ES2788660T3 - Derivados de 2,3-dihidro-1h-inden-1-ona como antagonistas de receptores huérfanos relacionados con el ácido retinoico gamma (ROR gamma) para el tratamiento de la esclerosis múltiple - Google Patents
Derivados de 2,3-dihidro-1h-inden-1-ona como antagonistas de receptores huérfanos relacionados con el ácido retinoico gamma (ROR gamma) para el tratamiento de la esclerosis múltiple Download PDFInfo
- Publication number
- ES2788660T3 ES2788660T3 ES14844892T ES14844892T ES2788660T3 ES 2788660 T3 ES2788660 T3 ES 2788660T3 ES 14844892 T ES14844892 T ES 14844892T ES 14844892 T ES14844892 T ES 14844892T ES 2788660 T3 ES2788660 T3 ES 2788660T3
- Authority
- ES
- Spain
- Prior art keywords
- compound
- mmol
- trifluoromethyl
- dihydro
- inden
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 201000006417 multiple sclerosis Diseases 0.000 title claims description 67
- 238000011282 treatment Methods 0.000 title claims description 21
- QNXSIUBBGPHDDE-UHFFFAOYSA-N indan-1-one Chemical class C1=CC=C2C(=O)CCC2=C1 QNXSIUBBGPHDDE-UHFFFAOYSA-N 0.000 title description 11
- 102000016978 Orphan receptors Human genes 0.000 title description 6
- 108070000031 Orphan receptors Proteins 0.000 title description 6
- SHGAZHPCJJPHSC-YCNIQYBTSA-N all-trans-retinoic acid Chemical compound OC(=O)\C=C(/C)\C=C\C=C(/C)\C=C\C1=C(C)CCCC1(C)C SHGAZHPCJJPHSC-YCNIQYBTSA-N 0.000 title description 6
- 229930002330 retinoic acid Natural products 0.000 title description 6
- 229960001727 tretinoin Drugs 0.000 title description 6
- 239000002464 receptor antagonist Substances 0.000 title description 2
- 229940044551 receptor antagonist Drugs 0.000 title description 2
- 150000001875 compounds Chemical class 0.000 claims abstract description 279
- -1 (trifluoromethyl)thio Chemical group 0.000 claims abstract description 60
- 150000003839 salts Chemical class 0.000 claims abstract description 44
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 claims abstract description 17
- 125000004178 (C1-C4) alkyl group Chemical group 0.000 claims abstract description 13
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims abstract description 13
- 125000003226 pyrazolyl group Chemical group 0.000 claims abstract description 9
- 125000004076 pyridyl group Chemical group 0.000 claims abstract description 9
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 claims abstract description 6
- 125000000876 trifluoromethoxy group Chemical group FC(F)(F)O* 0.000 claims abstract description 6
- 125000001475 halogen functional group Chemical group 0.000 claims abstract 11
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims description 31
- 229910052739 hydrogen Inorganic materials 0.000 claims description 31
- 239000001257 hydrogen Substances 0.000 claims description 30
- 229910052757 nitrogen Inorganic materials 0.000 claims description 30
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 27
- 201000010099 disease Diseases 0.000 claims description 27
- 239000008194 pharmaceutical composition Substances 0.000 claims description 15
- 125000001424 substituent group Chemical group 0.000 claims description 13
- 206010028980 Neoplasm Diseases 0.000 claims description 10
- 208000023275 Autoimmune disease Diseases 0.000 claims description 9
- 229910052799 carbon Inorganic materials 0.000 claims description 9
- 239000000546 pharmaceutical excipient Substances 0.000 claims description 9
- 201000011510 cancer Diseases 0.000 claims description 7
- 201000004681 Psoriasis Diseases 0.000 claims description 6
- 229910052760 oxygen Inorganic materials 0.000 claims description 6
- 201000010915 Glioblastoma multiforme Diseases 0.000 claims description 4
- 230000001684 chronic effect Effects 0.000 claims description 4
- 208000005017 glioblastoma Diseases 0.000 claims description 4
- 208000032839 leukemia Diseases 0.000 claims description 4
- 229910052717 sulfur Inorganic materials 0.000 claims description 4
- 238000002560 therapeutic procedure Methods 0.000 claims description 4
- 230000001154 acute effect Effects 0.000 claims description 3
- 150000002431 hydrogen Chemical group 0.000 claims description 3
- 206010005949 Bone cancer Diseases 0.000 claims description 2
- 208000018084 Bone neoplasm Diseases 0.000 claims description 2
- 206010006187 Breast cancer Diseases 0.000 claims description 2
- 208000026310 Breast neoplasm Diseases 0.000 claims description 2
- 208000006168 Ewing Sarcoma Diseases 0.000 claims description 2
- 208000000172 Medulloblastoma Diseases 0.000 claims description 2
- 208000034578 Multiple myelomas Diseases 0.000 claims description 2
- 206010035226 Plasma cell myeloma Diseases 0.000 claims description 2
- 208000029742 colonic neoplasm Diseases 0.000 claims description 2
- 231100000844 hepatocellular carcinoma Toxicity 0.000 claims description 2
- 206010073071 hepatocellular carcinoma Diseases 0.000 claims description 2
- 206010005003 Bladder cancer Diseases 0.000 claims 1
- 201000009030 Carcinoma Diseases 0.000 claims 1
- 206010009944 Colon cancer Diseases 0.000 claims 1
- 206010033128 Ovarian cancer Diseases 0.000 claims 1
- 206010061535 Ovarian neoplasm Diseases 0.000 claims 1
- 208000005718 Stomach Neoplasms Diseases 0.000 claims 1
- 208000007097 Urinary Bladder Neoplasms Diseases 0.000 claims 1
- 210000003443 bladder cell Anatomy 0.000 claims 1
- 210000000481 breast Anatomy 0.000 claims 1
- 206010017758 gastric cancer Diseases 0.000 claims 1
- 201000011549 stomach cancer Diseases 0.000 claims 1
- 201000005112 urinary bladder cancer Diseases 0.000 claims 1
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 426
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 294
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 291
- 238000006243 chemical reaction Methods 0.000 description 190
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 163
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 147
- 239000012044 organic layer Substances 0.000 description 118
- 239000000741 silica gel Substances 0.000 description 102
- 229910002027 silica gel Inorganic materials 0.000 description 102
- 239000007787 solid Substances 0.000 description 102
- 238000003818 flash chromatography Methods 0.000 description 101
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 88
- 239000000243 solution Substances 0.000 description 80
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 59
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 53
- 229910052938 sodium sulfate Inorganic materials 0.000 description 53
- 235000011152 sodium sulphate Nutrition 0.000 description 53
- 238000005160 1H NMR spectroscopy Methods 0.000 description 50
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 50
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 46
- 239000000203 mixture Substances 0.000 description 30
- 125000000217 alkyl group Chemical group 0.000 description 28
- 230000000694 effects Effects 0.000 description 28
- 239000003814 drug Substances 0.000 description 24
- 125000000623 heterocyclic group Chemical group 0.000 description 22
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 21
- 125000005843 halogen group Chemical group 0.000 description 20
- 125000001072 heteroaryl group Chemical group 0.000 description 20
- 125000003118 aryl group Chemical group 0.000 description 19
- 229940079593 drug Drugs 0.000 description 18
- 239000011541 reaction mixture Substances 0.000 description 18
- 241000699670 Mus sp. Species 0.000 description 17
- 238000012360 testing method Methods 0.000 description 17
- 238000000034 method Methods 0.000 description 16
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 15
- 239000005557 antagonist Substances 0.000 description 15
- 210000004027 cell Anatomy 0.000 description 14
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 12
- 125000004432 carbon atom Chemical group C* 0.000 description 12
- 125000001188 haloalkyl group Chemical group 0.000 description 12
- 229920006395 saturated elastomer Polymers 0.000 description 12
- UOPZFPWKIZTDRV-UHFFFAOYSA-N 5-bromo-6-methoxy-2,3-dihydroinden-1-one Chemical compound C1=C(Br)C(OC)=CC2=C1CCC2=O UOPZFPWKIZTDRV-UHFFFAOYSA-N 0.000 description 11
- 108050003558 Interleukin-17 Proteins 0.000 description 11
- 102000013691 Interleukin-17 Human genes 0.000 description 11
- 210000001744 T-lymphocyte Anatomy 0.000 description 11
- 238000003556 assay Methods 0.000 description 11
- CYPYTURSJDMMMP-WVCUSYJESA-N (1e,4e)-1,5-diphenylpenta-1,4-dien-3-one;palladium Chemical compound [Pd].[Pd].C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1.C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1.C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1 CYPYTURSJDMMMP-WVCUSYJESA-N 0.000 description 10
- MUALRAIOVNYAIW-UHFFFAOYSA-N binap Chemical compound C1=CC=CC=C1P(C=1C(=C2C=CC=CC2=CC=1)C=1C2=CC=CC=C2C=CC=1P(C=1C=CC=CC=1)C=1C=CC=CC=1)C1=CC=CC=C1 MUALRAIOVNYAIW-UHFFFAOYSA-N 0.000 description 10
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 description 10
- 229910000024 caesium carbonate Inorganic materials 0.000 description 10
- 238000009472 formulation Methods 0.000 description 10
- 230000001404 mediated effect Effects 0.000 description 10
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 10
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 10
- 206010039073 rheumatoid arthritis Diseases 0.000 description 10
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 9
- 201000002491 encephalomyelitis Diseases 0.000 description 9
- 230000005764 inhibitory process Effects 0.000 description 9
- 238000003756 stirring Methods 0.000 description 9
- 102000004127 Cytokines Human genes 0.000 description 8
- 108090000695 Cytokines Proteins 0.000 description 8
- 210000000068 Th17 cell Anatomy 0.000 description 8
- 239000003795 chemical substances by application Substances 0.000 description 8
- WMAZDKMPJNHNIV-UHFFFAOYSA-N COc1cc2C(=O)C(Cc3cc(ccc3F)C(F)(F)F)Cc2cc1N1CCOCC1 Chemical compound COc1cc2C(=O)C(Cc3cc(ccc3F)C(F)(F)F)Cc2cc1N1CCOCC1 WMAZDKMPJNHNIV-UHFFFAOYSA-N 0.000 description 7
- 239000002775 capsule Substances 0.000 description 7
- 239000000969 carrier Substances 0.000 description 7
- 230000002757 inflammatory effect Effects 0.000 description 7
- 239000003112 inhibitor Substances 0.000 description 7
- 230000024949 interleukin-17 production Effects 0.000 description 7
- 230000004770 neurodegeneration Effects 0.000 description 7
- 238000000746 purification Methods 0.000 description 7
- 208000024891 symptom Diseases 0.000 description 7
- OPAXHVNZGUGCKW-UHFFFAOYSA-N 4-(trifluoromethylsulfanyl)benzaldehyde Chemical compound FC(F)(F)SC1=CC=C(C=O)C=C1 OPAXHVNZGUGCKW-UHFFFAOYSA-N 0.000 description 6
- 0 CCC(CC(*OBr)=Cc1cccc(C(F)(F)F)c1)=C Chemical compound CCC(CC(*OBr)=Cc1cccc(C(F)(F)F)c1)=C 0.000 description 6
- PPWUKBPSDPHSDR-UHFFFAOYSA-N COc1cc2C(=O)C(Cc3c(Cl)cccc3C(F)(F)F)Cc2cc1N1CCOCC1 Chemical compound COc1cc2C(=O)C(Cc3c(Cl)cccc3C(F)(F)F)Cc2cc1N1CCOCC1 PPWUKBPSDPHSDR-UHFFFAOYSA-N 0.000 description 6
- HBEFBXHJZMYUFM-UHFFFAOYSA-N COc1cc2C(=O)C(Cc3ccc(nc3)C(F)(F)F)Cc2cc1N1CCOCC1 Chemical compound COc1cc2C(=O)C(Cc3ccc(nc3)C(F)(F)F)Cc2cc1N1CCOCC1 HBEFBXHJZMYUFM-UHFFFAOYSA-N 0.000 description 6
- KPXUQRNTSMAVRN-UHFFFAOYSA-N COc1cc2C(=O)C(Cc3cncc(c3)C(O)(C(F)(F)F)C(F)(F)F)Cc2cc1N1CCOCC1 Chemical compound COc1cc2C(=O)C(Cc3cncc(c3)C(O)(C(F)(F)F)C(F)(F)F)Cc2cc1N1CCOCC1 KPXUQRNTSMAVRN-UHFFFAOYSA-N 0.000 description 6
- 108010010803 Gelatin Proteins 0.000 description 6
- 102000007399 Nuclear hormone receptor Human genes 0.000 description 6
- 108020005497 Nuclear hormone receptor Proteins 0.000 description 6
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 6
- 230000002411 adverse Effects 0.000 description 6
- 125000003545 alkoxy group Chemical group 0.000 description 6
- 239000008346 aqueous phase Substances 0.000 description 6
- 125000003710 aryl alkyl group Chemical group 0.000 description 6
- 239000006184 cosolvent Substances 0.000 description 6
- 238000011161 development Methods 0.000 description 6
- 230000018109 developmental process Effects 0.000 description 6
- 206010012601 diabetes mellitus Diseases 0.000 description 6
- 231100000673 dose–response relationship Toxicity 0.000 description 6
- 229960000556 fingolimod Drugs 0.000 description 6
- KKGQTZUTZRNORY-UHFFFAOYSA-N fingolimod Chemical compound CCCCCCCCC1=CC=C(CCC(N)(CO)CO)C=C1 KKGQTZUTZRNORY-UHFFFAOYSA-N 0.000 description 6
- 229920000159 gelatin Polymers 0.000 description 6
- 239000008273 gelatin Substances 0.000 description 6
- 235000019322 gelatine Nutrition 0.000 description 6
- 235000011852 gelatine desserts Nutrition 0.000 description 6
- VBZWSGALLODQNC-UHFFFAOYSA-N hexafluoroacetone Chemical compound FC(F)(F)C(=O)C(F)(F)F VBZWSGALLODQNC-UHFFFAOYSA-N 0.000 description 6
- 230000002209 hydrophobic effect Effects 0.000 description 6
- 238000001727 in vivo Methods 0.000 description 6
- 238000002347 injection Methods 0.000 description 6
- 239000007924 injection Substances 0.000 description 6
- 239000000463 material Substances 0.000 description 6
- 229920001223 polyethylene glycol Polymers 0.000 description 6
- 239000000651 prodrug Substances 0.000 description 6
- 229940002612 prodrug Drugs 0.000 description 6
- 150000003384 small molecules Chemical class 0.000 description 6
- 239000000126 substance Substances 0.000 description 6
- 239000000725 suspension Substances 0.000 description 6
- 230000001225 therapeutic effect Effects 0.000 description 6
- IGMVUYYIXHDEHX-UHFFFAOYSA-N 5-(1,1,1,3,3,3-hexafluoro-2-hydroxypropan-2-yl)pyridine-3-carbaldehyde Chemical compound FC(F)(F)C(C(F)(F)F)(O)C1=CN=CC(C=O)=C1 IGMVUYYIXHDEHX-UHFFFAOYSA-N 0.000 description 5
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical class [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 5
- CZYNTCAZXZSKGD-UHFFFAOYSA-N COc1cc2C(=O)C(Cc3ccc(Cl)cc3F)Cc2cc1N1CCOCC1 Chemical compound COc1cc2C(=O)C(Cc3ccc(Cl)cc3F)Cc2cc1N1CCOCC1 CZYNTCAZXZSKGD-UHFFFAOYSA-N 0.000 description 5
- 208000006545 Chronic Obstructive Pulmonary Disease Diseases 0.000 description 5
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 5
- 208000032928 Dyslipidaemia Diseases 0.000 description 5
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 5
- 208000017170 Lipid metabolism disease Diseases 0.000 description 5
- DUTRZNVJIOCNQX-UHFFFAOYSA-N OC(c1cccc(C=O)n1)(C(F)(F)F)C(F)(F)F Chemical compound OC(c1cccc(C=O)n1)(C(F)(F)F)C(F)(F)F DUTRZNVJIOCNQX-UHFFFAOYSA-N 0.000 description 5
- 208000008589 Obesity Diseases 0.000 description 5
- 208000001132 Osteoporosis Diseases 0.000 description 5
- 208000002193 Pain Diseases 0.000 description 5
- 102000001708 Protein Isoforms Human genes 0.000 description 5
- 108010029485 Protein Isoforms Proteins 0.000 description 5
- 229920002472 Starch Polymers 0.000 description 5
- 208000006673 asthma Diseases 0.000 description 5
- 210000004369 blood Anatomy 0.000 description 5
- 239000008280 blood Substances 0.000 description 5
- 239000003153 chemical reaction reagent Substances 0.000 description 5
- 206010009887 colitis Diseases 0.000 description 5
- 238000002868 homogeneous time resolved fluorescence Methods 0.000 description 5
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 5
- 208000015122 neurodegenerative disease Diseases 0.000 description 5
- 235000020824 obesity Nutrition 0.000 description 5
- 230000036470 plasma concentration Effects 0.000 description 5
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 5
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 5
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 5
- 229910000027 potassium carbonate Inorganic materials 0.000 description 5
- 230000003389 potentiating effect Effects 0.000 description 5
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 5
- 125000006413 ring segment Chemical group 0.000 description 5
- 235000000346 sugar Nutrition 0.000 description 5
- 239000003981 vehicle Substances 0.000 description 5
- UNILWMWFPHPYOR-KXEYIPSPSA-M 1-[6-[2-[3-[3-[3-[2-[2-[3-[[2-[2-[[(2r)-1-[[2-[[(2r)-1-[3-[2-[2-[3-[[2-(2-amino-2-oxoethoxy)acetyl]amino]propoxy]ethoxy]ethoxy]propylamino]-3-hydroxy-1-oxopropan-2-yl]amino]-2-oxoethyl]amino]-3-[(2r)-2,3-di(hexadecanoyloxy)propyl]sulfanyl-1-oxopropan-2-yl Chemical compound O=C1C(SCCC(=O)NCCCOCCOCCOCCCNC(=O)COCC(=O)N[C@@H](CSC[C@@H](COC(=O)CCCCCCCCCCCCCCC)OC(=O)CCCCCCCCCCCCCCC)C(=O)NCC(=O)N[C@H](CO)C(=O)NCCCOCCOCCOCCCNC(=O)COCC(N)=O)CC(=O)N1CCNC(=O)CCCCCN\1C2=CC=C(S([O-])(=O)=O)C=C2CC/1=C/C=C/C=C/C1=[N+](CC)C2=CC=C(S([O-])(=O)=O)C=C2C1 UNILWMWFPHPYOR-KXEYIPSPSA-M 0.000 description 4
- NHBKXEKEPDILRR-UHFFFAOYSA-N 2,3-bis(butanoylsulfanyl)propyl butanoate Chemical compound CCCC(=O)OCC(SC(=O)CCC)CSC(=O)CCC NHBKXEKEPDILRR-UHFFFAOYSA-N 0.000 description 4
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 4
- SOMJOILNRPKGID-UHFFFAOYSA-N CCS(=O)(=O)Oc1cc2C(=O)C(Cc3ncccc3C(F)(F)F)Cc2cc1OC Chemical compound CCS(=O)(=O)Oc1cc2C(=O)C(Cc3ncccc3C(F)(F)F)Cc2cc1OC SOMJOILNRPKGID-UHFFFAOYSA-N 0.000 description 4
- KCDZEXQCCQLLTP-UHFFFAOYSA-N COc1cc2C(=O)C(Cc3cc(Cl)cc(c3)C(F)(F)F)Cc2cc1N1CCOCC1 Chemical compound COc1cc2C(=O)C(Cc3cc(Cl)cc(c3)C(F)(F)F)Cc2cc1N1CCOCC1 KCDZEXQCCQLLTP-UHFFFAOYSA-N 0.000 description 4
- JBGVMTGKCRQJFA-UHFFFAOYSA-N COc1cc2C(=O)C(Cc3cc(Cl)ccc3C(F)(F)F)Cc2cc1N1CCOCC1 Chemical compound COc1cc2C(=O)C(Cc3cc(Cl)ccc3C(F)(F)F)Cc2cc1N1CCOCC1 JBGVMTGKCRQJFA-UHFFFAOYSA-N 0.000 description 4
- PYKNHIHZPHUBLX-UHFFFAOYSA-N COc1cc2C(=O)C(Cc3ccc(SC(F)(F)F)cc3)Cc2cc1N1CCOCC1 Chemical compound COc1cc2C(=O)C(Cc3ccc(SC(F)(F)F)cc3)Cc2cc1N1CCOCC1 PYKNHIHZPHUBLX-UHFFFAOYSA-N 0.000 description 4
- HKBQTDQANIQUJZ-UHFFFAOYSA-N COc1cc2C(=O)C(Cc3cccc(OC(F)(F)F)c3)Cc2cc1N1CCOCC1 Chemical compound COc1cc2C(=O)C(Cc3cccc(OC(F)(F)F)c3)Cc2cc1N1CCOCC1 HKBQTDQANIQUJZ-UHFFFAOYSA-N 0.000 description 4
- GFGOVWLFOVHFFG-UHFFFAOYSA-N COc1cc2C(=O)C(Cc3ncccc3C(F)(F)F)Cc2cc1N1CCN(C)CC1 Chemical compound COc1cc2C(=O)C(Cc3ncccc3C(F)(F)F)Cc2cc1N1CCN(C)CC1 GFGOVWLFOVHFFG-UHFFFAOYSA-N 0.000 description 4
- 102000006386 Myelin Proteins Human genes 0.000 description 4
- 108010083674 Myelin Proteins Proteins 0.000 description 4
- 102000001253 Protein Kinase Human genes 0.000 description 4
- 239000004480 active ingredient Substances 0.000 description 4
- 125000002252 acyl group Chemical group 0.000 description 4
- 125000004429 atom Chemical group 0.000 description 4
- 239000000872 buffer Substances 0.000 description 4
- 238000004113 cell culture Methods 0.000 description 4
- 210000003169 central nervous system Anatomy 0.000 description 4
- 125000004093 cyano group Chemical group *C#N 0.000 description 4
- 230000006378 damage Effects 0.000 description 4
- 238000001514 detection method Methods 0.000 description 4
- 230000004069 differentiation Effects 0.000 description 4
- LDCRTTXIJACKKU-ONEGZZNKSA-N dimethyl fumarate Chemical compound COC(=O)\C=C\C(=O)OC LDCRTTXIJACKKU-ONEGZZNKSA-N 0.000 description 4
- FRYHCSODNHYDPU-UHFFFAOYSA-N ethanesulfonyl chloride Chemical compound CCS(Cl)(=O)=O FRYHCSODNHYDPU-UHFFFAOYSA-N 0.000 description 4
- 239000000499 gel Substances 0.000 description 4
- 125000004438 haloalkoxy group Chemical group 0.000 description 4
- 125000004446 heteroarylalkyl group Chemical group 0.000 description 4
- 125000000592 heterocycloalkyl group Chemical group 0.000 description 4
- 238000001990 intravenous administration Methods 0.000 description 4
- 239000007788 liquid Substances 0.000 description 4
- IUYHWZFSGMZEOG-UHFFFAOYSA-M magnesium;propane;chloride Chemical compound [Mg+2].[Cl-].C[CH-]C IUYHWZFSGMZEOG-UHFFFAOYSA-M 0.000 description 4
- 210000005012 myelin Anatomy 0.000 description 4
- SGIWFELWJPNFDH-UHFFFAOYSA-N n-(2,2,2-trifluoroethyl)-n-{4-[2,2,2-trifluoro-1-hydroxy-1-(trifluoromethyl)ethyl]phenyl}benzenesulfonamide Chemical compound C1=CC(C(O)(C(F)(F)F)C(F)(F)F)=CC=C1N(CC(F)(F)F)S(=O)(=O)C1=CC=CC=C1 SGIWFELWJPNFDH-UHFFFAOYSA-N 0.000 description 4
- 239000013642 negative control Substances 0.000 description 4
- 108090000629 orphan nuclear receptors Proteins 0.000 description 4
- 102000004164 orphan nuclear receptors Human genes 0.000 description 4
- 238000004806 packaging method and process Methods 0.000 description 4
- 239000013641 positive control Substances 0.000 description 4
- 235000015320 potassium carbonate Nutrition 0.000 description 4
- 238000002360 preparation method Methods 0.000 description 4
- 108060006633 protein kinase Proteins 0.000 description 4
- 150000003254 radicals Chemical class 0.000 description 4
- 108090000064 retinoic acid receptors Proteins 0.000 description 4
- 102000003702 retinoic acid receptors Human genes 0.000 description 4
- 239000003381 stabilizer Substances 0.000 description 4
- 235000019698 starch Nutrition 0.000 description 4
- 230000000638 stimulation Effects 0.000 description 4
- 150000008163 sugars Chemical class 0.000 description 4
- 238000003786 synthesis reaction Methods 0.000 description 4
- 231100000419 toxicity Toxicity 0.000 description 4
- 230000001988 toxicity Effects 0.000 description 4
- GLGNXYJARSMNGJ-VKTIVEEGSA-N (1s,2s,3r,4r)-3-[[5-chloro-2-[(1-ethyl-6-methoxy-2-oxo-4,5-dihydro-3h-1-benzazepin-7-yl)amino]pyrimidin-4-yl]amino]bicyclo[2.2.1]hept-5-ene-2-carboxamide Chemical compound CCN1C(=O)CCCC2=C(OC)C(NC=3N=C(C(=CN=3)Cl)N[C@H]3[C@H]([C@@]4([H])C[C@@]3(C=C4)[H])C(N)=O)=CC=C21 GLGNXYJARSMNGJ-VKTIVEEGSA-N 0.000 description 3
- PVOAHINGSUIXLS-UHFFFAOYSA-N 1-Methylpiperazine Chemical compound CN1CCNCC1 PVOAHINGSUIXLS-UHFFFAOYSA-N 0.000 description 3
- WYFCZWSWFGJODV-MIANJLSGSA-N 4-[[(1s)-2-[(e)-3-[3-chloro-2-fluoro-6-(tetrazol-1-yl)phenyl]prop-2-enoyl]-5-(4-methyl-2-oxopiperazin-1-yl)-3,4-dihydro-1h-isoquinoline-1-carbonyl]amino]benzoic acid Chemical compound O=C1CN(C)CCN1C1=CC=CC2=C1CCN(C(=O)\C=C\C=1C(=CC=C(Cl)C=1F)N1N=NN=C1)[C@@H]2C(=O)NC1=CC=C(C(O)=O)C=C1 WYFCZWSWFGJODV-MIANJLSGSA-N 0.000 description 3
- UZOFELREXGAFOI-UHFFFAOYSA-N 4-methylpiperidine Chemical compound CC1CCNCC1 UZOFELREXGAFOI-UHFFFAOYSA-N 0.000 description 3
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 3
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 3
- ZPQOLAWSQKXORY-UHFFFAOYSA-N COc1cc2C(=O)C(Cc3c(F)cccc3F)Cc2cc1N1CCOCC1 Chemical compound COc1cc2C(=O)C(Cc3c(F)cccc3F)Cc2cc1N1CCOCC1 ZPQOLAWSQKXORY-UHFFFAOYSA-N 0.000 description 3
- DLIZMPQHTHUYKX-UHFFFAOYSA-N COc1cc2C(=O)C(Cc3cc(ccc3C(O)(C(F)(F)F)C(F)(F)F)C(F)(F)F)Cc2cc1N1CCOCC1 Chemical compound COc1cc2C(=O)C(Cc3cc(ccc3C(O)(C(F)(F)F)C(F)(F)F)C(F)(F)F)Cc2cc1N1CCOCC1 DLIZMPQHTHUYKX-UHFFFAOYSA-N 0.000 description 3
- AVBJQXGJDUEIMB-UHFFFAOYSA-N COc1cc2C(=O)C(Cc3cc(ccc3Cl)C(F)(F)F)Cc2cc1N1CCOCC1 Chemical compound COc1cc2C(=O)C(Cc3cc(ccc3Cl)C(F)(F)F)Cc2cc1N1CCOCC1 AVBJQXGJDUEIMB-UHFFFAOYSA-N 0.000 description 3
- GRQXXAVLHRDADV-UHFFFAOYSA-N COc1cc2C(=O)C(Cc3ccc(Cl)c(c3)C(F)(F)F)Cc2cc1N1CCOCC1 Chemical compound COc1cc2C(=O)C(Cc3ccc(Cl)c(c3)C(F)(F)F)Cc2cc1N1CCOCC1 GRQXXAVLHRDADV-UHFFFAOYSA-N 0.000 description 3
- LHHWRRVPYHPYMU-UHFFFAOYSA-N COc1cc2C(=O)C(Cc3ccc(Cl)cc3C(F)(F)F)Cc2cc1N1CCOCC1 Chemical compound COc1cc2C(=O)C(Cc3ccc(Cl)cc3C(F)(F)F)Cc2cc1N1CCOCC1 LHHWRRVPYHPYMU-UHFFFAOYSA-N 0.000 description 3
- DSBWPZVJSRCINU-UHFFFAOYSA-N COc1cc2C(=O)C(Cc3ccc(F)cc3F)Cc2cc1N1CCOCC1 Chemical compound COc1cc2C(=O)C(Cc3ccc(F)cc3F)Cc2cc1N1CCOCC1 DSBWPZVJSRCINU-UHFFFAOYSA-N 0.000 description 3
- RDWQVEQATLCJRL-UHFFFAOYSA-N COc1cc2C(=O)C(Cc3ccc(SC(F)(F)F)cc3)Cc2cc1N1CCN(C)CC1 Chemical compound COc1cc2C(=O)C(Cc3ccc(SC(F)(F)F)cc3)Cc2cc1N1CCN(C)CC1 RDWQVEQATLCJRL-UHFFFAOYSA-N 0.000 description 3
- OMPWBZAKNFBVQC-UHFFFAOYSA-N COc1cc2C(=O)C(Cc3cccc(c3)C(F)(F)F)Cc2cc1N1CCN(C)CC1 Chemical compound COc1cc2C(=O)C(Cc3cccc(c3)C(F)(F)F)Cc2cc1N1CCN(C)CC1 OMPWBZAKNFBVQC-UHFFFAOYSA-N 0.000 description 3
- KDAZYRXSLFIIRV-UHFFFAOYSA-N COc1cc2C(=O)C(Cc3cccc(n3)C(O)(C(F)(F)F)C(F)(F)F)Cc2cc1N1CCOCC1 Chemical compound COc1cc2C(=O)C(Cc3cccc(n3)C(O)(C(F)(F)F)C(F)(F)F)Cc2cc1N1CCOCC1 KDAZYRXSLFIIRV-UHFFFAOYSA-N 0.000 description 3
- IWNZTDZHSAPELD-UHFFFAOYSA-N COc1cc2C(=O)C(Cc3cncc(c3)C(O)(C(F)(F)F)C(F)(F)F)Cc2cc1N1CCN(C)CC1 Chemical compound COc1cc2C(=O)C(Cc3cncc(c3)C(O)(C(F)(F)F)C(F)(F)F)Cc2cc1N1CCN(C)CC1 IWNZTDZHSAPELD-UHFFFAOYSA-N 0.000 description 3
- RPGCXXUKTLPAPD-UHFFFAOYSA-N COc1cc2C(=O)C(Cc3ncc(cc3C(F)(F)F)C(O)(C(F)(F)F)C(F)(F)F)Cc2cc1N1CCOCC1 Chemical compound COc1cc2C(=O)C(Cc3ncc(cc3C(F)(F)F)C(O)(C(F)(F)F)C(F)(F)F)Cc2cc1N1CCOCC1 RPGCXXUKTLPAPD-UHFFFAOYSA-N 0.000 description 3
- COYSSAHTTZOPKS-UHFFFAOYSA-N COc1cc2C(=O)C(Cc3sc(nc3Cl)N3CCOCC3)Cc2cc1N1CCOCC1 Chemical compound COc1cc2C(=O)C(Cc3sc(nc3Cl)N3CCOCC3)Cc2cc1N1CCOCC1 COYSSAHTTZOPKS-UHFFFAOYSA-N 0.000 description 3
- OYTGWRRNOBNNRA-UHFFFAOYSA-N COc1ccc2C(=O)C(Cc3ccc(SC(F)(F)F)cc3)Cc2c1OC Chemical compound COc1ccc2C(=O)C(Cc3ccc(SC(F)(F)F)cc3)Cc2c1OC OYTGWRRNOBNNRA-UHFFFAOYSA-N 0.000 description 3
- 239000004215 Carbon black (E152) Substances 0.000 description 3
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 3
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 3
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 3
- 241000282412 Homo Species 0.000 description 3
- 102000015696 Interleukins Human genes 0.000 description 3
- 108010063738 Interleukins Proteins 0.000 description 3
- 239000002139 L01XE22 - Masitinib Substances 0.000 description 3
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 3
- 108010029279 Member 3 Group F Nuclear Receptor Subfamily 1 Proteins 0.000 description 3
- 102000001691 Member 3 Group F Nuclear Receptor Subfamily 1 Human genes 0.000 description 3
- 241000699666 Mus <mouse, genus> Species 0.000 description 3
- 208000001388 Opportunistic Infections Diseases 0.000 description 3
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 3
- 239000002202 Polyethylene glycol Substances 0.000 description 3
- OFOBLEOULBTSOW-UHFFFAOYSA-N Propanedioic acid Natural products OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 3
- KAESVJOAVNADME-UHFFFAOYSA-N Pyrrole Chemical compound C=1C=CNC=1 KAESVJOAVNADME-UHFFFAOYSA-N 0.000 description 3
- RWRDLPDLKQPQOW-UHFFFAOYSA-N Pyrrolidine Chemical compound C1CCNC1 RWRDLPDLKQPQOW-UHFFFAOYSA-N 0.000 description 3
- 239000007868 Raney catalyst Substances 0.000 description 3
- NPXOKRUENSOPAO-UHFFFAOYSA-N Raney nickel Chemical compound [Al].[Ni] NPXOKRUENSOPAO-UHFFFAOYSA-N 0.000 description 3
- 229910000564 Raney nickel Inorganic materials 0.000 description 3
- 241000700159 Rattus Species 0.000 description 3
- 208000007400 Relapsing-Remitting Multiple Sclerosis Diseases 0.000 description 3
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 3
- 125000004453 alkoxycarbonyl group Chemical group 0.000 description 3
- 125000003282 alkyl amino group Chemical group 0.000 description 3
- 125000004414 alkyl thio group Chemical group 0.000 description 3
- VREFGVBLTWBCJP-UHFFFAOYSA-N alprazolam Chemical compound C12=CC(Cl)=CC=C2N2C(C)=NN=C2CN=C1C1=CC=CC=C1 VREFGVBLTWBCJP-UHFFFAOYSA-N 0.000 description 3
- 238000010171 animal model Methods 0.000 description 3
- 230000001363 autoimmune Effects 0.000 description 3
- 230000015572 biosynthetic process Effects 0.000 description 3
- 210000004556 brain Anatomy 0.000 description 3
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 3
- 239000001913 cellulose Substances 0.000 description 3
- 229920002678 cellulose Polymers 0.000 description 3
- 235000010980 cellulose Nutrition 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- 238000000576 coating method Methods 0.000 description 3
- 229940125758 compound 15 Drugs 0.000 description 3
- 239000013078 crystal Substances 0.000 description 3
- 125000000753 cycloalkyl group Chemical group 0.000 description 3
- 230000007423 decrease Effects 0.000 description 3
- 125000004663 dialkyl amino group Chemical group 0.000 description 3
- 238000011979 disease modifying therapy Methods 0.000 description 3
- 208000035475 disorder Diseases 0.000 description 3
- 239000003937 drug carrier Substances 0.000 description 3
- 239000000839 emulsion Substances 0.000 description 3
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 3
- 238000002474 experimental method Methods 0.000 description 3
- 210000002443 helper t lymphocyte Anatomy 0.000 description 3
- 231100000304 hepatotoxicity Toxicity 0.000 description 3
- 229930195733 hydrocarbon Natural products 0.000 description 3
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 3
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Natural products C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 description 3
- 238000000338 in vitro Methods 0.000 description 3
- 238000011534 incubation Methods 0.000 description 3
- SNWQUNCRDLUDEX-UHFFFAOYSA-N inden-1-one Chemical class C1=CC=C2C(=O)C=CC2=C1 SNWQUNCRDLUDEX-UHFFFAOYSA-N 0.000 description 3
- 230000002401 inhibitory effect Effects 0.000 description 3
- 229940125425 inverse agonist Drugs 0.000 description 3
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 3
- 239000008101 lactose Substances 0.000 description 3
- 239000003446 ligand Substances 0.000 description 3
- DLEDOFVPSDKWEF-UHFFFAOYSA-N lithium butane Chemical compound [Li+].CCC[CH2-] DLEDOFVPSDKWEF-UHFFFAOYSA-N 0.000 description 3
- 210000004185 liver Anatomy 0.000 description 3
- 238000004519 manufacturing process Methods 0.000 description 3
- WJEOLQLKVOPQFV-UHFFFAOYSA-N masitinib Chemical compound C1CN(C)CCN1CC1=CC=C(C(=O)NC=2C=C(NC=3SC=C(N=3)C=3C=NC=CC=3)C(C)=CC=2)C=C1 WJEOLQLKVOPQFV-UHFFFAOYSA-N 0.000 description 3
- 229960004655 masitinib Drugs 0.000 description 3
- MZRVEZGGRBJDDB-UHFFFAOYSA-N n-Butyllithium Substances [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 description 3
- 231100000417 nephrotoxicity Toxicity 0.000 description 3
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 description 3
- XIPFMBOWZXULIA-UHFFFAOYSA-N pivalamide Chemical compound CC(C)(C)C(N)=O XIPFMBOWZXULIA-UHFFFAOYSA-N 0.000 description 3
- 239000000843 powder Substances 0.000 description 3
- 108090000765 processed proteins & peptides Proteins 0.000 description 3
- 230000000770 proinflammatory effect Effects 0.000 description 3
- 210000002966 serum Anatomy 0.000 description 3
- 239000000377 silicon dioxide Substances 0.000 description 3
- 239000000600 sorbitol Substances 0.000 description 3
- 239000008107 starch Substances 0.000 description 3
- 125000003107 substituted aryl group Chemical group 0.000 description 3
- PXQLVRUNWNTZOS-UHFFFAOYSA-N sulfanyl Chemical class [SH] PXQLVRUNWNTZOS-UHFFFAOYSA-N 0.000 description 3
- 239000004094 surface-active agent Substances 0.000 description 3
- 238000013268 sustained release Methods 0.000 description 3
- 239000012730 sustained-release form Substances 0.000 description 3
- 239000003826 tablet Substances 0.000 description 3
- KZPYGQFFRCFCPP-UHFFFAOYSA-N 1,1'-bis(diphenylphosphino)ferrocene Chemical compound [Fe+2].C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1 KZPYGQFFRCFCPP-UHFFFAOYSA-N 0.000 description 2
- KQZLRWGGWXJPOS-NLFPWZOASA-N 1-[(1R)-1-(2,4-dichlorophenyl)ethyl]-6-[(4S,5R)-4-[(2S)-2-(hydroxymethyl)pyrrolidin-1-yl]-5-methylcyclohexen-1-yl]pyrazolo[3,4-b]pyrazine-3-carbonitrile Chemical class ClC1=C(C=CC(=C1)Cl)[C@@H](C)N1N=C(C=2C1=NC(=CN=2)C1=CC[C@@H]([C@@H](C1)C)N1[C@@H](CCC1)CO)C#N KQZLRWGGWXJPOS-NLFPWZOASA-N 0.000 description 2
- ONBQEOIKXPHGMB-VBSBHUPXSA-N 1-[2-[(2s,3r,4s,5r)-3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]oxy-4,6-dihydroxyphenyl]-3-(4-hydroxyphenyl)propan-1-one Chemical compound O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1OC1=CC(O)=CC(O)=C1C(=O)CCC1=CC=C(O)C=C1 ONBQEOIKXPHGMB-VBSBHUPXSA-N 0.000 description 2
- HZAXFHJVJLSVMW-UHFFFAOYSA-N 2-Aminoethan-1-ol Chemical compound NCCO HZAXFHJVJLSVMW-UHFFFAOYSA-N 0.000 description 2
- XQODIRIVQXQUFN-UHFFFAOYSA-N 4,5-dimethoxy-2,3-dihydroinden-1-one Chemical compound COC1=CC=C2C(=O)CCC2=C1OC XQODIRIVQXQUFN-UHFFFAOYSA-N 0.000 description 2
- RSIWALKZYXPAGW-NSHDSACASA-N 6-(3-fluorophenyl)-3-methyl-7-[(1s)-1-(7h-purin-6-ylamino)ethyl]-[1,3]thiazolo[3,2-a]pyrimidin-5-one Chemical compound C=1([C@@H](NC=2C=3N=CNC=3N=CN=2)C)N=C2SC=C(C)N2C(=O)C=1C1=CC=CC(F)=C1 RSIWALKZYXPAGW-NSHDSACASA-N 0.000 description 2
- KDCGOANMDULRCW-UHFFFAOYSA-N 7H-purine Chemical compound N1=CNC2=NC=NC2=C1 KDCGOANMDULRCW-UHFFFAOYSA-N 0.000 description 2
- UJOBWOGCFQCDNV-UHFFFAOYSA-N 9H-carbazole Chemical compound C1=CC=C2C3=CC=CC=C3NC2=C1 UJOBWOGCFQCDNV-UHFFFAOYSA-N 0.000 description 2
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 2
- LGSYLIZFPQYCET-WUXMJOGZSA-N C1c2cc(Br)c(OC)cc2C(=O)\C1=C\c1ccc(SC(F)(F)F)cc1 Chemical compound C1c2cc(Br)c(OC)cc2C(=O)\C1=C\c1ccc(SC(F)(F)F)cc1 LGSYLIZFPQYCET-WUXMJOGZSA-N 0.000 description 2
- SHQQFTRPYZXFFB-LFYBBSHMSA-N C1c2cc(Br)c(OC)cc2C(=O)\C1=C\c1cccc(C(F)(F)F)c1 Chemical compound C1c2cc(Br)c(OC)cc2C(=O)\C1=C\c1cccc(C(F)(F)F)c1 SHQQFTRPYZXFFB-LFYBBSHMSA-N 0.000 description 2
- AHXUZAMLGCJOQS-UHFFFAOYSA-N CCS(=O)(=O)Oc1cc2C(=O)C(Cc3ccc(SC(F)(F)F)cc3)Cc2cc1OC Chemical compound CCS(=O)(=O)Oc1cc2C(=O)C(Cc3ccc(SC(F)(F)F)cc3)Cc2cc1OC AHXUZAMLGCJOQS-UHFFFAOYSA-N 0.000 description 2
- DKIPHARGQOTYBK-UHFFFAOYSA-N COc1cc2C(=O)C(Cc3ccc(SC(F)(F)F)cc3)Cc2cc1Cl Chemical compound COc1cc2C(=O)C(Cc3ccc(SC(F)(F)F)cc3)Cc2cc1Cl DKIPHARGQOTYBK-UHFFFAOYSA-N 0.000 description 2
- HVANMQXKNUWHQJ-UHFFFAOYSA-N COc1cc2C(=O)C(Cc3ccc(SC(F)(F)F)cc3)Cc2cc1N1CCNCC1 Chemical compound COc1cc2C(=O)C(Cc3ccc(SC(F)(F)F)cc3)Cc2cc1N1CCNCC1 HVANMQXKNUWHQJ-UHFFFAOYSA-N 0.000 description 2
- XZYDXDCEOFIFTK-UHFFFAOYSA-N COc1cc2C(=O)C(Cc3ccc(c(Cl)c3)C(F)(F)F)Cc2cc1N1CCOCC1 Chemical compound COc1cc2C(=O)C(Cc3ccc(c(Cl)c3)C(F)(F)F)Cc2cc1N1CCOCC1 XZYDXDCEOFIFTK-UHFFFAOYSA-N 0.000 description 2
- WCXHEKCJEKNBEO-UHFFFAOYSA-N COc1cc2C(=O)C(Cc3ncccc3C(F)(F)F)Cc2cc1-c1ccc(OC(F)(F)F)cc1F Chemical compound COc1cc2C(=O)C(Cc3ncccc3C(F)(F)F)Cc2cc1-c1ccc(OC(F)(F)F)cc1F WCXHEKCJEKNBEO-UHFFFAOYSA-N 0.000 description 2
- VRNDNTBVUYAHPN-UHFFFAOYSA-N COc1cc2C(=O)C(Cc3ncccc3C(F)(F)F)Cc2cc1Cl Chemical compound COc1cc2C(=O)C(Cc3ncccc3C(F)(F)F)Cc2cc1Cl VRNDNTBVUYAHPN-UHFFFAOYSA-N 0.000 description 2
- PODOMJWPFFTFFR-UHFFFAOYSA-N COc1cc2C(=O)C(Cc3ncccc3C(F)(F)F)Cc2cc1N1CCN(CCO)CC1 Chemical compound COc1cc2C(=O)C(Cc3ncccc3C(F)(F)F)Cc2cc1N1CCN(CCO)CC1 PODOMJWPFFTFFR-UHFFFAOYSA-N 0.000 description 2
- AOYDJHGSLOSBFH-UHFFFAOYSA-N COc1cc2C(=O)CCc2cc1N1CCN(C)CC1 Chemical compound COc1cc2C(=O)CCc2cc1N1CCN(C)CC1 AOYDJHGSLOSBFH-UHFFFAOYSA-N 0.000 description 2
- OVEKJSWHOTUQCM-UHFFFAOYSA-N COc1cc2C(=O)CCc2cc1N1CCOCC1 Chemical compound COc1cc2C(=O)CCc2cc1N1CCOCC1 OVEKJSWHOTUQCM-UHFFFAOYSA-N 0.000 description 2
- SWPFOGVOYDIINM-UHFFFAOYSA-N COc1ccc2C(=O)C(Cc3ccc(SC(F)(F)F)cc3)Cc2c1 Chemical compound COc1ccc2C(=O)C(Cc3ccc(SC(F)(F)F)cc3)Cc2c1 SWPFOGVOYDIINM-UHFFFAOYSA-N 0.000 description 2
- GJYFVIBZDVDKOJ-UHFFFAOYSA-N COc1ccc2C(=O)C(Cc3ncccc3C(F)(F)F)Cc2c1 Chemical compound COc1ccc2C(=O)C(Cc3ncccc3C(F)(F)F)Cc2c1 GJYFVIBZDVDKOJ-UHFFFAOYSA-N 0.000 description 2
- KMOMLSIYNWRKMH-UHFFFAOYSA-N COc1ccc2C(=O)C(Cc3ncccc3C(F)(F)F)Cc2c1OC Chemical compound COc1ccc2C(=O)C(Cc3ncccc3C(F)(F)F)Cc2c1OC KMOMLSIYNWRKMH-UHFFFAOYSA-N 0.000 description 2
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 2
- VTYYLEPIZMXCLO-UHFFFAOYSA-L Calcium carbonate Chemical compound [Ca+2].[O-]C([O-])=O VTYYLEPIZMXCLO-UHFFFAOYSA-L 0.000 description 2
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 2
- BVKZGUZCCUSVTD-UHFFFAOYSA-L Carbonate Chemical compound [O-]C([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-L 0.000 description 2
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 2
- 208000016192 Demyelinating disease Diseases 0.000 description 2
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 2
- LTMHDMANZUZIPE-AMTYYWEZSA-N Digoxin Natural products O([C@H]1[C@H](C)O[C@H](O[C@@H]2C[C@@H]3[C@@](C)([C@@H]4[C@H]([C@]5(O)[C@](C)([C@H](O)C4)[C@H](C4=CC(=O)OC4)CC5)CC3)CC2)C[C@@H]1O)[C@H]1O[C@H](C)[C@@H](O[C@H]2O[C@@H](C)[C@H](O)[C@@H](O)C2)[C@@H](O)C1 LTMHDMANZUZIPE-AMTYYWEZSA-N 0.000 description 2
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 2
- 238000002965 ELISA Methods 0.000 description 2
- 102000004190 Enzymes Human genes 0.000 description 2
- 108090000790 Enzymes Proteins 0.000 description 2
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 2
- YLQBMQCUIZJEEH-UHFFFAOYSA-N Furan Chemical compound C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 2
- 108010072051 Glatiramer Acetate Proteins 0.000 description 2
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 2
- 108010005716 Interferon beta-1a Proteins 0.000 description 2
- 108010005714 Interferon beta-1b Proteins 0.000 description 2
- 241001465754 Metazoa Species 0.000 description 2
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 2
- YNAVUWVOSKDBBP-UHFFFAOYSA-N Morpholine Chemical compound C1COCCN1 YNAVUWVOSKDBBP-UHFFFAOYSA-N 0.000 description 2
- 101000998145 Mus musculus Interleukin-17A Proteins 0.000 description 2
- 101100091494 Mus musculus Rorc gene Proteins 0.000 description 2
- 208000008238 Muscle Spasticity Diseases 0.000 description 2
- 238000005481 NMR spectroscopy Methods 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 2
- GLUUGHFHXGJENI-UHFFFAOYSA-N Piperazine Chemical compound C1CNCCN1 GLUUGHFHXGJENI-UHFFFAOYSA-N 0.000 description 2
- 102000016202 Proteolipids Human genes 0.000 description 2
- 108010010974 Proteolipids Proteins 0.000 description 2
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 2
- SMWDFEZZVXVKRB-UHFFFAOYSA-N Quinoline Chemical compound N1=CC=CC2=CC=CC=C21 SMWDFEZZVXVKRB-UHFFFAOYSA-N 0.000 description 2
- 108091008773 RAR-related orphan receptors γ Proteins 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 2
- YTPLMLYBLZKORZ-UHFFFAOYSA-N Thiophene Chemical compound C=1C=CSC=1 YTPLMLYBLZKORZ-UHFFFAOYSA-N 0.000 description 2
- GWEVSGVZZGPLCZ-UHFFFAOYSA-N Titan oxide Chemical compound O=[Ti]=O GWEVSGVZZGPLCZ-UHFFFAOYSA-N 0.000 description 2
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 2
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Chemical compound NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 2
- DPXJVFZANSGRMM-UHFFFAOYSA-N acetic acid;2,3,4,5,6-pentahydroxyhexanal;sodium Chemical compound [Na].CC(O)=O.OCC(O)C(O)C(O)C(O)C=O DPXJVFZANSGRMM-UHFFFAOYSA-N 0.000 description 2
- 239000002253 acid Substances 0.000 description 2
- 230000004913 activation Effects 0.000 description 2
- 239000000443 aerosol Substances 0.000 description 2
- 239000000556 agonist Substances 0.000 description 2
- 125000001931 aliphatic group Chemical group 0.000 description 2
- 125000003342 alkenyl group Chemical group 0.000 description 2
- 125000000304 alkynyl group Chemical group 0.000 description 2
- PNEYBMLMFCGWSK-UHFFFAOYSA-N aluminium oxide Inorganic materials [O-2].[O-2].[O-2].[Al+3].[Al+3] PNEYBMLMFCGWSK-UHFFFAOYSA-N 0.000 description 2
- 238000004458 analytical method Methods 0.000 description 2
- 230000003042 antagnostic effect Effects 0.000 description 2
- 239000007864 aqueous solution Substances 0.000 description 2
- 210000003050 axon Anatomy 0.000 description 2
- 239000002585 base Substances 0.000 description 2
- HUMNYLRZRPPJDN-UHFFFAOYSA-N benzaldehyde Chemical compound O=CC1=CC=CC=C1 HUMNYLRZRPPJDN-UHFFFAOYSA-N 0.000 description 2
- 230000027455 binding Effects 0.000 description 2
- 239000011230 binding agent Substances 0.000 description 2
- 238000004166 bioassay Methods 0.000 description 2
- 230000033228 biological regulation Effects 0.000 description 2
- 230000037396 body weight Effects 0.000 description 2
- 239000011575 calcium Substances 0.000 description 2
- 229910052791 calcium Inorganic materials 0.000 description 2
- 239000001506 calcium phosphate Substances 0.000 description 2
- 229910000389 calcium phosphate Inorganic materials 0.000 description 2
- 235000011010 calcium phosphates Nutrition 0.000 description 2
- 239000001768 carboxy methyl cellulose Substances 0.000 description 2
- 230000024245 cell differentiation Effects 0.000 description 2
- 230000001413 cellular effect Effects 0.000 description 2
- 239000000460 chlorine Substances 0.000 description 2
- 229910052801 chlorine Inorganic materials 0.000 description 2
- 229940126142 compound 16 Drugs 0.000 description 2
- 229940125782 compound 2 Drugs 0.000 description 2
- 235000009508 confectionery Nutrition 0.000 description 2
- 239000013058 crude material Substances 0.000 description 2
- 125000001316 cycloalkyl alkyl group Chemical group 0.000 description 2
- 230000002939 deleterious effect Effects 0.000 description 2
- UREBDLICKHMUKA-CXSFZGCWSA-N dexamethasone Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1C[C@@H](C)[C@@](C(=O)CO)(O)[C@@]1(C)C[C@@H]2O UREBDLICKHMUKA-CXSFZGCWSA-N 0.000 description 2
- 229960003957 dexamethasone Drugs 0.000 description 2
- 239000008121 dextrose Substances 0.000 description 2
- LTMHDMANZUZIPE-PUGKRICDSA-N digoxin Chemical group C1[C@H](O)[C@H](O)[C@@H](C)O[C@H]1O[C@@H]1[C@@H](C)O[C@@H](O[C@@H]2[C@H](O[C@@H](O[C@@H]3C[C@@H]4[C@]([C@@H]5[C@H]([C@]6(CC[C@@H]([C@@]6(C)[C@H](O)C5)C=5COC(=O)C=5)O)CC4)(C)CC3)C[C@@H]2O)C)C[C@@H]1O LTMHDMANZUZIPE-PUGKRICDSA-N 0.000 description 2
- 229960005156 digoxin Drugs 0.000 description 2
- LTMHDMANZUZIPE-UHFFFAOYSA-N digoxine Natural products C1C(O)C(O)C(C)OC1OC1C(C)OC(OC2C(OC(OC3CC4C(C5C(C6(CCC(C6(C)C(O)C5)C=5COC(=O)C=5)O)CC4)(C)CC3)CC2O)C)CC1O LTMHDMANZUZIPE-UHFFFAOYSA-N 0.000 description 2
- 239000003085 diluting agent Substances 0.000 description 2
- 229960004419 dimethyl fumarate Drugs 0.000 description 2
- 239000002552 dosage form Substances 0.000 description 2
- 238000009510 drug design Methods 0.000 description 2
- 238000010828 elution Methods 0.000 description 2
- BJXYHBKEQFQVES-NWDGAFQWSA-N enpatoran Chemical compound N[C@H]1CN(C[C@H](C1)C(F)(F)F)C1=C2C=CC=NC2=C(C=C1)C#N BJXYHBKEQFQVES-NWDGAFQWSA-N 0.000 description 2
- 150000002148 esters Chemical class 0.000 description 2
- CCIVGXIOQKPBKL-UHFFFAOYSA-M ethanesulfonate Chemical compound CCS([O-])(=O)=O CCIVGXIOQKPBKL-UHFFFAOYSA-M 0.000 description 2
- GWNFQAKCJYEJEW-UHFFFAOYSA-N ethyl 3-[8-[[4-methyl-5-[(3-methyl-4-oxophthalazin-1-yl)methyl]-1,2,4-triazol-3-yl]sulfanyl]octanoylamino]benzoate Chemical compound CCOC(=O)C1=CC(NC(=O)CCCCCCCSC2=NN=C(CC3=NN(C)C(=O)C4=CC=CC=C34)N2C)=CC=C1 GWNFQAKCJYEJEW-UHFFFAOYSA-N 0.000 description 2
- 238000011156 evaluation Methods 0.000 description 2
- 239000010685 fatty oil Substances 0.000 description 2
- 229910052731 fluorine Inorganic materials 0.000 description 2
- 239000011737 fluorine Substances 0.000 description 2
- 239000012634 fragment Substances 0.000 description 2
- RWSXRVCMGQZWBV-WDSKDSINSA-N glutathione Chemical compound OC(=O)[C@@H](N)CCC(=O)N[C@@H](CS)C(=O)NCC(O)=O RWSXRVCMGQZWBV-WDSKDSINSA-N 0.000 description 2
- 125000004475 heteroaralkyl group Chemical group 0.000 description 2
- 125000005842 heteroatom Chemical group 0.000 description 2
- 125000004476 heterocycloamino group Chemical group 0.000 description 2
- 125000002768 hydroxyalkyl group Chemical group 0.000 description 2
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 2
- AWJUIBRHMBBTKR-UHFFFAOYSA-N isoquinoline Chemical compound C1=NC=CC2=CC=CC=C21 AWJUIBRHMBBTKR-UHFFFAOYSA-N 0.000 description 2
- 210000004731 jugular vein Anatomy 0.000 description 2
- 239000010410 layer Substances 0.000 description 2
- 230000000670 limiting effect Effects 0.000 description 2
- 239000002502 liposome Substances 0.000 description 2
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 2
- 231100000053 low toxicity Toxicity 0.000 description 2
- 239000007937 lozenge Substances 0.000 description 2
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 2
- 230000014759 maintenance of location Effects 0.000 description 2
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 2
- 239000001630 malic acid Substances 0.000 description 2
- 239000003550 marker Substances 0.000 description 2
- 210000003205 muscle Anatomy 0.000 description 2
- 210000003007 myelin sheath Anatomy 0.000 description 2
- 230000023105 myelination Effects 0.000 description 2
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 2
- 230000001537 neural effect Effects 0.000 description 2
- 230000007935 neutral effect Effects 0.000 description 2
- 231100000862 numbness Toxicity 0.000 description 2
- 239000003921 oil Substances 0.000 description 2
- 235000019198 oils Nutrition 0.000 description 2
- 210000004248 oligodendroglia Anatomy 0.000 description 2
- 239000003960 organic solvent Substances 0.000 description 2
- 238000007911 parenteral administration Methods 0.000 description 2
- 230000037361 pathway Effects 0.000 description 2
- VLTRZXGMWDSKGL-UHFFFAOYSA-N perchloric acid Chemical compound OCl(=O)(=O)=O VLTRZXGMWDSKGL-UHFFFAOYSA-N 0.000 description 2
- 239000000825 pharmaceutical preparation Substances 0.000 description 2
- 239000012071 phase Substances 0.000 description 2
- WVDDGKGOMKODPV-ZQBYOMGUSA-N phenyl(114C)methanol Chemical compound O[14CH2]C1=CC=CC=C1 WVDDGKGOMKODPV-ZQBYOMGUSA-N 0.000 description 2
- 239000006187 pill Substances 0.000 description 2
- 229920003023 plastic Polymers 0.000 description 2
- 239000004033 plastic Substances 0.000 description 2
- 239000000244 polyoxyethylene sorbitan monooleate Substances 0.000 description 2
- 235000010482 polyoxyethylene sorbitan monooleate Nutrition 0.000 description 2
- 229940068968 polysorbate 80 Drugs 0.000 description 2
- 229920000053 polysorbate 80 Polymers 0.000 description 2
- 238000012545 processing Methods 0.000 description 2
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 2
- QJZUKDFHGGYHMC-UHFFFAOYSA-N pyridine-3-carbaldehyde Chemical compound O=CC1=CC=CN=C1 QJZUKDFHGGYHMC-UHFFFAOYSA-N 0.000 description 2
- 238000011002 quantification Methods 0.000 description 2
- 125000001453 quaternary ammonium group Chemical group 0.000 description 2
- 102000005962 receptors Human genes 0.000 description 2
- 108020003175 receptors Proteins 0.000 description 2
- 230000008439 repair process Effects 0.000 description 2
- 230000004044 response Effects 0.000 description 2
- YGSDEFSMJLZEOE-UHFFFAOYSA-N salicylic acid Chemical compound OC(=O)C1=CC=CC=C1O YGSDEFSMJLZEOE-UHFFFAOYSA-N 0.000 description 2
- 238000000926 separation method Methods 0.000 description 2
- 235000019812 sodium carboxymethyl cellulose Nutrition 0.000 description 2
- 229920001027 sodium carboxymethylcellulose Polymers 0.000 description 2
- 239000002904 solvent Substances 0.000 description 2
- 208000018198 spasticity Diseases 0.000 description 2
- 238000001228 spectrum Methods 0.000 description 2
- 210000000278 spinal cord Anatomy 0.000 description 2
- 238000013222 sprague-dawley male rat Methods 0.000 description 2
- 230000000087 stabilizing effect Effects 0.000 description 2
- 239000007858 starting material Substances 0.000 description 2
- KDYFGRWQOYBRFD-UHFFFAOYSA-N succinic acid Chemical compound OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 2
- 239000000454 talc Substances 0.000 description 2
- 229910052623 talc Inorganic materials 0.000 description 2
- 235000012222 talc Nutrition 0.000 description 2
- 229940121136 tecfidera Drugs 0.000 description 2
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 2
- 229940124597 therapeutic agent Drugs 0.000 description 2
- 238000002877 time resolved fluorescence resonance energy transfer Methods 0.000 description 2
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 2
- QORWJWZARLRLPR-UHFFFAOYSA-H tricalcium bis(phosphate) Chemical compound [Ca+2].[Ca+2].[Ca+2].[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O QORWJWZARLRLPR-UHFFFAOYSA-H 0.000 description 2
- LNAZSHAWQACDHT-XIYTZBAFSA-N (2r,3r,4s,5r,6s)-4,5-dimethoxy-2-(methoxymethyl)-3-[(2s,3r,4s,5r,6r)-3,4,5-trimethoxy-6-(methoxymethyl)oxan-2-yl]oxy-6-[(2r,3r,4s,5r,6r)-4,5,6-trimethoxy-2-(methoxymethyl)oxan-3-yl]oxyoxane Chemical compound CO[C@@H]1[C@@H](OC)[C@H](OC)[C@@H](COC)O[C@H]1O[C@H]1[C@H](OC)[C@@H](OC)[C@H](O[C@H]2[C@@H]([C@@H](OC)[C@H](OC)O[C@@H]2COC)OC)O[C@@H]1COC LNAZSHAWQACDHT-XIYTZBAFSA-N 0.000 description 1
- WWTBZEKOSBFBEM-SPWPXUSOSA-N (2s)-2-[[2-benzyl-3-[hydroxy-[(1r)-2-phenyl-1-(phenylmethoxycarbonylamino)ethyl]phosphoryl]propanoyl]amino]-3-(1h-indol-3-yl)propanoic acid Chemical compound N([C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)O)C(=O)C(CP(O)(=O)[C@H](CC=1C=CC=CC=1)NC(=O)OCC=1C=CC=CC=1)CC1=CC=CC=C1 WWTBZEKOSBFBEM-SPWPXUSOSA-N 0.000 description 1
- IXRAQYMAEVFORF-UTLNTRLCSA-N (3S,8S,9S,10R,13S,14S,17R)-10,13-dimethyl-17-[(2R)-6-methylheptan-2-yl]-2,3,4,7,8,9,11,12,14,15,16,17-dodecahydro-1H-cyclopenta[a]phenanthrene-3,16-diol Chemical class C1C=C2C[C@@H](O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC(O)[C@H]([C@H](C)CCCC(C)C)[C@@]1(C)CC2 IXRAQYMAEVFORF-UTLNTRLCSA-N 0.000 description 1
- UDQTXCHQKHIQMH-KYGLGHNPSA-N (3ar,5s,6s,7r,7ar)-5-(difluoromethyl)-2-(ethylamino)-5,6,7,7a-tetrahydro-3ah-pyrano[3,2-d][1,3]thiazole-6,7-diol Chemical compound S1C(NCC)=N[C@H]2[C@@H]1O[C@H](C(F)F)[C@@H](O)[C@@H]2O UDQTXCHQKHIQMH-KYGLGHNPSA-N 0.000 description 1
- WCGUUGGRBIKTOS-GPOJBZKASA-N (3beta)-3-hydroxyurs-12-en-28-oic acid Chemical compound C1C[C@H](O)C(C)(C)[C@@H]2CC[C@@]3(C)[C@]4(C)CC[C@@]5(C(O)=O)CC[C@@H](C)[C@H](C)[C@H]5C4=CC[C@@H]3[C@]21C WCGUUGGRBIKTOS-GPOJBZKASA-N 0.000 description 1
- MPDDTAJMJCESGV-CTUHWIOQSA-M (3r,5r)-7-[2-(4-fluorophenyl)-5-[methyl-[(1r)-1-phenylethyl]carbamoyl]-4-propan-2-ylpyrazol-3-yl]-3,5-dihydroxyheptanoate Chemical compound C1([C@@H](C)N(C)C(=O)C2=NN(C(CC[C@@H](O)C[C@@H](O)CC([O-])=O)=C2C(C)C)C=2C=CC(F)=CC=2)=CC=CC=C1 MPDDTAJMJCESGV-CTUHWIOQSA-M 0.000 description 1
- STPKWKPURVSAJF-LJEWAXOPSA-N (4r,5r)-5-[4-[[4-(1-aza-4-azoniabicyclo[2.2.2]octan-4-ylmethyl)phenyl]methoxy]phenyl]-3,3-dibutyl-7-(dimethylamino)-1,1-dioxo-4,5-dihydro-2h-1$l^{6}-benzothiepin-4-ol Chemical compound O[C@H]1C(CCCC)(CCCC)CS(=O)(=O)C2=CC=C(N(C)C)C=C2[C@H]1C(C=C1)=CC=C1OCC(C=C1)=CC=C1C[N+]1(CC2)CCN2CC1 STPKWKPURVSAJF-LJEWAXOPSA-N 0.000 description 1
- OIIOPWHTJZYKIL-PMACEKPBSA-N (5S)-5-[[[5-[2-chloro-3-[2-chloro-3-[6-methoxy-5-[[[(2S)-5-oxopyrrolidin-2-yl]methylamino]methyl]pyrazin-2-yl]phenyl]phenyl]-3-methoxypyrazin-2-yl]methylamino]methyl]pyrrolidin-2-one Chemical compound C1(=C(N=C(C2=C(C(C3=CC=CC(=C3Cl)C3=NC(OC)=C(N=C3)CNC[C@H]3NC(=O)CC3)=CC=C2)Cl)C=N1)OC)CNC[C@H]1NC(=O)CC1 OIIOPWHTJZYKIL-PMACEKPBSA-N 0.000 description 1
- DEVSOMFAQLZNKR-RJRFIUFISA-N (z)-3-[3-[3,5-bis(trifluoromethyl)phenyl]-1,2,4-triazol-1-yl]-n'-pyrazin-2-ylprop-2-enehydrazide Chemical compound FC(F)(F)C1=CC(C(F)(F)F)=CC(C2=NN(\C=C/C(=O)NNC=3N=CC=NC=3)C=N2)=C1 DEVSOMFAQLZNKR-RJRFIUFISA-N 0.000 description 1
- JYEUMXHLPRZUAT-UHFFFAOYSA-N 1,2,3-triazine Chemical compound C1=CN=NN=C1 JYEUMXHLPRZUAT-UHFFFAOYSA-N 0.000 description 1
- DDMOUSALMHHKOS-UHFFFAOYSA-N 1,2-dichloro-1,1,2,2-tetrafluoroethane Chemical compound FC(F)(Cl)C(F)(F)Cl DDMOUSALMHHKOS-UHFFFAOYSA-N 0.000 description 1
- WKXVETMYCFRGET-UHFFFAOYSA-N 1,3-thiazole-2-sulfonamide Chemical class NS(=O)(=O)C1=NC=CS1 WKXVETMYCFRGET-UHFFFAOYSA-N 0.000 description 1
- WZZBNLYBHUDSHF-DHLKQENFSA-N 1-[(3s,4s)-4-[8-(2-chloro-4-pyrimidin-2-yloxyphenyl)-7-fluoro-2-methylimidazo[4,5-c]quinolin-1-yl]-3-fluoropiperidin-1-yl]-2-hydroxyethanone Chemical compound CC1=NC2=CN=C3C=C(F)C(C=4C(=CC(OC=5N=CC=CN=5)=CC=4)Cl)=CC3=C2N1[C@H]1CCN(C(=O)CO)C[C@@H]1F WZZBNLYBHUDSHF-DHLKQENFSA-N 0.000 description 1
- SGIBOXBBPQRZDM-UHFFFAOYSA-N 1-benzylpiperidine-4-carbaldehyde Chemical compound C1CC(C=O)CCN1CC1=CC=CC=C1 SGIBOXBBPQRZDM-UHFFFAOYSA-N 0.000 description 1
- 125000004973 1-butenyl group Chemical group C(=CCC)* 0.000 description 1
- 125000004972 1-butynyl group Chemical group [H]C([H])([H])C([H])([H])C#C* 0.000 description 1
- IXPNQXFRVYWDDI-UHFFFAOYSA-N 1-methyl-2,4-dioxo-1,3-diazinane-5-carboximidamide Chemical compound CN1CC(C(N)=N)C(=O)NC1=O IXPNQXFRVYWDDI-UHFFFAOYSA-N 0.000 description 1
- ALOCUZOKRULSAA-UHFFFAOYSA-N 1-methylpiperidin-4-amine Chemical compound CN1CCC(N)CC1 ALOCUZOKRULSAA-UHFFFAOYSA-N 0.000 description 1
- 125000006023 1-pentenyl group Chemical group 0.000 description 1
- SIJBDWPVNAYVGY-UHFFFAOYSA-N 2,2-dimethyl-1,3-dioxolane Chemical compound CC1(C)OCCO1 SIJBDWPVNAYVGY-UHFFFAOYSA-N 0.000 description 1
- 125000006069 2,3-dimethyl-2-butenyl group Chemical group 0.000 description 1
- FEYDZHNIIMENOB-UHFFFAOYSA-N 2,6-dibromopyridine Chemical compound BrC1=CC=CC(Br)=N1 FEYDZHNIIMENOB-UHFFFAOYSA-N 0.000 description 1
- XWGMPMPFEXRKQO-UHFFFAOYSA-N 2-(5-bromopyridin-3-yl)-1,1,1,3,3,3-hexafluoropropan-2-ol Chemical compound FC(F)(F)C(C(F)(F)F)(O)C1=CN=CC(Br)=C1 XWGMPMPFEXRKQO-UHFFFAOYSA-N 0.000 description 1
- FVAAOCYFPRQZNQ-UHFFFAOYSA-N 2-(6-bromopyridin-2-yl)-1,1,1,3,3,3-hexafluoropropan-2-ol Chemical compound FC(F)(F)C(C(F)(F)F)(O)C1=CC=CC(Br)=N1 FVAAOCYFPRQZNQ-UHFFFAOYSA-N 0.000 description 1
- TVTJUIAKQFIXCE-HUKYDQBMSA-N 2-amino-9-[(2R,3S,4S,5R)-4-fluoro-3-hydroxy-5-(hydroxymethyl)oxolan-2-yl]-7-prop-2-ynyl-1H-purine-6,8-dione Chemical compound NC=1NC(C=2N(C(N(C=2N=1)[C@@H]1O[C@@H]([C@H]([C@H]1O)F)CO)=O)CC#C)=O TVTJUIAKQFIXCE-HUKYDQBMSA-N 0.000 description 1
- LDLCZOVUSADOIV-UHFFFAOYSA-N 2-bromoethanol Chemical compound OCCBr LDLCZOVUSADOIV-UHFFFAOYSA-N 0.000 description 1
- 125000004974 2-butenyl group Chemical group C(C=CC)* 0.000 description 1
- 125000000069 2-butynyl group Chemical group [H]C([H])([H])C#CC([H])([H])* 0.000 description 1
- OZZOJJJYKYKBNH-UHFFFAOYSA-N 2-chloro-5-(trifluoromethyl)benzaldehyde Chemical compound FC(F)(F)C1=CC=C(Cl)C(C=O)=C1 OZZOJJJYKYKBNH-UHFFFAOYSA-N 0.000 description 1
- XDMZVNQKVMTCSP-UHFFFAOYSA-N 2-fluoro-3-(trifluoromethyl)benzaldehyde Chemical compound FC1=C(C=O)C=CC=C1C(F)(F)F XDMZVNQKVMTCSP-UHFFFAOYSA-N 0.000 description 1
- IDLNLGMUINCSGS-UHFFFAOYSA-N 2-fluoro-5-(trifluoromethyl)benzaldehyde Chemical compound FC1=CC=C(C(F)(F)F)C=C1C=O IDLNLGMUINCSGS-UHFFFAOYSA-N 0.000 description 1
- HZLCGUXUOFWCCN-UHFFFAOYSA-N 2-hydroxynonadecane-1,2,3-tricarboxylic acid Chemical compound CCCCCCCCCCCCCCCCC(C(O)=O)C(O)(C(O)=O)CC(O)=O HZLCGUXUOFWCCN-UHFFFAOYSA-N 0.000 description 1
- 125000006029 2-methyl-2-butenyl group Chemical group 0.000 description 1
- 125000006024 2-pentenyl group Chemical group 0.000 description 1
- 125000004105 2-pyridyl group Chemical group N1=C([*])C([H])=C([H])C([H])=C1[H] 0.000 description 1
- SOSPMXMEOFGPIM-UHFFFAOYSA-N 3,5-dibromopyridine Chemical compound BrC1=CN=CC(Br)=C1 SOSPMXMEOFGPIM-UHFFFAOYSA-N 0.000 description 1
- NMTUHPSKJJYGML-UHFFFAOYSA-N 3-(trifluoromethyl)benzaldehyde Chemical compound FC(F)(F)C1=CC=CC(C=O)=C1 NMTUHPSKJJYGML-UHFFFAOYSA-N 0.000 description 1
- FIIIEUQXYKDPRD-UHFFFAOYSA-N 3-(trifluoromethyl)pyridine-2-carbaldehyde Chemical compound FC(F)(F)C1=CC=CN=C1C=O FIIIEUQXYKDPRD-UHFFFAOYSA-N 0.000 description 1
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 description 1
- OXBHKEYDKAWFLS-UHFFFAOYSA-N 3-bromo-2,6-difluorobenzaldehyde Chemical compound FC1=CC=C(Br)C(F)=C1C=O OXBHKEYDKAWFLS-UHFFFAOYSA-N 0.000 description 1
- ALIVUZVZCLVJQN-UHFFFAOYSA-N 3-bromo-5-(trifluoromethoxy)benzaldehyde Chemical compound FC(F)(F)OC1=CC(Br)=CC(C=O)=C1 ALIVUZVZCLVJQN-UHFFFAOYSA-N 0.000 description 1
- PCRLZGCXLNNMFL-UHFFFAOYSA-N 3-bromo-5-(trifluoromethyl)benzaldehyde Chemical compound FC(F)(F)C1=CC(Br)=CC(C=O)=C1 PCRLZGCXLNNMFL-UHFFFAOYSA-N 0.000 description 1
- UTSBZTCYCRVHCK-UHFFFAOYSA-N 3-chloro-4-(trifluoromethyl)benzaldehyde Chemical compound FC(F)(F)C1=CC=C(C=O)C=C1Cl UTSBZTCYCRVHCK-UHFFFAOYSA-N 0.000 description 1
- NWSKKQLZBXTZTP-UHFFFAOYSA-N 3-chloro-5-(trifluoromethyl)benzaldehyde Chemical compound FC(F)(F)C1=CC(Cl)=CC(C=O)=C1 NWSKKQLZBXTZTP-UHFFFAOYSA-N 0.000 description 1
- 125000006027 3-methyl-1-butenyl group Chemical group 0.000 description 1
- 125000002103 4,4'-dimethoxytriphenylmethyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C(*)(C1=C([H])C([H])=C(OC([H])([H])[H])C([H])=C1[H])C1=C([H])C([H])=C(OC([H])([H])[H])C([H])=C1[H] 0.000 description 1
- BEOBZEOPTQQELP-UHFFFAOYSA-N 4-(trifluoromethyl)benzaldehyde Chemical compound FC(F)(F)C1=CC=C(C=O)C=C1 BEOBZEOPTQQELP-UHFFFAOYSA-N 0.000 description 1
- QGHDLJAZIIFENW-UHFFFAOYSA-N 4-[1,1,1,3,3,3-hexafluoro-2-(4-hydroxy-3-prop-2-enylphenyl)propan-2-yl]-2-prop-2-enylphenol Chemical group C1=C(CC=C)C(O)=CC=C1C(C(F)(F)F)(C(F)(F)F)C1=CC=C(O)C(CC=C)=C1 QGHDLJAZIIFENW-UHFFFAOYSA-N 0.000 description 1
- AHFINSWGYAZBOZ-UHFFFAOYSA-N 4-chloro-2-(trifluoromethyl)benzaldehyde Chemical compound FC(F)(F)C1=CC(Cl)=CC=C1C=O AHFINSWGYAZBOZ-UHFFFAOYSA-N 0.000 description 1
- NIHMMULLFBKTOK-UHFFFAOYSA-N 4-chloro-3-(trifluoromethyl)benzaldehyde Chemical compound FC(F)(F)C1=CC(C=O)=CC=C1Cl NIHMMULLFBKTOK-UHFFFAOYSA-N 0.000 description 1
- RNDMOLOBKVJEMH-UHFFFAOYSA-N 4-chloro-5-methoxy-2,3-dihydroinden-1-one Chemical compound COC1=CC=C2C(=O)CCC2=C1Cl RNDMOLOBKVJEMH-UHFFFAOYSA-N 0.000 description 1
- VKLKXFOZNHEBSW-UHFFFAOYSA-N 5-[[3-[(4-morpholin-4-ylbenzoyl)amino]phenyl]methoxy]pyridine-3-carboxamide Chemical compound O1CCN(CC1)C1=CC=C(C(=O)NC=2C=C(COC=3C=NC=C(C(=O)N)C=3)C=CC=2)C=C1 VKLKXFOZNHEBSW-UHFFFAOYSA-N 0.000 description 1
- CVRLMVFONQYBOC-UHFFFAOYSA-N 5-chloro-2-(trifluoromethyl)benzaldehyde Chemical compound FC(F)(F)C1=CC=C(Cl)C=C1C=O CVRLMVFONQYBOC-UHFFFAOYSA-N 0.000 description 1
- XIUZEIYKEBEXIB-UHFFFAOYSA-N 5-chloro-6-methoxy-2,3-dihydroinden-1-one Chemical compound C1=C(Cl)C(OC)=CC2=C1CCC2=O XIUZEIYKEBEXIB-UHFFFAOYSA-N 0.000 description 1
- CZAZDIRRZHGKBI-UHFFFAOYSA-N 5-methyl-1h-pyrazole-3-carbaldehyde Chemical compound CC=1C=C(C=O)NN=1 CZAZDIRRZHGKBI-UHFFFAOYSA-N 0.000 description 1
- MRPAGRCGPAXOGS-UHFFFAOYSA-N 6-(trifluoromethyl)pyridine-3-carbaldehyde Chemical compound FC(F)(F)C1=CC=C(C=O)C=N1 MRPAGRCGPAXOGS-UHFFFAOYSA-N 0.000 description 1
- FYPURVLMVKTZAS-UHFFFAOYSA-N 6-methoxy-5-morpholin-4-yl-2-[[3-(trifluoromethyl)phenyl]methyl]-2,3-dihydroinden-1-one Chemical compound C1C=2C=C(N3CCOCC3)C(OC)=CC=2C(=O)C1CC1=CC=CC(C(F)(F)F)=C1 FYPURVLMVKTZAS-UHFFFAOYSA-N 0.000 description 1
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 1
- 229920001817 Agar Polymers 0.000 description 1
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 1
- 241000416162 Astragalus gummifer Species 0.000 description 1
- 201000004569 Blindness Diseases 0.000 description 1
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 1
- SSNZGGJPQCBMQU-UHFFFAOYSA-L C1(=CC=CC=C1)C.C([O-])([O-])=O.[Cs+].[Cs+] Chemical compound C1(=CC=CC=C1)C.C([O-])([O-])=O.[Cs+].[Cs+] SSNZGGJPQCBMQU-UHFFFAOYSA-L 0.000 description 1
- YVMCDMPXSZSUPR-DHZHZOJOSA-N C1c2c(Cl)c(OC)ccc2C(=O)\C1=C\c1ccc(SC(F)(F)F)cc1 Chemical compound C1c2c(Cl)c(OC)ccc2C(=O)\C1=C\c1ccc(SC(F)(F)F)cc1 YVMCDMPXSZSUPR-DHZHZOJOSA-N 0.000 description 1
- YKJMJVXTFROCOW-FMIVXFBMSA-N C1c2c(OC)c(OC)ccc2C(=O)\C1=C\c1ccc(SC(F)(F)F)cc1 Chemical compound C1c2c(OC)c(OC)ccc2C(=O)\C1=C\c1ccc(SC(F)(F)F)cc1 YKJMJVXTFROCOW-FMIVXFBMSA-N 0.000 description 1
- KIDTUPIWCJSIRX-VCHYOVAHSA-N C1c2cc(N3CCC(C)CC3)c(OC)cc2C(=O)\C1=C\c1cc(C(F)(F)F)ccc1 Chemical compound C1c2cc(N3CCC(C)CC3)c(OC)cc2C(=O)\C1=C\c1cc(C(F)(F)F)ccc1 KIDTUPIWCJSIRX-VCHYOVAHSA-N 0.000 description 1
- UMCGGGWVXLNODY-WOJGMQOQSA-N C1c2cc(N3CCC(C)CC3)c(OC)cc2C(=O)\C1=C\c1ccc(SC(F)(F)F)cc1 Chemical compound C1c2cc(N3CCC(C)CC3)c(OC)cc2C(=O)\C1=C\c1ccc(SC(F)(F)F)cc1 UMCGGGWVXLNODY-WOJGMQOQSA-N 0.000 description 1
- TVMLQWKBJHALKJ-LICLKQGHSA-N C1c2cc(N3CCN(C)CC3)c(OC)cc2C(=O)\C1=C\c1cc(C(F)(F)F)ccc1 Chemical compound C1c2cc(N3CCN(C)CC3)c(OC)cc2C(=O)\C1=C\c1cc(C(F)(F)F)ccc1 TVMLQWKBJHALKJ-LICLKQGHSA-N 0.000 description 1
- ZNNSORQCNQLQMO-GZTJUZNOSA-N C1c2cc(N3CCN(C)CC3)c(OC)cc2C(=O)\C1=C\c1ccc(SC(F)(F)F)cc1 Chemical compound C1c2cc(N3CCN(C)CC3)c(OC)cc2C(=O)\C1=C\c1ccc(SC(F)(F)F)cc1 ZNNSORQCNQLQMO-GZTJUZNOSA-N 0.000 description 1
- GKLPOFJGBYFDNK-LICLKQGHSA-N C1c2cc(N3CCN(C)CC3)c(OC)cc2C(=O)\C1=C\c1cnccc1 Chemical compound C1c2cc(N3CCN(C)CC3)c(OC)cc2C(=O)\C1=C\c1cnccc1 GKLPOFJGBYFDNK-LICLKQGHSA-N 0.000 description 1
- BXHWYRNMUHYCOS-HZHRSRAPSA-N C1c2cc(N3CCOCC3)c(OC)cc2C(=O)\C1=C\C(CC1)CCN1Cc1ccccc1 Chemical compound C1c2cc(N3CCOCC3)c(OC)cc2C(=O)\C1=C\C(CC1)CCN1Cc1ccccc1 BXHWYRNMUHYCOS-HZHRSRAPSA-N 0.000 description 1
- STTWBVFGPGJOHB-NTEUORMPSA-N C1c2cc(N3CCOCC3)c(OC)cc2C(=O)\C1=C\c1[nH]nc(C)c1 Chemical compound C1c2cc(N3CCOCC3)c(OC)cc2C(=O)\C1=C\c1[nH]nc(C)c1 STTWBVFGPGJOHB-NTEUORMPSA-N 0.000 description 1
- SNMSKFIVZFRAQF-OQLLNIDSSA-N C1c2cc(N3CCOCC3)c(OC)cc2C(=O)\C1=C\c1c(C(F)(F)F)ccc(Cl)c1 Chemical compound C1c2cc(N3CCOCC3)c(OC)cc2C(=O)\C1=C\c1c(C(F)(F)F)ccc(Cl)c1 SNMSKFIVZFRAQF-OQLLNIDSSA-N 0.000 description 1
- UEQFNSJYUCWBTC-OQLLNIDSSA-N C1c2cc(N3CCOCC3)c(OC)cc2C(=O)\C1=C\c1c(Cl)ccc(C(F)(F)F)c1 Chemical compound C1c2cc(N3CCOCC3)c(OC)cc2C(=O)\C1=C\c1c(Cl)ccc(C(F)(F)F)c1 UEQFNSJYUCWBTC-OQLLNIDSSA-N 0.000 description 1
- GORPAEDFVNAVGN-UKTHLTGXSA-N C1c2cc(N3CCOCC3)c(OC)cc2C(=O)\C1=C\c1c(F)c(Br)ccc1F Chemical compound C1c2cc(N3CCOCC3)c(OC)cc2C(=O)\C1=C\c1c(F)c(Br)ccc1F GORPAEDFVNAVGN-UKTHLTGXSA-N 0.000 description 1
- CZGXDHNLVLAYHI-OQLLNIDSSA-N C1c2cc(N3CCOCC3)c(OC)cc2C(=O)\C1=C\c1c(F)c(C(F)(F)F)ccc1 Chemical compound C1c2cc(N3CCOCC3)c(OC)cc2C(=O)\C1=C\c1c(F)c(C(F)(F)F)ccc1 CZGXDHNLVLAYHI-OQLLNIDSSA-N 0.000 description 1
- QOCMZDIOYUNHGH-OVCLIPMQSA-N C1c2cc(N3CCOCC3)c(OC)cc2C(=O)\C1=C\c1c(F)cc(Cl)cc1 Chemical compound C1c2cc(N3CCOCC3)c(OC)cc2C(=O)\C1=C\c1c(F)cc(Cl)cc1 QOCMZDIOYUNHGH-OVCLIPMQSA-N 0.000 description 1
- NKNHLPUSZGXPAW-OVCLIPMQSA-N C1c2cc(N3CCOCC3)c(OC)cc2C(=O)\C1=C\c1c(F)cc(F)cc1 Chemical compound C1c2cc(N3CCOCC3)c(OC)cc2C(=O)\C1=C\c1c(F)cc(F)cc1 NKNHLPUSZGXPAW-OVCLIPMQSA-N 0.000 description 1
- UPMHKRZWQVVIKA-OQLLNIDSSA-N C1c2cc(N3CCOCC3)c(OC)cc2C(=O)\C1=C\c1c(F)ccc(C(F)(F)F)c1 Chemical compound C1c2cc(N3CCOCC3)c(OC)cc2C(=O)\C1=C\c1c(F)ccc(C(F)(F)F)c1 UPMHKRZWQVVIKA-OQLLNIDSSA-N 0.000 description 1
- UARSRVGXDIIYEA-OVCLIPMQSA-N C1c2cc(N3CCOCC3)c(OC)cc2C(=O)\C1=C\c1cc(C(F)(F)F)c(Cl)cc1 Chemical compound C1c2cc(N3CCOCC3)c(OC)cc2C(=O)\C1=C\c1cc(C(F)(F)F)c(Cl)cc1 UARSRVGXDIIYEA-OVCLIPMQSA-N 0.000 description 1
- ISEJMLBQGSVNKU-GIDUJCDVSA-N C1c2cc(N3CCOCC3)c(OC)cc2C(=O)\C1=C\c1cc(C(F)(F)F)cc(Br)c1 Chemical compound C1c2cc(N3CCOCC3)c(OC)cc2C(=O)\C1=C\c1cc(C(F)(F)F)cc(Br)c1 ISEJMLBQGSVNKU-GIDUJCDVSA-N 0.000 description 1
- PIDDZVQMONYFPY-GIDUJCDVSA-N C1c2cc(N3CCOCC3)c(OC)cc2C(=O)\C1=C\c1cc(C(F)(F)F)cc(Cl)c1 Chemical compound C1c2cc(N3CCOCC3)c(OC)cc2C(=O)\C1=C\c1cc(C(F)(F)F)cc(Cl)c1 PIDDZVQMONYFPY-GIDUJCDVSA-N 0.000 description 1
- JSKXICGHTMSIID-GIDUJCDVSA-N C1c2cc(N3CCOCC3)c(OC)cc2C(=O)\C1=C\c1cc(C(O)(C(F)(F)F)C(F)(F)F)cnc1 Chemical compound C1c2cc(N3CCOCC3)c(OC)cc2C(=O)\C1=C\c1cc(C(O)(C(F)(F)F)C(F)(F)F)cnc1 JSKXICGHTMSIID-GIDUJCDVSA-N 0.000 description 1
- SFJFEVGRJHHGPD-OVCLIPMQSA-N C1c2cc(N3CCOCC3)c(OC)cc2C(=O)\C1=C\c1cc(Cl)c(C(F)(F)F)cc1 Chemical compound C1c2cc(N3CCOCC3)c(OC)cc2C(=O)\C1=C\c1cc(Cl)c(C(F)(F)F)cc1 SFJFEVGRJHHGPD-OVCLIPMQSA-N 0.000 description 1
- KHARBQPPSSJFDP-GIDUJCDVSA-N C1c2cc(N3CCOCC3)c(OC)cc2C(=O)\C1=C\c1cc(OC(F)(F)F)cc(Br)c1 Chemical compound C1c2cc(N3CCOCC3)c(OC)cc2C(=O)\C1=C\c1cc(OC(F)(F)F)cc(Br)c1 KHARBQPPSSJFDP-GIDUJCDVSA-N 0.000 description 1
- ONKIEGJUXCTZAG-MHWRWJLKSA-N C1c2cc(N3CCOCC3)c(OC)cc2C(=O)\C1=C\c1ccc(C(F)(F)F)cc1 Chemical compound C1c2cc(N3CCOCC3)c(OC)cc2C(=O)\C1=C\c1ccc(C(F)(F)F)cc1 ONKIEGJUXCTZAG-MHWRWJLKSA-N 0.000 description 1
- UQUQBEXMIQJTEB-MHWRWJLKSA-N C1c2cc(N3CCOCC3)c(OC)cc2C(=O)\C1=C\c1ccc(SC(F)(F)F)cc1 Chemical compound C1c2cc(N3CCOCC3)c(OC)cc2C(=O)\C1=C\c1ccc(SC(F)(F)F)cc1 UQUQBEXMIQJTEB-MHWRWJLKSA-N 0.000 description 1
- KSKKNDKECMXTQO-OVCLIPMQSA-N C1c2cc(N3CCOCC3)c(OC)cc2C(=O)\C1=C\c1cnc(C(F)(F)F)cc1 Chemical compound C1c2cc(N3CCOCC3)c(OC)cc2C(=O)\C1=C\c1cnc(C(F)(F)F)cc1 KSKKNDKECMXTQO-OVCLIPMQSA-N 0.000 description 1
- MQHUTKHSIMPLAE-CXUHLZMHSA-N C1c2cc(N3CCOCC3)c(OC)cc2C(=O)\C1=C\c1cnccc1 Chemical compound C1c2cc(N3CCOCC3)c(OC)cc2C(=O)\C1=C\c1cnccc1 MQHUTKHSIMPLAE-CXUHLZMHSA-N 0.000 description 1
- YJKVDRVTUSRFQE-GXDHUFHOSA-N C1c2cc(N3CCOCC3)c(OC)cc2C(=O)\C1=C\c1nc(C(O)(C(F)(F)F)C(F)(F)F)ccc1 Chemical compound C1c2cc(N3CCOCC3)c(OC)cc2C(=O)\C1=C\c1nc(C(O)(C(F)(F)F)C(F)(F)F)ccc1 YJKVDRVTUSRFQE-GXDHUFHOSA-N 0.000 description 1
- OGOSBSHYIKBBBT-UHFFFAOYSA-N CC(C)(C)C(N[IH]C)=O Chemical compound CC(C)(C)C(N[IH]C)=O OGOSBSHYIKBBBT-UHFFFAOYSA-N 0.000 description 1
- KCBAMQOKOLXLOX-BSZYMOERSA-N CC1=C(SC=N1)C2=CC=C(C=C2)[C@H](C)NC(=O)[C@@H]3C[C@H](CN3C(=O)[C@H](C(C)(C)C)NC(=O)CCCCCCCCCCNCCCONC(=O)C4=C(C(=C(C=C4)F)F)NC5=C(C=C(C=C5)I)F)O Chemical compound CC1=C(SC=N1)C2=CC=C(C=C2)[C@H](C)NC(=O)[C@@H]3C[C@H](CN3C(=O)[C@H](C(C)(C)C)NC(=O)CCCCCCCCCCNCCCONC(=O)C4=C(C(=C(C=C4)F)F)NC5=C(C=C(C=C5)I)F)O KCBAMQOKOLXLOX-BSZYMOERSA-N 0.000 description 1
- JGLMVXWAHNTPRF-CMDGGOBGSA-N CCN1N=C(C)C=C1C(=O)NC1=NC2=CC(=CC(OC)=C2N1C\C=C\CN1C(NC(=O)C2=CC(C)=NN2CC)=NC2=CC(=CC(OCCCN3CCOCC3)=C12)C(N)=O)C(N)=O Chemical compound CCN1N=C(C)C=C1C(=O)NC1=NC2=CC(=CC(OC)=C2N1C\C=C\CN1C(NC(=O)C2=CC(C)=NN2CC)=NC2=CC(=CC(OCCCN3CCOCC3)=C12)C(N)=O)C(N)=O JGLMVXWAHNTPRF-CMDGGOBGSA-N 0.000 description 1
- 208000011597 CGF1 Diseases 0.000 description 1
- SCJNYBYSTCRPAO-LXBQGUBHSA-N CN(C)C\C=C\C(=O)NC1=CC=C(N=C1)C(=O)N[C@@]1(C)CCC[C@H](C1)NC1=NC(C2=CNC3=CC=CC=C23)=C(Cl)C=N1 Chemical compound CN(C)C\C=C\C(=O)NC1=CC=C(N=C1)C(=O)N[C@@]1(C)CCC[C@H](C1)NC1=NC(C2=CNC3=CC=CC=C23)=C(Cl)C=N1 SCJNYBYSTCRPAO-LXBQGUBHSA-N 0.000 description 1
- XRYGCVVVDCEPRL-UHFFFAOYSA-N CNC1CCN(C)CC1 Chemical compound CNC1CCN(C)CC1 XRYGCVVVDCEPRL-UHFFFAOYSA-N 0.000 description 1
- LXDIZNCSHGUWMV-UHFFFAOYSA-N COc1cc2C(=O)C(CC3CCN(Cc4ccccc4)CC3)Cc2cc1N1CCN(C)C(C)C1 Chemical compound COc1cc2C(=O)C(CC3CCN(Cc4ccccc4)CC3)Cc2cc1N1CCN(C)C(C)C1 LXDIZNCSHGUWMV-UHFFFAOYSA-N 0.000 description 1
- JKMMTHNRTOGOPH-UHFFFAOYSA-N COc1cc2C(=O)C(CC3CCN(Cc4ccccc4)CC3)Cc2cc1N1CCOCC1 Chemical compound COc1cc2C(=O)C(CC3CCN(Cc4ccccc4)CC3)Cc2cc1N1CCOCC1 JKMMTHNRTOGOPH-UHFFFAOYSA-N 0.000 description 1
- AXSSIGOXFCNTIR-UHFFFAOYSA-N COc1cc2C(=O)C(Cc3cc(C)n[nH]3)Cc2cc1N1CCOCC1 Chemical compound COc1cc2C(=O)C(Cc3cc(C)n[nH]3)Cc2cc1N1CCOCC1 AXSSIGOXFCNTIR-UHFFFAOYSA-N 0.000 description 1
- WDSGSXUIZDOYOM-UHFFFAOYSA-N COc1cc2C(=O)C(Cc3ccc(SC(F)(F)F)cc3)Cc2cc1Br Chemical compound COc1cc2C(=O)C(Cc3ccc(SC(F)(F)F)cc3)Cc2cc1Br WDSGSXUIZDOYOM-UHFFFAOYSA-N 0.000 description 1
- BQDSUUIMMOHYIW-UHFFFAOYSA-N COc1cc2C(=O)C(Cc3ccc(SC(F)(F)F)cc3)Cc2cc1N1CCC(C)CC1 Chemical compound COc1cc2C(=O)C(Cc3ccc(SC(F)(F)F)cc3)Cc2cc1N1CCC(C)CC1 BQDSUUIMMOHYIW-UHFFFAOYSA-N 0.000 description 1
- ZSIBRXLQDSBQKX-UHFFFAOYSA-N COc1cc2C(=O)C(Cc3ccc(SC(F)(F)F)cc3)Cc2cc1NC1CCN(C)CC1 Chemical compound COc1cc2C(=O)C(Cc3ccc(SC(F)(F)F)cc3)Cc2cc1NC1CCN(C)CC1 ZSIBRXLQDSBQKX-UHFFFAOYSA-N 0.000 description 1
- TYIDHENENWDKDO-UHFFFAOYSA-N COc1cc2C(=O)C(Cc3ccc(cc3)C(F)(F)F)Cc2cc1N1CCOCC1 Chemical compound COc1cc2C(=O)C(Cc3ccc(cc3)C(F)(F)F)Cc2cc1N1CCOCC1 TYIDHENENWDKDO-UHFFFAOYSA-N 0.000 description 1
- VYXRJGDQGOGPLT-UHFFFAOYSA-N COc1cc2C(=O)C(Cc3cccc(c3)C(F)(F)F)Cc2cc1N1CCC(C)CC1 Chemical compound COc1cc2C(=O)C(Cc3cccc(c3)C(F)(F)F)Cc2cc1N1CCC(C)CC1 VYXRJGDQGOGPLT-UHFFFAOYSA-N 0.000 description 1
- BJIWWYPOGLBEHP-UHFFFAOYSA-N COc1cc2C(=O)C(Cc3cccc(c3F)C(F)(F)F)Cc2cc1N1CCOCC1 Chemical compound COc1cc2C(=O)C(Cc3cccc(c3F)C(F)(F)F)Cc2cc1N1CCOCC1 BJIWWYPOGLBEHP-UHFFFAOYSA-N 0.000 description 1
- IBJUWDIIWMLCAC-UHFFFAOYSA-N COc1cc2C(=O)C(Cc3cccnc3)Cc2cc1N1CCN(C)CC1 Chemical compound COc1cc2C(=O)C(Cc3cccnc3)Cc2cc1N1CCN(C)CC1 IBJUWDIIWMLCAC-UHFFFAOYSA-N 0.000 description 1
- NNMIGKXQLFIUDJ-UHFFFAOYSA-N COc1cc2C(=O)C(Cc3cccnc3)Cc2cc1N1CCOCC1 Chemical compound COc1cc2C(=O)C(Cc3cccnc3)Cc2cc1N1CCOCC1 NNMIGKXQLFIUDJ-UHFFFAOYSA-N 0.000 description 1
- IGCWZHIGORPXQM-UHFFFAOYSA-N COc1cc2C(=O)C(Cc3ncccc3C(F)(F)F)Cc2cc1-c1ccccc1OC(F)(F)F Chemical compound COc1cc2C(=O)C(Cc3ncccc3C(F)(F)F)Cc2cc1-c1ccccc1OC(F)(F)F IGCWZHIGORPXQM-UHFFFAOYSA-N 0.000 description 1
- JRRDWFDHSJLTSR-UHFFFAOYSA-N COc1cc2C(=O)C(Cc3ncccc3C(F)(F)F)Cc2cc1N1CCNCC1 Chemical compound COc1cc2C(=O)C(Cc3ncccc3C(F)(F)F)Cc2cc1N1CCNCC1 JRRDWFDHSJLTSR-UHFFFAOYSA-N 0.000 description 1
- IFJFXSYTSXTOIB-UHFFFAOYSA-N COc1cc2C(=O)CCc2cc1N1CCC(C)CC1 Chemical compound COc1cc2C(=O)CCc2cc1N1CCC(C)CC1 IFJFXSYTSXTOIB-UHFFFAOYSA-N 0.000 description 1
- JIWGAIFWYHYATD-UHFFFAOYSA-N COc1cc2C(=O)CCc2cc1N1CCNCC1 Chemical compound COc1cc2C(=O)CCc2cc1N1CCNCC1 JIWGAIFWYHYATD-UHFFFAOYSA-N 0.000 description 1
- PKMUHQIDVVOXHQ-HXUWFJFHSA-N C[C@H](C1=CC(C2=CC=C(CNC3CCCC3)S2)=CC=C1)NC(C1=C(C)C=CC(NC2CNC2)=C1)=O Chemical compound C[C@H](C1=CC(C2=CC=C(CNC3CCCC3)S2)=CC=C1)NC(C1=C(C)C=CC(NC2CNC2)=C1)=O PKMUHQIDVVOXHQ-HXUWFJFHSA-N 0.000 description 1
- KXDHJXZQYSOELW-UHFFFAOYSA-M Carbamate Chemical compound NC([O-])=O KXDHJXZQYSOELW-UHFFFAOYSA-M 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 1
- KZBUYRJDOAKODT-UHFFFAOYSA-N Chlorine Chemical compound ClCl KZBUYRJDOAKODT-UHFFFAOYSA-N 0.000 description 1
- PTOAARAWEBMLNO-KVQBGUIXSA-N Cladribine Chemical compound C1=NC=2C(N)=NC(Cl)=NC=2N1[C@H]1C[C@H](O)[C@@H](CO)O1 PTOAARAWEBMLNO-KVQBGUIXSA-N 0.000 description 1
- 229920002261 Corn starch Polymers 0.000 description 1
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 1
- 230000004568 DNA-binding Effects 0.000 description 1
- 229920002307 Dextran Polymers 0.000 description 1
- 239000004338 Dichlorodifluoromethane Substances 0.000 description 1
- 206010061818 Disease progression Diseases 0.000 description 1
- 102100025027 E3 ubiquitin-protein ligase TRIM69 Human genes 0.000 description 1
- 238000008157 ELISA kit Methods 0.000 description 1
- LVGKNOAMLMIIKO-UHFFFAOYSA-N Elaidinsaeure-aethylester Natural products CCCCCCCCC=CCCCCCCCC(=O)OCC LVGKNOAMLMIIKO-UHFFFAOYSA-N 0.000 description 1
- 241000792859 Enema Species 0.000 description 1
- 101100322888 Escherichia coli (strain K12) metL gene Proteins 0.000 description 1
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 1
- 206010016654 Fibrosis Diseases 0.000 description 1
- 108010024636 Glutathione Proteins 0.000 description 1
- 102000003886 Glycoproteins Human genes 0.000 description 1
- 108090000288 Glycoproteins Proteins 0.000 description 1
- 238000010268 HPLC based assay Methods 0.000 description 1
- 241000238631 Hexapoda Species 0.000 description 1
- 101000830203 Homo sapiens E3 ubiquitin-protein ligase TRIM69 Proteins 0.000 description 1
- 101000998146 Homo sapiens Interleukin-17A Proteins 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- 206010061218 Inflammation Diseases 0.000 description 1
- 108010002350 Interleukin-2 Proteins 0.000 description 1
- JVTAAEKCZFNVCJ-UHFFFAOYSA-M Lactate Chemical compound CC(O)C([O-])=O JVTAAEKCZFNVCJ-UHFFFAOYSA-M 0.000 description 1
- 240000007472 Leucaena leucocephala Species 0.000 description 1
- 235000010643 Leucaena leucocephala Nutrition 0.000 description 1
- 108060001084 Luciferase Proteins 0.000 description 1
- 239000005089 Luciferase Substances 0.000 description 1
- 229930195725 Mannitol Natural products 0.000 description 1
- FQISKWAFAHGMGT-SGJOWKDISA-M Methylprednisolone sodium succinate Chemical compound [Na+].C([C@@]12C)=CC(=O)C=C1[C@@H](C)C[C@@H]1[C@@H]2[C@@H](O)C[C@]2(C)[C@@](O)(C(=O)COC(=O)CCC([O-])=O)CC[C@H]21 FQISKWAFAHGMGT-SGJOWKDISA-M 0.000 description 1
- 241001529936 Murinae Species 0.000 description 1
- HSHXDCVZWHOWCS-UHFFFAOYSA-N N'-hexadecylthiophene-2-carbohydrazide Chemical compound CCCCCCCCCCCCCCCCNNC(=O)c1cccs1 HSHXDCVZWHOWCS-UHFFFAOYSA-N 0.000 description 1
- MBBZMMPHUWSWHV-BDVNFPICSA-N N-methylglucamine Chemical compound CNC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO MBBZMMPHUWSWHV-BDVNFPICSA-N 0.000 description 1
- 238000012565 NMR experiment Methods 0.000 description 1
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 1
- ZCQWOFVYLHDMMC-UHFFFAOYSA-N Oxazole Chemical compound C1=COC=N1 ZCQWOFVYLHDMMC-UHFFFAOYSA-N 0.000 description 1
- 229910019142 PO4 Inorganic materials 0.000 description 1
- 229920002642 Polysorbate 65 Polymers 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 1
- 206010053395 Progressive multiple sclerosis Diseases 0.000 description 1
- WTKZEGDFNFYCGP-UHFFFAOYSA-N Pyrazole Chemical compound C=1C=NNC=1 WTKZEGDFNFYCGP-UHFFFAOYSA-N 0.000 description 1
- CZPWVGJYEJSRLH-UHFFFAOYSA-N Pyrimidine Chemical compound C1=CN=CN=C1 CZPWVGJYEJSRLH-UHFFFAOYSA-N 0.000 description 1
- 108091008680 RAR-related orphan receptors Proteins 0.000 description 1
- 108091008778 RORγ2 Proteins 0.000 description 1
- OZBSSKGBKHOLGA-UHFFFAOYSA-N SR 1001 Chemical compound S1C(NC(=O)C)=NC(C)=C1S(=O)(=O)NC1=CC=C(C(O)(C(F)(F)F)C(F)(F)F)C=C1 OZBSSKGBKHOLGA-UHFFFAOYSA-N 0.000 description 1
- 208000034189 Sclerosis Diseases 0.000 description 1
- 229920002684 Sepharose Polymers 0.000 description 1
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 1
- 229920002125 Sokalan® Polymers 0.000 description 1
- 229930182558 Sterol Natural products 0.000 description 1
- 229930006000 Sucrose Natural products 0.000 description 1
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 1
- FZWLAAWBMGSTSO-UHFFFAOYSA-N Thiazole Chemical compound C1=CSC=N1 FZWLAAWBMGSTSO-UHFFFAOYSA-N 0.000 description 1
- 229920001615 Tragacanth Polymers 0.000 description 1
- 102000040945 Transcription factor Human genes 0.000 description 1
- 108091023040 Transcription factor Proteins 0.000 description 1
- 206010044565 Tremor Diseases 0.000 description 1
- GSEJCLTVZPLZKY-UHFFFAOYSA-N Triethanolamine Chemical compound OCCN(CCO)CCO GSEJCLTVZPLZKY-UHFFFAOYSA-N 0.000 description 1
- 239000007983 Tris buffer Substances 0.000 description 1
- 241000700605 Viruses Species 0.000 description 1
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 1
- SPXSEZMVRJLHQG-XMMPIXPASA-N [(2R)-1-[[4-[(3-phenylmethoxyphenoxy)methyl]phenyl]methyl]pyrrolidin-2-yl]methanol Chemical compound C(C1=CC=CC=C1)OC=1C=C(OCC2=CC=C(CN3[C@H](CCC3)CO)C=C2)C=CC=1 SPXSEZMVRJLHQG-XMMPIXPASA-N 0.000 description 1
- YQMMVZVQAHYXFZ-UHFFFAOYSA-N [2-fluoro-4-(trifluoromethoxy)phenyl]boronic acid Chemical compound OB(O)C1=CC=C(OC(F)(F)F)C=C1F YQMMVZVQAHYXFZ-UHFFFAOYSA-N 0.000 description 1
- 238000002835 absorbance Methods 0.000 description 1
- 238000010521 absorption reaction Methods 0.000 description 1
- FHEAIOHRHQGZPC-KIWGSFCNSA-N acetic acid;(2s)-2-amino-3-(4-hydroxyphenyl)propanoic acid;(2s)-2-aminopentanedioic acid;(2s)-2-aminopropanoic acid;(2s)-2,6-diaminohexanoic acid Chemical compound CC(O)=O.C[C@H](N)C(O)=O.NCCCC[C@H](N)C(O)=O.OC(=O)[C@@H](N)CCC(O)=O.OC(=O)[C@@H](N)CC1=CC=C(O)C=C1 FHEAIOHRHQGZPC-KIWGSFCNSA-N 0.000 description 1
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 239000008272 agar Substances 0.000 description 1
- 235000010419 agar Nutrition 0.000 description 1
- 229940040563 agaric acid Drugs 0.000 description 1
- 229960000548 alemtuzumab Drugs 0.000 description 1
- 235000010443 alginic acid Nutrition 0.000 description 1
- 239000000783 alginic acid Substances 0.000 description 1
- 229920000615 alginic acid Polymers 0.000 description 1
- 229960001126 alginic acid Drugs 0.000 description 1
- 150000004781 alginic acids Chemical class 0.000 description 1
- 229910001413 alkali metal ion Inorganic materials 0.000 description 1
- 125000000278 alkyl amino alkyl group Chemical group 0.000 description 1
- 125000002947 alkylene group Chemical group 0.000 description 1
- IYABWNGZIDDRAK-UHFFFAOYSA-N allene Chemical group C=C=C IYABWNGZIDDRAK-UHFFFAOYSA-N 0.000 description 1
- 229910052782 aluminium Inorganic materials 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- 150000001408 amides Chemical class 0.000 description 1
- 125000004103 aminoalkyl group Chemical group 0.000 description 1
- 125000002178 anthracenyl group Chemical group C1(=CC=CC2=CC3=CC=CC=C3C=C12)* 0.000 description 1
- 230000003110 anti-inflammatory effect Effects 0.000 description 1
- 229940058307 antinematodal tetrahydropyrimidine derivative Drugs 0.000 description 1
- 238000013459 approach Methods 0.000 description 1
- 239000008135 aqueous vehicle Substances 0.000 description 1
- 230000002238 attenuated effect Effects 0.000 description 1
- 229940003504 avonex Drugs 0.000 description 1
- 230000004888 barrier function Effects 0.000 description 1
- PQNFLJBBNBOBRQ-UHFFFAOYSA-N benzocyclopentane Natural products C1=CC=C2CCCC2=C1 PQNFLJBBNBOBRQ-UHFFFAOYSA-N 0.000 description 1
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 1
- 229940021459 betaseron Drugs 0.000 description 1
- 229920000249 biocompatible polymer Polymers 0.000 description 1
- 230000008827 biological function Effects 0.000 description 1
- 239000012267 brine Substances 0.000 description 1
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 1
- 229910052794 bromium Inorganic materials 0.000 description 1
- 229910000019 calcium carbonate Inorganic materials 0.000 description 1
- AXCZMVOFGPJBDE-UHFFFAOYSA-L calcium dihydroxide Chemical compound [OH-].[OH-].[Ca+2] AXCZMVOFGPJBDE-UHFFFAOYSA-L 0.000 description 1
- 238000000500 calorimetric titration Methods 0.000 description 1
- 239000007963 capsule composition Substances 0.000 description 1
- 239000004202 carbamide Substances 0.000 description 1
- 239000001569 carbon dioxide Substances 0.000 description 1
- 229910002092 carbon dioxide Inorganic materials 0.000 description 1
- 229960004424 carbon dioxide Drugs 0.000 description 1
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 1
- 125000004181 carboxyalkyl group Chemical group 0.000 description 1
- 125000002843 carboxylic acid group Chemical group 0.000 description 1
- 230000003915 cell function Effects 0.000 description 1
- 238000004587 chromatography analysis Methods 0.000 description 1
- 229960002436 cladribine Drugs 0.000 description 1
- 239000011248 coating agent Substances 0.000 description 1
- 229940110456 cocoa butter Drugs 0.000 description 1
- 235000019868 cocoa butter Nutrition 0.000 description 1
- 238000004440 column chromatography Methods 0.000 description 1
- 229940126208 compound 22 Drugs 0.000 description 1
- 229940125833 compound 23 Drugs 0.000 description 1
- 229940125851 compound 27 Drugs 0.000 description 1
- 229940125877 compound 31 Drugs 0.000 description 1
- 229940125936 compound 42 Drugs 0.000 description 1
- 229940127271 compound 49 Drugs 0.000 description 1
- 229940126545 compound 53 Drugs 0.000 description 1
- 229940126179 compound 72 Drugs 0.000 description 1
- 229940038717 copaxone Drugs 0.000 description 1
- 239000008120 corn starch Substances 0.000 description 1
- 229940099112 cornstarch Drugs 0.000 description 1
- 230000001186 cumulative effect Effects 0.000 description 1
- 238000005520 cutting process Methods 0.000 description 1
- 125000004966 cyanoalkyl group Chemical group 0.000 description 1
- 125000004122 cyclic group Chemical group 0.000 description 1
- 125000006310 cycloalkyl amino group Chemical group 0.000 description 1
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 1
- 229960002806 daclizumab Drugs 0.000 description 1
- 230000002950 deficient Effects 0.000 description 1
- 239000003405 delayed action preparation Substances 0.000 description 1
- 238000013461 design Methods 0.000 description 1
- 125000004985 dialkyl amino alkyl group Chemical group 0.000 description 1
- PXBRQCKWGAHEHS-UHFFFAOYSA-N dichlorodifluoromethane Chemical compound FC(F)(Cl)Cl PXBRQCKWGAHEHS-UHFFFAOYSA-N 0.000 description 1
- 235000019404 dichlorodifluoromethane Nutrition 0.000 description 1
- 229940087091 dichlorotetrafluoroethane Drugs 0.000 description 1
- 235000014113 dietary fatty acids Nutrition 0.000 description 1
- ZBCBWPMODOFKDW-UHFFFAOYSA-N diethanolamine Chemical compound OCCNCCO ZBCBWPMODOFKDW-UHFFFAOYSA-N 0.000 description 1
- IJKVHSBPTUYDLN-UHFFFAOYSA-N dihydroxy(oxo)silane Chemical compound O[Si](O)=O IJKVHSBPTUYDLN-UHFFFAOYSA-N 0.000 description 1
- 230000009266 disease activity Effects 0.000 description 1
- 230000005750 disease progression Effects 0.000 description 1
- 239000002270 dispersing agent Substances 0.000 description 1
- 238000009826 distribution Methods 0.000 description 1
- 239000008298 dragée Substances 0.000 description 1
- 239000013583 drug formulation Substances 0.000 description 1
- 239000003596 drug target Substances 0.000 description 1
- 239000000975 dye Substances 0.000 description 1
- 238000000921 elemental analysis Methods 0.000 description 1
- 230000001804 emulsifying effect Effects 0.000 description 1
- 206010014599 encephalitis Diseases 0.000 description 1
- 239000007920 enema Substances 0.000 description 1
- 229940079360 enema for constipation Drugs 0.000 description 1
- 238000005516 engineering process Methods 0.000 description 1
- LAUNICFMAJGVPI-UHFFFAOYSA-N ethyl 4-[2-oxo-3-(3-oxopiperazin-1-yl)imidazolidin-1-yl]piperazine-1-carboxylate Chemical compound C1CN(C(=O)OCC)CCN1N1C(=O)N(N2CC(=O)NCC2)CC1 LAUNICFMAJGVPI-UHFFFAOYSA-N 0.000 description 1
- LVGKNOAMLMIIKO-QXMHVHEDSA-N ethyl oleate Chemical compound CCCCCCCC\C=C/CCCCCCCC(=O)OCC LVGKNOAMLMIIKO-QXMHVHEDSA-N 0.000 description 1
- 229940093471 ethyl oleate Drugs 0.000 description 1
- 125000005469 ethylenyl group Chemical group 0.000 description 1
- 238000013401 experimental design Methods 0.000 description 1
- 229940077362 extavia Drugs 0.000 description 1
- 239000000194 fatty acid Substances 0.000 description 1
- 229930195729 fatty acid Natural products 0.000 description 1
- 230000004761 fibrosis Effects 0.000 description 1
- 239000000945 filler Substances 0.000 description 1
- 238000002866 fluorescence resonance energy transfer Methods 0.000 description 1
- 239000011888 foil Substances 0.000 description 1
- 230000037406 food intake Effects 0.000 description 1
- 125000002485 formyl group Chemical group [H]C(*)=O 0.000 description 1
- 239000012458 free base Substances 0.000 description 1
- 230000006870 function Effects 0.000 description 1
- 239000000417 fungicide Substances 0.000 description 1
- 230000002496 gastric effect Effects 0.000 description 1
- 239000007903 gelatin capsule Substances 0.000 description 1
- 239000011521 glass Substances 0.000 description 1
- 229960003776 glatiramer acetate Drugs 0.000 description 1
- 229960003180 glutathione Drugs 0.000 description 1
- 125000005456 glyceride group Chemical group 0.000 description 1
- 239000008187 granular material Substances 0.000 description 1
- 238000000227 grinding Methods 0.000 description 1
- 229910052736 halogen Inorganic materials 0.000 description 1
- 150000002367 halogens Chemical class 0.000 description 1
- 125000005553 heteroaryloxy group Chemical group 0.000 description 1
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 238000004128 high performance liquid chromatography Methods 0.000 description 1
- 230000013632 homeostatic process Effects 0.000 description 1
- 229920001600 hydrophobic polymer Polymers 0.000 description 1
- 239000001866 hydroxypropyl methyl cellulose Substances 0.000 description 1
- 235000010979 hydroxypropyl methyl cellulose Nutrition 0.000 description 1
- 229920003088 hydroxypropyl methyl cellulose Polymers 0.000 description 1
- 230000002519 immonomodulatory effect Effects 0.000 description 1
- 230000001900 immune effect Effects 0.000 description 1
- 230000036039 immunity Effects 0.000 description 1
- 230000003053 immunization Effects 0.000 description 1
- 238000002649 immunization Methods 0.000 description 1
- 239000002955 immunomodulating agent Substances 0.000 description 1
- 229940121354 immunomodulator Drugs 0.000 description 1
- 230000001024 immunotherapeutic effect Effects 0.000 description 1
- 230000001771 impaired effect Effects 0.000 description 1
- 238000002513 implantation Methods 0.000 description 1
- 230000006698 induction Effects 0.000 description 1
- 208000015181 infectious disease Diseases 0.000 description 1
- 230000004054 inflammatory process Effects 0.000 description 1
- 238000002329 infrared spectrum Methods 0.000 description 1
- 238000001802 infusion Methods 0.000 description 1
- 229940047124 interferons Drugs 0.000 description 1
- 230000000968 intestinal effect Effects 0.000 description 1
- 238000007918 intramuscular administration Methods 0.000 description 1
- 238000010255 intramuscular injection Methods 0.000 description 1
- 239000007927 intramuscular injection Substances 0.000 description 1
- 238000007912 intraperitoneal administration Methods 0.000 description 1
- 238000007913 intrathecal administration Methods 0.000 description 1
- 238000007914 intraventricular administration Methods 0.000 description 1
- PNDPGZBMCMUPRI-UHFFFAOYSA-N iodine Chemical compound II PNDPGZBMCMUPRI-UHFFFAOYSA-N 0.000 description 1
- 229910052740 iodine Inorganic materials 0.000 description 1
- 239000011630 iodine Substances 0.000 description 1
- INQOMBQAUSQDDS-UHFFFAOYSA-N iodomethane Chemical compound IC INQOMBQAUSQDDS-UHFFFAOYSA-N 0.000 description 1
- 239000003456 ion exchange resin Substances 0.000 description 1
- 229920003303 ion-exchange polymer Polymers 0.000 description 1
- 150000002500 ions Chemical class 0.000 description 1
- 125000005468 isobutylenyl group Chemical group 0.000 description 1
- FZWBNHMXJMCXLU-BLAUPYHCSA-N isomaltotriose Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@@H]1OC[C@@H]1[C@@H](O)[C@H](O)[C@@H](O)[C@@H](OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C=O)O1 FZWBNHMXJMCXLU-BLAUPYHCSA-N 0.000 description 1
- 125000001972 isopentyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])C([H])([H])* 0.000 description 1
- 238000002372 labelling Methods 0.000 description 1
- 239000004922 lacquer Substances 0.000 description 1
- 229940001447 lactate Drugs 0.000 description 1
- 229960004577 laquinimod Drugs 0.000 description 1
- GKWPCEFFIHSJOE-UHFFFAOYSA-N laquinimod Chemical compound OC=1C2=C(Cl)C=CC=C2N(C)C(=O)C=1C(=O)N(CC)C1=CC=CC=C1 GKWPCEFFIHSJOE-UHFFFAOYSA-N 0.000 description 1
- 229940047834 lemtrada Drugs 0.000 description 1
- 230000003902 lesion Effects 0.000 description 1
- 229940057995 liquid paraffin Drugs 0.000 description 1
- 230000005923 long-lasting effect Effects 0.000 description 1
- 208000018769 loss of vision Diseases 0.000 description 1
- 231100000864 loss of vision Toxicity 0.000 description 1
- 239000000314 lubricant Substances 0.000 description 1
- 238000003468 luciferase reporter gene assay Methods 0.000 description 1
- 238000012792 lyophilization process Methods 0.000 description 1
- 210000002540 macrophage Anatomy 0.000 description 1
- 159000000003 magnesium salts Chemical class 0.000 description 1
- 235000019359 magnesium stearate Nutrition 0.000 description 1
- 229940049920 malate Drugs 0.000 description 1
- 239000011976 maleic acid Substances 0.000 description 1
- BJEPYKJPYRNKOW-UHFFFAOYSA-N malic acid Chemical compound OC(=O)C(O)CC(O)=O BJEPYKJPYRNKOW-UHFFFAOYSA-N 0.000 description 1
- 239000000594 mannitol Substances 0.000 description 1
- 235000010355 mannitol Nutrition 0.000 description 1
- 238000001819 mass spectrum Methods 0.000 description 1
- 238000002483 medication Methods 0.000 description 1
- 238000002844 melting Methods 0.000 description 1
- 230000008018 melting Effects 0.000 description 1
- 230000003340 mental effect Effects 0.000 description 1
- 239000002207 metabolite Substances 0.000 description 1
- 229910052751 metal Inorganic materials 0.000 description 1
- 239000002184 metal Substances 0.000 description 1
- 229910021645 metal ion Inorganic materials 0.000 description 1
- 229940098779 methanesulfonic acid Drugs 0.000 description 1
- 229920000609 methyl cellulose Polymers 0.000 description 1
- YMCAVGXTSCNFDE-BBACVFHCSA-N methyl n-[(2s)-1-[(2s)-2-[5-[4-[4-[2-[(8s)-7-[(2s)-2-(methoxycarbonylamino)-3-methylbutanoyl]-1,4-dioxa-7-azaspiro[4.4]nonan-8-yl]-1h-imidazol-5-yl]phenyl]phenyl]-1h-imidazol-2-yl]pyrrolidin-1-yl]-3-methyl-1-oxobutan-2-yl]carbamate Chemical compound COC(=O)N[C@@H](C(C)C)C(=O)N1CCC[C@H]1C1=NC=C(C=2C=CC(=CC=2)C=2C=CC(=CC=2)C=2NC(=NC=2)[C@H]2N(CC3(C2)OCCO3)C(=O)[C@@H](NC(=O)OC)C(C)C)N1 YMCAVGXTSCNFDE-BBACVFHCSA-N 0.000 description 1
- 239000001923 methylcellulose Substances 0.000 description 1
- 235000010981 methylcellulose Nutrition 0.000 description 1
- 125000001570 methylene group Chemical group [H]C([H])([*:1])[*:2] 0.000 description 1
- 229960004584 methylprednisolone Drugs 0.000 description 1
- 150000007522 mineralic acids Chemical class 0.000 description 1
- KKZJGLLVHKMTCM-UHFFFAOYSA-N mitoxantrone Chemical compound O=C1C2=C(O)C=CC(O)=C2C(=O)C2=C1C(NCCNCCO)=CC=C2NCCNCCO KKZJGLLVHKMTCM-UHFFFAOYSA-N 0.000 description 1
- 229960001156 mitoxantrone Drugs 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- 125000002950 monocyclic group Chemical group 0.000 description 1
- 238000010172 mouse model Methods 0.000 description 1
- 230000035772 mutation Effects 0.000 description 1
- YCJZWBZJSYLMPB-UHFFFAOYSA-N n-(2-chloropyrimidin-4-yl)-2,5-dimethyl-1-phenylimidazole-4-carboxamide Chemical compound CC=1N(C=2C=CC=CC=2)C(C)=NC=1C(=O)NC1=CC=NC(Cl)=N1 YCJZWBZJSYLMPB-UHFFFAOYSA-N 0.000 description 1
- 125000001280 n-hexyl group Chemical group C(CCCCC)* 0.000 description 1
- 125000000740 n-pentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000001624 naphthyl group Chemical group 0.000 description 1
- 239000006199 nebulizer Substances 0.000 description 1
- 230000014399 negative regulation of angiogenesis Effects 0.000 description 1
- 230000007302 negative regulation of cytokine production Effects 0.000 description 1
- 230000004112 neuroprotection Effects 0.000 description 1
- 230000000324 neuroprotective effect Effects 0.000 description 1
- 229910017604 nitric acid Inorganic materials 0.000 description 1
- 125000004433 nitrogen atom Chemical group N* 0.000 description 1
- 125000006574 non-aromatic ring group Chemical group 0.000 description 1
- 108020004017 nuclear receptors Proteins 0.000 description 1
- 229950005751 ocrelizumab Drugs 0.000 description 1
- 238000001543 one-way ANOVA Methods 0.000 description 1
- 238000003305 oral gavage Methods 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- 150000007530 organic bases Chemical class 0.000 description 1
- 229920000620 organic polymer Polymers 0.000 description 1
- 239000003791 organic solvent mixture Substances 0.000 description 1
- 229940046781 other immunosuppressants in atc Drugs 0.000 description 1
- 230000002611 ovarian Effects 0.000 description 1
- 235000006408 oxalic acid Nutrition 0.000 description 1
- 125000004043 oxo group Chemical group O=* 0.000 description 1
- FJKROLUGYXJWQN-UHFFFAOYSA-N papa-hydroxy-benzoic acid Natural products OC(=O)C1=CC=C(O)C=C1 FJKROLUGYXJWQN-UHFFFAOYSA-N 0.000 description 1
- QNGNSVIICDLXHT-UHFFFAOYSA-N para-ethylbenzaldehyde Natural products CCC1=CC=C(C=O)C=C1 QNGNSVIICDLXHT-UHFFFAOYSA-N 0.000 description 1
- 230000007170 pathology Effects 0.000 description 1
- 125000001147 pentyl group Chemical group C(CCCC)* 0.000 description 1
- 229940127557 pharmaceutical product Drugs 0.000 description 1
- 230000000144 pharmacologic effect Effects 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 1
- 239000010452 phosphate Substances 0.000 description 1
- 239000002504 physiological saline solution Substances 0.000 description 1
- 239000000049 pigment Substances 0.000 description 1
- 239000013612 plasmid Substances 0.000 description 1
- 239000004014 plasticizer Substances 0.000 description 1
- 125000003367 polycyclic group Chemical group 0.000 description 1
- 229940068886 polyethylene glycol 300 Drugs 0.000 description 1
- 229920000642 polymer Polymers 0.000 description 1
- 239000001816 polyoxyethylene sorbitan tristearate Substances 0.000 description 1
- 235000010988 polyoxyethylene sorbitan tristearate Nutrition 0.000 description 1
- 229920001282 polysaccharide Polymers 0.000 description 1
- 239000005017 polysaccharide Substances 0.000 description 1
- 150000004804 polysaccharides Chemical class 0.000 description 1
- 229940099511 polysorbate 65 Drugs 0.000 description 1
- 229910052700 potassium Inorganic materials 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- 235000011181 potassium carbonates Nutrition 0.000 description 1
- 229920001592 potato starch Polymers 0.000 description 1
- 229940116317 potato starch Drugs 0.000 description 1
- 229960004618 prednisone Drugs 0.000 description 1
- XOFYZVNMUHMLCC-ZPOLXVRWSA-N prednisone Chemical compound O=C1C=C[C@]2(C)[C@H]3C(=O)C[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 XOFYZVNMUHMLCC-ZPOLXVRWSA-N 0.000 description 1
- 239000000955 prescription drug Substances 0.000 description 1
- 239000003755 preservative agent Substances 0.000 description 1
- 230000002335 preservative effect Effects 0.000 description 1
- 230000002265 prevention Effects 0.000 description 1
- 230000008569 process Effects 0.000 description 1
- 239000003380 propellant Substances 0.000 description 1
- 238000011321 prophylaxis Methods 0.000 description 1
- QQONPFPTGQHPMA-UHFFFAOYSA-N propylene Natural products CC=C QQONPFPTGQHPMA-UHFFFAOYSA-N 0.000 description 1
- 125000004805 propylene group Chemical group [H]C([H])([H])C([H])([*:1])C([H])([H])[*:2] 0.000 description 1
- 125000005470 propylenyl group Chemical group 0.000 description 1
- 125000002568 propynyl group Chemical group [*]C#CC([H])([H])[H] 0.000 description 1
- 238000000159 protein binding assay Methods 0.000 description 1
- 230000029983 protein stabilization Effects 0.000 description 1
- 108090000623 proteins and genes Proteins 0.000 description 1
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 1
- HNJBEVLQSNELDL-UHFFFAOYSA-N pyrrolidin-2-one Chemical compound O=C1CCCN1 HNJBEVLQSNELDL-UHFFFAOYSA-N 0.000 description 1
- 125000000168 pyrrolyl group Chemical group 0.000 description 1
- 238000004445 quantitative analysis Methods 0.000 description 1
- 229940038850 rebif Drugs 0.000 description 1
- 230000000306 recurrent effect Effects 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 210000003289 regulatory T cell Anatomy 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 229910052702 rhenium Inorganic materials 0.000 description 1
- 229940100486 rice starch Drugs 0.000 description 1
- 229960004889 salicylic acid Drugs 0.000 description 1
- 238000005070 sampling Methods 0.000 description 1
- 238000003118 sandwich ELISA Methods 0.000 description 1
- 230000037390 scarring Effects 0.000 description 1
- 230000002784 sclerotic effect Effects 0.000 description 1
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 230000028327 secretion Effects 0.000 description 1
- 230000035807 sensation Effects 0.000 description 1
- 238000012163 sequencing technique Methods 0.000 description 1
- 235000011803 sesame oil Nutrition 0.000 description 1
- 239000008159 sesame oil Substances 0.000 description 1
- 108010085082 sigma receptors Proteins 0.000 description 1
- 239000002002 slurry Substances 0.000 description 1
- 230000004039 social cognition Effects 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- 239000011734 sodium Substances 0.000 description 1
- 235000010413 sodium alginate Nutrition 0.000 description 1
- 239000000661 sodium alginate Substances 0.000 description 1
- 229940005550 sodium alginate Drugs 0.000 description 1
- 239000011780 sodium chloride Substances 0.000 description 1
- KSAVQLQVUXSOCR-UHFFFAOYSA-M sodium lauroyl sarcosinate Chemical compound [Na+].CCCCCCCCCCCC(=O)N(C)CC([O-])=O KSAVQLQVUXSOCR-UHFFFAOYSA-M 0.000 description 1
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 1
- 239000007901 soft capsule Substances 0.000 description 1
- 239000012439 solid excipient Substances 0.000 description 1
- 241000894007 species Species 0.000 description 1
- 210000004988 splenocyte Anatomy 0.000 description 1
- 239000007921 spray Substances 0.000 description 1
- 238000005507 spraying Methods 0.000 description 1
- 206010041823 squamous cell carcinoma Diseases 0.000 description 1
- 238000010186 staining Methods 0.000 description 1
- 239000008223 sterile water Substances 0.000 description 1
- 150000003432 sterols Chemical class 0.000 description 1
- 235000003702 sterols Nutrition 0.000 description 1
- 238000007920 subcutaneous administration Methods 0.000 description 1
- 125000000547 substituted alkyl group Chemical group 0.000 description 1
- 239000000758 substrate Substances 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-L succinate(2-) Chemical compound [O-]C(=O)CCC([O-])=O KDYFGRWQOYBRFD-UHFFFAOYSA-L 0.000 description 1
- 239000001384 succinic acid Substances 0.000 description 1
- 239000005720 sucrose Substances 0.000 description 1
- 239000000829 suppository Substances 0.000 description 1
- 239000002511 suppository base Substances 0.000 description 1
- 230000004083 survival effect Effects 0.000 description 1
- 239000000375 suspending agent Substances 0.000 description 1
- 238000010189 synthetic method Methods 0.000 description 1
- 239000006188 syrup Substances 0.000 description 1
- 235000020357 syrup Nutrition 0.000 description 1
- 238000007910 systemic administration Methods 0.000 description 1
- 238000004885 tandem mass spectrometry Methods 0.000 description 1
- 239000011975 tartaric acid Substances 0.000 description 1
- 235000002906 tartaric acid Nutrition 0.000 description 1
- 229940095064 tartrate Drugs 0.000 description 1
- 238000003419 tautomerization reaction Methods 0.000 description 1
- 229960000331 teriflunomide Drugs 0.000 description 1
- UTNUDOFZCWSZMS-YFHOEESVSA-N teriflunomide Chemical compound C\C(O)=C(/C#N)C(=O)NC1=CC=C(C(F)(F)F)C=C1 UTNUDOFZCWSZMS-YFHOEESVSA-N 0.000 description 1
- CWXPZXBSDSIRCS-UHFFFAOYSA-N tert-butyl piperazine-1-carboxylate Chemical compound CC(C)(C)OC(=O)N1CCNCC1 CWXPZXBSDSIRCS-UHFFFAOYSA-N 0.000 description 1
- 125000001712 tetrahydronaphthyl group Chemical group C1(CCCC2=CC=CC=C12)* 0.000 description 1
- 125000001412 tetrahydropyranyl group Chemical group 0.000 description 1
- 150000005326 tetrahydropyrimidines Chemical class 0.000 description 1
- 150000003536 tetrazoles Chemical class 0.000 description 1
- 125000004001 thioalkyl group Chemical group 0.000 description 1
- 229930192474 thiophene Natural products 0.000 description 1
- 210000001541 thymus gland Anatomy 0.000 description 1
- 210000001519 tissue Anatomy 0.000 description 1
- 239000004408 titanium dioxide Substances 0.000 description 1
- 231100000440 toxicity profile Toxicity 0.000 description 1
- 238000012546 transfer Methods 0.000 description 1
- 230000035903 transrepression Effects 0.000 description 1
- 238000011269 treatment regimen Methods 0.000 description 1
- 150000003626 triacylglycerols Chemical class 0.000 description 1
- 150000003852 triazoles Chemical class 0.000 description 1
- CYRMSUTZVYGINF-UHFFFAOYSA-N trichlorofluoromethane Chemical compound FC(Cl)(Cl)Cl CYRMSUTZVYGINF-UHFFFAOYSA-N 0.000 description 1
- 229940029284 trichlorofluoromethane Drugs 0.000 description 1
- 125000004044 trifluoroacetyl group Chemical group FC(C(=O)*)(F)F 0.000 description 1
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 1
- 229960000281 trometamol Drugs 0.000 description 1
- 229940079023 tysabri Drugs 0.000 description 1
- 241000701447 unidentified baculovirus Species 0.000 description 1
- 125000004417 unsaturated alkyl group Chemical group 0.000 description 1
- 229940096998 ursolic acid Drugs 0.000 description 1
- PLSAJKYPRJGMHO-UHFFFAOYSA-N ursolic acid Natural products CC1CCC2(CCC3(C)C(C=CC4C5(C)CCC(O)C(C)(C)C5CCC34C)C2C1C)C(=O)O PLSAJKYPRJGMHO-UHFFFAOYSA-N 0.000 description 1
- 238000010200 validation analysis Methods 0.000 description 1
- 235000015112 vegetable and seed oil Nutrition 0.000 description 1
- 239000008158 vegetable oil Substances 0.000 description 1
- 230000004393 visual impairment Effects 0.000 description 1
- 238000005406 washing Methods 0.000 description 1
- 229940100445 wheat starch Drugs 0.000 description 1
- 229910052725 zinc Inorganic materials 0.000 description 1
- 239000011701 zinc Substances 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/535—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines
- A61K31/5375—1,4-Oxazines, e.g. morpholine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/12—Ketones
- A61K31/122—Ketones having the oxygen directly attached to a ring, e.g. quinones, vitamin K1, anthralin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/21—Esters, e.g. nitroglycerine, selenocyanates
- A61K31/255—Esters, e.g. nitroglycerine, selenocyanates of sulfoxy acids or sulfur analogues thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/445—Non condensed piperidines, e.g. piperocaine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/445—Non condensed piperidines, e.g. piperocaine
- A61K31/451—Non condensed piperidines, e.g. piperocaine having a carbocyclic group directly attached to the heterocyclic ring, e.g. glutethimide, meperidine, loperamide, phencyclidine, piminodine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/445—Non condensed piperidines, e.g. piperocaine
- A61K31/4523—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems
- A61K31/4545—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems containing a six-membered ring with nitrogen as a ring hetero atom, e.g. pipamperone, anabasine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/496—Non-condensed piperazines containing further heterocyclic rings, e.g. rifampin, thiothixene or sparfloxacin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/535—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines
- A61K31/5375—1,4-Oxazines, e.g. morpholine
- A61K31/5377—1,4-Oxazines, e.g. morpholine not condensed and containing further heterocyclic rings, e.g. timolol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/06—Antipsoriatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C323/00—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups
- C07C323/22—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups containing thio groups and doubly-bound oxygen atoms bound to the same carbon skeleton
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D211/00—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings
- C07D211/04—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D211/06—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D211/08—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon or substituted hydrocarbon radicals directly attached to ring carbon atoms
- C07D211/10—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon or substituted hydrocarbon radicals directly attached to ring carbon atoms with radicals containing only carbon and hydrogen atoms attached to ring carbon atoms
- C07D211/14—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon or substituted hydrocarbon radicals directly attached to ring carbon atoms with radicals containing only carbon and hydrogen atoms attached to ring carbon atoms with hydrocarbon or substituted hydrocarbon radicals attached to the ring nitrogen atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D211/00—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings
- C07D211/04—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D211/06—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D211/36—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D211/38—Halogen atoms or nitro radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D211/00—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings
- C07D211/04—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D211/06—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D211/36—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D211/56—Nitrogen atoms
- C07D211/58—Nitrogen atoms attached in position 4
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/24—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with substituted hydrocarbon radicals attached to ring carbon atoms
- C07D213/44—Radicals substituted by doubly-bound oxygen, sulfur, or nitrogen atoms, or by two such atoms singly-bound to the same carbon atom
- C07D213/46—Oxygen atoms
- C07D213/50—Ketonic radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D231/00—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings
- C07D231/02—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings
- C07D231/10—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D231/12—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D241/00—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings
- C07D241/02—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings not condensed with other rings
- C07D241/04—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings not condensed with other rings having no double bonds between ring members or between ring members and non-ring members
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D277/00—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings
- C07D277/02—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings
- C07D277/20—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D277/32—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D277/38—Nitrogen atoms
- C07D277/42—Amino or imino radicals substituted by hydrocarbon or substituted hydrocarbon radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D295/00—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms
- C07D295/04—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms with substituted hydrocarbon radicals attached to ring nitrogen atoms
- C07D295/10—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms with substituted hydrocarbon radicals attached to ring nitrogen atoms substituted by doubly bound oxygen or sulphur atoms
- C07D295/112—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms with substituted hydrocarbon radicals attached to ring nitrogen atoms substituted by doubly bound oxygen or sulphur atoms with the ring nitrogen atoms and the doubly bound oxygen or sulfur atoms separated by carbocyclic rings or by carbon chains interrupted by carbocyclic rings
- C07D295/116—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms with substituted hydrocarbon radicals attached to ring nitrogen atoms substituted by doubly bound oxygen or sulphur atoms with the ring nitrogen atoms and the doubly bound oxygen or sulfur atoms separated by carbocyclic rings or by carbon chains interrupted by carbocyclic rings with the doubly bound oxygen or sulfur atoms directly attached to a carbocyclic ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D295/00—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms
- C07D295/04—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms with substituted hydrocarbon radicals attached to ring nitrogen atoms
- C07D295/12—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms with substituted hydrocarbon radicals attached to ring nitrogen atoms substituted by singly or doubly bound nitrogen atoms
- C07D295/135—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms with substituted hydrocarbon radicals attached to ring nitrogen atoms substituted by singly or doubly bound nitrogen atoms with the ring nitrogen atoms and the substituent nitrogen atoms separated by carbocyclic rings or by carbon chains interrupted by carbocyclic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/10—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a carbon chain containing aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/10—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings linked by a carbon chain containing aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2602/00—Systems containing two condensed rings
- C07C2602/02—Systems containing two condensed rings the rings having only two atoms in common
- C07C2602/04—One of the condensed rings being a six-membered aromatic ring
- C07C2602/08—One of the condensed rings being a six-membered aromatic ring the other ring being five-membered, e.g. indane
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Epidemiology (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Emergency Medicine (AREA)
- Immunology (AREA)
- Dermatology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Thiazole And Isothizaole Compounds (AREA)
- Hydrogenated Pyridines (AREA)
- Pyridine Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
Abstract
Un compuesto de acuerdo con la Fórmula I: **(Ver fórmula)** o una sal aceptable farmacéuticamente de este, en donde: X es O; R1 es **(Ver fórmula)** o R1 es alquilo C1-4 opcionalmente sustituido con 1-6 sustituyentes halo independientes; o R1 es fenilo, piridinilo o pirazolilo, cada uno opcionalmente sustituido con 1-5 sustituyentes halo, trifluorometilo, (trifluorometil)tio, trifluorometoxi, 1,1,1,3,3,3,-hexafluoro-2-hidroxipropan-2-ilo **(Ver fórmula)** o independientes; m es 0 o 1; la línea punteada indica un doble enlace opcional cuando m=1; R2 es halo, -OH, -CN, -OCH3, -OS(O)2CH2CH3, **(Ver fórmula)** o fenilo opcionalmente sustituido con 0-5 sustituyentes halo, trifluorometoxi independientes; R3 es **(Ver fórmula)** n es una cadena de 2 o 3 carbonos; R4 es H, OH, OCH3, o -OS(O)2CH2CH3;
Description
DESCRIPCIÓN
Derivados de 2,3-dihidro-1h-inden-1-ona como antagonistas de receptores huérfanos relacionados con el ácido retinoico gamma (ROR gamma) para el tratamiento de la esclerosis múltiple
Campo de la invención
La presente invención se dirige a compuestos, su síntesis y su uso como antagonistas, agonistas inversos, moduladores y/o inhibidores del Receptor nuclear huérfano yt relacionado con el ácido retinoico (RORYt)/RORy. En particular, la presente invención se dirige a compuestos 2, 3-dihidro-1H-inden-1-ona sustituida que modulan (RORyt)/RORy. Los compuestos de la presente invención son útiles para modular la actividad de (RORyt)/RORy y para su uso en el tratamiento de enfermedades o afecciones mediadas por (RORyt)/RORy tales como, por ejemplo, estados de enfermedad asociados con la inmunopatología de enfermedades autoinmunitarias humanas tales como Esclerosis múltiple (EM), Artritis reumatoide (AR), Colitis inflamatoria, Psoriasis, Enfermedad pulmonar obstructiva crónica (EPOC), Dolor, Obesidad, Diabetes, Dislipidemia, Osteoporosis, Asma, Enfermedades neurodegenerativas y Cáncer.
Antecedentes de la invención
La esclerosis múltiple (EM) es una enfermedad desmielinizante inflamatoria autoinmunitaria del SNC (sistema nervioso central) que daña las vainas de mielina grasa alrededor de los axones del cerebro y la médula espinal. Este daño, destrucción y pérdida o cicatrización de las vainas de mielina (esclerosis o placas) da como resultado un amplio espectro de síntomas. La EM es una enfermedad crónica e incapacitante que afecta a aproximadamente 400000 personas en los Estados Unidos y a casi 2,5 millones de personas en todo el mundo. La EM afecta de manera sustancial y adversa la calidad de vida de cada individuo, con síntomas que incluyen pérdida de control y fuerza muscular, fatiga, debilidad, pérdida de visión, espasticidad, equilibrio, sensación, problemas de vejiga e intestinos, entumecimiento, pérdida de visión, temblores, y función mental adversa tal como la depresión. Solo en los Estados Unidos se estima que la atención de la EM cuesta casi $ 13 mil millones por año.
Las citocinas inflamatorias y sus receptores tienen un papel importante en la progresión de las lesiones de EM, y se ha encontrado que los niveles de citocinas pro y antiinflamatorias se correlacionan con los cambios en la actividad de la enfermedad de EM. Actualmente, los tratamientos disponibles generalmente se centran en estrategias para tratar las recaídas (prednisona oral y metilprednisolona i.v.), controlar los síntomas y reducir el progreso de la enfermedad con Fármacos modificadores de la enfermedad (FME). Incluye fármacos para la EM recurrente-remitente (EMRR) tales como los agentes terapéuticos inmunomoduladores (p-interferones) (Avonex, Betaseron, Rebif y Extavia), Anticuerpos monoclonales (Tysabri, Lemtrada), otros inmunosupresores (Mitoxantrona), Copaxona (inyección de acetato de glatiramer), y otros agentes orales; Teriflunomida, Fingolimod pero ninguno de estos medicamentos es una cura o previene los síntomas recurrentes. Además de los FME orales existentes tales como Fingolimod y Tecfidera recientemente aprobada (BG-12 o Fumarato de dimetilo) y otros agentes experimentales que aún no se han aprobado, se encuentran en fase III de desarrollo clínico tal como Laquinimod, y se ha informado que Masitinib (inmunomoduladores orales) provoca infecciones oportunistas, estimulación de anticuerpos, toxicidades hepáticas y renales. Los inhibidores o antagonistas específicos a la diana deben promover la mielinización, la reparación neuronal y la neuroprotección. Inhibir la enfermedad y detener la neurodegeneración debería eliminar muchos de estos eventos adversos.
En la actualidad, se ha informado que los fármacos orales DMT tales como Fingolimod, Cladribine y Tecfidera - así como también aproximadamente 46 agentes experimentales adicionales en diversas etapas de desarrollo clínico, tales como Laquinimode y Masitinib - provocan eventos adversos graves incluidas infecciones oportunistas, estimulación de anticuerpos, y toxicidades hepáticas y renales. Por lo tanto, existe una continua necesidad de fármacos efectivos para la EM con mejores perfiles de toxicidad. Los inhibidores o antagonistas específicos a la diana deben promover la mielinización, la reparación neuronal, detener la neurodegeneración y deberían eliminar muchos de estos eventos adversos.
La evidencia que respalda los perfiles de riesgo-beneficio para los agentes disponibles incluidos los DMT de AcM emergentes (Alemtuzumab, Ocrelizumab y Daclizumab) aún no ha surgido por completo. Muchos de estos agentes aprobados y experimentales aún por aprobar crean eventos adversos graves que incluyen infecciones oportunistas, estimulación de anticuerpos y toxicidades hepáticas y renales. Por lo tanto, la EM sigue siendo una diana importante para las terapias innovadoras que pueden tener efectos inmunoterapéuticos y/o neuroprotectores sobre la enfermedad. Esta oportunidad ha fortalecido el enfoque de investigación sobre RORyt clínicamente relevante, como una diana atractiva para el tratamiento de la EM. RORyt como un receptor de hormona nuclear que es un regulador clave de la diferenciación de células T auxiliares tipo 17 (Th17). Las células Th17 se producen normalmente en respuesta a la infección, pero se han relacionado con el desarrollo de enfermedades autoinmunitarias. Se han informado algunos agonistas inversos de RORyt y RORy en etapa pre-clínica. Por ejemplo, los fungicidas de tipo azol, T0901317, SR1001, ácido ursólico, VPR-66, digoxina y sulfamoil tiazoles sustituidos con hexafluoro tuvieron eficacia en modelos de ratones con encefalomielitis autoinmunitaria (EAE). El documento WO 2012/158784 A2 describe moduladores de moléculas pequeñas de receptores huérfanos relacionados con el receptor de ácido retinoico, tales como RORa, RORp o RORy. El documento EP 1468684 A1 describe el aglutinante del receptor sigma que contiene el derivado de indanona.
Al menos una docena de empresas; Lycera/Merck, Karo Bio/Pfizer, Phenex/J&J, Orphagen/JT (OR-1050/T0901317, 5 ^M), Tempero/GSK (TMP-778, GSK-805), Exelixis/BMS, Teijin/Amgen, Cognoci (COG112), Innovimmune (INV-17), Visionary, 4SC Discovery y Genentech, tienen programas de RORYt de molécula pequeña en etapa preclínica de desarrollo. La identificación de antagonistas específicos de RORYt y penetrantes cerebrales que rescatan la destrucción de mielina, restauran los axones del cerebro y la médula espinal después de la administración oral será un enfoque significativo para el desarrollo de los agentes terapéuticos de la EM. La clase 2, 3-dihidro-1H-inden-1-ona sustituida de compuestos reivindicados en la Fórmula I se descubrió como un potente antagonista de RORYt oralmente disponible y penetrante cerebral, y un compuesto candidato de molécula pequeña como nuevas entidades farmacológicas para el tratamiento de tales enfermedades reivindicadas.
La presente invención incluye antagonistas de moléculas pequeñas que se dirigen al receptor nuclear huérfano Yt relacionado con ácido retinoico (RORYt)/RORY. RORYt es el factor de transcripción clave y es el regulador maestro de las células Th17 (T auxiliares 17) humanas, un subconjunto único de células T CD4+. RORYt controla la diferenciación celular, la función y la liberación de Interleucina IL-17 (linfocitos T auxiliares productores de IL-17) por las células Th17 y ayuda a mediar la inmunopatología de enfermedades autoinmunitarias humanas tales como Esclerosis múltiple (EM), Artritis reumatoide (AR), Colitis inflamatoria, Psoriasis, EPOC, Dolor, Obesidad, Diabetes, Dislipidemia, Osteoporosis, Asma, Enfermedades neurodegenerativas y Cáncer.
Además, los antagonistas de receptores nucleares huérfanos Yt (RORYt)/RORY relacionados con el ácido retinoico, el 2, 3-dihidro-1H-inden-1-ona sustituida de la presente invención pueden usarse para el tratamiento de múltiples cánceres, incluidos los gástricos, los cánceres de colon, la leucemia mielogénica crónica (LMC), la leucemia mielogénica aguda (LMA), carcinomas de células escamosas y de vejiga, meduloblastoma, carcinoma hepatocelular, mieloma múltiple, vejiga, glioblastoma multiforme (GBM), cánceres de mama y de ovario, sarcoma de Ewing y enfermedades asociadas al cáncer de huesos. Los métodos de tratamiento con los compuestos de Fórmula I comprenden administrar una dosis segura y efectiva de los compuestos de acuerdo con la Fórmula I o una sal farmacéuticamente aceptable, formulaciones de los mismos para pacientes humanos.
La inducción de afecciones de EM autoinmunitaria en ratones mediante el uso de inmunización con Glucoproteína del oligodendrocito asociada a la mielina (MOG) o Proteína proteolipídica (PLP) provoca la activación de RORYt y la diferenciación de las células Th17 que reclutan citocinas proinflamatorias IL-17A (IL-17), IL-17F, IL-21 e IL-22 conduce a mielina esclerótica y oligodendrocitos dañados. Los ratones con células T deficientes en RORYt han atenuado la enfermedad y carecen de células Th17 infiltrantes de tejido. Por lo tanto, RORYt es un regulador clave de la homeostasis inmunitaria y es una diana terapéutica potencial para la esclerosis múltiple. Mediante el uso de la tecnología interna basada en el diseño de fármacos basado en fragmentos (FFDD) propiedad de la compañía y ensayos de isoforma RORYt específicos de diseño exclusivo, una novedosa y potente molécula pequeña de la serie de 2, 3-dihidro-1H-inden-1-ona sustituida es antagonista específico de RORYt (agonista inverso) y demostraron su actividad de RORYt en un ensayo Prom/LUCPorter de IL-17A activada por RORYt en células HEK 293, en la liberación de IL-17 de ensayos de células T CD4+, así como también la inhibición de la producción de IL-17 in vivo en experimentos con ratones BALB/cy el efecto de la serie de 2, 3-dihidro-1H-inden-1-ona sustituida de compuestos de Fórmula I en MOG35-55 inducido en ratones C57/BL6 o BALB/c, PLPi39-i5i (Proteína proteolipídica) inducida en ratones SJL/J y la enfermedad desmielinizante inducida por el virus de la encefalitis murina de Theiler (Tm EV-IDD) en ratones BALB/c hembras inducidos. El modelo de EAE aguda/recurrente en ratones SJL/J hembra se describe en esta solicitud.
Resumen de la invención
La presente invención se refiere a compuestos activos en la superfamilia de receptores de hormonas nucleares, específicamente RORYt y en general, incluyendo RORa, -P y - y (NR1F1-3 o RORA-C), sus dos isoformas, y1 y y2 (RORYt/ROR-YT) y cualquier mutación de esta superfamilia de receptores de hormonas nucleares y dichos compuestos para su uso en el tratamiento de enfermedades y afecciones asociadas con la regulación de la actividad de esta superfamilia de receptores de hormonas nucleares. Más específicamente, la invención se refiere a compuestos de la Fórmula I y Ia como se describe más abajo. Por lo tanto, la invención proporciona compuestos para usar en métodos terapéuticos que implican la inhibición y/o modulación de RORa, -P y - y específicamente RORYt como nuevos compuestos que pueden ser para usar en los métodos terapéuticos que implican la modulación de la inmunopatología de enfermedades autoinmunitarias humanas tales como Esclerosis múltiple (EM), Artritis reumatoide (AR), Colitis inflamatoria, Psoriasis, EPOC, Dolor, Obesidad, Diabetes, Dislipidemia, Osteoporosis, Asma, Enfermedades neurodegenerativas y Cáncer.

La presente invención se refiere a compuestos de acuerdo con las Fórmulas I y la: a una composición farmacéuticamente aceptable, sus sales, su síntesis y los compuestos para usar como inhibidores de RORYt y RORy, incluidos para usar en el tratamiento de diversas inmunopatologías de enfermedades autoinmunitarias humanas tales como la Esclerosis múltiple (MS), Artritis reumatoide (AR), Colitis inflamatoria, Psoriasis, EPOC, Dolor, Obesidad, Diabetes, Dislipidemia, Osteoporosis, Asma, Enfermedades neurodegenerativas y Cáncer.
Breve descripción de las figuras
Figura 1: Estructura cristalina de rayos X de RORy en el complejo con el antagonista de RORYt que contiene 2, 3-dihidro-1H-inden-1-ona sustituida.
Figura 2A: Efecto de los antagonistas de RORg que contienen las series de 2, 3-dihidro-1H-inden-1-ona sustituida sobre la actividad del promotor de IL-17A mediada por RORYt.
Figura 2B: Curva de respuesta a dosis de IL-17A mediada por RORYt.
Figura 3A: Efecto de los antagonistas de RORg que contienen las series de 2, 3-dihidro-1H-inden-1-ona sustituida sobre la actividad del promotor de IL-17A mediada por RORYt.
Figura 3B: Curva de respuesta a dosis de IL-17A mediada por RORYt.
Figura 4: Inhibición de la citocina IL-17 por las series de 2, 3-dihidro-1H-inden-1-ona sustituida.
Figura 5: Efecto del compuesto de prueba sobre EAE inducida por MOG35-55 en ratones C57/BL6 hembras.
Figura 6: Efecto de la administración del compuesto de prueba y de Fingolimod sobre el peso corporal de ratones EAE (C57/BL6 hembras).
Descripción detallada de la invención
Los compuestos de la presente invención se describen por Fórmula I:

o sales farmacéuticamente aceptables de éste, en donde:
X es O;
R1 es

o
R1 es alquilo C i-4 opcionalmente sustituido con 1-6 sustituyentes halo independientes; o
R1 es fenilo, piridinilo o pirazolilo, cada uno opcionalmente sustituido con 1-5 sustituyentes halo, trifluorometilo, (trifluorometil)tio, trifluorometoxi, 1,1,1,3,3,3,-hexafluoro-2-hidroxipropan-2-ilo,

independientes;
m es 0 o 1;
la línea punteada indica un doble enlace opcional cuando m=1;
R2 es halo, -OH, -CN, -OCH3, -OS(O)2CH2CHs,



o fenilo opcionalmente sustituido con 0-5 sustituyentes halo, trifluorometoxi independientes;
R3 es
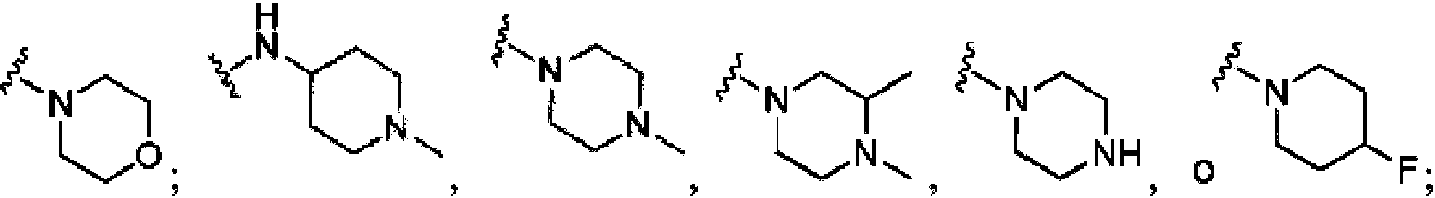
n es una cadena de 2 o 3 carbonos; y
R4 es H, OH, OCH3, o -OS(O)2CH2CH3.
Los compuestos de la presente invención pueden prepararse mediante el uso de los compuestos

En una modalidad del aspecto de la invención, los compuestos de la presente invención se describen por la Fórmula (I) y sus sales farmacéuticamente aceptables, en donde X es O, R1 es

y las otras variables son como se definieron anteriormente para la Fórmula (I).
En otra modalidad del aspecto de la invención, los compuestos de la presente invención se describen por la Fórmula (I) y sus sales farmacéuticamente aceptables, en donde X es O, R1 es

y las otras variables son como se definieron anteriormente para la Fórmula (I).
En otra modalidad más del aspecto de la invención, los compuestos de la presente invención se describen por la Fórmula (I) y sus sales farmacéuticamente aceptables, en donde X es O, R1 es alquilo C1-4 opcionalmente sustituido con 1-6 sustituyentes halo independientes y las otras variables son como se definieron anteriormente para la Fórmula (I).
Aún en otra modalidad del aspecto de la invención, los compuestos de la presente invención se describen por la Fórmula (I) y sus sales farmacéuticamente aceptables, en donde X es O, R1 es fenilo, opcionalmente sustituido con 1-5 sustituyentes halo, trifluorometilo, (trifluorometil)tio independientes

y las otras variables son como se definieron anteriormente para la Fórmula (I).
En otra modalidad del aspecto de la invención, los compuestos de la presente invención se describen por la Fórmula (I) y sus sales farmacéuticamente aceptables, en donde X es O, R1 es piridinilo, opcionalmente sustituido con 1-5 sustituyentes halo, trifluorometilo, (trifluorometil)tio independientes

y las otras variables son como se definieron anteriormente para la Fórmula (I).
Aún en otra modalidad del aspecto de la invención, los compuestos de la presente invención se describen por la Fórmula (I) y sus sales farmacéuticamente aceptables, en donde X es O, R1 es pirazolilo opcionalmente sustituido con 1-5 sustituyentes halo, trifluorometilo, (trifluorometil)tio independientes

y las otras variables son como se definieron anteriormente para la Fórmula (I).
En un aspecto de la presente invención, los compuestos de la presente invención se describen por la fórmula (Ia) y sus sales farmacéuticamente aceptables,

en donde
X es O o S;
R1a es

o
R1a es alquilo C1-4 opcionalmente sustituido con 1-6 sustituyentes halo independientes; o
R1a es fenilo, piridinilo o pirazolilo, cada uno opcionalmente sustituido con 1-5 sustituyentes halo, trifluorometilo, (trifluorometil)tio,

independientes;
la línea punteada indica un doble enlace opcional;
X1 es C o N; y X2 se selecciona de C, N u O; en donde al menos uno de X1 y X2 no es C;
R5 está ausente, es halo, hidrógeno o alquilo C1-4;
y R6 es halo, hidrógeno o alquilo C1-4.
En la presente descripción se describen compuestos descritos por la Fórmula (Ib) y sus sales farmacéuticamente aceptables,

en donde
X es O o S;
R1 es

o
R1 es alquilo C1-4 opcionalmente sustituido con 1-6 sustituyentes halo independientes; o
R1 es fenilo, piridinilo o pirazolilo, cada uno opcionalmente sustituido con 1-5 sustituyentes halo, trifluorometilo, (trifluorometil)tio,

independientes;
la línea punteada indica un doble enlace opcional
X1 y X 2 son independientemente C, N u O; en donde al menos uno no es C;
R5 está ausente, halo o alquilo C0-4.
y R6 es halo o alquilo C0-4.
En la presente descripción se describen (pero no de acuerdo con la invención) compuestos de la Fórmula (Ic) y sus sales farmacéuticamente aceptables,
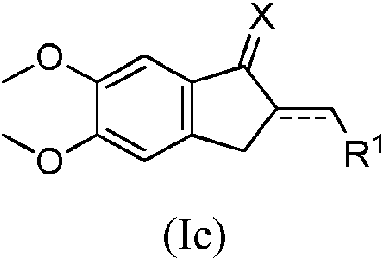
en donde
X es O o S;
R1 es

o
R1 es alquilo C1-4 opcionalmente sustituido con 1-6 sustituyentes halo independientes; o
R1 es fenilo, piridinilo o pirazolilo, cada uno opcionalmente sustituido con 1-5 sustituyentes halo, trifluorometilo, (trifluorometil)tio,

independientes;
la línea punteada indica un doble enlace opcional. Los compuestos de la presente invención incluyen:






A menos que se indique de cualquier otra forma los siguientes términos usados en la descripción y las reivindicaciones tienen los significados que se exponen más abajo:
"Alquilo" se refiere a un radical hidrocarburo lineal o ramificado saturado de uno a seis átomos de carbono, preferentemente de uno a cuatro átomos de carbono, por ejemplo, metilo, etilo, propilo, 2-propilo, n-butilo, isobutilo, tercbutilo, pentilo, hexilo y similares, preferentemente metilo, etilo, propilo o 2-propilo. Los alquilos de cadena lineal saturados representativos incluyen metilo, etilo, n-propilo, n-butilo, n-pentilo, n-hexilo y similares; mientras que los alquilos ramificados saturados incluyen isopropilo, sec-butilo, isobutilo, ferc-butilo, isopentilo y similares. Los alquilos cíclicos se denominan en la presente descripción como un "cicloalquilo".
Los alquilos insaturados contienen al menos un enlace doble o triple entre átomos de carbono adyacentes (denominados "alquenilo" o "alquinilo", respectivamente). Los alquenilos de cadena lineal y ramificada representativos incluyen etilenilo, propilenilo, 1-butenilo, 2-butenilo, isobutilenilo, 1-pentenilo, 2-pentenilo, 3-metil-1-butenilo, 2-metil-2-butenilo, 2,3-dimetil-2-butenilo y similares; mientras que los alquinilos de cadena lineal y ramificada representativos incluyen acetilenilo, propinilo, 1 -butinilo, 2-butinilo, 1 -pentinilo, 2-pentinilo, 3-metil-1-butiniloysimilares.
"alquilo C0-4" se refiere a un alquilo con 0, 1, 2, 3 o 4 átomos de carbono. alquilo C0-4 con 0 átomos de carbono es un átomo de hidrógeno cuando es terminal y es un enlace directo cuando se une.
"Alquileno" significa un radical hidrocarburo divalente saturado lineal de uno a seis átomos de carbono o un radical hidrocarburo divalente saturado ramificado de tres a seis átomos de carbono, por ejemplo, metileno, etileno, 2,2-dimetiletileno, propileno, 2-metilpropileno, butileno, pentileno, y similares, preferentemente metileno, etileno o propileno. "Cicloalquilo" se refiere a un radical hidrocarburo cíclico saturado de tres a ocho átomos de carbono, por ejemplo, ciclopropilo, ciclobutilo, ciclopentilo o ciclohexilo.
"Alcoxi" significa un radical -ORa donde Ra es un alquilo como se definió anteriormente, por ejemplo, metoxi, etoxi, propoxi, butoxi y similares.
"Halo" significa flúor, cloro, bromo, o yodo, preferentemente, fluoro y cloro.
"Haloalquilo" significa alquilo sustituido con uno o más, preferentemente uno, dos o tres, átomos halo iguales o diferentes, por ejemplo, -CH2C -CF3, -CH2CF3, -CH2CCl3, y similares.
"Haloalcoxi" significa un radical -ORb donde Rb es un haloalquilo como se definió anteriormente, por ejemplo, trifluorometoxi, tricloroetoxi, 2,2-dicloropropoxi ysimilares.
"Acilo" significa un radical -C(O)Rc donde Rc es hidrógeno, alquilo o haloalquilo como se define en la presente descripción, por ejemplo, formilo, acetilo, trifluoroacetilo, butanoiloysimilares.
"Arilo" se refiere a un grupo monocíclico o policíclico de anillo fusionado de carbono (es decir, anillos que comparten pares adyacentes de átomos de carbono) d e 6 a 12 átomos de carbono quetienen un sistema de electrones pi completamente
conjugados. Los ejemplos, sin limitación, de grupos arilo son fenilo, naftilo y antracenilo. El grupo arilo puede ser sustituido o no sustituido. A menos que se indique específicamente de cualquier otra manera, "arilo sustituido" se refiere al grupo arilo que está sustituido con uno o más, con mayor preferencia uno, dos o tres, aún con mayor preferencia uno o dos sustituyentes seleccionados independientemente del grupo que consiste en alquilo (en donde el alquilo puede ser opcionalmente sustituido con uno o dos sustituyentes), haloalquilo, halo, hidroxi, alcoxi, mercapto, alquiltio, ciano, acilo, nitro, fenoxi, heteroarilo, heteroariloxi, haloalquilo, haloalcoxi, carboxi, alcoxicarbonilo, amino, alquilamino dialquilamino, arilo, heteroarilo, carbociclo o heterociclo (en donde el arilo, heteroarilo, carbociclo o heterociclo pueden estar opcionalmente sustituidos).
"Heteroarilo" se refiere a un anillo monocíclico o fusionado (es decir, anillos que comparten un par de átomos adyacentes) de 5 a 12 átomos de anillo que contienen uno, dos, tres o cuatro heteroátomos del anillo seleccionados de N, O, o S, los átomos del anillo restantes son C, y, adicionalmente, tienen un sistema de electrones pi completamente conjugado. Los ejemplos, sin limitación, de grupos heteroarilo no sustituidos son pirrol, furano, tiofeno, imidazol, oxazol, tiazol, pirazol, piridina, pirimidina, quinoleina, isoquinolina, purina, triazol, tetrazol, triazina, y carbazol. El grupo heteroarilo puede estar no sustituido o sustituido, tal como, por ejemplo, 5-metiltiazolilo. A menos que se indique específicamente de cualquier otra manera, "heteroarilo sustituido" se refiere al grupo heteroarilo que está sustituido con uno o más, con mayor preferencia uno, dos o tres, aún con mayor preferencia uno o dos sustituyentes seleccionados independientemente del grupo que consiste en alquilo (en donde el alquilo puede ser opcionalmente sustituido con uno o dos sustituyentes), haloalquilo, halo, hidroxi, alcoxi, mercapto, alquiltio, ciano, acilo, nitro, haloalquilo, haloalcoxi, carboxi, alcoxicarbonilo, amino, alquilamino dialquilamino, arilo, heteroarilo, carbociclo o heterociclo (en donde el arilo, heteroarilo, carbociclo o heterociclo pueden estar opcionalmente sustituidos).
"Carbociclo" se refiere a un sistema de anillo saturado, insaturado o aromático que tiene de 3 a 14 átomos de carbono del anillo. El término "carbociclo", ya sea saturado o parcialmente insaturado, se refieren también a anillos que están opcionalmente substituidos. El término "carbociclo" incluye arilo. El término "carbociclo" incluyen también anillos alifáticos que están fusionados a uno o más anillos aromáticos o no aromáticos, tales como en un decahidronaftilo o tetrahidronaftilo, donde el radical o punto de unión está sobre el anillo alifático. El grupo carbociclo puede ser sustituido o no sustituido. A menos que se indique específicamente de cualquier otra manera, "carbociclo sustituido" se refiere al grupo carbociclo que está sustituido con uno o más, con mayor preferencia uno, dos o tres, aún con mayor preferencia uno o dos sustituyentes seleccionados independientemente del grupo que consiste en alquilo (en donde el alquilo puede ser opcionalmente sustituido con uno o dos sustituyentes), haloalquilo, halo, hidroxi, alcoxi, mercapto, alquiltio, ciano, acilo, nitro, haloalquilo, haloalcoxi, carboxi, alcoxicarbonilo, amino, alquilamino dialquilamino, arilo, heteroarilo, carbociclo o heterociclo (en donde el arilo, heteroarilo, carbociclo o heterociclo pueden estar opcionalmente sustituidos).
"Heterociclilo" se refiere a un sistema de anillos cíclicos saturados, insaturados o aromáticos que tienen 3 a 14 átomos del anillo en el que uno, dos o tres átomos del anillo son heteroátomos seleccionados de N, O, o S(O)m (donde m es un número entero de 0 a 2), los átomos restantes del anillo son C, donde uno o dos átomos de C pueden remplazarse, opcionalmente, por un grupo carbonilo. El término "heterociclo" incluye heteroarilo. A menos que se indique específicamente de cualquier otra manera, "heterociclilo sustituido" se refiere al anillo heterociclilo que está sustituido independientemente con uno o más, preferentemente uno, dos o tres sustituyentes seleccionados de alquilo (en donde el alquilo puede estar opcionalmente sustituido con uno o dos sustituyentes), haloalquilo, cicloalquilamino, cicloalquilo o cicloalquil-alquilo, cicloalquilo-aminoalquilo, cicloalquil-alquilo amino-alquilo, cianoalquilo, halo, nitro, ciano, hidroxi, alcoxi, amino, alquilamino, dialquilamino, hidroxialquilo, carboxialquilo, aminoalquilo, alquilaminoalquilo, dialquilaminoalquilo, aralquilo, heteroaralquilo, arilo, heteroarilo, carbociclo, heterociclo (en donde el arilo, heteroarilo, carbociclo o heterociclo puede estar opcionalmente sustituido), aralquilo, heteroaralquilo, heterocicloamino saturado o insaturado, heterocicloaminoalquilo saturado o insaturado, y -CORd (donde Rd es alquilo). Más específicamente, el término heterociclilo incluye, pero no se limita a, tetrahidropiranilo, 2,2-dimetil-1,3-dioxolano, piperidino, N-metilpiperidin-3-ilo, piperazino, N-metilpirrolidin-3-ilo, pirrolidino, morfolino, 4-ciclopropilmetilpiperazino, tiomorfolino, tiomorfolino-1-óxido, tiomorfolino-1,1-dióxido, 4-etiloxicarbonilpiperazino, 3-oxopiperazino, 2-imidazolidona, 2-pirrolidinona, 2-oxohomopiperazino, tetrahidropirimidin-2-ona y sus derivados, que incluyen 2-metil-4,5,6,7-tetrahidro-1H-pirrolo[2,3-c]piridinilo. En ciertas modalidades, el grupo heterociclo está opcionalmente sustituido con uno o dos sustituyentes seleccionados independientemente de halo, alquilo, alquilo sustituido con carboxi, éster, hidroxilo, alquilamino, heterocicloamino saturado o insaturado, heterocicloaminoalquilo saturado o insaturado, o dialquilamino.
"Opcional" u "opcionalmente" significa que el evento o circunstancia descrito subsecuentemente puede producirse pero no es necesario que se produzca, y que la descripción incluye los casos donde el evento o circunstancia se produce y los casos en los que no se produce. Por ejemplo, "grupo heterocíclico opcionalmente sustituido con un grupo alquilo" significa que el alquilo podría pero no necesita estar presente, y la descripción incluye situaciones donde el grupo heterociclo está sustituido con un grupo alquilo y situaciones donde el grupo heterociclo no está sustituido con el grupo alquilo.
Por último, a menos que se indique específicamente de cualquier otra manera, el término "sustituido" como se usa en la presente descripción significa cualquiera de los grupos anteriores (por ejemplo, alquilo, arilo, heteroarilo, carbociclo, heterociclo, etc.) en donde al menos un átomo de hidrógeno se reemplaza con un sustituyente. En el caso de un sustituyente oxo ("=O") se reemplazan dos átomos de hidrógeno. Los "sustituyentes" dentro del contexto de esta invención, si no se especifican, incluyen halógeno, hidroxi, oxo, ciano, nitro, amino, alquilamino, dialquilamino, alquilo, alcoxi, tioalquilo, haloalquilo (por ejemplo, -CF3), hidroxialquilo, arilo, arilo sustituido, arilalquilo, arilalquilo sustituido,
heteroarilo, heteroarilo sustituido, heteroarilalquilo, heteroarilalquilo sustituido, heterociclo, heterociclo sustituido, heterocicloalquilo, heterocicloalquilo sustituido, -NReRf, -NReC(=O)Rf, -NReC(=O)NReRf, -NReC(=O)ORf-NReSO2Rf, -Oe,-C(=O)Re-C(=O)ORe, -C(=O)NReRf, -OC(=O)NReRf, -SH, -SRe, -SORe, -S(=O)2Re, -OS(=O)2Re, -S(=O)2ORe, en donde Re y Rf son iguales o diferentes e independientemente hidrógeno, alquilo, haloalquilo, alquilo sustituido, arilo, arilo sustituido, arilalquilo, arilalquilo sustituido, heteroarilo, heteroarilo sustituido, heteroarilalquilo, heteroarilalquilo sustituido, heterociclo, heterociclo sustituido, heterocicloalquilo o heterocicloalquilo sustituido.
Los compuestos que tienen la misma fórmula molecular pero difieren en la naturaleza o secuencia de unión de sus átomos o la disposición de sus átomos en el espacio se denominan 'isómeros'. Los isómeros que difieren en la disposición de sus átomos en el espacio se denominan "estereoisómeros". Los estereoisómeros que no son imágenes espejo entre sí se denominan 'diastereómeros' y aquellos que son imágenes espejo no superponibles entre sí se denominan 'enantiómeros'. Cuando un compuesto tiene un centro asimétrico, por ejemplo, está unido a cuatro grupos diferentes, un par de enantiómeros es posible. Un enantiómero puede caracterizarse por la configuración absoluta de su centro asimétrico y se describe por las reglas de secuenciación R- y S- de Cahn y Prelog (Cahn, R., Ingold, C., y Prelog, V. Angew. Chem.
78:413-47, 1966; Angew. Chem. Internat. Ed. Eng. 5:385-415, 511, 1966), o por la manera en la cual la molécula rota el plano de luz polarizada y se designa como dextrógiro o levógiro (es decir, como isómeros (+) o (-) respectivamente). Un compuesto quiral puede existir como enantiómero individual o como una mezcla de este. Una mezcla que contiene proporciones iguales de los enantiómeros se denomina una 'mezcla racémica'.
Los compuestos de esta invención pueden poseer uno o más centro asimétricos; dichos compuestos pueden producirse por lo tanto como estereoisómeros individuales (R)- o (S)- o como mezclas de estos. A menos que se indique de cualquier otra forma, la descripción o mención de un compuesto particular en la descripción y reivindicaciones pretende incluir los enantiómeros individuales y mezclas, racémico o de cualquier otra forma, de este. Los métodos para la determinación de la estereoquímica y la separación de estereoisómeros son bien conocidos en la técnica (ver el análisis en el Capítulo 4 de ADVANCED ORGANIC CHEMISTRY, 4ta edición, March, J., John Wiley and Sons, Ciudad de Nueva York, 1992).
Los compuestos de la presente invención pueden exhibir los fenómenos de tautomerismo. Esta invención abarca cualquier forma tautomérica que posea la capacidad de modular la actividad de (RORYt)/RORY y no se limita a ninguna forma tautomérica.
Se contempla que un compuesto de la presente invención podría ser metabolizado por enzimas en el cuerpo del organismo tal como el ser humano, para generar un metabolito que pueda modular la actividad de las proteínas quinasas. Un compuesto de la presente invención o su sal farmacéuticamente aceptable puede administrarse como tal a un paciente humano o puede administrarse en composiciones farmacéuticas en las que los materiales anteriores se mezclan con portadores o excipientes adecuados. Las técnicas para la formulación y la administración de fármacos pueden encontrarse, por ejemplo, en REMINGTON'S PHARMa Co LOGICAL SCIENc Es , Mack Publishing Co., Easton, PA, última edición.
Una "composición farmacéutica" se refiere a una mezcla de uno o más de los compuestos descritos en la presente descripción o sus sales farmacéuticamente aceptables, con otros componentes químicos, tales como excipientes farmacéuticamente aceptables. El propósito de una composición farmacéutica es facilitar la administración de un compuesto a un organismo.
"Excipiente farmacéuticamente aceptable" se refiere a una sustancia inerte añadida a una composición farmacéutica para facilitar aún más la administración de un compuesto. Los ejemplos, sin limitación, de excipientes incluyen carbonato de calcio, fosfato de calcio, diversos azúcares y tipos de almidón, derivados de celulosa, gelatina, aceites vegetales y polietilenglicoles.
"Sal farmacéuticamente aceptable" se refiere a aquellas sales que retienen la efectividad biológica y las propiedades del compuesto original. Tales sales pueden incluir: (1) sal de adición de ácido que se obtiene mediante la reacción de la base libre del compuesto original con ácidos inorgánicos tales como ácido clorhídrico, ácido bromhídrico, ácido nítrico, ácido fosfórico, ácido sulfúrico y ácido perclórico y similares, o con ácidos orgánicos tales como ácido acético, ácido oxálico, ácido (D) o (L)-málico, ácido maleico, ácido metanosulfónico, ácido etanosulfónico, ácido p-toluenosulfónico, ácido salicílico, ácido tartárico, ácido cítrico, ácido succínico o ácido malónico y similares, preferentemente ácido clorhídrico o ácido (L)-málico; o (2) sales formadas cuando un protón ácido presente en el compuesto original se reemplaza por un ion metálico, por ejemplo, un ion de metal alcalino, un ion alcalinotérreo o un ion de aluminio; o coordina con una base orgánica tal como etanolamina, dietanolamina, trietanolamina, trometamina, N-metilglucamina y similares.
El compuesto de la presente invención también puede actuar, o estar diseñado para actuar, como un profármaco. Un "profármaco" se refiere a un agente, que se convierte en el fármaco original in vivo. Los profármacos son frecuentemente útiles porque, en algunas situaciones, son más fáciles de administrar que el fármaco original. Pueden, por ejemplo, ser biodisponible por vía oral, mientras que el fármaco original no lo es. El profármaco puede tener, además, solubilidad mejorada en composiciones farmacéuticas sobre el fármaco madre. Un ejemplo, sin limitación, de un profármaco sería un compuesto de la presente invención, que se administra como un éster (el "profármaco"), fosfato, amida, carbamato o urea.
"Cantidad terapéuticamente efectiva" se refiere a la cantidad del compuesto que se administra que aliviará en alguna medida uno o más de los síntomas del trastorno que se trata. En referencia al tratamiento de la EM, una cantidad terapéuticamente efectiva se refiere a esa cantidad que tiene el efecto de: (1) mejorar la pérdida de control y fuerza muscular, (2) disminuir la fatiga o debilidad, (3) estabilizar la pérdida de visión, (4) disminuir la espasticidad, (5) aumentar el equilibrio, (6) disminuir los problemas en la vejiga y los intestinos, (7) disminuir el entumecimiento o aliviar uno o más síntomas asociados con la EM.
El término "enfermedad", como se usa en la presente descripción, significa cualquier enfermedad u otra afección perjudicial en la que se sabe que el receptor nuclear huérfano y (RORyt)/RORy relacionado con el ácido retinoico desempeña un papel. El término "enfermedad" también significa aquellas enfermedades o afecciones que se alivian mediante el tratamiento con moduladores (RORyt)/RORy.
La frase "afección mediada por la actividad de (RORyt)/RORy" o "enfermedad", como se usa en la presente descripción, significa cualquier enfermedad u otra afección perjudicial en la que se sabe que la actividad de (RORyt)/RORy desempeña un papel. El término "afección mediada por la actividad de (RORyt)/RORy" también significa aquellas enfermedades o afecciones que se alivian mediante el tratamiento con un inhibidor de (RORyt)/RORy.
Como se usa en la presente descripción, "administrar" o "administración" se refiere al suministro de un compuesto inventivo o de una sal farmacéuticamente aceptable de este o de una composición farmacéutica que contiene un compuesto inventivo o una sal farmacéuticamente aceptable de este de esta invención a un organismo para el propósito de prevención o tratamiento de un trastorno relacionado con (RORyt)/RORy.
Las vías de administración adecuadas pueden incluir, sin limitación, administración oral, rectal, transmucosal o intestinal o inyecciones intramusculares, subcutáneas, intratecales, intraventriculares directa, intravenosas, intravítreas, intraperitoneales, intranasales o intraoculares. En ciertas modalidades, las vías de administración preferidas son orales e intravenosas.
Las composiciones farmacéuticas de la presente invención pueden fabricarse mediante procesos bien conocidos en la técnica, por ejemplo, por medio de procesos convencionales de mezclado, disolución, granulación, producción de grajeas, levigación, emulsión, encapsulado, atrapamiento o liofilización.
Las composiciones farmacéuticas para su uso de acuerdo con la presente invención, pueden formularse de cualquier manera convencional mediante el uso de uno o más portadores fisiológicamente aceptables que comprenden excipientes y auxiliares los cuales facilitan el procesamiento de los compuestos activos en las preparaciones que pueden usarse farmacéuticamente. La formulación propia depende de la ruta de administración escogida.
Para inyección, los compuestos de la invención pueden formularse en soluciones acuosas, preferentemente, en tampones fisiológicamente compatibles tales como solución de Hanks, solución de Ringer, o tampón de solución salina fisiológica. Para la administración transmucosal, se usan penetrantes adecuados en la formulación para la barrera a permear. Dichos penetrantes generalmente se conocen en la técnica.
Para la administración oral, los compuestos se pueden formular mediante la combinación de los compuestos activos con portadores farmacéuticamente aceptables bien conocidos en la técnica. Dichos portadores permiten que los compuestos de la invención se formulen como tabletas, píldoras, pastillas, grageas, cápsulas, líquidos, geles, jarabes, lechadas, suspensiones y similares, para la ingestión oral por un paciente. Las preparaciones farmacéuticas para uso oral pueden prepararse mediante el uso de un excipiente sólido, opcionalmente triturando una mezcla resultante y procesando la mezcla de gránulos, después de añadir otros auxiliares adecuados, si se desea, para producir el núcleo de las tabletas o confites. Los excipientes útiles son, en particular, rellenos tales como azúcares, que incluyen lactosa, sacarosa, manitol, o sorbitol, preparaciones de celulosa tales como, por ejemplo, almidón de maíz, almidón de trigo, almidón de arroz y almidón de papa y otros materiales tales como gelatina, goma de tragacanto, metilcelulosa, hidroxipropilmetilcelulosa, carboximetilcelulosa de sodio y/o polivinilpirrolidona (PVP). Si se desea, se pueden añadir agentes desintegrantes, tales como polivinilpirrolidona reticulado, agar, o ácido algínico. También puede usarse una sal tal como el alginato de sodio.
Se proporcionan núcleos de grageas con revestimientos adecuados. Para este propósito, pueden usarse soluciones concentradas de azúcar que opcionalmente pueden contener goma arábiga, talco, polivinilpirrolidona, gel de carbopol, polietilenglicol, y/o dióxido de titanio, soluciones de laca y solventes orgánicos adecuados o mezclas de solventes. Los tintes o pigmentos se pueden adicionar a las tabletas o a los recubrimientos de los confites para la identificación o para caracterizar diferentes combinaciones de las dosis del compuesto activo.
Las composiciones farmacéuticas que pueden usarse oralmente incluyen cápsulas de ajuste a presión que se preparan de gelatina, así como también cápsulas blandas, selladas que se preparan de gelatina y un plastificante, tal como glicerol o sorbitol. Las cápsulas de ajuste a presión pueden contener los ingredientes activos en una mezcla con relleno tal como lactosa, un aglutinante tal como almidón, y/o un lubricante tal como talco o estearato de magnesio y, opcionalmente, estabilizadores. En cápsulas blandas, los compuestos activos pueden disolverse o suspenderse en líquidos adecuados, tales como aceites grasos, parafina líquida, o glicoles de polietileno líquidos. También pueden añadirse estabilizadores en estas formulaciones. Las composiciones farmacéuticas que también pueden usarse incluyen cápsulas de gelatina dura.
Las cápsulas o píldoras pueden empaquetarse en botellas de vidrio o plástico marrón para proteger el compuesto activo de la luz. Los recipientes que contienen la formulación de la cápsula del compuesto activo se almacenan preferentemente a temperatura ambiente controlada (15-30 °C).
Para la administración por inhalación, los compuestos para el uso de acuerdo con la presente invención pueden suministrarse convenientemente en la forma de una atomización de aerosol mediante el uso de empaques a presión o un nebulizador y un propulsor adecuado, por ejemplo, sin limitación, diclorodifluorometano, triclorofluorometano, diclorotetrafluoroetano o dióxido de carbono. En el caso de un aerosol presurizado, la unidad de dosificación puede controlarse mediante una válvula para suministrar una cantidad medida. Las cápsulas y cartuchos, por ejemplo, de gelatina para usar en un inhalador o insuflador pueden formularse para que contengan una mezcla en polvo del compuesto y una base de polvo adecuada tal como lactosa o almidón.
Los compuestos pueden formularse además para la administración parenteral, por ejemplo, por inyección en bolo o infusión continua. Las formulaciones para inyección pueden presentarse en forma de dosis unitarias, por ejemplo, en ampollas o en recipientes de dosis múltiples, con adición de un conservante. Las composiciones pueden tomar tales formas de suspensiones, soluciones o emulsiones en vehículos oleosos o acuosos, y pueden contener materiales de formulación tales como agentes de suspensión, estabilización y/o dispersión.
Las composiciones farmacéuticas para administración parenteral incluyen soluciones acuosas de una forma soluble en agua, tal como, sin limitación, una sal, del compuesto activo. Adicionalmente, las suspensiones de los compuestos activos pueden prepararse en un vehículo lipofílico. Los vehículos lipofílicos adecuados incluyen aceites grasos tales como el aceite de sésamo, ésteres sintéticos de ácidos grasos tales como oleato de etilo y triglicéridos, o materiales tales como liposomas. Las suspensiones acuosas para inyección pueden contener sustancias que aumentan la viscosidad de la suspensión, tales como carboximetilcelulosa sódica, sorbitol, o dextrano. Opcionalmente, la suspensión también puede contener estabilizadores adecuados y/o agentes que aumentan la solubilidad de los compuestos para permitir preparaciones con soluciones de alta concentración.
Alternativamente, el ingrediente activo puede estar en forma de polvo para constitución con un vehículo adecuado, por ejemplo, agua esterilizada libre de pirógenos, antes de su uso.
Los compuestos también pueden formularse en composiciones rectales tales como supositorios o enemas de retención, mediante el uso, por ejemplo de bases de supositorio convencionales tales como manteca de cacao u otros glicéridos.
Además de las formulaciones que se describieron anteriormente, los compuestos también pueden formularse como preparaciones de depósito. Tales formulaciones de larga duración se pueden administrar por implantación (por ejemplo, por vía subcutánea o intramuscular) o por inyección intramuscular. Un compuesto de esta invención puede formularse para esta vía de administración con materiales poliméricos o hidrófobos adecuados (por ejemplo, en una emulsión con un aceite farmacológicamente aceptable), con resinas de intercambio iónico o como un derivado escasamente soluble tal como, sin limitación, una sal escasamente soluble.
Un ejemplo no limitante de un portador farmacéutico para los compuestos hidrófobos de la invención es un sistema de cosolventes que comprende alcohol bencílico, un surfactante no polar, un polímero orgánico miscible en agua y una fase acuosa tal como el sistema de cosolventes de VPD. VPD es una solución de alcohol bencílico al 3 % p/v, polisorbato surfactante no polar 80 al 8 % p/v y polietilenglicol 300 al 65 % p/v, hasta el volumen en etanol absoluto. El sistema de cosolventes de VPD (VPD: D5W) consiste en VPD diluida 1:1 con una solución de dextrosa al 5 % en agua. Este sistema de cosolventes disuelve bien los compuestos hidrófobos, y en sí mismo produce baja toxicidad tras la administración sistémica. Naturalmente, las proporciones de tal sistema de cosolventes pueden variar considerablemente sin destruir sus características de solubilidad y toxicidad. Además, la identidad de los componentes cosolventes puede variarse: por ejemplo, pueden usarse otros surfactantes no polares de baja toxicidad en lugar de polisorbato 80, puede variarse el tamaño de la fracción de polietilenglicol, otros polímeros biocompatibles pueden reemplazar al polietilenglicol, por ejemplo, polivinilpirrolidona y otros azúcares o polisacáridos pueden sustituir a la dextrosa.
Alternativamente, pueden usarse otros sistemas de suministro para compuestos farmacéuticos hidrófobos. Los liposomas y las emulsiones son ejemplos bien conocidos de vehículos o portadores de suministro para fármacos hidrófobos. Además, también pueden emplearse ciertos solventes orgánicos tal como el dimetilsulfóxido, aunque frecuentemente a costa de una mayor toxicidad.
Adicionalmente, los compuestos pueden suministrarse mediante el uso de un sistema de liberación sostenida, tales como matrices semipermeables de polímeros hidrofóbicos sólidos que contienen el agente terapéutico. Varios materiales de liberación sostenida se han establecido y se conocen bien por aquellos con experiencia en la técnica. Las cápsulas de liberación sostenida, en dependencia de su naturaleza química, pueden liberar los compuestos durante algunas semanas hasta aproximadamente 100 días. En dependencia de la naturaleza química y la estabilidad biológica del reactivo terapéutico, pueden usarse estrategias adicionales para la estabilización de las proteínas.
Las composiciones farmacéuticas en la presente descripción pueden comprender además portadores o excipientes en fase sólida o de gel adecuados. Los ejemplos de tales portadores o excipientes incluyen, pero no se limitan, a carbonato
de calcio, fosfato de calcio, varios azúcares, almidones, derivados de celulosa, gelatina y polímeros tales como polietilenglicoles.
Muchos de los compuestos moduladores (RORYt)/RORy de la invención pueden proporcionarse como sales fisiológicamente aceptables en donde el compuesto reivindicado puede formar la especie cargada negativa o positivamente. Los ejemplos de sales en las que el compuesto forma el resto cargado positivamente incluyen, sin limitación, amonio cuaternario (definido en otra parte en la presente descripción), sales tales como clorhidrato, sulfato, carbonato, lactato, tartrato, malato, maleato, succinato en donde el átomo de nitrógeno del grupo de amonio cuaternario es un nitrógeno del compuesto seleccionado de esta invención que ha reaccionado con el ácido apropiado. Las sales en las que un compuesto de esta invención forma las especies cargadas negativamente incluyen, sin limitación, las sales de sodio, potasio, calcio y magnesio formadas por la reacción de un grupo de ácido carboxílico en el compuesto con una base apropiada (por ejemplo, hidróxido de sodio (NaOH), hidróxido de potasio (KOH), hidróxido de calcio (Ca(OH)2), etc.).
Las composiciones farmacéuticas adecuadas para usar en la presente invención incluyen composiciones en donde los ingredientes activos están contenidos en una cantidad suficiente para lograr el propósito pretendido, por ejemplo, la modulación de la actividad de la proteína quinasa y/o el tratamiento o prevención de un trastorno relacionado con la proteína quinasa.
Más específicamente, una cantidad terapéuticamente efectiva significa una cantidad de compuesto efectiva para evitar, aliviar o mejorar los síntomas de la enfermedad o prolongar la sobrevivencia del sujeto que se trata.
La determinación de una cantidad terapéuticamente efectiva está bien aceptada dentro de la capacidad de los expertos en la técnica, especialmente a la luz de la descripción detallada proporcionada en la presente descripción.
Para cualquier compuesto usado en el método en la presente descripción, la cantidad o dosis terapéuticamente efectiva puede estimarse inicialmente a partir de ensayos de cultivos celulares. A continuación, la dosificación puede formularse para usar en modelos animales con el fin de lograr un intervalo de concentración circulante que incluya la IC50 como se determina en el cultivo celular (es decir, la concentración del compuesto de prueba que logra una inhibición semimáxima de (RORyt)/RORy, o la actividad del marcador sustituto). Tal información puede usarse después para determinar las dosis útiles en humanos con mayor precisión.
La toxicidad y la eficacia terapéutica de los compuestos descritos en la presente descripción pueden determinarse mediante procedimientos farmacéuticos estándar en cultivos celulares o animales experimentales, por ejemplo, determinando la IC50 y la LD50 (las cuales se analizan en otra parte en la presente descripción) para un compuesto sujeto. Los datos obtenidos de esos ensayos de cultivo de células y estudios en animales pueden usarse en la formulación de un intervalo de dosificación para su uso en humanos. La dosificación puede variar dependiendo de la forma de dosificación empleada y la vía de administración utilizada. La formulación exacta, la vía de administración y dosificación puede ser elegida por el médico individual teniendo en cuenta la afección del paciente. (Ver, por ejemplo, GOODMAN & GILMAN'S THE PHARMACOLOGICAL BASIS OF THERAPEUTICS, Cap. 3, 9a ed., Ed. por Hardman, J. y Limbard, L., McGraw-Hill, Nueva York, 1996, p.46.)
La cantidad y el intervalo de dosificación pueden ajustarse individualmente para proporcionar niveles plasmáticos de las especies activas que son suficientes para mantener los efectos terapéuticos deseados. Estos niveles plasmáticos se denominan concentraciones mínimas efectivas (MEC). La MEC variará para cada compuesto, pero puede estimarse a partir de los datos in vitro, por ejemplo, la concentración necesaria para lograr una inhibición del 50-90 % de (RORyt)/RORy, o el marcador sustituto pueden determinarse mediante el uso de los ensayos descritos en la presente descripción. Las dosificaciones necesarias para lograr la MEC dependerán de las características individuales y de la vía de administración. Los ensayos por HPLC o bioensayos pueden usarse para determinar las concentraciones plasmáticas.
Los intervalos de dosificación también pueden determinarse mediante el uso del valor de MEC. Los compuestos deben administrarse mediante el uso de un régimen que mantenga niveles plasmáticos por encima de la MEC durante 10-90 % del tiempo, preferentemente entre 30-90 % y con la máxima preferencia entre 50-90 %.
En la actualidad, las cantidades terapéuticamente efectivas de los compuestos de la presente invención pueden variar de aproximadamente 2,5 mg/m2 a 1500 mg/m2 por día. Las cantidades ilustrativas adicionales varían de 0,2-1000 mg/qid, 2 500 mg/qid y 20-250 mg/qid.
En casos de administración local o absorción selectiva, la concentración local efectiva del fármaco puede no relacionarse con la concentración plasmática y otros procedimientos conocidos en la técnica pueden emplearse para determinar la cantidad y el intervalo de dosificación correctos.
La cantidad de una composición administrada dependerá, por supuesto, del sujeto que se trata, la severidad de la afección, la manera de administración, el criterio del médico responsable, etcétera.
Las composiciones, si se desea, pueden presentarse en un empaque o dispositivo dispensador, tal como un kit aprobado por la FDA, que puede contener una o más formas de dosificación unitarias que contienen el ingrediente activo. El
empaque puede comprender, por ejemplo, una lámina de metal o plástico, tal como un empaque tipo burbuja. El empaque o dispositivo dispensador puede acompañarse con las instrucciones para la administración. El empaque o dispensador puede estar acompañado además de un aviso asociado con el contenedor en una forma prescrita por una agencia gubernamental que regula la fabricación, uso o venta de los productos farmacéuticos, cuyo aviso es reflejo de la aprobación por la agencia de la forma de las composiciones o de administración humana o veterinaria. Tal aviso, por ejemplo, puede ser del etiquetado aprobado por la Administración de fármacos y alimentos de los Estados Unidos para la prescripción de fármacos o de un prospecto del producto aprobado. Las composiciones que comprenden un compuesto de la invención formulado en un portador farmacéutico compatible también pueden prepararse, colocarse en un contenedor apropiado, y etiquetarse para el tratamiento de una afección indicada. Las afecciones adecuadas indicadas en la etiqueta pueden incluir tratamiento de un tumor, inhibición de angiogénesis, tratamiento de fibrosis, diabetes y similares.
Como se mencionó anteriormente, los compuestos y composiciones de la invención encontrarán utilidad en una amplia gama de enfermedades y afecciones mediadas por proteínas quinasas, incluidas enfermedades y afecciones mediadas por la actividad de (RORYt)/RORY. Tales enfermedades pueden incluir a manera de ejemplo y sin limitación, Esclerosis múltiple (EM), Artritis reumatoide (AR), Colitis inflamatoria, Psoriasis, EPOC, Dolor, Obesidad, Diabetes, Dislipidemia, Osteoporosis, Asma, Enfermedades neurodegenerativas y Cáncer.
La invención se comprenderá aún más después de considerar los siguientes ejemplos no limitantes.
Tabla 1a: Antagonistas de RORy que contienen 2, 3-dihidro-1H-inden-1-ona sustituida





Tabla 1b: Antagonistas de RORy que contienen 2, 3-dihidro-1H-inden-1-ona sustituida




Tabla 2: Lista de abreviaturas y significado usos en esta solicitud


Métodos de preparación de los compuestos
En ciertas modalidades, los Ejemplos más abajo son compuestos preparados de acuerdo con los procedimientos generales dados en las siguientes secciones. Aunque los métodos y esquemas sintéticos representan las síntesis de ciertos compuestos de la presente invención, los métodos y otros métodos conocidos por un experto en la técnica pueden aplicarse a todos los compuestos del género, la subclase de género y las especies de cada uno de estos compuestos como se describe en la presente descripción. Los aspectos de esta invención pueden entenderse a partir de los siguientes esquemas generales 1 y 2.

Detalles experimentales y ejemplos
Los puntos de fusión se determinaron en un aparato de Polmon digital MP-96. Los espectros de NMR1H y NMR 13C se registraron a RT en CDCh o DMSO-d6 en el espectrofotómetro de RMN a 400 MHz de Jeol mediante el uso de picos de solventes para CDCh: 7,27 y DMSO-d62,50 (D) como referencias internas. La asignación de desplazamientos químicos se basa en experimentos de NMR estándar (1H, 13C). Los espectros de masas se registraron en un espectrómetro LCMS LC-210EV de Shimadzu con una fuente de ionización API-ES. Jasco-FTIR-4100 se usó para registrar los espectros IR. Los análisis de TLC se realizaron en sílice F254 y la detección por luz UV a 254 nm, o por pulverización con reactivo de
tinción fosfomolibdic-H2SO4, KMNO4 o yodo. La cromatografía en columna se realizó sobre gel de sílice 60 (230 mesh). Las purificaciones y separaciones se realizaron en un sistema de cromatografía instantánea de sílice estándar. La pureza de las muestras se ha determinado mediante HPLC para el % de área del pico correspondiente a la retención de compuesto y el análisis elemental para C, H, N y O se llevó a cabo mediante el uso del analizador elemental Perkin-Elmer 2400 y el análisis de cloruro realizado mediante valoración calorimétrica en el Intertek USA Inc., QTI.
Metodología general de síntesis
Los compuestos de esta invención se preparan en general mediante métodos tales como los representados en los Esquemas más abajo, y los ejemplos preparativos que siguen.
Preparación de los ejemplos
Ejemplo 1: 6-metoxi-5-morfol¡no-2, 3-dihidro-1H-inden-1-ona (13)

Etapa 1: A una solución de 5-bromo-6-metoxi-2,3-dihidro-1H-inden-1-ona (11) (1 g, 4,149 mmol) y morfolina (0,36 g, 4,149 mmol) en 15 ml de tolueno se añadió carbonato de cesio (2,69 g, 8,298 mmol). La reacción se desgasificó y se purgó con nitrógeno durante 10 minutos. Se añadieron, desgasificaron y purgaron Pd2(dba)3 (189,3 mg, 0,207 mmol) y BINAP (64,5 mg, 0,103 mmol) con nitrógeno durante otros 10 minutos. La reacción se calentó a 110 °C durante la noche en un vial de microondas sellado. Después de la terminación del material de partida, la reacción se diluyó con cloroformo y se filtró a través de lecho de celite. La capa orgánica se concentró para producir el compuesto bruto 13 y el bruto resultante se purificó mediante cromatografía instantánea mediante el uso de gel de sílice de 100-200 mesh eluyendo el compuesto en acetato de etilo al 25 % en hexano como un sólido de color amarillo pálido 6-metoxi-5-morfolino-2,3-dihidro-1H-inden-1-ona 13. 1HNMR (400 MHz, CDCls) 6 ppm 7,70 (s, 1H), 7,20 (s, 1H), 3,93 (s, 3H), 3,06 (m, 2H), 2,72 (m, 2H); MS (ESI) m/z 247,9 (M+H).
Ejemplo 2: 2-(4-cloro-2-fluorobencil)-6-metoxi-5-morfolino-2, 3-dihidro-1H-inden-1-ona (16)

Etapa 2: A una solución del compuesto 13 (150 mg, 0,607 mmol) en 15 ml de tolueno se añadió 14 (86,2 mg, 0,607 mmol). Se añadió ácido p-toluenosulfónico (PTSA) (230,9 mg, 1,214 mmol) a la mezcla de reacción, y se agitó a 120 °C durante 6 h. La masa de reacción se diluyó con acetato de etilo y se lavó con agua (3 x 25 ml). La capa orgánica se secó sobre sulfato de sodio y se concentró para producir el compuesto bruto 15. El compuesto bruto 15 se purificó mediante cromatografía instantánea mediante el uso de gel de sílice de 100-200 mesh. El compuesto (E)-2-(4-cloro-2-fluorobenciliden)-6-metoxi-5-morfolino-2,3-dihidro-1H-inden-1-ona 15 se eluyó en acetato de etilo al 28 % en hexano para proporcionar un sólido de color amarillo.
Etapa 3: El compuesto 15 (45 mg, 0,121 mmol) se disolvió en metanol y se añadieron 20 mg de Pd/C y se agitó la reacción en globo de hidrógeno durante 6 h. La reacción se filtró a través de un lecho de celite y se lavó con metanol en exceso. La capa orgánica se concentró para producir el compuesto bruto 16. El bruto 16 se purificó mediante cromatografía instantánea mediante el uso de gel de sílice de 100-200 mesh. El compuesto 16 se eluyó con acetato de etilo al 23 % en hexano como 2-(4-cloro-2-fluorobencil)-6-metoxi-5-morfolino-2,3-dihidro-1H-inden-1-ona sólido de color semiblanco 16.
1HNMR (400 MHz, CDCla) 6 ppm 7,18 (m, 2H), 6,82 (m, 3H), 3,88 (m, 3H), 3,86 (m, 4H), 3,31 (m, 1H), 3,16 (m, 4H), 3,06 (m, 1H), 2,95 (m, 1H), 2,73 (m, 2H); MS (ESI) m/z 389,8 (M+H).
Ejemplo 3: 2-(4-doro-2-fluorobencil)-5-(3,4-dimetilpiperazin-1-il)-6-metoxi-2,3-dihidro-1H-inden-1-ona (20)

Ejemplo 4: 2-((1-benc¡lp¡perid¡n-4-¡l)met¡l)-6-metox¡-5-morfol¡no-2,3-d¡h¡dro-1H-¡nden-1-ona (23)

Etapa 1: A una solución de 13 (150 mg, 0,607 mmol) en 15 ml de tolueno se añadió 1-bencilpiperidina-4-carbaldehído 21 (123,4 mg, 0,607 mmol). Se añadió ácido p-toluenosulfónico (PTSA) (230,9 mg, 1,214 mmol) a la mezcla de reacción, y se agitó a 120 °C durante 6 h. La masa de reacción se diluyó con acetato de etilo y se lavó con agua (3 x 25 ml). La capa orgánica se secó sobre sulfato de sodio y se concentró para proporcionar el material bruto, que se purificó mediante cromatografía instantánea mediante el uso gel de sílice de 100-200 mesh. La elución en acetato de etilo al 30 % en hexano proporcionó un sólido de color amarillo (E)-2-((1-bencilpiperidin-4-il)metileno)-6-metoxi-5-morfolino-2, 3-dihidro-1H-inden-1-ona 22.
Etapa 2: El compuesto 22 (85 mg, 0,195 mmol) se disolvió en 25 ml de metanol, se añadieron 40 mg de Pd/C y se agitó bajo un globo de hidrógeno durante 6 h. La reacción se filtró a través de un lecho de celite y se lavó con metanol en exceso. La capa orgánica se concentró para producir el compuesto bruto 23. El material bruto se purificó mediante cromatografía rápida mediante el uso de gel de sílice de 100-200 mesh. El compuesto se eluyó con acetato de etilo al 20 % en hexano para producir el compuesto sólido de color semiblanco 2-((1-bencilpiperidin-4-il)metil)-6-metoxi-5-morfolino-2,3-dihidro-1H-inden-1-ona 23. 1HNMR (400 MHz, CDCla) 6 ppm 7,31 (m, 5H), 7,15 (bs, 1H), 6,85 (bs, 1H), 3,88 (m, 7H), 3,51 (bs, 2H), 3,22 (m, 4H), 2,90 (m, 2H), 2,65 (m, 2H), 1,90 (m, 3H), 1,64 (m, 2H), 1,58 (bs, 4H); MS (ESI) m/z 435,2 (M+H).
Ejemplo 5: 2-((1-bencilpiperidin-4-il)metil)-5-(3,4-dimetilpiperazin-1-il)-6-metoxi-2, 3-dihidro-1H-inden-1-ona (25)

Ejemplo 6: 2-(2,4-difluorobencil)-6-metoxi-5-morfolino-2,3-dihidro-1H-inden-1-ona (28)

Etapa 1: A una solución del compuesto 13 (150 mg, 0,607 mmol) en 15 ml de tolueno se añadió 26 (86,2 mg, 0,607 mmol), a continuación se añadió PTSA (230,9 mg, 1,214 mmol) y se agitó a 120 °C durante 6 h. La mezcla resultante se diluyó con acetato de etilo y se lavó con agua (3 x 25 ml). La capa orgánica se secó sobre sulfato de sodio y se concentró para producir el compuesto bruto 27, que se purificó mediante cromatografía instantánea mediante el uso de gel de sílice de 100-200 mesh. La elución al acetato de etilo al 28 % en hexano proporcionó el compuesto sólido de color amarillo (E)-2-(2,4-difluorobencilideno)-6-metoxi-5-morfolino-2,3-dihidro-1H-inden-1-ona 27.
Etapa 2: A 27 (45 mg, 0,121 mmol), disuelto en metanol, se añadieron 20 mg de Pd/C y se agitó en un globo de hidrógeno durante 6 h. La reacción se filtró a través de un lecho de celite y se lavó con metanol en exceso. La capa orgánica se concentró para producir el bruto, que se purificó mediante cromatografía instantánea mediante el uso de gel de sílice de 100-200 mesh. El compuesto se eluyó con acetato de etilo al 23 % en hexano como un compuesto sólido de color semiblanco 2-(2,4-difluorobencil)-6-metoxi-5-morfolino-2,3-dihidro-1H-inden-1-ona 28. 1HNMR (400 MHz, CDCl3) 8 ppm 7,18 (m, 2H), 6,82 (m, 3H), 3,88 (m, 3H), 3,86 (m, 4H), 3,31 (m, 1H), 3,16 (m, 4H), 3,06 (m, 1H), 2,95 (m, 1H), 2,73 (m, 2H); MS (ESI) m/z 373,9 (M+H).
Ejemplo 7: 6-metoxi-5-morfolino-2-(3-(trifluorometoxi)bencil)-2,3-dihidro-1H-inden-1-ona (31)

A una solución del compuesto 13 (150 mg, 0,607 mmol) en 15 ml de tolueno se añadió 3-bromo-5-(trifluorometoxi)benzaldehído 29 (115,4 mg, 0,607 mmol). Se añadió PTSA (230,9 mg, 1,214 mmol) a la mezcla de reacción, a continuación se agitó a 120 °C durante 6 h. La mezcla de reacción se diluyó con acetato de etilo y se lavó con agua (3 x 25 ml). La capa orgánica se secó sobre sulfato de sodio y se concentró para producir el compuesto bruto (E)-2-(3-bromo-5-(trifluorometoxi)bencilideno)-6-metoxi-5-morfolino-2, 3-dihidro-1H-inden-1-ona 30, que se purificó mediante cromatografía instantánea mediante el uso de gel de sílice de 100-200 mesh. El compuesto 30 se eluyó en acetato de etilo al 25 % en hexano para proporcionar un sólido de color amarillo. A 30 (45 mg, 0,107 mmol), disuelto en 20 ml de metanol, se añadió 20 mg de Pd/C y se agitó bajo un globo de hidrógeno durante 6 h. La reacción se filtró a través de un lecho de celite y se lavó con metanol en exceso. La capa orgánica se concentró para producir el compuesto desbromado en bruto 31. El bruto 31 se purificó mediante cromatografía instantánea mediante el uso de gel de sílice de 100-200 mesh. Compuesto 31 se eluyó como compuesto desbromado al acetato de etilo al 25 % en hexano como compuesto sólido de color semiblanco 6-metoxi-5-morfolino-2-(3-(trifluorometoxi)bencil)-2,3-dihidro-1H-inden-1-ona 31. 1HNMR (400 MHz, CDCla) 8 ppm 7,43 (m, 4H), 7,18 (s, 1H), 6,81 (s, 1H), 3,89 (m, 3H), 3,87 (m, 4H), 3,43 (m, 1H), 3,17 (m, 4H), 3,06 (m, 1H), 2,97 (m, 1H), 2,69 (m, 2H); MS (ESI) m/z 422,1 (M+H).
Ejemplo 8: 6-metox¡-5-morfol¡no-2-(3-(tr¡fluorometil)benc¡l)-2,3-d¡h¡dro-1H-¡nden-1-ona (37)

A una solución de 13 (450 mg, 1,819 mmol) en 10 ml de tolueno se añadió 3-bromo-5-(trifluorometil)benzaldehído 35 (316,8 mg, 1,819 mmol). Se añadió PTSA (692,2 mg, 3,639 mmol) a la mezcla de reacción, a continuación se agitó a 120 °C durante 6 h. La masa de reacción se diluyó con acetato de etilo y se lavó con agua (3 x 25 ml). La capa orgánica se secó sobre sulfato de sodio y se concentró para producir el bruto 36, que se purificó mediante cromatografía instantánea mediante el uso de gel de sílice de 100-200 mesh. El compuesto (E)-2-(3-bromo-5-(trifluorometil)bencilideno)-6-metoxi-5-morfolino-2,3-dihidro-1Hinden-1-ona 36 se eluyó en acetato de etilo al 30 % en hexano para proporcionar un sólido de color amarillo.
El 36 (570 mg, 1,413 mmol) se disolvió en metanol y se añadió Pd/C (350 mg), y se agitó bajo un globo de hidrógeno durante 6 h. La reacción se filtró a través de un lecho de celite y se lavó con metanol en exceso. La capa orgánica se concentró para producir el bruto 37, que se purificó mediante cromatografía instantánea mediante el uso de gel de sílice de 100-200 mesh. El compuesto 6-metoxi-5-morfolino-2-(3-(trifluorometil)bencil)-2,3-dihidro-1H-inden-1-ona 37 se eluyó como compuesto desbromado en acetato de etilo al 28 % en hexano como un sólido de color semiblanco. 1HNMR (400 MHz, CDCla) 8 ppm 7,47 (m, 4H), 7,18 (s, 1H), 6,81 (s, 1H), 3,89 (m, 3H), 3,87 (m, 3H), 3,40 (m, 1H), 3,17 (m, 4H), 3,06 (m, 1H), 2,97 (m, 1H), 2,69 (m, 2H); MS (ESI) m/z 406,0 (M+H).
Ejemplo 9: 2-(2,6-difluorobencil)-6-metoxi-5-morfolino-2,3-dihidro-1H-inden-1-ona (42)

A una solución de 13 (150 mg, 0,607 mmol) en 15 ml de tolueno se añadió 3-bromo-2, 6-difluorobenzaldehído 40 (81,4 mg, 0,607 mmol) y subsecuentemente se añadió PTSA (230,9 mg, 1,214 mmol) y se agitó a 120 °C durante 6 h. La masa de reacción se diluyó con acetato de etilo y se lavó con agua (3 x 25 ml). La capa orgánica se secó sobre sulfato de sodio y se concentró para producir el compuesto bruto 41. El bruto 41 se purificó mediante cromatografía instantánea mediante el uso de gel de sílice de 100-200 mesh. El compuesto (E)-2-(3-bromo-2,6-difluorobenciliden)-6-metoxi-5-morfolino-2,3-dihidro-1H-inden-1-ona 41 se eluyó en acetato de etilo al 32 % en hexano para proporcionar un sólido de color amarillo.
El 41 (40 mg, 0,088 mmol) se disolvió en 15 ml de metanol y se añadieron 20 mg de Pd/C y se agitó bajo un globo de hidrógeno durante 6 h. La reacción se filtró a través de lecho de élite y se lavó con metanol en exceso. La capa orgánica se concentró para producir el compuesto bruto 42, que se purificó mediante cromatografía instantánea mediante el uso de gel de sílice de 100-200 mesh. El compuesto 2-(2,6-difluorobencil)-6-metoxi-5-morfolino-2,3-dihidro-1H-inden-1-ona 42 se eluyó como compuesto desbromado en acetato de etilo al 26 % en hexano como un sólido de color semiblanco. 1HNMR (400 MHz, CDCla) 8 ppm 7,17 (m, 2H), 6,89 (m, 3H), 3,89 (m, 7H), 3,35 (m, 1H), 3,19 (m, 4H), 3,06 (m, 2H), 2,76 (m, 2H); MS (ESI) m/z 374,0 (M+H).
Ejemplo 10: 2-((4-cloro-2-morfolinotiazol-5-il)metil)-6-metoxi-5-morfolino-2,3-dihidro-1H-inden-1-ona (47)

Ejemplo 11: 2-(3-cloro-5-(trifluorometil)bencil)-6-metoxi-5-morfolino-2,3-dihidro-1H-inden-1-ona (50)

A una solución de 13 (100 mg, 0,404 mmol) en 5 ml de tolueno se añadió 3-cloro-5-(trifluorometil) benzaldehído 48 (84,4 mg, 0,404 mmol). Se añadió PTSA (153,9 mg, 0,809 mmol) a la masa de reacción, a continuación se agitó a 120 °C durante 6 h. La masa de reacción se diluyó con acetato de etilo y se lavó con agua (3 x 25 ml). La capa orgánica se secó sobre sulfato de sodio y se concentró para producir el compuesto bruto 49, que se purificó mediante cromatografía instantánea mediante el uso de gel de sílice de 100-200 mesh. El compuesto (E)-2-(3-cloro-5-(trifluorometil)bencilideno)
6-metoxi-5-morfolino-2,3-dihidro-1H-inden-1-ona 49 se eluyó en acetato de etilo al 35 % en hexano para proporcionar un sólido de color amarillo.
El 49 (30 mg, 0,068 mmol) se disolvió en 20 ml de metanol, se añadieron 10 mg de Pd/C y se agitó bajo un globo de hidrógeno durante 6 h. La reacción se filtró a través de un lecho de celite y se lavó con metanol en exceso. La capa orgánica se concentró para producir el compuesto bruto 50. El bruto 50 se purificó mediante cromatografía instantánea mediante el uso de gel de sílice de 100-200 mesh. El compuesto 2-(3-cloro-5-(trifluorometil)bencil)-6-metoxi-5-morfolino-2,3-dihidro-1H-inden-1-ona 50 se eluyó en acetato de etilo al 28 % en hexano como un sólido de color semiblanco. 1HNMR (400 MHz, CDCla) 8 ppm 7,43 (m, 3H), 7,18 (s, 1H), 6,82 (s, 1H), 3,90 (m, 3H), 3,87(m, 4H), 3,37(m, 1H), 3,17(m, 4H), 3,09 (m, 1H), 2,96 (m, 1H), 2,70 (m, 2H); MS (ESI) m/z 440,0 (M+H).
Ejemplo 12: 2-(5-cloro-2-(trifluorometil)bencil)-6-metoxi-5-morfolino-2, 3-dihidro-1H-inden-1-ona (53)

A una solución de 13 (100 mg, 0404 mmol) en 5 ml de tolueno se añadió 5-cloro-2-(trifluorometil)benzaldehído 51 (84,4 mg, 0,404 mmol) y PTSA (153,9 mg, 0,809 mmol). La reacción se agitó a 120 °C durante 6 h. La mezcla de reacción se diluyó con acetato de etilo y se lavó con agua (3 x 25 ml). La capa orgánica se secó sobre sulfato de sodio y se concentró para producir el compuesto bruto 52. El bruto 52 se purificó mediante cromatografía instantánea mediante el uso de gel de sílice de 100-200 mesh. El compuesto (E)-2-(5-cloro-2-(trifluorometil)bencilideno)-6-metoxi-5-morfolino-2,3-dihidro-1H-inden-1-ona 52 se eluyó en acetato de etilo al 28 % en hexano para proporcionar un sólido de color amarillo.
El compuesto 52 (50 mg, 0,114 mmol) se disolvió en 30 ml de metanol, se añadieron 17 mg de Pd/C y la reacción se agitó bajo un globo de hidrógeno durante 6 h. La reacción se filtró a través de un lecho de celite y se lavó con metanol en exceso. La capa orgánica se concentró para producir el bruto, que se purificó mediante cromatografía instantánea mediante el uso de gel de sílice de 100-200 mesh. El compuesto 53 se eluyó en acetato de etilo al 24 % en hexano como compuesto sólido de color semiblanco 2-(5-cloro-2-(trifluorometil)bencil)-6-metoxi-5-morfolino-2,3-dihidro-1H-inden-1-ona 53. 1HNMR (400 MHz, CDCla) 8 ppm 7,43 (m, 3H), 7,18 (s, 1H), 6,82 (s, 1H), 3,90 (m, 3H), 3,87 (m, 4H), 3,48 (d, 1H), 3,38 (m, 1H), 3,17 (m, 4H), 3,09 (m, 1H), 2,95 (m, 1H), 2,67 (m, 2H); MS (ESI) m/z 440,0 (M+H).
Ejemplo 13: 2-(4-cloro-2-(trifluorometil) bencil)-6-metoxi-5-morfolino-2, 3-dihidro-1H-inden-1-ona (56)

A una solución de 13 (100 mg, 0404 mmol) en 5 ml de tolueno se añadió el compuesto 4-cloro-2-(trifluorometil)benzaldehído 54 (84,4 mg, 0,404 mmol). Se añadió PTSA (153,9 mg, 0,809 mmol) a la masa de reacción y se agitó a 120 °C durante 6 h. La masa de reacción se diluyó con acetato de etilo y se lavó con agua (3 x 25 ml). La capa orgánica se secó sobre sulfato de sodio y se concentró para producir el compuesto bruto 55, que se purificó mediante cromatografía instantánea mediante el uso de gel de sílice de 100-200 mesh. El compuesto 55 se eluyó en acetato de etilo al 28 % en hexano para proporcionar un sólido de color amarillo.
El compuesto 55 (40 mg, 0,104 mmol) se disolvió en 30 ml de metanol, se añadieron 14 mg de Pd/C y la reacción se agitó bajo un globo de hidrógeno durante 6 h. La reacción se filtró a través de un lecho de celite y se lavó con metanol en exceso. La capa orgánica se concentró para producir el bruto 56. El cual se purificó mediante cromatografía instantánea mediante el uso de gel de sílice de 100-200 mesh. El compuesto se eluyó en acetato de etilo al 24 % en hexano como 2-(4-cloro-2-(trifluorometil)bencil)-6-metoxi-5-morfolino-2,3-dihidro-1H-inden-1-ona sólido de color semiblanco 56. 1HNMR (400 MHz, CDCh) 8 pp 7,64 (m, 1H), 7,44 (m, 2H), 7,19 (m, 2H), 6,81 (s, 1H), 4,00 (bs, 2H), 3,48 (d, 1H), 3,89 (m, 7H), 3,48 (m, 4H), 3,17 (m, 5H), 2,90 (m, 1H), 2,64 (m, 1H).
Ejemplo 14: 2-(2-cloro-6-(trifluorometil) bencil)-6-metoxi-5-morfolino-2,3-dihidro-1H-inden-1-ona (59)

Ejemplo 15: 2-((2-doro-5-(trifluorometil)piridin-3-il)metil)-6-metoxi-5-morfolino-2, 3-dihidro-1H-inden-1-ona (62)

Ejemplo 16: N-(2-((6-metoxi-5-morfolino-1-oxo-2, 3-dihidro-1H-inden-2-il)metM)-4-(trifluorometM)fenM) pivalamida (65)

Ejemplo 17: 2-(2-cloro-6-(trifluorometil) bencil)-6-metoxi-5-morfolino-2,3-dihidro-1H-inden-1-ona (68)

A una solución de 13 (250 mg, 1,010 mmol) en 10 ml de tolueno se añadió 3-(trifluorometil) picolinaldehído (177,0 mg, 1,010 mmol). Se añadió PTSA (384,5 mg, 2,021 mmol) a la mezcla de reacción y se agitó a 120 °C durante 6 h. La masa de reacción se diluyó con acetato de etilo y se lavó con agua (3 x 25 ml). La capa orgánica se secó sobre sulfato de sodio y se concentró para producir el compuesto bruto (E)-6-metoxi-5-morfolino-2-((3-(tnfluorometil)piridin-2-il)metileno)-2,3-dihidro-1H-inden-1-ona 67. El compuesto 67 se purificó mediante cromatografía instantánea mediante el uso de gel de sílice de 100-200 mesh. El compuesto se eluyó en acetato de etilo al 34 % en hexano para proporcionar un sólido de color amarillo.
El compuesto 67 (80 mg, 0,197 mmol) se disolvió en 15 ml de metanol, se añadieron 50 mg de Pd/C, y la reacción se agitó bajo un globo de hidrógeno durante 6 h. La reacción se filtró a través de un lecho de celite y se lavó con metanol en exceso. La capa orgánica se concentró para producir el bruto. El bruto se purificó mediante cromatografía instantánea mediante el uso de gel de sílice de 100-200 mesh. El compuesto se eluyó con acetato de etilo al 26 % en hexano como compuesto sólido de color semiblanco 68. 1HNMR (400 MHz, CDCla) 8 ppm 8,64 (d, 1H), 7,91 (d, 1H), 7,22 (d, 2H), 6,83 (s, 1H), 3,89 (m, 7H), 3,65 (dd, 1H), 3,44 (m, 1H), 3,16 (m, 6H), 2,72 (dd, 1H); MS (ESI) m/z 407,0 (M+H).
Ejemplo 18: 6-metoxi-5-morfolino-2-((6-(trifluorometil)piridin-3-il) metil)-2,3-dihidro-1H-inden-1-ona (71)

A una solución de 13 (100 mg, 0,404 mmol) en 5 ml de tolueno se añadió 6-(trifluorometil) nicotinaldehído 69 (70,8 mg, 0,404 mmol). Se añadió PTSA (154 mg, 0,808 mmol) y la mezcla de reacción se agitó a 120 °C durante 6 h. La masa de reacción se diluyó con acetato de etilo y se lavó con agua (3 x 25 ml). La capa orgánica se secó sobre sulfato de sodio y se concentró para producir el compuesto bruto (E)-6-metoxi-5-morfolino-2-((6-(trifluorometil)piridin-3-il)metileno)-2,3-dihidro-1H-inden-1-ona 70. El bruto 70 se purificó mediante cromatografía instantánea mediante el uso de gel de sílice de 100-200 mesh. El compuesto se eluyó en acetato de etilo al 32 % en hexano para proporcionar un sólido de color amarillo 70.
El compuesto 70 (55 mg, 0,136 mmol) se disolvió en 20 ml de metanol, se añadieron 35 mg de Pd/C, y la reacción se agitó bajo un globo de hidrógeno durante 6 h. La reacción se filtró a través de un lecho de celite y se lavó con metanol en exceso. La capa orgánica se concentró para producir el compuesto bruto 71. El bruto 71 se purificó mediante cromatografía instantánea mediante el uso de gel de sílice de 100-200 mesh. El compuesto se eluyó en acetato de etilo al 28 % en hexano como compuesto sólido de color semiblanco 6-metoxi-5-morfolino-2-((6-(trifluorometil)piridin-3-il)metil)-2,3-dihidro-1H-inden-1-ona 71. 1HNMR (400 MHz, CDCla) 8 ppm 8,61 (s, 1H), 7,74 (d, 1H), 7,59 (d, 1H), 7,17 (s, 1H), 6,80 (s, 1H), 3,89 (m, 3H), 3,86 (m, 4H), 3,39 (dd, 1H), 3,17 (m, 4H), 3,11 (m, 1H), 2,98 (m, 1H), 2,83 (m, 1H), 2,71 (m, 1H); MS (ESI) m/z 407,0 (M+H).
Ejemplo 19: 6-metoxi-5-morfolino-2-(4-((trifluorometil) tio)bencil)-2, 3-dihidro-1H-inden-1-ona (74)

A una solución de 13 (1,2 g, 4,858 mmol) en 40 ml de tolueno se añadió 4-((trifluorometil)tio)benzaldehído 72 (1 g, 4,858 mmol). Se añadió PTSA (1,84 g, 9,716 mmol) y la mezcla de reacción se agitó a 120 °C durante 6 h. La masa de reacción se diluyó con acetato de etilo y se lavó con agua (3 x 25 ml). La capa orgánica se secó sobre sulfato de sodio y se concentró para producir el compuesto bruto 73, que se purificó mediante cromatografía instantánea mediante el uso de gel de sílice de 100-200 mesh. El compuesto (E)-6-metoxi-5-morfolino-2-(4-((trifluorometil)tio)bencilideno)-2,3-dihidro-1H-inden-1-ona 73 se eluyó en acetato de etilo al 37 % en hexano para proporcionar un sólido de color amarillo.
El compuesto 73 (1,0 g, 2,296 mmol) se disolvió en 250 ml de metanol, se añadieron 400 mg de Pd/C y la reacción se agitó bajo un globo de hidrógeno durante 6 h. El resultado se filtró mediante un lecho de celite y se lavó con metanol en exceso. La capa orgánica se concentró para producir el compuesto bruto 74, que se purificó mediante cromatografía instantánea mediante el uso de gel de sílice de 100-200 mesh. El compuesto se eluyó en acetato de etilo al 30 % en hexano como un sólido de color semiblanco 6-metoxi-5-morfolino-2-(4-((trifluorometil)tio)bencil)-2,3-dihidro-1H-inden-1-ona 74. 1HNMR (400 MHz, CDCla) 8 ppm 7,56 (d, 2H), 7,28 (d, 2H), 7,18 (s, 1H), 6,81 (s, 1H), 3,89 (m, 3H), 3,87 (m, 4H), 3,37 (dd, 1H), 3,15 (m, 4H), 3,06 (m, 1H), 2,96 (m, 1H), 2,66 (m, 2H); MS (ESI) m/z 438,0 (M+H).
Ejemplo 20: 2-(2-fluoro-5-(trifluorometil) bencil)-6-metoxi-5-morfolino-2, 3-dihidro-1H-inden-1-ona (77)

A una solución de 13 (100 mg, 0,404 mmol) en 10 ml de tolueno se añadió 2-fluoro-5-(trifluorometil) benzaldehído 75 (77,7 mg, 0404 mmol). Se añadió PTSA (153,8 mg, 0809 mmol) y la mezcla de reacción se agitó a 120 °C durante 6 h. La masa de reacción se diluyó con acetato de etilo y se lavó con agua (3 x 25 ml). La capa orgánica se secó sobre sulfato de sodio y se concentró para producir el compuesto bruto 76 que se purificó mediante cromatografía instantánea mediante el uso de gel de sílice de 100-200 mesh. El compuesto (E)-2-(2-fluoro-5-(trifluorometil)bencilideno)-6-metoxi-5-morfolino-2,3-dihidro-1H-inden-1-ona 76 se eluyó en acetato de etilo al 34 % en hexano para proporcionar un sólido de color amarillo.
El compuesto 76 (50 mg, 0,118 mmol) se disolvió en 20 ml de metanol, se añadieron 30,6 mg de Pd/C, y la reacción se agitó bajo un globo de hidrógeno durante 6 h. El resultado se filtró mediante un lecho de celite y se lavó con metanol en exceso. La capa orgánica se concentró para producir el bruto que se purificó mediante cromatografía instantánea mediante el uso de gel de sílice de 100-200 mesh. El compuesto se eluyó en acetato de etilo al 26 % en hexano como compuesto sólido de color semiblanco 2-(2-fluoro-5-(trifluorometil)bencil)-6-metoxi-5-morfolino-2,3-dihidro-1H-inden-1-ona 77.
1HNMR (400 MHz, CDCls) 8 ppm 7,51 (dd, 1H), 7,48 (m, 1H), 7,15 (m, 2H), 6,82 (s, 1H), 3,89 (m, 3H), 3,87 (m, 4H), 3,40 (m, 1H), 3,17 (m, 4H), 3,11 (m, 1H), 3,02 (m, 1H), 2,70 (m, 2H); MS (ESI) m/z 424,0 (M+H).
Ejemplo 21: 2-(2-fluoro-5-(trifluorometil) bencil)-6-metoxi-5-morfolino-2, 3-dihidro-1H-inden-1-ona (80)

A una solución de 13 (100 mg, 0,404 mmol) en 10 ml de tolueno se añadió 2-fluoro-3-(trifluorometil) benzaldehído 78 (77,7 mg, 0,404 mmol). Se añadió PTSA (153,8 mg, 0,809 mmol) a la mezcla de reacción y se agitó a 120 °C durante 6 h. La masa de reacción se diluyó con acetato de etilo y se lavó con agua (3 x 25 ml). La capa orgánica se secó sobre sulfato de sodio y se concentró para producir el compuesto bruto 79 que se purificó mediante cromatografía instantánea mediante el uso de gel de sílice de 100-200 mesh. El compuesto (E)-2-(2-fluoro-3-(trifluorometil)bencilideno)-6-metoxi-5-morfolino-2,3-dihidro-1H-inden-1-ona 79 se eluyó en acetato de etilo al 34 % en hexano para proporcionar el color amarillo.
El compuesto 79 (40 mg, 0,094 mmol) se disolvió en 15 ml de metanol, se añadieron 25 mg de Pd/C, y la reacción se agitó bajo un globo de hidrógeno durante 6 h. La reacción se filtró a través de un lecho de celite y se lavó con metanol en exceso. La capa orgánica se concentró para producir el bruto 80 que se purificó mediante cromatografía instantánea mediante el uso de gel de sílice de 100-200 mesh. El compuesto se eluyó en acetato de etilo al 30 % en hexano como compuesto sólido de color semiblanco 2-(2-fluoro-3-(trifluorometil)bencil)-6-metoxi-5-morfolino-2,3-dihidro-1H-inden-1-ona 80. 1HNMR (400 MHz, CDCla) 8 ppm 7,48 (m, 2H), 7,33 (m, 1H), 7,15 (m, 1H), 6,82 (s, 1H), 3,88 (m, 3H), 3,86 (m, 4H), 3,47 (dd, 1H), 3,14 (m, 4H), 3,08 (m, 1H), 2,83 (m, 1H), 2,55 (m, 2H); MS (ESI) m/z 424,0 (M+H).
Ejemplo 22: 6-metoxi-5-morfolino-2-((6-(trifluorometil)piridin-3-il) metil)-2, 3-dihidro-1H-inden-1-ona (83)
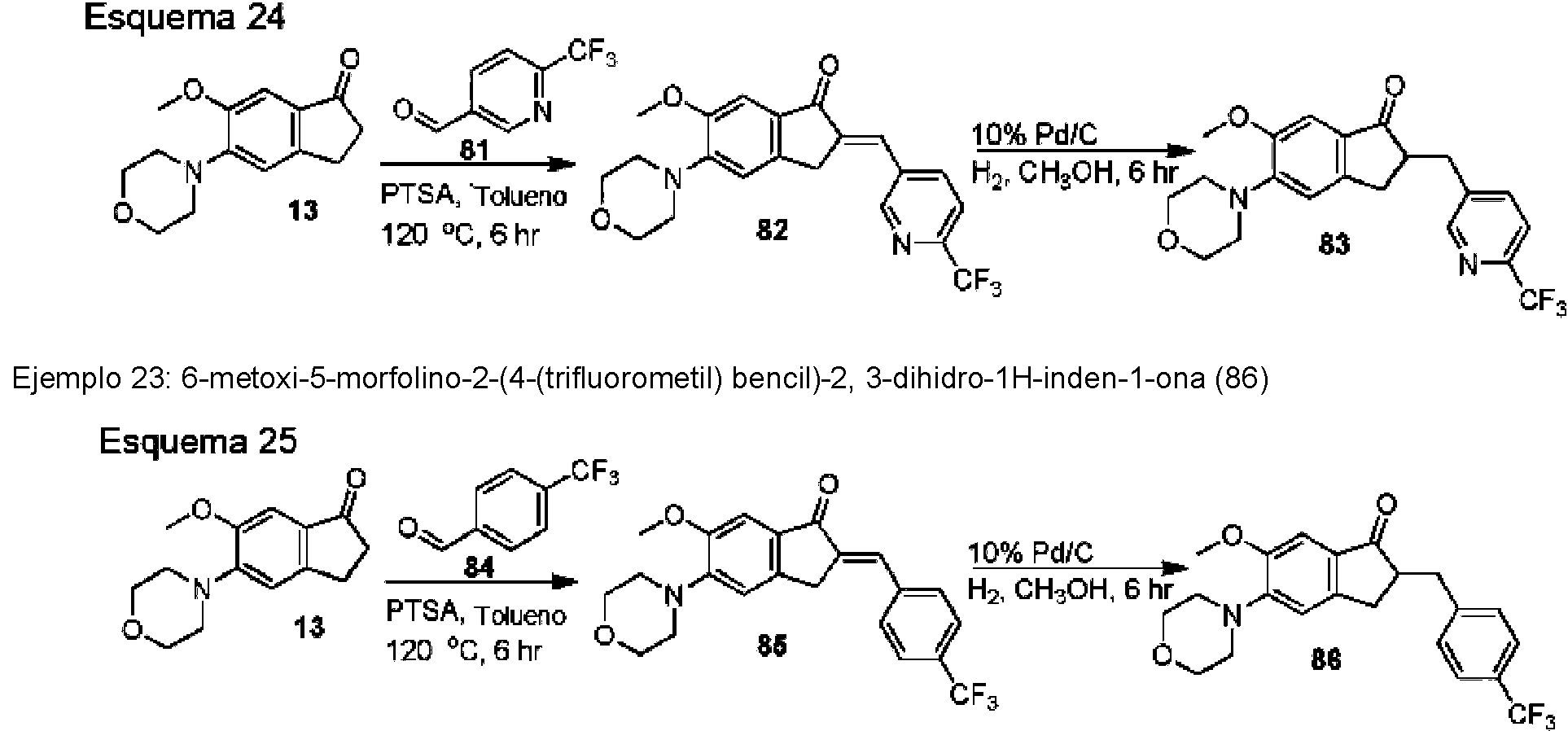
A una solución de 13 (100 mg, 04048 mmol) en 10 ml de tolueno se añadió 4-(trifluorometil) benzaldehído 84 (70,4 mg, 0,404 mmol). Se añadió PTSA (153,9 mg, 0,809 mmol) a la masa de reacción, a continuación se agitó a 120 °C durante 6 h. La masa de reacción se diluyó con acetato de etilo y se lavó con agua (3 x 25 ml). La capa orgánica se secó sobre sulfato de sodio y se concentró para producir el bruto 85 que se purificó mediante cromatografía instantánea mediante el uso de gel de sílice de 100-200 mesh. El compuesto 85 se eluyó en acetato de etilo al 28 % en hexano para proporcionar el compuesto sólido de color amarillo (E)-6-metoxi-5-morfolino-2-(4-(trifluorometil)bencilideno)-2,3-dihidro-1H-inden-1-ona 85.
El 85 (50 mg, 0,123 mmol) se disolvió en 20 ml de metanol, se añadieron 20 mg de Pd/C, y la reacción se agitó bajo un globo de hidrógeno durante 6 h, a continuación se filtró a través de lecho de celite y se lavó con un metanol en exceso. La capa orgánica se concentró para producir el bruto 86 que se purificó mediante cromatografía instantánea mediante el uso de gel de sílice de 100-200 mesh. El compuesto 86 se eluyó en acetato de etilo al 25 % en hexano como compuesto sólido de color semiblanco 6-metoxi-5-morfolino-2-(4-(trifluorometil)bencil)-2,3-dihidro-1H-inden-1-ona 86. 1HNMR (400 MHz, CDCla) 8 ppm 7,53 (d, 2H), 7,34 (d, 2H), 7,18 (s, 1H), 6,80 (s, 1H), 3,89 (m, 3H), 3,86 (m, 4H), 3,38 (m, 1H), 3,15 (m, 4H), 3,08 (m, 1H), 2,96 (m, 1H), 2,68 (m, 2H); MS (ESI) m/z 406,0 (M+H).
Ejemplo 24: 2-(4-cloro-3-(trifluorometil) bencil)-6-metoxi-5-morfolino-2, 3-dihidro-1H-inden-1-ona (89)

A una solución de 13 (100 mg, 0,404 mmol) en 10 ml de tolueno se añadió 4-cloro-3-(trifluorometil) benzaldehído 87 (84,4 mg, 0,404 mmol). Se añadió PTSA (153,8 mg, 0,808 mmol) y la reacción se agitó a 120 °C durante 6 h. La masa de reacción se diluyó con acetato de etilo y se lavó con agua (3 x 25 ml). La capa orgánica se secó sobre sulfato de sodio y se concentró para producir el bruto 88 que se purificó mediante cromatografía instantánea mediante el uso de gel de sílice de 100-200 mesh. El compuesto (E)-2-(4-cloro-3-(trifluorometil)bencilideno)-6-metoxi-5-morfolino-2,3-dihidro-1H-inden-1-ona 88 se eluyó en acetato de etilo al 31 % en hexano para proporcionar un sólido de color amarillo.
El 88 (65 mg, 0,148 mmol) se disolvió en 50 ml de acetato de etilo, se añadieron 15 mg de Pd/C, y la reacción se agitó bajo un globo de hidrógeno durante 6 h, a continuación se filtró a través de lecho de celite y se lavó con metanol en exceso. La capa orgánica se concentró para producir el compuesto bruto 89. El bruto se purificó mediante cromatografía instantánea mediante el uso de gel de sílice de 100-200 mesh. El compuesto 89 se eluyó en acetato de etilo al 25 % en hexano como compuesto sólido de color semiblanco 2-(4-cloro-3-(trifluorometil)bencil)-6-metoxi-5-morfolino-2,3-dihidro-1H-inden-1-ona 89. 1HNMR (400 MHz, CDCla) 8 ppm 7,55 (d, 1H), 7,41 (d, 1H), 7,34 (dd, 1H), 7,17 (s, 1H), 6,81 (s, 1H), 3,87 (m, 7H), 3,33 (dd, 1H), 3,17 (m, 4H), 3,06 (m, 1H), 2,93 (m, 1H), 2,66 (m, 2H); MS (ESI) m/z 439,9 (M+H).
Ejemplo 25: 2-(3-cloro-4-(trifluorometil) bencil)-6-metoxi-5-morfolino-2,3-dihidro-1H-inden-1-ona (92)

A una solución de 13 (550 mg, 2,226 mmol) en 25 ml de tolueno se añadió 3-cloro-4-(trifluorometil) benzaldehído 90 (464,2 mg, 2,226 mmol). Se añadió PTSA (846,7 mg, 4,452 mmol) a la reacción, y se agitó a 120 °C durante 6 h, a continuación se diluyó con acetato de etilo y se lavó con agua (3 x 25 ml). La capa orgánica se secó sobre sulfato de sodio y se concentró para producir el compuesto bruto 91 que se purificó mediante cromatografía instantánea mediante el uso de gel de sílice de 100-200 mesh. El compuesto se eluyó en acetato de etilo al 30 % en hexano para proporcionar el compuesto sólido de color amarillo (E)-2-(3-cloro-4-(trifluorometil)bencilideno)-6-metoxi-5-morfolino-2,3-dihidro-1H-inden-1-ona 91.
El 91 (220 mg, 0,502 mmol) se disolvió en 150 ml de acetato de etilo, se añadieron 100 mg de Pd/C, y la reacción se agitó bajo un globo de hidrógeno durante 6 h, a continuación se filtró a través de lecho de celite y se lavó con metanol en exceso. La capa orgánica se concentró para producir el bruto 92 que se purificó mediante cromatografía instantánea
mediante el uso de gel de sílice de 100-200 mesh. El compuesto se eluyó con acetato de etilo al 28 % en hexano como compuesto sólido de color semiblanco 2-(3-doro-4-(trifluorometil)bencil)-6-metoxi-5-morfolino-2,3-dihidro-1H-inden-1-ona 92. 1HNMR (400 MHz, CDCls) 8 ppm 7,59 (d, 1H), 7,49 (d, 1H), 7,31 (m, 1H), 7,19 (s, 1H), 6,83 (s, 1H), 3,89 (m, 3H), 3,87 (m, 4H), 3,53 (m, 1H), 3,17 (m, 4H), 3,07 (m, 2H), 2,91 (m, 1H), 2,73 (m, 1H); MS (ESI) m/z 440,0 (M+H).
Ejemplo 26: 6-metoxi-2-((3-met¡l-1H-p¡razol-5-¡l) metil)-5-morfolino-2, -dihidro-1H-inden-1-ona (95)

A una solución de 13 (100 mg, 0,404 mmol) en 10 ml de tolueno se añadió 3-metil-1H-pirazol-5-carbaldehído 93 (70,8 mg, 0,404 mmol). Se añadió PTSA (153,8 mg, 0,808 mmol) a la masa de reacción, a continuación se agitó a 120 °C durante 6 h, se diluyó con acetato de etilo y se lavó con agua (3 x 25 ml). La capa orgánica se secó sobre sulfato de sodio y se concentró para producir el bruto 94 y se purificó mediante cromatografía instantánea mediante el uso de gel de sílice de 100-200 mesh eluyendo en MeOH al 2 % en DCM para proporcionar el compuesto sólido de color amarillo (E)-6-metoxi-2-((3-metil-1H-pirazol-5-il) metileno)-5-morfolino-2, 3-dihidro-1H-inden-1-ona 94.
El 94 (85 mg, 0,250 mmol) se disolvió en 50 ml de metanol, se añadieron 55 mg de Pd/C, y la reacción se agitó bajo un globo de hidrógeno durante 6 h, se filtró a través de lecho de celite y se lavó con metanol en exceso. La capa orgánica se concentró para producir el bruto 95 y se purificó mediante cromatografía instantánea mediante el uso de gel de sílice de 100-200 mesh. El compuesto 95 se eluyó en MeOH al 2 % en DCM como compuesto sólido de color semiblanco 6-metoxi-2-((3-metil-1H-pirazol-5-il)metil)-5-morfolino-2,3-dihidro-1H-inden-1-ona 95.1HNMR(400 MHz, CDCla) 8 ppm 7,17 (s, 1H), 6,82 (s, 1H), 5,86 (s, 1H), 3,88 (m, 3H), 3,86 (m, 4H), 3,18 (m, 6H), 2,97 (m, 1H), 2,81 (m, 1H), 2,77 (m, 1H); MS (ESI) m/z 342,0 (M+H).
Ejemplo 29: 6-metoxi-5-(4-metilpiperazin-1-il)-2-(4-((trifluorometil) tio)bencil)-2,3-dihidro-1H-inden-1-ona (106)

A una solución de 5-bromo-6-metoxi-2, 3-dihidro-1H-inden-1-ona 11 (250 mg, 1,04 mol) y N-metilpiperazina (125 mg, 1,248 mol) en 15 ml de tolueno se añadió carbonato de cesio (677 mg, 2,08 mol). La reacción se desgasificó y se purgó con nitrógeno durante 10 minutos. Se añadieron Pd2(dba)3 (4,7 mg, 0,052 mol) y BINAP (6,4 mg, 0,104 mol) y nuevamente se desgasificaron y purgaron con nitrógeno durante otros 10 minutos. La reacción se calentó a 110 °C durante la noche en condiciones selladas. La reacción se diluyó con cloroformo y se filtró a través de un lecho de celite. La capa orgánica se concentró para producir el compuesto bruto AS-3061 y se purificó mediante cromatografía instantánea mediante el uso de gel de sílice de 100-200 mesh eluyendo el compuesto en metanol al 2 % en cloroformo como compuesto sólido de color amarillo pálido 6-metoxi-5-(4-metilpiperazin-1-il)-2,3-dihidro-1H-inden-1-ona AS-3061. 1HNMR (400 MHz, CDCh) 8 ppm 7,24 (s, 1H), 7,16 (s, 1H), 6,90 (s, 1H), 4,06 (m, 1H), 3,92 (s, 3H), 3,89 (s, 1H), 3,70 (s, 3H), 3,50 (m, 2H), 3,29 (s, 4H), 3,01 (m, 4H), 2,62 (m, 6H), 2,42 (s, 3H); MS (ESI) m/z 260,9 (M+H).
A una solución de 11 (250 mg, 1,04 mol) 15 ml de tolueno se añadió 4-((trifluorometil) tio) benzaldehído 72 (235 mg, 1,144 mol). Se añadió PTSA (357 mg, 2,08 mol) a la masa de reacción, que se agitó a 120 °C durante 6 h, se diluyó con acetato de etilo y se lavó con agua (3 x 25 ml). La capa orgánica se secó sobre sulfato de sodio y se concentró para producir el compuesto bruto 104 que se purificó mediante cromatografía instantánea mediante el uso de gel de sílice de 100-200
mesh. El compuesto 104 se eluyó en acetato de etilo al 10 % en hexano para proporcionar un sólido de color amarillo (E)-5-bromo-6-metoxi-2-(4-((trifluorometil)tio)bencilideno)-2,3-dihidro-1H-inden-1-ona 104.
El 104 (125 mg, 0,467 mol) se disolvió en 25 ml de metanol, se añadieron 10 mg de Pt/C y la reacción se agitó bajo un globo de hidrógeno durante 6 h, a continuación se filtró a través de lecho de celite y se lavó con un metanol en exceso. La capa orgánica se concentró para producir el compuesto bruto (E)-6-metoxi-5-(4-metilpiperazin-1-il)-2-(4-((trifluorometil) tio) bencilideno)-2, 3-dihidro-1H-inden-1-ona 105 y se purificó mediante cromatografía instantánea mediante el uso de gel de sílice de 100-200 mesh. El compuesto se eluyó con acetato de etilo al 12 % en hexano como compuesto sólido de color semiblanco 105.
A una solución de 105 (75 mg, 0,174 mol) y N-metil piperazina (20,9 mg, 0,209 mol) en 15 ml de tolueno se añadió carbonato de cesio (113,3 mg, 0,348 mol). La reacción se desgasificó y se purgó con nitrógeno durante 10 minutos. Se añadieron Pd2(dba)3 (7,96 mg, 0,0087 mol) y BINAP (10,8 mg, 0,0174 mol) y nuevamente se desgasificaron y purgaron con nitrógeno durante otros 10 minutos. La reacción se calentó a 110 °C durante la noche en condiciones selladas. La reacción se diluyó con cloroformo y se filtró a través de un lecho de celite. La capa orgánica se concentró para producir el compuesto bruto 106 y se purificó mediante cromatografía instantánea mediante el uso de gel de sílice de 100-200 mesh eluyendo el compuesto en metanol al 5 % en cloroformo como un sólido de color marrón 6-metoxi-5-(4-metilpiperazin-1-il)-2-(4-((trifluorometil)tio)bencil)-2,3-dihidro-1H-inden-1-ona 106. 1HNMR (400 MHz, CDCh) 8 ppm 7,57 (d, 2H), 7,28 (bs, 2H), 7,20 (s, 1H), 6,90 (s, 1H), 3,91 (s, 3H), 3,70 (bs, 4H), 3,35 (d, 2H), 2,99 (bs, 5H), 2,73 (bs, 3H); MS (ESI) m/z 451,0 (M+H).
Ejemplo 31: 6-metoxi-5-(4-metilpiperazin-1-il)-2-((3-(trifluorometil) piridin-2-il) metil)-2, 3-dihidro-1H-inden-1-ona (113)

Ejemplo 32: 6-metoxi-5-(piperazin-1-il)-2-(4-((trifluorometil)tio)bencil)-2,3-dihidro-1H-inden-1-ona (117)
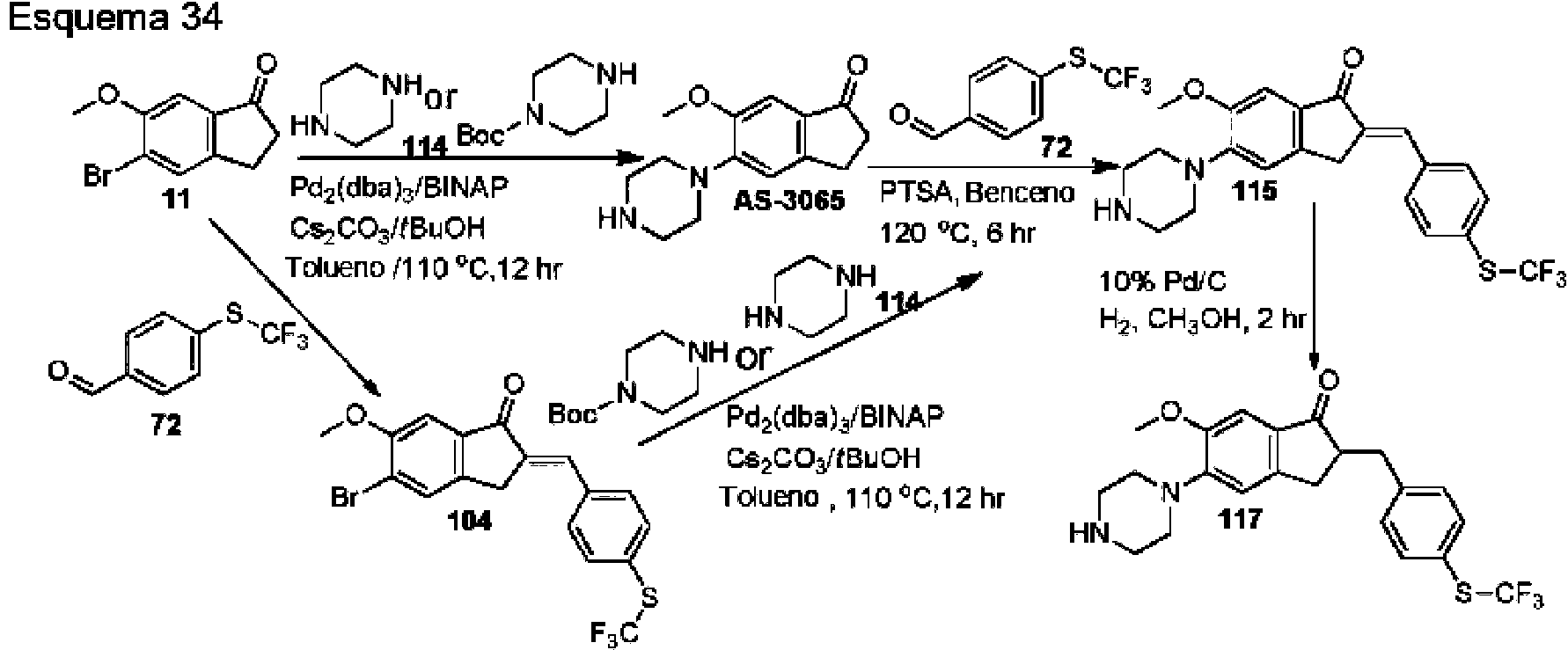
A una solución de 5-bromo-6-metoxi-2,3-dihidro-1H-inden-1-ona 11 (250 mg, 1,04 mol), piperazina (107 mg, 1,248 mol) y/o Boc-piperazina en 15 ml de tolueno se añadió carbonato de cesio (677 mg, 2,08 mol). La reacción se desgasificó y se purgó con nitrógeno durante 10 minutos. Se añadieron Pd2(dba)3 (4,7 mg, 0,052 mol) y BINAP (6,4 mg, 0,104 mol) y nuevamente se desgasificaron y purgaron con nitrógeno durante otros 10 minutos. La mezcla de reacción se calentó a 110 °C durante la noche en condiciones selladas. Después de la terminación, la reacción se diluyó con cloroformo y se filtró a través de lecho de celite. La capa orgánica se concentró para producir el compuesto bruto y se purificó mediante cromatografía ultrarrápida mediante el uso de gel de sílice de 100-200 mesh eluyendo el compuesto AS-3065 en metanol
al 4 % en cloroformo como compuesto sólido de color amarillo pálido 6-metoxi-5-(piperazin-1-il)-2,3-dihidro-1H-inden-1-ona AS-3065. 1HNMR (400 MHz, CDCl3) 8 ppm 7,17 (s, 1H), 6,90 (s, 1H), 3,89 (s, 3H), 3,21 (m, 4H), 3,15 (m, 4H), 3,035 (m, 2H), 2,67 (m, 2H).
Ejemplo 41: 6-metoxi-5-(4-metMpiperidin-1-il)-2-(4-((trifluorometil)tio)bencil)-2, 3-dihidro-1H-inden-1-ona (140)
Es 
A una solución del compuesto 5-bromo-6-metoxi-2, 3-dihidro-1H-inden-1-ona 11 (250 mg, 1,04 mol) y 4-metilpiperidina 129 (120 mg, 1,248 mol) en 15 ml de tolueno se añadió carbonato de cesio (677 mg, 2,08 mol). La reacción se desgasificó y se purgó con nitrógeno durante 10 minutos, se añadieron Pd2(dba)3 (4,7 mg, 0,052 mol) y BINAP (6,7 mg, 0,104 mol) y nuevamente se desgasificó y purgó con nitrógeno durante otros 10 minutos. La reacción se calentó a 110 °C durante la noche en condiciones selladas. Después de la terminación, la reacción se diluyó con cloroformo y se filtró a través de lecho de celite. La capa orgánica se concentró para producir el compuesto bruto AS-3070 que se purificó mediante cromatografía instantánea mediante el uso de gel de sílice de 100-200 mesh eluyendo el compuesto en acetato de etilo al 25 % en hexano como un sólido de color amarillo pálido 6-metoxi-5-(4-metilpiperidin-1-il)-2,3-dihidro-1H-inden-1-ona AS-3070. 1HNMR (400 MHz, CDCla) 8 ppm 7,14 (s, 1H), 6,90 (s, 1H), 3,88 (s, 3H), 3,60 (d, 2H), 3,00 (m, 2H), 2,64 (m, 4H), 1,73 (m, 2H), 1,41 (m, 3H), 0,99 (m, 3H).
A una solución de 5-bromo-6-metoxi-2, 3-dihidro-1H-inden-1-ona 11 (250 mg, 0,1037 mmol) en benceno se añadió 4-((trifluorometil) tio) benzaldehído 72 (213 mg, 0,103 mmol). Se añadió PTSA (395 mg, 0,207 mmol), la masa de reacción se agitó a 120 °C durante 6 h, a continuación se diluyó con acetato de etilo y se lavó con agua (3 x 25 ml). La capa orgánica se secó sobre sulfato de sodio y se concentró para producir el compuesto bruto 104 y se purificó mediante cromatografía instantánea mediante el uso de gel de sílice de 100-200 mesh. El compuesto 104 se eluyó en acetato de etilo al 30 % en hexano para proporcionar un sólido de color amarillo.
A una solución de 104 (150 mg, 0,348 mmol) y 4-metilpiperidina (69 mg, 0,696 mmol) en tolueno y fBuOH (8:2, 10 ml) se añadió carbonato de cesio 228 mg, 0,696). La reacción se desgasificó y se purgó con nitrógeno durante 10 minutos. Se añadieron Pd2(dba)3 (15,9 mg, 0,0174 mmol) y BINAP (32,4 mg, 0,15 mmol) y nuevamente se desgasificaron y purgaron con nitrógeno durante otros 10 minutos. La reacción se calentó a 110 °C durante la noche en condiciones selladas. La reacción se diluyó con cloroformo y se filtró a través de un lecho de celite. La capa orgánica se concentró para producir el compuesto bruto 139 que se purificó mediante cromatografía instantánea mediante el uso de gel de sílice de 100-200 mesh eluyendo el compuesto en acetato de etilo al 10 % en hexano como un sólido de color amarillo pálido (E)-6-metoxi-5-(4-metilpiperidin-1-il)-2-(4-((trifluorometil)tio)bencilideno)-2,3-dihidro-1H-inden-1-ona 139.
El 139 (80 mg, 0,178 mmol) se disolvió en 50 ml de metanol y se añadió níquel Raney (8 mg) y la reacción se agitó bajo un globo de hidrógeno durante 2 h. La reacción se filtró a través de un lecho de celite y se lavó con metanol en exceso. La capa orgánica se concentró para producir el compuesto bruto 140 y se purificó mediante cromatografía instantánea mediante el uso de gel de sílice de 100-200 mesh. El compuesto 140 se eluyó en acetato de etilo al 8 % en hexano como compuesto sólido de color semiblanco 6-metoxi-5-(4-metilpiperidin-1-il)-2-(4-((trifluorometil)tio)bencil)-2,3-dihidro-1H-inden-1-ona 140. 1HNMR (400 MHz, CDCl3) 8 ppm 7,27 (m, 3H), 7,11 (m, 4H), 6,81 (s, 1H), 3,89 (s, 3H), 3,60 (bs, 2H), 3,37 (dd, 1H), 2,94 (m, 2H), 2,59 (m, 4H), 0,94 (m, 3H), 0,86 (m, 4H); MS (ESI) m/z 450,1 (M+H).
E je m p lo 42 : 6 -m e to x i-5 -(4 -m e t i lp ip e r id in -1 - i l) -2 -( (3 -( t r i f lu o ro m e ti l) p ir id in -2 - il) m e til) -2 , 3 -d ih id ro -1 H - in d e n -1 -o n a (142 )

Ejemplo 43: 6-metoxi-5-((1-metilpiperidin-4-il)amino)-2-(4-((trifluorometil)tio) bencil)-2, 3-dihidro-1H-inden-1-ona (144)

A una solución de 11 (250 mg, 1,04 mol) 15 mi de tolueno se añadió 4-((trifluorometil) tio) benzaldehído 72 (235 mg, 1,144 mol). Se añadió PTSA (357 mg, 2,08 mol), la mezcla de reacción se agitó a 120 °C durante 6 h, se diluyó con acetato de etilo y se lavó con agua (3 x 25 ml). La capa orgánica se secó sobre sulfato de sodio y se concentró para producir el compuesto bruto 104 y se purificó mediante cromatografía instantánea mediante el uso de gel de sílice de 100-200 mesh. El compuesto resultante (E)-5-bromo-6-metoxi-2-(4-((trifluorometil) tio)bencilideno)-2,3-dihidro-1H-inden-1-ona 104 se eluyó en acetato de etilo al 10 % en hexano para proporcionar un sólido de color amarillo.
El 104 (125 mg, 0,467 mol) se disolvió en 25 ml de metanol, se añadieron 10 mg de Pt/C, la reacción se agitó bajo un globo de hidrógeno durante 6 h, se filtró a través de un lecho de celite y se lavó con metanol en exceso. La capa orgánica se concentró para producir el compuesto bruto 143 y se purificó mediante cromatografía instantánea mediante el uso de gel de sílice de 100-200 mesh. El compuesto 5-bromo-6-metoxi-2-(4-((trifluorometil)tio)bencil)-2,3-dihidro-1H-inden-1-ona 143 se eluyó en acetato de etilo al 12 % en hexano como un sólido de color semiblanco.
A una solución de 143 (75 mg, 0,174 mol) y 1-metilpiperidin-4-amina 134 (23,8 mg, 0,0208 mol) en 15 ml de tolueno se añadió carbonato de cesio (113,3 mg, 0,348 mol). La reacción se desgasificó y se purgó con nitrógeno durante 10 minutos. Se añadieron Pd2(dba)3 (7,96 mg, 0,0087 mol) y BINAP (10,8 mg, 0,0174 mol) y nuevamente se desgasificaron y purgaron con nitrógeno durante otros 10 minutos. La reacción se calentó a 110 °C durante la noche en condiciones selladas. La reacción se diluyó con cloroformo y se filtró a través de un lecho de celite. La capa orgánica se concentró para producir el compuesto bruto 144 y se purificó mediante cromatografía instantánea mediante el uso de gel de sílice de 100-200 mesh eluyendo el compuesto con metanol al 7,5 % en cloroformo como un sólido de color marrón 6-metoxi-5-((1-metilpiperidin-4-il)amino)-2-(4-((trifluorometil) tio) bencil)-2, 3-dihidro-1H-inden-1-ona 144. 1HNMR (400 MHz, CDCh) ó ppm 7,55 (bs, 2H), 7,28 (bs, 2H), 7,08 (s, 1H), 6,42 (s, 1H), 4,99 (s, 1H), 3,89 (s, 3H), 3,37 (m, 4H), 2,84 (m, 6H), 2,66 (m, 3H), 2,09 (m, 4H); MS (ESI) m/z 465,0 (M+H).
E je m p lo 47 : 6 -m e to x i-5 -(4 -m e t i lp ip e r id in -1 - i l) -2 -(3 -( t r i f lu o ro m e t i l) b e n c il) -2 , 3 -d ih id ro -1 H - in d e n -1 -o n a (154 )
Esquema 49
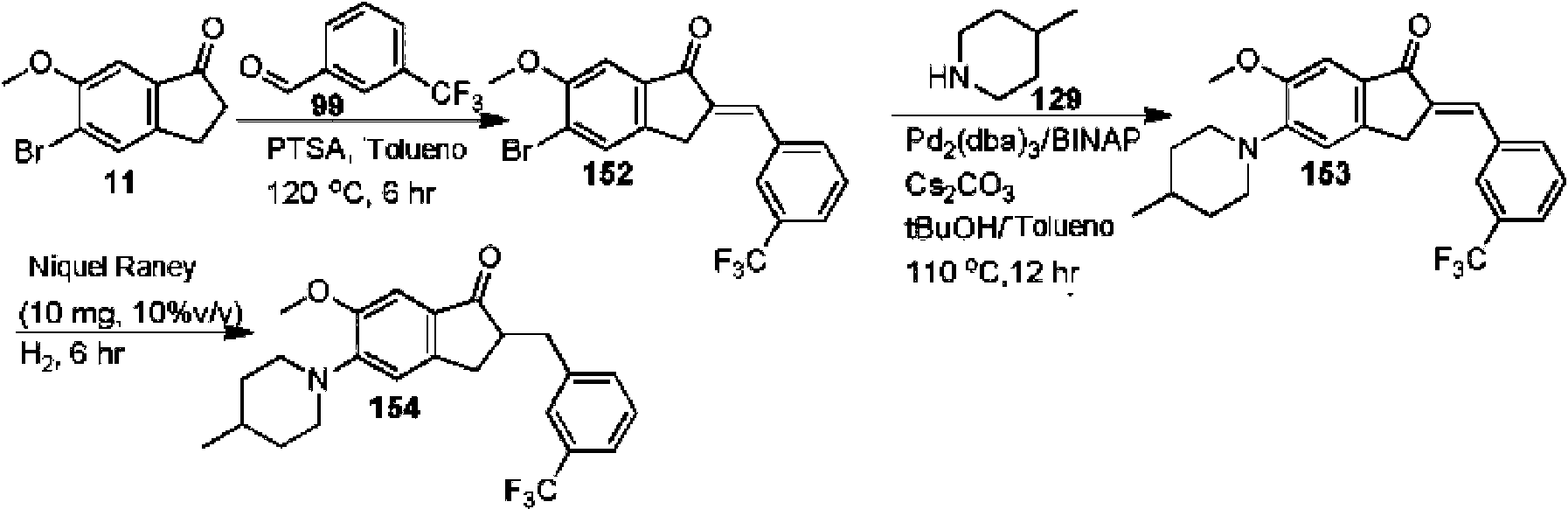
A una solución de 11 (200 mg, 0,829 mmol) en tolueno se añadió 3-(trifluorometM) benzaldehído 99 (158,86 mg, 0,9128 mmol). Se añadió PTSA (285,5 mg, 172,20 mmol) a la masa de reacción, a continuación se agitó a 120 °C durante 6 h, se diluyó con acetato de etilo y se lavó con agua (3 x 25 ml). La capa orgánica se secó sobre sulfato de sodio y se concentró para producir el compuesto bruto 152 que se purificó mediante cromatografía instantánea mediante el uso de gel de sílice de 100-200 mesh. El compuesto (E)-5-bromo-6-metoxi-2-(3-(trifluorometil)bencilideno)-2,3-dihidro-1H-inden-1-ona 152 se eluyó en acetato de etilo al 30 % en hexano para proporcionar un sólido de color amarillo.
A una solución de 152 (120 mg, 0,3007 mmol) y 4-metilpiperidina 129 (60,2 mg, 0,6015 mmol) en tolueno se añadió carbonato de cesio (197,2 mg, 0,6015 mmol). La reacción se desgasificó y se purgó con nitrógeno durante 10 minutos. Se añadieron Pd2(dba)3 (13,7 mg, 0,0150 mmol) y BINAP (28,1 mg, 0,0451 mmol) y nuevamente se desgasificaron y purgaron con nitrógeno durante otros 10 minutos. La reacción se calentó a 110 °C durante la noche en condiciones selladas. La reacción se diluyó con cloroformo y se filtró a través de un lecho de celite. La capa orgánica se concentró para producir el compuesto bruto 153 que se purificó mediante cromatografía instantánea mediante el uso de gel de sílice de 100-200 mesh eluyendo el compuesto (E)-6-metoxi-5-(4-metilpiperidin-1-il)-2-(3-(trifluorometil)bencilideno)-2,3-dihidro-1H-inden-1-ona 153 en acetato de etilo al 25 % en hexano como un sólido de color amarillo pálido.
El 153 (100 mg, 0,240 mmol) se disolvió en metanol y se añadió níquel Raney (10 mg, 10 % v/v) y la reacción se agitó bajo un globo de hidrógeno durante 6 h. La reacción se filtró a través de un lecho de celite y se lavó con metanol en exceso. La capa orgánica se concentró para producir el compuesto bruto 154 que se purificó mediante cromatografía instantánea mediante el uso de gel de sílice de 100-200 mesh. El compuesto 154 se eluyó en acetato de etilo al 20 % en hexano como compuesto sólido de color semiblanco 6-metoxi-5-(4-metilpiperidin-1-il)-2-(3-(trifluorometil) bencil)-2,3-dihidro-1H-inden-1-ona 154. 1HNMR (400 MHz, CDCla) 8 ppm 7,40 (m, 4H), 7,15 (s, 1H), 6,82 (s, 1H), 3,89 (s, 3H), 3,60 (s, 2H), 3,41 (dd, 1H), 2,94 (m, 2H), 2,62 (m, 4H), 0,92 (m, 4H), 0,85 (m, 3H); MS (ESI) m/z 418,0 (M+H).
Ejemplo 48: 6-metoxi-5-(4-metilpiperazin-1-il)-2-(3-(trifluorometil) bencil)-2, 3-dihidro-1H-inden-1-ona (156)

Una solución de 11 (200 mg, 0,829 mmol) en tolueno se añadió 3-(trifluorometil) benzaldehído 99 (158,86 mg, 0,9128 mmol). Se añadió PTSA (285,5 mg, 172,20 mmol) a la masa de reacción. La reacción se agitó a 120 °C durante 6 h. La masa de reacción se diluyó con acetato de etilo y se lavó con agua (3 x 25 ml). La capa orgánica se secó sobre sulfato de sodio y se concentró para producir el compuesto bruto 152 que se purificó mediante cromatografía instantánea mediante el uso de gel de sílice de 100-200 mesh. El compuesto (E)-5-bromo-6-metoxi-2-(3-(trifluorometil)bencilideno)-2,3-dihidro-1H-inden-1-ona 152 se eluyó en acetato de etilo al 30 % en hexano para proporcionar un sólido de color amarillo.
A una solución de 152 (120 mg, 0,3009 mmol) y 4-metilpiperazina (60,1 mg, 0,6105 mmol) en tolueno se añadió carbonato de cesio (197,2 mg, 0,6015 mmol). La reacción se desgasificó y se purgó con nitrógeno durante 10 minutos. Se añadieron Pd2(dba)3 (13,7 mg, 0,0150 mmol) y BINAP (28,0 mg, 0,0451 mmol) y nuevamente se desgasificaron y purgaron con nitrógeno durante otros 10 minutos. La reacción se calentó a 110 °C durante la noche en condiciones selladas. La reacción se diluyó con cloroformo y se filtró a través de un lecho de celite. La capa orgánica se concentró para producir el compuesto bruto 155 que se purificó mediante cromatografía instantánea mediante el uso de gel de sílice de 100-200 mesh eluyendo el compuesto en acetato de etilo al 25 % en hexano como un sólido de color amarillo pálido (E)-6-metoxi-5-(4-metilpiperazin-1-il)-2-(3-(trifluorometil)bencilideno)-2,3-dihidro-1H inden-1-ona 155.
El 155 (100 mg, 0,241 mmol) se disolvió en metanol y se añadió níquel Raney (10 mg, 10 % v/v) y la reacción se agitó en un globo de hidrógeno durante 6 h. La reacción se filtró a través de un lecho de celite y se lavó con metanol en exceso. La capa orgánica se concentró para producir el producto bruto 156 que se purificó mediante cromatografía instantánea mediante el uso de gel de sílice de 100-200 mesh. El compuesto 156 se eluyó en acetato de etilo al 20 % en hexano como sólido de color semiblanco 6-metoxi-5-(4-metilpiperazin-1-il)-2-(3-(trifluorometil)bencil)-2,3-dihidro-1H-inden-1-ona 156.
1HNMR (400 MHz, CDCla) 8 ppm 7,42 (m, 4H), 7,17 (s, 1H), 6,83 (s, 1H), 3,41 (dd, 1H), 3,21 (bs, 4H), 3,03 (m, 1H), 2,95 (m, 1H), 2,68 (m, 2H), 2,61 (bs, 4H), 2,35 (s, 3H); MS (ESI) m/z 419,0 (M+H).
Ejemplo 49: 2-(2-cloro-5-(trifluorometil) bencil)-6-metoxi-5-morfolino-2,3-dihidro-1H-inden-1-ona (158)

A una solución de 6-metoxi-5-morfolino-2, 3-dihidro-1H-inden-1-ona 13 (0,120 mg, 0,485 mmol) en tolueno (10 ml) se añadió 2-cloro-5-(trifluorometil) benzaldehído 75 (0,101 mg, 0,485 mmol). Se añadió PTSA (0,184 mg, 0,971 mmol) a la masa de reacción, la reacción se agitó a 100 °C durante 6 h, se diluyó con acetato de etilo y se lavó con agua (3 x 25 ml). La capa orgánica se secó sobre sulfato de sodio y se concentró para producir el compuesto bruto 157 que se purificó mediante cromatografía instantánea mediante el uso de gel de sílice de 100-200 mesh. El compuesto se eluyó en acetato de etilo al 15 % en hexano para proporcionar un sólido de color amarillo (E)-2-(2-cloro-5-(trifluorometil) bencilideno)-6-metoxi-5-morfolino-2, 3-dihidro-1H-inden-1-ona 157.
El 157 (100 mg, 0,228 mmol) se disolvió en metanol y se añadió Pd/C (40 mg), la reacción se agitó bajo un globo de hidrógeno durante 6 h, se filtró a través de lecho de celite y se lavó con metanol en exceso. La capa orgánica se concentró para producir el compuesto bruto 158 que se purificó mediante cromatografía instantánea mediante el uso de gel de sílice de 100-200 mesh. El compuesto se eluyó en acetato de etilo al 10 % en hexano como 2-(2-cloro-5-(trifluorometil)bencil)-6-metoxi-5-morfolino-2,3-dihidro-1H-inden-1-ona sólido de color semiblanco 158. 1HNMR (400 MHz, CDCh) 8 ppm 7,43 (m, 3H), 7,20 (s, 1H), 6,84 (s, 1H), 3,89 (m, 7H), 3,54 (dd, 1H), 3,14 (m, 4H), 3,07 (m, 2H), 2,75 (m, 2H); MS (ESI) m/z 440,0 (M+H).
Ejemplo 50: 6-metoxi-5-(piperazin-1-il)-2-((3-(trifluorometil)piridin-2-il) metil)-2, 3-dihidro-1H-inden-1-ona (164)

Ejemplo 51: 4,5-dimetoxi-2-(4-((trifluorometil)tio)bencil)-2,3-dihidro-1H-inden-1-ona (167)
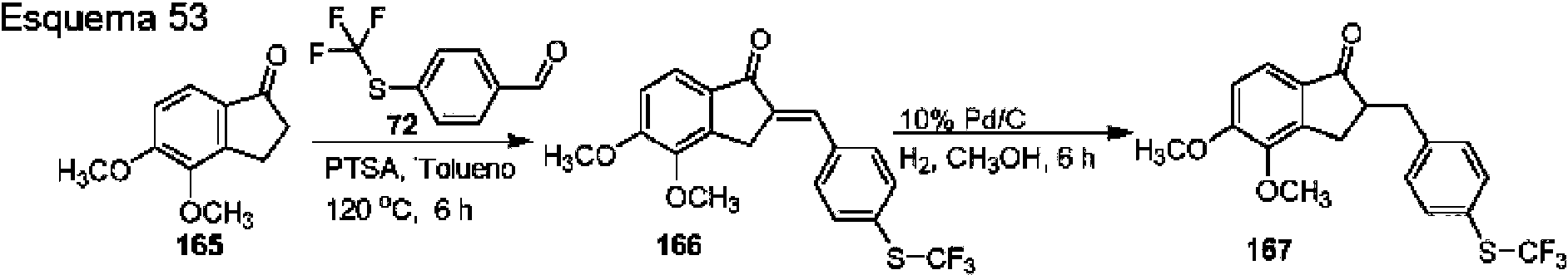
A una solución de 4,5-dimetoxi-2,3-dihidro-1H-inden-1-ona (165) (100 mg, 0,520 mmol) en 15 ml de tolueno se añadió 4-((trifluorometil)tio)benzaldehído (72) (118,4 mg, 0,572 mmol) y PTSA (178,9 mg, 1,414 mmol). La reacción se agitó a 120 °C durante 6 h. La mezcla de reacción resultante se diluyó con acetato de etilo y se lavó con (3x 25 ml). La capa orgánica se secó sobre sulfato de sodio y se concentró para producir el bruto, que se purificó mediante cromatografía instantánea mediante el uso de gel de sílice de 100-200 mesh. El compuesto se eluyó con acetato de etilo al 25 % en hexano para proporcionar un sólido de color amarillo del producto intermedio (E)-4,5-dimetoxi-2-(4-((trifluorometil)tio)bencilideno)-2,3-dihidro-1H-inden-1-ona 166.
El 166 (85 mg, 0.223 mmol) se disolvió en 25 ml de metanol. Se añadieron 20 mg de Pd/C y la reacción se agitó bajo globo de H2 durante 6 h. La reacción se filtró a través de un lecho de Celite y se lavó con metanol en exceso. La capa orgánica se concentró para producir el compuesto bruto 4,5-dimetoxi-2-(4-((trifluorometil)tio)bencil)-2,3-dihidro-1H-inden-1-ona 167, que se purificó mediante cromatografía instantánea mediante el uso de gel de sílice de 100-200 mesh. El compuesto 167 resultante se eluyó en acetato de etilo al 20 % en hexano como un sólido de color semiblanco. 1H NMR (400 MHz, CDCla) 8 ppm 7,56 (m, 3H, 7,29 (m, 2H), 6,96 (d, 1H), 3,94 (s, 3H), 3,85 (s, 3H), 3,37 (m, 1H), 3,17 (m, 1H), 2,95 (m, 1H), 2,71 (m, 2H); MS (ESI) m/z 382,9 (M+H).
Ejemplo 52: 4,5-dimetoxi-2-((3-(trifluorometil)piridin-2-il)metil)-2,3-dihidro-1H-inden-1-ona (169)

A una solución de 165 (100 mg, 0,518 mmol) en 15 ml de tolueno se añadió 66 (99,7 mg, 0569 mmol). Se añadió PTSA (196,8 mg, 1,03 mmol) a la masa de reacción, que a continuación se agitó a 120 °C durante 12 h. La masa de reacción se diluyó con acetato de etilo y se lavó con agua (3 x 25 ml). La capa orgánica se secó sobre sulfato de sodio y se concentró para producir el bruto, que se usó en la siguiente etapa sin purificación adicional.
El 168 (100 mg) se disolvió en 25 ml de metanol y se añadieron 15 mg de Pd/C, y la reacción se agitó bajo globo de H2 durante 6 h. La reacción se filtró a través de un lecho de celite y se lavó con metanol en exceso. La capa orgánica se concentró para producir el bruto 169 y se purificó mediante cromatografía instantánea mediante el uso de gel de sílice de 100-200 mesh. El compuesto se eluyó en acetato de etilo al 20 % en hexano como un sólido de color semiblanco 168. 1H NMR (400 MHz, CDCla) 8 ppm 8,64 (d, 1H), 7,91 (d, 1H), 7,56 (d, 1H), 6,97 (d, 1H), 7,11 (d, 1H), 3,86 (s, 6H), 3,66 (d, 1H), 3,43 (m, 1H), 3,32 (m, 1H), 3,17 (m, 1H), 2,74 (dd, 1H); MS-ES+351,9.
Ejemplo 53: 5-cloro-6-metoxi-2-(4-((trifluorometil)tio)bencil)-2,3-dihidro-1H-inden-1-ona (172)

A una solución de 5-cloro-6-metoxi-2,3-dihidro-1H-inden-1-ona 170 (100 mg, 0,510 mmol) en 15 ml de tolueno se añadió 4-((trifluorometil)tio)benzaldehído 72 (98,4 mg, 0,561 mmol). Se añadió PTSA (175,9 mg, 1,2 mmol) a la masa de reacción, que se agitó a 120 °C durante 6 h. La masa de reacción se diluyó con acetato de etilo y se lavó con agua (3 x 25 ml). La capa orgánica se secó sobre sulfato de sodio y se concentró para producir el (E)-5-cloro-6-metoxi-2-(4-((trifluorometil)tio)bencilideno)-2,3-dihidro-1H-inden-1-ona bruto (171) que se purificó mediante cromatografía instantánea
mediante el uso de gel de sílice de 100-200 mesh. El compuesto se eluyó en acetato de etilo al 22 % en hexano para proporcionar un sólido de color amarillo.
El compuesto 171 (85 mg, 0,195 mmol) se disolvió en 25 ml de metanol y Pt/C (40 mg) y la reacción se agitó bajo globo de H2 durante 6 h. La reacción se filtró a través de un lecho de celite y se lavó con metanol en exceso. La capa orgánica se concentró para producir el bruto. El bruto se purificó mediante cromatografía instantánea mediante el uso de gel de sílice de 100-200 mesh. El compuesto se eluyó en acetato de etilo al 25 % en hexano como un sólido de color semiblanco 172. 1H NMR (400 MHz, CDCls) 8 ppm 7,57 (d, 2H), 7,43 (s, 1H), 7,27(d, 2H), 7,25 (s, 1H), 3,94 (s, 3H), 3,37 (dd, 1H), 3,11 (m, 1H), 2,99 (m, 1H), 2,72 (m, 2H); MS-ES+386,8.
Ejemplo 54: 5-cloro-6-metoxi-2-((3-(trifluorometil)piridin-2-il)metil)-2,3-dihidro-1H-inden-1-ona (174)

A una solución de 170 (150 mg, 0,7769 mmol) en 15 ml de tolueno se añadió 66 (203,9 mg, 1,165 mmol). Se añadió PTSA (443,06 mg, 0,2337 mmol) a la masa de reacción, que se agitó a 120 °C durante 6 h. La masa de reacción se diluyó con acetato de etilo y se lavó con agua (3 x 25 ml). La capa orgánica se secó sobre sulfato de sodio y se concentró para producir el bruto, usado para la siguiente etapa sin purificación adicional.
El 173 (50 mg) se disolvió en 25 ml de metanol. Se añadieron 10 mg de Pt/C y la reacción se agitó bajo globo de H2 durante 6 h. La reacción se filtró a través de un lecho de celite y se lavó con metanol en exceso. La capa orgánica se concentró para producir el bruto. El bruto se purificó mediante cromatografía instantánea mediante el uso de gel de sílice de 100-200 mesh. El compuesto 174 se eluyó en acetato de etilo al 20 % en hexano como un sólido de color semiblanco 174. 1H NMR (400 MHz, CDCla) 8 ppm 8,57 (d, 1H), 7,91 (d, 1H), 7,43 (s, 1H), 7,29 (s, 1H), 7,23 (d, 1H), 3,95 (s, 3H), 3,64 (dd, 1H), 3,40 (m, 1H), 3,23 (m, 2H), 2,75 (dd, 1H); MS-ES+357,8.
Ejemplo 55: 4-cloro-5-metox¡-2-(4-((tr¡fluorornet¡l)t¡o)benc¡l)-2,3-d¡h¡dro-1H-inden-1-ona (177)

A una solución de 4-cloro-5-metoxi-2,3-dihidro-1H-inden-1-ona 175 (100 mg, 0,510 mmol) en 15 ml de tolueno se añadió el compuesto 72 (123,4 mg, 0,607 mmol). Se añadió PTSA (230,9 mg, 1,214 mmol) a la masa de reacción, y se agitó a 120 °C durante 6 h. La masa de reacción se diluyó con acetato de etilo y se lavó con agua (3 x 25 ml). La capa orgánica se secó sobre sulfato de sodio y se concentró para producir el bruto 176, que se purificó mediante cromatografía instantánea mediante el uso de gel de sílice de 100-200 mesh. El compuesto se eluyó en acetato de etilo al 24 % en hexano para proporcionar un sólido de color amarillo.
El (E)-4-cloro-5-metoxi-2-(4-((trifluorometil)tio)bencilideno)-2,3-dihidro-1H-inden-1-ona 176 (75 mg, 0,195 mmol) se disolvió en 25 ml de metanol a los que se añadió 40 mg de Pt/c y la reacción se agitó bajo globo de H2 durante 6 h. La reacción se filtró a través de un lecho de celite y se lavó con metanol en exceso. La capa orgánica se concentró para producir el bruto, que se purificó mediante cromatografía instantánea mediante el uso de gel de sílice de 100-200 mesh. El compuesto final 177 se eluyó en acetato de etilo al 20 % en hexano como un sólido de color semiblanco. 1H NMR (400 MHz, CDCls) 8 ppm 7,69 (d, 1H), 7,58 (d, 2H), 7,29 (d, 2H), 6,98 (d, 1H), 3,99 (s, 3H), 3,38 (dd, 1H), 3,18 (dd, 1H), 2,99 (m, 1H), 2,73 (m, 1H); MS-ES+386,8.
E je m p lo 56 : 4 -c lo ro -5 -m e to x i-2 -( (3 -( t r i f lu o ro m e t i l)p ir id in -2 - i l)m e t i l) -2 ,3 -d ih id ro -1 H - in d e n -1 -o n a (179 )

A una solución de 175 (100 mg, 0,776 mmol) en 15 ml de tolueno se añadió 66 (203,9 mg, 1,16 mmol). Se añadió PTSA (452 mg, 2,303 mmol) a la masa de reacción y se agitó a 120 °C durante 6 h. La masa de reacción se diluyó con acetato de etilo y se lavó con agua (3 x 25 ml). La capa orgánica se secó sobre sulfato de sodio y se concentró para producir el bruto 178, usado en la siguiente etapa sin purificación adicional
El 178 (70 mg) se disolvió en 25 ml de metanol, se añadieron 10 mg de Pt/C y la reacción se agitó bajo globo de H2 durante 6 h. La reacción se filtró a través de un lecho de celite y se lavó con metanol en exceso. La capa orgánica se concentró para producir el bruto, que se purificó mediante cromatografía instantánea mediante el uso de gel de sílice de 100-200 mesh. El compuesto 179 se eluyó en acetato de etilo al 20 % en hexano como un sólido de color semiblanco 179. 1H NMR (400 MHz, CDCls) 8 ppm 8,60 (d, 1H), 7,91 (d, 1H), 7,72 (d, 1H), 7,24 (d, 1H), 6,99 (d, 1H), 4,00 (s, 3H), 3,66 (dd, 1H), 3,44 (m, 1H), 3,24 (m, 2H), 2,75 (dd, 1H); MS-ES+355,8.
Ejemplo 57: 5-metoxi-2-(4-((trifluorometil)tio)bencil)-2,3-dihidro-1H-inden-1-ona (182)

A una solución de 180 (100 mg, 0,617 mmol) en 15 ml de tolueno se añadió 72 (139,4 mg, 0,6787 mmol). Se añadió PTSA (212,9 mg, 1,234 mmol) a la masa de reacción y se agitó a 120 °C durante 6 h. La masa de reacción se diluyó con acetato de etilo y se lavó con agua (3 x 25 ml). La capa orgánica se secó sobre sulfato de sodio y se concentró para producir el bruto, que se purificó mediante cromatografía instantánea mediante el uso de gel de sílice de 100-200 mesh. El compuesto se eluyó en acetato de etilo al 15 % en hexano para proporcionar 181 como un sólido amarillo.
El 181 (90 mg, 0,270 mmol) se disolvió en 25 ml de metanol, se añadieron 15 mg de Pd/C y la reacción se agitó bajo globo de H2 durante 6 h. La reacción se filtró a través de un lecho de celite y se lavó con metanol en exceso. La capa orgánica se concentró para producir el bruto, que se purificó mediante cromatografía instantánea mediante el uso de gel de sílice de 100-200 mesh. El compuesto se eluyó en acetato de etilo al 20 % en hexano como un sólido de color semiblanco 182.
1H NMR (400 MHz, CDCla) 8 ppm 7,70 (d, 1H), 7,56 (d, 2H), 7,28 (d, 2H), 6,89 (m, 1H), 6,83 (m, 1H), 3,86 (s, 3H), 3,38 (dd, 1H), 3,13 (dd, 1H), 2,96 (m, 1H), 2,70 (m, 2H); MS-ES+352,9.
Ejemplo 58: 5-metoxi-2-((3-(trifluorometil)piridin-2-il)metil)-2,3-dihidro-1 H-inden-1-ona (184)

A una solución de 180 (100 mg, 0,617 mmol) en 15 ml tolueno se añadió 66 (151,2 mg, 0,864 mmol). Se añadió PTSA (234,5 mg, 1,234 mmol) a la masa de reacción. La reacción se agitó a 120 °C durante 6 h. La masa de reacción se diluyó con acetato de etilo y se lavó con agua (3 x 25 ml). La capa orgánica se secó sobre sulfato de sodio y se concentró para producir el bruto 183, usado en la siguiente etapa sin purificación adicional.
El 183 (100 mg) se disolvió en 25 ml de metanol, se añadieron 10 mg de Pd/C y la reacción se agitó bajo globo de H2 durante 6 h. La reacción se filtró a través de un lecho de celite y se lavó con metanol en exceso. La capa orgánica se concentró para producir el bruto, que se purificó mediante cromatografía instantánea mediante el uso de gel de sílice de 100-200 mesh. El compuesto 184 se eluyó en acetato de etilo al 12 % en hexano como un sólido de color semiblanco 184. 1H NMR (400 MHz, CDCls) 8 ppm 8,64 (d, 1H), 7,91 (d, 1H), 7,72 (d, 1H), 6,90 (d, 1H), 6,85 (s, 1H), 3,87 (s, 3H), 3,65 (dd, 1H), 3,42 (m, 1H), 3,27 (m, 1H), 3,15 (m, 1H), 2,79 (dd, 1H); MS-ES+321,8.
Ejemplo 59: Etanosulfonato de 6-metoxi-3-oxo-2-(4-((trifluorometil)tio)bencil)-2,3-dihidro-1H-inden-5-ilo (188)

A una solución de 185 (50 mg, 0,280 mmol) en 15 ml de tolueno se añadió 72 (34,4 mg, 0,280 mmol). Se añadió PTSA (94,129 mg, 0,561 mmol) a la masa de reacción y se agitó a 120 °C durante 6 h. La masa de reacción se diluyó con acetato de etilo y se lavó con agua (3 x 25 ml). La capa orgánica se secó sobre sulfato de sodio y se concentró para producir el bruto, que se purificó mediante cromatografía instantánea mediante el uso de gel de sílice de 100-200 mesh. El compuesto 186 se eluyó en acetato de etilo al 30 % en hexano para proporcionar un sólido de color amarillo.
El 186 (70 mg 0,195 mmol) se disolvió en 25 ml de metanol, se añadieron 30 mg de Pd/C y la reacción se agitó bajo globo de H2 durante 6 h. La reacción se filtró a través de un lecho de celite y se lavó con metanol en exceso. La capa orgánica se concentró para producir el bruto 187 que se purificó mediante cromatografía instantánea mediante el uso de gel de sílice de 100-200 mesh. El compuesto 187 se eluyó en acetato de etilo al 20 % en hexano como un sólido de color semiblanco. 1H NMR (400 MHz, CDCls) 8 ppm 7,56 (d, 2H), 7,28 (d, 2H), 6,80 (s, 1H), 5,67 (s, 1H), 3,96 (s, 3H), 3,36 (dd, 1H), 3,07 (dd, 1H), 2,94 (m, 1H), 2,70 (m, 2H); MS-ES+368,9.
El 187 (50 mg, 0,1366 mmol) se disolvió en acetona y se añadió K2CO3 (37,7 mg, 0,2732 mmol) seguido de cloruro de etanosulfonilo (50 mg, 0,1366 mmol). La reacción se agitó durante 12 h a 70 °C, después se diluyó con acetato de etilo y se lavó con agua y salmuera (3x50 ml). La capa orgánica se concentró para producir el bruto 188 que se purificó mediante cromatografía instantánea mediante el uso de gel de sílice 100-200 mesh. El compuesto se eluyó en acetato de etilo al 15 % en hexano como un sólido de color semiblanco 188. 1H NMR (400 MHz, CDCla) 8 ppm 7,65 (s, 1H), 7,57 (d, 2H), 7,27 (d, 2H), 6,94 (s, 1H), 3,95 (s, 3H), 3,35 (m, 3H), 3,14 (m, 1H), 2,99 (m, 1H), 2,71 (m, 2H), 1,57 (m, 3H); MS-ES+460,8.
Ejemplo 60: Etanosulfonato de 6-metoxi-3-oxo-2-((3-(trifluorometil)piridin-2-il)metil)-2,3-dihidro-1H-inden-5-ilo (191)

A una solución de 185 (50 mg, 0,2808 mmol) en 15 ml de tolueno se añadió 66 (68,8 mg, 0,393 mmol). Se añadió PTSA (106,7 mg, 0,561 mmol) a la masa de reacción y se agitó a 120 °C durante 6 h. La masa de reacción se diluyó con acetato de etilo y se lavó con agua (3 x 25ml). La capa orgánica se secó sobre sulfato de sodio y se concentró para producir el bruto 189, que se usó en la siguiente etapa sin purificación adicional.
El 189 (50 mg) se disolvió en 25 ml de metanol, se añadieron 10 mg de Pd/C y la reacción se agitó bajo globo de H2 durante 6 h. La reacción se filtró a través de un lecho de celite y se lavó con metanol en exceso. La capa orgánica se concentró para producir el bruto 190, que se purificó mediante cromatografía instantánea mediante el uso de gel de sílice de 100-200 mesh. El compuesto se eluyó en acetato de etilo al 20 % en hexano como un sólido de color semiblanco 190.
1H NMR (400 MHz, CDCls) 8 ppm 8,64 (d, 1H), 7,91 (d, 1H), 7,29 (s, 1H), 6,82 (s, 1H), 3,97 (s, 3H), 3,64 (dd, 1H), 3,43 (m, 1H), 3,13 (m, 2H), 2,74 (dd, 1H); MS-ES+337,8.
El 190 (40 mg, 0,108 mmol) se disolvió en acetona, K2CO3 (29,9 mg, 0,217 mmol) añadido, seguido de cloruro de etanosulfonilo (16,6 mg, 0,103 mmol). La reacción se agitó durante aproximadamente 12 h a 70 °C. La masa de reacción se diluyó con acetato de etilo y se lavó con agua (3 x 50 ml). La capa orgánica se concentró para producir el bruto 191, que se purificó mediante cromatografía instantánea mediante el uso de gel de sílice de 100-200 mesh. El compuesto se eluyó en acetato de etilo al 14 % en hexano como un sólido de color semiblanco 191. 1H NMR (400 MHz, CDCh) 8 ppm 8,60 (d, 1H), 7,91 (d, 1H), 7,67 (s, 1H), 7,27 (s, 1H), 6,96 (s, 1H), 3,95 (s, 3H), 3,65 (dd, 1H), 3,42 (m, 2H), 3,20 (m, 2H), 2,82 (dd, 1H), 1,57 (m, 2H), 1,20 (m, 3H); MS-ES+ 429,8.
Ejemplo 61: Etanosulfonato de 1-oxo-2-(4-((trifluorometil)tio)bencil)-2,3-dihidro-1H-inden-5-ilo (194)

A una solución de 191 (250 mg, 1,688 mmol) en 15 ml de tolueno se añadió 72 (382,5 mg, 1,858 mmol). Se añadió PTSA (641,7 mg, 3,376 mmol) a la masa de reacción, a continuación se agitó a 120 °C durante 6 h, se diluyó con acetato de etilo y se lavó con agua (3 x 25 ml). La capa orgánica se secó sobre sulfato de sodio y se concentró para producir el bruto, que se purificó mediante cromatografía instantánea mediante el uso de gel de sílice de 100-200 mesh. El compuesto se eluyó con acetato de etilo al 30 % en hexano para proporcionar un sólido de color amarillo 192.
El 192 (250 mg, 0,744 mmol) se disolvió en 25 ml de metanol, se añadieron 10 mg de Pd/C y la reacción se agitó bajo globo de H2 durante 6 h. La reacción se filtró a través de un lecho de celite y se lavó con metanol en exceso. La capa orgánica se concentró para producir el bruto, que se purificó mediante cromatografía instantánea mediante el uso de gel de sílice de 100-200 mesh. El compuesto se eluyó en acetato de etilo al 20 % en hexano como un sólido de color semiblanco 193. 1H NMR (400 MHz, CDCla) 8 ppm 7,69 (d, 1H), 7,56 (d, 2H), 7,28 (d, 2H), 6,79 (d, 2H), 5,46 (s, 1H), 3,38 (dd, 1H), 3,11 (dd, 1H), 2,97 (m, 1H), 2,70 (m, 2H); MS-ES+336,9.
El 193 (100 mg, 0,295 mmol) se disolvió en acetona y K2CO3 (62 mg, 0,442 mmol) añadido, seguido de cloruro de etanosulfonilo (42 mg, 0,324 mmol). La reacción se agitó durante aproximadamente 12 h a 70 °C. La masa de reacción se diluyó con acetato y se lavó con agua (3 x 50 ml). La capa orgánica se concentró para producir el bruto, que se purificó mediante cromatografía instantánea mediante el uso de gel de sílice de 100-200 mesh. El compuesto se eluyó en acetato de etilo al 20 % en hexano como un sólido de color semiblanco 194. 1HNMR (400 MHz, CDCla) 8 ppm 7,81 (d, 1H), 7,58 (d, 2H), 7,34 (s, 1H) 7,26 (m, 3H), 3,33 (m, 3H), 3,21 (m, 2H), 3,02 (m, 1H), 2,75 (m, 2H), 1,56 (m, 4H), 1,28 (m, 3H); MS-ES+429,0.
Ejemplo 62: Etanosulfonato de 6-metoxi-3-oxo-2-((3-(trifluorometil)piridin-2-il)metil)-2,3-dihidro-1H-inden-5-ilo (197)

A una solución de 185 (100 mg, 0,6754 mmol) en 15 ml de tolueno se añadió 66 (130,09 mg, 0,742 mmol). PTSA durante 6 h. La masa de reacción se diluyó con acetato de etilo y se lavó con agua (3 x 25 ml). La capa orgánica se secó sobre sulfato de sodio y se concentró para producir el bruto 195, que se usó en la siguiente etapa sin purificación adicional.
El 195 (100 mg) se disolvió en 25 ml de metanol, se añadieron 20 mg de Pd/C y la reacción se agitó bajo globo de H2 durante 6 h. La reacción se filtró a través de un lecho de celite y se lavó con metanol en exceso. La capa orgánica se concentró para producir el bruto 196, que se purificó mediante cromatografía instantánea mediante el uso de gel de sílice de 100-200 mesh. El compuesto 196 se eluyó en acetato de etilo al 20 % en hexano como un sólido de color semiblanco 196. 1HNMR (400 MHz, CDCls) 8 ppm 10,4 (s, 1H), 8,74 (d, 1H), 8,14 (d, 1H), 7,49 (m, 2H), 6,81 (m, 2H), 3,45 (dd, 1H), 3,18 (m, 2H), 3,01 (m, 1H), 2,77 (dd, 1H); MS-ES+ 307,8.
El 196 (40 mg, 0,1299 mmol) se disolvió en acetona, K2CO3 (35,7 mg, 0,259 mmol) añadido, seguido de cloruro de etanosulfonilo (33,1 mg, 0,259 mmol). La reacción se agitó durante aproximadamente 12 h a 70 °C. La masa de reacción se diluyó con acetato y se lavó con agua (3x50 ml). La capa orgánica se concentró para producir el bruto 197, que se purificó mediante cromatografía instantánea mediante el uso de gel de sílice de 100-200 mesh. El compuesto se eluyó en acetato de etilo al 20 % en hexano como un sólido de color semiblanco 197. 1HNMR (400 MHz, CDCla) 8 ppm 8,56 (d, 1H), 7,91 (d, 1H), 7,83 (d, 1H), 7,35 (s, 1H), 7,27 (s, 1H), 3,64 (dd, 1H), 3,41 (m, 1H), 3,31 (m, 4H), 2,88 (m, 1H), 1,57 (m, 4H), 1,23 (m 3H); MS-ES+ 399,9.
Ejemplo 63: 6-metoxi-5-(2-(trifluorometoxi)fenil)-2-((3 -(trifluorometil)piridin-2-il)metil)-2,3-dihidro-1H-inden-1-ona (213)

acetonitrilo se añadió carbonato de cesio (271 mg, 0,826 mmol). La reacción se desgasificó y se purgó con N2 durante 10 min. Se añadió Pd(dppf)Cl2 (16,88 mg, 0,02 mmol) a la reacción, a continuación se desgasificó y se purgó con nitrógeno durante otros 10 minutos. La reacción se calentó a 90 °C en condiciones selladas durante la noche. La mezcla de reacción se dejó enfriar a rt y se diluyó con cloroformo. La capa orgánica se filtró a través de un tapón de celite y se concentró para producir el bruto, que se purificó mediante cromatografía instantánea mediante el uso de gel de sílice de 100-200 mesh. El compuesto 211 se eluyó en acetato de etilo al 5 % en hexano como sólido semiblanco 211.
A una solución de 211 (150 mg, 0,465 mmol) y 66 (122 mg, 0,698 mmol) en 15 ml de tolueno se añadió PTSA (177 mg, 0,9314 mmol). La reacción se agitó a 120 °C durante 6 h, se diluyó con acetato de etilo y se lavó con agua (3 x 25 ml). La capa orgánica se secó sobre sulfato de sodio y se concentró para producir el bruto 212 , que se purificó mediante cromatografía instantánea mediante el uso de gel de sílice de 100-200 mesh. El compuesto 212 se eluyó en acetato de etilo al 10 % en hexano para proporcionar un sólido de color amarillo.
El 212 (70 mg, 0,145 mmol) se disolvió en 25 ml de metanol y se añadieron 10 mg de Pd/C. La reacción se agitó bajo globo de H2 durante 6 h, a continuación se filtró a través de lecho de celite y se lavó con metanol en exceso. La capa orgánica se concentró para producir el bruto 213 que se purificó mediante cromatografía instantánea mediante el uso de gel de sílice de 100-200 mesh. El compuesto 213 se eluyó en acetato de etilo al 12 % en hexano como un sólido de color semiblanco. 1HNMR (400 MHz, CDCls) 8 ppm 8,16 (d, 1H), 7,92 (d, 1H), 7,41 (m, 1H), 7,34 (m, 4H), 7,24 (m, 2H), 3,82 (s, 3H), 3,68 (dd, 1H), 3,49 (m, 1H), 3,26 (m, 1H), 2,79 (dd, 1H); MS-ES+ 482,0.
Ejemplo 64: 5-(2-fluoro-4-(trifluorometoxi)fenil)-6-metoxi-2-((3-(trifluorometil)piridin-2-il)metil)-2,3-dihidro-1H-inden-1-ona (216)

A una solución de 203 (100 mg, 0,414 mmol) y ácido (2-fluoro-4-(trifluorometoxi)fenil)borónico (101,8 mg, 0,456 mmol) en acetonitrilo se añadió carbonato de cesio (272,1 mg, 0,829 mmol). La reacción se desgasificó y se purgó con nitrógeno durante 10 minutos. Se añadió Pd(dppf)Ch (16,9 mg, 0,020 mmol) a la reacción, que a continuación se desgasificó y se purgó con nitrógeno durante otros 10 minutos. La reacción se calentó a 90 °C en condiciones selladas durante la noche, a continuación se dejó enfriar a rt y se diluyó con cloroformo. La capa orgánica se filtró a través de un tapón de celite y se concentró para producir el bruto 214, que se purificó mediante cromatografía instantánea mediante el uso de gel de sílice de 100-200 mesh. El compuesto se eluyó en acetato de etilo al 5 % en hexano como sólido semiblanco 214.
A una solución de 214 (110 mg, 0,322 mmol)) en 15 ml de tolueno se añadió 66 (84,9 mg, 0,483 mmol). Se añadió PTSA (122,4 mg, 0,644 mmol) a la masa de reacción, que se agitó a 120 °C durante 6 h. La masa de reacción se diluyó con acetato de etilo y se lavó con agua (3 x 25 ml). La capa orgánica se secó sobre sulfato de sodio y se concentró para producir el bruto 215, que se purificó mediante cromatografía instantánea mediante el uso de gel de sílice de 100-200 mesh. El compuesto se eluyó con acetato de etilo al 10 % en hexano para proporcionar un sólido de color amarillo 215.
El 215 (65 mg, 0,130 mmol) se disolvió en 25 ml de metanol, se añadieron 10 mg de Pd/C, y la reacción se agitó bajo globo de H2 durante 6 h, filtrado a través de lecho de celite y lavado con metanol en exceso. La capa orgánica se concentró para producir el bruto 216, que se purificó mediante cromatografía instantánea mediante el uso de gel de sílice de 100 200 mesh. El compuesto 216 se eluyó en acetato de etilo al 12 % en hexano como un sólido de color semiblanco. 1HNMR (400 MHz, CDCla) 8 ppm 8,63 (d, 1H), 7,92 (d, 1H), 7,36 (m, 2H), 7,28 (m, 2H), 7,06 (m, 2H), 3,86 (s, 3H), 3,68 (dd, 1H), 3,48 (m, 1H), 3,26 (m, 2H), 2,80 (dd, 1H); MS-ES+ 500,1.
Ejemplo 65: 6-metoxi-5-(4-metilpiperazin-1-il)-2-((3-(trifluorometil)piridin-2-il)metil)-2,3-dihidro-1H-inden-1-ona (250)

A una solución de 203 (1,0 g, 0,0041 mmol) y piperazina-1-carboxilato de terc-butilo (960 mg, 0,00518 mmol) en 15 ml de tolueno se añadió carbonato de cesio (2,69 g, 0,0082 mmol). La reacción se desgasificó y se purgó con N2 durante 10 min. Se añadieron Pd2(dba)3 (112 mg, 0,000123 mmol) y BINAP (153 mg, 0,000246 mmol) y nuevamente se desgasificaron y purgaron con nitrógeno durante otros 10 minutos. La reacción se calentó a 110 °C durante la noche en condiciones selladas, a continuación se diluyó con cloroformo y se filtró a través de lecho de celite. La capa orgánica se concentró para producir el bruto 246, que se purificó mediante cromatografía instantánea mediante el uso de gel de sílice de 100-200 mesh eluyendo el compuesto en acetato de etilo al 25 % en hexano como un sólido de color amarillo pálido 246.
A una solución de 246 (0,42 g, 0,0012 mmol) en MeOH/H2O se añadió 66 (233 mg, 0,00133 mmol) e hidróxido de sodio (96 mg, 0,0024 mmol). La reacción se agitó a RT durante 6 h, se diluyó con cloroformo y se lavó con agua (3 x 25 ml). La capa orgánica se secó sobre sulfato de sodio y se concentró para producir el bruto, que se purificó mediante cromatografía instantánea mediante el uso de gel de sílice de 100-200 mesh. El compuesto 247 se eluyó en acetato de etilo al 30 % en hexano para proporcionar un sólido de color amarillo 247.
El 247 (400 mg, 0,0009 mmol) se disolvió en 25 ml de metanol y se añadieron 40 mg de Pd/C y la reacción se agitó bajo globo de H2 durante 6 h. La reacción se filtró a través de un lecho de celite y se lavó con metanol en exceso. La capa orgánica se concentró para producir el bruto 248, que se purificó mediante cromatografía instantánea mediante el uso de gel de sílice de 100-200 mesh. El compuesto se eluyó en acetato de etilo al 20 % en hexano como un sólido de color semiblanco 248.
A una solución 248 (300 mg, 0,0059 mmol) en DCM se añadió ácido trifluoroacético 3 ml y se mantuvo en agitación durante 4 horas a RT. Después de la terminación de la reacción, el solvente se evaporó y el resto se diluyó con agua. La capa acuosa se lavó con acetato de etilo y se mantuvo a un lado. El pH de la capa acuosa se ajustó a 9-10, a continuación se extrajo con cloroformo dos veces. La capa orgánica combinada se secó sobre sulfato de sodio y se concentró para producir el bruto 249, que se tituló con hexano a 5 °C para producir un sólido de color amarillo pálido 249.
Una solución de 249 (0,15 g, 0,00037 mmol) se disolvió en DCM. Se añadió a la reacción trietilamina (56 mg, 0,00055 mmol) seguido de yoduro de metilo (50 mg, 0,00044 mmol) y se mantuvo la agitación a RT durante 4 h. Después de la terminación de la reacción, se diluyó con DCM y se lavó con agua dos veces. La capa orgánica se secó sobre sulfato de sodio y se concentró para producir el bruto 250, que se purificó mediante cromatografía ultrarrápida mediante el uso de alúmina neutra eluyendo el compuesto en MeOH al 2 % en cloroformo como un sólido espeso y pegajoso 250. 1HNMR (400 MHz, CDCla) 8 ppm 8,65 (d, 1H), 7,91 (d, 1H), 7,27 (d, 1H), 7,20 (s, 1H), 6,90 (s, 1H), 3,90 (m, 4H), 3,64 (dd, 1H), 3,42 (m, 1H), 3,03 (m, 4H), 2,65 (m, 4H), 2,36 (m, 4H); MS-ES+420,0.
Ejemplo 66: 5-(4-(2-hidroxietil)piperazin-1-il)-6-metoxi-2-((3-(trifluorometil)piridin-2-il)metil)-2,3-dihidro-1H-inden-1-ona (251)
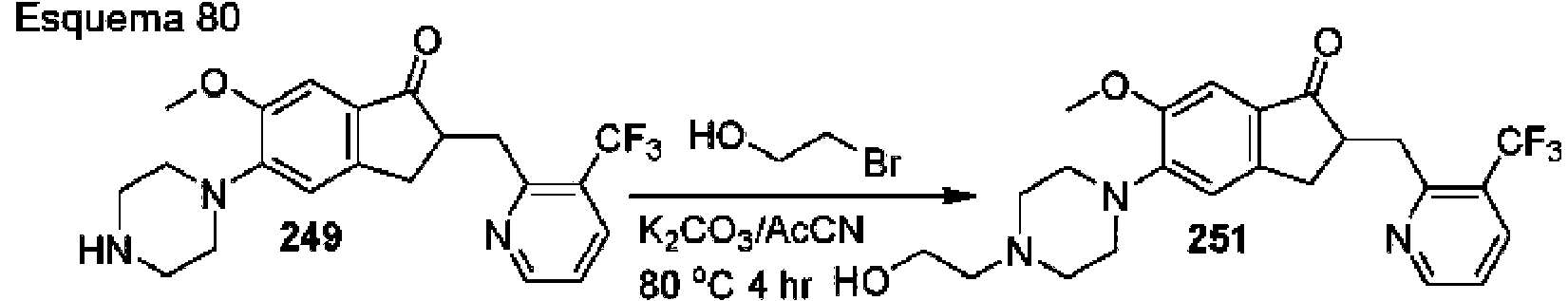
A una solución agitada de 249 (150 mg, 0,00037 mmol) en acetonitrilo se añadió carbonato de potasio (100 mg, 0,00074 mmol) y a continuación 2-bromo etanol (57 mg, 0,00046 mmol). La mezcla de reacción agitada se calentó a 80 °C durante 4 h. Después de la terminación de la reacción, se filtró a través de lecho de celite y se lavó con cloroformo. La capa orgánica se concentró para producir el bruto 251, que se purificó mediante cromatografía instantánea mediante el uso de alúmina neutra eluyendo el compuesto con metanol al 2 % en cloroformo para producir el compuesto 251. 1HNMR (400 MHz, CDCla) 8 ppm 8,65 (d, 1H), 7,91 (d, 1H), 7,20 (m, 2H), 6,84 (s, 1H), 3,89 (m, 5H), 3,66 (m, 4H), 3,43 (m, 1H), 3,12 (m, 4H), 3,03 (m, 1H), 2,72 (m, 4H), 2,63 (m, 3H); MS-ES+450,0.
Ejemplo 67: 2-((5-(1,1,1,3,3,3-hexafluoro-2-hidroxipropan-2-il)piridin-3-il)metil)-6-metoxi-5-morfolino-2,3-dihidro-1H-inden-1-ona (245)
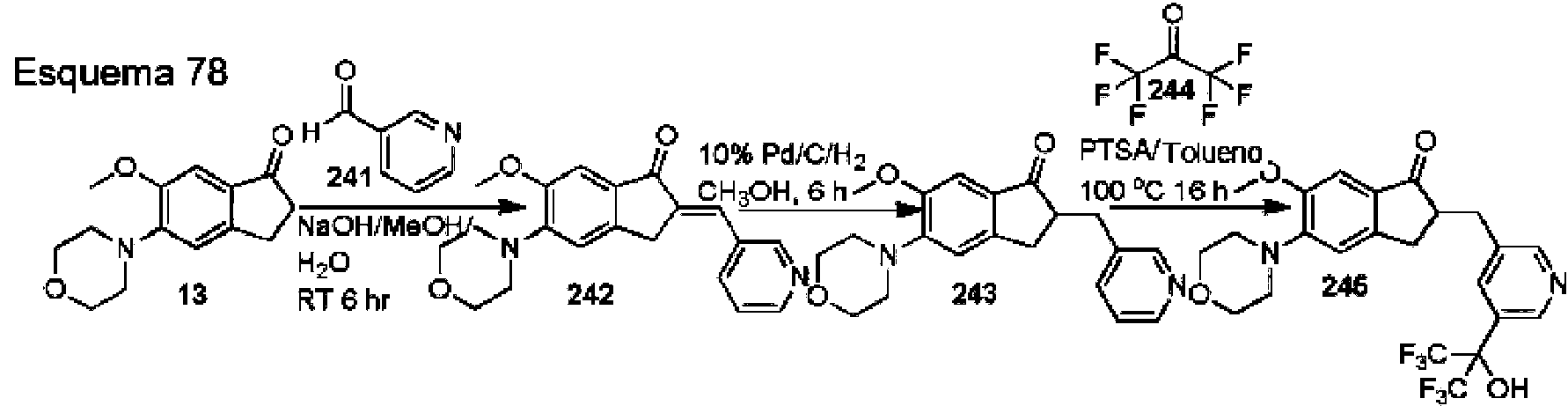
A una solución de 13 (0,1 g, 0,404 mmol) en MeOH/H2O (1:1) se añadió nicotinaldehído 241 (0,052 g, 0,484 mmol) y NaOH (0,032 g, 0,808 mmol). La reacción se agitó a RT durante 6 h, se diluyó con cloroformo y se lavó con agua (3 x 25 ml). La capa orgánica se secó sobre sulfato de sodio y se concentró para producir el bruto, que se purificó mediante cromatografía instantánea mediante el uso de gel de sílice de 100-200 mesh. El compuesto 242 se eluyó en acetato de etilo al 30 % en hexano para proporcionar un sólido de color amarillo (E)-6-metoxi-5-morfolino-2-(piridin-3-ilmetilen)-2,3-dihidro-1H-inden-1-ona 242.
El 242 (0,1 g 0,297 mmol) se disolvió en 25 ml de metanol y se añadieron 40 mg de Pd/C. La reacción se agitó bajo globo de H2 durante 6 h. La reacción se filtró a través de un lecho de celite y se lavó con metanol en exceso. La capa orgánica se concentró para producir el bruto que se purificó mediante cromatografía instantánea mediante el uso de gel de sílice de 100-200 mesh. El compuesto 243 se eluyó en acetato de etilo al 20 % en hexano como sólido de color semiblanco de 6-metoxi-5-morfolino-2-(piridin-3-ilmetil)-2,3-dihidro-1H-inden-1-ona 243. 1HNMR (400 MHz, CDCh) 8 ppm 8,47 (m, 2H), 7,56 (d, 1H), 7,22 (m, 1H), 7,17 (s, 1H), 6,84 (s, 1H), 3,89 (m, 3H), 3,87 (m, 4H), 3,30 (dd, 1H), 3,07 (m, 4H), 2,99 (m, 1H), 2,95 (m, 1H), 2,70 (m, 2H).
A una solución agitada de 243 (0,08 g, 0,236 mmol) en tolueno se añadió PTSA (0,090 g, 0,472 mmol) y se mantuvo en agitación a 120 °C. Se añadió hexafluoroacetona 244 (0,129 g, 0,590 mmol) a la reacción y se calentó a 100 °C durante 16 h. La reacción se enfrió a TA, se diluyó con agua, se extrajo con acetato de etilo dos veces, la capa orgánica se secó sobre sulfato de sodio y se concentró para producir el bruto, que se purificó mediante cromatografía instantánea mediante el uso de gel de sílice de 100-200 mesh eluyendo el compuesto en acetato de etilo al 30 % en hexano como un sólido espeso y pegajoso 2-((5-(1,1,1,3,3,3-hexafluoro-2-hidroxipropan-2-il)piridin-3-il)metil)-6-metoxi-5-morfolino-2,3-dihidro-1H-inden-1-ona 245. 1HNMR (400 MHz, CDCla) 8 ppm 7,06 (m, 2H), 6,88 (s,1H), 6,90(s, 1H), 6,123 (s, 1H), 3,88 (m, 7H), 3,49 (m, 1H), 3,08 (m, 2H), 3,03 (m, 6H), 2,29 (m, 2H).
Ejemplo 68: 2-((5-(1,1,1,3,3,3-hexafluoro-2-hidroxipropan-2-il)piridin-3-il)metil)-6-metoxi-5-(4-metilpiperazin-1-il)-2,3-dihidro-1H-inden-1-ona (256)

A una solución de 6-metoxi-5-(4-metilpiperazin-1-il)-2,3-dihidro-1H-inden-1-ona 253 (0,1 g, 0,384 mmol) en MeOH/H2O (1: 1) se añadió nicotinaldehído (0,049 g, 0,461 mmol) y se añadió NaOH (0,03 g, 0,768 mmol) a la masa de reacción, que a continuación se agitó a RT durante 6 h. La masa de reacción se diluyó con cloroformo y se lavó con agua (3 x 25 ml). La capa orgánica se secó sobre sulfato de sodio y se concentró para producir el bruto, que se purificó mediante cromatografía instantánea mediante el uso de gel de sílice de 100-200 mesh. El compuesto se eluyó en acetato de etilo al 30 % en hexano para proporcionar un sólido de color amarillo (E)-6-metoxi-5-(4-metilpiperazin-1-il)-2-(piridin-3-ilmetilen)-2,3-dihidro-1H-inden-1-ona 254.
El 254 (0,08 g, 0,229 mmol) se disolvió en 25 ml de metanol, se añadieron 20 mg de Pd/C y la reacción se agitó bajo globo de H2 durante 6 h. La reacción se filtró a través de un lecho de celite y se lavó con metanol en exceso. La capa orgánica se concentró para producir el bruto, que se purificó mediante cromatografía instantánea mediante el uso de gel de sílice de 100-200 mesh. El compuesto se eluyó en acetato de etilo al 20 % en hexano como sólido de color semiblanco de 6-metoxi-5-(4-metilpiperazin-1-il)-2-(piridin-3-ilmetil)-2,3-dihidro-1H-inden-1-ona 255.
A una solución agitada de 255 (0,05 g, 0,142 mmol) en tolueno se añadió PTSA (0,054 g, 0,284 mmol) y se mantuvo en agitación a 120 °C. Se añadió hexafluoroacetona 244 (0,078 g, 0,356 mmol) a la reacción y se mantuvo a temperatura y se agitó durante 16 h. La reacción se enfrió a continuación a RT, se diluyó con agua y se extrajo con acetato de etilo dos veces. La capa orgánica se secó sobre sulfato de sodio y se concentró para producir el bruto, que se purificó mediante cromatografía instantánea mediante el uso de gel de sílice de 100-200 mesh y eluyendo el compuesto en acetato de etilo al 30 % en hexano como un sólido espeso y pegajoso de 2-((5-(1,1,1,3,3,3-hexafluoro-2-hidroxipropan-2-il)piridin-3-il)metil)-6-metoxi-5-(4-metilpiperazin-1-il)-2,3-dihidro-1H-inden-1-ona 256.
Ejemplo 69: 5-(1,1,1,3,3,3-hexafluoro-2-hidroxipropan-2-il)nicotinaldehído (259)

A una solución agitada de 3,5-dibromopiridina 257 (1,09 g, 4,60 mmol) en THF seco enfriado a 0 °C, se añadió cloruro de isopropilmagnesio (2,4 ml, 4,8 mmol) gota a gota mientras se agitaba durante 10 minutos. Se añadió hexafluoroacetona (CF3-CO-CF3) 244 a la reacción resultante y se continuó agitando durante otras 2 h. Después de la terminación de los materiales de partida, la reacción se interrumpió con una solución saturada de cloruro de amonio. La capa orgánica se separó y la fase acuosa se extrajo nuevamente con éter dietílico, la capa orgánica combinada se secó sobre sulfato de sodio y se concentró para producir el bruto 2-(5-bromopiridin-3-il)-1,1,1,3,3,3-hexafluoropropan-2-ol 258. El bruto se purificó a través de gel de sílice mediante el uso de gel de sílice de 100-200 mesh eluyendo el compuesto en de acetato de etilo 20:5 en hexano como un aceite espeso del compuesto 258.
A una solución agitada de 258 (0,347 g, 1,07 mmol) en THF seco, enfriado a 0 °C, se añadió cloruro de isopropilmagnesio (1,2 ml, 2,4 mmol) gota a gota y se agitó durante 10 minutos seguido de la adición de DMF (0,015 ml, 1,94 mmol) a la reacción y agitado durante 2 h adicionales. Después de la terminación de la reacción, se apagó con solución de cloruro de amonio saturada. La capa orgánica se separó y la fase acuosa se extrajo nuevamente con éter dietílico, la capa orgánica combinada se secó sobre sulfato de sodio y se concentró para producir el compuesto bruto 5-(1,1,1,3,3,3-hexafluoro-2-hidroxipropan-2-il)nicotinaldehído 259 usado directamente para continuar con la siguiente etapa.
Ejemplo 70: 6-(1,1,1,3,3,3-hexafluoro-2-hidroxipropan-2-il)picolinaldehído (262)

A una solución agitada de 2,6-dibromo-piridina 260 (2,37 g, 10 mmol) en THF seco, enfriado a 0 °C, se añadió n-BuLi 1,4 mmol gota a gota en hexano (7,9 ml, 11 mmol) y se agitó durante 10 minutos. A continuación se añadió hexafluoroacetona (CF3-CO-CF3) 244 a la mezcla de reacción y se continuó agitando durante otras 2 h. Después de la terminación de la reacción, se apagó con solución de cloruro de amonio saturada. La capa orgánica se separó y la fase acuosa se extrajo nuevamente con éter dietílico, la capa orgánica combinada se secó sobre sulfato de sodio y se concentró para producir el compuesto bruto 2-(6-bromopiridin-2-il)-1,1,1,3,3,3-hexafluoropropan-2-ol 261, que se purificó a través de gel de sílice de 100-200 mesh eluyendo el compuesto en acetato de etilo al 15 % en hexano para producir el compuesto oleoso espeso 261.
A una solución agitada de 261 (0,5 g, 1,54 mmol) en THF seco, enfriado a 0 °C, se añadió n-BuLi (0,118 g, 1,85 mmol) gota a gota y se continuó agitando durante 10 minutos. A continuación se añadió DMF (0,224 g, 3,08 mmol) a la mezcla de reacción mientras se agitaba durante otras 2 h. Después de la terminación de la reacción, se apagó con solución de cloruro de amonio saturada. La capa orgánica se separó y la fase acuosa se extrajo nuevamente con éter dietílico, la capa orgánica combinada se secó sobre sulfato de sodio y se concentró para producir el compuesto bruto 6-(1,1,1,3,3,3-hexafluoro-2-hidroxipropan-2-il)picolinaldehído 262.
Ejemplo 71: 2-((5-(1,1,1,3,3,3-hexafluoro-2-hidroxipropan-2-il)piridin-3-il)metil)-6-metoxi-5-morfolino-2,3-dihidro-1H-inden-1-ona (264)

A una solución de 13 (0,1 g, 0,40 mmol)), MeOH/H2O (1:1), se añadió 5-(1,1,1,3,3,3-hexafluoro-2-hidroxipropan-2-il)nicotinaldehído 259 (0,132 g, 0,485 mmol) y NaOH (0,032 g, 0,80 mmol). La reacción se agitó a RT durante 6 h. La masa de reacción se diluyó con cloroformo y se lavó con agua (3 x 25 ml). La capa orgánica se secó sobre sulfato de sodio y se concentró para producir el bruto 263, que se purificó mediante cromatografía instantánea mediante el uso de gel de sílice de 100-200 mesh. El compuesto se eluyó en acetato de etilo al 30 % en hexano para proporcionar un sólido de color amarillo (E)-2-((5-(1,1,1,3,3,3-hexafluoro-2-hidroxipropan-2-il)piridin-3-il)metilen)-6-metoxi-5-morfolino-2,3-dihidro-1H-inden-1-ona 263.
El 263 (50 mg, 0,090 mmol) se disolvió en 25 ml de metanol, se añadieron 40 mg de Pd/C y la reacción se agitó bajo globo de H2 durante 6 h. La reacción se filtró a través de un lecho de celite y se lavó con metanol en exceso. La capa orgánica se concentró para producir el bruto, que se purificó mediante cromatografía instantánea mediante el uso de gel de sílice de 100-200 mesh. El compuesto bruto 264 se eluyó en acetato de etilo al 20 % en hexano como un sólido de color
semiblanco 2-((5-(1,1,1,3,3,3-hexafluoro-2-hidroxipropan-2-il)piridin-3-il)metil)-6-metoxi-5-morfolino-2,3-dihidro-1H-inden-1-ona 264.
Ejemplo 72: 2-((6-(1,1,1,3,3,3-hexafluoro-2-hidroxipropan-2-il)piridin-2-il)metil)-6-metoxi-5-morfolino-2,3-dihidro-1H-inden-1-ona (266)
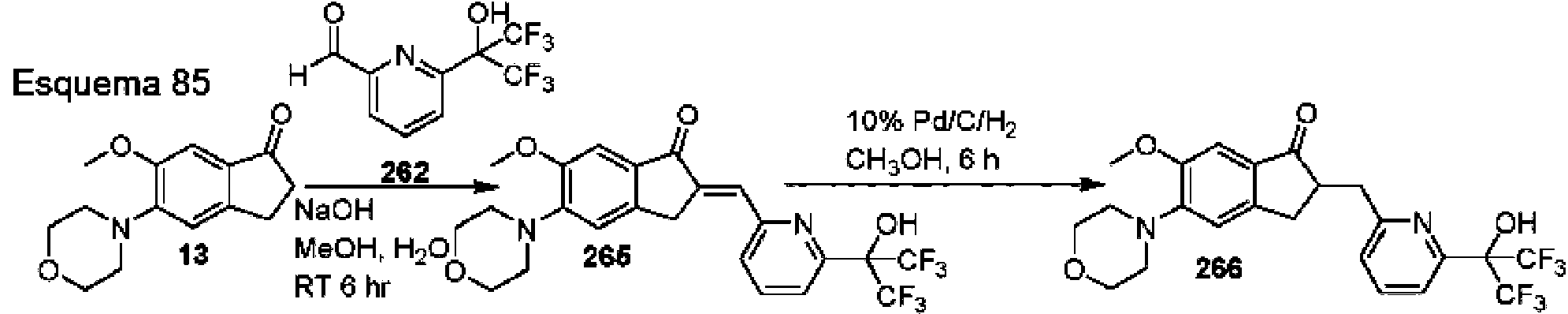
A una solución de 13 (0,1 g, 0,40 mmol)) en MeOH/H2O (1:1) se añadió 6-(1,1,1,3,3,3-hexafluoro-2-hidroxipropan-2-il)picolinaldehído 262 (0,132 g, 0,485 mmol), NaOH (0,032 g, 0,80 mmol), y la reacción se agitó a RT durante 6 h. La masa de reacción se diluyó con cloroformo y se lavó con agua (3 x 25 ml). La capa orgánica se secó sobre sulfato de sodio y se concentró para producir el bruto, que se purificó mediante cromatografía instantánea mediante el uso de gel de sílice de 100-200 mesh. El compuesto 265 se eluyó en acetato de etilo al 30 % en hexano para proporcionar un sólido de color amarillo (E)-2-((6-(1,1,1,3,3,3-hexafluoro-2-hidroxipropan-2-il)piridin-2-il)metilen)-6-metoxi-5-morfolino-2,3-dihidro-1H-inden-1-ona 265.
El 265 (50 mg 0,1 mmol) se disolvió en 25 ml de metanol, se añadieron 40 mg de Pd/C y la reacción se agitó bajo globo de H2 durante 6 h. La reacción se filtró a través de un lecho de celite y se lavó con metanol en exceso. La capa orgánica se concentró para producir el bruto 266 y se purificó mediante cromatografía instantánea mediante el uso de gel de sílice de 100-200 mesh. El compuesto 266 se eluyó en acetato de etilo al 20 % en hexano como un sólido de color semiblanco 2-((6-(1,1,1,3,3,3-hexafluoro-2-hidroxipropan-2-il)piridin-2-il)metil)-6-metoxi-5-morfolino-2,3-dihidro-1H-inden-1-ona 266.
Ejemplo 73: 2-((5-(1,1,1,3,3,3-hexafluoro-2-hidroxipropan-2-il)-3-(trifluorometil)piridin-2-il)metil)-6-metoxi-5-morfolino-2,3-dihidro-1H-inden-1-ona (268)

A una solución agitada de 267 (0,1 g, 0,245 mmol) en tolueno se añadió PTSA (0,094 g, 0,490 mmol) y se mantuvo en agitación a 120 °C durante 1 hora. Después de la adición de hexafluoroacetona 244 (0,134 g, 0,614 mmol), la reacción se calentó a 100 °C durante 16 h. Después de la terminación de la reacción, se enfrió a RT, se diluyó con agua y se extrajo con acetato de etilo dos veces. La capa orgánica se secó sobre sulfato de sodio y se concentró para producir el compuesto bruto 268, que se purificó mediante cromatografía ultrarrápida mediante el uso de gel de sílice de 100-200 mesh eluyendo el compuesto en acetato de etilo al 30 % en hexano como compuesto sólido espeso y pegajoso 2-((5-(1,1,1,3,3,3-hexafluoro-2-hidroxipropan-2-il)-3-(trifluorometil)piridin-2-il)metil)-6-metoxi-5-morfolino-2,3-dihidro-1H-inden-1-ona 268.
Ejemplo 74: 2-(2-(1,1,1,3,3,3-hexafluoro-2-hidroxipropan-2-il)-5-(trifluorometil)bencil)-6-metoxi-5-morfolino-2,3-dihidro-1H-inden-1-ona (270)

A una solución agitada de 2-(2-doro-5-(trifluorometil)bencil)-6-metoxi-5-morfolino-2,3-dihidro-1H-inden-1-ona 269 (0,1 g, 0,226 mmol) en THF seco, enfriado a 0 °C, se añadió n-BuLi (0,021 g, 0,339 mmol) gota a gota y la solución se agitó durante 10 minutos. A continuación se añadió hexafluoroacetona 244 (0,10 g, 0,453 mmol) a la reacción y se agitó durante otras 2 h. Después de la terminación de la reacción, se apagó con solución de cloruro de amonio saturada. La capa orgánica se separó y la fase acuosa se extrajo nuevamente con éter dietílico, la capa orgánica combinada se secó sobre sulfato de sodio y se concentró para producir el bruto 270, que se purificó a través de gel de sílice de 100-200 mesh eluyendo con acetato de etilo al 15 % en hexano para producir 20 mg del compuesto 2-(2-(1,1,1,3,3,3-hexafluoro-2-hidroxipropan-2-il)-5-(trifluorometil)bencil)-6-metoxi-5-morfolino-2,3-dihidro-1H-inden-1-ona 270.
Ejemplos adicionales del método de uso
Ejemplo 75:
Modelado estructural de RORYt: Los compuestos de la Fórmula I proporcionada en las TABLAS 1A y 1B fueron diseñados mediante el uso de estructuras cristalinas de RORYt. La superfamilia de receptores de hormonas nucleares RORa/Y contiene un motivo de unión de ADN de dedo de zinc tipo II característico y un bolsillo hidrófobo de unión a ligando. Tres subtipos; RORa, -p y -y (NR1F1-3 o RORA-C), se han identificado con distribuciones tisulares y función biológica únicas. En el timo, RORa y dos isoformas, Yl y Y2 (RORyt/ROR-yT), se han identificado con RORyt distinto de la isoforma RORyI porque carece del terminal amino de RORy1. Una secuencia de búsqueda ROR-Yt (NP_001001523, 1-497) en rcsb.org proporcionó las 6 plantillas homólogas de estructura cristalina de rayos X y 3B0W se selecciona para la estrategia FFDD conducen al descubrimiento de 2, 3-dihidro-1H-inden-1-ona sustituida que contiene antagonistas de RORy representados en la Figura 1 y sus análogos.
Ensayos de inhibición in vitro
Ejemplo 76:
Ensayo de unión de RORYt por TR-FRET: RORy-LBD contiene un péptido etiqueta 6-His y GST de SRC1-2 y se empleó biotinil-CPSSHSSLTERHKlLHRLLQEGSPS aminoterminal y se usó el RORy-LBD de 6-His-(GST) que se expresó y purificó de células de insecto Sf-21 infectadas con baculovirus de Invitrogen y purificado con glutatión sepharose. El protocolo de ensayo incluía 50 |jl de tampón de TR-FRET KCI 50 mM, TRIS 50 mM, Na-EDTA 1 mM, DTT 0,1 mM (pH 7,0); 10 nM de 6-His(GST)RORgamma-LBD, Anti-6-His-Eu 1 nM, 0,1 jM de péptido SRC1_2 y Strep-APC 10nM. El péptido que se une debido a las curvas de dosis de los compuestos se añadió al DMSO al 0,1 %. Se leyó fluorescencia a una longitud de onda de 340 nm en cada pocilio de placas de 94 pocilios mediante el uso de un lector de fluorescencia Envision en modo de resolución temporal después de una incubación durante la noche a 4 °C. Los datos de respuesta a la dosis y los valores de IC50 se proporcionan en la Tabla 3.
Ejemplo 77:
Efectos de los compuestos de prueba sobre la IL-17A activada por RORyt
Células HEK293 Prom/LUCPorter™: Se analizaron los efectos antagonistas (o agonistas inversos) de los compuestos de prueba sobre las células HEK 293 Prom/LUCPorter™ con IL-17A activada por RORYt y se evaluó la IC50 de cada compuesto. La línea celular HEK 293 Prom/LUCPorter™ de IL-17A (IMGENeX, IML-301) se sembró en placas blancas de 96 pocilios a 0,75 x 105 células por pocilio durante la noche. Las células se transfectaron con el plásmido de RORYt (IMGENEX, IMP-122) durante 6 h. Las células se trataron con una serie de concentraciones de 8 puntos de cada compuesto (100, 33,33, 11,11, 3,70, 1,23, 0,41, 0,14 y 0,0457 jM ) durante 16 h. La actividad de luciferasa se analizó mediante el uso del reactivo de ensayo reportero de luciferasa (IMGENEX, LS100). Los datos se analizaron mediante el uso del software Excel y Prism. El por ciento de actividad se definió como 100 x (1 - (Pocilio - Control A)/(Control B -Control A)), donde el Control A eran los pocilios que contenían células transfectadas con RORyt y el Control B eran los pocilios que contenían células no transfectadas. Ambos controles A y B fueron tratados con vehículo solo.
La evaluación de IC50 del compuesto seleccionado de la TABLA 1 sobre la actividad del promotor de IL-17A mediada por RORYt y su curva de respuesta a dosis de 10 puntos se da en la Figura 2 y 3.
Ejemplo 78:
Estimación de la citocina IL-17 por HTRF: El ensayo se diseñó para evaluar la potencia de los compuestos de prueba en la liberación de citocinas IL-17 a partir de las células T CD4+. Para ayudar en la identificación de inhibidores potentes, se determina la IC50 para los compuestos de prueba en la liberación de IL-17 a partir de células T CD4+ a través de la generación de curvas de respuesta a dosis de 10 puntos (DRC). Los métodos de ensayo usados para cuantificar la secreción de IL-17 es HTRF (fluorescencia resuelta en el tiempo homogénea). La IL-17 secretada se detecta mediante AcM anti-IL-17 marcado con XL665, el segundo AcM marcado con Criptato, que se une a epítopos específicos de IL-17 humana. Al acercarse tanto el donante como el aceptor se produce la transferencia de energía (FRET). Esta técnica se ha mejorado aún más mediante el uso de marcadores de larga duración combinados con la detección sobre una base de fluorescencia resuelta en el tiempo, lo que permite la maximización de las interferencias de fluorescencia rápidas.
El ensayo se realizó de acuerdo con las instrucciones del fabricante. Todos los reactivos se reconstituyeron de acuerdo con las instrucciones proporcionadas. Los reactivos se dispensaron en una placa de 96 pocillos de media área en el siguiente orden: 10 j l de estándar o muestra, AcMXL665 contra IL-17 y 5 j l de IL-17 Criptato. Para el control negativo, el estándar o muestra se reemplaza por 10 j l de diluyente. La placa se cubre con un sellador de placa y se incuba a rt durante 2 h. Después de la incubación, se retira el sellador de placas y se lee la placa mediante el uso del protocolo de HTRF en el lector de microplacas Envision. Todos los análisis de datos se llevaron a cabo mediante el uso de Microsoft Excel (2010) y se determinó la IC50 mediante el uso del software GraphPad Prism 4. El % de inhibición de los compuestos de prueba se determinó mediante el uso de la siguiente fórmula:
% de inhibición = 100-( 100*Recuentos de compuesto de prueba promedio - Recuentos de control negativo promedio/ (Recuentos de control positivo promedio - Recuentos de control negativo promedio)). Dosis-Los datos de respuesta a dosis y los valores de CI50 se proporcionan en la Tabla 3.
Tabla 3: Lista de compuestos que contienen 2, 3-dihidro-1H-inden-1-ona sustituida y resultados correspondientes de la actividad antagonista contra RORyt, celular y de inhibición de IL-17*



Experimento con modelos in vivo
Ejemplo 79:
Eficacia de compuestos que contienen 2, 3-dihidro-1-H-inden-1-ona sustituida seleccionados sobre la inhibición de la producción de citocina-IL-17 en células T CD4+ inducida por anticuerpos anti-CD3e en ratones BALB/c machos
Citocina IL-17 en ratones in vivo mediante el método de ELISA: Se realizó un análisis cuantitativo de ELISA tipo sándwich de IL-17 de ratón a partir de las muestras de suero recolectadas (kit ELISA Ready-DuoSet ELISA de IL-17 de ratón, de R&D Systems, Estados Unidos). Las muestras de suero probadas se recolectaron de ratones después de 1,5 h de desafío de anticuerpos anti-CD3e, antes del desafío de anticuerpos, los ratones se trataron con diferentes dosis de compuestos de prueba o dexametasona como se mencionó anteriormente en el diseño experimental. El anticuerpo de captura se diluyó en tampón de recubrimiento (IX PBS) y se transfirió a cada pocillo. La placa se selló y se incubó durante la noche a RT. Al día siguiente, los pocillos de la placa se aspiraron y se lavaron (3 veces), permitiendo un tiempo de remojo de un minuto en cada etapa. La placa se bloqueó mediante el uso de BSA al 1 % en PBS, se selló y se incubó en condiciones ambientales durante no menos de una hora. Después del período de incubación, la placa se lavó como se describió anteriormente, y los estándares y las muestras de suero se añadieron a los pocillos apropiados y se incubaron durante dos horas a RT. Las muestras se diluyeron en una relación de 1:10 con BSA al 1 % en Pb S (diluyente reactivo). Después de dos horas, los pocillos se lavaron como se mencionó anteriormente tres veces y se añadió el anticuerpo de detección y se incubó durante dos horas. Después del lavado (como se mencionó anteriormente), la enzima HRP se añade e incuba durante 20 minutos y se lava nuevamente y se añade sustrato de acuerdo con las instrucciones del fabricante y se incuba durante 30 minutos/hasta que se desarrolle un color azul. La placa se leyó a 450/570 nm mediante el uso del lector de absorbancia de microplacas (SpectraMax; M3). Estadísticas: Se realizó un ANOVA unidireccional seguido de la prueba
de comparación múltiple de Dunnett. El porcentaje de inhibición de la producción de citocinas se calculó para todos los grupos en comparación con el control positivo.
Producción de IL-17: La administración intravenosa de 5 pg/animal de anticuerpo anti-CD3e a cada ratón dio como resultado una elevación significativa de la producción de lL-17 en el punto de tiempo de 1,5 h en comparación con el grupo de control negativo. La administración oral de 10 mg/kg de los compuestos de prueba, 20 minutos antes de la administración de anticuerpos dio como resultado un 50 % respectivamente. El tratamiento con dexametasona mostró una reducción del 94 %, respectivamente, en la producción de IL-17 en comparación con el grupo de control positivo. Esto se muestra en la Figura 4.
Ejemplo 80:
Experimentos farmacocinéticos: La biodisponibilidad y farmacocinética de algunos compuestos de la presente invención se examinaron en ratas macho Sprague Dawley. Se usaron un total de 6 ratas macho en el estudio. El estudio se realizó mediante el uso de diseño paralelo (n=3) con muestreo en serie.
Las formulaciones de dosis se prepararon el día de la dosificación. Las muestras de sangre se recolectaron a los 0,083 (solo IV), 0,25, 0,5, 1, 2, 4, 8 y 24 h después de la dosificación. En cada punto de tiempo, se extrajeron aproximadamente 0,2 ml de sangre de cada rata canulada a través de la vena yugular y se transfirieron a un tubo de microcentrífuga previamente marcado que contenía 20 pl de K2EDTA de 200 mm por ml de sangre. Después de la recolección de la muestra de sangre, se purgó un volumen igual de solución salina heparinizada en la vena yugular de la rata. Las muestras de sangre se centrifugaron a 5000 g durante 5 minutos a 4 ± 2 °C. El plasma se separó dentro de los 30 minutos del tiempo programado y se almacenó por debajo de -60 °C hasta el bioanálisis. Las muestras de plasma se analizaron para los EJEMPLOS de prueba seleccionados mediante el uso de un método de detección por espectrometría de masas en tándem cromatográfico líquido (LC-MS/MS) con un límite inferior de cuantificación de 2,21 ng/ml. Los parámetros farmacocinéticos para los EJEMPLOS seleccionados se calcularon mediante el uso de la herramienta de análisis no compartimental del software WinNonlin® validado (Versión 5.2).
Los parámetros farmacocinéticos (media ± DE; n = 3) de 7 después de la administración intravenoso en bolo y por sonda oral de 7 soluciones en ratas Sprague Dawley macho se muestran en la TABLA 4 más abajo:
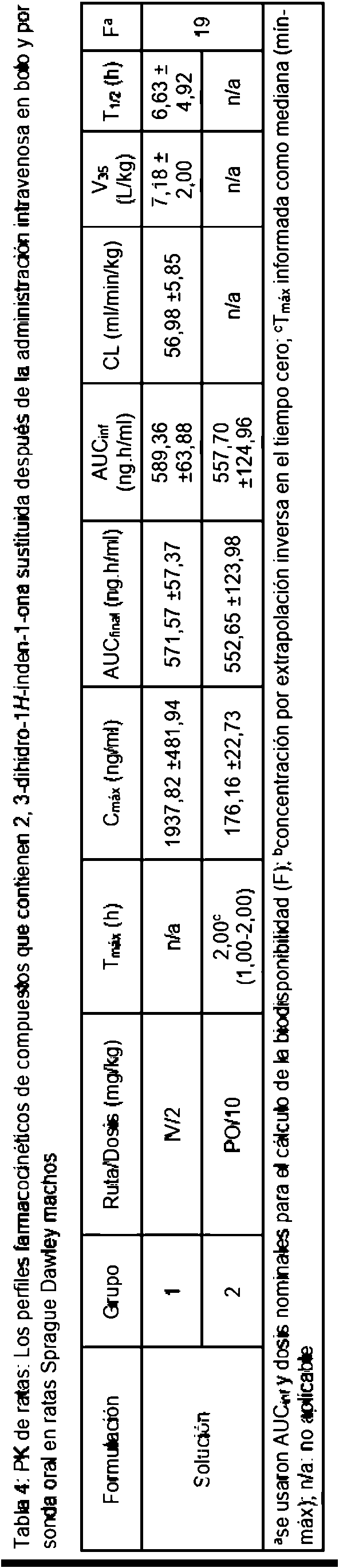
Ejemplo 81: Evaluación de la potencia de los compuestos de prueba sobre la liberación de IL-17 de las células T CD4+: Se ha establecido un ensayo de HTRF basado en células in vitro robusto para la activación y estimulación de linfocitos T CD4+ para la diferenciación de células T auxiliares 17 (Th17) y la producción de IL-17. Los compuestos de prueba son inhibidores selectivos de moléculas pequeñas de RORYt, se encontró que inhiben la producción de IL-17, con una IC50 de 220 nM a 1,2 pM. La cuantificación del ensayo se realizó mediante el uso de un Dex estándar y se informó que SB203589 inhibe la IL-17, con una IC50 de 14 y 37 nM, respectivamente. Estas series de compuestos inhiben de forma dependiente de dosis el conjunto de células T CD4+y agotan la IL-17 liberada de los esplenocitos de ratón.
Ejemplo 82: Eficacia de los análogos de la clase dihidro-1H-inden-1-ona en EAE inducida por MOG35-55 en ratones C57BL/6 hembras: Para establecer la validación del concepto in vivo en el modelo de EAE crónica por MOG de ratón, los análogos principales de la clase dihidro-1H-inden-1-ona se administraron por vía oral QD durante 28 días como un tratamiento profiláctico donde la gravedad de la enfermedad se controló con éxito con los análogos de la clase de dihidro-1H-inden-1-ona y Fingolimod control (Figura 5). Las puntuaciones acumulativas de EAE (Escala: 0-3,5) de 0,2, 0,0, 0,4, 0. 7. 0,1 en los días 9-18 (p=0,001) llegaron a 0 en el día 18 hasta el día 28 (p=0,001) con los análogos de la clase de dihidro-1H-inden-1-ona y puntuaciones similares se observaron para Fingolimod también (Figura 6). Los resultados son sorprendentes para este estudio, mientras que el control positivo (ratones enfermos) tuvo una puntuación de EAE de >3,0 (>95 % de incidencia de la enfermedad). Los ratones tratados con la clase dihidro-1H-inden-1-ona toleraron bien el estudio con condiciones corporales óptimas y no tuvieron signos de cambios en la química clínica o eventos de toxicidad observados durante todo el estudio. Todos los ratones en los grupos de tratamiento tienen pesos corporales similares a los del grupo tratado con vehículo. El tratamiento con los análogos de la clase de dihidro-1-H-inden-1-ona de este estudio proporcionó una evidencia convincente de su actividad antagonista contra RORYt, que si se traslada a la eficacia celular en un sistema in vivo apoya aún más el uso de esta serie de compuestos para la e M y otras indicaciones reivindicadas.
Referencias
1. Kipp, M. van der Valk, P. Amor, S. (2012). Pathology of multiple sclerosis. CNS Neurol Disord Drug Targets 11(5): 506 517. PMID: 12858058.
2. Kieseier, B. C. y H. P. Hartung (2003). Multiple paradigm shifts in multiple sclerosis. Curr Opin Neurol 16(3): 247-252. PMID: 22583433.
3. Steinman, L. (2013). Multiple sclerosis: a coordinated immunological attack against myelin in the central nervous system. Cell 85, 299-302. PMID: 8616884
4. Pottgen, J. Dziobek, I. Reh, S. Heesen, C. Gold, S. M. (2013). Impaired social cognition in multiple sclerosis." JNeurol Neurosurg Psychiatry 84(5): 523-528. PMID: 23315621.
5. Kallaur, A. P. Oliveira, S. R. Colado Simao, A. N. Delicato de Almeida, E. R. Kaminami Morimoto, H. Lopes, J. de Carvalho Jennings Pereira, W. L. Marques Andrade, R. Muliterno Pelegrino, L. Donizete Borelli, S. Kaimen-Maciel, D. R. Reiche, E. M. 2013). Cytokine profile in relapsing remitting multiple sclerosis patients and the association between progression and activity of the disease. Mol Med Rep 7(3): 1010-1020. PMID: 23292766
6. Glass, C. K. y K. Saijo (2010). Nuclear receptor transrepression pathways that regulate inflammation in macrophages and T cells. Nat Rev Immunol 10(5): 365-376. PMID: 20414208
7. Lopez-Diego, R. S. y H. L. Weiner (2008). Novel therapeutic strategies for multiple sclerosis--a multifaceted adversary. Nat Rev Drug Discov 7(11): 909-925. PMID: 18974749.
8. Ali, R. Nicholas, R. S. Muraro, P. A. (2013). Drugs in development for relapsing multiple sclerosis. Drugs 73(7): 625 650. PMID: 23609782
9. The MS Disease-Modifying Medications, General Information. 2013 Multiple Sclerosis Society and mscoalition.org/EmergingTherapies.
10. Vermersch, P. Benrabah, R. Schmidt, N. Zephir, H. Clavelou, P. Vongsouthi, C. Dubreuil, P. Moussy, A. Hermine, O. (2012). Masitinib treatment in patients with progressive multiple sclerosis: a randomized pilot study. BMC Neurol 12: 36. PMID:22691628
11. Ivanov,, II, McKenzie, B. S. Zhou, L. Tadokoro, C. E. Lepelley, A. Lafaille, J. J. Cua, D. J. Littman, D. R. (2006). The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell 126(6): 1121-1133. PMID: 16990136
12. Wang, Y. Kumar, N. Solt, L. A. Richardson, T. I. Helvering, L. M. Crumbley, C. Garcia-Ordonez, R. D. Stayrook, K. R. Zhang, X. Novick, S. Chalmers, M. J. Griffin, P. R. Burris, T. P. (2010). Modulation of retinoic acid receptor-related orphan receptor alpha and gamma activity by 7-oxygenated sterol ligands. Journal of Biological Chemistry 285(7): 5013-5025. PMID: 19965867
13. Kryczek, I, Wei, S, Zou, L, Altuwaijri, S, Szeliga, W, Kolls, J, Chang, A, Zou, W. (2007). Cutting edge: Th17 and regulatory T cell dynamics and the regulation by IL-2 in the tumor microenvironment. J. Immunol. 178, 6730-6733. PMID: 17513719
14. Hu, Y. Shen, F. Crellin, N. K. Ouyang, W. (2011). The IL-17 pathway as a major therapeutic target in autoimmune diseases. Ann N Y Acad Sci 1217: 60-76. PMID: 21155836
15. Tesmer, L. A. Lundy, S. K. Sarkar, S. Fox, D. A. (2008). Th17 cells in human disease. Immunol Rev 223: 87-113. PMID: 18613831
16. Rauen, T. Juang, Y. T. Hedrich, C. M. Kis-Toth, K. Tsokos, G. C. (2012). A novel isoform of the orphan receptor RORgammat suppresses IL-17 production in human T cells. Genes Immun 13(4): 346-350. PMID: 22237416
17. Kamenecka, T. M, Lyda, B, Ra, M, Chang, M. R, Griffin, P.R. (2013) Synthetic modulators of the retinoic acid receptorrelated orphan receptors. Med. Chem. Commun. 4, 764-776
18. Fragment-Field Drug Design (FFDD) (2011), Proprietary empirical driven fragment fields, Arrien Pharmaceuticals, USA.
19. www.dinicaltrials.gov/multiple sclerosis
20. (a) Kumar, N. Lyda, B. Chang, M. R. Lauer, J. L. Solt, L. A. Burris, T. P. Kamenecka, T. M. Griffin, P. R. (2012). Identification of SR2211: a potent synthetic RORgamma-selective modulator. ACS Chem Biol 7(4): 672-677. PMID: 22292739. (b) Kumar, N. Solt, L. A. Conkright, J. J. Wang, Y. Istrate, M. A. Busby, S. A. Garcia-Ordonez, R. D. Burris, T. P. Griffin, P. R. (2010). The benzenesulfoamide T0901317 [N-(2,2,2-trifluoroethyl)-N-[4-[2,2,2-trifluoro-1-hydroxy-1-(trifluoromethyl)ethyl]phenyl] benzenesulfonamide] is a novel retinoic acid receptor-related orphan receptor-alpha/gamma inverse agonist. Molecular Pharmacology 77(2): 228-236. PMID: 19887649. (c) Khan, P. M. El-Gendy Bel, D. Kumar, N. Garcia-Ordonez, R. Lin, L. Ruiz, C. H. Cameron, M. D. Griffin, P. R. Kamenecka, T. M. (2013). Small molecule amides as potent ROR-gamma selective modulators. Bioorganic & Medicinal Chemistry Letters 23(2): 532-536. PMID: 23232056 (d) Huh, J.R, Englund, E.E, Wang, H, Huang, R, Huang, P, Rastinejad, F, Inglese, J, Austin, C.P, Ronald L. Johnson, Huang, W, Littman, D. R. (2013) Identification of Potent and Selective Diphenylpropanamide RORy Inhibitors. ACS Med. Chem. Lett. 4 (1), 79-84.
21. Sheridan, C. (2013). Footrace to clinic heats up for T-cell nuclear receptor inhibitors. Nature Biotechnology 31(5): 370. PMID: 23657373
22. Solt, L. A., Kumar, N., Nuhant, P., Wang, Y., Lauer, J. L., Liu, J., Istrate, M. A., Kamenecka, T. M., Roush, W. R., Vidovic, D., Schurer, S. C., Xu, J., Wagoner, G., Drew, P. D., Griffin, P. R., y Burris, T. P. (2011) Suppression of TH17 differentiation and autoimmunity by a synthetic ROR ligand. Nature 472, 491-494. PMID: 21499262
23. Ransohoff, R. M. (2012). Animal models of multiple sclerosis: the good, the bad and the bottom line. Nat Neurosci 15(8): 1074-1077. PMID: 22837037
24. (a) Jin, L. Martynowski, D. Zheng, S. Wada, T. Xie, W. Li, Y. (2010). Structural basis for hydroxycholesterols as natural ligands of orphan nuclear receptor RORgamma. Mol Endocrinol 24(5): 923-929.
25. Fujita-Sato, S., Ito, S., Isobe, T., Ohyama, T., Wakabayashi, K., Morishita, K., Ando, O., e Isono, F. (2011) Structural basis of digoxin that antagonizes RORgamma t receptor activity and suppresses Th17 cell differentiation and interleukin (IL)-17 production. J. Biol. Chem. 286, 31409-31417. PMID: 21733845
26. Xiao S, Yosef N, Yang J, Wang Y, Zhou L, Zhu C, Wu C, Baloglu E, Schmidt D, Ramesh R, Lobera M, Sundrud M. S, Tsai P. Y, Xiang Z, Wang J, Xu Y, Lin X, Kretschmer K, Rahl P. B, Young R. A, Zhong Z, Hafler D. A, Regev A, Ghosh S, Marson A, Kuchroo V. K. (2014) Small-molecule RORyt antagonists inhibit T helper 17 cell transcriptional network by divergent mechanisms. Immunity. 40 (4): 477-489. PMiD: 24745332
27. Wingerchuk D. M, Carter J. L. (2014) Multiple sclerosis: current and emerging disease-modifying therapies and treatment strategies. Mayo Clin Proc. 89 (2):225-240. PMID: 24485135
28. Zhang W, Zhang J, Fang L, Zhou L, Wang S, Xiang Z, Li Y, Wisely B, Zhang G, An G, Wang Y, Leung S, Zhong Z. (2012) Increasing human Th17 differentiation through activation of orphan nuclear receptor retinoid acid-related orphan receptor y (RORy) by a class of aryl amide compounds. Mol Pharmacol. 82 (4):583-590. PMID: 22700697
29. Jacob S. L, Daniel J. Cua. (2014) The Emerging Landscape of RORyt Biology. Immunity, 40 (4), 17, 451-452.
30. Skepner J, Ramesh R, Trocha M, Schmidt D, Baloglu E, Lobera M, Carlson T, Hill J, Orband-Miller LA, Barnes A, Boudjelal M, Sundrud M, Ghosh S, Yang J. (2014) Pharmacologic inhibition of RORyt regulates Th17 signature gene expression and suppresses cutaneous inflammation in vivo. J Immunol.192(6):2564-2575. PMID: 24516292
31. Isono F, Fujita-Sato S, Ito S. (2014) Inhibiting RORYt/Th17 axis for autoimmune disorders. Drug Discov Today. 19 (8): 1205-1211. PMID: 24792721
Claims (1)
- REIVINDICACIONESUn compuesto de acuerdo con la Fórmula I:
 o una sal aceptable farmacéuticamente de este, en donde:X es O;R1 es
o una sal aceptable farmacéuticamente de este, en donde:X es O;R1 es oR1 es alquilo C1-4 opcionalmente sustituido con 1-6 sustituyentes halo independientes; oR1 es fenilo, piridinilo o pirazolilo, cada uno opcionalmente sustituido con 1-5 sustituyentes halo, trifluorometilo, (trifluorometil)tio, trifluorometoxi, 1,1,1,3,3,3,-hexafluoro-2-hidroxipropan-2-ilo
oR1 es alquilo C1-4 opcionalmente sustituido con 1-6 sustituyentes halo independientes; oR1 es fenilo, piridinilo o pirazolilo, cada uno opcionalmente sustituido con 1-5 sustituyentes halo, trifluorometilo, (trifluorometil)tio, trifluorometoxi, 1,1,1,3,3,3,-hexafluoro-2-hidroxipropan-2-ilo o
o independientes;m es 0 o 1;la línea punteada indica un doble enlace opcional cuando m=1;R2 es halo, -OH, -CN, -OCH3, -OS(O)2CH2CHs,
independientes;m es 0 o 1;la línea punteada indica un doble enlace opcional cuando m=1;R2 es halo, -OH, -CN, -OCH3, -OS(O)2CH2CHs,

 o fenilo opcionalmente sustituido con 0-5 sustituyentes halo, trifluorometoxi independientes;R3 es
o fenilo opcionalmente sustituido con 0-5 sustituyentes halo, trifluorometoxi independientes;R3 es R4 es H, OH, OCH3, o -OS(O)2CH2CH3;Un compuesto de acuerdo con la Fórmula Ia:
R4 es H, OH, OCH3, o -OS(O)2CH2CH3;Un compuesto de acuerdo con la Fórmula Ia: o una sal aceptable farmacéuticamente de este, en donde:X es O o S;R1a es alquilo C1-4 opcionalmente sustituido con 1-6 sustituyentes halo independientes; o un fenilo, piridinilo o pirazolilo, cada uno opcionalmente sustituido con 1-5 sustituyentes halo, trifluorometilo, (trifluorometil)tio;o
o una sal aceptable farmacéuticamente de este, en donde:X es O o S;R1a es alquilo C1-4 opcionalmente sustituido con 1-6 sustituyentes halo independientes; o un fenilo, piridinilo o pirazolilo, cada uno opcionalmente sustituido con 1-5 sustituyentes halo, trifluorometilo, (trifluorometil)tio;o independientes; o es
independientes; o es la línea punteada indica un doble enlace opcional;X1 es C o N; y X2 se selecciona de C, N u O; en donde al menos uno de X1 y X2 no es C;R5 está ausente, halo, hidrógeno o alquilo C1-4; yR6 es halo, hidrógeno, o alquilo C1-4.3. El compuesto de acuerdo con la reivindicación 2, o una sal farmacéuticamente aceptable de este, en donde R1a es alquilo C1-4 opcionalmente sustituido con 1-6 sustituyentes halo independientes.4. El compuesto de acuerdo con la reivindicación 2, o una sal farmacéuticamente aceptable de este, en donde R1a es fenilo, opcionalmente sustituido con 1-5 sustituyentes halo, trifluorometilo, (trifluorometil)tio,
la línea punteada indica un doble enlace opcional;X1 es C o N; y X2 se selecciona de C, N u O; en donde al menos uno de X1 y X2 no es C;R5 está ausente, halo, hidrógeno o alquilo C1-4; yR6 es halo, hidrógeno, o alquilo C1-4.3. El compuesto de acuerdo con la reivindicación 2, o una sal farmacéuticamente aceptable de este, en donde R1a es alquilo C1-4 opcionalmente sustituido con 1-6 sustituyentes halo independientes.4. El compuesto de acuerdo con la reivindicación 2, o una sal farmacéuticamente aceptable de este, en donde R1a es fenilo, opcionalmente sustituido con 1-5 sustituyentes halo, trifluorometilo, (trifluorometil)tio, independientes.5. El compuesto de acuerdo con la reivindicación 2, o una sal farmacéuticamente aceptable de este, en donde R1a es piridinilo, opcionalmente sustituido con 1-5 sustituyentes halo, trifluorometilo o (trifluorometil)tio,
independientes.5. El compuesto de acuerdo con la reivindicación 2, o una sal farmacéuticamente aceptable de este, en donde R1a es piridinilo, opcionalmente sustituido con 1-5 sustituyentes halo, trifluorometilo o (trifluorometil)tio, independientes.6. El compuesto de acuerdo con la reivindicación 2, o una sal farmacéuticamente aceptable de este, en donde R1a es pirazolilo, opcionalmente sustituido con 1-5 sustituyentes halo, trifluorometilo, (trifluorometil)tio independientes,
independientes.6. El compuesto de acuerdo con la reivindicación 2, o una sal farmacéuticamente aceptable de este, en donde R1a es pirazolilo, opcionalmente sustituido con 1-5 sustituyentes halo, trifluorometilo, (trifluorometil)tio independientes, independientes.7. El compuesto, de acuerdo con la reivindicación 2 , en donde el compuesto de acuerdo con la fórmula Ia se selecciona de:
independientes.7. El compuesto, de acuerdo con la reivindicación 2 , en donde el compuesto de acuerdo con la fórmula Ia se selecciona de:

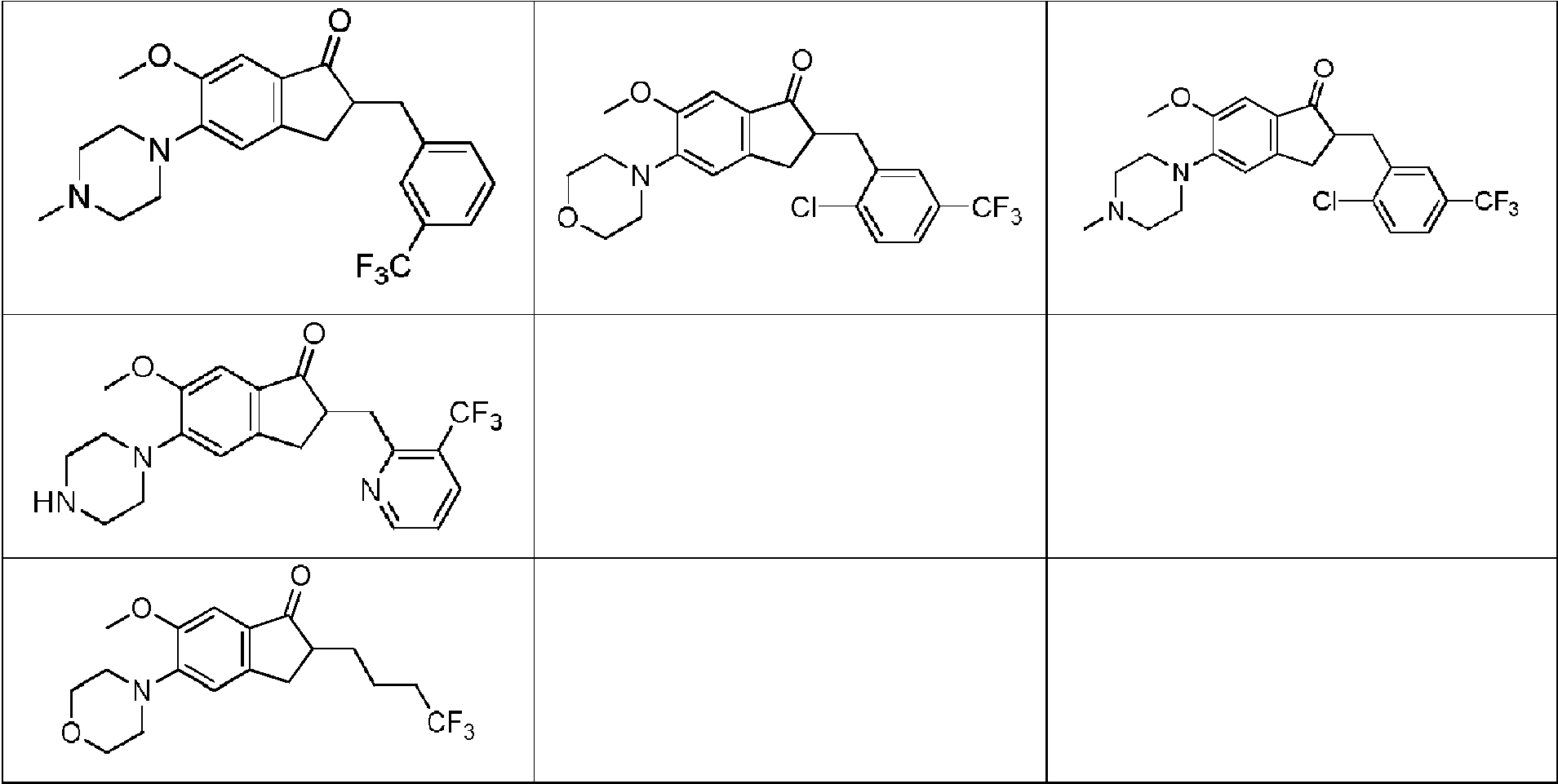 o una sal farmacéuticamente aceptable de estos.8. El compuesto, de acuerdo con la reivindicación 2, en donde el compuesto de acuerdo con la fórmula la se selecciona de:
o una sal farmacéuticamente aceptable de estos.8. El compuesto, de acuerdo con la reivindicación 2, en donde el compuesto de acuerdo con la fórmula la se selecciona de: o una sal farmacéuticamente aceptable de este.9. Un compuesto seleccionado del grupo que consiste en:
o una sal farmacéuticamente aceptable de este.9. Un compuesto seleccionado del grupo que consiste en:


 na sal farmacéuticamente aceptable de estos.compuesto como se definió en cualquiera de las reivindicaciones 1 a 9, o una sal farmacéuticamente este, para usar en terapia.11. Un compuesto como se definió en cualquiera de las reivindicaciones 1 a 9, o una sal farmacéuticamente aceptable de este, para usar en el tratamiento de una enfermedad autoinmunitaria o cáncer.12. El compuesto, o la sal farmacéuticamente aceptable de este, para usar de acuerdo con la reivindicación 11, en donde la enfermedad autoinmunitaria es esclerosis múltiple (EM) o psoriasis.13. El compuesto, o una sal farmacéuticamente aceptable de este, para usar de acuerdo con la reivindicación 11, en donde el cáncer es cáncer gástrico, cáncer de colon, leucemia mielogénica crónica (LMC), leucemia mielogénica aguda (LMA), carcinomas de células escamosas o de vejiga, meduloblastoma, hepatocelular carcinoma, mieloma múltiple, cáncer de vejiga, glioblastoma multiforme (GBM), cáncer de mama y de ovario, sarcoma de Ewing o enfermedades asociadas al cáncer de huesos.14. Una composición farmacéutica que comprende un compuesto como se define en cualquiera de las reivindicaciones 1 a 9, o una sal farmacéuticamente aceptable de este, y un excipiente farmacéuticamente aceptable.15. La composición farmacéutica de acuerdo con la reivindicación 14, en donde el compuesto es:
na sal farmacéuticamente aceptable de estos.compuesto como se definió en cualquiera de las reivindicaciones 1 a 9, o una sal farmacéuticamente este, para usar en terapia.11. Un compuesto como se definió en cualquiera de las reivindicaciones 1 a 9, o una sal farmacéuticamente aceptable de este, para usar en el tratamiento de una enfermedad autoinmunitaria o cáncer.12. El compuesto, o la sal farmacéuticamente aceptable de este, para usar de acuerdo con la reivindicación 11, en donde la enfermedad autoinmunitaria es esclerosis múltiple (EM) o psoriasis.13. El compuesto, o una sal farmacéuticamente aceptable de este, para usar de acuerdo con la reivindicación 11, en donde el cáncer es cáncer gástrico, cáncer de colon, leucemia mielogénica crónica (LMC), leucemia mielogénica aguda (LMA), carcinomas de células escamosas o de vejiga, meduloblastoma, hepatocelular carcinoma, mieloma múltiple, cáncer de vejiga, glioblastoma multiforme (GBM), cáncer de mama y de ovario, sarcoma de Ewing o enfermedades asociadas al cáncer de huesos.14. Una composición farmacéutica que comprende un compuesto como se define en cualquiera de las reivindicaciones 1 a 9, o una sal farmacéuticamente aceptable de este, y un excipiente farmacéuticamente aceptable.15. La composición farmacéutica de acuerdo con la reivindicación 14, en donde el compuesto es: o una sal farmacéuticamente aceptable de este.
o una sal farmacéuticamente aceptable de este.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201361876099P | 2013-09-10 | 2013-09-10 | |
| PCT/US2014/053227 WO2015038350A2 (en) | 2013-09-10 | 2014-08-28 | Substituted 2, 3-dihydro-1h-inden-1-one retinoic acid-related orphan nuclear receptor antagonists for treating multiple sclerosis |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2788660T3 true ES2788660T3 (es) | 2020-10-22 |
Family
ID=52626170
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES14844892T Active ES2788660T3 (es) | 2013-09-10 | 2014-08-28 | Derivados de 2,3-dihidro-1h-inden-1-ona como antagonistas de receptores huérfanos relacionados con el ácido retinoico gamma (ROR gamma) para el tratamiento de la esclerosis múltiple |
Country Status (17)
| Country | Link |
|---|---|
| US (4) | US9359315B2 (es) |
| EP (2) | EP3044223B1 (es) |
| JP (2) | JP6437560B2 (es) |
| AU (1) | AU2014318178B2 (es) |
| CA (1) | CA2916419C (es) |
| CY (1) | CY1123133T1 (es) |
| DK (1) | DK3044223T3 (es) |
| ES (1) | ES2788660T3 (es) |
| HR (1) | HRP20200686T1 (es) |
| HU (1) | HUE051458T2 (es) |
| LT (1) | LT3044223T (es) |
| PL (1) | PL3044223T3 (es) |
| PT (1) | PT3044223T (es) |
| RS (1) | RS60342B1 (es) |
| SI (1) | SI3044223T1 (es) |
| SM (1) | SMT202000310T1 (es) |
| WO (1) | WO2015038350A2 (es) |
Families Citing this family (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9359315B2 (en) * | 2013-09-10 | 2016-06-07 | Arrien Pharmaceuticals Llc | Substituted 2,3-dihydro-1H-inden-1-one retinoic acid-related orphan nuclear receptor antagonists for treating multiple sclerosis |
| AR110481A1 (es) | 2016-12-05 | 2019-04-03 | Lead Pharma Holding Bv | MODULADORES DE ROR g (RORg) |
| US11447444B1 (en) | 2019-01-18 | 2022-09-20 | Centrexion Therapeutics Corporation | Capsaicinoid prodrug compounds and their use in treating medical conditions |
| US20220202811A1 (en) * | 2019-02-20 | 2022-06-30 | The Regents Of The University Of California | Treatment for retinoic acid receptor-related orphan receptor Ɣ (rorƔ)-dependent cancers |
| WO2023232870A1 (en) | 2022-05-31 | 2023-12-07 | Immunic Ag | Rorg/rorgt modulators for the treatment of virus infections like covid-19 |
| WO2024012534A1 (zh) * | 2022-07-13 | 2024-01-18 | 武汉人福创新药物研发中心有限公司 | 杂环并苯环类化合物及其制备方法和用途 |
| WO2025122714A1 (en) * | 2023-12-06 | 2025-06-12 | University Of Virginia Patent Foundation | Haloperidol derivatives, pharmaceutical composition comprising said derivatives, and therapeutic use thereof |
| CN119912357B (zh) * | 2025-01-07 | 2026-02-06 | 广西民族大学 | α-对氨基亚苄基-5,6-二甲氧基-1-茚酮类衍生物及其制备方法与应用 |
Family Cites Families (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| FI95572C (fi) * | 1987-06-22 | 1996-02-26 | Eisai Co Ltd | Menetelmä lääkeaineena käyttökelpoisen piperidiinijohdannaisten tai sen farmaseuttisen suolan valmistamiseksi |
| JP2777159B2 (ja) * | 1988-12-22 | 1998-07-16 | エーザイ株式会社 | 環状アミン誘導体を含有する医薬 |
| DE4439822A1 (de) * | 1994-11-08 | 1996-08-29 | Bayer Ag | Verfahren zur Herstellung von Benzyl-piperidylmethyl-indanonen |
| EP1047674B1 (en) | 1998-01-16 | 2005-03-30 | Eisai Co., Ltd. | Process for production of donepezil derivatives |
| JP4331913B2 (ja) * | 2000-02-14 | 2009-09-16 | メルク エンド カムパニー インコーポレーテッド | エストロゲンレセプターモデュレーター |
| EP1311272B1 (en) * | 2000-03-03 | 2006-11-22 | Eisai Co., Ltd. | Novel methods using cholinesterase inhibitors |
| WO2003061658A1 (en) * | 2002-01-22 | 2003-07-31 | Eisai Co., Ltd. | Sigma receptor binder containing indanone derivative |
| CA2585655A1 (en) * | 2004-11-01 | 2006-05-11 | Merck & Co., Inc. | Synthesis of 1,5-disubstituted-2-hydroxy-gibbatetraen-6-ones |
| AR051597A1 (es) * | 2004-11-01 | 2007-01-24 | Merck & Co Inc | Moduladores de los receptores de estrogeno |
| HU227474B1 (en) | 2005-12-20 | 2011-07-28 | Richter Gedeon Nyrt | Process for industrial scale production of high purity donepezil hydrochloride polymorph i. |
| WO2008057309A2 (en) * | 2006-11-02 | 2008-05-15 | Merck & Co., Inc. | Estrogen receptor modulators |
| WO2012158784A2 (en) * | 2011-05-16 | 2012-11-22 | Theodore Mark Kamenecka | Modulators of the nuclear hormone receptor ror |
| US9359315B2 (en) * | 2013-09-10 | 2016-06-07 | Arrien Pharmaceuticals Llc | Substituted 2,3-dihydro-1H-inden-1-one retinoic acid-related orphan nuclear receptor antagonists for treating multiple sclerosis |
-
2014
- 2014-08-27 US US14/469,900 patent/US9359315B2/en active Active
- 2014-08-28 LT LTEP14844892.1T patent/LT3044223T/lt unknown
- 2014-08-28 SI SI201431560T patent/SI3044223T1/sl unknown
- 2014-08-28 JP JP2016542000A patent/JP6437560B2/ja active Active
- 2014-08-28 CA CA2916419A patent/CA2916419C/en active Active
- 2014-08-28 EP EP14844892.1A patent/EP3044223B1/en active Active
- 2014-08-28 AU AU2014318178A patent/AU2014318178B2/en active Active
- 2014-08-28 PL PL14844892T patent/PL3044223T3/pl unknown
- 2014-08-28 HU HUE14844892A patent/HUE051458T2/hu unknown
- 2014-08-28 PT PT148448921T patent/PT3044223T/pt unknown
- 2014-08-28 DK DK14844892.1T patent/DK3044223T3/da active
- 2014-08-28 ES ES14844892T patent/ES2788660T3/es active Active
- 2014-08-28 EP EP20158172.5A patent/EP3680237A1/en active Pending
- 2014-08-28 RS RS20200523A patent/RS60342B1/sr unknown
- 2014-08-28 SM SM20200310T patent/SMT202000310T1/it unknown
- 2014-08-28 WO PCT/US2014/053227 patent/WO2015038350A2/en not_active Ceased
- 2014-08-28 HR HRP20200686TT patent/HRP20200686T1/hr unknown
-
2016
- 2016-03-31 US US15/087,811 patent/US10172866B2/en active Active
- 2016-03-31 US US15/087,807 patent/US9446047B2/en active Active
-
2018
- 2018-06-15 JP JP2018114836A patent/JP6574289B2/ja active Active
-
2019
- 2019-01-04 US US16/239,869 patent/US10682358B2/en active Active
-
2020
- 2020-05-06 CY CY20201100413T patent/CY1123133T1/el unknown
Also Published As
| Publication number | Publication date |
|---|---|
| HUE051458T2 (hu) | 2021-03-01 |
| US20150072980A1 (en) | 2015-03-12 |
| SI3044223T1 (sl) | 2020-08-31 |
| PL3044223T3 (pl) | 2020-09-21 |
| JP6437560B2 (ja) | 2018-12-12 |
| HRP20200686T1 (hr) | 2020-10-02 |
| US9359315B2 (en) | 2016-06-07 |
| EP3044223A4 (en) | 2017-04-26 |
| LT3044223T (lt) | 2020-07-10 |
| EP3680237A1 (en) | 2020-07-15 |
| WO2015038350A2 (en) | 2015-03-19 |
| US10172866B2 (en) | 2019-01-08 |
| JP2018184408A (ja) | 2018-11-22 |
| US20160213676A1 (en) | 2016-07-28 |
| CY1123133T1 (el) | 2021-10-29 |
| PT3044223T (pt) | 2020-05-06 |
| JP2016531146A (ja) | 2016-10-06 |
| DK3044223T3 (da) | 2020-05-04 |
| JP6574289B2 (ja) | 2019-09-11 |
| WO2015038350A3 (en) | 2015-05-28 |
| RS60342B1 (sr) | 2020-07-31 |
| US10682358B2 (en) | 2020-06-16 |
| US20160213627A1 (en) | 2016-07-28 |
| US9446047B2 (en) | 2016-09-20 |
| EP3044223B1 (en) | 2020-04-01 |
| CA2916419A1 (en) | 2015-03-19 |
| US20190209574A1 (en) | 2019-07-11 |
| CA2916419C (en) | 2019-06-18 |
| AU2014318178A8 (en) | 2016-01-14 |
| EP3044223A2 (en) | 2016-07-20 |
| SMT202000310T1 (it) | 2020-07-08 |
| AU2014318178A1 (en) | 2015-03-19 |
| AU2014318178B2 (en) | 2016-09-08 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2788660T3 (es) | Derivados de 2,3-dihidro-1h-inden-1-ona como antagonistas de receptores huérfanos relacionados con el ácido retinoico gamma (ROR gamma) para el tratamiento de la esclerosis múltiple | |
| US11254681B2 (en) | Optionally fused heterocyclyl-substituted derivatives of pyrimidine useful for the treatment of inflammatory, metabolic, oncologic and autoimmune diseases | |
| ES2816382T3 (es) | Compuestos de alquinilbenceno heterocíclicos, y composiciones médicas y usos de los mismos | |
| CA2974853C (en) | 4h-pyrrolo[3,2-c]pyridin-4-one derivatives | |
| CA3204823A1 (en) | Prmts inhibitors | |
| US7557112B2 (en) | Aromatic-ring-fused pyrimidine derivative | |
| CN112312904A (zh) | 螺环化合物 | |
| CN107922431A (zh) | Hpk1抑制剂及其使用方法 | |
| CA3176325A1 (en) | Halogenated-heteroaryl and other heterocyclic kinase inhibitors, and uses thereof | |
| WO2021060453A1 (ja) | 架橋型光学活性2級アミン誘導体 | |
| CA2885476A1 (en) | Urea and amide derivatives of aminoalkylpiperazines and use thereof | |
| CN108463222B (zh) | 用于治疗疾病的杂环化合物 | |
| WO2018187294A1 (en) | Pyrimido-pyridazinone compound combinations, methods, kits and formulations thereof | |
| TW591031B (en) | Thienopyridine derivatives, their production and use | |
| ES2953139T3 (es) | Inhibidores de secreción de proteínas basados en triaciclododecansulfonamida ("TCD") | |
| Class et al. | Patent application title: Substituted 2, 3-dihydro-1H-inden-1-one Retinoic acid-related orphan nuclear receptor Antagonists for Treating Multiple Sclerosis | |
| RU2795572C2 (ru) | Соединения и композиции для ингибирования ire1 | |
| WO2023001045A9 (zh) | 一种外用消炎偶联化合物药物及其制法和应用 | |
| CN121627720A (zh) | 大环化合物的制备及其应用 |