ES2784477T3 - Acidos carboxílicos heterocíclicos como activadores de guanilato ciclasa soluble - Google Patents
Acidos carboxílicos heterocíclicos como activadores de guanilato ciclasa soluble Download PDFInfo
- Publication number
- ES2784477T3 ES2784477T3 ES15747311T ES15747311T ES2784477T3 ES 2784477 T3 ES2784477 T3 ES 2784477T3 ES 15747311 T ES15747311 T ES 15747311T ES 15747311 T ES15747311 T ES 15747311T ES 2784477 T3 ES2784477 T3 ES 2784477T3
- Authority
- ES
- Spain
- Prior art keywords
- mmol
- mixture
- reduced pressure
- under reduced
- solution
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- -1 Heterocyclic carboxylic acids Chemical class 0.000 title claims abstract description 83
- 102000007637 Soluble Guanylyl Cyclase Human genes 0.000 title description 73
- 108010007205 Soluble Guanylyl Cyclase Proteins 0.000 title description 73
- 239000012190 activator Substances 0.000 title description 21
- 150000001875 compounds Chemical class 0.000 claims abstract description 177
- 150000003839 salts Chemical class 0.000 claims abstract description 38
- 125000000623 heterocyclic group Chemical group 0.000 claims abstract description 30
- 229910052757 nitrogen Inorganic materials 0.000 claims abstract description 16
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Natural products C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 claims abstract description 15
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 claims abstract description 15
- 125000001072 heteroaryl group Chemical group 0.000 claims abstract description 13
- 125000003718 tetrahydrofuranyl group Chemical group 0.000 claims abstract description 13
- 125000001412 tetrahydropyranyl group Chemical group 0.000 claims abstract description 13
- 229910052731 fluorine Inorganic materials 0.000 claims abstract description 12
- 125000003566 oxetanyl group Chemical group 0.000 claims abstract description 12
- 229910052721 tungsten Inorganic materials 0.000 claims abstract description 12
- 229910052799 carbon Inorganic materials 0.000 claims abstract description 8
- 229910052801 chlorine Inorganic materials 0.000 claims abstract description 7
- KYQCOXFCLRTKLS-UHFFFAOYSA-N Pyrazine Chemical compound C1=CN=CC=N1 KYQCOXFCLRTKLS-UHFFFAOYSA-N 0.000 claims abstract description 6
- 229910052727 yttrium Inorganic materials 0.000 claims abstract description 6
- 125000000842 isoxazolyl group Chemical group 0.000 claims abstract description 4
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims abstract description 4
- 125000004076 pyridyl group Chemical group 0.000 claims abstract description 4
- PCNDJXKNXGMECE-UHFFFAOYSA-N Phenazine Natural products C1=CC=CC2=NC3=CC=CC=C3N=C21 PCNDJXKNXGMECE-UHFFFAOYSA-N 0.000 claims abstract description 3
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims description 40
- 208000035475 disorder Diseases 0.000 claims description 22
- 230000004913 activation Effects 0.000 claims description 20
- 201000010099 disease Diseases 0.000 claims description 18
- 230000003176 fibrotic effect Effects 0.000 claims description 12
- 208000010228 Erectile Dysfunction Diseases 0.000 claims description 9
- XEEYBQQBJWHFJM-UHFFFAOYSA-N Iron Chemical compound [Fe] XEEYBQQBJWHFJM-UHFFFAOYSA-N 0.000 claims description 9
- 239000003814 drug Substances 0.000 claims description 9
- 201000001881 impotence Diseases 0.000 claims description 9
- 208000017169 kidney disease Diseases 0.000 claims description 9
- 239000008194 pharmaceutical composition Substances 0.000 claims description 8
- 206010012601 diabetes mellitus Diseases 0.000 claims description 7
- 210000004185 liver Anatomy 0.000 claims description 7
- 208000014001 urinary system disease Diseases 0.000 claims description 6
- 208000027866 inflammatory disease Diseases 0.000 claims description 5
- 208000024172 Cardiovascular disease Diseases 0.000 claims description 4
- 208000012902 Nervous system disease Diseases 0.000 claims description 4
- 208000025966 Neurological disease Diseases 0.000 claims description 4
- 208000018737 Parkinson disease Diseases 0.000 claims description 4
- 230000000747 cardiac effect Effects 0.000 claims description 4
- 208000004296 neuralgia Diseases 0.000 claims description 4
- 208000021722 neuropathic pain Diseases 0.000 claims description 4
- 206010053219 non-alcoholic steatohepatitis Diseases 0.000 claims description 4
- 208000024827 Alzheimer disease Diseases 0.000 claims description 3
- 206010004446 Benign prostatic hyperplasia Diseases 0.000 claims description 3
- 206010020853 Hypertonic bladder Diseases 0.000 claims description 3
- 208000009722 Overactive Urinary Bladder Diseases 0.000 claims description 3
- 208000004403 Prostatic Hyperplasia Diseases 0.000 claims description 3
- 239000003937 drug carrier Substances 0.000 claims description 3
- 208000008338 non-alcoholic fatty liver disease Diseases 0.000 claims description 3
- 208000020629 overactive bladder Diseases 0.000 claims description 3
- 230000002685 pulmonary effect Effects 0.000 claims description 3
- 206010012289 Dementia Diseases 0.000 claims description 2
- 208000007342 Diabetic Nephropathies Diseases 0.000 claims description 2
- 208000010412 Glaucoma Diseases 0.000 claims description 2
- 208000033679 diabetic kidney disease Diseases 0.000 claims description 2
- 201000006938 muscular dystrophy Diseases 0.000 claims description 2
- 239000000546 pharmaceutical excipient Substances 0.000 claims 1
- 239000000203 mixture Substances 0.000 description 317
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 281
- 230000002829 reductive effect Effects 0.000 description 255
- 239000000243 solution Substances 0.000 description 242
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 179
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 159
- 238000003818 flash chromatography Methods 0.000 description 147
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 142
- 239000000741 silica gel Substances 0.000 description 142
- 229910002027 silica gel Inorganic materials 0.000 description 142
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 129
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 93
- 235000019439 ethyl acetate Nutrition 0.000 description 89
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 85
- 229910001868 water Inorganic materials 0.000 description 84
- 238000006243 chemical reaction Methods 0.000 description 71
- 239000011541 reaction mixture Substances 0.000 description 65
- 239000000543 intermediate Substances 0.000 description 62
- 239000012267 brine Substances 0.000 description 55
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 55
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 53
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 53
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 48
- 238000000034 method Methods 0.000 description 43
- 239000012044 organic layer Substances 0.000 description 43
- 238000003756 stirring Methods 0.000 description 43
- 239000012043 crude product Substances 0.000 description 42
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 39
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 39
- 239000000284 extract Substances 0.000 description 39
- 239000007787 solid Substances 0.000 description 39
- MWUXSHHQAYIFBG-UHFFFAOYSA-N Nitric oxide Chemical compound O=[N] MWUXSHHQAYIFBG-UHFFFAOYSA-N 0.000 description 38
- 239000000706 filtrate Substances 0.000 description 35
- 238000002360 preparation method Methods 0.000 description 35
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 34
- 239000012074 organic phase Substances 0.000 description 32
- 229920006395 saturated elastomer Polymers 0.000 description 32
- UGFAIRIUMAVXCW-UHFFFAOYSA-N Carbon monoxide Chemical compound [O+]#[C-] UGFAIRIUMAVXCW-UHFFFAOYSA-N 0.000 description 29
- 229910002091 carbon monoxide Inorganic materials 0.000 description 29
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 29
- 239000002904 solvent Substances 0.000 description 29
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 29
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 28
- 239000003153 chemical reaction reagent Substances 0.000 description 28
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 26
- 239000012298 atmosphere Substances 0.000 description 25
- 238000010898 silica gel chromatography Methods 0.000 description 24
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 23
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 22
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 22
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 20
- LVEYOSJUKRVCCF-UHFFFAOYSA-N 1,3-Bis(diphenylphosphino)propane Substances C=1C=CC=CC=1P(C=1C=CC=CC=1)CCCP(C=1C=CC=CC=1)C1=CC=CC=C1 LVEYOSJUKRVCCF-UHFFFAOYSA-N 0.000 description 19
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical compound [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 19
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 19
- 238000001914 filtration Methods 0.000 description 18
- 238000011282 treatment Methods 0.000 description 18
- 235000019270 ammonium chloride Nutrition 0.000 description 17
- 239000013058 crude material Substances 0.000 description 17
- 239000012071 phase Substances 0.000 description 17
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 16
- 239000008346 aqueous phase Substances 0.000 description 16
- OCXGTPDKNBIOTF-UHFFFAOYSA-N dibromo(triphenyl)-$l^{5}-phosphane Chemical compound C=1C=CC=CC=1P(Br)(C=1C=CC=CC=1)(Br)C1=CC=CC=C1 OCXGTPDKNBIOTF-UHFFFAOYSA-N 0.000 description 16
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 15
- 239000002253 acid Substances 0.000 description 15
- 235000019253 formic acid Nutrition 0.000 description 15
- 239000010410 layer Substances 0.000 description 15
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 14
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 14
- 230000000694 effects Effects 0.000 description 14
- 239000003921 oil Substances 0.000 description 14
- WSFSSNUMVMOOMR-UHFFFAOYSA-N Formaldehyde Chemical compound O=C WSFSSNUMVMOOMR-UHFFFAOYSA-N 0.000 description 13
- IJGRMHOSHXDMSA-UHFFFAOYSA-N nitrogen Substances N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 13
- 229910000029 sodium carbonate Inorganic materials 0.000 description 13
- 235000017550 sodium carbonate Nutrition 0.000 description 13
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 12
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 12
- 239000005909 Kieselgur Substances 0.000 description 12
- 125000000217 alkyl group Chemical group 0.000 description 12
- 238000010992 reflux Methods 0.000 description 12
- 235000017557 sodium bicarbonate Nutrition 0.000 description 12
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 12
- 125000003118 aryl group Chemical group 0.000 description 11
- 230000015572 biosynthetic process Effects 0.000 description 11
- 229950002128 cinaciguat Drugs 0.000 description 11
- WPYWMXNXEZFMAK-UHFFFAOYSA-N cinaciguat Chemical compound C=1C=C(C(O)=O)C=CC=1CN(CCCCC(=O)O)CCC1=CC=CC=C1OCC(C=C1)=CC=C1CCC1=CC=CC=C1 WPYWMXNXEZFMAK-UHFFFAOYSA-N 0.000 description 11
- 238000001816 cooling Methods 0.000 description 11
- ZOOGRGPOEVQQDX-KHLHZJAASA-N cyclic guanosine monophosphate Chemical compound C([C@H]1O2)O[P@](O)(=O)O[C@@H]1[C@H](O)[C@H]2N1C(N=C(NC2=O)N)=C2N=C1 ZOOGRGPOEVQQDX-KHLHZJAASA-N 0.000 description 11
- 239000000725 suspension Substances 0.000 description 11
- 239000007864 aqueous solution Substances 0.000 description 10
- 239000002585 base Substances 0.000 description 10
- 239000000460 chlorine Substances 0.000 description 10
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 10
- 235000019341 magnesium sulphate Nutrition 0.000 description 10
- 239000000463 material Substances 0.000 description 10
- 238000000746 purification Methods 0.000 description 10
- 239000011734 sodium Substances 0.000 description 10
- 238000003786 synthesis reaction Methods 0.000 description 10
- PQHLRGARXNPFCF-UHFFFAOYSA-N 5-chloro-2-[(5-chlorothiophen-2-yl)sulfonylamino]-n-(4-morpholin-4-ylsulfonylphenyl)benzamide Chemical compound S1C(Cl)=CC=C1S(=O)(=O)NC1=CC=C(Cl)C=C1C(=O)NC1=CC=C(S(=O)(=O)N2CCOCC2)C=C1 PQHLRGARXNPFCF-UHFFFAOYSA-N 0.000 description 9
- 206010016654 Fibrosis Diseases 0.000 description 9
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical class Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 9
- 241000699670 Mus sp. Species 0.000 description 9
- 241000700159 Rattus Species 0.000 description 9
- 229950007878 ataciguat Drugs 0.000 description 9
- 239000002552 dosage form Substances 0.000 description 9
- 239000012299 nitrogen atmosphere Substances 0.000 description 9
- 239000000523 sample Substances 0.000 description 9
- 239000012279 sodium borohydride Substances 0.000 description 9
- 229910000033 sodium borohydride Inorganic materials 0.000 description 9
- WGQKYBSKWIADBV-UHFFFAOYSA-N benzylamine Chemical compound NCC1=CC=CC=C1 WGQKYBSKWIADBV-UHFFFAOYSA-N 0.000 description 8
- 125000004432 carbon atom Chemical group C* 0.000 description 8
- 230000004761 fibrosis Effects 0.000 description 8
- 230000002209 hydrophobic effect Effects 0.000 description 8
- 206010020772 Hypertension Diseases 0.000 description 7
- 101100030361 Neurospora crassa (strain ATCC 24698 / 74-OR23-1A / CBS 708.71 / DSM 1257 / FGSC 987) pph-3 gene Proteins 0.000 description 7
- 102000008299 Nitric Oxide Synthase Human genes 0.000 description 7
- 108010021487 Nitric Oxide Synthase Proteins 0.000 description 7
- 150000001412 amines Chemical group 0.000 description 7
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical class OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 7
- 150000003278 haem Chemical class 0.000 description 7
- 239000008241 heterogeneous mixture Substances 0.000 description 7
- 238000004128 high performance liquid chromatography Methods 0.000 description 7
- 229910052739 hydrogen Inorganic materials 0.000 description 7
- 239000003112 inhibitor Substances 0.000 description 7
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 7
- 229940127296 soluble guanylate cyclase stimulator Drugs 0.000 description 7
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 6
- WMFOQBRAJBCJND-UHFFFAOYSA-M Lithium hydroxide Chemical compound [Li+].[OH-] WMFOQBRAJBCJND-UHFFFAOYSA-M 0.000 description 6
- BZLVMXJERCGZMT-UHFFFAOYSA-N Methyl tert-butyl ether Chemical compound COC(C)(C)C BZLVMXJERCGZMT-UHFFFAOYSA-N 0.000 description 6
- 238000003556 assay Methods 0.000 description 6
- 210000004027 cell Anatomy 0.000 description 6
- 230000002440 hepatic effect Effects 0.000 description 6
- 239000001257 hydrogen Substances 0.000 description 6
- 230000001900 immune effect Effects 0.000 description 6
- 238000011534 incubation Methods 0.000 description 6
- 238000002347 injection Methods 0.000 description 6
- 239000007924 injection Substances 0.000 description 6
- 239000012280 lithium aluminium hydride Substances 0.000 description 6
- 229910052763 palladium Inorganic materials 0.000 description 6
- NFHFRUOZVGFOOS-UHFFFAOYSA-N palladium;triphenylphosphane Chemical compound [Pd].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 NFHFRUOZVGFOOS-UHFFFAOYSA-N 0.000 description 6
- 239000000047 product Substances 0.000 description 6
- 150000003254 radicals Chemical class 0.000 description 6
- BNRNXUUZRGQAQC-UHFFFAOYSA-N sildenafil Chemical compound CCCC1=NN(C)C(C(N2)=O)=C1N=C2C(C(=CC=1)OCC)=CC=1S(=O)(=O)N1CCN(C)CC1 BNRNXUUZRGQAQC-UHFFFAOYSA-N 0.000 description 6
- 229910052717 sulfur Inorganic materials 0.000 description 6
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 5
- NKBWMBRPILTCRD-UHFFFAOYSA-N 2-Methylheptanoic acid Chemical compound CCCCCC(C)C(O)=O NKBWMBRPILTCRD-UHFFFAOYSA-N 0.000 description 5
- 229940127280 BAY 41-2272 Drugs 0.000 description 5
- ATOAHNRJAXSBOR-UHFFFAOYSA-N BAY 41-2272 Chemical compound NC1=NC(C=2C3=CC=CN=C3N(CC=3C(=CC=CC=3)F)N=2)=NC=C1C1CC1 ATOAHNRJAXSBOR-UHFFFAOYSA-N 0.000 description 5
- XTHFKEDIFFGKHM-UHFFFAOYSA-N Dimethoxyethane Chemical compound COCCOC XTHFKEDIFFGKHM-UHFFFAOYSA-N 0.000 description 5
- 241000282414 Homo sapiens Species 0.000 description 5
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 5
- 229910010084 LiAlH4 Inorganic materials 0.000 description 5
- 229940123333 Phosphodiesterase 5 inhibitor Drugs 0.000 description 5
- 229960000583 acetic acid Drugs 0.000 description 5
- 239000002671 adjuvant Substances 0.000 description 5
- 230000008901 benefit Effects 0.000 description 5
- 239000000872 buffer Substances 0.000 description 5
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 description 5
- 229910000024 caesium carbonate Inorganic materials 0.000 description 5
- 230000002950 deficient Effects 0.000 description 5
- 239000003480 eluent Substances 0.000 description 5
- 238000010438 heat treatment Methods 0.000 description 5
- 125000005842 heteroatom Chemical group 0.000 description 5
- 239000013067 intermediate product Substances 0.000 description 5
- 230000001404 mediated effect Effects 0.000 description 5
- 230000002503 metabolic effect Effects 0.000 description 5
- 150000004702 methyl esters Chemical class 0.000 description 5
- 229910052760 oxygen Inorganic materials 0.000 description 5
- 239000003208 petroleum Substances 0.000 description 5
- 239000002244 precipitate Substances 0.000 description 5
- 208000002815 pulmonary hypertension Diseases 0.000 description 5
- 230000002441 reversible effect Effects 0.000 description 5
- 239000000126 substance Substances 0.000 description 5
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 4
- 201000001320 Atherosclerosis Diseases 0.000 description 4
- 102000004190 Enzymes Human genes 0.000 description 4
- 108090000790 Enzymes Proteins 0.000 description 4
- 206010019280 Heart failures Diseases 0.000 description 4
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 description 4
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 4
- 208000027418 Wounds and injury Diseases 0.000 description 4
- 239000000654 additive Substances 0.000 description 4
- 238000004458 analytical method Methods 0.000 description 4
- ZADPBFCGQRWHPN-UHFFFAOYSA-N boronic acid Chemical compound OBO ZADPBFCGQRWHPN-UHFFFAOYSA-N 0.000 description 4
- 239000003054 catalyst Substances 0.000 description 4
- 230000004087 circulation Effects 0.000 description 4
- 239000012230 colorless oil Substances 0.000 description 4
- 230000006378 damage Effects 0.000 description 4
- 208000037765 diseases and disorders Diseases 0.000 description 4
- 238000010828 elution Methods 0.000 description 4
- 238000006460 hydrolysis reaction Methods 0.000 description 4
- 238000001727 in vivo Methods 0.000 description 4
- 208000014674 injury Diseases 0.000 description 4
- 229910052742 iron Inorganic materials 0.000 description 4
- 210000003734 kidney Anatomy 0.000 description 4
- 239000002245 particle Substances 0.000 description 4
- 239000002590 phosphodiesterase V inhibitor Substances 0.000 description 4
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 4
- 230000008569 process Effects 0.000 description 4
- 239000000651 prodrug Substances 0.000 description 4
- 229940002612 prodrug Drugs 0.000 description 4
- 230000011664 signaling Effects 0.000 description 4
- 210000003491 skin Anatomy 0.000 description 4
- MNWBNISUBARLIT-UHFFFAOYSA-N sodium cyanide Chemical compound [Na+].N#[C-] MNWBNISUBARLIT-UHFFFAOYSA-N 0.000 description 4
- 239000012321 sodium triacetoxyborohydride Substances 0.000 description 4
- FPGGTKZVZWFYPV-UHFFFAOYSA-M tetrabutylammonium fluoride Chemical compound [F-].CCCC[N+](CCCC)(CCCC)CCCC FPGGTKZVZWFYPV-UHFFFAOYSA-M 0.000 description 4
- VZGDMQKNWNREIO-UHFFFAOYSA-N tetrachloromethane Chemical compound ClC(Cl)(Cl)Cl VZGDMQKNWNREIO-UHFFFAOYSA-N 0.000 description 4
- 230000001225 therapeutic effect Effects 0.000 description 4
- WJKHJLXJJJATHN-UHFFFAOYSA-N triflic anhydride Chemical compound FC(F)(F)S(=O)(=O)OS(=O)(=O)C(F)(F)F WJKHJLXJJJATHN-UHFFFAOYSA-N 0.000 description 4
- 238000004704 ultra performance liquid chromatography Methods 0.000 description 4
- KZPYGQFFRCFCPP-UHFFFAOYSA-N 1,1'-bis(diphenylphosphino)ferrocene Chemical compound [Fe+2].C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1 KZPYGQFFRCFCPP-UHFFFAOYSA-N 0.000 description 3
- FEYDZHNIIMENOB-UHFFFAOYSA-N 2,6-dibromopyridine Chemical compound BrC1=CC=CC(Br)=N1 FEYDZHNIIMENOB-UHFFFAOYSA-N 0.000 description 3
- APIXJSLKIYYUKG-UHFFFAOYSA-N 3 Isobutyl 1 methylxanthine Chemical compound O=C1N(C)C(=O)N(CC(C)C)C2=C1N=CN2 APIXJSLKIYYUKG-UHFFFAOYSA-N 0.000 description 3
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 3
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 3
- 206010009900 Colitis ulcerative Diseases 0.000 description 3
- 208000011231 Crohn disease Diseases 0.000 description 3
- 206010064911 Pulmonary arterial hypertension Diseases 0.000 description 3
- RWRDLPDLKQPQOW-UHFFFAOYSA-N Pyrrolidine Chemical compound C1CCNC1 RWRDLPDLKQPQOW-UHFFFAOYSA-N 0.000 description 3
- 206010063837 Reperfusion injury Diseases 0.000 description 3
- 208000006011 Stroke Diseases 0.000 description 3
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical group [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 3
- 201000006704 Ulcerative Colitis Diseases 0.000 description 3
- 230000003213 activating effect Effects 0.000 description 3
- 230000000996 additive effect Effects 0.000 description 3
- 150000001299 aldehydes Chemical class 0.000 description 3
- 150000008064 anhydrides Chemical class 0.000 description 3
- 230000003502 anti-nociceptive effect Effects 0.000 description 3
- 229910052786 argon Inorganic materials 0.000 description 3
- 230000009286 beneficial effect Effects 0.000 description 3
- 229910052796 boron Inorganic materials 0.000 description 3
- UWTDFICHZKXYAC-UHFFFAOYSA-N boron;oxolane Chemical compound [B].C1CCOC1 UWTDFICHZKXYAC-UHFFFAOYSA-N 0.000 description 3
- 229910052794 bromium Inorganic materials 0.000 description 3
- 208000019425 cirrhosis of liver Diseases 0.000 description 3
- 230000003247 decreasing effect Effects 0.000 description 3
- 239000008367 deionised water Substances 0.000 description 3
- 229910021641 deionized water Inorganic materials 0.000 description 3
- 230000001419 dependent effect Effects 0.000 description 3
- BNIILDVGGAEEIG-UHFFFAOYSA-L disodium hydrogen phosphate Chemical compound [Na+].[Na+].OP([O-])([O-])=O BNIILDVGGAEEIG-UHFFFAOYSA-L 0.000 description 3
- 229910000397 disodium phosphate Inorganic materials 0.000 description 3
- 235000019800 disodium phosphate Nutrition 0.000 description 3
- 239000006185 dispersion Substances 0.000 description 3
- 229940079593 drug Drugs 0.000 description 3
- 239000002158 endotoxin Substances 0.000 description 3
- 150000002148 esters Chemical class 0.000 description 3
- 230000006870 function Effects 0.000 description 3
- 239000007789 gas Substances 0.000 description 3
- 230000007062 hydrolysis Effects 0.000 description 3
- 239000007788 liquid Substances 0.000 description 3
- 210000004072 lung Anatomy 0.000 description 3
- 238000004519 manufacturing process Methods 0.000 description 3
- 210000001589 microsome Anatomy 0.000 description 3
- 208000010125 myocardial infarction Diseases 0.000 description 3
- 210000000651 myofibroblast Anatomy 0.000 description 3
- 229910000489 osmium tetroxide Inorganic materials 0.000 description 3
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 3
- LJCNRYVRMXRIQR-OLXYHTOASA-L potassium sodium L-tartrate Chemical compound [Na+].[K+].[O-]C(=O)[C@H](O)[C@@H](O)C([O-])=O LJCNRYVRMXRIQR-OLXYHTOASA-L 0.000 description 3
- 230000003389 potentiating effect Effects 0.000 description 3
- 125000003373 pyrazinyl group Chemical group 0.000 description 3
- 230000004044 response Effects 0.000 description 3
- 229960003310 sildenafil Drugs 0.000 description 3
- 235000011006 sodium potassium tartrate Nutrition 0.000 description 3
- 239000012453 solvate Substances 0.000 description 3
- 239000011593 sulfur Chemical group 0.000 description 3
- JYEBYYKITMDTSA-UHFFFAOYSA-N tert-butyl 6-(bromomethyl)-5-fluoro-8-methoxy-3,4-dihydro-1H-isoquinoline-2-carboxylate Chemical compound COC1=CC(CBr)=C(F)C2=C1CN(CC2)C(=O)OC(C)(C)C JYEBYYKITMDTSA-UHFFFAOYSA-N 0.000 description 3
- 238000012360 testing method Methods 0.000 description 3
- 230000009424 thromboembolic effect Effects 0.000 description 3
- 210000001519 tissue Anatomy 0.000 description 3
- ITMCEJHCFYSIIV-UHFFFAOYSA-M triflate Chemical compound [O-]S(=O)(=O)C(F)(F)F ITMCEJHCFYSIIV-UHFFFAOYSA-M 0.000 description 3
- RKMGAJGJIURJSJ-UHFFFAOYSA-N 2,2,6,6-tetramethylpiperidine Chemical compound CC1(C)CCCC(C)(C)N1 RKMGAJGJIURJSJ-UHFFFAOYSA-N 0.000 description 2
- LTMRRSWNXVJMBA-UHFFFAOYSA-L 2,2-diethylpropanedioate Chemical compound CCC(CC)(C([O-])=O)C([O-])=O LTMRRSWNXVJMBA-UHFFFAOYSA-L 0.000 description 2
- SYPZKXFUCNTLLN-UHFFFAOYSA-N 2-(2-chloropyrimidin-4-yl)-6-methylphenol Chemical compound ClC1=NC=CC(=N1)C1=C(C(=CC=C1)C)O SYPZKXFUCNTLLN-UHFFFAOYSA-N 0.000 description 2
- AUWDOZOUJWEPBA-UHFFFAOYSA-N 2-(4-methoxyphenyl)ethanol Chemical compound COC1=CC=C(CCO)C=C1 AUWDOZOUJWEPBA-UHFFFAOYSA-N 0.000 description 2
- OTUYBYTUBWJBLO-UHFFFAOYSA-N 3-bromo-5-methylphenol Chemical compound CC1=CC(O)=CC(Br)=C1 OTUYBYTUBWJBLO-UHFFFAOYSA-N 0.000 description 2
- LTPVSOCPYWDIFU-UHFFFAOYSA-N 4-methoxyphenylethylamine Chemical compound COC1=CC=C(CCN)C=C1 LTPVSOCPYWDIFU-UHFFFAOYSA-N 0.000 description 2
- DTLMHPLXFNMMBY-UHFFFAOYSA-N 6-(bromomethyl)-5-methyl-3,4-dihydro-1H-isoquinoline-2-carboxylic acid Chemical compound BrCC=1C(=C2CCN(CC2=CC=1)C(=O)O)C DTLMHPLXFNMMBY-UHFFFAOYSA-N 0.000 description 2
- ATRRKUHOCOJYRX-UHFFFAOYSA-N Ammonium bicarbonate Chemical compound [NH4+].OC([O-])=O ATRRKUHOCOJYRX-UHFFFAOYSA-N 0.000 description 2
- 229910000013 Ammonium bicarbonate Inorganic materials 0.000 description 2
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 2
- 206010002388 Angina unstable Diseases 0.000 description 2
- 206010003225 Arteriospasm coronary Diseases 0.000 description 2
- ZOXJGFHDIHLPTG-UHFFFAOYSA-N Boron Chemical compound [B] ZOXJGFHDIHLPTG-UHFFFAOYSA-N 0.000 description 2
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 description 2
- IMTAMTUGILGMPL-UHFFFAOYSA-N CC1C2=CC=CC=C2CN1C(=O)O Chemical compound CC1C2=CC=CC=C2CN1C(=O)O IMTAMTUGILGMPL-UHFFFAOYSA-N 0.000 description 2
- 239000004215 Carbon black (E152) Substances 0.000 description 2
- 208000006545 Chronic Obstructive Pulmonary Disease Diseases 0.000 description 2
- OCUCCJIRFHNWBP-IYEMJOQQSA-L Copper gluconate Chemical class [Cu+2].OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C([O-])=O.OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C([O-])=O OCUCCJIRFHNWBP-IYEMJOQQSA-L 0.000 description 2
- 208000003890 Coronary Vasospasm Diseases 0.000 description 2
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 2
- XKMLYUALXHKNFT-UUOKFMHZSA-N Guanosine-5'-triphosphate Chemical compound C1=2NC(N)=NC(=O)C=2N=CN1[C@@H]1O[C@H](COP(O)(=O)OP(O)(=O)OP(O)(O)=O)[C@@H](O)[C@H]1O XKMLYUALXHKNFT-UUOKFMHZSA-N 0.000 description 2
- 241000282412 Homo Species 0.000 description 2
- QIGBRXMKCJKVMJ-UHFFFAOYSA-N Hydroquinone Chemical compound OC1=CC=C(O)C=C1 QIGBRXMKCJKVMJ-UHFFFAOYSA-N 0.000 description 2
- 208000004454 Hyperalgesia Diseases 0.000 description 2
- 206010061218 Inflammation Diseases 0.000 description 2
- 206010022489 Insulin Resistance Diseases 0.000 description 2
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 2
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 2
- 241000699666 Mus <mouse, genus> Species 0.000 description 2
- 102100028452 Nitric oxide synthase, endothelial Human genes 0.000 description 2
- 208000002193 Pain Diseases 0.000 description 2
- 208000005764 Peripheral Arterial Disease Diseases 0.000 description 2
- 208000030831 Peripheral arterial occlusive disease Diseases 0.000 description 2
- 102000004861 Phosphoric Diester Hydrolases Human genes 0.000 description 2
- 108090001050 Phosphoric Diester Hydrolases Proteins 0.000 description 2
- 201000004681 Psoriasis Diseases 0.000 description 2
- DWAQJAXMDSEUJJ-UHFFFAOYSA-M Sodium bisulfite Chemical compound [Na+].OS([O-])=O DWAQJAXMDSEUJJ-UHFFFAOYSA-M 0.000 description 2
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 2
- 208000007718 Stable Angina Diseases 0.000 description 2
- ZSJLQEPLLKMAKR-UHFFFAOYSA-N Streptozotocin Natural products O=NN(C)C(=O)NC1C(O)OC(CO)C(O)C1O ZSJLQEPLLKMAKR-UHFFFAOYSA-N 0.000 description 2
- 201000009594 Systemic Scleroderma Diseases 0.000 description 2
- 206010042953 Systemic sclerosis Diseases 0.000 description 2
- 102000009618 Transforming Growth Factors Human genes 0.000 description 2
- 108010009583 Transforming Growth Factors Proteins 0.000 description 2
- 208000007814 Unstable Angina Diseases 0.000 description 2
- 208000035868 Vascular inflammations Diseases 0.000 description 2
- XJLXINKUBYWONI-DQQFMEOOSA-N [[(2r,3r,4r,5r)-5-(6-aminopurin-9-yl)-3-hydroxy-4-phosphonooxyoxolan-2-yl]methoxy-hydroxyphosphoryl] [(2s,3r,4s,5s)-5-(3-carbamoylpyridin-1-ium-1-yl)-3,4-dihydroxyoxolan-2-yl]methyl phosphate Chemical compound NC(=O)C1=CC=C[N+]([C@@H]2[C@H]([C@@H](O)[C@H](COP([O-])(=O)OP(O)(=O)OC[C@@H]3[C@H]([C@@H](OP(O)(O)=O)[C@@H](O3)N3C4=NC=NC(N)=C4N=C3)O)O2)O)=C1 XJLXINKUBYWONI-DQQFMEOOSA-N 0.000 description 2
- XBJFCYDKBDVADW-UHFFFAOYSA-N acetonitrile;formic acid Chemical compound CC#N.OC=O XBJFCYDKBDVADW-UHFFFAOYSA-N 0.000 description 2
- 230000002378 acidificating effect Effects 0.000 description 2
- 230000002411 adverse Effects 0.000 description 2
- 150000001347 alkyl bromides Chemical class 0.000 description 2
- 230000029936 alkylation Effects 0.000 description 2
- 238000005804 alkylation reaction Methods 0.000 description 2
- 125000002947 alkylene group Chemical group 0.000 description 2
- 229910052782 aluminium Inorganic materials 0.000 description 2
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 2
- VSCWAEJMTAWNJL-UHFFFAOYSA-K aluminium trichloride Chemical compound Cl[Al](Cl)Cl VSCWAEJMTAWNJL-UHFFFAOYSA-K 0.000 description 2
- 235000012538 ammonium bicarbonate Nutrition 0.000 description 2
- 239000001099 ammonium carbonate Substances 0.000 description 2
- 238000010171 animal model Methods 0.000 description 2
- 206010003246 arthritis Diseases 0.000 description 2
- 208000006673 asthma Diseases 0.000 description 2
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical group [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 2
- 125000002047 benzodioxolyl group Chemical group O1OC(C2=C1C=CC=C2)* 0.000 description 2
- 125000004619 benzopyranyl group Chemical group O1C(C=CC2=C1C=CC=C2)* 0.000 description 2
- AGEZXYOZHKGVCM-UHFFFAOYSA-N benzyl bromide Chemical compound BrCC1=CC=CC=C1 AGEZXYOZHKGVCM-UHFFFAOYSA-N 0.000 description 2
- 230000017531 blood circulation Effects 0.000 description 2
- 230000036772 blood pressure Effects 0.000 description 2
- UORVGPXVDQYIDP-UHFFFAOYSA-N borane Chemical compound B UORVGPXVDQYIDP-UHFFFAOYSA-N 0.000 description 2
- 230000002612 cardiopulmonary effect Effects 0.000 description 2
- 210000001715 carotid artery Anatomy 0.000 description 2
- 239000000679 carrageenan Substances 0.000 description 2
- 235000010418 carrageenan Nutrition 0.000 description 2
- 229920001525 carrageenan Polymers 0.000 description 2
- 229940113118 carrageenan Drugs 0.000 description 2
- 239000001913 cellulose Substances 0.000 description 2
- 229920002678 cellulose Polymers 0.000 description 2
- 239000003795 chemical substances by application Substances 0.000 description 2
- 238000004587 chromatography analysis Methods 0.000 description 2
- 229960004106 citric acid Drugs 0.000 description 2
- 238000004440 column chromatography Methods 0.000 description 2
- 201000011634 coronary artery vasospasm Diseases 0.000 description 2
- 210000004351 coronary vessel Anatomy 0.000 description 2
- 230000007812 deficiency Effects 0.000 description 2
- 238000010511 deprotection reaction Methods 0.000 description 2
- 238000004807 desolvation Methods 0.000 description 2
- NKLCNNUWBJBICK-UHFFFAOYSA-N dess–martin periodinane Chemical compound C1=CC=C2I(OC(=O)C)(OC(C)=O)(OC(C)=O)OC(=O)C2=C1 NKLCNNUWBJBICK-UHFFFAOYSA-N 0.000 description 2
- 238000011833 dog model Methods 0.000 description 2
- 230000002526 effect on cardiovascular system Effects 0.000 description 2
- 230000002500 effect on skin Effects 0.000 description 2
- 239000000839 emulsion Substances 0.000 description 2
- 210000002950 fibroblast Anatomy 0.000 description 2
- 239000012065 filter cake Substances 0.000 description 2
- 239000011888 foil Substances 0.000 description 2
- 206010061989 glomerulosclerosis Diseases 0.000 description 2
- 239000008103 glucose Substances 0.000 description 2
- 239000003119 guanylate cyclase activator Substances 0.000 description 2
- 125000005843 halogen group Chemical group 0.000 description 2
- 230000000004 hemodynamic effect Effects 0.000 description 2
- 235000009200 high fat diet Nutrition 0.000 description 2
- 150000004678 hydrides Chemical class 0.000 description 2
- 229930195733 hydrocarbon Natural products 0.000 description 2
- 238000000338 in vitro Methods 0.000 description 2
- 238000011065 in-situ storage Methods 0.000 description 2
- 230000002757 inflammatory effect Effects 0.000 description 2
- 230000004054 inflammatory process Effects 0.000 description 2
- 238000001802 infusion Methods 0.000 description 2
- 230000002401 inhibitory effect Effects 0.000 description 2
- 201000004332 intermediate coronary syndrome Diseases 0.000 description 2
- 201000006334 interstitial nephritis Diseases 0.000 description 2
- 208000001286 intracranial vasospasm Diseases 0.000 description 2
- 238000001990 intravenous administration Methods 0.000 description 2
- 208000028867 ischemia Diseases 0.000 description 2
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 2
- 150000002576 ketones Chemical class 0.000 description 2
- 230000003907 kidney function Effects 0.000 description 2
- DLEDOFVPSDKWEF-UHFFFAOYSA-N lithium butane Chemical compound [Li+].CCC[CH2-] DLEDOFVPSDKWEF-UHFFFAOYSA-N 0.000 description 2
- 210000005265 lung cell Anatomy 0.000 description 2
- 239000002207 metabolite Substances 0.000 description 2
- VNWKTOKETHGBQD-UHFFFAOYSA-N methane Chemical compound C VNWKTOKETHGBQD-UHFFFAOYSA-N 0.000 description 2
- XALGDQALSLWQRH-UHFKNVDDSA-N methyl (1R,2R)-2-methyl-3-azabicyclo[3.1.0]hexane-1-carboxylate Chemical compound COC(=O)[C@]12[C@H](NCC2C1)C XALGDQALSLWQRH-UHFKNVDDSA-N 0.000 description 2
- SBYYTPILEYEXAK-WUMONGPASA-N methyl (1R,2S)-3-(6-bromopyridin-2-yl)-2-(methoxymethyl)-3-azabicyclo[3.1.0]hexane-1-carboxylate Chemical compound COC(=O)[C@]12[C@H](N(CC2C1)C1=NC(=CC=C1)Br)COC SBYYTPILEYEXAK-WUMONGPASA-N 0.000 description 2
- 239000002480 mineral oil Substances 0.000 description 2
- 235000010446 mineral oil Nutrition 0.000 description 2
- 238000010172 mouse model Methods 0.000 description 2
- 238000002552 multiple reaction monitoring Methods 0.000 description 2
- 201000006417 multiple sclerosis Diseases 0.000 description 2
- MZRVEZGGRBJDDB-UHFFFAOYSA-N n-Butyllithium Substances [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 description 2
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical class CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 2
- 229930027945 nicotinamide-adenine dinucleotide Natural products 0.000 description 2
- 239000002840 nitric oxide donor Substances 0.000 description 2
- 150000007524 organic acids Chemical class 0.000 description 2
- CTSLXHKWHWQRSH-UHFFFAOYSA-N oxalyl chloride Chemical compound ClC(=O)C(Cl)=O CTSLXHKWHWQRSH-UHFFFAOYSA-N 0.000 description 2
- JMJRYTGVHCAYCT-UHFFFAOYSA-N oxan-4-one Chemical compound O=C1CCOCC1 JMJRYTGVHCAYCT-UHFFFAOYSA-N 0.000 description 2
- 230000003647 oxidation Effects 0.000 description 2
- 238000007254 oxidation reaction Methods 0.000 description 2
- 239000001301 oxygen Substances 0.000 description 2
- 230000037361 pathway Effects 0.000 description 2
- XHXFXVLFKHQFAL-UHFFFAOYSA-N phosphoryl trichloride Chemical compound ClP(Cl)(Cl)=O XHXFXVLFKHQFAL-UHFFFAOYSA-N 0.000 description 2
- 239000000843 powder Substances 0.000 description 2
- 230000002035 prolonged effect Effects 0.000 description 2
- 102000004169 proteins and genes Human genes 0.000 description 2
- 108090000623 proteins and genes Proteins 0.000 description 2
- XKMLYUALXHKNFT-UHFFFAOYSA-N rGTP Natural products C1=2NC(N)=NC(=O)C=2N=CN1C1OC(COP(O)(=O)OP(O)(=O)OP(O)(O)=O)C(O)C1O XKMLYUALXHKNFT-UHFFFAOYSA-N 0.000 description 2
- 230000035484 reaction time Effects 0.000 description 2
- 230000009467 reduction Effects 0.000 description 2
- 238000006722 reduction reaction Methods 0.000 description 2
- 238000006268 reductive amination reaction Methods 0.000 description 2
- 208000037803 restenosis Diseases 0.000 description 2
- 230000000717 retained effect Effects 0.000 description 2
- 238000012552 review Methods 0.000 description 2
- 238000005096 rolling process Methods 0.000 description 2
- 238000005070 sampling Methods 0.000 description 2
- 210000002966 serum Anatomy 0.000 description 2
- WRIKHQLVHPKCJU-UHFFFAOYSA-N sodium bis(trimethylsilyl)amide Chemical compound C[Si](C)(C)N([Na])[Si](C)(C)C WRIKHQLVHPKCJU-UHFFFAOYSA-N 0.000 description 2
- QDRKDTQENPPHOJ-UHFFFAOYSA-N sodium ethoxide Chemical compound [Na+].CC[O-] QDRKDTQENPPHOJ-UHFFFAOYSA-N 0.000 description 2
- 235000010267 sodium hydrogen sulphite Nutrition 0.000 description 2
- JQWHASGSAFIOCM-UHFFFAOYSA-M sodium periodate Chemical compound [Na+].[O-]I(=O)(=O)=O JQWHASGSAFIOCM-UHFFFAOYSA-M 0.000 description 2
- 241000894007 species Species 0.000 description 2
- ZSJLQEPLLKMAKR-GKHCUFPYSA-N streptozocin Chemical compound O=NN(C)C(=O)N[C@H]1[C@@H](O)O[C@H](CO)[C@@H](O)[C@@H]1O ZSJLQEPLLKMAKR-GKHCUFPYSA-N 0.000 description 2
- 229960001052 streptozocin Drugs 0.000 description 2
- 150000003467 sulfuric acid derivatives Chemical class 0.000 description 2
- 230000002459 sustained effect Effects 0.000 description 2
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 2
- HJUGFYREWKUQJT-UHFFFAOYSA-N tetrabromomethane Chemical compound BrC(Br)(Br)Br HJUGFYREWKUQJT-UHFFFAOYSA-N 0.000 description 2
- WROMPOXWARCANT-UHFFFAOYSA-N tfa trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F.OC(=O)C(F)(F)F WROMPOXWARCANT-UHFFFAOYSA-N 0.000 description 2
- 238000002560 therapeutic procedure Methods 0.000 description 2
- 238000004809 thin layer chromatography Methods 0.000 description 2
- LEIMLDGFXIOXMT-UHFFFAOYSA-N trimethylsilyl cyanide Chemical compound C[Si](C)(C)C#N LEIMLDGFXIOXMT-UHFFFAOYSA-N 0.000 description 2
- 208000001072 type 2 diabetes mellitus Diseases 0.000 description 2
- 238000001946 ultra-performance liquid chromatography-mass spectrometry Methods 0.000 description 2
- 229910052720 vanadium Inorganic materials 0.000 description 2
- 230000003612 virological effect Effects 0.000 description 2
- 239000003039 volatile agent Substances 0.000 description 2
- 239000003643 water by type Substances 0.000 description 2
- 239000011701 zinc Substances 0.000 description 2
- VNDYJBBGRKZCSX-UHFFFAOYSA-L zinc bromide Chemical compound Br[Zn]Br VNDYJBBGRKZCSX-UHFFFAOYSA-L 0.000 description 2
- UHVMMEOXYDMDKI-JKYCWFKZSA-L zinc;1-(5-cyanopyridin-2-yl)-3-[(1s,2s)-2-(6-fluoro-2-hydroxy-3-propanoylphenyl)cyclopropyl]urea;diacetate Chemical compound [Zn+2].CC([O-])=O.CC([O-])=O.CCC(=O)C1=CC=C(F)C([C@H]2[C@H](C2)NC(=O)NC=2N=CC(=CC=2)C#N)=C1O UHVMMEOXYDMDKI-JKYCWFKZSA-L 0.000 description 2
- HZSPBSVPQUWUPN-QPUJVOFHSA-N (1R,6S)-3-(6-bromopyridin-2-yl)-3-azabicyclo[4.1.0]heptane-6-carboxylic acid Chemical compound BrC1=CC=CC(=N1)N1C[C@@H]2C[C@@]2(CC1)C(=O)O HZSPBSVPQUWUPN-QPUJVOFHSA-N 0.000 description 1
- OGSOZDUKUQRPIT-KPZWWZAWSA-N (1R,6S)-3-[6-(2-hydroxyphenyl)pyridin-2-yl]-3-azabicyclo[4.1.0]heptane-6-carboxylic acid Chemical compound OC1=C(C=CC=C1)C1=CC=CC(=N1)N1C[C@@H]2C[C@@]2(CC1)C(=O)O OGSOZDUKUQRPIT-KPZWWZAWSA-N 0.000 description 1
- AOFUBOWZWQFQJU-SNOJBQEQSA-N (2r,3s,4s,5r)-2,5-bis(hydroxymethyl)oxolane-2,3,4-triol;(2s,3r,4s,5s,6r)-6-(hydroxymethyl)oxane-2,3,4,5-tetrol Chemical compound OC[C@H]1O[C@](O)(CO)[C@@H](O)[C@@H]1O.OC[C@H]1O[C@H](O)[C@H](O)[C@@H](O)[C@@H]1O AOFUBOWZWQFQJU-SNOJBQEQSA-N 0.000 description 1
- VIJSPAIQWVPKQZ-BLECARSGSA-N (2s)-2-[[(2s)-2-[[(2s)-2-[[(2s)-2-[[(2s)-2-[[(2s)-2-acetamido-5-(diaminomethylideneamino)pentanoyl]amino]-4-methylpentanoyl]amino]-4,4-dimethylpentanoyl]amino]-4-methylpentanoyl]amino]propanoyl]amino]-5-(diaminomethylideneamino)pentanoic acid Chemical compound NC(=N)NCCC[C@@H](C(O)=O)NC(=O)[C@H](C)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC(C)(C)C)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCCNC(N)=N)NC(C)=O VIJSPAIQWVPKQZ-BLECARSGSA-N 0.000 description 1
- JEQDSBVHLKBEIZ-REOHCLBHSA-N (2s)-2-chloropropanoyl chloride Chemical compound C[C@H](Cl)C(Cl)=O JEQDSBVHLKBEIZ-REOHCLBHSA-N 0.000 description 1
- FEWIGMWODIRUJM-HWKANZROSA-N (E)-4-hexen-3-one Chemical compound CCC(=O)\C=C\C FEWIGMWODIRUJM-HWKANZROSA-N 0.000 description 1
- CTKINSOISVBQLD-GSVOUGTGSA-N (R)-Glycidol Chemical compound OC[C@@H]1CO1 CTKINSOISVBQLD-GSVOUGTGSA-N 0.000 description 1
- CTKINSOISVBQLD-VKHMYHEASA-N (S)-Glycidol Chemical compound OC[C@H]1CO1 CTKINSOISVBQLD-VKHMYHEASA-N 0.000 description 1
- UWYVPFMHMJIBHE-OWOJBTEDSA-N (e)-2-hydroxybut-2-enedioic acid Chemical class OC(=O)\C=C(\O)C(O)=O UWYVPFMHMJIBHE-OWOJBTEDSA-N 0.000 description 1
- QPFMBZIOSGYJDE-UHFFFAOYSA-N 1,1,2,2-tetrachloroethane Chemical compound ClC(Cl)C(Cl)Cl QPFMBZIOSGYJDE-UHFFFAOYSA-N 0.000 description 1
- ZWHFQQSDBWEIEL-UHFFFAOYSA-N 1-(bromomethyl)-2-fluoro-3-methoxy-5-methylbenzene Chemical compound BrCC1=C(C(=CC(=C1)C)OC)F ZWHFQQSDBWEIEL-UHFFFAOYSA-N 0.000 description 1
- GIGRWGTZFONRKA-UHFFFAOYSA-N 1-(bromomethyl)-4-methoxybenzene Chemical compound COC1=CC=C(CBr)C=C1 GIGRWGTZFONRKA-UHFFFAOYSA-N 0.000 description 1
- HXKKHQJGJAFBHI-UHFFFAOYSA-N 1-aminopropan-2-ol Chemical compound CC(O)CN HXKKHQJGJAFBHI-UHFFFAOYSA-N 0.000 description 1
- XVNQVSGOIUYOPB-UHFFFAOYSA-N 1-bromo-3-fluoro-5-methoxybenzene Chemical compound COC1=CC(F)=CC(Br)=C1 XVNQVSGOIUYOPB-UHFFFAOYSA-N 0.000 description 1
- AOEVRCZZWJWKPG-UHFFFAOYSA-N 1-bromo-3-methoxy-5-methylbenzene Chemical compound COC1=CC(C)=CC(Br)=C1 AOEVRCZZWJWKPG-UHFFFAOYSA-N 0.000 description 1
- IIZPXYDJLKNOIY-JXPKJXOSSA-N 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCC\C=C/C\C=C/C\C=C/C\C=C/CCCCC IIZPXYDJLKNOIY-JXPKJXOSSA-N 0.000 description 1
- LZMHWZHOZLVYDL-UHFFFAOYSA-N 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one Chemical compound C1=CC=C2N3C(=O)ON=C3C=NC2=C1 LZMHWZHOZLVYDL-UHFFFAOYSA-N 0.000 description 1
- YBYIRNPNPLQARY-UHFFFAOYSA-N 1H-indene Natural products C1=CC=C2CC=CC2=C1 YBYIRNPNPLQARY-UHFFFAOYSA-N 0.000 description 1
- BTTNYQZNBZNDOR-UHFFFAOYSA-N 2,4-dichloropyrimidine Chemical compound ClC1=CC=NC(Cl)=N1 BTTNYQZNBZNDOR-UHFFFAOYSA-N 0.000 description 1
- OZAIFHULBGXAKX-UHFFFAOYSA-N 2-(2-cyanopropan-2-yldiazenyl)-2-methylpropanenitrile Chemical compound N#CC(C)(C)N=NC(C)(C)C#N OZAIFHULBGXAKX-UHFFFAOYSA-N 0.000 description 1
- OBTZDIRUQWFRFZ-UHFFFAOYSA-N 2-(5-methylfuran-2-yl)-n-(4-methylphenyl)quinoline-4-carboxamide Chemical compound O1C(C)=CC=C1C1=CC(C(=O)NC=2C=CC(C)=CC=2)=C(C=CC=C2)C2=N1 OBTZDIRUQWFRFZ-UHFFFAOYSA-N 0.000 description 1
- BPXKZEMBEZGUAH-UHFFFAOYSA-N 2-(chloromethoxy)ethyl-trimethylsilane Chemical compound C[Si](C)(C)CCOCCl BPXKZEMBEZGUAH-UHFFFAOYSA-N 0.000 description 1
- GVNVAWHJIKLAGL-UHFFFAOYSA-N 2-(cyclohexen-1-yl)cyclohexan-1-one Chemical compound O=C1CCCCC1C1=CCCCC1 GVNVAWHJIKLAGL-UHFFFAOYSA-N 0.000 description 1
- UHOIFEOHBGCPHE-UHFFFAOYSA-N 2-[(2-methylpropan-2-yl)oxycarbonyl]-3,4-dihydro-1h-isoquinoline-6-carboxylic acid Chemical compound OC(=O)C1=CC=C2CN(C(=O)OC(C)(C)C)CCC2=C1 UHOIFEOHBGCPHE-UHFFFAOYSA-N 0.000 description 1
- YSUIQYOGTINQIN-UZFYAQMZSA-N 2-amino-9-[(1S,6R,8R,9S,10R,15R,17R,18R)-8-(6-aminopurin-9-yl)-9,18-difluoro-3,12-dihydroxy-3,12-bis(sulfanylidene)-2,4,7,11,13,16-hexaoxa-3lambda5,12lambda5-diphosphatricyclo[13.2.1.06,10]octadecan-17-yl]-1H-purin-6-one Chemical compound NC1=NC2=C(N=CN2[C@@H]2O[C@@H]3COP(S)(=O)O[C@@H]4[C@@H](COP(S)(=O)O[C@@H]2[C@@H]3F)O[C@H]([C@H]4F)N2C=NC3=C2N=CN=C3N)C(=O)N1 YSUIQYOGTINQIN-UZFYAQMZSA-N 0.000 description 1
- LIHCOUDNHILORI-UHFFFAOYSA-N 2-fluoro-3-methoxybenzaldehyde Chemical compound COC1=CC=CC(C=O)=C1F LIHCOUDNHILORI-UHFFFAOYSA-N 0.000 description 1
- DKIQXHIAEMGZGO-UHFFFAOYSA-N 2-fluoro-5-methoxybenzaldehyde Chemical compound COC1=CC=C(F)C(C=O)=C1 DKIQXHIAEMGZGO-UHFFFAOYSA-N 0.000 description 1
- UPHOPMSGKZNELG-UHFFFAOYSA-N 2-hydroxynaphthalene-1-carboxylic acid Chemical class C1=CC=C2C(C(=O)O)=C(O)C=CC2=C1 UPHOPMSGKZNELG-UHFFFAOYSA-N 0.000 description 1
- CDJJKCZKTVLVEQ-UHFFFAOYSA-N 3,4-dihydro-1h-isoquinoline-2,6-dicarboxylic acid Chemical compound OC(=O)C1=CC=C2CN(C(=O)O)CCC2=C1 CDJJKCZKTVLVEQ-UHFFFAOYSA-N 0.000 description 1
- QBWKPGNFQQJGFY-QLFBSQMISA-N 3-[(1r)-1-[(2r,6s)-2,6-dimethylmorpholin-4-yl]ethyl]-n-[6-methyl-3-(1h-pyrazol-4-yl)imidazo[1,2-a]pyrazin-8-yl]-1,2-thiazol-5-amine Chemical compound N1([C@H](C)C2=NSC(NC=3C4=NC=C(N4C=C(C)N=3)C3=CNN=C3)=C2)C[C@H](C)O[C@H](C)C1 QBWKPGNFQQJGFY-QLFBSQMISA-N 0.000 description 1
- JPCISVSOTKMFPG-UHFFFAOYSA-N 3-methoxy-2-methylbenzoic acid Chemical compound COC1=CC=CC(C(O)=O)=C1C JPCISVSOTKMFPG-UHFFFAOYSA-N 0.000 description 1
- 229960000549 4-dimethylaminophenol Drugs 0.000 description 1
- SLHGBFQBNFLRBH-UHFFFAOYSA-N 6-(bromomethyl)-3,4-dihydro-1H-isoquinoline-2-carboxylic acid Chemical compound BrCC=1C=C2CCN(CC2=CC=1)C(=O)O SLHGBFQBNFLRBH-UHFFFAOYSA-N 0.000 description 1
- KGSDFAQFMVJHTF-UHFFFAOYSA-N 6-(bromomethyl)-8-methyl-3,4-dihydro-1H-isoquinoline-2-carboxylic acid Chemical compound BrCC=1C=C2CCN(CC2=C(C=1)C)C(=O)O KGSDFAQFMVJHTF-UHFFFAOYSA-N 0.000 description 1
- 125000004939 6-pyridyl group Chemical group N1=CC=CC=C1* 0.000 description 1
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 1
- OZAIFHULBGXAKX-VAWYXSNFSA-N AIBN Substances N#CC(C)(C)\N=N\C(C)(C)C#N OZAIFHULBGXAKX-VAWYXSNFSA-N 0.000 description 1
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical compound [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 description 1
- USFZMSVCRYTOJT-UHFFFAOYSA-N Ammonium acetate Chemical compound N.CC(O)=O USFZMSVCRYTOJT-UHFFFAOYSA-N 0.000 description 1
- 208000003017 Aortic Valve Stenosis Diseases 0.000 description 1
- 102000013918 Apolipoproteins E Human genes 0.000 description 1
- 108010025628 Apolipoproteins E Proteins 0.000 description 1
- 206010003210 Arteriosclerosis Diseases 0.000 description 1
- 208000037260 Atherosclerotic Plaque Diseases 0.000 description 1
- AQYFUZRYBJBAGZ-UHFFFAOYSA-N BAY-41-8543 Chemical compound NC1=NC(C=2C3=CC=CN=C3N(CC=3C(=CC=CC=3)F)N=2)=NC(N)=C1N1CCOCC1 AQYFUZRYBJBAGZ-UHFFFAOYSA-N 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-M Bicarbonate Chemical class OC([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-M 0.000 description 1
- 108010006654 Bleomycin Proteins 0.000 description 1
- 108010017384 Blood Proteins Proteins 0.000 description 1
- 102000004506 Blood Proteins Human genes 0.000 description 1
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 1
- 201000006474 Brain Ischemia Diseases 0.000 description 1
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 1
- COVZYZSDYWQREU-UHFFFAOYSA-N Busulfan Chemical compound CS(=O)(=O)OCCCCOS(C)(=O)=O COVZYZSDYWQREU-UHFFFAOYSA-N 0.000 description 1
- 0 CC(C)(C)OC(*(CC1)Cc2c1c(N)c(COC=C)cc2OC)=O Chemical compound CC(C)(C)OC(*(CC1)Cc2c1c(N)c(COC=C)cc2OC)=O 0.000 description 1
- ATINASUKCFAKRP-UHFFFAOYSA-N CC(C)(C)OC(N1CC(C)(C)c2cc(CBr)ccc2CC1)=O Chemical compound CC(C)(C)OC(N1CC(C)(C)c2cc(CBr)ccc2CC1)=O ATINASUKCFAKRP-UHFFFAOYSA-N 0.000 description 1
- HXERJDLBQXDUTR-XHDPSFHLSA-N CCOC([C@@](C1)(CCC2)[C@@H]1CN2c1nc(Br)ccc1)=O Chemical compound CCOC([C@@](C1)(CCC2)[C@@H]1CN2c1nc(Br)ccc1)=O HXERJDLBQXDUTR-XHDPSFHLSA-N 0.000 description 1
- NNMGVRVQUSRDRO-IONNQARKSA-N CCOC([C@]1(C2)[C@@H]2CNCC1)=O Chemical compound CCOC([C@]1(C2)[C@@H]2CNCC1)=O NNMGVRVQUSRDRO-IONNQARKSA-N 0.000 description 1
- KIJDWKUJPWXBAZ-RCOVLWMOSA-N CNC(N(C[C@@H]1C2)C[C@]12C(OC)=O)=N Chemical compound CNC(N(C[C@@H]1C2)C[C@]12C(OC)=O)=N KIJDWKUJPWXBAZ-RCOVLWMOSA-N 0.000 description 1
- 101100074846 Caenorhabditis elegans lin-2 gene Proteins 0.000 description 1
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 1
- 206010007556 Cardiac failure acute Diseases 0.000 description 1
- 206010007558 Cardiac failure chronic Diseases 0.000 description 1
- 206010008120 Cerebral ischaemia Diseases 0.000 description 1
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 1
- 208000026151 Chronic thromboembolic pulmonary hypertension Diseases 0.000 description 1
- 101150065749 Churc1 gene Proteins 0.000 description 1
- YASYEJJMZJALEJ-UHFFFAOYSA-N Citric acid monohydrate Chemical compound O.OC(=O)CC(O)(C(O)=O)CC(O)=O YASYEJJMZJALEJ-UHFFFAOYSA-N 0.000 description 1
- 102000004654 Cyclic GMP-Dependent Protein Kinases Human genes 0.000 description 1
- 108010003591 Cyclic GMP-Dependent Protein Kinases Proteins 0.000 description 1
- XDTMQSROBMDMFD-UHFFFAOYSA-N Cyclohexane Chemical compound C1CCCCC1 XDTMQSROBMDMFD-UHFFFAOYSA-N 0.000 description 1
- 201000003883 Cystic fibrosis Diseases 0.000 description 1
- 101100407341 Drosophila melanogaster Pde9 gene Proteins 0.000 description 1
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical class OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 1
- 208000005189 Embolism Diseases 0.000 description 1
- PIICEJLVQHRZGT-UHFFFAOYSA-N Ethylenediamine Chemical compound NCCN PIICEJLVQHRZGT-UHFFFAOYSA-N 0.000 description 1
- CWYNVVGOOAEACU-UHFFFAOYSA-N Fe2+ Chemical compound [Fe+2] CWYNVVGOOAEACU-UHFFFAOYSA-N 0.000 description 1
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 1
- 206010018364 Glomerulonephritis Diseases 0.000 description 1
- 206010018372 Glomerulonephritis membranous Diseases 0.000 description 1
- 102000051325 Glucagon Human genes 0.000 description 1
- 108060003199 Glucagon Proteins 0.000 description 1
- CTKINSOISVBQLD-UHFFFAOYSA-N Glycidol Chemical compound OCC1CO1 CTKINSOISVBQLD-UHFFFAOYSA-N 0.000 description 1
- AEMRFAOFKBGASW-UHFFFAOYSA-N Glycolic acid Chemical class OCC(O)=O AEMRFAOFKBGASW-UHFFFAOYSA-N 0.000 description 1
- 108010078321 Guanylate Cyclase Proteins 0.000 description 1
- 102000014469 Guanylate cyclase Human genes 0.000 description 1
- 239000007821 HATU Substances 0.000 description 1
- 208000032759 Hemolytic-Uremic Syndrome Diseases 0.000 description 1
- 208000035154 Hyperesthesia Diseases 0.000 description 1
- 201000009794 Idiopathic Pulmonary Fibrosis Diseases 0.000 description 1
- 208000010159 IgA glomerulonephritis Diseases 0.000 description 1
- 206010021263 IgA nephropathy Diseases 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- 206010061216 Infarction Diseases 0.000 description 1
- 208000022559 Inflammatory bowel disease Diseases 0.000 description 1
- 102000004310 Ion Channels Human genes 0.000 description 1
- 150000000994 L-ascorbates Chemical class 0.000 description 1
- 235000019687 Lamb Nutrition 0.000 description 1
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 1
- 239000012448 Lithium borohydride Substances 0.000 description 1
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 1
- 241000124008 Mammalia Species 0.000 description 1
- ZRVUJXDFFKFLMG-UHFFFAOYSA-N Meloxicam Chemical compound OC=1C2=CC=CC=C2S(=O)(=O)N(C)C=1C(=O)NC1=NC=C(C)S1 ZRVUJXDFFKFLMG-UHFFFAOYSA-N 0.000 description 1
- 239000012359 Methanesulfonyl chloride Substances 0.000 description 1
- 101100497386 Mus musculus Cask gene Proteins 0.000 description 1
- 208000009525 Myocarditis Diseases 0.000 description 1
- 208000034827 Neointima Diseases 0.000 description 1
- 208000028389 Nerve injury Diseases 0.000 description 1
- 102000008052 Nitric Oxide Synthase Type III Human genes 0.000 description 1
- 108010075520 Nitric Oxide Synthase Type III Proteins 0.000 description 1
- XSUXDJHVNPNNFJ-UHFFFAOYSA-N OBOC=C Chemical compound OBOC=C XSUXDJHVNPNNFJ-UHFFFAOYSA-N 0.000 description 1
- HBQNZYGYTVVZQK-UHFFFAOYSA-N OCC=1C=NC=2CN(CCC=2C=1)C(=O)O Chemical compound OCC=1C=NC=2CN(CCC=2C=1)C(=O)O HBQNZYGYTVVZQK-UHFFFAOYSA-N 0.000 description 1
- 102000008212 P-Selectin Human genes 0.000 description 1
- 108010035766 P-Selectin Proteins 0.000 description 1
- 229910019142 PO4 Inorganic materials 0.000 description 1
- 229910019213 POCl3 Inorganic materials 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- XBDQKXXYIPTUBI-UHFFFAOYSA-N Propionic acid Chemical class CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 1
- 102100038239 Protein Churchill Human genes 0.000 description 1
- 108010029485 Protein Isoforms Proteins 0.000 description 1
- 102000001708 Protein Isoforms Human genes 0.000 description 1
- 102100028255 Renin Human genes 0.000 description 1
- 108090000783 Renin Proteins 0.000 description 1
- 241000219061 Rheum Species 0.000 description 1
- 206010050207 Skin fibrosis Diseases 0.000 description 1
- 208000001871 Tachycardia Diseases 0.000 description 1
- 229920002253 Tannate Polymers 0.000 description 1
- FZWLAAWBMGSTSO-UHFFFAOYSA-N Thiazole Chemical compound C1=CSC=N1 FZWLAAWBMGSTSO-UHFFFAOYSA-N 0.000 description 1
- 208000001435 Thromboembolism Diseases 0.000 description 1
- 208000007536 Thrombosis Diseases 0.000 description 1
- 206010048302 Tubulointerstitial nephritis Diseases 0.000 description 1
- SECKRCOLJRRGGV-UHFFFAOYSA-N Vardenafil Chemical compound CCCC1=NC(C)=C(C(N=2)=O)N1NC=2C(C(=CC=1)OCC)=CC=1S(=O)(=O)N1CCN(CC)CC1 SECKRCOLJRRGGV-UHFFFAOYSA-N 0.000 description 1
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 1
- 150000001242 acetic acid derivatives Chemical class 0.000 description 1
- PBCJIPOGFJYBJE-UHFFFAOYSA-N acetonitrile;hydrate Chemical compound O.CC#N PBCJIPOGFJYBJE-UHFFFAOYSA-N 0.000 description 1
- WETWJCDKMRHUPV-UHFFFAOYSA-N acetyl chloride Chemical compound CC(Cl)=O WETWJCDKMRHUPV-UHFFFAOYSA-N 0.000 description 1
- 239000012346 acetyl chloride Substances 0.000 description 1
- 150000008043 acidic salts Chemical class 0.000 description 1
- 150000007513 acids Chemical class 0.000 description 1
- 239000004480 active ingredient Substances 0.000 description 1
- 125000002015 acyclic group Chemical group 0.000 description 1
- 125000002252 acyl group Chemical group 0.000 description 1
- 125000001931 aliphatic group Chemical group 0.000 description 1
- 150000001447 alkali salts Chemical class 0.000 description 1
- 125000003342 alkenyl group Chemical group 0.000 description 1
- 125000003545 alkoxy group Chemical group 0.000 description 1
- 125000004453 alkoxycarbonyl group Chemical group 0.000 description 1
- 125000000304 alkynyl group Chemical group 0.000 description 1
- PNEYBMLMFCGWSK-UHFFFAOYSA-N aluminium oxide Inorganic materials [O-2].[O-2].[O-2].[Al+3].[Al+3] PNEYBMLMFCGWSK-UHFFFAOYSA-N 0.000 description 1
- CEGOLXSVJUTHNZ-UHFFFAOYSA-K aluminium tristearate Chemical compound [Al+3].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O CEGOLXSVJUTHNZ-UHFFFAOYSA-K 0.000 description 1
- 229940063655 aluminum stearate Drugs 0.000 description 1
- 210000002821 alveolar epithelial cell Anatomy 0.000 description 1
- 230000001668 ameliorated effect Effects 0.000 description 1
- 125000003368 amide group Chemical group 0.000 description 1
- 150000005005 aminopyrimidines Chemical class 0.000 description 1
- QGZKDVFQNNGYKY-UHFFFAOYSA-N ammonia Natural products N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 1
- SWLVFNYSXGMGBS-UHFFFAOYSA-N ammonium bromide Chemical compound [NH4+].[Br-] SWLVFNYSXGMGBS-UHFFFAOYSA-N 0.000 description 1
- 239000000908 ammonium hydroxide Substances 0.000 description 1
- 125000002178 anthracenyl group Chemical group C1(=CC=CC2=CC3=CC=CC=C3C=C12)* 0.000 description 1
- 239000000427 antigen Substances 0.000 description 1
- 108091007433 antigens Proteins 0.000 description 1
- 102000036639 antigens Human genes 0.000 description 1
- 206010002906 aortic stenosis Diseases 0.000 description 1
- 238000010936 aqueous wash Methods 0.000 description 1
- 239000012300 argon atmosphere Substances 0.000 description 1
- 208000037849 arterial hypertension Diseases 0.000 description 1
- 208000011775 arteriosclerosis disease Diseases 0.000 description 1
- 235000010323 ascorbic acid Nutrition 0.000 description 1
- 125000004429 atom Chemical group 0.000 description 1
- YEJAJYAHJQIWNU-UHFFFAOYSA-N azelastine hydrochloride Chemical compound Cl.C1CN(C)CCCC1N1C(=O)C2=CC=CC=C2C(CC=2C=CC(Cl)=CC=2)=N1 YEJAJYAHJQIWNU-UHFFFAOYSA-N 0.000 description 1
- 125000002393 azetidinyl group Chemical group 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-N benzenesulfonic acid Chemical class OS(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-N 0.000 description 1
- 125000000499 benzofuranyl group Chemical group O1C(=CC2=C1C=CC=C2)* 0.000 description 1
- 150000001558 benzoic acid derivatives Chemical class 0.000 description 1
- 125000001164 benzothiazolyl group Chemical group S1C(=NC2=C1C=CC=C2)* 0.000 description 1
- 125000004541 benzoxazolyl group Chemical group O1C(=NC2=C1C=CC=C2)* 0.000 description 1
- 125000002619 bicyclic group Chemical group 0.000 description 1
- 230000004071 biological effect Effects 0.000 description 1
- 230000033228 biological regulation Effects 0.000 description 1
- SIPUZPBQZHNSDW-UHFFFAOYSA-N bis(2-methylpropyl)aluminum Chemical compound CC(C)C[Al]CC(C)C SIPUZPBQZHNSDW-UHFFFAOYSA-N 0.000 description 1
- 230000000740 bleeding effect Effects 0.000 description 1
- 229960001561 bleomycin Drugs 0.000 description 1
- OYVAGSVQBOHSSS-UAPAGMARSA-O bleomycin A2 Chemical compound N([C@H](C(=O)N[C@H](C)[C@@H](O)[C@H](C)C(=O)N[C@@H]([C@H](O)C)C(=O)NCCC=1SC=C(N=1)C=1SC=C(N=1)C(=O)NCCC[S+](C)C)[C@@H](O[C@H]1[C@H]([C@@H](O)[C@H](O)[C@H](CO)O1)O[C@@H]1[C@H]([C@@H](OC(N)=O)[C@H](O)[C@@H](CO)O1)O)C=1N=CNC=1)C(=O)C1=NC([C@H](CC(N)=O)NC[C@H](N)C(N)=O)=NC(N)=C1C OYVAGSVQBOHSSS-UAPAGMARSA-O 0.000 description 1
- 210000004369 blood Anatomy 0.000 description 1
- 239000008280 blood Substances 0.000 description 1
- 229910000085 borane Inorganic materials 0.000 description 1
- MCQRPQCQMGVWIQ-UHFFFAOYSA-N boron;methylsulfanylmethane Chemical compound [B].CSC MCQRPQCQMGVWIQ-UHFFFAOYSA-N 0.000 description 1
- 229940098773 bovine serum albumin Drugs 0.000 description 1
- 210000004556 brain Anatomy 0.000 description 1
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 1
- 150000001649 bromium compounds Chemical class 0.000 description 1
- GZUXJHMPEANEGY-UHFFFAOYSA-N bromomethane Chemical class BrC GZUXJHMPEANEGY-UHFFFAOYSA-N 0.000 description 1
- 125000005997 bromomethyl group Chemical group 0.000 description 1
- 238000011095 buffer preparation Methods 0.000 description 1
- 239000011575 calcium Substances 0.000 description 1
- 229910052791 calcium Inorganic materials 0.000 description 1
- 239000007894 caplet Substances 0.000 description 1
- 239000002775 capsule Substances 0.000 description 1
- 125000002837 carbocyclic group Chemical group 0.000 description 1
- 150000004649 carbonic acid derivatives Chemical class 0.000 description 1
- 150000001735 carboxylic acids Chemical group 0.000 description 1
- 230000009787 cardiac fibrosis Effects 0.000 description 1
- 238000007675 cardiac surgery Methods 0.000 description 1
- 230000001101 cardioplegic effect Effects 0.000 description 1
- 239000000969 carrier Substances 0.000 description 1
- 150000001768 cations Chemical class 0.000 description 1
- 230000010261 cell growth Effects 0.000 description 1
- 210000003169 central nervous system Anatomy 0.000 description 1
- 206010008118 cerebral infarction Diseases 0.000 description 1
- 210000004978 chinese hamster ovary cell Anatomy 0.000 description 1
- 150000001805 chlorine compounds Chemical class 0.000 description 1
- IJOOHPMOJXWVHK-UHFFFAOYSA-N chlorotrimethylsilane Chemical compound C[Si](C)(C)Cl IJOOHPMOJXWVHK-UHFFFAOYSA-N 0.000 description 1
- 230000001684 chronic effect Effects 0.000 description 1
- 208000020832 chronic kidney disease Diseases 0.000 description 1
- 230000007882 cirrhosis Effects 0.000 description 1
- 150000001860 citric acid derivatives Chemical class 0.000 description 1
- 229960002303 citric acid monohydrate Drugs 0.000 description 1
- 238000002648 combination therapy Methods 0.000 description 1
- 230000000295 complement effect Effects 0.000 description 1
- 229940125846 compound 25 Drugs 0.000 description 1
- 239000012141 concentrate Substances 0.000 description 1
- 235000008504 concentrate Nutrition 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- 208000029078 coronary artery disease Diseases 0.000 description 1
- 210000005226 corpus cavernosum Anatomy 0.000 description 1
- 230000009260 cross reactivity Effects 0.000 description 1
- NKKMVIVFRUYPLQ-NSCUHMNNSA-N crotononitrile Chemical compound C\C=C\C#N NKKMVIVFRUYPLQ-NSCUHMNNSA-N 0.000 description 1
- 210000000805 cytoplasm Anatomy 0.000 description 1
- 230000007423 decrease Effects 0.000 description 1
- 238000013461 design Methods 0.000 description 1
- ZBCBWPMODOFKDW-UHFFFAOYSA-N diethanolamine Chemical class OCCNCCO ZBCBWPMODOFKDW-UHFFFAOYSA-N 0.000 description 1
- 230000004069 differentiation Effects 0.000 description 1
- 125000005046 dihydronaphthyl group Chemical group 0.000 description 1
- 239000003085 diluting agent Substances 0.000 description 1
- 238000010790 dilution Methods 0.000 description 1
- 239000012895 dilution Substances 0.000 description 1
- 150000002009 diols Chemical class 0.000 description 1
- 125000000532 dioxanyl group Chemical group 0.000 description 1
- YWWZCHLUQSHMCL-UHFFFAOYSA-N diphenyl diselenide Chemical compound C=1C=CC=CC=1[Se][Se]C1=CC=CC=C1 YWWZCHLUQSHMCL-UHFFFAOYSA-N 0.000 description 1
- 239000001177 diphosphate Substances 0.000 description 1
- XPPKVPWEQAFLFU-UHFFFAOYSA-J diphosphate(4-) Chemical class [O-]P([O-])(=O)OP([O-])([O-])=O XPPKVPWEQAFLFU-UHFFFAOYSA-J 0.000 description 1
- 235000011180 diphosphates Nutrition 0.000 description 1
- 238000004090 dissolution Methods 0.000 description 1
- 231100000673 dose–response relationship Toxicity 0.000 description 1
- 230000003828 downregulation Effects 0.000 description 1
- 238000012377 drug delivery Methods 0.000 description 1
- 229940048820 edetates Drugs 0.000 description 1
- 239000003792 electrolyte Substances 0.000 description 1
- 230000003511 endothelial effect Effects 0.000 description 1
- 210000003038 endothelium Anatomy 0.000 description 1
- 230000007705 epithelial mesenchymal transition Effects 0.000 description 1
- AFAXGSQYZLGZPG-UHFFFAOYSA-L ethanedisulfonate group Chemical class C(CS(=O)(=O)[O-])S(=O)(=O)[O-] AFAXGSQYZLGZPG-UHFFFAOYSA-L 0.000 description 1
- YGKGOUNPUCQPKB-UHFFFAOYSA-N ethenylboronic acid;pyridine Chemical compound OB(O)C=C.C1=CC=NC=C1 YGKGOUNPUCQPKB-UHFFFAOYSA-N 0.000 description 1
- NNMGVRVQUSRDRO-YOXFSPIKSA-N ethyl (1S)-3-azabicyclo[4.1.0]heptane-6-carboxylate Chemical compound C(C)OC(=O)C12CCNC[C@H]2C1 NNMGVRVQUSRDRO-YOXFSPIKSA-N 0.000 description 1
- RQCOPVYEHVOEMR-UHFFFAOYSA-N ethyl 4-ethoxy-3-oxobutanoate Chemical compound CCOCC(=O)CC(=O)OCC RQCOPVYEHVOEMR-UHFFFAOYSA-N 0.000 description 1
- 125000004494 ethyl ester group Chemical group 0.000 description 1
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 1
- PQVSTLUFSYVLTO-UHFFFAOYSA-N ethyl n-ethoxycarbonylcarbamate Chemical compound CCOC(=O)NC(=O)OCC PQVSTLUFSYVLTO-UHFFFAOYSA-N 0.000 description 1
- 150000002171 ethylene diamines Chemical class 0.000 description 1
- 238000011156 evaluation Methods 0.000 description 1
- 238000000105 evaporative light scattering detection Methods 0.000 description 1
- 238000002474 experimental method Methods 0.000 description 1
- 231100000573 exposure to toxins Toxicity 0.000 description 1
- 239000011737 fluorine Substances 0.000 description 1
- 201000005206 focal segmental glomerulosclerosis Diseases 0.000 description 1
- 150000004675 formic acid derivatives Chemical class 0.000 description 1
- VZCYOOQTPOCHFL-OWOJBTEDSA-L fumarate(2-) Chemical class [O-]C(=O)\C=C\C([O-])=O VZCYOOQTPOCHFL-OWOJBTEDSA-L 0.000 description 1
- 230000009760 functional impairment Effects 0.000 description 1
- 125000002541 furyl group Chemical group 0.000 description 1
- 239000000499 gel Substances 0.000 description 1
- 238000007429 general method Methods 0.000 description 1
- 239000012362 glacial acetic acid Substances 0.000 description 1
- 230000001434 glomerular Effects 0.000 description 1
- MASNOZXLGMXCHN-ZLPAWPGGSA-N glucagon Chemical compound C([C@@H](C(=O)N[C@H](C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H]([C@@H](C)O)C(O)=O)C(C)C)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](C)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CO)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CO)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@@H](NC(=O)CNC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CO)NC(=O)[C@@H](N)CC=1NC=NC=1)[C@@H](C)O)[C@@H](C)O)C1=CC=CC=C1 MASNOZXLGMXCHN-ZLPAWPGGSA-N 0.000 description 1
- 229960004666 glucagon Drugs 0.000 description 1
- 150000002306 glutamic acid derivatives Chemical class 0.000 description 1
- 239000008187 granular material Substances 0.000 description 1
- 229910052736 halogen Inorganic materials 0.000 description 1
- 150000002367 halogens Chemical class 0.000 description 1
- 230000036541 health Effects 0.000 description 1
- 230000004217 heart function Effects 0.000 description 1
- 150000002391 heterocyclic compounds Chemical class 0.000 description 1
- 239000000833 heterodimer Substances 0.000 description 1
- FEWIGMWODIRUJM-UHFFFAOYSA-N hex-4-en-3-one Natural products CCC(=O)C=CC FEWIGMWODIRUJM-UHFFFAOYSA-N 0.000 description 1
- 150000004677 hydrates Chemical class 0.000 description 1
- 150000003840 hydrochlorides Chemical class 0.000 description 1
- 150000002431 hydrogen Chemical class 0.000 description 1
- 206010020718 hyperplasia Diseases 0.000 description 1
- 230000001631 hypertensive effect Effects 0.000 description 1
- 125000002883 imidazolyl group Chemical group 0.000 description 1
- 125000003392 indanyl group Chemical group C1(CCC2=CC=CC=C12)* 0.000 description 1
- 125000003453 indazolyl group Chemical group N1N=C(C2=C1C=CC=C2)* 0.000 description 1
- 125000003454 indenyl group Chemical group C1(C=CC2=CC=CC=C12)* 0.000 description 1
- 125000001041 indolyl group Chemical group 0.000 description 1
- 230000007574 infarction Effects 0.000 description 1
- 230000005764 inhibitory process Effects 0.000 description 1
- 229910052500 inorganic mineral Inorganic materials 0.000 description 1
- 208000036971 interstitial lung disease 2 Diseases 0.000 description 1
- 210000001955 intestinal smooth muscle cell Anatomy 0.000 description 1
- 150000004694 iodide salts Chemical class 0.000 description 1
- 229910052740 iodine Inorganic materials 0.000 description 1
- 239000011630 iodine Substances 0.000 description 1
- 150000002500 ions Chemical class 0.000 description 1
- 208000037906 ischaemic injury Diseases 0.000 description 1
- 230000000302 ischemic effect Effects 0.000 description 1
- 210000004153 islets of langerhan Anatomy 0.000 description 1
- 125000000904 isoindolyl group Chemical group C=1(NC=C2C=CC=CC12)* 0.000 description 1
- 125000002183 isoquinolinyl group Chemical group C1(=NC=CC2=CC=CC=C12)* 0.000 description 1
- 150000003893 lactate salts Chemical class 0.000 description 1
- 239000000787 lecithin Substances 0.000 description 1
- 235000010445 lecithin Nutrition 0.000 description 1
- 229940067606 lecithin Drugs 0.000 description 1
- 210000000265 leukocyte Anatomy 0.000 description 1
- 230000000670 limiting effect Effects 0.000 description 1
- 238000004811 liquid chromatography Methods 0.000 description 1
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 1
- 229910052744 lithium Inorganic materials 0.000 description 1
- YNESATAKKCNGOF-UHFFFAOYSA-N lithium bis(trimethylsilyl)amide Chemical compound [Li+].C[Si](C)(C)[N-][Si](C)(C)C YNESATAKKCNGOF-UHFFFAOYSA-N 0.000 description 1
- GLXDVVHUTZTUQK-UHFFFAOYSA-M lithium hydroxide monohydrate Substances [Li+].O.[OH-] GLXDVVHUTZTUQK-UHFFFAOYSA-M 0.000 description 1
- 229940040692 lithium hydroxide monohydrate Drugs 0.000 description 1
- WGOPGODQLGJZGL-UHFFFAOYSA-N lithium;butane Chemical compound [Li+].CC[CH-]C WGOPGODQLGJZGL-UHFFFAOYSA-N 0.000 description 1
- 210000001853 liver microsome Anatomy 0.000 description 1
- 239000007937 lozenge Substances 0.000 description 1
- 206010025135 lupus erythematosus Diseases 0.000 description 1
- 239000011777 magnesium Substances 0.000 description 1
- 229910052749 magnesium Inorganic materials 0.000 description 1
- 230000014759 maintenance of location Effects 0.000 description 1
- 150000002688 maleic acid derivatives Chemical class 0.000 description 1
- 150000004701 malic acid derivatives Chemical class 0.000 description 1
- 238000004949 mass spectrometry Methods 0.000 description 1
- 238000005259 measurement Methods 0.000 description 1
- 229960001929 meloxicam Drugs 0.000 description 1
- 201000008350 membranous glomerulonephritis Diseases 0.000 description 1
- 231100000855 membranous nephropathy Toxicity 0.000 description 1
- 230000004060 metabolic process Effects 0.000 description 1
- 229910052751 metal Inorganic materials 0.000 description 1
- 239000002184 metal Substances 0.000 description 1
- AFVFQIVMOAPDHO-UHFFFAOYSA-M methanesulfonate group Chemical class CS(=O)(=O)[O-] AFVFQIVMOAPDHO-UHFFFAOYSA-M 0.000 description 1
- 229940098779 methanesulfonic acid Drugs 0.000 description 1
- QARBMVPHQWIHKH-UHFFFAOYSA-N methanesulfonyl chloride Chemical compound CS(Cl)(=O)=O QARBMVPHQWIHKH-UHFFFAOYSA-N 0.000 description 1
- SJFNDMHZXCUXSA-UHFFFAOYSA-M methoxymethyl(triphenyl)phosphanium;chloride Chemical compound [Cl-].C=1C=CC=CC=1[P+](C=1C=CC=CC=1)(COC)C1=CC=CC=C1 SJFNDMHZXCUXSA-UHFFFAOYSA-M 0.000 description 1
- QGBPKJFJAVDUNC-UHFFFAOYSA-N methyl 4-methoxy-3-oxobutanoate Chemical compound COCC(=O)CC(=O)OC QGBPKJFJAVDUNC-UHFFFAOYSA-N 0.000 description 1
- RWKGYDMSJZUVNO-UHFFFAOYSA-N methyl 5,6,7,8-tetrahydro-1,7-naphthyridine-3-carboxylate Chemical compound C1NCCC2=CC(C(=O)OC)=CN=C21 RWKGYDMSJZUVNO-UHFFFAOYSA-N 0.000 description 1
- LRMHVVPPGGOAJQ-UHFFFAOYSA-N methyl nitrate Chemical class CO[N+]([O-])=O LRMHVVPPGGOAJQ-UHFFFAOYSA-N 0.000 description 1
- 150000005451 methyl sulfates Chemical class 0.000 description 1
- KTMKRRPZPWUYKK-UHFFFAOYSA-N methylboronic acid Chemical compound CB(O)O KTMKRRPZPWUYKK-UHFFFAOYSA-N 0.000 description 1
- 230000004089 microcirculation Effects 0.000 description 1
- 230000003228 microsomal effect Effects 0.000 description 1
- 230000005012 migration Effects 0.000 description 1
- 238000013508 migration Methods 0.000 description 1
- 235000010755 mineral Nutrition 0.000 description 1
- 239000011707 mineral Substances 0.000 description 1
- 150000007522 mineralic acids Chemical class 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- 125000002757 morpholinyl group Chemical group 0.000 description 1
- 210000003205 muscle Anatomy 0.000 description 1
- 208000037891 myocardial injury Diseases 0.000 description 1
- 208000031225 myocardial ischemia Diseases 0.000 description 1
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000001624 naphthyl group Chemical group 0.000 description 1
- 239000006199 nebulizer Substances 0.000 description 1
- 230000008692 neointimal formation Effects 0.000 description 1
- 238000013059 nephrectomy Methods 0.000 description 1
- 230000008764 nerve damage Effects 0.000 description 1
- 230000007971 neurological deficit Effects 0.000 description 1
- 210000000440 neutrophil Anatomy 0.000 description 1
- 150000002823 nitrates Chemical class 0.000 description 1
- 150000002825 nitriles Chemical class 0.000 description 1
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 1
- 235000015097 nutrients Nutrition 0.000 description 1
- 125000005060 octahydroindolyl group Chemical group N1(CCC2CCCCC12)* 0.000 description 1
- 230000003287 optical effect Effects 0.000 description 1
- OIPPWFOQEKKFEE-UHFFFAOYSA-N orcinol Chemical compound CC1=CC(O)=CC(O)=C1 OIPPWFOQEKKFEE-UHFFFAOYSA-N 0.000 description 1
- 125000001715 oxadiazolyl group Chemical group 0.000 description 1
- BLUAEWKJZIBBFF-UHFFFAOYSA-N oxazepine-4-carboxylic acid Chemical compound OC(=O)C1=CC=CON=C1 BLUAEWKJZIBBFF-UHFFFAOYSA-N 0.000 description 1
- 125000002971 oxazolyl group Chemical group 0.000 description 1
- 230000036542 oxidative stress Effects 0.000 description 1
- LXNAVEXFUKBNMK-UHFFFAOYSA-N palladium(II) acetate Substances [Pd].CC(O)=O.CC(O)=O LXNAVEXFUKBNMK-UHFFFAOYSA-N 0.000 description 1
- YJVFFLUZDVXJQI-UHFFFAOYSA-L palladium(ii) acetate Chemical compound [Pd+2].CC([O-])=O.CC([O-])=O YJVFFLUZDVXJQI-UHFFFAOYSA-L 0.000 description 1
- 230000004963 pathophysiological condition Effects 0.000 description 1
- 239000013610 patient sample Substances 0.000 description 1
- 239000008188 pellet Substances 0.000 description 1
- 235000019371 penicillin G benzathine Nutrition 0.000 description 1
- 230000000144 pharmacologic effect Effects 0.000 description 1
- 125000001792 phenanthrenyl group Chemical group C1(=CC=CC=2C3=CC=CC=C3C=CC12)* 0.000 description 1
- 235000021317 phosphate Nutrition 0.000 description 1
- 150000003013 phosphoric acid derivatives Chemical class 0.000 description 1
- 229910052698 phosphorus Inorganic materials 0.000 description 1
- 125000004193 piperazinyl group Chemical group 0.000 description 1
- 125000003386 piperidinyl group Chemical group 0.000 description 1
- 230000007505 plaque formation Effects 0.000 description 1
- 230000010118 platelet activation Effects 0.000 description 1
- 231100000683 possible toxicity Toxicity 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- 229910052700 potassium Inorganic materials 0.000 description 1
- SCVFZCLFOSHCOH-UHFFFAOYSA-M potassium acetate Chemical compound [K+].CC([O-])=O SCVFZCLFOSHCOH-UHFFFAOYSA-M 0.000 description 1
- IUBQJLUDMLPAGT-UHFFFAOYSA-N potassium bis(trimethylsilyl)amide Chemical compound C[Si](C)(C)N([K])[Si](C)(C)C IUBQJLUDMLPAGT-UHFFFAOYSA-N 0.000 description 1
- 229910000027 potassium carbonate Inorganic materials 0.000 description 1
- LPNYRYFBWFDTMA-UHFFFAOYSA-N potassium tert-butoxide Chemical compound [K+].CC(C)(C)[O-] LPNYRYFBWFDTMA-UHFFFAOYSA-N 0.000 description 1
- 238000001556 precipitation Methods 0.000 description 1
- 238000012746 preparative thin layer chromatography Methods 0.000 description 1
- 238000012545 processing Methods 0.000 description 1
- 230000035755 proliferation Effects 0.000 description 1
- TVDSBUOJIPERQY-UHFFFAOYSA-N prop-2-yn-1-ol Chemical compound OCC#C TVDSBUOJIPERQY-UHFFFAOYSA-N 0.000 description 1
- 210000002307 prostate Anatomy 0.000 description 1
- 208000005069 pulmonary fibrosis Diseases 0.000 description 1
- 125000000561 purinyl group Chemical group N1=C(N=C2N=CNC2=C1)* 0.000 description 1
- 125000004309 pyranyl group Chemical group O1C(C=CC=C1)* 0.000 description 1
- 125000003226 pyrazolyl group Chemical group 0.000 description 1
- 125000000714 pyrimidinyl group Chemical group 0.000 description 1
- 125000000719 pyrrolidinyl group Chemical group 0.000 description 1
- 125000000168 pyrrolyl group Chemical group 0.000 description 1
- 125000002294 quinazolinyl group Chemical group N1=C(N=CC2=CC=CC=C12)* 0.000 description 1
- 125000002943 quinolinyl group Chemical group N1=C(C=CC2=CC=CC=C12)* 0.000 description 1
- HDACQVRGBOVJII-JBDAPHQKSA-N ramipril Chemical compound C([C@@H](C(=O)OCC)N[C@@H](C)C(=O)N1[C@@H](C[C@@H]2CCC[C@@H]21)C(O)=O)CC1=CC=CC=C1 HDACQVRGBOVJII-JBDAPHQKSA-N 0.000 description 1
- 229960003401 ramipril Drugs 0.000 description 1
- 239000012429 reaction media Substances 0.000 description 1
- 239000012048 reactive intermediate Substances 0.000 description 1
- 108020003175 receptors Proteins 0.000 description 1
- 102000005962 receptors Human genes 0.000 description 1
- 238000001953 recrystallisation Methods 0.000 description 1
- 239000012088 reference solution Substances 0.000 description 1
- 201000002793 renal fibrosis Diseases 0.000 description 1
- 230000010410 reperfusion Effects 0.000 description 1
- WXXSNCNJFUAIDG-UHFFFAOYSA-N riociguat Chemical compound N1=C(N)C(N(C)C(=O)OC)=C(N)N=C1C(C1=CC=CN=C11)=NN1CC1=CC=CC=C1F WXXSNCNJFUAIDG-UHFFFAOYSA-N 0.000 description 1
- 229960000529 riociguat Drugs 0.000 description 1
- 229940125526 sGC activator Drugs 0.000 description 1
- 150000003873 salicylate salts Chemical class 0.000 description 1
- 239000012047 saturated solution Substances 0.000 description 1
- 230000020341 sensory perception of pain Effects 0.000 description 1
- 238000000926 separation method Methods 0.000 description 1
- 239000000377 silicon dioxide Substances 0.000 description 1
- 238000009097 single-agent therapy Methods 0.000 description 1
- 210000000329 smooth muscle myocyte Anatomy 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- 239000011780 sodium chloride Substances 0.000 description 1
- UKLNMMHNWFDKNT-UHFFFAOYSA-M sodium chlorite Chemical compound [Na+].[O-]Cl=O UKLNMMHNWFDKNT-UHFFFAOYSA-M 0.000 description 1
- 229960002218 sodium chlorite Drugs 0.000 description 1
- 229910052938 sodium sulfate Inorganic materials 0.000 description 1
- 235000011152 sodium sulphate Nutrition 0.000 description 1
- AKHNMLFCWUSKQB-UHFFFAOYSA-L sodium thiosulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=S AKHNMLFCWUSKQB-UHFFFAOYSA-L 0.000 description 1
- 235000019345 sodium thiosulphate Nutrition 0.000 description 1
- 239000011343 solid material Substances 0.000 description 1
- 208000010110 spontaneous platelet aggregation Diseases 0.000 description 1
- 238000013097 stability assessment Methods 0.000 description 1
- 239000007858 starting material Substances 0.000 description 1
- 239000011550 stock solution Substances 0.000 description 1
- 239000000758 substrate Substances 0.000 description 1
- 150000003890 succinate salts Chemical class 0.000 description 1
- 125000000475 sulfinyl group Chemical group [*:2]S([*:1])=O 0.000 description 1
- 239000000829 suppository Substances 0.000 description 1
- 230000001629 suppression Effects 0.000 description 1
- 230000004083 survival effect Effects 0.000 description 1
- 238000001308 synthesis method Methods 0.000 description 1
- 239000006188 syrup Substances 0.000 description 1
- 235000020357 syrup Nutrition 0.000 description 1
- 239000003826 tablet Substances 0.000 description 1
- 230000006794 tachycardia Effects 0.000 description 1
- 150000003892 tartrate salts Chemical class 0.000 description 1
- DYHSDKLCOJIUFX-UHFFFAOYSA-N tert-butoxycarbonyl anhydride Chemical compound CC(C)(C)OC(=O)OC(=O)OC(C)(C)C DYHSDKLCOJIUFX-UHFFFAOYSA-N 0.000 description 1
- ACQUKYSUICAHTK-UHFFFAOYSA-N tert-butyl 2-(bromomethyl)-7,8-dihydro-5H-1,6-naphthyridine-6-carboxylate Chemical compound C(C)(C)(C)OC(=O)N1CC=2C=CC(=NC=2CC1)CBr ACQUKYSUICAHTK-UHFFFAOYSA-N 0.000 description 1
- ROUYFJUVMYHXFJ-UHFFFAOYSA-N tert-butyl 4-oxopiperidine-1-carboxylate Chemical compound CC(C)(C)OC(=O)N1CCC(=O)CC1 ROUYFJUVMYHXFJ-UHFFFAOYSA-N 0.000 description 1
- UWOVCEFGMXDUDK-UHFFFAOYSA-N tert-butyl 6-(bromomethyl)-5,8-dimethyl-3,4-dihydro-1h-isoquinoline-2-carboxylate Chemical compound C1CN(C(=O)OC(C)(C)C)CC2=C1C(C)=C(CBr)C=C2C UWOVCEFGMXDUDK-UHFFFAOYSA-N 0.000 description 1
- TZRQZPMQUXEZMC-UHFFFAOYSA-N tert-butyl n-(2-bromoethyl)carbamate Chemical compound CC(C)(C)OC(=O)NCCBr TZRQZPMQUXEZMC-UHFFFAOYSA-N 0.000 description 1
- DSPYCWLYGXGJNJ-UHFFFAOYSA-N tert-butyl n-prop-2-ynylcarbamate Chemical compound CC(C)(C)OC(=O)NCC#C DSPYCWLYGXGJNJ-UHFFFAOYSA-N 0.000 description 1
- MHYGQXWCZAYSLJ-UHFFFAOYSA-N tert-butyl-chloro-diphenylsilane Chemical compound C=1C=CC=CC=1[Si](Cl)(C(C)(C)C)C1=CC=CC=C1 MHYGQXWCZAYSLJ-UHFFFAOYSA-N 0.000 description 1
- BCNZYOJHNLTNEZ-UHFFFAOYSA-N tert-butyldimethylsilyl chloride Chemical compound CC(C)(C)[Si](C)(C)Cl BCNZYOJHNLTNEZ-UHFFFAOYSA-N 0.000 description 1
- DPKBAXPHAYBPRL-UHFFFAOYSA-M tetrabutylazanium;iodide Chemical compound [I-].CCCC[N+](CCCC)(CCCC)CCCC DPKBAXPHAYBPRL-UHFFFAOYSA-M 0.000 description 1
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 1
- 125000001712 tetrahydronaphthyl group Chemical group C1(CCCC2=CC=CC=C12)* 0.000 description 1
- 125000003831 tetrazolyl group Chemical group 0.000 description 1
- 125000001113 thiadiazolyl group Chemical group 0.000 description 1
- 125000000335 thiazolyl group Chemical group 0.000 description 1
- 230000008719 thickening Effects 0.000 description 1
- 125000001544 thienyl group Chemical group 0.000 description 1
- 125000004568 thiomorpholinyl group Chemical group 0.000 description 1
- FYSNRJHAOHDILO-UHFFFAOYSA-N thionyl chloride Chemical compound ClS(Cl)=O FYSNRJHAOHDILO-UHFFFAOYSA-N 0.000 description 1
- 125000000464 thioxo group Chemical group S=* 0.000 description 1
- 201000005665 thrombophilia Diseases 0.000 description 1
- LBLYYCQCTBFVLH-UHFFFAOYSA-M toluenesulfonate group Chemical group C=1(C(=CC=CC1)S(=O)(=O)[O-])C LBLYYCQCTBFVLH-UHFFFAOYSA-M 0.000 description 1
- 230000009466 transformation Effects 0.000 description 1
- 230000009261 transgenic effect Effects 0.000 description 1
- 230000001052 transient effect Effects 0.000 description 1
- 230000007704 transition Effects 0.000 description 1
- 238000011269 treatment regimen Methods 0.000 description 1
- 125000004306 triazinyl group Chemical group 0.000 description 1
- 125000001425 triazolyl group Chemical group 0.000 description 1
- 230000001960 triggered effect Effects 0.000 description 1
- ZKEFILOOQGVKNY-UHFFFAOYSA-N triphenylphosphane;dihydrobromide Chemical compound Br.Br.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 ZKEFILOOQGVKNY-UHFFFAOYSA-N 0.000 description 1
- 210000003708 urethra Anatomy 0.000 description 1
- 210000003932 urinary bladder Anatomy 0.000 description 1
- 229960002381 vardenafil Drugs 0.000 description 1
- 210000003462 vein Anatomy 0.000 description 1
- 238000010792 warming Methods 0.000 description 1
- 239000011995 wilkinson's catalyst Substances 0.000 description 1
- UTODFRQBVUVYOB-UHFFFAOYSA-P wilkinson's catalyst Chemical compound [Cl-].C1=CC=CC=C1P(C=1C=CC=CC=1)(C=1C=CC=CC=1)[Rh+](P(C=1C=CC=CC=1)(C=1C=CC=CC=1)C=1C=CC=CC=1)P(C=1C=CC=CC=1)(C=1C=CC=CC=1)C1=CC=CC=C1 UTODFRQBVUVYOB-UHFFFAOYSA-P 0.000 description 1
- BPICBUSOMSTKRF-UHFFFAOYSA-N xylazine Chemical compound CC1=CC=CC(C)=C1NC1=NCCCS1 BPICBUSOMSTKRF-UHFFFAOYSA-N 0.000 description 1
- 229960001600 xylazine Drugs 0.000 description 1
- 229910052725 zinc Inorganic materials 0.000 description 1
- XOOUIPVCVHRTMJ-UHFFFAOYSA-L zinc stearate Chemical class [Zn+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O XOOUIPVCVHRTMJ-UHFFFAOYSA-L 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/14—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing three or more hetero rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4427—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems
- A61K31/444—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems containing a six-membered ring with nitrogen as a ring heteroatom, e.g. amrinone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/47—Quinolines; Isoquinolines
- A61K31/472—Non-condensed isoquinolines, e.g. papaverine
- A61K31/4725—Non-condensed isoquinolines, e.g. papaverine containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/16—Drugs for disorders of the alimentary tract or the digestive system for liver or gallbladder disorders, e.g. hepatoprotective agents, cholagogues, litholytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/08—Drugs for disorders of the urinary system of the prostate
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/10—Drugs for disorders of the urinary system of the bladder
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
- A61P15/10—Drugs for genital or sexual disorders; Contraceptives for impotence
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P21/00—Drugs for disorders of the muscular or neuromuscular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/04—Centrally acting analgesics, e.g. opioids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
- A61P25/16—Anti-Parkinson drugs
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
- A61P27/06—Antiglaucoma agents or miotics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Medicinal Chemistry (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Life Sciences & Earth Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Biomedical Technology (AREA)
- Urology & Nephrology (AREA)
- Endocrinology (AREA)
- Diabetes (AREA)
- Epidemiology (AREA)
- Pain & Pain Management (AREA)
- Reproductive Health (AREA)
- Ophthalmology & Optometry (AREA)
- Dermatology (AREA)
- Gynecology & Obstetrics (AREA)
- Cardiology (AREA)
- Obesity (AREA)
- Psychiatry (AREA)
- Psychology (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Physical Education & Sports Medicine (AREA)
- Rheumatology (AREA)
- Hospice & Palliative Care (AREA)
- Heart & Thoracic Surgery (AREA)
- Hematology (AREA)
- Pulmonology (AREA)
- Gastroenterology & Hepatology (AREA)
- Emergency Medicine (AREA)
Abstract
Un compuesto de fórmula I **(Ver fórmula)** en donde: X es un enlace; Y es C o N; W es C o N, con la condición de que Y y W no son ambos N; V es -C(R11)(R12)- o -OCH2-, con la condición de que si V es -OCH2, entonces Z es -CH2-, Y y W son ambos C; Z es -CH2-, -C(R10)2CH2- o -C(O)-; R1 es H, Me o -CH2OMe; R2 y R3 junto con los carbonos a los que están unidos para formar un anillo de 3 miembros condensado; B es **(Ver fórmula)** o R5 y R6 se seleccionan independientemente entre H, Me, F, Cl y CF3; R7 es H, Me, Et, -OMe, CN, F o -CH2OMe o no está presente cuando Y es N; R8 es H, Me o F o no está presente cuando W es N; R9 es -(CH2)n heterociclilo, en donde el heterociclilo se selecciona entre tetrahidropiranilo, tetrahidrofuranilo, oxetanilo y [1,4]-dioxanilo o -CH(R10)heteroarilo, en donde el heteroarilo se selecciona entre el grupo que consiste en pirazina, imidazol, piridilo e isoxazolilo y en donde el heteroarilo está opcionalmente sustituido con un grupo metilo; cada R10 es independientemente H o Me; R11 es H o Me; R12 es H o Me; m es 0 o 1, con la condición de que si m es 0, Z es -CH2-, V es -C(R11)(R12)- y R11 y R12 son ambos H; y n es 0 o 1; o una sal del mismo.
Description
DESCRIPCIÓN
Ácidos carboxílicos heterocíclicos como activadores de guanilato ciclasa soluble
Campo de la invención
La presente invención proporciona nuevos compuestos que activan o potencian a la guanilato ciclasa soluble (sGC) y por tato son útiles para el tratamiento de una diversidad de enfermedades y trastornos que están mediados o sostenidos por la reducción o disminución de la actividad de la guanilato ciclasa soluble, incluyendo enfermedades cardiovasculares, enfermedad renal, diabetes, trastornos fibróticos, trastornos urológicos, trastornos neurológicos y trastornos inflamatorios. La presente invención también se refiere a composiciones farmacéuticas que comprenden estos compuestos y a estos compuestos para su uso en el tratamiento de diversas enfermedades y trastornos.
Antecedentes
La guanilato ciclasa soluble (sGC) es un receptor del óxido nítrico (NO) que se encuentra en el citoplasma de muchos tipos de células. En seres humanos, la sGC funcional es un heterodímero compuesto por una subunidad alfa 1 o alfa 2 combinada con la subunidad beta 1 que tiene un grupo hemo prostético. En condiciones no fisiopatológicas, la unión del NO al grupo hemo de la sGC, activa la enzima para catalizar la conversión de guanosina-5'-trifosfato (GTP) en guanosín monofosfato cíclico (cGMP). El cGMP es un segundo mensajero que ejerce efectos modulando isoformas de proteína cinasa dependiente de cGMP (PKG), fosfodiesterasas y canales de iones activados por cGMP. Al hacerlo, se ha demostrado que la sGC modula numerosas rutas asociadas a enfermedades que incluyen hipertensión arterial, hipertensión pulmonar, ateroesclerosis, insuficiencia cardíaca, cirrosis hepática, fibrosis renal y disfunción eréctil (O. Evgenov et al., Nature Reviews, 2006, 5, 755-768 and Y. Wang-Rosenke et al., Curr. Med. Chem., 2008, 15, 1396-1406).
En condiciones normales, el hierro presente en la sGC se encuentra en estado ferroso que puede unirse al NO y al monóxido de carbono (CO). Sin embargo, en condiciones de estrés oxidativo, que pueden producirse en diversas enfermedades, los informes publicados indican que ni el NO ni el CO pueden activar al hierro del grupo hemo que se oxida al estado férrico. Se establecido la hipótesis de que la incapacidad del NO para señalizar a través de la sGC con un hierro del grupo hemo oxidado, contribuye a los procesos de la enfermedad. Recientemente, se han descrito dos clases nuevas de compuestos que potencian la actividad de la sGC de una manera dependiente del grupo hemo (estimuladores de sGC) e independiente del grupo hemo (activadores de sGC). La actividad de los estimuladores de la sGC se sinergiza con el NO para aumentar la producción de cGMP, mientras que los activadores de sGC son solo aditivos con NO para aumentar los niveles de cGMP (O. Evgenov et al., Nature Reviews, 2006, 5, 755-768). Tanto los estimuladores como los activadores de sGC han demostrado ser beneficiosos en modelos animales de enfermedad. Los activadores de sGC proporcionan la ventaja de poder dirigirse preferentemente a la forma enferma, no funcional de la enzima. Los activadores de sGC incluyen BAY 58-2667 (cinaciguat) (JP Stasch et al., Brit J. Pharmacol., 2002, 136, 773-783) and HMR-1766 (ataciguat) (U. Schindler et al., 2006, Mol. Pharmacol., 69, 1260 1268).
El NO tiene un papel importante en el mantenimiento de la función celular y tisular normal. Sin embargo, la señalización adecuada en la ruta del NO se puede interrumpir en diversas etapas. La señalización del NO puede verse afectada por niveles reducidos de enzimas de óxido nítrico sintasa (NOS), por la actividad de la NOS, la biodisponibilidad del NO, niveles de sGC y actividad de sGC. Los activadores de sGC posibilitan evitar el impedimento funcional producido por todas estas deficiencias. Dado que la activación de la sGC se produce en etapas posteriores de la síntesis de NO o según la disponibilidad de NO, estas deficiencias no afectarán a la actividad de los activadores de la sGC. Como se ha descrito anteriormente, la actividad de la sGC, cuya función se interrumpe por la oxidación del hierro del grupo hemo, se corregirá con los activadores de sGC. Por tanto, los activadores de sGC posibilitan proporcionar beneficios en muchas enfermedades producidas por una señalización defectuosa en la ruta del NO.
La activación de la sGC posibilita proporcionar beneficio terapéutico en la aterosclerosis y arteriosclerosis. Se ha demostrado que el tratamiento con cinaciguat impide la hiperplasia de la neoíntima después de la denudación endotelial por lesión por alambre de la arteria carótida en ratas (K. Hirschberg et al., Cardiovasc. Res., 2010, 87, Suppl. 1, S100, Resumen 343). Ataciguat inhibió la formación de placa aterosclerótica en ratones ApoE-/-alimentados con una dieta rica en grasas (M. van Eickels, BMC Pharmacology, 2007, 7, Suppl. 1, s4). La disminución de la producción de NO en ratones deficientes en óxido nítrico sintetasa endotelial (eNOS, endothelial nitric oxide synthase) aumentó la inflamación vascular y la resistencia a la insulina en respuesta a un exceso de nutrientes. En el mismo estudio, sildenafil, un inhibidor de la fosfodiesterasa 5 (PDE5), redujo la inflamación vascular y la resistencia a la insulina en ratones alimentados con una dieta rica en grasas (N. Rizzo et al., Arterioscler. Thromb. Vasc. Biol., 2010, 30, 758-765). En un modelo de reperfusión e isquemia cerebral, los ratones deficientes para la subunidad alfa1 tenían un volumen de infarto más grande y mayores déficits neurológicos que los ratones de tipo silvestre (D. Atochin et al., Stroke 2010, 41, 1815-1819). Por último, después de una lesión por globo de las arterias carótidas de rata in vivo, un estimulador de sGC (YC-1) inhibió la formación de la neoíntima (C. Wu, J. Pharmacol. Sci., 2004, 94, 252-260).
Las complicaciones de la diabetes pueden reducirse a través de la activación de la sGC. La supresión inducida por glucosa de la liberación de glucagón se pierde en los islotes pancreáticos que carecen de PKG, lo que sugiere un papel de la producción de cGMP mediada por sGC en la regulación de la glucosa (V. Leiss et al., BMC Pharmacology, 2009, 9, Suppl. 1, P40).
Desde el punto de vista clínico, está bien establecido que la elevación de cGMP mediante el tratamiento con inhibidores de PDE5, es eficaz para el tratamiento de la disfunción eréctil (DE). Sin embargo, un 30 % de los pacientes con DE son resistentes al tratamiento con inhibidores de PDE5 (S. Gur et al., Curr. Pharm. Des., 2010, 16, 1619-1633). El estimulador de sGC, BAY-41-2272, puede relajar el músculo del cuerpo cavernoso de una manera dependiente de sGC, sugiriendo por tanto que una mayor actividad de sGC podría proporcionar beneficios en pacientes con DE (C. Teixeira et al., J. Pharmacol. & Exp. Ther., 2007, 322, 1093-1102). Asimismo, los estimuladores de sGC y los activadores de sGC utilizados individualmente o en combinación con el inhibidor de PDE5, pudieron tratar la DE en modelos animales (documento WO 10/081647).
Existen pruebas de que la activación de la sGC puede ser útil para prevenir la fibrosis tisular, incluyendo la del pulmón, hígado, piel y riñón. Se cree que los procesos de transición epitelio-mesénquima (TEM) y conversión de fibroblastos a miofibroblastos, contribuyen a la fibrosis tisular. Cuando se combinó cincaciguat o bAy 41-2272 con sildenafil, se inhibió la conversión de fibroblastos a miofibroblastos en el pulmón (T. Dunkern et al., Eur. J. Pharm., 2007, 572, 12-22). El NO puede inhibir la TEM de las células epiteliales alveolares (S. Vyas-Read et al., Am. J. Physiol. Lung Cell Mol. Physiol., 2007, 293, 1212-1221), lo que sugiere que la activación de la sGC está implicada en este proceso. También se ha demostrado que el NO inhibe la señalización del TGF (factor de crecimiento transformante) beta glomerular (E. Dreieicher et al., J. Am. Soc. Nephrol., 2009, 20, 1963-1974) lo que indica que la activación de la sGC puede inhibir la esclerosis glomerular. En un modelo de suero porcino y en un modelo de fibrosis hepática con tetracloruro de carbono, un activador de sGC (BAY 60-2260) fue eficaz para inhibir la fibrosis (A. Knorr et al., Arzneimittel-Forschung, 2008, 58, 71-80), lo que sugiere que el aumento de la actividad de la sGC puede usarse para tratar la esteatohepatitis no alcohólica (NASH, siglas del inglés non-alcoholic steatohepatitis). En los modelos de fibrosis dérmica inducida por bleomicina y de fibrosis cutánea de ratón Tsk-1 (del inglés tight skin, piel tensa), el estimulador de sGC, BAY 41-2272, pudo inhibir el engrosamiento dérmico y la diferenciación de miofibroblastos (C. Beyer et al., Ann. Rheum. Dis., 2012, 71, 1019-1026), lo que indica que la activación de la sGC puede ser útil para el tratamiento de la esclerosis sistémica.
Los estudios clínicos han demostrado eficacia utilizando el activador de sGC, cinaciguat, para el tratamiento de la insuficiencia cardíaca aguda descompensada (H. Lapp et al., Circulation, 2009, 119, 2781-2788). Esto es compatible con los resultados obtenidos de un modelo canino de insuficiencia cardíaca inducida por taquicardia, en el que la infusión intravenosa breve de cinaciguat pudo producir descarga cardíaca (G. Boerrigter et al., Hypertension, 2007, 49, 1128-1133). En un modelo de insuficiencia cardíaca crónica inducida por infarto de miocardio en la rata, HMR 1766 mejoró la función cardíaca y redujo la fibrosis cardíaca que adicionalmente se potenció con ramipril (F. Daniela, Circulation, 2009, 120, Suppl. 2, S852-S853).
Los activadores de sGC pueden usarse para tratar la hipertensión. Esto se ha demostrado claramente en estudios clínicos en los que la dosis de cinaciguat se ajusta en función de la magnitud de la reducción de la presión sanguínea obtenida (H. Lapp et al., Circulation, 2009, 119, 2781-2788). Estudios preclínicos usando cinaciguat, ya habían demostrado con anterioridad, la capacidad de activación de sGC para reducir la presión sanguínea (J.-P. Stasch et al., 2006, J. Clin. Invest., 116, 2552-2561). Se han comunicado hallazgos similares usando también HMR 1766, el activador de sGC (U. Schindler et al., 2006, Mol. Pharmacol., 69, 1260-1268).
La activación de la sGC posibilita reducir la inflamación debido a los efectos sobre el endotelio. BAY 41-2272 y un donador de NO, inhibieron la adhesión y el rodamiento leucocitario en ratones deficientes en eNOS. Se demostró que esto estaba mediado por la regulación negativa de la expresión de la molécula de adhesión P selectina (A. Ahluwalla et al., Proc. Natl. Acad. Sci. USA, 2004, 101, 1386-1391). Se demostró que los inhibidores de NOS y sGc aumentaban la expresión de ICAM inducida por endotoxina (LPS) en los vasos de microcirculación mesentérica. Esto fue reducido por un donador de NO de una manera dependiente de cGMP. El tratamiento de ratones con inhibidores de NOS o sGC aumentó la migración, el rodamiento y la adhesión de neutrófilos inducida por LPS o carragenina (D. Dal Secco, Nitric Oxide, 2006, 15, 77-86).
Se ha demostrado que la activación de sGC produce protección frente a lesión por isquemia y reperfusión utilizando BAY 58-2667 en un modelo de corazón tanto aislado como in vivo (T. Krieg et al., Eur. Heart J., 2009, 30, 1607 6013). Se obtuvieron resultados similares usando el mismo compuesto en un modelo canino de parada cardiopléjica y circulación extracorpórea (T. Radovits et al., Eur J. Cardiothorac. Surg., 2010).
La capacidad de activación de la sGC para inhibir in vitro el crecimiento de células de músculo liso intestinal (A.-M. Pelletier et al., Am. J. Physiol. Gastrointest. Liver Physiol. 2010, 298, G896-G907) es compatible con un papel en las enfermedades inflamatorias intestinales, incluida la colitis ulcerosa y la enfermedad de Crohn.
En algunos estudios se ha indicado la posibilidad de que la activación de la sGC tenga efectos antinociceptivos. En
modelos de nocicepción en ratones (ensayo de retorcimiento) y ratas (hiperalgesia en la pata) de diabetes inducida con estreptozotocina, la elevación de los niveles de cGMP mediante la administración de sildenafil bloqueó la respuesta al dolor, que a su vez se anuló con inhibidor de NOS o sGC (C. Patil et al., Pharm., 2004, 72, 190-195). Se ha demostrado que el inhibidor de sGC, 1H-1,2,4-oxadiazolo4,2-a.quinoxalin-1-ona (ODQ) bloquea los efectos antinociceptivos de diversos agentes, incluidos meloxicam y diselenuro de difenilo en un modelo de dolor inducido con formalina (P Aguirre-Banuelos et al., Eur. J. Pharmacol., 2000, 395, 9-13 y L. Savegnago et al., J. Pharmacy Pharmacol., 2008, 60, 1679-1686) y xilazina en un modelo de presión de pata (T. Romero et al., Eur. J. Pharmacol., 2009, 613, 64-67). Asimismo, ataciguat fue antinociceptivo en el modelo de hiperalgesia térmica desencadenada por inflamación inducida por carragenina y en el modelo de dolor neuropático por lesión nerviosa preservada en ratones (documento WO 09/043495).
Se ha demostrado que la inhibición de PDE9, una fosfodiesterasa específica para cGMP expresada en el cerebro, mejora la potenciación prolongada (F. van der Staay et al., Neuropharmacol. 2008, 55, 908-918). En el sistema nervioso central, la sGC es la enzima primaria que cataliza la formación de cGMP (K. Domek-Lopacinska et al., Mol. Neurobiol., 2010, 41, 129-137). Por tanto, la activación de sGC puede ser beneficiosa en el tratamiento de la enfermedad de Alzheimer y Parkinson.
En un estudio clínico de fase II, el estimulador de sGC, riociguat, fue eficaz en el tratamiento de la hipertensión pulmonar tromboembólica crónica y la hipertensión arterial pulmonar (H. Ghofrani et al., Eur. Respir. J., 2010, 36, 792-799). Estos hallazgos amplían los estudios preclínicos en los que BAY 41-2272 y cinaciguat redujeron la hipertensión pulmonar en modelos de ratón (R. Dumitrascu et al., Circulation, 2006, 113, 286-295) y cordero (O. Evgenov et al., 2007, Am. J. Respir. Crit. Care Med., 176, 1138-1145). Se obtuvieron resultados similares usando HMR 1766 en un modelo de ratón de hipertensión pulmonar (N. Weissmann et al., 2009, Am. J. Physiol. Lung Cell. Mol. Physiol., 297, L658-665).
La activación de la sGC posibilita tratar enfermedades renales crónicas. Tanto BAY 58-2667 como HMR 1766, mejoraron la función y estructura renal en un modelo de nefrectomía subtotal de enfermedad renal en rata (P. Kalk et al., 2006, Brit. J. Pharmacol., 148, 853-859 y K. Benz et al., 2007, Kidney Blood Press. Res., 30, 224-233). El tratamiento con BAY 58-2667, mejoró la función renal y la supervivencia en ratas transgénicas con renina hipertensiva (ratas TG(mRen2)27) tratadas con un inhibidor de NOS (J.-P. Stasch et al., 2006, J. Clin. Invest., 116, 2552-2561). El tratamiento con BAY 41-2272 preservó la estructura y la función renal en un modelo crónico de enfermedad renal en ratas inducida por uninefrectomía y tratamiento con anticuerpos anti-thyl (Y. Wang et al., 2005, Kidney Intl., 68, 47-61).
Las enfermedades causadas por coagulación sanguínea excesiva pueden tratarse con activadores de sGC. La activación de sGC usando BAY 58-2667 pudo inhibir la agregación plaquetaria inducida por diversos estímulos ex vivo. Además, este compuesto inhibió la formación de trombos in vivo en ratones y el tiempo de hemorragia prolongada (J.-P. Stasch et al., 2002, Brit. J. Pharmacol., 136, 773-783). En otro estudio usando HMR 1766, se inhibió la activación plaquetaria in vivo en ratas tratadas con estreptozotocina (A. Schafer et al., 2006, Arterioscler. Thromb. Vasc. Biol., 2006, 26, 2813-2818).
La activación de sGC también puede ser beneficiosa en el tratamiento de trastornos urológicos (documento WO/08138483). Estudios clínicos que utilizan el inhibidor de PDE5, vardenafil, confirman esto (C. Stief et al., 2008, Eur. Urol., 53, 1236-1244). El estimulador de la guanilato ciclasa soluble, BAY 41-8543, pudo inhibir la proliferación de células de músculo liso de próstata, vejiga y uretra, utilizando muestras de pacientes (B. Fibbi et al., 2010, J. Sex. Med., 7, 59-69), proporcionando así más pruebas que confirman la utilidad del tratamiento de trastornos urológicos con activadores de sGC.
Los estudios anteriores proporcionan pruebas sobre el uso de activadores de sGC para tratar enfermedades cardiovasculares, incluida la hipertensión, ateroesclerosis, arteriopatía periférica, restenosis, infarto de miocardio, ictus, insuficiencia cardíaca, vasoespasmo coronario, vasoespasmo cerebral, lesión por isquemia/reperfusión, hipertensión pulmonar tromboembólica, hipertensión arterial pulmonar, angina estable e inestable, trastornos tromboembólicos. Además, los activadores de sGC posibilitan tratar enfermedades renales, diabetes, trastornos fibróticos, incluidos los de la piel, hígado, riñón y pulmones, trastornos urológicos que incluyen vejiga hiperactiva, hiperplasia prostática benigna, disfunción eréctil y trastornos neurológicos, incluida la enfermedad de Alzheimer, la enfermedad de Parkinson, así como el dolor neuropático. El tratamiento con activadores de sGC también puede proporcionar beneficios en trastornos inflamatorios tales como psoriasis, esclerosis múltiple, artritis, asma, colitis ulcerosa, Enfermedad de Crohn y enfermedad pulmonar obstructiva crónica.
En el documento WO 2012/122340, se desvelan compuestos heterocíclicos útiles como activadores de sGC.
Breve sumario de la invención
La presente invención proporciona nuevos compuestos que activan o potencian la sGC y son por lo tanto útiles para tratar una diversidad de enfermedades y trastornos que se pueden aliviar mediante la activación o potenciación de la sGC incluyendo enfermedades cardiovasculares, inflamatorias y renales.
En consecuencia, la invención proporciona nuevos compuestos para su uso como medicamentos, más específicamente para su uso en el tratamiento de una enfermedad o trastorno que se puede mejorar mediante la activación o potenciación de la sGC.
Esta invención se refiere también a composiciones farmacéuticas que comprenden estos compuestos y divulga métodos para usar estos compuestos en el tratamiento de diversas enfermedades y trastornos, procesos para preparar estos compuestos e intermedios útiles en estos procesos.
En un aspecto más, la presente invención proporciona activadores de la guanilato ciclasa soluble que tienen propiedades de solubilidad consistentes con propiedades farmacocinéticas aceptables. Tal como se conoce en la técnica, los compuestos poco solubles pueden sufrir una pobre exposición al ser humano. Se esperaría que los compuestos de la presente invención tengan propiedades de exposición consistentes con ser un fármaco adecuado. En un aspecto más, la presente invención proporciona compuestos con propiedades de estabilidad metabólica consistentes con propiedades farmacocinéticas aceptables. Tal como se conoce en la técnica, los compuestos que tienen una pobre estabilidad metabólica pueden no conseguir fácilmente niveles terapéuticos deseados. Se esperaría que los compuestos del presente documento tengan propiedades de estabilidad metabólica consistentes con ser un fármaco adecuado.
Descripción detallada de la invención
En una realización (1), se proporcionan compuestos de fórmula I

en donde:
X es un enlace;
Y es C o N;
W es C o N, con la condición de que Y y W no son ambos N;
V es -C(R11)(R12)- o -OCH2-, con la condición de que si V es -OCH2 , entonces Z es -CH2- e Y y W son ambos C; Z es -CH2-, -C(R10)2CH2- o -C(O)-;
R1 es H, Me o -CH2Oalquilo C1-2;
R2 y R3 junto con los carbonos a los que están unidos para formar un anillo de 3 miembros condensado;
B es

R5 y R6 se seleccionan independientemente entre H, Me, F, Cl y CF3;
R7 es H, Me, Et, -OMe, CN, F o -CH2OMe o no está presente cuando Y es N;
R8 es H, Me o F o no está presente cuando W es N;
R9 es -(CH2)n heterociclilo, en donde el heterociclilo se selecciona entre tetrahidropiranilo, tetrahidrofuranilo,
oxetanilo y [1,4]-dioxanilo o -CH(R10)heteroarilo, en donde el heteroarilo se selecciona entre el grupo que consiste en pirazina, imidazol, piridilo e isoxazolilo y en donde el heteroarilo está opcionalmente sustituido con un grupo metilo; cada R10 es independientemente H o Me;
R11 es H o Me;
R12 es H o Me;
m es 0 o 1, con la condición de que si m es 0, Z es -CH2-, V es -C(R11)(R12)- y R11 y R12 son ambos H y n es 0 o 1 o una sal del mismo.
En una segunda realización (2), se proporcionan compuestos tal como se describen en la realización (1) anteriormente en donde:
X es un enlace;
Y es C o N;
W es C;
V es -C(R11)(R12);
Z es -CH2-, -C(R10)2CH2- o -C(O)-;
R1 es H, Me o -CH2OMe;
R2 y R3 junto con los carbonos a los que están unidos para formar un anillo de 3 miembros condensado;
B es

R7 es H, Me, Et, -OMe, CN o F o no está presente cuando Y es N;
R8 es H, Me o F;
R9 es -(CH2)n heterociclilo, en donde el heterociclilo se selecciona entre tetrahidropiranilo, tetrahidrofuranilo, oxetanilo y [1,4]-dioxanilo;
cada R10 es independientemente H o Me;
R11 es H o Me;
R12 es H o Me;
m es 1 y
n es 0 o 1;
o una sal del mismo.
En otra realización (3), se proporcionan compuestos tal como se describen en cualquiera de las realizaciones (1) o (2) anteriores en donde:
Y es C;
W es C;
V es -C(R11)(R12)-;
Z es -CH2- o -C(R10)2CH2- y
R9 es -(CH2)n heterociclilo, en donde el heterociclilo se selecciona entre tetrahidropiranilo, tetrahidrofuranilo, oxetanilo y [1,4]-dioxanilo;
o una sal del mismo.
En otra realización (4), se proporcionan compuestos tal como se describen en una cualquiera de las realizaciones (1) a (3) anteriores, en donde:
X es un enlace;
R1 es H, Me o -CH2OMe; y
R2 y R3 junto con los carbonos a los que están unidos para formar un anillo de 3 miembros condensado; o una sal del mismo.
En otra realización (5), se proporcionan compuestos tal como se describen en una cualquiera de las realizaciones (1) a (4) anteriores en donde:
Z es -CH2-; o una sal del mismo.
En otra realización (6), se proporcionan compuestos tal como se describen en una cualquiera de las realizaciones (1) a (5) anteriores en donde:
Z es -C(R10)2CH2- y
R10 es H
o una sal del mismo.
En otra realización (7), se proporcionan compuestos tal como se describen en una cualquiera de las realizaciones (1) a (6) anteriores, en donde:
B es

o una sal del mismo.
En otra realización (8), se proporcionan compuestos tal como se describen en una cualquiera de las realizaciones (1) a (7) anteriores, en donde:
B es

o una sal del mismo.
En otra realización (9), se proporcionan compuestos tal como se describen en una cualquiera de las realizaciones (1) a (8) anteriores, en donde:
B es

o una sal del mismo.
En otra realización (10), se proporcionan compuestos tal como se describen en una cualquiera de las realizaciones (1) a (9) anteriores, en donde:
R9 se selecciona entre -(CH2)n heterociclilo, en donde el heterociclilo se selecciona entre tetrahidropiranilo, tetrahidrofuranilo, oxetanilo y [1,4]-dioxanilo; o una sal del mismo.
En otra realización (11), se proporcionan compuestos tal como se describen en una cualquiera de las realizaciones (1) a (10) anteriores, en donde:
X es un enlace;
Y es C;
W es -C(R11)(R12)-;
V es C;
Z es -CH2- o -C(R10)2CH2 ;
R1 es H, Me o -CH2OMe;
R2 y R3 junto con los carbonos a los que están unidos para formar un anillo de 3 miembros condensado; B es

R7 es H, Me, Et, -OMe, CN, F o -CH2OMe;
R8 es H, Me o F;
R9 es -(CH2)n heterociclilo, en donde el heterociclilo se selecciona entre tetrahidropiranilo, tetrahidrofuranilo, oxetanilo y [1,4]-dioxanilo;
R11 es H;
R12 es H;
n es 0 y
m es 1;
o una sal del mismo.
En otra realización (12), se proporcionan compuestos tal como se describen en una cualquiera de las realizaciones (1) a (11) anteriores, en donde:
R9 se selecciona entre

o una sal del mismo.
En otra realización (13), se proporcionan compuestos tal como se describen en una cualquiera de las realizaciones (1) a (12) anteriores, en donde:
R5 y R6 se seleccionan independientemente entre H y Me; o una sal del mismo.
En otra realización (14), se proporcionan compuestos tal como se describen en una cualquiera de las realizaciones (1) a (13) anteriores, en donde:
R10 es H
R11 es H y
R12 es H
o una sal del mismo.
En otro aspecto de la invención, se proporciona un compuesto de la fórmula general I de acuerdo con una cualquiera de las realizaciones (1) a (14) anteriores o una sal farmacéuticamente aceptable del mismo para su uso en un método terapéutico tal como de describe en el presente documento anteriormente y a continuación.
Los siguientes son compuestos representativos de la invención que se pueden fabricar por los esquemas sintéticos generales, los ejemplos y los métodos conocidos en la técnica.

















En una realización, la invención se refiere a uno cualquiera de los compuestos representados en la tabla 1 anterior y las sales farmacéuticamente aceptables del mismo.
En otra realización la invención se refiere al grupo de compuestos representados en la tabla 1 que consiste en los compuestos 20-63, 135-136, 138-159, 161-164, 188-191 y 198.
En otra realización la invención se refiere al grupo de compuestos representados en la tabla 1 que consiste en los compuestos 198, 212-213, 215-227, 237, 243, 262-263, 268, 279-281 y 314-318.
En otra realización la invención se refiere al grupo de compuestos representados en la tabla 1 que consiste en los compuestos 43, 44, 45, 46, 47, 48, 49, 53, 55, 56, 57, 58, 59, 60, 61, 161, 162, 163, 164, 212, 213, 215, 216, 217, 218, 219, 220, 221, 222, 223, 224, 225, 226, 227, 237, 243, 262, 263, 268, 279, 280, 281, 314, 316, 317 y 318.
A menos que se indique específicamente, a través de la memoria descriptiva y las reivindicaciones adjuntas, una fórmula o nombre químico dado abarcará tautómeros y todos los estereoisómeros, isómeros ópticos y geométricos (por ejemplo, enantiómeros, diastereómeros, isómeros E/Z, etc.) y racematos de los mismos así como mezclas en diferentes proporciones de los enantiómeros separados, mezclas de diastereómeros o mezclas de cualquiera de las formas anteriores en las que dichos isómeros y enantiómeros existen, así como sales, incluyendo sales farmacéuticamente aceptables de los mismos y solvatos de los mismos tales como, por ejemplo, hidratos que incluyen solvatos de los compuestos libres o solvatos de una sal del compuesto.
Algunos de los compuestos de fórmula I pueden existir en más de una forma tautomérica. La invención incluye métodos para usar todos estos tautómeros.
La invención incluye derivados farmacéuticamente aceptables de los compuestos de fórmula I. Un "derivado farmacéuticamente aceptable" se refiere a cualquier sal o éster farmacéuticamente aceptable o cualquier otro compuesto que, tras la administración a un paciente, es capaz de proporcionar (directa o indirectamente) un compuesto útil para la invención o un metabolito farmacológicamente activo o un residuo farmacológicamente activo del mismo. Debe entenderse que un metabolito farmacológicamente activo significa cualquier compuesto de la invención capaz de ser metabolizado enzimática o químicamente. Esto incluye, por ejemplo, compuestos derivados hidroxilados u oxidados de la fórmula I.
Tal como se usa en el presente documento, "sales farmacéuticamente aceptables" se refiere a derivados de los compuestos divulgados en donde el compuesto precursor se modifica fabricando sales ácidas o básicas del mismo. Los ejemplos de sales farmacéuticamente aceptables incluyen, pero no se limitan a, sales de ácidos minerales u orgánicos de restos básicos tales como aminas; sales alcalinas u orgánicas de restos ácidos tales como ácidos carboxílicos y similares. Por ejemplo, dichas sales incluyen acetatos, ascorbatos, bencenosulfonatos, benzoatos, besilatos, bicarbonatos, bitartratos, bromuros/bromhidratos, edetatos, camsilatos, carbonatos, cloruros/clorhidratos, citratos, edisilatos, etano disulfonatos, estolatos esilatos, fumaratos, gluceptatos, gluconatos, glutamatos, glicolatos, glicolilarsnilatos, hexilresorcinatos, hidrabaminas, hidroximaleatos, hidroxinaftoatos, yoduros, isotionatos, lactatos, lactobionatos, malatos, maleatos, mandelatos, metanosulfonatos, metilbromuros, metilnitratos, metilsulfatos, mucatos, napsilatos, nitratos, oxalatos, pamoatos, pantotenatos, fenilacetatos, fosfatos/difosfatos, poligalacturonatos, propionatos, salicilatos, estearatos, subacetatos, succinatos, sulfamidas, sulfatos, tanatos, tartratos, teoclatos, toluenosulfonatos, trietyoduros, amonio, benzatinas, cloroprocaínas, colinas, dietanolaminas, etilendiaminas, megluminas y procaínas. Otras sales farmacéuticamente aceptables se pueden formar con cationes de metales tales como aluminio, calcio, litio, magnesio, potasio, sodio, cinc y similares. (véase también Pharmaceutical salts, Birge, S. M. et al., J. Pharm. Sci., (1977), 66, 1-19).
Las sales farmacéuticamente aceptables de la presente invención se pueden sintetizar por métodos químicos convencionales a partir del compuesto precursor que contiene un resto básico o ácido. En general, dichas sales se pueden preparar haciendo reaccionar las formas de ácido o base libre de estos compuestos con una cantidad suficiente de la base o el ácido apropiado en agua o en un diluyente orgánico como éter, acetato de etilo, etanol, isopropanol o acetonitrilo o una mezcla de los mismos.
Las sales de otros ácidos distintos a los mencionados anteriormente que por ejemplo son útiles para purificar o aislar los compuestos de la presente invención (por ejemplo, sales de trifluoro acetato) comprenden también una parte de la invención.
Además, el uso de profármacos de los compuestos de fórmula I está dentro del alcance de la invención. Los profármacos incluyen aquellos compuestos que, mediante una transformación química simple, se modifican para producir los compuestos de la invención. Las transformaciones químicas simples incluyen hidrólisis, oxidación y reducción. De manera específica, cuando se administra un profármaco a un paciente, el profármaco se puede transformar en un compuesto divulgado anteriormente en el presente documento, impartiendo de este modo el efecto farmacológico deseado.
Los compuestos de la invención son solamente aquellos que se contempla que son "químicamente estables" tal
como apreciarán los expertos en la técnica. Por ejemplo, un compuesto que pudiera tener una 'valencia colgante' o un 'carbanión' no son compuestos contemplados por los métodos de la invención divulgados en el presente documento.
En cuanto a todos los compuestos divulgados en el presente documento en esta solicitud, en el caso en el que la nomenclatura sea contraria a la estructura, se entenderá que el compuesto se define mediante la estructura.
Todos los términos, tal como se usan en el presente documento, en esta memoria descriptiva, a menos que se indique otra cosa, se entenderán en su significado habitual, tal como se conoce en la técnica. Por ejemplo, "Alquilo C1-4" es un radical hidrocarburo monovalente, alifático, saturado, que contiene 1-4 carbonos tal como metilo, etilo, npropilo, 1 -metiletilo (isopropilo), n-butilo o f-butilo; "Alcoxi C1-4" es un alquilo C1-4 con un oxígeno terminal, tal como metoxi, etoxi, propoxi, butoxi. Debe entenderse que todos los grupos alquilo, alquenilo y alquinilo son lineales o ramificados, ciclados o no ciclados, donde sea estructuralmente posible y a menos que no se especifique de otra manera.
Otras definiciones más específicas son como siguen:
La expresión "alquilo C1-n", en donde n es un número entero de 2 a n, ya sea solo o en combinación con otro radical denota un radical hidrocarburo lineal o ramificado, saturado, acíclico, con de 1 a n átomos de C. Por ejemplo la expresión alquilo C1-5 abarca los radicales H3C-, H3C-CH2-, H3C-CH2-CH2-, H3C-CH(CH3)-, H3C-CH2-CH2-CH2-, H3C-CH2-CH(CH3)-, H3C-CH(CH3)-CH2-, H3C-C(CH3)2-, H3C-CH2-CH2-CH2-CH2-, H3C-CH2-CH2-CH(CH3)-, H3C-CH2-CH(CH3)-CH2-, H3C-CH(CH3)-CH2-CH2-, H3C-CH2-C(CH3)2-, H3C-C(CH3)2-CH2-, H3C-CH(CH3)-CH(CH3)- y H3C-CH2-CH(CH2CH3)-.
El término "-alquileno C1-n" en donde n es un número entero de 1 a n, ya sea solo o en combinación con otro radical, representa un radical alquilo divalente, de cadena lineal o ramificada, acíclico, que contiene de 1 a n átomos de carbono. Por ejemplo el término alquileno C1-4-incluye -(CH2)-, -(CH2-CH2)-, -(CH(CH3))-, -(CH2-CH2-CH2)-, -(C(CH3)2)-, -(CH(CH2CH3))-, -(CH(CH3)-CH2)-, -(CH2-CH(CH3))-, -(CH2-CH2-CH2-CH2)-, -(CH2-CH2-CH(CH3))-, -(CH(CH3)-CH2-CH2)-, -(CH2-CH(CH3)-CH2)-, -(CH2-C(CH3)2)-, -(C(CH3)2-CH2)-, -(CH(CH3)-CH(CH3))-, -(CH2-CH(CH2CH3 ))-, -(CH(CH2CH3 )-CH2)-, -(CH(CH2CH2CH3 ))-, -(CHCH(CH3 )2)- y - C(CH3 )(CH2CH3 )-.
Debe entenderse que el término "heteroátomo", tal como se usa en el presente documento, significa átomos distintos de carbono, tales como O, N, S y P.
En todos los grupos o cadenas de carbono alquilo, opcionalmente se puede sustituir uno o más átomos de carbono por heteroátomos: O, S o N, debe entenderse que si N no está sustituido entonces es NH, debe entenderse también que los heteroátomos se pueden sustituir tanto por átomos de carbono terminales como por átomos de carbono internos dentro de una cadena de carbono ramificada o sin ramificar. Dichos grupos se pueden sustituir tal como se ha descrito anteriormente en el presente documento por grupos tales como oxo para dar como resultado definiciones tales como, pero sin limitación: alcoxicarbonilo, acilo, amido y tioxo.
El término "arilo", tal como se usa en el presente documento, ya sea solo o en combinación con otro radical, representa un grupo monocíclico aromático carbocíclico que contiene 6 átomos de carbono que además se puede condensar a un segundo grupo carbocíclico de 5 o 6 miembros que puede ser aromático, saturado o insaturado. Arilo incluye, pero sin limitación, fenilo, indanilo, indenilo, naftilo, antracenilo, fenantrenilo, tetrahidronaftilo y dihidronaftilo.
El término "heteroarilo" significa un anillo heteroarilo monocíclico de 5 a 6 miembros aromático o heteroarilo bicíclico de 7 a 11 aromático, en donde al menos uno de los anillos es aromático, en donde el anillo heteroarilo contiene 1-4 heteroátomos tales como N, O y S. Los ejemplos no limitantes de anillos heteroarilo monocíclicos de 5 a 6 miembros incluyen furanilo, oxazolilo, isoxazolilo, oxadiazolilo, tiazolilo, pirazolilo, pirrolilo, imidazolilo, tetrazolilo, triazolilo, tienilo, tiadiazolilo, piridinilo, pirimidinilo, piridazinilo, pirazinilo, triazinilo y purinilo. Los ejemplos no limitantes de anillos heteroarilo bicíclicos de 7 a 11 miembros incluyen benzoimidazolilo, quinolinilo, dihidro-2H-quinolinilo, isoquinolinilo, quinazolinilo, indazolilo, tieno[2,3-d]pirimidinilo, indolilo, isoindolilo, benzofuranilo, benzopiranilo, benzodioxolilo, benzoxazolilo y benzotiazolilo.
El término "heterociclilo" significa un radical heterocíclico monocíclico de 4-8 miembros, no aromático, estable o un radical heterocíclico bicíclico condensado, bicíclico puenteado o espirocíclico, de 6 a 11 miembros, no aromático, estable. El heterociclo de 5 a 11 miembros consta de átomos de carbono y uno o más, preferentemente de uno a cuatro heteroátomos elegidos entre nitrógeno, oxígeno y azufre. El heterociclo puede estar saturado o parcialmente insaturado. Los ejemplos no limitantes de radicales heterocíclicos monocíclicos de 4-8 miembros, no aromáticos, incluyen tetrahidrofuranilo, azetidinilo, pirrolidinilo, piranilo, tetrahidropiranilo, dioxanilo, tiomorfolinilo, 1,1-dioxo-1A6-tiomorfolinilo, morfolinilo, piperidinilo, piperazinilo y azepinilo. Los ejemplos no limitantes de radicales bicíclicos condensados de 6 a 11 miembros, no aromáticos, incluyen octahidroindolilo, octahidrobenzofuranilo y octahidrobenzotiofenilo. Los ejemplos no limitantes de radicales bicíclicos puenteados de 6 a 11 miembros, no aromáticos, incluyen 2-azabiciclo[2.2.1]heptanilo, 3-azabiciclo[3.1.0]hexanilo y 3-azabiciclo[3.2.1]octanilo. Los ejemplos no limitantes de radicales espirocíclicos de 6 a 11 miembros, no aromáticos, incluyen 7-azaespiro[3,3]heptanilo, 7-espiro[3,4]octanilo y 7-aza-espiro[3,4]octanilo. El término "heterociclilo" pretende incluir todas
las formas isoméricas posibles.
Se debe entender que el término "halógeno", tal como se usa en la presente memoria descriptiva, significa bromo, cloro, flúor o yodo. Las definiciones "halogenado", "parcial o completamente halogenado"; parcial o completamente fluorado; "sustituido por uno o más átomos de halógeno", incluyen, por ejemplo, mono, di o tri halo derivados en uno o más átomos de carbono. Para alquilo, un ejemplo no limitante sería -CH2CHF2 , -CF3 , etc.
Cada alquilo, heterociclo, arilo o heteroarilo o los análogos de los mismos, descritos en el presente documento se entenderá que está opcionalmente parcial o completamente halogenado.
Tal como se usa en el presente documento, "nitrógeno" o N y "azufre" o S incluye una forma oxidada de nitrógeno o azufre y la forma cuaternizada de cualquier nitrógeno básico. Por ejemplo, para un radical -S-alquilo C1-6, a menos que se indique de otra manera, se entenderá que incluye -S(O)-alquilo C1-6 y -S(O)2-alquilo C1-6, asimismo, -S-Ra se puede representar como fenil-S(O)m- cuando Ra es fenilo y donde m es 0, 1 o 2.
MÉTODOS DE SÍNTESIS GENERALES
Los compuestos de la invención se pueden preparar por los métodos generales y los ejemplos presentados a continuación y los métodos conocidos por los expertos habituales en la técnica. Las condiciones de reacción y los tiempos de reacción óptimos pueden variar dependiendo de los reactivos particulares usados. A menos que se indique de otra manera, los disolventes, temperaturas, presiones y otras condiciones de reacción se pueden seleccionar fácilmente por un experto habitual en la técnica. Los procedimientos específicos se proporcionan en el apartado de Ejemplos de síntesis. Los intermedios usados en las síntesis siguientes o están disponibles en el mercado o se pueden preparar fácilmente mediante los métodos conocidos por los expertos en la técnica. El progreso de la reacción se puede controlar por métodos convencionales tales como cromatografía de capa fina (TLC) cromatografía líquida de alta presión-espectrometría de masas (HPLC-MS). Los intermedios y productos se pueden purificar por métodos conocidos en la técnica, incluyendo cromatografía en columna, HPLC, TLC preparativa o recristalización.
Los métodos descritos a continuación y en la sección de Ejemplos de síntesis se pueden usar para preparar los compuestos de fórmula I.
Los compuestos de fórmula I se pueden preparar como se describe en el esquema 1

Tal como se ilustra en el esquema I anterior, la amina II (X = enlace, R = Me, Et o tere-butilo) y el heterociclo dihalo (B = 2,6 piridilo o 2,5-tiazol; Hal = Cl o Br) III se someten a reflujo en un disolvente adecuado tal como N,N-dimetilformamida con una base adecuada tal como carbonato potásico (K2CO3) proporcionando el heteroarilo IV. El compuesto IV se acopla con especies de boro, V, en presencia de un catalizador de paladio tal como tetraquis(trifenil)fosfina (0) y una base adecuada tal como Na2CO3 en 1,4-dioxano acuoso a aproximadamente 80 °C para proporcionar VI. Alquilación del producto intermedio de fenol, VI con bromuro de alquilo VII, usando una base tal como carbonato de cesio (Cs2CO3) en un disolvente tal como acetona. La posterior desprotección del grupo Boc con un ácido adecuado tal como ácido trifluoroacético (TFA) proporciona el compuesto VIII. La aminación reductiva de la amina, VIII, con la cetona o el aldehído deseado usando una fuente de hidruro apropiada tal como NaBH3CN en un disolvente tal como MeOH que contiene un ácido orgánico tal como AcOH a aproximadamente 50 °C, seguido de hidrólisis in situ con una base tal como LiOH acuoso (donde R = Me o Et) o usando ácido fórmico (donde R = tere-butilo), proporciona el compuesto de fórmula I deseado.
Esquema II




Como alternativa, tal como se ilustra en el esquema II, cuando B es un anillo de pirimidina 2,6 disustituido, la 2,6-dihalopirimidina (Cl o Br) de partida se acopla con especies de boro, V, en presencia de un catalizador de paladio tal como tetraquis(trifenil)fosfina (0) y una base adecuada tal como Na2CO3 en 1,4-dioxano acuoso a 80 °C para proporcionar IX. La amina II (X = enlace, R = Me, Et o tere-butilo) y IX se someten a reflujo en un disolvente adecuado tal como W,W-dimetilformamida con una base adecuada tal como carbonato potásico (K2CO3) proporcionando la aminopirimidina X. Esto se sigue de alquilación del producto intermedio de fenol, X con bromuro de alquilo VII, usando una base tal como carbonato de cesio (Cs2CO3 ) en un disolvente tal como acetona. La posterior desprotección del grupo Boc con un ácido adecuado tal como ácido trifluoroacético (TFA) proporciona el compuesto XI. La aminación reductiva de la amina, XI, con la cetona o el aldehído deseado usando una fuente de hidruro apropiada tal como NaBH3CN en un disolvente tal como MeOH que contiene un ácido orgánico tal como AcOH a aproximadamente 50 °C, seguido de hidrólisis in situ con una base tal como LiOH acuoso (donde R = Me o Et) o usando ácido fórmico (donde R = tere-butilo), proporciona el compuesto de fórmula I deseado.
Métodos de UPLC/MS
Los tiempos de retención (TR) indicados para los compuestos en la sección de Ejemplos de síntesis se obtuvieron por UPLC/MS usando uno de los métodos siguientes:
Para cada uno de los métodos, los siguientes son idénticos:
Componentes del sistema UPLC/MS- Acquity UPLC con detectores PDA, SQ y ELS.
Detección- condiciones PDA: 210 a 400 nm. Frecuencia de muestreo: 20 pts/s. Respuesta de filtro: rápida.
Condiciones ELSD- ganancia: 1000. Frecuencia de muestreo: 20 pts/s. Temperatura del tubo de deriva: 55 °C. Modo nebulizador: enfriamiento. Presión de gas: 282,685 kPa (41 psi).
Condiciones de MS- Instrumento: Acquity SQD con fuente de ESCi. Modo de ionización: ESI+/-. Tensión de capilaridad: 3,5 kV. Tensión del cono: 5 V. Extractor: 1,3 V. Temperatura de la fuente: 150 °C. Temperatura de desolvatación: 350 °C. Gas de desolvatación: 800 l/h. Gas del cono: 50 l/h.
Las condiciones específicas para cada método son como siguen
Método A1
Columna- Waters BEH C18, 2,1x50 mm, diámetro de partículas de 1,7 um.
Descripción y gradiente: método de gradiente rápido medio polar. ESI+/- modo iónico 80-1000 Da. Gradiente: A al 90 % a B al 95 % en 1,19 min, parada al 95 % de B a 1,70 min. Caudal 0,8 ml/min. A=(agua al 95 % acetonitrilo al 5 % ácido fórmico al 0,05 %) B=(acetonitrilo ácido fórmico al 0,05 %).
Volumen de inyección de muestra: 1 ul
Método A2
Columna: HSS T32,1x100 mm, diámetro de partículas de 1,8 um.
Descripción y gradiente: método de gradiente polar. ESI+/- modo iónico 80-1000 Da. Gradiente: A al 95 % a B al 95 % en 3,65 min parada al 95 % de B a 4,95 min. Caudal 0,6 ml/min. A=(agua al 95 % acetonitrilo al 5 % ácido fórmico al 0,05 %) B=(acetonitrilo ácido fórmico al 0,05 %).
Volumen de inyección de muestra: 1 ul
Método A3
Columna: BEH 2,1x50 mm C18, diámetro de partículas de 1,7 um.
Descripción y gradiente: método de gradiente largo medio polar. ESI+/- modo iónico 80-1000 Da. Gradiente: A al 90 % a B al 95 % en 4,45 min parada al 95 % de B a 4,58 min. Caudal 0,8 ml/min. A=(agua al 95 % acetonitrilo 5 % ácido fórmico al 0,05 %) B=(Acetonitrilo+ácido fórmico al 0,05 %)
Volumen de inyección de muestra: 1 ul
Método A4
Columna: BEH 2,1x50 mm C18, diámetro de partículas de 1,7 um.
Descripción y gradiente: Método de gradiente rápido medio polar tamponado básico. ESI+/- modo iónico 80 1000 Da. Gradiente: A al 90 % a B al 95 % en 1,19 min parada al 95 % de B a 1,70 min. Caudal 0,8 ml/min. A=(agua al 95 % acetonitrilo al 5 % bicarbonato de amonio 2,5 mM) B=(Acetonitrilo).
Volumen de inyección de muestra: 1 ul
Se usa el método A1 para todos los compuestos excepto cuando se indica.
Ejemplos de síntesis
Los compuestos finales se denominan mediante números de compuestos que corresponden a los números de compuesto de la tabla 1. Los intermedios reciben números con guiones que corresponden a las figuras y números que se muestran en el esquema para cada ejemplo.
Síntesis de los productos intermedios:
Ejemplo de referencia 1: Preparación del producto intermedio éster etílico del ácido (S)-3-aza-biciclo[4.1.0]heptan-6-carboxílico (A-1)

A una solución en agitación de NaOEt al 21 % en EtOH (53,3 ml, 148,2 mmol) en EtOH anhidro (100 ml), a 0 °C, se le añade lentamente dietilmalonato (25 g, 156 mmol). Después de la adición, la mezcla de reacción se calienta a temperatura ambiente y se deja en agitación hasta que se disuelven los materiales sólidos. Después, se añade gota a gota (RJ-epi-clorohidrina (10,8 ml, 140,5 mmol) en EtOH (5 ml). La mezcla se calienta a reflujo durante 36 h y después se deja enfriar a temperatura ambiente y se diluye con agua. Los orgánicos volátiles se eliminan a presión reducida y el residuo resultante se extrae con EtOAc. Los extractos orgánicos combinados se secan (Na2SO4), se filtran y se concentran a presión reducida. El residuo se purifica primero por cromatografía ultrarrápida sobre gel de sílice para proporcionar el producto en bruto que se recoge en tolueno (100 ml) y se trata con K2CO3 (5,1 g, 37,0 mmol). La mezcla se calienta a 110 °C durante 3 h. La mezcla de reacción se enfría a temperatura ambiente, se filtra a través de un lecho de tierra de diatomeas y se aclara con DCM. El filtrado se concentra a presión reducida para proporcionar A-1-1 (18 g, 75 % de rendimiento).
A una solución de A-1-1 (17,1 g, 100,3 mmol) en EtOH (150 ml) se le añade borohidruro sódico (2,85 g, 75,4 mmol). La mezcla resultante se agita a temperatura ambiente durante 2 h y después se añade HCl 1 N (40 ml) y la mezcla se concentra a presión reducida. El residuo se extrae con EtOAc y los extractos orgánicos combinados se lavan con salmuera, se secan sobre Na2SO4 anhidro y se concentran a presión reducida. El residuo se purifica por cromatografía ultrarrápida sobre gel de sílice para proporcionar A-1-2 (9,8 g, 56 % de rendimiento).
A una solución en agitación de A-1-2 (7,3 g, 41,9 mmol) en DMF (200 ml), a -40 °C, se le añade imidazol (5,71 g, 83,8 mmol). La solución se agita a esta temperatura durante 1,5 h y después, se añade gota a gota una solución de TBDPS-Cl (11,3 ml, 44,0 mmol) en DMF (70 ml). La solución resultante se deja calentar lentamente a temperatura ambiente y se agita durante una noche. La mezcla de reacción se diluye con MeOH y los volátiles orgánicos se eliminan a presión reducida. El residuo se diluye con EtOAc y se lava con agua seguido de salmuera. La fase orgánica se concentra a presión reducida y el residuo se purifica por cromatografía ultrarrápida sobre gel de sílice para proporcionar A-1-3 (12,2 g, 71 % de rendimiento).
A una solución en agitación de A-1-3 (11,4 g, 27,6 mmol) en DCM (150 ml), a 0 °C, se le añade peryodinano de Dess-Martin (14,1 g, 33,2 mmol). Después de 30 min a 0 °C, el baño de refrigeración se retira y la agitación se mantiene durante una hora más. El medio de reacción se neutraliza con NaHCO3 acuoso. La mezcla se diluye con EtOAc, se lava con agua, seguido de salmuera, después se concentra a presión reducida. El residuo se purifica por cromatografía ultrarrápida sobre gel de sílice para proporcionar A-1-4 (10,5 g, 93 % de rendimiento).
Una suspensión de cloruro de metoximetiltrifenilfosfina (10,4 g, 30,4 mmol) y ferc-butóxido de potasio (3,4 g, 30,4 mmol) en THF (120 ml) se agita a 0 °C durante 30 min.
A esta suspensión se le añade, gota a gota, una solución de A-1-4 (10,4 g, 25,3 mmol) en THF (20 ml). La mezcla de reacción se agita a 0 °C durante 1 h, después se calienta a temperatura ambiente y se agita durante una noche. La mezcla de reacción se diluye con agua y se extrae con EtOAc. La fase orgánica se lava con salmuera, después se concentra a presión reducida. El residuo se purifica por cromatografía ultrarrápida sobre gel de sílice para proporcionar A-1-5 en forma de una mezcla de dos isómeros (10,0 g, 90 % de rendimiento).
A una solución en agitación de A-1-5 (10,0 g, 22,8 mmol) en THF (100 ml), enfriada a 0 °C, se le añade TBAF (27,4 ml, 27,4 mmol). La solución se calienta a temperatura ambiente y se agita durante 1,5 h. La mezcla se concentra a presión reducida y el residuo se purifica por cromatografía ultrarrápida sobre gel de sílice para proporcionar A-1-6 en forma de una mezcla de dos isómeros (4,2 g, 92 % de rendimiento).
A una solución en agitación de A-1-6 (2,8 g, 14,0 mmol) en DCM (30 ml), enfriada a 0 °C, se le añade TEA (5,3 ml, 42.0 mmol) seguido de metano sulfónico anhídrido (3,7 g, 21,0 mmol). La solución se calienta a temperatura ambiente y se agita durante 2 h. A la mezcla se le añade una solución acuosa saturada de NaHCO3 (100 ml). Las fases se separan y la capa acuosa se extrae con DCM. Los extractos orgánicos combinados se secan sobre MgSO4 anhidro, se filtran y se concentran a presión reducida para proporcionar A-1-7 (3,8 g, 98 % de rendimiento).
A una solución de A-1-7 (3,8 g, 13,7 mmol) en ACN (30 ml) se le añade bencil amina (2,2 ml, 20,5 mmol), seguido de K2CO3 (5,7 g, 41,0 mmol). La mezcla se calienta a 80 °C durante una noche, después se enfría a temperatura ambiente. Se forma un precipitado que se elimina por filtración y la almohadilla del filtro se aclara a fondo con ACN. El filtrado se concentra a presión reducida y el residuo se purifica por cromatografía ultrarrápida sobre gel de sílice para proporcionar A-1-8 (2,6 g, 66 % de rendimiento).
A una solución en agitación de A-1-8 (2,6 g, 9,0 mmol) en THF (30 ml), a 0 °C, se le añade HCl 6 N (4,5 ml, 27 mmol). Después de 20 min, el baño de refrigeración se retira y la mezcla de reacción se deja en agitación durante 5 h más. La mezcla de reacción se neutraliza después con una solución acuosa saturada de Na2CO3 y se extrae con EtOAc. La fase orgánica se lava con salmuera y se concentra a presión reducida para dar A-1-9.
A una solución en agitación de A-1-9 en DCE (50 ml), 0 °C, se le añade triacetoxiborohidruro sódico (3,6 g, 17.1 mmol). La mezcla de reacción se agita a 0 °C durante 2 h, después se consume el exceso de los reactivos mediante la adición de una solución acuosa saturada de Na2CO3. La mezcla se extrae con EtOAc y la fase orgánica se lava con salmuera y se concentra a presión reducida. El residuo se purifica por cromatografía ultrarrápida sobre gel de sílice para proporcionar A-1-10 (1,4 g, 62 % de rendimiento a partir de A-1-8).
Se carga un matraz con paladio al 10 % sobre carbono (0,25 g, 0,23 mmol) y la atmósfera se vacía y se vuelve a llenar con argón tres veces. A esto se le añade una solución de A-1-10 (1,00 g, 3,86 mmol) en EtOH (40 ml). La mezcla de reacción se coloca en atmósfera de hidrógeno, se agita a temperatura ambiente durante tres días, después se filtra a través de tierra de diatomeas y se concentra a presión reducida para proporcionar A-1 (0,72 g, cuant.).
Ejemplo 2: Preparación del producto intermedio éster metílico del ácido (Rj-3-aza-biciclo[3.1.0]hexan-1-carboxílico (A-2)

A una solución de solución al 21 % de NaOEt (53 ml, 150 mmol) en EtOH (100 ml), enfriada a 0 °C, se le añade dietilmalonato (25 g, 16 mmol). Cuando la mezcla se espesa, se añade más EtOH (50 ml) y la mezcla se calienta a temperatura ambiente y se mantiene en agitación hasta que todos los sólidos se disuelven. A la mezcla se le añade, gota a gota, una solución de RJ-epiclorohidrina (10,8 ml, 140 mmol) en EtOH (5 ml). Después de la adición, la mezcla se calienta a reflujo durante 36 h y después, se enfría a temperatura ambiente y se diluye con agua. La solución se extrae con EtOAc y los extractos combinados se secan sobre Na2SO4 anhidro y se concentra a presión reducida. El residuo se purifica por cromatografía ultrarrápida sobre gel de sílice para proporcionar A-2-1(13,5 g, 51 % de rendimiento).
A una solución de A-2-1 (11,5 g, 68 mmol) en EtOH (150 ml) se le añade borohidruro sódico (1,9 g, 51 mmol). La mezcla resultante se agita a temperatura ambiente durante 2 h. Los reactivos en exceso se consumen mediante la adición de una solución 1 N de HCl (40 ml). La mezcla se concentra a presión reducida después se diluye con agua y se extrae con EtOAc. Los extractos combinados se lavan con salmuera, se secan sobre Na2SO4 anhidro y se concentran a presión reducida. El residuo se purifica por cromatografía ultrarrápida sobre gel de sílice para proporcionar A-2-2 (8,5 g, 72 % de rendimiento).
A una solución de A-2-2 (8.2 g, 47 mmol) en DCM (100 ml), enfriada a 0 °C, se le añade TEA (25 ml, 190 mmol) seguido de anhídrido sulfónico (20 g, 120 mmol). Después de la adición, la solución se calienta a temperatura ambiente y se agita durante 2 h. A la mezcla se le añade una solución acuosa saturada de NaHCO3 (100 ml). Las fases se separan y la capa acuosa se extrae con DCM. Los extractos orgánicos combinados se secan sobre MgSO4 anhídrido y se concentran a presión reducida para proporcionar A-2-3 (15,5 g, 96 % de rendimiento).
Una solución de A-2-3 (15,5 g, 45 mmol), bencil amina (7,7 ml, 70 mmol) y K2CO3 (19 g, 140 mmol) en ACN (150 ml) se calienta a 80 °C durante 36 h. Después de enfriar a temperatura ambiente se separa por filtración un precipitado y la almohadilla del filtro se aclara con ACN. El filtrado se concentra a presión reducida. El residuo se purifica por cromatografía ultrarrápida sobre gel de sílice para proporcionar A-2-4 (8,4 g, 73 % de rendimiento).
Una mezcla de amina A-2-4 (1,4 g, 6,0 mmol), Boc2O (2,0 g, 9,0 mmol) y Pd al 5 %/C (200 mg) en MeOH (60 ml) se agita en atmósfera de hidrógeno durante 3 h. La mezcla se filtra y el filtrado se concentra a presión reducida. El residuo se purifica por cromatografía ultrarrápida sobre gel de sílice para proporcionar A-2-5 (1,5 g, 100 % de rendimiento).
A una solución de A-2-5 (1,4 g, 5,8 mmol) en DCM (15 ml), a 0 °C, se le añade TFA (4,4 ml, 58 mmol). El baño de hielo se retira inmediatamente después de la adición de TFA y la reacción se mantiene a temperatura ambiente durante 3 h. Después se eliminan los disolventes a presión reducida y el residuo se diluye con DCM. La mezcla se enfría a 0 °C y se neutraliza con una solución acuosa saturada de NaHCO3. La mezcla heterogénea resultante se deja calentar a temperatura ambiente y se agita durante 1 h. La mezcla se filtra a través de un separador de fases y la fase acuosa retenida se lava a fondo con DCM. El disolvente se concentra a presión reducida para proporcionar A-2 (0,85 g, 98 % de rendimiento).
Ejemplo 3: Preparación del producto intermedio éster metílico del ácido (1R,2R)-2-metil-3-aza-biciclo[3.1.0]hexan-1-carboxílico (A-3) y éster metílico del ácido (1R,2S)-2-metil-3-aza-biciclo[3.1.0]hexan-1-carboxílico (A-4)

Una solución de bencil amina (11,2 g, 104 mmol) y crotonitrilo (7,0 g, 110 mmol) en EtOH (125 ml) se calienta a reflujo durante 24 h. La mezcla se enfría a temperatura ambiente y se concentra a presión reducida. El residuo se purifica por cromatografía ultrarrápida sobre gel de sílice para proporcionar A-3-1 (13 g, 73 % de rendimiento). Una solución de A-3-1 (13 g, 74 mmol) y (R)-glicidol (11 g, 150 mmol) en EtOH (100 ml) se calienta a reflujo durante 2 días. La mezcla se enfría a temperatura ambiente y se concentra a presión reducida. El residuo se purifica por cromatografía ultrarrápida sobre gel de sílice para proporcionar A-3-2 (13 g, 71 % de rendimiento).
A una solución del diol A-3-2 (11,5 g, 46,0 mmol) en DCM (150 ml), enfriada a 0 °C, se le añade TEA (29 ml, 232 mmol) seguido de anhídrido sulfónico (24 g, 140 mmol). La solución se calienta a temperatura ambiente y se agita durante 2 h. A la mezcla se le añade una solución acuosa saturada de NaHCO3 (100 ml). La mezcla se separa y la capa acuosa se extrae con DCM. Los extractos orgánicos combinados se secan sobre MgSO4 anhidro y se concentran a presión reducida para proporcionar A-3-3 (18,5 g, 100 % de rendimiento).
A un matraz que contiene THF (250 ml) se le añade una solución 1 N de NaHMDS (100 ml, 100 mmol) en THF. La solución se enfría a 0 °C y se añade gota a gota una solución de A-3-3 (18,5 g, 46 mmol) en THF (50 ml). La mezcla se agita a 0 °C durante 10 min, después se elimina el baño de refrigeración y la agitación continúa a temperatura ambiente durante 2 h. Los reactivos en exceso se consumen mediante la adición de una solución acuosa saturada de NaHCO3. La mezcla se extrae con EtOAc y los extractos orgánicos combinados se lavan con salmuera y se concentran a presión reducida. El residuo se purifica por cromatografía ultrarrápida sobre gel de sílice para proporcionar el compuesto A-3-4 (2,4 g, 24 % de rendimiento) y el compuesto A-4-1 (2,7 g, 28 % de rendimiento). La mezcla de nitrilo A-3-4 (2,4 g, 11 mmol) y Ba(OH)2^8 H2O (5,3 g, 17 mmol) en agua (100 ml) se calienta a reflujo durante 5 días. La mezcla se enfría a temperatura ambiente y la solución se acidifica mediante la adición de una solución 6 N de HCl. La mezcla se concentra a presión reducida y el residuo se suspende en EtOH. La mezcla se filtra y el filtrado se concentra a presión reducida. El residuo se purifica por cromatografía ultrarrápida sobre gel de sílice para proporcionar A-3-5 (2,6 g, 99 % de rendimiento).
A una solución de A-3-5 (2,6 g, 11 mmol) en MeOH (50 ml), enfriada a 0 °C, se le añade TMSCHN2 hasta que persiste un color amarillento. La mezcla se agita a 0 °C durante 30 min y después, el exceso de reactivos se
consume mediante la adición de ácido acético. La solución se concentra a presión reducida y el residuo se purifica por cromatografía ultrarrápida sobre gel de sílice para proporcionar el A-3-6 (1,8 g, 65 % de rendimiento).
Una mezcla de A-3-6 (1,8 g, 7,3 mmol) y Pd al 5 %/C (0,50 g) en MeOH (20 ml) se agita a temperatura ambiente durante una noche en atmósfera de hidrógeno. La mezcla se filtra a través de un lecho de tierra de diatomeas y la almohadilla del filtro se aclara con MeOH. El filtrado se concentra a presión reducida para proporcionar A-3 (1,1 g, 97 % de rendimiento).
El intermedio A-4 se puede preparar de un modo similar a partir de A-4-1.
Ejemplo 4: Preparación del producto intermedio éster metílico del ácido (1R,2S)-2-metoximetil-3-azabiciclo[3.1.0]hexan-1-carboxílico (A-5) y éster metílico del ácido (1R,2R)-2-metoximetil-3-aza-biciclo[3.1.0]hexan-1-carboxílico (A-6)

Una solución de éster metílico del ácido 4-metoxi-3-oxo-butírico (25 g, 170 mmol), bencil amina (18,3 ml, 171 mmol) y ácido acético (0,50 ml, 8,5 mmol) en tolueno (120 ml) se calienta a 60 °C durante 5 h, después se enfría a temperatura ambiente. La mezcla se concentra a presión reducida y el residuo se destila azeotrópicamente con tolueno 2 veces para proporcionar A-5-1 (40 g, 100 % de rendimiento).
A una solución de A-5-1 (20 g, 85 mmol) en DCE (200 ml), enfriada a 0 °C, se le añade ácido acético (25 ml, 420 mmol) y triacetoxiborohidruro sódico (54 g, 250 mmol). La mezcla se agita a 0 °C durante 2 h, después se calienta a temperatura ambiente y se agita durante 2 h más. La mezcla se concentra a presión reducida y el residuo se diluye con EtOAc. La mezcla se alcaliniza mediante la adición de una solución acuosa saturada de Na2CO3. La fase orgánica se separa y la fase acuosa se extrae con EtOAc. Los extractos combinados se secan sobre Na2SO4 anhidro y se concentran a presión reducida. El residuo se purifica por cromatografía ultrarrápida sobre gel de sílice para proporcionar A-5-2 (18 g, 89 % de rendimiento).
La solución de A-5-2 (18 g, 75 mmol) y ^Rj-(+)-glicidol (11 g, 150 mmol) en MeOH (100 ml) se calienta a reflujo durante 2 días, después se enfría a temperatura ambiente y se concentra a presión reducida. El residuo se purifica por cromatografía ultrarrápida sobre gel de sílice para proporcionar A-5-3 (14 g, 59 % de rendimiento).
A una solución de A-5-3 (10 g, 32 mmol) en DCM (100 ml), enfriada a 0 °C, se le añade TEA (20 ml, 160 mmol), seguido de sulfónico anhídrido (17 g, 96 mmol). La solución se calienta a temperatura ambiente y se agita durante 2 h. La mezcla se diluye con una solución acuosa saturada de NaHCO3 (100 ml) y se extrae con EtOAc. Los extractos orgánicos combinados se lavan con salmuera, se secan sobre MgSO4 anhidro y se concentran a presión reducida para proporcionar A-5-4 (15 g, 100 % de rendimiento).
Se carga un matraz con THF (150 ml) seguido de una solución 1 N de NaHMDS en THF (70 ml, 70 mmol). La solución se enfría a -20 °C, después se añade gota a gota una solución de A-5-4 (15 g, 32 mmol) en THF (30 ml). La mezcla se agita a -20 °C durante 1 h, después se deja calentar lentamente a temperatura ambiente y en agitación
durante 2 h más. La reacción se interrumpe mediante la adición de una solución acuosa saturada de NaHCO3, se extrae con EtOAc. Los extractos combinados se lavan con salmuera y se concentran a presión reducida. El residuo se purifica por cromatografía ultrarrápida sobre gel de sílice para proporcionar A-5-5 (1 g, 11 % de rendimiento) y A-6-1 (2,1 g, 21 % de rendimiento).
Una mezcla de A-5-5 (1,6 g, 5,8 mmol) y Pd al 5 %/C (0,50 g) en MeOH (10 ml) se agita durante una noche a temperatura ambiente en atmósfera de hidrógeno. La mezcla se filtra a través de un lecho de tierra de diatomeas y la almohadilla del filtro se aclara con MeOH. El filtrado se concentra a presión reducida para proporcionar A-5 (1,1 g, 100 % de rendimiento)
El intermedio A-6 se puede preparar de un modo similar a partir de A-6-1.
Los intermedios A-14 y A-15 se pueden preparar como se describe para los intermedios A-5 y A-6 usando éster etílico del ácido 4-etoxi-3-oxo-butírico.

El intermedio racémico A-20 se puede preparar como se describe para los intermedios A-5 usando glicidol racémico.

Ejemplo 7: Preparación del producto intermedio éster metílico del ácido (1S,2S)-2-metil-3-aza-biciclo[3.1.0]hexan-1-carboxílico (A-9) y éster metílico del ácido (1S,2R)-2-metil-3-aza-biciclo[3.1.0]hexan-1-carboxílico (A-10)

Los intermedios A-9 y A-10 se pueden preparar como se describe para los ejemplos A-3 y A-4 en el ejemplo 3 usando ('Sj-(-)-glicidol.
Ejemplo 8: Preparación del producto intermedio éster metílico del ácido (1S,2R)-2-metoximetil-3-azabiciclo[3.1.0]hexan-1-carboxílico (A-11) y éster metílico del ácido (1S,2S)-2-metoximetil-3-aza-biciclo[3.1.0]hexan-1-carboxílico (A-12)

Los intermedios A-11 y A-12 se pueden preparar como se describe para los intermedios A-5 y A-6 en el ejemplo 4 usando (S)-(-}-glicidol.
Ejemplo de referencia 9: Preparación del producto intermedio éster etílico del ácido (1R,6S)-3-(6-bromo-piridin-2-il)-3-aza-biciclo[4.1.0]heptan-6-carboxílico (B-1)

A una solución de A-1 (2,01 g, 11,8 mmol), en DMA (30 ml), se le añade 2,6-dibromopiridina (3,65 g, 15,4) seguido de carbonato de cesio (8,11 g, 24,9 mmol). La mezcla de reacción se calienta a 100 °C durante una noche y después, se enfría a temperatura ambiente y se diluye con agua y MTBE. La fase orgánica se separa y la fase acuosa se extrae con MTBE. Los extractos orgánicos combinados se lavan con salmuera, se secan sobre Na2SO4 anhidro, se filtran y se concentran a presión reducida. El material en bruto se purifica mediante cromatografía ultrarrápida sobre gel de sílice para proporcionar B-1 (2,6 g, 68 % de rendimiento).
Los productos intermedios s reactivos apropiados.

Ejemplo 10: Preparación del producto intermedio éster metílico del ácido (1R,2S)-3-(6-bromo-piridin-2-il)-2-metoximetil-3-aza-biciclo[3.1.0]hexan-1-carboxílico (B-3)

Una suspensión de A-5 (1,1 g, 5,8 mmol) y 2,6-dibromopiridina (4,2 g, 17 mmol) en 2,2,6,6-tetrametilpiperidina (3,5 ml, 17 mmol) se calienta a 130 °C durante 48 h. La mezcla de reacción se enfría a temperatura ambiente y se diluye con EtOAc. La mezcla se lava con una solución acuosa saturada de NaHCO3 seguida de salmuera y después, se concentra a presión reducida. El residuo se purifica por cromatografía ultrarrápida sobre gel de sílice para proporcionar B-3 (1,7 g, 84 % de rendimiento).
Los productos intermedios siguientes se pueden preparar de una manera similar usando los reactivos apropiados. El intermedio B-26 es racémico, generado a partir del producto intermedio racémico A-20.

Ejemplo 11: Preparación del producto intermedio 2-(2-cloro-pirimidin-4-il)-6-metil-fenol (B-16)

A una solución de 3,0 g (20 mmol) de 2,4-dicloro-pirimidina en una mezcla 10:1 de dioxano:agua (220 ml) se le añaden 3,3 g (22 mmol) de ácido 2-hidroxi-3-metil-fenil borónico seguido de 6,0 g (57 mmol) de carbonato sódico. Se burbujea argón a través de la solución durante 15 min, después se añaden 2,4 g (2,1 mmol) de tetraquis(trifenilfosfina)paladio (0). La reacción se calienta durante una noche a 100 °C, después se enfría a temperatura ambiente y se filtra a través de tierra de diatomeas. La almohadilla del filtro se lava con EtOAc, después se coloca sobre un matraz receptor nuevo se lava con una mezcla 1:1 de MeOH:DCM. El filtrado se concentra a presión reducida hasta B-16 (1,60 g, 36,0 %)
Los productos intermedios siguientes se pueden preparar de una manera similar usando los reactivos apropiados.

Ejemplo de referencia 13: Preparación del producto intermedio ácido (1R,6S)-3-[6-(2-hidroxi-fenil)-piridin-2-il]-3-azabiciclo[4.1.0]heptan-6-carboxílico (C-1)

Una solución de B-1 (0,204 g, 0,628 mmol) y ácido 2-hidroxfenilborónico (0,113 g, 0,816 mmol) en 1,2-DME (12 ml) se rocía con nitrógeno durante aproximadamente 10 min. Después, se introduce tetraquis(trifenilfosfina)paladio (0,849 g, 0,0735 mmol), seguido de una solución acuosa al 20 % de carbonato sódico (1,0 ml, 2,0 mmol). La mezcla
de reacción se rocía con nitrógeno durante 15 min más y después, se calienta en un reactor de microondas a 125 °C durante 30 min. El material en bruto se purifica por cromatografía ultrarrápida en columna sobre gel de sílice para proporcionar (0,20 g, 96 % de rendimiento) de C-1.
Los productos intermedios siguientes se pueden preparar de una manera similar usando los reactivos apropiados.

(continuación)

Ejemplo de referencia 15: Preparación del producto intermedio éster etílico del ácido (1 R,6S)-3-[4-(2-h idroxi-fenil)-pirimidin-2-il]-3-aza-biciclo[4.1.0]heptan-6-carboxílico (C-29)

A una solución de 0,200 g (0,968 mmol) de B-14 en DMF (5 ml) se le añaden 0,35 g (2,1 mmol) de A-14 seguido de 1,0 g (7,2 mmol) de carbonato potásico. La mezcla se calienta a 80 °C durante 2 días, después se enfría a temperatura ambiente y se diluye con agua. La mezcla se extrae con EtOAc. El extracto orgánico combinado se lava con agua seguida de salmuera, después se seca sobre sulfato sódico anhidro y se concentra a presión reducida. El residuo se purifica por cromatografía ultrarrápida sobre gel de sílice para proporcionar C-29 (0,221 g, 67,3 %).
El intermedio siguiente se puede preparar a partir de B-14 y A-2 de una manera similar usando los reactivos apropiados.

El intermedio siguiente se puede preparar a partir de B-16 y A-2 de una manera similar usando los reactivos apropiados.

Ejemplo 16: Preparación del producto intermedio ferc-butil éster del ácido 6-bromometil-3,4-dihidro-1H-isoquinolin-2-carboxílico (D-1)

A una solución de 2-ferc-butil éster del ácido 3,4-dihidro-1H-isoquinolin-2,6-dicarboxílico (12,50 g, 45,08 mmol) en THF seco (125,0 ml), en atmósfera de nitrógeno a 25 °C, se le añade complejo de borano y THF mediante una jeringa (99,17 ml, 99,17 mmol). La mezcla se agita a 25 °C durante 16 h, después se añade lentamente agua (10,0 ml) seguida de Na2CO32,0 M (15,0 ml). Esta mezcla se agita durante 15 min y después se diluye con EtOAc y se recogen las capas orgánicas. Las capas orgánicas se aclaran con HCl 1 M, se secan sobre MgSO4 y se concentran a presión reducida para proporcionar un aceite. El aceite se purifica por cromatografía sobre gel de sílice para producir D-1 -1 (11,8 g, 99,3 % de rendimiento), en forma de un sólido de color blanco.
A una solución de alcohol, D-1-1, (9,50 g, 36,1 mmol) y N,N-diisopropiletilamina (9,43 ml, 54,1 mmol) en diclorometano (200,0 ml) se le añade dibromuro de trifenilfosfina (23,79 g, 54,11 mmol) a 0 °C. La reacción se agita durante 1 h, después se concentra a presión reducida. El residuo resultante se purifica por cromatografía sobre gel de sílice para producir D-1 (8,74 g, 74 % de rendimiento), en forma de un sólido de color blanco.
Los productos intermedios siguientes se sintetizan de una manera similar a partir de reactivos apropiados:

Ejemplo 17: Preparación del producto intermedio ferc-butil éster del ácido 6-bromometil-5-metil-3,4-dihidro-1H-isoquinolin-2-carboxílico (D-3)

Se añade una solución de ácido 3-metoxi-2-metil-benzoico (350 g, 2,10 mol) en THF (1,4 l) a una suspensión de LAH (95,9 g, 1,40 mol) en THF (2,5 l) a 0 °C. La mezcla se agita a temperatura ambiente durante 0,5 h, después se calienta a reflujo durante 1 h. Después, la mezcla se enfría a 0 °C y se inactiva lentamente mediante la adición de solución acuosa saturada de cloruro de amonio. Se añade un gran exceso de Na2SO4 sólido y EtOAc, después los sólidos se recogen por filtración. El filtrado se concentra a presión reducida para proporcionar D-3-1 en bruto (350 g, rendimiento cuant.) que se usa directamente en la etapa siguiente.
A una solución del compuesto D-3-1 (294,0 g, 1,90 mol) en diclorometano (2,2 l) a -10 °C se le añade cloruro de tionilo (SOCl2) (460,0 g, 3,90 mol). Después, la mezcla de reacción se calienta a reflujo durante 1 h, seguido de concentración a presión reducida para proporcionar D-3-2 en bruto (298 g, 92 % de rendimiento) que se usa directamente en la etapa siguiente.
Una mezcla del compuesto D-3-2 (298,0 g, 1,8 mol) y NaCN (154,5 g, 2,1 mol) en DMF (1,2 l) se agita a temperatura ambiente durante 12 h, después se extrae con EtOAc y H2O. La capa orgánica se seca sobre Na2SO4, se filtra y se concentra a presión reducida. El residuo se purifica por cromatografía sobre gel de sílice (éter de petróleo:EtOAc = 50:1) para proporcionar el producto intermedio D-3-3 (230,0 g, 79 % de rendimiento).
Una mezcla del compuesto D-3-3 (180,0 g, 1,10 mol), Ni Raney (40,0 g) y amoniaco acuoso (250,0 ml) en MeOH (1,0 l) se agita en atmósfera de H2 (344,738 kPa (50 psi)) a temperatura ambiente durante 5 h. Después, la mezcla se filtra y se concentra para dar el compuesto D-3-4 (165,0 g) que se usa directamente en la etapa siguiente.
Una solución del compuesto D-3-4 (165,0 g, 1,0 mol) y formaldehído acuoso (HCHO) (37 % en peso, 30 g, 1,0 mol) en ácido fórmico (HCO2H) (1,5 l) se agita a 50 °C durante una noche. El disolvente se elimina a presión reducida para proporcionar el compuesto D-3-5 (150,0 g) que se usa directamente en la etapa siguiente.
El compuesto D-3-5 (150,0 g, 847 mmol) se suspende en HBr acuoso (48 %, 1,0 l), después calentó a 100 °C durante una noche. La eliminación del disolvente a presión reducida proporciona el compuesto D-3-6 (195,0 g) que
se usa directamente en la etapa siguiente.
A una solución del compuesto D-3-6 (195,0 g, 799 mmol) en THF (1,0 l) y H2O (1,0 l) se le añade Et3N (242,0 g, 2,4 mol) y Boc2O (174,0 g, 799 mmol). La mezcla resultante se agita a temperatura ambiente durante una noche, después se extrae con EtOAc. Las fases orgánicas combinadas se lavan con salmuera, se secan sobre Na2SO4 , se filtran y se concentran a presión reducida. El producto en bruto se purifica por cromatografía sobre gel de sílice (usando 10:1 éter de petróleo:EtOAc) para proporcionar el compuesto D-3-7 (100,0 g, 56 % de rendimiento en 4 etapas).
A una solución del compuesto D-3-7 (100 g, 380 mmol) y Et3N (76,8 g, 760 mmol) en diclorometano (1,5 l), enfriada a 0 °C, se le añade anhídrido tríflico (Tf2O) (107,0 g, 380 mmol) mediante un embudo de adición. Una vez completada la adición de Tf2O la solución se calienta después a temperatura ambiente durante 5 h. La mezcla de reacción se trata después con H2O y diclorometano y se separa la fase orgánica. La fase orgánica se lava con salmuera y se seca sobre Na2SO4 anhídrido. Después, la mezcla se filtra y se concentra a presión reducida. El residuo se purifica por cromatografía ultrarrápida sobre gel de sílice (usando 20:1 éter de petróleo:EtOAc) para proporcionar el compuesto D-3-8 (105 g, 70 % de rendimiento).
el compuesto D-3-8 (50,0 g, 127 mmol) se combina con acetato de paladio (II) (5,0 g), dppp (5,0 g) y Et3N (25,7 g, 254 mmol) en EtOH (1,0 l). Después, la mezcla se agita a 80 °C durante una noche en atmósfera de CO a una presión de 4 MPa. La mezcla se enfría a temperatura ambiente y los sólidos se eliminan por filtración. El filtrado se concentra a presión reducida y el residuo se purifica por cromatografía ultrarrápida sobre gel de sílice (usando 20:1 éter de petróleo:EtOAc) para proporcionar el compuesto D-3-9 (25,0 g, 62 % de rendimiento).
A una solución de LAH (12,5 g, 330 mmol) en THF (400 ml), enfriada a -30 °C, se le añade, gota a gota durante 30 min, una solución del compuesto D-3-9 (35,0 g, 110 mmol) en THF (400 ml). Después de la adición, la mezcla de reacción se agita a 0 °C durante 30 min, después se trata con H2O y diclorometano. La fase orgánica es: separada, lavada con salmuera, secada sobre Na2SO4 anhidro, filtrada y concentrada a presión reducida. El producto en bruto se purifica por cromatografía ultrarrápida sobre gel de sílice (usando 10:1 éter de petróleo:EtOAc) para proporcionar el producto intermedio D-3-10 deseado (21,1 g, 69 % de rendimiento).
A una solución del alcohol D-3-10, (6,00 g, 21,6 mmol) y W,W-diisopropiletilamina (5,65 ml, 32,5 mmol) en diclorometano (200 ml) se le añade dibromuro de trifenilfosfina (14,3 g, 32,5 mmol) a 0 °C. La reacción se agita durante 1 h y después, se concentra a presión reducida. El residuo resultante se purifica por cromatografía ultrarrápida sobre gel de sílice para proporcionar D-3 (6,60 g, 90 % de rendimiento), en forma de un sólido de color blanco.
Ejemplo 18: Preparación del producto intermedio ferc-butil éster del ácido 6-bromometil-8-metil-3,4-dihidro-1H-isoquinolin-2-carboxílico (D-4).

A una mezcla de 3-bromo-5-metil-fenol (185 g; 0,940 mol) y K2CO3 (437 g, 3,17 mol) en acetona (2 l) se le añade Mel (424 g, 2,99 mol). La mezcla se agita a 40 °C durante 16 h. La mezcla se enfría a temperatura ambiente, se filtra y se concentra a presión reducida. Después de la filtración, la mezcla se purifica por cromatografía ultrarrápida sobre gel de sílice para proporcionar 1-bromo-3-metoxi-5-metil-benceno, D-4-1 (189 g, rendimiento cuant.) en forma de un aceite de color amarillo claro.
A una mezcla de D-4-1 (200 g, 0,995 mol) en THF seco (1,7 l), a -70 °C, se le añade gota a gota una solución de n-BuLi en hexanos (438 ml; 1,09 mol). Después de agitar durante 1 h a -70 °C, se añade gota a gota DMF seca (76,3 g, 1,04 mol) a -70 °C. Después de esto, la mezcla se agita durante 1 h a -70 °C. La mezcla se vierte en NH4Cl (1 l) y se extrae con EtOAc. Los extractos combinados se lavan con salmuera, se secan sobre Na2SO4 y se concentran a presión reducida para proporcionar D-4-2 (147 g, 98 % de rendimiento) en forma de un aceite de color amarillo.
La mezcla de D-4-2 (150 g, 0,999 mol) y NH4OAc (30,8 g, 0,40 mol) en MeNO2 (1,5 l) se somete a reflujo durante 16 h. La mezcla se concentra, después se diluye con EtOAc (1000 ml) y se lava secuencialmente con agua (1 l), seguida de salmuera (100 ml). La fase orgánica se seca sobre Na2SO4 anhidro y se concentra a presión reducida. La mezcla se tritura con PE:EtOAc = 10:1 durante 10 min y el sólido se recoge por filtración para proporcionar D-4-3 (80 g, 42 % de rendimiento) en forma de un sólido de color amarillo.
A una mezcla de LiAlH4 (78,6 g, 2,00 mol) en THF seco (1 l), a 0 °C, se le añade una solución de D-4-3 (78 g, 0,404 mol) en THF (200 ml). La mezcla se calienta a 70 °C y se agita durante 16 h. La mezcla se enfría a 0 °C, se inactiva lentamente con agua (78 ml), seguida de una solución al 15 % en peso de NaOH (78 ml) y después con más agua (235 ml). Después de la filtración, la mezcla se concentra a presión reducida para proporcionar D-4-4 (40 g, 60 % de rendimiento) en forma de un aceite de color amarillo claro.
La mezcla del compuesto D-4-4 ( 66 g, 0,40 mol) y ácido fórmico (73,5 g, 1,60 mol) en dioxano (600 ml) se agita durante 16 h a 90 °C. La mezcla se concentra a presión reducida para proporcionar D-4-5 (77 g, 90 % de rendimiento) en forma de un sólido de color amarillo.
A una solución de D-4-5 (76,0 g, 0,354 mol) en diclorometano (2,5 l), a 15 °C, se le añade POCl3 (155 g, 1,01 mol). Después de la adición, la mezcla se somete a reflujo durante 3 h, después se enfría a temperatura ambiente. La solución se concentra a presión reducida. Al residuo se le añade agua (1,5 l), tolueno (1,5 l) y NaOH al 20 % (500 ml). La mezcla se somete después a reflujo durante 1 h, después se enfría a temperatura ambiente. La mezcla se diluye con EtOAc y se lava con agua, seguida de salmuera. La fase orgánica combinada se seca sobre Na2SO4 anhidro y después, se concentra a presión reducida. El residuo se purifica mediante una columna ultrarrápida sobre gel de sílice (PE:EtOAc = 10:1) para proporcionar D-4-6 (58,5 g, 94 % de rendimiento) en forma de un aceite de color pardo.
A una solución de D-4-6 (58,5 g, 0,334 mol) en MeOH (500 ml), a 0 °C, se le añade NaBHU (63,3 g, 1,67 mol). La mezcla se mantiene a 0 °C durante 4 h. La solución se inactiva con HCl 1 N (100 ml). El pH se ajusta a pH 8 mediante la adición de NaHCO3. La mezcla se extrae con DCM. Los extractos orgánicos combinados se secan sobre Na2SO4 anhidro y se concentran para proporcionar D-4-7 en forma de un aceite de color pardo.
Una solución de D-4-7 en bruto (83 g, 0,47 mol) en una solución de HBr (al 40 % en agua, 500 ml) se calienta a 90 °C durante 12 h. La solución se concentra a presión reducida para obtener D-4-8 que se usa directamente en la reacción siguiente.
A una solución de D-4-8 en bruto en DCM (1 l) se le añade Boc2O (72 g, 0,33 mol) y trietilamina (63 g, 0,62 mol). La mezcla resultante se agita durante 12 h a 15 °C, después se diluye con DCM (1500 ml) y agua (100 ml). La capa orgánica se separa y se lava con HCl 0,5 N (100 ml) seguido de salmuera (100 ml). La fase orgánica se seca sobre Na2SO4 anhidro y se concentra a presión reducida. El residuo se purifica por cromatografía ultrarrápida en columna sobre gel de sílice para proporcionar D-4-9 (33,4 g, 34 % de rendimiento a partir de D-4-6) en forma de un sólido de color blanco.
A una solución de D-4-9 (33 g; 0,113 mol) y piridina (20,1 g, 0,254 mol) en diclorometano seco (300 ml), a -30 °C, se le añade Tf2O (39,4 g, 0,139 mol) gota a gota. La mezcla se agita durante 1 h a -30 °C, después se calienta a 15 °C y se agita durante 8 h. La mezcla se diluye con diclorometano (500 ml) y agua (100 ml). La fase orgánica se concentra a presión reducida y el residuo se purifica por cromatografía ultrarrápida sobre gel de sílice para proporcionar D-4-10 (43 g, 96 % de rendimiento) en forma de un sólido de color blanco.
Una solución de D-4-10 (43 g, 0,109 mol), EtsN (33,0 g, 0,327 mol), DPPP (4,53 g) y Pd(OAc)2 (5 g) en MeOH (500 ml) se agita a una presión de 3 MPa de CO a 90 °C durante 2 días. Después de la filtración y la concentración el residuo se purifica por cromatografía sobre gel de sílice (PE:EtOAc =50:1) para proporcionar el 2-ferc-butil éster 6-metil éster del ácido 8-metil-3,4-dihidro-1H-isoquinolin-2,6-dicarboxílico, D-4-11 (21 g, 64 % de rendimiento) en forma de un aceite incoloro.
A una solución de D-4-11 (21 g, 0,0693 mol) en THF seco (500 ml), a -50 °C, se le añadió LiAlH4 (7,4 g, 208 mmol). La mezcla se agita a -50 °C durante 1 h y después se calienta a 0 °C y se agita durante 30 min más. La reacción se inactiva lentamente con agua (7,4 ml), NaOH al 15 % (7,4 ml) y más agua (22,2 ml). La mezcla se filtra y el filtrado se concentra a presión reducida. El residuo se purifica por HPLc prep. El eluyente se concentra a presión reducida para eliminar los orgánicos volátiles. El resto de la mezcla acuosa restante se extrae con diclorometano. Los extractos orgánicos combinados se secan sobre Na2SO4 y se concentran a presión reducida para proporcionar D-4-12 (14,8 g, 77 % de rendimiento) en forma de un aceite incoloro.
A una solución de D-4-12 (13,4 g, 0,0485 mol) y DIEA (11,8 ml, 0,679 mol) en diclorometano (200 ml), a -30 °C, se le añade dibromuro de trifenilfosfina (26,6 g, 0,606 mol). La mezcla resultante se agita 1 h, tiempo durante el cual el baño frío se deja calentar a -10 °C. Los volátiles se eliminan de la mezcla a -10 °C, el residuo se suspende en diclorometano (50 ml) y el filtrado se purifica por cromatografía ultrarrápida sobre gel de sílice para proporcionar D-4 (16,2 g, rendimiento cuant.) en forma de un sólido de color blanco.
Ejemplo 19: Preparación del producto intermedio terc-butil éster del ácido 6-bromometil-5,8-dimetil-3,4-dihidro-1H-isoquinolin-2-carboxílico (D-5).

Una solución de boc-4-piperidinona (14,0 g, 70,3 mmol) y pirrolidina (8,71 ml, 106 mmol) en tolueno (60 ml) se somete a reflujo en condiciones de Dean Stark durante 24 h. La reacción se concentra después a presión reducida. El residuo resultante se disuelve en tolueno (60 ml) y se trata con 4-hexen-3-ona (8,32 ml, 70,3 mmol) e hidroquinona (0,080 g, 0,73 mmol). La solución se calienta a reflujo durante 24 h, después se enfría a temperatura ambiente. La mezcla se diluye con EtOAc y se lava con HCl 1 N. Los extractos orgánicos combinados se secan y se concentran a presión reducida para proporcionar un aceite viscoso. El material se purifica por cromatografía ultrarrápida sobre gel de sílice para proporcionar D-5-1 en forma de un sólido de color amarillo (11,7 g, 60 % de rendimiento).
Se añade gota a gota una solución 1,0 M de LiHMDS en THF (43 ml) a una solución de D-5-1 (10,00 g, 35,79 mmol) en THF (50,0 ml) a -78 °C. Esta mezcla se agita a -78 °C durante 30 min, después se añade gota a gota TMS-Cl (5,45 ml, 42,9 mmol). La mezcla se agita a -78 °C durante 2 h más, después se calienta a temperatura ambiente y se diluye con éter dietílico (200 ml). Esta mezcla se añade a una solución saturada de Na2CO3 y las fases se separan. Los extractos orgánicos combinados se secan y se concentran a presión reducida. El residuo se disuelve en MeCN (50,0 ml) y se añade Pd(OAc)2 (8,04 g, 35,8 mmol). La mezcla resultante se enfría en un baño de agua para mantener la temperatura de la reacción por debajo de 35 °C y se agita durante una noche. La reacción se filtra a través de tierra de diatomeas y el filtrado se concentra a presión reducida. El residuo se recoge en 200 ml de EtOAc, después se trata con solución 1,0 M de TBAF (50,0 ml). Esta mezcla se agita durante 30 min y después se lava secuencialmente con una solución de HCl 1 N y tiosulfato sódico al 10 %. Los extractos orgánicos se secan y se concentran. El material se purifica por cromatografía sobre gel de sílice para proporcionar D-5-2 en forma de un sólido de color blanquecino (6,11 g, 62 % de rendimiento).
A una solución de D-5-2 (1,50 g, 5,41 mmol) en diclorometano (25 ml) a temperatura ambiente se le añade piridina (0,87 ml, 11 mmol). La solución se enfría a -30 °C y se añade gota a gota Tf2O (1,00 ml, 5,95 mmol). La reacción se agita a -30 °C durante 1 h y después se calienta a temperatura ambiente. La mezcla se concentra a presión reducida y el residuo se diluye con EtOAc, después se lava secuencialmente con solución de HCl 1 N, NaHCO3 saturado y después salmuera. La mezcla se seca sobre MgSO4 anhidro y se concentra a presión reducida. El material resultante se purifica por cromatografía ultrarrápida sobre gel de sílice para proporcionar D-5-3 en forma de un sólido
de color blanco (1,61 g, 73 % de rendimiento).
El triflato D-5-3 (1,00 g, 2,44 mmol) se combina con el boronato (0,647 g, 2,69 mmol) y Pd(PPh3)4 (0,144 g, 0,124 mmol) en una mezcla de DME (15,0 ml) y Na2CO32,0 M (1,27 ml). La reacción se irradia en un reactor de microondas a 120 °C durante 40 min. La mezcla se concentra a presión reducida y el residuo se purifica por cromatografía ultrarrápida sobre gel de sílice para proporcionar D-5-4, un sólido de color blanco (0,662 g, 94 % de rendimiento).
El sustrato D-5-4 (1,03 g, 3,58 mmol), NalO4 (2,34 g, 10,9 mmol), OsO4 al 2,5 % en peso en f-BuOH (1,0 ml), THF (12,4 ml) y H2O (2,4 ml) se combinan a temperatura ambiente. La mezcla se agita durante una noche en oscuridad, después se diluye con agua y diclorometano. Las fases se separan usando una frita hidrófoba. La fase orgánica se seca sobre Na2SO4, se filtra y se concentra a presión reducida. El residuo se purifica por cromatografía ultrarrápida sobre gel de sílice para proporcionar D-5-5 en forma de un aceite de color ámbar (0,786 g, 76 % de rendimiento). El aldehído D-5-5 (0,785 g, 2,71 mmol) se disuelve en THF (5,0 ml) y MeOH (5,0 ml). La mezcla se enfría a 0 °C y se añade NaBH4 (0,156 g, 4,07 mmol). La reacción se agita a temperatura ambiente durante 30 min. Los reactivos en exceso se consumen mediante la adición de una solución acuosa de NH4Cl y la mezcla se agita a temperatura ambiente durante 10 min. La mezcla se extrae con EtOAc y la fase orgánica se lava con una solución de NH4Cl seguido de salmuera. La fase orgánica se seca después sobre MgSO4 anhidro y se concentra a presión reducida. El material resultante se purifica por cromatografía ultrarrápida sobre gel de sílice para proporcionar D-5-6 (0,626 g, 79 % de rendimiento) en forma de un sólido de color blanco.
A una solución del alcohol D-5-6 (0,300 g, 1,03 mmol) y W,W-diisopropiletilamina (0,269 ml, 1,54 mmol) en diclorometano (10,0 ml), a 0 °C, se le añade dibromuro de trifenilfosfina (0,679 g, 1,54 mmol). La reacción se agita durante 2 h y se concentra a presión reducida. El residuo resultante se purifica por cromatografía sobre gel de sílice para proporcionar D-5 (0,338 g, 93 % de rendimiento) en forma de un sólido de color blanco.
Ejemplo 20: Preparación del intermedio 8-etil-6(bromometil)-3,4-dihidroisoquinolin-2(1H)-carboxilato de ferc-butilo (D-6)

D-6-1 D-6-2 D-6-3
n 2


J-D
A la mezcla de 3-bromo-5-met¡lfenol (300 g, 1,60 mol) y K2CO3 (665 g, 4,8 mol) en DMF (2000 ml) a temperatura ambiente se le añade MeI (250 g, 1,8 mol) gota a gota. La mezcla se agita durante una noche después se diluye con H2O y se extrae con EtOAc. La capa orgánica se seca sobre sulfato sódico anhidro y se concentra a presión reducida. El residuo se purifica por cromatografía ultrarrápida sobre gel de sílice para proporcionar D-6-1 (165 g, 52,0 % de rendimiento).
Una mezcla de D-6-1 (100 g, 497 mmol), NBS (88,5 g, 497 mmol) y AIBN (10 g, 50 mmol) en CCl4 (700 ml) se calienta a reflujo durante 12 h. La mezcla se enfría a temperatura ambiente, se diluye con H2O y se extrae con EtOAc. La capa orgánica se separa, se seca sobre sulfato sódico anhidro y se concentra a presión reducida. El residuo se purifica por cromatografía ultrarrápida sobre gel de sílice para proporcionar D-6-2 (48 g, 42 % de rendimiento).
Se agita una solución del compuesto D-6-2 (80,0 g, 286 mmol) y TMSCN (28,2 g, 286 mmol) en MeCN (600 ml) a temperatura ambiente durante 0,5 h. La mezcla se enfría a 0 °C y se añade TBAF (74,6 g, 286 mmol). La mezcla se agita durante 12 h después se diluye con agua y se extrae con EtOAc. La capa orgánica se separa, se seca sobre sulfato sódico anhidro y se concentra a presión reducida. El residuo se purifica por cromatografía ultrarrápida sobre gel de sílice para proporcionar D-6-3 (39 g, 60 % de rendimiento).
Una solución de D-6-3 (12 g, 53 mmol) y Ni(s) (10 g) en una mezcla de MeOH (80 ml) e hidróxido de amonio (80 ml)
se agita a temperatura ambiente durante 5 horas en atmósfera de 344,738 kPa (50 psi) de hidrógeno. La mezcla se filtra y el filtrado se concentra a presión reducida para proporcionar D-6-4 (8 g), que se usa directamente en la etapa siguiente.
Una mezcla de D-6-4 (75 g, 330 mmol) y formaldehído (8,8 g, 290 mmol) en ácido fórmico (500 ml) se agita durante una noche en atmósfera de N2 a 50 °C. El disolvente se elimina a presión reducida y el residuo se purifica por cromatografía ultrarrápida sobre gel de sílice para proporcionar D-6-5 (54 g, 64 % de rendimiento en 2 etapas). Una mezcla de D-6-5 (45 g, 186 mmol) en una solución acuosa de HBr (400 ml) se agita a 90 °C durante 12 h. El disolvente se elimina a presión reducida y el residuo se purifica por cromatografía sobre gel de sílice para proporcionar D-6-6 (21 g, 53 % de rendimiento).
Una mezcla de D-6-6 (20 g, 88 mmol), Boc2O (19,1 g, 87,7 mmol) y TEA (17,7 g, 175 mmol) en una mezcla 1:1 de THF:agua (200 ml) se agita a temperatura ambiente durante 3 h. La mezcla se diluye con agua y se extrae con EtOAc. La capa orgánica se seca sobre sulfato sódico anhidro y se concentra a presión reducida. El residuo se purifica por cromatografía ultrarrápida sobre gel de sílice para proporcionar D-6-7 (20 g, 70 % de rendimiento).
Una mezcla de D-6-7 (14 g, 43 mmol), K2CO3 (17,7 g, 128 mmol), Pd(dppf)Cl2 (2,5 g), Pd (PPha)4 (2,5 g) y el éster vinil borónico (7,22 g, 46,9 mmol) en DMF (150 ml) se agita a reflujo durante una noche. La mezcla se filtra y el filtrado se concentra a presión reducida. El residuo se purifica por cromatografía ultrarrápida sobre gel de sílice para proporcionar D-6-8 (7,2 g, 61 % de rendimiento).
Una mezcla de D-6-8 (7,2 g, 26,2 mmol) y 10% Pd-C (2 g) en MeOH (100 ml) se agita a temperatura ambiente en atmósfera de 344,738 kPa (50 psi) de H2 durante 12 h. La mezcla se filtra a través de tierra de diatomeas y el filtrado se concentra para dar el producto en bruto que se purifica por cromatografía ultrarrápida sobre gel de sílice para proporcionar D-6-9 (5,8 g, 80 % de rendimiento).
Una mezcla de D-6-9 (5,8 g, 20,9 mmol), Tf2O (5,9 g, 20,9 mmol) y TEA (6,3 g, 62,7 mmol) en DCM (70 ml) se agita a temperatura ambiente durante 3 h. La reacción se diluye con H2O y se extrae con EtOAc. La capa orgánica se seca sobre Na2SO4 y se concentra a presión reducida. El residuo se purifica por cromatografía ultrarrápida sobre gel de sílice para proporcionar D-6-10 (7,0 g, 82 % de rendimiento).
Una mezcla de D-6-10 (7,0 g, 17 mmol), Pd(OAc)2 (1,4 g), dppp (1,4 g) y Et3N (5,2 g, 51,3 mmol) en MeOH (80 ml) se agita durante 2 días a 80 °C en atmósfera de 3 MPa de CO. La mezcla se filtra a través de tierra de diatomeas y el filtrado se concentra a presión reducida. El residuo se purifica por cromatografía ultrarrápida sobre gel de sílice para proporcionar D-6-11 (4,8 g, 88 % de rendimiento).
A una solución de LiAlH4 (1,1 g, 30,1 mmol) en THF (10 ml), a -50 °C, se le añade, gota a gota durante un periodo de 30 minutos, una solución de D-6-11 (4,8 g, 15 mmol) en THF (50 ml). Después de la adición, la mezcla de reacción se agita a 0 °C durante 2,5 h después se diluye con H2O seguida de DCM. La capa orgánica se separa, se lava con salmuera después se seca sobre Na2SO4 y se concentra a presión reducida. El residuo se purifica por cromatografía ultrarrápida sobre gel de sílice para proporcionar D-6-12 (4,1 g, 92 % de rendimiento).
A una solución de alcohol, D-6-12, (3,12 g, 10,7 mmol) y W,W-diisopropiletilamina (2,80 ml, 16,1 mmol) en diclorometano (57 ml), a 0 °C, se le añade dibromuro de trifenilfosfina (6,92 g, 16,1 mmol). La reacción se agita a 0 °C durante 2 h, después se concentra a presión reducida. El residuo resultante se purifica por cromatografía sobre gel de sílice para producir D-6 (2,90 g, 76 % de rendimiento).
Ejemplo 21: Preparación de intermedio 8-ciano-6-(bromometil)-3,4-dihidroisoquinolin-2(1H)-carboxilato de ferc-butilo (D-7)
ZnCN

D-7-3 D-7-4 D-7
Una solución del compuesto D-6-7 (11 g, 35 mmol), Pd(dppf)Cl2 (2,5 g), Pd (PPh3)4 (2,5 g), ZnCN (2,8 g, 31,3 mmol), Zn (1,1 g, 17,4 mmol) en DMF (110 ml) se agita a reflujo durante una noche. La mezcla se filtra a través de tierra de diatomeas y el filtrado se concentra a presión reducida. El residuo se purifica por cromatografía ultrarrápida sobre gel de sílice para proporcionar D-7-1 (6,5 g, 71 % de rendimiento).
Una solución de D-7-1 (12 g, 44 mmol), Tf2O (12 g, 44 mmol) y TEA (13,3 g, 131 mmol) en DCM (120 ml) se agita a temperatura ambiente durante 3 h. La reacción se diluye con H2O y se extrae con EtOAc. La capa orgánica se seca sobre Na2SO4 anhidro, se filtra y se concentra a presión reducida. El residuo se purifica por cromatografía ultrarrápida sobre gel de sílice para proporcionar D-7-2 (9,0 g, 51 % de rendimiento).
Una mezcla de D-7-2 (9,5 g, 23,4 mmol), Pd(OAc)2 (1,9 g), dppp (1,9 g) y Et3N (7,1 g, 70,1 mmol) en MeOH (90 ml) se agita a 80 °C en una atmósfera de 3 MPa de CO durante 2 d. El sólido se elimina por filtración y el filtrado se concentra a presión reducida. El residuo se purifica por cromatografía ultrarrápida sobre gel de sílice para proporcionar D-7-3 (6,0 g, 80 % de rendimiento).
A una solución de LiAlH4 (1,4 g, 38 mmol) en THF (10 ml), a -50 °C, se le añade durante 30 min, una solución de D-7-3 (6,0 g, 19 mmol) en THF (50 ml). Después de la adición, la mezcla de reacción se agita a -20 °C durante 4,5 h, después se trata con H2O seguido de DCM. La capa orgánica se separa, se lava con salmuera, se seca sobre Na2SO4 y se concentra a presión reducida. El residuo se purifica por cromatografía ultrarrápida sobre gel de sílice sobre gel de sílice para proporcionar D-7-4 (4,1 g, 74 % de rendimiento).
A una solución de alcohol, D-7-4, (1,00 g, 3,47 mmol) y W,W-diisopropiletilamina (1,00 ml, 5,74 mmol) en diclorometano (50 ml), a 0 °C, se le añade dibromuro de trifenilfosfina (2,50 g, 5,69 mmol). La reacción se agita durante 1 h, después se concentra a presión reducida. El residuo resultante se purifica por cromatografía sobre gel de sílice para producir D-7 (0,900 g, 74 % de rendimiento).
Ejemplo 22: Preparación del intermedio ferc-butil éster del ácido 6-bromometil-8-metoxi-3,4-dihidro-1H-isoquinolin-2-carboxílico (D-8)

Una solución de D-8-3 (22,5 g, 68,6 mmol) y K2CO3 (28,4 g, 205,7 mmol) en DMF (200 ml) se agita a temperatura ambiente durante 2 h. Después se añade BnBr (11,7 g, 68,6 mmol) a la mezcla de reacción.
La mezcla se agita a temperatura ambiente durante una noche. La reacción se diluye con agua y se extrae con EtOAc. La capa orgánica se seca, se filtra y se concentra a presión reducida. El producto en bruto se purifica por cromatografía ultrarrápida sobre gel de sílice para proporcionar D-8-1 (20 g, 70 % de rendimiento).
Una solución de D-8-1 (10 g, 24 mmol), B2Pin2 (7,2 g, 29 mmol), KOAc (7,0 g, 71 mmol) y Pd(dppf)Cl2 (2 g) en dioxano (100 ml) se agita a 90 °C durante una noche. Después de la filtración, el filtrado se concentra a presión reducida y el residuo se purifica por cromatografía ultrarrápida sobre gel de sílice para proporcionar D-8-2 (5,4 g, 67 % de rendimiento).
Una solución de D-8-2 (15 g, 32 mmol), NH4Cl (1,7 g, 120 mmol) y H2O2 (11 g, 30%, 97 mmol) en THF/H2O=1:1 (150 ml) se agita a temperatura ambiente durante 12 h. La reacción se interrumpe mediante la adición de una solución acuosa de NaS2O4 y se extrae con EtOAc. La capa orgánica se seca, se filtra y se concentra a presión reducida. El residuo se purifica por cromatografía ultrarrápida sobre gel de sílice para proporcionar D-8-3 (9,0 g, 79 % de rendimiento).
A una mezcla de D-8-3 (9 g, 25,3 mmol) y K2CO3 (10,5 g, 76,0 mmol) en DMF (80 ml) se le añade MeI (3,6 g, 25 mmol) a temperatura ambiente. La mezcla se agita durante una noche a temperatura ambiente, después se diluye con H2O y se extrae con EtOAc. La capa orgánica se seca, se filtra y se concentra a presión reducida. El producto en bruto se purifica por cromatografía ultrarrápida sobre gel de sílice para proporcionar D-8-4 (7,5 g, 80 % de rendimiento).
Una mezcla de D-8-4 (12 g, 32 mmol) y Pd-C (12 g) en MeOH (100 ml) se agita en atmósfera de 344,738 kPa (50 psi) de H2 a temperatura ambiente durante 12 h. La mezcla se filtra a través de tierra de diatomeas y el filtrado se concentra a presión reducida. El producto en bruto se purifica por cromatografía ultrarrápida sobre gel de sílice para proporcionar D-8-5 (7,6 g, 85 % de rendimiento).
Una solución de D-8-5 (7,0 g, 25 mmol), Tf2O (7,1 g, 25 mmol) y TEA (7,6 g, 75 mmol) en DCM (70 ml) se agita a temperatura ambiente durante 3 h. La reacción se diluye con H2O y se extrae con EtOAc. La capa orgánica se seca,
se filtra y se concentra a presión reducida. El producto en bruto se purifica por cromatografía ultrarrápida sobre gel de sílice para proporcionar D-8-6 (7,3 g, 73 % de rendimiento).
Una mezcla de D-8-6 (9 g, 22 mmol), Pd(OAc)2 (1,8 g), dppp (1,8 g) y Et3N (6,6 g, 65,6 mmol) en MeOH (80 ml) se agita durante una noche a 80 °C en atmósfera de 3 MPa de CO. El sólido se elimina por filtración y el filtrado se concentra a presión reducida. El residuo se purifica por cromatografía ultrarrápida sobre gel de sílice para proporcionar D-8-7 (6,8 g, 85 % de rendimiento).
A una solución de LiAlH4 (1,6 g, 42 mmol) en THF (10 ml) a -50 °C se le añade, gota a gota durante 30 min, una solución de D-8-7 (6,8 g, 21 mmol) en THF (70 ml). Después de la adición, la mezcla de reacción se agita a 0 °C durante 2,5 h, después se trata con H2O y DCM. La capa orgánica se separa, se lava con salmuera, se seca sobre Na2SO4 y se concentra a presión reducida. El residuo se purifica por cromatografía ultrarrápida sobre gel de sílice para proporcionar D-8-8 (5,9 g, 90 % de rendimiento).
A una solución de alcohol, D-8-8, (6,37 g, 21,7 mmol) y N,N-diisopropiletilamina (5,30 ml, 30,4 mmol) en diclorometano (ml), enfriado a -45 °C, se le añade dibromuro de trifenilfosfina (11,9 g, 27,1 mmol). La reacción se calienta a 0 °C, se agita durante 3 h, después se concentra a presión reducida. El residuo resultante se purifica por cromatografía sobre gel de sílice para producir D-8 (6,58 g, 85 % de rendimiento).
Ejemplo 23: Preparación del intermedio ferc-butil éster del ácido 6-bromometil-8-fluoro-3,4-dihidro-1H-isoquinolin-2-carboxílico (D-9)

Una mezcla de 1-bromo-3-fluoro-5-metoxi-benceno (80 g, 0,39 mol), TEA (118 g, 1,17 mol), Pd(OAc)2 (16 g, 20 %) y DPPP (16 g, 20 %) en MeOH (800 ml) se agita en atmósfera de 3 Mpa de CO durante 2 días. La mezcla se filtra y el filtrado se concentra a presión reducida. El producto en bruto se purifica por cromatografía ultrarrápida sobre gel de sílice para proporcionar el compuesto D-9-1 (42 g, 59 % de rendimiento).
A una solución de D-9-1 (250 g, 1,4 mol) en THF (2000 ml), enfriada a -50 °C, se le añade LAH (77 g, 2,0 mol). La mezcla se calienta lentamente a 0 °C y se agita durante 3 h. Los reactivos en exceso se consumen mediante la adición de una solución acuosa de NH4Cl y la mezcla se extrae con EtOAc. La fase orgánica se seca sobre sulfato sódico y se concentra a presión reducida. El producto en bruto se purifica por cromatografía ultrarrápida sobre gel de sílice para proporcionar D-9-2 (100 g, 47 % de rendimiento).
Una mezcla de D-9-2 (50 g, 320 mmol), solución acuosa de HBr (200 ml) y tolueno (200 ml) se agita a temperatura ambiente durante 1 día. La reacción se diluye con H2O y se extrae con DCM. Las capas orgánicas se secan sobre sulfato sódico anhidro y se concentran a presión reducida. El producto en bruto se purifica por cromatografía ultrarrápida sobre gel de sílice para proporcionar D-9-3 (45 g, 64 % de rendimiento).
Una solución de D-9-3 (60 g, 270 mmol) y TMSCN (28 g, 300 mmol) en ACN (600 ml) se agita a temperatura ambiente durante 0,5 h. A esto se le añade TBAF (79 g, 300 mmol) y la mezcla de reacción se agita a temperatura ambiente durante una noche. La reacción se diluye con H2O y se extrae con DCM. La capa orgánica se seca sobre sulfato sódico anhidro y se concentra a presión reducida. El producto en bruto se purifica por cromatografía ultrarrápida sobre gel de sílice para proporcionar D-9-4 (36 g, 78 % de rendimiento).
Una mezcla de D-9-4 (12 g, 73 mmol), Ni (12 g), NH3.H2O (80 ml) y MeOH (80 ml) se agita en atmósfera de 206,843 kPa (30 Psi) de H2 a temperatura ambiente durante 5 h. La mezcla se filtra y el filtrado se concentra a presión reducida para proporcionar el producto en bruto que contiene D-9-5, que se usa directamente sin purificación adicional.
Una mezcla del producto en bruto que contiene D-9-5 (45 g, 270 mmol) y HCHO (7,25 g, 239 mmol) en HCO2H (500 ml) se agita a 50 °C durante una noche. El disolvente se elimina a presión reducida y el producto en bruto se purifica por cromatografía ultrarrápida sobre gel de sílice para proporcionar D-9-6 (40 g, 62 % de rendimiento en 2 etapas). Una solución de D-9-6 (40 g, 220 mmol) en una solución acuosa de HBr (400 ml) se agita a 90 °C durante 2 días. El disolvente se elimina a presión reducida y el producto en bruto se recoge en una solución acuosa saturada de NaHCO3, después se extrae con DCM. Las capas orgánicas se concentran a presión reducida y el producto en bruto se purifica por cromatografía ultrarrápida sobre gel de sílice para proporcionar D-9-7 (19 g, 52 % de rendimiento). Una mezcla de D-9-7 (38 g, 230 mmol), TEA (46 g, 450 mmol) y Boc2O (49,1 g, 227 mmol) en THF/H2O (1:1) (400 ml) se agita a temperatura ambiente durante 3 h. La reacción se diluye con H2O y se extrae con DCM. La capa orgánica se seca sobre sulfato sódico anhidro, se filtra y se concentra a presión reducida. El producto en bruto se purifica por cromatografía ultrarrápida sobre gel de sílice para proporcionar D-9-8 (28 g, 46 % de rendimiento).
Una mezcla de D-9-8 (14 g, 52 mmol), Tf2O (14,8 g, 52,4 mmol) y TEA (15,8 g, 157 mmol) en DCM (60 ml) se agita a temperatura ambiente durante 3 h. La reacción se diluye con H2O y se extrae con DCM. La capa orgánica se seca sobre sulfato sódico anhidro, se filtra y se concentra a presión reducida. El producto en bruto se purifica por cromatografía ultrarrápida sobre gel de sílice para proporcionar D-9-9 (10 g, 59 % de rendimiento).
Una mezcla de D-9-9 (18 g, 45 mmol), TEA (13,6 g, 135 mmol), Pd(OAc)2 (3,6 g) y DPPP (3,6 g) en MeOH (150 ml) se agita en atmósfera de 3 MPa de CO a 90 °C durante 2 días. La mezcla se filtra y el filtrado se concentra a presión reducida. El producto en bruto se purifica por cromatografía ultrarrápida sobre gel de sílice para proporcionar D-9-10 (11,8 g, 85 % de rendimiento).
A una solución de D-9-10 (11,8 g, 38,2 mmol) en THF (100 ml), enfriada a -50 °C, se le añade LAH (2,17 g, 57,3 mmol). Después, la mezcla de reacción se agita a 0 °C durante 3 h. Los reactivos en exceso se consumen mediante la adición de una solución acuosa saturada de NH4Cl. La mezcla se extrae con DCM y la capa orgánica se seca sobre sulfato sódico anhidro, se filtra y se concentra a presión reducida. El producto en bruto se purifica por cromatografía ultrarrápida sobre gel de sílice para proporcionar D-9-11 (9,8 g, 92 % de rendimiento).
A una solución de alcohol, D-9-11, (4,00 g, mmol) y piridina (2,25 ml, 21,3 mmol) en diclorometano (66 ml), a 0 °C, se le añade dibromuro de trifenilfosfina (9,00 g, 21,3 mmol). La reacción se agita durante 3 h, después se concentra a presión reducida. El residuo resultante se purifica por cromatografía sobre gel de sílice para producir D-9 (2,49 g, 49 % de rendimiento).
Ejemplo 24: Preparación del intermedio ferc-butil éster del ácido 6-bromometil-5-fluoro-8-metil-3,4-dihidro-1H-isoquinolin-2-carboxílico (D-10)

1-bromometil-2-fluoro-3-metoxi-5-metil-benceno (1,3 g, 5,4 mmol) y NaCN (0,29 g, 5,9 mmol) se combinan en DMF (15 ml), después se agitan a 45 °C durante 2 h. La mezcla se diluye con EtOAc/agua (100 ml/200 ml) y las capas se separan. La fase orgánica se seca sobre sulfato sódico anhidro, después se filtra y se concentra a presión reducida. El material en bruto se purifica por cromatografía ultrarrápida sobre gel de sílice para proporcionar D-10-1 (0,926 g, 96 % de rendimiento).
A una solución de D-10-1 (0,92 g, 5,2 mmol) en THF se le añade, gota a gota mediante una jeringa, una solución de complejo de borano-THF (1,0 M, 11 ml, 11 mmol). Una vez completada la adición, la mezcla se calienta a 55 °C y se agita durante una noche. La mezcla resultante se enfría a temperatura ambiente y los reactivos en exceso se consumen mediante la adición de agua (3 ml). Después de 5 min, se añade HCl conc. (3 ml). Después de agitar durante 1 h, se añaden agua (10 ml) y NaOH sólido hasta que la mezcla se hace alcalina. Después se añade DCM (50 ml) y las capas se separan con una frita hidrófoba. La fase orgánica se vuelve a secar sobre sulfato sódico anhidro, se filtra y se concentra a presión reducida. El residuo en bruto se purifica por cromatografía ultrarrápida de fase inversa usando una mezcla de MeCN/agua con ácido fórmico al 0,1 %. El eluyente se elimina a presión reducida y el producto aislado se destila azeotrópicamente con MTBE para proporcionar D-10-2 (0,777 g, 66 %) en forma de la sal de formiato.
Una mezcla de D-10-2 (0,775 g, 3,49 mmol) y CH2O (37% en H2O, 0,26 ml, 3,5 mmol) en HCOOH (10 ml) se agita a 60 °C durante 16 horas. El disolvente se elimina a presión reducida y el sólido en bruto se destila azeotrópicamente con tolueno para proporcionar el D-10-3 en bruto que no se purifica sino que se usa inmediatamente en la reacción siguiente.
El D-10-3 en bruto se suspende en una solución acuosa al 48 % de HBr (15 ml) y después se calienta a 95 °C y se agita durante una noche. La mezcla se enfría a temperatura ambiente, se concentra a presión reducida y después se destila azeotrópicamente con tolueno para proporcionar el D-10-4 en bruto, que no se purifica más, sino que se usa inmediatamente en la reacción siguiente.
El D-10-4 en bruto se suspende a temperatura ambiente en una 4:1 mezcla de DCM/DMF (25 ml) que contiene 4-DMAP (0,040 g, 0,3 mmol) y Et3N (2,1 ml, 15 mmol). A esta mezcla se le añade Boc2O (0,665 g, 3,04 mmol) en una porción. La mezcla se agita durante una noche, después se añade una solución saturada de NH4Cl (50 ml) y las capas se separan con una frita hidrófoba. La fase orgánica se concentra a presión reducida y el producto en bruto se purifica por cromatografía ultrarrápida sobre gel de sílice para proporcionar D-10-5. También se aísla una cantidad adicional del producto protegido N,O-Diboc. Este material se trata con LiOH (100 mg) en una mezcla de THF/MeOH/H2O (2:1:1,10 ml). La reacción de hidrólisis se concentra y se purifica por cromatografía ultrarrápida sobre gel de sílice para proporcionar más D-10-5. Las fracciones de producto combinadas se combinan para proporcionar D-10-5 (0,290 g, 30 %).
Una mezcla de D-10-5 (0,290 g, 1,03 mmol), 4-DMAP (13 mg, 0,11 mmol) y Et3N en DCM (8 ml) se enfría a 0 C y después se trata con Tf2O (0,21 ml, 1,2 mmol). La mezcla se deja calentar a temperatura ambiente y se agita durante una noche. La mezcla se diluye con una solución acuosa saturada de NaHCO3 (10 ml). Las capas se separan usando una frita hidrófoba y la fase orgánica se concentra a presión reducida. El residuo en bruto se purifica por cromatografía ultrarrápida sobre gel de sílice para proporcionar D-10-6 (0,35 g, 81 %).
Una mezcla del D-10-6 (0,29 g, 0,70 mmol), complejo ácido vinilborónico-piridina (0,18 g, 0,75 mmol) y una solución 2,0 M de Na2CO3 (0,70 ml, 1,4 mmol) en 1,2-Dm E (4 ml) se carga con Pd[P(Ph3)4] y después se irradia en un reactor de microondas a 120 °C durante 40 min. La mezcla se diluye con agua (5 ml) y DCM (15 ml). Después de un mezclado vigoroso, las capas se separan usando una frita hidrófoba. La capa orgánica se concentra a presión reducida y el producto en bruto se purifica por cromatografía ultrarrápida sobre gel de sílice para proporcionar D-10-7 que se usa inmediatamente en la reacción siguiente.
Una mezcla de D-10-7 y NalO4 (0,55 g, 2,6 mmol) en una mezcla 4:1 de THF:H2O (20 ml) se trata con una solución acuosa al 4 % en peso de OsO4 (0,34 ml, 0,04 mmol). La suspensión resultante se agita durante una noche a temperatura ambiente en ausencia de luz. La suspensión se filtra y el filtrado se concentran a presión reducida para eliminar los orgánicos volátiles. La fase acuosa restante se diluye con DCM (20 ml) y después se reparte usando una frita hidrófoba. La mezcla se concentra a presión reducida para proporcionar el D-10-8 en bruto que no se modifica sino que se usa inmediatamente en la reacción siguiente.
El D-10-8 en bruto se disuelve en una mezcla 1:1 de THF:MeOH (20 ml) y se trata con NaBH4 sólido (50 mg, 1,3 mmol). La mezcla se agita a temperatura ambiente durante 30 min, después se concentra a presión reducida. El residuo se diluye con DCM (20 ml) y una solución acuosa saturada de NH4Cl (40 ml). La solución se agita vigorosamente durante 15 min y después se separan las fases usando una frita hidrófoba. La fase orgánica se concentra a presión reducida y el residuo se purifica por cromatografía ultrarrápida sobre gel de sílice para proporcionar D-10-9 (0,174 g, 69 % en 3 etapas).
Una mezcla de D-10-9 (0,174 g, 0,589 mmol) y W,W-isopropiletilamina (0,18 ml, 1,0 mmol) en DCM (15 ml), enfriada a 0 °C, se trata con dibromotrifenolfosforano (0,39 g, 0,89 mmol) en una porción. La mezcla se agita a temperatura ambiente durante 1 hora, después se concentra a presión reducida. El residuo en bruto se purifica por cromatografía ultrarrápida sobre gel de sílice para proporcionar D-10 (0,210 g, 100 %).
Ejemplo 25: Preparación del intermedio ferc-butil éster del ácido 7-bromometil-6-metil-1,2,4,5-tetrahidrobenzo[d]azepin-3-carboxílico (D-11]

Una mezcla de D-3-3 (1608 g, 9,975 mol) y KOH (1117 g, 19,95 mol) en EtOH_(15 l) se calienta a reflujo durante 5 h. El disolvente se elimina a presión reducida. El pH del residuo se ajusta a pH 1. La mezcla se filtra y la torta de filtro se seca para producir D-11 -1 (1474 g, 86 % de rendimiento).
A una solución en agitación de (COCl)2 (8,18 mol) y DMF (70,000 ml) en DCM (7,5 l) se le añade D-11-1 (737 g, 4,09 mol). La mezcla se agita a temperatura ambiente durante 2 h, después se concentra a presión reducida. El residuo se añade a una solución en agitación de 2,2-dimetoxietil-1-amina (430 g, 4,09 mol) y t Ea (454 g, 4,50 mol) en DCM (1000 ml). La mezcla se agita a temperatura ambiente durante 2 h, después se concentra a presión reducida. El residuo se purifica por cromatografía ultrarrápida en columna sobre gel de sílice para proporcionar D-11 -2 (1474 g, 96 % de rendimiento).
Una solución de D-11-2 (1053 g, 3,939 mol) en una mezcla de AcOH (2 l) y ácido clorhídrico concentrado (2 l) se agita a temperatura ambiente durante 16 h. La mezcla se concentra a presión reducida. El residuo se cristaliza, se lava con agua y EtOH, se recoge por filtración y se seca para proporcionar D-11-3 (358 g, 45 % de rendimiento). Una mezcla de Pd/C (4 g) y D-11-3 (40,0 g, 0,197 mol) en AcOH (2 l) se agita a temperatura ambiente en atmósfera de H2 durante 16 h. La mezcla se filtra a través de tierra de diatomeas y se concentra a presión reducida. El residuo se recristaliza en EtOH y el sólido formado se recoge por filtración y se seca para dar D-11-4 (37 g, 92 % de rendimiento).
A una solución en agitación de D-11-4 (130 g, 0,633 mol) en THF (1300 ml) se le añade BMS_(127 ml, 1,27 mol), lentamente en atmósfera de N2 , mientras la temperatura se mantiene por debajo de -5 °C. La mezcla de reacción se agita durante 16 h. Los reactivos en exceso se consumen con más ácido clorhídrico concentrado y la mezcla se somete a reflujo durante 2 h. El disolvente se elimina a presión reducida y el residuo se diluye con agua y se lava con DCM. La fase acuosa se ajusta a pH = 9 y el sólido formado se recoge por filtración y se seca para proporcionar D-11-5 (37 g, 92 % de rendimiento).
Una solución de D-11-5 (220 g, 1,15 mol) en una solución acuosa al 48 % de HBr (1800 ml) se agita a 110 °C durante 4 h en atmósfera de N2. La mezcla se concentra a presión reducida para proporcionar el D-11-6 en bruto que se usa sin más purificación.
Una mezcla de D-11-6 (267 g, 1,51 mol), B0 C2O (492 g, 2,26 mol) y TEA (380 g, 3,77 mol) en diclorometano (2670 ml) se agita a temperatura ambiente durante 2 h. La mezcla de reacción se concentra a presión reducida y el residuo se purifica por cromatografía ultrarrápida en columna sobre gel de sílice para proporcionar D-11-7 (230 g, 64 % a partir del rendimiento de D-11-5).
Una mezcla del compuesto D-11-7 (267 g, 0,963 mol) y Tf2O (271 g, 0,963 mol) en DCM (2670 ml) se agita a temperatura ambiente durante 2 h en atmósfera de N2. La mezcla de reacción se concentra a presión reducida y el residuo se purifica por cromatografía ultrarrápida en columna sobre gel de sílice para proporcionar D-11-8 (220 g, 56 % de rendimiento).
Una mezcla de D-11-8 (20 g, 0,049 mol), dppp (2,0 g), Pd(OAc)2 (2,0 g) y TEA (9,9 g, 0,098 mol) en EtOH (400,000 ml) se agita a 80 °C durante 12 h en atmósfera de CO. La mezcla de reacción se concentra a presión reducida y el residuo se purifica por cromatografía ultrarrápida en columna sobre gel de sílice para proporcionar D-11-9 (8 g, 50 % de rendimiento).
A una solución en agitación de D-11-9 (22 g, 0,066 mol) en THF (300 ml), enfriada a -40 °C, se le añade lentamente LAH (2,5 g, 0,066 mol). Después de completar la adición, la mezcla se agita a temperatura ambiente durante 2 h. Los reactivos en exceso se consumen mediante la adición de agua. La mezcla se concentra a presión reducida y el residuo se vuelve a recoger en agua y se extrae con DCM. La fase orgánica se seca sobre sulfato sódico anhidro y se concentra a presión reducida. El residuo se purifica por cromatografía ultrarrápida en columna sobre gel de sílice para proporcionar D-11-10 (14 g, 71 % de rendimiento).
A una solución de alcohol, D-11-10, (19,0 g, 65,2 mmol) y N,N-diisopropiletilamina (13,0 ml, 74,6 mmol) en diclorometano (340 ml), a 0 °C, se le añade dibromuro de trifenilfosfina (30,0 g, 68,2 mmol). La reacción se agita durante 1 h, después se concentra a presión reducida. El residuo resultante se purifica por cromatografía sobre gel de sílice para producir D-11 (22,4 g, 97 % de rendimiento).
Ejemplo 26: Preparación del intermedio ferc-butil éster del ácido 3-hidroximetil-5,8-dihidro-6H-[1,7]naftiridin-7-carboxílico (D-12)

A una solución del éster metílico del ácido 5,6,7,8-tetrahidro-[1,7]naftiridin-3-carboxílico (232,8 mg, 1,018 mmol) y Boc anhídrido (379,4 mg, 1,738 mmol) a 0 °C en THF (3,4 ml) se le añade TEA (0,500 ml). Se añade DCM (1,0 ml) y la mezcla de reacción se agita a temperatura ambiente durante una noche. La mezcla de reacción se diluye con agua y la fase acuosa se extrae con EtOAc. Los extractos orgánicos combinados se lavan con salmuera, se secan sobre sulfato sódico anhidro y se concentran a presión reducida. El material en bruto se purifica por cromatografía ultrarrápida en columna sobre gel de sílice para proporcionar D-12-1 (0,169 g, 57 % de rendimiento).
A una solución a 0 °C del éster de partida (0,169 g, 0,576 mmol) en THF (5 ml) se le añade DiBAl-H 1,0 M en tolueno (3,4 ml, 3,4 mmol) durante el lapso de 15 min. El baño de hielo se retira aproximadamente 2 h después. La mezcla de reacción se deja calentar de manera gradual a temperatura ambiente y se mantiene a temperatura ambiente durante las 2 h siguientes. Finalmente, la mezcla de reacción se enfría a 0 °C y se introduce sal de Rochelle (6 ml). La mezcla heterogénea resultante se deja calentar a temperatura ambiente y se agita a esta temperatura (durante el periodo del fin de semana). Después, la mezcla se diluye con agua y EtOAc. La fase acuosa se extrae con EtOAc (x3). Los extractos orgánicos combinados se lavan con salmuera, se secan (Na2SO4 ), se filtran y se concentran a presión reducida para dar D-12-2, que no se purifica sino que se usa directamente en la reacción siguiente.
A una solución a 0 °C de D-12-2 (0,261 mg, 0,987 mmol) en DCM se le añade NBS (0,211 g, 1,19 mmol), seguido de PPh3 (0,311 g, 1,19 mmol). La mezcla de reacción se deja en agitación a 0 °C durante 1 h, después se concentra a
presión reducida sin calentamiento. El material en bruto se purifica por cromatografía ultrarrápida sobre gel de sílice para proporcionar D-12 (0,22 g, 68 % de rendimiento) en forma de un sólido de color blanco.
Ejemplo 27: Preparación del intermedio 1-oxo-2-(tetrahidro-piran-4-il)-1,2,3,4-tetrahidro-isoquinolin-6-ilmetil éster del ácido metanosulfónico (D-13)

A un matraz cargado con etanol (180 ml), enfriado a 0 °C, se le añade 4,0 ml (56 mmol) de cloruro de acetilo. La mezcla se agita a 0 °C durante 30 min, después se añaden 5,00 g (18,0 mmol) de 2-íerc-butil éster del ácido 3,4-dihidro-1H-isoquinolin-2,6-dicarboxílico. La mezcla se calienta a 70 °C y se agita durante 2 días. La mezcla se enfría a temperatura ambiente y se filtra a través de tierra de diatomeas. El filtrado se concentra a presión reducida para proporcionar D-13-1 (3,47 g, 79,6 %) en forma de un polvo de color blanco.
A una solución de 3,45 g (14,3 mmol) de D-13-1 en DCM (150 ml) se le añade 2,0 g (20 mmol) de tetrahidro-piran-4-ona. La mezcla se agita a temperatura ambiente durante 30 min, después se añaden 12 g (56 mmol) de triacetoxiborohidruro sódico. La mezcla se agita a temperatura ambiente durante 4 días, después se diluye con una solución acuosa saturada de bicarbonato sódico. La mezcla se separa y la fase acuosa se extrae con DCM. La fase orgánica combinada se seca sobre sulfato sódico anhidro y se concentra a presión reducida. El residuo se purifica por cromatografía ultrarrápida sobre gel de sílice para proporcionar D-13-2 (2,51 g, 45 %).
A una solución de 2,51 g (8,67 mmol) de D-13-2 en una mezcla 4:1 de 1,1,2,2,-tetracloroetano:agua se le añade 2,4 g (26 mmol) de clorito de sodio. La mezcla se calienta durante una noche a 55 °C, después se enfría a temperatura ambiente. Los reactivos en exceso se consumen mediante la adición de una solución de bisulfito sódico al 10 %. La mezcla se diluye con agua y se extrae con DCM. La fase orgánica combinada se lava con una solución 2 N de HCl, se seca sobre sulfato sódico anhidro y se concentra a presión reducida. El residuo se purifica por cromatografía ultrarrápida sobre gel de sílice para proporcionar D-13-3 (0,64 g, 24 %).
A una solución de 0,640 g (2,11 mmol) de D-13-3 en THF (20 ml) se le añade 2,5 ml (5,0 mmol) de borohidruro de litio en forma de una solución 2 M en THF. La mezcla se agita durante una noche a temperatura ambiente, después los reactivos en exceso se consumen mediante la adición lenta de agua. La mezcla se diluye con agua y se extrae con EtOAc. La fase orgánica combinada se lava con salmuera, se seca sobre sulfato sódico anhidro y se concentra a presión reducida. El residuo se purifica por cromatografía ultrarrápida sobre gel de sílice para proporcionar D-13-4 (0,090 g, 16 %)
A una solución de 0,090 g (0,34 mmol) de D-13-4 en DCM (5 ml) se le añade 0,070 g (0,40 mmol) de metanosulfónico anhídrido seguido de 0,075 ml (0,43 mmol) de DIEA. La mezcla se agita durante una noche a
temperatura ambiente, después se lava con agua, se seca sobre sulfato sódico anhidro y se concentra a presión reducida para proporcionar D-13 0,16 g (100 %) en forma de un aceite transparente que se usa directamente sin más purificación.
Ejemplo 28. Preparación de los intermedios ferc-butil éster del ácido 5-bromometil-4-metil-1,3-dihidro-isoindolo-2-carboxílico (D-14) y ferc-butil éster del ácido 6-bromometil-4-metil-1,3-dihidro-isoindolo-2-carboxílico (D-15)

A una solución en agitación de Boc-propargilamina (2,00 g, 12,9 mmol) en THF (30,0 ml) y yoduro de tetrabutilamonio (0,476 g, 1,29 mmol) se le añade una solución 0,5 M de KHMDS (25,8 ml, 12,9 mmol) en THF y la mezcla se agita durante 30 min a temperatura ambiente. El bromuro (1,69 ml, 19,3 mmol) se añade gota a gota y la mezcla se agita durante 30 min a temperatura ambiente y después se somete a reflujo durante 2 h. La reacción se diluye con NH4Cl saturado y se extrae con EtOAc. Los extractos orgánicos combinados se secan con MgSO4 y se concentran a presión reducida. El material en bruto se purifica por cromatografía ultrarrápida sobre gel de sílice para proporcionar D-14-1 (2,13 g) en forma de un aceite incoloro.
Se añade alcohol propargílico (2,39 ml, 41,1 mmol) gota a gota a 0 °C a una solución de D-14-1 (2,13 g, 10,28 mmol) en etanol anhidro (50,0 ml). Se añade catalizador de Wilkinson (0,95 g, 1,0 mmol) y la mezcla se agita durante una noche a temperatura ambiente. La mezcla de reacción en bruto se concentra a presión reducida y el residuo se purifica por cromatografía ultrarrápida sobre gel de sílice para proporcionar una mezcla de D-14-2 y D-15-1 (1,93 g). La mezcla no se separa sino que se lleva a la etapa siguiente.
A una solución de la mezcla que contiene D-14-2 y D-15-1 (1,93 g, 7,33 mmol) y N,N-diisopropiletilamina (1,91 ml, 11,0 mmol) en diclorometano (50,0 ml), a 0 °C, se le añade dibromuro de trifenilfosfina (4,73 g, 11,0 mmol). La reacción se agita durante 2 h, después se concentra a presión reducida. El residuo se purifica por cromatografía ultrarrápida sobre gel de sílice para producir la mezcla de regioisómeros D-14 y D-15 (2,12 g) en forma de un sólido de color blanco.
Ejemplo 29: Preparación de los intermedios ferc-butil éster del ácido (R)-8-bromometil-1-metil-1,2,4,5-tetrahidrobenzo[d]azepin-3-carboxílico (D-17) y ferc-butil éster del ácido (S)-8-bromometil-1-metil-1,2,4,5-tetrahidrobenzo[d]azepin-3-carboxílico (D-18).

A una solución de alcohol de 4-metoxifenetilo (2,50 g, 16,4 mmol) en DCM (20,0 ml) se le añade Et3N (2,75 ml, 19,7 mmol) seguido de cloruro de metanosulfonilo (1,53 ml, 19,7 mmol). La mezcla se agita a temperatura ambiente durante una noche, después se extrae con DCM, se lava con salmuera, se seca sobre MgSO4 y se concentra a presión reducida para proporcionar 17-1 (3,75). El material se lleva más adelante sin más purificación.
El 17-1 en bruto (3,75 g, 16,3 mmol) se trata con 1-amino-2-propanol puro (20 ml) y se calienta a reflujo durante 3 h. La mezcla se diluye con agua (50 ml) y se extrae con EtOAc. Los orgánicos combinados se lavan con salmuera, se secan con Na2SO4, se filtran y se concentran a presión reducida. La amina en bruto se disuelve en DCM (20 ml) y se añade HCl 2,0 M en éter (5 ml, 10 mmol) para formar un precipitado de color blanco. El sólido formado es el que se recoge por filtración y se seca en la almohadilla del filtro para proporcionar 17-2 (2,63 g), que se usa sin más purificación.
A una solución de 17-2, (2,63 g, 12,5 mmol) en DCM (60 ml), a 0 °C, se le añade dimetil formamida (0,49 ml, 6,3 mmol) seguida de bromuro de tionilo (1,26 ml, 16,3 mmol). La mezcla se agita durante 14 h mientras se calienta a 20 °C. Se añade éter dietílico frío (30 ml, 0 °C) y la reacción se enfría a 0 °C causando que un sólido precipite de la solución. El sólido formado se recoge por filtración y se seca en la almohadilla del filtro para producir 17-3 en forma de un sólido de color blanquecino (3,31 g).
A un matraz que contiene 17-3 (1,00 g, 3,67 mmol) se le añade cloruro de aluminio (0,882 g, 4,40 mmol). La mezcla se calienta a 150 °C durante 20 h. Mientras la reacción está aún templada, se añade agua (20 ml), después de 5 min
se añade EtOAc:DCM y la reacción se deja enfriar a 20 °C con agitación. A esto se le añade NaHCO3 saturado (25 ml) para dar una emulsión. Las capas se separan. A la capa acuosa se le añade tetrahidrofurano (50 ml) y ditercbutildicarbonato y la mezcla se agita durante una noche. La reacción se reparte entre EtOAc y ácido cítrico saturado. Las capas se separan y las orgánicas se lavan con salmuera, se secan sobre MgSO4, se filtran y se concentran a presión reducida para formar 17-4 en forma de un sólido de color blanco (0,625 g).
A una solución de 17-4 (0,625 g, 2,25 mmol) en DCM (20,0 ml) a temperatura ambiente se le añade piridina (0,36 ml, 4,5 mmol). La solución se enfría a -30 °C y se añade anhídrido trifluorometanosulfónico (0,42 ml, 2,5 mmol) gota a gota. La reacción se agita a -30 °C durante 1 hora, después se deja calentar a temperatura ambiente. Se concentra a presión reducida. El residuo se diluye con EtOAc y se lava con HCl 1 N seguido de NaHCO3 saturado y salmuera. La mezcla se seca sobre MgSO4 y se concentra a presión reducida. El material resultante se purifica por cromatografía ultrarrápida sobre gel de sílice para proporcionar 17-5 (0,850 g).
El triflato (0,850 g, 2,08 mmol) se combina con el boronato (0,600 g, 2,49 mmol) y Pd(PPh3)4 (0,12 g, 0,11 mmol) en una mezcla de DME (15,0 ml) y Na2CO32,0 M (1,09 ml). La reacción se calienta en un reactor de microondas a 120 °C durante 40 minutos. La reacción se concentra y se purifica por cromatografía ultrarrápida sobre gel de sílice para proporcionar 17-6 en forma de un aceite (0,519 g).
A una solución de 17-6 (0,519 g, 1,81 mmol) en una mezcla de THF (7,0 ml) y H2O (1,50 ml) se le añade NalO4 (1,18 g, 5,52 mmol). La mezcla se agita a temperatura ambiente durante una noche en la oscuridad, después se diluye con una mezcla de agua y DCM. Las capas se separan con una frita hidrófoba y la orgánica se seca sobre MgSO4, después se filtra y se concentra a presión reducida. El residuo se purifica por cromatografía ultrarrápida sobre gel de sílice para proporcionar 17-7 en forma de un aceite oscuro (0,390 g).
A una solución de 17-7 (0,390 g, 1,35 mmol) en una mezcla de THF (5 ml) y MeOH (5 ml), enfriada a 0 °C, se le añade NaBH4 (0,077 g, 2,0 mmol). La reacción se calienta a temperatura ambiente y se agita durante 30 min. La reacción se diluye con una solución ac. de NH4Cl y se agita durante 10 minutos. La mezcla se extrae con EtOAc y la fase orgánica combinada se lava con NH4Cl seguido de salmuera, después se seca sobre MgSO4 y se concentra a presión reducida. El material resultante se purifica por cromatografía ultrarrápida sobre gel de sílice para proporcionar 17-8 (0,326 g).
El 17-8 racémico se resuelve en una ChiralCel 10 u (300 x 50 mm) usando IPA al 20 % en CO2 supercrítico a 200 ml/min a 10 MPA (100 bar) de presión a 38 °C para proporcionar 17-9 (primer pico de elución) y 18-1 (segundo pico de elución). La estereoquímica absoluta no se establece y las estructuras se dibujan de manera arbitraria.
A una solución de 17-9 (1,58 g, 5,44 mmol) en DCM (30 ml), enfriado a 0 °C, se le añade N,N-diisopropiletilamina (1,42 ml, 8,16 mmol) seguida de dibromuro de trifenilfosfina (3,514 g, 8,159 mmol). La reacción se agita durante 2 h y se concentra a presión reducida. El residuo resultante se purifica por cromatografía ultrarrápida sobre gel de sílice para proporcionar el compuesto del título D-17 (1,79 g).
A una solución de 18-1 (1,64 g, 5,64 mmol) en DCM (30 ml), enfriado a 0 °C, se le añade N,N-diisopropiletilamina (1,47 ml, 8,45 mmol) seguido de dibromuro de trifenilfosfina (3,64 g, 8,45 mmol) a 0 °C. La reacción se agita durante 2 h y se concentra a presión reducida. El residuo resultante se purifica por cromatografía ultrarrápida sobre gel de sílice para proporcionar el compuesto del título D-18 (1,86 g).
El intermedio siguiente se sintetiza de una manera similar a partir de los reactivos apropiados:

Ejemplo 30: Preparación del intermedio ferc-butil éster del ácido 7-bromometil-6-fluoro-1,2,4,5-tetrahidrobenzo[d]azepin-3-carboxílico (D-20).

A una solución de 2-fluoro-3-metoxibenzaldehído (20,0 g, 130 mmol) en una mezcla de THF (100 ml) y MeOH (50 ml), enfriada a 0 °C, se le añade NaBH4 (7,40 g, 195 mmol). La reacción se calienta a temperatura ambiente y se agita durante 30 minutos. La mezcla se diluye con NH4Cl ac. y se extrae con EtOAc. Los extractos combinados se lavan con NH4Cl, salmuera, se secan sobre MgSO4 y se concentran a presión reducida. El material resultante se purifica por cromatografía ultrarrápida sobre gel de sílice para proporcionar D-20-1 (21 g).
A una solución en agitación y enfriada (-10 °C) de D-20-1 (20,0 g, 128 mmol) en DCM (140 ml) se le añade SOCl2 (18,5 ml, 256 mmol). Después de la adición, la solución se calienta a reflujo durante 6 h, después se concentra para proporcionar D-20-2 (23 g), que se usa directamente en la etapa siguiente sin más purificación.
A una solución de D-20-2 (22 g, 126 mmol) en DMF (80 ml) se le añade NaCN (7,4 g, 150 mmol) y la mezcla se agita a temperatura ambiente durante una noche. La mezcla se diluye con H2O y se extrae con EtOAc. Los extractos combinados se lavan con H2O seguida de salmuera, después se concentran a presión reducida. El residuo se purifica dos veces por cromatografía ultrarrápida para proporcionar D-20-3 (15,5 g).
A una solución de D-20-3 (15,4 g, 93 mmol) en EtOH (100 ml) se le añade KOH (12,3 g, 186 mmol) y la mezcla se somete a reflujo durante una noche. El disolvente se evapora y el residuo se diluye con H2O. La mezcla se acidifica con HCl concentrado a pH = 1 causando que se forme un precipitado. El precipitado se recoge por filtración. El sólido recogido se cristaliza por _. El sólido se recoge por filtración y la torta de filtro se lava con H2O fría y se seca al vacío a 40 °C durante una noche para proporcionar D-20-4 (11,5 g).
A una solución en agitación y enfriada (0 °C) de DMF (0,50 ml) en DCM (50 ml) se le añade, gota a gota, cloruro de oxalilo (4,6 ml, 54 mmol). El baño de refrigeración se retira después de la adición y la agitación continúa durante 10 min. A esta mezcla se le añade, en múltiples porciones, D-20-4 (5,0 g, 27 mmol). La agitación continúa durante 2,5 h más, después el disolvente se evapora para proporcionar D-20-5 (5,7 g), que se usa directamente en la etapa siguiente.
A una solución en agitación de D-20-5 (2,50 g, 13,6 mmol) en DMF (50 ml) se le añade sucesivamente DIEA (5,9 ml, 34 mmol), HATU (6,4 g, 16 mmol) y amina (1,7 ml, 16 mmol). La mezcla se agita a temperatura ambiente durante una noche. El disolvente se elimina a presión reducida y el residuo se purifica por cromatografía ultrarrápida sobre gel de sílice para proporcionar D-20-6 (2,1 g).
Una mezcla de D-20-6 (1,6 g, 5,9 mmol) en H2SO4 concentrado (6,60 ml, 118 mmol) se agita a temperatura ambiente durante 1 h, después se vierte sobre hielo y se neutraliza con Na2CO3. La mezcla se extrae con EtOAc y los extractos combinados se concentran a presión reducida. El residuo se purifica por cromatografía ultrarrápida sobre gel de sílice para proporcionar D-20-7 (0,530 g).
Una mezcla de D-20-7 (2,4 g, 11 mmol) y Pd al 10 %/C (0,200 g) en ácido acético (10 ml) se agita durante una noche en atmósfera de hidrógeno. La mezcla se filtra a través de Celite y el filtrado se concentra a presión reducida para proporcionar D-20-8 (2,5 g) que se usa directamente en la etapa siguiente sin más purificación.
A una solución en agitación y enfriada (0 °C) de D-20-8 (2,4 g, 11 mmol) en THF (40 ml) se le añade, gota a gota, una solución de borano en THF (11 ml, 2,0 M, 22 mmol). Después de la adición, la solución se agita durante 15 h y después la solución se calienta a reflujo durante 2 h. La solución se enfría a temperatura ambiente y se añade lentamente una solución al 10 % de HCl (20 ml). La mezcla se somete a reflujo durante 2 h más, después se enfría a temperatura ambiente. El disolvente se concentra a presión reducida y el residuo se lava con éter, después el pH se ajusta a pH 9 mediante la adición de una solución al 10 % de NaOH. La mezcla se extrae con DCM, los extractos combinados se secan (Na2SO4) y se concentran a presión reducida para proporcionar D-20-9 (1,7 g), que se usa directamente en la etapa siguiente.
Una mezcla de D-20-9 (1,5 g, 7,7 mmol) en HBr al 48 % se calienta a 100 °C durante 3 h. Después de enfriar a temperatura ambiente, el disolvente se concentra a presión reducida para proporcionar D-20-10 (2,1 g) que se usa directamente en la etapa siguiente.
A una solución en agitación y enfriada (0 °C) de D-20-10 (2,1 g, 12 mmol) en DCM se le añade sucesivamente DIEA (6,4 ml, 35 mmol) y Boc anhídrido (3,0 g, 14 mmol). La mezcla se agita durante 3 h, después el disolvente se concentra a presión reducida y el residuo se purifica por cromatografía ultrarrápida sobre gel de sílice cromatografía para proporcionar D-20-11 (1,2 g).
A una solución en agitación y enfriada (0 °C) de D-20-11 (1,0 g, 3,5 mmol) en DCM (10 ml) se le añade sucesivamente TEA (1,2 ml, 8,9 mmol) y Tf2O (0,7 ml, 4,3 mmol). La mezcla se agita a 0 °C durante 2 h después se diluye con una solución saturada de NaHCO3 y se extrae con EtOAc. Los extractos combinados se lavan con NaHCO3 saturado, seguido de salmuera, después se secan (Na2SO4) y se concentran a presión reducida para proporcionar D-20-12, que se usa directamente en la etapa siguiente.
Una mezcla del D-20-12 en bruto, Pd(OAc)2 (0,082 g, 0,37 mmol), dppp (0,15 g, 0,36 mmol) en una mezcla de MeOH (6,0 ml) y DMSO (9,0 ml) se lava abundantemente con CO durante 5 min. A esta mezcla se le añade TEA (1,5 ml, 11 mmol). La mezcla se calienta a 70 °C durante una noche en atmósfera de CO. La mezcla se enfría a temperatura ambiente y los orgánicos volátiles se eliminan a presión reducida. El residuo se purifica por cromatografía ultrarrápida sobre gel de sílice para proporcionar D-20-13 (0,710 g).
A una solución en agitación y enfriada (-78 °C) de D-20-13 (0,71 g, 2,2 mmol) en DCM (20 ml) se le añade una solución de Dibal-H (6,6 ml, 1,0 M, 6,6 mmol). Después de 20 min de agitación, el baño de refrigeración se retira y la agitación continúa durante 3 h. A esta mezcla se le añade MeOH seguido de Na2SO4.12H2O. La agitación continúa durante 2 h, después la mezcla se filtra a través de un lecho de Celite y la almohadilla del filtro se aclara con MeOH/DCM al 10 %. El filtrado se concentra a presión reducida. El residuo se purifica por cromatografía ultrarrápida sobre gel de sílice para proporcionar D-20-14 (0,325 g).
Una mezcla de D-20-14 (0,32 g, 1,1 mmol) y DIEA (0,28 ml, 1,6 mmol) en DCM (10 ml) se enfría a -30 °C. A esta se le añade Ph3PBr2 (0,595 g, 1,30 mmol) en una porción. Después de agitar durante 1 h a esta temperatura la solución se calienta lentamente hasta 0 °C durante 1 h. La reacción se concentra a presión reducida y el residuo sólido se diluye con DCM para dar una suspensión, que se purifica por cromatografía ultrarrápida sobre gel de sílice para proporcionar el compuesto del título D-20 (0,351 g).
El intermedio siguiente se sintetiza de una manera similar a partir de los reactivos apropiados:

E je m p lo 31: P rep a ra c ión de l in te rm e d io fe rc -bu til és te r de l ác ido 6 -b ro m o m e til-8 -m e to x im e til-3 ,4 -d ih id ro -1 H -is o q u in o lin -2 -ca rb o x ílico (D -21).

A una mezcla de D-6-6 (60,0 g, 183 mmol) y TEA (55 g, 550 mmol) en DCM (600 ml) se le añade Tf2O (51,6 g, 183 mmol). La mezcla se agita a temperatura ambiente durante 3 h, después se diluye con H2O y se extrae con DCM. La capa orgánica se seca sobre Na2SO4, se filtra y se concentra a presión reducida. El residuo se purifica por cromatografía ultrarrápida sobre gel de sílice para proporcionar D-21-1 (60 g).
Una mezcla de D-21-1 (60 g, 130 mmol), TEA (39,5 g, 391 mmol), Pd(OAc)2 (12 g) y DPPP (12 g) en MeOH seco (600 ml) se agita en atmósfera de 344,738 kPa (50 psi) de CO a 65 °C durante 4 h. La mezcla se filtra y el filtrado se concentra a presión reducida. El residuo se purifica por cromatografía ultrarrápida sobre gel de sílice para proporcionar D-21-2 (40 g).
A una solución de D-21-2 (40.0 g, 108 mmol) en THF (400 ml), enfriada a -50 °C, se le añade LAH (6,1 g, 160 mmol). La mezcla se agita a -50 °C durante 3 h. Los reactivos en exceso se consumen mediante la adición de NH4Cl acuoso saturado. La mezcla se extrae con EtOAc. La capa orgánica se seca, se filtra y se concentra a presión reducida. El residuo se purifica por cromatografía ultrarrápida sobre gel de sílice para proporcionar D-21-3 (25 g).
A una solución de D-21-3 (25 g, 73 mmol) en DCM (250 ml), a 0 °C, se le añade NaH (4,38 g, 110 mmol, dispersión al 60 % en aceite mineral). A esta mezcla se le añade PMBBr (16,1 g, 80,4 mmol). La mezcla se calienta a temperatura ambiente y se agita durante 1 h. Los reactivos en exceso se consumen mediante la adición de solución acuosa saturada de NH4Cl. La mezcla se extrae con DCM. La capa orgánica se seca, se filtra y se concentra a presión reducida. El residuo se purifica por cromatografía ultrarrápida sobre gel de sílice para proporcionar D-21-4 (27 g).
Una mezcla de D-21-4 (27 g, 58 mmol), TEA (17,7 g, 175 mmol), Pd(OAc)2 (5,4 g) y DPPP (5,4 g) en MeOH seco (300 ml) se agita en atmósfera de 344,738 kPa (50 psi) de CO a 65 °C durante 3 días. La mezcla se filtra y el filtrado se concentra a presión reducida. El residuo se purifica por cromatografía ultrarrápida sobre gel de sílice para proporcionar D-21-5 (20 g).
A una solución de LAH (2,6 g, 68 mmol) en THF (60 ml), a -50 °C, se le añade gota a gota una solución de D-21-5 (20,0 g, 45,3 mmol) en THF (130 ml) durante 30 min. Después de la adición, la mezcla de reacción se agita a 0 °C durante 4,5 h. La mezcla de reacción se trata con una mezcla de NH4Cl acuoso saturado y DCM. La capa orgánica se separa, se lava con salmuera, se seca sobre Na2SO4, se filtra y se concentra a presión reducida. El residuo se purifica por cromatografía ultrarrápida sobre gel de sílice para proporcionar D-21-6 (14 g).
A una suspensión de NaH (0,43 g, dispersión al 60 % en aceite mineral, 10,6 mmol) en DMF (15 ml) se le añade D
21-6 (4,0 g, 9,7 mmol), seguido de Mel (0,80 ml, 13 mmol). La mezcla se agita a 20 °C durante 16 horas. Se añade agua y la mezcla se extrae con EtOAc. Las capas orgánicas se lavan con salmuera, se secan (Na2SO4) y se concentran a presión reducida. El residuo se purifica por cromatografía ultrarrápida sobre gel de sílice para proporcionar D-21-7 (2,7 g).
Una mezcla de D-21-7 (3,8 g, 8,9 mmol) y TFA (6,7 ml, 89 mmol) en DCM (20 ml) se agita a 0 °C durante 3 h. La reacción se diluye con NaHCO3 acuoso y se extrae con EtOAc. La fase orgánica combinada se seca (Na2SO4) y se concentra a presión reducida para proporcionar D-21-8, que se usa directamente en la etapa siguiente.
A una solución del D-21-8 en bruto en DCM (20 ml), a 0 °C, se le añade DIEA (2,5 ml, 14 mmol) seguida de Boc2O (1,9 g, 9 mmol). La solución se agita a 0 °C durante 2 h, después se concentra a presión reducida. El residuo se purifica por cromatografía ultrarrápida sobre gel de sílice para proporcionar D-21-9 (1,1 g)
A una solución de D-21-9 (1,1 g, 4,0 mmol) en DCM, enfriada a -30 °C, se le añade DIEA (0,90 ml, 5,4 mmol) seguida de Ph3PBr2 (2,0 g, 4,5 mmol) en una porción. La mezcla se agita a -30 °C durante 1 h, después se calienta durante un periodo de 1 hora hasta 0 °C. La mezcla se concentra a presión reducida y el residuo resultante se diluye con DCM para dar una suspensión que se purifica por cromatografía ultrarrápida sobre gel de sílice para proporcionar el compuesto del título D-21 (1,1 g).
Ejemplo 32: Preparación del intermedio ferc-butil éster del ácido 8-bromometil-6-metil-2,3-dihidro-5H-benzo[f][1,4]oxazepin-4-carboxílico (D-22).

A la mezcla del compuesto 5-metil-bencen-1,3-diol (200 g, 1,61 mol) y K2CO3 (448 g, 3,22 mol) en DMF (2000 ml) se le añade BnBr (248 g, 1,45 mol) gota a gota a temperatura ambiente. La mezcla se agita durante 12 h después se diluye con H2O y se extrae con EtOAc. La capa orgánica se secó, se filtró y se evaporó a presión reducida. El producto en bruto se purifica por cromatografía sobre gel de sílice para proporcionar D-22-1 (137,5 g).
A una solución del compuesto D-22-1 (220 g, 1,02 mol) en DCM (2200 ml), a -50 °C en atmósfera de N2, se le añade
NBS (146 g, 0,82 mol). La mezcla se agita durante 0,5 h a -50 °C después se diluye con H2O y se extrae con DCM. La capa orgánica se secó, se filtró y se evaporó a presión reducida. El producto en bruto se purifica por cromatografía sobre gel de sílice para proporcionar D-22-2 (108 g).
A la solución de D-22-2 (108 g, 0,37 mol) y DIPEA (143 g, 1,10 mol) en DCM (1000 ml), a 0 °C, se le añade SEMCl (74 g, 0,44 mol) gota a gota. La mezcla se agita a temperatura ambiente durante 3 h, después se diluye con H2O y se extrae con DCM. La capa orgánica se seca, se filtra y se evapora a presión reducida. El producto en bruto se purifica por cromatografía sobre gel de sílice para proporcionar D-22-3 (101 g).
A una solución de D-22-3 (110 g, 0,24 mol) en THF (1000 ml), a -78 °C en atmósfera de N2 , se le añade n-BuLi (120 ml, 0,29 mol) gota a gota. La mezcla se agita durante 0,5 h a -78 °C y después se añade DMF (26 g, 0,36 mol) a la mezcla gota a gota. La mezcla se agita durante 1,5 h después se diluye con solución ac. NH4Cl y se extrae con EtOAc. La capa orgánica se seca, se filtra y se evapora a presión reducida. El producto en bruto se purifica por cromatografía sobre gel de sílice para proporcionar D-22-4 (53 g).
Una solución de D-22-4 (106,5 g, 0,30 mol) y CBr4 (100 g, 0,30 mol) en /-PrOH (1000 ml) se agita a 80 °C durante 3 h. El disolvente se elimina a presión reducida y el producto en bruto se purifica por cromatografía sobre gel de sílice para proporcionar D-22-5 (52 g).
Una solución de D-22-5 (50 g, 0,21 mol), ferc-butil éster del ácido (2-bromo-etil)-carbámico (46 g, 0,21 mol), y Cs2COa (203 g, 0,63 mol) en DMF (500 ml) se agita a temperatura ambiente durante 10 min en atmósfera de N2. Después, la mezcla se agita a 80 °C durante 12 h. El disolvente se elimina a presión reducida y el producto en bruto se purifica por cromatografía sobre gel de sílice para proporcionar D-22-6 (60,5 g).
A una solución de D-22-6 (60 g, 0,16 mol) en DCM (600 ml) se le añade TFA (100 g) gota a gota en atmósfera de N2. La mezcla se agita a temperatura ambiente durante 1 h después se diluye con H2O y se extrae con EtOAc. La capa orgánica se secó, se filtró y se evaporó a presión reducida. El producto en bruto se purifica por cromatografía sobre gel de sílice para proporcionar D-22-7 (28,3 g).
Una mezcla de D-22-7 (28,3 g, 0,11 mol) y Pd-C (seco, 5 g) en MeOH (250 ml) se agita a temperatura ambiente en atmósfera de H2 (344,738 kPa (50 psi)) durante 8 h. La reacción se filtra y el disolvente se evapora a presión reducida para proporcionar D-22-8 (19 g), que se usa directamente sin más purificación.
Una solución de D-22-8 (19 g, 0,087 mol), TEA (26,4 g, 0,26 mol) y Boc2O (15,6 g, 0,087 mol) en DCM (200 ml) se agita a temperatura ambiente durante 0,5 h. La reacción se diluye con H2O y se extrae con DCM. La capa orgánica se seca, se filtra y se evapora a presión reducida. El producto en bruto se purifica por cromatografía sobre gel de sílice para proporcionar D-22-9 (13,3 g).
A una solución de D-22-9 (11 g, 39 mmol) y TEA (11,9 g, 118 mmol) en DCM (110 ml) se le añade Tf2O (11,1 g, 39 mmol) gota a gota a temperatura ambiente en atmósfera de N2. La mezcla se agita a temperatura ambiente durante 3 h, después se diluye con H2O y se extrae con DCM. La capa orgánica se seca, se filtra y se evapora a presión reducida. El producto en bruto se purifica por cromatografía sobre gel de sílice para proporcionar D-22-10 (13,1 g).
Una mezcla de D-22-10 (13,1 g, 32 mmol), TEA (9,7 g, 96 mmol), Pd(OAc)2 (2,6 g, 20%) y DPPP (2,6 g, 20%) en MeOH (130 ml) se agita en atmósfera de CO (3 MPa) a 90 °C durante 2 días. La mezcla se filtra y el filtrado se evapora a presión reducida. El producto en bruto se purifica por cromatografía sobre gel de sílice para proporcionar D-22-11 (9,2 g).
A una solución de LAH (1,6 g, 43 mmol) en THF (46 ml), a -50 °C, se le añade una solución de D-22-11 (9,2 g, 29 mmol) en THF (46 ml) gota a gota durante 30 min. Después de la adición, la mezcla de reacción se agita a 0 °C durante 4,5 h. La mezcla de reacción se diluye con H2O y se extrae con DCM. La capa orgánica se seca, se filtra y se evapora a presión reducida. El producto en bruto se purifica por cromatografía sobre gel de sílice para proporcionar D-22-12 (6,7 g).
A una solución de D-22-12 (1,61 g, 5,48 mmol) en diclorometano (30 ml), a 0 °C, se le añade DIEA (1,4 ml, 8,2 mmol). A esta solución se le añade dibromuro de trifenilfosfina (3,54 g, 8,21 mmol) en lotes (x4) durante el lapso de 10 minutos. La reacción se mantiene a 0 °C durante aproximadamente 2 h, después el baño de hielo se retira y la mezcla de reacción se deja calentar a temperatura ambiente durante el lapso de 1,5 h más. La mezcla de reacción se concentra a presión reducida y el residuo se purifica por cromatografía ultrarrápida sobre gel de sílice para proporcionar el compuesto del título, D-22 (1,73 g) en forma de un sólido de color blanco.
E je m p lo 33: P rep a ra c ión de l in te rm ed io fe rc -bu til é s te r del ác ido 3 -b ro m o m e til-5 ,6 ,8 ,9 -te tra h id ro -p ir id o [2 ,3 -d ]a z e p in -7 -ca rb o x ílico (D -23).

A una solución del triflato de partida (0,523 g, 1,15 mmol) en DMF (20 ml) se le añade Pd(PPh3)4 (0,200 g, 0,173 mmol) y DIEA (0,650 ml, 3,73 mmol) seguida de ácido fórmico (0,065 ml, 1,7 mmol). La mezcla resultante se calienta a 60 °C durante 3,5 h, después se enfría a temperatura ambiente. Se añaden agua y EtOAc a la mezcla de reacción. La fase acuosa se separa de la fase orgánica y después, se extrae con EtOAc (x3). Los extractos orgánicos combinados se lavan con salmuera, se secan (Na2SO4), se filtran y se concentran a presión reducida. El material en bruto se purifica mediante cromatografía ultrarrápida en columna sobre gel de sílice para proporcionar D-23-1 (0,3627 g).
A una solución a 0 °C de D-23-1 (0,4507 g, 1,471 mmol) en THF (15 ml) se le añade una solución 1,0 M de DiBAl-H en tolueno (4,6 ml, 4,6 mmol) durante el lapso de 5 minutos. La mezcla de reacción se mantiene a 0 °C durante un total de 2 h y 45 minutos. La mezcla de reacción, aún a 0 °C, se trata con sal de Rochelle (15 ml). La mezcla heterogénea resultante se deja calentar a temperatura ambiente y se agita a esta temperatura durante una noche. La mezcla se diluye con agua y EtOAc. La fase acuosa se extrae con EtOAc (x4). Los extractos orgánicos combinados se lavan con salmuera, se secan (Na2SO4), se filtran y se concentran a presión reducida. El material en bruto se purifica por cromatografía ultrarrápida sobre gel de sílice para proporcionar D-23-2 (0,2784 mg).
A una solución a 0 °C de D-23-2 (0,2062 g, 0,7408 mmol) en DCM (5 ml) se le añade NBS (0,1726 g, 0,9698 mmol), seguida de PPh3 (0,255 g, 0,972 mmol). La mezcla de reacción se deja en agitación a 0° durante 1,5 h. La mezcla de reacción se concentra parcialmente a presión reducida (sin calentamiento). El material en bruto se purifica por cromatografía ultrarrápida en columna sobre gel de sílice para proporcionar el compuesto del título D-23 (0,1844 g).
Ejemplo 34: Preparación del intermedio ferc-butil éster del ácido 2-bromometil-7,8-dihidro-5H-[1,6]naftiridin-6-carboxílico (D-24).

A una solución a 0 °C del éster de partida (1,06 g, 3,63 mmol) en THF (33 ml) se le añade DiBAl-H 1,0 M en tolueno (11 ml, 11 mmol). La mezcla de reacción se deja en agitación a 0 °C durante 2,5 h, después la mezcla de reacción se trata con sal de Rochelle (35 ml). La mezcla heterogénea resultante se deja calentar a temperatura ambiente y se agita a esta temperatura durante 2 días. La mezcla se diluye con agua y EtOAc y la fase acuosa se extrae con EtOAc (x4). Los extractos orgánicos combinados se lavan con salmuera, se secan (Na2SO4), se filtran y se concentran a presión reducida. El material en bruto se purifica por cromatografía ultrarrápida sobre gel de sílice para proporcionar D-24-1 (0,459 g).
A una solución a 0 °C de D-24-1 (0,459 g, 1,74 mmol) en DCM (12 ml) se le añade NBS (0,371 g, 2,08 mmol), seguida de PPh3 (0,557 g, 2,12 mmol). La mezcla de reacción se deja en agitación a 0° durante 1 h y 40 minutos. La mezcla de reacción se concentra parcialmente a presión reducida (sin calentamiento) y el residuo se purifica por cromatografía ultrarrápida en columna sobre gel de sílice para proporcionar el compuesto del título, D-24 (0,422 g).
E je m p lo 35: P rep a ra c ión de l in te rm ed io fe rc -bu til é s te r de l ác ido 8 -b ro m o m e til-6 -m e til-1 ,2 ,4 ,5 -te tra h id ro -b e n zo [d ]a ze p in -3 -ca rb o x ílico (D -25).

Una mezcla heterogénea del ácido borónico de partida (4,98 g, 17,9 mmol) y NIS (8,06 g, 35,8 mmol) en acetonitrilo (103 ml) se calienta a 80 °C en una corriente de N2 durante aproximadamente 19 h. Después, la mezcla de reacción se diluye con salmuera y DCM. La fase acuosa se extrae con DCM (x3). Los extractos orgánicos combinados se lavan con NaHSO3 1,0 M, seguido de una segunda porción de salmuera, se secan (Na2SO4), se filtran y se concentran a presión reducida hasta un volumen reducido. La solución restante se vierte directamente sobre una columna para purificación. El material en bruto se purifica cromatografía ultrarrápida sobre gel de sílice para proporcionar D-25-1 (6,24 g).
Una mezcla de D-25-1 (5,01 g, 13,9 mmol), ácido metilborónico (2,18 g, 36,5 mmol) y K3PO4 (7,49 g, 35,3 mmol) en 1,2-DME (135 ml) se rocía con N2 durante 15 minutos antes de la adición del catalizador de Pd (aproximadamente 1,2 g, 1,5 mmol). La mezcla de reacción resultante se rocía con N2 durante 15 minutos más. Después, la mezcla de reacción se calienta en un matraz a presión de 350 ml en un bloque de calentamiento a 100 °C durante 24 h. Después, se deja enfriar a temperatura ambiente y se diluye con EtOAc y agua. La fase acuosa se extrae con EtOAc (x3). Los extractos orgánicos combinados se lavan con salmuera, se secan (Na2SO4), se filtran y se concentran a presión reducida para dar el material en bruto. El material en bruto se purifica por cromatografía ultrarrápida sobre gel de sílice para proporcionar D-25-2 (2,46 g).
Una solución de D-25-2 (2,96 g, 11,9 mmol) en MeOH (70 ml) y THF (30 ml) se hidrogena sobre Pd/C a temperatura ambiente y presión atmosférica durante aproximadamente 24 h. La mezcla de reacción se filtra a través de un lecho de Celite y se lava vigorosamente con MeOH. La solución resultante se concentra a presión reducida para proporcionar D-25-3 (2,52 g), que se usó como es habitual en la transformación siguiente.
Una solución a 0 °C de H2SO4 concentrado (10 ml) en H2O (38 ml) se le añade a D-25-3 (2,52 g, 11,5 mmol) a 0 °C. Aproximadamente 20 minutos después, se añade una solución de NaNO2 (0,800 g, 11,6 mmol) en H2O (13 ml) gota a gota durante el lapso de 35 minutos. Después de 40 minutos a esta temperatura, la mezcla de reacción se calienta a temperatura ambiente y se mantiene a esta temperatura durante 1,5 h. Después, se añade H2O (40 ml) y la solución resultante se calienta a reflujo durante aproximadamente 2 h, después se enfría a temperatura ambiente. La solución se satura con NaCl y se extrae con EtOAc (x4). Los extractos orgánicos combinados se secan (Na2SO4), se filtran y se concentran a presión reducida para dar un aceite pegajoso. El material en bruto se purifica por cromatografía ultrarrápida sobre gel de sílice para proporcionar D-25-4 (1,63 g) en forma de un sólido de color amarillo pálido.
Una mezcla de D-25-4 (0,524 g, 2,39 mmol) y bromuro de amonio (0,265 g, 2,70 mmol) en etilen diamina (0,800 ml, 12,0 mmol) se calienta a 100 °C durante aproximadamente 4 d. La mezcla de reacción se enfría a temperatura ambiente y se diluye con una pequeña cantidad de agua y después se acidifica a pH 6 con ácido acético glacial. Este material se purifica por cromatografía en columna de fase inversa ultrarrápida C18 usando un eluyente de agua y acetonitrilo con aditivo de TFA al 0,1 %. La separación del producto del material de partida sin reaccionar no se logra y la mezcla que contiene D-25-5 se usa tal cual en la reacción siguiente.
A una mezcla a 0 °C de la mezcla que contiene D-25-5 en DCM (50 ml) se le añade exceso de N,N-diisopropiletilamina (aproximadamente 7,0 ml, 40,2 mmol), seguido de exceso de (Boc)2O (5,48 g, 25,1 mmol). El baño de hielo se retira inmediatamente después de la adición y la mezcla de reacción se mantiene a temperatura ambiente durante aproximadamente 48 h. La mezcla de reacción se concentra a presión reducida hasta un volumen reducido y después, se purifica por cromatografía ultrarrápida sobre gel de sílice para proporcionar D-25-6 (0,439 g). A una solución a 0 °C de D-25-6 en DCM (7 ml) se le añade TEA (0,600 ml, 4,30 mmol) seguida de Tf2O (0,320 ml, 1,90 mmol) durante el lapso de 5 minutos. La mezcla de reacción se deja en agitación a 0 °C durante 2,5 h. La mezcla se diluye con NaHCO3 acuoso saturado (10 ml). La fase acuosa se extrae con EtOAc (x3). Los extractos orgánicos combinados se lavan con NaHCO3 acuoso saturado, salmuera, se secan (Na2SO4 ) y se concentran a presión reducida para proporcionar D-25-7 que se usó sin más purificación.
Una solución de D-25-7 (0,686 g, 1,68 mmol) y el boronato de partida (0,520 g, 2,16 mmol) en DME (12 ml) se rocía con N2 durante 10 minutos antes de la adición del catalizador de Pd (0,206 g, 0,178 mmol) y Na2CO3 acuoso (2,0 M, 2,1 ml). La mezcla de reacción se rocía con N2 durante 5 minutos más antes de calentarla en un reactor de microondas a 120 °C durante 40 minutos. La mezcla de reacción se diluye con agua y EtOAc. La fase acuosa se extrae con EtOAc (x3). Los extractos orgánicos combinados se lavan con salmuera, se secan (Na2SO4 ), se filtran y se concentran a presión reducida. El material en bruto se purifica por cromatografía ultrarrápida en columna sobre gel de sílice para proporcionar D-25-8 (0,2804 g) en forma de un aceite incoloro.
A una solución de D-25-8 en THF (10 ml) y agua (3 ml) se le añade peryodato sódico (0,661 g, 3,09 mmol). La mezcla heterogénea se deja en agitación durante 10 minutos antes de la introducción de tetraóxido de osmio (4 % en peso en agua, aproximadamente 0,4 ml). La suspensión muy espesa se agita vigorosamente durante una noche (20 h). El matraz de reacción se envuelve con papel de aluminio para excluir la luz. La mezcla de reacción se diluye con DCM (40 ml) y agua (40 ml). La mezcla heterogénea se deja en agitación vigorosa durante 45 minutos, después se pasa a través de un separador de fases. La fase acuosa retenida se lava en profundidad con DCM. La fase orgánica se seca (Na2SO4), se filtra y se concentra a presión reducida. El material en bruto se purifica por cromatografía ultrarrápida en columna sobre gel de sílice para proporcionar D-25-9 (0,2308 g).
A una solución a 0 °C de D-25-9 en una mezcla de THF (4 ml) y MeOH (4 ml) se le añade NaBH4 (0,0504 g, 1,33 mmol) en un único lote. El baño de hielo se retira aproximadamente 10 minutos después y la mezcla de reacción se mantiene a temperatura ambiente durante 1 h y 15 minutos. Se añade más NaBH4 (0,0281 g) a la mezcla de reacción a 0 °C 1 h y 25 minutos después del inicio de la reacción para conducir la reacción a su finalización. Finalmente, la mezcla de reacción se inactiva con NH4Cl acuoso saturado (2 h y 40 minutos, tiempo de reacción total). La mezcla de reacción se deja en agitación a temperatura ambiente durante aproximadamente 1 se extrae con EtOAc (x3). Los extractos orgánicos combinados se lavan con NH4Cl acuoso saturado, salmuera, se secan (MgSO4), se filtran y se concentran a presión reducida. El material en bruto se purifica por cromatografía ultrarrápida en columna sobre gel de sílice para proporcionar D-25-10 (0,208 g).
A una solución a 0 °C de D-25-10 (0,208 g, 0,715 mmol) y DIEA (0,200 ml, 1,15 mmol) se le añade dibromuro de trifenilfosfina (0,480 g, 1,14 mmol) en lotes (x3) durante el intervalo de 5 minutos. La solución incolora, transparente, se vuelve de color amarillo después de la adición del dibromuro. La mezcla de reacción se agita a temperatura ambiente durante 1 hora, después se concentra hasta un volumen reducido. La solución restante se purifica por cromatografía ultrarrápida en columna sobre gel de sílice para proporcionar el compuesto del título, D-25 (0,2185 g). Ejemplo 36: Preparación del intermedio 6-(bromometil)-5-fluoro-8-metoxi-3,4-dihidro-1H-isoquinolin-2-carboxilato de ferc-butilo (D-28)
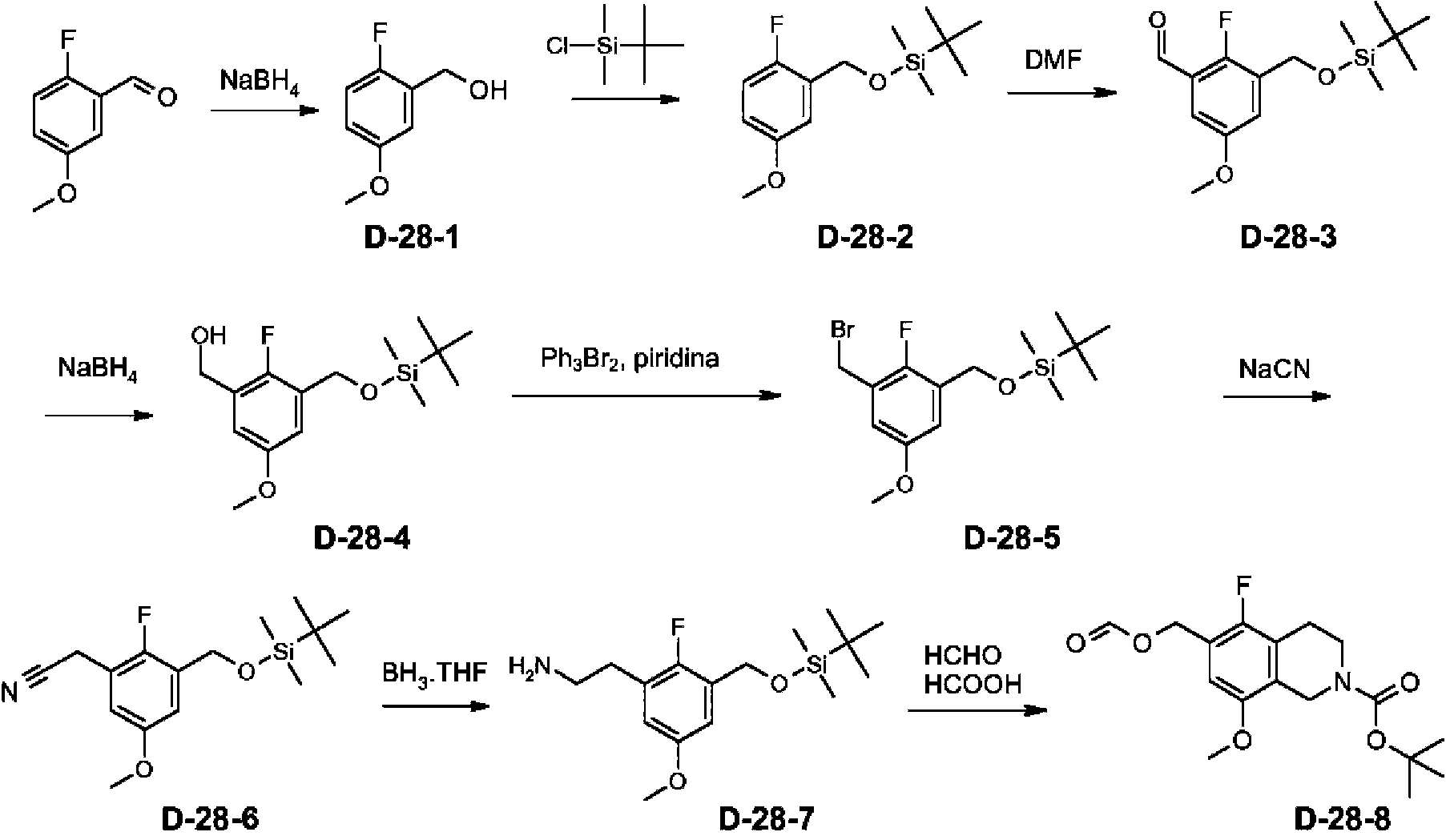
Na PhjBrí, pindma2XO P 3 

A una solución de 2-fluoro-5-metoxibenzaldehído (5,00 g, 32,4 mmol) en MeOH (90 ml) se le añade NaBH (1,93 g, 50,9 mmol). La mezcla se agita a temperatura ambiente durante 1 h, después se añade agua (50 ml). La mezcla resultante se agita durante 15 min y se concentra a presión reducida. El residuo se disuelve en agua (50 ml) y se extrae con diclorometano (2 x 50 ml). Las capas orgánicas combinadas se lavan con salmuera (50 ml), se pasan a través de un separador de fases y se concentran a presión reducida para proporcionar el compuesto D-28-1 (4,88 g). Una mezcla de D-28-1 (4,88 g, 31,2 mmol), cloruro de t-butildimetilsililo (7,06 g, 46,8 mmol), imidazol (4,25 g, 62,44 mmol) y THF (130 ml) se agita a temperatura ambiente durante 16 h, después se concentra a presión reducida. El residuo se disuelve en agua (50 ml). La mezcla se extrae con MTBE (50 ml). La capa orgánica se lava de manera secuencial con una solución acuosa 1 N de HCl (50 ml) y salmuera (50 ml), después se seca sobre Na2SO4 y se concentra a presión reducida. El residuo se purifica por cromatografía ultrarrápida para proporcionar el compuesto D-28-2 (8,23 g).
A una solución de D-28-2 (8,23 g, 30,4 mmol) en THF (150 ml) enfriada en atmósfera de argón a - 78 °C se le añade una solución de s-BuLi 1,4 M en ciclohexano (ml, mmol). La mezcla se agita a -78 °C durante 1,5 h, después se añade DMF (5,16 ml, 67,0 mmol). La mezcla resultante se agita a -78 °C durante 30 min, después a temperatura ambiente durante 45 min. Se añade agua (50 ml), las capas se separan y la capa acuosa se extrae con EtOAc (2 x 100 ml). Las capas orgánicas combinadas se lavan con salmuera (100 ml) y después se concentran a presión reducida. El producto en bruto se purifica por cromatografía ultrarrápida sobre gel de sílice para proporcionar el compuesto D-28-3 (4,67 g).
A una solución del compuesto D-28-3 (4,67 g) en MeOH (60 ml) se le añade NaBH4 (0,93 g, 24,6 mmol). La mezcla se agita a temperatura ambiente durante 1 h, después se añade agua (50 ml). La mezcla resultante se agita durante 20 min, después se concentra a presión reducida. El residuo se disuelve en agua (50 ml) y se extrae con diclorometano (2 x 55 ml). Las capas orgánicas combinadas se pasan a través de un separador de fases y se concentran a presión reducida. El producto en bruto se purifica por cromatografía ultrarrápida sobre gel de sílice para proporcionar el compuesto D-28-4 (2,15 g).
A una solución de D-28-4 (2,15 g, 7,16 mmol) y piridina (0,94 ml, 8,95 mmol) en diclorometano (35 ml) se le añade dibromotrifenilfosforano (3,47 g, 8,23 mmol) a 0 °C. La mezcla se agita a 0 °C durante 1,5 h, después se concentra a
presión reducida. El residuo se tritura con EtOAc al 20 % en heptano (100 ml) y se filtra. El filtrado se concentra a presión reducida y el producto en bruto se purifica por cromatografía ultrarrápida sobre gel de sílice para proporcionar el compuesto D-28-5 (2,20 g).
Una mezcla de D-28-5 (2,20 g, 6,05 mmol) y NaCN (0,33 g, 6,7 mmol) en DMF (16 ml) se agita a 45 °C durante 2 h y se diluye con MTBE (75 ml) y agua (100 ml). La capa acuosa se extrae con MTBE (75 ml). Las capas orgánicas combinadas se lavan con agua (2 x 75 ml), después salmuera (75 ml) y se concentran a presión reducida. El producto en bruto se purifica por cromatografía ultrarrápida sobre gel de sílice para proporcionar el compuesto D-28-6 (1,71 g).
A una solución del compuesto D-28-6 (1,71 g, 5,53 mmol) en THF (22 ml) se le añade una solución 1,0 M de complejo de borano-THF en THF (12,16 ml, 12,16 mmol) en atmósfera de argón a temperatura ambiente. La mezcla se calienta a 55 °C durante 1 h, después se enfría a temperatura ambiente. Se añade agua (10 ml) y la mezcla resultante se agita durante 15 min, después se concentra a presión reducida. El producto en bruto se purifica por cromatografía ultrarrápida de fase inversa C18 para proporcionar el compuesto D-28-7 (1,10 g).
Una mezcla del compuesto D-28-7 (1,10 g, 3,06 mmol), formaldehído al 15 % en peso en agua (0,25 ml) y ácido fórmico (8,80 ml) se agita a 60 °C durante 5,5 h. Después, la mezcla se concentra a presión reducida y el residuo se destila azeotrópicamente con tolueno (2 x 50 ml). El residuo se recoge en DCM (16,6 ml) y a este se le añade DMAP (37,4 mg, 0,31 mmol), trietilamina (1,90 ml, 13,52 mmol) y Boc2O (667,8 mg, 3,06 mmol). La mezcla se agita a temperatura ambiente durante 2 h, después se concentra a presión reducida. El producto en bruto se purifica por cromatografía ultrarrápida sobre gel de sílice para proporcionar el compuesto D-28-8 (0,26 g).
Una mezcla de D-28-8 (0,26 g, 0,77 mmol) y Na2CO3 (438,5 mg, 4,14 mmol) en MeOH (6,20 ml) se agita a temperatura ambiente durante una noche. El disolvente se evapora a presión reducida. El residuo se disuelve en agua (20 ml) y la mezcla se extrae con DCM (3 x 20 ml). Las capas orgánicas combinadas se concentran a presión reducida y el producto en bruto se purifica por cromatografía ultrarrápida sobre gel de sílice para proporcionar el compuesto D-28-9 (0,2307 g).
A una solución de D-28-9 (0,230 g, 0,74 mmol) y piridina (0,09 ml, 0,85 mmol) en DCM (7,6 ml), a 0 °C, se le añade dibromotrifenilfosforano (0,3586 g, 0,85 mmol). La mezcla se agita a 0 °C durante 1 h, después se concentra a presión reducida. El residuo se tritura con EtOAc al 20 % en heptano (50 ml) y se filtra. El filtrado se concentra a presión reducida y el producto en bruto se purifica por cromatografía ultrarrápida sobre gel de sílice para proporcionar el compuesto del título D-28 (0,2107 g).
Ejemplo 37: Preparación de los intermedios ferc-butil éster del ácido (R)-7-bromometil-1-metil-1,2,4,5-tetrahidrobenzo[d]azepin-3-carboxílico (D-29) y ferc-butil éster del ácido (R)-7-bromometil-1-metil-1,2,4,5-tetrahidrobenzo[d]azepin-3-carboxílico (D-30).

Se añade cloruro de 2-cloro-propionilo (109 g, 0,860 mol) gota a gota a una solución en agitación de 2-(4-metoxifen¡l)-et¡lamina (130 g, 0,860 mol) y TEA (174 g, 1,72 mol) en ACN (2 l) a 0 °C en atmósfera de N2. La solución se calienta a 20 °C durante 2 h, después se evapora y se extrae con EtOAc. Los extractos orgánicos combinados se lavan con salmuera, se secan con Na2SÜ4 anhidro, se filtran y se concentran para dar D-29-1 (190 g).
Una mezcla de D-29-1 (100,00 g, 413,71 mmol) y AlCb (165 g, 1,24 mol) se calienta a 150 °C en atmósfera de N2 durante 12 h. La reacción se enfría a temperatura ambiente, se diluye con agua y se extrae con EtOAc. Los extractos orgánicos combinados se lavan con salmuera, se secan sobre Na2SO4 anhidro, se filtran y se concentran a presión reducida. El residuo se purifica por cromatografía ultrarrápida en columna sobre gel de sílice para producir D-29-2 (55,0 g).
A una mezcla de D-29-2 (77,0 g, 0,403 mol) en THF (770 ml) se le añade lentamente dimetilsulfuro de borano (10 M, 89 ml) a temperatura ambiente en atmósfera de N2. La mezcla se agita durante 10 min y después se calienta a 65 °C durante 16 h. La mezcla se enfría a temperatura ambiente y se inactiva con HCl (10 %) y se agita durante 20 min. El pH de la mezcla se basifica mediante la adición de Na2CO3. A esto se le añade (Boc)2O (88 g, 0,403 mol) y la reacción se agita a temperatura ambiente durante 16 h. La mezcla se extrae con EtOAc, se lava con salmuera, se seca con Na2SO4 anhidro, se filtra y se concentra a presión reducida. El residuo se purifica por cromatografía ultrarrápida en columna sobre gel de sílice para proporcionar D-29-3 (57,0 g) en forma de un sólido de color amarillo. A una mezcla de D-29-3 (135 g, 0,487 mol) y piridina (77 g, 0,97 mol) en DCM (1350 ml), a -50 °C en atmósfera de N2 , se le añade Tf2O (151 g, 0,535 mol) gota a gota durante 10 min. La mezcla de reacción se deja calentar a temperatura ambiente durante 2 h y después se concentra a presión reducida. El residuo resultante se diluye con EtOAc, se lava con HCl 1 N, seguido de NaHCO3 saturado y salmuera, después se seca sobre Na2SO4 y se concentra a presión reducida. Este residuo se purifica por cromatografía ultrarrápida en columna sobre gel de sílice para proporcionar D-29-4 (165 g).
Una mezcla de D-29-4 (140 g, 0,342 mol), dppp (14 g), Pd(OAc)2 (14 g), TEA (69 g, 0,684 mol) en EtOH (2800 ml) se agita a 80 °C en atmósfera de CO (4 MPa) durante 12 h. La mezcla de reacción se enfría a temperatura ambiente y se concentra a presión reducida. El residuo resultante se purifica por cromatografía ultrarrápida en columna sobre gel de sílice para dar D-29-5 (108 g).
A una solución en agitación de D-29-5 (5,00 g, 15,0 mmol) en THF (100 ml), a -40 °C, se le añade lentamente hidruro de litio y aluminio (0,597 g, 15,7 mmol) manteniendo la temperatura a -40 °C. Después de completar la
adición, la mezcla se calienta a temperatura ambiente y se agita durante 2 h. El disolvente se elimina a presión reducida y el residuo se separa con diclorometano y H2O. La fase orgánica se seca sobre Na2SO4 anhidro y se concentra a presión reducida. El residuo se purifica por cromatografía ultrarrápida en columna sobre gel de sílice para dar D-29-6 (4,0 g) en forma de un aceite.
El D-29-6 racémico se resuelve en una LUX 5 u celulosa (30 x 250 mm) usando IPA al 10 % en CO2 supercrítico a 85 g/min a una presión de 14 Mpa (140 bar) a 40 °C para proporcionar D-29-7 (primer pico de elución, 0,980 g) y D-30-1 (segundo pico de elución, 1,118 g). La estereoquímica absoluta no se estableció y las estructuras dibujadas se asignas de manera arbitraria.
A una solución del alcohol D-29-7 (0,980 g, 3,36 mmol) y N,N-diisopropiletilamina (0,879 ml, 5,04 mmol) en DCM (30,0 ml), a 0 °C, se le añade dibromuro de trifenilfosfina (2,173 g, 5,045 mmol). La reacción se agita durante 2 h, después se concentra a presión reducida. El residuo resultante se purifica por cromatografía ultrarrápida en columna sobre gel de sílice para proporcionar el compuesto del título D-29 (0,786 g).
A una solución del alcohol D-30-1 (1,118 g, 3,837 mmol) y N,N-diisopropiletilamina (1,003 ml, 5,755 mmol) en DCM (30,0 ml), a 0 °C, se le añade dibromuro de trifenilfosfina (2,479 g, 5,755 mmol). La reacción se agita durante 2 h, después se concentra a presión reducida. El residuo resultante se purifica por cromatografía ultrarrápida en columna sobre gel de sílice para proporcionar el compuesto del título D-30 (0,948 g).
Ejemplo de referencia 38: Preparación del intermedio ferc-butil éster del ácido 6-{2-[6-((1R,6S)-6-carboxi-3-azabiciclo[4.1.0]hept-3-il)-piridin-2-il]-fenoximetil}-3,4-dihidro-1H-isoquinolin-2-carboxílico (E-1)

A una solución de 0,109 g (0,322 mmol) de C-1 en acetona (15 ml) se le añade 0,11 g (0,34 mmol) de D-1 seguido de 0,35 g (1,1 mmol) de carbonato de cesio. La mezcla se agita a temperatura ambiente durante 4 días, después se filtra para eliminar los inorgánicos no solubles y se concentra a presión reducida. El residuo se purifica por cromatografía ultrarrápida sobre gel de sílice para proporcionar E-1 (0,065 g, 35 % de rendimiento).
Los productos intermedios siguientes se pueden preparar a partir del producto intermedio C-1 de una manera similar usando los reactivos apropiados.

continuación
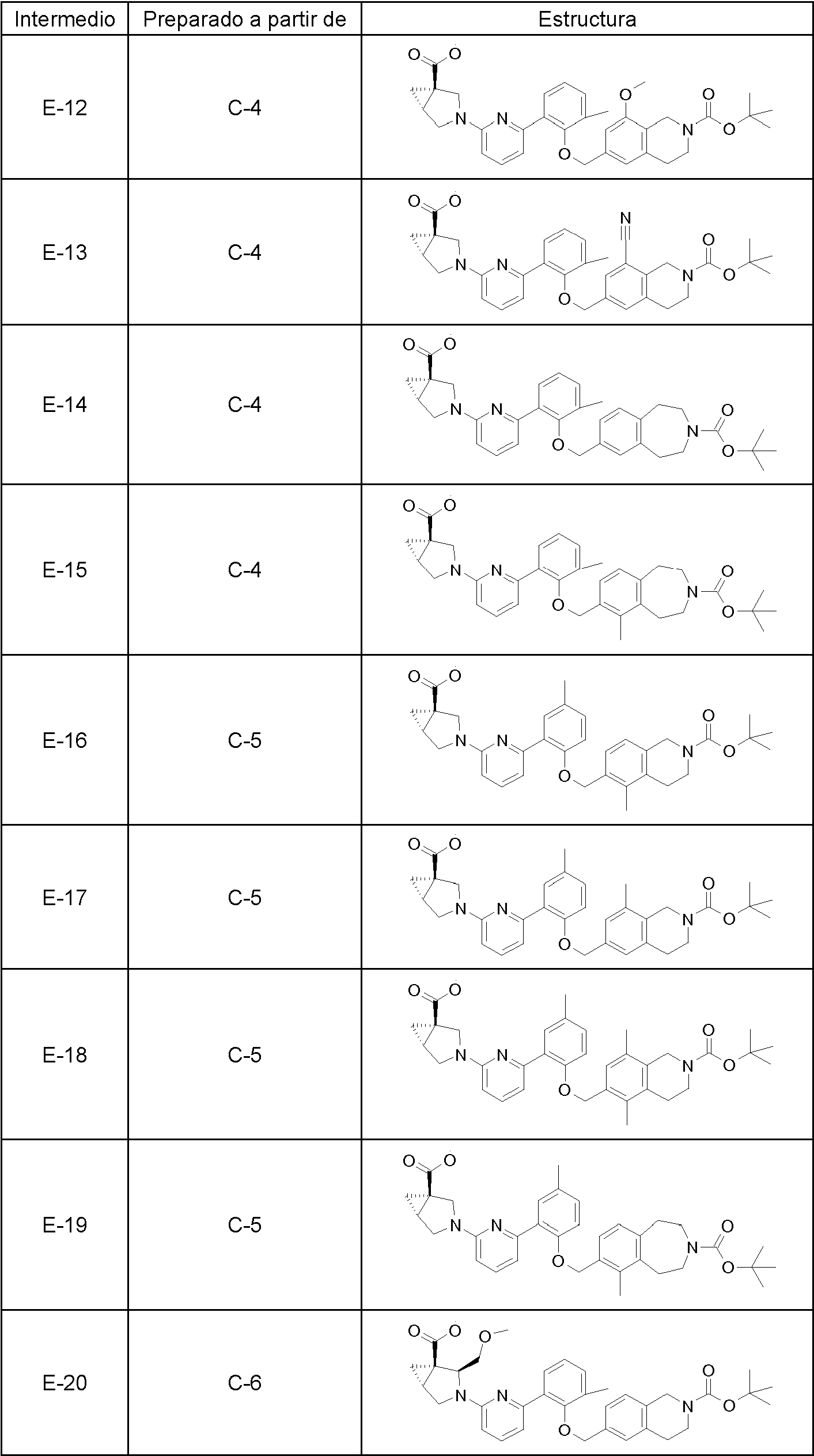
continuación

continuación

continuación

continuación

continuación
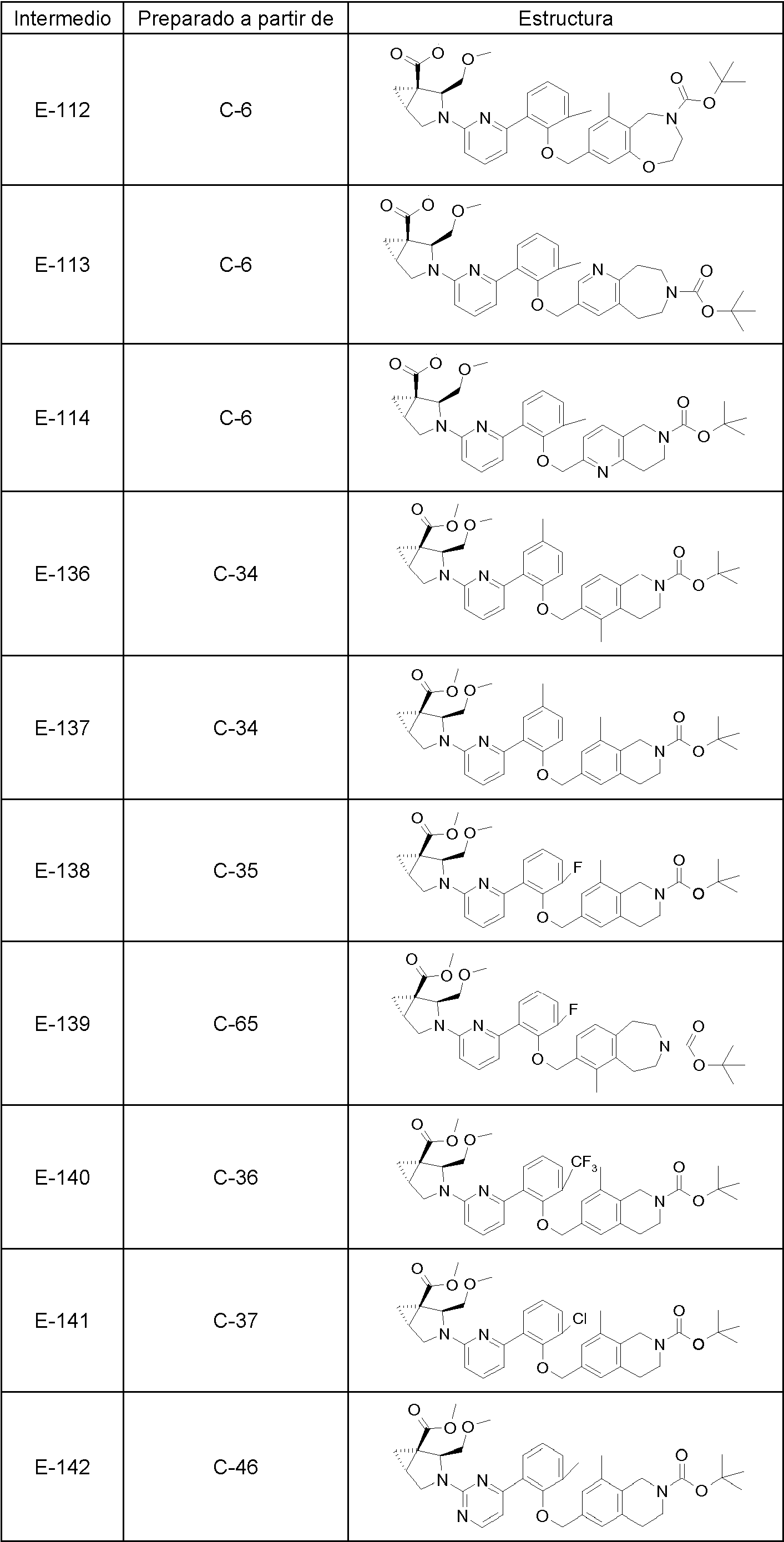
continuación

Ejemplo de referencia 41: Preparación del intermedio éster etílico del ácido (1R,6S)-3-{6-[2-(1,2,3,4-tetrahidroisoquinolin-6-ilmetoxi)-fenil]-piridin-2-il}-3-aza-biciclo[4.1.0]heptan-6-carboxílico sal del ácido trifluoroacético (F-1)

A una solución de 0,065 g (0,11 mmol) de E-1 en DCM (1 ml) se le añade 0,15 g (0,67 mmol) de dibromuro de cinc. La mezcla se agita durante una noche a temperatura ambiente, después se filtra y se concentra a presión reducida. El residuo se purifica mediante cromatografía ultrarrápida de fase inversa con aditivo de TFA al 0,1 %. El eluyente se concentra a presión reducida para proporcionar F-1, que se usa directamente en la secuencia de reacción siguiente. Los productos intermedios siguientes se pueden preparar de una manera similar usando los reactivos apropiados.

continuación

continuación

continuación

continuación

Síntesis de los compuestos finales:
Ejemplo de referencia 43: Preparación de ácido (1R,6S)-3-(6-{2-[2-(tetrahidro-piran-4-il)-1,2,3,4-tetrahidroisoquinolin-6-ilmetoxi]-fenil}-piridin-2-il)-3-aza-biciclo[4.1,0]heptan-6-carboxílico (1)

A una solución de F-1 en DCM (10 ml) se le añade 0,050 ml (0,54 mmol) de tetrahidro-piran-4-ona seguido de 0,10 g (0,42 mmol) de triacetoxiborohidruro sódico. La mezcla se agita durante una noche a temperatura ambiente después se diluye con metanol y se concentra a presión reducida. El residuo se purifica por cromatografía ultrarrápida sobre
gel de sílice para proporcionar 0,020 g (32 % en dos etapas) de 1-1.
A una solución de 0,020 g (0,035 mmol) de 1-1 en una mezcla 1:1:1 de agua:MeOH:THF (15 ml) se le añade 0,020 g (0,48 mmol) de hidróxido de litio monohidrato. La mezcla se agita durante una noche a temperatura ambiente, después se concentra a presión reducida. El residuo se purifica mediante cromatografía ultrarrápida de fase inversa con aditivo de TFA al 0,1 %. El eluyente se elimina a presión reducida para proporcionar 1 (0,020 g, 74) en forma de la sal TFA. MS, electronebulización, m/z = 540,4[M+H], TR 1,11 min.
Los siguientes compuestos se pueden preparar a partir del producto intermedio F- indicado de una manera similar usando los reactivos apropiados.

continuación

continuación

Los siguientes compuestos de la tabla 1 se preparan de una manera similar al procedimiento descrito en el ejemplo de referencia 43, usando el intermedio F-11 y los reactivos apropiados. Los diastereómeros se separan antes de la etapa de síntesis final. La configuración absoluta del centro diastereomérico no se determina:
Compuesto 25: MS, electronebulización, m/z = 540,4 [M+H], TR 0,27 min;
Compuesto 26: MS, electronebulización, m/z = 540,5 [m +h ], TR 0,27 min;
Los siguientes compuestos de la tabla 1 se preparan de una manera similar al procedimiento descrito en el ejemplo de referencia 43, usando el intermedio F-22 y los reactivos apropiados. Los diastereómeros se separan antes de la etapa de síntesis final. La configuración absoluta del centro diastereomérico no se determina:
Compuesto 51: MS, electronebulización, m/z = 584,4 [M+H], TR 0,52 min;
Compuesto 52: MS, electronebulización, m/z = 584,4 [m +h ], TR 0,54 min;
EVALUACIÓN DE LA ACTIVIDAD BIOLÓGICA
Ensayo celular
El ensayo con el activador celular de sGC se realizó con y sin suero humano (SH) al 50 % usando células de ovario de hámster chino que se habían transfectado de manera estable para expresar las subunidades alfa 1 y beta 1 de la guanilato ciclasa soluble (sGC) humana. Las células se preincubaron con 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-ona (ODQ) 40 microM, un inhibidor de sGC, durante una hora en tampón que contenía albúmina de suero bovino al 0,1 % y 3-isobutil-1-metilxantina (IBMX). Se prepararon curvas de respuesta a la concentración para los compuestos de ensayo en DMSO. Se realizó una dilución intermedia de los compuestos en tampón que contenía IBMX o IBMX que contenía SH de tipo AB. Los compuestos diluidos se añadieron a las células y se incubaron a temperatura ambiente durante treinta minutos. El cGMP se midió usando un kit de fluorescencia homogénea con resolución temporal de CisBio y se calculó la CE50 de cada compuesto.
Los compuestos representativos de la presente invención se ensayaron para determinar la actividad del ensayo anterior. Los compuestos preferidos tenían una CE50 <1000 nM en el ensayo anterior y los compuestos más preferidos tenían una CE50 <200 nM. Como ejemplos, los datos de los compuestos representativos de la Tabla 1 se muestran en la Tabla 2.
Tabla 2

continuación

EVALUACIÓN DE LA SOLUBILIDAD
La solubilidad se midió mediante el siguiente método.
1. Preparación de las muestras:
En un formato de placa de 96 pocillos, se prepararon 100 pl de solución de reserva de DMSO 10 mM de cada compuesto. El experimento se realizó en una sola determinación a 3 valores de pH (2,2, 4,5 y 6,8). Para cada pH y una referencia, se necesitaron 40 pl de cada compuesto.
Preparación del tampón:
McIlvaine pH 2,2: A 2,076 g de ácido cítrico monohidratado y 0,043 g de Na2HPO4 x 2 H2O se añadieron 100 ml de agua desionizada.
McIlvaine pH 4,5: A 1,166 g de ácido cítrico monohidratado y 1,585 g de Na2HPO4 x 2 H2O se añadieron 100 ml de agua desionizada.
McIlvaine pH 6,8: A 0,476 g de ácido cítrico monohidratado y 2,753 g de Na2HPO4 x 2 H2O se añadieron 100 ml de agua desionizada.
Con un dispositivo de manipulación de líquidos (Multipette® o un manipulador de líquidos) adecuado, a cada uno de los pocillos de una placa de profundidad de 96 pocillos, se añadieron 390 pl de cada solución tampón y 10 pl de compuesto. Las placas se cubrieron firmemente y se agitaron durante 24 h en un agitador elevado (a 54 RPM) a temperatura ambiente. El contenido de DMSO en el tampón final era del 2,5 % v/v.
Después de 24 h, las placas se centrifugaron para eliminar las gotículas de la tapa antes de abrirlas (durante ~ 5 min a 2500 RPM).
La filtración se realizó al vacío con una placa de filtro Millipore de 96 pocillos. El filtrado se recogió en una placa de profundidad con pocillos y se transfirió a una placa adecuada para el análisis mediante UPLC.
La placa de referencia se preparó añadiendo 10 pl de compuesto a 390 pl de acetonitrilo/agua 50:50 en una placa de profundidad de 96 pocillos y se transfirió a una placa adecuada para el análisis mediante UPLC. Los pocillos se comprobaron visualmente para determinar la precipitación y cualquier presencia observada quedó registrada en los comentarios de los resultados comunicados.
2. Medición de las muestras
Las muestras se midieron con UPLC-UV usando el método cromatográfico descrito a continuación.

Para generar conjuntos de muestras (de acuerdo con el diseño de la placa) se utilizo el programa informático Waters Empower®2, Métodos de conjuntos de muestras y métodos instrumentales.
Un conjunto de muestras comprende los métodos para tres placas de 96 pocillos (una placa de referencia y dos placas de muestra, e incluye un método de conjunto de muestras y un método instrumental).
3. Procesamiento y análisis de datos
Los cromatogramas UV recogidos a 254 nm se integraron y procesaron.
Se suponía que el compuesto estaba completamente disuelto en la solución de referencia acetonitrilo/agua al 50:50) Los datos de solubilidad (pg/ml) de los compuestos de la Tabla 1 se muestran en la Tabla 3 a continuación.
Tabla 3

continuación

EVALUACION DE LA ESTABILIDAD METABOLICA
Objetivo
El ensayo de estabilidad metabólica en microsomas hepáticos humanos (MHH) de alto rendimiento de 5 puntos temporales, está diseñado para determinar el metabolismo de los compuestos in vitro. Los compuestos se incubaron con los MHH a una concentración de 1 |j M, a 37°°C, durante un total de 60 min. El porcentaje de compuesto restante a los 5, 15, 30 y 60 min se usó para calcular la t1/2 (min), CLint (ml/min/kg), CLh (ml/min/kg) y % de Qh. El ensayo se basaba en un formato de 96 pocillos y podía admitir hasta 92 compuestos por placa (n = 1).
Incubación
Usando el cabezal multicanal de 96 pocillos, el Biomek FX, equipado con un bloque/agitador térmico Peltier, se programó para realizar las siguientes etapas:
1. Traspasar con una pipeta 175 j l de microsomas de 1,15 mg/ml a cada uno de los 96 insertos cónicos (Analytical Sales and Products, número de catálogo 96PL05) que encajan en la placa del bloque/agitador térmico Peltier (la placa de incubación)
2. Añadir 5 pl de los compuestos de la placa de ensayo a los microsomas y agitar la mezcla a 600 rpm a 42,1° C durante 10 min (se requiere un ambiente de 42,1° C en el Peltier para incubar las muestras a 37° C)
3. Después de 10 min, solicitar al usuario que añada la placa de NADPH a la plataforma y añada 20 pl de la placa de NADPH a la placa de incubación para comenzar la reacción
4. Al minuto 0, a los 5 minutos, 15 minutos, 30 minutos y 60 minutos, añadir a la placa de "enfriamiento", 215 pl de acetonitrilo al 100 % enfriado que contenía uno o más patrones internos
5. Al cabo de 0 min, 5 min, 15 min, 30 min y 60 min de incubación, aspirar 12 j l de la mezcla de incubación y añadirlos a la solución de enfriamiento para detener la reacción
6. Añadir, a cada uno de los pocillos de las placas de enfriamiento a los 0, 5, 15, 30 y 60 minutos, 185 j l de agua
de calidad HPLC (cromatografía líquida de alto rendimiento, siglas del inglés High Performance Liquid Chromatography) para diluir los compuestos a la concentración adecuada para el espectrómetro de masas Después de recopilar todos los puntos temporales, las placas de enfriamiento se sellaron con alfombrillas de placa perforables de 96 pocillos o con una lámina termosellada y se centrifugan a 3000 rpm durante 15 min para sedimentar los microsomas.
Análisis
Las placas se analizaron usando CL/EM/EM (cromatografía líquida y espectrometría de masas) con ionización por electropulverización (IEP) y las transiciones MMR (monitorización de múltiples reacciones) determinadas anteriormente. El método de CL incluye los siguientes parámetros:
Volumen de inyección: 5 pl
Fases móviles: Ácido fórmico al 0,1 % en agua (A) y ácido fórmico al 0,1 % en acetonitrilo (B) (calidad HPLC) Temperatura a la izquierda y a la derecha: 35 °C
Tiempo de ejecución: 4,0 min
Columna: Thermo Scientific, Aquasil C18, 50 x 2,1 mm, 5 p, número de pieza 77505-052130, o equivalente Gradiente de bomba de CL:

Si la forma del pico es pobre y no puede integrarse adecuadamente, puede utilizarse el siguiente método de CL: Volumen de inyección: 5 pl
Fases móviles: Bicarbonato de amonio 2,5 mM (A) y acetonitrilo al 100 % (calidad HPLC) (B)
Lavado acuoso: Agua al 90 %, Acetonitrilo al 10 % (calidad HPLC)
Lavado Orgánico: Acetonitrilo al 90 %, Agua al 10 % (calidad HPLC)
Temperatura a la izquierda y a la derecha: 35 °C
Tiempo de ejecución: 4,5 min
Columna: Phenomex Luna 3u C18(2) 100A, 50 x 2,00 mm
Gradiente de bomba de CL:

Usando una plantilla de Excel en la plataforma Activitybase, las áreas de pico correspondientes a 5, 15, 30 y 60 min se compararon con el área de pico a los 0 min para calcular el porcentaje de compuesto restante usando la siguiente ecuación:
Porcentaje de compuesto restante = (ABC en el tiempo t min/ABC en el tiempo 0 min) x 100 donde t = 0, 5, 15, 30 o 60 min.
El tiempo (min) se representa frente al el logaritmo natural (Ln) del porcentaje del compuesto restante para determinar la pendiente. La pendiente se usa para calcular la t1/2 (min) usando la ecuación, t1/2 = 0,693/pendiente. Clint, Aclaramiento (Cl del inglés clearance) intrínseco
• 0,693/t1/2*peso prom. hígado en g/peso prom. corporal en kg * f(u)/concentración de proteína en incubación en mg/ml* mg de proteína microsomal/g de hígado
• 0,693/t1/2 * 26 g/kg * 1/1,0 mg/ml * 45 mg/g
Clh, Aclaramiento hepático
• Flujo hepático * f(u) * Clint/(flujo hepático f (u) * Clint)
Qh, % de flujo sanguíneo hepático
• (Clh/flujo hepático) * 100
Los datos de estabilidad metabólica (% de Qh) de los compuestos de la Tabla 1 se muestran a continuación en la Tabla 4. Los compuestos preferidos tienen valores de % de Qh inferiores a 24.
Tabla 4

MÉTODOS DE USO TERAPÉUTICO
Los compuestos divulgados en el presente documento activan de manera eficaz la guanilato ciclasa soluble. La activación o potenciación de la guanilato ciclasa soluble es un medio atractivo para prevenir y tratar una diversidad de enfermedades y afecciones asociadas con activación deficiente de la sGC. Por lo tanto, en una realización de la invención, se proporcionan métodos para tratar enfermedades que se pueden aliviar mediante la activación o potenciación de la sGC. Estas incluyen:
Enfermedades cardiovasculares y relacionadas incluyendo hipertensión, ateroesclerosis, arteriopatía periférica, eventos cardíacos adversos mayores (MACE), infarto de miocardio, reestenosis, estenosis de la válvula aórtica, apoplejía, insuficiencia cardíaca, vasoespasmo coronario, vasoespasmo cerebral, lesión por isquemia/reperfusión, hipertensión pulmonar tromboembólica, hipertensión arterial pulmonar, angina estable e inestable y trastornos tromboembólicos;
Enfermedades inflamatorias incluyendo psoriasis, esclerosis múltiple, artritis, asma y enfermedad pulmonar obstructiva crónica;
Trastornos fibróticos de la piel incluyendo, pero sin limitación, esclerosis sistémica;
Trastornos fibróticos hepáticos incluyendo, pero sin limitación, cirrosis de cualquier etiología incluyendo esteatohepatitis no alcohólica o fibrosis en áreas específicas del hígado tal como fibrosis periportal que puede estar causada por lesión inmunológica, efectos hemodinámicos y/u otras causas;
Trastornos inflamatorios del intestino incluyendo, pero sin limitación, colitis ulcerosa y enfermedad de Crohn; Trastornos fibróticos renales incluyendo, pero sin limitación, glomeruloesclerosis, glomeruloesclerosis focal, fibrosis mesangial, fibrosis intersticial debida a lesión inmunológica, efectos hemodinámicos, diabetes (tipos I y 2), nefropatía diabética, nefropatía IgA, nefropatía por lupus, nefropatía membranosa, hipertensión, síndrome urémico hemolítico, glomerulonefritidas múltiples, nefritis intersticial, nefritis tubulointersticial de nuevo por causas inmunológicas y no inmunológicas;
Trastornos fibróticos pulmonares, tanto difusos como localizados, debidos a causas inmunológicas y no inmunológicas, incluyendo, pero sin limitación, fibrosis pulmonar idiopática, fibrosis pulmonar debida a exposición a toxinas, químicos, fármacos y fibrosis quística;
Trastornos fibróticos cardíacos debidos a causas inmunológicas y no inmunológicas incluyendo cardiopatía isquémica (arteriopatía coronaria) y disminución del flujo sanguíneo transitoria y/o sostenida en uno o más de los vasos coronarios incluyendo la posiblemente relacionada con intervenciones en las arterias o venas coronarias, la asociada con cirugía cardíaca y/o el uso de procedimientos de derivación cardiopulmonar y miocarditis debida a causas víricas y no víricas, así como lesión miocárdica inmunológicamente relacionada potencialmente debida a la reactividad cruzada con otros antígenos a los cuales está expuesto el cuerpo humano;
Otras enfermedades mediadas al menos parcialmente por la disminución o reducción de la actividad de la guanilato ciclasa soluble, tales como enfermedad renal, diabetes, glaucoma, distrofia muscular, trastornos urológicos incluyendo vejiga hiperactiva, hiperplasia prostática benigna, disfunción eréctil y trastornos neurológicos incluyendo enfermedad de Alzheimer, demencia, enfermedad de Parkinson y dolor neuropático. Estos trastornos han sido bien caracterizados en el hombre, aunque también existen con una etiología similar en otros mamíferos y se pueden tratar mediante las composiciones farmacéuticas de la presente invención.
En consecuencia, se puede usar un compuesto de fórmula I de acuerdo con cualquiera de las realizaciones descritas en el presente documento o una sal farmacéuticamente aceptable del mismo para la preparación de un medicamento para tratar una enfermedad o trastorno mediado por la activación deficiente de la sGC, incluyendo todas las enfermedades o trastornos mencionados anteriormente.
Para uso terapéutico, los compuestos de la invención se pueden administrar mediante una composición farmacéutica en cualquier forma de dosificación farmacéutica convencional de cualquier manera convencional. Las formas de dosificación convencionales habitualmente incluyen un vehículo farmacéuticamente aceptable adecuado a la forma de dosificación particular seleccionada. Las vías de administración incluyen, pero no se limitan a, por vía intravenosa, por vía intramuscular, por vía subcutánea, por vía intrasinovial, por infusión, por vía sublingual, por vía transdérmica, por vía oral, por vía tópica o por inhalación. Los modos de administración preferidos son oral e intravenoso.
Los compuestos de esta invención se pueden administrar solos o en combinación con adyuvantes que potencian la estabilidad de los inhibidores, facilitan la administración de las composiciones farmacéuticas que contienen los mismos en determinadas realizaciones, proporcionan mayor disolución o dispersión, incrementan la actividad inhibidora, proporcionan terapias complementarias y similares, incluyendo otros ingredientes activos. En una
realización, por ejemplo, se pueden administrar múltiples compuestos de la presente invención. De manera ventajosa, dichas terapias de combinación utilizan dosis menores de los productos terapéuticos convencionales, evitando de este modo la posible toxicidad y los efectos secundarios adversos que incurren cuando estos agentes se usan en forma de mono terapias. Los compuestos de la invención se pueden combinar físicamente con los productos terapéuticos convencionales o con otros adyuvantes en una única composición farmacéutica. De manera ventajosa, los compuestos se pueden administrar después en una única forma de dosificación. En algunas realizaciones, las composiciones farmacéuticas que comprenden dichas combinaciones de compuestos contienen al menos aproximadamente el 5 %, pero más preferentemente al menos aproximadamente el 20 %, de un compuesto de fórmula I (p/p) o una combinación del mismo. El porcentaje óptimo (p/p) de un compuesto de la invención puede variar y está dentro del alcance de los expertos en la técnica. Como alternativa, los compuestos de la presente invención y los productos terapéuticos convencionales u otros adyuvantes se pueden administrar por separado (ya sea en serie o en paralelo). La dosificación separada permite mayor flexibilidad en el régimen de dosificación.
Tal como se ha mencionado anteriormente, las formas de dosificación de los compuestos de esta invención pueden incluir vehículos y adyuvantes farmacéuticamente aceptables conocidos por los expertos habituales en la técnica y adecuados a la forma de dosificación. Estos vehículos y adyuvantes incluyen, por ejemplo, intercambiadores de iones, alúmina, estearato de aluminio, lecitina, proteínas séricas, sustancias tamponantes, agua, sales o electrolitos y sustancias basadas en celulosa. Las formas de dosificación preferidas incluyen comprimido, cápsula, comprimido oblongo, líquido, solución, suspensión, emulsión, pastillas para chupar, jarabe, polvo reconstituible, gránulo, supositorio y parche transdérmico. Se conocen métodos para preparar dichas formas de dosificación (véase, por ejemplo, H.C. Ansel y N. G. Popovish, Pharmaceutical Dosage Forms and Drug Delivery Systems, 5a ed., Lea y Febiger (1990)). Los expertos habituales en la técnica pueden seleccionar los niveles y requerimientos de dosificación para los compuestos de la presente invención a partir de los métodos y técnicas disponibles adecuados para un paciente particular. En algunas realizaciones, los niveles de dosificación varían de aproximadamente 1 1000 mg/dosis para un paciente de 70 kg. Aunque puede ser suficiente una dosis diaria, se pueden administrar hasta 5 dosis al día. Para dosis orales, se pueden necesitar hasta 2000 mg/día. Tal como apreciarán los expertos en la técnica, se pueden requerir dosis menores o mayores dependiendo de factores particulares. Por ejemplo, las dosis específicas y los regímenes de tratamiento dependerán de factores tales como el perfil de salud general del paciente, la gravedad y el curso del trastorno del paciente o su disposición del mismo y el juicio de médico a cargo.
Claims (13)
1. Un compuesto de fórmula I

en donde:
X es un enlace;
Y es C o N;
W es C o N, con la condición de que Y y W no son ambos N;
V es -C(R11)(R12)- o -OCH2-, con la condición de que si V es -OCH2 , entonces Z es -CH2-, Y y W son ambos C; Z es -CH2-, -C(R10)2CH2- o -C(O)-;
R1 es H, Me o -CH2OMe;
R2 y R3 junto con los carbonos a los que están unidos para formar un anillo de 3 miembros condensado;
B es

R5 y R6 se seleccionan independientemente entre H, Me, F, Cl y CF3;
R7 es H, Me, Et, -OMe, CN, F o -CH2OMe o no está presente cuando Y es N;
R8 es H, Me o F o no está presente cuando W es N;
R9 es -(CH2)n heterociclilo, en donde el heterociclilo se selecciona entre tetrahidropiranilo, tetrahidrofuranilo, oxetanilo y [1,4]-dioxanilo o -CH(R10)heteroarilo, en donde el heteroarilo se selecciona entre el grupo que consiste en pirazina, imidazol, piridilo e isoxazolilo y en donde el heteroarilo está opcionalmente sustituido con un grupo metilo;
cada R10 es independientemente H o Me;
R11 es H o Me;
R12 es H o Me;
m es 0 o 1, con la condición de que si m es 0, Z es -CH2-, V es -C(R11)(R12)- y R11 y R12 son ambos H; y n es 0 o 1;
o una sal del mismo.
2. El compuesto de acuerdo con la reivindicación 1, en donde:
W es C;
V es -C(R11)(R12)-;
R5 y R6 se seleccionan independientemente entre H, Me, F y Cl;
R7 es H, Me, Et, -OMe, CN o F o no está presente cuando Y es N;
R8 es H, Me o F;
R9 es -(CH2)n heterociclilo, en donde el heterociclilo se selecciona entre tetrahidropiranilo, tetrahidrofuranilo, oxetanilo y [1,4]-dioxanilo; y
m es 1 o una sal del mismo.
3. El compuesto de acuerdo con la reivindicación 1 o 2, en donde:
Y es C;
Z es -CH2- o -C(R10)2CH2- y
R9 es -(CH2)n heterociclilo, en donde el heterociclilo se selecciona entre tetrahidropiranilo, tetrahidrofuranilo, oxetanilo y [1,4]-dioxanilo;
o una sal del mismo.
4. El compuesto de acuerdo con una cualquiera de las reivindicaciones 1 a 3, en donde:
Z es -CH2-; y
R9 es -(CH2)n heterociclilo, en donde el heterociclilo se selecciona entre tetrahidropiranilo, tetrahidrofuranilo, oxetanilo y [1,4]-dioxanilo;
o una sal del mismo.
5. El compuesto de acuerdo con una cualquiera de las reivindicaciones 1 a 3, en donde:
Z es -C(R10)2CH2-;
R10 es H; y
R9 es -(CH2)n heterociclilo, en donde el heterociclilo se selecciona entre tetrahidropiranilo, tetrahidrofuranilo, oxetanilo y [1,4]-dioxanilo;
o una sal del mismo.
6. El compuesto de acuerdo con una cualquiera de las reivindicaciones 1 a 3, en donde
Y es C;
W es C;
Y es -C(R11)(R12)-;
Z es -CH2- o -C(R10)2CH2 ;
B es

R7 es H, Me, Et, -OMe, CN, F o -CH2OMe;
R8 es H, Me o F;
R9 es -(CH2)n heterociclilo, en donde el heterociclilo se selecciona entre tetrahidropiranilo, tetrahidrofuranilo, oxetanilo y [1,4]-dioxanilo;
R11 es H;
R12 es H;
n es 0 y
m es 1;
o una sal del mismo.
7. El compuesto de acuerdo con una cualquiera de las reivindicaciones 1 a 6, seleccionado entre el grupo que consiste en


8. Una composición farmacéutica que comprende un compuesto de acuerdo con una cualquiera de las reivindicaciones 1 a 7 y un excipiente o vehículo farmacéuticamente aceptable.
9. Un compuesto de acuerdo con una cualquiera de las reivindicaciones 1 a 7 para su uso como un medicamento.
10. Un compuesto de acuerdo con una cualquiera de las reivindicaciones 1 a 7 para su uso en el tratamiento de una enfermedad o trastorno que se puede aliviar mediante potenciación o activación de la sGC en donde la enfermedad o trastorno se selecciona entre una enfermedad cardiovascular, enfermedad inflamatoria, trastorno fibrótico hepático, trastorno fibrótico cutáneo, trastorno fibrótico renal, trastorno fibrótico pulmonar y trastorno fibrótico cardíaco.
11. Un compuesto de acuerdo con una cualquiera de las reivindicaciones 1 a 7 para su uso de acuerdo con la reivindicación 10 en donde la enfermedad se selecciona entre enfermedad renal, diabetes, glaucoma, distrofia muscular, trastornos urológicos incluyendo vejiga hiperactiva, hiperplasia prostática benigna, disfunción eréctil y trastornos neurológicos incluyendo enfermedad de Alzheimer, demencia, enfermedad de Parkinson y dolor neuropático.
12. Un compuesto de acuerdo con una cualquiera de las reivindicaciones 1 a 7 para su uso de acuerdo con la reivindicación 10 en donde la enfermedad es nefropatía diabética.
13. Un compuesto de acuerdo con una cualquiera de las reivindicaciones 1 a 7 para su uso de acuerdo con la reivindicación 10 en donde la enfermedad es esteatohepatitis no alcohólica.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201462027376P | 2014-07-22 | 2014-07-22 | |
| PCT/US2015/041245 WO2016014463A1 (en) | 2014-07-22 | 2015-07-21 | Heterocyclic carboxylic acids as activators of soluble guanylate cyclase |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2784477T3 true ES2784477T3 (es) | 2020-09-28 |
Family
ID=53783363
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES15747311T Active ES2784477T3 (es) | 2014-07-22 | 2015-07-21 | Acidos carboxílicos heterocíclicos como activadores de guanilato ciclasa soluble |
Country Status (26)
| Country | Link |
|---|---|
| US (1) | US9353090B2 (es) |
| EP (1) | EP3172202B1 (es) |
| JP (1) | JP6602840B2 (es) |
| KR (1) | KR20170035993A (es) |
| CN (1) | CN106459017B (es) |
| AP (1) | AP2016009615A0 (es) |
| AR (1) | AR101265A1 (es) |
| AU (1) | AU2015292833B2 (es) |
| BR (1) | BR112017000943A2 (es) |
| CA (1) | CA2955937A1 (es) |
| CL (1) | CL2017000071A1 (es) |
| CO (1) | CO2017000438A2 (es) |
| DK (1) | DK3172202T3 (es) |
| EA (1) | EA032972B1 (es) |
| ES (1) | ES2784477T3 (es) |
| HU (1) | HUE049231T2 (es) |
| IL (1) | IL249587A0 (es) |
| MX (1) | MX378717B (es) |
| PE (1) | PE20170249A1 (es) |
| PH (1) | PH12017500111A1 (es) |
| PL (1) | PL3172202T3 (es) |
| PT (1) | PT3172202T (es) |
| SG (2) | SG11201700507XA (es) |
| TW (1) | TWI711615B (es) |
| UY (1) | UY36226A (es) |
| WO (1) | WO2016014463A1 (es) |
Families Citing this family (22)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| AU2017216429B2 (en) * | 2016-02-01 | 2022-10-06 | Cyclerion Therapeutics, Inc. | Use of sGC stimulators for the treatment of Nonalcoholic Steatohepatitis (NASH) |
| EP3512825B1 (en) | 2016-09-14 | 2020-10-21 | Genomatica, Inc. | 1,3-fatty diol compounds and derivatives thereof |
| CN109890379A (zh) | 2016-10-11 | 2019-06-14 | 拜耳制药股份公司 | 包含sGC活化剂和盐皮质激素受体拮抗剂的组合产品 |
| CN106495999B (zh) * | 2016-10-18 | 2019-08-16 | 亳州学院 | 一种芳香醛及其合成方法 |
| US12491261B2 (en) | 2016-10-26 | 2025-12-09 | Acuitas Therapeutics, Inc. | Lipid nanoparticle formulations |
| AR112392A1 (es) | 2017-07-14 | 2019-10-23 | Glaxosmithkline Ip Dev Ltd | Compuesto tricíclico de pirazol, su uso para fabricar un medicamento y composición farmaceutica que lo comprende |
| EP4501322A3 (en) * | 2017-08-17 | 2025-04-16 | Acuitas Therapeutics, Inc. | Lipids for use in lipid nanoparticle formulations |
| WO2019081456A1 (en) | 2017-10-24 | 2019-05-02 | Bayer Aktiengesellschaft | USE OF SGC ACTIVATORS AND STIMULATORS COMPRISING A BETA2 SUBUNIT |
| EP3498298A1 (en) | 2017-12-15 | 2019-06-19 | Bayer AG | The use of sgc stimulators and sgc activators alone or in combination with pde5 inhibitors for the treatment of bone disorders including osteogenesis imperfecta (oi) |
| EP3784238A4 (en) * | 2018-04-27 | 2021-12-29 | Hetero Labs Ltd. | Process for preparation of ((3r,11br)-1,3,4,6,7,11b-hexahydro-9,10-di(methoxy-d)-3-(2-methylpropyl)-2h-benzo[a]quinolizin-2-one |
| EP3787610A1 (en) | 2018-04-30 | 2021-03-10 | Bayer Aktiengesellschaft | The use of sgc activators and sgc stimulators for the treatment of cognitive impairment |
| EP3793553A1 (en) | 2018-05-15 | 2021-03-24 | Bayer Aktiengesellschaft | 1,3-thiazol-2-yl substituted benzamides for the treatment of diseases associated with nerve fiber sensitization |
| EP3574905A1 (en) | 2018-05-30 | 2019-12-04 | Adverio Pharma GmbH | Method of identifying a subgroup of patients suffering from dcssc which benefits from a treatment with sgc stimulators and sgc activators in a higher degree than a control group |
| CA3116576A1 (en) | 2018-10-18 | 2020-04-23 | Acuitas Therapeutics, Inc. | Lipids for lipid nanoparticle delivery of active agents |
| US12234217B2 (en) | 2018-12-03 | 2025-02-25 | Fmc Corporation | Method for preparing N-phenylpyrazole-1-carboxamides |
| US20220128561A1 (en) | 2019-01-17 | 2022-04-28 | Bayer Aktiengesellschaft | Methods to determine whether a subject is suitable of being treated with an agonist of soluble gyanylyl cyclase (sgc) |
| EP4010333A1 (en) | 2019-08-09 | 2022-06-15 | Kalvista Pharmaceuticals Limited | Plasma kallikrein inhibitors |
| ES3054438T3 (en) | 2020-07-16 | 2026-02-03 | Acuitas Therapeutics Inc | Cationic lipids for use in lipid nanoparticles |
| CN114957087A (zh) * | 2022-04-13 | 2022-08-30 | 湖南复瑞生物医药技术有限责任公司 | 一种帕罗韦德中间体制备方法 |
| EP4536205A1 (en) | 2022-06-09 | 2025-04-16 | Bayer Aktiengesellschaft | Soluble guanylate cyclase activators for use in the treatment of heart failure with preserved ejection fraction in women |
| CN117384082A (zh) * | 2022-07-20 | 2024-01-12 | 杭州国瑞生物科技有限公司 | 一种6,6-二甲基-3-氮杂双环[3.1.0]己烷的合成方法 |
| CN120225203A (zh) | 2022-11-04 | 2025-06-27 | 勃林格殷格翰国际有限公司 | 用于治疗系统性硬化症的可溶性鸟苷酸环化酶活化剂 |
Family Cites Families (24)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20040067938A1 (en) | 2000-09-29 | 2004-04-08 | Penglie Zhang | Quaternary amines and related inhibitors of factor xa |
| AU2006272088A1 (en) | 2005-07-18 | 2007-01-25 | Bayer Schering Pharma Aktiengesellschaft | Novel use of activators and stimulators of soluble guanylate cyclase for the prevention or treatment of renal disorders |
| US20080045560A1 (en) | 2006-08-15 | 2008-02-21 | Wyeth | Pyrrolidine and related derivatives useful as PR modulators |
| AU2008250643A1 (en) | 2007-05-12 | 2008-11-20 | Bayer Schering Pharma Aktiengesellschaft | sGC stimulators, sGC activators and combinations for the treatment of urological disorders |
| BRPI0816382A2 (pt) | 2007-09-06 | 2015-02-24 | Merck Sharp & Dohme | Composto, composição, e, métodos para ativar a guanilato ciclase solúvel e para tratar ou previnir doenças |
| WO2009043495A1 (en) | 2007-10-05 | 2009-04-09 | Sanofi-Aventis Deutschland Gmbh | Use of sulfonyl-substituted 2-sulfonylaminobenzoic acid n-phenylamides in the treatment of pain |
| WO2009068652A1 (en) | 2007-11-30 | 2009-06-04 | Smithkline Beecham Corporation | 2, 6-disubstituted pyridines and 2, 4-disubstituted pyrimidines as soluble guanylate cyclase activators |
| TW200938529A (en) | 2007-12-03 | 2009-09-16 | Smithkline Beecham Corp | Compounds |
| CA2720343A1 (en) | 2008-04-04 | 2009-10-08 | Takeda Pharmaceutical Company Limited | Heterocyclic derivative and use thereof |
| WO2010015652A2 (en) | 2008-08-07 | 2010-02-11 | Smithkline Beecham Corporation | Thiazole compounds as activators of soluble guanylate cyclase |
| WO2010015653A1 (en) | 2008-08-07 | 2010-02-11 | Smithkline Beecham Corporation | Pyrimidine derivatives as activators of soluble guanylate cyclase |
| AU2009322836B2 (en) | 2008-11-25 | 2013-04-04 | Merck Sharp & Dohme Corp. | Soluble guanylate cyclase activators |
| JP5780430B2 (ja) | 2009-01-17 | 2015-09-16 | アドヴェリオ・ファーマ・ゲゼルシャフト・ミット・ベシュレンクテル・ハフツング | 勃起不全の処置用のPDE5阻害剤と組み合わせたsGC刺激剤またはsGC活性化剤 |
| KR20110133034A (ko) | 2009-02-26 | 2011-12-09 | 머크 샤프 앤드 돔 코포레이션 | 가용성 구아닐레이트 시클라제 활성화제 |
| CN102770133A (zh) | 2010-02-05 | 2012-11-07 | 拜耳知识产权有限责任公司 | 用于治疗囊性纤维化的单独和与PDE5抑制剂组合的sGC刺激剂或sGC激活剂 |
| WO2011095553A1 (en) | 2010-02-05 | 2011-08-11 | Bayer Schering Pharma Aktiengesellschaft | Sgc stimulators or sgc activators in combination with pde5 inhbitors for the treatment of erectile dysfunction |
| HRP20150987T1 (hr) | 2010-05-26 | 2015-10-23 | Adverio Pharma Gmbh | UPORABA sGC STIMULATORA, sGC AKTIVATORA, SAMOSTALNO I U KOMBINACIJAMA S INHIBITORIMA PDE5 ZA LIJEÄŚENJE SISTEMSKE SKLEROZE (SSc) |
| EP2687210A1 (de) | 2010-06-25 | 2014-01-22 | Bayer Intellectual Property GmbH | Verwendung von Stimulatoren und Aktivatoren der löslichen Guanylatzyklase zur Behandlung von Sichelzellanämie und Konservierung von Blutersatzstoffen |
| EP2632551B1 (en) | 2010-10-28 | 2016-07-06 | Merck Sharp & Dohme Corp. | Soluble guanylate cyclase activators |
| JP5715713B2 (ja) * | 2011-03-10 | 2015-05-13 | ベーリンガー インゲルハイム インターナショナル ゲゼルシャフト ミット ベシュレンクテル ハフツング | 可溶性グアニル酸シクラーゼ活性化因子 |
| US8815857B2 (en) | 2011-08-12 | 2014-08-26 | Boehringer Ingelheim International Gmbh | Soluble guanylate cyclase activators |
| EP2594270A3 (en) | 2011-11-18 | 2013-07-31 | BIP Patents | The use of sGC stimulators, sGC activators, alone and combinations with PDE5 inhibitors for the treatment of systemic sclerosis (SSc) |
| HRP20191755T1 (hr) * | 2012-09-07 | 2019-12-27 | Boehringer Ingelheim International Gmbh | Alkoksipirazoli kao aktivatori topljive gvanilat ciklaze |
| WO2015095515A1 (en) | 2013-12-20 | 2015-06-25 | Novartis Ag | Sgc activators for the treatment of glaucoma |
-
2015
- 2015-07-21 PL PL15747311T patent/PL3172202T3/pl unknown
- 2015-07-21 AP AP2016009615A patent/AP2016009615A0/en unknown
- 2015-07-21 ES ES15747311T patent/ES2784477T3/es active Active
- 2015-07-21 CA CA2955937A patent/CA2955937A1/en not_active Abandoned
- 2015-07-21 CN CN201580033515.4A patent/CN106459017B/zh active Active
- 2015-07-21 TW TW104123596A patent/TWI711615B/zh not_active IP Right Cessation
- 2015-07-21 SG SG11201700507XA patent/SG11201700507XA/en unknown
- 2015-07-21 AU AU2015292833A patent/AU2015292833B2/en not_active Ceased
- 2015-07-21 DK DK15747311.7T patent/DK3172202T3/da active
- 2015-07-21 JP JP2017503554A patent/JP6602840B2/ja active Active
- 2015-07-21 AR ARP150102304A patent/AR101265A1/es unknown
- 2015-07-21 US US14/804,363 patent/US9353090B2/en active Active
- 2015-07-21 PT PT157473117T patent/PT3172202T/pt unknown
- 2015-07-21 SG SG10201806565SA patent/SG10201806565SA/en unknown
- 2015-07-21 EA EA201790246A patent/EA032972B1/ru not_active IP Right Cessation
- 2015-07-21 PE PE2017000087A patent/PE20170249A1/es unknown
- 2015-07-21 KR KR1020177004761A patent/KR20170035993A/ko not_active Withdrawn
- 2015-07-21 MX MX2017000926A patent/MX378717B/es unknown
- 2015-07-21 BR BR112017000943A patent/BR112017000943A2/pt not_active Application Discontinuation
- 2015-07-21 WO PCT/US2015/041245 patent/WO2016014463A1/en not_active Ceased
- 2015-07-21 HU HUE15747311A patent/HUE049231T2/hu unknown
- 2015-07-21 EP EP15747311.7A patent/EP3172202B1/en active Active
- 2015-07-22 UY UY0001036226A patent/UY36226A/es unknown
-
2016
- 2016-12-15 IL IL249587A patent/IL249587A0/en unknown
-
2017
- 2017-01-11 CL CL2017000071A patent/CL2017000071A1/es unknown
- 2017-01-18 PH PH12017500111A patent/PH12017500111A1/en unknown
- 2017-01-19 CO CONC2017/0000438A patent/CO2017000438A2/es unknown
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2784477T3 (es) | Acidos carboxílicos heterocíclicos como activadores de guanilato ciclasa soluble | |
| ES2748798T3 (es) | Alcoxi pirazoles como activadores de la guanilato ciclasa soluble | |
| CA2930414C (en) | Tetrahydroquinoline compositions as bet bromodomain inhibitors | |
| US12486276B2 (en) | 5-(2,5-difluorophenyl)pyrrolidin-1-yl)-3-(1H-pyrazol-1-yl)pyrazolo[1,5-a]pyrimidine derivatives and related compounds as Trk kinase inhibitors for treating cancer | |
| WO2021088945A1 (zh) | 作为shp2抑制剂的化合物及其应用 | |
| WO2018228275A1 (zh) | 作为mnk抑制剂的杂环化合物 | |
| CN106661034A (zh) | 作为tlr7激动剂的吡咯并嘧啶化合物 | |
| EP3036238A1 (en) | Furo- and thieno-pyridine carboxamide compounds useful as pim kinase inhibitors | |
| JP7282743B2 (ja) | 複素環化合物 | |
| KR20190006567A (ko) | 브로모도메인 억제제로서의 피리딘 디카르복사미드 유도체 | |
| WO2026030722A1 (en) | Substituted imidazolyl dihydropyrimidine-2,4-dione and substituted imidazolyl piperidine-2,6-dione derivatives and uses thereof | |
| HK40070408B (en) | 5-(2-(2,5-difluorophenyl)pyrrolidin-1-yl)-3-(1h-pyrazol-1-yl)pyrazolo[1,5-a]pyrimidine derivatives and related compounds as trk kinase inhibitors for treating cancer | |
| EA043792B1 (ru) | ПРОИЗВОДНЫЕ 5-(2-(2,5-ДИФТОРФЕНИЛ)ПИРРОЛИДИН-1-ИЛ)-3-(1H-ПИРАЗОЛ-1-ИЛ)ПИРАЗОЛО[1,5-а]ПИРИМИДИНА И РОДСТВЕННЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ TRK КИНАЗЫ ДЛЯ ЛЕЧЕНИЯ РАКА |