ES2826992T3 - Evaluación de la estabilidad de un producto biológico en jeringas precargadas - Google Patents
Evaluación de la estabilidad de un producto biológico en jeringas precargadas Download PDFInfo
- Publication number
- ES2826992T3 ES2826992T3 ES17793966T ES17793966T ES2826992T3 ES 2826992 T3 ES2826992 T3 ES 2826992T3 ES 17793966 T ES17793966 T ES 17793966T ES 17793966 T ES17793966 T ES 17793966T ES 2826992 T3 ES2826992 T3 ES 2826992T3
- Authority
- ES
- Spain
- Prior art keywords
- container
- formula
- medical use
- alkyl
- alkenyl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 238000011156 evaluation Methods 0.000 title description 8
- 150000001875 compounds Chemical class 0.000 claims abstract description 49
- 238000000034 method Methods 0.000 claims abstract description 42
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 claims abstract description 22
- 230000015556 catabolic process Effects 0.000 claims abstract description 22
- 238000006731 degradation reaction Methods 0.000 claims abstract description 22
- 239000000243 solution Substances 0.000 claims abstract description 21
- 125000003118 aryl group Chemical group 0.000 claims abstract description 18
- 125000000882 C2-C6 alkenyl group Chemical group 0.000 claims abstract description 17
- 125000003601 C2-C6 alkynyl group Chemical group 0.000 claims abstract description 16
- 125000006239 protecting group Chemical group 0.000 claims abstract description 16
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 claims abstract description 13
- 238000004811 liquid chromatography Methods 0.000 claims abstract description 12
- 239000007864 aqueous solution Substances 0.000 claims abstract description 11
- 125000004454 (C1-C6) alkoxycarbonyl group Chemical group 0.000 claims abstract description 9
- 108090000765 processed proteins & peptides Proteins 0.000 claims description 30
- 229920001971 elastomer Polymers 0.000 claims description 15
- 229940071643 prefilled syringe Drugs 0.000 claims description 14
- 239000005060 rubber Substances 0.000 claims description 10
- WFKWXMTUELFFGS-UHFFFAOYSA-N tungsten Chemical compound [W] WFKWXMTUELFFGS-UHFFFAOYSA-N 0.000 claims description 10
- 229910052721 tungsten Inorganic materials 0.000 claims description 10
- 239000010937 tungsten Substances 0.000 claims description 10
- 239000000853 adhesive Substances 0.000 claims description 8
- 230000001070 adhesive effect Effects 0.000 claims description 8
- 238000002144 chemical decomposition reaction Methods 0.000 claims description 7
- 239000011521 glass Substances 0.000 claims description 6
- 239000004033 plastic Substances 0.000 claims description 5
- 229920003023 plastic Polymers 0.000 claims description 5
- 102000004196 processed proteins & peptides Human genes 0.000 claims description 5
- 239000002904 solvent Substances 0.000 claims description 5
- VYZAMTAEIAYCRO-UHFFFAOYSA-N Chromium Chemical compound [Cr] VYZAMTAEIAYCRO-UHFFFAOYSA-N 0.000 claims description 4
- 229910052782 aluminium Inorganic materials 0.000 claims description 4
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 claims description 4
- 150000001413 amino acids Chemical class 0.000 claims description 4
- 229910052751 metal Inorganic materials 0.000 claims description 4
- 239000002184 metal Substances 0.000 claims description 4
- 229920002545 silicone oil Polymers 0.000 claims description 4
- ZOXJGFHDIHLPTG-UHFFFAOYSA-N Boron Chemical compound [B] ZOXJGFHDIHLPTG-UHFFFAOYSA-N 0.000 claims description 3
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 claims description 3
- XUIMIQQOPSSXEZ-UHFFFAOYSA-N Silicon Chemical compound [Si] XUIMIQQOPSSXEZ-UHFFFAOYSA-N 0.000 claims description 3
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 claims description 3
- 229910052796 boron Inorganic materials 0.000 claims description 3
- 229910052749 magnesium Inorganic materials 0.000 claims description 3
- 239000011777 magnesium Substances 0.000 claims description 3
- 230000003647 oxidation Effects 0.000 claims description 3
- 238000007254 oxidation reaction Methods 0.000 claims description 3
- 239000000546 pharmaceutical excipient Substances 0.000 claims description 3
- 229910052710 silicon Inorganic materials 0.000 claims description 3
- 239000010703 silicon Substances 0.000 claims description 3
- 239000004094 surface-active agent Substances 0.000 claims description 3
- 239000011701 zinc Substances 0.000 claims description 3
- 229910052725 zinc Inorganic materials 0.000 claims description 3
- 239000002253 acid Substances 0.000 claims description 2
- 150000007513 acids Chemical class 0.000 claims description 2
- 239000002585 base Substances 0.000 claims description 2
- 239000006172 buffering agent Substances 0.000 claims description 2
- 239000002738 chelating agent Substances 0.000 claims description 2
- 230000009615 deamination Effects 0.000 claims description 2
- 238000006481 deamination reaction Methods 0.000 claims description 2
- 238000004949 mass spectrometry Methods 0.000 claims description 2
- 238000005259 measurement Methods 0.000 claims description 2
- 239000003755 preservative agent Substances 0.000 claims description 2
- 150000003839 salts Chemical class 0.000 claims description 2
- 235000000346 sugar Nutrition 0.000 claims description 2
- 150000008163 sugars Chemical class 0.000 claims description 2
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 claims description 2
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 abstract description 9
- 125000003342 alkenyl group Chemical group 0.000 abstract description 2
- 125000000217 alkyl group Chemical group 0.000 abstract 2
- 125000000304 alkynyl group Chemical group 0.000 abstract 1
- 239000000047 product Substances 0.000 description 35
- -1 polyethylene Polymers 0.000 description 32
- 239000012632 extractable Substances 0.000 description 11
- 239000012633 leachable Substances 0.000 description 11
- 238000001195 ultra high performance liquid chromatography Methods 0.000 description 9
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 8
- 239000000203 mixture Substances 0.000 description 8
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 7
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 6
- 238000012545 processing Methods 0.000 description 6
- 235000018102 proteins Nutrition 0.000 description 6
- 102000004169 proteins and genes Human genes 0.000 description 6
- 108090000623 proteins and genes Proteins 0.000 description 6
- 238000006243 chemical reaction Methods 0.000 description 5
- 239000007857 degradation product Substances 0.000 description 5
- 239000000806 elastomer Substances 0.000 description 5
- 229920000840 ethylene tetrafluoroethylene copolymer Polymers 0.000 description 5
- 230000003993 interaction Effects 0.000 description 5
- 238000004519 manufacturing process Methods 0.000 description 5
- 239000000463 material Substances 0.000 description 5
- 238000003860 storage Methods 0.000 description 5
- 125000002777 acetyl group Chemical class [H]C([H])([H])C(*)=O 0.000 description 4
- 239000008186 active pharmaceutical agent Substances 0.000 description 4
- 238000013459 approach Methods 0.000 description 4
- 230000015572 biosynthetic process Effects 0.000 description 4
- 239000000872 buffer Substances 0.000 description 4
- 125000004432 carbon atom Chemical group C* 0.000 description 4
- 238000011161 development Methods 0.000 description 4
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 4
- 230000014759 maintenance of location Effects 0.000 description 4
- 239000000825 pharmaceutical preparation Substances 0.000 description 4
- 239000000126 substance Substances 0.000 description 4
- 229920002725 thermoplastic elastomer Polymers 0.000 description 4
- USFZMSVCRYTOJT-UHFFFAOYSA-N Ammonium acetate Chemical compound N.CC(O)=O USFZMSVCRYTOJT-UHFFFAOYSA-N 0.000 description 3
- 239000005695 Ammonium acetate Substances 0.000 description 3
- 102100027508 RANBP2-like and GRIP domain-containing protein 5/6 Human genes 0.000 description 3
- 229940024606 amino acid Drugs 0.000 description 3
- 235000001014 amino acid Nutrition 0.000 description 3
- 229940043376 ammonium acetate Drugs 0.000 description 3
- 235000019257 ammonium acetate Nutrition 0.000 description 3
- 229960000074 biopharmaceutical Drugs 0.000 description 3
- 229910052804 chromium Inorganic materials 0.000 description 3
- 239000011651 chromium Substances 0.000 description 3
- LOKCTEFSRHRXRJ-UHFFFAOYSA-I dipotassium trisodium dihydrogen phosphate hydrogen phosphate dichloride Chemical compound P(=O)(O)(O)[O-].[K+].P(=O)(O)([O-])[O-].[Na+].[Na+].[Cl-].[K+].[Cl-].[Na+] LOKCTEFSRHRXRJ-UHFFFAOYSA-I 0.000 description 3
- 239000003814 drug Substances 0.000 description 3
- 238000005755 formation reaction Methods 0.000 description 3
- 238000009472 formulation Methods 0.000 description 3
- 150000002430 hydrocarbons Chemical group 0.000 description 3
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 3
- 125000004184 methoxymethyl group Chemical group [H]C([H])([H])OC([H])([H])* 0.000 description 3
- 238000012986 modification Methods 0.000 description 3
- 230000004048 modification Effects 0.000 description 3
- 239000002953 phosphate buffered saline Substances 0.000 description 3
- 235000010482 polyoxyethylene sorbitan monooleate Nutrition 0.000 description 3
- 229920000053 polysorbate 80 Polymers 0.000 description 3
- 238000011160 research Methods 0.000 description 3
- 229910001220 stainless steel Inorganic materials 0.000 description 3
- 239000010935 stainless steel Substances 0.000 description 3
- 238000003786 synthesis reaction Methods 0.000 description 3
- 125000002221 trityl group Chemical group [H]C1=C([H])C([H])=C([H])C([H])=C1C([*])(C1=C(C(=C(C(=C1[H])[H])[H])[H])[H])C1=C([H])C([H])=C([H])C([H])=C1[H] 0.000 description 3
- YQTCQNIPQMJNTI-UHFFFAOYSA-N 2,2-dimethylpropan-1-one Chemical group CC(C)(C)[C]=O YQTCQNIPQMJNTI-UHFFFAOYSA-N 0.000 description 2
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 2
- 239000004812 Fluorinated ethylene propylene Substances 0.000 description 2
- 101000579956 Homo sapiens RANBP2-like and GRIP domain-containing protein 5/6 Proteins 0.000 description 2
- XEEYBQQBJWHFJM-UHFFFAOYSA-N Iron Chemical compound [Fe] XEEYBQQBJWHFJM-UHFFFAOYSA-N 0.000 description 2
- DCXYFEDJOCDNAF-REOHCLBHSA-N L-asparagine Chemical compound OC(=O)[C@@H](N)CC(N)=O DCXYFEDJOCDNAF-REOHCLBHSA-N 0.000 description 2
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 2
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 2
- ZDXPYRJPNDTMRX-VKHMYHEASA-N L-glutamine Chemical compound OC(=O)[C@@H](N)CCC(N)=O ZDXPYRJPNDTMRX-VKHMYHEASA-N 0.000 description 2
- KDXKERNSBIXSRK-YFKPBYRVSA-N L-lysine Chemical compound NCCCC[C@H](N)C(O)=O KDXKERNSBIXSRK-YFKPBYRVSA-N 0.000 description 2
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 2
- 241001465754 Metazoa Species 0.000 description 2
- PXHVJJICTQNCMI-UHFFFAOYSA-N Nickel Chemical compound [Ni] PXHVJJICTQNCMI-UHFFFAOYSA-N 0.000 description 2
- 239000004698 Polyethylene Substances 0.000 description 2
- 239000004743 Polypropylene Substances 0.000 description 2
- 229920001328 Polyvinylidene chloride Polymers 0.000 description 2
- 239000000654 additive Substances 0.000 description 2
- 150000001412 amines Chemical group 0.000 description 2
- 238000004458 analytical method Methods 0.000 description 2
- 238000003556 assay Methods 0.000 description 2
- 125000003236 benzoyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C(*)=O 0.000 description 2
- 125000001584 benzyloxycarbonyl group Chemical group C(=O)(OCC1=CC=CC=C1)* 0.000 description 2
- IKZZIQXKLWDPCD-UHFFFAOYSA-N but-1-en-2-ol Chemical compound CCC(O)=C IKZZIQXKLWDPCD-UHFFFAOYSA-N 0.000 description 2
- 230000008859 change Effects 0.000 description 2
- 235000013870 dimethyl polysiloxane Nutrition 0.000 description 2
- 229940079593 drug Drugs 0.000 description 2
- 230000000694 effects Effects 0.000 description 2
- 125000005448 ethoxyethyl group Chemical group [H]C([H])([H])C([H])([H])OC([H])([H])C([H])([H])* 0.000 description 2
- 238000000105 evaporative light scattering detection Methods 0.000 description 2
- 125000005843 halogen group Chemical group 0.000 description 2
- 238000004128 high performance liquid chromatography Methods 0.000 description 2
- 230000007774 longterm Effects 0.000 description 2
- 239000000314 lubricant Substances 0.000 description 2
- 238000001819 mass spectrum Methods 0.000 description 2
- 150000002739 metals Chemical class 0.000 description 2
- 229920009441 perflouroethylene propylene Polymers 0.000 description 2
- 229940127557 pharmaceutical product Drugs 0.000 description 2
- 230000000144 pharmacologic effect Effects 0.000 description 2
- 229920000435 poly(dimethylsiloxane) Polymers 0.000 description 2
- 229920002492 poly(sulfone) Polymers 0.000 description 2
- 229920000573 polyethylene Polymers 0.000 description 2
- 229920000139 polyethylene terephthalate Polymers 0.000 description 2
- 239000005020 polyethylene terephthalate Substances 0.000 description 2
- 229920001155 polypropylene Polymers 0.000 description 2
- 229920001343 polytetrafluoroethylene Polymers 0.000 description 2
- 239000004810 polytetrafluoroethylene Substances 0.000 description 2
- 238000012216 screening Methods 0.000 description 2
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 2
- 125000001412 tetrahydropyranyl group Chemical group 0.000 description 2
- 229930195735 unsaturated hydrocarbon Natural products 0.000 description 2
- 239000003643 water by type Substances 0.000 description 2
- MTCFGRXMJLQNBG-REOHCLBHSA-N (2S)-2-Amino-3-hydroxypropansäure Chemical compound OC[C@H](N)C(O)=O MTCFGRXMJLQNBG-REOHCLBHSA-N 0.000 description 1
- 125000004191 (C1-C6) alkoxy group Chemical group 0.000 description 1
- 125000003088 (fluoren-9-ylmethoxy)carbonyl group Chemical group 0.000 description 1
- 125000000453 2,2,2-trichloroethyl group Chemical group [H]C([H])(*)C(Cl)(Cl)Cl 0.000 description 1
- HJKLEAOXCZIMPI-UHFFFAOYSA-N 2,2-diethoxyethanamine Chemical class CCOC(CN)OCC HJKLEAOXCZIMPI-UHFFFAOYSA-N 0.000 description 1
- SMZOUWXMTYCWNB-UHFFFAOYSA-N 2-(2-methoxy-5-methylphenyl)ethanamine Chemical compound COC1=CC=C(C)C=C1CCN SMZOUWXMTYCWNB-UHFFFAOYSA-N 0.000 description 1
- 125000003821 2-(trimethylsilyl)ethoxymethyl group Chemical group [H]C([H])([H])[Si](C([H])([H])[H])(C([H])([H])[H])C([H])([H])C(OC([H])([H])[*])([H])[H] 0.000 description 1
- NIXOWILDQLNWCW-UHFFFAOYSA-N 2-Propenoic acid Natural products OC(=O)C=C NIXOWILDQLNWCW-UHFFFAOYSA-N 0.000 description 1
- 125000003903 2-propenyl group Chemical group [H]C([*])([H])C([H])=C([H])[H] 0.000 description 1
- 125000002774 3,4-dimethoxybenzyl group Chemical group [H]C1=C([H])C(=C([H])C(OC([H])([H])[H])=C1OC([H])([H])[H])C([H])([H])* 0.000 description 1
- 125000004172 4-methoxyphenyl group Chemical group [H]C1=C([H])C(OC([H])([H])[H])=C([H])C([H])=C1* 0.000 description 1
- FIHBHSQYSYVZQE-UHFFFAOYSA-N 6-prop-2-enoyloxyhexyl prop-2-enoate Chemical compound C=CC(=O)OCCCCCCOC(=O)C=C FIHBHSQYSYVZQE-UHFFFAOYSA-N 0.000 description 1
- SGODNSMVTKYTGP-UHFFFAOYSA-N 8-chloro-2-methylocta-1,3-diene Chemical compound CC(=C)C=CCCCCCl SGODNSMVTKYTGP-UHFFFAOYSA-N 0.000 description 1
- 239000004475 Arginine Substances 0.000 description 1
- DCXYFEDJOCDNAF-UHFFFAOYSA-N Asparagine Natural products OC(=O)C(N)CC(N)=O DCXYFEDJOCDNAF-UHFFFAOYSA-N 0.000 description 1
- 239000002028 Biomass Substances 0.000 description 1
- DCERHCFNWRGHLK-UHFFFAOYSA-N C[Si](C)C Chemical compound C[Si](C)C DCERHCFNWRGHLK-UHFFFAOYSA-N 0.000 description 1
- KXDHJXZQYSOELW-UHFFFAOYSA-M Carbamate Chemical compound NC([O-])=O KXDHJXZQYSOELW-UHFFFAOYSA-M 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-L Carbonate Chemical compound [O-]C([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-L 0.000 description 1
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical compound [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 description 1
- 229920002943 EPDM rubber Polymers 0.000 description 1
- 244000043261 Hevea brasiliensis Species 0.000 description 1
- 101000579953 Homo sapiens RANBP2-like and GRIP domain-containing protein 2 Chemical group 0.000 description 1
- 101000579954 Homo sapiens RanBP2-like and GRIP domain-containing protein 3 Proteins 0.000 description 1
- 101001096534 Homo sapiens Regulator of G-protein signaling 3 Proteins 0.000 description 1
- XUJNEKJLAYXESH-REOHCLBHSA-N L-Cysteine Chemical compound SC[C@H](N)C(O)=O XUJNEKJLAYXESH-REOHCLBHSA-N 0.000 description 1
- ONIBWKKTOPOVIA-BYPYZUCNSA-N L-Proline Chemical compound OC(=O)[C@@H]1CCCN1 ONIBWKKTOPOVIA-BYPYZUCNSA-N 0.000 description 1
- ODKSFYDXXFIFQN-BYPYZUCNSA-P L-argininium(2+) Chemical compound NC(=[NH2+])NCCC[C@H]([NH3+])C(O)=O ODKSFYDXXFIFQN-BYPYZUCNSA-P 0.000 description 1
- HNDVDQJCIGZPNO-YFKPBYRVSA-N L-histidine Chemical compound OC(=O)[C@@H](N)CC1=CN=CN1 HNDVDQJCIGZPNO-YFKPBYRVSA-N 0.000 description 1
- FFEARJCKVFRZRR-BYPYZUCNSA-N L-methionine Chemical compound CSCC[C@H](N)C(O)=O FFEARJCKVFRZRR-BYPYZUCNSA-N 0.000 description 1
- COLNVLDHVKWLRT-QMMMGPOBSA-N L-phenylalanine Chemical compound OC(=O)[C@@H](N)CC1=CC=CC=C1 COLNVLDHVKWLRT-QMMMGPOBSA-N 0.000 description 1
- AYFVYJQAPQTCCC-GBXIJSLDSA-N L-threonine Chemical compound C[C@@H](O)[C@H](N)C(O)=O AYFVYJQAPQTCCC-GBXIJSLDSA-N 0.000 description 1
- QIVBCDIJIAJPQS-VIFPVBQESA-N L-tryptophane Chemical compound C1=CC=C2C(C[C@H](N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-VIFPVBQESA-N 0.000 description 1
- OUYCCCASQSFEME-QMMMGPOBSA-N L-tyrosine Chemical compound OC(=O)[C@@H](N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-QMMMGPOBSA-N 0.000 description 1
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 1
- 241000721701 Lynx Species 0.000 description 1
- 239000004472 Lysine Substances 0.000 description 1
- CERQOIWHTDAKMF-UHFFFAOYSA-N Methacrylic acid Chemical compound CC(=C)C(O)=O CERQOIWHTDAKMF-UHFFFAOYSA-N 0.000 description 1
- 229920000459 Nitrile rubber Polymers 0.000 description 1
- 229920006169 Perfluoroelastomer Polymers 0.000 description 1
- 239000004952 Polyamide Substances 0.000 description 1
- 229920002614 Polyether block amide Polymers 0.000 description 1
- 101710136728 Probable UDP-arabinopyranose mutase 1 Proteins 0.000 description 1
- ONIBWKKTOPOVIA-UHFFFAOYSA-N Proline Natural products OC(=O)C1CCCN1 ONIBWKKTOPOVIA-UHFFFAOYSA-N 0.000 description 1
- 102100027505 RANBP2-like and GRIP domain-containing protein 1 Human genes 0.000 description 1
- 102100027511 RANBP2-like and GRIP domain-containing protein 2 Human genes 0.000 description 1
- 102100027510 RanBP2-like and GRIP domain-containing protein 3 Human genes 0.000 description 1
- 102100027509 RanBP2-like and GRIP domain-containing protein 4 Human genes 0.000 description 1
- 101710140404 Regulator of G-protein signaling 4 Proteins 0.000 description 1
- MTCFGRXMJLQNBG-UHFFFAOYSA-N Serine Natural products OCC(N)C(O)=O MTCFGRXMJLQNBG-UHFFFAOYSA-N 0.000 description 1
- 101000665501 Solanum tuberosum Probable UDP-arabinopyranose mutase 2 Chemical group 0.000 description 1
- 229910000831 Steel Inorganic materials 0.000 description 1
- AYFVYJQAPQTCCC-UHFFFAOYSA-N Threonine Natural products CC(O)C(N)C(O)=O AYFVYJQAPQTCCC-UHFFFAOYSA-N 0.000 description 1
- 239000004473 Threonine Substances 0.000 description 1
- RTAQQCXQSZGOHL-UHFFFAOYSA-N Titanium Chemical compound [Ti] RTAQQCXQSZGOHL-UHFFFAOYSA-N 0.000 description 1
- QIVBCDIJIAJPQS-UHFFFAOYSA-N Tryptophan Natural products C1=CC=C2C(CC(N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-UHFFFAOYSA-N 0.000 description 1
- DHKHKXVYLBGOIT-UHFFFAOYSA-N acetaldehyde Diethyl Acetal Natural products CCOC(C)OCC DHKHKXVYLBGOIT-UHFFFAOYSA-N 0.000 description 1
- 239000002671 adjuvant Substances 0.000 description 1
- 238000004220 aggregation Methods 0.000 description 1
- 230000002776 aggregation Effects 0.000 description 1
- 238000013019 agitation Methods 0.000 description 1
- 125000003545 alkoxy group Chemical group 0.000 description 1
- HSFWRNGVRCDJHI-UHFFFAOYSA-N alpha-acetylene Natural products C#C HSFWRNGVRCDJHI-UHFFFAOYSA-N 0.000 description 1
- 150000001408 amides Chemical class 0.000 description 1
- 229910052787 antimony Inorganic materials 0.000 description 1
- WATWJIUSRGPENY-UHFFFAOYSA-N antimony atom Chemical compound [Sb] WATWJIUSRGPENY-UHFFFAOYSA-N 0.000 description 1
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 1
- 235000009697 arginine Nutrition 0.000 description 1
- 125000002029 aromatic hydrocarbon group Chemical group 0.000 description 1
- 229910052785 arsenic Inorganic materials 0.000 description 1
- RQNWIZPPADIBDY-UHFFFAOYSA-N arsenic atom Chemical compound [As] RQNWIZPPADIBDY-UHFFFAOYSA-N 0.000 description 1
- 229960001230 asparagine Drugs 0.000 description 1
- 235000009582 asparagine Nutrition 0.000 description 1
- 229940009098 aspartate Drugs 0.000 description 1
- 229910052788 barium Inorganic materials 0.000 description 1
- DSAJWYNOEDNPEQ-UHFFFAOYSA-N barium atom Chemical compound [Ba] DSAJWYNOEDNPEQ-UHFFFAOYSA-N 0.000 description 1
- 238000003339 best practice Methods 0.000 description 1
- 229920005557 bromobutyl Polymers 0.000 description 1
- 125000004369 butenyl group Chemical group C(=CCC)* 0.000 description 1
- 229920005549 butyl rubber Polymers 0.000 description 1
- 125000000480 butynyl group Chemical group [*]C#CC([H])([H])C([H])([H])[H] 0.000 description 1
- 125000003636 chemical group Chemical group 0.000 description 1
- 238000007385 chemical modification Methods 0.000 description 1
- 239000003153 chemical reaction reagent Substances 0.000 description 1
- 229910052801 chlorine Inorganic materials 0.000 description 1
- 239000000460 chlorine Substances 0.000 description 1
- 125000001309 chloro group Chemical group Cl* 0.000 description 1
- 238000004587 chromatography analysis Methods 0.000 description 1
- 238000003776 cleavage reaction Methods 0.000 description 1
- 239000011248 coating agent Substances 0.000 description 1
- 238000000576 coating method Methods 0.000 description 1
- 239000010941 cobalt Substances 0.000 description 1
- 229910017052 cobalt Inorganic materials 0.000 description 1
- GUTLYIVDDKVIGB-UHFFFAOYSA-N cobalt atom Chemical compound [Co] GUTLYIVDDKVIGB-UHFFFAOYSA-N 0.000 description 1
- 230000001427 coherent effect Effects 0.000 description 1
- 229920000891 common polymer Polymers 0.000 description 1
- 229910052802 copper Inorganic materials 0.000 description 1
- 239000010949 copper Substances 0.000 description 1
- XUJNEKJLAYXESH-UHFFFAOYSA-N cysteine Natural products SCC(N)C(O)=O XUJNEKJLAYXESH-UHFFFAOYSA-N 0.000 description 1
- 235000018417 cysteine Nutrition 0.000 description 1
- 238000003066 decision tree Methods 0.000 description 1
- 238000012217 deletion Methods 0.000 description 1
- 238000001514 detection method Methods 0.000 description 1
- 238000003745 diagnosis Methods 0.000 description 1
- JBSLOWBPDRZSMB-FPLPWBNLSA-N dibutyl (z)-but-2-enedioate Chemical compound CCCCOC(=O)\C=C/C(=O)OCCCC JBSLOWBPDRZSMB-FPLPWBNLSA-N 0.000 description 1
- 201000010099 disease Diseases 0.000 description 1
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 1
- 238000001647 drug administration Methods 0.000 description 1
- 238000012377 drug delivery Methods 0.000 description 1
- 238000012362 drug development process Methods 0.000 description 1
- 229940088679 drug related substance Drugs 0.000 description 1
- 230000009881 electrostatic interaction Effects 0.000 description 1
- 150000002081 enamines Chemical class 0.000 description 1
- 150000002148 esters Chemical class 0.000 description 1
- QHSJIZLJUFMIFP-UHFFFAOYSA-N ethene;1,1,2,2-tetrafluoroethene Chemical group C=C.FC(F)=C(F)F QHSJIZLJUFMIFP-UHFFFAOYSA-N 0.000 description 1
- HQQADJVZYDDRJT-UHFFFAOYSA-N ethene;prop-1-ene Chemical group C=C.CC=C HQQADJVZYDDRJT-UHFFFAOYSA-N 0.000 description 1
- 125000003754 ethoxycarbonyl group Chemical group C(=O)(OCC)* 0.000 description 1
- 239000005038 ethylene vinyl acetate Substances 0.000 description 1
- 125000002534 ethynyl group Chemical group [H]C#C* 0.000 description 1
- 230000001747 exhibiting effect Effects 0.000 description 1
- 238000000605 extraction Methods 0.000 description 1
- 238000011049 filling Methods 0.000 description 1
- 238000005429 filling process Methods 0.000 description 1
- 239000012530 fluid Substances 0.000 description 1
- 229920001973 fluoroelastomer Polymers 0.000 description 1
- 229920005560 fluorosilicone rubber Polymers 0.000 description 1
- 125000002485 formyl group Chemical group [H]C(*)=O 0.000 description 1
- 231100000024 genotoxic Toxicity 0.000 description 1
- 230000001738 genotoxic effect Effects 0.000 description 1
- 229930195712 glutamate Natural products 0.000 description 1
- ZDXPYRJPNDTMRX-UHFFFAOYSA-N glutamine Natural products OC(=O)C(N)CCC(N)=O ZDXPYRJPNDTMRX-UHFFFAOYSA-N 0.000 description 1
- 235000004554 glutamine Nutrition 0.000 description 1
- 229920005555 halobutyl Polymers 0.000 description 1
- 125000006038 hexenyl group Chemical group 0.000 description 1
- 125000005980 hexynyl group Chemical group 0.000 description 1
- HNDVDQJCIGZPNO-UHFFFAOYSA-N histidine Natural products OC(=O)C(N)CC1=CN=CN1 HNDVDQJCIGZPNO-UHFFFAOYSA-N 0.000 description 1
- 235000014304 histidine Nutrition 0.000 description 1
- 230000002209 hydrophobic effect Effects 0.000 description 1
- 229920002681 hypalon Polymers 0.000 description 1
- 150000002466 imines Chemical class 0.000 description 1
- 230000005847 immunogenicity Effects 0.000 description 1
- 239000004615 ingredient Substances 0.000 description 1
- 238000002347 injection Methods 0.000 description 1
- 239000007924 injection Substances 0.000 description 1
- 229910052742 iron Inorganic materials 0.000 description 1
- 239000002085 irritant Substances 0.000 description 1
- 231100000021 irritant Toxicity 0.000 description 1
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 238000002372 labelling Methods 0.000 description 1
- 238000002386 leaching Methods 0.000 description 1
- 238000002025 liquid chromatography-photodiode array detection Methods 0.000 description 1
- 229910052744 lithium Inorganic materials 0.000 description 1
- WPBNNNQJVZRUHP-UHFFFAOYSA-L manganese(2+);methyl n-[[2-(methoxycarbonylcarbamothioylamino)phenyl]carbamothioyl]carbamate;n-[2-(sulfidocarbothioylamino)ethyl]carbamodithioate Chemical compound [Mn+2].[S-]C(=S)NCCNC([S-])=S.COC(=O)NC(=S)NC1=CC=CC=C1NC(=S)NC(=O)OC WPBNNNQJVZRUHP-UHFFFAOYSA-L 0.000 description 1
- 229930182817 methionine Natural products 0.000 description 1
- 125000001160 methoxycarbonyl group Chemical group [H]C([H])([H])OC(*)=O 0.000 description 1
- GPKUICFDWYEPTK-UHFFFAOYSA-N methoxycyclohexatriene Chemical compound COC1=CC=C=C[CH]1 GPKUICFDWYEPTK-UHFFFAOYSA-N 0.000 description 1
- 125000004092 methylthiomethyl group Chemical group [H]C([H])([H])SC([H])([H])* 0.000 description 1
- 230000003278 mimic effect Effects 0.000 description 1
- 230000000116 mitigating effect Effects 0.000 description 1
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000001280 n-hexyl group Chemical group C(CCCCC)* 0.000 description 1
- 125000000740 n-pentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000001624 naphthyl group Chemical group 0.000 description 1
- 229920003052 natural elastomer Polymers 0.000 description 1
- 229920001194 natural rubber Polymers 0.000 description 1
- 229910052759 nickel Inorganic materials 0.000 description 1
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 description 1
- 230000000269 nucleophilic effect Effects 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- 230000001590 oxidative effect Effects 0.000 description 1
- 125000004430 oxygen atom Chemical group O* 0.000 description 1
- 238000004806 packaging method and process Methods 0.000 description 1
- 239000002245 particle Substances 0.000 description 1
- 125000002255 pentenyl group Chemical group C(=CCCC)* 0.000 description 1
- 125000005981 pentynyl group Chemical group 0.000 description 1
- COLNVLDHVKWLRT-UHFFFAOYSA-N phenylalanine Natural products OC(=O)C(N)CC1=CC=CC=C1 COLNVLDHVKWLRT-UHFFFAOYSA-N 0.000 description 1
- UYWQUFXKFGHYNT-UHFFFAOYSA-N phenylmethyl ester of formic acid Natural products O=COCC1=CC=CC=C1 UYWQUFXKFGHYNT-UHFFFAOYSA-N 0.000 description 1
- 125000003170 phenylsulfonyl group Chemical group C1(=CC=CC=C1)S(=O)(=O)* 0.000 description 1
- 229920001200 poly(ethylene-vinyl acetate) Polymers 0.000 description 1
- 229920005559 polyacrylic rubber Polymers 0.000 description 1
- 229920002647 polyamide Polymers 0.000 description 1
- 239000004417 polycarbonate Substances 0.000 description 1
- 229920000515 polycarbonate Polymers 0.000 description 1
- 229920000728 polyester Polymers 0.000 description 1
- 229920000642 polymer Polymers 0.000 description 1
- 238000002360 preparation method Methods 0.000 description 1
- 230000002265 prevention Effects 0.000 description 1
- 125000004368 propenyl group Chemical group C(=CC)* 0.000 description 1
- 125000002568 propynyl group Chemical group [*]C#CC([H])([H])[H] 0.000 description 1
- 238000012797 qualification Methods 0.000 description 1
- 238000010948 quality risk assessment Methods 0.000 description 1
- 230000006340 racemization Effects 0.000 description 1
- 239000002994 raw material Substances 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 238000006722 reduction reaction Methods 0.000 description 1
- 238000012502 risk assessment Methods 0.000 description 1
- 229920006395 saturated elastomer Polymers 0.000 description 1
- 229930195734 saturated hydrocarbon Natural products 0.000 description 1
- 230000007017 scission Effects 0.000 description 1
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 230000035945 sensitivity Effects 0.000 description 1
- 231100000202 sensitizing Toxicity 0.000 description 1
- 230000001235 sensitizing effect Effects 0.000 description 1
- 229920002379 silicone rubber Polymers 0.000 description 1
- 239000004945 silicone rubber Substances 0.000 description 1
- 125000003808 silyl group Chemical group [H][Si]([H])([H])[*] 0.000 description 1
- 238000001228 spectrum Methods 0.000 description 1
- 238000007619 statistical method Methods 0.000 description 1
- 239000010959 steel Substances 0.000 description 1
- 229920003048 styrene butadiene rubber Polymers 0.000 description 1
- 125000001424 substituent group Chemical group 0.000 description 1
- 238000006467 substitution reaction Methods 0.000 description 1
- 229940124530 sulfonamide Drugs 0.000 description 1
- 150000003456 sulfonamides Chemical class 0.000 description 1
- 229920003051 synthetic elastomer Polymers 0.000 description 1
- ILMRJRBKQSSXGY-UHFFFAOYSA-N tert-butyl(dimethyl)silicon Chemical group C[Si](C)C(C)(C)C ILMRJRBKQSSXGY-UHFFFAOYSA-N 0.000 description 1
- 125000001981 tert-butyldimethylsilyl group Chemical group [H]C([H])([H])[Si]([H])(C([H])([H])[H])[*]C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 125000005931 tert-butyloxycarbonyl group Chemical group [H]C([H])([H])C(OC(*)=O)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 238000012360 testing method Methods 0.000 description 1
- 125000003718 tetrahydrofuranyl group Chemical group 0.000 description 1
- 230000001225 therapeutic effect Effects 0.000 description 1
- 229920001169 thermoplastic Polymers 0.000 description 1
- 239000004416 thermosoftening plastic Substances 0.000 description 1
- 239000010936 titanium Substances 0.000 description 1
- 229910052719 titanium Inorganic materials 0.000 description 1
- 125000002088 tosyl group Chemical group [H]C1=C([H])C(=C([H])C([H])=C1C([H])([H])[H])S(*)(=O)=O 0.000 description 1
- 231100000331 toxic Toxicity 0.000 description 1
- 230000002588 toxic effect Effects 0.000 description 1
- 238000012546 transfer Methods 0.000 description 1
- 125000004665 trialkylsilyl group Chemical group 0.000 description 1
- 125000000026 trimethylsilyl group Chemical group [H]C([H])([H])[Si]([*])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- OUYCCCASQSFEME-UHFFFAOYSA-N tyrosine Natural products OC(=O)C(N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-UHFFFAOYSA-N 0.000 description 1
- 229960005486 vaccine Drugs 0.000 description 1
- 238000010200 validation analysis Methods 0.000 description 1
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 1
- 239000002699 waste material Substances 0.000 description 1
Classifications
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N21/00—Investigating or analysing materials by the use of optical means, i.e. using sub-millimetre waves, infrared, visible or ultraviolet light
- G01N21/75—Systems in which material is subjected to a chemical reaction, the progress or the result of the reaction being investigated
- G01N21/77—Systems in which material is subjected to a chemical reaction, the progress or the result of the reaction being investigated by observing the effect on a chemical indicator
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K7/00—Peptides having 5 to 20 amino acids in a fully defined sequence; Derivatives thereof
- C07K7/04—Linear peptides containing only normal peptide links
- C07K7/06—Linear peptides containing only normal peptide links having 5 to 11 amino acids
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N30/00—Investigating or analysing materials by separation into components using adsorption, absorption or similar phenomena or using ion-exchange, e.g. chromatography or field flow fractionation
- G01N30/02—Column chromatography
- G01N30/04—Preparation or injection of sample to be analysed
- G01N30/06—Preparation
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N30/00—Investigating or analysing materials by separation into components using adsorption, absorption or similar phenomena or using ion-exchange, e.g. chromatography or field flow fractionation
- G01N30/02—Column chromatography
- G01N30/62—Detectors specially adapted therefor
- G01N30/72—Mass spectrometers
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N30/00—Investigating or analysing materials by separation into components using adsorption, absorption or similar phenomena or using ion-exchange, e.g. chromatography or field flow fractionation
- G01N30/02—Column chromatography
- G01N30/62—Detectors specially adapted therefor
- G01N30/72—Mass spectrometers
- G01N30/7233—Mass spectrometers interfaced to liquid or supercritical fluid chromatograph
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/68—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving proteins, peptides or amino acids
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N30/00—Investigating or analysing materials by separation into components using adsorption, absorption or similar phenomena or using ion-exchange, e.g. chromatography or field flow fractionation
- G01N30/02—Column chromatography
- G01N2030/022—Column chromatography characterised by the kind of separation mechanism
- G01N2030/027—Liquid chromatography
Landscapes
- Life Sciences & Earth Sciences (AREA)
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Immunology (AREA)
- Biochemistry (AREA)
- General Health & Medical Sciences (AREA)
- Engineering & Computer Science (AREA)
- Physics & Mathematics (AREA)
- Analytical Chemistry (AREA)
- General Physics & Mathematics (AREA)
- Pathology (AREA)
- Molecular Biology (AREA)
- Biomedical Technology (AREA)
- Hematology (AREA)
- Urology & Nephrology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Medicinal Chemistry (AREA)
- Organic Chemistry (AREA)
- Biotechnology (AREA)
- Microbiology (AREA)
- Cell Biology (AREA)
- Food Science & Technology (AREA)
- Genetics & Genomics (AREA)
- Biophysics (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Plasma & Fusion (AREA)
- Peptides Or Proteins (AREA)
- Investigating Or Analysing Biological Materials (AREA)
- Medical Preparation Storing Or Oral Administration Devices (AREA)
- Investigating Or Analyzing Non-Biological Materials By The Use Of Chemical Means (AREA)
- Infusion, Injection, And Reservoir Apparatuses (AREA)
Abstract
Método para determinar el porcentaje de degradación de un compuesto de fórmula (I) mediante el contacto con parte de un recipiente para uso médico, que comprende las etapas sucesivas siguientes: i) preparar una disolución acuosa que comprende un compuesto de fórmula (I) a continuación: **(Ver fórmula)** en la que: R1, R2, R4 y R5 representan cada uno independientemente de otro H o un grupo alquilo (C1-C6), alquenilo (C2-C6) o alquinilo (C2-C6); X representa H o un grupo alquilo (C1-C6), alquenilo (C2-C6), alquinilo (C2-C6), alcoxicarbonilo (C1-C6), arilo o un grupo protector; n y m representan cada uno independientemente de otro un número entero de 0 a 10; R3 representa -(CH2)2-COOH, -CH2-COOH, -(CH2)4-COOH, -CH2-OH, -C(OH)-CH3, -CH2-CO-NH2, 20 CH2-SH, -(CH2)4-NH2, -(CH2)2-CO-NH2, -CH2-CH2-S-CH3, -(CH2)3-NH-C(NH)-NH2; **(Ver fórmula)** o R3 representa un grupo de fórmula (II) a continuación: **(Ver fórmula)** con R6, R7, R8 y R9 representando cada uno independientemente de otro H o un grupo alquilo (C1-C6), alquenilo (C2-C6) o alquinilo (C2-C6); X representa H o un grupo alquilo (C1-C6), alquenilo (C2-C6), alquinilo (C2-C6), alcoxicarbonilo (C1-C6), arilo o un grupo protector; r y p representan cada uno independientemente de otro un número entero de 0 a 10; ii) añadir a la disolución preparada en la etapa i) parte de un recipiente para uso médico durante un periodo de tiempo de 1 hora a 2 meses, a una temperatura en un intervalo de 5 a 80ºC; iii) analizar mediante cromatografía líquida la disolución obtenida en la etapa ii); y iv) determinar el porcentaje de degradación.
Description
DESCRIPCIÓN
Evaluación de la estabilidad de un producto biológico en jeringas precargadas
Introducción/campo de la invención
El dominio técnico de la invención es la evaluación de la estabilidad de un producto biológico en jeringas precargadas (PFS).
La invención se refiere a un método para determinar el riesgo de degradación química de un producto biológico; la degradación se produce principalmente por contacto, en particular contacto a largo plazo, con parte de un recipiente para uso médico, por ejemplo una jeringa, y el producto biológico que comprende por lo menos un aminoácido seleccionado de entre metionina, tirosina, triptófano, histidina, glutamina, asparagina, cisteína, arginina, aspartato, glutamato, lisina, prolina, serina, treonina y fenilalanina.
Es bien conocido que el desarrollo y la preparación de un producto de medicamento farmacéutico son complicados debido a los requisitos legales, de seguridad y de calidad. En particular, el riesgo de contaminar pacientes con compuestos contaminantes debe gestionarse y controlarse para evaluar la gestión del riesgo de la calidad.
El riesgo de compuestos contaminantes se incrementa en el caso de que el producto de medicamento farmacéutico se encuentre en disolución en un recipiente para uso médico, siendo el típico una jeringa precargada que contiene, por ejemplo, una vacuna. Los compuestos contaminantes son inevitables al considerar todas las superficies poliméricas con las que una sustancia farmacológica o producto farmacéutico entra en contacto directo y/o indirecto durante la fabricación, llenado, envasado y etiquetado, almacenamiento y transporte. Son fuentes de compuestos contaminantes partes del recipiente (son ejemplos el cilindro de vidrio, la aguja hipodérmica de acero inoxidable, el protector de caucho de la aguja, los lubricantes de aceite de silicona y el tapón de caucho del émbolo) y otras fuentes inesperadas son los residuos de las herramientas de procesamiento y los aditivos para unir la aguja al cilindro, y en términos generales, la parte de un recipiente para el uso médico.
Los compuestos contaminantes pueden potenciar la inmunogenicidad, modificando químicamente el producto biológico terapéutico o presentando una actividad adyuvante inmunitaria directa.
En consecuencia, existe una necesidad de predecir el riesgo de liberación de compuestos contaminantes a partir de un recipiente para uso médico en el que el producto biológico se ha almacenado durante un tiempo prolongado. En consecuencia, los proveedores de dispositivos de administración farmacológica y fabricantes farmacéuticos han llevado a cabo estudios de extracción y lixiviación a fin de predecir y evaluar la naturaleza de dichos compuestos contaminantes que pueden lixiviar en el recipiente y entrar en contacto con el producto biológico. El Product Quality Research Institute (PQRI) ha contribuido significativamente a las mejores prácticas en las evaluaciones de extraíbles y lixiviables (E+L) en productos farmacéuticos nasales de inhalación oral OINDP) y actualmente se está concentrando en los productos farmacéuticos parenterales y oftálmicos (PODP) para clasificar los lixiviables en genotóxicos, irritantes, sensibilizadores y otros tóxicos, con un umbral propuesto para cada clase. Supeditada a la validación de dicho esquema de clasificación y a una base de datos suficientemente grande para el análisis estadístico, un compuesto contaminante será considerado para la cualificación basándose en sus umbrales respectivos.
Sin embargo, existe una necesidad de un marco científico coherente, capaz de comprender el potencial de un compuesto contaminante de interactuar con el producto biológico y causar degradación química.
Con el fin de evaluar la idoneidad de un sistema de jeringa precargada (SJP) para el desarrollo de un producto de anticuerpo monoclonal (mAb), se ha desarrollado un enfoque experimental conocido que consiste en la evaluación siguiente de la integridad de un producto biológico entero al ponerlo en contacto con componentes de la jeringa precargada durante un estudio acelerado de estabilidad de mínimo 6 meses. Dicho enfoque subraya el potencial de los lixiviables de interactuar con productos biológicos, aunque la naturaleza única de cada proteína provoca que dicho estudio sea muy específico y restringe a los fabricantes farmacéuticos en la realización de este tipo de estudio, que es largo y caro, para cada producto biológico en desarrollo.
Se ha desarrollado otro enfoque experimental conocido que muestra un importante interés en productos biológicos que son susceptibles a modificaciones estructurales inducidas por lixiviados y proponen un programa holístico de extraíbles y lixiviables para el producto biotecnológico. El método identifica los compuestos que pueden formar potencialmente modificaciones covalentes de proteínas y han establecido un árbol de decisiones para el análisis científico y de riesgos del perfil de extraíbles y lixiviables. Sin embargo, dicho enfoque requiere encontrar, identificar y cuantificar todos los potenciales extraíbles y lixiviables a partir de cualquier material procedente de parte de un recipiente para uso médico. Dicha información se encuentra protegida principalmente como 'know-how' por el proveedor de dispositivos de administración farmacéutica y, en consecuencia, no se encuentra fácilmente disponible. Con el fin de obtener una lista exhaustiva, deben llevarse a cabo estudios de extraíbles/lixiviables con un panel de técnicas analíticas y métodos de cribado exhaustivos y conocimientos expertos asociados en la identificación de estructuras químicas.
Hoofnagle A.N. et al., CLINICAL CHEMISTRY, 62:1, páginas 48 a 69, 2016, dan a conocer recomendaciones para la generación, cuantificación, almacenamiento y manipulación de péptidos utilizados para ensayos basados en espectrometría de masas, incluyendo un ensayo CPTAC para evaluar la estabilidad peptídica. El almacenamiento a corto plazo, de menos de 3 meses, se describe a una temperatura comprendida en el intervalo de -20°C a 4°C. Majumdar S. et al., JOURNAL OF PHARMACEUTICAL SCIENCES, vol. 100, n° 7, páginas 2563 a 2573, 2011, dan a conocer un método para evaluar el efecto de las superficies de jeringa siliconadas y modificadas sobre la estabilidad de las formulaciones de proteínas en términos de agregación, utilizando el análisis de partículas subvisibles mediante obtención de imágenes de microflujos.
Akala E.O. et al., Pharmaceutical Manufacturing Handbook: Regulations and Quality, páginas 641 a 686, 2008, es una revisión que investiga el efecto del envasado sobre el producto farmacéutico (no peptídico), que da a conocer principalmente un ensayo de HPLC después del almacenamiento a 40°C durante 6 meses.
Raulfs M.D. et al., JOURNAL OF THE AMERICAN SOCIETY FOR MASS SPECTROMETRY, ELSEVIER SCIENCE INC, US, vol. 25, n° 10, páginas 1705 a 1715, 2014, estudian la fragmentación de péptidos AAX(X)AA en la espectrometría de masas.
Alston R.W. et al., BIOPHYSICAL JOURNAL, vol. 94, n° 6, páginas 2288 a 2296, 2008, estudian la estabilidad de las proteínas, de las proteínas desnaturalizadas y de los péptidos AAWAA.
Yoshioka S.: "Stability of Dosage Forms", en: "Stability of Drugs and Dosage Forms", Kluwer Academic Publishers, Boston, 2002, es una obra sobre la estabilidad de los fármacos y formas de administración. El tiempo de almacenamiento se comenta a partir de los estudios acelerados de temperatura. No se mencionan datos de los ensayos.
De esta manera, existe una necesidad continua en la técnica de optimización de los métodos científicos de reducción del riesgo de la estabilidad a largo plazo de productos biológicos en un recipiente para uso médico, tal como jeringas precargadas.
Inesperadamente, los inventores han encontrado que la utilización de un péptido de modelo mimético podría cumplir dicho requisito. Mediante la utilización de un péptido mimético especialmente diseñado, los inventores han desarrollado un método para mimetizar y evaluar la degradación química de un producto biológico que se encuentra en contacto con parte de un recipiente para uso médico. Dicho método asimismo es capaz de predecir la susceptibilidad a la degradación de un producto biológico en disolución en un recipiente para uso médico. En el presente contexto, la invención se refiere a un método para determinar el porcentaje de degradación de un compuesto de fórmula (I) mediante el contacto con parte de un recipiente para uso médico, que comprende las etapas sucesivas siguientes:
i) preparar una disolución acuosa que comprende un compuesto de fórmula (I) a continuación:

Ri, R2, R4 y R5 representan, cada uno independientemente, H o un grupo alquilo (C1-C6), alquenilo (C2-C6) o alquinilo (C2-C6),
X representa H o un grupo alquilo (C1-C6), alquenilo (C2-C6), alquinilo (C2-C6), alcoxicarbonilo (C1-C6), arilo o un grupo protector,
n y m representan, cada uno independientemente, un número entero entre 0 y 10,
Rs representa -(CH2)2-COOH, -CH2-COOH, -(CH2)4-COOH, -CH2-OH, -C(OH)-CHs, -CH2-CO-NH2 , CH2-SH, -(CH2)4-NH2, -(CH2)2-CO-NH2, -CH2-CH2-S-CH3, -(CH2)3-NH-C(NH)-NH2,

o R3 representa un grupo de fórmula (II), a continuación:
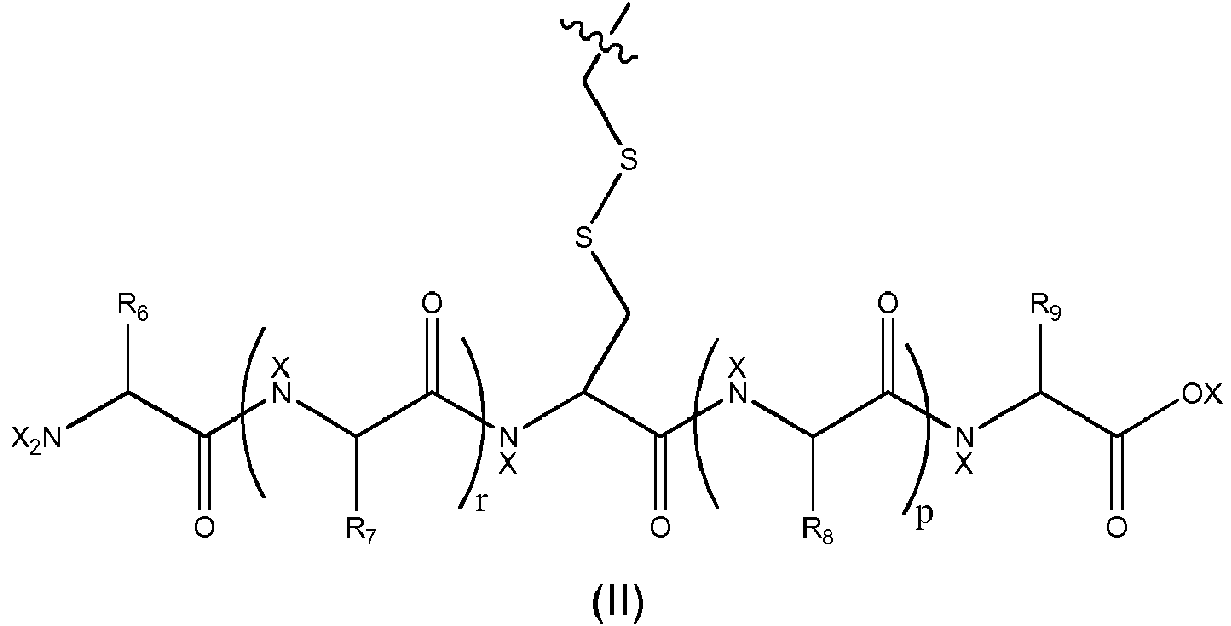
en la que R6, R7, R8y R9 representan, cada uno independientemente, H o un grupo alquilo (C1-C6), alquenilo (C2-C6) o alquinilo (C2 -C6 ),
X representa H o un grupo alquilo (C1-C6), alquenilo (C2-C6), alquinilo (C2-C6), alcoxicarbonilo (C1-C6), arilo o un grupo protector,
r y p representan, cada uno independientemente, un número entero entre 0 y 10,
ii) añadir a la disolución preparada en la etapa i) parte de un recipiente para uso médico durante un periodo de tiempo de 1 hora a 2 meses, a una temperatura comprendida en el intervalo de entre 5°C y 80°C, iii) analizar mediante cromatografía líquida la disolución obtenida en la etapa ii), y
iv) determinar el porcentaje de degradación.
La presente invención pretende proporcionar un nuevo método con una o más de las características siguientes:
• el método según la invención permite mimetizar los sitios sensibles a la degradación de productos biológicos;
• el método según la invención permite evaluar la susceptibilidad de los sitios sensibles de productos biológicos en la interacción con parte de un recipiente para uso médico, tal como componentes de jeringas precargadas;
• el método según la invención permite ayudar a los fabricantes biotecnológicos/farmacéuticos a realizar la selección de menor riesgo del recipiente para uso médico y garantizar un tiempo de comercialización predecible;
• el método según la invención permite evaluar la eficiencia de los aditivos farmacéuticos para proteger la integridad farmacológica;
• el método según la invención permite desarrollar metodologías y herramientas para la evaluación de cribado rápido de las interacciones;
• el método según la invención permite proporcionar un estudio acelerado de estabilidad con disoluciones
modelo de contacto directo con parte de un recipiente para uso médico, residuos del procedimiento, mezcla de extraíbles o extraíbles individuales;
• el método según la invención permite predecir el riesgo de degradación de productos biológicos;
• el método según la invención permite ayudar a los fabricantes biotecnológicos/farmacéuticos a cribar y seleccionar adicionalmente la parte adecuada de los recipientes en el procedimiento de desarrollo temprano de medicamentos;
• el método según la invención permite ayudar a los fabricantes biotecnológicos/farmacéuticos en la selección de la parte óptimamente adecuada de los recipientes para la configuración de uso médico;
• el método según la invención permite comprender los problemas de interacción de productos biológicos en recipientes precargados para uso médico para investigaciones posteriores a la comercialización;
• el método según la invención permite evaluar la parte nueva de un recipiente;
• el método según la invención permite seleccionar la parte óptimamente adecuada de un recipiente;
• el método según la invención permite controlar y evaluar las consecuencias de los cambios de una parte de un recipiente.
Definiciones
La expresión “parte de un recipiente” utilizada en la expresión “parte de un recipiente para uso médico” se refiere al recipiente completo mismo, a una parte del recipiente, a un elemento del recipiente o a la materia prima utilizada para fabricar el recipiente, o incluso a compuestos contaminantes. Un recipiente utilizado en la presente invención no se encuentra limitado por el material de fabricación, e incluye muchos materiales, tales como vidrio, metales (por ejemplo, acero, acero inoxidable, aluminio, etc.), polímeros (por ejemplo, termoplásticos, elastómeros, termoplásticos-elastómeros) y lubricantes de aceite de silicona. Entre los polímeros comunes se incluyen polietileno (PE), polipropileno (PP), policarbonato (PC), etileno-propileno fluorado (FEP), etileno tetrafluoroetileno (ETFE) y politetrafluoroetileno (PTFe ). Entre los elastómeros comunes se incluyen elastómeros saturados e insaturados: elastómero de bromobutilo, mezcla de clorobutilo-isopreno, caucho natural y caucho de poliisopreno sintético, caucho de butilo y caucho de butilo halogenado, caucho de estireno-butadieno, caucho de nitrilo, caucho de etileno-propileno (EPM) y caucho de etileno-propileno-dieno, caucho poliacrílico, caucho de silicona, caucho de fluorosilicona, fluoroelastómeros, perfluoroelastómeros, amidas en bloque de poliéter, polietileno clorosulfonado, etileno-acetato de vinilo, etc.
Se entiende que parte de un recipiente para uso médico puede contener compuestos contaminantes.
Otros materiales son poliéster, dicloruro de polivinilideno (PVDC), alcohol etilvinílico (EVOH), copolímero de poliamida (PA), tereftalato de polietileno (PET), polidimetilsiloxanos (PDMS) y polisulfona (PS).
Entre los metales comunes se incluyen cromo, cobre, hierro, manganeso, níquel, tungsteno y cinc, litio, boro, magnesio, aluminio, silicio, titanio, cromo, cobalto, arsénico, antimonio, bario y sus derivados oxidativos.
La expresión “recipiente para uso médico” se refiere a cualesquiera medios que se utilizan para “contener”, “alojar”, “mezclar”, “combinar”, “dispensar”, “inyectar”, “transferir”, “nebulizar”, etc. un producto biológico durante la investigación, procesamiento, desarrollo, formulación, fabricación, almacenamiento y/o administración. Por ejemplo, el recipiente para uso médico en la presente invención incluye, aunque sin limitación, material de vidrio de laboratorio general, matraces, vasos, probetas graduadas, fermentadores, biorreactores, tubos, tuberías, bolsas, bolsas de biomasa de un solo uso, jarras, viales, cierres de vial (por ejemplo, tapones de goma, rosca sobre capuchón), ampollas, jeringas, jeringas precargadas, jeringas precargables, autoinyectores, plumas inyectoras, tapones, tapones de jeringa, émbolos de jeringa, cierres de goma, cierres de plástico, cierres de vidrio, cilindros, capuchones de punta, capuchones, agujas, émbolos, tapones de émbolo, vástagos de émbolo, cilindros de vidrio, agujas hipodérmicas de acero inoxidable, coberturas de aguja de goma, tapón de émbolo de goma recubierta, etiquetas adhesivas, etiquetas de tinta, etiquetas de recubrimiento y similares. Pueden encontrarse recipientes adicionales para uso médico contemplados en la presente invención en catálogos publicados de proveedores y fabricantes de equipos de laboratorio, tales como VWR(TM) (West Chester, PA), BD Biosciences (Franklin Lakes, NJ), Fisher Scientific International Inc. (Hampton, NH) y Sigma-Aldrich (St. Louis, MO).
La expresión “jeringa precargada” se refiere a una jeringa ya cargada con un fluido, asimismo abreviada como PFS.
La expresión “jeringa precargable” se refiere a una jeringa vacía que puede llenarse.
El término “jeringa” se refiere a una jeringa precargada o precargable. La expresión “proveedor de dispositivo de administración farmacéutica” se refiere a una parte proveedora de un recipiente para uso médico.
Tal como se define en la presente memoria, un “producto biológico” utilizado en la presente invención incluye péptidos, proteínas, conjugados de polisacárido-proteína, producto biofarmacéutico a base de proteínas, productos farmacéuticos para uso humano y/o animal. La mayor parte del tiempo, el producto biológico podría ser un principio farmacéutico activo (API). Un API está destinado a proporcionar actividad farmacológica u otro efecto directo en el diagnóstico, curación, mitigación, tratamiento o prevención de una enfermedad o a afectar la estructura o cualquier función del cuerpo de seres humanos o animales.
El término “degradación” o “degradación química” se refiere a una modificación química de productos biológicos debido a la reacción con componentes en el medio, en particular por compuestos contaminantes. Generalmente, dichas modificaciones se producen con las cadenas laterales más reactivos y son predominantemente oxidaciones, reducciones y sustituciones nucleófilas y electrófilas. Entre las degradaciones se incluyen escisiones de enlaces peptídicos, racemizaciones, eliminaciones p y formaciones de productos mediante la reacción de proteínas con compuestos químicos añadidos. Las degradaciones excluyen el cambio conformacional natural, el cambio en la estructura nativa, tales como interacciones electrostáticas, interacciones hidrofóbicas, etc.
El término “estabilidad” se refiere a estabilidad química, ya que no se produce degradación química del producto biológico. La estructura primaria del producto biológico se mantiene sin cambios durante el almacenamiento en el recipiente para uso médico o durante el contacto con parte de un recipiente para uso médico.
Tal como se indica en la presente memoria, la expresión “compuestos contaminantes” pretende cubrir los compuestos contaminantes que incluyen extraíbles, lixiviables o residuos del procesamiento.
Los extraíbles son compuestos que pueden extraerse de parte de un recipiente para uso médico, tales como componentes elastoméricos o plásticos, extraídos en presencia de un solvente, en particular bajo condiciones duras. Los extraíbles pueden ser mezclas complejas que consisten principalmente en oligómeros y aditivos de propiedades físicas y químicas diversas y con frecuencia se encuentran presentes a concentraciones mucho más bajas que cualquier otro ingrediente del producto biológico, dificultando la detección de su presencia. La American Food & Drug Administration (FDA) requiere la evaluación de los extraíbles para su impacto sobre la seguridad y eficacia de los productos biológicos.
Los lixiviables son compuestos que se lixivian hacia la formulación procedentes de parte de un recipiente para uso médico, tales como componentes elastoméricos o plásticos, en particular bajo condiciones de uso real. Entre los ejemplos de lixiviables se incluyen, aunque sin limitación, ácido acrílico, ácido metacrílico, diacrilato de 1,6-hexanodiol y maleato de dibutilo. El impacto de los lixiviables sobre el producto biológico puede estar relacionado con la degradación, agregación, formación de partículas y/o problemas de calidad del producto, tales como la reacción con la formulación o proteínas.
La expresión “residuos de procesamiento” se refiere a compuestos químicos procedentes del recipiente para uso médico o procedentes de los procedimientos de fabricación o llenado del producto farmacéutico inyectable. A título de ejemplo de residuos de procesamiento podrían indicarse: tensioactivos, tungsteno, ácidos orgánicos, sin limitarse a esta lista.
El término “alquilo (C1-C6)”, tal como se utiliza en la presente invención, se refiere a una cadena de hidrocarburo saturado monovalente ramificado que contiene 1 a 6 átomos de carbono, incluyendo, aunque sin limitación, metilo, etilo, n-propilo, isopropilo, n-butilo, isobutilo, sec-butilo, t-butilo, n-pentilo, n-hexilo y similares.
El término “alquenilo (C2-C6)”, tal como se utiliza en la presente invención, se refiere a una cadena de hidrocarburo insaturado monovalente lineal o ramificado que contiene 2 a 6 átomos de carbono y que comprende por lo menos un doble enlace, incluyendo, aunque sin limitación, etenilo, propenilo, butenilo, pentenilo, hexenilo y similares.
El término “alquinilo (C2-C6)”, tal como se utiliza en la presente invención, se refiere a una cadena de hidrocarburo insaturado monovalente lineal o ramificado que contiene 2 a 6 átomos de carbono y que comprende por lo menos un triple enlace, incluyendo, aunque sin limitación, etinilo, propinilo, butinilo, pentinilo, hexinilo y similares.
El término “alcoxi Ci-Ca”, tal como se utiliza en la presente invención, se refiere a un grupo alquilo (C1-C6) tal como se ha definido anteriormente, unido a la molécula mediante un átomo de oxígeno, incluyendo, aunque sin limitación, metoxi, etoxi, n-propoxi, isopropoxi, n-butoxi, isobutoxi, sec-butoxi, t-butoxi, n-pentoxi, n-hexoxi y similares.
El término “alcoxicarbonilo (C1-C6)”, tal como se utiliza en la presente invención, se refiere a un grupo alcoxi C1-C6 tal como se ha definido anteriormente, unido a la molécula mediante un grupo -C(=O)-, incluyendo, aunque sin limitación, etoxicarbonilo, metoxicarbonilo y t-butiloxicarbonilo (Boc).
El término “arilo”, tal como se utiliza en la presente invención, se refiere a un grupo hidrocarburo aromático que comprende preferentemente 6 a 10 átomos de carbono y que comprende uno o más anillos fusionados, tales como, por ejemplo, un grupo fenilo o naftilo. Ventajosamente, es un grupo fenilo.
La expresión “grupo protector”, tal como se utiliza en la presente invención, se refiere a un grupo químico que bloquea selectivamente un sitio reactivo en un compuesto multifuncional de manera que se lleva a cabo selectivamente una reacción química en otro sitio reactivo no protegido.
La expresión “grupo protector de O”, tal como se utiliza en la presente invención, se refiere a un sustituyente que protege los grupos hidroxilo (OH) de los compuestos de fórmula (I) frente a reacciones no deseables durante los procedimientos sintéticos, tales como los grupos protectores de O dados a conocer en Greene's Protective Groups In Organic Synthesis", 4a edición, 2007, John Wiley & Sons, Hoboken, New Jersey. Habitualmente, el grupo Ox de los compuestos de fórmula (I) se cubre con dicho grupo protector a fin de proteger la función hidroxilo. Un grupo hidroxilo protegido por un grupo protector de O puede ser, por ejemplo, un éter, un éster, un carbonato, un acetal y similares. En particular, los grupos protectores de O pueden ser un alquilo (C1-C6) sustituido opcionalmente con uno o varios (particularmente 1 a 3) átomos de halógeno (tales como átomos de cloro), tales como metilo, etilo, terc-butilo o 2,2,2-tricloroetilo, un aril-alquilo (C1-C6), tal como bencilo, estando la fracción arilo sustituida opcionalmente con uno o varios grupos metoxi, tales como bencilo (Bn) o p-metoxibencilo (PMB), un derivado tritilo de fórmula -CAr1Ar2Ar3, tal como trifenilmetilo (asimismo denominado tritilo, - Tr), (4-metoxifenil)difenilmetilo (también denominado metoxitritilo, - NMT) o bis-(4-metoxifenil)fenilmetilo (asimismo denominado dimetoxitritilo -DMT); un grupo metilo sustituido de fórmula CH2ORGP2 o CH2SRGP2 (en particular, CH2ORGP2 ), por ejemplo, metoximetilo (MOM), benciloximetilo, 2-metoxietoximetilo (MEM), 2-(trimetilsilil)etoximetilo o metiltiometilo; un grupo etilo sustituido de fórmula CH2CH2ORGP2 o -CH2CH2SRGP2 (en particular, - CH2CH2ORGP2), por ejemplo, etoxietilo (EE); un grupo sililo de fórmula - SiRGP3RGP4RGP5, por ejemplo, trimetilsililo (TMS), trietilsililo (t Es ), t-butildimetilsililo (TBS o TBDMS) y t-butildifenilsililo (TBDPS); un grupo carbonilado de fórmula CO-RGP6, tal como acetilo (Ac), pivaloilo (Piv o Pv) o benzoilo (Bz) o de fórmula -CO2-RGP7 , tal como aliloxicarbonilo (Alloc) o 9-fluorenilmetiloxicarbonilo (Fmoc); o un grupo tetrahidropiranilo (THP) o tetrahidrofuranilo,
en los que Ar1, Ar2 y Ar3 representan, independientemente unos de otros, un arilo, tal como fenilo, sustituido opcionalmente con uno o varios grupos metoxi; RGP2 representa un alquilo (C1-C6) (tal como metilo o etilo) sustituido opcionalmente con un arilo (tal como fenilo), un alcoxi C1-C6 (tal como metoxi) o un grupo trialquilsililo (tal como SiMe3); RGP3, RGP4 y RGP5 que representan, independientemente unos de otros, un grupo alquilo (C1-C6) o arilo (tal como fenilo), y RGP6 y RGP7 representan, independientemente uno de otro, un alquilo (C1-C6), un alquenilo (C2-C6), un arilo, un aril-alquilo (C1-C6) o un grupo 9-fluorenilmetilo.
En particular, será un grupo bencilo, acetilo o metoximetilo.
La expresión “grupo protector de N”, tal como se utiliza en la presente invención, sirve principalmente para controlar la síntesis del compuesto de fórmula (I). Con este propósito, todas las fracciones típicamente utilizadas en la química de péptidos, como grupos protectores en general, resultan adecuadas. Habitualmente, el grupo X2N de los compuestos de fórmula (I) se cubre con dicho grupo protector a fin de proteger la función amina frente a reacciones no deseables durante los procedimientos sintéticos. Los grupos protectores de N utilizados comúnmente se dan a conocer en "Greene's Protective Groups In Organic Synthesis", 4a edición, 2007, John Wiley & Sons, Hoboken, New Jersey. La función amina X2N protegida con un grupo protector de N puede ser un carbamato, una amida, una sulfonamida, un derivado de N-alquilo, un derivado de aminoacetal, un derivado N-bencilo, un derivado imina, un derivado enamina o un derivado de N-heteroátomo. En particular, los grupos protectores de N pueden ser formilo; un arilo, tal como un fenilo, sustituido opcionalmente con uno o varios grupos metoxi, tales como p-metoxifenilo (PMP); un aril-alquilo (C1-C6), tal como un bencilo, estando la fracción arilo sustituida opcionalmente con uno o varios grupos metoxi, tales como bencilo (Bn), p-metoxibencilo (PMB) o 3,4-dimetoxibencilo (DMPM); CO-RGP1, tal como acetilo (Ac), pivaloilo (Piv o Pv), benzoilo (Bz) o p-metoxibencilcarbonilo (Moz); -CO2-RGP1 , tal como t-butiloxicarbonilo (Boc), tricloroetoxicarbonilo (TROC), aliloxicarbonilo (Alloc), benciloxicarbonilo (Cbz o Z) o 9-fluorenilmetiloxicarbonilo (Fmoc); -SO2-RGP1 , tal como fenilsulfonilo, tosilo (Ts o Tos) o 2 nitrobencenosulfonilo (asimismo denominado nosilo - Nos o Ns), y similares,
en el que RGP1 representa un alquilo (C1-C6) sustituido opcionalmente con uno o varios átomos de halógeno, tales como F o Cl; un alquenilo (C2-C6), tal como un alilo; un arilo, tal como fenilo, sustituido opcionalmente con uno o varios grupos seleccionados de OMe (metoxi) y NO2 (nitro); un aril-alquilo (C1-C6), tal como un bencilo, estando la fracción arilo sustituida opcionalmente con uno o varios grupos metoxi, o un grupo 9-fluorenilmetilo.
En particular, puede ser un grupo terc-butiloxicarbonilo (Boc), benciloxicarbonilo (Cbz) o 9-fluorenilmetiloxicarbonilo (Fmoc).
La expresión “cromatografía líquida”, tal como se utiliza en la presente invención, se refiere a técnicas analíticas utilizando un aparato de cromatografía líquida utilizado para separar compuestos acoplado con un detector utilizado para detectar los productos de degradación de fórmula (I) y la presencia de compuestos contaminantes. El aparato de cromatografía líquida podría seleccionarse de HPLC (cromatografía líquida de alto rendimiento),
UPLC (cromatografía líquida de ultra-alto rendimiento), UHPLC (cromatografía líquida de rendimiento ultraelevado) y RRLC (cromatografía líquida de resolución rápida). El detector podría seleccionarse de entre detector de UV, detector de matriz de diodos (DAD), espectrómetro de masas (EM), detector de matriz de fotodiodos (PDA) combinado con un espectrómetro de masas (PDA/EM o DAD/EM), detección evaporativa de dispersión lumínica (ELSD) y detector corona CAD.
Breve descripción de los dibujos
La figura 1 es un cromatograma de UPLC/DAD del pentapéptido AAWAA tras 15 o 30 días a 25°C/60% de humedad relativa (HR) en contacto directo con un tapón de émbolo recubierto y con uno no recubierto.
La figura 2 son los espectros de masas en modo de electropulverización positiva (ESI+) de: (a) péptido AAWAA intacto (tiempo de retención: 17,2 min) y (b) forma oxidada de AAWAA (tiempo de retención: 17,1 min) detectado tras 30 días (25°C) de contacto entre el tapón de émbolo no recubierto y la disolución de péptido AAWAA. La figura 3 es el cromatograma de ULPC/DAD del péptido AAHAA tras 3 días a 70°C en contacto directo con dos componentes de jeringa: (a) adhesivo y (b) tungsteno insoluble, (c) es AAHAA de referencia.
La figura 4 es la comparación entre el perfil de degradación de los péptidos AAMAA en el caso de que se almacenen en contacto directo con capuchón de punta de elastómero termoplástico (TPE) durante 3 días a 70°C.
Descripción de la invención
La invención se refiere a un método para determinar el porcentaje de degradación de un compuesto de fórmula (I) mediante el contacto con parte de un recipiente para uso médico, que comprende las etapas sucesivas siguientes: i) preparar una disolución acuosa que comprende un compuesto de fórmula (I) a continuación:

Ri, R2, R4 y R5 representan, cada uno independientemente, H o un grupo alquilo (C1-C6), alquenilo (C2-C6) o alquinilo (C2-C6),
X representa H o un grupo alquilo (C1-C6), alquenilo (C2-C6), alquinilo (C2-C6), alcoxicarbonilo (C1-C6), arilo o un grupo protector,
n y m representan, cada uno independientemente, un número entero entre 0 y 10,
Rs representa -(CH2)2-COOH, -CH2-COOH, -(CH2)4-COOH, -CH2-OH, -C(OH)-CHs, -CH2-CO-NH2 , CH2-SH, -(CH2)4-NH2, -(CH2)2-CO-NH2 , -CH2-CH2-S-CH3 , -(CH2)3-NH-C(NH)-NH2,


en la que R6, R7, R8y R9 representan, cada uno independientemente, H o un grupo alquilo (C1-C6), alquenilo (C2-C6) o alquinilo (C2-C6),
X representa H o un grupo alquilo (C1-C6), alquenilo (C2-C6), alquinilo (C2-C6), alcoxicarbonilo (C1-C6), arilo o un grupo protector,
r y p representan, cada uno independientemente, un número entero entre 0 y 10,
ii) añadir a la disolución preparada en la etapa i) parte de un recipiente para uso médico durante un periodo de tiempo de 1 hora a 2 meses, a una temperatura comprendida en el intervalo de entre 5°C y 80°C, iii) analizar mediante cromatografía líquida la disolución obtenida en la etapa ii), y
iv) determinar el porcentaje de degradación.
Según el método de la invención, el contacto con parte de un recipiente para uso médico puede ser directo o indirecto. La expresión contacto directo se refiere a que el producto biológico se encuentra en contacto con parte del recipiente sin ningún intermediario. La expresión contacto indirecto se refiere a que el producto biológico en ningún momento entra en contacto con parte del recipiente, aunque entra en contacto con otro compuesto/intermediario que ha estado en contacto directo con parte de dicho recipiente.
Etapa (i):
En la etapa i) se prepara una disolución acuosa. La disolución acuosa preparada en la etapa i) comprende preferentemente 30% a 100% de agua, más preferentemente 75% a 100%, todavía más preferentemente de 85% a 100%, porcentaje en masa.
Según una variante, la disolución acuosa preparada en la etapa i) puede contener además excipientes, agentes amortiguadores, ácidos, bases, sales, conservantes, solubilizadores, tensioactivos, agentes quelantes, azúcares, aminoácidos, péptidos, solventes y combinaciones de los mismos.
La disolución acuosa preparada en la etapa i) comprende preferentemente por lo menos 5 ^g/ml de compuesto de fórmula (I), más preferentemente por lo menos 15 ^g/ml. La cantidad de compuesto de fórmula (I) utilizada en la etapa i) depende de la sensibilidad del aparato detector utilizado en la etapa iii), habitualmente se utilizan 45 ^g/ml en la etapa i) a fin de detectar los productos de degradación peptídica sintetizados.
En una forma de realización preferida de la invención, el compuesto utilizado en la etapa i) es de fórmula (I) tal como se ha definido anteriormente, en la que X=H.
En una forma de realización preferida de la invención, el compuesto utilizado en la etapa i) es de fórmula (I) tal como se ha definido anteriormente, en la que n=1 y m=1.
En una forma de realización preferida de la invención, el compuesto utilizado en la etapa i) es de fórmula (I) tal como se ha definido anteriormente, en la que Ri, R2, R4 yd R5 representan, cada uno, CH3 y X=H, n=1 y m=1. En una forma de realización preferida de la invención, el compuesto utilizado en la etapa i) es de fórmula (I) tal como se ha definido anteriormente, en la que R1, R2, R4 y R5 representan, cada uno, CH3 y X=H, n=1, m=1 y R3 es uno de entre:

En una forma de realización preferida de la invención, el compuesto utilizado en la etapa i) es de fórmula (I) tal como se ha definido anteriormente, en la que R1, R2, R4 y R5 representan, cada uno, CH3, y X=H, n=1, m=1 y R3 es uno de entre:
-CH2-CH2-S-CH3 (Met), -(CH2)3-NH-C(NH)-NH2 (Arg), -(CH2)4-NH2 (Lys), -CH2-CO-NH2 (Asn), -CH2-COOH (Asp), CH2-CH2-CO-NH2 (Gln), -(CH2)2-COOH (Glu).
En una forma de realización preferida de la invención, el compuesto utilizado en la etapa i) es de fórmula (I) tal como se ha definido anteriormente, en la que R1, R2, R4y R5 representan, cada uno, CH3, y X=H, n=1, m=1 y R3 es uno de-CH2-CH2-S-CH3.

Etapa (ii):
Durante la etapa ii), parte de un recipiente para uso médico se añade a la disolución preparada en la etapa i) durante un periodo de tiempo de 1 hora a 2 meses, a una temperatura comprendida en el intervalo de entre 5°C y 80°C.
Preferentemente, la etapa ii) se lleva a cabo bajo agitación parcial y/o en una cámara climática.
Preferentemente, la parte del recipiente para uso médico es una jeringa, una jeringa precargable, parte de una jeringa precargable, una jeringa precargada o parte de una jeringa precargada (ver las definiciones, anteriormente). Preferentemente, la parte del recipiente para uso médico es un cilindro, un capuchón de punta, un capuchón, una aguja o un tapón de émbolo, todos de una jeringa, una jeringa precargable o una jeringa precargada.
Preferentemente, la parte del recipiente para uso médico es de vidrio, caucho, tungsteno o sus derivados, adhesivo, aceite de silicona, plástico o metal (aluminio, boro, silicio, magnesio, cinc o cromo).
La duración preferente de la etapa ii) se encuentra comprendida en el intervalo de 4 horas a 2 meses. Más preferentemente, la duración de la etapa ii) se encuentra comprendida en el intervalo de 7 días a 45 días.
La temperatura de la etapa ii) del método según la invención preferentemente se encuentra en el intervalo de 10°C a 80°C, más preferentemente de 15°C a 75°C, todavía más preferentemente de 18°C a 70°C, todavía más preferentemente de 18°C a 30°C. La temperatura de la etapa ii) del método según la invención es preferentemente 25°C, 45°C o 70°C.
Etapa (iii):
En la etapa iii), la disolución obtenida en la etapa ii) se analiza mediante cromatografía líquida.
La cromatografía preferida es la cromatografía líquida de ultra-alto rendimiento (UPLC). El detector preferido se selecciona de entre espectrómetro de masas (EM), detector de matriz de fotodiodos (PDA) y detector de matriz de fotodiodos en combinación con un espectrómetro de masas (PDA/EM). Preferentemente, la cromatografía líquida es cromatografía líquida UPLC/detector de matriz de fotodiodos en combinación con un espectrómetro de masas. Se obtiene un cromatograma de PDA a fin de permitir la evaluación del rendimiento de degradación del péptido mimético sintetizado (compuesto de fórmula (I)) y la detección de los productos de degradación relacionados. Se obtiene el espectro de EM a fin de permitir la identificación de los productos de degradación de los compuestos de fórmula (I).
Etapa (iv):
La etapa iv) es la determinación del porcentaje de degradación del compuesto de fórmula (I).
Etapa (v):
El método según la invención puede comprender, además, después de la etapa iv), una etapa de medición v) mediante espectrometría de masas y una etapa vi) de identificación de la degradación química, tal como una oxidación o una desaminación. El detector de Em asimismo puede utilizarse para la determinación del porcentaje de degradación del compuesto de fórmula (I) en el caso de que el compuesto de fórmula (I) coeluya con un producto contaminante o cualesquiera compuestos presentes en el producto biológico.
En la presente descripción, incluyendo las reivindicaciones adjuntas, a menos que se especifique lo contrario, los porcentajes son en masa.
Se proporcionan los ejemplos a continuación para la invención únicamente a título ilustrativo y no limitativo. Ejemplos
Materiales:
Se sintetizaron pentapéptidos AAXAA en GeneCust (Luxemburgo) con una pureza >99%. Se adquirió PBS (solución salina amortiguada con fosfato) y amortiguador de acetato amónico de Sigma-Aldrich (St. Louis, MO). Todos los demás reactivos eran de grado analítico.
El recubrimiento se refiere a ETFE (etileno-tetrafluoroetileno).
El péptido n° 1 presentaba la fórmula siguiente: Ala-Ala-Trp-Ala-Ala (AAWAA)
El péptido n° 2 presentaba la fórmula siguiente: Ala-Ala-His-Ala-Ala (AAHAA)
El péptido n° 3 presentaba la fórmula siguiente: Ala-Ala-Met-Ala-Ala (AAMAA)
La parte del recipiente utilizada fue el tapón del émbolo y el capuchón de punta de TPE de diferentes fabricantes de caucho (tales como Becton Dickinson, Aptar Stelmi, West, Sumitomo), el adhesivo utilizado para unir la aguja y tungsteno insoluble, residuos potenciales del procedimiento de formación de la punta.
Aparato:
La cromatografía líquida utilizada era de Waters.
Se analizaron las disoluciones de péptidos mediante CL/DAD/EM con el espectrómetro de masas Quattro Premier XE Mass, de Waters.
El software utilizado era Mass Lynx.
Ejemplo 1: ensayos de un tapón de émbolo con péptido mimético n°1
El péptido AAWAA se disolvió en amortiguador de acetato de amonio pH 4 (20 m) Tween-80 al 0,02% hasta una concentración final de 45 pg/ml. Las disoluciones de péptido AAWAA obtenidas se almacenaron en contacto directo con un tapón de émbolo no recubierto y uno recubierto con ETFE durante 30 días a 25°C/60% de HR.
Se presentan los resultados en las figuras 1 y 2.
La figura 1 es un cromatograma de UPLC/DAD de pentapéptico AAWAA en contacto directo con un tapón de émbolo recubierto y uno no recubierto tras 15 días (c, d) y tras 30 días (a, b) de almacenamiento a 25°C/60% de
HR. Los cromatogramas e y f son disoluciones de AAWAA de referencia (sin ningún contacto directo con tapones de émbolo).
La figura 2 son los espectros de masas en modo de electropulverización positiva (ESI+) de: (a) péptido AAWAA intacto (tiempo de retención: 17,2 min) y (b) forma oxidada de AAWAA (tiempo de retención: 17,1 min) detectado tras 30 días (25°C) de contacto entre el tapón de émbolo no recubierto y la disolución de péptido AAWAA.
Tal como se observa en la figura 1 (a y b), el rendimiento de degradación de AAWAA es más importante en el caso de que el péptido se encuentre en contacto directo con el tapón de émbolo no recubierto. En la figura 1b se muestran productos de degradación de AAWAA. El rendimiento de degradación más elevado de AAWAA al almacenarlo en contacto directo con el tapón no recubierto subraya los mayores riesgos del aminoácido W y las interacciones lixiviables.
Ejemplo 2: ensayos de residuos de adhesivo y de tungsteno con el péptido mimético n° 2 a temperatura elevada y tiempo de duración corto
El péptido AAHAA se disolvió en amortiguador de acetato de amonio pH 4 (20 mM) Tween-80 al 0,02% hasta una concentración final de 45 pg/ml. La disolución de péptido AAHAA se almacenó en contacto directo con el adhesivo utilizado para unir la aguja a la jeringa y con tungsteno insoluble, un potencial residuo de procesamiento, durante 3 días a 70°C.
Se presentan los resultados en la figura 3.
La figura 3 son cromatogramas de ULPC/DAD del péptido AAHAA tras 3 días a 70°C en contacto directo con dos componentes de jeringa: (a) adhesivo y (b) tungsteno insoluble; (c) son cromatogramas de referencia.
Tal como se observa en la figura 3, los péptidos AAHAA reaccionan con el tungsteno insoluble tras 3 días a 70°C.
Ejemplo 3: ensayos de un capuchón de punta con péptido mimético n° 3 a temperatura elevada y tiempo de duración corto
Las disoluciones de péptido AAWAA se disolvieron en amortiguador de PBS pH 7 (20 mM) Tween-80 (al 0,02%) hasta una concentración final de 45 pg/ml. Las disoluciones de péptido AAMAA obtenidas se almacenaron en contacto directo con un capuchón de punta de TPE durante 3 días a 70°C.
Claims (11)
1. Método para determinar el porcentaje de degradación de un compuesto de fórmula (I) mediante el contacto con parte de un recipiente para uso médico, que comprende las etapas sucesivas siguientes:
i) preparar una disolución acuosa que comprende un compuesto de fórmula (I) a continuación:

Ri, R2, R4y R5 representan cada uno independientemente de otro H o un grupo alquilo (C1-C6), alquenilo (C2-C6) o alquinilo (C2-C6);
X representa H o un grupo alquilo (C1-C6), alquenilo (C2-C6), alquinilo (C2-C6), alcoxicarbonilo (C1-C6), arilo o un grupo protector;
n y m representan cada uno independientemente de otro un número entero de 0 a 10;
R3 representa -(CH2)2-COOH, -CH2-COOH, -(CH2)4-COOH, -CH2-OH, -C(OH)-CH3, -CH2-CO-NH2, CH2-SH, -(CH2)4-NH2, -(CH2)2-CO-NH2, -CH2-CH2-S-CH3, -(CH2)3-NH-C(NH)-NH2;

con R6, R7, R8 y R9 representando cada uno independientemente de otro H o un grupo alquilo (C1-C6), alquenilo (C2-C6) o alquinilo (C2-C6);
X representa H o un grupo alquilo (C1-C6), alquenilo (C2-C6), alquinilo (C2-C6), alcoxicarbonilo (C1-C6), arilo o un grupo protector;
r y p representan cada uno independientemente de otro un número entero de 0 a 10;
ii) añadir a la disolución preparada en la etapa i) parte de un recipiente para uso médico durante un periodo de tiempo de 1 hora a 2 meses, a una temperatura en un intervalo de 5 a 80°C;
iii) analizar mediante cromatografía líquida la disolución obtenida en la etapa ii); y
iv) determinar el porcentaje de degradación.
2. Método según la reivindicación 1, que comprende además después de la etapa iv), una etapa de medición v) mediante espectrometría de masas y una etapa vi) de identificación de la degradación química, como una oxidación o una desaminación.
3. Método según cualquiera de las reivindicaciones anteriores, en el que R1, R2, R4 y R5 representan cada uno CH3, y X=H, n=1 y m=1.
4. Método según cualquiera de las reivindicaciones anteriores, en el que la disolución acuosa preparada en la etapa i) comprende de 30 a 100% de agua, porcentaje en masa.
5. Método según cualquiera de las reivindicaciones anteriores, en el que la disolución acuosa preparada en la etapa i) contiene además excipientes, agentes amortiguadores, ácidos, bases, sales, conservantes, solubilizadores, tensioactivos, agentes quelantes, azúcares, aminoácidos, péptidos, solventes y combinaciones de los mismos.
6. Método según cualquiera de las reivindicaciones anteriores, en el que la disolución acuosa preparada en la etapa i) comprende por lo menos 5 pg/ml de compuesto de fórmula (I).
7. Método según cualquiera de las reivindicaciones anteriores, en el que la parte del recipiente para uso médico es una jeringa, una jeringa precargable, parte de una jeringa precargable, una jeringa precargada o parte de una jeringa precargada.
8. Método según cualquiera de las reivindicaciones anteriores, en el que la parte del recipiente para uso médico es un cilindro, un capuchón de punta, un capuchón, una aguja o un tapón de émbolo, todos de una jeringa, una jeringa precargable o una jeringa precargada.
9. Método según cualquiera de las reivindicaciones anteriores, en el que la parte del recipiente para uso médico es vidrio, caucho, tungsteno o sus derivados, adhesivo, aceite de silicona, plástico, metal o (aluminio, boro, silicio, magnesio, cinc o cromo).
10. Método según cualquiera de las reivindicaciones anteriores, en el que la duración de la etapa ii) se encuentra en el intervalo de 4 horas a 2 meses, preferentemente de 7 días a 45 días.1
11. Método según cualquiera de las reivindicaciones anteriores, en el que la temperatura de la etapa ii) se encuentra en el intervalo de 15 a 75°C, preferentemente de 18 a 70°C.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP16306490 | 2016-11-15 | ||
| PCT/EP2017/078452 WO2018091314A1 (en) | 2016-11-15 | 2017-11-07 | Assessment of stability of biological product in prefilled syringes |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2826992T3 true ES2826992T3 (es) | 2021-05-19 |
Family
ID=57570024
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES17793966T Active ES2826992T3 (es) | 2016-11-15 | 2017-11-07 | Evaluación de la estabilidad de un producto biológico en jeringas precargadas |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US11156558B2 (es) |
| EP (1) | EP3542163B1 (es) |
| CN (1) | CN110121648B (es) |
| ES (1) | ES2826992T3 (es) |
| RU (1) | RU2751155C2 (es) |
| WO (1) | WO2018091314A1 (es) |
Family Cites Families (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN1014228B (zh) * | 1987-09-30 | 1991-10-09 | 上海市血液中心 | 医用深低温保存容器 |
| JPH0687883B2 (ja) * | 1991-10-14 | 1994-11-09 | ファイザー製薬株式会社 | 医療用薬剤供給包装体 |
| RU2333663C2 (ru) * | 2002-07-29 | 2008-09-20 | Аминотек Ас | Способ получения пептидов/аминокислот из сырья, содержащего белок, продукты, получаемые таким способом, и применение этих продуктов |
| KR100999705B1 (ko) * | 2003-04-23 | 2010-12-08 | 가부시키가이샤 오츠까 세이야꾸 고죠 | 약액 충전 플라스틱 앰플 및 그의 제조 방법 |
| DE60315955T2 (de) * | 2003-05-19 | 2008-06-05 | Euro-Celtique S.A. | Trockene liposomale PVP-Jod Zubereitungen |
| GB0503411D0 (en) * | 2005-02-18 | 2005-03-30 | Shimadzu Res Lab Europe Ltd | Mass spectrometry precursor ion selection |
| JP4710006B2 (ja) * | 2005-03-28 | 2011-06-29 | 国立大学法人岩手大学 | 生体用金属の生体適合性評価方法 |
| IN2014MN00844A (es) * | 2011-10-07 | 2015-05-01 | Mitsubishi Gas Chemical Co | |
| FR2995213A1 (fr) * | 2012-09-12 | 2014-03-14 | Lfb Biotechnologies | Seringue contenant une composition, notamment pharmaceutique, comprenant des immunoglobulines, son procede de fabrication et son utilisation |
| EP2965907B1 (en) * | 2013-03-06 | 2017-04-19 | Mitsubishi Gas Chemical Company, Inc. | Oxygen-absorbing medical multiwall container and biopharmaceutical storage method |
| WO2016135050A1 (en) * | 2015-02-23 | 2016-09-01 | Stevanato Group International A. S. | Screening method for assessing the delamination propensity of glass |
-
2017
- 2017-11-07 ES ES17793966T patent/ES2826992T3/es active Active
- 2017-11-07 WO PCT/EP2017/078452 patent/WO2018091314A1/en not_active Ceased
- 2017-11-07 RU RU2019115810A patent/RU2751155C2/ru active
- 2017-11-07 CN CN201780070502.3A patent/CN110121648B/zh active Active
- 2017-11-07 US US16/349,761 patent/US11156558B2/en active Active
- 2017-11-07 EP EP17793966.7A patent/EP3542163B1/en active Active
Also Published As
| Publication number | Publication date |
|---|---|
| RU2751155C2 (ru) | 2021-07-08 |
| US11156558B2 (en) | 2021-10-26 |
| RU2019115810A (ru) | 2020-12-17 |
| CN110121648A (zh) | 2019-08-13 |
| EP3542163A1 (en) | 2019-09-25 |
| WO2018091314A1 (en) | 2018-05-24 |
| CN110121648B (zh) | 2022-07-26 |
| EP3542163B1 (en) | 2020-07-22 |
| US20190383749A1 (en) | 2019-12-19 |
| RU2019115810A3 (es) | 2021-01-29 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US20220202965A1 (en) | Radiotracer compositions and methods | |
| Xu et al. | Drug conjugates based on a monovalent affibody targeting vector can efficiently eradicate HER2 positive human tumors in an experimental mouse model | |
| WO2012022676A1 (en) | Peptide radiotracer compositions | |
| SA516370893B1 (ar) | مشتق جديد من نظير إنسولين | |
| Zhang et al. | Comparison of 18F-labeled CXCR4 antagonist peptides for PET imaging of CXCR4 expression | |
| HRP20201489T1 (hr) | Imunoglobulini konjugirani s cys80 | |
| Kagdi et al. | Determination of holdup volume and transient contact compatibility of closed system transfer devices for a reconstituted lyophilized drug product | |
| Effendi et al. | Development of radiogallium-labeled peptides for platelet-derived growth factor receptor β (PDGFR β) imaging: Influence of different linkers | |
| Matalińska et al. | Novel NK1R-targeted 68Ga-/177Lu-radioconjugates with potential application against glioblastoma multiforme: Preliminary exploration of structure–activity relationships | |
| ES2826992T3 (es) | Evaluación de la estabilidad de un producto biológico en jeringas precargadas | |
| Nock et al. | Nonpeptidic Z360-analogs tagged with trivalent radiometals as anti-CCK2R cancer theranostic agents: a preclinical study | |
| Larkina et al. | Comparative preclinical evaluation of peptide-based chelators for the labeling of DARPin G3 with 99mTc for radionuclide imaging of HER2 expression in cancer | |
| Nsereko et al. | Innovative peptide therapeutics in the pipeline: transforming cancer detection and treatment | |
| Collado Camps et al. | Conjugation to a cell-penetrating peptide drives the tumour accumulation of the GLP1R antagonist exendin (9-39) | |
| ES2675102T3 (es) | Método de radioconjugación | |
| Witkowska et al. | Bifunctional opioid/melanocortin peptidomimetics for use in neuropathic pain: variation in the type and length of the linker connecting the two pharmacophores | |
| US10548995B2 (en) | Radiolabelling method | |
| US20160303262A1 (en) | Purification method and compositions | |
| George et al. | Scavenging strategy for specific activity improvement: application to a new CXCR4‐specific cyclopentapeptide positron emission tomography tracer | |
| Vavere et al. | Synthesis of L-[methyl-11C] methionine ([11C] MET) | |
| Tatsi et al. | Bis (Disulfide)-Bridged Somatostatin-14 Analogs and Their [111In] In-Radioligands: Synthesis and Preclinical Profile | |
| Aldrich et al. | Optimization of the Macrocyclic Tetrapeptide [D-Trp] CJ-15,208 to Prevent Stress-Induced Relapse of Cocaine-Seeking Behavior | |
| Sahithi et al. | Stability indicating RP-HPLC method for the simultaneous estimation of Trastuzumab and Pembrolizumab in pharmaceutical dosage forms |