ES2839523T3 - Derivado de quinazolinona, procedimiento de preparación del mismo, composición farmacéutica y aplicaciones - Google Patents
Derivado de quinazolinona, procedimiento de preparación del mismo, composición farmacéutica y aplicaciones Download PDFInfo
- Publication number
- ES2839523T3 ES2839523T3 ES17738201T ES17738201T ES2839523T3 ES 2839523 T3 ES2839523 T3 ES 2839523T3 ES 17738201 T ES17738201 T ES 17738201T ES 17738201 T ES17738201 T ES 17738201T ES 2839523 T3 ES2839523 T3 ES 2839523T3
- Authority
- ES
- Spain
- Prior art keywords
- compound
- formula
- pharmaceutically acceptable
- stereoisomer
- solvate
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 0 CC(N1C(*)(CCC(N2)=O)C2=O)=Nc2cc(F)cc(NC)c2C1=O Chemical compound CC(N1C(*)(CCC(N2)=O)C2=O)=Nc2cc(F)cc(NC)c2C1=O 0.000 description 16
- NTMZVULXIMYDOB-UHFFFAOYSA-N CC(N1C(CCC(N2)=O)C2=O)=Nc2cc(C#N)cc(N)c2C1=O Chemical compound CC(N1C(CCC(N2)=O)C2=O)=Nc2cc(C#N)cc(N)c2C1=O NTMZVULXIMYDOB-UHFFFAOYSA-N 0.000 description 1
- LGHCPGQNUCQKFK-UHFFFAOYSA-N CC(N1C(CCC(N2)=O)C2=O)=Nc2cc(F)cc(NC)c2C1=O Chemical compound CC(N1C(CCC(N2)=O)C2=O)=Nc2cc(F)cc(NC)c2C1=O LGHCPGQNUCQKFK-UHFFFAOYSA-N 0.000 description 1
- NTMZVULXIMYDOB-NSHDSACASA-N CC(N1[C@@H](CCC(N2)=O)C2=O)=Nc2cc(C#N)cc(N)c2C1=O Chemical compound CC(N1[C@@H](CCC(N2)=O)C2=O)=Nc2cc(C#N)cc(N)c2C1=O NTMZVULXIMYDOB-NSHDSACASA-N 0.000 description 1
- LGHCPGQNUCQKFK-NSHDSACASA-N CC(N1[C@@H](CCC(N2)=O)C2=O)=Nc2cc(F)cc(NC)c2C1=O Chemical compound CC(N1[C@@H](CCC(N2)=O)C2=O)=Nc2cc(F)cc(NC)c2C1=O LGHCPGQNUCQKFK-NSHDSACASA-N 0.000 description 1
- JHEZLGFIEHDZHQ-UHFFFAOYSA-N NCC(CCCCCC(O)=O)=O Chemical compound NCC(CCCCCC(O)=O)=O JHEZLGFIEHDZHQ-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09B—ORGANIC DYES OR CLOSELY-RELATED COMPOUNDS FOR PRODUCING DYES, e.g. PIGMENTS; MORDANTS; LAKES
- C09B57/00—Other synthetic dyes of known constitution
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/506—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/517—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with carbocyclic ring systems, e.g. quinazoline, perimidine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/16—Drugs for disorders of the alimentary tract or the digestive system for liver or gallbladder disorders, e.g. hepatoprotective agents, cholagogues, litholytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/04—Centrally acting analgesics, e.g. opioids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/20—Hypnotics; Sedatives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/04—Immunostimulants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/08—Antiallergic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
- A61P5/14—Drugs for disorders of the endocrine system of the thyroid hormones, e.g. T3, T4
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B59/00—Introduction of isotopes of elements into organic compounds ; Labelled organic compounds per se
- C07B59/002—Heterocyclic compounds
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Public Health (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Pharmacology & Pharmacy (AREA)
- Medicinal Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Immunology (AREA)
- Epidemiology (AREA)
- Neurology (AREA)
- Biomedical Technology (AREA)
- Neurosurgery (AREA)
- Oncology (AREA)
- Pulmonology (AREA)
- Diabetes (AREA)
- Endocrinology (AREA)
- Hematology (AREA)
- Pain & Pain Management (AREA)
- Urology & Nephrology (AREA)
- Gastroenterology & Hepatology (AREA)
- Rheumatology (AREA)
- Anesthesiology (AREA)
- Transplantation (AREA)
- Reproductive Health (AREA)
- Virology (AREA)
- Communicable Diseases (AREA)
- Ophthalmology & Optometry (AREA)
- Dermatology (AREA)
- Vascular Medicine (AREA)
- Cardiology (AREA)
- Heart & Thoracic Surgery (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
Un compuesto de fórmula I, o una sal, solvato, polimorfo, cocristal, estereoisómero o compuesto isotópico farmacéuticamente aceptable del mismo: **(Ver fórmula)** en la que, X se selecciona del grupo que consiste en flúor, cloro, bromo, yodo y ciano; **(Ver fórmula)** Z es **(Ver fórmula)**en el que el carbono marcado con * es un centro asimétrico; R1 es -NR1'R2'; en el que R1' y R2' se selecciona cada uno independientemente del grupo que consiste en H, D, metilo y etilo; R2, R3, R5, R6, R7, R8, R9 y R10 son cada uno independientemente H o D; R4 es CH3, CH2D, CHD2 o CD3.
Description
DESCRIPCIÓN
Derivado de quinazolinona, procedimiento de preparación del mismo, composición farmacéutica y aplicaciones
La presente solicitud reivindica prioridad de la solicitud de patente china CN201610023840.9, presentada el 14 de febrero de 2016.
Campo de la invención
Se proporcionan un derivado de quinazolinona, un proceso de preparación, una composición farmacéutica y derivados de quinazolinona y composiciones farmacéuticas para su uso en el tratamiento o la prevención de una enfermedad, trastorno o afección.
Antecedentes de la invención
El factor de necrosis tumoral a (TNF-a) es un tipo de citocina proinflamatoria que desempeña un papel importante en homeostasis inmunitaria, inflamación y defensa del huésped. Se ha demostrado que el TNF-a es uno de los principales mediadores de la inflamación. El TNF-a también puede producirse por tumores. Si bien es capaz de promover la formación de tumores, el TNF-a también puede causar la muerte programada de las células tumorales. Además, el TNF-a también afecta a procesos tales como la apoptosis, la necrosis, la angiogénesis, la activación de células inmunitarias, la diferenciación y la migración celular, todos los cuales desempeñan un papel importante en la tumorigénesis y la progresión tumoral.
La actividad incontrolada de TNF-a o la sobreproducción de TNF-a se asocian con la patología de diversas enfermedades, incluidas, pero sin limitación, cánceres, tales como cáncer de colon, recto, mama, cerebro e intestino; y enfermedades inflamatorias, especialmente inflamación asociada al cáncer. La desregulación de TNF-a también puede provocar enfermedades autoinmunitarias, síndrome de choque tóxico, caquexia, artritis, psoriasis, infección por VIH y SIDA, enfermedades del sistema nervioso y enfermedades del sistema nervioso central, septicemia, insuficiencia cardiaca congestiva, rechazo de trasplante e infecciones víricas. Por lo tanto, la reducción del nivel de TNF-a, o la regulación de la actividad de TNF-a, es una estrategia prometedora en el tratamiento de muchas enfermedades inmunológicas, inflamatorias y malignas (por ejemplo, cánceres e inflamación). Como tal, Sethi et al. Front. Bíoscí. (2008) 13, 5094-5107 y Results Prob. Cell Differ. (2009) 49, 1-15.
La lenalidomida (3-(4-amino-1,3-dihidro-1-oxo-2H-isoindol-2-il)-piperidina-2,6-diona) es un regulador inmunitario de molécula pequeña. Se ha demostrado que la lenalidomida puede inhibir la secreción de TNF-a y otras citocinas proinflamatorias, y aumentar la secreción de citocinas antinflamatorias. La lenalidomida se aprobó para el tratamiento del mieloma múltiple (en 2006), el síndrome mielodisplásico (en 2005) y el linfoma de células del manto (en 2013). Además, en ensayos clínicos, la lenalidomida sola o en combinación con otros agentes terapéuticos puede tratar linfoma no de Hodgkin, carcinoma de tiroides papilar y folicular, leucemia linfocítica crónica, leucemia mielógena crónica, amiloidosis, síndrome de dolor regional complejo de tipo I, melanoma maligno, radiculopatía, mielofibrosis, glioblastoma, gliosarcoma, glioma maligno, leucemia mieloide, tumor de células plasmáticas refractario, leucemia mielomonocítica crónica, linfoma folicular, melanoma de cuerpo ciliar y crónico, melanoma irídico, melanoma interocular recurrente, melanoma con diseminación extraocular, tumor sólido, linfoma de linfocito T, linfoma eritroide, leucemia monoblástica y monocítica; leucemia mieloide y tumores cerebrales, meningioma, tumor de médula espinal, cáncer de tiroides, linfoma de células del manto, cáncer de pulmón de células no pequeñas, cáncer de ovario, carcinoma de células renales, linfoma de Burkitt, linfoma de Hodgkin, linfoma de células grandes y macroglobulinemia (véase el documento WO 2012/015986).
El documento WO 2008/039489 describe compuestos que tienen un núcleo de 2-(2,6-dioxopiperidin-3-il)-1-oxo-4-H-quinazolina que actúan contra el cáncer y otras enfermedades, en particular enfermedades inflamatorias, mediante la inhibición del TNF-a. El documento WO 2014/039421 describe isotopólogos deuterados de 3-(5-amino-2-metil-4-oxoquinazolin-3(4H)-il)-piperidina-2,6-diona, y su uso para el tratamiento o la prevención de enfermedades y afecciones que incluyen enfermedades inflamatorias, enfermedades autoinmunitarias y cánceres. El documento WO 2012/125475 divulga procedimientos de uso de compuestos y composiciones para modular la actividad linfocítica. El compuesto I es 3-(5-amino-2-metil-4-oxoquinazolin-3(4H)-il)piperidin-2,6-diona. El documento WO 2009/042177 describe compuestos que tienen un núcleo de 2-(2,6-dioxopiperidin-3-il)-1-oxo-4-H-quinazolina que actúan contra el cáncer y otras enfermedades, en particular enfermedades inflamatorias, mediante la inhibición de TNF-a. El documento WO 2014/152833 describe 3-(4-oxoquinazolin-3(4H)-ilo sustituido en 6, 7 u 8)-piperidina-2,6-dionas enriquecidas con deuterio en la posición 3.
No obstante, la lenalidomida tiene muchos efectos secundarios. De hecho, la información de prescripción de lenalidomide indica claramente que el medicamento tiene un riesgo de mielosupresión, trombosis venosa profunda, embolia pulmonar y teratogénesis. Durante los ensayos clínicos, la mayor parte de los pacientes que toman lenalidomida necesitan una reducción de la dosis debido a la toxicidad hematológica. Por lo tanto, aunque la lenalidomida tiene una actividad útil, su eficacia está limitada por la aparición significativa de efectos secundarios. Por
lo tanto, es deseable en el campo poseer derivados de lenalidomida con estructuras mejoradas para optimizar su rendimiento.
Resumen de la divulgación
La presente invención se define en y mediante las reivindicaciones adjuntas. La divulgación se refiere a derivados de quinazolinona, a procesos de preparación, a composiciones farmacéuticas y a los compuestos y las composiciones de la divulgación para su uso en el tratamiento o la prevención de una enfermedad, trastorno o afección. El derivado de quinazolinona de la divulgación puede regular la generación o la actividad de citocinas tales como TNF-a, por lo que puede utilizarse de forma eficaz para el tratamiento del cáncer y de una enfermedad inflamatoria.
En el presente documento se divulga un compuesto de fórmula I, o una sal, un solvato, un polimorfo, un cocristal, un estereoisómero o un compuesto isotópico farmacéuticamente aceptable del mismo.

en el que,
X se selecciona del grupo que consiste en flúor, cloro, bromo, yodo y ciano;

R1 es -NR1'R2'; en el que R1' y R2' se seleccionan cada uno independientemente del grupo que consiste en H, D, metilo y etilo;
R2, R3, R5, R6, R7, R8, R9y R10 son cada uno independientemente H o D;
R4 es CH3 , CH2 D, CHD2 o CD3.
En una forma de realización de la divulgación, el centro asimétrico es preferentemente un átomo de carbono configurado en S o un átomo de carbono racémico.
En una forma de realización de la divulgación, Z se selecciona de cualquiera de las estructuras siguientes:


En un aspecto de la divulgación, R1 es -NRr R2'.
En un aspecto de la divulgación, R1' y R2' se seleccionan cada uno independientemente del grupo que consiste en H, D, metilo y etilo.
En una forma de realización de la divulgación, X es flúor, cloro, bromo o yodo, R1 es NH2 , NHD o ND2 ; R2, R3, R5, R6, R7, R8, R9 y R10 son independientemente H o D; R4 es CH3 , CH2 D, CHD2 o CD3.
Preferentemente, el compuesto de fórmula I se selecciona de cualquiera de las estructuras siguientes:


En un aspecto adicional, la divulgación también se refiere a procesos para preparar el compuesto de fórmula I, que puede sintetizarse mediante procesos conocidos utilizando materiales de partida disponibles comercialmente. La divulgación da preferencia particular a cualquiera de los siguientes.
El proceso A comprende la etapa siguiente:
reducir o desproteger el compuesto A1 para dar el compuesto de fórmula I;

R1b'
en los que R1a es nitro, azida o l N - R 1b"; R1b' y R1b" son independientemente H, D o un grupo protector de amino que se selecciona del grupo que consiste en benciloxicarbonilo (Cbz), terc-butoxicarbonilo (Boc), fluorenilmetiloxicarbonilo (Fmoc), p-metoxibencilo (PMB), bencilo (Bn), siempre que R1b' y R1b” no son simultáneamente H o D. Las definiciones de R1, R2, R3, R4, X y Z son tal como se han definido anteriormente.
El proceso B-1 comprende las etapas siguientes:
desproteger el compuesto B3 para dar el compuesto B2; y después someter el compuesto B2 a amidación para dar el compuesto de fórmula I;

El proceso B-2 comprende las etapas siguientes:
someter el compuesto B3 a reacción de ciclación para dar el compuesto de fórmula I;

En el proceso B-1 y el proceso B-2, uno de Ra y Rb es 4“ NRa Rb y e| otro es ” f~o tB u , “ f -0611 0 - f-O M e . uno ^ pa' y Rb es ^~NRa Rb y e| otro es “ i"O H ; R a " y R b " son cac|a uno independientemente H o D. Las definiciones de R1, R2, R3, R4, R5, R6, R7, R8, R9, X y Z son tal como se han definido anteriormente.
El proceso C-1 comprende las etapas siguientes:
hacer reaccionar el compuesto C1 y el compuesto Z-NH2 tal como se muestra a continuación para dar el compuesto de fórmula I;

en los que las definiciones de R1, R2, R3, R4, X y Z son tal como se han definido anteriormente.
Paralelamente, en el proceso C-1, el grupo Z en el compuesto Z-NH2 puede reemplazarse por

R1a (es decir, C1-1 ) y se llevan a cabo las reacciones correspondientes para dar el compuesto intermedio B3 o B2 o A1. Asimismo, en el proceso A, el grupo Z del compuesto A1 se reemplaza por

) y se lleva a cabo la reacción correspondiente para dar el compuesto intermedio B3 o
B2; en el que R1a, Ra, Rb, Ra', Rb', R2, R3, R4, R5, R6, R7, R8 y R9, X y Z son tal como se han definido anteriormente.
Las condiciones y las etapas aplicadas en las reacciones químicas involucradas en los procesos anteriores se pueden realizar haciendo referencia a las condiciones y etapas rutinarias para este tipo de reacción en la técnica, y el compuesto obtenido mediante el proceso anterior puede modificarse adicionalmente en las posiciones periféricas para obtener otros compuestos diana de la divulgación.
También se proporciona un compuesto intermedio de fórmulas A1, A1-1, A1 -2, B2, B3, C1, C1-1:

en las que l como se han definido a En un aspecto adicional, la divulgación también se refiere a composiciones farmacéuticas que comprenden el compuesto de fórmula I, o la sal, solvato, polimorfo, cocristal, estereoisómero o compuesto isotópico farmacéuticamente aceptable del mismo, y uno o más excipientes farmacéuticamente aceptables.
l como se han definido a En un aspecto adicional, la divulgación también se refiere a composiciones farmacéuticas que comprenden el compuesto de fórmula I, o la sal, solvato, polimorfo, cocristal, estereoisómero o compuesto isotópico farmacéuticamente aceptable del mismo, y uno o más excipientes farmacéuticamente aceptables.
Los excipientes farmacéuticamente aceptables pueden ser los que se utilizan ampliamente en la fabricación de fármacos. Los excipientes se utilizan principalmente para proporcionar una composición farmacéutica segura, estable y funcionalizada, y también pueden proporcionar un procedimiento que haga que el ingrediente activo se disuelva a la velocidad deseada o facilite la absorción eficaz del ingrediente activo después de su administración a un sujeto. El excipiente puede ser un material de carga inerte o uno que proporcione algunas funciones, tales como estabilizar el valor de pH general de la composición o prevenir la degradación del ingrediente activo de la composición. El excipiente farmacéuticamente aceptable puede comprender uno o más excipientes seleccionados del grupo que consiste en aglutinante, agente de suspensión, emulsionante, diluyente, material de carga, agente de granulación, adhesivo, agente disgregante, lubricante, agente anti-adhesivo, deslizante, agente humectante, agente gelificante, retardante de la absorción, inhibidor o agente reforzante de la disolución, adsorbente, tampón, agente quelante, conservante, colorante, corrector y agente edulcorante.
La composición farmacéutica de la divulgación se puede preparar basándose en el contenido divulgado en el presente documento según cualquier procedimiento conocido por un experto en la técnica. Por ejemplo, la composición farmacéutica se puede preparar mezclando el compuesto de fórmula I, o la sal, solvato, polimorfo, cocristal, estereoisómero o compuesto isotópico farmacéuticamente aceptable del mismo, con uno o más excipientes farmacéuticamente aceptables, basándose en la tecnología de preparación común para medicamentos. Las tecnologías incluyen, pero sin limitación, procesos convencionales de mezclado, disolución, granulación, emulsión, levigación, envoltura, embebido o liofilización.
La composición farmacéutica según la divulgación puede formularse para su administración por cualquier vía, incluyendo inyección (intravenosa), administración mucosa, administración oral (preparación sólida y líquida), inhalación, administración ocular, administración rectal, tópica o parenteral (infusión, inyección, implante, administración subcutánea, venosa, arterial, intramuscular). La composición farmacéutica de la divulgación también puede adoptar formas farmacéuticas de liberación controlada o liberación retardada. Los ejemplos de preparación sólida de uso oral incluyen, pero sin limitación, polvos, cápsula, comprimido oblongo, cápsula blanda o comprimido. Los ejemplos de preparación líquida para administración oral o mucosa incluyen, pero sin limitación, suspensión, emulsión, elixir y solución. Los ejemplos de preparación tópica incluyen, pero sin limitación, emulsión, gel, pomada, crema, parche, pasta, espuma, loción, gotas o preparación de suero. Los ejemplos de preparación para administración parenteral incluyen, pero sin limitación, solución inyectable, preparación seca que puede disolverse o suspenderse en un vehículo farmacéuticamente aceptable, suspensión inyectable y emulsión inyectable. Los ejemplos de otras preparaciones adecuadas de la composición farmacéutica incluyen, pero sin limitación, gotas para los ojos y otras preparaciones oftálmicas; aerosol, tal como pulverización nasal o inhalación; formas farmacéuticas líquidas adecuadas para administración parenteral; supositorio y pastilla.
La composición farmacéutica de la divulgación puede comprender además uno o más agentes terapéuticos adicionales. A continuación se divulga más información sobre los agentes terapéuticos adicionales que pueden estar incluidos en la composición farmacéutica de la divulgación. La cantidad y el tipo de los agentes terapéuticos adicionales dependen de la enfermedad, trastorno o afección que se va a tratar o prevenir, la gravedad de la enfermedad, trastorno o afección y los factores del sujeto al que se administrará la composición, tales como la edad, el peso, la condición física y la vía de administración.
La cantidad terapéutica o profiláctica del compuesto de fórmula I, o la sal, solvato, polimorfo, cocristal, estereoisómero o compuesto isotópico farmacéuticamente aceptable del mismo, cualquier composición farmacéutica o preparación del mismo, puede administrarse a un sujeto a lo largo de un periodo (ciclo de administración del fármaco), seguido de un periodo exento de compuesto (ciclo de no administración del fármaco). El ciclo de administración del fármaco y el ciclo de no administración del fármaco se pueden repetir durante el tiempo requerido. La duración y el tiempo requeridos del ciclo de administración del fármaco y del ciclo de no administración del fármaco dependen del tipo y/o la gravedad de la enfermedad, trastorno o afección que se está tratando o previniendo, y el sexo, la edad, el peso del sujeto y otros parámetros (por ejemplo, las condiciones biológicas, físicas y fisiológicas del sujeto). Un experto en la técnica puede determinar de forma adecuada una duración y un tiempo adecuados para el ciclo de administración del fármaco y el ciclo de no administración del fármaco basándose en los contenidos divulgados en el presente documento.
También se divulga un procedimiento para regular la generación o la actividad de TNF-a, que comprende administrar a un sujeto con necesidad de ello una cantidad terapéuticamente eficaz del compuesto de fórmula I, o la sal, solvato, polimorfo, cocristal, estereoisómero o compuesto isotópico farmacéuticamente aceptable del mismo, o la composición farmacéutica del mismo.
También se divulga el uso de los compuestos de fórmula I, o la sal, solvato, polimorfo, cocristal, estereoisómero o compuesto isotópico farmacéuticamente aceptable del mismo en la fabricación de un regulador de la generación o la actividad de TNF-a.
En una forma de realización, cuando se utiliza el término "regular" para describir la actividad o la generación de una molécula específica, se refiere a inhibir la actividad o la generación de la molécula. En otra forma de realización, cuando se utiliza el término "regular" para describir la actividad o la generación de una molécula específica, se refiere a aumentar o potenciar la actividad o la generación de la molécula. Sin embargo, en otra forma de realización, cuando se utiliza el término "regular" para describir la actividad o la generación de una molécula específica, se refiere a disminuir o aumentar la actividad o la generación de la molécula.
En un aspecto adicional, la divulgación se refiere a compuestos de fórmula I, o la sal, solvato, estereoisómero o compuesto isotópico farmacéuticamente aceptable del mismo para su uso en el tratamiento o la prevención de una enfermedad, trastorno o afección. Los ejemplos de la enfermedad, trastorno o afección que se va a tratar o prevenir incluyen, pero sin limitación, trastornos asociados con TNF-a, cánceres, enfermedades y trastornos asociados con angiogénesis no deseada, dolores, síndrome de degeneración macular (DM), enfermedades de la piel, queratosis, enfermedad del sistema respiratorio (tal como enfermedades pulmonares), enfermedades de inmunodeficiencia, enfermedades del sistema nervioso central (SNC), enfermedades autoinmunitarias, aterosclerosis, herencia, alergia, virus, trastornos del sueño y síndrome asociado, enfermedades inflamatorias, enfermedades asociadas a PDE-4 o enfermedades asociadas a IL-2. Los ejemplos bien conocidos de la enfermedad, trastorno o afección en el campo incluyen, pero sin limitación, los descritos en las publicaciones de patente PCT WO2012015986 y WO2006018182 y la publicación de patente de Estados Unidos US20100204227.
Los ejemplos de enfermedad asociada a TNF-a de la divulgación incluyen, pero sin limitación, aquellas enfermedades o trastornos descritos en el documento WO9803502. Los ejemplos específicos incluyen, pero sin limitación, inflamación; cánceres; endotoxemia o síndrome de choque tóxico; caquexia; síndrome de dificultad respiratoria del adulto; enfermedades de resorción ósea tales como artritis; hipercalcemia; reacción de injerto contra huésped; enfermedad de tipo cerebral; enfermedad inflamatoria pulmonar crónica; lesión por reperfusión; infarto de miocardio;
apoplejía; choque circulatorio; artritis reumatoide; enfermedad de Crohn; infección por VIH y SIDA; otras enfermedades tales como espondilitis reumatoide, osteoartritis, artritis psoriásica, choque séptico, septicemia, enfermedad degenerativa, colitis ulcerosa, esclerosis múltiple, lupus eritematoso sistémico; asma; enfermedades autoinmunitarias; daño por radiación; lesión alveolar hiperóxica; infecciones víricas tales como las provocadas por el virus del herpes; conjuntivitis viral o dermatitis atópica.
En una forma de realización preferida, la enfermedad, trastorno o afección asociada a TNF-a de la divulgación se selecciona de entre síndrome mielodisplásico, mieloma múltiple, linfoma de células del manto, linfoma difuso de linfocitos B grandes, linfoma del sistema nervioso central, linfoma no de Hodgkin; carcinoma de tiroides papilar y folicular; cáncer de mama, leucemia linfocítica crónica, leucemia mielógena crónica, amiloidosis, síndrome de dolor regional complejo de tipo I, melanoma maligno, radiculopatía, mielofibrosis, glioblastoma, gliosarcoma, glioma maligno, tumor de células plasmáticas refractario, leucemia mielomonocítica crónica, linfoma folicular, melanoma de cuerpos ciliares y crónico, melanoma irídico, melanoma interocular recurrente, melanoma con diseminación extraocular, tumor sólido, linfoma de linfocitos T, linfoma eritroide, leucemia monoblástica y monocítica; leucemia mieloide, tumor cerebral, meningioma, tumor de médula espinal, cáncer de tiroides, cáncer de pulmón de células no pequeñas, cáncer de ovario, carcinoma de células renales, linfoma de Burkitt, linfoma de Hodgkin, linfoma de células grandes, astrocitoma, carcinoma hepatocelular, macroglobulinemia primaria (macroglobulinemia de Waldenstrom). En una forma de realización, el cáncer es metastásico. En otra forma de realización, el cáncer es refractario o no responde al tratamiento con quimioterapia o radioterapia.
El procedimiento para tratar o prevenir una enfermedad, trastorno o afección de la divulgación comprende administrar el compuesto de fórmula I, o la sal, solvato, estereoisómero o compuesto isotópico farmacéuticamente aceptable del mismo a un sujeto por cualquier medio adecuado, tal como inyección, administración mucosa, administración oral, inhalación, administración ocular, administración rectal, implante de acción prolongada, liposoma, emulsión o procedimiento de liberación mantenida.
Un experto en la técnica comprenderá que la cantidad terapéuticamente eficaz o profilácticamente eficaz de los compuestos para el uso de la divulgación puede variar con factores para un sujeto específico, tales como la edad, la dieta, la salud, la gravedad, la complicación y el tipo de enfermedad, trastorno o afección que se va a tratar o prevenir, y la preparación utilizada. Basándose en las divulgaciones de la divulgación, un experto en la técnica puede determinar fácilmente la cantidad terapéuticamente eficaz o profilácticamente eficaz del compuesto que hay que administrar al sujeto para inducir la respuesta biológica o médica deseada en el sujeto.
En cualquier aplicación descrita en la divulgación, el compuesto de fórmula I, o la sal, solvato, polimorfo, cocristal, estereoisómero o compuesto isotópico farmacéuticamente aceptable del mismo, puede utilizarse solo o en combinación con, por ejemplo, radioterapia o radioinmunoterapia, y puede utilizarse en combinación adicional con uno o más agentes terapéuticos que tienen actividad farmacéutica (en lo sucesivo, "agente(s) terapéutico(s) adicional(es)").
En una forma de realización de la divulgación, el o los agente(s) terapéutico(s) adicional(es) pueden ser un compuesto natural, semisintético o sintético. En otra forma de realización, el o los agente(s) terapéutico(s) adicional(es) pueden ser una molécula pequeña, tal como una molécula sintética orgánica o inorgánica, o una molécula o biomolécula más grande, tal como proteínas o ácidos nucleicos con actividad farmacológica. En otra forma de realización, el o los agente(s) terapéutico(s) adicional(es) pueden ser agentes anti-angiogénicos, agentes inmunorreguladores, agentes inmunoterapéuticos, agentes quimioterapéuticos o compuestos hormonales.
En una forma de realización de la divulgación, una composición que comprende el compuesto de fórmula I, o la sal, solvato, polimorfo, cocristal, estereoisómero o compuesto isotópico farmacéuticamente aceptable del mismo, y un agente terapéutico adicional pueden administrarse a un sujeto simultáneamente. En otra forma de realización, el compuesto de fórmula I, o la sal, solvato, polimorfo, cocristal, estereoisómero o compuesto isotópico farmacéuticamente aceptable del mismo, y el agente terapéutico adicional pueden administrarse secuencialmente. En otra forma de realización, el compuesto de fórmula I, o la sal, solvato, estereoisómero o compuesto isotópico farmacéuticamente aceptable del mismo, y el agente terapéutico adicional pueden administrarse por separado. El agente terapéutico adicional puede administrarse antes, consecutivamente o después de la administración del compuesto de fórmula I, o la sal, solvato, polimorfo, cocristal, estereoisómero o compuesto isotópico farmacéuticamente aceptable del mismo.
Según la divulgación, uno o más agentes terapéuticos adicionales que pueden administrarse en combinación con el compuesto de fórmula I, o la sal, solvato, polimorfo, cocristal, estereoisómero o compuesto isotópico farmacéuticamente aceptable del mismo, dependen de una diversidad de factores, tales como la enfermedad, trastorno o afección que se va a tratar o prevenir. Un experto en la técnica puede determinar fácilmente (un) agente(s) terapéutico(s) adicional(es) adecuado(s) para su administración en combinación con el compuesto de fórmula I, o la sal, solvato, polimorfo, cocristal, estereoisómero o compuesto isotópico farmacéuticamente aceptable del mismo, basándose en la contenidos divulgados en el presente documento.
La cantidad terapéuticamente eficaz del agente terapéutico adicional utilizado en la aplicación de la divulgación es conocida por un experto en la técnica, y la guía de administración puede referirse a las patentes y solicitudes
publicadas citadas en el presente documento, y Wells et al., eds., Pharmacotherapy Handbook, 2a edición, Appleton y Lange, Stamford, Conn. (2000); PDR Pharmacopoeia, Tarascon Pocket Pharmacopoeia 2000, Deluxe Edition, Tarascon Publishing, Loma Linda, Calif. (2000) y otras publicaciones médicas citadas en el presente documento. No obstante, un experto en la técnica es capaz de determinar el intervalo de dosis óptimo del agente terapéutico adicional.
Según una forma de realización de la divulgación, cuando se administra en combinación con (un) agente(s) terapéutico(s) adicional(es), la cantidad terapéuticamente eficaz del compuesto de fórmula I, o la sal, solvato, polimorfo, cocristal, estereoisómero, compuesto isotópico farmacéuticamente aceptable del mismo es inferior a la cantidad terapéuticamente eficaz requerida del compuesto de fórmula I, o la sal, solvato, polimorfo, cocristal, estereoisómero o compuesto isotópico farmacéuticamente aceptable del mismo no administrado en combinación con (un) agente(s) terapéutico(s) adicional(es). En otra forma de realización, la cantidad terapéuticamente eficaz del o de los agente(s) terapéutico(s) adicional(es) es inferior a cuando la administración se realiza sin el compuesto de fórmula I, o la sal, solvato, polimorfo, cocristal, estereoisómero o compuesto isotópico farmacéuticamente aceptable del mismo. De esta forma, se pueden minimizar los efectos secundarios asociados con dosis altas de cualquiera de los fármacos. Otras ventajas potenciales, por ejemplo, mejorar el régimen de administración y/o reducir el coste de los fármacos, son obvias para un experto en la técnica.
Según una forma de realización de la divulgación, cuando el compuesto de fórmula I, o la sal, solvato, polimorfo, cocristal, estereoisómero o compuesto isotópico farmacéuticamente aceptable del mismo, y el o los agente(s) terapéutico(s) adicional(es) se administran a un sujeto para su uso en el tratamiento o la prevención de una enfermedad, trastorno o afección, el compuesto de fórmula I, o la sal, solvato, polimorfo, cocristal, estereoisómero o compuesto isotópico farmacéuticamente aceptable del mismo, y el o los agente(s) terapéutico(s) adicional(es) se pueden administrar por la misma o por diferentes vías. El o los agente(s) terapéutico(s) adicional(es) se pueden administrar por cualquier vía descrita en el presente documento, incluidas, pero sin limitación, oral, inhalación, inyección, ocular, mucosa, rectal, emulsión, liposoma, implante de acción prolongada o procedimiento de liberación mantenida. La vía de administración específica del o de los agente(s) terapéutico(s) adicional(es) depende del agente en sí y de la preparación, y de la enfermedad, trastorno o afección que se va a prevenir o tratar. De acuerdo con las divulgaciones del presente documento, un experto en la técnica es capaz de determinar la vía de administración del o de los agente(s) terapéutico(s) adicional(es).
La presente solicitud cita o describe una diversidad de publicaciones, artículos y patentes; el propósito de citar o describir estas referencias o discutir estas referencias es ilustrar los antecedentes de la invención más que admitir que el contenido de estas referencias contribuye a una parte de la técnica anterior de la invención.
A menos que se definan de otra forma, los términos técnicos y científicos utilizados en el presente documento tienen los mismos significados que los entendidos comúnmente por un experto en la técnica. En caso contrario, determinados términos utilizados en el presente documento tienen los significados especificados en la presente descripción. Cabe señalar que, a menos que se indique lo contrario explícitamente en el contexto, la forma singular utilizada en el presente documento y en las reivindicaciones adjuntas abarca el significado plural.
Tal como se utiliza en el presente documento, cuando la sal, la composición y el excipiente específicos se describen como "farmacéuticamente aceptables", significa que la sal, la composición o el excipiente son generalmente no tóxicos, seguros y adecuados para su administración a un sujeto, preferentemente a un mamífero, de forma más preferida a un ser humano.
El término "sal farmacéuticamente aceptable" utilizado en el presente documento se refiere a una sal orgánica o inorgánica farmacéuticamente aceptable. Los ejemplos de la sal incluyen, pero sin limitación, sulfato, citrato, acetato, oxalato, cloruro, bromuro, yoduro, nitrato, hidrosulfato, fosfato, fosfato ácido, isonicotinato, lactato, salicilato, citrato ácido, tartrato, oleato, tanato, pantotenato, bitartrato, ascorbato, succinato, maleato, gentisinato, fumarato, gluconato, glucuronato, sacarato, formiato, benzoato, glutamato, metanosulfonato, etanosulfonato, benzosulfonato, ptoluenosulfonato y embonato (es decir, 1-1-metilen-bis(2-hidroxilo-3-naftoato)). Los compuestos de la divulgación pueden utilizarse para formar sales farmacéuticamente aceptables con varios aminoácidos. La sal alcalina adecuada incluye, pero sin limitación, sal de aluminio, sal de calcio, sal de litio, sal de magnesio, sal de potasio, sal de sodio, sal de zinc, sal de bismuto y sal de dietanolamina. La revisión con respecto a las sales farmacéuticamente aceptables hace referencia a Handbook of Pharmaceutical Salts: Properties, Selection, and Use (P. Heinrich Stahl y Camille G. Wermuth, ed., Wiley-VCH, 2002).
Tal como se utiliza en el presente documento, el término "metabolito" se refiere a una sustancia activa producida por una molécula de fármaco que ha sufrido cambios de estructura química in vivo, siendo la sustancia activa generalmente un derivado de la molécula de fármaco mencionada anteriormente y pudiendo estar también modificada químicamente.
Tal como se utiliza en el presente documento y a menos que se especifique lo contrario, el término "polimorfo" se refiere a uno o más tipos de estructura cristalina formados mediante diferentes disposiciones de moléculas en el espacio reticular cuando cristalizan.
Tal como se utiliza en el presente documento, el término "cocristal" se refiere a un sistema de múltiples componentes que comprende una o más moléculas de API (ingrediente farmacéutico activo) y una o más moléculas objeto (o ligando). En el cocristal, las moléculas de API y las moléculas de objeto (o ligando) están presentes como sólidos a temperatura ambiente cuando se utilizan en su forma pura solas (para distinguir el cocristal del solvato o el hidrato). De esta definición particular se excluyen las sales en las que se produce un intercambio de protones significativo o completo entre las moléculas de API y las moléculas huésped. En el cocristal, el API y los ligandos interactúan a través de enlaces de hidrógeno y otras posibles interacciones no covalentes. Cabe señalar que el propio cocristal puede formar solvatos, incluidos hidratos.
Tal como se utiliza en el presente documento, el término "solvato" se refiere a una forma cristalina del compuesto de fórmula I, o la sal, polimorfo, cocristal, estereoisómero o compuesto isotópico farmacéuticamente aceptable del mismo, que además tiene una o más moléculas de disolvente incorporadas en la estructura cristalina. El solvato puede incluir una cantidad estequiométrica o una cantidad no estequiométrica de disolvente, y la molécula de disolvente en el disolvente puede estar presente en una disposición ordenada o no ordenada. El solvato que contiene una cantidad no estequiométrica de moléculas de disolvente puede formarse perdiendo al menos una molécula de disolvente (pero no todas) del solvato. En una forma de realización particular, un solvato se refiere a un hidrato, lo que significa que el cristal del compuesto incluye además una molécula de agua, y se utiliza agua como disolvente.
Los compuestos de la presente divulgación pueden formar profármacos.
Tal como se utiliza en el presente documento y a menos que se especifique lo contrario, el término "profármaco" se refiere a un derivado del compuesto que comprende un grupo funcional biológicamente reactivo, en el que el grupo funcional biológicamente reactivo puede escindirse del compuesto o hacerse reaccionar de otras formas para dar el compuesto en condiciones biológicas (in vivo o in vitro). Normalmente, el profármaco es inactivo, o al menos tiene una actividad inferior que el compuesto, lo que hace que el compuesto muestre su actividad después de escindirse del grupo funcional biológicamente reactivo. El grupo funcional biológicamente reactivo se puede hidrolizar u oxidar en condiciones biológicas para dar el compuesto. Por ejemplo, el profármaco puede contener un grupo biológicamente hidrolizable. Los ejemplos del grupo biológicamente hidrolizable incluyen, pero sin limitación, un fosfato biológicamente hidrolizable, un éster biológicamente hidrolizable, una amida biológicamente hidrolizable, un éster carbónico biológicamente hidrolizable, un carbamato biológicamente hidrolizable y un ureido biológicamente hidrolizable. La revisión con respecto al profármaco hace referencia a documentos tales como J. Rautio et al., Nature Reviews Drug Discovery (2008) 7, 255-270 y Prodrugs: Challenges and Rewards (V. Stella et al. Ed., Springer, 2007).
El compuesto de fórmula I de la divulgación, la sal, solvato, polimorfo, cocristal, estereoisómero o compuesto isotópico farmacéuticamente aceptable del mismo, puede contener uno o más centros asimétricos ("estereoisómero"). Tal como se utiliza en el presente documento, el término "estereoisómero" se refiere a todos los estereoisómeros, incluidos enantiómeros, diastereoisómeros, epímeros, isómeros endo-exo, atropisómeros, regioisómeros, isómeros cis y trans. El "estereoisómero" también incluye en el presente documento "estereoisómero puro" y "estereoisómero enriquecido" o "isómero racémico" de los diversos estereoisómeros mencionados anteriormente. Estos estereoisómeros pueden prepararse según un proceso de síntesis asimétrico, o pueden separarse, purificarse y enriquecerse mediante un proceso de separación quiral (que incluye, pero sin limitación, cromatografía en capa fina, cromatografía rotatoria, cromatografía en columna, cromatografía de gases, cromatografía líquida de alta presión), así como obtenerse por separación quiral mediante unión (tal como unión química) o salificación (tal como unión física) con otro(s) compuesto(s) quiral(es). El término "estereoisómero puro" en el presente documento se refiere a que el contenido en masa de un estereoisómero del compuesto no es inferior al 95% con respecto otros estereoisómeros del compuesto. El término "estereoisómero enriquecido" en el presente documento se refiere a que el contenido en masa de un estereoisómero del compuesto no es inferior al 50% con respecto a otros estereoisómeros del compuesto. El término "isómero racémico" en el presente documento se refiere a que el contenido en masa de un estereoisómero del compuesto es igual al de otro estereoisómero del compuesto.
El término "compuesto isotópico" utilizado en el presente documento se refiere a que hay uno o más isótopos atómicos con abundancia natural o no natural contenidos en el compuesto de fórmula I, o la sal, solvato, polimorfo, cocristal o estereoisómero farmacéuticamente aceptable del mismo. Los isótopos atómicos con abundancia no natural incluyen, pero sin limitación, deuterio (2H o D), tritio (3H o T), yodo-125 (125I), fósforo-32 (32P), carbono-13 (13C) o carbono-14 (14C). El compuesto isotópico mencionado anteriormente también se puede utilizar como un agente terapéutico o de diagnóstico (es decir, un agente de desarrollo interno) o una herramienta de investigación. Todas las variantes isotópicas del compuesto de la divulgación, ya sean radiactivas o no, están incluidas en el alcance de la divulgación.
El término "isotópicamente enriquecido" utilizado en este documento se refiere a que hay uno o más isótopos atómicos con abundancia no natural contenidos en el compuesto de fórmula I, o la sal, solvato, polimorfo, cocristal, estereoisómero o compuesto isotópico farmacéuticamente aceptable del mismo. El término "isotópicamente enriquecido" también se refiere a que el compuesto de fórmula I, o la sal, solvato, polimorfo, cocristal, estereoisómero o compuesto isotópico farmacéuticamente aceptable del mismo, contiene al menos un átomo isotópico con abundancia no natural.
Tal como se utiliza en este documento, el término "paciente" o "sujeto" se refiere a cualquier animal que se va a tratar o se ha tratado con el compuesto o la composición según una forma de realización de la divulgación, prefiriéndose un mamífero y siendo el más preferido un ser humano. El término "mamífero" utilizado en el presente documento incluye cualquier mamífero. Los ejemplos de mamíferos incluyen, pero sin limitación, ganado vacuno, caballos, ovejas, cerdos, gatos, perros, ratones, ratas, conejos, cobayas, monos, seres humanos, siendo los más preferidos los seres humanos. Los términos "sujeto" y "paciente" se utilizan indistintamente en el presente documento.
En una forma de realización, los términos "tratar" y "tratamiento" se refieren a una mejora, la prevención o la reversión de una enfermedad o un trastorno o al menos uno de sus síntomas identificables, tales como el tratamiento del cáncer mediante la reducción o la estabilización de los síntomas del cáncer o una enfermedad. En otra forma de realización, "tratar" o "tratamiento" se refiere a una mejora, la prevención o la reversión de al menos un parámetro corporal medible de una enfermedad o trastorno que se está tratando, pudiendo la enfermedad o trastorno no identificarse en mamíferos. Sin embargo, en otra forma de realización, el término "tratar" o "tratamiento" se refiere a ralentizar el progreso de una enfermedad o trastorno, en físico, tal como estabilizar síntomas identificables, o en fisiológico, tal como estabilizar parámetros físicos, o en ambos. En otra forma de realización, el término "tratar" o "tratamiento" se refiere a retrasar la aparición de una enfermedad o trastorno.
En algunas formas de realización, el compuesto se administra con fines preventivos. Tal como se utiliza en el presente documento, "prevenir" o "prevención" se refiere a una reducción del riesgo de padecer una enfermedad o síntoma determinado. En una forma de realización preferida, el compuesto designado se administra a un sujeto con un propósito de prevención, tal como a un sujeto con antecedentes familiares o tendencia a padecer cáncer o una enfermedad autoinmunitaria.
Tal como se utiliza en el presente documento, "cantidad terapéuticamente eficaz" se refiere a una cantidad del compuesto o la composición que puede provocar una respuesta biológica o médica (que buscan investigadores, veterinarios, médicos u otros clínicos) para un sistema tisular, un animal o una persona, que puede incluir el alivio de los síntomas de la enfermedad o síntoma que se está tratando. En una forma de realización preferida, la cantidad terapéuticamente eficaz es una cantidad que es suficiente para tratar eficazmente, tratar de forma mejorada o prevenir el cáncer, el síntoma o el trastorno asociado con una angiogénesis no deseada o TNF-a.
El término "cantidad profilácticamente eficaz" se refiere a una cantidad de un compuesto o agente activo (buscado por investigadores, veterinarios, médicos u otros clínicos), que puede inhibir la aparición de una enfermedad en un sujeto. Una cantidad profilácticamente eficaz de un compuesto se refiere a una cantidad de un agente terapéutico que se utiliza solo o en combinación con otro compuesto activo, que puede proporcionar un beneficio terapéutico para tratar o prevenir la enfermedad, trastorno o afección. A menos que se especifique lo contrario, la forma singular del término utilizado en el presente documento, "un" o "una", también incluye un significado plural.
A menos que se especifique lo contrario, los términos "o" o "y" utilizados en el presente documento se refieren a "y/o".
A menos que se especifique lo contrario, el "Jm¡" o en el grupo específico del presente documento se refiere a una posición de conexión.
El término "opcional" u "opcionalmente" significa que el evento o la circunstancia que se describe a continuación puede suceder o no. Este término engloba los casos en que el evento o la circunstancia puede suceder o no. Por ejemplo, "sustitución opcional" o "opcionalmente sustituido" abarca los casos que no están sustituidos o que están sustituidos.
El deuterio (D o 2H) es un isótopo estable no radiactivo de hidrógeno, su peso atómico es 2,0144. El hidrógeno existe en forma de una mezcla isotópica de H (hidrógeno o protio), D (2H o deuterio) y T (3H o tritio) en la naturaleza, en la que la abundancia de deuterio es del 0,0156%. Según el conocimiento técnico común en el campo, de todos los compuestos cuyas estructuras contienen átomos de hidrógeno naturales, el átomo de hidrógeno en realidad representa una mezcla de H, D y T. Por lo tanto, si un compuesto contiene deuterio cuya abundancia es superior a su abundancia natural del 0,0156% en cualquier posición, estos compuestos deben considerarse no naturales o enriquecidos en deuterio y, por tanto, estos compuestos son novedosos con respecto a sus análogos no enriquecidos.
En la divulgación, el compuesto "enriquecido en deuterio" se refiere a un compuesto de fórmula I, o la sal, solvato, polimorfo, cocristal, estereoisómero o compuesto isotópico farmacéuticamente aceptable del mismo, en el que la abundancia de deuterio es superior a su abundancia natural en cualquier posición relevante. Por lo tanto, en el compuesto "enriquecido con deuterio", la abundancia de deuterio en cualquiera de las posiciones relevantes probablemente se encuentre entre más del 0,0156% y el 100%. La posición enriquecida con deuterio está representada por D, mientras que la posición no enriquecida con deuterio está representada por H. Según el conocimiento técnico común en el campo, el símbolo H puede no representarse en la posición enriquecida sin deuterio. Un ejemplo de un proceso para preparar un compuesto enriquecido en deuterio es reemplazar el hidrógeno por deuterio o emplear material de partida enriquecido en deuterio para sintetizar el compuesto.
En la divulgación, el porcentaje de deuterio en el deuterio enriquecido o la abundancia de deuterio se refiere al porcentaje molar.
En la divulgación, no enriquecido con deuterio se refiere al hidrógeno en forma natural, que está en forma de una mezcla de isótopos H (hidrógeno o protio), D (2H o deuterio) y T (3H o tritio).
Cada una de las condiciones preferidas mencionadas anteriormente se puede combinar de cualquier forma sin apartarse del conocimiento común en la técnica y formando así varias formas de realización preferidas de la divulgación.
Los reactivos y materiales de partida utilizados en el presente documento están todos disponibles comercialmente. Los efectos positivos logrados por los compuestos de la divulgación son que pueden regular la generación y/o la actividad de citocinas (por ejemplo, TNF-a) a fin de utilizarlos eficazmente para el tratamiento del cáncer y enfermedades inflamatorias.
Descripción detallada de las formas de realización
La divulgación se ilustrará adicionalmente mediante los ejemplos siguientes, pero no se debe considerar que la divulgación se limita al alcance de los ejemplos. Los procedimientos experimentales que no se especifican en detalle en los ejemplos siguientes son aquellos según procedimientos y condiciones convencionales, o según los manuales del producto.
Ejemplo 1: Síntesis del compuesto K101
Esquema de síntesis

A un disolvente de K101-a (60 g, 38,7 mmol) en H2SO4 con. (150 ml) se añadió gota a gota una mezcla de HNO3 con. (36 ml) y H2SO4 con. (200 ml) a lo largo de un periodo de 2 horas a 0 °C controlando mientras la temperatura de la mezcla de reacción a 0 °C - 15 °C. La mezcla de reacción se agitó durante otra hora y se inactivó vertiéndola en hielo triturado. Después, la capa acuosa se extrajo con DCM (100 ml x 2). La solución orgánica combinada se secó sobre Na2SO4 anhidro, se filtró y se concentró para proporcionar un producto bruto, que se purificó mediante cromatografía en columna en gel de sílice para proporcionar el producto K101-b (16 g, 21%).
RMN de 1H (CDCl3, 300 MHz): 57,78 (d, J = 6,9 Hz, 2 H), 2,55 (s, 3 H).
Etapa 2: Síntesis del compuesto K101-c
A una solución de K101-b (9,0 g, 45,0 mmol) en EtOH (100 ml) se añadió gota a gota una solución de Na2S (16,2 g, 67,5 mmol) en H2O (50 ml) durante 30 min a 25 °C. La mezcla se agitó durante 4 horas y después se concentró para proporcionar un producto bruto. El producto bruto se diluyó con H2O (200 ml) y se extrajo con EtOAc (100 ml x 2), la
capa orgánica combinada se secó sobre Na2SÜ4 anhidro, se filtró y se concentró, el residuo se purificó mediante cromatografía en columna (PE/EtOAc = 10/1) para proporcionar el producto K101 -c (5,0 g, 65%).
RMN de 1H (DMSO-afe, 300 MHz): 56,84 (dd, J = 6,3, 1,8 Hz, 1 H), 6,68 (dd, J = 8,4, 1,8 Hz, 1 H), 5,90 (s ancho, 2 H), 2,03 (s, 3 H).
Etapa 3: Síntesis del compuesto K101-e
A una solución de K101-c (1,60 g, 9,40 mmol) en THF (150 ml) se añadió (Boc)2O (2,25, 10,0 mmol) y DMAP (1,15 g, 9,40 mmol). La mezcla se agitó durante 18 horas a 25 °C y se concentró para eliminar el THF. El residuo se diluyó con EtOAc (200 ml), después se lavó con HCl 1 N (100 ml x 2) y se secó sobre Na2SO4 anhidro, se filtró y se concentró para proporcionar un producto bruto (1,7 g). El producto bruto se añadió a una mezcla de piridina (30 ml) y H2O (15 ml). La mezcla se calentó a 80 °C, después se añadió KMnO4 (3,2 g, 19,8 mmol) en 4 lotes a lo largo de un periodo de 2 horas (un lote cada 30 minutos). La mezcla resultante se agitó durante la noche. La solución de reacción se filtró y la torta se lavó con agua caliente. El filtrado se extrajo con DCM (150 ml x 3). La fase orgánica combinada se secó sobre Na2SO4, se filtró y se concentró para proporcionar un producto bruto. El producto bruto se purificó mediante cromatografía en columna en gel de sílice (EtOAc/PE = 1/5) para proporcionar el producto K101-e (1,0 g, 30% para 2 etapas).
RMN de 1H (CDCla, 300 MHz): 5 10,53 (s ancho, 1H), 8,01 (dd, J = 11,4, 2,4 Hz, 1 H), 7,46 (dd, J = 7,8, 2,4 Hz, 1 H), 1,45 (s, 9 H).
Etapa 4: Síntesis del compuesto K101 -g
A una solución de HCl 4 N en 1,4-dioxano (80 ml) se añadió K101-e (1,0 g, 3,3 mmol). La mezcla se agitó durante 2 horas a 25 °C y se concentró para proporcionar un producto bruto (800 mg). Una mezcla del producto bruto y Ac2O (10 ml) se calentó a reflujo y se agitó durante 4 horas. La solución de reacción se concentró y el residuo se agitó con (EtOAc/Et2O = 1/2, 30 ml) durante 30 min. Las impurezas sólidas se eliminaron por filtración. El filtrado se concentró para proporcionar el producto K101-g (670 mg, 91% para 2 etapas).
RMN de 1H (DMSO-afe, 300 MHz): 58,14 (dd, J = 8,1,2,4 Hz, 1 H), 7,74 (dd, J = 9,0, 2,4 Hz, 1 H), 2,42 (s, 3 H). Etapa 5: Síntesis del compuesto K101 -h
A una mezcla de K101 -g (500 mg, 2,23 mmol) en MeCN (25 ml) se añadió clorhidrato de 4,5-diamino-5-oxopentanoato de (S)-terc-butilo (640 mg, 2,68 mmol), imidazol (334 mg, 4,91 mmol) y fosfito de trifenilo (832 mg, 2,68 mmol). La solución de reacción se agitó a reflujo durante 16 horas. Esta mezcla se concentró y se diluyó con H2O (150 ml) y se extrajo con EtOAc (100 ml x 2). La capa orgánica combinada se secó sobre Na2SO4 anhidro, se filtró y se concentró. El residuo se purificó mediante cromatografía en columna sobre gel de sílice (EtOAc/PE = 1/3) para proporcionar el producto K101-h (600 mg, 66%).
RMN de 1H (DMSO-afe, 300 MHz): 57,98 (dd, J = 8,7, 2,4 Hz, 1 H), 7,68 (dd, J = 9,9, 2,4 Hz, 1 H), 7,42-7,49 (m, 1 H), 7,13-7,21 (m, 1 H), 4,68-4,92 (m, 1 H), 2,54 (s, 3 H), 2,05-2,43 (m, 4 H) ), 1,28 (s, 9 H).
Etapa 6: Síntesis del compuesto K101-j
A una solución de K101 -h (600 mg, 1,47 mmol) en EtOH (60 ml) se añadió solución ac. saturada de NH4Cl (20 ml). La mezcla se calentó a 80 °C y se añadió polvo de Fe (600 mg, 10,7 mmol). La mezcla de reacción se calentó con agitación durante otras 3 horas, se filtró y se concentró para eliminar la mayor parte del EtOH. La mezcla restante se extrajo con EtOAc (150 ml x 2). La capa orgánica combinada se secó y se concentró para proporcionar el producto K101 -j (540 mg, 97%).
RMN de 1H (DMSO-afe, 300 MHz): 56,97-7,50 (m, 4 H), 6,30-6,33 (m, 2 H), 4,56-4,73 (m, 1 H), 2,44 (s, 3 H), 2,06-2,32 (m, 4 H), 1,32 (s, 9 H).
Etapa 7: Síntesis del compuesto K101-k
A una solución de HCl 4 N en 1,4-dioxano (20 ml) se añadió K101 -j (540 mg, 1,43 mmol). Esta mezcla se agitó a 25 °C durante 2 horas, después se concentró para proporcionar el producto K101-k (492 mg).
RMN 1H (DMSO-afe, 300 MHz): 57,12-7,56 (m, 4H), 6,64 (d, J = 6,0 Hz, 1 H), 6,51 (d, J = 6,0 Hz, 1 H), 4,80 (s ancho, 1 H), 2,76 (s, 3 H), 1,98-2,38 (m, 4 H).
Etapa 8: Síntesis del compuesto K101
A una solución de K101 -k (400 mg, 1,24 mmol) en MeCN se añadió CDI (400 mg, 2,48 mmol). La solución de reacción se calentó a 95 °C y se agitó durante la noche, después se concentró para proporcionar un producto bruto. El producto bruto se purificó por HPLC para proporcionar el producto K101 (210 mg, 56%).
RMN de 1H (DMSO-rá, 300 MHz): 5 10,99 (s, 1 H), 7,32 (s ancho, 2 H), 6,34 (d, J = 10,8 Hz, 2 H), 5,13-5,19 (m, 1 H), 2,82-2,88 (m, 1 H), 2,58-2,78 (m, 2 H), 2,53 (s, 3 H), 2,11-2,18 (m, 1 H). CL-EM: 305,1 ([M 1]+).
Ejemplo 2: Síntesis del compuesto K105

Etapa 1: Síntesis del compuesto K105-b
A una mezcla de K101-g (800mg, 3,57 mmol) en CH3CN (30 ml), K105-a (726 mg, 3,57 mmol) se añadieron imidazol (533 mg, 7,85 mmol) y trifenilfosfito (1,33 g, 4,28 mmol). La solución de reacción se agitó a reflujo durante 16 horas. Esta mezcla se concentró y se diluyó con EtOAc (500 ml), se lavó sucesivamente con agua, solución ac. saturada de NaHCO3 y salmuera. La capa orgánica se secó sobre Na2SO4, se filtró y se concentró. El residuo se purificó mediante cromatografía en columna en gel de sílice (EtOAc/PE = 1/1) para proporcionar el producto K105-b (480 mg, rendimiento: 33%).
RMN de 1H (DMSO-cfe, 300 MHz): 58,00 (dd, J = 8,1,2,4 Hz, 1 H), 7,70 (dd, J = 9,6, 2,1 Hz, 1 H), 7,21-7,38 (m, 2 H), 2,57 (s, 3 H), 2,24-2,51 (m, 2 H), 2,08-2,18 (m, 2 H), 1,31 (s, 9 H).
Etapa 2: Síntesis del compuesto K105-c
A una solución de K105-b (480 mg, 1,17 mmol) en EtOH (60 ml) se añadió solución ac. saturada de NH4Cl (20 ml). La mezcla se calentó a 80 °C y se añadió polvo de Fe (6570 mg, 11,72 mmol) a la solución de reacción. La mezcla se agitó durante 3 horas a 80 °C, después se enfrió a temperatura ambiente, se filtró y se concentró a presión reducida para eliminar la mayor parte del EtOH. La capa acuosa restante se extrajo con EtOAc (100 ml x 3). La capa orgánica combinada se secó, se filtró y se concentró. El residuo se purificó mediante cromatografía en columna en gel de sílice (EtOAc/PE = 1/1) para proporcionar el producto K105-c (437 mg, rendimiento: 98%).
RMN de 1H (DMSO-cfe, 300 MHz): 57,07-7,49 (m, 4 H), 6,30-6,35 (m, 2 H), 2,45 (s, 3 H), 2,07-2,34 (m, 4 H), 1,33 (s, 9 H).
Etapa 3: Síntesis del compuesto K105
A una solución de HCl 6 N en 1,4-dioxano (30 ml) se añadió K105-c (437 mg, 1,15 mmol). Esta mezcla se agitó a 25 °C durante 2 horas y después se concentró. El residuo se disolvió en DMF (3 ml) y DCM (30 ml). La mezcla se enfrió a -40 °C y se añadió SOCl2 (685 mg, 5,76 mmol) en DCM (2 ml) gota a gota. Después, la mezcla se hizo reaccionar a -40~-30 °C durante 1,5 horas. Se añadió piridina (912 mg, 11,52 mmol) en DCM (2 ml) y la mezcla se agitó a -40~-30 °C durante 1 hora. Se añadió Et3N (237 mg, 2,7 mmol) en DCM (1 ml) y la mezcla se agitó a -40~-30 °C durante 1 hora. Después se añadió H2O (10 ml) para inactivar la reacción. La mezcla se concentró a presión reducida para proporcionar un producto bruto. El producto bruto se purificó mediante Prep-HPLC para proporcionar K105 (68 mg).
RMN de 1H (DMSO-C6, 400 MHz): 510,94 (s ancho, 1 H), 7,35 (s ancho, 2 H), 6,36 (s, 1 H), 6,33 (s, 1 H), 5,14-5,19 (m, 0,21 H), 2,87-2,77 (m, 1 H), 2,63-2,54 (m, 2 H), 2,51 (s, 3 H), 2,11-2,16 (m, 1 H). EM: 306,1 ([M 1]+).
Ejemplo 3: Síntesis del compuesto K102
El compuesto K102 se sintetizó mediante un procedimiento similar al descrito para K105 en el ejemplo 2 excepto que se utilizó el sustrato correspondiente en lugar del compuesto K105-a en la etapa 1.
RMN de 1H (DMSO-afe, 400 MHz): 511,01 (s, 1 H), 7,32 (s ancho, 2 H), 6,32-6,36 (m, 2 H), 5,16 (dd, J = 11,6, 5,6 Hz, 1 H), 2,78-2,83 (m, 1 H), 2,57-2,66 (m, 2 H), 2,54 (s, 3 H), 2,08-2,17 (m, 1 H). EM: 305,1 ([M 1]+).
Ejemplo 6: Síntesis del compuesto K104

El compuesto K104 se sintetizó mediante un procedimiento similar al del compuesto K105 descrito en el ejemplo 2,
excepto que se utilizó el sustrato correspondiente lugar de K105-a en la etapa 1.
lugar de K105-a en la etapa 1.
RMN de 1H (400 MHz, DMSO-d6) 511,01 (s, 1 H), 7,33 (s ancho, 1 H), 6,33-6,36 (m, 2 H), 2,79-2,83 (m, 1 H), 2,57 2,62 (m, 2 H, 2,53 (s, 3 H), 2,12-2,16 (m, 1 H). CL-EM: 306,1 ([M 1] ).
Ejemplo 7: Síntesis del compuesto K501

Etapa 1: Síntesis del compuesto K501 -B
A la solución de compuesto K501-A (14,0 g, 51,1 mmol) en THF (90 ml) se añadió LiOH (6,4 g, 153 mmol) y H2O (30 ml). La mezcla de reacción se agitó a 25 °C durante la noche y después se concentró. El líquido remanente se diluyó con Et2O (60 ml) y agua (100 ml). Se separó la capa orgánica. La capa acuosa se ajustó con HCl 2 N a pH = 2, se extrajo con EtOAc (150 ml). Las capas orgánicas se lavaron con salmuera saturada (200 ml), se secaron, se filtraron y se concentraron para proporcionar K501-B (12,9 g) como un sólido amarillo.
RMN 1H (400 MHz, DMSO-ds): 58,30 (d, J = 2,0 Hz, 1 H), 8,14 (d, J = 2,0 Hz, 1 H), 2,45 (s, 3 H).
Etapa 2: Síntesis del compuesto K501-C
A la solución de K501-B (12,9 g, 49,61 mmol) en t-BuOH (200 ml) se añadió éster difenílico de ácido fosforazídico (20,5 g, 74,42 mmol) y Et3N (7,5 g, 74,4 mmol). La mezcla se agitó a 80 °C durante la noche. La solución de reacción se concentró y el líquido restante se diluyó con EtOAc (300 ml) y agua (200 ml), la capa orgánica se lavó con salmuera saturada (200 ml), se secó, se filtró y se concentró. El residuo sólido se purificó mediante cromatografía en columna sobre gel de sílice PE:EA (10:1) para proporcionar K501 -C (15,3 g) como un sólido amarillo.
RMN de 1H (400 MHz, CDCh) 58,34 (s, 1 H), 7,64 (d, J = 2,0 Hz, 1 H), 6,46 (s, 1 H), 2,29 (s, 3 H), 1,53 (s, 9 H).
Etapa 3: Síntesis del compuesto K501-D
A una mezcla de piridina (300 ml) y H2O (150 ml) se añadió K501-C (15,3 g, 46,2 mmol). Esta mezcla se calentó a 80 °C, KMnO4 (29,2 g, 184,8 mmol) en 6 lotes durante 3 horas (un lote cada 30 minutos). La mezcla resultante se agitó durante la noche y después se filtró la solución de reacción. La torta del filtro se lavó con EtOAc (800 ml) y agua caliente (200 ml). El filtrado combinado se concentró y se ajustó con HCl 1 N a pH = 2, se extrajo con EtOAc (800 ml). La capa orgánica combinada se secó sobre Na2SO4, después se filtró y se concentró para proporcionar el residuo sólido. El residuo se purificó mediante cromatografía en columna sobre gel de sílice (PE:EA 30:1 ~5:1) para proporcionar K501-D (9,8 g) como un sólido amarillo.
RMN de 1H (400 MHz, DMSO-rá) 59,55 (s, 1 H), 8,29 (d, J = 2,0 Hz, 1 H), 7,91 (d, J = 2,0 Hz, 1 H), 1,47 (s, 9 H). Etapa 4: Síntesis del compuesto K501-E
A una solución de K501-D en DCM (100 ml) se añadió CF3COOH a 0 °C. La mezcla se hizo reaccionar durante la noche a 25 °C y después se concentró. Se añadió HCl en 1,4-dioxano (30 ml). La mezcla se agitó durante 20 min a 25 °C, después se concentró para proporcionar K501-E (6,7 g).
RMN de 1H (400 MHz, DMSO-cfe) 57,19-7,24 (m, 1H), 7,11-7,13 (m, 1H).
Etapa 5: Síntesis del compuesto K501-F
Una solucion de K501-E (2,8 g) en Ac2O (20 ml) y HOAc (60 ml) se calentó a reflujo y se agitó durante 3 horas. La solución de reacción se concentró y el residuo se agitó y se suspendió en EtOAc:PE (2:1, 15 ml) durante 1 hora, después se filtró para proporcionar K501-F (2,3 g) como un sólido amarillo.
RMN de 1H (400 MHz, DMSO-cfe) 58,36 (d, J = 1,6 Hz, 1 H), 8,08 (d, J = 2,0 Hz, 1 H), 2,42 (s, 3 H).
Etapa 6: Síntesis del compuesto K501-G
A una mezcla de K501-F (500 mg, 1,75 mmol), clorhidrato de 3-aminopiperidin-2,6-diona (433 mg, 2,63 mmol) en CH3CN (20 ml) se añadió imidazol (262 mg, 3,86 mmol), (PhO)3P (816 mg, 2,63 mmol). La mezcla se agitó durante 16 horas a 85 °C. Una vez completada la reacción, se eliminó el disolvente al vacío. Al residuo se añadieron 9 ml de EtOAc y 9 ml de H2O, la mezcla se agitó y se suspendió durante 1 hora y se filtró para proporcionar K501-G (382 mg, bruto) como un sólido gris.
Etapa 7: Síntesis del compuesto K501
Una mezcla de K501-G en HOAc (15 ml) se calentó a 80 °C y después se añadió polvo de Fe (965 mg, 17,3 mmol). La mezcla se hizo reaccionar durante 2 horas, después se filtró para eliminar el polvo de Fe. E1HOAc se eliminó al vacío para proporcionar un producto bruto. El producto bruto se purificó mediante cromatografía en columna en gel de sílice (CH3CN:DCM 1:1) para dar un producto que se purificó adicionalmente mediante prep-HPLC para proporcionar K501 (180 mg).
RMN de 1H (400 MHz, DMSO-C6): 5 11,00 (s, 1 H), 7,24 (s, 2 H), 6,74 (dd, J = 13,2, 1,6 Hz, 2H), 5,15-5,19 (m, 1H), 2,78-2,87 (m, 1H), 2,59-2,65 (m, 2H), 2,58 (s, 3H), 2,14-2,18 (m, 1H) CL-EM: 367,0 ([M 2]+).
Ejemplo 8: Síntesis del compuesto K401
El compuesto K401 se sintetizó mediante un procedimiento similar al compuesto K501 descrito en el ejemplo 7 excepto que se utilizó el material de partida correspondiente, ácido 5-cloro-2-metil-3-nitrobenzoico, en lugar del compuesto K501-B.

RMN de 1H (400 MHz, DMSO-C6): 511,00 (s, 1H), 7,24 (s ancho, 2H), 6,76 (d, J = 1,6 Hz, 1 H), 6,73 (d, J = 1,6 Hz, 1 H) , 5,15-5,19 (m, 1 H), 2,82-2,83 (m, 1 H), 2,58-2,62 (m, 2 H), 2,54 (s, 3 H), 2,07-2,16 (m, 1 H). CL-EM: 321,0 ([M I ] ).
Ejemplo 12: Síntesis del compuesto K700

Etapa 1: Síntesis del compuesto K700-A
A una solución de K501 -F (500 g, 1,754 mmol), 4,5-diamino-5-oxopentanoato de terc-butilo (433 mg, 2,631 mmol) en CH3CN (40 ml) se añadió imidazol (525 mg, 7,717 mmol), (PhO)3P (1,3 g, 4,209 mmol). La mezcla de reacción se calentó a 85 °C y se hizo reaccionar durante 16 horas. Cuando se completó la reacción, se eliminó el disolvente al vacío. Al residuo se añadió EtOAc (100 ml) y H2O (50 ml). La fase orgánica se separó y se lavó con una solución acuosa sat. de NaHCO3 (50 ml), se secó y se concentró para proporcionar un producto bruto. El producto bruto se purificó mediante cromatografía en columna sobre gel de sílice (PE:EtOAc 3:1 ~ 1:1) para proporcionar K700-A (1,29 g) como un sólido amarillo.
RMN de 1H (400 MHz, DMSO-da) 58,19 (d, J = 2,0 Hz, 1 H), 8,07 (d, J= 2,0 Hz, 1 H), 7,47 (s ancho, 1 H), 7,19 (s ancho, 1 H), 4,83 (s ancho, 1 H), 2,56 (s, 3 H), 2,27-2,47 (m, 2 H), 2,20-2,23 (m, 1 H), 2,07-2,09 (m, 1 H), 1,23 (s, 9 H).
Etapa 2: Síntesis del compuesto K700-B
A una solución de K700-A (1,29 g, 2,76 mmol) en EtOH (180 ml), se añadió solución ac. sat. de NH4Cl (60 ml). La mezcla se calentó a 80 °C y se añadió polvo de Fe (1,54 g, 27,6 mmol). La mezcla se hizo reaccionar durante 3 horas. Después, la solución de reacción se filtró para eliminar el polvo de Fe. Se eliminó el EtOH al vacío. El residuo se extrajo con EtOAc (150 ml), se repartió, se secó, se concentró y se purificó mediante cromatografía en columna sobre gel de sílice (PE:EtOAc 1:1~1:5) para proporcionar la K700-B (994 mg) como un sólido amarillo.
RMN de 1H (300 MHz, DMSO-da) 57,37-7,42 (m, 1H), 7,22-7,33 (m, 2H), 7,02-7,06 (m, 1H), 6,69-6,73 (m, 2H), 4,70 (s ancho, 1H), 2,44 (s, 3H), 2,02-2,37 (m, 4H), 1,32 (s, 9H).
Etapa 3: Síntesis del compuesto K700-C
A una solución de K700-B (894 mg, 2,04 mmol) en dioxano (50 ml), se añadió bis(pinacolato)diboro (1,03 g, 4,07 mmol), CH3CO2K (399 mg, 4,07 mmol) y Pd(dppf)Cl2 (156 mg, 0,20 mmol). La mezcla se calentó a 100 °C en Ar y se hizo reaccionar durante 3 horas. La solución de reacción se filtró y se concentró para proporcionar un producto bruto, que se purificó mediante cromatografía en columna sobre gel de sílice DCM: MeOH (20:1) para proporcionar el K700-C (1,26 g).
Etapa 4: Síntesis del compuesto K700-D
A una solución de K700-C (1,45 g, 2,98 mmol) en THF (30 ml), se añadió NH4Cl (159 mg, 2,98 mmol) en H2O (15 ml) y se añadió gota a gota H2O2 (22,5 ml) a 25 °C. La mezcla se agitó durante la noche. La mezcla se lavó con solución ac. de Na2SO3 se extrajo con EtOAc (150 ml x 3). La capa orgánica combinada se secó, se concentró y se purificó mediante prep-HPLC para proporcionar K700-D (437mg) como un sólido amarillo.
RMN de 1H (300 MHz, DMSO-da) 59,86 (s, 1 H), 7,31-7,36 (m, 1 H), 6,97-7,03 (m, 3 H), 5,99 (s, 2 H), 4,56 (s ancho, 1 H), 2,39 (s, 3 H), 2,05 -2,27 (m, 4 H), 1,34 (s, 9 H).
Etapa 5: Síntesis del compuesto K700-E
A una solución de HCl 8 N en 1,4-dioxano (20 ml) se añadió K700-D (300 mg, 0,80 mmol). La mezcla se agitó durante 2 horas a 40 °C y después se concentró para proporcionar el producto bruto K700-E (307 mg).
Etapa 6: Síntesis del compuesto K700
A una mezcla de K700-E (307 mg, 0,96 mmol) en CH3CN (20 ml) se añadió CDI (466 mg, 2,88 mmol) a 25 °C. La mezcla se calentó a 85 °C y se hizo reaccionar durante la noche. A la solución de reacción se añadió H2O (20 ml). La mezcla se calentó a 60 °C y se hizo reaccionar durante 3 horas, se concentró y se purificó mediante HPLC preparativa para proporcionar un producto bruto, que después se agitó y se suspendió en CH3CN (5 ml) durante 1 hora para proporcionar K700 (119 mg) como un sólido amarillo.
RMN de 1H (400 MHz, DMSO-cfe) 610,91 (s, 1 H), 9,92 (s, 1 H), 6,93 (s, 2 H), 5,99-6,01 (m, 2 H), 5,04-5,08 (m, 1 H), 2,76-2,81 (m, 1 H), 2,55-2,61 (m, 2 H), 2,48 (s, 3 H), 2,009-2,13 (m, 1 H). CL-EM: 303,0 ([M 1]+).
Ejemplo 13: Síntesis del compuesto K613

Etapa 1: Síntesis del compuesto K613-B
Se disolvió K101-e (3,4 g, 11,32 mmol) en 30 ml de DMF a 25 °C y se añadió Cs2CO3 (9,23 g, 28,31 mmol). La mezcla se agitó durante 30 min. Se añadió CH3 I (2,1 ml, 34,0 mmol). La mezcla se agitó a 25 °C durante la noche, se diluyó con 200 ml de EtOAc, se lavó sucesivamente con agua y salmuera sat., se secó sobre Na2SO4 anhidro, se concentró a sequedad para proporcionar K613-B (3,5 g).
RMN de 1H (DMSO-cfe, 300 MHz): 6 8,09 (d, J = 9,3 Hz, 1 H), 7,92 (d, J = 9,6 Hz, 1 H), 3,80 (s, 3 H), 3,10 (s, 3 H), 1,28 (s, 9 H).
Etapa 2: Síntesis de K613-C
A una solución de K613-B (3,5 g, 10,66 mmol) en 100 ml de MeOH se añadió Pd/C al 10% (700 mg, al 50% en agua). La mezcla se agitó durante la noche en atmósfera de H2 a 50 psi. El Pd/C se eliminó por filtración, el filtrado se concentró a sequedad para proporcionar el producto K613-C (3,0 g).
RMN de 1H (DMSO-cfe, 300 MHz): 66,43-6,48 (m, 1 H), 6,38 (s, 2 H), 6,29-6,33 (m, 1 H), 3,72 (s, 3 H), 3,03 (s, 3 H), 1,24 (s, 9 H).
Etapa 3: Síntesis del compuesto K613-D
A una solución de K613-C (3,0 g, 10 mmol) en 60 ml de MeOH y 20 ml de H2O se añadió UOH.H2O (2,11 g, 50,2 mmol). La mezcla se calentó a 70 °C y se hizo reaccionar durante 5 horas, después se enfrió a 25 °C y se añadieron 50 ml de H2O. La mezcla se concentró para eliminar MeOH y después se enfrió con agua helada, se ajustó con HCl 2 N a pH = 2. Se formó una precipitación sólida. Se realizó una filtración. El sólido se lavó con agua fría y éter de petróleo, después se secó para proporcionar el producto K613-D (2,8 g).
RMN de 1H (DMSO-cfe, 300 MHz): 66,40-6,45 (m, 1H), 6,27 (dd, J = 9,6, 2,4 Hz, 1 H), 3,03 (s, 3 H), 1,26 (s, 9 H). Etapa 4: Síntesis del compuesto K613-E
Se disolvió K613-D (2,8 g, 9,85 mmol) en 50 ml de Ac2O. La mezcla se calentó a 50 °C y se hizo reaccionar durante 5 horas, después se enfrió a 25 °C y se concentró a sequedad para proporcionar el producto bruto K613-E (3,0 g), que se utilizó directamente en la etapa siguiente.
Etapa 5: Síntesis del compuesto K613-F
Se disolvió K613-E (3,0 g, 9,85 mmol) en 50 ml de CH3CN. A la mezcla se añadió inmediatamente clorhidrato de 3-aminopiperidin-2,6-diona (2,43 g, 14,78 mmol), trifenilfosfito (6,72 g, 21,67 mmol) e imidazol (2,01 g, 29,55 mmol). La mezcla se sometió a reflujo durante la noche, después se enfrió a 25 °C y se concentró a sequedad. Se añadieron 50 ml de agua helada y 30 ml de éter de petróleo/EtOAc (1:1). La mezcla se agitó durante 30 min, se filtró, se lavó sucesivamente con agua helada y éter de petróleo:EtOAc (1: 1) y se secó para proporcionar el producto K613-F (2,8 g).
Etapa 6: Síntesis del compuesto K613
Se disolvió K613-F (2,8 g) en 50 ml de DCM, se enfrió con agua helada y se añadieron gota a gota 50 ml de TFA. Después, la solución de reacción se agitó a 25 °C durante 2 horas y se concentró a sequedad. Después se añadieron 20 ml de agua helada y la mezcla se basificó con NaHCO3 sat. Se formó una precipitación sólida. Se realizó una filtración. El sólido se lavó con agua helada y éter de petróleo, se secó y se purificó mediante HPLC preparativa para proporcionar K613 (719 mg) como un sólido blanco.
RMN de 1H (400 MHz, DMSO-rá) 511,01 (s, 1 H), 8,47 (s, 1 H), 6,38 (dd, J = 10,0, 2,4 Hz, 1 H), 6,26 (dd, J = 12,8, 2,4 Hz, 1 H), 5,19 (dd, J = 11,2, 6,0 Hz, 1 H), 2,80-2,84 (m, 4 H), 2,56-2,65 (m, 5 H), 2,13-2,19 (m, 1 H). CL-EM: ([M 1]+) = 319,2
Ejemplo 19: Síntesis del compuesto K724

Etapa 1: Síntesis del compuesto K724-A
El compuesto K720-A se sintetizó mediante un procedimiento similar al del compuesto K700-B descrito en el ejemplo 12 excepto que se utilizó clorhidrato de (S)-4,5-diamino-5-oxopentanoato de terc-butilo en lugar del 4,5-diamino-5-oxopentanoato de terc-butilo.
Un disolvente de K720-A (1,0 g, 2,28 mmol) y CuCN (1,02 g, 11,4 mmol) en DMF (30 ml) se hizo reaccionar a 140 °C en atmósfera de N2 durante la noche. Se añadió H2O (200 ml). La mezcla se extrajo con EtOAc (100 ml x 2), se lavó con salmuera sat. (100 ml x 2), se secó, se concentró y se purificó mediante cromatografía en columna sobre gel de sílice (PE:EtOAc 1:3) para proporcionar el producto K724-A (0,24 g) como un sólido amarillo.
RMN de 1H (400 MHz, DMSO-cfe) 57,05-7,44 (m, 4H), 6,90 (d, J = 1,6 Hz, 1 H), 6,83 (d, J = 1,6 Hz, 1 H), 4,71 (s ancho, 1 H), 2,16-2,48 (m, 7 H), 1,32 (s, 9 H).
Etapa 2: Síntesis del compuesto K724-B
A una solución de compuesto K724-A (0,22 g, 0,57 mmol) en DCM (10 ml) se añadió TFA (10 ml). La mezcla se agitó a 20 °C durante 4 horas, después se concentró y se purificó utilizando una columna C18 (CH3CN:H2O 10:90) para proporcionar el producto K724-B (0,18 g) como un sólido amarillo.
Etapa 3: Síntesis del compuesto K724
A una solución de K724-B (180 mg, 0,547 mmol) en CH3 se añadió CN (40 ml), CDI (133 mg, 0,82 mmol). La solución de reacción se calentó a 80 °C y se agitó durante la noche, después se concentró y se purificó por HPLC prep. K724 (45 mg).
RMN de 1H (400 MHz, DMSO-cfe) 511,05 (s, 1 H), 7,42 (s, 2 H), 6,93 (d, J= 1,6 Hz, 1 H), 6,85 (d, J = 2,0 Hz, 1 H), 5,22 (dd, J = 11,2, 5,6 Hz, 1 H), 2,79-2,88 (m, 1 H), 2,57-2,65 (m, 5 H), 2,14-2,20 (m, 1 H). CL-EM: ([M 1] ) = 312.0.
Ejemplo 20: Síntesis del compuestos K402, K404, K405 y compuestos K502, K504, K505

Estos compuestos se sintetizaron mediante un proce
excepto que se utilizaron los compuestos de partida
K101-g en la etapa 1, y se utilizaron los sustratos correspondientes para reemplazar K105-a. El K401-F se sintetizó mediante un procedimiento similar al del compuesto K501-F descrito en el ejemplo 7.
Ejemplo de eficacia 1: Ensayo de inhibición de la actividad de TNF-a
Reactivos para el ensayo
DPBS (10X): Invitrogen, N.° de catálogo 14190
RPMI 1640: Medio RPMI 1640 (IX), líquido, GIBCO, N.° de catálogo 22400-105
FBS inactivado térmicamente: Invitrogen, N.° de catálogo 10100147
DMSO: dimetilsulfóxido, Sigma, N.° de catálogo D8418
LPS: Sigma, N.° de catálogo L6529
Pen/Estrep. (100X): Gibco, N.° de catálogo 15140
hPBMC: CTL, N.° de catálogo CTL-UP1
Lavado CTL anti-agregados 20X: CTL, N.° de catálogo CTL-AA-005
Conjunto de ELISA de TNF humano: BD, N.° de catálogo 555212
Etapas de recuperación y plaqueado de células PBMC
1. Recuperación de células
1) La agitación se realizó de forma continua en un baño de agua a 37 °C para descongelar rápidamente las células.
2) Las células se añadieron suavemente a un tubo de centrífuga de 15 ml, al que después se añadieron cuidadosamente 10 ml de medio de recuperación precalentado nuevo y después se realizó una centrifugación a 1000 rpm durante 10 min.
3) Se descartó el medio sobrenadante y se realizó la resuspensión con 10 ml de medio completo RPMI 1640 precalentado recién preparado.
2. Plaqueado en placa de 96 pocillos
1) Se calculó el número total de células necesarias para el experimento y se ajustó a la concentración celular apropiada por ml. 100 ul y 105 células por pocillo.
2) La suspensión celular se diluyó con un volumen apropiado de medio de cultivo celular.
3) La suspensión celular se añadió a un pocillo de muestras estéril desechable.
4) Se añadieron 100 ul de suspensión celular a cada pocillo de una placa de 96 pocillos.
5) La placa se incubó en una incubadora a 37 °C, 5% de CO2 , durante 2 horas.
Etapa de preparación del compuesto
1. LPS: La solución madre de 1 mg/ml se diluyó con agua, se dividió en partes alícuotas y se almacenó a -80 °C. Antes de cada ensayo, la solución de trabajo de LPS se diluyó a partir de la solución madre con medio RPMI 1640 carente de suero.
2. Compuesto de ensayo
Se disolvió la solución madre 20 mM en DMSO y se comprobó la solubilidad del compuesto, se dividió en partes alícuotas y se almacenó a -80 °C.
Preparación de gradiente de compuesto 8X:
Se diluyó una serie de gradiente de concentración de compuesto con DMSO: se obtuvieron concentraciones 10 mM, 2 mM, 0,4 mM, 80 uM, 16 uM, 3,2 uM, 0,64 uM, 0,128 uM y después los compuestos se diluyeron 125 veces con medio RPMI 1640 carente de suero hasta un final 8X. La concentración final de DMSO en cultivo celular fue del 0,1%. Procedimientos experimentales de procesamiento de compuestos y recolección de sobrenadantes
1. Plaqueado de células: se plaquearon células nuevas en placas de cultivo celular de 96 pocillos según el procedimiento anterior, 100 ul y 105 células por pocillo, y después se incubaron en una incubadora a 37 °C, 5% de CO2 durante 2 horas.
2. Preparación del compuesto: antes del ensayo, se añadieron compuestos a las placas según la descripción anterior. Se preparó una dosis de compuesto en concentración 8X con medio RPMI 1640 carente de suero y se añadieron todos los gradientes de solución a la placa de compuesto.
3. Adición de compuesto: se añadieron 16,7 ul de solución de compuesto en concentración de trabajo a cada pocillo de la placa de cultivo celular. La placa se incubó en una incubadora a 37 °C, 5% de CO2 durante 1 hora.
4. Se añadieron 16,7 ul de LPS 8X por pocillo (la concentración final de LPS es CE80, se necesitaba determinar la cantidad de cada PBMC). La placa se incubó durante 18 horas en una incubadora a 37 °C, 5% de CO2.
5. Se recogieron 80 ul de sobrenadante por pocillo y después se sometieron a un ensayo ELISA de TNF-a. El sobrenadante recogido se puede almacenar a -80 °C. El sobrenadante necesitaba diluirse en varias proporciones para asegurar que la dosis experimental no excediera el intervalo lineal de la curva estándar de TNF-a, dependiendo de la cantidad de TNF-a liberada en diferentes donantes. Típicamente, se diluyeron 20-100 pl de sobrenadante a 200 pl y después se utilizaron para experimentos ELISA.
Etapa de ELISA de TNF-a
El procedimiento de ensayo de ELISA de TNF-a se remitió al procedimiento experimental del kit de ELISA de TNF-a humano BD.
Diseño experimental
Cuatro compuestos por placa. Se realizó una dilución de 5 veces, a partir de 10 uM, mediante 8 gradientes, y se produjeron pocillos paralelos. Se analizaron un total de 16 compuestos.
Se añadió el patrón de TNF-a a cada placa. (1er pocillo, partiendo de 500 pg/ml, dilución doble, 7 gradientes).
ZPE (inhibición del 0%) utilizó 15 pg/ml de LPS DMSO al 0,1%, mientras que HPE (inhibición del 100%) utilizó solo DMSO al 0,1%.
Se calcularon las estadísticas de la tasa de inhibición. La tasa de inhibición (%) = [1-(Max-Min)/(Ensayo cpd-Min)]*100%. Se utilizó la CI50 para evaluar la concentración del compuesto de ensayo (nM) al 50% de inhibición. Los dos resultados experimentales se muestran en la tabla 1 y la tabla 1 -1.
En el presente ejemplo de eficacia y en el ejemplo de eficacia 2, las estructuras correspondientes a los códigos para el compuesto de la divulgación son todas tal como se han descrito anteriormente. Los códigos y las estructuras de los compuestos de referencia se resumen en la tabla N.
Tabla N

Tabla 1

Tabla 1-1


Ejemplo de eficacia 2: procedimiento experimental de proliferación de células CTG
Se inocularon células MM.1S (células de mieloma) (ATCC, número de catálogo CRL-2974), células DOHH2 (células de linfoma de células del manto) (DSMZ, número de catálogo ACC-47), células NCI-H929 (células de mieloma) (ATCC, número de catálogo CRL-9068), o células WSU-DLCL-2 (células de linfoma difuso de células B grandes) (DSMZ, número de catálogo ACC-575), células Namalwa.CSN/70 (células de linfoma no de Hodgkin (DSMZ, número de catálogo ACC-70) como (1,8-15) x 103 por pocillo de pared blanca, placa de 96 pocillos de fondo transparente que contenía medio específico (Corning, número de catálogo CLS3903), que se cultivó en una incubadora a 37 °C, 5% de CO2 durante 24 horas. Los compuestos se formularon en DMSO (Sigma, Cat. N° 276855) como una solución madre 150 mM, que se diluyó a la concentración requerida (la concentración final de DMSO es del 0,2%) en medio de cultivo y se añadió a cada pocillo, 2 pocillos/concentración. La placa se incubó en una incubadora a 37 °C, 5% de CO2 durante 72-120 horas. Posteriormente, se añadieron 100 pl de reactivo de ensayo de actividad celular CellTiter-Glo® (Promega, n° de cat. G7570) a cada pocillo, que se mezclaron en un agitador de placas durante 10 minutos para inducir la lisis celular. La placa de 96 pocillos se dejó reposar a temperatura ambiente durante 10 minutos para estabilizar la señal de luminiscencia. Se adhirió una película de base blanca en la parte inferior de la placa y se utilizó EnSpire para leer la placa. El procesamiento de datos se realizó con el programa informático XLfit para obtener los valores de CI50. Los datos experimentales específicos para varios lotes se muestran en la tabla 2, tabla 3 y tabla 4.
Tabla 2

Tabla 4

Claims (11)
1. Un compuesto de fórmula I, o una sal, solvato, polimorfo, cocristal, estereoisómero o compuesto isotópico farmacéuticamente aceptable del mismo:

en la que,
X se selecciona del grupo que consiste en flúor, cloro, bromo, yodo y ciano;

R1 es -NR1'R2'; en el que R1' y R2' se selecciona cada uno independientemente del grupo que consiste en H, D, metilo y etilo;
R2, R3, R5, R6, R7, R8, R9 y R10 son cada uno independientemente H o D;
R4 es CH3 , CH2 D, CHD2 o CD3.
2. El compuesto de fórmula I según la reivindicación 1, o la sal, solvato, polimorfo, cocristal, estereoisómero o compuesto isotópico farmacéuticamente aceptable del mismo, en el que
Z se selecciona del grupo que consiste en cualquiera de las estructuras siguientes:


3. El compuesto de fórmula I según la reivindicación 1, o la sal, solvato, polimorfo, cocristal, estereoisómero o compuesto isotópico farmacéuticamente aceptable del mismo, en el que

4. El compuesto de fórmula I según la reivindicación 1, o la sal, solvato, polimorfo, cocristal, estereoisómero o compuesto isotópico farmacéuticamente aceptable del mismo, en el que
X es flúor, cloro, bromo o yodo,
R1 es NH2 , NHD o ND2 ;
R2, R3, R5, R6, R7, R8, R9 y R10 son independientemente H o D; y
R4 es CH3 , CH2 D, CHD2 o CD3.
5. El compuesto de fórmula I según la reivindicación 1, o la sal, solvato, polimorfo, cocristal, estereoisómero o compuesto isotópico farmacéuticamente aceptable del mismo, en el que el carbono marcado con * es un centro asimétrico, y el centro asimétrico es un átomo de carbono configurado en S o átomo de carbono racémico.
6. El compuesto de fórmula I según la reivindicación 1, o la sal, solvato, polimorfo, cocristal, estereoisómero o compuesto isotópico farmacéuticamente aceptable del mismo, en el que el compuesto de fórmula I se selecciona de cualquiera de las estructuras siguientes:
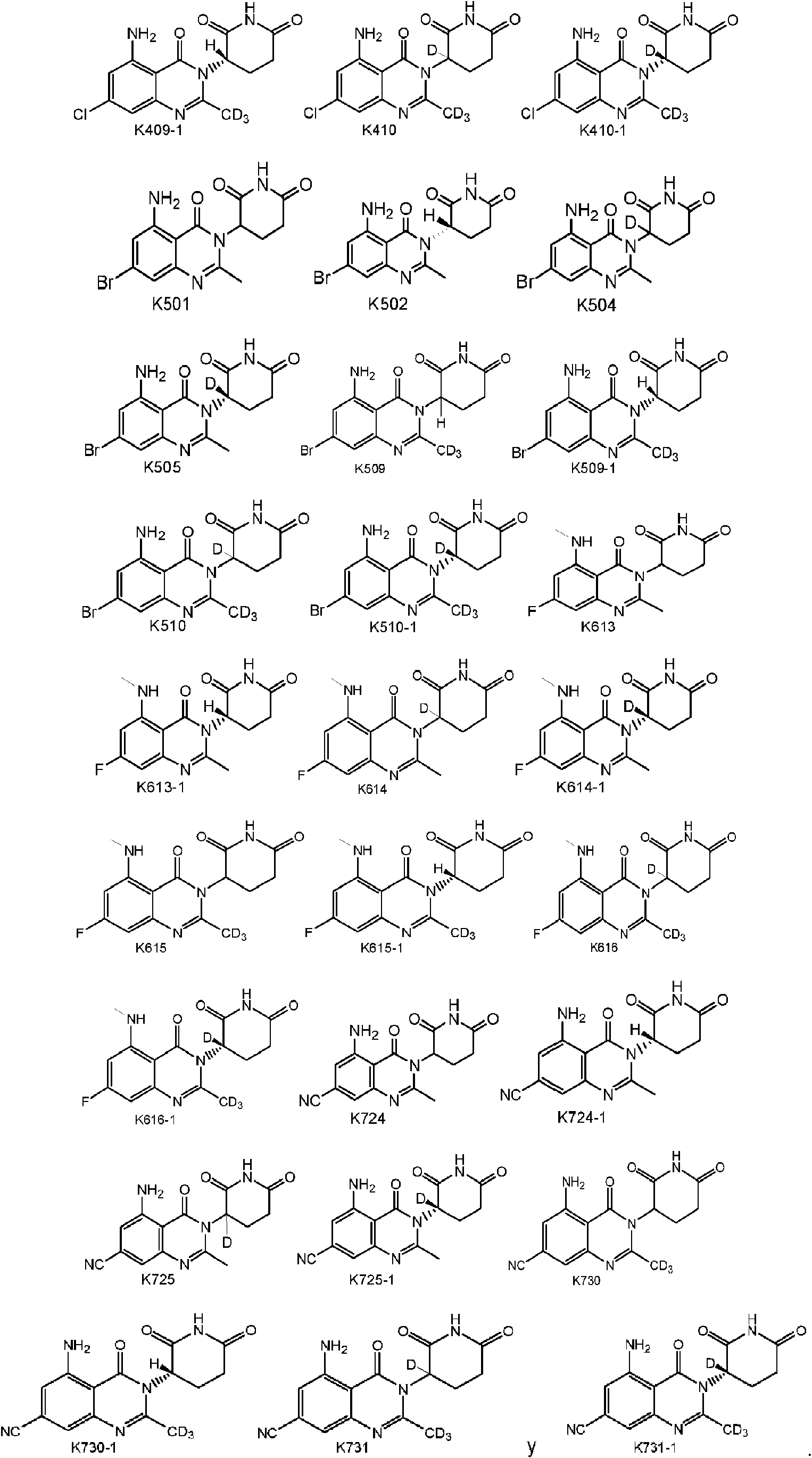
7. Un proceso para preparar el compuesto de fórmula I según una cualquiera de las reivindicaciones 1 a 6, o la sal, solvato, polimorfo, cocristal, estereoisómero o compuesto isotópico farmacéuticamente aceptable del mismo, que se selecciona del grupo que consiste en uno cualquiera de los siguientes:
El proceso A que comprende la etapa siguiente:
reducir o desproteger el compuesto A1 para dar el compuesto de fórmula I;

que se selecciona del grupo que consiste en benciloxicarbonilo (Cbz), terc-butoxicarbonilo (Boc), fluorenilmetiloxicarbonilo (Fmoc), p-metoxibencilo (PMB), bencilo (Bn), siempre que R1b' y R1b” no sean simultáneamente H o D. Las definiciones de R1, R2, R3, R4, X y Z son tal como se han definido en una cualquiera de las reivindicaciones 1 a 6.
El proceso B-1 que comprende las etapas siguientes:
desproteger el compuesto B3 para dar el compuesto B2; y después someter el compuesto B2 a amidación para dar el compuesto de fórmula I;

someter el compuesto B3 a reacción de ciclación para dar el compuesto de fórmula I;

R1, R2, R3, R4, R5, R6, R7, R8, R9, X y Z son tal como se han definido en una cualquiera de las reivindicaciones 1 a 6. El proceso C-1 comprende las etapas siguientes:
hacer reaccionar el compuesto C1 y el compuesto Z-NH2 tal como se muestra a continuación para dar el compuesto de fórmula I;

en los que las definiciones de R1, R2, R3, R4, X y Z son tal como se han definido en una cualquiera de las reivindicaciones 1 a 6.
8. Un compuesto intermedio de fórmula A1, A1-1, A1 -2, B2, B3, C1, o C1-1:

en los que R1a, Ra, Rb, Ra', Rb', R1, R2, R3, R4, R5, R6, R7, R8, R9, X y Z son tal como se han definido en una cualquiera de las reivindicaciones 1 a 7.
9. Una composición farmacéutica que comprende el compuesto de fórmula I según cualquiera de las reivindicaciones 1 a 6, o la sal, solvato, polimorfo, cocristal, estereoisómero o compuesto isotópico farmacéuticamente aceptable del mismo, y uno o más excipientes farmacéuticamente aceptables.
10. El compuesto de fórmula I según una cualquiera de las reivindicaciones 1 a 6, o la sal, solvato, polimorfo, cocristal, estereoisómero o compuesto isotópico farmacéuticamente aceptable del mismo, o
la combinación del compuesto de fórmula I según una cualquiera de las reivindicaciones 1 a 6, o la sal, solvato, polimorfo, cocristal, estereoisómero o compuesto isotópico farmacéuticamente aceptable del mismo, y uno o más de otros agentes terapéuticos seleccionados del grupo que consiste en agente anti-angiogénico, agente inmunorregulador, agente inmunoterapéutico, agente quimioterapéutico o compuesto hormonal,
para su uso en el tratamiento o la prevención de una enfermedad, trastorno o afección;
en el que la enfermedad, trastorno o afección se selecciona del grupo que consiste en: trastornos asociados con TNF-a, cánceres, enfermedades y trastornos asociados con angiogénesis no deseada, dolores, síndrome de degeneración macular, enfermedades de la piel, queratosis, enfermedad del sistema respiratorio, enfermedades de inmunodeficiencia, enfermedades del sistema nervioso central, enfermedades autoinmunitarias, aterosclerosis, herencia, alergia, virus, trastornos del sueño y síndrome asociado, enfermedades inflamatorias, enfermedades asociadas a PDE-4 o enfermedades asociadas a IL-2.
11. El compuesto de fórmula I según la reivindicación 10, o la sal, solvato, polimorfo, cocristal, estereoisómero o compuesto isotópico farmacéuticamente aceptable del mismo, o la combinación según la reivindicación 10, para su uso en el tratamiento o prevención de una enfermedad, trastorno o afección;
en el que la enfermedad, trastorno o afección se selecciona del grupo que consiste en: síndrome mielodisplásico, mieloma múltiple, linfoma de células del manto, linfoma difuso de células B grandes, linfoma del sistema nervioso central, linfoma no de Hodgkin; carcinoma de tiroides papilar y folicular; cáncer de mama, leucemia linfocítica crónica, leucemia mielógena crónica, amiloidosis, síndrome de dolor regional complejo tipo I, melanoma maligno, radiculopatía, mielofibrosis, glioblastoma, gliosarcoma, glioma maligno, tumor de células plasmáticas refractario, leucemia mielomonocítica crónica, linfoma folicular, melanoma de cuerpo ciliar y crónico, melanoma irídico, melanoma interocular recurrente, melanoma de extensión extraocular, tumor sólido, linfoma de linfocitos T, linfoma eritroide, leucemia monoblástica y monocítica; leucemia mieloide, tumor cerebral, meningioma, tumor espinal, cáncer de tiroides, cáncer de pulmón de células no pequeñas, cáncer de ovario, carcinoma de células renales, linfoma de Burkitt, linfoma de Hodgkin, linfoma de células grandes, astrocitoma, carcinoma hepatocelular o macroglobulinemia primaria.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201610023840 | 2016-01-14 | ||
| PCT/CN2017/071147 WO2017121388A1 (zh) | 2016-01-14 | 2017-01-13 | 喹唑啉酮衍生物、其制备方法、药物组合物及应用 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2839523T3 true ES2839523T3 (es) | 2021-07-05 |
Family
ID=59310818
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES17738201T Active ES2839523T3 (es) | 2016-01-14 | 2017-01-13 | Derivado de quinazolinona, procedimiento de preparación del mismo, composición farmacéutica y aplicaciones |
Country Status (13)
| Country | Link |
|---|---|
| US (2) | US11001566B2 (es) |
| EP (1) | EP3404024B1 (es) |
| JP (1) | JP6930748B2 (es) |
| KR (1) | KR102331013B1 (es) |
| CN (1) | CN108473461B (es) |
| AU (1) | AU2017206382C1 (es) |
| CA (1) | CA3011254C (es) |
| DK (1) | DK3404024T3 (es) |
| ES (1) | ES2839523T3 (es) |
| PL (1) | PL3404024T3 (es) |
| RU (1) | RU2730500C2 (es) |
| WO (1) | WO2017121388A1 (es) |
| ZA (1) | ZA201804692B (es) |
Families Citing this family (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR102331013B1 (ko) * | 2016-01-14 | 2021-11-26 | 강푸 바이오파마슈티칼즈 리미티드 | 퀴나졸리논 유도체, 이의 제조방법, 약학 조성물 및 적용 |
| FI3784663T3 (fi) * | 2018-04-23 | 2023-10-06 | Celgene Corp | Substituoituja 4-aminoisoindoliini-1,3-dioniyhdisteitä ja niiden käyttö lymfooman hoitamiseksi |
| CN108654689B (zh) * | 2018-06-22 | 2021-08-03 | 南京大学 | 一种低共熔非腐蚀酸性催化剂、制备方法及其用途 |
| CN112840476A (zh) | 2018-10-12 | 2021-05-25 | 雅宝公司 | 包含硅和锂的颗粒 |
| WO2020210630A1 (en) | 2019-04-12 | 2020-10-15 | C4 Therapeutics, Inc. | Tricyclic degraders of ikaros and aiolos |
| WO2021080931A1 (en) | 2019-10-21 | 2021-04-29 | Celgene Corporation | Solid forms comprising (s)-2-(2,6-dioxopiperidin-3-yl)-4-((2-fluoro-4-((3-morpholinoazetidin-1-yl)methyl)benzyl)amino)isoindoline-1,3-dioneand salts thereof, and compositions comprising the same and their use |
| EP4096713A1 (en) * | 2020-01-31 | 2022-12-07 | Seagen Inc. | Anti-cd30 antibody-drug conjugates and their use for the treatment of non-hodgkin lymphoma |
| WO2021168314A1 (en) * | 2020-02-21 | 2021-08-26 | Plexium, Inc. | Quinazolinone compounds and related compounds |
| KR20230066554A (ko) * | 2020-07-24 | 2023-05-16 | 이니팜, 인크. | 퀴나졸리논 hsd17b13 억제제 및 이의 용도 |
| EP4198030A4 (en) * | 2020-08-14 | 2024-01-10 | Shanghaitech University | IMMUNO-REGULATORY COMPOUND AND ITS ANTITUMOR APPLICATION |
| AU2021377248B2 (en) | 2020-11-13 | 2025-12-18 | Inipharm, Inc. | Dichlorophenol hsd17b13 inhibitors and uses thereof |
Family Cites Families (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| ME02420B (me) * | 2006-09-26 | 2016-09-20 | Celgene Corp | 5-supstituirani derivati kinazolinona kao sredstva protiv raka |
| CA2704710C (en) * | 2007-09-26 | 2016-02-02 | Celgene Corporation | 6-, 7-, or 8-substituted quinazolinone derivatives and compositions comprising the same |
| PH12013501837A1 (en) * | 2011-03-11 | 2013-11-18 | Celgene Corp | Use of 3-(5-amino-2-methyl-4-oxoquinazolin-3(4h)-yl)piperidine-2,6-dione in treatment of immune-related and inflammatory diseases |
| EP2892887B1 (en) | 2012-09-04 | 2020-07-15 | Celgene Corporation | Isotopologues of 3-(5-amino-2-methyl-4-oxoquinazolin-3(4h)-yl) piperidine-2-6-dione and methods of preparation thereof |
| JP6359563B2 (ja) | 2013-01-14 | 2018-07-18 | デュートルクス・リミテッド・ライアビリティ・カンパニーDeuteRx, LLC | 3−(5置換−4−オキソキナゾリン−3(4h)−イル)−3−ジュウテロピペリジン−2,6−ジオン誘導体 |
| CA2941560A1 (en) * | 2013-03-14 | 2014-09-25 | Deuterx, Llc | 3-(substituted-4-oxo-quinazolin-3(4h)-yl)-3-deutero-piperidine-2,6-dione derivatives |
| WO2016105518A1 (en) | 2014-12-23 | 2016-06-30 | Dana-Farber Cancer Institute, Inc. | Methods to induce targeted protein degradation through bifunctional molecules |
| KR102331013B1 (ko) * | 2016-01-14 | 2021-11-26 | 강푸 바이오파마슈티칼즈 리미티드 | 퀴나졸리논 유도체, 이의 제조방법, 약학 조성물 및 적용 |
-
2017
- 2017-01-13 KR KR1020187022763A patent/KR102331013B1/ko active Active
- 2017-01-13 WO PCT/CN2017/071147 patent/WO2017121388A1/zh not_active Ceased
- 2017-01-13 DK DK17738201.7T patent/DK3404024T3/da active
- 2017-01-13 AU AU2017206382A patent/AU2017206382C1/en active Active
- 2017-01-13 RU RU2018129344A patent/RU2730500C2/ru active
- 2017-01-13 JP JP2018555808A patent/JP6930748B2/ja active Active
- 2017-01-13 US US16/069,026 patent/US11001566B2/en active Active
- 2017-01-13 EP EP17738201.7A patent/EP3404024B1/en active Active
- 2017-01-13 CN CN201780006458.XA patent/CN108473461B/zh active Active
- 2017-01-13 ES ES17738201T patent/ES2839523T3/es active Active
- 2017-01-13 CA CA3011254A patent/CA3011254C/en active Active
- 2017-01-13 PL PL17738201T patent/PL3404024T3/pl unknown
-
2018
- 2018-07-13 ZA ZA2018/04692A patent/ZA201804692B/en unknown
-
2021
- 2021-03-24 US US17/211,203 patent/US11702399B2/en active Active
Also Published As
| Publication number | Publication date |
|---|---|
| EP3404024A4 (en) | 2019-07-31 |
| AU2017206382A1 (en) | 2018-08-02 |
| US11001566B2 (en) | 2021-05-11 |
| JP6930748B2 (ja) | 2021-09-01 |
| AU2017206382C1 (en) | 2020-12-24 |
| WO2017121388A1 (zh) | 2017-07-20 |
| EP3404024B1 (en) | 2020-11-18 |
| RU2730500C2 (ru) | 2020-08-24 |
| NZ744387A (en) | 2021-07-30 |
| RU2018129344A (ru) | 2020-02-17 |
| JP2019501977A (ja) | 2019-01-24 |
| US11702399B2 (en) | 2023-07-18 |
| PL3404024T3 (pl) | 2021-04-19 |
| EP3404024A1 (en) | 2018-11-21 |
| US20200039950A1 (en) | 2020-02-06 |
| RU2018129344A3 (es) | 2020-02-21 |
| AU2017206382B2 (en) | 2020-08-20 |
| KR20180100211A (ko) | 2018-09-07 |
| CN108473461B (zh) | 2021-06-15 |
| CA3011254A1 (en) | 2017-07-20 |
| CN108473461A (zh) | 2018-08-31 |
| ZA201804692B (en) | 2020-03-25 |
| DK3404024T3 (da) | 2021-01-11 |
| HK1255062A1 (zh) | 2019-08-02 |
| US20210206743A1 (en) | 2021-07-08 |
| KR102331013B1 (ko) | 2021-11-26 |
| CA3011254C (en) | 2022-03-15 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2839523T3 (es) | Derivado de quinazolinona, procedimiento de preparación del mismo, composición farmacéutica y aplicaciones | |
| ES2902052T3 (es) | Nuevo derivado de isoindolina, y composición farmacéutica y aplicación del mismo | |
| ES2883285T3 (es) | Antagonistas de tlr7/8 y sus usos | |
| ES2812877T3 (es) | Derivado de isoindolina, producto intermedio, método de preparación, composición farmacéutica y uso del mismo | |
| ES2841912T3 (es) | Inhibidores de CXCR2 | |
| CA3124130A1 (en) | Substituted 3-((3-aminophenyl)amino)piperidine-2,6-dione compounds, compositions thereof, and methods of treatment therewith | |
| JP6616412B2 (ja) | Nik阻害剤としての新規のピラゾロピリミジン誘導体 | |
| JP6603713B2 (ja) | Nik阻害剤としての新規のチエノピリミジン誘導体 | |
| CA3150701A1 (en) | Alkynyl quinazoline compounds | |
| ES3008927T3 (en) | Process for preparing modulators of p300 and/or cbp | |
| CN101676266B (zh) | 氘代的ω-二苯基脲及衍生物以及包含该化合物的药物组合物 | |
| BR112015017963A2 (pt) | composto de fenil amino pirimidina deuterado, método para preparar a composição farmacêutica, composição farmacêutica e uso do composto | |
| CN104557871B (zh) | 具有螺环取代基的芳基吗啉类化合物,其制备方法和用途 | |
| ES3053012T3 (en) | Condensed pyridine derivatives substitued by amide functions as acss2 inhibitors | |
| AU2024271907A1 (en) | Hdac1/2 selective inhibitor and use thereof | |
| ES3042173T3 (en) | Cgrp antagonist compounds | |
| CN116425670A (zh) | 作为tead抑制剂的新型杂环化合物 | |
| NZ744387B2 (en) | Quinazolinone derivative, preparation method therefor, pharmaceutical composition, and applications | |
| WO2025016439A1 (zh) | 用作cdk2/4蛋白激酶抑制剂的化合物及其应用 | |
| JP2023535932A (ja) | 三環式のヘテロ環 | |
| HK1255062B (zh) | 喹唑啉酮衍生物、其制备方法、药物组合物及应用 |