ES2802412T3 - Compuestos de sulfonamida inversa a base de sulfuro, alquilo y piridilo para el tratamiento del VHB - Google Patents
Compuestos de sulfonamida inversa a base de sulfuro, alquilo y piridilo para el tratamiento del VHB Download PDFInfo
- Publication number
- ES2802412T3 ES2802412T3 ES15864386T ES15864386T ES2802412T3 ES 2802412 T3 ES2802412 T3 ES 2802412T3 ES 15864386 T ES15864386 T ES 15864386T ES 15864386 T ES15864386 T ES 15864386T ES 2802412 T3 ES2802412 T3 ES 2802412T3
- Authority
- ES
- Spain
- Prior art keywords
- mmol
- compound
- alkyl
- cycloalkyl
- fluorophenyl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- QXUXCFWMAXGLJA-UHFFFAOYSA-N CC(C)(C)c1ccc(CSNc2nccc(C(Nc(cc3Cl)ccc3F)=O)c2)cc1 Chemical compound CC(C)(C)c1ccc(CSNc2nccc(C(Nc(cc3Cl)ccc3F)=O)c2)cc1 QXUXCFWMAXGLJA-UHFFFAOYSA-N 0.000 description 1
- PCLNDBWBEMQCAO-UHFFFAOYSA-N CC(C)(CC1)CCC1Sc1cc(C(Nc(cc2)cc(Cl)c2F)=O)ccc1F Chemical compound CC(C)(CC1)CCC1Sc1cc(C(Nc(cc2)cc(Cl)c2F)=O)ccc1F PCLNDBWBEMQCAO-UHFFFAOYSA-N 0.000 description 1
- KDCKIAYAAQYERU-UHFFFAOYSA-N CCC(C1)(CC1Sc1cc(C(Nc(cc2Cl)ccc2F)=O)ccc1F)C(O)=O Chemical compound CCC(C1)(CC1Sc1cc(C(Nc(cc2Cl)ccc2F)=O)ccc1F)C(O)=O KDCKIAYAAQYERU-UHFFFAOYSA-N 0.000 description 1
- DLERJUHVAMJNHY-UHFFFAOYSA-N CCc1ccc(CSNc2cc(C(Nc(cc3Cl)ccc3F)=O)ccn2)cc1 Chemical compound CCc1ccc(CSNc2cc(C(Nc(cc3Cl)ccc3F)=O)ccn2)cc1 DLERJUHVAMJNHY-UHFFFAOYSA-N 0.000 description 1
- SLBBNPIFAHQVTB-UHFFFAOYSA-N COc(ccc(C(Nc(cc1F)ccc1F)=O)c1)c1SC(CC1)CCC1O Chemical compound COc(ccc(C(Nc(cc1F)ccc1F)=O)c1)c1SC(CC1)CCC1O SLBBNPIFAHQVTB-UHFFFAOYSA-N 0.000 description 1
- HLEIUDJVVVLVFV-UHFFFAOYSA-N CS(NC(C(CC1)CCC1C(SC1C=C(CC(Nc(cc2F)cc(F)c2F)=O)C=CC1F)=N)=O)(=O)=O Chemical compound CS(NC(C(CC1)CCC1C(SC1C=C(CC(Nc(cc2F)cc(F)c2F)=O)C=CC1F)=N)=O)(=O)=O HLEIUDJVVVLVFV-UHFFFAOYSA-N 0.000 description 1
- BKBJEDQVEUIDOG-UHFFFAOYSA-N CS(Nc1nccc(C(Nc(cc2Cl)ccc2F)=O)c1)(=O)=O Chemical compound CS(Nc1nccc(C(Nc(cc2Cl)ccc2F)=O)c1)(=O)=O BKBJEDQVEUIDOG-UHFFFAOYSA-N 0.000 description 1
- FQYBBTLAMSZAEI-UHFFFAOYSA-N Cc(c(SC(CC1)CCC1O)c1)ccc1C(Nc(cc1Cl)ccc1F)=O Chemical compound Cc(c(SC(CC1)CCC1O)c1)ccc1C(Nc(cc1Cl)ccc1F)=O FQYBBTLAMSZAEI-UHFFFAOYSA-N 0.000 description 1
- MWXJGVDMTVXASE-UHFFFAOYSA-N Cc(c(SC(CC1)CCC1O)c1)ccc1C(Nc(cc1F)ccc1F)=O Chemical compound Cc(c(SC(CC1)CCC1O)c1)ccc1C(Nc(cc1F)ccc1F)=O MWXJGVDMTVXASE-UHFFFAOYSA-N 0.000 description 1
- GQABQHOMFCRJHK-UHFFFAOYSA-N O=C(c1cc(F)nc(NS(c2ccccc2)(=O)=O)c1)Nc(cc1Cl)ccc1F Chemical compound O=C(c1cc(F)nc(NS(c2ccccc2)(=O)=O)c1)Nc(cc1Cl)ccc1F GQABQHOMFCRJHK-UHFFFAOYSA-N 0.000 description 1
- YWJVDYFFLQZOLF-UHFFFAOYSA-N O=C(c1cc(SCC2CCCCC2)ccc1)Nc(cc1Cl)ccc1F Chemical compound O=C(c1cc(SCC2CCCCC2)ccc1)Nc(cc1Cl)ccc1F YWJVDYFFLQZOLF-UHFFFAOYSA-N 0.000 description 1
- OOVUFWNYJJEABN-UHFFFAOYSA-N O=C(c1cccc(NS(N(CC2)CCC2OCc2ccccc2)(=O)=O)c1)Nc(cc1Cl)ccc1F Chemical compound O=C(c1cccc(NS(N(CC2)CCC2OCc2ccccc2)(=O)=O)c1)Nc(cc1Cl)ccc1F OOVUFWNYJJEABN-UHFFFAOYSA-N 0.000 description 1
- FOXHQTZPYWVEEB-UHFFFAOYSA-N O=C(c1ccnc(NS(C2CC2)(=O)=O)c1)Nc(cc1F)ccc1F Chemical compound O=C(c1ccnc(NS(C2CC2)(=O)=O)c1)Nc(cc1F)ccc1F FOXHQTZPYWVEEB-UHFFFAOYSA-N 0.000 description 1
- XLFFJJMORJWJHC-UHFFFAOYSA-N O=C(c1ccnc(NS(CC2OCCCC2)(=O)=O)c1)Nc(cc1Cl)ccc1F Chemical compound O=C(c1ccnc(NS(CC2OCCCC2)(=O)=O)c1)Nc(cc1Cl)ccc1F XLFFJJMORJWJHC-UHFFFAOYSA-N 0.000 description 1
- BUYIOVZGQCXDAA-UHFFFAOYSA-N O=C(c1ccnc(NSCCc2ncccc2)c1)Nc(cc1Cl)ccc1F Chemical compound O=C(c1ccnc(NSCCc2ncccc2)c1)Nc(cc1Cl)ccc1F BUYIOVZGQCXDAA-UHFFFAOYSA-N 0.000 description 1
- MVLYQRSPNREREG-UHFFFAOYSA-N OC(CC(CC1)CCC1Sc1cc(C(Nc(cc2Cl)ccc2F)=O)ccc1F)=O Chemical compound OC(CC(CC1)CCC1Sc1cc(C(Nc(cc2Cl)ccc2F)=O)ccc1F)=O MVLYQRSPNREREG-UHFFFAOYSA-N 0.000 description 1
- KLPDBIPBAHBHQW-NVAANYKZSA-N OC(c(cc1SC(CCC2)C[C@H]2C(O)=O)ccc1F)Nc(cc1F)cc(F)c1F Chemical compound OC(c(cc1SC(CCC2)C[C@H]2C(O)=O)ccc1F)Nc(cc1F)cc(F)c1F KLPDBIPBAHBHQW-NVAANYKZSA-N 0.000 description 1
- BTQLXKZVYQWYRB-UHFFFAOYSA-N OCCCSc1cc(C(Nc(cc2F)cc(F)c2F)=O)ccc1F Chemical compound OCCCSc1cc(C(Nc(cc2F)cc(F)c2F)=O)ccc1F BTQLXKZVYQWYRB-UHFFFAOYSA-N 0.000 description 1
- WQSFCIQQHNJBJR-UHFFFAOYSA-N OCCSc1cc(C(Nc(cc2Cl)ccc2F)=O)ccc1 Chemical compound OCCSc1cc(C(Nc(cc2Cl)ccc2F)=O)ccc1 WQSFCIQQHNJBJR-UHFFFAOYSA-N 0.000 description 1
- BIHGTHYHFQVPTF-KWCCSABGSA-N O[C@H](CCCC1)CC1Sc1cc(C(Nc(cc2F)cc(F)c2F)=O)ccc1F Chemical compound O[C@H](CCCC1)CC1Sc1cc(C(Nc(cc2F)cc(F)c2F)=O)ccc1F BIHGTHYHFQVPTF-KWCCSABGSA-N 0.000 description 1
- BIHGTHYHFQVPTF-KGLIPLIRSA-N O[C@H](CCCC1)C[C@H]1Sc1cc(C(Nc(cc2F)cc(F)c2F)=O)ccc1F Chemical compound O[C@H](CCCC1)C[C@H]1Sc1cc(C(Nc(cc2F)cc(F)c2F)=O)ccc1F BIHGTHYHFQVPTF-KGLIPLIRSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C321/00—Thiols, sulfides, hydropolysulfides or polysulfides
- C07C321/24—Thiols, sulfides, hydropolysulfides, or polysulfides having thio groups bound to carbon atoms of six-membered aromatic rings
- C07C321/28—Sulfides, hydropolysulfides, or polysulfides having thio groups bound to carbon atoms of six-membered aromatic rings
- C07C321/30—Sulfides having the sulfur atom of at least one thio group bound to two carbon atoms of six-membered aromatic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/185—Acids; Anhydrides, halides or salts thereof, e.g. sulfur acids, imidic, hydrazonic or hydroximic acids
- A61K31/19—Carboxylic acids, e.g. valproic acid
- A61K31/192—Carboxylic acids, e.g. valproic acid having aromatic groups, e.g. sulindac, 2-aryl-propionic acids, ethacrynic acid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/16—Amides, e.g. hydroxamic acids
- A61K31/165—Amides, e.g. hydroxamic acids having aromatic rings, e.g. colchicine, atenolol, progabide
- A61K31/167—Amides, e.g. hydroxamic acids having aromatic rings, e.g. colchicine, atenolol, progabide having the nitrogen of a carboxamide group directly attached to the aromatic ring, e.g. lidocaine, paracetamol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/16—Amides, e.g. hydroxamic acids
- A61K31/18—Sulfonamides
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/335—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin
- A61K31/34—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin having five-membered rings with one oxygen as the only ring hetero atom, e.g. isosorbide
- A61K31/341—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin having five-membered rings with one oxygen as the only ring hetero atom, e.g. isosorbide not condensed with another ring, e.g. ranitidine, furosemide, bufetolol, muscarine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/38—Heterocyclic compounds having sulfur as a ring hetero atom
- A61K31/381—Heterocyclic compounds having sulfur as a ring hetero atom having five-membered rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/397—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having four-membered rings, e.g. azetidine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/415—1,2-Diazoles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/445—Non condensed piperidines, e.g. piperocaine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/445—Non condensed piperidines, e.g. piperocaine
- A61K31/4453—Non condensed piperidines, e.g. piperocaine only substituted in position 1, e.g. propipocaine, diperodon
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/535—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines
- A61K31/5375—1,4-Oxazines, e.g. morpholine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/16—Drugs for disorders of the alimentary tract or the digestive system for liver or gallbladder disorders, e.g. hepatoprotective agents, cholagogues, litholytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/20—Antivirals for DNA viruses
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C311/00—Amides of sulfonic acids, i.e. compounds having singly-bound oxygen atoms of sulfo groups replaced by nitrogen atoms, not being part of nitro or nitroso groups
- C07C311/01—Sulfonamides having sulfur atoms of sulfonamide groups bound to acyclic carbon atoms
- C07C311/02—Sulfonamides having sulfur atoms of sulfonamide groups bound to acyclic carbon atoms of an acyclic saturated carbon skeleton
- C07C311/03—Sulfonamides having sulfur atoms of sulfonamide groups bound to acyclic carbon atoms of an acyclic saturated carbon skeleton having the nitrogen atoms of the sulfonamide groups bound to hydrogen atoms or to acyclic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C311/00—Amides of sulfonic acids, i.e. compounds having singly-bound oxygen atoms of sulfo groups replaced by nitrogen atoms, not being part of nitro or nitroso groups
- C07C311/22—Sulfonamides, the carbon skeleton of the acid part being further substituted by singly-bound oxygen atoms
- C07C311/23—Sulfonamides, the carbon skeleton of the acid part being further substituted by singly-bound oxygen atoms having the sulfur atoms of the sulfonamide groups bound to acyclic carbon atoms
- C07C311/24—Sulfonamides, the carbon skeleton of the acid part being further substituted by singly-bound oxygen atoms having the sulfur atoms of the sulfonamide groups bound to acyclic carbon atoms of an acyclic saturated carbon skeleton
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C311/00—Amides of sulfonic acids, i.e. compounds having singly-bound oxygen atoms of sulfo groups replaced by nitrogen atoms, not being part of nitro or nitroso groups
- C07C311/22—Sulfonamides, the carbon skeleton of the acid part being further substituted by singly-bound oxygen atoms
- C07C311/29—Sulfonamides, the carbon skeleton of the acid part being further substituted by singly-bound oxygen atoms having the sulfur atom of at least one of the sulfonamide groups bound to a carbon atom of a six-membered aromatic ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C321/00—Thiols, sulfides, hydropolysulfides or polysulfides
- C07C321/24—Thiols, sulfides, hydropolysulfides, or polysulfides having thio groups bound to carbon atoms of six-membered aromatic rings
- C07C321/28—Sulfides, hydropolysulfides, or polysulfides having thio groups bound to carbon atoms of six-membered aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C323/00—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups
- C07C323/50—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups containing thio groups and carboxyl groups bound to the same carbon skeleton
- C07C323/62—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups containing thio groups and carboxyl groups bound to the same carbon skeleton having the sulfur atom of at least one of the thio groups bound to a carbon atom of a six-membered aromatic ring of the carbon skeleton
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D211/00—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings
- C07D211/04—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D211/06—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D211/36—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D211/40—Oxygen atoms
- C07D211/44—Oxygen atoms attached in position 4
- C07D211/46—Oxygen atoms attached in position 4 having a hydrogen atom as the second substituent in position 4
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/60—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D213/78—Carbon atoms having three bonds to hetero atoms, with at the most one bond to halogen, e.g. ester or nitrile radicals
- C07D213/81—Amides; Imides
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D231/00—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings
- C07D231/02—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings
- C07D231/10—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D231/14—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D231/18—One oxygen or sulfur atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D295/00—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms
- C07D295/16—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms acylated on ring nitrogen atoms
- C07D295/18—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms acylated on ring nitrogen atoms by radicals derived from carboxylic acids, or sulfur or nitrogen analogues thereof
- C07D295/182—Radicals derived from carboxylic acids
- C07D295/185—Radicals derived from carboxylic acids from aliphatic carboxylic acids
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D305/00—Heterocyclic compounds containing four-membered rings having one oxygen atom as the only ring hetero atoms
- C07D305/02—Heterocyclic compounds containing four-membered rings having one oxygen atom as the only ring hetero atoms not condensed with other rings
- C07D305/04—Heterocyclic compounds containing four-membered rings having one oxygen atom as the only ring hetero atoms not condensed with other rings having no double bonds between ring members or between ring members and non-ring members
- C07D305/06—Heterocyclic compounds containing four-membered rings having one oxygen atom as the only ring hetero atoms not condensed with other rings having no double bonds between ring members or between ring members and non-ring members with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to the ring atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D307/00—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom
- C07D307/02—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom not condensed with other rings
- C07D307/04—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom not condensed with other rings having no double bonds between ring members or between ring members and non-ring members
- C07D307/10—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom not condensed with other rings having no double bonds between ring members or between ring members and non-ring members with substituted hydrocarbon radicals attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D307/00—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom
- C07D307/02—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom not condensed with other rings
- C07D307/04—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom not condensed with other rings having no double bonds between ring members or between ring members and non-ring members
- C07D307/18—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom not condensed with other rings having no double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D309/00—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only ring hetero atom, not condensed with other rings
- C07D309/02—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only ring hetero atom, not condensed with other rings having no double bonds between ring members or between ring members and non-ring members
- C07D309/04—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only ring hetero atom, not condensed with other rings having no double bonds between ring members or between ring members and non-ring members with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D333/00—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom
- C07D333/02—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings
- C07D333/04—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings not substituted on the ring sulphur atom
- C07D333/26—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings not substituted on the ring sulphur atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D333/00—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom
- C07D333/02—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings
- C07D333/04—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings not substituted on the ring sulphur atom
- C07D333/26—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings not substituted on the ring sulphur atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D333/30—Hetero atoms other than halogen
- C07D333/34—Sulfur atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D333/00—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom
- C07D333/02—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings
- C07D333/46—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings substituted on the ring sulfur atom
- C07D333/48—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings substituted on the ring sulfur atom by oxygen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/12—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2601/00—Systems containing only non-condensed rings
- C07C2601/02—Systems containing only non-condensed rings with a three-membered ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2601/00—Systems containing only non-condensed rings
- C07C2601/04—Systems containing only non-condensed rings with a four-membered ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2601/00—Systems containing only non-condensed rings
- C07C2601/06—Systems containing only non-condensed rings with a five-membered ring
- C07C2601/08—Systems containing only non-condensed rings with a five-membered ring the ring being saturated
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2601/00—Systems containing only non-condensed rings
- C07C2601/12—Systems containing only non-condensed rings with a six-membered ring
- C07C2601/14—The ring being saturated
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2601/00—Systems containing only non-condensed rings
- C07C2601/18—Systems containing only non-condensed rings with a ring being at least seven-membered
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2602/00—Systems containing two condensed rings
- C07C2602/02—Systems containing two condensed rings the rings having only two atoms in common
- C07C2602/14—All rings being cycloaliphatic
- C07C2602/18—All rings being cycloaliphatic the ring system containing six carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2602/00—Systems containing two condensed rings
- C07C2602/02—Systems containing two condensed rings the rings having only two atoms in common
- C07C2602/14—All rings being cycloaliphatic
- C07C2602/20—All rings being cycloaliphatic the ring system containing seven carbon atoms
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Epidemiology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Virology (AREA)
- Communicable Diseases (AREA)
- Oncology (AREA)
- Engineering & Computer Science (AREA)
- Pain & Pain Management (AREA)
- Biotechnology (AREA)
- Gastroenterology & Hepatology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Molecular Biology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Plural Heterocyclic Compounds (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Furan Compounds (AREA)
- Epoxy Compounds (AREA)
- Pyridine Compounds (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Hydrogenated Pyridines (AREA)
- Pyrane Compounds (AREA)
Abstract
compuesto de Fórmula IIIa: **(Ver fórmula)** o una sal farmacéuticamente aceptable del mismo; en la que cada R1 se selecciona independientemente de H, halo y alquilo de C1-6; R2 se selecciona de halo, OH, CN, -alquilo de C1-6, -alcoxi de C1-6, halo-(alquilo de C1-6), di-halo-(alquilo de C1-6) y tri-halo-(alquilo de C1-6); R3 es halo; y R4 se selecciona de -alquilo de C1-6, -heteroalquilo de C1-6, -cicloalquilo de C3-7, -heterocicloalquilo de C3-7, (alquilo de C1-6)-(cicloalquilo de C3-7), -(cicloalquilo de C3-7)-(alquilo de C1-6), cicloalquilo de C7-8 con puente, - (alquilo de C1-6)-C(O)O-(alquilo de C1-6) y -(alquilo de C1-6)-(heterocicloalquilo de C3-7), cada uno de los cuales puede estar sustituido con 0, 1 o 2 grupos seleccionados independientemente de (=O), -OH, -alquilo de C1-6, - cicloalquilo de C3-7, halo-(alquilo de C1-6), di-halo-(alquilo de C1-6), tri-halo-(alquilo de C1-6), C(O)OH, fenilo y halo; o R4 se selecciona de (cicloalquilo de C3-7)-C(O)R5 y -(cicloalquilo de C7-8 con puente)-C(O)R5, y R5 se selecciona de N(CH3)2, NS(O)2CH3, y heterocicloalquilo de C3-7, en los que: "alquilo", por sí mismo o como parte de otro sustituyente, significa, a menos que se señale de otro modo, un hidrocarburo que tiene el número de átomos de carbono designado (es decir, C1-6 significa uno a seis átomos de carbono), e incluye grupos sustituyentes de cadena lineal, ramificada, o cíclicos; y "cicloalquilo" se refiere a un radical no aromático monocíclico o policíclico, en el que cada uno de los átomos que forman el anillo (es decir, átomos esqueléticos) es un átomo de carbono, y/o se refiere a grupos "carbociclilo no aromático insaturado" o "carbociclilo insaturado no aromático", opcionalmente en el que el grupo cicloalquilo comprende un anillo aromático condensado.
Description
DESCRIPCIÓN
Compuestos de sulfonamida inversa a base de sulfuro, alquilo y piridilo para el tratamiento del VHB SOLICITUDES RELACIONADAS
Esta solicitud reivindica la prioridad de la Solicitud de Patente Provisional U.S. n° 62/086.323, presentada el 2 de diciembre de 2014, y de la Solicitud de Patente Provisional U.S. n° 62/097.854, presentada el 30 de diciembre de 2014.
ANTECEDENTES DE LA INVENCIÓN
La infección por el virus de la hepatitis B crónica (VHB) es un problema de salud global significativo, que afecta a alrededor del 5% de la población mundial (alrededor de 350 millones de personas en todo el mundo y 1,25 millones de personas en los Estados Unidos de América).
A pesar de la disponibilidad de una vacuna profiláctica contra el VHB, la carga de la infección crónica por el VHB sigue siendo un problema médico mundial insatisfecho importante, debido a las opciones de tratamiento subóptimas y las tasas sostenidas de nuevas infecciones en la mayor parte del mundo en desarrollo. Los tratamientos actuales no proporcionan una cura, y se limitan a solo dos clases de agentes (interferón y análogos/inhibidores de nucleósidos de la polimerasa viral); los problemas de resistencia a los fármacos, baja eficacia y tolerabilidad limitan su impacto. Las bajas tasas de curación del VHB se atribuyen al menos en parte a la presencia y persistencia de ADN circular covalentemente cerrado (ADNccc) en el núcleo de los hepatocitos infectados. Sin embargo, la supresión persistente del ADN del VHB ralentiza la progresión de la enfermedad hepática y ayuda a prevenir el carcinoma hepatocelular. Los objetivos actuales de la terapia para pacientes infectados con VHB están dirigidos a reducir el ADN del VHB en suero a niveles bajos o indetectables, y en última instancia, a reducir o prevenir el desarrollo de cirrosis y carcinoma hepatocelular.
Existe la necesidad en la técnica de nuevos agentes terapéuticos que traten, mejoren o prevengan la infección por VHB. La administración de estos agentes terapéuticos a un paciente infectado con VHB, ya sea como monoterapia o en combinación con otros tratamientos contra el VHB o tratamientos auxiliares, conducirá a un pronóstico significativamente mejorado, una progresión disminuida de la enfermedad, o tasas de seroconversión mejoradas. El documento WO2014106019 tiene un resumen, que señala “la presente invención se refiere a nuevos compuestos y nuevos métodos de uso de dichos compuestos de fórmula (I), útiles como inhibidores de ensamblaje de la nucleocápside para el tratamiento de virus, especialmente, pero no exclusivamente, incluyendo inhibidores de la encapsidación de ARN pregenómico del VHB para el tratamiento de la infección por el virus de la hepatitis B (VHB) y afecciones relacionadas. (Fórmula (I)) Incluyendo hidratos, solvatos, sales farmacéuticamente aceptables, profármacos y complejos de los mismos, en la que A, R1, R2, R3, R4, R5, R6, R7, y R8 se definen aquí”.
SUMARIO DE LA INVENCIÓN
Aquí se proporcionan compuestos útiles para el tratamiento de la infección por VHB en un sujeto que lo necesita. En un aspecto, se proporcionan aquí compuestos de Fórmula IIIa:

o una sal farmacéuticamente aceptable del mismo, como se define en la reivindicación 1.
En otro aspecto, se proporcionan aquí compuestos de Fórmula IVa:

IVa
o una sal farmacéuticamente aceptable del mismo, como se define en la reivindicación 4.
En otro aspecto, se proporcionan aquí composiciones que comprenden un compuesto proporcionado aquí (también denominado aquí “un compuesto de la invención”), o una sal, solvato o N-óxido del mismo, que además comprende al menos un vehículo farmacéuticamente aceptable.
En otro aspecto, se proporciona aquí un compuesto o composición para uso en el tratamiento de una infección por VHB en un individuo que lo necesita.
También se describe aquí un método para reducir la carga viral asociada con una infección por VHB en un individuo que lo necesita, que comprende administrar al individuo una cantidad terapéuticamente eficaz de un compuesto de la invención.
También se describe aquí un método para reducir la recurrencia de una infección por VHB en un individuo que lo necesita, que comprende administrar al individuo una cantidad terapéuticamente eficaz de un compuesto de la invención.
También se describen aquí métodos para reducir un impacto fisiológico adverso de una infección por VHB en un individuo que lo necesita, que comprende administrar al individuo una cantidad terapéuticamente eficaz de un compuesto de la invención.
También se describe aquí un método para inducir la remisión de una lesión hepática por una infección por VHB en un individuo que lo necesita, que comprende administrar al individuo una cantidad terapéuticamente eficaz de un compuesto de la invención.
También se describe aquí un método para reducir el impacto fisiológico de la terapia antiviral a largo plazo para la infección por VHB en un individuo que lo necesita, que comprende administrar al individuo una cantidad terapéuticamente eficaz de un compuesto de la invención.
También se describe aquí un método de tratamiento profiláctico de una infección por VHB en un individuo que lo necesita, en el que el individuo padece una infección por VHB latente, que comprende administrar al individuo una cantidad terapéuticamente eficaz de un compuesto de la invención.
En el compuesto o composición anterior para uso, el uso puede comprender además la administración al individuo de al menos un agente terapéutico adicional. En una realización, el agente terapéutico adicional puede seleccionarse de, pero no se limita a, el grupo que consiste en un inhibidor de la VHB polimerasa, agentes inmunomoduladores, interferón pegilado, inhibidor de la entrada viral, inhibidor de la maduración viral, modulador del ensamblaje de la cápside, inhibidor de la transcriptasa inversa, un inhibidor de ciclofilina/TNF, un agonista de TLR, una vacuna contra el VHB, y agentes de mecanismos distintos o desconocidos, y una combinación de los mismos.
En otra realización, el al menos un agente terapéutico adicional se selecciona del grupo que consiste en una vacuna contra el VHB, un inhibidor de la VHB polimerasa, interferón, interferón pegilado, inhibidor de la entrada viral, inhibidor de la maduración viral, modulador del ensamblaje de la cápside, inhibidor de la transcriptasa inversa, un agonista de TLR, y agentes de mecanismo distinto o desconocido, y una combinación de los mismos.
En aún otra realización, el agente terapéutico adicional se selecciona del grupo que consiste en un inhibidor de la VHB polimerasa, interferón, inhibidor de la entrada viral, inhibidor de la maduración viral, modulador del ensamblaje de la cápside, inhibidor de la transcriptasa inversa, un agonista de TLR, y agentes de mecanismos distintos o desconocidos, y una combinación de los mismos.
En otra realización, el interferón pegilado es interferón alfa (IFN-a) pegilado, interferón lambda (IFN- l ) pegilado o interferón gamma (IFN-g) pegilado.
En aún otra realización, el inhibidor de la transcriptasa inversa es al menos uno de Zidovudina, Didanosina, Zalcitabina, ddA (2’,3’-didesoxiadenosina), Estavudina, Lamivudina, Abacavir, Emtricitabina, Entecavir, Apricitabina, Atevirapina,
ribavirina, aciclovir, famciclovir, valaciclovir, ganciclovir, valganciclovir, Tenofovir, Adefovir, cidofovir, Efavirenz, Nevirapina, Delavirdina o Etravirina.
En aún otra realización, el compuesto y el al menos un agente terapéutico adicional se formulan conjuntamente.
En todavía otra realización, el compuesto y el al menos un agente terapéutico adicional son para administración conjunta.
BREVE DESCRIPCIÓN DE LAS FIGURAS
La Figura 1 muestra un esquema general utilizado para preparar compuestos seleccionados de la invención.
La Figura 2 muestra un segundo esquema general utilizado para preparar compuestos seleccionados de la invención.
La Figura 3 muestra los intermedios A-F, que se usan durante la preparación de compuestos seleccionados de la invención.
La Figura 4 muestra los intermedios H-V, que se usan durante la preparación de compuestos seleccionados de la invención.
La Figura 5 muestra un tercer esquema general utilizado para preparar compuestos seleccionados de la invención.
La Figura 6 muestra un cuarto esquema general utilizado para preparar compuestos seleccionados de la invención.
La Figura 7 muestra un quinto esquema general utilizado para preparar compuestos seleccionados de la invención.
DESCRIPCIÓN DETALLADA DE LA INVENCIÓN
Se proporcionan aquí compuestos que son útiles en el tratamiento y prevención del VHB en el hombre. En un aspecto no limitante, estos compuestos modulan o interrumpen la replicación viral del VHB para proporcionar partículas virales defectuosas con una virulencia enormemente reducida. Los compuestos de la invención tienen una potente actividad antiviral, exhiben perfiles metabólicos, de distribución de tejidos, de seguridad y farmacéuticos favorables, y son adecuados para uso en el hombre.
El VHB, un agente etiológico de la hepatitis aguda/crónica, consiste en un ADN circular de 3,2 kb parcialmente bicatenario a partir del cual se sintetizan cuatro proteínas: el núcleo, la polimerasa, el antígeno de superficie, y el producto del gen X.
Se han identificado cuatro promotores con funciones únicas en el genoma del VHB. El promotor pregenómico/central dirige la síntesis de ARNm de 3,6 kb que contiene toda la información genética codificada por el virus. Este ARN sirve como intermedio de la replicación y como molde para la síntesis de núcleo y polimerasa. El promotor S y el promotor pre-S dirigen la síntesis de ARN de 2,1 y 2,4 kb utilizados para la generación de proteínas pre-S1, pre-S2 y S. El promotor X dirige la transcripción de ARN de 0,9 kb específico para la síntesis del producto del gen X. La utilización específica de hígado y específica de estado de diferenciación de estos promotores está regulada por los dos elementos potenciadores, es decir, potenciador I (ENI) y potenciador II (ENII). Estos potenciadores, junto con el elemento de unión a HNF-1 (factor nuclear de hepatocito-1), son en gran parte responsables del tropismo restringido del VHB a los hepatocitos.
El mecanismo de replicación del VHB difiere del de otros virus de ADN por cuanto, como los retrovirus, está involucrado la etapa de transcripción inversa. Tras la infección de los hepatocitos, un genoma parcialmente bicatenario se convierte en un ADN superenrollado circular bicatenario completo. Empleando esto como molde, se transcribe el ARN de 3,6 kb, que se denomina el pregenoma. El pregenoma se empaqueta en una nucleocápside y se transcribe inversamente utilizando polimerasa como cebador de iniciación para generar el ADN monocatenario de cadena negativa. La polimerización de la segunda cadena sigue hasta que se sintetiza aproximadamente la mitad del genoma, lo que da como resultado la generación de un genoma circular parcialmente bicatenario, que está recubierto y es segregado por las células infectadas.
Los compuestos de la invención son útiles en el tratamiento del VHB al interrumpir, acelerar, reducir, retrasar o inhibir la replicación viral normal, induciendo así una replicación viral aberrante y dando lugar a efectos antivirales tales como la interrupción del ensamblaje o desmontaje del virión, la maduración del virión, o la salida del virus.
En una realización, los compuestos de la invención interrumpen la replicación viral cuando el virión es inmaduro. En otra realización, los compuestos de la invención interrumpen la replicación viral cuando el virión está maduro. En aún otra realización, los compuestos de la invención interrumpen la replicación viral durante la infectividad viral. En aún otra realización, la interrupción de la replicación viral atenúa la infectividad viral del VHB o reduce la carga viral. En
aún otra realización, la interrupción, inhibición, retraso o reducción de la replicación viral erradica el virus del organismo hospedante. En aún otra realización, la erradicación del VHB de un hospedante evita ventajosamente la necesidad de una terapia crónica a largo plazo o reduce la duración de la terapia a largo plazo.
En una realización, los compuestos descritos aquí son adecuados para monoterapia, y son eficaces contra cepas de VHB naturales o nativas y contra cepas de VHB resistentes a fármacos conocidos actualmente. En otra realización, los compuestos descritos aquí son adecuados para uso en terapia de combinación.
En otra realización, los compuestos de la invención pueden usarse en métodos para modular (por ejemplo, inhibir, interrumpir o acelerar) la actividad, estabilidad, función y propiedades de replicación viral del ADNccc del VHB. En aún otra realización, los compuestos de la invención pueden usarse en métodos para disminuir o prevenir la formación de ADNccc del VHB.
En otra realización, los compuestos de la invención pueden usarse en métodos para modular (por ejemplo, inhibir, interrumpir o acelerar) la actividad del ADNccc del VHB. En aún otra realización, los compuestos de la invención pueden usarse en métodos para disminuir o prevenir la formación de ADNccc del VHB.
Definiciones
Como se usa aquí, cada uno de los siguientes términos tiene el significado asociado con él en esta sección.
A menos que se defina lo contrario, todos los términos técnicos y científicos utilizados aquí generalmente tienen el mismo significado que comúnmente entiende un experto en la técnica a la que pertenece esta invención. Generalmente, la nomenclatura utilizada aquí y los procedimientos de laboratorio en cultivo celular, genética molecular, química orgánica, y química de péptidos son los bien conocidos y comúnmente empleados en la técnica.
Como se usan aquí, los artículos “un” y “una” se refieren a uno o más de uno (es decir, al menos uno) del objeto gramatical del artículo. A modo de ejemplo, “un elemento” significa un elemento o más de un elemento. Además, el uso del término “incluyendo”, así como otras formas, tales como “incluir”, “incluye”, e “incluido”, no es limitante.
Como se usa aquí, la expresión “alrededor de” será entendida por las personas con conocimientos normales en la técnica, y variará en cierta medida en el contexto en el que se usa. Como se usa aquí, cuando se hace referencia a un valor medible tal como una cantidad, una duración temporal, y similar, la expresión “alrededor de” pretende abarcar variaciones de ±20% o ±10%, más preferiblemente ±5%, incluso más preferiblemente ±1 %, y aún más preferiblemente ±0,1% del valor especificado, ya que tales variaciones son apropiadas para realizar los métodos descritos.
Como se usa aquí, la expresión “modulador del ensamblaje de la cápside” se refiere a un compuesto que interrumpe y/o acelera y/o inhibe y/o dificulta y/o retrasa y/o reduce y/o modifica el ensamblaje de la cápside normal (por ejemplo, durante la maduración) y/o el desmontaje normal de la cápside (por ejemplo, durante la infectividad) y/o perturba la estabilidad de la cápside, lo que induce de ese modo una morfología y función aberrantes de la cápside. En una realización, un modulador del ensamblaje de la cápside acelera el ensamblaje y/o desmontaje de la cápside, induciendo así una morfología aberrante de la cápside. En otra realización, un modulador del ensamblaje de la cápside interactúa (por ejemplo, se une en un sitio activo, se une en un sitio alostérico, modifica y/o dificulta el plegamiento, y similar) con la proteína principal de ensamblaje de la cápside (CA), interrumpiendo así el ensamblaje y/o desmontaje de la cápside. En aún otra realización, un modulador del ensamblaje de la cápside provoca una perturbación en la estructura y/o función de CA (por ejemplo, la capacidad de CA para ensamblarse, desensamblarse, unirse a un sustrato, plegarse en una conformación adecuada, o similar), lo que atenúa la infectividad del virus y/o es letal para el virus.
Como se usa aquí, la expresión “modulador del ensamblaje de la cápside descrito en la bibliografía” se refiere a un modulador del ensamblaje de la cápside que no es un compuesto de la presente invención.
Como se usa aquí, el término “tratamiento” o “tratar” se define como la aplicación o administración de un agente terapéutico, es decir, un compuesto de la invención (solo o en combinación con otro agente farmacéutico), a un paciente, o la aplicación o administración de un agente terapéutico a un tejido o línea celular aislada de un paciente (por ejemplo, para diagnóstico o aplicaciones ex vivo), que tiene infección por VHB, un síntoma de infección por VHB, o el potencial de desarrollar infección por VHB, con el propósito de sanar, mitigar, aliviar, alterar, remediar, mejorar, corregir o afectar la infección por VHB, los síntomas de la infección por VHB, o el potencial de desarrollar infección por VHB. Dichos tratamientos pueden adaptarse o modificarse específicamente, según el conocimiento obtenido en el campo de la farmacogenómica.
Como se usa aquí, el término “prevenir” o “prevención” significa que no se produjo ningún trastorno o desarrollo de la enfermedad, si no se había producido ninguno, o ningún otro trastorno o desarrollo de la enfermedad, si ya se había producido el trastorno o enfermedad. También se considera la capacidad de alguien para prevenir algunos o todos los síntomas asociados con el trastorno o la enfermedad.
Como se usa aquí, el término “paciente”, “individuo” o “sujeto” se refiere a un mamífero humano o no humano. Los mamíferos no humanos incluyen, por ejemplo, ganado y mascotas, tales como mamíferos ovinos, bovinos, porcinos, caninos, felinos y murinos. Preferiblemente, el paciente, sujeto o individuo es un ser humano.
Como se usa aquí, las expresiones “cantidad eficaz”, “cantidad farmacéuticamente eficaz” y “cantidad terapéuticamente eficaz” se refieren a una cantidad no tóxica pero suficiente de un agente para proporcionar el resultado biológico deseado. Ese resultado puede ser la reducción y/o el alivio de los signos, síntomas o causas de una enfermedad, o cualquier otra alteración deseada de un sistema biológico. Un experto en la técnica puede determinar una cantidad terapéutica apropiada en cualquier caso individual utilizando experimentación de rutina.
Como se usa aquí, la expresión “sales farmacéuticamente aceptables” se refiere a derivados de los compuestos descritos en los que el compuesto original se modifica convirtiendo un resto ácido o base existente en su forma de sal. Los ejemplos de sales farmacéuticamente aceptables incluyen, pero no se limitan a, sales de ácidos minerales u orgánicos de restos básicos tales como aminas; sales alcalinas u orgánicas de restos ácidos tales como ácidos carboxílicos; y similares. Las sales farmacéuticamente aceptables de la presente invención incluyen las sales no tóxicas convencionales del compuesto original formado, por ejemplo, a partir de ácidos inorgánicos u orgánicos no tóxicos. Las sales farmacéuticamente aceptables de la presente invención se pueden sintetizar a partir del compuesto original que contiene un resto básico o ácido por métodos químicos convencionales. Generalmente, tales sales pueden prepararse haciendo reaccionar las formas de ácido o base libres de estos compuestos con una cantidad estequiométrica de la base o ácido apropiado en agua o en un disolvente orgánico, o en una mezcla de los dos; generalmente, se prefieren medios no acuosos como éter, acetato de etilo, etanol, isopropanol o acetonitrilo. Las listas de sales adecuadas se encuentran en Remington’s Pharmaceutical Sciences, 17a ed., Mack Publishing Company, Easton, Pa., 1985, p. 1418 y Journal of Pharmaceutical Science, 66, 2 (1977).
Como se usa aquí, la expresión “farmacéuticamente aceptable” se refiere a un material, tal como un vehículo o diluyente, que no anula la actividad biológica o las propiedades del compuesto, y es relativamente no tóxico, es decir, el material puede administrarse a un individuo sin causar efectos biológicos indeseables o interactuar de manera nociva con ninguno de los componentes de la composición en la que está contenido.
Como se usa aquí, la expresión “vehículo farmacéuticamente aceptable” significa un material, composición o vehículo farmacéuticamente aceptable, tal como una carga líquida o sólida, estabilizador, agente dispersante, agente de suspensión, diluyente, excipiente, agente espesante, disolvente, o material encapsulante, involucrado en portar o transportar un compuesto útil dentro de la invención dentro o hacia el paciente de modo que pueda realizar su función prevista. Típicamente, tales constructos se portan o transportan desde un órgano, o porción del cuerpo, a otro órgano, o porción del cuerpo. Cada vehículo debe ser “aceptable” en el sentido de ser compatible con los otros ingredientes de la formulación, incluyendo el compuesto útil dentro de la invención, y no perjudicial para el paciente. Algunos ejemplos de materiales que pueden servir como vehículos farmacéuticamente aceptables incluyen: azúcares, tales como lactosa, glucosa y sacarosa; almidones, tales como almidón de maíz y almidón de patata; celulosa, y sus derivados, tales como carboximetilcelulosa de sodio, etilcelulosa y acetato de celulosa; tragacanto en polvo; malta; gelatina; talco; excipientes, tales como manteca de cacao y ceras para supositorios; aceites, tales como aceite de cacahuete, aceite de semilla de algodón, aceite de cártamo, aceite de sésamo, aceite de oliva, aceite de maíz y aceite de soja; glicoles, tales como propilenglicol; polioles, tales como glicerina, sorbitol, manitol y polietilenglicol; ésteres, tales como oleato de etilo y laurato de etilo; agar; agentes amortiguadores, tales como hidróxido de magnesio e hidróxido de aluminio; agentes tensioactivos; ácido algínico; agua libre de pirógenos; disolución salina isotónica; disolución de Ringer; alcohol etílico; disoluciones amortiguadoras de fosfato; y otras sustancias compatibles no tóxicas empleadas en formulaciones farmacéuticas. Como se usa aquí, “vehículo farmacéuticamente aceptable” también incluye cualquiera y todos los recubrimientos, agentes antibacterianos y antifúngicos, y agentes retardadores de la absorción, y similares, que son compatibles con la actividad del compuesto útil dentro de la invención, y son fisiológicamente aceptables para el paciente. Los compuestos activos suplementarios también se pueden incorporar en las composiciones. La expresión “vehículo farmacéuticamente aceptable” puede incluir además una sal farmacéuticamente aceptable del compuesto útil dentro de la invención. Otros ingredientes adicionales que pueden incluirse en las composiciones farmacéuticas usadas en la práctica de la invención son conocidos en la técnica, y se describen, por ejemplo, en Remington’s Pharmaceutical Sciences (Genaro, Ed., Mack Publishing Co., 1985, Easton, PA).
Como se usa aquí, el término “composición” o “composición farmacéutica” se refiere a una mezcla de al menos un compuesto útil dentro de la invención con un vehículo farmacéuticamente aceptable. La composición farmacéutica facilita la administración del compuesto a un paciente o sujeto. Existen múltiples técnicas de administración de un compuesto en la técnica que incluyen, pero no se limitan a, administración intravenosa, oral, aerosol, parenteral, oftálmica, pulmonar, y tópica.
Como se usa aquí, el término “alquilo”, por sí mismo o como parte de otro sustituyente, significa, a menos que se señale de otro modo, un hidrocarburo de cadena lineal o ramificada que tiene el número de átomos de carbono designado (es decir, C1-6 significa uno a seis átomos de carbono), e incluye grupos sustituyentes de cadena lineal o ramificada o cíclicos. Los ejemplos incluyen metilo, etilo, propilo, isopropilo, butilo, isobutilo, terc-butilo, pentilo, neopentilo, hexilo y ciclopropilmetilo. El más preferido es alquilo (C1-C6), particularmente etilo, metilo, isopropilo, isobutilo, n-pentilo, n-hexilo y ciclopropilmetilo.
Como se usa aquí, el término “heteroalquilo”, por sí mismo o en combinación con otro término, significa, a menos que se señale de otro modo, un grupo alquilo estable de cadena lineal o ramificada que consiste en el número indicado de átomos de carbono y uno o dos heteroátomos seleccionados del grupo que consiste en O, N y S, y en el que los átomos de nitrógeno y azufre pueden estar opcionalmente oxidados, y el heteroátomo de nitrógeno puede estar opcionalmente cuaternizado. El o los heteroátomos se pueden colocar en cualquier posición del grupo heteroalquilo, incluso entre el resto del grupo heteroalquilo y el fragmento al que está unido, así como unido al átomo de carbono más distante en el grupo heteroalquilo. Los ejemplos incluyen: -O-CH2-CH2-CH3 , -CH2-CH2-CH2-OH, -CH2-CH2-NH-CH3 , -CH2-S-CH2-CH3 , y -CH2CH2-S(=O)-CH3. Hasta dos heteroátomos pueden ser consecutivos, tales como, por ejemplo, -CH2-NH-OCH3 , o -CH2-CH2-S-S-CH3. Los grupos heteroalquilo preferidos tienen 1-10 carbonos.
Como se usa aquí, el término “alcoxi”, empleado solo o en combinación con otros términos, significa, a menos que se señale de otro modo, un grupo alquilo que tiene el número designado de átomos de carbono, como se definió anteriormente, conectado al resto de la molécula a través de un átomo de oxígeno, tal como, por ejemplo, metoxi, etoxi, 1-propoxi, 2-propoxi (isopropoxi), y los homólogos e isómeros superiores. Los preferidos son alcoxi (C1 -C3), particularmente etoxi y metoxi.
Como se usa aquí, el término “halo” o “halógeno”, solo o como parte de otro sustituyente, significa, a menos que se señale de otro modo, un átomo de flúor, cloro, bromo o yodo, preferiblemente, flúor, cloro o bromo, más preferiblemente, flúor o cloro.
Como se usa aquí, el término “cicloalquilo” se refiere a un radical no aromático monocíclico o policíclico, en el que cada uno de los átomos que forman el anillo (es decir, átomos esqueléticos) es un átomo de carbono. En una realización, el grupo cicloalquilo está saturado o parcialmente insaturado. En otra realización, el grupo cicloalquilo está condensado con un anillo aromático. Los grupos cicloalquilo incluyen grupos que tienen de 3 a 10 átomos anulares. Los ejemplos ilustrativos de grupos cicloalquilo incluyen, pero no se limitan a, los siguientes restos:

Los cicloalquilos monocíclicos incluyen, pero no se limitan a, ciclopropilo, ciclobutilo, ciclopentilo, ciclohexilo, cicloheptilo, y ciclooctilo. Los cicloalquilos dicíclicos incluyen, pero no se limitan a, tetrahidronaftilo, indanilo, y tetrahidropentaleno. Los cicloalquilos policíclicos incluyen adamantina y norbornano. El término cicloalquilo incluye grupos “carbociclilo no aromático insaturado” o “carbociclilo insaturado no aromático”, los cuales se refieren a un carbociclo no aromático como se define aquí, que contiene al menos un doble enlace carbono o un triple enlace carbono.
Como se usa aquí, el término “heterocicloalquilo” o “heterociclilo” se refiere a un grupo heteroalicíclico que contiene uno a cuatro heteroátomos anulares, cada uno seleccionado de O, S y N. En una realización, cada grupo heterocicloalquilo tiene de 4 a 10 átomos en su sistema anular, con la condición de que el anillo de dicho grupo no contenga dos átomos O o S adyacentes. En otra realización, el grupo heterocicloalquilo está condensado con un anillo aromático. En una realización, los heteroátomos de nitrógeno y azufre pueden estar opcionalmente oxidados, y el átomo de nitrógeno puede estar opcionalmente cuaternizado. El sistema heterocíclico puede estar unido, a menos que se señale de otro modo, en cualquier heteroátomo o átomo de carbono que proporcione una estructura estable. Un heterociclo puede ser de naturaleza aromática o no aromática. En una realización, el heterociclo es un heteroarilo.
Un ejemplo de un grupo heterocicloalquilo de 3 miembros incluye, y no se limita a, aziridina. Los ejemplos de grupos heterocicloalquilo de 4 miembros incluyen, y no se limitan a, azetidina y una beta lactama. Los ejemplos de grupos heterocicloalquilo de 5 miembros incluyen, y no se limitan a, pirrolidina, oxazolidina y tiazolidindiona. Los ejemplos de grupos heterocicloalquilo de 6 miembros incluyen, y no se limitan a, piperidina, morfolina y piperazina. Otros ejemplos no limitantes de grupos heterocicloalquilo son:


Los ejemplos de heterociclos no aromáticos incluyen grupos monocíclicos tales como aziridina, oxirano, tiirano, azetidina, oxetano, tietano, pirrolidina, pirrolina, pirazolidina, imidazolina, dioxolano, sulfolano, 2,3-dihidrofurano, 2,5-dihidrofurano, tetrahidrofurano, tiofano, piperidina, 1,2,3,6-tetrahidropiridina, 1,4-dihidropiridina, piperazina, morfolina, tiomorfolina, pirano, 2,3-dihidropirano, tetrahidropirano, 1,4-dioxano, 1,3-dioxano, homopiperazina, homopiperidina, 1,3-dioxepano, 4,7-dihidro-1,3-dioxepina, y óxido de hexametileno.
Como se usa aquí, el término “aromático” se refiere a un carbociclo o heterociclo con uno o más anillos poliinsaturados y que tiene un carácter aromático, es decir, que tiene (4n 2) electrones p (pi) deslocalizados, en el que n es un número entero.
Como se usa aquí, el término “arilo”, empleado solo o en combinación con otros términos, significa, a menos que se señale de otro modo, un sistema aromático carbocíclico que contiene uno o más anillos (típicamente uno, dos o tres anillos), en el que dichos anillos pueden estar unidos juntos de manera colgante, tal como un bifenilo, o pueden estar condensados, tal como naftaleno. Los ejemplos de grupos arilo incluyen fenilo, antracilo y naftilo. Ejemplos preferidos son fenilo y naftilo, el más preferido es fenilo.
Como se usa aquí, la expresión “cicloalquilo de Cn-n' con puente” se refiere a un sistema anular condensado que comprende n-n' carbonos, un sistema anular bicíclico con puente que comprende n-n' carbonos, o un sistema anular espiro que comprende n-n' carbonos, en el que n es 6, 7 u 8, y n' es 8, 9, 10, 11, 12, 13 o 14. Los ejemplos de n-n' incluyen, pero no se limitan a, 6-14, 6-10, 6-8, 7-14, 7-10, 7-8, 8-14 u 8-10.
Como se usa aquí, el término “heteroarilo” o “heteroaromático” se refiere a un heterociclo que tiene carácter aromático. Un heteroarilo policíclico puede incluir uno o más anillos que están parcialmente saturados. Los ejemplos incluyen los siguientes restos:

Los ejemplos de grupos heteroarilo también incluyen piridilo, pirazinilo, pirimidinilo (particularmente 2- y 4-pirimidinilo), piridazinilo, tienilo, furilo, pirrolilo (particularmente 2-pirrolilo), imidazolilo, tiazolilo, oxazolilo, pirazolilo (particularmente 3- y 5-pirazolilo), isotiazolilo, 1,2,3-triazolilo, 1,2,4-triazolilo, 1,3,4-triazolilo, tetrazolilo, 1,2,3-tiadiazolilo, 1,2,3 oxadiazolilo, 1,3,4-tiadiazolilo y 1,3,4-oxadiazolilo.
Los ejemplos de heterociclos y heteroarilos policíclicos incluyen indolilo (particularmente 3-, 4-, 5-, 6- y 7-indolilo), indolinilo, quinolilo, tetrahidroquinolilo, isoquinolilo (particularmente 1- y 5-isoquinolilo), 1,2,3,4-tetrahidroisoquinolilo, cinolinilo, quinoxalinilo (particularmente 2- y 5-quinoxalinilo), quinazolinilo, ftalazinilo, 1,8-naftiridinilo, 1,4-benzodioxanilo, cumarina, dihidrocumarina, 1,5-naftiridinilo, benzofurilo (particularmente 3-, 4-, 5-, 6- y 7-benzofurilo), 2,3-dihidrobenzofurilo, 1,2-bencisoxazolilo, benzotienilo (particularmente 3-, 4-, 5-, 6-, y 7-benzotienilo), benzoxazolilo, benzotiazolilo (particularmente 2-benzotiazolilo y 5-benzotiazolilo), purinilo, bencimidazolilo (particularmente 2-bencimidazolilo), benzotriazolilo, tioxantinilo, carbazolilo, carbolinilo, acridinilo, pirrolizidinilo, y quinolizidinilo.
Compuestos de la invención
La presente invención se refiere al descubrimiento de compuestos que son útiles en el tratamiento y prevención de la infección por VHB en el hombre. En un aspecto, los compuestos de la invención son útiles en el tratamiento del VHB al interrumpir, acelerar, reducir, retrasar o inhibir la replicación viral normal del VHB, induciendo así replicación viral aberrante y conduciendo a efectos antivirales tales como la interrupción del ensamblaje o desensamblaje del virión, o la maduración del virión, o la salida del virus.
Los disruptores de replicación viral descritos aquí pueden usarse como monoterapia o en nuevos regímenes de combinación de clase cruzada para tratar la infección por VHB en el hombre. La terapia de combinación con medicamentos que exhiben diferentes mecanismos de acción (MOA) que actúan en diferentes etapas del ciclo de vida del virus puede brindar una mayor eficacia debido a los efectos antivirales aditivos o sinérgicos. Los regímenes de tratamiento del VIH evaluados clínicamente han demostrado que la terapia de combinación mejora la eficacia de la reducción de la carga viral, y reduce drásticamente la aparición de resistencia antiviral. La terapia de combinación para el tratamiento de la infección por el virus de la hepatitis C (VHC) también ha dado como resultado una mejora significativa en la respuesta antiviral sostenida y las tasas de erradicación. De este modo, es probable que el uso de los inhibidores de la replicación viral del VHB de la presente invención, en combinación con, por ejemplo, fármacos NA, produzca un efecto antiviral más profundo y mayores tasas de erradicación de la enfermedad que los estándares de atención actuales.
En un aspecto, la resistencia a fármacos representa una amenaza importante para las terapias actuales para la infección crónica por el VHB, y la terapia de combinación de clase cruzada es una estrategia comprobada para retrasar la aparición de cepas de resistencia a fármacos. Los disruptores de la replicación viral de la presente invención pueden, cuando se administran solos o en combinación con otra terapia de VHB, ofrecer mejores perfiles de resistencia a fármacos y un mejor manejo del VHB crónico.
Los compuestos útiles dentro de la invención pueden sintetizarse usando técnicas bien conocidas en la técnica de la síntesis orgánica. Los materiales de partida e intermedios requeridos para la síntesis pueden obtenerse de fuentes comerciales o sintetizarse de acuerdo con métodos conocidos por los expertos en la técnica.
En un aspecto, el compuesto de la invención es un compuesto de Fórmula IIIa:

o una sal farmacéuticamente aceptable del mismo;
en la que
cada R1 se selecciona independientemente de H, halo y alquilo de C1-6;
R2 se selecciona de halo, OH, CN, alquilo de C1-6, alcoxi de C1-6, halo-(alquilo de C1-6), di-halo-(alquilo de C1-6) y tri-halo-(alquilo de C1-6);
R3 es halo; y
R4 se selecciona de alquilo de C1-6, heteroalquilo de C1-6, cicloalquilo de C3-7, heterocicloalquilo de C3-7, (alquilo de C1-6)-(cicloalquilo de C3-7), (cicloalquilo de C3-7)-(alquilo de C1-6), cicloalquilo de C7-8 con puente, (alquilo de C1-6)-C(O)O-(alquilo de C1-6) y (alquilo de C1-6)-(heterocicloalquilo de C3-7), cada uno de los cuales puede estar sustituido con uno o dos grupos seleccionados independientemente de (=O), OH, alquilo de C1-6, cicloalquilo de C3-7, halo-(alquilo de C1-6), di-halo-(alquilo de C1-6), tri-halo-(alquilo de C1-6), C(O)OH, fenilo y halo;
o R4 se selecciona de (cicloalquilo de C3-7)-C(O)R5 y (cicloalquilo de C7-8 con puente)-C(O)R5 , en el que R5 se selecciona de N(CH3)2 , NS(O)2CH3 , y heterocicloalquilo de C3-7,
en los que:
“alquilo”, por sí mismo o como parte de otro sustituyente, significa, a menos que se señale de otro modo, un hidrocarburo que tiene el número de átomos de carbono designado (es decir, C1-6 significa uno a seis átomos de carbono), e incluye grupos sustituyentes de cadena lineal o ramificada o cíclicos; y
“cicloalquilo” se refiere a un radical no aromático monocíclico o policíclico, en el que cada uno de los átomos que forman el anillo (es decir, átomos esqueléticos) es un átomo de carbono, y/o se refiere a grupos “carbociclilo no aromático insaturado” o “carbociclilo insaturado no aromático”, opcionalmente en el que el grupo cicloalquilo comprende un anillo aromático condensado.
En una realización de Fórmula IIIa, R2 se selecciona de halo, OH, CN, alquilo de C1-6, alcoxi de C1-6, y CF3.
En otra realización, R4 se selecciona de alquilo de C1-6, heteroalquilo de C1-6, cicloalquilo de C3-7, heterocicloalquilo de C3-7, (alquilo de C^Mcicloalquilo de C3-7), y (alquilo de C^Mheterocicloalquilo de C3-7), cada uno de los cuales puede estar sustituido con uno o dos grupos seleccionados independientemente de (=O), OH, alquilo de C1-6, C(O)OH, y halo.
En aún otra realización, cada R1 se selecciona independientemente de halo y alquilo de C1-6.
En todavía otra realización, cada R1 se selecciona independientemente de H y halo.
En otra realización, cada R1 se selecciona independientemente de halo.
En aún otra realización de Fórmula IIIa, R2 se selecciona de halo, CN, alquilo de C1-6, alcoxi C1-6, y tri-halo-(alquilo de C1-6).
En todavía otra realización, R2 se selecciona de halo, CN, CH3 , OCH3 , y CF3.
En otra realización, R2 es halo.
En aún otra realización, R2 es F.
En todavía otra realización, R3 es F.
En otra realización de Fórmula IIIa, R4 se selecciona de cicloalquilo de C3-7, heterocicloalquilo de C3-7, (alquilo de C 1-6)-(cicloalquilo de C3-7), y (alquilo de C1-6)-(heterocicloalquilo de C3-7), cada uno de los cuales puede estar sustituido con uno o dos grupos seleccionados independientemente de (=O), OH, alquilo de C1-6, C(O)OH, fenilo y halo; o R4 se selecciona de (cicloalquilo de C3-7)-C(O)R5 , en el que R5 se selecciona de N(CH3)2 , NS(O)2CH3 , y heterocicloalquilo de C3-7.
En aún otra realización, R4 se selecciona de cicloalquilo de C3-7, heterocicloalquilo de C3-7, (alquilo de C1-6)-(cicloalquilo de C3-7), y (alquilo de C^Mheterocicloalquilo de C3-7), todos los cuales pueden estar opcionalmente sustituidos independientemente con uno o dos grupos seleccionados de (=O), OH, alquilo de C1-6, C(O)OH, fenilo, y halo; o R4 se selecciona de (cicloalquilo de C3-7)-C(O)R5 , en el que R5 se selecciona de N(CH3)2 , NS(O)2CH3 , y heterocicloalquilo de C3-7.
En todavía otra realización, R4 es cicloalquilo de C3-7 o (alquilo de C1-e)-(cicloalquilo de C3-7), cada uno de los cuales está sustituido con (=O), OH, alquilo de C1-6, C(O)OH, o halo;
o R4 se selecciona de (cicloalquilo de C3-7)-C(O)R5 , en el que R5 es N(CH3)2 , o heterocicloalquilo de C3-7.
En otra realización, R4 es cicloalquilo de C3-7 o (alquilo de C1-6)-(cicloalquilo de C3-7), cada uno de los cuales es C(O)OH sustituido;
o R4 se selecciona de (cicloalquilo de C3-7)-C(O)R5 , en el que R5 es heterocicloalquilo de C3-7.
En aún otra realización, R4 se selecciona de (cicloalquilo de C3-7)-C(O)R5 , en el que R5 es heterocicloalquilo de C3-7. En otra realización de Fórmula IIIa, o una sal farmacéuticamente aceptable de la misma, cada R1 es halo; R2 es halo; R3 es halo; y R4 es cicloalquilo de C3-7 o (alquilo de C1-6)-(cicloalquilo de C3-7), cada uno de los cuales puede estar sustituido independientemente con C(O)OH; o R4 es (cicloalquilo de C3-7)-C(O)R5 , y R5 es heterocicloalquilo de C3-7. En otra realización, R4 es cicloalquilo de C3-7 o (alquilo de C1-6)-(cicloalquilo de C3-7), cada uno de los cuales puede estar sustituido independientemente con C(O)OH; o R4 es (cicloalquilo de C3-7)-C(O)R5 , y R5 es morfolinilo.
En otra realización de Fórmula IIIa, o una sal farmacéuticamente aceptable de la misma, el compuesto se selecciona del grupo que consiste en:

Se apreciará que la descripción de la presente invención aquí debe interpretarse en congruencia con las leyes y principios del enlace químico. En algunos casos, puede ser necesario eliminar un átomo de hidrógeno para acomodar un sustituyente en cualquier lugar dado.
Las realizaciones preferidas de Fórmula IIIa, que incluyen sales farmacéuticamente aceptables de la misma, se muestran a continuación en la Tabla 1, y también se consideran “compuestos de la invención”. Los compuestos que no entran dentro del alcance de las reivindicaciones se muestran junto con estos compuestos. Algunos compuestos de la Tabla 1 no incluyen hidrógenos en grupos hidroxilo; se entiende que “-O” indica un sustituyente hidroxilo en estas posiciones.









En otro aspecto, el compuesto de la invención es un compuesto de Fórmula IVa:

o una sal farmacéuticamente aceptable del mismo;
en la que
R1 es halo;
R3 es halo; y
R4 se selecciona de alquilo de Ci -6 , cicloalquilo de C3-7, (alquilo de Ci -6)-(cicloalquilo de C3-7), y (alquilo de Cí ñ anlo, en el que los grupos cicloalquilo de C3-7 y (alquilo de C1-6)-arilo están opcionalmente sustituidos con alquilo de C1-6 ;
en los que:
“alquilo”, por sí mismo o como parte de otro sustituyente, significa, a menos que se señale de otro modo, un hidrocarburo que tiene el número de átomos de carbono designado (es decir, C1-6 significa uno a seis átomos de carbono), e incluye grupos sustituyentes de cadena lineal o ramificada o cíclicos; y
“cicloalquilo” se refiere a un radical no aromático monocíclico o policíclico, en el que cada uno de los átomos que forman el anillo (es decir, átomos esqueléticos) es un átomo de carbono, y/o se refiere a grupos “carbociclilo no aromático insaturado” o “carbociclilo insaturado no aromático”, opcionalmente en el que el grupo cicloalquilo comprende un anillo aromático condensado.
En una realización de Fórmula IVa, R3 es F.
En otra realización, R4 se selecciona de alquilo de C2-6, cicloalquilo de C3-7, (alquilo de C1-6)-(cicloalquilo de C3-7), y (alquilo de C1-6)-arilo, en el que los grupos cicloalquilo de C3-7 y (alquilo de C1-6)-arilo están opcionalmente sustituidos con alquilo de C1-6.
En otra realización, R4 se selecciona de grupos alquilo de C1-6, cicloalquilo de C3-7, (alquilo de C1-6)-(cicloalquilo de C3-7), y (alquilo de C1-e)-arilo, en el que los grupos cicloalquilo de C3-7 y (alquilo de C1-6)-arilo están sustituidos con alquilo de C1-6.
En otra realización, R4 se selecciona de alquilo de C2-6, cicloalquilo de C3-7, (alquilo de C1-6)-(cicloalquilo de C3-7), y (alquilo de C1-6)-arilo, en el que los grupos cicloalquilo de C3-7 y (alquilo de C1-6)-arilo están sustituidos con alquilo de C1-6.
En otra realización de Fórmula IVa, o una sal farmacéuticamente aceptable de la misma, el compuesto se selecciona del grupo que consiste en:


Las realizaciones preferidas de Fórmula IVa, que incluyen sales farmacéuticamente aceptables de la misma, se muestran a continuación en la Tabla 2, y también se consideran “compuestos de la invención”. Los compuestos que no entran dentro del alcance de las reivindicaciones se muestran junto con estos compuestos. Algunos compuestos de la Tabla 2 no incluyen hidrógenos en grupos hidroxilo; se entiende que “-O” indica un sustituyente hidroxilo en estas posiciones.
Tabla 2.







La invención incluye además una composición que comprende un compuesto de acuerdo con la Fórmula Illa, o Fórmula IVa, o una sal, solvato, o N-óxido del mismo, que comprende además al menos un vehículo farmacéuticamente aceptable.
Los compuestos descritos aquí también incluyen compuestos marcados isotópicamente en los que uno o más átomos se reemplazan por un átomo que tiene el mismo número atómico, pero una masa atómica o número másico diferente de la masa atómica o número másico que se encuentra generalmente en la naturaleza. Los ejemplos de isótopos adecuados para su inclusión en los compuestos descritos aquí incluyen, y no se limitan a, 2H, 3H, 11C, 13C, 14C, 36Cl, 18F, 123I, 125I, 13N, 15N, 15O, 17O, 18O, 32P, y 35S. En una realización, los compuestos marcados isotópicamente son útiles en estudios de distribución de fármacos y/o tejidos de sustrato. En otra realización, la sustitución por isótopos más pesados tales como el deuterio proporciona una mayor estabilidad metabólica (por ejemplo, una vida media in vivo aumentada o requisitos de dosificación reducidos). En aún otra realización, la sustitución con isótopos emisores de positrones, tales como 11C, 18F, 15O y 13N, es útil en estudios de topografía por emisión de positrones (PET) para examinar la ocupación del receptor de sustrato. Los compuestos marcados isotópicamente se preparan por cualquier método adecuado o por procedimientos que usan un reactivo marcado isotópicamente apropiado en lugar del reactivo no marcado empleado de otro modo.
En una realización, los compuestos descritos aquí se marcan por otros medios, que incluyen, pero no se limitan a, el uso de cromóforos o restos fluorescentes, marcadores bioluminiscentes, o marcadores quimioluminiscentes.
Los compuestos descritos aquí y otros compuestos relacionados que tienen diferentes sustituyentes, se sintetizan usando técnicas y materiales descritos aquí y como se describe, por ejemplo, en Fieser and Fieser’s Reagents for Organic Synthesis, Volúmenes 1-17 (John Wiley and Sons, 1991); Rodd’s Chemistry of Carbon Compounds, Volúmenes 1-5 y Suplementos (Elsevier Science Publishers, 1989); Organic Reactions, Volúmenes 1-40 (John Wiley and Sons, 1991), Larock’s Comprehensive Organic Transformations (VCH Publishers Inc., 1989), March, Advanced Organic Chemistry 4a Ed., (Wiley 1992); Carey y Sundberg, Advanced Organic Chemistry 4a Ed., Vols. A and B (Plenum 2000, 2001), y Green y Wuts, Protective Groups in Organic Synthesis 3a Ed., (Wiley 1999). Los métodos generales para la preparación del compuesto como se describe aquí se modifican mediante el uso de reactivos y condiciones apropiados, para la introducción de los diversos restos encontrados en la fórmula como se proporciona aquí.
Los compuestos descritos aquí se sintetizan usando cualquier procedimiento adecuado a partir de compuestos que están disponibles de fuentes comerciales, o se preparan usando los procedimientos descritos aquí.
Métodos de tratamiento
Se describe aquí un método para tratar una infección por VHB en un individuo que lo necesita, que comprende administrar al individuo una cantidad terapéuticamente eficaz de un compuesto de la invención.
También se describe aquí un método para reducir la carga viral asociada con una infección por VHB en un individuo que lo necesita, que comprende administrar al individuo una cantidad terapéuticamente eficaz de un compuesto de la invención.
También se describe aquí un método para reducir la reaparición de una infección por VHB en un individuo que lo necesita, que comprende administrar al individuo una cantidad terapéuticamente eficaz de un compuesto de la invención.
También se describe aquí un método para reducir un impacto fisiológico adverso de una infección por VHB en un individuo que lo necesita, que comprende administrar al individuo una cantidad terapéuticamente eficaz de un compuesto de la invención.
También se describe aquí un método para inducir la remisión de una lesión hepática por una infección por VHB en un individuo que lo necesita, que comprende administrar al individuo una cantidad terapéuticamente eficaz de un compuesto de la invención.
También se describe aquí un método para reducir el impacto fisiológico de la terapia antiviral a largo plazo para la infección por VHB en un individuo que lo necesita, que comprende administrar al individuo una cantidad terapéuticamente eficaz de un compuesto de la invención.
También se describe aquí un método de tratamiento profiláctico de una infección por VHB en un individuo que lo necesita, en el que el individuo padece una infección latente por VHB, que comprende administrar al individuo una cantidad terapéuticamente eficaz de un compuesto de la invención.
En un aspecto, se proporciona aquí un compuesto o composición para uso en el tratamiento de una infección por VHB en un individuo que lo necesita.
En una realización, el uso descrito aquí comprende administrar al individuo al menos un agente terapéutico adicional seleccionado del grupo que consiste en una vacuna contra el VHB, un inhibidor de la VHB polimerasa, interferón, interferón pegilado, un inhibidor de la entrada viral, un inhibidor de la maduración viral, un modulador del ensamblaje de la cápside, un inhibidor de la transcriptasa inversa, un agonista de TLR, y agentes de mecanismo distinto o desconocido, y una combinación de los mismos. En otra realización, el interferón pegilado es interferón alfa pegilado (IFN-a), interferón lambda pegilado (IFN-l), o interferón gamma pegilado (IFN-g). En todavía otra realización, el inhibidor de la transcriptasa inversa es al menos uno de Zidovudina, Didanosina, Zalcitabina, ddA, Estavudina, Lamivudina, Abacavir, Emtricitabina, Entecavir, Apricitabina, Atevirapina, ribavirina, aciclovir, famciclovir, valaciclovir, ganciclovir, valganciclovir, Tenofovir, Adefovir, cidofovir, Efavirenz, Nevirapina, Delavirdina, o Etravirina. En aún otra realización, el compuesto y el al menos un agente terapéutico adicional se formulan conjuntamente. En todavía otra realización, el compuesto y el al menos un agente terapéutico adicional son para administración conjunta.
También se describe aquí un método para tratar una infección por VHB en un individuo que lo necesita, que comprende administrar al individuo una cantidad terapéuticamente eficaz del compuesto de Fórmula I, o una sal farmacéuticamente aceptable del mismo. También se describe aquí un método para tratar una infección por VHB en un individuo que lo necesita, que comprende administrar al individuo una cantidad terapéuticamente eficaz del compuesto de Fórmula II, o una sal farmacéuticamente aceptable del mismo. También se describe aquí un método para tratar una infección por VHB en un individuo que lo necesita, que comprende administrar al individuo una cantidad terapéuticamente eficaz del compuesto de Fórmula III, o una sal farmacéuticamente aceptable del mismo. También se describe aquí un método para tratar una infección por VHB en un individuo que lo necesita, que comprende administrar al individuo una cantidad terapéuticamente eficaz del compuesto de Fórmula IIIa, o una sal farmacéuticamente aceptable del mismo.
También se describe aquí un método para tratar una infección por VHB en un individuo que lo necesita, que comprende administrar al individuo una cantidad terapéuticamente eficaz del compuesto 110, o una sal farmacéuticamente aceptable del mismo.
También se describe aquí un método para tratar una infección por VHB en un individuo que lo necesita, que comprende administrar al individuo una cantidad terapéuticamente eficaz del compuesto 112A, o una sal farmacéuticamente aceptable del mismo.
También se describe aquí un método para tratar una infección por VHB en un individuo que lo necesita, que comprende administrar al individuo una cantidad terapéuticamente eficaz del compuesto 112B, o una sal farmacéuticamente aceptable del mismo.
También se describe aquí un método para tratar una infección por VHB en un individuo que lo necesita, que comprende administrar al individuo una cantidad terapéuticamente eficaz del compuesto 116, o una sal farmacéuticamente aceptable del mismo.
También se describe aquí un método para tratar una infección por VHB en un individuo que lo necesita, que comprende administrar al individuo una cantidad terapéuticamente eficaz del compuesto de Fórmula IV, o una sal farmacéuticamente aceptable del mismo. También se describe aquí un método para tratar una infección por VHB en un individuo que lo necesita, que comprende administrar al individuo una cantidad terapéuticamente eficaz del compuesto de Fórmula IVa, o una sal farmacéuticamente aceptable del mismo. También se describe aquí un método para tratar una infección por VHB en un individuo que lo necesita, que comprende administrar al individuo una cantidad terapéuticamente eficaz del compuesto de Fórmula V, o una sal farmacéuticamente aceptable del mismo. También se describe aquí un método para tratar una infección por VHB en un individuo que lo necesita, que comprende administrar al individuo una cantidad terapéuticamente eficaz del compuesto de Fórmula VI, o una sal farmacéuticamente aceptable del mismo.
También se describe aquí un método para tratar una infección por VHB en un individuo que lo necesita, que comprende administrar al individuo una cantidad terapéuticamente eficaz del compuesto 124, o una sal farmacéuticamente aceptable del mismo.
También se describe aquí un método para tratar una infección por VHB en un individuo que lo necesita, que comprende administrar al individuo una cantidad terapéuticamente eficaz del compuesto 128, o una sal farmacéuticamente aceptable del mismo.
También se describe aquí un método para tratar una infección por VHB en un individuo que lo necesita, que comprende administrar al individuo una cantidad terapéuticamente eficaz del compuesto 130, o una sal farmacéuticamente aceptable del mismo.
También se describe aquí un método para tratar una infección por VHB en un individuo que lo necesita, que comprende administrar al individuo una cantidad terapéuticamente eficaz del compuesto 131, o una sal farmacéuticamente aceptable del mismo.
También se describe aquí un método para tratar una infección por VHB en un individuo que lo necesita, que comprende administrar al individuo una cantidad terapéuticamente eficaz del compuesto 133, o una sal farmacéuticamente aceptable del mismo.
También se describe aquí un método para tratar una infección por VHB en un individuo que lo necesita, que comprende administrar al individuo una cantidad terapéuticamente eficaz del compuesto 150, o una sal farmacéuticamente aceptable del mismo.
También se describe aquí un método para tratar una infección por VHB en un individuo que lo necesita, que comprende administrar al individuo una cantidad terapéuticamente eficaz del compuesto 163, o una sal farmacéuticamente aceptable del mismo.
También se describe aquí un método para tratar una infección por VHB en un individuo que lo necesita, que comprende administrar al individuo una cantidad terapéuticamente eficaz del compuesto 180, o una sal farmacéuticamente aceptable del mismo.
También se describe aquí un método para tratar una infección por VHB en un individuo que lo necesita, que comprende administrar al individuo una cantidad terapéuticamente eficaz del compuesto 181, o una sal farmacéuticamente aceptable del mismo.
También se describe aquí un método para tratar una infección por VHB en un individuo que lo necesita, que comprende administrar al individuo una cantidad terapéuticamente eficaz del compuesto 188, o una sal farmacéuticamente aceptable del mismo.
También se describe aquí un método para tratar una infección por VHB en un individuo que lo necesita, que comprende administrar al individuo una cantidad terapéuticamente eficaz del compuesto 189, o una sal farmacéuticamente aceptable del mismo.
También se describe aquí un método para tratar una infección por VHB en un individuo que lo necesita, que comprende administrar al individuo una cantidad terapéuticamente eficaz del compuesto 194, o una sal farmacéuticamente aceptable del mismo.
También se describe aquí un método para tratar una infección por VHB en un individuo que lo necesita, que comprende administrar al individuo una cantidad terapéuticamente eficaz del compuesto 195, o una sal farmacéuticamente aceptable del mismo.
También se describe aquí un método para tratar una infección por VHB en un individuo que lo necesita, que comprende administrar al individuo una cantidad terapéuticamente eficaz del compuesto 197, o una sal farmacéuticamente aceptable del mismo.
También se describe aquí un método para tratar una infección por VHB en un individuo que lo necesita, que comprende administrar al individuo una cantidad terapéuticamente eficaz del compuesto 198, o una sal farmacéuticamente aceptable del mismo.
También se describe aquí un método para tratar una infección por VHB en un individuo que lo necesita, que comprende administrar al individuo una cantidad terapéuticamente eficaz del compuesto 199, o una sal farmacéuticamente aceptable del mismo.
También se describe aquí un método para tratar una infección por VHB en un individuo que lo necesita, que comprende administrar al individuo una cantidad terapéuticamente eficaz del compuesto 200, o una sal farmacéuticamente aceptable del mismo.
También se describe aquí un método para tratar una infección por VHB en un individuo que lo necesita, que comprende administrar al individuo una cantidad terapéuticamente eficaz del compuesto 201, o una sal farmacéuticamente aceptable del mismo.
También se describe aquí un método para tratar una infección por VHB en un individuo que lo necesita, que comprende administrar al individuo una cantidad terapéuticamente eficaz del compuesto 202, o una sal farmacéuticamente aceptable del mismo.
También se describe aquí un método para tratar una infección por VHB en un individuo que lo necesita, que comprende administrar al individuo una cantidad terapéuticamente eficaz del compuesto 203, o una sal farmacéuticamente aceptable del mismo.
También se describe aquí un método para tratar una infección por VHB en un individuo que lo necesita, que comprende administrar al individuo una cantidad terapéuticamente eficaz del compuesto 204, o una sal farmacéuticamente aceptable del mismo.
También se describe aquí un método para tratar una infección por VHB en un individuo que lo necesita, que comprende administrar al individuo una cantidad terapéuticamente eficaz del compuesto 205, o una sal farmacéuticamente aceptable del mismo.
También se describe aquí un método para tratar una infección por VHB en un individuo que lo necesita, que comprende administrar al individuo una cantidad terapéuticamente eficaz del compuesto 206, o una sal farmacéuticamente aceptable del mismo.
También se describe aquí un método para tratar una infección por VHB en un individuo que lo necesita, que comprende administrar al individuo una cantidad terapéuticamente eficaz del compuesto 207, o una sal farmacéuticamente aceptable del mismo.
Terapias de combinación
Se pretende que los compuestos de la presente invención sean útiles en combinación con uno o más compuestos adicionales útiles para tratar la infección por VHB. Estos compuestos adicionales pueden comprender compuestos de la presente invención o compuestos conocidos para tratar, prevenir o reducir los síntomas o efectos de la infección por VHB. Dichos compuestos incluyen, pero no se limitan a, inhibidores de la VHB polimerasa, interferones, inhibidores de la entrada viral, inhibidores de la maduración viral, moduladores del ensamblaje de la cápside descritos en la bibliografía, y otros agentes con mecanismos distintos o desconocidos que afectan el ciclo de vida del VHB y/o las consecuencias de infección por VHB.
En ejemplos no limitantes, los compuestos de la invención pueden usarse en combinación con uno o más fármacos (o una sal, solvato o profármaco de los mismos) seleccionados del grupo que consiste en:
inhibidores de la transcriptasa inversa del VHB, e inhibidores de la ADN y ARN polimerasa, incluyendo, pero sin limitarse a: lamivudina (3TC, Zeffix, Heptovir, Epivir, y Epivir-HBV), entecavir (Baraclude, Entavir), adefovir dipivoxil (Hepsara, Preveon, bis-POM PMEA), tenofovir disoproxil fumarato (Viread, TDF o PMPA);
interferones, que incluyen, pero sin limitarse a, interferón alfa (IFN-a), interferón lambda (IFN-l), e interferón gamma (IFN-g);
inhibidores de la entrada viral;
inhibidores de la maduración viral;
moduladores del ensamblaje de la cápside descritos en la bibliografía, tales como, pero sin limitarse a, BAY 41 4109;
compuestos de mecanismo distinto o desconocido, tales como, pero sin limitarse a, AT-61 ((E)-N-(1-cloro-3-oxo-1 -fenil-3-(piperidin-1 -il)prop-1 -en-2-il)benzamida), AT-130 ((E)-N-(1 -bromo-1 -(2-metoxifenil)-3-oxo-3-(piperidin-1-il)prop-1-en-2-il)-4-nitrobenzamida), y análogos similares.
En otra realización, el agente terapéutico adicional seleccionado de terapias inmunomoduladoras o inmunoestimuladoras, que incluye agentes biológicos que pertenecen a la clase de interferones, tales como interferón alfa 2a o 2b o interferones modificados tales como interferón pegilado, alfa 2a, alfa 2b, lambda; o moduladores de TLR tales como agonistas de TLR-7 o agonistas de TLR-9, o agentes antivirales que bloquean la entrada o maduración viral o se dirigen a la VHB polimerasa tales como inhibidores de la nucleósido o nucleótido o no nucleósido/no nucleótido polimerasa, y agentes de mecanismo distinto o desconocido, incluyendo agentes que interrumpen la función de otras proteínas virales esenciales o proteínas hospedantes requeridas para la replicación o persistencia del VHB.
En una realización de la terapia de combinación, el inhibidor de la transcriptasa inversa y/o el inhibidor de la ADN y/o ARN polimerasa incluye Zidovudina, Didanosina, Zalcitabina, ddA, Estavudina, Lamivudina, Abacavir, Emtricitabina, Entecavir, Apricitabina, Atevirapina, ribavirina, aciclovir, famciclovir, valaciclovir, ganciclovir, valganciclovir, Tenofovir, Adefovir, cidofovir, Efavirenz, Nevirapina, Delavirdina, o Etravirina.
En otra realización de la terapia de combinación, el agonista de TLR-7 se selecciona del grupo que consiste en SM360320 (9-bencil-8-hidroxi-2-(2-metoxi-etoxi)adenina) y AZD 8848 ([3-({[3-(6-amino-2-butoxi-8-oxo-7,8-dihidro-9H-purin-9-il)propil][3-(4-morfolinil)propil]amino}metil)fenil]acetato de metilo).
Se puede calcular un efecto sinérgico, por ejemplo, utilizando métodos adecuados tales como, por ejemplo, la ecuación de Emax sigmoidea (Holford y Scheiner, 1981, Clin. Pharmacokinet. 6: 429-453), la ecuación de la aditividad de Loewe (Loewe & Muischnek, 1926, Arch. Exp. Pathol Pharmacol. 114: 313-326), y la ecuación de efecto mediana (Chou y Talalay, 1984, Adv. Enzyme Regul. 22: 27-55). Cada ecuación mencionada anteriormente se puede aplicar a los datos experimentales para generar un gráfico correspondiente para ayudar a evaluar los efectos de la combinación de
fármacos. Los gráficos correspondientes asociados con las ecuaciones mencionadas anteriormente son la curva de efecto frente a la concentración, la curva de isobolograma, y la curva de índice de combinación, respectivamente.
Administración/dosificación/formulaciones
El régimen de administración puede afectar a lo que constituye una cantidad eficaz. Las formulaciones terapéuticas pueden administrarse al paciente antes o después del inicio de una infección por VHB. Además, varias dosis divididas, así como las dosis escalonadas, pueden administrarse diaria o secuencialmente, o la dosis puede infundirse continuamente, o puede ser una inyección en bolo. Además, las dosificaciones de las formulaciones terapéuticas pueden aumentarse o disminuirse proporcionalmente según lo indiquen las exigencias de la situación terapéutica o profiláctica.
La administración de las composiciones de la presente invención a un paciente, preferiblemente un mamífero, más preferiblemente un ser humano, puede llevarse a cabo usando procedimientos conocidos, a dosis y durante períodos de tiempo eficaces para tratar la infección por VHB en el paciente. Una cantidad eficaz del compuesto terapéutico necesaria para lograr un efecto terapéutico puede variar según factores tales como el estado de la enfermedad o trastorno en el paciente; la edad, el sexo y el peso del paciente; y la capacidad del compuesto terapéutico para tratar la infección por VHB en el paciente. Los regímenes de dosificación pueden ajustarse para proporcionar la respuesta terapéutica óptima. Por ejemplo, se pueden administrar diariamente varias dosis divididas, o la dosis se puede reducir proporcionalmente según lo indiquen las exigencias de la situación terapéutica. Un ejemplo no limitativo de un intervalo de dosis eficaz para un compuesto terapéutico de la invención es de alrededor de 1 y 5.000 mg/kg de peso corporal/por día. Un experto habitual en la técnica podría estudiar los factores relevantes y determinar la cantidad eficaz del compuesto terapéutico sin experimentación excesiva.
Los niveles de dosificación reales de los ingredientes activos en las composiciones farmacéuticas de esta invención pueden variarse para obtener una cantidad del ingrediente activo que sea eficaz para lograr la respuesta terapéutica deseada para un paciente, composición y modo de administración particulares, sin ser tóxico para el paciente.
En particular, el nivel de dosificación seleccionado dependerá de una variedad de factores, que incluyen la actividad del compuesto particular empleado, el tiempo de administración, la velocidad de excreción del compuesto, la duración del tratamiento, otros fármacos, compuestos o materiales utilizados en combinación con el compuesto, la edad, el sexo, el peso, la condición, la salud general, y el historial médico previo del paciente que se está tratando, y factores similares bien conocidos en las técnicas médicas.
Un médico, por ejemplo médico o veterinario, que tenga una habilidad normal en la técnica, puede determinar y prescribir fácilmente la cantidad eficaz de la composición farmacéutica requerida. Por ejemplo, el médico o veterinario podría comenzar las dosis de los compuestos de la invención empleados en la composición farmacéutica a niveles inferiores a los requeridos para lograr el efecto terapéutico deseado, y aumentar gradualmente la dosis hasta que se logre el efecto deseado.
En realizaciones particulares, es especialmente ventajoso formular el compuesto en forma de unidad de dosificación para facilitar la administración y la uniformidad de la dosificación. La forma de unidad de dosificación como se usa aquí se refiere a unidades físicamente discretas adecuadas como dosificaciones unitarias para los pacientes a tratar; conteniendo cada unidad una cantidad predeterminada de compuesto terapéutico, calculada para producir el efecto terapéutico deseado en asociación con el vehículo farmacéutico requerido. Las formas de unidad de dosificación de la invención están dictadas por y dependen directamente de (a) las características únicas del compuesto terapéutico y el efecto terapéutico particular que se va a lograr, y (b) las limitaciones inherentes en la técnica de la composición/formulación de dicho compuesto terapéutico para el tratamiento de la infección por VHB en un paciente.
En una realización, las composiciones de la invención se formulan usando uno o más excipientes o vehículos farmacéuticamente aceptables. En una realización, las composiciones farmacéuticas de la invención comprenden una cantidad terapéuticamente eficaz de un compuesto de la invención y un vehículo farmacéuticamente aceptable.
El vehículo puede ser un disolvente o medio de dispersión que contiene, por ejemplo, agua, etanol, poliol (por ejemplo, glicerol, propilenglicol, y polietilenglicol líquido, y similares), mezclas adecuadas de los mismos, y aceites vegetales. La fluidez adecuada puede mantenerse, por ejemplo, mediante el uso de un recubrimiento tal como la lecitina, mediante el mantenimiento del tamaño de partícula requerido en el caso de dispersión, y mediante el uso de tensioactivos. La prevención de la acción de los microorganismos puede lograrse mediante diversos agentes antibacterianos y antifúngicos, por ejemplo parabenos, clorobutanol, fenol, ácido ascórbico, timerosal, y similares. En muchos casos, será preferible incluir agentes isotónicos, por ejemplo azúcares, cloruro de sodio, o polialcoholes tales como manitol y sorbitol, en la composición. La absorción prolongada de las composiciones inyectables puede lograrse incluyendo en la composición un agente que retrase la absorción, por ejemplo monoestearato de aluminio o gelatina. En una realización, el vehículo farmacéuticamente aceptable no es DMSO solo.
Las composiciones de la invención pueden administrarse al paciente en dosis que oscilan de una a cinco veces por día o más. Las composiciones de la invención pueden administrarse al paciente en un intervalo de dosis que incluye, pero no se limita a, una vez al día, cada dos días, cada tres días a una vez a la semana, y una vez cada dos semanas. Será evidente para un experto en la técnica que la frecuencia de administración de las diversas composiciones de
combinación de la invención variará de un individuo a otro dependiendo de muchos factores, que incluyen, pero no se limitan a, edad, enfermedad o trastorno a tratar, género, salud general, y otros factores. De este modo, la invención no debe interpretarse como limitada a ningún régimen de dosificación particular, y la dosificación precisa y la composición que se administrará a cualquier paciente serán determinadas por el médico a cargo, teniendo en cuenta todos los demás factores sobre el paciente.
Los compuestos de la invención para administración pueden estar en el intervalo de alrededor de 1 mg a alrededor de 10.000 mg, alrededor de 20 mg a alrededor de 9.500 mg, alrededor de 40 mg a alrededor de 9.000 mg, alrededor de 75 mg a alrededor de 8.500 mg, alrededor de 150 mg a alrededor de 7.500 mg, alrededor de 200 mg a alrededor de 7.000 mg, alrededor de 3050 mg a alrededor de 6,000 mg, alrededor de 500 mg a alrededor de 5.000 mg, alrededor de 750 mg a alrededor de 4.000 mg, alrededor de 1 mg a alrededor de 3.000 mg, alrededor de 10 mg a alrededor de 2.500 mg, alrededor de 20 mg a alrededor de 2.000 mg, alrededor de 25 mg a alrededor de 1.500 mg, alrededor de 30 mg a alrededor de 1.000 mg, alrededor de 40 mg a alrededor de 900 mg, alrededor de 50 mg a alrededor de 800 mg, alrededor de 60 mg a alrededor de 750 mg, alrededor de 70 mg a alrededor de 600 mg, alrededor de 80 mg a alrededor de 500 mg, y cualquiera y todos los incrementos completos o parciales entre ellos.
En algunas realizaciones, la dosis de un compuesto de la invención es de alrededor de 1 mg a alrededor de 2.500 mg. En algunas realizaciones, una dosis de un compuesto de la invención utilizado en las composiciones descritas aquí es menor que alrededor de 10.000 mg, o menor que alrededor de 8.000 mg, o menor que alrededor de 6.000 mg, o menor que alrededor de 5.000 mg, o menor que alrededor de 3.000 mg, o menor que alrededor de 2.000 mg, o menor que alrededor de 1.000 mg, o menor que alrededor de 500 mg, o menor que alrededor de 200 mg, o menor que alrededor de 50 mg. De manera similar, en algunas realizaciones, una dosis de un segundo compuesto (es decir, un fármaco usado para tratar la enfermedad de Parkinson) como se describe aquí es menor que alrededor de 1.000 mg, o menor que alrededor de 800 mg, o menor que alrededor de 600 mg, o menor que alrededor de 500 mg, o menor que alrededor de 400 mg, o menor que alrededor de 300 mg, o menor que alrededor de 200 mg, o menor que alrededor de 100 mg, o menor que alrededor de 50 mg, o menor que alrededor de 40 mg, o menor que alrededor de 30 mg, o menor que alrededor de 25 mg, o menor que alrededor de 20 mg, o menor que alrededor de 15 mg, o menor que alrededor de 10 mg, o menor que alrededor de 5 mg, o menor que alrededor de 2 mg, o menor que alrededor de 1 mg, o menor que alrededor de 0,5 mg, y cualquiera y todos los incrementos completos o parciales de los mismos.
En una realización, la presente invención se refiere a una composición farmacéutica envasada que comprende un recipiente que contiene una cantidad terapéuticamente eficaz de un compuesto de la invención, solo o en combinación con un segundo agente farmacéutico; e instrucciones para usar el compuesto para tratar, prevenir o reducir uno o más síntomas de infección por VHB en un paciente.
Las formulaciones pueden emplearse en mezclas con excipientes convencionales, es decir, sustancias portadoras orgánicas o inorgánicas farmacéuticamente aceptables adecuadas para administración oral, parenteral, nasal, intravenosa, subcutánea, entérica, o cualquier otro modo de administración adecuado, conocidas en la técnica. Las preparaciones farmacéuticas pueden esterilizarse y, si se desea, mezclarse con agentes auxiliares, por ejemplo lubricantes, conservantes, estabilizantes, agentes humectantes, emulsionantes, sales para influir en la presión osmótica, amortiguadores, sustancias colorantes, saborizantes y/o aromáticas, y similares. También se pueden combinar, cuando se desee, con otros agentes activos, por ejemplo otros agentes analgésicos.
Las vías de administración de cualquiera de las composiciones de la invención incluyen oral, nasal, rectal, intravaginal, parenteral, bucal, sublingual o tópica. Los compuestos para uso en la invención pueden formularse para la administración por cualquier vía adecuada, tal como para la administración oral o parenteral, por ejemplo transdérmica, transmucosal (por ejemplo, sublingual, lingual, (trans)bucal, (trans)uretral, vaginal (por ejemplo, trans- y perivaginalmente), (intra)nasal y (trans)rectal), intravesical, intrapulmonar, intraduodenal, intragástrica, intratecal, subcutánea, intramuscular, intradérmica, intraarterial, intravenosa, intrabronquial, por inhalación, y tópica.
Las composiciones y formas de dosificación adecuadas incluyen, por ejemplo, comprimidos, cápsulas, comprimidos oblongos, pastillas, cápsulas de gel, trociscos, dispersiones, suspensiones, disoluciones, jarabes, gránulos, perlas, parches transdérmicos, geles, polvos, peletes, magmas, pastillas para chupar, cremas, pastas, apósitos, lociones, discos, supositorios, aerosoles líquidos para administración nasal u oral, formulaciones en polvo seco o en aerosol para inhalación, composiciones y formulaciones para administración intravesical, y similares. Debe entenderse que las formulaciones y composiciones que serían útiles en la presente invención no están limitadas a las formulaciones y composiciones particulares que se describen aquí.
Dosificación
La cantidad o dosis terapéuticamente eficaz de un compuesto de la presente invención dependerá de la edad, el sexo y el peso del paciente, la condición médica actual del paciente, y la progresión de la infección por VHB en el paciente que está siendo tratado. El experto en la técnica podrá determinar las dosis apropiadas, dependiendo de estos y otros factores.
Una dosis adecuada de un compuesto de la presente invención puede estar en el intervalo de alrededor de 0,01 mg a alrededor de 5.000 mg por día, tal como de alrededor de 0,1 mg a alrededor de 1.000 mg, por ejemplo, de alrededor
de 1 mg a alrededor de 500 mg, tal como alrededor de 5 mg a alrededor de 250 mg por día. La dosis puede administrarse en una dosis única o en dosis múltiples, por ejemplo de 1 a 4 o más veces por día. Cuando se usan dosis múltiples, la cantidad de cada dosis puede ser igual o diferente. Por ejemplo, se puede administrar una dosis de 1 mg por día como dos dosis de 0,5 mg, con un intervalo de alrededor de 12 horas entre dosis.
Se entiende que la cantidad de compuesto dosificado por día puede administrarse, en ejemplos no limitantes, todos los días, en días alternos, cada 2 días, cada 3 días, cada 4 días, o cada 5 días. Por ejemplo, con la administración en días alternos, se puede iniciar una dosis de 5 mg por día el lunes, con una primera dosis subsiguiente de 5 mg por día administrada el miércoles, una segunda dosis subsiguiente de 5 mg por día administrada el viernes, y así sucesivamente.
Una vez que ha mejorado la condición del paciente, se administra una dosis de mantenimiento si es necesario. Posteriormente, la dosis o la frecuencia de administración, o ambas, se reduce, en función de la carga viral, hasta un nivel en el que se retiene la enfermedad mejorada. En una realización, los pacientes requieren tratamiento intermitente a largo plazo ante cualquier recurrencia de síntomas y/o infección.
Los compuestos para uso en el método de la invención pueden formularse en forma de dosificación unitaria. La expresión “forma de dosificación unitaria” se refiere a unidades físicamente discretas adecuadas como dosificación unitaria para pacientes sometidos a tratamiento, conteniendo cada unidad una cantidad predeterminada de material activo, calculada para producir el efecto terapéutico deseado, opcionalmente en asociación con un vehículo farmacéutico adecuado. La forma de dosificación unitaria puede ser para una sola dosis diaria o una de múltiples dosis diarias (por ejemplo, alrededor de 1 a 4 o más veces por día). Cuando se usan múltiples dosis diarias, la forma de dosificación unitaria puede ser igual o diferente para cada dosis.
La toxicidad y la eficacia terapéutica de dichos regímenes terapéuticos se determinan opcionalmente en cultivos celulares o animales experimentales, incluyendo, pero sin limitarse a, la determinación de la LD50 (la dosis letal para el 50% de la población) y la ED50 (la dosis terapéuticamente eficaz en el 50% de la población). La relación de dosis entre los efectos tóxicos y terapéuticos es el índice terapéutico, que se expresa como la relación entre LD50 y ED50. Se prefieren los inhibidores del ensamblaje de la cápside que exhiben altos índices terapéuticos. Los datos obtenidos de ensayos de cultivo celular y estudios en animales se usan opcionalmente para formular un intervalo de dosificación para uso en seres humanos. La dosificación de tales inhibidores del ensamblaje de la cápside se encuentra preferiblemente dentro de un intervalo de concentraciones circulantes que incluyen la ED50 con toxicidad mínima. La dosificación opcionalmente varía dentro de este intervalo dependiendo de la forma de dosificación empleada y la vía de administración utilizada.
Los expertos en la técnica reconocerán, o podrán determinar usando no más que la experimentación habitual, numerosos equivalentes a los procedimientos, realizaciones, reivindicaciones, y ejemplos específicos descritos aquí. Tales equivalentes se consideraron dentro del alcance de esta invención y cubiertos por las reivindicaciones adjuntas a la misma. Por ejemplo, debe entenderse que las modificaciones en las condiciones de reacción, que incluyen, pero no se limitan a, los tiempos de reacción, el tamaño/volumen de reacción, y los reactivos experimentales, tales como disolventes, catalizadores, presiones, condiciones atmosféricas, por ejemplo atmósfera de nitrógeno, y agentes reductores/oxidantes, con alternativas reconocidas en la técnica y que no utilizan más que la experimentación habitual, están dentro del alcance de la presente solicitud.
Debe entenderse que, siempre que se proporcionen aquí valores e intervalos, todos los valores e intervalos abarcados por estos valores e intervalos deben estar comprendidos dentro del alcance de la presente invención. Además, todos los valores que caen dentro de estos intervalos, así como los límites superior o inferior de un intervalo de valores, también están contemplados por la presente solicitud.
Los siguientes ejemplos ilustran adicionalmente aspectos de la presente invención. Sin embargo, de ninguna manera son una limitación de las enseñanzas o descripción de la presente invención como se establece aquí.
EJEMPLOS
La invención se describe ahora con referencia a los siguientes Ejemplos. Estos Ejemplos se proporcionan solo con fines ilustrativos, y la invención no se limita a estos Ejemplos, sino que más bien abarca todas las variaciones que son evidentes como resultado de las enseñanzas proporcionadas aquí. Los compuestos según las reivindicaciones se exponen a continuación en los siguientes Ejemplos. Los compuestos que no entran en las reivindicaciones son compuestos de referencia.
Ejemplo: preparación de compuestos de la invención
Las Figuras 1 y 2 muestran esquemas generales usados para preparar compuestos seleccionados de la invención. Las Figuras 3 y 4 muestran intermedios usados en la preparación de compuestos seleccionados de la invención.
Intermedio A: A una disolución en DMF (15 ml) de ácido 4-fluoro-3-yodobenceno (2,0 g, 7,52 mmoles), diisopropiletilamina (2 ml, 11,3 mmoles) y 3-cloro-4-fluoroanilina (1,1 g, 7,52 mmoles). La mezcla se agitó a TA durante 1 hora antes de añadir HATU (3,54 g, 9,33 mmoles). La disolución resultante se agitó a TA durante la
noche, se paralizó con agua, y se extrajo con EtOAc. Los orgánicos combinados se lavaron adicionalmente con agua y salmuera, se secaron sobre Na2SO4, se filtró, y el filtrado se concentró a vacío. La purificación por cromatografía en columna (SiO2 , 0-50% EtOAc/hexanos proporcionó un sólido blanco (2,8 g, 96% de rendimiento). LC-MS: 394 (M+H)+.
Intermedio B: Preparado de manera análoga al Intermedio A pero usando 3,4-difluoroanilina. LC-MS: 378 (M+H)+.
Intermedio C: Preparado de manera análoga al Intermedio A, usando 3,4,5-trifluoroanilina y DMAP. La mezcla de reacción también se calentó a 50°C durante 13 h. LC-MS: 395 (M+H)+.
Intermedio D: Preparado de manera análoga al intermedio A pero usando 3-metil-4-fluoroanilina. LC-MS: 374 (M+H)+.
Intermedio E: Etapa 1: Una disolución en dioxano (20 ml) de intermedio C (620 mg, 1,78 mmoles) y diisopropiletilamina (0,450 ml, 2,67 mmoles) se desoxigenó enérgicamente mediante purga debajo de la superficie con nitrógeno durante 15 minutos. Después, se añadieron tris(dibencilidenacetona)dipaladio (0) (98 mg, 0,097 mmoles), xantfos (103 mg, 0,178 mmoles) y 3-mercaptopropanoato de 2-etilhexilo (0,450 ml, 1,95 mmoles), y la vasija de reacción se selló y se calentó a 100°C durante 13 h. Después de enfriar hasta TA, la mezcla de reacción se diluyó con éter, y se lavó con agua y salmuera. El extracto orgánico se secó (Na2SO4), se filtró, y el filtrado se concentró a vacío. La purificación del producto bruto así obtenido mediante cromatografía en columna (SiO2 , 0-20% EtOAc/Hexanos) proporcionó el intermedio deseado (757 mg, 87% de rendimiento).
Etapa 2: Una disolución en THF (15 ml) de 3-((2-fluoro-5-((3,4,5-trifluorofenil)carbamoil)fenil)tio)propanoato de 2-etilhexilo (757mg, 1,56 mmoles) se enfrió hasta 0°C antes de que se añadiera KOtBu (384 mg, 3,43 mmoles) en una porción. La mezcla resultante se dejó calentar hasta TA. La mezcla de reacción se diluyó con éter, y se lavó con agua y salmuera. El extracto orgánico se secó sobre Na2SO4, se filtró, y el filtrado se concentró a vacío. La purificación del producto bruto así obtenido mediante cromatografía en columna (SiO2 , 0-100% EtOAc/Hexanos) proporcionó el compuesto del título (400 mg, 85% de rendimiento). LC-MS: 302 (M+H)+.
Intermedio F: A una disolución en DMF (20 ml) de ácido 3-yodobenzoico (2,2 g, 8,8 mmoles), diisopropilamina (10 ml, 13,2 mmoles) y 3-cloro-4-fluoroanilina (1,3 g, 8,8 mmoles). La mezcla se agitó a TA durante 1 hora antes de añadir HATU (4 g, 10,56 mmoles). La disolución resultante se agitó a TA durante la noche, después de lo cual la mezcla de reacción se paralizó con agua y se extrajo con EtOAc. Los orgánicos combinados se lavaron adicionalmente con agua y salmuera, se secaron sobre Na2SO4, se filtraron, y el filtrado se concentró a vacío. La purificación por cromatografía en columna (SiO2 , 0-60% EtOAc/Hexanos) proporcionó un sólido blanco (3 g, 94% de rendimiento). LC-MS: 375 (M+H)+.
Ejemplo 1: Ácido 2-(1-(((2-fluoro-5-((3,4,5-trifluorofenil)carbamoil)fenil)tio)metil)ciclopropil)acético

A una disolución toluénica (5 ml) de intermedio C (90 mg, 0,259 mmoles) se añadió diisopropiletilamina (0,090 ml, 0,518 mmoles), y se desoxigenó vigorosamente mediante purga debajo de la superficie con nitrógeno durante 15 minutos. Después, se añadió tris(dibencilidenacetona)dipaladio (0) (6,5 mg, 0,0064 mmoles), xantfos (8 mg, 0,0129 mmoles) y ácido 1-(mercaptometil)ciclopropilacético (38 mg, 0,259 mmoles), y la vasija de reacción se selló y se calentó a 102°C durante 13 h. Después de enfriar hasta TA, la mezcla de reacción se diluyó con éter, y se lavó secuencialmente con HCl 1N, agua y salmuera. El extracto orgánico se secó entonces sobre Na2SO4, se filtró, y el filtrado se concentró a vacío. La purificación del producto bruto así obtenido mediante cromatografía en columna (SiO2 , 0-100% EtOAc/Hexanos) proporcionó el producto deseado (92 mg, 86% de rendimiento). LC-MS: 414 (M+H)+.
Ejemplo 2: 3-(Ciclohexiltio)-4-fluoro-W-(3,4,5-trifluorofenil)benzamida

Preparada de manera análoga al Ejemplo 1, pero usando en su lugar ciclohexanotiol. LC-MS: 384 (M+H)+. Ejemplo 3: 3-(Ciclopentiltio)-4-fluoro-W-(3,4,5-trifluorofenil)benzamida

Preparada de manera análoga al Ejemplo 1, pero usando en su lugar ciclopentanotiol. LC-MS: 370 (M+H)+. Ejemplo 4: 4-Fluoro-3-((tetrahidro-2W-piran-4-il)tio)-W-(3,4,5-trifluorofenil)benzamida

Preparada de manera análoga al Ejemplo 1, pero usando en su lugar oxano-4-tiol. LC-MS: 386 (M+H)+.
Ejemplo 5: 3-(terc-Butiltio)-4-fluoro-W-(3,4,5-trifluorofenil)benzamida

P re p a ra d a d e m a n e ra a n á lo g a a l E je m p lo 1, p e ro u s a n d o e n su lu g a r 2 -m e t i l-2 -p ro p a n o t io l. L C -M S : 358 (M H )+ .
Ejemplo 6: 3-(Benciltio)-4-fluoro-W-(3,4,5-trifluorofenil)benzamida

Preparada de manera análoga al Ejemplo 1, pero usando en su lugar bencilmercaptano. LC-MS: 392 (M+H)+.
Ejemplo 7: 4-Fluoro-3-((2-hidroxietil)tio)-W-(3,4,5-trifluorofenil)benzamida

Preparada de manera análoga al Ejemplo 1, pero usando en su lugar 2-mercaptoetanol. LC-MS: 346 (M+H)+.
Ejemplo 8: 3-(Ciclohexiltio)-4-fluoro-W-(4-fluoro-3-metilfenil)benzamida

Preparada de manera análoga al Ejemplo 1, pero usando en su lugar el Intermedio D y ciclohexanotiol. LC-MS: 362 (M+H)+.
Ejemplo 9: 3-(Ciclopentiltio)-4-fluoro-W-(4-fluoro-3-metilfenil)benzamida

Preparada de manera análoga al Ejemplo 1, pero usando en su lugar el Intermedio D y ciclopentanotiol. LC-MS: 348 (M+H)+.
Ejemplo 10: (c/'s/írans)-4-Fluoro-3-((4-hidroxiciclohexil)tio)-W-(3,4,5-trifluorofenil)benzamida

Etapa 1: El 4-mercaptociclohexanol se preparó de acuerdo con el procedimiento descrito en E.J. Corey et al. J. Org. Chem. 1966, 31, 1663.
Etapa 2: Preparada de manera análoga al Ejemplo 1, pero usando en su lugar 4-mercaptociclohexanol. LC-MS: 400 (M+H)+.
Ejemplo 11: (c/'s/frans)-4-Fluoro-W-(4-fluoro-3-metilfenil)-3-((4-hidroxiciclohexil)tio)benzamida

Etapa 1: Preparada de manera análoga al Ejemplo 10, pero usando en su lugar el Intermedio D. LC-MS: 378 (M+H)+.
Ejemplo 12: (±)-4-Fluoro-3-((3-oxocicloheptil)tio)-W-(3,4,5-trifluorofenil)benzamida

A una disolución en THF (15 ml) de intermedio E (40 mg, 0,133 mmoles) y trietilamina (0,03 ml, 0,199 mmoles) se añadió ciclohep-2-en-1-ona (0,016 ml, 0,146 mmoles). La mezcla resultante se agitó a TA durante 3 h. La mezcla de reacción se diluyó con éter, y se lavó con agua y salmuera. El extracto orgánico se secó entonces sobre Na2SO4, se filtró, y el filtrado se concentró a vacío. El producto bruto así obtenido se purificó entonces mediante HPLC preparativa para proporcionar el compuesto del título. LC-MS: 412 (M+H)+.
Ejemplo 13: (±)-W-(3-Cloro-4-fluorofenil)-4-fluoro-3-((3-oxocicloheptil)tio)benzamida
Etapa 1: A una disolución agitada de ácido 3-clorosulfonil-4-fluoro-benzoico (6,2 g, 26,0 mmoles) se añadió HCL concentrado (20 ml), y cloruro de estaño (II) dihihdratado (18 g, 91 mmoles), y se calentó a 100°C durante 13 h. Después de enfriar hasta TA, la mezcla de reacción se diluyó con agua, y se basificó con NaHCO3 sat., y se filtró. El filtrado se acidificó con HCl 1N, y se extrajo con DCM dos veces. Los orgánicos combinados se lavaron entonces con salmuera, y la fase orgánica se secó entonces sobre Na2SO4, se filtró, y el filtrado se concentró a vacío. No fue necesaria una purificación adicional para proporcionar ácido 4-fluoro-3-mercaptobenzoico (3,5 g, 80% de rendimiento)
Etapa 2: A una disolución en THF (15 ml) de ácido 4-fluoro-3-mercaptobenzoico (48 mg, 0,279 mmoles) y trietilamina (0,056 ml, 0,307 mmoles), se añadió ciclohept-2-en-1-ona (0,035 ml, 0,307 mmoles). La mezcla resultante se agitó a TA durante 3 h, o hasta que se formó el intermedio, ácido 4-fluoro-3-((3-oxocicloheptil)tio)benzoico. En la misma vasija de reacción, se añadieron 3-cloro-4-fluoroanilina (41 mg, 0,279 mmoles) y trietilamina (0,056 ml, 0,307 mmoles), y se agitó a TA durante 1 h antes de añadir HATU (127 mg, 0,334 mmoles). La mezcla resultante se agitó a TA durante 13 h, después de lo cual la mezcla de reacción se lavó con agua y EtOAc. Los orgánicos combinados se lavaron entonces con salmuera, y las fases orgánicas se secaron sobre Na2SO4, se filtraron, y el filtrado se concentró a vacío. El producto bruto así obtenido se purificó entonces mediante HPLC preparativa para proporcionar el compuesto del título. LC-MS: 10 (M+H)+.
Ejemplos 14A: (±)-4-Fluoro-3-(((1S,3fí)-3-hidroxicicloheptil)tio)-W-(3,4,5-trifluorofenil)benzamida y 14B: (±)-4-Fluoro-3-(((1fí,3fl)-3-hidroxicicloheptil)tio)-W-(3,4,5-trifluorofenil)benzamida

Una disolución en MeOH (10 ml) del Ejemplo 12 (350 mg, 0,855) se enfrió hasta 0°C antes de que se añadiera NaBH4 (50 mg, 1,28 mmoles). La mezcla resultante se dejó calentar hasta TA durante 13 h. La mezcla de reacción se paralizó con HCl 1N, y se extrajo con EtOAC dos veces. Los orgánicos combinados se lavaron entonces con salmuera, y el extracto orgánico se secó entonces sobre Na2SO4, se filtró, y el filtrado se concentró a vacío. El producto bruto así obtenido se purificó entonces mediante HPLC preparativa para proporcionar los 2 diastereómeros aislados como sólidos blancos, cis (35 mg, 10% de rendimiento) y trans (79 mg, 25%) LC-MS: 414 (M+H)+.
Ejemplo 15: (±)-4-Fluoro-3-((3-fluorocicloheptil)tio)-W-(3,4,5-trifluorofenil)benzamida

Una disolución en DCM (5 ml) del Ejemplo 14 (240 mg, 0,581 mmoles) se enfrió hasta -78°C antes de añadir DAST (0,077 ml, 0,581 mmoles). La mezcla resultante se dejó calentar hasta TA durante 13 h. La mezcla de reacción se enfrió con NaHCO3 sat., y extrajo con DCM. Los orgánicos combinados se lavaron entonces con agua y salmuera, se secaron sobre Na2SO4, se filtraron, y el filtrado se concentró a vacío. La purificación del producto bruto así obtenido mediante cromatografía en columna (SiO2 , 0-50% EtOAc/Hexanos) proporcionó el compuesto del título (98 mg, 3% de rendimiento). LC-MS: 416 (M+H)+.
Ejemplo 16: (±)-4-Fluoro-3-(((1fl,3S)-3-hidroxi-3-metilcicloheptil)tio)-W-(3,4,5-trifluorofenil)benzamida

A una disolución en THF (5 ml) del Ejemplo 12 (86 mg, 0,209 mmoles) enfriada hasta 0°C se añadió bromuro de metilmagnesio (3 M, 0,210 ml, 0,63 mmoles). Después de agitar a 0°C durante 30 minutos, la mezcla se dejó calentar hasta temperatura ambiente, después de lo cual se lavó secuencialmente con HCl 1N, NaOH 1N, y se extrajo con EtOAC dos veces. Los orgánicos combinados se lavaron entonces con agua y salmuera, se secaron sobre Na2SO4, se filtraron, y el filtrado se concentró a vacío. El producto bruto así obtenido se purificó entonces mediante HPLC preparativa para proporcionar el compuesto del título. LC-MS: 428 (M+H)+.
Ejemplo 17: 4-Fluoro-3-((3-hidroxipropil)tio)-W-(3,4,5-trifluorofenil)benzamida

Preparada de manera análoga al Ejemplo 1, pero usando en su lugar 3-mercapto-1-propanol. LC-MS: 360 (M+H)+.
Ejemplo 18: (±)-4-Fluoro-3-((3-hidroxibutil)tio)-W-(3,4,5-trifluorofenil)benzamida

Etapa 1: A una disolución en THF (5 ml) del Ejemplo 17 (200 mg, 0,557 mmoles) se añadió periodinano de Dess-Martin (236 mg, 0,557 mmoles), y la mezcla se agitó a TA durante 16 h. La mezcla de reacción se lavó con NaHCO3 sat., y se extrajo con DCM y luego se lavó adicionalmente con agua y salmuera. El extracto orgánico se secó entonces sobre Na2SO4, se filtró, y el filtrado se concentró a vacío. La purificación del producto bruto así obtenido mediante cromatografía en columna (SiO2 , 0-100% EtOAc/Hexanos) proporcionó el aldehído (80 mg, 42% de rendimiento).
Etapa 2: Preparada de manera análoga al Ejemplo 16, pero usando como intermedio el aldehído en la etapa anterior. LC-MS: 374 (M+H)+.
Ejemplo 19: 4-Fluoro-3-((3-hidroxi-3-metilbutil)tio)-W-(3,4,5-trifluorofenil)benzamida

Preparada de manera análoga al Ejemplo 18, pero usando el Ejemplo 18 como el intermedio. LC-MS: 388 (M+H)+.
Ejemplo 20: (±)WL(3-Cloro-4-fluorofenil)-4-fluoro-3-((3-oxociclohexil)tio)benzamida

Preparada de manera análoga al Ejemplo 13, pero usando en su lugar 2-ciclohexen-1-ona. LC-MS: 396 (M+H)+.
Ejemplo 21: (±)-W-(3-Cloro-4-fluorofenil)-4-fluoro-3-(((1 fl,3fl)-3-hidroxiciclohexil)tio)benzamida

Preparada de manera análoga al Ejemplo 14, pero usando el Ejemplo 20 como el intermedio. LC-MS: 398 (M+H)+.
Ejemplo 22: (±)-W-(3-Cloro-4-fluorofenil)-4-fluoro-3-((3-oxociclopentil)tio)benzamida

P re p a ra d a d e m a n e ra a n á lo g a a l E je m p lo 13, p e ro u s a n d o e n su lu g a r c ic lo p e n t-2 -e n -o n a . L C -M S : 396 (M H )+ .
Ejemplo 23: 4-Fluoro-3-((4-hidroxibutil)tio)-W-(3,4,5-trifluorofenil)benzamida

Preparada de manera análoga al Ejemplo 1, pero usando en su lugar 4-mercapto-1-butanol. LC-MS: 374 (M+H)+.
Ejemplo 24: (c/'s/frans)-W-(3-Cloro-4-fluorofenil)-4-fluoro-3-((4-hidroxiciclohexil)tio)benzamida

Preparada de manera análoga al Ejemplo 10, pero usando en su lugar el Intermedio A. LC-MS: 398 (M+H)+. Ejemplo 25: (±)-W-(3-Cloro-4-fluorofenil)-4-fluoro-3-(((1 S,3S)-3-hidroxiciclopentil)tio)benzamida

Preparada de manera análoga al Ejemplo 21, pero usando el Ejemplo 22 como el intermedio. LC-MS: 384 (M+H)+.
Ejemplo 26: W-(3-Cloro-4-fluorofenil)-4-fluoro-3-((2-hidroxietil)tio)benzamida
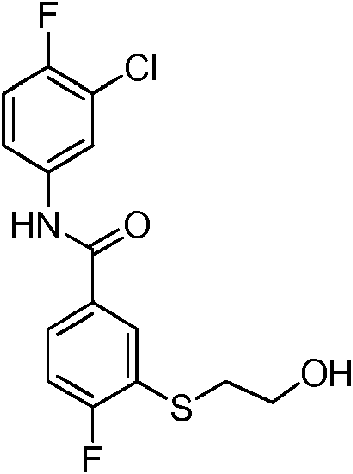
P re p a ra d a d e m a n e ra a n á lo g a a l E je m p lo 7, p e ro u s a n d o e n su lu g a r e l In te rm e d io A . L C -M S : 344 (M H )+ .
Ejemplo 27 W-(3-Cloro-4-fluorofenil)-4-fluoro-3-((3-hidroxipropil)tio)benzamida

Preparada de manera análoga al Ejemplo 17, pero usando en su lugar el Intermedio A. LC-MS: 358 (M+H)+.
Ejemplo 28A: (±)-W-(3-Cloro-4-fluorofenil)-4-fluoro-3-(((1 S,3fí)-3-hidroxicicloheptil)tio)benzamida

Preparada de manera análoga al Ejemplo 14A, pero usando en su lugar el Intermedio A. LC-MS: 412 (M+H)+. Ejemplo 28B: (±)-W-(3-Cloro-4-fluorofenil)-4-fluoro-3-(((1 S,3S)-3-hidroxicicloheptil)tio)benzamida

Preparada de manera análoga al Ejemplo 14B, pero usando en su lugar el Intermedio A. LC-MS: 412 (M+H)+. Ejemplo 29: (±)-4-Fluoro-3-((4-hidroxipentil)tio)-W-(3,4,5-trifluorofenil)benzamida

Preparada de manera análoga al Ejemplo 18, pero usando en su lugar el Ejemplo 23 como el intermedio. LC-MS: 388 (M+H)+.
Ejemplo 30: 4-Fluoro-3-((4-oxopentil)tio)-W-(3,4,5-trifluorofenil)benzamida

Preparada de manera análoga a la etapa 1 del Ejemplo 18, pero usando en su lugar el Ejemplo 29 como material de partida. LC-MS: 385 (M+H)+.
Ejemplo 31: 4-Fluoro-3-((4-hidroxi-4-metilpentil)tio)-W-(3,4,5-trifluorofenil)benzamida

Preparada de manera análoga a la etapa 1 de Ejemplo 19, pero usando en su lugar el Ejemplo 30 como material de partida. LC-MS: 402 (M+H)+.
Ejemplo 32 N-(3-Cloro-4-fluorofenil)-4-fluoro-3-(((1-(2-hidroxietil)ciclopropil)metil)tio)benzamida

Etapa 1: A una disolución en THF (5 ml) de ácido 2-(1-(((5-(((3-cloro-4-fluorofenil)carbamoil)-2-fluorofenil)tio)metil)ciclopropil)acético (preparado en una ruta análoga a la del Ejemplo 1 usando el Intermedio A; 177 mg, 0,428 mmoles), enfriada hasta 0°C, se añadió trietilamina (0,115 ml, 0,642 mmoles) y cloroformiato de etilo (0,050 ml, 0,514 mmoles), y la disolución resultante se dejó calentar hasta TA durante un período de 13 h. Se añadieron MeOH (2 ml) y NaBH4 (97 mg, 2,57 mmoles) a la mezcla de reacción, y se agitó a TA durante 6 h. La mezcla de reacción se lavó con HCl 1N, y se extrajo con EtOAc. Las fases orgánicas combinadas se lavaron entonces con agua y salmuera, luego se secaron sobre Na2SO4, se filtraron, y el filtrado se concentró a vacío. La purificación del producto bruto así obtenido mediante cromatografía en columna (SiO2 , 0-50% EtOAc/Hexanos) proporcionó el éster (163 mg, 87% de rendimiento).
Etapa 2: A una disolución en THF (5 ml) del éster (17 mg, 0,038 mmoles) se añadió LiBH4 (9 mg, 0,38 mmoles), y la disolución resultante se agitó a TA durante 2 h. La mezcla de reacción se lavó con HCl 1N, y se extrajo con EtOAc. Los orgánicos combinados se lavaron entonces con agua y salmuera, luego se secaron sobre Na2SO4, se filtraron, y el filtrado se concentró a vacío. La purificación del producto bruto así obtenido mediante cromatografía en columna (SiO2 , 0-50% EtOAc/hexanos) proporcionó el compuesto del título (10 mg, 67% de rendimiento). LC-MS: 400 (M+H)+.
Ejemplo 33: 4-Fluoro-3-(((1-(2-hidroxietil)dclopropil)metil)tio)-W-(3,4,5-trifluorofenil)benzamida

Preparada de manera análoga al Ejemplo 32, pero usando en su lugar el intermedio C. LC-MS: 400 (M+H)+.
Ejemplo 34: (c/s/frans)-W-(3-Cloro-4-fluorofenil)-3-((4-hidroxiciclohexil)tio)benzamida
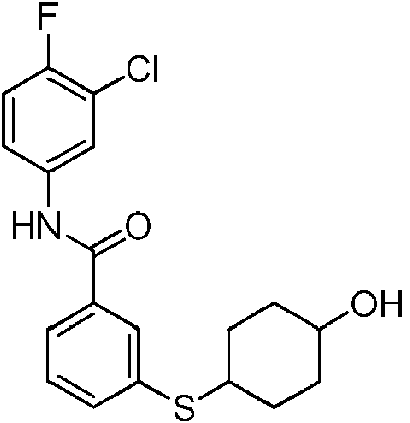
Preparada de manera análoga al Ejemplo 10, pero usando en su lugar el Intermedio F. LC-MS: 380 (M+H)+.
Ejemplo 35: W-(3-Cloro-4-fluorofenil)-3-(ciclohexiltio)benzamida

Preparada de manera análoga al Ejemplo 2, pero usando en su lugar el Intermedio F. LC-MS: 364 (M+H)+.
Ejemplo 36: W-(3-Cloro-4-fluorofenil)-3-((2-hidroxietil)tio)benzamida

P re p a ra d a d e m a n e ra a n á lo g a a l E je m p lo 7, p e ro u s a n d o e n su lu g a r e l In te rm e d io F. L C -M S : 326 (M H )+ .
Ejemplo 37: W-(3-Cloro-4-fluorofenil)-3-(ciclopentiltio)benzamida
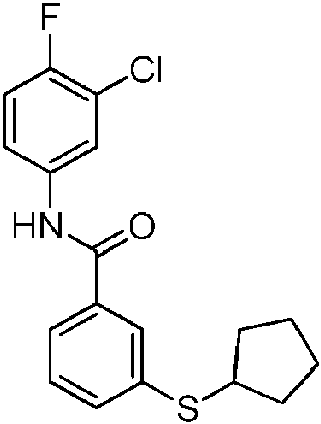
Preparada de manera análoga al Ejemplo 3, pero usando en su lugar el Intermedio F. LC-MS: 350 (M+H)+.
Ejemplo 38: (±)-W-(3-Cloro-4-fluorofenil)-3-((3-oxocicloheptil)tio)benzamida

Preparada de manera análoga al Ejemplo 12, pero usando en su lugar el Intermedio F. LC-MS: 392 (M+H)+.
Ejemplo 39A: (±)-W-(3-Cloro-4-fluorofenil)-3-(((1 S,3fl)-3-hidroxicicloheptil)tio)benzamida

Preparada de manera análoga al Ejemplo 14A, pero usando en su lugar el Intermedio F. LC-MS: 394 (M+H)+.
Ejemplo 39B: (±)-W-(3-Cloro-4-fluorofenil)-3-(((1 fí,3fí)-3-hidroxicicloheptil)tio)benzamida

Preparada de manera análoga al Ejemplo 14B, pero usando en su lugar el Intermedio F. LC-MS: 394 (M+H)+.
Ejemplo 40: W-(3-Cloro-4-fluorofenil)-3-((3-hidroxipropil)tio)benzamida

Preparada de manera análoga al Ejemplo 17, pero usando en su lugar el Intermedio F. LC-MS: 340 (M+H)+.
Ejemplo 41: 3-((2-Aminoetil)tio)-W-(3-cloro-4-fluorofenil)-4-fluorobenzamida

Preparada de manera análoga al Ejemplo 26, pero usando en su lugar 2-aminoetanotiol. LC-MS: 343 (M+H)+. Ejemplo 42: W-(3-Cloro-4-fluorofenil)-4-fluoro-3-((3,3,3-trifluoropropil)tio)benzamida

Preparada de manera análoga al Ejemplo 26, pero usando en su lugar 3,3,3-trifluoropropilmercaptano. LC-MS: 396 (M+H)+.
Ejemplo 43: W-(3-Cloro-4-fluorofenil)-3-((ciclohexilmetil)tio)benzamida

P re p a ra d a d e m a n e ra a n á lo g a a l E je m p lo 35 , p e ro u s a n d o e n su lu g a r c ic lo h e x a n o m e ta n o t io l. L C -M S : 378 (M H )+ .
Ejemplo 44: W-(3-Cloro-4-fluorofenil)-3-((2-morfolinoetil)tio)benzamida

Preparada de manera análoga al Ejemplo 35, pero usando en su lugar 2-morfolin-4-iletanotiol. LC-MS: 395 (M+H)+.
Ejemplo 45: (±)-W-(3-Cloro-4-fluorofenil)-4-fluoro-3-(((tetrahidrofuran-2-il)metil)tio)benzamida

A una disolución en DMSO/glicol (4 ml/0,1 ml) del Intermedio A (116 mg, 0,295 mmoles) se añadió carbonato de cesio (144 mg, 0,442 mmoles), y la disolución se desoxigenó vigorosamente mediante purga debajo de la superficie con nitrógeno durante 15 minutos. Después, se añadieron [1,1’-Bis(difenilfosfino)ferroceno]dicloropaladio (21 mg, 0,0295 mmoles), 1,1’-Bis(difenilfosfino)ferroceno (8 mg, 0,0147 mmoles), tiosulfato de sodio (116 mg, 0,737 mmoles) y cloruro de tetrahidrofurfurilo (0,479 ml, 4,43 mmoles), y la vasija de reacción se selló y se calentó a 120°C durante 13 h. Después de enfriar hasta TA, la mezcla de reacción se diluyó con éter, y se lavó con NH4Cl sat., agua y salmuera. Los extractos orgánicos se secaron entonces sobre Na2SO4, se filtraron, y el filtrado se concentró a vacío. El producto bruto así obtenido se purificó entonces mediante HPLC preparativa para proporcionar el compuesto del título (16 mg, 14% de rendimiento). LC-MS: 384 (M+H)+.
Ejemplo 46: (±)-W-(3-Cloro-4-fluorofenil)-4-fluoro-3-(((tetrahidrofuran-3-il)metil)tio)benzamida

Preparada de manera análoga al Ejemplo 45, pero usando en su lugar 3-(clorometil)tetrahidrofurano. LC-MS: 384 (M+H)+.
Ejemplo 47: M-(3-Cloro-4-fluorofenil)-4-fluoro-3-(((tetrahidro-2H-piran-4-il)metil)tio)benzamida
Preparada de manera análoga al Ejemplo 45, pero usando en su lugar 4-(clorometil)tetrahidropirano. LC-MS: 398 (M+H)+.
Ejemplo 48: 5-(Ciclopentiltio)-6-oxo-W-(3,4,5-trifluorofenil)-1,6-dihidropiridin-3-carboxamida

Etapa 1: Una suspensión agitada de ácido 5-bromo-6-oxo-1,6-dihidropiridin-3-carboxílico (992 mg, 4,55 mmoles) y piridina (1 ml) en DCM (10 ml) se enfrió hasta 0°C, se trató con cloruro de tionilo (0,397 ml, 5,46 mmoles), se calentó a TA, y se agitó durante 30 minutos. La mezcla de reacción se enfrió nuevamente hasta 0°C, y se trató con una disolución de 3,4,5-trifluoroanilina (709 mg, 5,00 mmoles), TEA (2,2 ml, 16 mmoles) y DMAP (12 mg, 0,46 mmoles) en DCM (5 ml) antes de agitar a TA durante 18 h. La mezcla de reacción se diluyó con EtOAc, se lavó con NaHCO3 sat., HCl 1N y salmuera. El extracto orgánico se secó entonces sobre MgSO4, se filtró, y el filtrado se concentró a vacío. La purificación por cromatografía en columna (SiO2 , 0-100% EtOAc/Hexanos) dio 5-bromo-6-oxo-W-(3,4,5-trifluorofenil)-1,6-dihidropiridin-3-carboxamida (1,18 g, 75% de rendimiento).
Etapa 2: Se añadieron tris(dibencilidenacetona)dipaladio (0) (32 mg, 0,035 mmoles), xantfos (40 mg, 0,070 mmoles) y ciclopentanototiol (0,082 ml, 0,768 mmoles) a una disolución en dioxano (5 ml) de 5-bromo-6-oxo-N-(3,4,5-trifluorofenil)-1,6-dihidropiridin-3-carboxamida (242 mg, 0,698 mmoles) y diisopropiletilamina (0,182 ml, 1,05 mmoles). La disolución se desoxigenó vigorosamente mediante purga debajo de la superficie con nitrógeno durante 15 minutos, y la vasija de reacción se selló y se calentó a 100°C durante 15 h. Después de enfriar hasta TA, la mezcla de reacción se diluyó con EtOAc y se lavó con HCl 1N, NaHCO3 sat. y salmuera. El extracto orgánico se secó entonces sobre MgSO4, se filtró, y el filtrado se concentró a vacío. La purificación por cromatografía en columna (SiO2 , 0-100% EtOAc/Hexanos) proporcionó el compuesto del título. (165 mg, 64% de rendimiento) LC-MS: 369 (M+H)+.
Ejemplo 49: Ácido 2-(1-(((5-((3-cloro-4-fluorofenil)carbamoil)-2-fluorofenil)tio)metil)ciclopropil)acético

Preparado de manera análoga al Ejemplo 1, pero usando en su lugar el Intermedio A. LC-MS: 412 (M+H)+.
Ejemplo 50: 2-((5-((3-Cloro-4-fluorofenil)carbamoil)-2-fluorofenil)tio)-2-metilpropanoato de etilo
Etapa 1: A una disolución en acetona (10 ml) del Ejemplo 13, etapa 1 (100 mg, 0,58 mmoles), se añadió carbonato de potasio (120 mg, 0,87 mmoles) y 2-bromo-2-metilpropanoato de etilo (0,100 ml, 0,7 mmoles), y la disolución resultante se calentó a 60°C durante 13 h. Los volátiles se eliminaron a vacío, y el residuo resultante se repartió entre éter y HCl ac. al 10%. Los orgánicos combinados se lavaron entonces con agua y salmuera, y entonces se secaron sobre Na2SÜ4, se filtraron, y el filtrado se concentró a vacío. No fue necesaria más purificación.
Etapa 2: A una disolución en DMF (15 ml) del ácido benzoico aislado (166 mg, 0,58 mmoles) se añadió diisopropilamina (2 ml, 11,3 mmoles) y 3-cloro-4-fluoroanilina (93 mg, 0,64 mmoles). La mezcla se agitó a TA durante 1 hora antes de añadir HATU (243 mg, 0,64 mmoles). La disolución resultante se agitó a TA durante la noche. La mezcla de reacción se paralizó con agua y se extrajo con EtOAc, y los orgánicos combinados se lavaron adicionalmente con agua y salmuera, después se secaron sobre Na2SÜ4, se filtraron, y el filtrado se concentró a vacío. La purificación por cromatografía en columna (SiÜ2 , 0-70% EtOAc/hexanos) para proporcionar el compuesto del título como un sólido blanco (90 mg, 37% de rendimiento). LC-MS: 414 (M+h )+.
Ejemplo 51: Ácido 2-((5-((3-cloro-4-fluorofenil)carbamoil)-2-fluorofenil)tio)-2-metilpropanoico

A una disolución en THF/MeOH (3 ml/3 ml) del Ejemplo 50 (84 mg, 0,20 mmoles) se añadió LiÜH 2N (0,300 ml, 0,6 mmoles). La mezcla resultante se agitó a TA durante 13 h, después de lo cual se diluyó con EtÜAc y se lavó secuencialmente con HCl 1N, agua y salmuera, y los volátiles se eliminaron a vacío. La mezcla resultante se sometió a purificación mediante cromatografía en columna (SiÜ2 , 0-50% de acetona/hexanos para proporcionar una espuma blanca (60 mg, 85% de rendimiento). LC-MS: 386 (M+H)+.
Ejemplo 52: W-(3-cloro-4-fluorofenil)-3-(cicloheptiltio)-4-fluorobenzamida

Preparada de manera análoga al Ejemplo 50, pero usando en su lugar bromocicloheptano en la etapa 1. LC-MS: 396 (M+H)+.
Ejemplo 53: Ácido (±)-3-((5-((3-cloro-4-fluorofenil)carbamoil)-2-fluorofenil)tio)ciclopentanocarboxílico

Etapa 1: Preparado como en el Ejemplo 14, pero usando 3-oxociclopentanocarboxilato de metilo como material de partida.
Etapa 2: A una disolución en DCM (15 ml) del alcohol (1,29 g, 8,9 mmoles) se añadió trietilamina (1,8 ml, 13,0 mmoles). La disolución se enfrió hasta 0°C antes de que se añadiera gota a gota cloruro de metanosulfonilo (0,93 ml, 12,0 mmoles). La mezcla resultante se dejó calentar hasta TA durante 13 h. La reacción se diluyó con d Cm y se lavó con agua, la fase orgánica se separó, se secó (Na2SO4), se filtró, y el filtrado se concentró a vacío. No fue necesaria una purificación adicional para proporcionar 3-((metilsulfonil)oxi)ciclopentanocarboxilato de metilo. Etapa 3: A una disolución en THF (8 ml) de 5-bromo-2-fluorobencenotiol (336 mg, 1,6 mmoles), bajo purga de N2 , se añadió NaH (65 mg, 1,6 mmoles) a TA en una porción. Después de 45 min, esta mezcla se añadió al mesilato como una disolución en THF (3 ml), gota a gota durante 3 minutos. La suspensión resultante se selló entonces y se calentó a 50°C durante 4,5 h. Después de enfriar hasta TA, la mezcla de reacción se diluyó con éter y se paralizó con NaHCO3sat., y después se lavó adicionalmente con agua y salmuera. El extracto orgánico se secó sobre Na2SO4, se filtró, y el filtrado se concentró a vacío. La purificación por cromatografía en columna (SiO2 , 0-50% EtOAc/hexanos proporcionó 3-((5-bromo-2-fluorofenil)tio)ciclopentanocarboxilato de metilo como un aceite incoloro (310 mg, 88% de rendimiento).
Etapa 4: Se combinaron 3-((5-bromo-2-fluorofenil)tio)ciclopentanocarboxilato de metilo (103 mg, 0,309 mmoles), carbonato de sodio (49 mg, 0,464 mmoles), acetato de paladio (II) (2 mg, 0,0093 mmoles) y xantfos (5 mg, 0,0093 mmoles), y la vasija se volvió a llenar con monóxido de carbono por medio de un globo. Después, se añadió 3-cloro-4-fluoroanilina (67 mg, 0,464 mmoles) como una disolución toluénica (5 ml), y la vasija de reacción se selló entonces y se calentó a 80°C durante 13 h. Después de enfriar hasta TA, la mezcla de reacción se diluyó con éter, y se lavó con agua y salmuera. El extracto orgánico se secó entoncs sobre Na2SO4, se filtró, y el filtrado se concentró a vacío. El producto bruto así obtenido se purificó entonces mediante HPLC preparativa para proporcionar 3-((5-((3-cloro-4-fluorofenil)carbamoil)-2-fluorofenil)tio)ciclopentanocarboxilato de metilo (12 mg, 10% de rendimiento).
Etapa 5: A una disolución en THF/MeOH (3 ml/3 ml) del éster mencionado anteriormente (12 mg, 0,0281 mmoles) se añadió LiOH 2N (2 ml). La mezcla resultante se agitó a TA durante 13 h, después de lo cual se diluyó con EtOAc y se lavó secuencialmente con HCl 1N, agua y salmuera, y los volátiles se eliminaron a vacío. La mezcla resultante se sometió a purificación por medio de HPLC preparativa para proporcionar el compuesto del título como un sólido blanco (6,5 mg, 60% de rendimiento). LC-MS: 412 (M+H)+.
Ejemplo 54: Ácido (±)-3-((2-fluoro-5-((3,4,5-trifluorofenil)carbamoil)fenil)tio)ciclopentanocarboxílico

Preparado de manera análoga al Ejemplo 53, pero usando en su lugar 3,4,5-trifluoroanilina en la etapa 4. LC-MS: 414 (M+H)+.
Ejemplo 55: Ácido (±)-3-((5-((3,4-difluorofenil)carbamoil)-2-fluorofenil)tio)ciclopentanocarboxílico

Preparado de manera análoga al Ejemplo 53, pero usando en su lugar 3,4,-difluoroanilina en la etapa 4. LC-MS: 396 (M+H)+.
Ejemplo 56: Ácido 1-((5-((3-cloro-4-fluorofenil)carbamoil)-2-fluorofenil)tio)ciclobutanocarboxílico
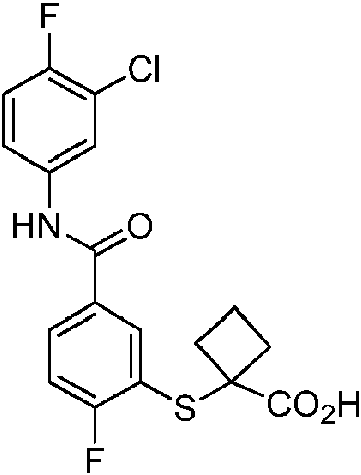
Etapa 1: A una disolución en DMSO (2,5 ml) de 5-bromo-2-fluorobencenotiol (207 mg, 1,0 mmoles) se añadió 1-bromociclobutanocarboxilato de etilo (0,240 ml, 1,5 mmoles) y carbonato de cesio (490 mg, 1,5 mmoles). La suspensión resultante se selló entonces y se calentó a 100°C durante 72 h. Después de enfriar hasta TA, la mezcla de reacción se diluyó con éter, y se lavó secuencialmente con agua y salmuera. El extracto orgánico se secó sobre Na2SO4, se filtró, y el filtrado se concentró a vacío. La purificación por cromatografía en columna (SiO2 , 0-50% EtOAc/Hexanos) produjo 1-((5-((3-cloro-4-fluorofenil)carbamoil)-2-fluorofenil)tio)ciclobutanocarboxilato de etilo como un líquido amarillo pálido (308 mg, 92% de rendimiento) .
Etapa 2: Se combinaron 1-((5-bromo-2-fluorofenil)tio)ciclobutanocarboxilato de etilo (150 mg, 0,450 mmoles), carbonato de sodio (71 mg, 0,675 mmoles), acetato de paladio (II) (30 mg, 0,135 mmoles) y xantfos (78 mg, 0,135 mmoles), y la vasija se volvió a llenar con monóxido de carbono a través de un globo. Después, se añadió 3-cloro-4-fluoroanilina (98 mg, 0,675 mmoles) como una disolución toluénica (5 ml), y la vasija de reacción se selló entonces y se calentó a 80°C durante 13 h. Después de enfriar hasta TA, la mezcla de reacción se diluyó con éter, y se lavó con agua y salmuera. El extracto orgánico se secó sobre Na2SO4, se filtró, y el filtrado se concentró a vacío. El producto bruto así obtenido se purificó entonces mediante HPLC preparativa para proporcionar 1-((5-((3-cloro-4-fluorofenil)carbamoil)-2-fluorofenil)tio)ciclobutanocarboxilato de etilo (60 mg, 31% de rendimiento).
Etapa 3: A una disolución en THF/MeOH (3 ml/3 ml) del éster (60 mg, 0,141 mmoles) se añadió LiOH 2N (2 ml). La mezcla resultante se agitó a TA durante 13 h, después de lo cual se diluyó con EtOAc y se lavó secuencialmente con HCl 1N, agua y salmuera, y los volátiles se eliminaron a vacío. La mezcla resultante se sometió a purificación mediante HPLC preparativa para proporcionar el compuesto del título como un sólido blanco (35 mg, 64% de rendimiento). LC-MS: 398 (M+H)+.
Ejemplo 57: Ácido 1-((5-((3,4-difluorofenil)carbamoil)-2-fluorofenil)tio) ciclobutanocarboxílico

Preparado de manera análoga al Ejemplo 56, pero usando 3,4-difluoroanilina en la etapa 2. LC-MS: 382 (M+H)+. Ejemplo 58: Ácido (c/'s/frans)-3-((5-((3-cloro-4-fluorofenil)carbamoil)-2-fluorofenil)tio)ciclobutanocarboxílico

Etapa 1: A una disolución en DMSO (3 ml) de 5-bromo-2-fluorobencenotiol (250 mg, 1,2 mmoles) se añadió 3-clorociclobutanocarboxilato de metilo (270 mg, 1,8 mmoles), carbonato de cesio (490 mg, 1,5 mmoles) y yoduro
de sodio ( 18 mg, 0,12 mmoles). La suspensión resultante se selló entonces y se calentó a 100°C durante 16 h. Después de enfriar hasta TA, la mezcla de reacción se diluyó con éter y se lavó secuencialmente con agua y salmuera. El extracto orgánico se secó entonces sobre Na2SO4, se filtró, y el filtrado se concentró a vacío. La purificación por cromatografía en columna (SiO2 , 0-50% EtOAc/Hexanos) produjo 3-((5-bromo-2-fluorofenil)tio)ciclobutanocarboxilato de metilo como un líquido amarillo pálido (160 mg, 42% de rendimiento). Etapa 2: Se combinaron 3-((5-bromo-2-fluorofenil)tio)ciclobutanocarboxilato de metilo (85 mg, 0,266 mmoles), carbonato de sodio (42 mg, 0,399 mmoles), acetato de paladio (II) (12 mg, 0,053 mmoles) y xantfos (31 mg, 0,053 mmoles), y la vasija se volvió a llenar con monóxido de carbono a través de un globo. Después, se añadió 3-cloro-4-fluoroanilina (58 mg, 0,399 mmoles) como una disolución toluénica (5 ml), y la vasija de reacción se selló entonces y se calentó a 80°C durante 13 h. Después de enfriar hasta TA, la mezcla de reacción se diluyó con éter, y se lavó con agua y salmuera. El extracto orgánico se secó sobre Na2SO4, se filtró, y el filtrado se concentró a vacío. El producto bruto así obtenido se purificó entonces mediante HPLC preparativa para proporcionar 3-((5-((3-cloro-4-fluorofenil)carbamoil)-2-fluorofenil)tio)ciclobutanocarboxilato de etilo (90 mg, 82% de rendimiento).
Etapa 3: A una disolución en THF/MeOH (3 ml/3 ml) del éster (90 mg, 0,218 mmoles) se añadió LiOH 2N (2 ml). La mezcla resultante se agitó a TA durante 13 h, después de lo cual se diluyó con EtOAc y se lavó secuencialmente con HCl 1N, agua y salmuera, y los volátiles se eliminaron a vacío. La mezcla resultante se sometió a purificación por medio de HPLC preparativa para proporcionar el compuesto del título como un sólido blanco (8 mg, 93% de rendimiento). LC-MS: 398 (M+H)+.
Ejemplo 59: Ácido (c/'s/frans)-3-((5-((3,4-difluorofenil)carbamoil)-2-fluorofenil)tio) ciclobutanocarboxílico

Preparado de manera análoga al Ejemplo 59, pero usando 3,4-difluoroanilina en la etapa 2. LC-MS: 382 (M+H)+. Ejemplo 60: W-(3-Cloro-4-fluorofenil)-4-fluoro-3-((oxetan-3-ilmetil)tio)benzamida

Preparada de manera análoga al Ejemplo 50, pero usando 3-(clorometil)oxetano en la etapa 1. LC-MS: 370 (M+H)+.
Ejemplo 61: Ácido (c/'s/frans)-4-((5-((3-cloro-4-fluorofenil)carbamoil)-2-fluorofenil)tio)ciclohexanocarboxílico Etapa 1: A una disolución en MeOH (40 ml) de 4-oxociclohexanocarboxilato de metilo (5 g, 32 mmoles) enfriada hasta 0°C se añadió NaBH4 (1,2 g, 32 mmoles). La mezcla resultante se dejó calentar hasta TA durante 13 h. La mezcla de reacción se paralizó con HCl 1N y se extrajo con EtOAc dos veces. Los orgánicos combinados se lavaron entonces con salmuera, se secaron sobre Na2SO4, se filtraron, y el filtrado se concentró a vacío. La purificación por cromatografía en columna (SiO2 , 0-50% EtOAc/hexanos) produjo 4-hidroxiciclohexanocarboxilato de metilo como un aceite incoloro (3,78 g, 75% de rendimiento).
Etapa 2: A una disolución en DCM (45 ml) del alcohol (3,78 g, 24 mmoles) se añadió trietilamina (4,9 ml, 35 mmoles). La disolución se enfrió hasta 0°C antes de que se añadiera gota a gota cloruro de metanosulfonilo (2,5 ml, 32 mmoles). La mezcla resultante se dejó calentar hasta TA durante 13 h, se diluyó con DCM, y se lavó con agua. Las fases orgánicas se separaron, se secaron sobre Na2SO4, se filtraron, y el filtrado se concentró a vacío.
No fue necesaria una purificación adicional para proporcionar 4-((metilsulfonil)oxi)ciclohexanocarboxilato de metilo. Etapa 3: A una disolución en DMSO (15 ml) de 5-bromo-2-fluorobencenotiol (3,70 ml, 3,0 mmoles) se añadió 4-((metilsulfonil)oxi)ciclohexanocarboxilato de metilo (1,06 g, 4,5 mmoles) y carbonato de cesio (1,47 g, 4,5 mmoles). La suspensión resultante se selló entonces y se calentó a 100°C durante 72 h. Después de enfriar hasta TA, la mezcla de reacción se diluyó con éter y se lavó secuencialmente con agua y salmuera. El extracto orgánico se secó sobre Na2SO4, se filtró, y el filtrado se concentró a vacío. La purificación por cromatografía en columna (SiO2 , 0-50% EtOAc/hexanos) produjo 4-((5-bromo-2-fluorofenil)tio)ciclohexanocarboxilato de metilo como un aceite amarillo pálido (746 mg, 72% de rendimiento).
Etapa 4: Se combinaron 4-((5-bromo-2-fluorofenil)tio)ciclohexanocarboxilato de metilo (250 mg, 0,720 mmoles), carbonato de sodio (106 mg, 1,0 mmoles), acetato de paladio (II) (32 mg, 0,144 mmoles) y xantfos (83 mg, 0,144 mmoles), y la vasija se purgó con monóxido de carbono a través de un globo. Después, se añadió 3-cloro-4-fluoroanilina (145 mg, 1,0 mmoles) como una disolución toluénica (5 ml), y la vasija de reacción se selló entonces y se calentó a 80°C durante 13 h bajo una atmósfera de monóxido de carbono. Después de enfriar hasta TA, la mezcla de reacción se diluyó con éter, y se lavó con agua y salmuera. El extracto orgánico se secó sobre Na2SO4, se filtró, y el filtrado se concentró a vacío. El producto bruto así obtenido se purificó entonces mediante HPLC preparativa para proporcionar 4-((5-((3-cloro-4-fluorofenil)carbamoil)-2-fluorofenil)tio)ciclohexanocarboxilato de metilo (200 mg, 64% de rendimiento).
Etapa 5: A una disolución en THF/MeOH (3 ml/3 ml) del éster (200 mg, 0,455 mmoles) se añadió LiOH 2N (2 ml). La mezcla resultante se agitó a TA durante 13 h, después de lo cual se diluyó con EtOAc y se lavó secuencialmente con HCl 1N, agua y salmuera, y los volátiles se eliminaron a vacío. La mezcla resultante se sometió a purificación mediante HPLC preparativa para proporcionar el compuesto del título (100 mg, 52% de rendimiento). LC-MS: 426 (M+H)+.
Ejemplo 62: Ácido (c/s/frans)-4-((2-fluoro-5-((3,4,5-trifluorofenil)carbamoil)fenil)tio)ciclohexanocarboxílico

Preparado de manera análoga al Ejemplo 61, pero usando 3,4,5-trifluoroanilina en la etapa 4. LC-MS: 428 (M+H)+.
Ejemplo 63: Ácido (cis/trans)-4-((5-((3,4-difluorofenil)carbamoil)-2-fluorofenil)tio)ciclohexanocarboxílico Preparado de manera análoga al Ejemplo 61, pero usando 3,4,-difluoroanilina en la etapa 4. LC-MS: 410 (M+H)+.
Ejemplo 64: (±)-W-(3-Cloro-4-fluorofenil)-4-fluoro-3-((tetrahidro-2H-piran-3-il)tio)benzamida

Etapa 1: A una disolución en DMSO (15 ml) de 5-bromo-2-fluorobencenotiol (335 mg, 1,62 mmoles) se añadió 3 bromotetrahidro-2H-pirano (400 mg, 2,43 mmoles) y carbonato de cesio (790 mg, 2,43 mmoles). La suspensión resultante se selló entonces y se calentó a 100°C durante 16 h. Después de enfriar hasta TA, la mezcla de reacción se diluyó con éter y se lavó secuencialmente con agua y salmuera. El extracto orgánico se secó sobre Na2SO4, se filtró, y el filtrado se concentró a vacío. La purificación por cromatografía en columna (SiO2 , 0-30% EtOAc/Hexanos) dio 3-((5-bromo-2-fluorofenil)tio)tetrahidro-2H-pirano como un aceite incoloro (292 mg, 62% de rendimiento).
Etapa 2: Se combinaron 3-((5-bromo-2-fluorofenil)tio)tetrahidro-2H-pirano (165 mg, 0,567 mmoles), carbonato de sodio (90 mg, 0,850 mmoles), acetato de paladio (II) (25 mg, 0,113 mmoles) y xantfos (65 mg, 0,113 mmoles), y la vasija se volvió a llenar con monóxido de carbono a través del globo. Después, se añadió 3-cloro-4-fluoroanilina (123 mg, 0,850 mmoles) como una disolución toluénica (5 ml), y la vasija de reacción se selló entonces y se calentó a 80°C durante 13 h bajo una atmósfera de CO. Después de enfriar hasta TA, la mezcla de reacción se diluyó con éter, y se lavó con agua y salmuera. El extracto orgánico se secó sobre Na2SO4, se filtró, y el filtrado se concentró a vacío. El producto bruto así obtenido se purificó entonces mediante HPLC preparativa para proporcionar el compuesto del título (100 mg, 46% de rendimiento). LC-MS: 384 (M+H)+.
Ejemplo 65: W-(3-Cloro-4-fluorofenil)-4-fluoro-3-(oxetan-3-iltio)benzamida

Preparada de manera análoga al Ejemplo 64, pero usando 3-bromooxetano en la etapa 2. LC-MS: 356 (M+H)+.
Ejemplo 66: (±)-W-(3-Cloro-4-fluorofenil)-4-fluoro-3-((tetrahidrofuran-3-il)tio)benzamida

Preparada de manera análoga al Ejemplo 64, pero usando 3-bromotetrahidrofurano en la etapa 2. LC-MS: 370 (M+H)+.

Intermedio G: (2-Bromo-W-(3,4,5-trifluorofenil)isonicotinamida): A una disolución anhidra en DMF (150 ml) de ácido 4-bromo-isonicotínico (8,0 g, 39,6 mmoles) y 2,3,4-trifluoroanilina (6,1 g, 7,52 mmoles) en atmósfera de nitrógeno se añadió secuencialmente HATU (18,0 g, 47,5 mmoles), diisopropiletilamina (21,0 ml, 120 mmoles) y una cantidad catalítica de DMAP (488 mg, 4 mmoles). La disolución resultante se agitó a TA durante la noche. La mezcla de reacción se paralizó con disolución acuosa de HCl 0,5N, y se extrajo con EtOAc. Las fases orgánicas combinadas se lavaron adicionalmente con agua y disolución de NaHCO3 sat., agua y salmuera, se secaron sobre MgSO4 anhidro, se filtraron, y el filtrado se concentró a vacío. El residuo se trituró en DCM (40 ml) durante 15 minutos y se filtró. La torta del filtro se lavó con DCM (10 ml) y se secó a vacío. Purificación por cromatografía en columna (SiO2 , 0-50% EtOAc/hexanos proporcionó un sólido blanquecino (6,96 g, 50% de rendimiento). LC-MS: 332 (M+H)+.
Ejemplo 67: (c/s/frans)-2-((4-Hidroxiciclohexil)tio)-W-(3,4,5-trifluorofenil)isonicotinamida

A una disolución en 1,4-dioxano (3 ml) de Intermedio G (2-bromo-W-(3,4,5-trifluorofenil)isonicotinamida) (53,0 mg, 0,15 mmoles) y 4-mercaptociclohexanol (0,02 ml, 0,16 mmoles) se añadió secuencialmente tris(dibencilidenacetona)dipaladio (0) (7,0 mg, 0,008 mmoles), xantfos (5,0mg, 0,008 mmoles) y diisopropilamina (0,052 ml, 0,30 mmoles). La disolución resultante se desoxigenó mediante purga debajo de la superficie con nitrógeno durante 5 minutos, y la vasija de reacción se selló después y se calentó a 100°C durante 16 horas. Después de enfriar hasta TA, la mezcla de reacción se diluyó con éter, se lavó secuencialmente con HCl 1N, agua y salmuera. El extracto orgánico se secó entonces sobre Na2SO4, se filtró, y el filtrado se concentró a vacío. La purificación del producto bruto así obtenido mediante cromatografía en columna (SiO2 , 0-100% EtOAc/Hexanos) proporcionó el producto deseado (43,8 mg, 76% de rendimiento). LC-MS: 383 (M+H)+.
Intermedio H: ((3-Yodo-4-metil-W-(3-cloro-4-fluorofenil)benzamida): Preparada de manera análoga al intermedio G, pero usando la 3-cloro-4-fluoroanilina y el ácido 3-yodo-4-metilbenzoico. LC-MS: 390 (M+H)+.
Intermedio I: ((3-Yodo-4-metil-W-(3,4-difluorofenil)benzamida): Preparada de manera análoga al intermedio G, pero usando la 3,4-difluoroanilina y el ácido 3-yodo-4-metilbenzoico. LC-MS: 374 (M+H)+.
Intermedio J: ((3-Yodo-4-metil-W-(3,4,5-trifluorofenil)benzamida): Preparada de manera análoga al intermedio G, pero usando la 3,4-5-trifluoroanilina y el ácido 3-yodo-4-metilbenzoico. LC-MS: 392 (M+H)+.
Intermedio K: ((3-Bromo-4-metoxi-W-(3-cloro-4-fluorofenil)benzamida): Preparada de manera análoga al intermedio G, pero usando la 3-cloro-4-fluoroanilina y el ácido 3-bromo-4-metoxibenzoico. LC-MS: 359 (M+H)+.
Intermedio L: ((3-Bromo-4-metoxi-W-(3,4-difluorofenil)benzamida): Preparada de manera análoga al intermedio G, pero usando la 3,4-difluoroanilina y el ácido 3-bromo-4-metoxibenzoico. LC-MS: 343 (M+H)+.
Intermedio M: ((3-Bromo-4-metoxi-W-(3,4,5-trifluorofenil)benzamida): Preparada de manera análoga al intermedio G, pero usando la 3,4-5-trifluoroanilina y el ácido 3-bromo-4-metoxibenzoico. LC-MS: 361 (M+H)+.
Intermedio N: ((3-Yodo-4-cloro-W-(3-cloro-4-fluorofenil)benzamida): Preparada de manera análoga al intermedio G, pero usando la 3-cloro-4-fluoroanilina y el ácido 4-cloro-3-yodobenzoico. LC-MS: 411 (M+H)+.
Intermedio O: ((3-Yodo-4-cloro-W-(3,4-difluorofenil)benzamida): Preparada de manera análoga al intermedio G, pero usando la 3,4-difluoroanilina y el ácido 4-cloro-3-yodobenzoico. LC-MS: 394 (M+H)+.
Intermedio P: ((3-Yodo-4-cloro-W-(3,4,5-trifluorofenil)benzamida): Preparada de manera análoga al intermedio G, pero usando la 3,4-5-trifluoroanilina y el ácido 4-cloro-3-yodobenzoico. LC-MS: 412 (M+H)+.
Intermedio Q: ((3-Bromo-4-trifluorometil-W-(3-cloro-4-fluorofenil)benzamida): Preparada de manera análoga al intermedio G, pero usando la 3-cloro-4-fluoroanilina y el ácido 3-bromo-4-trifluorometilbenzoico. LC-MS: 397 (M+H)+. Intermedio R: ((3-Bromo-4-trifluorometil-W-(3,4-difluorofenil)benzamida): Preparada de manera análoga al intermedio G, pero usando el ácido 3,4-difluoroanilina 3-bromo-4-trifluorometilbenzoico. LC-MS: 381 (M+H)+.
Intermedio S: ((3-Bromo-4-trifluorometil-W-(3,4,5-trifluorofenil)benzamida): Preparada de manera análoga al intermedio G, pero usando la 3,4-5-trifluoroanilina y el ácido 3-bromo-4-trifluorometilbenzoico. LC-MS: 399 (M+H)+. Intermedio T: ((3-Bromo-4-ciano-W-(3-cloro-4-fluorofenil)benzamida): Preparada de manera análoga al intermedio G, pero usando la 3-cloro-4-fluoroanilina y el ácido 3-bromo-4-cianobenzoico. LC-MS: 355 (M+H)+.
Intermedio U: ((3-Bromo-4-ciano-W-(3,4-difluorofenil)benzamida): Preparada de manera análoga al intermedio G, pero usando el ácido 3,4-difluoroanilina 3-bromo-4-cianobenzoico. lC-MS: 338 (M+H)+.
Intermedio V: ((3-Bromo-4-ciano-W-(3,4,5-trifluorofenil)benzamida): Preparada de manera análoga al intermedio G, pero usando la 3,4-5-trifluoroanilina y el ácido 3-bromo-4-cianobenzoico. LC-MS: 356 (M+H)+.
Ejemplo 68: N-(3-Cloro-4-fluorofenil)-3-(ciclohexiltio)-4-metilbenzamida

Preparada de manera análoga al Ejemplo 67, pero usando el Intermedio H y ciclohexanotiol. LC-MS: 377 (M-H)-. Ejemplo 69: N-(3-Cloro-4-fluorofenil)-3-(ciclohexiltio)-4-metoxibenzamida

Preparada de manera análoga al Ejemplo 67, pero usando el Intermedio K y ciclohexanotiol. LC-MS: 394 (M+H)+. Ejemplo 70: (c/s/frans)-W-(3-Cloro-4-fluorofenil)-3-((4-hidroxiciclohexil)tio)-4-metoxibenzamida

Preparada de manera análoga al Ejemplo 67, pero usando el Intermedio K y 4-hidroxiciclohexanotiol. LC-MS: 410 (M+H)+.
Ejemplo 71: 4-Cloro-W-(3-cloro-4-fluorofenil)-3-(ciclohexiltio)benzamida

Preparada de manera análoga al Ejemplo 67, pero usando el Intermedio N y ciclohexanotiol. LC-MS: 397 (M-H)-. Ejemplo 72: (±)-W-(3-Cloro-4-fluorofenil)-4-fluoro-3-((2-fluorociclohexil)tio)benzamida

Preparada de manera análoga al Ejemplo 64, pero usando 1-bromo-2-fluorociclohexano en la etapa 1. LC-MS: 400 (M+H)+.
Ejemplo 73: W-(3-Cloro-4-fluorofenil)-3-((4,4-dimetilciclohexil)tio)-4-fluorobenzamida

Etapa 1: A una disolución en DCM (45 ml) del 4,4-dimetilciclohexanol (200 mg, 1,56 mmoles) se añadió trietilamina (4,9 ml, 35 mmoles). La disolución se enfrió hasta 0°C antes de que se añadiera gota a gota cloruro de metanosulfonilo (0,324 ml, 2,34 mmoles). La mezcla resultante se dejó calentar hasta TA durante 13 h. La reacción se diluyó con d Cm y se lavó con agua, la fase orgánica se secó (Na2SÜ4), se filtró, y el filtrado se concentró a vacío. No fue necesaria una purificación adicional para proporcionar metanosulfonato de 4,4-dimetilciclohexilo.
Etapa 2: A una disolución en DMSO (15 ml) de 5-bromo-2-fluorobencenotiol (130 mg, 0,62 mmoles) se añadieron metanosulfonato de 4,4-dimetilciclohexilo (190 mg, 0,94 mmoles) y carbonato de cesio (306 mg, 0,94 mmoles). La suspensión resultante se sellóentonces y se calentó a 100°C durante 72 h. Después de enfriar hasta TA, la mezcla de reacción se diluyó con éter y se lavó secuencialmente con agua y salmuera. El extracto orgánico se secó entonces sobre Na2SO4, se filtró, y el filtrado se concentró a vacío. La purificación por cromatografía en columna (SiO2 , 0-100% EtOAc/Hexanos) proporcionó (5-bromo-2-fluorofenil)(4,4-dimetilciclohexil)sulfano (131 mg, 67% de rendimiento).
Etapa 3: Se añadieron (5-bromo-2-fluorofenil)(4,4-dimetilciclohexil)sulfano (131 mg, 0,413 mmoles), carbonato de sodio (66 mg, 0,620 mmoles), acetato de paladio (II) (2 mg, 0,0083 mmoles) y xantfos (5 mg, 0,0083 mmoles), y la vasija se volvió a llenar con monóxido de carbono a través de un globo. Después, se añadió 3-cloro-4-fluoroanilina (90 mg, 0,620 mmoles) como una disolución toluénica (5 ml), y la vasija de reacción se selló entonces y se calentó a 80°C durante 13 h en una atmósfera de monóxido de carbono. Después de enfriar hasta TA, la mezcla de reacción se diluyó con éter, y se lavó con agua y salmuera. El extracto orgánico se secó sobre Na2SO4, se filtró, y el filtrado se concentró a vacío. El producto bruto así obtenido se purificó entonces mediante HPLC preparativa para proporcionar el compuesto del título (55 mg, 33% de rendimiento). LC-MS: 410 (M+H)+.
Ejemplo 74: (c/s/frans)-W-(3-Cloro-4-fluorofenil)-3-((4-hidroxiciclohexil)tio)-4-metilbenzamida

Preparada de manera análoga al Ejemplo 67, pero usando el Intermedio H y 4-hidroxiciclohexanotiol. LC-MS: 394 (M+H)+.
Ejemplo 75: (cis/trans)-4-Cloro-W-(3-cloro-4-fluorofenil)-3-((4-hidroxiciclohexil)tio)benzamida

Preparada de manera análoga al Ejemplo 67, pero usando el Intermedio N y 4-hidroxiciclohexanotiol. LC-MS: 415 (M+H)+.
Ejemplo 76: 3-(Ciclohexiltio)-4-metil-W-(3,4,5-trifluorofenil)benzamida

Preparada de manera análoga al Ejemplo 67, pero usando el Intermedio J y ciclohexanotiol. LC-MS: 380 (M+H)+. Ejemplo 77: (c/s/frans)-3-((4-Hidroxiciclohexil)tio)-4-metil-W-(3,4,5-trifluorofenil)benzamida

Preparada de manera análoga al Ejemplo 67, pero usando el Intermedio J y 4-hidroxiciclohexanotiol. LC-MS: 396 (M+H)+.
Ejemplo 78: 3-(Ciclohexiltio)-4-metoxi-W-(3,4,5-trifluorofenil)benzamida

Preparada de manera análoga al Ejemplo 67, pero usando el Intermedio M y ciclohexanotiol. LC-MS: 396 (M+H)+.
Ejemplo 79: 4-Cloro-3-(ciclohexiltio)-W-(3,4,5-trifluorofenil)benzamida

Preparada de manera análoga al Ejemplo 67, pero usando el Intermedio P y ciclohexanotiol. LC-MS: 398 (M-H)-.
Ejemplo 80: 3-(Ciclohexiltio)-W-(3,4-difluorofenil)-4-metoxibenzamida

Preparada de manera análoga al Ejemplo 67, pero usando el Intermedio L y ciclohexanotiol. LC-MS: 378 (M+H)+.
Ejemplo 81: (c/s/frans)-4-Cloro-3-((4-hidroxiciclohexil)tio)-W-(3,4,5-trifluorofenil)benzamida

Preparada de manera análoga al Ejemplo 67, pero usando el Intermedio P y 4-hidroxiciclohexanotiol. LC-MS: 414 (M-1).
Ejemplo 82: (±)-W-(3-Cloro-4-fluorofenil)-3-((3,3-dimetilciclohexil)tio)-4-fluorobenzamida

Etapa 1: A una disolución en MeOH (40 ml) de 3,3-dimetilciclohexanona (450 mg, 3,57 mmoles) enfriada a 0°C se añadió NaBH4 (200m g, 5,35 mmoles). La mezcla resultante se dejó calentar hasta TA durante 13 h. La mezcla de reacción se paralizó con HCl 1N y se extrajo con EtOAc dos veces, y los orgánicos combinados se lavaron entonces adicionalmente con salmuera, se secaron sobre Na2SO4, se filtraron, y el filtrado se concentró a vacío. No fue necesaria una purificación adicional para proporcionar 3,3-dimetilciclohexanol como un aceite incoloro (450 mg, 98% de rendimiento).
Etapa 2: A una disolución en DCM (45 ml) del alcohol (450 mg, 3,51 mmoles) se añadió trietilamina (0,730 ml, 35 mmoles). La disolución se enfrió hasta 0°C antes de que se añadiera gota a gota cloruro de metanosulfonilo (0,460 ml, 5,27 mmoles). La mezcla resultante se dejó calentar hasta TA durante 13 h. La reacción se diluyó con DCM y se lavó con agua, las fases orgánicas se separaron, se secaron (Na2SO4), se filtraron, y el filtrado se concentró a vacío. No fue necesaria una purificación adicional para proporcionar metanosulfonato de 3,3-dimetilciclohexilo.
Etapa 3: A una disolución en DMSO (15 ml) de 5-bromo-2-fluorobencenotiol (150 mg, 0,72 mmoles), se añadieron metanosulfonato de 3,3-dimetilciclohexilo (227 mg, 1,08 mmoles) y carbonato de cesio (353 mg, 1,68 mmoles). La suspensión resultante se selló entonces y se calentó a 100°C durante 72 h. Después de enfriar hasta TA, la mezcla de reacción se diluyó con éter y se lavó secuencialmente con agua y salmuera. El extracto orgánico se secó sobre Na2SO4, se filtró, y el filtrado se concentró a vacío. La purificación por cromatografía en columna (SiO2 , 0-30% EtOAc/hexanos) proporcionó (5-bromo-2-fluorofenil)(3,3-dimetilciclohexil)sulfano como un aceite incoloro (100 mg, 44% de rendimiento).
Etapa 4: Se combinaron (5-bromo-2-fluorofenil)(3,3-dimetilciclohexil)sulfano (100 mg, 0,315 mmoles), carbonato de sodio (50 mg, 0,472 mmoles), acetato de paladio (II) (2 mg, 0,0063 mmoles) y xantfos (4 mg, 0,0063 mmoles), y la vasija se volvió a llenar con monóxido de carbono a través de un globo. Después, se añadió 3-cloro-4-fluoroanilina (68 mg, 0,472 mmoles) como una disolución toluénica (5 ml), y la vasija de reacción se selló entonces y se calentó a 80°C durante 13 h en una atmósfera de monóxido de carbono. Después de enfriar hasta TA, la mezcla de reacción se diluyó con éter, y se lavó con agua y salmuera. El extracto orgánico se secó sobre Na2SO4, se filtró, y el filtrado se concentró a vacío. El producto bruto así obtenido se purificó entonces mediante HPLC preparativa para proporcionar 4-((5-((3-cloro-4-fluorofenil)carbamoil)-2-fluorofenil)tio)ciclohexanocarboxilato de metilo (6,5 mg, 5% de rendimiento). LC-MS: 410 (M+H)+.
Ejemplo 83: N-(3-Cloro-4-fluorofenil)-3-((4,4-difluorociclohexil)tio)-4-fluorobenzamida

Preparada de manera análoga al Ejemplo 64, pero usando en su lugar 4-bromo-1,1 -difluorociclohexano como material de partida en la etapa 1. LC-MS: 418 (M+H)+.
Ejemplo 84: (c/s/frans)-3-((4-Hidroxiciclohexil)tio)-4-metoxi-W-(3,4,5-trifluorofenil)benzamida

Preparada de manera análoga al Ejemplo 67, pero usando el Intermedio M y 4-hidroxiciclohexanotiol. LC-MS: 412 (M+H)+.
Ejemplo 85: 3-(Ciclohexiltio)-W-(3,4-difluorofenil)-4-metilbenzamida

Preparada de manera análoga al Ejemplo 67, pero usando el Intermedio I y ciclohexanotiol. LC-MS: 362 (M+H)+. Ejemplo 86: 4-Cloro-3-(ciclohexiltio)-W-(3,4-difluorofenil)benzamida

Preparada de manera análoga al Ejemplo 67, pero usando el Intermedio O y ciclohexanotiol. LC-MS: 382 (M+H)+.
Ejemplo 87: (c/s/frans)-4-Cloro-W-(3,4-difluorofenil)-3-((4-hidroxiciclohexil)tio)benzamida

Preparada de manera análoga al Ejemplo 67, pero usando el Intermedio O y 4-hidroxiciclohexanotiol. LC-MS: 398 (M+H)+.
Ejemplo 88: (c/s/frans)-W-(3,4-Difluorofenil)-3-((4-hidroxiciclohexil)tio)-4-metilbenzamida

Preparada de manera análoga al Ejemplo 67, pero usando el Intermedio I y 4-hidroxiciclohexanotiol. LC-MS: 378 (M+H)+.
Ejemplo 89: (c/s/frans)-W-(3,4-Difluorofenil)-3-((4-hidroxiciclohexil)tio)-4-metoxibenzamida
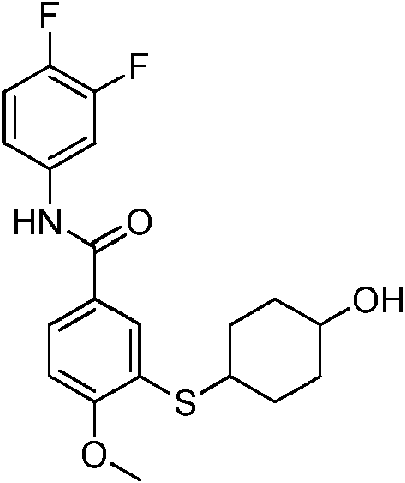
Preparada de manera análoga al Ejemplo 67, pero usando el Intermedio L y 4-hidroxiciclohexanotiol. LC-MS: 394 (M+H)+.
Ejemplo 90: (c/s/frans)-3-((4-Hidroxiciclohexil)tio)-4-(trifluorometil)-W-(3,4,5-trifluorofenil)benzamida Preparada de manera análoga al Ejemplo 67, pero usando el Intermedio S y 4-hidroxiciclohexanotiol. LC-MS: 448 (M-H)-.
Ejemplo 91: (c/s/frans)-W-(3-Cloro-4-fluorofenil)-3-((4-hidroxiciclohexil)tio)-4-(trifluorometil)benzamida

Preparada de manera análoga al Ejemplo 67, pero usando el Intermedio Q y 4-hidroxiciclohexanotiol. LC-MS: 446 (M-1).
Ejemplo 92: (c/s/frans)-W-(3,4-Difluorofenil)-4-hidroxi-3-((4-hidroxiciclohexil)tio)benzamida

A la disolución en DCM (1 ml) del compuesto en el Ejemplo 89 (10,0 mg, 0,025 mmoles) se añadió gota a gota una disolución de tribromuro de boro (2M en DCM; 1,0 ml) a temperatura ambiente bajo atmósfera de nitrógeno. El matraz de reacción se selló y se agitó durante la noche a TA. El tratamiento acuoso, seguido de la purificación del residuo bruto por medio de cromatografía en columna (SiO2 , 0-100% EtOAc/hexanos proporcionó un sólido blanquecino (4,2 mg, 44% de rendimiento). LC-MS: 380 (M+H)+.
Ejemplo 93: (c/s/frans)-4-Ciano-W-(3,4-difluorofenil)-3-((4-hidroxiciclohexil)tio)benzamida


Preparada de manera análoga al Ejemplo 67, pero usando el Intermedio V y 4-hidroxiciclohexanotiol. LC-MS: 405 (M-H)-.
Ejemplo 95: (c/s/írans)-W-(3-Cloro-4-fluorofenil)-4-ciano-3-((4-hidroxiciclohexil)tio)benzamida

Preparada de manera análoga al Ejemplo 67, pero usando el Intermedio T y 4-hidroxiciclohexanotiol. LC-MS: 405 (M+H)+.
Ejemplo 96A: Ácido (1S,3fl)-3-((5-((3-cloro-4-fluorofenil)carbamoil)-2-fluorofenil)tio)-1-etilciclobutanocarboxílico

Etapa 1: A un recipiente lavado con nitrógeno que contenía NaH (82 mg, 3,4 mmoles) se añadió THF (6 ml), seguido de la adición gota a gota de 3-hidroxiciclobutanocarboxilato de metilo (0,360 ml, 3,4 mmoles). La suspensión resultante se dejó agitar a 0°C durante 35 minutos, y después a temperatura ambiente durante 45 min. En un matraz de fondo redondo separado, enjuagado con nitrógeno, se enfrió diisopropilamina (0,560 ml, 4 mmoles) en THF (5 ml) a -78°C antes de añadir gota a gota n-BuLi (2,0 M en hexanos, 2 ml, 4 mmoles) a lo largo de 5 min. La disolución resultante se dejó agitar a -78°C durante 30 minutos, y después a 0°C durante 30 min. El LDA así generado se añadió entonces gota a gota a la suspensión de alcóxido de sodio a -78°C. Finalmente, se añadió yoduro de etilo (0,320 ml, 4 mmoles), y se agitó a 0°C durante 15 minutos y después a TA durante 2,5 horas. La mezcla bruta se paralizó con NH4Cl sat., y se extrajo en éter. Los orgánicos combinados se lavaron entonces adicionalmente con salmuera, se secaron sobre Na2Sü4, se filtraron, y el filtrado se concentró a vacío.
La purificación por cromatografía en columna (SiÜ2 , 0-50% EtOAc/hexanos) dio 1 -etil-3-hidroxiciclobutanocarboxilato de metilo (277 mg, 51% de rendimiento).
Etapa 2: A una disolución en DCM (4 ml) del alcohol (135 mg, 0,84 mmoles) se añadió trietilamina (0,164 ml, 1,2 mmoles). La disolución se enfrió hasta 0°C antes de que se añadiera gota a gota cloruro de metanosulfonilo (0,085 ml, 1,1 mmoles). La mezcla resultante se dejó calentar hasta TA durante 13 h. La reacción se diluyó con DCM y se lavó con agua, después se secó sobre Na2SÜ4, se filtró, y el filtrado se concentró a vacío. No fue necesaria una purificación adicional para proporcionar 1-etil-3-((metilsulfonil)oxi)ciclobutanocarboxilato de metilo.
Etapa 3: A una disolución en DMSO (5 ml) de ácido 4-fluoro-3-mercaptobenzoico (80 mg, 0,45 mmoles), se añadieron 1-etil-3-((metilsulfonil)oxi)ciclobutanocarboxilato de metilo (146 mg, 0,54 mmoles) y carbonato de cesio (222 mg, 0,68 mmoles). La suspensión resultante se selló entonces y se calentó a 100°C durante 72 h. Después de enfriar hasta TA, la mezcla de reacción se diluyó con éter y se lavó secuencialmente con agua y salmuera. El extracto orgánico se secó entonces sobre Na2SO4, se filtró, y el filtrado se concentró a vacío. La purificación por cromatografía en columna (SiO2 , 0-20% EtOAc/hexanos) proporcionó ácido 3-((3-etil-3-(metoxicarbonil)ciclobutil)tio)-4-fluorobenzoico como un aceite naranja oscuro (44 mg, 31% de rendimiento).
Etapa 4: A una disolución en DMF (5 ml) del ácido benzoico (44 mg, 0,141 mmoles) se añadió diisopropiletilamina (0,038 ml, 0,211 mmoles) y 3-cloro-4-fluoroanilina (21 mg, 0,141 mmoles). La mezcla de reacción se agitó a TA durante 1 hora antes de añadir HATU (64 mg, 0,141 mmoles). La disolución resultante se agitó a TA durante la noche. La mezcla de reacción se paralizó con agua, y se extrajo con EtOAc. Los orgánicos combinados se lavaron adicionalmente con agua y salmuera, se secaron sobre Na2SO4, se filtraron, y el filtrado se concentró a vacío . La purificación por HPLC preparativa proporcionó la separación de los 2 diastereoisómeros.
Etapa 5: A una disolución en THF/MeOH (3 ml/3 ml) del éster (50 mg, 0,113 mmoles) se añadió LiOH 2N (2 ml). La mezcla resultante se agitó a TA durante 13 h, después de lo cual se diluyó con EtOAc y se lavó secuencialmente con HCl 1N, agua y salmuera, y los volátiles se eliminaron a vacío. La mezcla resultante se sometió a purificación por HPLC preparativa para proporcionar el compuesto del título (25 mg, 52% de rendimiento). LC-MS: 426 (M+H)+.
Ejemplo 96B: Ácido (1fl,3S)-3-((5-((3-cloro-4-fluorofenil)carbamoil)-2-fluorofenil)tio)-1-etilciclobutanocarboxílico

Preparado de manera análoga al Ejemplo 96A. LC-MS: 426 (M+H)+.
Ejemplo 97: Ácido (±)-4-((5-((3-cloro-4-fluorofenil)carbamoil)-2-fluorofenil)tio)cicloheptanocarboxílico

Preparado de manera análoga al Ejemplo 82, pero usando en su lugar 4-oxocicloheptanocarboxilato de metilo como material de partida en la etapa 1. lC-MS: 442 (m H)+.

Intermedio W: (Acido 4-fluoro-3-mercaptobenzoico): Se añadió HCl concentrado (120,0 ml) a una mezcla pura de ácido 3-clorosulfonil-4-fluorobenzoico (32,0 g, 134,0 mmoles) y dihidrato de cloruro de estaño (90,0 g, 400 mmoles). La disolución de reacción se calentó a reflujo a 100°C durante la noche, se enfrió hasta TA, se diluyó con agua destilada (200 ml), y después se basificó hasta un pH de 10 usando disolución acuosa de hidróxido de sodio 1,0 N. Los sólidos resultantes se separaron por filtración, y el filtrado se acidificó hasta pH 1 usando una disolución de HCl 1N. Se separó
un sólido blanco por filtración, y se lavó con agua y hexanos para proporcionar el compuesto del título (23,2 g, rendimiento cuantitativo). LC-MS: 171 (M-H)-.
Ejemplo 98: Ácido (c/'s/frans)-3-((2-fluoro-5-((3,4,5-trifluorofenil)carbamoil)fenil)tio)biciclo[3.1.0]hexano-6-carboxílico

Etapa 1: A una disolución en DMSO (6,0 ml) del intermedio W (172,0 mg, 1 mmol) y 3-((metilsulfonil)oxi)biciclo[3.1.0]hexano-6-carboxilato de etilo (248,3 mg, 1 mmol) se añadió carbonato de cesio anhidro (986,4 mg, 3 mmoles), y la disolución de reacción fue desoxigenado mediante purga debajo de la superficie con nitrógeno durante 5 minutos. El matraz se selló y se calentó a 60°C durante 4 horas, se enfrió hasta TA, y se repartió entre HCl 1N y EtOAc. Las fases orgánicas combinadas se lavaron con agua y salmuera, se secaron sobre MgSO4 anhidro, se filtraron y se concentraron a vacío para proporcionar el aducto bruto, ácido 3-((6-(etoxicarbonil)biciclo[3.1.0]hexan-3-il)tio)-4-fluorobenzoico). LC-MS: 323 (M-H)-.
Etapa 2: El acoplamiento HATU de ácido 3-((6-(etoxicarbonil)biciclo[3.1.0]hexan-3-il)tio)-4-fluorobenzoico bruto y 3,4,5-trifluoranilina, ejemplificado en ejemplos anteriores, proporcionó 3-((2-fluoro-5-((3,4,5-trifluorofenil)carbamoil)fenil)tio)biciclo[3.1.0]hexano-6-carboxilato) de etilo bruto. LC-MS: 452 (M-H)-.
Etapa 3: A una disolución en THF/MeOH (15 ml/15 ml) del 3-((2-fluoro-5-((3,4,5-trifluorofenil)carbamoil)fenil)tio)biciclo[3.1.0]hexano-6-carboxilato) de etilo bruto se añadió LiOH 2N (10 ml). La mezcla resultante se agitó a TA durante 13 h, después de lo cual se diluyó con EtOAc y se lavó secuencialmente con HCl 1N, agua y salmuera, y los volátiles se eliminaron a vacío. La mezcla resultante se sometió a purificación por HPLC preparativa para proporcionar el compuesto del título como un sólido blanquecino (106,4 mg, 25% de rendimiento global en 3 etapas). LC-MS: 424 (M-H)-.
Ejemplo 99: (c/'s/írans)-3-((2-Fluoro-5-((3,4,5-trifluorofenil)carbamoil)fenil)tio)-W-(metilsulfonil)biciclo[3.1.0]hexano-6-carboxamida

A una disolución en DCM (30 ml) del Ejemplo 98, ácido (3-((2-fluoro-5-((3,4,5-trifluorofenil)carbamoil)fenil)tio)biciclo[3.1.0]hexano-6-carboxílico (555,0 mg, 1,30 mmoles), se añadió sucesivamente DMAP (318,0 mg, 2,6 mmoles), EDCl (404,0 mg, 2,6 mmoles) y metanosulfonamida (247,0 mg 2,6 mmoles) bajo atmósfera de N2. El matraz de reacción se selló y se agitó a TA durante la noche. La concentración de la mezcla de reacción a vacío, y la purificación del residuo mediante HPLC preparativa (acetonitrilo al 30-100% en agua que contiene TFA al 0,1%) proporcionó el compuesto del título como un sólido blanquecino (136,0 mg, rendimiento del 21%). LC-MS: 501 (M-H)-.
Ejemplo 100: 4-Fluoro-3-(((1S,4S)-4-((metilsulfonil)carbamoil)ciclohexil)tio)-W-(3,4,5-tnfluorofenil)benzamida

Preparada de manera análoga a los Ejemplos 98 y 99, pero usando f/-ans-4-((tosilsulfonil)oxi)ciclohexanocarboxilato de etilo como material de partida para proporcionar el compuesto del título como un sólido blanquecino. LC-MS: 503 (M-H)-.
Ejemplo 101: Ácido (±)-2-(3-((2-fluoro-5-((3,4,5-trifluorofenil)carbamoil)fenil)tio)ciclopentil)acético

Etapa 1: Preparado según el procedimiento descrito en House, H. O. et.al. J. Org. Chem 1983, 48, 1643
Etapa 2: A una disolución en MeOH (15 ml) de 2-oxabiciclo[3.2.1]octan-3-ona, se añadió HCl concentrado (1 ml) y se calentó a reflujo durante 3 h. Después de enfriar hasta TA, la mezcla de reacción se diluyó con EtOAc y se lavó secuencialmente con agua y salmuera, se secó sobre Na2SO4, se filtró, y el filtrado se concentró a vacío. No fue necesaria una purificación adicional para proporcionar 2-(3-hidroxiciclopentil)acetato de metilo.
Etapa 3: A una disolución en DCM (4 ml) del alcohol (806 mg, 5,10 mmoles) se añadió trietilamina (1,0 ml, 7,65 mmoles). La disolución se enfrió hasta 0°C antes de añadir gota a gota cloruro de metanosulfonilo (0,590 ml, 7,65 mmoles). La mezcla resultante se dejó calentar hasta TA durante 13 h. La reacción se diluyó con DCM y se lavó con agua, se secó (Na2SO4), se filtró, y el filtrado se concentró a vacío. No fue necesaria ninguna purificación adicional para proporcionar 2-(3-((metilsulfonil)oxi)ciclopentil)acetato de metilo.
Etapa 4: A una disolución en DMSO (5 ml) de ácido 4-fluoro-3-mercaptobenzoico (150 mg, 0,872 mmoles), se añadieron 2-(3-((metilsulfonil)oxi)ciclopentil)acetato de metilo (246 mg, 1,04 mmoles) y carbonato de cesio (425 mg, 1,31 mmoles) y yoduro de sodio (cat.). La suspensión resultante se selló entonces y se calentó a 80°C durante 16 h. Después de enfriar hasta TA, la mezcla de reacción se diluyó con éter y se lavó secuencialmente con agua y salmuera. El extracto orgánico se secó entonces sobre Na2SO4, se filtró, y el filtrado se concentró a vacío. La purificación por HPLC preparativa proporcionó ácido 4-fluoro-3-((3-(metoxicarbonil)ciclopentil)tio)benzoico como un sólido blanco (52 mg, 20% de rendimiento).
Etapa 4: A una disolución en DMF (5 ml) de ácido 4-fluoro-3-((3-(metoxicarbonil)ciclopentil)tio)benzoico (52 mg, 0,166 mmoles), diisopropiletilamina (0,045 ml, 0,249 mmoles) y 3,4,5-trifluoroanilina (25 mg, 0,166 mmoles). La mezcla se agitó a TA durante 1 hora antes de que se añadieran HATU (95 mg, 0,249 mmoles) y DMAP (20 mg, 0,166 mmoles). La disolución resultante se agitó a TA durante la noche, se paralizó con agua y se extrajo con EtOAc. Los orgánicos combinados se lavaron adicionalmente con agua y salmuera, se secaron sobre Na2SO4, se filtraron, y el filtrado se concentró a vacío. La purificación por cromatografía en columna (SiO2 , 0-100% EtOAc/hexanos) dio 3-((2-fluoro-5-((3,4,5-trifluorofenil)carbamoil)fenil)tio)ciclopentanocarboxilato de metilo (22 mg, 73% de rendimiento).
Etapa 6: A una disolución en THF/MeOH (3 ml/3 ml) del éster (22 mg, 0,0498 mmoles) se añadió LiOH 2N (2 ml). La mezcla resultante se agitó a TA durante 13 h, después de lo cual se diluyó con EtOAc y se lavó secuencialmente con HCl 1N, agua y salmuera, y los volátiles se eliminaron a vacío. La mezcla resultante se sometió a purificación por medio de HPLC preparativa para proporcionar el compuesto del título (15 mg, 72% de rendimiento). LC-MS: 428 (M+H)+.
Ejemplo 102: Ácido (±)-2-(3-((5-((3-cloro-4-fluorofenil)carbamoil)-2-fluorofenil)tio)ciclopentil)acético

Preparado de manera análoga al Ejemplo 102, pero usando en su lugar 3-cloro-4-fluoroanilina en la etapa 4. LC-MS: 426 (M+H)+.
Ejemplo 103: (c/s/frans)-3-((4-(Dimetilcarbamoil)ciclohexil)tio)-4-fluoro-W-(3,4,5-trifluorofenil)benzamida

A una disolución en DMF (5 ml) del Ejemplo 62 (35 mg, 0,082 mmoles) se añadieron diisopropiletilamina (0,022 ml, 0,123 mmoles) y dimetilamina (0,041 ml, 0,082 mmoles). La mezcla de reacción se agitó a TA durante 1 hora antes de añadir HATU (37 mg, 0,0984 mmoles). La disolución resultante se agitó a TA durante 13 h, se paralizó con agua, y se extrajo con EtOAc. Los orgánicos combinados se lavaron adicionalmente con agua y salmuera, se secaron (Na2SO4), se filtraron, y el filtrado se concentró a vacío. La purificación por HPLC preparativa proporcionó el compuesto del título (25 mg, 67% de rendimiento). LC-MS: 455 (M+H)+.
Ejemplo 104: (c/s/frans)-4-Fluoro-3-((4-(piperidin-1-carbonil)ciclohexil)tio)-W-(3,4,5-trifluorofenil)benzamida

Preparada de manera análoga al Ejemplo 103, pero usando piperidina. LC-MS: 495 (M+H)+.
Ejemplo 105: Ácido (±)-2-(3-((5-((3,4-difluorofenil)carbamoil)-2-fluorofenil)tio)ciclopentil)acético Preparado de manera análoga al Ejemplo 102, pero usando 3,4-difluoroanilina en la etapa 4. LC-MS: 410 (M+H)+. Ejemplo 106: Ácido (c/s/frans)-2-(4-((5-((3-cloro-4-fluorofenil)carbamoil)-2-fluorofenil)tio)ciclohexil)acético

Etapa 1; A una disolución de 2-(4-hidroxiciclohexil)acetato de metilo (1,00 g, 5,81 mmoles) en DCM (20 ml) a 0°C se añadió TEA (1,21 ml, 8,72 mmoles), dimetilaminopiridina (0,035 g, 0,29 mmoles) y cloruro de 4-metilbenceno-1 -sulfonilo (1,22 g, 6,39 mmoles). La mezcla resultante se dejó calentar lentamente hasta TA, y se agitó durante 48 h. La reacción se evaporó, después se diluyó con EtOAc, y se añadió una disolución ac. al 20% de ácido cítrico monohidratado. Los orgánicos se separaron, se lavaron con agua, y se concentraron a vacío. El material bruto resultante se purificó sobre gel de sílice, eluyendo con un gradiente de disolvente de 0% a 50% de EtOAc en hexanos, para proporcionar 2-(4-(tosiloxi)ciclohexil)acetato de metilo puro.
Etapa 2; Se agitaron, a 0°C, ácido 4-fluoro-3-mercaptobenzoico (0,40 g, 2,32 mmol, intermedio W) y DMF (23 ml) mientras se purgaba con nitrógeno subsuperficial. A la disolución fría resultante se añadió Cs2CO3 (1,51 g, 8,72 mmoles) y 2-(4-hidroxiciclohexil)acetato de metilo (0,83 g, 2,55 mmoles). El baño de hielo se retiró, y la reacción se agitó a 60°C durante 16 h. La reacción se diluyó con EtOAc y una disolución ac. al 20% de ácido cítrico monohidratado. Los orgánicos se separaron, se lavaron con una disolución ac. al 10% de ácido cítrico monohidratado, dos veces con agua, se concentraron a vacío, y se evaporaron conjuntamente con heptanos. El material bruto resultante se purificó en gel de sílice, eluyendo con un gradiente de disolvente de 10% a 70% de acetona en hexanos, para proporcionar ácido 4-fluoro-3-((4-(2-metoxi-2-oxoetil)ciclohexil)tio)benzoico puro. Etapa 3; A una disolución agitada de ácido 4-fluoro-3-((4-(2-metoxi-2-oxoetil)ciclohexil)tio)benzoico (0,19 g, 0,58 mmoles) en DMF anhidra (6 ml) se añadió HATU (0,27 g, 0,70 mmoles), 3-cloro-4-fluoroanilina (0,093 g, 0,64 mmoles), DIPEA (0,31 ml, 1,74 mmoles) y una cantidad catalítica de DMAP. La mezcla resultante se agitó a temperatura ambiente durante 16 h. La reacción se diluyó con EtOAc y HCl 0,5 M. Los orgánicos se lavaron entonces una vez más con HCl 0,5 M, dos veces con agua, se concentraron a vacío, y se evaporaron conjuntamente con EtOAc. El material bruto resultante se purificó sobre gel de sílice, eluyendo con un gradiente de disolvente de 0% a 40% de EtOAc en hexanos, para proporcionar 2-(4-((5-((3-cloro-4-fluorofenil)carbamoil)-2-fluorofenil)tio)ciclohexil)acetato de metilo.
Etapa 4; 2-(4-((5-((3-Cloro-4-fluorofenil)carbamoil)-2-fluorofenil)tio)ciclohexil)acetato de metilo (0,16 g, 0,35 mmoles), Th F (4 ml), MeOH (4 ml), LiOH 1 M (2,82 ml, 2,82 mmoles) y agua (1 ,2 ml) se agitaron a TA durante 16 h. La reacción se concentró y se diluyó con EtOAc y una disolución ac. al 20% de ácido cítrico monohidratado. Los orgánicos se separaron, se lavaron tres veces con agua, y se concentraron a vacío. El sólido resultante proporcionó el compuesto del título en forma pura. LC-MS: 440 (M+H)+.
Ejemplo 107: Ácido (c/'s/írans)-2-(4-((5-((3,4,5-trifluorofenil)carbamoil)-2-fluorofenil)tio)ciclohexil)acético
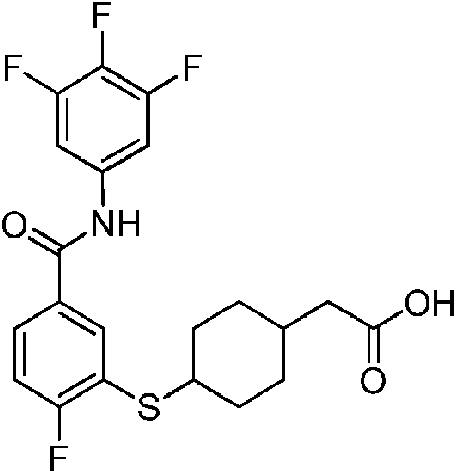
El compuesto del título se preparó de manera análoga al Ejemplo 106 usando 3,4,5-trifluoroanilina en la etapa 3. LC-MS: 442 (M+H)+.
Ejemplo 108: (c/'s/frans)-4-Fluoro-3-((4-(piperazin-1-carbonil)ciclohexil)tio)-W-(3,4,5-trifluorofenil)benzamida

Etapa 1: A una disolución en DMF (5 ml) del Ejemplo 62 (35 mg, 0,082 mmoles) se añadió diisopropiletilamina (0,022 ml, 0,123 mmoles) y 1-piperazin-carboxilato de terc-butilo (15 mg, 0,082 mmoles). La mezcla se agitó a TA durante 1 hora antes de añadir HATU (37 mg, 0,0984 mmoles). La disolución resultante se agitó a TA durante 13 h, se paralizó con agua, y se extrajo con EtOAc. Los orgánicos combinados se lavaron adicionalmente con agua y salmuera, se secaron sobre Na2SO4, se filtraron, y el filtrado se concentró a vacío. La purificación por HPLC preparativa proporcionó 4-(4-((2-fluoro-5-((3,4,5-trifluorofenil)carbamoil)fenil)tio)ciclohexanocarbonil)-piperazin-1-carboxilato de terc-butilo (20 mg, 42% de rendimiento). LC-MS: 455 (M+H)+.
Etapa 2: A una disolución en DCM (5 ml) de la amida (20 mg, 0,033) se añadió ácido trifluoroacético (1 ml), y se agitó a TA durante 2 h. La disolución resultante se concentró a vacío, y luego se diluyó con EtOAc y se lavó con disolución de NaHCO3 sat. Los orgánicos combinados se lavaron adicionalmente con agua y salmuera, se secaron sobre Na2SO4, se filtraron, y el filtrado se concentró a vacío para proporcionar el compuesto del título como un sólido blanco (15 mg, 94% de rendimiento). LC-MS: 496 (M+H)+.
Ejemplo 109: Ácido (c/s/írans)-4-(((5-((3-cloro-4-fluorofenil)carbamoil)-2-fluorofenil)tio)metil)ciclohexanocarboxílico

El compuesto del título se preparó de manera análoga al Ejemplo 106 usando 4-(hidroximetil)ciclohexanocarboxilato de metilo en la etapa 1. LC-m S: 440 (M+H)+.
Ejemplo 110: Ácido (c/s/írans)-4-(((5-((3,4,5-trifluorofenil)carbamoil)-2-fluorofenil)tio)metil)ciclohexanocarboxílico

El compuesto del título se preparó de manera análoga al Ejemplo 106 usando 4-(hidroximetil)ciclohexanocarboxilato de metilo en la etapa 1 y 3,4,5-trifluoroanilina en la etapa 3. LC-MS: 442 (M+H)+.
Ejemplo 111: Ácido (c/'s/frans)-4-((2-fluoro-5-((3,4,5-trifluorofenil)carbamoil)fenil)tio)-1-hidroxiciclohexanocarboxílico

Etapa 1: A una disolución en DMSO (3,0 ml) del intermedio E (4-fluoro-3-mercapto-W-(3,4,5-trifluorofenil)benzamida) (100,0 mg, 0,33 mmoles) y 1 -hidroxi-4-(tosiloxi)ciclohexanocarboxilato de metilo (110 mg, 0,33 mmoles) se añadió carbonato de cesio anhidro (325 mg, 1,0 mmoles). La disolución resultante se desoxigenó mediante purga debajo de la superficie con nitrógeno durante 5 minutos, y la vasija de reacción se selló entonces y se calentó a 60°C durante la noche. Después de enfriar hasta TA, la disolución se acidificó con HCl 0,5 N y se extrajo con EtOAc. La fase orgánica se lavó con agua y salmuera, se secó sobre sulfato de sodio anhidro, se filtró y se concentró a vacío para proporcionar el éster bruto, 4-((2-fluoro-5-((3,4,5-trifluorofenil)carbamoil)fenil)tio)-1 -hidroxiciclohexanocarboxilato de metilo. LC-MS: 456 (M-H)-.
Etapa 2: A una disolución en THF/MeOH (3 ml/3 ml) del residuo bruto en la etapa 1, 4-((2-fluoro-5-((3,4,5-trifluorofenil)carbamoil)fenil)tio)-1-hidroxiciclohexanocarboxilato de metilo, se añadió LiOH 2N (3 ml). La mezcla resultante se agitó a TA durante 13 h, después de lo cual se diluyó con EtOAc y se lavó secuencialmente con HCl 1N, agua y salmuera, y los volátiles se eliminaron a vacío. La mezcla resultante se sometió a purificación por medio de HPLC preparativa para proporcionar el compuesto del título como un color blanquecino (25,0 mg, rendimiento global del 17% en 2 etapas). LC-MS: 442 (M-H)-.
Ejemplo 112A: Ácido c/'s-4-((2-fluoro-5-((3,4,5-trifluorofenil)carbamoil)fenil)tio)ciclohexanocarboxílico

Preparado de manera análoga al Ejemplo 111, pero usando írans-4-((tosilsulfonil)oxi)ciclohexanocarboxilato de etilo como material de partida.). LC-MS: 428 (M-H)-.
Ejemplo 112B: Ácido frans-4-((2-fluoro-5-((3,4,5-trifluorofenil)carbamoil)fenil)tio)ciclohexanocarboxílico

Preparado de manera análoga al Ejemplo 111, pero usando cis-4-((tosilsulfonil)oxi)ciclohexanocarboxilato de etilo como material de partida). LC-MS: 428 (M-H)-.
Ejemplo 113: Ácido (±)-3-((2-fluoro-5-((3,4,5-trifluorofenil)carbamoil)fenil)tio)biciclo[4.1.0]heptano-7-carboxílico

Etapa 1; A una disolución de 9-BBN (disolución 0,5 M en THF, 40,00 ml, 20,00 mmoles) a 0°C se añadió lentamente ciclohexa-1,4-dieno (1,88 ml, 20,00 mmoles). La mezcla resultante se dejó calentar lentamente a TA y se agitó durante 16 h. A la disolución agitada, a TA, se añadió NaOH 3 M (6 ml), seguido de la adición gota a gota de una disolución ac. al 30% de peróxido de hidrógeno. La mezcla de reacción se calentó a reflujo durante 1 hora, después se enfrió hasta TA y se diluyó con Et2O y salmuera. Los orgánicos se separaron, se lavaron con agua y se concentraron a vacío. El material bruto resultante se purificó sobre gel de sílice, eluyendo con un gradiente de disolvente de 0% al 60% de EtOAc en hexanos, para proporcionar ciclohex-3-enol.
Etapa 2; Se agitó ciclohex-3-enol (0,90 g, 9,17 mmoles) en DCM (20 ml) a 0°C, y se añadió TEA (1,53 ml, 11,00 mmoles) y dimetilaminopiridina (1,06 g, 8,71 mmoles). La mezcla resultante se agitó fría durante 5 minutos, después se añadió terc-butilclorodimetilsilano (1,38 g, 9,17 mmoles). La disolución se dejó calentar lentamente a TA y se agitó durante 16 h. La reacción se evaporó, después se diluyó con Et2O y HCl 1 M. Los orgánicos se separaron, se lavaron una vez más con HCl 1 M, con agua, y se secaron sobre Na2SO4, se filtraron, y se concentraron a vacío. El aceite bruto resultante que contiene íerc-butil(ciclohex-3-en-1-iloxi)dimetilsilano se llevó a la siguiente etapa sin purificación adicional.
Etapa 3; A una disolución agitada de íerc-butil(ciclohex-3-en-1-iloxi)dimetilsilano (1,00 g, 4,71 mmoles) en DCM (40 ml) se añadió tetraacetato de di-rodio (0,042 g, 0,094 mmoles), seguido de adición muy lenta (durante 6 h, vía una bomba de jeringa) de 2-diazoacetato de etilo (disolución al 87% en DCM, 0,69 ml, 5,65 mmoles). La mezcla resultante se agitó a TA durante 16 h. La reacción se filtró a través de papel, y se concentró a vacío. El material bruto resultante se purificó sobre gel de sílice, eluyendo con un gradiente de disolvente de 0% al 40% de EtOAc en hexanos, para proporcionar 3-((íe/'c-butildimetilsilil)oxi)biciclo[4.1.0]heptano-7-carboxilato de etilo.
Etapa 4; A una disolución de 3-((íerc-butildimetilsilil)oxi)biciclo[4.1.0]heptano-7-carboxilato de etilo (0,80 g, 2,68 mmoles) en THF (30 ml) se añadió TBAF (disolución 1,0 M en THF; 5,36 ml, 5,36 mmoles). La disolución se agitó entonces a TA durante 48 h. La reacción se concentró y se diluyó con EtOAc y agua. Los orgánicos se lavaron una vez más con agua, se concentraron a vacío, y se evaporaron conjuntamente con EtOAc. El material bruto resultante que contenía 3-hidroxibiciclo[4.1.0]heptano-7-carboxilato de etilo se usó sin purificación adicional.
Etapa 5; El compuesto del título se preparó de manera análoga al Ejemplo 106 usando 3-hidroxibiciclo[4.1.0]heptano-7-carboxilato de etilo en la etapa 1. LC-MS: 440 (M+H)+.
Ejemplo 114: (c/s/frans)-3-((4-((Ciclopropilsulfonil)carbamoil)ciclohexil)tio)-4-fluoro-W-(3,4,5-trifluorofenil)benzamida

A una disolución en DCM (10 ml) del Ejemplo 62 (34 mg, 0,086 mmoles), se añadieron ciclopropilsulfonamida (14 mg, 0,119 mmoles), DMAP (15 mg, 0,119 mmoles) y EDCI (23 mg, 0,119 mmoles), y se agitó a TA durante 16 h. La mezcla de reacción se concentró a vacío, y el residuo se purificó por HPLC preparativa para proporcionar el compuesto del título (25 mg, 45% de rendimiento). LC-MS: 531 (M+H)+.
Ejemplo 115: (c/s/frans)-3-((4-(Etilcarbamoil)ciclohexil)tio)-4-fluoro-W-(3,4,5-trifluorofenil)benzamida

Preparada de manera análoga al Ejemplo 104, pero usando etilamina. LC-MS: 455 (M+H)+.
Ejemplo 116: (c/s/frans)-4-Fluoro-3-((4-(morfolin-4-carbonil)ciclohexil)tio)-W-(3,4,5-trifluorofenil)benzamida

Preparada de manera análoga al Ejemplo 104, pero usando morfolina. LC-MS: 497 (M+H)+.
Ejemplo 117: (c/s/frans)-3-((4-(2,6-Diazaspiro[3.3]heptano-2-carbonil)ciclohexil)tio)-4-fluoro-W-(3,4,5-trifluorofenil)benzamida

Preparada de manera análoga al Ejemplo 10, pero usando en su lugar 2,6-diazaspiro[3.3]heptano-2-carboxilato de terc-butil en la etapa 1. LC-MS: 508 (M+H)+.
Ejemplo 118: Ácido (c is /tran s)-1 -etil-4-((2-fluoro-5-((3,4,5-trifluorofenil)carbamoil)fenil)tio)ciclohexanocarboxílico
Etapa 1: Preparada de manera análoga al Ejemplo 111 (Etapa 1), pero usando 1 -etil-4-(tosiloxi)ciclohexanocarboxilato de etilo, para proporcionar el éster intermedio, (1-etil-4-((2-fluoro-5-((3,4,5-trifluorofenil)carbamoil)fenil)tio)ciclohexanocarboxilato) de etilo. LC-MS: 484 (M+H)+.
Etapa 2: A una disolución en 1,4-dioxano/MeOH (0,5 ml/0,5 ml) del residuo bruto en la etapa 1, (1-etil-4-((2-fluoro-5-((3,4,5-trifluorofenil)carbamoil)fenil)tio)ciclohexanocarboxilato de etilo (36,0 mg, 0,074 mmoles), se añadió disolución acuosa de NaOH 12,5N (3 ml, 37,5 mmoles). La disolución de reacción se agitó a 50°C durante 16 h, se enfrió hasta TA, y se acidificó con HCl 1N, y luego se extrajo con EtOAc. Las fases orgánicas se lavaron con agua y salmuera, se secaron sobre sulfato de sodio anhidro, se filtraron, y se concentraron a vacío, y se purificaron por HPLC para proporcionar el compuesto del título como un sólido blanquecino (10,1 mg, 30% de rendimiento). LC-MS: 456 (M+H)+.
Ejemplo 119: Ácido (c/'s/frans)-4-((5-((3-cloro-4-fluorofenil)carbamoil)-2-fluorofenil)tio)-1-metilciclohexanocarboxílico

Etapa 1: A una disolución en MeOH (40 ml) de 1-metil-4-oxociclohexanocarboxilato de metilo (1,38 g, 8,11 mmoles), enfriada hasta 0°C, se añadió NaBH4 (450 mg, 12,16 mmoles). La mezcla resultante se dejó calentar hasta TA durante 13 h. La mezcla de reacción se paralizó con HCl 1N, y se extrajo con EtOAc dos veces. Los orgánicos combinados se lavaron entonces con salmuera, se secaron sobre Na2SO4, se filtraron, y el filtrado se concentró a vacío. No fue necesaria una purificación adicional para proporcionar 4-hidroxi-1-metilciclohexanocarboxilato de metilo.
Etapa 2: A una disolución en DCM (45 ml) del alcohol (610 mg, 3,55 mmoles) se añadió trietilamina (0,736 ml, 5,32 mmoles). La disolución se enfrió hasta 0°C antes de que se añadiera gota a gota cloruro de metanosulfonilo (0,410 ml, 5,32 mmoles). La mezcla resultante se dejó calentar hasta TA durante 13 h. La reacción se diluyó con DCM y se lavó con agua, después se secó sobre Na2SO4, se filtró, y el filtrado se concentró a vacío. No fue necesaria una purificación adicional para proporcionar 1-metil-4-((metilsulfonil)oxi)ciclohexanocarboxilato de metilo.
Etapa 3: A una disolución en acetona (80 ml) del mesilato (812 mg, 3,24 mmoles) se añadió yoduro de sodio (4,8 g, 32,4 mmoles), y la disolución resultante se agitó a 80°C en la oscuridad durante 16 h. Los volátiles se evaporaron a continuación a vacío, y después se repartieron con EtOAc y agua. Los orgánicos combinados se lavaron adicionalmente con Na2S2O3 ac. al 10% y salmuera, después se secaron sobre Na2SO4, se filtraron, y el filtrado se concentró a vacío. No fue necesaria una purificación adicional para proporcionar 4-yodo-1-metilciclohexanocarboxilato de metilo.
Etapa 4: A una disolución en DMSO (15 ml) de ácido 4-fluoro-3-mercaptobenzoico (55 mg, 0,319 mmoles), se añadieron 4-yodo-1-metilciclohexanocarboxilato de metilo (111 mg, 0,382 mmoles) y carbonato de cesio (156 mg, 0,479 mmoles). La suspensión resultante se selló entonces y se calentó a 60°C durante 13 h. Después de enfriar hasta TA, la mezcla de reacción se diluyó con éter, y se lavó secuencialmente con HCl 1N, agua y salmuera. La fase orgánica se secó entonces sobre Na2SO4, se filtró, y el filtrado se concentró a vacío. La purificación por cromatografía en columna (SiO2 , 0-60% EtOAc/hexanos) proporcionó ácido 4-fluoro-3-((4-(metoxicarbonil)-4-metilciclohexil)tio)benzoico.
Etapa 5: A una disolución en DMF (5 ml) del ácido benzoico (30 mg, 0,0920 mmoles) se añadió diisopropiletilamina (0,020 ml, 0,138 mmoles) y 3-cloro-4-fluoroanilina (13 mg, 0,0920 mmoles). La mezcla de reacción se agitó a TA durante 1 hora antes de añadir HATU (42 mg, 0,110 mmoles). La disolución resultante se agitó a TA durante la noche, se paralizó con agua, y se extrajo con EtOAc. Los orgánicos combinados se lavaron adicionalmente con agua y salmuera, se secaron sobre Na2SO4, se filtraron, y el filtrado se concentró a vacío. La purificación por cromatografía en columna (SiO2 , 0-100% EtOAc/hexanos) produjo 4-((5-((3-cloro-4-fluorofenil)carbamoil)-2-fluorofenil)tio)-1-metilciclohexanocarboxilato de metilo. (30 mg, 62% de rendimiento).
Etapa 6: A una disolución en THF/MeOH (3 ml/3 ml) del éster (30 mg, 0,066 mmoles) se añadió LiOH 2N (2 ml). La mezcla resultante se agitó a TA durante 13 h, después de lo cual se diluyó con EtOAc y se lavó secuencialmente con HCl 1N, agua y salmuera, y los volátiles se eliminaron a vacío. La mezcla resultante se sometió a purificación por HPLC preparativa para proporcionar el compuesto del título (20 mg, 68% de rendimiento). LC-MS: 440 (M+H)+.
Ejemplo 120: Ácido (±)-3-((2-fluoro-5-((3,4,5-trifluorofenil)carbamoil)fenil)tio)-1 -metilciclopentanocarboxílico

Etapa 1: A una vasija lavada con nitrógeno que contenía NaH (158 mg, 6,59 mmoles) se añadió THF (6 ml), seguido de la adición gota a gota de 3-hidroxiciclopentanocarboxilato de metilo (950 mg, 6,59 mmoles). La suspensión resultante se dejó agitar a 0°C durante 35 minutos, y después a TA durante 45 min. En un matraz de fondo redondo separado, enjuagado con nitrógeno, se enfrió diisopropilamina 1,1 ml, 7,91 mmoles) en THF (10 ml) a -78°C antes de añadir n-BuLi (2,5 M en hexanos, 3,1 ml, 7,91 mmoles) gota a gota durante 5 minutos. La disolución resultante se dejó agitar a -78°C durante 30 minutos, y después a 0°C durante 30 min. El LDA así generado se añadió entonces gota a gota a la suspensión de alcóxido de sodio a -78°C. Finalmente, se añadió yoduro de metilo (0,492 ml, 7,91 mmoles), y se agitó a 0°C durante 15 minutos, y después a TA durante 2,5 horas. La mezcla bruta se paralizó con disolución de NH4Cl sat., y se extrajo en éter. Los orgánicos combinados se lavaron entonces adicionalmente con salmuera, se secaron sobre Na2SÜ4, se filtraron, y el filtrado se concentró a vacío. La purificación por cromatografía en columna (SiÜ2, 0-100% EtOAc/hexanos) dio 3-hidroxi-1-metilciclopentanocarboxilato de metilo (200 mg, 20% de rendimiento).
Etapa 2: A una disolución en piridina (10 ml) del alcohol (102 mg, 0,645 mmoles) se añadió gota a gota cloruro de p-toluenosulfonilo (184 mg, 1,2 mmoles) como una disolución en piridina (5 ml). La mezcla resultante se dejó calentar hasta TA durante 13 h. La reacción se diluyó con DCM y se lavó con disolución de NaHCÜ3 sat., agua y salmuera. Los orgánicos separados se secaron entonces sobre Na2SÜ4, se filtraron, y el filtrado se concentró a vacío. No fue necesaria una purificación adicional para proporcionar 1-metil-3-(tosiloxi)ciclopentanocarboxilato de metilo.
Etapa 3: A una disolución en DMSÜ (5 ml) de intermedio E (67 mg, 0,222 mmoles), se añadieron 1 -metil-3-(tosiloxi)ciclopentanocarboxilato de metilo (100 mg, 0,333 mmoles) y carbonato de cesio (144 mg, 0,444 mmoles). La suspensión resultante se selló entonces y se calentó a 60°C durante 16 h. Después de enfriar hasta TA, la mezcla de reacción se diluyó con éter y se lavó secuencialmente con agua y salmuera. El extracto orgánico se secó entonces sobre Na2SÜ4, se filtró, y el filtrado se concentró a vacío. La purificación por HPLC preparativa proporcionó 3-((2-fluoro-5-((3,4,5-trifluorofenil)carbamoil)fenil)tio)-1-metilciclopentanocarboxilato de metilo (70 mg, 72% de rendimiento).
Etapa 5: A una disolución en THF/MeÜH (3 ml/3 ml) del éster (70 mg, 0,158 mmoles) se añadió LiÜH 2N (2 ml). La mezcla resultante se agitó a TA durante 13 h, después de lo cual se diluyó con EtÜAc y se lavó secuencialmente con HCl 1N, agua y salmuera, y los volátiles se eliminaron a vacío. La mezcla resultante se sometió a purificación por HPLC preparativa para proporcionar el compuesto del título (40 mg, 60% de rendimiento). LC-MS: 428 (M+H)+.
Ejemplo 121A: Ácido (±)-(1S,3fl)-3-((5-((3-cloro-4-fluorofenil)carbamoil)-2-fluorofenil)tio)ciclohexanocarboxílico

Etapa 1; A una disolución de ácido 3-hidroxiciclohexanocarboxílico (1,25 g, 8,67 mmoles) en MeÜH (20 ml) a TA se añadió cloruro de tionilo (1,26 ml, 17,34 mmoles). La mezcla resultante estaba a 70°C durante 16 h. La reacción se enfrió hasta TA, se evaporó, y se coevaporó secuencialmente una vez con MeÜH y dos veces con EtÜAc para proporcionar 3-hidroxiciclohexanocarboxilato de metilo puro.
Etapa 2; El compuesto del título se preparó de manera análoga al Ejemplo 106 usando 3-hidroxiciclohexanocarboxilato de metilo en la etapa 1 y separación cromatográfica de los diastereoisómeros. LC-MS: 426 (M+H)+.
Ejemplo 121B: Ácido (±)-(1fl,3fl)-3-((5-((3-cloro-4-fluorofenil)carbamoil)-2-fluorofenil)tio)ciclohexanocarboxílico

El compuesto del título se preparó de manera análoga al Ejemplo 106 usando 3-hidroxiciclohexanocarboxilato de metilo en la etapa 1 y separación cromatográfica de diastereoisómeros en la etapa 3. LC-MS: 426 (M+H)+.
Ejemplo 122A: Ácido (±)-(1 S,3fí)-3-((2-fluoro-5-((3,4,5-trifluorofenil)carbamoil)fenil)tio)ciclohexanocarboxílico

El compuesto del título se preparó de manera análoga al Ejemplo 106 usando 3-hidroxiciclohexanocarboxilato de metilo en la etapa 1 y 3,4,5-trifluoroanilina en la etapa 3. La separación cromatográfica de diastereoisómeros se logró en la etapa 3. LC-MS: 426 (M+H)+.
Ejemplo 122B: Ácido (±)-(1 R,3R)-3-((2-fluoro-5-((3,4,5-trifluorofenil)carbamoil)fenil)tio)ciclohexanocarboxílico

El compuesto del título se preparó de manera análoga al Ejemplo 106 usando 3-hidroxiciclohexanocarboxilato de metilo en la etapa 1 y 3,4,5-trifluoroanilina en la etapa 3. La separación cromatográfica de los diastereoisómeros se logró en la etapa 3. LC-MS: 426 (M+H)+.
Las Figuras 5-7 muestran esquemas generales usados para preparar compuestos seleccionados de la invención. Intermedio G: 2-Bromo-W-(3-cloro-4-fluorofenil)isonicotinamida

A una disolución agitada de ácido 2-bromoisonicotínico (6,00 g, 29,70 mmoles) en DMF anhidra (100 ml) se añadió HATU (13,55 g, 35,64 mmoles), 3-cloro-4-fluoroanilina (4,54 g, 31,19 mmoles), base de Hünigs (15,56 ml, 89,10 mmoles) y una cantidad catalítica de DMAP. La mezcla resultante se agitó a TA durante 16 h. La reacción se diluyó con EtOAc y salmuera. La fase orgánica se lavó entonces una vez más con salmuera, HCl 0,5 M (2X), con agua, y se concentró a vacío. El sólido bruto resultante se suspendió en DCM, se agitó durante 20 minutos, y se filtró. El sólido posterior proporcionó el compuesto del título en forma pura. LC-MS: 330 (M+H)+.
Intermedio H: 2-Bromo-W-(3,4,5-trifluorofenil)isonicotinamida

El compuesto del título se preparó de manera análoga al intermedio G usando 3,4,5-trifluoroanilina. LC-MS: 332 (M+H)+.
Intermedio I: 2-Bromo-W-(3,4-difluorofenil)isonicotinamida

El compuesto del título se preparó de manera análoga al intermedio G usando 3,4-difluoroanilina. LC-MS: 314 (M+H)+. Intermedio J: 2-Amino-W-(3-cloro-4-fluorofenil)isonicotinamida

A una disolución agitada de ácido 2-aminoisonicotínico (5,00 g, 36,2 mmoles) en DMF anhidra (100 ml) se añadió HATU (16,5 g, 35,64 mmoles), 3-cloro-4-fluoroanilina (5,27 g, 36,2 mmoles), diisopropiletilamina (19 ml, 110 mmoles) y dimetilaminopiridina (750 mg, 6,1 mmoles). La mezcla resultante se agitó a TA durante 16 horas, después se diluyó con EtOAc y NaHCO3 acuoso saturado. Los orgánicos se lavaron entonces con salmuera, se secaron sobre MgSO4, y se concentraron parcialmente a vacío. El sólido resultante que se desprendió se filtró, y se encontró que era el compuesto del título puro. El filtrado se concentró y se purificó por cromatografía en gel de sílice c18, eluyendo con
un gradiente de 5% a 100% de MeCN en agua que contenía 0,5% de ácido fórmico. El producto que contenía fracciones se concentró, se diluyó con NaHCO3 acuoso saturado, y se extrajo con EtOAc. Los orgánicos se secaron entonces sobre MgSO4 y se concentraron a vacío para proporcionar compuesto de título adicional. LC-MS: 266 (M+H)+.
Intermedio K: 2-Amino-W-(3,4-difluorofenil)isonicotinamida

Preparada de manera análoga al Intermedio D usando 3,4-difluoroanilina.
Intermedio L: Ácido 2-(ciclopropanosulfonamido)isonicotínico

Etapa 1; A una disolución de 2-aminoisonicotinato de metilo (0,408 g, 2,68 mmoles) en piridina anhidra (10 ml) se añadió dimetilaminopiridina (0,01 g, 0,08 mmoles) y cloruro de ciclopropanosulfonilo (0,27 ml, 2,7 mmoles). La mezcla resultante se calentó hasta 60°C durante 16 h. Los volátiles se eliminaron a vacío, y el residuo se diluyó con EtOAc y HCl 1N. Los orgánicos se lavaron con HCl 1N (2X), con salmuera, se secaron sobre MgSO4, se filtraron, y se concentraron a vacío. El material bruto resultante se purificó sobre gel de sílice, eluyendo con un gradiente de disolvente de 0% a 100% de EtOAc en hexanos, para proporcionar 2-(ciclopropanosulfonamido)isonicotinato de metilo.
Etapa 2; A una disolución de 2-(ciclopropanosulfonamido)isonicotinato de metilo (0,400 g, 1,56 mmoles) en metanol (10 ml) y tetrahidrofurano (20 ml) se añadió NaOH 3 N (10 ml), y la mezcla resultante se agitó durante 1 h. La mezcla de reacción se acidificó con HCl 1N y se extrajo con EtOAc (2X). Los orgánicos combinados se lavaron con salmuera, se secaron sobre MgSO4, y los volátiles se eliminaron a vacío para proporcionar el compuesto del título. LC-MS: 243 (M+H)+.
Intermedio M: Ácido 5-(ciclopropanosulfonamido)nicotínico

El compuesto del título se preparó de manera análoga al intermedio L usando 5-aminonicotinato de metilo como material de partida. LC-MS: 243 (M+H)+.
Intermedio N: Ácido 6-(ciclopropanosulfonamido)picolínico
El compuesto del título se preparó de manera análoga al intermedio L usando 6-aminopicolinato de metilo como material de partida. LC-MS: 243 (M+H)+.
Intermedio O: 3-Amino-N-(3-cloro-4-fluorofenil)benzamida

A una disolución de ácido 3-((terc-butoxicarbonil)amino)benzoico (1 g, 4,21 mmoles) en DMF (10 ml) se añadió 3-cloro-4-fluoroanilina (613 mg, 4,21 mmoles), W,N-diisopropiletilamina (1,47 ml, 8,42 mmoles) y HATU (1,76 g, 4,63 mmoles). Se añadió DMAP catalítica (~20 mg), y la mezcla de reacción se agitó a temperatura ambiente durante 16 h. La disolución se diluyó con EtOAc y se lavó con HCl 1N, con salmuera, y la fase orgánica se separó, se secó (MgSO4), se filtró, y se evaporó a vacío. El sólido aislado se purificó usando cromatografía sobre gel de sílice, eluyendo con 0 a 40% de EtOAc en hexanos, para proporcionar el intermedio (3-((3-cloro-4-fluorofenil)carbamoil)fenil)carbamato de terc-butilo como un sólido incoloro. Éste se disolvió en diclorometano (15 ml), al que se añadió TFA (5 ml), y la mezcla de reacción se agitó a temperatura ambiente hasta que el análisis por LCMS determinó la reacción completa. La disolución se diluyó entonces con EtOAc, se paralizó con la adición de NaHCO3 acuoso saturado, la fase orgánica se separó, se secó (MgSO4), se filtró y se evaporó a vacío para proporcionar el compuesto del título.
Compuesto 123: N-(3-Cloro-4-fluorofenil)-6-(ciclopropanosulfonamido)picolinamida

El compuesto del título se preparó de manera análoga al Compuesto 124 usando el Intermedio H como material de partida. LC-MS: 370 (M+H)+.
Compuesto 124: N-(3-Cloro-4-fluorofenil)-2-(ciclopropanosulfonamido)isonicotinamida

A una mezcla de yoduro de cobre (I) (0,23 g, 1,21 mmoles), carbonato de potasio (1,68 g, 12,14 mmoles) y ciclopropilsulfonamida (0,809 g, 6,67 mmoles) en un RBF (matraz de fondo redondo), en atmósfera de nitrógeno, se añadió DMF (60 ml), trans-(1 R,2R)-N,N’-bismetil-1,2-ciclohexanodiamina (0,344 g, 2,42 mmoles) y 2-bromo-N-(3-cloro-4-fluorofenil)isonicotinamida (Intermedio G) (2,00 g, 6,07 mmoles). La mezcla resultante se purgó por debajo de la superficie con gas nitrógeno durante 10 minutos antes de calentar hasta 100°C durante 16 h. La reacción se enfrió hasta TA y se diluyó con EtOAc y HCl 0,5M. Los orgánicos se lavaron dos veces con HCl 0,5 M, con agua (2X), y se concentraron a vacío. El sólido bruto resultante se suspendió en DCM, se agitó durante 1 hora, y se filtró. El sólido resultante proporcionó el compuesto del título en forma pura. LC-MS: 370 (M+H)+.
C o m p u e s to 125 : W -(3 -C lo ro -4 - f lu o ro fe n il) -5 -(c ic lo p ro p a n o s u lfo n a m id o )n ic o t in a m id a

A una disolución agitada de Intermedio G (0,077 g, 0,32 mmoles) en DMF anhidra (3 ml) se añadió HATU (0,150 g, 0,38 mmoles), 3-cloro-4-fluoroanilina (0,046 g, 0,32 mmoles), diisopropiletilamina (0,17 ml, 0,95 mmoles) y dimetilaminopiridina (0,01 g, 0,08 mmoles). La mezcla resultante se agitó a TA durante 4 h, después se diluyó con EtOAc y agua. La capa orgánica se lavó con salmuera, se secó sobre MgSO4, y se concentró a vacío. El residuo se purificó por cromatografía sobre gel de sílice, eluyendo con un gradiente de 0% al 100% de EtOAc en hexanos, para proporcionar el compuesto del título. LC-MS: 370 (M+H)+.
Compuesto 126: W-(3-Cloro-4-fluorofenil)-4-(ciclopropanosulfonamido)picolinamida

El compuesto del título se preparó de manera análoga al Compuesto 136 usando 2-bromopiridin-4-amina como material de partida. LC-MS: 370 (M+H)+.
Compuesto 127: 2-(Ciclopropanosulfonamido)-W-(3,4,5-trifluorofenil)isonicotinamida

El compuesto del título se preparó de manera análoga al Compuesto 125 usando 3,4,5-trifluoroanilina e intermedio F como materiales de partida. LC-MS: 372 (M+H)+.
Compuesto 128: 2-(Ciclopropanosulfonamido)-W-(3,4-difluorofenil)isonicotinamida
El compuesto del título se preparó de manera análoga al Compuesto 125 usando 3,4-difluoroanilina e intermedio F como materiales de partida. lC-MS: 354 (M+H)+.
Compuesto 129: W-(3-Ciano-4-fluorofenil)-2-(ciclopropanosulfonamido)isonicotinamida

El compuesto del título se preparó de manera análoga al Compuesto 125 usando 5-amino-2-fluorobenzonitrilo e intermedio F como materiales de partida. LC-MS: 361 (M+H)+.
Compuesto 130: W-(3-Cloro-4-fluorofenil)-2-(1 -metilciclopropanosulfonamido)isonicotinamida

A una disolución de Intermedio D (0,150 g, 0,57 mmoles) en piridina anhidra (5 ml) se añadió dimetilaminopiridina (0,01 g, 0,08 mmoles) y cloruro de 1-metilciclopropano-1-sulfonilo (0,081 ml, 0,68 mmoles). La mezcla resultante se calentó hasta 60°C durante 16 h. Los volátiles se eliminaron a vacío, y el residuo se diluyó con EtOAc y HCl 1N. Los orgánicos se lavaron con HCl 1N (2X), con salmuera, se secaron sobre MgSO4, se filtraron, y se concentraron a vacío.
El material bruto resultante se purificó sobre gel de sílice, eluyendo con un gradiente de disolvente de 0% a 100% de EtOAc en hexanos, para proporcionar el compuesto del título. LC-MS: 384 (M+H)+.
Compuesto 131: W-(3-Cloro-4-fluorofenil)-2-(1 -metilciclopropanosulfonamido)isonicotinamida

El compuesto del título se preparó de manera análoga al Compuesto 130 usando cloruro de etanosulfonilo como material de partida. LC-MS: 358 (M+H)+.
C o m p u e s to 132 : W -(3 -C lo ro -4 - f lu o ro fe n il) -2 -(3 ,3 ,3 - tr i f lu o ro p ro p ils u lfo n a m id o ) is o n ic o t in a m id a

El compuesto del título se preparó de manera análoga al Compuesto 130 usando cloruro de 3,3,3-trifluoropropano-1-sulfonilo como material de partida. LC-MS: 426 (M+H)+.
Compuesto 133: W-(3-Cloro-4-fluorofenil)-2-(propilsulfonamido)isonicotinamida

El compuesto del título se preparó de manera análoga al Compuesto 130 usando cloruro de propano-1 -sulfonilo como material de partida. LC-m S: 372 (M+H)+.
Compuesto 134: W-(3-Cloro-4-fluorofenil)-2-(ciclohexanosulfonamido)isonicotinamida

El compuesto del título se preparó de manera análoga al Compuesto 130 usando cloruro de ciclohexanosulfonilo como material de partida. LC-m S: 412 (M+H)+.
Compuesto 135: W-(3-Cloro-4-fluorofenil)-2-(4-metilfenilsulfonamido)isonicotinamida

El compuesto del título se preparó de manera análoga al Compuesto 130 usando cloruro de p-toluenosulfonilo como material de partida. LC-MS: 420 (M+H)+.
C o m p u e s to 136 : W -(3 -C lo ro -4 - f lu o ro fe n il) -2 -( c ic lo p ro p a n o s u lfo n a m id o ) -6 -m e t il is o n ic o t in a m id a

Etapa 1; A una disolución de 4-cloro-6-metilpiridin-2-amina (0,33 g, 2,34 mmoles) en piridina anhidra (6 ml), a 0°C, se añadió dimetilaminopiridina (0,029 g, 0,23 mmoles) y cloruro de ciclopropanosulfonilo (0,41 ml, 3,97 mmoles). La mezcla resultante se agitó fría durante 5 minutos, y después se calentó hasta 60°C durante 16 h. La reacción se enfrió hasta TA, y se diluyó con EtOAc y HCl 1 M/salmuera 1:1. Los orgánicos se lavaron dos veces con HCl 1 M, con salmuera (2X), se secaron sobre Na2SO4, se filtraron, y se concentraron a vacío. El material bruto resultante se purificó sobre gel de sílice, eluyendo con un gradiente de disolvente de 0% al 70% de EtOAc en hexanos, para proporcionar N-(4-cloro-6-metilpiridin-2-il)ciclopropanosulfonamida.
Etapa 2; Se combinaron N-(4-cloro-6-metilpiridin-2-il)ciclopropanosulfonamida (0,18 g, 0,73 mmoles), carbonato de sodio (0,15 g, 1,46 mmoles), 3-cloro-4-fluoroanilina (0,16 g, 1,10 mmoles), tolueno (10 ml), xantfos (0,018 g, 0,03 mmoles) y Pd(OAc)2 (0,007 g, 0,03 mmoles) en un RBF, que se agitó y purgó con monóxido de carbono gaseoso. La mezcla resultante se agitó bajo una atmósfera de monóxido de carbono, y se calentó a 90°C durante 16 h. La reacción se enfrió hasta TA y se diluyó con EtOAc y salmuera. Los orgánicos se lavaron con agua, se secaron sobre Na2SO4, se filtraron, y se concentraron a vacío. El material bruto resultante se purificó sobre gel de sílice, eluyendo con un gradiente de disolvente de 10% a 100% de EtOAc en hexanos, para proporcionar el compuesto del título con impurezas. El residuo así obtenido se purificó adicionalmente usando una placa de TLC preparatoria, usando metanol al 7% en DCM como eluyente, para proporcionar el compuesto del título. LC-MS: 384 (M+H)+.
Compuesto 137: W-(3-Cloro-4-fluorofenil)-2-(ciclopropanosulfonamido)-6-fluoroisonicotinamida

Etapa 1; A una disolución de ácido 2,6-difluoroisonicotínico (1,0 g, 6,3 mmoles) en DMF (15 ml) se añadió HATU (2,87 g, 7,5 mmoles), seguido de W,W-diisopropiletilamina (3,3 ml, 18,9 mmoles). Después de agitar a temperatura ambiente durante 18 h, la mezcla de reacción se paralizó con disolución ac. sat. de NH4Cl, se extrajo con EtOAc, se lavó con agua, se secó (MgSO4), se filtró, y se evaporó a vacío. El material resultante se trituró a partir de una mezcla de EtOAc y hexanos para proporcionar un sólido incoloro aislado por filtración, se secó a vacío, y se usó sin purificación adicional.
Etapa 2; A una disolución de la W-(3-cloro-4-fluorofenil)-2,6-difluoroisonicotinamida aislada (400 mg, 1,40 mmoles) en DMSO anhidro (5 ml) se añadió K2CO3 (400 mg, 2,9 mmoles) en un vial sellable, que entonces se cerró y se calentó a 110°C durante 18 h. Después de enfriar, la mezcla se diluyó con disolución ac. sat. de NH4Cl, se extrajo con EtOAc, la fase orgánica se separó, se lavó con agua, se secó (MgSO4), se filtró, y se evaporó a vacío. La purificación por cromatografía sobre gel de sílice proporcionó el compuesto del título como un sólido incoloro. LC-MS: 388 (M+H)+.
C o m p u e s to 138 : N -(3 -C lo ro -4 - f lu o ro fe n il) -2 - f lu o ro -6 -( fe n ils u lfo n a m id o ) is o n ic o t in a m id a

Preparada de manera análoga a Compuesto 137 usando bencenosulfonamida en la etapa 2. LC-MS: 388 (M+H)+ 424. Compuesto 139: N-(3-Cloro-4-fluorofenil)-2-(fenilmetilsulfonamido)isonicotinamida

El compuesto del título se preparó de manera análoga al Compuesto 130 usando cloruro de fenilmetanosulfonilo como material de partida. LC-m S: 420 (M+H)+.
Compuesto 140: W-(3-Cloro-4-fluorofenil)-2-(2-metoxietilsulfonamido)isonicotinamida

A una disolución de 2-amino-W-(3-cloro-4-fluorofenil)isonicotinamida (Intermedio J) (0,15 g, 0,57 mmoles) en dioxano anhidro (5 ml) se añadió trietilamina (0,16 ml, 1,13 mmoles) y cloruro de 2-metoxietanosulfonilo (0,11 ml, 0,903 mmoles). La mezcla resultante se agitó y se calentó hasta 80°C durante 48 h. La reacción se enfrió hasta TA y se evaporó a vacío. Los orgánicos se lavaron dos veces con HCl 1 M, con salmuera (2X), se secaron sobre Na2SÜ4, se filtraron, y se concentraron a vacío. El material bruto resultante se purificó sobre gel de sílice, eluyendo con un gradiente de disolvente de 10% al 100% de EtOAc en hexanos, para proporcionar el compuesto del título. LC-MS: 386 (M+H)+.
Compuesto 141: W-(3-Cloro-4-fluorofenil)-2-(tiofeno-2-sulfonamido)isonicotinamida
A una disolución de 2-amino-N-(3-cloro-4-fluorofenil)isonicotinamida (Intermedio J) (0,15 g, 0,57 mmoles) en piridina anhidra (5 ml) se añadió cloruro de tiofeno-2-sulfonilo (0,17 g, 0,903 mmoles). La mezcla resultante se agitó a TA durante 16 h. La reacción se diluyó con EtOAc y HCl 1M. Los orgánicos se lavaron dos veces con salmuera, con agua, se coevaporaron con heptanos, y se concentraron a vacío. El producto se obtuvo a partirde 10% de MeOH/DCM. El sólido filtrado resultante proporcionó el compuesto del título. lC-MS: 412 (M+H)+.
Compuesto 142: W-(3-Cloro-4-fluorofenil)-2-(ciclohexilmetilsulfonamido)isonicotinamida

A una disolución de 2-amino-N-(3-cloro-4-fluorofenil)isonicotinamida (Intermedio J) (0,15 g, 0,57 mmoles) en piridina anhidra (5 ml) y cloruro de ciclohexilmetanosulfonilo (0,13 g, 0,79 mmoles). La mezcla resultante se agitó a TA durante 16 h. La reacción se diluyó con EtOAc y HCl 1M. Los orgánicos se lavaron dos veces con salmuera, con agua, y se concentraron a vacío. El material bruto resultante se purificó sobre gel de sílice, eluyendo con un gradiente de disolvente de 10% al 100% de EtOAc en hexanos, para proporcionar el compuesto del título. LC-MS: 426 (M+H)+. Compuesto 143: W-(3-Cloro-4-fluorofenil)-2-(2-feniletilsulfonamido)isonicotinamida

El compuesto del título se preparó de manera análoga al Compuesto 130 usando cloruro de 2-feniletanosulfonilo. LC-MS: 434 (M+H)+.
Compuesto 144: N-(3-Cloro-4-fluorofenil)-2-(ciclopropanosulfonamido)-5-fluoroisonicotinamida

Etapa 1: A una disolución de ácido 2-bromo-5-fluoroisonicotínico (3,0 g, 13,6 mmoles) en una mezcla de MeOH (10 ml) y benceno (20 ml), enfriada a 0°C, se añadió (trimetilsilil)diazometano (disolución 2,0M en hexanos; 14 ml, 28 mmoles) durante un período de 10 minutos. Después de agitar a temperatura ambiente durante 1,5 horas, la disolución se evaporó hasta sequedad, y el residuo se purificó sobre gel de sílice, eluyendo con un gradiente de EtOAc del 0 al 40% en hexanos, para proporcionar 2-bromo-5-fluoroisonicotinato de metilo como un sólido incoloro. Etapa 2: Se mezclaron ciclopropanosulfonamida (310 mg, 2,56 mmoles), carbonato de cesio (1,1 g, 3,38 mmoles), Pd2(dba)3 (40 mg, 0,044 mmoles), Xantphos (51 mg, 0,09 mmoles) y 2-bromo-5-fluoroisonicotinato de metilo (500 mg, 2,14 mmoles) en un vial sellable. A esta mezcla se añadió p-dioxano (10 ml), y la suspensión se desgasificó rociando por debajo de la superficie con gas nitrógeno durante 5 minutos. El vial se selló entonces, y se calentó a 100°C durante 3 horas, después de lo cual la mezcla de reacción puso verde a roja. Después de enfriar, la
disolución se repartió entre agua y EtOAc, la disolución acuosa se acidificó con HCl 1N y se extrajo con EtOAc (2X). Las fases orgánicas se combinaron, se secaron (MgSO4), se filtraron, y se evaporaron a vacío. El análisis de RMN 1H confirmó el aislamiento de 2-(ciclopropanosulfonamido)-5-fluoroisonicotinato de metilo como un sólido incoloro.
Etapa 3: El compuesto del título se preparó de acuerdo con el mismo procedimiento descrito para el Compuesto 125, usando el ácido previamente aislado y purificando la amida resultante por trituración en acetona/hexanos. LC-MS: 388 (M+H)+.
Compuesto 143: N-(3-Cloro-4-fluorofenil)-2-(2-ciclopropiletilsulfonamido)isonicotinamida

El compuesto del título se preparó de manera análoga al Compuesto 130 usando cloruro de 2-ciclopropiletanosulfonilo como material de partida. LC-MS: 398 (M+H)+.
Compuesto 146: W-(3-Cloro-4-fluorofenil)-2-((1 -cianociclopropil) metilsulfonamido)isonicotinamida

El compuesto del título se preparó de manera análoga al Compuesto 130 usando cloruro de (1 cianociclopropil)metanosulfonilo como material de partida. LC-MS: 409 (M+H)+.
Compuesto 147: W-(3-Cloro-4-fluorofenil)-2-(3-metilbutilsulfonamido)isonicotinamida

El compuesto del título se preparó de manera análoga al Compuesto 130 usando cloruro de 3-metilbutano-1-sulfonilo como material de partida. LC-MS: 400 (M+H)+.
C o m p u e s to 148 : W -(3 -C lo ro -4 - f lu o ro fe n il) -2 -( c ic lo p ro p a n o s u lfo n a m id o ) -5 -m e to x is o n ic o t in a m id a

Etapa 1: A una disolución del éster metílico aislado del Compuesto 144, etapa 1 (1,0 g, 4,27 mmoles), en MeOH (10 ml), se añadió NaOMe (disolución al 25% en peso: 5 ml). Después de agitar a TA durante 1,5 horas, la reacción se paralizó mediante adición de disolución ac. sat. de NH4Cl. La mezcla resultante se extrajo con EtOAc, la fase orgánica se separó, se secó (MgSO4), se filtró, y se evaporó a vacío. La purificación por cromatografía sobre gel de sílice proporcionó 2-bromo-5-metoxisonicotinato de metilo como un sólido incoloro.
Etapa 2: se mezclaron carbamato de terc-butilo (300 mg, 2,56 mmoles), carbonato de cesio (1,1 g, 3,38 mmoles), Pd2(dba)3 (40 mg, 0,044 mmoles), Xantphos (51 mg, 0,09 mmoles) y 2-bromo-5-metoxiisonicotinato de metilo (527 mg, 2,14 mmoles) en un vial sellable. A esta mezcla se añadió p-dioxano (10 ml), y la suspensión se desgasificó rociando por debajo de la superficie con gas nitrógeno durante 5 minutos. Después, el vial se selló y se calentó a 100°C durante 5 horas, después de lo cual la mezcla de reacción se puso verde a roja. Después de enfriar, la disolución se repartió entre agua y EtOAc, la disolución acuosa se acidificó con HCl 1N y se extrajo con EtOAc (2X). Las fases orgánicas se combinaron, se secaron (MgSO4), se filtraron, y se evaporaron a vacío. La purificación del residuo aislado en gel de sílice, eluyendo con 0 a 40% de EtOAc en hexanos, proporcionó 2-((terc- butoxicarbonil)amino)-5-metoxisonicotinato de metilo como un sólido incoloro
Etapa 3: 2-((terc-butoxicarbonil)amino)-5-metoxisonicotinato de metilo (300 mg; 1,06 mmoles) se disolvió en 8 ml de HCl 4N en dioxanos, y se agitó a TA durante 18 horas. La mezcla de reacción se evaporó hasta sequedad a vacío para proporcionar 2-amino-5-metoxisonicotinato de metilo, usado sin más análisis o purificación.
Etapa 4: A una disolución del 2-amino-5-metoxisonicotinato de metilo aislado (1,06 mmoles) en piridina (4 ml) se añadió cloruro de ciclopropanosulfonilo (110 ml, 1,07 mmoles), y la mezcla se agitó durante 18 horas. La mezcla se diluyó con EtOAc, se lavó con agua, se secó (MgSO4), se filtró, y se evaporó a vacío. La purificación del residuo sobre gel de sílice proporcionó 2-(ciclopropanosulfonamido)-5-metoxisonicotinato de metilo como un sólido incoloro.
Etapa 5: Se disolvió 2-(ciclopropanosulfonamido)-5-metoxisonicotinato de metilo (190 mg, 0,66 mmoles) en una mezcla de THF (3 ml) y agua (1 ml), a la que se añadió UOH-H2O (80 mg, 1,9 mmoles). Después de agitar a TA durante 12 horas, la disolución se acidificó con HCl 1N, se extrajo con EtOAc, la fase orgánica se separó, se secó (MgSO4), se filtró, y se evaporó a vacío. El ácido 2-(ciclopropanosulfonamido)-5-metoxisonicotínico proporcionado se usó en la etapa final sin purificación adicional.
Etapa 6: El ácido 2-(ciclopropanosulfonamido)-5-metoxisonicotínico (80,0 mg, 0,294 mmoles) se disolvió en DMF (3 ml), al que se añadió HATU (125,0 mg, 0,324 mmoles), seguido de la base de Hünigs (0,3 ml, 1,7 mmoles). Después de agitar durante 1 hora, se añadió 3-cloro-4-fluoroanilina (52 mg, 0,357 mmoles), y la mezcla de reacción se mantuvo a TA durante 18 horas. La disolución se diluyó entonces con EtOAc y HCl 1N, la fase orgánica se separó, se lavó con agua, se secó (MgSO4), se filtró, y se evaporó a vacío. El residuo se trituró con una mezcla de EtOAc y hexanos para proporcionar el compuesto del título como un sólido incoloro. LC-MS: 400 (M+H)+.
Compuesto 149: 5-Cloro-W-(3-cloro-4-fluorofenil)-2-(ciclopropanosulfonamido)isonicotinamida
Etapa 1; A una disolución de 2-amino-5-cloroisonicotinato de metilo (0,50 g, 2,68 mmoles) en piridina anhidra (10 ml) a 0°C se añadió dimetilaminopiridina catalítica y cloruro de ciclopropanosulfonilo (0,44 ml, 4,29 mmoles). La mezcla resultante se calentó a 35°C durante 16 h. La reacción se enfrió hasta TA y se diluyó con EtOAc y salmuera. Los orgánicos se lavaron con HCl 1 M, salmuera y agua, se coevaporaron con heptanos, y se concentraron a vacío. El material bruto resultante se purificó en una columna C18, eluyendo con un gradiente de disolvente de 20% a 100% de acetonitrilo en agua, para proporcionar 5-cloro-2-(ciclopropanosulfonamido)isonicotinato de metilo. Etapa 2; Se combinaron 5-cloro-2-(ciclopropanosulfonamido)isonicotinato de metilo (0,23 g, 0,77 mmoles), THF (6,2 ml), MeOH (6,2 ml) y LiOH 1 M (6,19 ml, 6,19 mmoles), y se agitaron a TA durante 16 h. La reacción se concentró, y se diluyó con EtOAc y HCl 1M. Los orgánicos se lavaron con agua, y se concentraron a vacío. El material bruto resultante que contenía ácido 5-cloro-2-(ciclopropanosulfonamido)isonicotínico se usó como tal en la etapa siguiente.
Etapa 3; El compuesto del título se preparó de manera análoga al Intermedio G usando condiciones de acoplamiento de HATU y ácido 5-cloro-2-(ciclopropanosulfonamido)isonicotínico. LC-MS: 405 (M+H)+.
Compuesto 150: W-(3-Cloro-4-fluorofenil)-2-(ciclopentilmetilsulfonamido)isonicotinamida

El compuesto del título se preparó de manera análoga al Compuesto 130 usando cloruro de ciclopentilmetanosulfonilo. LC-MS: 412 (M+H)+.
Compuesto 151: W-(3-Cloro-4-fluorofenil)-2-((4-fluorofenil)metilsulfonamido)isonicotinamida

El compuesto del título se preparó de manera análoga al Compuesto 130 usando cloruro de (4-fluorofenil)metanosulfonilo como material de partida. LC-MS: 438 (M+H)+.
Compuesto 152: W-(3-Cloro-4-fluorofenil)-2-(p-tolilmetilsulfonamido)isonicotinamida
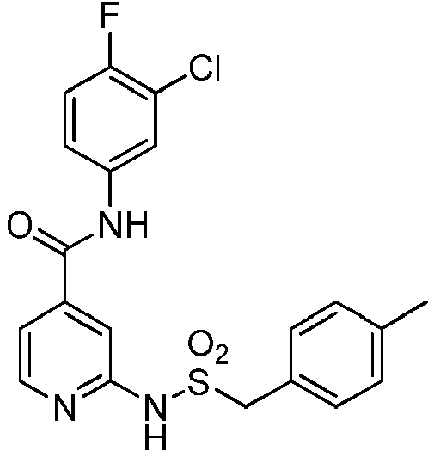
El c o m p u e s to d e l t í tu lo s e p re p a ró d e m a n e ra a n á lo g a a l C o m p u e s to 130 u s a n d o c lo ru ro d e (p - to l i lm e ta n o s u lfo n ilo c o m o m a te r ia l d e p a r tid a . L C -M S : 434 (M H )+.
C o m p u e s to 153 : W -(3 ,4 -D if lu o ro fe n il) -2 -( (4 - f lu o ro fe n il)m e t ils u lfo n a m id o ) is o n ic o t in a m id a

El compuesto del título se preparó de manera análoga al Compuesto 130 usando cloruro de (p-tolilmetanosulfonilo e Intermedio D como material de partida. LC-MS: 434 (M+H)+.
Compuesto 154: W-(3-Cloro-4-fluorofenil)-2-(1,1-dimetiletilsulfonamido)isonicotinamida

El compuesto del título se preparó de manera análoga al Compuesto 124 usando 2-metilpropano-2-sulfonamida como material de partida. LC-mS: 386 (M+H)+.
Compuesto 155: W-(3-Cloro-4-fluorofenil)-2-((1 -hidroxiciclobutil)metilsulfonamido)isonicotinamida

Etapa 1; A una disolución de bencilamina (4,00 g, 37,33 mmoles) en DCM (50 ml) a 0°C se añadió gota a gota cloruro de metanosulfonilo (1,44 ml, 18,66 mmoles). La mezcla resultante se agitó fría durante 10 minutos, y después se diluyó con agua. Los orgánicos se lavaron con agua, con HCl 1M, se secaron sobre Na2SÜ4, se filtraron y se concentraron a vacío. El material bruto resultante que contenía W-bencilmetanosulfonamida se usó como tal en la siguiente etapa.
Etapa 2; A una disolución de W-bencilmetanosulfonamida (1,00 g, 5,39 mmoles) en THF anhidro (10 ml) a -78°C en atmósfera de nitrógeno se añadió gota a gota nBuLi (2,5M en hexanos, 4,32 ml, 10,78 mmoles). Después de 5 minutos de agitación a -78°C, se añadió gota a gota ciclobutanona (1,92 ml, 21,56). La disolución resultante se agitó a -78°C durante 2 h, a TA 1 h, y se paralizó con 1 ml de ácido acético. La reacción se evaporó, y el residuo se diluyó con EtOAc y una disolución saturada de NaHCÜ3. Los orgánicos se lavaron una vez más con una disolución saturada de NaHCÜ3, con salmuera y agua, y se concentraron a vacío. El material bruto resultante se purificó sobre gel de sílice, eluyendo con un gradiente de disolvente de 0% a 9% de MeÜH en DCM, para proporcionar W-bencil-1-(1-hidroxiciclobutil)metanosulfonamida, que estaba contaminada con impurezas. El sólido resultante se trituró en EtOAc/heptanos 1:1, y se filtró para obtener material puro.
Etapa 3; Se combinaron W-bencil-1 -(1 -hidroxiciclobutil)metanosulfonamida (0,34 g, 1,33 mmoles), Pd(OH)2 (70 mg, 20% p/p), MeÜH (6,2 ml), y se agitaron bajo una atmósfera de hidrógeno a 60°C durante 16 h. La reacción se filtró a través de una almohadilla de celite, y se concentró a vacío para obtener (1 -hidroxiciclobutil)metanosulfonamida, que se usó como tal en la etapa siguiente.
Etapa 4; El compuesto del título se preparó de manera análoga al Compuesto 124 usando (1-hidroxiciclobutil)metanosulfonamida y condiciones de acoplamiento de CuI. LC-MS: 414 (M+H)+.
Compuesto 156: W-(3-Cloro-4-fluorofenil)-2-(2-hidroxi-2-metilpropilsulfonamido)isonicotinamida

El compuesto del título se preparó de manera análoga al Compuesto 155 usando acetona en la etapa 2. LC-MS: 402 (M+H)+.
Compuesto 157: N-(3-Cloro-4-fluorofenil)-2-(metilsulfonamido)isonicotinamida

El compuesto del título se preparó de manera análoga al Compuesto 124 usando metanosulfonamida y condiciones de acoplamiento de CuI. lC-MS: 402 (M+H)+.
Compuesto 158: W-(3-Cloro-4-fluorofenil)-2-(ciclopropilmetilsulfonamido)isonicotinamida

El compuesto del título se preparó de manera análoga al Compuesto 130 usando cloruro de ciclopropilmetanosulfonilo. LC-MS: 384 (M+H)+.
Compuesto 159: W-(3-Cloro-4-fluorofenil)-2-((3-metiloxetan-3-il)metilsulfonamido)isonicotinamida
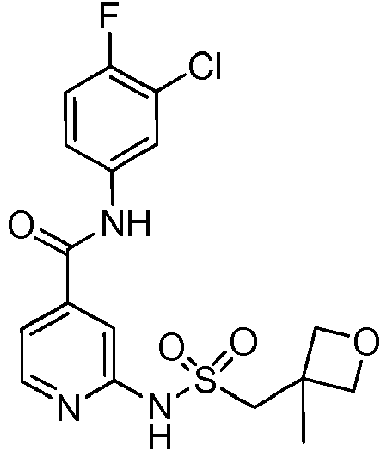
El c o m p u e s to d e l t í tu lo s e p re p a ró d e m a n e ra a n á lo g a a l C o m p u e s to 130 u s a n d o c lo ru ro d e (3 -m e ti lo x e ta n -3 -il)m e ta n o s u lfo n ilo . L C -M S : 414 (M H )+ .
C o m p u e s to 160 : W -(3 -C lo ro -4 - f lu o ro fe n il) -2 -( te t ra h id ro -2 H -p ira n -4 -s u lfo n a m id o ) is o n ic o t in a m id a
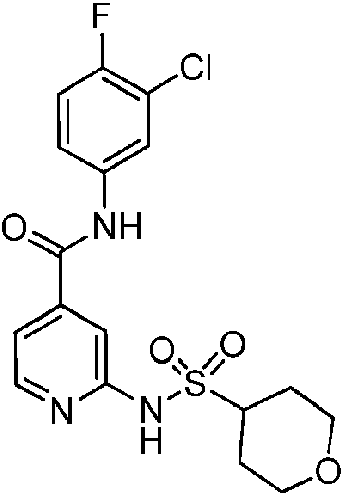
El compuesto del título se preparó de manera análoga al Compuesto 130 usando cloruro de tetrahidro-2H-piran-4-sulfonilo. LC-MS: 414 (M+H)+.
Compuesto 161: (±)-W-(3-Cloro-4-fluorofenil)-2-(tetrahidrofuran-3-sulfonamido)isonicotinamida

El compuesto del título se preparó de manera análoga al Compuesto 130 usando cloruro de tetrahidrofurano-3-sulfonilo. LC-MS: 400 (M+H)+.
Compuesto 162: (±)-W-(3-cloro-4-fluorofenil)-2-(3-hidroxi-2-metilpropilsulfonamido)isonicotinamida

El compuesto del título se preparó de manera análoga al Compuesto 130 usando 3-(clorosulfonil)-2-metilpropanoato de metilo, seguido de reducción con LiBH4. LC-MS: 402 (M+H)+.
Compuesto 163: W-(3-Cloro-4-fluorofenil)-2-(1 -metiletilsulfonamido)isonicotinamida

E l c o m p u e s to d e l t í tu lo s e p re p a ró d e m a n e ra a n á lo g a a l C o m p u e s to 124 u s a n d o p ro p a n o -2 -s u lfo n a m id a c o m o m a te r ia l d e p a r t id a . L C -M S : 372 (M H )+ .
C o m p u e s to 164 : W -(3 -C lo ro -4 - f lu o ro fe n il) -2 -(2 ,2 -d im e t ilp ro p ils u lfo n a m id o ) is o n ic o t in a m id a

El compuesto del título se preparó de manera análoga al Compuesto 124 usando 2,2-dimetilpropano-1-sulfonamida como material de partida. LC-MS: 400 (M+H)+.
Compuesto 165: W-(3,4-Difluorofenil)-2-(1-metiletilsulfonamido)isonicotinamida

El compuesto del título se preparó de manera análoga al Compuesto 124 usando propano-2-sulfonamida e Intermedio C como materiales de partida. LC-MS: 356 (M+H)+.
Compuesto 166: W-(3,4-Difluorofenil)-2-(tetrahidro-2H-piran-4-sulfonamido)isonicotinamida

El compuesto del título se preparó de manera análoga al Compuesto 130 usando cloruro de tetrahidro-2H-piran-4-sulfonilo e Intermedio G. LC-MS: 398 (M+H)+.
Compuesto 167: (±)-W-(3,4-Difluorofenil)-2-(tetrahidrofurano-3-sulfonamido)isonicotinamida

E l c o m p u e s to d e l t í tu lo s e p re p a ró d e m a n e ra a n á lo g a a l C o m p u e s to 130 u s a n d o c lo ru ro d e te tra h id ro fu ra n o -3 -s u lfo n ilo e In te rm e d io G . L C -M S : 384 (m H )+
C o m p u e s to 168 : (± ) -W -(3 -C lo ro -4 - f lu o ro fe n il) -2 -( ( te t ra h id ro fu ra n o -2 - il)m e t i ls u lfo n a m id o ) is o n ic o t in a m id a

El compuesto del título se preparó de manera análoga al Compuesto 130 usando cloruro de (tetrahidrofurano-2-il)metanosulfonilo. LC-MS: 414 (M+H)+.
Compuesto 169: (±)-W-(3-Cloro-4-fluorofenil)-2-((tetrahidro-2H-piran-2-il)metilsulfonamido)isonicotinamida

El compuesto del título se preparó de manera análoga al Compuesto 130 usando cloruro de (tetrahidro-2H-piran-2-il)metanosulfonilo. LC-MS: 428 (M+H)+.
Compuesto 170: (±)-W-(3-Cloro-4-fluorofenil)-2-((tetrahidro-2H-piran-3-il)metilsulfonamido)isonicotinamida

El compuesto del título se preparó de manera análoga al Compuesto 124 usando (tetrahidro-2H-piran-3-il)metanosulfonamida. LC-MS: 428 (M+H)+.
Compuesto 171: (c/s/f/"ans)-W-(3-Cloro-4-fluorofenil)-2-(4-hidroxiciclohexanosulfonamido)isonicotinamida

Etapa 1; A un RBF en un baño de hielo a 0°C se añadió gota a gota clorito de sodio (1,03 g, 11,34 mmoles) y acetonitrilo anhidro (10 ml), seguido de HCl concentrado (2,27 ml). A la mezcla agitada fría se añadió gota a gota 4-mercaptociclohexanol (0,50 g, 3,78 mmol, disuelto en 3 ml de acetonitrilo). El baño de hielo se retiró, y la mezcla
resultante se agitó a TA durante 2 h, después se diluyó con EtOAc y agua. Los orgánicos se lavaron secuencialmente con agua, salmuera, se secaron sobre Na2SO4, se filtraron, y se concentraron a vacío. El aceite bruto resultante que contenía cloruro de 4-hidroxiciclohexano-1-sulfonilo se usó como tal en la etapa siguiente. Etapa 2; Se disolvió cloruro de 4-hidroxiciclohexano-1-sulfonilo (0,70 g, bruto) en DCM (15 ml) y se agitó a -78°C. A la mezcla agitada fría se añadió amoníaco líquido condensado (15 ml), y la mezcla resultante se agitó a -78°C durante 2 h, se calentó a reflujo a 0°C durante 2 h, después se dejó calentar hasta TA durante 16 h. Los disolventes se evaporaron a vacío, y el sólido bruto resultante que contenía 4-hidroxiciclohexano-1-sulfonamida se usó como tal en la etapa siguiente.
Etapa 3; El compuesto del título se preparó de manera análoga al Compuesto 124 usando 4-hidroxiciclohexano-1-sulfonamida y condiciones de acoplamiento Cul. LC-MS: 428 (M+H)+.
Compuesto 172: W-(3-Cloro-4-fluorofenil)-2-(2-(piridin-2-il)etilsulfonamido)isonicotinamida

El compuesto del título se preparó de manera análoga al Compuesto 130 usando hidrocloruro de cloruro de 2-(piridin-2-il)etanosulfonilo como material de partida. LC-MS: 428 (M+H)+.
Compuesto 173: W-(3-Cloro-4-fluorofenil)-2-(2-(4-metoxifenil)etilsulfonamido)isonicotinamida

El compuesto del título se preparó de manera análoga al Compuesto 130 usando cloruro de 2-(4-metoxifenil)etanosulfonilo como material de partida. LC-MS: 464 (M+H)+.
Compuesto 174: W-(3,4-Difluorofenil)-2-(2-(piridin-2-il)etilsulfonamido)isonicotinamida

El compuesto del título se preparó de manera análoga al Compuesto 130 usando hidrocloruro de cloruro de 2-(piridin-2-il)etanosulfonilo e Intermedio E como materiales de partida. LC-MS: 419 (M+H)+.
C o m p u e s to 175 : N -(3 ,4 -D if lu o ro fe n il) -2 -(2 -(4 -m e to x ife n il)e t i ls u lfo n a m id o ) is o n ic o t in a m id a

El compuesto del título se preparó de manera análoga al Compuesto 130 usando cloruro de 2-(4-metoxifenil)etanosulfonilo e Intermedio E como materiales de partida. LC-MS: 448 (M+H)+.
Compuesto 176: W-(3-Cloro-4-fluorofenil)-2-((4-cianofenil)metilsulfonamido)isonicotinamida

El compuesto del título se preparó de manera análoga al Compuesto 130 usando cloruro de (4-cianofenil)metanosulfonilo como material de partida. LC-MS: 445 (M+H)+.
Compuesto 177: 2-((4-Cianofenil)metilsulfonamido)-W-(3,4-difluorofenil)isonicotinamida

El compuesto del título se preparó de manera análoga al Compuesto 130 usando cloruro de (4-cianofenil)metanosulfonilo e Intermedio E como materiales de partida. LC-MS: 429 (M+H)+.
Compuesto 178: W-(3-Cloro-4-fluorofenil)-2-((tetrahidro-2H-piran-4-il)metilsulfonamido)isonicotinamida

El c o m p u e s to d e l t í tu lo s e p re p a ró d e m a n e ra a n á lo g a a l C o m p u e s to 124 u s a n d o ( te t ra h id ro -2 H -p ira n -4 -i l )m e ta n o s u lfo n a m id a . L C -M S : 428 (M H )+ .
C o m p u e s to 179 : (± ) -W -(3 -C lo ro -4 - f lu o ro fe n il) -2 -( te tra h id ro -2 H -p ira n -3 -s u lfo n a m id o ) is o n ic o t in a m id a

El compuesto del título se preparó de manera análoga al Compuesto 124 usando tetrahidro-2H-piran-3-sulfonamida. LC-MS: 414 (M+H)+.
Compuesto 180: W-(3-Cloro-4-fluorofenil)-2-((4-etilfenil)metilsulfonamido)isonicotinamida
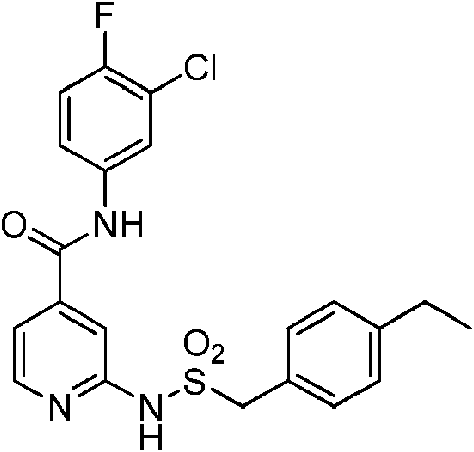
El compuesto del título se preparó de manera análoga al Compuesto 124 usando (4-etilfenil)metanosulfonamida. LC-MS: 448 (M+H)+.
Compuesto 181: W-(3,4-Difluorofenil)-2-((4-etilfenil)metilsulfonamido)isonicotinamida

El compuesto del título se preparó de manera análoga al Compuesto 124 usando (4-etilfenil)metanosulfonamida e Intermedio C. LC-MS: 432 (M+H)+.
Compuesto 182: (±)-W-(3-Cloro-4-fluorofenil)-2-(3-hidroxiciclohexanosulfonamido)isonicotinamida

Etapa 1; A una disolución de ciclohex-2-enona (2,00 g, 20,81 mmoles) en DCE (100 ml) a TA se añadió cloruro de indio (0,23 g, 1,04 mmoles) y ácido tioacético (2,38 ml, 31,21 mmoles). La mezcla resultante se agitó a 60°C durante 16 h, después se diluyó con EtOAc y salmuera. Los orgánicos se lavaron con agua, con salmuera, se
secaron sobre Na2SÜ4, se filtraron, y se concentraron a vacío. El material bruto resultante se purificó sobre gel de sílice, eluyendo con un gradiente de disolvente de 0% al 40% de EtOAc en hexanos, para proporcionar de S-(3-oxociclohexil)etanotioato puro como un aceite.
Etapa 2; A una disolución de S-(3-oxociclohexil)etanotioato (1,00 g, 5,81 mmoles) en MeÜH (50 ml) a 0°C se añadió NaBH4 (0,27 g, 6,97 mmoles) La mezcla resultante se agitó a 0°C durante 1,5 h, después se diluyó con EtOAc y con agua después de reducir el volumen de MeÜH con una corriente de nitrógeno gaseoso. Los orgánicos se lavaron con agua dos veces y se concentraron a vacío. El material bruto resultante que contenía S-(3-hidroxiciclohexilo)etanotioato se llevó como tal a la etapa siguiente.
Etapa 3; Se combinaron S-(3-hidroxiciclohexil)etanotioato (0,36 g, 2,07 mmoles), THF (7,5 ml), MeÜH (7,5 ml), agua (4,2 ml) y NaÜH 5 M (3,31 ml, 16,53 mmoles), y se agitaron a TA durante 48 h. La reacción se paralizó con una disolución ac. al 20% de ácido cítrico monohidratado, los orgánicos se evaporaron parcialmente, y la disolución restante se diluyó con EtÜAc y salmuera. Los orgánicos se lavaron dos veces con agua y se concentraron a vacío.
El bruto resultante se coevaporó con EtÜAc para proporcionar 3-mercaptociclohexanol como un aceite, y se usó como tal en la etapa siguiente.
Etapa 4; A un RBF en un baño de hielo a 0°C se añadió gota a gota clorito de sodio (0,67 g, 7,37 mmoles) y acetonitrilo anhidro (10 ml), seguido de HCl concentrado (1,50 ml). A la mezcla agitada fría se añadió gota a gota 3-mercaptociclohexanol (0,33 g, 2,46 mmol, disuelto en 3 ml de acetonitrilo). El baño de hielo se retiró, y la mezcla resultante se agitó a TA durante 2 h, después se diluyó con EtÜAc y agua. Los orgánicos se lavaron secuencialmente con agua, salmuera, se secaron sobre Na2SÜ4, se filtraron, y se concentraron a vacío. El aceite bruto inodoro resultante que contenía cloruro de 3-hidroxiciclohexano-1-sulfonilo se usó como tal en la etapa siguiente.
Etapa 5; Se disolvió cloruro de 3-hidroxiciclohexano-1 -sulfonilo (~0,40 g, bruto) en DCM (10 ml), y se agitó a -78°C. A la mezcla agitada fría se añadió amoníaco líquido condensado (10 ml), y la mezcla resultante se agitó a -78°C durante 2 h, se calentó a reflujo a 0°C durante 2 h, después se dejó calentar hasta TA durante 16 h. Los disolventes se evaporaron a vacío, se coevaporaron con MeÜH/EtÜAc 1:1, y el sólido bruto resultante que contenía 3-hidroxiciclohexano-1-sulfonamida se usó como tal en la etapa siguiente.
Etapa 6; El compuesto del título se preparó de manera análoga al Compuesto 124 usando 3-hidroxiciclohexano-1 -sulfonamida y condiciones de acoplamiento Cul. LC-MS: 428 (M+H)+.
Compuesto 183: (±)-W-(3-Cloro-4-fluorofenil)-2-(3-hidroxicicloheptanosulfonamido)isonicotinamida

El compuesto del título se preparó de manera análoga al Compuesto 182 usando ciclohept-2-enona en la etapa 1. LC-MS: 442 (M+H)+.
Compuesto 184: 2-(1 -Bencilciclopropanosulfonamido)-W-(3-cloro-4-fluorofenil)isonicotinamida

Etapa 1: A una disolución de terc-butilamina (29,6 ml, 0,282 mol) en THF anhidro (250 ml), enfriada a -20°C, se añadió cloruro de 3-cloropropilsulfonilo (25 g, 0,141 mol) durante un período de 10 minutos. La mezcla de reacción
se agitó vigorosamente a temperatura ambiente durante 16 horas, después de lo cual se filtró a vacío. La concentración del filtrado proporcionó un aceite incoloro que se disolvió en DCM, se lavó secuencialmente con HCl 1N y después agua, la fase orgánica se secó (MgSO4), se filtró, y se evaporó a vacío. El sólido resultante se trituró a partir de una mezcla de DCM/Hexanos para proporcionar W-(te/-c-butil)-3-cloropropano-1-sulfonamida como un sólido incoloro después de filtrar y secar a vacío.
Etapa 2: A una disolución de W-(íerc-butil)-3-cloropropano-1-sulfonamida (2,5 g, 11,7 mmoles) en THF anhidro (100 ml) enfriada a -78°C se añadió n-butil-litio (2,5M en hexanos; 10 ml, 25,0 mmoles). La disolución resultante se dejó calentar hasta temperatura ambiente, se agitó durante 1,5 horas, y despuésse volvió a enfriar hasta -78°C, después de lo cual se realizó una nueva adición de n-butil-litio (2,5M en hexanos; 5 ml, 12,5 mmoles). Después de calentar adicoinalmente hasta temperatura ambiente, la disolución se volvió a enfriar hasta -78°C, y se añadió bromuro de bencilo (1,5 ml, 12,6 mmoles). La disolución final se agitó a temperatura ambiente durante 12 horas, después de lo cual se añadió disolución de ac. sat. de NH4Cl, y la mezcla resultante se extrajo con EtOAc (2X), las fases orgánicas se secaron (MgSO4), se filtraron y se evaporaron a vacío. La trituración del residuo con hexanos y la filtración dieron 1-bencilo-W-(te/"c-butil)ciclopropano-1-sulfonamida como un sólido incoloro.
Etapa 3: A 1-bencilo-W-(íerc-butil)ciclopropano-1-sulfonamida (2,0 g, 7,5 mmoles) se añadió ácido trifluoroacético (30 ml). Después de agitar a temperatura ambiente durante 16 horas, la mezcla de reacción se evaporó hasta sequedad, y el sólido resultante se trituró a partir de una mezcla mínima de EtOAc/hexano para proporcionar 1-bencilciclopropano-1-sulfonamida como un sólido incoloro.
Etapa 4: El compuesto del título se preparó de manera análoga al Compuesto 124 usando la 1 -bencilciclopropano-1 -sulfonamida preparada. LC-MS: 460 (M+H)+.
Compuesto 185: 2-(1 -Bencilciclopropanosulfonamido)-W-(3,4-difluorofenil)isonicotinamida

El compuesto del título se preparó de manera análoga al Compuesto 124 usando la 1-bencilciclopropano-1-sulfonamida preparada y el Intermedio C. LC-MS: 444 (M+H)+.
Compuesto 186: (±)-W-(3-Cloro-4-fluorofenil)-2-(3-hidroxiciclopentanosulfonamido)isonicotinamida

El compuesto del título se preparó de manera análoga al Compuesto 182 usando ciclopent-2-enona en la etapa 1. LC-MS: 414 (M+H)+.
C o m p u e s to 187 : Á c id o 3 -(W (4 -( (3 -c lo ro -4 - f lu o ro fe n il)c a rb a m o il)p ir id in -2 - il) s u lfa m o il)b e n z o ic o

El compuesto del título se preparó de manera análoga al Compuesto 130 usando cloruro de (4-cianofenil)metanosulfonilo como material de partida y después condiciones de hidrólisis de éster estándar para proporcionar el ácido. LC-MS: 448 (M-H)-.
Compuesto 188: W-(3-Cloro-4-fluorofenil)-2-((4-isopropilfenil) metilsulfonamido)isonicotinamida

El compuesto del título se preparó de manera análoga al Compuesto 124 usando (4-isopropilfenil)metanosulfonamida. LC-MS: 462 (M+H)+.
Compuesto 189: W-(3,4-Difluorofenil)-2-((4-isopropilfenil)metilsulfonamido)isonicotinamida

El compuesto del título se preparó de manera análoga al Compuesto 124 usando (4-isopropilfenil)metanosulfonamida e Intermedio C como materiales de partida. LC-MS: 446 (M+H)+.
Compuesto 190: N-(3-Cloro-4-fluorofenil)-2-(naftalen-2-ilmetilsulfonamido)isonicotinamida

El c o m p u e s to d e l t í tu lo s e p re p a ró d e m a n e ra a n á lo g a a l C o m p u e s to 130 u s a n d o c lo ru ro d e n a fta le n -2 -i lm e ta n o s u lfo n ilo c o m o m a te r ia l d e p a r t id a . L C -M S : 470 (M H )+ .
C o m p u e s to 191 : W -(3 ,4 -D if lu o ro fe n il) -2 -(n a f ta le n -2 - i lm e t ils u lfo n a m id o ) is o n ic o t in a m id a

El compuesto del título se preparó de manera análoga al Compuesto 130 usando cloruro de naftalen-2-ilmetanosulfonilo e Intermedio E como materiales de partida. LC-MS: 470 (M+H)+.
Compuesto 192: W-(3-Cloro-4-fluorofenil)-2-((4-clorofenil)metilsulfonamido)isonicotinamida

El compuesto del título se preparó de manera análoga al Compuesto 130 usando cloruro de (4-clorofenil)metanosulfonilo como material de partida. LC-MS: 454 (M+H)+.
Compuesto 193: 2-((4-Clorofenil)metilsulfonamido)-W-(3,4-difluorofenil)isonicotinamida

El compuesto del título se preparó de manera análoga al Compuesto 130 usando cloruro de (4-clorofenil)metanosulfonilo e Intermedio E como materiales de partida. LC-MS: 438 (M+H)+.
Compuesto 194: 2-((4-(íerc-Butil)fenil)metilsulfonamido)-W-(3-cloro-4-fluorofenil)isonicotinamida

Etapa 1: A una disolución de 1 -(te/c-butil)-4-(clorometil)benceno (1,0 g, 5,47 mmoles) en DMSO (10 ml) se añadió 3-metoxi-3-oxopropano-1-sulfinato de sodio (véase Baskin, J. M.; Wang, Z. Tet. Lett. 2002, 43, 8479; 1,14 g, 6,55 mmoles), y se agitó vigorosamente durante 18 h. Se añadió NaOMe (25% en peso; 1,5 ml) y, después de agitar durante 30 minutos, la disolución se enfrió en un baño de hielo y se añadió una disolución premezclada de ácido hidroxilamina-O-sulfónico (3,1 g, 27,4 mmoles), NaOAc (1,7 g, 20,7 mmoles) en agua (25 ml). La disolución
resultante se agitó a temperatura ambiente durante 18 horas, después de lo cual se extrajo con EtOAc, la fase orgánica se separó, se secó (MgSO4), se filtró, y se evaporó a vacío. El residuo se purificó entonces usando cromatografía sobre gel de sílice para proporcionar (4-(terc-butil)fenil)metanosulfonamida como un sólido incoloro. Etapa 2: El compuesto del título se preparó de manera análoga al Compuesto 124 usando (4-(tercbutil)fenil)metanosulfonamida como material de partida. LC-MS: 476 (M+H)+.
Compuesto 195: 2-((4-(terc-Butil)fenil)metilsulfonamido)-W-(3,4-difluorofenil)isonicotinamida
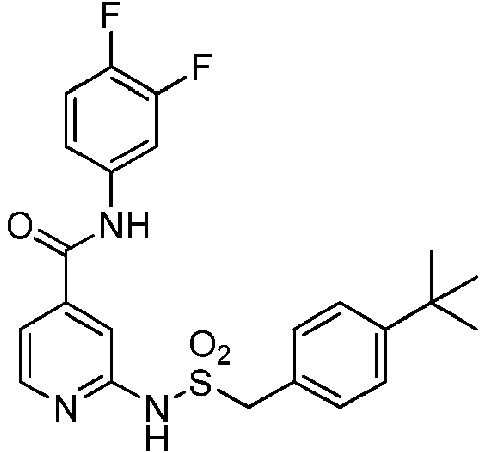
El compuesto del título se preparó de manera análoga al Compuesto 124 usando (4-(tercbutil)fenil)metanosulfonamida e Intermedio C como materiales de partida. LC-MS: 460 (M+H)+.
Compuesto 196: W-(3-Cloro-4-fluorofenil)-2-((4-(2-hidroxipropan-2-il)fenil)metilsulfonamido)isonicotinamida

Etapa 1: Se disolvió 2-(p-tolil)propan-2-ol (90% Tech; 3,0 g, 19,9 mmoles) en CCl4, y se añadió W-bromosuccinimida (3,48 g, 19,6 mmoles) y peróxido de benzoilo (100 mg, cat.). La mezcla de reacción se calentó a reflujo durante 4 horas, se enfrió y se filtró. El filtrado se evaporó, y el residuo se purificó sobre gel de sílice, eluyendo con 0 a 10% de EtOAc en hexanos, para proporcionar 2-(4-(bromometil)fenil)propan-2-ol como un aceite incoloro.
Etapa 2: La (4-(2-hidroxipropan-2-il)fenil)metanosulfonamida se prepara de manera idéntica a la Etapa 1 como se describe para el Compuesto 194.
Etapa 3: El compuesto del título se preparó de manera análoga al Compuesto 124 usando (4-(2-hidroxipropan-2-il)fenil)metanosulfonamida como material de partida. LC-MS: 478 (M+H)+.
Compuesto 197: 4-fluoro-W-(4-fluoro-3-metilfenil)-3-(1 -metil-1 H-pirazol-3-sulfonamido)benzamida

Etapa 1: A una disolución de ácido 3-amino-4-fluorobenzoico (2,0 g, 7,7 mmoles) en diclorometano (20 ml) se añadió 4-fluoro-3-metilanilina (968 mg, 7,7 mmoles), diisopropiletilamina (2,68 ml, 15,4 mmoles) y HATU (3,22 g, 8,47 mmoles). La mezcla de reacción se mantuvo a temperatura ambiente durante 12 horas, después de lo cual se añadió agua y se diluyó con diclorometano. La suspensión resultante se filtró, y los sólidos se lavaron con diclorometano y se secaron a vacío para proporcionar 1,39 g de 3-amino-4-fluoro-W-(4-fluoro-3metilfenil)benzamida como un sólido incoloro. La purificación del filtrado usando cromatografía en columna, eluyendo con 0 al 100% de EtOAc en , proporcionó 1,0 g adicionales de la amida deseada.
Etapa 2: A una disolución de 3-amino-4-fluoro-W-(4-fluoro-3-metilfenil)benzamida (110 mg, 0,42 mmoles) en piridina (3 ml) enfriada a 0°C se añadió cloruro de 1 -metil-1 H-pirazol-3-sulfonilo (90 mg, 0,50 mmoles) y DMAP catalítica (10 mg). Después de agitar a temperatura ambiente durante 16 horas, la disolución se diluyó con EtOAc y agua, la fase orgánica se separó, se lavó con salmuera, se secó (MgSO4), se filtró, y se evaporó a vacío. La purificación del residuo usando HPLC preparativa proporcionó el compuesto del título.
Compuesto 198: N-(3-cloro-4-fluorofenil)-3-(2-metoxietilsulfonamido)benzamida

Preparada de manera idéntica al Compuesto 75, etapa 2, usando el Intermedio O como material de partida y cloruro de 2-metoxietanosulfonilo.
Compuesto 199: W-(3-cloro-4-fluorofenil)-3-(tiofeno-2-sulfonamido)benzamida

Preparada de manera idéntica al Compuesto 75, etapa 2, usando el Intermedio O como material de partida y cloruro de tiofeno-2-sulfonilo.
Compuesto 200: W-(3-cloro-4-fluorofenil)-3-(1 H-pirazol-4-sulfonamido)benzamida

Preparada de manera idéntica al Compuesto 75, etapa 2, usando el Intermedio O como material de partida y cloruro de 1 H-pirazol-4-sulfonilo.
C o m p u e s to 201 : W -(3 -c lo ro -4 - f lu o ro fe n il) -3 -(3 ,5 -d im e ti l-1 H -p ira z o l-4 -s u lfo n a m id o )b e n z a m id a

Preparada de manera idéntica al Compuesto 75, etapa 2, usando el Intermedio O como material de partida y cloruro de 3,5-dimetil-1 H-pirazol-4-sulfonilo.
Compuesto 202: (±)-W-(3-cloro-4-fluorofenil)-3-(tetrahidrofurano-3-sulfonamido)benzamida

Preparada de manera idéntica al Compuesto 75, etapa 2, usando el Intermedio O como material de partida y cloruro de tetrahidrofurano-3-sulfonilo.
Compuesto 203: (±)-W-(3-cloro-4-fluorofenil)-3-((tetrahidrofurano-2-il)metilsulfonamido)benzamida

Preparada de manera idéntica al Compuesto 75, etapa 2, usando el Intermedio O como material de partida y cloruro de (tetrahidrofurano-2-il)metanosulfonilo.
Compuesto 204: (±)-W-(3-cloro-4-fluorofenil)-3-(2-metoxi-1 -metiletilsulfonamido)benzamida

Preparada de manera idéntica al Compuesto 75, etapa 2, usando el Intermedio O como material de partida y cloruro de 1-metoxipropano-2-sulfonilo.
C o m p u e s to 205 : (± ) -W -(3 -c lo ro -4 - f lu o ro fe n il) -3 -(2 -m e t ilte t ra h id ro fu ra n o -3 -s u lfo n a m id o )b e n z a m id a

Preparada de manera idéntica al Compuesto 75, etapa 2, usando el Intermedio O como material de partida y cloruro de 2-metiltetrahidrofurano-3-sulfonilo.
Compuesto 206: (±)-W-(3-cloro-4-fluorofenil)-3-(4-hidroxi-1,1-dioxidotetrahidrotiofen-3-sulfonamido)benzamida

Preparada de manera idéntica al Compuesto 75, etapa 2, usando el Intermedio O como material de partida y 1,1-dióxido de cloruro de 4-hidroxitetrahidrotiofen-3-sulfonilo.
Compuesto 207: 3-(4-(benciloxi)piperidin-1 -sulfonamido)-W-(3-cloro-4-fluorofenil)benzamida

Etapa 1: Preparación de cloruro de 4-(benciloxi)piperidin-1 -sulfonilo; A una disolución enfriada con hielo de hidrocloruro de 4-(benciloxi)piperidina (0,5 g, 2,2 mmoles) en DCM (10 ml) se añadió trietilamina (920 ml 6,6 mmoles), seguido de la adición gota a gota de ácido clorosulfónico (146 ml, 2,2 mmoles). La disolución resultante se agitó a temperatura ambiente durante 16 horas y se concentró, el residuo se lavó con Et2O, y se secó a vacío.
El residuo bruto se suspendió en benceno (7 ml), se añadió PCl5 (458 mg, 2,2 mmoles), y se calentó a reflujo durante 2 horas. Después de enfriar, la mezcla se diluyó en EtOAc y se lavó con disolución de ácido cítrico al 5%, bicarbonato saturado y salmuera. Después de secar con MgSO4, el material bruto se concentró para proporcionar cloruro de 4-(benciloxi)piperidin-1 -sulfonilo como un aceite usado sin purificación adicional.
Etapa 2: Preparada de manera idéntica al Compuesto 75, etapa 2, usando el Intermedio O como material de partida y cloruro de 4-(benciloxi)piperidin-1 -sulfonilo.
Los compuestos de la invención pueden poseer uno o más estereocentros, y cada estereocentro puede existir independientemente en la configuración R o S. En una realización, los compuestos descritos aquí están presentes en formas ópticamente activas o racémicas. Debe entenderse que los compuestos descritos aquí abarcan formas racémicas, ópticamente activas, regioisoméricas y estereoisoméricas, o combinaciones de las mismas, que poseen las propiedades terapéuticamente útiles descritas aquí.
La preparación de formas ópticamente activas se logra de cualquier manera adecuada, incluso a modo de ejemplo no limitativo, por resolución de la forma racémica con técnicas de recristalización, síntesis a partir de materiales de partida ópticamente activos, síntesis quiral, o separación cromatográfica usando una fase estacionaria quiral. En una realización, se utiliza una mezcla de uno o más isómeros como el compuesto terapéutico descrito aquí. En otra realización, los compuestos descritos aquí contienen uno o más centros quirales. Estos compuestos se preparan por cualquier medio, incluyendo síntesis estereoselectiva, síntesis enantioselectiva, y/o separación de una mezcla de enantiómeros y/o diastereómeros. La resolución de los compuestos e isómeros de los mismos se logra por cualquier medio, incluyendo, a modo de ejemplo no limitativo, procedimientos químicos, procedimientos enzimáticos, cristalización fraccionada, destilación, y cromatografía.
Los métodos y formulaciones descritos aquí incluyen el uso de N-óxidos (si corresponde), formas cristalinas (también conocidas como polimorfos), solvatos, fases amorfas y/o sales farmacéuticamente aceptables de compuestos que tienen la estructura de cualquier compuesto de la invención, así como los metabolitos y metabolitos activos de estos compuestos que tienen el mismo tipo de actividad. Los solvatos incluyen solvatos de agua, éter (por ejemplo, tetrahidrofurano, metil terc-butil éter) o alcohol (por ejemplo, etanol), acetatos, y similares. En una realización, los compuestos descritos aquí existen en formas solvatadas con disolventes farmacéuticamente aceptables tales como agua y etanol. En otra realización, los compuestos descritos aquí existen en forma no solvatada.
En una realización, los compuestos de la invención pueden existir como tautómeros. Todos los tautómeros están incluidos dentro del alcance de los compuestos presentados aquí.
Materiales:
A menos que se señale de otro modo, todos los materiales de partida y resinas se obtuvieron de proveedores comerciales y se usaron sin purificación.
Ejemplo: Ensayo de transferencia de punto de la inhibición de la replicación del VHB
Los compuestos activos en el ensayo del ensamblaje del VHB se evaluaron para determinar su actividad y toxicidad en el ensayo celular. En el primer ensayo antiviral, se evalúa la capacidad de los compuestos para inhibir la replicación del VHB en una línea celular de hepatoma que produce VHB, utilizando el método de transferencia de punto.
Brevemente, monocapas confluentes de células HepG2-2.2.15 se incuban con medio completo que contiene diversas concentraciones de un compuesto de ensayo. Tres días después, el medio de cultivo se reemplaza por medio nuevo que contiene el compuesto de ensayo diluido apropiadamente. Seis días después de la administración inicial del compuesto de ensayo, se recoge el sobrenadante del cultivo celular, y se realiza la lisis celular. Las muestras se aplican sobre membranas Nylos, y el ADN se inmoviliza a la membrana mediante reticulación con UV. Después de la prehibridación, se añade la sonda de VHB, y la hibridación se realiza durante la noche. Las membranas se exponen a las películas KODAK; la actividad antiviral se calcula a partir de la reducción en los niveles de ADN del VHB (EC50). La EC50 para la actividad antiviral se calcula a partir de las curvas de respuesta a la dosis de los compuestos activos. El comportamiento del ensayo a lo largo del tiempo se monitoriza mediante el uso de los compuestos de control positivo estándar ETV, BAY 41 -4109 y HAP-1.
La citotoxidad de los compuestos (TC50) se mide en esta misma línea celular HepG2-2.2.15 utilizando un ensayo de citotoxicidad basado en CELLTITER BLUE empleado según lo recomendado por el fabricante (Promega). Para confirmar y expandir estos resultados, se realiza un segundo ensayo antiviral en compuestos activos utilizando la línea celular estable de VHB HepG2.2.15 y midiendo la potencia anti-VHB mediante PCR en tiempo real y la citotoxicidad mediante CELLTITER BLUE. En este ensayo, 24 horas después de la siembra celular, las células HepG2-2.2.15 se incuban con medio completo que contiene diversas concentraciones de un compuesto de ensayo, con BAY 41-4109 y HAP-1 como controles positivos. Después de tres días, el medio de cultivo se reemplaza por medio nuevo que contiene el compuesto de ensayo adecuadamente diluido. El cultivo celular se recoge seis días después de la administración inicial del compuesto de ensayo, seguido de extracción de ADN del VHB usando QIAamp 96 DNA Blood Kit (Qiagen). El ADN del VHB extraído se diluye y analiza mediante PCR en tiempo real. Se genera una curva estándar trazando el valor de Ct frente a la cantidad de patrón de plásmido de VHB. La citotoxicidad se determina de manera similar al método descrito anteriormente mediante la aplicación de un método de absorción de colorante (kit CELLTITER BLUE, Promega).
Los compuestos seleccionados se evalúan para determinar su actividad y toxicidad en un ensayo celular. En el primer ensayo antiviral, se evalúa la capacidad de los compuestos para inhibir la replicación del VHB en una línea celular de hepatoma que produce VHB, utilizando el método de transferencia de punto.
Las monocapas confluentes de células HepG2-2.2.15 se incuban con medio completo que contiene diversas concentraciones de un compuesto de ensayo. Tres días después, el medio de cultivo se reemplaza por medio nuevo que contiene el compuesto de ensayo diluido apropiadamente. Seis días después de la administración inicial del compuesto de ensayo, se recoge el sobrenadante del cultivo celular, y se realiza la lisis celular. Las muestras se aplican sobre membranas Nylos, y el ADN se inmoviliza a la membrana mediante reticulación con UV. Después de la prehibridación, se añade la sonda de VHB, y la hibridación se realiza durante la noche. Las membranas se exponen a las películas KODAK; la actividad antiviral se calcula a partir de la reducción en los niveles de ADN del VHB (EC50).
La EC50 para la actividad antiviral se calcula a partir de las curvas de respuesta a la dosis de los compuestos activos. El comportamiento del ensayo a lo largo del tiempo se monitoriza mediante el uso de los compuestos de control positivo estándar ETV, BAY 41-4109 y HAP-1. Los resultados para compuestos seleccionados de la invención se ilustran en la Tabla 3.
La citotoxidad (CC50) se mide en esta misma línea celular HepG2-2.2.15 utilizando un ensayo de citotoxicidad basado en CELLTITER BLUE empleado según lo recomendado por el fabricante (Promega).
Ejemplo: Ensayo de inhibición de la replicación del VHB
La inhibición de la replicación del VHB por los compuestos de esta invención se pudo determinar en células infectadas o transfectadas con VHB, o células con VHB integrado de manera estable, tales como células HepG2.2.15 (Sells et al. 1987). En este ejemplo, las células HepG2.2.15 se mantienen en medio de cultivo celular que contiene 10% de suero fetal bovino (FBS), geneticina, L-glutamina, penicilina y estreptomicina. Las células HepG2.2.15 se pudieron sembrar en placas de 96 pocillos a una densidad de 40.000 células/pocillo, y se pudieron tratar con compuestos diluidos en serie a una concentración final de DMSO de 0,5%, ya sea solos o en combinación añadiendo fármacos en un formato de casilla de verificación. Las células se incuban con compuestos durante tres días, después de lo cual se elimina el medio y se añade a las células medio nuevo que contiene compuestos, y se incuban durante otros tres días. En el día 6 , se elimina el sobrenadante, y se trata con DNasa a 37°C durante 60 minutos, seguido de inactivación enzimática a 75°C durante 15 minutos. El ADN del VHB encapsulado se libera de los viriones y se une covalentemente a la VHB polimerasa incubando en amortiguador de lisis (Affymetrix QS0010) que contiene 2,5 mg de proteinasa K a 50°C durante 40 minutos. El ADN del VHB se desnaturaliza mediante la adición de NaOH 0,2 M y se detecta usando un kit de análisis QuantiGene de ADN ramificado (ADNb) de acuerdo con las recomendaciones del fabricante (Affymetrix).
Los niveles de ADN del VHB también se pudieron cuantificar usando qPCR, basado en la amplificación de la extracción de ADN del VHB encapsulado con disolución QUICKEXTRACT (Epicentre Biotechnologies) y la amplificación del ADN del VHB utilizando sondas de PCR específicas del VHB que pueden hibridarse con el ADN del VHB y una sonda marcada fluorescentemente para cuantificación. Además, la viabilidad celular de las células HepG2.2.15 incubadas con compuestos de ensayo, solos o en combinación, se determina usando el reactivo CELLTITER-GLO de acuerdo con el protocolo del fabricante (Promega). La señal de fondo media de los pocillos que contienen solo medio de cultivo se resta de todas las demás muestras, y el porcentaje de inhibición en cada concentración de compuesto se calcula normalizando a señales de células HepG2.2.15 tratadas con DMSO al 0,5% usando la ecuación E1.
E1: % de inhibición = (DMSOave - Xi)/(DMSOave x 100%
en la que DMSOave es la señal media calculada a partir de los pocillos que se tratan con el control de DMSO (control de inhibición al 0%), y Xi es la señal medida a partir de los pocillos individuales. Los valores de EC50, concentraciones eficaces que lograron un 50% de efecto inhibidor, se determinan mediante un ajuste no lineal utilizando el software Graphpad Prism (San Diego, CA) y la ecuación E2.
E2: Y = Ymin (Ymax - Ymin)/(1+10(LogEC50-X) x pendiente de Hill)
en la que Y representa los valores del porcentaje de inhibición, y X representa el logaritmo de las concentraciones de compuestos.
Los compuestos seleccionados de la invención se analizaron en el ensayo de replicación del VHB, como se describió anteriormente, y en la Tabla 4 se muestra un grupo representativo de estos compuestos activos.
Tabla 3.



(en la que los compuestos según realizaciones de la solicitud son como se definen en las reivindicaciones anejas).
Tabla 4.


(en la que los compuestos según realizaciones de la solicitud son como se definen en las reivindicaciones anejas).
Claims (11)
1. Un compuesto de Fórmula Illa:

o una sal farmacéuticamente aceptable del mismo;
en la que
cada R1 se selecciona independientemente de H, halo y alquilo de C1-6;
R2 se selecciona de halo, OH, CN, -alquilo de C1-6, -alcoxi de C1-6, halo-(alquilo de C1-6), di-halo-(alquilo de C1-6) y tri-halo-(alquilo de C1-6);
R3 es halo;
y R4 se selecciona de -alquilo de C1-6, -heteroalquilo de C1-6, -cicloalquilo de C3-7, -heterocicloalquilo de C3-7, (alquilo de C1-6)-(cicloalquilo de C3-7), -(cicloalquilo de C3-7)-(alquilo de C1-6), cicloalquilo de C7-8 con puente, -(alquilo de C1-6)-C(O)O-(alquilo de C1-6) y -(alquilo de C1-6)-(heterocicloalquilo de C3-7), cada uno de los cuales puede estar sustituido con 0, 1 o 2 grupos seleccionados independientemente de (=O), -OH, -alquilo de C1-6, -cicloalquilo de C3-7, halo-(alquilo de C1-6), di-halo-(alquilo de C1-6), tri-halo-(alquilo de C1-6), C(O)OH, fenilo y halo; o
R4 se selecciona de (cicloalquilo de C3-7)-C(O)R5 y -(cicloalquilo de C7-8 con puente)-C(O)R5, y R5 se selecciona de N(CH3)2, NS(O)2CH3, y heterocicloalquilo de C3-7,
en los que:
“alquilo”, por sí mismo o como parte de otro sustituyente, significa, a menos que se señale de otro modo, un hidrocarburo que tiene el número de átomos de carbono designado (es decir, C1-6 significa uno a seis átomos de carbono), e incluye grupos sustituyentes de cadena lineal, ramificada, o cíclicos; y
“cicloalquilo” se refiere a un radical no aromático monocíclico o policíclico, en el que cada uno de los átomos que forman el anillo (es decir, átomos esqueléticos) es un átomo de carbono, y/o se refiere a grupos “carbociclilo no aromático insaturado” o “carbociclilo insaturado no aromático”, opcionalmente en el que el grupo cicloalquilo comprende un anillo aromático condensado.
2. El compuesto de la reivindicación 1, o una sal farmacéuticamente aceptable del mismo, en el que R4 es (cicloalquilo de C3-7)-C(O)R5, y R5 es morfolinilo.
3. El compuesto de la reivindicación 1 o la reivindicación 2, o una sal farmacéuticamente aceptable del mismo, en el que el compuesto se selecciona del grupo que consiste en:





4. Un compuesto de Fórmula IVa:

o una sal farmacéuticamente aceptable del mismo;
en la que
R1 es halo;
R3 es halo; y
R4 se selecciona de alquilo de C1-6, cicloalquilo de C3-7, (alquilo de C1-6)-(cicloalquilo de C3-7), y (alquilo de C1-6)-arilo, en el que los grupos cicloalquilo de C3-7 y (alquilo de C1-6)-arilo están opcionalmente sustituidos con alquilo de C1-6;
en los que:
“alquilo”, por sí mismo o como parte de otro sustituyente, significa, a menos que se señale de otro modo, un hidrocarburo que tiene el número de átomos de carbono designado (es decir, C1-6 significa uno a seis átomos de carbono), e incluye grupos sustituyentes de cadena lineal, ramificada, o cíclicos; y
“cicloalquilo” se refiere a un radical no aromático monocíclico o policíclico, en el que cada uno de los átomos que forman el anillo (es decir, átomos esqueléticos) es un átomo de carbono, y/o se refiere a grupos “carbociclilo no aromático insaturado” o “carbociclilo insaturado no aromático”, opcionalmente en el que el grupo cicloalquilo comprende un anillo aromático condensado.
5. El compuesto de la reivindicación 4, o una sal farmacéuticamente aceptable del mismo, en el que el compuesto se selecciona del grupo que consiste en:


6. Una composición que comprende un compuesto de acuerdo con una cualquiera de las reivindicaciones 1 -5, o una sal, solvato o N-óxido del mismo, que comprende además al menos un vehículo farmacéuticamente aceptable.
7. Un compuesto de acuerdo con una cualquiera de las reivindicaciones 1-5, o la composición de la reivindicación 6, para uso en el tratamiento de una infección por VHB en un individuo que lo necesita.
8. El compuesto o composición para uso de acuerdo con la reivindicación 7, en el que dicho uso comprende además la administración de al menos un agente terapéutico adicional seleccionado del grupo que consiste en una vacuna contra el VHB, un inhibidor de la VHB polimerasa, interferón, interferón pegilado, un inhibidor de la entrada viral, un inhibidor de la maduración viral, BAY 41-4109, un inhibidor de la transcriptasa inversa, un agonista de TLR, AT-61 ((E)-N-(1 -cloro-3-oxo-1 -fenil-3-(piperidin-1 -il)prop-1 -en-2-il)benzamida), y AT-130 ((E)-N-(1 -bromo-1 -(2-metoxifenil)-3-oxo-3-(piperidin-1-il)prop-1-en-2-il)-4-nitrobenzamida), y una combinación de los mismos.
9. El compuesto o composición para uso de acuerdo con la reivindicación 8, en el que el interferón pegilado es interferón alfa pegilado (IFN-a), interferón lambda pegilado (IFN-l) o interferón gamma pegilado (IFN-g).
10. E l c o m p u e s to o c o m p o s ic ió n p a ra u s o d e a c u e rd o c o n la re iv in d ic a c ió n 8, en e l q u e e l in h ib id o r d e la t ra n s c r ip ta s a in v e rs a e s a l m e n o s u n o d e Z id o v u d in a , D id a n o s in a , Z a lc ita b in a , d d A , E s ta v u d in a , L a m iv u d in a , A b a c a v ir ,
Emtricitabina, Entecavir, Apricitabina, Atevirapina, ribavirina, aciclovir, famciclovir, valaciclovir, ganciclovir, valganciclovir, Tenofovir, Adefovir, cidofovir, Efavirenz, Nevirapina, Delavirdina, o Etravirina.
11. El compuesto o composición para uso de acuerdo con la reivindicación 8, en el que el agonista de TLR se selecciona del grupo que consiste en SM360320 (9-bencil-8-hidroxi-2-(2-metoxi-etoxi)adenina) y AZD 8848 ([3-({[3-(6-amino-2-butoxi-8-oxo-7,8-dihidro-9H-purin-9-il)propil][3-(4-morfolinil)propil]amino}metil)fenil]acetato de metilo).
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201462086323P | 2014-12-02 | 2014-12-02 | |
| US201462097854P | 2014-12-30 | 2014-12-30 | |
| PCT/US2015/063417 WO2016089990A1 (en) | 2014-12-02 | 2015-12-02 | Sulfide alkyl and pyridyl reverse sulfonamide compounds for hbv treatment |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2802412T3 true ES2802412T3 (es) | 2021-01-19 |
Family
ID=56092385
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES15864386T Active ES2802412T3 (es) | 2014-12-02 | 2015-12-02 | Compuestos de sulfonamida inversa a base de sulfuro, alquilo y piridilo para el tratamiento del VHB |
Country Status (8)
| Country | Link |
|---|---|
| EP (1) | EP3227262B1 (es) |
| JP (1) | JP2018504373A (es) |
| CN (1) | CN107406378A (es) |
| AU (1) | AU2015358561A1 (es) |
| CA (1) | CA2969557A1 (es) |
| DK (1) | DK3227262T3 (es) |
| ES (1) | ES2802412T3 (es) |
| WO (1) | WO2016089990A1 (es) |
Families Citing this family (49)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| BR112014015197A8 (pt) | 2011-12-21 | 2017-06-13 | Novira Therapeutics Inc | agentes antivirais de hepatite b |
| KR102271574B1 (ko) | 2012-08-28 | 2021-07-01 | 얀센 사이언시즈 아일랜드 언리미티드 컴퍼니 | 설파모일-아릴아미드 및 b형 간염 치료제로서의 그 용도 |
| WO2014131847A1 (en) | 2013-02-28 | 2014-09-04 | Janssen R&D Ireland | Sulfamoyl-arylamides and the use thereof as medicaments for the treatment of hepatitis b |
| MX353412B (es) | 2013-04-03 | 2018-01-10 | Janssen Sciences Ireland Uc | Derivados de n-fenil-carboxamida y su uso como medicamentos para el tratamiento de la hepatitis b. |
| TWI733288B (zh) | 2013-05-17 | 2021-07-11 | 愛爾蘭商健生科學愛爾蘭無限公司 | 胺磺醯基吡咯醯胺衍生物及其作為用於治療b型肝炎藥物的用途 |
| DK2997019T3 (en) | 2013-05-17 | 2018-12-03 | Janssen Sciences Ireland Uc | SULFAMOYLTHIOPHENAMIDE DERIVATIVES AND USE THEREOF AS MEDICINES FOR TREATING HEPATITIS B |
| HRP20180791T1 (hr) | 2013-07-25 | 2018-09-07 | Janssen Sciences Ireland Uc | Glioksamid supstituirani derivati pirolamida i njihova uporaba kao lijekova za liječenje hepatitisa b |
| CA2923712C (en) | 2013-10-23 | 2021-11-02 | Janssen Sciences Ireland Uc | Carboxamide derivatives and the use thereof as medicaments for the treatment of hepatitis b |
| US9169212B2 (en) | 2014-01-16 | 2015-10-27 | Novira Therapeutics, Inc. | Azepane derivatives and methods of treating hepatitis B infections |
| US10392349B2 (en) | 2014-01-16 | 2019-08-27 | Novira Therapeutics, Inc. | Azepane derivatives and methods of treating hepatitis B infections |
| CA2936947A1 (en) | 2014-02-05 | 2015-08-13 | Novira Therapeutics, Inc. | Combination therapy for treatment of hbv infections |
| TW201620893A (zh) | 2014-02-06 | 2016-06-16 | 健生科學愛爾蘭無限公司 | 磺醯胺基吡咯醯胺衍生物及其用作治療b型肝炎之醫藥品的用途 |
| US9765050B2 (en) * | 2014-12-30 | 2017-09-19 | Novira Therapeutics, Inc. | Pyridyl reverse sulfonamides for HBV treatment |
| JP2018510159A (ja) | 2015-03-19 | 2018-04-12 | ノヴィラ・セラピューティクス・インコーポレイテッド | アゾカン及びアゾナン誘導体及びb型肝炎感染症の治療法 |
| US10875876B2 (en) | 2015-07-02 | 2020-12-29 | Janssen Sciences Ireland Uc | Cyclized sulfamoylarylamide derivatives and the use thereof as medicaments for the treatment of hepatitis B |
| TW201718496A (zh) | 2015-09-29 | 2017-06-01 | 諾維拉治療公司 | B型肝炎抗病毒劑之晶型 |
| KR102398439B1 (ko) * | 2016-03-07 | 2022-05-16 | 이난타 파마슈티칼스, 인코포레이티드 | B형 간염 항바이러스제 |
| AU2017248828A1 (en) | 2016-04-15 | 2018-11-01 | Janssen Sciences Ireland Uc | Combinations and methods comprising a capsid assembly inhibitor |
| SG11201902944TA (en) | 2016-11-07 | 2019-05-30 | Arbutus Biopharma Corp | Substituted pyridinone-containing tricyclic compounds, and methods using same |
| CA3056563C (en) * | 2017-03-17 | 2023-03-14 | Meiji Seika Pharma Co., Ltd. | Plant disease control agent |
| MA49014A (fr) | 2017-03-21 | 2020-02-05 | Arbutus Biopharma Corp | Dihydroindène-4-carboxamides substitués, leurs analogues et procédés d'utilisation correspondant |
| SG11202001685TA (en) | 2017-08-28 | 2020-03-30 | Enanta Pharm Inc | Hepatitis b antiviral agents |
| EA202091113A1 (ru) | 2017-11-02 | 2020-08-28 | Айкурис Гмбх Унд Ко. Кг | Новые высокоактивные аминотиазолзамещенные индол-2-карбоксамиды, активные в отношении вируса гепатита b (hbv) |
| KR20200083552A (ko) | 2017-11-02 | 2020-07-08 | 아이쿠리스 게엠베하 운트 코. 카게 | B형 간염 바이러스 (hbv)에 활성인 신규 고활성 피라졸로-피페리딘 치환된 인돌-2-카르복스아미드 |
| CN109942537B (zh) * | 2018-03-03 | 2023-11-17 | 中国人民解放军第二军医大学 | 一类aldh2激动剂、制备方法及其用途 |
| MX2020009531A (es) | 2018-03-14 | 2020-10-05 | Janssen Sciences Ireland Unlimited Co | Regimen posologico del modulador del ensamblaje de la capside. |
| EP3597637A1 (en) | 2018-07-19 | 2020-01-22 | Irbm S.P.A. | Inhibitors of hepatitis b virus |
| EP3597653A1 (en) | 2018-07-19 | 2020-01-22 | Irbm S.P.A. | Cyclic inhibitors of hepatitis b virus |
| TWI826492B (zh) | 2018-07-27 | 2023-12-21 | 加拿大商愛彼特生物製藥公司 | 經取代四氫環戊[c]吡咯、經取代二氫吡咯,其類似物及使用其之方法 |
| EP3608326A1 (en) | 2018-08-10 | 2020-02-12 | Irbm S.P.A. | Tricyclic inhibitors of hepatitis b virus |
| AR116946A1 (es) | 2018-11-02 | 2021-06-30 | Aicuris Gmbh & Co Kg | Derivados de urea 6,7-dihidro-4h-pirazolo[4,3-c]piridinas activas contra el virus de la hepatitis b (vhb) |
| AR117189A1 (es) | 2018-11-02 | 2021-07-21 | Aicuris Gmbh & Co Kg | Derivados de 6,7-dihidro-4h-pirazolo[1,5-a]pirazin indol-2-carboxamidas activos contra el virus de la hepatitis b (vhb) |
| PY1991603A (es) | 2018-11-02 | 2020-09-17 | Aicuris Gmbh & Co Kg | Nueva 6,7-dihidro-4h-pirazolo [1,5-a] pirazina indole -2-carboxamidas activas contra el virus de la hepatitis b (vhb) |
| PY1991611A (es) | 2018-11-02 | 2022-03-14 | Aicuris Gmbh & Co Kg | Nueva urea 6,7-dihidro-4h-pirazolo [1,5-a] pirazinas activas contra el virus de la hepatitis b (vhb) |
| UY38437A (es) | 2018-11-02 | 2020-05-29 | Aicuris Gmbh & Co Kg | Nuevas urea 6,7-dihidro-4h-pirazolo[1,5-a]pirazinas activas contra el virus de la hepatitis b (hbv) |
| WO2020089460A1 (en) | 2018-11-02 | 2020-05-07 | Aicuris Gmbh & Co. Kg | Novel urea 6,7-dihydro-4h-thiazolo[5,4-c]pyridines active against the hepatitis b virus (hbv) |
| IL283190B2 (en) | 2018-11-21 | 2025-08-01 | Enanta Pharm Inc | Functionalized heterocycles as antiviral agents |
| TW202415643A (zh) | 2018-12-12 | 2024-04-16 | 加拿大商愛彼特生物製藥公司 | 經取代之芳基甲基脲類及雜芳基甲基脲類、其類似物及其使用方法 |
| WO2020169784A1 (en) | 2019-02-22 | 2020-08-27 | Janssen Sciences Ireland Unlimited Company | Amide derivatives useful in the treatment of hbv infection or hbv-induced diseases |
| BR112021021564A2 (pt) | 2019-04-30 | 2022-01-04 | Aicuris Gmbh & Co Kg | Indol-2-carboxamidas inovadoras ativas contra o vírus da hepatite b (hbv) |
| WO2020221824A1 (en) | 2019-04-30 | 2020-11-05 | Aicuris Gmbh & Co. Kg | Novel indolizine-2-carboxamides active against the hepatitis b virus (hbv) |
| US20220227785A1 (en) | 2019-04-30 | 2022-07-21 | Aicuris Gmbh & Co. Kg | Novel phenyl and pyridyl ureas active against the hepatitis b virus (hbv) |
| JP2022533008A (ja) | 2019-04-30 | 2022-07-21 | アイクリス ゲゼルシャフト ミット ベシュレンクテル ハフツング ウント コンパニー コマンディトゲゼルシャフト | B型肝炎ウイルス(hbv)に対し活性な新規のオキサリルピペラジン |
| AR119732A1 (es) | 2019-05-06 | 2022-01-05 | Janssen Sciences Ireland Unlimited Co | Derivados de amida útiles en el tratamiento de la infección por vhb o de enfermedades inducidas por vhb |
| EP3741762A1 (en) | 2019-05-23 | 2020-11-25 | Irbm S.P.A. | Oxalamido-substituted tricyclic inhibitors of hepatitis b virus |
| UY38705A (es) | 2019-05-23 | 2020-12-31 | Irbm S P A | Inhibidores tricíclicos sustituidos con oxalamido del virus de hepatitis b |
| EP3825318A1 (en) | 2019-11-25 | 2021-05-26 | Promidis S.r.l. | Oxalamido-substituted tricyclic inhibitors of hepatitis b virus |
| WO2021188414A1 (en) | 2020-03-16 | 2021-09-23 | Enanta Pharmaceuticals, Inc. | Functionalized heterocyclic compounds as antiviral agents |
| CN111825582B (zh) * | 2020-08-12 | 2022-04-08 | 江西理工大学 | 一种以芳基磺酰氯为硫源合成β-硫代羰基化合物的方法 |
Family Cites Families (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS5916871A (ja) * | 1982-07-20 | 1984-01-28 | Shionogi & Co Ltd | スルホンアミド系ベンズアミド類 |
| AU567140B2 (en) * | 1984-01-06 | 1987-11-12 | Shionogi & Co., Ltd. | Sulphonamido-benzamide derivatives |
| JP2001526255A (ja) * | 1997-12-23 | 2001-12-18 | ワーナー−ランバート・カンパニー | 炎症性疾患およびアテローム性動脈硬化症を治療または予防するチオ尿素およびベンズアミド化合物、組成物並びに方法 |
| BR112014015197A8 (pt) * | 2011-12-21 | 2017-06-13 | Novira Therapeutics Inc | agentes antivirais de hepatite b |
| BR112015015584A2 (pt) * | 2012-12-27 | 2017-12-12 | Baruch S Blumberg Inst | novos agentes antivirais contra infecção por hbv |
| WO2014131847A1 (en) * | 2013-02-28 | 2014-09-04 | Janssen R&D Ireland | Sulfamoyl-arylamides and the use thereof as medicaments for the treatment of hepatitis b |
-
2015
- 2015-12-02 CN CN201580075330.XA patent/CN107406378A/zh active Pending
- 2015-12-02 DK DK15864386.6T patent/DK3227262T3/da active
- 2015-12-02 CA CA2969557A patent/CA2969557A1/en not_active Abandoned
- 2015-12-02 JP JP2017529364A patent/JP2018504373A/ja active Pending
- 2015-12-02 AU AU2015358561A patent/AU2015358561A1/en not_active Abandoned
- 2015-12-02 ES ES15864386T patent/ES2802412T3/es active Active
- 2015-12-02 EP EP15864386.6A patent/EP3227262B1/en active Active
- 2015-12-02 WO PCT/US2015/063417 patent/WO2016089990A1/en not_active Ceased
Also Published As
| Publication number | Publication date |
|---|---|
| EP3227262B1 (en) | 2020-05-13 |
| EP3227262A4 (en) | 2018-07-11 |
| JP2018504373A (ja) | 2018-02-15 |
| WO2016089990A1 (en) | 2016-06-09 |
| EP3227262A1 (en) | 2017-10-11 |
| CN107406378A (zh) | 2017-11-28 |
| DK3227262T3 (da) | 2020-08-10 |
| AU2015358561A1 (en) | 2017-06-08 |
| CA2969557A1 (en) | 2016-06-09 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2802412T3 (es) | Compuestos de sulfonamida inversa a base de sulfuro, alquilo y piridilo para el tratamiento del VHB | |
| ES2797123T3 (es) | Agentes antivirales para la hepatitis B | |
| US10556904B2 (en) | Derivatives and methods of treating hepatitis B infections | |
| US9205079B2 (en) | Hepatitis B antiviral agents | |
| US10428041B2 (en) | Pyridyl reverse sulfonamides for HBV treatment | |
| US10710959B2 (en) | Sulfide alkyl compounds for HBV treatment | |
| HK1246774A1 (en) | Sulfide alkyl and pyridyl reverse sulfonamide compounds for hbv treatment | |
| NZ724493B2 (en) | Hepatitis B Antiviral Agents | |
| HK1197550A (en) | Hepatitis b antiviral agents |