ES2857152T3 - Composición farmacéutica y administraciones de la misma - Google Patents
Composición farmacéutica y administraciones de la misma Download PDFInfo
- Publication number
- ES2857152T3 ES2857152T3 ES17204189T ES17204189T ES2857152T3 ES 2857152 T3 ES2857152 T3 ES 2857152T3 ES 17204189 T ES17204189 T ES 17204189T ES 17204189 T ES17204189 T ES 17204189T ES 2857152 T3 ES2857152 T3 ES 2857152T3
- Authority
- ES
- Spain
- Prior art keywords
- weight
- pharmaceutical composition
- solid dispersion
- compound
- composition
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 239000008194 pharmaceutical composition Substances 0.000 title claims abstract description 363
- 239000007962 solid dispersion Substances 0.000 claims abstract description 338
- 229940125904 compound 1 Drugs 0.000 claims abstract description 241
- 239000006185 dispersion Substances 0.000 claims abstract description 85
- DBMJMQXJHONAFJ-UHFFFAOYSA-M Sodium laurylsulphate Chemical compound [Na+].CCCCCCCCCCCCOS([O-])(=O)=O DBMJMQXJHONAFJ-UHFFFAOYSA-M 0.000 claims abstract description 77
- 235000019333 sodium laurylsulphate Nutrition 0.000 claims abstract description 77
- ZUAAPNNKRHMPKG-UHFFFAOYSA-N acetic acid;butanedioic acid;methanol;propane-1,2-diol Chemical compound OC.CC(O)=O.CC(O)CO.OC(=O)CCC(O)=O ZUAAPNNKRHMPKG-UHFFFAOYSA-N 0.000 claims abstract description 73
- PURKAOJPTOLRMP-UHFFFAOYSA-N ivacaftor Chemical compound C1=C(O)C(C(C)(C)C)=CC(C(C)(C)C)=C1NC(=O)C1=CNC2=CC=CC=C2C1=O PURKAOJPTOLRMP-UHFFFAOYSA-N 0.000 claims abstract description 32
- 229920001577 copolymer Polymers 0.000 claims abstract description 4
- 239000000203 mixture Substances 0.000 claims description 285
- 238000000034 method Methods 0.000 claims description 121
- AICOOMRHRUFYCM-ZRRPKQBOSA-N oxazine, 1 Chemical compound C([C@@H]1[C@H](C(C[C@]2(C)[C@@H]([C@H](C)N(C)C)[C@H](O)C[C@]21C)=O)CC1=CC2)C[C@H]1[C@@]1(C)[C@H]2N=C(C(C)C)OC1 AICOOMRHRUFYCM-ZRRPKQBOSA-N 0.000 claims description 108
- 239000000314 lubricant Substances 0.000 claims description 92
- 239000004094 surface-active agent Substances 0.000 claims description 92
- 239000007884 disintegrant Substances 0.000 claims description 91
- 239000011230 binding agent Substances 0.000 claims description 90
- 229920000639 hydroxypropylmethylcellulose acetate succinate Polymers 0.000 claims description 62
- 239000000945 filler Substances 0.000 claims description 59
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 claims description 56
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 claims description 51
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 claims description 46
- 239000008101 lactose Substances 0.000 claims description 46
- 229920000168 Microcrystalline cellulose Polymers 0.000 claims description 38
- 235000019813 microcrystalline cellulose Nutrition 0.000 claims description 38
- 201000003883 Cystic fibrosis Diseases 0.000 claims description 37
- 238000000576 coating method Methods 0.000 claims description 35
- 239000011248 coating agent Substances 0.000 claims description 33
- 229920002785 Croscarmellose sodium Polymers 0.000 claims description 31
- DPXJVFZANSGRMM-UHFFFAOYSA-N acetic acid;2,3,4,5,6-pentahydroxyhexanal;sodium Chemical compound [Na].CC(O)=O.OCC(O)C(O)C(O)C(O)C=O DPXJVFZANSGRMM-UHFFFAOYSA-N 0.000 claims description 31
- 229960001681 croscarmellose sodium Drugs 0.000 claims description 31
- 235000010947 crosslinked sodium carboxy methyl cellulose Nutrition 0.000 claims description 31
- 239000008108 microcrystalline cellulose Substances 0.000 claims description 31
- 229940016286 microcrystalline cellulose Drugs 0.000 claims description 31
- 229940075614 colloidal silicon dioxide Drugs 0.000 claims description 29
- 108010079245 Cystic Fibrosis Transmembrane Conductance Regulator Proteins 0.000 claims description 28
- 235000019359 magnesium stearate Nutrition 0.000 claims description 28
- 230000035772 mutation Effects 0.000 claims description 12
- 235000012054 meals Nutrition 0.000 claims description 4
- 235000011888 snacks Nutrition 0.000 claims description 4
- 238000002560 therapeutic procedure Methods 0.000 claims description 3
- 102100023419 Cystic fibrosis transmembrane conductance regulator Human genes 0.000 claims 6
- IYKJEILNJZQJPU-UHFFFAOYSA-N acetic acid;butanedioic acid Chemical compound CC(O)=O.OC(=O)CCC(O)=O IYKJEILNJZQJPU-UHFFFAOYSA-N 0.000 abstract 1
- 235000010979 hydroxypropyl methyl cellulose Nutrition 0.000 abstract 1
- UFVKGYZPFZQRLF-UHFFFAOYSA-N hydroxypropyl methyl cellulose Chemical compound OC1C(O)C(OC)OC(CO)C1OC1C(O)C(O)C(OC2C(C(O)C(OC3C(C(O)C(O)C(CO)O3)O)C(CO)O2)O)C(CO)O1 UFVKGYZPFZQRLF-UHFFFAOYSA-N 0.000 abstract 1
- 229920003088 hydroxypropyl methyl cellulose Polymers 0.000 abstract 1
- 239000001866 hydroxypropyl methyl cellulose Substances 0.000 abstract 1
- ZWEHNKRNPOVVGH-UHFFFAOYSA-N 2-Butanone Chemical compound CCC(C)=O ZWEHNKRNPOVVGH-UHFFFAOYSA-N 0.000 description 69
- 239000004615 ingredient Substances 0.000 description 65
- 239000007921 spray Substances 0.000 description 54
- 239000002904 solvent Substances 0.000 description 47
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 44
- WHNWPMSKXPGLAX-UHFFFAOYSA-N N-Vinyl-2-pyrrolidone Chemical compound C=CN1CCCC1=O WHNWPMSKXPGLAX-UHFFFAOYSA-N 0.000 description 36
- 229920000642 polymer Polymers 0.000 description 34
- 239000003086 colorant Substances 0.000 description 32
- 239000007787 solid Substances 0.000 description 27
- 238000002156 mixing Methods 0.000 description 26
- 239000000976 ink Substances 0.000 description 25
- 239000003814 drug Substances 0.000 description 22
- 102000012605 Cystic Fibrosis Transmembrane Conductance Regulator Human genes 0.000 description 21
- 239000000546 pharmaceutical excipient Substances 0.000 description 21
- 239000003795 chemical substances by application Substances 0.000 description 20
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 20
- 238000004090 dissolution Methods 0.000 description 20
- 239000000047 product Substances 0.000 description 20
- 239000002245 particle Substances 0.000 description 19
- 229940079593 drug Drugs 0.000 description 17
- 230000008569 process Effects 0.000 description 17
- 239000012748 slip agent Substances 0.000 description 17
- 239000007916 tablet composition Substances 0.000 description 16
- 230000032258 transport Effects 0.000 description 15
- 239000008186 active pharmaceutical agent Substances 0.000 description 14
- 201000010099 disease Diseases 0.000 description 14
- 108090000623 proteins and genes Proteins 0.000 description 14
- 210000004027 cell Anatomy 0.000 description 13
- 239000000843 powder Substances 0.000 description 12
- 230000000694 effects Effects 0.000 description 11
- -1 glidant Substances 0.000 description 11
- 239000004203 carnauba wax Substances 0.000 description 10
- 235000013869 carnauba wax Nutrition 0.000 description 10
- 201000009266 primary ciliary dyskinesia Diseases 0.000 description 10
- 239000000126 substance Substances 0.000 description 10
- 108091006146 Channels Proteins 0.000 description 9
- 230000004071 biological effect Effects 0.000 description 9
- 150000001875 compounds Chemical class 0.000 description 9
- 239000003623 enhancer Substances 0.000 description 9
- MVPICKVDHDWCJQ-UHFFFAOYSA-N ethyl 3-pyrrolidin-1-ylpropanoate Chemical compound CCOC(=O)CCN1CCCC1 MVPICKVDHDWCJQ-UHFFFAOYSA-N 0.000 description 9
- 229940045902 sodium stearyl fumarate Drugs 0.000 description 9
- 238000001694 spray drying Methods 0.000 description 9
- 239000000725 suspension Substances 0.000 description 9
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 8
- 208000025678 Ciliary Motility disease Diseases 0.000 description 8
- 102100026383 Vasopressin-neurophysin 2-copeptin Human genes 0.000 description 8
- 238000000149 argon plasma sintering Methods 0.000 description 8
- 201000010064 diabetes insipidus Diseases 0.000 description 8
- 239000007789 gas Substances 0.000 description 8
- 230000001965 increasing effect Effects 0.000 description 8
- 239000001993 wax Substances 0.000 description 8
- 229920003084 Avicel® PH-102 Polymers 0.000 description 7
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 7
- 238000007792 addition Methods 0.000 description 7
- 150000001450 anions Chemical class 0.000 description 7
- 239000002775 capsule Substances 0.000 description 7
- 238000001035 drying Methods 0.000 description 7
- 239000003205 fragrance Substances 0.000 description 7
- 239000000463 material Substances 0.000 description 7
- 210000004379 membrane Anatomy 0.000 description 7
- 239000012528 membrane Substances 0.000 description 7
- 239000013557 residual solvent Substances 0.000 description 7
- 239000000377 silicon dioxide Substances 0.000 description 7
- 235000012239 silicon dioxide Nutrition 0.000 description 7
- 239000011734 sodium Substances 0.000 description 7
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 6
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 6
- 229930006000 Sucrose Natural products 0.000 description 6
- 239000004480 active ingredient Substances 0.000 description 6
- 235000010980 cellulose Nutrition 0.000 description 6
- 229920002678 cellulose Polymers 0.000 description 6
- 238000007906 compression Methods 0.000 description 6
- 230000006835 compression Effects 0.000 description 6
- 230000007812 deficiency Effects 0.000 description 6
- 239000000975 dye Substances 0.000 description 6
- 239000007888 film coating Substances 0.000 description 6
- 238000009501 film coating Methods 0.000 description 6
- 239000000796 flavoring agent Substances 0.000 description 6
- 235000019634 flavors Nutrition 0.000 description 6
- 239000012530 fluid Substances 0.000 description 6
- 238000004519 manufacturing process Methods 0.000 description 6
- 238000003825 pressing Methods 0.000 description 6
- 230000001698 pyrogenic effect Effects 0.000 description 6
- 239000011343 solid material Substances 0.000 description 6
- 238000005507 spraying Methods 0.000 description 6
- 239000005720 sucrose Substances 0.000 description 6
- 238000012360 testing method Methods 0.000 description 6
- 230000004584 weight gain Effects 0.000 description 6
- 235000019786 weight gain Nutrition 0.000 description 6
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 5
- 108010067035 Pancrelipase Proteins 0.000 description 5
- 208000035475 disorder Diseases 0.000 description 5
- 239000012738 dissolution medium Substances 0.000 description 5
- 229960001855 mannitol Drugs 0.000 description 5
- 238000000634 powder X-ray diffraction Methods 0.000 description 5
- 238000012545 processing Methods 0.000 description 5
- 102000004169 proteins and genes Human genes 0.000 description 5
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 4
- 206010006474 Bronchopulmonary aspergillosis allergic Diseases 0.000 description 4
- 208000006545 Chronic Obstructive Pulmonary Disease Diseases 0.000 description 4
- 206010067265 Heterotaxia Diseases 0.000 description 4
- 208000015439 Lysosomal storage disease Diseases 0.000 description 4
- 229930195725 Mannitol Natural products 0.000 description 4
- 206010033645 Pancreatitis Diseases 0.000 description 4
- 208000031733 Situs inversus totalis Diseases 0.000 description 4
- 208000006778 allergic bronchopulmonary aspergillosis Diseases 0.000 description 4
- 239000003242 anti bacterial agent Substances 0.000 description 4
- 210000000981 epithelium Anatomy 0.000 description 4
- 238000001914 filtration Methods 0.000 description 4
- 235000013305 food Nutrition 0.000 description 4
- 238000009472 formulation Methods 0.000 description 4
- RWSXRVCMGQZWBV-WDSKDSINSA-N glutathione Chemical compound OC(=O)[C@@H](N)CCC(=O)N[C@@H](CS)C(=O)NCC(O)=O RWSXRVCMGQZWBV-WDSKDSINSA-N 0.000 description 4
- 239000000594 mannitol Substances 0.000 description 4
- 235000010355 mannitol Nutrition 0.000 description 4
- 229910052757 nitrogen Inorganic materials 0.000 description 4
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 4
- 230000028327 secretion Effects 0.000 description 4
- 208000008797 situs inversus Diseases 0.000 description 4
- 239000012064 sodium phosphate buffer Substances 0.000 description 4
- 239000011877 solvent mixture Substances 0.000 description 4
- 229940124597 therapeutic agent Drugs 0.000 description 4
- OOUGLTULBSNHNF-UHFFFAOYSA-N 3-[5-(2-fluorophenyl)-1,2,4-oxadiazol-3-yl]benzoic acid Chemical compound OC(=O)C1=CC=CC(C=2N=C(ON=2)C=2C(=CC=CC=2)F)=C1 OOUGLTULBSNHNF-UHFFFAOYSA-N 0.000 description 3
- PNEYBMLMFCGWSK-UHFFFAOYSA-N Alumina Chemical compound [O-2].[O-2].[O-2].[Al+3].[Al+3] PNEYBMLMFCGWSK-UHFFFAOYSA-N 0.000 description 3
- 229920002261 Corn starch Polymers 0.000 description 3
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 3
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 3
- 201000000101 Hyperekplexia Diseases 0.000 description 3
- 206010058271 Hyperexplexia Diseases 0.000 description 3
- 208000019693 Lung disease Diseases 0.000 description 3
- 229920002472 Starch Polymers 0.000 description 3
- 235000021355 Stearic acid Nutrition 0.000 description 3
- 229920006125 amorphous polymer Polymers 0.000 description 3
- 229940088710 antibiotic agent Drugs 0.000 description 3
- 229940124630 bronchodilator Drugs 0.000 description 3
- XAAHAAMILDNBPS-UHFFFAOYSA-L calcium hydrogenphosphate dihydrate Chemical compound O.O.[Ca+2].OP([O-])([O-])=O XAAHAAMILDNBPS-UHFFFAOYSA-L 0.000 description 3
- 239000001506 calcium phosphate Substances 0.000 description 3
- 235000011010 calcium phosphates Nutrition 0.000 description 3
- 210000000170 cell membrane Anatomy 0.000 description 3
- 239000001913 cellulose Substances 0.000 description 3
- 239000000460 chlorine Substances 0.000 description 3
- 208000015532 congenital bilateral absence of vas deferens Diseases 0.000 description 3
- 238000001816 cooling Methods 0.000 description 3
- 239000008120 corn starch Substances 0.000 description 3
- 229940099112 cornstarch Drugs 0.000 description 3
- 230000007547 defect Effects 0.000 description 3
- 230000002950 deficient Effects 0.000 description 3
- 239000008367 deionised water Substances 0.000 description 3
- 229910021641 deionized water Inorganic materials 0.000 description 3
- 238000012217 deletion Methods 0.000 description 3
- 230000037430 deletion Effects 0.000 description 3
- 235000019700 dicalcium phosphate Nutrition 0.000 description 3
- 229940095079 dicalcium phosphate anhydrous Drugs 0.000 description 3
- 238000010438 heat treatment Methods 0.000 description 3
- 229920003063 hydroxymethyl cellulose Polymers 0.000 description 3
- 229940031574 hydroxymethyl cellulose Drugs 0.000 description 3
- 150000002500 ions Chemical class 0.000 description 3
- 210000004072 lung Anatomy 0.000 description 3
- 239000002609 medium Substances 0.000 description 3
- 235000016709 nutrition Nutrition 0.000 description 3
- OQCDKBAXFALNLD-UHFFFAOYSA-N octadecanoic acid Natural products CCCCCCCC(C)CCCCCCCCC(O)=O OQCDKBAXFALNLD-UHFFFAOYSA-N 0.000 description 3
- 239000003921 oil Substances 0.000 description 3
- 229920003109 sodium starch glycolate Polymers 0.000 description 3
- 229940079832 sodium starch glycolate Drugs 0.000 description 3
- 239000008109 sodium starch glycolate Substances 0.000 description 3
- 239000000600 sorbitol Substances 0.000 description 3
- 235000010356 sorbitol Nutrition 0.000 description 3
- 229910001220 stainless steel Inorganic materials 0.000 description 3
- 239000010935 stainless steel Substances 0.000 description 3
- 235000019698 starch Nutrition 0.000 description 3
- 239000008117 stearic acid Substances 0.000 description 3
- 229960004793 sucrose Drugs 0.000 description 3
- 235000000346 sugar Nutrition 0.000 description 3
- 150000008163 sugars Chemical class 0.000 description 3
- 239000000454 talc Substances 0.000 description 3
- 229910052623 talc Inorganic materials 0.000 description 3
- 229960000707 tobramycin Drugs 0.000 description 3
- NLVFBUXFDBBNBW-PBSUHMDJSA-N tobramycin Chemical compound N[C@@H]1C[C@H](O)[C@@H](CN)O[C@@H]1O[C@H]1[C@H](O)[C@@H](O[C@@H]2[C@@H]([C@@H](N)[C@H](O)[C@@H](CO)O2)O)[C@H](N)C[C@@H]1N NLVFBUXFDBBNBW-PBSUHMDJSA-N 0.000 description 3
- QORWJWZARLRLPR-UHFFFAOYSA-H tricalcium bis(phosphate) Chemical class [Ca+2].[Ca+2].[Ca+2].[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O QORWJWZARLRLPR-UHFFFAOYSA-H 0.000 description 3
- VBICKXHEKHSIBG-UHFFFAOYSA-N 1-monostearoylglycerol Chemical compound CCCCCCCCCCCCCCCCCC(=O)OCC(O)CO VBICKXHEKHSIBG-UHFFFAOYSA-N 0.000 description 2
- 102100034452 Alternative prion protein Human genes 0.000 description 2
- 208000024827 Alzheimer disease Diseases 0.000 description 2
- 102100032187 Androgen receptor Human genes 0.000 description 2
- 208000009575 Angelman syndrome Diseases 0.000 description 2
- WZPBZJONDBGPKJ-UHFFFAOYSA-N Antibiotic SQ 26917 Natural products O=C1N(S(O)(=O)=O)C(C)C1NC(=O)C(=NOC(C)(C)C(O)=O)C1=CSC(N)=N1 WZPBZJONDBGPKJ-UHFFFAOYSA-N 0.000 description 2
- 206010002961 Aplasia Diseases 0.000 description 2
- 201000009144 Bartter disease type 3 Diseases 0.000 description 2
- 208000037245 Bartter syndrome type 3 Diseases 0.000 description 2
- 206010006458 Bronchitis chronic Diseases 0.000 description 2
- 101150029409 CFTR gene Proteins 0.000 description 2
- 208000031976 Channelopathies Diseases 0.000 description 2
- 208000010693 Charcot-Marie-Tooth Disease Diseases 0.000 description 2
- 206010010356 Congenital anomaly Diseases 0.000 description 2
- 201000002200 Congenital disorder of glycosylation Diseases 0.000 description 2
- 206010010774 Constipation Diseases 0.000 description 2
- 208000024940 Dent disease Diseases 0.000 description 2
- 208000003556 Dry Eye Syndromes Diseases 0.000 description 2
- 206010013883 Dwarfism Diseases 0.000 description 2
- 206010014561 Emphysema Diseases 0.000 description 2
- XRHVZWWRFMCBAZ-UHFFFAOYSA-L Endothal-disodium Chemical compound [Na+].[Na+].C1CC2C(C([O-])=O)C(C(=O)[O-])C1O2 XRHVZWWRFMCBAZ-UHFFFAOYSA-L 0.000 description 2
- 208000024720 Fabry Disease Diseases 0.000 description 2
- 201000011240 Frontotemporal dementia Diseases 0.000 description 2
- 108010024636 Glutathione Proteins 0.000 description 2
- DCXXMTOCNZCJGO-UHFFFAOYSA-N Glycerol trioctadecanoate Natural products CCCCCCCCCCCCCCCCCC(=O)OCC(OC(=O)CCCCCCCCCCCCCCCCC)COC(=O)CCCCCCCCCCCCCCCCC DCXXMTOCNZCJGO-UHFFFAOYSA-N 0.000 description 2
- 208000019683 Gorham-Stout disease Diseases 0.000 description 2
- 208000005139 Hereditary Angioedema Types I and II Diseases 0.000 description 2
- 208000033981 Hereditary haemochromatosis Diseases 0.000 description 2
- 101000775732 Homo sapiens Androgen receptor Proteins 0.000 description 2
- 101000736368 Homo sapiens PH and SEC7 domain-containing protein 4 Proteins 0.000 description 2
- 208000000563 Hyperlipoproteinemia Type II Diseases 0.000 description 2
- 208000000038 Hypoparathyroidism Diseases 0.000 description 2
- 208000026350 Inborn Genetic disease Diseases 0.000 description 2
- UQSXHKLRYXJYBZ-UHFFFAOYSA-N Iron oxide Chemical compound [Fe]=O UQSXHKLRYXJYBZ-UHFFFAOYSA-N 0.000 description 2
- 208000003892 Kartagener syndrome Diseases 0.000 description 2
- 102100024640 Low-density lipoprotein receptor Human genes 0.000 description 2
- 208000007466 Male Infertility Diseases 0.000 description 2
- 208000029725 Metabolic bone disease Diseases 0.000 description 2
- 208000002678 Mucopolysaccharidoses Diseases 0.000 description 2
- 108010021466 Mutant Proteins Proteins 0.000 description 2
- 102000008300 Mutant Proteins Human genes 0.000 description 2
- 208000010316 Myotonia congenita Diseases 0.000 description 2
- 206010049088 Osteopenia Diseases 0.000 description 2
- 208000001132 Osteoporosis Diseases 0.000 description 2
- 102100036232 PH and SEC7 domain-containing protein 4 Human genes 0.000 description 2
- 208000035467 Pancreatic insufficiency Diseases 0.000 description 2
- 208000018737 Parkinson disease Diseases 0.000 description 2
- 208000000609 Pick Disease of the Brain Diseases 0.000 description 2
- 108091000054 Prion Proteins 0.000 description 2
- 208000024777 Prion disease Diseases 0.000 description 2
- 201000005660 Protein C Deficiency Diseases 0.000 description 2
- 208000021386 Sjogren Syndrome Diseases 0.000 description 2
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 2
- 208000035954 Thomsen and Becker disease Diseases 0.000 description 2
- GWEVSGVZZGPLCZ-UHFFFAOYSA-N Titan oxide Chemical compound O=[Ti]=O GWEVSGVZZGPLCZ-UHFFFAOYSA-N 0.000 description 2
- 206010045261 Type IIa hyperlipidaemia Diseases 0.000 description 2
- 208000006269 X-Linked Bulbo-Spinal Atrophy Diseases 0.000 description 2
- 208000004622 abetalipoproteinemia Diseases 0.000 description 2
- 238000009825 accumulation Methods 0.000 description 2
- 239000000654 additive Substances 0.000 description 2
- 239000000443 aerosol Substances 0.000 description 2
- 150000001413 amino acids Chemical class 0.000 description 2
- 206010002026 amyotrophic lateral sclerosis Diseases 0.000 description 2
- 239000002260 anti-inflammatory agent Substances 0.000 description 2
- 229940121363 anti-inflammatory agent Drugs 0.000 description 2
- 208000006673 asthma Diseases 0.000 description 2
- 229960003995 ataluren Drugs 0.000 description 2
- 229960003644 aztreonam Drugs 0.000 description 2
- WZPBZJONDBGPKJ-VEHQQRBSSA-N aztreonam Chemical compound O=C1N(S([O-])(=O)=O)[C@@H](C)[C@@H]1NC(=O)C(=N/OC(C)(C)C(O)=O)\C1=CSC([NH3+])=N1 WZPBZJONDBGPKJ-VEHQQRBSSA-N 0.000 description 2
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid group Chemical group C(C1=CC=CC=C1)(=O)O WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 2
- 235000013361 beverage Nutrition 0.000 description 2
- 230000003115 biocidal effect Effects 0.000 description 2
- 239000000038 blue colorant Substances 0.000 description 2
- 239000001045 blue dye Substances 0.000 description 2
- 206010006451 bronchitis Diseases 0.000 description 2
- 208000007451 chronic bronchitis Diseases 0.000 description 2
- 210000004081 cilia Anatomy 0.000 description 2
- 230000001886 ciliary effect Effects 0.000 description 2
- MYSWGUAQZAJSOK-UHFFFAOYSA-N ciprofloxacin Chemical compound C12=CC(N3CCNCC3)=C(F)C=C2C(=O)C(C(=O)O)=CN1C1CC1 MYSWGUAQZAJSOK-UHFFFAOYSA-N 0.000 description 2
- 238000004040 coloring Methods 0.000 description 2
- 206010012601 diabetes mellitus Diseases 0.000 description 2
- 239000003085 diluting agent Substances 0.000 description 2
- 206010015037 epilepsy Diseases 0.000 description 2
- 210000002919 epithelial cell Anatomy 0.000 description 2
- 239000003172 expectorant agent Substances 0.000 description 2
- 201000001386 familial hypercholesterolemia Diseases 0.000 description 2
- 208000016361 genetic disease Diseases 0.000 description 2
- 229960003180 glutathione Drugs 0.000 description 2
- 238000013038 hand mixing Methods 0.000 description 2
- 208000012770 hereditary angioedema type 1 Diseases 0.000 description 2
- 208000013746 hereditary thrombophilia due to congenital protein C deficiency Diseases 0.000 description 2
- 150000002632 lipids Chemical class 0.000 description 2
- 239000007788 liquid Substances 0.000 description 2
- 208000019423 liver disease Diseases 0.000 description 2
- 201000001441 melanoma Diseases 0.000 description 2
- 235000013336 milk Nutrition 0.000 description 2
- 239000008267 milk Substances 0.000 description 2
- 210000004080 milk Anatomy 0.000 description 2
- 230000000510 mucolytic effect Effects 0.000 description 2
- 229940066491 mucolytics Drugs 0.000 description 2
- 206010028093 mucopolysaccharidosis Diseases 0.000 description 2
- 210000003097 mucus Anatomy 0.000 description 2
- 208000000638 myeloperoxidase deficiency Diseases 0.000 description 2
- 230000004770 neurodegeneration Effects 0.000 description 2
- 208000015122 neurodegenerative disease Diseases 0.000 description 2
- 229940045258 pancrelipase Drugs 0.000 description 2
- HYAFETHFCAUJAY-UHFFFAOYSA-N pioglitazone Chemical compound N1=CC(CC)=CC=C1CCOC(C=C1)=CC=C1CC1C(=O)NC(=O)S1 HYAFETHFCAUJAY-UHFFFAOYSA-N 0.000 description 2
- 108010040003 polyglutamine Proteins 0.000 description 2
- 229920000155 polyglutamine Polymers 0.000 description 2
- 239000000244 polyoxyethylene sorbitan monooleate Substances 0.000 description 2
- 235000010482 polyoxyethylene sorbitan monooleate Nutrition 0.000 description 2
- 229920000136 polysorbate Polymers 0.000 description 2
- 229920000053 polysorbate 80 Polymers 0.000 description 2
- 238000010926 purge Methods 0.000 description 2
- 230000001105 regulatory effect Effects 0.000 description 2
- 150000003839 salts Chemical class 0.000 description 2
- 238000012216 screening Methods 0.000 description 2
- BNRNXUUZRGQAQC-UHFFFAOYSA-N sildenafil Chemical compound CCCC1=NN(C)C(C(N2)=O)=C1N=C2C(C(=CC=1)OCC)=CC=1S(=O)(=O)N1CCN(C)CC1 BNRNXUUZRGQAQC-UHFFFAOYSA-N 0.000 description 2
- 201000009890 sinusitis Diseases 0.000 description 2
- 239000000779 smoke Substances 0.000 description 2
- 239000011780 sodium chloride Substances 0.000 description 2
- 208000011580 syndromic disease Diseases 0.000 description 2
- 230000001225 therapeutic effect Effects 0.000 description 2
- 210000001519 tissue Anatomy 0.000 description 2
- 230000000007 visual effect Effects 0.000 description 2
- 229920003169 water-soluble polymer Polymers 0.000 description 2
- SFWLDKQAUHFCBS-AOEYGKNYSA-N (1S,4S,13S,16S,19S,22S,25S,28R,31S,37S,40S,41S,44R,47S,50S,53S,56R,65S,70S)-44-amino-47-(4-aminobutyl)-4,16,22-tribenzyl-31-[(R)-carboxy(hydroxy)methyl]-2,5,14,17,20,23,26,29,32,35,38,45,48,51,54,57,67-heptadecahydroxy-37-(2-hydroxy-2-iminoethyl)-50-(3-hydroxy-3-iminopropyl)-41,70-dimethyl-8-oxo-25-propan-2-yl-42,69,72-trithia-3,6,9,15,18,21,24,27,30,33,36,39,46,49,52,55,58,60,66-nonadecazapentacyclo[38.18.9.319,56.328,53.09,13]triheptaconta-2,5,14,17,20,23,26,29,32,35,38,45,48,51,54,57,66-heptadecaene-65-carboxylic acid Chemical compound CC(C)[C@@H]1\N=C(O)/[C@H](Cc2ccccc2)\N=C(O)/[C@@H]2\N=C(O)/[C@H](Cc3ccccc3)\N=C(O)/[C@@H]3CCCN3C(=O)C\N=C(O)/[C@H](Cc3ccccc3)\N=C(O)/[C@@H]3CNCCCC[C@H](\N=C(O)\[C@@H]4\N=C(O)\[C@H](CC(O)=N)\N=C(O)\C\N=C(O)/[C@@H](\N=C(O)\[C@H](CSC[C@@H](N=C(O)[C@H](CCC(O)=N)N=C(O)[C@H](CCCCN)N=C(O)[C@@H](N)CS[C@H]4C)C(O)=N[C@@H](CS[C@H]2C)C(O)=N3)\N=C1\O)[C@@H](O)C(O)=O)C(O)=O SFWLDKQAUHFCBS-AOEYGKNYSA-N 0.000 description 1
- DVSZKTAMJJTWFG-SKCDLICFSA-N (2e,4e,6e,8e,10e,12e)-docosa-2,4,6,8,10,12-hexaenoic acid Chemical compound CCCCCCCCC\C=C\C=C\C=C\C=C\C=C\C=C\C(O)=O DVSZKTAMJJTWFG-SKCDLICFSA-N 0.000 description 1
- JNYAEWCLZODPBN-JGWLITMVSA-N (2r,3r,4s)-2-[(1r)-1,2-dihydroxyethyl]oxolane-3,4-diol Chemical compound OC[C@@H](O)[C@H]1OC[C@H](O)[C@H]1O JNYAEWCLZODPBN-JGWLITMVSA-N 0.000 description 1
- QSIRXSYRKZHJHX-TWXHAJHVSA-N (2s)-6-amino-2-[[(2s)-2-[[(2s)-2-[[(2s)-2-[[(2s)-2-[[(2s)-6-amino-2-[[(2s)-2-[[(2s)-2-[[(2s)-2-[[(2s)-2-[[(2s)-6-amino-2-[[(2s)-2-[[(2s)-2-[[(2s)-2-[[(2s)-2-[[(2s)-6-amino-2-[[(2s)-2-[[(2s)-2-[[(2s)-2-[[(2s)-2-[[(2s)-2,6-diaminohexanoyl]amino]-4-methylpen Chemical compound NCCCC[C@H](N)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCCN)C(O)=O QSIRXSYRKZHJHX-TWXHAJHVSA-N 0.000 description 1
- HPKHMTMYHNDKJH-UHFFFAOYSA-N 1-(2,4-ditert-butyl-5-hydroxyphenyl)quinolin-4-one Chemical compound C1=C(O)C(C(C)(C)C)=CC(C(C)(C)C)=C1N1C2=CC=CC=C2C(=O)C=C1 HPKHMTMYHNDKJH-UHFFFAOYSA-N 0.000 description 1
- GZJLLYHBALOKEX-UHFFFAOYSA-N 6-Ketone, O18-Me-Ussuriedine Natural products CC=CCC=CCC=CCC=CCC=CCC=CCCCC(O)=O GZJLLYHBALOKEX-UHFFFAOYSA-N 0.000 description 1
- SDDSJMXGJNWMJY-BRHAQHMBSA-N 7-[(2r,4ar,5r,7ar)-2-[(3s)-1,1-difluoro-3-methylpentyl]-2-hydroxy-6-oxo-3,4,4a,5,7,7a-hexahydrocyclopenta[b]pyran-5-yl]heptanoic acid Chemical compound O1[C@](C(F)(F)C[C@@H](C)CC)(O)CC[C@@H]2[C@@H](CCCCCCC(O)=O)C(=O)C[C@H]21 SDDSJMXGJNWMJY-BRHAQHMBSA-N 0.000 description 1
- 102000005416 ATP-Binding Cassette Transporters Human genes 0.000 description 1
- 108010006533 ATP-Binding Cassette Transporters Proteins 0.000 description 1
- 108700028369 Alleles Proteins 0.000 description 1
- 108091006515 Anion channels Proteins 0.000 description 1
- 102000037829 Anion channels Human genes 0.000 description 1
- 206010003591 Ataxia Diseases 0.000 description 1
- 102000014461 Ataxins Human genes 0.000 description 1
- 108010078286 Ataxins Proteins 0.000 description 1
- SGHZXLIDFTYFHQ-UHFFFAOYSA-L Brilliant Blue Chemical compound [Na+].[Na+].C=1C=C(C(=C2C=CC(C=C2)=[N+](CC)CC=2C=C(C=CC=2)S([O-])(=O)=O)C=2C(=CC=CC=2)S([O-])(=O)=O)C=CC=1N(CC)CC1=CC=CC(S([O-])(=O)=O)=C1 SGHZXLIDFTYFHQ-UHFFFAOYSA-L 0.000 description 1
- 108010078791 Carrier Proteins Proteins 0.000 description 1
- 206010008025 Cerebellar ataxia Diseases 0.000 description 1
- 102000011045 Chloride Channels Human genes 0.000 description 1
- 108010062745 Chloride Channels Proteins 0.000 description 1
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 1
- 206010008631 Cholera Diseases 0.000 description 1
- 206010062264 Congenital hyperthyroidism Diseases 0.000 description 1
- 206010012735 Diarrhoea Diseases 0.000 description 1
- 206010061818 Disease progression Diseases 0.000 description 1
- 102000004190 Enzymes Human genes 0.000 description 1
- 108090000790 Enzymes Proteins 0.000 description 1
- 208000028782 Hereditary disease Diseases 0.000 description 1
- 241000282412 Homo Species 0.000 description 1
- 101000907783 Homo sapiens Cystic fibrosis transmembrane conductance regulator Proteins 0.000 description 1
- 101000851058 Homo sapiens Neutrophil elastase Proteins 0.000 description 1
- 208000015178 Hurler syndrome Diseases 0.000 description 1
- 206010020850 Hyperthyroidism Diseases 0.000 description 1
- 206010051125 Hypofibrinogenaemia Diseases 0.000 description 1
- HEFNNWSXXWATRW-UHFFFAOYSA-N Ibuprofen Chemical compound CC(C)CC1=CC=C(C(C)C(O)=O)C=C1 HEFNNWSXXWATRW-UHFFFAOYSA-N 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- 206010061218 Inflammation Diseases 0.000 description 1
- 102000004310 Ion Channels Human genes 0.000 description 1
- 108090000862 Ion Channels Proteins 0.000 description 1
- COLNVLDHVKWLRT-QMMMGPOBSA-N L-phenylalanine Chemical compound OC(=O)[C@@H](N)CC1=CC=CC=C1 COLNVLDHVKWLRT-QMMMGPOBSA-N 0.000 description 1
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 1
- 206010056886 Mucopolysaccharidosis I Diseases 0.000 description 1
- 206010068871 Myotonic dystrophy Diseases 0.000 description 1
- 208000012902 Nervous system disease Diseases 0.000 description 1
- 208000025966 Neurological disease Diseases 0.000 description 1
- HSVIINWFPKEOEQ-NSHDSACASA-N Ofloxacin methyl ester Chemical compound C([C@H](C)N1C=C(C(C(=C11)C=C2F)=O)C(=O)OC)OC1=C2N1CCN(C)CC1 HSVIINWFPKEOEQ-NSHDSACASA-N 0.000 description 1
- 206010031243 Osteogenesis imperfecta Diseases 0.000 description 1
- QSXMZJGGEWYVCN-UHFFFAOYSA-N Pirbuterol acetate Chemical compound CC(O)=O.CC(C)(C)NCC(O)C1=CC=C(O)C(CO)=N1 QSXMZJGGEWYVCN-UHFFFAOYSA-N 0.000 description 1
- 238000012300 Sequence Analysis Methods 0.000 description 1
- GIIZNNXWQWCKIB-UHFFFAOYSA-N Serevent Chemical compound C1=C(O)C(CO)=CC(C(O)CNCCCCCCOCCCCC=2C=CC=CC=2)=C1 GIIZNNXWQWCKIB-UHFFFAOYSA-N 0.000 description 1
- 208000009415 Spinocerebellar Ataxias Diseases 0.000 description 1
- 102000003673 Symporters Human genes 0.000 description 1
- 108090000088 Symporters Proteins 0.000 description 1
- PGAVKCOVUIYSFO-XVFCMESISA-N UTP Chemical compound O[C@@H]1[C@H](O)[C@@H](COP(O)(=O)OP(O)(=O)OP(O)(O)=O)O[C@H]1N1C(=O)NC(=O)C=C1 PGAVKCOVUIYSFO-XVFCMESISA-N 0.000 description 1
- 240000008042 Zea mays Species 0.000 description 1
- 235000005824 Zea mays ssp. parviglumis Nutrition 0.000 description 1
- 235000002017 Zea mays subsp mays Nutrition 0.000 description 1
- 238000010521 absorption reaction Methods 0.000 description 1
- 239000002253 acid Substances 0.000 description 1
- 230000004913 activation Effects 0.000 description 1
- 230000002411 adverse Effects 0.000 description 1
- NDAUXUAQIAJITI-UHFFFAOYSA-N albuterol Chemical compound CC(C)(C)NCC(O)C1=CC=C(O)C(CO)=C1 NDAUXUAQIAJITI-UHFFFAOYSA-N 0.000 description 1
- 229960004821 amikacin Drugs 0.000 description 1
- LKCWBDHBTVXHDL-RMDFUYIESA-N amikacin Chemical compound O([C@@H]1[C@@H](N)C[C@H]([C@@H]([C@H]1O)O[C@@H]1[C@@H]([C@@H](N)[C@H](O)[C@@H](CO)O1)O)NC(=O)[C@@H](O)CCN)[C@H]1O[C@H](CN)[C@@H](O)[C@H](O)[C@H]1O LKCWBDHBTVXHDL-RMDFUYIESA-N 0.000 description 1
- 229960005475 antiinfective agent Drugs 0.000 description 1
- 239000004599 antimicrobial Substances 0.000 description 1
- 239000003125 aqueous solvent Substances 0.000 description 1
- 201000004562 autosomal dominant cerebellar ataxia Diseases 0.000 description 1
- 229960004099 azithromycin Drugs 0.000 description 1
- MQTOSJVFKKJCRP-BICOPXKESA-N azithromycin Chemical compound O([C@@H]1[C@@H](C)C(=O)O[C@@H]([C@@]([C@H](O)[C@@H](C)N(C)C[C@H](C)C[C@@](C)(O)[C@H](O[C@H]2[C@@H]([C@H](C[C@@H](C)O2)N(C)C)O)[C@H]1C)(C)O)CC)[C@H]1C[C@@](C)(OC)[C@@H](O)[C@H](C)O1 MQTOSJVFKKJCRP-BICOPXKESA-N 0.000 description 1
- 230000002146 bilateral effect Effects 0.000 description 1
- 239000013060 biological fluid Substances 0.000 description 1
- 230000033228 biological regulation Effects 0.000 description 1
- 229920001222 biopolymer Polymers 0.000 description 1
- 230000015572 biosynthetic process Effects 0.000 description 1
- 235000021152 breakfast Nutrition 0.000 description 1
- 239000004161 brilliant blue FCF Substances 0.000 description 1
- 235000012745 brilliant blue FCF Nutrition 0.000 description 1
- 239000000168 bronchodilator agent Substances 0.000 description 1
- 230000001413 cellular effect Effects 0.000 description 1
- 230000008859 change Effects 0.000 description 1
- 230000005465 channeling Effects 0.000 description 1
- 229910052801 chlorine Inorganic materials 0.000 description 1
- 150000001805 chlorine compounds Chemical group 0.000 description 1
- 210000000349 chromosome Anatomy 0.000 description 1
- 230000001684 chronic effect Effects 0.000 description 1
- 229960003405 ciprofloxacin Drugs 0.000 description 1
- 230000015271 coagulation Effects 0.000 description 1
- 238000005345 coagulation Methods 0.000 description 1
- 229950005980 cobiprostone Drugs 0.000 description 1
- 239000008119 colloidal silica Substances 0.000 description 1
- 238000009500 colour coating Methods 0.000 description 1
- 238000002648 combination therapy Methods 0.000 description 1
- 238000005056 compaction Methods 0.000 description 1
- 235000005822 corn Nutrition 0.000 description 1
- 229940092125 creon Drugs 0.000 description 1
- 239000002178 crystalline material Substances 0.000 description 1
- 230000003247 decreasing effect Effects 0.000 description 1
- 230000018044 dehydration Effects 0.000 description 1
- 238000006297 dehydration reaction Methods 0.000 description 1
- 230000001079 digestive effect Effects 0.000 description 1
- 230000005750 disease progression Effects 0.000 description 1
- 235000020669 docosahexaenoic acid Nutrition 0.000 description 1
- KAUVQQXNCKESLC-UHFFFAOYSA-N docosahexaenoic acid (DHA) Natural products COC(=O)C(C)NOCC1=CC=CC=C1 KAUVQQXNCKESLC-UHFFFAOYSA-N 0.000 description 1
- 108010067396 dornase alfa Proteins 0.000 description 1
- 239000003792 electrolyte Substances 0.000 description 1
- 230000002708 enhancing effect Effects 0.000 description 1
- 229940088598 enzyme Drugs 0.000 description 1
- 235000020937 fasting conditions Nutrition 0.000 description 1
- 229940083280 fd&c blue #1 aluminum lake Drugs 0.000 description 1
- 230000035558 fertility Effects 0.000 description 1
- 230000020764 fibrinolysis Effects 0.000 description 1
- 238000011049 filling Methods 0.000 description 1
- 239000010419 fine particle Substances 0.000 description 1
- 230000004907 flux Effects 0.000 description 1
- 229960000308 fosfomycin Drugs 0.000 description 1
- YMDXZJFXQJVXBF-STHAYSLISA-N fosfomycin Chemical compound C[C@@H]1O[C@@H]1P(O)(O)=O YMDXZJFXQJVXBF-STHAYSLISA-N 0.000 description 1
- 230000004927 fusion Effects 0.000 description 1
- 230000002496 gastric effect Effects 0.000 description 1
- 239000008187 granular material Substances 0.000 description 1
- 230000005484 gravity Effects 0.000 description 1
- 125000001475 halogen functional group Chemical group 0.000 description 1
- 102000056427 human CFTR Human genes 0.000 description 1
- 102000052502 human ELANE Human genes 0.000 description 1
- 150000004677 hydrates Chemical class 0.000 description 1
- XXSMGPRMXLTPCZ-UHFFFAOYSA-N hydroxychloroquine Chemical compound ClC1=CC=C2C(NC(C)CCCN(CCO)CC)=CC=NC2=C1 XXSMGPRMXLTPCZ-UHFFFAOYSA-N 0.000 description 1
- 229960004171 hydroxychloroquine Drugs 0.000 description 1
- 229960001680 ibuprofen Drugs 0.000 description 1
- 238000010191 image analysis Methods 0.000 description 1
- 230000001771 impaired effect Effects 0.000 description 1
- 239000005414 inactive ingredient Substances 0.000 description 1
- KHLVKKOJDHCJMG-QDBORUFSSA-L indigo carmine Chemical compound [Na+].[Na+].N/1C2=CC=C(S([O-])(=O)=O)C=C2C(=O)C\1=C1/NC2=CC=C(S(=O)(=O)[O-])C=C2C1=O KHLVKKOJDHCJMG-QDBORUFSSA-L 0.000 description 1
- 239000004179 indigotine Substances 0.000 description 1
- 235000012738 indigotine Nutrition 0.000 description 1
- 208000015181 infectious disease Diseases 0.000 description 1
- 208000021267 infertility disease Diseases 0.000 description 1
- 230000004054 inflammatory process Effects 0.000 description 1
- 208000019715 inherited Creutzfeldt-Jakob disease Diseases 0.000 description 1
- 239000003112 inhibitor Substances 0.000 description 1
- 230000003834 intracellular effect Effects 0.000 description 1
- 229960001375 lactose Drugs 0.000 description 1
- 229950004331 lancovutide Drugs 0.000 description 1
- 238000002356 laser light scattering Methods 0.000 description 1
- 230000004807 localization Effects 0.000 description 1
- 239000011777 magnesium Substances 0.000 description 1
- 229910052749 magnesium Inorganic materials 0.000 description 1
- 238000000691 measurement method Methods 0.000 description 1
- 230000007246 mechanism Effects 0.000 description 1
- 230000001404 mediated effect Effects 0.000 description 1
- 238000002844 melting Methods 0.000 description 1
- 230000008018 melting Effects 0.000 description 1
- 230000000813 microbial effect Effects 0.000 description 1
- 238000000465 moulding Methods 0.000 description 1
- 230000003274 myotonic effect Effects 0.000 description 1
- 239000002105 nanoparticle Substances 0.000 description 1
- 239000006199 nebulizer Substances 0.000 description 1
- 230000002988 nephrogenic effect Effects 0.000 description 1
- 230000000926 neurological effect Effects 0.000 description 1
- 239000002773 nucleotide Substances 0.000 description 1
- 125000003729 nucleotide group Chemical group 0.000 description 1
- 239000003960 organic solvent Substances 0.000 description 1
- 230000003204 osmotic effect Effects 0.000 description 1
- 230000011164 ossification Effects 0.000 description 1
- 238000012856 packing Methods 0.000 description 1
- 229940124531 pharmaceutical excipient Drugs 0.000 description 1
- COLNVLDHVKWLRT-UHFFFAOYSA-N phenylalanine Natural products OC(=O)C(N)CC1=CC=CC=C1 COLNVLDHVKWLRT-UHFFFAOYSA-N 0.000 description 1
- 230000026731 phosphorylation Effects 0.000 description 1
- 238000006366 phosphorylation reaction Methods 0.000 description 1
- 239000000049 pigment Substances 0.000 description 1
- 229960005095 pioglitazone Drugs 0.000 description 1
- 229960004994 pirbuterol acetate Drugs 0.000 description 1
- 239000011148 porous material Substances 0.000 description 1
- 230000003389 potentiating effect Effects 0.000 description 1
- 239000002244 precipitate Substances 0.000 description 1
- 230000000750 progressive effect Effects 0.000 description 1
- 201000002212 progressive supranuclear palsy Diseases 0.000 description 1
- 230000020978 protein processing Effects 0.000 description 1
- 230000002685 pulmonary effect Effects 0.000 description 1
- 229940107568 pulmozyme Drugs 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 230000000241 respiratory effect Effects 0.000 description 1
- 210000001533 respiratory mucosa Anatomy 0.000 description 1
- 208000023504 respiratory system disease Diseases 0.000 description 1
- 229960002052 salbutamol Drugs 0.000 description 1
- 229960004017 salmeterol Drugs 0.000 description 1
- 230000003248 secreting effect Effects 0.000 description 1
- 238000005029 sieve analysis Methods 0.000 description 1
- 229960003310 sildenafil Drugs 0.000 description 1
- 229910052710 silicon Inorganic materials 0.000 description 1
- 239000010703 silicon Substances 0.000 description 1
- 108010081062 sinapultide Proteins 0.000 description 1
- 229960000802 sinapultide Drugs 0.000 description 1
- 229940126586 small molecule drug Drugs 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- 239000008275 solid aerosol Substances 0.000 description 1
- 238000003756 stirring Methods 0.000 description 1
- 238000003786 synthesis reaction Methods 0.000 description 1
- WMOVHXAZOJBABW-UHFFFAOYSA-N tert-butyl acetate Chemical compound CC(=O)OC(C)(C)C WMOVHXAZOJBABW-UHFFFAOYSA-N 0.000 description 1
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- PASYJVRFGUDDEW-WMUGRWSXSA-J tetrasodium;[[(2r,3s,5r)-5-(4-amino-2-oxopyrimidin-1-yl)-3-hydroxyoxolan-2-yl]methoxy-oxidophosphoryl] [[[(2r,3s,4r,5r)-5-(2,4-dioxopyrimidin-1-yl)-3,4-dihydroxyoxolan-2-yl]methoxy-oxidophosphoryl]oxy-oxidophosphoryl] phosphate Chemical compound [Na+].[Na+].[Na+].[Na+].O=C1N=C(N)C=CN1[C@@H]1O[C@H](COP([O-])(=O)OP([O-])(=O)OP([O-])(=O)OP([O-])(=O)OC[C@@H]2[C@H]([C@@H](O)[C@@H](O2)N2C(NC(=O)C=C2)=O)O)[C@@H](O)C1 PASYJVRFGUDDEW-WMUGRWSXSA-J 0.000 description 1
- 239000004408 titanium dioxide Substances 0.000 description 1
- 229940054369 ultrase Drugs 0.000 description 1
- 210000001177 vas deferen Anatomy 0.000 description 1
- 239000011800 void material Substances 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/2004—Excipients; Inactive ingredients
- A61K9/2022—Organic macromolecular compounds
- A61K9/205—Polysaccharides, e.g. alginate, gums; Cyclodextrin
- A61K9/2054—Cellulose; Cellulose derivatives, e.g. hydroxypropyl methylcellulose
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/47—Quinolines; Isoquinolines
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
- A61K9/16—Agglomerates; Granulates; Microbeadlets ; Microspheres; Pellets; Solid products obtained by spray drying, spray freeze drying, spray congealing,(multiple) emulsion solvent evaporation or extraction
- A61K9/1605—Excipients; Inactive ingredients
- A61K9/1629—Organic macromolecular compounds
- A61K9/1635—Organic macromolecular compounds obtained by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyvinyl pyrrolidone, poly(meth)acrylates
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
- A61K9/16—Agglomerates; Granulates; Microbeadlets ; Microspheres; Pellets; Solid products obtained by spray drying, spray freeze drying, spray congealing,(multiple) emulsion solvent evaporation or extraction
- A61K9/1605—Excipients; Inactive ingredients
- A61K9/1629—Organic macromolecular compounds
- A61K9/1652—Polysaccharides, e.g. alginate, cellulose derivatives; Cyclodextrin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/2004—Excipients; Inactive ingredients
- A61K9/2022—Organic macromolecular compounds
- A61K9/2027—Organic macromolecular compounds obtained by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyvinyl pyrrolidone, poly(meth)acrylates
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/28—Dragees; Coated pills or tablets, e.g. with film or compression coating
- A61K9/2806—Coating materials
- A61K9/282—Organic compounds, e.g. fats
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/10—Laxatives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/16—Drugs for disorders of the alimentary tract or the digestive system for liver or gallbladder disorders, e.g. hepatoprotective agents, cholagogues, litholytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/18—Drugs for disorders of the alimentary tract or the digestive system for pancreatic disorders, e.g. pancreatic enzymes
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/02—Nasal agents, e.g. decongestants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/04—Drugs for disorders of the respiratory system for throat disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/06—Antiasthmatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/16—Central respiratory analeptics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/02—Drugs for disorders of the urinary system of urine or of the urinary tract, e.g. urine acidifiers
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
- A61P15/08—Drugs for genital or sexual disorders; Contraceptives for gonadal disorders or for enhancing fertility, e.g. inducers of ovulation or of spermatogenesis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/04—Drugs for skeletal disorders for non-specific disorders of the connective tissue
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/08—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/08—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease
- A61P19/10—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease for osteoporosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P21/00—Drugs for disorders of the muscular or neuromuscular system
- A61P21/02—Muscle relaxants, e.g. for tetanus or cramps
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
- A61P25/16—Anti-Parkinson drugs
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/22—Anxiolytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/06—Antihyperlipidemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/08—Antiallergic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
- A61P5/06—Drugs for disorders of the endocrine system of the anterior pituitary hormones, e.g. TSH, ACTH, FSH, LH, PRL, GH
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
- A61P5/14—Drugs for disorders of the endocrine system of the thyroid hormones, e.g. T3, T4
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
- A61P5/18—Drugs for disorders of the endocrine system of the parathyroid hormones
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
- A61P5/48—Drugs for disorders of the endocrine system of the pancreatic hormones
- A61P5/50—Drugs for disorders of the endocrine system of the pancreatic hormones for increasing or potentiating the activity of insulin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/10—Antioedematous agents; Diuretics
Landscapes
- Health & Medical Sciences (AREA)
- Engineering & Computer Science (AREA)
- Chemical & Material Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Pharmacology & Pharmacy (AREA)
- Veterinary Medicine (AREA)
- Life Sciences & Earth Sciences (AREA)
- Medicinal Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Organic Chemistry (AREA)
- Diabetes (AREA)
- Epidemiology (AREA)
- Pulmonology (AREA)
- Physical Education & Sports Medicine (AREA)
- Neurology (AREA)
- Biomedical Technology (AREA)
- Neurosurgery (AREA)
- Endocrinology (AREA)
- Hematology (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Rheumatology (AREA)
- Obesity (AREA)
- Reproductive Health (AREA)
- Otolaryngology (AREA)
- Urology & Nephrology (AREA)
- Hospice & Palliative Care (AREA)
- Gastroenterology & Hepatology (AREA)
- Gynecology & Obstetrics (AREA)
- Psychiatry (AREA)
- Immunology (AREA)
- Ophthalmology & Optometry (AREA)
- Emergency Medicine (AREA)
- Psychology (AREA)
- Pain & Pain Management (AREA)
- Pregnancy & Childbirth (AREA)
Abstract
Una composición farmacéutica que comprende una dispersión sólida, en donde la dispersión sólida comprende: a) 80% en peso de N-[2,4-Bis(1,1-dimetiletil)-5-hidroxifenil]-1,4-dihidro-4-oxoquinolin-3-carboxamida (Compuesto 1) amorfa o sustancialmente amorfa por peso de la dispersión, en donde el Compuesto 1 sustancialmente amorfo comprende menos del 15% de Compuesto 1 cristalino, b) 19,5% en peso de succinato de acetato de hidroxipropilmetilcelulosa (HPMCAS) o copolímero de vinilpirrolidona/acetato de vinilo (PVP/VA) por peso de la dispersión, y c) 0,5% en peso de lauril sulfato de sodio (SLS) por peso de la dispersión.
Description
DESCRIPCIÓN
Composición farmacéutica y administraciones de la misma
REFERENCIA CRUZADA A SOLICITUDES RELACIONADAS
Esta solicitud reivindica prioridad de la USSN 61/088.704 presentada el 13 de agosto de 2008, USSN 61/088.801, presentada el 14 de agosto de 2008, USSN 61/090.096, presentada el 19 de agosto de 2008, USSN 61/146.163, presentada el 21 de enero de 2009, USSN 61/181.527, presentada el 27 de mayo de 2009 y USSN 61/183.345, presentada el 2 de junio de 2009.
CAMPO DE LA INVENCIÓN
La presente invención se refiere a composiciones farmacéuticas que comprenden una dispersión sólida de N-[2,4-Bis(1,1-dimetiletilo)-5-hidroxifenilo]-1,4-dihidro-4-oxoquinolina-3-carboxamida, y métodos de fabricación y administración de composiciones farmacéuticas que comprenden N-[2,4-Bis(1,1-dimetiletilo)-5-hidroxifenilo]-1,4-dihidro-4-oxoquinolina-3-carboxamida.
ANTECEDENTES
La fibrosis quística (FQ) es una enfermedad genética recesiva que afecta a aproximadamente 30,000 niños y adultos en los Estados Unidos y aproximadamente 30,000 niños y adultos en Europa. A pesar del progreso en el tratamiento de la FQ, no hay cura.
La FQ es causada por mutaciones en el gen regulador de la conductancia transmembrana de la fibrosis quística (CFTR) que codifica un canal de ion de cloruro epitelial responsable de ayudar en la regulación de la absorción y secreción de agua y sal en diversos tejidos. Los fármacos de molécula pequeña, conocidos como potenciadores que aumentan la probabilidad de apertura del canal de CFTR, representan una estrategia terapéutica potencial para tratar la FQ.
Específicamente, CFTR es un canal de aniones mediado por cAMP/ATP que se expresa en una variedad de tipos de células, incluyendo absorción y células epiteliales secretoras, donde regula el flujo de aniones a través de la membrana, así como la actividad de otros canales iónicos y proteínas En las células del epitelio, el funcionamiento normal del CFTR es fundamental para el mantenimiento del transporte de electrolitos en todo el cuerpo, incluidos los tejidos respiratorios y digestivos. CFTR está compuesto por aproximadamente 1480 aminoácidos que codifican una proteína compuesta por una repetición en tándem de dominios transmembrana, cada uno con seis hélices de transmembrana y un dominio de unión a nucleótidos. Los dos dominios de transmembrana están unidos por un dominio regulatorio (R) grande y polar con múltiples sitios de fosforilación que regulan la actividad del canal y el tráfico celular.
El gen que codifica CFTR se ha identificado y secuenciado (Véase Gregory, R.J. et al (1990) Nature 347: 382-386; Rich, DP et al (1990) Nature 347: 358-362), (Riordan, J.R. y et al (1989) Science 245:1066 - 1073). Un defecto en este gen causa mutaciones en CFTR que producen fibrosis quística ("FQ"), la enfermedad genética mortal más común en los seres humanos. La fibrosis quística afecta aproximadamente a uno de cada 2.500 bebés en los Estados Unidos. Dentro de la población general de los Estados Unidos, hasta 10 millones de personas portan una sola copia del gen defectuoso sin aparentes efectos negativos. Por el contrario, las personas con dos copias del gen asociado a la FQ padecen los efectos debilitantes y fatales de la FQ, incluida la enfermedad pulmonar crónica.
En pacientes con FQ, las mutaciones en CFTR expresadas endógenamente en el epitelio respiratorio conducen a reducción de la secreción de aniones apical causando un desequilibrio en transporte de iones y fluidos. La disminución resultante en el transporte de aniones contribuye a una mayor acumulación de moco en el pulmón y las infecciones microbianas que las acompañan y que finalmente causan la muerte en pacientes con FQ. Además de la enfermedad respiratoria, los pacientes con FQ suelen padecer problemas gastrointestinales e insuficiencia pancreática que, si no se tratan, ocasionan la muerte. Además, la mayoría de los hombres con fibrosis quística son infértiles y la fertilidad se disminuye entre las mujeres con fibrosis quística. En contraste con los efectos severos de dos copias del gen asociado a FQ, los individuos con una copia única del gen asociado a FQ exhiben una mayor resistencia al cólera y a la deshidratación resultante de la diarrea, quizás explicando la frecuencia relativamente alta del gen de FQ en la población .
El análisis de secuencia del gen CFTR de cromosomas de FQ ha revelado una variedad de mutaciones causantes de enfermedades (Cutting, G. R. et al (1990) Nature 346:366-369; Dean, M. et al (1990) Cell 61:863:870; y Kerem, BS y col., (1989) Science 245:1073 - 1080, Kerem, B-S et al (1990) Proc. Natl. Acad. Sci. EE.UU. 87:8447 - 8451). Hasta la fecha, se han identificado más de 1000 mutaciones que causan enfermedades en el gen de la FQ (http://www.genet.sickkids.on.ca/cftr/). La mutación más prevalente es una deleción de fenilalanina en la posición 508 de la secuencia de ácido CFTR amino, y se conoce comúnmente como AF508-CFTR. Esta mutación ocurre en
aproximadamente el 70% de los casos de fibrosis quística y se asocia con una enfermedad grave.
La supresión de residuo 508 en AF508-CFTR impide que la proteína naciente se pliegue correctamente. Esto da como resultado la incapacidad de la proteína mutante para salir de la sala de emergencias y el tráfico a la membrana plasmática. Como resultado, el número de canales presentes en la membrana es mucho menor que el observado en las células que expresan CFTR de tipo salvaje. Además del tráfico deteriorado, la mutación resulta en un canal defectuoso. Juntos, la reducción del número de canales en la membrana y la activación defectuosa conducen a un transporte aniónico reducido a través de los epitelios, lo que produce un transporte defectuoso de iones y fluidos. (Quinton, P.M. (1990), FASEB J. 4:2709 - 2727). Los estudios han demostrado, sin embargo, que los números reducidos de AF508-CFTR en la membrana son funcionales, aunque menos que CFTR de tipo salvaje. (Dalemans et al., (1991), Nature Lond., 354:526 - 528, Denning et al., Supra, Pasyk y Foskett (1995), J. Cell, Biochem., 270:12347 - 50). Además de AF508-CFTR, otras mutaciones causantes de enfermedades en CFTR que dan como resultado un tráfico defectuoso, síntesis y/o canalización pueden regularse hacia arriba o hacia abajo para alterar la secreción de aniones y modificar la progresión y/o gravedad de la enfermedad.
Aunque CFTR transporta una variedad de moléculas, además de aniones, es evidente que este papel (el transporte de aniones) representa un elemento en un importante mecanismo de transporte de iones y agua a través del epitelio. Los otros elementos incluyen el canal epitelial Na+, ENaC, Na+/2Cl-/K+ co-transportador, bomba Na+-K -ATPasa y los canales de la membrana basolateral K+, que son responsables de la absorción de cloruro en la célula.
Estos elementos trabajan juntos para conseguir el transporte direccional a través del epitelio a través de su expresión selectiva y la localización dentro de la célula. La absorción de cloruro tiene lugar por la actividad coordinada de ENaC y CFTR presente en la membrana apical y la bomba Na+-K+-ATPasa y los canales iónicos Cl expresados en la superficie basolateral de la célula. El transporte secundario activo de cloruro desde el lado luminal conduce a la acumulación de cloruro intracelular, que luego puede salir pasivamente de la célula a través de canales de Cl, dando como resultado un transporte vectorial. La disposición del transportador Na+/2Cl-/K+, bomba Na+-K+-ATPasa y los canales K+ de la membrana basolateral en la superficie basolateral y CFTR en el lado luminal coordinan la secreción de cloruro a través de CFTR en el lado luminal. Debido a que el agua probablemente nunca se transporta activamente, su flujo y epitelio depende de pequeños gradientes osmóticos transepiteliales generados por el flujo masivo de sodio y cloruro.
Como se discutió anteriormente, se cree que la supresión de residuo 508 en AF508-CFTR impide que la proteína naciente se pliegue correctamente, lo que resulta en la incapacidad de esta proteína mutante para salir del ER, y el tráfico a la membrana plasmática. Como resultado, cantidades insuficientes de la proteína madura están presentes en la membrana plasmática y el transporte de cloruro dentro de los tejidos epiteliales se reduce significativamente. De hecho, este fenómeno celular del procesamiento ER defectuoso de los transportadores ABC por la maquinaria ER ha demostrado ser la base subyacente no solo para la enfermedad de la FQ, sino también para una amplia gama de otras enfermedades aisladas y hereditarias.
N-[2,4-Bis(1,1-dimetiletilo)-5-hidroxifenilo]-1,4-dihidro-4-oxoquinolina-3-carboxamida es un potenciador de CFTR selectivo y potente de formas de tipo silvestre y mutantes (incluyendo, por ejemplo, AF508, R117H, y G551D) de CFTR humano. N-[2,4-Bis(1,1-dimetiletilo)-5-hidroxiefenilo]-1,4-dihidro-4-oxoquinolina-3-carboxamida es útil para el tratamiento de pacientes adultos con fibrosis quística y al menos un Alelo G551D-CFTR.
Por consiguiente, existe la necesidad de composiciones farmacéuticas biodisponibles estables de N-[2,4-Bis(1,1-dimetiletilo)-5-hidroxifenilo]-1,4-dihidro-4-oxoquinolina-3-carboxamida útil para tratar pacientes que padecen de FQ y métodos para su administración.
RESUMEN DE LA INVENCIÓN
La invención se refiere a composiciones farmacéuticas que comprenden una dispersión sólida de N-[2,4-bis(1,1-dimetiletil)-5-hidroxifenil]-1,4-dihidro-4-oxoquinolin-3-carboxamida ("Compuesto 1" ) como se define en las reivindicaciones. Las composiciones farmacéuticas también pueden incluir uno o más de los siguientes excipientes: una carga, un disgregante, un deslizante, un lubricante, un aglutinante y un surfactante.
En una realización, la presente invención proporciona una composición farmacéutica que comprende una dispersión sólida del compuesto amorfo 1, en el que la dispersión sólida comprende aproximadamente 25 mg del compuesto amorfo 1. En ciertas realizaciones, la dispersión sólida comprende aproximadamente 25 mg de Compuesto 1sustancialmente amorfo.
En una realización, la presente invención proporciona una composición farmacéutica que comprende una dispersión sólida del compuesto amorfo 1, en el que la dispersión sólida comprende aproximadamente 50 mg del compuesto amorfo 1. En ciertas realizaciones, la dispersión sólida comprende aproximadamente 50 mg de Compuesto 1 sustancialmente amorfo.
En una realización, la presente invención proporciona una composición farmacéutica que comprende una dispersión sólida del compuesto amorfo 1, en el que la dispersión sólida comprende aproximadamente 75 mg del compuesto amorfo 1. En ciertas realizaciones, la dispersión sólida comprende aproximadamente 75 mg de Compuesto 1 sustancialmente amorfo.
En una realización, la presente invención proporciona una composición farmacéutica que comprende una dispersión sólida del compuesto amorfo 1, en el que la dispersión sólida comprende aproximadamente 100 mg del compuesto amorfo 1. En ciertas realizaciones, la dispersión sólida comprende aproximadamente 100 mg de Compuesto 1 sustancialmente amorfo.
En una realización, la presente invención proporciona una composición farmacéutica que comprende una dispersión sólida del compuesto amorfo 1, en el que la dispersión sólida comprende aproximadamente 150 mg del compuesto amorfo 1. En ciertas realizaciones, la dispersión sólida comprende aproximadamente 150 mg de Compuesto 1 sustancialmente amorfo.
En una realización, la presente invención proporciona una composición farmacéutica que comprende una dispersión sólida del compuesto amorfo 1, en el que la dispersión sólida comprende aproximadamente 250 mg de compuesto amorfo 1. En ciertas realizaciones, la dispersión sólida comprende aproximadamente 250 mg de Compuesto 1 sustancialmente amorfo.
En un aspecto, la forma sólida del Compuesto 1 en la composición farmacéutica es una dispersión sólida que comprende el Compuesto 1 sustancialmente amorfo o amorfo y un polímero de acetato succinato de hidroxipropilmetilcelulosa (HPMCAS) o copolímero de vinilpirrolidona/acetato de vinilo (PVP/VA). Las realizaciones de este aspecto incluyen una o más de las siguientes: La dispersión sólida es un polvo que tiene un diámetro de partícula medio mayor de aproximadamente 5 pm o la dispersión sólida tiene una densidad aparente de aproximadamente 0,10 g/cc o mayor. 1 g/cc = 1 g/cm3.
En otras realizaciones más, las composiciones farmacéuticas también comprenden una carga (por ejemplo, lactosa, sorbitol, celulosas, fosfatos de calcio, almidones, azúcares (por ejemplo, manitol, sacarosa o similares) o cualquier combinación de los mismos) en concentraciones de por lo menos aproximadamente el 10% en peso en peso de la composición; un disgregante (por ejemplo, croscarmelosa de sodio, glicolato de almidón de sodio o una combinación de los mismos) en concentraciones de aproximadamente el 10% en peso o menos en peso de la composición; un surfactante (por ejemplo, laurilsulfato de sodio, estearilfumarato de sodio (SSF), monooleato de polioxietilen 20 sorbitán o cualquier combinación de los mismos) en concentraciones de aproximadamente el 10% en peso o menos en peso de la composición; un aglutinante (por ejemplo, celulosa microcristalina, fosfato de calcio dibásico, sacarosa, almidón de maíz, celulosa modificada (por ejemplo, hidroximetilcelulosa), o cualquier combinación de los mismos) en concentraciones de por lo menos aproximadamente el 1% en peso en peso de la composición; un deslizante (por ejemplo, dióxido de silicio coloidal, talco o una combinación de los mismos) en concentraciones de aproximadamente el 2% en peso o menos en peso de la composición; y un lubricante (por ejemplo, estearato de magnesio, ácido esteárico, aceite hidrogenado, estearilfumarato de sodio o cualquier combinación de los mismos) en concentraciones de aproximadamente el 2% en peso o menos en peso de la composición.
Dichas composiciones farmacéuticas pueden comprender opcionalmente uno o más colorantes, fragancias, y/o sabores para mejorar su atractivo visual, el gusto y olor.
Otro aspecto de la presente invención proporciona una composición farmacéutica que consiste en una tableta que comprende una dispersión sólida, una carga, un disgregante, un agente surfactante, un aglutinante, un deslizante y un lubricante, en el que la tableta tiene una disolución de al menos aproximadamente 50% en aproximadamente 30 minutos, y la dispersión sólida comprende Compuesto 1 sustancialmente amorfo. Como se observa a continuación, la disolución se mide con un aparato estándar USP tipo II que emplea un medio de disolución de sulfato de laurilo sódico al 0,6% disuelto en 900 mL de agua DI (o un volumen de medio que tiene la misma proporción de agua SLS a DI) a una temperatura de aproximadamente 37°C. Se ensaya una única tableta experimental en cada recipiente de prueba del aparato. La disolución también se puede medir con un aparato USP Tipo II estándar que emplea un medio de disolución de laurilo de sulfato sódico al 0,7% disuelto en 900 mL de 50 mM de tampón de fosfato sódico (pH 6,8) a una temperatura de aproximadamente 37°C. La disolución también se puede medir con un aparato USP Tipo II estándar que emplea un medio de disolución de laurilo de sulfato sódico al 0,5% disuelto en 900 mL de 50 mM de tampón de fosfato sódico (pH 6,8) a una temperatura de aproximadamente 37°C. Se ensaya una única tableta experimental en cada recipiente de prueba del aparato.
Otro aspecto de la presente invención proporciona una composición farmacéutica que consiste de un comprimido que comprende una dispersión sólida que comprende el compuesto 1 amorfo o sustancialmente amorfo y HPMCAS o PVP/VA y, una carga, un disgregante, un surfactante, un aglutinante, un deslizante, y un lubricante, en donde el comprimido tiene una dureza de por lol menos aproximadamente 5 Kp. 1 Kp = 9.8 N.
En otro aspecto más, las tabletas descritas aquí están recubiertas.
En otro aspecto, los comprimidos recubiertos descritos en este documento son de color.
En otro aspecto más, las tabletas recubiertas de color incluyen texto o imágenes. Por ejemplo, el texto o las imágenes pueden imprimirse en la tableta recubierta de color.
En otros aspectos más, las tabletas revestidas de color incluyen aproximadamente 3% en peso de un revestimiento de película que comprende un colorante azul, tal como OPADRY® II. En algunas realizaciones, las tabletas de color pueden etiquetarse con un logotipo y texto que indique la intensidad del ingrediente activo en la tableta usando una tinta negra, tal como Opacode® Wb o Opacode® S-1-17823. En todavía otras realizaciones, los comprimidos recubiertos de color se recubren con un colorante, se enceran y luego se marcan con un logotipo, otra imagen y/o texto usando una tinta adecuada. En algunas realizaciones, las tabletas se recubren con aproximadamente 3% en peso de colorante, y se enceran con polvo de cera de Carnauba pesado en la cantidad de aproximadamente 0,01% p/p del peso del núcleo de la tableta de partida. Las tabletas enceradas se pueden etiquetar con un logotipo y texto que indique la intensidad del ingrediente activo en la tableta utilizando una tinta adecuada.
Otro aspecto de la presente invención proporciona un método de producción de una composición farmacéutica formada por las etapas de proporcionar una mezcla de una dispersión sólida del compuesto amorfo 1, un aglutinante, un agente deslizante, un agente surfactante, un lubricante, un disgregante, y una carga, y comprimir la mezcla en una tableta que tiene una disolución de al menos aproximadamente 50% en aproximadamente 30 minutos. En un ejemplo, la mezcla se comprime hasta una dureza de al menos aproximadamente 5 Kp.
Otro aspecto de la presente invención proporciona un método de producción de una composición farmacéutica formada por las etapas de proporcionar una mezcla de una dispersión sólida del compuesto amorfo 1, un aglutinante, un agente deslizante, un agente surfactante, un lubricante, un disgregante, y una carga, y comprimir la mezcla en una tableta que tiene una disolución de al menos aproximadamente 70% en aproximadamente 30 minutos.
Otro aspecto de la presente invención proporciona un método de administración de una composición farmacéutica mediante la administración oral a un paciente al menos una vez por día de al menos un comprimido que comprende una dispersión sólida de Compuesto 1 amorfo o sustancialmente amorfo, una carga, un aglutinante, un deslizante, un desintegrante, un surfactante y un lubricante, en el que la dispersión sólida comprende al menos aproximadamente 25 mg de Compuesto 1 amorfo o sustancialmente amorfo. En algunas realizaciones, la tableta se administra oralmente al paciente una vez al día. Otros comprimidos útiles en este método comprenden una dispersión sólida que contiene al menos aproximadamente 50 mg de Compuesto 1 amorfo o sustancialmente amorfo. Algunos comprimidos útiles en este método comprenden una dispersión sólida que contiene al menos aproximadamente 75 mg de Compuesto 1 amorfo o sustancialmente amorfo. Otras tabletas útiles en este método comprenden una dispersión sólida que contiene al menos aproximadamente 100 mg de Compuesto 1 amorfo o sustancialmente amorfo. Aún otros comprimidos útiles en este método comprenden una dispersión sólida que contiene al menos aproximadamente 150 mg de Compuesto 1 amorfo o sustancialmente amorfo. En otro método , la administración comprende administrar oralmente a un paciente al menos una vez al día al menos una tableta que comprende una dispersión sólida de Compuesto 1 amorfo o sustancialmente amorfo, una carga, un aglutinante, un deslizante, un desintegrante, un agente surfactante y un lubricante, en el que la dispersión sólida contiene al menos aproximadamente 250 mg de Compuesto 1 amorfo o sustancialmente amorfo.
En algunas realizaciones, la presente invención proporciona un método de administración oral de las composiciones farmacéuticas descritas aquí al menos una vez al día. En otras realizaciones, la presente invención proporciona un método de administración oral de la composición farmacéutica descrita aquí una vez al día. En algunas realizaciones, la presente invención proporciona un método de administración oral de la composición farmacéutica descrita aquí dos veces al día.
En un aspecto, la invención también proporciona un método para tratar o disminuir la gravedad de una enfermedad en un paciente que comprende administrar a dicho paciente una de las composiciones tal como se definen en el presente documento, y que dicha enfermedad se selecciona de fibrosis quística, el asma, la EPOC inducida por humo, bronquitis crónica, rinosinusitis, estreñimiento, pancreatitis, insuficiencia pancreática, infertilidad masculina causada por ausencia congénita bilateral del conducto deferente (CBAVD), enfermedad pulmonar leve, pancreatitis idiopática, aspergilosis broncopulmonar alérgica (ABPA), enfermedad hepática, enfisema hereditario, hemocromatosis hereditaria, deficiencias de coagulación y fibrinólisis, como deficiencia de proteína C, angioedema hereditario de tipo 1, deficiencias de procesamiento de lípidos, como hipercolesterolemia familiar, quilomicronemia tipo 1, abetalipoproteinemia, enfermedades de almacenamiento lisosómico, como enfermedad de células I/pseudo-Hurler, mucopolisacaridosis, Sandhof/Tay-Sachs, Crigler-Najjar tipo II, poliendocrinopatía/hiperinsulemia, diabetes mellitus, enanismo de Laron, deficiencia de mieloperoxidasa, hipoparatiroidismo primario, melanoma, glicanosis CDG tipo 1, hipertiroidismo congénito, osteogénesis imperfecta, hipofibrinogenemia hereditaria, deficiencia de ACT, diabetes insípida (DI), DI neurofisaria, DI neprogénica, síndrome de Charcot-Marie Tooth , Enfermedad de Perlizaeus-Merzbacher, enfermedades neurodegenerativas como la enfermedad de Alzheimer, enfermedad de Parkinson, esclerosis lateral amiotrófica, plasma supranuclear progresivo, enfermedad de Pick, varios trastornos
neurológicos de la poliglutamina como Huntington, ataxia espinocerebular tipo I, atrofia muscular espinal y bulbar, distrofia pallidoluysiana dentatorubal y miotónica, así como encefalopatías espongiformes, como la enfermedad hereditaria de Creutzfeldt-Jakob (debido a un defecto en el procesamiento de la proteína priónica), enfermedad de Fabry, síndrome de Straussler-Scheinker, EPOC, enfermedad del ojo seco, enfermedad de Sjogren, Osteoporosis, Osteopenia, síndrome de Gorham, canalopatías de cloro, como miotonía congenita (formas de Thomson y Becker), síndrome de Bartter tipo III, enfermedad de Dent, hiperekplexia, epilepsia, hipereplexia, enfermedad de almacenamiento lisosomal, síndrome de Angelman y discinesia ciliar primaria (PCD), un término para trastornos hereditarios de la estructura y/o función de los cilios, incluida la PCD con situs inversus (también conocido como síndrome de Kartagener), PCD sin situs inversus y aplasia ciliar.
BREVE DESCRIPCIÓN DE LAS FIGURAS
La Figura 1 presenta una ilustración gráfica de los perfiles de disolución de tabletas ejemplares de acuerdo con la presente invención.
Esta figura se presenta a modo de ejemplo y no pretende ser limitante.
DESCRIPCIÓN DETALLADA
La presente invención proporciona una composición farmacéutica que comprende una dispersión sólida de N-[2,4-Bis(1,1-dimetiletilo)-5-hidroxifenilo]-1,4-dihidro-4-oxoquinolina-3-carboxamida, un método de fabricación de una composición farmacéutica que comprende N-[2,4-Bis(1,1-dimetiletilo)-5-hidroxifenilo]-1,4-dihidro-4-oxoquinolina-3-carboxamida, y un método para administrar una composición farmacéutica que comprende una forma sólida de N-[2,4-Bis(1,1 -dimetiletilo)-5-hidroxifenilo]-1,4-dihidro-4-oxoquinolina-3-carboxamida.
I. DEFINICIONES
Tal como se utiliza aquí, el término "ingrediente farmacéutico activo" o "API" se refiere a un compuesto biológicamente activo. Las API ejemplares incluyen un potenciador de FQ (por ejemplo, N-[2,4-Bis(1,1-dimetiletilo)-5-hidroxifenilo]-1,4-dihidro-4-oxoquinolina-3-carboxamida).
Como se usa en el presente documento, el término "compuesto 1" se usa indistintamente con "N-[2,4-Bis(1,1-dimetiletilo)-5-hidróxidos yphenyl]-1,4-dihidro-4-oxoquinolina-3-carboxamida ", que tiene la siguiente estructura:

"Compuesto 1" también significa las formas tautoméricas tales como:

Tal como se utiliza aquí, el término "amorfo" se refiere a un material sólido que no tiene orden de largo alcance en la posición de sus moléculas. Los sólidos amorfos generalmente son líquidos sobreenfriados en los que las moléculas están dispuestas de manera aleatoria, de modo que no hay una disposición bien definida, por ejemplo, empaquetamiento molecular, y sin orden de largo alcance. Los sólidos amorfos generalmente son isotrópicos, es decir, exhiben propiedades similares en todas las direcciones y no tienen puntos de fusión definidos. Por ejemplo, un material amorfo es un material sólido que no tiene pico(s) cristalino(s) característico(s) agudo(s) en su patrón de
difracción de potencia de rayos X (XRPD) (es decir, no es cristalino como se determina por XRPD). En cambio, uno o varios picos amplios (p. ej., halos) aparecen en su patrón XRPD. Los picos anchos son característicos de un sólido amorfo. Véase, US 2004/0006237 para una comparación de XRPDs de un material amorfo y material cristalino.
Tal como se utiliza aquí, el término "sustancialmente amorfo" se refiere a un material sólido que tiene poca o ninguna orden de largo alcance en la posición de sus moléculas. Por ejemplo, los materiales sustancialmente amorfos tienen menos de aproximadamente 15% de cristalinidad (por ejemplo, menos de aproximadamente 10% de cristalinidad o menos de aproximadamente 5% de cristalinidad). También se observa que el término "sustancialmente amorfo" incluye el descriptor, "amorfo", que se refiere a materiales que no tienen (0%) cristalinidad.
Como se usa en este documento, el término "dispersión" se refiere a un sistema disperso en el que se distribuye una sustancia, la fase dispersa, en unidades discretas, a lo largo de una segunda sustancia (la fase continua o vehículo). El tamaño de la fase dispersa puede variar considerablemente (por ejemplo, moléculas individuales, partículas coloidales de dimensión nanométrica, hasta micras de tamaño múltiple). En general, las fases dispersas pueden ser sólidas, líquidas o gases. En el caso de una dispersión sólida, las fases dispersa y continua son ambas sólidas. En aplicaciones farmacéuticas, una dispersión sólida puede incluir: un fármaco amorfo en un polímero amorfo; un fármaco amorfo en polímero cristalino; un fármaco cristalino en un polímero amorfo; o un fármaco cristalino en un polímero cristalino. En esta invención, una dispersión sólida puede incluir un fármaco amorfo en un polímero amorfo o un fármaco amorfo en un polímero cristalino. En algunas realizaciones, una dispersión sólida incluye el polímero que constituye la fase dispersa, y el fármaco constituye la fase continua. O bien, una dispersión sólida incluye el fármaco que constituye la fase dispersa, y el polímero constituye la fase continua.
Tal como se utiliza aquí, el término "dispersión sólida" se refiere en general a una dispersión sólida de dos o más componentes, por lo general uno o más fármacos (por ejemplo, un fármaco (por ejemplo, el Compuesto 1)) y polímero, pero que contiene posiblemente otros componentes tales como surfactantes u otros excipientes farmacéuticos, donde el fármaco (por ejemplo, Compuesto 1) es sustancialmente amorfo (por ejemplo, que tiene aproximadamente 15% o menos (por ejemplo, aproximadamente 10% o menos, o aproximadamente 5% o menos)) de medicamento cristalino (por ejemplo, N-[2,4-Bis(1,1-dimetiletilo)-5-hidroxifenilo]-1,4-dihidro-4-oxoquinolina-3-carboxamida) o amorfo (es decir, que no tiene fármaco cristalino), y la estabilidad física y/o la disolución y/o la solubilidad del fármaco amorfo o sustancialmente amorfo se potencia por los otros componentes. Las dispersiones sólidas incluyen típicamente un compuesto dispersado en un medio portador apropiado, tal como un portador de estado sólido. Por ejemplo, un vehículo comprende un polímero (por ejemplo, un polímero soluble en agua o un polímero parcialmente soluble en agua) y puede incluir excipientes opcionales tales como excipientes funcionales (por ejemplo, uno o más surfactantes) o excipientes no funcionales (por ejemplo, uno o más rellenos). Otra dispersión sólida a modo de ejemplo es un coprecipitado o una co-fusión de N-[2,4-Bis(1,1-dimetiletilo)-5-hidroxifenilo]-1,4-dihidro-4-oxoquinolina-3-carboxamida con al menos un polímero.
Un "Co-precipitado" es un producto después de la disolución de un fármaco y un polímero en un disolvente o mezcla de disolvente, seguido de la eliminación del disolvente o mezcla de disolvente. A veces, el polímero puede suspenderse en el disolvente o la mezcla de disolventes. El disolvente o la mezcla de disolventes incluye disolventes orgánicos y fluidos supercríticos. Una "co-fusión" es un producto tras calentamiento de un fármaco y de un polímero para fusión, opcionalmente en presencia de un solvente o mezcla de solventes, seguido por mezcla, eliminación de al menos una porción del solvente si corresponde y enfriamiento a la temperatura ambiente a una velocidad seleccionada.
Como se usa en este documento, "cristalinidad" se refiere al grado de orden estructural en un sólido. Por ejemplo, el Compuesto 1, que es sustancialmente amorfo, tiene menos de aproximadamente 15% de cristalinidad, o su estructura de estado sólido es menos de aproximadamente 15% de cristalina. En otro ejemplo, el Compuesto 1, que es amorfo, tiene una cristalinidad cero (0%).
Como se usa en este documento, un " potenciador de FQ" se refiere a un compuesto que exhibe actividad biológica caracterizado por incrementar la funcionalidad de gating de la proteína CFTR mutante presente en la superficie celular a niveles aproximadamente de tipo salvaje.
Como se usa en este documento, un "excipiente" es un ingrediente inactivo en una composición farmacéutica. Los ejemplos de excipientes incluyen cargas o diluyentes, surfactantes, aglutinantes, deslizantes, lubricantes, disgregantes y similares.
Como se usa en este documento, un "disgregante" es un excipiente que hidrata una composición farmacéutica y ayuda en la dispersión de la tableta. Los ejemplos de desintegrantes incluyen croscarmelosa sódica y/o glicolato sódico de almidón.
Como se usa en este documento, un "diluyente" o "relleno" es un excipiente que agrega volumen a una composición farmacéutica. Los ejemplos de cargas incluyen lactosa, sorbitol, celulosas, fosfatos de calcio, almidones, azúcares (por ejemplo, manitol, sacarosa, o similar) o cualquier combinación de los mismos.
Como se usa en este documento, un "surfactante" es un excipiente que imparte composiciones farmacéuticas con solubilidad y/o humectabilidad mejorada. Los ejemplos de surfactantes incluyen laurilo de sulfato de sodio (SLS), estearilfumarato de sodio (SSF), monooleato de polioxietileno y sorbitán (por ejemplo, Tween™), o cualquier combinación de los mismos.
Como se usa en este documento, un "aglutinante" es un excipiente que confiere una composición farmacéutica con una mejora de la cohesión o resistencia a la tracción (por ejemplo, dureza). Los ejemplos de aglutinantes incluyen fosfato de calcio dibásico, sacarosa, almidón de maíz (maíz), celulosa microcristalina y celulosa modificada (por ejemplo, hidroximetilcelulosa).
Como se usa en este documento, un "agente deslizante" es un excipiente que imparte composiciones farmacéuticas con propiedades de flujo mejoradas. Los ejemplos de deslizantes incluyen sílice coloidal y/o talco.
Como se usa en este documento, un "colorante" es un excipiente que confiere una composición farmacéutica con un color deseado. Los ejemplos de colorantes incluyen pigmentos disponibles comercialmente tales como FD&C Blue n° 1 Aluminum Lake, FD&C Blue n° 2, otros colores FD&C Blue, dióxido de titanio, óxido de hierro y/o combinaciones de los mismos.
Como se usa en este documento, un "lubricante" es un excipiente que se añade a composiciones farmacéuticas que se presionan en comprimidos. El lubricante ayuda a la compactación de gránulos en tabletas y a la eyección de una tableta de una composición farmacéutica a partir de una prensa de moldeo. Los ejemplos de lubricantes incluyen estearato de magnesio, ácido esteárico (estearina), aceite hidrogenado, estearilfumarato de sodio o cualquier combinación de los mismos.
Como se usa en este documento, "friabilidad" se refiere a la propiedad de un comprimido para permanecer intacto y retener su forma a pesar de una fuerza externa de presión. La friabilidad se puede cuantificar usando la expresión matemática presentada en la ecuación 1:
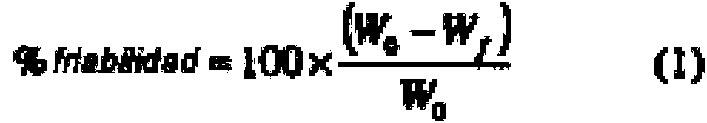
donde Wo es el peso original de la tableta y Wt es el peso final de la tableta después de que se pasa a través del friabilador.
La friabilidad se mide usando un aparato de prueba de USP estándar que hace girar las tabletas experimentales durante 100 revoluciones. Algunas tabletas de la presente invención tienen una friabilidad de menos de aproximadamente 1% (por ejemplo, menos de aproximadamente 0,75%, menos de aproximadamente 0,50% o menos de aproximadamente 0,30%)
Como se usa en este documento, "diámetro medio de partícula" es el diámetro medio de partícula como se mide utilizando técnicas de medición tales como dispersión de luz láser, análisis de imágenes, o el análisis de tamiz.
Como se usa en la presente memoria, "densidad aparente" es la masa de partículas de material dividido por el volumen total que ocupan las partículas. El volumen total incluye el volumen de partícula, el volumen de vacío entre partículas y el volumen de poro interno. La densidad aparente no es una propiedad intrínseca de un material; puede cambiar dependiendo de cómo se procesa el material.
II. COMPOSICIÓN FARMACÉUTICA
En un aspecto, la presente invención proporciona una composición farmacéutica que comprende un potenciador FQ de API (por ejemplo, una dispersión sólida de Compuesto 1).
En una realización, la presente invención proporciona una composición farmacéutica que comprende una dispersión sólida de Compuesto 1 sustancialmente amorfo, en el que la dispersión sólida comprende aproximadamente 25 mg de Compuesto 1 sustancialmente amorfo.
En una realización, la presente invención proporciona una composición farmacéutica que comprende una dispersión sólida de Compuesto 1 sustancialmente amorfo, en el que la dispersión sólida comprende aproximadamente 50 mg de Compuesto 1 sustancialmente amorfo.
En una realización, la presente invención proporciona una composición farmacéutica que comprende una dispersión sólida de Compuesto 1 sustancialmente amorfo, en el que la dispersión sólida comprende
aproximadamente 75 mg de Compuesto 1 sustancialmente amorfo.
En una realización, la presente invención proporciona una composición farmacéutica que comprende una dispersión sólida de Compuesto 1 sustancialmente amorfo, en el que la dispersión sólida comprende aproximadamente 100 mg de Compuesto 1 sustancialmente amorfo.
En una realización, la presente invención proporciona una composición farmacéutica que comprende una dispersión sólida de Compuesto 1 sustancialmente amorfo, en el que la dispersión sólida comprende aproximadamente 150 mg de Compuesto 1 sustancialmente amorfo.
En una realización, la presente invención proporciona una composición farmacéutica que comprende una dispersión sólida de Compuesto 1 sustancialmente amorfo, en el que la dispersión sólida comprende aproximadamente 250 mg de Compuesto 1 sustancialmente amorfo.
En una realización, la presente invención proporciona una composición farmacéutica que comprende una dispersión sólida del Compuesto 1 amorfo, en el que la dispersión sólida comprende aproximadamente 25 mg del Compuesto 1 amorfo.
En una realización, la presente invención proporciona una composición farmacéutica que comprende una dispersión sólida del Compuesto 1 amorfo, en el que la dispersión sólida comprende aproximadamente 50 mg del Compuesto 1 amorfo.
En una realización, la presente invención proporciona una composición farmacéutica que comprende una dispersión sólida del Compuesto 1 amorfo, en el que la dispersión sólida comprende aproximadamente 75 mg del Compuesto 1 amorfo.
En una realización, la presente invención proporciona una composición farmacéutica que comprende una dispersión sólida del Compuesto 1 amorfo, en el que la dispersión sólida comprende aproximadamente 100 mg del Compuesto 1 amorfo.
En una realización, la presente invención proporciona una composición farmacéutica que comprende una dispersión sólida del Compuesto 1 amorfo, en el que la dispersión sólida comprende aproximadamente 150 mg de Compuesto 1 amorfo.
En una realización, la presente invención proporciona una composición farmacéutica que comprende una dispersión sólida del Compuesto 1 amorfo, en el que comprende de dispersión sólida aproximadamente 250 mg del Compuesto 1 amorfo.
Otro aspecto de la presente invención proporciona una composición farmacéutica que comprende una dispersión sólida de Compuesto 1 en la que la dispersión sólida comprende un polímero.
En una realización, la presente invención proporciona una composición farmacéutica que comprende una dispersión sólida de Compuesto 1 sustancialmente amorfo y HPMCAS, en el que la dispersión sólida comprende aproximadamente 25 mg de Compuesto 1 sustancialmente amorfo.
En una realización, la presente invención proporciona una composición farmacéutica que comprende una dispersión sólida de Compuesto 1 sustancialmente amorfo y HPMCAS, en el que la dispersión sólida comprende aproximadamente 50 mg de Compuesto 1 sustancialmente amorfo.
En una realización, la presente invención proporciona una composición farmacéutica que comprende una dispersión sólida de Compuesto 1 sustancialmente amorfo y HPMCAS, en el que la dispersión sólida comprende aproximadamente 75 mg de Compuesto 1 sustancialmente amorfo.
En una realización, la presente invención proporciona una composición farmacéutica que comprende una dispersión sólida de Compuesto 1 sustancialmente amorfo y HPMCAS, en el que la dispersión sólida comprende aproximadamente 100 mg de Compuesto 1 sustancialmente amorfo.
En una realización, la presente invención proporciona una composición farmacéutica que comprende una dispersión sólida de Compuesto 1 sustancialmente amorfo y HPMCAS, en el que la dispersión sólida comprende aproximadamente 150 mg de Compuesto 1 sustancialmente amorfo.
En una realización, la presente invención proporciona una composición farmacéutica que comprende una dispersión sólida de Compuesto 1 sustancialmente amorfo y HPMCAS, en el que la dispersión sólida comprende aproximadamente 250 mg de Compuesto 1 sustancialmente amorfo.
En una realización, la presente invención proporciona una composición farmacéutica que comprende una dispersión sólida del Compuesto 1 amorfo y HPMCAS, en el que la dispersión sólida comprende aproximadamente 25 mg del Compuesto 1 amorfo.
En una realización, la presente invención proporciona una composición farmacéutica que comprende una dispersión sólida del Compuesto 1 amorfo y HPMCAS, en el que la dispersión sólida comprende aproximadamente 50 mg del Compuesto 1 amorfo.
En una realización, la presente invención proporciona una composición farmacéutica que comprende una dispersión sólida del Compuesto 1 amorfo y HPMCAS, en el que la dispersión sólida comprende aproximadamente 75 mg del Compuesto 1 amorfo.
En una realización, la presente invención proporciona una composición farmacéutica que comprende una dispersión sólida del Compuesto 1 amorfo y HPMCAS, en el que la dispersión sólida comprende aproximadamente 100 mg del Compuesto 1 amorfo.
En una realización, la presente invención proporciona una composición farmacéutica que comprende una dispersión sólida del Compuesto 1 amorfo y HPMCAS, en el que la dispersión sólida comprende aproximadamente 150 mg de Compuesto 1 amorfo.
En una realización, la presente invención proporciona una composición farmacéutica que comprende una dispersión sólida del Compuesto 1 amorfo y HPMCAS, en el que la dispersión sólida comprende aproximadamente 250 mg del Compuesto 1 amorfo.
En una realización, la presente invención proporciona una composición farmacéutica que comprende una dispersión sólida de Compuesto 1 sustancialmente amorfo y PVP/VA, en donde la dispersión sólida comprende aproximadamente 25 mg de Compuesto 1 sustancialmente amorfo.
En una realización, la presente invención proporciona una composición farmacéutica que comprende una dispersión sólida de Compuesto 1 sustancialmente amorfo y PVP/VA, en donde la dispersión sólida comprende aproximadamente 50 mg de Compuesto 1 sustancialmente amorfo.
En una realización, la presente invención proporciona una composición farmacéutica que comprende una dispersión sólida de Compuesto 1 sustancialmente amorfo y PVP/VA, en donde la dispersión sólida comprende aproximadamente 75 mg de Compuesto 1 sustancialmente amorfo.
En una realización, la presente invención proporciona una composición farmacéutica que comprende una dispersión sólida de Compuesto 1 sustancialmente amorfo y PVP/VA, en donde la dispersión sólida comprende aproximadamente 100 mg de Compuesto 1 sustancialmente amorfo.
En una realización, la presente invención proporciona una composición farmacéutica que comprende una dispersión sólida de Compuesto 1 sustancialmente amorfo y PVP/VA, en donde la dispersión sólida comprende aproximadamente 150 mg de Compuesto 1 sustancialmente amorfo.
En una realización, la presente invención proporciona una composición farmacéutica que comprende una dispersión sólida de Compuesto 1 sustancialmente amorfo y PVP/VA, en donde la dispersión sólida comprende aproximadamente 250 mg de Compuesto 1 sustancialmente amorfo.
En una realización, la presente invención proporciona una composición farmacéutica que comprende una dispersión sólida de Compuesto 1 amorfo y PVP/VA, en donde la dispersión sólida comprende aproximadamente 25 mg de Compuesto 1 amorfo.
En una realización, la presente invención proporciona una composición farmacéutica que comprende una dispersión sólida de Compuesto 1 amorfo y PVP/VA, en donde la dispersión sólida comprende aproximadamente 50 mg de Compuesto 1 amorfo.
En una realización, la presente invención proporciona una composición farmacéutica que comprende una dispersión sólida de Compuesto 1 amorfo y PVP/VA, en donde la dispersión sólida comprende aproximadamente 75 mg de Compuesto 1 amorfo.
En una realización, la presente invención proporciona una composición farmacéutica que comprende una dispersión sólida de Compuesto 1 amorfo y PVP/VA, en donde la dispersión sólida comprende aproximadamente 100 mg de Compuesto 1 amorfo.
En una realización, la presente invención proporciona una composición farmacéutica que comprende una
dispersión sólida de Compuesto 1 amorfo y PVP/VA, en donde la dispersión sólida comprende aproximadamente 150 mg de Compuesto 1 amorfo.
En una realización, la presente invención proporciona una composición farmacéutica que comprende una dispersión sólida de Compuesto 1 amorfo y PVP/VA, en donde la dispersión sólida comprende aproximadamente 250 mg de Compuesto 1 amorfo.
Un aspecto de la presente invención proporciona una composición farmacéutica que comprende un API potenciador de la FQ (por ejemplo, una dispersión sólida del Compuesto 1) y otros excipientes (por ejemplo, una carga, un disgregante, un surfactante, un aglutinante, un deslizante, un colorante, un lubricante, o cualquier combinación de los mismos).
En una realización, la presente invención proporciona una composición farmacéutica que comprende: a. una dispersión sólida de la reivindicación 1;
b. una carga
c. un disgregante;
d. un surfactante;
e. un aglutinante
f. un deslizante; y
g. un lubricante,
en donde la dispersión sólida comprende aproximadamente 25 mg de Compuesto 1 sustancialmente amorfo.
En una realización, la presente invención proporciona una composición farmacéutica que comprende: a. una dispersión sólida de la reivindicación 1;
b. una carga
c. un disgregante;
d. un surfactante;
e. un aglutinante
f. un deslizante; y
g. un lubricante,
en donde la dispersión sólida comprende aproximadamente 50 mg de Compuesto 1 sustancialmente amorfo.
En otra realización, la presente invención proporciona una composición farmacéutica que comprende: a. una dispersión sólida de la reivindicación 1;
b. una carga
c. un disgregante;
d. un surfactante;
e. un aglutinante
f. un deslizante; y
g. un lubricante,
en donde la dispersión sólida comprende aproximadamente 75 mg de Compuesto 1 sustancialmente amorfo.
En una realización, la presente invención proporciona una composición farmacéutica que comprende: a. una dispersión sólida de la reivindicación 1;
b. una carga
c. un disgregante;
d. un surfactante;
e. un aglutinante
f. un deslizante; y
g. un lubricante,
en donde la dispersión sólida comprende aproximadamente 100 mg de Compuesto 1 sustancialmente amorfo.
En una realización, la presente invención proporciona una composición farmacéutica que comprende: a. una dispersión sólida de la reivindicación 1;
b una carga
c. un disgregante;
d. un surfactante;
e. un aglutinante
f. un deslizante; y
g. un lubricante,
en donde la dispersión sólida comprende aproximadamente 150 mg de Compuesto 1 sustancialmente amorfo.
En otra realización, la presente invención proporciona una composición farmacéutica que comprende: a. una dispersión sólida de la reivindicación 1;
b. una carga
c. un disgregante;
d. un surfactante;
e. un aglutinante
f. un deslizante; y
g. un lubricante,
en donde la dispersión sólida comprende aproximadamente 250 mg de Compuesto 1 sustancialmente amorfo.
En una realización, la presente invención proporciona una composición farmacéutica que comprende: a. una dispersión sólida de la reivindicación 1;
b. una carga
c. un disgregante;
d. un surfactante;
e. un aglutinante
f. un deslizante; y
g. un lubricante,
en donde la dispersión sólida comprende aproximadamente 25 mg de Compuesto 1 amorfo.
En una realización, la presente invención proporciona una composición farmacéutica que comprende: a. una dispersión sólida de la reivindicación 1;
b. una carga
c. un disgregante;
d. un surfactante;
e. un aglutinante
f. un deslizante; y
g. un lubricante,
en donde la dispersión sólida comprende aproximadamente 50 mg de Compuesto 1 amorfo.
En otra realización, la presente invención proporciona una composición farmacéutica que comprende: a. una dispersión sólida de la reivindicación 1;
b. una carga
c. un disgregante;
d. un surfactante;
e. un aglutinante
f. un deslizante; y
g. un lubricante,
en donde la dispersión sólida comprende aproximadamente 75 mg de Compuesto 1 amorfo.
En una realización, la presente invención proporciona una composición farmacéutica que comprende: a. una dispersión sólida de la reivindicación 1;
b. una carga
c. un disgregante;
d. un surfactante;
e. un aglutinante
f. un deslizante; y
g. un lubricante,
en donde la dispersión sólida comprende aproximadamente 100 mg de Compuesto 1 amorfo.
En una realización, la presente invención proporciona una composición farmacéutica que comprende: a. una dispersión sólida de la reivindicación 1;
b. una carga
c. un disgregante;
d. un surfactante;
e. un aglutinante
f. un deslizante; y
g. un lubricante,
en donde la dispersión sólida comprende aproximadamente 150 mg de Compuesto 1 amorfo.
En otra realización, la presente invención proporciona una composición farmacéutica que comprende: a. una dispersión sólida de la reivindicación 1;
b. una carga
c. un disgregante;
d. un surfactante;
e. un aglutinante
f. un deslizante; y
g. un lubricante,
en donde la dispersión sólida comprende aproximadamente 250 mg de Compuesto 1 amorfo.
En una realización, la composición farmacéutica comprende una dispersión sólida de la reivindicación 1, una carga, un disgregante, un surfactante, un aglutinante, un deslizante y un lubricante, en donde la dispersión sólida comprende el Compuesto 1 y un polímero.
En algunas realizaciones, la composición farmacéutica comprende una dispersión sólida, una carga, un disgregante, un surfactante, un aglutinante, un deslizante y un lubricante, en donde la dispersión sólida comprende el 80% en peso del Compuesto 1 en peso de la dispersión y un polímero, como se define en la reivindicación 1.
Las dispersiones que comprenden el Compuesto 1, es decir, N-[2,4-bis(1,1-dimetiletil)-5-hidroxifenil]-1,4-dihidro-4-oxoquinolin-3-carboxamida se describen en la publicación de PCT N° WO 2007/079139.
En una realización, la composición farmacéutica de la presente invención comprende una dispersión sólida del compuesto 1. Por ejemplo, la dispersión sólida comprende Compuesto 1 sustancialmente amorfo, donde el compuesto 1 es menor que aproximadamente 15% (por ejemplo, menor que aproximadamente 10 % o menor que aproximadamente 5% cristalino), y al menos un polímero. En otro ejemplo, la dispersión sólida comprende Compuesto 1 amorfo, es decir, el Compuesto 1 tiene aproximadamente 0% de cristalinidad. La concentración del Compuesto 1 en la dispersión sólida depende de varios factores tales como la cantidad de composición farmacéutica que se necesita para proporcionar una cantidad deseada del Compuesto 1 y el perfil de disolución deseado de la composición farmacéutica.
Los polímeros útiles en estas dispersiones sólidas son polímeros inertes farmacéuticamente aceptables que son por lo menos parcialmente solubles en agua o fluidos biológicos. La dispersión sólida comprende N-[2,4-bis(1,1-dimetiletil)-5-hidroxifenil]-1,4-dihidro-4-oxoquinolin-3-carboxamida sustancialmente amorfa o amorfa y HPMCAS o PVP/VA.
En otra realización, la composición farmacéutica comprende una dispersión sólida que contiene Compuesto 1 sustancialmente amorfo y HPMCAS o PVP/VA, en la que la dispersión sólida tiene un diámetro medio de partículas, medido mediante dispersión de luz (por ejemplo, usando un Malvern Mastersizer disponible de Malvern Instruments en Inglaterra) de más de aproximadamente 5 gm (por ejemplo, mayor de aproximadamente 6 gm, mayor de aproximadamente 7 gm, mayor de aproximadamente 8 gm, o mayor de aproximadamente 10 gm). Por ejemplo, la composición farmacéutica comprende una dispersión sólida que contiene el Compuesto 1 amorfo y HPMCAS o PVP/VA, en la que la dispersión sólida tiene un diámetro de partícula medio, medido por dispersión de luz, de más de aproximadamente 5 gm (por ejemplo, mayor de aproximadamente 6 gm, mayor de aproximadamente 7 gm, mayor de aproximadamente 8 gm, o mayor de aproximadamente 10 gm). En otro ejemplo, la composición farmacéutica comprende una dispersión sólida que comprende sustancialmente Compuesto 1 amorfo y HPMCAS, en la que la dispersión sólida tiene un diámetro de partícula medio, medido por dispersión de luz, de aproximadamente 7 gm a aproximadamente 25 gm. Por ejemplo, la composición farmacéutica comprende una dispersión sólida que comprende el Compuesto 1 amorfo y HPMCAS, en la que la dispersión sólida tiene un diámetro de partícula medio, medido por dispersión de luz, de aproximadamente 7 gm a aproximadamente 25 gm.
En aún otro ejemplo, la composición farmacéutica comprende una dispersión sólida que comprende sustancialmente Compuesto 1 amorfo y HPMCAS, en la que la dispersión sólida tiene un diámetro de partícula medio, medido por dispersión de luz, de aproximadamente 10 pm a aproximadamente 35 pm. Por ejemplo, la composición farmaceútica comprende una dispersión sólida que comprende el Compuesto 1 amorfo y HPMCAS, en la que la dispersión sólida tiene un diámetro de partícula medio, medido por dispersión de luz, de aproximadamente 10 pm a aproximadamente 35 pm. En otro ejemplo, la composición farmacéutica comprende una dispersión sólida que comprende sustancialmente Compuesto 1 amorfo y HPMCAS o PVP/VA, en la que la dispersión sólida tiene una densidad aparente de aproximadamente 0,10 g/cc o mayor (por ejemplo, 0,15 g/cc o mayor, 0,17 g/cc o mayor). Por ejemplo, la composición farmacéutica que comprende una dispersión sólida que comprende el Compuesto 1 amorfo y HPMCAS o PVP/VA, en la que la dispersión sólida tiene una densidad aparente de aproximadamente 0,10 g/cc o mayor (por ejemplo, 0,15 g/cc o mayor, 0,17 g/cc o mayor). En otro ejemplo, la composición farmacéutica comprende una dispersión sólida que comprende Compuesto 1 sustancialmente amorfo y HPMCAS o PVP/VA, en la que la dispersión sólida tiene una densidad aparente de aproximadamente 0,10 g/cc a aproximadamente 0,45 g/cc (por ejemplo, de aproximadamente 0,15 g/cc a aproximadamente 0,42 g/cc, o de aproximadamente 0,17 g/cc a aproximadamente 0,40 g/cc). En todavía otro ejemplo, la composición farmacéutica comprende una dispersión sólida que incluye el Compuesto 1 amorfo y HPMCAS o PVP/VA, en la que la dispersión sólida tiene una densidad aparente de aproximadamente 0,10 g/cc a aproximadamente 0,45 g/cc (por ejemplo, de aproximadamente 0,15 g/cc a aproximadamente 0,42 g/cc, o de aproximadamente 0,17 g/cc a aproximadamente 0,40 g/cc). En otro ejemplo, la composición farmacéutica comprende una dispersión sólida que comprende Compuesto 1 sustancialmente amorfo y HPMCAS, en la que la dispersión sólida tiene una densidad aparente de aproximadamente 0,10 g/cc a aproximadamente 0,45 g/cc (por ejemplo, de aproximadamente 0,15 g/cc a aproximadamente 0,42 g/cc, o de aproximadamente 0,17 g/cc a aproximadamente 0,40 g/cc). Por ejemplo, la composición farmacéutica incluye una dispersión sólida que comprende el Compuesto 1 amorfo y HPMCAS, en la que la dispersión sólida tiene una densidad aparente de aproximadamente 0,10 g/cc a aproximadamente 0,45 g/cc (por ejemplo, de aproximadamente 0,15 g/cc a aproximadamente 0,42 g/cc, o de aproximadamente 0,17 g/cc a aproximadamente 0,40 g/cc).
La dispersión sólida contiene el 80% en peso de Compuesto 1 sustancialmente amorfo o amorfo, el 19,5% en peso de HPMCAS o PVP/VA y el 0,5% en peso de SLS.
Además de la dispersión sólida del Compuesto 1, las composiciones farmacéuticas de la presente invención también comprenden uno o más excipientes como cargas, disgregantes, surfactantes, aglutinantes, deslizantes, lubricantes, colorantes o fragancias.
Las cargas adecuadas para la presente invención son compatibles con los ingredientes de la composición farmacéutica, es decir, no reducen sustancialmente la solubilidad, la dureza, la estabilidad química, la estabilidad física o la actividad biológica de la composición farmacéutica. Las cargas ejemplares incluyen lactosa, sorbitol, celulosas, fosfatos de calcio, almidones, azúcares (por ejemplo, manitol, sacarosa o similares) o cualquier combinación de los mismos. En una realización, la composición farmacéutica comprende por lo menos una carga en una cantidad de por lo menos aproximadamente el 10% en peso (por ejemplo, por lo menos aproximadamente el 20% en peso, por lo menos aproximadamente el 25% en peso, o por lo menos aproximadamente el 27% en peso) en peso de la composición. Por ejemplo, la composición farmacéutica comprende de aproximadamente el 10% en peso a aproximadamente el 60% en peso (por ejemplo, de aproximadamente el 20% en peso a aproximadamente el 55% en peso, de aproximadamente el 25% en peso a aproximadamente el 50% en peso, o de aproximadamente el 27% en peso a aproximadamente el 45% en peso) de carga, en peso de la composición. En otro ejemplo, la composición farmacéutica comprende por lo menos aproximadamente el 20% en peso (por ejemplo, por lo menos el 25% en peso o por lo menos el 27% en peso) de lactosa, en peso de la composición. En otro ejemplo más, la composición farmacéutica comprende de aproximadamente el 20% en peso a aproximadamente el 60% en peso (por ejemplo, de aproximadamente el 25% en peso a aproximadamente el 55% en peso o de aproximadamente el 27% en peso a aproximadamente el 45% en peso) de lactosa, en peso de la composición.
Los disgregantes adecuados para la presente invención mejoran la dispersión de la composición farmacéutica y son compatibles con los ingredientes de la composición farmacéutica, es decir, no reducen sustancialmente la estabilidad química, la estabilidad física, la dureza o la actividad biológica de la composición farmacéutica. Los disgregantes ejemplares incluyen croscarmelosa de sodio, glicolato de almidón de sodio o una combinación de los mismos. En una realización, la composición farmacéutica comprende disgregante en una cantidad de aproximadamente el 10% en peso o menos (por ejemplo, aproximadamente el 7% en peso o menos, aproximadamente el 6% en peso o menos, o aproximadamente el 5% en peso o menos) en peso de la composición. Por ejemplo, la composición farmacéutica comprende de aproximadamente el 1% en peso a aproximadamente el 10% en peso (por ejemplo, de aproximadamente el 1,5% en peso a aproximadamente el 7,5% en peso o de aproximadamente el 2,5% en peso a aproximadamente el 6% en peso) de disgregante, en peso de la composición. En otro ejemplo, la composición farmacéutica comprende aproximadamente el 10% en peso o menos (por ejemplo, el 7% en peso o menos, el 6% en peso o menos, o el 5% en peso o menos) de croscarmelosa de sodio, en peso de la composición. En otro ejemplo más, la composición farmacéutica comprende de aproximadamente el 1% en peso a aproximadamente el 10% en peso (por ejemplo, de aproximadamente el 1,5% en peso a aproximadamente el 7,5% en peso o de aproximadamente el 2,5% en peso a aproximadamente el 6% en
peso) de croscarmelosa de sodio, en peso de la composición. En algunos ejemplos, la composición farmacéutica comprende de aproximadamente el 0,1% a aproximadamente el 10% en peso (por ejemplo, de aproximadamente el 0,5% en peso a aproximadamente el 7,5% en peso o de aproximadamente el 1,5% en peso a aproximadamente el 6% en peso) de disgregante, en peso de la composición. En otros ejemplos más, la composición farmacéutica comprende de aproximadamente el 0,5% a aproximadamente el 10% en peso (por ejemplo, de aproximadamente el 1,5% en peso a aproximadamente el 7,5% en peso o de aproximadamente el 2,5% en peso a aproximadamente el 6% en peso) de disgregante, en peso de la composición.
Los surfactantes adecuados para la presente invención mejoran la solubilidad de la composición farmacéutica y son compatibles con los ingredientes de la composición farmacéutica, es decir, no reducen sustancialmente la estabilidad química, la estabilidad física, la dureza o la actividad biológica de la composición farmacéutica. Los surfactantes ejemplares incluyen lauril sulfato de sodio (SLS), estearilfumarato de sodio (SSF), monooleato de polioxietilen 20 sorbitán (por ejemplo, Tween™), cualquier combinación de los mismos, o similares. En una realización, la composición farmacéutica comprende un surfactante en una cantidad de aproximadamente el 10% en peso o menos (por ejemplo, aproximadamente el 5% en peso o menos, aproximadamente el 2% en peso o menos, aproximadamente el 1% en peso o menos, aproximadamente el 0,8% en peso o menos, o aproximadamente el 0,6% en peso o menos) en peso de la composición. Por ejemplo, la composición farmacéutica incluye de aproximadamente el 10% en peso a aproximadamente el 0,1% en peso (por ejemplo, de aproximadamente el 5% en peso a aproximadamente el 0,2% en peso o de aproximadamente el 2% en peso a aproximadamente el 0,3% en peso) de surfactante, en peso de la composición. En otro ejemplo, la composición farmacéutica comprende el 10% en peso o menos (por ejemplo, aproximadamente el 5% en peso o menos, aproximadamente el 2% en peso o menos, aproximadamente el 1% en peso o menos, aproximadamente el 0,8% en peso o menos, o aproximadamente el 0,6% en peso o menos) de lauril sulfato de sodio, en peso de la composición. En otro ejemplo más, la composición farmacéutica comprende de aproximadamente el 10% en peso a aproximadamente el 0,1% en peso (por ejemplo, de aproximadamente el 5% en peso a aproximadamente el 0,2% en peso o de aproximadamente el 2% en peso a aproximadamente el 0,3% en peso) de lauril sulfato de sodio, por peso de la composición.
Los aglutinantes adecuados para la presente invención mejoran la resistencia del comprimido de la composición farmacéutica y son compatibles con los ingredientes de la composición farmacéutica, es decir, no reducen sustancialmente la estabilidad química, la estabilidad física o la actividad biológica de la composición farmacéutica. Los aglutinantes ejemplares incluyen celulosa microcristalina, fosfato de calcio dibásico, sacarosa, almidón de maíz, celulosa modificada (por ejemplo, hidroximetilcelulosa) o cualquier combinación de los mismos. En una realización, la composición farmacéutica comprende un aglutinante en una cantidad de por lo menos aproximadamente el 1% en peso (por ejemplo, por lo menos aproximadamente el 10% en peso, por lo menos aproximadamente el 15% en peso, por lo menos aproximadamente el 20% en peso, o por lo menos aproximadamente el 22% en peso) en peso de la composición. Por ejemplo, la composición farmacéutica comprende de aproximadamente el 5% en peso a aproximadamente el 50% en peso (por ejemplo, de aproximadamente el 10% en peso a aproximadamente el 45% en peso o de aproximadamente el 20% en peso a aproximadamente el 45% en peso) de aglutinante, en peso de la composición. En otro ejemplo, la composición farmacéutica comprende por lo menos aproximadamente el 1% en peso (por ejemplo, por lo menos aproximadamente el 10% en peso, por lo menos aproximadamente el 15% en peso, por lo menos aproximadamente el 20% en peso, o por lo menos aproximadamente el 22% en peso) de celulosa microcristalina, en peso de la composición. En otro ejemplo más, la composición farmacéutica comprende de aproximadamente el 5% en peso a aproximadamente el 50% en peso (por ejemplo, de aproximadamente el 10% en peso a aproximadamente el 45% en peso o de aproximadamente el 20% en peso a aproximadamente el 45% en peso) de celulosa microcristalina, en peso de la composición.
Los deslizantes adecuados para la presente invención mejoran las propiedades de flujo de la composición farmacéutica y son compatibles con los ingredientes de la composición farmacéutica, es decir, no reducen sustancialmente la solubilidad, la dureza, la estabilidad química, la estabilidad física o la actividad biológica de la composición farmacéutica. Los deslizantes ejemplares incluyen dióxido de silicio coloidal, talco o una combinación de los mismos. En una realización, la composición farmacéutica comprende un deslizante en una cantidad del 2% en peso o menos (por ejemplo, el 1,75% en peso, el 1,25% en peso o menos, o el 1,00% en peso o menos) en peso de la composición. Por ejemplo, la composición farmacéutica comprende de aproximadamente el 2% en peso a aproximadamente el 0,05% en peso (por ejemplo, de aproximadamente el 1,5% en peso a aproximadamente el 0,07% en peso o de aproximadamente el 1,0% en peso a aproximadamente el 0,09% en peso) de deslizante, en peso de la composición. En otro ejemplo, la composición farmacéutica comprende el 2% en peso o menos (por ejemplo, el 1,75% en peso, el 1,25% en peso o menos, o el 1,00% en peso o menos) de dióxido de silicio coloidal, en peso de la composición. En otro ejemplo más, la composición farmacéutica comprende de aproximadamente el 2% en peso a aproximadamente el 0,05% en peso (por ejemplo, de aproximadamente el 1,5% en peso a aproximadamente el 0,07% en peso o de aproximadamente el 1,0% en peso a aproximadamente el 0,09% en peso) de dióxido de silicio coloidal, en peso de la composición.
Los lubricantes adecuados para la presente invención mejoran la compresión y expulsión de composiciones
farmacéuticas comprimidas de una prensa de matriz y son compatibles con los ingredientes de la composición farmacéutica, es decir, no reducen sustancialmente la solubilidad, la dureza o la actividad biológica de la composición farmacéutica. Los lubricantes ejemplares incluyen estearato de magnesio, ácido esteárico (estearina), aceite hidrogenado, estearilfumarato de sodio o cualquier combinación de los mismos. En una realización, la composición farmacéutica comprende un lubricante en una cantidad del 2% en peso o menos (por ejemplo, el 1,75% en peso, el 1,25% en peso o menos, o el 1,00% en peso o menos) en peso de la composición. Por ejemplo, la composición farmacéutica comprende de aproximadamente el 2% en peso a aproximadamente el 0,10% en peso (por ejemplo, de aproximadamente el 1,5% en peso a aproximadamente el 0,15% en peso o de aproximadamente el 1,3% en peso a aproximadamente el 0,30% en peso) de lubricante, en peso de la composición. En otro ejemplo, la composición farmacéutica comprende el 2% en peso o menos (por ejemplo, el 1,75% en peso, el 1,25% en peso o menos, o el 1,00% en peso o menos) de estearato de magnesio, en peso de la composición. En otro ejemplo más, la composición farmacéutica comprende de aproximadamente el 2% en peso a aproximadamente el 0,10% en peso (por ejemplo, de aproximadamente el 1,5% en peso a aproximadamente el 0,15% en peso o de aproximadamente el 1,3% en peso a aproximadamente el 0,30% en peso) de estearato de magnesio, en peso de la composición.
Las composiciones farmacéuticas de la presente invención pueden comprender opcionalmente uno o más colorantes, sabores, y/o fragancias para mejorar el atractivo visual, el sabor y/o aroma de la composición. Los colorantes adecuados, sabores, o fragancias son compatibles con los ingredientes de la composición farmacéutica, es decir, que no reducen sustancialmente la solubilidad, la estabilidad química, la estabilidad física, la dureza, o la actividad biológica de la composición farmacéutica. En una realización, la composición farmacéutica comprende un colorante, un sabor, y/o una fragancia. Por ejemplo, la composición farmacéutica comprende menos de aproximadamente 1% en peso (por ejemplo, menos de aproximadamente 0,75% en peso o menos de aproximadamente 0,5% en peso) de cada ingrediente opcionalmente, es decir, colorante, sabor y/o fragancia, en peso de la composición. En otro ejemplo, la composición farmacéutica comprende menos de aproximadamente 1% en peso (por ejemplo, menos de aproximadamente 0,75% en peso o menos de aproximadamente 0,5% en peso) de un colorante. En todavía otro ejemplo, la composición farmacéutica comprende menos de aproximadamente 1% en peso (por ejemplo, menos de aproximadamente 0,75% en peso o menos de aproximadamente 0,5% en peso) de un colorante azul (por ejemplo, FD&C Blue n° 1 y/o FD&C Blue n° 2 Aluminum Lake, comercialmente disponible de Colorcon, Inc., de West Point, PA.)
En algunas realizaciones, la composición farmacéutica se puede convertir en tabletas y las tabletas se pueden revestir con un colorante y opcionalmente marcado con un logo, otra imagen y/o texto utilizando una tinta adecuada. En todavía otras realizaciones, la composición farmacéutica se puede convertir en tabletas y las tabletas se pueden revestir con un colorante, encerado, y opcionalmente marcado con un logo, otra imagen y/o texto utilizando una tinta adecuada. Colorantes y tintas adecuadas son compatibles con los ingredientes de la composición farmacéutica, es decir, que no reducen sustancialmente la solubilidad, la estabilidad química, la estabilidad física, la dureza, o la actividad biológica de la composición farmacéutica. Los colorantes y tintas adecuadas pueden ser de cualquier color y son a base de agua o disolvente basado. En una realización, los comprimidos preparados a partir de la composición farmacéutica se recubren con un colorante y se marcan a continuación con un logotipo, otra imagen, y/o el texto usando una tinta adecuada. Por ejemplo, comprimidos que comprenden composición farmacéutica como se describe aquí pueden ser revestidos con aproximadamente 3% en peso (por ejemplo, menos de aproximadamente 6% en peso o menos de aproximadamente 4% en peso) de recubrimiento de película que comprende un colorante. Los comprimidos de color se pueden marcar con un logotipo y texto que indica la fuerza del ingrediente activo en el comprimido usando una tinta adecuada. En otro ejemplo, comprimidos que comprenden composición farmacéutica como se describe aquí pueden ser revestidos con aproximadamente 3% en peso (por ejemplo, menos de aproximadamente 6% en peso o menos de aproximadamente 4% en peso) de un recubrimiento de película que comprende un colorante azul (por ejemplo, OPADRY® II, disponible comercialmente en Colorcon, Inc., de West Point, PA.). Los comprimidos de color se pueden marcar con un logotipo y texto que indica la fuerza del ingrediente activo en el comprimido usando una tinta de color negro (por ejemplo, Opacode® WB, disponible comercialmente de Colorcon, Inc., de West Point, PA.). En otra realización, los comprimidos preparados a partir de la composición farmacéutica se recubren con un colorante, encerado, y después se marcan con un logotipo, otra imagen, y/o el texto usando una tinta adecuada. Por ejemplo, comprimidos que comprenden composición farmacéutica como se describe aquí pueden ser revestidos con aproximadamente 3% en peso (por ejemplo, menos de aproximadamente 6% en peso o menos de aproximadamente 4% en peso) de recubrimiento de película que comprende un colorante. Los comprimidos de color pueden ser encerados con polvo de cera de Carnauba pesado en una cantidad de aproximadamente 0,01% p/p del peso inicial del núcleo del comprimido. Las tabletas enceradas se pueden marcar con un logotipo y texto que indica la fuerza del ingrediente activo en el comprimido usando una tinta adecuada. En otro ejemplo, comprimidos que comprenden composición farmacéutica como se describe aquí pueden ser revestidos con aproximadamente 3% en peso (por ejemplo, menos de aproximadamente 6% en peso o menos de aproximadamente 4% en peso) de un recubrimiento de película que comprende un colorante azul (por ejemplo, OPADRY® II, disponible comercialmente en Colorcon, Inc., de West Point, PA.). Los comprimidos de color pueden ser encerados con cera de Carnauba en polvo pesada en una cantidad de aproximadamente 0,01% p/p del peso inicial del núcleo del comprimido. Las tabletas enceradas se pueden marcar con un logotipo y texto que indica la fuerza del ingrediente activo en el comprimido usando una tinta de color negro (por ejemplo, Opacode® S-1-17823 - una tinta a base de disolvente,
disponible comercialmente de Colorcon, Inc., de West Point, PA.).
Una composición farmacéutica ejemplar comprende de aproximadamente el 5% en peso a aproximadamente el 50% en peso (por ejemplo, de aproximadamente el 5% en peso a aproximadamente el 25% en peso, de aproximadamente el 15% en peso a aproximadamente el 40% en peso, o de aproximadamente el 30% en peso a aproximadamente el 50% en peso) de una dispersión sólida, por peso de la composición, que comprende de aproximadamente el 40% en peso a aproximadamente el 60% en peso de Compuesto 1 sustancialmente amorfo, en peso de la dispersión, y de aproximadamente el 60% en peso a aproximadamente el 40% en peso de un polímero, en peso de la dispersión; de aproximadamente el 25% en peso a aproximadamente el 50% en peso de una carga; de aproximadamente el 1% en peso a aproximadamente el 10% en peso de un disgregante; de aproximadamente el 2% en peso a aproximadamente el 0,3% en peso de un surfactante; de aproximadamente el 5% en peso a aproximadamente el 50% en peso de un aglutinante; de aproximadamente el 2% en peso a aproximadamente el 0,05% en peso de un deslizante; y de aproximadamente el 2% en peso a aproximadamente el 0,1% en peso de un lubricante. O, la composición farmacéutica comprende de aproximadamente el 5% en peso a aproximadamente el 50% en peso (por ejemplo, de aproximadamente el 5% en peso a aproximadamente el 25% en peso, de aproximadamente el 15% en peso a aproximadamente el 40% en peso, o de aproximadamente el 30% en peso a aproximadamente el 50% en peso) de una dispersión sólida, por peso de la composición, que comprende de aproximadamente el 40% en peso a aproximadamente el 60% en peso de Compuesto 1 amorfo, en peso de la dispersión, y de aproximadamente el 60% en peso a aproximadamente el 40% en peso de un polímero, en peso de la dispersión; de aproximadamente el 25% en peso a aproximadamente el 50% en peso de una carga; de aproximadamente el 1% en peso a aproximadamente el 10% en peso de un disgregante; de aproximadamente el 2% en peso a aproximadamente el 0,3% en peso de un surfactante; de aproximadamente el 5% en peso a aproximadamente el 50% en peso de un aglutinante; de aproximadamente el 2% en peso a aproximadamente el 0,05% en peso de un deslizante; y de aproximadamente el 2% en peso a aproximadamente el 0,1% en peso de un lubricante.
Otra composición farmacéutica ejemplar comprende de aproximadamente el 5% en peso a aproximadamente el 50% en peso (por ejemplo, de aproximadamente el 5% en peso a aproximadamente el 25% en peso, de aproximadamente el 15% en peso a aproximadamente el 40% en peso, o de aproximadamente el 30% en peso a aproximadamente el 50% en peso) de una dispersión sólida, por peso de la composición, que comprende de aproximadamente el 70% en peso a aproximadamente el 90% en peso de Compuesto 1 sustancialmente amorfo, en peso de la dispersión, y de aproximadamente el 30% en peso a aproximadamente el 10% en peso de un polímero, en peso de la dispersión; de aproximadamente el 25% en peso a aproximadamente el 50% en peso de una carga; de aproximadamente el 1% en peso a aproximadamente el 10% en peso de un disgregante; de aproximadamente el 2% en peso a aproximadamente el 0,3% en peso de un surfactante; de aproximadamente el 5% en peso a aproximadamente el 50% en peso de un aglutinante; de aproximadamente el 2% en peso a aproximadamente el 0,05% en peso de un deslizante; y de aproximadamente el 2% en peso a aproximadamente el 0,1% en peso de un lubricante. O, la composición farmacéutica comprende de aproximadamente el 5% en peso a aproximadamente el 50% en peso (por ejemplo, de aproximadamente el 5% en peso a aproximadamente el 25% en peso, de aproximadamente el 15% en peso a aproximadamente el 40% en peso, o de aproximadamente el 30% en peso a aproximadamente el 50% en peso) de una dispersión sólida, por peso de la composición, que comprende de aproximadamente el 70% a aproximadamente el 90% en peso de Compuesto 1 amorfo, en peso de la dispersión, y de aproximadamente el 30% en peso a aproximadamente el 10% en peso de un polímero, en peso de la dispersión; de aproximadamente el 25% en peso a aproximadamente el 50% en peso de una carga; de aproximadamente el 1% en peso a aproximadamente el 10% en peso de un disgregante; de aproximadamente el 2% en peso a aproximadamente el 0,3% en peso de un surfactante; de aproximadamente el 5% en peso a aproximadamente el 50% en peso de un aglutinante; de aproximadamente el 2% en peso a aproximadamente el 0,05% en peso de un deslizante; y de aproximadamente el 2% en peso a aproximadamente el 0,1% en peso de un lubricante.
Otra composición farmacéutica de la presente invención comprende aproximadamente el 40% en peso de una dispersión sólida por peso de la composición, en donde la dispersión comprende el 80% en peso del Compuesto 1 sustancialmente amorfo por peso de la dispersión, el 19,5% en peso de PVP/VA por peso de la dispersión y el 0,5% en peso de SLS por peso de la dispersión; aproximadamente el 27% en peso de celulosa microcristalina por peso de la composición; aproximadamente el 27% en peso de lactosa por peso de la composición; aproximadamente el 3% en peso de croscarmelosa de sodio por peso de la composición; aproximadamente el 0,5% en peso de SLS por peso de la composición; aproximadamente el 1% en peso de dióxido de silicio coloidal por peso de la composición; aproximadamente el 1% en peso de estearato de magnesio por peso de la composición y aproximadamente el 0,4% en peso de colorante por peso de la composición. O, la composición farmacéutica de la presente invención comprende aproximadamente el 40% en peso de una dispersión sólida por peso de la composición, en donde la dispersión comprende el 80% en peso del Compuesto amorfo 1 en peso de la dispersión, el 19,5% en peso de PVP/VA por peso del dispersión y el 0,5% en peso de SLS por peso de la dispersión; aproximadamente el 27% en peso de celulosa microcristalina por peso de la composición; aproximadamente el 27% en peso de lactosa por peso de la composición; aproximadamente el 3% en peso de croscarmelosa de sodio por peso de la composición; aproximadamente el 0,5% en peso de SLS por peso
de la composición; aproximadamente el 1% en peso de dióxido de silicio coloidal por peso de la composición; aproximadamente el 1% en peso de estearato de magnesio por peso de la composición y aproximadamente el 0,4% en peso de colorante por peso de la composición.
Otra composición farmacéutica de la presente invención comprende aproximadamente el 40% en peso de una dispersión sólida por peso de la composición, en donde la dispersión comprende el 80% en peso de Compuesto
1 sustancialmente amorfo por peso de la dispersión, el 19,5% en peso de HPMCAS por peso de la dispersión y el 0,5% en peso de SLS por peso de la dispersión; aproximadamente el 27% en peso de celulosa microcristalina por peso de la composición; aproximadamente el 27% en peso de lactosa por peso de la composición; aproximadamente el 3% en peso de croscarmelosa de sodio por peso de la composición; aproximadamente el 0,5% en peso de SLS por peso de la composición; aproximadamente el 1% en peso de dióxido de silicio coloidal por peso de la composición; aproximadamente el 1% en peso de estearato de magnesio por peso de la composición y aproximadamente el 0,4% en peso de colorante por peso de la composición. O, la composición farmacéutica de la presente invención comprende aproximadamente el 40% en peso de una dispersión sólida por peso de la composición, en donde la dispersión comprende el 80% en peso del Compuesto amorfo 1 por peso de la dispersión, el 19,5% en peso de HPMCAS por peso de la dispersión, y el 0,5% en peso de SLS por peso de la dispersión; aproximadamente el 27% en peso de celulosa microcristalina por peso de la composición; aproximadamente el 27% en peso de lactosa por peso de la composición; aproximadamente el 3% en peso de croscarmelosa de sodio por peso de la composición; aproximadamente el 0,5% en peso de SLS por peso de la composición; aproximadamente el 1% en peso de dióxido de silicio coloidal por peso de la composición; aproximadamente el 1% en peso de estearato de magnesio por peso de la composición y aproximadamente el 0,4% en peso de colorante por peso de la composición.
En otra composición farmacéutica más de la presente invención comprende aproximadamente el 34,5% en peso de una dispersión sólida por peso de la composición, en donde la dispersión comprende el 80% en peso de Compuesto 1 sustancialmente amorfo por peso de la dispersión, el 19,5% en peso de HPMCAS por peso de la dispersión y el 0,5% en peso de SLS por peso de la dispersión; aproximadamente el 30% en peso de celulosa microcristalina por peso de la composición; aproximadamente el 30% en peso de lactosa por peso de la composición; aproximadamente el 3% en peso de croscarmelosa de sodio por peso de la composición; aproximadamente el 0,5% en peso de SLS por peso de la composición; aproximadamente el 1% en peso de dióxido de silicio coloidal por peso de la composición; aproximadamente el 1% en peso de estearato de magnesio por peso de la composición.
En otra composición farmacéutica más de la presente invención, una composición de comprimido farmacéutico en forma de comprimido oblongo que tiene una dureza de 9,5 Kp ± 15 por ciento comprende aproximadamente el 34% en peso de una dispersión sólida por peso de la composición, en donde la dispersión comprende el 80% en peso de Compuesto 1 substancialmente amorfo por peso de la dispersión, el 19,5% en peso de HPMCAS por peso de la dispersión y el 0,5% en peso de SLS por peso de la dispersión; aproximadamente el 30% en peso de celulosa microcristalina por peso de la composición; aproximadamente el 30% en peso de lactosa por peso de la composición; aproximadamente el 3% en peso de croscarmelosa de sodio por peso de la composición; aproximadamente el 0,5% en peso de SLS por peso de la composición; aproximadamente el 1% en peso de dióxido de silicio coloidal por peso de la composición; y aproximadamente el 1% en peso de estearato de magnesio por peso de la composición. En ciertas realizaciones, el comprimido farmacéutico con forma de comprimido oblongo contiene 150 mg del Compuesto 1. En ciertas realizaciones, el comprimido farmacéutico con forma de comprimido oblongo contiene 100 mg del Compuesto 1.
En otra composición farmacéutica más de la presente invención, una composición de comprimido farmacéutico con forma de comprimido oblongo que tiene una dureza inicial de 11 Kp ± 20 por ciento comprende aproximadamente el 40% en peso de una dispersión sólida por peso de la composición, en donde la dispersión comprende el 80% en peso del Compuesto 1 substancialmente amorfo por peso de la dispersión, el 19,5% en peso de HPMCAS por peso de la dispersión y el 0,5% en peso de SLS por peso de la dispersión; aproximadamente el 30% en peso de celulosa microcristalina por peso de la composición; aproximadamente el 30% en peso de lactosa por peso de la composición; aproximadamente el 3% en peso de croscarmelosa de sodio por peso de la composición; aproximadamente el 0,5% en peso de SLS por peso de la composición; aproximadamente el 1% en peso de dióxido de silicio coloidal por peso de la composición; y aproximadamente el 1% en peso de estearato de magnesio por peso de la composición. En ciertas realizaciones, el comprimido farmacéutico con forma de comprimido oblongo contiene 150 mg del Compuesto 1.
En otra composición farmacéutica más de la presente invención, una composición de comprimido farmacéutico con forma de comprimido oblongo que tiene una dureza inicial de 11 Kp ± 20 por ciento comprende aproximadamente el 34,1% en peso de una dispersión sólida por peso de la composición, en donde la dispersión comprende el 80% en peso de Compuesto 1 substancialmente amorfo por peso de la dispersión, el 19,5% en peso de HPMCAS por peso de la dispersión y el 0,5% en peso de SLS por peso de la dispersión; aproximadamente el 30% en peso de celulosa microcristalina por peso de la composición; aproximadamente el 30,4% en peso de lactosa por peso de la composición; aproximadamente el 3% en peso de croscarmelosa de sodio por peso de la
composición; aproximadamente el 0,5% en peso de SLS por peso de la composición; aproximadamente el 1% en peso de dióxido de silicio coloidal por peso de la composición; y aproximadamente el 1% en peso de estearato de magnesio por peso de la composición. En algunos aspectos, la composición de comprimido farmacéutico con forma de comprimido oblongo contiene 100 mg de Compuesto 1. En otros aspectos, la composición de comprimido farmacéutico con forma de comprimido oblongo incluye un recubrimiento colorante y un logotipo o texto impreso. En algunas realizaciones de este aspecto, la composición de comprimido farmacéutico con forma de comprimido oblongo incluye un recubrimiento de azul de OPADRY® II y un logotipo o texto de tinta a base de agua o solvente. En ciertas realizaciones, el comprimido farmacéutico con forma de comprimido oblongo contiene 150 mg de Compuesto 1.
En otra composición farmacéutica de la presente invención, una composición de comprimido farmacéutico con forma de comprimido oblongo que tiene una dureza inicial de entre aproximadamente 6 y 16 Kp comprende aproximadamente el 34,1% en peso de una dispersión sólida por peso de la composición, en donde la dispersión comprende el 80% en peso del Compuesto 1 substancialmente amorfo por peso de la dispersión, el 19,5% en peso de HPMCAS por peso de la dispersión y el 0,5% en peso de SLS por peso de la dispersión; aproximadamente el 30,5% en peso de celulosa microcristalina por peso de la composición; aproximadamente el 30,4% en peso de lactosa por peso de la composición; aproximadamente el 3% en peso de croscarmelosa de sodio por peso de la composición; aproximadamente el 0,5% en peso de SLS por peso de la composición; aproximadamente el 0,5% en peso de dióxido de silicio coloidal por peso de la composición; y aproximadamente el 1% en peso de estearato de magnesio por peso de la composición. En algunos aspectos, la composición de comprimido farmacéutico con forma de comprimido oblongo contiene 100 mg de Compuesto 1. En algunos aspectos adicionales, la composición de comprimido farmacéutico con forma de comprimido oblongo comprende un recubrimiento de colorante, un recubrimiento de cera y un logotipo o texto impreso. En algunas realizaciones de este aspecto, el comprimido farmacéutico con forma de comprimido oblongo incluye un recubrimiento de azul de OPADRY® II y un logotipo o texto de tinta a base de agua o solvente. En algunos casos, el recubrimiento colorante es azul de OPADRY® II. En algunos casos, el recubrimiento de cera comprende cera de Carnauba. En ciertos aspectos, la tinta para el logo o texto impreso es una tinta a base de solvente. En algunos aspectos, la composición de comprimido farmacéutico con forma de comprimido oblongo contiene 150 mg de Compuesto 1.
En otra composición farmacéutica más de la presente invención, una composición de comprimido farmacéutico que tiene una dureza inicial de entre aproximadamente 9 y 21 Kp comprende aproximadamente el 34,1% en peso de una dispersión sólida por peso de la composición, en donde la dispersión comprende el 80% en peso de Compuesto 1 sustancialmente amorfo por peso de la dispersión, el 19,5% en peso de HPMCAS por peso de la dispersión y el 0,5% en peso de SLS por peso de la dispersión; aproximadamente el 30,5% en peso de celulosa microcristalina por peso de la composición; aproximadamente el 30,4% en peso de lactosa por peso de la composición; aproximadamente el 3% en peso de croscarmelosa de sodio por peso de la composición; aproximadamente el 0,5% en peso de SLS por peso de la composición; aproximadamente el 0,5% en peso de dióxido de silicio coloidal por peso de la composición; y aproximadamente el 1% en peso de estearato de magnesio por peso de la composición. En algunas realizaciones, la composición de comprimido farmacéutico con forma de comprimido oblongo contiene 150 mg de Compuesto 1. En algunos aspectos, la composición de comprimido farmacéutico con forma de comprimido oblongo comprende además un recubrimiento de colorante, una capa de cera y un logotipo o texto impreso. En algunos casos, el comprimido incluye un recubrimiento de azul de OPADRY® II y un logotipo o texto de tinta a base de agua o solvente. En otros casos más, el recubrimiento de cera comprende cera de Carnauba. En algunas realizaciones, la tinta para el logotipo o texto impreso es una tinta a base de solvente. En algunos aspectos, la composición de comprimido farmacéutico con forma de comprimido oblongo contiene 100 mg de Compuesto 1.
En otra composición farmacéutica más de la presente invención, una composición farmacéutica comprende aproximadamente el 34% en peso de una dispersión sólida por peso de la composición, en donde la dispersión comprende el 80% en peso de Compuesto 1 sustancialmente amorfo por peso de la dispersión, el 19,5% en peso de HPMCAS por peso de la dispersión, y el 0,5% en peso de SLS por peso de la dispersión; aproximadamente el 30% en peso de celulosa microcristalina por peso de la composición; aproximadamente el 30% en peso de lactosa por peso de la composición; aproximadamente el 3% en peso de croscarmelosa de sodio por peso de la composición; aproximadamente el 0,5% en peso de SLS por peso de la composición; aproximadamente el 1% en peso de dióxido de silicio coloidal por peso de la composición; y aproximadamente el 1% en peso de estearato de magnesio por peso de la composición. En ciertas realizaciones, la composición farmacéutica contiene 150 mg de Compuesto 1. En otras realizaciones, la composición farmacéutica contiene 100 mg de Compuesto 1.
En otra composición farmacéutica más de la presente invención, una composición farmacéutica comprende aproximadamente el 40% en peso de una dispersión sólida por peso de la composición, en donde la dispersión comprende el 80% en peso del Compuesto 1 sustancialmente amorfo por peso de la dispersión, el 19,5% en peso de HPMCAS por peso de la dispersión y el 0,5% en peso de SLS por peso de la dispersión; aproximadamente el 30% en peso de celulosa microcristalina por peso de la composición; aproximadamente el 30% en peso de lactosa por peso de la composición; aproximadamente el 3% en peso de croscarmelosa de sodio por peso de la composición; aproximadamente el 0,5% en peso de SLS por peso de la composición; aproximadamente el 1% en
peso de dióxido de silicio coloidal por peso de la composición; y aproximadamente el 1% en peso de estearato de magnesio por peso de la composición. En ciertas realizaciones, la composición farmacéutica contiene 150 mg de Compuesto 1. En otras realizaciones, la composición farmacéutica contiene 100 mg de Compuesto 1.
En otra composición farmacéutica más de la presente invención, una composición farmacéutica comprende aproximadamente el 34,1% en peso de una dispersión sólida por peso de la composición, en donde la dispersión comprende el 80% en peso de Compuesto 1 sustancialmente amorfo por peso de la dispersión, el 19,5% en peso de HPMCAS por peso de la dispersión y el 0,5% en peso de SLS por peso de la dispersión; aproximadamente el 30% en peso de celulosa microcristalina por peso de la composición; aproximadamente el 30,4% en peso de lactosa por peso de la composición; aproximadamente el 3% en peso de croscarmelosa de sodio por peso de la composición; aproximadamente el 0,5% en peso de SLS por peso de la composición; aproximadamente el 1% en peso de dióxido de silicio coloidal por peso de la composición; y aproximadamente el 1% en peso de estearato de magnesio por peso de la composición. En algunos aspectos, la composición farmacéutica contiene 100 mg de Compuesto 1. En otras realizaciones, la composición farmacéutica contiene 150 mg del Compuesto 1. En otros aspectos, la composición farmacéutica se forma como una composición de comprimido que incluye un recubrimiento colorante y un logotipo o texto impreso. En algunas realizaciones de este aspecto, la composición del comprimido farmacéutico incluye un recubrimiento de azul de OPADRY® II y un logotipo o texto de tinta a base de agua o solvente.
En otra composición farmacéutica de la presente invención, una composición farmacéutica comprende aproximadamente el 34,1% en peso de una dispersión sólida en peso de la composición, en donde la dispersión comprende de aproximadamente 80% en peso de Compuesto 1 sustancialmente amorfo en peso de la dispersión, sobre 19,5% en peso de HPMCAS en peso de la dispersión, y aproximadamente 0,5% en peso de SLS en peso de la dispersión; aproximadamente 30,5% en peso de celulosa microcristalina en peso de la composición; aproximadamente 30,4% en peso de lactosa en peso de la composición; alrededor de 3% en peso de croscarmelosa de sodio en peso de la composición; aproximadamente 0,5% en peso de SLS en peso de la composición; aproximadamente 0,5% en peso de dióxido de silicio coloidal en peso de la composición; y aproximadamente 1% en peso de estearato de magnesio en peso de la composición. En algunos aspectos, la tableta farmacéutica contiene 100 mg de compuesto 1. En otras realizaciones, la composición farmacéutica contiene 150 mg de Compuesto 1. En algunos aspectos adicionales, la composición farmacéutica se forma como un comprimido y comprende un colorante recubierto, un recubrimiento de cera, y un logotipo impreso o texto. En algunas realizaciones de este aspecto, el comprimido farmacéutico incluye un revestimiento OPADRY® II azul y un logo de tinta o texto a base de agua o disolvente. En algunos casos, el revestimiento colorante es OPADRY® II azul. En algunos casos, la capa de cera comprende cera de carnauba. En ciertos aspectos, la tinta para el logotipo impreso o texto es una tinta a base de disolvente.
También se observa que las composiciones farmacéuticas de la presente invención se pueden procesar en forma de tableta, forma de cápsula, o suspensión que se adapta para administración oral o se pueden reconstituir en un disolvente acuoso (por ejemplo, agua DI o solución salina) para administración oral, IV, o inhalación (por ejemplo, nebulizador).
Otro aspecto de la presente invención proporciona una composición farmacéutica que consiste en una tableta que incluye un potenciador FQ de API (por ejemplo, una dispersión sólida de N-[2,4-Bis(1,1-dimetiletilo)-5-hidroxifenilo]-1,4-dihidro-4- oxoquinolina-3-carboxamida) y otros excipientes (por ejemplo, una carga, un disgregante, un surfactante, un aglutinante, un agente deslizante, un colorante, un lubricante, o cualquier combinación de los mismos), cada uno de los cuales se describe anteriormente y en los ejemplos a continuación, en el que la tableta tiene una disolución de al menos aproximadamente 50% (por ejemplo, al menos aproximadamente 60%, al menos aproximadamente 70%, al menos aproximadamente 80%, al menos aproximadamente 90%, o al menos aproximadamente 99%) en unos 30 minutos. En un ejemplo, la composición farmacéutica se compone de una tableta que incluye un potenciador FQ de API (por ejemplo, una dispersión sólida de Compuesto 1) y otros excipientes (por ejemplo, una carga, un disgregante, un surfactante, un aglutinante, un agente deslizante, un colorante, un lubricante, o cualquier combinación de los mismos), cada uno de los cuales se describe anteriormente y en los Ejemplos a continuación, en el que la tableta tiene una disolución de aproximadamente 50% a aproximadamente 100% (por ejemplo, de aproximadamente 55% a aproximadamente 95% o de aproximadamente 60% a aproximadamente 90%) en unos 30 minutos. En otro ejemplo, la composición farmacéutica consiste en un comprimido que comprende una dispersión sólida que comprende Compuesto 1 amorfo o sustancialmente amorfo y HPMCAS o PVP/VA; y, una carga, un disgregante, un agente surfactante, un aglutinante, un deslizante y un lubricante, en el que la tableta tiene una disolución de al menos aproximadamente 50% (por ejemplo, al menos aproximadamente 60%, al menos aproximadamente 70%, al menos aproximadamente el 80%, al menos aproximadamente 90%, o al menos aproximadamente 99%) en unos 30 minutos. En todavía otro ejemplo, la composición farmacéutica consiste en un comprimido que comprende una dispersión sólida que comprende Compuesto 1 amorfo o sustancialmente amorfo y HPMCAS o PVP/VA; y, una carga, un disgregante, un agente surfactante, un aglutinante, un deslizante y un lubricante, en el que la tableta tiene una disolución de aproximadamente 50% a aproximadamente 100% (por ejemplo, de aproximadamente 55% a aproximadamente 95% o de aproximadamente 60% a aproximadamente 90%) en unos 30 minutos.
En una realización, el comprimido comprende una dispersión sólida que comprende por lo menos aproximadamente 25 mg (por ejemplo, por lo menos aproximadamente 30 mg, por lo menos aproximadamente 40 mg, o por los menos aproximadamente 50 mg) del Compuesto 1 amorfo o sustancialmente amorfo; y PVP/VA y SLS. En otra realización, el comprimido comprende una dispersión sólida que comprende por lo menos aproximadamente 25 mg (por ejemplo, por lo menos aproximadamente 30 mg, por lo menos aproximadamente 40 mg, por lo menos aproximadamente 50 mg, por lo menos aproximadamente 100 mg o por lo menos 150 mg) de Compuesto 1 sustancialmente amorfo o amorfo; y HPMCAS y SLS.
La disolución se puede medir con un aparato USP estándar de Tipo II que emplea un medio de disolución de 0,6% de sulfato de laurilo sódico disuelto en 900 mL de agua DI, agitación a aproximadamente 50-75 rpm a una temperatura de aproximadamente 37°C. Una tableta experimental única se ensaya en cada recipiente de ensayo del aparato. La disolución también se puede medir con un aparato USP estándar Tipo II que emplea un medio de disolución de 0,7% de sulfato de laurilo sódico disuelto en 900 mL de 50 mM de tampón de fosfato de sodio (pH 6,8), agitación a aproximadamente 65 rpm a una temperatura de aproximadamente 37°C. Una tableta experimental única se ensaya en cada recipiente de ensayo del aparato. La disolución también se puede medir con un aparato USP estándar Tipo II que emplea un medio de disolución de 0,5% de sulfato de laurilo sódico disuelto en 900 mL de 50 mM de tampón de fosfato de sodio (pH 6,8), agitación a aproximadamente 65 rpm a una temperatura de aproximadamente 37°C. Una tableta experimental única se ensaya en cada recipiente de ensayo del aparato.
Otro aspecto de la presente invención proporciona una composición farmacéutica que consiste en una tableta que comprende un potenciador FQ de API (por ejemplo, una dispersión sólida de Compuesto 1) y otros excipientes (por ejemplo, una carga, un disgregante, un agente surfactante, un aglutinante, un deslizante, un colorante, un lubricante, o cualquier combinación de los mismos), cada uno de los cuales se describe anteriormente y en los Ejemplos a continuación, en el que el comprimido tiene una dureza de al menos aproximadamente 5 Kp. En un ejemplo, la composición farmacéutica se compone de una tableta que comprende un potenciador FQ de API (por ejemplo, una dispersión sólida de Compuesto 1) y otros excipientes (por ejemplo, una carga, un disgregante, un surfactante, un aglutinante, un agente deslizante, un colorante, un lubricante, o cualquier combinación de los mismos), cada uno de los cuales se describe anteriormente y en los Ejemplos a continuación, en el que el comprimido tiene una dureza de al menos aproximadamente 5 Kp (por ejemplo, al menos aproximadamente 5,5, al menos aproximadamente 6 Kp, o por lo menos aproximadamente 7 Kp).
III. MÉTODO DE PRODUCIR UNA COMPOSICIÓN FARMACÉUTICA
Otro aspecto de la presente invención proporciona un método para producir una composición farmacéutica que proporciona una mezcla de una dispersión sólida de N-[2,4-Bis(acetato de 1, 1 -dimetilo)- 5-hidroxifenilo]-1,4-dihidro-4-oxoquinolina-3-carboxamida amorfa o sustancialmente amorfa, un aglutinante, un agente deslizante, un agente surfactante, un lubricante, un desintegrante, y una carga, y comprimiendo la mezcla en un comprimido que tiene una disolución de al menos aproximadamente 50% dentro de unos 30 minutos.
Cada uno de los ingredientes de esta mezcla se describe anteriormente y en los Ejemplos a continuación. Además, la mezcla puede comprender aditivos opcionales, tales como uno o más colorantes, uno o más sabores, y/o una o más fragancias como se describe anteriormente y en los Ejemplos a continuación. Y, también se presentan las concentraciones relativas (por ejemplo, % en peso) de cada uno de estos ingredientes (y cualquier aditivo opcional) en la mezcla anteriormente y en los Ejemplos a continuación. Los ingredientes que constituyen la mezcla se pueden proporcionar de forma secuencial o en cualquier combinación de las adiciones; y, los ingredientes o combinación de ingredientes se pueden proporcionar en cualquier orden. En una realización el lubricante es el último componente añadido a la mezcla.
En una realización, la mezcla comprende una dispersión sólida de Compuesto 1 sustancialmente amorfo, un aglutinante, un agente deslizante, un agente surfactante, un lubricante, un disgregante, y una carga, en el que cada uno de estos ingredientes se proporciona en forma de polvo (por ejemplo, proporcionado como partículas que tienen un diámetro medio, medido por dispersión de luz, de 250 pm o menos (por ejemplo, 150 pm o menos, 100 pm o menos, 50 pm o menos, 45 pm o menos, 40 pm o menos, o 35 pm o menos)). Por ejemplo, la mezcla comprende una dispersión sólida del Compuesto 1 amorfo, un aglutinante, un agente deslizante, un agente surfactante, un lubricante, un disgregante, y una carga, en el que cada uno de estos ingredientes se proporciona en forma de polvo (por ejemplo, como partículas que tienen un diámetro medio, medido por dispersión de luz, de 250 pm o menos (por ejemplo, 150 pm o menos, 100 pm o menos, 50 pm o menos, 45 pm o menos, 40 pm o menos, o 35 pm o menos)).
En otra realización, la mezcla comprende una dispersión sólida de Compuesto 1 sustancialmente amorfo, un aglutinante, un agente deslizante, un agente surfactante, un lubricante, un disgregante, y una carga, en el que cada uno de estos ingredientes es sustancialmente libre de agua. Cada uno de los ingredientes comprende menos de 5% en peso (por ejemplo, menos de 2% en peso, menos de 1% en peso, menos de 0,75% en peso, menos de 0,5% en peso, o menos de 0,25% en peso) de agua en peso del ingrediente. Por ejemplo, la mezcla comprende una dispersión sólida del Compuesto 1 amorfo, un aglutinante, un agente deslizante, un agente surfactante, un
lubricante, un disgregante, y una carga, en donde cada uno de estos ingredientes es sustancialmente libre de agua. A saber, cada uno de los ingredientes comprende menos de 5% en peso (por ejemplo, menos de 2% en peso, menos de 1% en peso, menos de 0,75% en peso, menos de 0,5% en peso, o menos de 0,25% en peso) de agua por peso del ingrediente.
En otra realización, la compresión de la mezcla en un comprimido se lleva a cabo al rellenar una forma (por ejemplo, un molde) con la mezcla y al aplicar presión a la mezcla. Esto se puede lograr usando una prensa de troquel u otro aparato similar. También se observó que la aplicación de presión a la mezcla en la forma puede repetirse utilizando la misma presión durante cada compresión o el uso de diferentes presiones durante las compresiones. En otro ejemplo, la mezcla se comprime utilizando una prensa de troquel que aplica presión suficiente para formar un comprimido que tiene una disolución de aproximadamente 50% o más en aproximadamente 30 minutos (por ejemplo, aproximadamente 55% o más en aproximadamente 30 minutos o aproximadamente 60% o más en aproximadamente 30 minutos). Por ejemplo, la mezcla se comprime utilizando una prensa de troquel para producir una dureza de comprimido de al menos aproximadamente 5 Kp (al menos aproximadamente 5,5 Kp, al menos aproximadamente 6 Kp, al menos aproximadamente 7 Kp, al menos aproximadamente 11 Kp, o al menos 21Kp). En algunos casos, la mezcla se comprime para producir una dureza de comprimido de entre aproximadamente 6 y 21 Kp.
En algunas realizaciones, los comprimidos que comprenden una composición farmacéutica como se describe en el presente documento se pueden recubrir con aproximadamente 3,0% en peso de un recubrimiento de película que comprende un colorante en peso de la tableta. En ciertos casos, la suspensión colorante o solución usada para recubrir los comprimidos comprende aproximadamente 20% p/p de sólidos en peso de la suspensión de colorante o solución. En aún otros ejemplos, los comprimidos recubiertos se pueden marcar con un logotipo, otra imagen o texto.
En otra realización, el método de producción de una composición farmacéutica que comprende proporcionar una mezcla de una dispersión sólida del Compuesto 1 sustancialmente amorfo, un aglutinante, un agente deslizante, un agente surfactante, un lubricante, un disgregante y una carga; mezclando la mezcla hasta que la mezcla esté sustancialmente homogénea, y comprimiendo la mezcla en un comprimido como se describe anteriormente o en los ejemplos siguientes. O bien, el método de producción de una composición farmacéutica que comprende proporcionar una mezcla de una dispersión sólida del Compuesto 1 amorfo, un aglutinante, un agente deslizante, un agente surfactante, un lubricante, un disgregante y una carga; mezclando la mezcla hasta que la mezcla esté sustancialmente homogénea, y comprimiendo la mezcla en un comprimido como se describe anteriormente o en los ejemplos siguientes. Por ejemplo, la mezcla se mezcla por agitación, o similar usando el mezclado manual, un mezclador, una licuadora, cualquier combinación de los mismos, o similares. Cuando se añaden los ingredientes o combinaciones de ingredientes secuencialmente, la mezcla puede producirse entre adiciones sucesivas, de forma continua durante toda la adición de ingredientes, después de la adición de todos los ingredientes o combinaciones de ingredientes, o cualquier combinación de los mismos. La mezcla se mezcla hasta que tenga una composición sustancialmente homogénea.
IV. ADMINISTRACIÓN DE UNA FORMULACIÓN FARMACÉUTICA
En otro aspecto, la invención también proporciona un método para tratar o disminuir la gravedad de una enfermedad en un paciente que comprende administrar a dicho paciente una de las composiciones tal como se definen en el presente documento, y que dicha enfermedad se selecciona de fibrosis quística, el asma, el humo EPOC inducida, bronquitis crónica, rinosinusitis, estreñimiento, pancreatitis, insuficiencia pancreática, infertilidad masculina causada por la ausencia bilateral congénita de los vasos deferentes (CBAVD), enfermedad leve pulmonar, pancreatitis idiopática, aspergilosis broncopulmonar alérgica (ABPA), enfermedad hepática, enfisema hereditario, la hemocromatosis hereditaria, deficiencias de la coagulación-fibrinolisis, tales como deficiencia de proteína C, angioedema hereditaria de Tipo 1, las deficiencias de procesamiento de lípidos, tales como hipercolesterolemia familiar, quilomicronemia de Tipo 1, abetalipoproteinemia, enfermedades de almacenamiento lisosomal, tales como enfermedad de célula I/pseudo-Hurler, mucopolisacaridosis, Sandhof/Tay-Sachs, Crigler-Najjar Tipo II, poliendocrinopatía/hiperinsulemia, Diabetes mellitus, enanismo de Laron, deficiencia de mieloperoxidasa, hipoparatiroidismo primario, melanoma, glucanosis CDG Tipo 1, el hipertiroidismo congénito, osteogénesis imperfecta, hipofibrinogenemia hereditaria, deficiencia de ACT, diabetes insípida (DI), DI neurofiseal, DI nefrogénica, síndrome de Charcot-Marie Tooth, enfermedad de Perlizaeus-Merzbacher, enfermedades neurodegenerativas tales como la enfermedad de Alzheimer, enfermedad de Parkinson, esclerosis lateral amiotrófica, parálisis supranuclear progresiva, enfermedad de Pick, varios trastornos neurológicos de poliglutamina como la de Huntington, ataxia espinocerebelar tipo I, atrofia muscular espinal y bulbar, palidoluisiana dentatorubal, y distrofia miotónica, así como encefalopatías espongiformes, tales como la enfermedad hereditaria de Creutzfeldt-Jakob (debido a defecto en el procesamiento de proteínas de prion), enfermedad de Fabry, síndrome de Straussler-Scheinker, EPOC, enfermedad del ojo seco, o enfermedad de Sjogren, osteoporosis, osteopenia, síndrome de Gorham, canalopatías de cloruro tales como miotonía congénita (formas Thomson y Becker), síndrome de Bartter de tipo III, enfermedad de Dent, hiperekplexia, epilepsia, hiperekplexia, enfermedad de almacenamiento lisosomal, síndrome de Angelman, y discinesia ciliar primaria (PCD), un término para trastornos hereditarios de la estructura y/o
la función de los cilios, incluyendo PCD con situs inversus (también conocido como síndrome de Kartagener), PCD sin situs inversus y aplasia ciliar.
Otro aspecto de la presente invención proporciona un método de administración de una composición farmacéutica mediante la administración oral a un paciente al menos una vez por día de la composición que comprende una dispersión sólida de Compuesto 1 amorfo o sustancialmente amorfo, en el que la dispersión sólida comprende al menos aproximadamente 25 mg de Compuesto 1 amorfo o sustancialmente amorfo.
Otro aspecto de la presente invención proporciona un método de administración de una composición farmacéutica mediante la administración oral a un paciente al menos una vez por día de la composición que comprende una dispersión sólida de Compuesto 1 amorfo o sustancialmente amorfo, en el que la dispersión sólida comprende al menos aproximadamente 50 mg de Compuesto 1 amorfo o sustancialmente amorfo.
Otro aspecto de la presente invención proporciona un método de administración de una composición farmacéutica mediante la administración oral a un paciente al menos una vez por día de la composición que comprende una dispersión sólida de Compuesto 1 amorfo o sustancialmente amorfo, en el que la dispersión sólida comprende al menos aproximadamente 75 mg de Compuesto 1 amorfo o sustancialmente amorfo.
Otro aspecto de la presente invención proporciona un método de administración de una composición farmacéutica mediante la administración oral a un paciente al menos una vez por día de la composición que comprende una dispersión sólida de Compuesto 1 amorfo o sustancialmente amorfo, en el que la dispersión sólida comprende al menos aproximadamente 100 mg de Compuesto 1 amorfo o sustancialmente amorfo.
Otro aspecto de la presente invención proporciona un método de administración de una composición farmacéutica mediante la administración oral a un paciente al menos una vez por día de la composición que comprende una dispersión sólida de Compuesto 1 amorfo o sustancialmente amorfo, en la que la dispersión sólida comprende al menos aproximadamente 150 mg de Compuesto 1 amorfo o sustancialmente amorfo.
Otro aspecto de la presente invención proporciona un método de administración de una composición farmacéutica mediante la administración oral a un paciente al menos una vez por día de la composición que comprende una dispersión sólida de Compuesto 1 amorfo o sustancialmente amorfo, en el que la dispersión sólida comprende al menos aproximadamente 250 mg de Compuesto 1 amorfo o sustancialmente amorfo.
Otro aspecto de la presente invención proporciona un método de administración de una composición farmacéutica mediante la administración por vía oral a un paciente dos veces por día de la composición que comprende una dispersión sólida del Compuesto 1 amorfo o sustancialmente amorfo, en el que la dispersión sólida comprende al menos aproximadamente 25 mg de Compuesto 1 amorfo o sustancialmente amorfo.
Otro aspecto de la presente invención proporciona un método de administración de una composición farmacéutica mediante la administración por vía oral a un paciente dos veces al día de la composición que comprende una dispersión sólida de Compuesto 1 amorfo o sustancialmente amorfo, en el que la dispersión sólida comprende al menos aproximadamente 50 mg de Compuesto 1 amorfo o sustancialmente amorfo.
Otro aspecto de la presente invención proporciona un método de la administración de una composición farmacéutica mediante la administración oral a un paciente dos veces al día de la composición que comprende una dispersión sólida de Compuesto 1 amorfo o sustancialmente amorfo, en el que la dispersión sólida comprende al menos aproximadamente 75 mg de Compuesto 1 amorfo o sustancialmente amorfo.
Otro aspecto de la presente invención proporciona un método de administración de una composición farmacéutica mediante la administración oral a un paciente dos veces al día de la composición que comprende una dispersión sólida de Compuesto 1 amorfo o sustancialmente amorfo, en el que la dispersión sólida comprende al menos aproximadamente 100 mg de Compuesto 1 amorfo o sustancialmente amorfo.
Otro aspecto de la presente invención proporciona un método de administración de una composición farmacéutica mediante la administración oral a un paciente dos veces al día de la composición que comprende una dispersión sólida de Compuesto 1 amorfo o sustancialmente amorfo, en el que la dispersión sólida comprende al menos aproximadamente 150 mg de Compuesto 1 amorfo o sustancialmente amorfo.
Otro aspecto de la presente invención proporciona un método de administración de una composición farmacéutica mediante la administración oral a un paciente dos veces al día de la composición que comprende una dispersión sólida de Compuesto 1 amorfo o sustancialmente amorfo, en el que la dispersión sólida comprende al menos aproximadamente 250 mg de Compuesto 1 amorfo o substancialmente amorfo.
Otro aspecto de la presente invención proporciona un método de administración de una composición farmacéutica mediante la administración oral a un paciente una vez cada 12 horas. La composición comprende una
dispersión sólida de Compuesto 1 amorfo o sustancialmente amorfo, en el que la dispersión sólida comprende al menos aproximadamente 25 mg de Compuesto 1 amorfo o sustancialmente amorfo.
Otro aspecto de la presente invención proporciona un método de administración de una composición farmacéutica mediante la administración oral a un paciente una vez cada 12 horas. La composición comprende una dispersión sólida de Compuesto 1 amorfo o sustancialmente amorfo, en el que la dispersión sólida comprende al menos aproximadamente 50 mg de Compuesto 1 amorfo o sustancialmente amorfo.
Otro aspecto de la presente invención proporciona un método de administración de una composición farmacéutica mediante la administración oral a un paciente una vez cada 12 horas. La composición que comprende una dispersión sólida de Compuesto 1 amorfo o sustancialmente amorfo, en el que la dispersión sólida comprende al menos aproximadamente 75 mg de Compuesto 1 amorfo o sustancialmente amorfo.
Otro aspecto de la presente invención proporciona un método de administración de una composición farmacéutica mediante la administración oral a un paciente una vez cada 12 horas. La composición que comprende una dispersión sólida de Compuesto 1 amorfo o sustancialmente amorfo, en el que la dispersión sólida comprende al menos aproximadamente 100 mg de Compuesto 1 amorfo o sustancialmente amorfo.
Otro aspecto de la presente invención proporciona un método de administración de una composición farmacéutica mediante la administración oral a un paciente una vez cada 12 horas. La composición que comprende una dispersión sólida de Compuesto 1 amorfo o sustancialmente amorfo, en el que la dispersión sólida comprende al menos aproximadamente 150 mg de Compuesto 1 amorfo o sustancialmente amorfo.
Otro aspecto de la presente invención proporciona un método de administración de una composición farmacéutica mediante la administración oral a un paciente una vez cada 12 horas. La composición que comprende una dispersión sólida de Compuesto 1 amorfo o sustancialmente amorfo, en el que la dispersión sólida comprende al menos aproximadamente 250 mg de Compuesto 1 amorfo o sustancialmente amorfo.
En todavía otros aspectos de la presente invención, una composición farmacéutica como se describe en el presente documento se administra por vía oral a un paciente una vez cada 24 horas.
Otro aspecto de la presente invención proporciona un método de administración de una composición farmacéutica mediante la administración oral a un paciente una vez al día de la composición que comprende una dispersión sólida de Compuesto 1 amorfo o sustancialmente amorfo, en el que la dispersión sólida comprende al menos aproximadamente 25 mg del Compuesto 1 amorfo o sustancialmente amorfo.
Otro aspecto de la presente invención proporciona un método de administración de una composición farmacéutica mediante la administración oral a un paciente una vez al día de la composición que comprende una dispersión sólida de Compuesto 1 amorfo o sustancialmente amorfo, en el que la dispersión sólida comprende al menos aproximadamente 50 mg de Compuesto 1 amorfo o sustancialmente amorfo.
Otro aspecto de la presente invención proporciona un método de administración de una composición farmacéutica mediante la administración oral a un paciente una vez al día de la composición que comprende una dispersión sólida de Compuesto 1 amorfo o sustancialmente amorfo, en la que la dispersión sólida comprende al menos aproximadamente 75 mg de Compuesto 1 amorfo o sustancialmente amorfo.
Otro aspecto de la presente invención proporciona un método de administración de una composición farmacéutica mediante la administración oral a un paciente una vez al día de la composición que comprende una dispersión sólida de Compuesto 1 amorfo o sustancialmente amorfo, en el que la dispersión sólida comprende al menos aproximadamente 100 mg de Compuesto 1 amorfo o sustancialmente amorfo.
Otro aspecto de la presente invención proporciona un método de administración de una composición farmacéutica mediante la administración por vía oral a un paciente una vez al día de la composición que comprende una dispersión sólida de Compuesto 1 amorfo o sustancialmente amorfo, en el que la dispersión sólida comprende al menos aproximadamente 150 mg de Compuesto 1 amorfo o sustancialmente amorfo.
Otro aspecto de la presente invención proporciona un método de administración de una composición farmacéutica mediante la administración por vía oral a un paciente una vez al día de la composición que comprende una dispersión sólida de Compuesto 1 amorfo o sustancialmente amorfo, en el que la dispersión sólida comprende al menos aproximadamente 250 mg de Compuesto 1 amorfo o sustancialmente amorfo.
Otro aspecto de la presente invención proporciona un método de la administración de una composición farmacéutica que comprende administrar oralmente a un paciente al menos una vez por día al menos un comprimido que comprende una composición farmacéutica que contiene una dispersión sólida de Compuesto 1 sustancialmente amorfo, una carga, un aglutinante, un agente deslizante, un desintegrante, un surfactante, y un lubricante, cada uno
de los cuales se describe anteriormente y en los ejemplos a continuación, en el que la dispersión sólida comprende al menos 25 mg (por ejemplo, al menos 35 mg, al menos 40 mg, o al menos 45 mg) del Compuesto 1 sustancialmente amorfo.
Las composiciones farmacéuticas de la invención pueden usarse en métodos de administración de una composición farmacéutica que comprenden administrar por vía oral a un paciente por lo menos una comprimido que comprende:
a. una dispersión sólida de la reivindicación 1 que comprende aproximadamente 25 mg de Compuesto 1 sustancialmente amorfo o amorfo;
b. una carga
c. un disgregante;
d. un surfactante;
e. un aglutinante
f. un deslizante; y
g. un lubricante.
Las composiciones farmacéuticas de la invención pueden usarse en métodos de administración de una composición farmacéutica que comprenden administrar por vía oral a un paciente por lo menos un comprimido que comprende:
a. una dispersión sólida de la reivindicación 1 que comprende aproximadamente 50 mg de Compuesto 1 sustancialmente amorfo o amorfo;
b. una carga
c. un disgregante;
d. un surfactante;
e. un aglutinante
f. un deslizante; y
g. un lubricante.
Las composiciones farmacéuticas de la invención pueden usarse en métodos de administración de una composición farmacéutica que comprenden administrar por vía oral a un paciente por lo menos un comprimido que comprende:
a. una dispersión sólida de la reivindicación 1 que comprende aproximadamente 75 mg de Compuesto 1 sustancialmente amorfo o amorfo;
b. una carga
c. un disgregante;
d. un surfactante;
e. un aglutinante
f. un deslizante; y
g. un lubricante.
Las composiciones farmacéuticas de la invención pueden usarse en métodos de administración de una composición farmacéutica que comprenden administrar por vía oral a un paciente por lo menos un comprimido que comprende:
a. una dispersión sólida de la reivindicación 1 que comprende aproximadamente 100 mg de Compuesto 1 sustancialmente amorfo o amorfo;
b. una carga
c. un disgregante;
d. un surfactante;
e. un aglutinante
f. un deslizante; y
g. un lubricante.
Las composiciones farmacéuticas de la invención pueden usarse en métodos de administración de una composición farmacéutica que comprenden administrar por vía oral a un paciente por lo menos un comprimido que comprende:
a. una dispersión sólida de la reivindicación 1 que comprende aproximadamente 150 mg de Compuesto 1 sustancialmente amorfo o amorfo;
b. una carga
c. un disgregante;
d. un surfactante;
e. un aglutinante
f. un deslizante; y
g. un lubricante.
Las composiciones farmacéuticas de la invención pueden usarse en métodos de administración de una composición farmacéutica que comprenden administrar por vía oral a un paciente por lo menos un comprimido que comprende:
a. una dispersión sólida de la reivindicación 1 que comprende aproximadamente 250 mg de Compuesto 1 sustancialmente amorfo o amorfo;
b. una carga
c. un disgregante;
d. un surfactante;
e. un aglutinante
f. un deslizante; y
g. un lubricante.
Las composiciones farmacéuticas de la invención pueden usarse en métodos de administración de una composición farmacéutica que comprenden administrar por vía oral a un paciente por lo menos un comprimido que comprende:
a. una dispersión sólida de la reivindicación 1 que comprende aproximadamente 25 mg de Compuesto 1 sustancialmente amorfo o amorfo y PVP/VA;
b. una carga
c. un disgregante;
d. un surfactante;
e. un aglutinante
f. un deslizante; y
g. un lubricante.
Las composiciones farmacéuticas de la invención pueden usarse en métodos de administración de una composición farmacéutica que comprenden administrar por vía oral a un paciente por lo menos un comprimido que comprende:
a. una dispersión sólida de la reivindicación 1 que comprende aproximadamente 50 mg de Compuesto 1 sustancialmente amorfo o amorfo y PVP/VA;
b. una carga
c. un disgregante;
d. un surfactante;
e. un aglutinante
f. un deslizante; y
g. un lubricante.
Las composiciones farmacéuticas de la invención pueden usarse en métodos de administración de una composición farmacéutica que comprenden administrar por vía oral a un paciente por lo menos un comprimido que comprende:
a. una dispersión sólida de la reivindicación 1 que comprende aproximadamente 75 mg de Compuesto 1 sustancialmente amorfo o amorfo y PVP/VA;
b. una carga
c. un disgregante;
d. un surfactante;
e. un aglutinante
f. un deslizante; y
g. un lubricante.
Las composiciones farmacéuticas de la invención pueden usarse en métodos de administración de una composición farmacéutica que comprenden administrar por vía oral a un paciente por lo menos un comprimido que comprende:
a. una dispersión sólida de la reivindicación 1 que comprende aproximadamente 100 mg de Compuesto 1 sustancialmente amorfo o amorfo y PVP/VA;
b. una carga
c. un disgregante;
d. un surfactante;
e. un aglutinante
f. un deslizante; y
g. un lubricante.
Las composiciones farmacéuticas de la invención pueden usarse en métodos de administración de una composición farmacéutica que comprenden administrar por vía oral a un paciente por lo menos un comprimido que comprende:
a. una dispersión sólida de la reivindicación 1 que comprende aproximadamente 150 mg de Compuesto 1 sustancialmente amorfo o amorfo y PVP/VA;
b. una carga
c. un disgregante;
d. un surfactante;
e. un aglutinante
f. un deslizante; y
g. un lubricante.
Las composiciones farmacéuticas de la invención pueden usarse en métodos de administración de una composición farmacéutica que comprenden administrar por vía oral a un paciente por lo menos un comprimido que comprende:
a. una dispersión sólida de la reivindicación 1 que comprende aproximadamente 250 mg de Compuesto 1 sustancialmente amorfo o amorfo y PVP/VA;
b. una carga
c. un disgregante;
d. un surfactante;
e. un aglutinante
f. un deslizante; y
g. un lubricante.
Las composiciones farmacéuticas de la invención pueden usarse en métodos de administración de una composición farmacéutica que comprenden administrar por vía oral a un paciente por lo menos un comprimido que comprende:
a. una dispersión sólida de la reivindicación 1 que comprende aproximadamente 25 mg de Compuesto 1 sustancialmente amorfo o amorfo y HPMCAS;
b. una carga
c. un disgregante;
d. un surfactante;
e. un aglutinante
f. un deslizante; y
g. un lubricante.
Las composiciones farmacéuticas de la invención pueden usarse en métodos de administración de una composición farmacéutica que comprenden administrar por vía oral a un paciente por lo menos un comprimido que comprende:
a. una dispersión sólida de la reivindicación 1 que comprende aproximadamente 50 mg de Compuesto 1 sustancialmente amorfo o amorfo y HPMCAS;
b. una carga
c. un disgregante;
d. un surfactante;
e. un aglutinante
f. un deslizante; y
g. un lubricante.
Las composiciones farmacéuticas de la invención pueden usarse en métodos de administración de una composición farmacéutica que comprenden administrar por vía oral a un paciente por lo menos un comprimido que comprende:
a. una dispersión sólida de la reivindicación 1 que comprende aproximadamente 75 mg de Compuesto 1 sustancialmente amorfo o amorfo y HPMCAS;
b. una carga
c. un disgregante;
d. un surfactante;
e. un aglutinante
f. un deslizante; y
g. un lubricante.
Las composiciones farmacéuticas de la invención pueden usarse en métodos de administración de una composición farmacéutica que comprenden administrar por vía oral a un paciente por lo menos un comprimido que comprende:
a. una dispersión sólida de la reivindicación 1 que comprende aproximadamente 100 mg de Compuesto 1 sustancialmente amorfo o amorfo y HPMCAS;
b. una carga
c. un disgregante;
d. un surfactante;
e. un aglutinante
f. un deslizante; y
g. un lubricante.
Las composiciones farmacéuticas de la invención pueden usarse en métodos de administración de una composición farmacéutica que comprenden administrar por vía oral a un paciente por lo menos un comprimido que comprende:
a. una dispersión sólida de la reivindicación 1 que comprende aproximadamente 150 mg de Compuesto 1 sustancialmente amorfo o amorfo y HPMCAS;
b. una carga
c. un disgregante;
d. un surfactante;
e. un aglutinante
f. un deslizante; y
g. un lubricante.
Las composiciones farmacéuticas de la invención pueden usarse en métodos de administración de una composición farmacéutica que comprenden administrar por vía oral a un paciente por lo menos un comprimido que comprende:
a. una dispersión sólida de la reivindicación 1 que comprende aproximadamente 250 mg de Compuesto 1 sustancialmente amorfo o amorfo y HPMCAS;
b. una carga
c. un disgregante;
d. un surfactante;
e. un aglutinante
f. un deslizante; y
g. un lubricante.
En algunas realizaciones, la presente invención proporciona un método para administrar por vía oral la composición farmacéutica descrita en este documento una vez al día. En otras realizaciones, la presente invención proporciona un método de administración por vía oral de la composición farmacéutica descrita en la presente dos veces al día.
Otro aspecto de la presente invención proporciona un método de administración de una composición farmacéutica mediante la administración por vía oral a un paciente por lo menos una vez al día de por lo menos un comprimido que comprende una dispersión sólida de Compuesto 1 sustancialmente amorfo o amorfo, una carga, un aglutinante, un deslizante, un disgregante, un surfactante y un lubricante, en el que la dispersión sólida comprende por lo menos aproximadamente 25 mg de Compuesto 1 sustancialmente amorfo o amorfo. En algunas realizaciones, el comprimido se administra por vía oral al paciente una vez al día. En otro método, la administración comprende administrar por vía oral a un paciente dos veces al día por lo menos un comprimido que comprende una dispersión sólida del Compuesto 1 sustancialmente amorfo o amorfo, una carga, un aglutinante, un deslizante, un disgregante, un surfactante y un lubricante, en el que la dispersión sólida contiene por lo menos aproximadamente 25 mg del Compuesto 1 sustancialmente amorfo o amorfo. Algunos comprimidos útiles en este método comprenden una dispersión sólida que contiene por lo menos aproximadamente 50 mg de Compuesto 1 sustancialmente amorfo o amorfo. En otro método, la administración comprende administrar por vía oral a un paciente dos veces al día por lo menos un comprimido que comprende una dispersión sólida del Compuesto 1 sustancialmente amorfo o amorfo, una carga, un aglutinante, un deslizante, un disgregante, un surfactante y un lubricante, en el que la dispersión sólida contiene por lo menos aproximadamente 50 mg del Compuesto 1 sustancialmente amorfo o amorfo. Algunos comprimidos útiles en este método comprenden una dispersión sólida que contiene por lo menos aproximadamente 75 mg de Compuesto 1 sustancialmente amorfo o amorfo. En otro método, la administración comprende administrar
por vía oral a un paciente dos veces al día por lo menos un comprimido que comprende una dispersión sólida del Compuesto 1 sustancialmente amorfo o amorfo, una carga, un aglutinante, un deslizante, un disgregante, un surfactante y un lubricante, en el que la dispersión sólida contiene por lo menos aproximadamente 75 mg del Compuesto 1 sustancialmente amorfo o amorfo. Otro aspecto de la presente invención proporciona un método de administración de una composición farmacéutica administrando por vía oral a un paciente por lo menos una vez al día por lo menos un comprimido que comprende una dispersión sólida del Compuesto 1 sustancialmente amorfo o amorfo, una carga, un aglutinante, un deslizante, un disgregante, un surfactante y un lubricante, en el que la dispersión sólida contiene por lo menos aproximadamente 100 mg del Compuesto 1 sustancialmente amorfo o amorfo. En algunas realizaciones, el comprimido se administra por vía oral al paciente una vez al día. En otro método, la administración comprende administrar por vía oral a un paciente dos veces al día por lo menos un comprimido que comprende una dispersión sólida de Compuesto 1 sustancialmente amorfo o amorfo, una carga, un aglutinante, un deslizante, un disgregante, un surfactante y un lubricante, en el que la dispersión sólida contiene por lo menos aproximadamente 100 mg de Compuesto 1 sustancialmente amorfo o amorfo. Otros comprimidos útiles en este método comprenden una dispersión sólida que contiene por lo menos aproximadamente 150 mg de Compuesto 1 sustancialmente amorfo o amorfo. En otro método, la administración incluye administrar por vía oral a un paciente dos veces al día por lo menos un comprimido que comprende una dispersión sólida de un compuesto sustancialmente amorfo o amorfo 1, una carga, un aglutinante, un deslizante, un disgregante, un surfactante y un lubricante, en el que la dispersión sólida contiene por lo menos aproximadamente 150 mg de Compuesto 1 sustancialmente amorfo o amorfo. En otro método, la administración incluye administrar por vía oral a un paciente por lo menos una vez al día por lo menos un comprimido que comprende una dispersión sólida de Compuesto 1 sustancialmente amorfo o amorfo, una carga, un aglutinante, un deslizante, un disgregante, un surfactante y un lubricante, en el que la dispersión sólida contiene por lo menos aproximadamente 250 mg de Compuesto 1 sustancialmente amorfo o amorfo. En otro método, la administración incluye administrar por vía oral a un paciente una vez al día por lo menos un comprimido que comprende una dispersión sólida de Compuesto 1 sustancialmente amorfo o amorfo, una carga, un aglutinante, un deslizante, un disgregante, un surfactante, y un lubricante, en el que la dispersión sólida contiene por lo menos aproximadamente 250 mg de Compuesto 1 sustancialmente amorfo o amorfo. En otro método, la administración incluye administrar por vía oral a un paciente dos veces al día por lo menos un comprimido que comprende una dispersión sólida de Compuesto 1 sustancialmente amorfo o amorfo, una carga, un aglutinante, un deslizante, un disgregante, un surfactante y un lubricante, en el que la dispersión sólida contiene por lo menos aproximadamente 250 mg de Compuesto 1 sustancialmente amorfo o amorfo. En otro método, la administración incluye administrar por vía oral a un paciente dos veces al día por lo menos un comprimido que comprende una dispersión sólida de Compuesto 1 sustancialmente amorfo o amorfo, una carga, un aglutinantes, un deslizante, un disgregante, un surfactante y un lubricante, en el que la dispersión sólida contiene por lo menos aproximadamente 250 mg de Compuesto 1 sustancialmente amorfo o amorfo.
En una realización, el método de administración de una composición farmacéutica incluye administrar por vía oral a un paciente por lo menos una vez al día por lo menos un comprimido que incluye una composición farmacéutica que contiene una dispersión sólida del Compuesto 1 amorfo, una carga, un aglutinante, un deslizante, un disgregante , un surfactante y un lubricante, cada uno de los cuales se describe con anterioridad y en los ejemplos siguientes, en donde la dispersión sólida comprende por lo menos 25 mg (por ejemplo, por lo menos 35 mg, por lo menos 40 mg o por lo menos 45 mg) de Compuesto amorfo 1.
En una realización, el método de administración de una composición farmacéutica incluye administrar por vía oral a un paciente por lo menos una vez al día por lo menos un comprimido que comprende una composición farmacéutica que contiene una dispersión sólida del Compuesto 1 sustancialmente amorfo, una carga, un aglutinante, un deslizante, un disgregante, un surfactante y un lubricante, en donde la dispersión sólida comprende de aproximadamente 30 mg a aproximadamente 300 mg (por ejemplo, de aproximadamente 40 mg a aproximadamente 280 mg o de aproximadamente 45 mg a aproximadamente 260 mg, o de aproximadamente 50 mg a aproximadamente 200 mg) de Compuesto 1 sustancialmente amorfo. O, el método de administración de una composición farmacéutica incluye administrar por vía oral a un paciente por lo menos una vez al día por lo menos un comprimido que comprende una composición farmacéutica que contiene una dispersión sólida de Compuesto 1 amorfo, una carga, un aglutinante, un deslizante, un disgregante, un surfactante y un lubricante, en donde la dispersión sólida comprende de aproximadamente 30 mg a aproximadamente 300 mg (por ejemplo, de aproximadamente 40 mg a aproximadamente 280 mg o de aproximadamente 45 mg a aproximadamente 260 mg, o de aproximadamente 50 mg a aproximadamente 200 mg ) del Compuesto amorfo 1.
En otra realización, el método de administración de una composición farmacéutica incluye administrar por vía oral a un paciente una vez al día por lo menos un comprimido que comprende una composición farmacéutica que contiene una dispersión sólida del Compuesto 1, una carga, un aglutinante, un deslizante, un disgregante, un surfactante, y un lubricante, cada uno de los cuales se describe anteriormente y en los Ejemplos siguientes, en donde la dispersión sólida comprende por lo menos 25 mg (por ejemplo, por lo menos 35 mg, por lo menos 40 mg, por lo menos 45 mg, por lo menos 75 mg, por lo menos aproximadamente 100 mg, por lo menos aproximadamente 150 mg, o por lo menos 250 mg) de Compuesto 1 sustancialmente amorfo o Compuesto 1 amorfo. Por ejemplo, el método de administración de una composición farmacéutica incluye administrar por vía oral a un paciente una vez al día un comprimido que comprende una composición farmacéutica que contiene una dispersión sólida del Compuesto
1, una carga, un aglutinante, un deslizante, un disgregante, un surfactante y un lubricante, en donde la dispersión sólida comprende por lo menos 75 mg (por ejemplo, por lo menos 100 mg, por lo menos 125 mg, por lo menos 140 mg, por lo menos 150 mg, o por lo menos 250 mg) de Compuesto 1 sustancialmente amorfo o Compuesto 1 amorfo. En otro ejemplo, el método de administración de una composición farmacéutica incluye administrar por vía oral a un paciente una vez al día una pluralidad de comprimidos (por ejemplo, dos comprimidos, tres comprimidos, cuatro o cinco comprimidos), en donde cada comprimido comprende una composición farmacéutica que comprende una dispersión sólida de Compuesto 1 sustancialmente amorfo o Compuesto 1 amorfo, una carga, un aglutinante, un deslizante, un disgregante, un surfactante y un lubricante, en donde la dispersión sólida comprende por lo menos 25 mg (por ejemplo, por lo menos 35 mg, por lo menos 40 mg, por lo menos 45 mg, por lo menos 75 mg, por lo menos aproximadamente 150 mg, o por lo menos 250 mg,) de Compuesto 1 sustancialmente amorfo o Compuesto 1 amorfo.
En otra realización, el método de administración de una composición farmacéutica incluye administrar por vía oral a un paciente dos veces al día por lo menos un comprimido que comprende una composición farmacéutica que contiene una dispersión sólida del Compuesto 1, una carga, un aglutinante, un deslizante, un disgregante, un surfactante, y un lubricante, cada uno de los cuales se describe anteriormente y en los Ejemplos siguientes, en donde la dispersión sólida comprende por lo menos 25 mg (por ejemplo, por lo menos 35 mg, por lo menos 40 mg, por lo menos 45 mg, por lo menos 50 mg, por lo menos por lo menos 75 mg, por lo menos aproximadamente 150 mg, o por lo menos 250 mg) de Compuesto 1 sustancialmente amorfo o Compuesto 1 amorfo. Por ejemplo, el método de administración de una composición farmacéutica incluye administrar por vía oral a un paciente dos veces al día un comprimido que comprende un composición farmacéutica que contiene una dispersión sólida del Compuesto 1, una carga, un aglutinante, un deslizante, un disgregante, un surfactante y un lubricante, en donde la dispersión sólida comprende por lo menos 75 mg (por ejemplo, por lo menos 100 mg, por lo menos 125 mg, por lo menos 140 mg, por lo menos 150 mg, o por lo menos 250 mg) de Compuesto 1 sustancialmente amorfo o Compuesto 1 amorfo. En otro ejemplo, el método de administración de una composición farmacéutica incluye administrar por vía oral a un paciente dos veces al día una pluralidad de comprimidos (por ejemplo, dos comprimidos, tres comprimidos, cuatro o cinco comprimidos), en donde cada comprimido comprende una composición farmacéutica que comprende una dispersión sólida de Compuesto 1 sustancialmente amorfo o Compuesto 1 amorfo, una carga, un aglutinante, un deslizante, un disgregante, un surfactante y un lubricante, en donde la dispersión sólida comprende por lo menos 25 mg (por ejemplo, por lo menos 35 mg, por lo menos 40 mg, por lo menos 45 mg, por lo menos 50 mg, por lo menos 75 mg, por lo menos aproximadamente 150 mg, o por lo menos 250 mg) de Compuesto 1 sustancialmente amorfo o Compuesto 1 amorfo.
Se observa que los métodos de administración de la presente invención pueden incluir opcionalmente la administración oral de una bebida (agua, leche, o similares), la alimentación, y/o composiciones farmacéuticas adicionales que incluyen APIs adicionales. Cuando el método de administración incluye la administración por vía oral de una bebida (agua, leche, o similares), alimentos (incluyendo una comida o un aperitivo FQ estándar alto en grasa y alta en calorías), y/o composiciones farmacéuticas adicionales incluyendo APIs adicionales, la administración oral de la bebida, comida, y/o API adicional puede ocurrir simultáneamente con la administración oral del comprimido, antes de la administración oral del comprimido, y/o después de la administración del comprimido. Por ejemplo, en un ejemplo, el método de administración de una composición farmacéutica que incluye la administración oral a un paciente al menos una vez por día de al menos un comprimido que comprende una composición farmacéutica que contiene una dispersión sólida de Compuesto 1 sustancialmente amorfo 1 o Compuesto 1 amorfo, una carga, un aglutinante, un agente deslizante, un disgregante, un agente surfactante, un lubricante, y un segundo API. En otro ejemplo, el método de administración de una composición farmacéutica que incluye la administración oral a un paciente al menos una vez por día de al menos un comprimido que comprende una composición farmacéutica que comprende una dispersión sólida de Compuesto 1 amorfo o Compuesto 1 sustancialmente amorfo, una carga, un aglutinante, un deslizante, un disgregante, un surfactante, y un lubricante, en el que la dispersión sólida comprende al menos 25 mg (por ejemplo, al menos 35 mg, al menos 45 mg, o al menos 50 mg) de Compuesto 1 amorfo o Compuesto 1 sustancialmente amorfo, y la administración oral a un paciente al menos una vez por día de una segunda composición farmacéutica que comprende un segundo API. En todavía otros ejemplos, el método de administración de una composición farmacéutica que incluye la administración oral a un paciente cada 12 horas de al menos un comprimido que comprende una composición farmacéutica como se describe en el presente documento, en los que se administra el comprimido aproximadamente 30 minutos después de consumir una comida o aperitivo de FQ alto en grasas, alto en calorías.
También se apreciará que el compuesto y las composiciones farmacéuticamente aceptables de la presente invención se pueden emplear en terapias de combinación, es decir, el compuesto y las composiciones de farmacéuticamente aceptables pueden administrarse simultáneamente con, antes de, o posteriormente a, uno o más otros agentes terapéuticos o procedimientos médicos deseados. La combinación particular de terapias (agentes terapéuticos o procedimientos) a emplear en un régimen de combinación tendrá en cuenta la compatibilidad de los agentes terapéuticos deseados y/o procedimientos y el efecto terapéutico que se desea alcanzar. También se apreciará que las terapias empleadas pueden lograr un efecto deseado para el mismo trastorno (por ejemplo, un compuesto de la invención puede administrarse simultáneamente con otro agente usado para tratar el mismo trastorno), o pueden conseguir efectos diferentes (por ejemplo, control de cualquier efecto adverso). Tal como se
usa en el presente documento, los agentes terapéuticos adicionales que se administran normalmente para tratar o prevenir una enfermedad particular, o condición, son conocidos como "apropiados para la enfermedad, o afección, que se está tratando".
En una realización, el agente adicional se selecciona de un agente mucolítico, broncodilatador, un anti biótico, un agente anti-infeccioso, un agente anti-inflamatorio, un modulador de CFTR otro que el Compuesto 1 de la presente invención, o un agente nutricional.
En una realización, el agente adicional es un antibiótico. Antibióticos ejemplares útiles en este documento incluyen tobramicina, incluyendo tobramicina inhalada en polvo (TIP), azitromicina, aztreonam, incluyendo la forma de aerosol de aztreonam, amicacina, incluyendo formulaciones liposomales de los mismos, ciprofloxacina, incluyendo formulaciones de los mismos adecuadas para la administración por inhalación, levoflaxacina, incluyendo formulaciones de aerosol de los mismos, y combinaciones de dos antibióticos, por ejemplo, fosfomicina y tobramicina.
En otra realización, el agente adicional es un mucolítico. Ejemplos de mucolíticos útiles en esta invención incluyen Pulmozyme®.
En otra realización, el agente adicional es un broncodilatador. Broncodilatadores ejemplares incluyen albuterol, sulfato de metaprotenerol, acetato de pirbuterol, salmeterol, o sulfato de tetrabulina.
En otra realización, el agente adicional es eficaz en la restauración de líquido de la superficie de la vía aérea pulmonar. Tales agentes mejoran el movimiento de la sal en y fuera de las células, permitiendo que el moco en las vías respiratorias del pulmón se hidraten en mayor medida y, por lo tanto, se aclaren con más facilidad. Los ejemplos de tales agentes incluyen la solución salina hipertónica, tetrasodio de denufosol ([[(3S, 5R)-5-(4-amino-2-oxopirimidina-1 -ilo)-3-hidroxioxolano-2-ilo]metoxi-hidroxifosforilo] [[[(2R,3S,4R,5R)-5-(2,4-dioxopirimidina-1 -ilo)-3, 4-dihidroxioxolano-2-ilo]metoxi-hidroxifosforilo]oxi-hidroxifosforilo] fosfato de hidrógeno), o bronchitol (formulación inhalada de manitol).
En otra realización, el agente adicional es un agente anti-inflamatorio, es decir, un agente que puede reducir la inflamación en los pulmones. Los ejemplos de tales agentes útiles en esta invención incluyen ibuprofeno, ácido docosahexanoico (DHA), el sildenafilo, el glutatión inhalado, pioglitazona, hidroxicloroquina, o simavastatina.
En otra realización, el agente adicional es un modulador CFTR distinto de compuesto 1, es decir, un agente que tiene el efecto de modular la actividad de CFTR. Los ejemplos de tales agentes incluyen ataluren ("PTC124®"; ácido 3-[5-(2-fluorofenilo)-1,2,4-oxadiazol-3-ilo]benzoico), sinapultida, lancovutida, depelestat (un inhibidor recombinante de elastasa neutrófila humana), cobiprostona (7-{(2R,4aR,5R,7aR)-2-[(3S)-1,1-difluoro-3-metilpentilo]-2-hidroxi-6-oxooctahidrociclopenta[b]pirano-5-ilo}heptanoico), o (ácido 3-(6-(1 -(2,2-difluorobenzo[d][1,3]dioxol-5-ilo)ciclopropanocarboxamido)-3-metilo-piridina-2-ilo)benzoico. En otra realización, el agente adicional es de (3-(6-(1-(2,2-difluorobenzo[d][1,3]dioxol-5-ilo)ciclopropanocarboxamido)-3-metilpiridina-2-ilo)benzoico.
En otra realización, el agente adicional es un agente nutricional. Los ejemplos de tales agentes incluyen pancrelipasa (reemplazo de enzima pancreática), incluyendo Pancrease®, Pancreacarb®, Ultrase®, o Creon®, Liprotomase® (anteriormente Trizytek®), Aquadeks®, o inhalación de glutatión. En una realización, el agente nutricional adicional es pancrelipasa.
VI. EJEMPLOS
A fin de que la invención descrita aquí pueda entenderse más completamente, se exponen los siguientes ejemplos. Debe entenderse que estos ejemplos son sólo para fines ilustrativos y no se deben interpretar como limitantes de esta invención de ninguna manera.
A. Fabricación de comprimidos
Intermedio A (Composición de referencia)
Un sistema de disolvente de cetona de metiletilo (MEK) y agua DI, formulado de acuerdo con la relación de 90% en peso de MEK/10% en peso de agua DI, se añadió a un reactor equipado con un agitador magnético y un circuito térmico. En este sistema disolvente, polímero de succinato de acetato de hipromelosa (grado HG, disponible comercialmente de Biddle Sawyer Corporation en Nueva York, Nueva York o Shina-Etsu Chemical Co. en Tokio, Japón), sulfato de laurilo sódico (SLS), y N-[2,4-Bis(1,1-dimetilo-etilo)-5-hidroxifenilo]-1,4-dihidro-4-oxoquinolina-3-carboxamida se añadieron de acuerdo con la relación de 49,5% en peso de succinato de acetato de hipromelosa / 0,5% en peso de sulfato de laurilo sódico (SLS) / 50% en peso de N-[2,4-Bis(1,1-dimetiletilo)-5-hidroxifenilo]-1,4-dihidro-4-oxoquinolina-3-carboxamida. La mezcla resultante contenía 20% en peso de sólidos disueltos. Las cantidades reales de los ingredientes y las cantidades de los disolventes utilizados para generar esta mezcla se
recitan en la Tabla A1, a continuación:
Tabla A1: Ingredientes de dispersión de aerosol sólidos para Intermedio A

___ ___
La mezcla se mezcló a temperatura ambiente hasta estar sustancialmente homogénea y todos los componentes se disolvieron sustancialmente.
Un secador de pulverización, Niro Mobile Minor Spray Dryer con cámara extendida, equipado con un atomizador de dos fluidos de 1,3 mm situado aproximadamente 5 cm de la parte superior del recipiente de secado por pulverización se aplicó de acuerdo con los parámetros secos de pulverización de la Tabla A2.
Tabla A2: Parámetros del proceso de pulverización en seco usados para generar Intermedio A.

Un ciclón inercial separa el producto del gas de proceso y vapores de disolvente, y una bolsa de filtro recoge las partículas finas no separadas por el ciclón. El producto resultante se transfirió a un secador de bandejas al vacío para el secado para reducir disolventes residuales a un nivel de menos de aproximadamente 5.000 ppm y para generar Intermedio A seco.
Intermedio B
Un sistema de disolvente de MEK, agua DI, y acetona, formulado de acuerdo con la relación de 65% en peso de MEK / 9% en peso de agua DI / 26% en peso de acetona, se calentó a una temperatura de 20 - 30°C en un reactor de equipado con un agitador magnético y un circuito térmico. En este sistema disolvente, un copolímero de vinilpirrolidona y vinilacetatopolivinilpirrolidona (PVP VA-64 comercialmente disponible de Shanghai Lite Chemical Technology Co., Ltd. Shanghai, China), SLS, y N-[2,4-Bis(1,1-dimetiletilo)-5-hidroxifenilo]-1,4-dihidro-4-oxoquinolina-3-carboxamida se añadieron de acuerdo con la relación de 19,5% en peso PVPVA-64 / 0,5% en peso de sulfato de laurilo sódico / 80% en peso de N-[2,4-Bis(1,1-dimetiletilo)-5-hidroxifenilo]-1,4-dihidro-4-oxoquinolina-3-carboxamida. La mezcla resultante contenía 11,5% en peso de sólidos. Las cantidades reales de ingredientes y disolventes usados para generar esta mezcla se recitan en la Tabla B1, a continuación:
Tabla B1: Ingredientes de dispersión de pulverización sólidos para el Intermedio B

_______
La mezcla se mantuvo a una temperatura de 20 - 30°C y se mezcló hasta estar sustancialmente homogénea y todos los componentes se disolvieron sustancialmente.
Un secador de pulverización, Niro Production Minor Spray Dryer, equipado con boquillas de presión (serie Spray Systems Maximum Passage SK-MFP tiene un tamaño de orificio de 72), se usó en el modo de secado por pulverización normal, siguiendo los parámetros del proceso de pulverización en seco recitados en Tabla B2, a continuación. La boquilla de pulverización se encuentra a unos 5 cm de la parte superior del recipiente de secado por pulverización.
Tabla B2: Parámetros del proceso de pulverización en seco usados para generar el Intermedio B.

Un ciclón de alta eficiencia separó el producto húmedo del gas de pulverización y vapores de disolvente. El producto húmedo se transfiere a un secador de vacío de bandeja para el secado para reducir disolventes residuales a un nivel de menos de aproximadamente 5.000 ppm y para generar Intermedio B seco.
Intermedio C:
Un sistema de disolvente de MEK y agua DI, formulado de acuerdo con la relación de 90% en peso de MEK/10% en peso de agua DI, se calentó a una temperatura de 20 - 30°C en un reactor, equipado con un agitador magnético y circuito térmico. En este sistema disolvente, polímero de succinato de acetato de hipromelosa (HPMCAS) (grado HG), SLS, y N-[2,4-Bis(1,1-dimetiletilo)-5-hidroxifenilo]-1,4-dihidro-4-oxoquinolina-3-carboxamida se añadieron de acuerdo con la relación de 19,5% en peso de succinato de acetato de hipromelosa /0,5% en peso de SLS / 80% en peso de N-[2,4-Bis(1,1-dimetiletilo)-5-hidroxifenilo]-1,4-dihidro-4-oxoquinolina-3-carboxamida. La mezcla resultante contenía 12,5% en peso de sólidos. Las cantidades reales de ingredientes y disolventes usados para generar esta mezcla se recitan en la Tabla C1, a continuación:
Tabla C1: Ingredientes de dispersión de pulverización sólidos para el intermedio C.

_______
La mezcla se mantuvo a una temperatura de 20 - 30°C y se mezcló hasta estar sustancialmente homogénea y todos los componentes se disolvieron sustancialmente.
Un secador de pulverización, Niro Producción Minor Spray Dryer, equipado con boquillas de presión (serie Spray Systems Maximum Passage SK-MFP que tienen un tamaño de orificio de 72), se usó en el modo de secado por pulverización normal, siguiendo los parámetros del proceso de pulverización en seco recitados en la Tabla C2, a continuación. La boquilla de pulverización se encuentra a unos 5 cm de la parte superior del recipiente de secado por pulverización.
Tabla C2: Parámetros del proceso de pulverización en seco usados para generar Intermedio C.

Un ciclón de alta eficiencia separó el producto húmedo a partir del gas de pulverización y vapores de disolvente. El producto húmedo se transfiere a un secador de vacío de bandeja para el secado para reducir disolventes residuales a un nivel de menos de aproximadamente 5.000 ppm y para generar Intermedio C seco. Intermedio D:
Un sistema de disolvente de MEK y agua DI, formulado de acuerdo con la relación de 90% en peso de MEK / 10% en peso de agua DI, se calentó a una temperatura de 20 - 30°C en un reactor, equipado con un agitador magnético y circuito térmico. En este sistema disolvente, polímero de succinato de acetato de hipromelosa (HPMCAS) (grado HG), SLS, y N-[2,4-Bis(1,1-dimetiletilo)-5-hidroxifenilo]-1,4-dihidro-4-oxoquinolina-3-carboxamida se añadieron de acuerdo con la relación de 19,5% en peso de succinato de acetato de hipromelosa /0,5% en peso de SLS/80% en peso de N-[2,4-Bis(1,1-dimetiletilo)-5-hidroxifenilo]-1,4-dihidro-4-oxoquinolina-3-carboxamida. La mezcla resultante contenía 12,5% en peso de sólidos. Las cantidades reales de ingredientes y disolventes usados para generar esta mezcla se recitan en la Tabla D1, a continuación:
Tabla D1: Ingredientes de dispersión de pulverización sólidos para el intermedio D.

____ ____
La mezcla se mantuvo a una temperatura de 20 - 30°C y se mezcló hasta estar sustancialmente homogénea y se disolvieron sustancialmente todos los componentes.
Un secador de pulverización, Niro Mobil Minor Spray Dryer equipado con una boquilla de dos fluidos de 1,0 mm, fue utilizado en el modo de secado por pulverización normal, siguiendo los parámetros del proceso de pulverización en seco recitados en la Tabla D2, a continuación.
Tabla D2: Parámetros del proceso de pulverización en seco usados para generar Intermedio D.

Un ciclón de alta eficiencia separó el producto húmedo del gas de pulverización y vapores de disolvente. El producto húmedo contenida 6,3% MEK y 0,7% de agua y tenía un tamaño medio de partícula de 7um y una densidad aparente de 0,23 g/cc. El producto húmedo se transfiere a un secador de vacío de bandeja para el secado para reducir disolventes residuales a un nivel de menos de aproximadamente 5.000 ppm y para generar intermedio D seco. El Intermedio D seco contenía <0,5% MEK y 0,3% de agua.
Intermedio E:
Un sistema de disolvente de MEK y agua DI, formulado de acuerdo con la relación de 90% en peso de MEK / 10% en peso de agua DI, se calentó a una temperatura de 20 - 30°C en un reactor, equipado con un agitador magnético y circuito térmico. En este sistema disolvente, polímero de succinato de acetato de hipromelosa (HPMCAS) (grado HG), SLS, y N-[2,4-Bis(1,1-dimetiletilo)-5-hidroxifenilo]-1,4-dihidro-4-oxoquinolina-3-carboxamida se añadieron de acuerdo con la relación de 19,5% en peso de succinato de acetato de hipromelosa /0,5% en peso de SLS / 80% en peso de N-[2,4-Bis(1,1-dimetiletilo)-5-hidroxifenilo]-1,4-dihidro-4-oxoquinolina-3-carboxamida. La mezcla resultante contenía 10,5% en peso de sólidos. Las cantidades reales de ingredientes y disolventes usados para generar esta mezcla se recitan en la Tabla E1, a continuación:
Tabla E1: Ingredientes de dispersión de pulverización sólidos para el Intermedio E.

_______
La temperatura de la mezcla se ajustó a un intervalo de 30 - 45°C y se mezcló hasta estar sustancialmente homogénea y todos los componentes se disolvieron sustancialmente.
Un secador de pulverización, Niro PSD4 Comercial Spray Dryer, equipado con toberas de presión (serie Spray Systems Maximum Passage SK-MFP teniendo un tamaño de orificio/núcleo 54/21, 53/21 o 52/21) equipado con tapa contra la formación de rebabas, fue utilizado en el modo de secado por pulverización normal, siguiendo los parámetros del proceso de pulverización en seco recitados en la Tabla E2, a continuación.
Tabla E2: Parámetros del proceso de pulverización en seco usados para generar Intermedio E.

Un ciclón de alta eficiencia separó el producto húmedo a partir del gas de pulverización y vapores de disolvente. El producto húmedo contenía 8,8 a 12.5% en peso. MEK/Agua un tamaño medio de partícula de 16 -24um y una densidad aparente de 0,28 - 0,36 g/cc. El producto húmedo se transfiere a un secador de vacío de doble cono de acero inoxidable de 350L para el secado para reducir disolventes residuales a un nivel de menos de aproximadamente 5.000 ppm y para generar Intermedio E seco. El Intermedio E seco contenía <0,3% MEK y 0,8% de agua.
Intermedio F:
Un sistema de disolvente de MEK y agua DI, formulado de acuerdo con la relación de 90% en peso de MEK/10% en peso de agua DI, se calentó a una temperatura de 20 - 30°C en un reactor, equipado con un agitador magnético y circuito térmico. En este sistema disolvente, el polímero de succinato de acetato de hipromelosa (HPMCAS) (grado HG), SLS, y N-[2,4-Bis(1,1-dimetiletilo)-5-hidroxifenilo]-1,4-dihidro-4-oxoquinolina-3-carboxamida se añadieron de acuerdo con la relación de 19,5% en peso de succinato de acetato de hipromelosa /0,5% en peso de SLS/80% en peso de N-[2,4-Bis(1,1-dimetiletilo)-5-hidroxifenilo]-1,4-dihidro-4-oxoquinolina-3-carboxamida. La mezcla resultante contenía 10,5% en peso de sólidos. Las cantidades reales de ingredientes y disolventes usados para generar esta mezcla se recitan en la Tabla F1, a continuación:
Tabla F1: Ingredientes de dispersión de pulverización sólidos para el intermedio F.

La temperatura de la mezcla se ajustó a un intervalo de 20 - 45°C y se mezcló hasta estar sustancialmente homogénea y todos los componentes se disolvieron sustancialmente.
Un secador de pulverización, Niro PSD4 Commercial Spray Dryer, equipado con boquilla de presión (serie Spray Systems Maximum Passage SK-MFP que tiene un tamaño de orificio/núcleo 54/21) equipado con una tapa contra la formación de rebabas, fue utilizado en el modo de secado por pulverización normal, siguiendo los parámetros del proceso de pulverización en seco recitados en la Tabla F2, a continuación.
Tabla F2: parámetros del proceso de pulverización en seco usados para generar Intermedio F.

Un ciclón de alta eficiencia separó el producto húmedo del gas de pulverización y vapores de disolvente. El producto húmedo contenía 8,5 a 9,7% MEK y 0,56 - 0,83% de agua y tenía un tamaño medio de partícula de 17 -19um y una densidad aparente de 0,27 - 0,33 g/cc. El producto húmedo se transfiere a un secador de vacío de doble cono de acero inoxidable 4.000L para el secado para reducir disolventes residuales a un nivel de menos de aproximadamente 5.000 ppm y para generar intermedio F seco. El intermedio F seco contenía <0,03% MEK y 0,3% de agua.
Intermedio G:
Un sistema de disolvente de MEK y agua DI, formulado de acuerdo con la relación de 90% en peso de MEK/10% en peso de agua DI, se calentó a una temperatura de 20 - 30°C en un reactor, equipado con un agitador magnético y circuito térmico. En este sistema disolvente, polímero de succinato de acetato de hipromelosa (HPMCAS) (grado HG), SLS, y N-[2,4-Bis(1,1-dimetiletilo)-5-hidroxifenilo]-1,4-dihidro-4-oxoquinolina-3-carboxamida se añadieron de acuerdo con la relación de 19,5% en peso de succinato de acetato de hipromelosa /0,5% en peso de SLS/80% en peso de N-[2,4-Bis(1,1-dimetiletilo)-5-hidroxifenilo]-1,4-dihidro-4-oxoquinolina-3-carboxamida. La mezcla resultante contenía 10,5% en peso de sólidos. Las cantidades reales de ingredientes y disolventes usados para generar esta mezcla se recitan en la Tabla G1, a continuación:
Tabla G1: Ingredientes de dispersión de pulverización sólidos para el Intermedio G.

_ ______
La temperatura de la mezcla se ajustó a un intervalo de 20 - 45°C y se mezcló hasta estar sustancialmente homogénea y todos los componentes se disolvieron sustancialmente.
Un secador de pulverización, Niro Production Minor Spray Dryer, equipado con boquilla de presión (serie Spray Systems Maximum Passage SK-MFP tiene un tamaño de orificio de 72) fue utilizado en el modo de secado por pulverización normal, siguiendo los parámetros del proceso de pulverización en seco recitados en la Tabla G2, abajo.
G2 Tabla: Parámetros del proceso de pulverización en seco usados para generar Intermedio G.

Un ciclón de alta eficiencia separó el producto húmedo del gas de pulverización y vapores de disolvente. El producto húmedo contenía 10,8% de MEK y 0,7% de agua y tenía un tamaño medio de partícula de 19um y una densidad aparente de 0,32 g/cc. El producto húmedo se transfiere a un secador de vacío de doble cono de acero inoxidable 4.000L para el secado para reducir disolventes residuales a un nivel de menos de aproximadamente 5.000 ppm y para generar Intermedio seco. El Intermedio G seco contenía <0,05% MEK y 0,7% de agua.
Ejemplo 1: Tableta ejemplar 1 (formulada para tener 25 mg de Compuesto 1)
Un lote de núcleo redondo de comprimidos de 3/8" se formula para que tenga aproximadamente 25 mg de Compuesto 1 por comprimido utilizando las cantidades de los ingredientes enumerados en la Tabla 1, a continuación.
Tabla 1: Ingredientes para Tableta Ejemplar 1.

Intermedio A, celulosa microcristalina (FMC MCC Avicel® PH102, disponible comercialmente de FMC BioPolimer Corporation de Filadelfia, PA), lactosa (Foremost FastFlo® Lactosa n° 316 disponible comercialmente de Foremost Farms USA de Baraboo, WI), croscarmelosa sódica (FMC Ac-Di-Sol®, disponible comercialmente de FMC BioPolymer Corporation de Filadelfia, PA), SLS, y dióxido de silicio coloidal (Cabot Cab-O-Sil® M-5P Fumed Silicon Dioxide, disponible comercialmente de Cabot Corporation de Alpharetta, GA) se tamizaron a través de un tamiz de malla 20 para eliminar grumos.
Cada uno de los ingredientes tamizados se añadió a un mezclador en V cuarto 16 en el siguiente orden: 1) lactosa;
2) SLS;
3) croscarmelosa sódica;
4) dióxido de silicio coloidal;
5) Intermedio A; y
6) celulosa microcristalina de PH101
La mezcla se mezcló durante 25 minutos en un mezclador en V a 20-24 rpm. El estearato de magnesio se tamizó a través de un tamiz de malla 30 para eliminar grumos, y se añadió a la mezcla, que se mezcló durante otros
3 minutos.
Una vez que se ha completado la mezcla final, la mezcla se transfirió a Piccola B-Tooling, prensa de comprimidos rotativa de estación 10 (medio equipada) para la compresión. Prensar la mezcla en comprimidos generó comprimidos redondos de 3/8" que tiene aproximadamente 25 mg de N-[2,4-Bis(1,1-dimetiletilo)-5-hidroxifenilo]-1,4-dihidro-4-oxoquinolina-3-carboxamida.
Ejemplo 2: Tableta Ejemplar 2 (Formulada para tener 50 mg de Compuesto 1)
Un lote de núcleo de comprimidos redondos de 3/8" se formula para tener aproximadamente 50 mg de Compuesto 1 por comprimido utilizando las cantidades de los ingredientes enumerados en la Tabla 2, a continuación.
Tabla 2: Ingredientes para Ejemplar Tablet 2.

Intermedio A, celulosa microcristalina, lactosa, croscarmelosa sódica, SLS, y dióxido de silicio coloidal se tamizaron a través de un tamiz de malla 20 para eliminar grumos, y cada uno de los ingredientes tamizados se añadió a un mezclador en V de cuarto 16 en el siguiente orden:
1) lactosa;
2) SLS;
3) croscarmelosa sódica;
4) dióxido de silicio coloidal;
5) Intermedio A; y
6) celulosa microcristalina PH101
La mezcla se mezcló durante 25 minutos en un mezclador en V a 20-24 rpm. El estearato de magnesio se tamizó a través de un tamiz de malla 30 para eliminar grumos, y se añadió a la mezcla, que se mezcló durante otros 3 minutos.
Una vez que se ha completado la mezcla final, la mezcla se transfirió a Piccola B-Tooling, prensa de comprimidos rotativa de estación 10 (medio equipado) para la compresión. Prensar la mezcla en comprimidos generó comprimidos redondos de 3/8” que tiene aproximadamente 50 mg de N-[2,4-Bis(1,1-dimetiletilo)-5-hidroxifeni lo]-1,4-dihidro-4-oxoquinolina-3-carboxamida.
Ejemplo 3: Tableta Ejemplar 3 (Formulada con Polímero PVP/VA para tener 150 mg del Compuesto 1)
Un lote de comprimidos con forma de comprimido oblongo se formuló para tener aproximadamente 150 mg de Compuesto 1 por comprimido utilizando las cantidades de los ingredientes enumerados en la Tabla 3, a continuación.
Tabla 3: Ingredientes para Tableta Ejemplar 3.

Una mezcla deslizante de dióxido de silicio coloidal (Dióxido de silicio pirógeno Cabot Cab-O-Sil® M-5P) y SLS fue producido por la mezcla de estos dos ingredientes, en las cantidades dadas en la Tabla 3, y filtrando la mezcla resultante por un tamiz de malla de 70. Una mezcla de color de colorante (Colorcon azul n° 1, Aluminum Lake n° 5516) y croscarmelosa sódica (FMC Ac-Di-Sol®) fue producida por la mezcla a mano de estos dos ingredientes, en las cantidades dadas en la Tabla 3, y filtrando la mezcla resultante a través de un tamiz de malla 70. La mezcla de agente de deslizamiento y la mezcla de color se mezclaron a mano y se añadieron a un recipiente de mezcla de 2 L. Se añadió el intermedio B a esta mezcla en el recipiente de mezcla de 2 L, y los contenidos de contenedor de mezcla de 2 L se mezclaron a mano y se filtraron a través de un tamiz de malla 30. La mezcla resultante se mezcló en un mezclador Turbula durante 20 minutos a una velocidad de 22 rpm.
La celulosa microcristalina (FMC MCC Avicel® PH102) y lactosa (Foremost FastFlo® Lactose n° 316) fueron se filtró a través de un tamiz de malla 30 y se añadió al recipiente de mezcla. La mezcla resultante se mezcló en un mezclador Turbula durante 20 minutos a una velocidad de 22 rpm.
El estearato de magnesio se filtró a través de un tamiz de malla 70 y se añadió a la mezcla en el recipiente de mezcla, y la mezcla resultante se mezcló durante 5 minutos a una velocidad de 22 rpm.
La mezcla resultante se comprimió en comprimidos usando una bota alimentada por gravedad equipada con utillaje de cápsula de tipo B 0,64" x 0,32" configurada para producir un comprimido que tiene una dureza inicial de aproximadamente 8 Kp 15%.
Ejemplo 4: Tableta Ejemplar 4 (Formulada con polímero HPMCAS para tener 150 mg del Compuesto 1)
Un lote de comprimidos con forma de comprimido oblongo se formula para tener aproximadamente 150 mg de Compuesto 1 por comprimido utilizando las cantidades de los ingredientes enumerados en la Tabla 4, a continuación.
Tabla 4: Ingredientes para Tableta Ejemplar 4.

Una mezcla deslizante de dióxido de silicio coloidal (Dióxido de silicio pirógeno Cabot Cab-O-Sil® M-5P) y SLS fue producida por la mezcla de estos dos ingredientes, en las cantidades dadas en la Tabla 4, y filtrando la mezcla resultante a mano por un tamiz de malla 70. Una mezcla de color incluyendo colorante (Colorcon azul n° 1, Aluminum Lake n° 5516) y croscarmelosa sódica (FMC Ac-Di-Sol®) fue producida por la mezcla de estos dos
ingredientes, en las cantidades dadas en la Tabla 4, y el filtrado de la mezcla resultante a través de un tamiz de malla 70. La mezcla de agente de deslizamiento y la mezcla de color se mezclaron a mano y se añadieron a un recipiente de mezcla 2 L. El Intermedio C se añadió a esta mezcla en el recipiente de mezcla de 2 L, y los contenidos de contenedores de mezcla de 2 L se mezclaron y se filtraron mano a través de un tamiz de malla 30. La mezcla resultante se mezcló en un mezclador Turbula durante 20 minutos a una velocidad de 22 rpm.
La celulosa microcristalina (FMC MCC Avicel® PH102) y lactosa (Foremost FastFlo® Lactose n° 316) se filtró a través de un tamiz de malla 30 y se añade al recipiente de mezcla. La mezcla resultante se mezcló en un mezclador Turbula durante 20 minutos a una velocidad de 22 rpm.
El estearato de magnesio se filtró a través de un tamiz de malla 70 y se añade a la mezcla en el recipiente de mezcla, y la mezcla resultante se mezcló durante 5 minutos a una velocidad de 22 rpm.
La mezcla resultante se comprimió en comprimidos usando una prensa de comprimidos equipado con utillaje de cápsula de tipo B 0,64" x 0,32" configurada para producir un comprimido que tiene una dureza inicial de aproximadamente 9,5 Kp 15%.
Ejemplo 5: Tableta Ejemplar 5 (Formulada con polímero HPMCAS para tener 150 mg del Compuesto 1)
Un lote de comprimidos con forma de comprimido oblongo se formula para tener aproximadamente 150 mg de Compuesto 1 por comprimido utilizando las cantidades de los ingredientes enumerados en la Tabla 5, a continuación.
Tabla 5: Ingredientes para Tableta Ejemplar 5.

Una mezcla de dióxido de silicio coloidal (Dióxido de silicio pirógeno Cabot Cab-O-Sil® M-5P), SLS, croscarmelosa sódica (FMC Ac-Di-Sol®), y aproximadamente 10% de la lactosa (Foremost FastFlo® lactosa n° 316) dada en la Tabla 5 se produjo mezclando estos ingredientes en un mezclador en V para proporcionar aproximadamente 125 inversiones. Esta mezcla, premezcla 1, se molió por cono a través de un tamiz de malla 40, se recogió y se almacenó para uso posterior.
Aproximadamente el 20% de la lactosa (Foremost FastFlo® Lactose n° 316) dado en la Tabla 5 se molió por cono a través de un tamiz de malla 30, se recogió y almacenó para su uso posterior como una premezcla 2. El Intermedio G se filtró a través de una malla 30, se recogió y se almacenó para su uso posterior como premezcla 3. La celulosa microcristalina (FMC MCC Avicel® PH102) se filtró a través de un tamiz de malla 30, se recogió y se almacenó para su uso posterior como premezcla 4.
Un mezclador en V, se cargó con la premezcla 2, el 70% restante de la lactosa (Foremost FastFlo® Lactose n° 316) dado en la Tabla 3, Premezcla 3, Premezcla 1 y Premezcla 4, en ese orden, y se mezcló durante aproximadamente 500 inversiones. La mezcla mezclada se ensayó para determinar la uniformidad.
El estearato de magnesio se filtró a través de un tamiz de malla 70 y se añadió a la mezcla en el recipiente de mezcla, y la mezcla resultante se mezcló para proporcionar aproximadamente 125 inversiones.
La mezcla resultante se comprimió en comprimidos usando una prensa Killian T100 equipado con utillaje de cápsula de tipo B 0,64" x 0,32" configurado para producir un comprimido que tiene una dureza inicial de aproximadamente 11 Kp 20%.
Ejemplo 6: Tableta Ejemplar 6 (Formulada con polímero de HPMCAS para tener 100 mg del Compuesto 1)
Un lote de comprimidos con forma de comprimido oblongo se formula para tener aproximadamente 150 mg de Compuesto 1 por comprimido utilizando las cantidades de los ingredientes enumerados en la Tabla 6, a continuación.
Tabla 6: Ingredientes para Tableta Ejemplar 6.

Una mezcla de dióxido de silicio coloidal (Dióxido de silicio pirógeno Cabot Cab-O-Sil® M-5P), SLS, croscarmelosa sódica (FMC Ac-Di-Sol®), y aproximadamente 10% de la lactosa (Foremost FastFlo® Lactose n° 316) dada en la Tabla 6 fue producida por la mezcla de estos ingredientes en un mezclador en V para proporcionar aproximadamente 125 inversiones. Esta mezcla, premezcla 1, se molió por cono a través de un tamiz de malla 40, se recogió y se almacenó para uso posterior.
Aproximadamente el 20% de la lactosa (Foremost FastFlo® Lactose n° 316) dado en la Tabla 6 se molió por cono a través de un tamiz de malla 30, se recogió y se almacenó para su uso posterior como premezcla 2. El Intermedio G se filtró a través de una malla 30, se recogió y se almacenó para su uso posterior como premezcla 3. La celulosa microcristalina (FMC MCC Avicel® PH102) se filtró a través de un tamiz de malla 30, se recogió y se almacenó para su uso posterior como premezcla 4.
Un mezclador en V se cargó con la premezcla 2, el 70% restante de la lactosa (Foremost FastFlo® Lactose n° 316) dado en la Tabla 3, premezcla 3, premezcla 1 y premezcla 4, en ese orden, y se mezcló durante aproximadamente 500 inversiones. La mezcla mezclada se ensayó para determinar la uniformidad.
El estearato de magnesio se filtró a través de un tamiz de malla 70 y se añadió a la mezcla en el recipiente de mezcla, y la mezcla resultante se mezcló para proporcionar aproximadamente 125 inversiones.
La mezcla resultante se comprimió en comprimidos usando una prensa Killian T100 equipada con utillaje de cápsula de tipo B 0,64" x 0,32" configurado para producir un comprimido que tiene una dureza inicial de aproximadamente 11 Kp 20%.
Ejemplo 7: Tabletas ejemplares 7 y 8 (Tableta 5 y 6 con revestimiento por pulverización
Un lote de comprimidos con forma de comprimido oblongo del Ejemplo 5 y 6 se revistió por pulverización con OPADRY® II (Azul, Colorcon) a una ganancia de peso de aproximadamente 3,0% usando un recipiente de recubrimiento 24" configurado con los parámetros en la Tabla 7 seguido por el logotipo de la impresión mediante Opacode® WB (Negro, Colorcon).
Tabla 7: Parámetros del proceso de recubrimiento por pulverización

La suspensión de OPADRY® II se preparó mediante la medición de una cantidad de agua desionizada que cuando se combina con OPADRY® II produciría un contenido total de sólidos de 20% p/p. El agua se mezcla a un vórtice seguido de adición de OPADRY® II durante un período de aproximadamente 5 minutos. Una vez que se humedeció el polvo de OPADRY® II, se continuó la mezcla para asegurar que todo el material sólido se dispersara bien. La suspensión se cargó entonces en un instrumento de revestimiento de bandeja Thomas 24" usando condiciones de recubrimiento descritas en la Tabla 7.
Los núcleos de comprimidos se colocan en la bandeja de revestimiento y se pre-calentó. La temperatura de entrada se aumentó de temperatura ambiente a aproximadamente 55°C y después se aumentó a medida que fuera necesaria para proporcionar la temperatura de escape en la Tabla 7. El procedimiento de revestimiento se llevó a cabo con dispersión de revestimiento al 20% p/p de OPADRY® II (Serie Azul 85) para obtener una ganancia de peso diana del 3%. Las tabletas recubiertas se dejaron secar durante aproximadamente 2 minutos y sin pulverización. Se dejó que la temperatura del lecho se enfriara a aproximadamente 35°C.
Una vez recubiertos con OPADRY® II, los comprimidos se marcaron a continuación utilizando una prensa de tableta Hartnett Delta cargada de Opacode® WB.
Ejemplo 8: Tableta Ejemplar 9 (Formulada con polímero HPMCAS para tener 100 mg de Compuesto 1)
Un lote de comprimidos con forma de comprimido oblongo se formula para tener aproximadamente 100 mg de Compuesto 1 por comprimido utilizando las cantidades de los ingredientes enumerados en la Tabla 8, a continuación.
Tabla 8: Ingredientes para Tableta Ejemplar 9.

El dióxido de silicio coloidal (Dióxido de silicio pirógeno Cabot Cab-O-Sil® M-5P) y la celulosa microcristalina (FMC MCC Avicel® PH102) se pasaron a través de un tamiz de malla 30.
La croscarmelosa sódica (FMC Ac-Di-Sol®), SLS, Intermedio F, y la lactosa (Foremost FastFlo® Lactose n° 316) también se pasaron, de forma individual en el orden anterior, a través de la misma pantalla de malla 30. Se utilizó una purga de nitrógeno durante el cribado del Intermedio F. Los componentes tamizados se cargaron en un mezclador en V de 10 pies cúbicos, que se purgó con nitrógeno, y se mezcló durante aproximadamente 180 (+/- 10)
inversiones.
El estearato de magnesio se filtró a través de un tamiz de malla 40 en el recipiente de mezcla y se mezcló para proporcionar aproximadamente 54 inversiones.
La mezcla resultante se comprime en comprimidos usando una prensa totalmente equipada 36 Fette 2090 con utillaje de cápsula de tipo B de 0,568" x 0,2885" configurado para producir un comprimido que tiene una dureza diana inicial de aproximadamente 10 Kp 20%.
Ejemplo 9: Tableta Ejemplar 10 (Tableta 9 con revestimiento por pulverización)
Un lote de comprimidos con forma de comprimido oblongo del Ejemplo 8 se revistió por pulverización con OPADRY® II (azul, Colorcon) a una ganancia de peso de aproximadamente 3,0% usando un recipiente de 24" de revestimiento configurado con los parámetros de la Tabla 9, seguido de recubrimiento de cera y luego prensado utilizando Opacode® S-1-17823 (Negro a base de disolvente, Colorcon).
Tabla 9: Parámetros del proceso de recubrimiento por proyección

La suspensión de OPADRY® II se preparó mediante la medición de una cantidad de agua desionizada que cuando se combina con OPADRY® II produciría un contenido de sólidos total de 20% p/p. El agua se mezcla a un vórtice seguido de adición de OPADRY® II durante un período de aproximadamente 5 minutos. Una vez que se humedeció el polvo OPADRY® II, se continuó la mezcla para asegurar que todo el material sólido se dispersara bien. La suspensión se cargó entonces en un instrumento de revestimiento de bandeja Thomas 24" usando condiciones de recubrimiento descritas en la Tabla 9.
Las tabletas no recubiertas se colocan en la bandeja de revestimiento y se pre-calientan. La entrada se aumentó de temperatura ambiente a aproximadamente 55°C y después se aumentó a medida que fuera necesario para proporcionar la temperatura de escape en la Tabla 9. El proceso de recubrimiento se realizó con dispersión de revestimiento al 20% p/p de OPADRY® II (Serie Azul 85) para obtener una ganancia de peso diana del 3%. Las tabletas recubiertas se dejaron secar durante aproximadamente 2 minutos y sin pulverización. Se dejó que la temperatura del lecho se enfriara a aproximadamente 35°C.
Después de enfriarse, el polvo de cera de carnauba se pesó en una cantidad de aproximadamente 0,01% p/p del peso inicial del núcleo del comprimido. Con el flujo de aire detenido, el polvo de cera de carnauba fue rociado uniformemente sobre el lecho de comprimidos. La cama del bombo se enciende a la velocidad indicada en la Tabla 9. Después de 5 minutos, el flujo de aire se enciende (sin calentamiento) a la configuración indicada en la Tabla 9. Después de aproximadamente un minuto, se apagaron el flujo de aire y el recipiente.
Una vez recubiertos con OPADRY® II, los comprimidos después se marcaron usando una prensa de tableta Delta Hartnett cargada de Opacode® S-1-17.823.
Ejemplo 10: Tableta Ejemplar 11 (Formulada con polímero de HPMCAS para tener 150 mg de Compuesto 1) Un lote de comprimidos con forma de comprimido oblongo fue formulado para tener aproximadamente 100 mg de Compuesto 1 por comprimido utilizando las cantidades de ingredientes enumerados en la Tabla 11, a continuación.
Tabla 10: Ingredientes para Tableta Ejemplar 11.

El dióxido de silicio coloidal (Dióxido de silicio pirógeno Cabot Cab-O-Sil® M-5P) y la celulosa microcristalina (FMC MCC Avicel® PH102) se pasaron a través de un tamiz de malla 30.
La croscarmelosa sódica (FMC Ac-Di-Sol®), SLS, Intermedio F, y la lactosa (Foremost FastFlo® Lactose n° 316) también se pasaron, de forma individual en el orden anterior, a través de la misma pantalla de malla 30. Se utilizó una purga de nitrógeno durante el cribado de Intermedio F. Los componentes tamizados se cargaron en un mezclador en V de 10 pies cúbicos, que se purgó con nitrógeno, y se mezcló durante aproximadamente 180 (+/- 10) inversiones.
El estearato de magnesio se filtró a través de un tamiz de malla 40 en el recipiente de mezcla y se mezcló para proporcionar aproximadamente 54 inversiones.
La mezcla resultante se comprime en comprimidos usando una prensa totalmente equipada 36 Fette 2090 con utillaje de cápsula de tipo B de 0,568" x 0,2885" configurado para producir un comprimido que tiene una dureza inicial diana de aproximadamente 10 Kp 620%.
Ejemplo 11: Tablera Ejemplar 12 (Tableta 11 con revestimiento de pulverización)
Un lote de comprimidos con forma de comprimido oblongo del Ejemplo 10 se revistieron por pulverización con OPADRY® II (azul, Colorcon) a una ganancia de peso de aproximadamente 3,0% usando un recipiente de 24" de revestimiento configurado con los parámetros de la Tabla 11, seguido de recubrimiento de cera y después prensado utilizando Opacode® S-1 -17823 (Negro a base de disolvente, Colorcon).
Tabla 11: Parámetros del proceso de recubrimiento por pulverización

La suspensión de OPADRY® II se preparó mediante la medición de una cantidad de agua desionizada que cuando se combina con OPADRY® II produciría un contenido de sólidos total de 20% p/p. El agua se mezcla a un vórtice seguido de adición de OPADRY® II durante un período de aproximadamente 5 minutos. Una vez que se humedeció el polvo OPADRY® II, se continuó la mezcla para asegurar que todo el material sólido se dispersara bien. La suspensión se cargó entonces en un instrumento de revestimiento de recipiente Thomas 24" usando
condiciones de recubrimiento descritas en la Tabla 11.
Comprimidos no recubiertos se colocan en el recipiente de revestimiento y se pre-calientan. La entrada se aumentó de temperatura ambiente a aproximadamente 55°C y después se aumentó a medida que fuera necesario para proporcionar la temperatura de escape en la Tabla 9. El proceso de recubrimiento se realizó con dispersión de revestimiento al 20% p/p de OPADRY® II (Serie Azul 85) para obtener una ganancia de peso diana del 3%. Las tabletas recubiertas se dejaron secar durante aproximadamente 2 minutos y sin pulverización. Se dejó que la temperatura del lecho se enfriara a aproximadamente 35°C.
Después de enfriarse, el polvo de cera de carnauba se pesó en una cantidad de aproximadamente 0,01% p/p del peso inicial del núcleo del comprimido. Con el flujo de aire detenido, el polvo de cera de carnauba fue rociado uniformemente sobre el lecho de comprimidos. La cama del bombo se enciende a la velocidad indicada en la Tabla 11. Después de 5 minutos, el flujo de aire se enciende (sin calentamiento) a la configuración indicada en la Tabla 11. Después de aproximadamente un minuto, se apagaron el flujo de aire y el recipiente.
Una vez recubiertos con OPADRY® II, los comprimidos después se marcaron usando una prensa de tableta Delta Hartnett cargada de Opacode® S-1-17.823.
B. Administración de formulaciones farmacéuticas
Ejemplo 12: Administración ejemplar A
Se administra a los pacientes humanos por vía oral una formulación farmacéutica de acuerdo con la Tabla 12:
Tabla 12: Administración ejemplar A de las formulaciones farmacéuticas de la presente invención.

Las formulaciones farmacéuticas se administran a sujetos entre las 7:00 h y las 9:00 h, y la formulación farmacéutica se da en aproximadamente el mismo tiempo (dentro de una ventana de 1 horas) en cada ocasión de dosificación. Para las administraciones que se producen en condiciones de ayuno del paciente, la comida se deja 4 horas después de administrarse la formulación farmacéutica. Para las administraciones que permiten la alimentación, se da el desayuno unos 30 minutos antes de la hora de dosificación y se consume en unos 25 minutos. En cada una de estas administraciones, se pide al paciente que no se acueste durante 4 horas después de tomar el fármaco del estudio.
Ejemplo 13: Administración ejemplar B
Se administra a los pacientes humanos por vía oral una formulación farmacéutica de acuerdo con la Tabla 13:
Tabla 13: Administración ejemplar B de formulaciones farmacéuticas de la presente invención.
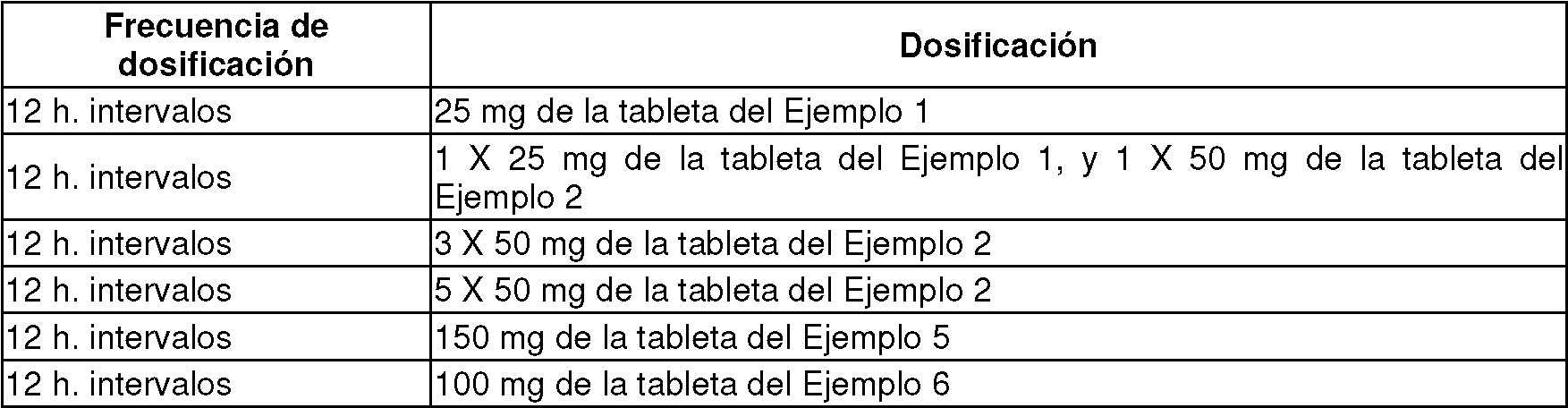
Las formulaciones farmacéuticas se administran a los pacientes aproximadamente cada 12 horas.
Ejemplo 14: Perfil de disolución de varios comprimidos ejemplares
Haciendo referencia a la Figura 1, los perfiles de disolución de varios comprimidos ejemplares se ilustran gráficamente. Se hace notar que cada uno de los comprimidos de ejemplo ilustrados en la Figura 1 están disueltos a al menos 50% en aproximadamente 30 minutos.
Claims (1)
- REIVINDICACIONES1. Una composición farmacéutica que comprende una dispersión sólida, en donde la dispersión sólida comprende: a) 80% en peso de N-[2,4-Bis(1,1-dimetiletil)-5-hidroxifenil]-1,4-dihidro-4-oxoquinolin-3-carboxamida (Compuesto 1) amorfa o sustancialmente amorfa por peso de la dispersión, en donde el Compuesto 1 sustancialmente amorfo comprende menos del 15% de Compuesto 1 cristalino,b) 19,5% en peso de succinato de acetato de hidroxipropilmetilcelulosa (HPMCAS) o copolímero de vinilpirrolidona/acetato de vinilo (PVP/VA) por peso de la dispersión, yc) 0,5% en peso de lauril sulfato de sodio (SLS) por peso de la dispersión.2. La composición farmacéutica de la reivindicación 1, en donde la composición farmacéutica comprende además del 20% en peso al 45% en peso de celulosa microcristalina por peso de la composición farmacéutica.3. La composición farmacéutica de la reivindicación 1 o la reivindicación 2, en donde la composición farmacéutica comprende además del 27% en peso al 45% en peso de lactosa por peso de la composición farmacéutica.4. La composición farmacéutica de cualquiera de las reivindicaciones 1 a 3, en donde la composición farmacéutica comprende además del 2,5% en peso al 6% en peso de croscarmelosa de sodio por peso de la composición farmacéutica.5. La composición farmacéutica de cualquiera de las reivindicaciones 1 a 4, en donde la composición farmacéutica comprende además del 0,3% en peso al 2% en peso de SLS por peso de la composición farmacéutica.6. La composición farmacéutica de cualquiera de las reivindicaciones 1 a 5, en donde la composición farmacéutica comprende además del 0,09% en peso al 1,0% en peso de dióxido de silicio coloidal por peso de la composición farmacéutica.7. La composición farmacéutica de cualquiera de las reivindicaciones 1 a 6, en donde la composición farmacéutica comprende además del 0,30% en peso al 1,3% en peso de estearato de magnesio por peso de la composición farmacéutica.8. La composición farmacéutica de cualquiera de las reivindicaciones 1 a 7, en donde la dispersión sólida comprende 150 mg de Compuesto 1 amorfo o sustancialmente amorfo.9. La composición farmacéutica de cualquiera de las reivindicaciones 1 a 7, en donde la dispersión sólida comprende 100 mg de Compuesto 1 amorfo o sustancialmente amorfo.10. La composición farmacéutica de cualquiera de las reivindicaciones 1 a 9, que comprende el 34,1% de la dispersión sólida por peso de la composición farmacéutica.11. Una composición farmacéutica de acuerdo con la reivindicación 1, que comprende además:a. una dispersión sólida de acuerdo con la reivindicación 1 en una cantidad que varía del 30% al 50% por peso de la composición farmacéutica;b. una carga, en una cantidad que varía del 25% en peso al 50% en peso por peso de la composición farmacéutica;c. un disgregante, en una cantidad que varía del 1% en peso al 10% en peso por peso de la composición farmacéutica;d. un surfactante, en una cantidad que varía del 0,3% en peso al 2% en peso por peso de la composición farmacéutica;e. un aglutinante, en una cantidad que varía del 5% en peso al 50% en peso por peso de la composición farmacéutica;f. un deslizante, en una cantidad que varía del 0,05% en peso al 2% en peso por peso de la composición farmacéutica; yg. un lubricante, en una cantidad que varía del 0,1% en peso al 2% en peso por peso de la composición farmacéutica.12. La composición farmacéutica de cualquiera de las reivindicaciones 1 a 10, que comprende:a. el 34,1% en peso de una dispersión sólida de acuerdo con la reivindicación 1;b. el 30,5% de celulosa microcristalina por peso de la composición;c. el 30,4% de lactosa por peso de la composición;d. el 3% de croscarmelosa de sodio por peso de la composición;e. el 0,5% de SLS por peso de la composición;f. el 0,5% de dióxido de silicio coloidal por peso de la composición; yg. el 1,0% de estearato de magnesio por peso de la composición.13. La composición farmacéutica de cualquiera de las reivindicaciones 1 a 12, en donde el Compuesto 1 sustancialmente amorfo comprende menos del 5% de Compuesto 1 cristalino.14. La composición farmacéutica de cualquiera de las reivindicaciones 1 a 13, en donde la composición farmacéutica está en forma de comprimido.15. La composición farmacéutica de la reivindicación 14, en donde el comprimido comprende además un recubrimiento.16. La composición farmacéutica de la reivindicación 14, en donde el comprimido tiene una dureza de por lo menos 49 N (5 kp).17. La composición farmacéutica de cualquiera de las reivindicaciones 1 a 16 para su uso en un método para tratar o reducir la gravedad de la fibrosis quística en un paciente con necesidad de ello.18. La composición farmacéutica para su uso de acuerdo con la reivindicación 17, en donde dicho paciente tiene un regulador de la conductancia transmembrana de la fibrosis quística (CFTR) con una mutación AF508.19. La composición farmacéutica para su uso de acuerdo con la reivindicación 17, en donde dicho paciente tiene un regulador de la conductancia transmembrana de la fibrosis quística (CFTR) con una mutación R117H.20. La composición farmacéutica para su uso de acuerdo con la reivindicación 17, en donde dicho paciente tiene un regulador de la conductancia transmembrana de la fibrosis quística (CFTR) con una mutación G551D.21. La composición farmacéutica para su uso de acuerdo con la reivindicación 17, en donde el método comprende administrar por vía oral la composición farmacéutica al paciente una vez al día.22. La composición farmacéutica para su uso de acuerdo con la reivindicación 17, en donde el método comprende administrar por vía oral la composición farmacéutica al paciente cada 12 horas.23. La composición farmacéutica para su uso de acuerdo con la reivindicación 17, en donde el método comprende administrar por vía oral la composición farmacéutica concurrentemente con una comida CF o aperitivo con alto contenido de grasas y calorías altas.24. La composición farmacéutica para su uso de acuerdo con la reivindicación 17, en donde el método comprende administrar por vía oral la composición farmacéutica 30 minutos después de administrar una comida CF o aperitivo con alto contenido de grasas y calorías altas.25. La composición farmacéutica para su uso de acuerdo con la reivindicación 17, en donde el método comprende administrar la composición farmacéutica concurrentemente, antes o después de uno o más de otros procedimientos terapéuticos o médicos deseados.
Applications Claiming Priority (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US8870408P | 2008-08-13 | 2008-08-13 | |
| US8880108P | 2008-08-14 | 2008-08-14 | |
| US9009608P | 2008-08-19 | 2008-08-19 | |
| US14616309P | 2009-01-21 | 2009-01-21 | |
| US18152709P | 2009-05-27 | 2009-05-27 | |
| US18334509P | 2009-06-02 | 2009-06-02 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2857152T3 true ES2857152T3 (es) | 2021-09-28 |
Family
ID=41206223
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES17204189T Active ES2857152T3 (es) | 2008-08-13 | 2009-08-13 | Composición farmacéutica y administraciones de la misma |
| ES09789125.3T Active ES2660143T3 (es) | 2008-08-13 | 2009-08-13 | Composición farmacéutica de N-[2,4-Bis(1,1-dimetiletilo)-5-hidroxifenilo]-1,4-dihidro-4-oxoquinolina-3carboxamida y administración de la misma |
Family Applications After (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES09789125.3T Active ES2660143T3 (es) | 2008-08-13 | 2009-08-13 | Composición farmacéutica de N-[2,4-Bis(1,1-dimetiletilo)-5-hidroxifenilo]-1,4-dihidro-4-oxoquinolina-3carboxamida y administración de la misma |
Country Status (27)
| Country | Link |
|---|---|
| US (1) | US20120220625A1 (es) |
| EP (3) | EP3842037A1 (es) |
| JP (2) | JP5575768B2 (es) |
| KR (5) | KR20190143497A (es) |
| CN (1) | CN102231990B (es) |
| AU (2) | AU2009282419B2 (es) |
| BR (1) | BRPI0916877A2 (es) |
| CA (1) | CA2733908C (es) |
| CY (2) | CY1119945T1 (es) |
| DK (2) | DK2328618T3 (es) |
| EA (1) | EA021706B1 (es) |
| ES (2) | ES2857152T3 (es) |
| HR (2) | HRP20180328T1 (es) |
| HU (2) | HUE035931T2 (es) |
| IL (3) | IL211203A (es) |
| LT (2) | LT3345625T (es) |
| ME (1) | ME03019B (es) |
| MX (3) | MX2011001782A (es) |
| NO (1) | NO2328618T3 (es) |
| NZ (1) | NZ591535A (es) |
| PL (2) | PL2328618T3 (es) |
| PT (2) | PT2328618T (es) |
| RS (2) | RS56894B1 (es) |
| SI (2) | SI3345625T1 (es) |
| SM (2) | SMT201800074T1 (es) |
| WO (1) | WO2010019239A2 (es) |
| ZA (1) | ZA201101097B (es) |
Families Citing this family (119)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| ZA200602755B (en) | 2003-09-06 | 2007-06-27 | Vertex Pharma | Modulators of ATP-binding cassette transporters |
| US7977322B2 (en) | 2004-08-20 | 2011-07-12 | Vertex Pharmaceuticals Incorporated | Modulators of ATP-binding cassette transporters |
| LT2502911T (lt) | 2004-06-24 | 2017-09-11 | Vertex Pharmaceuticals Incorporated | Atp surišančios kasetės transporterių moduliatoriai |
| ATE512145T1 (de) | 2005-08-11 | 2011-06-15 | Vertex Pharma | Modulatoren des cystic fibrosis transmembrane conductance regulators |
| EP1945632B1 (en) | 2005-11-08 | 2013-09-18 | Vertex Pharmaceuticals Incorporated | Heterocyclic modulators of atp-binding cassette transporters |
| WO2007079139A2 (en) | 2005-12-28 | 2007-07-12 | Vertex Pharmaceuticals, Inc. | Solid forms of n-[2,4-bis(1,1-dimethylethyl)-5-hydroxyphenyl]-1,4-dihydro-4-oxoquinoline-3-carboxamide |
| AU2006336504C9 (en) | 2005-12-28 | 2015-05-14 | Vertex Pharmaceuticals Incorporated | 1-(benzo [D] [1,3] dioxol-5-yl) -N- (phenyl) cyclopropane- carboxamide derivatives and related compounds as modulators of ATP-Binding Cassette transporters for the treatment of Cystic Fibrosis |
| US7645789B2 (en) | 2006-04-07 | 2010-01-12 | Vertex Pharmaceuticals Incorporated | Indole derivatives as CFTR modulators |
| NZ571803A (en) | 2006-04-07 | 2011-12-22 | Vertex Pharma | Amide indole derivatives as modulators of ATP-binding cassette transporters |
| USRE50453E1 (en) | 2006-04-07 | 2025-06-10 | Vertex Pharmaceuticals Incorporated | Indole derivatives as CFTR modulators |
| US10022352B2 (en) | 2006-04-07 | 2018-07-17 | Vertex Pharmaceuticals Incorporated | Modulators of ATP-binding cassette transporters |
| US8563573B2 (en) | 2007-11-02 | 2013-10-22 | Vertex Pharmaceuticals Incorporated | Azaindole derivatives as CFTR modulators |
| US8969386B2 (en) | 2007-05-09 | 2015-03-03 | Vertex Pharmaceuticals Incorporated | Modulators of CFTR |
| HUE028426T2 (en) | 2007-08-24 | 2016-12-28 | Vertex Pharma | Isothiazolopyridinones for (inter alia) treating cystic fibrosis |
| SI2217572T1 (sl) | 2007-11-16 | 2014-05-30 | Vertex Pharmaceuticals Incorporated | Izokinolinski modulatorji prenaĺ alcev z atp-vezavno kaseto |
| EP2639224B1 (en) | 2007-12-07 | 2016-08-24 | Vertex Pharmaceuticals Incorporated | Process for producing cycloalkylcarboxiamido-pyridine benzoic acids |
| SI2225230T1 (sl) | 2007-12-07 | 2017-03-31 | Vertex Pharmaceuticals Incorporated | Trdne oblike 3-(6-(1-2,2-difluorobenzo(d)(1,3)dioxol-5-il)ciklopropan- karboksamido)-3-metilpiridin-2-il) benzojske kisline |
| WO2009076141A2 (en) * | 2007-12-07 | 2009-06-18 | Vertex Pharmaceuticals Incorporated | Formulations of 3-(6-(1-(2,2-difluorobenzo[d][1,3]dioxol-5-yl) cycklopropanecarboxamido)-3-methylpyridin-2-yl)benzoic acid |
| US20100036130A1 (en) | 2007-12-07 | 2010-02-11 | Vertex Pharmaceuticals Incorporated | Processes for producing cycloalkylcarboxamido-pyridine benzoic acids |
| NZ720282A (en) | 2008-02-28 | 2017-12-22 | Vertex Pharma | Heteroaryl derivatives as cftr modulators |
| EP2271621B1 (en) | 2008-03-31 | 2013-11-20 | Vertex Pharmaceuticals Incorporated | Pyridyl derivatives as cftr modulators |
| US12458635B2 (en) | 2008-08-13 | 2025-11-04 | Vertex Pharmaceuticals Incorporated | Pharmaceutical composition and administrations thereof |
| US20100074949A1 (en) | 2008-08-13 | 2010-03-25 | William Rowe | Pharmaceutical composition and administration thereof |
| HRP20180328T1 (hr) | 2008-08-13 | 2018-04-20 | Vertex Pharmaceuticals Inc. | FARMACEUTSKI PRIPRAVAK N-[2,4-bis(1,1-DIMETILETIL)-5-HIDROKSIFENIL]-1,4-DIHIDRO-4-OKSOKINOLIN-3-KARBOKSAMIDA I NJEGOVO DAVANJE |
| JP2012504143A (ja) * | 2008-09-29 | 2012-02-16 | バーテックス ファーマシューティカルズ インコーポレイテッド | 3−(6−(1−(2,2−ジフルオロベンゾ[d][1,3]ジオキソール−5−イル)シクロプロパンカルボキサミド)−3−メチルピリジン−2−イル)安息香酸の用量単位 |
| NZ592685A (en) | 2008-10-23 | 2013-04-26 | Vertex Pharma | Modulators of cystic fibrosis transmembrane conductance regulator |
| SI2408750T1 (sl) | 2009-03-20 | 2015-11-30 | Vertex Pharmaceuticals Incorporated | Postopek za izdelavo modulatorjev cistično-fibroznega transmembranskega regulatorja prevodnosti |
| US8802868B2 (en) | 2010-03-25 | 2014-08-12 | Vertex Pharmaceuticals Incorporated | Solid forms of (R)-1(2,2-difluorobenzo[D][1,3]dioxo1-5-yl)-N-(1-(2,3-dihydroxypropyl-6-fluoro-2-(1-hydroxy-2-methylpropan2-yl)-1H-Indol-5-yl)-Cyclopropanecarboxamide |
| HRP20211752T1 (hr) | 2010-04-07 | 2022-02-18 | Vertex Pharmaceuticals Incorporated | Farmaceutski pripravci 3-(6-(1-(2,2-difluorobenzo[d][1,3]dioksol-5-il)ciklopropankarboksamido)-3-metilpiridin-2-il)benzojeve kiseline i njihova primjena |
| HRP20160682T1 (hr) | 2010-04-07 | 2016-07-29 | Vertex Pharmaceuticals Incorporated | Čvrsti oblici 3-(6-(1-(2,2-difluorobenzo[d][1,3]dioksol-5-il)ciklopropankarboksiamido)-3-metilpiridin-2-il)benzojeve kiseline |
| ES2608474T3 (es) | 2010-04-22 | 2017-04-11 | Vertex Pharmaceuticals Incorporated | Proceso de producción de compuestos indol cycloalkylcarboxamido |
| US8563593B2 (en) | 2010-06-08 | 2013-10-22 | Vertex Pharmaceuticals Incorporated | Formulations of (R)-1-(2,2-difluorobenzo[D] [1,3] dioxol-5-yl)-N-(1-(2,3-dihydroxypropyl)-6-fluoro-2-(1-hydroxy-2-methylpropan-2-yl)-1H-indol-5-yl)cyclopropanecarboxamide |
| RU2013113627A (ru) | 2010-08-27 | 2014-10-10 | Вертекс Фармасьютикалз Инкорпорейтед | Фармацевтическая композиция и ее введения |
| HUE047354T2 (hu) | 2011-05-18 | 2020-04-28 | Vertex Pharmaceuticals Europe Ltd | Ivacaftor deuterizált származékai |
| JP6063455B2 (ja) | 2011-05-18 | 2017-01-18 | コンサート ファーマシューティカルズ インコーポレイテッド | 重水素化されたイバカフトルの誘導体 |
| EP2567959B1 (en) | 2011-09-12 | 2014-04-16 | Sanofi | 6-(4-hydroxy-phenyl)-3-styryl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| IN2014KN00885A (es) | 2011-11-08 | 2015-10-02 | Vertex Pharma | |
| WO2013112804A1 (en) | 2012-01-25 | 2013-08-01 | Vertex Pharmaceuticals Incorporated | Formulations of 3-(6-(1-(2.2-difluorobenzo[d][1,3]dioxol-5-yl) cyclopropanecarboxamido)-3-methylpyridin-2-yl)benzoic acid |
| EP2819670A1 (en) | 2012-02-27 | 2015-01-07 | Vertex Pharmaceuticals Incorporated | Pharmaceutical composition and administration thereof |
| HK1206995A1 (en) * | 2012-04-06 | 2016-01-22 | The Uab Research Foundation | Methods for increasing cftr activity |
| WO2014014841A1 (en) | 2012-07-16 | 2014-01-23 | Vertex Pharmaceuticals Incorporated | Pharmaceutical compositions of (r)-1-(2,2-diflurorbenzo[d][1,3]dioxol-5-yl)-n-(1-(2,3-dihydroxypropyl)-6-fluoro-2-(1-hydroxy-2-methylpropan-2-yl)-1h-indol-5-yl) cyclopropanecarboxamide and administration thereof |
| HK1206991A1 (en) | 2012-07-20 | 2016-01-22 | Otsuka Pharmaceutical Co., Ltd. | Tablet having dry-ink film on surface thereof, and ink for inkjet printer |
| LT3470063T (lt) | 2012-11-02 | 2025-02-10 | Vertex Pharmaceuticals Incorporated | Farmacinės kompozicijos, skirtos cftr nulemtų ligų gydymui |
| MY183582A (en) | 2012-11-19 | 2021-02-26 | Vertex Pharmaceuticals Europe Ltd | Deuterated cftr potentiators |
| EP2968323A4 (en) | 2013-03-13 | 2016-12-14 | Flatley Discovery Lab | PYRIDAZINONE COMPOUNDS AND METHODS FOR THE TREATMENT OF CYSTIC FIBROSIS |
| PT3038601T (pt) * | 2013-08-27 | 2020-06-30 | Gilead Pharmasset Llc | Formulação combinada de dois compostos antivirais |
| JP6963896B2 (ja) | 2013-11-12 | 2021-11-10 | バーテックス ファーマシューティカルズ インコーポレイテッドVertex Pharmaceuticals Incorporated | Cftr媒介性疾患の処置のための医薬組成物を調製する方法 |
| EP3091981B1 (en) * | 2014-01-09 | 2021-03-10 | Verastem, Inc. | Compositions and methods for treatment of abnormal cell growth |
| NZ723859A (en) * | 2014-03-14 | 2023-01-27 | Servier Lab | Pharmaceutical compositions of therapeutically active compounds and their uses |
| ES2885181T3 (es) | 2014-04-15 | 2021-12-13 | Vertex Pharma | Composiciones farmacéuticas para el tratamiento de enfermedades mediadas por el regulador de la conductancia transmembrana de fibrosis quística |
| GB201415381D0 (en) | 2014-08-29 | 2014-10-15 | Algipharma As | Inhalable powder formulations of alginate oligomers |
| TWI735416B (zh) | 2014-10-06 | 2021-08-11 | 美商維泰克斯製藥公司 | 囊腫纖維化症跨膜傳導調節蛋白之調節劑 |
| KR20170063954A (ko) | 2014-10-07 | 2017-06-08 | 버텍스 파마슈티칼스 인코포레이티드 | 낭성 섬유증 막횡단 전도도 조절자의 조정제의 공-결정 |
| PT3221692T (pt) * | 2014-11-18 | 2021-09-10 | Vertex Pharma | Processo de realização de testagem de alta produtividade por cromatografia líquida de alta eficiência |
| GB201504878D0 (en) | 2015-03-23 | 2015-05-06 | Algipharma As | Use of alginate oligomers and CFTR modulators in the treatment of conditions associated with CFTR dysfuntion |
| UA124619C2 (uk) | 2015-09-21 | 2021-10-20 | Вертекс Фармасьютікалз (Юроп) Лімітед | Введення дейтерованих підсилювачів cftr |
| HK1258473A1 (zh) | 2015-09-25 | 2019-11-15 | Vertex Pharmaceuticals (Europe) Limited | 氘代cftr增效剂 |
| GB201517639D0 (en) | 2015-10-06 | 2015-11-18 | Algipharma As | Use of alginate oligomers to treat or prevent microbial overgrowth in the intestinal tract |
| CA3019394A1 (en) * | 2016-04-08 | 2017-10-12 | X4 Pharmaceuticals, Inc. | Methods for treating cancer |
| FR3055800B1 (fr) | 2016-09-15 | 2020-06-26 | Unither Pharmaceuticals | Composition solide a ingestion rapide et deglutition facilitee, sous forme de particules solides non agglomerees, comprenant deux differents types de particules |
| SI3519401T1 (sl) | 2016-09-30 | 2022-01-31 | Vertex Pharmaceuticals Incorporated | Modulator regulatorja transmembranske prevodnosti pri cistični fibrozi, farmacevtski sestavki, postopki zdravljenja in proces za izdelavo modulatorja |
| CN110267948B (zh) | 2016-12-09 | 2023-12-08 | 弗特克斯药品有限公司 | 囊性纤维化跨膜传导调控剂的调节剂、药物组合物、治疗方法和制备所述调节剂的方法 |
| AU2018279646B2 (en) | 2017-06-08 | 2023-04-06 | Vertex Pharmaceuticals Incorporated | Methods of treatment for cystic fibrosis |
| CA3068609A1 (en) | 2017-07-01 | 2019-01-10 | Vertex Pharmaceuticals Incorporated | Compositions and methods for treatment of cystic fibrosis |
| US20200171015A1 (en) | 2017-07-17 | 2020-06-04 | Vertex Pharmaceuticals Incorporated | Methods of treatment for cystic fibrosis |
| MA49631A (fr) | 2017-07-17 | 2020-05-27 | Vertex Pharma | Méthodes de traitement de la fibrose kystique |
| TWI799435B (zh) | 2017-08-02 | 2023-04-21 | 美商維泰克斯製藥公司 | 製備化合物之製程 |
| US10654829B2 (en) | 2017-10-19 | 2020-05-19 | Vertex Pharmaceuticals Incorporated | Crystalline forms and compositions of CFTR modulators |
| CA3082444A1 (en) | 2017-12-01 | 2019-06-06 | Vertex Pharmaceuticals Incorporated | Processes for making modulators of cystic fibrosis transmembrane conductance regulator |
| WO2019113089A1 (en) | 2017-12-04 | 2019-06-13 | Vertex Pharmaceuticals Incorporated | Compositions for treating cystic fibrosis |
| KR102716244B1 (ko) | 2017-12-08 | 2024-10-14 | 버텍스 파마슈티칼스 인코포레이티드 | 낭포성 섬유증 막횡단 전도 조절자의 조정제의 제조 방법 |
| TWI810243B (zh) | 2018-02-05 | 2023-08-01 | 美商維泰克斯製藥公司 | 用於治療囊腫纖化症之醫藥組合物 |
| AU2019222758B2 (en) | 2018-02-15 | 2022-07-07 | Vertex Pharmaceuticals Incorporated | Macrocycles as modulators of cystic fibrosis transmembrane conductance regulator, pharmaceutical compositions thereof, their use in the treatment of cystic fibrosis, and process for making them |
| US20210139514A1 (en) | 2018-04-05 | 2021-05-13 | Vertex Pharmaceuticals Incorporated | Modulators of cystic fibrosis transmembrane conductance regulator |
| EP3774825A1 (en) | 2018-04-13 | 2021-02-17 | Vertex Pharmaceuticals Incorporated | Modulators of cystic fibrosis transmembrane conductance regulator, pharmaceutical compositions, methods of treatment, and process for making the modulator |
| PT3880197T (pt) | 2018-11-14 | 2023-05-09 | Vertex Pharma | Métodos de tratamento para a fibrose quística |
| TWI848092B (zh) | 2019-04-03 | 2024-07-11 | 美商維泰克斯製藥公司 | 囊腫纖維化跨膜傳導調節蛋白調節劑 |
| WO2020214921A1 (en) | 2019-04-17 | 2020-10-22 | Vertex Pharmaceuticals Incorporated | Solid forms of modulators of cftr |
| WO2020242935A1 (en) | 2019-05-29 | 2020-12-03 | Vertex Pharmaceuticals Incorporated | Methods of treatment for cystic fibrosis |
| CA3150162A1 (en) | 2019-08-14 | 2021-02-18 | Vertex Pharmaceuticals Incorporated | Crystalline forms of cftr modulators |
| TWI899097B (zh) | 2019-08-14 | 2025-10-01 | 美商維泰克斯製藥公司 | 製備cftr調節劑之方法 |
| CN114585628B (zh) | 2019-08-14 | 2024-03-26 | 弗特克斯药品有限公司 | 囊性纤维化跨膜传导调节因子的调节剂 |
| TWI867024B (zh) | 2019-08-14 | 2024-12-21 | 美商維泰克斯製藥公司 | 囊腫纖維化跨膜傳導調節蛋白之調節劑 |
| MX2022007580A (es) * | 2019-12-20 | 2022-07-19 | Intervet Int Bv | Una composicion farmaceutica de un compuesto de pirazol dispersado en una matriz polimerica. |
| EP4164618A4 (en) * | 2020-06-10 | 2024-06-19 | Board of Regents, The University of Texas System | 3D LASER SINTERING PROCESS FOR IMPROVED ACTIVE INGREDIENT RELEASE |
| WO2022194399A1 (en) | 2020-07-13 | 2022-09-22 | Idorsia Pharmaceuticals Ltd | Macrocycles as cftr modulators |
| WO2022013360A1 (en) | 2020-07-17 | 2022-01-20 | Synthon B.V. | Pharmaceutical composition comprising ivacaftor |
| CR20230120A (es) | 2020-08-07 | 2023-09-01 | Vertex Pharma | Moduladores del regulador de la conductancia transmembrana de la fibrosis quística |
| IL300413A (en) | 2020-08-13 | 2023-04-01 | Vertex Pharma | Crystal forms of CFTR modulators |
| CA3191084A1 (en) * | 2020-09-09 | 2022-03-17 | Gerald Burke | Formulations of a somatostatin modulator |
| EP4225447A1 (en) | 2020-10-07 | 2023-08-16 | Vertex Pharmaceuticals Incorporated | Modulators of cystic fibrosis transmembrane conductance regulator |
| US20230373935A1 (en) | 2020-10-07 | 2023-11-23 | Vertex Pharmaceuticals Incorporated | Modulators of cystic fibrosis transmembrane conductance regulator |
| EP4225765A2 (en) | 2020-10-07 | 2023-08-16 | Vertex Pharmaceuticals Incorporated | Modulators of cystic fibrosis transmembrane conductance regulator |
| WO2022076629A1 (en) | 2020-10-07 | 2022-04-14 | Vertex Pharmaceuticals Incorporated | Modulators of cystic fibrosis transmembrane conductance regulator |
| US20230374038A1 (en) | 2020-10-07 | 2023-11-23 | Vertex Pharmaceuticals Incorporated | Modulators of cystic fibrosis transmembrane conductance regulator |
| EP4225446A1 (en) | 2020-10-07 | 2023-08-16 | Vertex Pharmaceuticals Incorporated | Modulators of cystic fibrosis transmembrane conductance regulator |
| WO2022076628A1 (en) | 2020-10-07 | 2022-04-14 | Vertex Pharmaceuticals Incorporated | Modulators of cystic fibrosis transmembrane conductance regulator |
| EP4225762A1 (en) | 2020-10-07 | 2023-08-16 | Vertex Pharmaceuticals Incorporated | Modulators of cystic fibrosis transmembrane conductance regulator |
| WO2022076626A1 (en) | 2020-10-07 | 2022-04-14 | Vertex Pharmaceuticals Incorporated | Modulators of cystic fibrosis transmembrane conductance regulator |
| MX2023004074A (es) | 2020-10-07 | 2023-07-05 | Vertex Pharma | Moduladores del regulador de la conductancia transmembrana de la fibrosis quistica. |
| US12324802B2 (en) | 2020-11-18 | 2025-06-10 | Vertex Pharmaceuticals Incorporated | Modulators of cystic fibrosis transmembrane conductance regulator |
| AU2021397294A1 (en) | 2020-12-10 | 2023-07-06 | Vertex Pharmaceuticals Incorporated | Methods of treatment for cystic fibrosis |
| IL314449A (en) | 2022-02-03 | 2024-09-01 | Vertex Pharma | Methods of treatment for cystic fibrosis |
| JP2025505577A (ja) | 2022-02-03 | 2025-02-28 | バーテックス ファーマシューティカルズ インコーポレイテッド | (6a,12a)-17-アミノ-12-メチル-6,15-ビス(トリフルオロメチル)-13,19-ジオキサ-3,4,18-トリアザトリシクロ[12.3.1.12,5]ノナデカ-1(18),2,4,14,16-ペンタエン-6-オールの調製方法及び結晶形態 |
| WO2023154291A1 (en) | 2022-02-08 | 2023-08-17 | Vertex Pharmaceuticals Incorporated | Modulators of cystic fibrosis transmembrane conductance regulator |
| IL315715A (en) | 2022-04-06 | 2024-11-01 | Vertex Pharma | Modulators of cystic fibrosis transmembrane conductance regulation |
| CA3253977A1 (en) | 2022-05-16 | 2023-11-23 | Vertex Pharmaceuticals Incorporated | SOLID FORMS OF MACROCYCLIC COMPOUNDS AS CFTR MODULATORS AND THEIR PREPARATION |
| EP4525858A1 (en) | 2022-05-16 | 2025-03-26 | Vertex Pharmaceuticals Incorporated | Methods of treatment for cystic fibrosis |
| US20260034064A1 (en) | 2022-08-04 | 2026-02-05 | Vertex Pharmaceuticals Incorporated | Compositions for the treatment of cftr-mediated diseases |
| EP4335434A1 (en) | 2022-08-17 | 2024-03-13 | Sanovel Ilac Sanayi Ve Ticaret A.S. | Pharmaceutical compositions comprising ivacaftor |
| KR20250084132A (ko) | 2022-09-15 | 2025-06-10 | 이도르시아 파마슈티컬스 리미티드 | 마크로시클릭 cftr 조절제 |
| TW202421102A (zh) | 2022-09-15 | 2024-06-01 | 瑞士商愛杜西亞製藥有限公司 | 巨環cftr調節劑與cftr校正子及/或cftr增效劑之組合 |
| CA3267791A1 (en) | 2022-09-15 | 2024-03-21 | Idorsia Pharmaceuticals Ltd | CRYSTALLINE FORM OF (3S,7S,10R,13R)-13-BENZYL-20-FLUORO-7-ISOBUTYL-N-(2-(3-METHOXY-1,2,4-OXADIAZOL-5-YL)ETHYL)-6,9-DIMETHYL-1,5,8,11-TETRAOXO-10-(2,2,2-TRIFLUOROETHYL)-1,2,3,4,5,6,7,8,9,10,11,12,13,14-TETRADECAHYDRO-[1]OXA[4,7,10,14]TETRAAZACYCLOHEPTADECINO [16,17-F]QUINOLINE-3-CARBOXAMIDE |
| WO2025076240A1 (en) | 2023-10-04 | 2025-04-10 | Vertex Pharmaceuticals Incorporated | Solid forms of modulators of cystic fibrosis transmembrane conductance regulator |
| WO2025076235A1 (en) | 2023-10-04 | 2025-04-10 | Vertex Pharmaceuticals Incorporated | Modulators of cystic fibrosis transmembrane conductance regulator |
| EP4570241A1 (en) | 2023-12-12 | 2025-06-18 | Genepharm S.A. | Compositions of ivacaftor |
| EP4574138A1 (en) | 2023-12-20 | 2025-06-25 | Sanovel Ilac Sanayi Ve Ticaret Anonim Sirketi | A tablet composition comprising ivacaftor |
| WO2025166342A1 (en) | 2024-02-04 | 2025-08-07 | Vertex Pharmaceuticals Incorporated | Combination of vanzacaftor, tezacaftor, deutivacaftor for use in treating cystic fibrosis |
| WO2025186214A1 (en) | 2024-03-05 | 2025-09-12 | Idorsia Pharmaceuticals Ltd | Macrocyclic cftr modulators |
Family Cites Families (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| ES2234451T1 (es) | 2001-11-14 | 2005-07-01 | Teva Pharmaceutical Industries Ltd. | Formas cristalinas y amorfas de losartan potasio y procedimiento para su preparacion. |
| CN100563658C (zh) * | 2003-11-14 | 2009-12-02 | 味之素株式会社 | 苯丙氨酸衍生物的固体分散体或固体分散体医药制剂 |
| JP2006206612A (ja) * | 2004-05-27 | 2006-08-10 | Ono Pharmaceut Co Ltd | 固形製剤用組成物 |
| WO2007079139A2 (en) * | 2005-12-28 | 2007-07-12 | Vertex Pharmaceuticals, Inc. | Solid forms of n-[2,4-bis(1,1-dimethylethyl)-5-hydroxyphenyl]-1,4-dihydro-4-oxoquinoline-3-carboxamide |
| MX2009006806A (es) | 2006-12-22 | 2009-08-27 | Vertex Pharma | Secado por rocio fluidizado. |
| HRP20180328T1 (hr) | 2008-08-13 | 2018-04-20 | Vertex Pharmaceuticals Inc. | FARMACEUTSKI PRIPRAVAK N-[2,4-bis(1,1-DIMETILETIL)-5-HIDROKSIFENIL]-1,4-DIHIDRO-4-OKSOKINOLIN-3-KARBOKSAMIDA I NJEGOVO DAVANJE |
| US8314239B2 (en) * | 2008-10-23 | 2012-11-20 | Vertex Pharmaceutical Incorporated | Modulators of cystic fibrosis transmembrane conductance regulator |
-
2009
- 2009-08-13 HR HRP20180328TT patent/HRP20180328T1/hr unknown
- 2009-08-13 ME MEP-2018-54A patent/ME03019B/me unknown
- 2009-08-13 SI SI200932107T patent/SI3345625T1/sl unknown
- 2009-08-13 LT LTEP17204189.9T patent/LT3345625T/lt unknown
- 2009-08-13 JP JP2011522994A patent/JP5575768B2/ja active Active
- 2009-08-13 ES ES17204189T patent/ES2857152T3/es active Active
- 2009-08-13 NO NO09789125A patent/NO2328618T3/no unknown
- 2009-08-13 PL PL09789125T patent/PL2328618T3/pl unknown
- 2009-08-13 PT PT97891253T patent/PT2328618T/pt unknown
- 2009-08-13 BR BRPI0916877A patent/BRPI0916877A2/pt not_active Application Discontinuation
- 2009-08-13 KR KR1020197037948A patent/KR20190143497A/ko not_active Ceased
- 2009-08-13 PT PT172041899T patent/PT3345625T/pt unknown
- 2009-08-13 EP EP20211291.8A patent/EP3842037A1/en active Pending
- 2009-08-13 DK DK09789125.3T patent/DK2328618T3/en active
- 2009-08-13 AU AU2009282419A patent/AU2009282419B2/en active Active
- 2009-08-13 DK DK17204189.9T patent/DK3345625T3/da active
- 2009-08-13 WO PCT/US2009/004629 patent/WO2010019239A2/en not_active Ceased
- 2009-08-13 MX MX2011001782A patent/MX2011001782A/es unknown
- 2009-08-13 KR KR1020227014317A patent/KR20220057663A/ko not_active Ceased
- 2009-08-13 MX MX2018015847A patent/MX373230B/es unknown
- 2009-08-13 NZ NZ591535A patent/NZ591535A/xx unknown
- 2009-08-13 CA CA2733908A patent/CA2733908C/en active Active
- 2009-08-13 ES ES09789125.3T patent/ES2660143T3/es active Active
- 2009-08-13 KR KR1020117005734A patent/KR20110042356A/ko not_active Ceased
- 2009-08-13 RS RS20180161A patent/RS56894B1/sr unknown
- 2009-08-13 KR KR1020247015082A patent/KR20240066199A/ko active Pending
- 2009-08-13 EP EP09789125.3A patent/EP2328618B1/en active Active
- 2009-08-13 EP EP17204189.9A patent/EP3345625B1/en not_active Revoked
- 2009-08-13 RS RS20210129A patent/RS61408B1/sr unknown
- 2009-08-13 SM SM20180074T patent/SMT201800074T1/it unknown
- 2009-08-13 SM SM20210077T patent/SMT202100077T1/it unknown
- 2009-08-13 HU HUE09789125A patent/HUE035931T2/hu unknown
- 2009-08-13 CN CN2009801354496A patent/CN102231990B/zh active Active
- 2009-08-13 SI SI200931803T patent/SI2328618T1/en unknown
- 2009-08-13 KR KR1020177016117A patent/KR20170072950A/ko not_active Ceased
- 2009-08-13 HU HUE17204189A patent/HUE053357T2/hu unknown
- 2009-08-13 LT LTEP09789125.3T patent/LT2328618T/lt unknown
- 2009-08-13 EA EA201170330A patent/EA021706B1/ru unknown
- 2009-08-13 MX MX2014008855A patent/MX366161B/es unknown
- 2009-08-13 PL PL17204189T patent/PL3345625T3/pl unknown
-
2011
- 2011-02-10 ZA ZA2011/01097A patent/ZA201101097B/en unknown
- 2011-02-13 IL IL211203A patent/IL211203A/en active IP Right Grant
-
2012
- 2012-02-10 US US13/370,540 patent/US20120220625A1/en not_active Abandoned
-
2014
- 2014-03-12 JP JP2014048523A patent/JP2014111656A/ja not_active Withdrawn
-
2016
- 2016-08-16 AU AU2016216569A patent/AU2016216569B2/en active Active
-
2017
- 2017-05-21 IL IL252421A patent/IL252421B/en active IP Right Grant
-
2018
- 2018-02-26 CY CY20181100229T patent/CY1119945T1/el unknown
-
2020
- 2020-07-05 IL IL275854A patent/IL275854A/en unknown
-
2021
- 2021-02-05 HR HRP20210208TT patent/HRP20210208T1/hr unknown
- 2021-02-22 CY CY20211100146T patent/CY1123901T1/el unknown
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2857152T3 (es) | Composición farmacéutica y administraciones de la misma | |
| US11564916B2 (en) | Pharmaceutical composition and administrations thereof | |
| US20140315948A1 (en) | Pharmaceutical Composition and Administrations Thereof | |
| JP2011530598A5 (es) | ||
| US12458635B2 (en) | Pharmaceutical composition and administrations thereof | |
| HK40051819A (en) | Pharmaceutical composition and administrations thereof | |
| HK1256805B (en) | Pharmaceutical composition and administrations thereof | |
| HK1151738A (en) | Pharmaceutical composition of n-[2,4-bis(1,1-dimethylethyl)-5-hydroxyphenyl]-1,4-dihydro-4-oxoquinoline-3-carboxamide and administration thereof | |
| HK1151738B (en) | Pharmaceutical composition of n-[2,4-bis(1,1-dimethylethyl)-5-hydroxyphenyl]-1,4-dihydro-4-oxoquinoline-3-carboxamide and administration thereof | |
| HK1161140B (en) | Pharmaceutical composition of n-[2,4-bis(1,1-dimethylethyl)-5-hydroxyphenyl]-1,4-dihydro-4-oxoquinoline-3-carboxamide and administration thereof |