ES2808004T3 - Métodos para clasificar pacientes con un cáncer sólido - Google Patents
Métodos para clasificar pacientes con un cáncer sólido Download PDFInfo
- Publication number
- ES2808004T3 ES2808004T3 ES17725195T ES17725195T ES2808004T3 ES 2808004 T3 ES2808004 T3 ES 2808004T3 ES 17725195 T ES17725195 T ES 17725195T ES 17725195 T ES17725195 T ES 17725195T ES 2808004 T3 ES2808004 T3 ES 2808004T3
- Authority
- ES
- Spain
- Prior art keywords
- cancer
- cells
- density
- biological markers
- tumor
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6876—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes
- C12Q1/6883—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes for diseases caused by alterations of genetic material
- C12Q1/6886—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes for diseases caused by alterations of genetic material for cancer
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6844—Nucleic acid amplification reactions
- C12Q1/6851—Quantitative amplification
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6844—Nucleic acid amplification reactions
- C12Q1/6862—Ligase chain reaction [LCR]
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/53—Immunoassay; Biospecific binding assay; Materials therefor
- G01N33/575—Immunoassay; Biospecific binding assay; Materials therefor for cancer
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/53—Immunoassay; Biospecific binding assay; Materials therefor
- G01N33/575—Immunoassay; Biospecific binding assay; Materials therefor for cancer
- G01N33/57535—Immunoassay; Biospecific binding assay; Materials therefor for cancer of the large intestine, e.g. colon, rectum or anus
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/53—Immunoassay; Biospecific binding assay; Materials therefor
- G01N33/575—Immunoassay; Biospecific binding assay; Materials therefor for cancer
- G01N33/5758—Immunoassay; Biospecific binding assay; Materials therefor for cancer involving compounds serving as markers for tumours, cancers or neoplasias, e.g. cellular determinants, receptors, heat shock/stress proteins, A-protein, oligosaccharides or metabolites
-
- G—PHYSICS
- G16—INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR SPECIFIC APPLICATION FIELDS
- G16H—HEALTHCARE INFORMATICS, i.e. INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR THE HANDLING OR PROCESSING OF MEDICAL OR HEALTHCARE DATA
- G16H50/00—ICT specially adapted for medical diagnosis, medical simulation or medical data mining; ICT specially adapted for detecting, monitoring or modelling epidemics or pandemics
- G16H50/20—ICT specially adapted for medical diagnosis, medical simulation or medical data mining; ICT specially adapted for detecting, monitoring or modelling epidemics or pandemics for computer-aided diagnosis, e.g. based on medical expert systems
-
- G—PHYSICS
- G16—INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR SPECIFIC APPLICATION FIELDS
- G16H—HEALTHCARE INFORMATICS, i.e. INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR THE HANDLING OR PROCESSING OF MEDICAL OR HEALTHCARE DATA
- G16H50/00—ICT specially adapted for medical diagnosis, medical simulation or medical data mining; ICT specially adapted for detecting, monitoring or modelling epidemics or pandemics
- G16H50/30—ICT specially adapted for medical diagnosis, medical simulation or medical data mining; ICT specially adapted for detecting, monitoring or modelling epidemics or pandemics for calculating health indices; for individual health risk assessment
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q2600/00—Oligonucleotides characterized by their use
- C12Q2600/118—Prognosis of disease development
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q2600/00—Oligonucleotides characterized by their use
- C12Q2600/158—Expression markers
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N2800/00—Detection or diagnosis of diseases
- G01N2800/50—Determining the risk of developing a disease
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N2800/00—Detection or diagnosis of diseases
- G01N2800/52—Predicting or monitoring the response to treatment, e.g. for selection of therapy based on assay results in personalised medicine; Prognosis
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N2800/00—Detection or diagnosis of diseases
- G01N2800/60—Complex ways of combining multiple protein biomarkers for diagnosis
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Engineering & Computer Science (AREA)
- Chemical & Material Sciences (AREA)
- Immunology (AREA)
- Biomedical Technology (AREA)
- Molecular Biology (AREA)
- Pathology (AREA)
- General Health & Medical Sciences (AREA)
- Urology & Nephrology (AREA)
- Hematology (AREA)
- Analytical Chemistry (AREA)
- Organic Chemistry (AREA)
- Biotechnology (AREA)
- Biochemistry (AREA)
- Microbiology (AREA)
- Physics & Mathematics (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Zoology (AREA)
- Wood Science & Technology (AREA)
- Medical Informatics (AREA)
- Public Health (AREA)
- Cell Biology (AREA)
- Medicinal Chemistry (AREA)
- Food Science & Technology (AREA)
- General Physics & Mathematics (AREA)
- Genetics & Genomics (AREA)
- Biophysics (AREA)
- Bioinformatics & Cheminformatics (AREA)
- General Engineering & Computer Science (AREA)
- Primary Health Care (AREA)
- Epidemiology (AREA)
- Databases & Information Systems (AREA)
- Data Mining & Analysis (AREA)
- Oncology (AREA)
- Hospice & Palliative Care (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Measuring Or Testing Involving Enzymes Or Micro-Organisms (AREA)
- Investigating Or Analysing Biological Materials (AREA)
- Medicines Containing Antibodies Or Antigens For Use As Internal Diagnostic Agents (AREA)
Abstract
Un método in vitro para el pronóstico del tiempo de supervivencia de un paciente que padece un cáncer sólido, cuyo método comprende los siguientes pasos: a) detectar y cuantificar dos o más marcadores biológicos indicativos del estado de la respuesta inmune de dicho paciente contra dicho cáncer, en donde cada marcador biológico indicativo del estado de la respuesta inmune se detecta in vitro y se cuantifica en una muestra tumoral obtenida de dicho paciente; b) comparar cada valor obtenido en la etapa a) para dichos dos o más marcadores biológicos con una distribución de valores obtenidos para cada uno de dichos dos o más marcadores biológicos de un grupo de referencia de pacientes que padecen dicho cáncer; c) determinar para cada valor obtenido en el paso a) para dichos dos o más marcadores biológicos el percentil de la distribución a la que corresponden los valores obtenidos en el paso a); d) calcular el valor medio aritmético o el valor medio de los percentiles de los marcadores biológicos determinados en el paso c); y e) comparar el valor medio aritmético o el valor medio de percentiles obtenidos en el paso d) con un valor medio aritmético de referencia predeterminado o un valor medio predeterminado de percentiles, cuyo valor de referencia predeterminado se correlaciona con el tiempo de supervivencia.
Description
DESCRIPCIÓN
Métodos para clasificar pacientes con un cáncer sólido
Campo de la invención
La presente invención se refiere a métodos para clasificar pacientes que padecen un cáncer sólido, particularmente a métodos para el pronóstico del tiempo de supervivencia de un paciente que padece un cáncer sólido y/o métodos para evaluar la capacidad de respuesta de un paciente que padece un cáncer sólido frente al tratamiento antitumoral.
Antecedentes de la invención
Según lo explicado detalladamente en Galon et al. (Cancer classification using the Immunoscore: a worldwide task force, J Transl Med. 2012; 10: 205), la predicción del resultado clínico en el cáncer generalmente se logra mediante una evaluación histopatológica de las muestras de tejido obtenidas durante la resección quirúrgica del tumor primario. La estadificación tradicional del tumor (clasificación AJCC/UICC-TNM) resume los datos sobre la carga tumoral (T), la presencia de células cancerosas en el drenaje y los ganglios linfáticos regionales (N) y la evidencia de metástasis (M). Sin embargo, en la actualidad se reconoce que el resultado clínico puede variar significativamente entre los pacientes dentro que están en el mismo estado de evolución del cáncer. La clasificación actual proporciona cierta información pronóstica limitada sin predecir la respuesta que va a surgir frente al tratamiento. La bibliografía reciente ha aludido a la importancia del sistema inmunitario del huésped en el control de la progresión tumoral. Por lo tanto, la evidencia apoya la noción de incluir biomarcadores inmunológicos, implementados como una herramienta para la predicción del pronóstico y la respuesta a la terapia. La acumulación de datos, recopilados de grandes cohortes de cánceres humanos, ha demostrado el impacto de la clasificación inmune, que tiene un valor pronóstico que puede aumentar la importancia de la clasificación TNM AJCC/UICC. Por lo tanto, es imprescindible comenzar a incorporar el 'Immunoscore' en la clasificación tradicional, proporcionando así una herramienta de pronóstico esencial y potencialmente predictiva. La introducción de este parámetro como biomarcador para clasificar los cánceres, como parte de la evaluación diagnóstica y pronóstica de tumores de rutina, facilitará la toma de decisiones clínicas, incluida la estratificación racional del tratamiento de pacientes.
Numerosas solicitudes de patentes han descrito métodos para el pronóstico del tiempo de supervivencia de pacientes que padecen cáncer sólido y/o métodos para evaluar la capacidad de respuesta de pacientes que padecen cánceres sólidos frente al tratamiento antitumoral, mediante la medición de biomarcadores inmunológicos. Se pueden citar, por ejemplo: los documentos WO2015007625, WO2014023706, WO2014009535, WO2013186374, WO2013107907, WO2013107900, WO2012095448, WO2012072750 y WO2007045996. Todos estos métodos proporcionan buenos resultados. Como se explica en la revisión de Galon et al. Immunity 39, 2013, 11 -26, las firmas inmunes pronósticas y predictivas se superponen en gran medida.
El documento WO2010/120814 describe, entre otros, métodos que se basan en un análisis estadístico del cuartil para el pronóstico de pacientes que padecen la enfermedad inflamatoria intestinal (EII).
Los presentes inventores han logrado aumentar aún más la precisión de los métodos para el pronóstico del tiempo de supervivencia de un paciente que padece un cáncer sólido y de los métodos para evaluar la capacidad de respuesta de un paciente que padece un cáncer sólido frente al tratamiento antitumoral.
Sumario y descripción detallada de la invención
El alcance de la presente invención se define en las reivindicaciones y cualquier información que no se encuentre dentro de las reivindicaciones se proporciona únicamente a título informativo.
La presente invención se refiere a un método in vitro para el pronóstico del tiempo de supervivencia de un paciente que padece un cáncer sólido, cuyo método comprende los siguientes pasos:
a) detectar y cuantificar dos o más marcadores biológicos indicativos del estado de la respuesta inmune de dicho paciente contra dicho cáncer, en el que cada marcador biológico indicativo del estado de la respuesta inmune se cuantifica en una muestra tumoral obtenida de dicho paciente;
b) comparar cada valor obtenido en la etapa a) para dichos dos o más marcadores biológicos con una distribución de valores obtenidos para cada uno de dichos dos o más marcadores biológicos de un grupo de referencia de pacientes que padecen dicho cáncer;
c) determinar para cada valor obtenido en el paso a) para dichos dos o más marcadores biológicos cuyo percentil de la distribución corresponde a los valores obtenidos en el paso a);
d) calcular el valor medio aritmético o el valor medio del percentil; y
e) comparar el valor medio aritmético o el valor medio del percentil obtenido en la etapa d) con un valor medio aritmético de referencia predeterminado o un valor medio predeterminado del percentil, cuyo valor de referencia predeterminado se correlaciona con el tiempo de supervivencia.
Por lo general, el tiempo de supervivencia es la supervivencia libre de enfermedad (DFS), la supervivencia libre de progresión (PFS), la supervivencia específica de la enfermedad (DSS) o la supervivencia general (OS).
La presente invención también se refiere a un método in vitro para evaluar la capacidad de respuesta de un paciente que padece un cáncer sólido frente a un tratamiento antitumoral, cuyo método comprende los siguientes pasos: a) detectar y cuantificar dos o más marcadores biológicos indicativos del estado de la respuesta inmune de dicho paciente contra dicho cáncer, en el que cada marcador biológico indicativo del estado de la respuesta inmune se detecta cuantificado in vitro en una muestra tumoral obtenida de dicho paciente;
b) comparar cada valor obtenido en el paso a) para dichos dos o más marcadores biológicos con una distribución de valores obtenidos para cada uno de dichos dos o más marcadores biológicos a partir de un grupo de referencia de pacientes que padecen dicho cáncer;
c) determinar para cada valor obtenido en el paso a) para dichos dos o más marcadores biológicos el percentil de la distribución a la que corresponden los valores obtenidos en el paso a);
d) calcular el valor medio aritmético o el valor medio del percentil; y
e) comparar el valor medio aritmético o el valor medio del percentil obtenido en la etapa d) con un valor medio aritmético de referencia predeterminado o un valor medio predeterminado del percentil, cuyo valor de referencia predeterminado se correlaciona con la respuesta frente a dicho tratamiento antitumoral.
La expresión "valor medio aritmético" debe entenderse ampliamente y abarca la media aritmética "clásica" de una muestra, que es la suma de los valores muestreados dividido por el número de elementos en la muestra y, también, la media aritmética ponderada:

Los pesos wi pueden representar una medida de la fiabilidad de la influencia sobre la media por los valores respectivos.
El valor medio se adapta particularmente cuando se cuantifican muchos marcadores biológicos (típicamente más de 5, 10, 15, 200).
Los métodos de la invención tienen ventajas únicas y múltiples:
Dichos métodos funcionan de manera muy significativa y tienen un fuerte poder discriminatorio porque los biomarcadores considerados en estos métodos tienen características específicas importantes.
Los biomarcadores considerados son biomarcadores inmunes. En particular, los biomarcadores son biomarcadores inmunes adaptativos. Una de sus características esenciales es que estos biomarcadores inmunes están asociados con el tiempo de recurrencia, con el tiempo antes de la muerte, con el tiempo antes de la respuesta al tratamiento, con la amplitud de la respuesta al tratamiento y con el tiempo prolongado durante el cual el paciente está respondiendo al tratamiento.
Una de las ventajas de los métodos de la invención es que el nivel de intensidad, expresión, densidad y cantidad de cada biomarcador está asociado con la longitud, el tiempo de supervivencia, la supervivencia prolongada y la respuesta prolongada frente al tratamiento.
Los métodos de la invención se aplican porque cuanto más altos o más bajos sean los niveles de los biomarcadores, más larga o más corta será la supervivencia y mejor o peor será entonces la respuesta frente al tratamiento.
Uno de los elementos novedosos esenciales asociados con el gran rendimiento de los métodos es que los métodos son independientes de los datos clínicos y el seguimiento de los pacientes. El percentil medio se puede calcular en función de los datos brutos y normalizados del marcador biológico, independientemente del resultado de los pacientes.
Una reacción inmune adaptativa integral se caracteriza por un conjunto de biomarcadores que, cuando se enriquecen conjuntamente, los biomarcadores mejoran la estratificación de los pacientes en categorías de supervivencia prolongada y una mejor respuesta frente al tratamiento.
La media de los valores percentiles de todos los biomarcadores, ponderados para cada biomarcador, permite una estratificación muy precisa de los pacientes en categorías de riesgo.
Típicamente, el grupo de referencia de pacientes que padecen dicho cáncer comprende al menos: 100, 200, 300, 400, 500, 1000 o 2000 pacientes.
Típicamente, los métodos de la invención se aplican a diversos órganos de origen canceroso (tales como mama, colon, recto, pulmón, cabeza y cuello, vejiga, ovario, próstata) y, también, a varios tipos de células cancerosas (adenocarcinoma, carcinoma de células escamosas, cáncer de células grandes, melanoma, etc.
Típicamente, el paciente sometido al método anterior puede sufrir un cáncer sólido seleccionado del grupo que consiste en cáncer cortical suprarrenal, cáncer anal, cáncer del conducto biliar (por ejemplo, cáncer periférico, cáncer del conducto biliar distal, cáncer del conducto biliar intrahepático), cáncer de vejiga, cáncer de hueso (p. ej., osteoblastoma, osteocrondroma, hemangioma, fibroma condromixoide, osteosarcoma, condrosarcoma, fibrosarcoma, histiocitoma fibroso maligno, tumor de células gigantes del hueso, cordoma, mieloma múltiple), cáncer del cerebro y del sistema nervioso central (p. ej., meningioma, astocitoma, oligodendrogliomas, ependimoma, gliomas, meduloblastoma, ganglioglioma, schwannoma, germinoma, craneofaringioma), cáncer de mama (p. ej., carcinoma ductal in situ, carcinoma ductal infiltrante, carcinoma lobular infiltrante, carcinoma lobular in situ, ginecomastia), cáncer cervical, cáncer colorrectal, cáncer de endometrio (por ejemplo, cáncer de endometrio, adenocarcinoma, adenocantoma, adenocarcinoma seroso papilar, células claras), cáncer de esófago, cáncer de vesícula biliar (adenocarcinoma mucinoso, carcinoma de células pequeñas), tumores carcinoides gastrointestinales (p. ej. coriocarcinoma, corioadenoma destrueno), sarcoma de Kaposi, cáncer de riñón (por ejemplo, cáncer de células renales), cáncer de laringe e hipofaringe, cáncer de hígado (por ejemplo, hemangioma, adenoma hepático, hiperplasia nodular focal, carcinoma hepatocelular), cáncer de pulmón (por ejemplo, cáncer de pulmón de células pequeñas, cáncer de pulmón de células no pequeñas), mesotelioma, plasmacitoma, cáncer de cavidad nasal y seno paranasal (p. ej., estesioneuroblastoma, granuloma de línea media), cáncer de nasofaringe, neuroblastoma, cáncer de la cavidad oral y orofaríngeo, cáncer de ovario, cáncer de páncreas, cáncer de pene, cáncer de pituitaria, cáncer de próstata, retinoblastoma, rabdomiosarcoma (p. ej., rabdomiosarcoma embrionario, rabdomiosarcoma alveolar, rabdomiosarcoma pleomórfico), cáncer de glándulas salivales, cáncer de piel (p. ej. melanoma, cáncer de piel no melanoma), cáncer de estómago, cáncer testicular (p. ej. seminoma, cáncer de células germinales no seminoma), cáncer de timo, cáncer de tiroides (por ejemplo, carcinoma folicular, carcinoma anaplásico, carcinoma poco diferenciado, carcinoma de tiroides medular), cáncer vaginal, cáncer vulvar y cáncer uterino (p. ej. leiomiosarcoma uterino).
En una realización preferida, el cáncer es un cáncer colorrectal.
Como se usa en el presente documento, la expresión "muestra de tejido tumoral" significa cualquier muestra de tumor de tejido derivada del paciente. Dicha muestra de tejido se obtiene con el propósito de una evaluación in vitro. En algunas realizaciones, la muestra de tumor puede provenir del tumor resecado del paciente. En algunas realizaciones, la muestra de tumor puede provenir de una biopsia realizada en el tumor primario del paciente o realizada en una muestra metastásica distante del tumor primario del paciente. Por ejemplo, una biopsia endoscópica realizada en el intestino del paciente afectado por un cáncer colorrectal. Típicamente, la muestra de tejido tumoral se fija en formalina y se incrusta en un fijador rígido, como la parafina (cera) o el epoxi, que se coloca en un molde y luego se endurece para producir un bloque que se corte fácilmente. Se pueden preparar después finas rodajas de material utilizando un microtomo, colocarlas en un portaobjetos de vidrio y enviarlas p. ej. a un análisis inmunohistoquímico (usando un autómata IHC como BenchMark® XT, para obtener portaobjetos teñidos). La muestra de tejido tumoral se puede usar en micromatrices, llamadas micromatrices de tejido (TMA). Las TMA consisten en bloques de parafina en los que se ensamblan hasta 1000 núcleos de tejido separados en forma de matriz para permitir un análisis histológico múltiple. Esta tecnología permite la visualización rápida de dianas moleculares en muestras de tejido a la vez, ya sea a nivel de ADN, ARN o proteína. La tecnología TMA se describe en los documentos WO2004000992, US8068988, Olli et al 2001 Human Molecular Genetics, Tzankov et al 2005, Elsevier; Kononen et al. 1198; Nature Medicine.
En algunas realizaciones, la muestra de tejido tumoral abarca (i) un tumor primario global (como un todo), (ii) una muestra de tejido del centro del tumor, (iii) una muestra de tejido del tejido que rodea directamente al tumor, cuyo tejido puede denominarse más específicamente el "margen invasivo" del tumor, (iv) islotes linfoides cercanos al tumor, (v) los ganglios linfáticos ubicados en la proximidad más cercana del tumor, (vi) una muestra de tejido tumoral recolectada antes de la cirugía (para el seguimiento de pacientes después del tratamiento, por ejemplo), y (vii) una metástasis a distancia.
Como se usa en el presente documento, el "margen invasivo" tiene su significado general en la técnica y se refiere al entorno celular que rodea el tumor. En algunas realizaciones, la muestra de tejido tumoral, independientemente de si se deriva del centro del tumor, del margen invasivo del tumor o de los ganglios linfáticos más cercanos, abarca piezas o cortes de tejido que se han extraído del centro del tumor del margen invasivo que rodea el tumor, incluso después de una resección quirúrgica del tumor o después de la recolección de una muestra de tejido para la biopsia, para una cuantificación adicional de uno o varios marcadores biológicos, especialmente a través de métodos de histología o inmunohistoquímica, a través de métodos de citometría de flujo y métodos de análisis de expresión de
genes o proteínas, incluido el análisis genómico y proteómico. La muestra de tejido tumoral puede, por supuesto, someterse a una variedad de técnicas preparativas y de almacenamiento posteriores a la recolección reconocidas (por ejemplo, fijación, almacenamiento, congelación, etc.). La muestra puede estar recién preparada, congelada, fijada (p. Ej., fijada con formalina) o embebida (p. ej., embebida en parafina). En algunas realizaciones, cuando la cuantificación del número de ganglios linfáticos que drenan el tumor se realiza en el tumor reseccionado, la muestra de tejido tumoral proviene de dicho tumor reseccionado y abarca el centro del tumor y, opcionalmente, el margen invasivo del tumor. En dichas realizaciones, la cuantificación del marcador de la respuesta inmune adaptativa se realiza típicamente por inmunohistoquímica (IHC) como se describe más adelante. En algunas realizaciones, cuando la cuantificación del número de ganglios linfáticos que drenan el tumor se realiza mediante imágenes, la muestra de tejido tumoral proviene de una biopsia. En dichas realizaciones, la cuantificación del marcador de la respuesta inmune adaptativa se realiza típicamente determinando el nivel de expresión de al menos un gen.
Típicamente, la muestra de tumor puede seleccionarse del grupo que consiste en (i) un tumor primario global (como un todo), (ii) una muestra de tejido del centro del tumor, (iii) una muestra de tejido del tejido que rodea directamente el tumor cuyo tejido puede denominarse más específicamente el "margen invasivo" del tumor, (iv) los ganglios linfáticos ubicados en la proximidad más cercana del tumor o una estructura linfoide terciaria inducida por el tumor, (v) una biopsia tumoral realizada en cualquier momento y, típicamente, cirugía previa, y (vi) una metástasis a distancia.
En una realización preferida, los dos o más marcadores biológicos se cuantifican en el centro del tumor y/o en el margen invasivo del tumor.
En una realización preferida, los dos o más marcadores biológicos se cuantifican en el centro del tumor y/o en el margen invasivo del tumor.
La muestra puede estar recién preparada, congelada, fijada (p. ej., fijada con formalina) o embebida (p. ej., embebida en parafina). En una realización particular, la muestra de tumor proviene de una biopsia realizada en un tumor del paciente.
Un ejemplo es una biopsia endoscópica realizada en el intestino del paciente que padece cáncer colorrectal o que se sospecha que padece cáncer colorrectal.
Como se pretende en este documento, un "marcador biológico" consiste en cualquier parámetro detectable, medible y cuantificable que sea indicativo del estado de la respuesta inmune del paciente con cáncer contra el tumor.
Los marcadores biológicos incluyen la presencia o el número o la densidad de células del sistema inmune en el sitio del tumor.
Los marcadores biológicos también incluyen la presencia o la cantidad de proteínas específicamente producidas por las células del sistema inmune en el sitio del tumor.
Los marcadores biológicos también incluyen la presencia o la cantidad de cualquier material biológico que sea indicativo del nivel de expresión de genes relacionados con el aumento de una respuesta inmune específica del huésped en el sitio del tumor. Por lo tanto, los marcadores biológicos incluyen la presencia o la cantidad de un ARN mensajero (ARNm) transcrito a partir de ADN genómico que codifica proteínas que son producidas específicamente por células del sistema inmune en el sitio del tumor.
Por lo tanto, los marcadores biológicos incluyen antígenos de superficie expresados específicamente por células del sistema inmune, incluidos linfocitos B, linfocitos T, células dendríticas de monocitos/macrófagos, células NK, células NKT y células NK-DC, que se reclutan dentro del tejido tumoral o en su proximidad, incluso dentro del margen invasivo del tumor y en los ganglios linfáticos más cercanos o, alternativamente, el ARNm que codifica dichos antígenos de superficie.
Ilustrativamente, los antígenos de superficie de interés utilizados como marcadores biológicos incluyen CD3, CD4, CD8 y CD45RO que se expresan por células T o subconjuntos de células T.
Por ejemplo, si la expresión del antígeno CD3, o la expresión del ARNm del mismo, se usa como marcador biológico, la cuantificación de este marcador biológico, en la etapa a) del método de acuerdo con la invención, es indicativa del nivel de la respuesta inmune adaptativa del paciente que involucra a todos los linfocitos T y células NKT.
Por ejemplo, si la expresión del antígeno CD8, o la expresión del ARNm del mismo, se usa como marcador biológico, la cuantificación de este marcador biológico, en la etapa a) del método de acuerdo con la invención, es indicativa del nivel de la respuesta inmune adaptativa del paciente con linfocitos T citotóxicos.
Por ejemplo, si la expresión del antígeno CD45RO, o la expresión del ARNm del mismo, se usa como marcador biológico, la cuantificación de este marcador biológico, en la etapa a) del método de acuerdo con la invención, es
indicativa del nivel de la respuesta inmune adaptativa del paciente con linfocitos T de memoria o linfocitos T efectores de memoria.
Sin embargo, de manera ilustrativa, las proteínas utilizadas como marcadores biológicos también incluyen proteínas citolíticas producidas específicamente por las células del sistema inmune, como la perforina, la granulisina y, también, la granzima-B.
Numerosas solicitudes de patentes han descrito una gran cantidad de marcadores biológicos indicativos del estado de la respuesta inmune que podrían usarse en los métodos de la invención.
Típicamente, se pueden usar los marcadores biológicos indicativos del estado de la respuesta inmune descritos en los documentos WO2015007625, WO2014023706, WO2014009535, WO2013186374, WO2013107907, WO2013107900, WO2012095448, WO2012072750 y WO2007045996.
Típicamente, se puede cuantificar una combinación de 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21,22, 23, 24,25, 26, 27, 28, 29, 30, 31,32, 33, 34, 35, 36, 37, 38, 39, 40, 41,42, 43, 44, 45, 46, 47, 48, 49 y 50 de marcadores biológicos distintos, preferiblemente una combinación de 2, 3, 4, 5, 6, 7, 8, 9 o 10 marcadores biológicos y, más preferiblemente, una combinación de 2, 3, 4, 5 o 6, marcadores biológicos
En una realización preferida, los marcadores biológicos indicativos del estado de la respuesta inmune son los descritos en el documento WO2007045996.
Típicamente, los marcadores biológicos que pueden usarse son la densidad celular de las células del sistema inmune.
En una realización preferida, los dos o más marcadores biológicos comprenden la densidad de las células CD3+, la densidad de las células CD8+, la densidad de las células CD45RO+, la densidad de las células GZM-B+ y/o la densidad de las células B.
En una realización más preferida, los dos o más marcadores biológicos comprenden la densidad de las células CD3+ y la densidad de las células CD8+, la densidad de las células CD3+ y la densidad de las células CD45RO+, la densidad de las células CD3+, la densidad de las células GZM-B+, la densidad de células CD8+ y la densidad de células CD45RO+, la densidad de células CD8+ y la densidad de células GZM-B+; o la densidad de las células CD45RO+ y la densidad de las células GZM-B+.
La densidad de las células B también se puede medir (véase los documentos WO2013107900 y WO2013107907). La densidad de las células DC también se puede medir (véase el documento WO2013107907).
En una realización preferida, la densidad de células del sistema inmune se cuantifica en el centro del tumor y/o en el margen invasivo del tumor.
En una realización más preferida, los dos o más marcadores biológicos comprenden la densidad de células CD3+ en el centro del tumor, la densidad de células CD8+ en el centro del tumor, la densidad de células CD3+ en el margen invasivo y la densidad de células CD8+ en el margen invasivo.
La densidad se puede medir en el "punto frío", es decir, en las regiones de la muestra tumoral donde la densidad es la más baja, o en los 2, 3, 4, 5, 6, 7, 8, 9, 10 "puntos fríos", correspondientes al área de 2 a 10 con las densidades más bajas.
La densidad también se puede medir en el "punto caliente", es decir, en las regiones donde la densidad es la más alta, o en los 2, 3, 4, 5, 6, 7, 8, 9, 10 "puntos calientes", correspondiente al área de 2 a 10 con las mayores densidades.
También se puede determinar la densidad media en toda la muestra tumoral.
Típicamente, el método descrito en el documento WO2013186374 puede usarse para cuantificar las células inmunes en la muestra de tumor.
Como se usa en el presente documento, la expresión "marcador" consiste en cualquier parámetro detectable, medible o cuantificable que sea indicativo del estado de la respuesta inmune adaptativa del sujeto. Un marcador se convierte en un "marcador biológico" con el propósito de llevar a cabo el método de la presente invención cuando se encuentra una buena correlación estadística entre (i) un aumento o una disminución del valor de cuantificación para dicho marcador y (ii) el tiempo de supervivencia en realidad observado dentro de los pacientes. Para calcular los valores de correlación para cada marcador probado y determinar así la relevancia estadística de dicho marcador como un "marcador biológico" de acuerdo con la invención, se puede usar cualquiera de los métodos estadísticos conocidos por los expertos en la materia. De manera ilustrativa, se pueden utilizar métodos estadísticos que utilizan curvas de Kaplan-Meier y/o análisis univariados que utilizan la prueba de log-rank y/o un modelo de riesgos proporcionales de Cox, como se muestra en los ejemplos de este documento. Cualquier marcador para el cual un valor de P menor que 0,05, e incluso preferiblemente menor que 10-3, 10-4, 10-5, 10-6 o 10-7 (según el análisis
univariante y multivariante (por ejemplo, la prueba de log-rank y la prueba de Cox, respectivamente) se determina que consiste en un "marcador biológico" utilizable en el método de pronóstico del cáncer de la invención. En algunas realizaciones, el marcador incluye la presencia o el número o la densidad de células del sistema inmune. En algunas realizaciones, el marcador incluye la presencia o la cantidad de proteínas específicamente producidas por las células del sistema inmune. En algunas realizaciones, el marcador incluye la presencia o la cantidad de cualquier material biológico que sea indicativo del nivel de genes relacionados con el aumento de una respuesta inmune específica del huésped. Por lo tanto, en algunas realizaciones, el marcador incluye la presencia o la cantidad del ARN mensajero (ARNm) transcrito a partir de ADN genómico que codifica proteínas, que son producidas específicamente por las células del sistema inmune. En algunas realizaciones, el marcador incluye antígenos de superficie que se expresan específicamente por células del sistema inmune, incluidos linfocitos B, linfocitos T, células dendríticas de monocitos/macrófagos, células NK, células NKT y células NK-DC o, alternativamente, la codificación de ARNm para dichos antígenos de superficie. Cuando se realiza el método de la presente invención con más de un marcador biológico, el número de marcadores biológicos distintos que se cuantifican en la etapa a) son generalmente de menos de 100 marcadores distintos y, en la mayoría de las realizaciones, de menos de 50 marcadores distintos. El número de marcadores biológicos distintos que es necesario para obtener un pronóstico preciso y confiable, utilizando el método de la presente invención, puede variar notablemente según el tipo de técnica utilizada para la cuantificación. Ilustrativamente, se puede encontrar una alta significación estadística con una combinación de un pequeño número de marcadores biológicos, cuando el método de la presente invención se realiza mediante la detección inmunohistoquímica in situ de marcadores de proteínas de interés, en particular cuando se realiza una cuantificación separada de dichos marcadores, tanto en el centro del tumor (CT) como en el margen invasivo (IM). Ilustrativamente, se obtuvo una alta significación estadística con solo un marcador o una combinación de dos marcadores, como se describe en los EJEMPLOS. Más ilustrativamente, también se encontró una alta significación estadística con un pequeño número de marcadores biológicos, cuando el método de la presente invención se realizaba mediante análisis de expresión génica de marcadores génicos de interés. Sin desear limitarse a ninguna teoría en particular, los inventores creen que se alcanza una relevancia altamente estadística (valor P inferior a 10-3) cuando se realiza el método de la presente invención, usando un análisis de expresión génica para la cuantificación de marcadores biológicos, y mediante el uso de una combinación de diez marcadores biológicos distintos y, más preferiblemente, una combinación de quince marcadores biológicos distintos, lo más preferiblemente veinte marcadores biológicos distintos, o más. En algunas realizaciones, se determina el nivel de 1; 2; 3; 4; 5; 6; 7; 8; 9; 10; 11; 12; 13; 14; 15; 16; 17; 18; 19; 20; 21; 22; 23; 24; 25; 26; 27; 28; 29; 30; 31; 32; 33; 34; 35; 36; 37; 38; 39; 40; 41; 42; 43; 44; 45; 46; 47; 48; 49; o 50 marcadores. En la presente memoria descriptiva, el nombre de cada uno de los diversos marcadores biológicos de interés se refiere al nombre reconocido internacionalmente del gen correspondiente, tal como se encuentra en las secuencias de genes reconocidas internacionalmente y en las bases de datos de secuencias de proteínas, incluida la base de datos del Comité de Nomenclatura de Genes HUGO. En la presente memoria descriptiva, el nombre de cada uno de los diversos marcadores biológicos de interés también puede referirse al nombre internacionalmente reconocido del gen correspondiente, como se encuentra en la base de datos de secuencias de genes y secuencias de proteínas Genbank reconocida internacionalmente. A través de estas bases de datos de secuencias reconocidas internacionalmente, el experto en la técnica puede recuperar las secuencias de ácido nucleico y de aminoácidos correspondientes a cada uno de los marcadores biológicos de interés descritos en este documento.
Los marcadores biológicos indicativos del estado de la respuesta inmune pueden comprender el nivel de expresión de uno o más genes o proteínas correspondientes enumerados en la Tabla 9 del documento WO2007045996 que son: 18s, ACE, ACTB, AGTR1, AGTR2, APC, APOA1, ARF1, AXIN1, BAX, BCL2, BCL2L1, CXCR5, BMP2, BRCA1, BTLA, C3, CASP3, CASP9, CCL1, CCL11, CCL13, CCL16, CCL17, CCL18, CCL19, CCL2, CCL20, CCL21, CCL22, CCL23, CCL24, CCL25, CCL26, CCL27, CCL28, CCL3, CCL5, CCL7, CCL8, CCNB1, CCND1, CCNE1, CCR1, CCR10, CCR2, CCR3, CCR4, CCR5, CCR6, CCR7, CCR8, CCR9, CCRL2, CD154, CD19, CD1a, CD2, CD226, CD244, PDCD1LG1, CD28, CD34, CD36, CD38, CD3E, CD3G, CD3Z, CD4, CD40LG, CD5, CD54, CD6, CD68, CD69, CLIP, CD80, CD83, SLAMF5, CD86, CD8A, CDH1, CDH7, CDK2, CDK4, CDKN1A, CDKN1B, CDKN2A, CDKN2B, CEACAM1, COL4A5, CREBBP, CRLF2, CSF1, CSF2, CSF3, CTLA4, CTNNB1, CTSC, CX3CL1, CX3CR1, CXCL1, CXCL10, CXCL11, CXCL12, CXCL13, CXCL14, CXCL16, CXCL2, CXCL3, CXCL5, CXCL6, CXCL9, CXCR3, CXCR4, CXCR6, CYP1A2, CYP7A1, DCC, DCN, DEFA6, DICER1, DKK1, Dok-1, Dok-2, DOK6, DVL1, E2F4, EBI3, ECE1, ECGF1, EDN1, EGF, EGFR, EIF4E, CD105, ENPEP, ERBB2, EREG, FCGR3A„ CGR3B, FN1, FOXP3, FYN, FZD1, GAPD, GLI2, GNLY, GOLPH4, GRB2, GSK3B, GSTP1, GUSB, GZMA, GZMB, GZMH, GZMK, HLA-B, HLA- C, HLA-, MA, HLA-DMB, HLA-DOA, HLA-DOB, HLA-DPA1, HLA-DQA2, HLA-DRA, HLX1, HMOX1, HRAS, HSPB3, HUWE1, ICAM1, ICAM-2, ICOS, ID1, ifna1, ifna17, ifna2, ifna5, ifna6, ifna8, IFNAR1, IFNAR2, IFNG, IFNGR1, IFNGR2, IGF1, IHH, IKBKB, IL10, IL12A, IL12B, IL12RB1, IL12RB2, IL13, IL13RA2, IL15, IL15RA, IL17, IL17R, IL17RB, IL18, IL1A, IL1B, IL1R1, IL2, IL21, IL21R, IL23A, IL23R, IL24, IL27, IL2RA, IL2RB, IL2RG, IL3, IL31RA, IL4, IL4RA, IL5, IL6, IL7, IL7RA, IL8, CXCR1, CXCR2, IL9, IL9R, IRF1, ISGF3G, ITGA4, ITGA7, integrina alfa E (antígeno CD103, linfocito de la mucosa humana, antígeno 1; polipéptido alfa), Gen hCG33203, ITGB3, JAK2, JAK3, KLRB1, KLRC4, KLRF1, KLRG1, KRAS, LAG3, LAIR2, LEF1, LGALS9, LILRB3, LRP2, LTA, SLAMF3, MADCAM1, MADH3, MADH7,MAF, MAP2K1, MDM2, MICA, MICB, MKI67, MMP12, MMP9, MTA1, MTSS1, MYC, MYD88, MYH6, NCAM1, NFATC1, NKG7, NLK, NOS2A, P2X7, PDCD1, PECAM-„ CXCL4, PGK1, PIAS1, PIAS2, PIAS3, PIAS4, PLAT, PML, PP1A, CXCL7, PPP2CA, PRF1, PROM1, PSMB5, PTCH, PTGS2, PTP4A3, PTPN6, PTPRC, RAB23, RAC/RHO, RAC2, RAF, RB1, RBL1, REN, Drosha, SELE, SELL, SELP, SERPINE1, SFRP1, SIRP beta 1, SKI, SLAMF1, SLAMF6, SLAMF7, SLAMF8, SMAD2, SMAD4, SMO, SMOH,
SMURF1, SOCS1, SOCS2, SOCS3, SOCS4, SOCS5, SOCS6, SOCS7, SOD1, SOD2, SOD3, SOS1, SOX17, CD43, ST14, STAM, STAT1, STAT2, STAT3, STAT4, STAT5A, STAT5B, STAT6, STK36, TAP1, TAP2, TBX21, TCF7, TERT, TFRC, TGFA, TGFB1, TGFBR1, TGFBR2, TIM-3, TLR1, TLR10, TLR2, TLR3, TLR4, TLR5, TLR6, TLR7, TLR8, TLR9, TNF, TNFRSF10A, TNFRSF11A, TNFRSF18, TNFRSF1A, TNFRSF1B, OX-40, TNFRSF5, TNFRSF6, TNFRSF7, TNFRSF8, TNFRSF9, TNFSF10, TNFSF6, TOB1, TP53, TSLP, VCAM1, VEGF, WIF1, WNT1, WNT4, XCL1, XCR1, ZAP70 y ZIC2.
En la presente memoria descriptiva, el nombre de cada uno de los genes de interés se refiere al nombre internacionalmente reconocido del gen correspondiente, tal como se encuentra en las secuencias de genes y bases de datos de secuencias de proteínas reconocidas internacionalmente, incluida la base de datos del Comité de Nomenclatura de Genes HUGO. En la presente memoria descriptiva, el nombre de cada uno de los genes de interés también puede referirse al nombre reconocido internacionalmente del gen correspondiente, como se encuentra en la base de datos de secuencias de genes Genbank reconocida internacionalmente. A través de estas bases de datos de secuencias reconocidas internacionalmente, todo experto en la técnica puede recuperar el ácido nucleico de cada uno de los genes de interés descritos en este documento.
En una realización preferida, los marcadores biológicos indicativos del estado de la respuesta inmune son los descritos en el documento WO2014023706:
En esta realización, se evalúa un nivel de expresión EL1 de un solo gen representativo de la respuesta inmune adaptativa humana y de un solo gen representativo de la respuesta inmunosupresora humana (un par de genes) en el método de la invención. Preferiblemente se usan de uno a tres genes de cada uno y, más preferiblemente, de uno o dos genes de cada uno. Un número limitado de genes de cada tipo proporciona resultados buenos y confiables y es fácil de implementar. Particularmente, un solo valor de referencia es suficiente para cada uno de los dos genes. Cuanto mayor es el número de genes, más sofisticado es el valor de referencia. A continuación se dan ejemplos de determinación de valores de referencia.
El uso de más genes que uno o dos pares de genes es más difícil de implementar y más costoso y requiere mucho tiempo, pero ofrece otras ventajas. Por ejemplo, si la evaluación del nivel de expresión de un gen es errónea, el resultado general se compensa con la reserva de los otros genes del mismo tipo (respuesta inmune adaptativa humana o respuesta inmunosupresora).
Como se usa en el presente documento, la expresión "gen representativo de la respuesta inmune adaptativa" se refiere a cualquier gen que sea expresado por una célula que es actora de la respuesta inmune adaptativa en el tumor o que contribuya al establecimiento de la respuesta inmune adaptativa en el tumor. La respuesta inmune adaptativa, también llamada "respuesta inmune adquirida", comprende la estimulación dependiente de antígeno de los subtipos de células T, la activación de células B y la producción de anticuerpos. Por ejemplo, las células de la respuesta inmune adaptativa incluyen, entre otras, células T citotóxicas, células T de memoria, células Th1 y Th2, macrófagos activados y células dendríticas activadas, células NK y células NKT. Por consiguiente, un gen representativo de la respuesta inmune adaptativa puede seleccionarse típicamente del grupo de genes co modulados para la inmunidad adaptativa Th1, para la respuesta citotóxica o para la respuesta de memoria, y puede codificar un marcador de superficie de células Th1, una interleucina (o un receptor de interleucina), o una quimiocina o (un receptor de quimiocina).
En una realización particular, el gen representativo de la respuesta inmune adaptativa se selecciona del grupo que consiste en:
- la familia de quimiocinas y receptores de quimiocinas que consisten en: CXCL13, CXCL9, CCL5, CCR2, CXCL10, CXCL11, CXCR3, CCL2 y CX3CL1,
- la familia de las citocinas que consta de: IL15,
- la familia TH1 compuesta por: IFNG, IRF1, STAT1, STAT4 y TBX21
- la familia de receptores de membrana de linfocitos que consiste en: ITGAE, CD3D, CD3E, CD3G, CD8A, CD247, CD69 e ICOS,
- la familia de moléculas citotóxicas que consiste en: GNLY, GZMH, GZMA, GZMB, GZMK, GZMM y PRF1, y la quinasa LTK.
Los genes preferidos o las proteínas correspondientes, por proporcionar los mejores resultados para la respuesta de un paciente frente al tratamiento como se muestra a continuación en la tabla 5, se muestran en la tabla 1:

Tabla 1
Como se usa en el presente documento, la expresión "gen representativo de la respuesta inmunosupresora" se refiere a cualquier gen que es expresado por una célula que es un actor de la respuesta inmunosupresora en el tumor o que contribuye al establecimiento de la respuesta inmunosupresora en el tumor. Por ejemplo, la respuesta inmunosupresora comprende:
- co-inhibición de la estimulación dependiente de antígeno de los subtipos de células T: genes CD276, CTLA4, PDCD1, CD274, TIM-3 o VTCN1 (B7H4),
- inactivación de macrófagos y células dendríticas e inactivación de células NK: genes TSLP, CD1A, o VEGFA - expresión del marcador de células madre cancerosas, diferenciación y/u oncogénesis: PROM1, IHH
- expresión de proteínas inmunosupresoras producidas en el entorno tumoral: genes PF4, REN, VEGFA.
Por ejemplo, las células de la respuesta inmunosupresora incluyen células dendríticas inmaduras (CD1A), células T reguladoras (células Treg) y células Th17 que expresan el gen IL17A.
En consecuencia, un gen representativo de la respuesta inmune adaptativa puede seleccionarse típicamente del grupo de los genes inmunes adaptativos co-modulados, mientras que los genes inmunosupresores pueden ser representativos de la inactivación de las células inmunes (por ejemplo, células dendríticas) y pueden contribuir a la inducción de una respuesta inmunosupresora.
En una realización particular, el gen o las proteínas correspondientes representativas de la respuesta inmunosupresora se seleccionan del grupo que consiste en los genes mostrados en la Tabla 2 a continuación:

Tabla 2
Dichos genes son preferidos porque proporcionan los mejores resultados para la respuesta de un paciente frente al tratamiento.
En condiciones preferidas para implementar la invención, se selecciona un gen representativo de la respuesta inmune adaptativa del grupo que consiste en GNLY, CXCL13, CX3CL1, CXCL9, ITGAE, CCL5, GZMH, IFNG, CCR2, CD3D, CD3E, CD3G, CD8A, CXCL10, CXCL11, GZMA, GZMB, GZMK, GZMM, IL15, IRF1, LTK, PRF1, STAT1, CD69, CD247, IcOs, CXCR3, StAt4, CCL2 y TBX21 y un gen representativo de la respuesta inmunosupresora se selecciona del grupo que consiste en PF4, REN, VEGFA, TSLP, IL17A, PROM1 IHH, CD1A, CTLA4, PDCD1, CD276, CD274, TIM-3 y VTCN1 (B7H4).
Debido a que algunos genes se encuentran con mayor frecuencia cuando se combina un gen adaptativo y un gen inmunosupresor, los genes más preferidos son:
- genes representativos de la respuesta inmune adaptativa: CD3G, CD8A, CCR2 y GZMA
- genes representativos de la respuesta inmunosupresora: REN, IL17A, CTLA4 y PDCD1.
En condiciones preferidas adicionales para implementar la invención, un gen representativo de la respuesta inmune adaptativa y un gen representativo de la respuesta inmunosupresora se seleccionan respectivamente de los grupos que consisten en los genes de las Tablas 1 y 2 anteriores.
Las combinaciones preferidas de dos pares de genes (total de 4 genes) son:
- CCR2, CD3G, IL17A y REN y
- CD8A, CCR2, REN y PDCD1.
La elección precisa de los genes seleccionados para su uso en el presente proceso puede depender del tipo de tratamiento contemplado para el paciente. Por ejemplo, se contempla para un paciente los genes seleccionados del grupo que consiste en CX3CL1 IL15, CD247, CD3G, CD8A, PRF1, CcL5 y TBX21 para la respuesta inmunosupresora, preferiblemente CX3CL1 e IL15 y el gen CTLA4 para la respuesta inmune adaptativa se preferirán cuando se use un medicamento, tal como un anticuerpo monoclonal que funcione activando el sistema inmune como Ipilimumab, también conocido como MDX-010 o MDX-101, comercializado como Yervoy®.
Los genes seleccionados del grupo que consiste en IL15 y GZMA para la respuesta inmune adaptativa, y el gen VEGFA para la respuesta inmunosupresora se preferirán cuando sea contemplado para un paciente un tratamiento tal como un anticuerpo que inhiba el factor de crecimiento endotelial vascular A (VEGF-A) como el bevacizumab comercializado como Avastin®.
Consideraciones similares se aplican, por ejemplo, para el par de genes GZMA - PDCD1 (también designado como CD279), cuando se contempla para un paciente en un tratamiento como un anticuerpo cuya diana es PD-1 como BMS-936558.
En una realización preferida, los marcadores biológicos indicativos del estado de la respuesta inmune son los descritos en el documento WO2014009535:
Los marcadores biológicos indicativos del estado de la respuesta inmune pueden comprender el nivel de expresión de uno o más genes del grupo que consiste en CCR2, CD3D, CD3E, CD3G, CD8A, CXCL10, CXCL11, GZMA, GZMB, GZMK, GZMM, IL15, IRF1, PRF1, STAT1, CD69, ICOS, CXCR3, STAT4, CCL2 y TBX21.
En una realización preferida, los marcadores biológicos indicativos del estado de la respuesta inmune son los descritos en el documento WO2012095448:
Los marcadores biológicos indicativos del estado de la respuesta inmune pueden comprender el nivel de expresión de uno o más genes del grupo que consiste en GZMH, IFNG, CXCL13, GNLY, LAG3, ITGAE, CCL5, CXCL9, PF4, IL17A, TSLP, REN, IHH, PROM1 y VEGFA.
En una realización preferida, los marcadores biológicos indicativos del estado de la respuesta inmune son los descritos en el documento WO2012072750:
Los marcadores biológicos indicativos del estado de la respuesta inmune pueden comprender el nivel de expresión de un grupo de miARN que comprende: miR.609, miR.518c, miR.520f, miR.220a, miR.362, miR.29a, miR.660, miR.603, miR.558, miR519b, miR.494, miR.130a o miR.639.
Métodos generales para cuantificar marcadores biológicos.
Cualquiera de los métodos conocidos por los expertos en la técnica para cuantificar los tipos celulares, un marcador biológico de tipo proteína o ácido nucleico incluido en el presente documento pueden usarse para realizar el método pronóstico de cáncer de la invención. Por lo tanto, cualquiera de las técnicas estándar y no estándar (emergentes) reconocidas en la técnica para detectar y cuantificar una proteína o un ácido nucleico en una muestra puede aplicarse fácilmente.
La expresión de un marcador biológico usado en la invención puede evaluarse mediante cualquiera de una amplia variedad de métodos reconocidos para detectar la expresión de un ácido nucleico o proteína transcrita. Los ejemplos no limitativos de tales métodos incluyen métodos inmunológicos para la detección de proteínas secretadas, de la superficie celular, citoplasmáticas o nucleares, métodos de purificación de proteínas, ensayos de función o actividad de proteínas, métodos de hibridación de ácidos nucleicos, métodos de transcripción inversa de ácidos nucleicos y métodos de amplificación de ácidos nucleicos.
En una realización preferida, la expresión de un marcador se evalúa usando un anticuerpo (por ejemplo, un anticuerpo radiomarcado, marcado con cromóforo, marcado con fluoróforo, polímero-esqueleto o anticuerpo marcado con enzima), un derivado de anticuerpo (por ejemplo, un anticuerpo conjugado con un sustrato o con la proteína o ligando de un par proteína-ligando {por ejemplo, biotina-estreptavidina}), o un fragmento de anticuerpo (por ejemplo, un anticuerpo de cadena sencilla, un dominio hipervariable de anticuerpo aislado, etc.) que se une específicamente con una proteína marcadora o fragmento de la misma, que incluye una proteína marcadora que ha sufrido la totalidad o una parte de su modificación postraduccional normal.
En ciertas realizaciones, puede cuantificarse un marcador biológico, o un conjunto de marcadores biológicos, con cualquiera de los métodos de inmunohistoquímica conocidos en la técnica.
Típicamente, para un análisis posterior, una sección delgada del tumor se incuba primero con anticuerpos marcados dirigidos contra un marcador biológico de interés. Después del lavado, los anticuerpos marcados que se unen a dicho marcador biológico de interés se revelan mediante la técnica apropiada, dependiendo del tipo de marcador que lleve el anticuerpo marcado, p. ej. un marcador radiactivo, fluorescente o enzimático. El marcado múltiple se puede realizar simultáneamente.
La inmunohistoquímica típicamente incluye los siguientes pasos: i) fijar la muestra de tejido tumoral con formalina, ii) incrustar dicha muestra de tejido tumoral en parafina, iii) cortar dicha muestra de tejido tumoral en secciones para su tinción, iv) incubar dichas secciones con la pareja de unión específica para la proteína de punto de control inmune de interés, v) enjuagar dichas secciones, vi) incubar dicha sección con un anticuerpo secundario típicamente biotinilado y vii) revelar el complejo antígeno-anticuerpo típicamente con el complejo avidina-biotina-peroxidasa. En consecuencia, la muestra de tejido tumoral se incuba en primer lugar con las parejas de unión que son de interés para la proteína de punto de control inmune. Después del lavado, los anticuerpos marcados que se unen a la proteína de punto de control inmune de interés se revelan mediante la técnica apropiada, dependiendo del tipo de marcador que lleve el anticuerpo marcado, p. ej. un marcador radiactivo, fluorescente o enzimático. Un marcado múltiple se puede realizar simultáneamente. Alternativamente, el método de la presente invención puede usar un anticuerpo secundario acoplado a un sistema de amplificación (para intensificar la señal de tinción) y moléculas enzimáticas. Dichos anticuerpos secundarios acoplados están disponibles comercialmente, p. ej. de Dako, EnVision System. Se puede usar una contratinción, p. ej. Hematoxilina y Eosina, DAPI, Hoechst. Se pueden lograr otros métodos de tinción usando cualquier método o sistema adecuado como sería evidente para todo experto en la materia, incluidos sistemas automatizados, semiautomatizados o manuales.
Por ejemplo, uno o más marcadores se pueden unir al anticuerpo, lo que permite la detección de la proteína diana (es decir, los marcadores biológicos). Los ejemplos de marcadores incluyen isótopos radiactivos, fluoróforos, ligandos, agentes quimioluminiscentes, enzimas y combinaciones de los mismos. Los ejemplos no limitantes de marcadores que pueden conjugarse con ligandos de afinidad primarios y/o secundarios incluyen tintes fluorescentes o metales (por ejemplo, fluoresceína, rodamina, ficoeritrina, fluorescamina), tintes cromóforos (por ejemplo, rodopsina), compuestos quimioluminiscentes (por ejemplo, luminal, imidazol) y proteínas bioluminiscentes (p. ej.,
luciferina, luciferasa), haptenos (p. ej., biotina). Una variedad de otros fluorescentes y cromóforos útiles se describen en Stryer L (1968) Science 162: 526-533 y Brand L y Gohlke J R (1972) Annu. Rev. Biochem. 41:843-868. Los ligandos de afinidad también pueden marcarse con enzimas (por ejemplo, peroxidasa de rábano picante, fosfatasa alcalina, beta-lactamasa), radioisótopos (p. ej. 3H, 14C, 32P, 35S o 125I) y partículas (por ejemplo, oro). Los diferentes tipos de marcadores pueden conjugarse con un ligando de afinidad utilizando diversos agentes químicos, p. ej. la reacción de amina o la reacción de tiol. Sin embargo, se pueden usar otros grupos reactivos que las aminas y los tioles, p. ej. aldehídos, ácidos carboxílicos y glutamina. Se conocen varios métodos de tinción enzimática en la técnica para detectar las proteínas de interés. Por ejemplo, las interacciones enzimáticas se pueden visualizar utilizando diferentes enzimas como la peroxidasa, la fosfatasa alcalina o diferentes cromógenos como DAB, AEC o Fast Red. En algunas realizaciones, el marcador es un punto cuántico. Por ejemplo, los puntos cuánticos (Qdots) se están volviendo cada vez más útiles en una lista creciente de aplicaciones que incluyen inmunohistoquímica, citometría de flujo y ensayos basados en placas y, por lo tanto, se pueden usar junto con esta invención. Los nanocristales Qdot tienen propiedades ópticas únicas que incluyen una señal extremadamente brillante para detectar la sensibilidad y la cuantificación; alta fotoestabilidad para imágenes y análisis. Se necesita una única fuente de excitación, y una gama creciente de conjugados los hace útiles en una amplia gama de aplicaciones basadas en células. Los bioconjugados Qdot se caracterizan por rendimientos cuánticos comparables a los tintes tradicionales más brillantes disponibles. Además, estos fluoróforos basados en puntos cuánticos absorben 10-1000 veces más luz que los tintes tradicionales. La emisión de los puntos cuánticos Qdot subyacentes es estrecha y simétrica, lo que significa que la superposición con otros colores se minimiza, lo que resulta en un sangrado mínimo en los canales de detección adyacentes y una diafonía atenuada, a pesar del hecho de que se pueden usar muchos más colores simultáneamente. En otros ejemplos, el anticuerpo puede conjugarse con péptidos o proteínas que pueden detectarse mediante una pareja o anticuerpo de unión marcado. En un ensayo indirecto de IHC, es necesario que el anticuerpo secundario o segunda pareja de unión detecte la unión de la primera pareja de unión, ya que no está marcada.
En algunas realizaciones, las muestras teñidas resultantes se fotografían cada una usando un sistema para visionar la señal detectable y adquiriendo una imagen, tal como una imagen digital de la tinción. Los métodos para la adquisición de imágenes son habituales para los expertos en la materia. Por ejemplo, una vez que la muestra se ha teñido, se puede usar cualquier dispositivo de imagen óptico o no óptico para detectar la mancha o el marcador del biomarcador, como, por ejemplo, microscopios ópticos verticales o invertidos, escaneo con microscopios confocales, cámaras, microscopios electrónicos de barrido o de túnel, microscopios de sonda de barrido y detectores infrarrojos de imágenes. En algunos ejemplos, la imagen se puede capturar digitalmente. Las imágenes obtenidas se pueden usar para determinar cuantitativa o semicuantitativamente la cantidad de proteína de punto de control inmune en la muestra, o el número absoluto de células positivas para el fabricante de interés, o la superficie de las células positivas para el fabricante de interés. Varios sistemas automatizados de procesamiento, barrido y análisis de muestras adecuados para su uso con IHC están disponibles en la técnica. Dichos sistemas pueden incluir tinción automática y barrido por microscópico, análisis computarizado de imágenes, comparación de secciones en serie (para controlar la variación en la orientación y el tamaño de una muestra), generación de informes digitales y archivo y seguimiento de muestras (como portaobjetos en las que las secciones de tejido están situadas). Los sistemas de visualización celular están disponibles comercialmente y combinan microscopios de luz convencionales con sistemas de procesamiento de imágenes digitales para realizar análisis cuantitativos en células y tejidos, incluidas las muestras inmunoteñidas. Ver, por ejemplo, el sistema CAS-200 (Becton, Dickinson & Co.). En particular, la detección puede hacerse manualmente o mediante técnicas de procesamiento de imágenes que involucran procesadores informáticos y software. Usando dicho software, por ejemplo, las imágenes se pueden configurar, calibrar, estandarizar y/o validar en función de factores que incluyen, por ejemplo, la calidad de la mancha o la intensidad de la mancha, utilizando procedimientos conocidos por los expertos en la materia (ver, por ejemplo, la Publicación de Patente de EE. UU. Publicada No. US20100136549). La imagen puede analizarse cuantitativa o semicuantitativamente y puntuarse en función de la intensidad de tinción de la muestra. La histoquímica cuantitativa o semicuantitativa se refiere al método de barrido y calificación de muestras que se han sometido a la histoquímica, para identificar y cuantificar la presencia del biomarcador especificado (es decir, la proteína del punto de control inmune). Los métodos cuantitativos o semicuantitativos pueden emplear un software de visualización para detectar densidades de tinción o cantidad de tinción o métodos para detectar la tinción por el ojo humano, en los que un operador capacitado clasificaría los resultados numéricamente. Por ejemplo, las imágenes pueden analizarse cuantitativamente utilizando algoritmos de recuento de píxeles y patrones de reconocimiento de tejidos (p. ej. el Software Aperio Spectrum, plataforma de análisis cuantitativo automatizado (plataforma AQUA®) o Tribvn con el software Ilastic y Calopix) y otros métodos estándar que miden o cuantifican o semicuantifican el grado de tinción; véase, por ejemplo, la patente de EE.UU. N° 8.023.714; Pat. de EE.UU. N° 7.257.268; Pat. de EE.UU. N° 7.219.016; Pat. de EE.UU. N° 7.646.905; Publicación de Patente de los Estados Unidos N° US20100136549 y 20110111435; Camp et al. (2002) Nature Medicine, 8: 1323-1327; Bacus et al. ( 1997) Analyt Quant Cyto1Histol, 19: 316-328). Se puede calcular y puntuar una relación entre la mancha positiva fuerte (como la mancha marrón) y la suma del área manchada total. La cantidad del biomarcador detectado (es decir, la proteína del punto de control inmune) se cuantifica y se da como un porcentaje de píxeles positivos y/o una puntuación. Por ejemplo, la cantidad puede cuantificarse como un porcentaje de píxeles positivos. En algunos ejemplos, la cantidad se cuantifica como el porcentaje de área manchada, por ejemplo, el porcentaje de píxeles positivos. Por ejemplo, una muestra puede tener al menos, o aproximadamente al menos, o aproximadamente 0, 1%, 2%, 3%, 4%, 5%, 6%, 7%, 8%, 9%, 10%, 11%, 12%, 13%, 14%, 15%, 16%, 17%, 18%, 19%, 20%, 21%, 22%, 23%, 24%, 25%, 26%, 27%, 28% , 29%, 30%,
31%, 32%, 33%, 34%, 35%, 40%, 45%, 50%, 55%, 60%, 65%, 70%, 75%, 80%, 85 %, 90%, 95% o más píxeles positivos en comparación con el área de tinción total. Por ejemplo, la cantidad puede cuantificarse como un número absoluto de células positivas para el fabricante de interés. En algunas realizaciones, se da una puntuación a la muestra que es una representación numérica de la intensidad o cantidad de la tinción histoquímica de la muestra, y representa la cantidad de biomarcador diana (por ejemplo, la proteína del punto de control inmune) presente en la muestra. La densidad óptica o los valores de porcentaje de área pueden recibir una puntuación escalada, por ejemplo en una escala entera.
Por lo tanto, en algunas realizaciones, el método de la presente invención comprende los pasos que consisten en i) proporcionar una o más rebanadas de tejido de inmunotinción obtenidas por un sistema automatizado de tinción de portaobjetos utilizando un pareja de unión capaz de interactuar selectivamente con el marcador biológico, ii) proceder a la digitalización de los portaobjetos del paso i) mediante la captura de un barrido de alta resolución, iii) detectar el corte de la sección de tejido en la imagen digital, iv) proporcionar una cuadrícula de referencia de tamaño con unidades distribuidas uniformemente que tengan una misma superficie, siendo dicha cuadrícula adaptada al tamaño de la sección del tejido a analizar, y v) detectar, cuantificar y medir la intensidad o el número absoluto de células teñidas en cada unidad.
Las técnicas de análisis múltiples de tejido son particularmente útiles para cuantificar varias proteínas de punto de control inmune en la muestra de tejido tumoral. Dichas técnicas deben permitir medir al menos cinco, o al menos diez o más biomarcadores a partir de una sola muestra de tejido tumoral. Además, es ventajoso para la técnica preservar la localización del biomarcador y ser capaz de distinguir la presencia de biomarcadores en células cancerosas y no cancerosas. Dichos métodos incluyen la inmunohistoquímica en capas (L-IHC), barrido de expresión en capas (LES) o la inmunotransferencia de tejido múltiple (MTI) que se enseña, por ejemplo, en las patentes de EE.UU. Nos. 6.602.661, 6.969.615, 7.214.477 y 7.838.222; Publ. de EE. UU. No. 2011/0306514; y en Chung & Hewitt, Meth Mol Biol, Prot Blotting Detect, Kurlen & Scofield, eds. 536: 139-148, 2009, cada referencia enseñando que se pueden usar hasta 8, hasta 9, hasta 10, hasta 11 o más imágenes de una sección de tejido en membranas, papeles, filtros y otros elementos similares. Las membranas recubiertas útiles para llevar a cabo el proceso L-IHC/MTI están disponibles en 20/20 GeneSystems, Inc. (Rockville, MD).
En algunas realizaciones, el método L-IHC se puede realizar en cualquiera de una variedad de muestras de tejido, ya sean recién preparadas o conservadas. Las muestras incluyeron biopsias con aguja gruesa que se fijaron habitualmente en formalina normal tamponada al 10% y se procesaron en el departamento de patología. Se cortaron secciones estándar de tejido de cinco pm de espesor de los bloques de tejido en portaobjetos cargados que se usaron para L-IHC. Por lo tanto, L-IHC permite el análisis de múltiples marcadores en una sección de tejido al obtener copias de moléculas transferidas desde la sección de tejido a membranas plurales recubiertas de bioafinidad para producir esencialmente copias de "imágenes" de tejido. En el caso de una sección de parafina, la sección de tejido se desparafinará como se conoce en la técnica, por ejemplo, exponiendo la sección a xileno o un sustituto de xileno como NEO-CLEAR® y soluciones graduadas de etanol. La sección se puede tratar con una proteinasa, como papaína, tripsina, proteinasa K y similares. Luego, una pila de un sustrato de membrana que comprende, por ejemplo, varias láminas de una estructura polimérica recubierta de 10 pm de grosor con poros de 0,4 pm de diámetro canaliza moléculas de tejido, tal como proteínas, a través de la pila, para después colocarlas en la sección de tejido. El movimiento de las moléculas de fluido y tejido está configurado para ser esencialmente perpendicular a la superficie de la membrana. El sándwich de la sección, las membranas, los papeles espaciadores, los papeles absorbentes, el peso, etc. pueden exponerse al calor para facilitar el movimiento de las moléculas del tejido al apilamiento de la membrana. Una porción de las proteínas del tejido se captura en cada una de las membranas recubiertas de bioafinidad de la pila (disponible en 20/20 GeneSystems, Inc., Rockville, MD). Por lo tanto, cada membrana comprende una copia del tejido y puede sondearse para un biomarcador diferente utilizando técnicas de inmunotransferencia estándar, lo que permite la expansión abierta de un perfil marcador como se realiza en una sola sección de tejido. Como la cantidad de proteína puede ser menor en las membranas, más distal en la pila del tejido, lo que puede surgir, por ejemplo, en diferentes cantidades de moléculas en la muestra de tejido, se pueden incluir en el procedimiento diferente movilidad de las moléculas liberadas de la muestra de tejido, diferente afinidad de unión de las moléculas a las membranas, la duración de la transferencia, etc., la normalización de los valores, la ejecución de los controles, la evaluación de los niveles transferidos de las moléculas de tejido, etc., para corregir los cambios que ocurren dentro y con las membranas y entre ellas mismas, y permitir una comparación directa de la información dentro, con y entre las membranas. Por lo tanto, la proteína total se puede determinar por membrana utilizando, por ejemplo, cualquier medio para cuantificar proteínas, como biotinilando moléculas disponibles, como por ej., proteínas, utilizando un reactivo y método estándar, y luego revelando la biotina unida exponiendo la membrana a una avidina o estreptavidina marcada; una mancha de proteína, como Blot fastStain, Ponceau Red, manchas azules brillantes, etc., como se conoce en la técnica.
En algunas realizaciones, los presentes métodos utilizan la tecnología de Impresión Múltiple de Tejido (MTI) para medir biomarcadores, en la que el método conserva un tejido de biopsia concreto al permitir múltiples biomarcadores, en algunos casos al menos seis biomarcadores.
En algunas realizaciones, existen sistemas de análisis de tejido múltiple alternativos que también pueden emplearse como parte de la presente invención. Una de estas técnicas es el sistema de ensayo de Monitoreo de Reacción
Seleccionada (SRM) basado en la espectrometría de masas ("Liquid Tissue" disponible de OncoPlexDx (Rockville, MD)). Esa técnica se describe en la patente de EE.UU. No. 7.473.532.
En algunas realizaciones, el método de la presente invención utilizó la técnica múltiple IHC desarrollada por GE Global Research (Niskayuna, NY). Esa técnica se describe en las Pub. de EE.UU. Nos. 2008/0118916 y 2008/0118934. Allí, el análisis secuencial se realiza en muestras biológicas que contienen múltiples dianas, incluidos los pasos de unir una sonda fluorescente a la muestra seguido de la detección de la señal, luego la inactivación de la sonda seguido de la unión de la sonda a otra diana, la detección e inactivación, y la continuación de este proceso hasta que todas las dianas hayan sido detectadas.
En algunas realizaciones, se pueden realizar imágenes de tejido múltiple cuando se usa fluorescencia (por ejemplo, fluoróforos o puntos cuánticos) en las que la señal se puede medir con un sistema de visualización multiespectral. La visualización multiespectral es una técnica en la que se recopila información espectroscópica en cada píxel de una imagen y los datos resultantes se analizan con un software de procesamiento de imágenes espectrales. Por ejemplo, el sistema puede tomar una serie de imágenes a diferentes longitudes de onda que se pueden seleccionar de forma electrónica y continua y luego utilizar con un programa de análisis diseñado para manejar dichos datos. De este modo, el sistema puede obtener información cuantitativa de múltiples colorantes simultáneamente, incluso cuando los espectros de los colorantes se superponen mucho o cuando se localizan conjuntamente, o se producen en el mismo punto de la muestra, siempre que las curvas espectrales sean diferentes. Muchos materiales biológicos emiten fluorescencia automáticamente o emiten luz de baja energía cuando son excitados por una luz de mayor energía. Esta señal puede dar como resultado imágenes y datos de menor contraste. Las cámaras de alta sensibilidad sin capacidad de visualización de imágenes multiespectrales solo aumentan la señal de autofluorescencia junto con la señal de fluorescencia. Las imágenes multiespectrales pueden desmezclar o separar la autofluorescencia del tejido y, por lo tanto, aumentar la relación señal/ruido alcanzable. Brevemente, la cuantificación se puede realizar mediante los siguientes pasos: i) proporcionar una micromatriz de tejido tumoral (TMA) obtenido del paciente, ii) las muestras de TMA se tiñen con anti-anticuerpos que tienen especificidad de las proteínas de punto de control inmunitario de interés, iii ) el portaobjetos de TMA se tiñe aún más con un marcador de células epiteliales para ayudar en la segmentación automática del tumor y el estroma, iv) el portaobjetos de TMA se escanea usando un sistema de visualización multiespectral, v) las imágenes escaneadas se procesan usando un software de análisis de imagen automatizado (p. ej. Perkin Elmer Technology) que permite la detección, cuantificación y segmentación de tejidos específicos a través de potentes algoritmos de reconocimiento de patrones. El algoritmo de aprendizaje automático generalmente fue entrenado previamente para segmentar el tumor del estroma e identificar las células marcadas.
La determinación de un nivel de expresión de un gen en una muestra de tumor obtenida de un paciente puede implementarse mediante un panel de técnicas habituales en la técnica.
Típicamente, el nivel de expresión del gen se evalúa determinando la cantidad de ARNm producida por ese gen.
Los métodos para determinar la cantidad de ARNm son habituales en la técnica. Por ejemplo, el ácido nucleico contenido en las muestras (por ejemplo, células o tejidos preparados del paciente) se extrae primero de acuerdo con métodos estándar, por ejemplo usando enzimas líticas o soluciones químicas o se extrae mediante resinas de unión a ácido nucleico siguiendo las instrucciones del fabricante. El ARNm extraído de este modo se detecta por hibridación (p. ej., análisis de transferencia Northern) y/o amplificación (por ejemplo, RT-PCR). Preferiblemente, se prefiere RT-PCR cuantitativa o semicuantitativa. La RT-PCR cuantitativa o semicuantitativa en tiempo real es particularmente ventajosa.
Otros métodos de amplificación incluyen la reacción en cadena de la ligasa (LCR), la amplificación mediada por transcripción (TMA), la amplificación por desplazamiento de cadena (SDA) y la amplificación basada en secuencias de ácido nucleico (NASBA), secuenciación cuantitativa de nueva generación de ARN (NGS).
Los ácidos nucleicos que comprenden al menos 10 nucleótidos y que exhiben complementariedad de secuencia u homología con el ARNm de interés en este documento encuentran utilidad como sondas de hibridación o cebadores de amplificación. Se entiende que tales ácidos nucleicos no necesitan ser completamente idénticos, pero típicamente son al menos aproximadamente 80% idénticos con una región homóloga de tamaño comparable, más preferiblemente 85% idénticos e incluso más preferiblemente 90-95% idénticos. En ciertas realizaciones, será ventajoso usar ácidos nucleicos en combinación con medios apropiados, tales como un marcador detectable, para detectar la hibridación. Se conoce una amplia variedad de indicadores apropiados en la técnica que incluyen ligandos fluorescentes, radiactivos, enzimáticos u otros (por ejemplo, avidina/biotina).
Las sondas comprenden típicamente ácidos nucleicos monocatenarios de entre 10 y 1000 nucleótidos de longitud, por ejemplo de entre 10 y 800, más preferiblemente de entre 15 y 700, típicamente de entre 20 y 500 nucleótidos. Los cebadores son típicamente ácidos nucleicos monocatenarios más cortos, de entre 10 y 25 nucleótidos de longitud, diseñados para coincidir perfectamente o casi perfectamente con el ácido nucleico de interés a amplificar. Las sondas y los cebadores son "específicos" para los ácidos nucleicos con los que hibridan, es decir, preferiblemente hibridan en condiciones de hibridación de alta rigurosidad (correspondiente a la temperatura de fusión más alta Tm, por ejemplo, 50% de formamida, 5x o 6x SCC. SCC es NaCl 0,15 M, citrato de Na 0,015 M).
Los ácidos nucleicos que pueden usarse como cebadores o sondas en el método de amplificación y detección anterior pueden ensamblarse como un kit. Tal kit incluye cebadores de consenso y sondas moleculares. Un kit preferido también incluye los componentes necesarios para determinar si se ha producido la amplificación. Un kit también puede incluir, por ejemplo, tampones de PCR y enzimas; secuencias de control positivas, cebadores de control de reacción; e instrucciones para amplificar y detectar las secuencias específicas.
En una realización particular, la expresión de un marcador biológico usado en la invención puede evaluarse marcando el biomarcador (en su ADN, ARN o proteína) con un código de barras digital de oligonucleótidos, y para medir o contar el número de códigos de barras.
En una realización particular, los métodos de la invención comprenden los pasos de proporcionar ARN totales extraídos de células de cúmulo y someter los ARN a amplificación e hibridación a sondas específicas, más particularmente por medio de una RT-PCR cuantitativa o semicuantitativa.
Las sondas realizadas con los métodos descritos pueden usarse para la detección de ácido nucleico, como los procedimientos de hibridación in situ (ISH) (por ejemplo, hibridación fluorescente in situ (FISH), hibridación in situ cromogénica (CISH) e hibridación in situ de plata (SISH)) o hibridación genómica comparativa (CGH).
La hibridación in situ (ISH) implica poner en contacto una muestra que contiene la secuencia de ácido nucleico diana (p. ej., la secuencia de ácido nucleico genómica diana) en el contexto de una preparación de cromosoma metafásico o interfásico (como una muestra de células o tejidos montada en un portaobjetos) con una marcador sonda específicamente hibridable o específico para la secuencia de ácido nucleico diana (por ejemplo, la secuencia de ácido nucleico genómico diana). Los portaobjetos se pretratan opcionalmente, por ejemplo, para eliminar la parafina u otros materiales que puedan interferir con la hibridación uniforme. La muestra y la sonda se tratan, por ejemplo, calentando para desnaturalizar los ácidos nucleicos bicatenarios. La sonda (formulada en un tampón de hibridación adecuado) y la muestra se combinan, bajo condiciones y durante un tiempo suficiente que permita que se produzca la hibridación (típicamente para alcanzar el equilibrio). La preparación del cromosoma se lava para eliminar el exceso de sonda, y la detección del marcado específico de la diana del cromosoma se realiza utilizando técnicas estándar.
Por ejemplo, se puede detectar una sonda biotinilada usando avidina marcada con fluoresceína o avidina-fosfatasa alcalina. Para la detección de fiuorocromo, el fiuorocromo se puede detectar directamente, o las muestras se pueden incubar, por ejemplo, con avidina conjugada con isotiocianato de fluoresceína (FITC). La amplificación de la señal de FITC puede efectuarse, si fuera necesario, mediante incubación con anticuerpos antiavidina de cabra conjugados con biotina, el lavado y una segunda incubación con avidina conjugada con FITC. Para la detección por actividad enzimática, las muestras pueden incubarse, por ejemplo, con estreptavidina, lavarse, incubarse con fosfatasa alcalina conjugada con biotina, lavarse nuevamente y preequilibrarse (por ejemplo, en tampón de fosfatasa alcalina (AP)). Para una descripción general de los procedimientos de hibridación in situ, ver, por ejemplo, la patente de EE.UU. No. 4.888.278.
Se conocen numerosos procedimientos para FISH, CISH y SISH en la técnica. Por ejemplo, los procedimientos para realizar FISH se describen en las patentes de EE.UU. Nos. 5.447.841; 5.472.842; y 5.427.932; y, por ejemplo, en Pinkel et al., Proc. Natl. Acad. Sci. 83:2934-2938, 1986; Pinkel et al., Proc. Natl. Acad. Sci. 85: 9138-9142, 1988; y Lichter et al., Proc. Natl. Acad. Sci. 85:9664-9668, 1988. CISH se describe, por ejemplo, en Tanner et al., Am. J. Pathol. 157: 1467-1472, 2000 y la patente de EE.UU. No. 6.942.970. Se proporcionan métodos de detección adicionales en la patente de EE.Uu. No. 6.280.929.
Se pueden emplear numerosos reactivos y esquemas de detección junto con los procedimientos FISH, CISH y SISH para mejorar la sensibilidad, la resolución u otras propiedades deseables. Como se discutió anteriormente, las sondas marcadas con fluoróforos (incluidos los tintes fluorescentes y QUANTUM DOTS®) pueden detectarse directamente de forma óptica cuando se realiza FISH. Alternativamente, la sonda puede marcarse con una molécula no fluorescente, como un hapteno (como los siguientes ejemplos no limitantes: biotina, digoxigenina, DNP y varios oxazoles, pirrazoles, tiazoles, nitroarilos, benzofurazanos, triterpenos, ureas, tioureas, rotenonas, cumarina, compuestos a base de courmarina, podofilotoxina, compuestos a base de podofilotoxina y combinaciones de los mismos), ligando u otros restos indirectamente detectables. Las sondas marcadas con tales moléculas no fluorescentes (y las secuencias de ácido nucleico diana a las que se unen) se pueden detectar al poner en contacto la muestra (por ejemplo, la muestra de células o tejidos a la que se une la sonda) con un reactivo de detección marcado, como un anticuerpo (o receptor u otro pareja de unión específica) específico para el hapteno o ligando elegido. El reactivo de detección puede marcarse con un fluoróforo (por ejemplo, QUANTUM DOT®) o con otro resto detectable indirectamente, o puede ponerse en contacto con uno o más agentes de unión específicos adicionales (por ejemplo, anticuerpos secundarios o específicos) que pueden marcarse con un fluoróforo.
En otros ejemplos, la sonda o el agente de unión específico (como un anticuerpo, por ejemplo, un anticuerpo primario, receptor u otro agente de unión) se marca con una enzima que es capaz de convertir una composición fluorogénica o cromogénica en una señal fluorescente detectable, coloreada o detectable de otra manera (por ejemplo, como en el depósito de partículas metálicas detectables en SISH). Como se indicó anteriormente, la enzima se puede unir directa o indirectamente a través de un enlazante a la sonda relevante o al reactivo de
detección. Ejemplos de reactivos adecuados (p. ej., reactivos de unión) y productos químicos (p. ej., agentes químicos enlazantes y de unión) se describen en las publicaciones de solicitud de patente de EE. UU. Nos.
2006/0246524; 2006/0246523 y 2007/0117153.
Los expertos en la materia apreciarán que al seleccionar adecuadamente los pares de agentes de unión específicos de sonda marcadas, se pueden producir esquemas de detección múltiples para facilitar la detección de múltiples secuencias de ácidos nucleicos diana (por ejemplo, secuencias de ácidos nucleicos diana genómico) en un solo ensayo (por ejemplo, en una sola muestra de células o tejidos o en más de una muestra de células o tejidos). Por ejemplo, una primera sonda que corresponde a una primera secuencia diana puede marcarse con un primer hapteno, como biotina, mientras que una segunda sonda que corresponde a una segunda secuencia diana puede marcarse con un segundo hapteno, como DNP. Después de la exposición de la muestra a las sondas, las sondas unidas se pueden detectar poniendo en contacto la muestra con un primer agente de unión específico (en este caso, avidina marcada con un primer fluoróforo, por ejemplo, un primer QUANTUM DOT® espectralmente distinto, por ejemplo, que emita a 585 mn) y un segundo agente de unión específico (en este caso un anticuerpo anti-DNP, o fragmento de anticuerpo, marcado con un segundo fluoróforo (por ejemplo, un segundo QUANTUM DOT® espectralmente distinto, por ejemplo, que emita a 705 mn). Se pueden agregar otros pares de sondas/agentes de unión al esquema de detección múltiple usando otros fluoróforos espectralmente distintos. Se pueden prever numerosas variaciones directas e indirectas (un paso, dos pasos o más), todas las cuales son adecuadas en el contexto de las sondas y ensayos descritos.
Las sondas comprenden típicamente ácidos nucleicos monocatenarios de entre 10 y 1000 nucleótidos de longitud, por ejemplo de entre 10 y 800, más preferiblemente de entre 15 y 700, típicamente de entre 20 y 500. Los cebadores son típicamente ácidos nucleicos monocatenarios más cortos, de entre 10 y 25 nucleótidos de longitud, diseñados para coincidir perfectamente o casi perfectamente con un ácido nucleico de interés para amplificar. Las sondas y los cebadores son "específicos" para los ácidos nucleicos con los que hibridan, es decir, preferiblemente se hibridan en condiciones de hibridación de alta rigurosidad (correspondiente a la temperatura de fusión más alta Tm, por ejemplo, 50% de formamida, 5x o 6x SCC. SCC es NaCl 0,15 M, citrato de Na 0,015 M).
Los cebadores o sondas de ácido nucleico utilizados en el método de amplificación y detección anterior pueden ensamblarse como un kit. Tal kit incluye cebadores de consenso y sondas moleculares. Un kit preferido también incluye los componentes necesarios para determinar si se ha producido la amplificación. El kit también puede incluir, por ejemplo, tampones de PCR y enzimas; secuencias de control positivo, cebadores de control de reacción; e instrucciones para amplificar y detectar las secuencias específicas.
En una realización particular, los métodos de la invención comprenden los pasos de proporcionar ARN totales extraídos de células de cúmulo y someter los ARN a amplificación e hibridación a sondas específicas, más particularmente por medio de una RT-PCR cuantitativa o semicuantitativa.
En otra realización preferida, el nivel de expresión se determina mediante análisis de chips de ADN. Tales chips de ADN o micromatrices de ácido nucleico consisten en diferentes sondas de ácido nucleico que están unidas químicamente a un sustrato, que puede ser un microchip, un portaobjetos de vidrio o una cuenta del tamaño de una microesfera. Un microchip puede estar constituido por polímeros, plásticos, resinas, polisacáridos, sílice o materiales a base de sílice, carbono, metales, vidrios inorgánicos o nitrocelulosa. Las sondas comprenden ácidos nucleicos tales como ADNc u oligonucleótidos que pueden tener de aproximadamente 10 a aproximadamente 60 pares de bases. Para determinar el nivel de expresión, una muestra de un sujeto de prueba, opcionalmente sometida primero a una transcripción inversa, se marca y se pone en contacto con la micromatriz en condiciones de hibridación, lo que conduce a la formación de complejos entre ácidos nucleicos diana que son complementarios con las secuencias de sonda unidas a la superficie de micromatrices. Los complejos hibridados marcados se detectan y pueden cuantificarse o semicuantificarse. El marcado se puede lograr por varios métodos, p. ej. mediante el uso de un marcado radiactivo o fluorescente. Muchas variantes de la tecnología de hibridación de micromatrices están disponibles para los expertos en la materia (véase, por ejemplo, la revisión de Hoheisel, Nature Reviews, Genetics, 2006, 7: 200-210).
El nivel de expresión de un gen puede expresarse como nivel de expresión absoluto o nivel de expresión normalizado. Ambos tipos de valores pueden usarse en el presente método. El nivel de expresión de un gen se expresa preferiblemente como el nivel de expresión normalizado cuando se usa PCR cuantitativa como método de evaluación del nivel de expresión porque pequeñas diferencias al comienzo de un experimento podrían proporcionar grandes diferencias después de varios ciclos.
Típicamente, los niveles de expresión se normalizan corrigiendo el nivel de expresión absoluto de un gen y comparando su expresión con la expresión de un gen que no es relevante para determinar la etapa de cáncer del paciente, por ejemplo, un gen de mantenimiento que se exprese constitutivamente. Los genes adecuados para la normalización incluyen genes de mantenimiento como el gen actina ACTB, el gen ribosómico 18S, GUSB, PGK1 y TFRC. Esta normalización permite comparar el nivel de expresión de una muestra, por ejemplo, una muestra de paciente con el nivel de expresión de otra muestra, o comparar muestras de diferentes fuentes.
En la presente memoria descriptiva, el nombre de cada uno de los genes de interés se refiere al nombre internacionalmente reconocido del gen correspondiente, tal como se encuentra en las bases de datos de secuencias de genes y secuencias de proteínas reconocidas internacionalmente, incluida la base de datos del Comité de Nomenclatura de Genes HUGO. En la presente memoria descriptiva, el nombre de cada uno de los genes de interés también puede referirse al nombre reconocido internacionalmente del gen correspondiente, como se encuentra en la base de datos de secuencias de genes Genbank reconocida internacionalmente. A través de estas bases de datos de secuencias reconocidas internacionalmente, cada experto en la técnica puede recuperar el ácido nucleico de cada uno de los genes de interés descritos en este documento.
El método de pronóstico del cáncer de la invención se puede realizar con una combinación de genes siempre que la combinación comprenda al menos un gen representativo de la respuesta inmune adaptativa y al menos un gen representativo de la respuesta inmunosupresora. El número de genes que pueden usarse en el presente método solo está limitado por el número de genes biológicos distintos de interés que están prácticamente disponibles en el momento de llevar a cabo el método. En consecuencia, en una realización, una combinación de 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21,22,23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41,42, 43, 44, 45, 46, 47,48, 49 y 50 genes distintos se cuantifican, preferiblemente una combinación de 2, 3, 4, 5, 6, 7, 8, 9 o 10 genes y, más preferiblemente, una combinación de 2, 3, 4, 5 o 6 genes. Sin embargo, el número de genes combinados que se requieren para alcanzar una alta relevancia estadística (p. ej. el valor P inferior a 10-3) dependerá de la técnica utilizada para cuantificar la combinación de genes. El número de genes utilizados como genes representativos de la respuesta inmune adaptativa y el número de genes utilizados como genes representativos de la respuesta inmunosupresora pueden ser iguales o diferentes.
En las condiciones preferidas de implementación de la invención, se usa un número aproximadamente proporcionado de genes de cada tipo (respuesta inmune adaptativa y respuesta inmunosupresora), por ejemplo 2 de cada uno, o tres de cada uno, o 5 de un tipo y 6 del otro tipo.
La determinación de un nivel de expresión de un gen en una muestra de tumor obtenida de un paciente puede implementarse mediante un panel de técnicas habituales en la técnica.
Típicamente, el nivel de expresión de un gen se evalúa determinando la cantidad de ARNm producido por este gen. Por lo tanto, un objeto de la presente solicitud es un método para detectar pacientes con un cáncer definido anteriormente que comprende determinar el nivel de expresión ELA de uno o varios genes representativos de la respuesta inmune adaptativa humana o el nivel de expresión ELI de uno o varios genes representativos de la respuesta inmunosupresora humana determinando la cantidad de ARNm correspondiente a dichos genes.
Los métodos para determinar una cantidad de ARNm son habituales en la técnica. Por ejemplo, el ácido nucleico contenido en las muestras (por ejemplo, células o tejidos preparados del paciente) se extrae primero de acuerdo con métodos estándar, por ejemplo usando enzimas líticas o soluciones químicas o se extrae mediante resinas de unión a ácidos nucleicos siguiendo las instrucciones del fabricante. El ARNm extraído de este modo se detecta por hibridación (p. ej., análisis de transferencia Northern) y/o amplificación (por ejemplo, RT-PCR). Preferiblemente se prefiere RT-PCR cuantitativa o semicuantitativa. La RT-PCR cuantitativa o semicuantitativa a tiempo real es particularmente ventajosa.
Otros métodos de amplificación incluyen la reacción en cadena de la ligasa (LCR), la amplificación mediada por transcripción (TMA), la amplificación por desplazamiento de cadena (SDA) y la amplificación basada en secuencias de ácido nucleico (NASBA), la secuenciación cuantitativa de nueva generación de ARN (NGS).
Los ácidos nucleicos que comprenden al menos 10 nucleótidos y que exhiben complementariedad de secuencia u homología con el ARNm de interés en el presente documento encuentran utilidad como sondas de hibridación o cebadores de amplificación. Se entiende que tales ácidos nucleicos no necesitan ser completamente idénticos, pero típicamente son al menos aproximadamente 80% idénticos con la región homóloga de tamaño comparable, más preferiblemente 85% idénticos e incluso más preferiblemente 90-95% idénticos. En ciertas realizaciones, será ventajoso usar ácidos nucleicos en combinación con medios apropiados, tales como un marcador detectable, para detectar la hibridación. Se conoce una amplia variedad de indicadores apropiados en la técnica que incluyen ligandos fluorescentes, radiactivos, enzimáticos u otros (por ejemplo, avidina/biotina).
Las sondas comprenden típicamente ácidos nucleicos monocatenarios de entre 10 y 1000 nucleótidos de longitud, por ejemplo de entre 10 y 800, más preferiblemente de entre 15 y 700, típicamente de entre 20 y 500 nucleótidos. Los cebadores son típicamente ácidos nucleicos monocatenarios más cortos, de entre 10 y 25 nucleótidos de longitud, diseñados para coincidir perfectamente o casi perfectamente con un ácido nucleico de interés para amplificar. Las sondas y los cebadores son "específicos" para los ácidos nucleicos con los que se hibridan, es decir, preferiblemente se hibridan en condiciones de hibridación de alta rigurosidad (correspondientes a la temperatura de fusión más alta Tm, por ejemplo, 50% de formamida, 5x o 6x SCC. SCC es NaCl 0,15 M, citrato de Na 0,015 M). Los ácidos nucleicos que pueden usarse como cebadores o sondas en el método de amplificación y detección anterior pueden ensamblarse como un kit. Tal kit incluye cebadores de consenso y sondas moleculares. Un kit preferido también incluye los componentes necesarios para determinar si se ha producido la amplificación. Un kit
también puede incluir, por ejemplo, tampones de PCR y enzimas; secuencias de control positivo, cebadores de control de reacción; e instrucciones para amplificar y detectar las secuencias específicas.
En una realización particular, los métodos de la invención comprenden los pasos de proporcionar ARN totales extraídos de células de cúmulo y someter los ARN a amplificación e hibridación a sondas específicas, más particularmente por medio de una RT-PCR cuantitativa o semicuantitativa.
Las sondas realizadas con los métodos descritos pueden usarse para la detección de ácido nucleico, como los procedimientos de hibridación in situ (ISH) (por ejemplo, hibridación fluorescente in situ (FISH), hibridación in situ cromogénica (CISH) e hibridación in situ de plata (SISH) ) o hibridación genómica comparativa (CGH).
La hibridación in situ (ISH) implica poner en contacto una muestra que contiene la secuencia de ácido nucleico diana (p. ej., la secuencia de ácido nucleico genómica diana) en el contexto de una preparación de cromosoma metafásico o interfásico (como una muestra de células o tejidos montada en un portaobjetos) con una marcador sonda específicamente hibridable o específico para la secuencia de ácido nucleico diana (por ejemplo, secuencia de ácido nucleico genómica diana). Los portaobjetos se pretratan opcionalmente, por ejemplo, para eliminar la parafina u otros materiales que puedan interferir con la hibridación uniforme. La muestra y la sonda se tratan, por ejemplo, calentando para desnaturalizar los ácidos nucleicos bicatenarios. La sonda (formulada en un tampón de hibridación adecuado) y la muestra se combinan, bajo condiciones y durante el tiempo suficiente para permitir que se produzca la hibridación (típicamente para alcanzar el equilibrio). La preparación del cromosoma se lava para eliminar el exceso de sonda, y la detección del marcado específico de la diana del cromosoma se realiza utilizando técnicas estándar.
Por ejemplo, se puede detectar una sonda biotinilada usando avidina marcada con fluoresceína o avidina-fosfatasa alcalina. Para la detección del fluorocromo, el fluorocromo se puede detectar directamente o las muestras se pueden incubar, por ejemplo, con avidina conjugada con fluoresceína isotiocianato (FITC). La amplificación de la señal de FITC puede efectuarse, si fuera necesario, mediante incubación con anticuerpos antiavidina de cabra conjugados con biotina, lavado y una segunda incubación con avidina conjugada con FITC. Para la detección por actividad enzimática, las muestras pueden incubarse, por ejemplo, con estreptavidina, lavarse, incubarse con fosfatasa alcalina conjugada con biotina, lavarse nuevamente y preequilibrarse (por ejemplo, en tampón de fosfatasa alcalina (AP)). Para una descripción general de los procedimientos de hibridación in situ, ver, por ejemplo, la patente de EE.UU. No. 4.888.278.
Se conocen numerosos procedimientos para FISH, CISH y SISH en la técnica. Por ejemplo, los procedimientos para realizar FISH se describen en las patentes de EE.UU. Nos. 5.447.841; 5.472.842; y 5.427.932; y, por ejemplo, en Pinkel et al., Proc. Natl. Acad. Sci. 83:2934-2938, 1986; Pinkel et al., Proc. Natl. Acad. Sci. 85: 9138-9142, 1988; y Lichter et al., Proc. Natl. Acad. Sci. 85:9664-9668, 1988. CISH se describe en, por ejemplo, Tanner et al., Am. J. Pathol. 157: 1467-1472, 2000 y la patente de EE.UU. No. 6.942.970. Se proporcionan métodos de detección adicionales en la patente de EE.UU. No. 6.280.929.
Se pueden emplear numerosos reactivos y esquemas de detección junto con los procedimientos FISH, CISH y SISH para mejorar la sensibilidad, la resolución u otras propiedades deseables. Como se discutió anteriormente, las sondas marcadas con fluoróforos (incluidos los tintes fluorescentes y QUANTUM DOTS®) pueden detectarse directamente de forma óptica cuando se realiza FISH. Alternativamente, la sonda puede marcarse con una molécula no fluorescente, como un hapteno (como los siguientes ejemplos no limitantes: biotina, digoxigenina, DNP y varios oxazoles, pirrazoles, tiazoles, nitroarilos, benzofurazanos, triterpenos, ureas, tioureas, rotenonas, cumarina, compuestos a base de cumarina, podofilotoxina, compuestos a base de podofilotoxina y combinaciones de los mismos), ligando u otro resto indirectamente detectable. Las sondas marcadas con tales moléculas no fluorescentes (y las secuencias de ácido nucleico diana a las que se unen) se pueden detectar al poner en contacto la muestra (por ejemplo, la muestra de células o tejidos a la que se une la sonda) con un reactivo de detección marcado, como p. ej. un anticuerpo (o receptor u otro pareja de unión específica) específico para el hapteno o ligando elegido. El reactivo de detección puede marcarse con un fluoróforo (por ejemplo, QUANTUM DOT®) o con otro resto detectable indirectamente, o puede ponerse en contacto con uno o más agentes de unión específicos adicionales (por ejemplo, anticuerpos secundarios o específicos), que pueden marcarse con un fluoróforo.
En otros ejemplos, la sonda o el agente de unión específico (como un anticuerpo, por ejemplo, un anticuerpo primario, receptor u otro agente de unión) se marca con una enzima capaz de convertir una composición fluorogénica o cromogénica en una señal fluorescente detectable, coloreada o detectable de otra manera (por ejemplo, como en el depósito de partículas metálicas detectables en SISH). Como se indicó anteriormente, la enzima se puede unir directa o indirectamente a través de un enlazante a la sonda relevante o al reactivo de detección. Se describen ejemplos de reactivos adecuados (por ejemplo, reactivos de unión) y productos químicos (por ejemplo, productos químicos de unión y enlazantes) en las Publicaciones de Solicitud de Patentes de los Estados Unidos Nos. 2006/0246524; 2006/0246523 y 2007/0117153.
Los expertos en la materia apreciarán que, al seleccionar adecuadamente los pares de agentes de unión específicos de sondas marcadas, se pueden producir esquemas de detección múltiples para facilitar la detección de múltiples secuencias de ácidos nucleicos diana (por ejemplo, secuencias de ácido nucleico diana genómico) en un solo
ensayo (por ejemplo, en una sola muestra de células o tejidos o en más de una muestra de células o tejidos). Por ejemplo, una primera sonda que corresponda a una primera secuencia diana puede marcarse con un primer hapteno, como la biotina, mientras que una segunda sonda que corresponda a una segunda secuencia diana puede marcarse con un segundo hapteno, como DNP. Después de la exposición de la muestra a las sondas, las sondas unidas se pueden detectar poniendo en contacto la muestra con un primer agente de unión específico (en este caso, avidina marcada con un primer fluoróforo, por ejemplo, un primer QUANTUM DOT® espectralmente distinto, por ejemplo, que emita a 585 mn) y un segundo agente de unión específico (en este caso un anticuerpo anti-DNP, o fragmento de anticuerpo, marcado con un segundo fluoróforo (por ejemplo, un segundo QUANTUM DOT® espectralmente distinto, por ejemplo, que emita a 705 mn). Se pueden agregar pares de sondas/agentes de unión adicionales al esquema de detección múltiple usando otros fluoróforos espectralmente distintos. Se pueden prever numerosas variaciones directas e indirectas (un paso, dos pasos o más), todas las cuales son adecuadas en el contexto de las sondas y ensayos descritos.
Las sondas comprenden típicamente ácidos nucleicos monocatenarios de entre 10 y 1000 nucleótidos de longitud, por ejemplo de entre 10 y 800, más preferiblemente de entre 15 y 700, típicamente de entre 20 y 500. Los cebadores son típicamente ácidos nucleicos monocatenarios más cortos, de entre 10 y 25 nucleótidos de longitud, diseñados para coincidir perfectamente o casi perfectamente con un ácido nucleico de interés para amplificar. Las sondas y los cebadores son "específicos" para los ácidos nucleicos con los que se hibridan, es decir, preferiblemente se hibridan en condiciones de hibridación de alta rigurosidad (correspondientes a la temperatura de fusión más alta Tm, por ejemplo, 50% de formamida, 5x o 6x SCC. SCC es NaCl 0,15 M, citrato de Na 0,015 M).
Los cebadores o sondas de ácido nucleico utilizados en el método de amplificación y detección anterior pueden ensamblarse como un kit. Tal kit incluye cebadores de consenso y sondas moleculares. Un kit preferido también incluye los componentes necesarios para determinar si se ha producido la amplificación. El kit también puede incluir, por ejemplo, tampones de PCR y enzimas; secuencias de control positivo, cebadores de control de reacción; e instrucciones para amplificar y detectar las secuencias específicas.
En una realización particular, los métodos de la invención comprenden los pasos de proporcionar ARN totales extraídos de células de cúmulo y someter los ARN a la amplificación e hibridación en sondas específicas, más particularmente por medio de una RT-PCR cuantitativa o semicuantitativa.
En otra realización preferida, el nivel de expresión se determina mediante el análisis de un chip de ADN. Tal chip de ADN o micromatriz de ácido nucleico consiste en diferentes sondas de ácido nucleico que están unidas químicamente a un sustrato, que puede ser un microchip, un portaobjetos de vidrio o una cuenta del tamaño de una microesfera. Un microchip puede estar constituido por polímeros, plásticos, resinas, polisacáridos, sílice o materiales a base de sílice, carbono, metales, vidrios inorgánicos o nitrocelulosa. Las sondas comprenden ácidos nucleicos tales como ADNc u oligonucleótidos que pueden tener de aproximadamente 10 a aproximadamente 60 pares de bases. Para determinar el nivel de expresión, una muestra de un sujeto de prueba, opcionalmente sometida primero a una transcripción inversa, se marca y se pone en contacto con la micromatriz en condiciones de hibridación, lo que conduce a la formación de complejos entre ácidos nucleicos diana que son complementarios con las secuencias de sonda unidas a la superficie de micromatrices. Los complejos hibridados marcados se detectan y pueden cuantificarse o semicuantificarse. El marcado se puede lograr por varios métodos, p. ej. mediante el uso de un marcado radiactivo o fluorescente. Muchas variantes de la tecnología de hibridación de micromatrices están disponibles para los expertos en la materia (véase, por ejemplo, la revisión de Hoheisel, Nature Reviews, Genetics, 2006, 7: 200-210).
El nivel de expresión de un gen puede expresarse como un nivel de expresión absoluto o nivel de expresión normalizado. Ambos tipos de valores pueden usarse en el presente método. El nivel de expresión de un gen se expresa preferiblemente como el nivel de expresión normalizado cuando se usa PCR cuantitativa como método de evaluación del nivel de expresión porque pequeñas diferencias al comienzo de un experimento podrían proporcionar grandes diferencias después de varios ciclos.
Típicamente, los niveles de expresión se normalizan corrigiendo el nivel de expresión absoluto de un gen y comparando su expresión con la expresión de un gen que no es relevante para determinar la etapa de cáncer del paciente, por ejemplo, un gen de mantenimiento que se expresa constitutivamente. Los genes adecuados para la normalización incluyen genes de mantenimiento como el gen de actina ACTB, el gen ribosómico 18S, GUSB, PGK1 y TFRC. Esta normalización permite comparar el nivel de expresión de una muestra, por ejemplo, una muestra de paciente, con el nivel de expresión de otra muestra, o comparar muestras de diferentes fuentes.
Tratamientos antitumorales
Por lo tanto, el presente método permite definir, entre otras cosas, un nuevo grupo de pacientes que nunca se habían identificado hasta ahora, es decir, pacientes cuyo cáncer se tratará con éxito mediante un tratamiento contra el cáncer.
Un tratamiento contra el cáncer puede consistir en radioterapia, quimioterapia o inmunoterapia. El tratamiento puede consistir en una terapia adyuvante (es decir, el tratamiento realizado después de la resección quirúrgica del tumor
primario) de una terapia neoadyuvante (es decir, el tratamiento realizado antes de la resección quirúrgica del tumor primario).
Por lo tanto, la presente invención se refiere a un agente quimioterapéutico, un agente radioterapéutico o un agente inmunoterapéutico, preferiblemente el último, para su uso en el tratamiento de un paciente con cáncer en estadio I-III que ha sido considerado como una buena guía sensible al tratamiento antitumoral de acuerdo con el método anterior de la invención.
La expresión "agente quimioterapéutico" se refiere a compuestos químicos que son efectivos para inhibir el crecimiento tumoral. Los ejemplos de agentes quimioterapéuticos incluyen agentes alquilantes tales como tiotepa y ciclofosfamida; alquil sulfonatos tales como busulfano, improsulfán y piposulfán; aziridinas tales como benzodopa, carboquona, meturedopa y uredopa; etileniminas y metilamelaminas, que incluyen altretamina, trietilenomelamina, trietilenfosforamida, trietilentofosfatofano y trimetilolomelamina; acetogeninas (especialmente bullatacina y bullatacinona); una carnptotecina (que incluye el análogo sintético topotecan); briostatina; callistatina; CC-1065 (incluyendo sus análogos sintéticos adozelesin, carzelesin y bizelesin); criptoficinas (particularmente criptoficina1 y criptoficina8); dolastatina; duocarmicina (incluidos los análogos sintéticos, KW-2189 y CBI-TMI); eleuterobina; pancratistatina; una sarcodictina; espongistatina; mostazas nitrogenadas tales como clorambucilo, clornafazina, colofosfamida, estrarnustina, ifosfamida, mecloretamina, clorhidrato de óxido de mecloretamina, melfalan, novembichina, fenesterina, prednimustina, trifosfamida, mostaza de uracilo; nitrosoureas tales como carmustina, clorozotocina, fotemustina, lomustina, nimustina, ranimustina; antibióticos como los antibióticos enediyne (por ejemplo, caliqueamicina, especialmente caliqueamicina (11 y caliqueamicina 211, ver, por ejemplo, Agnew Chem Intl. Ed. Engl. 33: 183-186 (1994); dinemicina, que incluye dinemicina A; una esperamicina; así como un cromóforo de neocarzinostatina y cromóforos antibióticos relacionados de la cromoproteína enediyna), aclacinomisinas, actinomicina, autramicina, azaserina, bleomicinas, cactinomicina, carabicina, canninomicina, carzinofilina, cromomicinas, dactinomicina, daunorrubicina, detorrubicina, 6-diazo-5-oxo-L-norleucina, doxorubicina (incluyendo morfolino-doxorubicina, cianomorfolino-doxorrubicina, 2-pirrolino-doxorrubicina y desoxidoxorrubicina), epirubicina, esorubicina, idanrbicina, marcellomicina, mitomicinas, ácido micofenólico, nogalamicina, olivomicinas, peplomicina, potfiromicina, puromicina, quelamicina, rodorrubicina, estreptomgrina, estreptozocina, tubercidina, ubenimex, zinostatina, zorubicina; anti-metabolitos como el metotrexato y 5-fluorouracilo (5-FU); análogos de ácido fólico como denopterina, metotrexato, pteropterina, trimetrexato; análogos de purina como el fludarabina, 6-mercaptopurina, tiamiprina, tioguanina; análogos de pirimidina como ancitabina, azacitidina, 6-azauridina, carmofur, citarabina, didesoxiuridina, doxifluridina, enocitabina, floxuridina, 5-FU; andrógenos como calusterona, propionato de dromostanolona, epitiostanol, mepitiostano, testolactona; anti-adrenales como aminoglutetimida, mitotano, trilostano; reabastecedor de ácido fólico como el ácido frolínico; aceglatona; aldofosfamida glicósido; ácido aminolevulínico; amsacrina; bestrabucil; bisantreno; edatraxato; defofamina; demecolcina; diaziquona; elfornitina; acetato de elliptinio; una epotilona; etoglucid; nitrato de galio; hidroxiurea; lentinan; lonidamina; maitansinoides como maitansina y ansamitocinas; mitoguazona; mitoxantrona; mopidamol; nitracrina; pento estatina; fenamet; pirarubicina; ácido podofilínico; 2-etilhidrazida; procarbazina; PSKOR; razoxana; rizoxina; sizofiran; espirogennanio; ácido tenuazónico; triaziquona; 2,2',2"-triclorotrietilamina; tricotecenos (especialmente toxina T-2, verracurin A, roridin A y anguidina); uretano; vindesina; dacarbazina; mannomustina; mitobromtol; mitolactol; pipobroman; gacitosina; arabinosida ("Ara-C"); ciclofosfamida; tiotepa; taxoides, p. ej. paclitaxel (TAXOLOR , Bristol-Myers Squibb Oncology, Princeton, N.].) y doxetaxel (TAXOTERE®, Rhone-Poulenc Rorer, Antony, Francia); clorambucilo; gemcitabina; 6-tioguanina; mercaptopurina; metotrexato; análogos de platino como cisplatina y carboplatin; vinblastina; platino; etopósido (VP-16); ifosfamida; mitomicina C; mitoxantrona; vincristina; vinorelbina; navelbina; novantrona; tenipósido; daunomicina; aminopterina; xeloda; ibandronato; CPT-11; inhibidor de topoisomerasa RFS 2000; difluorometilornitina (DMFO); ácido retinoico; capecitabina; y sales, ácidos o derivados farmacéuticamente aceptables de cualquiera de los anteriores. También se incluyen en esta definición los agentes antihormonales que actúan para regular o inhibir la acción de la honnona en tumores como los antiestrógenos, que incluyen, por ejemplo, tamoxifeno, raloxifeno, inhibidores de la aromatasa 4(5)-imidazoles, 4-hidroxitamoxifeno, trioxifeno, keoxifeno, LY117018, onapristona y toremifeno (Fareston); y antiandrógenos tales como flutamida, nilutamida, bicalutamida, leuprolida y goserelina; y sales, ácidos o derivados farmacéuticamente aceptables de cualquiera de los anteriores.
La expresión "agente inmunoterapéutico", como se usa en el presente documento, se refiere a un compuesto, composición o tratamiento que mejora, estimula o aumenta indirecta o directamente la respuesta inmune del cuerpo contra las células cancerosas y/o que disminuye los efectos secundarios de otras terapias contra el cáncer. Por lo tanto, la inmunoterapia es una terapia que estimula o mejora directa o indirectamente las respuestas del sistema inmunitario frente a las células cancerosas y/o disminuye los efectos secundarios que otros agentes anticancerígenos puedan haber causado. La inmunoterapia también se conoce en la técnica como terapia inmunológica, terapia biológica, terapia de modificación de respuesta biológica y bioterapia. Los ejemplos de agentes inmunoterapéuticos comunes conocidos en la técnica incluyen, entre otros, citocinas, vacunas contra el cáncer, anticuerpos monoclonales y adyuvantes que no son citocinas. Alternativamente, el tratamiento inmunoterapéutico puede consistir en administrar al paciente una cantidad de células inmunes (células T, NK, células, células dendríticas, células B, etc.).
Los agentes inmunoterapéuticos pueden ser inespecíficos, es decir, estimular el sistema inmunitario en general para que el cuerpo humano se vuelva más efectivo en la lucha contra el crecimiento y/o la propagación de las células
cancerosas, o pueden ser específicos, es decir, dirigidos a las células cancerosas en sí. Los regímenes inmunoterapéuticos pueden combinar el uso de agentes inmunoterapéuticos no específicos y específicos.
Los agentes inmunoterapéuticos no específicos son sustancias que estimulan o mejoran indirectamente el sistema inmunitario. Los agentes inmunoterapéuticos no específicos se han utilizado solos como terapia principal para el tratamiento del cáncer, así como además de una terapia principal, en cuyo caso el agente inmunoterapéutico no específico funciona como un adyuvante para mejorar la efectividad de otras terapias (por ejemplo, vacunas contra el cáncer). Los agentes inmunoterapéuticos no específicos también pueden funcionar en este último contexto para reducir los efectos secundarios de otras terapias, por ejemplo, la supresión de la médula ósea inducida por ciertos agentes quimioterapéuticos. Los agentes inmunoterapéuticos no específicos pueden actuar sobre las células clave del sistema inmunitario y causar respuestas secundarias, como una mayor producción de citocinas e inmunoglobulinas. Alternativamente, los propios agentes pueden comprender citocinas. Los agentes inmunoterapéuticos no específicos se clasifican generalmente como citocinas o adyuvantes no citocinas.
Varias citocinas han encontrado aplicación en el tratamiento del cáncer como inmunoterapias generales no específicas diseñadas para estimular el sistema inmunitario o como adyuvantes provistos con otras terapias. Las citocinas adecuadas incluyen, entre otras, interferones, interleucinas y factores estimuladores de colonias.
Los interferones (IFN) contemplados por la presente invención incluyen los tipos comunes de IFN, IFN-alfa (IFN-a), IFN-beta (IFN-b) e IFN-gamma (IFN-y). Los IFN pueden actuar directamente sobre las células cancerosas, por ejemplo, ralentizando su crecimiento, promoviendo su desarrollo en células con un comportamiento más normal y/o aumentando su producción de antígenos, lo que hace que las células cancerosas sean más fáciles de reconocer y destruir para el sistema inmune. Los IFN también pueden actuar indirectamente sobre las células cancerosas, por ejemplo, al desacelerar la angiogénesis, aumentar el sistema inmunitario y/o estimular las células asesinas naturales (NK), las células T y los macrófagos. El IFN-alfa recombinante está disponible comercialmente como Roferon (Roche Pharmaceuticals) e Intron A (Schering Corporation). El uso de IFN-alfa, solo o en combinación con otros inmunoterapéuticos o con quimioterapéuticos, ha demostrado eficacia en el tratamiento de varios tipos de cáncer, incluido el melanoma (incluido el melanoma metastásico), el cáncer renal (incluido el cáncer renal metastásico), el cáncer de mama, el cáncer de próstata, y el cáncer cervical (incluyendo el cáncer cervical metastásico).
Las interleucinas contempladas por la presente invención incluyen IL-2, IL-4, IL-11 e IL-12. Los ejemplos de interleucinas recombinantes disponibles comercialmente incluyen Proleukin® (IL-2; Chiron Corporation) y Neumega® (IL-12; Wyeth Pharmaceuticals). Zymogenetics, Inc. (Seattle, Washington) está probando actualmente una forma recombinante de IL-21, que también se contempla para su uso en las combinaciones de la presente invención. Las interleucinas, solas o en combinación con otros agentes inmunoterapéuticos o con agentes quimioterapéuticos, han demostrado eficacia en el tratamiento de varios tipos de cáncer, incluido el cáncer renal (incluido el cáncer renal metastásico), el melanoma (incluido el melanoma metastásico), el cáncer de ovario (incluido el cáncer de ovario recurrente), el cáncer cervical (incluido el cáncer cervical metastásico), cáncer de mama, cáncer colorrectal, cáncer de pulmón, cáncer de cerebro y cáncer de próstata.
Las interleucinas también han mostrado una buena actividad en combinación con IFN-alfa en el tratamiento de varios tipos de cáncer (Negrier et al, Ann Oncol. 2002 13(9): 1460-8; Touranietal, J. Clin. Oncol 2003 21 (21):398794).
Los factores estimulantes de colonias (CSF) contemplados por la presente invención incluyen el factor estimulante de colonias de granulocitos (G-CSF o filgrastim), el factor estimulante de colonias de granulocitos-macrófagos (GM-CSF o sargramostim) y la eritropoyetina (epoetina alfa, darbepoyetina). El tratamiento con uno o más factores de crecimiento puede ayudar a estimular la generación de nuevas células sanguíneas en pacientes sometidos a quimioterapia tradicional. En consecuencia, el tratamiento con CSF puede ser útil para disminuir los efectos secundarios asociados con la quimioterapia y puede permitir el uso de dosis más altas de agentes quimioterapéuticos. Varios factores estimulantes de colonias recombinantes están disponibles comercialmente, por ejemplo, Neupogen® (G-CSF; Amgen), Neulasta (pelfilgrastim; Amgen), Leukina (GM-CSF; Berlex), Procrit (eritropoyetina; Ortho Biotech), Epogen (eritropoyetina; Amgen), Arnesp (eritropoyetina). Los factores estimulantes de colonias han demostrado eficacia en el tratamiento del cáncer, incluido el melanoma, el cáncer colorrectal (incluido el cáncer colorrectal metastásico) y el cáncer de pulmón.
Los adyuvantes que no son citoquinas adecuados para usar en las combinaciones de la presente invención incluyen, entre otros, Levamisol, hidróxido de alumbre (alumbre), bacilo de Calmette-Guerin (ACG), adyuvante de Freund incompleto (IFA), QS-21, DETOX, hemocianina de lapa californiana (KLH) y dinitrofenilo (DNP). Los adyuvantes que no son citoquinas en combinación con otros agentes inmuno y/o quimioterapéuticos han demostrado eficacia contra diversos tipos de cáncer, incluidos, por ejemplo, el cáncer de colon y el cáncer colorrectal (Levimasol); melanoma (BCG y QS-21); cáncer renal y cáncer de vejiga (BCG).
Además de disponer de dianas específicas o no específicas, los agentes inmunoterapéuticos pueden ser activos, es decir, estimular la propia respuesta inmune del cuerpo, o pueden ser pasivos, es decir, comprender componentes del sistema inmunitario que se generaron externamente al cuerpo.
La inmunoterapia pasiva específica generalmente implica el uso de uno o más anticuerpos monoclonales que son específicos para un antígeno particular que se encuentra en la superficie de una célula cancerosa o que son específicos para un factor de crecimiento celular particular. Los anticuerpos monoclonales se pueden usar en el tratamiento del cáncer de varias maneras, por ejemplo, para mejorar la respuesta inmune de un sujeto frente a un tipo específico de cáncer, para interferir con el crecimiento de las células cancerosas al atacar factores de crecimiento celular específicos, como aquellos involucrados en la angiogénesis, o al mejorar la entrega de otros agentes anticancerígenos a las células cancerosas cuando se unen o se conjugan con agentes tales como agentes quimioterapéuticos, partículas radioactivas o toxinas.
Los anticuerpos monoclonales utilizados actualmente como agentes inmunoterapéuticos contra el cáncer que son adecuados para su inclusión en las combinaciones de la presente invención incluyen, entre otros, rituximab (Rituxan®), trastuzumab (Herceptin®), ibritumomab tiuxetan (Zevalin®), tositumomab (Bexxar®), cetuximab (C-225, Erbitux®), bevacizumab (Avastin®), gemtuzumab ozogamicina (Mylotarg®), alemtuzumab (Campath®) y BL22. Los anticuerpos monoclonales se usan en el tratamiento de una amplia gama de cánceres, incluido el cáncer de mama (incluido el cáncer de mama metastásico avanzado), el cáncer colorrectal (incluido el cáncer colorrectal avanzado y/o metastásico), el cáncer de ovario, cáncer de pulmón, cáncer de próstata, cáncer cervical, melanoma y tumores cerebrales. Otros ejemplos incluyen anticuerpos anti-CTLA4 (p. ej. Ipilimumab), anticuerpos anti-PDl, anticuerpos anti-PDLl, anticuerpos anti-TIM3, anticuerpos anti-LAG3, anticuerpos anti-B7H3, anticuerpos anti-B7H4 o anticuerpos anti-B7H6.
Los anticuerpos monoclonales se pueden usar solos o en combinación con otros agentes inmunoterapéuticos o agentes quimioterapéuticos.
La inmunoterapia específica activa generalmente implica el uso de vacunas contra el cáncer. Se han desarrollado vacunas contra el cáncer que comprenden células cancerosas completas, partes de células cancerosas o uno o más antígenos derivados de células cancerosas. Las vacunas contra el cáncer, solas o en combinación con uno o más agentes inmuno o quimioterapéuticos, se están investigando en el tratamiento de varios tipos de cáncer, incluidos melanoma, cáncer renal, cáncer de ovario, cáncer de mama, cáncer colorrectal y cáncer de pulmón. Los agentes inmunoterapéuticos no específicos son útiles en combinación con vacunas contra el cáncer para mejorar la respuesta inmune del cuerpo.
El tratamiento inmunoterapéutico puede consistir en una inmunoterapia adoptiva según lo descrito por Nicholas P. Restifo, Mark E. Dudley y Steven A. Rosenberg "Adoptive immunotherapy for cancer: harnessing the T cell response”, Nature Reviews Immunology, Volumen 12, abril de 2012). En la inmunoterapia adoptiva, los linfocitos circulantes del paciente, o linfocitos infiltrados por tumores, se aíslan in vitro, se activan mediante linfocinas como IL-2 o se transmutan con genes para la necrosis tumoral y se readministran (Rosenberg et al, 1988; 1989). Los linfocitos activados son preferiblemente las propias células del paciente que se aislaron previamente de una muestra de sangre o tumor y se activaron (o "expandieron") in vitro. Esta forma de inmunoterapia ha producido varios casos de regresión de melanoma y carcinoma renal.
La expresión "agente radioterapéutico", como se usa en el presente documento, pretende referirse a cualquier agente radioterapéutico conocido por los expertos en la materia que sea eficaz para tratar o mejorar el cáncer, sin limitación. Por ejemplo, el agente radioterapéutico puede ser un agente como los administrados en braquiterapia o terapia con radionúclidos. Tales métodos pueden comprender opcionalmente además la administración de una o más terapias adicionales contra el cáncer, tales como, entre otras, quimioterapias y/u otra radioterapia.
En una realización preferida, el tratamiento antitumoral es un tratamiento con un agente de inmunoterapia contra el cáncer de bloqueo del punto de control.
Como se usa en el presente documento, la expresión "agente de inmunoterapia contra el cáncer del bloqueo del punto de control" o "inhibidor del punto de control inmunitario" (ambas expresiones se usarán indistintamente) tiene su significado general en la técnica y se refiere a cualquier compuesto que inhiba la función de una proteína de punto de control inmunitario inhibidora. La inhibición incluye la reducción de la función y el bloqueo completo. Los inhibidores del punto de control inmunitario preferidos son anticuerpos que reconocen específicamente las proteínas del punto de control inmunitario. Se conocen varios inhibidores del punto de control inmunitario y, en analogía con estos inhibidores de la proteína del punto de control inmunitario conocidos, se pueden desarrollar inhibidores del punto de control inmunitario alternativos en el futuro (próximo). Los inhibidores del punto de control inmunitario incluyen péptidos, anticuerpos, moléculas de ácido nucleico y moléculas pequeñas. En particular, el inhibidor del punto de control inmunitario usado en la presente invención se administra para mejorar la proliferación, migración, persistencia y/o actividad citotóxica de las células T CD8+ en el sujeto y, en particular, la infiltración tumoral de las células T CD8+ del sujeto. Como se usa en el presente documento, "células T CD8+" tiene su significado general en la técnica y se refiere a un subconjunto de células T que expresan CD8 en su superficie. Están restringidos por el MHC de clase I y funcionan como células T citotóxicas. Las "células T CD8+" también se denominan células T CD8+ y se denominan linfocitos T citotóxicos (CTL), células T-asesinas, células T citolíticas, células T CD8+ o células T asesinas. Los antígenos CD8 son miembros de la familia de supergenes de inmunoglobulina y son elementos de reconocimiento asociativo en las principales interacciones restringidas de clase I del complejo de histocompatibilidad principal. La capacidad del inhibidor del punto de control inmunitario para mejorar la actividad de destrucción de
células T CD8 puede determinarse mediante cualquier ensayo habitual en la técnica. Típicamente, dicho ensayo es un ensayo in vitro en el que las células T CD8+ se ponen en contacto con células diana (por ejemplo, células diana que son reconocidas y/o lisadas por las células T CD8+). Por ejemplo, el inhibidor del punto de control inmunitario usado en la presente invención puede seleccionarse por la capacidad de aumentar la lisis específica por las células T CD8+ en más de aproximadamente un 20%, preferiblemente con al menos aproximadamente 30%, al menos aproximadamente 40%, al menos aproximadamente 50%, o más de la lisis específica obtenida en la misma relación de efector: células diana con células T CD8+ o líneas de células T CD8 que se ponen en contacto con el inhibidor del punto de control inmunitario usado en la presente invención. Los ejemplos de protocolos para ensayos de citotoxicidad clásicos son convencionales.
Por lo general, el agente de inmunoterapia contra el cáncer del bloqueo del punto de control es un agente que bloquea un receptor inmunosupresor expresado por los linfocitos T activados, como la proteína 4 asociada a los linfocitos T citotóxicos (CTLA4) y la muerte celular programada 1 (PDCD1, mejor conocida como PD-1), o por células NK, como varios miembros de la familia del receptor tipo inmunoglobulina de células asesinas (KIR), o un agente que bloquea los ligandos principales de estos receptores, como el ligando PD-1 CD274 (mejor conocido como PD-L1 o B7- H1).
Típicamente, el agente de inmunoterapia del cáncer de bloqueo del punto de control es un anticuerpo.
En algunas realizaciones, el agente de inmunoterapia contra el cáncer del bloqueo del punto de control es un anticuerpo seleccionado del grupo que consiste en anticuerpos anti-CTLA4, anticuerpos anti-PDl, anticuerpos anti-PDL1, anticuerpos anti-PDL2, anticuerpos anti-TIM-3, anticuerpos anti-LAG3, anticuerpos anti-IDOl, anticuerpos anti-TIGIT, anticuerpos anti-B7H3, anticuerpos anti-B7H4, anticuerpos anti-BTLA y anticuerpos anti-B7H6.
Los ejemplos de anticuerpos anti-CTLA-4 se describen en las patentes de EE. UU. Nos: 5.811.097; 5.811.097; 5.855.887; 6.051.227; 6.207.157; 6.682.736; 6.984.720; y 7.605.238. Un anticuerpo anti-CDLA-4 es tremelimumab (ticilimumab, CP-675,206). En algunas realizaciones, el anticuerpo anti-CTLA-4 es ipilimumab (también conocido como 10D1, MDX-D010), un anticuerpo IgG monoclonal completamente humano que se une a CTLA-4.
Se describen ejemplos de anticuerpos PD-1 y PD-L1 en las patentes de los Estados Unidos Nos. 7.488.802; 7.943.743; 8.008.449; 8.168.757; 8.217.149 y las solicitudes de patente publicadas PCT Nos: WO03042402, WO2008156712, WO2010089411, WO2010036959, WO2011066342, WO2011159877, WO2011082400 y WO2011161699. En algunas realizaciones, los bloqueadores PD-1 incluyen anticuerpos anti-PD-Ll. En ciertas otras realizaciones, los bloqueadores de PD-1 incluyen anticuerpos anti-PD-1 y proteínas de unión similares como nivolumab (MDX 1106, BMS 936558, ONO 4538), un anticuerpo IgG4 completamente humano que se une y bloquea la activación de PD-1 por sus ligandos PD-L1 y PD-L2; lambrolizumab (MK-3475 o SCH 900475), un anticuerpo IgG4 monoclonal humanizado contra PD-1; CT-011 un anticuerpo humanizado que se une a PD-1; AMP-224 es una proteína de fusión de B7-DC; una porción de anticuerpo Fc; BMS-936559 (MDX- 1105-01) para el bloqueo de PD-L1 (B7-H1).
Otros inhibidores del punto de control inmunitario incluyen los inhibidores del gen 3 de activación de linfocitos (LAG-3), como IMP321, una proteína de fusión de Ig soluble (Brignone et al, 2007, J. Immunol. 179:4202-4211).
Otros inhibidores del punto de control inmunitario incluyen los inhibidores B7, como los inhibidores B7-H3 y B7-H4. En particular, el anticuerpo anti-B7-H3 MGA271 (Loo et al, 2012, Clin. Cancer Res. 15 de julio (18) 3834).
También se incluyen los inhibidores TIM3 (dominio de inmunoglobulina de células T y dominio de mucina 3) (Fourcade et al, 2010, J. Exp. Med. 207: 2175-86 y Sakuishi et al, 2010, J. Exp. Med. 207:2187-94). Como se usa en el presente documento, el término "TIM-3'' tiene su significado general en la técnica y se refiere a la inmunoglobulina de células T y la molécula 3 que contiene el dominio de mucina. El ligando natural de TIM-3 es la galectina 9 (Gal9). En consecuencia, la expresión "inhibidor de TIM-3", como se usa en el presente documento, se refiere a un compuesto, sustancia o composición que puede inhibir la función de TIM-3. Por ejemplo, el inhibidor puede inhibir la expresión o actividad de TIM-3, modular o bloquear la ruta de señalización de TIM-3 y/o bloquear la unión de TIM-3 a la galectina-9. Los anticuerpos que tienen especificidad por TIM-3 son habituales en la técnica y típicamente los descritos en los documentos WO2011155607, WO2013006490 y WO2010117057. En algunas realizaciones, el inhibidor del punto de control inmune es un inhibidor de la 2,3-dioxigenasa de Indoleamina (IDO), preferiblemente un inhibidor IDOl. Se describen ejemplos de inhibidores de IDO en el documento WO 2014150677. Los ejemplos de inhibidores de IDO incluyen, sin limitación, 1 -metil-triptófano (IMT), p-(3-benzofuranil)-alanina, p-(3-benzo(b)tienil)-alanina), 6-nitro-triptófano, 6-flúor-triptófano, 4-metil-triptófano, 5-metil-triptófano, 6-metil-triptófano, 5-metoxitriptófano, 5-hidroxi-triptófano, indol 3-carbinol, 3,3'-di-indolilmetano, galato de epigalocatequina, 5-Br-4-Cl-indoxil 1,3-diacetato, 9-vinilcarbazol, acemetacina, 5-bromo-triptofano, 5-bromoindoxil diacetato, ácido 3-amino-naftoico, pirrolidina ditiocarbamato, 4-fenilimidazol, un derivado de brasinina, un derivado de tiohidantoína, un derivado de pcarbolina o un derivado de brasilexina. Preferiblemente, el inhibidor de IDO se selecciona de 1 -metil-triptófano, p-(3-benzofuranil)-alanina, 6-nitro-L-triptófano, ácido 3-amino-naftoico y p-[3-benzo(b)tienil]-alanina o un derivado o profármaco del mismo.
En algunas realizaciones, el inhibidor del punto de control inmunitario es un anticuerpo anti-TIGIT (inmunoglobina de células T y dominio ITIM).
En una realización preferida, el agente de inmunoterapia para el cáncer del bloqueo del punto de control es un anticuerpo de bloqueo CTLA4, como Ipilimumab, o un anticuerpo de bloqueo PD-1, como Nivolumab o Pembrolizumab, o una combinación de los mismos.
La invención se ilustrará adicionalmente mediante las siguientes figuras y ejemplos. Estos ejemplos y figuras no deben interpretarse de ninguna manera como limitantes del alcance de la presente invención.
Figuras
La figura 1 es una ilustración esquemática de un sistema para llevar a cabo la invención.
La Figura 2 es un diagrama de flujo que ilustra un ejemplo de un procedimiento para implementar la invención; La Figura 3 representa una curva de Kaplan Meir para 5 valores medios aritméticos de referencia predeterminados de percentil.
EJEMPLOS
Un grupo de referencia de pacientes con cáncer colorrectal en fase I/II/III (n = 539) se analizó de acuerdo con el método de puntuación algorítmica. Los tumores completos de los portaobjetos se tiñeron con anticuerpos CD3 y CD8. Mediante el uso de un software de análisis de imágenes y patología digital, se determinaron el margen invasivo (IM) del tumor y el núcleo (centro) (CT) del tumor. Se cuantificaron las densidades de CD3 y CD8 en las regiones CT e IM (células/mm2). La distribución de todas las densidades o todos los marcadores se traza y se indica en una tabla. Se derivaron los percentiles correspondientes (es decir, con un intervalo de pasos del 5%). La media sobre los valores de los biomarcadores de 4 densidades se calculó con un peso de 1 para cada biomarcador, en el ejemplo ilustrado en la tabla a continuación. Las curvas de Kaplan Meier para dicha cohorte se ilustran en la Figura 3, con 5 categorías de grupos (I0, I1, I2, I3, I4), correspondientes a los valores del percentil medio de 0-10%, 10-25%, 25-75%, 75-95%, 95-100%, respectivamente.
En las siguientes tablas hay una ilustración de un ejemplo de cálculo del valor medio aritmético de percentiles para un paciente hipotético:

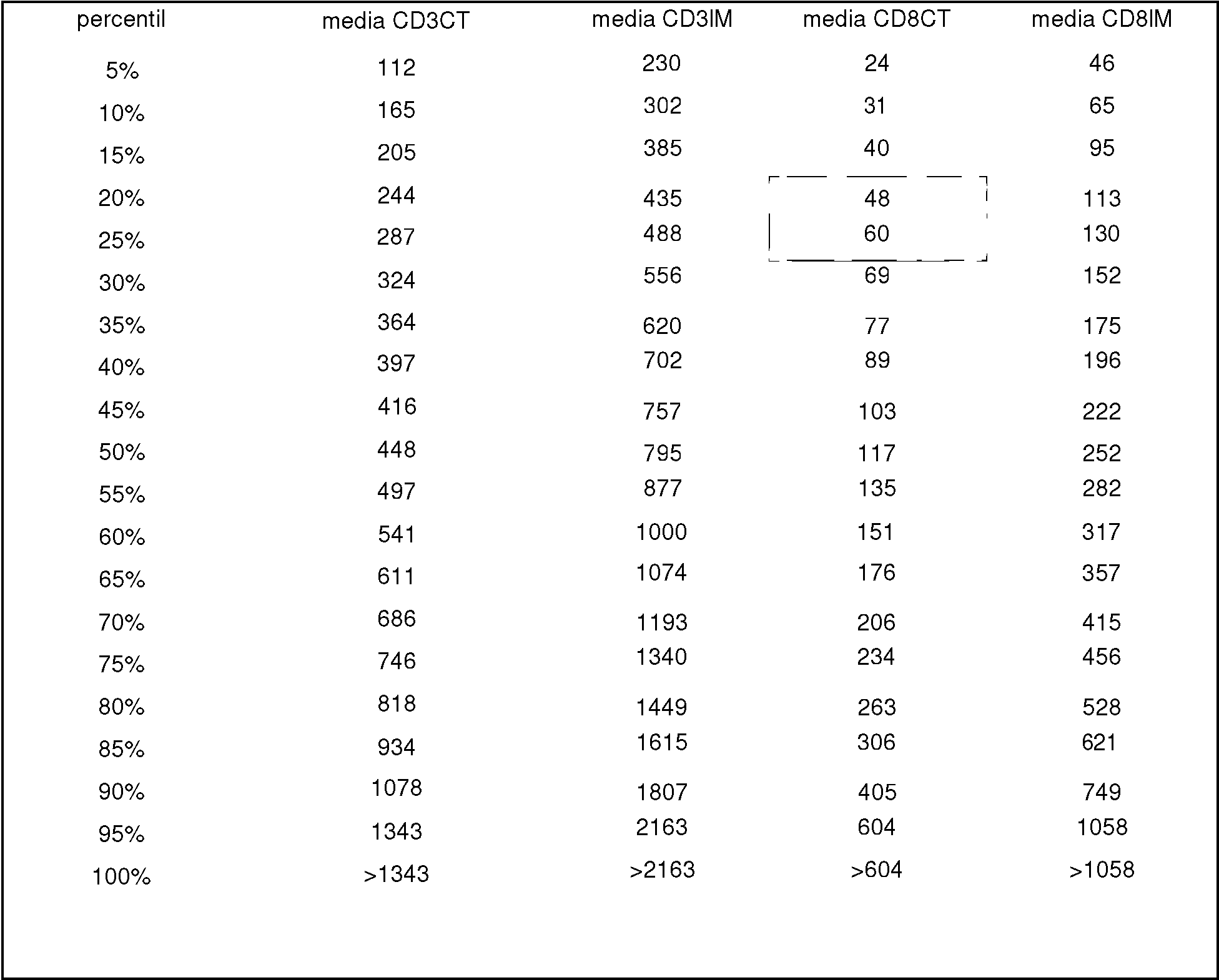
El valor medio aritmético del percentil para el paciente hipotético sería aproximadamente 47.5. El paciente hipotético se clasificaría como perteneciente al grupo 12.
Se ha realizado el mismo tipo de experimentos con los 21 marcadores biológicos que son los niveles de expresión de los 21 genes del grupo que consiste en CCR2, CD3D, CD3E, CD3G, CD8A, CXCL10, CXCL11, GZMA, GZMB, GZMK, GZMM, IL15 , IRF1, PRF1, STAT1, CD69, ICOS, CXCR3, STAT4, CCL2 y TBX21 y con los 15 marcadores biológicos que son los niveles de expresión de los 15 genes del grupo que consiste en GZMH, IFNG, CXCL13, GNLY, LAG3, ITGAE , CCL5, CXCL9, PF4, IL17A, TSLP, REN, IHH, PROM1 y VEGFA.
Se han obtenido resultados muy precisos calculando el valor medio del percentil.
Referencias
A lo largo de esta solicitud, varias referencias describen el estado de la técnica a la que pertenece esta invención.
Claims (19)
1. Un método in vitro para el pronóstico del tiempo de supervivencia de un paciente que padece un cáncer sólido, cuyo método comprende los siguientes pasos:
a) detectar y cuantificar dos o más marcadores biológicos indicativos del estado de la respuesta inmune de dicho paciente contra dicho cáncer, en donde cada marcador biológico indicativo del estado de la respuesta inmune se detecta in vitro y se cuantifica en una muestra tumoral obtenida de dicho paciente;
b) comparar cada valor obtenido en la etapa a) para dichos dos o más marcadores biológicos con una distribución de valores obtenidos para cada uno de dichos dos o más marcadores biológicos de un grupo de referencia de pacientes que padecen dicho cáncer;
c) determinar para cada valor obtenido en el paso a) para dichos dos o más marcadores biológicos el percentil de la distribución a la que corresponden los valores obtenidos en el paso a);
d) calcular el valor medio aritmético o el valor medio de los percentiles de los marcadores biológicos determinados en el paso c); y
e) comparar el valor medio aritmético o el valor medio de percentiles obtenidos en el paso d) con un valor medio aritmético de referencia predeterminado o un valor medio predeterminado de percentiles, cuyo valor de referencia predeterminado se correlaciona con el tiempo de supervivencia.
2. Un método in vitro para evaluar la capacidad de respuesta de un paciente que padece un cáncer sólido frente a un tratamiento antitumoral, cuyo método comprende los siguientes pasos:
a) detectar y cuantificar dos o más marcadores biológicos indicativos del estado de la respuesta inmune de dicho paciente contra dicho cáncer, en donde cada marcador biológico indicativo del estado de la respuesta inmune se detecta in vitro y se cuantifica en una muestra tumoral obtenida de dicho paciente;
b) comparar cada valor obtenido en la etapa a) para dichos dos o más marcadores biológicos con una distribución de valores obtenidos para cada uno de dichos dos o más marcadores biológicos de un grupo de referencia de pacientes que padecen dicho cáncer;
c) determinar para cada valor obtenido en el paso a) para dichos dos o más marcadores biológicos el percentil de la distribución a la que corresponden los valores obtenidos en el paso a);
d) calcular el valor medio aritmético o el valor medio de los percentiles de los marcadores biológicos determinados en el paso c); y
e) comparar el valor medio aritmético o el valor medio de los percentiles obtenidos en el paso d) con un valor medio aritmético de referencia predeterminado o un valor medio predeterminado de percentiles, cuyo valor de referencia predeterminado se correlaciona con la respuesta frente a dicho tratamiento antitumoral.
3. El método de acuerdo con la reivindicación 1 o 2, en el que el cáncer sólido es un cáncer colorrectal, un cáncer de mama, un cáncer de pulmón, un cáncer de cabeza y cuello, cáncer de vejiga, un cáncer de ovario o un cáncer de próstata.
4. El método según la reivindicación 3, en el que el cáncer sólido es un cáncer colorrectal.
5. El método de acuerdo con cualquiera de las reivindicaciones 1 a 4, en el que los dos o más marcadores biológicos comprenden la densidad celular de las células del sistema inmune.
6. El método de acuerdo con la reivindicación 5, en el que los dos o más marcadores biológicos comprenden la densidad de las células CD3+, la densidad de las células CD8+, la densidad de las células CD45RO+, la densidad de las células GZM-B+, la densidad de las células B y/o la densidad de las células DC.
7. El método de acuerdo con la reivindicación 6, en el que los dos o más marcadores biológicos comprenden la densidad de células CD3+ y la densidad de células CD8+, la densidad de células CD3+ y la densidad de células CD45RO+, la densidad de células CD3+ y la densidad de células GZM-B+, la densidad de las células CD8+ y la densidad de las células CD45RO+, la densidad de las células CD8+ y la densidad de las células GZM-B+; o la densidad de las células CD45RO+ y la densidad de las células GZM-B+.
8. El método según cualquiera de las reivindicaciones 5 a 7, en el que la densidad de células del sistema inmune se cuantifica en el centro del tumor y/o en el margen invasivo del tumor.
9. El método de acuerdo con la reivindicación 8, en el que los dos o más marcadores biológicos comprenden la densidad de células CD3+ en el centro del tumor, la densidad de células CD8+ en el centro del tumor, la densidad de células CD3+ en el margen invasivo, y la densidad de células CD8+ en el margen invasivo.
10. El método según cualquiera de las reivindicaciones 5 a 9, en el que la densidad se mide en una región de la muestra tumoral donde la densidad es la más baja.
11. El método según cualquiera de las reivindicaciones 5 a 9, en el que la densidad se mide en una región de la muestra tumoral donde la densidad es la más alta.
12. El método según cualquiera de las reivindicaciones 1 a 11, en el que los dos o más marcadores biológicos comprenden el nivel de expresión de uno o más genes del grupo que consiste en CCR2, CD3D, CD3E, CD3G, CD8A, CXCL10, CXCL11, GZMA, GZMB, GZMK, GZMM, IL15, IRF1, PRF1, STAT1, CD69, ICOS, CXCR3, STAT4, CCL2 y TBX21.
13. El método según cualquiera de las reivindicaciones 1 a 12, en el que los dos o más marcadores biológicos comprenden el nivel de expresión de uno o más genes del grupo que consiste en GZMH, IFNG, CXCL13, GNLY, LAG3, ITGAE, CCL5, CXCL9, PF4, IL17A, TSLP, REN, IHH, PROM1 y VEGFA.
14. El método según cualquiera de las reivindicaciones 1 a 12, en el que los dos o más marcadores biológicos comprenden un nivel de expresión de al menos un gen representativo de la respuesta inmune adaptativa humana y un nivel de expresión de al menos un gen representativo de la respuesta inmunosupresora humana.
15. El método de acuerdo con la reivindicación 14, en el que el al menos un gen representativo de la respuesta inmune adaptativa humana se selecciona del grupo que consiste en:

y dicho al menos un gen representativo de la respuesta inmunosupresora humana se selecciona del grupo que consiste en:


16. El método de acuerdo con una cualquiera de las reivindicaciones anteriores, en el que cada marcador biológico es un ácido nucleico.
17. El método de acuerdo con una cualquiera de las reivindicaciones anteriores, en el que cada marcador biológico se detecta por hibridación y/o amplificación de ARNm.
18. El método de acuerdo con una cualquiera de las reivindicaciones 1 a 15, en el que cada marcador biológico es una proteína.
19. El método de acuerdo con una cualquiera de las reivindicaciones 1 a 15 y 18, en el que cada marcador biológico se detecta por inmunohistoquímica.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP16305536 | 2016-05-09 | ||
| PCT/EP2017/061089 WO2017194556A1 (en) | 2016-05-09 | 2017-05-09 | Methods for classifying patients with a solid cancer |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2808004T3 true ES2808004T3 (es) | 2021-02-25 |
Family
ID=56014938
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES17725195T Active ES2808004T3 (es) | 2016-05-09 | 2017-05-09 | Métodos para clasificar pacientes con un cáncer sólido |
Country Status (13)
| Country | Link |
|---|---|
| US (1) | US20190309369A1 (es) |
| EP (1) | EP3455631B1 (es) |
| JP (3) | JP7281903B2 (es) |
| KR (1) | KR102245021B1 (es) |
| CN (2) | CN109690314B (es) |
| AU (1) | AU2017261685B2 (es) |
| ES (1) | ES2808004T3 (es) |
| MX (1) | MX393482B (es) |
| PL (1) | PL3455631T3 (es) |
| RU (1) | RU2745730C2 (es) |
| SG (1) | SG11201809317VA (es) |
| WO (1) | WO2017194556A1 (es) |
| ZA (1) | ZA201807020B (es) |
Families Citing this family (21)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB202209539D0 (en) | 2017-06-13 | 2022-08-10 | Bostongene Corp | Systems and methods for identifying cancer treatments from normalized biomarker scores |
| EP3659110A1 (en) | 2017-07-24 | 2020-06-03 | Ventana Medical Systems, Inc. | Methods and systems for evaluation of immune cell infiltrate in tumor samples |
| ES2965647T3 (es) | 2018-01-31 | 2024-04-16 | Ventana Med Syst Inc | Procedimientos y sistemas para la evaluación de infiltrado de células inmunitarias en cáncer colorrectal en estadio III |
| FR3079617B1 (fr) | 2018-03-29 | 2023-12-22 | Office National Detude Et De Rech Aerospatiales Onera | Methode de detection de cellules presentant au moins une anomalie dans un echantillon cytologique |
| SG11202112712RA (en) * | 2019-06-03 | 2021-12-30 | Inst Nat Sante Rech Med | Methods for modulating a treatment regimen |
| CN110988324B (zh) * | 2019-11-29 | 2021-08-24 | 广州市雷德医学检验实验室有限公司 | 免疫状态确定系统、方法、装置及存储介质 |
| CN111257563B (zh) * | 2020-01-22 | 2022-08-23 | 广州泛恩生物科技有限公司 | Cxcl13检测剂在制备预测免疫治疗效果的试剂盒中的用途 |
| CN111999503B (zh) * | 2020-05-28 | 2022-05-20 | 首都医科大学附属北京地坛医院 | 一组用于预测急性病毒性呼吸道传染病重症化的标志物及其应用和试剂盒 |
| EP4172628A1 (en) * | 2020-06-30 | 2023-05-03 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Methods for predicting the risk of recurrence and/or death of patients suffering from a solid cancer after preoperative adjuvant therapy and radical surgery |
| CN113174439B (zh) * | 2021-03-30 | 2022-06-28 | 中国医学科学院肿瘤医院 | 一种基于免疫基因对评分体系在预测非小细胞肺癌患者免疫治疗效果中的应用 |
| WO2022229136A1 (en) * | 2021-04-27 | 2022-11-03 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Methods for predicting the risk of lymph node metastasis and/or recurrence of patients suffering from a t1 cancer treated by endoscopic resection |
| CA3218832A1 (en) * | 2021-05-13 | 2022-11-17 | CentricsBio, Inc. | Combined therapy using anti-cd300c antibody |
| US20250035635A1 (en) | 2022-03-17 | 2025-01-30 | Institut National De La Sante Et De La Recherche Medicale | Methods for predicting response to an immunotherapeutic treatment in a patient with a cancer |
| JP2025520176A (ja) * | 2022-06-03 | 2025-07-01 | ウニヴェルシタ・デグリ・ストゥディ・ディ・ローマ・ラ・サピエンツァ | 転移性腎癌患者における予後スコアの決定法 |
| CN115058504B (zh) * | 2022-07-14 | 2025-07-04 | 广州医科大学 | 膀胱癌th17 cd4+t细胞亚群及其特征基因与应用 |
| CN115747331B (zh) * | 2022-09-22 | 2023-08-25 | 中山大学肿瘤防治中心(中山大学附属肿瘤医院、中山大学肿瘤研究所) | 用于预测鼻咽癌预后的三级淋巴结构成分标志物组合、系统及应用 |
| TWI826081B (zh) * | 2022-10-28 | 2023-12-11 | 臺北醫學大學 | 癌症進展評估方法及其系統 |
| CN117476097B (zh) * | 2023-10-25 | 2024-06-07 | 中山大学附属第六医院 | 一种基于三级淋巴结构特征基因的结直肠癌预后和治疗反应预测模型及其构建方法和应用 |
| CN119685476A (zh) * | 2024-05-29 | 2025-03-25 | 深圳湾实验室 | 一种与癌症预后相关的基因标志物及其应用 |
| CN119291200A (zh) * | 2024-10-16 | 2025-01-10 | 华中科技大学同济医学院附属协和医院 | 一种用于检测cxcr4+cd8+t细胞用于预测肿瘤免疫治疗的检测试剂盒 |
| CN120259410B (zh) * | 2025-06-05 | 2025-09-23 | 北京大橡科技有限公司 | 基于双Panel多重生物标志物分析的肿瘤免疫响应可能性的评分方法 |
Family Cites Families (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN1771334A (zh) * | 2003-03-07 | 2006-05-10 | 施瑞修德制药公司 | 用于确定肿瘤对抗肿瘤药剂的治疗的敏感性的方法 |
| AR053272A1 (es) * | 2005-05-11 | 2007-04-25 | Hoffmann La Roche | Determinacion de responsivos a la quimioterapia |
| NZ562237A (en) * | 2007-10-05 | 2011-02-25 | Pacific Edge Biotechnology Ltd | Proliferation signature and prognosis for gastrointestinal cancer |
| WO2010120814A1 (en) * | 2009-04-14 | 2010-10-21 | Prometheus Laboratories Inc. | Inflammatory bowel disease prognostics |
| PL2619576T3 (pl) * | 2010-09-24 | 2021-07-05 | Niels Grabe | Środki i sposoby do prognozowania odpowiedzi na leczenie u pacjenta chorego na nowotwór złośliwy |
| GB201021289D0 (en) | 2010-12-15 | 2011-01-26 | Immatics Biotechnologies Gmbh | Novel biomarkers for a prediction of the outcome of an immunotherapy against cancer |
| EP2805165B1 (en) * | 2012-01-20 | 2018-03-14 | INSERM - Institut National de la Santé et de la Recherche Médicale | Methods for predicting the survival time of a patient suffering from a solid cancer based on density of b cells |
| ES2902879T3 (es) | 2012-06-14 | 2022-03-30 | Inst Nat Sante Rech Med | Método de cuantificación de células inmunitarias en tejidos tumorales y sus aplicaciones |
| EP2872646B1 (en) * | 2012-07-12 | 2017-08-30 | Institut National de la Sante et de la Recherche Medicale (INSERM) | Methods for predicting the survival time and treatment responsiveness of a patient suffering from a solid cancer with a signature of at least 7 genes |
| EP2880180B1 (en) | 2012-08-06 | 2018-10-03 | Institut National de la Sante et de la Recherche Medicale (INSERM) | Methods and kits for screening patients with a cancer |
| WO2014151606A2 (en) * | 2013-03-15 | 2014-09-25 | Oncomed Pharmaceuticals, Inc. | Methods of treating pancreatic cancer |
| US9851357B2 (en) * | 2013-07-15 | 2017-12-26 | Inserm (Institute National De La Sante Et De La Recherche Medicale | Method for the prognosis of survival time of a patient suffering from a solid cancer |
-
2017
- 2017-05-09 EP EP17725195.6A patent/EP3455631B1/en active Active
- 2017-05-09 CN CN201780028301.7A patent/CN109690314B/zh active Active
- 2017-05-09 WO PCT/EP2017/061089 patent/WO2017194556A1/en not_active Ceased
- 2017-05-09 MX MX2018013744A patent/MX393482B/es unknown
- 2017-05-09 RU RU2018143409A patent/RU2745730C2/ru active
- 2017-05-09 ES ES17725195T patent/ES2808004T3/es active Active
- 2017-05-09 AU AU2017261685A patent/AU2017261685B2/en active Active
- 2017-05-09 JP JP2018559227A patent/JP7281903B2/ja active Active
- 2017-05-09 SG SG11201809317VA patent/SG11201809317VA/en unknown
- 2017-05-09 CN CN202210976807.3A patent/CN115198018A/zh active Pending
- 2017-05-09 PL PL17725195T patent/PL3455631T3/pl unknown
- 2017-05-09 KR KR1020187035300A patent/KR102245021B1/ko active Active
- 2017-05-09 US US16/099,451 patent/US20190309369A1/en active Pending
-
2018
- 2018-10-22 ZA ZA2018/07020A patent/ZA201807020B/en unknown
-
2021
- 2021-11-19 JP JP2021188647A patent/JP7654318B2/ja active Active
-
2023
- 2023-12-14 JP JP2023211166A patent/JP2024019551A/ja active Pending
Also Published As
| Publication number | Publication date |
|---|---|
| JP2024019551A (ja) | 2024-02-09 |
| JP7654318B2 (ja) | 2025-04-01 |
| JP7281903B2 (ja) | 2023-05-26 |
| AU2017261685B2 (en) | 2023-09-07 |
| RU2018143409A3 (es) | 2020-06-29 |
| JP2019516979A (ja) | 2019-06-20 |
| BR112018072993A8 (pt) | 2022-11-22 |
| MX393482B (es) | 2025-03-24 |
| EP3455631B1 (en) | 2020-06-24 |
| JP2022027788A (ja) | 2022-02-14 |
| CN109690314A (zh) | 2019-04-26 |
| BR112018072993A2 (pt) | 2019-03-06 |
| US20190309369A1 (en) | 2019-10-10 |
| CN109690314B (zh) | 2022-08-02 |
| KR102245021B1 (ko) | 2021-04-26 |
| PL3455631T3 (pl) | 2021-04-06 |
| RU2745730C2 (ru) | 2021-03-31 |
| CN115198018A (zh) | 2022-10-18 |
| WO2017194556A1 (en) | 2017-11-16 |
| EP3455631A1 (en) | 2019-03-20 |
| RU2018143409A (ru) | 2020-06-10 |
| AU2017261685A1 (en) | 2018-11-15 |
| SG11201809317VA (en) | 2018-11-29 |
| ZA201807020B (en) | 2020-05-27 |
| MX2018013744A (es) | 2019-08-16 |
| KR20190016025A (ko) | 2019-02-15 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP7654318B2 (ja) | 固形腫瘍を有する患者をクラス分けするための方法 | |
| AU2013296233B2 (en) | Biomarker associated with risk of melanoma reoccurrence | |
| JP6995091B2 (ja) | 癌患者をスクリーニングするための方法及びキット | |
| JP7741831B2 (ja) | 術前補助療法及び根治手術後の固形がんを患っている患者の再発及び/又は死亡のリスクを予測するための方法 | |
| US20180216193A1 (en) | Methods for predicting the survival time and treatment responsiveness of a patient suffering from a solid cancer | |
| JP7554779B2 (ja) | 処置レジメンを調節する方法 | |
| HK40007449A (en) | Methods for classifying patients with a solid cancer | |
| HK40007449B (en) | Methods for classifying patients with a solid cancer | |
| BR112018072993B1 (pt) | Método in vitro para o prognóstico do tempo de sobrevivência de um paciente que sofre de um câncer sólido e método in vitro para avaliar a capacidade de resposta de um paciente que sofre de um câncer sólido a um tratamento antitumoral | |
| US20250320564A1 (en) | Methods and kits for treatment of cancer patients who are good responders to antitumoral treatment | |
| JP2020509354A (ja) | がんの重症度を評価するためのtim−3 |