ES2838007T3 - Composiciones de liberación retardada de linaclotida - Google Patents
Composiciones de liberación retardada de linaclotida Download PDFInfo
- Publication number
- ES2838007T3 ES2838007T3 ES14824655T ES14824655T ES2838007T3 ES 2838007 T3 ES2838007 T3 ES 2838007T3 ES 14824655 T ES14824655 T ES 14824655T ES 14824655 T ES14824655 T ES 14824655T ES 2838007 T3 ES2838007 T3 ES 2838007T3
- Authority
- ES
- Spain
- Prior art keywords
- composition
- cases
- linaclotide
- weight
- composition comprises
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 239000000203 mixture Substances 0.000 title claims abstract description 755
- KXGCNMMJRFDFNR-WDRJZQOASA-N linaclotide Chemical compound C([C@H](NC(=O)[C@@H]1CSSC[C@H]2C(=O)N[C@H]3CSSC[C@H](N)C(=O)N[C@H](C(N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC=4C=CC(O)=CC=4)C(=O)N2)=O)CSSC[C@H](NC(=O)[C@H](C)NC(=O)[C@@H]2CCCN2C(=O)[C@H](CC(N)=O)NC3=O)C(=O)N[C@H](C(NCC(=O)N1)=O)[C@H](O)C)C(O)=O)C1=CC=C(O)C=C1 KXGCNMMJRFDFNR-WDRJZQOASA-N 0.000 title claims abstract description 382
- 229960000812 linaclotide Drugs 0.000 title claims abstract description 381
- 108010024409 linaclotide Proteins 0.000 title claims abstract description 381
- 230000003111 delayed effect Effects 0.000 title claims abstract description 89
- 239000003826 tablet Substances 0.000 claims abstract description 145
- 239000004372 Polyvinyl alcohol Substances 0.000 claims abstract description 55
- 229920002451 polyvinyl alcohol Polymers 0.000 claims abstract description 55
- HNDVDQJCIGZPNO-UHFFFAOYSA-N histidine Natural products OC(=O)C(N)CC1=CN=CN1 HNDVDQJCIGZPNO-UHFFFAOYSA-N 0.000 claims abstract description 18
- 239000002662 enteric coated tablet Substances 0.000 claims abstract description 9
- 229920000642 polymer Polymers 0.000 claims description 77
- 208000002551 irritable bowel syndrome Diseases 0.000 claims description 64
- 206010010774 Constipation Diseases 0.000 claims description 53
- 238000000034 method Methods 0.000 claims description 51
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims description 38
- 208000002193 Pain Diseases 0.000 claims description 36
- 208000018522 Gastrointestinal disease Diseases 0.000 claims description 33
- 239000002552 dosage form Substances 0.000 claims description 30
- 239000002702 enteric coating Substances 0.000 claims description 27
- 238000009505 enteric coating Methods 0.000 claims description 27
- 201000006549 dyspepsia Diseases 0.000 claims description 16
- 210000003405 ileum Anatomy 0.000 claims description 14
- 210000001072 colon Anatomy 0.000 claims description 13
- 229920003088 hydroxypropyl methyl cellulose Polymers 0.000 claims description 13
- 208000004998 Abdominal Pain Diseases 0.000 claims description 12
- 208000009935 visceral pain Diseases 0.000 claims description 12
- 208000005615 Interstitial Cystitis Diseases 0.000 claims description 11
- 229940100467 polyvinyl acetate phthalate Drugs 0.000 claims description 11
- 208000010643 digestive system disease Diseases 0.000 claims description 10
- 201000010099 disease Diseases 0.000 claims description 10
- 208000007784 diverticulitis Diseases 0.000 claims description 10
- 208000018685 gastrointestinal system disease Diseases 0.000 claims description 10
- 229920003132 hydroxypropyl methylcellulose phthalate Polymers 0.000 claims description 10
- 229940031704 hydroxypropyl methylcellulose phthalate Drugs 0.000 claims description 10
- 229920002744 polyvinyl acetate phthalate Polymers 0.000 claims description 10
- 230000001684 chronic effect Effects 0.000 claims description 9
- 239000011241 protective layer Substances 0.000 claims description 9
- 229920002907 Guar gum Polymers 0.000 claims description 8
- 208000021302 gastroesophageal reflux disease Diseases 0.000 claims description 8
- 235000010417 guar gum Nutrition 0.000 claims description 8
- 239000000665 guar gum Substances 0.000 claims description 8
- 229960002154 guar gum Drugs 0.000 claims description 8
- 208000022559 Inflammatory bowel disease Diseases 0.000 claims description 7
- 201000009273 Endometriosis Diseases 0.000 claims description 6
- 206010054048 Postoperative ileus Diseases 0.000 claims description 6
- 208000003728 Vulvodynia Diseases 0.000 claims description 6
- 206010069055 Vulvovaginal pain Diseases 0.000 claims description 6
- 229920003145 methacrylic acid copolymer Polymers 0.000 claims description 6
- 230000001681 protective effect Effects 0.000 claims description 6
- 206010009900 Colitis ulcerative Diseases 0.000 claims description 5
- 208000011231 Crohn disease Diseases 0.000 claims description 5
- 201000006704 Ulcerative Colitis Diseases 0.000 claims description 5
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 claims description 5
- IXPNQXFRVYWDDI-UHFFFAOYSA-N 1-methyl-2,4-dioxo-1,3-diazinane-5-carboximidamide Chemical compound CN1CC(C(N)=N)C(=O)NC1=O IXPNQXFRVYWDDI-UHFFFAOYSA-N 0.000 claims description 4
- 208000014540 Functional gastrointestinal disease Diseases 0.000 claims description 4
- 206010021518 Impaired gastric emptying Diseases 0.000 claims description 4
- 208000000450 Pelvic Pain Diseases 0.000 claims description 4
- 235000021355 Stearic acid Nutrition 0.000 claims description 4
- 229920002301 cellulose acetate Polymers 0.000 claims description 4
- 208000014797 chronic intestinal pseudoobstruction Diseases 0.000 claims description 4
- 208000001288 gastroparesis Diseases 0.000 claims description 4
- 235000010979 hydroxypropyl methyl cellulose Nutrition 0.000 claims description 4
- 239000001866 hydroxypropyl methyl cellulose Substances 0.000 claims description 4
- IWVKTOUOPHGZRX-UHFFFAOYSA-N methyl 2-methylprop-2-enoate;2-methylprop-2-enoic acid Chemical compound CC(=C)C(O)=O.COC(=O)C(C)=C IWVKTOUOPHGZRX-UHFFFAOYSA-N 0.000 claims description 4
- OQCDKBAXFALNLD-UHFFFAOYSA-N octadecanoic acid Natural products CCCCCCCC(C)CCCCCCCCC(O)=O OQCDKBAXFALNLD-UHFFFAOYSA-N 0.000 claims description 4
- 235000010413 sodium alginate Nutrition 0.000 claims description 4
- 239000000661 sodium alginate Substances 0.000 claims description 4
- 229940005550 sodium alginate Drugs 0.000 claims description 4
- 239000008117 stearic acid Substances 0.000 claims description 4
- 201000005078 Colonic Pseudo-Obstruction Diseases 0.000 claims description 3
- 206010068620 Post procedural constipation Diseases 0.000 claims description 3
- IYKJEILNJZQJPU-UHFFFAOYSA-N acetic acid;butanedioic acid Chemical compound CC(O)=O.OC(=O)CCC(O)=O IYKJEILNJZQJPU-UHFFFAOYSA-N 0.000 claims description 3
- 229920001577 copolymer Polymers 0.000 claims description 3
- 208000024798 heartburn Diseases 0.000 claims description 3
- UFVKGYZPFZQRLF-UHFFFAOYSA-N hydroxypropyl methyl cellulose Chemical compound OC1C(O)C(OC)OC(CO)C1OC1C(O)C(O)C(OC2C(C(O)C(OC3C(C(O)C(O)C(CO)O3)O)C(CO)O2)O)C(CO)O1 UFVKGYZPFZQRLF-UHFFFAOYSA-N 0.000 claims description 3
- 229940127240 opiate Drugs 0.000 claims description 3
- PGJKTKAYCLVULZ-UHFFFAOYSA-N 2-methylbut-2-enoic acid prop-2-enoic acid Chemical compound OC(=O)C=C.CC=C(C)C(O)=O PGJKTKAYCLVULZ-UHFFFAOYSA-N 0.000 claims 1
- 239000000047 product Substances 0.000 description 111
- 239000011324 bead Substances 0.000 description 107
- 238000000576 coating method Methods 0.000 description 98
- 230000000087 stabilizing effect Effects 0.000 description 97
- 150000001768 cations Chemical class 0.000 description 87
- 239000011248 coating agent Substances 0.000 description 85
- 239000000243 solution Substances 0.000 description 69
- 239000002775 capsule Substances 0.000 description 58
- 150000003141 primary amines Chemical class 0.000 description 49
- 238000009472 formulation Methods 0.000 description 47
- 150000001413 amino acids Chemical group 0.000 description 45
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 44
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 44
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 43
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 41
- ROHFNLRQFUQHCH-YFKPBYRVSA-N L-leucine Chemical compound CC(C)C[C@H](N)C(O)=O ROHFNLRQFUQHCH-YFKPBYRVSA-N 0.000 description 40
- ROHFNLRQFUQHCH-UHFFFAOYSA-N Leucine Natural products CC(C)CC(N)C(O)=O ROHFNLRQFUQHCH-UHFFFAOYSA-N 0.000 description 40
- 229960003136 leucine Drugs 0.000 description 40
- 230000007062 hydrolysis Effects 0.000 description 39
- 238000006460 hydrolysis reaction Methods 0.000 description 39
- 108090000765 processed proteins & peptides Proteins 0.000 description 37
- 229940024606 amino acid Drugs 0.000 description 36
- 235000001014 amino acid Nutrition 0.000 description 36
- 230000003647 oxidation Effects 0.000 description 36
- 238000007254 oxidation reaction Methods 0.000 description 36
- 230000021736 acetylation Effects 0.000 description 35
- 238000006640 acetylation reaction Methods 0.000 description 35
- -1 PlasAcryl® Polymers 0.000 description 34
- 150000003839 salts Chemical class 0.000 description 34
- 229910052751 metal Inorganic materials 0.000 description 32
- 239000002184 metal Substances 0.000 description 32
- 238000005469 granulation Methods 0.000 description 30
- 230000003179 granulation Effects 0.000 description 30
- 210000000214 mouth Anatomy 0.000 description 29
- 208000035475 disorder Diseases 0.000 description 28
- 229940079593 drug Drugs 0.000 description 28
- 239000003814 drug Substances 0.000 description 28
- 239000000654 additive Substances 0.000 description 27
- 239000004067 bulking agent Substances 0.000 description 27
- 239000007921 spray Substances 0.000 description 25
- XUJNEKJLAYXESH-REOHCLBHSA-N L-Cysteine Chemical compound SC[C@H](N)C(O)=O XUJNEKJLAYXESH-REOHCLBHSA-N 0.000 description 24
- 229920003134 Eudragit® polymer Polymers 0.000 description 23
- 239000012530 fluid Substances 0.000 description 23
- 229920003143 Eudragit® FS 30 D Polymers 0.000 description 21
- AGPKZVBTJJNPAG-WHFBIAKZSA-N L-isoleucine Chemical compound CC[C@H](C)[C@H](N)C(O)=O AGPKZVBTJJNPAG-WHFBIAKZSA-N 0.000 description 21
- AGPKZVBTJJNPAG-UHFFFAOYSA-N isoleucine Natural products CCC(C)C(N)C(O)=O AGPKZVBTJJNPAG-UHFFFAOYSA-N 0.000 description 21
- 229960000310 isoleucine Drugs 0.000 description 21
- 229920000573 polyethylene Polymers 0.000 description 21
- QNAYBMKLOCPYGJ-REOHCLBHSA-N L-alanine Chemical compound C[C@H](N)C(O)=O QNAYBMKLOCPYGJ-REOHCLBHSA-N 0.000 description 20
- HNDVDQJCIGZPNO-YFKPBYRVSA-N L-histidine Chemical compound OC(=O)[C@@H](N)CC1=CN=CN1 HNDVDQJCIGZPNO-YFKPBYRVSA-N 0.000 description 20
- FFEARJCKVFRZRR-BYPYZUCNSA-N L-methionine Chemical compound CSCC[C@H](N)C(O)=O FFEARJCKVFRZRR-BYPYZUCNSA-N 0.000 description 20
- 208000021017 Weight Gain Diseases 0.000 description 20
- 229960003767 alanine Drugs 0.000 description 20
- 235000004279 alanine Nutrition 0.000 description 20
- 210000001035 gastrointestinal tract Anatomy 0.000 description 20
- 229930182817 methionine Natural products 0.000 description 20
- 229960004452 methionine Drugs 0.000 description 20
- 230000004584 weight gain Effects 0.000 description 20
- 235000019786 weight gain Nutrition 0.000 description 20
- 239000004698 Polyethylene Substances 0.000 description 19
- 239000000725 suspension Substances 0.000 description 19
- 238000004090 dissolution Methods 0.000 description 18
- 229960002885 histidine Drugs 0.000 description 18
- 238000013268 sustained release Methods 0.000 description 18
- 239000012730 sustained-release form Substances 0.000 description 18
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 17
- ZZSNKZQZMQGXPY-UHFFFAOYSA-N Ethyl cellulose Chemical compound CCOCC1OC(OC)C(OCC)C(OCC)C1OC1C(O)C(O)C(OC)C(CO)O1 ZZSNKZQZMQGXPY-UHFFFAOYSA-N 0.000 description 16
- 235000014304 histidine Nutrition 0.000 description 16
- 210000003750 lower gastrointestinal tract Anatomy 0.000 description 16
- 238000002156 mixing Methods 0.000 description 16
- 235000010987 pectin Nutrition 0.000 description 16
- 229920001277 pectin Polymers 0.000 description 16
- 239000001814 pectin Substances 0.000 description 16
- 239000008187 granular material Substances 0.000 description 15
- 238000002360 preparation method Methods 0.000 description 15
- 230000002496 gastric effect Effects 0.000 description 14
- 239000010410 layer Substances 0.000 description 14
- 239000007788 liquid Substances 0.000 description 14
- 239000000463 material Substances 0.000 description 13
- 238000005507 spraying Methods 0.000 description 13
- 238000003756 stirring Methods 0.000 description 13
- 102100034605 Atrial natriuretic peptide receptor 3 Human genes 0.000 description 12
- 102000000820 Enterotoxin Receptors Human genes 0.000 description 12
- 108010001687 Enterotoxin Receptors Proteins 0.000 description 12
- WSFSSNUMVMOOMR-UHFFFAOYSA-N Formaldehyde Chemical compound O=C WSFSSNUMVMOOMR-UHFFFAOYSA-N 0.000 description 12
- 101000924488 Homo sapiens Atrial natriuretic peptide receptor 3 Proteins 0.000 description 12
- 239000003795 chemical substances by application Substances 0.000 description 12
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 12
- 238000004519 manufacturing process Methods 0.000 description 12
- 239000008380 degradant Substances 0.000 description 11
- 239000000945 filler Substances 0.000 description 11
- 238000005550 wet granulation Methods 0.000 description 11
- OUYCCCASQSFEME-QMMMGPOBSA-N L-tyrosine Chemical compound OC(=O)[C@@H](N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-QMMMGPOBSA-N 0.000 description 10
- 241001465754 Metazoa Species 0.000 description 10
- 239000002253 acid Substances 0.000 description 10
- 238000007906 compression Methods 0.000 description 10
- 230000006835 compression Effects 0.000 description 10
- 238000007908 dry granulation Methods 0.000 description 10
- 230000001965 increasing effect Effects 0.000 description 10
- 239000004014 plasticizer Substances 0.000 description 10
- 239000000454 talc Substances 0.000 description 10
- 229910052623 talc Inorganic materials 0.000 description 10
- 235000012222 talc Nutrition 0.000 description 10
- MTCFGRXMJLQNBG-REOHCLBHSA-N (2S)-2-Amino-3-hydroxypropansäure Chemical compound OC[C@H](N)C(O)=O MTCFGRXMJLQNBG-REOHCLBHSA-N 0.000 description 9
- 229920000168 Microcrystalline cellulose Polymers 0.000 description 9
- QIVBCDIJIAJPQS-UHFFFAOYSA-N Tryptophan Natural products C1=CC=C2C(CC(N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-UHFFFAOYSA-N 0.000 description 9
- 239000007950 delayed release tablet Substances 0.000 description 9
- 229920000639 hydroxypropylmethylcellulose acetate succinate Polymers 0.000 description 9
- 239000012729 immediate-release (IR) formulation Substances 0.000 description 9
- 230000000968 intestinal effect Effects 0.000 description 9
- 235000019813 microcrystalline cellulose Nutrition 0.000 description 9
- 239000008108 microcrystalline cellulose Substances 0.000 description 9
- 229940016286 microcrystalline cellulose Drugs 0.000 description 9
- 230000028327 secretion Effects 0.000 description 9
- 208000024891 symptom Diseases 0.000 description 9
- VTYYLEPIZMXCLO-UHFFFAOYSA-L Calcium carbonate Chemical compound [Ca+2].[O-]C([O-])=O VTYYLEPIZMXCLO-UHFFFAOYSA-L 0.000 description 8
- 229920001661 Chitosan Polymers 0.000 description 8
- WHUUTDBJXJRKMK-UHFFFAOYSA-N Glutamic acid Natural products OC(=O)C(N)CCC(O)=O WHUUTDBJXJRKMK-UHFFFAOYSA-N 0.000 description 8
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical compound NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 description 8
- ONIBWKKTOPOVIA-BYPYZUCNSA-N L-Proline Chemical compound OC(=O)[C@@H]1CCCN1 ONIBWKKTOPOVIA-BYPYZUCNSA-N 0.000 description 8
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 8
- KZSNJWFQEVHDMF-BYPYZUCNSA-N L-valine Chemical compound CC(C)[C@H](N)C(O)=O KZSNJWFQEVHDMF-BYPYZUCNSA-N 0.000 description 8
- 239000002202 Polyethylene glycol Substances 0.000 description 8
- DOOTYTYQINUNNV-UHFFFAOYSA-N Triethyl citrate Chemical compound CCOC(=O)CC(O)(C(=O)OCC)CC(=O)OCC DOOTYTYQINUNNV-UHFFFAOYSA-N 0.000 description 8
- 229910052782 aluminium Inorganic materials 0.000 description 8
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 8
- 239000011230 binding agent Substances 0.000 description 8
- 239000000872 buffer Substances 0.000 description 8
- 150000001875 compounds Chemical class 0.000 description 8
- 229960002433 cysteine Drugs 0.000 description 8
- XUJNEKJLAYXESH-UHFFFAOYSA-N cysteine Natural products SCC(N)C(O)=O XUJNEKJLAYXESH-UHFFFAOYSA-N 0.000 description 8
- 235000018417 cysteine Nutrition 0.000 description 8
- 238000001035 drying Methods 0.000 description 8
- 235000013305 food Nutrition 0.000 description 8
- 229940068196 placebo Drugs 0.000 description 8
- 239000000902 placebo Substances 0.000 description 8
- 229920001223 polyethylene glycol Polymers 0.000 description 8
- 229960002429 proline Drugs 0.000 description 8
- 239000008213 purified water Substances 0.000 description 8
- 229960001153 serine Drugs 0.000 description 8
- 210000002784 stomach Anatomy 0.000 description 8
- 238000011282 treatment Methods 0.000 description 8
- 239000001069 triethyl citrate Substances 0.000 description 8
- VMYFZRTXGLUXMZ-UHFFFAOYSA-N triethyl citrate Natural products CCOC(=O)C(O)(C(=O)OCC)C(=O)OCC VMYFZRTXGLUXMZ-UHFFFAOYSA-N 0.000 description 8
- 235000013769 triethyl citrate Nutrition 0.000 description 8
- 229960004441 tyrosine Drugs 0.000 description 8
- 239000004475 Arginine Substances 0.000 description 7
- DCXYFEDJOCDNAF-UHFFFAOYSA-N Asparagine Natural products OC(=O)C(N)CC(N)=O DCXYFEDJOCDNAF-UHFFFAOYSA-N 0.000 description 7
- 229920000623 Cellulose acetate phthalate Polymers 0.000 description 7
- 239000001856 Ethyl cellulose Substances 0.000 description 7
- 229920003141 Eudragit® S 100 Polymers 0.000 description 7
- 108010010803 Gelatin Proteins 0.000 description 7
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 7
- 241000282412 Homo Species 0.000 description 7
- ODKSFYDXXFIFQN-BYPYZUCNSA-P L-argininium(2+) Chemical compound NC(=[NH2+])NCCC[C@H]([NH3+])C(O)=O ODKSFYDXXFIFQN-BYPYZUCNSA-P 0.000 description 7
- DCXYFEDJOCDNAF-REOHCLBHSA-N L-asparagine Chemical compound OC(=O)[C@@H](N)CC(N)=O DCXYFEDJOCDNAF-REOHCLBHSA-N 0.000 description 7
- ZDXPYRJPNDTMRX-VKHMYHEASA-N L-glutamine Chemical compound OC(=O)[C@@H](N)CCC(N)=O ZDXPYRJPNDTMRX-VKHMYHEASA-N 0.000 description 7
- KDXKERNSBIXSRK-YFKPBYRVSA-N L-lysine Chemical compound NCCCC[C@H](N)C(O)=O KDXKERNSBIXSRK-YFKPBYRVSA-N 0.000 description 7
- AYFVYJQAPQTCCC-GBXIJSLDSA-N L-threonine Chemical compound C[C@@H](O)[C@H](N)C(O)=O AYFVYJQAPQTCCC-GBXIJSLDSA-N 0.000 description 7
- QIVBCDIJIAJPQS-VIFPVBQESA-N L-tryptophane Chemical compound C1=CC=C2C(C[C@H](N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-VIFPVBQESA-N 0.000 description 7
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 7
- 239000004472 Lysine Substances 0.000 description 7
- ONIBWKKTOPOVIA-UHFFFAOYSA-N Proline Natural products OC(=O)C1CCCN1 ONIBWKKTOPOVIA-UHFFFAOYSA-N 0.000 description 7
- MTCFGRXMJLQNBG-UHFFFAOYSA-N Serine Natural products OCC(N)C(O)=O MTCFGRXMJLQNBG-UHFFFAOYSA-N 0.000 description 7
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 7
- AYFVYJQAPQTCCC-UHFFFAOYSA-N Threonine Natural products CC(O)C(N)C(O)=O AYFVYJQAPQTCCC-UHFFFAOYSA-N 0.000 description 7
- 239000004473 Threonine Substances 0.000 description 7
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 7
- 229960003121 arginine Drugs 0.000 description 7
- 235000009697 arginine Nutrition 0.000 description 7
- 229960001230 asparagine Drugs 0.000 description 7
- 235000009582 asparagine Nutrition 0.000 description 7
- 229920002678 cellulose Polymers 0.000 description 7
- 229940081734 cellulose acetate phthalate Drugs 0.000 description 7
- 230000001419 dependent effect Effects 0.000 description 7
- 239000012153 distilled water Substances 0.000 description 7
- 235000019325 ethyl cellulose Nutrition 0.000 description 7
- 229920001249 ethyl cellulose Polymers 0.000 description 7
- 239000010408 film Substances 0.000 description 7
- 239000008273 gelatin Substances 0.000 description 7
- 229920000159 gelatin Polymers 0.000 description 7
- 229940014259 gelatin Drugs 0.000 description 7
- 235000019322 gelatine Nutrition 0.000 description 7
- 235000011852 gelatine desserts Nutrition 0.000 description 7
- 229960002989 glutamic acid Drugs 0.000 description 7
- 235000013922 glutamic acid Nutrition 0.000 description 7
- 239000004220 glutamic acid Substances 0.000 description 7
- ZDXPYRJPNDTMRX-UHFFFAOYSA-N glutamine Natural products OC(=O)C(N)CCC(N)=O ZDXPYRJPNDTMRX-UHFFFAOYSA-N 0.000 description 7
- 229960002743 glutamine Drugs 0.000 description 7
- 235000004554 glutamine Nutrition 0.000 description 7
- 235000018977 lysine Nutrition 0.000 description 7
- 229960003646 lysine Drugs 0.000 description 7
- 239000008194 pharmaceutical composition Substances 0.000 description 7
- 229960002898 threonine Drugs 0.000 description 7
- 229960004799 tryptophan Drugs 0.000 description 7
- OUYCCCASQSFEME-UHFFFAOYSA-N tyrosine Natural products OC(=O)C(N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-UHFFFAOYSA-N 0.000 description 7
- 210000002438 upper gastrointestinal tract Anatomy 0.000 description 7
- 229960004295 valine Drugs 0.000 description 7
- SERLAGPUMNYUCK-DCUALPFSSA-N 1-O-alpha-D-glucopyranosyl-D-mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO[C@H]1O[C@H](CO)[C@@H](O)[C@H](O)[C@H]1O SERLAGPUMNYUCK-DCUALPFSSA-N 0.000 description 6
- 206010012735 Diarrhoea Diseases 0.000 description 6
- 229920003137 Eudragit® S polymer Polymers 0.000 description 6
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 6
- COLNVLDHVKWLRT-QMMMGPOBSA-N L-phenylalanine Chemical compound OC(=O)[C@@H](N)CC1=CC=CC=C1 COLNVLDHVKWLRT-QMMMGPOBSA-N 0.000 description 6
- TWRXJAOTZQYOKJ-UHFFFAOYSA-L Magnesium chloride Chemical compound [Mg+2].[Cl-].[Cl-] TWRXJAOTZQYOKJ-UHFFFAOYSA-L 0.000 description 6
- 241000124008 Mammalia Species 0.000 description 6
- 229920002125 Sokalan® Polymers 0.000 description 6
- KZSNJWFQEVHDMF-UHFFFAOYSA-N Valine Natural products CC(C)C(N)C(O)=O KZSNJWFQEVHDMF-UHFFFAOYSA-N 0.000 description 6
- 238000010521 absorption reaction Methods 0.000 description 6
- ZUAAPNNKRHMPKG-UHFFFAOYSA-N acetic acid;butanedioic acid;methanol;propane-1,2-diol Chemical compound OC.CC(O)=O.CC(O)CO.OC(=O)CCC(O)=O ZUAAPNNKRHMPKG-UHFFFAOYSA-N 0.000 description 6
- 229960005261 aspartic acid Drugs 0.000 description 6
- 235000003704 aspartic acid Nutrition 0.000 description 6
- OQFSQFPPLPISGP-UHFFFAOYSA-N beta-carboxyaspartic acid Natural products OC(=O)C(N)C(C(O)=O)C(O)=O OQFSQFPPLPISGP-UHFFFAOYSA-N 0.000 description 6
- 229920002988 biodegradable polymer Polymers 0.000 description 6
- 239000004621 biodegradable polymer Substances 0.000 description 6
- 239000001913 cellulose Substances 0.000 description 6
- 239000002274 desiccant Substances 0.000 description 6
- 239000007884 disintegrant Substances 0.000 description 6
- 210000000936 intestine Anatomy 0.000 description 6
- 239000000905 isomalt Substances 0.000 description 6
- 235000010439 isomalt Nutrition 0.000 description 6
- HPIGCVXMBGOWTF-UHFFFAOYSA-N isomaltol Natural products CC(=O)C=1OC=CC=1O HPIGCVXMBGOWTF-UHFFFAOYSA-N 0.000 description 6
- 235000019359 magnesium stearate Nutrition 0.000 description 6
- 239000011159 matrix material Substances 0.000 description 6
- 239000006186 oral dosage form Substances 0.000 description 6
- 239000000546 pharmaceutical excipient Substances 0.000 description 6
- COLNVLDHVKWLRT-UHFFFAOYSA-N phenylalanine Natural products OC(=O)C(N)CC1=CC=CC=C1 COLNVLDHVKWLRT-UHFFFAOYSA-N 0.000 description 6
- 229960005190 phenylalanine Drugs 0.000 description 6
- 230000008569 process Effects 0.000 description 6
- 102000004196 processed proteins & peptides Human genes 0.000 description 6
- 239000003381 stabilizer Substances 0.000 description 6
- 239000004474 valine Substances 0.000 description 6
- 239000002699 waste material Substances 0.000 description 6
- ZOOGRGPOEVQQDX-UUOKFMHZSA-N 3',5'-cyclic GMP Chemical compound C([C@H]1O2)OP(O)(=O)O[C@H]1[C@@H](O)[C@@H]2N1C(N=C(NC2=O)N)=C2N=C1 ZOOGRGPOEVQQDX-UUOKFMHZSA-N 0.000 description 5
- UXVMQQNJUSDDNG-UHFFFAOYSA-L Calcium chloride Chemical compound [Cl-].[Cl-].[Ca+2] UXVMQQNJUSDDNG-UHFFFAOYSA-L 0.000 description 5
- 206010009944 Colon cancer Diseases 0.000 description 5
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 5
- 102000004190 Enzymes Human genes 0.000 description 5
- 108090000790 Enzymes Proteins 0.000 description 5
- 229920003139 Eudragit® L 100 Polymers 0.000 description 5
- 229920003135 Eudragit® L 100-55 Polymers 0.000 description 5
- 241000282414 Homo sapiens Species 0.000 description 5
- 229920002153 Hydroxypropyl cellulose Polymers 0.000 description 5
- 229930195725 Mannitol Natural products 0.000 description 5
- 206010028980 Neoplasm Diseases 0.000 description 5
- 229920003171 Poly (ethylene oxide) Polymers 0.000 description 5
- 229920002472 Starch Polymers 0.000 description 5
- 230000003187 abdominal effect Effects 0.000 description 5
- DPXJVFZANSGRMM-UHFFFAOYSA-N acetic acid;2,3,4,5,6-pentahydroxyhexanal;sodium Chemical compound [Na].CC(O)=O.OCC(O)C(O)C(O)C(O)C=O DPXJVFZANSGRMM-UHFFFAOYSA-N 0.000 description 5
- 238000004458 analytical method Methods 0.000 description 5
- 230000015572 biosynthetic process Effects 0.000 description 5
- 229960002713 calcium chloride Drugs 0.000 description 5
- 239000001110 calcium chloride Substances 0.000 description 5
- 229910001628 calcium chloride Inorganic materials 0.000 description 5
- 235000011148 calcium chloride Nutrition 0.000 description 5
- 201000011510 cancer Diseases 0.000 description 5
- ZOOGRGPOEVQQDX-UHFFFAOYSA-N cyclic GMP Natural products O1C2COP(O)(=O)OC2C(O)C1N1C=NC2=C1NC(N)=NC2=O ZOOGRGPOEVQQDX-UHFFFAOYSA-N 0.000 description 5
- 235000010977 hydroxypropyl cellulose Nutrition 0.000 description 5
- 239000001863 hydroxypropyl cellulose Substances 0.000 description 5
- 206010020718 hyperplasia Diseases 0.000 description 5
- 210000003767 ileocecal valve Anatomy 0.000 description 5
- 239000002346 layers by function Substances 0.000 description 5
- 239000000314 lubricant Substances 0.000 description 5
- 239000011777 magnesium Substances 0.000 description 5
- 239000000594 mannitol Substances 0.000 description 5
- 235000010355 mannitol Nutrition 0.000 description 5
- 229920000609 methyl cellulose Polymers 0.000 description 5
- 235000010981 methylcellulose Nutrition 0.000 description 5
- 239000001923 methylcellulose Substances 0.000 description 5
- 239000003755 preservative agent Substances 0.000 description 5
- 239000002994 raw material Substances 0.000 description 5
- 230000002829 reductive effect Effects 0.000 description 5
- 238000010561 standard procedure Methods 0.000 description 5
- 235000019698 starch Nutrition 0.000 description 5
- VBICKXHEKHSIBG-UHFFFAOYSA-N 1-monostearoylglycerol Chemical compound CCCCCCCCCCCCCCCCCC(=O)OCC(O)CO VBICKXHEKHSIBG-UHFFFAOYSA-N 0.000 description 4
- 229920001817 Agar Polymers 0.000 description 4
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 4
- 229920002785 Croscarmellose sodium Polymers 0.000 description 4
- 229920002307 Dextran Polymers 0.000 description 4
- 229920003138 Eudragit® L 30 D-55 Polymers 0.000 description 4
- 206010017943 Gastrointestinal conditions Diseases 0.000 description 4
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 4
- 239000004471 Glycine Substances 0.000 description 4
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 4
- 229920000881 Modified starch Polymers 0.000 description 4
- WHNWPMSKXPGLAX-UHFFFAOYSA-N N-Vinyl-2-pyrrolidone Chemical compound C=CN1CCCC1=O WHNWPMSKXPGLAX-UHFFFAOYSA-N 0.000 description 4
- PXHVJJICTQNCMI-UHFFFAOYSA-N Nickel Chemical compound [Ni] PXHVJJICTQNCMI-UHFFFAOYSA-N 0.000 description 4
- 206010036772 Proctalgia Diseases 0.000 description 4
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 4
- 235000010419 agar Nutrition 0.000 description 4
- 238000000889 atomisation Methods 0.000 description 4
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 4
- 239000011575 calcium Substances 0.000 description 4
- 229960005069 calcium Drugs 0.000 description 4
- 229910052791 calcium Inorganic materials 0.000 description 4
- 229910000019 calcium carbonate Inorganic materials 0.000 description 4
- 125000002091 cationic group Chemical group 0.000 description 4
- 235000010980 cellulose Nutrition 0.000 description 4
- HGAZMNJKRQFZKS-UHFFFAOYSA-N chloroethene;ethenyl acetate Chemical compound ClC=C.CC(=O)OC=C HGAZMNJKRQFZKS-UHFFFAOYSA-N 0.000 description 4
- 239000003086 colorant Substances 0.000 description 4
- CVSVTCORWBXHQV-UHFFFAOYSA-N creatine Chemical compound NC(=[NH2+])N(C)CC([O-])=O CVSVTCORWBXHQV-UHFFFAOYSA-N 0.000 description 4
- 229960001681 croscarmellose sodium Drugs 0.000 description 4
- 235000010947 crosslinked sodium carboxy methyl cellulose Nutrition 0.000 description 4
- 230000006240 deamidation Effects 0.000 description 4
- 238000006731 degradation reaction Methods 0.000 description 4
- 239000003085 diluting agent Substances 0.000 description 4
- 239000002270 dispersing agent Substances 0.000 description 4
- 239000007903 gelatin capsule Substances 0.000 description 4
- 229960001031 glucose Drugs 0.000 description 4
- 229960002449 glycine Drugs 0.000 description 4
- 238000010438 heat treatment Methods 0.000 description 4
- 229910052749 magnesium Inorganic materials 0.000 description 4
- WPBNNNQJVZRUHP-UHFFFAOYSA-L manganese(2+);methyl n-[[2-(methoxycarbonylcarbamothioylamino)phenyl]carbamothioyl]carbamate;n-[2-(sulfidocarbothioylamino)ethyl]carbamodithioate Chemical compound [Mn+2].[S-]C(=S)NCCNC([S-])=S.COC(=O)NC(=S)NC1=CC=CC=C1NC(=S)NC(=O)OC WPBNNNQJVZRUHP-UHFFFAOYSA-L 0.000 description 4
- 230000002981 neuropathic effect Effects 0.000 description 4
- XOAAWQZATWQOTB-UHFFFAOYSA-N taurine Chemical compound NCCS(O)(=O)=O XOAAWQZATWQOTB-UHFFFAOYSA-N 0.000 description 4
- 238000012546 transfer Methods 0.000 description 4
- 230000001960 triggered effect Effects 0.000 description 4
- 229920001285 xanthan gum Polymers 0.000 description 4
- 235000010493 xanthan gum Nutrition 0.000 description 4
- 239000000230 xanthan gum Substances 0.000 description 4
- 229940082509 xanthan gum Drugs 0.000 description 4
- 239000011701 zinc Substances 0.000 description 4
- 229910052725 zinc Inorganic materials 0.000 description 4
- HDTRYLNUVZCQOY-UHFFFAOYSA-N α-D-glucopyranosyl-α-D-glucopyranoside Natural products OC1C(O)C(O)C(CO)OC1OC1C(O)C(O)C(O)C(CO)O1 HDTRYLNUVZCQOY-UHFFFAOYSA-N 0.000 description 3
- FHVDTGUDJYJELY-UHFFFAOYSA-N 6-{[2-carboxy-4,5-dihydroxy-6-(phosphanyloxy)oxan-3-yl]oxy}-4,5-dihydroxy-3-phosphanyloxane-2-carboxylic acid Chemical compound O1C(C(O)=O)C(P)C(O)C(O)C1OC1C(C(O)=O)OC(OP)C(O)C1O FHVDTGUDJYJELY-UHFFFAOYSA-N 0.000 description 3
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 3
- 206010002153 Anal fissure Diseases 0.000 description 3
- 208000016583 Anus disease Diseases 0.000 description 3
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 3
- 229920000858 Cyclodextrin Polymers 0.000 description 3
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 3
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 3
- 229920003136 Eudragit® L polymer Polymers 0.000 description 3
- 229920003161 Eudragit® RS 30 D Polymers 0.000 description 3
- 208000009531 Fissure in Ano Diseases 0.000 description 3
- 206010017993 Gastrointestinal neoplasms Diseases 0.000 description 3
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 3
- CERQOIWHTDAKMF-UHFFFAOYSA-M Methacrylate Chemical compound CC(=C)C([O-])=O CERQOIWHTDAKMF-UHFFFAOYSA-M 0.000 description 3
- SJRJJKPEHAURKC-UHFFFAOYSA-N N-Methylmorpholine Chemical compound CN1CCOCC1 SJRJJKPEHAURKC-UHFFFAOYSA-N 0.000 description 3
- MBBZMMPHUWSWHV-BDVNFPICSA-N N-methylglucamine Chemical compound CNC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO MBBZMMPHUWSWHV-BDVNFPICSA-N 0.000 description 3
- 208000030053 Opioid-Induced Constipation Diseases 0.000 description 3
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 3
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 3
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 3
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 3
- 229930006000 Sucrose Natural products 0.000 description 3
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 3
- 206010043345 Testicular pain Diseases 0.000 description 3
- HDTRYLNUVZCQOY-WSWWMNSNSA-N Trehalose Natural products O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@@H]1O[C@@H]1[C@H](O)[C@@H](O)[C@@H](O)[C@@H](CO)O1 HDTRYLNUVZCQOY-WSWWMNSNSA-N 0.000 description 3
- ZOIORXHNWRGPMV-UHFFFAOYSA-N acetic acid;zinc Chemical compound [Zn].CC(O)=O.CC(O)=O ZOIORXHNWRGPMV-UHFFFAOYSA-N 0.000 description 3
- 239000013543 active substance Substances 0.000 description 3
- 229940072056 alginate Drugs 0.000 description 3
- 229920000615 alginic acid Polymers 0.000 description 3
- 235000010443 alginic acid Nutrition 0.000 description 3
- HDTRYLNUVZCQOY-LIZSDCNHSA-N alpha,alpha-trehalose Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@@H]1O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 HDTRYLNUVZCQOY-LIZSDCNHSA-N 0.000 description 3
- 239000003963 antioxidant agent Substances 0.000 description 3
- 238000003556 assay Methods 0.000 description 3
- 239000006172 buffering agent Substances 0.000 description 3
- 229960003563 calcium carbonate Drugs 0.000 description 3
- 229940045110 chitosan Drugs 0.000 description 3
- 239000007931 coated granule Substances 0.000 description 3
- 208000029742 colonic neoplasm Diseases 0.000 description 3
- 238000013270 controlled release Methods 0.000 description 3
- 230000001276 controlling effect Effects 0.000 description 3
- 229960000913 crospovidone Drugs 0.000 description 3
- LOKCTEFSRHRXRJ-UHFFFAOYSA-I dipotassium trisodium dihydrogen phosphate hydrogen phosphate dichloride Chemical compound P(=O)(O)(O)[O-].[K+].P(=O)(O)([O-])[O-].[Na+].[Na+].[Cl-].[K+].[Cl-].[Na+] LOKCTEFSRHRXRJ-UHFFFAOYSA-I 0.000 description 3
- 230000000694 effects Effects 0.000 description 3
- 230000002708 enhancing effect Effects 0.000 description 3
- 210000002919 epithelial cell Anatomy 0.000 description 3
- 238000002474 experimental method Methods 0.000 description 3
- 239000011888 foil Substances 0.000 description 3
- 235000011187 glycerol Nutrition 0.000 description 3
- 238000009434 installation Methods 0.000 description 3
- 238000004811 liquid chromatography Methods 0.000 description 3
- 229910001629 magnesium chloride Inorganic materials 0.000 description 3
- 150000002892 organic cations Chemical class 0.000 description 3
- 239000006179 pH buffering agent Substances 0.000 description 3
- 239000002953 phosphate buffered saline Substances 0.000 description 3
- 229920001983 poloxamer Polymers 0.000 description 3
- 239000004584 polyacrylic acid Substances 0.000 description 3
- 229920000136 polysorbate Polymers 0.000 description 3
- 235000013809 polyvinylpolypyrrolidone Nutrition 0.000 description 3
- 229920000523 polyvinylpolypyrrolidone Polymers 0.000 description 3
- 239000011591 potassium Substances 0.000 description 3
- 229910052700 potassium Inorganic materials 0.000 description 3
- 201000007094 prostatitis Diseases 0.000 description 3
- 102000005962 receptors Human genes 0.000 description 3
- 108020003175 receptors Proteins 0.000 description 3
- 230000002441 reversible effect Effects 0.000 description 3
- HFHDHCJBZVLPGP-UHFFFAOYSA-N schardinger α-dextrin Chemical compound O1C(C(C2O)O)C(CO)OC2OC(C(C2O)O)C(CO)OC2OC(C(C2O)O)C(CO)OC2OC(C(O)C2O)C(CO)OC2OC(C(C2O)O)C(CO)OC2OC2C(O)C(O)C1OC2CO HFHDHCJBZVLPGP-UHFFFAOYSA-N 0.000 description 3
- 239000000377 silicon dioxide Substances 0.000 description 3
- 239000011734 sodium Substances 0.000 description 3
- 235000015424 sodium Nutrition 0.000 description 3
- 229910052708 sodium Inorganic materials 0.000 description 3
- 239000007787 solid Substances 0.000 description 3
- 239000002904 solvent Substances 0.000 description 3
- 235000010356 sorbitol Nutrition 0.000 description 3
- 239000000600 sorbitol Substances 0.000 description 3
- 238000001694 spray drying Methods 0.000 description 3
- 229910001220 stainless steel Inorganic materials 0.000 description 3
- 239000010935 stainless steel Substances 0.000 description 3
- 239000008107 starch Substances 0.000 description 3
- 229940032147 starch Drugs 0.000 description 3
- 239000005720 sucrose Substances 0.000 description 3
- 239000004094 surface-active agent Substances 0.000 description 3
- 238000012360 testing method Methods 0.000 description 3
- 229940074410 trehalose Drugs 0.000 description 3
- 229920003169 water-soluble polymer Polymers 0.000 description 3
- 238000005303 weighing Methods 0.000 description 3
- 239000004246 zinc acetate Substances 0.000 description 3
- 235000013904 zinc acetate Nutrition 0.000 description 3
- HZAXFHJVJLSVMW-UHFFFAOYSA-N 2-Aminoethan-1-ol Chemical compound NCCO HZAXFHJVJLSVMW-UHFFFAOYSA-N 0.000 description 2
- MSWZFWKMSRAUBD-IVMDWMLBSA-N 2-amino-2-deoxy-D-glucopyranose Chemical compound N[C@H]1C(O)O[C@H](CO)[C@@H](O)[C@@H]1O MSWZFWKMSRAUBD-IVMDWMLBSA-N 0.000 description 2
- WRMNZCZEMHIOCP-UHFFFAOYSA-N 2-phenylethanol Chemical compound OCCC1=CC=CC=C1 WRMNZCZEMHIOCP-UHFFFAOYSA-N 0.000 description 2
- 229910002016 Aerosil® 200 Inorganic materials 0.000 description 2
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 2
- 229920000856 Amylose Polymers 0.000 description 2
- PLTGTJAZQRGMPP-FXQIFTODSA-N Asn-Pro-Ala Chemical compound OC(=O)[C@H](C)NC(=O)[C@@H]1CCCN1C(=O)[C@@H](N)CC(N)=O PLTGTJAZQRGMPP-FXQIFTODSA-N 0.000 description 2
- BVKZGUZCCUSVTD-UHFFFAOYSA-M Bicarbonate Chemical compound OC([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-M 0.000 description 2
- QFOHBWFCKVYLES-UHFFFAOYSA-N Butylparaben Chemical compound CCCCOC(=O)C1=CC=C(O)C=C1 QFOHBWFCKVYLES-UHFFFAOYSA-N 0.000 description 2
- 241000283707 Capra Species 0.000 description 2
- VYZAMTAEIAYCRO-UHFFFAOYSA-N Chromium Chemical compound [Cr] VYZAMTAEIAYCRO-UHFFFAOYSA-N 0.000 description 2
- 208000015943 Coeliac disease Diseases 0.000 description 2
- 208000001333 Colorectal Neoplasms Diseases 0.000 description 2
- ISWAQPWFWKGCAL-ACZMJKKPSA-N Cys-Cys-Glu Chemical compound [H]N[C@@H](CS)C(=O)N[C@@H](CS)C(=O)N[C@@H](CCC(O)=O)C(O)=O ISWAQPWFWKGCAL-ACZMJKKPSA-N 0.000 description 2
- ALNKNYKSZPSLBD-ZDLURKLDSA-N Cys-Thr-Gly Chemical compound [H]N[C@@H](CS)C(=O)N[C@@H]([C@@H](C)O)C(=O)NCC(O)=O ALNKNYKSZPSLBD-ZDLURKLDSA-N 0.000 description 2
- 201000003883 Cystic fibrosis Diseases 0.000 description 2
- 229920001353 Dextrin Polymers 0.000 description 2
- 239000004375 Dextrin Substances 0.000 description 2
- NIQCNGHVCWTJSM-UHFFFAOYSA-N Dimethyl phthalate Chemical compound COC(=O)C1=CC=CC=C1C(=O)OC NIQCNGHVCWTJSM-UHFFFAOYSA-N 0.000 description 2
- 239000004386 Erythritol Substances 0.000 description 2
- UNXHWFMMPAWVPI-UHFFFAOYSA-N Erythritol Natural products OCC(O)C(O)CO UNXHWFMMPAWVPI-UHFFFAOYSA-N 0.000 description 2
- 229920003157 Eudragit® RL 30 D Polymers 0.000 description 2
- 229920003152 Eudragit® RS polymer Polymers 0.000 description 2
- 208000001640 Fibromyalgia Diseases 0.000 description 2
- 229930091371 Fructose Natural products 0.000 description 2
- 239000005715 Fructose Substances 0.000 description 2
- RFSUNEUAIZKAJO-ARQDHWQXSA-N Fructose Chemical compound OC[C@H]1O[C@](O)(CO)[C@@H](O)[C@@H]1O RFSUNEUAIZKAJO-ARQDHWQXSA-N 0.000 description 2
- 208000017228 Gastrointestinal motility disease Diseases 0.000 description 2
- 206010017999 Gastrointestinal pain Diseases 0.000 description 2
- 241000206672 Gelidium Species 0.000 description 2
- 208000032177 Intestinal Polyps Diseases 0.000 description 2
- XEEYBQQBJWHFJM-UHFFFAOYSA-N Iron Chemical compound [Fe] XEEYBQQBJWHFJM-UHFFFAOYSA-N 0.000 description 2
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 2
- BAPJBEWLBFYGME-UHFFFAOYSA-N Methyl acrylate Chemical compound COC(=O)C=C BAPJBEWLBFYGME-UHFFFAOYSA-N 0.000 description 2
- VVQNEPGJFQJSBK-UHFFFAOYSA-N Methyl methacrylate Chemical compound COC(=O)C(C)=C VVQNEPGJFQJSBK-UHFFFAOYSA-N 0.000 description 2
- DATAGRPVKZEWHA-YFKPBYRVSA-N N(5)-ethyl-L-glutamine Chemical compound CCNC(=O)CC[C@H]([NH3+])C([O-])=O DATAGRPVKZEWHA-YFKPBYRVSA-N 0.000 description 2
- 241000283973 Oryctolagus cuniculus Species 0.000 description 2
- 241001494479 Pecora Species 0.000 description 2
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 2
- RVGRUAULSDPKGF-UHFFFAOYSA-N Poloxamer Chemical compound C1CO1.CC1CO1 RVGRUAULSDPKGF-UHFFFAOYSA-N 0.000 description 2
- 229920000148 Polycarbophil calcium Polymers 0.000 description 2
- 229920000954 Polyglycolide Polymers 0.000 description 2
- 241000283984 Rodentia Species 0.000 description 2
- 229920001800 Shellac Polymers 0.000 description 2
- DBMJMQXJHONAFJ-UHFFFAOYSA-M Sodium laurylsulphate Chemical compound [Na+].CCCCCCCCCCCCOS([O-])(=O)=O DBMJMQXJHONAFJ-UHFFFAOYSA-M 0.000 description 2
- 208000024799 Thyroid disease Diseases 0.000 description 2
- CGDZGRLRXPNCOC-SRVKXCTJSA-N Tyr-Cys-Cys Chemical compound SC[C@@H](C(O)=O)NC(=O)[C@H](CS)NC(=O)[C@@H](N)CC1=CC=C(O)C=C1 CGDZGRLRXPNCOC-SRVKXCTJSA-N 0.000 description 2
- 230000003213 activating effect Effects 0.000 description 2
- 230000004913 activation Effects 0.000 description 2
- 230000000996 additive effect Effects 0.000 description 2
- 239000008272 agar Substances 0.000 description 2
- 229940023476 agar Drugs 0.000 description 2
- 230000000845 anti-microbial effect Effects 0.000 description 2
- 239000004599 antimicrobial Substances 0.000 description 2
- 108010010430 asparagine-proline-alanine Proteins 0.000 description 2
- 229910052788 barium Inorganic materials 0.000 description 2
- DSAJWYNOEDNPEQ-UHFFFAOYSA-N barium atom Chemical compound [Ba] DSAJWYNOEDNPEQ-UHFFFAOYSA-N 0.000 description 2
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 2
- MSWZFWKMSRAUBD-UHFFFAOYSA-N beta-D-galactosamine Natural products NC1C(O)OC(CO)C(O)C1O MSWZFWKMSRAUBD-UHFFFAOYSA-N 0.000 description 2
- 235000013361 beverage Nutrition 0.000 description 2
- VSGNNIFQASZAOI-UHFFFAOYSA-L calcium acetate Chemical compound [Ca+2].CC([O-])=O.CC([O-])=O VSGNNIFQASZAOI-UHFFFAOYSA-L 0.000 description 2
- 239000001639 calcium acetate Substances 0.000 description 2
- 235000011092 calcium acetate Nutrition 0.000 description 2
- 229960005147 calcium acetate Drugs 0.000 description 2
- 235000010216 calcium carbonate Nutrition 0.000 description 2
- OSGAYBCDTDRGGQ-UHFFFAOYSA-L calcium sulfate Chemical compound [Ca+2].[O-]S([O-])(=O)=O OSGAYBCDTDRGGQ-UHFFFAOYSA-L 0.000 description 2
- 239000007963 capsule composition Substances 0.000 description 2
- 239000001768 carboxy methyl cellulose Substances 0.000 description 2
- 230000015556 catabolic process Effects 0.000 description 2
- RZEKVGVHFLEQIL-UHFFFAOYSA-N celecoxib Chemical compound C1=CC(C)=CC=C1C1=CC(C(F)(F)F)=NN1C1=CC=C(S(N)(=O)=O)C=C1 RZEKVGVHFLEQIL-UHFFFAOYSA-N 0.000 description 2
- WZNRVWBKYDHTKI-UHFFFAOYSA-N cellulose, acetate 1,2,4-benzenetricarboxylate Chemical compound OC1C(O)C(O)C(CO)OC1OC1C(CO)OC(O)C(O)C1O.OC1C(O)C(O)C(CO)OC1OC1C(CO)OC(O)C(O)C1O.CC(=O)OCC1OC(OC(C)=O)C(OC(C)=O)C(OC(C)=O)C1OC1C(OC(C)=O)C(OC(C)=O)C(OC(C)=O)C(COC(C)=O)O1.CC(=O)OCC1OC(OC(C)=O)C(OC(C)=O)C(OC(C)=O)C1OC1C(OC(C)=O)C(OC(C)=O)C(OC(C)=O)C(COC(C)=O)O1.OC(=O)C1=CC(C(=O)O)=CC=C1C(=O)OCC1C(OC2C(C(OC(=O)C=3C(=CC(=CC=3)C(O)=O)C(O)=O)C(OC(=O)C=3C(=CC(=CC=3)C(O)=O)C(O)=O)C(COC(=O)C=3C(=CC(=CC=3)C(O)=O)C(O)=O)O2)OC(=O)C=2C(=CC(=CC=2)C(O)=O)C(O)=O)C(OC(=O)C=2C(=CC(=CC=2)C(O)=O)C(O)=O)C(OC(=O)C=2C(=CC(=CC=2)C(O)=O)C(O)=O)C(OC(=O)C=2C(=CC(=CC=2)C(O)=O)C(O)=O)O1 WZNRVWBKYDHTKI-UHFFFAOYSA-N 0.000 description 2
- 238000006243 chemical reaction Methods 0.000 description 2
- OSASVXMJTNOKOY-UHFFFAOYSA-N chlorobutanol Chemical compound CC(C)(O)C(Cl)(Cl)Cl OSASVXMJTNOKOY-UHFFFAOYSA-N 0.000 description 2
- OEYIOHPDSNJKLS-UHFFFAOYSA-N choline Chemical compound C[N+](C)(C)CCO OEYIOHPDSNJKLS-UHFFFAOYSA-N 0.000 description 2
- 229960001231 choline Drugs 0.000 description 2
- 229910052804 chromium Inorganic materials 0.000 description 2
- 239000011651 chromium Substances 0.000 description 2
- 239000008199 coating composition Substances 0.000 description 2
- 229920001688 coating polymer Polymers 0.000 description 2
- 229910017052 cobalt Inorganic materials 0.000 description 2
- 239000010941 cobalt Substances 0.000 description 2
- GUTLYIVDDKVIGB-UHFFFAOYSA-N cobalt atom Chemical compound [Co] GUTLYIVDDKVIGB-UHFFFAOYSA-N 0.000 description 2
- 230000000112 colonic effect Effects 0.000 description 2
- 229940111134 coxibs Drugs 0.000 description 2
- 229960003624 creatine Drugs 0.000 description 2
- 239000006046 creatine Substances 0.000 description 2
- 239000003255 cyclooxygenase 2 inhibitor Substances 0.000 description 2
- 125000000151 cysteine group Chemical group N[C@@H](CS)C(=O)* 0.000 description 2
- 108010069495 cysteinyltyrosine Proteins 0.000 description 2
- 230000003247 decreasing effect Effects 0.000 description 2
- 235000019425 dextrin Nutrition 0.000 description 2
- 239000008121 dextrose Substances 0.000 description 2
- DOIRQSBPFJWKBE-UHFFFAOYSA-N dibutyl phthalate Chemical compound CCCCOC(=O)C1=CC=CC=C1C(=O)OCCCC DOIRQSBPFJWKBE-UHFFFAOYSA-N 0.000 description 2
- FLKPEMZONWLCSK-UHFFFAOYSA-N diethyl phthalate Chemical compound CCOC(=O)C1=CC=CC=C1C(=O)OCC FLKPEMZONWLCSK-UHFFFAOYSA-N 0.000 description 2
- 230000029087 digestion Effects 0.000 description 2
- 208000024558 digestive system cancer Diseases 0.000 description 2
- 239000006185 dispersion Substances 0.000 description 2
- 239000012738 dissolution medium Substances 0.000 description 2
- 239000003937 drug carrier Substances 0.000 description 2
- 238000009506 drug dissolution testing Methods 0.000 description 2
- 210000001198 duodenum Anatomy 0.000 description 2
- 239000003792 electrolyte Substances 0.000 description 2
- UNXHWFMMPAWVPI-ZXZARUISSA-N erythritol Chemical compound OC[C@H](O)[C@H](O)CO UNXHWFMMPAWVPI-ZXZARUISSA-N 0.000 description 2
- 235000019414 erythritol Nutrition 0.000 description 2
- 229940009714 erythritol Drugs 0.000 description 2
- MMXKVMNBHPAILY-UHFFFAOYSA-N ethyl laurate Chemical compound CCCCCCCCCCCC(=O)OCC MMXKVMNBHPAILY-UHFFFAOYSA-N 0.000 description 2
- 238000009501 film coating Methods 0.000 description 2
- 239000007888 film coating Substances 0.000 description 2
- 239000000796 flavoring agent Substances 0.000 description 2
- 235000019634 flavors Nutrition 0.000 description 2
- 239000006260 foam Substances 0.000 description 2
- 201000010231 gastrointestinal system cancer Diseases 0.000 description 2
- 229960002442 glucosamine Drugs 0.000 description 2
- 239000008103 glucose Substances 0.000 description 2
- 229940075507 glyceryl monostearate Drugs 0.000 description 2
- 238000004128 high performance liquid chromatography Methods 0.000 description 2
- 239000003906 humectant Substances 0.000 description 2
- 238000001727 in vivo Methods 0.000 description 2
- 238000011534 incubation Methods 0.000 description 2
- 230000008991 intestinal motility Effects 0.000 description 2
- 230000003834 intracellular effect Effects 0.000 description 2
- 210000001630 jejunum Anatomy 0.000 description 2
- 239000008101 lactose Substances 0.000 description 2
- 238000011068 loading method Methods 0.000 description 2
- VQHSOMBJVWLPSR-WUJBLJFYSA-N maltitol Chemical compound OC[C@H](O)[C@@H](O)[C@@H]([C@H](O)CO)O[C@H]1O[C@H](CO)[C@@H](O)[C@H](O)[C@H]1O VQHSOMBJVWLPSR-WUJBLJFYSA-N 0.000 description 2
- 235000010449 maltitol Nutrition 0.000 description 2
- 239000000845 maltitol Substances 0.000 description 2
- 229940035436 maltitol Drugs 0.000 description 2
- 239000013563 matrix tablet Substances 0.000 description 2
- 238000005259 measurement Methods 0.000 description 2
- 238000002483 medication Methods 0.000 description 2
- 239000000155 melt Substances 0.000 description 2
- KBOPZPXVLCULAV-UHFFFAOYSA-N mesalamine Chemical compound NC1=CC=C(O)C(C(O)=O)=C1 KBOPZPXVLCULAV-UHFFFAOYSA-N 0.000 description 2
- 229960004963 mesalazine Drugs 0.000 description 2
- 230000006510 metastatic growth Effects 0.000 description 2
- IQSHMXAZFHORGY-UHFFFAOYSA-N methyl prop-2-enoate;2-methylprop-2-enoic acid Chemical compound COC(=O)C=C.CC(=C)C(O)=O IQSHMXAZFHORGY-UHFFFAOYSA-N 0.000 description 2
- LXCFILQKKLGQFO-UHFFFAOYSA-N methylparaben Chemical compound COC(=O)C1=CC=C(O)C=C1 LXCFILQKKLGQFO-UHFFFAOYSA-N 0.000 description 2
- 239000011859 microparticle Substances 0.000 description 2
- 230000004048 modification Effects 0.000 description 2
- 238000012986 modification Methods 0.000 description 2
- 239000001788 mono and diglycerides of fatty acids Substances 0.000 description 2
- 230000004899 motility Effects 0.000 description 2
- 229910052759 nickel Inorganic materials 0.000 description 2
- 229940005483 opioid analgesics Drugs 0.000 description 2
- 239000011049 pearl Substances 0.000 description 2
- 229960000292 pectin Drugs 0.000 description 2
- 239000000049 pigment Substances 0.000 description 2
- 229960000502 poloxamer Drugs 0.000 description 2
- 229920000744 poly(arginines) Polymers 0.000 description 2
- 229920001606 poly(lactic acid-co-glycolic acid) Polymers 0.000 description 2
- 229920000058 polyacrylate Polymers 0.000 description 2
- 229920001610 polycaprolactone Polymers 0.000 description 2
- 239000004632 polycaprolactone Substances 0.000 description 2
- 229950005134 polycarbophil Drugs 0.000 description 2
- 239000004633 polyglycolic acid Substances 0.000 description 2
- 239000000843 powder Substances 0.000 description 2
- 230000002265 prevention Effects 0.000 description 2
- 238000012545 processing Methods 0.000 description 2
- 230000002035 prolonged effect Effects 0.000 description 2
- QELSKZZBTMNZEB-UHFFFAOYSA-N propylparaben Chemical compound CCCOC(=O)C1=CC=C(O)C=C1 QELSKZZBTMNZEB-UHFFFAOYSA-N 0.000 description 2
- LXNHXLLTXMVWPM-UHFFFAOYSA-N pyridoxine Chemical compound CC1=NC=C(CO)C(CO)=C1O LXNHXLLTXMVWPM-UHFFFAOYSA-N 0.000 description 2
- 239000000018 receptor agonist Substances 0.000 description 2
- 229940044601 receptor agonist Drugs 0.000 description 2
- 230000009467 reduction Effects 0.000 description 2
- 239000004208 shellac Substances 0.000 description 2
- 229940113147 shellac Drugs 0.000 description 2
- ZLGIYFNHBLSMPS-ATJNOEHPSA-N shellac Chemical compound OCCCCCC(O)C(O)CCCCCCCC(O)=O.C1C23[C@H](C(O)=O)CCC2[C@](C)(CO)[C@@H]1C(C(O)=O)=C[C@@H]3O ZLGIYFNHBLSMPS-ATJNOEHPSA-N 0.000 description 2
- 235000013874 shellac Nutrition 0.000 description 2
- 235000012239 silicon dioxide Nutrition 0.000 description 2
- 235000019333 sodium laurylsulphate Nutrition 0.000 description 2
- 241000894007 species Species 0.000 description 2
- ATHGHQPFGPMSJY-UHFFFAOYSA-N spermidine Chemical compound NCCCCNCCCN ATHGHQPFGPMSJY-UHFFFAOYSA-N 0.000 description 2
- PFNFFQXMRSDOHW-UHFFFAOYSA-N spermine Chemical compound NCCCNCCCCNCCCN PFNFFQXMRSDOHW-UHFFFAOYSA-N 0.000 description 2
- 239000000126 substance Substances 0.000 description 2
- 235000001508 sulfur Nutrition 0.000 description 2
- 238000009492 tablet coating Methods 0.000 description 2
- 239000002700 tablet coating Substances 0.000 description 2
- 229960003080 taurine Drugs 0.000 description 2
- 238000002560 therapeutic procedure Methods 0.000 description 2
- MGSRCZKZVOBKFT-UHFFFAOYSA-N thymol Chemical compound CC(C)C1=CC=C(C)C=C1O MGSRCZKZVOBKFT-UHFFFAOYSA-N 0.000 description 2
- 208000021510 thyroid gland disease Diseases 0.000 description 2
- URAYPUMNDPQOKB-UHFFFAOYSA-N triacetin Chemical compound CC(=O)OCC(OC(C)=O)COC(C)=O URAYPUMNDPQOKB-UHFFFAOYSA-N 0.000 description 2
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 2
- 229960000281 trometamol Drugs 0.000 description 2
- JIAARYAFYJHUJI-UHFFFAOYSA-L zinc dichloride Chemical compound [Cl-].[Cl-].[Zn+2] JIAARYAFYJHUJI-UHFFFAOYSA-L 0.000 description 2
- OMDQUFIYNPYJFM-XKDAHURESA-N (2r,3r,4s,5r,6s)-2-(hydroxymethyl)-6-[[(2r,3s,4r,5s,6r)-4,5,6-trihydroxy-3-[(2s,3s,4s,5s,6r)-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl]oxyoxan-2-yl]methoxy]oxane-3,4,5-triol Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@@H]1OC[C@@H]1[C@@H](O[C@H]2[C@H]([C@@H](O)[C@H](O)[C@@H](CO)O2)O)[C@H](O)[C@H](O)[C@H](O)O1 OMDQUFIYNPYJFM-XKDAHURESA-N 0.000 description 1
- GGTYBZJRPHEQDG-WCCKRBBISA-N (2s)-2,5-diaminopentanoic acid hydrochloride Chemical compound Cl.NCCC[C@H](N)C(O)=O GGTYBZJRPHEQDG-WCCKRBBISA-N 0.000 description 1
- SLPUVFBNQHVEEU-WCCKRBBISA-N (2s)-2,5-diaminopentanoic acid;2-oxopentanedioic acid Chemical compound NCCC[C@H](N)C(O)=O.OC(=O)CCC(=O)C(O)=O SLPUVFBNQHVEEU-WCCKRBBISA-N 0.000 description 1
- PGRNZHOQVAPMFX-WCCKRBBISA-N (2s)-2-amino-5-(diaminomethylideneamino)pentanoic acid;2-oxopentanedioic acid Chemical compound OC(=O)CCC(=O)C(O)=O.OC(=O)[C@@H](N)CCCN=C(N)N PGRNZHOQVAPMFX-WCCKRBBISA-N 0.000 description 1
- PHIQHXFUZVPYII-ZCFIWIBFSA-O (R)-carnitinium Chemical compound C[N+](C)(C)C[C@H](O)CC(O)=O PHIQHXFUZVPYII-ZCFIWIBFSA-O 0.000 description 1
- JCIIKRHCWVHVFF-UHFFFAOYSA-N 1,2,4-thiadiazol-5-amine;hydrochloride Chemical compound Cl.NC1=NC=NS1 JCIIKRHCWVHVFF-UHFFFAOYSA-N 0.000 description 1
- LDVVTQMJQSCDMK-UHFFFAOYSA-N 1,3-dihydroxypropan-2-yl formate Chemical compound OCC(CO)OC=O LDVVTQMJQSCDMK-UHFFFAOYSA-N 0.000 description 1
- UBCHPRBFMUDMNC-UHFFFAOYSA-N 1-(1-adamantyl)ethanamine Chemical compound C1C(C2)CC3CC2CC1(C(N)C)C3 UBCHPRBFMUDMNC-UHFFFAOYSA-N 0.000 description 1
- OWEGMIWEEQEYGQ-UHFFFAOYSA-N 100676-05-9 Natural products OC1C(O)C(O)C(CO)OC1OCC1C(O)C(O)C(O)C(OC2C(OC(O)C(O)C2O)CO)O1 OWEGMIWEEQEYGQ-UHFFFAOYSA-N 0.000 description 1
- KMZHZAAOEWVPSE-UHFFFAOYSA-N 2,3-dihydroxypropyl acetate Chemical compound CC(=O)OCC(O)CO KMZHZAAOEWVPSE-UHFFFAOYSA-N 0.000 description 1
- CHHHXKFHOYLYRE-UHFFFAOYSA-M 2,4-Hexadienoic acid, potassium salt (1:1), (2E,4E)- Chemical compound [K+].CC=CC=CC([O-])=O CHHHXKFHOYLYRE-UHFFFAOYSA-M 0.000 description 1
- QTWJRLJHJPIABL-UHFFFAOYSA-N 2-methylphenol;3-methylphenol;4-methylphenol Chemical compound CC1=CC=C(O)C=C1.CC1=CC=CC(O)=C1.CC1=CC=CC=C1O QTWJRLJHJPIABL-UHFFFAOYSA-N 0.000 description 1
- QCDWFXQBSFUVSP-UHFFFAOYSA-N 2-phenoxyethanol Chemical compound OCCOC1=CC=CC=C1 QCDWFXQBSFUVSP-UHFFFAOYSA-N 0.000 description 1
- WADSJYLPJPTMLN-UHFFFAOYSA-N 3-(cycloundecen-1-yl)-1,2-diazacycloundec-2-ene Chemical compound C1CCCCCCCCC=C1C1=NNCCCCCCCC1 WADSJYLPJPTMLN-UHFFFAOYSA-N 0.000 description 1
- SQDAZGGFXASXDW-UHFFFAOYSA-N 5-bromo-2-(trifluoromethoxy)pyridine Chemical compound FC(F)(F)OC1=CC=C(Br)C=N1 SQDAZGGFXASXDW-UHFFFAOYSA-N 0.000 description 1
- 206010000060 Abdominal distension Diseases 0.000 description 1
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 1
- 102000009027 Albumins Human genes 0.000 description 1
- 108010088751 Albumins Proteins 0.000 description 1
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 1
- 240000002470 Amphicarpaea bracteata Species 0.000 description 1
- AKGWUHIOEVNNPC-LURJTMIESA-N Arg-OEt Chemical compound CCOC(=O)[C@@H](N)CCCNC(N)=N AKGWUHIOEVNNPC-LURJTMIESA-N 0.000 description 1
- 241000416162 Astragalus gummifer Species 0.000 description 1
- 241000271566 Aves Species 0.000 description 1
- 208000023514 Barrett esophagus Diseases 0.000 description 1
- 208000023665 Barrett oesophagus Diseases 0.000 description 1
- 239000005711 Benzoic acid Substances 0.000 description 1
- 241000283690 Bos taurus Species 0.000 description 1
- 241000282465 Canis Species 0.000 description 1
- 241000282472 Canis lupus familiaris Species 0.000 description 1
- 229920002134 Carboxymethyl cellulose Polymers 0.000 description 1
- 241000206601 Carnobacterium mobile Species 0.000 description 1
- 229920001287 Chondroitin sulfate Polymers 0.000 description 1
- ODKSFYDXXFIFQN-SCSAIBSYSA-N D-arginine Chemical compound OC(=O)[C@H](N)CCCNC(N)=N ODKSFYDXXFIFQN-SCSAIBSYSA-N 0.000 description 1
- 229930028154 D-arginine Natural products 0.000 description 1
- WQZGKKKJIJFFOK-QTVWNMPRSA-N D-mannopyranose Chemical compound OC[C@H]1OC(O)[C@@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-QTVWNMPRSA-N 0.000 description 1
- 239000004287 Dehydroacetic acid Substances 0.000 description 1
- PYGXAGIECVVIOZ-UHFFFAOYSA-N Dibutyl decanedioate Chemical compound CCCCOC(=O)CCCCCCCCC(=O)OCCCC PYGXAGIECVVIOZ-UHFFFAOYSA-N 0.000 description 1
- 235000019739 Dicalciumphosphate Nutrition 0.000 description 1
- 238000002965 ELISA Methods 0.000 description 1
- 229920003119 EUDRAGIT E PO Polymers 0.000 description 1
- LVGKNOAMLMIIKO-UHFFFAOYSA-N Elaidinsaeure-aethylester Natural products CCCCCCCCC=CCCCCCCCC(=O)OCC LVGKNOAMLMIIKO-UHFFFAOYSA-N 0.000 description 1
- 241000196324 Embryophyta Species 0.000 description 1
- 241000792859 Enema Species 0.000 description 1
- 101710146739 Enterotoxin Proteins 0.000 description 1
- 241000283073 Equus caballus Species 0.000 description 1
- PIICEJLVQHRZGT-UHFFFAOYSA-N Ethylenediamine Chemical compound NCCN PIICEJLVQHRZGT-UHFFFAOYSA-N 0.000 description 1
- 241000282324 Felis Species 0.000 description 1
- 241000282326 Felis catus Species 0.000 description 1
- 206010017367 Frequent bowel movements Diseases 0.000 description 1
- 229920000926 Galactomannan Polymers 0.000 description 1
- 241000287828 Gallus gallus Species 0.000 description 1
- 239000004348 Glyceryl diacetate Substances 0.000 description 1
- 108010078321 Guanylate Cyclase Proteins 0.000 description 1
- 102000014469 Guanylate cyclase Human genes 0.000 description 1
- 229920000084 Gum arabic Polymers 0.000 description 1
- 229920000663 Hydroxyethyl cellulose Polymers 0.000 description 1
- 239000004354 Hydroxyethyl cellulose Substances 0.000 description 1
- HEFNNWSXXWATRW-UHFFFAOYSA-N Ibuprofen Chemical compound CC(C)CC1=CC=C(C(C)C(O)=O)C=C1 HEFNNWSXXWATRW-UHFFFAOYSA-N 0.000 description 1
- 206010061218 Inflammation Diseases 0.000 description 1
- 229920001202 Inulin Polymers 0.000 description 1
- AHLPHDHHMVZTML-BYPYZUCNSA-N L-Ornithine Chemical compound NCCC[C@H](N)C(O)=O AHLPHDHHMVZTML-BYPYZUCNSA-N 0.000 description 1
- ODKSFYDXXFIFQN-BYPYZUCNSA-N L-arginine Chemical compound OC(=O)[C@@H](N)CCCN=C(N)N ODKSFYDXXFIFQN-BYPYZUCNSA-N 0.000 description 1
- 229930064664 L-arginine Natural products 0.000 description 1
- 235000014852 L-arginine Nutrition 0.000 description 1
- RHGKLRLOHDJJDR-BYPYZUCNSA-N L-citrulline Chemical compound NC(=O)NCCC[C@H]([NH3+])C([O-])=O RHGKLRLOHDJJDR-BYPYZUCNSA-N 0.000 description 1
- DWPCPZJAHOETAG-IMJSIDKUSA-N L-lanthionine Chemical compound OC(=O)[C@@H](N)CSC[C@H](N)C(O)=O DWPCPZJAHOETAG-IMJSIDKUSA-N 0.000 description 1
- 229930182821 L-proline Natural products 0.000 description 1
- 240000007472 Leucaena leucocephala Species 0.000 description 1
- 235000010643 Leucaena leucocephala Nutrition 0.000 description 1
- 206010058467 Lung neoplasm malignant Diseases 0.000 description 1
- 229920002774 Maltodextrin Polymers 0.000 description 1
- 239000005913 Maltodextrin Substances 0.000 description 1
- GUBGYTABKSRVRQ-PICCSMPSSA-N Maltose Natural products O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@@H]1O[C@@H]1[C@@H](CO)OC(O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-PICCSMPSSA-N 0.000 description 1
- 240000003183 Manihot esculenta Species 0.000 description 1
- 235000016735 Manihot esculenta subsp esculenta Nutrition 0.000 description 1
- ZRVUJXDFFKFLMG-UHFFFAOYSA-N Meloxicam Chemical compound OC=1C2=CC=CC=C2S(=O)(=O)N(C)C=1C(=O)NC1=NC=C(C)S1 ZRVUJXDFFKFLMG-UHFFFAOYSA-N 0.000 description 1
- 206010027476 Metastases Diseases 0.000 description 1
- 239000004368 Modified starch Substances 0.000 description 1
- 241000699670 Mus sp. Species 0.000 description 1
- FSVCELGFZIQNCK-UHFFFAOYSA-N N,N-bis(2-hydroxyethyl)glycine Chemical compound OCCN(CCO)CC(O)=O FSVCELGFZIQNCK-UHFFFAOYSA-N 0.000 description 1
- CMWTZPSULFXXJA-UHFFFAOYSA-N Naproxen Natural products C1=C(C(C)C(O)=O)C=CC2=CC(OC)=CC=C21 CMWTZPSULFXXJA-UHFFFAOYSA-N 0.000 description 1
- RHGKLRLOHDJJDR-UHFFFAOYSA-N Ndelta-carbamoyl-DL-ornithine Natural products OC(=O)C(N)CCCNC(N)=O RHGKLRLOHDJJDR-UHFFFAOYSA-N 0.000 description 1
- AHLPHDHHMVZTML-UHFFFAOYSA-N Orn-delta-NH2 Natural products NCCCC(N)C(O)=O AHLPHDHHMVZTML-UHFFFAOYSA-N 0.000 description 1
- UTJLXEIPEHZYQJ-UHFFFAOYSA-N Ornithine Natural products OC(=O)C(C)CCCN UTJLXEIPEHZYQJ-UHFFFAOYSA-N 0.000 description 1
- 208000018737 Parkinson disease Diseases 0.000 description 1
- 235000019483 Peanut oil Nutrition 0.000 description 1
- 241000286209 Phasianidae Species 0.000 description 1
- 229920002732 Polyanhydride Polymers 0.000 description 1
- 229920001710 Polyorthoester Polymers 0.000 description 1
- 239000004743 Polypropylene Substances 0.000 description 1
- 208000037062 Polyps Diseases 0.000 description 1
- 102100032709 Potassium-transporting ATPase alpha chain 2 Human genes 0.000 description 1
- HCBIBCJNVBAKAB-UHFFFAOYSA-N Procaine hydrochloride Chemical compound Cl.CCN(CC)CCOC(=O)C1=CC=C(N)C=C1 HCBIBCJNVBAKAB-UHFFFAOYSA-N 0.000 description 1
- 206010036774 Proctitis Diseases 0.000 description 1
- 102100038280 Prostaglandin G/H synthase 2 Human genes 0.000 description 1
- 108050003267 Prostaglandin G/H synthase 2 Proteins 0.000 description 1
- 108010083204 Proton Pumps Proteins 0.000 description 1
- 239000004373 Pullulan Substances 0.000 description 1
- 229920001218 Pullulan Polymers 0.000 description 1
- 241000700159 Rattus Species 0.000 description 1
- 208000015815 Rectal disease Diseases 0.000 description 1
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 1
- 239000004288 Sodium dehydroacetate Substances 0.000 description 1
- ABBQHOQBGMUPJH-UHFFFAOYSA-M Sodium salicylate Chemical compound [Na+].OC1=CC=CC=C1C([O-])=O ABBQHOQBGMUPJH-UHFFFAOYSA-M 0.000 description 1
- 244000061456 Solanum tuberosum Species 0.000 description 1
- 235000002595 Solanum tuberosum Nutrition 0.000 description 1
- 239000004376 Sucralose Substances 0.000 description 1
- 241000282887 Suidae Species 0.000 description 1
- 235000019486 Sunflower oil Nutrition 0.000 description 1
- 241000282898 Sus scrofa Species 0.000 description 1
- 206010042674 Swelling Diseases 0.000 description 1
- 239000005844 Thymol Substances 0.000 description 1
- ATJFFYVFTNAWJD-UHFFFAOYSA-N Tin Chemical compound [Sn] ATJFFYVFTNAWJD-UHFFFAOYSA-N 0.000 description 1
- 229920001615 Tragacanth Polymers 0.000 description 1
- 239000007983 Tris buffer Substances 0.000 description 1
- TVXBFESIOXBWNM-UHFFFAOYSA-N Xylitol Natural products OCCC(O)C(O)C(O)CCO TVXBFESIOXBWNM-UHFFFAOYSA-N 0.000 description 1
- 235000010489 acacia gum Nutrition 0.000 description 1
- 239000000205 acacia gum Substances 0.000 description 1
- CMWRPTYOFZMJLC-UHFFFAOYSA-N acetic acid;n,n'-bis(3-aminopropyl)butane-1,4-diamine Chemical compound CC(O)=O.NCCCNCCCCNCCCN CMWRPTYOFZMJLC-UHFFFAOYSA-N 0.000 description 1
- 230000000397 acetylating effect Effects 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 239000008186 active pharmaceutical agent Substances 0.000 description 1
- 230000002411 adverse Effects 0.000 description 1
- 239000000443 aerosol Substances 0.000 description 1
- 239000000556 agonist Substances 0.000 description 1
- WQZGKKKJIJFFOK-PHYPRBDBSA-N alpha-D-galactose Chemical compound OC[C@H]1O[C@H](O)[C@H](O)[C@@H](O)[C@H]1O WQZGKKKJIJFFOK-PHYPRBDBSA-N 0.000 description 1
- DKNWSYNQZKUICI-UHFFFAOYSA-N amantadine Chemical compound C1C(C2)CC3CC2CC1(N)C3 DKNWSYNQZKUICI-UHFFFAOYSA-N 0.000 description 1
- 229960003805 amantadine Drugs 0.000 description 1
- 150000001412 amines Chemical class 0.000 description 1
- 239000000908 ammonium hydroxide Substances 0.000 description 1
- 238000010171 animal model Methods 0.000 description 1
- 239000002518 antifoaming agent Substances 0.000 description 1
- 230000003078 antioxidant effect Effects 0.000 description 1
- 230000001580 bacterial effect Effects 0.000 description 1
- 229960000686 benzalkonium chloride Drugs 0.000 description 1
- UREZNYTWGJKWBI-UHFFFAOYSA-M benzethonium chloride Chemical compound [Cl-].C1=CC(C(C)(C)CC(C)(C)C)=CC=C1OCCOCC[N+](C)(C)CC1=CC=CC=C1 UREZNYTWGJKWBI-UHFFFAOYSA-M 0.000 description 1
- 229960001950 benzethonium chloride Drugs 0.000 description 1
- 235000010233 benzoic acid Nutrition 0.000 description 1
- 229960004365 benzoic acid Drugs 0.000 description 1
- CADWTSSKOVRVJC-UHFFFAOYSA-N benzyl(dimethyl)azanium;chloride Chemical compound [Cl-].C[NH+](C)CC1=CC=CC=C1 CADWTSSKOVRVJC-UHFFFAOYSA-N 0.000 description 1
- GUBGYTABKSRVRQ-QUYVBRFLSA-N beta-maltose Chemical compound OC[C@H]1O[C@H](O[C@H]2[C@H](O)[C@@H](O)[C@H](O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@@H]1O GUBGYTABKSRVRQ-QUYVBRFLSA-N 0.000 description 1
- 230000004071 biological effect Effects 0.000 description 1
- 229920001222 biopolymer Polymers 0.000 description 1
- 230000000903 blocking effect Effects 0.000 description 1
- 235000021152 breakfast Nutrition 0.000 description 1
- 201000008275 breast carcinoma Diseases 0.000 description 1
- 230000005587 bubbling Effects 0.000 description 1
- 230000003139 buffering effect Effects 0.000 description 1
- 229940067596 butylparaben Drugs 0.000 description 1
- LLSDKQJKOVVTOJ-UHFFFAOYSA-L calcium chloride dihydrate Chemical compound O.O.[Cl-].[Cl-].[Ca+2] LLSDKQJKOVVTOJ-UHFFFAOYSA-L 0.000 description 1
- 229940052299 calcium chloride dihydrate Drugs 0.000 description 1
- AXCZMVOFGPJBDE-UHFFFAOYSA-L calcium dihydroxide Chemical compound [OH-].[OH-].[Ca+2] AXCZMVOFGPJBDE-UHFFFAOYSA-L 0.000 description 1
- 239000000920 calcium hydroxide Substances 0.000 description 1
- 229910001861 calcium hydroxide Inorganic materials 0.000 description 1
- 229940095643 calcium hydroxide Drugs 0.000 description 1
- OLOZVPHKXALCRI-UHFFFAOYSA-L calcium malate Chemical compound [Ca+2].[O-]C(=O)C(O)CC([O-])=O OLOZVPHKXALCRI-UHFFFAOYSA-L 0.000 description 1
- 239000001506 calcium phosphate Substances 0.000 description 1
- 239000000378 calcium silicate Substances 0.000 description 1
- 229910052918 calcium silicate Inorganic materials 0.000 description 1
- 235000012241 calcium silicate Nutrition 0.000 description 1
- CJZGTCYPCWQAJB-UHFFFAOYSA-L calcium stearate Chemical compound [Ca+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O CJZGTCYPCWQAJB-UHFFFAOYSA-L 0.000 description 1
- 235000013539 calcium stearate Nutrition 0.000 description 1
- 239000008116 calcium stearate Substances 0.000 description 1
- OYACROKNLOSFPA-UHFFFAOYSA-N calcium;dioxido(oxo)silane Chemical compound [Ca+2].[O-][Si]([O-])=O OYACROKNLOSFPA-UHFFFAOYSA-N 0.000 description 1
- 238000005251 capillar electrophoresis Methods 0.000 description 1
- 235000010948 carboxy methyl cellulose Nutrition 0.000 description 1
- 239000008112 carboxymethyl-cellulose Substances 0.000 description 1
- 229960004203 carnitine Drugs 0.000 description 1
- 235000010418 carrageenan Nutrition 0.000 description 1
- 229920001525 carrageenan Polymers 0.000 description 1
- 239000000679 carrageenan Substances 0.000 description 1
- 229940113118 carrageenan Drugs 0.000 description 1
- 239000000969 carrier Substances 0.000 description 1
- 239000005018 casein Substances 0.000 description 1
- BECPQYXYKAMYBN-UHFFFAOYSA-N casein, tech. Chemical compound NCCCCC(C(O)=O)N=C(O)C(CC(O)=O)N=C(O)C(CCC(O)=N)N=C(O)C(CC(C)C)N=C(O)C(CCC(O)=O)N=C(O)C(CC(O)=O)N=C(O)C(CCC(O)=O)N=C(O)C(C(C)O)N=C(O)C(CCC(O)=N)N=C(O)C(CCC(O)=N)N=C(O)C(CCC(O)=N)N=C(O)C(CCC(O)=O)N=C(O)C(CCC(O)=O)N=C(O)C(COP(O)(O)=O)N=C(O)C(CCC(O)=N)N=C(O)C(N)CC1=CC=CC=C1 BECPQYXYKAMYBN-UHFFFAOYSA-N 0.000 description 1
- 235000021240 caseins Nutrition 0.000 description 1
- 239000004359 castor oil Substances 0.000 description 1
- 235000019438 castor oil Nutrition 0.000 description 1
- 230000003197 catalytic effect Effects 0.000 description 1
- 210000004534 cecum Anatomy 0.000 description 1
- 229940047495 celebrex Drugs 0.000 description 1
- 229960000590 celecoxib Drugs 0.000 description 1
- 229920001727 cellulose butyrate Polymers 0.000 description 1
- YMKDRGPMQRFJGP-UHFFFAOYSA-M cetylpyridinium chloride Chemical compound [Cl-].CCCCCCCCCCCCCCCC[N+]1=CC=CC=C1 YMKDRGPMQRFJGP-UHFFFAOYSA-M 0.000 description 1
- 229960001927 cetylpyridinium chloride Drugs 0.000 description 1
- 239000007910 chewable tablet Substances 0.000 description 1
- 229940068682 chewable tablet Drugs 0.000 description 1
- 235000013330 chicken meat Nutrition 0.000 description 1
- 229960004926 chlorobutanol Drugs 0.000 description 1
- 229940059329 chondroitin sulfate Drugs 0.000 description 1
- 229960002173 citrulline Drugs 0.000 description 1
- 235000013477 citrulline Nutrition 0.000 description 1
- 229940075614 colloidal silicon dioxide Drugs 0.000 description 1
- 238000002648 combination therapy Methods 0.000 description 1
- 238000012777 commercial manufacturing Methods 0.000 description 1
- 230000008602 contraction Effects 0.000 description 1
- 235000005687 corn oil Nutrition 0.000 description 1
- 239000002285 corn oil Substances 0.000 description 1
- 235000012343 cottonseed oil Nutrition 0.000 description 1
- 239000002385 cottonseed oil Substances 0.000 description 1
- 229930003836 cresol Natural products 0.000 description 1
- 229940013361 cresol Drugs 0.000 description 1
- PAFZNILMFXTMIY-UHFFFAOYSA-N cyclohexylamine Chemical compound NC1CCCCC1 PAFZNILMFXTMIY-UHFFFAOYSA-N 0.000 description 1
- 230000007547 defect Effects 0.000 description 1
- 230000000593 degrading effect Effects 0.000 description 1
- 235000019258 dehydroacetic acid Nutrition 0.000 description 1
- 229940061632 dehydroacetic acid Drugs 0.000 description 1
- JEQRBTDTEKWZBW-UHFFFAOYSA-N dehydroacetic acid Chemical compound CC(=O)C1=C(O)OC(C)=CC1=O JEQRBTDTEKWZBW-UHFFFAOYSA-N 0.000 description 1
- PGRHXDWITVMQBC-UHFFFAOYSA-N dehydroacetic acid Natural products CC(=O)C1C(=O)OC(C)=CC1=O PGRHXDWITVMQBC-UHFFFAOYSA-N 0.000 description 1
- 239000008367 deionised water Substances 0.000 description 1
- 229910021641 deionized water Inorganic materials 0.000 description 1
- 238000001514 detection method Methods 0.000 description 1
- 229940096516 dextrates Drugs 0.000 description 1
- 229940099371 diacetylated monoglycerides Drugs 0.000 description 1
- NEFBYIFKOOEVPA-UHFFFAOYSA-K dicalcium phosphate Chemical compound [Ca+2].[Ca+2].[O-]P([O-])([O-])=O NEFBYIFKOOEVPA-UHFFFAOYSA-K 0.000 description 1
- 229940038472 dicalcium phosphate Drugs 0.000 description 1
- 229910000390 dicalcium phosphate Inorganic materials 0.000 description 1
- 229960001259 diclofenac Drugs 0.000 description 1
- DCOPUUMXTXDBNB-UHFFFAOYSA-N diclofenac Chemical compound OC(=O)CC1=CC=CC=C1NC1=C(Cl)C=CC=C1Cl DCOPUUMXTXDBNB-UHFFFAOYSA-N 0.000 description 1
- 229960000616 diflunisal Drugs 0.000 description 1
- HUPFGZXOMWLGNK-UHFFFAOYSA-N diflunisal Chemical compound C1=C(O)C(C(=O)O)=CC(C=2C(=CC(F)=CC=2)F)=C1 HUPFGZXOMWLGNK-UHFFFAOYSA-N 0.000 description 1
- 230000001079 digestive effect Effects 0.000 description 1
- FBSAITBEAPNWJG-UHFFFAOYSA-N dimethyl phthalate Natural products CC(=O)OC1=CC=CC=C1OC(C)=O FBSAITBEAPNWJG-UHFFFAOYSA-N 0.000 description 1
- 229960001826 dimethylphthalate Drugs 0.000 description 1
- ZPWVASYFFYYZEW-UHFFFAOYSA-L dipotassium hydrogen phosphate Chemical compound [K+].[K+].OP([O-])([O-])=O ZPWVASYFFYYZEW-UHFFFAOYSA-L 0.000 description 1
- 235000019797 dipotassium phosphate Nutrition 0.000 description 1
- 229910000396 dipotassium phosphate Inorganic materials 0.000 description 1
- 208000037765 diseases and disorders Diseases 0.000 description 1
- 238000007922 dissolution test Methods 0.000 description 1
- 210000004921 distal colon Anatomy 0.000 description 1
- 125000002228 disulfide group Chemical group 0.000 description 1
- 206010060865 duodenogastric reflux Diseases 0.000 description 1
- 238000010828 elution Methods 0.000 description 1
- 239000003995 emulsifying agent Substances 0.000 description 1
- 238000005538 encapsulation Methods 0.000 description 1
- 239000007920 enema Substances 0.000 description 1
- 229940079360 enema for constipation Drugs 0.000 description 1
- 239000000147 enterotoxin Substances 0.000 description 1
- 231100000655 enterotoxin Toxicity 0.000 description 1
- 230000007613 environmental effect Effects 0.000 description 1
- 210000000981 epithelium Anatomy 0.000 description 1
- 238000011067 equilibration Methods 0.000 description 1
- 229940031098 ethanolamine Drugs 0.000 description 1
- BEFDCLMNVWHSGT-UHFFFAOYSA-N ethenylcyclopentane Chemical compound C=CC1CCCC1 BEFDCLMNVWHSGT-UHFFFAOYSA-N 0.000 description 1
- 229960001617 ethyl hydroxybenzoate Drugs 0.000 description 1
- LVGKNOAMLMIIKO-QXMHVHEDSA-N ethyl oleate Chemical compound CCCCCCCC\C=C/CCCCCCCC(=O)OCC LVGKNOAMLMIIKO-QXMHVHEDSA-N 0.000 description 1
- 229940093471 ethyl oleate Drugs 0.000 description 1
- 239000004403 ethyl p-hydroxybenzoate Substances 0.000 description 1
- 235000010228 ethyl p-hydroxybenzoate Nutrition 0.000 description 1
- 229940012017 ethylenediamine Drugs 0.000 description 1
- NUVBSKCKDOMJSU-UHFFFAOYSA-N ethylparaben Chemical compound CCOC(=O)C1=CC=C(O)C=C1 NUVBSKCKDOMJSU-UHFFFAOYSA-N 0.000 description 1
- 229960005293 etodolac Drugs 0.000 description 1
- XFBVBWWRPKNWHW-UHFFFAOYSA-N etodolac Chemical compound C1COC(CC)(CC(O)=O)C2=N[C]3C(CC)=CC=CC3=C21 XFBVBWWRPKNWHW-UHFFFAOYSA-N 0.000 description 1
- 229960004945 etoricoxib Drugs 0.000 description 1
- MNJVRJDLRVPLFE-UHFFFAOYSA-N etoricoxib Chemical compound C1=NC(C)=CC=C1C1=NC=C(Cl)C=C1C1=CC=C(S(C)(=O)=O)C=C1 MNJVRJDLRVPLFE-UHFFFAOYSA-N 0.000 description 1
- 238000001704 evaporation Methods 0.000 description 1
- 230000008020 evaporation Effects 0.000 description 1
- 210000003608 fece Anatomy 0.000 description 1
- 239000012467 final product Substances 0.000 description 1
- 235000013355 food flavoring agent Nutrition 0.000 description 1
- 235000003599 food sweetener Nutrition 0.000 description 1
- 229920001002 functional polymer Polymers 0.000 description 1
- 229930182830 galactose Natural products 0.000 description 1
- 239000000499 gel Substances 0.000 description 1
- 238000001502 gel electrophoresis Methods 0.000 description 1
- 150000004676 glycans Chemical class 0.000 description 1
- ZEMPKEQAKRGZGQ-XOQCFJPHSA-N glycerol triricinoleate Natural products CCCCCC[C@@H](O)CC=CCCCCCCCC(=O)OC[C@@H](COC(=O)CCCCCCCC=CC[C@@H](O)CCCCCC)OC(=O)CCCCCCCC=CC[C@H](O)CCCCCC ZEMPKEQAKRGZGQ-XOQCFJPHSA-N 0.000 description 1
- 235000019443 glyceryl diacetate Nutrition 0.000 description 1
- 125000003976 glyceryl group Chemical group [H]C([*])([H])C(O[H])([H])C(O[H])([H])[H] 0.000 description 1
- 239000001087 glyceryl triacetate Substances 0.000 description 1
- 235000013773 glyceryl triacetate Nutrition 0.000 description 1
- 150000002334 glycols Chemical class 0.000 description 1
- 150000002337 glycosamines Chemical class 0.000 description 1
- 239000003979 granulating agent Substances 0.000 description 1
- 230000012010 growth Effects 0.000 description 1
- 239000001307 helium Substances 0.000 description 1
- 229910052734 helium Inorganic materials 0.000 description 1
- SWQJXJOGLNCZEY-UHFFFAOYSA-N helium atom Chemical compound [He] SWQJXJOGLNCZEY-UHFFFAOYSA-N 0.000 description 1
- 229940088597 hormone Drugs 0.000 description 1
- 239000005556 hormone Substances 0.000 description 1
- 239000001257 hydrogen Substances 0.000 description 1
- 229910052739 hydrogen Inorganic materials 0.000 description 1
- 239000008172 hydrogenated vegetable oil Substances 0.000 description 1
- 229920001477 hydrophilic polymer Polymers 0.000 description 1
- 230000002209 hydrophobic effect Effects 0.000 description 1
- 235000019447 hydroxyethyl cellulose Nutrition 0.000 description 1
- 229960003943 hypromellose Drugs 0.000 description 1
- 229960001680 ibuprofen Drugs 0.000 description 1
- 150000002466 imines Chemical class 0.000 description 1
- 238000003018 immunoassay Methods 0.000 description 1
- 238000000338 in vitro Methods 0.000 description 1
- 230000004054 inflammatory process Effects 0.000 description 1
- 239000003112 inhibitor Substances 0.000 description 1
- 230000002401 inhibitory effect Effects 0.000 description 1
- 230000005764 inhibitory process Effects 0.000 description 1
- 230000003871 intestinal function Effects 0.000 description 1
- 229940029339 inulin Drugs 0.000 description 1
- JYJIGFIDKWBXDU-MNNPPOADSA-N inulin Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)OC[C@]1(OC[C@]2(OC[C@]3(OC[C@]4(OC[C@]5(OC[C@]6(OC[C@]7(OC[C@]8(OC[C@]9(OC[C@]%10(OC[C@]%11(OC[C@]%12(OC[C@]%13(OC[C@]%14(OC[C@]%15(OC[C@]%16(OC[C@]%17(OC[C@]%18(OC[C@]%19(OC[C@]%20(OC[C@]%21(OC[C@]%22(OC[C@]%23(OC[C@]%24(OC[C@]%25(OC[C@]%26(OC[C@]%27(OC[C@]%28(OC[C@]%29(OC[C@]%30(OC[C@]%31(OC[C@]%32(OC[C@]%33(OC[C@]%34(OC[C@]%35(OC[C@]%36(O[C@@H]%37[C@@H]([C@@H](O)[C@H](O)[C@@H](CO)O%37)O)[C@H]([C@H](O)[C@@H](CO)O%36)O)[C@H]([C@H](O)[C@@H](CO)O%35)O)[C@H]([C@H](O)[C@@H](CO)O%34)O)[C@H]([C@H](O)[C@@H](CO)O%33)O)[C@H]([C@H](O)[C@@H](CO)O%32)O)[C@H]([C@H](O)[C@@H](CO)O%31)O)[C@H]([C@H](O)[C@@H](CO)O%30)O)[C@H]([C@H](O)[C@@H](CO)O%29)O)[C@H]([C@H](O)[C@@H](CO)O%28)O)[C@H]([C@H](O)[C@@H](CO)O%27)O)[C@H]([C@H](O)[C@@H](CO)O%26)O)[C@H]([C@H](O)[C@@H](CO)O%25)O)[C@H]([C@H](O)[C@@H](CO)O%24)O)[C@H]([C@H](O)[C@@H](CO)O%23)O)[C@H]([C@H](O)[C@@H](CO)O%22)O)[C@H]([C@H](O)[C@@H](CO)O%21)O)[C@H]([C@H](O)[C@@H](CO)O%20)O)[C@H]([C@H](O)[C@@H](CO)O%19)O)[C@H]([C@H](O)[C@@H](CO)O%18)O)[C@H]([C@H](O)[C@@H](CO)O%17)O)[C@H]([C@H](O)[C@@H](CO)O%16)O)[C@H]([C@H](O)[C@@H](CO)O%15)O)[C@H]([C@H](O)[C@@H](CO)O%14)O)[C@H]([C@H](O)[C@@H](CO)O%13)O)[C@H]([C@H](O)[C@@H](CO)O%12)O)[C@H]([C@H](O)[C@@H](CO)O%11)O)[C@H]([C@H](O)[C@@H](CO)O%10)O)[C@H]([C@H](O)[C@@H](CO)O9)O)[C@H]([C@H](O)[C@@H](CO)O8)O)[C@H]([C@H](O)[C@@H](CO)O7)O)[C@H]([C@H](O)[C@@H](CO)O6)O)[C@H]([C@H](O)[C@@H](CO)O5)O)[C@H]([C@H](O)[C@@H](CO)O4)O)[C@H]([C@H](O)[C@@H](CO)O3)O)[C@H]([C@H](O)[C@@H](CO)O2)O)[C@@H](O)[C@H](O)[C@@H](CO)O1 JYJIGFIDKWBXDU-MNNPPOADSA-N 0.000 description 1
- 150000002500 ions Chemical class 0.000 description 1
- 229910052742 iron Inorganic materials 0.000 description 1
- 239000012948 isocyanate Substances 0.000 description 1
- FZWBNHMXJMCXLU-BLAUPYHCSA-N isomaltotriose Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@@H]1OC[C@@H]1[C@@H](O)[C@H](O)[C@@H](O)[C@@H](OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C=O)O1 FZWBNHMXJMCXLU-BLAUPYHCSA-N 0.000 description 1
- 238000006317 isomerization reaction Methods 0.000 description 1
- 150000002576 ketones Chemical group 0.000 description 1
- 210000003734 kidney Anatomy 0.000 description 1
- 239000000832 lactitol Substances 0.000 description 1
- 235000010448 lactitol Nutrition 0.000 description 1
- VQHSOMBJVWLPSR-JVCRWLNRSA-N lactitol Chemical compound OC[C@H](O)[C@@H](O)[C@@H]([C@H](O)CO)O[C@@H]1O[C@H](CO)[C@H](O)[C@H](O)[C@H]1O VQHSOMBJVWLPSR-JVCRWLNRSA-N 0.000 description 1
- 229960003451 lactitol Drugs 0.000 description 1
- 229960001375 lactose Drugs 0.000 description 1
- 238000003475 lamination Methods 0.000 description 1
- 239000003446 ligand Substances 0.000 description 1
- 108020001756 ligand binding domains Proteins 0.000 description 1
- 229940059904 light mineral oil Drugs 0.000 description 1
- 230000000670 limiting effect Effects 0.000 description 1
- 210000004185 liver Anatomy 0.000 description 1
- 229960000994 lumiracoxib Drugs 0.000 description 1
- KHPKQFYUPIUARC-UHFFFAOYSA-N lumiracoxib Chemical compound OC(=O)CC1=CC(C)=CC=C1NC1=C(F)C=CC=C1Cl KHPKQFYUPIUARC-UHFFFAOYSA-N 0.000 description 1
- 210000004072 lung Anatomy 0.000 description 1
- 201000005202 lung cancer Diseases 0.000 description 1
- 208000020816 lung neoplasm Diseases 0.000 description 1
- 239000011654 magnesium acetate Substances 0.000 description 1
- UEGPKNKPLBYCNK-UHFFFAOYSA-L magnesium acetate Chemical compound [Mg+2].CC([O-])=O.CC([O-])=O UEGPKNKPLBYCNK-UHFFFAOYSA-L 0.000 description 1
- 235000011285 magnesium acetate Nutrition 0.000 description 1
- 229940069446 magnesium acetate Drugs 0.000 description 1
- 229960002337 magnesium chloride Drugs 0.000 description 1
- 235000011147 magnesium chloride Nutrition 0.000 description 1
- VTHJTEIRLNZDEV-UHFFFAOYSA-L magnesium dihydroxide Chemical compound [OH-].[OH-].[Mg+2] VTHJTEIRLNZDEV-UHFFFAOYSA-L 0.000 description 1
- 239000000347 magnesium hydroxide Substances 0.000 description 1
- 229910001862 magnesium hydroxide Inorganic materials 0.000 description 1
- 229960000816 magnesium hydroxide Drugs 0.000 description 1
- HCWCAKKEBCNQJP-UHFFFAOYSA-N magnesium orthosilicate Chemical compound [Mg+2].[Mg+2].[O-][Si]([O-])([O-])[O-] HCWCAKKEBCNQJP-UHFFFAOYSA-N 0.000 description 1
- 239000000391 magnesium silicate Substances 0.000 description 1
- 229910052919 magnesium silicate Inorganic materials 0.000 description 1
- 235000019792 magnesium silicate Nutrition 0.000 description 1
- 229940049920 malate Drugs 0.000 description 1
- BJEPYKJPYRNKOW-UHFFFAOYSA-N malic acid Chemical compound OC(=O)C(O)CC(O)=O BJEPYKJPYRNKOW-UHFFFAOYSA-N 0.000 description 1
- 229940035034 maltodextrin Drugs 0.000 description 1
- 229960001855 mannitol Drugs 0.000 description 1
- 230000001404 mediated effect Effects 0.000 description 1
- 239000002609 medium Substances 0.000 description 1
- 229960003194 meglumine Drugs 0.000 description 1
- 229960001929 meloxicam Drugs 0.000 description 1
- HEBKCHPVOIAQTA-UHFFFAOYSA-N meso ribitol Natural products OCC(O)C(O)C(O)CO HEBKCHPVOIAQTA-UHFFFAOYSA-N 0.000 description 1
- DWPCPZJAHOETAG-UHFFFAOYSA-N meso-lanthionine Natural products OC(=O)C(N)CSCC(N)C(O)=O DWPCPZJAHOETAG-UHFFFAOYSA-N 0.000 description 1
- 239000004292 methyl p-hydroxybenzoate Substances 0.000 description 1
- 235000010270 methyl p-hydroxybenzoate Nutrition 0.000 description 1
- 229960002216 methylparaben Drugs 0.000 description 1
- 230000000813 microbial effect Effects 0.000 description 1
- 239000003094 microcapsule Substances 0.000 description 1
- 239000004005 microsphere Substances 0.000 description 1
- 239000002480 mineral oil Substances 0.000 description 1
- 235000010446 mineral oil Nutrition 0.000 description 1
- 230000000116 mitigating effect Effects 0.000 description 1
- 235000019426 modified starch Nutrition 0.000 description 1
- 238000000465 moulding Methods 0.000 description 1
- 210000004877 mucosa Anatomy 0.000 description 1
- 229960002009 naproxen Drugs 0.000 description 1
- CMWTZPSULFXXJA-VIFPVBQESA-N naproxen Chemical compound C1=C([C@H](C)C(O)=O)C=CC2=CC(OC)=CC=C21 CMWTZPSULFXXJA-VIFPVBQESA-N 0.000 description 1
- 229920001206 natural gum Polymers 0.000 description 1
- 239000000041 non-steroidal anti-inflammatory agent Substances 0.000 description 1
- 229940021182 non-steroidal anti-inflammatory drug Drugs 0.000 description 1
- 238000005935 nucleophilic addition reaction Methods 0.000 description 1
- 239000002674 ointment Substances 0.000 description 1
- 239000004006 olive oil Substances 0.000 description 1
- 235000008390 olive oil Nutrition 0.000 description 1
- 239000000014 opioid analgesic Substances 0.000 description 1
- 229940100691 oral capsule Drugs 0.000 description 1
- 210000000056 organ Anatomy 0.000 description 1
- 229960003104 ornithine Drugs 0.000 description 1
- 229940075858 ornithine alpha-ketoglutarate Drugs 0.000 description 1
- 210000001672 ovary Anatomy 0.000 description 1
- 238000010525 oxidative degradation reaction Methods 0.000 description 1
- 238000012856 packing Methods 0.000 description 1
- 210000000496 pancreas Anatomy 0.000 description 1
- 239000002245 particle Substances 0.000 description 1
- 238000003921 particle size analysis Methods 0.000 description 1
- 239000000312 peanut oil Substances 0.000 description 1
- 230000002351 pectolytic effect Effects 0.000 description 1
- 230000002572 peristaltic effect Effects 0.000 description 1
- 150000002978 peroxides Chemical class 0.000 description 1
- WRLGYAWRGXKSKG-UHFFFAOYSA-M phenobarbital sodium Chemical compound [Na+].C=1C=CC=CC=1C1(CC)C(=O)NC([O-])=NC1=O WRLGYAWRGXKSKG-UHFFFAOYSA-M 0.000 description 1
- 229960003742 phenol Drugs 0.000 description 1
- 229960005323 phenoxyethanol Drugs 0.000 description 1
- WVDDGKGOMKODPV-ZQBYOMGUSA-N phenyl(114C)methanol Chemical compound O[14CH2]C1=CC=CC=C1 WVDDGKGOMKODPV-ZQBYOMGUSA-N 0.000 description 1
- 229940067107 phenylethyl alcohol Drugs 0.000 description 1
- 229940096826 phenylmercuric acetate Drugs 0.000 description 1
- PDTFCHSETJBPTR-UHFFFAOYSA-N phenylmercuric nitrate Chemical compound [O-][N+](=O)O[Hg]C1=CC=CC=C1 PDTFCHSETJBPTR-UHFFFAOYSA-N 0.000 description 1
- 239000008363 phosphate buffer Substances 0.000 description 1
- 239000008055 phosphate buffer solution Substances 0.000 description 1
- 230000035479 physiological effects, processes and functions Effects 0.000 description 1
- 229960000540 polacrilin potassium Drugs 0.000 description 1
- 229940071745 poly(sodium vinyl sulfonate) Drugs 0.000 description 1
- 229920002401 polyacrylamide Polymers 0.000 description 1
- 239000004626 polylactic acid Substances 0.000 description 1
- 229920005862 polyol Polymers 0.000 description 1
- 150000003077 polyols Chemical class 0.000 description 1
- 229920001155 polypropylene Polymers 0.000 description 1
- 229920001282 polysaccharide Polymers 0.000 description 1
- 239000005017 polysaccharide Substances 0.000 description 1
- 229950008882 polysorbate Drugs 0.000 description 1
- 229920000915 polyvinyl chloride Polymers 0.000 description 1
- 239000004800 polyvinyl chloride Substances 0.000 description 1
- 229940093932 potassium hydroxide Drugs 0.000 description 1
- 239000008057 potassium phosphate buffer Substances 0.000 description 1
- 239000004302 potassium sorbate Substances 0.000 description 1
- 235000010241 potassium sorbate Nutrition 0.000 description 1
- 229940069338 potassium sorbate Drugs 0.000 description 1
- WVWZXTJUCNEUAE-UHFFFAOYSA-M potassium;1,2-bis(ethenyl)benzene;2-methylprop-2-enoate Chemical compound [K+].CC(=C)C([O-])=O.C=CC1=CC=CC=C1C=C WVWZXTJUCNEUAE-UHFFFAOYSA-M 0.000 description 1
- ASHGTUMKRVIOLH-UHFFFAOYSA-L potassium;sodium;hydrogen phosphate Chemical compound [Na+].[K+].OP([O-])([O-])=O ASHGTUMKRVIOLH-UHFFFAOYSA-L 0.000 description 1
- 229940069328 povidone Drugs 0.000 description 1
- 230000002335 preservative effect Effects 0.000 description 1
- MFDFERRIHVXMIY-UHFFFAOYSA-N procaine Chemical compound CCN(CC)CCOC(=O)C1=CC=C(N)C=C1 MFDFERRIHVXMIY-UHFFFAOYSA-N 0.000 description 1
- 229960004919 procaine Drugs 0.000 description 1
- 229960001309 procaine hydrochloride Drugs 0.000 description 1
- 239000004405 propyl p-hydroxybenzoate Substances 0.000 description 1
- 235000010232 propyl p-hydroxybenzoate Nutrition 0.000 description 1
- 235000013772 propylene glycol Nutrition 0.000 description 1
- 229960003415 propylparaben Drugs 0.000 description 1
- 210000002307 prostate Anatomy 0.000 description 1
- 230000002797 proteolythic effect Effects 0.000 description 1
- 235000019423 pullulan Nutrition 0.000 description 1
- 230000000541 pulsatile effect Effects 0.000 description 1
- 238000000746 purification Methods 0.000 description 1
- 235000008160 pyridoxine Nutrition 0.000 description 1
- 239000011677 pyridoxine Substances 0.000 description 1
- 230000001698 pyrogenic effect Effects 0.000 description 1
- 238000003127 radioimmunoassay Methods 0.000 description 1
- 230000009257 reactivity Effects 0.000 description 1
- 238000003259 recombinant expression Methods 0.000 description 1
- 239000006215 rectal suppository Substances 0.000 description 1
- 210000000664 rectum Anatomy 0.000 description 1
- 230000001105 regulatory effect Effects 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 229960000888 rimantadine Drugs 0.000 description 1
- 229960000371 rofecoxib Drugs 0.000 description 1
- RZJQGNCSTQAWON-UHFFFAOYSA-N rofecoxib Chemical compound C1=CC(S(=O)(=O)C)=CC=C1C1=C(C=2C=CC=CC=2)C(=O)OC1 RZJQGNCSTQAWON-UHFFFAOYSA-N 0.000 description 1
- 238000012216 screening Methods 0.000 description 1
- 230000035945 sensitivity Effects 0.000 description 1
- 230000001953 sensory effect Effects 0.000 description 1
- 239000008159 sesame oil Substances 0.000 description 1
- 235000011803 sesame oil Nutrition 0.000 description 1
- 238000007873 sieving Methods 0.000 description 1
- 230000019491 signal transduction Effects 0.000 description 1
- 238000009097 single-agent therapy Methods 0.000 description 1
- 210000000813 small intestine Anatomy 0.000 description 1
- WXMKPNITSTVMEF-UHFFFAOYSA-M sodium benzoate Chemical compound [Na+].[O-]C(=O)C1=CC=CC=C1 WXMKPNITSTVMEF-UHFFFAOYSA-M 0.000 description 1
- 239000004299 sodium benzoate Substances 0.000 description 1
- 235000010234 sodium benzoate Nutrition 0.000 description 1
- 229960003885 sodium benzoate Drugs 0.000 description 1
- 235000019812 sodium carboxymethyl cellulose Nutrition 0.000 description 1
- 229920001027 sodium carboxymethylcellulose Polymers 0.000 description 1
- 239000011780 sodium chloride Substances 0.000 description 1
- 235000019259 sodium dehydroacetate Nutrition 0.000 description 1
- 229940079839 sodium dehydroacetate Drugs 0.000 description 1
- 238000002415 sodium dodecyl sulfate polyacrylamide gel electrophoresis Methods 0.000 description 1
- 229940083608 sodium hydroxide Drugs 0.000 description 1
- JXKPEJDQGNYQSM-UHFFFAOYSA-M sodium propionate Chemical compound [Na+].CCC([O-])=O JXKPEJDQGNYQSM-UHFFFAOYSA-M 0.000 description 1
- 239000004324 sodium propionate Substances 0.000 description 1
- 235000010334 sodium propionate Nutrition 0.000 description 1
- 229960003212 sodium propionate Drugs 0.000 description 1
- 229960004025 sodium salicylate Drugs 0.000 description 1
- 229920003109 sodium starch glycolate Polymers 0.000 description 1
- 239000008109 sodium starch glycolate Substances 0.000 description 1
- 229940079832 sodium starch glycolate Drugs 0.000 description 1
- DSOWAKKSGYUMTF-GZOLSCHFSA-M sodium;(1e)-1-(6-methyl-2,4-dioxopyran-3-ylidene)ethanolate Chemical compound [Na+].C\C([O-])=C1/C(=O)OC(C)=CC1=O DSOWAKKSGYUMTF-GZOLSCHFSA-M 0.000 description 1
- 235000010199 sorbic acid Nutrition 0.000 description 1
- 239000004334 sorbic acid Substances 0.000 description 1
- 229940075582 sorbic acid Drugs 0.000 description 1
- 239000003549 soybean oil Substances 0.000 description 1
- 235000012424 soybean oil Nutrition 0.000 description 1
- 229940063673 spermidine Drugs 0.000 description 1
- 229940063675 spermine Drugs 0.000 description 1
- 230000002269 spontaneous effect Effects 0.000 description 1
- 239000007858 starting material Substances 0.000 description 1
- WPLOVIFNBMNBPD-ATHMIXSHSA-N subtilin Chemical compound CC1SCC(NC2=O)C(=O)NC(CC(N)=O)C(=O)NC(C(=O)NC(CCCCN)C(=O)NC(C(C)CC)C(=O)NC(=C)C(=O)NC(CCCCN)C(O)=O)CSC(C)C2NC(=O)C(CC(C)C)NC(=O)C1NC(=O)C(CCC(N)=O)NC(=O)C(CC(C)C)NC(=O)C(NC(=O)C1NC(=O)C(=C/C)/NC(=O)C(CCC(N)=O)NC(=O)C(CC(C)C)NC(=O)C(C)NC(=O)CNC(=O)C(NC(=O)C(NC(=O)C2NC(=O)CNC(=O)C3CCCN3C(=O)C(NC(=O)C3NC(=O)C(CC(C)C)NC(=O)C(=C)NC(=O)C(CCC(O)=O)NC(=O)C(NC(=O)C(CCCCN)NC(=O)C(N)CC=4C5=CC=CC=C5NC=4)CSC3)C(C)SC2)C(C)C)C(C)SC1)CC1=CC=CC=C1 WPLOVIFNBMNBPD-ATHMIXSHSA-N 0.000 description 1
- 235000019408 sucralose Nutrition 0.000 description 1
- BAQAVOSOZGMPRM-QBMZZYIRSA-N sucralose Chemical compound O[C@@H]1[C@@H](O)[C@@H](Cl)[C@@H](CO)O[C@@H]1O[C@@]1(CCl)[C@@H](O)[C@H](O)[C@@H](CCl)O1 BAQAVOSOZGMPRM-QBMZZYIRSA-N 0.000 description 1
- 229960004793 sucrose Drugs 0.000 description 1
- 235000000346 sugar Nutrition 0.000 description 1
- 150000008163 sugars Chemical class 0.000 description 1
- 229910052717 sulfur Inorganic materials 0.000 description 1
- 125000004434 sulfur atom Chemical group 0.000 description 1
- 229960000894 sulindac Drugs 0.000 description 1
- MLKXDPUZXIRXEP-MFOYZWKCSA-N sulindac Chemical compound CC1=C(CC(O)=O)C2=CC(F)=CC=C2\C1=C/C1=CC=C(S(C)=O)C=C1 MLKXDPUZXIRXEP-MFOYZWKCSA-N 0.000 description 1
- 239000002600 sunflower oil Substances 0.000 description 1
- 239000006228 supernatant Substances 0.000 description 1
- 239000000375 suspending agent Substances 0.000 description 1
- 239000007939 sustained release tablet Substances 0.000 description 1
- 239000003765 sweetening agent Substances 0.000 description 1
- 230000008961 swelling Effects 0.000 description 1
- 230000002195 synergetic effect Effects 0.000 description 1
- 238000003786 synthesis reaction Methods 0.000 description 1
- 229920001059 synthetic polymer Polymers 0.000 description 1
- 230000008685 targeting Effects 0.000 description 1
- YBRBMKDOPFTVDT-UHFFFAOYSA-N tert-butylamine Chemical compound CC(C)(C)N YBRBMKDOPFTVDT-UHFFFAOYSA-N 0.000 description 1
- 230000002381 testicular Effects 0.000 description 1
- 229940026510 theanine Drugs 0.000 description 1
- 238000011285 therapeutic regimen Methods 0.000 description 1
- 229960000790 thymol Drugs 0.000 description 1
- 229910052718 tin Inorganic materials 0.000 description 1
- 239000011135 tin Substances 0.000 description 1
- 235000010487 tragacanth Nutrition 0.000 description 1
- 239000000196 tragacanth Substances 0.000 description 1
- 229940116362 tragacanth Drugs 0.000 description 1
- 102000027257 transmembrane receptors Human genes 0.000 description 1
- 108091008578 transmembrane receptors Proteins 0.000 description 1
- 229960002622 triacetin Drugs 0.000 description 1
- WEAPVABOECTMGR-UHFFFAOYSA-N triethyl 2-acetyloxypropane-1,2,3-tricarboxylate Chemical compound CCOC(=O)CC(C(=O)OCC)(OC(C)=O)CC(=O)OCC WEAPVABOECTMGR-UHFFFAOYSA-N 0.000 description 1
- 125000005591 trimellitate group Chemical group 0.000 description 1
- 210000003932 urinary bladder Anatomy 0.000 description 1
- 229960002004 valdecoxib Drugs 0.000 description 1
- LNPDTQAFDNKSHK-UHFFFAOYSA-N valdecoxib Chemical compound CC=1ON=C(C=2C=CC=CC=2)C=1C1=CC=C(S(N)(=O)=O)C=C1 LNPDTQAFDNKSHK-UHFFFAOYSA-N 0.000 description 1
- 229920002554 vinyl polymer Polymers 0.000 description 1
- 230000009278 visceral effect Effects 0.000 description 1
- 229940011671 vitamin b6 Drugs 0.000 description 1
- 238000004017 vitrification Methods 0.000 description 1
- 230000001755 vocal effect Effects 0.000 description 1
- 239000003643 water by type Substances 0.000 description 1
- 238000009736 wetting Methods 0.000 description 1
- 239000000080 wetting agent Substances 0.000 description 1
- 239000000811 xylitol Substances 0.000 description 1
- 235000010447 xylitol Nutrition 0.000 description 1
- HEBKCHPVOIAQTA-SCDXWVJYSA-N xylitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)CO HEBKCHPVOIAQTA-SCDXWVJYSA-N 0.000 description 1
- 229960002675 xylitol Drugs 0.000 description 1
- 229960000314 zinc acetate Drugs 0.000 description 1
- 239000011592 zinc chloride Substances 0.000 description 1
- 235000005074 zinc chloride Nutrition 0.000 description 1
- 229960001939 zinc chloride Drugs 0.000 description 1
- XOOUIPVCVHRTMJ-UHFFFAOYSA-L zinc stearate Chemical compound [Zn+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O XOOUIPVCVHRTMJ-UHFFFAOYSA-L 0.000 description 1
- UHVMMEOXYDMDKI-JKYCWFKZSA-L zinc;1-(5-cyanopyridin-2-yl)-3-[(1s,2s)-2-(6-fluoro-2-hydroxy-3-propanoylphenyl)cyclopropyl]urea;diacetate Chemical compound [Zn+2].CC([O-])=O.CC([O-])=O.CCC(=O)C1=CC=C(F)C([C@H]2[C@H](C2)NC(=O)NC=2N=CC(=CC=2)C#N)=C1O UHVMMEOXYDMDKI-JKYCWFKZSA-L 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/04—Peptides having up to 20 amino acids in a fully defined sequence; Derivatives thereof
- A61K38/10—Peptides having 12 to 20 amino acids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
- A61K9/16—Agglomerates; Granulates; Microbeadlets ; Microspheres; Pellets; Solid products obtained by spray drying, spray freeze drying, spray congealing,(multiple) emulsion solvent evaporation or extraction
- A61K9/1605—Excipients; Inactive ingredients
- A61K9/1611—Inorganic compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
- A61K9/16—Agglomerates; Granulates; Microbeadlets ; Microspheres; Pellets; Solid products obtained by spray drying, spray freeze drying, spray congealing,(multiple) emulsion solvent evaporation or extraction
- A61K9/1605—Excipients; Inactive ingredients
- A61K9/1617—Organic compounds, e.g. phospholipids, fats
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
- A61K9/16—Agglomerates; Granulates; Microbeadlets ; Microspheres; Pellets; Solid products obtained by spray drying, spray freeze drying, spray congealing,(multiple) emulsion solvent evaporation or extraction
- A61K9/1605—Excipients; Inactive ingredients
- A61K9/1629—Organic macromolecular compounds
- A61K9/1635—Organic macromolecular compounds obtained by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyvinyl pyrrolidone, poly(meth)acrylates
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
- A61K9/16—Agglomerates; Granulates; Microbeadlets ; Microspheres; Pellets; Solid products obtained by spray drying, spray freeze drying, spray congealing,(multiple) emulsion solvent evaporation or extraction
- A61K9/1605—Excipients; Inactive ingredients
- A61K9/1629—Organic macromolecular compounds
- A61K9/1652—Polysaccharides, e.g. alginate, cellulose derivatives; Cyclodextrin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
- A61K9/16—Agglomerates; Granulates; Microbeadlets ; Microspheres; Pellets; Solid products obtained by spray drying, spray freeze drying, spray congealing,(multiple) emulsion solvent evaporation or extraction
- A61K9/167—Agglomerates; Granulates; Microbeadlets ; Microspheres; Pellets; Solid products obtained by spray drying, spray freeze drying, spray congealing,(multiple) emulsion solvent evaporation or extraction with an outer layer or coating comprising drug; with chemically bound drugs or non-active substances on their surface
- A61K9/1676—Agglomerates; Granulates; Microbeadlets ; Microspheres; Pellets; Solid products obtained by spray drying, spray freeze drying, spray congealing,(multiple) emulsion solvent evaporation or extraction with an outer layer or coating comprising drug; with chemically bound drugs or non-active substances on their surface having a drug-free core with discrete complete coating layer containing drug
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/2004—Excipients; Inactive ingredients
- A61K9/2009—Inorganic compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/2004—Excipients; Inactive ingredients
- A61K9/2013—Organic compounds, e.g. phospholipids, fats
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/2004—Excipients; Inactive ingredients
- A61K9/2013—Organic compounds, e.g. phospholipids, fats
- A61K9/2018—Sugars, or sugar alcohols, e.g. lactose, mannitol; Derivatives thereof, e.g. polysorbates
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/2004—Excipients; Inactive ingredients
- A61K9/2022—Organic macromolecular compounds
- A61K9/2027—Organic macromolecular compounds obtained by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyvinyl pyrrolidone, poly(meth)acrylates
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/2004—Excipients; Inactive ingredients
- A61K9/2022—Organic macromolecular compounds
- A61K9/205—Polysaccharides, e.g. alginate, gums; Cyclodextrin
- A61K9/2054—Cellulose; Cellulose derivatives, e.g. hydroxypropyl methylcellulose
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/28—Dragees; Coated pills or tablets, e.g. with film or compression coating
- A61K9/2806—Coating materials
- A61K9/2833—Organic macromolecular compounds
- A61K9/284—Organic macromolecular compounds obtained by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyvinyl pyrrolidone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/28—Dragees; Coated pills or tablets, e.g. with film or compression coating
- A61K9/2806—Coating materials
- A61K9/2833—Organic macromolecular compounds
- A61K9/284—Organic macromolecular compounds obtained by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyvinyl pyrrolidone
- A61K9/2846—Poly(meth)acrylates
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/28—Dragees; Coated pills or tablets, e.g. with film or compression coating
- A61K9/2886—Dragees; Coated pills or tablets, e.g. with film or compression coating having two or more different drug-free coatings; Tablets of the type inert core-drug layer-inactive layer
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/48—Preparations in capsules, e.g. of gelatin, of chocolate
- A61K9/4808—Preparations in capsules, e.g. of gelatin, of chocolate characterised by the form of the capsule or the structure of the filling; Capsules containing small tablets; Capsules with outer layer for immediate drug release
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/48—Preparations in capsules, e.g. of gelatin, of chocolate
- A61K9/4816—Wall or shell material
- A61K9/4825—Proteins, e.g. gelatin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/48—Preparations in capsules, e.g. of gelatin, of chocolate
- A61K9/4841—Filling excipients; Inactive ingredients
- A61K9/485—Inorganic compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/48—Preparations in capsules, e.g. of gelatin, of chocolate
- A61K9/4841—Filling excipients; Inactive ingredients
- A61K9/4858—Organic compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/48—Preparations in capsules, e.g. of gelatin, of chocolate
- A61K9/4841—Filling excipients; Inactive ingredients
- A61K9/4866—Organic macromolecular compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/48—Preparations in capsules, e.g. of gelatin, of chocolate
- A61K9/4891—Coated capsules; Multilayered drug free capsule shells
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/48—Preparations in capsules, e.g. of gelatin, of chocolate
- A61K9/50—Microcapsules having a gas, liquid or semi-solid filling; Solid microparticles or pellets surrounded by a distinct coating layer, e.g. coated microspheres, coated drug crystals
- A61K9/5005—Wall or coating material
- A61K9/5021—Organic macromolecular compounds
- A61K9/5026—Organic macromolecular compounds obtained by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyvinyl pyrrolidone, poly(meth)acrylates
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/48—Preparations in capsules, e.g. of gelatin, of chocolate
- A61K9/50—Microcapsules having a gas, liquid or semi-solid filling; Solid microparticles or pellets surrounded by a distinct coating layer, e.g. coated microspheres, coated drug crystals
- A61K9/5005—Wall or coating material
- A61K9/5021—Organic macromolecular compounds
- A61K9/5036—Polysaccharides, e.g. gums, alginate; Cyclodextrin
- A61K9/5042—Cellulose; Cellulose derivatives, e.g. phthalate or acetate succinate esters of hydroxypropyl methylcellulose
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/04—Drugs for disorders of the alimentary tract or the digestive system for ulcers, gastritis or reflux esophagitis, e.g. antacids, inhibitors of acid secretion, mucosal protectants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/10—Laxatives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/14—Prodigestives, e.g. acids, enzymes, appetite stimulants, antidyspeptics, tonics, antiflatulents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K2300/00—Mixtures or combinations of active ingredients, wherein at least one active ingredient is fully defined in groups A61K31/00 - A61K41/00
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Epidemiology (AREA)
- Organic Chemistry (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Inorganic Chemistry (AREA)
- Molecular Biology (AREA)
- Biophysics (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Immunology (AREA)
- Gastroenterology & Hepatology (AREA)
- Nutrition Science (AREA)
- Medicinal Preparation (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Peptides Or Proteins (AREA)
- Endocrinology (AREA)
- Zoology (AREA)
- Pain & Pain Management (AREA)
- Rheumatology (AREA)
Abstract
Composición de liberación retardada que comprende un comprimido con recubrimiento entérico, en la que el comprimido comprende: linaclotida; Ca2+; histidina; y alcohol polivinílico (PVA).
Description
DESCRIPCIÓN
Composiciones de liberación retardada de linaclotida
CAMPO DE LA INVENCIÓN
[0001] La presente invención se refiere a composiciones farmacéuticas de liberación retardada que comprenden linaclotida o sales farmacéuticamente aceptables de la misma, así como a diversos métodos y procedimientos para la preparación y el uso de las composiciones.
ANTECEDENTES DE LA INVENCIÓN
[0002] Se han usado diversas técnicas de formulación para desarrollar composiciones de liberación retardada para agentes farmacéuticamente activos, que incluyen el uso de recubrimientos entéricos o polímeros sensibles al pH. Sin embargo, los componentes específicos de estas composiciones varían enormemente y dependen significativamente del agente farmacéuticamente activo particular y las propiedades deseadas. Por ejemplo, la formulación debe ser compatible con el agente farmacéuticamente activo y asimismo proporcionar el comportamiento de disolución y las propiedades de estabilidad necesarios.
[0003] Las patentes de EE. UU. 7304036 y 7371 727 describen péptidos que actúan como agonistas del receptor de la guanilato ciclasa C (GC-C) para el tratamiento de trastornos gastrointestinales (GI). Un péptido particular descrito es la linaclotida, que consiste en la secuencia de aminoácidos siguiente: Cys Cys Glu Tyr Cys Cys Asn Pro Ala Cys Thr Gly Cys Tyr. La linaclotida tiene la estructura química de:

[0004] La linaclotida se administra por vía oral y ha sido aprobada en EE. UU. por la FDA para el tratamiento del síndrome del intestino irritable con estreñimiento (SII-e) y del estreñimiento idiopático crónico (EIC). En seres humanos, se ha demostrado que la linaclotida afecta a la fisiología del tracto GI, entre otros, reduciendo el dolor visceral, reduciendo la distensión y aumentando el tránsito GI, lo que puede conducir a frecuencia de defecación aumentada y consistencia de las heces mejorada. La linaclotida administrada por vía oral actúa localmente uniéndose a, y activando, los receptores de la GC-C en la superficie luminal del intestino. El receptor de la GC-C es un regulador clave de la función intestinal en los mamíferos y se encuentra en toda la superficie luminal del tracto GI. El receptor de la GC-C responde a las hormonas endógenas, guanilina y uroguanilina, y a los péptidos bacterianos entéricos de la familia de las enterotoxinas termoestables (péptido ST). Cuando la linaclotida se une al receptor de la GC-C, hay una elevación del segundo mensajero, GMP cíclico (c-GMP), y un aumento en la secreción de cloruro y bicarbonato, lo que da como resultado un aumento en la secreción de fluidos intestinales y la reducción del dolor.
[0005] Según lo aprobado por la FDA, la linaclotida se administra en una formulación de cápsulas orales, sólidas y de liberación inmediata fabricada encapsulando perlas estratificadas con fármaco en cápsulas de gelatina. Debido a la alta expresión de receptores de la GC-C en todo el tracto GI, la linaclotida de una formulación de liberación inmediata activa el receptor de la GC-C comenzando desde el tracto GI superior, lo que da como resultado una cantidad significativa de fluido intestinal que es llevado al tracto GI inferior. Para reducir o mitigar este efecto, se necesitan composiciones de liberación retardada que presenten liberación de linaclotida dirigida al segmento distal o inferior del tracto gastrointestinal. El direccionamiento al tracto GI inferior para la liberación de linaclotida puede ayudar a evitar la secreción excesiva de fluidos, pero al mismo tiempo a mantener o mejorar la eficacia de la linaclotida para tratar los síntomas abdominales e intestinales de los trastornos GI.
[0006] Se estima que, en EE. UU., 13 millones de adultos padecen SII-e y 35 millones de adultos padecen EIC. Asimismo, se estima que 15 millones de adultos padecen SII-m. Actualmente, 6,3 millones de adultos solicitan atención médica y no están satisfechos con los tratamientos actuales para el SII-m. El dolor abdominal o visceral es un síntoma predominante descrito por los pacientes que padecen SII, EIC y otros trastornos gastrointestinales. Como resultado, hay una necesidad de terapias que proporcionen alivio mejorado del dolor visceral y tolerabilidad superior para los adultos que padecen SII-e, SII-m y e Ic .
[0007] Se ha demostrado anteriormente que la linaclotida reduce el dolor visceral, que se cree que está mediado por el aumento del cGMP en el tracto GI. Los estudios en animales han demostrado que la linaclotida administrada por vía oral puede tratar el dolor visceral que se origina en el tracto GI inferior. Sin embargo, debido al entorno reductor del tracto intestinal, se cree que gran parte de la dosis oral de linaclotida es degradada antes de
alcanzar el colon distal. En voluntarios humanos tratados con linaclotida, solo se encuentra aproximadamente 5 % de la dosis oral en las heces. Las composiciones de liberación retardada (“LR”) de linaclotida que se dirigen al tracto GI inferior pueden mejorar la eficacia de la linaclotida en el alivio del dolor asociado a diversos trastornos GI permitiendo la administración de una dosis superior de linaclotida al colon. Tales composiciones LR de linaclotida tendrían potencial para liberar linaclotida predominantemente (o completamente) en el tracto GI inferior. Como resultado, por ejemplo, la formulación o composición LR tendría una capacidad aumentada para tratar trastornos asociados al tracto G i inferior. Asimismo, puede tener capacidad para provocar incidencias de eventos adversos (tales como diarrea) inferiores a la forma farmacéutica de liberación inmediata, p. ej., porque provocaría una secreción general de fluidos intestinales inferior al no activar los receptores de la GC-C en el tracto GI superior. Esto se produciría a la vez que se mantiene o incluso mejora la eficacia de la linaclotida para tratar los síntomas de trastornos GI, tales como dolor.
[0008] A pesar de la necesidad de composiciones de liberación retardada de linaclotida, existen dificultades para preparar tales formulaciones debido a la inestabilidad química intrínseca de la linaclotida, por ejemplo, por reacciones de degradación inducidas por la humedad tales como hidrólisis, desamidación, isomerización y multimerización. Estas dificultades se pueden exacerbar cuando se producen formulaciones que tienen dosis inferiores de linaclotida.
[0009] Por consiguiente, hay una necesidad actual y continua de composiciones de liberación retardada que proporcionen una administración estable y fiable de linaclotida a áreas objetivo del tracto gastrointestinal.
[0010] El documento US 2011/059903 se refiere a formulaciones estables que comprenden linaclotida. El documento WO 2013/025969 describe péptidos, composiciones y métodos para tratar trastornos gastrointestinales.
RESUMEN
[0011] Según la presente invención, se proporcionan composiciones de liberación retardada y materia relacionada como se definen en las reivindicaciones adjuntas. Cualquier materia que no esté dentro del alcance de las reivindicaciones se proporciona únicamente con fines informativos.
[0012] En particular, la invención se refiere a una composición de liberación retardada que comprende un comprimido con recubrimiento entérico, donde el comprimido comprende:
linaclotida;
Ca2+;
histidina; y
alcohol polivinílico (PVA).
[0013] La invención también se refiere a una forma de dosificación unitaria que comprende la composición anterior y a esta composición para uso en un método para tratar un trastorno gastrointestinal en un paciente, así como en un método para tratar o aliviar el dolor en un paciente.
[0014] Más generalmente, la presente descripción se refiere a formas farmacéuticas orales, sólidas y estables de linaclotida que presentan liberación retardada de linaclotida al tracto gastrointestinal inferior. Además, la presente descripción se refiere a métodos para tratar afecciones administrando estas composiciones de liberación retardada. Las composiciones de liberación retardada de la presente descripción se pueden usar para tratar diversas afecciones, pero son particularmente adecuadas para tratar trastornos gastrointestinales, tales como síndrome del intestino irritable (“SU”) (por ejemplo, SII con estreñimiento “SII-e”, SII con diarrea “SII-d” o SII mixto, con estreñimiento y diarrea, “SII-m”), estreñimiento (por ejemplo, estreñimiento idiopático crónico), cáncer de colon, diverticulitis, cistitis intersticial y dolor abdominal o visceral.
[0015] Según algunos casos de la presente descripción, se describen métodos para el tratamiento de un trastorno o dolor gastrointestinal que comprenden administrar a un paciente con necesidad de la misma una composición descrita en esta memoria.
BREVE DESCRIPCIÓN DE LAS FIGURAS
[0016]
La figura 1 muestra el perfil de liberación a diversos pH para composiciones de comprimidos de liberación retardada que comprenden 100 |jg de linaclotida.
La figura 2 muestra el perfil de liberación a diversos pH para composiciones de comprimidos de liberación retardada que comprenden 25 jg de linaclotida. La figura 3 muestra un perfil de disolución para una cápsula de linaclotida resistente a los ácidos no recubierta y recubierta.
DESCRIPCIÓN DETALLADA
[0017] Como se mencionó anteriormente, según la presente invención, se proporcionan composiciones de liberación retardada y materia relacionada como se definen en las reivindicaciones adjuntas. Cualquier materia que no esté dentro del alcance de las reivindicaciones se proporciona únicamente con fines informativos.
[0018] Más generalmente, en esta memoria se describen formas farmacéuticas orales de liberación retardada de linaclotida (colectivamente, “LR”). Hasta ahora, la única formulación de linaclotida aprobada es una cápsula que presenta liberación inmediata (“LI”). Estas formas farmacéuticas LI liberan la mayor parte o la totalidad de la linaclotida contenida en las mismas en el tracto GI superior. Esto, a su vez, provoca la activación del receptor de la GC-C y la secreción de fluidos tanto en el tracto GI superior como, en menor medida, en el tracto GI inferior. La diferencia entre la activación del tracto GI superior e inferior y la secreción de fluidos por la forma farmacéutica LI se debe, en parte, al hecho de que la linaclotida (una vez liberada desde la forma farmacéutica) experimenta digestión proteolítica y pierde parte o toda la capacidad de activar los receptores de la GC-C, particularmente en el momento en que alcanza el tracto GI inferior (tal como el íleon, el íleon terminal, la válvula ileocecal o el colon).
[0019] Las formas farmacéuticas LR descritas en esta memoria liberan la mayor parte o la totalidad de la linaclotida contenida en las mismas en el tracto GI inferior, tal como en las proximidades de la válvula ileocecal o en el colon (y con menos o ninguna liberación en el estómago, duodeno y/o yeyuno). Por lo tanto, las formas farmacéuticas descritas tienen capacidad para conseguir una secreción general de fluidos inferior a las formas farmacéuticas LI en el tracto GI superior, a la vez que mejoran o siguen manteniendo una eficacia excelente para tratar el SII-e, EIC y para modular la transducción de señales sensoriales de dolor gastrointestinal. Los pacientes con SII describen dolor abdominal en el cuadrante inferior izquierdo como síntoma de su trastorno, por lo que se cree que el dolor del SII se origina en el colon. Asimismo, se cree que las formas farmacéuticas LR son ideales para tratar enfermedades y trastornos asociados al tracto GI inferior. Como las formas farmacéuticas LR no liberarán ninguna (o un pequeño porcentaje) de su linaclotida en el estómago y el tracto GI superior (lo que puede provocar la digestión rápida de la linaclotida), algunos casos preferidos de la forma farmacéutica LR incorporarán dosis bajas de linaclotida (en comparación con las cantidades de la forma LI aprobada), pero mantendrán los mismos niveles de eficacia que la LI en el tratamiento de los síntomas del SII-e y EIC.
[0020] En general, el receptor de la guanilato ciclasa C (CG-C) es un receptor transmembranario que está ubicado sobre la superficie apical de las células epiteliales en el estómago e intestino. El receptor tiene un dominio extracelular de unión al ligando, una región transmembranaria única y un dominio de guanilil ciclasa en el extremo C. Cuando un ligando se une al dominio extracelular de la GC-C, el dominio catalítico intracelular cataliza la producción de cGMP a partir de GTP. In vivo, este aumento en el cGMP intracelular inicia una cascada de acontecimientos que conduce a secreción aumentada de cloruro y bicarbonato en la luz intestinal, pH luminal aumentado, absorción luminal de sodio reducida, secreción de fluidos aumentada y aceleración del tránsito intestinal. El cGMP es secretado bidireccionalmente desde el epitelio a la mucosa y la luz. Normalmente, el pH del tracto GI aumenta gradualmente del estómago (pH 1,5-3) al íleon terminal (pH 7-8) antes de caer en el colon a pH 5,5-7,0.
[0021] La linaclotida se une al receptor de la GC-C intestinal, que es un regulador del equilibrio de fluidos y electrolitos en el intestino. La linaclotida es un péptido que consiste en la secuencia de aminoácidos Cys1 Cys2 Glu3 Tyr4 Cys5 Cys6 Asnz Pros Alag Cysi0 Thr-n Glyi2 Cysi3 Tyr-M. Se puede usar cualquier forma deseada de linaclotida en la composición, por ejemplo, cualquier sal o hidrato farmacéuticamente aceptable del péptido, cualquier forma aislada y/o purificada del mismo o cualquier forma disulfuro del mismo. La linaclotida tiene enlaces disulfuro entre Cysi y Cys6, Cys2 y Cys-m, y Cys5 y Cysi3.
[0022] Las composiciones LR que contienen linaclotida se pueden usar para tratar un abanico de trastornos. En diversos casos, el paciente padece un trastorno gastrointestinal; el paciente padece un trastorno seleccionado del grupo que consiste en: trastornos de la motilidad gastrointestinal, seudoobstrucción intestinal crónica, seudoobstrucción colónica, enfermedad inflamatoria intestinal, enfermedad de Crohn, reflujo duodenogástrico, dispepsia, dispepsia funcional, dispepsia no ulcerosa, un trastorno gastrointestinal funcional, ardor gástrico funcional, enfermedad por reflujo gastroesofágico (ERGE), gastroparesia, síndrome del intestino irritable (p. ej., síndrome del intestino irritable con predominio de diarrea (SII-d), síndrome del intestino irritable con predominio de estreñimiento (SII-e) y/o síndrome del intestino irritable alternativo o mixto (SII-m)), íleo posoperatorio, colitis ulcerosa, estreñimiento crónico, estreñimiento, cistitis intersticial, prostatitis, dolor testicular, síndrome de vejiga dolorosa, endometriosis, vulvodinia, dolor rectal, enfermedad o dolor diverticular, dolor asociado a trastornos GI y trastornos y afecciones asociados a estreñimiento (p. ej., estreñimiento asociado al uso de analgésicos opiáceos, estreñimiento posquirúrgico y estreñimiento asociado a trastornos neuropáticos, así como otras afecciones y trastornos descritos en esta memoria). El paciente puede ser diagnosticado con un trastorno gastrointestinal funcional (p. ej., SII-e) según los Criterios de Roma (p. ej., Roma II).
[0023] En algunos casos, la composición LR comprende perlas con recubrimiento entérico que comprenden un recubrimiento polimérico sensible al pH que se puede aplicar a perlas centrales que se han recubierto con linaclotida y una potencial subcapa polimérica. En casos adicionales, las perlas con recubrimientos entéricos y de subcapa protectora se podrían encapsular además en cápsulas para conseguir las concentraciones de las dosis unitarias
deseadas. En algunos casos, el polímero actúa como recubrimiento tanto protector como estabilizante, o como agente formador de película dentro de la composición de liberación retardada. En otros casos, la composición LR comprende comprimidos con recubrimiento entérico que comprenden un núcleo de comprimido de liberación inmediata y que contienen una dosis unitaria de linaclotida que se disuelve solo en las condiciones de pH del segmento distal del intestino. En otros casos, la composición LR comprende una cápsula de gelatina o HPMC con recubrimiento entérico, consiguiéndose la disgregación de la cápsula en el tracto GI inferior. En algunos casos, el recubrimiento entérico o funcional se selecciona de entre copolímeros de acrilato de metilo-ácido metacrílico (p. ej., Eudragit®); acetatosuccinato de celulosa (CAS); ftalato de hidroxipropilmetilcelulosa (HPMCP); PVA; PVP; PVP-LP, acetato-succinato de hidroxipropilmetilcelulosa (HPMCAS); acetato-ftalato de polivinilo (PVAP). copolímeros de metacrilato de metilo-ácido metacrílico; alginato de sodio y ácido esteárico; goma guar; y carbómeros. En casos adicionales, el recubrimiento entérico se selecciona de entre Eudragit® FS30D, PlasAcryl®, Eudragit® S100, Eudragit® L100, Eudragit® L100-55, Eudragit® L30D-55, Eudragit® S, Eudragit® RL30D, Eudragit® RS30D, Eudragit® RS, Eudragit® EC o una mezcla de los mismos.
[0024] Las composiciones de liberación retardada pueden incluir cualquier cantidad eficaz de linaclotida. En algunos casos, por ejemplo, la composición comprende de 0,05 |jg a 6 mg de linaclotida. En algunos casos, por ejemplo, la composición comprende de 1 jg a 2 mg de linaclotida. En algunos casos, la composición comprende de 25 jg a 2 mg de linaclotida, por ejemplo, de 50 jg a 1 mg de linaclotida. En algunos casos, por ejemplo, la composición comprende de 0,1 jg a 90 jg de linaclotida. En algunos casos, por ejemplo, la composición comprende de 0,1 jg a 45 jg de linaclotida. En algunos casos, por ejemplo, la composición comprende de 0,1 jg a 25 jg de linaclotida. En algunos casos, por ejemplo, la composición comprende de 36 jg a 290 jg de linaclotida. En algunos casos, la composición comprende 0,05 jg , 0,1 jg , 0,15 jg , 0,25 jg , 0,5 jg , 0,75 jg , 1 jg , 1,5 jg , 2 jg , 2,5 jg , 3 jg , 3,5 jg , 4 jg , 4,5 jg , 5 jg , 7,5 jg , 9 jg , 10 jg , 15 jg , 20 jg , 25 jg , 30 jg , 35 jg , 36 jg , 40 jg , 45 jg , 50 jg , 60 jg , 72 jg , 75 jg , 90 jg , 100 jg , 145 jg , 150 jg , 200 jg , 250 jg , 290 jg , 300 jg , 350 jg , 400 jg , 450 jg , 500 jg , 550 jg , 579 jg , 600 jg , 650 jg , 700 jg , 750 jg , 800 jg , 850 jg , 900 jg , 950 jg o 1 jg de linaclotida. En algunos casos, la composición comprende de 100 jg a 600 jg de linaclotida. En algunos casos, la composición comprende 25 jg , 50 jg , 75 jg , 100 jg , 150 jg , 290 jg , 300 jg , 400 jg , 500 jg o 600 jg de linaclotida. En algunos casos, la composición comprende 9 jg , 10 jg , 15 jg , 36 jg , 72 jg , 75 jg , 145 jg , 290 jg , 579 jg o 600 jg de linaclotida.
[0025] En algunos casos, la composición comprende 9 jg de linaclotida. En algunos casos, la composición comprende 10 jg de linaclotida. En algunos casos, la composición comprende 25 jg de linaclotida. En algunos casos, la composición comprende 50 jg de linaclotida. En algunos casos, la composición comprende 75 jg de linaclotida. En algunos casos, la composición comprende 100 jg de linaclotida. En algunos casos, la composición comprende 150 jg de linaclotida. En algunos casos, la composición comprende 145 jg de linaclotida. En algunos casos, la composición comprende 290 jg de linaclotida.
[0026] Se ha encontrado que, en algunos casos, la estabilidad de las composiciones de liberación retardada de linaclotida se puede aumentar o mejorar incluyendo en las composiciones una cantidad adecuada de un componente de amina primaria impedida estéricamente (p. ej., aminoácido), un componente catiónico (p. ej., catión metálico) y/o un componente polimérico. Estos componentes aumentan o mejoran la estabilidad de las composiciones de liberación retardada de linaclotida, por ejemplo, impidiendo, disminuyendo y/o reduciendo la degradación de la linaclotida dentro de la composición (por ejemplo, debido a reacciones de degradación impulsadas por la humedad, p. ej., reacciones de hidrólisis, desamidación y/o multimerización). Por ejemplo, se ha encontrado que, en algunos casos, la adición o inclusión de una cantidad adecuada de un catión (p. ej., Mg2+, Ca2+, Zn2+) en la composición aumenta la estabilidad de la composición frente a la degradación oxidativa de la linaclotida. Asimismo, se ha encontrado que, en algunos casos, la inclusión de una cantidad adecuada de una amina primaria impedida estéricamente, por ejemplo, en forma de un aminoácido (p. ej., histidina), en la composición aumenta la estabilidad de la composición frente a, por ejemplo, la adición nucleofílica de formaldehído o un equivalente al formaldehído al extremo N de la linaclotida, p. ej., actuando como antioxidante y/o tamponando la composición. Asimismo, se ha encontrado que, en algunos casos, la inclusión de tanto una amina primaria impedida estéricamente (p. ej., histidina) como un catión (p. ej., Ca2+) en cantidades adecuadas en la composición aumenta la estabilidad de la composición frente a la formación de productos de la hidrólisis y de formaldehído (Cys1-IMD) de la linaclotida. También se ha encontrado que, en algunos casos, la inclusión de una cantidad adecuada de un polímero (p. ej., polivinilpirrolidona o alcohol polivinílico) en la composición de liberación retardada aumenta la estabilidad de la composición, por ejemplo, reduciendo la movilidad y/o reactividad de la linaclotida dentro de la composición, p. ej., formando un complejo o una matriz (por ejemplo, una matriz vítrea y/o rígida) con la linaclotida (p. ej., mediante reacción de vitrificación), impidiendo o disminuyendo la formación de enlaces de hidrógeno entre la linaclotida y las moléculas de agua y/o mejorando la integridad estructural tridimensional de la linaclotida.
[0027] A este respecto, se ha encontrado que, en algunos casos, combinar la linaclotida en una composición farmacéutica de liberación retardada con concentraciones o relaciones molares del catión y la amina primaria impedida estéricamente específicas provoca una potenciación o mejora sinérgica en la estabilidad de la linaclotida dentro de la composición, por ejemplo, en comparación con composiciones similares que no contienen el catión y/o la amina primaria impedida estéricamente y/o las mismas concentraciones de estos componentes. En algunos casos, la composición puede comprender cualquier cantidad estabilizante de un componente de amina primaria impedida
estéricamente. En otros casos, la composición comprende una relación molar de amina primaria impedida estéricamente a linaclotida entre 400:1 y 1:1. En casos adicionales, la composición comprende una relación molar de amina primaria impedida estéricamente a linaclotida entre 200:1 y 50:1. En otros casos, la composición puede comprender una relación molar de amina primaria impedida estéricamente (p. ej., aminoácido) a linaclotida entre 100:1 y 1:100. En algunos casos, la composición comprende una relación molar de amina primaria impedida estéricamente a linaclotida entre 100:1 y 1:1. En algunos casos, la composición comprende una relación molar de amina primaria impedida estéricamente a linaclotida entre 90:1 y 2:1. En algunos casos, la composición comprende una relación molar de amina primaria impedida estéricamente a linaclotida entre 80:1 y 5:1. En algunos casos, la composición comprende una relación molar de amina primaria impedida estéricamente a linaclotida entre 70:1 y 10:1. En algunos casos, la composición comprende una relación molar de amina primaria impedida estéricamente a linaclotida entre 60:1 y 20:1. En algunos casos, la composición comprende una relación molar de amina primaria impedida estéricamente a linaclotida entre 50:1 y 30:1. En algunos casos, la composición comprende una relación molar de amina primaria impedida estéricamente a linaclotida entre 40:1 y 20:1. En algunos casos, la composición comprende una relación molar de amina primaria impedida estéricamente a linaclotida entre 100:1 y 20:1. En algunos casos, la composición comprende una relación molar de amina primaria impedida estéricamente a linaclotida entre 100:1 y 25:1. En algunos casos, la composición comprende una relación molar de amina primaria impedida estéricamente a linaclotida entre 100:1 y 30:1. En algunos casos, la composición comprende una relación molar de amina primaria impedida estéricamente a linaclotida entre 100:1 y 40:1. En algunos casos, la composición comprende una relación molar de amina primaria impedida estéricamente a linaclotida entre 100:1 y 50:1. En algunos casos, la composición comprende una relación molar de amina primaria impedida estéricamente a linaclotida entre 100:1 y 60:1. En algunos casos, la composición comprende una relación molar de amina primaria impedida estéricamente a linaclotida entre 100:1 y 70:1. En algunos casos, la composición comprende una relación molar de amina primaria impedida estéricamente a linaclotida de al menos 5:1. En algunos casos, la composición comprende una relación molar de amina primaria impedida estéricamente a linaclotida de al menos 10:1. En algunos casos, la composición comprende una relación molar de amina primaria impedida estéricamente a linaclotida de al menos 20:1. En algunos casos, la composición comprende una relación molar de amina primaria impedida estéricamente a linaclotida de al menos 25:1. En algunos casos, la composición comprende una relación molar de amina primaria impedida estéricamente a linaclotida de al menos 30:1. En algunos casos, la composición comprende una relación molar de amina primaria impedida estéricamente a linaclotida de al menos 40:1.
[0028] Las aminas primarias impedidas estéricamente adecuadas para su inclusión en la composición de liberación retardada son, por ejemplo, aminoácidos de origen natural (p. ej., alanina, arginina, asparagina, ácido aspártico, cisteína, ácido glutámico, glutamina, glicina, histidina, isoleucina, leucina, lisina, meglumina, metionina, fenilalanina, prolina, serina, treonina, triptófano, tirosina, valina), aminoácidos sintéticos (p. ej., lantionina, teanina o 1 aminociclohexano), aminoazúcares (p. ej., quitosano o glucosamina) o combinación o mezclas de los mismos. En algunos casos, la composición comprende un aminoácido seleccionado de entre alanina, arginina, asparagina, ácido aspártico, cisteína, ácido glutámico, glutamina, histidina, isoleucina, leucina, lisina, metionina, fenilalanina, prolina, serina, treonina, triptófano, tirosina, valina o una mezcla de los mismos. En algunos casos, la composición comprende un aminoácido seleccionado de entre leucina, isoleucina, asparagina, glutamina, ácido glutámico, histidina, cisteína, alanina, serina, treonina, tirosina, prolina, triptófano o una combinación o mezcla de los mismos. En algunos casos, la composición comprende un aminoácido seleccionado de entre leucina, isoleucina, metionina, alanina o una combinación o mezcla de los mismos. En algunos casos, la composición comprende un aminoácido seleccionado de entre leucina, isoleucina, alanina o una combinación o mezcla de los mismos. En algunos casos, la composición comprende un aminoácido seleccionado de entre leucina, isoleucina, metionina o una combinación o mezcla de los mismos. En algunos casos, la composición comprende un aminoácido seleccionado de entre leucina, metionina, alanina o una combinación o mezcla de los mismos. En algunos casos, la composición comprende leucina, metionina o una mezcla de las mismas. En algunos casos, la composición comprende leucina, isoleucina o una mezcla de las mismas. En algunos casos, la composición comprende leucina, alanina o una mezcla de las mismas. En algunos casos, la composición comprende leucina. En algunos casos, la composición comprende isoleucina. En algunos casos, la composición comprende metionina. En algunos casos, la composición comprende alanina. En algunos casos, la composición comprende histidina.
[0029] La composición de liberación retardada puede comprender cualquier cantidad estabilizante de un catión (p. ej., catión metálico). En algunos casos, la composición comprende una relación molar de catión a linaclotida entre 300:1 y 1:1. En casos adicionales, la composición comprende una relación molar de catión a linaclotida entre 250:1 y 30:1. En otros casos, la composición puede comprender una relación molar de catión a linaclotida entre 100:1 y 1:100. En algunos casos, la composición comprende una relación molar de catión a linaclotida entre 100:1 y 1:1. En algunos casos, la composición comprende una relación molar de catión a linaclotida entre 90:1 y 2:1. En algunos casos, la composición comprende una relación molar de catión a linaclotida entre 80:1 y 5:1. En algunos casos, la composición comprende una relación molar de catión a linaclotida entre 70:1 y 10:1. En algunos casos, la composición comprende una relación molar de catión a linaclotida entre 60:1 y 20:1. En algunos casos, la composición comprende una relación molar de catión a linaclotida entre 50:1 y 30:1. En algunos casos, la composición comprende una relación molar de catión a linaclotida entre 40:1 y 20:1. En algunos casos, la composición comprende una relación molar de catión a linaclotida entre 100:1 y 20:1. En algunos casos, la composición comprende una relación molar de catión a linaclotida entre 100:1 y 25:1. En algunos casos, la composición comprende una relación molar de catión a linaclotida entre 100:1
y 30:1. En algunos casos, la composición comprende una relación molar de catión a linaclotida entre 100:1 y 40:1. En algunos casos, la composición comprende una relación molar de catión a linaclotida entre 100:1 y 50:1. En algunos casos, la composición comprende una relación molar de catión a linaclotida entre 100:1 y 60:1. En algunos casos, la composición comprende una relación molar de catión a linaclotida entre 100:1 y 70:1. En algunos casos, la composición comprende una relación molar de catión a linaclotida de al menos 5:1. En algunos casos, la composición comprende una relación molar de catión a linaclotida de al menos 10:1. En algunos casos, la composición comprende una relación molar de catión a linaclotida de al menos 20:1. En algunos casos, la composición comprende una relación molar de catión a linaclotida de al menos 25:1. En algunos casos, la composición comprende una relación molar de catión a linaclotida de al menos 30:1. En algunos casos, la composición comprende una relación molar de catión a linaclotida de al menos 40:1. En algunos casos, la composición comprende una relación molar de catión a linaclotida de al menos 60:1.
[0030] Cualquier catión o cationes adecuados se pueden incluir en la composición, por ejemplo, cualquier catión metálico o catión orgánico adecuado. En algunos casos, la composición comprende un catión metálico seleccionado de entre calcio, potasio, magnesio, zinc, aluminio, hierro, estaño, manganeso, cromo, cobalto, níquel, bario, sodio o una combinación o mezcla de los mismos. En algunos casos, la composición comprende un catión metálico seleccionado de entre calcio, potasio, magnesio, zinc, aluminio, manganeso, cromo, cobalto, níquel, bario, sodio o una combinación o mezcla de los mismos. En algunos casos, la composición comprende un catión metálico seleccionado de entre aluminio, calcio, potasio, sodio, magnesio, manganeso, zinc o una combinación o mezcla de los mismos. En algunos casos, la composición comprende un catión metálico seleccionado de entre calcio, magnesio, manganeso, zinc o una combinación o mezcla de los mismos. En algunos casos, la composición comprende un catión metálico divalente. En algunos casos, la composición comprende un catión metálico divalente seleccionado de entre Al3+, Ca2+, Mg2+, Zn2+, Mn2+ o una combinación o mezcla de los mismos. En algunos casos, la composición comprende Mg2+. En algunos casos, la composición comprende Ca2+. En algunos casos, la composición comprende Zn2+. En algunos casos, la composición comprende aluminio.
[0031] Asimismo, el catión metálico se puede añadir a la composición en cualquier forma adecuada, por ejemplo, cualquier sal farmacéuticamente aceptable con cualquier contraión adecuado. Las sales metálicas adecuadas incluyen, por ejemplo, cloruro de calcio, carbonato de calcio, acetato de calcio, cloruro de magnesio, acetato de magnesio, acetato de zinc, cloruro de zinc o mezclas de los mismos. En algunos casos, la composición comprende cloruro de calcio, cloruro de magnesio, acetato de zinc o cualquier combinación o mezcla de los mismos. En algunos casos, la composición comprende cloruro de calcio. En algunos casos, la composición comprende cloruro de magnesio. En algunos casos, la composición comprende acetato de zinc. Los cationes orgánicos adecuados incluyen, por ejemplo, hidróxido de amonio, D-arginina, L-arginina, t-butilamina, acetato de calcio hidratado, carbonato de calcio, DL-malato de calcio, hidróxido de calcio, colina, etanolamina, etilendiamina, glicina, L-histidina, L-lisina, hidróxido de magnesio, N-metil-D-glucamina, clorhidrato de L-ornitina, hidróxido de potasio, clorhidrato de procaína, L-prolina, piridoxina, L-serina, hidróxido de sodio, DL-triptófano, trometamina, L-tirosina, L-valina, carnitina, taurina, malato de creatina, alfa-cetoglutarato de arginina, alfa-cetoglutarato de ornitina, acetato de espermina, cloruro de espermidina o combinaciones o mezclas de los mismos. En algunos casos, el catión orgánico se selecciona del grupo que consiste en N-metil D-glucamina, colina, arginina, lisina, procaína, trometamina (TRIS), espermina, N-metilmorfolina, glucosamina, N,N-bis(2-hidroxietil)glicina, diazabicicloundeceno, creatina, éster etílico de arginina, amantadina, rimantadina, ornitina, taurina, citrulina o una combinación o mezcla de los mismos.
[0032] La composición puede contener cualquier cantidad estabilizante de un polímero. En algunos casos, la composición comprende entre 1 y 25 % en peso de un polímero, con respecto al peso total de la composición. En algunos casos, la composición comprende entre 1 y 10 % en peso de un polímero, con respecto al peso total de la composición.
[0033] En algunos casos, la composición comprende entre 2 y 4 % en peso de un polímero, con respecto al peso total de la composición. En algunos casos, la composición comprende entre 0,1 y 75 % en peso de un polímero. En algunos casos, la composición comprende entre 0,1 y 55% en peso de un polímero. En algunos casos, la composición comprende entre 0,1 y 35 % en peso de un polímero. En algunos casos, la composición comprende entre 0,1 y 30 % en peso de un polímero. En algunos casos, la composición comprende entre 0,1 y 25 % en peso de un polímero. En algunos casos, la composición comprende entre 1 y 25 % en peso de un polímero. En algunos casos, la composición comprende entre 5 y 25 % en peso de un polímero. En algunos casos, la composición comprende entre 10 y 25 % en peso de un polímero. En algunos casos, la composición comprende entre 15 y 25 % en peso de un polímero. En algunos casos, la composición comprende entre 0,1 y 22 % en peso de un polímero. En algunos casos, la composición comprende entre 1 y 22 % en peso de un polímero. En algunos casos, la composición comprende entre 5 y 22 % en peso de un polímero. En algunos casos, la composición comprende entre 10 y 22 % en peso de un polímero. En algunos casos, la composición comprende entre 0,1 y 20 % en peso de un polímero. En algunos casos, la composición comprende entre 1 y 20 % en peso de un polímero. En algunos casos, la composición comprende entre 5 y 20 % en peso de un polímero. En algunos casos, la composición comprende entre 10 y 20 % en peso de un polímero. En algunos casos, la composición comprende entre 0,01 y 15 % en peso de un polímero. En algunos casos, la composición comprende entre 0,01 y 10 % en peso de un polímero. En algunos casos, la composición comprende entre 0,01 y 5 % en peso de un polímero. En algunos casos, la composición comprende entre 0,1 y 95 % en peso, por
ejemplo, entre 5 y 95 % en peso, entre 15 y 95 % en peso, entre 25 y 95 % en peso, entre 35 y 95 % en peso, entre 45 y 95 % en peso, entre 0,1 y 85 % en peso, entre 1 y 85 % en peso, entre 5 y 85 % en peso, entre 15 y 85 % en peso, entre 25 y 85 % en peso, entre 35 y 85 % en peso, entre 0,1 y 80 % en peso, entre 1 y 80 % en peso, entre 5 y 80 % en peso, entre 15 y 80 % en peso, entre 25 y 80 % en peso, entre 35 y 80 % en peso, entre 0,1 y 75 % en peso, entre 1 y 75 % en peso, entre 5 y 75 % en peso, entre 15 y 75 % en peso, entre 25 y 75 % en peso, entre 35 y 75 % en peso, entre 0,1 y 65 % en peso, entre 1 y 65 % en peso, entre 5 y 65 % en peso, entre 15 y 65 % en peso, entre 25 y 65 % en peso, entre 35 y 65 % en peso, entre 0,1 y 60 % en peso, entre 1 y 60 % en peso, entre 5 y 60 % en peso, entre 15 y 60 % en peso, entre 25 y 60 % en peso o entre 35 y 60 % en peso de un polímero.
[0034] En algunos casos, el polímero actúa como recubrimiento tanto protector como estabilizante, o como agente formador de película dentro de la composición de liberación retardada. En algunos casos, la composición de liberación retardada comprende una relación molar de polímero (p. ej., PVP o PVA) a linaclotida entre 80:1 y 300:1, por ejemplo, entre 100:200:1, entre 110:1 y 190:1, o incluso entre 120:1 y 180:1. En algunos casos, la composición de liberación retardada comprende una relación molar de polímero (p. ej., PVP o PVA) a linaclotida superior a aproximadamente 80:1, por ejemplo, superior a aproximadamente 100:1, o incluso superior a aproximadamente 120:1. En algunos casos, la composición de liberación retardada comprende una relación en peso de polímero (p. ej., PVP o PVA) a linaclotida entre 10:1 y 300:1, por ejemplo, entre 80:1 y 200:1, entre 100:1 y 180:1, o incluso entre 110:1 y 150:1. En algunos casos, la composición de liberación retardada comprende una relación en peso de polímero (p. ej., PVP o PVA) a linaclotida entre 100:1 y 500:1, por ejemplo, entre 200:1 y 400:1, entre 250:1 y 350:1, o incluso entre 300:1 y 350:1.
[0035] Los polímeros adecuados para su inclusión en las composiciones de liberación retardada son, por ejemplo, polivinilpirrolidona (PVP), alcohol polivinílico (PVA), alcohol polivinílico con índice de peróxidos bajo (PVA-LP), hidroxilpropilmetilcelulosa (HPMC), hidroxilpropilcelulosa (HPC), metilcelulosa, polímeros de metacrilato, ciclodextrina, dextrina, dextrano, ácido poliacrílico, quitosano, goma guar, goma xantana, óxido de polietileno (p. ej., óxido de polietileno polipropileno), poli(vinilsulfonato de sodio), polietilenglicol, poli(arginina), policarbófilo, polivinilpirrolidona-co-acetato de vinilo, un poloxámero (p. ej., productos Pluronic® disponibles de BASF), alginato, trehalosa, sucrosa, inulina o una combinación o mezcla de los mismos. En algunos casos, la composición comprende un polímero seleccionado de entre PVP, PVA, polímeros de metacrilato, ciclodextrina, dextrano, ácido poliacrílico, quitosano, goma guar, goma xantana, óxido de polietileno, polietilenglicol, poli(arginina), policarbófilo, polivinilpirrolidona-co-acetato de vinilo, un poloxámero o una combinación o mezcla de los mismos En algunos casos, la composición comprende PVP, PVA, óxido de polietileno o una mezcla de los mismos. En algunos casos, la composición comprende PVP, PVA o una mezcla de los mismos. En algunos casos, la composición comprende PVP. En algunos casos, la composición comprende PVA.
[0036] En algunos casos, la composición de liberación retardada comprende dos o más agentes estabilizantes. Por ejemplo, la composición puede incluir una cantidad estabilizante de un polímero y una cantidad estabilizante de una amina primaria impedida estéricamente. Asimismo, la composición puede incluir una cantidad estabilizante de un polímero y una cantidad estabilizante de un catión (p. ej., catión metálico). Además, la composición puede incluir una cantidad estabilizante de una amina primaria impedida estéricamente y una cantidad estabilizante de un catión (p. ej., catión metálico). En algunos casos, la composición comprende una cantidad estabilizante de un polímero, una cantidad estabilizante de una amina primaria impedida estéricamente y una cantidad estabilizante de un catión (p. ej., catión metálico).
[0037] En algunos casos, la composición de liberación retardada comprende una cantidad estabilizante de PVP y una cantidad estabilizante de un aminoácido seleccionado de entre histidina, alanina, arginina, asparagina, ácido aspártico, cisteína, ácido glutámico, glutamina, glicina, histidina, isoleucina, leucina, lisina, metionina, fenilalanina, prolina, serina, treonina, triptófano, tirosina, valina o una mezcla de los mismos. En algunos casos, la composición comprende una cantidad estabilizante de PVP y una cantidad estabilizante de un aminoácido seleccionado de entre alanina, arginina, asparagina, ácido aspártico, cisteína, ácido glutámico, glutamina, histidina, isoleucina, leucina, lisina, metionina, fenilalanina, prolina, serina, treonina, triptófano, tirosina, valina o una mezcla de los mismos. En algunos casos, la composición comprende una cantidad estabilizante de PVP y una cantidad estabilizante de leucina, isoleucina, metionina, alanina, o una combinación o mezcla de las mismas. En algunos casos, la composición comprende una cantidad estabilizante de PVP y una cantidad estabilizante de leucina.
[0038] En algunos casos, la composición de liberación retardada comprende una cantidad estabilizante de PVA y una cantidad estabilizante de un aminoácido seleccionado de entre alanina, arginina, asparagina, ácido aspártico, cisteína, ácido glutámico, glutamina, glicina, histidina, isoleucina, leucina, lisina, metionina, fenilalanina, prolina, serina, treonina, triptófano, tirosina, valina o una mezcla de los mismos. En algunos casos, la composición comprende una cantidad estabilizante de PVA y una cantidad estabilizante de un aminoácido seleccionado de entre alanina, arginina, asparagina, ácido aspártico, cisteína, ácido glutámico, glutamina, histidina, isoleucina, leucina, lisina, metionina, fenilalanina, prolina, serina, treonina, triptófano, tirosina, valina o una mezcla de los mismos. En algunos casos, la composición comprende una cantidad estabilizante de PVA y una cantidad estabilizante de leucina, isoleucina, metionina, alanina, o una combinación o mezcla de las mismas. En algunos casos, la composición comprende una cantidad estabilizante de PVA y una cantidad estabilizante de leucina.
[0039] En algunos casos, la composición de liberación retardada comprende una cantidad estabilizante de PVP y una cantidad estabilizante de un catión (p. ej., catión metálico). En algunos casos, la composición comprende una cantidad estabilizante de PVP y una cantidad estabilizante de un catión metálico divalente. En algunos casos, la composición comprende una cantidad estabilizante de PVP y una cantidad estabilizante de Mg2+, Ca2+, Zn2+ o una sal de los mismos o una combinación o mezcla de los mismos. En algunos casos, la composición comprende una cantidad estabilizante de PVP y una cantidad estabilizante de Ca2+ o una sal del mismo. En algunos casos, la composición comprende una cantidad estabilizante de PVP y una cantidad estabilizante de Mg2+ o una sal del mismo. En algunos casos, la composición comprende una cantidad estabilizante de PVP y una cantidad estabilizante de Zn2+ o una sal del mismo.
[0040] En algunos casos, la composición de liberación retardada comprende una cantidad estabilizante de PVA y una cantidad estabilizante de un catión (p. ej., catión metálico). En algunos casos, la composición comprende una cantidad estabilizante de PVA y una cantidad estabilizante de un catión metálico divalente. En algunos casos, la composición comprende una cantidad estabilizante de PVA y una cantidad estabilizante de Mg2+, Ca2+, Zn2+ o una sal de los mismos o una combinación o mezcla de los mismos. En algunos casos, la composición comprende una cantidad estabilizante de PVA y una cantidad estabilizante de Ca2+ o una sal del mismo. En algunos casos, la composición comprende una cantidad estabilizante de PVA y una cantidad estabilizante de Mg2+ o una sal del mismo. En algunos casos, la composición comprende una cantidad estabilizante de PVA y una cantidad estabilizante de Zn2+ o una sal del mismo.
[0041] En algunos casos, la composición de liberación retardada comprende una cantidad estabilizante de un aminoácido seleccionado de entre leucina, isoleucina, metionina, alanina; y una cantidad estabilizante de un catión metálico divalente seleccionado de entre Mg2+, Ca2+, Zn2+ o una sal de los mismos o una combinación o mezcla de los mismos. En algunos casos, la composición comprende una cantidad estabilizante de un aminoácido seleccionado de entre leucina e isoleucina; y una cantidad estabilizante de un catión metálico divalente seleccionado de entre Mg2+, Ca2+ o una sal de los mismos o una combinación o mezcla de los mismos. En algunos casos, la composición comprende una cantidad estabilizante de un aminoácido seleccionado de entre leucina o metionina; y una cantidad estabilizante de un catión metálico divalente seleccionado de entre Ca2+, Zn2+ o una sal de los mismos o una combinación o mezcla de los mismos. En algunos casos, la composición comprende una cantidad estabilizante de leucina y una cantidad estabilizante de Ca2+ o una sal del mismo. En algunos casos, la composición comprende una cantidad estabilizante de un catión y una cantidad estabilizante de una amina primaria impedida estéricamente. En algunos casos, la composición comprende un catión y una amina primaria impedida estéricamente en una relación molar de catión:amina primaria impedida estéricamente (p. ej., Ca2+:leucina) de al menos 1,5:1, p. ej., al menos 2:1, al menos 2,5:1, al menos 3:1, al menos 4:1 o incluso al menos 5:1 (por ejemplo, una relación molar entre 1,5:1 y 5:1, p. ej., entre 2:1 y 4:1).
[0042] En algunos casos, la composición de liberación retardada comprende (i) una cantidad estabilizante de PVP o PVA, (ii) una cantidad estabilizante de leucina, isoleucina, metionina, alanina y (iii) una cantidad estabilizante de Mg2+, Ca2+, Zn2+ o una sal de los mismos o una combinación o mezcla de los mismos. En algunos casos, la composición comprende una cantidad estabilizante de PVP, una cantidad estabilizante de leucina y una cantidad estabilizante de un catión metálico. En algunos casos, la composición comprende una cantidad estabilizante de PVA, una cantidad estabilizante de histidina y una cantidad estabilizante de Ca2+ o una sal del mismo. En algunos casos, la composición comprende una cantidad estabilizante de PVP, una cantidad estabilizante de leucina y una cantidad estabilizante de Mg2+ o una sal del mismo. En algunos casos, la composición comprende una cantidad estabilizante de PVP, una cantidad estabilizante de leucina y una cantidad estabilizante de Zn2+ o una sal del mismo. En algunos casos, la composición comprende una cantidad estabilizante de PVA, una cantidad estabilizante de leucina y una cantidad estabilizante de Ca2+ o una sal del mismo. En algunos casos, la composición comprende una cantidad estabilizante de PVA, una cantidad estabilizante de leucina y una cantidad estabilizante de Mg2+ o una sal del mismo. En algunos casos, la composición comprende una cantidad estabilizante de PVA, una cantidad estabilizante de leucina y una cantidad estabilizante de Zn2+ o una sal del mismo.
[0043] En algunos casos, la composición comprende (i) entre 0,1 y 30 % en peso de un polímero, (ii) una amina primaria impedida estéricamente (p. ej., un aminoácido) en una relación molar de amina primaria a linaclotida entre 100:1 y 10:1 y (iii) un catión (p. ej., un catión metálico) en una relación molar de catión a linaclotida entre 100:1 y 40:1. En algunos casos, la composición comprende (i) entre 5 y 25 % en peso de un polímero, (ii) una amina primaria impedida estéricamente (p. ej., un aminoácido) en una relación molar de amina primaria a linaclotida 100:1 y 30:1 (p. ej., entre 60:1 y 30:1 o incluso entre 50:1 y 30:1) y (iii) un catión (p. ej., un catión metálico) en una relación molar de catión a linaclotida entre 100:1 y 60:1. En algunos casos, la composición comprende (i) entre 0,1 y 30 % en peso de un polímero seleccionado de entre PVP y PVA, (ii) un aminoácido seleccionado de entre leucina, isoleucina, alanina y metionina en una relación molar de aminoácido a linaclotida entre 100:1 y 10:1 y (iii) un catión metálico seleccionado de entre Ca2+, Mg2+ y Zn2+ en una relación molar de catión a linaclotida entre 100:1 y 40:1. En algunos casos, la composición comprende (i) entre 5 y 25 % en peso de un polímero seleccionado de entre PVP y PVA, (ii) un aminoácido seleccionado de entre leucina, isoleucina, alanina y metionina en una relación molar de aminoácido a linaclotida 100:1 y 30:1 (p. ej., entre 60:1 y 30:1) y (iii) un catión metálico seleccionado de entre Ca2+, Mg2+ y Zn2+ en
una relación molar de catión a linaclotida entre 100:1 y 60:1. En algunos casos, la composición comprende (i) entre 0,1 y 30 % en peso (p. ej., entre 5 y 25 % en peso) de PVP o PVA, (ii) leucina en una relación molar de leucina a linaclotida entre 100:1 y 30:1 (p. ej., entre 60:1 y 30:1 o incluso entre 50:1 y 30:1) y (iii) Ca2+ en una relación molar de Ca2+ a linaclotida entre 100:1 y 60:1.
[0044] En algunos casos, la composición comprende (i) entre 45 y 99 % en peso de un polímero, (ii) una amina primaria impedida estéricamente (p. ej., un aminoácido) en una relación molar de amina primaria a linaclotida entre 100:1 y 10:1, y (iii) un catión (p. ej., un catión metálico) en una relación molar de catión a linaclotida entre 100:1 y 40:1. En algunos casos, la composición comprende (i) entre 45 y 70 % en peso de un polímero, (ii) una amina primaria impedida estéricamente (p. ej., un aminoácido) en una relación molar de amina primaria a linaclotida 100:1 y 30:1 (p. ej. entre 60:1 y 30:1 o incluso entre 50:1 y 30:1) y (iii) un catión (p. ej., un catión metálico) en una relación molar de catión a linaclotida entre 100:1 y 60:1. En algunos casos, la composición comprende (i) entre 45 y 99 % en peso de un polímero seleccionado de entre PVP y PVA, (ii) un aminoácido seleccionado de entre leucina, isoleucina, alanina y metionina en una relación molar de aminoácido a linaclotida entre 100:1 y 10:1 y (iii) un catión metálico seleccionado de entre Ca2+, Mg2+ y Zn2+ en una relación molar de catión a linaclotida entre 100:1 y 40:1. En algunos casos, la composición comprende (i) entre 45 y 70 % en peso de un polímero seleccionado de entre PVP y PVA, (ii) un aminoácido seleccionado de entre leucina, isoleucina, alanina y metionina en una relación molar de aminoácido a linaclotida 100:1 y 30:1 (p. ej., entre 60:1 y 30:1) y (iii) un catión metálico seleccionado de entre Ca2+, Mg2+ y Zn2+ en una relación molar de catión a linaclotida entre 100:1 y 60:1. En algunos casos, la composición comprende (i) entre 45 y 99 % en peso (p. ej., entre 45 y 70 % en peso) de PVP o PVA, (ii) leucina en una relación molar de leucina a linaclotida entre 100:1 y 30:1 (p. ej., entre 60:1 y 30:1 o incluso entre 50:1 y 30:1) y (iii) Ca2+ en una relación molar de Ca2+ a linaclotida entre 100:1 y 60:1.
[0045] La composición de liberación retardada (p. ej., comprimido de liberación retardada) también puede comprender uno o más agentes de carga. Los agentes de carga adecuados incluyen, pero no se limitan a, almidón, carbonato de calcio, sulfato de calcio, hidroxilpropilmetilcelulosa, fructosa, metilcelulosa, dextratos, dextrosa, dextrano, lactitol, maltosa, sucrosa, sorbitol, isomalt, almidón pregelatinizado, fosfato dicálcico, celulosa microcristalina, manitol, gelatina, trehalosa, eritritol, maltitol, lactosa, glucosa o una combinación de los mismos, o una mezcla de los mismos. En algunos casos, el agente de carga es isomalt. En algunos casos, el agente de carga es gelatina. En algunos casos, el agente de carga es manitol. En algunos casos, el agente de carga es almidón pregelatinizado. En algunos casos, el agente de carga es celulosa microcristalina.
[0046] La composición de liberación retardada (p. ej., comprimido de liberación retardada) puede comprender cualquier concentración adecuada de agente de carga. En algunos casos, por ejemplo, la composición comprende uno o más agentes de carga en una concentración de 0,1-99 % en peso, con respecto al peso total de la composición. En algunos casos, por ejemplo, la composición comprende uno o más agentes de carga en una concentración de 1 95 % en peso del uno o más agentes de carga, con respecto al peso total de la composición. En algunos casos, por ejemplo, la composición comprende uno o más agentes de carga en una concentración de 10-90 % en peso del uno o más o agentes de carga, con respecto al peso total de la composición. En algunos casos, por ejemplo, la composición comprende uno o más agentes de carga en una concentración de 20-90 % en peso del uno o más o agentes de carga, con respecto al peso total de la composición. En algunos casos, por ejemplo, la composición comprende uno o más agentes de carga en una concentración de 25-85 % en peso del uno o más agentes de carga, con respecto al peso total de la composición. En algunos casos, por ejemplo, la composición comprende uno o más agentes de carga en una concentración de 30-80 % en peso del uno o más agentes de carga, con respecto al peso total de la composición. En algunos casos, por ejemplo, la composición comprende uno o más agentes de carga en una concentración de 40 70 % en peso del uno o más agentes de carga, con respecto al peso total de la composición. En algunos casos, por ejemplo, la composición comprende uno o más agentes de carga en una concentración de 10-60 % en peso del uno o más agentes de carga, con respecto al peso total de la composición. En algunos casos, por ejemplo, la composición comprende uno o más agentes de carga en una concentración de 20-50 % en peso del uno o más agentes de carga, con respecto al peso total de la composición. En algunos casos, la composición comprende uno o más agentes de carga en una concentración de al menos 20 % en peso, por ejemplo, al menos 40 % en peso, al menos 60 % en peso, al menos 70 % en peso, al menos 80 % en peso o al menos 90 % en peso, con respecto al peso total de la composición.
[0047] En algunos casos, la composición de liberación retardada (p. ej., película de liberación retardada) comprende uno o más plastificantes. Los plastificantes adecuados incluyen, pero no se limitan a, polietilenglicol, propilenglicol, glicerina, glicerol, monoacetina, diacetina, triacetina, ftalato de dimetilo, ftalato de dietilo, ftalato de dibutilo, sebacato de dibutilo, titrato de trietilo, citrato de tributilo, citrato de trietilo, citrato de trietilacetilo, aceite de ricino, monoglicéridos acetilados, sorbitol o combinaciones de los mismos. En casos ejemplares, la concentración del plastificante en la formulación puede ser de aproximadamente 0 a aproximadamente 30 % en peso, por ejemplo, aproximadamente 1 a aproximadamente 20% en peso, aproximadamente 0 a aproximadamente 10% en peso, aproximadamente 1 a aproximadamente 5 % en peso o incluso de aproximadamente 0 a aproximadamente 4 % en peso.
[0048] En algunos casos, la composición de liberación retardada comprende un agente formador de película, un polímero hidrosoluble, un polímero sensible al pH, un polímero biodegradable o una combinación de los mismos.
Los polímeros hidrosolubles, sensibles al pH o biodegradables que se pueden usar en las formulaciones de disolución por vía oral de la presente descripción incluyen, pero no se limitan a, derivados de celulosa, polímeros sintéticos, poliacrilatos y gomas naturales. Por ejemplo, los polímeros hidrosolubles usados en las formulaciones de disolución por vía oral de la presente descripción pueden incluir, pero no se limitan a, metilcelulosa, hidroxipropilcelulosa, hidroxipropilmetilcelulosa, etilcelulosa, hidroxietilcelulosa, hidroxipropilcelulosa, carboximetilcelulosa, acetato-ftalato de celulosa, acetato-butirato de celulosa, amilosa, dextrano, caseína, pululano, gelatina, pectina, agar, carragenano, goma xantana, tragacanto, goma guar, goma acacia, goma arábiga, polietilenglicol, óxido de polietileno, polivinilpirrolidona, alcohol polivinílico, ciclodextrina, polímeros de carboxivinilo, alginato de sodio, ácido poliacrílico, metacrilato de metilo o mezclas de los mismos. En casos ejemplares, la concentración del polímero hidrosoluble en la formulación puede ser de aproximadamente 20% a aproximadamente 90% (en peso), preferentemente entre aproximadamente 40 % y aproximadamente 80 % (en peso).
[0049] En algunos casos, el polímero sensible al pH es Eudragit® L100 que tiene un pH umbral de 6,0. En algunos casos, el polímero sensible al pH es Eudragit® S100 que tiene un pH umbral de 7,0. En algunos casos, el polímero sensible al pH es Eudragit® L-30D que tiene un pH umbral de 5,6. En algunos casos, el polímero sensible al pH es Eudragit® FS 30D que tiene un pH umbral de 6,8. En algunos casos, el polímero sensible al pH es Eudragit® L00-55 que tiene un pH umbral de 5,5. En algunos casos, el polímero sensible al pH es acetato-ftalato de polivinilo que tiene un pH umbral de 5,0. En algunos casos, el polímero sensible al pH es ftalato de hidroxipropilmetilcelulosa que tiene un pH umbral de 4,5-4,8. En algunos casos, el polímero sensible al pH es ftalato de hidroxipropilmetilcelulosa 50 que tiene un pH umbral de 5,2. En algunos casos, el polímero sensible al pH es ftalato de hidroxipropilmetilcelulosa 55 que tiene un pH umbral de 5,4. En algunos casos, el polímero sensible al pH es acetato-trimelitato de celulosa que tiene un pH umbral de 4,8. En algunos casos, el polímero sensible al pH es acetato-ftalato de celulosa que tiene un pH umbral de 5,0.
[0050] Un experto en la materia, con la ayuda de esta descripción, entenderá que se pueden incluir otros componentes para mejorar una o más propiedades de la composición de liberación retardada. En algunos casos, por ejemplo, las composiciones de liberación retardada pueden incluir uno o más disgregantes, lubricantes, aditivos antiaglomerantes, agentes antimicrobianos, agentes antiespumantes, emulsionantes, tensioactivos, agentes tamponadores y/o agentes colorantes.
[0051] Los disgregantes adecuados incluyen, por ejemplo, agar-agar, carbonato de calcio, celulosa microcristalina, croscarmelosa de sodio, crospovidona, povidona, polacrilin potasio, glicolato sódico de almidón, almidón de patata o tapioca, otros almidones, almidón pregelatinizado, arcillas, otras alginas, otras celulosas, gomas y mezclas de los mismos. En algunos casos, el disgregante es crospovidona. En algunos casos, el disgregante es croscarmelosa de sodio.
[0052] Los lubricantes adecuados incluyen, por ejemplo, estearato de calcio, estearato de magnesio, aceite mineral, aceite mineral ligero, glicerina, sorbitol, manitol, polietilenglicol, otros glicoles, ácido esteárico, laurilsulfato de sodio, talco, aceite vegetal hidrogenado (p. ej., aceite de cacahuete, aceite de semilla de algodón, aceite de girasol, aceite de sésamo, aceite de oliva, aceite de maíz y aceite de soja), estearato de zinc, oleato de etilo, laurato de etilo, agar, gel de sílice Syloid (AEROSIL® 200, W.R. Grace Co., Baltimore, MD, EE. UU.), un aerosol coagulado de sílice sintética (Evonik Degussa Co., Plano, TX, EE. UU.), un dióxido de silicio pirogénico (CAB-O-SIL, Cabot Co., Boston, MA, EE. UU.) y mezclas de los mismos.
[0053] Los aditivos antiaglomerantes adecuados incluyen, por ejemplo, silicato de calcio, silicato de magnesio, dióxido de silicio, dióxido de silicio coloidal, talco, glicerilo y mezclas de los mismos.
[0054] Los aditivos antimicrobianos adecuados que se pueden usar, p. ej., como conservante para las composiciones de linaclotida incluyen, por ejemplo, cloruro de benzalconio, cloruro de bencetonio, ácido benzoico, alcohol bencílico, butilparabeno, cloruro de cetilpiridinio, cresol, clorobutanol, ácido deshidroacético, etilparabeno, metilparabeno, fenol, alcohol feniletílico, fenoxietanol, acetato fenilmercúrico, nitrato fenilmercúrico, sorbato de potasio, propilparabeno, benzoato de sodio, deshidroacetato de sodio, propionato de sodio, ácido sórbico, timersol, timol y mezclas de los mismos.
[0055] La composición también puede comprender cualquier vehículo o medio farmacéuticamente aceptable adecuado. Los vehículos farmacéuticamente aceptables adecuados incluyen, por ejemplo, cualquier disolvente, dispersante, agente tamponador del pH, recubrimiento, agente potenciador de la absorción, agente de liberación controlada y uno o más excipientes inertes (p. ej., agentes de carga, almidones, polioles, agentes granulación, celulosa microcristalina, diluyentes, lubricantes, aglutinantes, agentes disgregantes) o similares. Además, las composiciones pueden contener cualquier componente, aditivo y/o especie adicional deseados, por ejemplo, aditivos tensioactivos, aditivos dispersantes, humectantes, agentes de suspensión, solubilizantes, agentes tamponadores, disgregantes, conservantes, colorantes, saborizantes y similares. En algunos casos, la composición comprende una o más especies iónicas que interactúan con la linaclotida.
[0056] La composición también puede comprender cualquier agente tamponador del pH adecuado. En algunos
casos, el agente tamponador del pH está presente en la composición en una cantidad suficiente para conseguir el punto isoeléctrico de la linaclotida. A este respecto, la composición puede tener cualquier pH deseado. En algunos casos, la composición tiene un pH de 2 a 5 (por ejemplo, un pH de 2 a 4,5, un pH de 2 a 4, un pH de 2,5 a 4, un pH de 2,5 a 3,5, un pH de 2,5 a 3 o incluso un pH de 3).
[0057] En algunos casos, la composición comprende linaclotida y un producto de la hidrólisis, p. ej., un producto de la hidrólisis que comprende o tiene una estructura de:

[0058] La composición puede contener cualquier concentración deseada del producto de la hidrólisis. En algunos casos, la composición comprende menos de 10 % en peso del producto de la hidrólisis. En algunos casos, la composición comprende menos de 7 % en peso del producto de la hidrólisis. En algunos casos, la composición comprende menos de 6 % en peso del producto de la hidrólisis. En algunos casos, la composición comprende menos de 5 % en peso del producto de la hidrólisis. En algunos casos, la composición comprende menos de 4 % en peso del producto de la hidrólisis. En algunos casos, la composición comprende menos de 3 % en peso del producto de la hidrólisis. En algunos casos, la composición comprende menos de 2 % en peso del producto de la hidrólisis. En algunos casos, la composición comprende menos de 1 % en peso del producto de la hidrólisis. En algunos casos, la composición comprende entre 0,01 y 10 % en peso del producto de la hidrólisis. En algunos casos, la composición comprende entre 0,1 y 7 % en peso del producto de la hidrólisis. En algunos casos, la composición comprende entre 0,1 y 5 % en peso del producto de la hidrólisis. En algunos casos, la composición comprende entre 0,5 y 5 % en peso del producto de la hidrólisis. En algunos casos, la composición comprende entre 1 y 5 % en peso del producto de la hidrólisis. En algunos casos, la composición comprende entre 0,1 y 4 % en peso del producto de la hidrólisis. En algunos casos, la composición comprende entre 0,5 y 4 % en peso del producto de la hidrólisis. En algunos casos, la composición comprende entre 1 y 4 % en peso del producto de la hidrólisis. En algunos casos, la composición comprende entre 0,1 y 3 % en peso del producto de la hidrólisis. En algunos casos, la composición comprende entre 0,5 y 3 % en peso del producto de la hidrólisis. En algunos casos, la composición comprende entre 1 y 3 % en peso del producto de la hidrólisis. En algunos casos, la composición comprende entre 0,1 y 2,5 % en peso del producto de la hidrólisis. En algunos casos, la composición comprende entre 0,5 y 2,5 % en peso del producto de la hidrólisis. En algunos casos, la composición comprende entre 1 y 2,5 % en peso del producto de la hidrólisis. En algunos casos, la composición comprende entre 0,1 y 2% en peso del producto de la hidrólisis. En algunos casos, la composición comprende entre 0,5 y 2 % en peso del producto de la hidrólisis. En algunos casos, la composición comprende entre 1 y 2 % en peso del producto de la hidrólisis. En algunos casos, la composición comprende entre 0,1 y 1,5 % en peso del producto de la hidrólisis. En algunos casos, la composición comprende entre 0,5 y 1,5 % en peso del producto de la hidrólisis. En algunos casos, la composición comprende entre 0,1 y 1 % en peso del producto de la hidrólisis. En algunos casos, la composición comprende entre 0,5 y 1 % en peso del producto de la hidrólisis.
[0059] En algunos casos, la composición comprende linaclotida y un producto de la oxidación, p. ej., un producto de la oxidación que comprende o tiene una estructura de:

[0060] Alternativamente, o además, la composición comprende linaclotida y un producto de la oxidación que tiene la estructura representada, pero donde la oxidación se produce en uno cualquiera o más de los seis azufres del cisteinilo representados. La composición puede contener cualquier concentración deseada del producto de la oxidación. En algunos casos, la composición comprende menos de 10 % en peso del producto de la oxidación. En algunos casos, la composición comprende menos de 7 % en peso del producto de la oxidación. En algunos casos, la composición comprende menos de 6 % en peso del producto de la oxidación. En algunos casos, la composición comprende menos de 5 % en peso del producto de la oxidación. En algunos casos, la composición comprende menos de 4 % en peso del producto de la oxidación. En algunos casos, la composición comprende menos de 3 % en peso del producto de la oxidación. En algunos casos, la composición comprende menos de 2 % en peso del producto de la oxidación. En algunos casos, la composición comprende menos de 1 % en peso del producto de la oxidación. En algunos casos, la composición comprende entre 0,01 y 10 % en peso del producto de la oxidación. En algunos casos, la composición comprende entre 0,1 y 7 % en peso del producto de la oxidación. En algunos casos, la composición comprende entre 0,1 y 5 % en peso del producto de la oxidación. En algunos casos, la composición comprende entre 0,5 y 5 % en peso del producto de la oxidación. En algunos casos, la composición comprende entre 1 y 5 % en peso
del producto de la oxidación. En algunos casos, la composición comprende entre 0,1 y 4 % en peso del producto de la oxidación. En algunos casos, la composición comprende entre 0,5 y 4 % en peso del producto de la oxidación. En algunos casos, la composición comprende entre 1 y 4 % en peso del producto de la oxidación. En algunos casos, la composición comprende entre 0,1 y 3 % en peso del producto de la oxidación. En algunos casos, la composición comprende entre 0,5 y 3 % en peso del producto de la oxidación. En algunos casos, la composición comprende entre 1 y 3 % en peso del producto de la oxidación. En algunos casos, la composición comprende entre 0,1 y 2,5 % en peso del producto de la oxidación. En algunos casos, la composición comprende entre 0,5 y 2,5 % en peso del producto de la oxidación. En algunos casos, la composición comprende entre 1 y 2,5 % en peso del producto de la oxidación. En algunos casos, la composición comprende entre 0,1 y 2 % en peso del producto de la oxidación. En algunos casos, la composición comprende entre 0,5 y 2% en peso del producto de la oxidación. En algunos casos, la composición comprende entre 1 y 2 % en peso del producto de la oxidación. En algunos casos, la composición comprende entre 0,1 y 1,5% en peso del producto de la oxidación. En algunos casos, la composición comprende entre 0,5 y 1,5% en peso del producto de la oxidación. En algunos casos, la composición comprende entre 0,1 y 1 % en peso del producto de la oxidación. En algunos casos, la composición comprende entre 0,5 y 1 % en peso del producto de la oxidación.
[0061] En algunos casos, la composición comprende linaclotida y un producto de la acetilación, p. ej. un producto de la acetilación que comprende o tiene:

[0062] La composición puede contener cualquier concentración deseada del producto de la acetilación. En algunos casos, la composición comprende menos de 10 % en peso del producto de la acetilación. En algunos casos, la composición comprende menos de 7 % en peso del producto de la acetilación. En algunos casos, la composición comprende menos de 6 % en peso del producto de la acetilación. En algunos casos, la composición comprende menos de 5 % en peso del producto de la acetilación. En algunos casos, la composición comprende menos de 4 % en peso del producto de la acetilación. En algunos casos, la composición comprende menos de 3 % en peso del producto de la acetilación. En algunos casos, la composición comprende menos de 2 % en peso del producto de la acetilación. En algunos casos, la composición comprende menos de 1 % en peso del producto de la acetilación. En algunos casos, la composición comprende entre 0,01 y 10 % en peso del producto de la acetilación. En algunos casos, la composición comprende entre 0,1 y 7 % en peso del producto de la acetilación. En algunos casos, la composición comprende entre 0,1 y 5 % en peso del producto de la acetilación. En algunos casos, la composición comprende entre 0,5 y 5 % en peso del producto de la acetilación. En algunos casos, la composición comprende entre 1 y 5 % en peso del producto de la acetilación. En algunos casos, la composición comprende entre 0,1 y 4 % en peso del producto de la acetilación. En algunos casos, la composición comprende entre 0,5 y 4 % en peso del producto de la acetilación. En algunos casos, la composición comprende entre 1 y 4 % en peso del producto de la acetilación. En algunos casos, la composición comprende entre 0,1 y 3 % en peso del producto de la acetilación. En algunos casos, la composición comprende entre 0,5 y 3 % en peso del producto de la acetilación. En algunos casos, la composición comprende entre 1 y 3% en peso del producto de la acetilación. En algunos casos, la composición comprende entre 0,1 y 2,5 % en peso del producto de la acetilación. En algunos casos, la composición comprende entre 0,5 y 2,5 % en peso del producto de la acetilación. En algunos casos, la composición comprende entre 1 y 2,5 % en peso del producto de la acetilación. En algunos casos, la composición comprende entre 0,1 y 2 % en peso del producto de la acetilación. En algunos casos, la composición comprende entre 0,5 y 2 % en peso del producto de la acetilación. En algunos casos, la composición comprende entre 1 y 2 % en peso del producto de la acetilación. En algunos casos, la composición comprende entre 0,1 y 1,5 % en peso del producto de la acetilación. En algunos casos, la composición comprende entre 0,5 y 1,5 % en peso del producto de la acetilación. En algunos casos, la composición comprende entre 0,1 y 1 % en peso del producto de la acetilación. En algunos casos, la composición comprende entre 0,5 y 1 % en peso del producto de la acetilación.
[0063] En algunos casos, se proporciona una composición farmacéutica que comprende linaclotida y uno o más péptidos seleccionados de entre:
i. un péptido (“Cys1-IMD”) o una sal farmacéuticamente aceptable del mismo, donde el péptido comprende la estructura de aminoácidos de:

ii. un péptido de hidrólisis (“Asp7”) o una sal farmacéuticamente aceptable del mismo, donde el péptido comprende la estructura de aminoácidos de:

iii. un péptido de acetilación (Cys1-N-acetilo) o una sal farmacéuticamente aceptable del mismo, donde el péptido comprende la estructura de aminoácidos de:

iv. un péptido de trisulfuro de linaclotida o una sal farmacéuticamente aceptable del mismo, donde el péptido comprende la secuencia de aminoácidos de Cys Cys Glu Tyr Cys Cys Asn Pro Ala Cys Thr Gly Cys Tyr donde se puede fijar un átomo de azufre adicional a uno cualquiera de los seis azufres del cisteinilo;
v. un péptido (“Des-Tyr14”) o una sal farmacéuticamente aceptable del mismo, donde el péptido comprende la estructura de aminoácidos de:

o
vi. un péptido (Cys1-a-cetona) o una sal farmacéuticamente aceptable del mismo, donde el péptido comprende la estructura de aminoácidos de:

[0064] En algunos casos, el péptido Cys1-a-cetona puede estar presente en su forma hidratada o una sal farmacéuticamente aceptable de la misma, donde el péptido comprende una estructura de aminoácidos de:

[0065] Un experto en la materia reconocerá que el péptido Cys1-a-cetona se convertirá fácilmente entre su forma hidratada y de cetona.
[0066] En algunos casos, el péptido Cys1-a-cetona comprende menos de aproximadamente 15 % en peso de la composición, menos de aproximadamente 10 % en peso de la composición, menos de aproximadamente 7 % en peso de la composición, menos de aproximadamente 5 % en peso de la composición, menos de aproximadamente 4 % en peso de la composición, menos de aproximadamente 3 % en peso de la composición, menos de aproximadamente 2 % en peso de la composición, menos de aproximadamente 1,5 % en peso de la composición o menos de aproximadamente 1 % en peso de la composición. En otros ejemplos de casos, el péptido Cys1-a-cetona comprende de aproximadamente 0,01 % a aproximadamente 15% en peso de la composición, aproximadamente 0,05 % a aproximadamente 10 % en peso de la composición, aproximadamente 0,05 % a aproximadamente 7 % en peso de la composición o aproximadamente 0,05 % a aproximadamente 5 % en peso de la composición.
[0067] En algunos casos, el péptido Cys1-IMD comprende menos de aproximadamente 15 % en peso de la composición, menos de aproximadamente 10 % en peso de la composición, menos de aproximadamente 7 % en peso de la composición, menos de aproximadamente 5 % en peso de la composición, menos de aproximadamente 4 % en peso de la composición, menos de aproximadamente 3,5 % en peso de la composición, menos de aproximadamente 3 % en peso de la composición, menos de aproximadamente 2 % en peso de la composición o menos de aproximadamente 1 % en peso de la composición. En otros ejemplos, el péptido Cys1-IMD comprende de aproximadamente 0,01% a aproximadamente 15% en peso de la composición, aproximadamente 0,05% a aproximadamente 10 % en peso de la composición, aproximadamente 0,05 % a aproximadamente 7 % en peso de la composición o aproximadamente 0,05 % a aproximadamente 5 % en peso de la composición.
[0068] En algunos casos, el péptido de hidrólisis (“Asp7”) comprende menos de aproximadamente 15% en peso de la composición, menos de aproximadamente 10 % en peso de la composición, menos de aproximadamente 7 % en peso de la composición, menos de aproximadamente 5 % en peso de la composición, menos de aproximadamente 4 % en peso de la composición, menos de aproximadamente 3,5 % en peso de la composición, menos de aproximadamente 3 % en peso de la composición, menos de aproximadamente 2 % en peso de la composición o menos de aproximadamente 1 % en peso de la composición. En otros casos ejemplares, el péptido de hidrólisis (“Asp7”) comprende de aproximadamente 0,01 % a aproximadamente 15% en peso de la composición, aproximadamente 0,05% a aproximadamente 10% en peso de la composición, aproximadamente 0,05% a aproximadamente 7 % en peso de la composición o aproximadamente 0,05 % a aproximadamente 5 % en peso de la composición.
[0069] En algunos casos, el péptido de acetilación (“Cys1-N-acetilo”) comprende menos de aproximadamente 15% en peso de la composición, menos de aproximadamente 10% en peso de la composición, menos de aproximadamente 7 % en peso de la composición, menos de aproximadamente 5 % en peso de la composición, menos de aproximadamente 4 % en peso de la composición, menos de aproximadamente 3,5 % en peso de la composición, menos de aproximadamente 3 % en peso de la composición, menos de aproximadamente 2 % en peso de la composición o menos de aproximadamente 1 % en peso de la composición. En otros casos ejemplares, el péptido de acetilación (“Cys1-N-acetilo”) comprende de aproximadamente 0,01 % a aproximadamente 15% en peso de la composición, aproximadamente 0,05% a aproximadamente 10% en peso de la composición, aproximadamente 0,05 % a aproximadamente 7 % en peso de la composición o aproximadamente 0,05 % a aproximadamente 5 % en peso de la composición.
[0070] En algunos casos, el péptido de trisulfuro de linaclotida comprende menos de aproximadamente 15 % en peso de la composición, menos de aproximadamente 10 % en peso de la composición, menos de aproximadamente 7 % en peso de la composición, menos de aproximadamente 5 % en peso de la composición, menos de aproximadamente 4 % en peso de la composición, menos de aproximadamente 3,5 % en peso de la composición, menos de aproximadamente 3 % en peso de la composición, menos de aproximadamente 2 % en peso de la composición o menos de aproximadamente 1 % en peso de la composición. En otros casos ejemplares, el péptido de trisulfuro de linaclotida comprende de aproximadamente 0,01 % a aproximadamente 15 % en peso de la composición, aproximadamente 0,05% a aproximadamente 10% en peso de la composición, aproximadamente 0,05% a aproximadamente 7 % en peso de la composición o aproximadamente 0,05 % a aproximadamente 5 % en peso de la composición.
[0071] En algunos casos, el péptido Des-Tyr14 comprende menos de aproximadamente 15 % en peso de la composición, menos de aproximadamente 10 % en peso de la composición, menos de aproximadamente 7 % en peso de la composición, menos de aproximadamente 5 % en peso de la composición, menos de aproximadamente 4 % en peso de la composición, menos de aproximadamente 3,5 % en peso de la composición, menos de aproximadamente 3 % en peso de la composición, menos de aproximadamente 2 % en peso de la composición o menos de aproximadamente 1 % en peso de la composición. En otros casos ejemplares, el péptido Des-Tyr14 comprende de aproximadamente 0,01% a aproximadamente 15% en peso de la composición, aproximadamente 0,05% a aproximadamente 10 % en peso de la composición, aproximadamente 0,05 % a aproximadamente 7 % en peso de la composición o aproximadamente 0,05 % a aproximadamente 5 % en peso de la composición.
[0072] En algunos casos, la composición comprende linaclotida y cualquier concentración deseada de multímeros. En algunos casos, la composición comprende menos de 10% en peso de uno o más multímeros. En algunos casos, la composición comprende entre 0,5 y 1 % en peso de uno o más multímeros.
[0073] En algunos casos, la composición comprende una cantidad eficaz de linaclotida y cualquier cantidad deseada de linaclotida en forma reducida. Como se emplea en esta memoria, el término “linaclotida en forma reducida” se refiere a linaclotida que no tiene enlaces disulfuro entre los aminoácidos de cisteína. En algunos casos, la composición comprende menos de 10 % en peso de linaclotida en forma reducida. En algunos casos, la composición comprende entre 0,5 y 1 % en peso de linaclotida en forma reducida.
[0074] En algunos casos, la composición comprende una cantidad eficaz de linaclotida y cualquier cantidad deseada de linaclotida en forma aleatorizada. Como se emplea en esta memoria, el término “linaclotida en forma aleatorizada” se refiere a linaclotida que tiene enlaces disulfuro entre Cys1 y Cys-iü, entre Cys1 y Cys-i3, entre Cys1 y Cys5, entre Cys1 y Cys2, entre Cys2 y Cys6, entre Cys2 y Cys-i3, entre Cys2 y Cys5, entre Cys5 y Cys6 y/o entre Cys5 y Cysm En algunos casos, la composición comprende entre 0,5 y 1 % en peso de linaclotida en forma aleatorizada. En algunos casos, la composición comprende menos de 10 % en peso de linaclotida en forma aleatorizada.
[0075] En algunos casos, la composición comprende una concentración total de degradante inferior a aproximadamente 10 % en peso. En algunos casos, la composición comprende una concentración total de degradante inferior a aproximadamente 8 % en peso. En algunos casos, la composición comprende una concentración total de degradante inferior a aproximadamente 7 % en peso. En algunos casos, la composición comprende una concentración total de degradante inferior a aproximadamente 6,5 % en peso. En algunos casos, la composición comprende una concentración total de degradante inferior a aproximadamente 6 % en peso. En algunos casos, la composición comprende una concentración total de degradante inferior a aproximadamente 5,5 % en peso. En algunos casos, la composición comprende una concentración total de degradante inferior a aproximadamente 5 % en peso. En algunos casos, la composición comprende una concentración total de degradante inferior a aproximadamente 4 % en peso. En algunos casos, la composición comprende una concentración total de degradante inferior a aproximadamente 3 % en peso. En algunos casos, la composición comprende una concentración total de degradante inferior a aproximadamente 2,5 % en peso. En algunos casos, la composición comprende una concentración total de degradante inferior a aproximadamente 2 % en peso. En algunos casos, la composición comprende una concentración total de degradante inferior a aproximadamente 1 % en peso.
[0076] En algunos casos, las composiciones se pueden preparar mediante secado por pulverización, que es una técnica usada para preparar micropartículas (p. ej., microcápsulas o microesferas) de fármacos. Los péptidos secados por pulverización generalmente conservan su actividad biológica tras la disolución y pueden tener características físicas útiles, que incluyen un tamaño de las partículas uniforme y una forma esférica. Además, las micropartículas preparadas mediante secado por pulverización a menudo fluyen libremente, lo que es útil para procedimientos de fabricación farmacéuticos tales como el moldeo de comprimidos y el encapsulamiento en cápsulas. Los procedimientos de secado por pulverización también son útiles porque se pueden utilizar fácilmente a gran escala para la fabricación clínica y comercial. En un caso, el tampón de pulverización comprende HCl, histidina, 1,5% de PVA y 0,6 % de talco. Esta formulación se puede usar para producir intervalos de administración inferiores, entre 36 290 |jg.
[0077] La composición, cuando se administra, se disolverá para liberar linaclotida en áreas objetivo del tracto gastrointestinal. La formulación puede liberar la linaclotida durante un período de tiempo que se determina mediante varios factores diferentes. Estos factores incluyen las dimensiones de la formulación, la concentración de la linaclotida y cómo se dispersa la linaclotida en la formulación. Por ejemplo, variando el espesor y la superficie de las formulaciones, se puede ajustar la velocidad de disolución. Una formulación espesa se disolverá más lentamente que una formulación fina similar y puede ser deseable administrar dosis altas de linaclotida.
[0078] En algunos casos, la composición de liberación retardada tiene una velocidad de disgregación inferior a aproximadamente 60 minutos en las condiciones de pH objetivo. En algunos casos, la composición de liberación retardada tiene una velocidad de disgregación inferior a aproximadamente 30 minutos en las condiciones de pH objetivo. En algunos casos, la composición de liberación retardada tiene una velocidad de disgregación inferior a
aproximadamente 25 minutos. En algunos casos, la composición de liberación retardada tiene una velocidad de disgregación inferior a aproximadamente 20 minutos. En algunos casos, la composición de liberación retardada tiene una velocidad de disgregación inferior a aproximadamente 15 minutos. En algunos casos, la composición de liberación retardada tiene una velocidad de disgregación inferior a aproximadamente 10 minutos. En algunos casos, la composición de liberación retardada se disgrega en menos de aproximadamente 30 minutos después de entrar a un entorno objetivo. En algunos casos, la composición de liberación retardada se disgrega en menos de aproximadamente 25 minutos después de entrar a un entorno objetivo. En algunos casos, la composición de liberación retardada se disgrega en menos de aproximadamente 20 minutos después de entrar a un entorno objetivo. En algunos casos, la composición de liberación retardada se disgrega en menos de aproximadamente 15 minutos después de entrar a un entorno objetivo.
[0079] En algunos casos, la composición de liberación retardada libera al menos aproximadamente 75 % de la linaclotida contenida en la misma en un plazo de 60 minutos tras entrar en un entorno objetivo. En algunos casos, la composición de liberación retardada libera al menos aproximadamente 75 % de la linaclotida contenida en la misma en un plazo de 30 minutos tras entrar en un entorno objetivo. En algunos casos, la composición de liberación retardada libera al menos aproximadamente 80 % de la linaclotida contenida en la misma en un plazo de 30 minutos tras entrar en un entorno objetivo. En algunos casos, la composición de liberación retardada libera al menos aproximadamente 85 % de la linaclotida contenida en la misma en un plazo de 30 minutos tras entrar en un entorno objetivo. En algunos casos, la composición de liberación retardada libera al menos aproximadamente 90 % de la linaclotida contenida en la misma en un plazo de 30 minutos tras entrar en un entorno objetivo. En algunos casos, la composición de liberación retardada libera al menos aproximadamente 95 % de la linaclotida contenida en la misma en un plazo de 30 minutos tras entrar en un entorno objetivo. En algunos casos, la composición de liberación retardada libera al menos aproximadamente 99 % de la linaclotida contenida en la misma en un plazo de 30 minutos tras entrar en un entorno objetivo.
[0080] En algunos casos, la composición de liberación retardada libera al menos aproximadamente 40 % de la linaclotida contenida en la misma en un plazo de 15 minutos tras entrar en un entorno objetivo. En algunos casos, la composición de liberación retardada libera al menos aproximadamente 50 % de la linaclotida contenida en la misma en un plazo de 15 minutos tras entrar en un entorno objetivo. En algunos casos, la composición de liberación retardada libera al menos aproximadamente 60 % de la linaclotida contenida en la misma en un plazo de 15 minutos tras entrar en un entorno objetivo. En algunos casos, la composición de liberación retardada libera al menos aproximadamente 70 % de la linaclotida contenida en la misma en un plazo de 15 minutos tras entrar en un entorno objetivo. En algunos casos, la composición de liberación retardada libera al menos aproximadamente 80 % de la linaclotida contenida en la misma en un plazo de 15 minutos tras entrar en un entorno objetivo. En algunos casos, la composición de liberación retardada libera al menos aproximadamente 85 % de la linaclotida contenida en la misma en un plazo de 15 minutos tras entrar en un entorno objetivo. En algunos casos, la composición de liberación retardada libera al menos aproximadamente 90 % de la linaclotida contenida en la misma en un plazo de 15 minutos tras entrar en un entorno objetivo. En algunos casos, la composición de liberación retardada libera al menos aproximadamente 95 % de la linaclotida contenida en la misma en un plazo de 15 minutos tras entrar en un entorno objetivo.
[0081] En algunos casos, la composición de liberación retardada libera al menos aproximadamente 80 % de la linaclotida contenida en la misma entre aproximadamente 2 y aproximadamente 2 horas tras entrar en un entorno objetivo.
[0082] En algunos casos, la composición de liberación retardada libera al menos aproximadamente 75 % de la linaclotida contenida en la misma en un plazo de 30 minutos tras entrar en contacto con un pH superior a 5. En algunos casos, la composición de liberación retardada libera al menos aproximadamente 80 % de la linaclotida contenida en la misma en un plazo de 30 minutos tras entrar en contacto con un pH superior a 5. En algunos casos, la composición de liberación retardada libera al menos aproximadamente 85 % de la linaclotida contenida en la misma en un plazo de 30 minutos tras entrar en contacto con un pH superior a 5. En algunos casos, la composición de liberación retardada libera al menos aproximadamente 90 % de la linaclotida contenida en la misma en un plazo de 30 minutos tras entrar en contacto con un pH superior a 5. En algunos casos, la composición de liberación retardada libera al menos aproximadamente 95 % de la linaclotida contenida en la misma en un plazo de 30 minutos tras entrar en contacto con un pH superior a 5. En algunos casos, la composición de liberación retardada libera al menos aproximadamente 99 % de la linaclotida contenida en la misma en un plazo de 30 minutos tras entrar en contacto con un pH superior a 5.
[0083] En algunos casos, la composición de liberación retardada libera al menos aproximadamente 75 % de la linaclotida contenida en la misma en un plazo de 30 minutos tras entrar en contacto con un pH superior a 7. En algunos casos, la composición de liberación retardada libera al menos aproximadamente 80 % de la linaclotida contenida en la misma en un plazo de 30 minutos tras entrar en contacto con un pH superior a 7. En algunos casos, la composición de liberación retardada libera al menos aproximadamente 85 % de la linaclotida contenida en la misma en un plazo de 30 minutos tras entrar en contacto con un pH superior a 7. En algunos casos, la composición de liberación retardada libera al menos aproximadamente 90 % de la linaclotida contenida en la misma en un plazo de 30 minutos tras entrar en contacto con un pH superior a 7. En algunos casos, la composición de liberación retardada libera al menos aproximadamente 95 % de la linaclotida contenida en la misma en un plazo de 30 minutos tras entrar en contacto con
un pH superior a 7. En algunos casos, la composición de liberación retardada libera al menos aproximadamente 99 % de la linaclotida contenida en la misma en un plazo de 30 minutos tras entrar en contacto con un pH superior a 7.
[0084] En algunos casos, las composiciones de linaclotida se pueden formular combinando composiciones LI y LR de linaclotida en una forma farmacéutica. Por ejemplo, se puede fabricar una cápsula LR de linaclotida encapsulando perlas LI y perlas LR en una cápsula. En función del requisito para el perfil de liberación del fármaco, la cantidad de perlas LI y l R en tal cápsula puede variar de 1 a 99 % (% en peso). En algunos casos, por ejemplo, la cápsula LR comprende al menos aproximadamente 50 % de perlas LR con respecto a las perlas LI. En algunos casos, la cápsula comprende a menos aproximadamente 55 % de perlas LR con respecto a las perlas LI. En algunos casos, la cápsula comprende a menos aproximadamente 60 % de perlas LR con respecto a las perlas LI. En algunos casos, la cápsula comprende a menos aproximadamente 65 % de perlas LR con respecto a las perlas LI. En algunos casos, la cápsula comprende a menos aproximadamente 70 % de perlas LR con respecto a las perlas LI. En algunos casos, la cápsula comprende a menos aproximadamente 75 % de perlas LR con respecto a las perlas LI. En algunos casos, la cápsula comprende a menos aproximadamente 80 % de perlas LR con respecto a las perlas LI. En algunos casos, la cápsula comprende a menos aproximadamente 85 % de perlas LR con respecto a las perlas LI. En algunos casos, la cápsula comprende a menos aproximadamente 90 % de perlas LR con respecto a las perlas LI. En algunos casos, la cápsula comprende a menos aproximadamente 92 % de perlas LR con respecto a las perlas LI. En algunos casos, la cápsula comprende a menos aproximadamente 94 % de perlas LR con respecto a las perlas LI. En algunos casos, la cápsula comprende a menos aproximadamente 96 % de perlas LR con respecto a las perlas LI. En algunos casos, la cápsula comprende a menos aproximadamente 98 % de perlas LR con respecto a las perlas LI.
[0085] En algunos casos, las composiciones LR de linaclotida se formulan para la administración de linaclotida al íleon, íleon terminal o colon.
[0086] En algunos casos, las composiciones LR de linaclotida se formulan para la administración de linaclotida al colon.
[0087] La composición se puede usar para tratar otras enfermedades, trastornos o afecciones que son sensibles al tratamiento con agonistas del receptor de la GC-C. La composición se puede usar para tratar cualquier trastorno y/o afección gastrointestinal en un paciente (p. ej., mamífero o ser humano) o la inflamación o el dolor asociados a los mismos. Las composiciones de liberación retardada de la presente descripción se pueden usar para tratar diversas afecciones, pero son particularmente adecuadas para tratar trastornos gastrointestinales, tales como síndrome del intestino irritable (“SII”) (por ejemplo, SII con estreñimiento “SII-e”, SII con diarrea “SII-d” o SII mixto, con estreñimiento y diarrea, “SII-m”), estreñimiento (por ejemplo, estreñimiento idiopático crónico), cáncer de colon, diverticulitis, cistitis intersticial y dolor abdominal o visceral.
[0088] Tales trastornos y afecciones gastrointestinales adecuados también incluyen, pero no se limitan a, síndrome del intestino irritable, síndrome del intestino irritable con estreñimiento, cáncer de colon, dispepsia (que incluye dispepsia funcional o dispepsia no ulcerosa), trastornos de la motilidad gastrointestinal, trastornos gastrointestinales funcionales, enfermedad por reflujo gastroesofágico (ERGE), enfermedad de Crohn, colitis ulcerosa, enfermedad inflamatoria intestinal, ardor gástrico funcional, gastroparesia, seudoobstrucción intestinal crónica (o seudoobstrucción colónica) y trastornos y afecciones asociados al estreñimiento, por ejemplo, estreñimiento crónico, estreñimiento inducido por opioides, estreñimiento posquirúrgico (íleo posoperatorio) y estreñimiento asociado a trastornos neuropáticos o una combinación de síntomas de los mismos (tal como una combinación de síndrome del intestino irritable y estreñimiento crónico), estreñimiento asociado a trastornos neuropáticos (p. ej., estreñimiento asociado a enfermedad de Parkinson), estreñimiento asociado a fibrosis quística o enfermedad tiroidea. En algunos casos, se proporciona un método para tratar trastornos gastrointestinales en un paciente (p. ej., mamífero o ser humano) diagnosticado con uno o más trastornos o afecciones gastrointestinales, donde el método comprende administrar una cantidad eficaz de la composición al paciente.
[0089] En algunos casos, se proporciona un método para tratar trastornos gastrointestinales que comprende administrar a un paciente con necesidad de la misma una cantidad terapéuticamente eficaz de composiciones descritas en esta memoria. El trastorno gastrointestinal se selecciona del grupo que consiste en: síndrome del intestino irritable (SII), estreñimiento, un trastorno gastrointestinal funcional, enfermedad por reflujo gastroesofágico, ardor gástrico funcional, enfermedad inflamatoria intestinal, dispepsia, dolor por diverticulitis, dolor visceral o dolor abdominal, prostatitis, dolor testicular, inflamación abdominal o visceral, síndrome de vejiga dolorosa, endometriosis, vulvodinia o dolor rectal. En algunos casos, el estreñimiento es estreñimiento crónico, estreñimiento idiopático, estreñimiento idiopático crónico, estreñimiento debido a íleo posoperatorio o estreñimiento provocado por el uso de opiáceos. En otros casos, el síndrome del intestino irritable es síndrome del intestino irritable con estreñimiento (SII-e), síndrome del intestino irritable con diarrea (SII-d) o síndrome del intestino irritable mixto (SII-m).
[0090] En algunos casos, se proporciona un método para tratar un trastorno que comprende administrar a un paciente con necesidad de la misma una cantidad terapéuticamente eficaz de la composición descrita en esta memoria. En algunos casos, el trastorno es cáncer seleccionado de entre cáncer colorrectal/colorrectal con metástasis local, pólipos intestinales, esófago de Barrett, cáncer del tracto gastrointestinal, cáncer de pulmón, cáncer o
crecimientos precancerosos o crecimientos metastásicos de células epiteliales, pólipos, carcinoma de mama, colorrectal, de pulmón, ovario, páncreas, próstata, riñón, estómago, vejiga, hígado, esofágico y testicular.
[0091] En algunos casos, se proporcionan métodos para prevenir un cáncer o hiperplasia del tracto gastrointestinal o prevenir la reaparición de cáncer o hiperplasia del tracto gastrointestinal en un paciente con necesidad de lo mismo que comprenden administrar una cantidad eficaz de la composición o la forma farmacéutica oral al paciente. En algunos casos, el cáncer o hiperplasia es cáncer colorrectal, pólipos intestinales o crecimientos precancerosos o crecimientos metastásicos de células epiteliales gastrointestinales. En algunos casos, la composición o forma farmacéutica oral se administra simultánea o secuencialmente con una cantidad eficaz de un inhibidor de la COX-2. Los ejemplos de inhibidores de la COX-2 muy selectivos y selectivos incluyen etoricoxib, rofecoxib, lumiracoxib, valdecoxib, celecoxib (Celebrex®), sulindaco, diclofenaco, meloxicam y etodolaco. Los AINE no selectivos que inhiben la COX-2 incluyen naproxeno, ibuprofeno, salicilato de sodio y diflunisal. Como se emplea en esta memoria, los términos “prevenir” o “prevención” significan detener, retardar la aparición (es decir, el período anterior a la manifestación clínica de una enfermedad) o la reaparición de cáncer o hiperplasia y/o reducir el riesgo de desarrollar cáncer o hiperplasia en comparación con un paciente que no ha sido tratado con una composición descrita en esta memoria.
[0092] En algunos casos, se proporcionan métodos para tratar trastornos gastrointestinales en pacientes pediátricos con las composiciones y formas farmacéuticas orales descritas en esta memoria. En algunos casos, se proporcionan métodos para tratar trastornos gastrointestinales en un paciente pediátrico diagnosticado con uno o más trastornos o afecciones gastrointestinales, donde el método comprende administrar una cantidad eficaz de la composición o la forma farmacéutica oral al paciente. En algunos casos, se proporcionan métodos para usar las composiciones y formas farmacéuticas orales para tratar trastornos gastrointestinales que incluyen, pero no se limitan a, trastornos de la motilidad GI, síndrome del intestino irritable, síndrome del intestino irritable con predominio de estreñimiento (SII-e), síndrome del intestino irritable de tipo mixto (SII-m), estreñimiento crónico, estreñimiento idiopático crónico, estreñimiento inducido por opioides, estreñimiento posquirúrgico (íleo posoperatorio), estreñimiento asociado a trastornos neuropáticos, estreñimiento asociado a fibrosis quística o enfermedad tiroidea, dispepsia (que incluye dispepsia funcional o dispepsia no ulcerosa), gastroparesia, trastornos de la motilidad gastrointestinal, trastornos gastrointestinales funcionales, enfermedad por reflujo gastroesofágico (ERGE), enfermedad inflamatoria intestinal, enfermedad de Crohn, colitis ulcerosa, ardor gástrico funcional, seudoobstrucción intestinal crónica (o seudoobstrucción colónica), dolor visceral, dolor abdominal, dolor pélvico, dolor por fisura anal, dolor asociado a vulvodinia, dolor asociado a endometriosis, dolor asociado a fibromialgia, dolor abdominal funcional, dolor por cistitis intersticial, diverticulitis, dolor asociado a diverticulitis y dolor asociado a esprúe celíaco. En algunos casos, se proporcionan métodos para tratar el SII-e, SII-m o el estreñimiento crónico (p. ej., estreñimiento idiopático crónico) en pacientes pediátricos con las composiciones y formas farmacéuticas orales descritas en esta memoria. En algunos casos, se proporcionan métodos para tratar el SII-e en un paciente pediátrico con necesidad de lo mismo. En algunos casos, se proporcionan métodos para tratar el estreñimiento idiopático crónico en un paciente pediátrico con necesidad de lo mismo.
[0093] En algunos casos, un método es para tratar o aliviar el dolor, que comprende administrar a un paciente con necesidad de la misma una cantidad terapéuticamente eficaz de la composición descrita en esta memoria. En algunos casos, el dolor se selecciona de entre dolor visceral; dolor abdominal; dolor pélvico; o dolor asociado a trastornos gastrointestinales, enfermedades venéreas, síndrome de vejiga dolorosa, dolor por diverticulitis, prostatitis, dolor testicular, endometriosis, vulvodinia, dolor rectal o cistitis intersticial. En algunas realizaciones, el dolor se selecciona de entre dolor pélvico, dolor asociado a proctitis, dolor por fisura anal, dolor asociado a vulvodinia, dolor asociado a endometriosis, dolor asociado a fibromialgia, dolor abdominal funcional, dolor por cistitis intersticial, dolor asociado a enfermedad venérea, diverticulitis, dolor asociado a diverticulitis y dolor asociado a esprúe celíaco.
[0094] En otro caso, el método es para aumentar la motilidad intestinal en un paciente con necesidad de lo mismo, que comprende administrar una cantidad eficaz de la composición al paciente. La motilidad intestinal implica distensiones y contracciones espontáneas coordinadas del estómago, los intestinos, el colon y el recto para mover los alimentos a través del tracto gastrointestinal durante el proceso digestivo. En algunos casos, el trastorno es íleo posoperatorio o estreñimiento provocado por el uso de opiáceos.
[0095] En casos ejemplares, los métodos pueden comprender administrar una cantidad terapéuticamente eficaz de la composición farmacéutica a un paciente con necesidad de la misma. Una cantidad eficaz de una composición que comprende linaclotida o una sal farmacéuticamente aceptable de la misma necesaria para conseguir los resultados deseados (tales como el tratamiento y/o alivio deseados de los síntomas) de un sujeto depende de varios factores conocidos, tales como la identidad y gravedad del trastorno que se está tratando, así como la edad, el peso, etc., del paciente que se está tratando.
[0096] En algunos casos, la composición o forma farmacéutica oral se administra a un paciente pediátrico con necesidad de la misma como un comprimido, una cápsula o un sobre. En algunos casos, se abre un sobre que comprende la composición y el contenido se espolvorea sobre, o se agita en, alimentos, tales como compota de manzana, o en una bebida, tal como agua. En algunos casos, se traga una cápsula entera con líquido, tal como agua,
o se abre y se esparce sobre, o se agitar en, alimentos o una bebida. Los comprimidos se pueden tragar enteros, triturar y agitar en alimentos o una bebida, o se pueden formular como un comprimido masticable.
[0097] Un sujeto o paciente en el que la administración de la composición farmacéutica es una pauta terapéutica eficaz para una enfermedad o un trastorno es preferentemente un ser humano, pero puede ser cualquier animal, lo que incluye un animal de laboratorio en el contexto de un ensayo clínico o un experimento de cribado o actividad. Por tanto, como puede apreciar fácilmente un experto en la materia, los métodos, los compuestos y las composiciones descritos en esta memoria son particularmente adecuados para la administración a cualquier animal, particularmente un mamífero, lo que incluye, pero no se limita a, seres humanos, roedores y no roedores, tales como sujetos felinos o caninos, animales de granja, tales como, pero no limitados a, sujetos bovinos, equinos, caprinos, ovinos y porcinos, animales salvajes (ya sea en libertad o en un zoológico), animales de investigación, tales como ratones, ratas, conejos, cabras, ovejas, cerdos, perros, gatos, etc., especies aviares, tales como pollos, pavos, pájaros cantores, etc., p. ej., para uso médico veterinario.
[0098] En algunos casos, la composición de linaclotida se puede formular como una forma farmacéutica rectal para administración rectal. Las formas farmacéuticas rectales incluyen, sin limitación, supositorios rectales, espumas o aerosoles rectales, enemas, geles rectales y pomadas rectales. En algunos casos, la forma farmacéutica rectal se puede administrar a un paciente con necesidad de la misma. En algunos casos, la forma farmacéutica rectal se puede administrar a un paciente para tratar dolor abdominal o dolor rectal, dolor por fisuras anales, colitis ulcerosa, enfermedad de Crohn o enfermedad inflamatoria intestinal. En algunos casos, la forma farmacéutica rectal se puede administrar a un paciente pediátrico o geriátrico.
[0099] En algunos casos, el intervalo de dosis eficaces de linaclotida para seres humanos adultos es de 25 |jg a 6 mg al día por vía oral. En algunos casos, el intervalo de dosis es 15 jg a 2 mg al día por vía oral. En algunos casos, el intervalo de dosis para seres humanos adultos es 50 jg a 1 mg al día por vía oral (por ejemplo, 15 jg , 36 jg , 50 jg , 72 jg , 100 jg , 145 jg , 150 jg , 200 jg , 250 jg , 290 jg , 300 jg , 350 jg , 400 jg , 450 jg , 500 jg , 550 jg , 579 jg , 600 jg , 650 jg , 700 jg , 750 jg , 800 jg , 850 jg , 900 jg , 950 jg o 1 mg). En algunos casos, el intervalo de dosis para seres humanos adultos es 36 jg a 290 jg al día por vía oral. En algunos casos, el intervalo de dosis es 100 jg a 600 jg al día por vía oral. En algunos casos, la dosis es 50 jg , 100 jg , 150 jg , 200 jg , 300 jg , 400 jg , 500 jg o 600 jg de linaclotida al día por vía oral. En algunos casos, la dosis es 50 jg de linaclotida al día por vía oral. En algunos casos, la dosis es 100 jg de linaclotida al día por vía oral. En algunos casos, la dosis es 145 jg de linaclotida al día por vía oral. En algunos casos, la dosis es 200 jg de linaclotida al día por vía oral. En algunos casos, la dosis es 290 jg de linaclotida al día por vía oral. En algunos casos, la dosis es 400 jg de linaclotida al día por vía oral. En algunos casos, la dosis es 500 jg de linaclotida al día por vía oral. En algunos casos, la dosis es 600 jg de linaclotida al día por vía oral.
[0100] En algunos casos, el intervalo de dosis pediátricas eficaces de linaclotida es de 0,05 jg a 2 mg al día por vía oral. En algunos casos, el intervalo de dosis pediátricas eficaces de linaclotida es de 0,05 mg a 100 jg al día por vía oral. En algunos casos, el intervalo de dosis pediátricas eficaces de linaclotida es de 0,1 mg a 90 jg al día por vía oral. En algunos casos, el intervalo de dosis pediátricas eficaces de linaclotida es de 0,1 mg a 50 jg al día por vía oral. En algunos casos, el intervalo de dosis pediátricas eficaces de linaclotida es de 0,1 mg a 25 jg al día por vía oral. En algunos casos, el intervalo de dosis pediátricas eficaces de linaclotida es de 0,1 mg a 10 jg al día por vía oral. En algunos casos, el intervalo de dosis pediátricas eficaces de linaclotida es de 0,1 mg a 5 jg al día por vía oral. En algunos casos, el intervalo de dosis pediátricas eficaces de linaclotida es de 0,1 mg a 1 jg al día por vía oral. En algunos casos, el intervalo de dosis pediátricas eficaces de linaclotida es de 0,1 mg a 0,5 jg al día por vía oral. En algunos casos, el intervalo de dosis pediátricas eficaces de linaclotida es 0,1 jg al día por vía oral. En algunos casos, el intervalo de dosis pediátricas eficaces de linaclotida es 0,15 jg al día por vía oral. En algunos casos, el intervalo de dosis pediátricas eficaces de linaclotida es 0,25 jg al día por vía oral. En algunos casos, el intervalo de dosis pediátricas eficaces de linaclotida es 0,5 jg al día por vía oral. En algunos casos, el intervalo de dosis pediátricas eficaces de linaclotida es 3,5 jg al día por vía oral. En algunos casos, el intervalo de dosis pediátricas eficaces de linaclotida es 15 jg al día por vía oral. En algunos casos, el intervalo de dosis pediátricas eficaces de linaclotida es 36 jg al día por vía oral. En algunos casos, el intervalo de dosis pediátricas eficaces de linaclotida es 45 jg al día por vía oral. En algunos casos, el intervalo de dosis pediátricas eficaces de linaclotida es 60 jg al día por vía oral. En algunos casos, el intervalo de dosis pediátricas eficaces de linaclotida es 90 jg al día por vía oral. En algunos casos, la forma de dosificación unitaria y la dosis diaria son equivalentes.
[0101] En algunos casos, la forma de dosificación unitaria se administra con alimentos en cualquier momento del día, sin alimentos en cualquier momento del día, con alimentos después de ayuno durante la noche (p. ej., con el desayuno). En algunos casos, la forma de dosificación unitaria se administra una vez al día, dos veces al día o tres veces al día. En algunos casos, una, dos o tres formas farmacéuticas unitarias contendrán la dosis oral diaria de linaclotida. La cantidad precisa de compuesto administrada a un paciente será responsabilidad del médico responsable del tratamiento. Sin embargo, la dosis empleada dependerá de varios factores, que incluyen la edad y el sexo del paciente, el trastorno preciso que se está tratando y su gravedad.
[0102] En algunos casos, las composiciones se administran como una monoterapia. En algunos casos, la
composición consiste esencialmente en una cantidad eficaz de linaclotida. En algunos casos, la composición consiste en una cantidad eficaz de linaclotida.
[0103] En algunos casos, las composiciones se administran directamente a un paciente, por ejemplo, en forma de un comprimido de liberación retardada o una cápsula de liberación retardada. En algunos casos, las composiciones se disuelven, disgregan y/o mezclan en, o dentro de, alimentos o bebidas antes de la administración a los pacientes (p. ej., pacientes ancianos o pediátricos). En algunos casos, la composición se disuelve o disgrega en un líquido, una solución o un fluido que contiene opcionalmente uno o más agentes estabilizantes, uno o más conservantes, uno o más edulcorantes o similares, etc., antes de la administración a un paciente (p. ej., paciente anciano o pediátrico). En algunos casos, la composición es una composición de múltiples dosis, es decir, que contiene dos, tres, cinco, siete, diez, quince, veinte, veinticinco, treinta, cuarenta, cincuenta, sesenta, setenta, ochenta, noventa o más dosis diarias de linaclotida.
[0104] En otros casos, las composiciones se administran como parte de una politerapia. Por ejemplo, una composición se puede usar en combinación con otros fármacos o terapias que se usan en el tratamiento, la prevención, la inhibición y/o la mejora de las enfermedades o afecciones para las que los compuestos de la descripción son útiles. La linaclotida se puede coadministrar o coformular con otros medicamentos. En un caso, la composición de linaclotida se puede coadministrar con otros medicamentos usados para tratar trastornos gastrointestinales que incluyen, pero no se limitan a, agentes inhibidores de los ácidos tales como agonistas del receptor de histamina-2 (AH2) y/o inhibidores de la bomba de protones (IBP). En un caso, la composición de linaclotida se puede coadministrar con otros medicamentos usados para tratar trastornos gastrointestinales que incluyen 5-ASA tales como mesalamina.
[0105] Tal fármaco o fármacos distintos se pueden administrar por una vía y en una cantidad usadas habitualmente, por lo tanto, simultánea o secuencialmente con un compuesto de la descripción. Cuando un compuesto de la presente descripción se usa simultáneamente con uno o más fármacos distintos, se puede emplear una forma de dosificación unitaria que contiene estos otros fármacos además del compuesto de la descripción. Por consiguiente, las composiciones farmacéuticas de la presente descripción incluyen aquellas que también contienen uno o más componentes activos distintos, además de un compuesto de descripción.
[0106] Se pueden usar varios métodos para evaluar la bioactividad de la composición de linaclotida, que incluyen, pero no se limitan a, inmunoanálisis (p. ej., enzimoinmunoanálisis de adsorción), radioinmunoanálisis, análisis inmunoradiométricos, electroforesis en gel (p. ej., SDS-PAGE), cromatografía líquida de alta resolución (CLAR) y/o electroforesis capilar de alta resolución (ECAR). En algunos casos, la bioactividad de la composición se evalúa mediante un método que comprende fijar la linaclotida, incubar la linaclotida con guanilato ciclasa C (GCC), incubar la linaclotida unida a la GCC con anticuerpos contra la GCC, incubar la linaclotida unida a los anticuerpos contra la GCC con anticuerpos marcados fluorescentemente contra los anticuerpos contra la GCC y detectar la linaclotida unida a los anticuerpos contra la GCC midiendo la intensidad de fluorescencia con un lector de placas. La concentración del fármaco se puede calcular a continuación en base a la lectura de fluorescencia de la solución.
[0107] Por ejemplo, la bioactividad de las composiciones de linaclotida se puede evaluar y cuantificar usando el método siguiente, aunque hay otros métodos disponibles. La composición se añade a un matraz aforado que contiene 60 ml de tampón fosfato que tiene un pH de 4,5 y el matraz se agita durante 60 minutos. A continuación, se retiran 0,2 ml del sobrenadante y se añaden en uno o más pocillos de una placa de 96 pocillos que está recubierta con receptores de la GC-C. La placa se sella y se incuba a 37 °C durante 2 horas. Al final de la incubación, se retira la muestra y la placa se lava con solución salina tamponada con fosfato (PBS). La linaclotida unida se incuba a continuación durante 1 hora, a temperatura ambiente, con GC-C (tal como está disponible en Sigma-Aldrich Inc.) marcada con isocianato de fluoresceína (FITC) en tampón de bloqueo. Después de la incubación, el pocillo se lava con PBS. La intensidad de fluorescencia del producto final se detecta, por ejemplo, usando un lector de placas. La concentración de linaclotida se calcula a continuación en base a la lectura de fluorescencia de la solución.
Definiciones
[0108] Como se emplea en esta memoria, a menos que se indique de otro modo, el término “liberación retardada” significa que la composición se disuelve, funde, disgrega, licúa, etc., en un área objetivo del tracto gastrointestinal de tal manera que ya no permanece sustancialmente nada de la linaclotida en una formulación, composición o forma farmacéutica. Las composiciones de liberación retardada incluyen composiciones de liberación lenta, composiciones gastrorretentivas, composiciones de liberación dirigida (p. ej., composiciones de liberación colónica, o composiciones que se dirigen a la válvula ileocecal, etc.), composiciones de liberación prolongada y/o combinaciones de las mismas.
[0109] Como se emplea en esta memoria, a menos que se indique de otro modo, el término “composición de liberación retardada” (“LR”) significa que la composición es una forma farmacéutica que libera linaclotida en un momento distinto al inmediatamente posterior a la administración oral.
[0110] Como se emplea en esta memoria, a menos que se indique de otro modo, el término “composición de
liberación prolongada” significa que la composición es una forma farmacéutica que libera linaclotida a lo largo de un período de tiempo prolongado después de la administración. Esto permite una reducción en la frecuencia de administración en comparación con las composiciones de liberación inmediata.
[0111] Como se emplea en esta memoria, a menos que se indique de otro modo, “disgregación” y “ liberación” se usan en esta memoria para indicar que la cápsula, la película, la perla o el comprimido que comprende linaclotida se disuelve, funde, disgrega, licúa, etc., en el entorno de una cavidad oral de tal manera que sustancialmente toda la linaclotida deja de permanecer en una forma de formulación, p. ej., un pH superior a 5 o 7, o en una solución de tampón fosfato y mantenida a 37 ±1 °C.
[0112] El término “liberada desde”, cuando se refiere a la liberación de linaclotida desde la composición, a menos que se indique de otro modo, se usa en esta memoria para indicar que la linaclotida ya no permanece en una forma de la composición.
[0113] Como se emplea en esta memoria, a menos que se indique de otro modo, el término “entrada en un entorno objetivo” significa el contacto de la composición en el interior de un paciente en un órgano objetivo o segmento del mismo, o en el interior de un segmento del tracto GI destinado a la liberación de linaclotida, p. ej., que tiene un pH superior a 5 o 7.
[0114] Como se emplea en esta memoria, a menos que se indique de otro modo, el término “tracto gastrointestinal inferior (GI)” significa el segmento distal del tracto gastrointestinal, por ejemplo, el íleon, el íleon terminal, la válvula ileocecal o el colon.
[0115] Como se emplea en esta memoria, a menos que se indique de otro modo, el término “tracto gastrointestinal (GI) superior” significa el segmento proximal del tracto gastrointestinal, por ejemplo, el estómago, duodeno y/o yeyuno.
[0116] Como se emplea en esta memoria, a menos que se indique lo contrario, “agente estabilizante” se refiere a un componente polimérico, de amina primaria impedida estéricamente (p. ej., aminoácido) o catiónico (p. ej., catión metálico) de la composición que está incluido en la composición en una cantidad estabilizante. Por ejemplo, un agente estabilizante polimérico es un polímero que está incluido en la composición en una cantidad estabilizante. Similarmente, un agente estabilizante de amina primaria impedida estéricamente es una amina primaria impedida estéricamente que está incluida en la composición en una cantidad estabilizante. Asimismo, un agente estabilizante catiónico es un catión que está incluido en la composición en una cantidad estabilizante.
[0117] Como se emplea en esta memoria, a menos que se indique de otro modo, “cantidad estabilizante” se refiere a una concentración, dentro de la composición, de un componente polimérico, de amina primaria impedida estéricamente (p. ej., aminoácido) o catiónico metálico en el que el componente aumenta la estabilidad de la linaclotida en la composición, en comparación con una composición similar que no tiene una cantidad estabilizante del mismo componente.
[0118] Como se emplea en esta memoria, a menos que se indique de otro modo, el término “sustancialmente toda” significa al menos aproximadamente 90 %, por ejemplo, al menos aproximadamente 95 % o incluso al menos aproximadamente 99 %.
[0119] Como se emplea en esta memoria, a menos que se indique de otro modo, el término “aislado y purificado” significa al menos 95 por ciento puro (por ejemplo, al menos 96 % puro, al menos 97 % puro, al menos 98 % puro o incluso al menos 99 % puro), como se mide, por ejemplo, mediante pureza cromatográfica usando CLAR.
[0120] Como se emplea en esta memoria, a menos que se indique de otro modo, “cantidad terapéuticamente eficaz” significa la cantidad de una linaclotida o una sal farmacéuticamente aceptable de la misma que, cuando se administra a un mamífero para tratar un estado, un trastorno o una afección, es suficiente para lograr un tratamiento (como se define más adelante). La “cantidad terapéuticamente eficaz” variará en función del compuesto, la enfermedad y su gravedad y la edad, el sexo, el peso, el estado físico y la sensibilidad del mamífero que se va a tratar. Por ejemplo, una cantidad terapéuticamente eficaz de linaclotida, o su sal o hidrato farmacéuticamente aceptable, puede ser una cantidad eficaz para tratar trastornos gastrointestinales, que incluyen síndrome del intestino irritable, síndrome del intestino irritable con predominio de estreñimiento, estreñimiento crónico, estreñimiento inducido por opioides y/o dispepsia.
[0121] Como se emplea en esta memoria, a menos que se indique de otro modo, “farmacéuticamente aceptable” significa biológica o farmacológicamente compatible para uso in vivo en animales o seres humanos, y preferentemente significa aprobado por una agencia reguladora del gobierno federal o estatal o enumerado en la Farmacopea de los Estados Unidos u otra farmacopea generalmente reconocida para uso en animales, y más particularmente en seres humanos.
[0122] Como se emplea en esta memoria, a menos que se indique de otro modo, el término “tratar”, en todas sus formas verbales, se emplea en esta memoria para indicar mitigar, aliviar, prevenir y/o controlar al menos un síntoma de un trastorno en un sujeto, incluyendo el trastorno, por ejemplo, un trastorno gastrointestinal, tal como síndrome del intestino irritable, síndrome del intestino irritable con predominio de estreñimiento, estreñimiento crónico, estreñimiento inducido por opioides, dispepsia o una combinación de síntomas de los mismos. Con el significado de la presente descripción, el término “tratar” también simboliza, detener, retardar la aparición (es decir, el período previo a la manifestación clínica de una enfermedad) y/o reducir el riesgo de desarrollar o empeorar una enfermedad. El término “tratamiento” significa el acto de “tratar” como se definió anteriormente.
[0123] Como se emplea en esta memoria, a menos que se indique de otro modo, el término “aditivos” se refiere a un aditivo farmacéuticamente aceptable. Los aditivos farmacéuticamente aceptables incluyen, sin limitación, aglutinantes, disgregantes, aditivos dispersantes, lubricantes, deslizantes, antioxidantes, aditivos de recubrimiento, diluyentes, tensioactivos, aditivos saborizantes, humectantes, aditivos potenciadores de la absorción, aditivos de liberación controlada, aditivos antiaglomerantes, agentes antimicrobianos (p. ej., conservantes), colorantes, desecantes, plastificantes y pigmentos.
[0124] Como se emplea en esta memoria, a menos que se indique de otro modo, un “excipiente” es cualquier aditivo, carga, aglutinante o agente farmacéuticamente aceptable.
[0125] Como se emplea en esta memoria, a menos que se indique de otro modo, “condiciones rigurosas” se refiere a 40 °C y 75 % de humedad relativa (HR).
[0126] Como se emplea en esta memoria, a menos que se indique de otro modo, los términos “aproximadamente” y “alrededor de” significan dentro de un intervalo de error aceptable para el valor particular como es determinado por un experto en la materia, que dependerá, en parte, de cómo se mide o determina el valor, es decir, de las limitaciones del sistema de medición. Por ejemplo, “aproximadamente” puede significar comprendido en 1 o más de 1 desviaciones estándar, según la práctica en la técnica. Alternativamente, “aproximadamente”, con respecto a las composiciones, puede significar más o menos un intervalo de hasta 20%, preferentemente hasta 10%. Alternativamente, particularmente con respecto a sistemas o procesos biológicos, el término puede significar dentro de un orden de magnitud, preferentemente dentro de 5 veces, y más preferentemente dentro de 2 veces, de un valor. Los valores particulares se describen en la solicitud y las reivindicaciones, a menos que se indique de otro modo, el término “aproximadamente” significa dentro de un intervalo de error aceptable para el valor particular.
[0127] Todos los porcentajes en peso (es decir, “% en peso” y “% p” y p/p) a los que se hace referencia en esta memoria, a menos que se indique de otro modo, se miden con respecto al peso total de la composición farmacéutica.
[0128] El término “que consiste esencialmente en”, y variantes del mismo, cuando se usan para hacer referencia a la composición, se emplean en esta memoria para indicar que la composición incluye linaclotida y otros aditivos, excipientes y/o componentes farmacéuticamente inactivos deseados (p. ej., polímeros, aminas primarias impedidas estéricamente, cationes, agentes de carga, aglutinantes, vehículos, excipientes, diluyentes, aditivos disgregantes, lubricantes, disolventes, dispersantes, aditivos de recubrimiento, aditivos potenciadores de la absorción, productos de la hidrólisis, productos de formaldehído imina, productos de la oxidación, productos de la acetilación, productos de la desamidación, multímeros, aditivos de liberación controlada, aditivos antiaglomerantes, aditivos antimicrobianos, conservantes, aditivos edulcorantes, colorantes, saborizantes, desecantes, plastificantes, pigmentos o similares), y ningún otro ingrediente farmacéutico activo.
EJEMPLOS
[0129] Los ejemplos siguientes son meramente ilustrativos y no se deben interpretar como limitantes del alcance de la invención de ninguna manera, ya que muchas variaciones y equivalentes que están abarcados por la presente invención como se define en las reivindicaciones adjuntas resultarán evidentes para los expertos en la materia tras leer la presente descripción.
[0130] Se usan varias estrategias de formulación para dirigir la linaclotida al tracto GI inferior. (i) Las perlas con recubrimiento entérico que comprenden un recubrimiento polimérico sensible al pH se pueden aplicar a perlas centrales que se han recubierto con linaclotida y potencialmente una subcapa. Estas perlas se podrían encapsular además en cápsulas para conseguir las concentraciones deseadas de las dosis unitarias. (ii) Los comprimidos con recubrimiento entérico que comprenden un comprimido de liberación inmediata central que contiene una dosis unitaria de linaclotida se pueden recubrir con recubrimientos que se disuelven solo en las condiciones de pH del segmento distal del intestino, de manera que la linaclotida se liberará en el tracto GI inferior. (iii) Los recubrimientos entéricos se pueden aplicar a una cápsula de gelatina o HPMC, haciendo que la cápsula se desintegre y disuelva solo en el tracto GI inferior. Las perlas o los comprimidos cargados con linaclotida se pueden encapsular en la cápsula para conseguir la liberación retardada. (iv) Adicionalmente, se pueden encapsular perlas de liberación inmediata a la concentración de la dosis apropiada en una cápsula en la que la cubierta de la cápsula está fabricada de materiales poliméricos entéricos. Esta cubierta de la cápsula entérica se puede usar como tal o recubrir adicionalmente con polímeros
sensibles al pH para conseguir el perfil de liberación y el punto de liberación al pH deseado.
[0131] La linaclotida o una sal farmacéuticamente aceptable de la misma se puede producir y purificar usando técnicas convencionales conocidas en la técnica, p. ej., síntesis química o expresión recombinante seguida de purificación usando técnicas convencionales.
[0132] Preparación de la solución de recubrimiento de linaclotida para perlas: se mezclan aproximadamente 32 g a 42 g de agua purificada con ácido clorhídrico para crear una solución con un pH entre 1,5 y 2,0. El catión, si se usa, se añade a la solución en una cantidad adecuada para proporcionar la concentración deseada y la solución se mezcla durante el tiempo suficiente para producir una solución transparente. La amina primaria impedida estéricamente, si se usa, se añade a la solución en una cantidad adecuada para proporcionar la concentración deseada y la solución se mezcla durante el tiempo suficiente para producir una solución transparente. A continuación, se añaden otros aditivos, tales como antioxidantes, si se desea. Se analiza el pH de la solución y se añade ácido clorhídrico, si es necesario, para producir una solución que tiene un pH entre 1,5 y 2,0. A continuación, se añade el aglutinante a la solución y la mezcla se agita a continuación durante el tiempo suficiente para conseguir una solución transparente. La cantidad deseada de linaclotida se añade a la solución y se mezcla durante 30-100 minutos para proporcionar la solución de recubrimiento.
[0133] En un caso, la solución de recubrimiento comprende linaclotida, histidina, 1,5% de PVA y 0,6% de talco. Esta formulación se puede usar para producir intervalos de administración entre 36-290 |jg.
[0134] Preparación de las perlas activas: se añaden aproximadamente 30-36 g de perlas de celulosa microcristalina secas a una máquina de recubrimiento de lecho fluido de minicolumna. Las perlas de celulosa microcristalina se fluidifican y se calientan antes de la estratificación. A continuación, la solución de recubrimiento se estratifica sobre las perlas. La temperatura de pulverización se controla entre 24 °C y 55 °C controlando la temperatura de entrada, la velocidad de pulverización, la presión de atomización y el volumen de aire. Después de que toda la solución de recubrimiento está estratificada sobre las perlas, las perlas se secan. El producto de este procedimiento se denomina perlas activas.
[0135] Preparación de perlas activas con recubrimiento protector: se añaden aproximadamente 35 g de perlas activas a una máquina de recubrimiento de lecho fluido de minicolumna. Las perlas activas se fluidifican y calientan antes de recubrir con Aquacoat® (p. ej., dispersión acuosa de etilcelulosa Aquacoat®, 15% p/p, biopolímero FMC, ECD-30), Eudragit® (p. ej., Eudragit® E PO PE-EL, Roehm Pharma Polymers) u Opadry® (p. ej., dispersión AMB Opadry®, 20 % p/p, Colorcon). A continuación, la solución de recubrimiento se estratifica sobre las perlas. La temperatura de pulverización se controla entre 24 °C y 55 °C controlando la temperatura de entrada, la velocidad de pulverización, la presión de atomización y el volumen de aire. Después de que toda la solución de recubrimiento está estratificada sobre las perlas, las perlas se secan.
Ejemplo 1 (Ejemplo de referencia)
Perlas de linaclotida de liberación retardada
[0136] Las perlas de liberación retardada se pueden fabricar de varias maneras:
A. Perlas con recubrimiento entérico
[0137] Las perlas de liberación retardada comprenden recubrimientos que se pueden diseñar para que sean resistentes al pH bajo del estómago, pero se descompongan rápidamente y liberen linaclotida activa solo por debajo del pH del tracto GI inferior (pH > 5). Los polímeros funcionales de esta categoría incluyen: copolímeros de acrilato de metilo-ácido metacrílico (p. ej., Eudragit®); acetato-succinato de celulosa (CAS); ftalato de hidroxipropilmetilcelulosa (HPMCP); acetato-succinato de hidroxipropilmetilcelulosa (HPMCAS); acetato-ftalato de polivinilo (PVAP). copolímeros de metacrilato de metilo-ácido metacrílico; alginato de sodio y ácido esteárico; goma guar; y carbómeros.
[0138] La solución/suspensión de recubrimiento entérico puede comprender un polímero de disolución entérica o una combinación de un plastificante, material humectante, agente antiaglomerante y diluyente (acuoso u orgánico). Normalmente se añaden plastificantes al recubrimiento polimérico para ayudar a la formación de la capa de película. Se podría aplicar una subcapa a las perlas estratificadas con linaclotida antes del recubrimiento entérico para separar la linaclotida y el polímero entérico para mejorar la estabilidad.
Ejemplo 2 (Ejemplo de referencia)
Perlas de linaclotida LR recubiertas con Eudragit® FS30D
[0139]
T l 1 m i i n ^ rl n n l lin l i

T l 2 m i i n rl LR lin l i r i r n E r i F D

Procedimiento de fabricación:
A. Preparación de solución de recubrimiento
[0140] El recubrimiento de las perlas comienza con la preparación de la solución de recubrimiento. Se pesa PlasACRYL™ en un recipiente y se agita con una mezcladora estacionaria. Se pesa Eudragit® FS30D y se añade lentamente al recipiente de PlasACRYL™ con agitación. Se pesa agua purificada y se añade lentamente a la suspensión de PlasACRYL™/Eudragit® con agitación. La suspensión se mezcla durante 10 min y a continuación se pasa a través de un tamiz de 0,5 mm. La suspensión polimérica está lista para el recubrimiento de las perlas.
B. Recubrimiento de perlas
[0141] Un lecho fluido del Wurster se calienta a 40 °C. Las perlas centrales de linaclotida se pesan y se añaden al Wurster. Las perlas se calientan a 35 °C. El recubrimiento de las perlas continúa con la suspensión de Eudragit® FS 30 D manteniendo la temperatura de 28° a 32 °C y la presión del aire de atomización a 3 bar. Al final del recubrimiento, las perlas descargadas se colocan en un horno de aire circulado y se secan durante 2 h a 40 °C.
Ejemplo 3 (Ejemplo de referencia)
Perlas de linaclotida LR recubiertas con Eudragit® FS30D con subcapa de PVP
[0142]
Tabla 3 Composición de perlas de liberación retardada de linaclotida recubiertas con Eudragit® FS30D con subcapa de PVP

Procedimiento de fabricación:
A. Preparación de solución de subcapa
[0143] En un recipiente, se pesa agua purificada hasta 1000 g y se añade al PVP pesado previamente con agitación. La solución se mezcla hasta que sea transparente.
B. Recubrimiento de perlas con subcapa
[0144] Un lecho fluido del Wurster se calienta a 60 °C. Las perlas centrales de linaclotida se pesan y se añaden al Wurster. Las perlas se calientan a 50 °C antes de comenzar el recubrimiento de las perlas. Durante el recubrimiento, la temperatura del producto se controla de 45° a 50 °C. Una vez terminado el recubrimiento, las perlas se secan durante 30 min a temperaturas de producto de 45° a 50 °C.
C. Recubrimiento de Eudragit® FS30D
[0145] Las etapas de fabricación para perlas de linaclotida recubiertas con Eudragit® FS30D se proporcionan en el Ejemplo 1.
[0146] Los materiales que se pueden usar como subcapa incluyen polímeros tales como alcohol polivinílico (PVA), polivinilpirrolidona (PVP), hipromelosa (HPMC), hidroxipropilcelulosa (HPC), metilcelulosa, almidón, gelatina, dextrina, maltodextrina, poloxámero, polidextrosa, goma laca, quitosano, alginato, pectina, polietilenglicol, goma guar, albúmina y azúcares tales como manitol, lactosa, isomalt, sorbital, xilitol, maltitol, sucrosa, trehalosa, fructosa, glucosa, dextrosa, eritritol, sucralosa, etc., o sus combinaciones. El nivel de subcapa varía de 1 a 60 % en peso en la fórmula de perlas.
Ejemplo 4 (Ejemplo de referencia)
Perlas de linaclotida recubiertas con Acril-EZEO (Eudragit® L100-55)
[0147]

continuación

Procedimiento de fabricación:
A. Preparación de solución de PVP
[0148] En un recipiente de acero inoxidable, se pesa agua hasta 2000 g. Se añade PVP con agitación. La solución se mezcla hasta que sea transparente. La leucina y el cloruro de calcio se pesan y se añaden a la solución de PVP con agitación. La solución se mezcla hasta que sea transparente. La solución se ajusta a pH 2, a continuación se añade linaclotida y se mezcla hasta obtener otra vez una solución transparente.
B. Preparación de suspensión de Acryl-EZE®
[0149] En un recipiente de acero inoxidable, se pesa agua destilada hasta 8000 g. Se pesa Acryl-EZE® y se añade al agua. La suspensión se mezcla con una mezcladora estacionaria durante 20 min. La suspensión se pasa a través de un filtro de malla 60.
C. Recubrimiento
[0150] Pesar perlas de isomalt y añadir al Wurster de un lecho fluido calentado a 50 °C. Comenzar a pulverizar la solución de fármaco, con una temperatura del producto de alrededor de 40 °C. Tras terminar de pulverizar la solución de fármaco, secar las perlas durante 30 min con una temperatura de producto de alrededor de 40 °C. Comenzar a pulverizar la capa Acryl-EZE®, mantener la temperatura del producto a ~30 °C. Al final del recubrimiento de Acryl-EZE®, secar las perlas durante 30 min con la temperatura de producto de ~30 °C.
Disolución de perlas LR de linaclotida recubiertas con Acryl-EZE®
[0151]

[0152] Para conseguir la disolución del fármaco en diferentes pH intestinales, se puede sustituir Acryl-EZE® (Eudragit® L100-55, se disuelve a pH superior a 5,5) por otros polímeros de metacrilato, tales como Eudragit® L (se disuelve a pH 6), Eudragit® S y Eudragit® FS30D (se disuelven a pH 7) o sus combinaciones. Por ejemplo, (i) Forma 1: disolución del fármaco a pH > 5,5 usando perlas recubiertas con Acryl-EZE®; (ii) Forma 2: liberaciones de fármaco a pH > 6,5 usando recubrimiento de Eudragit® L/S (50:50); (iii) Forma 4: liberaciones de fármaco a pH 7 recubriendo las perlas con Eudragit® FS30D. Además, se pueden colocar dos o más de estos tipos de perlas en una cápsula para proporcionar administración pulsátil en diferentes regiones del tracto GI. Por ejemplo, en una cápsula se combinan perlas LR con perlas de la Forma 1, o usando una combinación de perlas de la Forma 1 y la Forma 2, o una combinación de perlas de la Forma 2 y la Forma 3, o una combinación de perlas de la Forma 1, la Forma 2 y la Forma 3, etc.
Ejemplo 5 (Ejemplo de referencia)
Perlas LR de linaclotida recubiertas con HPMCAS
[0153]

Ejemplo 6 (Ejemplo de referencia)
Perlas LR de linaclotida recubiertas con etilcelulosa
[0154]
Tabl m i i n rl li r i n r r LR lin l i r i r n il l losa

[0155] Similarmente, se pueden recubrir perlas de linaclotida con otros polímeros entéricos tales como acetatoftalato de celulosa (CAP), acetato-trimelitato de celulosa (CAT), acetato-succinato de celulosa (CAS), acetato-ftalato de polivinilo (PVAP) y otro copolímero. Para ayudar a la formación de la película polimérica, se añaden plastificantes tales como citrato de trietilo (TEC), polietilenglicol (PEG), monoglicérido acetilado (AMG) o sus combinaciones a la solución de recubrimiento polimérico. Además, se añaden tensioactivos tales como laurilsulfato de sodio, polisorbato, a la solución de recubrimiento como agentes humectantes. Para garantizar la liberación entérica del fármaco, el recubrimiento polimérico se aplica en un porcentaje en peso de 5 a 80 % en función del tamaño y la fabricación de las perlas y las propiedades de la subcapa.
Ejemplo 7
Comprimido de linaclotida de liberación retardada
[0156] La linaclotida se puede formular en un comprimido para liberación retardada del fármaco. En comparación con un volumen igual de perlas, los comprimidos tienen una superficie específica mucho menor, lo que los hace potencialmente menos propensos a la degradación inducida por factores ambientales tales como la humedad, oxidación, desamidación, etc. Además, la menor superficie del comprimido puede resultar ventajosa cuando se necesita un recubrimiento entérico, ya que se necesita mucho menos material de recubrimiento para cubrir la superficie de la forma farmacéutica.
Comprimido con recubrimiento entérico
[0157] Los recubrimientos entéricos se pueden aplicar en un bombo de recubrimiento de comprimidos, y los recubrimientos que se usan para perlas de liberación retardada se pueden usar para que los comprimidos formen comprimidos de liberación retardada. La cantidad de polímero de recubrimiento en el comprimido puede variar de 5 a 60 % (ganancia de peso) en función del tamaño, la forma y las propiedades superficiales del comprimido. Se puede aplicar una subcapa a los comprimidos para separar la linaclotida del recubrimiento entérico o funcional.
Ejemplo 8 (Ejemplo de referencia)
[0158]
Tabla 7 Com ^ ragit® FS30D

Procedimiento de fabricación:
A. Comprimido
[0159] La solución de granulación se puede preparar disolviendo PVP, histidina y cloruro de calcio en agua, ajustando el pH de la solución a 2 y disolviendo linaclotida. La granulación se realiza en un lecho fluido pulverizando la solución de granulación sobre la carga isomalt. Al final de la granulación, secar los gránulos durante 30 min. Los gránulos se mezclan a continuación con los componentes del comprimido, que incluyen isomalt, crospovidona, estearato de Mg y talco, hasta conseguir uniformidad y se comprimen para obtener comprimidos.
B. Recubrimiento entérico
[0160] La suspensión de Eudragit® FS30D se puede preparar como se describe en el Ejemplo 1. Para el recubrimiento de comprimidos, los comprimidos de núcleo de linaclotida se colocan en un bombo de recubrimiento y se calientan hasta 35 °C. Comenzar el recubrimiento de los comprimidos con suspensión de Eudragit® FS 30 D, mantener la temperatura del producto de 28° a 32 °C y la presión del aire de atomización a 3 bar. Al final del
recubrimiento, descargar los comprimidos, colocarlos en un horno de aire circulado y secar durante 2 h a 40 °C.
[0161] Similarmente, también se pueden aplicar otros recubrimientos entéricos tales como Eudragit® L, S, etilcelulosa, HPMCAS, PVAP, CAP, c As , etc., para formar comprimidos de liberación retardada con diversas ganancias de peso.
Gránulos con recubrimiento entérico
[0162] Los recubrimientos entéricos también se pueden aplicar a gránulos de fármaco y los comprimidos de liberación retardada se pueden fabricar comprimiendo los gránulos con recubrimiento entérico para obtener comprimidos. Los gránulos tienen una superficie aumentada y necesitan cantidades mayores de recubrimiento para cubrir completamente la superficie de los gránulos. Una vez que el comprimido se disgrega, los gránulos con recubrimiento entérico liberan linaclotida de una manera más uniforme en comparación con los comprimidos. Un fallo de recubrimiento en uno o algunos gránulos tendrá un impacto menor en el comportamiento general de la forma farmacéutica de linaclotida.
Ejemplo 9 (Ejemplo de referencia)
[0163]
Tabla m i i n ^ r n l li r i n r r LR lin l i r i r n E r i FS30D

Comprimidos de matriz LR
[0164] El comprimido de matriz también se puede formular para conseguir la liberación retardada del fármaco. Los polímeros para recubrimiento entérico se pueden usar indistintamente como carga para gránulos de linaclotida o como matriz (carga) para el comprimido.
Ejemplo 10 (Ejemplo de referencia)
[0165]
Tabla 9 Com rimido de matriz LR de linaclotida fabricado con ranulos LR

Ejemplo 11 (Ejemplo de referencia)
[0166]
T l 1 m rimi m riz LR lin l i

[0167] En los dos ejemplos anteriores, se puede sustituir Eudragit® L100 por otros polímeros de disolución entérica (Eudragit® S, Eudragit® FS30D, Acryl-EZE®, HPMCAS, HPMCP, CAP, CAS, PVAP, etc.) en un porcentaje o ganancia en peso de 5 a 90 % en un comprimido.
Ejemplo 12 (Ejemplo de referencia)
Cápsulas con recubrimiento entérico para liberación retardada de linaclotida
[0168] Aparte de aplicados directamente a perlas, gránulos o comprimidos, los recubrimientos entéricos también se pueden aplicar a cápsulas. El recubrimiento de cápsulas se puede realizar sobre cápsulas llenas. Primero se retirará el polvo de las cápsulas para crear una superficie limpia y uniforme. Para evitar defectos de recubrimiento alrededor de la conexión entre la tapa y el cuerpo de las cápsulas, las cápsulas limpias se cierran antes del recubrimiento. Las cápsulas cerradas se recubren con un recubrimiento entérico en un bombo de recubrimiento. Los recubrimientos entéricos se podrían aplicar indistintamente a cápsulas de gelatina o HPMC y, en función del tamaño y la propiedad del recubrimiento de las cápsulas, se pueden aplicar con una ganancia de peso de 5 a 60 %.
Ejemplo 13
Composiciones de liberación retardada que comprenden linaclotida
(solo comprimidos con recubrimiento entérico son según la invención)
[0169] Las cápsulas de liberación retardada que comprenden linaclotida se pueden formular para que se dirijan al íleon o al colon. La composición se formula para que incluya una liberación desencadenada por el pH en base al recubrimiento entérico de un comprimido, una cápsula o perlas recubiertas con linaclotida contenidas en una cápsula de gelatina dura. La composición se puede formular para que comprenda además aditivos estabilizantes tales como un catión divalente y un aminoácido. Se puede usar PVA como aglutinante y como capa protectora entre la linaclotida y el recubrimiento entérico. La linaclotida o la linaclotida con capa protectora de PVA (como perlas, cápsula o comprimido) se puede recubrir con un recubrimiento entérico adicional (p. ej., Eudragit® FS30D, Eudragit® S100, Eudragit® L100, Eudragit® L100-55, Eudragit® L 30D-55) que se disuelve de manera dependiente del pH para liberar al pH apropiado de 7 en el íleon del tracto GI. Los recubrimientos entéricos pueden consistir en mezclas que combinan diferentes tipos de Eudragit®-Eudragit® S100/Eudragit® L100 en relaciones diferentes; Eudragit® S100/Eudragit® L100-55 en diversas relaciones; Eudragit® FS30D/Eudragit® L 30D-55, Eudragit® FS30D Eudragit® S/Eudragit® RS o EC en diversas relaciones. Las composiciones pueden comprender además otros excipientes que incluyen agentes plastificantes tales como citrato de trietilo. Los recubrimientos pueden comprender además disgregantes como sólidos suspendidos para acelerar la liberación desencadenada por el pH relevante, lo que da como resultado sistemas mixtos como croscarmelosa de sodio/Eudragit® S. Por facilidad de procesamiento, se puede usar un agente antiadherente (p. ej., talco, Aerosil® 200 o PlasAcryl™) para evitar que las perlas se adhieran.
[0170] Adicionalmente, se pueden aplicar dos recubrimientos de Eudragit® para garantizar la liberación rápida una vez que se alcanza la región de pH deseada en el tracto GI, lo que incluye sistemas de recubrimiento parcialmente neutralizados. Se pueden incluir agentes tamponadores tales como hidrogenofosfato de potasio en una de las dos películas de Eudragit®. Los recubrimientos de película dependientes del pH alternativos distintos de Eudragit® incluyen acetato-succinato de hidroxipropilmetilcelulosa (HPMCAS, p. ej., Aqoat® AS-HF), acetato-ftalato de celulosa (CAP, p. ej., Aquateric®) o goma laca.
Ejemplo 14 (Ejemplo de referencia)
Composiciones de linaclotida con gránulos/perlas LR y gránulos/perlas LI
[0171] El comprimido LR se puede formular comprimiendo gránulos LI y gránulos LR para obtener un comprimido. Alternativamente, la formulación LI y LR se puede integrar en una formulación para conseguir la cinética de disolución deseada. Por ejemplo, se puede aplicar una capa de fármaco de liberación inmediata como una capa exterior o una subcapa sobre perlas/gránulos que contienen la porción LR, como se ilustra a continuación.
T l 11 m i i n rl LR lin l i ^ n x ri r LR

T l 11 m i i n rl LR lin l i ^ n x ri r LR

Ejemplo 15 (Ejemplo de referencia)
Formulación de liberación lenta (LL) de linaclotida
[0172] En una formulación de liberación lenta, la linaclotida se libera de modo lento y constante a lo largo de un período de horas a días para mantener el nivel de fármaco eficaz en la circulación sanguínea a lo largo de un período de tiempo prolongado y aumentar el cumplimiento terapéutico del paciente reduciendo las frecuencias de administración. La estrategia usada más frecuentemente para mantener la liberación del fármaco es a través de la formación de un sistema de matriz en el que se incrusta el fármaco activo. El sistema de matriz a menudo implica usar materiales de control de la liberación de las categorías siguientes: 1 ) polímeros hidrófilos con capacidad de gelificación alta tales como hidroxipropilmetilcelulosa (HPMC), hidroxipropilcelulosa (HPC), metilcelulosa, carboximetilcelulosa de sodio, carbopol, goma guar, agar-agar, alginato, manosa, galactosa, goma xantana, quitosano, almidón modificado, poliacrilamida, polivinilpirrolidona reticulada, alcohol polivinílico reticulado, metilacrilato de polihidroxietilo (PHEMA), etc.; 2 ) materiales hidrófobos tales como polietileno, cloruro de polivinilo, etilcelulosa, polímeros de acrilato y sus copolímeros; 3) polímeros biodegradables tales como ácido poliláctico (PLA), ácido poliglicólico (PGA), ácido poli(láctico-co-glicólico) (PLGA), policaprolactona (PCL), polianhídrido, poliortoésteres, etc.
Ejemplo 16
Composiciones de liberación mantenida y dependiente del pH que comprenden linaclotida
(solo comprimidos con recubrimiento entérico son según la invención)
[0173] Las composiciones de liberación retardada que combinan un recubrimiento entérico (liberación dependiente del pH) con un recubrimiento de liberación mantenida se podrían aplicar a un comprimido o una cápsula de linaclotida o a perlas recubiertas de linaclotida contenidas en una cápsula de gelatina dura. Las composiciones se podrían formular con aditivos estabilizantes tales como un catión divalente y un aminoácido. El PVA se puede usar como aglutinante, así como capa protectora entre la linaclotida y el recubrimiento entérico.
[0174] La hidroxipropilcelulosa se puede usar como polímero de liberación mantenida o como una capa protectora sobre la linaclotida o la linaclotida recubierta con una capa protectora adicional de PVA. Las composiciones se pueden recubrir con un recubrimiento entérico de liberación dependiente del pH (p. ej., Eudragit®). Se podrían aplicar dos recubrimientos de Eudragit® diferentes con un recubrimiento exterior de Eudragit® FS que se disuelve a pH 6 , 8 (para liberación en el área del íleon y la válvula ileocecal). La fórmula del recubrimiento para el recubrimiento interior con Eudragit® FS podría contener: Eudragit® FS30D, Eudragit® L 30D-55, monoestearato de glicerilo, Tween® 80 y agua. Se podría aplicar una capa interior de Eudragit® RL-RS para permitir la liberación lenta del fármaco en el colon. Ambas capas se aplicarían como se describió anteriormente sobre perlas recubiertas con linaclotida o con linaclotida con una capa protectora de PVA. La fórmula del recubrimiento para el recubrimiento interior con Eudragit® RL-RS puede comprender Eudragit® RL30D, Eudragit® RS30D, monoestearato de glicerilo, citrato de trietilo, Tween® 80 y agua.
Ejemplo 17
Composiciones de liberación desencadenada microbianamente y en función del pH que comprenden linaclotida
(solo comprimidos con recubrimiento entérico son según la invención)
[0175] Las composiciones de liberación retardada de linaclotida se podrían formular con una combinación de un recubrimiento entérico para liberación dependiente del pH y una capa de liberación desencadenada microbianamente (p. ej., polímero biodegradable) que se puede aplicar a un comprimido o una cápsula de linaclotida o a perlas recubiertas de linaclotida contenidas en una cápsula de gelatina dura. Las composiciones pueden comprender además aditivos estabilizantes tales como un catión divalente o un aminoácido. El PVA se puede usar como aglutinante, así como capa protectora entre la linaclotida y el recubrimiento entérico. Un polímero biodegradable se puede usar como agente de liberación como una capa protectora sobre la linaclotida o la linaclotida recubierta con una capa protectora adicional de PVA. El polímero biodegradable podría ser un recubrimiento de película a base de polisacáridos vegetales, animales o microbianos de origen natural tales como pectina, quitosano, sulfato de condroitina, galactomanano y amilosa. Estas composiciones pueden incluir recubrimientos mixtos o combinaciones de recubrimientos, por ejemplo, con HPMC/pectina, etilcelulosa/pectina, acetato de celulosa/pectina, quitosano/pectina o pectina/ Eudragit® (p. ej., Eudragit® RS 30D). Las composiciones pueden incluir excipientes adicionales tales como plastificantes. El pVa se puede usar como polímero formador de película, por ejemplo, en una combinación con quitosano. Estas composiciones pueden incluir además un recubrimiento entérico dependiente del pH adicional (p. ej., Eudragit®) para la liberación más eficaz de la linaclotida al íleon.
Ejemplo 18
Medición del contenido y la pureza de péptidos ejemplares
[0176] El contenido y la pureza de la linaclotida se pueden determinar mediante cromatografía líquida en gradiente de fase inversa usando un sistema de CL Agilent Series 1100 con software Chemstation Rev A.09.03 o equivalente. Se usa una columna YMC Pro™ C18 (dimensiones: 3,0 x 150 mm, 3,5 um, 120 A; Waters Corp., Milford, MA) o equivalente y se mantiene a 40 °C. La fase móvil A (FMA) consiste en agua con 0,1 % de ácido trifluoroacético, mientras que la fase móvil B (FMB) consiste en 95 % de acetonitrilo:5 % de agua con 0,1 % de ácido trifluoroacético. La elución de la linaclotida se logra con un gradiente de 0 % a 47 % de FMB en 28 minutos, seguido de una rampa a 100 % de FMB en 4 minutos con una retención de 5 minutos a 100 % de FMB para lavar la columna. El reequilibrado de la columna se realiza volviendo a 0 % de FMB en 1 minuto, seguido de una retención de 10 minutos a 100 % de FMA. El caudal es 0,6 mL/min y la detección se logra mediante UV a 220 nm.
Ejemplo 19
Ensayos de liberación de fármaco in vitro
[0177] La liberación de linaclotida desde los comprimidos recubiertos se puede evaluar mediante ensayos de disolución usando un aparato de disolución de paletas USP XXIV tipo II (modelo PTW, Pharma Test, Hainburg, Alemania). Los ensayos se realizan por triplicado, a una velocidad de las paletas de 100 rpm en 900 ml de medio de disolución mantenido a 37,0 ± 0,5 °C. Los comprimidos se ensayan primero en ácido clorhídrico 0,1 N (pH 1,2) durante 30 minutos o 2 horas para simular la residencia gástrica y a continuación durante 6 horas en medios de tampón de pH y composición iónica variables, similares a las condiciones de pH del intestino delgado. La liberación de linaclotida se puede evaluar a pH 6,8-7,4 en dos medios de tampón compendiales: tampón mixto de fosfato de sodio y potasio 0,067 M (Sorensen) y tampón de fosfato de potasio 0,05 M, así como en un tampón de solución salina multielectrolítica (Hanks) de pH 7,4 que es similar en composición iónica al fluido intestinal. Todos los tampones se preparan al momento con agua desionizada y se desairean burbujeando con helio antes de su uso. La cantidad de linaclotida liberada desde la forma farmacéutica se puede determinar mediante cromatografía líquida en gradiente de fase inversa
como se describe en el ejemplo 16.
Ejemplo 20 (Ejemplo de referencia)
Estudios de disolución con perlas recubiertas con pectina y etilcelulosa
[0178] Las formulaciones de tres capas que contienen cantidades diferentes de pectina y Surelease® se pueden preparar y recubrir sobre las perlas de linaclotida a espesores de película diferentes. La formulación de la capa uno (F1) se prepara mezclando una solución de 2 % (p/v) de pectina USP en agua destilada con Surelease® y agua destilada para dar como resultado una relación de pectina (P) a Surelease® (S) de 1:12 en peso. La formulación de la capa dos (F2) se prepara de manera similar, pero usando una solución de 3 % (p/v) de pectina USP en agua destilada con Surelease® y agua destilada para dar como resultado una relación de pectina (P) a Surelease® (S) de 1 :6 en peso. La formulación de la capa tres (F3) se prepara mezclando una solución de 5 % (p/v) de pectina USP en agua destilada con Surelease® y agua destilada para dar como resultado una relación de pectina (P) a Surelease® (S) de 1:3 en peso. El recubrimiento se realiza utilizando una máquina de recubrimiento de lecho fluidizado Aeromatic Strea-1 de laboratorio (Niro-Aeromatic, Columbia, MD). El recubrimiento se realiza a la temperatura de entrada de 50 °C y la solución de recubrimiento se aplicó a través de una boquilla de pulverización de 1 ,0 mm a una velocidad de pulverización de 2 ml/min usando una bomba peristáltica Rabbit (Rainin Instrument Co. Inc., Woburn, MA) y 172,4 kPa (25 psi) de presión del aire de atomización. Las soluciones de recubrimiento se recubren sobre perlas de linaclotida para dar como resultado espesores de recubrimiento diferentes. El espesor de la película se expresa como el porcentaje teórico del peso ganado (GPT) usado con respecto al peso de las perlas no recubiertas. Las perlas se recubrieron con ganancias de peso de 9 %, 14 %, 16 % y 18 % para la formulación de capa F1, 12 %, 25 %, 30 % y 35 % para la formulación de capa F2, y ganancias de peso de 25 %, 35 %, 45 % y 55 % para la formulación de capa F3. Las perlas recubiertas se curaron durante 30 min a 50 °C.
[0179] La liberación de linaclotida desde perlas recubiertas se puede evaluar en una máquina de ensayos de disolución (estación de ensayos de disolución VK 7000, Vankel Industries, Inc., NJ), siguiendo el método de cesta de la USP. Todos los ensayos se realizan en 900 ml de medio de disolución mantenido a 37 ± 0,5 °C con una velocidad de rotación de las paletas de 50 rpm. Los estudios de disolución se llevan a cabo en condiciones que simulan el pH y los tiempos que es probable encontrar durante el tránsito en el tracto GI. Los ensayos se llevan a cabo usando fluido gástrico simulado (FGS) durante 2 h a pH 1,4, seguido de fluido del intestino delgado simulado (FIDS) durante 4 h a pH 7,4, seguido de fluido cecal simulado (FCS) a pH 6 durante al menos 6 h (o hasta liberación del fármaco de 100 %) con o sin la adición de 3 ml de enzimas. El tampón a pH 6 se usó para llegar a un compromiso entre el pH medio del ciego y el pH óptimo para la actividad de las enzimas. La cantidad de linaclotida liberada desde la forma farmacéutica se determina mediante cromatografía líquida en gradiente de fase inversa como se describe en el Ejemplo 16. Para cada experimento de disolución, se realiza un duplicado al mismo tiempo en las mismas condiciones. Después de las 4 h en FIDS, se añaden enzimas pectinolíticas a uno de los recipientes de disolución, pero no al otro. Por tanto, uno es un estudio de liberación con enzimas y el otro es un estudio de liberación sin enzimas. Cada experimento de disolución se repite 3 veces (n = 3).
Ejemplo 21
Perfiles de liberación de comprim idos de linaclotida
[0180] Los perfiles de liberación de comprimidos de linaclotida se han ensayado a diversos pH para tres formulaciones de liberación retardada. La muestra 181-64B contiene una dosis de 100 mg de linaclotida, una subcapa de Opadry® II y una capa funcional de Eudragit® FS 30D y talco. La muestra 181-64C contiene una dosis de 100 mg de linaclotida, una subcapa de Opadry II y una capa funcional de Eudragit® FS 30D y Eudragit® con PlasAcryl™. Los perfiles de liberación para el lote 181-64B y el lote 181-64C se proporcionan en la figura 1. El perfil de liberación de una dosis de 25 mg de linaclotida se proporciona en la figura 2.
Ejemplo 22
Preparación de comprim idos de linaclotida
[0181] Los comprimidos de liberación retardada se pueden preparar preparando primero los componentes del comprimido central siguientes: una base de placebo, una base de 750 pg/225 mg de linaclotida y cargas pregranuladas.
Procedimiento de fabricación de granulaciones:
[0182] Los componentes de los comprimidos se pueden preparar en granulaciones independientes para su mezcla antes de la compresión de los comprimidos. El uso de componentes independientes de los comprimidos, tales como la base de placebo y la base de carga pregranulada proporcionaron, entre otras cosas, propiedades ventajosas para los perfiles de estabilidad y liberación de los comprimidos. Por ejemplo, todos los componentes de los
comprimidos enumerados en la Tabla 12 se podrían preparar independientemente mediante granulación en estado húmedo y mezclar antes de la compresión, o mezclar entre sí y procesar como una mezcla para granulación en estado húmedo. En otro procedimiento, los componentes de los comprimidos enumerados en la Tabla 12 se podrían preparar independientemente mediante granulación en estado seco y mezclar antes de la compresión, o mezclar entre sí y procesar como una mezcla para granulación en estado seco. En otro procedimiento, los componentes de los comprimidos se mezclan directamente para la compresión. En un procedimiento preferido, la base de carga pregranulada y/o la base de placebo se preparan mediante granulación en estado húmedo y se secan antes de mezclar con la base de linaclotida de 750 pg/225 mg. La base de linaclotida se podría preparar mediante procedimientos de granulación en estado húmedo o mediante el procedimiento de recubrimiento Wurster. Este procedimiento preferido, presentó ganancias de estabilidad adicionales para el comprimido reduciendo la exposición a la humedad de la linaclotida durante el procesamiento y minimizando la humedad residual en el núcleo del comprimido.
T l 12: m n n r iv r n n r i n ^ m rimi

[0183] A continuación, comprimir las mezclas anteriores en una prensa de comprimidos adecuada para conseguir un peso de comprimido central de 225 mg. En un bombo de recubrimiento perforado, añadir una subcapa (OPADRY® II) con una ganancia de peso de 4 % p/p. Las condiciones de recubrimiento se deben ajustar y supervisar para que la absorción de humedad durante el recubrimiento se mantenga al mínimo. Cuando se miden mediante pérdida por desecación (LOD), los comprimidos con subcapa no deben tener más de 1,5 % de LOD. En un bombo de recubrimiento perforado, añadir una capa funcional sobre los comprimidos con subcapa. La capa funcional es indistintamente Eudragit® FS30D o Eudragit® S100. Aplicar la capa funcional a 5 mg de peso del polímero/cm2 de la superficie del comprimido. Esto viene a ser aproximadamente 4,5 % de ganancia de peso total del polímero durante el recubrimiento funcional. Las condiciones de recubrimiento se deben ajustar y supervisar para que la absorción de humedad durante el recubrimiento se mantenga al mínimo. Cuando se mide mediante pérdida por desecación, el comprimido recubierto funcionalmente no debe tener más de 2,0 % de LOD.
Preparación de base de placebo:
[0184] La Tabla 13 representa la formulación para la granulación de base de placebo:
T l 1 : F rm l i n l r n l i n l

[0185] La preparación de base de placebo se puede preparar proporcionando primero las materias primas de la Tabla 14.
Tabl 14: M ri rim r l r r i n l cebo

[0186] Tarar el recipiente de mezcla y añadir 2682,1 ± 5,0 g de agua tratada en el recipiente. Instalar una mezcladora y comenzar a agitar el agua. Añadir el EMPROVE® al agua mientras se agita y encender el temporizador. Cubrir y calentar la solución a 70 °C mientras se agita y mantener la temperatura hasta que el material esté visualmente disuelto.
[0187] Ajustar el pH de la solución a 1,5 con ácido clorhídrico. Añadir cloruro de calcio dihidratado a la solución mientras se agita. Mezclar hasta que se disuelva. Añadir L-histidina a la solución mientras se agita. Agitar durante aproximadamente 15 minutos. Registrar el pH inicial. Ajustar el pH de la solución a 5,0 con ácido clorhídrico. Registrar el pH final de la solución y la adición de ácido clorhídrico. Mezclar hasta que todo el material esté disuelto. Mientras se mezcla, ajustar el pH de la solución a 2,5 con ácido clorhídrico. Registrar el pH final de la solución y la adición de ácido clorhídrico. Garantizar que el granulador supercortante esté configurado correctamente para granular con el recipiente de 25 L, la cuchilla de mezcla y el sistema de cuchillas que giran en el plano vertical. Pasar celulosa microcristalina a través de un tamiz de malla 16 al recipiente del granulador. Calcular el peso neto de la solución de granulación que se va a añadir. Bombear la solución de granulación al granulador a una velocidad de aproximadamente 300 g/min mientras se mezcla con los parámetros siguientes: velocidad del impulsor 1 (290 rpm, velocidad de la punta de 5,5 m/s), velocidad del sistema de cuchillas 1 (1760 rpm). Detener el granulador y raspar los lados y el fondo del recipiente. Mezclar durante 3 minutos adicionales según los parámetros siguientes: velocidad del impulsor 1 (290 rpm, velocidad de la punta de 5,5 m/s), velocidad del sistema de cuchillas 1 (1760 rpm). Tarar una bolsa de polietileno y descargar la granulación húmeda finalizada en ella. Pesar la granulación. Transferir la granulación húmeda al lecho fluido FLM-3 para el secado. Secar la granulación usando los parámetros aproximados siguientes. Secar hasta que la LOD de la granulación no sea superior a 1,2 % de humedad. Descargar la granulación seca en una bolsa de polietileno tarada.

[0188] Los parámetros son solo parámetros sugeridos y se pueden ajustar para el secado óptimo.
[0189] Filtrar la granulación seca a través de un tamiz de malla n.° 30. Tarar una bolsa de polietileno y descargar la granulación seca en ella. Pesar la granulación. Envasar la granulación seca en bolsas selladas con papel de aluminio con desecante.
Preparación de base de linaclotida (es decir, 750 ug-225 mg)
[0190] La Tabla 15 representa la formulación para la granulación de base de 750 ug/225 mg:
T l 1 : F rm l i n r l r n l i n 7 22 m

continuación

[0191] La granulación de base de 750 pg/225 mg se puede preparar proporcionando primero las materias primas de la Tabla 16.
Ta l 1 : M ri rim l r n l i n lin l tida

[0192] Mientras se mezcla, añadir la linaclotida a la solución de granulación. Mezclar hasta que se disuelva. Garantizar que el granulador supercortante esté configurado correctamente para granular con el recipiente de 25 L, la cuchilla de mezcla y el sistema de cuchillas que giran en el plano vertical. Pasar celulosa microcristalina a través de un tamiz de malla 16 al recipiente del granulador. Bombear la solución de granulación en el granulador a una velocidad de aproximadamente 300 g/min mientras se mezcla con los parámetros que se indican a continuación: velocidad del impulsor 1 (290 rpm, velocidad de la punta de 5,5 m/s), velocidad del sistema de cuchillas 1 (1760 rpm). Tarar una bolsa de polietileno y descargar la granulación húmeda finalizada en ella. Pesar la granulación. Transferir la granulación húmeda al lecho fluido FLM-3 para el secado. Secar la granulación usando los parámetros aproximados siguientes. Secar hasta que la LOD de la granulación no sea superior a 1,2 % de humedad. Descargar la granulación seca en una bolsa de polietileno tarada.

[0193] Nota: los parámetros son solo parámetros sugeridos y se pueden ajustar para un secado óptimo.
[0194] Filtrar la granulación seca a través de un tamiz de malla n.° 30. Tarar una bolsa de polietileno y descargar la granulación seca en ella. Pesar la granulación. Envasar la granulación seca en bolsas selladas con papel de aluminio con desecante.
Preparación de cargas pregranuladas:
[0195] La Tabla 17 representa la formulación para las cargas pregranuladas.
T l 17: F rm l i n l r n l i n r

continuación

[0196] La preparación de cargas se puede realizar proporcionando primero las materias primas de la Tabla 18.
T l 1 : M ri rim r l r r i n r n l i n r

[0197] A continuación, registrar el peso de tara del recipiente de acero inoxidable. Tarar el recipiente y pesar la cantidad necesaria de agua tratada en el recipiente. Transferir el agua a una caldera con encamisado. Instalar la mezcladora y comenzar a agitar el agua en la caldera. Añadir el EMPROVE® (alcohol polivinílico) al agua mientras agita y encender el temporizador. Cubrir y calentar la solución a 70 °C mientras se agita y mantener la temperatura hasta que el material esté visualmente disuelto. Calcular el peso de la pérdida de agua debida a la evaporación durante el calentamiento. Añadir esta cantidad de agua tratada a la solución. Añadir una bolsa de cada uno de celulosa microcristalina, Ac-Di-Sol (es decir, croscarmelosa de sodio) y Pearlitol 100SD (es decir, manitol) a un recipiente del granulador supercortante. Mezclar durante aproximadamente 2 minutos según los parámetros siguientes: velocidad del impulsor 1 (290 rpm, velocidad de la punta de 5,5 m/s), velocidad del sistema de cuchillas 1 (1760 rpm). Bombear 2217 ± 5 g de la solución de granulación al granulador a una velocidad de aproximadamente 300 g/min mientras se mezcla con los parámetros siguientes: velocidad del impulsor 1 (290 rpm, velocidad de la punta de 5,5 m/s), velocidad del sistema de cuchillas 1 (1760 rpm). Detener el granulador y raspar los lados y el fondo del recipiente. Mezclar durante 30 segundos a 1 minuto adicionales según los parámetros siguientes: velocidad del impulsor 1 (290 rpm, velocidad de la punta de 5,5 m/s), velocidad del sistema de cuchillas 1 (1760 rpm). Tarar una bolsa de polietileno y descargar la granulación húmeda finalizada en ella. Pesar la granulación. Pasar la granulación húmeda a través del molino Comil con filtro 2A375Q03763 con potencia de 5-10 %. Transferir la granulación húmeda al lecho fluido FLM-3 para el secado. Secar la granulación usando los parámetros aproximados siguientes. Secar hasta que la LOD de la granulación no sea superior a 1,0 % de humedad. Descargar la granulación seca en una bolsa de polietileno tarada.

[0198] Los parámetros son solo parámetros sugeridos y se pueden ajustar para el secado óptimo.
[0199] Moler la granulación con el molino Comil, impulsor redondo, filtro 2A045R03137. Realizar el análisis del tamaño de las partículas con un dispositivo de tamizado sónico usando los tamaños de filtro (en micrómetros) siguientes: 38, 75, 106, 150, 180, 250. Tarar una bolsa de polietileno y descargar la granulación seca y molida en ella. Pesar la granulación. Envasar la granulación seca en bolsas selladas con papel de aluminio con desecante.
Ejemplo 23
Mezcla y compresión de comprim idos de 25 pg
[0200] Según el procedimiento del Ejemplo 22, se puede preparar un comprimido de linaclotida de dosis de 25 |jg con la formulación de la Tabla 19.
Tabla 19: Formulación de un^ com rimido de linaclotida de 25 |jg:

[0201] Primero, proporcionar las materias primas de la Tabla 20.
Tabla 20: M ri rim r l r r i n n m rimi lin l i a de 25 jg

[0202] Premezcla: instalar una mezcladora de 7,5 L ( 8 qt) y añadir la base de 750 jg/225 mg y la base de placebo. Cerrar las tapas y mezclar durante 10 minutos. Tarar una bolsa de polietileno y descargar la premezcla en ella.
[0203] Submezcla A: añadir 650 g de la premezcla a una mezcladora en V de 15 L (16 qt). Añadir las cargas pregranuladas para la submezcla A a la mezcladora en V de 15 L (16 qt). Cerrar las tapas y mezclar durante 10 minutos. Pasar el estearato de magnesio para la submezcla A a través del filtro de malla 40. Añadir a la mezcladora en V de 15 L (16 qt). Cerrar las tapas y mezclar durante 3 minutos. Tarar una bolsa de polietileno y descargar la mezcla en ella.
[0204] Submezcla B: añadir 650 g de la premezcla a una mezcladora en V de 15 L (16 qt). Añadir las cargas pregranuladas para la submezcla B a la mezcladora en V de 15 L (16 qt). Cerrar las tapas y mezclar durante 10 minutos. Pasar el estearato de magnesio para la submezcla B a través del filtro de malla 40. Añadir a la mezcladora en V de 15 L (16 qt). Cerrar las tapas y mezclar durante 3 minutos. Tarar una bolsa de polietileno y descargar la mezcla en ella.
[0205] Compresión: garantizar que una prensa de comprimidos Korsch XL 100 con accesorios sc de 0,8 cm (0,32") esté configurada según el SOP E-59 con accesorios cóncavos redondos de 0,8 cm (0,32") (10 estaciones deseadas), alimentador de paletas, y que todos los demás componentes estén sujetos correctamente. Rotar
manualmente la mesa de moldes al menos una revolución completa para garantizar la instalación correcta. Confirmar visualmente la presencia de todos los moldes y troqueles, y que los moldes están nivelados con la mesa de moldes. Verificar que las especificaciones de la mesa cumplen las especificaciones de los comprimidos en proceso de la Tabla 21.
T l 21: E ifi i n l m rimi n r

[0206] Cargar la mezcla en la tolva. Ajustar la velocidad de la torreta a 20 RPM. Ajustar la cantidad de llenado del molde y los parámetros de compresión para producir un comprimido con un peso objetivo de 225 mg y con una dureza objetivo de 0,08 kN-0,12 kN (8-12 kP). Todo el material de desecho se debe recoger en la bolsa de polietileno de residuos de la fabricación de comprimidos. Encender el temporizador y comprimir los comprimidos.
Ejemplo 24
Mezcla y compresión de comprim idos de 100 pg
[0207] Según el procedimiento del Ejemplo 22, se puede preparar un comprimido de linaclotida de dosis de 100 |jg con la formulación de la Tabla 22.
Tabla 22: Formulación de un^ com rimido de linaclotida de 100 jg :

[0208] Primero, proporcionar las materias primas de la Tabla 23.
Tabla 23: M ri rim r l r r i n m rimi lin l i ^de 100 jg

[0209] Premezcla: instalar una mezcladora de 15 L (16 qt) y añadir la base de 750 jg/225 mg y la base de placebo. Cerrar las tapas y mezclar durante 10 minutos. Tarar una bolsa de polietileno y descargar la premezcla en ella.
[0210] Añadir 2500 g de la premezcla a una mezcladora en V de 0,03 metros cúbicos (1 pie cúbico). Añadir las
cargas pregranuladas a la mezcladora en V de 0,03 metros cúbicos (1 pie cúbico). Cerrar las tapas y mezclar durante 10 minutos. Pasar el estearato de magnesio a través del filtro de malla 40. Añadir a la mezcladora en V de 0,08 metros cúbicos (1 pie cúbico). Cerrar las tapas y mezclar durante 3 minutos. Tarar una bolsa de polietileno y descargar la mezcla en ella.
[0211] Compresión: garantizar que una prensa de comprimidos Korsch XL 100 con accesorios sc de 0,8 cm (0,32") esté configurada según el SOP E-59 con accesorios cóncavos redondos de 0,8 cm (0,32") (10 estaciones deseadas), alimentador de paletas, y que todos los demás componentes estén sujetos correctamente. Rotar manualmente la mesa de moldes al menos una revolución completa para garantizar la instalación correcta. Confirmar visualmente la presencia de todos los moldes y troqueles, y que los moldes están nivelados con la mesa de moldes. Verificar que las especificaciones de la mesa cumplen las especificaciones de los comprimidos en proceso de la Tabla 24.
T l 24: E ifi i n l m rimi n r

[0212] Cargar la mezcla en la tolva. Ajustar la velocidad de la torreta a 20 RPM. Ajustar la cantidad de llenado del molde y los parámetros de compresión para producir un comprimido con un peso objetivo de 225 mg y con una dureza objetivo de 0,08 kN-0,12 kN (8-12 kP). Todo el material de desecho se debe recoger en la bolsa de polietileno de residuos de la fabricación de comprimidos. Encender el temporizador y comprimir los comprimidos.
Ejemplo 25
Mezcla y compresión de comprim idos de 290 pg
[0213] Según el procedimiento del Ejemplo 22, se puede preparar un comprimido de linaclotida de dosis de 290 |jg con la formulación de la Tabla 25.
Tabla 25: Formulación de un com rimido de linaclotida de 290 |jg:

[0214] Primero, proporcionar las materias primas de la Tabla 26.
Tabla 26: M ri rim r l r r i n n m rimi lin l i de 290 jg

continuación

[0215] Submezcla A: añadir las cargas pregranuladas y la base de linaclotida para la submezcla A a una mezcladora en V de 15 L (16 qt). Cerrar las tapas y mezclar durante 10 minutos. Pasar el estearato de magnesio para la submezcla A a través del filtro de malla 40. Añadir a la mezcladora en V de 15 L (16 qt). Cerrar las tapas y mezclar durante 3 minutos. Tarar una bolsa de polietileno y descargar la mezcla en ella.
[0216] Submezcla B: añadir las cargas pregranuladas y la base de linaclotida para la submezcla B a una mezcladora en V de 15 L (16 qt). Cerrar las tapas y mezclar durante 10 minutos. Pasar el estearato de magnesio para la submezcla B a través del filtro de malla 40. Añadir a la mezcladora en V de 15 L (16 qt). Cerrar las tapas y mezclar durante 3 minutos. Tarar una bolsa de polietileno y descargar la mezcla en ella.
[0217] Compresión: garantizar que una prensa de comprimidos Korsch XL 100 con accesorios sc de 0,8 cm (0,32") esté configurada según el SOP E-59 con accesorios cóncavos redondos de 0,8 cm (0,32") (10 estaciones deseadas), alimentador de paletas, y que todos los demás componentes estén sujetos correctamente. Rotar manualmente la mesa de moldes al menos una revolución completa para garantizar la instalación correcta. Confirmar visualmente la presencia de todos los moldes y troqueles, y que los moldes están nivelados con la mesa de moldes. Verificar que las especificaciones de la mesa cumplen las especificaciones de los comprimidos en proceso de la Tabla 27.
T l 27: E ifi i n l m rimi n r

[0218] Cargar la mezcla en la tolva. Ajustar la velocidad de la torreta a 20 RPM. Ajustar la cantidad de llenado del molde y los parámetros de compresión para producir un comprimido con un peso objetivo de 225 mg y con una dureza objetivo de 0,08 kN-0,12 kN (8-12 kP). Todo el material de desecho se debe recoger en la bolsa de polietileno de residuos de la fabricación de comprimidos. Encender el temporizador y comprimir los comprimidos.
Ejemplo 26
Subrecubrim iento de comprim idos de 25 pg de linaclotida
[0219] Los comprimidos de 25 |jg del Ejemplo 23 se pueden recubrir con un subrecubrimiento de Opadry® II. La fórmula de la Tabla 28 representa las cantidades necesarias para preparar el material de recubrimiento, que se prepara en exceso para tener en cuenta las pérdidas en el procedimiento de recubrimiento.
Tabla 28: F rm l r rimi n r m rimi de 25 |jg

[0220] Añadir 1600 g de agua purificada en un recipiente de dimensiones adecuadas. Añadir 400 g de Opadry® II en un recipiente de dimensiones adecuadas. Añadir Opadry® al agua. Calcular la cantidad teórica de solución necesaria para conseguir una ganancia de peso de 4,0 %, con una eficacia teórica de 85 %. Preparar una bolsa de polietileno para los comprimidos residuales recogidos durante el procedimiento de recubrimiento. Garantizar que el Compu-Lab se haya configurado con el bombo de 48 cm (19 pulgadas) y la cámara. Verificar que los conductos de alimentación de líquido sean tubos Tygon 17. Verificar que la pistola esté instalada en el bombo. La pistola de pulverización debe consistir en 1 pistola de pulverización JAU de % equipada con una boquilla de pulverización de líquidos 40100 AB y un soporte para la boquilla de aire compatible. La pistola se debe instalar tan lejos como sea posible del lecho de comprimidos, con el ángulo de pulverización perpendicular al tercio superior del lecho. Verificar el peso inicial de los comprimidos pesando 100 comprimidos sin recubrimiento. Calcular el peso promedio de un único comprimido dividiendo el peso de los comprimidos por 100. Analizar la humedad inicial de los comprimidos triturando aproximadamente 1 gramo de comprimidos y ejecutando un análisis de LOD durante 10 minutos a 105 °C. Ajustar la bomba para que el caudal de líquido sea aproximadamente 10 g/min. Cebar los conductos situados después de las pistolas y verificar que no haya fugas en los conductos o la pistola. Cargar los comprimidos en el bombo de recubrimiento y comenzar a calentar durante 20 minutos con una temperatura de entrada de 50 °C y un flujo de aire de 0,16 m3/s (350 CFM). Mover de vez en cuando durante el calentamiento.
[0221] Una vez que se ha alcanzado la temperatura objetivo del lecho, comenzar a pulverizar la suspensión de recubrimiento según los parámetros de proceso objetivo descritos en la Tabla 29 más adelante. Analizar y presentar el contenido de humedad de los comprimidos periódicamente según se desee. Una vez que se ha aplicado la cantidad teórica de solución, comprobar la ganancia de peso de los comprimidos. Cuando se haya conseguido una ganancia de peso de 4 %, parar de pulverizar y secar los comprimidos durante 30 minutos con una temperatura de entrada de 50 °C, reduciendo la velocidad del bombo al mínimo o moviendo. Descargar los comprimidos y determinar el nuevo peso de los comprimidos recubiertos.
T l 2 : P r m r r iv

Ejemplo 27
Subrecubrim iento de comprim idos de linaclotida de 100 pg
[0222] Los comprimidos de 100 |jg del Ejemplo 24 se pueden recubrir con un subrecubrimiento de Opadry® II. La fórmula de la Tabla 30 representa las cantidades necesarias para preparar el material de recubrimiento, que se prepara en exceso para tener en cuenta las pérdidas en el procedimiento de recubrimiento.
Tabla 30: F rm l r rimi n r ^ m rimi e 100 jg

[0223] Añadir 4000 g de agua purificada en un recipiente de dimensiones adecuadas. Añadir 1000 g de Opadry® II en un recipiente de dimensiones adecuadas. Añadir Opadry® al agua. Calcular la cantidad teórica de solución necesaria para conseguir una ganancia de peso de 4,0 %, con una eficacia teórica de 85 %. Garantizar que
el Compu-Lab se haya configurado con el bombo de 61 cm (24 pulgadas) y la cámara. Verificar que los conductos de alimentación de líquido sean tubos Tygon 17. Verificar que la pistola esté instalada en el bombo. La pistola de pulverización debe consistir en 2 pistolas de pulverización JAU de % equipadas con una boquilla de pulverización de líquidos 40100 AB y un soporte para la boquilla de aire compatible. La pistola se debe instalar tan lejos como sea posible del lecho de comprimidos, con el ángulo de pulverización perpendicular al tercio superior del lecho. Verificar el peso inicial de los comprimidos pesando 100 comprimidos sin recubrimiento. Calcular el peso promedio de un único comprimido dividiendo el peso de los comprimidos por 100. Analizar la humedad inicial de los comprimidos triturando aproximadamente 1 gramo de comprimidos y ejecutando un análisis de LOD durante 10 minutos a 105 °C. Ajustar la bomba para que el caudal de líquido sea aproximadamente 20 g/min. Cebar los conductos situados después de las pistolas y verificar que no haya fugas en los conductos o la pistola. Cargar los comprimidos en el bombo de recubrimiento y comenzar a calentar durante 20 minutos con una temperatura de entrada de 50 °C y un flujo de aire de 0,16 m3/s (400 CFM). Mover de vez en cuando durante el calentamiento.
[0224] Una vez que se ha alcanzado la temperatura objetivo del lecho, comenzar a pulverizar la suspensión de recubrimiento según los parámetros de proceso objetivo descritos en la Tabla 31 más adelante. Analizar y presentar el contenido de humedad de los comprimidos periódicamente según se desee. Una vez que se ha aplicado la cantidad teórica de solución, comprobar la ganancia de peso de los comprimidos. Cuando se haya conseguido una ganancia de peso de 4 %, parar de pulverizar y secar los comprimidos durante 30 minutos con una temperatura de entrada de 50 °C, reduciendo la velocidad del bombo al mínimo o moviendo. Descargar los comprimidos y determinar el nuevo peso de los comprimidos recubiertos.
T l 1: P r m r r iv

Ejemplo 28
Subrecubrim iento de comprim idos de linaclotida de 290 pg
[0225] Los comprimidos de 290 |jg del Ejemplo 25 se pueden recubrir con un subrecubrimiento de Opadry® II. La fórmula de la Tabla 32 representa las cantidades necesarias para preparar el material de recubrimiento, que se prepara en exceso para tener en cuenta las pérdidas en el procedimiento de recubrimiento.
Tabla 32: F rm l r rimi n r ^ m rimi e 100 jg

[0226] Añadir 1600 g de agua purificada en un recipiente de dimensiones adecuadas. Añadir 400 g de Opadry® II en un recipiente de dimensiones adecuadas. Añadir Opadry® al agua. Calcular la cantidad teórica de solución necesaria para conseguir una ganancia de peso de 4,0 %, con una eficacia teórica de 85 %. Garantizar que el Compu-Lab se haya configurado con el bombo de 48 cm (19 pulgadas) y la cámara. Verificar que los conductos de alimentación de líquido sean tubos Tygon 17. Verificar que la pistola esté instalada en el bombo. La pistola de pulverización debe consistir en 1 pistola de pulverización JAU de % equipada con una boquilla de pulverización de líquidos 40100 AB y un soporte para la boquilla de aire compatible. La pistola se debe instalar tan lejos como sea posible del lecho de comprimidos, con el ángulo de pulverización perpendicular al tercio superior del lecho. Verificar
el peso inicial de los comprimidos pesando 100 comprimidos sin recubrimiento. Calcular el peso promedio de un único comprimido dividiendo el peso de los comprimidos por 100. Analizar la humedad inicial de los comprimidos triturando aproximadamente 1 gramo de comprimidos y ejecutando un análisis de LOD durante 10 minutos a 105 °C. Ajustar la bomba para que el caudal de líquido sea aproximadamente 10 g/min. Cebar los conductos situados después de las pistolas y verificar que no haya fugas en los conductos o la pistola. Cargar los comprimidos en el bombo de recubrimiento y comenzar a calentar durante 20 minutos con una temperatura de entrada de 50 °C y un flujo de aire de 0,16 m3/s (350 CFM). Mover de vez en cuando durante el calentamiento.
[0227] Una vez que se ha alcanzado la temperatura objetivo del lecho, comenzar a pulverizar la suspensión de recubrimiento según los parámetros de proceso objetivo descritos en la Tabla 33 más adelante. Analizar y presentar el contenido de humedad de los comprimidos periódicamente según se desee. Una vez que se ha aplicado la cantidad teórica de solución, comprobar la ganancia de peso de los comprimidos. Cuando se haya conseguido una ganancia de peso de 4 %, parar de pulverizar y secar los comprimidos durante 30 minutos con una temperatura de entrada de 50 °C, reduciendo la velocidad del bombo al mínimo o moviendo. Descargar los comprimidos y determinar el nuevo peso de los comprimidos recubiertos.
T l : P r m r r iv

Ejemplo 29
Recubrimiento funcional o entérico de comprim idos de linaclotida
[0228] Los comprimidos de los ejemplos anteriores se pueden preparar con un recubrimiento funcional. La formulación de la Tabla 34 representa las cantidades necesarias para preparar el material de recubrimiento, que se prepara en exceso para tener en cuenta las pérdidas en el procedimiento de recubrimiento:
Tabla 34: F rm l i n r l r imi n r rimi n ^ funcional
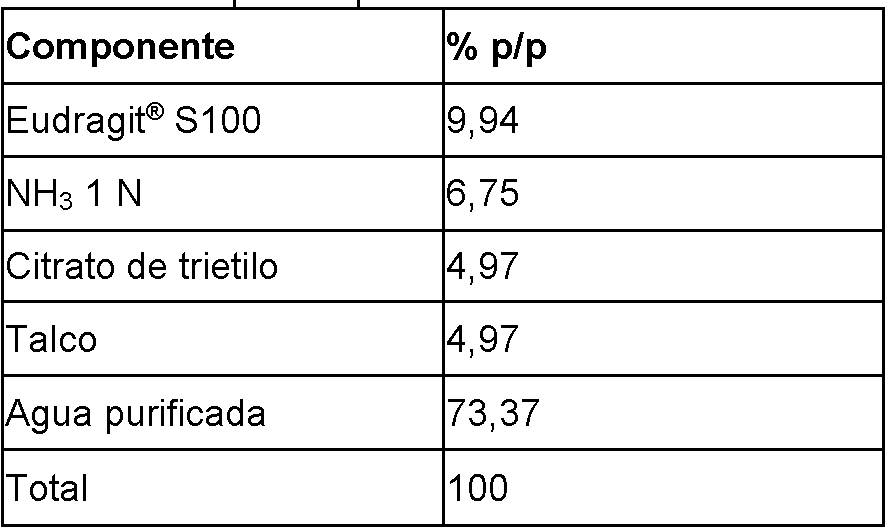
[0229] Para preparar el recubrimiento funcional, añadir la cantidad necesaria de Eudragit® S100 en un recipiente de dimensiones adecuadas. Añadir 2/3 de la cantidad necesaria de agua purificada en un recipiente de dimensiones adecuadas. Comenzar la agitación del agua hasta que se consiga un vórtice. Añadir S100 lentamente al agua y mezclar hasta que el polvo esté completamente humedecido y la formación de grumos o espuma se haya disipado (aproximadamente 5 minutos).
[0230] Añadir la cantidad necesaria de NH3 1 N en un recipiente de dimensiones adecuadas. Añadir el NH3 1 N lentamente a la suspensión de Eudragit® y mezclar durante un mínimo de 60 minutos. Añadir la cantidad necesaria
de citrato de trietilo en un recipiente de dimensiones adecuadas. Añadir el citrato de trietilo a la suspensión de Eudragit® y mezclar durante un mínimo de 60 minutos. Añadir la cantidad necesaria de talco en un recipiente de dimensiones adecuadas. Homogeneizar el talco en el 1/3 restante de agua purificada durante 10 minutos (o hasta homogeneidad) usando un homogeneizador Silverson. Verter la suspensión de talco en la suspensión de Eudragit® mientras se mezcla. Mezclar durante como máximo 5 minutos. Filtrar la suspensión de recubrimiento a través de un filtro de malla n.° 30.
[0231] Calcular la cantidad teórica de solución necesaria para conseguir una ganancia de peso de 9,0 %, con una eficacia teórica de 90 %. Garantizar que el Compu-Lab se haya configurado con el bombo de 48 cm (19 pulgadas) y la cámara. Verificar que los conductos de alimentación de líquido sean tubos Tygon 17. Verificar que la pistola esté instalada en el bombo. La pistola de pulverización debe consistir en 1 pistola de pulverización JAU de % equipada con una boquilla de pulverización de líquidos 40100 AB y un soporte para la boquilla de aire compatible. La pistola se debe instalar tan lejos como sea posible del lecho de comprimidos, con el ángulo de pulverización perpendicular al tercio superior del lecho. Cargar los comprimidos en el bombo de recubrimiento. Calentar los comprimidos con una temperatura de entrada de 30 grados. Garantizar que la temperatura del lecho alcance aproximadamente 30 grados antes de continuar con la siguiente etapa. Ajustar la bomba para que el caudal de líquido sea aproximadamente 12 g/min. Cebar los conductos situados después de las pistolas y verificar que no haya fugas en los conductos o la pistola. Analizar la humedad inicial de los comprimidos triturando aproximadamente 2 gramos de comprimidos y ejecutando un análisis de LOD durante 10 minutos a 105 °C.
[0232] Una vez que se ha alcanzado la temperatura objetivo del lecho, comenzar a pulverizar la suspensión de recubrimiento según los parámetros de proceso objetivo descritos en la Tabla 35 más adelante. Analizar y presentar el contenido de humedad de los comprimidos periódicamente según se desee. Una vez que se ha aplicado la cantidad teórica de solución, comprobar la ganancia de peso de los comprimidos. Cuando se haya conseguido una ganancia de peso de 9 %, parar de pulverizar y secar los comprimidos durante 5-10 minutos con una temperatura de entrada de 40 °C, reduciendo la velocidad del bombo al mínimo o moviendo.
Tabla 35: Parám r r iv r l r imi n r rimi n por pulverización

[0233] Analizar la humedad final de los comprimidos triturando aproximadamente 2 gramos de comprimidos y ejecutando un análisis de LOD durante 10 minutos a 105 °C. La humedad debe ser < la humedad inicial de los comprimidos. Descargar los comprimidos y determinar el nuevo peso de los comprimidos recubiertos. Secar los comprimidos durante al menos 2 horas en un horno de convección mecánica con la temperatura programada a 40 °C. Permitir que los comprimidos alcancen 25-30 °C antes de envasar a granel. Envasar a granel los comprimidos en una bolsa de aluminio con desecantes y almacenar a 5 °C.
Ejemplo 30 (Ejemplo de referencia)
Formulación de cápsulas de linaclotida resistentes a ácidos y cápsulas resistentes a ácidos recubiertas
[0234] Las cápsulas de linaclotida resistentes a ácidos y las cápsulas recubiertas se pueden preparar para liberación dirigida. Una formulación de una cápsula de linaclotida resistente a ácidos se proporciona en la Tabla 36.
Tabla 36: Formulación de una cá sula de linaclotida resistente a ácidos

continuación

[0235] Las cápsulas resistentes a ácidos también pueden incluir un recubrimiento. En la Tabla 35 se proporciona una formulación de cápsulas de linaclotida resistentes a ácidos. La figura 3 muestra un perfil de disolución de cápsulas resistentes a ácidos no recubiertas y recubiertas en medios ácidos.

OTROS CASOS
[0236] La presente invención no debe estar limitada en su alcance por los casos específicos descritos en esta memoria. De hecho, diversas modificaciones de la invención, además de las descritas en esta memoria, resultarán evidentes para los expertos en la materia a partir de la descripción anterior y las figuras adjuntas. Tales modificaciones también están destinadas a estar comprendidas dentro del alcance de las reivindicaciones adjuntas. Además, se debe entender que todos los valores son aproximados y se proporcionan con fines descriptivos.
Claims (21)
1. Composición de liberación retardada que comprende un comprimido con recubrimiento entérico, en la que el comprimido comprende:
linaclotida;
Ca2+;
histidina; y
alcohol polivinílico (PVA).
2. Composición de la reivindicación 1, en la que el recubrimiento entérico comprende copolímeros de acrilato de metiloácido metacrílico; acetato-succinato de celulosa (CAS); ftalato de hidroxipropilmetilcelulosa (HPMCP); acetatosuccinato de hidroxipropilmetilcelulosa (HPMCAS); acetato-ftalato de polivinilo (PVAP); copolímeros de metacrilato de metilo-ácido metacrílico; alginato de sodio y ácido esteárico; goma guar; o mezclas de los mismos.
3. Composición de la reivindicación 1 o 2, en la que el recubrimiento entérico se selecciona de entre copolímeros de metacrilato de metilo-ácido metacrílico.
4. Composición de cualquiera de las reivindicaciones 1-3, que comprende además una película polimérica o un subrecubrimiento protectores.
5. Composición de la reivindicación 4, en la que la composición comprende PVA adicional como capa protectora entre el comprimido y el recubrimiento entérico.
6. Composición de cualquiera de las reivindicaciones 1-5, en la que la composición libera al menos 70% de la linaclotida a un pH superior a 5 o 7.
7. Composición de cualquiera de las reivindicaciones 1-5, en la que la composición libera al menos 80% de la linaclotida a un pH superior a 5 o 7.
8. Composición de cualquiera de las reivindicaciones 1-7, en la que la composición tiene una velocidad de disgregación inferior a 30 segundos a un pH superior a 5 o 7.
9. Composición de cualquiera de las reivindicaciones 1-8, en la que la composición libera linaclotida en el íleon, el íleon terminal o el colon.
10. Composición de cualquiera de las reivindicaciones 1-9, en la que la composición se disgrega en el íleon o el colon.
11. Composición de la reivindicación 1, en la que la composición comprende un polímero sensible al pH.
12. Forma de dosificación unitaria que comprende la composición de cualquiera de las reivindicaciones 1-11.
13. Forma de dosificación unitaria de la reivindicación 12, en la que la linaclotida está presente en la composición en una cantidad entre 1 |jg y 300 |jg.
14. Composición de cualquiera de las reivindicaciones 1-11 para uso en un método para tratar un trastorno gastrointestinal en un paciente.
15. Composición para uso de la reivindicación 14, en la que el trastorno gastrointestinal se selecciona del grupo que consiste en: síndrome del intestino irritable (SII), estreñimiento, un trastorno gastrointestinal funcional, enfermedad por reflujo gastroesofágico, ardor gástrico funcional, dispepsia, diverticulitis, dolor visceral, dolor abdominal, gastroparesia, seudoobstrucción intestinal crónica, seudoobstrucción colónica, enfermedad de Crohn, colitis ulcerosa y enfermedad inflamatoria intestinal.
16. Composición para uso de la reivindicación 15, en la que el trastorno gastrointestinal es estreñimiento.
17. Composición para uso de la reivindicación 16, en la que el estreñimiento es estreñimiento crónico, estreñimiento idiopático, estreñimiento idiopático crónico, estreñimiento debido a íleo posoperatorio o estreñimiento provocado por el uso de opiáceos.
18. Composición para uso de la reivindicación 15, en la que el trastorno gastrointestinal es síndrome del intestino irritable (SII).
19. Composición para su uso de la reivindicación 18, en la que el síndrome del intestino irritable es síndrome del intestino irritable con predominio de estreñimiento (SII-e), síndrome del intestino irritable con predominio de diarrea
(SII-d) o síndromes del intestino irritable mixtos (SII-m).
20. Composición de cualquiera de las reivindicaciones 1-11 para uso en un método para tratar o aliviar el dolor en un paciente.
21. Composición para uso de la reivindicación 20, en la que el dolor se selecciona de entre dolor visceral; dolor por diverticulitis; dolor pélvico; dolor abdominal; o dolor asociado a trastornos gastrointestinales, enfermedades venéreas, endometriosis, vulvodinia, síndrome de vejiga dolorosa o cistitis intersticial.
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201361914952P | 2013-12-11 | 2013-12-11 | |
| US201361914951P | 2013-12-11 | 2013-12-11 | |
| PCT/US2014/069838 WO2015089326A1 (en) | 2013-12-11 | 2014-12-11 | Delayed release compositions of linaclotide |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2838007T3 true ES2838007T3 (es) | 2021-07-01 |
Family
ID=52293217
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES14824655T Active ES2838007T3 (es) | 2013-12-11 | 2014-12-11 | Composiciones de liberación retardada de linaclotida |
Country Status (29)
| Country | Link |
|---|---|
| US (7) | US20160310559A1 (es) |
| EP (2) | EP3079669B1 (es) |
| JP (4) | JP6964380B2 (es) |
| KR (3) | KR102337809B1 (es) |
| CN (3) | CN115089556A (es) |
| AU (3) | AU2014362220B2 (es) |
| CA (1) | CA2933587C (es) |
| CL (1) | CL2016001424A1 (es) |
| CY (1) | CY1123953T1 (es) |
| DK (1) | DK3079669T3 (es) |
| EA (1) | EA201691216A1 (es) |
| EC (2) | ECSP16059106A (es) |
| ES (1) | ES2838007T3 (es) |
| HR (1) | HRP20201753T1 (es) |
| HU (1) | HUE052981T2 (es) |
| IL (2) | IL246155A0 (es) |
| LT (1) | LT3079669T (es) |
| MX (2) | MX386584B (es) |
| NZ (2) | NZ721952A (es) |
| PE (2) | PE20161373A1 (es) |
| PH (1) | PH12016501122B1 (es) |
| PL (1) | PL3079669T3 (es) |
| PT (1) | PT3079669T (es) |
| RS (1) | RS61133B1 (es) |
| SG (2) | SG10201804817TA (es) |
| SI (1) | SI3079669T1 (es) |
| SM (1) | SMT202000686T1 (es) |
| UA (1) | UA119335C2 (es) |
| WO (2) | WO2015089335A1 (es) |
Families Citing this family (19)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| UA119335C2 (uk) * | 2013-12-11 | 2019-06-10 | Айронвуд Фармасьютикалз, Інк. | Композиції лінаклотиду з затриманим вивільненням |
| TW201618783A (zh) | 2014-08-07 | 2016-06-01 | 艾森塔製藥公司 | 以布魯頓(Bruton)氏酪胺酸激酶(BTK)佔據和BTK再合成速率為基礎之治療癌症、免疫和自體免疫疾病及發炎性疾病之方法 |
| CA2936740C (en) | 2014-10-31 | 2017-10-10 | Purdue Pharma | Methods and compositions particularly for treatment of attention deficit disorder |
| WO2016125064A1 (en) * | 2015-02-02 | 2016-08-11 | Aurobindo Pharma Ltd | Stable compositions comprising linaclotide |
| EP3302440B1 (en) * | 2015-06-05 | 2021-01-06 | Ironwood Pharmaceuticals, Inc. | Modified or targeted release formulations of linaclotide |
| WO2017156214A1 (en) * | 2016-03-11 | 2017-09-14 | Gateway Pharmaceutical LLC | Pharmaceutical compositions for colon-specific delivery |
| US10588864B2 (en) | 2016-03-11 | 2020-03-17 | Gateway Pharmaceuticals LLC | Pharmaceutical compositions for colon-specific delivery |
| JP6957610B2 (ja) * | 2016-10-06 | 2021-11-02 | スキャンポ・アーゲーSucampo AG | 医薬品用途のための多層ビーズ |
| MX2019007574A (es) * | 2016-12-21 | 2019-09-04 | Ironwood Pharmaceuticals Inc | Metodos de tratamiento de sindrome de intestino irritable con formulaciones modificadas o de liberacion retardada de linaclotida. |
| KR102227486B1 (ko) * | 2017-06-30 | 2021-03-12 | 롯데정밀화학 주식회사 | 프로톤 펌프 저해제를 포함하는 경구용 고형제제 조성물, 이를 포함하는 경구용 고형제제 및 그 제조방법 |
| CA3095036A1 (en) * | 2018-03-23 | 2019-09-26 | Palatin Technologies, Inc. | Melanocortin receptor-specific peptide formulations and methods for gastrointestinal tract-specific delivery |
| US10722473B2 (en) | 2018-11-19 | 2020-07-28 | Purdue Pharma L.P. | Methods and compositions particularly for treatment of attention deficit disorder |
| WO2020251923A1 (en) * | 2019-06-10 | 2020-12-17 | Ironwood Pharmaceuticals, Inc. | Treatment of abdominal pain associated with diarrhea-predominant irritable bowel syndrome |
| KR102104507B1 (ko) * | 2019-08-23 | 2020-04-24 | 브릿지바이오테라퓨틱스(주) | 팔미토일-l-프롤릴-l-프롤릴-글리실-l-타이로신 나트륨을 포함하는 약제학적 제제 및 이의 제조방법 |
| CN110935007B (zh) * | 2019-12-12 | 2023-06-23 | 烟台大学 | 利那洛肽复方组合物、制剂及其用途和制备方法 |
| GB202017863D0 (en) * | 2020-11-12 | 2020-12-30 | Intract Pharma Ltd | Novel compositions |
| WO2022245650A1 (en) * | 2021-05-19 | 2022-11-24 | Alberto Paz | Orally administered compositions for cancer treatment |
| AU2023356242A1 (en) * | 2022-10-03 | 2025-04-24 | Brim Biotechnology, Inc. | Compositions comprising pedf-derived short peptides (pdsp) and uses thereof |
| JPWO2024143501A1 (es) * | 2022-12-28 | 2024-07-04 |
Family Cites Families (18)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7304036B2 (en) | 2003-01-28 | 2007-12-04 | Microbia, Inc. | Methods and compositions for the treatment of gastrointestinal disorders |
| US7371727B2 (en) | 2003-01-28 | 2008-05-13 | Microbia, Inc. | Methods and compositions for the treatment of gastrointestinal disorders |
| CN201252259Y (zh) * | 2008-07-24 | 2009-06-03 | 富士康(昆山)电脑接插件有限公司 | 电连接器 |
| PE20110543A1 (es) * | 2008-08-15 | 2011-08-04 | Ironwood Pharmaceuticals Inc | Formulacion solida oral que comprende linaclotida, cation de calcio y leucina |
| US20100221329A1 (en) * | 2008-12-03 | 2010-09-02 | Synergy Pharmaceuticals, Inc. | Formulations of guanylate cyclase c agonists and methods of use |
| US20130012454A1 (en) * | 2009-07-06 | 2013-01-10 | Ironwood Pharmaceuticals, Inc. | Orally Disintegrating Compositions of Linaclotide |
| CA2770077A1 (en) * | 2009-08-06 | 2011-02-10 | Ironwood Pharmaceuticals, Inc. | Formulations comprising linaclotide |
| MX2012001691A (es) * | 2009-08-12 | 2012-02-29 | Forest Lab Holdings Ltd | Composiciones de linaclotida que se desintegran oralmente. |
| WO2011020054A1 (en) * | 2009-08-13 | 2011-02-17 | Ironwood Pharmaceuticals Inc. | Method for modulating the pharmacodynamic effect of orally administered guanylate cyclase receptor agonists |
| UA108636C2 (xx) * | 2010-02-17 | 2015-05-25 | Пептид | |
| EP2603232B1 (en) * | 2010-08-11 | 2019-09-25 | Ironwood Pharmaceuticals, Inc. | Stable formulations of linaclotide |
| CA2811001A1 (en) * | 2010-09-11 | 2012-03-15 | Ironwood Pharmaceuticals, Inc. | Treatment of constipation-predominant irritable bowel syndrome |
| JP2012155108A (ja) * | 2011-01-26 | 2012-08-16 | Kyocera Document Solutions Inc | 現像ローラ、現像装置および画像形成装置 |
| MX347354B (es) * | 2011-08-17 | 2017-04-24 | Ironwood Pharmaceuticals Inc | Tratamientos para trastornos gastrointestinales. |
| WO2013047795A1 (ja) * | 2011-09-30 | 2013-04-04 | アステラス製薬株式会社 | 粒子状医薬組成物 |
| JP2016521249A (ja) * | 2012-07-12 | 2016-07-21 | フォレスト ラボラトリーズ ホールディングス リミテッド | リナクロチド組成物 |
| WO2014131024A2 (en) * | 2013-02-25 | 2014-08-28 | Synergy Pharmaceuticals Inc. | Agonists of guanylate cyclase and their uses |
| UA119335C2 (uk) * | 2013-12-11 | 2019-06-10 | Айронвуд Фармасьютикалз, Інк. | Композиції лінаклотиду з затриманим вивільненням |
-
2014
- 2014-11-12 UA UAA201607434A patent/UA119335C2/uk unknown
- 2014-12-11 CN CN202210517834.4A patent/CN115089556A/zh active Pending
- 2014-12-11 KR KR1020167018606A patent/KR102337809B1/ko active Active
- 2014-12-11 MX MX2016007625A patent/MX386584B/es unknown
- 2014-12-11 DK DK14824655.6T patent/DK3079669T3/da active
- 2014-12-11 EA EA201691216A patent/EA201691216A1/ru unknown
- 2014-12-11 SM SM20200686T patent/SMT202000686T1/it unknown
- 2014-12-11 SI SI201431715T patent/SI3079669T1/sl unknown
- 2014-12-11 WO PCT/US2014/069851 patent/WO2015089335A1/en not_active Ceased
- 2014-12-11 SG SG10201804817TA patent/SG10201804817TA/en unknown
- 2014-12-11 PE PE2016000804A patent/PE20161373A1/es unknown
- 2014-12-11 US US15/103,616 patent/US20160310559A1/en not_active Abandoned
- 2014-12-11 CN CN201480075352.1A patent/CN106659687A/zh active Pending
- 2014-12-11 NZ NZ721952A patent/NZ721952A/en unknown
- 2014-12-11 NZ NZ759512A patent/NZ759512A/en unknown
- 2014-12-11 PT PT148246556T patent/PT3079669T/pt unknown
- 2014-12-11 JP JP2016538745A patent/JP6964380B2/ja active Active
- 2014-12-11 CA CA2933587A patent/CA2933587C/en active Active
- 2014-12-11 US US15/103,634 patent/US20160310560A1/en not_active Abandoned
- 2014-12-11 EP EP14824655.6A patent/EP3079669B1/en active Active
- 2014-12-11 KR KR1020237008167A patent/KR20230039764A/ko not_active Ceased
- 2014-12-11 SG SG11201604729QA patent/SG11201604729QA/en unknown
- 2014-12-11 HR HRP20201753TT patent/HRP20201753T1/hr unknown
- 2014-12-11 WO PCT/US2014/069838 patent/WO2015089326A1/en not_active Ceased
- 2014-12-11 EP EP20198666.8A patent/EP3821881A1/en active Pending
- 2014-12-11 PL PL14824655T patent/PL3079669T3/pl unknown
- 2014-12-11 CN CN202011156517.1A patent/CN112569199A/zh active Pending
- 2014-12-11 RS RS20201435A patent/RS61133B1/sr unknown
- 2014-12-11 PE PE2021000748A patent/PE20211976A1/es unknown
- 2014-12-11 PH PH1/2016/501122A patent/PH12016501122B1/en unknown
- 2014-12-11 HU HUE14824655A patent/HUE052981T2/hu unknown
- 2014-12-11 ES ES14824655T patent/ES2838007T3/es active Active
- 2014-12-11 LT LTEP14824655.6T patent/LT3079669T/lt unknown
- 2014-12-11 KR KR1020217039962A patent/KR102509291B1/ko active Active
- 2014-12-11 AU AU2014362220A patent/AU2014362220B2/en active Active
-
2016
- 2016-06-09 IL IL246155A patent/IL246155A0/en unknown
- 2016-06-10 CL CL2016001424A patent/CL2016001424A1/es unknown
- 2016-06-10 MX MX2021011906A patent/MX2021011906A/es unknown
- 2016-07-08 EC ECIEPI201659106A patent/ECSP16059106A/es unknown
-
2017
- 2017-02-17 US US15/435,701 patent/US20170157200A1/en not_active Abandoned
- 2017-09-28 US US15/718,075 patent/US20180015139A1/en not_active Abandoned
-
2019
- 2019-02-21 JP JP2019029432A patent/JP2019073559A/ja active Pending
- 2019-09-11 US US16/567,082 patent/US20200038476A1/en not_active Abandoned
-
2020
- 2020-04-01 AU AU2020202319A patent/AU2020202319B2/en active Active
- 2020-11-26 CY CY20201101128T patent/CY1123953T1/el unknown
-
2021
- 2021-05-06 IL IL283012A patent/IL283012A/en unknown
- 2021-05-06 JP JP2021078445A patent/JP7483656B2/ja active Active
- 2021-09-28 EC ECSENADI202172161A patent/ECSP21072161A/es unknown
- 2021-10-18 US US17/503,601 patent/US20220031802A1/en not_active Abandoned
-
2022
- 2022-03-18 AU AU2022201891A patent/AU2022201891A1/en not_active Abandoned
-
2023
- 2023-07-14 JP JP2023115832A patent/JP2023126475A/ja active Pending
- 2023-11-09 US US18/505,414 patent/US20240075094A1/en not_active Abandoned
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2838007T3 (es) | Composiciones de liberación retardada de linaclotida | |
| JP2022093472A (ja) | リナクロチドの改変放出製剤または遅延放出製剤による過敏性腸症候群の処置方法 | |
| ES2868873T3 (es) | Formulaciones de liberación modificada o dirigida de linaclotida | |
| US20230398173A1 (en) | Delayed Release Compositions of Linaclotide | |
| CA3140528A1 (en) | Treatment of abdominal pain associated with diarrhea-predominant irritable bowel syndrome | |
| HK40081509A (en) | Delayed release compositions of linaclotide | |
| HK40051190A (en) | Delayed release compositions of linaclotide | |
| BR112016013419B1 (pt) | Composições de liberação retardada compreendendo um comprimido com revestimento entérico, forma de dosagem unitária e uso | |
| HK40017265A (en) | Methods of treating irritable bowel syndrome with modified or delayed release formulations of linaclotide | |
| HK1237651A1 (en) | Delayed release compositions of linaclotide |