ES2854751T3 - Un procedimiento para producir nanopartículas híbridas amorfas estables que comprende al menos un inhibidor de la proteína cinasa y al menos un componente polimérico estabilizante y formador de matriz - Google Patents
Un procedimiento para producir nanopartículas híbridas amorfas estables que comprende al menos un inhibidor de la proteína cinasa y al menos un componente polimérico estabilizante y formador de matriz Download PDFInfo
- Publication number
- ES2854751T3 ES2854751T3 ES13736318T ES13736318T ES2854751T3 ES 2854751 T3 ES2854751 T3 ES 2854751T3 ES 13736318 T ES13736318 T ES 13736318T ES 13736318 T ES13736318 T ES 13736318T ES 2854751 T3 ES2854751 T3 ES 2854751T3
- Authority
- ES
- Spain
- Prior art keywords
- hybrid nanoparticles
- dissolution
- matrix
- experiments
- solution
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 238000000034 method Methods 0.000 title claims abstract description 205
- 230000000087 stabilizing effect Effects 0.000 title claims abstract description 90
- 229920000642 polymer Polymers 0.000 title claims abstract description 64
- 230000008569 process Effects 0.000 title claims abstract description 62
- 239000003909 protein kinase inhibitor Substances 0.000 title claims abstract description 47
- 229940043355 kinase inhibitor Drugs 0.000 title claims abstract description 43
- 239000002105 nanoparticle Substances 0.000 title description 276
- 239000002904 solvent Substances 0.000 claims abstract description 98
- 239000002245 particle Substances 0.000 claims abstract description 82
- 239000012530 fluid Substances 0.000 claims abstract description 69
- ZBNZXTGUTAYRHI-UHFFFAOYSA-N Dasatinib Chemical compound C=1C(N2CCN(CCO)CC2)=NC(C)=NC=1NC(S1)=NC=C1C(=O)NC1=C(C)C=CC=C1Cl ZBNZXTGUTAYRHI-UHFFFAOYSA-N 0.000 claims abstract description 37
- 239000002067 L01XE06 - Dasatinib Substances 0.000 claims abstract description 37
- 229960002448 dasatinib Drugs 0.000 claims abstract description 37
- 239000012296 anti-solvent Substances 0.000 claims abstract description 33
- 239000007962 solid dispersion Substances 0.000 claims abstract description 15
- 238000002156 mixing Methods 0.000 claims abstract description 11
- 239000011159 matrix material Substances 0.000 claims abstract description 6
- 150000003839 salts Chemical class 0.000 claims abstract description 5
- 239000012453 solvate Substances 0.000 claims abstract description 5
- 238000005507 spraying Methods 0.000 claims abstract description 3
- 229920002554 vinyl polymer Polymers 0.000 claims description 93
- 239000000203 mixture Substances 0.000 claims description 78
- 239000002202 Polyethylene glycol Substances 0.000 claims description 55
- 229920001223 polyethylene glycol Polymers 0.000 claims description 55
- 229920001577 copolymer Polymers 0.000 claims description 49
- 229920003132 hydroxypropyl methylcellulose phthalate Polymers 0.000 claims description 44
- -1 acetate-polyethylene Chemical group 0.000 claims description 40
- 229940100467 polyvinyl acetate phthalate Drugs 0.000 claims description 27
- 239000004359 castor oil Substances 0.000 claims description 25
- 235000019438 castor oil Nutrition 0.000 claims description 25
- ZEMPKEQAKRGZGQ-XOQCFJPHSA-N glycerol triricinoleate Natural products CCCCCC[C@@H](O)CC=CCCCCCCCC(=O)OC[C@@H](COC(=O)CCCCCCCC=CC[C@@H](O)CCCCCC)OC(=O)CCCCCCCC=CC[C@H](O)CCCCCC ZEMPKEQAKRGZGQ-XOQCFJPHSA-N 0.000 claims description 25
- 229920002153 Hydroxypropyl cellulose Polymers 0.000 claims description 22
- 235000010977 hydroxypropyl cellulose Nutrition 0.000 claims description 22
- 229920000036 polyvinylpyrrolidone Polymers 0.000 claims description 20
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 claims description 20
- WHNWPMSKXPGLAX-UHFFFAOYSA-N N-Vinyl-2-pyrrolidone Chemical compound C=CN1CCCC1=O WHNWPMSKXPGLAX-UHFFFAOYSA-N 0.000 claims description 19
- 229920001983 poloxamer Polymers 0.000 claims description 18
- 229920000623 Cellulose acetate phthalate Polymers 0.000 claims description 16
- 229940081734 cellulose acetate phthalate Drugs 0.000 claims description 16
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 claims description 14
- 229920003088 hydroxypropyl methyl cellulose Polymers 0.000 claims description 13
- 235000010979 hydroxypropyl methyl cellulose Nutrition 0.000 claims description 13
- 229940113116 polyethylene glycol 1000 Drugs 0.000 claims description 12
- 229920000639 hydroxypropylmethylcellulose acetate succinate Polymers 0.000 claims description 11
- 239000001863 hydroxypropyl cellulose Substances 0.000 claims description 9
- 229940031704 hydroxypropyl methylcellulose phthalate Drugs 0.000 claims description 9
- 229920002125 Sokalan® Polymers 0.000 claims description 8
- NIXOWILDQLNWCW-UHFFFAOYSA-N Acrylic acid Chemical compound OC(=O)C=C NIXOWILDQLNWCW-UHFFFAOYSA-N 0.000 claims description 7
- 229960001631 carbomer Drugs 0.000 claims description 7
- 229940099418 d- alpha-tocopherol succinate Drugs 0.000 claims description 7
- 239000003960 organic solvent Substances 0.000 claims description 7
- 239000002253 acid Substances 0.000 claims description 6
- 229920001519 homopolymer Polymers 0.000 claims description 6
- 239000003381 stabilizer Substances 0.000 claims description 6
- RHQDFWAXVIIEBN-UHFFFAOYSA-N Trifluoroethanol Chemical compound OCC(F)(F)F RHQDFWAXVIIEBN-UHFFFAOYSA-N 0.000 claims description 5
- 238000000634 powder X-ray diffraction Methods 0.000 claims description 5
- KDYFGRWQOYBRFD-UHFFFAOYSA-L succinate(2-) Chemical compound [O-]C(=O)CCC([O-])=O KDYFGRWQOYBRFD-UHFFFAOYSA-L 0.000 claims description 5
- 229920000663 Hydroxyethyl cellulose Polymers 0.000 claims description 4
- 239000004354 Hydroxyethyl cellulose Substances 0.000 claims description 4
- 229920003144 amino alkyl methacrylate copolymer Polymers 0.000 claims description 4
- 235000019447 hydroxyethyl cellulose Nutrition 0.000 claims description 4
- 239000001866 hydroxypropyl methyl cellulose Substances 0.000 claims description 4
- UFVKGYZPFZQRLF-UHFFFAOYSA-N hydroxypropyl methyl cellulose Chemical compound OC1C(O)C(OC)OC(CO)C1OC1C(O)C(O)C(OC2C(C(O)C(OC3C(C(O)C(O)C(CO)O3)O)C(CO)O2)O)C(CO)O1 UFVKGYZPFZQRLF-UHFFFAOYSA-N 0.000 claims description 4
- JJTUDXZGHPGLLC-UHFFFAOYSA-N lactide Chemical compound CC1OC(=O)C(C)OC1=O JJTUDXZGHPGLLC-UHFFFAOYSA-N 0.000 claims description 4
- 229920000609 methyl cellulose Polymers 0.000 claims description 4
- 239000001923 methylcellulose Substances 0.000 claims description 4
- 235000010981 methylcellulose Nutrition 0.000 claims description 4
- 239000007787 solid Substances 0.000 claims description 4
- 229920002678 cellulose Polymers 0.000 claims description 3
- 239000001913 cellulose Substances 0.000 claims description 3
- 229960000913 crospovidone Drugs 0.000 claims description 3
- 229920003145 methacrylic acid copolymer Polymers 0.000 claims description 3
- XNGIFLGASWRNHJ-UHFFFAOYSA-L phthalate(2-) Chemical compound [O-]C(=O)C1=CC=CC=C1C([O-])=O XNGIFLGASWRNHJ-UHFFFAOYSA-L 0.000 claims description 3
- 229920000523 polyvinylpolypyrrolidone Polymers 0.000 claims description 3
- 235000013809 polyvinylpolypyrrolidone Nutrition 0.000 claims description 3
- 239000001267 polyvinylpyrrolidone Substances 0.000 claims description 3
- 239000004743 Polypropylene Substances 0.000 claims 1
- HSFWRNGVRCDJHI-UHFFFAOYSA-N alpha-acetylene Natural products C#C HSFWRNGVRCDJHI-UHFFFAOYSA-N 0.000 claims 1
- 239000006185 dispersion Substances 0.000 claims 1
- GDCRSXZBSIRSFR-UHFFFAOYSA-N ethyl prop-2-enoate;2-methylprop-2-enoic acid Chemical compound CC(=C)C(O)=O.CCOC(=O)C=C GDCRSXZBSIRSFR-UHFFFAOYSA-N 0.000 claims 1
- 239000008101 lactose Substances 0.000 claims 1
- 229940117841 methacrylic acid copolymer Drugs 0.000 claims 1
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims 1
- 229920001155 polypropylene Polymers 0.000 claims 1
- 229960001295 tocopherol Drugs 0.000 claims 1
- 239000011732 tocopherol Substances 0.000 claims 1
- 238000002474 experimental method Methods 0.000 description 213
- 238000004090 dissolution Methods 0.000 description 189
- 239000000243 solution Substances 0.000 description 135
- 238000012369 In process control Methods 0.000 description 93
- 210000004544 dc2 Anatomy 0.000 description 93
- 238000004190 ion pair chromatography Methods 0.000 description 93
- 239000005536 L01XE08 - Nilotinib Substances 0.000 description 51
- HHZIURLSWUIHRB-UHFFFAOYSA-N nilotinib Chemical compound C1=NC(C)=CN1C1=CC(NC(=O)C=2C=C(NC=3N=C(C=CN=3)C=3C=NC=CC=3)C(C)=CC=2)=CC(C(F)(F)F)=C1 HHZIURLSWUIHRB-UHFFFAOYSA-N 0.000 description 51
- 229960001346 nilotinib Drugs 0.000 description 49
- 102100028675 DNA polymerase subunit gamma-2, mitochondrial Human genes 0.000 description 43
- 101000837415 Homo sapiens DNA polymerase subunit gamma-2, mitochondrial Proteins 0.000 description 43
- VTGGYCCJUPYZSX-UHFFFAOYSA-N 4-methyl-n-[3-(4-methylimidazol-1-yl)-5-(trifluoromethyl)phenyl]-3-[(4-pyridin-3-ylpyrimidin-2-yl)amino]benzamide;hydrochloride Chemical compound Cl.C1=NC(C)=CN1C1=CC(NC(=O)C=2C=C(NC=3N=C(C=CN=3)C=3C=NC=CC=3)C(C)=CC=2)=CC(C(F)(F)F)=C1 VTGGYCCJUPYZSX-UHFFFAOYSA-N 0.000 description 35
- 238000005259 measurement Methods 0.000 description 34
- 229940079593 drug Drugs 0.000 description 32
- 239000003814 drug Substances 0.000 description 32
- 230000006872 improvement Effects 0.000 description 30
- 230000000052 comparative effect Effects 0.000 description 28
- 230000000968 intestinal effect Effects 0.000 description 28
- 239000000843 powder Substances 0.000 description 28
- 230000003381 solubilizing effect Effects 0.000 description 28
- 229960003005 axitinib Drugs 0.000 description 27
- RITAVMQDGBJQJZ-FMIVXFBMSA-N axitinib Chemical compound CNC(=O)C1=CC=CC=C1SC1=CC=C(C(\C=C\C=2N=CC=CC=2)=NN2)C2=C1 RITAVMQDGBJQJZ-FMIVXFBMSA-N 0.000 description 27
- 239000005411 L01XE02 - Gefitinib Substances 0.000 description 26
- 239000003798 L01XE11 - Pazopanib Substances 0.000 description 26
- 239000002146 L01XE16 - Crizotinib Substances 0.000 description 26
- 229960005061 crizotinib Drugs 0.000 description 26
- KTEIFNKAUNYNJU-GFCCVEGCSA-N crizotinib Chemical compound O([C@H](C)C=1C(=C(F)C=CC=1Cl)Cl)C(C(=NC=1)N)=CC=1C(=C1)C=NN1C1CCNCC1 KTEIFNKAUNYNJU-GFCCVEGCSA-N 0.000 description 26
- 229960002584 gefitinib Drugs 0.000 description 26
- XGALLCVXEZPNRQ-UHFFFAOYSA-N gefitinib Chemical compound C=12C=C(OCCCN3CCOCC3)C(OC)=CC2=NC=NC=1NC1=CC=C(F)C(Cl)=C1 XGALLCVXEZPNRQ-UHFFFAOYSA-N 0.000 description 26
- 229960000639 pazopanib Drugs 0.000 description 26
- CUIHSIWYWATEQL-UHFFFAOYSA-N pazopanib Chemical compound C1=CC2=C(C)N(C)N=C2C=C1N(C)C(N=1)=CC=NC=1NC1=CC=C(C)C(S(N)(=O)=O)=C1 CUIHSIWYWATEQL-UHFFFAOYSA-N 0.000 description 26
- 229960003862 vemurafenib Drugs 0.000 description 26
- GPXBXXGIAQBQNI-UHFFFAOYSA-N vemurafenib Chemical compound CCCS(=O)(=O)NC1=CC=C(F)C(C(=O)C=2C3=CC(=CN=C3NC=2)C=2C=CC(Cl)=CC=2)=C1F GPXBXXGIAQBQNI-UHFFFAOYSA-N 0.000 description 26
- GTTBEUCJPZQMDZ-UHFFFAOYSA-N erlotinib hydrochloride Chemical compound [H+].[Cl-].C=12C=C(OCCOC)C(OCCOC)=CC2=NC=NC=1NC1=CC=CC(C#C)=C1 GTTBEUCJPZQMDZ-UHFFFAOYSA-N 0.000 description 25
- 238000009472 formulation Methods 0.000 description 24
- 239000002609 medium Substances 0.000 description 21
- 229920002744 polyvinyl acetate phthalate Polymers 0.000 description 20
- BCFGMOOMADDAQU-UHFFFAOYSA-N lapatinib Chemical compound O1C(CNCCS(=O)(=O)C)=CC=C1C1=CC=C(N=CN=C2NC=3C=C(Cl)C(OCC=4C=C(F)C=CC=4)=CC=3)C2=C1 BCFGMOOMADDAQU-UHFFFAOYSA-N 0.000 description 19
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 18
- 229920003083 Kollidon® VA64 Polymers 0.000 description 18
- 239000003112 inhibitor Substances 0.000 description 18
- 238000012360 testing method Methods 0.000 description 16
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 16
- 241000282472 Canis lupus familiaris Species 0.000 description 15
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 15
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 14
- 230000015572 biosynthetic process Effects 0.000 description 14
- 230000002496 gastric effect Effects 0.000 description 13
- 150000001875 compounds Chemical class 0.000 description 12
- 239000008194 pharmaceutical composition Substances 0.000 description 12
- 229960000487 sorafenib tosylate Drugs 0.000 description 12
- IVDHYUQIDRJSTI-UHFFFAOYSA-N sorafenib tosylate Chemical compound [H+].CC1=CC=C(S([O-])(=O)=O)C=C1.C1=NC(C(=O)NC)=CC(OC=2C=CC(NC(=O)NC=3C=C(C(Cl)=CC=3)C(F)(F)F)=CC=2)=C1 IVDHYUQIDRJSTI-UHFFFAOYSA-N 0.000 description 12
- MLDQJTXFUGDVEO-UHFFFAOYSA-N BAY-43-9006 Chemical compound C1=NC(C(=O)NC)=CC(OC=2C=CC(NC(=O)NC=3C=C(C(Cl)=CC=3)C(F)(F)F)=CC=2)=C1 MLDQJTXFUGDVEO-UHFFFAOYSA-N 0.000 description 11
- 239000005511 L01XE05 - Sorafenib Substances 0.000 description 11
- 239000002136 L01XE07 - Lapatinib Substances 0.000 description 11
- 238000010521 absorption reaction Methods 0.000 description 11
- 229960004891 lapatinib Drugs 0.000 description 11
- 238000011068 loading method Methods 0.000 description 11
- 238000001556 precipitation Methods 0.000 description 11
- 229960003787 sorafenib Drugs 0.000 description 11
- 239000005551 L01XE03 - Erlotinib Substances 0.000 description 10
- 229960001433 erlotinib Drugs 0.000 description 10
- AAKJLRGGTJKAMG-UHFFFAOYSA-N erlotinib Chemical compound C=12C=C(OCCOC)C(OCCOC)=CC2=NC=NC=1NC1=CC=CC(C#C)=C1 AAKJLRGGTJKAMG-UHFFFAOYSA-N 0.000 description 10
- 239000007788 liquid Substances 0.000 description 10
- 108060006633 protein kinase Proteins 0.000 description 10
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 9
- 102000001253 Protein Kinase Human genes 0.000 description 9
- 238000001727 in vivo Methods 0.000 description 9
- 230000007935 neutral effect Effects 0.000 description 9
- 230000036470 plasma concentration Effects 0.000 description 9
- 238000001990 intravenous administration Methods 0.000 description 8
- 239000003607 modifier Substances 0.000 description 8
- AZBFJBJXUQUQLF-UHFFFAOYSA-N n-(1,5-dimethylpyrrolidin-3-yl)pyrrolidine-1-carboxamide Chemical class C1N(C)C(C)CC1NC(=O)N1CCCC1 AZBFJBJXUQUQLF-UHFFFAOYSA-N 0.000 description 8
- MQHIQUBXFFAOMK-UHFFFAOYSA-N pazopanib hydrochloride Chemical compound Cl.C1=CC2=C(C)N(C)N=C2C=C1N(C)C(N=1)=CC=NC=1NC1=CC=C(C)C(S(N)(=O)=O)=C1 MQHIQUBXFFAOMK-UHFFFAOYSA-N 0.000 description 8
- 210000002784 stomach Anatomy 0.000 description 8
- 238000003860 storage Methods 0.000 description 8
- 230000002378 acidificating effect Effects 0.000 description 7
- 238000004458 analytical method Methods 0.000 description 7
- 210000004027 cell Anatomy 0.000 description 7
- 239000011521 glass Substances 0.000 description 7
- 229910052757 nitrogen Inorganic materials 0.000 description 7
- 239000006069 physical mixture Substances 0.000 description 7
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 6
- AOBORMOPSGHCAX-UHFFFAOYSA-N Tocophersolan Chemical compound OCCOC(=O)CCC(=O)OC1=C(C)C(C)=C2OC(CCCC(C)CCCC(C)CCCC(C)C)(C)CCC2=C1C AOBORMOPSGHCAX-UHFFFAOYSA-N 0.000 description 6
- 239000004480 active ingredient Substances 0.000 description 6
- 239000003795 chemical substances by application Substances 0.000 description 6
- GVJHHUAWPYXKBD-UHFFFAOYSA-N d-alpha-tocopherol Natural products OC1=C(C)C(C)=C2OC(CCCC(C)CCCC(C)CCCC(C)C)(C)CCC2=C1C GVJHHUAWPYXKBD-UHFFFAOYSA-N 0.000 description 6
- 230000001079 digestive effect Effects 0.000 description 6
- 230000009246 food effect Effects 0.000 description 6
- 210000003736 gastrointestinal content Anatomy 0.000 description 6
- 238000004128 high performance liquid chromatography Methods 0.000 description 6
- 239000000463 material Substances 0.000 description 6
- 239000000546 pharmaceutical excipient Substances 0.000 description 6
- 108090000623 proteins and genes Proteins 0.000 description 6
- 230000000717 retained effect Effects 0.000 description 6
- 239000000126 substance Substances 0.000 description 6
- 229920003211 cis-1,4-polyisoprene Polymers 0.000 description 5
- 229920002689 polyvinyl acetate Polymers 0.000 description 5
- 239000011118 polyvinyl acetate Substances 0.000 description 5
- 229920006316 polyvinylpyrrolidine Polymers 0.000 description 5
- 239000011164 primary particle Substances 0.000 description 5
- 102000004169 proteins and genes Human genes 0.000 description 5
- 206010028980 Neoplasm Diseases 0.000 description 4
- 108091000080 Phosphotransferase Proteins 0.000 description 4
- 229920002690 Polyoxyl 40 HydrogenatedCastorOil Polymers 0.000 description 4
- ATUOYWHBWRKTHZ-UHFFFAOYSA-N Propane Chemical compound CCC ATUOYWHBWRKTHZ-UHFFFAOYSA-N 0.000 description 4
- 239000013543 active substance Substances 0.000 description 4
- 229940045988 antineoplastic drug protein kinase inhibitors Drugs 0.000 description 4
- 238000001514 detection method Methods 0.000 description 4
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 4
- 239000012738 dissolution medium Substances 0.000 description 4
- 239000002552 dosage form Substances 0.000 description 4
- 230000037406 food intake Effects 0.000 description 4
- 238000012623 in vivo measurement Methods 0.000 description 4
- 238000001802 infusion Methods 0.000 description 4
- 229960001320 lapatinib ditosylate Drugs 0.000 description 4
- VNWKTOKETHGBQD-UHFFFAOYSA-N methane Chemical compound C VNWKTOKETHGBQD-UHFFFAOYSA-N 0.000 description 4
- 239000011859 microparticle Substances 0.000 description 4
- 102000020233 phosphotransferase Human genes 0.000 description 4
- 238000005086 pumping Methods 0.000 description 4
- 238000002411 thermogravimetry Methods 0.000 description 4
- 229960000984 tocofersolan Drugs 0.000 description 4
- AOBORMOPSGHCAX-DGHZZKTQSA-N tocofersolan Chemical compound OCCOC(=O)CCC(=O)OC1=C(C)C(C)=C2O[C@](CCC[C@H](C)CCC[C@H](C)CCCC(C)C)(C)CCC2=C1C AOBORMOPSGHCAX-DGHZZKTQSA-N 0.000 description 4
- 239000005483 tyrosine kinase inhibitor Substances 0.000 description 4
- 229940121358 tyrosine kinase inhibitor Drugs 0.000 description 4
- 239000002076 α-tocopherol Substances 0.000 description 4
- 235000004835 α-tocopherol Nutrition 0.000 description 4
- OTMSDBZUPAUEDD-UHFFFAOYSA-N Ethane Chemical compound CC OTMSDBZUPAUEDD-UHFFFAOYSA-N 0.000 description 3
- VGGSQFUCUMXWEO-UHFFFAOYSA-N Ethene Chemical compound C=C VGGSQFUCUMXWEO-UHFFFAOYSA-N 0.000 description 3
- 239000005977 Ethylene Substances 0.000 description 3
- CERQOIWHTDAKMF-UHFFFAOYSA-M Methacrylate Chemical compound CC(=C)C([O-])=O CERQOIWHTDAKMF-UHFFFAOYSA-M 0.000 description 3
- 229920003091 Methocel™ Polymers 0.000 description 3
- RVGRUAULSDPKGF-UHFFFAOYSA-N Poloxamer Chemical compound C1CO1.CC1CO1 RVGRUAULSDPKGF-UHFFFAOYSA-N 0.000 description 3
- 229920001244 Poly(D,L-lactide) Polymers 0.000 description 3
- 239000002775 capsule Substances 0.000 description 3
- 229910002092 carbon dioxide Inorganic materials 0.000 description 3
- 239000001569 carbon dioxide Substances 0.000 description 3
- 230000008859 change Effects 0.000 description 3
- 230000001186 cumulative effect Effects 0.000 description 3
- 235000013305 food Nutrition 0.000 description 3
- 235000012631 food intake Nutrition 0.000 description 3
- 210000001035 gastrointestinal tract Anatomy 0.000 description 3
- 239000000178 monomer Substances 0.000 description 3
- 230000003204 osmotic effect Effects 0.000 description 3
- 230000002018 overexpression Effects 0.000 description 3
- PNJWIWWMYCMZRO-UHFFFAOYSA-N pent‐4‐en‐2‐one Natural products CC(=O)CC=C PNJWIWWMYCMZRO-UHFFFAOYSA-N 0.000 description 3
- 230000026731 phosphorylation Effects 0.000 description 3
- 238000006366 phosphorylation reaction Methods 0.000 description 3
- 229960000502 poloxamer Drugs 0.000 description 3
- LQIAZOCLNBBZQK-UHFFFAOYSA-N 1-(1,2-Diphosphanylethyl)pyrrolidin-2-one Chemical compound PCC(P)N1CCCC1=O LQIAZOCLNBBZQK-UHFFFAOYSA-N 0.000 description 2
- JVTIXNMXDLQEJE-UHFFFAOYSA-N 2-decanoyloxypropyl decanoate 2-octanoyloxypropyl octanoate Chemical compound C(CCCCCCC)(=O)OCC(C)OC(CCCCCCC)=O.C(=O)(CCCCCCCCC)OCC(C)OC(=O)CCCCCCCCC JVTIXNMXDLQEJE-UHFFFAOYSA-N 0.000 description 2
- RFVNOJDQRGSOEL-UHFFFAOYSA-N 2-hydroxyethyl octadecanoate Chemical compound CCCCCCCCCCCCCCCCCC(=O)OCCO RFVNOJDQRGSOEL-UHFFFAOYSA-N 0.000 description 2
- IELOKBJPULMYRW-NJQVLOCASA-N D-alpha-Tocopheryl Acid Succinate Chemical compound OC(=O)CCC(=O)OC1=C(C)C(C)=C2O[C@@](CCC[C@H](C)CCC[C@H](C)CCCC(C)C)(C)CCC2=C1C IELOKBJPULMYRW-NJQVLOCASA-N 0.000 description 2
- JIGUQPWFLRLWPJ-UHFFFAOYSA-N Ethyl acrylate Chemical compound CCOC(=O)C=C JIGUQPWFLRLWPJ-UHFFFAOYSA-N 0.000 description 2
- 229920003139 Eudragit® L 100 Polymers 0.000 description 2
- 229920003155 Eudragit® RL 100 Polymers 0.000 description 2
- 229920003156 Eudragit® RL PO Polymers 0.000 description 2
- 229920003160 Eudragit® RS PO Polymers 0.000 description 2
- XPDWGBQVDMORPB-UHFFFAOYSA-N Fluoroform Chemical compound FC(F)F XPDWGBQVDMORPB-UHFFFAOYSA-N 0.000 description 2
- 241000282412 Homo Species 0.000 description 2
- 229920003085 Kollidon® CL Polymers 0.000 description 2
- VVQNEPGJFQJSBK-UHFFFAOYSA-N Methyl methacrylate Chemical compound COC(=O)C(C)=C VVQNEPGJFQJSBK-UHFFFAOYSA-N 0.000 description 2
- PXHVJJICTQNCMI-UHFFFAOYSA-N Nickel Chemical compound [Ni] PXHVJJICTQNCMI-UHFFFAOYSA-N 0.000 description 2
- GQPLMRYTRLFLPF-UHFFFAOYSA-N Nitrous Oxide Chemical compound [O-][N+]#N GQPLMRYTRLFLPF-UHFFFAOYSA-N 0.000 description 2
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 2
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 2
- ZUAAPNNKRHMPKG-UHFFFAOYSA-N acetic acid;butanedioic acid;methanol;propane-1,2-diol Chemical compound OC.CC(O)=O.CC(O)CO.OC(=O)CCC(O)=O ZUAAPNNKRHMPKG-UHFFFAOYSA-N 0.000 description 2
- 230000009471 action Effects 0.000 description 2
- 238000005054 agglomeration Methods 0.000 description 2
- 230000002776 aggregation Effects 0.000 description 2
- 150000001413 amino acids Chemical class 0.000 description 2
- 239000007864 aqueous solution Substances 0.000 description 2
- 239000000872 buffer Substances 0.000 description 2
- 238000010924 continuous production Methods 0.000 description 2
- 238000011161 development Methods 0.000 description 2
- 239000002270 dispersing agent Substances 0.000 description 2
- 238000001914 filtration Methods 0.000 description 2
- 230000009477 glass transition Effects 0.000 description 2
- 238000010438 heat treatment Methods 0.000 description 2
- 150000004677 hydrates Chemical class 0.000 description 2
- 238000000338 in vitro Methods 0.000 description 2
- 238000002347 injection Methods 0.000 description 2
- 239000007924 injection Substances 0.000 description 2
- 210000002429 large intestine Anatomy 0.000 description 2
- 230000000670 limiting effect Effects 0.000 description 2
- 238000001565 modulated differential scanning calorimetry Methods 0.000 description 2
- 230000035772 mutation Effects 0.000 description 2
- 238000010979 pH adjustment Methods 0.000 description 2
- 239000012071 phase Substances 0.000 description 2
- 239000002244 precipitate Substances 0.000 description 2
- 239000000047 product Substances 0.000 description 2
- 230000002062 proliferating effect Effects 0.000 description 2
- 239000001294 propane Substances 0.000 description 2
- 230000002829 reductive effect Effects 0.000 description 2
- 238000000926 separation method Methods 0.000 description 2
- 210000000813 small intestine Anatomy 0.000 description 2
- 239000007790 solid phase Substances 0.000 description 2
- 238000001179 sorption measurement Methods 0.000 description 2
- 239000007921 spray Substances 0.000 description 2
- 238000013112 stability test Methods 0.000 description 2
- 229940069905 tasigna Drugs 0.000 description 2
- 239000012085 test solution Substances 0.000 description 2
- 230000001225 therapeutic effect Effects 0.000 description 2
- 150000004917 tyrosine kinase inhibitor derivatives Chemical class 0.000 description 2
- 230000003827 upregulation Effects 0.000 description 2
- 238000005406 washing Methods 0.000 description 2
- MTCFGRXMJLQNBG-REOHCLBHSA-N (2S)-2-Amino-3-hydroxypropansäure Chemical compound OC[C@H](N)C(O)=O MTCFGRXMJLQNBG-REOHCLBHSA-N 0.000 description 1
- WLAMNBDJUVNPJU-UHFFFAOYSA-N 2-methylbutyric acid Chemical compound CCC(C)C(O)=O WLAMNBDJUVNPJU-UHFFFAOYSA-N 0.000 description 1
- DOUBAFNWVFAWEC-UHFFFAOYSA-N 3-hydroxypropyl acetate Chemical compound CC(=O)OCCCO DOUBAFNWVFAWEC-UHFFFAOYSA-N 0.000 description 1
- XXJWYDDUDKYVKI-UHFFFAOYSA-N 4-[(4-fluoro-2-methyl-1H-indol-5-yl)oxy]-6-methoxy-7-[3-(1-pyrrolidinyl)propoxy]quinazoline Chemical compound COC1=CC2=C(OC=3C(=C4C=C(C)NC4=CC=3)F)N=CN=C2C=C1OCCCN1CCCC1 XXJWYDDUDKYVKI-UHFFFAOYSA-N 0.000 description 1
- WLCZTRVUXYALDD-IBGZPJMESA-N 7-[[(2s)-2,6-bis(2-methoxyethoxycarbonylamino)hexanoyl]amino]heptoxy-methylphosphinic acid Chemical compound COCCOC(=O)NCCCC[C@H](NC(=O)OCCOC)C(=O)NCCCCCCCOP(C)(O)=O WLCZTRVUXYALDD-IBGZPJMESA-N 0.000 description 1
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 1
- 101100248253 Arabidopsis thaliana RH40 gene Proteins 0.000 description 1
- 101100180402 Caenorhabditis elegans jun-1 gene Proteins 0.000 description 1
- 229910002483 Cu Ka Inorganic materials 0.000 description 1
- 230000004544 DNA amplification Effects 0.000 description 1
- GWCPMNRTISDVKH-UHFFFAOYSA-N F.F.F.F.F.F.S Chemical compound F.F.F.F.F.F.S GWCPMNRTISDVKH-UHFFFAOYSA-N 0.000 description 1
- 108010072039 Histidine kinase Proteins 0.000 description 1
- 108090000144 Human Proteins Proteins 0.000 description 1
- 102000003839 Human Proteins Human genes 0.000 description 1
- AYFVYJQAPQTCCC-GBXIJSLDSA-N L-threonine Chemical compound C[C@@H](O)[C@H](N)C(O)=O AYFVYJQAPQTCCC-GBXIJSLDSA-N 0.000 description 1
- OUYCCCASQSFEME-QMMMGPOBSA-N L-tyrosine Chemical compound OC(=O)[C@@H](N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-QMMMGPOBSA-N 0.000 description 1
- 239000005517 L01XE01 - Imatinib Substances 0.000 description 1
- 239000002147 L01XE04 - Sunitinib Substances 0.000 description 1
- 239000002118 L01XE12 - Vandetanib Substances 0.000 description 1
- 239000002145 L01XE14 - Bosutinib Substances 0.000 description 1
- 239000002144 L01XE18 - Ruxolitinib Substances 0.000 description 1
- 241001465754 Metazoa Species 0.000 description 1
- CERQOIWHTDAKMF-UHFFFAOYSA-N Methacrylic acid Chemical compound CC(=C)C(O)=O CERQOIWHTDAKMF-UHFFFAOYSA-N 0.000 description 1
- 239000005462 Mubritinib Substances 0.000 description 1
- 229920002685 Polyoxyl 35CastorOil Polymers 0.000 description 1
- 229920002701 Polyoxyl 40 Stearate Polymers 0.000 description 1
- 108090000412 Protein-Tyrosine Kinases Proteins 0.000 description 1
- 102000004022 Protein-Tyrosine Kinases Human genes 0.000 description 1
- KAESVJOAVNADME-UHFFFAOYSA-N Pyrrole Chemical compound C=1C=CNC=1 KAESVJOAVNADME-UHFFFAOYSA-N 0.000 description 1
- MTCFGRXMJLQNBG-UHFFFAOYSA-N Serine Natural products OCC(N)C(O)=O MTCFGRXMJLQNBG-UHFFFAOYSA-N 0.000 description 1
- AYFVYJQAPQTCCC-UHFFFAOYSA-N Threonine Natural products CC(O)C(N)C(O)=O AYFVYJQAPQTCCC-UHFFFAOYSA-N 0.000 description 1
- 239000004473 Threonine Substances 0.000 description 1
- 229960001686 afatinib Drugs 0.000 description 1
- ULXXDDBFHOBEHA-CWDCEQMOSA-N afatinib Chemical compound N1=CN=C2C=C(O[C@@H]3COCC3)C(NC(=O)/C=C/CN(C)C)=CC2=C1NC1=CC=C(F)C(Cl)=C1 ULXXDDBFHOBEHA-CWDCEQMOSA-N 0.000 description 1
- 150000001447 alkali salts Chemical class 0.000 description 1
- 229910052782 aluminium Inorganic materials 0.000 description 1
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 1
- 230000000259 anti-tumor effect Effects 0.000 description 1
- 239000000063 antileukemic agent Substances 0.000 description 1
- 239000002246 antineoplastic agent Substances 0.000 description 1
- 239000008135 aqueous vehicle Substances 0.000 description 1
- 238000003556 assay Methods 0.000 description 1
- 230000006399 behavior Effects 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- 239000008280 blood Substances 0.000 description 1
- 210000004369 blood Anatomy 0.000 description 1
- 230000037396 body weight Effects 0.000 description 1
- 238000009835 boiling Methods 0.000 description 1
- 229960003736 bosutinib Drugs 0.000 description 1
- UBPYILGKFZZVDX-UHFFFAOYSA-N bosutinib Chemical compound C1=C(Cl)C(OC)=CC(NC=2C3=CC(OC)=C(OCCCN4CCN(C)CC4)C=C3N=CC=2C#N)=C1Cl UBPYILGKFZZVDX-UHFFFAOYSA-N 0.000 description 1
- 201000011510 cancer Diseases 0.000 description 1
- 239000007963 capsule composition Substances 0.000 description 1
- 229940075508 carbomer homopolymer type b Drugs 0.000 description 1
- 229910052799 carbon Inorganic materials 0.000 description 1
- 229960002412 cediranib Drugs 0.000 description 1
- 230000022131 cell cycle Effects 0.000 description 1
- 230000010261 cell growth Effects 0.000 description 1
- 230000004640 cellular pathway Effects 0.000 description 1
- 230000004715 cellular signal transduction Effects 0.000 description 1
- 230000005754 cellular signaling Effects 0.000 description 1
- AFYPFACVUDMOHA-UHFFFAOYSA-N chlorotrifluoromethane Chemical compound FC(F)(F)Cl AFYPFACVUDMOHA-UHFFFAOYSA-N 0.000 description 1
- 238000004140 cleaning Methods 0.000 description 1
- 229920001688 coating polymer Polymers 0.000 description 1
- 210000001072 colon Anatomy 0.000 description 1
- 239000000306 component Substances 0.000 description 1
- 239000013078 crystal Substances 0.000 description 1
- 238000002425 crystallisation Methods 0.000 description 1
- 230000008025 crystallization Effects 0.000 description 1
- 238000013480 data collection Methods 0.000 description 1
- 230000003247 decreasing effect Effects 0.000 description 1
- 230000003111 delayed effect Effects 0.000 description 1
- 230000001419 dependent effect Effects 0.000 description 1
- 230000002074 deregulated effect Effects 0.000 description 1
- 238000013461 design Methods 0.000 description 1
- 230000004069 differentiation Effects 0.000 description 1
- 201000010099 disease Diseases 0.000 description 1
- 239000007884 disintegrant Substances 0.000 description 1
- 238000001647 drug administration Methods 0.000 description 1
- 238000009506 drug dissolution testing Methods 0.000 description 1
- 230000008482 dysregulation Effects 0.000 description 1
- 230000000694 effects Effects 0.000 description 1
- 239000000839 emulsion Substances 0.000 description 1
- 238000005516 engineering process Methods 0.000 description 1
- 239000002702 enteric coating Substances 0.000 description 1
- 238000009505 enteric coating Methods 0.000 description 1
- 230000002255 enzymatic effect Effects 0.000 description 1
- 239000002532 enzyme inhibitor Substances 0.000 description 1
- 229940125532 enzyme inhibitor Drugs 0.000 description 1
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 1
- 238000001125 extrusion Methods 0.000 description 1
- 239000000945 filler Substances 0.000 description 1
- 239000010419 fine particle Substances 0.000 description 1
- 230000004907 flux Effects 0.000 description 1
- 229950005309 fostamatinib Drugs 0.000 description 1
- GKDRMWXFWHEQQT-UHFFFAOYSA-N fostamatinib Chemical compound COC1=C(OC)C(OC)=CC(NC=2N=C(NC=3N=C4N(COP(O)(O)=O)C(=O)C(C)(C)OC4=CC=3)C(F)=CN=2)=C1 GKDRMWXFWHEQQT-UHFFFAOYSA-N 0.000 description 1
- 239000007789 gas Substances 0.000 description 1
- 239000004083 gastrointestinal agent Substances 0.000 description 1
- 229940127227 gastrointestinal drug Drugs 0.000 description 1
- 125000005456 glyceride group Chemical group 0.000 description 1
- 238000005469 granulation Methods 0.000 description 1
- 230000003179 granulation Effects 0.000 description 1
- 238000000892 gravimetry Methods 0.000 description 1
- 238000000227 grinding Methods 0.000 description 1
- 230000012010 growth Effects 0.000 description 1
- 229940116364 hard fat Drugs 0.000 description 1
- 125000000487 histidyl group Chemical group [H]N([H])C(C(=O)O*)C([H])([H])C1=C([H])N([H])C([H])=N1 0.000 description 1
- 238000009474 hot melt extrusion Methods 0.000 description 1
- 230000002209 hydrophobic effect Effects 0.000 description 1
- 229960002411 imatinib Drugs 0.000 description 1
- KTUFNOKKBVMGRW-UHFFFAOYSA-N imatinib Chemical compound C1CN(C)CCN1CC1=CC=C(C(=O)NC=2C=C(NC=3N=C(C=CN=3)C=3C=NC=CC=3)C(C)=CC=2)C=C1 KTUFNOKKBVMGRW-UHFFFAOYSA-N 0.000 description 1
- 238000010874 in vitro model Methods 0.000 description 1
- 229910052738 indium Inorganic materials 0.000 description 1
- APFVFJFRJDLVQX-UHFFFAOYSA-N indium atom Chemical compound [In] APFVFJFRJDLVQX-UHFFFAOYSA-N 0.000 description 1
- 229910001867 inorganic solvent Inorganic materials 0.000 description 1
- 239000003049 inorganic solvent Substances 0.000 description 1
- 230000031891 intestinal absorption Effects 0.000 description 1
- 238000007918 intramuscular administration Methods 0.000 description 1
- 238000000111 isothermal titration calorimetry Methods 0.000 description 1
- 239000003446 ligand Substances 0.000 description 1
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 1
- 239000000314 lubricant Substances 0.000 description 1
- 238000004519 manufacturing process Methods 0.000 description 1
- 230000001404 mediated effect Effects 0.000 description 1
- 239000012528 membrane Substances 0.000 description 1
- 238000000386 microscopy Methods 0.000 description 1
- 239000004005 microsphere Substances 0.000 description 1
- 230000003278 mimic effect Effects 0.000 description 1
- 229950003968 motesanib Drugs 0.000 description 1
- RAHBGWKEPAQNFF-UHFFFAOYSA-N motesanib Chemical compound C=1C=C2C(C)(C)CNC2=CC=1NC(=O)C1=CC=CN=C1NCC1=CC=NC=C1 RAHBGWKEPAQNFF-UHFFFAOYSA-N 0.000 description 1
- 229950002212 mubritinib Drugs 0.000 description 1
- ZTFBIUXIQYRUNT-MDWZMJQESA-N mubritinib Chemical compound C1=CC(C(F)(F)F)=CC=C1\C=C\C1=NC(COC=2C=CC(CCCCN3N=NC=C3)=CC=2)=CO1 ZTFBIUXIQYRUNT-MDWZMJQESA-N 0.000 description 1
- 229940028444 muse Drugs 0.000 description 1
- 229910052759 nickel Inorganic materials 0.000 description 1
- 239000001272 nitrous oxide Substances 0.000 description 1
- 231100000957 no side effect Toxicity 0.000 description 1
- WWZKQHOCKIZLMA-UHFFFAOYSA-N octanoic acid Chemical compound CCCCCCCC(O)=O WWZKQHOCKIZLMA-UHFFFAOYSA-N 0.000 description 1
- QUANRIQJNFHVEU-UHFFFAOYSA-N oxirane;propane-1,2,3-triol Chemical compound C1CO1.OCC(O)CO QUANRIQJNFHVEU-UHFFFAOYSA-N 0.000 description 1
- 238000007911 parenteral administration Methods 0.000 description 1
- 230000037361 pathway Effects 0.000 description 1
- 229960003407 pegaptanib Drugs 0.000 description 1
- 230000035699 permeability Effects 0.000 description 1
- 125000002467 phosphate group Chemical group [H]OP(=O)(O[H])O[*] 0.000 description 1
- 229920000191 poly(N-vinyl pyrrolidone) Polymers 0.000 description 1
- 239000008389 polyethoxylated castor oil Substances 0.000 description 1
- 210000004896 polypeptide structure Anatomy 0.000 description 1
- 238000002360 preparation method Methods 0.000 description 1
- 239000003755 preservative agent Substances 0.000 description 1
- 230000002335 preservative effect Effects 0.000 description 1
- 230000002265 prevention Effects 0.000 description 1
- 238000012545 processing Methods 0.000 description 1
- 230000002035 prolonged effect Effects 0.000 description 1
- GMVPRGQOIOIIMI-DWKJAMRDSA-N prostaglandin E1 Chemical compound CCCCC[C@H](O)\C=C\[C@H]1[C@H](O)CC(=O)[C@@H]1CCCCCCC(O)=O GMVPRGQOIOIIMI-DWKJAMRDSA-N 0.000 description 1
- 238000010926 purge Methods 0.000 description 1
- 238000011002 quantification Methods 0.000 description 1
- 230000005855 radiation Effects 0.000 description 1
- 230000008707 rearrangement Effects 0.000 description 1
- 238000005057 refrigeration Methods 0.000 description 1
- 239000013557 residual solvent Substances 0.000 description 1
- 229960000215 ruxolitinib Drugs 0.000 description 1
- HFNKQEVNSGCOJV-OAHLLOKOSA-N ruxolitinib Chemical compound C1([C@@H](CC#N)N2N=CC(=C2)C=2C=3C=CNC=3N=CN=2)CCCC1 HFNKQEVNSGCOJV-OAHLLOKOSA-N 0.000 description 1
- 229950003647 semaxanib Drugs 0.000 description 1
- WUWDLXZGHZSWQZ-WQLSENKSSA-N semaxanib Chemical compound N1C(C)=CC(C)=C1\C=C/1C2=CC=CC=C2NC\1=O WUWDLXZGHZSWQZ-WQLSENKSSA-N 0.000 description 1
- 102000034285 signal transducing proteins Human genes 0.000 description 1
- 108091006024 signal transducing proteins Proteins 0.000 description 1
- 230000019491 signal transduction Effects 0.000 description 1
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 1
- 235000017557 sodium bicarbonate Nutrition 0.000 description 1
- 239000007909 solid dosage form Substances 0.000 description 1
- 239000006104 solid solution Substances 0.000 description 1
- 238000001694 spray drying Methods 0.000 description 1
- 125000003696 stearoyl group Chemical group O=C([*])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 238000007920 subcutaneous administration Methods 0.000 description 1
- 229910052717 sulfur Inorganic materials 0.000 description 1
- 229960001796 sunitinib Drugs 0.000 description 1
- WINHZLLDWRZWRT-ATVHPVEESA-N sunitinib Chemical compound CCN(CC)CCNC(=O)C1=C(C)NC(\C=C/2C3=CC(F)=CC=C3NC\2=O)=C1C WINHZLLDWRZWRT-ATVHPVEESA-N 0.000 description 1
- 239000006228 supernatant Substances 0.000 description 1
- 238000004441 surface measurement Methods 0.000 description 1
- 239000000375 suspending agent Substances 0.000 description 1
- 239000000725 suspension Substances 0.000 description 1
- 238000013268 sustained release Methods 0.000 description 1
- 239000012730 sustained-release form Substances 0.000 description 1
- 230000009885 systemic effect Effects 0.000 description 1
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 1
- 230000000699 topical effect Effects 0.000 description 1
- 231100000331 toxic Toxicity 0.000 description 1
- 230000002588 toxic effect Effects 0.000 description 1
- OUYCCCASQSFEME-UHFFFAOYSA-N tyrosine Natural products OC(=O)C(N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-UHFFFAOYSA-N 0.000 description 1
- 229960000241 vandetanib Drugs 0.000 description 1
- UHTHHESEBZOYNR-UHFFFAOYSA-N vandetanib Chemical compound COC1=CC(C(/N=CN2)=N/C=3C(=CC(Br)=CC=3)F)=C2C=C1OCC1CCN(C)CC1 UHTHHESEBZOYNR-UHFFFAOYSA-N 0.000 description 1
- 230000000007 visual effect Effects 0.000 description 1
- 235000012431 wafers Nutrition 0.000 description 1
- 239000000080 wetting agent Substances 0.000 description 1
- 229910052724 xenon Inorganic materials 0.000 description 1
- FHNFHKCVQCLJFQ-UHFFFAOYSA-N xenon atom Chemical compound [Xe] FHNFHKCVQCLJFQ-UHFFFAOYSA-N 0.000 description 1
- GVJHHUAWPYXKBD-IEOSBIPESA-N α-tocopherol Chemical compound OC1=C(C)C(C)=C2O[C@@](CCC[C@H](C)CCC[C@H](C)CCCC(C)C)(C)CCC2=C1C GVJHHUAWPYXKBD-IEOSBIPESA-N 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/4353—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems
- A61K31/437—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems the heterocyclic ring system containing a five-membered ring having nitrogen as a ring hetero atom, e.g. indolizine, beta-carboline
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4427—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems
- A61K31/4439—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems containing a five-membered ring with nitrogen as a ring hetero atom, e.g. omeprazole
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4427—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems
- A61K31/444—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems containing a six-membered ring with nitrogen as a ring heteroatom, e.g. amrinone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/445—Non condensed piperidines, e.g. piperocaine
- A61K31/4523—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems
- A61K31/4545—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems containing a six-membered ring with nitrogen as a ring hetero atom, e.g. pipamperone, anabasine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/506—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/517—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with carbocyclic ring systems, e.g. quinazoline, perimidine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/535—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines
- A61K31/5375—1,4-Oxazines, e.g. morpholine
- A61K31/5377—1,4-Oxazines, e.g. morpholine not condensed and containing further heterocyclic rings, e.g. timolol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/30—Macromolecular organic or inorganic compounds, e.g. inorganic polyphosphates
- A61K47/32—Macromolecular compounds obtained by reactions only involving carbon-to-carbon unsaturated bonds, e.g. carbomers, poly(meth)acrylates, or polyvinyl pyrrolidone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/30—Macromolecular organic or inorganic compounds, e.g. inorganic polyphosphates
- A61K47/36—Polysaccharides; Derivatives thereof, e.g. gums, starch, alginate, dextrin, hyaluronic acid, chitosan, inulin, agar or pectin
- A61K47/38—Cellulose; Derivatives thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/0053—Mouth and digestive tract, i.e. intraoral and peroral administration
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
- A61K9/16—Agglomerates; Granulates; Microbeadlets ; Microspheres; Pellets; Solid products obtained by spray drying, spray freeze drying, spray congealing,(multiple) emulsion solvent evaporation or extraction
- A61K9/1605—Excipients; Inactive ingredients
- A61K9/1629—Organic macromolecular compounds
- A61K9/1641—Organic macromolecular compounds obtained otherwise than by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyethylene glycol, poloxamers
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
- A61K9/16—Agglomerates; Granulates; Microbeadlets ; Microspheres; Pellets; Solid products obtained by spray drying, spray freeze drying, spray congealing,(multiple) emulsion solvent evaporation or extraction
- A61K9/1605—Excipients; Inactive ingredients
- A61K9/1629—Organic macromolecular compounds
- A61K9/1652—Polysaccharides, e.g. alginate, cellulose derivatives; Cyclodextrin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/48—Preparations in capsules, e.g. of gelatin, of chocolate
- A61K9/50—Microcapsules having a gas, liquid or semi-solid filling; Solid microparticles or pellets surrounded by a distinct coating layer, e.g. coated microspheres, coated drug crystals
- A61K9/51—Nanocapsules; Nanoparticles
- A61K9/5107—Excipients; Inactive ingredients
- A61K9/513—Organic macromolecular compounds; Dendrimers
- A61K9/5138—Organic macromolecular compounds; Dendrimers obtained by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyvinyl pyrrolidone, poly(meth)acrylates
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/48—Preparations in capsules, e.g. of gelatin, of chocolate
- A61K9/50—Microcapsules having a gas, liquid or semi-solid filling; Solid microparticles or pellets surrounded by a distinct coating layer, e.g. coated microspheres, coated drug crystals
- A61K9/51—Nanocapsules; Nanoparticles
- A61K9/5107—Excipients; Inactive ingredients
- A61K9/513—Organic macromolecular compounds; Dendrimers
- A61K9/5146—Organic macromolecular compounds; Dendrimers obtained otherwise than by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyethylene glycol, polyamines, polyanhydrides
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/48—Preparations in capsules, e.g. of gelatin, of chocolate
- A61K9/50—Microcapsules having a gas, liquid or semi-solid filling; Solid microparticles or pellets surrounded by a distinct coating layer, e.g. coated microspheres, coated drug crystals
- A61K9/51—Nanocapsules; Nanoparticles
- A61K9/5107—Excipients; Inactive ingredients
- A61K9/513—Organic macromolecular compounds; Dendrimers
- A61K9/5161—Polysaccharides, e.g. alginate, chitosan, cellulose derivatives; Cyclodextrin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/48—Preparations in capsules, e.g. of gelatin, of chocolate
- A61K9/50—Microcapsules having a gas, liquid or semi-solid filling; Solid microparticles or pellets surrounded by a distinct coating layer, e.g. coated microspheres, coated drug crystals
- A61K9/51—Nanocapsules; Nanoparticles
- A61K9/5192—Processes
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Veterinary Medicine (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Epidemiology (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Physics & Mathematics (AREA)
- Biomedical Technology (AREA)
- Nanotechnology (AREA)
- Optics & Photonics (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Organic Chemistry (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Inorganic Chemistry (AREA)
- Heart & Thoracic Surgery (AREA)
- Nutrition Science (AREA)
- Physiology (AREA)
- Cardiology (AREA)
- Oncology (AREA)
- Urology & Nephrology (AREA)
- Vascular Medicine (AREA)
- Hematology (AREA)
- Ophthalmology & Optometry (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Medicinal Preparation (AREA)
- Medicines Containing Material From Animals Or Micro-Organisms (AREA)
- Processes Of Treating Macromolecular Substances (AREA)
- Compositions Of Macromolecular Compounds (AREA)
Abstract
Procedimiento para producir partículas de dispersión sólida amorfas estables que comprende un inhibidor de la proteína cinasa seleccionado de entre dasatinib, una sal de dasatinib, un hidrato de dasatinib, un solvato de dasatinib o una combinación de los mismos y al menos un componente polimérico estabilizante y formador de matriz, dichas partículas de dispersión sólida tienen un grado de amorficidad del 100 %, el procedimiento comprende **(Ver fórmula)** a) proporcionar una primera corriente presurizada de dicho inhibidor de la proteína cinasa disuelto en un disolvente; b) proporcionar una segunda corriente presurizada de antidisolvente que comprende un fluido de CO2 supercrítico o subcrítico, donde dicho al menos un componente polimérico estabilizante y formador de matriz está presente en dicha primera o segunda corriente; y c) mezclar dicha primera y segunda corrientes, y pulverizar la corriente mixta a la salida de una boquilla, con lo que se forman dichas partículas de dispersión sólida; seguido de la recogida de dichas partículas de dispersión sólida.
Description
DESCRIPCIÓN
Un procedimiento para producir nanopartículas híbridas amorfas estables que comprende al menos un inhibidor de la proteína cinasa y al menos un componente polimérico estabilizante y formador de matriz
Campo de la invención
La presente invención se refiere al campo de los procedimientos para proporcionar componentes de composiciones farmacéuticas que comprenden fármacos poco solubles en agua. En particular, la presente invención se refiere a procedimientos para proporcionar nanopartículas híbridas de inhibidores de la proteína cinasa (IPC), para aumentar la velocidad de disolución y la biodisponibilidad resultante de dichos IPC, útiles en composiciones farmacéuticas. Antecedentes de la invención
Los componentes de las vías de transducción de señales celulares que regulan el crecimiento y la diferenciación de las células normales pueden, cuando se desregulan, llevar al desarrollo de trastornos proliferativos celulares y de cáncer. Las mutaciones en las proteínas de señalización celular pueden provocar que dichas proteínas se expresen o se activen a niveles inapropiados o en momentos inapropiados durante el ciclo celular, lo que a su vez puede llevar a un crecimiento celular descontrolado o a cambios en las propiedades de unión célula-célula.
Se ha demostrado que muchos trastornos proliferativos, tales como tumores y cánceres, están implicados en la sobreexpresión o regulación por incremento de la actividad de la proteína cinasa. Las proteínas cinasas son enzimas cinasas que modifican las proteínas añadiendo químicamente grupos fosfato (fosforilación). En general, la fosforilación da lugar a un cambio funcional de la proteína diana al cambiar la actividad enzimática, la ubicación celular o la asociación con otras proteínas. Los aminoácidos de la proteína diana pueden subdividir o caracterizar las proteínas cinasas, cuya fosforilación controlan: la mayoría de las cinasas actúan tanto sobre la serina como sobre la treonina, las tirosina cinasas actúan sobre la tirosina y un número (cinasas de especificidad doble) actúan sobre las tres. También hay proteínas cinasas que fosforilan otros aminoácidos, incluidas las histidina cinasas que fosforilan residuos de histidina. El genoma humano contiene aproximadamente 500 genes de la proteína cinasa, y las proteínas cinasas pueden modificar hasta un 30 % de todas las proteínas humanas. Se sabe que las cinasas regulan la mayoría de las vías celulares, especialmente las implicadas en la transducción de señales. La desregulación de las proteínas cinasas por mutación, reordenamiento de genes, amplificación de genes y sobreexpresión tanto del receptor como del ligando se ha implicado en el desarrollo y avance de cánceres en seres humanos. Los compuestos inhibidores de la proteína cinasa o los inhibidores de la proteína cinasa (IPC) son, por lo tanto, útiles para tratar enfermedades provocadas o agravadas por la sobreexpresión o regulación por incremento de las proteínas cinasas. Por ejemplo, se ha demostrado que los inhibidores de la tirosina cinasa (ITC también conocidos como tirfostinas) son agentes antitumorales y antileucémicos eficaces (Lowery A y col., Front Biosci. 1 de junio de 2011; 17: 1996-2007).
Un objetivo principal de la química de la formulación es mejorar la eficacia e inocuidad del fármaco, por ejemplo, mejorando la biodisponibilidad y estabilidad, así como la comodidad para el paciente. Biodisponibilidad significa la velocidad y el grado en que se absorbe una sustancia activa o terapéutica de una forma farmacéutica y se vuelve disponible en el sitio de acción. El procedimiento de administración más común y preferido debido a su comodidad, facilidad de ingestión y alto cumplimiento del tratamiento por parte del paciente es la vía oral de administración del fármaco. Sin embargo, para ciertos fármacos, la absorción del fármaco del tubo gastrointestinal está limitada por la poca solubilidad en agua y/o la poca permeabilidad de la membrana de las moléculas del fármaco.
En general, los IPC son bases débiles que se disuelven solo ligeramente a pH bajo (por ejemplo, 100-1000 mg/l) y son prácticamente insolubles a pH neutro (por ejemplo, 0,1-10 mg/l). Por lo tanto, es importante mejorar la solubilidad y la velocidad de disolución de los fármacos basados en IPC para mejorar la biodisponibilidad y la eficacia de la mayoría de estos fármacos. Los IPC típicos exhiben una estructura no polipeptídica y tienen pesos moleculares relativamente bajos, tales como <10000 dalton o <5000 dalton.
Se han documentado diversos procedimientos para mejorar las características de disolución de fármacos poco solubles en agua, que incluyen micronización, formación de sales o solvatos, complejos y microesferas. De forma adicional, se han hecho intentos para mejorar la biodisponibilidad proporcionada por las formas galénicas sólidas formando partículas que comprenden el fármaco o mezclando el fármaco poco soluble en agua con excipientes hidrófilos. Sin embargo, tradicionalmente, estos procedimientos conllevan limitaciones inherentes con respecto a la estabilidad física de las partículas durante el almacenamiento, problemas de trituración o dificultad de eliminación del disolvente frecuentemente tóxico. Además, es importante que el fármaco liberado de la fase sólida no se precipite en el tubo gastrointestinal, o que se precipite lo menos posible, sino que permanezca soluble en agua en los fluidos acuosos del tubo gastrointestinal, ya que dicha precipitación da lugar a una baja biodisponibilidad (véase, por ejemplo, Herve J. y col., Pharm Dev Technol., junio de 2011; 16(3):278-86).
La solubilidad dependiente del pH es un problema bien conocido para muchas formulaciones orales de sustancias poco solubles en agua, tales como los IPC, ya que la mayor parte de la absorción del fármaco se produce en el intestino delgado y grueso, donde el pH se encuentra cerca del neutro. Por tanto, existe una necesidad continua de
desarrollar y mejorar las características de disolución de las formas galénicas sólidas orales de fármacos basados en IPC. (Budha n R , Frymoyer A, Smelick GS, Jin JY, Yago MR, Dresser MJ, Holden SN, Benet LZ, Ware JA. Clin Pharmacol Ther. agosto de 2012;92(2):203-13). Por lo tanto, son muy deseables procedimientos para mejorar la disolución de fármacos basados en IPC, así como de otros fármacos poco solubles en agua, a pH neutro (intestinal). El documento US20090203709 describe una forma galénica farmacéutica que comprende un producto de dispersión sólida de al menos un inhibidor de la tirosina cinasa, al menos un polímero farmacéuticamente aceptable y al menos un solubilizante farmacéuticamente aceptable. Además, la referencia describe procedimientos para preparar la forma galénica farmacéutica mencionada anteriormente, que comprenden preparar el comprimido bucodispersable homogéneo de al menos un inhibidor de la tirosina cinasa, al menos un polímero farmacéuticamente aceptable y al menos un solubilizante farmacéuticamente aceptable, y permitir que el comprimido bucodispersable solidifique para obtener un producto de dispersión sólida.
El documento EP2105130 describe formulaciones farmacéuticas que comprenden una dispersión sólida o una solución sólida, que contienen un polímero y un agente activo en forma amorfa. Además, la formulación comprende un polímero externo para estabilizar la solución, de modo que el % en peso del polímero externo sea inferior a un 20 % del peso total de la formulación farmacéutica. De forma adicional, la referencia describe un procedimiento de extrusión por fusión en caliente para la producción de la formulación mencionada anteriormente. Otras técnicas anteriores relevantes incluyen los documentos WO 2008/008733 A2, WO 2008/055966 A1, WO 2011/159218 A1, WO 2007/133750 A2, WO 2009/072953 A1 y ARASALI SULAIMAN ZARENA Y COL.,: "Design of submicron and nanoparticle delivery systems using supercritical carbon dioxide-mediated processes: an overview (Diseño de sistemas de administración submicrónicos y de nanopartículas usando procedimientos supercríticos mediados por dióxido de carbono: una descripción general", THERAPEUTIC DELIVERY, vol. 2, n.° 2, 1 de febrero de 2011.
Resumen de la invención
La presente invención se refiere a procedimientos para producir nanopartículas híbridas amorfas estables, que comprenden dasatinib como inhibidor de la proteína cinasa y al menos un componente polimérico estabilizante y formador de matriz. Opcionalmente, se pueden añadir uno o más solubilizantes a las partículas, presentes por separado con respecto a las partículas o dentro de las partículas.
Breve descripción de los dibujos
La figura 1 proporciona un gráfico que muestra la solubilidad aparente de nanopartículas híbridas amorfas estables que comprenden nilotinib HCl producidas por los procedimientos de la invención. En el ejemplo 1, se encuentra experimentación adicional tanto con base de nilotinib como con nilotinib HCl. Los detalles de las partículas se describen en el ejemplo 1, tabla 1, para los experimentos 3, 30 y 37, respectivamente. En pocas palabras, el experimento 30 representa nanopartículas híbridas que comprenden nilotinib HCl y HPMCP HP55 y donde el copolímero solubilizante de polivinil caprolactama-acetato de polivinilo-polietilenglicol está presente por separado con respecto a las nanopartículas híbridas. El experimento 3 representa nilotinib HCl cristalino crudo y el experimento 37 representa nanopartículas híbridas de nilotinib HCl, HpMCP HP55 y el copolímero solubilizante de polivinil caprolactama-acetato de polivinilo-polietilenglicol, presente dentro de las nanopartículas híbridas. Los experimentos ilustrados en los gráficos se llevaron a cabo a pH 6,5, en FaSSIF.
La figura 2 proporciona un gráfico que muestra la solubilidad aparente de nanopartículas híbridas amorfas estables que comprenden erlotinib producidas por los procedimientos de la invención. En el ejemplo 2, se encuentra experimentación adicional con erlotinib. Los detalles de las nanopartículas híbridas se describen en el ejemplo 2, tabla 7, para los experimentos 58, 65 y 67, respectivamente. En pocas palabras, el experimento 65 representa nanopartículas híbridas con erlotinib HCl y HPMCP-AS, donde el copolímero solubilizante de polivinil caprolactamaacetato de polivinilo-polietilenglicol está presente por separado con respecto a las nanopartículas híbridas. El experimento 58 representa erlotinib HCl cristalino crudo y el experimento 67 representa nanopartículas híbridas de erlotinib HCl, HPMCP-AS y el copolímero solubilizante de polivinil caprolactama-acetato de polivinilo-polietilenglicol, presente dentro de las nanopartículas híbridas. Los experimentos ilustrados en los gráficos se llevaron a cabo a pH 6,5 en FaSSIF.
La figura 3 proporciona un gráfico que muestra la solubilidad aparente de nanopartículas híbridas amorfas estables que comprenden pazopanib producidas por los procedimientos de la invención. En el ejemplo 3, se encuentra experimentación adicional con pazopanib. Los detalles de las nanopartículas híbridas se describen en el ejemplo 3, tabla 13, para los experimentos 84, 91 y 93, respectivamente. En pocas palabras, el experimento 91 representa nanopartículas híbridas que comprenden pazopanib y PVP 90K y donde el copolímero solubilizante de polivinil caprolactama-acetato de polivinilo-polietilenglicol está presente por separado con respecto a las nanopartículas híbridas, el experimento 93 representa nanopartículas híbridas que comprenden pazopanib, PVP 90K y el copolímero solubilizante de polivinil caprolactama-acetato de polivinilo-polietilenglicol, presente dentro de las nanopartículas híbridas. El experimento 84 representa pazopanib cristalino crudo. Los experimentos ilustrados en los gráficos se llevaron a cabo a pH 6,5, en FaSSIF.
La figura 4 proporciona un gráfico que muestra la solubilidad aparente de nanopartículas híbridas amorfas estables que comprenden base de lapatinib producidas por los procedimientos de la invención. En el ejemplo 4, se encuentra experimentación adicional tanto con base de lapatinib como con sal de lapatinib ditosilato. Los detalles de las nanopartículas híbridas se describen en el ejemplo 4, tabla 19, para los experimentos 110, 122 y 126, respectivamente. En pocas palabras, el experimento 122 representa nanopartículas híbridas que comprenden base de lapatinib y HPC EF, donde el copolímero solubilizante de polivinil caprolactama-acetato de polivinilo-polietilenglicol está presente por separado con respecto a las nanopartículas híbridas. El experimento 110 representa base de lapatinib cruda y el experimento 126 representa nanopartículas híbridas de base de lapatinib, HPC LF y el copolímero solubilizante de polivinil caprolactama-acetato de polivinilo-polietilenglicol, presente dentro de las nanopartículas híbridas. Los experimentos ilustrados en los gráficos se llevaron a cabo a pH 6,5 en FaSSIF.
La figura 5 proporciona un gráfico que muestra la solubilidad aparente de nanopartículas híbridas amorfas estables que comprenden nilotinib producidas por los procedimientos de la invención. Los detalles de las nanopartículas híbridas se describen en el ejemplo 5, tabla 21, para los experimentos 127, 128 y 129, respectivamente. En pocas palabras, el experimento 129 representa una mezcla física de nilotinib HCl cristalino crudo, HPMCP HP55 y el copolímero solubilizante de polivinil caprolactama-acetato de polivinilo-polietilenglicol. El experimento 128 representa nanopartículas híbridas que comprenden nilotinib HCl y HPMCP HP55, donde el copolímero solubilizante de polivinil caprolactama-acetato de polivinilo-polietilenglicol está presente por separado con respecto a las nanopartículas híbridas. El experimento 127 representa nanopartículas híbridas de nilotinib HCl y HPMCP HP55. Los experimentos ilustrados en los gráficos se llevaron a cabo a pH 1,4 en SGF.
La figura 6 proporciona un gráfico que muestra la solubilidad aparente de nanopartículas híbridas amorfas estables que comprenden gefitinib producidas por los procedimientos de la invención. En el ejemplo 6, se encuentra experimentación adicional con gefitinib. Los detalles de las composiciones se describen en el ejemplo 6, tabla 22, para los experimentos 131, 133, 135 y 137, respectivamente. En pocas palabras, el experimento 131 representa gefitinib cristalino crudo. El experimento 133 representa una mezcla de gefitinib cristalino crudo, HPMCP HP55 y el copolímero solubilizante de polivinil caprolactama-acetato de polivinilo-polietilenglicol. El experimento 135 representa nanopartículas híbridas de gefitinib y HPMCP HP55. El experimento 137 representa nanopartículas híbridas de gefitinib y HPMCP HP55 donde el copolímero solubilizante de polivinil caprolactama-acetato de polivinilopolietilenglicol está presente por separado con respecto a las nanopartículas híbridas. Los experimentos ilustrados en los gráficos se llevaron a cabo a pH 6,5 en FaSSIF.
La figura 7 proporciona un gráfico que muestra la solubilidad aparente de nanopartículas híbridas amorfas estables que comprenden dasatinib producidas por los procedimientos de la invención. Los detalles de las nanopartículas híbridas se describen en el ejemplo 7, tabla 24, para los experimentos 138-141. En pocas palabras, el experimento 138 representa dasatinib cristalino crudo. El experimento 139 representa una mezcla de dasatinib cristalino crudo, Kollidon VA64 y el copolímero solubilizante de polivinil caprolactama-acetato de polivinilo-polietilenglicol. El experimento 140 representa nanopartículas híbridas de dasatinib y Kollidon VA64. El experimento 141 representa nanopartículas híbridas de dasatinib y Kollidon VA64 donde el copolímero solubilizante de polivinil caprolactamaacetato de polivinilo-polietilenglicol está presente por separado con respecto a las nanopartículas híbridas. Los experimentos ilustrados en los gráficos se llevaron a cabo a pH 6,5 en FaSSIF.
La figura 8 proporciona un gráfico que muestra la solubilidad aparente de nanopartículas híbridas amorfas estables que comprenden sorafenib producidas por los procedimientos de la invención. Los detalles de las nanopartículas híbridas se describen en el ejemplo 8, tabla 26, para los experimentos 142-145. En pocas palabras, el experimento 142 representa sorafenib tosilato cristalino crudo. El experimento 143 representa una mezcla de sorafenib tosilato cristalino crudo, HPMCP HP55 y el copolímero solubilizante de polivinil caprolactama-acetato de polivinilopolietilenglicol. El experimento 144 representa nanopartículas híbridas de sorafenib tosilato y HPMCP HP55. El experimento 145 representa nanopartículas híbridas de sorafenib tosilato y HPMCP HP55 donde el copolímero solubilizante de polivinil caprolactama-acetato de polivinilo-polietilenglicol está presente por separado con respecto a las nanopartículas híbridas. Los experimentos ilustrados en los gráficos se llevaron a cabo a pH 6,5 en FaSSIF.
La figura 9 proporciona un gráfico que muestra la solubilidad aparente de nanopartículas híbridas amorfas estables que comprenden crizotinib producidas por los procedimientos de la invención. En el ejemplo 10, se encuentra más experimentación con crizotinib. Los detalles de las composiciones se describen en el ejemplo 10, tabla 30, para los experimentos 150, 152, 153 y 156, respectivamente. En pocas palabras, el experimento 150 representa crizotinib cristalino crudo. El experimento 152 representa una mezcla de crizotinib cristalino crudo, PVP 30K y el solubilizante Cremophor RH40. El experimento 153 representa nanopartículas híbridas de crizotinib y PVP 30K. El experimento 156 representa nanopartículas híbridas de crizotinib y PVP 30K donde el solubilizante Cremophor RH40 está presente por separado con respecto a las nanopartículas híbridas. Los experimentos ilustrados en los gráficos se llevaron a cabo a pH 6,5 en FaSSIF.
La figura 10 proporciona un gráfico que muestra la solubilidad aparente de nanopartículas híbridas amorfas estables que comprenden axitinib producidas por los procedimientos de la invención. En el ejemplo 11, se encuentra más experimentación con axitinib. Los detalles de las composiciones se describen en el ejemplo 11, tabla 32, para los experimentos 157, 158, 160 y 162, respectivamente. En pocas palabras, el experimento 157 representa axitinib
cristalino crudo. El experimento 158 representa una mezcla de axitinib cristalino crudo, Kollidon VA64 y el copolímero solubilizante de polivinil caprolactama-acetato de polivinilo-polietilenglicol. El experimento 160 representa nanopartículas híbridas de axitinib y Kollidon VA64. El experimento 162 representa nanopartículas híbridas de axitinib y Kollidon VA64 donde el copolímero solubilizante de polivinil caprolactama-acetato de polivinilo-polietilenglicol está presente por separado con respecto a las nanopartículas híbridas. Los experimentos ilustrados en los gráficos se llevaron a cabo a pH 6,5 en FaSSIF.
La figura 11 proporciona un gráfico que muestra la solubilidad aparente de nanopartículas híbridas amorfas estables que comprenden vemurafenib producidas por los procedimientos de la invención. En el ejemplo 12, se encuentra más experimentación con vemurafenib. Los detalles de las composiciones se describen en el ejemplo 12, tabla 34, para los experimentos 164, 166, 168 y 170, respectivamente. En pocas palabras, el experimento 164 representa vemurafenib cristalino crudo. El experimento 166 representa una mezcla de vemurafenib cristalino crudo, CAP y el copolímero solubilizante de polivinil caprolactama-acetato de polivinilo-polietilenglicol. El experimento 168 representa nanopartículas híbridas de vemurafenib y CAP. El experimento 170 representa nanopartículas híbridas de vemurafenib y CAP donde el copolímero solubilizante de polivinil caprolactama-acetato de polivinilo-polietilenglicol está presente por separado con respecto a las nanopartículas híbridas. Los experimentos ilustrados en los gráficos se llevaron a cabo a pH 6,5 en FaSSIF.
La figura 12 proporciona un gráfico que muestra la velocidad de disolución de nanopartículas híbridas amorfas estables que comprenden base de nilotinib producidas por los procedimientos de la invención, medidas en estado de baja concentración. Los detalles se encuentran en los ejemplos 13 y 13.1, y en la tabla 36 para los experimentos 500 y 501. En pocas palabras, el experimento 500 representa nilotinib HCl crudo. El experimento 501 representa nanopartículas híbridas de base de nilotinib y HPMCp HP55. Los experimentos ilustrados en los gráficos se llevaron a cabo a pH 6,5 en FaSSIF.
La figura 13 proporciona un gráfico que muestra la velocidad de disolución de nanopartículas híbridas amorfas estables que comprenden erlotinib HCl producidas por los procedimientos de la invención, medidas en estado de baja concentración. Los detalles se encuentran en los ejemplos 13 y 13.2, tabla 37 para los experimentos 510 y 511. En pocas palabras, el experimento 510 representa erlotinib HCl crudo. El experimento 511 representa nanopartículas híbridas de erlotinib HCl y HPMC AS.
La figura 14 proporciona un gráfico que muestra la velocidad de disolución de nanopartículas híbridas amorfas estables que comprenden pazopanib HCl producidas por los procedimientos de la invención, medidas en estado de baja concentración. Los detalles se encuentran en los ejemplos 13 y 13.3, tabla 38 para los experimentos 520 y 521. En pocas palabras, el experimento 520 representa pazopanib HCl crudo El experimento 521 representa nanopartículas híbridas de pazopanib HCl y PVP90K.
La figura 15 proporciona un gráfico que muestra la velocidad de disolución de nanopartículas híbridas amorfas estables que comprenden base de lapatinib producidas por los procedimientos de la invención, medidas en estado de baja concentración. Los detalles se encuentran en los ejemplos 13 y 13.4, tabla 39 para los experimentos 530 y 531. En pocas palabras, el experimento 530 representa lapatinib ditosilato crudo. El experimento 531 representa nanopartículas híbridas de base de lapatinib y HPMCP If.
La figura 16 proporciona un gráfico que muestra la velocidad de disolución de nanopartículas híbridas amorfas estables que comprenden gefitinib producidas por los procedimientos de la invención, medidas en estado de baja concentración. Los detalles se encuentran en los ejemplos 13 y 13.5, tabla 40 para los experimentos 540 y 541. En pocas palabras, el experimento 540 representa gefitinib crudo. El experimento 541 representa nanopartículas híbridas de gefitinib y HPMCP HP55.
La figura 17 proporciona un gráfico que muestra la velocidad de disolución de nanopartículas híbridas amorfas estables que comprenden dasatinib producidas por los procedimientos de la invención, medidas en estado de baja concentración. Los detalles se encuentran en los ejemplos 13 y 13.6, tabla 41 para los experimentos 550 y 551. En pocas palabras, el experimento 550 representa dasatinib crudo. El experimento 551 representa nanopartículas híbridas de dasatinib y Kollidon VA64.
La figura 18 proporciona un gráfico que muestra la velocidad de disolución de nanopartículas híbridas amorfas estables que comprenden sorafenib tosilato producidas por los procedimientos de la invención, medidas en estado de baja concentración. Los detalles se encuentran en los ejemplos 13 y 13.7, tabla 42 para los experimentos 560 y 561. En pocas palabras, el experimento 560 representa sorafenib tosilato crudo. El experimento 561 representa nanopartículas híbridas de sorafenib tosilato y HPMCP HP55.
La figura 19 proporciona un gráfico que muestra la velocidad de disolución de nanopartículas híbridas amorfas estables que comprenden crizotinib producidas por los procedimientos de la invención, medidas en estado de baja concentración. Los detalles se encuentran en los ejemplos 13 y 13.8, tabla 43 para los experimentos 570 y 571. En pocas palabras, el experimento 570 representa crizotinib crudo. El experimento 571 representa nanopartículas híbridas de crizotinib y PVP 30K.
La figura 20 proporciona un gráfico que muestra la velocidad de disolución de nanopartículas híbridas amorfas estables que comprenden axitinib producidas por los procedimientos de la invención, medidas en estado de baja concentración. Los detalles se encuentran en los ejemplos 13 y 13.9, tabla 44 para los experimentos 580, 581 y 582. En pocas palabras, el experimento 580 representa axitinib crudo. El experimento 581 representa nanopartículas híbridas de axitinib y Kollidon VA64 y el experimento 582 representa nanopartículas híbridas de axitinib y HPMC AS.
La figura 21 proporciona un gráfico que muestra la velocidad de disolución de nanopartículas híbridas amorfas estables que comprenden vemurafenib producidas por los procedimientos de la invención, medidas en estado de baja concentración. Los detalles se encuentran en los ejemplos 13 y 13.10, tabla 45 para los experimentos 590, 591 y 592. En pocas palabras, el experimento 590 representa vemurafenib crudo. El experimento 591 representa nanopartículas híbridas de vemurafenib y Kollidon VA64 y el experimento 592 representa nanopartículas híbridas de vemurafenib y CAP.
La figura 22 proporciona gráficos que muestran la medición in vivo de las concentraciones plasmáticas después de la administración oral a perros beagle de composiciones, representadas por formulaciones que comprenden nanopartículas híbridas amorfas estables de base de nilotinib y los componentes poliméricos estabilizantes y formadores de matriz PVAP y HPMCP HP55, respectivamente (I/P), denominadas PVAP y HP55, así como donde se añadió el copolímero solubilizante de polivinil caprolactama-acetato de polivinilo-polietilenglicol (I/P+S), denominadas HP55 y PVAP, respectivamente. Los experimentos se llevaron a cabo en perros beagle pretratados para tuvieran un contenido estomacal neutro. Las nanopartículas híbridas se describen además en los experimentos 146 y 147 (ejemplo 9) y los detalles de los experimentos in vivo se exponen en el ejemplo 14. Los experimentos usaron una formulación comercializada que comprende nilotinib HCl ("Tasigna") como referencia.
La figura 23 proporciona gráficos que muestran la medición in vivo de las concentraciones plasmáticas después de la administración oral a perros beagle de composiciones, representadas por formulaciones que comprenden nanopartículas híbridas amorfas estables de base de nilotinib y los componentes poliméricos estabilizantes y formadores de matriz PVAP y HPMCP HP55, respectivamente (I/P), denominadas PVAP y HP55, producidas por los procedimientos de la invención, así como donde se añadió el copolímero solubilizante de polivinil caprolactamaacetato de polivinilo-polietilenglicol (I/P+S), denominadas PVAP y HP55, respectivamente. Los experimentos se llevaron a cabo en perros beagle pretratados para tuvieran un contenido estomacal ácido. Las nanopartículas híbridas se describen además en los experimentos 146 y 147 (ejemplo 9) y los detalles de los experimentos in vivo se exponen en el ejemplo 14. Los experimentos usaron una formulación comercializada que comprende nilotinib HCl ("Tasigna") como referencia.
La figura 24 proporciona gráficos que muestran la medición in vivo de las concentraciones plasmáticas después de la administración oral a perros beagle de composiciones, representadas por formulaciones que comprenden nanopartículas híbridas amorfas estables de base de nilotinib y los componentes poliméricos estabilizantes y formadores de matriz PVAP y HPMCP HP55, respectivamente (I/P), denominadas PVAP y HP55, producidas por los procedimientos de la invención, así como donde, después de la formación de las nanopartículas híbridas, se añadió el copolímero solubilizante de polivinil caprolactama-acetato de polivinilo-polietilenglicol (I/P+S), denominadas HP55 y PVAP, respectivamente. Los experimentos se llevaron a cabo en perros beagle pretratados para tuvieran un contenido estomacal ácido o neutro. Las nanopartículas híbridas se describen además en los experimentos 146 y 147 (ejemplo 9) y los detalles de los experimentos in vivo se exponen en el ejemplo 14.
La figura 25 proporciona gráficos que muestran la medición in vivo de las concentraciones plasmáticas después de la administración oral a perros beagle de composiciones, representadas por formulaciones que comprenden nanopartículas híbridas amorfas estables de base de nilotinib y los componentes poliméricos estabilizantes y formadores de matriz PVAP y HPMCP HP55, respectivamente (I/P), producidas por los procedimientos de la invención, denominadas PVAP y HP55. Los experimentos se llevaron a cabo en perros beagle pretratados para tuvieran un contenido estomacal ácido o neutro. Las nanopartículas híbridas se describen además en los experimentos 146 y 147 (ejemplo 9) y los detalles de los experimentos in vivo se exponen en el ejemplo 14.
La figura 26 proporciona un gráfico que muestra la solubilidad aparente para nanopartículas híbridas amorfas estables producidas por los procedimientos de la invención, antes y después de 11 meses de almacenamiento a temperatura ambiente. El experimento proporciona nanopartículas híbridas amorfas estables que comprenden base de nilotinib, HPMCP HP55 y la adición del copolímero solubilizante de polivinil caprolactama-acetato de polivinilo-polietilenglicol (I/P+S) como Exp 171 y Exp 172 con más detalles expuestos en el ejemplo 15.
La figura 27 proporciona patrones de difracción de rayos X en polvo (DRXP) superpuestos de nanopartículas híbridas estables al 40 % de carga de fármaco, I/P base de nilotinib/HPMCP HP55. Almacenamiento inicial (parte superior) y después de 12 meses a temperatura ambiente (parte inferior). Los patrones de DRXP están compensados para mejorar la comparación visual. En el ejemplo 15, se exponen más detalles.
Descripción detallada de la invención
Todas las patentes, solicitudes de patente y publicaciones mencionadas en esta solicitud se incorporan a esta solicitud en su totalidad a modo de referencia.
Como se usa aquí, la frase "nanopartículas híbridas" se refiere a un grupo de partículas, típicamente en el intervalo de tamaño promedio de 1 a 1000 nm, compuesto por al menos dos componentes, uno de los cuales es el IPC y el otro un componente polimérico estabilizante y formador de matriz. Las partículas pueden ser cristalinas o amorfas, o una mezcla de las mismas. Típicamente, en el sentido de la presente descripción, las partículas son "amorfas" o "esencialmente amorfas". Esto significa que casi todo, si no todo, el contenido de las partículas comprende un inhibidor de la proteína cinasa y un componente polimérico estabilizante y formador de matriz amorfos. El nivel o grado de amorficidad es al menos un 60 %, tal como un 70 %, tal como un 80 % o un 85 %, preferiblemente, al menos un 90 % y, más preferiblemente, >95 %, donde el 100 % representa que todo el material es amorfo en las partículas. La cuantificación de IPC cristalino o la ausencia de IPC cristalino se puede medir mediante procedimientos de difracción de rayos X en polvo como se describe en Saleki-Gerhardt A y col., Int J Pharm. 1994; 101:237-247) o por sorción de vapor de agua como se describe en Dash AK y col., J Pharm Sci. abril de 2002; 91(4):983-90.
La expresión "partículas de dispersión sólida" se refiere a "nanopartículas híbridas" como se definió anteriormente, sin embargo, las partículas de dispersión sólida son típicamente más grandes o de tamaño mucho mayor (típicamente |jm-mm, como se describe en Wu K. y col., J Pharm Sci., julio de 2009; 98(7):2422-3). El tamaño más pequeño de las nanopartículas híbridas contribuye a estabilizar además el IPC contra la cristalización. Típicamente, las nanopartículas híbridas se encuentran en el intervalo de tamaño promedio de 1 a 1000 nm, tal como por debajo de 500 nm, preferiblemente, por debajo de 250 nm.
El término "estable" se refiere al nivel de estabilidad de las partículas producidas por los procedimientos de la presente invención y se puede medir como la capacidad de las nanopartículas híbridas de permanecer en su estado físico durante 6-12 meses de almacenamiento a temperatura ambiente (por ejemplo, 20-25 °C). El nivel de estabilidad se puede medir mediante mediciones del ABC de la velocidad de disolución durante, por ejemplo, 80 minutos de las partículas, después de dicho almacenamiento.
La frase "inhibidor de la proteína cinasa" o "IPC" se refiere a un tipo de inhibidor enzimático que bloquea específicamente la acción de una o más proteínas cinasas. Los IPC incluyen, pero no se limitan a, inhibidores de la proteína cinasa e inhibidores de la tirosina cinasa, tales como axitinib, afatinib, bosutinib, crizotinib, cediranib, dasatinib, erlotinib, fostamatinib, gefitinib, imatinib, lapatinib, lenvatinib, lestaurtinib, motesanib, mubritinib, nilotinib, pazopanib, pegaptanib, ruxolitinib, sorafenib, semaxanib, sunitinib, tandunitib, tipifamib, vandetanib y vemurafenib; o sales o hidratos o solvatos de los mismos, o combinaciones de los mismos.
Por la frase "componente polimérico estabilizante y formador de matriz" se entiende el componente presente en las nanopartículas híbridas junto con el IPC. Típicamente, dicho componente polimérico estabilizante y formador de matriz exhibe una estructura polimérica, tal como, pero sin limitarse a, metilcelulosa, hidroxietilcelulosa, hidroxipropilcelulosa (por ejemplo, HPC ef, HPC If y HPC jf), hidroxipropilmetilcelulosa (por ejemplo, Methocel E3 y E15 y Pharmacoat), succinato de acetato de hidroxipropilmetilcelulosa (HPMC AS), ftalato de hidroxipropilmetilcelulosa (por ejemplo, HPMCP-HP55), polivinilpirrolidona (por ejemplo, PVP 30K y PVP 90K), ftalato de acetato de polivinilo ( P v A p ), copolividona (por ejemplo, Kollidon VA 64), crospovidona (por ejemplo, Kollidon CL), copolímero de ácido metacrílico y acrilato de etilo (por ejemplo, Kollicoat ME), copolímero de ácido metacrilato y metacrilato de metilo (por ejemplo, Eudragit L100), polietilenglicol (PEG), copolímero de DL lactida/glicólido, poli DL-lactida, ftalato de acetato de celulosa (CAP), copolímeros de aminoalquil metacrilato (por ejemplo, Eudragit RL100, RL PO o RS PO), homopolímero de carbómero tipo A (por ejemplo, Carbopol 971 P), homopolímero de carbómero tipo B (por ejemplo, Carbopol 974P) y poloxámeros (por ejemplo, Pluronics, Kolliphor).
El término "polímero" o "polimérico" se usa aquí para significar un compuesto que está hecho con monómeros conectados entre sí para formar una molécula más grande. En general, un polímero consiste en 20 o más monómeros conectados entre sí, sin embargo, aquí también se hace referencia a menos de 20 monómeros conectados entre sí como polímeros.
El término "solubilizante" se usa aquí para significar un compuesto que aumenta la solubilidad de una sustancia, tal como, pero sin limitarse a, copolímero de polivinil caprolactama-acetato de polivinilo-polietilenglicol (Soluplus), succinato de d-a-tocoferol polietilenglicol 1000 (TPGS), aceite de ricino hidrogenado PEG-40 (Cremophor RH40), aceite de ricino PEG-35 (Cremophor EL), estearato PEG-40 (MYRJ 540), grasa dura (por ejemplo, Gelucire 33/01), polioxilglicéridos (por ejemplo, Gelucire 44/14), polioxilglicéridos de estearoilo (por ejemplo, Gelucire 50/13), glicéridos caprílicos/cápricos PEG-8 (por ejemplo, Labrasol) y poloxámeros (por ejemplo, Pluronics, Kolliphor).
Como se usa en esta solicitud, la frase "partículas primarias" se refiere a las entidades de partículas más pequeñas formadas durante el proceso de precipitación. Los límites de las partículas se analizan mediante microscopía MEB. En función de los parámetros del procedimiento, las partículas primarias pueden formar juntas una red más o menos densa y porosa que forma partículas más grandes, aglomeradas o puente. Los parámetros que afectan la aglomeración son, por ejemplo, la temperatura que puede modificar la plasticidad de las partículas primarias; la relación disolvente/antidisolvente que afecta el tiempo de precipitación, la concentración de la solución del IPC; y la
naturaleza del agente o agentes poliméricos estabilizantes y formadores de matriz. El tamaño promedio de las partículas primarias se encuentra típicamente entre 1 y 1000 nm, preferiblemente, por debajo de 500 nm, más preferiblemente, por debajo de 250 nm.
Como se usa en esta solicitud, las frases "supercrítico" y "fluido supercrítico" se refieren a una sustancia química que se fija a una temperatura superior o igual a su temperatura crítica (Tc) y a una presión superior o igual a su presión crítica (Pc).
Como se usa en esta solicitud, las frases "subcrítico" y "fluido subcrítico" se refieren aquí a que una temperatura crítica (Tc) o presión crítica (Pc) se fija a una temperatura o presión superior a su temperatura crítica (Tc) o presión crítica (Pc), respectivamente, y la otra de temperatura crítica (Tc) o presión crítica (Pc) se fija a una temperatura o presión inferior a su temperatura crítica (Tc) o presión crítica (Pc), respectivamente.
La frase "área bajo la curva (ABC)" se refiere al área bajo la curva de concentración-tiempo, donde el eje x representa el tiempo y el eje y representa la concentración de fármaco solubilizado.
La frase "solubilidad aparente" se refiere a la concentración de material en el equilibrio aparente. Véase más información en la sección de ejemplos.
El término "sobresaturación" se usa aquí para significar que una solución contiene más sustancia disuelta de la que podría disolver el disolvente o el medio en circunstancias normales.
Como se usa en esta solicitud, el término "Soluplus" o "soluplus" se refiere a un copolímero de polivinil caprolactamaacetato de polivinilo-polietilenglicol.
Como se usa en esta solicitud, el término "TPGS" se refiere a succinato de d-a-tocoferol polietilenglicol 1000.
Como se usa en esta solicitud, la expresión "Chremophor RH40" se refiere a aceite de ricino hidrogenado PEG-40. Como se usa en esta solicitud, el término "PVAP" se refiere a ftalato de acetato de polivinilo.
Como se usa en esta solicitud, el término "PVP 90K" se refiere a polivinilpirrolidona K-90.
Como se usa en esta solicitud, el término "PVP 30K" se refiere a polivinilpirrolidona K-30.
Como se usa en esta solicitud, el término "HPMC-AS" se refiere a succinato de acetato de hidroxipropilmetilcelulosa. Como se usa en esta solicitud, el término "HPMC HP55" se refiere a ftalato de hidroxipropilmetilcelulosa.
Como se usa en esta solicitud, el término "HPC" se refiere a hidroxipropilcelulosa, tal como HPC EF y HPC LF. Como se usa en esta solicitud, el término "Kollidon VA64" se refiere a copolividona.
Como se usa en esta solicitud, el término "CAP" se refiere a ftalato de acetato de celulosa.
Los medios de disolución usados con el propósito de someter a prueba las nanopartículas híbridas producidas por los procedimientos de la presente invención incluyen fluido intestinal estimulado en estado de ayuno, denominado FaSSIF, fluido intestinal estimulado en estado alimentado, denominado FeSSIF y fluido gástrico simulado, denominado SGF. El medio FaSSIF está diseñado para representar un estado de ayuno y tiene un pH de aproximadamente 6,5, así como propiedades osmóticas particulares. El medio FeSSIF está diseñado para representar un estado de alimentado y tiene un pH de aproximadamente 5, así como propiedades osmóticas específicas. El medio SGF está diseñado para representar fluido gástrico y tiene un pH de aproximadamente 1,4, así como propiedades osmóticas particulares. Los medios FaSSIF, FeSSIF y SGF se utilizan en general en modelos in vitro para la disolución de fármacos poco solubles en agua. La elección del medio dependerá del lugar del tubo intestinal y en qué condiciones (en ayunas o alimentado) se desea que se disuelvan y absorban las partículas. Se describen más detalles con respecto a estos fluidos en, por ejemplo, Hervé J. y col, Pharm Dev Technol. junio de 2011; 16(3):278-86 y Jantratid, E. y Dressman, J. Dissolut. Technol. 20098, 21-25.
La frase "forma amorfa" significa una forma sólida no cristalina. La facilidad de disolución se puede atribuir al menos en parte a la cantidad de energía requerida para la disolución de los componentes de una fase sólida cristalina o amorfa. Las partículas amorfas requieren menos energía para la disolución en comparación con las partículas cristalinas del mismo compuesto.
Los procedimientos inventivos producen nanopartículas híbridas que comprenden un IPC o una combinación de dos o más IPC. Sin embargo, las partículas pueden comprender una combinación de uno o más IPC y al menos un principio activo adicional, tal como uno o más fármacos. Se pueden utilizar eficazmente varios tipos de IPC.
El término IPC (inhibidores de la proteína cinasa), como se usa en esta solicitud, pretende incluir también los hidratos, solvatos (alcoholatos), sales ácidas, sales básicas o cocristales farmacéuticamente aceptables de dichos compuestos inhibidores de la proteína cinasa.
Como se usa en esta solicitud, la expresión compuestos insolubles en agua o poco solubles en agua (o hidrófobos) se refiere a compuestos cuya solubilidad en agua a 25 °C es menor que 1 g/100 ml, especialmente menor que 0,1 g/100 ml en agua pura a pH neutro.
Las nanopartículas híbridas generadas por los procedimientos de la presente invención se encuentran típicamente en forma de partículas como se describe en otra parte de esta memoria descriptiva. Hay un número de procedimientos diferentes para la formación de partículas más grandes, por ejemplo, granulación, extrusión por fusión, secado por pulverización, precipitación, etc., todos los cuales abarcan en general comenzar con la formación de una mezcla entre el principio activo (PA) y el componente polimérico estabilizante y formador de matriz. Los procedimientos inventivos de la presente invención son procedimientos continuos para generar nanopartículas híbridas. En este contexto, los procedimientos continuos significan que la formación de partículas está en curso de forma continua mientras que al mismo tiempo se extraen/recogen/retienen de forma continua nanopartículas híbridas de la mezcla después de su formación. En los procedimientos preferidos, es decir, procedimientos de precipitación, esto significa que un fluido que es una solución del IPC, preferiblemente en forma de corriente de fluido, se mezcla con un fluido de antidisolvente, preferiblemente en forma de corriente de fluido de antidisolvente. El componente polimérico estabilizante y formador de matriz puede estar presente en uno o en ambos de los dos fluidos en función de sus características de solubilidad. La mezcla de los dos fluidos tiene lugar en una función de mezcla, por ejemplo, una cámara de mezcla. En el caso de que el procedimiento sea continuo, es decir, los dos fluidos son corrientes de fluido, la función de mezcla se asocia típicamente con una función de formación y separación de partículas donde la corriente de fluidos mixta puede pasar mientras retiene las nanopartículas híbridas. Los agentes que modifican las características de las partículas sin incorporarse a las partículas se pueden añadir a uno o ambos fluidos antes de la etapa de mezcla. Típicamente, los fluidos son líquidos convencionales o fluidos supercríticos, donde los fluidos supercríticos también incluyen fluidos subcríticos (es decir, fluidos para los que solo una de la presión y la temperatura está por encima de su valor supercrítico). Las combinaciones típicas son, a) líquidos convencionales (es decir, no supercríticos) tanto para la solución del PA como para el antidisolvente, b) solución supercrítica del PA combinada con líquido convencional para el antidisolvente, c) líquido convencional para la solución del PA combinada con fluido supercrítico para el antidisolvente, y d) fluidos supercríticos para ambos fluidos. En ciertas variantes, se puede omitir el antidisolvente. A continuación, se permite que una corriente de fluido, preferiblemente supercrítico, que contiene tanto el PA como el componente polimérico estabilizante y formador de matriz, se expanda hacia la función de formación de partículas. Se prefiere que al menos uno de los fluidos esté en un estado supercrítico en los procedimientos de precipitación descritos anteriormente. Estos tipos de procedimientos de precipitación se analizan en los documentos WO 2005061090 (Censdelivery AB), WO 2009072950 (XSpray Microparticles AB), WO 2009072953 (XSpray Microparticles AB), WO 2011159218 (XSpray Microparticles AB) y las referencias citadas en estas publicaciones.
El término "solución" abarca que el soluto es tanto un verdadero soluto como partículas diminutas de dimensiones coloidales (típicamente 1-1000 nm) y más pequeñas que las partículas que se van a producir.
Un sistema de formación de partículas preferido es el "Sistema Right Size" desarrollado por XSpray Microparticles AB, Suecia. Se puede encontrar una descripción detallada de la tecnología en las publicaciones WO proporcionadas en el párrafo anterior. Una característica importante del sistema es que las dos corrientes de fluido se deben fusionar dentro de una boquilla en un ángulo en el intervalo de 45-135°, con preferencia por aproximadamente 90°, y se debe pulverizar en una función de formación/separación de partículas. En principio, el sistema permite producir partículas de tamaño y/o morfología predeterminados. Aquí se describirán el sistema y el aparato Right Size usando el ejemplo no limitativo de un IPC como fármaco y CO2 como antidisolvente fluido supercrítico.
El sistema consiste en una configuración de bombeo para el IPC disuelto en un disolvente líquido, denominado solución del PA, y una configuración de bombeo para un antidisolvente, por ejemplo CO2 , sin embargo, también se pueden usar otros antidisolventes según sea apropiado. Cada configuración de bombeo incluye instrumentos tales como un fluxímetro y un medidor de presión que se usan para controlar las condiciones del procedimiento. Estas dos configuraciones de bombeo están conectadas de forma fluida a una boquilla de pulverización.
Se mezcla una corriente de solución líquida del PA con una corriente de CO2 en condiciones de flujo dentro de la boquilla de pulverización. El componente polimérico estabilizante y formador de matriz está presente en la solución del PA o en la corriente de CO2. Estas corrientes se pulverizan a la salida de la boquilla en un vaso de precipitados en condiciones controladas (típicamente presión y temperatura). El CO2 actúa como antidisolvente y hace que el PA se precipite junto con el componente polimérico estabilizante y formador de matriz en partículas finas. Las partículas se retienen en el vaso mediante un sistema de filtrado. Típicamente, se usa un regulador de contrapresión para controlar la presión dentro del vaso de precipitados.
Para preparar las nanopartículas híbridas de ciertos fármacos, por ejemplo, pero sin limitarse a, Pazopanib y Erlotinib, puede ser ventajoso tener una configuración de bombeo adicional para inyectar un disolvente adicional, denominado
modificador, en el CO2. Aquí se configura un control de configuración de bombeo para el modificador y el modificador se mezcla con el CO2 en una mezcladora antes de introducirlo en la boquilla.
Cuando se usa el sistema, el operador del sistema comienza equilibrando típicamente el sistema bombeando CO2 , una "solución similar al IPC" (una solución similar en composición a la solución del IPC pero que no contiene IPC ni excipiente) y el modificador (si se usa) a través del sistema hasta que los caudales, la presión y la temperatura hayan alcanzado el estado estable deseado. Los parámetros críticos para configurar el sistema son la composición de la solución del IPC, el caudal de la solución del IPC, el caudal de CO2 , la presión y temperatura del CO2, la naturaleza del modificador y el caudal del modificador, si se usa.
Después, la "solución similar al IPC" se cambia por la solución del IPC y las partículas se producen y retienen por debajo de la mezcla, por ejemplo, por debajo de la salida de la boquilla. Posteriormente, el sistema se limpia típicamente bombeando la "solución similar al IPC" a través del sistema. Las partículas se secan haciéndolas pasar el CO2 través de las partículas retenidas para extraer cualquier disolvente restante. A continuación, se despresuriza el vaso de precipitados y se pueden recoger las partículas.
La solución/disolvente y el antidisolvente son típicamente miscibles entre sí. La presión y la temperatura en la función de formación de partículas y/o anterior a esta función, tal como en la función de mezcla, proporcionan condiciones supercríticas o subcríticas con respecto al antidisolvente.
La concentración del IPC en la solución se encuentra típicamente por debajo de su concentración de saturación, tal como <50 %, tal como <60 %, tal como <75 %, tal como <85 % o tal como <95 % de la concentración de saturación. Las concentraciones adecuadas se encuentran típicamente en el intervalo <20 %, tal como <10 % o <5 % o <3 %, los límites inferiores son <0,005 % o un 0,1 % (todo en % p/v). El término "volátil" para disolventes significa típicamente puntos de ebullición de <200 °C, tales como <150 °C o <100 °C, a presión atmosférica. Son ejemplos los disolventes inorgánicos y los disolventes orgánicos con especial énfasis en dimetilsulfóxido y trifluoroetanol y mezclas de los mismos. El término disolvente incluye mezclas de líquidos que son miscibles entre sí. Las soluciones pueden contener agentes que mejoran o disminuyen la solubilidad del IPC, por ejemplo, componentes tampón ácidos, alcalinos y/u otros disolventes orgánicos.
Los fluidos ilustrativos que se pueden usar como antidisolvente son
a) gaseosos a temperatura ambiente y presiones atmosféricas, o
b) líquidos a temperatura ambiente y presión atmosférica.
El antidisolvente se selecciona típicamente por su capacidad para dispersarse fácilmente en pequeñas gotículas y por su capacidad para actuar como agente atomizador y antidisolvente contra el IPC presente en la solución.
Los compuestos/elementos según el grupo (a) se pueden seleccionar de entre dióxido de carbono (Pc = 74 bar y Tc = 31 °C) (preferido), óxido nitroso (Pc = 72 bar y Tc = 36 °C), hexafluoruro de azufre (Pc = 37 bar y Tc = 45 °C), etano (Pc = 48 bar y Tc = 32 ° C), etileno (Pc = 51 bar y Tc = 10 °C), xenón (Pc = 58 bar y Tc = 16 °C), trifluorometano (Pc = 47 bar y Tc = 26 °C), clorotrifluorometano (Pc = 39 bar y Tc = 29 °C) y nitrógeno (Pc = 34 bar y Tc = -147 °C) y mezclas que contienen estos compuestos/elementos. Pc significa presión crítica y Tc temperatura crítica. Los compuestos según el grupo (b) se seleccionan típicamente de entre líquidos convencionales de los mismos tipos generales que los analizados anteriormente para los disolventes, pero con la diferencia de que el IPC presente en la solución debe ser poco soluble en el antidisolvente. Los líquidos particulares del grupo (b) comprenden metanol, etanol, acetona, agua y mezclas que contienen uno o más de estos fluidos.
Los antidisolventes del grupo (a) anterior se usan típicamente a presiones y temperaturas que proporcionan i) condiciones supercríticas (fluido supercrítico) o ii) condiciones subcríticas (fluido subcrítico) en la función de formación de partículas y/o anterior a esta función, como en la función de mezcla y anterior a esta última función.
Variante (i) significa presiones y temperaturas que se encuentran típicamente por encima de la presión crítica Pc y la temperatura crítica Tc del antidisolvente usado. Para la presión, esto significa típicamente presiones en el intervalo (1,0-7,0) x Pc o en el intervalo >10 bar, de forma adecuada, >20 bar con preferencia por >30 bar, mayor que la Pc con límites superiores ilustrativos que son 100 bar, 200 bar y 300 bar superiores a la Pc. Para la temperatura, esto significa típicamente temperaturas dentro de (1,0-4,0) x Tc o en el intervalo de >5 °C, de forma adecuada, >10 °C con preferencia por >15 °C por encima de la Tc con límites superiores ilustrativos de 10 °C, 40 °C y 50 °C por encima de la Tc.
Variante (ii) significa que al menos uno de temperatura y presión, con preferencia solo para la temperatura, se encuentra/encuentran por debajo del valor crítico. (Tc y Pc, respectivamente). Por tanto, la temperatura se puede encontrar en el intervalo de (0,1-1) x Tc, tal como (0,5-1) x Tc o menos. Además, la temperatura puede ser baja, tal como -10 °C o -30 °C. Estas temperaturas se pueden combinar con las presiones definidas en el párrafo anterior o con presiones inferiores a la Pc del antidisolvente usado. Para el dióxido de carbono, esto significa que la temperatura en la función de formación de partículas es <+31 °C, tal como aproximadamente 25 °C o menos combinada con una
presión por encima o por debajo de 74 bar.
Los antidisolventes del grupo (b) anterior se usan típicamente en el estado subcrítico, es decir, como un fluido subcrítico.
Para los procedimientos de la invención que usan condiciones subcríticas en la cámara de formación de partículas. Esto incluye que la presión en la función de mezcla y en el fluido antidisolvente siempre es más alta que en la función de formación de partículas.
En un aspecto de la invención, se proporciona un procedimiento para producir nanopartículas híbridas amorfas estables que comprende al menos un inhibidor de la proteína cinasa y al menos un componente polimérico estabilizante y formador de matriz, que comprende
a) proporcionar una primera corriente, preferiblemente presurizada, de dicho inhibidor de la proteína cinasa disuelto en un disolvente;
b) proporcionar una segunda corriente de antidisolvente, preferiblemente presurizada; donde dicho al menos un componente polimérico estabilizante y formador de matriz está presente en dicha primera o segunda corriente; y c) mezclar dicha primera y segunda corrientes, y pulverizar la corriente mixta a la salida de una boquilla, con lo que se forman dichas nanopartículas híbridas; seguido de la recogida de dichas nanopartículas híbridas.
En una realización de este aspecto, dicho componente polimérico estabilizante y formador de matriz está presente en dicho disolvente.
En otra realización de este aspecto, dicho componente polimérico estabilizante y formador de matriz está presente en dicho antidisolvente.
En otra realización de este aspecto, al menos una de dichas corrientes de fluido es una corriente de fluido supercrítico, preferiblemente, una corriente de fluido supercrítico o subcrítico de CO2.
En otra realización de este aspecto, dicha corriente de antidisolvente es una corriente de fluido supercrítico o subcrítico de CO2.
En otra realización de este aspecto, dicha corriente de disolvente es una corriente de fluido supercrítico o subcrítico de CO2.
En otra realización de este aspecto, dicho al menos un componente polimérico estabilizante y formador de matriz está presente en dicha corriente de fluido supercrítico o subcrítico de CO2.
En otra realización de este aspecto, dicha corriente de disolvente es una corriente de fluido subcrítico de CO2. Dicha corriente de fluido supercrítico o subcrítico de CO2 se proporciona preferiblemente a aproximadamente 25 °C o menos, a una presión de aproximadamente 100 a aproximadamente 150 bar. Sin embargo, dicha corriente de disolvente puede comprender, por ejemplo, nitrógeno, metano, etano, propano, etileno, metanol, etanol, acetona, agua o una mezcla de estos compuestos tal como una mezcla de CO2 y nitrógeno. Asimismo, se puede añadir un disolvente adicional, tal como un disolvente orgánico (por ejemplo, metanol, acetona) o una solución acuosa (por ejemplo, agua) como modificador en un disolvente supercrítico/subcrítico.
En otra realización de este aspecto, dicha corriente de antidisolvente es una corriente de fluido supercrítico o subcrítico de CO2. Dicha corriente de fluido supercrítico o subcrítico de CO2 se proporciona preferiblemente a aproximadamente 25 °C o menos, a una presión de aproximadamente 100 a aproximadamente 150 bar. Sin embargo, dicha corriente de antidisolvente puede comprender, por ejemplo, nitrógeno, metano, etano, propano, etileno, metanol, etanol, acetona, agua o una mezcla de estos compuestos tal como una mezcla de CO2 y nitrógeno. Asimismo, se puede añadir un disolvente, tal como un disolvente orgánico (por ejemplo, metanol, acetona) o una solución acuosa (por ejemplo, agua) como modificador en un antidisolvente supercrítico o subcrítico.
El componente polimérico estabilizante y formador de matriz de la presente en los procedimientos de la invención incluye, pero no se limita a, metilcelulosa, hidroxietilcelulosa, hidroxipropilcelulosa (por ejemplo, HPC ef, HPC If y HPC jf), hidroxipropilmetilcelulosa (por ejemplo, Methocel E3 y E15 y Pharmacoat), succinato de acetato de hidroxipropilmetilcelulosa (HPMc AS), ftalato de hidroxipropilmetilcelulosa (por ejemplo, HPMC HP55), polivinilpirrolidona (por ejemplo, PVP 30K y PVP 90K), ftalato de acetato de polivinilo (PVAP), copolividona (por ejemplo, Kollidon VA 64), crospovidona (por ejemplo, Kollidon CL), copolímero de ácido metacrílico y acrilato de etilo (por ejemplo, Kollicoat ME), copolímero de ácido metacrilato y metacrilato de metilo (por ejemplo, Eudragit L100), polietilenglicol (PEG), copolímero de DL lactida/glicólido, poli DL-lactida, ftalato de acetato de celulosa (CAP), homopolímero de carbómero tipo A (Carbopol 971 P), homopolímero de carbómero tipo B (Carbopol 974P), copolímeros de aminoalquil metacrilato (por ejemplo, Eudragit RL100, RL PO o RS PO) y poloxámeros (por ejemplo, Pluronics, Kolliphor).
En consecuencia, en otra realización de este aspecto, dicho componente polimérico estabilizante y formador de matriz se selecciona de entre metilcelulosa, hidroxietilcelulosa, hidroxipropilcelulosa, hidroxipropilmetilcelulosa, succinato de acetato de hidroxipropilmetilcelulosa, ftalato de hidroxipropilmetilcelulosa, ftalato de acetato de polivinilo, copolvidona, crospovidona, copolímero de ácido metacrílico y acrilato de etilo, copolímero de metacrilato de ácido y metacrilato de metilo, polietilenglicol, copolímero de DL lactida/glicólido, poli DL-lactida, ftalato de acetato de celulosa, homopolímero de carbómero tipo A, homopolímero de carbómero tipo B, copolímeros de aminoalquil metacrilato y poloxámeros. Preferiblemente, dicho componente polimérico estabilizante y formador de matriz se selecciona de entre ftalato de hidroxipropilmetilcelulosa, hidroxipropilcelulosa, copolvidona, succinato de acetato de hidroxipropilmetilcelulosa, ftalato de acetato de polivinilo, ftalato de acetato de celulosa y polivinilpirrolidona.
En otra realización de este aspecto, se añade un solubilizante a las nanopartículas híbridas obtenidas en la etapa c. En este contexto, el solubilizante estará presente por separado con respecto a las partículas. Dicho solubilizante se puede seleccionar de entre copolímero de polivinil caprolactama-acetato de polivinilo-polietilenglicol, succinato de da-tocoferol polietilenglicol 1000 y un aceite de ricino hidrogenado, tal como aceite de ricino hidrogenado PEG-40 o aceite de ricino hidrogenado PEG-35. Además, dicho solubilizante puede ser un poloxámero.
Los procedimientos de la invención proporcionan nanopartículas híbridas amorfas estables que comprenden al menos un inhibidor de la proteína cinasa y al menos un componente polimérico estabilizante y formador de matriz, que muestran una mayor velocidad de disolución.
En consecuencia, en otra realización de este aspecto, se proporciona un procedimiento para producir nanopartículas híbridas amorfas estables que comprenden al menos un inhibidor de la proteína cinasa y al menos un componente polimérico estabilizante y formador de matriz, donde dichas nanopartículas híbridas muestran una velocidad de disolución aumentada de dicho inhibidor de la proteína cinasa, en comparación con la velocidad de disolución de dicho inhibidor de la proteína cinasa en forma cristalina cruda.
Típicamente, dicha velocidad de disolución se mide mediante un sistema de cubetas de lectura en condiciones de baja concentración, por ejemplo, según la Farmacopea de los Estados Unidos (USP4). La medición de la disolución en condiciones de baja concentración de nanopartículas híbridas se puede medir en un procedimiento que consiste en añadir la cantidad deseada de polvo en un sistema de cubetas de lectura (SOTAX, Allschwill, Suiza), montar la cubeta en su aparato y, a continuación, bombear el medio apropiado (típicamente FaSSIF, FeSSIF, SGF) a través del polvo. La temperatura del aparato se fija típicamente a 37 °C. La cantidad de polvo añadida a la cubeta depende de la carga de fármaco del polvo: la cantidad exacta de polvo se puede calcular a partir de los resultados obtenidos del análisis de la carga de fármaco de los polvos. El IPC se puede añadir a la cubeta de lectura y se bombea a través del polvo a un caudal de entre 5 y 25 ml de medio/min. Se recogen muestras de un ml del medio que pasa a través de la cubeta en momentos predeterminados y se analizan posteriormente mediante HPLC (por ejemplo, columna C18 Eclipse, 4,6 mm x 15 cm, 1 ml/min, detección de 254 a 400 nm). Las muestras se toman típicamente después de 0, 0,5, 1, 1,5, 2, 3, 4, 5, 6, 7, 8, 9, 10, 15, 20, 25, 30, 35 y 40 minutos desde el momento en que sale el medio de la cubeta de lectura. El % acumulado de solubilizado de la cantidad de sustancia activa añadida en la cubeta de lectura se puede calcular y representar frente al tiempo (min). La pendiente inicial ("velocidad de disolución inicial", que representa de 0 a 10 minutos) del gráfico se puede estimar y tomar como la velocidad de disolución del material en condiciones de baja concentración a 37 °C en el medio de disolución dado.
Preferiblemente, la velocidad de disolución se mide en los de 0 a 10 minutos iniciales de disolución.
La velocidad de disolución aumentada se mide preferiblemente en una solución como una relación de velocidad de disolución de dichas nanopartículas híbridas y dicho inhibidor de la proteína cinasa en forma cristalina cruda. Preferiblemente, dicha relación es de aproximadamente 1,5:1 a aproximadamente 500:1, tal como de aproximadamente 10:1 a aproximadamente 30:1.
Preferiblemente, la velocidad de disolución se mide en una solución con pH intestinal, tal como FaSSIF o FeSSIF o en una solución con pH gástrico, tal como SGF.
Típicamente, dicha velocidad de disolución se mide mediante un sistema de cubetas de lectura, por ejemplo, en condiciones de baja concentración. La medición de la disolución en condiciones de baja concentración de nanopartículas híbridas se puede medir en un procedimiento que consiste en añadir la cantidad deseada de polvo en un sistema de cubetas de lectura (SOTAX, Allschwill, Suiza), montar la cubeta en su aparato y, a continuación, bombear el medio apropiado (típicamente FaSSIF, FeSSIF, SGF) a través del polvo. La temperatura del aparato se fija típicamente a 37 °C. La cantidad de polvo que se añade a la cubeta depende de la carga de fármaco del polvo: La cantidad exacta de polvo se puede calcular a partir de los resultados obtenidos del análisis de la carga de fármaco de los polvos. El IPC se puede añadir a la cubeta de lectura y se bombea a través del polvo a un caudal de entre 5 y 25 ml de medio/min. Se recogen muestras de un ml del medio que pasa a través de la cubeta en momentos predeterminados y se analizan posteriormente mediante HPLC (por ejemplo, columna C18 Eclipse, 4,6 mm x 15 cm, 1 ml/min, detección de 254 a 400 nm). Las muestras se toman típicamente después de 0, 0,5, 1, 1,5, 2, 3, 4, 5, 6, 7, 8, 9, 10, 15, 20, 25, 30, 35 y 40 minutos desde el momento en que sale el medio de la cubeta de lectura. El % acumulado de solubilizado de la cantidad de sustancia activa añadida en la cubeta de lectura se puede calcular y
representar frente al tiempo (min). La pendiente inicial ("velocidad de disolución inicial", que representa de 0 a 10 minutos) del gráfico se puede estimar y tomar como la velocidad de disolución del material en condiciones de baja concentración a 37 °C en el medio de disolución dado.
En otra realización de este aspecto, se proporciona un procedimiento para producir nanopartículas híbridas amorfas estables que comprende al menos un inhibidor de la proteína cinasa y al menos un componente polimérico estabilizante y formador de matriz, que produce partículas que proporcionan un aumento en la solubilidad del inhibidor en una solución, dicho aumento medido como el área bajo la curva (ABC) durante de aproximadamente 40 minutos a aproximadamente 90 minutos, en dicha solución en comparación con el ABC del inhibidor en forma cristalina cruda. Preferiblemente, dicho aumento es de aproximadamente 2:1 a aproximadamente 10000:1, donde 1 representa el ABC del inhibidor en forma cristalina cruda. Preferiblemente, dicho aumento se mide en una solución con pH gástrico, tal como SGF, o se mide en una solución con pH intestinal, tal como FaSSIF o FeSSIF. Los procedimientos pueden producir partículas que proporcionan un aumento en la solubilidad del inhibidor en una solución hasta la sobresaturación, dicho aumento medido como el área bajo la curva (ABC) durante de aproximadamente 40 minutos a aproximadamente 90 minutos, en dicha solución en comparación con el ABC del inhibidor en forma cristalina cruda. En otra realización de este aspecto, se proporciona un procedimiento para producir nanopartículas híbridas amorfas estables que comprende al menos un inhibidor de la proteína cinasa y al menos un componente polimérico estabilizante y formador de matriz, caracterizado por la provisión de un patrón de difracción de rayos X en polvo amorfo.
En otra realización de este aspecto, se proporciona un procedimiento para producir nanopartículas híbridas amorfas estables que comprende al menos un inhibidor de la proteína cinasa y al menos un componente polimérico estabilizante y formador de matriz, donde la velocidad de disolución de dichas nanopartículas híbridas amorfas estables permanece estable hasta al menos aproximadamente un 90 %, después de 6 meses de almacenamiento o más, a temperatura ambiente.
En otra realización de este aspecto, dicho inhibidor de la proteína cinasa es dasatinib.
En otra realización de este aspecto, dichas nanopartículas híbridas tienen un tamaño promedio de diámetro de partícula de menos de aproximadamente 1000 nm, tal como menos de aproximadamente 500 nm, preferiblemente, menos de 250 nm.
En otra realización de este aspecto, dicho disolvente es un disolvente orgánico seleccionado de entre DMSO y trifluoroetanol, o una mezcla de estos disolventes, o una mezcla de estos disolventes con otro disolvente orgánico tal como DMSO/acetona, DMSO/tetrahidrofurano o trifluoroetanol/acetato de etilo. En otra realización de este aspecto, dichas partículas comprenden además un solubilizante dentro de las partículas. Dicho solubilizante puede ser copolímero de polivinil caprolactama-acetato de polivinilo-polietilenglicol, succinato de d-a-tocoferol polietilenglicol 1000 o un aceite de ricino hidrogenado, tal como aceite de ricino hidrogenado PEG-40 o aceite de ricino hidrogenado PEG-35.
En otra realización de este aspecto, el procedimiento comprende además formular las partículas retenidas en la etapa (c) como una composición farmacéutica que contiene las partículas y, opcionalmente, otros excipientes farmacéuticamente aceptables.
En otro aspecto de la invención, se proporcionan nanopartículas híbridas amorfas estables que comprenden al menos un inhibidor de la proteína cinasa y al menos un componente polimérico estabilizante y formador de matriz, obtenible por los procedimientos descritos anteriormente.
Las nanopartículas híbridas producidas por los procedimientos de la presente invención también se pueden disolver y el inhibidor de la proteína cinasa se puede absorber sistémica e independientemente del pH en el ambiente circundante, y típicamente aproximadamente en cantidades iguales, especialmente tanto a pH gástrico, tal como de aproximadamente pH 1,2 a aproximadamente pH 2,1, preferiblemente, de aproximadamente 1,7 como a un pH intestinal tal como de aproximadamente pH 4,5 a aproximadamente pH 8, preferiblemente, a un pH de aproximadamente 6. Por absorber sistémicamente, significa que el inhibidor de la proteína cinasa se libera de las nanopartículas híbridas y lo absorbe el torrente sanguíneo sistémico. Por lo tanto, en otra realización de este aspecto, se proporciona un procedimiento, donde dicho inhibidor de la proteína cinasa se absorbe sistémica e independientemente del pH. Típicamente, dicho inhibidor de la proteína cinasa se absorbe sistémicamente con cantidades aproximadamente iguales tanto a pH gástrico como a pH intestinal. Preferiblemente, dicho pH ácido es aproximadamente pH 1,4 y, preferiblemente, dicho pH neutro es aproximadamente pH 6,5.
Con cantidades aproximadamente iguales se entiende que la concentración de inhibidor de la proteína cinasa en el torrente sanguíneo, después de la exposición, es aproximadamente similar. Esto se puede ilustrar mediante una relación, donde la concentración de inhibidor de la proteína cinasa en el torrente sanguíneo se mide después de la administración en condiciones de pH gástrico (A) y se compara con la concentración de inhibidor de la proteína cinasa en el torrente sanguíneo medida después de la administración en condiciones de pH intestinal (N). Típicamente, la
relación A:N es de aproximadamente 0,75:1 a aproximadamente 1,5:1 y, preferiblemente, de aproximadamente 1:1 a aproximadamente 1,25:1. La medición de la concentración de inhibidor de la proteína cinasa en el torrente sanguíneo se puede llevar a cabo como área bajo la curva (ABC) durante 0-24 horas, la concentración máxima (Cmáx) o como biodisponibilidad.
En consecuencia, en otra realización de este aspecto, se proporciona un procedimiento para producir nanopartículas híbridas amorfas estables, que comprende al menos un inhibidor de la proteína cinasa y al menos un componente polimérico estabilizante y formador de matriz, donde la concentración de inhibidor de la proteína cinasa absorbido sistémicamente en condiciones de pH gástrico en comparación con la concentración de inhibidor de la proteína cinasa absorbido sistémicamente en condiciones de pH intestinal se encuentra en una relación de aproximadamente 0,75:1 a aproximadamente 1,5:1, preferiblemente, de aproximadamente 1:1 a aproximadamente 1,25:1. Típicamente, dicha condición de pH gástrico representa un pH de aproximadamente 1,4 y dicha condición de pH intestinal representa un pH de aproximadamente 6. Típicamente, la concentración se mide como área bajo la curva (ABC) durante 0-24 horas de exposición de la composición o como la concentración máxima (Cmáx).
Las cantidades de inhibidor de la proteína cinasa absorbido sistémicamente se pueden medir de varias maneras. En el ejemplo 14 de la presente descripción, se proporciona un procedimiento para medir los inhibidores de la proteína cinasa absorbidos sistémicamente a varios pH, es decir, en condiciones tanto ácidas como neutras.
En otra realización de este aspecto, se proporciona un procedimiento para producir nanopartículas híbridas amorfas estables que comprende al menos un inhibidor de la proteína cinasa y al menos un componente polimérico estabilizante y formador de matriz, que produce un aumento en la solubilidad del inhibidor en una solución hasta la sobresaturación, dicho aumento se mide como el área bajo la curva (ABC) durante aproximadamente 90 minutos, en dicha solución en comparación con el ABC del inhibidor en forma cristalina. Dicho aumento puede ser de aproximadamente 2:1 a aproximadamente 1000:1, donde 1 representa el ABC del inhibidor en forma cristalina. Para comprender cómo se disolverán in vivo las nanopartículas híbridas producidas por los procedimientos de la invención en los diferentes ambientes del estómago, intestino delgado, intestino grueso y colon, es importante elegir una solución apropiada para pruebas de disolución in vitro. Es fundamental que las condiciones de prueba in vitro imiten el entorno in vivo de la forma más parecida posible, por ejemplo, el pH y la osmolaridad. Típicamente, para la absorción intestinal, el pH se encuentra entre 6 y 7. Por lo tanto, la solución puede mantener un pH de aproximadamente pH 6 a aproximadamente pH 7, tal como aproximadamente pH 6,5.
Por lo tanto, en las realizaciones de la invención, la solución para la prueba tiene un pH de aproximadamente pH 4,5 a aproximadamente pH 8, tal como aproximadamente pH 6,5 o tal como aproximadamente pH 5. Las soluciones pueden representar fluido intestinal simulado en estado de ayuno (FaSSIF) o fluido intestinal simulado en estado alimentado (FeSSIF).
Típicamente, para la absorción gástrica, el pH se encuentra entre 1 y 2. Por lo tanto, la solución puede mantener un pH de aproximadamente pH 1 a aproximadamente pH 2, tal como aproximadamente pH 1,4. Por lo tanto, en las realizaciones de la invención, la solución para la prueba puede representar un fluido gástrico simulado (SGF).
La elección del medio dependerá del lugar del tubo intestinal y en qué condiciones (en ayunas o alimentado) se desea que se disuelva y absorba la composición. Las recetas y la preparación de estas soluciones se pueden obtener del fabricante (Biorelevant, Croydon, Reino Unido). También se describen más detalles en Jantratid, E. y Dressman, J. (2009) Dissolut. Technol. 8, 21-25).
En otra realización de este aspecto, se proporciona un procedimiento para producir nanopartículas híbridas amorfas estables, que comprende al menos un inhibidor de la proteína cinasa y al menos un componente polimérico estabilizante y formador de matriz y, además, un solubilizante seleccionado de entre copolímero de polivinil caprolactama-acetato de polivinilo-polietilenglicol, succinato de d-a-tocoferol polietilenglicol 1000, poloxámeros (por ejemplo, Pluronics, Kolliphor) y un aceite de ricino hidrogenado, tal como ricino hidrogenado PEG-40 aceite o aceite de ricino hidrogenado PEG-35. Preferiblemente, dicho solubilizante es copolímero de polivinil caprolactama-acetato de polivinilo-polietilenglicol o un aceite de ricino hidrogenado, tal como aceite de ricino hidrogenado PEG-40 o aceite de ricino hidrogenado PEG-35.
Puede ser además ventajoso usar diferentes agentes de dispersión y/u otros excipientes para lograr diferentes perfiles de disolución. Por ejemplo, el uso de polvo de Nilotinib HCl con HPC EF, HPMC (por ejemplo, Methocel) y poloxámero (por ejemplo, Lutrol® F127) da lugar a nanopartículas híbridas con diferentes perfiles de disolución.
La cantidad de IPC en las nanopartículas híbridas producidas por los procedimientos de la presente invención puede ser menor o mayor, tal como cuando la cantidad de IPC en las nanopartículas híbridas se encuentra de aproximadamente un 0,01 % en peso a aproximadamente un 99,9 % en peso.
En otra realización de este aspecto, se proporcionan nanopartículas híbridas producidas por los procedimientos de la presente invención, donde la cantidad de IPC en las nanopartículas híbridas se encuentra de aproximadamente un
10 % en peso a aproximadamente un 70 % en peso.
En otra realización de este aspecto, se proporcionan nanopartículas híbridas producidas por los procedimientos de la presente invención, donde la cantidad de IPC en las nanopartículas híbridas se encuentra de aproximadamente un 10 % en peso a aproximadamente un 50 % en peso.
En algunas realizaciones, puede ser ventajoso que la cantidad de IPC en las nanopartículas híbridas se encuentre de un 5 % en peso a aproximadamente un 50 % en peso, de un 10 % en peso a aproximadamente un 40 % en peso, de aproximadamente un 10 % en peso a aproximadamente un 30 % en peso, o de aproximadamente un 10 % en peso a aproximadamente un 20 % en peso.
El control de las características de las partículas puede ser conveniente para aplicaciones específicas. El tamaño de las partículas, la aglomeración de las partículas, la porosidad de las partículas y la elección y proporción del agente polimérico estabilizante y formador de matriz se podrían modificar para aumentar o disminuir la relación entre el área superficial y el volumen de las partículas o el comportamiento de las partículas en los fluidos gastrointestinales, lo que lleva a un aumento o disminución de la velocidad de disolución. En función de las características de disolución deseadas, se pueden adaptar dichas características de las partículas. Además, pueden estar presentes partículas con diferentes características en la misma composición farmacéutica para proporcionar una dosis inicial y una dosis prolongada o retardada de principio activo. De forma adicional, puede ser ventajoso proporcionar diferentes IPC y/u otro(s) principio(s) activo(s) en diferentes partículas primarias con diferentes características adaptadas para proporcionar velocidades de disolución deseadas para cada principio(s) activo(s).
Otras realizaciones de la invención proporcionan composiciones farmacéuticas que comprenden las nanopartículas híbridas obtenibles mediante los procedimientos de la invención. Dicha composición puede comprender además un solubilizante farmacéuticamente aceptable. Dicho solubilizante puede estar presente separado con respecto a las nanopartículas híbridas en la composición o entremezclarse aleatoriamente con las nanopartículas híbridas en la composición farmacéutica. La composición farmacéutica también puede estar en una forma galénica que consiste en varias capas, por ejemplo, comprimidos laminados o multicapa, de modo que las nanopartículas híbridas se separen del solubilizante. El solubilizante se puede seleccionar de entre copolímero de polivinil caprolactama-acetato de polivinilo-polietilenglicol, succinato de d-a-tocoferol polietilenglicol 1000 y un aceite de ricino hidrogenado, tal como aceite de ricino hidrogenado PEG-40 o aceite de ricino hidrogenado PEG-35. Dicho solubilizante también puede ser un poloxámero.
En otra realización de este aspecto, se proporcionan nanopartículas híbridas amorfas estables que comprenden al menos un inhibidor de la proteína cinasa y al menos un componente polimérico estabilizante y formador de matriz, obtenibles por los procedimientos de la presente invención.
En otra realización de este aspecto, se proporciona un procedimiento de la presente invención, que comprende además formular las partículas retenidas en la etapa (c) como una composición farmacéutica que contiene las partículas y, opcionalmente, otros excipientes farmacéuticamente aceptables.
Se apreciará que la cantidad de un inhibidor de la proteína cinasa en las nanopartículas híbridas producidas por los procedimientos de la presente invención requeridas para su uso en el tratamiento variará no solo con el inhibidor particular seleccionado, sino también con la vía de administración, la naturaleza de la afección para la que se requiere el tratamiento y la edad, peso y estado del paciente y, en última instancia, a criterio del médico responsable. En general, sin embargo, una dosis adecuada se puede encontrar en el intervalo de aproximadamente 0,005 a aproximadamente 30 mg/kg de peso corporal por día, preferiblemente, en el intervalo de 0,05 a 10 mg/kg/día.
La dosis deseada se presenta convenientemente en una dosis única o como dosis dividida administrada a intervalos apropiados, por ejemplo, de dos, tres, cuatro o más dosis al día. En función de la necesidad de tratamiento y/o prevención, la dosis deseada también puede ser, por ejemplo, una vez cada dos días, una vez cada tres días o, incluso, una vez a la semana.
La composición se puede administrar convenientemente en forma galénica unitaria, por ejemplo, que contiene de 0,5 a 1500 mg, convenientemente, de 1 a 1000 mg, más convenientemente, de 5 a 700 mg de principio activo por forma galénica unitaria. Normalmente, las composiciones de la invención se administrarán por vía oral, parenteral, intravenosa, intramuscular, subcutánea u otras vías inyectables, vía bucal, rectal, vaginal, transdérmica y/o nasal y/o vía inhalación, en una forma galénica farmacéuticamente aceptable. En función del trastorno y del paciente que se va a tratar y de la vía de administración, las composiciones se pueden administrar en dosis variables.
Las composiciones farmacéuticas incluyen, pero no se limitan a, aquellas adecuadas para administración oral, rectal, nasal, tópica (incluidas bucal y sublingual), transdérmica, vaginal o parenteral (incluidas intramuscular, subcutánea e intravenosa) o en una forma adecuada para administración por inhalación o insuflación. Según corresponda, las composiciones se pueden presentar convenientemente en unidades de dosificación discretas y se pueden preparar mediante cualquiera de los procedimientos bien conocidos en la técnica de la farmacia. Las composiciones farmacéuticas adecuadas para administración oral se presentan convenientemente como unidades discretas tales
como cápsulas, obleas o comprimidos, cada uno contiene una cantidad predeterminada del principio activo.
Los comprimidos y cápsulas para administración oral pueden contener excipientes convencionales tales como agentes aglutinantes, material de relleno, lubricantes, desintegrantes o agentes humectantes. Los comprimidos se pueden recubrir según los procedimientos bien conocidos en la técnica.
Las composiciones se pueden formular para administración parenteral (por ejemplo, por inyección, por ejemplo, inyección en bolo o infusión continua) y se pueden presentar en forma de dosis unitaria en ampollas, jeringas precargadas, infusión de pequeño volumen o en envases multidosis con un conservante añadido. Las composiciones pueden adoptar formas como suspensiones, soluciones o emulsiones en vehículos oleosos o acuosos y pueden contener agentes de formulación tales como agentes de suspensión, estabilización y/o dispersión.
Las composiciones descritas anteriormente se pueden adaptar para proporcionar una liberación mantenida del inhibidor activo.
Ejemplos
A continuación, sigue una serie de ejemplos no limitativos de nanopartículas híbridas producidas por los procedimientos de la presente invención. En las tablas, se aplican las siguientes abreviaturas de "composiciones": "I" representa el inhibidor de la proteína cinasa (IPC);
"P" representa el componente polimérico estabilizante y formador de matriz;
"S" representa el solubilizante;
"I+P" representa una mezcla física del inhibidor con el componente polimérico estabilizante y formador de matriz, es decir, sin procesamiento adicional;
"I+S" representa una mezcla física del inhibidor con el solubilizante;
"I+P+S" representa una mezcla física del inhibidor, el componente polimérico estabilizante y formador de matriz y el solubilizante;
"I/P" representa nanopartículas híbridas con el inhibidor y el componente polimérico estabilizante y formador de matriz;
"I/P+S" representa nanopartículas híbridas con el inhibidor y el componente polimérico estabilizante y formador de matriz y un solubilizante separado añadido;
"I/P/S" representa nanopartículas híbridas con el inhibidor y el componente polimérico estabilizante y formador de matriz y el solubilizante.
"Exp" representa el número de experimento.
Las nanopartículas híbridas se produjeron con IPC ejemplares, componentes poliméricos estabilizantes y formadores de matriz ("Polímeros"), solubilizantes, concentraciones de solución, relaciones, disolventes, antidisolventes, temperaturas y presiones como se expone a continuación y en la tabla A.
Se bombeó una solución de IPC/polímero al 3-6 % p/v en disolvente, con una relación IPC/polímero de aproximadamente un 20-70 % p/p, a través de una boquilla RightSize de XSpray a un caudal de 1 ml/min usando una bomba de cromatografía de líquidos de alta resolución, junto con una corriente de CO2 (supercrítica o subcrítica) de 100 g/min. La presión en la cámara de precipitación se fijó a aproximadamente 100-175 bar y la temperatura se fijó a de aproximadamente 10 a 50 °C. Ambas corrientes estaban en contacto dentro de la boquilla y las nanopartículas híbridas se formaron y, posteriormente, se recogieron en la partícula en la cámara de recogida. El CO2sc y el disolvente pasaron a través del sistema de filtrado de la cámara de recogida y se drenaron a través de la salida del regulador de contrapresión que mantiene la presión dentro de las cámaras de precipitación y de recogida. Después de bombear la solución de IPC/polímero y de limpiar los tubos con el mismo disolvente usado para preparar la solución IPC/polímero, los disolventes residuales que quedaron dentro de tanto la cámara precipitación como la de recogida se eliminaron mediante lavado de estas cámaras con CO2 puro. Después del proceso de lavado, el CO2 se drenó lentamente fuera de la cámara de recogida. Una vez que se eliminó por completo el CO2, se recogieron las partículas del sistema de filtrado para su análisis.
Para partículas de tipo I/P/S, se añade una cantidad definida de solubilizante y se disuelve en la solución IPC/polímero antes de bombear la solución a través de la boquilla para precipitación según los procedimientos descritos anteriormente.
Para partículas de tipo I/P+S, se añade una cantidad definida de solubilizante a las nanopartículas híbridas en un frasco de vidrio. El frasco de vidrio se gira lentamente para mezclar el solubilizante con las nanopartículas híbridas.
Tabla A Nanopartículas híbridas amorfas estables con IPC ejemplares, componentes poliméricos estabilizantes y formadores de matriz, disolventes, antidisolventes y condiciones

Descripción general del ensayo de medición de la disolución
El procedimiento consiste en añadir la cantidad deseada de polvo de nanopartículas híbridas en un frasco de vidrio y, a continuación, verter en él el medio apropiado (típicamente FaSSIF, FeSSIF o SGF). El medio se preparó de acuerdo con las instrucciones del fabricante. La cantidad de polvo añadida depende de la "concentración total de IPC" deseada. Para algunos experimentos en los que se sometieron a prueba y compararon polvos con altas cargas del fármaco, no se tuvo en cuenta la cantidad real de IPC en las nanopartículas híbridas. Para otros experimentos, primero, se estimó la carga de fármaco mediante HPLC y se calculó la cantidad de polvo para obtener la concentración de fármaco. Típicamente, el polvo se añadió en un frasco de vidrio de 8 ml y se añadieron 7 ml de solución (normalmente FaSSIF, FeSSIF o SGF). El frasco de vidrio se colocó en un agitador (aproximadamente 1 rotación por minuto) para su disolución. Se tomaron muestras de 500 pl después de diferentes momentos y, posteriormente, se centrifugaron a aproximadamente 15000 g durante 3 minutos. El sobrenadante resultante se analizó a continuación mediante HPLC (columna Ci8 Eclipse, 4,6 mm x 15 cm, 1 ml/min, detección a 254-400 nM. En general, las muestras se tomaron después de 5, 30 y 90 minutos y, por último, 150 minutos.
Comparativa
Ejemplo 1. Nanopartículas híbridas amorfas estables con nilotinib: solubilidad a pH 6,5 y pH 5
Se llevó a cabo un número de experimentos, donde base de nilotinib o nilotinib HCl representaba el inhibidor de la proteína cinasa. Los experimentos se llevaron a cabo midiendo la concentración de IPC solubilizado (mg/l) después de 5, 30 y 90 minutos de disolución en una solución a aproximadamente pH 6,5, a saber, FaSSIF (fluido intestinal simulado en estado de ayuno). Además, se llevaron a cabo experimentos en una solución alternativa a aproximadamente pH 5, a saber, FeSSIF (fluido intestinal simulado en estado alimentado). Se tomaron muestras de la solución a varios intervalos de tiempo y se midió la cantidad de inhibidor de la proteína cinasa mediante el ensayo de medición de la disolución descrito anteriormente.
Los resultados representativos en la solución FaSSIF se proporcionan a continuación en la tabla 1 y 2, donde la Tabla 1 proporciona datos de la concentración de nilotinib HCl (mg/l) después de 5, 30 y 90 minutos de disolución, mientras que la tabla 2 proporciona datos del % de nilotinib HCl solubilizado después de 30 minutos de disolución, el área bajo la curva (ABC: mg/min/l) durante 90 minutos de disolución y el aumento del ABC de las nanopartículas híbridas, en
comparación con nilotinib HCl en forma cristalina cruda añadido a la solución (experimentos 1-40). En las tablas 3 y 4, se proporcionan datos de disolución en la solución FeSSIF, presentados de manera similar a la tabla 1 y 2 (experimentos 41-55). La tabla 5 proporciona datos de un experimento comparativo con nanopartículas híbridas similares, llevado a cabo en FaSSIF y FeSSIF, respectivamente (experimentos 56-57). La tabla 6 presenta datos comparativos adicionales para experimentos llevados a cabo en FaSSIF y FeSSIF, respectivamente, con nanopartículas híbridas producidas por los procedimientos de la invención.
Tabla 1. Nilotinib: concentración de nilotinib HCl (mg/l) después de 5, 30 y 90 minutos de disolución en solución FaSSIF H 6,5



Tabla 2. Porcentaje de nilotinib HCl solubilizado después de 30 minutos de disolución, el área bajo la curva (ABC: mg/min/l) durante 90 minutos de disolución y el aumento del ABC de nanopartículas híbridas amorfas estables producidas por los procedimientos de la invención, en comparación con nilotinib en forma cristalina cruda añadido a la solución FaSSIF H 6,5



Tabla 3. Nilotinib: concentración de nilotinib HCl (mg/l) después de 5, 30 y 90 minutos de disolución en solución FeSSIF H 5


Tabla 4. Porcentaje de nilotinib HCl solubilizado después de 30 minutos de disolución, el área bajo la curva (ABC:
mg/min/l) durante 90 minutos de disolución y el aumento del ABC de nanopartículas híbridas amorfas estables producidas por los procedimientos de la invención, en comparación con nilotinib crudo en forma cristalina curda añadido a la solución FeSSIF H 5


Q- CO co X c co LU c o
LoO L rLa h LO- L 0L
Conclusiones Ejemplo 1
Los experimentos 17-23 muestran que se obtiene un aumento en la solubilidad con las nanopartículas híbridas amorfas estables producidas por los procedimientos de la invención con nilotinib HCl y un componente polimérico estabilizante y formador de matriz. Se logran mejoras particulares con los componentes poliméricos estabilizantes y formadores de matriz ftalato de hidroxipropilmetilcelulosa (HPMCP HP55) y ftalato de acetato de polivinilo (PVAP). Estas mejoras no se obtienen cuando se mezcla físicamente nilotinib HCl con un componente polimérico estabilizante y formador de matriz. Los experimentos 24-36 muestran claramente que se obtiene un aumento en la solubilidad con las nanopartículas híbridas producidas por los procedimientos de la invención con nilotinib HCl y un componente polimérico estabilizante y formador de matriz, donde se añade un solubilizante separado. Se logran mejoras particulares mediante la adición de un solubilizante separado tal como copolímero de polivinil caprolactama-acetato de polivinilo-polietilenglicol (Soluplus) o succinato de d-a-tocoferol polietilenglicol 1000 (TPGS). Estas mejoras no se obtuvieron cuando se mezcló físicamente nilotinib HCl un solubilizante y/o un componente polimérico estabilizante y formador de matriz (I+S o I+P+S). No se obtuvieron mejoras particulares con nanopartículas híbridas con nilotinib HCl, un componente polimérico estabilizante y formador de matriz y un solubilizante (I/P/S).
Los resultados llevados a cabo en FaSSIF y FeSSIF, respectivamente, indican que las nanopartículas híbridas amorfas estables producidas por los procedimientos de la invención proporcionan un aumento similar en la solubilidad. Un problema con la formulación de IPC es el efecto de los alimentos. Varios de los IPC están indicados para su administración en estado de ayuno a pesar de que los alimentos en la mayoría de los casos aumentan su biodisponibilidad. La baja biodisponibilidad podría explicar en parte los problemas digestivos asociados con los IPC. La velocidad de disolución similar en FaSSIF y FeSSIF indica que las nanopartículas híbridas producidas por los procedimientos de la invención (por ejemplo, experimentos 56/57) pueden reducir el efecto de los alimentos y los problemas digestivos del paciente por su mejora en la solubilidad que permite reducir la dosis. Por tanto, las nanopartículas híbridas producidas por los procedimientos de la invención se pueden administrar junto con la ingesta de alimentos.
Comparativa
Ejemplo 2. Nanopartículas híbridas amorfas estables con erlotinib HCl: solubilidad a pH 6,5 y pH 5
Se llevó a cabo un número de experimentos, donde erlotinib HCl representaba el IPC. Los experimentos se llevaron a cabo midiendo la concentración de IPC (mg/l) después de 5, 30 y 90 minutos de disolución en una solución a aproximadamente pH 6,5, a saber, FaSSIF (fluido intestinal simulado en estado de ayuno). Además, se llevaron a cabo experimentos en una solución alternativa a aproximadamente pH 5, a saber, FeSSIF (fluido intestinal simulado en estado alimentado). Se tomaron muestras de la solución a varios intervalos de tiempo y se midió la cantidad de IPC mediante el ensayo de medición de la disolución descrito anteriormente.
Los resultados representativos en la solución FaSSIF se proporcionan a continuación en la tabla 7 y 8, donde la Tabla 7 proporciona datos de concentración de erlotinib HCl (mg/l) después de 5, 30 y 90 minutos de disolución, mientras que la tabla 8 proporciona datos del % de erlotinib HCl solubilizado después de 30 minutos de disolución, el área bajo la curva (ABC: mg/min/l) durante 90 minutos de disolución y el aumento del ABC de las nanopartículas híbridas producidas por los procedimientos de la invención, en comparación con erlotinib HCl en forma cristalina cruda añadido a la solución (experimentos 58-68). En las tablas 9 y 10, se proporcionan datos de disolución en la solución FeSSIF, presentados de manera similar a la tabla 7 y 8 (experimentos 69-73). En la tabla 11, se presentan los datos de un experimento comparativo con nanopartículas híbridas similares producidas por los procedimientos de la invención, llevado a cabo en FaSSIF y FeSSIF, respectivamente (experimentos 74-83). La tabla 12 presenta datos comparativos adicionales para experimentos llevados a cabo en FaSSIF y FeSSIF, respectivamente, con nanopartículas híbridas amorfas estables producidas por los procedimientos de la invención.
Tabla 7. Erlotinib: concentración de erlotinib HCl (mg/l) después de 5, 30 y 90 minutos de disolución en solución FaSSIF H 6,5


Tabla 8. Porcentaje de erlotinib HCl solubilizado después de 30 minutos de disolución, el área bajo la curva (ABC: mg/min/l) durante 90 minutos de disolución y el aumento del ABC de nanopartículas híbridas amorfas estables producidas por los procedimientos de la invención, en comparación con erlotinib en forma cristalina cruda añadido a la solución FaSSIF H 6,5


Tabla 9. Erlotinib: concentración de erlotinib HCl (mg/l) después de 5, 30 y 90 minutos de disolución en solución FeSSIF H 5

Tabla 10. Porcentaje de erlotinib HCl solubilizado después de 30 minutos de disolución, el área bajo la curva (ABC: mg/min/l) durante 90 minutos de disolución y el aumento del ABC de nanopartículas híbridas amorfas estables producidas por los procedimientos de la invención, en comparación con erlotinib en forma cristalina cruda añadido a la solución FeSSIF H 5

Tabla 11. Erlotinib: concentración de erlotinib HCl después de 5, 30 y 90 minutos de disolución en solución FaSSIF y FeSSIF, res ectivamente


Tabla 12. Erlotinib: concentración de erlotinib HCl (mg/l) después de 5, 30 y 90 minutos de disolución en solución F IF F IF r ivm n r r n m m r iv


Conclusiones Ejemplo 2
Los experimentos muestran que se obtiene un aumento en la solubilidad con las nanopartículas híbridas amorfas estables producidas por los procedimientos de la invención con erlotinib HCl y un componente polimérico estabilizante y formador de matriz. Se logran mejoras particulares con el componente polimérico estabilizante y formador de matriz succinato de acetato de hidroxipropilmetilcelulosa (HPMC-AS). Los experimentos 65-66 y 72 muestran que se obtiene un aumento en la solubilidad con las nanopartículas híbridas producidas por los procedimientos de la invención con erlotinib HCl y un componente polimérico estabilizante y formador de matriz, donde se añade un solubilizante separado. Se logran mejoras mediante la adición de un solubilizante separado, donde dicho solubilizante se seleccionado de entre copolímero de polivinil caprolactama-acetato de polivinilo-polietilenglicol (Soluplus) y succinato de d-a-tocoferol polietilenglicol 1000 (TPGS). Esta mejora no se observó cuando se incorporó el solubilizante a las nanopartículas híbridas producidas por los procedimientos de la invención.
Las mezclas físicas de erlotinib HCl con un solubilizante y/o HPMC AS también mejoran la solubilidad en FaSSIF (experimentos 59, 60-61, 62-63) pero no en FeSSIF (experimento 69-72). Un problema con la formulación de IPC es el efecto de los alimentos. Varios de los IPC están indicados para su administración en estado de ayuno a pesar de que los alimentos en la mayoría de los casos aumentan su biodisponibilidad. La baja biodisponibilidad podría explicar en parte los problemas digestivos asociados con los IPC. Los datos indican que las nanopartículas híbridas producidas por los procedimientos de la invención pueden reducir el efecto de los alimentos y los problemas digestivos del paciente por su mejora igual en la solubilidad tanto en FaSSIF como en FeSSIF (experimentos 76/77 y 82/83) que además pueden permitir reducir potencialmente la dosis. Por tanto, las nanopartículas híbridas amorfas estables producidas por los procedimientos de la invención se pueden administrar junto con la ingesta de alimentos.
Comparativa
Ejemplo 3. Nanopartículas híbridas amorfas estables con pazopanib: solubilidad a pH 6,5 y pH 5
Se llevó a cabo un número de experimentos, donde pazopanib representaba el IPC. Los experimentos se llevaron a cabo midiendo la concentración de IPC (mg/l) después de 5, 30 y 90 minutos de disolución en una solución a aproximadamente pH 6,5, a saber, FaSSIF (fluido intestinal simulado en estado de ayuno). Además, se llevaron a cabo experimentos en una solución alternativa a aproximadamente pH 5, a saber, FeSSIF (fluido intestinal simulado en estado alimentado). Se tomaron muestras de la solución a varios intervalos de tiempo y se midió la cantidad de IPC mediante el ensayo de medición de la disolución descrito anteriormente.
Los resultados representativos en la solución FaSSIF se proporcionan a continuación en la tabla 13 y 14, donde la Tabla 13 proporciona datos de concentración de pazopanib (mg/l) después de 5, 30 y 90 minutos de disolución, mientras que la tabla 14 proporciona datos del % de pazopanib solubilizado después de 30 minutos de disolución, el área bajo la curva (ABC: mg/min/l) durante 90 minutos de disolución y el aumento del ABC con las nanopartículas híbridas producidas por los procedimientos de la invención, en comparación con pazopanib en forma cristalina cruda añadido a la solución (experimentos 84-93). En las tablas 15 y 16, se proporcionan datos de disolución en la solución FeSSIF, presentados de manera similar a la tabla 13 y 14 (experimentos 94-101). En la tabla 17, se presentan los datos de un experimento comparativo con nanopartículas híbridas similares producidas por los procedimientos de la invención, llevado a cabo en FaSSIF y FeSSIF, respectivamente (experimentos 102-109). La tabla 18 presenta datos comparativos adicionales para experimentos llevados a cabo en FaSSIF y FeSSIF, respectivamente, con nanopartículas híbridas producidas por los procedimientos de la invención.
Tabla 13. Pazopanib: concentración de pazopanib (mg/l) después de 5, 30 y 90 minutos de disolución en solución FaSSIF H 6,5


Tabla 14. Porcentaje de pazopanib solubilizado después de 30 minutos de disolución, el área bajo la curva (ABC: mg/min/l) durante 90 minutos de disolución y el aumento del ABC con nanopartículas híbridas amorfas estables producidas por los procedimientos de la invención, en comparación con pazopanib en forma cristalina cruda añadido a la solución FaSSIF H 6,5


Tabla 15. Pazopanib: concentración de pazopanib (mg/l) después de 5, 30 y 90 minutos de disolución en solución FeSSIF H 5

Tabla 16. Porcentaje de pazopanib solubilizado después de 30 minutos de disolución, el área bajo la curva (ABC: mg/min/l) durante 90 minutos de disolución y el aumento del ABC con nanopartículas híbridas amorfas estables producidas por los procedimientos de la invención, en comparación con pazopanib crudo en forma cristalina añadido a la solución FeSSIF H 5


Tabla 17. Pazopanib: concentración de pazopanib después de 5, 30 y 90 minutos de disolución en solución FaSSIF FeSSIF, resectivamente

Tabla 18. Pazopanib: concentración de pazopanib (mg/l) después de 5, 30 y 90 minutos de disolución en solución F IF F IF r ivm n r r n m m r iv


Conclusiones Ejemplo 3
Los experimentos muestran que se obtiene un aumento en la solubilidad con las nanopartículas híbridas amorfas estables producidas por los procedimientos de la invención con pazopanib y un componente polimérico estabilizante y formador de matriz. Se logran mejoras particulares con el componente estabilizante polimérico y formador de matriz polivinilpirrolidona K-90 (PVP 90K). Los experimentos 91-92 muestran que se obtiene un aumento en la solubilidad con las nanopartículas híbridas producidas por los procedimientos de la invención con pazopanib y un componente polimérico estabilizante y formador de matriz, donde se añade un solubilizante separado. Se logran mejoras mediante la adición de un solubilizante separado, donde dicho solubilizante se seleccionado de entre copolímero de polivinil caprolactama-acetato de polivinilo-polietilenglicol (Soluplus) y succinato de d-a-tocoferol polietilenglicol 1000 (TPGS). Esta mejora no se observó cuando se incorporó el solubilizante a las nanopartículas híbridas producidas por los procedimientos de la invención.
Los resultados llevados a cabo en FaSSIF y FeSSIF, respectivamente, indican que las nanopartículas híbridas producidas por los procedimientos de la invención proporcionan un aumento similar en la solubilidad. Un problema con la formulación de IPC es el efecto de los alimentos. Varios de los IPC están indicados para su administración en estado de ayuno a pesar de que los alimentos en la mayoría de los casos aumentan su biodisponibilidad. La baja biodisponibilidad podría explicar en parte los problemas digestivos asociados con los IPC. La velocidad de disolución similar en FaSSIF y FeSSIF indica que las nanopartículas híbridas producidas por los procedimientos de la invención pueden reducir el efecto de los alimentos y los problemas digestivos del paciente por su mejora igual en la solubilidad tanto en FaSSIF como en FeSSIF (experimentos 89/100 y 104/105) que además permiten reducir la dosis. Por tanto, las nanopartículas híbridas amorfas estables producidas por los procedimientos de la invención se pueden administrar junto con la ingesta de alimentos.
Comparativa
Ejemplo 4. Nanopartículas híbridas amorfas estables con lapatinib:
solubilidad a pH 6,5
Se llevó a cabo un número de experimentos, donde base de lapatinib o sal de lapatinib ditosilato representaba el IPC. Los experimentos se llevaron a cabo midiendo la concentración de IPC (mg/l) después de 5, 30 y 90 minutos de disolución en una solución a aproximadamente pH 6,5, a saber, FaSSIF (fluido intestinal simulado en estado de ayuno). Se tomaron muestras de la solución a varios intervalos de tiempo y se midió la cantidad de IPC mediante el ensayo de medición de la disolución descrito anteriormente.
Los resultados representativos en la solución FaSSIF se proporcionan a continuación en la tabla 19 y 20, donde la Tabla 19 proporciona datos de concentración de lapatinib (mg/l) después de 5, 30 y 90 minutos de disolución, mientras que la tabla 20 proporciona datos del % de lapatinib solubilizado después de 30 minutos de disolución, el área bajo la curva (ABC: mg/min/l) durante 90 minutos de disolución y el aumento del ABC con las nanopartículas híbridas producidas por los procedimientos de la invención, en comparación con sal de lapatinib ditosilato no formulada añadida
a la solución (experimentos 110-126).
Tabla 19. Lapatinib: concentración de lapatinib (mg/l) después de 5, 30 y 90 minutos de disolución en solución FaSSIF H 6,5


Tabla 20. Porcentaje de lapatinib solubilizado después de 30 minutos de disolución, el área bajo la curva (ABC: mg/min/l) durante 90 minutos de disolución y el aumento del ABC con nanopartículas híbridas amorfas estables producidas por los procedimientos de la invención, en comparación con sal de lapatinib ditosilato no formulada añadida a la solución FaSSIF H 6,5


Conclusiones Ejemplo 4
Los experimentos 122-125 muestran claramente que se obtiene un aumento en la solubilidad con nanopartículas híbridas amorfas estables producidas por los procedimientos de la invención, con lapatinib, en particular base de lapatinib y un componente polimérico estabilizante y formador de matriz, donde se añade un solubilizante separado a la composición. Se logran mejoras particulares con el componente estabilizante polimérico y formador de matriz hidroxipropilcelulosa EF e hidroxipropilcelulosa LF. Además, se logran mejoras mediante la adición de un solubilizante separado, donde dicho solubilizante se seleccionado de entre copolímero de polivinil caprolactama-acetato de polivinilo-polietilenglicol (Soluplus) y succinato de d-a-tocoferol polietilenglicol 1000 (TPGS).
Comparativa
Ejemplo 5. Nanopartículas híbridas amorfas estables con nilotinib HCl: solubilidad a pH 1,4
Se llevó a cabo un número de experimentos, donde nilotinib HCl representaba el IPC. Los experimentos se llevaron a cabo midiendo la concentración de IPC (mg/l) después de 5, 30 y 90 minutos de disolución en una solución a aproximadamente pH 1,4, a saber, SGF (fluido gástrico simulado). Se tomaron muestras de la solución a varios intervalos de tiempo y se midió la cantidad de IPC mediante el ensayo de medición de la disolución descrito anteriormente.
Los resultados representativos en la solución SGF se proporcionan a continuación en la tabla 21, que proporciona el porcentaje de nilotinib HCl solubilizado procedente tanto de una mezcla física con nilotinib HCl en forma cristalina cruda como de nanopartículas híbridas producidas por los procedimientos de la invención después de 5, 30 y 90 minutos de disolución. El nilotinib presente en la mezcla física de nilotinib HCl crudo con el componente polimérico estabilizante y formador de matriz PVAP y el solubilizante Soluplus (Exp.129) se disuelve completamente en 5 minutos en SGF, mientras que nilotinib solo se disuelve parcialmente después de 90 minutos en SGF con nanopartículas híbridas producidas por los procedimientos de la invención, donde los componentes están comprendidos como nanopartículas híbridas, con la adición de un solubilizante (Exp. 128) o sin la adición de un solubilizante (Exp. 127).
Conclusiones Ejemplo 5
Los experimentos 127-129 muestran que un nilotinib HCl en nanopartículas híbridas amorfas estables producidas por los procedimientos de la invención (exp 127 y 128) se solubilizan parcialmente a pH 1,4. Las nanopartículas híbridas amorfas estables producidas por los procedimientos con un polímero de recubrimiento entérico tal como PVAP están protegidas parcialmente del entorno ácido.
Comparativa
Ejemplo 6. Nanopartículas híbridas amorfas estables con gefitinib: solubilidad a pH 6,5
Se llevó a cabo un número de experimentos, donde gefitinib representaba el IPC. Los experimentos se llevaron a cabo midiendo la concentración de IPC (mg/l) después de 3, 40 y 80 minutos de disolución en una solución a aproximadamente pH 6,5, a saber, FaSSIF (fluido intestinal simulado en estado de ayuno). Se tomaron muestras de la solución a varios intervalos de tiempo y se midió la cantidad de IPC mediante el ensayo de medición de la disolución descrito anteriormente.
Los resultados representativos en la solución FaSSIF se proporcionan a continuación en la tabla 22 y 23, donde la Tabla 22 proporciona datos de concentración de gefitinib (mg/l) después de 3, 40 y 80 minutos de disolución, mientras que la tabla 23 proporciona datos del % de gefitinib solubilizado después de 40 minutos de disolución, el área bajo la curva (ABC: mg/min/l) durante 80 minutos de disolución y el aumento del ABC con las nanopartículas híbridas producidas por los procedimientos de la invención, en comparación con gefitinib no formulado añadido a la solución (experimentos 131-137).
Tabla 22. Gefitinib: concentración de gefitinib (mg/l) después de 3, 40 y 80 minutos de disolución en solución FaSSIF
H 6,5

Tabla 23. Porcentaje de gefitinib solubilizado después de 40 minutos de disolución, el área bajo la curva (ABC: mg/min/l) durante 80 minutos de disolución y el aumento del ABC con nanopartículas híbridas amorfas estables producidas por los procedimientos de la invención, en comparación con gefitinib no formulado añadido a la solución FaSSIF H 6,5


Los experimentos 131-137 muestran que se obtiene un aumento en la solubilidad con nanopartículas híbridas amorfas estables producidas por los procedimientos de la invención, con gefitinib, en particular gefitinib y un componente polimérico estabilizante y formador de matriz, donde se añade un solubilizante separado a la composición. Se logran mejoras particulares con el componente estabilizante polimérico y formador de matriz polivinilpirrolidona K-30 (PVP 30K) y ftalato de hidroxipropilmetilcelulosa (HPMCP HP55). Además, las mejoras se logran mediante la adición de un solubilizante separado añadido, donde dicho solubilizante es copolímero de polivinil caprolactama-acetato de polivinilo-polietilenglicol (Soluplus).
Ejemplo 7. Nanopartículas híbridas amorfas estables con dasatinib: solubilidad a pH 6,5
Se llevó a cabo un número de experimentos, donde dasatinib representaba el IPC. Los experimentos se llevaron a cabo midiendo la concentración de IPC (mg/l) después de 3, 40 y 80 minutos de disolución en una solución a aproximadamente pH 6,5, a saber, FaSSIF (fluido intestinal simulado en estado de ayuno). Se tomaron muestras de la solución a varios intervalos de tiempo y se midió la cantidad de IPC mediante el ensayo de medición de la disolución descrito anteriormente.
Los resultados representativos en la solución FaSSIF se proporcionan a continuación en la tabla 24 y 25, donde la Tabla 24 proporciona datos de concentración de dasatinib (mg/l) después de 3, 40 y 80 minutos de disolución, mientras que la tabla 25 proporciona datos del % de dasatinib solubilizado después de 40 minutos de disolución, el área bajo la curva (ABC: mg/min/l) durante 80 minutos de disolución y el aumento del ABC con las nanopartículas híbridas producidas por los procedimientos de la invención, en comparación con dasatinib no formulado añadido a la solución (experimentos 138-141).
Tabla 24. Dasatinib: concentración de dasatinib (mg/l) después de 3, 40 y 80 minutos de disolución en solución FaSSIF H 6,5

Tabla 25. Porcentaje de dasatinib solubilizado después de 40 minutos de disolución, el área bajo la curva (ABC: mg/min/l) durante 80 minutos de disolución y el aumento del ABC con nanopartículas híbridas amorfas estables producidas por los procedimientos de la invención, en comparación con dasatinib no formulado añadido a la solución FaSSIF H 6,5

Los experimentos 138-141 muestran que se obtiene un aumento en la solubilidad con nanopartículas híbridas amorfas estables producidas por los procedimientos de la invención, con dasatinib, en particular dasatinib y un componente polimérico estabilizante y formador de matriz, donde se añade un solubilizante separado a la composición. Se logran mejoras particulares con el componente polimérico estabilizante y formador de matriz copolividona (Kollidon VA64). Además, las mejoras se logran mediante la adición de un solubilizante separado añadido, donde dicho solubilizante es copolímero de polivinil caprolactama-acetato de polivinilo-polietilenglicol (Soluplus).
Comparativa
Ejemplo 8. Nanopartículas híbridas amorfas estables con sorafenib tosilato: solubilidad a pH 6,5
Se llevó a cabo un número de experimentos, donde sorafenib tosilato representaba el IPC. Los experimentos se llevaron a cabo midiendo la concentración de IPC (mg/l) después de 3, 40 y 80 minutos de disolución en una solución a aproximadamente pH 6,5, a saber, FaSSIF (fluido intestinal simulado en estado de ayuno). Se tomaron muestras de la solución a varios intervalos de tiempo y se midió la cantidad de IPC mediante el ensayo de medición de la disolución descrito anteriormente.
Los resultados representativos en la solución FaSSIF se proporcionan a continuación en la tabla 26 y 27, donde la Tabla 26 proporciona datos de concentración de sorafenib (mg/l) después de 3, 40 y 80 minutos de disolución, mientras que la tabla 27 proporciona datos del % de sorafenib solubilizado después de 40 minutos de disolución, el área bajo la curva (ABC: mg/min/l) durante 80 minutos de disolución y el aumento del ABC de las composiciones, en comparación con sorafenib tosilato no formulado añadido a la solución (experimentos 142-145).
Tabla 26. Sorafenib tosilato: concentración de sorafenib (mg/l) después de 3, 40 y 80 minutos de disolución en solución FaSSIF H 6,5

Tabla 27. Porcentaje de sorafenib solubilizado después de 40 minutos de disolución, el área bajo la curva (ABC: mg/min/l) durante 80 minutos de disolución y el aumento del ABC con nanopartículas híbridas amorfas estables producidas por los procedimientos de la invención, en comparación con sorafenib tosilato no formulado añadido a la solución FaSSIF H 6,5

Los experimentos 138-141 muestran que se obtiene un aumento en la solubilidad con nanopartículas híbridas amorfas estables producidas por los procedimientos de la invención, con dasatinib, en particular dasatinib y un componente polimérico estabilizante y formador de matriz, donde se añade un solubilizante separado a la composición. Se logran mejoras particulares con el componente estabilizante polimérico y formador de matriz ftalato de hidroxipropilmetilcelulosa (HPMCP HP55). Además, las mejoras se logran mediante la adición de un solubilizante separado añadido, donde dicho solubilizante es copolímero de polivinil caprolactama-acetato de polivinilopolietilenglicol (Soluplus).
Comparativa
Ejemplo 9. Nanopartículas híbridas amorfas estables con base de nilotinib: solubilidad a pH 6,5
Se llevó a cabo un número de experimentos, donde base de nilotinib representaba el IPC. Los experimentos se llevaron a cabo midiendo la concentración de IPC (mg/l) después de 3, 40 y 80 minutos de disolución en una solución a aproximadamente pH 6,5, a saber, FaSSIF (fluido intestinal simulado en estado de ayuno). Se tomaron muestras de la solución a varios intervalos de tiempo y se midió la cantidad de IPC mediante el ensayo de medición de la disolución descrito anteriormente.
Los resultados representativos en la solución FaSSIF se proporcionan a continuación en la tabla 28 y 29, donde la Tabla 28 proporciona datos de concentración de base de nilotinib (mg/l) después de 3, 40 y 80 minutos de disolución, mientras que la tabla 29 proporciona datos del % de base de nilotinib solubilizada después de 40 minutos de disolución, el área bajo la curva (ABC: mg/min/l) durante 80 minutos de disolución y el aumento del ABC de las composiciones, en comparación con base de nilotinib no formulada añadida la solución (experimentos 146-149).
Tabla 28. Base de nilotinib: concentración de base de nilotinib (mg/l) después de 3, 40 y 80 minutos de disolución en solución FaSSIF H 6,5


Tabla 29. Porcentaje de base de nilotinib solubilizada después de 40 minutos de disolución, el área bajo la curva (ABC: mg/min/l) durante 80 minutos de disolución y el aumento del ABC con nanopartículas híbridas amorfas estables producidas por los procedimientos de la invención, en comparación con base de nilotinib no formulada añadida a la solución FaSSIF H 6,5

Comparativa
Ejemplo 10. Nanopartículas híbridas amorfas estables con crizotinib: solubilidad a pH 6,5
Se llevó a cabo un número de experimentos, donde crizotinib representaba el IPC. Los experimentos se llevaron a cabo midiendo la concentración de IPC (mg/l) después de 3, 40 y 80 minutos de disolución en una solución a aproximadamente pH 6,5, a saber, FaSSIF (fluido intestinal simulado en estado de ayuno). Se tomaron muestras de la solución a varios intervalos de tiempo y se midió la cantidad de IPC mediante el ensayo de medición de la disolución descrito anteriormente. Los resultados representativos en la solución FaSSIF se proporcionan a continuación en la tabla 30 y 31, donde la Tabla 30 proporciona datos de concentración de crizotinib (mg/l) después de 3, 40 y 80 minutos de disolución, mientras que la tabla 31 proporciona datos del % de crizotinib solubilizado después de 40 minutos de disolución, el área bajo la curva (ABC: mg/min/l) durante 80 minutos de disolución y el aumento del ABC con las nanopartículas híbridas amorfas estables producidas por los procedimientos de la invención, en comparación con crizotinib no formulado añadido a la solución (experimentos 150-156).
Tabla 30. Crizotinib: concentración de crizotinib (mg/l) después de 3, 40 y 80 minutos de disolución en solución FaSSIF H 6,5


Tabla 31. Porcentaje de crizotinib solubilizado después de 40 minutos de disolución, el área bajo la curva (ABC: mg/min/l) durante 80 minutos de disolución y el aumento del ABC con nanopartículas híbridas amorfas estables producidas por los procedimientos de la invención, en comparación con crizotinib no formulado añadido a la solución FaSSIF H 6,5

Los experimentos 150-156 muestran que se obtiene un aumento en la solubilidad con nanopartículas híbridas amorfas estables producidas por los procedimientos de la invención, con crizotinib, en particular crizotinib y un componente polimérico estabilizante y formador de matriz, donde se añade un solubilizante separado a la composición. Se logran mejoras particulares con el componente polimérico estabilizante y formador de matriz polivinilpirrolidona K-30 (PVP 30K) y copolividona (Kollidon vA64). Además, se logran mejoras mediante la adición de un solubilizante separado, donde dicho solubilizante se selecciona de entre copolímero de polivinil caprolactama-acetato de polivinilopolietilenglicol (Soluplus) y aceite de ricino hidrogenado PEG-40 (Cremophor RH40).
Comparativa
Ejemplo 11. Nanopartículas híbridas amorfas estables con axitinib: solubilidad a pH 6,5
Se llevó a cabo un número de experimentos, donde axitinib representaba el IPC. Los experimentos se llevaron a cabo midiendo la concentración de IPC (mg/l) después de 3, 40 y 80 minutos de disolución en una solución a aproximadamente pH 6,5, a saber, FaSSIF (fluido intestinal simulado en estado de ayuno). Se tomaron muestras de la solución a varios intervalos de tiempo y se midió la cantidad de IPC mediante el ensayo de medición de la disolución descrito anteriormente. Los resultados representativos en la solución FaSSIF se proporcionan a continuación en la tabla 32 y 33, donde la Tabla 32 proporciona datos de concentración de axitinib (mg/l) después de 3, 40 y 80 minutos de disolución, mientras que la tabla 33 proporciona datos del % de axitinib solubilizado después de 40 minutos de disolución, el área bajo la curva (ABC: mg/min/l) durante 80 minutos de disolución y el aumento del ABC con las nanopartículas híbridas amorfas estables producidas por los procedimientos de la invención, en comparación con
axitinib no formulado añadido a la solución (experimentos 157-163).
Tabla 32. Axitinib: concentración de axitinib (mg/l) después de 3, 40 y 80 minutos de disolución en solución FaSSIF
H 6,5

Tabla 33. Porcentaje de axitinib solubilizado después de 40 minutos de disolución, el área bajo la curva (ABC: mg/min/l) durante 80 minutos de disolución y el aumento del ABC con nanopartículas híbridas amorfas estables producidas por los procedimientos de la invención, en comparación con axitinib no formulado añadido a la solución FaSSIF H 6,5

Los experimentos 157-163 muestran que se obtiene un aumento en la solubilidad con nanopartículas híbridas amorfas estables producidas por los procedimientos de la invención, con axitinib, en particular axitinib y un componente polimérico estabilizante y formador de matriz, donde se añade un solubilizante separado a la composición. Se logran mejoras particulares con el componente polimérico estabilizante y formador de matriz copolividona (Kollidon VA64) y succinato de acetato de hidroxipropilmetilcelulosa (HPMC AS). Además, las mejoras se logran mediante la adición de un solubilizante separado añadido, donde dicho solubilizante es copolímero de polivinil caprolactama-acetato de
polivinilo-polietilenglicol (Soluplus).
Comparativa
Ejemplo 12. Nanopartículas híbridas amorfas estables con vemurafenib: solubilidad a pH 6,5
Se llevó a cabo un número de experimentos, donde vemurafenib representaba el IPC. Los experimentos se llevaron a cabo midiendo la concentración de IPC (mg/l) después de 3, 40 y 80 minutos de disolución en una solución a aproximadamente pH 6,5, a saber, FaSSIF (fluido intestinal simulado en estado de ayuno). Se tomaron muestras de la solución a varios intervalos de tiempo y se midió la cantidad de IPC mediante el ensayo de medición de la disolución descrito anteriormente.
Los resultados representativos en la solución FaSSIF se proporcionan a continuación en la tabla 34 y 35, donde la Tabla 34 proporciona datos de concentración de vemurafenib (mg/l) después de 3, 40 y 80 minutos de disolución, mientras que la tabla 35 proporciona datos del % de vemurafenib solubilizado después de 40 minutos de disolución, el área bajo la curva (ABC: mg/min/l) durante 80 minutos de disolución y el aumento del ABC con las nanopartículas híbridas amorfas estables producidas por los procedimientos de la invención, en comparación con vemurafenib no formulado añadido a la solución (experimentos 164-170).
Tabla 34. Vemurafenib: concentración de vemurafenib (mg/l) después de 3, 40 y 80 minutos de disolución en solución FaSSIF H 6,5

Los experimentos 164-170 muestran que se obtiene un aumento en la solubilidad con nanopartículas híbridas amorfas estables producidas por los procedimientos de la invención, con vemurafenib, en particular vemurafenib y un componente polimérico estabilizante y formador de matriz, donde se añade un solubilizante separado a la composición. Se logran mejoras particulares con el componente polimérico estabilizante y formador de matriz copolividona (Kollidon VA64) y ftalato de acetato de celulosa (CAP). Además, las mejoras se logran mediante la adición de un solubilizante separado añadido, donde dicho solubilizante es copolímero de polivinil caprolactama-acetato de polivinilo-polietilenglicol (Soluplus).
Ejemplo 13. Medición de la velocidad de disolución en condiciones de baja concentración nanopartículas híbridas amorfas, estables, producidas por los procedimientos de la invención
La medición de la disolución en condiciones de baja concentración de nanopartículas híbridas amorfas estables producidas por los procedimientos de la invención se midieron en un procedimiento que consiste en añadir la cantidad deseada de polvo en un sistema de cubetas de lectura (SOTAX, Allschwill, Suiza), montar la cubeta en su aparato y, a continuación, bombear el medio apropiado (típicamente FaSSIF, FeSSIF, SGF) a través del polvo. La temperatura del aparato se fijó a 37 °C. La cantidad de polvo añadida a la cubeta depende de la carga de fármaco del polvo: la cantidad exacta de polvo se calculó a partir de los resultados obtenidos del análisis de la carga de fármaco de los polvos.
Típicamente, se añadieron de 3,5 a 7 mg del IPC a la cubeta de lectura y se bombeó a un caudal de entre 8 y 16 ml de medio/min (preferiblemente, aproximadamente 8 ml de medio/min) a través del polvo. Se recogieron muestras de un ml del medio que pasa a través la cubeta de lectura en momentos predeterminados. Estas muestras se analizaron mediante HPLC (por ejemplo, columna C18 Eclipse, 4,6 mm x 15 cm, 1 ml/min, detección 254-400 nm). Las muestras se tomaron después de 0, 0,5, 1, 1,5, 2, 3, 4, 5, 6, 7, 8, 9, 10, 15, 20, 25, 30, 35 y 40 minutos desde el momento en que sale el medio de la cubeta de lectura. El % acumulado de solubilizado de la cantidad de sustancia activa añadida en la cubeta de lectura se calculó y representó frente al tiempo (min). Se estimó la pendiente inicial ("velocidad de disolución inicial") del gráfico, según se midió durante 0-10 minutos, y se tomó la velocidad de disolución del material en condiciones de baja concentración a 37 °C en el medio de disolución dado.
Cada experimento comprende una comparación entre el IPC en forma bruta con nanopartículas híbridas amorfas estables producidas por los procedimientos de la invención con el inhibidor y un componente polimérico estabilizante y formador de matriz representativo.
Comparativa
Ejemplo 13,1. Medición de la velocidad de disolución en condiciones de baja concentración con nanopartículas híbridas amorfas, estables, producidas por los procedimientos de la invención, que comprenden nilotinib HCl
En los experimentos con nilotinib HCl, se pesaron 4 mg en la cubeta de lectura (experimento 500) y se compararon con nanopartículas híbridas amorfas estables producidas por los procedimientos de la invención con base de nilotinib y el componente polimérico estabilizante y formador de matriz HPMCP HP55 (experimento 501). Los resultados se representan en la tabla 36 a continuación.
T l . Nil ini H l n n i i n n nr i n n F IF


Los experimentos 500-501 muestran que la velocidad de disolución inicial de las nanopartículas híbridas amorfas estables producidas por los procedimientos de la invención, con base de nilotinib, es superior a la velocidad de disolución inicial de nilotinib HCl en forma cristalina cruda.
Comparativa
Ejemplo 13,2. Medición de la velocidad de disolución de condiciones de baja concentración con nanopartículas híbridas amorfas, estables, producidas por los procedimientos de la invención, que comprenden erlotinib HCl
En los experimentos con erlotinib HCl, se pesaron 3,5 mg en la cubeta de lectura (experimento 510) y se compararon con nanopartículas híbridas amorfas estables producidas por los procedimientos de la invención con erlotinib HCl y el componente polimérico estabilizante y formador de matriz HPMCP AS (experimento 511). Los resultados se representan en la tabla 37 a continuación.
T l 7. Erl ini H l n n i i n n nr i n n F IF


Los experimentos 510-511 muestran que la velocidad de disolución inicial de las nanopartículas híbridas amorfas estables producidas por los procedimientos de la invención, con erlotinib HCl, es superior a la velocidad de disolución inicial de erlotinib HCl en forma cristalina cruda.
Comparativa
Ejemplo 13,3. Medición de la velocidad de disolución de condiciones de baja concentración con nanopartículas híbridas amorfas, estables, producidas por los procedimientos de la invención, que comprenden pazopanib HCl
En los experimentos con pazopanib HCl, se pesaron 3,5 mg en la cubeta de lectura (experimento 520) y se compararon con nanopartículas híbridas amorfas estables producidas por los procedimientos de la invención con pazopanib HCl y el componente polimérico estabilizante y formador de matriz PVP90K (experimento 521). Los resultados se representan en la tabla 38 a continuación.
Tabla 38. Pazo anib HCl en condiciones de baa concentración en FaSSIF


Los experimentos 520-521 muestran que la velocidad de disolución inicial de las nanopartículas híbridas amorfas estables producidas por los procedimientos de la invención, con pazopanib HCl, es superior a la velocidad de disolución inicial de pazopanib HCl en forma cristalina cruda.
Comparativa
Ejemplo 13,4. Medición de la velocidad de disolución de condiciones de baja concentración con nanopartículas híbridas amorfas, estables, producidas por los procedimientos de la invención, que comprenden lapatinib ditosilato
En los experimentos con lapatinib ditosilato, se pesaron 4 mg en la cubeta de lectura (experimento 530) y se compararon con nanopartículas híbridas amorfas estables producidas por los procedimientos de la invención con base de lapatinib y el componente polimérico estabilizante y formador de matriz HPC If (experimento 531). Los resultados se representan en la tabla 39 a continuación.
T l . L ini i il n n i i n n nr i n n F IF


Los experimentos 530-531 muestran que la velocidad de disolución inicial de las nanopartículas híbridas amorfas estables producidas por los procedimientos de la invención, con base de lapatinib, es superior a la velocidad de disolución inicial de lapatinib ditosilato en forma cristalina cruda.
Comparativa
Ejemplo 13,5. Medición de la velocidad de disolución de condiciones de baja concentración con nanopartículas híbridas amorfas, estables, producidas por los procedimientos de la invención, que comprenden gefitinib
En los experimentos con gefitinib, se pesaron 3,5 mg en la cubeta de lectura (experimento 540) y se compararon con nanopartículas híbridas amorfas estables producidas por los procedimientos de la invención con gefitinib y el componente polimérico estabilizante y formador de matriz HPMCP HP55 (experimento 541). Los resultados se representan en la tabla 40 a continuación.
T l 4 . fiini n n i i n n nr i n n F IF

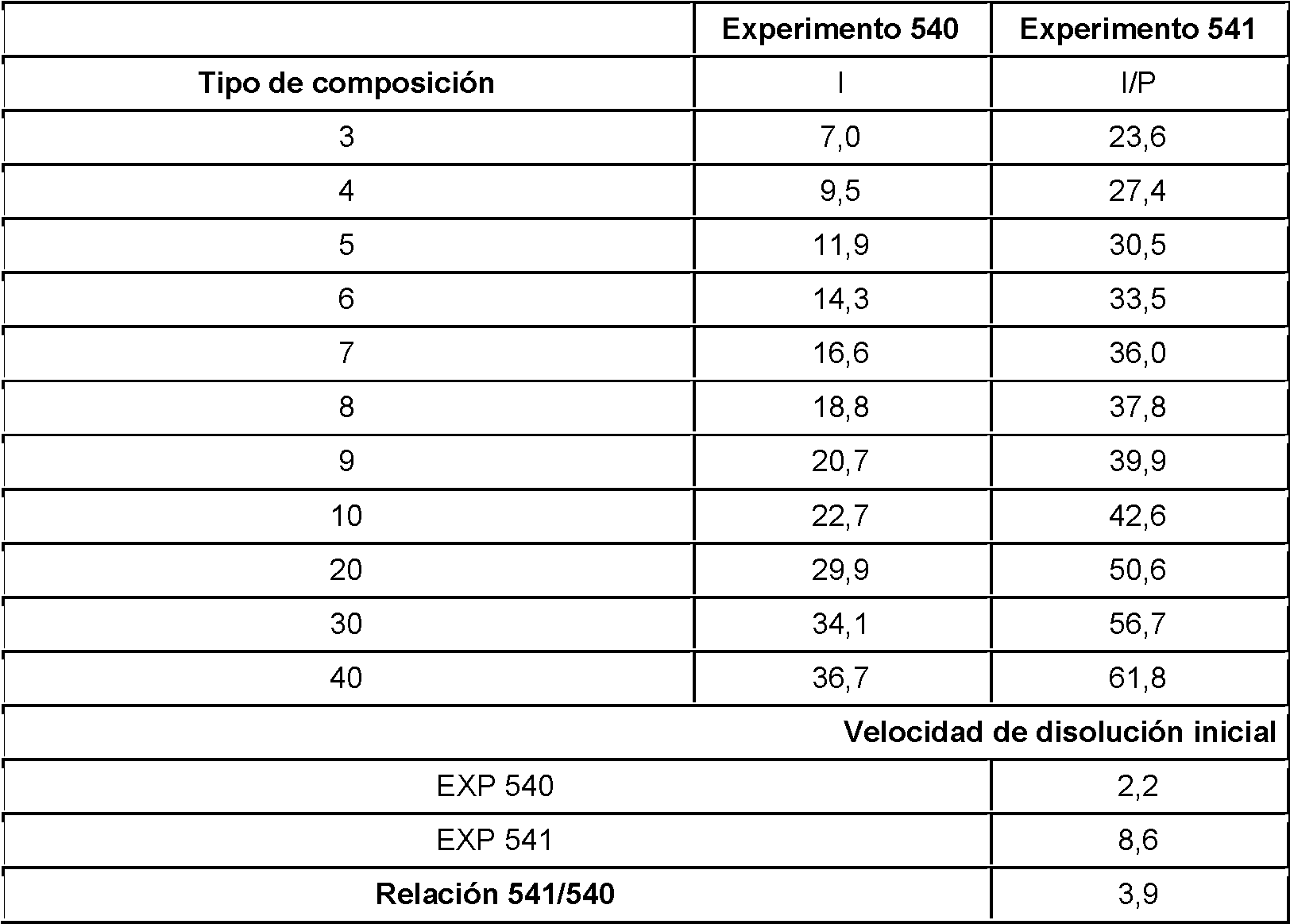
Los experimentos 540-541 muestran que la velocidad de disolución inicial de las nanopartículas híbridas amorfas estables producidas por los procedimientos de la invención, con gefitinib, es superior a la velocidad de disolución inicial de gefitinib en forma cristalina cruda.
Ejemplo 13,6. Medición de la velocidad de disolución de condiciones de baja concentración con nanopartículas híbridas amorfas, estables, producidas por los procedimientos de la invención, que comprenden dasatinib
En los experimentos con dasatinib, se pesaron 3,5 mg en la cubeta de lectura (experimento 550) y se compararon con nanopartículas híbridas amorfas estables producidas por los procedimientos de la invención con dasatinib y el componente polimérico estabilizante y formador de matriz copolividona Kollidon VA64 (experimento 551). Los resultados se representan en la tabla 41 a continuación.
T l 41. D ini n n i i n n nr i n n F IF


Los experimentos 550-551 muestran que la velocidad de disolución inicial de las nanopartículas híbridas amorfas estables producidas por los procedimientos de la invención, con dasatinib, es superior a la velocidad de disolución inicial de dasatinib en forma cristalina cruda.
Comparativa
Ejemplo 13,7. Medición de la velocidad de disolución de condiciones de baja concentración con nanopartículas híbridas amorfas, estables, producidas por los procedimientos de la invención, que comprenden sorafenib ditosilato
En los experimentos con sorafenib ditosilato, se pesaron 3,5 mg en la cubeta de lectura (experimento 560) y se compararon con nanopartículas híbridas amorfas estables producidas por los procedimientos de la invención con sorafenib ditosilato y el componente polimérico estabilizante y formador de matriz HPMCP HP55 (experimento 561). Los resultados se representan en la tabla 42 a continuación.
T l 42. r f ni i il n n i i n n nr i n n F IF


Los experimentos 560-561 muestran que la velocidad de disolución inicial de las nanopartículas híbridas amorfas estables producidas por los procedimientos de la invención, con sorafenib ditosilato, es superior a la velocidad de disolución inicial de sorafenib ditosilato en forma cristalina cruda.
Comparativa
Ejemplo 13,8. Medición de la velocidad de disolución de condiciones de baja concentración con nanopartículas híbridas amorfas, estables, producidas por los procedimientos de la invención, que comprenden crizotinib
En los experimentos con crizotinib, se pesaron 3,5 mg en la cubeta de lectura (experimento 570) y se compararon con nanopartículas híbridas amorfas estables producidas por los procedimientos de la invención con crizotinib y el componente polimérico estabilizante y formador de matriz PVP 30k (experimento 571). Los resultados se representan en la tabla 43 a continuación.
T l 4 . riz ini n n i i n n nr i n n F IF


Los experimentos 570-571 muestran que la velocidad de disolución inicial de las nanopartículas híbridas amorfas estables producidas por los procedimientos de la invención, con crizotinib, es superior a la velocidad de disolución inicial de crizotinib en forma cristalina cruda.
Comparativa
Ejemplo 13,9. Medición de la velocidad de disolución de condiciones de baja concentración con nanopartículas híbridas amorfas, estables, producidas por los procedimientos de la invención, que comprenden axitinib
En los experimentos con axitinib, se pesaron 3,5 mg en la cubeta de lectura (experimento 580) y se compararon con nanopartículas híbridas amorfas estables producidas por los procedimientos de la invención con axitinib y el componente polimérico estabilizante y formador de matriz Kollidon VA64 (experimento 581) o HPMCP AS (experimento 582). Los resultados se representan en la tabla 44 a continuación.
T l 44. Axiini n n i i n n nr i n n F IF


Los experimentos 580-582 muestran que la velocidad de disolución inicial de las nanopartículas híbridas amorfas estables producidas por los procedimientos de la invención, con axitinib, es superior a la velocidad de disolución inicial de axitinib en forma cristalina cruda.
Comparativa
Ejemplo 13.10. Medición de la velocidad de disolución en condiciones de baja concentración de nanopartículas híbridas amorfas, estables, producidas por los procedimientos de la invención que comprende vemurafenib
En los experimentos con vemurafenib, se pesaron 3,5 mg en la cubeta de lectura (experimento 590) y se compararon con nanopartículas híbridas amorfas estables producidas por los procedimientos de la invención con vemurafenib y el componente polimérico estabilizante y formador de matriz Kollidon VA64 (experimento 591) o CAP (experimento 592). Los resultados se representan en la tabla 45 a continuación.
Tabla 45. Vemurafenib en condiciones de baa concentración en FaSSIF

Los experimentos 590-592 muestran claramente que la velocidad de disolución inicial de las nanopartículas híbridas
amorfas estables producidas por los procedimientos de la invención, con vemurafenib, es superior a la velocidad de disolución inicial de vemurafenib en forma cristalina cruda.
Comparativa
Ejemplo 14. Medición
in vivo
de las concentraciones plasmáticas después de la administración oral de nanopartículas híbridas amorfas estables producidas por los procedimientos de la invención
Grupos de cuatro perros beagle macho recibieron dosis orales únicas (5 mg/kg) de composiciones en cápsulas que comprenden nanopartículas híbridas amorfas estables producidas por los procedimientos de la invención con base de nilotinib y cualquiera de los componentes poliméricos estabilizantes y formadores de matriz PVAP o HPMCP HP55, opcionalmente, con la adición del copolímero solubilizante de polivinil caprolactama-acetato de polivinilopolietilenglicol, y se compara con una formulación comercializada que comprende nilotinib HCl. Las nanopartículas híbridas amorfas estables sometidas a prueba son las que se establecen en los experimentos 146-149, en el ejemplo 9. El contenido del estómago de los perros se neutralizó con una solución de bicarbonato de sodio 5 min antes de la dosificación de la cápsula o se acidificó con un tampón HCl-KCl 10 min antes de la dosis. Un grupo de perros también recibió una dosis única intravenosa (1 mg/kg) de nilotinib. Se determinaron las concentraciones plasmáticas de nilotinib con un procedimiento de LC-MS/MS selectivo. No se observaron efectos secundarios en ningún animal estudiado. Resultados y conclusiones
Los perfiles de concentración plasmática-tiempo promedio ± SEM de base de nilotinib se muestran en las figuras 22 25, y los parámetros farmacocinéticos y los resultados se muestran en las tablas 46A y 46B.
Se calcularon los valores atípicos y se excluyeron en función de si un valor es un valor atípico significativo con respecto al resto a intervalos de confianza del 95 % (alfa = 5 %) según la prueba de Grubb. El valor crítico Z para la prueba de Grubb en el intervalo de confianza del 95 % con n = 4 es 1,48. Z = (Promedio - Valor cuestionable)/DT
Tabla 46A. Datos farmacocinéticos tras la administración oral única de diferentes composiciones de nilotinib que comprenden nanopartículas híbridas amorfas estables producidas por los procedimientos de la invención, en perros macho
de AP

Los valores se dan como promedio ± DT, excepto para T A de la formulación de nilotinib comercializada que se administra con un estómago demasiado ácido, donde solo se obtuvieron dos valores.
Los datos intravenosos (IV) se obtuvieron mediante infusión IV a velocidad constante de 1 mg/kg, de una solución de nilotinib a 0,2 mg/ml, en una HPpCD al 10 %, ajuste de pH a pH de 3,3 a 3,5. Co: 511 ± 46 ng/ml; TA: 3,3 ± 1,8 h; ABC0-24 h: 1000 ± 300 ng*h/ml.
Tabla 46B. Datos armacocin ticos tras la administraci n oral nica de dierentes ormulaciones de nilotinib que comprenden nanopartículas híbridas amorfas estables producidas por los procedimientos de la invención, en perros macho

Los valores se dan como promedio ± DT
Los datos intravenosos (IV) se obtuvieron mediante infusión IV a velocidad constante de 1 mg/kg, de una solución de nilotinib a 0,2 mg/ml, en una HPpCD al 10 %, ajuste de pH a pH de 3,3 a 3,5. Co: 511 ± 46 ng/ml; T / : 3,3 ± 1,8 h; ABC0-24h: 1000 ± 300 ng*h/ml.
La formulación de nilotinib comercializada administrada a un estómago acidificado mostró concentraciones plasmáticas aproximadamente 2 veces más altas que las de la misma formulación administrada a un estómago neutralizado. Ambas formulaciones que comprenden nanopartículas híbridas amorfas estables producidas por los procedimientos de la invención con base de nilotinib con PVAP y HPMCP HP55 como componentes poliméricos estabilizantes y formadores de matriz mostraron mejoras significativas en la exposición al plasma, con concentraciones plasmáticas aproximadamente 2 veces más altas que las de la formulación comercializada administrada a un estómago acidificado. Además, la combinación de nanopartículas híbridas producidas por los procedimientos de la invención podría dar una exposición al plasma que sea más o menos independiente del pH del estómago.
Se observaron mejoras adicionales en la disponibilidad oral cuando se combinaron formulaciones con nanopartículas híbridas amorfas estables producidas por los procedimientos de la invención con el copolímero solubilizante de polivinil caprolactama-acetato de polivinilo-polietilenglicol. Por tanto, las nanopartículas híbridas producidas por los procedimientos de la invención con base de nilotinib con PVAP y HPMCP HP55 como componentes poliméricos estabilizantes y formadores de matriz, donde se añadió y se administró el copolímero solubilizante de polivinil caprolactama-acetato de polivinilo-polietilenglicol a un estómago acidificado dio lugar a concentraciones plasmáticas de 2,3 a 3,1 veces más altas que las de la formulación comercializada. En este estudio, se logró una alta biodisponibilidad oral con las nanopartículas híbridas amorfas estables producidas por los procedimientos de la invención con base de nilotinib con HPMCP HP55 como componentes poliméricos estabilizantes y formadores de matriz, donde se añadió el copolímero solubilizante de polivinil caprolactama-acetato de polivinilo-polietilenglicol (I/P+S) y se administró al contenido del estómago neutralizado. En este caso, la exposición aumentó aproximadamente 7 veces con respecto a la de la formulación oral comercializada administrada en las mismas condiciones neutralizadas. En este estudio, la biodisponibilidad más alta, 36 ± 24 %, se logró cuando se administraron nanopartículas híbridas amorfas estables producidas por los procedimientos de la invención con base de nilotinib con PVAP como componentes poliméricos estabilizantes y formadores de matriz a un estómago neutralizado. Sin embargo, este grupo de estudio también vino acompañado de la desviación típica más alta del estudio.
Hubo una mejora en el rendimiento in vivo de las formulaciones innovadoras de nilotinib con las nanopartículas híbridas amorfas estables producidas por los procedimientos de la invención, que se basan en mejorar la absorción y biodisponibilidad mediante la optimización de la propiedades en estado sólido de la forma galénica. Los resultados del estudio in vivo en perros pueden predecir propiedades de absorción similares de nanopartículas híbridas producidas por los procedimientos de la invención, en pacientes, ya que parece haber una estrecha correlación en los procesos de absorción de fármacos gastrointestinales entre perros y humanos (Persson, E.M. y col., Pharm. Res. 2005, 22, 2141-2151). Las nanopartículas híbridas producidas por los procedimientos de la invención, con propiedades de absorción ventajosas, también predicen que las dosis orales usadas en la práctica clínica hoy en día se pueden reducir. Además, las nanopartículas híbridas amorfas estables producidas por los procedimientos de la invención pueden provocar una menor dependencia del pH en la absorción y biodisponibilidad de los IPC.
Comparativa
Ejemplo 15. Medición del grado/nivel de estabilidad de las nanopartículas híbridas amorfas estables producidas por los procedimientos de la invención
En pruebas de estabilidad de las nanopartículas híbridas amorfas estables producidas por los procedimientos de la
invención, se demostró que las partículas eran estables durante al menos 11 meses a temperatura ambiente (18 25 °C), según se miden mediante difracción de rayos X en polvo y velocidad de disolución por medición del ABC. En una serie de experimentos con nanopartículas híbridas que comprenden nilotinib y HPMCP HP55 producidas por los procedimientos de la invención, las partículas resultantes proporcionaron nanopartículas híbridas amorfas estables al 40 % de carga de fármaco (I/P base de nilotinib/HPMCP HP55: exp 146), medido por DRXP así como la velocidad de disolución por medición del ABC. El material mostró una temperatura de transición vítrea a aproximadamente 127 °C, lo que indica una única fase amorfa con estabilidad inherente. Los lotes parcialmente cristalinos también procesaron una estabilidad inherente similar. 6 meses de almacenamiento a temperatura ambiente (18-25 °C) de nanopartículas híbridas parcialmente cristalinas al 40 % de carga de fármaco, I/P base de nilotinib /HPMCP HP55 no mostraron ningún signo de inestabilidad física.
El análisis termogravimétrico proporcionó una pérdida de masa del 1,7 % de la temperatura ambiente a 120 °C. El análisis dinámico de absorción de vapor a 25 °C dio un aumento de masa relativo de aproximadamente un 7 % de un 0 a un 90 % de HR (tres ciclos de un 0 a 90 % de HR, no indujo un cambio de fase).
La alta temperatura de transición vítrea, la pérdida de masa del 1,7 % de la temperatura ambiente a 120 °C y la higrospopicidad moderada proponen una estabilidad inherente. Esto lo respaldan pruebas de estabilidad de varios lotes en varias condiciones. El punto de estabilidad más largo es de 12 meses a temperatura ambiente (18-25 °C). Ningún lote o condición ha mostrado signos de inestabilidad física (figura 27).
Calorimetría diferencial de barrido modulada (CDBm)
El análisis de calorimetría diferencial de barrido modulada (CDBm) se procesó en un TA Instruments modelo Q200 (New Castle, EE. UU.), equipado con un sistema de refrigeración refrigerado RC90 (Home Automation, Nueva Orleans, EE. UU.). Las muestras se pesaron a 7 ± 2 mg en recipientes de aluminio de baja masa Tzero y se sellaron con tapas Tzero. A continuación, se calentaron a una velocidad de calentamiento de 3 °C/min de 0 a 170 °C con una amplitud de temperatura de modulación convencional de 1 °C y un periodo de modulación de 40 segundos. Se utilizó nitrógeno de ultraalta pureza como gas de purga a un caudal de 50 ml/min. Todos los análisis de datos se llevaron a cabo utilizando el software TA Universal Analysis, versión 4.7A. Las calibraciones de temperatura y constante de cubeta se realizaron con el uso de un estándar de indio antes usar el instrumento. Los resultados de la CDB se evaluaron en términos de componentes directos e inversos del flujo de calor.
La termogravimetría (TG) se realizó en un Seiko TG/DTA 6200 y en Pt-pan abiertos de 90 pl con aproximadamente de 10 a 20 mg de muestra y un flujo de nitrógeno de 200 ml/min. El programa de temperatura fue de ambiente (20 °C) a 400 °C con una velocidad de calentamiento de 10 °C/min. Se sustrajo un blanco y se normalizaron los datos de la TG con respecto al tamaño de la muestra y se analizaron usando el software Muse Standard Analysis, versión 6.1 U.
Sorción dinámica de vapor (DVS)
La higroscopicidad de las muestras se estudió mediante gravimetría de absorción dinámica de vapor (DVS), usando un DVS-1 (Surface Measurement Ltd., Reino Unido). Se pesaron aproximadamente 10 mg de la sustancia en un vaso de vidrio. El peso relativo se registró en un intervalo de 20 segundos cuando la humedad relativa (HR) objetivo sobre la muestra aumentó gradualmente de un 0 % a un 90 % y, a continuación, se redujo de forma similar de nuevo a un 0 % de HR, con un 10 % de HR por etapa. Cada muestra se analizó en tres ciclos completos consecutivos. La condición para pasar al siguiente nivel de Hr fue un cambio de peso por debajo o igual a un 0,002 % en 15 minutos, con un tiempo total máximo por etapa de 24 horas. Debido al lento equilibrio en experimentos de este tipo, las cifras obtenidas se deben considerar como estimaciones más bajas de la absorción de agua. La temperatura se mantuvo a 25 °C.
Difracción de rayos X en polvo (DRXP)
Los experimentos de DRX se procesaron en un difractómetro X'Pert Pro (PANanalytical, Almelo, Países Bajos) configurado en geometría Bragg-Brentano. El difractómetro estaba equipado con un filtro de níquel de 20 pm y un detector X'Celerator RTMS con una longitud activa de 2,122 ° 20. Se colocó una muestra representativa sobre un soporte para muestras de cristal único de fondo de cero cuartos (Siltronix, Archamps, Francia). Los experimentos se procesaron usando radiación Cu Ka (45 kV y 40 mA) a temperatura y humedad ambiente. Las exploraciones se procesaron en modo continuo en el intervalo de 4,5-40 ° 20 utilizando rejillas de divergencia automática y antidifusoras con una longitud observada de 10 mm, un tiempo de recuento común de 299,72 segundos y un tamaño de paso de 0,0167 ° 20. La recolección de datos se realizó usando el software de aplicación X'Pert Data Collector V.2.2j y el software de control de instrumentos V.2.1E, mientras que el análisis de patrones se realizó usando X'Pert Data Viewer V.1.2c (todo los softwares son de PANanalytical, Almelo, Países Bajos).
Velocidad de disolución por medición del ABC
Las nanopartículas híbridas amorfas estables descritas en los Exp 171 y Exp172 (I/P), como se indica a continuación, se produjeron según el Exp 148, con base de nilotinib, HPMCP HP55 y se almacenaron a temperatura ambiente durante 11 meses. La velocidad de disolución en condiciones distintas a las de baja concentración se sometió a prueba en diferentes puntos temporales y los resultados se presentan en la tabla 47 y la figura 26. Se añadió copolímero de polivinil caprolactama-acetato de polivinilo-polietilenglicol para mejorar la solubilidad. Una comparación del ABC durante 80 minutos muestra claramente que el perfil de la velocidad de disolución de las partículas prácticamente no cambia después de 11 meses de almacenamiento, por ejemplo, la relación entre el ABC de las partículas producidas y sometidas a prueba, en comparación con las partículas producidas, sometidas a prueba y almacenadas durante 11 meses es más del 97 %.
T l 47.

Claims (12)
1. Procedimiento para producir partículas de dispersión sólida amorfas estables que comprende un inhibidor de la proteína cinasa seleccionado de entre dasatinib, una sal de dasatinib, un hidrato de dasatinib, un solvato de dasatinib o una combinación de los mismos y al menos un componente polimérico estabilizante y formador de matriz, dichas partículas de dispersión sólida tienen un grado de amorficidad del 100 %, el procedimiento comprende
a) proporcionar una primera corriente presurizada de dicho inhibidor de la proteína cinasa disuelto en un disolvente; b) proporcionar una segunda corriente presurizada de antidisolvente que comprende un fluido de CO2 supercrítico o subcrítico, donde dicho al menos un componente polimérico estabilizante y formador de matriz está presente en dicha primera o segunda corriente; y
c) mezclar dicha primera y segunda corrientes, y pulverizar la corriente mixta a la salida de una boquilla, con lo que se forman dichas partículas de dispersión sólida; seguido de la recogida de dichas partículas de dispersión sólida.
2. El procedimiento según la reivindicación 1, donde dicha segunda corriente presurizada de antidisolvente comprende un fluido de CO2 subcrítico con una presión por encima o por debajo de 74 bar y una temperatura de -30 °C a 31 °C.
3. El procedimiento según cualquiera de las reivindicaciones 1 o 2, donde la cantidad de inhibidor de la proteína cinasa es de un 10 a un 70 % en peso de las partículas de dispersión sólida formadas.
4. El procedimiento según cualquiera de las reivindicaciones anteriores, donde dicho al menos un componente polimérico estabilizante y formador de matriz está presente en dicho fluido de CO2 supercrítico o subcrítico.
5. El procedimiento según cualquiera de las reivindicaciones anteriores, donde dicho componente polimérico estabilizante y formador de matriz se selecciona de entre metilcelulosa, hidroxietilcelulosa, hidroxipropilcelulosa, hidroxipropilmetilcelulosa, succinato de acetato de hidroxipropilmetilcelulosa, ftalato de hidroxipropilmetilcelulosa, ftalato de acetato de polivinilo, copolvidona, crospovidona, copolímero de ácido metacrílico y acrilato de etilo, copolímero de metacrilato de ácido y metacrilato de metilo, polietilenglicol, copolímero de polivinil caprolactamaacetato de polivinilo-polietilenglicol, copolímero de DL lactida/glicólido, poli DL-lactida, ftalato de acetato de celulosa, homopolímero de carbómero tipo A, homopolímero de carbómero tipo B, copolímeros de aminoalquil metacrilato y poloxámeros.
6. El procedimiento según cualquiera de las reivindicaciones anteriores, donde dicho componente polimérico estabilizante y formador de matriz se selecciona de entre ftalato de hidroxipropilmetilcelulosa, hidroxipropilcelulosa, copolvidona, succinato de acetato de hidroxipropilmetilcelulosa, ftalato de acetato de polivinilo, ftalato de acetato de celulosa y polivinilpirrolidona.
7. El procedimiento según cualquiera de las reivindicaciones anteriores, donde se añade un solubilizante a las partículas de dispersión sólida obtenidas en la etapa c.
8. El procedimiento de la reivindicación 7, donde dicho solubilizante se selecciona de entre copolímero de polivinil caprolactama-acetato de polivinilo-polietilenglicol, succinato de d-a-tocoferol polietilenglicol 1000 y un aceite de ricino hidrogenado, tal como aceite de ricino hidrogenado PEG-40 o aceite de ricino hidrogenado PEG-35.
9. El procedimiento según cualquiera de las reivindicaciones anteriores, donde dichas partículas de dispersión sólida amorfas se caracterizan por proporcionar un patrón de difracción de rayos X de polvo amorfo.
10. El procedimiento según cualquiera de las reivindicaciones anteriores, donde dicho disolvente es un disolvente orgánico seleccionado de entre DMSO y trifluoroetanol (TFE), o una mezcla de los mismos.
11. El procedimiento según una cualquiera de las reivindicaciones anteriores, donde dichas partículas comprenden además un solubilizante, dentro de las partículas.
12. El procedimiento de la reivindicación 11, donde dicho solubilizante es copolímero de polivinil caprolactamaacetato de polivinilo-polietilenglicol, succinato de d-a-tocoferol polietilenglicol 1000 o un aceite de ricino hidrogenado, tal como aceite de ricino hidrogenado PEG-40 o aceite de ricino hidrogenado PEG-35.
Applications Claiming Priority (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201261586187P | 2012-01-13 | 2012-01-13 | |
| SE1250015 | 2012-01-13 | ||
| US201261713120P | 2012-10-12 | 2012-10-12 | |
| SE1251160 | 2012-10-12 | ||
| PCT/SE2013/050015 WO2013105894A1 (en) | 2012-01-13 | 2013-01-11 | A method for producing stable, amorphous hybrid nanoparticles comprising at least one protein kinase inhibitor and at least one polymeric stabilizing and matrix- forming component. |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2854751T3 true ES2854751T3 (es) | 2021-09-22 |
Family
ID=66441515
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES13736318T Active ES2854751T3 (es) | 2012-01-13 | 2013-01-11 | Un procedimiento para producir nanopartículas híbridas amorfas estables que comprende al menos un inhibidor de la proteína cinasa y al menos un componente polimérico estabilizante y formador de matriz |
Country Status (6)
| Country | Link |
|---|---|
| JP (1) | JP6828123B2 (es) |
| DK (1) | DK2802314T3 (es) |
| ES (1) | ES2854751T3 (es) |
| HR (1) | HRP20210237T1 (es) |
| IL (3) | IL276036B (es) |
| MX (3) | MX371297B (es) |
Families Citing this family (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| IL294928A (en) | 2020-01-24 | 2022-09-01 | Nanocopoeia Llc | Amorphous solid dispersions of dasatinib and uses thereof |
| EP4096791A1 (en) | 2020-01-31 | 2022-12-07 | Nanocopoeia LLC | Amorphous nilotinib microparticles and uses thereof |
| JP7431635B2 (ja) | 2020-03-26 | 2024-02-15 | 大王製紙株式会社 | 包装吸収性物品 |
| WO2021222739A1 (en) * | 2020-04-30 | 2021-11-04 | Nanocopoeia, Llc | Orally disintegrating tablet comprising amorphous solid dispersion of nilotinib |
| US11980619B2 (en) | 2021-07-28 | 2024-05-14 | Nanocopoeia, Llc | Pharmaceutical compositions and crushable tablets including amorphous solid dispersions of dasatinib and uses |
| WO2024214120A1 (en) * | 2023-04-12 | 2024-10-17 | Dr. Reddy's Laboratories Limited | Pharmaceutical compositions of nilotinib |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB9920558D0 (en) * | 1999-08-31 | 1999-11-03 | Bradford Particle Design Ltd | Methods for particle formation and their products |
| KR20090018864A (ko) * | 2006-06-13 | 2009-02-23 | 엘란 파마 인터내셔널 리미티드 | 나노입자형 키나제 억제제 제제 |
| JP2009543797A (ja) * | 2006-07-10 | 2009-12-10 | エラン ファーマ インターナショナル,リミティド | ナノ粒子ソラフェニブ製剤 |
| EP2089003A1 (en) * | 2006-11-09 | 2009-08-19 | Abbott GmbH & Co. KG | Pharmaceutical dosage form for oral administration of tyrosine kinase inhibitor |
| KR100924236B1 (ko) * | 2009-06-23 | 2009-10-29 | 충남대학교산학협력단 | 균일한 입도분포를 가지는 초미세입자의 신규한 제조방법 및 장치 |
-
2013
- 2013-01-11 MX MX2017005486A patent/MX371297B/es unknown
- 2013-01-11 MX MX2017005487A patent/MX371349B/es unknown
- 2013-01-11 MX MX2017005485A patent/MX371209B/es unknown
- 2013-01-11 ES ES13736318T patent/ES2854751T3/es active Active
- 2013-01-11 DK DK13736318.0T patent/DK2802314T3/da active
- 2013-01-11 HR HRP20210237TT patent/HRP20210237T1/hr unknown
-
2019
- 2019-12-03 JP JP2019218846A patent/JP6828123B2/ja not_active Expired - Fee Related
-
2020
- 2020-07-14 IL IL276036A patent/IL276036B/en unknown
-
2021
- 2021-07-21 IL IL285017A patent/IL285017B2/en unknown
- 2021-07-21 IL IL285016A patent/IL285016B2/en unknown
Also Published As
| Publication number | Publication date |
|---|---|
| HRP20210237T1 (hr) | 2021-03-19 |
| IL285017A (en) | 2021-08-31 |
| MX371209B (es) | 2020-01-22 |
| IL276036B (en) | 2021-09-30 |
| IL285017B2 (en) | 2023-02-01 |
| DK2802314T3 (da) | 2021-01-25 |
| IL285017B (en) | 2022-10-01 |
| MX371349B (es) | 2020-01-27 |
| IL285016A (en) | 2021-08-31 |
| JP6828123B2 (ja) | 2021-02-10 |
| JP2020055838A (ja) | 2020-04-09 |
| IL285016B2 (en) | 2023-02-01 |
| IL285016B (en) | 2022-10-01 |
| MX371297B (es) | 2020-01-24 |
| IL276036A (en) | 2020-08-31 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US12076314B2 (en) | Pharmaceutical compositions | |
| ES2854751T3 (es) | Un procedimiento para producir nanopartículas híbridas amorfas estables que comprende al menos un inhibidor de la proteína cinasa y al menos un componente polimérico estabilizante y formador de matriz |