ES2807200T3 - Composiciones antivirales tópicas y procedimientos de uso de las mismas - Google Patents
Composiciones antivirales tópicas y procedimientos de uso de las mismas Download PDFInfo
- Publication number
- ES2807200T3 ES2807200T3 ES15819451T ES15819451T ES2807200T3 ES 2807200 T3 ES2807200 T3 ES 2807200T3 ES 15819451 T ES15819451 T ES 15819451T ES 15819451 T ES15819451 T ES 15819451T ES 2807200 T3 ES2807200 T3 ES 2807200T3
- Authority
- ES
- Spain
- Prior art keywords
- composition
- topical composition
- nitric oxide
- time
- subject
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/0014—Skin, i.e. galenical aspects of topical compositions
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K33/00—Medicinal preparations containing inorganic active ingredients
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/02—Inorganic compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/06—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/06—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite
- A61K47/08—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite containing oxygen, e.g. ethers, acetals, ketones, quinones, aldehydes, peroxides
- A61K47/10—Alcohols; Phenols; Salts thereof, e.g. glycerol; Polyethylene glycols [PEG]; Poloxamers; PEG/POE alkyl ethers
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/06—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite
- A61K47/08—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite containing oxygen, e.g. ethers, acetals, ketones, quinones, aldehydes, peroxides
- A61K47/14—Esters of carboxylic acids, e.g. fatty acid monoglycerides, medium-chain triglycerides, parabens or PEG fatty acid esters
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/06—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite
- A61K47/24—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite containing atoms other than carbon, hydrogen, oxygen, halogen, nitrogen or sulfur, e.g. cyclomethicone or phospholipids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/30—Macromolecular organic or inorganic compounds, e.g. inorganic polyphosphates
- A61K47/36—Polysaccharides; Derivatives thereof, e.g. gums, starch, alginate, dextrin, hyaluronic acid, chitosan, inulin, agar or pectin
- A61K47/38—Cellulose; Derivatives thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/06—Ointments; Bases therefor; Other semi-solid forms, e.g. creams, sticks, gels
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/12—Keratolytics, e.g. wart or anti-corn preparations
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/20—Antivirals for DNA viruses
- A61P31/22—Antivirals for DNA viruses for herpes viruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Pharmacology & Pharmacy (AREA)
- Epidemiology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Engineering & Computer Science (AREA)
- Inorganic Chemistry (AREA)
- Oil, Petroleum & Natural Gas (AREA)
- Dermatology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Organic Chemistry (AREA)
- Virology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Molecular Biology (AREA)
- Communicable Diseases (AREA)
- Oncology (AREA)
- Biophysics (AREA)
- Biotechnology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicinal Preparation (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
Abstract
Una composición tópica para su uso en un procedimiento de tratamiento y/o prevención de una infección viral en un sujeto que lo necesite, comprendiendo dicho procedimiento: administrar la composición tópica a la piel de un sujeto, en la que la composición tópica comprende un ingrediente farmacéutico activo liberador de óxido nítrico que libera óxido nítrico a la piel del sujeto, y en la que la composición tópica mantiene una concentración en tiempo real de óxido nítrico de al menos aproximadamente 7 pmol de NO/mg de la composición durante al menos 1 hora después de la administración, medida por pruebas de liberación in vitro en tiempo real, tratando y/o evitando así infección viral en el sujeto, en la que el ingrediente farmacéutico activo que libera óxido nítrico comprende una partícula de sílice cocondensada que libera NO que tiene un grupo funcional de diolato de diazenio.
Description
DESCRIPCIÓN
Composiciones antivirales tópicas y procedimientos de uso de las mismas
Información de solicitud relacionada
Campo
La presente invención se refiere en general a composiciones antivirales tópicas para su uso en un procedimiento de tratamiento y/o prevención de una infección viral. La invención está estrictamente limitada al contenido de las reivindicaciones adjuntas.
Antecedentes
Los virus causan una serie de enfermedades que pueden tratarse tópicamente. Por ejemplo, las verrugas pueden ser causadas por el virus del papiloma humano y pueden tratarse tópicamente. Sin embargo, los virus pueden ser difíciles de tratar ya que invaden las células huésped y se replican. Además, han surgido nuevas cepas virales, incluidas las cepas resistentes a los antivirales. Véase, por ejemplo, WO 2011/085484 A1 o WO 03/095 398 A2 o WO 99/44622 A1 como representante de la técnica anterior.
Sumario
Se observa que los aspectos descritos con respecto a una realización pueden incorporarse en diferentes realizaciones, aunque no se describan específicamente en relación con la misma.
La invención se define como una composición tópica para su uso en un procedimiento de tratamiento y/o prevención de una infección viral en un sujeto que lo necesita, comprendiendo dicho procedimiento: administrar la composición tópica a la piel de un sujeto, en la que la composición tópica comprende un ingrediente farmacéutico activo liberador de óxido nítrico a la piel del sujeto, y en el que la composición tópica mantiene una concentración en tiempo real de óxido nítrico de al menos aproximadamente 7 pmol de NO/mg de la composición durante al menos 1 hora después de la administración, medida por pruebas de liberación in vitro en tiempo real, tratando y/o previniendo así la infección viral en el sujeto, en el que el ingrediente farmacéutico activo que libera óxido nítrico comprende una partícula de sílice condensada que libera NO y que tiene un grupo funcional de diolato de diazenio.
En algunas realizaciones, el procedimiento incluye administrar una composición tópica a la piel de un sujeto, en el que la composición tópica comprende un ingrediente farmacéutico activo que libera óxido nítrico en una cantidad de aproximadamente 0,5 % a aproximadamente 20 % en peso de la composición, tratando y o previniendo así la infección viral en el sujeto.
En algunas realizaciones, el procedimiento incluye administrar una composición tópica a la piel de un sujeto, en el que la composición tópica comprende un ingrediente farmacéutico activo liberador de óxido nítrico a la piel del sujeto, y en el que la composición tópica mantiene una concentración en tiempo real de óxido nítrico de al menos aproximadamente 7 pmol de NO/mg de la composición durante al menos 1 hora después de la administración, medida por pruebas de liberación in vitro en tiempo real, tratando y/o previniendo así la infección viral en el sujeto. En algunas realizaciones, el procedimiento incluye administrar una composición tópica a la piel de un sujeto, en el que la composición tópica comprende un ingrediente farmacéutico activo liberador de óxido nítrico a la piel del sujeto, y en el que la composición tópica mantiene una concentración en tiempo real de óxido nítrico de al menos aproximadamente 104 pmol de NO/cm2 durante un período de tiempo de al menos 1 hora después de la administración de la composición a la piel del sujeto, medida por pruebas de liberación in vitro en tiempo real tratando y/o previniendo así una infección viral en el sujeto.
Los aspectos anteriores y otros aspectos de la presente invención se describirán ahora con más detalle con respecto a otras realizaciones descritas en el presente documento.
Breve descripción de los dibujos
La Figura 1 muestra el esquema de infecciones experimentales en un conejo.
La Figura 2 muestra un gráfico de la media SEM de las mediciones del diámetro medio geométrico (GMD) de los papilomas de conejo inducidos por CRPV de conejos del Grupo A. Los papilomas se indujeron en 2 sitios con 5 |jg de preparación madre de plásmido wt-CRPV (•, ▼), y en 2 sitios con 5 |jg de preparación madre de plásmido mE8-CRPV (o, A). Los sitios izquierdos (L1 y L2) se trataron tópicamente con gel placebo (•, o) y los sitios derechos (R1 y R2) no se trataron (▼, A). Cada símbolo representa la media (± SEM) de GMD de las mediciones semanales.
La Figura 3 muestra un gráfico de la media SEM de mediciones GMD de papilomas de conejo inducidos por CRPV de conejos en el Grupo B. Se indujeron papilomas en 2 sitios con 5 jg de preparación madre de plásmido wt-CRPV (•, ▼), y en 2 sitios con 5 jg de preparación madre de plásmido mE8-CRPV (o, A). Los sitios
izquierdos (L1 y L2) se trataron tópicamente con Nitricil™ NVN1 al 1 % (•, o) y los sitios derechos (R1 y R2) no se trataron (▼, A). Cada símbolo representa la media (± SEM) de GMD de las mediciones semanales.
La Figura 4 muestra un gráfico de la media SEM de mediciones de GMD de papilomas de conejo inducidos por CRPV de conejos en el Grupo C. Se indujeron papilomas en 2 sitios con 5 |jg de preparación madre de plásmido wt-CRPV (•, ▼), y en 2 sitios con 5 jg de preparación madre de plásmido mE8-CRPV (o, A), los sitios izquierdos
(L1 y L2) se trataron tópicamente con Nitricil™ NVN4 al 1,6 % (•, o) y los sitios derechos (R1 y R2) no se trataron (▼, A). Cada símbolo representa la media (± SEM) de GMD de las mediciones semanales.
La Figura 5 muestra un gráfico de la media SEM de mediciones de GMD de papilomas de conejo inducidos por CRPV de conejos en el Grupo D. Se indujeron papilomas en 2 sitios con 5 jg de preparación madre de plásmido wt-CRPV (•, ▼), y en 2 sitios con 5 jg de preparación madre de plásmido mE8-CRPV (o, A). Los sitios izquierdos (L1 y L2) se trataron tópicamente con Nitricil™ NVN1 al 10 % (•, o) y los sitios derechos (R1 y R2) no se trataron (▼, A). Cada símbolo representa la media (± SEM) de GMD de las mediciones semanales.
La Figura 6 muestra un gráfico de la media SEM de mediciones GMD de papilomas de conejo inducidos por CRPV de conejos en el Grupo E. Se indujeron papilomas en 2 sitios con 5 jg de preparación madre de plásmido wt-CRPV (•, ▼), y en 2 sitios con 5 jg de preparación madre de plásmido mE8-CRPV (o, A) Los sitios izquierdos (L1 y L2) se trataron tópicamente con 16,3 % de Nitricil™ NVN4 (•, o) y los sitios derechos (R1 y R2) no se trataron (▼, A). Cada símbolo representa la media (±SEM) de GMD de las mediciones semanales.
La Figura 7 muestra un gráfico de la media SEM de mediciones GMD de papilomas de conejo inducidos por CRPV de conejos en el Grupo F. Se indujeron papilomas en 2 sitios con 5 jg de preparación madre de plásmido wt-CRPV (•, ▼), y en 2 sitios con 5 jg de preparación madre de plásmido mE8-CRPV (o, A). Los sitios izquierdos (L1 y L2) se trataron tópicamente con ungüento placebo (•, o) y los sitios derechos (R1 y R2) no se trataron (▼, A). Cada símbolo representa la media (±SEM) de GMD de las mediciones semanales.
La Figura 8 muestra un gráfico de la media ± SEM de mediciones GMD de papilomas de conejo inducidos por CRPV de conejos en el Grupo G. Se indujeron papilomas en 2 sitios con 5 jg de preparación madre de plásmido wt-CRPV (•, ▼), y en 2 sitios con 5 jg de preparación madre de plásmido mE8-CRPV (o, A). Los sitios izquierdos (L1 y l2) se trataron tópicamente con una sola fase, ungüento Nitricil™ NVN1 al 10 % (•, o) y los sitios derechos (R1 y R2) no se trataron (▼, A). Cada símbolo representa la media (± SEM) de GMD de las mediciones semanales.
La Figura 9 muestra un gráfico de la media SEM de mediciones de GMD de papilomas de conejo inducidos por CRPV de conejos en el Grupo H. Se indujeron papilomas en 2 sitios con 5 jg de preparación madre de plásmido wt-CRPV (•, ▼), y en 2 sitios con 5 jg de preparación madre de plásmido mE8-CRPV (o, A). Los sitios izquierdos (L1 y l2) se trataron tópicamente con 0,3 % de cidofovir en Cremophor (50 %) (•, o) y los sitios derechos (R1 y R2) no se trataron (▼, A). Cada símbolo representa la media (± SEM) de GMD de las mediciones semanales.
La Figura 10 muestra un gráfico del peso medio SEM del conejo (kg) para conejos en los Grupos A-H. Los pesos se trazan junto con barras de error SEM contra el tiempo después de la infección con CRPV.
La Figura 11 muestra un gráfico de la liberación acumulativa de óxido nítrico (NO) a lo largo del tiempo para las formulaciones utilizadas en los grupos B, D y G.
La Figura 12 muestra un gráfico de la liberación de NO en tiempo real a lo largo del tiempo para las formulaciones utilizadas en los Grupos B y D.
La Figura 13 muestra un gráfico de la liberación acumulativa de NO a lo largo del tiempo para las formulaciones utilizadas en los grupos C, E y G.
La Figura 14 muestra un gráfico de la liberación de NO en tiempo real a lo largo del tiempo para las formulaciones utilizadas en los Grupos C y E.
La Figura 15 muestra un gráfico de la liberación acumulativa de NO a lo largo del tiempo para las formulaciones utilizadas en los grupos B, C, D, E y G.
La Figura 16A muestra un gráfico de la liberación de NO en tiempo real en pmol/mg para las formulaciones utilizadas en los Grupos B, D, E y G con rectángulos que representan períodos de tiempo de 1 hora, 2 horas y 4 horas con rangos de liberación de NO según algunas realizaciones de la presente invención.
La Figura 16B es una versión ampliada de las primeras 1,5 horas de la Figura 16A con un rectángulo que representa el período de tiempo de 1 hora con intervalos de liberación de NO según algunas realizaciones de la presente invención.
La Figura 17 muestra un gráfico de la liberación de NO en tiempo real en pmol/cm2 para las formulaciones utilizadas en los Grupos D y E con rectángulos que representan períodos de tiempo de 1 hora, 2 horas y 4 horas con rangos de liberación de NO por cm2 según algunas realizaciones de la presente invención.
La Figura 18 muestra un gráfico del tamaño del papiloma (GMD) en mm a lo largo del tiempo para la formulación utilizada en el Grupo A. Se indujeron papilomas en 2 sitios con 5 |jg de preparación madre de plásmido wt-CRPV (•, ▼), y en 2 sitios con 5 jg de preparación madre de plásmido mE8-CRPV (O, A). Los sitios izquierdos (L1 y L2) se trataron tópicamente con el placebo (•, o) y los sitios derechos (R1 y R2) no se trataron (▼, A). Cada símbolo representa la media (± SEM) de GMD de las mediciones semanales.
La Figura 19 muestra un gráfico del tamaño del papiloma (GMD) en mm a lo largo del tiempo para la formulación utilizada en el Grupo B. Se indujeron papilomas en 2 sitios con 5 jg de preparación madre de plásmido wt-CRPV (•, ▼), y en 2 sitios con 5 jg de preparación madre de plásmido mE8-CRPV (O, A). Los sitios izquierdos (L1 y L2) se trataron tópicamente con la formulación de Nitricil™ NVN1 al 2 % (•, o) y los sitios derechos (R1 y R2) no se trataron (▼, A). Cada símbolo representa la media (± SEM) de GMD de las mediciones semanales.
La Figura 20 muestra un gráfico del tamaño del papiloma (GMD) en mm a lo largo del tiempo para la formulación utilizada en el Grupo C. Se indujeron papilomas en 2 sitios con 5 jg de preparación madre de plásmido wt-CRPV (•, ▼), y en 2 sitios con 5 jg de preparación madre de plásmido mE8-CRPV (o, A). Los sitios izquierdos (L1 y L2) se trataron tópicamente con la formulación de Nitricil™ NVN1 al 4 % (•, o) y los sitios derechos (R1 y R2) no se trataron (▼, A). Cada símbolo representa la media (± SEM) de GMD de las mediciones semanales.
La Figura 21 muestra un gráfico del tamaño del papiloma (GMD) en mm a lo largo del tiempo para la formulación utilizada en el Grupo D. Se indujeron papilomas en 2 sitios con 5 jg de preparación madre de plásmido wt-CRPV (•, ▼), y en 2 sitios con 5 jg de preparación madre de plásmido mE8-CRPV (o, A). Los sitios izquierdos (L1 y L2) se trataron tópicamente con la formulación de Nitricil™ NVN1 al 8 % (•, o) y los sitios derechos (R1 y R2) no se trataron (▼, A). Cada símbolo representa la media (± SEM) de GMD de las mediciones semanales.
La Figura 22 muestra un gráfico del tamaño del papiloma (GMD) en mm a lo largo del tiempo para la formulación utilizada en el Grupo E. Se indujeron papilomas en 2 sitios con 5 jg de preparación madre de plásmido wt-CRPV (•, ▼), y en 2 sitios con 5 jg de preparación madre de plásmido mE8-CRPV (o, A). Los sitios izquierdos (L1 y L2) se trataron tópicamente con la formulación de Nitricil™ NVN1 al 10 % (•, o) y los sitios derechos (R1 y R2) no se trataron (▼, A). Cada símbolo representa la media (± SEM) de GMD de las mediciones semanales.
La figura 23 muestra un gráfico del tamaño del papiloma (GMD) en mm a lo largo del tiempo para la formulación utilizada en el Grupo F. Se indujeron papilomas en 2 sitios con 5 jg de preparación madre de plásmido wt-CRPV (•, ▼), y en 2 sitios con 5 jg de preparación madre de plásmido mE8-CRPV (o, A). Los sitios izquierdos (L1 y L2) se trataron tópicamente con el control de imiquimod (•, o) y los sitios derechos (R1 y R2) no se trataron (▼, A). Cada símbolo representa la media (± SEM) de GMD de las mediciones semanales.
Descripción detallada
La presente invención se describirá ahora de forma más completa en lo que sigue. Sin embargo, esta invención puede realizarse de diferentes formas y no debe interpretarse como limitada a las realizaciones establecidas en este documento. Por el contrario, estas realizaciones se proporcionan de modo que esta divulgación sea exhaustiva y completa, y transmitirá completamente el alcance de la invención a los expertos en la materia.
La terminología utilizada en la descripción de la invención en el presente documento tiene el propósito de describir realizaciones particulares solamente y no pretende ser limitante de la invención. Como se usa en la descripción de la invención y las reivindicaciones adjuntas, las formas singulares "un", "una" y "el/la" están destinadas a incluir también las formas plurales, a menos que el contexto indique claramente otra cosa.
A menos que se defina otra cosa, todos los términos (incluidos los términos técnicos y científicos) utilizados en el presente documento tienen el mismo significado que comúnmente entiende un experto en la materia a la que pertenece esta invención. Se entenderá además que los términos, tales como los definidos en los diccionarios de uso común, deben interpretarse con un significado coherente con su significado en el contexto de la presente solicitud y la técnica relevante y no deben interpretarse de manera idealizada o en sentido excesivamente formal a menos que así se defina expresamente en el presente documento. La terminología utilizada en la descripción de la invención en este documento tiene el propósito de describir realizaciones particulares solamente y no pretende ser limitante de la invención. Todas las publicaciones, solicitudes de patente, patentes y otras referencias mencionadas en este documento se incorporan como referencia en su totalidad. En caso de conflicto en la terminología, la presente especificación es dominante.
Además, como se usa en el presente documento, "y/o" se refiere a y abarca todas y cada una de las combinaciones posibles de uno o más de los elementos listados asociados, así como la falta de combinaciones cuando se interpretan en la alternativa ("o").
A menos que el contexto indique otra cosa, se pretende específicamente que las diversas características de la invención descritas en el presente documento se puedan usar en cualquier combinación. Además, la presente invención también contempla que, en algunas realizaciones de la invención, cualquier característica o combinación de características expuestas en el presente documento puedan excluirse u omitirse. Como ilustración, si la especificación establece que un complejo comprende los componentes A, B y C, se pretende específicamente que cualquiera de A, B o C, o una combinación de los mismos, pueda omitirse y rechazarse.
Como se usa en este documento, la expresión de transición "que consiste esencialmente en" (y variantes gramaticales) debe interpretarse en el sentido de que abarca los materiales o pasos citados "y aquellos que no afecten materialmente las características básicas y novedosas" de la invención reivindicada. Véase, In re Herz, 537 F,2d 549, 551-52, 190 U.S.P.Q. 461, 463 (CCPA 1976) (énfasis en el original); véase también MPEP § 2111,03. Por lo tanto, el término "que consiste esencialmente en" como se usa en este documento no debe interpretarse como equivalente a "que comprende".
El término "aproximadamente", como se usa en el presente documento cuando se refiere a un valor medible, tal como una cantidad o concentración y similares, se refiere a variaciones de hasta ±20 % del valor especificado, tal como, pero no limitado a, ±10 %, ±5 %, ±1 %, ±0,5 %, o incluso ±0,1 % del valor especificado, así como el valor especificado. Por ejemplo, "aproximadamente X" donde X es el valor medible, debe incluir X así como variaciones de ±20 %, ±10 %, ±5 %, ±1 %, ±0,5 %, o incluso ±0,1 % de X. Un rango proporcionado aquí para un valor medible puede incluir cualquier otro rango y/o valor individual en el mismo.
De acuerdo con algunas realizaciones de la presente invención, en este documento se proporcionan procedimientos para tratar y/o prevenir una infección viral. Un procedimiento para tratar y/o prevenir una infección viral puede comprender la administración de una composición antiviral tópica (es decir, una composición de la presente invención) a la piel de un sujeto, tratando y/o previniendo así la infección viral en el sujeto. En algunas realizaciones, la composición antiviral tópica se puede administrar y/o aplicar a la piel del sujeto infectada por virus. En algunas realizaciones, un procedimiento de la presente invención puede suprimir y/o inhibir la replicación viral de un virus y/o mejorar la respuesta inmune local de un sujeto.
Las infecciones virales ejemplares incluyen, pero no se limitan a, una infección viral causada por citomegalovirus (CMV), virus de Epstein-Barr, virus de varicela zóster (VZV), virus vaccinia, virus de la viruela, virus de la viruela del mono, virus del herpes simple (HSV) 1+2), herpes zóster, virus del herpes humano 6 (HHV-6), virus del herpes humano 8 (HHV-8), virus del papiloma, molluscum contagiosum, orf, viruela y/o virus Coxsackie. En algunas realizaciones, la infección viral puede ser causada por un virus del papiloma, tal como un virus del papiloma humano. El virus del papiloma humano (HPV) puede ser HPV tipo 1, 2, 3, 4, 6, 10, 11, 16, 18, 31, 33, 35, 39, 45, 51, 52, 56, 58 y/o 59. En algunas realizaciones, la infección viral puede ser causada por un virus del herpes simple, como el herpes simple tipo 1 y/o el herpes simple tipo 2. En algunas realizaciones, la infección viral puede infectar la piel, incluidas las mucosas, del sujeto. En ciertas realizaciones, el virus puede ser un virus humano.
Según algunas realizaciones de la presente invención, se proporcionan en el presente documento procedimientos para tratar y/o prevenir afecciones cutáneas relacionadas con virus. Un procedimiento para tratar y/o prevenir una afección cutánea relacionada con virus puede comprender administrar una composición antiviral tópica (es decir, una composición de la presente invención) a la piel de un sujeto, por lo tanto, tratar y/o prevenir la afección cutánea relacionada con el virus en el sujeto. Las afecciones cutáneas relacionadas con el virus que pueden tratarse y/o prevenirse incluyen, entre otras, afecciones cutáneas asociadas con papulosis bowenoide, viruela de búfalo, verruga de carnicero, condylomata acuminate, viruela, citomegalovirus, herpes zóster diseminado, eczema herpeticum (erupción variceliforme de Kaposi), eczema vaccinatum, epidermodysplasia verruciformis, erythema infectiosum (quinta enfermedad, enfermedad de la mejilla abofeteada), viruela de corral, vaccinia generalizada, herpes genital (herpes genitalis, herpes progenitalis), tumor de Buschke-Lowenstein, enfermedad de mano-pie-boca (Coxsackie), Enfermedad de Heck (hiperplasia epitelial focal), herpangina, herpes gladiatorum (varicela), herpes simplex, queratoconjuntivitis herpética, sicosis herpética, uñero herpético, viruela del mono humana, infección por el virus linfotrópico T humano 1, tanapox humano, herpes simple intrauterino, sarcoma de Kaposi, úlcera de Lipschütz (ulcus vulvae acutum), nódulo de Milker, molluscum contagiosum, herpes simplex neonatal, zóster oftálmico, orf (dermatosis pustular contagiosa, ecthyma contagiosum, dermatitis labial infecciosa, viruela ovina), papilomatosis florida oral, leucoplakia pilosa oral (EBV), herpes orolabial (herpes labialis), vaccinia progresiva (vaccinia gangrenosum, vaccinia necrosum), pseudoviruela bovina, papilomatosis respiratoria recurrente (papilomatosis laríngea), viruela de foca, varicella (varicela), variola major (viruela), verruca plana (verrugas planas), verruca plantaris (verruga plantar), verruca vulgar (verruga), verruca palmares et plantares, y/o zóster (herpes zóster, culebrilla).
"Tratar", "que trata" o "tratamiento de" (y las variaciones gramaticales de los mismos) tal como se usan en el presente documento se refieren a cualquier tipo de tratamiento que imparta un beneficio a un sujeto y puede significar que la severidad de la afección del sujeto se reduce, al menos parcialmente mejora o se atenúa y/o que se logra algo de alivio, mitigación o disminución en al menos un síntoma clínico asociado con una infección viral y/o hay un retraso en la progresión de la infección y/o afección viral. En algunas realizaciones, la severidad de una infección viral (por ejemplo, una infección viral causada por el virus del papiloma humano) puede reducirse en un sujeto en comparación con la severidad de la infección viral en ausencia de un procedimiento de la presente invención. En
ciertas realizaciones, un procedimiento de la presente invención trata una infección viral en un sujeto, tal como una infección viral que ha afectado la piel del sujeto. En algunas realizaciones, un procedimiento de la presente invención puede tratar una infección viral eliminando y/o reduciendo el tamaño y/o la apariencia de al menos un síntoma clínico asociado con la infección viral (por ejemplo, una lesión benigna). En algunas realizaciones, un procedimiento de la presente invención puede tratar una infección viral eliminando al menos un síntoma clínico asociado con la infección viral (por ejemplo, una lesión benigna) durante un período de tiempo dado (por ejemplo, 1, 2, 3, 4, 5 o 6 días, o 1, 2, 3, 4 o más semanas, etc.).
En algunas realizaciones, una composición antiviral tópica de la presente invención se administra en una cantidad efectiva de tratamiento. Una cantidad "efectiva para el tratamiento" como se usa en el presente documento es una cantidad que es suficiente para tratar (como se define en el presente documento) a un sujeto. Los expertos en la materia apreciarán que los efectos terapéuticos no necesitan ser completos o curativos, siempre que se proporcione algún beneficio al sujeto. En algunas realizaciones, puede administrarse una cantidad efectiva de tratamiento de una composición antiviral tópica de la presente invención y puede incluir administrar una cantidad efectiva de tratamiento de un ingrediente farmacéutico activo que libera óxido nítrico. En algunas realizaciones, se puede administrar y/o aplicar una cantidad efectiva de tratamiento de óxido nítrico en un procedimiento de la presente invención. En algunas realizaciones, un procedimiento de la presente invención se lleva a cabo de tal manera que la administración de una composición antiviral tópica que comprende un ingrediente farmacéutico activo que libera óxido nítrico no produce efectos sistémicos por la administración de óxido nítrico, tal como, para ejemplo, en una cantidad efectiva de tratamiento.
Los términos "prevenir", "que previene" y "prevención" (y las variaciones gramaticales de los mismos) se refieren a evitar, reducir y/o retrasar el inicio de una infección viral y/o un síntoma clínico asociado con la misma en un sujeto y/o una reducción en la severidad del inicio de la infección viral y/o síntoma clínico en relación con lo que ocurriría en ausencia de un procedimiento de la presente invención. La prevención puede ser completa, por ejemplo, la ausencia total de infección viral y/o síntoma clínico. La prevención también puede ser parcial, de modo que la aparición de la infección viral y/o el síntoma clínico en el sujeto y/o la severidad del inicio es menor de lo que ocurriría en ausencia de un procedimiento de la presente invención. En ciertas realizaciones, un procedimiento de la presente invención previene una infección viral en un sujeto, tal como una infección viral que puede afectar la piel del sujeto.
En algunas realizaciones, una composición antiviral tópica de la presente invención se administra en una cantidad efectiva para la prevención. Una cantidad "efectiva para la prevención" como se usa en el presente documento es una cantidad que es suficiente para prevenir (como se define en el presente documento) la infección viral y/o el síntoma clínico en el sujeto. Los expertos en la materia apreciarán que el nivel de prevención no necesita ser completo, siempre que se proporcione algún beneficio al sujeto. En algunas realizaciones, puede administrarse una cantidad efectiva para la prevención de una composición antiviral tópica de la presente invención y puede incluir administrar una cantidad efectiva para la prevención de un ingrediente farmacéutico activo que libera óxido nítrico. En algunas realizaciones, se puede administrar y/o aplicar una cantidad de óxido nítrico efectiva para la prevención en un procedimiento de la presente invención. En algunas realizaciones, un procedimiento de la presente invención se lleva a cabo de tal manera que la administración de una composición antiviral tópica que comprende un ingrediente farmacéutico activo que libera óxido nítrico no produce efectos sistémicos a partir de la administración de óxido nítrico, tal como, para ejemplo, en una cantidad efectiva para la prevención.
La composición antiviral tópica se puede aplicar tópicamente a un sujeto usando cualquier procedimiento conocido por los expertos en la técnica. En algunas realizaciones, la composición puede aplicarse tópicamente al sujeto al menos 1,2, 3 o más veces por día. En algunas realizaciones, la composición puede aplicarse tópicamente al sujeto al menos 1, 2, 3, 4, 5, 6, 7, 8 o más veces por semana y/o mes. En ciertas realizaciones, la composición puede aplicarse tópicamente al sujeto una vez al día, dos veces al día, cada dos días, cada tercer día, una vez por semana o dos veces por semana. En algunas realizaciones, la composición se puede aplicar al menos una vez al día durante un período de tiempo prolongado (por ejemplo, una semana, un mes, 2 meses, etc.) y/o hasta que la infección viral y/o el síntoma clínico asociado a la misma hayan sido tratados y/o prevenidos. En algunas realizaciones, la composición puede aplicarse según sea necesario.
La presente invención encuentra uso tanto en aplicaciones veterinarias como médicas. Los sujetos adecuados de la presente invención incluyen, pero sin limitación, aves y mamíferos. El término "aviar" como se usa en el presente documento incluye, pero no se limita a, pollos, patos, gansos, codornices, pavos, faisanes, loros, periquitos, guacamayos, cacatúas, canarios y pinzones. El término "mamífero" como se usa en el presente documento incluye, pero no se limita a, primates (por ejemplo, simios y humanos), primates no humanos (por ejemplo, monos, babuinos, chimpancés, gorilas), bovinos, ovinos, caprinos, ungulados, porcinos, equinos, felinos, caninos, lagomorfos, pinnípedos, roedores (por ejemplo, ratas, hámsteres y ratones), etc. En algunas realizaciones de la presente invención, el sujeto es un mamífero y en ciertas realizaciones el sujeto es un humano. Los sujetos humanos incluyen tanto hombres como mujeres y sujetos de todas las edades, incluidos sujetos fetales, neonatales, lactantes, juveniles, adolescentes, adultos y geriátricos.
Los procedimientos de la presente invención también pueden llevarse a cabo en sujetos animales, particularmente sujetos mamíferos tales como ratones, ratas, perros, gatos, ganado y caballos con fines veterinarios, y/o con fines de criba y desarrollo de fármacos.
En algunas realizaciones, el sujeto "necesita" o "necesita de" un procedimiento de la presente invención, por ejemplo, el sujeto está en una población en riesgo (por ejemplo, el sujeto puede estar en riesgo de o más susceptible a una infección viral), el sujeto tiene hallazgos típicamente asociados con una infección viral, y/o se sospecha que el sujeto está o ha estado expuesto a un virus. En algunas realizaciones, un sujeto que lo necesita tiene un virus infección y/o un signo clínico o síntoma asociado con el mismo que puede tratarse con un procedimiento de la presente invención La presente invención puede ser particularmente adecuada para niños, adolescentes, adultos y/o sujetos geriátricos.
Una composición antiviral tópica de la presente invención puede administrarse y/o aplicarse tópicamente a cualquier porción de la piel de un sujeto, incluida la mucosa. Por ejemplo, la composición puede administrarse tópicamente a la mano, dedo, pie, dedo del pie, brazo, pierna, tronco, ano, genitales, cara, una membrana mucosa (incluida una cavidad corporal), uña, etc. de un sujeto. En algunas realizaciones, una composición antiviral de la presente invención se puede administrar tópicamente al menos a una porción de la mano, dedo, pie y/o dedo del pie de un sujeto. En algunas realizaciones, una composición antiviral de la presente invención puede administrarse tópicamente a al menos una porción del ano, genitales y/o membrana mucosa de un sujeto (por ejemplo, uretra, cuello uterino y/o vagina). En algunas realizaciones, una composición antiviral de la presente invención puede administrarse tópicamente a al menos una porción de la cara, labios y/o membrana mucosa de un sujeto (por ejemplo, fosas nasales, boca, lengua y/o faringe).
En algunas realizaciones, un procedimiento de la presente invención puede prevenir y/o reducir la apariencia y/o el tamaño de una lesión benigna. Las lesiones benignas ejemplares incluyen, pero no se limitan a, una verruga (por ejemplo, verrugas comunes (verruca vulgaris), verrugas planas, verrugas plantares, verrugas subungueales y/o periúngicas, verrugas anales/genitales, etc.), verrugas orales y/o papilomas laríngeos, condilomas de la mucosa anogenital, hiperplasia epitelial focal, papilomatosis florida oral, condyloma accuminata, papillomata, molluscum contagiosum, lesiones herpéticas, orf y/o viruela bovina. En algunas realizaciones, la lesión benigna puede ser una verruga genital externa y/o una verruga anal (por ejemplo, una verruga perianal). En algunas realizaciones, la lesión benigna puede ser una verruga no genital. En algunas realizaciones, la lesión benigna puede ser inducida y/o causada por un virus del papiloma, tal como un virus del papiloma humano.
Un procedimiento de la presente invención puede reducir la apariencia y/o el tamaño de una lesión benigna en al menos aproximadamente 5 %, 10 %, 15 %, 20 %, 25 %, 30 %, 35 %, 40 %, 45 %, 50 %, 55 %, 60 %, 65 %, 70 %, 75 %, 80 %, 85 %, 90 %, 95 %, 97 % o 100 % en comparación con la apariencia y/o tamaño de una lesión benigna previa para administrar una composición antiviral tópica de la presente invención. La apariencia de la lesión benigna se puede evaluar visualmente, por ejemplo, por el sujeto y/o un médico. El tamaño de la lesión benigna se puede determinar utilizando procedimientos conocidos por los expertos en la materia. En algunas realizaciones, un procedimiento de la presente invención puede prevenir y/o reducir la apariencia y/o el tamaño de una verruga.
En ciertas realizaciones, el sujeto puede ver una reducción en el tamaño y/o apariencia de una lesión benigna durante 1,2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, o más días y/o semanas. En algunas realizaciones, el procedimiento puede reducir el tamaño y/o la apariencia de una lesión benigna en la piel del sujeto con 12 semanas o menos, en algunas realizaciones, dentro de 8 semanas o menos, y en realizaciones adicionales, dentro de 4 semanas o menos. Un procedimiento de la presente invención puede reducir el número de lesiones benignas en al menos aproximadamente 5 %, 10 %, 15 %, 20 %, 25 %, 30 %, 35 %, 40 %, 45 %, 50 %, 55 %, 60 %, 65 %, 70 %, 75 %, 80 %, 85 %, 90 %, 95 %, 97 % o 100 % en comparación con el número de lesiones benignas antes de la administración de una composición antiviral tópica de la presente invención. El número de lesiones benignas puede evaluarse visualmente, por ejemplo, por el sujeto y/o un médico. El número de lesiones benignas se puede determinar utilizando procedimientos conocidos por los expertos en la materia. En algunas realizaciones, un procedimiento de la presente invención puede prevenir y/o reducir el número de verrugas.
Un procedimiento de la presente invención puede disminuir la tasa de recurrencia de una lesión benigna en un sujeto en al menos aproximadamente 5 %, 10 %, 15 %, 20 %, 25 %, 30 %, 35 %, 40 %, 45 %, 50 %, 55 %, 60 %, 65 %, 70 %, 75 %, 80 %, 85 %, 90 %, 95 %, 97 % o 100 % en comparación con la tasa de recurrencia del mismo tipo de lesión benigna en ausencia de la administración de una composición antiviral tópica de la presente invención. La tasa de recurrencia se puede determinar usando procedimientos conocidos por los expertos en la materia. Por ejemplo, después de un tratamiento y/o extirpación de una lesión benigna, el número de lesiones benignas puede determinarse visualmente después de un período de tiempo determinado para determinar la tasa de recurrencia. En algunas realizaciones, un procedimiento de la presente invención puede disminuir la tasa de recurrencia de las verrugas en un sujeto.
El procedimiento puede comprender administrar tópicamente y/o aplicar una composición antiviral tópica a la piel infectada por virus, incluida la mucosa, del sujeto que comprende una lesión benigna. En algunas realizaciones, la piel infectada por virus comprende una lesión y el procedimiento puede comprender además separar la lesión antes de administrar la composición tópica a la piel del sujeto. En otras realizaciones, la piel infectada por virus comprende una lesión y el procedimiento puede no comprender separar la lesión antes de administrar la composición tópica a la piel del sujeto. En algunas realizaciones, la lesión puede comprender una verruga.
En ciertas realizaciones, un procedimiento de la presente invención puede prevenir y/o reducir la apariencia y/o el tamaño de una lesión premaligna y/o una lesión maligna, tal como, por ejemplo, un tumor. La lesión premaligna y/o la lesión maligna pueden ser causadas y/o inducidas por una infección viral. En algunas realizaciones, una lesión premaligna y/o lesión maligna puede ser una lesión cutánea premaligna y/o maligna. En algunas realizaciones, la lesión premaligna y/o la lesión maligna puede deberse y/o ser causada por cáncer de cuello uterino, pene, ano y/o cavidad oral. En algunas realizaciones, la lesión premaligna y/o lesión maligna puede ser inducida y/o causada por un virus del papiloma, tal como un virus del papiloma humano. En algunas realizaciones, un procedimiento de la presente invención puede prevenir y/o reducir la apariencia y/o el tamaño de la neoplasia intraepitelial cervical. Un procedimiento de la presente invención puede reducir la apariencia y/o el tamaño de una lesión premaligna y/o maligna en al menos aproximadamente 5 %, 10 %, 15 %, 20 %, 25 %, 30 %, 35 %, 40 %, 45 %, 50 %, 55 %, 60 %, 65 %, 70 %, 75 %, 80 %, 85 %, 90 %, 95 %, 97 % o más en comparación con la apariencia y/o tamaño de una lesión premaligna y/o una lesión maligna antes de la administración de una composición antiviral tópica de la presente invención. La apariencia de la lesión premaligna y/o una lesión maligna puede evaluarse visualmente, por ejemplo, por el sujeto y/o el médico. El tamaño de la lesión premaligna y/o una lesión maligna se puede determinar usando procedimientos conocidos por los expertos en la materia.
Un procedimiento de la presente invención puede reducir el número de lesiones premalignas y/o lesiones malignas en al menos aproximadamente 5 %, 10 %, 15 %, 20 %, 25 %, 30 %, 35 %, 40 %, 45 %, 50 %, 55 %, 60 %, 65 %, 70 %, 75 %, 80 %, 85 %, 90 %, 95 %, 97 % o 100 % en comparación con el número de lesiones premalignas y/o malignas previas para administrar una composición antiviral tópica de la presente invención. El número de lesiones premalignas y/o lesiones malignas puede evaluarse visualmente, por ejemplo, por el sujeto y/o un médico. El número de lesiones premalignas y/o lesiones malignas se puede determinar utilizando procedimientos conocidos por los expertos en la materia.
Un procedimiento de la presente invención puede disminuir la tasa de recurrencia de una lesión premaligna y/o lesión maligna en un sujeto en al menos aproximadamente 5 %, 10 %, 15 %, 20 %, 25 %, 30 %, 35 %, 40 %, 45 %, 50 %, 55 %, 60 %, 65 %, 70 %, 75 %, 80 %, 85 %, 90 %, 95 %, 97 % o 100 % en comparación con la tasa de recurrencia del mismo tipo de lesión premaligna y/o lesión maligna en ausencia de la administración de una composición antiviral tópica de la presente invención. La tasa de recurrencia se puede determinar usando procedimientos conocidos por los expertos en la materia. Por ejemplo, después de un tratamiento y/o extirpación de una lesión premaligna y/o maligna, la cantidad de lesiones premalignas y/o malignas puede determinarse visualmente después de un período de tiempo determinado para determinar la tasa de recurrencia.
En algunas realizaciones, un procedimiento de la presente invención puede administrar óxido nítrico a la capa basal del epitelio de un sujeto. Un procedimiento de la presente invención puede administrar un tratamiento eficaz y/o una cantidad de óxido nítrico efectiva para la prevención a la capa basal del epitelio de un sujeto. En algunas realizaciones, el óxido nítrico puede administrarse a la membrana basal del epitelio de un sujeto. Es posible que las capas epiteliales superiores de la piel de un sujeto no necesiten separarse, exfoliarse y/o eliminarse para que el procedimiento administre óxido nítrico a la capa basal y/o la membrana basal.
En algunas realizaciones, un procedimiento de la presente invención puede administrar óxido nítrico a la piel de un sujeto. En algunas realizaciones, un procedimiento de la presente invención puede administrar óxido nítrico en una cantidad suficiente para inducir apoptosis u otro daño celular en células infectadas por virus. En algunas realizaciones, un procedimiento de la presente invención puede administrar óxido nítrico en una cantidad suficiente para inhibir y/o prevenir la replicación viral.
Se describe una composición antiviral tópica que comprende una partícula de sílice condensada que libera NO y que tiene un grupo funcional de diolato de diazenio. La composición antiviral tópica puede no comprender nitrito acidificado. "Nitrito acidificado", como se usa en el presente documento, se refiere a una composición de liberación de óxido nítrico donde el mecanismo principal de liberación de óxido nítrico es cuando un nitrito se reduce, en presencia de un ácido, a trióxido de dinitrógeno, que puede disociarse en óxido nítrico y óxido nitroso. En el procedimiento de tratamiento y/o prevención de una infección viral en un sujeto que lo necesita, el procedimiento puede administrar óxido nítrico a la piel de un sujeto sin manchar la piel del sujeto. Por ejemplo, el procedimiento puede administrar óxido nítrico a la piel de un sujeto sin manchar la piel del sujeto de amarillo, marrón y/o negro. "Ingrediente farmacéutico activo que libera óxido nítrico" y "API que libera NO", como se usa en este documento, se refieren a un compuesto u otra composición que proporciona óxido nítrico a la piel de un sujeto, pero no es óxido nítrico gaseoso. El API que libera NO incluye un compuesto que libera óxido nítrico, en lo sucesivo denominado "compuesto liberador de NO". Un compuesto liberador de NO incluye al menos un donante de NO, que es un grupo funcional que puede liberar óxido nítrico bajo ciertas condiciones.
El compuesto liberador de NO incluye un grupo funcional de diolato de diazenio como donador de NO. El grupo funcional de diolato de diazenio puede producir óxido nítrico bajo ciertas condiciones, como al exponerse al agua. Como otro ejemplo, en algunas realizaciones, el compuesto liberador de NO incluye un grupo funcional nitrosotiol como donante de NO. El donante de NO puede producir óxido nítrico bajo ciertas condiciones, como por ejemplo al exponerse a la luz. Ejemplos de otros grupos donantes de NO incluyen nitrosamina, hidroxilnitrosamina,
hidroxilamina e hidroxiurea. Cualquier combinación adecuada de donadores de NO y/o compuestos liberadores de NO también se puede usar en una segunda composición como se describe en el presente documento. Además, el donador de NO puede incorporarse a la molécula pequeña o macromolécula a través de interacciones covalentes y/o no covalentes.
Una macromolécula que libera NO puede estar en forma de una partícula que libera NO, como las descritas en la Patente de los Estados Unidos No. 8,282,967, la Patente de los Estados Unidos No 8,962,029 o la Patente de los Estados Unidos No. 8,956,658. Otros ejemplos no limitantes de compuestos liberadores de NO incluyen zeolitas liberadoras de NO como se describe en las Publicaciones de Patente de los Estados Unidos Nos. 2006/0269620 o 2010/0331968; armazones orgánicos metálicos (MOF) que liberan NO como se describe en las Publicaciones de Solicitud de Patente de los Estados Unidos Nos. 2010/0239512 o 2011/0052650; compuestos de donantes múltiples que liberan NO, como se describe en la Solicitud Internacional No. PCT/US2012/052350 titulada "Tunable Nitric Oxide-Releasing Macromolecules Having Multiple Nitric Oxide Donor Structures"; dendrímeros o estructuras metálicas que liberan NO, como se describe en la Publicación de los Estados Unidos No. 2009/0214618; recubrimientos que liberan óxido nítrico como se describe en la Publicación de los Estados Unidos No.
2011/0086234; y compuestos como se describe en la Publicación de los Estados Unidos No. 2010/0098733. Además, las macromoléculas liberadoras de NO se pueden fabricar como se describe en la Solicitud Internacional No. PCT/US2012/022048 titulada "Temperature Controlled Sol-Gel Cocondensation" presentada el 20 de enero de 2012.
El ingrediente farmacéutico activo que libera óxido nítrico puede incluir sílice precipitada cargada de NO. La sílice precipitada cargada de NO se puede formar a partir de monómeros de silano modificados con donantes de óxido nítrico en una red de siloxano cocondensada. En una realización de la presente invención, el donante de óxido nítrico puede ser un diolato de N-diazenio.
El donante de óxido nítrico puede formarse a partir de un aminoalcoxisilano mediante un procedimiento de precarga, y la red de siloxano cocondensada puede sintetizarse a partir de la condensación de una mezcla de silano que incluye un alcoxisilano y el aminoalcoxisilano para formar un donante de óxido nítrico red de siloxano cocondensada modificada. Como se usa en el presente documento, el "procedimiento de precarga" significa que el aminoalcoxisilano es "pretratado" o "precargado" con óxido nítrico antes de la condensación conjunta con alcoxisilano. En algunas realizaciones, el óxido nítrico precargado puede realizarse por procedimientos químicos. En otra realización, el procedimiento de "precarga" puede usarse para crear redes y materiales de siloxano cocondensados más funcionalizados con donantes de NO. En algunas realizaciones de la presente invención, el ingrediente farmacéutico activo que libera óxido nítrico puede comprender, consistir esencialmente o consistir en una red de sílice cocondensada sintetizada a partir de la condensación de una mezcla de silano que comprende un alcoxisilano y al menos un aminoalcoxisilano que tiene una amina sustituida por un diolato de diazenio (por ejemplo, un N-diolato de diazenio).
La red de siloxano cocondensada puede ser partículas de sílice con un tamaño uniforme, una colección de partículas de sílice con una variedad de tamaños, sílice amorfa, sílice pirógena, sílice nanocristalina, sílice cerámica, sílice coloidal, un recubrimiento de sílice, una película de sílice, sílice modificada orgánicamente, sílice mesoporosa, gel de sílice, vidrio bioactivo, o cualquier forma o estado adecuado de sílice.
En algunas realizaciones, el alcoxisilano es un tetraalcoxisilano que tiene la fórmula Si(OR)4, en la que R es un grupo alquilo. Los grupos R pueden ser iguales o diferentes. En algunas realizaciones, el tetraalcoxisilano se selecciona como ortosilicato de tetrametilo (TMOS) u ortosilicato de tetraetilo (TEOS). En algunas realizaciones, el aminoalcoxisilano tiene la fórmula: R"-(NH-R')n-Si(OR)3, en la que R es alquilo, R' es alquileno, alquileno ramificado o aralquileno, n es 1 o 2, y R" se selecciona del grupo que consiste en alquilo, cicloalquilo, arilo y alquilamino.
En algunas realizaciones, el aminoalcoxisilano puede seleccionarse de N-(6-aminohexil)aminopropiltrimetoxisilano (AHAP3); N-(2-aminoetil)-3-aminopropiltrimetoxisilano (AEAP3); (3-trimetoxisililpropil)di-etilentriamina (DET3); (aminoetilaminometil)fenetiltrimetoxisilano (AEMP3); [3-(metilamino)propil] trimetoxisilano (MAP3); N-butilaminopropiltrimetoxisilano (n-BAP3); t-butilamino-propiltrimetoxisilano (t-BAP3), N-etilaminoisobutiltrimetoxisilano (EAiB3); N-fenilamino-propiltrimetoxisilano (PAP3); y N-ciclohexilaminopropiltrimetoxisilano (cHAP3).
En algunas realizaciones, el aminoalcoxisilano tiene la fórmula: NH [R'-Si(OR)3]2, en la que R es alquilo y R' es alquileno. En algunas realizaciones, el aminoalcoxisilano puede seleccionarse entre bis(3-trietoxisililpropil)amina, bis-[3-(trimetoxisilil)propil]amina y bis-[(3-trimetoxisilil)propil]etilendiamina.
En algunas realizaciones, como se describe anteriormente en el presente documento, el aminoalcoxisilano está precargado para la liberación de NO y el grupo amino está sustituido con un dialato de diazenio. Por lo tanto, en algunas realizaciones, el aminoalcoxisilano tiene la fórmula: R"-N(NONO-X+)-R'-Si(OR)3, en la que R es alquilo, R' es alquileno o aralquileno, R" es alquilo o alquilamina, y X+ es un catión seleccionado del grupo que consiste en Na+, K+ y Li+.
La composición de la red de siloxano (por ejemplo, la cantidad o la composición química del aminoalcoxisilano) y las condiciones de carga de óxido nítrico (por ejemplo, el disolvente y la base) se pueden variar para optimizar la
cantidad y la duración de la liberación de óxido nítrico. Por lo tanto, en algunas realizaciones, la composición de las partículas de sílice puede modificarse para regular la vida media de liberación de NO de las partículas de sílice.
En otra realización, el grupo amino del aminoalcoxisilano está sustituido con un diolato de diazenio, y el aminoalcoxisilano tiene una fórmula de R"-N(NONO-X+)-R'-Si(OR)3, en la que: R es alquilo, R' es alquileno o aralquileno, R" es alquilo o alquilamina, y X+ es un catión seleccionado del grupo que consiste en Na+ y K+.
En ciertas realizaciones, la API de liberación de NO puede comprender una red de sílice cocondensada que comprende aminoetilaminopropil trimetoxisilano diolato de diazenio (AEAP3) y ortosilicato de tetrametilo (TMOS) y/o una red de sílice cocondensada que comprende aminoetilaminopropil trimetoxisilano diolato de diazenio (AEAP3) y ortosilicato de tetraetilo (TEOS). En algunas realizaciones, el API de liberación de NO puede comprender una red de sílice cocondensada que comprende metilaminopropil trimetoxisilano diolato de diazenio (MAP3) y tetrametilortosilicato (TMOS) y/o una red de sílice cocondensada que comprende metilaminopropil trimetoxisilano diolato de diazenio (MAP3) y ortosilicato de tetraetilo (TEOS).
En algunas realizaciones de la invención, el tamaño de partícula de un API de liberación de NO puede estar en un rango de aproximadamente 20 nm a aproximadamente 20 |jm o cualquier rango en el mismo, tal como, pero sin limitación, de aproximadamente 100 nm a aproximadamente 20 jm o aproximadamente 1 jm a aproximadamente 20 jm. El tamaño de partícula puede adaptarse para minimizar o prevenir la toxicidad y/o penetración a través de la epidermis (o dermis comprometida) y dentro de los vasos sanguíneos. En realizaciones particulares, el tamaño de partícula se distribuye alrededor de un tamaño medio de partícula de menos de 20 jm, o cualquier intervalo en el mismo, y el tamaño puede permitir que la partícula entre en un folículo. En algunas realizaciones, una API de liberación de NO puede tener un tamaño de partícula que se distribuye alrededor de un tamaño medio de partículas de aproximadamente 20, 19, 18, 17, 16, 15, 14, 13, 12, 11, 10, 9, 8, 7, 6, 5, 4, 3, 2 o 1 jm. En realizaciones adicionales, una API de liberación de NO puede tener un tamaño de partícula que se distribuye alrededor de un tamaño de partícula medio de menos de 10 jm, o cualquier intervalo en el mismo, tal como, pero sin limitación, de aproximadamente 2 jm a aproximadamente 10 jm o aproximadamente 4 jm a aproximadamente 8 jm. En otras realizaciones, el tamaño de partícula puede distribuirse alrededor de un tamaño medio de partícula mayor de 20 jm, o cualquier intervalo en el mismo, y el tamaño puede evitar que la partícula ingrese al folículo. En otras realizaciones adicionales, se puede proporcionar una mezcla de partículas con tamaños de partículas medios distribuidos alrededor de dos o más tamaños de partículas medios. Una API de liberación de NO puede ser micronizada (por ejemplo, molienda por bolas y/o chorro). Los procedimientos para proporcionar un tamaño de partícula y/o micronización deseados incluyen, pero no se limitan a, los descritos en la Publicación de Solicitud de Patente de los Estados Unidos No. 2013/0310533.
En algunas realizaciones, una API que libera NO puede estar presente en una composición antiviral tópica en una cantidad de aproximadamente 0,5 % a aproximadamente 25 % en peso de la composición. Por ejemplo, en algunas realizaciones, una API de liberación de NO puede estar presente en una composición de la presente invención en una cantidad de aproximadamente 0,5 % a aproximadamente 20 %, aproximadamente 0,5 % a aproximadamente 5 %, aproximadamente 1 % a aproximadamente 20 %, aproximadamente 1 % a aproximadamente 10 %, aproximadamente 1 % a aproximadamente 8 %, aproximadamente 1 % a aproximadamente 20 %, aproximadamente 5 % a aproximadamente 15 %, o aproximadamente 2 % a aproximadamente 6 % en peso de la composición. En ciertas realizaciones, un ingrediente farmacéutico activo que libera óxido nítrico puede estar presente en una composición de la presente invención en una cantidad de aproximadamente 0,5 %, 1 %, 2 %, 3 %, 4 %, 5 %, 6 %, 7 %, 8 %, 9 %, 10 %, 11 %, 12 %, 13 %, 14 %, 15 %, 16 %, 17 %, 18 %, 19 %, 20 %, 21 %, 22 %, 23 %, 24 %, o 25 % en peso de la composición.
Una composición de la presente invención puede comprender una API que libera NO y puede almacenar y/o liberar óxido nítrico en una cantidad de aproximadamente 0,05 % a aproximadamente 10 % en peso de la composición, tal como, pero sin limitarse a, aproximadamente 0,15 % a aproximadamente 2 %, aproximadamente 0,15 % a aproximadamente 1 %, aproximadamente 0,3 % a aproximadamente 1,2 %, aproximadamente 0,15 % a aproximadamente 6 %, aproximadamente 1 % a aproximadamente 10 %, aproximadamente 3 % a aproximadamente 6 %, o aproximadamente 1 % a aproximadamente 5 % en peso de la composición. En ciertas realizaciones, una composición de la presente invención puede comprender un producto farmacéutico activo que libera óxido nítrico y puede almacenar y/o liberar óxido nítrico en una cantidad de aproximadamente 0,15 %, 0,3 %, 0,6 %, 0,9 %, 1 %, 1.25 %, 1,5 %, 1,75 %, 2 %, 2,25 %, 2,5 %, 2,75 %, 3 %, 3,25 %, 3,5 %, 3,75 %, 4 %, 4,25 %, 4,5 %, 4,75 %, 5 %, 5.25 %, 5,5 %, 5,75 %, 6 %, 6,25 %, 6,5 %, 6,75 %, 7 %, 7,25 %, 7,5 %, 7,75 %, 8 %, 8,25 %, 8,5 %, 8,75 %, 9 %, 9.25 %, 9,5 %, 9,75 %, o 10 % en peso de la composición. La cantidad de óxido nítrico liberado puede determinarse usando pruebas de liberación in vitro en tiempo real. En algunas realizaciones, la liberación de óxido nítrico se puede determinar usando un analizador de óxido nítrico quimioluminiscente.
Una composición de la presente invención puede proporcionar y/o permitir un período prolongado de liberación de NO. En algunas realizaciones, una composición de la presente invención puede proporcionar y/o permitir una liberación continua de NO durante aproximadamente 1 hora o más, tal como, pero sin limitación, aproximadamente 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12 o más horas después de la administración de la composición tópica a un sujeto. En algunas realizaciones, la composición puede proporcionar una liberación continua de NO durante al menos aproximadamente 1, 2, 3, 4 o 5 horas después de la administración de la composición tópica a un sujeto.
En algunas realizaciones, una composición antiviral tópica de la presente invención puede proporcionar una velocidad de liberación de aproximadamente 1 a aproximadamente 5000 pmol de NO/mg/s de la composición en un período de tiempo definido después de la administración de la composición a un sujeto. Todas las liberaciones de óxido nítrico descritas en este documento, incluidas las descritas con respecto a un período de tiempo después de la administración a un sujeto, se mencionan con respecto a las pruebas de liberación in vitro en tiempo real. La liberación in vivo de óxido nítrico (es decir, la liberación de óxido nítrico cuando la composición antiviral tópica de la presente invención se aplica a un sujeto) puede variar con el sujeto al que se aplica la composición antiviral tópica. En algunas realizaciones, la liberación in vivo de óxido nítrico puede variar dependiendo de la realización particular de la composición antiviral tópica. Sin embargo, se cree que las diferencias en la liberación in vitro de composiciones antivirales tópicas según la presente invención se reflejarán en la liberación de óxido nítrico cuando la composición tópica se aplica a un sujeto. Por consiguiente, para mayor claridad, a menos que se indique específicamente que la liberación de óxido nítrico es cuando se aplica a un sujeto, las referencias a la liberación de óxido nítrico con respecto a las realizaciones de las composiciones antivirales tópicas de la presente invención serán con referencia a la liberación in vitro de la composición. El punto de tiempo cero o el punto de tiempo inicial de la prueba de liberación in vitro puede correlacionarse con el tiempo de administración a un sujeto con todos los puntos de tiempo real posteriores correspondientes a un cierto tiempo después de la administración.
En algunas realizaciones, la composición puede liberar aproximadamente 1 a aproximadamente 10, aproximadamente 1 a aproximadamente 100, aproximadamente 100 a aproximadamente 1000, aproximadamente 1000 a aproximadamente 4000, o aproximadamente 2500 a aproximadamente 5000 pmol de NO/mg de la composición a 1 hora, 45, 30, 15, 5, 4, 3, 2 o 1 minuto(s) medido por liberación in vitro. En algunas realizaciones, la composición puede liberar, en promedio, aproximadamente 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 20, 30, 40, 50, 100 o más pmol de NO/mg de la composición a las 24, 20, 15, 10, 5, 4, 3, 2 o 1 horas, medida por liberación in vitro.
En algunas realizaciones, los valores de liberación de NO proporcionados en este documento pueden incluir la cantidad de variación típicamente asociada con la fabricación de la API de liberación de NO. Por ejemplo, la variación en la liberación de NO se puede ver entre muestras en el mismo lote y/o lotes diferentes. En algunas realizaciones, la variación en la liberación de NO entre muestras en el mismo lote y/o lotes diferentes puede estar en un rango de ± aproximadamente 0 % a aproximadamente 15 % y esta variación puede incluirse en los valores de liberación de NO descritos aquí. En algunas realizaciones, la variación en la liberación de NO entre muestras en el mismo lote y/o lotes diferentes puede estar en un rango de ± aproximadamente 10 % a aproximadamente 15 % y esta variación puede incluirse en los valores de liberación de NO descritos aquí.
En algunas realizaciones, la composición puede liberar aproximadamente 1 a aproximadamente 10, aproximadamente 1 a aproximadamente 100, aproximadamente 100 a aproximadamente 1000, aproximadamente 1000 a aproximadamente 4000, o aproximadamente 2500 a aproximadamente 5000 pmol de NO/mg de la composición a 0,5, 1 hora, 45, 30, 15, 5, 4, 3, 2 o 1 minuto(s) después de la administración de la composición a un sujeto. En algunas realizaciones, la composición puede liberar, en promedio, aproximadamente 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 20, 30, 40, 50, 100 o más pmol de NO/mg de la composición a las 24, 20, 15, 10, 5, 4, 3, 2 o 1 horas después de la administración de la composición a un sujeto.
Una composición antiviral tópica de la presente invención puede proporcionar una liberación continua de NO durante al menos aproximadamente 0,5, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10 o más horas según se mide por liberación in vitro, y la composición puede tener una liberación de NO durante la liberación continua que, en promedio, está en un rango de aproximadamente 1 a aproximadamente 500 pmol de NO/mg de la composición, tal como, pero no limitado a, aproximadamente 10 a aproximadamente 50, aproximadamente 50 a aproximadamente 200, aproximadamente 100 a aproximadamente 500, aproximadamente 300 a aproximadamente 500, aproximadamente 1 a aproximadamente 10, o aproximadamente 1 a aproximadamente 3 pmol de NO/mg de la composición.
Una composición antiviral tópica de la presente invención puede proporcionar una liberación continua de NO durante al menos aproximadamente 0,5, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10 o más horas después de la administración. de la composición a un sujeto, y la composición puede tener una liberación de NO durante la liberación continua que, en promedio, está en un rango de aproximadamente 1 a aproximadamente 500 pmol de NO/mg de la composición, tal como, pero sin limitación aproximadamente 10 a aproximadamente 50, aproximadamente 50 a aproximadamente 200, aproximadamente 100 a aproximadamente 500, aproximadamente 300 a aproximadamente 500, aproximadamente 1 a aproximadamente 10, o aproximadamente 1 a aproximadamente 3 pmol de NO/mg de la composición.
En algunas realizaciones de la presente invención, una composición antiviral tópica de la presente invención mantiene una concentración en tiempo real de NO mayor de 5 pmol de NO/mg durante un período de al menos 4 horas, una concentración en tiempo real de NO de más de 6 pmol de NO/mg durante un período de al menos 2 horas, y/o una concentración en tiempo real de NO de más de 7 pmol de NO/mg durante un período de al menos 1 hora, medida por la liberación in vitro En algunas realizaciones, una composición antiviral tópica de la presente invención puede mantener una concentración de NO en tiempo real de al menos aproximadamente 5 pmol de NO/mg o más (por ejemplo, 10, 20, 30, 40, 50, 100, 150, 200, 250, 500, 1000, 2000 pmol de NO/mg de la composición o más) durante un período de al menos 1 hora o más (por ejemplo, 1, 1,5, 2, 2,5, 3, 3,5, 4, 4,5, 5, 5,5, 6, 6,5, 7, 7,5, 8, 8,5 horas o más) según lo medida por la liberación in vitro.
En realizaciones particulares de la presente invención, una composición antiviral tópica de la presente invención mantiene una concentración en tiempo real de NO en un intervalo de aproximadamente 5 pmol a aproximadamente 4000 pmol de NO/mg durante un período de al menos 4 horas, una concentración en tiempo real de NO en un rango de aproximadamente 6 a aproximadamente 4000 pmol de NO/mg durante un período de al menos 2 horas, y/o una concentración en tiempo real de NO en un rango de aproximadamente 7 a aproximadamente 4000 pmol de NO/mg durante un período de al menos 1 hora, medida por la liberación in vitro.
En algunas realizaciones de la presente invención, una composición antiviral tópica de la presente invención mantiene una concentración en tiempo real de NO mayor que 74 pmol de NO/cm2 durante un período de al menos 4 horas, una concentración en tiempo real de NO de más de 89 pmol de NO/cm2 por un período de al menos 2 horas, y/o una concentración en tiempo real de NO mayor de 104 pmol de NO/cm2 por un período de al menos 1 hora, medida por la liberación in vitro. En realizaciones particulares de la presente invención, una composición antiviral tópica de la presente invención mantiene una concentración en tiempo real de NO en un rango de aproximadamente 74 pmol de NO/cm2 a aproximadamente 59.520 pmol de NO/cm2 durante un período de al menos 4 horas, una concentración de NO en tiempo real en un rango de aproximadamente 89 a aproximadamente 59.520 pmol de NO/cm2 durante un período de al menos 2 horas, y/o una concentración de NO en tiempo real en un rango de aproximadamente 104 a 59.520 pmol de NO/cm2 durante un período de al menos 1 hora medida por la liberación in vitro.
En algunas realizaciones, una composición antiviral tópica de la presente invención puede proporcionar una liberación acumulativa de NO de al menos aproximadamente 10 nmol de NO/mg de la composición a las 24 horas o menos (por ejemplo, 24, 20, 15, 10, 5, 4, 2 o 1 horas) después de la administración de la composición a un sujeto. La composición antiviral tópica puede tener una liberación acumulativa de NO en un rango de aproximadamente 10 a aproximadamente 50, aproximadamente 10 a aproximadamente 100, aproximadamente 100 a aproximadamente 1000, aproximadamente 250 a aproximadamente 750, aproximadamente 500 a aproximadamente 750, aproximadamente 50 a aproximadamente 1000, aproximadamente 100 a aproximadamente 1500, aproximadamente 200 a aproximadamente 1000, aproximadamente 100 a aproximadamente 500, o aproximadamente 500 a aproximadamente 1000 nmol de NO/mg de la composición a las 24 horas o menos después de la administración de la composición a un sujeto.
En algunas realizaciones, una composición antiviral tópica de la presente invención proporciona una liberación acumulativa de NO en un intervalo de aproximadamente 180 nmol de NO/mg a aproximadamente 1000 nmol de NO/mg en 24 horas después de la administración de la composición a un sujeto. En algunas realizaciones de la presente invención, una composición antiviral tópica de la presente invención proporciona una liberación acumulativa de NO mayor de 180 nmol de NO/mg en 24 horas después de la administración de la composición a un sujeto mientras se mantiene una concentración de NO en tiempo real de más de 5 pmol de NO/mg durante un período de al menos 4 horas, medida por la liberación in vitro. En algunas realizaciones, una composición antiviral tópica de la presente invención proporciona una liberación acumulativa de NO en un intervalo de aproximadamente 90 nmol de NO/mg a aproximadamente 450 nmol de NO/mg en 4 horas después de la administración de la composición a un sujeto.
En algunas realizaciones, una composición antiviral tópica de la presente invención libera la mitad del NO liberado de la composición en aproximadamente 9 minutos o más en base a una liberación total de NO determinada a las 24 horas medida por la liberación in vitro. En algunas realizaciones, la composición antiviral tópica puede liberar la mitad del NO liberado de la composición en aproximadamente 10, 20, 30, 40, 50 o 60 minutos, o 2, 3, 4, 5, 6, 7 u 8 horas. o más con base en una liberación total de NO determinada a las 24 horas medida por la liberación in vitro. En algunas realizaciones, una composición antiviral tópica de la presente invención libera la mitad del NO liberado de la composición en un intervalo de aproximadamente 9 minutos a aproximadamente 8 horas en base a una liberación total de NO determinada a las 24 horas medida por liberación in vitro.
En algunas realizaciones, una composición antiviral tópica de la presente invención proporciona una concentración máxima (Cmáx) de NO liberado de más de 160 pmol de NO/mg con base en una liberación total de NO determinada a las 24 horas medida por liberación in vitro. En algunas realizaciones, la composición antiviral tópica de la presente invención puede proporcionar una Cmáx de NO liberado de aproximadamente 175, 200, 300, 400, 500, 600, 700, 800, 900, 1000, 1500, 2000, 2500, 3000, 3500 pmol de NO/mg o más con base en una liberación total de NO determinada a las 24 horas medida por la liberación in vitro. En algunas realizaciones, una composición antiviral tópica de la presente invención proporciona una concentración máxima de NO liberado en un intervalo de aproximadamente 160 pmol de NO/mg a aproximadamente 3500 pmol de NO/mg. En algunas realizaciones, una composición antiviral tópica de la presente invención proporciona una concentración máxima de NO liberado de más de 160 pmol de NO/mg y libera la mitad del NO liberado de la composición en aproximadamente 9 minutos o más en base a una liberación total de NO determinado a las 24 horas medido por liberación in vitro.
En realizaciones particulares de la presente invención, una composición antiviral tópica proporciona una concentración máxima de NO liberado de al menos aproximadamente 3000 pmol de NO/mg, libera al menos aproximadamente 900 nmol de NO/mg en 24 horas, y/o libera la mitad de la liberación de NO en aproximadamente 9 minutos en función de una liberación total de NO determinada a las 24 horas medida por la liberación in vitro. En realizaciones particulares de la presente invención, una composición antiviral tópica proporciona una concentración
máxima de NO liberado de al menos aproximadamente 13 pmol de NO/mg, libera al menos aproximadamente 300 nmol de NO/mg en 24 horas y/o libera la mitad de la liberación de NO en aproximadamente 420 minutos con base en una liberación total de NO determinada a las 24 horas medida por liberación in vitro. En realizaciones adicionales, una composición antiviral tópica tiene una concentración en tiempo real de NO de al menos 5 pmol de NO/mg a las 4 horas medida por la liberación in vitro. En realizaciones particulares de la presente invención, una composición antiviral tópica proporciona una concentración máxima de NO liberado en un intervalo de aproximadamente 12 pmol de NO/mg a aproximadamente 3200 pmol de NO/mg, una concentración en tiempo real de NO de al menos 5 pmol de NO/mg a las 4 horas, libera NO en un rango de aproximadamente 300 nmol de NO/mg a aproximadamente 1000 nmol de NO/mg en 24 horas, y/o libera la mitad de la liberación de NO en un rango de aproximadamente 9 minutos a aproximadamente 420 minutos con base en una liberación total de NO determinada a las 24 horas medida por liberación in vitro.
En algunas realizaciones, una composición antiviral tópica de la presente invención puede mantener una concentración en tiempo real de NO de al menos aproximadamente 70 pmol de NO/cm2 o más (por ejemplo, 75, 100, 150, 200, 250, 500, 1000, 2000, 3000, 4000, 5000, 6000, 7000 pmol de NO/cm2 o más) durante un período de al menos 0,5 horas o más (por ejemplo, 1, 1,5, 2, 2,5, 3, 3,5, 4, 4,5, 5, 5,5, 6, 6,5, 7, 7,5, 8, 8,5 horas o más) después de la administración de la composición a un sujeto, medida por la liberación in vitro. En algunas realizaciones de la presente invención, una composición antiviral tópica de la presente invención mantiene una concentración en tiempo real de NO mayor de 70 pmol de NO/cm2 durante un período de al menos 4 horas, medido por liberación in vitro.
En algunas realizaciones, una composición antiviral tópica de la presente invención puede proporcionar una liberación acumulativa de NO de al menos aproximadamente 4500 nmol de NO/cm2 (por ejemplo, 5000, 6000, 7000, 8000, 9000, 10000, 11000, 12000, 13.000, 14.000 nmol de NO/cm2 o más) a las 24 horas o menos (por ejemplo, 24, 20, 15, 10, 5, 4, 2 o 1 hora) después de la administración de la composición a un sujeto. En algunas realizaciones, una composición antiviral tópica de la presente invención proporciona una liberación acumulativa de NO en un intervalo de aproximadamente 4500 nmol de NO/cm2 a aproximadamente 14.000 nmol de NO/cm2 en 24 horas después de la administración de la composición a un sujeto. En algunas realizaciones de la presente invención, una composición antiviral tópica de la presente invención proporciona una liberación acumulativa de NO mayor de 4500 nmol de NO/cm2 en 24 horas después de la administración de la composición a un sujeto mientras se mantiene una concentración de NO en tiempo real de más de 70 pmol de NO/cm2 durante un período de al menos 4 horas, medida por la liberación in vitro. En algunas realizaciones, una composición antiviral tópica de la presente invención proporciona una liberación acumulativa de NO en un intervalo de aproximadamente 1300 nmol de NO/cm2 a aproximadamente 14.000 nmol de NO/cm2 en 4 horas después de la administración de la composición a un sujeto.
En algunas realizaciones, una composición antiviral tópica de la presente invención puede tener una concentración en tiempo real de NO de al menos aproximadamente 10 pmol de NO/cm2 o más (por ejemplo, 20, 30, 40, 50, 60, 70, 80, 90, 100, 200, 300, 400, 500, 600, 700, 800, 900, 1000 pmol de NO/cm2 o más) a las 0,5 horas o más (por ejemplo, 1, 1,5, 2, 2,5, 3, 3,5, 4, 4,5, 5, 5,5, 6, 6,5, 7, 7,5, 8, 8,5 horas o más) después de la administración de la composición a un sujeto. En algunas realizaciones, una composición antiviral tópica de la presente invención proporciona una concentración en tiempo real de NO de al menos 100 pmol de NO/cm2 a 0,5 horas, al menos 50 pmol de NO/cm2 a 1 hora, al menos 40 pmol de NO/cm2 a las 2 horas, al menos 25 pmol de NO/cm2 a las 3 horas, y/o al menos 20 pmol de NO/cm2 a las 4 horas, cada uno medido por liberación in vitro. En algunas realizaciones, una composición antiviral tópica de la presente invención proporciona una concentración en tiempo real de NO de al menos 130 pmol de NO/cm2 a 0,5 horas, al menos 115 pmol de NO/cm2 a 1 hora, al menos 90 pmol de NO/cm2 a las 2 horas, al menos 90 pmol de NO/cm2 a las 3 horas, y/o al menos 80 pmol de NO/cm2 a las 4 horas, cada uno medido por liberación in vitro.
En algunas realizaciones, una composición antiviral tópica de la presente invención proporciona una concentración en tiempo real de NO de al menos 800 pmol de NO/cm2 a 0,5 horas, al menos 500 pmol de NO/cm2 a 1 hora, al menos 200 pmol de NO/cm2 a las 2 horas, al menos 100 pmol de NO/cm2 a las 3 horas, y/o al menos 50 pmol de NO/cm2 a las 4 horas, cada uno medido por liberación in vitro.
En algunas realizaciones, una composición antiviral tópica de la presente invención proporciona una concentración máxima de NO liberado de más de 2400 pmol de NO/cm2. En algunas realizaciones, una composición antiviral tópica de la presente invención proporciona una concentración máxima de NO liberado en un intervalo de aproximadamente 2400 pmol de NO/cm2 a aproximadamente 47.000 pmol de NO/cm2. En algunas realizaciones, una composición antiviral tópica de la presente invención proporciona una concentración máxima de NO liberado de más de 2400 pmol de NO/cm2 y libera la mitad del NO liberado de la composición en 10 minutos o más en base a una liberación total de NO determinada a las 24 horas medido por liberación in vitro.
En realizaciones particulares de la presente invención, una composición antiviral tópica proporciona una concentración máxima de NO liberado de al menos aproximadamente 47.000 pmol de NO/cm2, libera al menos aproximadamente 13.000 nmol de NO/cm2 en 24 horas y/o libera la mitad de la liberación de NO en aproximadamente 9 minutos basada en una liberación total de NO determinada a las 24 horas medida por la liberación in vitro. En realizaciones particulares de la presente invención, una composición antiviral tópica proporciona una concentración máxima de NO liberado de al menos aproximadamente 190 pmol de NO/cm2, libera
al menos aproximadamente 4600 nmol de NO/cm2 en 24 horas y/o libera la mitad de NO en aproximadamente 420 minutos con base en una liberación total de NO determinada a las 24 horas medida por la liberación in vitro. En realizaciones adicionales, una composición antiviral tópica tiene una concentración en tiempo real de NO de al menos 70 pmol de NO/cm2 a las 4 horas medida por la liberación in vitro. En realizaciones particulares de la presente invención, una composición antiviral tópica proporciona una concentración máxima de liberación de NO en un intervalo de aproximadamente 190 pmol de NO/cm2 a aproximadamente 47.000 pmol de NO/cm2, una concentración en tiempo real de NO de al menos 70 pmol de NO/cm2 a las 4 horas, libera NO en un rango de aproximadamente 4600 nmol de NO/cm2 a aproximadamente 47.000 nmol de NO/cm2 en 24 horas y/o libera la mitad de la liberación de NO en un rango de aproximadamente 9 minutos a aproximadamente 420 minutos con base en una liberación total de NO determinada a las 24 horas medida por liberación in vitro.
La eficacia del producto liberador de óxido nítrico puede estar relacionada, no solo con la cantidad de liberación de óxido nítrico, sino también con la velocidad a la que se libera. Por consiguiente, los productos con una velocidad de liberación promedio durante aproximadamente 5 minutos y en algunas realizaciones, 4,7 minutos, de 600 nmol NO/mg hora05 o más, 900 nmol NO/mg hora05 o más, 2500 nmol NO/mg hora05 o más, 4000 nmol NO/mg hora05 o más o 4500 nmol NO/mg hora05 o más medido por liberación in vitro puede utilizarse de acuerdo con ciertas realizaciones de la presente invención.
El pH de una composición de la presente invención puede estar en un rango de aproximadamente 3 a aproximadamente 11. En algunas realizaciones, el pH de la composición puede estar en un rango de aproximadamente 3 a aproximadamente 5, aproximadamente 3,5 a aproximadamente 4,5, aproximadamente 5 a aproximadamente 7, aproximadamente 5,5 a aproximadamente 6,5, aproximadamente 3,5 a aproximadamente 6,5, aproximadamente 6 a aproximadamente 10, aproximadamente 6 a aproximadamente 7, aproximadamente 7 a aproximadamente 10, aproximadamente 7 a aproximadamente 9, aproximadamente 7 a aproximadamente 8, aproximadamente 7,5 a aproximadamente 8, o aproximadamente 8 a aproximadamente 9. En ciertas realizaciones, el pH de la composición puede ser aproximadamente 3, 4, 5, 6, 7, 8, 9, 10 u 11. El pH de la composición puede ser determinado antes de la administración a un sujeto y/o puede determinarse una vez aplicado a la piel del sujeto. En algunas realizaciones, donde una composición de la presente invención comprende dos o más partes y/o fases, el pH puede determinarse mediante la combinación y/o mezcla de las dos o más partes y/o fases antes y/o después de la administración a La piel del sujeto. En ciertas realizaciones, el pH de la composición se mide antes de la administración a la piel del sujeto, pero después de combinar todas las partes y/o fases de la composición.
El pH puede medirse usando procedimientos conocidos. El pH de una composición de la presente invención puede determinarse una vez que se alcanza un pH en estado estacionario antes y/o después de la administración y/o después de la combinación de una primera parte y una segunda parte de la composición. Alternativamente o, además, el pH de una composición de la presente invención puede determinarse después de un período de tiempo definido, tal como, pero no limitado a, después de aproximadamente 5, 10, 15, 20, 25, 30, 35, 40, 45, 50, 55, 60 minutos o más. En algunas realizaciones, el pH de una composición de la presente invención puede medirse in vitro. Alternativamente o, además, el pH de una composición de la presente invención puede medirse después de la administración a un sujeto, tal como, por ejemplo, un pH de la superficie de la piel puede medirse después de la administración de una composición de la presente invención a la piel de un sujeto.
Una composición de la presente invención puede comprender al menos dos partes. Las al menos dos partes pueden combinarse antes, durante y/o después de la administración a un sujeto para formar una composición antiviral de la presente invención. En algunas realizaciones, una composición de la presente invención comprende una primera parte que comprende una primera composición y una segunda parte que comprende una segunda composición. En algunas realizaciones, la primera y segunda composición se pueden combinar mezclando, agitando, mezclando, dispersando, moliendo, homogeneizando, aplicando a la misma área o región, y similares. En algunas realizaciones, la primera y segunda composición se pueden mezclar y/o mezclar antes, durante y/o después de la administración a la piel de un sujeto. En algunas realizaciones, la primera composición y la segunda composición se pueden combinar aplicando una o más capas de la segunda composición sobre un sujeto y luego aplicando una o más capas de la primera composición sobre un sujeto o viceversa para formar una composición antiviral tópica de la presente invención.
La segunda composición puede comprender una API que libera NO. En algunas realizaciones, una composición de la presente invención puede comprender una primera parte que comprende una primera composición que puede estar en forma de hidrogel. "Hidrogel", como se usa en el presente documento, se refiere a un gel hidrófilo que comprende una matriz de gel y agua. En algunas realizaciones, la primera composición puede comprender al menos un alcohol polihídrico, al menos un agente que aumenta la viscosidad y agua. En algunas realizaciones, la primera composición puede comprender al menos un alcohol polihídrico, al menos un agente que aumenta la viscosidad, agua, al menos un disolvente adicional (es decir, al menos un disolvente además del agua), un tampón y/o agente tamponante, un emoliente, y opcionalmente un conservante.
Alcoholes polihídricos ejemplares que pueden estar presentes en una primera composición incluyen, entre otros, glicerol, propilenglicol, polietilenglicol, polipropilenglicol, trietilenglicol, glicoles neopentales, butilenglicol, polietilenglicol, sorbitol, arabitol, eritritol, HSH, isomalt, lactitol maltitol, manitol, xilitol, treitol, ribitol, galactitol, fucitol,
iditol, inositol, volemitol y cualquier combinación de los mismos. En algunas realizaciones, la primera composición comprende glicerol, tal como, pero sin limitación, glicerol anhidro.
Un alcohol polihídrico puede estar presente en una primera composición en una cantidad de aproximadamente 1 % a aproximadamente 30 % en peso de la primera composición o cualquier rango y/o valor individual en la misma, tal como, pero sin limitarse a, aproximadamente 1 % a aproximadamente 20 %, aproximadamente 1 % a aproximadamente 10 %, aproximadamente 5 % a aproximadamente 10 %, o aproximadamente 5 % a aproximadamente 15 % en peso de la primera composición. En ciertas realizaciones, un alcohol polihídrico puede estar presente en la primera composición en una cantidad de aproximadamente 1 %, 2 %, 3 %, 4 %, 5 %, 6 %, 7 %, 8 %, 9 %, 10 %, 11 %, 12 %, 13 %, 14 %, 15 %, 16 %, 17 %, 18 %, 19 %, 20 %, 21 %, 22 %, 23 %, 24 %, 25 %, 26 %, 27 %, 28 %, 29 % o 30 % en peso de la primera composición o cualquier rango y/o valor individual en la misma.
Agentes que aumentan la viscosidad ejemplares que pueden estar presentes en una primera composición incluyen, pero no se limitan a, un carboxipolimetileno; un polímero poliacrílico tal como ácido poliacrílico, un polímero de poliacrilato, un polímero de poliacrilato reticulado, un ácido poliacrílico reticulado y mezclas de los mismos; un éter de celulosa tal como polímeros de hidroxialquil celulosa tales como hidroxipropil metil celulosa (HPMC), hidroxipropil celulosa, hidroxietil celulosa, metil celulosa, carboximetil celulosa y mezclas de los mismos; un metacrilato; una polivinilpirrolidona; polivinilpirrolidona reticulada; copolímero de polivinilpirrolidona-acetato de vinilo; alcohol de polivinilo; óxido de polietileno; polietilenglicol; copolímero de polivinilalquiléter-ácido maleico; un polímero de carboxivinilo; un polisacárido; una goma tal como alginato de sodio, carragenano, goma xantana, goma acacia, goma arábiga, goma guar, pululano, agar, quitina, quitosano, pectina, goma karaya, zeína, hordeína, gliadina, goma de algarrobilla, tragacanto y sus mezclas; una proteína tal como colágeno, aislado de proteína de suero, caseína, proteína de leche, proteína de soja, gelatina y mezclas de los mismos; un almidón como maltodextrina, amilosa, almidón rico en amilosa, almidón de maíz, almidón de patata, almidón de arroz, almidón de tapioca, almidón de guisantes, almidón de batata, almidón de cebada, almidón de trigo, almidón de maíz ceroso, almidón modificado (por ejemplo, almidón hidroxipropilado con alto contenido de amilosa), dextrina, levan, elsinan, gluten y mezclas de los mismos; bentonita; estearato de calcio; ceratonia dióxido de silicio coloidal; dextrina; hipromelosa policarbofilo; caolín; saponita; ésteres de sorbitán; sacarosa; aceite de sésamo; tragacanto; alginato de potasio; povidona; almidón; glicolato de sodio; fosfolípidos; y cualquier combinación de los mismos.
En algunas realizaciones, una primera composición puede comprender un carboxipolimetileno, tal como, entre otros, los disponibles comercialmente de Lubrizol Corporation de Wickliffe, Ohio, bajo el nombre comercial Carbopol®. Los polímeros Carbopol® ejemplares que pueden estar presentes en una primera composición incluyen, pero no se limitan a, polímero Carbopol® 974P NF, tal como Homopolímeros Tipo A, Tipo B y/o Tipo C; polímero Carbopol® Ultrez 10, 20, 21 NF; polímero Carbopol® 971P NF; polímero Carbopol® 980 Homopolymere tipo C, polímero Carbopol® 980 NF, polímero Carpobol® 980P, polímero Carbopol® ETD 2020 NF, polímero Carbopol® 71 G NF, polímero Carbopol® 981P NF, polímero Carbopol® 970P NF, polímero Carbopol® 981P NF, polímero Carbopol® 5984P NF, polímero Carbopol® 934P NF, polímero Carbopol® 940P NF, polímero Carbopol® 941P NF, polímero Carbopol® 13242 NF, polímero Carbopol® a A-1 USP NF, polímero Carbopol® TR1 NF, polímero Carbopol® TR2 NF, polímero Lubrizol Aqua CC y polímero SF-2, y cualquier combinación de los mismos.
En algunas realizaciones, una primera composición puede comprender una celulosa, tal como, pero sin limitación, una carboximetilcelulosa o una sal de la misma. En algunas realizaciones, una primera composición puede comprender carboximetilcelulosa sódica.
En algunas realizaciones, un agente que aumenta la viscosidad presente en la primera composición puede ser un polímero que comprende grupos ácidos, tales como, pero sin limitación, grupos de ácido carboxílico. Los grupos ácidos del polímero pueden neutralizarse parcialmente en la primera composición. En ciertas realizaciones, un agente que aumenta la viscosidad presente en la primera composición puede ser un carboxipolimetileno. En algunas realizaciones, un carboxipolimetileno presente en la primera composición puede neutralizarse parcialmente. Una primera composición puede comprender un carboxipolimetileno y tener un pH de aproximadamente 3 a aproximadamente 7, aproximadamente 3,5 a aproximadamente 6,5, aproximadamente 3,5 a aproximadamente 6 o aproximadamente 4 a aproximadamente 6. En ciertas realizaciones, una primera composición puede comprender un carboxipolimetileno y tener un pH de aproximadamente 3, 3,5, 4, 4,5, 5, 5,5, 6, 6,5 o 7.
Un agente que aumenta la viscosidad puede estar presente en la primera composición. En algunas realizaciones, una composición de la presente invención puede comprender al menos dos agentes que aumentan la viscosidad que pueden ser iguales o diferentes. En algunas realizaciones, un primer agente que aumenta la viscosidad puede estar presente en una primera composición en una cantidad de aproximadamente 0,01 % a aproximadamente 5 % en peso de la primera composición o cualquier rango y/o valor individual en la misma, tal como, pero sin limitarse a, aproximadamente 0,05 % a aproximadamente 3 %, aproximadamente 1 % a aproximadamente 5 %, aproximadamente 1 % a aproximadamente 3 %, o aproximadamente 0,1 % a aproximadamente 1,5 % en peso de la primera composición. En ciertas realizaciones, un primer agente que aumenta la viscosidad está presente en una primera composición en una cantidad de aproximadamente 0,01 %, 0,02 %, 0,03 %, 0,04 %, 0,05 %, 0,06 %, 0,07 %, 0,08 %, 0,09 %, 0,1 %, 0,2 %, 0,3 %, 0,4 %, 0,5 %, 0,6 %, 0,7 %, 0,8 %, 0,9 %, 1 %, 1,5 %, 2 %, 2,5 %, 3 %, 3,5 %, 4 %, 4,5 %, o 5 % en peso de la primera composición o cualquier rango y/o valor individual en la misma.
El agua puede estar presente en la primera composición en una cantidad de aproximadamente 55 % a aproximadamente 99 % en peso de la primera composición o cualquier rango y/o valor individual en la misma, tal como, pero no limitado a, aproximadamente 55 % a aproximadamente 75 %, aproximadamente 60 % a aproximadamente 95 %, aproximadamente 60 % a aproximadamente 80 %, aproximadamente 75 % a aproximadamente 95 %, o aproximadamente 80 % a aproximadamente 90 % en peso de la primera composición. En ciertas realizaciones, el agua está presente en una primera composición en una cantidad de aproximadamente 55 %, 56 %, 57 %, 58 %, 59 %, 60 %, 61 %, 62 %, 63 %, 64 %, 65 %, 66 %, 67 %, 68 %, 69 %, 70 %, 71 %, 72 %, 73 %, 74 %, 75 %, 76 %, 77 %, 78 %, 79 %, 80 %, 81 %, 82 %, 83 %, 84 %, 85 %, 86 %, 87 %, 88 %, 89 %, 90 %, 91 %, 92 %, 93 %, 94 %, 95 %, 96 %, 97 %, 98 % o 99 % en peso de la primera composición o cualquier rango y/o valor individual en la misma.
En algunas realizaciones, uno o más disolventes además del agua pueden estar presentes en una primera composición. Por ejemplo, la primera composición puede comprender agua y 1, 2, 3, 4, 5 o más disolventes adicionales. El uno o más disolventes además del agua pueden estar presentes en la primera composición en una cantidad de aproximadamente 0,5 % a aproximadamente 20 % en peso de la primera composición o cualquier rango y/o valor individual en la misma, como, pero no limitado a, aproximadamente 1 % a aproximadamente 15 %, aproximadamente 1 % a aproximadamente 10 %, aproximadamente 5 % a aproximadamente 15 %, o aproximadamente 1 % a aproximadamente 5 % en peso de la primera composición. En ciertas realizaciones, el uno o más disolventes además del agua pueden estar presentes en una primera composición en una cantidad de aproximadamente 0,5 %, 0,75 %, 1 %, 2 %, 3 %, 4 %, 5 %, 6 %, 7 %, 8 %, 9 %, 10 %, 11 %, 12 %, 13 %, 14 %, 15 %, 16 %, 17 %, 18 %, 19 % o 20 % en peso de la primera composición o cualquier rango y/o valor individual en el mismo. Los solventes de ejemplo además del agua que pueden estar presentes en una primera composición incluyen, pero no están limitados a un alcohol, tal como, por ejemplo, alcohol isopropílico o etanol.
En algunas realizaciones, una primera composición comprende, consiste esencialmente en, o consiste en al menos un alcohol polihídrico presente en una cantidad de aproximadamente 1 % a aproximadamente 30 % en peso de la primera composición, al menos un agente que aumenta la viscosidad presente en una cantidad de aproximadamente 0,1 % a aproximadamente 5 % en peso de la primera composición, y agua presente en una cantidad de aproximadamente 55 % a aproximadamente 99 % en peso de la primera composición. En ciertas realizaciones, el agente que aumenta la viscosidad puede ser un carboxipolimetileno o una carboximetilcelulosa o una sal del mismo. La primera composición puede ser un hidrogel.
Una primera composición puede comprender un conservante. Un conservante puede estar presente en una primera composición en una cantidad de aproximadamente 0,01 % a aproximadamente 1 % en peso de la primera composición o cualquier rango y/o valor individual en la misma, tal como, pero sin limitación, aproximadamente 0,01 % a aproximadamente 0,1 %, aproximadamente 0,05 % a aproximadamente 0,5 %, aproximadamente 0,05 % a aproximadamente 1 %, o aproximadamente 0,1 % a aproximadamente 1 % en peso de la primera composición. En ciertas realizaciones, un conservante está presente en una primera composición en una cantidad de aproximadamente 0,01 %, 0,02 %, 0,03 %, 0,04 %, 0,05 %, 0,06 %, 0,07 %, 0,08 %, 0,09 %, 0,1 %, 0,2 %, 0,3 %, 0,4 %, 0,5 %, 0,6 %, 0,7 %, 0,8 %, 0,9 %, o 1 % en peso de la primera composición o cualquier rango y/o valor individual en la misma. Los conservantes ejemplares que pueden estar presentes en una primera composición incluyen, entre otros, ácido sórbico, ácido benzoico, metilparaben, propilparaben, metilcloroisotiazolinona, metolisotiazolinona, diazolidinilurea, clorobutanol, triclosán, cloruro de bencetonio, p-hidroxibenzoato, clorhexidina, digluconato, bromuro de hexadeciltrimetilamonio, alcoholes, cloruro de benzalconio, ácido bórico, bronopol, butilparabeno, acetato de butileno cálcico, cloruro de calcio, lactato de calcio, dióxido de carbono, catiónico y bentonita, cetrimida, cloruro de cetilpiridinio, clorhexidina, clorobutanol, clorocresol, cloroxilenol, monohidrato de ácido cítrico, cresol, dimetil éter, etilparabeno, glicerina, hexetidina, imidurea, alcohol isopropílico, ácido láctico, monotioglicerol, ácido pentético, fenol, fenoxietanol, feniletil alcohol, acetato fenilmercúrico, borato fenilmercúrico, nitrato fenilmercúrico, benzoato de potasio, metabisulfito de potasio, sorbato de potasio, ácido propiónico, galato de propilo, propilenglicol, acetato de sodio, benzoato de sodio, borato de sodio, lactato de sodio, sulfito de sodio, propionato de sodio, metabisulfito de sodio, xilitol, dióxido de azufre, dióxido de carbono y cualquier combinación de los mismos.
Una primera composición puede comprender un agente neutralizante. Un agente neutralizante puede estar presente en una primera composición en una cantidad suficiente para proporcionar un pH deseado, tal como, pero no limitado a, un pH de aproximadamente 3 a aproximadamente 11, o cualquier rango y/o valor individual en el mismo, tal como, pero no limitado a, aproximadamente 3 a aproximadamente 8, aproximadamente 4 a aproximadamente 7, o aproximadamente 6 a aproximadamente 7, o aproximadamente 6 a 11.
En algunas realizaciones, un agente neutralizante puede estar presente en una primera composición en una cantidad suficiente para proporcionar a la primera composición un pH en un intervalo de aproximadamente 3 a aproximadamente 8.
En ciertas realizaciones, un agente neutralizante puede estar presente en una primera composición en una cantidad suficiente para proporcionar una composición de la presente invención con un pH deseado tras la combinación de la primera composición y una segunda parte (por ejemplo, una segunda composición) y/o tras la administración de la primera composición y/o la composición que comprende la primera composición y la segunda parte a la piel de un
sujeto. Un agente neutralizante puede estar presente en una primera composición en una cantidad suficiente para proporcionar una composición de la presente invención (por ejemplo, una composición que comprende la primera composición y una segunda composición) con un pH deseado, tal como, pero no limitado a, un pH de aproximadamente 3 a aproximadamente 11, o cualquier rango y/o valor individual en el mismo.
En algunas realizaciones, un agente neutralizante ajusta el pH de la primera composición y/o composición de la presente invención. En ciertas realizaciones de la presente invención, un agente neutralizante está presente en una primera composición en una cantidad suficiente para que la primera composición tenga un pH de aproximadamente 3, 3,5, 4, 4,5, 5, 5,5, 6, 6,5, 7, 7,5, o 8 o cualquier rango y/o valor individual en el mismo. En algunas realizaciones de la presente invención, un agente neutralizante está presente en una composición de la presente invención en una cantidad suficiente para que la composición tenga un pH de aproximadamente 3, 3,5, 4, 4,5, 5, 5,5, 6, 6,5, 7, 7,5, 8, 8,5, 9, 9,5, 10, 10,5 u 11, o cualquier rango y/o valor individual en el mismo.
Agentes neutralizantes ejemplares que pueden estar presentes en una primera composición incluyen, pero no se limitan a, bases tales como hidróxido de sodio, hidróxido de potasio y mezclas de los mismos; ácidos tales como ácido clorhídrico, ácido cítrico, ácido láctico, ácido glicólico, ácido acético y mezclas de los mismos; carbonato de sodio; trolamina; trometamina; aminometilpropanol; triisopropanolamina; aminometilpropanol; tetrahidroxipropil etilendiamina; EDTAtetrasódico; suttocida A; y cualquier combinación de los mismos.
Un agente neutralizante puede estar presente en una primera composición en una cantidad de aproximadamente 0,01 % a aproximadamente 1 % en peso de la primera composición o cualquier rango y/o valor individual en la misma, tal como, pero sin limitación, aproximadamente 0,01 % a aproximadamente 0,1 %, aproximadamente 0,05 % a aproximadamente 1 %, o aproximadamente 0,1 % a aproximadamente 1 % en peso de la primera composición. En ciertas realizaciones, un agente neutralizante puede estar presente en una primera composición en una cantidad de aproximadamente 0,01 %, 0,02 %, 0,03 %, 0,04 %, 0,05 %, 0,06 %, 0,07 %, 0,08 %, 0,09 %, 0,1 %, 0,2 %, 0,3 %, 0,4 %, 0,5 %, 0,6 %, 0,7 %, 0,8 %, 0,9 %, o 1 % en peso de la primera composición o cualquier rango y/o valor individual en la misma.
Una primera composición puede estar sin tampón o tamponada. En algunas realizaciones, una primera composición puede no estar tamponada. En otras realizaciones, una primera composición puede estar tamponada. Lostampones ejemplares que pueden estar presentes en una primera composición incluyen, pero sin limitación, tampones de ácido acético/acetato; tampones de ácido clorhídrico/citrato; tampones de citrofosfato; tampones de fosfato; tampones de ácido cítrico/citrato; tampones de ácido láctico; tampones de ácido tartárico; tampones de ácido málico; tampones de glicina/HCl; tampones salinos tales como solución salina tamponada con fosfato (PBS), solución salina tamponada con Tris (TBS), Tris-HCl, NaCl, solución salina tamponada con Tween (TNT), solución salina tamponada con fosfato, Triton X-100 (PBT) y mezclas de los mismos; tampones de cacodilato; tampones de barbital; tampones tris; y cualquier combinación de los mismos. En algunas realizaciones, el tampón puede ser un tampón de fosfato, tal como, por ejemplo, un fosfato de potasio dibásico y/o tampón monobásico de fosfato de potasio.
En algunas realizaciones, un tampón puede estar presente en una primera composición en una cantidad de aproximadamente 0,01 % a aproximadamente 20 % en peso de la primera composición o cualquier rango y/o valor individual en la misma, tal como, pero sin limitarse a, aproximadamente 0,1 % a aproximadamente 20 %, aproximadamente 1 % a aproximadamente 15 %, aproximadamente 5 % a aproximadamente 20 %, aproximadamente 10 % a aproximadamente 20 %, o aproximadamente 1 % a aproximadamente 10 % en peso de la primera composición. En ciertas realizaciones, un tampón está presente en una primera composición en una cantidad de aproximadamente 0,01 %, 0,02 %, 0,03 %, 0,04 %, 0,05 %, 0,06 %, 0,07 %, 0,08 %, 0,09 %, 0,1 %, 0,2 %, 0,3 %, 0,4 %, 0,5 %, 0,6 %, 0,7 %, 0,8 %, 0,9 %, 1 %, 2 %, 3 %, 4 %, 5 %, 6 %, 7 %, 8 %, 9 %, 10 %, 11 %, 12 %, 13 %, 14 %, 15 %, 16 %, 17 %, 18 %, 19 % o 20 % en peso de la primera composición o cualquier rango y/o valor individual en la misma.
En ciertas realizaciones, una primera composición puede comprender un agente tamponante. Agentes tamponantes ejemplares incluyen, pero no se limitan a, ácido cítrico, ácido acético, ácido láctico, ácido bórico, ácido succínico, ácido málico y cualquier combinación de los mismos. Un agente tamponante puede estar presente en una primera composición en una cantidad de aproximadamente 0,01 % a aproximadamente 4 % en peso de la primera composición o cualquier rango y/o valor individual en la misma, tal como, pero sin limitación, aproximadamente 0,01 % a aproximadamente 0,1 %, aproximadamente 0,05 % a aproximadamente 1 %, aproximadamente 0,1 % a aproximadamente 0,5 %, aproximadamente 1 % a aproximadamente 3 %, o aproximadamente 0,1 % a aproximadamente 2 % en peso de la primera composición. En ciertas realizaciones, un agente tamponante está presente en una primera composición en una cantidad de aproximadamente 0,01 %, 0,02 %, 0,03 %, 0,04 %, 0,05 %, 0,06 %, 0,07 %, 0,08 %, 0,09 %, 0,1 %, 0,2 %, 0,3 %, 0,4 %, 0,5 %, 0,6 %, 0,7 %, 0,8 %, 0,9 %, 1 %, 1,25 %, 1,5 %, 1,75 %, 2 %, 2,25 %, 2,5 %, 2,75 %, 3 %, 3,25 %, 3,5 %, 3,75 % o 4 % en peso de la primera composición o cualquier rango y/o valor individual en la misma.
En algunas realizaciones, un tampón y/o agente tamponante está presente en una primera composición en una cantidad suficiente para que la primera composición tenga un pH de aproximadamente 3 a aproximadamente 8 o cualquier intervalo y/o valor individual en el mismo, tal como, pero no limitado a, aproximadamente 3 a aproximadamente 6, aproximadamente 3 a aproximadamente 5, aproximadamente 4 a aproximadamente 7,
aproximadamente 5 a aproximadamente 7, o aproximadamente 6 a aproximadamente 7. En ciertas realizaciones de la presente invención, un tampón y/o el agente tamponante puede estar presente en una primera composición en una cantidad suficiente para que la primera composición tenga un pH de aproximadamente 3, 3,5, 4, 4,5, 5, 5,5, 6, 6,5, 7, 7,5 u 8, o cualquier rango y/o valor individual en el mismo.
En algunas realizaciones, un tampón y/o agente tamponante puede estar presente en una primera composición en una cantidad suficiente para proporcionar un pH deseado para una composición de la presente invención que comprende la primera composición y una segunda parte (por ejemplo, una segunda composición). Por ejemplo, una composición de la presente invención puede comprender una segunda composición y una primera composición que comprende un tampón y/o agente tamponante, en el que el tampón y/o agente tamponante está presente en una cantidad suficiente para proporcionar a la composición un pH de aproximadamente 3 a aproximadamente 11, tal como, pero no limitado a, aproximadamente 3 a aproximadamente 8, aproximadamente 7 a aproximadamente 11, aproximadamente 8 a aproximadamente 10, aproximadamente 3 a aproximadamente 5, aproximadamente 4 a aproximadamente 7, aproximadamente 5 a aproximadamente 7, o aproximadamente 6 a aproximadamente 7. En ciertas realizaciones de la presente invención, un tampón y/o agente tamponante puede estar presente en una primera composición en una cantidad suficiente para que la composición de la presente invención tenga un pH de aproximadamente 3, 3,5, 4, 4,5, 5, 5,5, 6, 6,5, 7, 7,5, 8, 8,5, 9, 9,5, 10, 10,5, u 11, o cualquier rango y/o valor individual en el mismo. En algunas realizaciones, el tampón y/o el agente tamponante pueden estar presentes en una primera composición en una cantidad suficiente para proporcionar un pH deseado tras la administración de una composición de la presente invención que comprende la primera composición y una segunda parte a la piel de un sujeto.
En algunas realizaciones, un tampón, agente tamponante y/o agente neutralizante puede estar presente en una primera composición en una cantidad suficiente para proporcionar una composición de la presente invención y/o una primera composición con un pH deseado.
En algunas realizaciones, se puede proporcionar un emoliente en una primera composición, tal como, pero sin limitación, siliconas, tales como, por ejemplo, ciclometicona, dimeticona, simeticona, alquil dimeticona C26-28, alquil meticona C26-28, polifenilsisquioxano, trimetilsiloxisilicato y polímeros cruzados de ciclopentasiloxano y dimeticona/viniltrimetilsiloxisilicato, y sus mezclas. En algunas realizaciones, una primera composición puede comprender ciclometicona. Puede estar presente un emoliente en la primera composición en una cantidad de aproximadamente 0,5 % a aproximadamente 10 % en peso de la primera composición o cualquier rango y/o valor individual en la misma, tal como, pero no limitado a, aproximadamente 1 % a aproximadamente 5 %, aproximadamente 0,5 % a aproximadamente 4 %, aproximadamente 1 % a aproximadamente 10 %, o aproximadamente 2 % a aproximadamente 8 % en peso de la primera composición. En ciertas realizaciones, un emoliente puede estar presente en la primera composición en una cantidad de aproximadamente 0,5 %, 1 %, 2 %, 3 %, 4 %, 5 %, 6 %, 7 %, 8 %, 9 % o 10 % en peso de la primera composición o cualquier rango y/o valor individual en la misma.
En ciertas realizaciones, una primera composición puede comprender al menos un alcohol polihídrico presente en una cantidad de aproximadamente 1 % a aproximadamente 30 % en peso de la primera composición, al menos un agente que aumenta la viscosidad presente en una cantidad de aproximadamente 0,01 % a aproximadamente 5 % en peso de la primera composición, agua presente en una cantidad de aproximadamente 55 % a aproximadamente 99 % en peso de la primera composición, y opcionalmente al menos un conservante en una cantidad de aproximadamente 0,01 % a aproximadamente 1 % en peso de la primera composición. La primera composición puede tener un pH en un intervalo de aproximadamente 3 a aproximadamente 8, aproximadamente 3 a aproximadamente 6, o aproximadamente 6 a aproximadamente 8 y puede estar tamponada. La primera composición puede ser un hidrogel.
En algunas realizaciones, una primera composición puede comprender, consistir esencialmente en, o consistir en un alcohol polihídrico en una cantidad de aproximadamente 1 % a aproximadamente 15 % en peso de la primera composición, un agente que aumenta la viscosidad en una cantidad de aproximadamente 0,1 % a aproximadamente 5 % en peso de la primera composición, agua en una cantidad de aproximadamente 55 % a aproximadamente 85 % en peso de la primera composición, opcionalmente un tampón en una cantidad de aproximadamente 0,1 % a aproximadamente 20 %, opcionalmente un tampón agente en una cantidad de aproximadamente 0,001 % a aproximadamente 2 % en peso de la primera composición, opcionalmente un conservante en una cantidad de aproximadamente 0,001 % a aproximadamente 1 % en peso de la primera composición, y opcionalmente un agente neutralizante en una cantidad de aproximadamente 0,001 % a aproximadamente 1 % en peso de la primera composición. La primera composición puede tener un pH en un rango de aproximadamente 3 a aproximadamente 5 o aproximadamente 5 a aproximadamente 7. En ciertas realizaciones, el agente que aumenta la viscosidad presente en la primera composición puede ser carboxipolimetileno o carboximetilcelulosa o una sal de la misma. En algunas realizaciones, la primera composición puede ser cosméticamente elegante. La primera composición puede ser un hidrogel.
En algunas realizaciones, una primera composición puede comprender, consistir esencialmente en, o consistir en un alcohol polihídrico en una cantidad de aproximadamente 1 % a aproximadamente 30 % en peso de la primera composición, un agente que aumenta la viscosidad en una cantidad de aproximadamente 0,01 % a
aproximadamente 5 % en peso de la primera composición, agua en una cantidad de aproximadamente 55 % a aproximadamente 99 % en peso de la primera composición, opcionalmente al menos un disolvente adicional (por ejemplo, un alcohol) en una cantidad de aproximadamente 0,5 % a aproximadamente 20 % en peso de la primera composición, opcionalmente un emoliente en una cantidad de aproximadamente 0,5 % a aproximadamente 10 % en peso de la primera composición, opcionalmente un tampón en una cantidad de aproximadamente 0,01 % a aproximadamente 20 % en peso de la primera composición, opcionalmente un conservante en una cantidad de aproximadamente 0,001 % a aproximadamente 1 % en peso de la primera composición, y opcionalmente un agente neutralizante en una cantidad de aproximadamente 0,001 % a aproximadamente 1 % en peso de la primera composición. En algunas realizaciones, el al menos un disolvente comprende un alcohol y la primera composición comprende un tampón, un emoliente y un conservante. La primera composición puede tener un pH en un intervalo de aproximadamente 3 a aproximadamente 5 o de aproximadamente 5 a aproximadamente 7. En algunas realizaciones, el pH puede ser de aproximadamente 4,5. En ciertas realizaciones, el agente que aumenta la viscosidad presente en la primera composición puede ser carboximetilcelulosa o una sal de la misma. En algunas realizaciones, la primera composición puede ser cosméticamente elegante. La primera composición puede ser un hidrogel.
Una composición de la presente invención puede comprender un ingrediente farmacéutico activo (API). Excepto el nitrito acidificado, cualquier API adecuada o combinaciones de API pueden incluirse en una composición de la presente invención. En algunas realizaciones, la API puede ser cualquier API adecuada que proporcione la liberación de óxido nítrico como se describe en este documento. Ejemplos de API incluyen, pero no se limitan a, agentes antimicrobianos, agentes antiacné, agentes antiinflamatorios, agentes analgésicos, agentes anestésicos, agentes antihistamínicos, agentes antisépticos, inmunosupresores, agentes antihemorrágicos, vasodilatadores, agentes de curación de heridas, agentes antibiopelícula y cualquier combinación de los mismos. Las API ejemplares incluyen, pero no se limitan a, las descritas en la Publicación de Solicitud Internacional No. WO 2013/006608.
En algunas realizaciones, una primera composición puede no comprender una API. En ciertas realizaciones, una primera composición no contiene un API que libera óxido nítrico (NO). En algunas realizaciones, una primera composición puede comprender al menos una API, pero la primera composición puede no comprender una API que libera NO. En algunas realizaciones, una primera composición comprende una API (por ejemplo, una API sensible a la humedad) y una segunda composición comprende una segunda API, tal como, por ejemplo, una API que libera NO.
En algunas realizaciones, la segunda composición puede ser una composición anhidra. "Anhidra", como se usa en el presente documento, significa que no hay adición directa de agua a la segunda composición cuando se está preparando. Sin embargo, los expertos en la materia reconocerán que el agua puede ser absorbida física y/o químicamente por la segunda composición y/o por uno o más ingredientes en la segunda composición en cualquier momento durante la preparación, almacenamiento y/o uso de la segunda composición (es decir, adición indirecta de agua a la segunda composición). En algunas realizaciones, el término "anhidra" significa que la segunda composición tiene un contenido de agua de menos del 5 % en peso de la segunda composición o cualquier rango y/o valor individual en la misma. Una segunda composición puede tener un contenido de agua de menos de 5, 4,5, 4, 3,5, 3, 2,5, 2, 1,5, 1 o 0,5 %, o cualquier rango en el mismo, en peso de la segunda composición. El contenido de agua puede medirse mediante procedimientos conocidos por los expertos en la técnica, tales como, pero sin limitarse a, valoración de Karl Fischer. En ciertas realizaciones, al entrar en contacto con una segunda composición, una composición de la presente invención agrega agua a la segunda composición y/o la segunda composición absorbe agua de una composición de la presente invención.
Las segundas composiciones ejemplares que pueden usarse y/o ponerse en contacto con una primera composición incluyen, pero no se limitan a, las descritas en la Publicación de Solicitud Internacional No. WO 2013/006608.
Una segunda composición ejemplar que puede usarse y/o ponerse en contacto con una primera composición para formar una composición antiviral tópica de la presente invención puede comprender una composición anhidra que comprende al menos un agente de aumento de la viscosidad presente en la segunda composición en un cantidad de aproximadamente 0,1 % a aproximadamente 30 % en peso de la segunda composición, al menos un disolvente orgánico presente en la segunda composición en una cantidad de aproximadamente 50 % a aproximadamente 90 en peso de la segunda composición, y al menos un humectante presente en la segunda composición en una cantidad de aproximadamente 2 % a aproximadamente 20 % en peso de la segunda composición. La segunda composición puede comprender además al menos un agente repelente al agua, también denominado repelente al agua.
Agentes que aumentan la viscosidad ejemplares para la segunda composición incluyen, pero no se limitan a, copolímeros de carboximetilcelulosa y ácido acrílico, N-vinilpirrolidona, polialquilenglicoles (por ejemplo, poli(etilenglicol)), óxidos de polialquileno (por ejemplo, óxido de polietileno), alcoholes polivinílicos, polivinilpirrolidona, polisiloxanos, poli(acetatos de vinilo), celulosa, celulosas derivadas, alginatos, copolímeros de los mismos y mezclas de los mismos. Un ejemplo específico de un agente de viscosidad para la segunda composición es una hidroxipropilcelulosa, como la hidroxipropilcelulosa Klucel® (por ejemplo, Grado Klucel® MF Pharm). Un agente que aumenta la viscosidad puede estar presente en la segunda composición en una cantidad de aproximadamente el 0,1 % a aproximadamente el 30 % en peso de la segunda composición o cualquier rango y/o valor individual en la misma, tal como, pero no limitado a, aproximadamente el 0,5 % a aproximadamente 20 %,
aproximadamente 0,1 % a aproximadamente 2 %, aproximadamente 0,5 % a aproximadamente 5 %, aproximadamente 1 % a aproximadamente 10 %, o aproximadamente 1 % a aproximadamente 5 % en peso de la segunda composición. En ciertas realizaciones, un agente que aumenta la viscosidad puede estar presente en la segunda composición en una cantidad de aproximadamente 0,1 %, 0,25 %, 0,5 %, 0,75 %, 1 %, 2 %, 3 %, 4 %, 5 %, 6 %, 7 %, 8 %, 9 %, 10 %, 11 %, 12 %, 13 %, 14 %, 15 %, 16 %, 17 %, 18 %, 19 %, 20 %, 21 %, 22 %, 23 %, 24 %, 25 %, 26 %, 27 %, 28 %, 29 % o 30 % en peso de la segunda composición o cualquier rango y/o valor individual en la misma.
Disolventes orgánicos ejemplares para la segunda composición incluyen, pero no se limitan a, acetona, alcohol metílico, etanol, isopropanol, alcohol butílico, acetato de etilo, dimetil isosorbida, propilenglicol, glicerol, etilenglicol, polietilenglicol, dietilenglicol monoetil éter o mezclas de los mismos. En algunas realizaciones de la presente invención, el disolvente orgánico en la segunda composición puede ser etanol y/o alcohol isopropílico. Puede estar presente un disolvente orgánico en la segunda composición en una cantidad de aproximadamente 40 % a aproximadamente 90 % en peso de la segunda composición o cualquier rango y/o valor individual en la misma, tal como, pero no limitado a, aproximadamente 40 % a aproximadamente 80 %, aproximadamente 50 % a aproximadamente 70 %, aproximadamente 50 % a aproximadamente 80 %, aproximadamente 60 % a aproximadamente 90 %, aproximadamente 70 % a aproximadamente 90 %, o aproximadamente 75 % a aproximadamente 85 % en peso de la segunda composición. En ciertas realizaciones, un disolvente orgánico puede estar presente en la segunda composición en una cantidad de aproximadamente 40 %, 41 %, 42 %, 43 %, 44 %, 45 %, 46 %, 47 %, 48 %, 49 %, 50 %, 51 %, 52 %, 53 %, 54 %, 55 %, 56 %, 57 %, 58 %, 59 %, 60 %, 61 %, 62 %, 63 %, 64 %, 65 %, 66 %, 67 %, 68 %, 69 %, 70 %, 71 %, 72 %, 73 %, 74 %, 75 %, 76 %, 77 %, 78 %, 79 %, 80 %, 81 %, 82 %, 83 %, 84 %, 85 %, 86 %, 87 %, 88 %, 89 % o 90 % en peso de la segunda composición o cualquier rango y/o valor individual en la misma.
Humectantes ejemplares para la segunda composición incluyen, pero no se limitan a, glicoles, tales como dietilenglicol monoetil éter; gliceroles; polioles de azúcar, tales como sorbitol, xilitol y maltitol; polioles tales como polidextrosas; quillaia, urea y mezclas de los mismos. En algunas realizaciones, el humectante en la segunda composición puede comprender un alquilenglicol, tal como, por ejemplo, hexilenglicol. Puede estar presente un humectante en la segunda composición en una cantidad de aproximadamente 2 % a aproximadamente 20 % en peso de la segunda composición o cualquier rango y/o valor individual en la misma, tal como, pero no limitado a, aproximadamente 2 % a aproximadamente 15 %, aproximadamente 5 % a aproximadamente 15 %, o aproximadamente 15 % a aproximadamente 20 % en peso de la segunda composición. En ciertas realizaciones, un humectante puede estar presente en la segunda composición en una cantidad de aproximadamente 2 %, 3 %, 4 %, 5 %, 6 %, 7 %, 8 %, 9 %, 10 %, 11 %, 12 %, 13 %, 14 %, 15 %, 16 %, 17 %, 18 %, 19 % o 20 % en peso de la segunda composición o cualquier rango y/o valor individual en la misma.
Los repelentes de agua ejemplares para la segunda composición incluyen, pero no se limitan a, siliconas, tales como ciclometicona, dimeticona, simeticona, alquil dimeticona C26-28, alquil meticona C26-28, polifenilsisquioxano, trimetilsiloxisilicato y polímeros cruzados de ciclopentasiloxano y dimeticona/viniltrimetilsiloxisilicato, y mezclas de los mismos. En algunas realizaciones, una segunda composición puede comprender ciclometicona. Un repelente de agua puede estar presente en la segunda composición en una cantidad de aproximadamente 0,5 % a aproximadamente 15 % en peso de la segunda composición o cualquier rango y/o valor individual en la misma, tal como, pero no limitado a, aproximadamente 0,5 % a aproximadamente 10 %, aproximadamente 1 % a aproximadamente 5 %, o aproximadamente 2 % a aproximadamente 5 % en peso de la segunda composición. En ciertas realizaciones, un repelente de agua puede estar presente en la segunda composición en una cantidad de aproximadamente 0,5 %, 1 %, 2 %, 3 %, 4 %, 5 %, 6 %, 7 %, 8 %, 9 %, 10 %, 11 %, 12 %, 13 %, 14 % o 15 % en peso de la segunda composición o cualquier rango y/o valor individual en la misma.
Por consiguiente, una composición antiviral tópica de la presente invención puede comprender al menos un alcohol polihídrico, un primer agente que aumenta la viscosidad, agua, un segundo agente que aumenta la viscosidad, al menos un disolvente orgánico, al menos un humectante, opcionalmente al menos un disolvente además de agua, opcionalmente un emoliente, opcionalmente un agente repelente al agua, opcionalmente al menos un conservante y opcionalmente al menos un agente tamponante y/o tampón. La composición puede estar tamponada a un pH de aproximadamente 3 a aproximadamente 11, tal como, pero no limitado a, aproximadamente 6 a aproximadamente 10, aproximadamente 7 a aproximadamente 10, o aproximadamente 7 a aproximadamente 9. En algunas realizaciones, la composición puede tener un pH de 7 o mayor, 7,5 o mayor, o 9,5 o mayor. En ciertas realizaciones, la composición puede comprender al menos un API, tal como, pero sin limitación, un ingrediente farmacéutico activo que libera óxido nítrico. En algunas realizaciones, la API que libera NO puede ser una macromolécula modificada con diolato de diazenio.
Una composición antiviral tópica de la presente invención puede comprender una primera composición y una segunda composición como se describe en el presente documento. Como reconocerán los expertos en la materia, la cantidad o concentración de componentes individuales en una composición de la presente invención puede variar dependiendo de la cantidad de la primera composición y la segunda composición presente en la composición (por ejemplo, la proporción de la primera composición y segunda composición presentes en la composición). En algunas realizaciones, la relación de una primera composición de la presente invención a una segunda composición en una composición de la presente invención puede ser aproximadamente 5:1 o menos, en realizaciones adicionales,
aproximadamente 4:1 o menos, aproximadamente 3:1 o menos, aproximadamente 2:1 o menos, aproximadamente 1:1 o menos, aproximadamente 0,5:1 o menos, o aproximadamente 0,2:1 o menos. En realizaciones particulares, la relación puede ser de aproximadamente 3:1. En realizaciones adicionales, la relación puede ser de aproximadamente 1:1.
En algunas realizaciones, una composición de la presente invención puede comprender, consistir esencialmente en, o consistir en un alcohol polihídrico en una cantidad de aproximadamente 0,5 % a aproximadamente 10 % en peso de la composición, un primer agente que aumenta la viscosidad en un cantidad de aproximadamente 0,01 % a aproximadamente 3 % en peso de la composición, agua en una cantidad de aproximadamente 30 % a aproximadamente 50 % en peso de la composición, al menos un disolvente además de agua en una cantidad de aproximadamente 0,5 % a aproximadamente 10 % en peso de la composición, un emoliente en una cantidad de aproximadamente 0,5 % a aproximadamente 5 % en peso de la composición, un tampón en una cantidad de aproximadamente 0,01 % a aproximadamente 10 % en peso de la composición, aumentando la segunda viscosidad agente en una cantidad de aproximadamente 0,01 % a aproximadamente 10 % en peso de la composición, un disolvente orgánico en una cantidad de aproximadamente 20 % a aproximadamente 45 % en peso de la composición, un humectante en una cantidad de aproximadamente 2 % a aproximadamente 10 % en peso de la composición, un agente repelente al agua en una cantidad de aproximadamente 0,1 % a aproximadamente 10 % en peso de la composición, una API que libera NO en una cantidad de aproximadamente 0,5 % a aproximadamente 25 % en peso de la composición, opcionalmente un agente tamponante en una cantidad de aproximadamente 0,001 % a aproximadamente 1 % en peso de la composición, opcionalmente un conservante en una cantidad de aproximadamente 0,001 % a aproximadamente 1 % en peso de la composición, y opcionalmente un agente neutralizante. El tampón, el agente tamponante y/o el agente neutralizante pueden estar presentes en una cantidad suficiente para proporcionar a la primera parte de la composición un pH de aproximadamente 3 a aproximadamente 8. La composición puede tener un pH de menos de aproximadamente 11, tal como, pero no limitado a, menos de aproximadamente 9,5, menos de aproximadamente 7 o menos de aproximadamente 6. En algunas realizaciones, la composición puede tener un pH de aproximadamente 4,5. El primer y segundo agentes que aumentan la viscosidad pueden ser iguales y/o diferentes. En ciertas realizaciones, el primer agente que aumenta la viscosidad puede ser un carboxipolimetileno y el segundo agente que aumenta la viscosidad puede ser una celulosa, tal como, pero sin limitación, hidroxipropilcelulosa. En algunas realizaciones, el primer agente que aumenta la viscosidad puede ser una carboximetilcelulosa o una sal de la misma y el segundo agente que aumenta la viscosidad puede ser una celulosa, tal como, pero sin limitación, hidroxipropilcelulosa. En algunas realizaciones, la composición puede ser cosméticamente elegante.
En algunas realizaciones, una composición de la presente invención puede comprender, consistir esencialmente en, o consistir en un alcohol polihídrico en una cantidad de aproximadamente 1 % a aproximadamente 7 % en peso de la composición, un primer agente que aumenta la viscosidad en un cantidad de aproximadamente 0,1 % a aproximadamente 3 % en peso de la composición, agua en una cantidad de aproximadamente 25 % a aproximadamente 40 % en peso de la composición, al menos un disolvente además de agua en una cantidad de aproximadamente 0,5 % a aproximadamente 10 % en peso de la composición, un emoliente en una cantidad de aproximadamente 0,5 % a aproximadamente 5 % en peso de la composición, un tampón en una cantidad de aproximadamente 0,01 % a aproximadamente 10 % en peso de la composición, aumentando la segunda viscosidad agente en una cantidad de aproximadamente 0,1 % a aproximadamente 2 % en peso de la composición, un disolvente orgánico en una cantidad de aproximadamente 20 % a aproximadamente 45 % en peso de la composición, un humectante en una cantidad de aproximadamente 2 % a aproximadamente 7 % en peso de la composición, un agente repelente al agua en una cantidad de aproximadamente 1 % a aproximadamente 5 % en peso de la composición, una API que libera NO en una cantidad de aproximadamente 5 % a aproximadamente 20 % en peso de la composición, opcionalmente un agente tamponante en una cantidad de aproximadamente 0,01 % a aproximadamente 0,2 % en peso de la composición, opcionalmente un conservante en una cantidad de aproximadamente 0,01 % a aproximadamente 0,3 % en peso de la composición, y opcionalmente un agente neutralizante. El tampón, el agente tamponante y/o el agente neutralizante pueden estar presentes en una cantidad suficiente para proporcionar a la primera parte de la composición un pH de aproximadamente 4 o aproximadamente 6. La composición puede tener un pH de menos de aproximadamente 11, tal como, pero sin limitación, menos de aproximadamente 9,5, menos de aproximadamente 7 o menos de aproximadamente 6. En algunas realizaciones, la composición puede tener un pH de aproximadamente 4,5. El primer y segundo agentes que aumentan la viscosidad pueden ser iguales y/o diferentes. En ciertas realizaciones, el primer agente que aumenta la viscosidad puede ser un carboxipolimetileno y el segundo agente que aumenta la viscosidad puede ser una celulosa, tal como, pero sin limitación, hidroxipropilcelulosa. En algunas realizaciones, el primer agente que aumenta la viscosidad puede ser una carboximetilcelulosa o una sal de la misma y el segundo agente que aumenta la viscosidad puede ser una celulosa, tal como, pero sin limitación, hidroxipropilcelulosa. En algunas realizaciones, la composición puede ser cosméticamente elegante. En ciertas realizaciones, la composición puede comprender una composición como se expone en la Tabla 2 y/o la Tabla 13.
En algunas realizaciones, una composición de la presente invención puede comprender, consistir esencialmente en, o consistir en un alcohol polihídrico en una cantidad de aproximadamente 1 % a aproximadamente 15 % en peso de la composición, un primer agente que aumenta la viscosidad en un cantidad de aproximadamente 0,01 % a aproximadamente 2,5 % en peso de la composición, agua en una cantidad de aproximadamente 25 % a
aproximadamente 50 % en peso de la composición, al menos un disolvente adicional (por ejemplo, un alcohol) en una cantidad de aproximadamente 0,5 % a aproximadamente 10 % en peso de la composición, un emoliente en una cantidad de aproximadamente 0,5 % a aproximadamente 5 % en peso de la composición, un tampón en una cantidad de aproximadamente 0,01 % a aproximadamente 10 % en peso de la composición, un segundo agente que aumenta la viscosidad en una cantidad de aproximadamente 0,01 % a aproximadamente 10 % en peso de la composición, un disolvente orgánico en una cantidad de aproximadamente 20 % a aproximadamente 50 % en peso de la composición, un humectante en una cantidad de aproximadamente 2 % a aproximadamente 10 % en peso de la composición, un agente repelente al agua en una cantidad de aproximadamente 0,1 % a aproximadamente 10 % en peso de la composición, una API de liberación de NO en una cantidad de aproximadamente 0,5 % a aproximadamente 25 % en peso de la composición, y opcionalmente un conservante en una cantidad de aproximadamente 0,001 % a aproximadamente 0,5 % en peso de la primera composición.
Una composición de la presente invención puede comprender al menos dos agentes diferentes de aumento de la viscosidad. Un agente que aumenta la viscosidad puede estar presente en la primera parte de una composición de la presente invención y el otro agente que aumenta la viscosidad puede estar presente en la segunda parte de la composición. En algunas realizaciones, una composición de la presente invención comprende un carboxipolimetileno y una celulosa, tal como, pero sin limitación, hidroxipropilcelulosa. El carboxipolimetileno puede estar presente en una primera composición de la presente invención y la celulosa puede estar presente en una segunda composición, que puede combinarse para formar una composición de la presente invención. En algunas realizaciones, una composición de la presente invención comprende carboximetilcelulosa sódica y una segunda celulosa, tal como, pero sin limitación, hidroxipropilcelulosa. La carboximetilcelulosa sódica puede estar presente en una primera composición de la presente invención y la segunda celulosa puede estar presente en una segunda composición, que puede combinarse para formar una composición de la presente invención. Una composición de la presente invención que comprende al menos dos agentes diferentes de aumento de la viscosidad puede proporcionar una composición cosméticamente elegante que comprende una API, tal como, pero sin limitación, una API en partículas y/o una API insoluble (por ejemplo, una solución acuosa y/o API insoluble en humedad, como, por ejemplo, peróxido de benzoilo).
Una composición de la presente invención puede comprender una API, carboximetilcelulosa sódica e hidroxipropilcelulosa, y puede ser una composición cosméticamente elegante. La composición puede no ser arenosa y/o puede tener una granulometría reducida en comparación con el API en ausencia de una composición de la presente invención. La composición puede no ser adherente (es decir, pegajosa) y/o puede tener una adherencia reducida (es decir, pegajosidad) en comparación con el API en ausencia de una composición de la presente invención. La composición puede tener una rigidez reducida y/o aumentada (es decir, dureza) y/o puede tener una mayor homogeneidad en comparación con el API en ausencia de una composición de la presente invención. En algunas realizaciones, una composición de la presente invención puede comprender una API y puede ser una composición cosméticamente elegante y homogénea.
Según las realizaciones de la presente invención, se puede proporcionar una composición antiviral tópica en un kit. En algunas realizaciones, un kit de la presente invención puede comprender una primera composición y una segunda composición como se describe en el presente documento que se puede combinar para formar la composición antiviral tópica. El kit puede almacenar por separado la primera y segunda composición. El kit puede configurarse para mezclar las dos composiciones tras la dispensación y/o para la aplicación a la piel de un sujeto en una proporción deseada (por ejemplo, 1:1, 2:1, etc.).
La presente invención se explica con mayor detalle en los siguientes ejemplos.
Ejemplos
Ejemplo 1
Grupos de conejos recibieron tratamientos antivirales en diversas dosis como se describe a continuación en la Tabla 1. Las formulaciones para los tratamientos antivirales en los Grupos A-G se proporcionan en las Tablas 2 y 3. Cada una de las formulaciones para los Grupos A-E incluía dos composiciones que se almacenaron por separado en una bomba de doble cámara. Antes de la aplicación, las dos composiciones se dispensaron y se mezclaron juntas en una proporción 1:1 para proporcionar una composición combinada que se aplicó al conejo. El pH objetivo para la composición combinada fue pH 8.
Tabla 1: Dosis de tratamiento antiviral.

Tabla 2: Formulaciones para los tratamientos antivirales en los Grupos A-E.

(continuación)

Tabla 3: Formulaciones para los tratamientos antivirales en los Grupos F y G

Los conejos se infectaron con el virus del papiloma de conejo de rabo blanco (wt CRPV) y el CRPV anulado E8 (mE8-CRPV) a partir del día 1 para un estudio de tratamiento antiviral terapéutico temprano. La Figura 1 muestra el esquema de las infecciones experimentales. Se incluyó el mE8-CRPV ya que este genoma genera papilomas más pequeños y de crecimiento más lento que son más clínicamente similares a las infecciones por el virus del papiloma humano.
Se adquirió un total de 32 conejos blancos adultos de Nueva Zelanda (incluidos ambos sexos) en Robinson, PA. y se usaron en el experimento. Los conejos fueron puestos en cuarentena y liberados (14 días). Cada conejo se inoculó con wt CRPV (en 2 sitios; 5 pg/sitio) y ADN viral de mE8-CRPV (en 2 sitios; 5 pg/sitio). El Ad N viral de CRPV se usó para generar papilomas y la infección se desarrolló mediante una técnica de escarificación tardía (Cladel N. M., et al., J Virol Methods 2008; 148(1-2): 34-39). De los dos sitios inoculados con uno de los virus, un sitio recibió tratamiento (es decir, el sitio izquierdo (L1 o L2)) y el otro sitio no fue tratado (es decir, el sitio derecho (R1 o R2)). Los conejos se colocaron en uno de los ocho grupos (Grupos A-H). Un grupo placebo (es decir, el Grupo A) sirvió como control para evaluar los efectos locales del tratamiento en los Grupos B-H tratados. Los grupos B-G representaron comparaciones de compuestos de prueba versus control negativo de placebo.
Los tratamientos para los Grupos A-H comenzaron en la semana dos en un momento en que los papilomas aún no eran visibles. Este punto de tiempo permitió evaluar los efectos sobre los papilomas subclínicos. El tratamiento fue 5 veces por semana (de lunes a viernes), durante cinco semanas con una dosis de 0,1 ml por cada sitio de aproximadamente 2,5 cm X 2,5 cm para tratamientos tópicos. Se tomaron pesos corporales semanalmente y se recolectaron los sueros sanguíneos al final del período de tratamiento para las químicas sanguíneas, según sea necesario.
La frecuencia y el tamaño de los papilomas se midieron semanalmente en 3 ejes (largo x ancho x alto) en mm. Los datos se ingresaron en una hoja de cálculo y se realizaron cálculos del diámetro medio geométrico de cada papiloma, media SEM para cada grupo, prueba t de Student entre cada grupo emparejado y gráficos hechos del tamaño del papiloma frente al tiempo. También se realizaron gráficos de cambios de peso.
Al finalizar, se recuperaron muestras de riñón e hígado para evaluación histológica y de toxicidad, según sea necesario. Los sitios de piel/papiloma fueron monitorizados fotográficamente y las biopsias fueron evaluadas para histología al final del experimento/tratamiento.
Las Figuras 2-9 muestran gráficas de la media (± SEM) de las mediciones de GMD para cada grupo de tratamiento representadas frente al tiempo después de la infección por CRPV. Para los cálculos de las GMD medias, no se utilizó el regresor espontáneo en el Grupo D. La Figura 10 muestra los pesos corporales medios para los grupos de tratamiento.
Como se ve en la Figura 2, el vehículo de gel sin una API de liberación de NO proporcionó poca o ninguna separación en el tamaño del papiloma entre los sitios tratados y no tratados para los sitios de virus de papiloma mutante de tipo salvaje o mE8-CRPV. En la Figura 3, el grupo Nitricil™ NVN1 al 1 % mostró cierta separación entre los sitios tratados y no tratados para la cepa mutante. Sin embargo, no se logró la inhibición completa del crecimiento del papiloma. En la Figura 4, el grupo Nitricil™ NVN4 al 1,6 % mostró poca o ninguna separación entre los sitios tratados y no tratados para el virus de tipo salvaje y la cepa mutante. La Figura 5 muestra que Nitricil™ NVN1 al 10 % fue eficaz en el tratamiento del virus de tipo salvaje y la cepa mutante con inhibición completa del crecimiento del papiloma en la cepa mutante y separación sustancial entre los sitios tratados y no tratados en el tipo salvaje de crecimiento más rápido. La Figura 6 muestra que el grupo con Nitricil™ NVN4 al 16,3 % fue efectivo para inhibir el crecimiento de la cepa mutante, pero no fue efectivo para inhibir el crecimiento del virus de tipo salvaje. Como se ve en la Figura 7, el vehículo de ungüento sin una API de liberación de NO proporcionó poca o ninguna separación en el tamaño del papiloma entre los sitios tratados y no tratados para los sitios de virus del papiloma de tipo salvaje o mutante. Como se ve en la Figura 8, el ungüento NVN1 al 10 % proporcionó poca o ninguna separación en el tamaño del papiloma entre los sitios tratados y no tratados para los sitios de virus del papiloma de tipo salvaje o mutante, lo que ilustra que la liberación de óxido nítrico de la composición afecta la efectividad de inhibiendo el crecimiento del papiloma, en oposición a la presencia del esqueleto de siloxano, ya que el ungüento NVN1 al 10 % y el gel NVN1 al 10 % contenían la misma cantidad del esqueleto de polisiloxano, pero lograron resultados marcadamente diferentes.
Ejemplo 2
Las pruebas de liberación in vitro se realizaron usando el analizador de óxido nítrico multicanal. Se usó una balanza analítica para pesar las formulaciones de prueba de los Grupos B-E y G descritos en el Ejemplo 1. Aproximadamente 20 mg de ambas fases de una formulación respectiva se transfirieron a una celda de medición de NO limpia y seca con una barra de agitación magnética. La liberación in vitro en tiempo real de óxido nítrico de las formulaciones se determinó mientras se mezclaba continuamente usando los siguientes parámetros instrumentales:
1. Caudal de nitrógeno húmedo: 112-115 ml/min
2. Temperatura de la muestra: 37°C
3. Detección: óxido nítrico por quimioluminiscencia
4. Frecuencia de adquisición de datos: 1 Hz, alternancia secuencial irregular
5. Duración: tiempo en el que la tasa de liberación de NO disminuye linealmente (NLT 8 h)
6. Software de adquisición: NovanWare
La conversión de partes por mil millones (PPB) NO a mol de óxido nítrico se logró midiendo el óxido nítrico generado a partir de una cantidad conocida de nitrito de sodio en una solución de yoduro de potasio para adquirir un factor de conversión de PPB a mol. Cualquier brecha en los datos de liberación de óxido nítrico en tiempo real resultante de la operación multicanal se llenó utilizando un programa de interpolación lineal. Para cualquier muestra que no se midió hasta el agotamiento del óxido nítrico, se realizó una extrapolación lineal a liberación cero de los últimos -5000 segundos de liberación. Los datos de liberación de óxido nítrico en tiempo real se integraron, dando como resultado una curva de acumulación de óxido nítrico total. Parámetros de liberación de óxido nítrico como Cmáx (es decir, la concentración máxima de NO liberado), Tmáx (es decir, el momento en que se alcanza la Cmáx), liberación acumulativa de óxido nítrico (es decir, la suma de todos los puntos de datos por unidad de tiempo) y tiempo hasta la mitad del total liberado (T50) (es decir, el tiempo en el que se libera el 50 % del NO acumulado) a partir de las curvas de liberación de óxido nítrico de acumulación total y en tiempo real. Todos los cálculos anteriores se realizaron automáticamente en un software de procesamiento de datos personalizado (NovanWare).
Los resultados de las pruebas de liberación in vitro, junto con el pH respectivo de las mezclas se resumen en la Tabla 4 a continuación.
Tabla 4: Datos de liberación de NO para las formulaciones probadas

(continuación)

Ejemplo 3
Los artículos de prueba en el Ejemplo 2 se aplicaron a los sitios de 2,5 cm x 2,5 cm de los conejos del Ejemplo 1. La aplicación de 0,1 ml de los artículos de prueba a 6,25 cm2 da como resultado una liberación del ensayo in vitro de NO/cm2 como se refleja en la Tabla 5.
Tabla 5: Datos de liberación de NO por unidad de área para las formulaciones probadas

(continuación)

Los perfiles de liberación de NO acumulativo y en tiempo real se ilustran en las Figuras 11 a 17 como se describe anteriormente. En base a la efectividad de las formulaciones para los grupos de tratamiento D y E contra las cepas mutantes, los parámetros para la liberación de NO en tiempo real que pueden ser antivirales pueden identificarse como se ilustra en las Figuras 16A, 16B y 17. Por consiguiente, en algunas realizaciones de la presente invención, la liberación de NO de una composición antiviral puede caer dentro de una o más de las ventanas ilustradas en las Figuras 16A y/o 17. La liberación de NO dentro de las ventanas definidas puede ocurrir en cualquier momento dentro del tiempo anticipado en el que se aplicará la composición. En consecuencia, la liberación de NO que cae dentro de la ventana puede ocurrir dentro de las primeras 1, 2 o 4 horas o puede comenzar a partir de otro momento durante el período de aplicación. Las ventanas de tiempo ilustradas en las Figuras 16A, 16B y 17 se muestran en la Tabla 6.
Tabla 6: ventanas de tiempo de liberación de NO.

Ejemplo 4
Los grupos de conejos recibieron tratamientos antivirales en varias dosis como se describe a continuación en la Tabla 7. Las formulaciones para los tratamientos antivirales en los Grupos A-F se proporcionan en la Tabla 8. Cada una de las formulaciones para los Grupos A-E incluía dos composiciones separadas que fueron almacenadas por separado en una bomba de doble cámara. Antes de la aplicación, las dos composiciones se dispensaron y se mezclaron juntas en una proporción 1:1 para proporcionar una composición combinada que se aplicó al conejo. El pH objetivo para la composición combinada fue pH 8.
Tabla 7: Dosis de tratamiento antiviral.
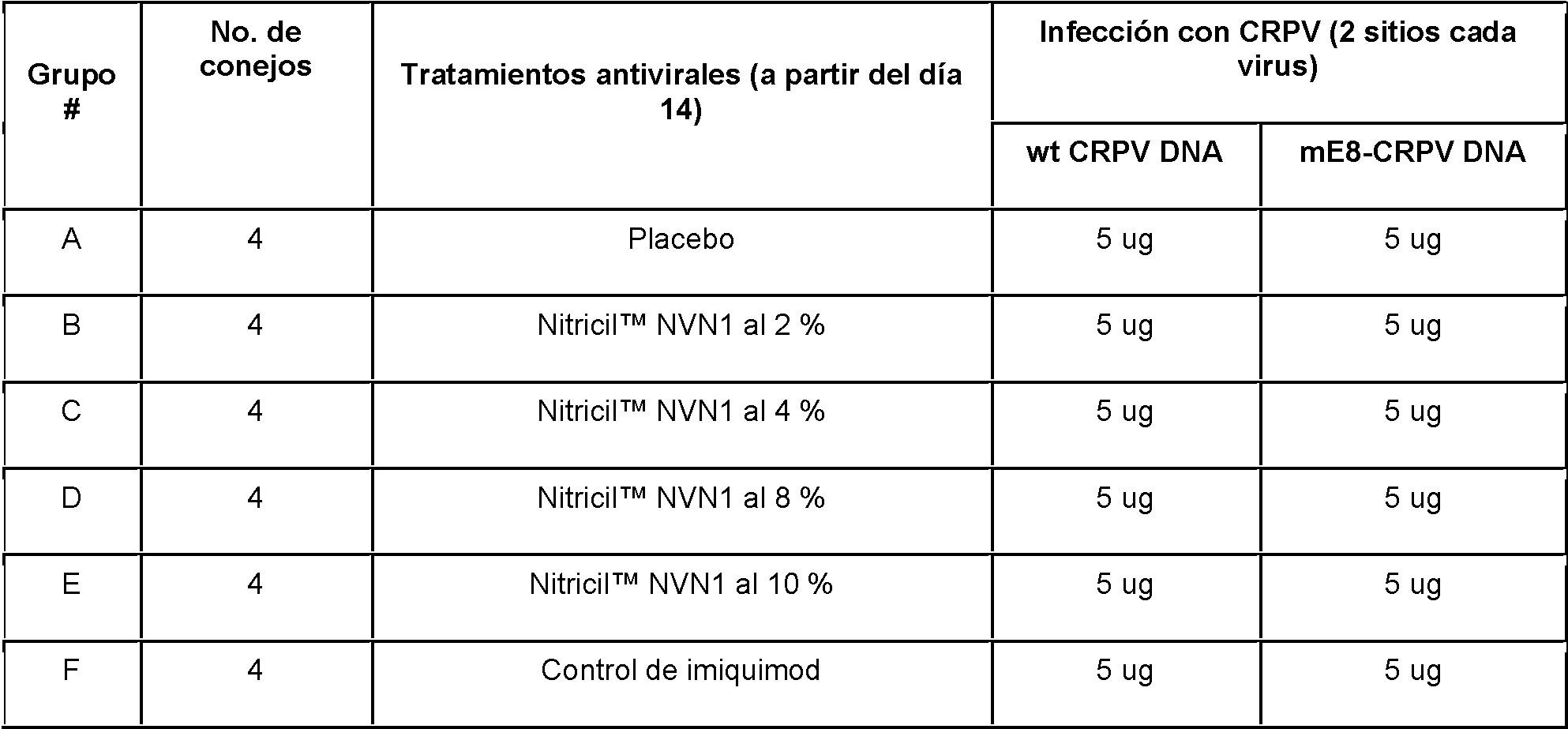
Tabla 8: Formulaciones para los tratamientos antivirales en los Grupos A-E

Tabla 9: Datos de liberación de NO para las formulaciones de la Tabla 8

(continuación)

Los conejos se infectaron con el virus del papiloma de conejo de rabo blanco (wt CRPV) y el CRPV anulado E8 (mE8-CRPV) a partir del día 1 para un estudio de tratamiento antiviral terapéutico temprano. La Figura 1 muestra el esquema de las infecciones experimentales. Se incluyó el mE8-CRPV ya que este genoma genera papilomas más pequeños y de crecimiento más lento que son más clínicamente similares a las infecciones por el virus del papiloma humano.
Se adquirió un total de 24 conejos blancos adultos de Nueva Zelanda (incluidos ambos sexos) en Robinson, PA. y se usaron en el experimento. Los conejos fueron puestos en cuarentena y despejados (14 días). Cada conejo fue inoculado con wt CRPV (en 2 sitios; 5 pg/sitio) y ADN viral mE8-CRPV (en 2 sitios; 5 pg/sitio). El Ad N viral de Cr PV se usó para generar papilomas y la infección se desarrolló mediante una técnica de escarificación retardada (Cladel N. M., et al., J Virol Methods 2008; 148(1-2): 34-39). De los dos sitios inoculados con uno de los virus, un sitio recibió tratamiento (es decir, el sitio izquierdo (L1 o L2)) y el otro sitio no fue tratado (es decir, el sitio derecho (R1 o R2)). Los conejos se colocaron en uno de seis grupos (Grupos A-F). Un grupo placebo (es decir, el Grupo A) sirvió como control para evaluar los efectos locales del tratamiento en los Grupos B-E tratados.
Los tratamientos para los grupos A-F comenzaron en la semana dos en un momento en que los papilomas aún no eran visibles. Este punto de tiempo permitió evaluar los efectos sobre los papilomas subclínicos. El tratamiento fue 5
veces por semana (de lunes a viernes), durante cinco semanas con una dosis de 0,1 ml por cada sitio de aproximadamente 2,5 cm x 2,5 cm para tratamientos tópicos. Se tomaron pesos corporales semanalmente y se recolectaron sueros sanguíneos al final del período de tratamiento para las químicas sanguíneas, según sea necesario.
La frecuencia y el tamaño de los papilomas se midieron semanalmente en 3 ejes (largo x ancho x alto) en mm. Los datos se ingresaron en una hoja de cálculo y se realizaron cálculos del diámetro medio geométrico de cada papiloma, media SEM para cada grupo, prueba t de Student entre cada grupo emparejado y gráficos hechos del tamaño del papiloma frente al tiempo. También se realizaron gráficos de cambios de peso.
Al finalizar, se recuperaron muestras de riñón e hígado para evaluación histológica y de toxicidad, según sea necesario. Los sitios de piel/papiloma fueron monitorizados fotográficamente y las biopsias fueron evaluadas para histología al final del experimento/tratamiento.
Las Figuras 18-23 muestran gráficos del tamaño del papiloma (GMD) en mm a lo largo del tiempo para las formulaciones en los Grupos A-F, respectivamente.
A la luz del régimen de dosificación de 5 días a la semana y una vez al día, las dosis que muestran una eficacia mínima podrían ser adecuadas para su uso con un programa de dosificación diferente, por ejemplo, dos veces al día, 7 días a la semana. Según la efectividad de las formulaciones para el tratamiento de los Grupos D y E contra las cepas mutantes, los parámetros para la liberación de NO en tiempo real pueden caer dentro de una o más de las ventanas ilustradas en las Figuras 16A y/o 17. Sin embargo, otras dosis también pueden ser eficaces en función de los grupos de tratamiento B y C, ya que la duración de la liberación de NO para estos grupos puede permitir una dosificación más frecuente. En consecuencia, un producto (por ejemplo, una composición tópica) con una liberación de NO que cae en una ventana de 30 minutos definida como medido en función del peso y que tiene una liberación instantánea mínima de 7 pmol/mg y una liberación instantánea máxima de 4000 pmol/mg puede ser adecuado. En algunas realizaciones, puede ser adecuado un producto con una liberación de NO dentro de una ventana de 30 minutos definida como medida con base en el peso y que tiene una liberación instantánea mínima de 15 pmol/mg y una liberación instantánea máxima de 4000 pmol/mg. La liberación de NO dentro de las ventanas definidas puede ocurrir en cualquier momento dentro del tiempo anticipado en el que el producto será aplicado y/o estará presente en la piel de un sujeto. En consecuencia, una liberación de NO que cae dentro de una o más de las ventanas ilustradas en las Figuras 16A y/o 17 pueden ocurrir dentro de las primeras 1, 2 o 4 horas después de la aplicación/administración o pueden comenzar a partir de otro momento después de la aplicación/administración. Ejemplo 5
Se obtuvieron muestras de tejido de conejos blancos de Nueva Zelanda infectados con el virus del papiloma de conejo de rabo blanco y tratados como se describe en el Ejemplo 4 al final del tratamiento. Las secciones de la piel se procesaron para portaobjetos de hematoxilina y eosina (H&E) y se sometieron a evaluación microscópica de forma ciega. Después de evaluar los portaobjetos, los resultados se resumieron en tres categorías principales: 1) papiloma, 2) hiperplasia de intensidad marcada y 3) hiperplasia de intensidad mínima a leve. La presencia de células inflamatorias se determinó cualitativamente. No se realizó un análisis cuantitativo. Sin embargo, la evaluación cualitativa reveló un nivel similar de inflamación entre las tres categorías, el que consistió en que la inflamación fue generalmente baja y comparable. La Tabla 10 proporciona los resultados histológicos de la tinción con H&E.
Tabla 10: Resultados de histología con H&E.

(continuación)

Los portaobjetos de tejido asignados a la Categoría #3 (hiperplasia de intensidad mínima a leve) incluyen las muestras de tejido obtenidas de animales tratados con Nitricil™ NVN1 al 8 % o Nitricil™ NVN1 al 10 %. No se observaron inclusiones virales basófilas de color oscuro a claro intranucleares en los portaobjetos de tejido asignados a la Categoría #3, lo que sugiere que el tratamiento con altas concentraciones de Nitricil™ NVN1, como, por ejemplo, con formulaciones de Nitricil™ NVN1 al 8 % o 10 %, suprime y/o inhibe la replicación viral de un virus sin alterar la infiltración local de las células inmunes inflamatorias.
Ejemplo 6
Se prepararon formulaciones adicionales y se usaron para determinar la eficacia en el tratamiento y/o prevención de afecciones cutáneas relacionadas con virus, tales como, por ejemplo, verrugas genitales. Estas formulaciones incluyeron Nitricil™ NVN1 en una cantidad de 4 %, 8 %, 12 % o 16 % junto con un placebo. Cada una de las formulaciones incluía un hidrogel que tenía un pH de 4,5 y una composición como se proporciona en la Tabla 11, y una segunda composición en forma de gel y que tenía una composición como se proporciona en la Tabla 12. Al mezclar el hidrogel y el gel, se combinó la composición se logró teniendo una composición como se proporciona en la Tabla 13.
Tabla 11: Composición del hidrogel pH 4,5

(continuación)

Tabla 12: Composición del gel

Tabla 13: Composición de la composición combinada

(continuación)

Claims (16)
1. Una composición tópica para su uso en un procedimiento de tratamiento y/o prevención de una infección viral en un sujeto que lo necesite, comprendiendo dicho procedimiento:
administrar la composición tópica a la piel de un sujeto,
en la que la composición tópica comprende un ingrediente farmacéutico activo liberador de óxido nítrico que libera óxido nítrico a la piel del sujeto, y
en la que la composición tópica mantiene una concentración en tiempo real de óxido nítrico de al menos aproximadamente 7 pmol de NO/mg de la composición durante al menos 1 hora después de la administración, medida por pruebas de liberación in vitro en tiempo real, tratando y/o evitando así infección viral en el sujeto, en la que el ingrediente farmacéutico activo que libera óxido nítrico comprende una partícula de sílice cocondensada que libera NO que tiene un grupo funcional de diolato de diazenio.
2. La composición tópica de la reivindicación 1, para el uso de la reivindicación 1, en el que la composición tópica mantiene una concentración en tiempo real de óxido nítrico de al menos aproximadamente 6 pmol de NO/mg de la composición durante al menos 2 horas después de la administración, medida mediante pruebas de liberación in vitro en tiempo real; y/o
en el que la composición tópica mantiene una concentración en tiempo real de óxido nítrico de al menos aproximadamente 5 pmol de NO/mg de la composición durante al menos 4 horas después de la administración, medida por pruebas de liberación in vitro en tiempo real.
3. La composición tópica de las reivindicaciones 1-2, para el uso de las reivindicaciones 1-2, en el que la composición tópica tiene una concentración máxima de óxido nítrico en un intervalo de aproximadamente 12 pmol de NO/mg de la composición a aproximadamente 3500 pmol de NO/mg de la composición, medida por pruebas de liberación in vitro en tiempo real.
4. La composición tópica de las reivindicaciones 1-3, para el uso de las reivindicaciones 1-3, en el que la composición tópica libera óxido nítrico en una cantidad acumulativa de aproximadamente 300 nmol de NO/mg de la composición a aproximadamente 1.000 nmol de NO/mg de la composición a las 24 horas después de la administración, medida por pruebas de liberación in vitro en tiempo real.
5. La composición tópica de las reivindicaciones 1-4, para el uso de las reivindicaciones 1-4, en el que la composición tópica tiene una vida media en un intervalo de aproximadamente 9 minutos a aproximadamente 420 minutos, medida por pruebas de liberación in vitro en tiempo real.
6. La composición tópica de las reivindicaciones 1-5, para el uso de las reivindicaciones 1-5,
en el que la composición tópica mantiene una concentración en tiempo real de óxido nítrico de al menos aproximadamente 104 pmol de NO/cm2 durante un período de tiempo de al menos 1 hora después de la administración de la composición a la piel del sujeto, medida por pruebas de liberación in vitro en tiempo real, tratando y/o previniendo, de este modo, una infección viral en el sujeto, y en el que la composición tópica mantiene una concentración en tiempo real de óxido nítrico de al menos aproximadamente 89 pmol de NO/cm2 durante un período de tiempo de al menos 2 horas después de la administración de la composición a la piel del sujeto, medida por pruebas de liberación in vitro en tiempo real,
en el que el ingrediente farmacéutico activo que libera óxido nítrico comprende un compuesto que libera óxido nítrico y en el que la composición tópica no comprende nitrito acidificado.
7. La composición tópica de la reivindicación 6, para el uso de la reivindicación 6, en el que la composición tópica mantiene una concentración en tiempo real de óxido nítrico de al menos aproximadamente 74 pmol de NO/cm2 durante un período de tiempo de al menos 4 horas después de la administración de la composición a la piel del sujeto, medida por pruebas de liberación in vitro en tiempo real.
8. La composición tópica de las reivindicaciones 6-7, para el uso de las reivindicaciones 6-7, en el que la composición tópica libera óxido nítrico en una cantidad acumulativa de aproximadamente 1.300 nmol de NO/cm2 a aproximadamente 14.000 nmol de NO/cm2 en un período de tiempo de aproximadamente 4 horas después de la administración de la composición a la piel del sujeto, medida por pruebas de liberación in vitro en tiempo real; y/o en el que la composición tópica libera óxido nítrico en una cantidad acumulativa de aproximadamente 4.500 nmol de NO/cm2 a aproximadamente 14.000 nmol de NO/cm2 en un período de tiempo de aproximadamente 24 horas después de la administración de la composición a la piel del sujeto, medida por pruebas de liberación in vitro en tiempo real.
9. La composición tópica de las reivindicaciones 6-8, para el uso de las reivindicaciones 6-8, en el que, después de la administración de la composición tópica a la piel del sujeto, la composición tópica tiene una concentración en
tiempo real de NO de al menos 100 pmol de NO/cm2 a las 0,5 horas después de la administración, al menos 50 pmol de NO/cm2 a la 1 hora después de la administración, al menos 40 pmol de NO/cm2 a las 2 horas después de la administración, al menos 25 pmol de NO/cm2 a las 3 horas después de la administración, y/o al menos 20 pmol de NO/cm2 a las 4 horas después de la administración, medida por pruebas de liberación in vitro en tiempo real.
10. La composición tópica de una cualquiera de las reivindicaciones 1-9, para el uso de una cualquiera de las reivindicaciones 1-9, en el que la infección viral es causada por citomegalovirus (CMV), virus de Epstein-Barr, virus de varicela zóster (VZV), virus vaccinia, virus de viruela bovina, virus de viruela del mono, virus de herpes simplex (HSV), herpes zóster, virus del herpes humano 6 (HHV-6), virus del herpes humano 8 (HHV-8), virus del papiloma, molluscum contagiosum, orf, variola y/o virus de Coxsackie.
11. La composición tópica de una cualquiera de las reivindicaciones 1-10, para el uso de una cualquiera de las reivindicaciones 1-10, en el que la infección viral es causada por un virus del papiloma.
12. La composición tópica de una cualquiera de las reivindicaciones 1-11, para el uso de una cualquiera de las reivindicaciones 1-11, en el que se administra una cantidad efectiva de tratamiento y/o una cantidad efectiva de óxido nítrico para la prevención a la capa basal y/o membrana basal del epitelio de un sujeto; y/o
en el que el óxido nítrico en una cantidad de aproximadamente 1 x 10-5 M a aproximadamente 1 x 10'7 M se administra a la capa basal y/o membrana basal del epitelio de un sujeto.
13. La composición tópica de una cualquiera de las reivindicaciones 1-12, para el uso de una cualquiera de las reivindicaciones 1-12, en el que la composición tópica induce apoptosis en células infectadas por virus.
14. La composición tópica de una cualquiera de las reivindicaciones 1-13, para el uso de una cualquiera de las reivindicaciones 1-13, en el que la composición tópica comprende el ingrediente farmacéutico activo que libera óxido nítrico en una cantidad de aproximadamente 0,5 % a aproximadamente 20 % en peso de la composición.
15. La composición tópica de una cualquiera de las reivindicaciones 1-14, para el uso de una cualquiera de las reivindicaciones 1-14, en el que la infección viral es causada por molluscum contagiosum.
16. La composición tópica de una cualquiera de las reivindicaciones 1-15, para el uso de una cualquiera de las reivindicaciones 1-15, en el que la composición tópica tiene una concentración máxima de óxido nítrico mayor que 160 pmol de NO/mg en base a una liberación total de NO determinada a las 24 horas después de la administración, medida por pruebas de liberación in vitro, opcionalmente en el que la composición tópica tiene una concentración máxima de óxido nítrico liberado en un intervalo de aproximadamente 160 pmol de NO/mg a aproximadamente 3.500 pmol de NO/mg, medida por pruebas de liberación in vitro.
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201462023587P | 2014-07-11 | 2014-07-11 | |
| US201562139176P | 2015-03-27 | 2015-03-27 | |
| PCT/US2015/039908 WO2016007834A1 (en) | 2014-07-11 | 2015-07-10 | Topical antiviral compositions and methods of using the same |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2807200T3 true ES2807200T3 (es) | 2021-02-22 |
Family
ID=55064948
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES15819451T Active ES2807200T3 (es) | 2014-07-11 | 2015-07-10 | Composiciones antivirales tópicas y procedimientos de uso de las mismas |
Country Status (10)
| Country | Link |
|---|---|
| US (1) | US10322081B2 (es) |
| EP (2) | EP3166593B1 (es) |
| JP (1) | JP6651499B2 (es) |
| KR (1) | KR102495101B1 (es) |
| CN (1) | CN106659675B (es) |
| AU (1) | AU2015287674B2 (es) |
| BR (1) | BR112017000456B1 (es) |
| CA (1) | CA2954061C (es) |
| ES (1) | ES2807200T3 (es) |
| WO (1) | WO2016007834A1 (es) |
Families Citing this family (18)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1871433B1 (en) | 2005-03-24 | 2009-04-22 | NOLabs AB | Cosmetic treatment with nitric oxide, device for performing said treatment and manufacturing method therefor |
| EP1888510A4 (en) | 2005-05-27 | 2013-01-16 | Univ North Carolina | NITROGEN OXIDE-RELATED PARTICLES FOR NITROGEN OXIDE THERAPEUTICS AND BIOMEDICAL APPLICATIONS |
| JP5548121B2 (ja) | 2007-05-14 | 2014-07-16 | リサーチ ファウンデーション オブ ステイト ユニバーシティ オブ ニューヨーク | バイオフィルム中の細菌細胞における生理学的分散応答の誘導 |
| ES2658897T3 (es) | 2011-08-24 | 2018-03-12 | Novan, Inc. | Macromoléculas de liberación de óxido nítrico ajustables que tienen múltiples estructuras donantes de óxido nítrico |
| ES2861439T3 (es) | 2012-03-14 | 2021-10-06 | Novan Inc | Composiciones farmacéuticas que liberan óxido nítrico |
| US11813284B2 (en) | 2013-08-08 | 2023-11-14 | Novan, Inc. | Topical compositions and methods of using the same |
| US10322082B2 (en) | 2014-07-11 | 2019-06-18 | Novan, Inc. | Topical antiviral compositions and methods of using the same |
| EP3177262A4 (en) | 2014-08-08 | 2018-04-18 | Novan Inc. | Topical emulsions |
| US10912743B2 (en) | 2016-03-02 | 2021-02-09 | Novan, Inc. | Compositions for treating inflammation and methods of treating the same |
| US11166980B2 (en) | 2016-04-13 | 2021-11-09 | Novan, Inc. | Compositions, systems, kits, and methods for treating an infection |
| WO2018236803A1 (en) | 2017-06-19 | 2018-12-27 | Novan, Inc. | Topical compositions and methods of using the same |
| WO2019169221A1 (en) | 2018-03-01 | 2019-09-06 | Novan, Inc. | Nitric oxide releasing suppositories and methods of use thereof |
| KR102831159B1 (ko) | 2018-04-16 | 2025-07-07 | 온퀄리티 파마슈티컬스 차이나 리미티드 | 암 치료의 부작용을 예방하거나 치료하기 위한 방법 |
| CN108355166B (zh) * | 2018-05-15 | 2021-02-19 | 黄冈师范学院 | 一种介孔生物活性玻璃/金属有机框架支架材料及制备方法 |
| US11541105B2 (en) | 2018-06-01 | 2023-01-03 | The Research Foundation For The State University Of New York | Compositions and methods for disrupting biofilm formation and maintenance |
| MX368750B (es) * | 2018-08-31 | 2019-10-15 | Cell Therapy And Tech S A De C V | Composiciones farmaceuticas semisolidas de base oleosa conteniendo pirfenidona para su aplicacion en la reparacion tisular. |
| US20230355662A1 (en) * | 2019-06-19 | 2023-11-09 | Zylo Therapeutics, Inc. | Sulfur functionalized monoliths and particles derived from the same as nitric oxide carriers for pharmaceutical and cosmetic applications |
| US20230108186A1 (en) * | 2020-02-07 | 2023-04-06 | Know Bio, Llc | Nitric oxide-releasing antibacterial compounds, formulations, and methods pertaining thereto |
Family Cites Families (104)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB9125712D0 (en) | 1991-12-03 | 1992-01-29 | Smithkline Beecham Plc | Skin care composition |
| DE4237453C1 (es) | 1992-11-06 | 1993-08-19 | Lts Lohmann Therapie-Systeme Gmbh & Co Kg, 5450 Neuwied, De | |
| JP3739100B2 (ja) | 1993-07-30 | 2006-01-25 | 救急薬品工業株式会社 | 低温架橋型ゲル剤 |
| US5840759A (en) | 1993-10-08 | 1998-11-24 | The United States Of America As Represented By The Department Of Health And Human Services | Use of nitric oxide releasing compounds to protect noncancerous cells from chemotherapeutic agents |
| US6709681B2 (en) | 1995-02-17 | 2004-03-23 | Aberdeen University | Acidified nitrite as an antimicrobial agent |
| GB9804469D0 (en) | 1998-03-02 | 1998-04-29 | Univ Aberdeen | Antiviral composition |
| US5968001A (en) | 1996-05-14 | 1999-10-19 | Bristol-Myers Squibb Company | Wound dressings with leak prevention seal |
| US5968528A (en) | 1997-05-23 | 1999-10-19 | The Procter & Gamble Company | Skin care compositions |
| US20020013304A1 (en) | 1997-10-28 | 2002-01-31 | Wilson Leland F. | As-needed administration of an androgenic agent to enhance female sexual desire and responsiveness |
| JP4139860B2 (ja) | 1997-11-10 | 2008-08-27 | ストラカン インターナショナル リミティッド | 浸透増強および刺激減少システム |
| US6103266A (en) | 1998-04-22 | 2000-08-15 | Tapolsky; Gilles H. | Pharmaceutical gel preparation applicable to mucosal surfaces and body tissues |
| US6103275A (en) | 1998-06-10 | 2000-08-15 | Nitric Oxide Solutions | Systems and methods for topical treatment with nitric oxide |
| US6465445B1 (en) | 1998-06-11 | 2002-10-15 | Endorecherche, Inc. | Medical uses of a selective estrogen receptor modulator in combination with sex steroid precursors |
| FR2780888B1 (fr) | 1998-07-09 | 2000-10-13 | Benac | Lotion nettoyante et traitante destinee au traitement des comedons |
| US6238683B1 (en) | 1998-12-04 | 2001-05-29 | Johnson & Johnson Consumer Companies, Inc. | Anhydrous topical skin preparations |
| US6479058B1 (en) | 1999-09-02 | 2002-11-12 | Mccadden Michael E. | Composition for the topical treatment of poison ivy and other forms of contact dermatitis |
| US6719997B2 (en) | 2000-06-30 | 2004-04-13 | Dermatrends, Inc. | Transdermal administration of pharmacologically active amines using hydroxide-releasing agents as permeation enhancers |
| US6475500B2 (en) | 2000-07-10 | 2002-11-05 | The Procter & Gamble Company | Anhydrous cosmetic compositions |
| GB0021317D0 (en) | 2000-08-30 | 2000-10-18 | Queen Mary & Westfield College | Transdermal pharmaceutical delivery composition |
| GB0022084D0 (en) | 2000-09-08 | 2000-10-25 | Univ Aberdeen | Treatment of multiply antibiotic-resistant organisms |
| WO2002041902A1 (en) | 2000-11-27 | 2002-05-30 | The University Of Akron | Treatment of disorders using polyethylenimine diazeniumdiolate |
| US6780849B2 (en) | 2000-12-21 | 2004-08-24 | Scimed Life Systems, Inc. | Lipid-based nitric oxide donors |
| GB0119011D0 (en) | 2001-08-03 | 2001-09-26 | Univ Aberdeen | Treatment of nail infections |
| US6703046B2 (en) | 2001-10-04 | 2004-03-09 | Medtronic Ave Inc. | Highly cross-linked, extremely hydrophobic nitric oxide-releasing polymers and methods for their manufacture and use |
| JP2003286153A (ja) | 2002-01-24 | 2003-10-07 | Shiseido Co Ltd | 皮膚外用組成物 |
| CA2485167A1 (en) * | 2002-05-07 | 2003-11-20 | The Government Of The United States Of America | Polydiazeniumdiolated cyclic polyamines with polyphasic nitric oxide release and related compounds, compositions comprising same and methods of using same |
| IL152486A0 (en) | 2002-10-25 | 2003-05-29 | Meir Eini | Alcohol-free cosmetic and pharmaceutical foam carrier |
| US7704518B2 (en) | 2003-08-04 | 2010-04-27 | Foamix, Ltd. | Foamable vehicle and pharmaceutical compositions thereof |
| US20080031907A1 (en) | 2002-10-25 | 2008-02-07 | Foamix Ltd. | Cosmetic and pharmaceutical foam |
| US20070292359A1 (en) | 2002-10-25 | 2007-12-20 | Foamix Ltd. | Polypropylene glycol foamable vehicle and pharmaceutical compositions thereof |
| US20070292461A1 (en) | 2003-08-04 | 2007-12-20 | Foamix Ltd. | Oleaginous pharmaceutical and cosmetic foam |
| US20080206161A1 (en) | 2002-10-25 | 2008-08-28 | Dov Tamarkin | Quiescent foamable compositions, steroids, kits and uses thereof |
| US9265725B2 (en) | 2002-10-25 | 2016-02-23 | Foamix Pharmaceuticals Ltd. | Dicarboxylic acid foamable vehicle and pharmaceutical compositions thereof |
| US9668972B2 (en) | 2002-10-25 | 2017-06-06 | Foamix Pharmaceuticals Ltd. | Nonsteroidal immunomodulating kit and composition and uses thereof |
| US20080317679A1 (en) | 2002-10-25 | 2008-12-25 | Foamix Ltd. | Foamable compositions and kits comprising one or more of a channel agent, a cholinergic agent, a nitric oxide donor, and related agents and their uses |
| US20080138296A1 (en) | 2002-10-25 | 2008-06-12 | Foamix Ltd. | Foam prepared from nanoemulsions and uses |
| US20050271596A1 (en) | 2002-10-25 | 2005-12-08 | Foamix Ltd. | Vasoactive kit and composition and uses thereof |
| US20040202684A1 (en) | 2003-04-08 | 2004-10-14 | David Djerassi | Personalized interactive natural cosmetics |
| NZ545001A (en) | 2003-07-03 | 2009-09-25 | Univ St Andrews | Zeolites for delivery of nitric oxide |
| WO2005004984A1 (en) | 2003-07-14 | 2005-01-20 | Power Paper Ltd. | Device and method for the treatment of pilosebaceous disorders |
| US20080069779A1 (en) | 2003-08-04 | 2008-03-20 | Foamix Ltd. | Foamable vehicle and vitamin and flavonoid pharmaceutical compositions thereof |
| US8795693B2 (en) | 2003-08-04 | 2014-08-05 | Foamix Ltd. | Compositions with modulating agents |
| US8486374B2 (en) | 2003-08-04 | 2013-07-16 | Foamix Ltd. | Hydrophilic, non-aqueous pharmaceutical carriers and compositions and uses |
| US20070166255A1 (en) | 2004-11-22 | 2007-07-19 | Gupta Shyam K | Treatment of Topical Discomforts Including Acne, Sunburn, Diaper Rash, Wound, Wrinkles and Dandruff/Hair Loss by Natural Lignans via Fatty Acid Desaturase Inhibition |
| SI1833443T1 (sl) | 2005-01-04 | 2012-03-30 | Teikoku Pharma Usa Inc | Hladilni pripravek v obliki topičnega obliža |
| EP1757278A1 (en) | 2005-08-23 | 2007-02-28 | NOLabs AB | Device, system, and method comprising microencapsulated liquid for release of nitric oxide from a polymer |
| ES2314882T3 (es) | 2005-02-11 | 2009-03-16 | Nolabs Ab | Dispositivo y metodo para el tratamiento de dermatomicosis y, en particular, de onicomicosis. |
| EP1690554A1 (en) | 2005-02-11 | 2006-08-16 | NOLabs AB | Device for treatment of infections, including dermatophytosis and onychomycosis |
| PL1846058T3 (pl) | 2005-02-11 | 2009-12-31 | Nolabs Ab | Produkt, sposób i zastosowanie go do leczenia neuropatii z wydzielaniem tlenku azotu |
| EP1704876A1 (en) * | 2005-03-24 | 2006-09-27 | NOLabs AB | Cosmetic treatment, device for performing said treatment and manufacturing method thereof |
| EP1871433B1 (en) | 2005-03-24 | 2009-04-22 | NOLabs AB | Cosmetic treatment with nitric oxide, device for performing said treatment and manufacturing method therefor |
| WO2007039825A2 (en) | 2005-05-09 | 2007-04-12 | Foamix Ltd. | Saccharide foamable compositions |
| EP1888510A4 (en) | 2005-05-27 | 2013-01-16 | Univ North Carolina | NITROGEN OXIDE-RELATED PARTICLES FOR NITROGEN OXIDE THERAPEUTICS AND BIOMEDICAL APPLICATIONS |
| US20080152596A1 (en) | 2005-07-19 | 2008-06-26 | Foamix Ltd. | Polypropylene glycol foamable vehicle and pharmaceutical compositions thereof |
| US9983874B2 (en) * | 2006-03-16 | 2018-05-29 | International Business Machines Corporation | Structure for a circuit function that implements a load when reservation lost instruction to perform cacheline polling |
| US7744928B2 (en) | 2006-04-14 | 2010-06-29 | Advanced Cardiovascular Systems, Inc. | Methods and compositions for treatment of lesioned sites of body vessels |
| EP2029106A2 (en) | 2006-06-07 | 2009-03-04 | Foamix Ltd. | Foamable vehicle comprising polypropylene glycol alkyl ether and pharmaceutical compositions thereof |
| US8333997B2 (en) | 2006-06-21 | 2012-12-18 | Albert Einstein College Of Medicine Of Yeshiva University | Compositions for sustained release of nitric oxide, methods of preparing same and uses thereof |
| WO2008110872A2 (en) | 2006-06-23 | 2008-09-18 | Foamix Ltd. | Foamable compositions and kits comprising one or more of a channel agent, a cholinergic agent, a nitric oxide donor, and related agents and their uses |
| WO2008038147A2 (en) | 2006-07-05 | 2008-04-03 | Foamix Ltd. | Foamable vehicle comprising dicarboxylic acid or dicarboxylic acid ester and pharmaceutical compositions thereof |
| GB0616350D0 (en) | 2006-08-17 | 2006-09-27 | Univ St Andrews | Adsorption and release of nitric oxide in metal organic frameworks |
| US20080166303A1 (en) | 2006-09-08 | 2008-07-10 | Dov Tamarkin | Colored or colorable foamable composition and foam |
| GB0618711D0 (en) | 2006-09-22 | 2006-11-01 | Univ Exeter | Agricultural treatment |
| US20080260655A1 (en) | 2006-11-14 | 2008-10-23 | Dov Tamarkin | Substantially non-aqueous foamable petrolatum based pharmaceutical and cosmetic compositions and their uses |
| WO2009007785A2 (en) | 2006-11-14 | 2009-01-15 | Foamix Ltd. | Stable non-alcoholic foamable pharmaceutical emulsion compositions with an unctuous emollient and their uses |
| EP2097065A2 (en) | 2006-11-29 | 2009-09-09 | Foamix Ltd. | Foamable waterless compositions with modulating agents |
| US20080292560A1 (en) | 2007-01-12 | 2008-11-27 | Dov Tamarkin | Silicone in glycol pharmaceutical and cosmetic compositions with accommodating agent |
| US20100178319A1 (en) | 2007-03-27 | 2010-07-15 | Lars Lindgren | Topical Dermal Delivery Device For Nitric Oxide Delivery |
| US8636982B2 (en) | 2007-08-07 | 2014-01-28 | Foamix Ltd. | Wax foamable vehicle and pharmaceutical compositions thereof |
| WO2009056991A2 (en) | 2007-09-04 | 2009-05-07 | Foamix Ltd. | Device for delivery of a foamable composition |
| US8399005B2 (en) | 2007-10-12 | 2013-03-19 | University Of North Carolina At Chapel Hill | Use of nitric oxide to enhance the efficacy of silver and other topical wound care agents |
| EP2219600A4 (en) | 2007-11-19 | 2012-09-12 | Stiefel Laboratories | TOPICAL COSMETIC SKIN COMPOSITIONS AND APPLICATION METHOD THEREFOR |
| US20090130029A1 (en) | 2007-11-21 | 2009-05-21 | Foamix Ltd. | Glycerol ethers vehicle and pharmaceutical compositions thereof |
| US9439857B2 (en) | 2007-11-30 | 2016-09-13 | Foamix Pharmaceuticals Ltd. | Foam containing benzoyl peroxide |
| WO2009072007A2 (en) | 2007-12-07 | 2009-06-11 | Foamix Ltd. | Carriers, formulations, methods for formulating unstable active agents for external application and uses thereof |
| CA2711703A1 (en) | 2008-01-08 | 2009-07-16 | Foamix Ltd. | Sensation modifying topical composition foam |
| FR2929278A1 (fr) | 2008-04-01 | 2009-10-02 | Centre Nat Rech Scient | Solide hybride cristallin poreux pour l'adsorption et la liberation de gaz a interet biologique. |
| US20120141384A1 (en) | 2008-05-06 | 2012-06-07 | Dov Tamarkin | Antibacterial conjugated boronic acids and pharmaceutical compositions thereof |
| WO2010044875A2 (en) | 2008-10-16 | 2010-04-22 | Novan, Inc. | Nitric oxide releasing particles for oral care applications |
| WO2010116467A1 (ja) * | 2009-03-30 | 2010-10-14 | Necディスプレイソリューションズ株式会社 | 画像表示装置、及び画像処理方法 |
| WO2011022652A1 (en) | 2009-08-21 | 2011-02-24 | Novan, Inc. | Topical gels |
| BR112012003804B1 (pt) | 2009-08-21 | 2019-02-19 | Novan, Inc. | Curativo para ferimentos, método para formar um curativo para ferimentos, e, kit de curativo para ferimento |
| WO2011047013A1 (en) | 2009-10-13 | 2011-04-21 | Novan, Inc. | Nitric oxide-releasing coatings |
| ES2541483T3 (es) | 2009-12-16 | 2015-07-21 | Shasun Pharmaceuticals Limited | Composición de hidrogel transdérmico de dexibuprofeno |
| WO2011085484A1 (en) * | 2010-01-13 | 2011-07-21 | Nitric Solutions Inc. | Antimicrobial nitric oxide compositions |
| EP2560634A1 (en) | 2010-04-23 | 2013-02-27 | Piramal Enterprises Limited | Nitric oxide releasing prodrugs of therapeutic agents |
| US8591876B2 (en) | 2010-12-15 | 2013-11-26 | Novan, Inc. | Methods of decreasing sebum production in the skin |
| EP2665763B1 (en) | 2011-01-20 | 2015-08-26 | Novan, Inc. | Temperature controlled sol-gel co-condensation |
| ES2695173T3 (es) | 2011-02-28 | 2019-01-02 | Novan Inc | Partículas de sílice modificadas con S-nitrosotiol que liberan óxido nítrico y procedimientos de fabricación de las mismas |
| WO2012153331A2 (en) | 2011-05-09 | 2012-11-15 | Topical Therapeutic Agent (Tta) Ltd. | Nitric oxide-sequestering topical formulations |
| WO2013006613A1 (en) | 2011-07-05 | 2013-01-10 | Novan, Inc. | Methods of manufacturing topical compositions and apparatus for same |
| ES2804263T3 (es) * | 2011-07-05 | 2021-02-05 | Novan Inc | Composiciones tópicas |
| ES2658897T3 (es) | 2011-08-24 | 2018-03-12 | Novan, Inc. | Macromoléculas de liberación de óxido nítrico ajustables que tienen múltiples estructuras donantes de óxido nítrico |
| WO2013063354A1 (en) | 2011-10-27 | 2013-05-02 | Novan, Inc. | Nitric oxide releasing bath compositions and methods of using the same |
| US20150111973A1 (en) | 2012-03-13 | 2015-04-23 | Novan, Inc. | Methods of modulating steroid hormone activity |
| ES2861439T3 (es) | 2012-03-14 | 2021-10-06 | Novan Inc | Composiciones farmacéuticas que liberan óxido nítrico |
| EP2885323B1 (en) | 2012-08-17 | 2023-08-30 | The University of North Carolina At Chapel Hill | Water soluble nitric oxide-releasing polyglucosamines and uses thereof |
| US9187501B2 (en) | 2012-08-28 | 2015-11-17 | The University Of North Carolina At Chapel Hill | Nitric oxide-releasing nanorods and their methods of use |
| US9855211B2 (en) | 2013-02-28 | 2018-01-02 | Novan, Inc. | Topical compositions and methods of using the same |
| US11813284B2 (en) | 2013-08-08 | 2023-11-14 | Novan, Inc. | Topical compositions and methods of using the same |
| WO2016160089A1 (en) | 2015-03-27 | 2016-10-06 | Novan, Inc. | Topical antiviral compositions, delivery systems, and methods of using the same |
| US10322082B2 (en) | 2014-07-11 | 2019-06-18 | Novan, Inc. | Topical antiviral compositions and methods of using the same |
| US10925689B2 (en) | 2014-07-14 | 2021-02-23 | Novan, Inc. | Nitric oxide releasing nail coating compositions, nitric oxide releasing nail coatings, and methods of using the same |
| EP3177262A4 (en) | 2014-08-08 | 2018-04-18 | Novan Inc. | Topical emulsions |
-
2015
- 2015-07-10 EP EP15819451.4A patent/EP3166593B1/en active Active
- 2015-07-10 BR BR112017000456-9A patent/BR112017000456B1/pt active IP Right Grant
- 2015-07-10 CA CA2954061A patent/CA2954061C/en active Active
- 2015-07-10 KR KR1020177001064A patent/KR102495101B1/ko active Active
- 2015-07-10 JP JP2017501173A patent/JP6651499B2/ja active Active
- 2015-07-10 AU AU2015287674A patent/AU2015287674B2/en active Active
- 2015-07-10 ES ES15819451T patent/ES2807200T3/es active Active
- 2015-07-10 US US15/324,332 patent/US10322081B2/en active Active
- 2015-07-10 CN CN201580048884.0A patent/CN106659675B/zh active Active
- 2015-07-10 WO PCT/US2015/039908 patent/WO2016007834A1/en not_active Ceased
- 2015-07-10 EP EP20168974.2A patent/EP3698775A1/en active Pending
Also Published As
| Publication number | Publication date |
|---|---|
| US10322081B2 (en) | 2019-06-18 |
| JP2017520594A (ja) | 2017-07-27 |
| CN106659675A (zh) | 2017-05-10 |
| JP6651499B2 (ja) | 2020-02-19 |
| CA2954061C (en) | 2023-01-24 |
| KR20170032301A (ko) | 2017-03-22 |
| US20170216197A1 (en) | 2017-08-03 |
| CA2954061A1 (en) | 2016-01-14 |
| KR102495101B1 (ko) | 2023-02-02 |
| AU2015287674A1 (en) | 2017-01-19 |
| EP3166593A4 (en) | 2018-02-07 |
| EP3166593B1 (en) | 2020-05-20 |
| AU2015287674B2 (en) | 2019-11-21 |
| EP3698775A1 (en) | 2020-08-26 |
| BR112017000456A2 (pt) | 2017-11-07 |
| WO2016007834A1 (en) | 2016-01-14 |
| EP3166593A1 (en) | 2017-05-17 |
| CN106659675B (zh) | 2023-07-04 |
| BR112017000456B1 (pt) | 2022-12-13 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2807200T3 (es) | Composiciones antivirales tópicas y procedimientos de uso de las mismas | |
| US12403087B2 (en) | Topical antiviral compositions, delivery systems, and methods of using the same | |
| JP7324244B2 (ja) | 感染症を治療するための組成物、システム、キットおよび方法 | |
| WO2016160089A1 (en) | Topical antiviral compositions, delivery systems, and methods of using the same | |
| ES2323395T3 (es) | Procedimiento y composicion para el tratamiento de irritaciones e infecciones epidermicas. | |
| US11285171B2 (en) | Nitric oxide releasing suppositories and methods of use thereof | |
| ES2986035T3 (es) | Composición para la aplicación nasal con estabilidad mejorada | |
| US10849864B2 (en) | Combinations and methods for the treatment and/or prevention of fungal infections | |
| CN103142461A (zh) | 一种阴道用凝胶剂及其制备方法 | |
| US20110070321A1 (en) | Method and Composition for Cutaneous Treatment of Herpes Simplex Infections | |
| CN110198720A (zh) | 用于治疗哺乳动物中的疱疹病毒的组合物和方法 | |
| HK40007303A (en) | Composition and method for treating herpesvirus in mammals | |
| BR112019018998A2 (pt) | tratamento de sintomas de herpes simples na pele e membrana mucosa de mamíferos |