8/00—Cosmetics or similar toiletry preparations
- A61K8/18—Cosmetics or similar toiletry preparations characterised by the composition
- A61K8/30—Cosmetics or similar toiletry preparations characterised by the composition containing organic compounds
- A61K8/64—Proteins; Peptides; Derivatives or degradation products thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K8/00—Cosmetics or similar toiletry preparations
- A61K8/18—Cosmetics or similar toiletry preparations characterised by the composition
- A61K8/30—Cosmetics or similar toiletry preparations characterised by the composition containing organic compounds
- A61K8/64—Proteins; Peptides; Derivatives or degradation products thereof
- A61K8/65—Collagen; Gelatin; Keratin; Derivatives or degradation products thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/0053—Mouth and digestive tract, i.e. intraoral and peroral administration
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/48—Preparations in capsules, e.g. of gelatin, of chocolate
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/48—Preparations in capsules, e.g. of gelatin, of chocolate
- A61K9/4841—Filling excipients; Inactive ingredients
- A61K9/485—Inorganic compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/48—Preparations in capsules, e.g. of gelatin, of chocolate
- A61K9/4841—Filling excipients; Inactive ingredients
- A61K9/4858—Organic compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/48—Preparations in capsules, e.g. of gelatin, of chocolate
- A61K9/4841—Filling excipients; Inactive ingredients
- A61K9/4866—Organic macromolecular compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/16—Drugs for disorders of the alimentary tract or the digestive system for liver or gallbladder disorders, e.g. hepatoprotective agents, cholagogues, litholytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/08—Drugs for disorders of the urinary system of the prostate
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/10—Drugs for disorders of the urinary system of the bladder
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/04—Antipruritics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/06—Antipsoriatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/10—Anti-acne agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/16—Emollients or protectives, e.g. against radiation
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/18—Antioxidants, e.g. antiradicals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/06—Antigout agents, e.g. antihyperuricemic or uricosuric agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/08—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P21/00—Drugs for disorders of the muscular or neuromuscular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/06—Antihyperlipidemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/02—Antithrombotic agents; Anticoagulants; Platelet aggregation inhibitors
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/10—Antioedematous agents; Diuretics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/06—Antiarrhythmics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/12—Antihypertensives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61Q—SPECIFIC USE OF COSMETICS OR SIMILAR TOILETRY PREPARATIONS
- A61Q19/00—Preparations for care of the skin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K2300/00—Mixtures or combinations of active ingredients, wherein at least one active ingredient is fully defined in groups A61K31/00 - A61K41/00
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Animal Behavior & Ethology (AREA)
- Veterinary Medicine (AREA)
- Chemical & Material Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Medicinal Chemistry (AREA)
- Organic Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Epidemiology (AREA)
- Dermatology (AREA)
- Diabetes (AREA)
- Hematology (AREA)
- Physical Education & Sports Medicine (AREA)
- Birds (AREA)
- Urology & Nephrology (AREA)
- Heart & Thoracic Surgery (AREA)
- Obesity (AREA)
- Cardiology (AREA)
- Rheumatology (AREA)
- Emergency Medicine (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Nutrition Science (AREA)
- Physiology (AREA)
- Pain & Pain Management (AREA)
- Neurology (AREA)
- Immunology (AREA)
- Inorganic Chemistry (AREA)
- Biochemistry (AREA)
- Oncology (AREA)
- Toxicology (AREA)
- Vascular Medicine (AREA)
- Biomedical Technology (AREA)
- Communicable Diseases (AREA)
Abstract
Una composición farmacéutica de ácidos grasos para su uso en el tratamiento de la dermatitis atópica, en la que la composición farmacéutica de ácidos grasos comprende DGLA encapsulado en una cubierta de cápsula, en la que DGLA representa al menos el 95 % en peso de todos los ácidos grasos en la composición farmacéutica de ácidos grasos y en la que el tratamiento de la dermatitis atópica comprende la administración de 1 g o 2 g de la composición farmacéutica de ácidos grasos al día.
Description
DESCRIPCIÓN
Composiciones farmacéuticas que comprenden DGLA y uso de las mismas
Campo técnico
La presente solicitud se refiere al uso de ciertas composiciones farmacéuticas que comprenden DGLA.
Antecedentes
El ácido dihomo gamma linolénico (DGLA) es un ácido graso esencial que se encuentra naturalmente en el cuerpo como producto de la elongación del ácido gamma linolénico (GLA). El GLA es, a su vez, un producto de desaturación del ácido linoleico. La encapsulación de gelatina blanda de DGLA es un desafío, ya que es propensa a la oxidación de aldehídos que pueden interaccionar con grupos amino en el polímero de gelatina en la cubierta de la cápsula. Esto puede causar una desaceleración en la liberación del fármaco debido a la reticulación de los polímeros de gelatina. El documento US 2008/108699 describe el uso de DGLA al 40 % en forma de triglicéridos en pruebas para el posible tratamiento de la dermatitis atópica, donde se comparan varias dietas de aceite de maíz con o sin DGLA, a niveles bajos o altos.
Breve descripción de los dibujos
Muchos aspectos de la presente divulgación pueden entenderse mejor con referencia a los siguientes dibujos. Los componentes en los dibujos no están necesariamente a escala. En cambio, se hace hincapié en ilustrar claramente los principios de la presente divulgación.
La figura 1 muestra el cambio en la presión arterial media (mm Hg) con dosis intravenosas de fenilefrina después de siete días consecutivos de sonda con aspirina a 10 mg/kg/día.
La figura 2 muestra el cambio en la presión arterial media (mmHg) con dosis intravenosas de fenilefrina después de siete días consecutivos de sonda con DGLA a 50 mg/kg aspirina a 10 mg/kg.
La figura 3 muestra el cambio en la presión arterial media (mm Hg) con dosis intravenosas de fenilefrina después de siete días consecutivos de sonda con DGLA a 500 mg/kg administrado de forma conjunta con aspirina a 10 mg/kg.
La figura 4 muestra la presión arterial media al inicio después de siete días consecutivos con seis grupos de sondas diferentes.
La figura 5 muestra la presión arterial media con una dosis intravenosa de fenilefrina a 20 pk/kg después de siete días consecutivos con seis grupos de sondas diferentes.
La figura 6 muestra la concentración media de DGLA libre en plasma (ng/ml, gráfico lineal), por cohorte de dosis (dosis única, población PK).
La figura 7 muestra la concentración media de DGLA libre en plasma (ng/ml, gráfico logarítmico-lineal), por cohorte de dosis (dosis única, población PK).
La figura 8 muestra la concentración media total de DGLA en plasma (ng/ml, gráfico lineal), por cohorte de dosis (dosis única, población PK).
La figura 9 muestra la concentración media total de DGLA en plasma (ng/ml, gráfico logarítmico-lineal), por cohorte de dosis (dosis única, población PK).
La figura 10 muestra la concentración media de DGLA libre en plasma (ng/ml, gráfico lineal), por cohorte de dosis (dosis múltiples, población PK).
La figura 11 muestra la concentración media de DGLA libre en plasma (ng/ml, gráfico logarítmico-lineal), por cohorte de dosis (dosis múltiples, población PK).
La figura 12 muestra la concentración media total de DGLA en plasma (ng/ml, gráfico lineal), por cohorte de dosis (dosis múltiples, población PK).
La figura 13 muestra la concentración media total de DGLA en plasma (ng/ml, gráfico logarítmico-lineal), por cohorte de dosis (dosis múltiples, población PK).
La figura 14 muestra la concentración media en el fluido de las ampollas de la piel de DGLA libre (ng/ml, gráfico
lineal), por cohorte de dosis (dosis múltiples, población PK).
La figura 15 muestra la concentración media en el fluido de las ampollas de la piel de DGLA libre (ng/ml, gráfico logarítmico-lineal), por cohorte de dosis (dosis múltiples, población PK).
La figura 16 muestra la concentración media en el fluido de las ampollas de la piel de DGLA total (ng/ml, gráfico lineal), por cohorte de dosis (dosis múltiples, población PK).
La figura 17 muestra la concentración media en el fluido de las ampollas de la piel de DGLA total (ng/ml, gráfico logarítmico-lineal), por cohorte de dosis (dosis múltiples, población PK)
La figura 18 muestra la concentración media de DGLA libre (ng/ml, gráfico lineal) en plasma y en el fluido de las ampollas de la piel, por cohorte de dosis (dosis múltiples, población PK).
La figura 19 muestra la concentración media de DGLA libre (ng/ml, gráfico logarítmico-lineal) en plasma y en el fluido de las ampollas de la piel, por cohorte de dosis (dosis múltiples, población PK).
La figura 20 muestra la concentración media total de DGLA (ng/ml, gráfico lineal) en plasma y en el fluido de las ampollas de la piel, por cohorte de dosis (dosis múltiples, población PK).
La figura 21 muestra la concentración media total de DGLA (ng/ml, gráfico logarítmico-lineal) en plasma y en el fluido de las ampollas de la piel, por cohorte de dosis (dosis múltiples, población PK).
La figura 22 muestra la concentración media de dihidrotestosterona en plasma (ng/ml, gráfico lineal), por cohorte de dosis (dosis múltiples, población PK).
La figura 23 muestra la concentración media de dihidrotestosterona en plasma (ng/ml, gráfico logarítmico-lineal), por cohorte de dosis (dosis múltiples, población PK).
Sumario
La presente invención proporciona una composición farmacéutica de ácidos grasos para su uso en el tratamiento de la dermatitis atópica, en la que la composición farmacéutica de ácidos grasos comprende DGLA encapsulado en una cubierta de cápsula. El DGLA representa al menos el 95 % en peso de todos los ácidos grasos en la composición farmacéutica de ácidos grasos. El tratamiento de la dermatitis atópica comprende la administración de 1 g o 2 g de la composición farmacéutica de ácidos grasos por día.
El tratamiento de la dermatitis atópica puede comprender la administración oral de 1 g o 2 g de dicha composición farmacéutica de ácidos grasos por día, en la que la composición farmacéutica de ácidos grasos comprende una cubierta de cápsula y el DGLA está encapsulado en la cubierta de la cápsula.
Opcionalmente, la composición farmacéutica comprende DGLA encapsulado en una cubierta de cápsula que comprende gelatina, d-sorbitol y alcoholes de azúcar 1,4-sorbitán. En una realización, la gelatina tiene una viscosidad en masa de gel de aproximadamente 9.500 a aproximadamente 11.000, por ejemplo de aproximadamente 9.775 o aproximadamente 10.500. En otra realización, la gelatina tiene una eclosión de aproximadamente 165 a aproximadamente 190, por ejemplo de aproximadamente 170 a aproximadamente 185. En otra realización, la gelatina tiene un porcentaje de cenizas > que aproximadamente 0,33.
Aspectos adicionales de la invención, descritos con más detalle a continuación, se exponen en las reivindicaciones adjuntas.
Descripción detallada
Aunque la presente invención puede encontrarse incorporada en diversas formas, la descripción siguiente de varias realizaciones se realiza sabiendo que la presente divulgación debe considerarse como ejemplo de la invención y no pretende limitar la invención a las realizaciones específicas ilustradas. Los encabezados se proporcionan solo por conveniencia y no se debe interpretar que limitan las reivindicaciones en modo alguno.
Las realizaciones ilustradas bajo cualquier encabezado se pueden combinar con realizaciones ilustradas bajo cualquier otro encabezado.
El uso de valores numéricos en los diversos valores cuantitativos especificados en esta solicitud, a menos que se indique expresamente lo contrario, se indican como aproximaciones como si los valores mínimo y máximo dentro de los intervalos indicados estuvieran ambos precedidos por la palabra "aproximadamente". De esta manera, se pueden usar ligeras variaciones de un valor indicado para conseguir sustancialmente los mismos resultados que el valor indicado. También, la divulgación de los intervalos está prevista como un intervalo continuo que incluye todos los valores entre los valores mínimo y máximo citados, así como cualquier intervalo que se puede formar mediante
dichos valores. En el presente documento se divulgan todas y cada una de las relaciones (e intervalos de cualquiera de dichas relaciones) que se pueden formar dividiendo un valor numérico citado en cualquier otro valor numérico citado. Por consiguiente, el experto apreciará que muchas de tales relaciones, intervalos e intervalos de las relaciones se pueden derivar de forma inequívoca a partir de los valores numéricos presentados en el presente documento y en todos los casos, tales relaciones, intervalos e intervalos de relaciones representan diversas realizaciones de la presente invención.
Composiciones
Las composiciones farmacéuticas administrables por vía oral pueden comprender DGLA o un derivado del mismo. El término DGLA en el presente documento se refiere a DGLA en forma de ácido libre. Los derivados de DGLA se pueden usar además de o en lugar de DGLA. Dichos derivados incluyen ésteres de alquilo, ésteres de alquilo inferior, tales como éster metílico o etílico de DGLA o DGLA en forma de triglicérido. La presente invención proporciona una composición farmacéutica que comprende DGLA encapsulado en una cubierta de cápsula. En una realización, se encapsulan aproximadamente 500 mg a aproximadamente 1 g de DGLA en la cubierta de la cápsula.
En una realización, la cubierta de la cápsula comprende gelatina, por ejemplo, Gelatina RXL o gelatina de hueso de lima con un peso molecular más bajo. En otra realización, la cubierta de la cápsula comprende Gelatina RXL que ha sido tratada con enzima proteolítica para cortar el patrón de gelatina y disminuir efectivamente su peso molecular. La composición farmacéutica puede comprender ésteres de DGLA de D-sorbitol y 1,4-sorbitán. En una realización, la cubierta de la cápsula comprende (a) gelatina y (b) plastificantes seleccionados de uno o más de d-sorbitol y 1,4-sorbitanos. En una realización, la gelatina es como se describe en el documento US 7,485,323.
En una realización, el plastificante comprende 1-4 sorbitanos en una cantidad de 20 %-30 %, por ejemplo, aproximadamente 24 % y 28 % (en seco) y un contenido de D-sorbitol de aproximadamente 30 %- 50 %, por ejemplo de aproximadamente 35 % a 45 % (en seco).
En algunas realizaciones, la cubierta de la cápsula comprende además glicerol, agua purificada, dióxido de titanio, triglicéridos de cadena media y lecitina.
El DGLA puede estar presente en una composición de la divulgación en una cantidad de aproximadamente 50 mg a aproximadamente 5.000 mg, de aproximadamente 75 mg a aproximadamente 2.500 mg o de aproximadamente 100 mg a aproximadamente 1.000 mg, por ejemplo aproximadamente 75 mg, aproximadamente 100 mg, aproximadamente 125 mg aproximadamente 150 mg, aproximadamente 175 mg, aproximadamente 200 mg, aproximadamente 225 mg aproximadamente 250 mg, aproximadamente 275 mg, aproximadamente 300 mg, aproximadamente 325 mg aproximadamente 350 mg, aproximadamente 375 mg, aproximadamente 400 mg, aproximadamente 425 mg aproximadamente 450 mg, aproximadamente 475 mg, aproximadamente 500 mg, aproximadamente 525 mg aproximadamente 550 mg, aproximadamente 575 mg, aproximadamente 600 mg, aproximadamente 625 mg aproximadamente 650 mg, aproximadamente 675 mg, aproximadamente 700 mg, aproximadamente 725 mg aproximadamente 750 mg, aproximadamente 775 mg, aproximadamente 800 mg, aproximadamente 825 mg aproximadamente 850 mg, aproximadamente 875 mg, aproximadamente 900 mg, aproximadamente 925 mg, aproximadamente 950 mg, aproximadamente 975 mg, aproximadamente 1.000 mg, aproximadamente 1025 mg, aproximadamente 1.050 mg, aproximadamente 1.075 mg, aproximadamente 1.100 mg, aproximadamente 1.025 mg, aproximadamente 1.050 mg, aproximadamente 1.075 mg, aproximadamente 1.200 mg, aproximadamente 1.225 mg, aproximadamente 1.250 mg, aproximadamente 1.275 mg, aproximadamente 1.300 mg, aproximadamente 1.325 mg, aproximadamente 1.350 mg, aproximadamente 1.375 mg, aproximadamente 1.400 mg, aproximadamente 1.425 mg, aproximadamente 1450 mg, aproximadamente 1.475 mg, aproximadamente 1.500 mg, aproximadamente 1.525 mg, aproximadamente 1.550 mg, aproximadamente 1.575 mg, aproximadamente 1.600 mg, aproximadamente 1.625 mg, aproximadamente 1.650 mg, aproximadamente 1.675 mg, aproximadamente 1.700 mg, aproximadamente 1.725 mg, aproximadamente 1.750 mg, aproximadamente 1.775 mg, aproximadamente 1.800 mg, aproximadamente 1.825 mg, aproximadamente 1.850 mg, aproximadamente 1.875 mg, aproximadamente 1900 mg, aproximadamente 1.925 mg, aproximadamente 1.950 mg, aproximadamente 1.975 mg, aproximadamente 2.000 mg, aproximadamente 2.025 mg, aproximadamente 2.050 mg, aproximadamente 2.075 mg, aproximadamente 2.100 mg, aproximadamente 2125 mg, aproximadamente 2.150 mg, aproximadamente 2.175 mg, aproximadamente 2.200 mg, aproximadamente 2.225 mg, aproximadamente 2.250 mg, aproximadamente 2.275 mg, aproximadamente 2.300 mg, aproximadamente 2.325 mg, aproximadamente 2.350 mg, aproximadamente 2.375 mg, aproximadamente 2.400 mg, aproximadamente 2.425 mg , aproximadamente 2.450 mg, aproximadamente 2.475 mg o aproximadamente 2.500 mg. En una realización de este tipo, la composición puede comprender además los ésteres DGLA de D-sorbitol y 1,4-sorbitano.
En una realización, una composición de la divulgación no contiene más de aproximadamente 5 %, no más de aproximadamente 4 %, no más de aproximadamente 3 %, no más de aproximadamente 2 %, no más de aproximadamente 1 % o no más de aproximadamente 0,5 %, en peso de ácidos grasos totales, de ácidos grasos distintos de DGLA.
En otra realización, DGLA representa al menos aproximadamente el 95 %, al menos aproximadamente el 97 %, al
menos aproximadamente el 98 %, al menos aproximadamente el 99 % o el 100 %, en peso, de todos los ácidos grasos presentes en una composición de la invención.
En una realización, una composición de la invención, cuando se coloca en una prueba de disgregación estándar, por ejemplo, como se establece en USP 2040 (Disgregación y disolución de suplementos dietéticos) con agua como medio, tiene una tasa de liberación de DGLA de menos de aproximadamente 60 minutos, menos de aproximadamente 50 minutos, menos de aproximadamente 40 minutos, menos de aproximadamente 30 minutos o menos de 20 minutos después del almacenamiento durante aproximadamente 1 mes, aproximadamente 2 meses o aproximadamente 3 meses a 40 °C/75 % de HR.
En una realización, después del almacenamiento durante aproximadamente 1 mes, aproximadamente 2 meses, aproximadamente 3 meses o aproximadamente 6 meses a 40 °C/75 % de HR, una composición de la invención comprende menos de aproximadamente 5 % en peso de ésteres de DGLA de todos los ácidos grasos, menos de aproximadamente 4 % de ésteres de DGLA en peso de todos los ácidos grasos, menos de aproximadamente 3 % de ésteres de DGLA en peso de todos los ácidos grasos, menos de aproximadamente 2 % de ésteres de DGLA en peso de todos los ácidos grasos o menos de aproximadamente 1 % de ésteres de DGLA en peso de todos los ácidos grasos.
Métodos
La composición de la invención, incluyendo composiciones descritas anteriormente en el presente documento o composiciones que pueden formularse combinando diversas realizaciones de la presente divulgación, se utiliza en el tratamiento o prevención de la dermatitis atópica.
El término "tratamiento" en relación con una enfermedad o trastorno dado, incluye, pero sin limitación, inhibir la enfermedad o trastorno, por ejemplo, detener el desarrollo de la enfermedad o trastorno; aliviar la enfermedad o trastorno, por ejemplo, causar la regresión de la enfermedad o trastorno; o aliviar una afección causada o resultante de la enfermedad o trastorno, por ejemplo, mitigar, prevenir o tratar los síntomas de la enfermedad o trastorno. El término "prevención" en relación con una enfermedad o trastorno dado significa: prevenir el inicio del desarrollo de la enfermedad si no se hubiera producido, evitar que aparezca la enfermedad o trastorno en un sujeto que puede estar predispuesto a sufrir el trastorno o enfermedad pero que aún no se ha diagnosticado que tiene el trastorno o enfermedad, y/o prevenir el desarrollo de una enfermedad/trastorno adicional si ya está presente.
En una realización, la invención proporciona una composición farmacéutica para su uso en el tratamiento de la dermatitis atópica, por ejemplo, la dermatitis atópica de leve a moderada, como se ha establecido anteriormente. En una realización, el DGLA se administra al sujeto diariamente durante un período de al menos aproximadamente 2 semanas, al menos aproximadamente 4 semanas o al menos aproximadamente 8 semanas. En una realización relacionada, tras el tratamiento de acuerdo con la presente invención, por ejemplo durante un periodo de aproximadamente 1 a aproximadamente 12 semanas, de aproximadamente 1 a aproximadamente 8 semanas o de aproximadamente 1 a aproximadamente 4 semanas, el sujeto o grupo de sujetos exhibe uno o más de los siguientes resultados:
(a) una reducción en el área del eccema y la puntuación del índice de gravedad (EASI) en relación con el momento basal o con el control con placebo;
(b) una reducción en el porcentaje del área de un sitio anatómico afectado por dermatitis atópica en relación con el momento basal o el control;
(c) una reducción en la puntuación de la evaluación global del investigador en relación con el momento basal o con el control con placebo;
(d) una reducción en la intensidad del eritema, edema/población, supuración/costras, excoriación, liquenificación y/o sequedad en relación con el momento basal o con el control con placebo;
(e) una reducción en el eritema, edema/población, supuración/costras, excoriación, liquenificación y/o sequedad en relación con el momento basal o con el control con placebo;
(f) una reducción en el área de superficie corporal (BSA) afectada por dermatitis atópica en relación con el momento basal o con el control con placebo;
(g) una reducción en la pérdida de sueño en relación con el momento basal o con el control con placebo;
(h) una reducción en la aparición de prurito (picor) en relación con el momento basal o con el control con placebo;
(i) una reducción en la gravedad del prurito como un promedio de los tres días y/o noches anteriores en una
escala analógica visual;
(j) una reducción en la puntuación SCORAD en relación con el momento basal o con el control con placebo; (k) una medida de eccema (POEM) orientada al paciente mejorada en comparación con el momento basal o con el control con placebo;
(l) una reducción en el número de días en la semana anterior en los que el sujeto informó que la piel tenía picor debido a eccema;
(m) una reducción en el número de días en la semana anterior en que el sujeto informó que su sueño se había visto perturbado debido a su eccema;
(n) una reducción en el número de días en la semana anterior en los que el sujeto experimentó sangrado de la piel;
(o) una reducción en el número de días en la semana anterior en los que el sujeto experimentó exudación en la piel o supuración de líquido transparente;
(p) una reducción en el número de días en la semana anterior en los que la piel del sujeto se agrietó;
(q) una reducción en el número de días en la semana anterior en los que la piel del sujeto se descamó;
(r) una reducción en el número de días en la semana anterior en los que el sujeto experimentó piel seca;
(s) un aumento en la pérdida de agua transepidérmica en comparación con el momento basal o el control con placebo;
(t) un incremento en el DGLA total y libre en plasma en comparación con el valor basal;
(u) un incremento en la relación DGLA:AA en comparación con el momento basal o el control con placebo; y/o (v) una reducción en la presión arterial en comparación con el momento basal o con el control con placebo. En una realización, el uso comprende medir los niveles basales de uno o más marcadores o parámetros establecidos en (a) -(v) anteriores antes de la dosificación al sujeto o grupo de sujetos. En otra realización, los métodos comprenden administrar una composición como se desvela en el presente documento al sujeto después de determinar los niveles basales de uno o más marcadores o parámetros establecidos en (a) -(v), y, posteriormente, tomar una medición adicional de dichos uno o más marcadores.
En otra realización, tras el tratamiento, con una composición de la presente invención, por ejemplo durante un periodo de aproximadamente 1 a aproximadamente 12 semanas, de aproximadamente 1 a aproximadamente 8 semanas o de aproximadamente 1 a aproximadamente 4 semanas, el sujeto o grupo de sujetos exhiben cualquiera de 2 o más de, cualquiera de 3 o más de, cualquiera de 4 o más de, cualquiera de 5 o más de, cualquiera de 6 o más de, cualquiera de 7 o más de, cualquiera de 8 o más de, cualquiera de 9 o más de, cualquiera de 10 o más de, cualquiera de 11 o más de, cualquiera de 12 o más de, cualquiera de 13 o más de, cualquiera de 14 o más de, cualquiera de 15 o más de, cualquiera de 16 o más de, cualquiera de 17 o más de, cualquiera de 18 o más de, cualquiera de 19 o más de, cualquiera de 20 o más de, 21 o más de los 22 resultados (a) - (v) descritos inmediatamente antes.
En otra realización, tras el tratamiento, con una composición de la presente invención, el sujeto o grupo de sujetos exhibe uno o más de los siguientes resultados:
(a) una reducción en el área del eccema y la puntuación del índice de gravedad (EASI) en relación con el momento basal o con el control con placebo de al menos aproximadamente 5 %, al menos aproximadamente el 10 %, al menos aproximadamente el 15 %, al menos aproximadamente el 20 %, al menos aproximadamente el 25 %, al menos aproximadamente el 30 %, al menos aproximadamente el 35 %, al menos aproximadamente el 40 %, al menos aproximadamente el 45 %, al menos aproximadamente el 50 %, al menos aproximadamente el 55 %, al menos aproximadamente el 60 %, al menos aproximadamente el 65 %, al menos aproximadamente el 70 %, al menos aproximadamente el 75 %, al menos aproximadamente el 80 %, al menos aproximadamente el 85 %, al menos aproximadamente el 90 % o al menos aproximadamente el 95 %;
(b) una reducción en el porcentaje del área de un sitio anatómico afectado por dermatitis atópica con respecto al valor basal o control de al menos aproximadamente el 5 %, al menos aproximadamente el 10%, al menos aproximadamente el 15 %, al menos aproximadamente el 20 %, al menos aproximadamente el 25 %, al menos aproximadamente el 30 %, al menos aproximadamente el 35 %, al menos aproximadamente el 40 %, al menos
aproximadamente el 45 %, al menos aproximadamente el 50 %, al menos aproximadamente el 55 % , al menos aproximadamente el 60 %, al menos aproximadamente el 65 %, al menos aproximadamente el 70 % , al menos aproximadamente el 75 %, al menos aproximadamente el 80 %, al menos aproximadamente el 85 % , al menos aproximadamente el 90 % o al menos aproximadamente el 95 %;
(c) una reducción en la puntuación de evaluación global del investigador en relación con el momento basal o el control con placebo de al menos aproximadamente el 5 %, al menos aproximadamente el 10%, al menos aproximadamente el 15 %, al menos aproximadamente el 20 %, al menos aproximadamente el 25 % , al menos aproximadamente el 30 %, al menos aproximadamente el 35 %, al menos aproximadamente el 40 % , al menos aproximadamente el 45 %, al menos aproximadamente el 50 %, al menos aproximadamente el 55 % , al menos aproximadamente el 60 %, al menos aproximadamente el 65 %, al menos aproximadamente el 70 % , al menos aproximadamente el 75 %, al menos aproximadamente el 80 %, al menos aproximadamente el 85 % , al menos aproximadamente el 90 % o al menos aproximadamente el 95 %;
(d) una reducción en la intensidad del eritema, edema/población, supuración/costras, excoriación, liquenificación y/o sequedad en relación con el momento basal o con el control con placebo de al menos aproximadamente el 5 %, al menos aproximadamente el 10%, al menos aproximadamente el 15%, al menos aproximadamente el 20 %, al menos aproximadamente el 25 %, al menos aproximadamente el 30 %, al menos aproximadamente el 35 %, al menos aproximadamente el 40 %, al menos aproximadamente el 45 %, al menos aproximadamente el 50 %, al menos aproximadamente el 55 %, al menos aproximadamente el 60 %, al menos aproximadamente el 65 %, al menos aproximadamente el 70 %, al menos aproximadamente el 75 %, al menos aproximadamente el 80 %, al menos aproximadamente el 85 %, al menos aproximadamente el 90 % o al menos aproximadamente el 95 %;
(e) una reducción en el eritema, edema/población, supuración/costras, excoriación, liquenificación y/o sequedad en relación con el momento basal o con el control con placebo de al menos aproximadamente el 5 %, al menos aproximadamente el 10 %, al menos aproximadamente el 15 %, al menos aproximadamente el 20 %, al menos aproximadamente el 25 %, al menos aproximadamente el 30 %, al menos aproximadamente el 35 %, al menos aproximadamente el 40 %, al menos aproximadamente el 45 %, al menos aproximadamente el 50 %, al menos aproximadamente el 55 %, al menos aproximadamente el 60 %, al menos aproximadamente el 65 %, al menos aproximadamente el 70 %, al menos aproximadamente el 75 %, al menos aproximadamente el 80 %, al menos aproximadamente el 85 %, al menos aproximadamente el 90 % o al menos aproximadamente el 95 %;
(f) una reducción en el área de superficie corporal (BSA) afectada por dermatitis atópica en relación con el momento basal o con el control con placebo de al menos aproximadamente el 5 %, al menos aproximadamente el 10 %, al menos aproximadamente el 15 %, al menos aproximadamente el 20 %, al menos aproximadamente el 25 %, al menos aproximadamente el 30 %, al menos aproximadamente el 35 %, al menos aproximadamente el 40 %, al menos aproximadamente el 45 %, al menos aproximadamente el 50 %, al menos aproximadamente el 55 %, al menos aproximadamente el 60 %, al menos aproximadamente el 65 %, al menos aproximadamente el 70 %, al menos aproximadamente el 75 %, al menos aproximadamente el 80 %, al menos aproximadamente el 85 %, al menos aproximadamente el 90 % o al menos aproximadamente el 95 %;
(g) una reducción en la pérdida de sueño en relación con el momento basal o el control con placebo de al menos aproximadamente el 5%, al menos aproximadamente el 10%, al menos aproximadamente el 15%, al menos aproximadamente el 20 %, al menos aproximadamente el 25 %, al menos aproximadamente el 30 %, al menos aproximadamente el 35 %, al menos aproximadamente el 40 %, al menos aproximadamente el 45 %, al menos aproximadamente el 50 %, al menos aproximadamente el 55 %, al menos aproximadamente el 60 %, al menos aproximadamente el 65 %, al menos aproximadamente el 70 %, al menos aproximadamente el 75 %, al menos aproximadamente el 80 %, al menos aproximadamente el 85 %, al menos aproximadamente el 90 % o al menos aproximadamente el 95 %;
(h) una reducción en la aparición de prurito (picor) en relación con el momento basal o el control con placebo de al menos aproximadamente el 5 %, al menos aproximadamente el 10 %, al menos aproximadamente el 15 %, al menos aproximadamente el 20 %, al menos aproximadamente el 25 %, al menos aproximadamente el 30 %, al menos aproximadamente el 35 %, al menos aproximadamente el 40 %, al menos aproximadamente el 45 %, al menos aproximadamente el 50 %, al menos aproximadamente el 55 %, al menos aproximadamente el 60 %, al menos aproximadamente el 65 %, al menos aproximadamente el 70 %, al menos aproximadamente el 75 %, al menos aproximadamente el 80 %, al menos aproximadamente el 85 %, al menos aproximadamente el 90 % o al menos aproximadamente el 95 %;
(i) una reducción en la gravedad del prurito como promedio de los tres días y/o noches anteriores en una escala analógica visual de al menos aproximadamente el 5 %, al menos aproximadamente el 10%, al menos aproximadamente el 15 %, al menos aproximadamente el 20 %, al menos aproximadamente el 25 %, al menos aproximadamente el 30 %, al menos aproximadamente el 35 %, al menos aproximadamente el 40 %, al menos aproximadamente el 45 %, al menos aproximadamente el 50 %, al menos aproximadamente el 55 %, al menos aproximadamente el 60 %, al menos aproximadamente el 65 %, al menos aproximadamente el 70 %, al menos
aproximadamente el 75 %, al menos aproximadamente el 80 %, al menos aproximadamente el 85 %, al menos aproximadamente el 90 % o al menos aproximadamente el 95 %;
(j) una reducción en la puntuación SCORAD en relación con el momento basal o el control con placebo de al menos aproximadamente el 5 %, al menos aproximadamente el 10%, al menos aproximadamente el 15%, al menos aproximadamente el 20 %, al menos aproximadamente el 25 %, al menos aproximadamente el 30 %, al menos aproximadamente el 35 %, al menos aproximadamente el 40 %, al menos aproximadamente el 45 %, al menos aproximadamente el 50 %, al menos aproximadamente el 55 %, al menos aproximadamente el 60 %, al menos aproximadamente el 65 %, al menos aproximadamente el 70 %, al menos aproximadamente el 75 %, al menos aproximadamente el 80 %, al menos aproximadamente el 85 %, al menos aproximadamente el 90 % o al menos aproximadamente el 95 %;
(k) una medida de eccema (POEM) orientada al paciente mejorada en comparación con el momento basal o el control con placebo de al menos aproximadamente el 5 %, al menos aproximadamente el 10 %, al menos aproximadamente el 15 %, al menos aproximadamente el 20 %, al menos aproximadamente el 25 %, al menos aproximadamente el 30 %, al menos aproximadamente el 35 %, al menos aproximadamente el 40 %, al menos aproximadamente el 45 %, al menos aproximadamente el 50 %, al menos aproximadamente el 55 %, al menos aproximadamente el 60 %, al menos aproximadamente el 65 %, al menos aproximadamente el 70 %, al menos aproximadamente el 75 %, al menos aproximadamente el 80 %, al menos aproximadamente el 85 %, al menos aproximadamente el 90 % o al menos aproximadamente el 95 %;
(l) una reducción en el número de días en la semana anterior en los que el sujeto informó que la piel le picaba debido a un eccema de al menos aproximadamente el 5 %, al menos aproximadamente el 10%, al menos aproximadamente el 15 %, al menos aproximadamente el 20 %, al menos aproximadamente el 25 %, al menos aproximadamente el 30 %, al menos aproximadamente el 35 %, al menos aproximadamente el 40 %, al menos aproximadamente el 45 %, al menos aproximadamente el 50 %, al menos aproximadamente el 55 %, al menos aproximadamente el 60 %, al menos aproximadamente el 65 %, al menos aproximadamente el 70 %, al menos aproximadamente el 75 %, al menos aproximadamente el 80 %, al menos aproximadamente el 85 %, al menos aproximadamente el 90 % o al menos aproximadamente el 95 %;
(m) una reducción en el número de días en la semana anterior en los que el sujeto informó que su sueño se había visto perturbado debido al eccema de al menos aproximadamente el 5 %, al menos aproximadamente el 10 %, al menos aproximadamente el 15 %, al menos aproximadamente el 20 %, al menos aproximadamente el 25 %, al menos aproximadamente el 30 %, al menos aproximadamente el 35 %, al menos aproximadamente el 40 %, al menos aproximadamente el 45 %, al menos aproximadamente el 50 %, al menos aproximadamente el 55 %, al menos aproximadamente el 60 %, al menos aproximadamente el 65 %, al menos aproximadamente el 70 %, al menos aproximadamente el 75 %, al menos aproximadamente el 80 %, al menos aproximadamente el 85 %, al menos aproximadamente el 90 % o al menos aproximadamente el 95 %;
(n) una reducción en el número de días en la semana anterior en los que el sujeto experimentó sangrado de la piel de al menos aproximadamente el 5 %, al menos aproximadamente el 10 %, al menos aproximadamente el 15 %, al menos aproximadamente el 20 %, al menos aproximadamente el 25 %, al menos aproximadamente el 30 %, al menos aproximadamente el 35 %, al menos aproximadamente el 40 %, al menos aproximadamente el 45 %, al menos aproximadamente el 50 %, al menos aproximadamente el 55 %, al menos aproximadamente el 60 %, al menos aproximadamente el 65 %, al menos aproximadamente el 70 %, al menos aproximadamente el 75 %, al menos aproximadamente el 80 %, al menos aproximadamente el 85 %, al menos aproximadamente el 90 % o al menos aproximadamente el 95 %;
(o) una reducción en el número de días en la semana anterior en los que el sujeto experimentó exudado en la piel o supuración de líquido transparente de al menos aproximadamente el 5 %, al menos aproximadamente el 10 %, al menos aproximadamente el 15 %, al menos aproximadamente el 20 %, al menos aproximadamente el 25 %, al menos aproximadamente el 30 %, al menos aproximadamente el 35 %, al menos aproximadamente el 40 %, al menos aproximadamente el 45 %, al menos aproximadamente el 50 %, al menos aproximadamente el 55 %, al menos aproximadamente el 60 %, al menos aproximadamente el 65 %, al menos aproximadamente el 70 %, al menos aproximadamente el 75 %, al menos aproximadamente el 80 %, al menos aproximadamente el 85 %, al menos aproximadamente el 90 % o al menos aproximadamente el 95 %;
(p) una reducción en el número de días en la semana anterior en los que la piel del sujeto se agrietó de al menos aproximadamente el 5 %, al menos aproximadamente el 10 %, al menos aproximadamente el 15 %, al menos aproximadamente el 20 %, al menos aproximadamente el 25 %, al menos aproximadamente el 30 %, al menos aproximadamente el 35 %, al menos aproximadamente el 40 %, al menos aproximadamente el 45 %, al menos aproximadamente el 50 %, al menos aproximadamente el 55 %, al menos aproximadamente el 60 %, al menos aproximadamente el 65 %, al menos aproximadamente el 70 %, al menos aproximadamente el 75 %, al menos aproximadamente el 80 %, al menos aproximadamente el 85 %, al menos aproximadamente el 90 % o al menos aproximadamente el 95 %;
(q) una reducción en el número de días en la semana anterior en los que la piel del sujeto se descamó de al menos aproximadamente el 5 %, al menos aproximadamente el 10 %, al menos aproximadamente el 15 %, al menos aproximadamente el 20 %, al menos aproximadamente el 25 %, al menos aproximadamente el 30 %, al menos aproximadamente el 35 %, al menos aproximadamente el 40 %, al menos aproximadamente el 45 %, al menos aproximadamente el 50 %, al menos aproximadamente el 55 %, al menos aproximadamente el 60 %, al menos aproximadamente el 65 %, al menos aproximadamente el 70 %, al menos aproximadamente el 75 %, al menos aproximadamente el 80 %, al menos aproximadamente el 85 %, al menos aproximadamente el 90 % o al menos aproximadamente el 95 %;
(r) una reducción en el número de días en la semana anterior en los que el sujeto experimentó piel seca de al menos aproximadamente el 5 %, al menos aproximadamente el 10%, al menos aproximadamente el 15%, al menos aproximadamente el 20 %, al menos aproximadamente el 25 %, al menos aproximadamente el 30 %, al menos aproximadamente el 35 %, al menos aproximadamente el 40 %, al menos aproximadamente el 45 %, al menos aproximadamente el 50 %, al menos aproximadamente el 55 %, al menos aproximadamente el 60 %, al menos aproximadamente el 65 %, al menos aproximadamente el 70 %, al menos aproximadamente el 75 %, al menos aproximadamente el 80 %, al menos aproximadamente el 85 %, al menos aproximadamente el 90 % o al menos aproximadamente el 95 %;
(s) un aumento en la pérdida de agua transepidérmica en comparación con el momento basal o el control con placebo de al menos aproximadamente el 5 %, al menos aproximadamente el 10 %, al menos aproximadamente el 15 %, al menos aproximadamente el 20 %, al menos aproximadamente el 25 %, al menos aproximadamente el 30 %, al menos aproximadamente el 35 %, al menos aproximadamente el 40 %, al menos aproximadamente el 45 %, al menos aproximadamente el 50 %, al menos aproximadamente el 55 %, al menos aproximadamente el 60 %, al menos aproximadamente el 65 %, al menos aproximadamente el 70 %, al menos aproximadamente el 75 %, al menos aproximadamente el 80 %, al menos aproximadamente el 85 %, al menos aproximadamente el 90 % o al menos aproximadamente el 95 %;
(t) un aumento en el DGLA total y libre en plasma en comparación con el momento basal de al menos aproximadamente el 5 %, al menos aproximadamente el 10 %, al menos aproximadamente el 15 %, al menos aproximadamente el 20 %, al menos aproximadamente el 25 %, al menos aproximadamente el 30 %, al menos aproximadamente el 35 %, al menos aproximadamente el 40 %, al menos aproximadamente el 45 %, al menos aproximadamente el 50 %, al menos aproximadamente el 55 %, al menos aproximadamente el 60 %, al menos aproximadamente el 65 %, al menos aproximadamente el 70 %, al menos aproximadamente el 75 %, al menos aproximadamente el 80 %, al menos aproximadamente el 85 %, al menos aproximadamente el 90 % o al menos aproximadamente el 95 %; y/o
(u) un aumento en la relación DGLAAA en comparación con el momento basal o el control con placebo de al menos aproximadamente el 5 %, al menos aproximadamente el 10%, al menos aproximadamente el 15%, al menos aproximadamente el 20 %, al menos aproximadamente el 25 %, al menos aproximadamente el 30 %, al menos aproximadamente el 35 %, al menos aproximadamente el 40 %, al menos aproximadamente el 45 %, al menos aproximadamente el 50 %, al menos aproximadamente el 55 %, al menos aproximadamente el 60 %, al menos aproximadamente el 65 %, al menos aproximadamente el 70 %, al menos aproximadamente el 75 %, al menos aproximadamente el 80 %, al menos aproximadamente el 85 %, al menos aproximadamente el 90 % o al menos aproximadamente el 95 %; y/o
(v) una reducción en la presión arterial media de al menos aproximadamente el 5 %, al menos aproximadamente el 10 %, al menos aproximadamente el 15 %, al menos aproximadamente el 20 %, al menos aproximadamente el 25 %, al menos aproximadamente el 30 %, al menos aproximadamente el 35 %, al menos aproximadamente el 40 %, al menos aproximadamente el 45 %, al menos aproximadamente el 50 %, al menos aproximadamente el 55 %, al menos aproximadamente el 60 %, al menos aproximadamente el 65 %, al menos aproximadamente el 70 %, al menos aproximadamente el 75 %, al menos aproximadamente el 80 %, al menos aproximadamente el 85 %, al menos aproximadamente el 90 % o al menos aproximadamente el 95 %.
En otra realización, tras el tratamiento con una composición de la presente invención después de la administración de una única dosis o administración de dosis múltiples, por ejemplo durante un periodo de aproximadamente 1 a aproximadamente 12 semanas, de aproximadamente 1 a aproximadamente 8 semanas o de aproximadamente 1 a aproximadamente 4 semanas, el sujeto o grupo de sujetos exhiben cualquiera de 2 o más de, cualquiera de 3 o más de, cualquiera de 4 o más de, cualquiera de 5 o más de, cualquiera de 6 o más de, cualquiera de 7 o más de, cualquiera de 8 o más de, cualquiera de 9 o más de, cualquiera de 10 o más de, cualquiera de 11 o más de, cualquiera de 12 o más de, cualquiera de 13 o más de, cualquiera de 14 o más de, cualquiera de 15 o más de, cualquiera de 16 o más de, cualquiera de 17 o más de, cualquiera de 18 o más de, cualquiera de 19 o más de, cualquiera de 20 o más de, 21 o más de los 22 resultados (a) -(v) descritos inmediatamente antes.
Tras el tratamiento de un sujeto o grupo de sujetos (alimentados o en ayunas) con una composición que comprende aproximadamente 200 mg de DGLA a aproximadamente 8.000 mg de DGLA (administrado como una o más unidades de dosificación, por ejemplo, como unidades de dosificación de 500 mg o 1 g equivalente a dosis diarias
totales de DGLA de aproximadamente 500 mg, aproximadamente 1.000 mg, aproximadamente 2.000 mg, aproximadamente 3.000 mg, aproximadamente 4.000 mg, aproximadamente 5.000 mg, aproximadamente 6.000 mg, aproximadamente 7.000 mg o aproximadamente 8.000 mg) y después de la administración de una dosis única o después de la administración de dosis múltiples, el sujeto o grupo de sujetos puede exhibir uno o más de los siguientes resultados:
(a) una Cmáx (o media o mediana de la Cmáx) de DGLA de aproximadamente 400 ng/ml a aproximadamente 4.500 ng/ml, de aproximadamente 500 ng/ml a aproximadamente 3.400 ng/ml, de aproximadamente 600 ng/ml a aproximadamente 3.300 ng/ml, de aproximadamente 700 ng/ml a aproximadamente 3.200 ng/ml, por ejemplo aproximadamente 900 ng/ml, aproximadamente 1.000 ng/ml, aproximadamente 1.100 ng/ml, aproximadamente 1.200 ng/ml, aproximadamente 1.300 ng/ml, aproximadamente 1.400 ng/ml, aproximadamente 1.500 ng/ml, aproximadamente 1.600 ng/ml, aproximadamente 1.700 ng/ml, aproximadamente 1.800 ng/ml, aproximadamente 1.900 ng/ml, aproximadamente 2.000 ng/ml, aproximadamente 2.100 ng/ml, aproximadamente 2.200 ng/ml, aproximadamente 2.300 ng/ml, aproximadamente 2.400 ng/ml, aproximadamente 2.500 ng/ml, aproximadamente 2.600 ng/ml, aproximadamente 2.700 ng/ml, aproximadamente 2.800 ng/ml, aproximadamente 2.900 ng/ml, aproximadamente 3.000 ng/ml, aproximadamente 3.100 ng/ml, aproximadamente 3.200 ng/ml, aproximadamente 3.300 ng/ml, aproximadamente 3.400 ng/ml, aproximadamente 3.500 ng/ml, aproximadamente 3.600 ng/ml, aproximadamente 3.700 ng/ml, aproximadamente 3.800 ng/ml, aproximadamente 3.900 ng/ml, aproximadamente 4.000 ng/ml, aproximadamente 4.100 ng/ml, aproximadamente 4.200 ng/ml, aproximadamente 4.300 ng/ml, aproximadamente 4.400 ng/ml o aproximadamente 4.500 ng/ml;
(b) una Cmáx/dosis de DGLA (o media o mediana de la Cmáx/dosis) de aproximadamente 0,5 (1/kl) a aproximadamente 3 (1/kl), de aproximadamente 0,6 (1/kl) a aproximadamente 2,5 (1/kl) o de aproximadamente 0,7 (1/kl) a aproximadamente 2 (1/kl), por ejemplo aproximadamente 0,7 (1/kl), aproximadamente 0,8 (1/kl), aproximadamente 0,9 (1/kl), aproximadamente 1 (1/kl), aproximadamente 1,5 (1/kl), aproximadamente 1,6 (1/kl), aproximadamente 1,7 (1/kl) o aproximadamente 1,8 (1/kl);
(c) una AUC0-24 de DGLA libre (o media o mediana de la AUC0-24) de aproximadamente 1.500 ng • h/ml a aproximadamente 12.000 ng • h/ml, de aproximadamente 2.000 ng • h/ml a aproximadamente 11.000 ng • h/ml o de aproximadamente 2.500 ng • h/ml a aproximadamente 10.000 ng • h/ml, por ejemplo, de aproximadamente 1.000 ng • h/ml, aproximadamente 1.500 ng • h/ml, aproximadamente 2.000 ng • h/ml, aproximadamente 2.500 ng • h/ml, aproximadamente 3.000 ng • h/ml, aproximadamente 3.500 ng • h/ml, aproximadamente 4.000 ng • h/ml, aproximadamente 4.500 ng • h/ml, aproximadamente 5.000 ng • h/ml, aproximadamente 5.500 ng • h/ml, aproximadamente 6.000 ng • h/ml, aproximadamente 6.500 ng • h/ml, aproximadamente 7.000 ng • h/ml, aproximadamente 7.500 ng • h/ml, aproximadamente 8.000 ng • h/ml, aproximadamente 8.500 ng • h/ml, aproximadamente 9.000 ng • h/ml, aproximadamente 9.500 ng • h/ml, aproximadamente 10.000 ng • h/ml, aproximadamente 10.500 ng • h/ml, aproximadamente 11.000 ng • h/ml, aproximadamente 11.500 ng • h/ml o aproximadamente 12.000 ng • h/ml.
(d) una AUCü-24/dosis de DGLA libre (o media o mediana de AUCüWdosis) de aproximadamente 1,5 ng • h/ml a aproximadamente 10 h/kl, de aproximadamente 1,7 a aproximadamente 8 h/kl o de aproximadamente 2 a aproximadamente 6 h/kl, por ejemplo, de aproximadamente 2 h/kl, aproximadamente 2,5 h/kl, aproximadamente 3 h/kl, aproximadamente 3,5 h/kl, aproximadamente 4 h/kl, aproximadamente 4,5 h/kl, aproximadamente 5 h/kl o aproximadamente 5,5 h/kl;
(e) un tmáx de DGLA libre (h) de aproximadamente 2 a aproximadamente 10 horas, de aproximadamente 3 a aproximadamente 8 horas, por ejemplo aproximadamente 3 horas, aproximadamente 4 horas, aproximadamente 5 horas, aproximadamente 6 horas, aproximadamente 7 horas o aproximadamente 8 horas;
(f) una Cmáx de DGLA total (o media o mediana de la Cmáx de DGLA total) de aproximadamente 4.000 ng/ml a aproximadamente 45.000 ng/ml, de aproximadamente 5.000 ng/ml a aproximadamente 34.000 ng/ml, de aproximadamente 6.000 ng/ml a aproximadamente 33.000 ng/ml o de aproximadamente 7.000 ng/ml a aproximadamente 32.000 ng/ml, por ejemplo, de aproximadamente 7.000 ng/ml, aproximadamente 7.200 ng/ml, aproximadamente 7.500 ng/ml, aproximadamente 8.000 ng/ml, aproximadamente 8.500 ng/ml, aproximadamente 9.000 ng/ml, aproximadamente 9.500 ng/ml, aproximadamente 10.000 ng/ml, aproximadamente 11.000 ng/ml, aproximadamente 12.000 ng/ml, aproximadamente 13.000 ng/ml, aproximadamente 14.000 ng/ml, aproximadamente 15.000 ng/ml, aproximadamente 16.000 ng/ml, aproximadamente 17.000 ng/ml, aproximadamente 18.000 ng/ml, aproximadamente 19.000 ng/ml, aproximadamente 20.000 ng/ml, aproximadamente 21.000 ng/ml, aproximadamente 22.000 ng/ml, aproximadamente 23.000 ng/ml, aproximadamente 24.000 ng/ml, aproximadamente 25.000 ng/ml, aproximadamente 26.000 ng/ml, aproximadamente 27.000 ng/ml, aproximadamente 28.000 ng/ml, aproximadamente 29.000 ng/ml, aproximadamente 30.000 ng/ml, aproximadamente 31.000 ng/ml, aproximadamente 32.000 ng/ml, aproximadamente 33.000 ng/ml, aproximadamente 34.000 ng/ml o aproximadamente 35.000 ng/ml;
(g) una Cmáx/dosis de DGLA total (o media o mediana de la Cmáx/dosis de DGLA total) de aproximadamente 2 (1/kl) a aproximadamente 25 (1/kl), de aproximadamente 4 (1/kl) a aproximadamente 20 (1/kl) o de
aproximadamente 5 (1/kl) a aproximadamente 17 (1/kl), por ejemplo aproximadamente 6 (1/kl), aproximadamente 9 (1/kl), aproximadamente 14 (1/kl) o aproximadamente 16 (l/kl);
(h) una AUC0-24 de DGLA total (o media o mediana de la AUC0-24 de DGLAtotal) de aproximadamente 15.000 ng • h/ml a aproximadamente 900.000 ng • h/ml, de aproximadamente 20.000 ng • h/ml a aproximadamente 250.000 ng • h/ml o de aproximadamente 25.000 ng • h/ml a aproximadamente 225.000 ng • h/ml, por ejemplo, de aproximadamente 40.000 ng • h/ml, aproximadamente 210.000 ng • h/ml, aproximadamente 215.000 ng • h/ml o aproximadamente 435.000 ng • h/ml;
(i) una AUCü-24/dosis de DGLA total (o media o mediana de la AUCüWdosis de DGLA total) de aproximadamente 50 a aproximadamente 400 h/kl, de aproximadamente 60 a aproximadamente 250 h/kl o de aproximadamente 70 a aproximadamente 225 h/kl, por ejemplo, de aproximadamente 80 h/kl, aproximadamente 100 h/kl, aproximadamente 110 h/kl o aproximadamente 215 h/kl;
(k) un tmáx de DGLA total (h) de aproximadamente 2 a aproximadamente 25 horas o de aproximadamente 3 a aproximadamente 20 horas, por ejemplo aproximadamente 8 horas, aproximadamente 10 horas o aproximadamente 18 horas;
(k) una relación entre la Cmáx de DGLA total y la Cmáx de DGLA libre de aproximadamente 5:1 a aproximadamente 12:1, de aproximadamente 6:1 a aproximadamente 10:1 o de aproximadamente 7:1 a aproximadamente 9:1, por ejemplo aproximadamente 7,7:1, aproximadamente 8,6:1, por ejemplo, aproximadamente 8.8:1 o aproximadamente 9,8:1;
(l) un nivel plasmático de DGLA libre en estado de equilibrio (Cprom) o la media o mediana del nivel plasmático de DGLA libre en estado de equilibrio (Cprom), después de 1 a aproximadamente 30, de 1 a aproximadamente 28, de 1 a aproximadamente 14 o de 1 a aproximadamente 10 días consecutivos de administración diaria, de hasta aproximadamente 2.000 ng/ml, hasta aproximadamente 750 ng/ml o hasta aproximadamente 700 ng/ml, por ejemplo, aproximadamente 385 ng/ml o aproximadamente 675 ng/ml;
(m) un nivel plasmático de DGLA total en estado de equilibrio (Cprom) o la media o mediana del nivel plasmático de DGLA total en estado de equilibrio (Cprom), después de 1 a aproximadamente 30, de 1 a aproximadamente 28, de 1 a aproximadamente 14 o de 1 a aproximadamente 10 días consecutivos de administración diaria, hasta 250.000 ng/ml, hasta 180.000 ng/ml, hasta 150.000 ng/ml, hasta 125.000 ng/ml o hasta 100.000 ng/ml; y/o
(n) una relación entre el DGLA libre en plasma y el en la piel (por ejemplo, como se mide en el fluido de las ampollas de la piel) de aproximadamente 0,2:1 a aproximadamente 5:1, de aproximadamente 0,5:1 a aproximadamente 2,5:1 o de aproximadamente 0,6:1 a aproximadamente 1,5:1. En otra realización, tras el tratamiento, con una composición de la presente invención, por ejemplo durante un periodo de aproximadamente 1 a aproximadamente 12 semanas, de aproximadamente 1 a aproximadamente 8 semanas o de aproximadamente 1 a aproximadamente 4 semanas, el sujeto o grupo de sujetos exhiben cualquiera de 2 o más de, cualquiera de 3 o más de, cualquiera de 4 o más de, cualquiera de 5 o más de, cualquiera de 6 o más de, cualquiera de 7 o más de, cualquiera de 8 o más de, cualquiera de 9 o más de, cualquiera de 10 o cualquiera de 11 o más de, cualquiera de 12 o más de, cualquiera de 13 o más de, cualquiera de 14 o más de, cualquiera de 15 o más de, cualquiera de 16 o más de, cualquiera de 17 o más de, cualquiera de 18 o más de, cualquiera de 19 o más de, cualquiera de 20 o más de, cualquier 21 más de los 22 resultados (a)- (n) descritos inmediatamente antes.
Tras el tratamiento de un sujeto en ayunas o alimentados o grupo de sujetos en ayunas o alimentados con una composición que comprende aproximadamente 200 mg de DGLA a aproximadamente 8.000 mg de DGLA (administrado como una o más unidades de dosificación, por ejemplo, como unidades de dosificación de 500 mg o 1 g equivalente a dosis diarias totales de DGLA de aproximadamente 500 mg, aproximadamente 1.000 mg, aproximadamente 2.000 mg, aproximadamente 3.000 mg, aproximadamente 4.000 mg, aproximadamente 5.000 mg, aproximadamente 6.000 mg, aproximadamente 7.000 mg o aproximadamente 8.000 mg) y después de la administración de una dosis única o después de la administración de dosis múltiples, el sujeto o grupo de sujetos puede exhibir uno o más de los siguientes resultados:
(a) una relación de Cmáx de DGLA libre en ayunas:alimentado entre aproximadamente 1:1 y aproximadamente 5:1, por ejemplo de aproximadamente 2,5:1, de aproximadamente 3:1 o aproximadamente 3,5:1;
(b) una relación entre la AUC0-24 de DGLA libre en ayunas:alimentado de entre aproximadamente 1:1 y aproximadamente 5:1, por ejemplo de aproximadamente 1,5:1, de aproximadamente 2:1 o de aproximadamente 2,5:1;
(c) una relación entre la Cmáx de DGLA total en ayunas:alimentado de entre aproximadamente 1:1 y aproximadamente 5:1, por ejemplo de aproximadamente 1:1, de aproximadamente 1,5:1 o aproximadamente 2:1; y/o
(d) una relación entre la AUC0-24 de DGLA total en ayunas: alimentado de entre aproximadamente 1:1 y aproximadamente 5:1, por ejemplo de aproximadamente 1,5:1, de aproximadamente 2:1 o de aproximadamente 2,5:1.
En una realización, una composición de la invención que contiene DGLA comprende la siguiente huella de ácidos grasos:

En una realización, una composición de la invención que contiene DGLA comprende la siguiente huella de ácidos grasos:

Una composición ilustrativa que contiene DGLA de la invención comprende la siguiente huella de ácidos grasos:

En una realización, una composición de la invención que contiene DGLA comprende la siguiente huella de ácidos grasos:


continuación

Ejemplos
Ejemplo 1
Se prepararon tres lotes de composiciones farmacéuticas que comprenden DGLA (con 2.000 pm de dl-alfa tocoferol) cargadas en cápsulas de gelatina como se muestra en la Tabla 1.
Tabla 1.

Las cubiertas de las cápsulas incluyeron los siguientes excipientes: gelatina, agua purificada, glicerol, dióxido de titanio y los auxiliares de procesamiento lecitina y triglicéridos de cadena media.
También se prepararon lotes adicionales de cápsulas de DGLA, incluyendo AAG DGLA (estabilizado con un dl-alfa tocoferol nominal de 2.000 ppm) en cápsulas que contenían gelatina, polisorb o mezcla de glicerol/polisorb, agua purificada, dióxido de titanio y los auxiliares de procesamiento lecitina y triglicéridos de cadena media como se muestra en la Tabla 2.
Tabla 2.

Las composiciones de la cubierta de cápsula para cada uno de los lotes se muestran a continuación en las Tablas 3 y 4.
Tabla 3.

Tabla 4.

Se realizó una prueba de estabilidad de las cápsulas anteriores. Las cápsulas de cada lote se mantuvieron durante hasta 6 meses y se evaluaron utilizando un protocolo de prueba de disgregación y disolución USP 2040 cualitativa o cuantitativa. Los resultados se muestran en las Tablas 5 - 7.

T l Pr n li ri D LA

Tabla 7.

continuación

Como se ha indicado anteriormente, hubo una ralentización de la velocidad de disolución en agua con el tiempo para las cápsulas formuladas con glicerol y gelatina bovina ácida estándar (E09726/01 y E09727). Hubo una tasa de liberación de DGLA de más de 30 minutos después de 6 meses a 40 °C/75 % de HR en fluido gástrico simulado (pH 1,2, pepsina).
Una tasa de liberación de DGLA de menos de 30 minutos después de 6 meses de 40 °C/75 % de HR solo se logró en fluido gástrico simulado (pH 1,2, pepsina) con cápsulas que contienen gelatina de hueso de lima con un peso molecular más bajo (Pm) (E09777/02).
Hubo un aumento significativo en la formación de glicéridos de DGLA con el tiempo en las cubiertas de las cápsulas de DGLA que contienen glicerol (Tabla 4). Esto dependía de la temperatura con las concentraciones más altas de DGLA formadas a 40 °C 75 % de HR.
Polisorb se usa habitualmente como plastificante hidrofílico para limitar el intercambio entre los medios de llenado de la cápsula y la cubierta. D-Sorbitol y 1,4-sorbitán tienen un PM más alto que el glicerol, lo que limita su movilidad a través de la cubierta de gelatina. A pesar de esto, todavía había interacción de D-sorbitol y 1,4-sorbitano para formar ésteres de AAG de DGLA en LOS lotes E09777 1/2 y 3.
No hubo reducción en el índice de acidez del DGLA para lotes formulados con D-sorbitol y 1,4-sorbitán (E09777 1/02/3) mientras que hubo una reducción en el índice de acidez para E09778 formulado con glicerol.
No hubo ralentización en la velocidad de disolución en agua con el tiempo para las cápsulas formuladas con D-sorbitol y 1,4-sorbitano (E09777/03). La velocidad de liberación de DGLA fue inferior a 30 minutos en agua después de 3 meses a 40 °C/75 % de HR.
Ejemplo 2
Se realizará un estudio aleatorizado, doble ciego, controlado con placebo y de fase II para evaluar la eficacia y la seguridad del DGLA administrado por vía oral a pacientes con dermatitis atópica de moderada a grave. En este estudio se incluirán aproximadamente 100 sujetos masculinos o femeninos, mayores de 18 años, con dermatitis atópica (DA) de moderada a grave.
La dermatitis atópica es un trastorno cutáneo inflamatorio crónico caracterizado por la presencia de prurito, pápulas, liquenificación, excoriaciones, xerosis y exudación. La prevalencia de la DA ha aumentado en las últimas décadas, y afecta hasta al 20 por ciento de los niños pequeños y la mayoría de los casos comienzan en niños menores de 5 años. La mayoría de los casos mejoran en la edad adulta.
La DA es una enfermedad multifactorial, siendo la genética, el entorno y la respuesta inmunológica deteriorada los factores predominantes. Las células dendríticas, los linfocitos T, los macrófagos, los queratinocitos, los mastocitos y los eosinófilos desempeñan todos un papel en la DA liberando citocinas y quimiocinas proinflamatorias que inducen la respuesta inflamatoria característica de las lesiones de dermatitis atópica.
La mayoría de los tratamientos actualmente aprobados para la DA son tópicos e incluyen corticosteroides, pimecrolimus y tacrolimus. Los corticosteroides han sido el pilar principal del tratamiento para la DA y se encuentran disponibles diferentes potencias y formulaciones. Los corticosteroides tópicos son efectivos para el tratamiento de la DA, pero su uso está limitado por el potencial de efectos secundarios locales como atrofia de la piel y estrías. La absorción sistémica de corticosteroides también puede inducir diabetes, cataratas, osteoporosis y supresión del eje
hipotálamo-hipofisario. Además, la mejora transitoria a menudo es seguida por un brote de rebote al suspender el tratamiento. Otros tratamientos disponibles incluyen inhibidores tópicos de la calcineurina (por ejemplo, pimecrolimus, tacrolimus) y preparaciones de alquitrán de hulla. Se han notificado casos de linfoma en pacientes tratados con inhibidores de la calcineurina. Los pacientes con enfermedad más grave son tratados con fototerapia ultravioleta B y A o agentes orales, tales como corticosteroides, ciclosporina, micofenolato mofetilo, metotrexato y azatioprina.
El ácido dihomo-gamma-linolénico (DGLA) es un ácido graso esencial que se encuentra naturalmente en el cuerpo como el producto de la elongación de 2 carbonos del ácido gamma-linolénico (GLA). El DGLA es metabolizado por las enzimas ciclooxigenasa (COX) y lipoxigenasa (LPO) para formar eicosanoides, prostaglandinas (PG) de la serie 1 y también ácido graso hidroxilo como ácido 15-hidroxieicosatrienoico DGLA (15 HETrE).
HIPÓTESIS Y OBJETIVOS DEL ESTUDIO DS107G es superior al placebo para mejorar la puntuación EASI en pacientes con dermatitis atópica de moderada a grave.
Objetivo principal: Para comparar la eficacia de dos dosis de cápsulas de DS107G administradas por vía oral frente al placebo, en el tratamiento de pacientes adultos con dermatitis atópica de moderada a grave.
Objetivo secundario: Para evaluar la seguridad de dos dosis de cápsulas de DS107G administradas por vía oral frente al placebo, en pacientes adultos con dermatitis atópica de moderada a grave.
CRITERIOS DE VALORACIÓN DEL ESTUDIO
Criterio de valoración principal
Proporción de pacientes que alcanzan una IGA (evaluación global del investigador) de 0 (claro) o 1 (casi claro) y una disminución de al menos 2 puntos en la IGA la semana 8.
Criterios de valoración secundarios
Cambio con respecto al momento basal en la IGA la semana 2, 4 y 8;
Cambio con respecto al momento basal en el EASI (área de eccema e índice de gravedad) las semanas 2, 4 y 8; Proporción de pacientes que logran una disminución de al menos 1 punto en el IGA la semana 8;
Cambio con respecto al momento basal en la medida de eccema orientada al paciente (POEM) las semanas 2, 4 y 8; Cambio con respecto al momento basal en la puntuación del Índice de calidad de vida dermatológica (DLQI) las semanas 2, 4 y 8;
Cambio con respecto al momento basal en el SCORAD las semanas 2, 4 y 8;
Cambio con respecto al valor basal en la puntuación del prurito de la Escala Visual Analógica (VAS) del paciente las semanas 2, 4 y 8;
Cambio con respecto al momento basal en el área de superficie corporal (BSA) las semanas 2, 4 y 8;
Número de acontecimientos adversos emergentes durante el tratamiento (TEAE) en cada grupo de tratamiento; Criterios de valoración exploratorios
Cambio con respecto al momento basal en la pérdida de agua transepidérmica (TEWL) las semanas 2, 4 y 8 (solo sitios seleccionados);
Concentraciones plasmáticas basales de DGLA total y libre, las semanas 4 y 8;
Perfil basal de ácidos grasos totales en plasma, la semana 4 y la semana 8 (la muestra se conservará y se analizará en una fecha posterior);
DISEÑO DEL ESTUDIO
En este estudio multicéntrico, doble ciego, controlado con placebo y de fase IIa se incluirán aproximadamente 100 pacientes con dermatitis atópica de moderada a grave. Todos los sujetos firmarán un consentimiento informado y se someterán a una selección para determinar la elegibilidad para el estudio. Los sujetos serán asignados al azar (1:1) en la visita basal para recibir 2 g de DS107G oral (cápsulas de DGLA proporcionadas como cápsulas de gelatina blanda ovales opacas que contienen 500 mg de DGLA), 1 g de DS107G o placebo una vez al día durante 8 semanas en ayunas. La inscripción se estratificará según la gravedad de la enfermedad (basada en IGA) y se reclutará un máximo del 30 % de los pacientes con dermatitis atópica leve (según lo definido por una puntuación de IGA de 2). Los sujetos acudirán a la clínica en 6 ocasiones: en el visita de selección, la visita basal, la semana 2, la semana 4, la semana 8 (finalización /finalización anticipada del tratamiento) y la semana 10 (seguimiento). Todos los sujetos saldrán del estudio en la visita de la semana 10. La variable de eficacia primaria será la proporción de pacientes que alcanzan una IGA de 0 (claro) o 1 (casi claro) y una disminución de al menos 2 puntos en la IGA la semana 8. Las
variables secundarias de eficacia incluirán una IGA en otras visitas, prurito (obtenido de la escala analógica visual SCORAD), EASI, BSA, POEM, DLQI, SCORAD y TEWL (solo para centros seleccionados). La seguridad se evaluará a través de los acontecimientos adversos, la exploración física, las constantes vitales y las pruebas de laboratorio de seguridad (incluidas pruebas de embarazo para mujeres en edad fértil). Las muestras para farmacocinética se obtendrán en las visitas basal (día 0), de la semana 4 y la semana 8 para medir los niveles mínimos plasmáticos de DGLA total y libre. Se retendrán muestras de plasma separadas para el análisis posterior del perfil de ácidos grasos totales y el perfil de interleucinas.
POBLACIÓN DEL ESTUDIO
En este estudio se pueden incluir aproximadamente 100 sujetos con dermatitis atópica de moderada a severa según la puntuación IGA y una BSA de, como mínimo, 10 %. Los sujetos serán varones o mujeres, de 18 años o más. Criterios de inclusión:
Sujeto masculino o femenino de 18 años o más el día de la firma del formulario de consentimiento informado (ICF). Diagnóstico clínicamente confirmado de dermatitis atópica activa según los criterios de Hanifin y Rajka (Apéndice G). Dermatitis atópica de moderada a grave al inicio del estudio según lo definido por una IGA de, como mínimo, 3 en la visita basal.
Dermatitis atópica que cubre como mínimo el 10 % del área de la superficie corporal al inicio del estudio.
El índice de masa corporal (IMC) está entre 18 y 35 kg/m2, ambos valores incluidos.
Las pacientes en edad fértil deben usar métodos anticonceptivos adecuados o tener una pareja esterilizada durante el estudio: anticonceptivos hormonales sistémicos, dispositivo intrauterino o método anticonceptivo de barrera junto con espermicida, o aceptar la abstinencia sexual. Los anticonceptivos hormonales deben estar en una dosis estable durante al menos un mes antes del inicio. Nota: Las mujeres que no están en edad fértil son:
mujeres que se han sometido a una esterilización quirúrgica (histerectomía u ooforectomía bilateral o ligadura de trompas);
mujeres mayores de 60 años de edad;
mujeres mayores de 40 años y menores de 60 años que han dejado de tener la menstruación durante al menos 12 meses y una prueba de hormona foliculoestimulante (FSH) que confirma la falta de potencial para tener hijos (FSH >40 mUI/ml) o la interrupción de la menstruación durante al menos 24 meses sin niveles de FSH confirmados.
Pacientes que pueden y desean suspender el tratamiento de la dermatitis atópica durante todo el estudio (excepto los emolientes permitidos);
Capaces y dispuestos a dar su consentimiento informado firmado y el consentimiento debe obtenerse antes de cualquier procedimiento relacionado con el estudio.
Criterios de exclusión
Pacientes de sexo femenino con prueba de embarazo positiva en la visita de selección o la visita del día 0 (basal) o mujeres en periodo de lactancia.
Cualquier condición médica clínicamente significativa controlada o no controlada o anomalía de laboratorio que, en opinión del investigador, podría poner al paciente en riesgo indebido o interferir con la interpretación de los resultados del estudio.
Deterioro clínicamente significativo de la función renal o hepática.
Otras afecciones de la piel que pueden interferir con el diagnóstico y/o la evaluación de la dermatitis atópica (tal como psoriasis o infecciones cutáneas virales, bacterianas y fúngicas actuales).
Antecedentes de hipersensibilidad a cualquier sustancia en cápsulas de DS107G o de placebo.
Uso de productos biológicos 3 meses antes del inicio del tratamiento/visita del día 0 (basal), o 5 semividas (lo que sea más larga).
Uso de tratamientos sistémicos (que no sean productos biológicos) que podrían afectar a la dermatitis atópica menos de 4 semanas antes de la visita basal (Día 0), por ejemplo, retinoides, inhibidores de calcineurina, metotrexato, ciclosporina, hidroxicarbamida (hidroxiurea), azatioprina y corticosteroides orales/inyectables; están permitidos los corticosteroides intranasales y los corticosteroides inhalados para afecciones médicas estables.
Tratamiento con cualquier fármaco experimental dentro de los 30 días previos a la visita del día 0 (basal) o 5 semividas (lo que sea más largo).
Exposición excesiva al sol, uso de cabinas de bronceado u otras fuentes de luz ultravioleta (UV) 4 semanas antes de la visita del Día 0 (basal) y/o está planeando un viaje a un clima soleado o usar cabinas de bronceado u otras fuentes de UV entre las visitas de selección y seguimiento.
Uso de cualquier tratamiento tópico para la dermatitis atópica 2 semanas antes del inicio del tratamiento/visita del día 0 (basal), incluyendo, pero sin limitación, corticosteroides tópicos, inhibidores de calcineurina, alquitranes, lejía, antimicrobianos y baños de lejía.
Uso de productos tópicos que contienen urea, ceramidas o ácido hialurónico 2 semanas antes del día 0.
Uso de antihistamínicos para la dermatitis atópica dentro de las 2 semanas anteriores al inicio.
Enfermedad cardiovascular no controlada significativo (antecedentes de anomalías en el ECG que son clínicamente significativas en opinión del investigador), neurológica, maligna, psiquiátrica respiratoria o hipertensiva, así como diabetes y artritis.
Historial médico de enfermedad infecciosa crónica (por ejemplo, hepatitis B, hepatitis C o infección con el virus de la inmunodeficiencia humana).
Antecedentes de abuso clínicamente significativo de drogas o alcohol el último año antes del Día 0 (basal).
Restricciones del estudio
Se preguntará a cada sujeto sobre los puntos específicos enumerados a continuación antes de la administración del fármaco. Si un sujeto admite un incumplimiento de estas restricciones, el investigador principal (o la persona designada) y/o el promotor decidirán si se permitirá o no que el sujeto permanezca en el estudio. El incumplimiento de estas restricciones se registrará.
Los sujetos recibirán instrucciones de abstenerse de planificar un viaje al clima soleado o el uso de equipos de bronceado entre las visitas de detección y seguimiento.
A los sujetos se les indicará que se abstengan de usar cualquier fármaco/tratamiento que pueda influir en la dermatitis atópica (consúltense los criterios de exclusión y la sección de terapias o procedimientos prohibidos) durante todo el estudio.
Los sujetos deberán comenzar a ayunar al menos 8 horas antes de la administración del fármaco al despertar. El ayuno continuará durante al menos 60 minutos después de la administración del fármaco, después de lo cual el sujeto podrá desayunar. Se permitirá beber agua en todo momento durante el período de ayuno, pero no se permitirán otros líquidos. El o los fármacos para otras afecciones que estén permitidos en el estudio se podrán tomar de la forma habitual.
Para las visitas basal (día 0), semana 4 y semana 8, se extraerá sangre para el análisis PK. Las muestras para PK se tomarán antes de la dosis; por tanto, el fármaco se administrará durante la visita del día 0 y las visitas de la semana 4. Debido a que la dosificación se realizará en la clínica el día 0 y la semana 4, los sujetos deberán ayunar durante al menos 8 horas antes de la administración del fármaco del estudio y se les permitirá comer 60 minutos después de la administración del fármaco del estudio.
Interrupciones
Los sujetos tienen derecho a retirarse del estudio en cualquier momento y por cualquier motivo sin penalización. El investigador también tiene derecho a retirar a los sujetos del estudio si considera que es lo mejor para el sujeto o si el sujeto no coopera o no cumple. Todos los interesados entienden que una tasa excesiva de retiradas puede hacer que el estudio no sea interpretable; por tanto, se debe evitar la retirada innecesaria de sujetos. Si un sujeto decide retirarse, se harán todos los esfuerzos posibles para completar e informar sobre las observaciones, particularmente la exploración de seguimiento, del modo más exhaustivo posible.
El investigador o uno de los miembros de su personal debe comunicarse con el sujeto, ya sea por teléfono o mediante una visita personal, para determinar lo más completamente posible el motivo d la retirada y registrar el
motivo en el documento fuente del sujeto y el CRF. Se debe realizar una evaluación final completa de finalización anticipada (semana 8) en el momento de la retirada del sujeto con una explicación de por qué el sujeto se retira del estudio. Si el motivo de la eliminación de un sujeto es un acontecimiento adverso o un resultado anormal de una prueba de laboratorio, se registrará el acontecimiento o prueba específica principal. Se pedirá a los sujetos que se retiren del estudio antes de la visita de la semana 8, si están de acuerdo, que acudan a una última evaluación (visita de finalización anticipada).
Las razones para la interrupción incluyen:
El investigador decide que el sujeto debe retirarse. Si esta decisión se toma debido a un acontecimiento adverso grave o persistente, anomalía de laboratorio o enfermedad intercurrente, se suspenderá el fármaco del estudio y se tomarán las medidas apropiadas. El investigador lo notificará al promotor o al representante de inmediato.
El sujeto o el médico encargado del tratamiento solicitan que el sujeto sea retirado del estudio.
El sujeto por cualquier motivo requiere tratamiento con otro agente terapéutico que haya demostrado ser efectivo para el tratamiento de la indicación del estudio. En este caso, la suspensión del estudio se produce inmediatamente después de la introducción del agente nuevo.
Se ha perdido al sujeto para el seguimiento, en este caso, se debe hacer un intento razonable de contactar con el sujeto y determinar su estado, y estos intentos deben documentarse.
Grave violación del protocolo, incluido el incumplimiento persistente.
El promotor o las autoridades reguladoras, por alguna razón, detienen el estudio. Todos los sujetos serán retirados del estudio y se les notificarán los motivos de la interrupción.
Embarazo en cualquier momento durante el estudio.
Otros: el sujeto puede retirarse del estudio por cualquier otro motivo, incluida la retirada del consentimiento.
TRATAMIENTO
Administración del tratamiento
Los sujetos que cumplen con todos los criterios de inclusión y ninguno de los criterios de exclusión pueden ser aceptados en el estudio. Cada sujeto debe leer y firmar un formulario de consentimiento informado antes de someterse a cualquier procedimiento de selección. Este estudio implica una comparación de DS107G (2 g) con placebo, administrado por vía oral una vez al día al despertar durante un total de 8 semanas. La última administración del fármaco del estudio debe realizarse el día anterior a la visita de la semana 8/visita de finalización anticipada (FA). Los sujetos serán asignados al azar a uno de los dos grupos de tratamiento en una proporción 1:1: Grupo de tratamiento A: 2 gramos de DS107G (4 cápsulas).
Grupo de tratamiento B: 2 gramos de cápsulas de placebo (4 cápsulas).
Los sujetos deberán comenzar a ayunar al menos 8 horas antes de la administración del fármaco al despertar. El ayuno continuará durante al menos 60 minutos después de la administración del fármaco, después de lo cual los sujetos pueden desayunar. Se permitirá beber agua en todo momento durante el período de ayuno, pero no se permitirán otros líquidos. El o los fármacos para otras afecciones que estén permitidos en el estudio se podrán tomar de la forma habitual.
Los envases de tipo blíster constarán de 7 filas de 4 cápsulas. Cada fila constituye una dosis diaria. Se indicará a los sujetos que tomen las 4 cápsulas de izquierda a derecha, de arriba a abajo.
Tratamiento de estudio
Las cápsulas DS107G se proporcionarán como cápsulas de gelatina blanda opacas ovaladas que contienen 500 mg de ácido graso libre DGLA (a Ag ).
Las cápsulas de placebo también se proporcionarán como cápsulas de gelatina blanda opacas ovaladas que contienen 500 mg de parafina líquida.
Las cápsulas de DS107G se suministrarán en forma fabricada (enmascaradas), envasadas en blíster de papel de aluminio de 28 unidades. El placebo se presentará en blíster y paquetes idénticos y se almacenará/empaquetará igual que las cápsulas de DS107G. El fármaco del estudio se etiquetará de acuerdo con las regulaciones de Ee .UU.
y Canadá.
El fármaco del estudio será proporcionado por el promotor al investigador y se conservará, en el centro, en una habitación o armario cerrado con acceso limitado. Las cápsulas de DS107G y placebo deben almacenarse a una temperatura ambiente controlada entre 15-30 °C y solo se suministrarán a los sujetos en el ensayo bajo la supervisión del investigador.
El centro del estudio dispensará el fármaco del estudio al sujeto en cada visita de estudio. Los sujetos deben devolver todos los paquetes de tipo blíster del fármaco del estudio (usados y sin usar) al centro del estudio. Las cápsulas dentro de los paquetes de tipo blíster se contarán antes de la dispensación y en el momento de la devolución, y los recuentos se registrarán en los documentos fuente y el eCRF. Cada sujeto debe recibir instrucciones sobre la importancia de devolver el fármaco del estudio en la próxima visita de estudio. Si un sujeto no devuelve el fármaco del estudio, se le indicará que lo devuelva lo antes posible.
El investigador es responsable de mantener registros precisos de la medicación del estudio recibida inicialmente, el fármaco del estudio dispensado/usado, la medicación devuelta por los sujetos y la medicación destruida o devuelta al promotor o su representante designado. Todos los formularios de contabilidad de fármacos del estudio y los registros de tratamiento deben conservarse en el archivo de estudio del investigador. Estos registros deben estar disponibles para inspección por parte del promotor, sus representantes designados o por agencias reguladoras en cualquier momento.
Las cajas de fármacos/paquetes de tipo blíster usadosse almacenarán de manera segura hasta su destrucción y el investigador deberá tenerlos en cuenta. El monitor del estudio realizará la contabilidad de todos los fármacos del estudio en el centro y ayudará a devolver el fármaco del estudio, incluido el fármaco de estudio usado y no usado para el promotor o el representante designado. Después de la verificación de la contabilidad del fármaco por parte del promotor, el investigador garantizará la destrucción o devolución adecuada del producto restante del estudio. Se contará todos los fármacos del estudio destruidos de forma accidental o deliberada. Se explicará cualquier discrepancia entre las cantidades entregadas y devueltas.
El inventario de fármacos y los registros de contabilidad se mantendrán en cada centro según las pautas de GCP/ICH. Aproximadamente 100 pacientes serán aleatorizados a grupos de tratamiento doble ciego en una proporción de 1:1 por un sistema interactivo de respuesta web (IWRS) o un sistema interactivo de respuesta de voz (IVRS), de la siguiente manera:
Grupo de tratamiento A: 2 gramos de DS107G (4 cápsulas).
Grupo de tratamiento B: 2 gramos de cápsulas de placebo (4 cápsulas).
Dignity Sciences o su representante designado generará una lista de aleatorización de bloques permutados y estratificada por centro. El programa de aleatorización con las asignaciones del fármaco del estudio se generará antes del inicio del estudio y será conocido solo por las personas responsables de etiquetar el fármaco del estudio. El IVRS o IWRS asignará un número de kit de fármaco del estudio a cada sujeto y el contenido se basará en el código de aleatorización.
En el centro de investigación, a cada sujeto se le asignará un número de identificación del paciente durante la selección que se utilizará en toda la documentación del paciente. El número de identificación del paciente contendrá el número del centro y el número del paciente asignado en orden numérico en la visita de selección (por ejemplo: 02 010 para el décimo paciente examinado en el centro n.° 02). Los números se asignarán en orden ascendente a partir de 001.
Justificación de la selección y la temporización de las dosis en el estudio
Todos los sujetos sanos toleraron bien las dosis de hasta 4 g. La dosis de 2 g se ha seleccionado como la dosis probada en el estudio actual según, principalmente, los resultados farmacocinéticos del ensayo de Fase I, que sugirió niveles de piel saturables de DGLA total con dosis orales repetidas de más de 2 g por día. Además, se consideraron los siguientes factores:
hubo casos gastrointestinales transitorios menos frecuentes registrados a una dosis de 2 g en comparación con 4 g diarios.
el número de cápsulas (4) administradas diariamente. Sería posible una dosis más alta, pero se considera menos deseable ya que demasiadas cápsulas pueden tener un impacto negativo en el cumplimiento del tratamiento por parte del paciente.
Apertura del enmascaramiento del estudio
En todo momento, la información sobre el tratamiento y la aleatorización se mantendrá confidencial y no se divulgará al investigador, el personal del estudio, el CRO o el equipo de estudio del promotor hasta que finalice el estudio. Los códigos de enmascaramiento solo deben abrirse en situaciones de emergencia por razones de seguridad del sujeto. El método será un proceso manual o electrónico. Cuando se ha abierto el enmascaramiento de un sujeto, el motivo debe estar completamente documentado. Cuando sea posible, el investigador debe contactar con el promotor o su representante designado antes de abrir el enmascaramiento. Si se abre el enmascaramiento, el investigador debe informar de inmediato al monitor médico. La documentación de la apertura del enmascaramiento debe registrarse con la fecha y la hora en que se abrió el enmascaramiento y los nombres del personal implicado.
El sujeto para el que se ha abierto el enmascaramiento será retirado del estudio y se someterá a los procedimientos de finalización anticipada (FA) (visita de la semana 8). En los casos en los que existan razones éticas para que el sujeto permanezca en el estudio, el investigador debe obtener la aprobación específica del promotor o su representante designado para que el sujeto continúe en el estudio.
Terapia Concomitante
Todos los medicamentos (incluidos los fármacos de venta libre, las vitaminas y los antiácidos) tomados < 4 semanas antes de la selección y durante todo el estudio deben registrarse. Se deben registrar todos los medicamentos tomados para la dermatitis atópica en los 2 meses previos a la selección.
Las entradas de medicamentos deben ser específicas para el nombre genérico. El nombre comercial puede usarse para los fármacos en combinación. Las entradas incluirán la dosis, la unidad y la frecuencia de administración, la vía de administración, la fecha de inicio, la fecha de suspensión y la indicación. Si se suspende el medicamento o se cambia la dosis, deberán registrarse dichos detalles.
El investigador debe evaluar cualquier procedimiento concomitante, medicamentos y suplementos dietéticos para aceptabilidad que no estén explícitamente prohibidos.
Terapias permitidas
Emolientes
Los sujetos pueden aplicar un emoliente suave de su elección sobre la piel, incluyendo lesiones de DA, siempre que el uso de emolientes se inicie al menos 2 semanas antes del Día 0 y continúe con la misma frecuencia y en las mismas zonas de la piel durante todo el estudio. Se solicitará a los sujetos que eviten el uso de emolientes que contengan cualquiera de los siguientes ingredientes:
(1) Urea
(2) Ceramida
(3) Ácido hialurónico
Se debe hacer todo lo posible para mantener el mismo emoliente durante todo el estudio. El nombre comercial del o los emolientes seleccionados se registrará en el documento fuente y el eCRF. No se pueden aplicar otros productos a las lesiones durante el estudio.
Otras terapias permitidas
Durante el estudio están permitidos los antihistamínicos no sedantes (por ejemplo, loratadina, fexofenadina) solo si se usan para tratar afecciones médicas distintas a la dermatitis atópica. Dichos medicamentos se permiten durante el estudio solo si el sujeto ha estado recibiendo una dosis estable durante al menos 2 semanas antes del Día 0 y continúa usando el mismo agente todos los días durante todo el estudio.
Se permiten los corticosteroides inhalados e intranasales para afecciones médicas estables.
Terapias o procedimientos prohibidos
Las siguientes terapias o procedimientos tópicos están prohibidos durante el estudio para todos los sujetos:
Tratamientos medicinales tópicos que podrían afectar a la dermatitis atópica, incluyendo, aunque sin limitaciones: corticosteroides tópicos
inhibidores de la calcineurina
alquitranes
lejía
baños de lejía con antimicrobianos
Cualquier producto tópico que contenga urea, ceramidas o ácido hialurónico
Terapia sistémica que podría afectar a la dermatitis atópica, por ejemplo, retinoides, inhibidores de calcineurina, metotrexato, ciclosporina, hidroxicarbamida (hidroxiurea), azatioprina y corticosteroides orales/inyectables Antihistamínicos (excepto antihistamínicos no sedantes)
Cualquier agente biológico
Fototerapia UVA o UVB
Terapia con psoralenos ultravioleta A (PUVA)
Exposición excesiva al sol o uso de cabina de bronceado
Cualquier agente en investigación
Evaluación del cumplimiento
El cumplimiento del tratamiento se evaluará en cada visita mediante preguntas directas, revisión del registro del cumplimiento del sujeto y recuento de cápsulas, y se basará en este último. Los sujetos recibirán un diario en papel en cada visita junto con la medicación del estudio. Los sujetos indicarán cualquier dosis olvidada en el diario, así como el momento de la última ingesta de alimentos antes de la administración del fármaco del estudio y la ingestión de alimentos después de la administración del fármaco del estudio. Se indicará a los sujetos que traigan todas las cápsulas y paquetes de tipo blíster (usados y sin usar) y el registro de cumplimiento a la próxima visita de estudio. Cualquier desviación del régimen de dosificación prescrito se registrará en el documento fuente y en el eCRF. Se asesorará a los sujetos que no cumplan de forma significativa.
PROCEDIMIENTOS DEL ESTUDIO
Consúltese el Apéndice A para ver un diagrama de flujo de los procedimientos a realizar en cada visita.
Selección, Visita 1 (Día -30 a -1)
La evaluación de selección solo se realizará después de que el sujeto haya aceptado participar y haya firmado y fechado el formulario de consentimiento informado. No se iniciará ningún tratamiento o procedimiento relacionado con el ensayo antes de firmar el consentimiento informado. La visita del día 0 debe realizarse, a más tardar, 30 días después de la visita de selección.
La evaluación de selección se realizará de acuerdo con los criterios de inclusión y exclusión. Si el sujeto cumple todos los criterios de inclusión y ninguno de los criterios de exclusión, el sujeto puede ser incluido en el estudio. Los siguientes procedimientos se realizarán en la visita de selección:
Consentimiento informado;
Revisión de los criterios de inclusión-exclusión, incluida la revisión de los criterios de Hanifin y Rajka (Apéndice G);
Asignación del número de identificación de sujeto (Número de centro - Número de sujeto);
Características demográficas;
Medicamentos concomitantes;
Historial médico/quirúrgico;
Exploración física;
Constantes vitales;
IMC;
Análisis clínicos de seguridad (bioquímica, coagulación, hematología y análisis de orina);
Prueba de embarazo en suero (solo en mujeres en edad fértil) y prueba de los niveles de FSH para mujeres mayores de 40 años y menores de 60 años que han dejado de menstruar durante al menos 12 meses pero menos de 24 meses;
Evaluación de BSA;
IGA;
Valor basal, Visita 2 (día 0)
Los sujetos deben ayunar durante al menos 8 horas antes de la administración del fármaco del estudio. Se les permitirá comer 60 minutos después de la administración del fármaco del estudio.
En esta visita se realizarán los siguientes procedimientos:
(1) Confirmar la elegibilidad con criterios de inclusión y exclusión
(2) Actualizar o confirmar el historial médico/quirúrgico
(3) Medicamentos concomitantes
(4) Constantes vitales
(5) IMC
(6) Análisis clínicos de seguridad y biomarcadores de inflamación (bioquímica, coagulación, hematología, análisis de orina y perfil de interleucinas).
(7) Prueba de embarazo en orina (solo mujeres en edad fértil)
(8) Extracción de sangre antes de la administración de la dosis para análisis farmacocinéticos
(9) Extracción de sangre para análisis del perfil de ácidos grasos totales
(10) Evaluación de BSA
(11) IGA
(12) Evaluación EASI
(13) Evaluación SCORAD (incluida la evaluación del prurito CON VAS)
(14) Cuestionario POEM
(15) Cuestionario DLQI
(16) Evaluación TEWL (solo para centros seleccionados)
(17) Aleatorizar a los sujetos en IVRS/IWRS
(18) Administración del fármaco del estudio
(19) Dispensación del fármaco del estudio
(20) Dispensación del registro del cumplimiento del sujeto
(21) Evaluación de acontecimientos adversos (después de la primera administración del fármaco del estudio) Semana 2, Visita 3 (día 14 ± 2 días)
En esta visita se realizarán los siguientes procedimientos:
(1) Constantes vitales
(2) Prueba de embarazo en orina (solo mujeres en edad fértil)
(3) Evaluación de BSA
(4) IGA
(5) Evaluación EASI
(6) Evaluación SCORAD (incluida la evaluación del prurito CON VAS)
(7) Cuestionario POEM
(8) Cuestionario DLQI
(9) Evaluación TEWL (solo para centros seleccionados)
(10) recopilación y dispensación de la revisión del fármaco del estudio, recopilación y dispensación del registro de cumplimiento del sujeto
(11) Recuento de cápsulas
(12) Medicamentos concomitantes
(13) Evaluación de acontecimientos adversos Semana 4, Visita 4 (Día 28 ± 2 días).
Los sujetos deben ayunar durante al menos 8 horas antes de la administración del fármaco del estudio. Se les permitirá comer 60 minutos después de la administración del fármaco del estudio.
En esta visita se realizarán los siguientes procedimientos:
(1) Constantes vitales
(2) Prueba de embarazo en orina (solo mujeres en edad fértil)
(3) Exploración física
(4) Análisis clínicos de seguridad (bioquímica, coagulación, hematología, análisis de orina y perfil de interleucinas)
(5) Extracción de sangre antes de la dosis para análisis farmacocinéticos (si la medicación del estudio se tomó antes de la visita, el sujeto debe volver al día siguiente)
(6) Extracción de sangre para análisis del perfil de ácidos grasos totales
(7) Evaluación de BSA
(8) IGA
(9) Evaluación EASI
(10) Evaluación SCORAD (incluida la evaluación del prurito CON VAS)
(11) Cuestionario POEM
(12) Cuestionario DLQI
(13) Evaluación TEWL (solo para centros seleccionados)
(14) Administración del fármaco del estudio (indique al sujeto que la última dosis debe producirse el día anterior a la visita de la semana 8)
(15) recopilación y dispensación del fármaco del estudio
(16) Revisión, recopilación y dispensación del registro de cumplimiento del sujeto
(17) Recuento de cápsulas
(18) Medicamentos concomitantes
(19) Evaluación de acontecimientos adversos.
Semana 8, Visita 5 (Día 56 ± 2 días) (Visita de fin del tratamiento/finalización anticipada).
En esta visita se realizarán los siguientes procedimientos:
(1) Revisión continua del historial médico
(2) Constantes vitales
(3) Exploración física
(4) IMC
(5) Análisis clínicos de seguridad (bioquímica, coagulación, hematología y análisis de orina)
(6) Prueba de embarazo en suero (solo mujeres en edad fértil)
(7) Extracción de sangre para análisis farmacocinéticos (si la medicación del estudio se tomó antes de la visita, el sujeto debe volver al día siguiente)
(8) Extracción de sangre para análisis del perfil de ácidos grasos totales
(9) Evaluación de BSA
(10) IGA
(11) Evaluación EASI
(12) Evaluación SCORAD (incluida la evaluación del prurito CON VAS)
(13) Cuestionario POEM
(14) Cuestionario DLQI
(15) Evaluación TEWL (solo para centros seleccionados)
(16) recopilación del fármaco del estudio
(17) Revisión y recopilación del registro de cumplimiento del sujeto
(18) Recuento de cápsulas
(19) Medicamentos concomitantes
(20) Evaluación de acontecimientos adversos
Seguimiento/Semana 10, Visita 6 (Día 70 ± 3 días).
En esta visita se realizarán los siguientes procedimientos:
(1) Revisión continua del historial médico
(2) Constantes vitales
(3) Exploración física
(4) Análisis clínicos de seguridad (bioquímica, coagulación, hematología y análisis de orina): solo si hay un cambio clínicamente significativo con respecto al valor basal en los resultados del análisis de seguridad la semana 8
(5) Prueba de embarazo en orina (solo mujeres en edad fértil)
(6) Evaluación de BSA
(7) IGA
(8) Evaluación EASI
(9) Evaluación SCORAD (incluida la evaluación del prurito CON VAS)
(10) Cuestionario POEM
(11) Cuestionario DLQI
(12) Evaluación TEWL (solo para centros seleccionados)
(13) Medicamentos concomitantes
(14) Evaluación de acontecimientos adversos
(15) Visita de finalización anticipada.
En el caso de que el sujeto termine el estudio antes de completarlo, deben completarse los procedimientos enumerados en la visita de la semana 8.
EVALUACIONES DE ESTUDIO
Evaluaciones de la eficacia
Las evaluaciones clínicas de la dermatitis atópica serán realizadas por un dermatólogo experimentado y cualificado (certificado por la junta o equivalente). Para asegurar la consistencia y reducir la variabilidad, el mismo evaluador debe realizar todas las evaluaciones sobre un sujeto determinado siempre que sea posible.
Evaluación global del investigador
La Evaluación global del investigador (IGA) de la gravedad de la enfermedad (Apéndice B) se evaluará en cada visita. La IGA es una evaluación global del estado actual de la enfermedad. Es una evaluación morfológica de 6 elementos de la gravedad general de la enfermedad y se determinará de acuerdo con las siguientes definiciones: 0 (claro), 1 (casi claro), 2 (leve), 3 (moderado), 4 (grave) y 5 (muy grave). Para ser elegibles, los sujetos deben tener una puntuación IGA > 3 en la visita basal (día 0).
Área de eccema e índice de gravedad (EASI)
El área de eccema y el índice de gravedad (EASI) se evaluarán en cada visita, excepto en la visita de selección. Cuantifica la gravedad de la dermatitis atópica de un sujeto según la gravedad de la lesión y el porcentaje de BSA afectado. El EASI es una puntuación compuesta que va de 0-72 que tiene en cuenta el grado de eritema, induración/papulación, excoriación y liquenificación (cada uno puntuado de 0 a 3 por separado) para cada una de las cuatro regiones del cuerpo, con ajuste para el porcentaje de BSA afectado para cada región del cuerpo y para la proporción de la región del cuerpo a todo el cuerpo. En el Apéndice C se proporciona un procedimiento detallado del cálculo de la puntuación EASI.
Área de superficie corporal (BSA)
La BSA general afectada por DA se evaluará (de 0 a 100 %) en cada visita. La palma de la mano de un paciente representa el 1 % de su BSA total. Para todas las visitas de estudio, excepto en la selección, la BSA de la piel afectada se medirá con la medición SCORAD (consúltese la descripción a continuación) y se evaluará como un criterio de valoración separado. Para ser elegibles, los sujetos deben tener una BSA de al menos 10 % en la visita basal (día 0).
La puntuación del Dermatitis atópica (SCORAD) SCORAD se medirá en cada visita, excepto en la visita de selección. El sistema de clasificación SCORAD fue desarrollado por el Grupo de trabajo europeo sobre dermatitis atópica (1993) y ha sido una herramienta estándar para evaluar la gravedad de la DA en estudios clínicos en Europa. Se seleccionarán seis puntos (eritema, edema/papulación, supuración/costras, excoriación, liquenificación y sequedad) para evaluar la gravedad de la DA. La BSA general afectada por DA se evaluará (de 0 a 100 %) y se incluirá en las puntuaciones de SCORAD. Los pacientes evaluarán la pérdida de sueño y el prurito en una escala analógica visual (0-10). La suma de estas medidas representa la SCORAD que puede variar de 0 a 103. El procedimiento detallado del cálculo de la puntuación SCORAD se proporciona en el Apéndice D.
Escala analógica visual del prurito
Para todas las visitas de estudio, excepto la de selección, la puntuación de gravedad del prurito se registrará con la medición SCORAD y esto se evaluará como un criterio de valoración separado. Esto se evaluará pidiendo a los sujetos que indiquen en la escala de 10 cm (0-10) del formulario de evaluación el punto correspondiente al valor promedio de los últimos tres días/noches.
Medida de eccema orientada por el paciente (POEM)
La medida de eccema orientada por el paciente (POEM) se evaluará en cada visita, excepto en la visita de selección. La POEM desarrollada por Charman et.al. es una autoevaluación de la gravedad de la enfermedad por parte del paciente. POEM tiene un valor máximo de veintiocho según la respuesta del paciente a siete preguntas calificadas según la siguiente escala:
Sin días = 0
1-2 días = 1
3-4 días = 2
5-6 días = 3
Todos los días = 4
En el Apéndice E se proporciona una descripción detallada de la evaluación POEM.
Cuestionario de calidad de vida dermatológica (DLQI)
El DLQI es un simple cuestionario validado de 10 preguntas que se completará en cada visita, excepto en la de selección. El cuestionario se proporciona en el Apéndice F.
Pérdida de agua transepidérmica (TEWL) (solo en centros seleccionados)
La gravedad clínica de la EA y el efecto asociado sobre la función de barrera de la piel se evaluarán en cada visita, excepto en la visita de selección. Esta evaluación se realizará en centros seleccionados que hayan demostrado experiencia previa con este dispositivo.
En la visita basal (Día 0), el investigador seleccionará tres áreas representativas de DA activa para cada sujeto; se registrará la ubicación de estos sitios. En visitas posteriores, las lecturas de TEWL para cada área de DA se tomarán en condiciones ambientales estándar (22-25 °C, 40-60 % de humedad relativa); la media de las mediciones TEWL se utilizará para los análisis.
Evaluaciones de seguridad
Constantes vitales
Las siguientes constantes vitales se registrarán en cada visita en posición de sedestación, después de haber estado sentado tranquilamente durante al menos 5 minutos: presión arterial sistólica y diastólica (mmHg), pulso (lpm), temperatura corporal (°C) y frecuencia respiratoria (respiraciones/min).
El peso (kg) y la altura (cm) se recopilarán para calcular el IMC y se registrarán en las visitas de selección, basal y de la semana 8. La altura solo se registrará una vez en la visita de selección y se utilizará el mismo valor para el cálculo del IMC en las visitas basal de la semana 8.
Cualquier hallazgo anormal relacionado con las constantes vitales que el investigador considere clínicamente significativo, debe registrarse como un AA.
Exploración física
Los siguientes centros/sistemas se incluirán en la exploración física. Cada sistema se puntuará como normal/anormal (no clínicamente significativo o clínicamente significativo). Los detalles pertinentes deben registrarse para cualquier hallazgo clínicamente significativo.
(1) Aspecto general
(2) Dermatológicos (excepto la dermatitis atópica)
(3) Cabeza, ojos, oídos, nariz, garganta (HEENT)
(4) Respiratorios
(5) Cardiovasculares
(6) Abdominales
(7) Neurológicos
(8) Musculoesqueléticos
(9) Linfáticos
Análisis de laboratorio clínico
Los análisis de laboratorio se realizarán en las visitas de selección, día 0, semana 4 y semana 8. Si los resultados de la semana 8 indican un cambio clínicamente significativo con respecto al valor basal, también se realizarán análisis de laboratorio en la semana 10. Los análisis incluirán análisis de orina, hematológicos con pruebas diferenciales y de coagulación, un panel de bioquímica estándar (la bioquímica incluye pruebas de la función hepática y colesterol), coagulación, prueba de embarazo en suero (visitas de selección y semana 8/de finalización anticipada) para mujeres en edad fértil (WOCBP). En las visitas basal (día 0) y de la semana 2, semana 4 y semana 10, se realizará una prueba de embarazo en orina para mujeres en edad fértil (realizada en el centro del investigador). En la visita de selección, se analizará los niveles de fSh en mujeres mayores de 40 años y menores de 60 años que hayan dejado de menstruar durante al menos 12 meses pero menos de 24 meses. Las pruebas específicas en estos paneles se enumeran a continuación en la Tabla 8:
Tabla 8. Análisis de laboratorio clínico

Niveles plasmáticos de DGLA total y libre
En las visitas basal y de la semana 4 y semana 8, se realizarán extracciones de sangre antes de la administración del fármaco del estudio (no se administrará el fármaco del estudio en la visita de la semana 8). Si un sujeto llega a la clínica después de tomar su dosis diaria de medicación del estudio, se pedirá al sujeto que vuelva al día siguiente para las extracciones de sangre para PK. Se medirán los niveles plasmáticos mínimos de DGLA total y DGLA libre. Se realizará una segunda extracción de sangre para una evaluación posterior del perfil de ácidos grasos totales en plasma. La extracción de sangre para el análisis de bioquímica sérica se dividirá en dos partes alícuotas para el análisis de bioquímica y la evaluación posterior de las interleucinas.
La fecha y hora de la última dosis del sujeto en el hogar antes de la visita se registrarán con precisión. El centro del estudio indicará a los sujetos que no tomen la dosis diaria del fármaco del estudio en su casa durante la visita de la semana 4. La dosificación se realizará en la clínica durante la visita de estudio. Se debe registrar la hora exacta de la obtención de la muestra.
Las muestras de sangre se procesarán lo antes posible, a más tardar 1 hora después de la extracción de sangre. El plasma obtenido se transferirá a tubos de polipropileno. Cada tubo se etiquetará para identificar el analito a analizar. Todas las muestras se congelarán en posición vertical. Las instrucciones detalladas para la obtención de muestras para PK, ácidos grasos totales e interleucina, procesamiento, almacenamiento y envío se proporcionarán en el manual del laboratorio central. Las etiquetas de cada tubo incluirán al menos la siguiente información:
(1) Protocolo de estudio
(2) Número de centro
(3) Número de identificación del sujeto
(4) Nombre de la visita
(5) Nombre del analito
(6) Primario (A) o Duplicado (B)
El envío de las muestras experimentales se enviará a ICON Central Laboratories.
Las instrucciones detalladas para el envío se proporcionarán en el manual del laboratorio central. A continuación, las muestras se enviarán a la instalación de análisis y se analizarán utilizando un método analítico validado de conformidad con sus procedimientos operativos estándar.
Las muestras de sangre para el análisis PK se mantendrán enmascaradas.
ACONTECIMIENTOS ADVERSOS
Un acontecimiento adverso es cualquier acontecimiento médico indeseable en un paciente al que se le administró un producto farmacéutico, sin tener en cuenta la posibilidad de una relación causal con este tratamiento.
Los investigadores son responsables de controlar la seguridad de los sujetos que participan en este estudio y de alertar al promotor de cualquier acontecimiento que parezca inusual, incluso si este acontecimiento puede considerarse un beneficio no anticipado para el sujeto. El investigador es responsable de la atención médica adecuada de los sujetos durante el estudio.
El investigador sigue siendo responsable de seguir a través de una opción de atención médica adecuada, acontecimientos adversos que son graves o que causaron la retirada del sujeto antes de completar el estudio. Se deje realizar el seguimiento del sujeto hasta que el acontecimiento se resuelva o sea estable. La frecuencia de seguimiento se deja a discreción del investigador.
La seguridad se evaluará mediante la recopilación de acontecimientos adversos, las constantes vitales, realizando exámenes físicos y evaluando los resultados de los análisis de laboratorio. Los acontecimientos adversos notificados se codificarán de acuerdo con la terminología de MedDRA.
Antes de la inscripción, el personal del centro del estudio tomará nota de la ocurrencia y la naturaleza de las afecciones médicas de cada sujeto) en la sección correspondiente del documento fuente y el CRF. Durante el estudio, el personal del centro volverá a anotar cualquier cambio en la o las afecciones y la ocurrencia y naturaleza de cualquier acontecimiento adverso.
Si un sujeto experimenta un acontecimiento adverso después de la primera dosis del fármaco del estudio, el acontecimiento se registrará como acontecimiento adverso en el documento fuente y el CRF. Todos los AA se describirán en los documentos fuente y en el CRF.
Causalidad de los acontecimientos adversos
El investigador establecerá la causalidad del AA con el tratamiento experimental. El investigador deberá tener en cuenta la historia del sujeto, los hallazgos más recientes de la exploración física y los medicamentos concomitantes. Las siguientes definiciones se utilizarán para determinar la causalidad de un AA:
No relacionado: la relación temporal del inicio del AA, en relación con el tratamiento experimental no es razonable u otra causa puede explicar la aparición del AA.
Relacionado: la relación temporal del inicio del AA, en relación con el tratamiento experimental es razonable, sigue un patrón de respuesta conocido al tratamiento y una causa alternativa es poco probable.
Gravedad de los acontecimientos adversos
La intensidad de un AA es una estimación de la gravedad relativa del acontecimiento realizada por el investigador en función de su experiencia clínica y su familiaridad con la literatura. Las siguientes definiciones se utilizarán para calificar la gravedad de un AA:
Leve: El sujeto apenas nota el síntoma y no influye sobre la realización de las actividades cotidianas. El tratamiento no está normalmente indicado.
Moderado: El síntoma es lo suficientemente grave como para incomodar al sujeto y afecta a la realización de las actividades cotidianas. El tratamiento puede ser necesario.
Grave: El síntoma causa graves molestias y las actividades cotidianas se ven significativamente afectadas o prevenidas. El tratamiento puede ser necesario.
Acontecimientos adversos graves
Si un paciente experimenta un acontecimiento adverso grave después de la primera dosis del fármaco del estudio, el acontecimiento se registrará como un acontecimiento adverso grave. Todos los AA se describirán en los documentos fuente y en el CRF.
Una reacción o acontecimiento adverso grave (experiencia)es cualquier acontecimiento médico desfavorable que, a cualquier dosis:
• da como resultado la muerte,
• es potencialmente mortal,
• requiere hospitalización del sujeto o prolongación de la hospitalización existente,
• da como resultado discapacidad/incapacidad persistente o significativa, o
• es una anomalía congénita/defecto de nacimiento.
El término "potencialmente mortal" en la definición de "grave" se refiere a un acontecimiento en el que el sujeto estaba en riesgo de muerte en el momento del acontecimiento; no se refiere a un acontecimiento que hipotéticamente podría haber causado la muerte si hubiera sido más grave.
El juicio médico y científico debe ejercerse al decidir si la notificación acelerada es apropiada en otras situaciones, tales como acontecimientos médicos importantes que pueden no poner en peligro la vida de inmediato o provocar la muerte o la hospitalización, pero pueden poner en peligro al sujeto o pueden requerir intervención para prevenir uno de los otros resultados enumerados en la definición anterior. Estos también deben considerarse serios.
Ejemplos de tales acontecimientos son el tratamiento intensivo en un departamento de urgencias o en el hogar para el broncoespasmo alérgico; discrasias sanguíneas o convulsiones que no resultan en hospitalización; o desarrollo de dependencia de fármacos o abuso de fármacos.
Informe de embarazo
Si una sujeto queda embarazada durante el estudio, la sujeto debe informar al centro del estudio lo antes posible. Tras la confirmación del embarazo, se debe suspender el fármaco del estudio en la sujeto, pero puede continuar participando en el estudio. El investigador debe completar un formulario de embarazo específico del estudio tras la confirmación de un embarazo y enviarlo a Innovaderm Research dentro de las 24 horas posteriores a la confirmación del embarazo. Innovaderm Research informará de todos los casos de embarazo al promotor de manera oportuna (la información de contacto que se utilizará es la misma que para los informes de AAG). El seguimiento posterior al tratamiento debe realizarse para garantizar la seguridad del sujeto. El embarazo no es en sí un AA o AAG; sin embargo, Las complicaciones o anomalías maternas/fetales se registrarán como AA o AAG, según sea apropiado. El investigador seguirá el embarazo hasta su finalización (o hasta la interrupción del embarazo) y notificará a Innovaderm Research el resultado como seguimiento del Formulario de embarazo inicial.
GARANTÍA DE CALIDAD DE DATOS/CONTROL DEL CENTRO
Durante el estudio, Se realizarán visitas de control a intervalos regulares. Las visitas de control se realizarán para garantizar el cumplimiento del protocolo, la calidad de los datos, la precisión de las entradas en el eCRF, la contabilidad del fármaco, el cumplimiento de los requisitos reglamentarios y adecuación continua del centro de investigación y sus instalaciones.
El centro puede ser auditado y/o controlado por un oficial de control de calidad nombrado por el promotor y/o las autoridades reguladoras pueden desear realizar auditorías en el centro. Se avisará al investigador antes de que ocurra una auditoría y se espera que coopere con cualquier auditoría, proporcione asistencia y documentación (incluidos los datos fuente) según lo solicitado.
RECOPILACIÓN Y RETENCIÓN DE DATOS
El personal del centro introducirá los datos de los sujetos utilizando Medrio eClinical Overnight, un sistema de captura y notificación de datos electrónicos (EDC) basado en la web. Esta aplicación se configurará para la entrada remota. Medrio Inc. son los desarrolladores y propietarios de Medrio eClinical Overnight. El software EDC ha sido completamente validado y cumple con los requisitos de 21 CFR Parte 11. El personal del centro del investigador no tendrá acceso al sistema EDC hasta que haya sido completamente formado por el promotor o el delegado. El personal investigador designado introducirá los datos requeridos por el protocolo en los eCRF utilizando esta aplicación basada en la web. Los programas de validación automática verifican las discrepancias de datos en los eCRF y, generando mensajes de error apropiados, permiten la modificación o verificación de los datos introducidos por el personal investigador antes de confirmar los datos. El investigador debe certificar que los datos son completos y exactos mediante la aplicación de una firma electrónica a los eCRF.
El investigador debe mantener los documentos fuente para cada sujeto en el estudio, que consiste en notas de casos y visitas (registros médicos clínicos) que contienen información demográfica y médica y los resultados de cualquier prueba o evaluación. Toda la información en los eCRF debe ser rastreable a estos documentos fuente en el archivo del sujeto. Los datos que no requieren un registro escrito o electrónico se definirán antes del inicio del estudio y se registrarán directamente en los eCRF, que se documentará como datos fuente.
Los datos recopilados serán codificados y almacenados electrónicamente en un sistema de base de datos. Los datos validados pueden transferirse posteriormente al promotor.
CONFIDENCIALIDAD DE DOCUMENTOS DE PRUEBA Y REGISTROS DE SUJETOS
El investigador debe asegurar que se mantendrá el anonimato de los sujetos y que sus identidades estén protegidas de partes no autorizadas. En los CRF u otro documento presentado al promotor, los sujetos no deben estar identificados por sus nombres, sino por un código de identificación. El investigador debe mantener un registro de inscripción de sujetos que relacione los códigos con los nombres de los sujetos. El investigador debe mantener documentos que no se presenten al promotor, por ejemplo, formularios de consentimiento por escrito de los sujetos, en la más estricta confidencialidad.
ARCHIVOS DEL INVESTIGADOR/RETENCIÓN DE DOCUMENTOS
El investigador debe mantener registros adecuados y precisos para permitir que la realización del estudio esté completamente documentada y que los datos del estudio se verifiquen posteriormente. Estos registros incluyen, aunque sin limitaciones, la identidad de todos los sujetos participantes, todos los documentos de consentimiento informado originales firmados, copias de todos los CRF, formularios de informes de seguridad, documentos fuente y registros detallados de la disposición del tratamiento y documentación adecuada de la correspondencia relevante. Estos documentos deben clasificarse en dos categorías diferentes: Archivo de estudio del investigador y documentos de origen clínico del sujeto.
El investigador debe conservar los registros de acuerdo con la Conferencia Internacional de Armonización (ICH), los reglamentos locales, o según lo especificado en el Acuerdo de ensayo clínico (CTA), lo que sea más largo.
TAMAÑO DE LA MUESTRA Y MÉTODOS ESTADÍSTICOS
Determinación del tamaño de la muestra
El criterio de valoración principal se puede traducir como un análisis de respuesta, en el que un sujeto se clasificará como Respondedor si logra una puntuación IGA de 0 (claro) o 1 (casi claro) en la semana 8, considerando una disminución de 2 puntos desde el momento basal. Un tamaño de muestra de 45 sujetos tendrá una potencia del 80 % para detectar una diferencia estadísticamente significativa del 25 % entre los respondedores del grupo tratado y del grupo de placebo, según una prueba de chi-cuadrado y un alfa de 0,05. Según la revisión de la literatura, se espera que el placebo alcance hasta el 7 %, por lo tanto, la proporción mínima esperada en el grupo tratado debe ser al menos del 32 %. Permitiendo un 10 % de abandono, en el estudio se deberá incluir un total de 100 sujetos. Planes estadísticos y analíticos
Las variables continuas se resumirán en tablas e incluirán el número de sujetos, la media, la desviación estándar, la mediana, el mínimo, el máximo y el rango intercuartílico. Las variables categóricas se presentarán en tablas como frecuencias y porcentajes.
Todas las pruebas estadísticas serán de dos colas y se realizarán con un nivel de significación de 0,05, a menos que se especifique de otro modo.
Disposición del sujeto
La eficacia se evaluará en función de la población ITT y los análisis se realizarán en función del tratamiento
aleatorizado y no del tratamiento recibido.
La población por protocolo (PP) incluirá a todos los sujetos que fueron asignados al azar sin desviaciones significativas del protocolo. Los criterios específicos para la población PP y la población ITT se detallarán en un plan de análisis estadístico separado.
La población de seguridad se definirá como todos los sujetos que recibieron al menos una dosis del fármaco. El análisis se realizará de acuerdo con el tratamiento real que recibieron.
Análisis de la eficacia
El criterio de valoración principal se puede traducir como un análisis de respuesta, en el que un sujeto se clasificará como Respondedor si logra una puntuación IGA de 0 (claro) o 1 (casi claro) en la semana 8, considerando una disminución de 2 puntos desde el momento basal. La comparación entre grupos para el criterio de valoración primario se realizará mediante una prueba de chi-cuadrado. Se realizará un análisis de sensibilidad para examinar la contribución del centro en las comparaciones utilizando una prueba de Cochran-Mantel-Haenszel donde el centro se utilizará como factor de estratificación. El análisis principal de la eficacia se realizará utilizando los valores observados y se realizará un análisis de apoyo con un análisis del punto de inflexión, una estrategia utilizada para evaluar el impacto de los valores perdidos. Los valores perdidos se establecerán en respondedor/no respondedor sucesivamente y se representarán las decisiones basadas en los valores de p. Los detalles de esta estrategia se presentarán en el plan de análisis estadístico. No habrá imputación por datos perdidos en otras visitas. Los análisis se realizarán utilizando la población ITT y servirán como análisis primario, mientras que el análisis del criterio de valoración primario utilizando la población PP se utilizará un análisis de sensibilidad.
Los criterios de valoración secundarios que implican un cambio con respecto al valor basal se analizarán utilizando un análisis de la covarianza (ANCOVA) que incluye el cambio desde el valor basal como dependiente, el centro y el grupo de tratamiento, y el centro como efectos fijos, y el valor basal como covariable. Las medias de Ls y el IC del 95 % se presentarán junto con el valor de p correspondiente de la comparación del tratamiento. Los criterios de valoración secundarios que implican la proporción se analizarán mediante una prueba de Cochran-Mantel-Haenszel estratificada por centro y se presentará el valor de p. Los análisis de los criterios de valoración secundarios se realizarán utilizando datos observados y no se utilizará ninguna imputación para la observación perdida.
Todos los detalles sobre los análisis estadísticos se incluirán en un plan de análisis estadístico separado.
Análisis de seguridad
Todos los acontecimientos adversos (AA) que se produzcan después de la administración del fármaco del primer estudio durante el estudio se clasificarán de acuerdo con la terminología del Diccionario médico para actividades reguladoras (MedDRA). Las descripciones de los AA incluirán la fecha de inicio, la fecha en que finalizó el AA (si se resolvió), la gravedad y seriedad del AA, la relación entre el AA y el fármaco del estudio, y el resultado. El enfoque en este protocolo se limitará a los acontecimientos adversos emergentes durante el tratamiento.
Los AA notificados se resumirán por la cantidad de sujetos que notifican los acontecimientos, así como por la Clasificación de Órganos y Sistemas, término preferente, gravedad, seriedad y relación con el fármaco del estudio. Para el resumen de los Aa por gravedad, cada paciente se contará solo una vez dentro de una Clasificación de órganos y sistemas o un Término preferente mediante el uso de los AA con la mayor intensidad dentro de cada categoría para cada análisis. Para el resumen de los AA por relación con la medicación del estudio, cada paciente se contará solo una vez dentro de una Clasificación de Órganos y Sistemas o un Término Preferente mediante el uso de los AA con la relación más alta notificada dentro de cada categoría. Para el resumen de los AA por relación con la medicación del estudio y la gravedad, cada paciente se contará solo una vez dentro de una Clasificación de Órganos y Sistemas o un Término Preferente (1) mediante el uso de la mayor relación notificada, seguido de (2) la intensidad más alta notificada.
Toda la información relativa a los AA observados durante el estudio se enumerará por paciente, detallando textualmente, Clasificación de Sistemas y Órganos, término preferente, fecha de inicio, fecha de finalización, intensidad, resultado y relación con el fármaco del estudio. El inicio del AA también se mostrará relativo (en número de días) al día de la administración del artículo de prueba. Los acontecimientos adversos graves (AAG) se tabularán por grupo de tratamiento, relación con el artículo de prueba y una referencia a la aparición de los AAG con el día relativo de dosificación.
Los fármacos concomitantes se codificarán con el Diccionario de fármacos de la OMS y se enumerarán por sujeto. Además, también se proporcionará una lista de sujetos que abandonaron el estudio y una lista de sujetos que experimentaron AAG.
Los resultados de los análisis de laboratorio y las constantes vitales se tabularán utilizando estadística descriptiva. El
valor en la visita, así como el cambio con respecto al valor basal se presentarán de manera descriptiva.
No se realizarán estadísticas inferenciales sobre variables de seguridad (TEAE, medicación concomitante, análisis de laboratorio y constantes vitales).
ASPECTOS ÉTICOS
Regulaciones locales/Declaración de Helsinki
Este estudio se llevará a cabo de acuerdo con los principios éticos que tienen su origen en la Declaración de Helsinki (2008) y que son consistentes con la directriz tripartita ICH de las "Buenas prácticas clínicas" (julio de 2002) y las leyes y regulaciones aplicables del país en que se realiza la investigación, lo que brinde la mayor protección al individuo.
Revisión ética
El promotor entiende que este protocolo (y cualquier enmienda), así como los procedimientos de consentimiento apropiados, será revisado y aprobado por una junta de ética de investigación/junta de revisión institucional (REB/IRB). Esta Junta debe operar de acuerdo con las regulaciones federales vigentes. El investigador enviará al promotor una carta o certificación de aprobación antes del inicio del estudio y también cada vez que se realicen modificaciones posteriores al protocolo.
Consentimiento informado
Es responsabilidad del investigador, o una persona designada por el investigador (si es aceptable por la regulación local), obtener el consentimiento informado por escrito de cada individuo que participa en este estudio después de una explicación adecuada de los objetivos, los métodos, objetivos y riesgos potenciales del estudio. También debe explicarse a los sujetos que son completamente libres de negarse a entrar en el estudio o retirarse de él en cualquier momento por cualquier motivo.
Si la nueva información de seguridad produce cambios significativos en la evaluación de riesgo/beneficio o cualquier información nueva que pueda afectar a la disposición a continuar participando, si es necesario, el formulario de consentimiento debe ser revisado y actualizado por la Junta de Ética de Investigación/Junta de Revisión Institucional. Todos los sujetos (incluidos los que ya están siendo tratados) deben ser informados de la nueva información, se les harán entregado una
en el estudio.
APÉNDICE A Diagrama de flujo del estudio

continuación

APÉNDICE B - Evaluación global del investigador

APÉNDICE C - Área de eccema e índice de gravedad (EASI)
Cuatro sitios anatómicos: cabeza, extremidades superiores, tronco y extremidades inferiores: se evalúan para determinar la existencia de eritema, induración (pápulas), excoriación y liquenificación como se ve el día de la
exploración. La gravedad de cada signo se evalúa mediante una escala de 4 puntos:
0 = Sin síntomas
1 = Ligero
2 = Moderado
3 = Marcado
El área afectada por la dermatitis atópica dentro de un sitio anatómico dado se estima como un porcentaje del área total de ese sitio anatómico y se le asigna un valor numérico de acuerdo con el grado de afectación de la dermatitis atópica de la siguiente manera:
0 = sin afectación
1 = < 10%
2 = 10 a <30 %
3 = 30 a <50 %
4 = 50 a <70 %
5 = 70 a <90 %
6 = 90 a 100 %
La puntuación EASI se obtiene usando la fórmula
EASI = 0,1 (Eh Ih Exh Lh) Ah 0,2 (Eu lu Exu Exu) Au 0,3 (Et It Ext Ext) At 0,4 (El II Exl Exl) Al
Donde E, I, Ex, L y A denotan eritema, induración, excoriación, liquenificación y área, respectivamente, y h, u, t e I denotan cabeza, extremidades superiores, tronco y extremidades inferiores, respectivamente.
APÉNDICE D - Puntuación de la dermatitis atópica (SCORAD)
Se seleccionan seis elementos (eritema, edema/papulación, supuración/costras, excoriación, liquenificación y sequedad) para evaluar la gravedad de la DA. La intensidad de cada elemento se califica utilizando una escala de 4 puntos:
0 = Sin síntomas
1 = Leve
2 = Moderado
3 = Grave
El área elegida para la calificación debe ser representativa (intensidad promedio) para cada elemento. A continuación se añadirán las clasificaciones de intensidad individuales para cada elemento (que van de 0 a 18) y se multiplicarán por 3,5, dando una puntuación máxima de 63.
La BSA global afectada por DA se evalúa (de 0 a 100 %) y se divide por 5. La palma de la mano de un paciente representa el 1 % de su BSA total. El máximo es 20.
Los elementos subjetivos incluyen la pérdida de sueño y la aparición de prurito. Estos se evalúan pidiendo a los pacientes que indiquen en la escala de 10 cm (0-10) del formulario de evaluación el punto correspondiente al valor promedio de los últimos tres días/noches. La puntuación máxima combinada de estos dos es 20.
La suma de las medidas anteriores representa el SCORAD que puede variar de 0 a 103. Si se excluyen las puntuaciones subjetivas de prurito y pérdida de sueño, la SCORAD se convierte en la SCORAD objetiva (intervalo de puntuación 0-83).
APÉNDICE E - Medida de eccema orientada por el paciente (POEM)
N. ° de ID del paciente:__ - ____ - Iniciales del paciente:____
Día de visita: Fecha de la visita (dd-mmm-aaaa):
Rodee con un círculo una respuesta para cada una de las siete preguntas siguientes sobre su eccema. Deje en blanco cualquier pregunta que no pueda responder.
Rodee con un círculo una respuesta para cada una de las siete preguntas siguientes sobre su eccema. Deje en blanco cualquier pregunta que no pueda responder.
1. En la última semana, ¿cuántos días le ha picado la piel debido al eccema?
Ningún día 1-2 días 3-4 días 5-6 días Todos los días
2. En la última semana, ¿cuántas noches se ha alterado su sueño debido al eccema?
Ningún día 1-2 días 3-4 días 5-6 días Todos los días
3. En la última semana, ¿cuántos días le ha sangrado la piel debido al eccema?
Ningún día 1-2 días 3-4 días 5-6 días Todos los días
4. En la última semana, ¿cuántos días ha supurado o exudado la piel un líquido claro debido al eccema? Ningún día 1-2 días 3-4 días 5-6 días Todos los días
5. En la última semana, ¿cuántos días se le ha agrietado la piel debido al eccema?
Ningún día 1-2 días 3-4 días 5-6 días Todos los días
6. En la última semana, ¿cuántos días se le ha descamado la piel debido al eccema?
Ningún día 1-2 días 3-4 días 5-6 días Todos los días
7. En la última semana, ¿cuántos días ha sentido la piel seca o áspera debido al eccema?
Ningún día 1-2 días 3-4 días 5-6 días Todos los días
© CR Charman, AJ Venn, HC Williams, diciembre de 2004.
APÉNDICE F - Índice de calidad de vida dermatológica (DLQI)
El objetivo de este cuestionario es medir cuánto ha afectado su problema en la piel a su vida EN LA ÚLTIMA SEMANA. Por favor, marque una casilla para cada pregunta.

continuación

El objetivo de este cuestionario es medir cuánto ha afectado su problema en la piel a su vida EN LA ÚLTIMA SEMANA. Por favor, marque una casilla para cada pregunta.
APÉNDICE G - Criterios de diagnóstico para la dermatitis atópica
Según el criterio de inclusión 2, un sujeto tendrá un diagnóstico clínico de dermatitis atópica de acuerdo con los criterios de Hanifin y Rajka. Los criterios son los siguientes:
Criterios principales (debe tener al menos tres)
Prurito
Morfología y distribución típica:
Adultos: liquenificación flexural o linealidad
Niños y bebés: afectación de las superficies faciales y extensoras
Dermatitis crónica o recurrente
Antecedentes personales o familiares de atopia
Criterios menores (debe tener al menos tres)
Xerosis
Ictiosis/queratosis pilar/hiperlinearidad palmar
Reactividad inmediata en la prueba cutánea (tipo 1)
IgE sérica elevada
Edad temprana al inicio
Tendencia a infecciones cutáneas (Staphylococcus aureus, herpes simple)/deterioro celular
Inmunidad
Dermatitis de manos/pies
Eccema en los pezones
Conjuntivitis
Pliegue de Dennie-Morgan
Queratocono
Catarata subcapsular anterior
Oscurecimiento orbital
Palidez/eritema facial
Pitiriasis alba
Pliegues anteriores del cuello
Picor al sudar
Intolerancia a la lana y a los disolventes lipídicos
Acentuación perifolicular
Intolerancia a alimentos
Evolución influenciada por factores ambientales/emocionales
Blanco demográfico/blanco diferido
Ejemplo 3
Se realizó una investigación in v ivo de la eficacia antihipertensora después de la administración conjunta de DGLA y aspirina en ratas con hipertensión espontánea. Este experimento investigó la eficacia del DGLA cuando se administró conjuntamente de forma crónica con aspirina para reducir la respuesta hipertensora inducida de forma aguda por la fenilefrina en ratas con hipertensión espontánea. En este ensayo se usaron ratas macho adultas con hipertensión espontánea (250-300 g), criadas por Charles River Laboratories. Los animales fueron identificados a su llegada según las pautas de CCPA. Los animales se estabularon por parejas por grupo (dosis baja aspirina, dosis
alta aspirina o solo aspirina) durante la duración de la sonda oral antes de la cirugía. Se registró todo el cuidado de los animales y el mantenimiento del animalario con documentos guardados en la instalación de las pruebas.
La rata es un modelo bien caracterizado y altamente sensible para la evaluación de los efectos de la tensión vascular y la evaluación de la eficacia después de la exposición crónica a un artículo de prueba. El diseño de este estudio se basa en las directrices tripartitas armonizadas de la Conferencia Internacional sobre Armonización (ICH) [ICH S7A] y en los procedimientos generalmente aceptados para la prueba de compuestos farmacéuticos. Este estudio de laboratorio no clínico fue diseñado como un estudio que no cumple con las GLP y no requirió la participación de QA.
El promotor seleccionó las concentraciones de DGLA a analizar (50 y 500 mg/kg), así como la concentración de aspirina (10 mg/kg) en función de la información disponible en el momento del diseño de este estudio. Se seleccionaron para cubrir un intervalo de concentraciones útiles para la identificación del mecanismo de acción del artículo de prueba.
Las concentraciones del control positivo (carvedilol 0,1 mg/kg) y el hipertensor (fenilefrina 3, 10 y 20 pg/kg) se seleccionaron según las referencias bibliográficas. Todas las preparaciones de soluciones se documentaron en un Formulario de Contenido de la Solución, que define la información del etiquetado y toda la información relevante sobre los reactivos: número de lote, condiciones de almacenamiento, contenido y fechas de preparación/caducidad. El artículo de prueba se formuló utilizando aceite de oliva como vehículo. Esta solución madre se administró después por sonda oral una vez al día durante siete días consecutivos. Se probaron una dosis baja y una dosis alta (50 mg/kg y 500 mg/kg).
Se preparó una solución madre de aspirina de 5 mg/ml disolviendo la cantidad apropiada de aspirina en DMSO al 100 % que luego se diluyó en agua. La concentración de DMSO fue inferior al 1 %. El volumen apropiado se administró por sonda oral a cada animal una vez al día durante siete días consecutivos. La solución madre se almacenó a temperatura ambiente y se consideró estable durante la duración de la sonda.
Se preparó una solución madre de 0,4 mg/ml de carvedilol en PBS (pH = 4,00 ± 0,05). El volumen apropiado se administró a cada animal mediante una inyección intravenosa al final del experimento el día de la cirugía. La solución madre se almacenó a 4 °C y se consideró estable durante la duración del experimento.
Se preparó una solución madre de fenilefrina de 1 mg/ml en PBS (pH = 7,4 ± 0,05). La solución madre se almacenó a 4 °C. La fecha de caducidad se estableció en 14 días después de la preparación.
Cualquier artículo de prueba restante se destruirá después de completar el estudio.
Las ratas macho con hipertensión espontánea que pesaban aproximadamente 300 g llegaron a las instalaciones al menos dos días antes de comenzar la sonda oral. Los animales fueron asignados a uno de tres grupos (dosis baja aspirina, dosis alta aspirina y aspirina), y se estabularon por parejas durante el período de aclimatación. Cada grupo (n = 4) recibió el compuesto apropiado por sonda oral usando una aguja de alimentación de 16G. La solución madre se administró una vez al día durante siete días consecutivos. Al séptimo día de administración, los animales fueron sometidos a cirugía y monitorización de la hipertensión.
Se anestesió a las ratas con una mezcla de isoflurano-O2 al 2,5-3,0 % en una caja de inducción. Cuando se extrajo a los animales de la caja de inducción, se conectaron a un cono nasal para mantener la anestesia durante la traqueotomía. Se realizó a los animales una traqueotomía con una sonda endotraqueal (7 cm de longitud desde el conector hasta la punta, hecha con un tubo PE205 de BD y una aguja 16G) para facilitar la respiración espontánea, estabilizar la hemodinámica y mantener a la rata anestesiada con una mezcla de isoflurano-O2. Se insertaron dos catéteres: uno en la arteria femoral derecha para medir la presión arterial sistémica (PAS) y uno en la vena femoral derecha para administrar el hipertensor y el control positivo. Las señales del ECG se adquirieron con contactos de ECG colocados en una configuración de Derivación-1 en el animal anestesiado y se colocó un pulsioxímetro en la pata trasera del animal para permitir el control continuo del estado general de la rata durante la cirugía. Después de las mediciones de referencia, se administraron tres dosis de fenilefrina (3, 10 y 20 ¿rg/kg) mediante una inyección en bolo i.v., dejando 5 minutos entre dosis. Se administró una dosis del control positivo, carvedilol (0,1 mg/kg), siguiendo la mayor concentración de fenilefrina. Después del control positivo, se administraron nuevamente 10 mg/kg de fenilefrina. La presión arterial, la presión arterial sistémica (PAS) y la frecuencia cardíaca se monitorizaron de forma continua durante un total de media hora. La hipertensión se detectó como un aumento de la presión arterial diastólica, sistólica o media. Al final del experimento, se sacrificó a los animales mediante exanguinación.
El software de análisis utilizado fue Clampfit 10.2.0.14 de Axon Instruments, instalado en ordenadores personales en red con Microsoft Windows. Clampfit 10.2.0.14 se ha validado completamente en el contexto conectado en el que se utiliza. El software de gráficos para ilustraciones es Microsoft Office Excel 2007 instalado en ordenadores personales en red que se ejecutan en Microsoft Windows. La presión arterial sistémica (PAS) registrada de forma continua se usó para calcular la presión arterial sistémica media (PASm) para cada condición. La frecuencia cardíaca se controló
de forma continua desde el momento en que se indujo la anestesia. Se analizaron ANOVA unidireccionales, comparando los parámetros previos y posteriores a la exposición entre los grupos experimentales. La significación estadística se confirmó en p < 0,05.
Tabla 9. Presión arterial y presión arterial sistémica media (mm Hg) después de siete días de sonda, antes de las dosis intravenosas de fenilefrina.
Tabla 9.

La Tabla 10 muestra el cambio en la presión arterial sistémica media con dosis intravenosas de fenilefrina después de siete días consecutivos de sonda con aspirina a 10 mg/kg/día.
Tabla 10.
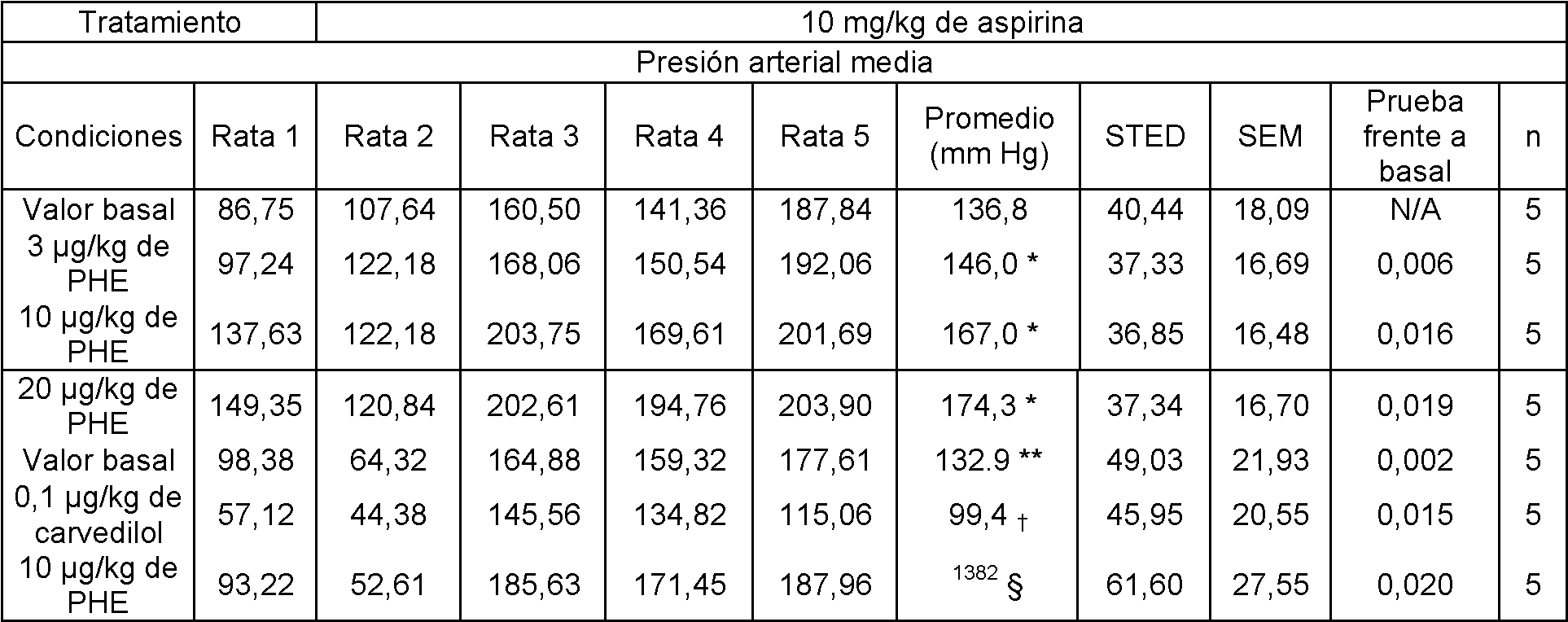
La figura 1 ilustra el cambio en la presión arterial sistémica media, en mm de Hg, después de la administración de fenilefrina. Cinco ratas recibieron dosis intravenosas de fenilefrina después de siete días consecutivos de sonda con aspirina a 10 mg/kg/día. La presión arterial sistémica media en cada una de las tres dosis de fenilefrina se comparó estadísticamente con la primera basal, mientras que la condición de carvedilol se comparó estadísticamente con la basal inmediatamente antes de la administración, y la dosis final de fenilefrina 10 ¿rg/kg) se comparó con la condición de control positivo directamente antes.
Estos resultados demuestran un aumento de la presión arterial sistémica media con dosis crecientes de fenilefrina. Todos los datos obtenidos fueron estadísticamente diferentes de los valores basales, lo que confirma el efecto hipertensor de la fenilefrina en el modelo de rata con hipertensión espontánea a la que se administró aspirina. El efecto de la fenilefrina fue completamente reversible; la presión arterial media durante el segundo valor basal (tras 20 prg/kg) fue, en realidad, bastante similar al valor basal original.
Carvedilol causó una disminución significativa cuando se añadió inmediatamente después de la fenilefrina, lo que confirma su conocida capacidad para reducir la presión arterial alta (el carvedilol es un beta-bloqueador /alfa-1-bloqueador no específico y bloquea la unión de la fenilefrina a los receptores alfa-1-adrenérgicos). La última adición de fenilefrina, después de carvedilol, causó un aumento limitado de la presión arterial sistémica media.
Figura 2. Cambio en la presión arterial media (mmHg) con dosis intravenosas de fenilefrina después de siete días consecutivos de sonda con DGLA a 50 mg/kg aspirina a 10 mg/kg.
Tabla 11. Cambio con respecto al valor basal en la presión arterial media con dosis intravenosas de fenilefrina después de siete días consecutivos de sonda con DGLA a 50 mg/kg aspirina a 10 mg/kg.
Tabla 11.

Los resultados presentados en la figura 2 demuestran cambios en la presión arterial media, en mm de Hg, después de dosis intravenosas de fenilefrina en cinco ratas con hipertensión espontánea después de siete días de alimentación oral con DGLA a 50 mg/kg aspirina a 10 mg/kg. Los resultados para el artículo de prueba se compararon con los datos obtenidos con el grupo de solo aspirina (10 mg/kg). El carvedilol se comparó con la segunda basal y la última dosis de fenilefrina se comparó estadísticamente con el carvedilol.
Los animales exhibieron un aumento de la presión arterial dependiente de la dosis a medida que aumentaba la concentración de fenilefrina. Las ratas a las que se les había administrado DGLA 50 mg/kg y aspirina exhibieron un aumento en la presión arterial para una dosis dada de fenilefrina que fue esencialmente la misma que la medida en las ratas que recibieron aspirina durante 10 días. Con las ratas a las que se administró DGLA a 50 mg/kg además de 10 mg/kg de aspirina, no hubo diferencias significativas en la presión arterial media en comparación con el grupo de aspirina en cualquiera de las concentraciones de fenilefrina. Por lo tanto, parece que la aspirina diaria previene los beneficios obtenidos de las dosis diarias de 50 mg/kg de DGLA.
Carvedilol causó una disminución significativa de la presión arterial en animales alimentados con DGLA aspirina. Debido a un problema con la anestesia, la rata n.° 3 no recibió la última dosis de fenilefrina (n = 4 para esta condición).
Tabla 12. Cambio en la presión arterial media con dosis intravenosas de fenilefrina después de siete días consecutivos de sonda con DGLA a 500 mg/kg coadministrado con aspirina a 10 mg/kg.
Tabla 12.

La figura 3 ilustra los cambios en la presión arterial media después de dosis intravenosas de fenilefrina después de siete días consecutivos de sonda con DGLA a 500 mg/kg coadministrado con aspirina a 10 mg/kg en cuatro ratas con hipertensión espontánea. Al igual que con la dosis más baja de DGLA aspirina, las condiciones de fenilefrina se compararon con las del grupo de solo aspirina. El carvedilol se comparó estadísticamente con el valor basal justo antes de la administración, mientras que la última dosis de fenilefrina (10 mg/kg) se comparó con el control positivo. A pesar de las presiones arteriales medias más bajas observadas, no hubo diferencia estadística significativa entre la presión arterial sistémica media de las ratas a las que se administró DGLA y aspirina y a las que se administró
solo aspirina. Por lo tanto, parecería que la aspirina nuevamente evitó el beneficio de la administración diaria de 500 mg/kg de DGLA. El control positivo logró disminuir estadísticamente la presión arterial de los animales, como se observó en los otros grupos. El aumento causado por la última dosis de fenilefrina no se consideró significativo. La Tabla 13 muestra la presión arterial media en el momento basal después de siete días consecutivos con seis grupos de sonda diferentes.
Tabla 13.
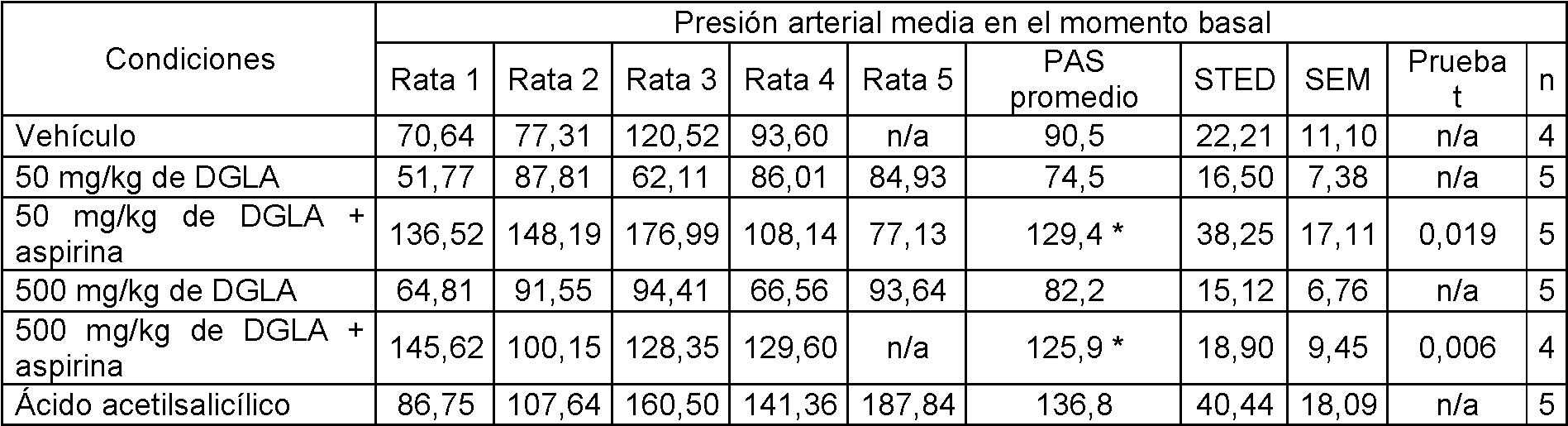
La figura 4 presenta las presiones arteriales medias e el momento basal de los seis grupos que fueron alimentados durante las dos partes de este estudio. La traqueotomía estabilizó la hemodinámica, pero también causó cambios en la presión arterial de las ratas. La presión arterial de las ratas que no recibieron aspirina (vehículo, DGLA 50 mg/kg y DGLA 500 mg/kg) se obtuvo durante la primera fase de este estudio (estudio n.° 20131022-1) y se registraron las de los otros grupos durante este presente estudio (diferentes lotes de animales de Charles River, pero la misma cepa y tamaño de rata). Se realizaron pruebas t estadísticas entre los grupos a los que se administró DGLA a 50 o 500 mg/kg y su equivalente añadido con aspirina a 10 mg/kg. Hubo un aumento estadísticamente significativo de la presión arterial media al inicio del estudio entre los 50 mg/kg de DGLA solo y los 50 mg/kg de DGLA coadministrado con aspirina a 10 mg/kg, así como entre el grupo de 500 mg/kg de DGLA en comparación con 500 mg/kg de DGLA con 10 mg/kg de aspirina (aumento significativo marcado con *).
Por lo tanto, parece que DGLA disminuye con éxito la presión arterial en ratas con hipertensión espontánea, un efecto que es eliminado por la aspirina.
La Tabla 14 muestra la presión arterial media con una dosis intravenosa de fenilefrina a 20 pk/kg después de siete días consecutivos con seis grupos de sonda diferentes.
Tabla 14.

La figura 5 presenta las presiones arteriales medias de todos los grupos analizados en los dos estudios que investigan DGLA. Estas presiones arteriales se obtuvieron después de una dosis intravenosa de fenilefrina a 20 pg/kg. Al igual que con los resultados adquiridos en el momento basal, cada dosis de DGLA se comparó estadísticamente con su condición equivalente en el grupo de aspirina solamente. Se observó un aumento significativo de la presión arterial media entre ambos grupos correspondientes (marcado con *).
Este estudio fue diseñado para investigar la eficacia de DGLA, solo y cuando se administra conjuntamente de forma crónica con aspirina, para reducir la presión arterial medida en ratas hipertensas, así como la reducción en la
respuesta hipertensora inducida de forma aguda por fenilefrina en la misma cepa de ratas con hipertensión espontánea.
La rata con hipertensión espontánea (SHR) es un modelo de uso habitual en la investigación de la hipertensión porque produce, dentro de cada colonia, cambios uniformes en respuesta a efectores directos e indirectos del sistema cardiovascular. Las ratas han sido seleccionadas y criadas de forma endogámica durante generaciones, definidas como hipertensas por una presión arterial sistólica de más de 150 mm Hg que persiste durante más de un mes.
El estudio reveló que la administración conjunta crónica de DGLA y aspirina impidió que DGLA redujera la presión arterial sistémica media (hasta en un 25 %) como se observó anteriormente en las ratas con hipertensión espontánea tratadas con DGLA.
Las dosis de DGLA probadas (50 y 500 mg/kg) combinadas con aspirina a 10 mg/kg no redujeron significativamente la elevación dependiente de la dosis de la presión arterial media después de la inyección de fenilefrina, en comparación con los animales a los que se administró solo aspirina. Las dos dosis de DGLA causaron efectos similares, lo que sugiere que los beneficios de mayores dosis de DGLA pueden ser mínimos si se administra conjuntamente aspirina. Cuando ambas dosis de DGLA se compararon con sus condiciones equivalentes de solo aspirina, la aspirina causó un claro aumento en la presión arterial media en la condición basal, así como cuando las ratas fueron expuestas a una dosis intravenosa de 20 pg/kg de fenilefrina.
En el presente estudio, se utilizó carvedilol como control positivo. Es un beta-bloqueador que se usa para tratar la insuficiencia cardíaca y la presión arterial alta. Actúa relajando los vasos sanguíneos y disminuyendo la frecuencia cardíaca, mejorando así la recarga ventricular, el flujo sanguíneo y disminuyendo la presión arterial. El efecto del carvedilol, cuando se inyecta por vía intravenosa en ratas con hipertensión espontánea, confirmó su capacidad para disminuir la presión arterial después de cada una de las tres dosis de fenilefrina y demostró la sensibilidad del sistema de prueba.
Ejemplo 4
Se realizó un estudio de fase uno en un solo centro, doble ciego, aleatorizado, controlado con placebo, de dos partes con sujetos sanos de ambos sexos, de 18 a 45 años, ambos incluidos. El objetivo principal de este estudio fue evaluar la seguridad y la farmacocinética (PK) plasmática de dosis únicas administradas por vía oral y dosis múltiples administradas por vía oral de cápsulas de DS107G (una vez al día durante 28 días) en sujetos sanos. Los objetivos secundarios de este estudio fueron evaluar lo siguiente:
El efecto de los alimentos sobre la PK de una dosis única administrada por vía oral de la cápsula de DS107G en sujetos sanos.
La PK de DGLA en la piel humana después de múltiples dosis de cápsula e DS107G administrada una vez al día durante 28 días a sujetos sanos.
Se inscribieron un total de 48 sujetos (incluidos 32 para el estudio de dosis única y 16 para el estudio de dosis múltiples). Los estudios de dosis única y múltiples consisten en 8 sujetos por cohorte (idealmente 4 varones y 4 mujeres, pero no menos de 3 por sexo).
Los sujetos fueron asignados al azar en una proporción de 3:1 de DG107G:placebo mediante aleatorización en bloque. La pare uno de dosis única del estudio comprendió tres cohortes de ocho sujetos cada una, con la adición de la cohorte cuatro basada en la evaluación de los datos de seguridad de la cohorte tres por el Comité de Monitorización de Seguridad (SMC). Se administró una dosis oral única de DS107G o placebo equivalente a sujetos en ayunas en las cohortes uno a tres (500, 1.000 y 2.000 mg, respectivamente) en paralelo; a los sujetos de la cohorte cuatro se les administró una dosis de 4.000 mg después de completar la cohorte tres. Los sujetos en la cohorte dos recibieron 1.000 mg o placebo equivalente en el estado alimentado > 14 días después de la primera dosis.
La parte dos de dosis múltiples del estudio comprendió dos cohortes (cohortes cinco y seis) de ocho sujetos cada una, a los que se administraron múltiples dosis del fármaco del estudio en ayunas durante 28 días. Los sujetos en las cohortes cinco y seis recibieron 2.000 o 4.000 mg de DS107G, respectivamente, o un placebo equivalente una vez al día durante 28 días. La cohorte seis se inició después de que el SMC evaluara los datos de seguridad de la cohorte cinco. No se planificaron análisis intermedios.
En la parte uno del estudio, se realizó un análisis PK plasmático de ácido dihomo-gamma-linolénico (DGLA) y ácido 15-hidroxienosatrienoico (15-HETrE), solo en sujetos en ayunas para las cohortes de una a cuatro hasta 312 horas después de la dosis los días 1 a 5, 8, 14 del estudio y la visita de seguimiento. En la parte dos, el análisis PK plasmático de DGLA se realizó para las cohortes cinco y seis hasta 168 horas después de la dosis los días 1, 2, 3, 5 y 8 del estudio. Las evaluaciones de seguridad se monitorizaron durante todo el estudio.
Se utilizó un diseño doble ciego, aleatorizado y controlado con placebo para minimizar el sesgo durante las evaluaciones de seguridad y tolerabilidad.
La parte uno del estudio siguió un diseño de dosis única ascendente con una escalada de dosis a 4.000 mg. Con la excepción de la dosis de 4.000 mg, todas las dosis en la parte uno del estudio se probaron previamente en seres humanos. No se observaron acontecimientos adversos relacionados con el tratamiento (TEAE) en los regímenes de dosis múltiples previamente probados, tal como 150 mg/día durante 28 días, 450 mg/día durante 28 días, 1.000 mg/día durante 14 días y 2.000 mg/día durante 10 días. La selección de dosis se basó en las dosis de DGLA oral probadas en estudios clínicos previos y la caracterización de la PK y la seguridad de las dosis previamente probadas y más altas se observaron como objetivo principal.
También se evaluó el efecto de los alimentos sobre la biodisponibilidad oral de DGLA.
Se incluyeron sujetos femeninos sanos en este estudio para evaluar un posible componente de género en el biomarcador di-hidrotestosterona ya que DGLA está implicado en el metabolismo de los esteroides.
La duración total de la participación de cada sujeto en la parte uno del estudio fue de aproximadamente 14 días, excluyendo el período de selección. En la Parte 2 del estudio, la duración fue de aproximadamente 42 días.
Los procedimientos de selección se realizaron para las partes una y dos antes del inicio del estudio el día -1 (basal) mediante la evaluación de las evaluaciones de seguridad. Las evaluaciones de seguridad incluyeron acontecimientos adversos (AA), pruebas de laboratorio clínico (hematología, bioquímica, virología [antígeno de superficie de hepatitis B, anticuerpos del virus de la inmunodeficiencia humana (VIH), anticuerpos contra la hepatitis C] y análisis de orina), resultados de la prueba de abuso de drogas (DOA) y prueba de embarazo para mujeres (gonadotropina p coriónica humana en orina [pHCG]), constantes vitales (presión arterial [BP], pulso, temperatura), Electrocardiogramas de 12 derivaciones (ECG), exploraciones físicas y evaluación de fármacos concomitantes (solo la parte dos del estudio).
Se requirió que todos los sujetos cumplieran con los criterios de inclusión y exclusión descritos a continuación. Sin embargo, el investigador y el promotor consideraron aceptables las desviaciones menores que eran clínicamente insignificantes y que no planteaban problemas de seguridad, consistente con el protocolo.
Todos los sujetos considerados para participar en el estudio debían cumplir los siguientes criterios de inclusión:
1. El sujeto era varón o mujer.
2. Sujeto femenino y pareja femenina de sujeto masculino:
• No debían estar embarazadas (las mujeres debían presentar una prueba de embarazo en orina negativa antes de entrar en el estudio).
• No debían estar en periodo de lactancia.
• No debían estar planificando un embarazo durante el período de estudio o dentro de los 3 meses posteriores al estudio.
• Debían usar una forma adecuada de anticoncepción antes de entrar en el estudio y durante 3 meses más después de la visita de seguimiento.
3. El sujeto tenía entre 18 y 45 años, ambos incluidos.
4. El sujeto había firmado el formulario de consentimiento informado.
5. El índice de masa corporal (IMC) del sujeto estaba entre 18.0 y 30.0 kg/m2, ambos incluidos.
6. Se consideró que el sujeto gozaba de buena salud en opinión del investigador al evaluar las evaluaciones de seguridad.
7. Los sujetos deben haber podido comunicarse bien con el investigador, para comprender y cumplir con los requisitos del estudio y para comprender y firmar el ICF escrito. Los sujetos fueron excluidos del estudio si había evidencia de alguno de los siguientes criterios: El sujeto tenía una enfermedad clínicamente significativa en las 4 semanas previas a la selección.
8. Mujeres en edad fértil y parejas femeninas de sujetos masculinos que no habían usado una medida anticonceptiva segura durante 3 meses antes del estudio y no estaban dispuestas a usar una medida anticonceptiva segura durante la duración del estudio; Entre los ejemplos de medidas anticonceptivas seguras se incluyen dispositivos intrauterinos o anticonceptivos orales, diafragma o preservativo si se usa en combinación con un espermicida.
9. El sujeto usó un fármaco recetado en las 2 semanas antes de la dosificación o utilizó preparaciones de venta libre (incluidas vitaminas y suplementos) durante 1 semana antes de la dosificación (con la excepción del paracetamol, que se permitió hasta 48 horas antes de la dosificación), y anticonceptivos hormonales.
10. El sujeto usó suplementos dietéticos ricos en ácidos grasos omega-3 u omega-6.
11. El sujeto tenía un historial significativo de abuso de drogas/disolventes o presentó resultados positivos en las pruebas de detección de DOA.
12. El sujeto tenía antecedentes de abuso de alcohol, en opinión del investigador, o bebía más de 28 unidades por semana (varones) o 21 unidades por semana (mujeres) en el momento de la selección.
13. El sujeto era, en opinión del investigador, no apto para participar en el estudio.
14. El sujeto había participado en otro estudio clínico con un fármaco/dispositivo de estudio dentro de los 3 meses anteriores al primer día de administración del fármaco del estudio.
15. El sujeto tuvo un resultado positivo en la prueba de anticuerpos contra el VIH, antígeno de superficie de hepatitis B o anticuerpos de hepatitis C en la selección.
16. El sujeto había tenido una reacción adversa grave o hipersensibilidad significativa a cualquier fármaco.
17. El sujeto había donado sangre o productos sanguíneos dentro de los 3 meses anteriores a la selección. 18. El sujeto tenía hipersensibilidad conocida a cualquiera de los ingredientes del fármaco del estudio.
Los sujetos eran libres de retirar el consentimiento del estudio en cualquier momento y por cualquier motivo. Además, el investigador podía retirar a un sujeto de la participación en el estudio si, en opinión del investigador, era para el mejor interés del sujeto. Se retiraría a un sujeto del estudio por cualquiera de las siguientes razones:
• Retirada de consentimiento en cualquier momento
• Desviaciones del protocolo
• Enfermedad secundaria
• Un AA (efecto adverso).
Aunque un sujeto no estaba obligado a dar una razón para una retirada prematura, el investigador debía hacer un esfuerzo razonable para obtener la razón respetando plenamente los derechos del sujeto. Si había una razón médica para la retirada del consentimiento, el sujeto debía permanecer bajo la supervisión del investigador hasta que el sujeto tuviera una salud satisfactoria; el investigador debía realizar una evaluación de seguimiento.
Si el investigador consideró que se consideraba en el mejor interés del bienestar del sujeto, el investigador debía informar al médico general del sujeto de la razón médica de la retirada del sujeto del estudio. Debía hacerse todo lo posible para contactar con un sujeto que regresó al centro para ninguna de las citas con el fin de garantizar que el sujeto tuviera una salud satisfactoria.
En la parte uno del estudio, los sujetos de las cohortes uno a tres recibieron una dosis única del fármaco del estudio (cápsulas de 500, 1.000 o 2.000 mg como 500 mg de DS107G o el placebo equivalente) en paralelo el día 1 del estudio después de al menos un ayuno de 8 horas, de acuerdo con la Tabla 15. La cohorte 4 se inició después de la revisión de los datos de seguridad de la cohorte 3 por el SMC.
El efecto de los alimentos se evaluó en la cohorte dos al menos 14 días después de la primera dosis, momento en el cual se administró una segunda dosis única. A los sujetos se les administró una dosis de 1.000 mg del fármaco del estudio con 240 ml de agua después de un período de ayuno de 10 horas y 30 minutos después de comenzar el consumo de una comida. A continuación, los sujetos luego se abstuvieron de la ingesta de alimentos durante al menos 4 horas después de la dosificación. El consumo de alimentos se estandarizó durante al menos 12 horas después de la dosificación utilizando una comida estandarizada con alto contenido de grasa (800 a 1.000 kcal con 500 a 600 kcal de grasa y 250 kcal de carbohidratos). Una comida típica de prueba estándar consistía en dos huevos fritos en mantequilla, dos tiras de bacon, dos rebanadas de pan tostado con mantequilla, 120 ml de patatas de tipo hash brown y 240 ml de leche entera.
En la parte dos del estudio (cohortes cinco y seis), a los sujetos se les administró el fármaco del estudio (cápsulas de DS107G de 500 mg o cápsulas de placebo equivalente) una vez al día en ayunas durante 28 días. Los sujetos de la cohorte cinco recibieron el fármaco del estudio primero y, si toleraron la dosis diaria de 2.000 mg durante los primeros 14 días, los sujetos de la cohorte seis comenzaron con la dosis diaria de 4.000 mg durante 28 días.
Tabla 15. Cohortes de tratamiento

continuación

El investigador principal decidía cualquier aumento en el nivel de dosis o el comienzo de una cohorte posterior. Se requirió un mínimo de 5 sujetos con datos de seguridad evaluables de la cohorte tres (dosis de 2.000 mg) antes de comenzar la cohorte cuatro (dosis de 4.000 mg). En la parte 2, se requirió un mínimo de 5 sujetos con 14 días de datos de seguridad evaluables de la cohorte 5 (2.000 mg/día) antes de comenzar la cohorte seis (4.000 mg/día). Se desarrolló una concentración de la cápsula DS107G de DGLA que contenía 500 mg de ácido graso libre DGLA (AAG). Las cápsulas incluyeron los siguientes excipientes: AAG DGLA (estabilizado con 2.000 ppm nominales de dlalfa-tocoferol). Todos los excipientes en la cubierta de la cápsula se usaron habitualmente en productos de gelatina blanda e incluyen una cubierta de gelatina certificada de encefalopatía espongiforme transmisible (TSE) que contenía los siguientes ingredientes: agua purificada, el plastificante glicerol, el colorante dióxido de titanio y los auxiliares de procesamiento lecitina y triglicéridos de cadena media. La cápsula de placebo (cápsula de placebo de DS107G) para estudios clínicos consistió en parafina líquida encapsulada en una cubierta de gelatina blanda y tenía un aspecto idéntico a la cápsula de DGLA.
Los sujetos que cumplían los criterios de elegibilidad fueron asignados aleatoriamente para recibir DGLA (dosis de 500, 1.000, 2.000, 4.000 mg) o cápsulas de placebo equivalente utilizando un programa de asignación al azar. La aleatorización fue aleatorización en bloque con una relación de tratamiento activo a placebo de 3:1. El programa de aleatorización fue generado por Planimeter utilizando SAS® 9.1.3 SP4.
A los sujetos no se les permitió usar fármacos de prescripción médica durante las 2 semanas previas a la dosificación o usar preparaciones de venta libre (incluidas vitaminas y suplementos) y anticonceptivos hormonales durante 1 semana antes de la dosificación, a excepción de paracetamol, que se permitió hasta 48 horas antes de la dosificación. Además, a los sujetos no se les permitió usar suplementos dietéticos ricos en ácidos grasos omega-3 u omega-6. A los sujetos no se les permitió consumir alcohol en exceso de 28 unidades por semana (sujetos masculinos) o 21 unidades por semana (sujetos femeninos).
Se aconsejó a los sujetos que evitaran consumir complementos alimenticios ricos en ácidos grasos omega-3 u omega-6 (por ejemplo, cápsulas de aceite de hígado de bacalao) durante 4 semanas antes de la selección y durante el estudio.
Se aconsejó a los sujetos que evitaran comer semillas de amapola y alimentos que contuvieran semillas de amapola durante al menos 24 horas antes de la recopilación de muestras de orina para analizar el DOA, ya que las semillas de amapola a veces pueden causar un resultado positivo.
A los sujetos de la cohorte dos sometidos a evaluación del efecto de los alimentos sobre las cápsulas orales de 1.000 mg de DGLA se les restringió el consumo de alimentos durante al menos 4 horas después de la administración. El consumo de alimentos se estandarizó durante al menos 12 horas después de la dosis utilizando una comida estandarizada rica en grasas.
Los sujetos debían abstenerse de tomar fármacos sistémicos o de venta libre (incluidas vitaminas y suplementos) durante el estudio, con la excepción de anticonceptivos hormonales. Se permitió el uso de paracetamol (a una dosis de hasta 4 g/día) hasta 48 horas antes de la dosificación. Los sujetos también debían abstenerse de consumir alcohol 48 horas antes de la primera administración del fármaco del estudio (Día 1) hasta la visita de seguimiento. No hubo restricciones en el consumo de cafeína o el consumo de tabaco antes o durante el estudio.
Se requirió que los sujetos evitaran el ejercicio y la actividad física extenuante durante al menos 3 a 4 horas antes de que se extrajera la sangre para la prueba de laboratorio clínico.
El análisis de las concentraciones plasmáticas de DGLA (libre y total) y 15-HETrE libre se realizó en muestras de sangre obtenidas de los sujetos. Además, se obtuvieron fluidos de ampollas de la piel los días 1, 7, 14 y 28 para el análisis de DGLA libre y total.
Las concentraciones plasmáticas de dihidroxitestosterona (DHT) se evaluaron en la Parte 2 como un biomarcador o
criterio de valoración de la eficacia exploratorio.
El análisis de todas las muestras de plasma y fluido de las ampollas de la piel se realizó utilizando métodos validados. Las concentraciones de DGLA libre y total se midieron en plasma y fluido de las ampollas de la piel por cromatografía de líquidos con espectrometría de masas en tándem (LC/MS/m S); el intervalo de cuantificación fue de 100 a 10.000 ng/ml para DGLA libre y de 5.000 a 500.000 ng/ml para DGLA total. Las concentraciones plasmáticas de 15-HETrE libre se midieron por LC/MS/MS con un intervalo de cuantificación de 100 a 10.000 ng/ml. Las concentraciones plasmáticas de DHT se midieron por LC/MS con un intervalo de cuantificación de 0,02 a 1,5 ng/ml. Los sujetos se monitorizaron durante todo el confinamiento en la unidad de Fase 1 para detectar reacciones adversas al fármaco y/o procedimientos del estudio.
Las evaluaciones farmacocinéticas utilizadas en este estudio fueron estándar para la evaluación de posibles agentes terapéuticos. Las evaluaciones de seguridad incluyeron métodos que se consideraron estándar para un estudio clínico de fase 1.
El estudio fue exploratorio y no se realizaron cálculos formales de potencia. El número de sujetos planeados para cada cohorte (8 sujetos) se consideró suficiente para permitir la evaluación de la seguridad y la exposición sistémica de la cápsula de DS107G.
El análisis de las poblaciones incluyó lo siguiente:
• La población por intención de tratar (ITT) consiste en todos los sujetos asignados al azar a los que se les ha administrado al menos 1 dosis del fármaco del estudio.
• La población por protocolo (PP) consiste en todos los sujetos de la población de ITT que no realizaron una violación importante del protocolo, como se define en el PAS.
• La población de PK contiene todos los sujetos incluidos en la población PP que tenían datos de PK evaluables derivados del plasma. Una observación de concentración plasmática se consideró una medición evaluable válida si los siguientes datos estuvieran disponibles: número de identificación del estudio, número de aleatorización, fecha y hora de la obtención de la muestra, dosis y concentración. Una serie de tales mediciones de la misma muestra se consideraron completas si cada concentración prescrita por protocolo era evaluable. Los datos de PK en plasma fueron evaluables por definición si los datos contenían una serie completa de observaciones. Cualquier observación de PK en plasma perdida daría como resultado datos de PK incompletos, por lo tanto, un sujeto con cualquier observación de PK plasmática perdida fue excluido de la población de PK. Los sujetos aleatorizados a recibir placebo también fueron excluidos de la población PK.
Las poblaciones definidas anteriormente se generaron por separado para los datos de la Parte 1 y LA Parte 2 del estudio. El análisis de seguridad se realizó en la población ITT.
Se realizaron análisis de seguridad en la población de seguridad tabulada por rama de tratamiento, cohorte y visita. Todos los datos de seguridad se caracterizaron por herramientas estadísticas descriptivas. No se estableció ninguna hipótesis para investigar en el protocolo de estudio. La evaluación de las evaluaciones de seguridad se realizó de manera descriptiva. Las variables continuas se caracterizaron por su media, desviación estándar (SD), mediana, valores mínimos y máximos; las variables discretas se caracterizaron por su distribución absoluta (frecuencia) y relativa (porcentaje [%]). Los grupos de tratamiento también fueron
los criterios de valoración primarios (características de PK derivadas de dosis orales únicas y múltiples de la cápsula de DS107G) se obtuvieron con la ayuda de modelos de PK no compartimentales. Los criterios de valoración secundarios consistieron en caracterizar el efecto de los alimentos sobre la PK de una dosis oral única de cápsula de DS107G y caracterizar la PK de DGLA en la piel humana después de múltiples dosis orales de la cápsula de DS107G).
Los criterios de valoración secundarios se notificaron utilizando las siguientes herramientas analíticas descriptivas: número de observaciones válidas, la media, la desviación estándar (SD), la mediana, los valores mínimos y máximos derivados de parámetros continuos agrupados por rama de tratamiento, visita y cohorte.
No se realizaron pruebas formales de hipótesis durante la evaluación de los datos del estudio. En la Parte 1 (dosis única ascendente), el grado de exposición fue de 1 día por definición (en caso de administración exitosa del fármaco del estudio). En la parte 2 (dosis múltiples ascendentes), el grado de exposición se calculó como el día de la última ingesta del fármaco del estudio menos el día de la primera ingesta del fármaco del estudio 1 (cuando no se documentó ninguna interrupción en la administración del fármaco del estudio). En caso de interrupción(es), el resultado de la fórmula anterior se redujo con el número de interrupción(es).
El análisis estadístico de los datos PK se realizó con el software SAS (Versión 9.1.3). No se realizó un análisis
farmacodinámico.
Los parámetros de PK en plasma de DGLA y de 15-HETrE se estimaron utilizando técnicas independientes del modelo (análisis no compartimental) e incluyeron: Cmáx, tmáx, Cúltima, Túltima, AUC0.24, AUCü.inf, AUCúltima, Az, Cl, V, y ti/2 para las partes 1 y 2, y tmín, cmín, CLss, Vss, Cprom y % de PTF solo para datos de la Parte 2 (estado en equilibrio). Todos los parámetros PK se resumieron descriptivamente, no se realizaron pruebas estadísticas formales debido a la naturaleza exploratoria del estudio.
En el contexto de dosis única (Parte 1), la interrupción del fármaco del estudio no fue un resultado potencial en las Cohortes uno, tres y cuatro; por tanto, no se requirió actividad de notificación para estas cohortes. Para la cohorte 2, los sujetos inscritos recibieron el fármaco del estudio dos veces: una dosis única administrada en estado de ayuno y una segunda dosis única administrada en estado de alimentación.
También se hicieron las siguientes enmiendas al protocolo.
• Tabla de estudios clínicos añadidos, para aclarar estudios clínicos previos con DGLA oral y sus conclusiones de seguridad.
• Se añadió una evaluación del riesgo/beneficio, para aclarar los riesgos y beneficios evaluados del ensayo propuesto para beneficio del Comité de Investigación y Ética.
• Se modificaron los criterios de inclusión, de modo que los sujetos que se incluyeron en la Parte 1 del estudio (cohortes de dosis única) pudieran volver a entrar y volver a inscribirse en la Parte 2 (cohortes de dosis múltiples), siempre que no tuvieran AA relacionados con el fármaco del estudio y tuvieron un período de lavado de al menos 14 días antes de comenzar el régimen de dosis múltiples. La razón de este cambio fue ayudar al reclutamiento sin comprometer la seguridad de los voluntarios.
• Se añadieron pruebas de laboratorio clínico para la coagulación (tiempo de protrombina y tiempo de protrombina parcial activada [APTT]) como evaluaciones en todos los puntos de tiempo de evaluación de laboratorio clínico en la Parte 2 del estudio (cohortes de dosis múltiples 5 y 6). Estas evaluaciones se añadieron para monitorizar cualquier cambio potencial en los factores de coagulación como un biomarcador exploratorio para futuros estudios.
• Se reemplazó la redacción ambigua para aclarar que se obtendrían registros de ECG individuales.
• 15-HETrE se eliminó como analito para pruebas en la Parte 2 (cohortes de dosis múltiples), dado que los datos preliminares de PK de las cohortes de la Parte 1 revelaron que todas las muestras para 15-HETrE estaban por debajo del límite de cuantificación (BLQ).
• Además del DGLA libre (no esterificado), se añadió la cuantificación de la DGLA "total" a todos los análisis, ya que el perfil de PK den plasma del "total" puede diferir del "libre".
• También, se incluyeron cambios en los análisis planificados para proporcionar análisis adicionales y resultados estadísticos.
RESULTADOS
Estudio de dosis única.
Se sometieron a selección a cuarenta sujetos; de esos 40, se excluyó a 4 sujetos por no cumplir los requisitos de inclusión/exclusión y 4 sujetos retiraron su consentimiento. La disposición de los 32 sujetos asignados al azar al fármaco del estudio se presenta en la Tabla 16.
T l 1 R m n l i i i n l - l i n ITT i ni
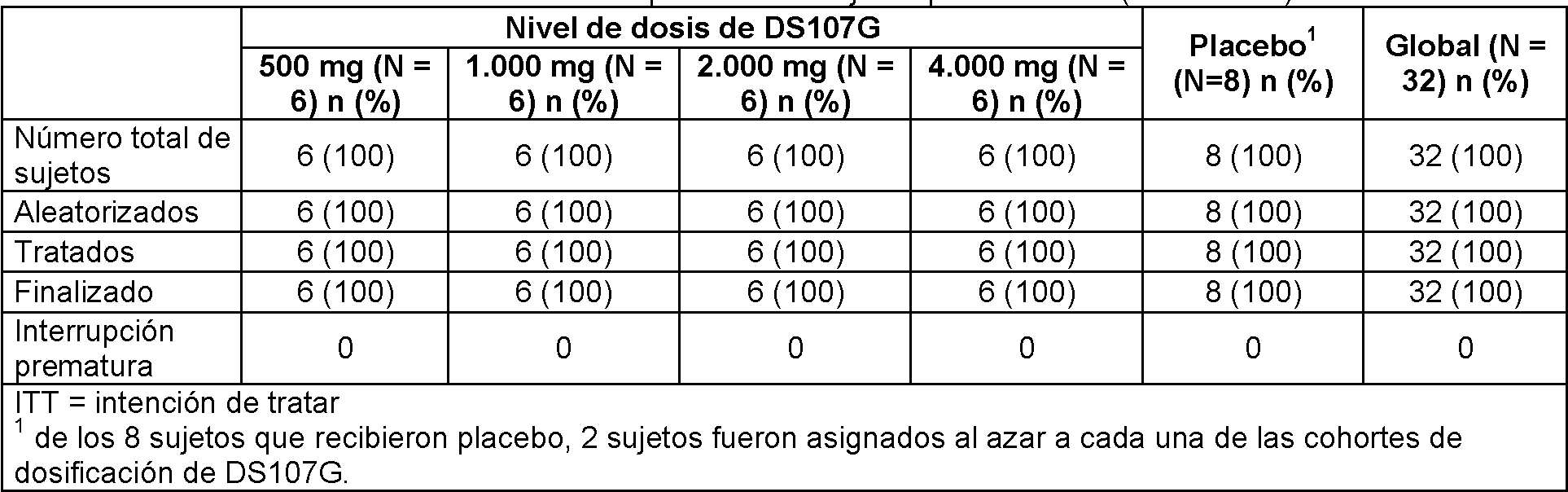
Concentraciones plasmáticas de DGLA libre y total en el momento basal, antes de la administración de DS107G, se resumieron en la Tabla 17. Estas concentraciones fueron generalmente muy variables.
T l 1 n nr i n l m i l D LA i ni l i n f rm in i

Las concentraciones plasmáticas medias después de una dosis única de DS107G se muestran gráficamente por cohorte de dosis para DGLA libre en la figura 6 (gráfico lineal) y la figura 7 (gráfico lineal), y para el DGLA total en la figura 8 (gráfico lineal) y la figura 9 ( diagrama logarítmico-lineal).
Después de una dosis única de cápsulas de DS107G (500, 1.000, 2.000 y 4.000 mg), la variabilidad entre sujetos (medida por SD) de DGLA libre y total fue alta tanto para las concentraciones plasmáticas como para los parámetros PK corregidos al inicio del estudio. En condiciones de ayuno, la Cmáx media corregida por el valor basal y la AUC0-24 para DGLA libre y total aumentaron de manera lineal (Tabla 18, Tabla 19). La mediana del tiempo hasta la concentración máxima (Tmáx) fue de 4 horas para DGLA libre (Tabla 18) y no fue consistente a través de la dosis, con valores de 8 horas a las 2 dosis más bajas y 18 horas a las 2 dosis más altas (Tabla 19). Aunque los parámetros PK de eliminación corregidos al inicio del estudio podrían determinarse para menos de la mitad de los sujetos en algunas cohortes, no hubo evidencia de farmacocinética no lineal en la semivida de eliminación o el aclaramiento para DGLA libre o total (Tabla 19 y Tabla 20). Aunque no se evalúa estadísticamente, la administración de una dosis única de 1.000 mg de DS107G en ayunas dio como resultado una tasa y un alcance aproximadamente 50 % más altos de la absorción total de DGLA en función de la Cmáx media corregida por el valor basal y la AUC0-24 (Tabla 19).
Tabla 18 Parámetros farmacocinéticos plasmáticos corregidos basales para DGLA libre después de la administración en a unas dosis única oblación farmacocinética

continuación

Tabla 19 Parámetros farmacocinéticos plasmáticos corregidos basales para DGLA total después de la administración en a unas dosis única oblación farmacocinética

continuación

El efecto de los alimentos sobre la PK corregida por el valor basal de dosis única de DGLA libre y total usando la dosis de 1.000 mg (Cohorte dos) se evaluó y se notificó en la Tabla 21. En resumen, la Cmáx media corregida por el valor basal de DGLA libre fue aproximadamente 3 veces mayor y ocurrió 1 hora (mediana) antes en ayunas que en condiciones de alimentación (Tabla 21). La AUC0-24 media corregida por el valor basal de DGLA libre en condiciones de ayuno fue aproximadamente 2 veces mayor que el valor en condiciones de alimentación. Por lo tanto, se observó una mayor tasa y extensión de absorción de DGLA en condiciones de ayuno. No hubo diferencias claras para las condiciones de ayuno frente a alimentación en la eliminación de DGLA libre (Tabla 20).
Para DGLA total, la Cmáx media corregida por el valor basal fue aproximadamente 1,5 veces mayor y el tmáx se produjo aproximadamente un 50 % antes (mediana, 8 frente a 15 h) en ayunas que en condiciones de alimentación (Tabla 20). La AUC0-24 media corregida por el valor basal en condiciones de ayuno fue aproximadamente 1,8 veces mayor que el valor en condiciones de alimentación. Poco menos de la mitad de los sujetos (2/6 en ayunas, 3/6 alimentados) tenía datos suficientes para estimar Az de DGLA total, t1/2, aclaramiento y volumen de distribución. Estos datos sugieren una mayor tasa y extensión de la absorción de DGLA total en ayunas. No hay conclusiones fiables con respecto a la eliminación de DGLA total o la distribución de volumen debido a la pequeña población de datos.
Según los parámetros PK que se muestran en la Tabla 20, la Cmáx media corregida por el valor basal fue ~ 10 veces (en ayunas) y ~ 20 veces (alimentados) más alta para DGLA total que para DGLA libre. La AUC0-24 media corregida por el valor basal fue ~ 54 veces (en ayunas) y ~ 56 veces (alimentados) más alta para DGLA total que para DGLA libre.
Tabla 20 Parámetros farmacocinéticos plasmáticos corregidos por el valor basal para DGLA, Después de la mini r i n n n fr n n lim n i ni l i n f rm in i

continuación

DSI07G fue bien tolerado como dosis única en cantidades de 500, 1.000, 2.000 o 4.000 mg por los voluntarios sanos. Los TEAE más comunes indicados fueron diarrea leve a moderada (término indicado: heces sueltas), por un porcentaje similar de sujetos entre los sujetos con tratamiento activo y con control con placebo (incidencia: 5/24 [20,8 %] sujetos tratados con tratamiento activo frente a 2/8 [25,0 %] sujetos tratados con control con placebo). Los acontecimientos de diarrea fueron de duración relativamente corta y el Investigador consideró que todos (incluidos los que se produjeron en sujetos controlados con placebo) estaban posiblemente relacionados con el fármaco del estudio. Cabe destacar que, los sujetos que recibieron una segunda dosis única de DS107G y que tenían TEAE de diarrea en ayunas no sufrieron recurrencia de la diarrea. Todos los demás TEAE solo se produjeron en 1 sujeto cada uno de ellos y solo en los grupos de tratamiento activo, incluidos: infección leve, dolor orofaríngeo y faringitis; y pirexia moderadamente intensa e infección del tracto urinario después de la dosificación en estado alimentado.
RESULTADOS DE DOSIS MÚLTIPLES - PARTE 2 DEL ESTUDIO
Las concentraciones plasmáticas medias y la concentración promedio en estado en equilibrio para la Parte 2 se muestran gráficamente mediante la cohorte de dosis para DGLA libre en la figura 10 (gráfico lineal) y la figura 11 (gráfico logarítmico-lineal), y para DGLA total en la figura 12 (gráfico lineal ) y la figura 13 (gráfico logarítmico-lineal). Los días 1 y 28, las concentraciones medias de DGLA libre alcanzaron su punto máximo aproximadamente 4 horas después de la dosificación, mientras que no fue evidente la concentración máxima promedio para DGLA total. Las concentraciones medias de DGLA libre y total aumentaron con el tiempo con dosificación repetida, aunque el aumento para DGLA total fue más pronunciado. Las concentraciones plasmáticas parecieron alcanzar el estado en equilibrio alrededor del día 14 para ambas dosis (2.000 y 4.000 mg diarios) y los analitos (DGLA libre y total) según la inspección visual de los gráficos de concentración media. Cuando la dosis se duplicó de 2.000 a 4.000 mg al día, la concentración promedio en estado en equilibrio aumentó 1,6 veces para DGLA libre pero solo 1,2 veces para DGLA total, lo que sugiere uno o más procesos saturables a la dosis más alta.
El parámetro PK se calculó después de corregir las concentraciones de DGLA administradas con las concentraciones basales de DGLA.
La farmacocinética corregida por el valor basal en plasma para DGLA libre se indica en la Tabla 21. En resumen, la Cmáx media corregida por el valor basal de DGLA libre y la AUC fueron mayores en la cohorte de dosis de DS107G más alta los dos días evaluados. La Cmáx media corregida por el valor basal para la dosis de 4.000 mg fue ~ 3 veces mayor que para la dosis de 2.000 mg el día 1 pero solo ~ 1,4 veces mayor el día 28. La AUC0-24 media corregida por el valor basal para la dosis de 4.000 mg fue ~ 2,5 veces mayor que para la dosis de 2.000 mg el día 1 y solo ~ 1,7 veces mayor el día 28. Los cambios con la dosis fueron lineales para la Cmáx y la AUC0-24 corregidas por el valor basal el día 1, pero solo para la AUC0-24 corregida por el valor basal el día 28. La alta variabilidad entre sujetos
podría haber causado esta inconsistencia. La mediana del tmáx fue similar para ambas cohortes de dosis el día 1 y el día 28, con valores de 4 o 5 horas. La mediana del t-i/2 de eliminación fue más larga el día 28 que el día 1, con el valor del día 28 dependiente del intervalo de tiempo evaluado. El aclaramiento medio disminuyó y el volumen de distribución medio aumentó con dosis múltiples.
Tabla 21 Parámetros farmacocinéticos corregidos por el valor basal en plasma para DGLA libre (dosis múltiples, l i n f rm in i

La farmacocinética corregida por el valor basal en plasma en estado en equilibrio para DGLA libre se indica en la Tabla 22. En resumen, las concentraciones plasmáticas de DGLA libre y total aumentaron con la administración repetida y alcanzaron el estado en equilibrio aproximadamente el día 14. En el estado en equilibrio, evaluado el día 28 (0-24 horas), la fluctuación valle máxima (pTf ) fue bastante alta para ambas cohortes de dosis (media, ~ 430 % y ~ 490 %; Tabla 9). La relación de acumulación (AR) media fue mayor para la cohorte de dosis de 2.000 mg que para la dosis de 4.000 mg tanto para la Cmáx como para la AUC (AR de - 2,8 y - 3,3 para 2.000 mg frente a - 1,4 y - 1,6 para 4.000 mg. Los datos sugieren una cinética saturable y/o la presencia de cambios en el volumen de distribución con la dosificación repetida de DGLA libre.
Tabla 22 Parámetros farmacocinéticos corregidos por el valor basal en plasma en estado en equilibrio para DGLA _________________________ libre (dosis múltiples, población farmacocinética)__________________________
Dí 2 24 h r i l i D 1 7G
= 6)


, ,
AR = relación de acumulación; Máx = máximo; media = media aritmética; mín = mínimo; N = número de sujetos que proporcionan datos; PTF = fluctuación valle máxima; SD = Desviación estándar
Nota: La concentración de DGLA previa a la dosis se restó de las concentraciones posteriores antes del cálculo del parámetro; los valores negativos fueron reemplazados por cero._____________________________
La farmacocinética corregida por el valor basal en plasma para DGLA total se indica en la Tabla 23. En resumen, la Cmáx media corregida por el valor basal de DGLA total y la AUC0-24 fueron mayores en la cohorte de dosis de DS107G más alta ambos dos días evaluados, como se esperaba. La Cmáx media corregida por el valor basal y la AUC0-24 para la dosis de 4.000 mg fueron -1,5 y -1,5 veces más altas, respectivamente, que para la dosis de 2.000 mg el día 1 pero solo -1,2 y -1,4 veces mayor que para la dosis de 2.000 mg el día 28.
Los cambios con la dosis en la Cmáx corregida por el valor basal y en la AUC4 no fueron lineales para DGLA total ninguno de los días evaluados. La alta variabilidad entre sujetos podría haber causado esta inconsistencia. La mediana de T se produjo antes con dosis múltiples (8-10 h) que con una dosis única (1018 h) en ambas cohortes de dosis. La mediana del t-i/2 de eliminación de DGLA total para la cohorte de dosis de 2.000 mg fue de 34,4 a 44,0 horas cuando se evaluó durante 24 horas (días 1 y 28, respectivamente), y 62,6 horas el día 28 cuando se evaluó durante de 0 a 168 horas. El aclaramiento medio y el volumen de distribución disminuyeron con dosis múltiples.

La farmacocinética corregida por el valor basal en plasma en estado en equilibrio para DGLA libre se indica en la Tabla 24. En resumen, en estado de equilibrio, evaluado el día 28 (0-24 horas), La fluctuación valle máxima (PTF) fue bastante alta para ambas cohortes de dosis (media, 62,5 % y 44,9 %). La AR media fue mayor para la cohorte de dosis de 2.000 mg que para la dosis de 4.000 mg para la Cmáx y la AUC. Los datos sugieren una cinética saturable y/o presencia de cambio en el volumen de distribución con dosis repetidas de DGLA total.
Tabla 24 Parámetros farmacocinéticos corregidos por el valor basal en plasma en estado en equilibrio para DGLA total dosis múlti les oblación farmacocinética

Las concentraciones medias de DGLA libre en el fluido de la ampolla de la piel se muestran por cohorte de dosis en la figura 14 (gráfico lineal) y la figura 15 (gráfico logarítmico-lineal). Las concentraciones medias se duplicaron aproximadamente con una duplicación de la dosis (según las concentraciones de los días 1, 8, 14 y 28) y se acumularon con dosis repetidas en ambos regímenes. Las concentraciones medias de DGLA libre el día 28 fueron aproximadamente 3 veces más altas que las del día 1 para 2.000 y 4.000 mg diarios.
Las concentraciones medias de DGLA total en el fluido de la ampolla de la piel se muestran por cohorte de dosis en la figura 16 (gráfico lineal) y la figura 17 (gráfico logarítmico-lineal). Las concentraciones medias de DGLA total aumentaron aproximadamente 1,4 veces con una duplicación de la dosis (según las concentraciones de los días 1, 8, 14 y 28). Las concentraciones medias de DGLA total el día 28 fueron aproximadamente 2,5 y 3 veces más altas que las del día 1 para 2.000 y 4.000 mg diarios, respectivamente.
Los perfiles de concentración en plasma y el fluido de las ampollas de la piel se superpusieron y las concentraciones medias de DGLA libre fueron algo similares en plasma y en el fluido de las ampollas de la piel para la misma dosis de DS107G el día 8 y el día 14 (pero no el día 28). La figura 18 [gráfico lineal] y la figura 19 [gráfico logarítmicolineal]), sugieren que el DGLA libre se distribuye en el plasma y la piel de manera similar.
Para DGLA total, las concentraciones medias fueron mucho más altas en plasma que en el fluido de las ampollas de la piel para la misma dosis de DS107G después del día 1 (Figura 20 [gráfico lineal] y Figura 21 [gráfico logarítmicolineal]), lo que indica que el DGLA total se encuentra más fácilmente en el plasma que en la piel. El mecanismo para la distribución limitada de DGLA total en la piel probablemente esté relacionado con la menor cantidad de lípidos en la piel en comparación con el plasma.
Las concentraciones plasmáticas de dihidrotestosterona (DHT) se cuantificaron como un criterio de valoración exploratorio o biomarcador de la eficacia. La variabilidad entre sujetos en los datos de concentración fue alta en función de la SD en la mayoría de los puntos temporales. Las concentraciones plasmáticas medias de DHT se muestran por cohorte de dosis en la figura 22 (lineal) y la figura 23 (logarítmico-lineal).
Ninguna de las muestras tenía concentraciones mensurables de 15-HETrE libre; todas las concentraciones estaban por debajo del LLOQ (100 ng/ml).
No hubo muertes para el estudio de dosis múltiples. Cuando se administró a voluntarios sanos como dosis de 2.000 o 4.000 mg una vez al día durante 28 días consecutivos, DSI07G fue bien tolerado, siendo el peor TEAE diarrea de leve a moderada (término indicado: heces sueltas) de duración relativamente corta. El investigador consideró que la mayoría de los acontecimientos de diarrea (incidencia 7/12 [43,8 %] en los sujetos tratados con tratamiento activo frente a 0/4 [0,0%] sujetos con control de placebo) posiblemente estaban relacionados con el fármaco del estudio. Una mayor proporción de sujetos notificó diarrea en el grupo de 4.000 mg (4/6 [66,7 %]) que en el grupo de 2.000 mg (3/6 [50 %]). La incidencia de TEAE entre los sujetos tratados con tratamiento activo fue mucho mayor que la de los sujetos tratados con control con placebo (11/12 [91,73 %, que notificaron un total de 52 TEAE frente a 1/4 [25,0 %] sujetos que notificaron un total de I TEAE, respectivamente). No hubo TEAE graves y, aparte de los TEAE distintos de diarrea, se consideró que todos los acontecimientos o no estaban relacionados con el fármaco del estudio o era improbable que lo estuvieran. Las náuseas fueron el siguiente TEAE más notificado (10 acontecimientos entre 4/6 [66,7 %] sujetos en el grupo de tratamiento con 4.000 mg); 9/10 de los acontecimientos de náuseas fueron leves y el otro fue de intensidad moderada. Aparte de la diarrea, todos los TEAE restantes se notificaron en 2 sujetos cada uno (bronquitis y nasofaringitis) o en 1 sujeto cada uno (dolor abdominal, astenia, pirexia, aumento de CPK en sangre, CRP aumentado, recuento de leucocitos disminuido, mareos, dolor de cabeza, tos y hematoma), la mayoría de los cuales fueron considerados por el investigador como no relacionados con el fármaco del estudio o de improbable relación con el mismo. Otros TEAE considerados posiblemente relacionados con el fármaco del estudio fueron dolor abdominal y astenia (término indicado "debilidad"), ambos tenían asociaciones temporales con acontecimientos de heces blandas.
No se observaron anomalías clínicamente significativas en las constantes vitales o el ECG en ningún paciente en el estudio de dosis múltiples.
Claims (9)
1. Una composición farmacéutica de ácidos grasos para su uso en el tratamiento de la dermatitis atópica, en la que la composición farmacéutica de ácidos grasos comprende DGLA encapsulado en una cubierta de cápsula, en la que DGLA representa al menos el 95 % en peso de todos los ácidos grasos en la composición farmacéutica de ácidos grasos y en la que el tratamiento de la dermatitis atópica comprende la administración de 1 g o 2 g de la composición farmacéutica de ácidos grasos al día.
2. Una composición farmacéutica de ácidos grasos para su uso en el tratamiento de la reivindicación 1, en la que la composición farmacéutica de ácidos grasos se administra por vía oral.
3. La composición farmacéutica de ácidos grasos para su uso en el tratamiento de la reivindicación 1 o 2, en la que la composición farmacéutica de ácidos grasos se administra como 1 a 8 cápsulas al día.
4. La composición farmacéutica de ácidos grasos para su uso en el tratamiento de la reivindicación 3, en la que cada cápsula comprende hasta 1 g de DGLA.
5. La composición farmacéutica de ácidos grasos para su uso en el tratamiento de la reivindicación 4, en la que cada cápsula comprende de 200 mg a 1 g de DGLA.
6. La composición farmacéutica de ácidos grasos para su uso en el tratamiento de cualquiera de las reivindicaciones precedentes, en la que la cubierta de la cápsula comprende gelatina y glicerol.
7. La composición farmacéutica de ácidos grasos para su uso en el tratamiento de la reivindicación 6, en la que la gelatina comprende gelatina de hueso de lima y la cubierta de la cápsula además comprende uno o más de: glicerol, agua purificada, dióxido de titanio, triglicéridos de cadena media y/o lecitina.
8. La composición farmacéutica de ácidos grasos para su uso en el tratamiento de cualquiera de las reivindicaciones precedentes, en la que la composición farmacéutica de ácidos grasos comprende no más del 5 % de cualquier ácido graso individual seleccionado de 20:2w6, 20:3u>3, 20:4u>6, 20:4u>3 y 20:5u>3, y no más del 2 % en peso de sustancias relacionadas no identificadas.
9. La composición farmacéutica de ácidos grasos para su uso en el tratamiento de cualquiera de las reivindicaciones precedentes, en la que la dermatitis atópica es dermatitis atópica grave.
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201462007752P | 2014-06-04 | 2014-06-04 | |
| US201462058469P | 2014-10-01 | 2014-10-01 | |
| PCT/EP2015/062518 WO2015185698A1 (en) | 2014-06-04 | 2015-06-04 | Pharmaceutical compositions comprising dgla and use of same |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2784240T3 true ES2784240T3 (es) | 2020-09-23 |
Family
ID=53298373
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES15727395T Active ES2784240T3 (es) | 2014-06-04 | 2015-06-04 | Composiciones farmacéuticas que comprenden DGLA y uso de las mismas |
Country Status (17)
| Country | Link |
|---|---|
| US (9) | US9682055B2 (es) |
| EP (2) | EP3692986A1 (es) |
| JP (3) | JP2017516823A (es) |
| KR (1) | KR102391827B1 (es) |
| CN (1) | CN107072959A (es) |
| AU (2) | AU2015270418B2 (es) |
| BR (1) | BR112016028518A2 (es) |
| CA (1) | CA2953633A1 (es) |
| ES (1) | ES2784240T3 (es) |
| IL (2) | IL249364B (es) |
| MA (1) | MA52644A (es) |
| MX (2) | MX375328B (es) |
| PH (1) | PH12016502530A1 (es) |
| RU (2) | RU2020101477A (es) |
| SG (2) | SG11201610207WA (es) |
| WO (1) | WO2015185698A1 (es) |
| ZA (2) | ZA201708681B (es) |
Families Citing this family (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB0907413D0 (en) | 2009-04-29 | 2009-06-10 | Equateq Ltd | Novel methods |
| EP3068757B1 (en) | 2013-11-15 | 2019-02-27 | DS Biopharma Limited | Lysine salt of 15-hydroxy-8(z),11(z),13(e)-eicosatrienoic acid |
| ES2784240T3 (es) * | 2014-06-04 | 2020-09-23 | Ds Biopharma Ltd | Composiciones farmacéuticas que comprenden DGLA y uso de las mismas |
| CN117402059A (zh) | 2015-05-13 | 2024-01-16 | Ds生物制药有限公司 | 包含15-氧代-epa或15-氧代-dgla的组合物及其制备和使用方法 |
| EP3399971A1 (en) * | 2016-01-07 | 2018-11-14 | DS Biopharma Limited | Pharmaceutical compositions comprising dgla and use of same |
| EP3624787A1 (en) * | 2017-05-19 | 2020-03-25 | DS Biopharma Limited | Pharmaceutical compositions comprising dgla and use of same |
| JP7500037B2 (ja) * | 2019-07-24 | 2024-06-17 | 国立大学法人 東京大学 | 血中尿酸値低下剤及び血中尿酸値低下用食品組成物 |
| US20210315851A1 (en) | 2020-04-03 | 2021-10-14 | Afimmune Limited | Compositions comprising 15-hepe and methods of treating or preventing hematologic disorders, and/or related diseases |
Family Cites Families (94)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS5264452A (en) | 1975-11-18 | 1977-05-27 | Nippon Suisan Kaisha Ltd | Method of producing processed fish meat |
| JPS5820575B2 (ja) | 1975-11-27 | 1983-04-23 | 日本水産株式会社 | スイサンカコウヒンノセイゾウホウ |
| US4273763A (en) | 1978-01-23 | 1981-06-16 | Efamol Limited | Pharmaceutical and dietary compositions |
| IE47777B1 (en) * | 1978-01-23 | 1984-06-13 | Efamol Ltd | Pharmaceutical and dietary composition comprising gamma-linolenic acids |
| US4444755A (en) | 1978-01-23 | 1984-04-24 | Efamol Limited | Treatment for skin disorders |
| DE2967049D1 (en) | 1978-04-11 | 1984-07-19 | Efamol Ltd | Pharmaceutical and dietary composition comprising gamma-linolenic acids |
| IE49783B1 (en) | 1979-05-18 | 1985-12-11 | Efamol Ltd | Pharmaceutical and dietary composition comprising epsilon-linolenic acids |
| IE51145B1 (en) | 1980-03-07 | 1986-10-15 | Efamol Ltd | Pharmaceutical and dietary compositions |
| IE53332B1 (en) | 1980-03-14 | 1988-10-26 | Efamol Ltd | Pharmaceutical compositions |
| US5324748A (en) | 1981-07-14 | 1994-06-28 | Efamol Limited | Method for enhancement of 1-series PG production |
| EP0087865B1 (en) | 1982-03-01 | 1986-10-01 | Efamol Limited | Pharmaceutical composition |
| EP0087863B1 (en) | 1982-03-01 | 1986-05-21 | Efamol Limited | Pharmaceutical and dietary composition |
| ATE22804T1 (de) | 1982-03-01 | 1986-11-15 | Efamol Ltd | Pharmazeutische zusammensetzung. |
| ATE22533T1 (de) | 1982-04-29 | 1986-10-15 | Efamol Ltd | Pharmazeutische zusammensetzung. |
| GB8302708D0 (en) | 1983-02-01 | 1983-03-02 | Efamol Ltd | Pharmaceutical and dietary composition |
| GB8319073D0 (en) | 1983-07-14 | 1983-08-17 | Efamol Ltd | Fatty acid compositions |
| GB8326130D0 (en) | 1983-09-29 | 1983-11-02 | Efamol Ltd | Topical preparations containing tars and fatty acids |
| GB8420771D0 (en) | 1984-08-15 | 1984-09-19 | Efamol Ltd | Treatment of skin disorders |
| GB8425006D0 (en) | 1984-10-03 | 1984-11-07 | Efamol Ltd | Composition of copper/fatty acids |
| GB8506027D0 (en) | 1985-03-08 | 1985-04-11 | Efamol Ltd | Pharmaceutical & dietary compositions |
| GB8522670D0 (en) | 1985-09-13 | 1985-10-16 | Efamol Ltd | Drug treatments |
| GB8524275D0 (en) * | 1985-10-02 | 1985-11-06 | Efamol Ltd | Pharmaceutical & dietary compositions |
| US5198468A (en) | 1987-06-24 | 1993-03-30 | Efamol Holdings Plc | Essential fatty acid composition |
| EP0309086A1 (en) | 1987-09-07 | 1989-03-29 | Efamol Holdings Plc | Treatment of male pattern baldness and of unwanted hair growth |
| GB8729153D0 (en) | 1987-12-14 | 1988-01-27 | Efamol Ltd | Fatty acid compositions |
| JPH04507397A (ja) | 1988-01-14 | 1992-12-24 | フリッツ,アンデルス | 湿疹治療用薬剤製造における必須脂肪酸の使用法 |
| GB8806737D0 (en) | 1988-03-22 | 1988-04-20 | Efamol Holdings | Therapeutic composition & method |
| CA1334002C (en) | 1988-09-28 | 1995-01-17 | Barry D. Sears | Essential fatty acid compositions and methods for the modulation of prostaglandin levels in mammals |
| US6140304A (en) | 1988-09-28 | 2000-10-31 | Eicotech Corporation | Method of and nutritional and pharmaceutical compositions for reduction of hyperinsulinemia |
| GB8916734D0 (en) * | 1989-07-21 | 1989-09-06 | Efamol Holdings | Pharmaceutical and dietary uses of fatty acids |
| GB8918294D0 (en) | 1989-08-10 | 1989-09-20 | Efamol Holdings | Pharmaceutical compositions |
| GB8920228D0 (en) | 1989-09-07 | 1989-10-18 | Efamol Holdings | Fatty acid therapy |
| JPH04290820A (ja) | 1991-03-18 | 1992-10-15 | Idemitsu Petrochem Co Ltd | 皮膚病犬の治療方法とその薬剤 |
| GB9111900D0 (en) * | 1991-06-03 | 1991-07-24 | Efamol Holdings | Fatty acid compositions |
| GB9112052D0 (en) | 1991-06-05 | 1991-07-24 | Efamol Holdings | Fatty acid treatment |
| JP3354581B2 (ja) | 1991-09-30 | 2002-12-09 | サントリー株式会社 | ジホモ−γ−リノレン酸及びこれを含有する脂質の製造方法 |
| GB9211229D0 (en) | 1992-05-27 | 1992-07-08 | Efamol Holdings | Fatty acid treatment |
| US5888541A (en) | 1992-08-21 | 1999-03-30 | Scotia Holdings Plc | Fatty acid treatment |
| GB9217780D0 (en) | 1992-08-21 | 1992-10-07 | Efamol Holdings | Fatty acid treatment |
| FI101039B (fi) | 1992-10-09 | 1998-04-15 | Eeva Kristoffersson | Menetelmä lääkepellettien valmistamiseksi |
| DE4238869C2 (de) | 1992-11-18 | 1994-09-08 | Wogepharm Gmbh | Mittel zur Behandlung des atopischen Ekzems und anderer entzündlicher Hautkrankheiten |
| GB9301446D0 (en) | 1993-01-26 | 1993-03-17 | Scotia Holdings Plc | Internal radiation damage |
| AU683027B2 (en) * | 1993-01-27 | 1997-10-30 | Scotia Holdings Plc | Triglycerides |
| CA2119000A1 (en) | 1993-03-19 | 1994-09-20 | David Frederick Horrobin | Formulation for use in smokers |
| JPH09505562A (ja) * | 1993-10-01 | 1997-06-03 | アール.ピー.シェーラー コーポレイション | 香料を小分けして提供する方法および組成物 |
| GB9403855D0 (en) * | 1994-03-01 | 1994-04-20 | Scotia Holdings Plc | Fatty acid derivatives |
| US5789441A (en) | 1996-02-15 | 1998-08-04 | Virocell Inc. | Leukotriene B4 as an antiviral and anti-neoplastic agent |
| GB9621373D0 (en) | 1996-10-14 | 1996-12-04 | Scotia Holdings Plc | Fatty acid treatment |
| GB9710351D0 (en) | 1997-05-20 | 1997-07-16 | Scotia Holdings Plc | Glucosamine fatty acids |
| GB9715444D0 (en) | 1997-07-22 | 1997-09-24 | Scotia Holdings Plc | Therapeutic and dietary compositions |
| JP2000191525A (ja) | 1998-12-25 | 2000-07-11 | Nof Corp | 皮膚外用剤組成物 |
| GB9916536D0 (en) | 1999-07-14 | 1999-09-15 | Scarista Limited | Nutritional or pharmaceutical compositions |
| EP1072198B1 (de) * | 1999-07-28 | 2008-05-14 | SWISS CAPS Rechte und Lizenzen AG | Präparat zur Verwendung als Medikament und/oder Nahrungsmittelergänzung |
| JP4932116B2 (ja) | 2000-02-16 | 2012-05-16 | ザ・ブリガーム・アンド・ウーメンズ・ホスピタル・インコーポレーテッド | アスピリン誘発脂質メディエータ |
| GB0016045D0 (en) | 2000-06-29 | 2000-08-23 | Laxdale Limited | Therapeutic combinations of fatty acids |
| JP2002047176A (ja) * | 2000-08-04 | 2002-02-12 | Idemitsu Technofine Co Ltd | IgE産生抑制剤 |
| US20020188024A1 (en) | 2000-08-23 | 2002-12-12 | Chilton Floyd H. | Fatty acid-containing emulsion with increased bioavailability |
| US20040043013A1 (en) | 2000-12-28 | 2004-03-04 | Mccleary Edward Larry | Metabolic uncoupling therapy |
| DE10121252A1 (de) | 2001-04-30 | 2002-11-07 | Christos C Zouboulis | Behandlung der Akne |
| SK14502003A3 (en) | 2001-05-30 | 2004-10-05 | Laxdale Ltd | Coenzyme Q and eicosapentaenoic acid |
| JP2003155233A (ja) | 2001-11-16 | 2003-05-27 | Idemitsu Technofine Co Ltd | 塗布剤 |
| AU2003299034A1 (en) | 2002-09-20 | 2004-04-08 | Griscom Bettle Iii | Transdermal compositions |
| AU2003284633B2 (en) | 2002-11-22 | 2008-07-10 | Nippon Suisan Kaisha, Ltd. | External composition containing polyunsaturated fatty acid or its salt or ester |
| US20050032757A1 (en) * | 2003-08-06 | 2005-02-10 | Cho Suk H. | Nutritional supplements |
| JP2005179211A (ja) | 2003-12-17 | 2005-07-07 | Idemitsu Kosan Co Ltd | 皮膚外用剤組成物 |
| US20070105954A1 (en) | 2003-12-31 | 2007-05-10 | Ingennus Limited | Formulation containing a carboxylic acid or an ester thereof |
| BRPI0512864B8 (pt) | 2004-07-01 | 2021-05-25 | Johnson & Johnson Vision Care | composição oftálmica, uso de pelo menos um ácido graxo ômega-6 e pelo menos um ácido graxo ômega-3 e método para preparar a referida composição |
| US7666447B2 (en) * | 2004-10-08 | 2010-02-23 | Pharmanutrients | Compositions including Krill extracts and conjugated linoleic acid and methods of using same |
| US7893106B2 (en) | 2004-11-19 | 2011-02-22 | Martek Biosciences, Corporation | Oxylipins from stearidonic acid and γ-linolenic acid and methods of making and using the same |
| JP5546087B2 (ja) | 2005-02-14 | 2014-07-09 | サントリーホールディングス株式会社 | 皮膚疾患経口治療または予防剤 |
| JP2006306812A (ja) | 2005-04-28 | 2006-11-09 | Suntory Ltd | 好酸球浸潤抑制剤 |
| WO2006085687A1 (ja) * | 2005-02-14 | 2006-08-17 | Suntory Limited | ジホモ−γ−リノレン酸(DGLA)を有効成分として含んで成る組成物 |
| US7485323B2 (en) | 2005-05-31 | 2009-02-03 | Gelita Ag | Process for making a low molecular weight gelatine hydrolysate and gelatine hydrolysate compositions |
| TW200711649A (en) | 2005-06-17 | 2007-04-01 | Combinatorx Inc | Combination therapy for the treatment of immunoinflammatory disorders |
| JP5101894B2 (ja) * | 2007-01-15 | 2012-12-19 | サントリーホールディングス株式会社 | 高度不飽和脂肪酸及びこれを含有する脂質の製造方法 |
| ITMI20070555A1 (it) | 2007-03-21 | 2007-06-20 | Giuliani Spa | Composizione provvista di attivita' di inibizione sulla 5 alfa-reduttasi |
| WO2009130291A2 (de) | 2008-04-25 | 2009-10-29 | Basf Plant Science Gmbh | Pflanzensamenöl |
| GB0907413D0 (en) | 2009-04-29 | 2009-06-10 | Equateq Ltd | Novel methods |
| CA2690488C (en) * | 2010-01-19 | 2013-06-11 | Accucaps Industries Limited | Pharmaceutical formulations of naproxen for soft gel encapsulation and combinations thereof |
| GB201001345D0 (en) | 2010-01-27 | 2010-03-17 | Equateq Ltd | Process for preparing and purifying fatty acids |
| CA2822263A1 (en) | 2010-12-21 | 2012-06-28 | Nestec S.A. | Methods and compositions for preventing and treating osteoarthritis |
| US8293790B2 (en) | 2011-10-19 | 2012-10-23 | Dignity Sciences Limited | Pharmaceutical compositions comprising DGLA and benzoyl peroxide and methods of use thereof |
| SG11201401629XA (en) | 2011-10-19 | 2014-05-29 | Dignity Sciences Ltd | Pharmaceutical compositions comprising dgla and/or 15-hetre and methods of use thereof |
| SG11201402653QA (en) | 2011-11-29 | 2014-06-27 | Dignity Sciences Ltd | Compositions comprising 20-carbon fatty acids and methods of making and using same |
| UA114615C2 (uk) * | 2012-01-06 | 2017-07-10 | Омтера Фармасьютікалз, Інк. | Збагачена дпк композиція омега-3 поліненасичених жирних кислот у формі вільної кислоти |
| US20120264705A1 (en) | 2012-01-26 | 2012-10-18 | Dignity Sciences Limited | Antimicrobial compositions comprising 15-hetre and methods of use thereof |
| US20130267598A1 (en) | 2012-02-23 | 2013-10-10 | Dignity Sciences Limited | Pharmaceutical compositions comprising dgla, 15-ohepa, and/or 15-hetre and methods of reducing sebum production using same |
| EP2833740B1 (en) * | 2012-04-04 | 2016-09-14 | Pronova BioPharma Norge AS | Compositions comprising omega-3 fatty acids and vitamin d for acne vulgaris and/or eczema, and methods and uses thereof |
| GB2504061A (en) * | 2012-05-25 | 2014-01-22 | Dignity Sciences Ltd | Omega-6 Enriched PUFA Phospholipids |
| WO2014022816A2 (en) | 2012-08-03 | 2014-02-06 | Dignity Sciences Limited | Pharmaceutical compositions comprising 15-hetre and methods of treating or preventing arthritis using same |
| GB201216385D0 (en) * | 2012-09-13 | 2012-10-31 | Chrysalis Pharma Ag | A pharmaceutical composition |
| ES2784240T3 (es) * | 2014-06-04 | 2020-09-23 | Ds Biopharma Ltd | Composiciones farmacéuticas que comprenden DGLA y uso de las mismas |
| CN117402059A (zh) | 2015-05-13 | 2024-01-16 | Ds生物制药有限公司 | 包含15-氧代-epa或15-氧代-dgla的组合物及其制备和使用方法 |
| EP3399971A1 (en) | 2016-01-07 | 2018-11-14 | DS Biopharma Limited | Pharmaceutical compositions comprising dgla and use of same |
-
2015
- 2015-06-04 ES ES15727395T patent/ES2784240T3/es active Active
- 2015-06-04 SG SG11201610207WA patent/SG11201610207WA/en unknown
- 2015-06-04 CA CA2953633A patent/CA2953633A1/en not_active Abandoned
- 2015-06-04 EP EP20153902.0A patent/EP3692986A1/en not_active Withdrawn
- 2015-06-04 AU AU2015270418A patent/AU2015270418B2/en not_active Ceased
- 2015-06-04 JP JP2016570986A patent/JP2017516823A/ja active Pending
- 2015-06-04 MX MX2016015961A patent/MX375328B/es active IP Right Grant
- 2015-06-04 KR KR1020177000244A patent/KR102391827B1/ko not_active Expired - Fee Related
- 2015-06-04 WO PCT/EP2015/062518 patent/WO2015185698A1/en not_active Ceased
- 2015-06-04 RU RU2020101477A patent/RU2020101477A/ru unknown
- 2015-06-04 CN CN201580039328.7A patent/CN107072959A/zh active Pending
- 2015-06-04 EP EP15727395.4A patent/EP3151825B1/en active Active
- 2015-06-04 SG SG10201912145VA patent/SG10201912145VA/en unknown
- 2015-06-04 BR BR112016028518A patent/BR112016028518A2/pt not_active IP Right Cessation
- 2015-06-04 US US14/730,818 patent/US9682055B2/en not_active Expired - Fee Related
- 2015-06-04 RU RU2016151719A patent/RU2714323C2/ru active
- 2015-06-04 MA MA052644A patent/MA52644A/fr unknown
-
2016
- 2016-11-04 US US15/343,689 patent/US20170071890A1/en not_active Abandoned
- 2016-12-02 MX MX2020009709A patent/MX2020009709A/es unknown
- 2016-12-04 IL IL249364A patent/IL249364B/en active IP Right Grant
- 2016-12-16 PH PH12016502530A patent/PH12016502530A1/en unknown
-
2017
- 2017-07-27 US US15/661,374 patent/US10105333B2/en active Active
- 2017-12-14 US US15/842,194 patent/US20180104208A1/en not_active Abandoned
- 2017-12-20 ZA ZA2017/08681A patent/ZA201708681B/en unknown
-
2018
- 2018-03-26 US US15/935,739 patent/US20180214408A1/en not_active Abandoned
- 2018-12-13 ZA ZA2018/08414A patent/ZA201808414B/en unknown
-
2019
- 2019-02-19 US US16/279,819 patent/US10537543B2/en not_active Expired - Fee Related
- 2019-02-19 US US16/279,794 patent/US20190269639A1/en not_active Abandoned
- 2019-11-08 US US16/678,431 patent/US10849870B2/en not_active Expired - Fee Related
-
2020
- 2020-01-14 JP JP2020003665A patent/JP2020090508A/ja not_active Withdrawn
- 2020-06-24 US US16/910,770 patent/US11478442B2/en active Active
- 2020-10-28 AU AU2020260433A patent/AU2020260433A1/en not_active Abandoned
-
2021
- 2021-02-07 IL IL280690A patent/IL280690A/en unknown
-
2022
- 2022-03-01 JP JP2022030554A patent/JP2022071061A/ja active Pending
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2784240T3 (es) | Composiciones farmacéuticas que comprenden DGLA y uso de las mismas | |
| ES2846176T3 (es) | Métodos para reducir el riesgo de un evento cardiovascular en un sujeto en tratamiento con estatinas usando éster etílico del ácido eicosapentaenoico | |
| CN114929209A (zh) | 肌萎缩侧索硬化的治疗 | |
| PT2275086E (pt) | Formulação de volume reduzido de acetato de glatirâmero e métodos de administração | |
| US20170196825A1 (en) | Pharmaceutical compositions comprising dgla and use of same | |
| Ertam et al. | The Turkish guideline for the diagnosis and management of atopic dermatitis-2018 | |
| JP2022505215A (ja) | 炎症性、線維性、および増殖性状態を治療するためのdglaおよび/または15-hetre | |
| CN110996937A (zh) | 包含dgla的药物组合物及其用途 | |
| HK40034640A (en) | Pharmaceutical compositions comprising dgla and use of same | |
| HK1236390A1 (en) | Pharmaceutical compositions comprising dgla and use of same | |
| HK1236390B (en) | Pharmaceutical compositions comprising dgla and use of same | |
| HK40081935A (en) | Treatment of amyotrophic lateral sclerosis | |
| Gold | Are Your Meds Making You Sick?: A Pharmacist's Guide to Avoiding Dangerous Drug Interactions, Reactions, and Side-Effects | |
| Bukhari | Pharmacy Registration Assessment Questions |