ES2864948T3 - Aparato de colocación de remaches ciegos y dispositivo de procesamiento de remaches ciegos con un aparato de colocación de remaches ciegos - Google Patents
Aparato de colocación de remaches ciegos y dispositivo de procesamiento de remaches ciegos con un aparato de colocación de remaches ciegos Download PDFInfo
- Publication number
- ES2864948T3 ES2864948T3 ES16204257T ES16204257T ES2864948T3 ES 2864948 T3 ES2864948 T3 ES 2864948T3 ES 16204257 T ES16204257 T ES 16204257T ES 16204257 T ES16204257 T ES 16204257T ES 2864948 T3 ES2864948 T3 ES 2864948T3
- Authority
- ES
- Spain
- Prior art keywords
- cyanopyrrolidin
- carboxamide
- methyl
- alkylene
- pyrazol
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Classifications
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B21—MECHANICAL METAL-WORKING WITHOUT ESSENTIALLY REMOVING MATERIAL; PUNCHING METAL
- B21J—FORGING; HAMMERING; PRESSING METAL; RIVETING; FORGE FURNACES
- B21J15/00—Riveting
- B21J15/02—Riveting procedures
- B21J15/04—Riveting hollow rivets mechanically
- B21J15/043—Riveting hollow rivets mechanically by pulling a mandrel
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B21—MECHANICAL METAL-WORKING WITHOUT ESSENTIALLY REMOVING MATERIAL; PUNCHING METAL
- B21J—FORGING; HAMMERING; PRESSING METAL; RIVETING; FORGE FURNACES
- B21J15/00—Riveting
- B21J15/10—Riveting machines
- B21J15/16—Drives for riveting machines; Transmission means therefor
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B21—MECHANICAL METAL-WORKING WITHOUT ESSENTIALLY REMOVING MATERIAL; PUNCHING METAL
- B21J—FORGING; HAMMERING; PRESSING METAL; RIVETING; FORGE FURNACES
- B21J15/00—Riveting
- B21J15/10—Riveting machines
- B21J15/16—Drives for riveting machines; Transmission means therefor
- B21J15/26—Drives for riveting machines; Transmission means therefor operated by rotary drive, e.g. by electric motor
Landscapes
- Engineering & Computer Science (AREA)
- Mechanical Engineering (AREA)
- Insertion Pins And Rivets (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
Aparato automático (1) de colocación de remaches ciegos con una carcasa (3), con un mecanismo (13) de tracción que puede moverse en relación con la carcasa (3) mediante un accionamiento (15), en donde el accionamiento (15) está configurado como un accionamiento eléctrico y, entre el accionamiento (15) y el mecanismo (13) de tracción, está dispuesto un engranaje (14) con una relación de transmisión modificable y con al menos dos etapas (24, 25) de engranaje entre el accionamiento (15) y el mecanismo (13) de tracción, y el engranaje (14) presenta un dispositivo (19) de conmutación que está conectado a un dispositivo (20) de mando, caracterizado por que está previsto un dispositivo de alimentación de remaches ciegos y el dispositivo (20) de mando selecciona una de las etapas (24, 25) de engranaje en función de una magnitud de funcionamiento del accionamiento eléctrico (15).
Description
DESCRIPCIÓN
Compuestos de 1-ciano-pirrolidina como inhibidores de USP30
La presente invención se refiere a nuevos compuestos y métodos para la fabricación de inhibidores de enzimas desubiquitinantes (DUB). En particular, la invención se refiere a la inhibición de ubiquitina C-terminal hidrolasa 30 (USP30). La invención se refiere adicionalmente al uso de inhibidores de DUB en el tratamiento de afecciones que implican disfunción mitocondrial y en el tratamiento del cáncer.
Antecedentes de la invención
La enumeración o discusión de un documento aparentemente publicado anteriormente en esta memoria descriptiva no debe tomarse necesariamente como un reconocimiento de que el documento es parte del estado de la técnica o es de conocimiento general común.
La ubiquitina es una pequeña proteína que consiste en 76 aminoácidos que es importante para la regulación de la función de las proteínas en la célula. La ubiquitinación y la desubiquitinación son procedimientos mediados enzimáticamente mediante los cuales la ubiquitina se une covalentemente o se escinde de una proteína diana mediante enzimas deubiquitinantes (DUB), de las cuales hay aproximadamente 95 DUB en células humanas, divididas en subfamilias según la homología de secuencia. La familia USP se caracteriza por sus cajas Cys e His comunes que contienen restos de Cys e His críticos para sus actividades DUB. Los procedimientos de ubiquitinación y desubiquitinación se han implicado en la regulación de muchas funciones celulares, incluyendo la progresión del ciclo celular, la apoptosis, la modificación de los receptores de la superficie celular, la regulación de la transcripción del ADN y la reparación del ADN. Por lo tanto, el sistema de ubiquitina se ha implicado en la patogenia de numerosos estados patológicos que incluyen inflamación, infección viral, disfunción metabólica, trastornos del SNC y oncogénesis (Clague et al., Physiol Rev 93:1289-1315, 2013).
La ubiquitina es un regulador maestro de la dinámica mitocondrial. Las mitocondrias son orgánulos dinámicos cuyos eventos de biogénesis, fusión y fisión están regulados por la regulación postraduccional a través de la ubiquitinación de muchos factores clave tales como las mitofusinas. Si bien se sabe que las ubiquitina ligasas tales como la parkina ubiquitinan una serie de proteínas mitocondriales, hasta hace poco, las enzimas deubiquitinantes seguían siendo esquivas. La USP30 es una proteína de 517 aminoácidos que se encuentra en la membrana externa mitocondrial (Nakamura et al., Mol Biol 19:1903-11, 2008). Es la única enzima desubiquitinante que lleva una señal de direccionamiento mitocondrial y se ha demostrado que desubiquitina varias proteínas mitocondriales. Se ha demostrado que USP30 se opone a la mitofagia mediada por parkina y que la reducción de la actividad de USP30 puede rescatar defectos mediados por parkina en la mitofagia (Bingol et al., Nature 510:370-5, 2014).
La disfunción mitocondrial se puede definir como una disminución del contenido mitocondrial (mitofagia o biogénesis mitocondrial), como una disminución de la actividad mitocondrial y la fosforilación oxidativa, pero también como una modulación de la generación de especies reactivas de oxígeno (ROS). De ahí un papel para las disfunciones mitocondriales en una gran cantidad de procedimientos y patologías de envejecimiento que incluyen, entre otras, enfermedades neurodegenerativas (p. ej., enfermedad de Parkinson (EP), enfermedad de Alzheimer, enfermedad de Huntington, esclerosis lateral amiotrófica (ELA), esclerosis muscular), cáncer, diabetes, trastornos metabólicos, enfermedades cardiovasculares, enfermedades psiquiátricas (p. ej., esquizofrenia) y osteoartritis.
Por ejemplo, la enfermedad de Parkinson afecta a alrededor de 10 millones de personas en todo el mundo (Parkinson’s Disease Foundation) y se caracteriza por la pérdida de neuronas dopaminérgicas en la sustancia negra. Los mecanismos exactos que subyacen a la EP no están claros; sin embargo, la disfunción mitocondrial se aprecia cada vez más como un determinante clave de la susceptibilidad neuronal dopaminérgica en la EP y es una característica de la enfermedad familiar y esporádica, así como del parkinsonismo inducido por toxinas. La parkina es una de varias proteínas que se han relacionado con la EP de inicio temprano. Si bien la mayoría de los casos de EP están relacionados con defectos en la alfa-sinucleína, 10% de los casos de Parkinson están relacionados con defectos genéticos específicos, uno de los cuales está en la ubiquitina E3 ligasa parkina. La parkina y la quinasa putativa 1 inducida por la proteína quinasa PTEN (PINK1) colaboran para ubiquitinar las proteínas de la membrana mitocondrial de las mitocondrias dañadas, lo que da como resultado mitofagia. La desregulación de la mitofagia da como resultado un aumento del estrés oxidativo, que se ha descrito como una característica de la EP. Por tanto, la inhibición de la USP30 podría ser una estrategia potencial para el tratamiento de la EP. Por ejemplo, los pacientes con EP con mutaciones de parkina que conducen a una actividad reducida podrían compensarse terapéuticamente mediante la inhibición de USP30.
Se ha informado de que el agotamiento de USP30 mejora el aclaramiento mitofágico de las mitocondrias y también mejora la muerte celular inducida por parkina (Liang et al., EMBO Reports 2015 DOI: 10,15252/embr.201439820). También se ha demostrado que USP30 regula la apoptosis dependiente de BAX/BAK independientemente de la expresión en exceso de parkina. El agotamiento de USP30 sensibiliza a las células cancerosas a miméticos de BH-3 tales como ABT-737, sin la necesidad de expresión en exceso de parkina. Por tanto, se ha demostrado un papel antiapoptótico para USP30 y, por tanto, la USP30 es un objetivo potencial para la terapia anticancerosa.
Hasta la fecha, no ha habido informes de inhibidores de DUB que hayan ingresado con éxito en la clínica. Por tanto,
existe la necesidad de compuestos y composiciones farmacéuticas para inhibir las DUB tales como USP30 para el tratamiento de indicaciones en las que se observa actividad de DUB, que incluyen, aunque no se limitan a, afecciones que implican disfunción mitocondrial y cáncer.
Lainé et al., Med Chem Lett. 2011, 2(2), 142-7 describen el compuesto N-[(3R)-1-ciano-3-pirrolidinil]-4-fluorobenzamida como inhibidor de la catepsina C. El documento WO2001/077073 describe los compuestos N-(1-ciano-3-pirrolidinil)-[1,1'-bifenil]-4-carboxamida y N-(1-ciano-3-piperidinil)-[1,1'-bifenil]-4-carboxamida como inhibidores de catepsina. El documento WO2009/129371 describe los compuestos N-[(3R)-1-ciano-3-pirrolidinil]-3-({[(3R)-1-ciano-3-pirrolidinil]amino}sulfonil)benzamida y N-[(3R)-1-ciano-3-pirrolidinil]-3-([(3R)-3-pirrolidinilamino]sulfonil)-benzamida como inhibidores de Catepsina C. El documento WO2016/021629 describe el compuesto 1-((3S,4R)-1-ciano-4-(3,4-difluorofenil)pirrolidin-3-il)-3-(1',4-dimetil-1-fenil-1H,1'H-[3,4'-bipirazol]-5-il)ureail)urea como inhibidor de TrkA. Estos compuestos pueden ser rechazados por las reivindicaciones adjuntas.
Falgueyret et al., J. Med. Chem. 2001,44, 94-104, y la Solicitud PCT WO 01/77073 se refieren a las cianopirrolidinas como inhibidores de las catepsinas K y L, con utilidad potencial en el tratamiento de la osteoporosis y otras afecciones relacionadas con la resorción ósea. El documento US 2012/077806 se refiere a una serie de acrilamidas como inhibidores potenciales del DUB USP9x.
Compendio de la invención
De acuerdo con un primer aspecto de la invención se proporciona:
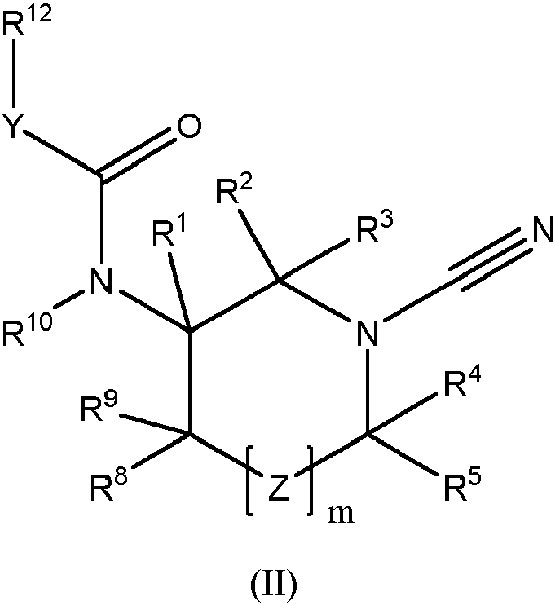
(i) un compuesto de fórmula (II)

un tautómero del mismo, o una sal farmacéuticamente aceptable de dicho compuesto o tautómero, en donde: m es 0 o 1;
cuando m es 1, Z es -C(R6)(R7)-;
R2 representa un átomo de hidrógeno, un grupo alquilo C1-C6, o alcoxi C1-C6 opcionalmente sustituidos;
R3, R4 y R5 representan cada uno independientemente un átomo de hidrógeno, un grupo alquilo C1-C3, o alcoxi C1-C3 opcionalmente sustituidos;
R1, R6, R7 y R8 representan cada uno independientemente un átomo de hidrógeno, un átomo de flúor, ciano, hidroxilo, un grupo alquilo C1-C3, o alcoxi C1-C3 opcionalmente sustituidos;
R9 representa un átomo de hidrógeno, un átomo de flúor, ciano, hidroxilo, alquilo C1-C6, un grupo alcoxi C1-C3, o un anillo de cicloalquilo de 3 a 8 miembros, o forma un anillo heterocíclico con R10;
R10 representa un átomo de hidrógeno, alquilo C1-C6 opcionalmente sustituido, o forma un anillo heterocíclico con R9, o forma un anillo monocíclico con R11;
Y representa un enlace covalente, -alquileno(C0-C3)-N(R11)-alquileno C0-C3 o alquileno C1-C3;
R11 representa un átomo de hidrógeno, alquilo C1-C6, o forma un anillo monocíclico con R10;
R12 representa un anillo de heteroarilo, heterociclilo o cicloalquilo de 3 a 14 miembros monocíclico sustituido, bicíclico opcionalmente sustituido o tricíclico opcionalmente sustituido;
en donde R12, cuando está sustituido, está sustituido con uno o más de -Q1-(R13)p, en donde:
p es 0 o 1;
Q1 representa un átomo de halógeno, ciano, oxo, hidroxilo, un enlace covalente, -alquilen(Co-C3)-NR14-, -alquilen(Co-C3)-NR14R15, -alquilen(C0-C3)-CONR14-, -alquilen(C0-C3)-NR14CO-, -alquilen(C0-C3)-NR14SO2-, un átomo de oxígeno, -alquilen(C0-C3)-CO-, -alquilen(C0-C3)-S(O)q-, -alquilen(C0-C3)-SO2NR14, -alquilen(C0-C3)-SO2NR14R15, -alcoxi C1-C6, haloalcoxi C1-C6, hidroxialquilo C1-C6, -alquilen(C0-C3)-SO2R14, -alquilen(C0-C3)-NR14COR15, -alquilen(C0-C3)-NR14CONR15R16, -alquilen(C0-C3)-NR14SO2NR15R16, -alquilen(C0-C3)-CONR14R15, -alquilen(C0-C3)-CO2R14, -alquilen(C0-C3)-NR14CO2R15, -alquilen(C0-C3)-SO2NR14R15, -alquilen(C0-C3)-C(O)R14 y alquilen(C1-C6)-NR14SO2R15, NO2, alquileno C1-C6, -alquenileno C2-C6, o un -grupo alquilo C1-C6 opcionalmente sustituido;
q es 0, 1 o 2;
R14, R15 y R16 representan cada uno independientemente un átomo de hidrógeno, alquilo C1-C6, o un grupo alquileno C1-C6;
cuando p es 1, R13 representa un anillo de heteroarilo, heterociclilo, arilo de 4 a 10 miembros o cicloalquilo de 3 a 8 miembros; en donde R13 puede estar opcionalmente sustituido con uno o más sustituyentes seleccionados entre halógeno, haloalquilo C1-C6, alcoxi C1-C6, haloalcoxi C1-C6, alquilo C1-C6, alquenilo C2-C6, alquinilo C2-C6, hidroxialquilo C1-C6, oxo, ciano, -Q2-R17, -Q2-NR17CONR18R19, -Q2-NR17R18, -Q2-COR17, -Q2-NR17COR18, -Q2-NR17CO2R18, -Q2-SO2R17, Q2-CONR17R18, -Q2-CO2R17, -Q2-SO2NR17R18, -Q2-NR17SO2R18, heterociclilo, cicloalquilo, heteroarilo y arilo; en donde dichos anillos de heterociclilo, cicloalquilo, heteroarilo y arilo pueden estar opcionalmente sustituidos con uno o más sustituyentes, que pueden ser iguales o diferentes, seleccionados entre Cl, F, OMe, Me, COCH3, CONH2, NHC(O)CH(CH3)2 , y CO2CH2CH3;
Q2 representa un enlace covalente, un átomo de oxígeno, -CO- o un grupo alquileno C1-C6 o alquenileno C2-C6; R16, R17, R18 representan cada uno independientemente hidrógeno, alquilo C1-C6, heterociclilo, heteroarilo, arilo o cicloalquilo; y
en donde los sustituyentes opcionales de alquilo de R1, R2 , R3, R4, R5, R6, R7 , R8 , R10 y Q1, que pueden ser iguales o diferentes, se seleccionan entre alcoxi C1-C3, halógeno, hidroxilo, tiol, ciano, amino, amido, nitro y SF5;
(ii) un compuesto de fórmula (II)
un tautómero del mismo, o una sal farmacéuticamente aceptable de dicho compuesto o tautómero, en donde: m es 0 o 1;
cuando m es 1, Z es -C(R6)(R7)-;
R2 representa un átomo de hidrógeno, un grupo alquilo C1-C6, o alcoxi C1-C6 opcionalmente sustituidos;
R3, R4 y R5 representan cada uno independientemente un átomo de hidrógeno, un grupo alquilo C1-C3, o alcoxi C1-C3 opcionalmente sustituidos;
R1, R6, R7 y R8 representan cada uno independientemente un átomo de hidrógeno, un átomo de flúor, ciano, hidroxilo, un grupo alquilo C1-C3, o alcoxi C1-C3 opcionalmente sustituidos;
R9 representa un átomo de hidrógeno, un átomo de flúor, ciano, hidroxilo, alquilo C1-C6, un grupo alcoxi C1-C3, o un anillo de cicloalquilo de 3 a 8 miembros, o forma un anillo heterocíclico con R10;
R10 representa un átomo de hidrógeno, alquilo C1-C6 opcionalmente sustituido, o forma un anillo heterocíclico con R9, o forma un anillo monocíclico con R11;
Y representa un enlace covalente, -alquileno(Co-C3)-N(R11)-alquileno C0-C3 o alquileno C1-C3;
R11 representa un átomo de hidrógeno, alquilo C1-C6, o forma un anillo monocíclico con R10;
R12 representa un anillo de arilo de 3 a 14 miembros monocíclico sustituido, bicíclico opcionalmente sustituido o tricíclico opcionalmente sustituido;
donde R12, cuando está sustituido, está sustituido con uno o más de -Q1-(R13)p, en donde:
p es 0 o 1;
Q1 representa un átomo de halógeno, ciano, oxo, hidroxilo, un enlace covalente, -alquilen(C0-C3)-NR14-, -alquilen(C0-C3)-NR14R15, -alquilen(C0-C3)-CONR14-, -alquilen(C0-C3)-NR14CO-, -alquilen(C0-C3)-NR14SO2-, un átomo de oxígeno, -alquilen(C0-C3)-CO-, -alquilen(C0-C3)-S(O)q-, -alquilen(C0-C3)-SO2NR14R15, -alcoxi C1-C6, haloalcoxi C1-C6, hidroxialquilo C1-C6, -alquilen(C0-C3)-SO2R14, -alquilen(C0-C3)-NR14COR15, -alquilen(C0-C3)-NR14CONR15R16, -alquilen(C0-C3)-NR14SO2NR15R16, -alquilen(C0-C3)-CONR14R15, -alquilen(C0-C3)-CO2R14, -alquilen(C0-C3)-NR14CO2R15, -alquilen(C0-C3)-SO2NR14R15, -alquilen(C0-C3)-C(O)R14 y alquilen(C1-C6)-NR14SO2R15, NO2, alquileno C1-C6, -alquenileno C2-C6, o un -grupo alquilo C1-C6 opcionalmente sustituido;
q es 0, 1 o 2;
R14, R15 y R16 representan cada uno independientemente un átomo de hidrógeno, alquilo C1-C6, o un grupo alquileno C1-C6; cuando p es 1, R13 representa un anillo de heteroarilo, heterociclilo de 4 a 10 miembros o cicloalquilo de 3 a 8 miembros; en donde R13 puede estar opcionalmente sustituido con uno o más sustituyentes seleccionados entre halógeno, haloalquilo C1-C6, alcoxi C1-C6, haloalcoxi C1-C6, alquilo C1-C6, alquenilo C2-C6, alquinilo C2-C6, hidroxialquilo C1-C6, oxo, ciano, -Q2-R17, -Q2-NR17CONR18R19, -Q2-NR17R18, -Q2-COR17, -Q2-NR17COR18, -Q2-NR17CO2R18, -Q2-SO2R17, Q2-CONR17R18, -Q2-CO2R17, -Q2-SO2NR17R18, -Q2-NR17SO2R18, heterociclilo, cicloalquilo, heteroarilo y arilo; en donde dichos anillos de heterociclilo, cicloalquilo, heteroarilo y arilo pueden estar opcionalmente sustituidos con uno o más sustituyentes, que pueden ser iguales o diferentes, seleccionados entre Cl, F, OMe, Me, COCH3, CONH2, NHC(O)CH(CH3)2 , y CO2CH2CH3 ;
Q2 representa un enlace covalente, un átomo de oxígeno, -CO- o un grupo alquileno C1-C6 o alquenileno C2-C6; R16, R17, R18 representan cada uno independientemente hidrógeno, alquilo C1-C6, heterociclilo, heteroarilo, arilo o cicloalquilo;
en donde los sustituyentes opcionales de alquilo de R1, R2, R3, R4, R5, R6, R7, R8, R10 y Q1, que pueden ser iguales o diferentes, se seleccionan entre alcoxi C1-C3, halógeno, hidroxilo, tiol, ciano, amino, amido, nitro y SF5; y donde el compuesto no tiene la fórmula:

y
(iii) un compuesto de fórmula (II)

un tautómero del mismo, o una sal farmacéuticamente aceptable de dicho compuesto o tautómero, en donde: m es 0 o 1;
cuando m es 1, Z es -C(R6)(R7)-;
R2 representa un átomo de hidrógeno, un grupo alquilo C1-C6, o alcoxi C1-C6 opcionalmente sustituidos;
R3, R4 y R5 representan cada uno independientemente un átomo de hidrógeno, un grupo alquilo C1-C3, o alcoxi C1-C3 opcionalmente sustituidos;
R1, R6, R7 y R8 representan cada uno independientemente un átomo de hidrógeno, un átomo de flúor, ciano, hidroxilo, un grupo alquilo C1-C3, o alcoxi C1-C3 opcionalmente sustituidos;
R9 representa un átomo de hidrógeno, un átomo de flúor, ciano, hidroxilo, alquilo C1-C6, un grupo alcoxi C1-C3, o un anillo de cicloalquilo de 3 a 8 miembros, o forma un anillo heterocíclico con R10;
R10 representa un átomo de hidrógeno, alquilo C1-C6 opcionalmente sustituido, o forma un anillo heterocíclico con R9, o forma un anillo monocíclico con R11;
Y representa un enlace covalente, -alquileno(C0-C3)-N(R11)-alquileno C0-C3 o alquileno C1-C3;
R11 representa un átomo de hidrógeno, alquilo C1-C6, o forma un anillo monocíclico con R10;
R12 representa un anillo de arilo monocíclico, bicíclico o tricíclico de 3 a 14 miembros;
donde R12 está sustituido con uno o más de -Q1-(R13)p, en donde:
p es 1;
Q1 representa ciano, oxo, hidroxilo, un enlace covalente, -alquilen(C0-C3)-NR14-, -alquilen(C0-C3)-NR14R15, -alquilen(C0-C3)-CONR14-, -alquilen(C0-C3)-NR14CO-, -alquilen(C0-C3)-NR14SO2-, un átomo de oxígeno, -alquilen(C0-C3)-CO-, -alquilen(C0-C3)-S(O)q-, -alquilen(C0-C3)-SO2NR14, -alquilen(C0-C3)-SO2NR14R15, -alcoxi C1-C6, haloalcoxi C1-C6, hidroxialquilo C1-C6, -alquilen(C0-C3)-SO2R14, -alquilen(C0-C3)-NR14COR15, -alquilen(C0-C3)-NR14CONR15R16, -alquilen(C0-C3)-NR14SO2NR15R16, -alquilen(C0-C3)-CONR14R15, -alquilen(C0-C3)-CO2R14, -alquilen(C0-C3)-NR14CO2R15, -alquilen(C0-C3)-SO2NR14R15, -alquilen(C0-C3)-C(O)R14 y alquilen(C1-C6)-NR14SO2R15, NO2, alquileno C1-C6, -alquenileno C2-C6, o un -grupo alquilo C1-C6 opcionalmente sustituido;
q es 0, 1 o 2;
R14, R15 y R16 representan cada uno independientemente un átomo de hidrógeno, alquilo C1-C6, o un grupo alquileno C1-C6;
cuando p es 1, R13 representa un anillo de arilo de 4 a 10 miembros; en donde R13 está sustituido con uno o más sustituyentes seleccionados entre haloalquilo C1-C6, alcoxi C1-C6, haloalcoxi C1-C6, alquilo C1-C6, alquenilo C2-C6, alquinilo C2-C6, hidroxialquilo C1-C6, oxo, ciano, -Q2-R17, -Q2-NR17CONR18R19, -Q2-NR17R18, -Q2-COR17, -Q2-NR17COR18, -Q2-NR17CO2R18, -Q2-SO2R17, Q2-CONR17R18, -Q2-CO2R17, -Q2-SO2NR17R18, -Q2-NR17SO2R18, heterociclilo, cicloalquilo, heteroarilo y arilo; en donde dichos anillos de heterociclilo, cicloalquilo, heteroarilo y arilo pueden estar opcionalmente sustituidos con uno o más sustituyentes, que pueden ser iguales o diferentes, seleccionados entre Cl, F, OMe, Me, COCH3, CONH2, NHC(O)CH(CH3)2, y CO2CH2CH3
Q2 representa un enlace covalente, un átomo de oxígeno, -CO- o un grupo alquileno C1-C6 o alquenileno C2-C6;
R16, R17, R18 representan cada uno independientemente hidrógeno, alquilo C1-C6, heterociclilo, heteroarilo, arilo o cicloalquilo; y
en donde los sustituyentes opcionales de alquilo de R1, R2, R3, R4, R5, R6, R7, R8, R10 y Q1, que pueden ser iguales o diferentes, se seleccionan entre alcoxi C1-C3, halógeno, hidroxilo, tiol, ciano, amino, amido, nitro y SF5.
Breve descripción de las figuras
La Figura 1 es un gráfico que muestra la actividad proteolítica de USP30 medida usando un ensayo de polarización de fluorescencia. Se incubaron varios volúmenes de USP30 purificada como se indica con un péptido marcado con TAMRA unido a ubiquitina mediante un enlace isopeptídico.
Descripción detallada de la invención
Las definiciones y explicaciones siguientes corresponden a los términos que se utilizan en toda la presente memoria, incluidas tanto la memoria descriptiva como las reivindicaciones. La referencia a los compuestos como se describe en la presente memoria (p. ej., un compuesto de fórmula I), incluye la referencia a la fórmula I, fórmula II y fórmula III, incluida cualquiera de sus realizaciones subgenéricas.
Cuando se ha hecho referencia a cualquier grupo de los compuestos de fórmula (I) como opcionalmente sustituido, este grupo puede estar sustituido como se describe en las reivindicaciones, o no sustituido. La sustitución puede ser con uno o más de los sustituyentes especificados que pueden ser iguales o diferentes. Se apreciará que el número y la naturaleza de los sustituyentes se seleccionarán para evitar cualquier combinación estéricamente indeseable.
En el contexto de la presente memoria descriptiva, a menos que se indique lo contrario, un grupo sustituyente alquilo, alquenilo o alquinilo o un radical alquilo o alquenilo en un grupo sustituyente puede ser lineal o ramificado. Las cadenas de alquilo y alquenilo también pueden incluir heteroátomos intermedios tales como oxígeno.
Alquilo C x -C y se refiere a un grupo hidrocarbonado alifático saturado que tiene x-y átomos de carbono que pueden ser lineales o ramificados. Por ejemplo, alquilo C1-C6 contiene de 1 a 6 átomos de carbono. "Ramificado" significa que está presente en el grupo al menos un punto de ramificación de carbono. Por ejemplo, terc-butilo e isopropilo son ambos grupos ramificados. Los ejemplos de los grupos alquilo C1-C6 incluyen metilo, etilo, propilo, 2-metil-1 -propilo, 2-metil-2-propilo, 2-metil-1 -butilo, 3-metil-1 -butilo, 2-metil-3-butilo, 2,2-dimetil-1 -propilo, 2-metil-pentilo, 3-metil-1-pentilo, 4-metil-1 -pentilo, 2-metil-2-pentilo, 3-metil-2-pentilo, 4-metil-2-pentilo, 2,2-dimetil-1 -butilo, 3,3-dimetil-1 -butilo, 2-etil-1 -butilo, n-butilo, isobutilo, terc-butilo, n-pentilo, isopentilo, neopentilo y n-hexilo.
Un grupo o radical alquileno C X -C y puede ser lineal o ramificado y se refiere a un grupo hidrocarbonado divalente que tiene un átomo de hidrógeno menos de alquilo CX-Cy como se definió anteriormente. Los ejemplos de grupos alquileno C1-C6 incluyen metileno, etileno, n-propileno, n-butileno, metilmetileno y dimetilmetileno.
Alquenilo C 2 -C 6 se refiere a un radical de cadena hidrocarbonada lineal o ramificada que contiene al menos dos átomos de carbono y al menos un doble enlace. Los ejemplos de grupos alquenilo incluyen etenilo, propenilo, 2-propenilo, 1-butenilo, 2-butenilo, 1-hexenilo, 2-metil-1-propenilo, 1,2-butadienilo, 1,3-pentadienilo, 1,4-pentadienilo y 1-hexadienilo.
Alquinilo C 2 -C 6 se refiere a un radical de cadena hidrocarbonada lineal o ramificada que contiene al menos dos átomos de carbono y al menos un triple enlace. Los ejemplos de grupos alquenilo incluyen etinilo, propinilo, 2-propinilo, 1 -butinilo, 2-butinilo y 1 -hexinilo.
Alcoxi C 1 -C 6 se refiere a un grupo o parte de un grupo que tiene un grupo alquilo -O-CX-Cy según la definición de alquilo CX-Cy anterior. Los ejemplos de alcoxi C1-C6 incluyen metoxi, etoxi, propoxi, isopropoxi, butoxi, pentoxi y hexoxi.
Haloalquilo C 1 -C 6 y haloalcoxi C 1 -C 6 se refieren a un grupo alquilo CX-Cy como se definió anteriormente en donde al menos un átomo de hidrógeno se reemplaza por un átomo de halógeno. Los ejemplos de grupos haloalquilo C1-C6 incluyen fluorometilo, difluorometilo, trifluorometilo, trifluoroetilo, pentafluoroetilo, fluorometoxi, difluorometoxi y trifluorometoxi.
Hidroxialquilo C 1 -C 6 se refiere a un grupo alquilo CX-Cy como se definió anteriormente en donde al menos un átomo de hidrógeno se reemplaza por un grupo hidroxi (-OH). Los ejemplos de grupos hidroxialquilo C1-6 incluyen hidroximetilo, hidroxietilo, dihidroxietilo, hidroxipropilo e hidroxiisopropilo.
Los términos "halógeno" o "halo" se refieren a átomos de cloro, bromo, flúor o yodo.
El término "oxo" significa =O.
Para evitar dudas, se entenderá que un anillo de heteroarilo, heterociclilo o arilo de 4 a 10 miembros, o un anillo de cicloalquilo de 3 a 8 miembros como se define según R2, R9, R11 o R13 o un anillo de heteroarilo, heterociclilo, cicloalquilo o arilo de 3 a 14 miembros como se define según R12 no incluye ninguna estructura anular inestable o, en el caso de sistemas anulares heteroarílicos y heterocíclicos, ningún enlace O-O, O-S o S-S. Los sistemas anulares
pueden ser monocíclicos, bicíclicos o tricíclicos cuando la definición lo permita. Los sistemas anulares bicíclicos y tricíclicos incluyen sistemas anulares puenteados, condensados y espiro, particularmente sistemas anulares fusionados. Un sustituyente, si está presente, puede estar anclado a cualquier átomo anular adecuado que puede ser un átomo de carbono o, en el caso de sistemas anulares heteroarílicos y heterocíclicos, un heteroátomo. La sustitución en un anillo de fenilo puede incluir un cambio en el átomo anular en la posición de sustitución de carbono a nitrógeno, que da como resultado un anillo de piridina.
"Cicloalquilo C x -C y " se refiere a un grupo hidrocarbonado cíclico no aromático de x-y átomos de carbono. Por ejemplo, cicloalquilo C3-C8 se refiere a un anillo hidrocarbonado que contiene de 3 a 8 átomos de carbono. Los ejemplos de cicloalquilo C3-C8 son ciclopropilo, ciclobutilo, ciclopentilo, ciclohexilo, ciclopentilo, ciclohexilo, cicloheptilo y ciclooctilo.
Un grupo/radical "arilo" se refiere a cualquier grupo hidrocarbonado monocíclico o bicíclico que comprende al menos un grupo aromático, por ejemplo, que tiene hasta 10 átomos de carbono miembros del anillo. Los ejemplos de grupos arilo incluyen fenilo, naftilo y tetrahidronaftilo.
Los grupos "heteroarilo" pueden ser monocíclicos, bicíclicos o tricíclicos. Los anillos bicíclicos pueden ser anillos aromáticos condensados donde ambos anillos son aromáticos o pueden ser anillos condensados donde uno de los anillos no es aromático. En el caso de R12, el anillo anclado al nitrógeno de la amida puede ser un anillo aromático, que puede fusionarse con un anillo aromático o no aromático adicional. Los anillos de heteroarilo comprenden 1, 2, 3 o 4 heteroátomos, en particular 1, 2 o 3 heteroátomos, seleccionados entre oxígeno, azufre y nitrógeno. Cuando el heteroátomo es nitrógeno, este puede oxidarse. Los ejemplos de grupos heteroarilo incluyen piridinilo, pirazinilo, pirimidinilo, piridazinilo, furilo, tiofenilo, pirrolilo, oxazolilo, tiazolilo, pirazolilo, triazolilo, tetrazolilo, indolilo, indolizinilo, isoindolilo, indolinilo, purinilo, furazanilo, imidazolilo, indazolilo, isotiazolilo, isoxazolilo, oxadiazolilo, oxazinanilo, tetrazolilo, tiadiazolilo, benzofuranilo, isobenzofuranilo, benzotiofenilo, isobenzotiofenilo, benzimidazolilo, benzotiazolilo, naftiridinilo, pteridinilo, pirazinilo, 4H-quinolizinilo, quinolinilo, isoquinolinilo, cinolinilo, ftalazinilo, quinazolinilo, imidazopiridinilo, pirazolopiridinilo, tiazolopiridinilo, isoindolinilo, triazinilo, piridazinilo, dihidropiridinilo, benzopirazolilo, quinoxalinilo, tetrahidropiridoindolilo, benzimidazolilo, pirrolopiridinilo, imidazopirimidinilo, pirazolopirimidinilo, pirrolopirimidinilo e imidazopirazinilo.
Los grupos "heterociclilo" también pueden ser monocíclicos o comprender dos o más anillos condensados que pueden ser saturados o parcialmente insaturados que comprenden 1, 2, 3 o 4 heteroátomos, en particular 1, 2 o 3 heteroátomos, seleccionados entre oxígeno, azufre y nitrógeno. Los ejemplos de grupos heterociclilo incluyen azetidinilo, pirrolidinilo, piperidinilo, azepanilo, diazepanilo, dihidrofuranilo (p. ej., 2,3-dihidrofuranilo, 2,5-dihidrofuranilo), 4,5-dihidro-1H-maleimido, dioxolanilo, morfolinilo, oxazolidinilo, piperazinilo, tetrahidrofuranilo, tiomorfolinilo, dihidropiranilo (p. ej., 3,4-dihidropiranilo, 3,6-dihidropiranilo), homopiperazinilo, dioxanilo, hexahidropirimidinilo, pirazolinilo, pirazolidinilo, piridazinilo, 4H-quinolizinilo, quinuclinilo, tetrahidropiranilo, tetrahidropiridinilo, tetrahidropirimidinilo, tetrahidrotiofenilo, tetrametilensulfóxido, tiazolidinilo, hidantoinilo, benzopiranilo, tetrahidrotiazolopiridinilo, tetrahidroquinolinilo, tetrahidroisoquinolinilo, benzomorfolinilo y dihidroisoquinolinilo.
"Opcionalmente sustituido" aplicado a cualquier grupo significa que dicho grupo puede, si se desea, estar sustituido con uno o más sustituyentes, que pueden ser iguales o diferentes. Los ejemplos de sustituyentes adecuados para radicales "sustituidos" y "opcionalmente sustituidos" incluyen halo, deuterio, alquilo C1-C6 o alquilo C1-C3, hidroxi, alcoxi C1-C6 o alcoxi C1-C3, ciano, amino, nitro o SF5 (un mimético conocido de NO2), arilo, heteroarilo, heterociclilo, cicloalquilo C3-C6, alquil(C1-C3)amino, alquenil(C2-C6)amino, dialquil(C1-C3)amino, acil(C1-C3)amino, diacil(C1-C3)amino, carboxi, alcoxi(C1-C3)carbonilo, carbamoilo, monocarbamoilo C1-C3, dicarbamoílo C1-C3 o cualquiera de los anteriores en los que un radical hidrocarbilo está sustituido a su vez con halo. En grupos que contienen un átomo de oxígeno tales como hidroxi y alcoxi, el átomo de oxígeno se puede reemplazar por azufre para formar grupos tales como tio (SH) y tio-alquilo (S-alquilo). Por tanto, los sustituyentes opcionales incluyen grupos tales como S-metilo. En los grupos tio-alquilo, el átomo de azufre se puede oxidar adicionalmente para formar un sulfóxido o una sulfona y, por lo tanto, los sustituyentes opcionales incluyen grupos tales como S(O)-alquilo y S(O)2-alquilo.
Los grupos sustituidos incluyen por ejemplo, Cl, F, OMe, Me, COCH3, CONH2, NHC(O)CH(CH3)2, CO2CH2CH3 etc. En el caso de grupos arilo, las sustituciones pueden ser en forma de anillos de átomos de carbono adyacentes en el anillo de arilo, por ejemplo, acetales cíclicos tales como O-CH2-O.
Los sustituyentes opcionales para cualquier grupo alquilo, alquenilo, alquinilo, alcoxi, alquileno o alquenileno descritos en la presente memoria se pueden seleccionar entre alcoxi C1-C3, halógeno, hidroxilo, tiol, ciano, amino, amido, nitro y SF5, en donde el alcoxi puede estar opcionalmente sustituido con halógeno. En particular, los sustituyentes opcionales se pueden seleccionar entre halógeno, hidroxilo, tiol, ciano, amino, amido, nitro y SF5, más concretamente flúor o hidroxilo.
Los términos "tratar" o "tratando" o "tratamiento" incluyen profilaxis y medios para mejorar, aliviar los síntomas, eliminar la causa de los síntomas, ya sea de forma temporal o permanente, o para prevenir o retrasar la aparición de los síntomas del trastorno o afección mencionados. Los compuestos de la invención son útiles en el tratamiento de seres humanos y animales no humanos.
La dosis del compuesto es aquella cantidad eficaz para prevenir la aparición de los síntomas del trastorno o para tratar algunos síntomas del trastorno que padece el paciente. Mediante "cantidad eficaz" o "cantidad terapéuticamente eficaz" o "dosis eficaz" se quiere significar la cantidad suficiente para provocar los efectos farmacológicos o terapéuticos deseados, produciendo así una prevención o tratamiento eficaz del trastorno. La prevención del trastorno se manifiesta retrasando la aparición de los síntomas del trastorno en un grado médicamente significativo. El tratamiento del trastorno se manifiesta por una disminución de los síntomas asociados con el trastorno o una mejora de la reaparición de los síntomas del trastorno.
Las sales farmacéuticamente aceptables de los compuestos de la invención incluyen, entre otras, sales de adición (p. ej., fosfatos, nitratos, sulfatos, boratos, acetatos, maleatos, citratos, fumaratos, succinatos, metanosulfonatos, benzoatos, salicilatos e hidrohaluros), sales derivadas de bases orgánicas (tales como litio, potasio y sodio), sales de aminoácidos (tales como glicina, alanina, valina, leucina, isoleucina, cisteína, metionina y prolina), bases inorgánicas (tales como trietilamina, hidróxido, colina, tiamina y N-N'-diacetiletilendiamina). Otras sales farmacéuticamente aceptables incluyen sales de amonio, sales de amonio sustituidas y sales de aluminio. Las sales farmacéuticamente aceptables adicionales incluyen sales de amonio cuaternario de los compuestos de fórmula (I) o fórmula (II).
Los expertos en la técnica conocen bien los métodos generales para la producción de sales. Tales sales se pueden formar por medios convencionales, por ejemplo, mediante la reacción de un ácido libre o una forma de base libre de un compuesto con uno o más equivalentes de un ácido o base apropiados, opcionalmente en un disolvente, o en un medio en donde la sal es insoluble, seguido de la eliminación de dicho disolvente, o dicho medio, utilizando técnicas convencionales (p. ej., a vacío, mediante liofilización o mediante filtración). Las sales también se pueden preparar intercambiando un contraión de un compuesto en forma de sal por otro contraión, por ejemplo, utilizando una resina de intercambio iónico adecuada.
Cuando los compuestos de la invención existen en diferentes formas enantioméricas y/o diastereoisoméricas, la invención se refiere a estos compuestos preparados como mezclas isoméricas o racematos, ya estén presentes en una forma ópticamente pura o como mezclas con otros isómeros. Los enantiómeros difieren solo en su capacidad para rotar la luz polarizada en el plano en cantidades iguales en direcciones opuestas y se denotan como formas (+)/(S) o (-)/(R respectivamente. Los enantiómeros o isómeros individuales pueden prepararse mediante métodos conocidos en la técnica, tales como resolución óptica de productos o intermedios (p. ej., separación cromatográfica quiral, por ejemplo, HPLC quiral o un enfoque de síntesis asimétrica). De manera similar, cuando los compuestos de la invención existen como formas tautoméricas alternativas, p. ej. ceto/enol, amida/ácido imídico, la invención se refiere a los tautómeros individuales aislados y a las mezclas de los tautómeros en todas las proporciones.
La divulgación incluye el compuesto según la fórmula (IB):

o una de sus sales farmacéuticamente aceptables, en donde n, X, R1, R2, R3, R8, R9, R10, R12 e Y se definen en la presente memoria para los compuestos de fórmula (I).
Se incluye en la presente memoria el compuesto según la fórmula (III):

o una de sus sales farmacéuticamente aceptables, en donde m, Z, R1, R2, R3, R8, R9, R10, R12 e Y se definen anteriormente para los compuestos de fórmula (II).
Isótopos
Los compuestos descritos en la presente memoria pueden contener una o más sustituciones isotópicas, y una referencia a un elemento particular incluye dentro de su alcance todos los isótopos del elemento. Por ejemplo, una referencia al hidrógeno incluye dentro de su alcance 1H, 2H(D) y 3H(T). De manera similar, las referencias al carbono y al oxígeno incluyen dentro de su alcance respectivamente 12C, 13C y 14C y 16O y 18O. Los ejemplos de isótopos incluyen 2H, 3H, 11C, 13C, 14C, 36Cl, 18F, 123I, 125I, 13N, 15N, 15O, 17O, 18O, 32P y 35S. De manera análoga, una referencia a un grupo funcional particular también incluye dentro de su alcance variaciones isotópicas, a menos que el contexto indique lo contrario. Por ejemplo, una referencia a un grupo alquilo tal como un grupo etilo también cubre variaciones en las que uno o más de los átomos de hidrógeno del grupo están en forma de un isótopo de deuterio o tritio, p. ej. como en un grupo etilo en donde los cinco átomos de hidrógeno están en la forma isotópica de deuterio (un grupo perdeuteroetilo).
Los isótopos pueden ser radiactivos o no radiactivos. En una realización, los compuestos no contienen isótopos radiactivos. Estos compuestos se prefieren para uso terapéutico. En otra realización, sin embargo, los compuestos pueden contener uno o más radioisótopos. Los compuestos que contienen tales radioisótopos pueden ser útiles en un contexto de diagnóstico.
Ciertos compuestos marcados isotópicamente de fórmula (I) o fórmula (II), por ejemplo, los que incorporan un isótopo radiactivo, son útiles en estudios de fármacos y/o distribución en tejidos sustrato. Los isótopos radiactivos, es decir, 3H y 14C son particularmente útiles para este propósito en vista de su facilidad de incorporación y medios fáciles de detección. La sustitución por isótopos más pesados, es decir, 2H, puede proporcionar ciertas ventajas terapéuticas resultantes de una mayor estabilidad metabólica, por ejemplo, aumento de semivida in vivo o reducción de los requisitos de dosificación y, por lo tanto, puede ser preferible en algunas circunstancias. La sustitución por isótopos emisores de positrones, tales como 11C, 18F, 15O y 13N, puede ser útil en estudios de topografía de emisión de positrones (PET) para examinar la ocupación del receptor. Los compuestos marcados isotópicamente de fórmula (I) o fórmula (II) generalmente se pueden preparar mediante mecanismos convencionales conocidos por los expertos en la técnica o mediante procedimientos análogos a los descritos en los ejemplos y preparaciones adjuntos utilizando un reactivo marcado isotópicamente apropiado en lugar del reactivo no etiquetado empleado previamente.
Formas cristalinas y amorfas
Los compuestos de fórmula (I) o fórmula (II) pueden existir en forma cristalina o amorfa y algunas de las formas cristalinas pueden existir como polimorfos, que se incluyen dentro del alcance de la presente invención. Las formas polimórficas de los compuestos de fórmula (I) o fórmula (II) se pueden caracterizar y diferenciar utilizando una serie de técnicas analíticas convencionales, que incluyen, pero no se limitan a, espectros infrarrojos, espectros Raman, difracción de rayos X de polvo, calorimetría de barrido diferencial, análisis termogravimétrico y resonancia magnética nuclear en estado sólido.
Por consiguiente, en realizaciones adicionales, la invención proporciona un compuesto según cualquiera de las realizaciones descritas en forma cristalina. El compuesto puede ser de 50% a 100% cristalino, y más concretamente es al menos 50% cristalino, o al menos 60% cristalino, o al menos 70% cristalino, o al menos 80% cristalino, o al menos 90% cristalino, o al menos 95% cristalino, o al menos 98% cristalino, o al menos 99% cristalino, o al menos 99,5% cristalino, o al menos 99,9% cristalino, por ejemplo, 100% cristalino. Alternativamente, el compuesto puede estar en forma amorfa.
La invención descrita en la presente memoria se refiere a todas las formas cristalinas, solvatos e hidratos de cualquiera de los compuestos descritos, independientemente de su preparación. En la medida en que cualquiera de los compuestos descritos en la presente memoria tenga centros ácidos o básicos tales como grupos carboxilato o amino, se incluyen en la presente memoria todas las formas de sal de dichos compuestos. En el caso de usos farmacéuticos, se debe considerar que la sal es una sal farmacéuticamente aceptable.
La invención se refiere a cualquier solvato de los compuestos y sus sales. Los solvatos preferidos son solvatos formados por la incorporación en la estructura en estado sólido (p. ej., estructura cristalina) de los compuestos de la invención de moléculas de un disolvente farmacéuticamente aceptable no tóxico (denominado a continuación disolvente solvatante). Los ejemplos de tales disolventes incluyen agua, alcoholes (tales como etanol, isopropanol y butanol) y dimetilsulfóxido. Los solvatos se pueden preparar recristalizando los compuestos de la invención con un disolvente o una mezcla de disolventes que contienen el disolvente solvatante. Se puede determinar si se ha formado o no un solvato en un caso dado sometiendo cristales del compuesto a análisis utilizando técnicas bien conocidas y convencionales tales como análisis termogravimétrico (TGE), calorimetría diferencial de barrido (DSC) y cristalografía de rayos X.
Los solvatos pueden ser solvatos estequiométricos o no estequiométricos. Los solvatos particulares pueden ser hidratos, y los ejemplos de hidratos incluyen hemihidratos, monohidratos y dihidratos. Para una discusión más detallada de los solvatos y los métodos utilizados para prepararlos y caracterizarlos, véase Bryn et al., Solid-State Chemistry of Drugs, Segunda edición, publicado por s Sc I, Inc de West Lafayette, IN, EE.UU., 1999, ISBN 0-967-06710-3.
La divulgación incluye derivados farmacéuticamente funcionales de compuestos como se definen en la presente memoria, incluidos derivados éster y/o derivados que tienen, o proporcionan, la misma función y/o actividad biológica que cualquier compuesto relevante de la invención. Por tanto, para los propósitos de esta descripción, el término también incluye profármacos de compuestos como se definen en la presente memoria.
El término "profármaco" de un compuesto relevante incluye cualquier compuesto que, después de la administración oral o parenteral, se metabolice in vivo para formar ese compuesto en una cantidad detectable experimentalmente, y dentro de un tiempo predeterminado (p. ej., dentro de un intervalo de dosificación de entre 6 y 24 horas (es decir, de una a cuatro veces al día).
Los profármacos de los compuestos se pueden preparar modificando grupos funcionales presentes en el compuesto de tal manera que las modificaciones se escinden, in vivo cuando tal profármaco se administra a un sujeto mamífero. Las modificaciones se logran típicamente sintetizando el compuesto original con un sustituyente de profármaco. Los profármacos incluyen compuestos en los que un grupo hidroxilo, amino, sulfhidrilo, carboxilo o carbonilo de un compuesto está unido a cualquier grupo que pueda escindirse in vivo para regenerar el grupo hidroxilo, amino, sulfhidrilo, carboxilo o carbonilo libre, respectivamente.
Los ejemplos de profármacos incluyen, pero no se limitan a, ésteres y carbamatos de grupos funcionales hidroxilo, grupos éster de grupos funcionales carboxilo, derivados N-acilo y bases de N-Mannich. Se puede encontrar información general sobre profármacos, p. ej. en Bundegaard, H. "Design of Prodrugs" pág. 1-92, Elsevier, Nueva York-Oxford (1985).
Los compuestos de la invención se pueden metabolizar in vivo. Los metabolitos de los compuestos de fórmula (I) y fórmula (II) también están dentro del alcance de la presente divulgación. El término "metabolitos" se refiere a todas las moléculas derivadas de cualquiera de los compuestos según la presente invención en una célula u organismo, preferiblemente mamífero. Preferiblemente, el término se refiere a moléculas que difieren de cualquier molécula que esté presente en cualquier célula u organismo en condiciones fisiológicas.
Un tratamiento definido en la presente memoria se puede aplicar como única terapia o puede implicar, además de los compuestos de la invención, cirugía convencional o radioterapia o quimioterapia. Además, los compuestos de fórmula (I) o fórmula (II) también se pueden utilizar combinados con agentes terapéuticos existentes para el tratamiento de afecciones asociadas con disfunción mitocondrial y cáncer, incluyendo terapias de moléculas pequeñas o terapias basadas en anticuerpos.
Los compuestos descritos en la presente memoria se caracterizan por un núcleo de cianopirrolidina o cianopiperidina.
La divulgación incluye compuestos que tienen la fórmula (I)

o una de sus sales farmacéuticamente aceptables, en donde:
n es 1 o 2 ;
cuando n es 1 , X es C R 4R5 y cuando n es 2 , X es C R 6R7C R 4 R5 (donde C R 4R5 es adyacente al átomo de N del heterociclo);
R2 representa un átomo de hidrógeno, ciano, un alquilo C1-C6 opcionalmente sustituido, un grupo alcoxi C1-C6 opcionalmente sustituido, un anillo de heteroarilo, heterociclilo, arilo de 4 a 10 miembros o cicloalquilo de 3 a 8 miembros opcionalmente sustituido;
R1, R3, R4 y R5 representan cada uno independientemente un átomo de hidrógeno, ciano, un grupo alquilo C1-C3 opcionalmente sustituido o alcoxi C1-C3 opcionalmente sustituido;
R6 , R7 y R8 representan cada uno independientemente un átomo de hidrógeno, un átomo de flúor, ciano, un grupo alquilo C1-C3 opcionalmente sustituido o alcoxi C1-C3 opcionalmente sustituido;
R9 representa un átomo de hidrógeno, un átomo de flúor, ciano, un alquilo C1-C6 opcionalmente sustituido, un grupo alcoxi C1-C3 opcionalmente sustituido, un heteroarilo, heterociclilo, arilo de 4 a 10 miembros o cicloalquilo de 3 a 8 miembros opcionalmente sustituidos, o forma un anillo heterocíclico opcionalmente sustituido con R 10 en donde el anillo comprende opcionalmente uno o más heteroátomos adicionales;
R10 representa un átomo de hidrógeno, alquilo C1-C6 o forma un anillo heterocíclico opcionalmente sustituido con R9 o R 11 en donde el anillo comprende opcionalmente uno o más heteroátomos adicionales;
Y representa un enlace covalente, NR11 o alquileno C1-C3 opcionalmente sustituido;
R11 representa un átomo de hidrógeno, un alquilo C1-C6 opcionalmente sustituido, un anillo de heteroarilo, heterociclilo, arilo de 4 a 10 miembros o cicloalquilo de 3 a 8 miembros, o forma un anillo heterocíclico opcionalmente sustituido con R10 en donde el anillo comprende opcionalmente uno o más heteroátomos adicionales;
R12 representa un anillo de heteroarilo, heterociclilo, arilo de 4 a 10 miembros o de cicloalquilo de 3 a 8 miembros sustituido.
En un primer aspecto, la presente invención proporciona un compuesto que tiene la fórmula (II)

un tautómero del mismo, o una sal farmacéuticamente aceptable de dicho compuesto o tautómero, en donde: m es 0 o 1 ;
cuando m es 1 , Z es -C(R6)(R 7)-;
R2 representa un átomo de hidrógeno, un grupo alquilo C1-C6, o alcoxi C1-C6 opcionalmente sustituidos;
R3, R4 y R5 representan cada uno independientemente un átomo de hidrógeno, un grupo alquilo C1-C3, o alcoxi C1-C3 opcionalmente sustituidos;
R1, R6, R7 y R8 representan cada uno independientemente un átomo de hidrógeno, un átomo de flúor, ciano, hidroxilo, un grupo alquilo C1-C3, o alcoxi C1-C3 opcionalmente sustituidos;
R9 representa un átomo de hidrógeno, un átomo de flúor, ciano, hidroxilo, alquilo C1-C6, un grupo alcoxi C1-C3, o un anillo de cicloalquilo de 3 a 8 miembros, o forma un anillo heterocíclico con R10;
R10 representa un átomo de hidrógeno, alquilo C1-C6 opcionalmente sustituido, o forma un anillo heterocíclico con R9, o forma un anillo monocíclico con R11;
Y representa un enlace covalente, -alquileno(C0-C3)-N(R11)-alquileno C0-C3 o alquileno C1-C3;
R11 representa un átomo de hidrógeno, alquilo C1-C6, o forma un anillo monocíclico con R10; y
en donde R12 se define como en los aspectos de la invención (i), (ii) y (iii), a continuación.
Según aspecto de la invención (i):
R12 representa un anillo de heteroarilo, heterociclilo o cicloalquilo de 3 a 14 miembros monocíclico sustituido, bicíclico opcionalmente sustituido o tricíclico opcionalmente sustituido;
en donde R12, cuando está sustituido, está sustituido con uno o más de -Q1-(R13)p, en donde:
p es 0 o 1;
Q1 representa un átomo de halógeno, ciano, oxo, hidroxilo, un enlace covalente, -alquilen(C0-C3)-NR14-, -alquilen(C0-C3)-NR14R15, -alquilen(C0-C3)-CONR14-, -alquilen(C0-C3)-NR14CO-, -alquilen(C0-C3)-NR14SO2-, un átomo de oxígeno, -alquilen(C0-C3)-CO-, -alquilen(C0-C3)-S(O)q-, -alquilen(C0-C3)-SO2NR14, -alquilen(C0-C3)-SO2NR14R15, -alcoxi C1-C6, haloalcoxi C1-C6, hidroxialquilo C1-C6, -alquilen(C0-C3)-SO2R14, -alquilen(C0-C3)-NR14COR15, -alquilen(C0-C3)-NR14CONR15R16, -alquilen(C0-C3)-NR14SO2NR15R16, -alquilen(C0-C3)-CONR14R15, -alquilen(C0-C3)-CO2R14, -alquilen(C0-C3)-NR14CO2R15, -alquilen(C0-C3)-SO2NR14R15, -alquilen(C0-C3)-C(O)R14 y alquilen(C1-C6)-NR14SO2R15, NO2, alquileno C1-C6, -alquenileno C2-C6, o un -grupo alquilo C1-C6 opcionalmente sustituido;
q es 0, 1 o 2;
R14, R15 y R16 representan cada uno independientemente un átomo de hidrógeno, alquilo C1-C6, o un grupo alquileno C1-C6;
cuando p es 1, R13 representa un anillo de heteroarilo, heterociclilo, arilo de 4 a 10 miembros o cicloalquilo de 3 a 8 miembros; en donde R13 puede estar opcionalmente sustituido con uno o más sustituyentes seleccionados entre halógeno, haloalquilo C1-C6, alcoxi C1-C6, haloalcoxi C1-C6, alquilo C1-C6, alquenilo C2-C6, alquinilo C2-C6, hidroxialquilo C1-C6, oxo, ciano, -Q2-R17, -Q2-NR17CONR18R19, -Q2-NR17R18, -Q2-COR17, -Q2-NR17COR18, -Q2-NR17CO2R18, -Q2-SO2R17, Q2-CONR17R18, -Q2-CO2R17, -Q2-SO2NR17R18, -Q2-NR17SO2R18, heterociclilo, cicloalquilo, heteroarilo y arilo; en donde dichos anillos de heterociclilo, cicloalquilo, heteroarilo y arilo pueden estar opcionalmente sustituidos con uno o más sustituyentes, que pueden ser iguales o diferentes, seleccionados entre Cl, F, OMe, Me, COCH3, CONH2, NHC(O)CH(CH3)2, y CO2CH2CH3 ;
Q2 representa un enlace covalente, un átomo de oxígeno, -CO- o un grupo alquileno C1-C6 o alquenileno C2-C6; y R16, R17, R18 representan cada uno independientemente hidrógeno, alquilo C1-C6, heterociclilo, heteroarilo, arilo o cicloalquilo.
Según aspecto de la invención (ii):
R12 representa un anillo de arilo de 3 a 14 miembros monocíclico sustituido, bicíclico opcionalmente sustituido o tricíclico opcionalmente sustituido;
donde R12, cuando está sustituido, está sustituido con uno o más de -Q1-(R13)p, en donde:
p es 0 o 1;
Q1 representa un átomo de halógeno, ciano, oxo, hidroxilo, un enlace covalente, -alquilen(C0-C3)-NR14-, -alquilen(C0-C3)-NR14R15, -alquilen(C0-C3)-CONR14-, -alquilen(C0-C3)-NR14CO-, -alquilen(C0-C3)-NR14SO2-, un átomo de oxígeno, -alquilen(C0-C3)-CO-, -alquilen(C0-C3)-S(O)q-, -alquilen(C0-C3)-SO2NR14R15, -alcoxi C1-C6, haloalcoxi C1-C6, hidroxialquilo C1-C6, -alquilen(C0-C3)-SO2R14, -alquilen(C0-C3)-NR14COR15, -alquilen(C0-C3)-NR14CONR15R16, -alquilen(C0-C3)-NR14SO2NR15R16, -alquilen(C0-C3)-CONR14R15, -alquilen(C0-C3)-CO2R14, -alquilen(C0-C3)
NR14CO2R15, -alquilen(C0-C3)-SO2NR14R15, -alquilen(C0-C3)-C(O)R14 y alquilen(Ci -C6)-NR14SO2R15, NO2, alquileno C1-C6, -alquenileno C2-C6, o un -grupo alquilo C1-C6 opcionalmente sustituido;
q es 0, 1 o 2;
R14, R15 y R16 representan cada uno independientemente un átomo de hidrógeno, alquilo C1-C6, o un grupo alquileno C1-C6;
cuando p es 1, R13 representa un anillo de heteroarilo de 4 a 10 miembros, heterociclilo o cicloalquilo de 3 a 8 miembros; en donde R13 puede estar opcionalmente sustituido con uno o más sustituyentes seleccionados entre halógeno, haloalquilo C1-C6, alcoxi C1-C6, haloalcoxi C1-C6, alquilo C1-C6, alquenilo C2-C6, alquinilo C2-C6, hidroxialquilo C1-C6, oxo, ciano, -Q2-R17, -Q2-NR17CONR18R19, -Q2-NR17R18, -Q2-COR17, -Q2-NR17COR18, -Q2-NR17CO2R18, -Q2-SO2R17, Q2-CONR17R18, -Q2-CO2R17, -Q2-SO2NR17R18, -Q2-NR17SO2R18, heterociclilo, cicloalquilo, heteroarilo y arilo; en donde dichos anillos de heterociclilo, cicloalquilo, heteroarilo y arilo pueden estar opcionalmente sustituidos con uno o más sustituyentes, que pueden ser iguales o diferentes, seleccionados entre Cl, F, OMe, Me, COCH3, CONH2, NHC(O)CH(CH3)2 , y CO2CH2CH3;
Q2 representa un enlace covalente, un átomo de oxígeno, -CO- o un grupo alquileno C1-C6 o alquenileno C2-C6; R16, R17, R18 representan cada uno independientemente hidrógeno, alquilo C1-C6, heterociclilo, heteroarilo, arilo o cicloalquilo; y
donde el compuesto no tiene la fórmula:

Según aspecto de la invención (iii):
R12 representa un anillo de arilo monocíclico, bicíclico o tricíclico de 3 a 14 miembros;
en donde R12 está sustituido con uno o más de -Q1-(R13)p, en donde:
p es 1;
Q1 representa ciano, oxo, hidroxilo, un enlace covalente, -alquilen(C0-C3)-NR14-, -alquilen(C0-C3)-NR14R15, -alquilen(C0-C3)-CONR14-, -alquilen(C0-C3)-NR14CO-, -alquilen(C0-C3)-NR14SO2-, un átomo de oxígeno, -alquilen(C0-C3)-CO-, -alquilen(C0-C3)-S(O)q-, -alquilen(C0-C3)-SO2NR14, -alquilen(C0-C3)-SO2NR14R15, -alcoxi C1-C6, haloalcoxi C1-C6 , hidroxialquilo C1-C6, -alquilen(C0-C3)-SO2R14, -alquilen(C0-C3)-NR14COR15, -alquilen(C0-C3)-NR14CONR15R16, -alquilen(C0-C3)-NR14SO2NR15R16, -alquilen(C0-C3)-CONR14R15, -alquilen(C0-C3)-CO2R14, -alquilen(C0-C3)-NR14CO2R15, -alquilen(C0-C3)-SO2NR14R15, -alquilen(C0-C3)-C(O)R14 y alquilen(CrC6)-NR14SO2R15, NO2, alquileno C1-C6, -alquenileno C2-C6, o un -grupo alquilo C1-C6 opcionalmente sustituido;
q es 0, 1 o 2;
R14, R15 y R16 representan cada uno independientemente un átomo de hidrógeno, alquilo C1-C6, o un grupo alquileno C1-C6;
cuando p es 1, R13 representa un anillo de arilo de 4 a 10 miembros; en donde R13 está sustituido con uno o más sustituyentes seleccionados entre haloalquilo C1-C6, alcoxi C1-C6, haloalcoxi C1-C6, alquilo C1-C6, alquenilo C2-C6, alquinilo C2-C6, hidroxialquilo C1-C6, oxo, ciano, -Q2-R17, -Q2-NR17CONR18R19, -Q2-NR17R18, -Q2-COR17, -Q2-NR17COR18, -Q2-NR17CO2R18, -Q2-SO2R17, Q2-CONR17R18, -Q2-CO2R17, -Q2-SO2NR17R18, -Q2-NR17SO2R18, heterociclilo, cicloalquilo, heteroarilo y arilo; en donde dichos anillos de heterociclilo, cicloalquilo, heteroarilo y arilo pueden estar opcionalmente sustituidos con uno o más sustituyentes, que pueden ser iguales o diferentes, seleccionados entre Cl, F, OMe, Me, COCH3, CONH2, NHC(O)CH(CH3)2, y CO2CH2CH3;
Q2 representa un enlace covalente, un átomo de oxígeno, -CO- o un grupo alquileno C1-C6 o alquenileno C2-C6; y R16, R17, R18 representan cada uno independientemente hidrógeno, alquilo C1-C6, heterociclilo, heteroarilo, arilo o
cicloalquilo.
Para los aspectos de la invención (i), (ii) y (iii), los sustituyentes opcionales de alquilo de R1, R2, R3, R4, R5, R6, R7, R8, R10 y Q1, que pueden ser iguales o diferentes, se seleccionan entre alcoxi C1-C3, halógeno, hidroxilo, tiol, ciano, amino, amido, nitro y SF5.
En una realización, R1 representa un átomo de hidrógeno. En otra realización, R1 representa metilo C1-C3, En otra realización, R1 representa metilo.
En una realización, R2 representa alquilo C1-C3, En otra realización R2 representa C1-C2 (p. ej., metilo o etilo). En otra realización, R2 representa metilo. En otra realización, R2 representa hidroxilo. En otra realización, R2 representa alquilo C1-C3, alquilo C1-C2 (p. ej., metilo o etilo) o hidroxilo y R1, R3, R4, R5, R8, R9 y R6 y R7 (si están presentes), representan cada uno independientemente un átomo de hidrógeno En otra realización R2 representa alquilo C1-C3 o alquilo C1-C2 (p. ej., metilo o etilo) y R1, R3, R4, R5, R8, R9 y R6 y R7 (si están presentes), representan cada uno independientemente un átomo de hidrógeno.
En una realización, R5 representa alquilo C1-C3, En otra realización R5 representa alquilo C1-C2 (p. ej., metilo o etilo). En otra realización, R5 representa metilo. En otra realización R5 representa alquilo C1-C3 o alquilo C1-C2 (p. ej., metilo o etilo) y R1, R2, R3, R4, R8, R9 y R6 y R7 (si están presentes) representan cada uno independientemente un átomo de hidrógeno.
En una realización, R6 y R7 cuando están presentes representan hidrógeno.
En una realización, R8 representa alquilo C1-C3, En otra realización R8 representa alquilo C1-C2 (p. ej., metilo o etilo). En otra realización, R8 representa metilo. En otra realización R8 representa alquilo C1-C3, alquilo C1-C2 (p. ej., metilo o etilo) o un átomo de flúor y R1, R2, R3, R4 , R5, R9 y R6 y R7 (si están presentes) representan cada uno independientemente un átomo de hidrógeno.
En una realización, R9 representa alquilo C1-C3, En otra realización R9 representa alquilo C1-C2 (p. ej., metilo o etilo). En otra realización, R9 representa metilo. En otra realización, R9 representa alcoxi C1-C3, En otra realización, R9 representa alcoxi C1-C2 (p. ej., metoxi o etoxi). En otra realización, R9 representa metoxi. En otra realización, R9 representa ciclopropilo. En otra realización, R9 representa un átomo de hidrógeno, un átomo de flúor, ciano, hidroxilo, un alquilo C1-C6 opcionalmente sustituido, un grupo alcoxi C1-C3 opcionalmente sustituido o un cicloalquilo C3-C4, En otra realización R9 representa alquilo C1-C3, alquilo C1-C2 (p. ej., metilo o etilo), un átomo de flúor o ciclopropilo y R1, R2, R3, R4, R5, R8 y R6 y R7 (si están presentes), representan cada uno independientemente un átomo de hidrógeno. En una realización, R9 no es fenilo, en particular, no es difluorofenilo.
El alquilo y alcoxi dentro de las definiciones de R1, R2, R3, R4, R5, R6, R7, R8 y R9 pueden estar opcionalmente sustituidos con uno o más sustituyentes seleccionados entre halógeno, hidroxilo, tiol, ciano, amino, nitro y SF5.
En una realización, R9 forma un anillo heterocíclico opcionalmente sustituido con R10 en donde el anillo comprende opcionalmente uno o más heteroátomos adicionales. En una realización, R9 forma un anillo heterocíclico de 5 miembros con R10- En otra realización, R9 forma un anillo heterocíclico de 6 miembros con R10 en donde el anillo comprende adicionalmente un heteroátomo de oxígeno. En otra realización, R9 forma un anillo heterocíclico opcionalmente sustituido con R10 donde el anillo comprende opcionalmente uno o más heteroátomos adicionales y R1, R2, R3, R4, R5, R8 y R6 y R7 (si están presentes) representan cada uno independientemente un átomo de hidrógeno. El anillo formado por R9 y R10 puede estar opcionalmente sustituido con uno o más de los sustituyentes definidos en la presente memoria. En una realización, los sustituyentes opcionales se seleccionan entre alquilo C1-C3, alcoxi C1-C3, halógeno, hidroxilo, tiol, ciano, amino, amido, nitro y SF5, en donde el alquilo y el alcoxi pueden estar opcionalmente sustituidos con halógeno.
En una realización, R10 representa un átomo de hidrógeno, alquilo C1-C6 opcionalmente sustituido o forma un anillo heterocíclico opcionalmente sustituido con R9 o R11 en donde el anillo comprende opcionalmente uno o más heteroátomos adicionales. En una realización, R10 representa un átomo de hidrógeno. En otra realización R10 representa alquilo C1-C3, En otra realización, R10 representa metilo. En otra realización, R10 representa etilo. En otra realización, el alquilo C1-C6 puede estar opcionalmente sustituido. Los sustituyentes opcionales para el alquilo se pueden seleccionar entre alcoxi C1-C3, halógeno, hidroxilo, tiol, ciano, amino, amido, nitro y SF5, en donde el alcoxi puede estar opcionalmente sustituido con halógeno, en particular flúor. En particular, el C1-C3 puede estar opcionalmente sustituido con alcoxi C1-C3, por ejemplo, metoxi. En una realización, R10 representa CH2CH2OCH3, En otra realización más, R10 forma un anillo heterocíclico de 5 miembros con R9' En otra realización, R10 forma un anillo heterocíclico de 6 miembros con R9 en donde el anillo comprende adicionalmente un heteroátomo de oxígeno.
En una realización, Y es un enlace covalente, -NR11- o alquileno C1-C3, En una realización, Y es un enlace covalente o alquileno C1-C3, En una realización, Y es un enlace covalente. En otra realización, Y representa alquileno C1-C2 (p. ej., metileno o etileno). En otra realización, Y es metileno. En otra realización, Y es -NH-.
R11 representa un átomo de hidrógeno, un alquilo C1-C6 opcionalmente sustituido, un anillo de heteroarilo, heterociclilo, arilo de 4 a 10 miembros o cicloalquilo de 3 a 8 miembros o forma un anillo heterocíclico monocíclico o bicíclico
opcionalmente sustituido con R10 con la condición de que cuando el anillo sea bicíclico no esté sustituido con NH2, En una realización, R11 es hidrógeno, alquilo C1-C6 o forma un anillo monocíclico de 5 o 6 miembros con R10
En una realización, R11 y R10 juntos forman un anillo de heterociclilo. El anillo puede ser monocíclico o bicíclico. En particular, cuando R11 y R10 juntos forman un anillo de heterociclilo, el anillo es un anillo monocíclico de 5 o 6 miembros.
En una realización, R11 y R10 juntos forman un anillo heterocíclico de 5 miembros. En otra realización, R11 y R10 juntos forman un anillo heterocíclico de 6 miembros. En una realización, cuando R11 y R10 juntos forman un anillo heterocíclico, el anillo no es dihidropurina.
Los compuestos de fórmula II pueden estar en la forma en la que m es 0 , es decir, en donde la estructura del núcleo es una cianopirrolidina. En tales casos, los compuestos pueden tener la fórmula:

o una de sus sales farmacéuticamente aceptables, en donde R1, R2, R3, R4, R5, R8, R9, R10, R12 e Y son como se definen en la presente memoria para los compuestos de fórmula II.
Alternativamente, los compuestos de fórmula II pueden estar en la forma en la que m es 1 , es decir, la estructura del núcleo es una cianopiperidina. En tales casos, los compuestos pueden tener la fórmula:

o una de sus sales farmacéuticamente aceptables, en donde R1, R2, R6, R7, R8, R se definen en la presente memoria para los compuestos de fórmula (II).
R6, R7, R8, R se definen en la presente memoria para los compuestos de fórmula (II).
Cuando m es 0 , los compuestos de fórmula II pueden estar en la forma en la que R9 y R10 juntos forman un anillo de heterociclilo de 5 miembros que se fusiona con el núcleo de cianopirrolidina para crear un anillo bicíclico de 8 miembros. En particular, Y puede ser un enlace covalente y R1, R2, R3, R4, R5 y R8 son cada uno de hidrógeno. En tales casos, los compuestos pueden tener la fórmula:

(IIC )
o una de sus sales farmacéuticamente aceptables, en donde R12 es como se ha definido en la presente memoria para los compuestos de fórmula (II).
En una realización adicional de la divulgación se proporciona un compuesto de fórmula IID:

(IID )
o una de sus sales farmacéuticamente aceptables, en donde R12 se ha definido anteriormente para los compuestos de fórmula (I) o fórmula (II).
Para los compuestos de fórmula (II), R 12 es un anillo de 3 a 14 miembros (p. ej., 3 , 4 , 5 , 6 , 7 , 8 , 9 , 1 0 , 11 , 12 , 13 o 14 miembros). Cuando R12 es un anillo monocíclico, el anillo debe estar sustituido. Cuando R12 es un anillo bicíclico o tricíclico, el anillo puede estar sustituido o no sustituido. En una realización, R 12 es un anillo sustituido.
Cuando R 12 es un anillo de heteroarilo o arilo sustituido, el anillo puede ser monocíclico, bicíclico o tricíclico y, en el caso de un anillo de heteroarilo, comprende uno o más (p. ej., 1 , 2 o 3 ) heteroátomos seleccionados entre nitrógeno, oxígeno y azufre, en particular nitrógeno.
Cuando R12 es un anillo de heteroarilo o arilo sustituido, el anillo puede ser monocíclico o bicíclico y, en el caso de un anillo de heteroarilo, comprende uno o más (p. ej., 1 , 2 o 3 ) heteroátomos seleccionados independientemente entre nitrógeno, oxígeno y azufre.
En una realización, R 12 se selecciona entre fenilo, pirrolidinilo, tiazolilo, piridinilo, isoxazolilo, oxazolilo, imidazolilo, pirazolilo, piridazinilo, pirimidinilo, indolilo, benzimidazolilo, quinolinilo, azetidinilo, indazolilo, pirazolopiridinilo, imidazopiridinilo, indolinilo, piperazinilo, morfolinilo, diazepanilo, tetrahidropiridoindolilo, benzomorfolinilo y pirrolopiridinilo. En una realización, R 12 se selecciona entre fenilo, pirrolidinilo, tiazolilo, piridinilo, isoxazolilo, oxazolilo, imidazolilo, pirazolilo, piridazinilo, pirimidinilo, indolilo, benzimidazolilo y quinolinilo.
Los ejemplos típicos de R12 incluyen fen-3 -ilo, fen-4 -ilo, pirrolidin-1 -ilo, tiazol-2 -ilo, tiazol-5 -ilo, piridin-2 -ilo, piridin-3 -ilo, piridin-4 -ilo, isoxazol -5 -ilo, oxazol-2 -ilo, pirazol-4 -ilo, pirazol-5 -ilo, pirimidin-2 -ilo, piraradazin-3 -ilo, imidazol-4 -ilo, indol-2 -ilo, bencimidazol-2 -il, quinolin-4 -ilo y quinolin-6 -ilo.
En particular, R 12 se puede seleccionar entre azetidinilo y pirrolidinilo. En una realización, R12 es azetidinilo. En otra realización, R 12 es pirrolidinilo. Cuando R12 es azetidinilo o pirrolidinilo, preferiblemente Y es un enlace covalente que está anclado al átomo de nitrógeno del anillo de azetidinilo o pirrolidinilo.
En una realización, cuando R 12 es piridinilo, el piridinilo es piridin-2 -ilo.
Los ejemplos de R 12 incluyen los que se muestran a continuación:


Donde * denota el punto de anclaje directo al núcleo de cianopirrolidina o cianopiperidina a través de -Y-C(O) N(R10)-. Los anillos monocíclicos están sustituidos con al menos un -Q1-(R13)p y los anillos bicíclicos y tricíclicos pueden estar no sustituidos o sustituidos con uno o más sustituyentes -Q1-(R13)p como se describe en la presente memoria. No se han mostrado los átomos de hidrógeno anclados a los átomos de nitrógeno del anillo. El experto en la técnica entenderá qué átomos de nitrógeno del anillo son adecuados para su sustitución y, cuando no esté sustituido, el nitrógeno se puede unir a un átomo de hidrógeno para completar su valencia, cuando sea apropiado.
Los ejemplos adicionales de R12 incluyen los que se muestran a continuación:


Cuando está sustituido, R12 puede estar sustituido con uno o más -Q1-(R13)p, en donde -Q1-(R13)p en cada caso puede ser igual o diferente.
p es 0 o 1 (cuando p es 1, Q1 es un enlace covalente o conector y R13 está presente, cuando p es 0, Q1 está presente y R13 está ausente).
Preferiblemente, p es 1.
Q1 representa un átomo de halógeno, ciano, oxo, hidroxilo, un enlace covalente, -alquilen(C0-C3)-NR14-, -alquilen(C0-C3)-NR14R15,-alquilen(C0-C3)-CONR14-, -alquilen(C0-C3)-NR14CO-, -alquilen(C0-C3)-NR14SO2-, -alquilen(C0-C3)-O-alquileno C0-C3, -alquilen(C0-C3)-CO-, -alquilen(C0-C3)-S(O)q-, -alquilen(C0-C3)-SO2NR14, -alcoxi C1-C6, haloalcoxi C1-C6, hidroxialquilo C1-C6, -alquilen(C0-C3)-SO2R14, -alquilen(C0-C3)-NR14COR15, -alquilen(C0-C3)-NR14CONR15R16, -alquilen(C0-C3)-NR14SO2NR15R16, -alquilen(C0-C3)-CONR14R15, -alquilen(C0-C3)-CO2R14, -alquilen(C0-C3)-NR14CO2R15, -alquilen(C0-C3)-SO2NR14R15, -alquilen(C0-C3)-CONR14, -alquilen(C0-C3)-C(O)R14 y alquilen(C1-C6)-NR14SO2R15, NO2, o un grupo alquileno C1-C6, -alquenileno C2-C6 o -alquilo C1-C6 opcionalmente sustituido; en donde q es 0, 1 o 2.
Q1 puede representar un átomo de halógeno, ciano, oxo, un enlace covalente, -NR14-, -NR14R15, -CONR14-, -NR14CO-, un átomo de oxígeno, -CO-, -S(O)q-, -SO2NR14, -alcoxi C1-C6, haloalcoxi C1-C6, hidroxialquilo C1-C6, -SO2R14, -NR14COR15, -NR14CONR15R16, -NR14SO2NR15R16 -CONR14R15, -CO2R14, -NR14CO2R15, -SO2NR14R15, -CONR14, -C(O)R14 y -NR14SO2R15, NO2, o un grupo alquileno C1-C6, -alquenileno C2-C6 o -alquilo C1-C6 opcionalmente sustituido; en donde
q es 0, 1 o 2.
En una realización, Q1 representa un átomo de halógeno, ciano, oxo, un enlace covalente, un átomo de oxígeno, un grupo -alquilen(C0-C3)-O-alquileno C0-C3, alcoxi C1-C6, alcoxi C1-C4, alcoxi C1-C2, haloalcoxi C1-C6, haloalcoxi C1-C4,
haloalcoxi C1-C2, -hidroxialquilo C1-C6, hidroxialquilo C1-C4, hidroxialquilo C1-C2, alquileno C1-C6, alquileno C1-C4, alquileno C1-C2, alquenileno C2-C6 o alquenileno C2-C4 que puede estar opcionalmente sustituido con hidroxi, un átomo de halógeno (p. ej., flúor, cloro o bromo), alquilo C1-C6, alquilo C1-C4, alquilo C1-C2, haloalquilo C1-C6, haloalquilo C1-C4, haloalquilo C1-C2, NR14-, -NR14R15-, -CONR14-, -NR14CO-, CO-, -S(O)q-, -SO2NR14,
-NR14SO2-, -SO2R14, -NR14COR15, -NR14CONR15R16, -NR14SO2NR15R16 -CONR14R15, -CO2R14, -NR14CO2R15, -SO2NR14R15, -CONR14, -C(O)R14, -NR14SO2R15, o NO2.
En una realización, Q1 representa un átomo de halógeno, ciano, oxo, un enlace covalente, un átomo de oxígeno, un grupo -O-metileno, -O-etileno, alcoxi C1-C6, alcoxi C1-C4, alcoxi C1-C2, haloalcoxi C1-C6, haloalcoxi C1-C4, haloalcoxi C1-C2, -hidroxialquilo C1-C6, hidroxialquilo C1-C4, hidroxialquilo C1-C2, alquileno C1-C6, alquileno C1-C4, alquileno C1-C2, alquenileno C2-C6 o alquenileno C2-C4 que puede estar opcionalmente sustituido con hidroxi, un átomo de halógeno (p. ej., flúor, cloro o bromo), alquilo C1-C6, alquilo C1-C4, alquilo C1-C2, haloalquilo C1-C6, haloalquilo C1-C4, haloalquilo C1-C2, NR14-, -NR14R15-, -CONR14-, -NR14CO-, CO-, -S(O)q-, -SO2NR14, -SO2R14,
-NR14COR15, -NR14CONR15R16, -NR14SO2NR15R16 -CONR14R15, -CO2R14, -NR14CO2R15, -SO2NR14R15, -CONR14, -C(O)R14, -NR14SO2R15, o NO2.
En otra realización, Q1 se selecciona entre halógeno, ciano, oxo, alquilo C1-C6 opcionalmente sustituido con flúor, alcoxi C1-C6 opcionalmente sustituido con flúor, -NR14COR15, un enlace covalente, un átomo de oxígeno, -alquilen(C0-C3)-O-alquileno C0-C3, -NR14-, alquileno C1-C6, -NR14SO2- y -NR14R15-.
En otra realización, Q1 se selecciona entre halógeno, oxo, un enlace covalente, -NR14R15-, un átomo de oxígeno, alcoxi C1-C6, alcoxi C1-C4, alcoxi C1-C2, -NR14COR15 o alquilo C1-C6, alquilo C1-C4 o alquilo C1-C2.
R14, R15 y R16 representan cada uno independientemente un átomo de hidrógeno o un grupo alquilo C1-C6 opcionalmente sustituido, o alquileno C1-C6 opcionalmente sustituido. El grupo alquilo o alquenileno puede estar opcionalmente sustituido con halógeno, hidroxilo, tiol, ciano, amino, amido, nitro y SF5.
En otra realización, Q1 se puede seleccionar entre un átomo de flúor, un átomo de cloro, un átomo de bromo, ciano, oxo, metilo, butilo, CF3, metoxi, OCF3, NMeC(O)CH(CH3)2, -NHCOCH(CH3)2, un enlace covalente, un átomo de oxígeno, -O-metileno, -NH-, alquileno C1-C2, -NMeS(O)2-, -OCH2- y -N(c H3)c H2-.
En otra realización, Q1 se puede seleccionar entre un átomo de flúor, un átomo de cloro, oxo, un enlace covalente, un átomo de oxígeno, metoxi, -NHCOCH(CH3)2 y -N(CH3)CH2-.
Cuando R12 es fenilo o piridinilo, el anillo de fenilo o piridinilo está preferiblemente sustituido con flúor en una de las posiciones orto del anillo. El anillo de fenilo o piridinilo puede estar sustituido adicionalmente con -Q1-(R13)p como se describió anteriormente.
R13 representa un anillo de heteroarilo de 4 a 10 miembros (p. ej., 4, 5, 6, 7, 8, 9 o 10 miembros), heterociclilo, arilo o cicloalquilo de 3 a 8 miembros (p. ej., 3, 4, 5, 6, 7 u 8 miembros) opcionalmente sustituido.
En una realización, R13 representa un anillo de heteroarilo de 4 a 10 miembros, heterociclilo, arilo o cicloalquilo de 3 a 8 miembros sustituido con uno o más sustituyentes seleccionados entre halógeno, haloalquilo C1-C6 opcionalmente sustituido, alcoxi C1-C6 opcionalmente sustituido, haloalcoxi C1-C6, alquilo C1-C6 opcionalmente sustituido, alquenilo C2-C6 opcionalmente sustituido, alquinilo C2-C6 opcionalmente sustituido, hidroxialquilo C1-C6, oxo, ciano, heterociclilo opcionalmente sustituido, cicloalquilo opcionalmente sustituido, heteroarilo opcionalmente sustituido, arilo opcionalmente sustituido, -Q2-R17, -Q2-NR17CONR18R19, -Q2-NR17R18, -Q2-COR17, -Q2-NR17COR18, -Q2-NR17CO2R18, -Q2-SO2R17, Q2-CONR17R18, -Q2-CO2R17, -Q2-SO2NR17R18 y -Q2-NR17SO2R18
Q2 representa un enlace covalente, un átomo de oxígeno, carbonilo o un grupo alquileno C1-C6 o alquenileno C2-C6. En una realización, Q2 se puede seleccionar entre un enlace covalente, un átomo de oxígeno, carbonilo o un alquileno C1-C6 opcionalmente sustituido (p. ej., alquileno C1-C3, alquileno C1-C4, alquileno C1-C2), alquenileno C2-C6 o alquenileno C2-C4, El alquileno y el alquenileno pueden estar opcionalmente sustituidos con halógeno, hidroxilo, tiol, ciano, amino, amido, nitro y SF5.
En otra realización Q2 se selecciona entre un enlace covalente, un átomo de oxígeno o un carbonilo. En particular, Q2 es un enlace covalente.
R17, R18 y R19 representan cada uno independientemente hidrógeno, alquilo C1-C6 opcionalmente sustituido, heterociclilo opcionalmente sustituido, heteroarilo opcionalmente sustituido, arilo opcionalmente sustituido o un cicloalquilo opcionalmente sustituido.
R17, R18 y R19 cada uno puede representar independientemente hidrógeno, alquilo C1-C6 o un anillo de heterociclilo, heteroarilo, arilo o cicloalquilo de 3 a 10 miembros, en particular de 3 a 6 miembros, en donde el anillo está
opcionalmente sustituido con uno o más sustituyentes seleccionados entre alquilo C1-C6, alcoxi C1-C6, halógeno, hidroxilo, tiol, ciano, amino, amido, nitro y SF5, en donde el alquilo o alcoxi están opcionalmente sustituidos con flúor.
R13 puede estar sustituido con halógeno, ciano, alquilo C1-C3 opcionalmente sustituido con flúor, alcoxi C1-C3 opcionalmente sustituido con flúor, o -Q2-R17, en donde Q2 representa un enlace covalente, un átomo de oxígeno, carbonilo o un grupo alquileno C1-C6 o alquenileno C2-C6 y R17 representa un anillo de heterociclilo, heteroarilo, arilo o cicloalquilo de 3 a 10 miembros opcionalmente sustituido, en donde los sustituyentes opcionales se seleccionan entre alquilo C1-C6, alcoxi C1-C6, halógeno, hidroxilo, tiol, ciano, amino, amido, nitro y SF5, en donde el alquilo o alcoxi están opcionalmente sustituidos con flúor.
En particular, R13 puede estar sustituido con flúor, cloro, ciano, metilo, CF3, etilo, metoxi o -Q2-R17, en donde Q2 es un enlace covalente, átomo de oxígeno o carbonilo y R17 se selecciona entre morfolinilo, ciclopropilo, fenilo o piridinilo opcionalmente sustituidos y los sustituyentes opcionales son uno o más átomos de flúor.
En una realización, R13 no está sustituido.
En una realización, R13 está sustituido con otros anillos de heteroarilo, heterociclilo, arilo o cicloalquilo de 3 a 8 miembros opcionalmente sustituidos, ya sea directamente anclados o a través de un grupo conector. El grupo conector puede ser un átomo de oxígeno o un carbonilo. El grupo conector puede ser un átomo de oxígeno o -CO-.
En una realización, R13 se selecciona entre fenilo, piridinilo, pirazolilo, imidazolilo, isoxazolilo, morfolinilo, piperidinilo, piperazinilo, quinolinilo, pirrolidinilo, benzopirazolilo, isoindolinilo, tetrahidroquinolinilo, homopiperazinilo, pirimidinilo, imidazopirimidinilo, imidazopiridinilo, indazolilo, pirrolopiridinilo, benzimidazolilo, piridazinilo, pirazolopirimidinilo, pirrolopirimidinilo, imidazopirazinilo y dihidroisoquinolinilo.
En una realización, R13 se selecciona entre fenilo, piridinilo, pirazolilo, imidazolilo, isoxazolilo, morfolinilo, piperidinilo, piperazinilo, quinolinilo, pirrolidinilo, benzopirazolilo, isoindolinilo, tetrahidroquinolinilo y homopiperazinilo.
En la presente invención, los compuestos de las fórmulas descritas en la presente memoria no incluyen compuestos de las siguientes estructuras:

Los compuestos de las fórmulas descritas en la presente memoria no incluyen los siguientes compuestos:
N-[(3R)-1-ciano-3-pirrolidinil]-4-fluoro-benzamida;
N-(1 -ciano-3-pirrolidinil)-[1,1'-bifenil]-4-carboxamida;
N-(1 -ciano-3-piperidinil)-[1,1'-bifenil]-4-carboxamida;
N-[(3R)-1-ciano-3-pirrolidinil]-3-({[(3R)-1-ciano-3-pirrolidinil]amino}sulfonil)benzamida;
N-[(3R)-1-ciano-3-pirrolidinil]-3-([(3R)-3-pirrolidinilamino]sulfonil)-benzamida; o
1 -((3S, 4R)-1 -ciano-4-(3,4-difluorofenil)pirrolidin-3-il)-3-(1 ',4-dimetil-1 -fenil-1 H,1'H-[3,4’-bipirazol]-5-il)ureail)urea. es decir, compuestos de las siguientes estructuras:

En la presente divulgación, los compuestos de fórmulas (I), (IB) y (IID), incluidas sus realizaciones subgenéricas, no incluyen compuestos de las siguientes estructuras:

Las realizaciones de la divulgación que se pueden mencionar incluyen compuestos de fórmulas (I), (IB) y (IID) en donde:
n, X, R1, R2, R3, R8 , R9, R10 e Y se definen anteriormente para los compuestos de fórmula (I);
R12 representa:
i) un anillo de heteroarilo, heterociclilo de 4 a 10 miembros o cicloalquilo de 3 a 8 miembros sustituido con uno o más de Q1-(R13)p;
ii) un anillo de arilo de 4 a 10 miembros sustituido con dos o más Q1-(R13)p;
iii) un anillo de arilo de 5, 7, 8, 9 o 10 miembros sustituido individualmente con Q1-(R13)p; o
iv) un anillo de arilo de 6 miembros sustituido individualmente con Q1-(R13')p;
en donde p es 0 o 1;
Q1 representa un átomo de halógeno, ciano, oxo, un enlace covalente, -NR14-, -NR14R15, -CONR14-, -NR14CO-, un átomo de oxígeno, -CO-, -S(O)q-, -SO2NR14-, alcoxi C1-C6, haloalcoxi C1-C6, hidroxialquilo C1-C6, -SO2R14, -NR14COR15, -NR14CONR15R16, -NR14SO2NR15R16-, -CONR14R15, -CO2R14, -NR14CO2R15, -SO2NR14R15, -CONR14, -C(O)R14 y -NR14SO2R15, NO2 o un grupo alquileno C1-C6 opcionalmente sustituido, -alquenileno C2-C6 o -alquilo C1-C6; Q1' representa un átomo de cloro o bromo, ciano, oxo, un enlace covalente, -NR14-, -NR14R15, -CONR14-, -NR14CO-,
un átomo de oxígeno, -CO-, -S(O)q-, alcoxi C1-C6, haloalcoxi C1-C6, hidroxialquilo C1-C6, -SO2R14, -NR14R15, -NR14COR15, -NR14CONR15R16, -CONR14R15, -CO2R145, -NR14CO2R15, -SO2NR14R15, -CONR14, -C(O)R14 y -NR14SO2R15 o un grupo alquileno C1-C6, -alquenileno C2-C6 o -alquilo C1-C6 opcionalmente sustituido;
q es 0, 1 o 2;
R14, R15 y R16 representan cada uno independientemente un átomo de hidrógeno o un grupo alquilo C1-C6 opcionalmente sustituido, o alquileno C1-C6 opcionalmente sustituido; y
cuando p es 1:
R13 representa un anillo de heteroarilo, heterociclilo, arilo de 4 a 10 miembros o cicloalquilo de 3 a 8 miembros (cuando p es 0, Q1 está presente y R13 está ausente), que está opcionalmente sustituido con uno o más sustituyentes seleccionados entre halógeno, haloalquilo C1-C6 opcionalmente sustituido, alcoxi C1-C6 opcionalmente sustituido, haloalcoxi C1-C6, alquilo C1-C6 opcionalmente sustituido, alquenilo C2-C6 opcionalmente sustituido, alquinilo C2-C6 opcionalmente sustituido, hidroxialquilo C1-C6, oxo, ciano, heterociclilo opcionalmente sustituido, cicloalquilo opcionalmente sustituido, heteroarilo opcionalmente sustituido, arilo opcionalmente sustituido, -Q2-R17, -Q2-NR17CONR18R19, -Q2-NR17R18, -Q2-COR17, -Q2-NR17COR18, -Q2-NR17CO2R18, -Q2-SO2R17, Q2-CONR17R18, -Q2-CO2R17, -Q2-SO2NR17R18 y -Q2-NR17SO2R18;
R13' representa un anillo de heteroarilo, heterociclilo de 4 a 10 miembros o cicloalquilo de 3 a 8 miembros opcionalmente sustituido, un anillo de arilo de 5, 7, 8, 9 o 10 miembros opcionalmente sustituido, o un anillo de 6 miembros sustituido (cuando p es 0, Q1' está presente y R13' está ausente) sustituido con uno o más sustituyentes seleccionados entre halógeno, haloalquilo C1-C6 opcionalmente sustituido, alcoxi C1-C6 opcionalmente sustituido, haloalcoxi C1-C6, alquilo C1-C6 opcionalmente sustituido, alquenilo C2-C6 opcionalmente sustituido, alquinilo C2-C6 opcionalmente sustituido, hidroxialquilo C1-C6, oxo, ciano, heterociclilo opcionalmente sustituido, cicloalquilo opcionalmente sustituido, heteroarilo opcionalmente sustituido, arilo opcionalmente sustituido, -Q2-R17, -Q2-NR17CONR18R19, -Q2-NR17R18, -Q2-COR17, -Q2-NR17COR18, -Q2-NR17CO2R18, -Q2-SO2R17, Q2-CONR17R18, -Q2-CO2R17, -Q2-SO2NR17R18 y -Q2-NR17SO2R18;
Q2 representa un enlace covalente, un átomo de oxígeno, carbonilo o un grupo alquileno C1-C6 o alquenileno C2-C6; y R17, R18, R19 representan cada uno independientemente hidrógeno, alquilo C1-C6 opcionalmente sustituido, heterociclilo opcionalmente sustituido, heteroarilo opcionalmente sustituido, arilo opcionalmente sustituido o un cicloalquilo opcionalmente sustituido.
En una realización particular de la divulgación, se proporciona un compuesto de fórmula (IID) en donde:
R12 se selecciona entre fenilo o piridinilo y está sustituido con uno o dos Q1(R13)p, en donde p es 1;
cada Q1 se selecciona independientemente de un enlace covalente, un átomo de flúor, alcoxi C1-C3 o alcoxi C1-C2 (p. ej., metoxi o etoxi); y
R13 se selecciona entre un heteroarilo o heterociclilo de 5 o 6 miembros que está opcionalmente sustituido con alquilo C1-C3.
Las realizaciones de la invención que se pueden mencionar incluyen compuestos de fórmulas (II) o (III) en donde: m, Z, R1, R2, R3, R4, R5, R8, R9, R10 e Y se definen anteriormente para los compuestos de fórmula (II);
R12 representa:
i) un anillo de heterociclilo o cicloalquilo monocíclico de 3 a 10 miembros sustituido con uno o más de Q1-(R13)p, o anillo de heterociclilo o cicloalquilo bicíclico opcionalmente sustituido con uno o más de Q1-(R13)p;
ii) un anillo de arilo monocíclico de 5 a 14 miembros sustituido con dos o más Q1-(R13)p, o anillo de arilo bicíclico o tricíclico opcionalmente sustituido con dos o más Q1-(R13)p;
iii) un anillo de arilo monocíclico de 5 o 7 a 14 miembros sustituido con uno o más Q1-(R13)p, o anillo de arilo bicíclico o tricíclico opcionalmente sustituido con uno o más Q1-(R13)p;
iv) un anillo de arilo de 6 miembros monosustituido con Q1'-(R13')p;
v) un anillo de heteroarilo de 5 a 14 miembros sustituido con uno o dos Q1-(R13)p;
vi) un anillo de heteroarilo de 6 a 14 miembros sustituido con uno o más Q1-(R13)p; o
vii) un anillo de heteroarilo de 5 miembros sustituido con uno o más Q1M-(R13")p.
en donde p es 0 o 1;
Q1 representa un átomo de halógeno, ciano, oxo, hidroxilo, un enlace covalente, -alquilen(C0-C3)-NR14-, -alquilen(Cü-C3)-NR14R15, -alquilen(C0-C3)-CONR14-, -alquilen(C0-C3)-NR14CO-, -alquilen(C0-C3)-NR14SO2-, -alquilen(C0-C3)-O-alquileno C0-C3, -alquilen(C0-C3)-CO-, -alquilen(C1-C6)-S(O)q-, -alquilen(C0-C3)-SO2NR14, -alcoxi C1-C6, haloalcoxi C1-C6, hidroxialquilo C1-C6, -alquilen(C0-C3)-SO2R14, -alquilen(C0-C3)-NR14COR15, -alquilen(C0-C3)-NR14CONR15R16,
-alquilen(C0-C3)-NR14SO2NR15R16, -alquilen(C0-C3)-CONR14R15, -alquilen(C0-C3)-CO2R14, -alquilen(C0-C3)-NR14CO2R15, -alquilen(C0-C3)-SO2NR14R15, -alquilen(C0-C3)-CONR14, -alquilen(C0-C3)-C(O)R14 y alquilen(C1-C6)-NR14SO2R15, NO2, o un grupo alquileno C1-C6, -alquenileno C2-C6 o -alquilo C1-C6 opcionalmente sustituido;
Q1' representa un átomo de cloro o bromo, ciano, oxo, hidroxilo, un enlace covalente, -alquilen(C0-C3)-NR14-, -alquilen(C0-C3)-NR14R15, -alquilen(C0-C3)-CONR14-, -alquilen(C0-C3)-NR14CO-, -alquilen(C0-C3)-NR14SO2-, -alquilen(C0-C3)-O-alquileno C0-C3, -alquilen(C0-C3)-CO-, -alquilen(C1-C6)-S(O)q-, -alquilen(C1-C6)-SO2NR14, -alcoxi C1-C6, haloalcoxi C1-C6, hidroxialquilo C1-C6, -alquilen(C0-C3)-SO2R14, -alquilen(C0-C3)-NR14COR15,
-alquilen(C0-C3)-NR14CONR15R16, -alquilen(C0-C3)-NR14SO2NR15R16, -alquilen(C0-C3)-CONR14R15, -alquilen(C0-C3)-CO2R14, -alquilen(C0-C3)-NR14CO2R15, -alquilen(C0-C3)-SO2NR14R15, -alquilen(C0-C3)-CONR14, -alquilen(C0-C3)-C(O)R14 y alquilen(C1-C6)-NR14SO2R15, NO2, o un grupo alquileno C1-C6, -alquenileno C2-C6 o -alquilo C1-C6 opcionalmente sustituido;
Q1" representa un átomo de halógeno, ciano, oxo, hidroxilo, un enlace covalente, -alquilen(C0-C3)-NR14-, -alquilen(C0-C3)-NR14R15, -alquilen(C0-C3)-CONR14-, -alquilen(C0-C3)-NR14CO-, -alquilen(C0-C3)-NR14SO2-, -alquilen(C0-C3)-O-alquileno C0-C3, -alquilen(C0-C3)-CO-, -alquilen(C1-C6)-S(O)q-, -alquilen(C0-C3)-SO2NR14, -alcoxi C1-C6, haloalcoxi C1-C6, hidroxialquilo C1-C6, -alquilen(C0-C3)-SO2R14, -alquilen(C0-C3)-NR14COR15, -alquilen(C0-C3)-NR14CONR15R16,
-alquilen(C0-C3)-NR14SO2NR15R16, -alquilen(C0-C3)-CONR14R15, -alquilen(C0-C3)-CO2R14, -alquilen(C0-C3)-NR14CO2R15, -alquilen(C0-C3)-SO2NR14R15, -alquilen(C0-C3)-CONR14, -alquilen(C0-C3)-C(O)R14 y alquilen(C1-C6)-NR14SO2R15, NO2, o un grupo alquileno C1-C6, -alquenileno C2-C6 o alquilo -C2-C6 opcionalmente sustituido;
q es 0, 1 o 2;
R14, R15 y R16 representan cada uno independientemente un átomo de hidrógeno o un grupo alquilo C1-C6 opcionalmente sustituido, o alquileno C1-C6 opcionalmente sustituido; y
cuando p es 1:
R13 representa un anillo de heteroarilo, heterociclilo, arilo de 4 a 10 miembros o cicloalquilo de 3 a 8 miembros, que está opcionalmente sustituido con uno o más sustituyentes seleccionados entre halógeno, haloalquilo C1-C6 opcionalmente sustituido, alcoxi C1-C6 opcionalmente sustituido, haloalcoxi C1-C6, alquilo C1-C6 opcionalmente sustituido, alquenilo C2-C6 opcionalmente sustituido, alquinilo C2-C6 opcionalmente sustituido, hidroxialquilo C1-C6, oxo, ciano, heterociclilo opcionalmente sustituido, cicloalquilo opcionalmente sustituido, heteroarilo opcionalmente sustituido, arilo opcionalmente sustituido, -Q2-R17, -Q2-NR17CONR18R19, -Q2-NR17R18, -Q2-COR17, -Q2-NR17COR18,
-Q2-NR17CO2R18, -Q2-SO2R17, Q2-CONR17R18, -Q2-CO2R17, -Q2-SO2NR17R18 y -Q2-NR17SO2R18;
R13' representa un anillo de heteroarilo, heterociclilo de 4 a 10 miembros o cicloalquilo de 3 a 8 miembros opcionalmente sustituido, un anillo de arilo de 5, 7, 8, 9 o 10 miembros opcionalmente sustituido, o un anillo de 6 miembros sustituido (cuando p es 0, Q1' está presente y R13' está ausente) sustituido con uno o más sustituyentes seleccionados entre halógeno, haloalquilo C1-C6 opcionalmente sustituido, alcoxi C1-C6 opcionalmente sustituido, haloalcoxi C1-C6, alquilo C1-C6 opcionalmente sustituido, alquenilo C2-C6 opcionalmente sustituido, alquinilo C2-C6 opcionalmente sustituido, hidroxialquilo C1-C6, oxo, ciano, heterociclilo opcionalmente sustituido, cicloalquilo opcionalmente sustituido, heteroarilo opcionalmente sustituido, arilo opcionalmente sustituido, -Q2-R17, -Q2-NR17CONR18R19, -Q2-NR17R18, -Q2-COR17, -Q2-NR17COR18, -Q2-NR17CO2R18, -Q2-SO2R17, Q2-CONR17R18, -Q2-CO2R17y -Q2-NR17SO2R18;
R13" representa un anillo de heteroarilo, arilo de 4 a 10 miembros o cicloalquilo de 3 a 8 miembros opcionalmente sustituido, un anillo de heteroarilo de 6 a 10 miembros opcionalmente sustituido o un anillo de heterociclilo de 5 miembros sustituido en donde los sustituyentes se seleccionan entre halógeno, haloalquilo C1-C6 opcionalmente sustituido, alcoxi C1-C6 opcionalmente sustituido, haloalcoxi C1-C6, alquilo C2-C6 opcionalmente sustituido, alquenilo C2-C6 opcionalmente sustituido, alquinilo C2-C6 opcionalmente sustituido, hidroxialquilo C1-C6, oxo, ciano, heterociclilo opcionalmente sustituido, cicloalquilo opcionalmente sustituido, heteroarilo opcionalmente sustituido, arilo opcionalmente sustituido, -Q2-R17, -Q2-NR17CONR18R19, -Q2-NR17R18, -Q2-COR17, -Q2-NR17COR18, -Q2-NR17CO2R18, -Q2-SO2R17, Q2-CONR17R18, -Q2-CO2R17y -Q2-NR17SO2R18
Q2 representa un enlace covalente, un átomo de oxígeno, carbonilo o un grupo alquileno C1-C6 o alquenileno C2-C6; y
R17, R18, R19 representan cada uno independientemente hidrógeno, alquilo C1-C6 opcionalmente sustituido, heterociclilo opcionalmente sustituido, heteroarilo opcionalmente sustituido, arilo opcionalmente sustituido o un cicloalquilo opcionalmente sustituido.
Los ejemplos de nuevos compuestos de fórmula (I) y/o fórmula (II) incluyen: (R)-N-(1 -cianopirrolidin-3-il)-5-fenMpicolinamida
(R)-N-(1 -cianopirrolidin-3-il)-3-metoxi-4-(1 -metil-1 H-pirazol-4-il)benzamida 2'-cloro-N-(1-cianopirrolidin-3-il)-[1,1'-bifenil]-4-carboxamida
6-(bencil(metil)amino)-N-(1-cianopirrolidin-3-il)nicotinamida
(R)-N-(1 -cianopirrolidin-3-il)-3-fenilazetidin-1 -carboxamida
N-((R)-1-cianopirrolidin-3-il)-4-((2S,6R)-2,6-dimetilmorfolino)-3-fluorobenzamida N-(1-cianopirrolidin-3-il)-4-feniltiazol-2-carboxamida
3- (3-clorofenil)-N-(1-cianopirrolidin-3-il)isoxazol-5-carboxamida
N-(1 -cianopirrolidin-3-il)-1 -fenil-1 H-imidazol-4-carboxamida
N-(1-cianopirrolidin-3-il)-1-(2,4-difluorobencil)-5-oxopirrolidin-3-carboxamida N-(1-cianopirrolidin-3-il)-5-oxo-1-fenilpirrolidin-3-carboxamida
N-(1-cianopirrolidin-3-il)-4-(3,5-dimetilisoxazol-4-il)benzamida
3'-cloro-N-(1-cianopirrolidin-3-il)-[1,1'-bifenil]-4-carboxamida
N-(1 -cianopirrolidin-3-il)-2'-metoxi-[1,1'-bifenil]-4-carboxamida
N-(1-cianopirrolidin-3-il)-4-fenoxibenzamida
2-([1,1'-bifenil]-4-il)-N-(1 -cianopirrolidin-3-il)acetamida
N-(1-cianopirrolidin-3-il)-2-fenilquinolin-4-carboxamida
6-(4-carbamoilpiperidin-1-il)-N-(1-cianopirrolidin-3-il)nicotinamida
N-(1-cianopirrolidin-3-il)-6-(4-(2,4-difluorofenil)piperazin-1-il)nicotinamida 4- (5-((1 -cianopirrolidin-3-il)carbamoil)piridin-2-il)piperazin-1 -carboxilato de etilo N-(1 -cianopirrolidin-3-il)-6-(2-(piridin-3-il)pirrolidin-1 -il)nicotinam ida
N-(1-cianopirrolidin-3-il)-6-(4-fenoxipiperidin-1-il)nicotinamida
N-(1 -cianopirrolidin-3-il)-6-(4-(piridin-4-il)piperidin-1 -il)nicotinam ida
6-(bencil(metil)amino)-N-(1-cianopirrolidin-3-il)picolinamida
N-(1-cianopirrolidin-3-il)-6-(3,4-dihidroisoquinolin-2(1 H)-il)picolinamida
N-(1-cianopirrolidin-3-il)-6-(4-fenoxipiperidin-1-il)picolinamida
N-(1-cianopirrolidin-3-il)-2-(3,4-dihidroisoquinolin-2(1 H)-il)isonicotinamida 2-(4-acetil-1,4-diazepan-1 -il)-N-(-(1 -cianopirrolidin-3-il)isonicotinamida
(R)-N-(1-cianopirrolidin-3-il)-6-fenilpicolinamida
(R)-N-(1-cianopirrolidin-3-il)-4-fenilpicolinamida
(R)-N-(1-cianopirrolidin-3-il)-4-morfolinobenzamida
(R)-N-(1-cianopirrolidin-3-il)-3-fluoro-4-morfolinobenzamida
(R)-N-(1-cianopirrolidin-3-il)-3-fenilisoxazol-5-carboxamida
(R)-N-(1-cianopirrolidin-3-il)-5-(piridin-4-il)isoxazol-3-carboxamida
(R)-N-(1 -cianopirrolidin-3-il)-[1,1'-bifenil]-4-carboxamida
(R)-6-(4-clorofenil)-N-(1-cianopirrolidin-3-il)nicotinamida
(R)-2-(2-clorofenil)-N-(1-cianopirrolidin-3-il)tiazol-5-carboxamida
(R)-4-(3-cloropiridin-4-il)-N-(1-cianopirrolidin-3-il)benzamida
(R)-4-(3-cloropiridin-4-il)-N-(1-cianopirrolidin-3-il)-3-metoxibenzamida
(R)-N-(1-cianopirrolidin-3-il)-3-metoxi-4-(2-metilpiridin-4-il)benzamida
(R)-N-(1-cianopirrolidin-3-il)-3-metoxi-4-(2-morfolinopiridin-4-il)benzamida
(R)-N-(1-cianopirrolidin-3-il)-4-fluoro-3-(piridin-4-il)benzamida
(R)-N-(1 -cianopirrolidin-3-il)-4-fluoro-3-(1 -metil-1 H-pirazol-4-il)benzamida
(R)-N-(1 -cianopirrolidin-3-il)-4-(1 -metil-1 H-pirazol-4-il)benzamida
(R) -N-(1 -cianopirrolidin-3-il)-5-(1 -metil-1 H-pirazol-4-il)pirimidin-2-carboxamida
N-((R)-1 -cianopirrolidin-3-il)-3-fenilpirrolidin-1 -carboxamida
(S) -N-(1-cianopirrolidin-3-il)-4-(piridin-4-il)benzamida
(S)-N-(1-cianopirrolidin-3-il)-6-fenilpicolinamida
(R)-4-(3-cloropiridin-4-il)-N-(1-cianopirrolidin-3-il)-N-metilbenzamida
(R)-1 -(1 -cianopirrolidin-3-il)-3-(imidazo[1,2-a]piridin-2-il)-1 -metilurea
(3aR,6aR)-1-([1,1'-bifenil]-3-carbonil)hexahidropirrolo[3,4-b]pirrolo-5(1 H)-carbonitrilo
(3aR,6aR)-1-(3-fenil-1 H-pirazol-5-carbonil)hexahidropirrolo[3,4-b]pirrolo-5(1 H)-carbonitrilo (3aR,6aR)-1-(3-fenilisoxazol-5-carbonil)hexahidropirrolo[3,4-b]pirrolo-5(1 H)-carbonitrilo (3aR,6aR)-1 -(1 -fenil-1 H-imidazol-4-carbonil)hexahidropirrolo[3,4-b]pirrolo-5(1 H)-carbonitrilo (3aR,6aR)-1-(3-(4-metoxifenil)-1 H-pirazol-5-carbonil)hexahidropirrolo[3,4-b]pirrolo-5(1 H)-carbonitrilo (3aR,6aR)-1-(3-(4-metoxifenil)isoxazol-5-carbonil)hexahidropirrolo[3,4-b]pirrolo-5(1 H)-carbonitrilo (3aR,6aR)-1-(4-fluoro-3-(piridin-4-il)benzoil)hexahidropirrolo[3,4-b]pirrolo-5(1 H)-carbonitrilo (3aR,6aR)-1 -(4-fluoro-3-(1 -metil-1 H-pirazol-4-il)benzoil)hexahidropirrolo[3,4-b]pirrolo-5(11 H)-carbonitrilo (3aR,6aR)-1-(4-(3-cloropiridin-4-il)-3-metoxibenzoil)hexahidropirrolo[3,4-b]pirrolo-5(1 H)-carbonitrilo (3aR,6aR)-1 -(3-metoxi-4-(1 -metil-1 H-pirazol-4-il)benzoil)hexahidropirrolo[3,4-b]pirrolo-5(1 H)-carbonitrilo (3aR,6aR)-1-(2-oxo-6-fenil-1,2-dihidropiridin-3-carbonil)hexahidropirrolo[3,4-b]pirrolo-5(1 H)-carbonitrilo (R)-N-(1-cianopirrolidin-3-il)-3-(N-metilisobutiramido)benzamida
(R)-N-(1-cianopirrolidin-3-il)-5-fenilpirimidin-2-carboxamida
(R)-N-(1-cianopirrolidin-3-il)-3-(piridin-4-il)isoxazol-5-carboxamida
(R)-N-(1-cianopirrolidin-3-il)-3-(piridin-3-il)isoxazol-5-carboxamida
(R)-N-(1-cianopirrolidin-3-il)-3-(piridin-2-il)isoxazol-5-carboxamida
N-(1-cianopirrolidin-3-il)-5-fenilpiridazin-3-carboxamida
N-(1 -cianopirrolidin-3-il)-N-metil-[1,1'-bifenil]-4-carboxamida
N-((3S,4R)-1-ciano-4-metilpirrolidin-3-il)-2-feniltiazol-5-carboxamida
N-((3R,4S)-1-ciano-4-metilpirrolidin-3-il)-2-feniltiazol-5-carboxamida
N-((3S,4R)-1-ciano-4-metilpirrolidin-3-il)-5-feniltiazol-2-carboxamida
N-((3R,4S)-1-ciano-4-metilpirrolidin-3-il)-5-feniltiazol-2-carboxamida
(R)-N-(1-cianopirrolidin-3-il)-2-(isoindolin-2-il)isonicotinamida
(R)-N-(1-cianopirrolidin-3-il)-2-(3,4-dihidroisoquinolin-2(1 H)-il)isonicotinamida
(R)-N-(1 -cianopirrolidin-3-il)-2-fluoro-4-(4-metil-1 H-imidazol-1 -il)benzamida
(R)-N-(1 -cianopirrolidin-3-il)-3-fluoro-4-(1 -metil-1 H-pirazol-4-il)benzamida
(R)-N-(1 -cianopirrolidin-3-il)-2-fluoro-4-(1 -metil-1 H-pirazol-4-il)benzamida
(R)-N-(1 -cianopirrolidin-3-il)-2,5-difluoro-4-(1 -metil-1 H-pirazol-4-il)benzamida
(R)-N-(1 -cianopirrolidin-3-il)-4-(1,3-dimetil-1 H-pirazol-4-il)-3-fluorobenzamida
(R)-N-(1 -cianopirrolidin-3-il)-4-(1,3-dimetil-1 H-pirazol-4-il)-2-fluorobenzamida
(R)-N-(1 -cianopirrolidin-3-il)-4-(1 -etil-1 H-pirazol-4-il)-2-fluorobenzamida
(R)-N-(1 -cianopirrolidin-3-il)-2-fluoro-4-(1 -(2-metoxietil)-1 H-pirazol-4-il)benzamida
(R)-N-(1 -cianopirrolidin-3-il)-6-(1 -metil-1 H-pirazol-4-il)-1 H-benzo[d]imidazol-2-carboxamida (R)-N-(1 -cianopirrolidin-3-il)-6-(1 -metil-1 H-pirazol-4-il)-1 H-indolo-2-carboxamida
(R)-N-(1-cianopirrolidin-3-il)-2-fluoro-4-(5-(trifluorometil)-1 H-pirazol-4-il)benzamida
(R)-N-(1 -cianopirrolidin-3-il)-2-fluoro-4-(1 -metil-1 H-indazol-5-il)benzamida
(R)-N-(1 -cianopirrolidin-3-il)-3-fluoro-N-metil-4-(1 -metil-1 H-pirazol-4-il)benzamida
(R)-N-(1 -cianopirrolidin-3-il)-N-metil-6-(1 -metil-1 H-pirazol-4-il)-1 H-benzo[d]imidazol-2-carboxamida (R)-N-(1-cianopirrolidin-3-il)-N-metil-3-fenoxiazetidin-1-carboxamida
(3aR,6aR)-5-ciano-N-(2-fluoro-4-(trifluorometil)fenil)hexahidropirrolo[3,4-b]pirrolo-1 (2H)-carboxamida (R)-N-(1-cianopirrolidin-3-il)-2-fluoro-4-(pirimidin-2-ilamino)benzamida
N-((3R,4S)-1-ciano-4-metilpirrolidin-3-il)-2-fluoro-4-((R)-3-metoxipirrolidin-1-il)benzamida
2-(2-clorofenil)-N-((3R,4R)-1-ciano-4-hidroxipirrolidin-3-il)tiazol-5-carboxamida
N-(1 -ciano-3-metilpirrolidin-3-il)-2-fluoro-4-(1 -metil-1 H-pirazol-4-il)benzamida
(R)-N-(1-cianopirrolidin-3-il)-5-(4-metoxifenil)-1 H-pirazol-3-carboxamida
(R)-N-(1-cianopirrolidin-3-il)-5-(piridin-2-il)-1 H-pirazol-3-carboxamida
(R)-N-(1-cianopirrolidin-3-il)-5-(2-metoxifenil)-1 H-pirazol-3-carboxamida
(R)-N-(1cianopirrolidin-3-il)-5-(2-fluorofenil)-1 H-pirazol-3-carboxamida
(R)-N-(1-cianopirrolidin-3-il)-6-morfolinonicotinamida
(R)-N-(1-cianopirrolidin-3-il)-3-(4-metoxifenil)isoxazol-5-carboxamida
(R)-N-(1 -cianopirrolidin-3-il)-1 H-indazol-3-carboxamida
(R)-N-(1-cianopirrolidin-3-il)-5-fenil-1 H-pirazol-3-carboxamida
(R)-N-(1-cianopirrolidin-3-il)-5-(piridin-3-il)-1 H-pirazol-3-carboxamida
(R)-N-(1 -cianopirrolidin-3-il)-2-fluoro-3-(1 -metil-1 H-pirazol-4-il)benzamida
(R)-N-(1-cianopirrolidin-3-il)-2-fluoro-4-(2-metilpirimidin-4-il)benzamida
(R)-N-(1 -cianopirrolidin-3-il)-5-(1 -metil-1 H-pirazol-4-il)picolinamida
(R)-N-(1 -cianopirrolidin-3-il)-2-fluoro-5-(1 -metil-1 H-pirazol-4-il)benzamida
(R)-N-(1-cianopirrolidin-3-il)-2-fluoro-4-(pirimidin-4-il)benzamida
(R)-N-(1-cianopirrolidin-3-il)-2-fluoro-4-(imidazo[1,2-a]pirimidin-6-il)benzamida
(R)-N-(1 -cianopirrolidin-3-il)-2-metoxi-4-(1 -metil-1 H-pirazol-4-il)benzamida
(R)-N-(1 -cianopirrolidin-3-il)-6-(1 -metil-1 H-pirazol-4-il)nicotinamida
(R)-N-(1 -cianopirrolidin-3-il)-3,5-difluoro-4-(1 -metil-1 H-pirazol-4-il)benzamida
(R)-N-(1 -cianopirrolidin-3-il)-2,6-difluoro-4-(1 -metil-1 H-pirazol-4-il)benzamida
(R)-N-(1 -cianopirrolidin-3-il)-6-(1 -metil-1 H-pirazol-4-il)pirazolo[1,5-a]piridin-3-carboxamida (R)-N-(1 -cianopirrolidin-3-il)-2-fluoro-3-metoxi-4-(1 -metil-1 H-pirazol-4-il)benzamida (R)-6-(3-cianofenil)-N-(1-cianopirrolidin-3-il)imidazo[1,2-a]piridin-2-carboxamida
(R)-6-(4-cianofenil)-N-(1-cianopirrolidin-3-il)imidazo[1,2-a]piridin-2-carboxamida
(R)-N-(1-cianopirrolidin-3-il)-2-fluoro-4-(imidazo[1,2-a]piridin-6-il)benzamida
(R)-N-(1-cianopirrolidin-3-il)-2-fluoro-4-(2-morfolinopiridin-4-il)benzamida
(R)-N-(1 -cianopirrolidin-3-il)-3-fluoro-5-(1 -metil-1 H-indazol-5-il)picolinamida
(R)-N-(1 -cianopirrolidin-3-il)-3-fluoro-5-(1 -metil-1 H-pirrolo[2,3-b]piridin-5-il)picolinamida (R)-N-(1 -cianopirrolidin-3-il)-5-(1,3-dimetil-1 H-pirazol-4-il)-3-fluoropicolinamida
(R)-3-cloro-N-(1-cianopirrolidin-3-il)-5-(4-fluorofenil)picolinamida
(R)-N-(1 -cianopirrolidin-3-il)-6-(1 -metil-1 H-pirazol-4-il)imidazo[1,2-a]piridin-2-carboxamida (R)-N-(1 -cianopirrolidin-3-il)-6-(1,3-dimetil-1 H-pirazol-4-il)imidazo[1,2-a]piridin-2-carboxamida (R)-N-(1 -cianopirrolidin-3-il)-5-(1 -metil-1 H-pirazol-4-il)-1 H-indazol-3-carboxamida
(R)-N-(1 -cianopirrolidin-3-il)-6-(1 -metil-1 H-pirazol-4-il)-1 H-indazol-3-carboxamida
(R)-N-(1-cianopirrolidin-3-il)-6-(3,5-dimetilisoxazol-4-il)-1 H-indolo-2-carboxamida
(R)-N-(1 -cianopirrolidin-3-il)-6-(1,3-dimetil-1 H-pirazol-4-il)-1 H-indolo-2-carboxamida (R)-N-(1 -cianopirrolidin-3-il)-5-(1 -metil-1 H-pirazol-4-il)-1 H-indolo-2-carboxamida
(R)-N-(1 -cianopirrolidin-3-il)-2,3-difluoro-4-(1 -metil-1 H-pirazol-4-il)benzamida
(R)-N-(1 -cianopirrolidin-3-il)-5-(1 -etil-1 H-pirazol-4-il)-4-metilpicolinamida
(R)-N-(1 -cianopirrolidin-3-il)-3-fenoxiazetidin-1 -carboxamida
(R)-3-(1 H-benzo[d]imidazol-2-il)-N-(1 -cianopirrolidin-3-il)azetidin-1 -carboxamida
(R)-N-(1 -cianopirrolidin-3-il)-4-fenilpiperazin-1 -carboxamida
N-((R)-1-cianopirrolidin-3-il)-2-fenilmorfoline-4-carboxamida
(R)-4-(2-cloro-6-fluorobencil)-N-(1 -cianopirrolidin-3-il)-1,4-diazepano-1 -carboxamida (R)-4-bencil-N-(1 -cianopirrolidin-3-il)-1,4-diazepano-1 -carboxamida
(R)-N-(1-cianopirrolidin-3-il)-1,3,4,9-tetrahidro-2H-pirido[3,4-b]indolo-2-carboxamida N-((R)-1-cianopirrolidin-3-il)-2-((2S,6R)-2,6-dimetilmorfolino)-5-fluoroisonicotinamida (R)-N-(1-cianopirrolidin-3-il)-5-fluoro-2-(isoindolin-2-il)isonicotinamida
(R)-N-(1-cianopirrolidin-3-il)-3-fluoro-4-(pirimidin-2-ilamino)benzamida
(R)-N-(1-cianopirrolidin-3-il)-2-fluoro-4-(pirrolidin-1-il)benzamida
(R)-N-(1-cianopirrolidin-3-il)-2,5-difluoro-4-morfolinobenzamida
(R)-N-(1-cianopirrolidin-3-il)-2,5-difluoro-4-(pirrolidin-1-il)benzamida
N-((R)-1-cianopirrolidin-3-il)-2-fluoro-4-((R)-3-metoxipirrolidin-1-il)benzamida
(R)-N-(1-cianopirrolidin-3-il)-3-metoxi-4-(pirimidin-2-ilamino)benzamida
(R)-N-(1-cianopirrolidin-3-il)-3-metoxi-4-((4-metilpirimidin-2-il)amino)benzamida
(R)-N-(1-cianopirrolidin-3-il)-2-fluoro-4-((4-metoxipirimidin-2-il)amino)benzamida
N-((R)-1 -cianopirrolidin-3-il)-5-metil-1 -(1 -feniletil)-1 H-pirazol-3-carboxamida
(R)-N-(1 -cianopirrolidin-3-il)-5-metil-1 -(piridin-2-ilmetil)-1 H-pirazol-3-carboxamida
(R)-N-(1-cianopirrolidin-3-il)-2-fluoro-4-(piridazin-4-il)benzamida
(R)-N-(1 -cianopirrolidin-3-il)-1 -isobutil-6-(1 -metil-1 H-pirazol-4-il)-1 H-indazol-3-carboxamida
(R)-N-(1 -cianopirrolidin-3-il)-6-(3,5-dimetilisoxazol-4-il)-1 -isobutil-1 H-indazol-3-carboxamida (R)-N-(1-cianopirrolidin-3-il)-1-(ciclopropilmetil)-6-(3,5-dimetilisoxazol-4-il)-1 H-indazol-3-carboxamida (R)-N-(1 -cianopirrolidin-3-il)-N-metil-6-(1 -metil-1 H-pirazol-4-il)imidazo[1,2-a]piridin-2-carboxamida (R)-N-(1 -cianopirrolidin-3-il)-N-metil-5-(1 -metil-1 H-pirazol-4-il)-1 H-indolo-2-carboxamida
(R)-N-(1-cianopirrolidin-3-il)-6-(3,5-dimetilisoxazol-4-il)-N-metil-1 H-benzo[d]imidazol-2-carboxamida (R)-N-(1 -cianopirrolidin-3-il)-7-(1 -metil-1 H-pirazol-4-il)imidazo[1,2-a]piridin-3-carboxamida
(R)-7-(3-cianofenil)-N-(1-cianopirrolidin-3-il)imidazo[1,2-a]piridin-3-carboxamida
(R)-N-(1-cianopirrolidin-3-il)-N-metil-7-(2-metilpiridin-4-il)imidazo[1,2-a]piridin-3-carboxamida (R)-N-(1-cianopirrolidin-3-il)-N-metil-7-(6-metilpiridin-3-il)imidazo[1,2-a]piridin-3-carboxamida (R)-N-(1 -cianopirrolidin-3-il)-7-(1,3-dimetil-1 H-pirazol-4-il)-N-metilimidazo[1,2-a]piridin-3-carboxamida (R)-N-(1-cianopirrolidin-3-il)-7-(2,6-dimetilpiridin-4-il)-N-metilimidazo[1,2-a]piridin-3-carboxamida (R)-N-(1-cianopirrolidin-3-il)-N-etil-7-(2-metilpiridin-4-il)imidazo[1,2-a]piridin-3-carboxamida
(R)-N-(1-cianopirrolidin-3-il)-7-morfolinoimidazo[1,2-a]piridin-3-carboxamida
(R)-6-(3-cianofenil)-N-(1-cianopirrolidin-3-il)-3-fluoroimidazo[1,2-a]piridin-2-carboxamida
(R)-N-(1 -cianopirrolidin-3-il)-3-fluoro-6-(1 -metil-1 H-pirazol-4-il)imidazo[1,2-a]piridin-2-carboxamida (R)-N-(1 -cianopirrolidin-3-il)-6-(1 -etil-1 H-pirazol-4-il)-3-fluoroimidazo[1,2-a]piridin-2-carboxamida (R)-N-(1 -cianopirrolidin-3-il)-6-(1,3-dimetil-1 H-pirazol-4-il)-3-fluoroimidazo[1,2-a]piridin-2-carboxamida (R)-N-(1-cianopirrolidin-3-il)-6-(3,5-dimetilisoxazol-4-il)-3-fluoroimidazo[1,2-a]piridin-2-carboxamida (R)-N-(1-cianopirrolidin-3-il)-2-fluoro-4-(pirazolo[1,5-a]pirimidin-5-il)benzamida
(R)-N-(1-cianopirrolidin-3-il)-5-(4-fluorofenil)picolinamida
N-((2R,3R)-1-ciano-2-metilpirrolidin-3-il)-5-(4-fluorofenil)picolinamida
3-cloro-N-((3R,4S)-1-ciano-4-metilpirrolidin-3-il)-4-morfolinobenzamida
N-((3R,4R)-1-ciano-4-fluoropirrolidin-3-il)-[1,1'-bifenil]-4-carboxamida
N-((3R,4R)-1 -ciano-4-ciclopropilpirrolidin-3-il)-3-fluoro-4-(1 -metil-1 H-pirazol-4-il)benzamida
N-((3S,4S)-1 -ciano-4-metoxipirrolidin-3-il)-N-metil-4-(1 -metil-1 H-pirazol-4-il)benzamida
(R)-N-(1 -cianopirrolidin-3-il)-5-(1,3-dimetil-1 H-pirazol-4-il)picolinamida
(R)-N-(1 -cianopirrolidin-3-il)-5-(2-metil-6-(trifluorometil)pirimidin-4-il)picolinam ida
(R)-N-(1-cianopirrolidin-3-il)-4-(2,6-dimetilpirimidin-4-il)-2-fluorobenzamida
(R)-N-(1-cianopirrolidin-3-il)-2-fluoro-4-(5-fluoro-2-metilpirimidin-4-il)benzamida
(R)-N-(1 -cianopirrolidin-3-il)-2-fluoro-4-(2-(trifluorometil)pirimidin-4-il)benzam ida
(R)-N-(1-cianopirrolidin-3-il)-2-fluoro-4-(2-metil-3H-pirrolo[2,3-d]pirimidin-4-il)benzamida
(R)-N-(1-cianopirrolidin-3-il)-2-fluoro-4-(imidazo[1,2-a]pirazin-3-il)benzamida
(R)-N-(1-cianopirrolidin-3-il)-3-fluoro-5-(pirazolo[1,5-a]pirimidin-5-il)picolinamida
(R)-N-(1-cianopirrolidin-3-il)-3-fluoro-5-(imidazo[1,2-a]piridin-6-il)picolinamida
(R)-N-(1-cianopirrolidin-3-il)-3-metoxi-3-fenilazetidin-1-carboxamida
(R)-N-(1-cianopirrolidin-3-il)-N-metil-3-fenilazetidin-1-carboxamida
(R)-N-(1-cianopirrolidin-3-il)-3-(4-metoxifenil)azetidin-1-carboxamida
(R)-3-(4-clorofenil)-N-(1-cianopirrolidin-3-il)azetidin-1-carboxamida
(R)-3-(3-clorofenil)-N-(1-cianopirrolidin-3-il)azetidin-1-carboxamida
(3aR,6aR)-1 -(3-fenilazetidin-1 -carbonil)hexahidropirrolo[3,4-b]pirrolo-5(1 H)-carbonitrilo
(R)-1 -(1 -cianopirrolidin-3-il)-1 -metil-3-(4-(1 -metil-1 H-pirazol-4-il)fenil)urea
(R)-1 -(1 -cianopirrolidin-3-il)-1 -metil-3-(4-(trifluorometil)fenil)urea
(3aR,6aR)-N-(4-cloro-2-fluorofenil)-5-cianohexahidropirrolo[3,4-b]pirrolo-1 (2H)-carboxamida (3aR,6aR)-5-ciano-N-(2-fluoro-4-(trifluorometoxi)fenil)hexahidropirrolo[3,4-b]pirrolo-1 (2H)-carboxamida (3aR,6aR)-5-ciano-N-(4-ciano-2-fluorofenil)hexahidropirrolo[3,4-b]pirrolo-1 (2H)-carboxamida (3aR,6aR)-5-ciano-N-(4-ciano-2,5-difluorofenil)hexahidropirrolo[3,4-b]pirrolo-1 (2H)-carboxamida (3aR,6aR)-N-(5-cloro-2-fluorofenil)-5-cianohexahidropirrolo[3,4-b]pirrolo-1 (2H)-carboxamida (3aR,6aR)-5-ciano-N-(2-fluoro-5-(trifluorometil)fenil)hexahidropirrolo[3,4-b]pirrolo-1 (2H)-carboxamida (3aR,6aR)-5-ciano-N-(5-fenilpiridin-2-il)hexahidropirrolo[3,4-b]pirrolo-1 (2H)-carboxamida
(3aR,6aR)-5-ciano-N-(4-(trifluorometil)fenil)hexahidropirrolo[3,4-b]pirrolo-1 (2H)-carboxamida
(R)-1 -(1 -cianopirrolidin-3-il)-1 -etil-3-(4-(trifluorometil)fenil)urea
1 -(1 -cianopirrolidin-3-il)-1 -(2-metoxietil)-3-(4-(trifluorometil)fenil)urea
(R)-N-(1 -cianopirrolidin-3-il)-N-etil-3-fluoro-4-(1 -metil-1 H-pirazol-4-il)benzamida
(R)-N-(1-cianopirrolidin-3-il)-N-etil-3-fenilazetidin-1-carboxamida
(R)-3-(2-oxo-3-(4-feniltiazol-2-il)imidazolidin-1 -il)pirrolidin-1 -carbonitrilo
(R)-3-(2-oxo-3-(4-feniltiazol-2-il)tetrahidropirimidin-1 (2H)-il)pirrolidin-1 -carbonitrilo
(R)-3-(3-(3-morfolinofenil)-2-oxoimidazolidin-1 -il)pirrolidin-1 -carbonitrilo
(R)-N-(1-cianopirrolidin-3-il)-4-(pirimidin-2-il)-3,4-dihidro-2H-benzo[b][1,4]oxazin-7-carboxamida
(R)-N-(1-cianopirrolidin-3-il)-4-(4-ciclopropilpirimidin-2-il)-3,4-dihidro-2H-benzo[b][1,4]oxazin-7-carboxamida (R)-N-(1-cianopirrolidin-3-il)-4-((4-ciclopropilpirimidin-2-il)amino)-3-fluorobenzamida
(R)-N-(1-cianopirrolidin-3-il)-4-((4-ciclopropilpirimidin-2-il)amino)-2,3-difluorobenzamida
(R)-N-(1-cianopirrolidin-3-il)-4-(N-metilisobutiramido)picolinamida
(R)-N-(1-cianopirrolidin-3-il)-[2,3'-bipiridino]-6'-carboxamida
(R)-N-(1-cianopirrolidin-3-il)-[2,4'-bipiridino]-2'-carboxamida
(R)-3-(4-clorofenil)-N-(1-cianopirrolidin-3-il)isoxazol-5-carboxamida
(R)-N-(1-cianopirrolidin-3-il)-3-(4-(trifluorometil)fenil)isoxazol-5-carboxamida
(R)-N-(1-cianopirrolidin-3-il)-3-(3,4-dimetoxifenil)isoxazol-5-carboxamida
(R)-N-(1-cianopirrolidin-3-il)-3-(3-metoxifenil)isoxazol-5-carboxamida
N-((R)-1 -cianopirrolidin-3-il)-1 -fenilpirrolidin-3-carboxam ida
(R)-N-(1 -cianopirrolidin-3-il)-3-fluoro-4-(4-metil-1 H-imidazol-1 -il)benzamida
(R)-N-(1 -cianopirrolidin-3-il)-3-fluoro-N-metil-4-(4-metil-1 H-imidazol-1 -il)benzamida
N-((R)-1 -cianopirrolidin-3-il)-3-(piridin-2-il)pirrolidin-1 -carboxamida
N-((R)-1 -cianopirrolidin-3-il)-3-(1 -metil-1 H-pirazol-4-il)pirrolidin-1 -carboxamida
(R)-N-(1-cianopirrolidin-3-il)-3-(2-metoxipiridin-4-il)-N-metilisoxazol-5-carboxamida
(R)-N-(1-cianopirrolidin-3-il)-2-fluoro-4-(N-metilfenilsulfonamido)benzamida
(R)-N-(1 -cianopirrolidin-3-il)-1 -metil-6-(1 -metil-1 H-pirazol-4-il)-1 H-indolo-2-carboxamida
(R)-N-(1 -cianopirrolidin-3-il)-1 -metil-5-(1 -metil-1 H-pirazol-4-il)-1 H-indolo-2-carboxamida
(R)-1 -(1 -cianopirrolidin-3-il)-3-(2-(isoindolin-2-il)piridin-4-il)-1 -metilurea
(R)-N-(1 -cianopirrolidin-3-il)-3-f luoro-1 -metil-5-(1 -metil-1 H-pirazol-4-il)-1 H-indolo-2-carboxamida
(R)-N-(1 -cianopirrolidin-3-il)-7-(1 -metil-1 H-pirazol-4-il)imidazo[1,5-a]piridin-3-carboxamida
(R)-N-(1 -cianopirrolidin-3-il)-3-(1 -fenil-1 H-pirazol-3-il)azetidin-1 -carboxamida
(R)-N-(1 -cianopirrolidin-3-il)-3-(1 -(pirazin-2-il)-1 H-pirazol-3-il)azetidin-1 -carboxamida
(R)-N-(1 -cianopirrolidin-3-il)-3-(2-fenilpirimidin-4-il)azetidin-1 -carboxam ida
(R)-3-(2-(4-clorofenil)pirimidin-4-il)-N-(1-cianopirrolidin-3-il)azetidin-1-carboxamida
(R)-3-(benciloxi)-N-(1-cianopirrolidin-3-il)-3-fenilazetidin-1-carboxamida
(R)-N-(1-cianopirrolidin-3-il)-1-(4-ciclopropilpirimidin-2-il)indolin-5-carboxamida
(R)-N-(1-cianopirrolidin-3-il)-1-(4-ciclopropilpirimidin-2-il)-N-metilindolin-5-carboxamida
(3aR,6aR)-5-ciano-N-(3-(2-metilpiridin-4-il)fenil)hexahidropirrolo[3,4-b]pirrolo-1 (2H)-carboxamida (3aR,6aR)-5-ciano-N-(4-(2-metilpiridin-4-il)fenil)hexahidropirrolo[3,4-b]pirrolo-1 (2H)-carboxamida (3aR,6aR)-5-ciano-N-(2-fluoro-4-(2-metilpiridin-4-il)fenil)hexahidropirrolo[3,4-b]pirrolo-1 (2H)-carboxamida (3aR,6aR)-5-ciano-N-(2'-metil-[3,4'-bipiridin]-6-il)hexahidropirrolo[3,4-b]pirrolo-1 (2H)-carboxamida (3aR,6aR)-5-ciano-N-(2-fluoro-4-(1-metil-1 H-pirazol-4-il)fenil)hexahidropirrolo[3,4-b]pirrolo-1 (2H)-carboxamida 1 -(3-fenil-1 H-pirazol-5-carbonil)hexahidropirrolo[3,4-b]pirrolo-5(1 H)-carbonitrilo
(3aR,6aR)-1 -(3-fenoxiazetidin-1 -carbonil)hexahidropirrolo[3,4-b]pirrolo-5(1 H)-carbonitrilo
N-(1-cianopiperidin-3-il)-[1,1'-bifenil]-3-carboxamida
1 -(3-bencilfenil)-3-(1 -cianopiperidin-3-il)urea
1 -(1 -cianopiperidin-3-il)-3-(3-fenoxifenil)urea
1 -(1 -cianopirrolidin-3-il)-3-(2,4-diclorofenil)urea
1 -(1 -cianopirrolidin-3-il)-3-(4-(trifluorometil)fenil)urea
1 -(3-bencilfenil)-3-(1 -cianopirrolidin-3-il)urea
1 -([1,1'-bifenil]-4-il)-3-(1 -cianopirrolidin-3-il)urea
1 -(1 -cianopirrolidin-3-il)-3-(3-fenoxifenil)urea
3-(3-bencilfenil)-1 -(1 -cianopirrolidin-3-il)-1 -metilurea
3-(3-clorofenil)-1 -(1 -cianopirrolidin-3-il)-1 -metilurea
1 -(1 -cianopirrolidin-3-il)-1 -metil-3-(3-fenoxifenil)urea
3-([1,1'-bifenil]-4-il)-1 -(1 -cianopirrolidin-3-il)-1 -metilurea
1 -(1 -cianopirrolidin-3-il)-3-(2,4-diclorofenil)-1 -metilurea
1 -(1 -cianopirrolidin-3-il)-1 -metil-3-(4-(trifluorometil)fenil)urea
(R)-N-(1-cianopirrolidin-3-il)-6-(3,5-dimetilisoxazol-4-il)-N-metil-1 H-indolo-2-carboxamida
(R)-N-(1 -cianopirrolidin-3-il)-N-metil-5-(1 -metil-1 H-pirazol-4-il)-1 H-pirrolo[2,3-c]piridin-2-carboxamida
N-((R)-1-cianopirrolidin-3-il)-N-metil-2-fenilmorfoline-4-carboxamida
(R)-N-(1 -cianopirrolidin-3-il)-N-metilindolin-1 -carboxamida
(R)-1 -(1 -cianopirrolidin-3-il)-1 -metil-3-(6-(trifluorometil)piridin-3-il)urea
(R)-3-(5-cloropiridin-2-il)-1 -(1 -cianopirrolidin-3-il)-1 -metilurea
(3aR,6aR)-1-(3-cloro-4-morfolinobenzoil)hexahidropirrolo[3,4-b]pirrolo-5(1 H)-carbonitrilo
(3aR,6aR)-1 -(indolin-1 -carbonil)hexahidropirrolo[3,4-b]pirrolo-5(1 H)-carbonitrilo
(R)-N-(1-cianopirrolidin-3-il)-3-(2-metilpiridin-4-il)isoxazol-5-carboxamida
(R)-N-(1-cianopirrolidin-3-il)-3-(3,4-dimetilfenil)isoxazol-5-carboxamida
(R)-N-(1-cianopirrolidin-3-il)-3-(2,4-difluorofenil)isoxazol-5-carboxamida
(R)-N-(1-cianopirrolidin-3-il)-N-metil-3-(2-metilpiridin-4-il)isoxazol-5-carboxamida
Cabe señalar que cada uno de los compuestos químicos enumerados anteriormente representa un aspecto particular e independiente de la invención.
En la presente memoria se describe un procedimiento para la preparación de un compuesto de fórmula (I) o una de sus sales farmacéuticamente aceptables que comprende las etapas de hacer reaccionar una amina de fórmula (IV) con un compuesto R12-Y-COOH para formar una amida:

Donde R1, R2, R3, R8, R9, R10, X y n, son como se definen en otra parte y PG es un grupo protector de amina. El grupo protector puede ser, pero no se limita a, BOC. Para un experto en la técnica resulta evidente combinar o ajustar tal grupo químico protector. Después del acoplamiento de R12-Y-COOH para formar una amida, el grupo protector se puede retirar para dejar la amina libre según la fórmula (V) que a continuación se puede tratar con bromuro de cianógeno para formar compuestos según la fórmula (I).
También se describe un procedimiento para la preparación de un compuesto de fórmula (I) o una de sus sales farmacéuticamente aceptables que comprende las etapas de hacer reaccionar una amina de fórmula (V) con bromuro de cianógeno para formar compuestos de N-CN:
Según un aspecto adicional de la divulgación, se proporciona un procedimiento para la preparación de un compuesto de fórmula (II) o una de sus sales farmacéuticamente aceptables que comprende las etapas de hacer reaccionar una amina de fórmula (IVA) con un compuesto R12-Y-COOH para formar una amida:

Donde R1, R2, R3, R4, R5, R8, R9, R10, R12, Z y m, son como se definen en otra parte para la fórmula II y PG es un grupo protector de amina. El grupo protector puede ser, pero no se limita a, BOC. Para un experto en la técnica resulta evidente combinar o ajustar tal grupo químico protector. Después del acoplamiento de R12-Y-COOH para formar una amida, el grupo protector se puede retirar para dejar la amina libre según la fórmula (VA) que a continuación se puede tratar con bromuro de cianógeno para formar compuestos según la fórmula (II).
Según un aspecto adicional de la divulgación, se proporciona un procedimiento para la preparación de un compuesto de fórmula (II) o una de sus sales farmacéuticamente aceptables que comprende las etapas de hacer reaccionar una amina de fórmula (VA) con bromuro de cianógeno para formar compuestos de N-CN:

Donde R1, R2, R3, R4, R5, R8, R9, R10, Z y m, son como se definen en otra parte para la fórmula II.
Según otro aspecto de la invención, se proporciona una composición farmacéutica que comprende un compuesto de fórmula (II).
Las composiciones farmacéuticas de esta invención comprenden cualquiera de los compuestos de la invención combinados con cualquier portador, adyuvante o vehículo farmacéuticamente aceptables. Los expertos en la técnica conocen ejemplos de portadores farmacéuticamente aceptables e incluyen, entre otros, agentes conservantes, cargas, agentes disgregantes, agentes humectantes, agentes emulsionantes, agentes de suspensión, agentes edulcorantes, agentes aromatizantes, agentes perfumantes, agentes antibacterianos, agentes antifúngicos, agentes lubricantes y agentes dispersantes, dependiendo de la naturaleza del modo de administración y formas de dosificación. Las composiciones pueden estar en forma de, por ejemplo, comprimidos, cápsulas, polvos, gránulos, elixires, pastillas, supositorios, jarabes y preparaciones líquidas que incluyen suspensiones y soluciones. El término "composición farmacéutica" en el contexto de esta invención significa una composición que comprende un agente activo y que comprende adicionalmente uno o más vehículos farmacéuticamente aceptables. La composición puede contener adicionalmente ingredientes seleccionados entre, por ejemplo, diluyentes, adyuvantes, excipientes, vehículos, agentes conservantes, cargas, agentes disgregantes, agentes humectantes, agentes emulsionantes, agentes de suspensión, agentes edulcorantes, agentes aromatizantes, agentes perfumantes, agentes antibacterianos, agentes antifúngicos, agentes lubricantes y agentes dispersantes, dependiendo de la naturaleza del modo de administración y formas de dosificación.
Según un aspecto adicional de la invención, se proporciona un compuesto de fórmula (II) o una composición farmacéutica del mismo para su uso en terapia.
También se describe un compuesto de fórmula (IA) o una composición farmacéutica del mismo para su uso en el tratamiento del cáncer y afecciones que implican disfunción mitocondrial.

o una de sus sales farmacéuticamente aceptables, en donde:
n es 1 o 2;
cuando n es 1, X es CR4R5 y cuando n es 2, X es CR6R7CR4R5 (donde CR4R5 es adyacente al átomo de N del heterociclo);
R2 representa un átomo de hidrógeno, ciano, un alquilo C1-C6 opcionalmente sustituido, un grupo alcoxi C1-C6 opcionalmente sustituido, un anillo de heteroarilo, heterociclilo, arilo de 4 a 10 miembros o cicloalquilo de 3 a 8 miembros opcionalmente sustituido;
R1, R3, R4 y R5 representan cada uno independientemente un átomo de hidrógeno, ciano, un grupo alquilo C1-C3 opcionalmente sustituido o alcoxi C1-C3 opcionalmente sustituido;
R6, R7 y R8 representan cada uno independientemente un átomo de hidrógeno, un átomo de flúor, ciano, un grupo alquilo C1-C3 opcionalmente sustituido o alcoxi C1-C3 opcionalmente sustituido;
R9 representa un átomo de hidrógeno, un átomo de flúor, ciano, un alquilo C1-C6 opcionalmente sustituido, un grupo alcoxi C1-C3 opcionalmente sustituido, un anillo de heteroarilo, heterociclilo, arilo de 4 a 10 miembros o cicloalquilo de 3 a 8 miembros opcionalmente sustituido, o forma un anillo heterocíclico opcionalmente sustituido con R10 en donde el anillo comprende opcionalmente uno o más heteroátomos adicionales;
R10 representa un átomo de hidrógeno, alquilo C1-C6, o forma un anillo heterocíclico opcionalmente sustituido con R9 o R11 en donde el anillo comprende opcionalmente uno o más heteroátomos adicionales;
Y representa un enlace covalente, NR11 o alquileno C1-C3 opcionalmente sustituido;
R11 representa un átomo de hidrógeno, un alquilo C1-C6 opcionalmente sustituido, un anillo de heteroarilo, heterociclilo, arilo de 4 a 10 miembros o cicloalquilo de 3 a 8 miembros, o forma un anillo heterocíclico opcionalmente sustituido con R10 en donde el anillo comprende opcionalmente uno o más heteroátomos adicionales;
R12 representa un anillo de heteroarilo, heterociclilo, arilo de 4 a 10 miembros o cicloalquilo de 3 a 8 miembros opcionalmente sustituido.
Las definiciones adicionales de R1, R2, R3, R4, R5, R6, R7, R8, R9, R10, R11, R12 e Y incluyen las descritas en las realizaciones anteriores para las fórmulas (I), (IB) e (IID).
Afecciones que implican disfunción mitocondrial
Los compuestos de la invención según las fórmulas (I), (IA) y (II) pueden utilizarse en el tratamiento de trastornos o enfermedades que tienen un componente relacionado con la disfunción mitocondrial, en particular trastornos o enfermedades vinculadas a la actividad de DUB. Más particularmente, los trastornos o enfermedades se relacionan con la actividad de USP30.
Los compuestos de fórmulas (I), (IA) y (II) como se describen en la presente memoria se pueden utilizar en la fabricación de un medicamento para el tratamiento de afecciones que implican disfunción mitocondrial.
En un aspecto adicional de la divulgación, se proporciona un método de tratamiento o prevención de una afección que implica disfunción mitocondrial, comprendiendo el método administrar una cantidad farmacéuticamente eficaz de un compuesto de fórmulas (I), (IA) y (II) o una composición farmacéutica del mismo a un individuo diagnosticado de una afección que implica disfunción mitocondrial.
Las disfunciones mitocondriales son el resultado de defectos de las mitocondrias, que son compartimentos especializados presentes en todas las células del organismo, excepto en los glóbulos rojos. Cuando las mitocondrias fallan, se genera cada vez menos energía dentro de la célula y se producirá una lesión celular o incluso la muerte celular. Si este procedimiento se repite en todo el organismo, la vida del sujeto en donde esto ocurre se ve seriamente comprometida. Las enfermedades de las mitocondrias aparecen con mayor frecuencia en órganos que requieren
mucha energía, como el cerebro, el corazón, el hígado, los músculos esqueléticos, los riñones y el sistema endocrino y respiratorio.
La afección que implica disfunción mitocondrial se puede seleccionar entre una afección que implica un defecto de mitofagia, una afección que implica una mutación en el ADN mitocondrial, una afección que implica estrés oxidativo mitocondrial, una afección que implica un defecto en el potencial de la membrana mitocondrial, biogénesis mitocondrial, una afección que implica un defecto en la forma o morfología mitocondrial, y una afección que implica un defecto de almacenamiento lisosómico.
En particular, la afección que implica la disfunción mitocondrial se puede seleccionar entre una enfermedad neurodegenerativa; síndrome de encefalopatía mitocondrial, acidosis láctica y episodios similares a apoplejía (MELAS); neuropatía óptica hereditaria de Leber (LHON); cáncer; neuropatía, ataxia, retinosis pigmentaria-síndrome de Leigh heredado de la madre (NARP-MILS); enfermedad de Danon; diabetes; trastornos metabólicos; cardiopatía isquémica que conduce a infarto de miocardio; enfermedades psiquiátricas, por ejemplo, esquizofrenia; deficiencia múltiple de sulfatasa (MSD); mucolipidosis II (ML II); mucolipidosis III (ML III); mucolipidosis IV (ML IV); gangliosidosis GM1 (GM1); lipofuscinosis ceroides neuronales (NCL1); enfermedad de Alpers; síndrome de Barth; defectos de betaoxidación; deficiencia de carnitina-acil-carnitina; deficiencia de carnitina; síndromes de deficiencia de creatina; deficiencia de coenzima Q10; deficiencia de complejo I; deficiencia de complejo II; deficiencia de complejo III; deficiencia de complejo IV; deficiencia de complejo V; deficiencia de COX; síndrome de oftalmoplejía externa progresiva crónica (CPEO); deficiencia de CPT I; deficiencia de CPT II; aciduria glutárica tipo II; síndrome de Kearns-Sayre; acidosis láctica; deficiencia de acil-CoA deshidrogenasa de cadena larga (LCHAD); enfermedad o síndrome de Leigh; miocardiopatía letal infantil (LIC); enfermedad de Luft; aciduria glutárica tipo II; deficiencia de acil-CoA deshidrogenasa de cadena media (MCAD); epilepsia mioclónica y síndrome de fibra roja irregular (MERRF); citopatía mitocondrial; síndrome de ataxia mitocondrial recesiva; síndrome de depleción del ADN mitocondrial; trastorno mioneurogastointestinal y encefalopatía; síndrome de Pearson; deficiencia de piruvato deshidrogenasa; deficiencia de piruvato carboxilasa; mutaciones POLG; deficiencia de 3-hidroxiacil-CoA deshidrogenasa de cadena media/corta (M/SCHAD); y deficiencia de acil-CoA deshidrogenasa de cadena muy larga (VLCAD).
La afección que implica la disfunción mitocondrial puede ser un trastorno del SNC, por ejemplo, una enfermedad neurodegenerativa.
Las enfermedades neurodegenerativas incluyen, pero no se limitan a, enfermedad de Parkinson, enfermedad de Alzheimer, esclerosis lateral amiotrófica (ELA), enfermedad de Huntington, isquemia, accidente cerebrovascular, demencia con cuerpos de Lewy y demencia frontotemporal.
En una realización particular, los compuestos de la invención son útiles en el tratamiento de la enfermedad de Parkinson, incluyendo, pero sin limitarse a, la EP relacionada con mutaciones en a-sinucleína, parkina y PINK1, enfermedad de Parkinson juvenil autosómica recesiva (AR-JP) donde la parkina está mutada.
Los compuestos de fórmulas (I), (IA) y (II) o las composiciones farmacéuticas de los mismos como se describen en la presente memoria se pueden combinar con uno o más agentes adicionales cuando se utilizan para el tratamiento de afecciones que implican disfunción mitocondrial. Los compuestos se pueden combinar con uno o más agentes adicionales seleccionados entre levodopa, un agonista de la dopamina, un inhibidor de la monoaminooxigenasa (MAO) B, un inhibidor de la catecol O-metiltransferasa (COMT), un anticolinérgico, riluzol, amantadina, un inhibidor de colinesterasa, memantina, tetrabenazina, un antipsicótico, diazepam, clonazepam, un antidepresivo y un anticonvulsivo.
Cáncer
Los compuestos de fórmulas (I) y (IA) también tienen uso en el tratamiento del cáncer y más concretamente en el tratamiento del cáncer relacionado con la actividad de DUB, especialmente la actividad de USP30.
Los compuestos de la invención según la fórmula (II) también tienen uso en el tratamiento del cáncer y más concretamente en el tratamiento del cáncer ligado a la actividad de DUB o la actividad de desumoilación. Los compuestos de la invención pueden ser útiles contra cualquier DUB o enzima desumoilante, incluyendo, pero sin limitarse a USP30 y USP10.
Los compuestos descritos en la presente memoria se pueden utilizar en la fabricación de un medicamento para el tratamiento del cáncer ligado a la actividad de DUB.
Los compuestos de fórmulas (I), (IB) y (II) como se describen en la presente memoria también se pueden utilizar en la fabricación de un medicamento para el tratamiento de un cáncer. En un aspecto adicional de la divulgación, se proporciona un método de tratamiento o prevención de un cáncer, comprendiendo el método administrar una cantidad farmacéuticamente eficaz de un compuesto de fórmulas (I), (IB) y (II) o una composición farmacéutica del mismo a un individuo que padece un cáncer.
Los compuestos de la invención también tienen uso en el tratamiento del cáncer relacionado con la disfunción mitocondrial.
En una realización, los compuestos según la invención tienen uso en el tratamiento del cáncer cuando las rutas apoptóticas están desreguladas y más concretamente cuando las proteínas de la familia BCL-2 están mutadas, o expresadas en exceso o insuficientemente expresadas.
Las referencias a "cáncer" o "tumor" incluyen, pero no se limitan, a cáncer de mama, ovario, próstata, pulmón, riñón, estómago, colon, testicular, cabeza y cuello, páncreas, cerebro, melanoma, hueso u otros cánceres de tejidos y órganos y cánceres de las células sanguíneas tales como linfomas y leucemias. Los cánceres particulares incluyen linfoma, mieloma múltiple, cáncer colorrectal y carcinoma de pulmón de células no pequeñas.
Los compuestos de fórmulas (I), (IA) y (II) o composiciones farmacéuticas de los mismos como se describen en la presente memoria se pueden combinar con uno o más agentes adicionales cuando se utilizan para el tratamiento del cáncer. Los compuestos se pueden combinar con un agente terapéutico antitumoral adicional, por ejemplo, fármacos quimioterapéuticos o inhibidores de otras proteínas reguladoras. En una realización, el agente terapéutico antitumoral adicional es un mimético de BH-3, En una realización adicional, los miméticos de BH-3 se pueden seleccionar entre, pero sin limitarse a, uno o más de ABT-737, ABT-199, ABT-263 y Obatoclax. En una realización adicional, el agente antitumoral adicional es un agente quimioterapéutico. Los agentes quimioterapéuticos se pueden seleccionar entre, pero sin limitarse a, olaparib, mitomicina C, cisplatino, carboplatino, oxaliplatino, radiación ionizante (IR), camptotecina, irinotecán, topotecán, temozolomida, taxanos, 5-fluoropirimidinas, gemcitabina y doxorrubicina.
Para tratar un trastorno de disfunción mitocondrial, las composiciones farmacéuticas de la invención se pueden diseñar para su administración por vía oral, parenteral o mucosa y la elección o la forma específica de composición dependen de la vía de administración. Así, para la administración oral, la composición puede estar en forma, por ejemplo, de comprimidos, pastillas, grageas, películas, polvos, elixires, jarabes, preparaciones líquidas que incluyen dispersiones, suspensiones, emulsiones, soluciones o aerosoles, sellos, gránulos, cápsulas, etc. Para la administración a las mucosas, la composición puede estar en forma de aerosoles, inhalantes, dispersiones, suspensiones, emulsiones, soluciones, geles, parches, películas, ungüentos, cremas, lociones, supositorios, etc. Para la administración parenteral, la composición está en forma de una preparación líquida tal como una solución, dispersión, emulsión o suspensión que incluye composiciones de liposomas.
Para tratar un trastorno del SNC, los compuestos de la invención deben tener la capacidad de atravesar la barrera hematoencefálica. En cuanto tal, tales compuestos tienen la capacidad de ingresar al sistema nervioso central de un paciente. Alternativamente, las composiciones farmacéuticas de la presente invención pueden eludir la barrera hematoencefálica mediante el uso de composiciones y métodos conocidos en la técnica para eludir la barrera hematoencefálica o se pueden inyectar directamente al cerebro. Las zonas adecuadas para la inyección incluyen la corteza cerebral, cerebelo, mesencéfalo, tronco encefálico, hipotálamo, médula espinal y tejido ventricular, y zonas del SNP que incluyen el cuerpo carotídeo y la médula suprarrenal. Otras formas de dosificación incluyen aquellas adecuadas para administración oral que incluyen, entre otras, comprimidos, grageas, polvos, elixires, jarabes, preparaciones líquidas que incluyen suspensiones, aerosoles, inhalantes, comprimidos, pastillas, emulsiones, soluciones, sellos, gránulos y cápsulas. Para la administración parenteral, las preparaciones incluyen soluciones, suspensiones y emulsiones acuosas, acuosas-orgánicas y orgánicas estériles.
Para tratar un cáncer, las composiciones farmacéuticas de la invención se pueden administrar de cualquier manera eficaz adecuada para dirigirse a las células cancerosas, por ejemplo, por vía oral en cualquier forma de dosificación aceptable por vía oral, incluidas, pero sin limitarse a, comprimidos, grageas, polvos, elixires, jarabes, líquidos. preparaciones que incluyen suspensiones, aerosoles, inhalantes, comprimidos, pastillas, emulsiones, soluciones, sellos, gránulos y cápsulas. Las preparaciones según la invención para administración parenteral incluyen soluciones, suspensiones y emulsiones acuosas, acuosas-orgánicas y orgánicas estériles.
Tales formas de dosificación se preparan según mecanismos conocidos en la técnica de la formulación farmacéutica. Cuando están en forma de aerosoles o inhalantes, las composiciones farmacéuticas se pueden administrar por vía nasal. Los expertos en la técnica conocen formulaciones adecuadas para este fin.
Las composiciones farmacéuticas de la invención se pueden administrar mediante inyección y pueden estar en forma de una preparación líquida estéril para su inyección, que incluye preparaciones de liposomas.
Las composiciones farmacéuticas de la invención también pueden estar en forma de supositorios para administración rectal. Estos se formulan de modo que la composición farmacéutica sea sólida a temperatura ambiente y líquida a la temperatura corporal para permitir la liberación del compuesto activo.
Las dosificaciones se pueden variar dependiendo de los requisitos del paciente, la gravedad de la afección que se esté tratando y el compuesto que se esté empleando. La determinación de la dosis adecuada para una situación particular es competencia del experto en la técnica. Generalmente, el tratamiento se inicia con dosis más pequeñas que son menores que la dosis óptima del compuesto. A partir de entonces, la dosis se aumenta en pequeños incrementos hasta que se alcanza el efecto óptimo conforme a las circunstancias.
La magnitud de una dosis eficaz de un compuesto variará, por supuesto, con la naturaleza de la gravedad de la afección que se vaya a tratar y con el compuesto concreto y su vía de administración. La selección de las dosis apropiadas está dentro de la capacidad de un experto medio en esta técnica, sin una carga excesiva. El intervalo de
dosis diaria es de aproximadamente 10 pg a aproximadamente 100 mg por kg de peso corporal de un animal humano y no humano y, en general, puede ser de aproximadamente 10 pg a 30 mg por kg de peso corporal por dosis. La dosis anterior se puede administrar de una a tres veces al día.
Metodologías sintéticas
Los compuestos de la invención se pueden preparar mediante una variedad de rutas sintéticas. A continuación, se muestran rutas ilustrativas para ciertos compuestos de la invención. Los compuestos representativos de la presente invención se pueden sintetizar según los métodos sintéticos generales descritos a continuación y se ilustran más concretamente en los esquemas que siguen. Dado que los esquemas son una ilustración, no se debe considerar que la invención está limitada por las reacciones químicas y las condiciones expresadas. La preparación de los diversos materiales de partida utilizados en los esquemas está dentro del conocimiento de las personas versadas en la técnica. Los expertos en la técnica apreciarán que, cuando sea apropiado, las transformaciones individuales dentro de un esquema se pueden completar en un orden diferente. Los siguientes esquemas describen métodos sintéticos generales mediante los cuales se pueden preparar compuestos intermedios y diana de la presente invención. Se pueden sintetizar compuestos y estereoisómeros, mezclas racémicas, diastereómeros y enantiómeros de los mismos representativos adicionales utilizando los intermedios preparados según los esquemas generales y otros materiales, compuestos y reactivos conocidos por los expertos en la técnica. Se pretende que todos estos compuestos, estereoisómeros, mezclas racémicas, diastereómeros y enantiómeros de los mismos estén incluidos dentro del alcance de la presente invención.
Los compuestos se caracterizaron mediante cromatografía liquida-espectroscopia de masas (LCMS) y/o RMN H1 Abreviaturas:
ac Acuoso
Ar Arilo
BOC Terc-butiloxicarbonilo
br Ancho (señal de RMN)
d Doblete (señal de RMN)
CDI Carbonildiimidazol
DCM Diclorometano
DCE 1,2-Dicloroetano
DIPEA Diisopropiletilamina
DMA Dimetilacetamida
DMAP Dimetilaminopiridina
DMF N,N-dimetilformamida
DMSO Dimetilsulfóxido
dppf 1,1'-Bis(difenilfosfino)ferroceno
EDC 1-Etil-3-(3-dimetilaminopropil)carbodiimida
ES Electronebulización
EtOAc Acetato de etilo
EtOH Etanol
Fmoc Fluorenilmetiloxicarbonilo
h Hora u horas
HATU Hexafluorofosfato de 1H-1,2,3-triazolo[4,5-b]piridinio-3-óxido de 1-[bis(dimetilamino)metileno] HBTU Hexafluorofosfato de O-benzotriazol-N,N,N',N'-tetrametil-uronio
HOAt 1 -Hidroxi-7-azabenzotriazol
HOBT 1-Hidroxibenzotriazol
IPA Alcohol isopropílico
LDA Diisopropilamida de litio
LiHMDS Hexametildisilazida de litio
m Multiplete (señal de RMN)
MeCN Acetonitrilo
MeOH Metanol
min Minutos)
NCS N-clorosuccinimida
PE Éter de petróleo
rt Temperatura ambiente
RT Tiempo de retención
s Singlete (señal de RMN)
t Triplete (señal de RMN)
T3P 2,4,6-T ripropil-1,3,5,2,4,6-trioxatrifosforinano-2,4,6-trióxido TBAI Yoduro de tetrabutilamonio
TBD 1,5,7-T riazabiciclo[4,4,0]dec-5-eno
TÉ Trietilamina
TFAA Anhídrido trifluoroacético
TFA Ácido trifluoroacético
THF Tetrahidrofurano
TLC Cromatografía en capa fina
Xantphos 4,5-Bis(difenilfosfino)-9,9-dimetilxanteno
X-Phos 2-Diciclohexilfosfino-2',4',6'-triisopropilbifenilo Métodos analíticos:















Reactivos y condiciones: a) EDC.HCl, HOBT, DIPEA, THF, rt, 15 h; b) TFA, DCM, 0°C y a continuación a 50°C, 12 h; c) bromuro de cianógeno, K2CO3, THF, 0°C a continuación a rt, 30 min
Método general B

Reactivos y condiciones: a) Pd (PPh3)4, Na2CO3, 1,4-dioxano:agua (2:1), 90°C, 8 h; b) UOH.H2O, THF, agua (1:1), rt, 3 h; c) HATU, DIPEA, THF, 0°C y a continuación a rt, 2 h; d) TFA, DCM, 0°C y a continuación a rt, 2 h; e) bromuro de cianógeno, K2CO3, THF, 0°C a continuación a rt, 1 h
Método general C

Reactivos y condiciones: a) T3P (50% en EtOAc), DIPEA, THF, 0°C y a continuación a rt, 1,5 h; b) ArB(OH)2, Pd(PPh3)4, K2CO3, 1,4-dioxano:agua (5:1), 80°C, 2 h; c) TFA, DCM, 0°C y a continuación a rt, 2 h; d) bromuro de cianógeno, K2CO3, THF, 0°C a continuación a rt, 20 min
Método general D
Reactivos y condiciones: a) HATU, DIPEA, DCM, 0°C a continuación rt, 16 h; b) R1R2NH, CS2CO3, DMF, 120°C, 16 h; c) HCl/EtOAc, rt, 2 h; d) bromuro de cianógeno, NaHCO3, EtOH, rt, 16 h
Método general E

Reactivos y condiciones: a) CDI, agua, (o THF o DCM), 0°C, 30 min y a continuación rt, 18 h (o trifosgeno, TEA, DCM, 0°C); b) TFA, DCM, rt, 3 h; c) bromuro de cianógeno, DIPEA, DCM, 0°C, 30 min
Método general F

Reactivos y condiciones: a) (opcionalmente sustituido) cis-2,6-dimetilmorfolina, NaOtBu, Xantphos, Pd2(dba)3, tolueno, 110°C, 1 h; b) LiOH.H2O, THF, agua, 50°C 4 h, a continuación, a rt, 15 h; c) HATU, DIPEA, DMF, rt, 2 h; d) TFA, DCM, rt, 1 h; e) bromuro de cianógeno, K2CO3, THF, rt, 30 min
Método general G

Reactivos y condiciones: a) HATU, DIPEA, THF, rt, 4 h; b) NaOtBu, DBU, BINAP, Pd2(dba)3, tolueno, 110°C, 1 h; c)
TFA, DCM, rt 1 h; d) bromuro de cianógeno, K2CO3, THF, rt, 30 min
Método general H
Reactivos y condiciones: a) HATU, DIPEA, THF, rt, 4 h; b) R1R2NH, NaOtBu o Cs2CO3, BINAP o Xantphos, Pd2(dba)3, tolueno o dioxano:agua, 110°C, 1 h; c) TFA, DCM, rt 1 h; d) bromuro de cianógeno, K2CO3, THF, rt, 30 min Ejemplo 1 (R)-N-(1-cianopirrolidin-3-il)-5-fenilpicolinamida
(Preparada según el método general A)
Etapa a. A una solución de (R)-3-amino-1N-BOC-pirrolidina (0,24 mmoles) en THF (10 ml) se le añadieron EDC.HCl (0,33 mmoles), HOBt (0,33 mmoles) y DIPEA (0,45 mmoles) a rt. La mezcla de reacción se agitó a rt durante 15 min. Se añadió ácido 5-fenilpiridin-2-carboxílico (0,22 mmoles) a la mezcla de reacción a rt y se agitó durante 15 h. La mezcla de reacción resultante se vertió en agua (50 ml) y se extrajo con EtOAc (3 x 10 ml). La fase orgánica combinada se recogió, se secó sobre Na2SO4 , se filtró y se concentró a presión reducida produciendo (R)-3-(5-fenilpicolinamido)pirrolidin-1 -carboxilato de terc-butilo (cuantitativo). Esta sustancia se utilizó directamente para la siguiente etapa sin purificación adicional. MS: ES+ 312,2 (M-tBu).
Etapa b. A una solución de (R)-3-(5-fenilpicolinamido)pirrolidin-1 -carboxilato de terc-butilo (0,27 mmoles) en DCM (10 ml) se le añadió TFA (1 ml) a 0°C. La mezcla de reacción se agitó a rt durante 3 h y a continuación se calentó a 50°C durante 12 h. La mezcla de reacción resultante se concentró a presión reducida. El residuo obtenido se destiló azeotrópicamente utilizando DCM (2 x 10 ml) y se secó a presión reducida produciendo la sal TFA de (R)-5-fenil-N-(pirrolidin-3-il) picolinamida (cuantitativo). Esta sustancia se utilizó directamente para la siguiente etapa sin purificación adicional. MS: ES+ 268,2
Etapa c. A una solución de sal de TFA de (R)-5-fenil-N-(pirrolidin-3-il)picolinamida (0,26 mmoles) en THF (15 ml) se le añadió K2CO3 (1,3 mmoles) a 0°C. La mezcla de reacción se agitó a 0°C durante 30 min. Se añadió bromuro de cianógeno (0,31 mmoles) a la mezcla de reacción a 0°C. A continuación, la mezcla de reacción se agitó a rt durante 30 min. La mezcla de reacción resultante se vertió en agua (50 ml) y se extrajo con EtOAc (3 x 10 ml). La fase orgánica combinada se recogió, se secó sobre Na2SO4, se filtró y se concentró a presión reducida. El resto resultante se purificó mediante cromatografía en columna (EtOAc al 40% en hexano) produciendo el compuesto del título (0,11 mmoles). LCMS: Método B, RT 3,68 min, MS: ES+ 293,5; RMN H1 (400 MHz, DMSO-de) 5 ppm 9,05 (d, J = 7,2 Hz, 1 H), 8,95 -8,96 (m, 1 H), 8,29 (dd, J = 1,2,4 Hz, 1 H), 8,10 (d, J = 8,4 Hz, 1 H), 7,81 (dd, J = 1,6, 6,8 Hz, 2 H), 7,55 - 7,57 (m, 2 H), 7,47 - 7,50 (m, 1 H), 4,53 - 4,58 (m, 1 H), 3,55 - 3,65 (m, 2 H), 3,40 - 3,49 (m, 2 H), 2,12 - 2,15 (m, 1 H), 2,05 - 2,10 (m, 1 H)
Ejemplo 2 (R)-N-( 1 -cianopirrolidin-3-il)-3-metoxi-4-( 1 -metil-1 H-pirazol-4-il)benzamida
(Preparada según el método general B)

Etapa a. A una solución de 4-bromo-3-metoxibenzoato de metilo (1,02 mmoles), se le añadió éster de pinacol de ácido 1-metilpirazol-4-borónico (1,52 mmoles) en 1,4-dioxano:agua (2:1) (6 ml) Na2CO3 (3,35 mmoles) a rt. La mezcla de reacción se purgó con nitrógeno durante 10 min. Se añadió Pd(PPh3)4 (0,04 mmoles) a la mezcla de reacción. La mezcla de reacción se calentó a 90°C durante 8 h. La mezcla de reacción resultante se vertió en agua (100 ml) y se extrajo con EtOAc (2 x 50 ml). La capa orgánica combinada se lavó con salmuera (100 ml), se secó sobre Na2SO4, se
filtró y se concentró a presión reducida produciendo 3-metoxi-4-(1-metil-1H-pirazol-4-il)benzoato de metilo (cuantitativo). Esta sustancia se utilizó para la siguiente etapa sin purificación adicional. MS: ES+ 247,1
Etapa b. A una solución de 3-metoxi-4-(1 -metil-1 H-pirazol-4-il)benzoato (3,65 mmoles) en THF:agua (1:1, 10 ml) se le añadió LiOH.H2O (14,60 mmoles) en porciones a rt. La mezcla de reacción se agitó a rt durante 3 h. La mezcla resultante se vertió en agua (50 ml) y se extrajo con EtOAc (2 x 50 ml). La capa acuosa resultante que contenía el producto se enfrió a 0°C y se neutralizó mediante la adición lenta de una solución acuosa diluida de HCl. La mezcla resultante se extrajo con EtOAc (2 x 50 ml). La fase orgánica combinada se secó sobre Na2SO4, se filtró y se concentró a presión reducida produciendo ácido 3-metoxi-4-(1-metil-1 H-pirazol-4-il)benzoico (1,63 mmoles). MS: ES+ 233,2
Etapa c. A una solución de (R)-3-amino-1N-BOC-pirrolidina (0,86 mmoles) y ácido 3-metoxi-4-(1-metil-1H-pirazol-4-il)benzoico (0,86 mmoles) en THF (7 ml) se le añadieron HATU (1,29 mmoles) y DIPEA (2,5 mmoles) a 0°C. La mezcla de reacción se agitó a rt durante 2 h. La mezcla de reacción resultante se vertió en agua (100 ml) y se extrajo con EtOAc (2 x 50 ml). La fase orgánica combinada se recogió y se lavó con una solución diluida de ácido cítrico (2 x 50 ml), salmuera (1 x 100 ml), se secó sobre Na2SO4, se filtró y se concentró a presión reducida produciendo (R)-3-(3-metoxi-4-(1-metil-1H-pirazol-4-il)benzamido)-pirrolidin-1-carboxilato de terc-butilo (0,9 mmoles). MS: ES+ 401,3
Etapa d. A una solución de (R)-3-(3-metoxi-4-(1 -metil-1 H-pirazol-4-il)-benzamido)pirrolidin-1 -carboxilato de terc-butilo (0,9 mmoles) en DCM (10 ml) se le añadió TFA (29,4 mmoles) a 0°C. La mezcla de reacción se agitó a rt durante 2 h. La mezcla de reacción resultante se concentró a presión reducida. El residuo obtenido se destiló azeotrópicamente con DCM (3 x 25 ml) y se secó para producir sal de TFA de (R)-3-metoxi-4-(1 -metil-1 H-pirazol-4-il)-N-(pirrolidin-3-il)benzamida (0,57 mmoles). MS: ES+ 301,24
Etapa e. A una solución de sal de TFA de (R)-3-metoxi-4-(1-metil-1H-pirazol-4-il)-N-(pirrolidin-3-il)benzamida (0,57 mmoles) en THF (10 ml) se le añadieron K2CO3 (4,7 mmoles) y bromuro de cianógeno (0,79 mmoles) a 0°C. La mezcla de reacción se agitó a rt durante 1 h. La mezcla de reacción se vertió en agua (50 ml) y se extrajo con acetato de etilo (2 x 50 ml). La capa orgánica combinada se lavó con salmuera (100 ml), se secó sobre Na2SO4, se filtró y se concentró a presión reducida. El residuo resultante se purificó mediante cromatografía ultrarrápida (MeOH al 1-5% en DCM) produciendo (R)-N-(1-cianopirrolidin-3-il)-3-metoxi-4-(1-metil-1H-pirazol-4-il)benzamida (0,29 mmoles). LCMS: Método B, RT 2,97 min, MS: ES+ 326,27; RMN H1 (400 MHz, DMSO-de) ó ppm 8,56 (d, J = 6,4 Hz, 1 H), 8,21 (s, 1 H), 7,98 (s, 1 H), 7,70 (d, J = 8,4 Hz, 1 H), 7,48 - 7,50 (m, 2 H), 4,45 - 4,53 (m, 1 H), 3,94 (s, 3 H), 3,88 (s, 3 H), 3,63 - 3,67 (m, 1 H), 3,54 - 3,60 (m, 1 H), 3,43 - 3,49 (m, 1 H), 3,30 - 3,33 (m, 1 H), 2,10 - 2,18 (m, 1 H), 1,94 - 2,00 (m, 1 H)
Ejemplo 3 2 '-cloro-N-( 1 -cianopirrolidin-3-il)-[ 1,1 '-bifenil]-4-carboxamida
(Preparada según el método general C)

Etapa a. A una solución de ácido 4-bromobenzoico (4,97 mmoles) en THF (20 ml) se le añadió T3P (50% en EtOAc) (14,9 mmoles) a 0°C. La mezcla de reacción se agitó a 0°C durante 20 min. Se añadieron 3-amino-1 N-BOC-pirrolidina (5,93 mmoles) y DIPEA (14,96 mmoles) a la mezcla de reacción a 0°C. La mezcla de reacción se agitó a rt durante 1,5 h. La mezcla resultante se vertió en una solución saturada de NaHCO3 (80 ml) y se extrajo con EtOAc (2 x 40 ml). La capa orgánica se lavó con HCl M (40 ml), se secó sobre Na2SO4, se filtró y se concentró a presión reducida. La mezcla resultante se purificó mediante trituración utilizando Et2O:hexano (1:1) produciendo 3-(4-bromobenzamido)pirrolidin-1-carboxilato de terc-butilo (1,90 mmoles). MS: ES+ 369,13
Etapa b. A una solución de 3-(4-bromobenzamido)pirrolidin-1-carboxilato de terc-butilo (0,67 mmoles) y ácido 2-clorofenilborónico (1,01 mmoles) en 1,4-dioxano:agua (5:1) (7,5 ml) se le añadió K2CO3 (2,03 mmoles) a rt. La mezcla de reacción se purgó con nitrógeno durante 30 min. Se añadió Pd(PPh3)4 (0,03 mmoles) a la mezcla de reacción en atmósfera de nitrógeno. La reacción se calentó a 80°C durante 2 h. La mezcla resultante se vertió en agua (20 ml) y se extrajo con EtOAc (3 x 20 ml). La capa orgánica combinada se secó sobre Na2SO4, se filtró y se concentró a presión reducida. El residuo resultante se purificó mediante cromatografía ultrarrápida (MeOH al 0,5% en DCM) produciendo 3-(2'-cloro-[1,1'-bifenil]-4-carboxamido)pirrolidin-1-carboxilato de terc-butilo (0,5 mmoles). MS: ES+ 401,18, RMN H1 (400 MHz, DMSO-de) ó ppm 8,65 (d, J = 6,8 Hz, 1 H), 7,94 (d, J = 8,4 Hz, 2 H), 7,65 - 7,58 (m, 2 H), 7,53 (d, J = 8,4 Hz, 2 H), 7,44 (dd, J = 2, 5,6 Hz, 2H), 4,44 - 4,45 (m, 1H), 3,51 - 3,60 (m, 1H), 3,16 - 3,30 (m, 1H), 2,08 - 2,10 (m, 2H), 1,92 - 1,93 (m, 2 H), 1,41 (s, 9 H).
Etapas c, d. El compuesto del título se sintetizó a partir del intermedio anterior utilizando un procedimiento similar al descrito para Ejemplo 2. LCMS: Método B, RT 4,01 min, MS: ES+ 326,23; RMN H1 (400 MHz, DMSO-d6) ó ppm 8,70 (d, J = 6,4 Hz, 1 H), 7,94 (d, J = 8,00 Hz, 2 H), 7,59 - 7,62 (m, 1 H), 7,54 (d, J = 8,00 Hz, 2 H), 7,42 - 7,46 (m, 3 H), 4,49 - 4,53 (m, 1 H), 3,60 - 3,68 (m, 1 H), 3,54 - 3,58 (m, 1 H), 3,45 -3,56 (m, 1 H), 3,34 - 3,44 (m, 1 H), 2,01 - 2,17 (m, 1 H), 1,95 - 2,01 (m, 1 H)
Ejemplo 4 6-(bencil(metil)amino)-N-(1-cianopirrolidin-3-il)nicotinamida
(Preparada según el método general D)

Etapa a. A una solución de 6-fluoropiridin-3-carboxílico (5,32 mmoles) en DCM (20 ml) se le añadió HATU (15,9 mmoles). La mezcla de reacción se agitó a 0°C durante 20 min. Se añadieron 3-amino-1N-BOC-pirrolidina (5,33 mmoles) y DIPEA (15,96 mmoles) a la mezcla de reacción a rt. La mezcla de reacción se agitó a rt durante 16 h. La mezcla resultante se vertió en una solución saturada de NaHCO3 (80 ml) y se extrajo con EtOAc (2 x 40 ml). La capa orgánica se lavó con HCl 1 M (40 ml), se secó sobre Na2SO4 anhidro, se filtró y se concentró a presión reducida. La mezcla resultante se purificó mediante cromatografía en columna (EtOAc de 0 a 20% en PE) produciendo 3-[(6-fluoropiridin-3-carbonil)amino]pirrolidin-1-carboxilato de terc-butilo (3,88 mmoles). MS: ES+ 310,10; RMN H1 (400 MHz, DMSO-d6) 5 ppm 8,75 (d, J = 6,0 Hz, 1 H), 8,71 (d, J = 2,0 Hz, 1 H), 8,35 - 8,46 (m, 1 H), 7,26 - 7,37 (m, 1 H), 4,34 - 4,51 (m, 1 H), 3,49 - 3,63 (m, 1 H), 3,33-3,43 (m, 2 H), 3,16 - 3,27 (m, 1 H), 2,05 - 2,18 (m, 1 H), 1,84 - 1,96 (m, 1 H), 1,37 - 1,46 (m, 9 H)
Etapa b. A una solución de 3-[(6-fluoropiridin-3-carbonil)amino]pirrolidin-1-carboxilato de terc-butilo (0,2 mmoles) y N-metil-1-fenil-metanamina (0,24 mmoles) en DMF (1 ml) se le añadió Cs2CO3 (0,6 mmoles) a rt. La reacción se calentó a 120°C durante 16 h. La mezcla resultante se concentró a presión reducida y el resto se purificó mediante TLC prep. (PE/EtOAc = 1: 2) produciendo 3-(6-(bencil(metil)amino)-nicotinamido)pirrolidin-1-carboxilato de terc-butilo. MS: ES+ 411,2
Etapas c, d. El compuesto del título se sintetizó a partir del intermedio anterior utilizando un procedimiento similar al descrito para Ejemplo 2 para proporcionar el compuesto del título (23,2 mg, 0,069 mmoles). LCMS: Método F, RT 2,39 min, MS: ES+ 336,2
Ejemplo 5 (R)-N-( 1 -cianopirrolidin-3-il)-3-fenilazetidin-1 -carboxamida
(Preparada según el método general E)

Etapa a. A una solución de (R)-3-amino-1 N-BOC-pirrolidina (1,34 mmoles) en agua (5 ml) se le añadió CDI (2,68 mmoles) a 0°C. La mezcla de reacción se agitó a 0°C durante 30 min. Se añadió 3-fenilazetidina (1,61 mmoles) a la mezcla de reacción a 0°C. La mezcla de reacción se agitó a rt durante 18 h. La mezcla de reacción resultante se vertió en agua (150 ml) y se extrajo con DCM (3 x 100 ml). La fase orgánica se recogió, se secó sobre Na2SO4, se filtró y se concentró a presión reducida. El residuo resultante se purificó mediante cromatografía ultrarrápida (MeOH al 1,5% en DCM) produciendo (R)-3-(3-fenilazetidin-1-carboxamido)pirrolidin-1-carboxilato de terc-butilo (0,60 mmoles). MS: ES+ 346,1
Etapa b. A una solución de (R)-3-(3-fenilazetidin-1-carboxamido)pirrolidin-1-carboxilato de terc-butilo (0,60 mmoles) en DCM (5 ml) se le añadió TFA (3,0 mmoles) a rt. La mezcla de reacción se agitó a rt durante 3 h. La mezcla de reacción resultante se concentró a presión reducida y se destiló azeotrópicamente con DCM (3 x 10 ml). El residuo resultante se purificó triturándolo con dietiléter (5 ml). El material obtenido se secó a presión reducida produciendo la sal de TFA de (R)-3-fenil-N-(pirrolidin-3-il)azetidin-1-carboxamida (0,25 mmoles). Esta sustancia se utilizó directamente para la siguiente etapa sin purificación adicional. MS: ES+ 246,53
Etapa c. A una solución de sal de TFA de (R)-3-fenil-N-(pirrolidin-3-il)azetidin-1-carboxamida (0,25 mmoles) en DCM (5 ml) se le añadió DIPEA (0,75 mmoles) a 0°C y se agitó durante 10 min. Se añadió bromuro de cianógeno (0,37 mmoles) a la mezcla de reacción a 0°C y se agitó durante 30 min adicionales. La mezcla de reacción resultante se vertió en agua helada (100 ml) y se extrajo con DCM (3 x 50 ml). La fase orgánica se recogió, se secó sobre Na2SO4, se filtró y se concentró a presión reducida. El residuo resultante se purificó mediante cromatografía ultrarrápida (MeOH al 2,0% en DCM) produciendo el compuesto del título (0,08 mmoles). LCMS: Método B, RT 3,26 min, MS: ES+ 271,38; RMN H1: (400 MHz, DMSO-de) 5 ppm 7,32 - 7,38 (m, 4 H), 7,23 - 7,27 (m, 1 H), 6,58 (d, J = 6,4 Hz, 1 H), 4,13 - 4,21 (m, 3 H), 3,74 - 3,81 (m, 3 H), 3,47 - 3,54 (m, 2 H), 3,36 - 3,45 (m, 1 H), 3,14 - 3,17 (m, 1 H), 1,96 - 2,05 (m, 1 H), 1,76 -1,84 (m, 1 H).
Ejemplo 6 N-((R)-1-cianopirrolidin-3-il)-4-((cis)-2,6-dimetilmorfolino)-3-fluorobenzamida
(Preparada según el método general F)

Etapa a. Una mezcla de 4-bromo-3-fluorobenzoato de metilo (0,42 mmoles), cis-2,6-dimetilmorfolina (0,42 mmoles) y NaOtBu (0,42 mmoles) en tolueno seco (2 ml) se agitó a rt en un tubo de vidrio. La mezcla de reacción se purgó con nitrógeno durante 10 min. Se añadieron Xantphos (0,021 mmoles) y Pd2(dba)3 (0,009 mmoles) a la mezcla de reacción y se selló el tubo de vidrio. La mezcla de reacción resultante se calentó a 110°C (temperatura externa) durante 1 h. Una vez completada, la mezcla de reacción se enfrió a rt y se diluyó con EtOAc (30 ml). La mezcla de reacción resultante se vertió en agua (40 ml). La mezcla se extrajo con EtOAc (2 x 20 ml). La capa orgánica combinada se lavó con salmuera (25 ml), se secó sobre Na2SO4, se filtró y se concentró a presión reducida. El residuo resultante se purificó mediante cromatografía ultrarrápida (EtOAc al 50% en hexano) produciendo 4-((cis)-2,6-dimetilmorfolino)-3-fluorobenzoato de metilo (0,37 mmoles). MS: ES+ 268,3.
Etapa b. A una solución de 4-((cis)-2,6-dimetilmorfolino)-3-fluorobenzoato de metilo (1,49 mmoles) en THF:agua (1:1, 8 ml) se le añadió LiOH(14,98 mmoles) a rt. La mezcla de reacción se agitó a 50°C durante 4 h y a continuación a rt durante 15 h. La mezcla de reacción resultante se ajustó a pH 4 utilizando una solución acuosa de HCl 1 M y la mezcla se extrajo con EtOAc (3 x 100 ml). La fase orgánica combinada se recogió, se secó sobre Na2SO4, se filtró y se concentró a presión reducida produciendo ácido 4-((cis)-2,6-dimetilmorfolino)-3-fluorobenzoico (0,95 mmoles). Esta sustancia se utilizó directamente para la siguiente etapa sin purificación adicional. MS: ES+ 254,26.
Etapa c. A una solución de ácido 4-((cis)-2,6-dimetilmorfolino)-3-fluorobenzoico (0,95 mmoles) en DMF (3 ml) se le añadió HATU (1,42 mmoles) a rt. La mezcla de reacción se agitó a rt durante 30 min. Se añadió una solución de (R)-3-aminopirrolidin-1-carboxilato de terc-butilo (0,85 mmoles) en DMF (1 ml) a la mezcla de reacción a rt. Se añadió DIPEA (2,85 mmoles) a la mezcla de reacción a rt. La mezcla de reacción resultante se agitó a rt durante 2 h. La mezcla de reacción resultante se vertió en agua (150 ml) y se extrajo con EtOAc (3 x 100 ml). La fase orgánica combinada se recogió, se lavó con una solución saturada de NaHCO3 (100 ml), salmuera (100 ml), se secó sobre Na2SO4, se filtró y se concentró a presión reducida produciendo (R)-3-(4-((cis)-2,6-dimetilmorfolino)-3-fluorobenzamido)pirrolidin-1-carboxilato de terc-butilo (cuantitativo). Esta sustancia se utilizó directamente para la siguiente etapa sin purificación adicional. MS: ES+ 422,4.
Etapas d, e. El compuesto del título se sintetizó a partir del intermedio anterior utilizando un procedimiento similar al descrito para Ejemplo 2 para proporcionar el compuesto del título (0,25 mmoles). LCMS: Método A, RT 3,87 min, MS: ES+ 346,98; RMN H1 (400 MHz, DMSO-d6) 5 ppm 8,48 (d, J = 6,4 Hz, 1 H), 7,64 - 7,67 (m, 2 H), 7,08 (t, J = 8,4 Hz, 1 H), 4,43 - 4,47 (m, 1 H), 3,71 - 3,75 (m, 2 H), 3,60 - 3,64 (m, 1 H), 3,51 - 3,55 (m, 1 H), 3,43 - 3,47 (m, 1 H), 3,27 - 3,38 (m, 3 H), 2,39 - 2,45 (m, 2 H), 2,11 - 2,15 (m, 1 H), 1,90 - 1,95 (m, 1 H), 1,13 (d, J = 6,4 Hz, 6 H)
Los compuestos de la Tabla 1 se sintetizaron utilizando los métodos generales A-F como se ilustra mediante los Ejemplos 1-6 utilizando (rac)-3-aminopirrolidin-1-carboxilato de terc-butilo (Número CAS 186550-13-0).

Tabla 1

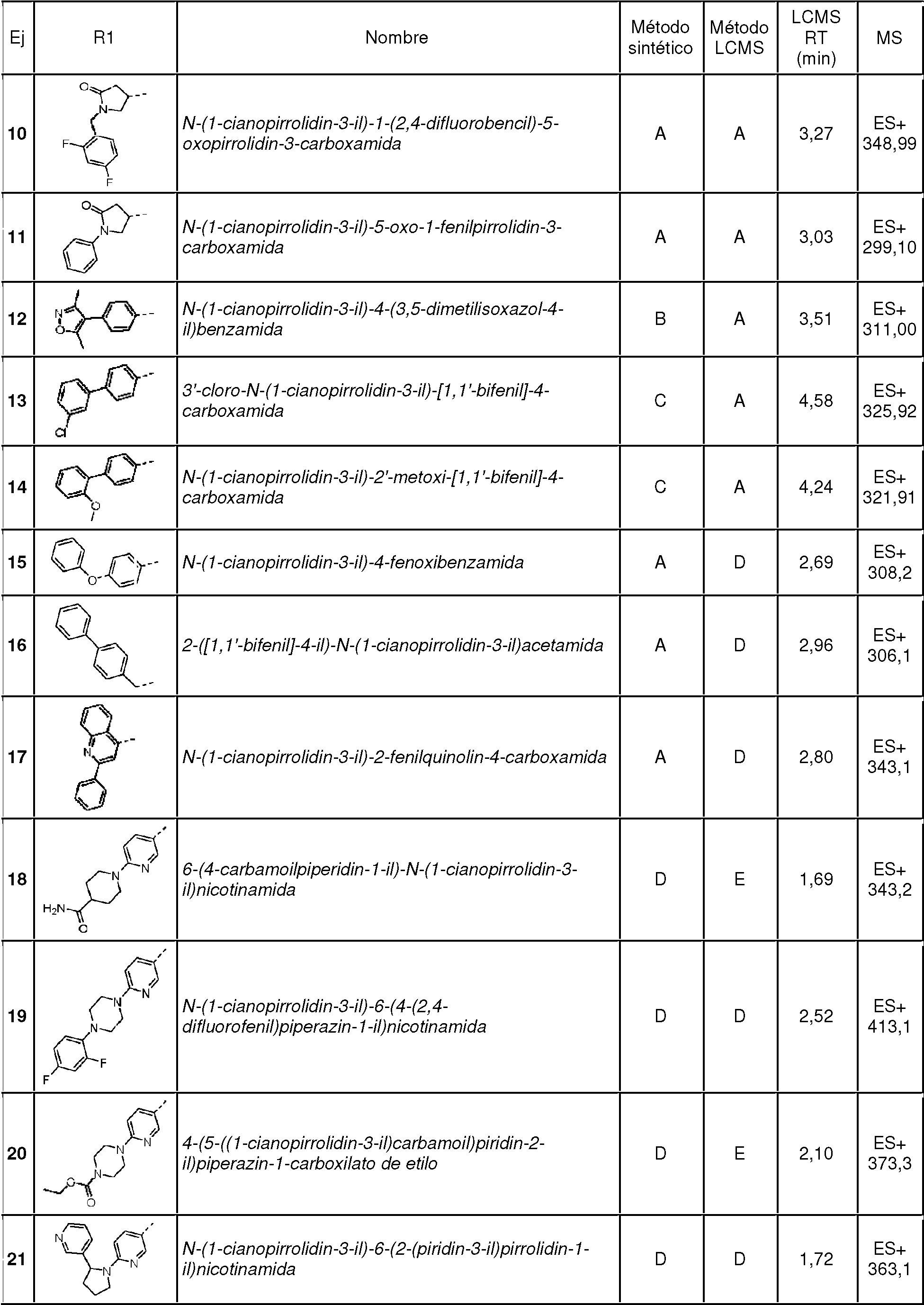

Los compuestos de la Tabla 2 se sintetizaron utilizando los métodos generales A-F como se ilustra mediante los Ejemplos 1-6 utilizando (R)-terc-butil 3-aminopirrolidin-1-carboxilato (Número CAS 147081-49-0).
Tabla 2


Los compuestos de la Tabla 3 se sintetizaron utilizando los métodos generales A-F como se ilustra mediante los Ejemplos 1-6 utilizando (S)-3-aminopirrolidin-1-carboxilato de terc-butilo (Número CAS 147081-44-5).
Tabla 3
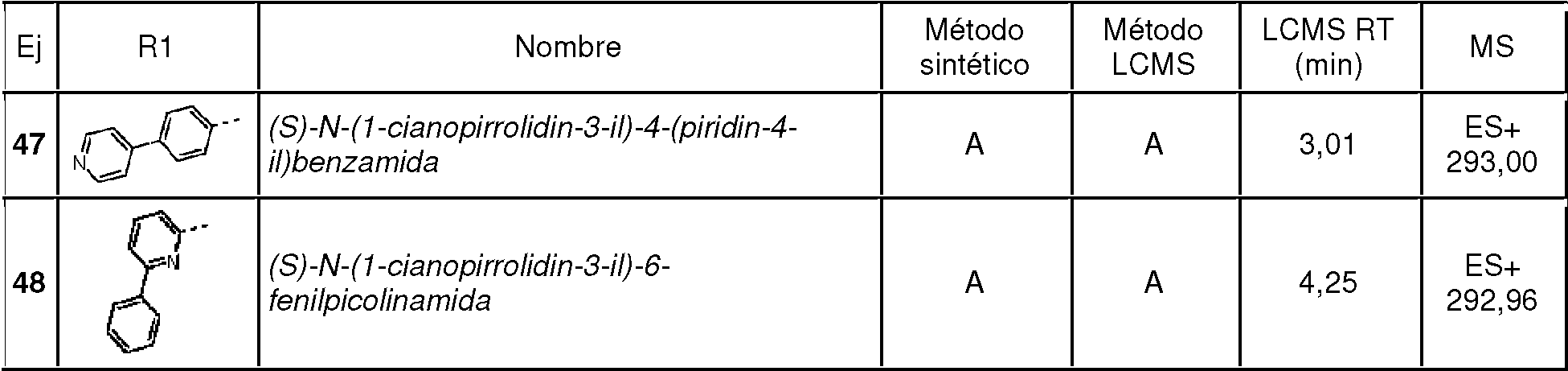
Los compuestos de la Tabla 4 se sintetizaron utilizando los métodos generales A-F como se ilustra mediante los Ejemplos 1-6 utilizando (R)-3-(metilamino)pirrolidin-1-carboxilato de terc-butilo (Número TarasconCAS 199336-83-9).
Tabla 4
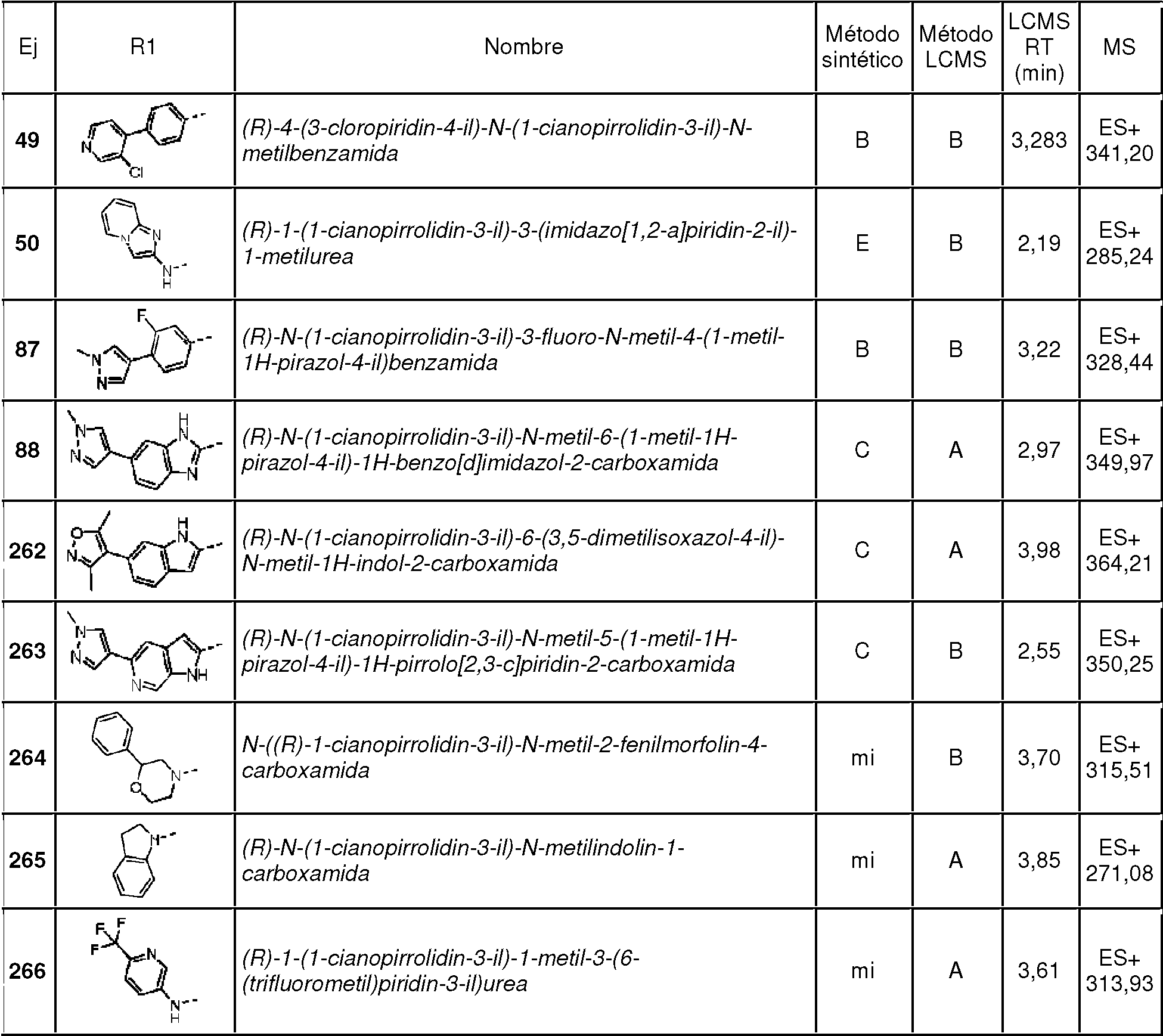

Los compuestos de la Tabla 5 se sintetizaron utilizando los métodos generales A-F como se ilustra mediante los Ejemplos 1-6 utilizando rac-(3aR, 6aR)-hexahidropirrolo[3,4-b]pirrol-5(1H)-carboxilato de terc-butilo (Número CAS 180975-51-3).

Tabla 5


Ejemplo 62 (R)-N-(1-cianopirrolidin-3-il)-3-(N-metilisobutiramido)benzamida

Etapa a. A una solución de 3-aminobenzoato de metilo (3,31 mmoles) en DCM (10 ml) se le añadió TEA (9,93 mmoles) a rt y la mezcla de reacción se agitó a rt durante 30 min. Se añadió cloruro de isobutirilo (4,96 mmoles) a la mezcla de reacción a 0°C. Después, la mezcla de reacción se agitó a rt durante 1 h. La mezcla de reacción resultante se vertió en agua (100 ml) y se extrajo con DCM (2 x 50 ml). La fase orgánica combinada se recogió, se secó sobre Na2SO4, se filtró y se concentró a presión reducida produciendo 3-isobutiramidobenzoato de metilo (cuantitativo). Esta sustancia se utilizó directamente para la siguiente etapa sin purificación adicional. MS: ES+ 222,2.
Etapa b. A una solución de 3-isobutiramidobenzoato de metilo (1,80 mmoles) en THF (10 ml) se le añadió NaH (aceite mineral al 60%, 3,61 mmoles) a 0°C. La mezcla de reacción se agitó a rt durante 30 min. Se añadió yoduro de metilo (3,61 mmoles) a la mezcla de reacción a rt. La mezcla de reacción se agitó a rt durante 2 h. La mezcla de reacción resultante se vertió en agua helada (50 ml) y se extrajo con EtOAc (3 x 20 ml). La fase orgánica combinada se recogió, se secó sobre Na2SO4, se filtró y se concentró a presión reducida produciendo 3-(N-metilisobutiramido)benzoato de metilo (1,65 mmoles). Esta sustancia se utilizó directamente para la siguiente etapa sin purificación adicional. MS: ES+ 236,6.
Etapa c. A una solución de 3-(N-metilisobutiramido)benzoato (1,61 mmoles) en THF:agua (10:2, 12 ml) se le añadió UOH.H2O (4,85 mmoles) a 0°C. La mezcla de reacción se agitó a rt durante 5 h. La mezcla de reacción resultante se ajustó a pH 3 mediante la adición lenta de una solución acuosa de ácido cítrico. La mezcla resultante se extrajo con EtOAc (3 x 20 ml). La fase orgánica combinada se recogió, se secó sobre Na2SO4, se filtró y se concentró a presión reducida produciendo ácido 3-(N-metilisobutiramido)benzoico (1,44 mmoles). Esta sustancia se utilizó directamente para la siguiente etapa sin purificación adicional. MS: ES+ 222,2
Etapa d. A una solución de ácido 3-(N-metilisobutiramido)benzoico (1,26 mmoles) en THF (15 ml) se le añadieron HATU (1,90 mmoles) y DIPEA (2,53 mmoles) a 0°C. La mezcla de reacción se agitó a rt durante 30 min. Se añadió (R)-3-amino-1N-BOC-pirrolidina (1,52 mmoles) a la mezcla de reacción a rt y se agitó durante 2 h. La mezcla de reacción resultante se vertió en agua (100 ml) y se extrajo con EtOAc (2 x 50 ml). La fase orgánica combinada se recogió, se secó sobre Na2SO4, se filtró y se concentró a presión reducida, produciendo (R)-3-(3-(N-metilisobutiramido)benzamido)pirrolidin-1-carboxilato de terc-butilo (cuantitativo). Esta sustancia se utilizó directamente para la siguiente etapa sin purificación adicional. MS: ES-388,6.
Etapa e. A una solución de (R)-3-(3-(N-metilisobutiramido)benzamido)pirrolidin-1-carboxilato de terc-butilo (1,02 mmoles) en DCM (15 ml) se le añadió TFA (4 ml) a 0°C. La mezcla de reacción se agitó a rt durante 1 h. La mezcla de reacción resultante se concentró a presión reducida produciendo la sal de TFA de (R)-3-(N-metilisobutiramido)-N-(pirrolidin-3-il)benzamida (0,99 mmoles). Esta sustancia se utilizó directamente para la siguiente etapa sin purificación adicional. MS: ES+ 290,4.
Etapa f. A una solución de sal de TFA de (R)-3-(N-metilisobutiramido)-N-(pirrolidin-3-il)benzamida (0,99 mmoles) en THF (15 ml) se le añadió K2CO3 (3,97 mmoles) a rt. La mezcla de reacción se agitó a rt durante 15 min. Se añadió bromuro de cianógeno (1,48 mmoles) a la mezcla de reacción a 0°C. La mezcla de reacción se agitó a rt durante 30 min. La mezcla de reacción resultante se vertió en agua (100 ml) y se extrajo con EtOAc (2 x 50 ml). La fase orgánica combinada se recogió, se secó sobre Na2SO4, se filtró y se concentró a presión reducida. El residuo resultante se purificó mediante cromatografía en columna (EtOAc al 77% en hexano) produciendo el compuesto del título (0,28 mmoles). LCMS: Método B, RT 3,15 min, MS: ES+ 315,1; RMN H1 (400 MHz, DMSO-d6) 5 ppm 8,64 (d, J = 6 Hz, 1 H), 7,81 - 7,86 (m, 2 H), 7,53 - 7,57 (m, 2 H), 4,46 - 4,49 (m, 1 H), 3,63 - 3,67 (m, 1 H), 3,52 - 3,58 (m, 1 H), 3,42 - 3,48 (m, 2 H), 3,17 (s, 3 H), 2,33 - 2,39 (m, 1 H), 2,09 - 2,18 (m, 1 H), 1,93 - 1,98 (m, 1 H), 0,88 - 1,0 (m, 6 H)
Ejemplo 63 (R)-N-(1-cianopirrolidin-3-il)-5-fenilpirim idin-2-carboxamida

Etapa a. A una solución de ácido 5-bromopirimidin-2-carboxílico (1,47 mmoles) en 1,4-dioxano:agua (3:1,7 ml) se le añadieron ácido fenilborónico (2,19 mmoles) y Na2CO3 (2,79 mmoles) a rt en un tubo de vidrio. La mezcla de reacción y se purgó con nitrógeno durante 10 min. Se añadió Pd(PPh3)4 (0,14 mol) a la mezcla de reacción en atmósfera de nitrógeno y el tubo de vidrio se selló. La mezcla de reacción se calentó a 100°C (temperatura externa) durante 2 h. La mezcla de reacción resultante se vertió en una solución de NaOH 1 M (50 ml) y se lavó con éter dietílico (50 ml). La capa acuosa resultante que contenía el producto se aciduló con HCl 1 M y se extrajo con EtOAc (3 x 20 ml). La fase orgánica combinada se secó sobre Na2SO4, se filtró y se concentró a presión reducida produciendo ácido 5-fenilpirimidin-2-carboxílico (1,25 mmoles). MS: ES+ 201,02, RMN H1 (400 MHz, DMSO-d6) 5 ppm 13,62 (s, 1 H), 9,30 (s, 2 H), 7,89 - 7,91 (m, 2 H), 7,51 - 7,60 (m, 3 H).
Etapa b. A una solución de ácido 5-fenilpirimidin-2-carboxílico (0,60 mmoles) en DCM (5 ml) se le añadieron HATU (0,90 mmoles), TEA (1,20 mmoles) y (R)-3-aminopirrolidin-1-carboxilato de terc-butilo. (0,60 mmoles) a rt. La mezcla de reacción se agitó a rt durante 16 h. La mezcla de reacción resultante se vertió en agua (50 ml) y se extrajo con EtOAc (3 x 20 ml). La fase orgánica combinada se recogió, se secó sobre Na2SO4, se filtró y se concentró a presión reducida produciendo (R)-3-(5-fenilpirimidin-2-carboxamido)pirrolidin-1-carboxilato de terc-butilo (0,35 mmoles). Esta sustancia se utilizó directamente para la siguiente etapa sin purificación adicional. MS: ES+ 369,40.
Etapa c. A una solución de (R)-3-(5-fenilpirimidin-2-carboxamido)pirrolidin-1-carboxilato de terc-butilo (0,33 mmoles) en DCM (5 ml) se le añadió TFA (0,5 ml) a 0°C. La mezcla de reacción se agitó a rt durante 6 h. La mezcla de reacción resultante se concentró a presión reducida. La sustancia bruta resultante se trituró con éter dietílico (5 ml) produciendo la sal de TFA de (R)-5-fenil-N-(pirrolidin-3-il)pirimidin-2-carboxamida (0,30 mmoles). Esta sustancia se utilizó directamente para la siguiente etapa sin purificación adicional. MS: ES+ 269,30.
Etapa d. A una solución de sal de TFA de (R)-5-fenil-N-(pirrolidin-3-il)pirimidin-2-carboxamida (0,27 mmoles) en DCM (5 ml) se le añadió K2CO3 (0,54 mmoles) a 0°C. Se añadió bromuro de cianógeno (0,40 mmoles) a la mezcla de reacción a 0°C. La mezcla de reacción se agitó a rt durante 2 h. La mezcla de reacción resultante se vertió en agua (50 ml) y se extrajo con DCM (3 x 20 ml). La fase orgánica combinada se recogió, se secó sobre Na2SO4, se filtró y se concentró a presión reducida. El residuo obtenido se trituró con éter dietílico (10 ml) produciendo el compuesto del título (0,15 mmoles). LCMS: Método A, RT 3,34 min, MS: ES+ 294,10; RMN H1 (400 MHz, DMSO-d6) 5 ppm 9,29 (s, 2 H), 9,22 (d, J = 7,20 Hz, 1 H), 7,89 - 7,91 (m, 2 H), 7,51 - 7,61 (m, 3 H), 4,52 - 4,57 (m, 1 H), 3,60 - 3,67 (m, 1 H), 3,54 - 3,58 (m, 1 H), 3,51 - 3,53 (m, 1 H), 3,40 - 3,45 (m, 1 H), 2,12 - 2,19 (m, 1 H), 1,99 - 2,08 (m, 1 H)
Ejemplo 64 (R)-N-(1-cianopirrolidin-3-il)-3-(piridin-4-il)isoxazol-5-carboxamida
Etapa a. A una solución de 4-piridincarboxaldehído (28,04 mmoles) en MeOH (30 ml) se le añadió NH2OH.HCI (55,94 mmoles) a rt. La mezcla de reacción se calentó a 60°C durante 30 min. Se observó precipitación en la mezcla de reacción. Los precipitados obtenidos se recogieron mediante filtración y se secaron a presión reducida para producir oxima de isonicotinaldehído (23,77 mmoles). Esta sustancia se utilizó directamente para la siguiente etapa sin purificación adicional. MS: ES+ 122,8; RMN H1 (400 MHz, DMSO-de) ó ppm 12,78 (s, 1 H), 8,89 (d, J = 6,40 Hz, 2 H), 8,42 (s, 1 H), 8,14 (d, J = 6,80 Hz, 2 H)
Etapa b. A una solución de oxima de isonicotinaldehído (22,95 mmoles) en DMF (30 ml) se le añadió NCS (34,36 mmoles) a rt y se agitó durante 1 h. La mezcla de reacción se enfrió a 0°C y se añadió una solución de propiolato de metilo (21,90 mmoles) en DCM (5 ml) a la mezcla de reacción a 0°C de una vez. Se añadió gota a gota TEA (43,56 mmoles) a la mezcla de reacción a 0°C. La mezcla de reacción se agitó a rt durante 3 h. La mezcla de reacción resultante se vertió en agua (100 ml) y se extrajo con EtOAc (3 x 30 ml). La fase orgánica combinada se recogió, se secó sobre Na2SO4, se filtró y se concentró a presión reducida. El residuo resultante se purificó mediante cromatografía ultrarrápida (EtOAc al 26% en hexano) produciendo 3-(piridin-4-il)isoxazol-5-carboxilato de metilo (2,45 mmoles). MS: ES+ 205,19.
Etapa c. A una solución de 3-(piridin-4-il)isoxazol-5-carboxilato de metilo (1,96 mmoles) en THF (5 ml) se le añadió TBD (3,79 mmoles) a rt. Se añadió (R)-3-aminopirrolidin-1-carboxilato de terc-butilo (1,96 mmoles) a la mezcla de reacción. La mezcla de reacción se agitó a rt durante 3 h. La mezcla de reacción resultante se vertió en agua (100 ml) y se extrajo con EtOAc (3 x 30 ml). La fase orgánica combinada se recogió, se secó sobre Na2SO4, se filtró y se concentró a presión reducida. El residuo resultante se purificó mediante cromatografía ultrarrápida (MeOH al 2% en DCM) produciendo el (R)-3-(3-(piridin-4-il)isoxazol-5-carboxamido)pirrolidin-1-carboxilato de terc-butilo (0,33 mmoles).). MS: ES+ 358,90.
Etapa d. A una solución de (R)-3-(3-(piridin-4-il)isoxazol-5-carboxamido)pirrolidin-1-carboxilato de terc-butilo (0,31 mmoles) en DCM (2 ml) se le añadió TFA (0,8 ml) a 0°C. La mezcla de reacción se agitó a rt durante 1 h. La mezcla de reacción resultante se concentró a presión reducida. La sustancia bruta resultante se trituró con éter dietílico (5 ml) produciendo la sal de TFA de (R)-3-(piridin-4-il)-N-(pirrolidin-3-il)isoxazol-5-carboxamida (0,20 mmoles). Esta sustancia se utilizó directamente para la siguiente etapa sin purificación adicional. MS: ES+ 259,20
Etapa e. A una solución de sal de TFA de (R)-3-(piridin-4-il)-N-(pirrolidin-3-il)isoxazol-5-carboxamida (0,18 mmoles) en DCM (2 ml) se le añadió K2CO3 (0,55 mmoles) a 0°C. Se añadió bromuro de cianógeno (0,27 mmoles) a la mezcla de reacción a 0°C. La mezcla de reacción se agitó a rt durante 2 h. La mezcla de reacción resultante se vertió en agua (50 ml) y se extrajo con DCM (3 x 20 ml). La fase orgánica combinada se recogió, se secó sobre Na2SO4, se filtró y se concentró a presión reducida. El residuo resultante se purificó mediante cromatografía ultrarrápida (MeOH al 2% en DCM) proporcionando el compuesto del título (0,07 mmoles). LCMS: Método A, RT 2,90 min, MS: ES+ 283,90; RMN H1 (400 MHz, DMSO-de) 5 ppm 9,37 (d, J = 6,80 Hz, 1 H), 8,77 (dd, J = 4,80, 1,60 Hz, 2 H), 7,92 (dd, J = 4,40, 1,60 Hz, 2 H), 7,82 (s, 1 H), 4,48 - 4,52 (m, 1 H), 3,63 - 3,67 (m, 1 H), 3,53 - 3,59 (m, 1 H), 3,44 - 3,49 (m, 1 H), 3,34 - 3,38 (m, 1 H), 2,11 - 2,17 (m, 1 H), 1,96 - 2,02 (m, 1 H)
Los compuestos de la Tabla 6 se sintetizaron utilizando un procedimiento similar al descrito para Ejemplo 64.

Tabla 6


Ejemplo 273 (R)-N-(1-cianopirrolidin-3-il)-N-metil-3-(2-metilpiridin-4-il)isoxazol-5-carboxamida

Sintetizada mediante un procedimiento similar al descrito para el Ejemplo 64. LCMS: Método A, RT 3,22 min, MS: ES+ 311,99; RMN H1 (400 MHz, MeOD) 5 ppm: 8,58 (d, J = 5,2 Hz, 1 H), 7,86 (s, 1 H), 7,77 (d, J = 5,2 Hz, 1 H),
7,45 - 7,48 (m, 1 H), 3,68 - 3,74 (m, 3 H), 3,53 - 3,59 (m, 2 H), 3,22 (s, 3 H), 3,65 (s, 3 H), 2,26 - 2,33 (m, 2 H).
Ejemplo 67 (R)-N-(1-cianopirrolidin-3-il)-.5-fenilpiridazin-3-carboxamida

Etapa a. Una solución de 5-cloropiridazin-3(2H)-ona (9,96 mmoles) y ácido fenilborónico (11,95 mmoles) se recogió en 1,4-dioxano (10 ml) en un tubo de vidrio. Se añadió Na2CO3 (19,92 mmoles) a la mezcla de reacción a rt en forma de una solución acuosa 2M. La mezcla de reacción se purgó con nitrógeno durante 10 min. Se añadió Pd(dppf)Cl2 (4,98 mmoles) a la mezcla de reacción a rt. El tubo de vidrio se selló herméticamente y se calentó a 110°C (temperatura externa) durante 24 h. La mezcla resultante se filtró a través de celite hyflow y se lavó con EtOAc (3 x 50 ml). Se añadió agua (2 x 50 ml) al producto filtrado. La fase de la capa orgánica se separó y se lavó con salmuera (100 ml), se secó sobre NaSO4 , se filtró y se concentró a presión reducida. El residuo resultante se purificó mediante cromatografía ultrarrápida (EtOAc al 27% en hexano) produciendo 5-fenilpiridazin-3 (2H)-ona (6,25 mmoles). MS: ES+ 172,79; RMN H1 (400 MHz, DMSO-de) 5 ppm 13,14 (s, 1 H), 8,31 (d, J = 2,4 Hz, 1 H), 7,81 - 7,84 (m, 2 H), 7,53 - 7,54 (m, 3 H), 7,14 (d, J = 2,4 Hz, 1 H)
Etapa b. Se añadió muy lentamente POCl3 (22 ml) a un matraz de fondo redondo de 100 ml que contenía 5-fenilpiridazin-3 (2H)-ona (12,19 mmoles) a rt. La mezcla de reacción se calentó a 90°C durante 1 h. La mezcla de reacción resultante se vertió cuidadosamente en agua enfriada con hielo (50 ml), se alcalinizó utilizando NaHCO3 sólido (pH ajustado de 8 a 9) y se extrajo con EtOAc (3 x 100 ml). La fase orgánica combinada se lavó con salmuera (100 ml), se secó sobre Na2SO4, se filtró y se concentró a presión reducida. El residuo resultante se trituró con hexano (3 x 20 ml) y se secó para producir 3-cloro-5-fenilpiridazina (12,06 mmoles). Esta sustancia se utilizó directamente para la siguiente etapa sin purificación adicional. MS: ES+ 192,1.
Etapa c. Una solución de 3-cloro-5-fenilpiridazina (2,63 mmoles) en MeOH:DMF (1:1, 10 ml) se recogió en un tubo de vidrio de 25 ml a rt. Se añadió TEA (3,95 mmoles) a la mezcla de reacción a rt y se agitó durante 5 min. La mezcla de reacción se trató con dppf (0,26 mmoles) y se purgó con nitrógeno durante 10 min. La mezcla de reacción resultante
se transfirió a un autoclave en atmósfera de nitrógeno. Se añadió Pd(OAc)2 (0,13 mmoles) a la mezcla de reacción a rt bajo atmósfera de nitrógeno. La mezcla de reacción se agitó en un autoclave a una presión de CO2 de 0,69 bar (10 psi) a 70°C durante 18 h. La mezcla de reacción resultante se filtró cuidadosamente a través de celite hyflow y se lavó con EtOAc (2 x 50 ml). Se añadió agua (2 x 50 ml) al producto filtrado. La fase orgánica se separó, se secó sobre Na2SO4, se filtró y se concentró a presión reducida. El residuo resultante se purificó mediante cromatografía ultrarrápida (EtOAc al 40% en hexano) produciendo 5-fenilpiridazin-3-carboxilato de metilo (0,65 mmoles). MS: ES+ 215,18; RMN H1 (400 MHz, DMSO-de) 5 ppm 9,86 (d, J = 2,4 Hz, 1 H), 8,44 (d, J = 2,4 Hz, 1 H), 7,99 - 8,04 (m, 2 H), 7,57 - 7,64 (m, 3 H), 4,00 (s, 3 H)
Etapa d. A una solución de 5-fenilpiridazin-3-carboxilato de metilo (0,51 mmoles) en THF (4 ml) se le añadió TBD (0,77 mmoles) a rt. Se añadió gota a gota una solución de (R)-3-amino-1N-BOC-pirrolidina (0,51 mmoles) en THF (1 ml) a la mezcla de reacción a rt. La mezcla de reacción se calentó a 70°C durante 12 h. La mezcla de reacción resultante se vertió en agua (50 ml), se extrajo con EtOAc (2 x 30 ml). La fase orgánica combinada se lavó con salmuera (50 ml), se secó sobre Na2SO4, se filtró y se concentró a presión reducida. El residuo resultante se purificó mediante cromatografía ultrarrápida (EtOAc al 30% en hexano) produciendo 3-(5-fenilpiridazin-3-carboxamido)pirrolidin-1-carboxilato de terc-butilo (0,19 mmoles). MS: ES+ 369,19.
Etapas e, f. El compuesto del título se sintetizó a partir del intermedio anterior utilizando un procedimiento similar al descrito para las etapas d, e del Ejemplo 64. LCMS: Método B, RT 3,25 min, MS: ES+ 294,27; RMN H1 (400 MHz, DMSO-de) 5 ppm 9,85 (d, J = 2,4 Hz, 1 H), 9,62 (d, J = 7,2 Hz, 1 H), 8,41 (d, J = 2 Hz, 1 H), 8,00 - 8,07 (m, 2 H),
7,56 - 7,61 (m, 3 H), 4,60 - 4,65 (m, 1 H), 3,57 - 3,69 (m, 2 H), 3,45 - 3,50 (m, 2 H), 2,07 - 2,21 (m, 2 H))
Ejemplo 68 N-(1-cianopirrolidin-3-il)-N-metil-[1,1’-bifenil]-4-carboxamida

Etapa a. A una solución de ácido 4-fenilbenzoico (2,52 mmoles) en THF (12,5 ml) se le añadió T3P (50% en EtOAc) (7,56 mmoles) a 0°C. La mezcla de reacción se agitó a 0°C durante 20 min. Se añadieron 3-aminopirrolidin-1-carboxilato de terc-butilo (2,52 mmoles) y DIPEA (7,56 mmoles) a la mezcla de reacción a rt. La mezcla de reacción se agitó a rt durante 16 h. La mezcla de reacción resultante se vertió en agua (50 ml) y se extrajo con EtOAc (3 x 30 ml). La capa orgánica combinada se lavó con HCl 1 M (30 ml), una solución acuosa de NaHCO3 (30 ml), salmuera (50 ml), se secó sobre Na2SO4, se filtró y se concentró a presión reducida. La mezcla resultante se purificó mediante cromatografía en columna (EtOAc al 25% en hexano) produciendo 3-([1,1'-bifenil]-4-carboxamido)pirrolidin-1-carboxilato de terc-butilo (1,03 mmoles) MS: ES+ 367,28.
Etapa b. A una solución de 3-([1,1 '-bifenil]-4-carboxamido)pirrolidin-1 -carboxilato de terc-butilo (0,96 mmoles) en DMF (9 ml) se le añadió NaH (aceite mineral al 60%, 1,95 mmoles) a 0°C. La mezcla de reacción se agitó a 0°C durante 15 min. Se añadió yoduro de metilo (1,45 mmoles) a la mezcla de reacción a 0°C. La mezcla de reacción se agitó a rt durante 10 min. La mezcla de reacción resultante se vertió en agua (100 ml) y se extrajo con EtOAc (3 x 30 ml). La capa orgánica combinada se lavó con salmuera (50 ml), se secó sobre Na2SO4, se filtró y se concentró a presión reducida produciendo 3-(N-metil-[1 ,r-bifenil]-4-carboxamido)pirrolidin-1-carboxilato de terc-butilo (1,18 mmoles) MS: ES+ 381,4
Etapas c, d. El compuesto del título se sintetizó a partir del intermedio anterior utilizando un procedimiento similar al descrito para las etapas b, c del Ejemplo 1. LCMS: Método A, RT 4,27 min, MS: ES+ 305,94; RMN H1 (400 MHz, DMSO-de) 5 ppm 7,63 - 7,68 (m, 2 H), 7,61 - 7,62 (m, 2 H), 7,47 -7,51 (m, 4 H), 7,40 -7,43 (m, 1 H), 5,13 (ancho s, 1 H), 3,63 - 3,68 (m, 2 H), 3,45 - 3,48 (m, 2 H), 3,03 (s, 3 H), 2,13 - 2,21 (m, 2 H)
Ejemplo 69 N-((3R,4S)-1-ciano-4-metilpirrolidin-3-il)-5-feniltiazol-2-carboxamida Ejemplo 70 N-((3S,4R)-1-ciano-4-metilpirrolidin-3-il)-5-feniltiazol-2-carboxamida

Etapa a. Una solución de crotonato de etilo (17,5 mmoles) y N-bencil-O-etil-N-((trimetilsilil)metil)hidroxilamina (19,2 mmoles) en tolueno (40 ml) se agitó a rt durante 5 min. Se añadió gota a gota TFA (17,5 mmoles) a la mezcla de reacción a rt. Después, la mezcla de reacción se calentó a 50°C durante 16 h. La mezcla de reacción resultante se vertió en agua (100 ml) y se alcalinizó con NaHCO3 sólido. La mezcla resultante se extrajo con EtOAc (2 x 180 ml). La fase orgánica combinada se recogió, se secó sobre Na2SO4 , se filtró y se concentró a presión reducida. El residuo resultante se purificó mediante cromatografía en columna (EtOAc al 0-9% en hexano) produciendo (±)-trans-1-bencil-4-metilpirrolidin-3-carboxilato de etilo (9,0 mmoles). MS: ES+ 248,33; RMN H1 (400 Mh z , CDCh) 5 ppm 7,24 - 7,36 (m, 5 H), 4,13 (q, J = 8,0, 5,2 Hz 2 H), 3,67 (d, J = 12,8 Hz, 1 H), 3,58 (d, J = 13,2 Hz, 1 H), 2,77 - 2,91 (m, 3 H),
2,47 - 2,59 (m, 2 H), 2,21 - 2,26 (m, 1 H), 1,27 (t, J = 7,2 Hz, 3 H), 1,16 (d, J = 6,71 Hz, 3 H).
Etapa b. A una solución de (±)-trans-1-bencil-4-metilpirrolidin-3-carboxilato de etilo (10 mmoles) en EtOH (30 ml) se le añadió polimetilhidrosiloxano (1,0 p/p), Pd(OH)2 al 20% sobre carbono (0,5 p/p) y anhídrido BOC (20 mmoles) a 0°C. La mezcla de reacción se agitó a rt durante 1,5 h. La mezcla de reacción resultante se filtró cuidadosamente a través de celite hyflow y el producto filtrado se concentró a presión reducida. El residuo resultante se purificó mediante cromatografía en columna (EtOAc al 0-10% en hexano) produciendo (±)-4-metilpirrolidin-1,3-dicarboxilato de 1-tercbutilo 3-etilo (8,5 mmoles). MS: ES+ 202,2 (M-tBu)
Etapa c. Se agitó una solución de (±)-4-metilpirrolidin-1,3-dicarboxilato de 1 -terc-butilo 3-etilo (8,5 mmoles) en THF (15 ml) a 0°C durante 5 min. Se añadió gota a gota una solución de NaOH (34,0 mmoles) en agua (15 ml) a la mezcla de reacción a 0°C. La mezcla de reacción se agitó a rt durante 16 h. La mezcla de reacción resultante se vertió en agua (200 ml) y se aciduló a pH 4,0 con HCl diluido. La mezcla obtenida se extrajo con EtOAc (2 x 150 ml). La fase orgánica combinada se recogió, se secó sobre Na2SO4, se filtró y se concentró a presión reducida produciendo ácido 1-[(terc-butoxi)carbonil]-(±)-trans-4-metilpirrolidin-3-carboxílico (7,1 mmoles). Esta sustancia se utilizó directamente para la siguiente etapa sin purificación adicional. MS: ES-228,28; RMN H1 (400 MHz, DMSO-d6) 5 ppm 12,51 (s ancho, 1 H), 3,47 - 3,56 (m, 2 H), 3,28 - 3,34 (m, 1 H), 2,78 - 2,86 (m, 1 H), 2,58-2,64 (m, 1 H), 2,27 - 2,34 (m, 1 H), 1,38 (s, 9 H), 1,04 (d, J = 4,8 Hz, 3 H).
Etapa d. A una solución de ácido 1-[(terc-butoxi)carbonil]-(±)-trans-4-metilpirrolidin-3-carboxílico (2,62 mmoles) en tolueno (7 ml) se le añadieron DIPEA (5,24 mmoles) y difenilfosforilazida (3,93 mmoles) gota a gota a 0°C. La mezcla de reacción se calentó a 80°C durante 3 h. La mezcla de reacción resultante se enfrió a rt seguido de la adición de NaOH 8 M (2 ml). La mezcla de reacción se calentó adicionalmente a 80°C durante 30 minutos. La mezcla de reacción resultante se vertió en agua (70 ml) y se extrajo con éter dietílico (2 x 70 ml) para eliminar las impurezas no polares. La capa acuosa resultante se extrajo adicionalmente con DCM (3 x 70 ml). La capa orgánica de DCM combinada se secó sobre Na2SO4 y se evaporó a presión reducida para producir 1-[(terc-butoxi)carbonil]-(+)-trans-3-amino-4-metilpirrolidina (1,17 mmoles, cuantitativo). Esta sustancia se utilizó directamente para la siguiente etapa sin purificación adicional. MS: ES+ 145,09 (M-tBu).
Etapas e, f, g. El compuesto del título se sintetizó como una mezcla racémica a partir del intermedio anterior utilizando un procedimiento similar al descrito para las etapas a, b, c de Ejemplo 1.
Etapa h. Los enantiómeros se separaron mediante HPLC quiral preparativa; fase móvil: (A) TFA al 0,1% en hexano (B) TFA al 0,1% en EtOH, columna: Chiralpak IB, 250 x 4,6 mm, 5 pm, caudal: 1 ml/min.
Ejemplo 69 N-((3R,4S)-1-ciano-4-metilpirrolidin-3-il)-5-feniltiazol-2-carboxamida
LCMS: Método B, RT 3,78 min, MS: ES+ 313,22; HPLC quiral: Método Y, RT 12,90 min; RMN H1 (400 MHz, DMSO
de) 5 ppm 8,82 (d, J = 7,2 Hz, 1 H), 8,49 (s, 1 H), 7,99 - 8,016 (m, 2 H), 7,52 - 7,55 (m, 3 H), 4,11 - 4,15 (m, 1 H), 3.73 - 3,77 (m, 1 H), 3,66 - 3,70 (m, 1 H), 3,25 - 3,29 (m, 1 H), 3,09 - 3,13 (m, 1 H), 2,26 - 2,33 (m, 1 H), 1,02 (d, J = 6.8 Hz, 3 H).
Ejemplo 70 N-((3S, 4R)-1-ciano-4-metilpirrolidin-3-il)-.5-feniltiazol-2-carboxamida
LCMS: Método B, RT 3,78 min, MS: ES+ 313,22; HPLC quiral: Método Y, RT 15,61 min; RMN H1 (400 MHz, DMSO-d6) 5 ppm 8,82 (d, J = 7,2 Hz, 1 H), 8,49 (s, 1 H), 7,99 - 8,016 (m, 2 H), 7,52 - 7,55 (m, 3 H), 4,11 - 4,15 (m, 1 H), 3.73 - 3,77 (m, 1 H), 3,66 - 3,70 (m, 1 H), 3,25 - 3,29 (m, 1 H), 3,09 - 3,13 (m, 1 H), 2,26 - 2,33 (m, 1 H), 1,02 (d, J = 6.8 Hz, 3 H).
Ejemplo 71 N-((3R,4S)-1-ciano-4-metilpirrolidin-3-il)-2-feniltiazol-5-carboxamida Ejemplo 72 N-((3S,4R)-1-ciano-4-metilpirrolidin-3-il)-2-feniltiazol-5-carboxamida

Los compuestos del título se sintetizaron como una mezcla racémica del intermedio anterior utilizando un procedimiento similar al descrito para el Ejemplos 69/70 y los enantiómeros se separaron mediante HPLC quiral preparativa; fase móvil: (A) TFA al 0,1% en hexano (B) TFA al 0,1% en IPA, columna: Chiralpak IB, 250 x 4,6 mm, 5 pm, caudal: 1 ml/min.
Ejemplo 71 N-((3R,4S)-1-ciano-4-metilpirrolidin-3-il)-2-feniltiazol-5-carboxamida
LCMS: Método B, RT 4,21 min, MS: ES+ 312,96; HPLC quiral: Método Z, RT 14,29 min; RMN H1 (400 MHz, DMSO-m 1 = 4 Hz 1 H 4 1 H = 2 Hz 2 H 42 - 2 m H 41 - 42 m 1 H,

Etapa a. A una solución de ácido 2-fluoropiridinc-4-carboxílico (0,5 g, 3,50 mmoles) en DCM (8 ml) se le añadieron HATU (2,01 g, 5,30 mmoles) y DIPEA (0,91 g, 7,08 mmoles) a 0°C. La mezcla de reacción se agitó a 0°C 20 min antes de añadir (R)-3-aminopirrolidin-1-carboxilato de terc-butilo (Número CAS 147081-49-0) (0,52 g, 2,83 mmoles). La mezcla de reacción se agitó a rt durante 4 h. La mezcla de reacción resultante se vertió en agua (100 ml) y se extrajo con DCM (3 x 50 ml). La fase orgánica combinada se recogió, se secó sobre Na2SO4, se filtró y se concentró a presión reducida produciendo (R)-3-(2-fluoroisonicotinamido)pirrolidin-1-carboxilato de terc-butilo (1,5 g, cuantitativo). MS: ES+ 254,2 (M-56).
Etapa b. A una solución de (R)-3-(2-fluoroisonicotinamido)pirrolidin-1-carboxilato terc-butilo (1,5 g, 4,84 mmoles) en DMF (4 ml) se le añadió K2CO3 (1,33 g, 9,6 mmoles) a rt y se agitó durante 10 min. Se añadió gota a gota a la mezcla de reacción a rt una solución de isoindolina (Número CAS 496-12-8) (0,63 g, 5,33 mmoles) en DMF (1 ml). La mezcla de reacción se calentó a 120°C durante 18 h. La mezcla de reacción resultante se vertió en agua (100 ml) y se extrajo con EtOAc (3 x 40 ml). La fase orgánica combinada se recogió, se lavó con salmuera (2 x 50 ml), se secó sobre Na2SO4, se filtró y se concentró a presión reducida. El residuo resultante se purificó mediante cromatografía ultrarrápida (EtOAc al 40% en hexano) produciendo (R)-3-(2-(isoindolin-2-il)isonicotinamido)pirrolidin-1-carboxilato de terc-butilo (0,35 g, 0,85 mmoles). MS: ES+ 409,3
Etapa c. A una solución de (R)-3-(2-(isoindolin-2-il)isonicotinamido)pirrolidin-1-carboxilato de terc-butilo (0,35 g, 0,85 mmoles) en DCM (6 ml) se le añadió TFA (2 ml) a 0°C. La mezcla de reacción se agitó a rt durante 2 h y a continuación se concentró a presión reducida. El residuo resultante se destiló azeotrópicamente utilizando DCM (2 x 20 ml). La sustancia resultante se trituró con n-pentano (2 x 20 ml), éter dietílico (2 x 20 ml) y finalmente se secó produciendo sal de TFA de (R)-2-(isoindolin-2-il)-N-(pirrolidin-3-il)isonicotinamida (0,18 g, 0,42 mmoles) MS: ES+ 309,3
Etapa d. A una solución de sal de TFA de (R)-2-(isoindolin-2-il)-N-(pirrolidin-3-il)isonicotinamida (0,18 g, 0,42 mmoles) en THF (6 ml) se le añadió K2CO3 (0,23 g, 1,70 mmoles) a 0°C. La mezcla de reacción se agitó a 0°C durante 20 min antes de añadir bromuro de cianógeno (0,045 g, 0,42 mmoles). La mezcla de reacción se agitó a rt durante 3 h. La mezcla de reacción resultante se vertió en agua (50 ml) y se extrajo con MeOH al 5% en DCM (2 x 50 ml). La capa orgánica combinada se secó sobre Na2SO4, se filtró y se concentró a presión reducida. La sustancia resultante se trituró con n-pentano (2 x 30 ml), éter dietílico (2 x 30 ml) y se purificó adicionalmente mediante TLC preparativa utilizando MeOH al 3% en DCM como fase móvil. El material obtenido se purificó adicionalmente mediante HPLC preparativa (fase móvil: ácido fórmico al 0,1% en agua/MeCN; columna: YMC ACTUS TRIART C18 (250x20 mm), 5 pm; caudal: 18 ml/min) produciendo el compuesto del título (0,033 g, 0,099 mmoles). LCMS: Método A, RT 3,92 min, MS: ES+ 334,01; RMN H1 (400 MHz, DMSO-de) 5 ppm 8,76 (d, J = 6,8 Hz, 1 H), 8,26 (d, J = 5,2 Hz, 1 H), 7,32 - 7, 44 (m, 4 H), 7,01 (d, J = 4,8 Hz, 1 H), 6,95 (s, 1 H), 4,8 (s, 4 H), 4,47 - 4,52 (m, 1 H), 3,64 - 3,68 (m, 1 H),
3,54 - 3,60 (m, 1 H), 3,34 - 3,49 (m, 1 H), 3,30 - 3,31 (m, 1 H), 2,13 - 2,18 (m, 1 H), 1,94 - 1,99 (m, 1 H).
Ejemplo 74 (R)-N-(1-cianopirrolidin-3-il)-2-(3,4-dihidroisoquinolin-2(1H)-il)isonicotinamida

Sintetizada utilizando un procedimiento similar al descrito para el Ejemplo 73 utilizando 1,2,3,4-tetrahidroisoquinolina (Número CAS 91-21-4). LCMS: Método B, RT 3,34 min, MS: ES+ 348,35
Ejemplo 75 (R)-N-(1-cianopirrolidin-3-il)-2-fluoro-4-(4-metil-1H-imidazol-1-il)benzamida

Etapa a. Una solución de anhídrido acético (0,89 g, 8,74 mmoles) en ácido fórmico (1,3 ml) se agitó a rt durante 30 min. Se añadió gota a gota una solución de éster metílico de ácido 4-amino-2-fluoro-benzoico (Número CAS 73792 08-2) (0,4 g, 2,36 mmoles) en THF (4 ml) y la mezcla de reacción se calentó a continuación a 60°C durante 16 h. La mezcla de reacción se vertió en agua (150 ml) y se agitó a rt durante 30 min. Los sólidos precipitados se recogieron mediante filtración a vacío, se lavaron con agua (2 x 25 ml) y finalmente se secaron a vacío produciendo 2-fluoro-4-formamidobenzoato de metilo (0,31 g, 1,56 mmoles) MS: e S+ 198,28
Etapa b. A una solución de 2-fluoro-4-formamidobenzoato de metilo (0,31 g, 1,56 mmoles) en DMF (4 ml) se le añadieron K2CO3 (0,32 g, 2,34 mmoles) y KI (0,025 g, 0,15 mmoles) a rt. Se añadió gota a gota cloroacetona (0,36 g, 3,90 mmoles) a la mezcla de reacción y se agitó a rt durante 16 h. La mezcla de reacción se vertió en agua (150 ml) y se extrajo con EtOAc (3 x 100 ml). La fase orgánica combinada se recogió, se lavó con salmuera (100 ml), se secó sobre Na2SO4, se filtró y se concentró a presión reducida produciendo 2-fluoro-4-(N-(2-oxopropil)formamido)benzoato de metilo (0,3 g, 1,18 mmoles) MS: ES+ 254,5.
Etapa c. A una solución de 2-fluoro-4-(N-(2-oxopropil)formamido)benzoato (0,3 g, 1,18 mmoles) en ácido acético glacial (4 ml) se le añadió acetato de amonio (0,54 g, 7,10 mmoles) a rt. La mezcla de reacción se calentó a 130°C durante 3 h. La mezcla de reacción resultante se dejó enfriar a rt y se alcalinizó utilizando hidróxido de amonio acuoso para ajustar a pH 7. La solución acuosa resultante se extrajo con EtOAc (3 x 120 ml). La fase orgánica combinada se recogió, se lavó con agua (100 ml), salmuera (150 ml), se secó sobre Na2SO4, se filtró y se concentró a presión reducida produciendo 2-fluoro-4-(4-metil-1H-imidazol-1-il)benzoato de metilo (0,32 g, 1,38 mmoles) MS: ES+ 235,2.
Etapa d. A una solución de 2-fluoro-4-(4-metil-1 H-imidazol-1 -il)benzoato de metilo (0,32 g, 1,38 mmoles) en THF:agua (1:1) se le añadió UOH.H2O (0,58 g, 13,80 mmoles) a rt. La mezcla de reacción se agitó a rt durante 8 h. La mezcla de reacción resultante se aciduló utilizando HCl 1 M para ajustar a pH 4. La solución acuosa resultante se extrajo con EtOAc (3 x 150 ml). El producto deseado permaneció en la capa acuosa que se evaporó. El producto deseado se
extrajo del residuo obtenido utilizando MeOH al 10% en DCM (40 ml). La capa orgánica obtenida se secó sobre Na2SÜ4, se filtró y se concentró a presión reducida produciendo ácido 2-fluoro-4-(4-metil-1H-imidazol-1-il)benzoico (0,29 g, 1,31 mmoles) MS: ES+ 221,14.
Etapa e. A una solución de ácido 2-fluoro-4-(4-metil-1 H-imidazol-1 -il)benzoico (0,29 g, 1,31 mmoles) en DMF (5 ml) se le añadió HATU (0,8 g, 2,1 mmoles) a rt. La mezcla de reacción se agitó a rt durante 30 min. Se añadió una solución de (R)-3-aminopirrolidin-1-carboxilato de terc-butilo (Número CAS 186550-13-0) (0,19 g, 1,05 mmoles) en DMF (1 ml) a la mezcla de reacción a rt seguido de la adición de DIPEA (0,51 g, 3,9 mmoles) a rt. La mezcla de reacción se agitó a rt durante 1 h. La mezcla de reacción resultante se vertió en agua helada (100 ml) y se extrajo con EtOAc (3 x 100 ml). La fase orgánica combinada se recogió y se lavó con una solución saturada de NaHCÜ3 (100 ml), salmuera (100 ml). La fase orgánica resultante se secó sobre Na2SÜ4, se filtró y se concentró a presión reducida produciendo (R)-3-(2-fluoro-4-(4-metil-1 H-imidazol-1-il)benzamido)pirrolidin-1-carboxilato de terc-butilo (0,3 g, 0,77 mmoles) MS: ES+ 389,4
Etapa f. A una solución de (R)-3-(2-fluoro-4-(4-metil-1 H-imidazol-1 -il)benzamido)-pirrolidin-1 -carboxilato de terc-butilo (0,3 g, 0,77 mmoles) en DCM (4 ml) se le añadió TFA (0,586 ml, 7,73 mmoles) a rt. La mezcla de reacción se agitó a rt durante 2 h. La mezcla de reacción resultante se concentró a presión reducida. El residuo obtenido se trituró con éter dietílico (3 x 25 ml) y se secó para producir sal de TFA de (R)-2-fluoro-4-(4-metil-1H-imidazol-1-il)-N-(pirrolidin-3-il)benzamida. (0,15 g, 0,37 mmoles). Esta sustancia se utilizó directamente para la siguiente etapa sin purificación adicional.
Etapa g. A una solución de sal de TFA de (R)-2-fluoro-4-(4-metil-1 H-imidazol-1-il)-N-(pirrolidin-3-il)benzamida (0,15 g, 0,37 mmoles) en THF (3 ml) se le añadieron K2CO3 (0,206 g, 1,49 mmoles) y bromuro de cianógeno (0,039 g, 0,37 mmoles) a rt. La mezcla de reacción se agitó a rt durante 30 min. La mezcla de reacción se vertió en agua (70 ml) y se extrajo con EtOAc (3 x 50 ml). La capa orgánica combinada se lavó con salmuera (50 ml), se secó sobre Na2SÜ4, se filtró y se concentró a presión reducida. El residuo resultante se purificó mediante cromatografía ultrarrápida (EtOAc al 100%) produciendo el compuesto del título (0,020 g, 0,06 mmoles) LCMS: Método A, RT 2,97 min, MS: ES+ 313,98;
RMN H1 (400 MHz, DMSO-d6) 5 ppm 8,68 (d, J = 6,8 Hz, 1 H), 8,33 (s, 1 H), 7,67 - 7,75 (m, 2 H), 7,57 - 7,60 (m, 2 H), 4,44 - 4,47 (m, 1 H), 3,61 - 3,65 (m, 1 H), 3,43 - 3,55 (m, 2 H), 3,28 - 3,31 (m, 1 H), 2,16 (s, 3 H), 2,08 - 2,13 (m, 1 H), 1,88 - 1,96 (m, 1 H).
Ejemplo 89 (R)-N-(1-cianopirrolidin-3-il)-N-metil-3-fenoxiazetidin-1-carboxamida
(Preparada según el método general E)

Etapa a. A una solución de (R)-3-(metilamino)pirrolidin-1-carboxilato de terc-butilo (Número CAS 199336-83-9) (0,22 g, 1,08 mmoles) y TEA (0,5 ml, 3,59 mmoles) en DCM (5 ml) se le añadió trifosgeno (0,1 g, 0,355 mmoles) a 0°C. La mezcla de reacción se agitó a 0°C durante 30 min. Se añadió una solución de hidrocloruro de 3-fenoxi-azetidina (Número CAS 301335-39-7) (0,2 g, 1,08 mmoles) y TEA (0,25 ml, 1,80 mmoles) a la mezcla de reacción a 0°C. La mezcla de reacción se dejó calentar a rt y se agitó durante 1 hora. La mezcla de reacción resultante se vertió en una solución saturada de NaHCO3 (50 ml) y se extrajo con DCM (3 x 25 ml). La capa orgánica combinada se lavó con salmuera (25 ml), se secó sobre Na2SO4, se filtró y se concentró a presión reducida. El residuo resultante se purificó mediante cromatografía ultrarrápida (MeOH al 5% en DCM) produciendo (R)-3-(N-metil-3-fenoxiazetidin-1 -carboxamido)pirrolidin-1 -carboxilato de terc-butilo (0,3 g, 0,80 mmoles). lCm S: Método C, RT 2,25 min, MS: ES+ 376,69.
Etapas b, c. El compuesto del título se sintetizó a partir del intermedio anterior utilizando un procedimiento similar al descrito para las etapas b, c del Ejemplo 5 para proporcionar el compuesto del título. LCMS: Método A, RT 3,84 min, MS: ES+ 301,21; RMN H1 (400 MHz, DMSO-de) 5 ppm 7,30 (t, J = 7,6 Hz, 2 H), 6,97 (t, 7,6 Hz, 1 H), 6,83 (d, J = 8,4 Hz, 2 H), 4,96 - 4,98 (m, 1 H), 4,56 - 4,60 (m, 1H), 4,31 - 4,38 (m, 2H), 3,83 - 3,89 (m, 2H), 3,37 - 3,52 (m, 3H), 3,24 -3,28 (m, 1H), 2,68 (s, 3H), 1,89 - 1,98 (m, 2 H).
Ejemplo 90 (3aR,6aR)-5-ciano-N-(2-fluoro-4-(trifluorometil)fenil)hexahidropirrolo[3,4-b]pirrol-1(2H)-
carboxamida

Sintetizada mediante un procedimiento similar al descrito para el Ejemplo 89 utilizando (3aR,6aR)-5-N-BOC-hexahidro-pirrolo[3,4-b]pirrol (Número CAS 370882-39-6) en la etapa a. LCMS: Método A, RT 4,19 min, MS: ES+ 343,05; RMN H1 (400 MHz, DMSO-de) 5 ppm 8,34 (s, 1 H), 8,46 (t, J = 8,0 Hz, 1 H), 7,68 (d, J = 10,8 Hz, 1 H), 7,52 (d, J = 7,6 Hz, 1 H), 4,37 (m, 1 H), 3,51 - 3,61 (m, 4 H), 3,41 - 3,45 (m, 1 H), 3,24 - 3,28 (m, 1 H), 2,96 - 2,98 (m, 1 H), 2,02 - 207 (m, 1 H), 1,80 - 1,86 (m, 1 H).
Ejemplo 91 (R)-N-(1-cianopirrolidin-3-il)-2-fluoro-4-(pirimidin-2-ilamino)benzamida
(Preparada según el método general G)

Etapa a. A una solución de ácido 4-amino-2-fluorobenzoico (0,4 g, 2,58 mmoles) en THF (10 ml) se le añadieron HATU (1,46 g, 3,868 mmoles) y DIPEA (1,3 ml, 7,74 mmoles) a rt y se agitó durante 20 min. Se añadió (R)-3-amino-1 N-BOC-pirrolidina (0,52 g, 2,84 mmoles) a la mezcla de reacción a rt y se agitó durante 4 h. La mezcla de reacción resultante se vertió en agua (30 ml) y se extrajo con EtOAc (2 x 50 ml). La fase orgánica combinada se recogió, se secó sobre Na2SO4, se filtró y se concentró a presión reducida produciendo (R)-3-(4-amino-2-fluorobenzamido)pirrolidin-1-carboxilato de terc-butilo (1,2 g, 3,71 mmoles). LCMS: Método C, RT 1,99 min, MS: ES+ 324,29.
Etapa b. Se preparó una mezcla de (R)-3-(4-amino-2-fluorobenzamido)pirrolidin-1-carboxilato de terc-butilo (0,7 g, 2,16 mmoles), 2-cloropirimidina (0,24 g, 2,16 mmoles), DBU (0,03 g, 1,73 mmoles) y terc-butóxido de sodio (0,31 g, 3,25 mmoles) en tolueno (15 ml) a rt. La mezcla de reacción se desgasificó durante 10 min a rt y a continuación se añadieron BINAP racémico (0,013 g, 0,021 mmoles) y Pd2(dba)3 (0,02 g, 0,021 mmoles) a la mezcla de reacción. La mezcla de reacción se calentó a 110°C durante 12 h. La mezcla de reacción resultante se dejó enfriar a rt y se vertió en agua (100 ml) y se extrajo con EtOAc (2 x 100 ml). La fase orgánica combinada se recogió, se secó sobre Na2SO4, se filtró y se concentró a presión reducida. El residuo resultante se purificó mediante cromatografía ultrarrápida (MeOH al 1,5% en DCM) produciendo (R)-3-(2-fluoro-4-(pirimidin-2-ilamino)benzamido)pirrolidin-1-carboxilato de terc-butilo (0,3 g, 0,75 mmoles). LCMS: Método C, RT 2,04 min, MS: ES+ 402,5.
Etapas c, d. El compuesto del título se sintetizó a partir del intermedio anterior utilizando un procedimiento similar al descrito para las etapas b, c del Ejemplo 1. LCMS: Método B, RT 2,98 min, MS: ES+ 305,94; RMN H1 (400 MHz, DMSO-de) ó ppm 10,14 (s, 1 H), 8,58 (d, J = 5 Hz, 2 H), 8,35 (d, J = 5 Hz, 1 H), 7,88 - 7,92 (m, 1 H), 7,54 - 7,57 (m, 2H), 6,97 (t, J = 5 Hz, 1H), 4,42 - 4, 46 (m, 1H), 3,60 - 3,64 (m, 1 H), 3,39 - 3,53 (m, 2H), 3,26 - 3,30 (m, 1 H), 2,09 -2,13 (m, 1H), 1,89 - 1,94 (m, 1H)
Ejemplo 92 N-((trans)-1-ciano-4-metilpirrolidin-3-il)-2-fluoro-4-((R)-3-metoxipirrolidin-1-il)benzamida
(Preparada según el método general H)
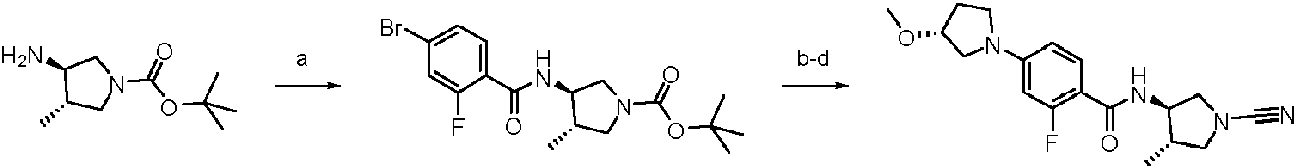
La etapa a se llevó a cabo utilizando un procedimiento similar al descrito para la etapa a de Ejemplo 91 utilizando 1-[(terc-butoxi) carbonil]-(±)-trans-3-amino-4-metilpirrolidina (descrita en la síntesis de los Ejemplos 69/70).
La etapa b se llevó a cabo utilizando un procedimiento similar a la etapa a del Ejemplo 6
Las etapas c-d se llevaron a cabo utilizando un procedimiento similar a las etapas c-d del Ejemplo 91. LCMS: Método B, RT 3,74 min, MS: ES+ 347,32; RMN H1 (400 MHz, DMSO-de) 5 ppm 7,86 - 7,89 (m, 1 H), 7,51 (t, J = 8,8 Hz, 1 H), 6,30 - 6,41 (m, 2 H), 4,11 - 4,15 (m, 2 H), 3,61 - 3,68 (m, 2 H), 3,43 - 3,45 (m, 1 H), 3,27 - 3,33 (m, 2 H), 3,25 (s, 3 H), 3,21 - 3,24 (m, 2 H), 3,05 - 3,10 (m, 1 H), 2,23 - 2,31 (m, 1 H), 2,04 - 2,09 (m, 2 H), 1,03 (d, J = 6,8 Hz, 3 H).
Ejemplo 93 2-(2-clorofenil)-N-((trans)-1-ciano-4-hidroxipirrolidin-3-il)tiazol-5-carboxamida

Sintetizada mediante un procedimiento similar al descrito para el Ejemplo 2, utilizando 2-bromotiazol-5-carboxilato de etilo (Número CAS 41731-83-3) en la etapa a y (3R,4R)-rel-3-amino-4-hidroxipirrolidin-1-carboxilato de terc-butilo (Número CAS 148214-90-8) en la etapa c. LCMS: Método A, RT 3,76 min, MS: ES+ 348,84; RMN H1 (400 MHz, DMSO-de) 5 ppm 8,86 (d, J = 6,8 Hz, 1 H), 8,60 (s, 1 H), 8,25- 8,27 (m, 1 H), 7,68 - 7,70 (m, 1 H), 7,50 - 7,61 (m, 2 H), 5,65 (dd, J = 16,4, 4,0 Hz, 1 H), 4,16 - 4,26 (m, 2 H), 3,75 - 3,79 (m, 1 H), 3,64 - 3,68 (m, 1 H), 3,40 - 3,43 (m, 1 H),
3,24 - 3,27 (m, 1 H).
Ejemplo 94 N-(1 -ciano-3-m etilpirrolidin-3-il)-2-fluoro-4-( 1 -metil- 1H-pirazol-4-il)benzamida

Sintetizada mediante un procedimiento similar al descrito para el Ejemplo 2 utilizando 3-amino-3-metilpirrolidin-1-carboxilato de terc-butilo (Número CAS 1158758-59-8) en la etapa c. LCMS: Método A, RT 3,43 min, MS: ES+ 328,15; RMN H1 (400 MHz, CDCta) 5 ppm 8,04 (t, J = 8,4 Hz, 1 H), 7,82 (s, 1 H), 7,71 (s, 1 H), 7,38 (d, J = 7,2 Hz, 1 H), 7,22 (d, J = 13,6 Hz, 1 H), 6,73 (d, J = 16 Hz, 1 H), 3,98 (s, 3 H), 3,89 (d, J = 10,4 Hz, 1 H), 3,58 - 3,66 (m, 2 H), 3,52 (d, J = 10 Hz, 1 H), 2,44 - 2,46 (m, 1 H), 2,00 - 2,04 (m, 1 H), 1,65 (s, 3 H).




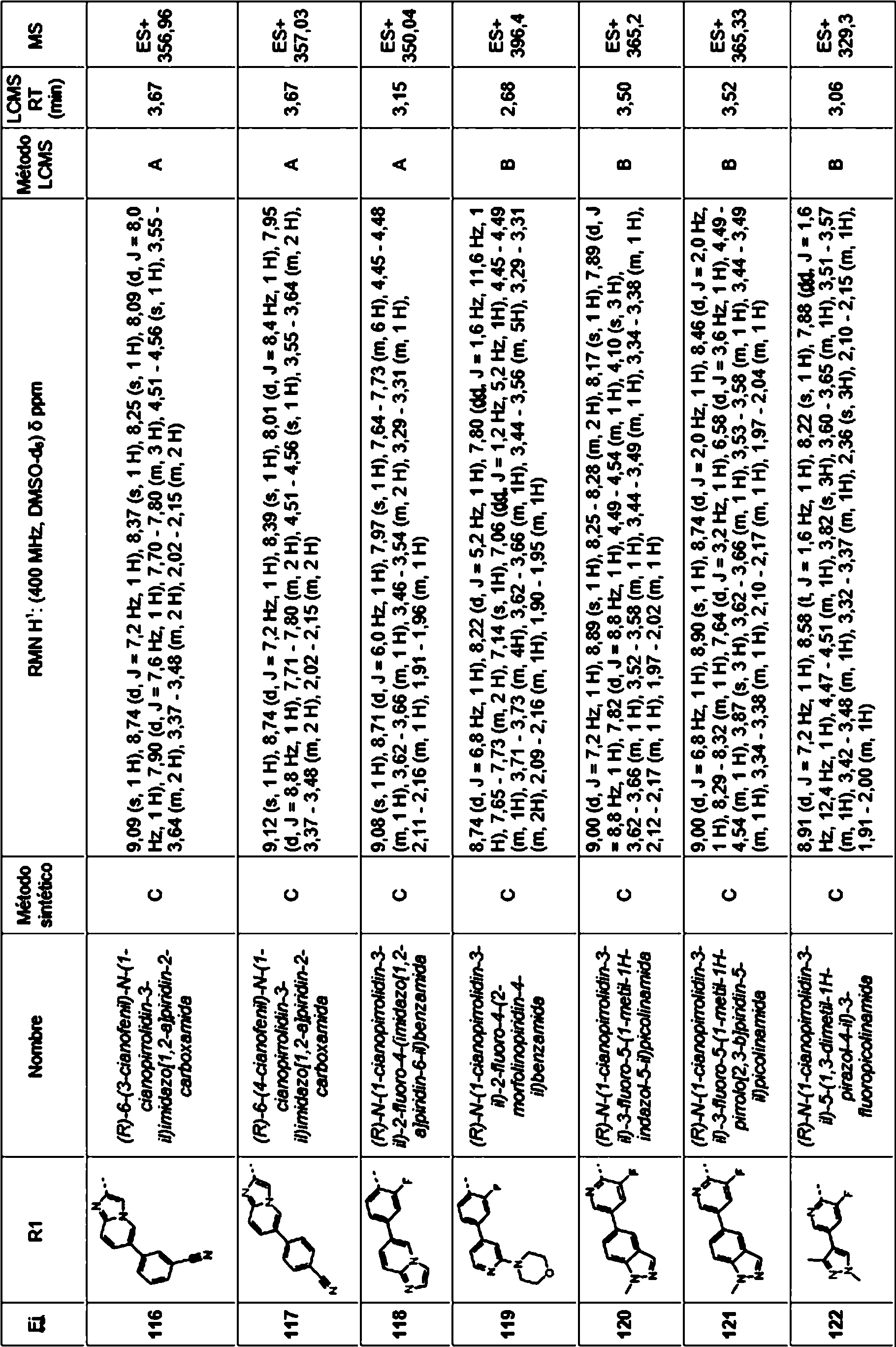

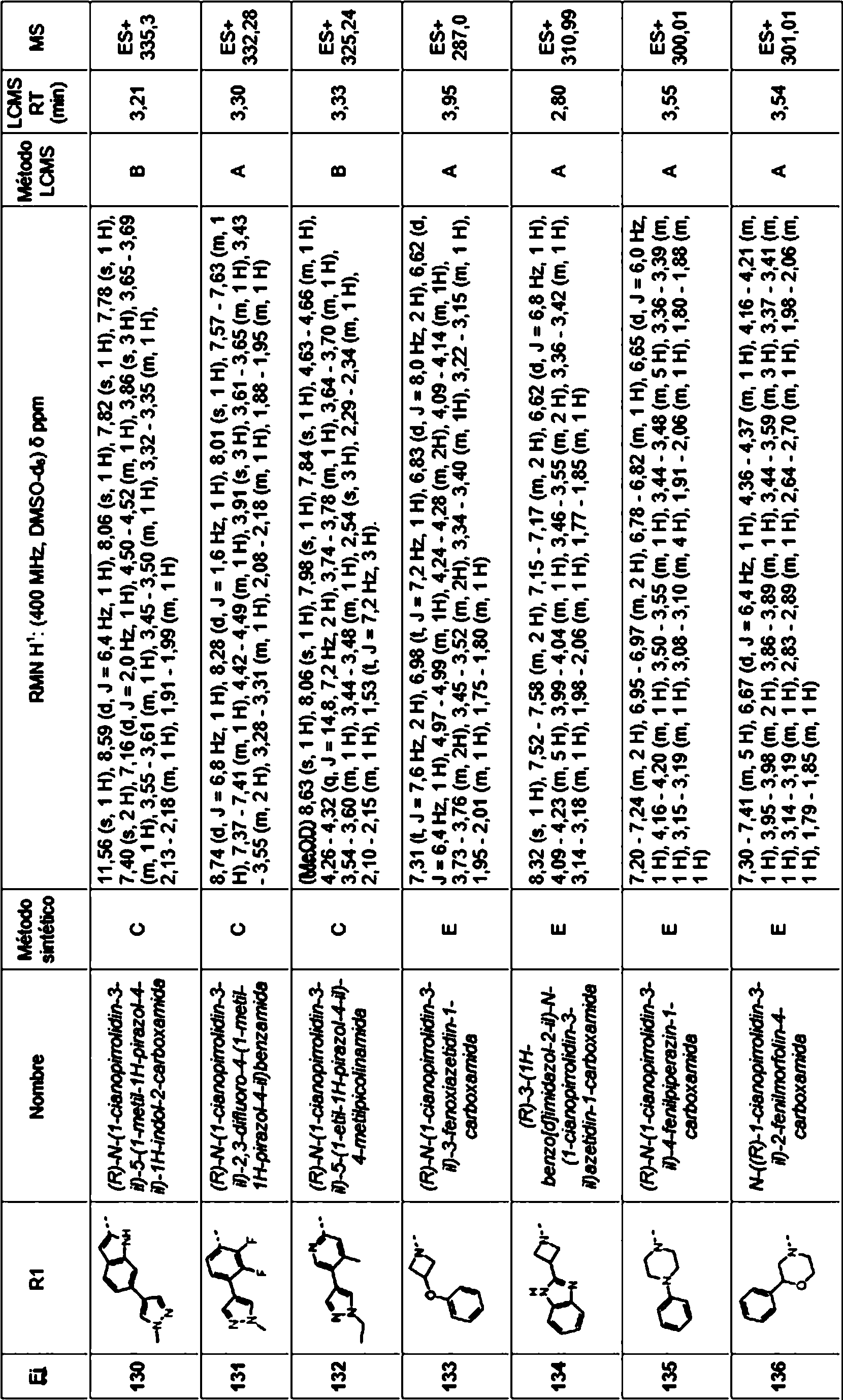


Ejemplo 150 N-((R)-1-cianopirrolidin-3-il)-5-metil-1-(1-feniletil)-1H-pirazol-3-carboxamida

Etapa a. A una solución agitada de 3-metil-1 H-pirazol-5-carboxilato de etilo (1,0 g, 6,49 mmoles) en THF (25 ml) se le añadió KOH (0,435 g, 7,78 mmoles) y se agitó a rt durante 15 min. La reacción se trató con (1-bromoetil)benceno (1,2 g, 6,49 mmoles) y se calentó a 80°C durante 8 h. La mezcla de reacción resultante se dejó enfriar a rt, se vertió rápidamente en agua (40 ml) y se extrajo con EtOAc (3 x 30 ml). La fase orgánica combinada se separó, se secó sobre Na2SO4, se filtró y se concentró a presión reducida. El residuo resultante se purificó mediante cromatografía en columna produciendo el regio-isómero no deseado 3-metil-1 -(1 -feniletil)-1 H-pirazol-5-carboxilato de etilo (0,12 g, 0,464 mmoles) en EtOAc al 5% en hexano y el regio-isómero 5-metil-1 -(1 -feniletil)-1 H-pirazol-3-carboxilato de etilo (0,8 g, 3,10 mmoles) en EtOAc al 12% en hexano. LCMS: Método C, rT 2,20 min, m S: ES+ 259,32; RMN H1 (400 MHz, CDCl3) 5 ppm: 7,26 - 7,34 (m, 2 H), 7,12 - 7,25 (m, 2 H), 6,60 (s, 1 H), 5,51 (q, J = 7,2 Hz, 1 H), 4,42 (q, J = 7,2 Hz, 2 H), 2,13 (s, 3 H), 1,99 (d, J = 6,8 Hz, 3 H), 1,41 (t, J = 6,8 Hz, 3 H).
Etapas b-e. El compuesto del título se sintetizó a partir del intermedio anterior utilizando un procedimiento similar al descrito para el Ejemplo 2 para proporcionar el compuesto del título. LCMS: Método B, RT 3,71 min, MS: ES+ 324,38; RMN H1 (400 MHz, DMSO-d6) 5 ppm 8,19 (d, J = 6,4 Hz, 1 H), 7,31 - 7,35 (m, 2 H), 7,26 - 7,27 (m, 1 H), 7,16 (d, J = 7,2 Hz, 2 H), 6,48 (s, 1 H), 5,64 - 5,66 (m, 1 H), 4,46 - 4,48 (m, 1 H), 3,58 - 3,62 (m, 2 H), 3,51 - 3,53 (m, 1 H), 3,41 -3,46 (m, 1 H), 2,16 (s, 3 H), 2,08 - 2,11 (m, 1 H), 1,95 -2,05 (m, 1 H), 1,83 (d, J = 6,8 Hz, 3 H)
Ejemplo 151 (R)-N-(1-cianopirrolidin-3-il)-5-metil-1-(piridin-2-ilmetil)-1H-pirazol-3-carboxamida

Sintetizada mediante un procedimiento similar al descrito para el Ejemplo 150. LCMS: Método B, RT 2,82 min, MS: ES+ 311,21; RMN H1 (400 MHz, DMSO-de) 5 ppm 8,53 (dd, J = 4,8, 0,8 Hz, 1 H), 8,32 (d, J = 7,2 Hz, 1 H), 7,76 - 7,81 (m, 1 H), 7,30 - 7,33 (m, 1 H), 6,98 (d, J = 7,6 Hz, 1 H), 6,52 (s, 1 H), 5,46 (s, 2 H), 4,41 - 4,46 (m, 1 H), 3,48 - 3,58 (m, 2 H), 3,38 - 3,42 (m, 1 H), 3,27 - 3,31 (m, 1 H), 2,26 (s, 3 H), 2,03 - 2,06 (m, 1 H), 1,91 - 1,96 (m, 1 H).
Ejemplo 152 (R)-N-(1-cianopirrolidin-3-il)-2-fluoro-4-(piridazin-4-il)benzamida
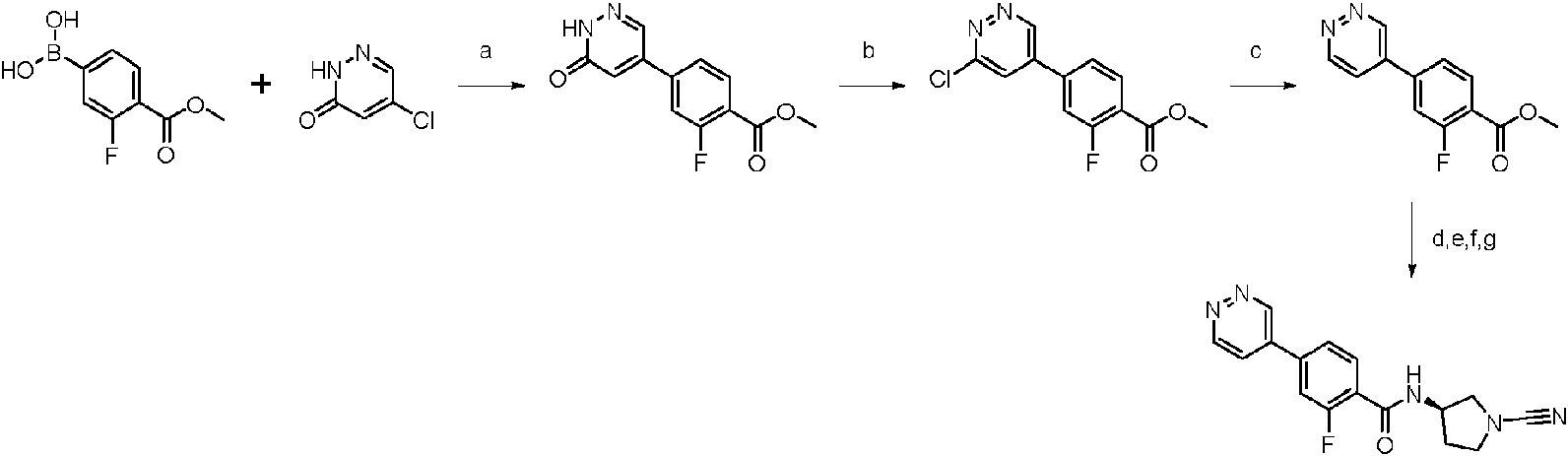
Etapa a. A una solución de 5-cloropiridazin-3(2H)-ona (0,50 g, 3,83 mmoles) y ácido 3-fluoro-4-(metoxicarbonil)fenil)borónico (0,91 g, 4,59 mmoles) en 1,4-dioxano:agua (9:1, 10 ml) se le añadió Na2CO3 (0,81 g, 7,66 mmoles) a rt. La mezcla de reacción resultante se desgasificó durante 20 min antes de añadir Pd(dppf)Cl2 (0,14 g, 0,19 mmoles) y la mezcla de reacción se calentó a 100°C durante 16 h. La mezcla de reacción resultante se vertió en agua (100 ml) y se extrajo con EtOAc (2 x 50 ml). La fase orgánica combinada se lavó con salmuera (100 ml). La fase orgánica combinada se secó sobre Na2SO4, se filtró y se concentró a presión reducida. El residuo obtenido se trituró con éter dietílico (2 x 10 ml) y se filtró a vacío produciendo 2-fluoro-4-(6-oxo-1,6-dihidropiridazin-4-il)benzoato de metilo (0,70 g, 2,82 mmoles). Esta sustancia se utilizó directamente para la siguiente etapa sin purificación adicional. LCMS: Método C, RT 1,66 min, MS: ES+ 249,22; RMN H1 (400 MHz, DMSO-de) 5 ppm 13,24 (s, 1 H), 8,35 (d, J = 1,60 Hz, 1 H), 7,99 (t, J = 7,60 Hz, 1 H), 7,91 (d, J = 12,40 Hz, 1 H), 7,80 (d, J = 8,00 Hz, 1 H), 7,31 (s, 1 H), 3,88 (s, 3 H).
Etapa b. Una solución de 2-fluoro-4-(6-oxo-1,6-dihidropiridazin-4-il)benzoato de metilo (0,90 g, 3,62 mmoles) en POCta (1,06 ml, 1,08 mmoles) se calentó a 100°C durante 2 h. La mezcla de reacción resultante se enfrió a rt y se vertió en agua helada (20 ml). El pH se ajustó a ~7-8 utilizando NaHCO3 sólido. La mezcla resultante se extrajo con EtOAc (3 x 100 ml). La fase orgánica combinada se lavó con salmuera (100 ml), se secó sobre Na2SO4, se filtró y se concentró a presión reducida produciendo 4-(6-cloropiridazin-4-il)-2-fluorobenzoato de metilo (1,1 g, cuantitativo). Esta sustancia se utilizó directamente para la siguiente etapa sin purificación adicional. LCMS: Método C, RT 1,96 min, MS: ES+ 267,26.
Etapa c. Se preparó en un autoclave una solución de 4-(6-cloropiridazin-4-il)-2-fluorobenzoato de metilo (1,10 g, 4,10 mmoles) en EtOAc:MeOH (1:1,20 ml). Se añadieron formiato de amonio (0,51 g, 8,21 mmoles) y Pd(OH)2 sobre carbono al 20% (0,5 g, 0,71 mmoles) a la mezcla de reacción a rt. La mezcla de reacción se calentó a 80°C durante 16 h. La mezcla de reacción resultante se enfrió a rt y se filtró cuidadosamente a través de celite hyflow. El lecho de celite se lavó cuidadosamente con MeOH (5 x 30 ml). El producto filtrado combinado se concentró a presión reducida. El residuo resultante se purificó mediante cromatografía en columna (MeOH al 2% en DCM) produciendo 2-fluoro-4-(piridazin-4-il)benzoato de metilo (0,17 g, 0,73 mmoles). LCMS: Método C, RT 1,69 min, MS: ES+ 233,26; RMN H1 (400 MHz, DMSO-d6) ó ppm 9,73 - 9,74 (m, 1 H), 9,36 - 9,37 (m, 1 H), 8,03 - 8,15 (m, 3 H), 7,93 - 7,94 (m, 1 H), 3,90 (s, 3 H)
Etapas d-g. El compuesto del título se sintetizó a partir del intermedio anterior utilizando un procedimiento similar al descrito para las etapas b-e del Ejemplo 2. LCm S: Método A, RT 2,68 min, MS: ES+ 312,32; RMN H1 (400 MHz, DMSO-d6) ó ppm 9,72 - 9,73 (m, 1 H), 9,34 (dd, J = 5,60, 1,20 Hz, 1 H), 8,82 (d, J = 6,40 Hz, 1 H), 8,11 (dd, J = 5,60, 2,80 Hz, 1 H), 7,99 (dd, J = 11,60, 1,60 Hz, 1 H), 7,89 (dd, J = 8,40, 2,00 Hz, 1 H), 7,74 (t, J = 7,60 Hz, 1 H), 4,44 -4,51 (m, 1 H), 3,63 - 3,70 (m, 1 H), 3,39 - 3,56 (m, 2 H), 3,28 - 3,32 (m, 1 H), 2,09 - 2,18 (m, 1 H), 1,89 - 1,97 (m, 1 H)
Ejemplo 153 (R)-N-(1-cianopirrolidin-3-il)-1-isobutil-6-(1-metil-1H-pirazol-4-il)-1H-indazol-3-carboxamida

Etapa a. A una mezcla de 6-bromoindolin-2,3-diona (5 g, 22,12 mmoles) en agua (55 ml) se le añadió NaOH (0,97 g, 24,3 mmoles) a rt. La mezcla de reacción se agitó a rt durante 30 min. Se añadió gota a gota una solución de NaNO2 (1,68 g, 24,3 mmoles) en 30 ml de agua a la mezcla de reacción a 5°C durante un período de 30 min. La mezcla de reacción se agitó a 5°C durante 20 min. La mezcla de reacción resultante se transfirió a un embudo de goteo y se añadió gota a gota a una solución de H2SO4 (4,5 ml) en agua (55 ml) a una temperatura inferior a 10°C durante un período de 25 min. La mezcla de reacción resultante se agitó a 10°C durante 20 min. Se añadió gota a gota una solución de cloruro de estaño(II) (10,06 g, 53,1 mmoles) en HCl concentrado (21 ml) a la mezcla de reacción a 5°C. La mezcla de reacción se agitó a 5°C durante 2 h. La mezcla de reacción resultante se filtró a vacío. Los sólidos deseados se lavaron con hexano (4 x 50 ml) y se secaron a alto vacío para producir ácido 6-bromo-1 H-indazol-3-carboxílico (5,95 g, 24,9 mmoles). LCMS: Método C, RT 1,66 min, MS: ES+ 239,20, 241,20
Etapa b. A una solución de ácido 6-bromo-1H-indazol-3-carboxílico (5,95 g, 24,9 mmoles) en MeOH (95 ml) se le añadió H2SO4 (5,8 ml, 58,0 mmoles) a rt. La mezcla de reacción se calentó a 90°C durante 3,5 h. La mezcla de reacción resultante se concentró a presión reducida. El residuo obtenido se vertió en una solución acuosa saturada de NaHCO3 (250 ml) y se extrajo con EtOAc (4 x 200 ml). La capa orgánica combinada se lavó con salmuera (2 x 150 ml), se secó sobre Na2SO4, se filtró y se concentró a presión reducida. El residuo resultante se purificó mediante cromatografía en columna (EtOAc al 11% en hexano) produciendo 6-bromo-1H-indazol-3-carboxilato de metilo (1,81 g, 7,98 mmoles). LCMS: Método C, RT 2,08 min, MS: ES+ 255,13, 257,10.
Etapa c. A una mezcla de ácido 6-bromo-1 H-indazol-3-carboxílico (0,25 g, 0,98 mmoles) en DMF (3 ml) y agua (3 ml) se le añadió 1-metil-4-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)-1H-pirazol (0,30 g, 2,07 mmoles) a rt, seguido de NaHCO3 (0,33 g, 3,92 mmoles). La mezcla de reacción se desgasificó con N2 durante 15 min, se añadió PdCl2(dppf) (0,071 g, 0,098 mmoles) a la mezcla de reacción a rt. La mezcla de reacción se calentó a 140°C durante 1,5 h. La mezcla de reacción resultante se vertió en agua fría (150 ml) y se extrajo con EtOAc (2 x 100 ml). La capa orgánica combinada se lavó con salmuera (100 ml), se secó sobre Na2SO4, se filtró y se concentró a presión reducida para producir 6-(1-metil-1H-pirazol-4-il)-1H-indazol-3-carboxilato de metilo (0,31 g, 1,21 mmoles). LCMS: Método A, RT 3,29 min, MS: ES+ 256,19.
Etapa d. A una solución de 6-(1-metil-1H-pirazol-4-il)-1H-indazol-3-carboxilato de metilo (0,31 g, 1,21 mmoles) en DMF (7 ml) se le añadió CS2CO3 (0,59 g, 1,81 mmoles) y 1 -yodo-2-metilpropano (0,16 ml, 1,45 mmoles) a rt. La mezcla de reacción se agitó a rt durante 2 h. La mezcla de reacción se vertió en agua fría (150 ml) y se extrajo con EtOAc (2 x 100 ml). La capa orgánica combinada se lavó con salmuera (100 ml), se secó sobre Na2SO4, se filtró y se concentró a presión reducida produciendo una mezcla de 1 -isobutil-6-(1 -metil-1 H-pirazol-4-il)-1 H-indazol-3-carboxilato de metilo (0,027 g, 0,08 mmoles) LCMS: Método C, RT 2,24 min, MS: ES+ 313,38, RMN H1 (400 MHz, DMSO-de) ó ppm 8,29 (s, 1 H), 8,03 (d, J = 4,8 Hz, 2 H), 8,0 (s, 1 H), 7,57 (dd, J = 1,2, 8,4 Hz, 1 H), 4,33 (d, J = 7,2 Hz, 2 H), 3,91 (s, 3 H), 3,89 (s, 3 H), 2,27 - 2,33 (m, 1 H), 0,88 (d, J = 6,4 Hz, 6 H) y 2-isobutil-6-(1 -metil-1 H-pirazol-4-il)-2H-indazol-3-carboxilato de metilo (0,007 g, 0,022 mmoles). LCMS: Método C, Rt 2,40 min, MS: ES+ 313,43.
Etapas e-h. El compuesto del título se sintetizó a partir del intermedio anterior utilizando un procedimiento similar al descrito para las etapas b-e del Ejemplo 2. LCMS: Método A, RT 3,98 min, MS: ES+ 392,23; RMN H1 (400 MHz,
DMSO-de) 5 ppm 8,60 (d, J = 7,2 Hz, 1 H), 8,28 (s, 1 H), 8,09 (d, J = 8,4 Hz, 1 H), 8,02 (s, 1 H), 7,97 (s, 1 H), 7,50 (d, J = 8,4 Hz, 1 H), 4,54 - 4,58 (m, 1 H), 4,30 (d, J = 7,2 Hz, 2 H), 3,89 (s, 3 H), 3,55 - 3,66 (m, 2 H), 3,39 - 3,47 (m, 2 H), 2,33 - 2,37 (m, 1 H), 2,11 - 2,16 (m, 1 H), 2,01 - 2,06 (m, 1 H), 0,90 (d, J = 6,4 Hz, 6 H).
Ejemplo 154 (R)-N-(1-cianopirrolidin-3-il)-6-(3,5-dimetilisoxazol-4-il)-1-isobutil-1H-indazol-3-carboxamida

Sintetizada mediante un procedimiento similar al descrito para el Ejemplo 153 utilizando ácido 3,5-dimetilisoxazol-4-borónico (Número CAS 16114-47-9) en la etapa c. LCMS: Método A, RT 4,54 min, MS: ES+ 407,07; RMN H1 (400 MHz, DMSO-d6) 5 ppm 8,67 (d, J = 6,8 Hz, 1 H), 8,21 (d, J = 8,0 Hz, 1 H), 7,82 (s, 1 H), 7,27 (dd, J = 8,8, J = 1,2 Hz, 1 H), 4,57 - 4,58 (m, 1 H), 4,35 (d, J = 7,2 Hz, 1 H), 3,55 - 3,66 (m, 2 H), 3,33 - 3,48 (m, 2 H), 2,44 - 2,46 (m, 4 H), 2,28 - 2,33 (m, 4 H), 1,99 - 2,15 (m, 2 H), 0,85 - 0,89 (m, 6 H).
Ejemplo 155 (R)-N-(1-cianopirrolidin-3-il)-1-(ciclopropilmetil)-6-(3,5-dimetilisoxazol-4-il)-1H-indazol-3-carboxamida

Sintetizada mediante un procedimiento similar al descrito para el Ejemplo 154 utilizando bromuro de ciclopropilmetilo (Número CAS 7051-34-5) en la etapa d. LCMS: Método A, RT 4,37 min, MS: ES+ 404,99; RMN H1 (400 MHz, DMSO-de) 5 ppm 8,68 (d, J = 7,2 Hz, 1 H), 8,22 (d, J = 8,4 Hz, 1 H), 7,83 (s, 1 H), 7,28 (dd, J = 8,4, 1,2 Hz, 1 H), 4,56 - 4,61 (m, 1 H), 4,43 - 4,45 (m, 2 H), 3,56 - 3,67 (m, 2 H), 3,39 - 3,48 (m, 2 H), 2,46 (s, 3 H), 2,29 (s, 3 H), 2,13 - 2,18 (m, 1 H), 2,03 - 2,11 (m, 1 H), 1,34 - 1,38 (m, 1 H), 0,44 - 0,54 (m, 4 H).
Ejemplo 156 (R)-N-(1-cianopirrolidin-3-il)-N-metil-6-(1-metil-1H-pirazol-4-il)imidazo[1,2-a]piridin-2-carboxamida

Sintetizada mediante un procedimiento similar al descrito para el Ejemplo 3 utilizando (R)-3-(metilamino)pirrolidin-1-carboxilato de terc-butilo (Número CAS 199336-83-9). LCMS: Método A, RT 2,87 min, MS: ES+ 350,17; r Mn H1 (400 MHz, DMSO-de) 5 ppm 8,82 (s, 1 H), 8,24 (s, 1 H), 8,19 (s, 1 H), 7,89 (s, 1 H), 7,65 (d, J = 9,2 Hz, 1 H), 7,58 (dd, J = 9,2, 1,2 Hz, 1 H), 3,89 (s, 3 H), 3,54 - 3,64 (m, 2 H), 3,43 - 3,47 (m, 3 H), 3,37 (s, 3 H), 2,07 - 2,17 (m; 2 H).
Ejemplo 157 (R)-N-(1-cianopirrolidin-3-il)-N-metil-5-(1-metil-1H-pirazol-4-il)-1H-indol-2-carboxamida

Sintetizada utilizando un procedimiento similar al descrito para el Ejemplo 3 utilizando (R)-3-(metilamino)pirrolidin-1-carboxilato de terc-butilo (Número CAS 199336-83-9). LCMS: Método A, RT 3,38 min, MS: ES+ 349,11; r Mn H1 (400 MHz, DMSO-d6) 5 ppm 11,56 (s, 1 H), 8,05 (s, 1 H), 7,81 (s, 1 H), 7,77 (s, 1 H), 7,41 (s, 2 H), 6,85 (s, 1 H), 5,16 (t, J = 8,0 Hz, 1 H), 3,86 (s, 3 H), 3,57 - 3,66 (m, 2 H), 3,40 - 3,49 (m, 2 H), 3,13 (s, 3 H), 2,08 - 2,17 (m, 2 H).
Ejemplo 158 (R)-N-(1-cianopirrolidin-3-il)-6-(3,5-dimetilisoxazol-4-il)-N-metil-1H-benzo[d]imidazol-2-carboxamida

Sintetizada mediante un procedimiento similar al descrito para el Ejemplo 3 utilizando (R)-3-(metilamino)pirrolidin-1-carboxilato de terc-butilo (Número CAS 199336-83-9). LCMS: Método B, RT 3,45 min, MS: e S+ 365,3; r Mn H1 (400 MHz, DMSO-d6) 5 ppm 13,02 (s, 1 H), 7,49 - 7,82 (m, 2 H), 7,28 (s, 1 H), 3,59 - 3,71 (m, 2 H), 3,44 - 3,54 (m, 3 H),
3,02 -3,09 (m, 3 H), 2,42 (s, 3 H), 2,24 (s, 3 H), 2,13 - 2,21 (m, 2 H).
Ejemplo 159 (R)-N-(1-cianopirrolidin-3-il)-7-(1-metil-1H-pirazol-4-il)imidazo[1,2-a]piridin-3-carboxamida

Etapa a. A una solución de éster etílico de ácido 7-bromo-imidazo[1,2-a]piridin-3-carboxílico (0,2 g, 0,743 mmoles) en THF:agua (1:1) (12 ml) se le añadió UOH.H2O (0,062 g, 1,49 mmoles) a 0°C. La mezcla de reacción se agitó a rt durante 6 h. La mezcla de reacción resultante se aciduló a pH 4 mediante la adición gota a gota de una solución de ácido cítrico al 10% y se agitó durante 10 min. Los sólidos resultantes se filtraron a vacío y se secaron para producir ácido 7-bromoimidazo[1,2-a]piridin-3-carboxílico (0,16 g, 0,666 mmoles). LCMS: Método C, RT 1,39 min, MS: ES+ 241,08, 242,99
Etapas b-e. El compuesto del título se sintetizó a partir del intermedio anterior utilizando un procedimiento similar al descrito para el Ejemplo 3 para proporcionar el compuesto del título. LCMS: Método B, RT 2,76 min, MS: ES+ 336,32; RMN H1 (400 MHz, DMSO-de) ó ppm 9,33 - 9,37 (m, 1 H), 8,54 (d, J = 6,4 Hz, 1 H), 8,38 (s, 1 H), 8,35 (s, 1 H), 8,10 (s, 1 H), 7,90 (d, J = 0,8 Hz, 1 H), 7,40 (dd, J = 2 Hz, 7,2 Hz, 1 H), 4,51 - 4,55 (m, 1 H), 3,89 (s, 3 H), 3,65 - 3,69 (m, 1 H), 3,54 - 3,60 (m, 1 H), 3,44 - 3,50 (m, 1 H), 3,31 - 3,35 (m, 1 H), 2,12 - 2,21 (m, 1 H), 1,94 - 2,01 (m, 1 H)
Ejemplo 160 (R)-7-(3-cianofenil)-N-(1-cianopirrolidin-3-il)imidazo[1,2-a]piridin-3-carboxamida
cedimiento similar al descrito para el Ejemplo 159. LCMS: Método A, RT 3,69 min, MS: Hz, DMSO-de) 5 ppm 9,48 (d, J = 7,6 Hz, 1 H), 8,68 (d, J = 6,8 Hz, 1 H), 8,44 (s, 1 H), 0 - 8,25 (m, 2H), 7,91 (d, J = 8 Hz, 1H), 7,73 (t, J = 7,6 Hz, 1H), 7,62 (dd, J = 2 Hz, 7,6 Hz, 7 - 3,71 (m, 1H), 3,55 - 3,61 (m, 1H), 3,47 - 3,51 (m, 1 H), 3,33 - 3,36 (m, 1 H), 2,15 - 2,20
pirrolidin-3-il)-N-metil-7-(2-metilpiridin-4-il)imidazo[1,2-a]piridin-3-carboxamida
cedimiento similar al descrito para el Ejemplo 159. LCMS: Método A, RT 3,04 min, MS: Hz, DMSO-de) 5 ppm 9,04 (d, J = 7,2 Hz, 1 H), 8,57 (d, J = 5,2 Hz, 1 H), 8,28 (s, 1 H), ,73 (d, J = 5,2 Hz, 1 H), 7,59 (dd, J = 2,0 Hz, 7,6 Hz, 1 H), 5,11 - 5,15 (m, 1 H), 3,57 - 3,69 ), 3,13 (s, 3 H), 2,57 (s, 3 H), 2,10 - 2,22 (m, 2 H)
pirrolidin-3-il)-N-metil-7-(6-metilpiridin-3-il)imidazo[1,2-a]piridin-3-carboxamida
cedimiento similar al descrito para el Ejemplo 159. LCMS: Método A, RT 3,15 min, MS: Hz, DMSO-d6) 5 ppm 9,03 (d, J = 7,2 Hz, 1 H), 8,97 (d, J = 2,0 Hz, 1 H), 8,14 - 8,20 (m, 2 Hz, 1 H), 7,40 (d, J = 8,4 Hz, 1 H), 5,12 - 5,15 (m, 1 H), 3,57 - 3,69 (m, 2 H), 3,40 - 3,50 (s, 3 H), 2,10 - 2,21 (m, 2 H)

Sintetizada mediante un procedimiento similar al descrito para el Ejemplo 159. LCMS: Método A, RT 3,06 min, MS: ES+ 364,15; RMN H1 (400 MHz, DMSO-d6) 5 ppm 8,94 (d, J = 7,2 Hz, 1 H), 8,19 (s, 1 H), 8,10 (s, 1 H), 7,67 (s, 1 H), 7,27 (dd, J = 7,2, 2 Hz, 1 H), 5,08 - 5,15 (m, 1 H), 3,81 (s, 3 H), 3,56 - 3,67 (m, 2 H), 3,39 - 3,47 (m, 2 H), 3,11 (s, 3 H), 2,41 (s, 3 H), 2,08 - 2,23 (m, 2 H).
Ejemplo 164 (R)-N-(1-cianopirrolidin-3-il)-7-(2,6-dimetilpiridin-4-il)-N-metilimidazo[1,2-a]piridin-3-carboxamida

Etapa a. A una solución de 5-bromopiridin-2-amina (5 g, 28,9 mmoles) en tolueno (30 ml) se le añadió 3,3,3-trifluoro-2-oxopropanoato de metilo (Número CAS 13089-11-7) (4,5 g, 28,9 mmoles) y piridina (4,65 ml, 57,8 mmoles) a rt. La mezcla de reacción se agitó a rt durante 5 min antes de añadir gota a gota SOCl2 (3,43 g, 28,9 mmoles) a la mezcla de reacción. La mezcla de reacción se agitó a rt durante 16 h. La mezcla de reacción resultante se vertió en agua fría (200 ml) y se alcalinizó con NaHCO3 sólido. La mezcla resultante se extrajo con EtOAc (3 x 100 ml). La capa orgánica combinada se secó sobre Na2SO4, se filtró y se concentró a presión reducida. El residuo resultante se purificó mediante cromatografía en columna (EtOAc al 1% en hexano) produciendo (Z)-2-((5-bromopiridin-2-il)imino)-3,3,3-trifluoropropanoato de metilo (5 g, 16,07 mmoles). LCMS: Método C, RT 2,75 min, m S: ES+ 313,10, 315,10
Etapa b. A una solución de (Z)-2-((5-bromopiridin-2-il)imino)-3,3,3-trifluoropropanoato de metilo (5 g, 16,1 mmoles) en MeCN (50 ml) se le añadió fosfito de trimetilo (2,99 g, 24,1 mmoles) a rt. La mezcla de reacción se calentó a 80°C durante 24 h. La mezcla de reacción resultante se enfrió a rt y se vertió en agua fría (150 ml) seguido de la adición de una solución de K2CO3 al 5% (100 ml). La mezcla resultante se extrajo con EtOAc (3 x 100 ml). La capa orgánica combinada se lavó con salmuera (100 ml), se secó sobre Na2SO4, se filtró y se concentró a presión reducida. El residuo resultante se purificó mediante cromatografía en columna (EtOAc al 25% en hexano) produciendo 6-bromo-3-fluoroimidazo[1,2-a]piridin-2-carboxilato de metilo (1,5 g, 5,49 mmoles). LCMS: Método C, RT 1,94 min, MS: ES+ 273,10, 275,13
Etapa c. Una mezcla de 6-bromo-3-fluoroimidazo[1,2-a]piridin-2-carboxilato de metilo (1,5 g, 5,49 mmoles) en HCl concentrado se calentó a 100°C durante 2,5 h. La mezcla de reacción resultante se concentró a presión reducida. El residuo obtenido se destiló azeotrópicamente con DCM (3 x 10 ml) y se secó a presión reducida para producir la sal de HCl de ácido 6-bromo-3-fluoroimidazo[1,2-a]piridin-2-carboxílico (1,2 g, 4,06 mmoles). LCMS: Método C, RT 1,68
min, MS: ES 259,30, 261,30
Etapas d-g. El compuesto del título se sintetizó a partir del intermedio anterior utilizando un procedimiento similar al descrito para el Ejemplo 3 para proporcionar el compuesto del título. LCMS: Método A, RT 3,80 min, MS: ES+ 374,99; RMN H1 (400 MHz, DMSO-d6) 5 ppm 8,83 (s, 1 H), 8,76 (d, J = 7,2 Hz, 1 H), 8,36 (s, 1 H), 8,16 (d, J = 8,0 Hz, 1 H), 7,90 (d, J = 7,6 Hz, 1 H), 7,80 (dd, J = 1,6 Hz, 9,6 Hz, 1 H), 7,72 (t, J = 8,0 Hz, 1 H), 7,66 (d, J = 9,2 Hz, 1 H), 4,50 -4,55 (m, 1 H), 3,54 - 3,63 (m, 2 H), 3,37 - 3,47 (m, 2 H), 2,01 - 2,14 (m, 2 H).
Los compuestos de la Tabla 8 se sintetizaron utilizando un procedimiento similar al descrito para el Ejemplo 167.

- - - - -168 O " 1 H-pirazol-4- (s, 1 H), 7,61 - 7,64 (m, 1 H), 7,54 - 7,57 (m, 1 H), A 2,98 ES+ /N—^
il imidazo 12- 4,49 - 4,54 (m, 1 H), 3,88 (s, 3 H), 3,55 - 3,62 (m, 2 H), 354,2


- -e - -prazo- s, , , , = , , , z, , , , = ,
169 N - ^< 4-il)-3- Hz, 1 H), 4,49 - 4,54 (m, 1 H), 4,14 - 4,19 (m, 2 H), B 3,22 ES+
ahr - , - - - J = 9,6, 1,2 Hz, 1 H),
170 pirazol-4-il)-3- (d, J = 9,6 Hz, 1 H), 7,47 (dd, A 3,11 ES+ N - “ luoroimidazo 12- 4,49 - 4,54 (m, 1 H), 3,81 (s, 3 H), 3,53 - 3,63 (m, 2 H), 368,1

- , -etilisoxazol- 4- Hz, 1 H), 7,40 (dd, J = 10,4, 1,6 Hz, 1 H), 4,50 - 4,55 (m,
171 íV dim B 3,34 ES+ fluoroimidazo[ 12- 1 H), 3,54 - 3,63 (m, 2 H), 3,36 - 3,47 (m, 2 H), 2,45 (s, 369,2 a]piridin-2-carboxamida 
Ejemplo 172 (R)-N-(1-cianopirrolidin-3-il)-2-fluoro-4-(pirazolo[1,5-a]pirim idin-5-il)benzamida

Etapa a. A una solución de ácido 3-fluoro-4-(metoxicarbonil)fenil)borónico (Número CAS 505083-04-5) (0,50 g, 2,50 mmoles) en THF:agua (5:2, 14 ml) se le añadió UOH.H2O (0,32 g, 7,57 mmoles) a 0°C. La mezcla de reacción se agitó a rt durante 24 h, a continuación, se calentó a 60°C durante 1 h. La mezcla de reacción resultante se enfrió a rt y se aciduló utilizando una solución de HCl 1M. La mezcla resultante se extrajo con EtOAc (3 x 50 ml). La fase orgánica combinada se secó sobre Na2SO4, se filtró y se concentró a presión reducida produciendo ácido 4-borono-2-fluorobenzoico (0,42 g, 2,31 mmoles). LCMS: Método C, RT 1,32 min, MS: ES-183,20; RMN H1 (400 MHz, DMSO-d6) 5 ppm 13,22 (s, 1 H), 8,42 (s, 2 H), 7,81 (t, J = 7,60 Hz, 1 H), 7,65 (d, J = 7,60 Hz, 1 H), 7,59 (d, J = 4,00 Hz, 1 H)
Etapas b-e. El compuesto del título se sintetizó a partir del intermedio anterior utilizando un procedimiento similar al
descrito para el Ejemplo 3 para proporcionar el compuesto del título. LCMS: Método A, RT 3,38 min, MS: ES+ 351,04; RMN H1 (400 MHz, DMSO-d6) 5 ppm 8,27 (d, J = 7,60 Hz, 1 H), 8,82 (d, J = 6,80 Hz, 1 H), 8,29 (d, J = 2,40 Hz, 1 H), 8,13 - 8,18 (m, 2 H), 7,75 - 7,77 (m, 2 H), 6,83 (d, J = 2,40 Hz, 1 H), 4,47 - 4,49 (m, 1 H), 3,63 - 3,67 (m, 1 H), 3,40 -3,56 (m, 2H), 3,28 - 3,30 (m, 1 H), 2,10 - 2,17 (m, 1 H), 1,91 - 1,96 (m, 1 H)
Ejemplo 173 (R)-N-(1-cianopirrolidin-3-il)-5-(4-fluorofenil)picolinamida

Etapa a. A una solución de 5-bromopicolinato de metilo (Número CAS 29682-15-3) (1 g, 4,62 mmoles) en THF (10 ml) se le añadieron DIPEA (1,79 g, 13,9 mmoles) y (R)-3-aminopirrolidin-1-carboxilato de terc-butilo (1,03 g, 5,55 mmoles) a rt seguido de trimetilaluminio (2 M en tolueno) (11,5 ml, 2,34 mmoles). La mezcla de reacción se calentó a 70°C durante 2 h. La mezcla de reacción resultante se vertió en una solución saturada de NH4Cl (150 ml) y la mezcla se filtró a través de celite hyflow. El lecho de celite se lavó con EtOAc (50 ml). El producto filtrado se extrajo con EtOAc (3 x 100 ml). La fase orgánica combinada se secó sobre Na2SO4, se filtró y se concentró a presión reducida. El residuo resultante se purificó mediante cromatografía en columna (EtOAc al 20% en hexano) produciendo (R)-3-(5-bromopicolinamido)pirrolidin-1-carboxilato de terc-butilo (1,2 g, 3,24 mmoles). LCMS: Método C, RT 2,34 min, MS: ES+ 314,18, 316,18 [M-56]; RMN H1 (400 MHz, DMSO-d6) 5 ppm 8,94 (d, J = 7,2 Hz, 1 H), 8,77 - 8,78 (m, 1 H), 8,26 (dd, J = 2, 8 Hz, 1 H), 7,96 (d, J = 8,4 Hz, 1 H), 4,42 - 4,45 (m, 1 H), 3,48 - 3,58 (m, 1 H), 3,37 - 3,39 (m, 1 H), 3,21 -3,29 (m, 2 H), 1,97 - 2,06 (m, 2 H), 1,40 (s, 9 H)
Etapas b-d. El compuesto del título se sintetizó a partir del intermedio anterior utilizando un procedimiento similar al descrito para las etapas b-d del Ejemplo 3 para proporcionar el compuesto del título. LCMS: Método A, RT 4,15 min, MS: ES+ 311,06; RMN H1 (400 MHz, DMSO-de) 5 ppm 9,06 (d, J = 7,2 Hz, 1 H), 8,94 (d, J = 2,0 Hz, 1 H), 8,28 (dd,
J = 2,4 Hz, 8,4 Hz, 1 H), 8,09 (d, J = 8,0 Hz, 1 H), 7,85 - 7,89 (m, 2 H), 7,39 (t, J = 8,8 Hz, 2 H), 4,53 - 4,57 (m, 1 H), 3,54 - 3,64 (m, 2 H), 3,39 - 3,48 (m, 2 H), 2,02 - 2,16 (m, 2 H)
Ejemplo 174 N-((cis)-1 -ciano-2-metilpirrolidin-3-il)-5-(4-fluorofenil)picolinamida

Sintetizada como una mezcla racémica utilizando un procedimiento similar al descrito para el Ejemplo 2 utilizando cis-3-amino-1 -BOC-2-metilpirrolidina (Número CAS 1374653-02-7) en la etapa c. LCMS: Método A, RT 4,35 min, MS: ES+ 324,96; RMN H1 (400 MHz, DMSO-de) 5 ppm 8,96 (s, 1 H), 8,76 (d, J = 8,0 Hz, 1 H), 8,29 (d, J = 8,0 Hz, 1 H), 8,11 (d, J = 8,0 Hz, 1 H), 7,85 - 7,89 (m, 2 H), 7,39 (t, J = 8,8 Hz, 2 H), 4,54 - 4,59 (m, 1 H), 3,82 - 3,87 (m, 1 H), 3,63 - 3,68 (m, 1 H), 3,37 - 3,43 (m, 1 H), 2,07 - 2,21 (m, 2 H), 1,08 (t, J = 6,4 Hz, 3 H).
Ejemplo 175 3-cloro-N-((trans)-1-ciano-4-metilpirrolidin-3-il)-4-morfolinobenzamida

Sintetizada como una mezcla racémica utilizando un procedimiento similar al descrito para el Ejemplos 69/70 utilizando ácido 3-cloro-4-morfolinobenzoico (Número CAS 26586-20-9) en la etapa e. LCMS: Método B, RT 3,66 min, MS: ES+ 349,10; RMN H1 (400 MHz, DMSO-de) 5 ppm 8,52 (d, J = 7,2 Hz, 1 H), 7,93 (d, J = 2,0, 1 H), 7,81 (dd, J = 8,0, 1,6 Hz, 1 H), 7,22 (d, J = 8,8 Hz, 1 H), 4,09 - 4,14 (m, 1 H), 3,75 (t, J = 4,8, 4 H), 3,63 - 3,72 (m, 2 H), 3,21 - 3,25 (m, 1 H), 3,06 - 3,11 (m, 1 H), 3,04 (t, J = 4,4, 4 H), 2,24 - 2,33 (m, 1 H), 1,99 (d, J = 6,8, 3 H).
Ejemplo 176 N-((trans)-1 -ciano-4-fluoropirrolidin-3-il)-[1,1 '-bifenil]-4-carboxamida

Etapa a. A una solución de ácido 4-fenilbenzoico (0,110 g, 0,555 mmoles) en THF (5 ml) se le añadieron DIPEA (0,110 g, 0,852 mmoles) y HATU (0,243 g, 0,639 mmoles) a rt y se agitó durante 30 min. Se añadió Trans-3-amino-4-fluoropirrolidin-1-carboxilato de terc-butilo (Número CAS 1363382-79-9) (0,100 g, 0,427 mmoles) a la mezcla de
reacción. La mezcla de reacción resultante se agitó a rt durante 1,5 h. La mezcla de reacción se vertió en una solución saturada de NaHCO3 (20 ml) y se extrajo con EtOAc (2 x 25 ml). La capa orgánica combinada se secó sobre Na2SO4, se filtró y se concentró a presión reducida. El residuo obtenido se purificó mediante cromatografía ultrarrápida (EtOAc al 10% en hexano) produciendo 3-([1,1'-bifenil]-4-carboxamido)-4-fluoropirrolidin-1-carboxilato de trans-terc-butilo (0,170 g, 0,443 mmoles). LCMS: Método C, RT 2,52 min, MS: ES-383,45; RMN H1 (400 MHz, DMSO-d6) 5 ppm 8,72 (d, J = 6,4 Hz, 1 H), 7,97 (d, J = 7,2 Hz, 2 H), 7,73 - 7,82 (m, 4 H), 7,48 - 7,52 (m, 2 H), 7,40 - 7,43 (m, 1 H), 4,52 - 4,54 (m, 1 H), 3,63 - 3,69 (m, 2 H), 3,52 - 3,58 (m, 1 H), 3,44 - 3,50 (m, 2 H), 1,44 (s, 9 H).
Etapa b. Se preparó a rt una solución de trans-3-([1,1'-bifenil]-4-carboxamido)-4-fluoropirrolidin-1-carboxilato de tercbutilo (0,150 g, 0,390 mmoles) en ácido fórmico (7,5 ml). La mezcla de reacción resultante se agitó a 50°C durante 3 h. La mezcla de reacción resultante se evaporó a presión reducida. El material obtenido se evaporó conjuntamente con DCM (2 x 30 ml) y se secó a vacío produciendo la sal fórmica de trans-N-(4-fluoropirrolidin-3-il)-[1,1'-bifenil]-4-carboxamida (0,140 g, cuantitativo). LCMS: Método C, RT 1,83 min, MS: ES+ 285,22.
Etapa c. El compuesto del título se sintetizó a partir del intermedio anterior utilizando un procedimiento similar al descrito para la etapa c del Ejemplo 1. LCMS: Método H, RT 26,23 min, MS: ES+ 309,92; RMN H1 (400 MHz, DMSO-d6) 5 ppm 8,75 (d, J = 6,4 Hz, 1 H), 7,97 (d, J = 8,4 Hz, 2 H), 7,97 (d, J = 8,1 Hz, 2 H), 7,73 - 7,75 (m, 2 H), 7,50 (t, J = 7,6 Hz, 2 H), 7,40 - 7,44 (m, 1 H), 5,12 - 5,24 (m, 1 H), 4,54 - 4,61 (m, 1 H), 3,82 - 3,89 (m, 1 H), 3,65 - 3,76 (m, 2 H), 3.56 - 3,59 (m, 1 H).
Ejemplo 177 N-((cis)-1-ciano-4-ciclopropilpirrolidin-3-il)-3-fluoro-4-(1-metil-1H-pirazol-4-il)benzamida Etapa a. Una solución de 6-oxa-3-azabiciclo[3,1,0]hexano-3-carboxilato de terc-butilo (Número CAS 114214-49-2) (1,00 g, 5,405 mmoles) en THF (40 ml) se enfrió a -30°C y se trató con un complejo de sulfuro de metilo y bromuro de cobre (I) (0,221 g, 1,078 mmoles). Se añadió gota a gota una solución 0,5 M de bromuro de ciclopropilmagnesio en THF (41 ml, 20,5 mmoles) a la mezcla de reacción a -30°C. Se dejó que la mezcla de reacción se calentara a -10°C y se agitó durante 1 h. La mezcla resultante se vertió en una solución saturada de NH4Cl (500 ml) y se extrajo con EtOAc (2 x 70 ml). La capa orgánica combinada se secó sobre Na2SO4, se filtró y se concentró a presión reducida. El residuo resultante se purificó mediante cromatografía en columna (EtOAc al 30% en hexano) produciendo 3-ciclopropil-4-hidroxipirrolidin-1 -carboxilato de trans-terc-butilo (1,70 g, 7,49 mmoles). LCMS: Método A, RT 3,91 min, MS: ES+ 228,00; RMN H1 (400 MHz, DMSO-ds) 5 ppm: 4,97 (d, J = 4,0 Hz, 1 H), 3,97 - 3,99 (m, 1 H), 3,59 - 3,72 (m, 1 H), 3,35 - 3,47 (m, 2 H), 3,02 - 3,07 (m, 2 H), 1,37 (s, 9 H), 0,55 - 0,60 (m, 1 H), 0,33- 0,45 (m, 2 H), 0,17 - 0,23 (m, 1 H), 0,06 - 0,12 (m, 1 H).
Etapa b. A una solución de trans-3-ciclopropil-4-hidroxipirrolidin-1 -carboxilato de terc-butilo (1,70 g, 7,49 mmoles) en THF (20 ml) se le añadió NaH (dispersión al 60% en aceite de parafina, 0,898 g, 37,42 mmoles) a rt. La mezcla de reacción se agitó a 50°C durante 1 h. La mezcla de reacción resultante se enfrió a rt. Se añadió gota a gota una solución de cloruro de p-toluenosulfonilo (2,800 g, 14,74 mmoles) en THF (10 ml) a la mezcla de reacción y se agitó durante 16 h. La mezcla de reacción resultante se vertió en agua (500 ml) y se extrajo con EtOAc (2 x 100 ml). La capa orgánica combinada se secó sobre Na2SO4, se filtró y se concentró a presión reducida. El residuo obtenido se purificó mediante cromatografía ultrarrápida (EtOAc al 17% en hexano) produciendo trans-3-ciclopropil-4-(tosiloxi)pirrolidin-1 -carboxilato de terc-butilo (1,500 g, 3,94 mmoles). LCMS: Método C, RT 2,65 min, MS: ES+ 326,30 (M-56).
Etapa c. A una solución de trans-3-ciclopropil-4-(tosiloxi)pirrolidin-1 -carboxilato de terc-butilo (1,40 g, 3,67 mmoles) en DMF (20 ml) se le añadió NaN3 (4,700 g, 72,31 mmoles) a rt. La mezcla de reacción se calentó a 60°C durante 16 h. La mezcla de reacción resultante se enfrió a rt y se vertió en agua fría (400 ml). La mezcla resultante se extrajo con EtOAc (2 x 70 ml). La capa orgánica combinada se secó sobre Na2SO4, se filtró y se concentró a presión reducida produciendo cis-3-azido-4-ciclopropilpirrolidin-1-carboxilato de terc-butilo (1,00 g, cuantitativo). Esta sustancia se utilizó directamente para la siguiente etapa sin purificación adicional.
Etapa d. A una solución de cis-3-azido-4-ciclopropilpirrolidin-1-carboxilato de terc-butilo (1,00 g, 3,97 mmoles) en MeOH (20 ml) se le añadió Pd al 10% sobre carbono (seco) (1,00 g) a rt. La mezcla de reacción se purgó con hidrógeno durante 2 h. La mezcla de reacción resultante se filtró cuidadosamente a través de celite hyflow y el producto filtrado obtenido se concentró a presión reducida produciendo cis-3-amino-4-ciclopropilpirrolidin-1-carboxilato de terc-butilo (0,570 g, 2,52 mmoles). LCMS: Método A, RT 3,75 min, MS: ES+ 227,00. Esta sustancia se utilizó directamente para la siguiente etapa sin purificación adicional.
Etapas e-g. El compuesto del título se sintetizó a partir del intermedio anterior utilizando un procedimiento similar al descrito para el Ejemplo 1. LCMS: Método B, RT 3,64 min, MS: ES+ 354,60; RMN H1 (400 MHz, DMSO-d6) 5 ppm 8.56 (d, J = 8,4 Hz, 1 H), 8,24 (d, J = 2,0 Hz, 1 H), 7,98 (s, 1 H), 7,84 (t, J = 8,0 Hz, 1 H), 7,72 - 7,78 (m, 2 H), 4,64 -4,68 (m, 1 H), 3,91 (s, 3 H), 3,68 - 3,72 (m, 1 H), 3,52 - 3,57 (m, 1 H), 3,37 - 3,47 (m, 2 H), 1,68 - 1,74 (m, 1 H), 0,69 -0,85 (m, 1 H), 0,39 - 0,42 (m, 2 H), 0,35 - 0,38 (m, 2 H).
Ejemplo 178 N-((trans)-1-ciano-4-metoxipirrolidin-3-il)-N-metil-4-(1-metil-1H-pirazol-4-il)benzamida

Etapa a. A una solución de 4-bromobenzoato de etilo (4,00 g, 17,47 mmoles) en 1,4-dioxano (10 ml) se le añadió 1-metil-4-(4,4,5,5-tetrametil-1,3,2- dioxaborolan-2-il)-1 H-pirazol (Número CAS 761446-44-0) (5,44 g, 26,20 mmoles) a rt. La mezcla de reacción resultante se desgasificó durante 30 min antes de la adición de Pd(PPh3)4 (0,201 g, 0,174 mmoles) y K2CO3 (4,82 g, 34,93 mmoles) a rt. La mezcla de reacción se calentó a 90°C durante 15 h. La mezcla de reacción resultante se enfrió a rt y se vertió en NaHCO3 acuoso saturado (10 ml). La mezcla resultante se extrajo con EtOAc (2 x 100 ml). La capa orgánica combinada se secó sobre Na2SO4, se filtró y se concentró a presión reducida produciendo 4-(1 -metil-1 H-pirazol-4-il)benzoato de etilo (4,00 g, 17,39 mmoles). LCMS: Método C, rT 2,124 min, MS: ES+ 231,10. Esta sustancia se utilizó directamente para la siguiente etapa sin purificación adicional.
Etapa b. A una solución de 4-(1-metil-1H-pirazol-4-il)benzoato de etilo (4,00 g, 17,39 mmoles) en THF (25 ml) se le añadió una solución de NaOH (1,40 g, 35,0 mmoles) en agua (10 ml) a rt. La mezcla de reacción se calentó a 80°C durante 15 h. La mezcla de reacción resultante se enfrió a rt y se vertió en agua (20 ml). La mezcla resultante se extrajo con EtOAc (100 ml). La capa acuosa resultante se aciduló con una solución diluida de HCl. Los sólidos obtenidos se filtraron y se lavaron con agua (20 ml). La sustancia sólida obtenida se secó a alto vacío produciendo ácido 4-(1-metil-1H-pirazol-4-il)benzoico (2,500 g, 12,376 mmoles). LCMS: Método C, RT 1,586, MS: ES+ 203,01. Esta sustancia se utilizó directamente para la siguiente etapa sin purificación adicional.
Etapa c. A una solución de trans-3-hidroxi-4-(metilamino)pirrolidin-1 -carboxilato de terc-butilo (Número CAS 203503-49-5) (0,800 g, 3,703 mmoles) en THF (10 ml) se le añadieron ácido 4-(1-metil-1H-pirazol-4-il)benzoico (0,90 g, 4,44 mmoles), HATU (2,80 g, 7,37 mmoles) y DIPEA (2 ml, 11,11 mmoles) a rt. La mezcla de reacción se agitó a rt durante 3 h. La mezcla de reacción resultante se vertió en agua (20 ml) y se extrajo con EtOAc (4 x 25 ml). La capa orgánica combinada se secó sobre Na2SO4, se filtró y se concentró a presión reducida. El residuo resultante se purificó mediante cromatografía ultrarrápida (MeOH al 10% en DCM) produciendo trans-3-hidroxi-4-(N-metil-4-(1 -metil-1 H-pirazol-4-il)benzamido)pirrolidin-1 -carboxilato de terc-butilo (0,170 g, 0,425 mmoles). LCMS: Método C, RT 1,90 min, MS: ES+ 401,65.
Etapa d. A una solución de trans-3-hidroxi-4-(N-metil-4-(1-metil-1H-pirazol-4-il)benzamido)pirrolidin-1-carboxilato de terc-butilo (0,250 g, 0,625 mmoles) en DMF (3 ml) se le añadió NaH (dispersión al 60% en aceite de parafina, 0,061 g, 1,54 mmoles) a 0°C. Se añadió yoduro de metilo (0,264 g, 1,86 mmoles) a la mezcla de reacción a 0°C. La mezcla de reacción se agitó a rt durante 3 h. La mezcla de reacción resultante se vertió en agua helada (10 ml) y se extrajo con EtOAc (2 x 50 ml). La capa orgánica combinada se secó sobre Na2SO4, se filtró y se concentró a presión reducida. El residuo resultante se purificó mediante cromatografía ultrarrápida (MeOH al 10% en DCM) produciendo trans-3-metoxi-4-(N-metil-4-(1-metil-1H-pirazol-4-il)benzamido)pirrolidin-1-carboxilato de terc-butilo (0,250 g, 0,603 mmoles). LCMS: Método C, RT 2,08 min, MS: ES+ 415,75.
Etapas e, f. El compuesto del título se sintetizó a partir del intermedio anterior utilizando un procedimiento similar al descrito para las etapas b, c del Ejemplo 1. LCMS: Método A, RT 3,17 min, MS: ES+ 340,06; RMN H1 (400 MHz, DMSO-d6) 5 ppm 8,22 (s, 1 H), 7,93 (s, 1 H), 7,63 (d, J = 8,0 Hz, 2 H), 7,39 (d, J = 7,6 Hz, 2 H), 4,09 - 4,13 (m, 1 H), 3,87 (s, 3 H), 3,67 - 3,76 (m, 3 H), 3,54 - 3,58 (m, 1 H), 3,42 - 3,45 (m, 1 H), 3,28 (s, 3 H), 2,91 (s, 3 H).
Ejemplo 179 (R)-N-(1-cianopirrolidin-3-il)-5-( 1,3-dimetil-1H-pirazol-4-il)picolinamida
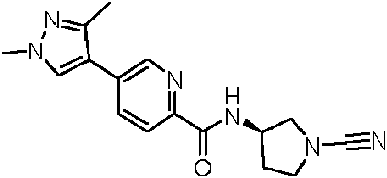
Sintetizada mediante un procedimiento similar al descrito para el Ejemplo 173. LCMS: Método A, RT 3,01 min, MS: ES+ 311,06; RMN H1 (400 MHz, DMSO-d6) 5 ppm 8,96 (d, J = 6,8 Hz, 1 H), 8,72 (s, 1 H), 8,15 (s, 1 H), 8,02 (s, 2 H), 4,51 - 4,54 (m, 1 H), 3,82 (s, 3 H), 3,53 - 3,64 (m, 2 H), 3,38 - 3,48 (m, 2 H), 2,35 (s, 3 H), 2,03 - 2,13 (m, 2 H)
Ejemplo 180 (R)-N-(1-cianopirrolidin-3-il)-5-(2-metil-6-(trifluorometil)pirimidin-4-il)picolinamida

Etapa a. A una solución de (R)-3-(5-bromopicolinamido)pirrolidin-1-carboxilato de terc-butilo (0,5 g, 1,35 mmoles) (preparada según el método descrito para Ejemplo 173 etapa a) en THF (15 ml) se le añadió bis(pinacolato)diboro (0,51 g, 2,02 mmoles) a rt. Se añadieron CH3COOK (0,26 g, 2,70 mmoles) y X-Phos (0,064 g, 0,13 mmoles) a la mezcla de reacción a rt. La mezcla de reacción se desgasificó con N2 durante 15 min antes de añadir Pd2(dba)3 (0,062 g, 0,067 mmoles). La mezcla de reacción y se calentó a 90°C durante 6 h. La mezcla resultante se vertió en agua (200 ml) y se extrajo con EtOAc (3 x 50 ml). La fase orgánica combinada se lavó con salmuera (200 ml) y se secó sobre Na2SO4, se filtró y se concentró a presión reducida. El residuo resultante se purificó mediante cromatografía en columna (MeOH al 10% en DCM) produciendo (R)-3-(5-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)picolinamido)pirrolidin-1-carboxilato de terc-butilo (0,2 g, 0,19 mmoles). LCMS: Método A, RT 2,10 min, MS: ES+ 336,07.
Etapas b-d. El compuesto del título se sintetizó a partir del intermedio anterior utilizando un procedimiento similar al descrito para las etapas b-d del Ejemplo 3 para proporcionar el compuesto del título. LCMS: Método B, RT 4,03 min, MS: ES+ 377,12; RMN H1 (400 MHz, DMSO-de) ó ppm 9,49 (s, 1 H), 9,18 (d, J = 7,2 Hz, 1 H), 8,86 (dd, J = 2,0 Hz, 8,4 Hz, 1 H), 8,59 (s, 1 H), 8,22 (d, J = 8,4 Hz, 1 H), 4,55 - 4,59 (m, 1 H), 3,55 - 3,66 (m, 2 H), 3,41 - 3,49 (m, 2 H), 2,84 (s, 3 H), 2,04 - 2,18 (m, 2 H)
Ejemplo 181 (R)-N-(1-cianopirrolidin-3-il)-4-(2,6-dimetilpirimidin-4-il)-2-fluorobenzamida

Etapa a. A una solución de ácido 4-bromo-2-fluorobenzoico (35,0 g, 159,81 mmoles) en THF (800 ml) se le añadió DIPEA (82,4 ml, 479 mmoles) a 0°C. Se añadió HATU (91,1 g, 240 mmoles) a la mezcla de reacción a 0°C. La mezcla de reacción se agitó a 0°C durante 45 min. Se añadió gota a gota (R)-3-aminopirrolidin-1-carboxilato de terc-butilo (29,7 g, 160 mmoles) a la mezcla de reacción a 0°C. La mezcla de reacción resultante se agitó durante 15 min a 0°C y a continuación a rt durante 2 h. La mezcla resultante se vertió en agua (1500 ml) y se extrajo con EtOAc (3 x 500 ml). La fase orgánica combinada se secó sobre Na2SO4, se filtró y se concentró a presión reducida. El residuo resultante se purificó mediante cromatografía en columna (EtOAc al 35% en hexano) produciendo (R)-3-(4-bromo-2-fluorobenzamido)pirrolidin-1 -carboxilato de terc-butilo (50,50 g, 130 mmoles). LCMS: Método C, RT 2,37 min, MS: ES+ 387,23; RMN H1 (400 MHz, DMSO-de) ó ppm 8,65 (d, J = 6,80 Hz, 1 H), 7,65 - 7,67 (m, 1 H), 7,48 - 7,54 (m, 2 H),
4,36 - 4,39 (m, 1 H), 3,47 - 3,56 (m, 1 H), 3,30 - 3,40 (m, 2 H), 3,14 - 3,18 (m, 1 H), 2,05 - 2,08 (m, 1 H), 1,83 - 1,86 (m, 1 H), 1,45 (s, 9 H).
Etapa b. A una solución de (R)-3-(4-bromo-2-fluorobenzamido)pirrolidin-1-carboxilato de terc-butilo (2,75 g, 8,33 mmoles) en DMF (10 ml) se le añadió bis(pinacolato)diboro (2,53 g, 9,99 mmoles) a rt seguido de CH3COOK (2,55 g, 26,0 mmoles). La mezcla de reacción resultante se desgasificó durante 10 min. Se añadió Pd(dppf)Cl2-CH2Cl2 (0,34 g, 0,41 mmoles) y la mezcla de reacción se calentó a 100°C durante 6,5 h. La mezcla de reacción se enfrió a rt. La mezcla de reacción resultante se vertió en agua helada (125 ml) y se extrajo con DCM (4 x 25 ml). La fase orgánica combinada se secó sobre Na2SO4, se filtró y se concentró a presión reducida. El residuo resultante se diluyó con agua (1200 ml) y se extrajo con DCM (4 x 100 ml). La fase orgánica combinada se secó sobre Na2SO4, se filtró y se concentró a presión reducida produciendo (R)-3-(2-fluoro-4-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)benzamido)pirrolidin-1-carboxilato de terc-butilo (2,20 g, 5,07 mmoles). Esta sustancia se utilizó directamente para la siguiente etapa sin purificación adicional. LCMS: Método C, RT 1,93 min, MS: ES+ 353,30
Etapa c. A una solución de (R)-3-(2-fluoro-4-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)benzamido)pirrolidin-1 -carboxilato de terc-butilo (0,250 g, 0,576 mmoles) en 1,4-dioxano:agua (8:2, 10 ml) se le añadió K2CO3 (0,238 g, 1,724 mmoles) seguido de 4-cloro-2,6-dimetilpirimidina (Número CAS 4472-45-1) (0,082 g, 0,576 mmoles) a rt. La mezcla de reacción se desgasificó durante 30 min. Se añadió PdCb(dppf) (0,042 g, 0,057 mmoles) a la mezcla de reacción a rt. La mezcla de reacción se calentó a 100°C durante 3 h. La mezcla de reacción resultante se enfrió a rt y se concentró a presión
reducida. La mezcla de reacción resultante se vertió en agua (25 ml) y se extrajo con EtOAc (2 x 25 ml). La fase orgánica combinada se secó sobre Na2SO4, se filtró y se concentró a presión reducida. El residuo obtenido se purificó mediante cromatografía ultrarrápida (EtOAc al 70% en hexano) produciendo (R)-3-(4-(2,6-dimetilpirimidin-4-il)-2-fluorobenzamido)pirrolidin-1-carboxilato de terc-butilo (0,110 g, 0,265 mmoles). LCm S: Método C, 2,18, MS: ES+ 415,38
Etapas d, e. El compuesto del título se sintetizó a partir del intermedio anterior utilizando un procedimiento similar al descrito para las etapas c, d del Ejemplo 3. LCMS: Método B, RT 3,64 min, MS: ES+ 340,10; RMN H1 (400 MHz, DMSO-d6) 5 ppm 8,79 (d, J = 6,80 Hz, 1 H), 8,06 - 8,12 (m, 2 H), 7,91 (s, 1 H), 7,71 - 7,75 (m, 1 H), 4,46 - 4,50 (m, 1 H), 3,63 - 3,67 (m, 1 H), 3,44 - 3,56 (m, 2 H), 3,27 - 3,32 (m, 1 H), 2,66 (s, 3 H), 2,51 (s, 3 H), 2,12 - 2,17 (m, 1 H), 1,91 - 1,96 (m, 1 H).
Los compuestos de la Tabla 9 se sintetizaron utilizando un procedimiento similar al descrito para el Ejemplo 181.

canoprro n- - - - , m, , , - , m, , , - , m,
183 1 m, 1 H), B 3,94 ES+ N^N fluoro-4-(2- H), 4,47 - 4,50 (m, 1 H), 3,66 - 3,67 (
tri luorometil irimidin- 342 - 351 m 3 H 214 - 217 m 1 H 191 - 380,11

Ejemplo 186 (R)-N-(1-cianopirrolidin-3-il)-3-fluoro-5-(pirazolo[1,5-a]pirim idin-5-il)picolinam ida

Sintetizada mediante un procedimiento similar al descrito para el Ejemplo 181, utilizando ácido 5-bromo-3-fluoropicolínico (Número CAS 669066-91-5) en la etapa a y 5-cloropirazolo[1,5-a]pirimidina (Número CAS 29274-24 6) en la etapa c. LCMS: Método A, RT 3,19 min, MS: ES+ 352,10; RMN H1 (400 MHz, DMSO-d6) 5 ppm 9,36 (d, J = 7,6 Hz, 1 H), 9,30 (s, 1 H), 9,13 (d, J = 6,8 Hz, 1 H), 8,63 (dd, J = 1,6 Hz, 11,6 Hz, 1 H), 8,34 (d, J = 2,4 Hz, 1 H), 7,85 (d, J = 7,6 Hz, 1 H), 6,89 - 6,90 (m, 1 H), 4,51 - 4,53 (m, 1 H), 3,63 - 3,67 (m, 1 H), 3,53 - 3,57 (m, 1 H), 3,38 - 3,46 (m, 1 H), 3,36 - 3,38 (m, 1 H), 2,13 - 2,18 (m, 1 H), 1,99 - 2,02 (m, 1 H)
Ejemplo 187 (R)-N-(1-cianopirrolidin-3-il)-3-fluoro-5-(imidazo[1,2-a]piridin-6-il)picolinamida
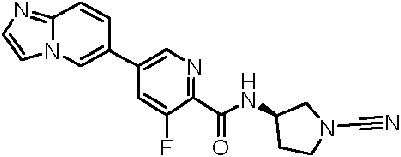
Sintetizada mediante un procedimiento similar al descrito para el Ejemplo 186. LCMS: Método A, RT 3,02 min, MS: ES+ 351,04; RMN H1 (400 MHz, DMSO-d6) 5 ppm 9,19 (s, 1 H), 9,02 (d, J = 6,4 Hz, 1 H), 8,88 (s, 1 H), 8,29 (d, J = 12,8 Hz, 1 H), 8,00 (s, 1 H), 7,73 (s, 2 H), 7,67 (s, 1 H), 4,05 - 4,54 (m, 1 H), 3,63 - 3,67 (m, 1 H), 3,46 - 3,58 (m, 2 H), 3,36 - 3,38 (m, 1 H), 2,12 - 2,33 (m, 1 H), 1,97 - 2,04 (m, 1 H)
Ejemplo 188 (R)-N-(1-cianopirrolidin-3-il)-3-metoxi-3-fenilazetidin-1-carboxamida

Etapa a. A una solución de 3-oxoazetidin-1 -carboxilato de terc-butilo (Número CAS 398489-26-4) (10 g, 58,41 mmoles) en THF (100 ml) se le añadió bromuro de fenil magnesio (1 M en THF) (64,2 ml, 64,25 mmoles) a 0°C. La mezcla de reacción se agitó a 0°C durante 2 h y a continuación a rt durante 16 h. La mezcla de reacción se vertió en una solución saturada de NH4Cl (150 ml) y se extrajo con EtOAc (3 x 30 ml). La capa orgánica combinada se lavó con agua (2 x 50 ml), salmuera (2 x 50 ml), se secó sobre Na2SO4, se filtró y se concentró a presión reducida. El residuo resultante se purificó mediante cromatografía en columna (EtOAc al 20% en hexano) produciendo 3-hidroxi-3-fenilazetidin-1-carboxilato de terc-butilo (11,92 g, 47,8 mmoles). LCMS: Método C, RT 2,22 min, MS: ES+ 194,1 [M-56], RMN H1 (400 MHz, DMSO-de) 5 ppm 7,49 (d, J = 7,2 Hz, 2 H), 7,38 (t, J = 8 Hz, 2 H), 7,27 - 7,31 (m, 1 H), 6,33 (s, 1 H), 4,03 (s, 4 H), 1,41 (s, 9 H)
Etapa b. A una solución de 3-hidroxi-3-fenilazetidin-1-carboxilato de terc-butilo (1 g, 4,01 mmoles) en MeCN (40 ml) se le añadió NaH (dispersión al 60% en aceite de parafina, 0,3 g, 7,63 mmoles) a rt. La mezcla de reacción se agitó a rt durante 30 min. Se añadió gota a gota una solución de yoduro de metilo (0,75 g, 5,28 mmoles) en MeCN (5 ml) a la mezcla de reacción a rt y se agitó durante 4 h. La mezcla resultante se vertió en agua (20 ml) y se extrajo con EtOAc (3 x 20 ml). La fase orgánica combinada se lavó con salmuera (2 x 20 ml), se secó sobre Na2SO4, se filtró y se concentró a presión reducida produciendo 3-metoxi-3-fenilazetidin-1-carboxilato de terc-butilo (1,13 g, 4,29 mmoles). LCMS: Método C, RT 2,39 min, MS: ES+ 208,01 [M-56], RMN H1 (400 MHz, DMSO-de) 5 ppm 7,34 - 7,50 (m, 5 H), 4,03 - 4,11 (m, 4 H), 2,96 (s, 1 H), 1,39 (s, 9 H)
Etapa c. A una solución de 3-metoxi-3-fenilazetidin-1-carboxilato de terc-butilo (1,12 g, 4,24 mmoles) en 1,4-dioxano (10 ml) se le añadió HCl 4 M en 1,4-dioxano (10 ml) gota a gota a rt. La mezcla de reacción se agitó a rt durante 3 h. La mezcla de reacción resultante se evaporó a presión reducida y el residuo obtenido se trituró con n-pentano (20 ml), éter dietílico (20 ml) y se secó a vacío para producir la sal de HCl de 3-metoxi-3-fenilazetidina (0,7 g, 3,5 mmoles). LCMS: Método C, RT 1,50 min, MS: ES+ 164,04
Etapas d-f. El compuesto del título se sintetizó a partir del intermedio anterior utilizando un procedimiento similar al descrito para el Ejemplo 89. LCMS: Método B, rT 3,38 min, MS: ES+ 301,20; RMN H1 (400 MHz, DMSO-d6) 5 ppm 7,34 - 7,46 (m, 5 H), 6,65 (d, J = 6,4 Hz, 1 H), 4,14 (t, J = 5,6 Hz, 1 H), 4,02 - 4,07 (m, 4 H), 3,44 - 3,52 (m, 2 H),
3,45 - 3,40 (m, 1 H), 3,13 - 3,16 (m, 1 H), 2,98 (s, 3 H), 1,95 - 2,07 (m, 1 H), 1,75 - 1,83 (m, 1 H).
Ejemplo 189 (R)-N-(1-cianopirrolidin-3-il)-N-metil-3-fenilazetidin-1-carboxamida

Etapa a. Se preparó una solución de ácido fenilborónico (0,65 g, 5,33 mmoles) en IPA (4,5 ml) en un tubo de vidrio apto para microondas. Se añadieron Nil2 (0,05 g, 0,15 mmoles) e hidrocloruro de rans-2-aminociclohexanol (0,024 g, 0,15 mmoles) a la mezcla de reacción a rt. Se añadió gota a gota bis(trimetilsilil)amida de sodio (1 M en THF) (5,3 ml, 5,28 mmoles) a la mezcla de reacción a rt. Se añadió N-BOC-3-yodoazetidina (Número CAS 254454-54-1) (0,75 g, 2,65 mmoles), se selló el tubo de vidrio y se sometió la mezcla de reacción a calentamiento por microondas a 80°C durante 50 min. La mezcla de reacción se enfrió a rt y se evaporó a presión reducida para producir un residuo de color
negro. El residuo resultante se purificó mediante cromatografía en columna (EtOAc al 6% en hexano) produciendo 3-fenilazetidin-1-carboxilato de terc-butilo (1,3 g, 5,57 mmoles). LCMS: Método C, RT 2,47 min, MS: eS+ 234,4 Etapa b. A una solución de 3-fenilazetidin-1 -carboxilato de terc-butilo (1,2 g, 5,15 mmoles) en DCM se le añadió TFA (3,6 ml) a 0°C. La mezcla de reacción se agitó a rt durante 3 h. La mezcla de reacción resultante se evaporó a presión reducida. El residuo se destiló azeotrópicamente con éter dietílico (10 ml) y se secó a vacío para producir la sal de TFA de 3-fenilazetidina (0,3 g, 1,21 mmoles). Esta sustancia se utilizó directamente para la siguiente etapa sin purificación adicional. LCMS: Método C, RT 1,08 min, MS: ES+ 134,19
Etapas c-e. El compuesto del título se sintetizó a partir del intermedio anterior utilizando un procedimiento similar al descrito para el Ejemplo 89 para proporcionar el compuesto del título. LCMS: Método A, RT 3,88 min, MS: ES+ 285,18; RMN H1 (400 MHz, DMSO-d6) 5 ppm 7,34 - 7,38 (m, 4 H), 7,24 - 7,27 (m, 1 H), 4,59 - 4,63 (m, 1 H), 4,26 - 4,32 (m, 2 H), 3,85 - 3,91 (m, 2 H), 3,75 - 3,79 (m, 1 H), 3,44 - 3,53 (m, 2 H), 3,25 - 3,40 (m, 2 H), 3,71 (s, 3 H), 1,91 - 2,01 (m, 2 H). Ejemplo 190 (R)-N-(1-cianopirrolidin-3-il)-3-(4-metoxifenil)azetidin-1-carboxamida
procedimiento similar al descrito para las etapas a, b del Ejemplo 189, utilizando ácido 4-ido de las etapas a-c del Ejemplo 5. LCMS: Método A, RT 3,44 min, MS: ES+ 301,08; RMN ppm 7,25 (d, J = 8,4 Hz, 2 H), 6,91 (d, J = 8,8 Hz, 2 H), 6,56 (d, J = 6,4 Hz, 1 H), 4,12 - 4,20 3 - 3,73 (m, 3 H), 3,45 - 3,53 (m, 2 H), 3,37 - 3,41 (m, 1 H), 3,13 - 3,17 (m, 1 H), 1,96 - 2,04 H).

Sintetizada utilizando un procedimiento similar al descrito para las etapas a, b del Ejemplo 189, utilizando ácido 4-clorofenilborónico, seguido de las etapas a-c del Ejemplo 5. LCMS: Método A, RT 3,86 min, MS: ES+ 304,94; RMN H1 (400 MHz, DMSO-de) 5 ppm 7,36-7,43 (m, 4 H), 6,59 (d, J = 6,4 Hz, 1 H), 4,12 - 4,22 (m, 3 H), 3,73 - 3,79 (m, 3 H), 3,45 - 3,77 (m, 2 H), 3,34 - 3,41 (m, 1 H), 3,13 - 3,17 (m, 1 H), 1,98 - 2,03 (m, 1 H), 1,78 - 1,82 (m, 1 H).
Ejemplo 192 (R)-3-(3-clorofenil)-N-(1-cianopirrolidin-3-il)azetidin-1-carboxamida
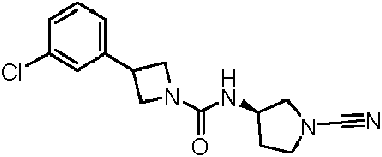
Sintetizada utilizando un procedimiento similar al descrito para las etapas a, b del Ejemplo 189, utilizando ácido 3-clorofenilborónico, seguido de las etapas a-c del Ejemplo 5. LCMS: Método B, RT 3,75 min, MS: ES+ 305,22; RMN H1 (400 MHz, DMSO-de) 5 ppm 7,38 - 7,41 (m, 2 H), 7,32 (d, J = 7,2 Hz, 2 H), 6,58 (d, J = 6,8 Hz, 1 H), 4,13 - 4,20 (m, 3 H), 3,77 - 3,78 (m, 3 H), 3,45 - 3,53 (m, 2 H), 3,35 - 3,39 (m, 1 H), 3,13 - 3,17 (m, 1 H), 1,96 - 2,09 (m, 1 H), 1,77 -1,83 (m, 1 H).
Ejemplo 193 (3aR,6aR)-1-(3-fenilazetidin-1-carbonil)hexahidropirrolo[3,4-b]pirrol-5(1H)-carbonitrilo

Sintetizado mediante un procedimiento similar al descrito para el Ejemplo 189. LCMS: Método A, RT 3,82 min, MS: ES+ 297,09; RMN H1 (400 MHz, DMSO-de) 5 ppm 7,35 - 7,39 (m, 4 H), 7,24 - 7,28 (m, 1 H), 4,35 - 4,40 (m, 1 H) 4,20 - 4,27 (m, 2 H), 3,92 - 3,96 (m, 1 H), 3,76 - 3,86 (m, 3 H), 3,50 - 3,55 (m, 2 H), 3,37 - 3,42 (m, 2 H), 3,16 - 3,23 (m, 1 H), 2,87 - 2,89 (m, 1 H), 1,74 - 1,78 (m, 1 H), 1,74 - 1,78 (m, 1 H).
Ejemplo 194 (R)-1-(1-cianopirrolidin-3-il)-1-metil-3-(4-(1-metil-1H-pirazol-4-il)fenil)urea
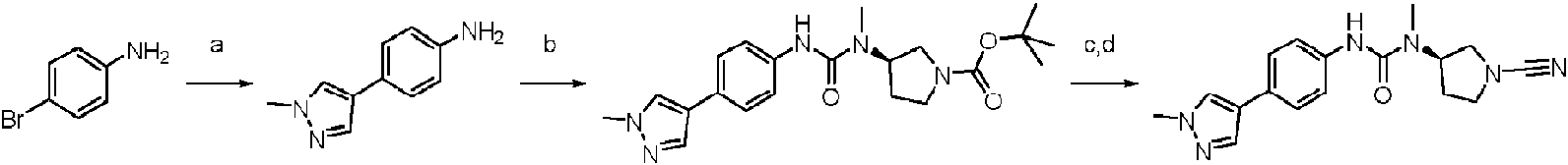
Etapa a. A una mezcla de 4-bromoanilina (3 g, 17,4 mmoles) en DMF:agua (8:2) (60 ml) se le añadieron 1 -metil-4-(4,4,5,5-tetrametil-1,3, 2-dioxaborolan-2-il)-1H-pirazol (Número CAS 761446-44-0) (3,62 g, 17,4 mmoles) y Na2CO3 (3,69 g, 34,8 mmoles) a rt. La mezcla de reacción se desgasificó con N2 durante 10 minutos antes de añadir PdCb(dppf) (1,27 g, 1,74 mmoles). La mezcla de reacción resultante se calentó a 110°C durante 2,5 h. La mezcla de reacción resultante se vertió en agua fría (300 ml) y se extrajo con EtOAc (3 x 300 ml). La capa orgánica combinada se secó sobre Na2SO4, se filtró y se concentró a presión reducida para producir 4-(1 -metil-1 H-pirazol-4-il)anilina (1,8 g, 10,39 mmoles). LCMS: Método A, RT 2,84 min, MS: ES+ 173,97, RMN 1H (400 MHz, DMSO-d6) 5 ppm 7,85 (s, 1 H), 7,63 (s, 1 H), 7,20 (d, J = 8,4 Hz, 2 H), 6,54 (d, J = 8,8 Hz, 2 H), 5,01 (s, 2 H), 3,81 (s, 3 H).
Etapa b. A una solución de 4-(1 -metil-1 H-pirazol-4-il)anilina (0,5 g, 2,89 mmoles) en DCM (10 ml) se le añadieron piridina (0,69 ml, 8,67 mmoles) y cloroformiato de 4-nitrofenilo (0,057 g, 4,33 mmoles) a rt. La mezcla de reacción se agitó a rt durante 2 h. Se añadió éster terc-butílico de ácido (R)-3-(metilamino)pirrolidin-1-carboxílico (0,693 g, 3,46 mmoles) a la mezcla de reacción a rt y se agitó durante 16 h más. La mezcla de reacción resultante se vertió en agua fría (100 ml) y se extrajo con DCM (3 x 100 ml). La capa orgánica combinada se lavó con ácido cítrico al 1 % (1 x 100 ml), salmuera (100 ml), se secó sobre Na2SO4, se filtró y se concentró a presión reducida para producir (R)-3-(1 -metil-3-(4-(1 -metil-1 H-pirazol-4-il)fenil)ureido)pirrolidin-1 -carboxilato de terc-butilo (0,6 g, 1,50 mmoles). LCMS: Método C, RT 2,07 min, MS: ES+ 400,40.
Etapas c, d. El compuesto del título se sintetizó a partir del intermedio anterior utilizando un procedimiento similar al descrito para las etapas b, c del Ejemplo 5 para proporcionar el compuesto del título. LCMS: Método A, RT 3,14 min, MS: ES+ 325,03; RMN H1 (400 MHz, DMSO-de) 5 ppm 8,35 (s, 1 H), 8,02 (s, 1 H), 7,77 (s, 1 H), 7,41 - 7,46 (m, 4 H), 4,88 - 4,92 (m, 1 H), 3,84 (s, 3 H), 3,50 - 3,56 (m, 2 H), 3,35 - 3,39 (m, 1 H), 3,27 - 3,31 (m, 1 H), 2,88 (s, 3 H), 1,94 -2,03 (m, 2 H).
Ejemplo 195 (R)-1-(1-cianopirrolidin-3-il)-1-metil-3-(4-(trifluorometil)fenil)urea

Etapa a. A una solución de 4-cloro-2-fluoroanilina (Número CAS 57946-56-2) (0,500 g, 3,434 mmoles) en cloroformo (10 ml) se le añadió DIPEA (0,891 g, 6,906 mmoles) a 0°C. Se añadió en porciones cloroformiato de 4-nitrofenilo (0,831 g, 4,122 mmoles) a la mezcla de reacción a 0°C. La mezcla de reacción se calentó a 60°C durante 2,5 h. La mezcla de reacción resultante se enfrió a rt y se vertió en agua (70 ml). La mezcla resultante se extrajo con DCM (3 x 40 ml). La fase orgánica combinada se secó sobre Na2SO4, se filtró y se concentró a presión reducida. El residuo resultante se purificó mediante cromatografía ultrarrápida (EtOAc al 11% en hexano) produciendo (4-cloro-2-fluorofenil)carbamato de 4-nitrofenil (0,230 g, 0,740 mmoles).
Etapa b. A una solución de (4-cloro-2-fluorofenil)carbamato de 4-nitrofenilo (0,220 g, 0,709 mmoles) en piridina (10 ml) se le añadió (3aR,6aR)-hexahidropirrolo[3,4-b]pirrol-5(1H)-carboxilato de terc-butilo (Número CAS 370882-39-6) (0,181 g, 0,852 mmoles) a rt. La mezcla de reacción se calentó a 130°C durante 8 h. La mezcla de reacción resultante se enfrió a rt y se vertió en agua (100 ml). La mezcla resultante se extrajo con EtOAc (3 x 50 ml). La fase orgánica combinada se lavó con ácido cítrico saturado (2 x 50 ml), se secó sobre Na2SO4, se filtró y se concentró a presión reducida. El residuo resultante se purificó mediante cromatografía ultrarrápida (EtOAc al 43% en hexano) produciendo (3aR,6aR)-1-((4-cloro-2-fluorofenil)carbamoil)hexahidropirrolo[3,4-b]pirrol-5(1H)-carboxilato de terc-butilo (0,097 g, 0,252 mmoles). LCMS: Método C, 2,263, MS: ES-382,59.
Etapas c, d. El compuesto del título se sintetizó a partir del intermedio anterior utilizando un procedimiento similar al
descrito para las etapas b, c del Ejemplo 5 para proporcionar el compuesto del título. LCMS: Método B, RT 3,59 min, MS: ES+ 309,42; RMN H1 (400 MHz, DMSO-d6) 5 ppm 8,15 (s, 1 H), 7,52 (t, J = 8,8 Hz, 1 H), 7,42 (dd, J = 2,4 Hz, 10,4 Hz, 1 H), 7,20 - 7,23 (m, 1 H), 4,31 - 4,34 (m, 1 H), 3,50 - 3,57 (m, 4 H), 3,39 - 3,42 (m, 1 H), 3,23 - 3,27 (m, 1 H), 2,93 - 2,98 (m, 1 H), 2,01 - 2,08 (m, 1 H), 1,79 - 1,84 (m, 1 H).
Los compuestos de la Tabla 10 se sintetizaron utilizando un procedimiento similar al método general E como se ilustra mediante el Ejemplo 5, Ejemplo 89 o Ejemplo 196.


Ejemplo 204 (R)-]-(1-cianopirrolidin-3-il)-1-etil-3-(4-(trifluorometil)fenil)urea

Etapa a. A una solución de (S)-3-hidroxipirrolidin-1 -carboxilato de terc-butilo (1 g, 5,34 mmoles) en DCM (2,5 ml) se le añadió TEA (1,8 ml, 13,3 mmoles) a 0°C seguido de cloruro de mesilo (0,62 ml, 8,01 mmoles). La mezcla de reacción se agitó a rt durante 1,5 h. La mezcla resultante se concentró a presión reducida para producir (S)-3-((metilsulfonil)oxi)pirrolidin-1 -carboxilato de terc-butilo (1,5 g, cuantitativo). Esta sustancia se utilizó directamente para la siguiente etapa sin purificación adicional. LCMS: Método C, RT 2,05 min, MS: ES+ 266,2
Etapa b. Una mezcla de (S)-3-hidroxipirrolidin-1 -carboxilato de terc-butilo (0,5 g, 1,88 mmoles) en etilamina acuosa (70% en agua) (10 ml) se calentó a 90°C durante 16 h. La mezcla resultante se concentró a presión reducida. El residuo obtenido se destiló azeotrópicamente con éter dietílico (3 x 5 ml) y se secó a alto vacío para producir (R)-3-(etilamino)pirrolidin-1 -carboxilato de terc-butilo (0,3 g, 1,39 mmoles). LCMS: Método C, RT 1,54 min, MS: ES+ 215,29
Etapas c-e. El compuesto del título se sintetizó a partir del intermedio anterior utilizando un procedimiento similar al descrito para las etapas b-d del Ejemplo 194 para proporcionar el compuesto del título. LCMS: Método A, RT 4,42 min, MS: ES+ 327,15; RMN H1 (400 MHz, DMSO-de) ó ppm 8,67 (s, 1 H), 7,70 (d, J = 8,8 Hz, 2 H), 7,59 (d, J = 8,8 Hz, 2 H), 4,61 - 4,65 (m, 1 H), 3,52 - 3,57 (m, 2 H), 3,31 - 3,43 (m, 3 H), 3,26 - 3,31 (m, 1 H), 2,00 - 2,08 (m, 2 H), 1,08 (t, J1 = 6,8, 3 H).
Ejemplo 205 1-(1-cianopirrolidin-3-il)-1-(2-metoxietil)-3-(4-(trifluorometil)fenil)urea

Sintetizada mediante un procedimiento similar al descrito para el Ejemplo 89 utilizando 3-((2-metoxietil)amino)pirrolidin-1 -carboxilato de terc-butilo (Número CAS 887587-33-9) en la etapa a. LCMS: Método A, RT 4,66 min, MS: ES+ 357,03; RMN H1 (400 MHz, DMSO-de) 5 ppm 8,91 (s, 1 H), 7,54 (d, J = 8,4 Hz, 2 H), 7,43 (d, J = 8,8 Hz, 2 H), 4,83 - 4,87 (m, 1 H), 3,57 - 3,79 (m, 4 H), 3,56 (s, 3 H), 3,37 - 3,53 (m, 3 H), 3,28 - 3,32 (m, 1 H), 2,19 -2,25 (m, 1 H), 1,97 - 2,05 (m, 1 H).
Ejemplo 206 (R)-N-(1-cianopirrolidin-3-il)-N-etil-3-fluoro-4-(1-metil-1H-pirazol-4-il)benzamida

Sintetizada mediante un procedimiento similar al descrito para el Ejemplo 63 utilizando (R)-3-(etilamino)pirrolidin-1-carboxilato de terc-butilo (descrito en la síntesis de Ejemplo 204) en la etapa b. LCMS: Método A, RT 3,53 min, MS: ES+ 342,06; RMN H1 (400 MHz, DMSO-de) 5 ppm 8,19 (d, J = 2,0 Hz, 1 H), 7,94 (s, 1 H), 7,78 (t, J = 8,0 Hz, 1 H), 7,30 (dd, J = 11,6, 1,6 Hz, 1 H), 7,21 (dd, J = 8,0, 1,6 Hz, 1 H), 4,39 - 4,43 (m, 1 H), 3,90 (s, 3 H), 3,51 - 3,59 (m, 2 H),
3,41 - 3,47 (m, 2 H), 3,28 - 3,33 (m, 2 H), 2,01 - 2,15 (m, 2 H), 1,04 - 1,10 (m, 3 H).
Ejemplo 207 (R)-N-(1-cianopirrolidin-3-il)-N-etil-3-fenilazetidin-1-carboxamida
Sintetizada mediante un procedimiento similar al descrito para el Ejemplo 189 utilizando (R)-3-(etilamino)pirrolidin-1-carboxilato de terc-butilo (descrito en la síntesis de Ejemplo 204) en la etapa c. LCMS: Método A, RT 4,07 min, MS: ES+ 299,08; RMN H1 (400 MHz, CDCh) 5 ppm 7,33 - 7,41 (m, 4 H), 7,29 - 7,31 (m, 1 H), 4,38 - 4,46 (m, 3 H), 4,02 -4,06 (m, 2 H), 3,77 - 3,81 (m, 1 H), 3,58 - 3,64 (m, 2 H), 3,36 - 3,47 (m, 2 H), 3,17 - 3,20 (m, 2 H), 2,11 - 2,15 (m, 2 H), 1,18 - 1,21 (m, 3 H).
Ejemplo 208 (R)-3-(2-oxo-3-(4-feniltiazol-2-il)imidazolidin-1-il)pirrolidin-1-carbonitrilo

Etapa a. A una solución agitada de (R)-3-amino-1N-BOC-pirrolidina (1,0 g, 5,376 mmoles) y DIPEA (1,04 g, 8,06 mmoles) en THF (15 ml) se le añadió trifosgeno (0,526 g, 1,774 mmoles) a 0°C. La mezcla de reacción se agitó a 0°C durante 2 h. Se añadió 4-feniltiazol-2-amina (0,95 g, 5,376 mmoles) a la mezcla de reacción a rt. La mezcla de reacción se calentó a 60°C durante 16 h. La mezcla de reacción resultante se enfrió a rt, se vertió rápidamente en agua (50 ml) y se extrajo con EtOAc (2 x 20 ml). La fase orgánica combinada se separó, se secó sobre Na2SO4, se filtró y se concentró a presión reducida produciendo (R)-3-(3-(4-feniltiazol-2-il)ureido)pirrolidin-1 -carboxilato de terc-butilo (1,0 g, 2,58 mmoles). Esta sustancia se utilizó directamente para la siguiente etapa sin purificación adicional. LCMS: Método C, RT 2,51 min, MS: ES+ 389,4
Etapa b. A una solución agitada de (R)-3-(3-(4-feniltiazol-2-il)ureido)pirrolidin-1 -carboxilato de terc-butilo (0,5 g, 1,29 mmoles) en DMF (10 ml) se le añadió K2CO3 (0,71 g, 5,15 mmoles) a rt. La mezcla de reacción se agitó a rt durante 15 min antes de añadir 1,2-dibromoetano (0,29 g, 1,55 mmoles). La mezcla de reacción se calentó a 100°C durante 2 h. La mezcla de reacción resultante se enfrió a rt, se vertió rápidamente en agua (50 ml) y se extrajo con EtOAc (2 x 20 ml). La fase orgánica combinada se separó, se secó sobre Na2SO4 , se filtró y se concentró a presión reducida. El residuo resultante se purificó mediante cromatografía en columna (EtOAc al 35% en hexano) produciendo (R)-3-(2-oxo-3-(4-feniltiazol-2-il)imidazolidin-1 -il)pirrolidin-1 -carboxilato de terc-butilo (0,07 g, 0,169 mmoles). LCMS: Método C, RT 2,80 min, MS: ES+ 415,4
Etapas c, d. El compuesto del título se sintetizó a partir del intermedio anterior utilizando un procedimiento similar al descrito para las etapas b, c del Ejemplo 5 para proporcionar el compuesto del título. LCMS: Método B, RT 4,47 min, MS: ES+ 340,28; RMN H1 (400 MHz, DMSO-d6) 5 ppm 7,89 (d, J = 7,2 Hz, 2 H), 7,56 (s, 1 H), 7,41 (t, J = 7,6 Hz, 2 H), 7,30 (t, J = 7,2 Hz, 1 H), 4,90 - 4,52 (m, 1 H), 4,07 - 4,12 (m, 2 H), 3,61 - 3,65 (m, 2 H), 3,41 - 3,58 (m, 4 H), 2,05 - 2,13 (m, 2 H).
Ejemplo 209 (R)-3-(2-oxo-3-(4-feniltiazol-2-il)tetrahidropirimidin-1(2H)-il)pirrolidin-1-carbonitrilo

Etapa a. A una solución agitada de (R)-3-amino-1 N-BOC-pirrolidina (1,0 g, 5,37 mmoles) en THF (12 ml) se le añadió isocianato de 2-cloroetilo (Número CAS 1943-83-5) (0,57 g, 5,37 mmoles) a 0°C. La mezcla de reacción se agitó a rt durante 1,5 h. Se añadió NaH (dispersión al 60% en aceite de parafina, 0,645 g, 16,12 mmoles) a la mezcla de reacción a 0°C. La mezcla de reacción se calentó a 50°C durante 16 h. La mezcla de reacción resultante se enfrió a rt, se vertió rápidamente en agua (50 ml) y se extrajo con EtOAc (4 x 50 ml). La fase orgánica combinada se separó, se secó sobre Na2SO4 , se filtró y se concentró a presión reducida. El residuo resultante se purificó mediante cromatografía ultrarrápida utilizando (MeOH al 8-9% en DCM) produciendo (R)-3-(2-oxoimidazolidin-1-il)pirrolidin-1-carboxilato de terc-butilo (1,03 g, 4,062 mmoles). LCMS: Método C, RT 1,67 min, m S: ES+ 256,32
Etapa b. A una solución agitada de (R)-3-(2-oxoimidazolidin-1-il)pirrolidin-1-carboxilato de terc-butilo (0,2 g, 0,829 mmoles) y 4-(3-bromofenil)morfolina (Número CAS 197846-82-5) (0,21 g, 0,83 mmoles) en tolueno (7 ml) se le añadió Cs2CO3 (0,81 g, 2,49 mmoles) a rt. La mezcla de reacción se desgasificó durante 30 min antes de la adición de Pd(OAc)2 (0,019 g, 0,083 mmoles) y BINAP (0,103 g, 0,166 mmoles). La mezcla de reacción se calentó a 90°C durante
10 h. La mezcla de reacción resultante se enfrió y se combinó con otros dos lotes preparados a la misma escala mediante un método idéntico. El exceso de disolvente se destiló a vacío y el resto resultante se purificó mediante cromatografía ultrarrápida (EtOAc al 54% en hexano) produciendo (R)-3-(3-(3-morfolinofenil)-2-oxoimidazolidin-1-il)pirrolidin-1 -carboxilato de terc-butilo (0,267 g, 0,461 mmoles). LCMS: Método C, RT 2,22 min, MS: ES+ 417,70
Etapas c, d. El compuesto del título se sintetizó a partir del intermedio anterior utilizando un procedimiento similar al descrito para las etapas b, c del Ejemplo 5 para proporcionar el compuesto del título. LCMS: Método B, RT 3,33 min, MS: ES+ 342,58; RMN H1 (400 MHz, DMSO-ds) 5 ppm 7,23 (s, 1 H), 7,15 (t, J = 8,0 Hz, 1 H), 6,94 (dd, J = 1,2 Hz, 8,0 Hz, 1 H), 6,61 (dd, J = 2,0 Hz, 8,4 Hz, 1 H), 4,42 - 4,49 (m, 1 H), 3,72 - 3,81 (m, 6 H), 3,34 - 3,56 (m, 6 H), 3,07 (t, J = 6,8 Hz, 4 H), 1,97 -2,12 (m, 2 H).
Ejemplo 211 (R)-N-(1-cianopirrolidin-3-il)-4-(pirimidin-2-il)-3,4-dihidro-2H-benzo[b][1,4]oxazin-7-carboxamida
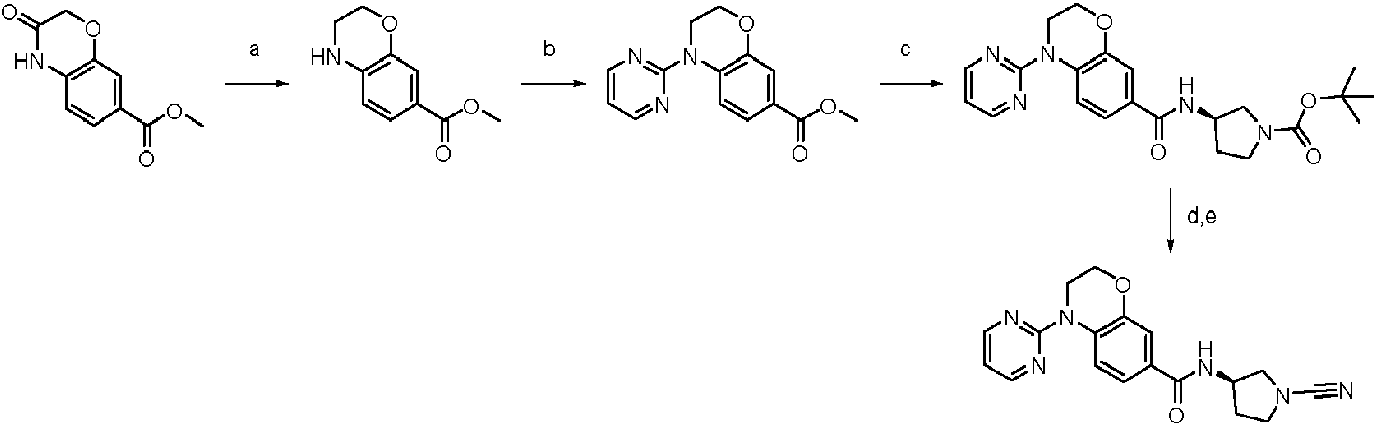
Etapa a. A una solución agitada de 3-oxo-3,4-dihidro-2H-benzo[b][1,4]oxazin-7-carboxilato de metilo (Número CAS 142166-00-5) (0,7 g, 3,38 mmoles) en THF (14 ml) se le añadió complejo de borano y dimetil sulfuro (0,513 g, 6,76 mmoles) a 0°C bajo nitrógeno. La mezcla de reacción se calentó a 60°C durante 3 h. La mezcla de reacción se enfrió a rt. Se añadió lentamente MeOH (2 ml) a la mezcla de reacción a 0°C y la mezcla de reacción resultante se calentó a 60°C durante 10 min. El exceso de disolvente se destiló a vacío. El material en bruto se purificó mediante cromatografía ultrarrápida. El residuo resultante se purificó mediante cromatografía ultrarrápida (EtOAc al 20% en hexano) produciendo 3,4-dihidro-2H-benzo[b][1,4]oxazin-7-carboxilato de metilo (0,55 g, 2,85 mmoles). LCMS: Método C, RT 1,96 min, MS: ES+ 194,1
Etapa b. A una solución agitada de 3,4-dihidro-2H-benzo[b][1,4]oxazin-7-carboxilato de metilo (0,44 g, 2,28 mmoles) y 2-cloropirimidina (0,782 g, 6,83 mmoles) en DMF (13,2 ml) se le añadió Cs2CO3 (2,23 g, 6,83 mmoles) a rt en atmósfera de nitrógeno. La mezcla de reacción se desgasificó durante 15 min a rt antes de la adición de Pd2(dba)3 (0,021 g, 0,023 mmoles) y xantphos (0,04 g, 0,683 mmoles). La mezcla de reacción se calentó a 140°C durante 15 h. La mezcla de reacción resultante se enfrió a rt, se vertió en agua (100 ml) y se extrajo con EtOAc (3 x 50 ml). La fase orgánica combinada se separó, se secó sobre Na2SO4, se filtró y se concentró a presión reducida. El residuo resultante se purificó mediante cromatografía ultrarrápida (EtOAc al 10% en hexano) produciendo 4-(pirimidin-2-il)-3,4-dihidro-2H-benzo[b][1,4]oxazin-7-carboxilato de metilo (0,29 g, 1,07 mmoles). LCm S: Método C, RT 2,25 min, MS: ES+ 272,18
Etapa c. A una solución agitada de 4-(pirimidin-2-il)-3,4-dihidro-2H-benzo[b][1,4]oxazin-7-carboxilato de metilo (0,29 g, 1,07 mmoles), (R)-3-amino-1N-BOC-pirrolidina (0,22 g, 1,18 mmoles) y DIPEA (0,276 g, 2,14 mmoles) en THF (5,8 ml) se le añadió TMA 2 M en tolueno (2,67 ml, 5,34 mmoles) a 0°C. La mezcla de reacción se calentó a 80°C durante 2 h. La mezcla de reacción resultante se enfrió a rt y se vertió rápidamente en una mezcla de EtOAc:agua (1:1, 100 ml). La mezcla de reacción se filtró a través de un lecho de celite, la fase orgánica se separó y la fase acuosa se extrajo de nuevo utilizando EtOAc (2 x 25 ml). La fase orgánica combinada se lavó con salmuera (25 ml), se separó, se secó sobre Na2SO4, se filtró y se concentró a presión reducida. El residuo resultante se purificó mediante cromatografía en columna (EtOAc al 70% en hexano) produciendo (R)-3-(4-(pirimidin-2-il)-3,4-dihidro-2H-benzo[b][1,4]oxazin-7-carboxamido)pirrolidin-1-carboxilato de terc-butilo (0,28 g, 0,66 mmoles). LCMS: Método C, RT 2,19 min, MS: ES+ 426,28
Etapas d, e. El compuesto del título se sintetizó a partir del intermedio anterior utilizando un procedimiento similar al descrito para las etapas d, e del Ejemplo 2 para proporcionar el compuesto del título. LCMS: Método A, RT 3,38 min, MS: ES+ 351,11; RMN H1 (400 MHz, DMSO-de) 5 ppm 8,58 (d, J = 4,4 Hz, 2 H), 8,47 (d, J = 6,4 Hz, 1 H), 8,13 (d, J = 8,4 Hz, 1 H), 7,37 - 7,43 (m, 2 H), 6,99 (t, J = 4,8 Hz, 1 H), 4,43 - 4,47 (m, 1 H), 4,30 - 4,32 (m, 2 H), 4,20 - 4,22 (m, 2 H), 3,60 - 3,64 (m, 1 H), 3,52 - 3,56 (m, 1 H), 3,42 - 3,47 (m, 1 H), 3,28 - 3,32 (m, 1 H), 2,08 - 2,13 (m, 1 H), 1,93 - 1,96 (m, 1 H).
Ejemplo 212 (R)-N-(1 -cianopirrolidin-3-il)-4-(4-ciclopropilpirimidin-2-il)-3,4-dihidro-2H-benzo[b][1,4]oxazin-7-carboxamida

Etapa a. A una solución agitada de 2,4-dicloropirimidina (4,0 g, 26,85 mmoles) y ácido ciclopropilborónico (2,54 g,
29,54 mmoles) en THF (80 ml) se le añadió K3PO4 (14,25 g, 67,13 mmoles) a rt. La mezcla de reacción se desgasificó durante 15 min a rt antes de la adición de Pd(dppf)Cl2 (1,965 g, 2,68 mmoles). La mezcla de reacción se calentó a 90°C durante 2 h. La mezcla de reacción se enfrió a rt, se vertió rápidamente en agua (150 ml) y se extrajo utilizando EtOAc (3 x 100 ml). La fase orgánica combinada se lavó con salmuera (100 ml), se separó, se secó sobre Na2SO4, se filtró y se concentró a presión reducida. El residuo resultante se purificó mediante cromatografía en columna (EtOAc al 20% en hexano) produciendo 2-cloro-4-ciclopropilpirimidina (1,0 g, 6,47 mmoles). LCMS: Método C, RT 2,01 min, MS: ES+ 155,15
Etapas b-e. El compuesto del título se sintetizó a partir del intermedio anterior utilizando un procedimiento similar al descrito para las etapas b-e del Ejemplo 211 para proporcionar el compuesto del título. LCMS: Método A, RT 4,22 min, MS: ES+ 391,16; RMN H1 (400 MHz, DMSO-d6) 5 ppm 8,45 (d, J = 6,8 Hz, 1 H), 8,35 (d, J = 4,8 Hz, 1 H), 8,10 (d, J = 8,4 Hz, 1 H), 7,37 - 7,41 (m, 2 H), 6,94 (d, J = 5,2 Hz, 1 H), 4,44 - 4,46 (m, 1 H), 4,27 - 4,29 (m, 2 H), 4,15 - 4,18 (m, 2 H), 3,60 - 3,64 (m, 1 H), 3,52 - 3,56 (m, 1 H), 3,41 - 3,47 (m, 1 H), 3,28 - 3,31 (m, 1 H), 2,03 - 2,13 (m, 2 H), 1,93 - 1,97 (m, 1 H), 1,00 (s, 4 H).
Ejemplo 213 (R)-N-(1-cianopirrolidin-3-il)-4-((4-ciclopropilpirimidin-2-il)amino)-3-fluorobenzamida

Sintetizada mediante un procedimiento similar al descrito para el Ejemplo 91 utilizando 2-cloro-4-ciclopropilpirimidina (como se describe para Ejemplo 212) en la etapa b. LCMS: Método B, RT 3,90 min, MS: ES+ 367,23; RMN H1 (400 MHz, DMSO-de) 5 ppm 9,06 (s, 1 H), 8,54 (d, J = 6,4 Hz, 1 H), 8,26 (d, J = 5,2 Hz, 1 H), 8,04 (d, J = 8,8 Hz, 1 H),
7,68 - 7,72 (m, 2 H), 6,88 (d, J = 5,2 Hz, 1 H), 4,45 - 4,49 (m, 1 H), 3,62 - 3,66 (m, 1 H), 3,53 - 3,59 (m, 1 H), 3,42 -3,48 (m, 1 H), 3,30 - 3,31 (m, 1 H), 2,10 - 2,15 (m, 1 H), 1,92 - 2,03 (m, 2 H), 1,01 - 1,03 (m, 4 H).
Ejemplo 214 (R)-N-(1-cianopirrolidin-3-il)-4-((4-ciclopropilpirimidin-2-il)amino)-2,3-difluorobenzamida

Sintetizada mediante un procedimiento similar al descrito para el Ejemplo 91 utilizando 2-cloro-4-ciclopropilpirimidina (como se describe para Ejemplo 212) en la etapa b. LCMS: Método A, RT 4,12 min, MS: ES+ 385,11; RMN H1 (400 MHz, DMSO-de) 5 ppm 9,35 (s, 1 H), 8,65 (d, J = 6,4 Hz, 1 H), 8,27 (d, J = 5,2 Hz, 1 H), 7,70 - 7,74 (m, 1 H), 7,33 -7,37 (m, 1 H), 6,89 (d, J = 5,2 Hz, 1 H), 4,43 - 4,47 (m, 1 H), 3,61 - 3,65 (m, 1 H), 3,43 - 3,55 (m, 2 H), 3,27 - 3,31 (m, 1 H), 2,08 - 2,14 (m, 1 H), 1,89 - 2,04 (m, 2 H), 0,97 - 1,05 (m, 4 H).
Ejemplo 215 (R)-N-(1-cianopirrolidin-3-il)-4-(N-metilisobutiramido)picolinamida

Etapas a, b. Siguiendo un procedimiento similar al descrito para las etapas a, b del Ejemplo 62, utilizando 4-aminopicolinato de metilo (Número CAS 71469-93-7) en la etapa a, para proporcionar 4-(N-metilisobutiramido)picolinato de metilo. LCMS: Método C, RT 1,72 min, MS: ES+ 237,00. Esta sustancia se utilizó directamente para la siguiente etapa sin purificación adicional.
Etapa c. A una solución de 4-(N-metilisobutiramido)picolinato de metilo (0,150 g, 0,635 mmoles) en THF (10 ml) se le añadió DIPEA (0,06 ml, 0,317 mmoles) a rt. La mezcla de reacción se enfrió a 0°C. Se añadió una solución de trimetilaluminio (2 M en tolueno) (1,5 ml, 3,177 mmoles) a la mezcla de reacción. La mezcla de reacción se agitó a 0°C durante 30 min y a continuación se trató con (R)-3-amino-1 N-BOC-pirrolidina (0,118 g, 0,633 mmoles). La mezcla de reacción se calentó a 90°C durante 2 h. La mezcla de reacción resultante se enfrió a rt y se vertió en una solución acuosa saturada de NaHCO3 (50 ml) y se extrajo con EtOAc (3 x 50 ml). La capa orgánica combinada se secó sobre Na2SO4, se filtró y se concentró a presión reducida produciendo (R)-3-(4-(N-metilisobutiramido)picolinamido)pirrolidin-1 -carboxilato de terc-butilo (0,220 g, 0,564 mmoles). LCMS: Método C, RT 2,097 min, MS: ES+ 391,50. Esta sustancia se utilizó directamente para la siguiente etapa sin purificación adicional.
Etapas d, e. El compuesto del título se sintetizó a partir del intermedio anterior utilizando un procedimiento similar al descrito para las etapas b, c del Ejemplo 1. LCMS: Método B, RT 3,07 min, MS: ES+ 316,10; RMN 1H (400 MHz, DMSO-de) 5 ppm 9,08 (d, J = 6,4 Hz, 1 H), 8,66 (d, J = 5,4 Hz, 1 H), 7,97 (d, J = 1,6 Hz, 1 H), 7,62 - 7,64 (m, 1 H),
4,51 - 4,55 (m, 1 H), 3,50 - 3,63 (m, 2 H), 3,38 - 3,47 (m, 2 H), 3,30 (s, 3 H), 2,74 - 2,78 (m, 1 H), 1,99 - 2,16 (m, 2 H), 1,01 (d, J = 6,4 Hz, 6 H).
Ejemplo 216 (R)-N-(1-cianopirrolidin-3-il)-[2,3'-bipiridin]-6'-carboxamida

Etapa a. A una solución de 5-bromopicolinato de metilo (Número CAS 29682-15-3) (0,500 g, 2,314 mmoles) en 1,4-dioxano (10 ml) se le añadió 2-(tributilestanil)piridina (Número CAS 17997-47-6) (1,00 g, 2,717 mmoles) a rt. La mezcla de reacción se desgasificó durante 10 min antes de la adición de Pd(PPh3)4 (0,132 g, 0,114 mmoles). La mezcla de reacción se calentó a 110°C durante 16 h. La mezcla de reacción resultante se vertió en agua (70 ml) y se extrajo con EtOAc (2 x 70 ml). La capa orgánica combinada se secó sobre Na2SO4, se filtró y se concentró a presión reducida produciendo [2,3'-bipiridin]-6'-carboxilato de metilo (0,300 g, 1,401 mmoles). LCMS: Método C, RT 1,75 min, MS: ES+ 215,19. Esta sustancia se utilizó directamente para la siguiente etapa sin purificación adicional.
Etapas b-e. El compuesto del título se sintetizó a partir del intermedio anterior utilizando un procedimiento similar al descrito para las etapas b-e del Ejemplo 2. LCm S: Método C, RT 1,76 min, MS: ES+ 294,32; RMN H1 (400 MHz, DMSO-d6) 5 ppm 9,33 - 9,34 (m, 1 H), 9,13 (d, J = 7,2 Hz, 1 H), 8,75 - 8,77 (m, 1 H), 8,64 (dd, J = 8,0, 2,0 Hz, 1 H), 8,14 - 8,17 (m, 2 H), 7,96 - 8,01 (m, 1 H), 7,47 - 7,50 (m, 1 H), 4,54 - 4,58 (m, 1 H), 3,55 - 3,65 (m, 2 H), 3,40 - 3,48 (m, 2 H), 2,03 - 2,17 (m, 2 H).
Ejemplo 217 (R)-N-(1-cianopirrolidin-3-il)-[2,4'-bipiridin]-2'-carboxamida

Sintetizada mediante un procedimiento similar al descrito para el Ejemplo 216 utilizando 4-bromopicolinato de metilo (Número CAS 29681 -42-3) en la etapa a. LCMS: Método B, RT 3,17 min, MS: ES+ 294,33; RMN H1 (400 MHz, DMSO-d6) 5 ppm 9,11 (d, J = 3,2 Hz, 1 H), 8,78 - 8,79 (m, 2 H), 8,71 (d, J = 1,2 Hz, 1 H), 8,29 (dd, J = 5,2, 1,6 Hz, 1 H), 8,20 (d, J = 8,0 Hz, 1 H), 7,98 - 8,03 (m, 1 H), 7,52 - 7,55 (m, 1 H), 4,54 - 4,62 (m, 1 H), 3,59 - 3,64 (m, 1 H), 3,55 - 3,59 (m, 1 H), 3,34 - 3,49 (m, 2 H), 2,12 - 2,19 (m, 1 H), 2,02 - 2,11 (m, 1 H).
Ejemplo 218 (R)-3-(4-clorofenil)-N-(1-cianopirrolidin-3-il)isoxazol-5-carboxamida

Etapa a. A una solución de ácido propiólico (0,800 g, 11,4 mmoles) en THF (20 ml) se le añadieron DIPEA (6,00 ml, 35,1 mmoles) y HATU (6,500 g, 17,105 mmoles). La mezcla de reacción se agitó a rt durante 30 min y a continuación se enfrió a 0°C. La mezcla de reacción se trató con (R)-3-amino-1N-BOC-pirrolidina (2,120 g, 11,4 mmoles) y a continuación se agitó a rt durante 15 h. La mezcla de reacción resultante se vertió en una solución saturada de NaHCO3 (50 ml) y se extrajo con EtOAc (3 x 30 ml). La capa orgánica combinada se lavó con salmuera (2 x 50 ml), se secó sobre Na2SO4, se filtró y se concentró a presión reducida produciendo (R)-3-propiolamidopirrolidin-1-carboxilato de terc-butilo (2,200 g, 9,24 mmoles). LCMS: Método C, RT 1,88 min, MS: ES+ 239,40. Esta sustancia se utilizó directamente para la siguiente etapa sin purificación adicional.
Etapa b. A una solución de 4-clorobenzaldehído (5,000 g, 35,5 mmoles) en EtOH (50 ml) se le añadió NH2OH.HG (2,500 g, 36,0 mmoles) a rt. Se añadió una solución de NaOH (4,300 g, 407 mmoles) en agua (30 ml) a la mezcla de reacción a rt. La mezcla de reacción se calentó a reflujo durante 2 h. La mezcla de reacción resultante se enfrió a rt y se aciduló utilizando una solución diluida de HCl para ajustar el pH ~ 3-4, Los precipitados resultantes se recogieron mediante filtración y se lavaron con agua (200 ml). La sustancia sólida resultante se disolvió en EtOAc (100 ml) y se lavó con una solución acuosa saturada de NaHCO3 (3 x 50 ml). La capa orgánica combinada se lavó con salmuera (2 x 70 ml), se secó sobre Na2SO4, se filtró y se concentró a presión reducida produciendo oxima de (E)-4-clorobenzaldehído (4,6 g, 29,49 mmoles). LCMS: Método C, RT 2,06 min, MS: ES 155,90; RMN H1 (400 MHz, DMSO-d6) 5 ppm 11,36 (s, 1 H), 8,15 (s, 1 H), 7,61 (d, J = 8,4 Hz, 2 H), 7,45 - 7,52 (m, 2 H). Esta sustancia se utilizó directamente para la siguiente etapa sin purificación adicional.
Etapas c, d. A una solución de oxima de (E)-4-clorobenzaldehído (0,700 g, 4,49 mmoles) en DCM (20 ml) se le añadió N-clorosuccinamida (0,900 g, 6,77 mmoles) a 0°C. La mezcla de reacción se agitó a rt durante 15 h. Después, la mezcla de reacción se enfrió a 0°C. Se añadió TEA (1,2 ml, 9,032 mmoles) a la mezcla de reacción y se agitó durante 5 min. Se añadió (R)-3-propiolamidopirrolidin-1-carboxilato de terc-butilo (1,300 g, 5,46 mmoles) a la mezcla de reacción a 0°C. La mezcla de reacción se agitó a rt durante 15 h. La mezcla de reacción resultante se vertió en agua (100 ml) y se extrajo con DCM (3 x 30 ml). La capa orgánica combinada se lavó con salmuera (2 x 40 ml), se secó sobre Na2SO4 , se filtró y se concentró a presión reducida. El residuo obtenido se purificó mediante cromatografía ultrarrápida (EtOAc al 12-15% en hexano) produciendo (R)-3-(3-(4-clorofenil)isoxazol-5-carboxamido)pirrolidin-1-carboxilato de terc-butilo (0,400 g, 1,023 mmoles). LCMS: Método C, RT 2,51 min, MS: ES-390,70.
Etapas e, f. El compuesto del título se sintetizó a partir del intermedio anterior utilizando un procedimiento similar al descrito para las etapas b, c del Ejemplo 1. LCMS: Método A, RT 4,30 min, MS: ES+ 316,91; RMN H1 (400 MHz, DMSO-d6) ó ppm 9,30 (d, J = 6,8 Hz, 1 H), 7,95 - 7,98 (m, 2 H), 7,71 (s, 1 H), 7,61 - 7,64 (m, 2 H), 4,46 - 4,53 (m, 1 H), 3,63 -3,67 (m, 1 H), 3,53 - 3,59 (m, 1 H), 3,43 - 3,49 (m, 1 H), 3,34 - 3,38 (m, 1 H), 2,10 - 2,19 (m, 1 H), 1,94 - 2,02 (m, 1 H).
Ejemplo 219 (R)-N-(1-cianopirrolidin-3-il)-3-(4-(trifluorometil)fenil)isoxazol-5-carboxamida

Etapa a. A una solución de ácido 2-metilensuccínico (Número CAS 97-65-4) (5000 g, 38,5 mmoles) en agua (70 ml) se le añadió anilina (3000 g, 32,3 mmoles) a rt. La mezcla de reacción se calentó a 110°C durante 30 h. La mezcla de reacción resultante se dejó enfriar a rt y se alcalinizó utilizando una solución de NaOH 1 M (100 ml). La mezcla obtenida se agitó durante 10 min a rt. Los precipitados resultantes se filtraron y el producto filtrado resultante se aciduló utilizando HCl concentrado. Los precipitados obtenidos se recogieron mediante filtración y se secaron al aire produciendo ácido 5-oxo-1-fenilpirrolidin-3-carboxílico (2,000 g, 9,76 mmoles). LCMS: Método C, RT 1,70 min, MS: ES+ 206,18; RMN H1 (400 MHz, DMSO-de) 5 ppm 12,78 (s, 1 H), 7,63 - 7,66 (m, 2 H), 7,36 - 7,40 (m, 2 H), 7,15 (t, J = 7,2 Hz, 1 H), 4,03 - 4,08 (m, 1 H), 3,95 - 3,99 (m, 1 H), 3,32 - 3,39 (m, 1 H), 2,67 - 2,82 (m, 2 H).
Etapa b. A una solución de ácido 5-oxo-1-fenilpirrolidin-3-carboxílico (1,000 g, 4,88 mmoles) en MeOH (10 ml) se le añadió lentamente SOCl2 (0,658 g, 5,82 mmoles) a 0°C durante un período de 30 min. La mezcla de reacción se agitó
a rt durante 1 h. La mezcla de reacción resultante se concentró a presión reducida produciendo 5-oxo-1 -fenilpirrolidin-3-carboxilato de metilo (0,900 g, 4,11 mmoles). LCMS: Método C, 1,90 min, MS: e S+ 220,50. Esta sustancia se utilizó directamente para la siguiente etapa sin purificación adicional.
Etapa c. A una solución de 5-oxo-1-fenilpirrolidin-3-carboxilato (1,500 g, 6,85 mmoles) en THF (40 ml) se le añadió 9-BBN (0,5 M en THF) (15 ml, 7,50 mmoles). La mezcla de reacción se agitó a rt durante 16 h. La mezcla de reacción resultante se evaporó a presión reducida y se purificó mediante cromatografía ultrarrápida (EtOAc al 40% en hexano) produciendo 1 -fenilpirrolidin-3-carboxilato de metilo (0,640 g, 3,12 mmoles). LCMS: Método C, RT 2,44 min. MS: ES+ 205,90.
Etapas d-g. El compuesto del título se sintetizó a partir del intermedio anterior utilizando un procedimiento similar al descrito para las etapas b-e del Ejemplo 2. LCMS: Método B, RT 3,55 min, MS: ES+ 285,28; RMN H1 (400 MHz, DMSO-de) 5 ppm 8,36 (d, J = 6,8 Hz, 1 H), 7,15 (t, J = 8,4 Hz, 2 H), 6,59 (t, J = 7,2 Hz, 1 H), 6,52 (d, J = 7,6 Hz, 2 H), 4,23 - 4,27 (m, 1 H), 3,37 - 3,55 (m, 4 H), 3,24 - 3,32 (m, 3 H), 3,14 - 3,22 (m, 1 H), 3,03 - 3,09 (m, 1 H), 2,01 - 2,17 (m, 3 H), 1,75 - 1,82 (m, 1 H).
Ejemplo 223 (R)-N-( 1 -cianopirrolidin-3-il)-3-fluoro-4-(4-metil-1 H-imidazol-1 -il)benzamida

Etapa a. A una solución de 3,4-difluorobenzaldehído (1,000 g, 7,04 mmoles) en DMF (10 ml) se le añadió 4-metil-1 H-imidazol (0,580 g, 7,07 mmoles) y K2CO3 (1,200 g, 8,70 mmoles) a rt. La mezcla de reacción se calentó a 110°C durante 16 h. La mezcla de reacción resultante se enfrió a rt y se vertió en una solución acuosa saturada de NaHCO3 (150 ml). La mezcla resultante se extrajo con EtOAc (3 x 50 ml). La fase orgánica combinada se secó sobre Na2SO4, se filtró y se concentró a presión reducida. El residuo resultante se purificó mediante cromatografía en columna (EtOAc al 30-50% en hexano) produciendo 3-fluoro-4-(4-metil-1H-imidazol-1-il)benzaldehído (1,300 g, 6,37 mmoles). LCMS: Método C, RT 1,38 min, MS: ES+ 205,19; RMN H1 (400 MHz, DMSO-de) 5 ppm 10,02 (s, 1 H), 8,07 (s, 1 H), 7,96 (d, J = 1,2 Hz, 1 H), 7,89 - 7,92 (m, 2 H), 7,40 (s, 1 H), 2,18 (s, 3 H).
Etapa b. A una solución de 3-fluoro-4-(4-metil-1H-imidazol-1-il)benzaldehído (1,300 g, 6,37 mmoles) en MeOH:agua (7:1, 16 ml) se le añadió KOH (1,420 g, 25,36 mmoles) a rt. La mezcla de reacción se calentó a 65°C. Se añadió lentamente a la mezcla de reacción una solución de H2O2 (30% p/p en agua) (5,60 ml, 49,41 mmoles) a 65°C y se agitó durante 16 h. La mezcla de reacción resultante se enfrió a rt y se concentró a presión reducida. La mezcla resultante se vertió en agua (200 ml), se aciduló utilizando una solución de HCl 1 M y se extrajo con EtOAc (100 ml). La fase orgánica combinada se secó sobre Na2SO4, se filtró y se concentró a presión reducida produciendo 3-fluoro-4-(4-metil-1H-imidazol-1-il)benzaldehído (0,550 g, 2,50 mmoles). LCMS: Método C, RT 1,25 min, MS: ES+ 221,19. Esta sustancia se utilizó directamente para la siguiente etapa sin purificación adicional.
Etapas c-e. El compuesto del título se sintetizó a partir del intermedio anterior utilizando un procedimiento similar al descrito para el Ejemplo 1. LCMS: Método A, RT 3,12 min, MS: ES+ 313,98; RMN H1 (400 MHz, DMSO-d6) 5 ppm 8,78 (d, J = 6,8 Hz, 1 H), 8,01 (s, 1 H), 7,92 - 7,96 (dd, J = 12,0, 1,6 Hz, 1 H), 7,83 - 7,85 (m, 1 H), 7,74 - 7,78 (m, 1 H), 7,36 (s, 1 H), 4,47 - 4,50 (m, 1 H), 3,63 - 3,67 (m, 1 H), 3,53 - 3,59 (m, 1 H), 3,45 - 3,49 (m, 1 H), 3,32 - 3,33 (m, 1 H), 2,18 (s, 3 H), 2,08 - 2,16 (m, 1 H), 1,93 - 2,00 (m, 1 H).
Ejemplo 224 (R)-N-(1-cianopirrolidin-3-il)-3-fluoro-N-metil-4-(4-metil-1H-imidazol-1-il)benzamida

Sintetizada mediante un procedimiento similar al descrito para el Ejemplo 223 utilizando 1-N-BOC-(3R)-3-(metilamino)pirrolidina en la etapa c. LCMS: Método A, RT 3,10 min, MS: ES+ 328,02; RMN H1 (400 MHz, DMSO-d6) 5 ppm 7,97 (s, 1 H), 7,69 (t, J = 8,0 Hz, 1 H), 7,55 - 7,62 (m, 1 H), 7,34 - 7,42 (m, 1 H), 7,32 (s, 1 H), 4,32 - 4,37 (m, 1 H), 3,52 - 3,54 (m, 2 H), 3,44 - 3,47 (m, 2 H), 2,89 (s, 3 H), 2,18 (s, 3 H), 2,02 - 2,09 (m, 2 H).
Ejemplo 225 N-((R)-1-cianopirrolidin-3-il)-3-(piridin-2-il)pirrolidin-1-carboxamida

Etapa a. A una solución de 2-vinilpiridina (Número CAS 100-69-6) (5,000 g, 47,62 mmoles) en DCM (30 ml) se le añadió TFA (0,542 g, 4,75 mmoles). La mezcla de reacción se agitó a rt durante 5 min y a continuación se trató gota a gota con una solución de N-bencil-1-metoxi-N-((trimetilsilil)metil)metanamina (Número CAS 93102-05-7) (16,92 g, 71,39 mmoles) en DCM (30 ml) durante un período de 45 min. La mezcla de reacción se agitó a rt durante 16 h. La mezcla de reacción se vertió en una solución acuosa saturada de NaHCO3 (250 ml) y se extrajo con DCM (3 x 100 ml). La fase orgánica combinada se secó sobre Na2SO4, se filtró y se concentró a presión reducida produciendo 2-(1 -bencilpirrolidin-3-il)piridina (8,00 g, 33,61 mmoles). LCMS: Método C, RT 1,52 min, MS: ES+ 239,25. Esta sustancia se utilizó directamente para la siguiente etapa sin purificación adicional.
Etapa b. A una solución de 2-(1-bencilpirrolidin-3-il)piridina (5,00 g, 21,01 mmoles) en EtOH (50 ml) se le añadió Pd(OH)2 al 20% sobre carbono (50% de contenido de humedad) (2,50 g) a rt. Se añadió polimetilhidroxilsilano (5,00 ml) a la mezcla de reacción a rt durante un período de 10 min. La mezcla de reacción resultante se agitó a rt durante 1 h antes de la adición de anhídrido BOC (9,150 g, 41,97 mmoles). La mezcla de reacción se agitó a rt durante 16 h. La mezcla de reacción resultante se filtró a través de un lecho de celite y se lavó con MeOH (50 ml). El producto filtrado combinado se concentró a presión reducida. El residuo resultante se purificó mediante cromatografía en columna (EtOAc al 10% en hexano) produciendo 3-(piridin-2-il)pirrolidin-1 -carboxilato de terc-butilo (2,50 g, 10,08 mmoles). LCMS: Método C, RT 1,84 min, MS: ES+ 249,40.
Etapa c. A una solución de 3-(piridin-2-il)pirrolidin-1 -carboxilato de terc-butilo (0,500 g, 2,016 mmoles) en DCM (4 ml) se le añadió TFA (0,459 g, 4,03 mmoles) a rt. La mezcla de reacción se agitó a rt durante 2 h. La mezcla de reacción resultante se evaporó a presión reducida. El residuo resultante se evaporó conjuntamente con DCM (3 x 10 ml) y se secó a alto vacío produciendo sal de TFA de 2-(pirrolidin-3-il)piridina (0,260 g, 0,984 mmoles). MS: ES+ 149,0. Esta sustancia se utilizó directamente para la siguiente etapa sin purificación adicional.
Etapas d-f. El compuesto del título se sintetizó a partir del intermedio anterior utilizando un procedimiento similar al descrito para el Ejemplo 5. LCMS: Método A, RT 2,84 min, MS: ES+ 285,98; RMN H1 (400 MHz, DMSO-d6) ó ppm 8,51 (d, J = 4,0 Hz, 1 H), 7,72 - 7,77 (m, 1 H), 7,34 (d, J = 7,6 Hz, 1 H), 7,24 - 7,27 (m, 1 H), 6,24 (d, J = 6,4 Hz, 1 H), 4,09 - 4,18 (m, 1 H), 3,68 - 3,73 (m, 1 H), 3,47 - 3,57 (m, 4 H), 3,36 - 3,45 (m, 1 H), 3,27 - 3,31 (m, 1 H), 3,16 - 3,19 (m, 2 H), 2,19 - 2,25 (m, 1 H), 1,95 - 2,11 (m, 2 H), 1,80 - 1,88 (m, 1 H)).
Ejemplo 226 N-((R)-1-cianopirrolidin-3-il)-3-(1-metil-1H-pirazol-4-il)pirrolidin-1-carboxamida

Sintetizada mediante un procedimiento similar al descrito para el Ejemplo 5 utilizando 1-metil-4-(pirrolidin-3-il)-1H-pirazol (Número CAS 1211542-11-8) en la etapa a. LCMS: Método H, r T 13,27 min, MS: ES+ 289,07; RMN H1 (400 MHz, DMSO-de) 5 ppm 7,54 (s, 1 H), 7,31 (s, 1 H), 6,18 (d, J = 6,4 Hz, 1 H), 4,11 - 4,14 (m, 1 H), 3,78 (s, 3 H), 3,60 -3,64 (m, 1 H), 3,45 - 3,52 (m, 2 H), 3,36 - 3,41 (m, 2 H), 3,21 - 3,28 (m, 1 H), 3,15 - 3,18 (m, 2 H), 3,04 - 3,08 (m, 1 H), 2,12 - 2,14 (m, 1 H), 1,95 - 2,02 (m, 1 H), 1,77 - 1,87 (m, 2 H).
Ejemplo 227 (R)-N-(1-cianopirrolidin-3-il)-3-(2-metoxipiridin-4-il)-N-metilisoxazol-5-carboxamida

Etapa a. A una solución de 2-metoxiisonicotinaldehído (Número CAS 72716-87-1) (0,500 g, 3,65 mmoles) en MeOH (7 ml) se le añadió NH2OH.HG (0,503 g, 7,29 mmoles) a rt. La mezcla de reacción se calentó a 70°C durante 1 h. La mezcla de reacción resultante se concentró a presión reducida produciendo oxima de (E)-2-metoxiisonicotinaldehído (1,20 g, cuantitativo). LCMS: Método A, RT 2,54 min, MS: ES+ 152,91. Esta sustancia se utilizó directamente para la siguiente etapa sin purificación adicional.
Etapa b. A una solución de oxima de (E)-2-metoxiisonicotinaldehído (0,600 g, 3,95 mmoles) en DMF (7 ml) se le añadió N-clorosuccinamida (0,787 g, 5,92 mmoles) a 0°C. La mezcla de reacción se agitó a rt durante 3 h. Se añadieron
1,8-diazabiciclo[5,4,0]undec-7-eno (0,900 g, 5,92 mmoles) y propiolato de metilo (0,500 g, 5,95 mmoles) a la mezcla de reacción y se agitó a rt durante 16 h. La mezcla resultante se vertió en agua fría (150 ml) y se extrajo con EtOAc (2 x 100 ml). La capa orgánica combinada se lavó con salmuera (100 ml), se secó sobre Na2SO4, se filtró y se concentró a presión reducida. El residuo resultante se purificó mediante cromatografía ultrarrápida (EtOAc al 70% en hexano) produciendo 3-(2-metoxipiridin-4-il)isoxazol-5-carboxilato de metilo (0,330 g, 1,410 mmoles). LCMS: Método C, RT 2,17 min, MS: ES+ 235,25.
Etapas c-f. El compuesto del título se sintetizó a partir del intermedio anterior utilizando un procedimiento similar al descrito para las etapas b-e del Ejemplo 2. LCMS: Método A, RT 3,68 min, MS: ES+ 328,02; RMN H1 (400 MHz, DMSO-de, 80°C) 5 ppm 8,34 (d, J = 5,2 Hz, 1 H), 7,59 (s, 1 H), 7,50 (dd, J = 5,6, 1,6 Hz, 1 H), 7,34 (s, 1 H), 4,50 - 5,10 (m, 1 H), 3,94 (s, 3 H), 3,56 - 3,65 (m, 2 H), 3,41 - 3,51 (m, 2 H), 3,04 (s, 3 H), 2,10 - 2,23 (m, 2 H).
Ejemplo 228 (R)-N-(1-cianopirrolidin-3-il)-2-fluoro-4-(N-metilfenilsulfonamido)benzamida

Etapa a. Una mezcla de 4-amino-2-fluorobenzoato de metilo (0,300 g, 1,77 mmoles) y formiato de etilo (10 ml) se calentó a 80°C durante 16 h. La mezcla de reacción resultante se enfrió a rt y se concentró a presión reducida produciendo 2-fluoro-4-formamidobenzoato de metilo (0,332 g, 1,685 mmoles). LCMS: Método C, Rt 1,79 min, MS: ES-196,12.
Etapa b. A una solución de 2-fluoro-4-formamidobenzoato de metilo (0,332 g, 1,68 mmoles) en THF (5 ml) se le añadió una solución 1 M de complejo de BH3.THF en THF (8,42 ml, 8,43 mmoles) a 0°C. La mezcla de reacción se agitó a rt durante 16 h. La mezcla de reacción resultante se enfrió a 0°C y se aciduló con HCl al 10% en MeOH (5 ml). La mezcla de reacción se calentó a 50°C durante 1 h. La mezcla de reacción resultante se enfrió a 0°C y se alcalinizó utilizando una solución acuosa saturada de NaHCO3. La mezcla de reacción resultante se concentró a presión reducida. El residuo obtenido se vertió en agua (30 ml) y se extrajo con EtOAc (2 x 20 ml). La capa orgánica combinada se secó sobre Na2SO4, se filtró y se concentró a presión reducida produciendo 2-fluoro-4-(metilamino)benzoato de metilo (0,331 g, cuantitativo). lCm S: Método C, RT 2,031 min. MS: ES+ 184,06. Esta sustancia se utilizó directamente para la siguiente etapa sin purificación adicional.
Etapa c. A una solución de 2-fluoro-4-(metilamino)benzoato de metilo (0,331 g, 1,81 mmoles) en piridina (3 ml) se le añadió cloruro de bencenosulfonilo (0,383 g, 2,17 mmoles) a 0°C y se agitó durante 1 h. La mezcla de reacción resultante se vertió en una solución acuosa saturada de ácido cítrico (20 ml) y se extrajo con EtOAc (30 ml). La capa orgánica se lavó con una solución acuosa saturada de ácido cítrico (2 x 20 ml). La capa orgánica se lavó con salmuera (20 ml), se secó sobre Na2SO4, se filtró y se concentró a presión reducida produciendo 2-fluoro-4-(N-metilfenilsulfonamido)benzoato de metilo (0,506 g, 1,56 mmoles). LCMS: Método C, RT 2,36 min, MS: ES+ 324,19. Esta sustancia se utilizó directamente para la siguiente etapa sin purificación adicional.
Etapas d-g. El compuesto del título se sintetizó a partir del intermedio anterior utilizando un procedimiento similar al descrito para las etapas b-e del Ejemplo 2. LCMS: Método A, RT 4,00 min, MS: ES+ 402,94; RMN H1 (400 MHz, DMSO-de) 5 ppm 8,68 (d, J = 6,4 Hz, 1 H), 7,72 - 7,75 (m, 1 H), 7,53 - 7,64 (m, 5 H), 7,09 - 7,15 (m, 2 H), 4,41 - 4,45 (m, 1 H), 3,60 - 3,64 (m, 1 H), 3,42 - 3,53 (m, 2 H), 3,26 - 3,29 (m, 1 H), 3,17 (s, 3 H), 2,06 - 2,15 (m, 1 H), 1,87 - 1,93 (m, 1 H). Ejemplo 229 (R)-N-(1-cianopirrolidin-3-il)-1-metil-6-(1-metil-1H-pirazol-4-il)-1H-indol-2-carboxamida

Etapa a. A una solución de ácido 6-bromo-1 H-indol-2-carboxílico (Número CAS 16732-65-3) (0,249 g, 1,04 mmoles)
en THF (8 ml) se le añadieron DIPEA (0,402 g, 3,112 mmoles) y HATU (0,394 g, 1,04 mmoles) a rt. La mezcla de reacción se agitó a rt durante 10 min. Se añadió gota a gota a la mezcla de reacción a rt una solución de (R)-3-amino-1 N-BOC-pirrolidina (Número CAS 147081 -49-0) (0,193 g, 1,03 mmoles) en THF (2 ml). La mezcla de reacción se agitó a rt durante 1 h. La mezcla de reacción resultante se vertió en agua (50 ml) y se extrajo con DCM (2 x 25 ml). La capa orgánica combinada se lavó con salmuera (20 ml), se secó sobre Na2SO4, se filtró y se concentró a presión reducida. El residuo resultante se purificó mediante cromatografía ultrarrápida (EtOAc al 40% en hexano) produciendo (R)-3-(6-bromo-1H-indol-2-carboxamido)pirrolidin-1-carboxilato de terc-butilo (0,400 g, 0,980 mmoles). LCMS: Método C, Rt 2,24 min, MS: ES+ 408,50, 410,50.
Etapa b. A una solución de (R)-3-(6-bromo-1-metil-1H-indol-2-carboxamido)pirrolidin-1-carboxilato de terc-butilo (0,460 g, 1,130 mmoles) en DMF (10 ml) se le añadieron Cs2CO3 (0,730 g, 2,239 mmoles) y yoduro de metilo (0,320 g, 2,254 mmoles). La mezcla de reacción se calentó a 100°C durante 2,5 h. La mezcla de reacción resultante se enfrió a rt y se vertió en agua (150 ml). La mezcla resultante se extrajo con DCM (2 x 25 ml). La fase orgánica combinada se lavó con salmuera (25 ml), se secó sobre Na2SO4, se filtró y se concentró a presión reducida produciendo (R)-3-(6-bromo-1-metil-1H-indol-2-carboxamido)pirrolidin-1-carboxilato de terc-butilo (0,400 g, 0,95 mmoles). LCMS: Método C, RT 2,61 min, MS: ES+ 422,30, 424,30. Esta sustancia se utilizó directamente para la siguiente etapa sin purificación adicional.
Etapas c-e. El compuesto del título se sintetizó a partir del intermedio anterior utilizando un procedimiento similar al descrito para las etapas b-d del Ejemplo 3. LCMS: Método A, RT 3,56 min, MS: ES+ 349,11; RMN H1 (400 MHz, DMSO-de) 5 ppm 8,62 (d, J = 6,8 Hz, 1 H), 8,18 (s, 1 H), 7,94 (s, 1 H), 7,72 (s, 1 H), 7,61 (d, J = 8,4 Hz, 1 H), 7,33 (d, J = 8,0 Hz, 1 H), 7,12 (s, 1 H), 4,47 - 4,50 (m, 1 H), 4,00 (s, 3 H), 3,88 (s, 3 H), 3,63 - 3,67 (m, 1 H), 3,56 - 3,58 (m, 1 H), 3,43 - 3,47 (m, 2 H), 2,11 - 2,14 (m, 1 H), 1,96 - 1,97 (m, 1 H).
Ejemplo 230 (R)-N(1-cianopirrolidin-3il)-1-metil-5-(1-metil-1Hpirazol-4il)-1H-indol-2-carboxamida

Etapa a. A una solución de 2-cloropiridin-4-amina (0,500 g, 3,91 mmoles) en tolueno (5 ml) se le añadieron isoindolina (0,557 g, 4,68 mmoles), terc-butóxido de potasio (2,070 g, 9,76 mmoles) y BINAP (0,243 g, 0,390 mmoles) a rt. La mezcla de reacción se desgasificó durante 5 min antes de la adición de Pd2(dba)3 (0,178 g, 0,194 mmoles) a rt. La mezcla de reacción se calentó a 110°C durante 4 h. La mezcla de reacción resultante se enfrió a rt y se vertió en agua (200 ml). La mezcla resultante se extrajo con DCM (3 x 100 ml). La fase orgánica combinada se secó sobre Na2SO4, se filtró y se concentró a presión reducida. El residuo resultante se purificó mediante cromatografía ultrarrápida (MeOH al 3% en DCM) produciendo 2-(isoindolin-2-il)piridin-4-amina (0,700 g, 3,317 mmoles). LCMS: Método C, Rt 1,68 min, MS: ES+ 212,13.
Etapas b-d. El compuesto del título se sintetizó a partir del intermedio anterior utilizando un procedimiento similar al descrito para las etapas b-d del Ejemplo 194. LCMS: Método A, RT 3,83 min, MS: ES+ 363,08; RMN H1 (400 MHz, DMSO-de) 5 ppm 8,60 (s, 1 H), 7,92 (d, J = 5,6 Hz, 1 H), 7,39 - 7,42 (m, 2 H), 7,30 - 7,33 (m, 2 H), 6,91 (d, J = 1,2 Hz, 1 H), 6,82 - 6,84 (m, 1 H), 4,85 - 4,93 (m, 1 H), 4,68 (s, 4 H), 3,50 - 3,58 (m, 2 H), 3,36 - 3,42 (m, 1 H), 3,30 - 3,32 (m, 1 H), 2,90 (s, 3 H), 1,95 - 2,07 (m, 2 H).
Ejemplo 232 (R)-N-(1-cianopirrolidin-3-il)-3-fluoro-1-metil-5-(1-metil-1H-pirazol-4-il)-1H-indol-2-carboxamida

Etapa a. A una solución de 5-bromo-1 H-indol-2-carboxilato de etilo (Número CAS 16732-70-0) (0,50 g, 1,86 mmoles) en DMF (5 ml) se le añadieron K2CO3 (0,521 g, 3,77 mmoles) y yoduro de metilo (0,536 g, 3,77 mmoles) a rt. La mezcla de reacción se calentó a 100°C durante 16 h. La mezcla de reacción resultante se enfrió a rt y se vertió en agua (50
ml). La mezcla resultante se extrajo con EtOAc (3 x 20 ml). La fase orgánica combinada se secó sobre Na2SÜ4, se filtró y se concentró a presión reducida produciendo 5-bromo-1-metil-1H-indol-2-carboxilato de etilo (0,426 g, 1,52 mmoles). LCMS: Método C, RT 2,83 min, MS: ES+ 282,10, 284,10. Esta sustancia se utilizó directamente para la siguiente etapa sin purificación adicional.
Etapa b. A una solución de 5-bromo-1 -metil-1 H-indol-2-carboxilato de etilo (0,200 g, 0,71 mmoles) en 1,2-dicloroetano (10 ml) se le añadió triflato de 1-fluoro-2,4,6-trimetilpiridinio (Número CAS 107264-00-6) (0,617 g, 2,13 mmoles) a rt. La mezcla de reacción se calentó a 100°C durante 16 h. La mezcla de reacción resultante se enfrió a rt y se vertió en agua (30 ml). La mezcla resultante se extrajo con EtOAc (3 x 10 ml). La fase orgánica combinada se lavó con salmuera (2 x 20 ml), se secó sobre Na2SÜ4, se filtró y se concentró a presión reducida. El residuo obtenido se purificó mediante cromatografía en columna (EtOAc al 2% en hexano) produciendo 5-bromo-3-fluoro-1 -metil-1 H-indol-2-carboxilato de etilo (0,135 g, 0,450 mmoles). LCMS: Método C, RT 2,96 min, MS: ES+ 300,20, 302,20.
Etapas c-g. El compuesto del título se sintetizó a partir del intermedio anterior utilizando un procedimiento similar al descrito para el Ejemplo 2. LCMS: Método A, RT 3,70 min, MS: ES+ 367,07; RMN H1 (400 MHz, DMSO-d6) ó ppm 8,64 (d, J = 6,0 Hz, 1 H), 8,17 (s, 1 H), 7,91 (s, 1 H), 7,79 (s, 1 H), 7,57 - 7,59 (m, 2 H), 4,51 - 4,52 (m, 1 H), 3,86 (s, 3 H), 3,83 (s, 3 H), 3,64 - 3,75 (m, 1 H), 3,39 - 3,56 (m, 3 H), 2,13 - 2,18 (m, 1 H), 1,94 - 1,99 (m, 1 H).
Ejemplo 233 (R)-N-(1-cianopirrolidin-3-il)-7-(1-metil-1H-pirazol-4-il)imidazo[1,5-a]piridin-3-carboxamida

Etapa a. A una solución de 4-bromopiridin-2-carbonitrilo (Número CAS 62150-45-2) (1,50 g, 8,20 mmoles) en 1,4-dioxano:agua (9:1, 20 ml) se le añadieron 1-metil-4-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)-1H-pirazol (1,870 g, 8,99 mmoles) y Cs2CO3 (8,010 g, 24,57 mmoles) a rt. La mezcla de reacción se desgasificó durante 10 min antes de la adición de PdCl2(dppf) (0,598 g, 0,816 mmoles). La mezcla de reacción se calentó a 80°C durante 1 h. La mezcla de reacción resultante se enfrió a rt y se vertió en agua (40 ml). La mezcla resultante se extrajo con EtOAc (3 x 75 ml). La fase orgánica combinada se lavó con salmuera (25 ml), se secó sobre Na2SO4, se filtró y se concentró a presión reducida. El residuo resultante se trituró con n-pentano (2 x 5 ml) y se secó a alto vacío produciendo 4-(1 -metil-1 H-pirazol-4-il)picolinonitrilo (2,090 g, 11,36 mmoles). LCMS: Método C, RT 1,66 min, MS: ES+ 185,19. Esta sustancia se utilizó directamente para la siguiente etapa sin purificación adicional.
Etapa b. A una solución de 4-(1 -metil-1 H-pirazol-4-il)picolinonitrilo (0,900 g, 4,891 mmoles) en THF (40 ml) se le añadió una solución 1 M de LiAlH4 en THF (4,88 ml, 4,891 mmoles) a -5°C. La mezcla de reacción se agitó a rt durante 1 h. La mezcla de reacción se diluyó con THF (50 ml), se trató con Na2SO4-10H2O (15,00 g) y se agitó durante 20 min. La mezcla de reacción resultante se filtró y se lavó con DCM (50 ml). El producto filtrado combinado se concentró a presión reducida. Al residuo obtenido se le añadió HCl 4 M en una solución de 1,4-dioxano para formar la sal de HCl correspondiente. Los precipitados obtenidos se recogieron mediante filtración bajo atmósfera de nitrógeno y se secaron bajo alto vacío produciendo la sal de HCl de (4-(1-metil-1H-pirazol-4-il)piridin-2-il)metanamina (1,28 g, cuantitativo). MS: ES+ 189,12. Esta sustancia se utilizó directamente para la siguiente etapa sin purificación adicional. Etapa c. A una solución de sal de HCl de (4-(1 -metil-1 H-pirazol-4-il)piridin-2-il)metanamina (0,500 g, 2,22 mmoles) en DCM (30 ml) se le añadió DIPEA (0,860 g, 6,65 mmoles) a 0°C. Se añadió gota a gota una solución de clorooxoacetato de etilo (0,300 g, 2,20 mmoles) en DCM (30 ml) a la mezcla de reacción a 0°C. La mezcla de reacción se agitó a rt durante 1 h. La mezcla de reacción resultante se vertió en agua (50 ml) y se extrajo con DCM (40 ml). La capa orgánica se lavó con salmuera (20 ml), se secó sobre Na2SO4, se filtró y se concentró a presión reducida produciendo 2-(((4-(1-metil-1H-pirazol-4-il)piridin-2-il)metil)amino)-2-oxoacetato de etilo (0,540 g, 1,87 mmoles). LCMS: Método F, RT 4,49 min, MS: ES+ 289,10. Esta sustancia se utilizó directamente para la siguiente etapa sin purificación adicional. Etapa d. Una mezcla de 2-(((4-(1-metil-1H-pirazol-4-il)piridin-2-il)metil)amino)-2-oxoacetato de etilo (0,540 g, 1,87 mmoles) y P2O5 (1,320 g, 9,295 mmoles) en POCh (11 ml) se calentó a 80°C durante 48 h. La mezcla de reacción resultante se enfrió a temperatura ambiente y se alcalinizó con solución una solución acuosa saturada de Na2CO3. La mezcla obtenida se extrajo con EtOAc (3 x 50 ml). La fase orgánica combinada se lavó con salmuera (40 ml), se secó sobre Na2SO4, se filtró y se concentró a presión reducida. El residuo obtenido se purificó mediante cromatografía ultrarrápida (EtOAc al 80% en hexano) produciendo 7-(1-metil-1H-pirazol-4-il)imidazo[1,5-a]piridin-3-carboxilato de etilo (0,070 g, 0,259 mmoles). LCMS: Método C, RT 1,78 min, MS: ES+ 271,40.
Etapas e-h. El compuesto del título se sintetizó a partir del intermedio anterior utilizando un procedimiento similar al
descrito para las etapas b-e del Ejemplo 2. LCMS: Método A, RT 3,22 min, MS: ES+ 336,07; RMN H1 (400 MHz, DMSO-de) 5 ppm 9,32 (d, J = 7,2 Hz, 1 H), 8,80 (d, J = 7,2 Hz, 1 H), 8,28 (s, 1 H), 8,01 (s, 1 H), 7,93 (s, 1 H), 7,51 (s, 1 H), 7,26 (d, J = 7,2 Hz, 1 H), 4,52 - 4,57 (m, 1 H), 3,88 (s, 3 H), 3,55 - 3,64 (m, 2 H), 3,38 - 3,51 (m, 2 H), 2,02 - 2,14 (m, 2 H).
Ejemplo 234 (R)-N-( 1 -cianopirrolidin-3-il)-3-( 1 -fen il-1 H-pirazol-3-il)azetidin-1 -carboxamida

Etapa a. A una solución de ácido 1-(terc-butoxicarbonil)azetidin-3-carboxílico (Número CAS 142253-55-2) (3,00 g, 14,92 mmoles) en THF (30 ml) se le añadió CDI (2,70 g, 16,67 mmoles) a 0°C. La mezcla de reacción se agitó a rt durante 2 h. Se añadió una solución de hidrocloruro de N,O-dimetilhidroxilamina (1,90 g, 19,48 mmoles) en MeCN (45 ml) a la mezcla de reacción seguido de la adición de TEA (3,10 ml, 22,39 mmoles). La mezcla de reacción se agitó a rt durante 16 h. La mezcla de reacción resultante se concentró a presión reducida y se vertió en agua (200 ml). La mezcla obtenida se extrajo con EtOAc (3 x 50 ml). La capa orgánica combinada se lavó con una solución de ácido cítrico (100 ml) y salmuera (100 ml). La fase orgánica combinada se secó sobre Na2SO4, se filtró y se concentró a presión reducida produciendo 3-(metoxi(metil)carbamoil)azetidin-1-carboxilato de terc-butilo (3,40 g, 13,93 mmoles). LCMS: Método C, RT 1,96 min, MS: ES+ 245,20. Esta sustancia se utilizó directamente para la siguiente etapa sin purificación adicional.
Etapa b. Una mezcla de CH3MgBr (3 M en éter dietílico) (9,25 ml, 27,75 mmoles) en tolueno:THF (7: 3, 48 ml) se enfrió a 0°C. Se añadió gota a gota una solución de 3-(metoxi(metil)carbamoil)azetidin-1-carboxilato de terc-butilo (3,40 g, 13,93 mmoles) en THF (20 ml) a la mezcla de reacción a 0°C. La mezcla de reacción se agitó a rt durante 1 h. La mezcla de reacción resultante se sofocó mediante la adición lenta de una solución de ácido cítrico (50 ml). La mezcla de reacción resultante se extrajo con EtOAc (3 x 50 ml). La capa orgánica combinada se lavó con salmuera (2 x 50 ml), se secó sobre Na2SO4, se filtró y se concentró a presión reducida produciendo 3-acetilazetidin-1-carboxilato de terc-butilo (2,150 g, 10,80 mmoles). LCMS: Método C, RT 1,98 min, MS: ES+ 144,10 (M-56). Esta sustancia se utilizó directamente para la siguiente etapa sin purificación adicional.
Etapa c. Una mezcla de 3-acetilazetidin-1-carboxilato de terc-butilo metilo (2,15 g, 10,80 mmoles) en dimetilacetal de N,N-dimetilformamida (16 ml, 120,6 mmoles) se calentó a 110°C durante 16 h. La mezcla de reacción resultante se concentró a presión reducida y el residuo obtenido se purificó mediante cromatografía ultrarrápida (EtOAc al 80-100% en hexano) produciendo (E)-3-(3-(dimetilamino)acriloil)azetidin-1-carboxilato de terc-butilo (1,900 g, 7,48 mmoles). LCMS: Método C, RT 1,91 min, m S: ES+ 255,20.
Etapa d. A una solución de (E)-3-(3-(dimetilamino)acriloil)azetidin-1-carboxilato de terc-butilo (0,500 g, 1,97 mmoles) en EtOH (6 ml) se le añadieron fenilhidrazina (0,280 g, 2,59 mmoles) y ácido acético (0,05 ml). La mezcla de reacción se calentó a 60°C durante 4 h. La mezcla de reacción resultante se enfrió a rt y se concentró a presión reducida. La sustancia bruta obtenido se purificó mediante cromatografía ultrarrápida para obtener 3-(1 -fenil-1 H-pirazol-3-il)azetidin-1-carboxilato de terc-butilo (EtOAc al 47%/hexano) (0,170 g, 0,57 mmoles) y 3-(1 -fenil-1 H-pirazol-5-il)azetidin-1-carboxilato de terc-butilo (EtOAc al 70%/hexano) (0,210 g, 0,70 mmoles). El 3-(1 -fenil-1 H-pirazol-3-il)azetidin-1-carboxilato de terc-butilo obtenido se utilizó para la siguiente etapa. LCMS: Método C, RT 2,58 min, MS: ES 300,30; RMN H1 (400 MHz, DMSO-d6) 5 ppm 8,46 (d, J = 2,4 Hz, 1 H), 7,81 (dd, J = 8,8, 1,2 Hz, 2 H), 7,49 (t, J = 5,6 Hz, 2 H), 7,29 (t, J = 7,6 Hz, 1 H), 6,56 (d, J = 2,4 Hz, 1 H), 4,23 (t, J = 8,0 Hz, 2 H), 3,89 - 3,96 (m, 2 H), 3,85 -3,89 (m, 1 H), 1,40 (s, 9 H).
Etapa e. A una solución de 3-(1 -fenil-1 H-pirazol-3-il)azetidin-1 -carboxilato de terc-butilo (0,270 g, 0,90 mmoles) en 1,4-dioxano (5 ml) se le añadió HCl 4 M en 1,4-dioxano (5 ml) a rt. La mezcla de reacción se agitó a rt durante 3 h. La mezcla de reacción resultante se concentró a presión reducida. El residuo obtenido se evaporó conjuntamente con DCM (3 x 10 ml) y finalmente se secó a alto vacío produciendo sal de HCl de 3-(azetidin-3-il)-1 -fenil-1 H-pirazol (0,270 g, cuantitativo). LCMS: Método C, RT 1,53 min, MS: ES+ 200,30. Esta sustancia se utilizó directamente para la siguiente etapa sin purificación adicional.
Etapas f-h. El compuesto del título se sintetizó a partir del intermedio anterior utilizando un procedimiento similar al descrito para el Ejemplo 89. LCMS: Método B, r T 3,54 min, MS: ES+ 337,22; RMN H1 (400 MHz, DMSO-d6) 5 ppm 8,46 (d, J = 2,4 Hz, 1 H), 7,81 (d, J = 8,0 Hz, 2 H), 7,48 (t, J = 8,0 Hz, 2 H), 7,29 (t, J = 7,6 Hz, 1 H), 6,54 - 6,57 (m, 2 H), 4,13 - 4,21 (m, 3 H), 3,84 - 3,94 (m, 3 H), 3,45 - 3,54 (m, 2 H), 3,35 - 3,41 (m, 1 H), 3,13 - 3,17 (m, 1 H), 1,98 - 2,03
(m, 1 H), 1,77 - 1,82 (m, 1 H).
Ejemplo 235 (R)-N-(1-cianopirrolidin-3-il)-3-(1-(pirazin-2-il)-1H-pirazol-3-il)azetidin-1-carboxamida
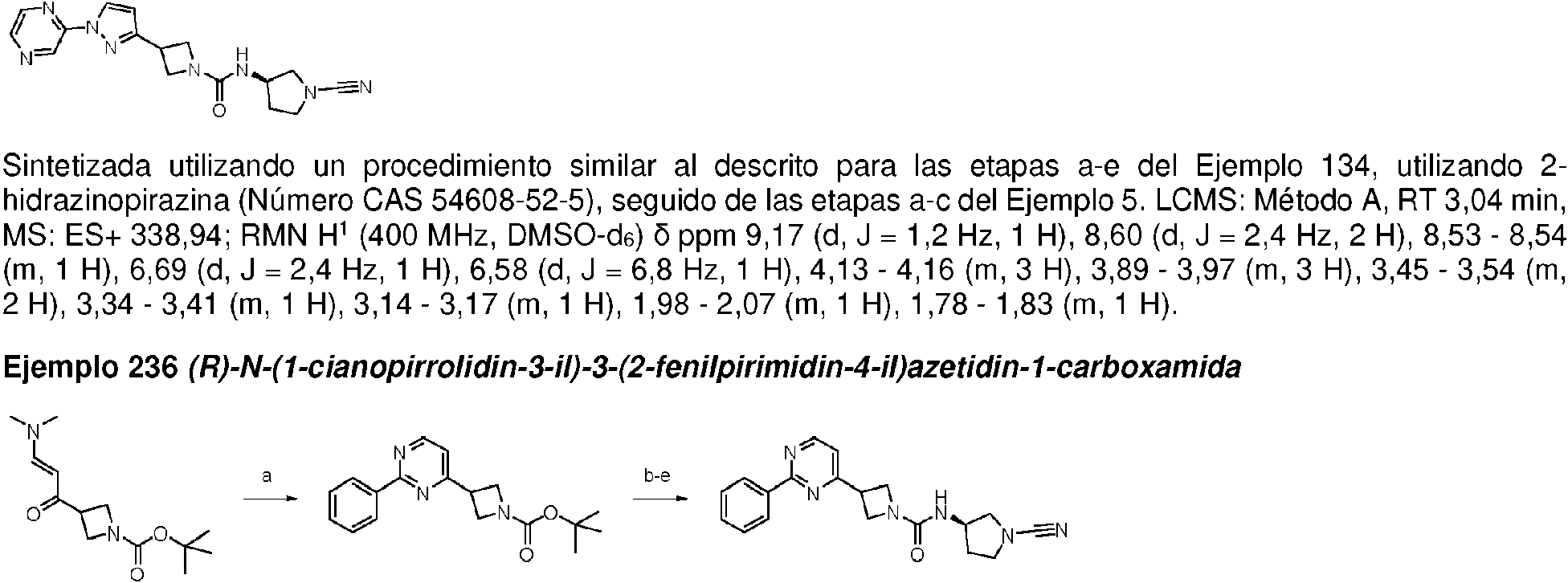
Etapa a. A una solución de (E)-3-(3-(dimetilamino) acriloil)azetidin-1-carboxilato de terc-butilo (descrito en las etapas a-c de Ejemplo 234) (0,900 g, 3,54 mmoles) en MeOH (10 ml) se le añadió NaOMe (0,480 g, 8,89 mmoles) a rt. Se añadió hidrato de hidrocloruro de benzamidina (0,700 g, 4,47 mmoles) a la mezcla de reacción a rt. La mezcla de reacción se calentó a reflujo durante 5 h. La mezcla de reacción resultante se enfrió a rt y se filtró a través de un lecho de celite, se lavó con MeOH (10 ml). El producto filtrado se concentró a presión reducida. El residuo obtenido se purificó mediante cromatografía en columna (EtOAc al 25% en hexano) produciendo 3-(2-fenilpirimidin-4-il)azetidin-1-carboxilato de terc-butilo (0,610 g, 1,96 mmoles). LCMS: Método C, RT 2,55 min, MS: eS+ 312,13; RMN H1 (400 MHz, DMSO-de) 5 ppm 8,83 (d, J = 5,2 Hz, 1 H), 8,42 - 8,44 (m, 2 H), 7,51 - 7,55 (m, 3 H), 7,39 (d, J = 5,2 Hz, 1 H), 4,23 -4,26 (m, 2 H), 4,13 - 4,10 (m, 2 H), 3,96 - 4,01 (m, 1 H), 1,42 (s, 9 H).
Etapas b-e. El compuesto del título se sintetizó utilizando un procedimiento similar al descrito para la etapa e de Ejemplo 134, seguido de las etapas a-c del Ejemplo 5. LCMS: Método A, RT 3,63 min, MS: ES+ 349,04; RMN H1 (400 MHz, DMSO-de) 5 ppm 8,83 - 8,84 (m, 1 H), 8,41 - 8,44 (m, 2 H), 7,52 - 7,56 (m, 3 H), 7,32 - 7,40 (m, 1 H), 6,63 (d, J = 6,4 Hz, 1 H), 4,17 - 4,24 (m, 3 H), 3,98 - 4,10 (m, 3 H), 3,46 - 3,55 (m, 2 H), 3,34 - 3,42 (m, 1 H), 3,15 - 3,17 (m, 1 H), 1,99 - 2,04 (m, 1 H), 1,81 - 1,84 (m, 1 H).
Ejemplo 237 (R)-3-(2-(4-clorofenil)pirimidin-4-il)-N-(1-cianopirrolidin-3-il)azetidin-1-carboxamida

Etapa a. A una solución de 3-hidroxi-3-fenilazetidin-1-carboxilato de terc-butilo (1,00 g, 4,016 mmoles) en DCM (50 ml) se le añadieron bromuro de tetrabutilamonio (0,129 g, 0,400 mmoles) y una solución de NaOH 4 M (40 ml). Se añadió gota a gota bromuro de bencilo (1,43 ml, 12,05 mmoles) a la mezcla de reacción a rt. La mezcla de reacción se calentó a 80°C durante 16 h. La mezcla de reacción resultante se enfrió a rt y se vertió en salmuera (50 ml). La mezcla resultante se extrajo con DCM (50 ml). La capa orgánica se lavó con salmuera (100 ml), se secó sobre Na2SO4, se filtró y se concentró a presión reducida produciendo 3-(benciloxi)-3-fenilazetidin-1 -carboxilato de terc-butilo (1,80 g, cuantitativo). LCMS: Método C, RT 2,88 min, MS: ES+ 340,30. Esta sustancia se utilizó directamente para la siguiente etapa sin purificación adicional.
Etapa b. A una solución de 3-(benciloxi)-3-fenilazetidin-1-carboxilato de terc-butilo (1,80 g, 5,31 mmoles) en DCM (50 ml) se le añadió HCl 4 M en 1,4-dioxano (18 ml) a 0°C. La mezcla de reacción se agitó a rt durante 1 h. La mezcla de reacción resultante se concentró a presión reducida. El residuo obtenido se evaporó conjuntamente con DCM (3 x 50 ml). El material obtenido se trituró con éter dietílico (3 x 40 ml) y finalmente se secó a alto vacío produciendo la sal de HCl de 3-(benciloxi)-3-fenilazetidina (1,17 g, 4,25 mmoles). lCMS: Método C, RT 1,72 min, MS: ES+ 240,23; RMN H1 (400 MHz, DMSO-d6) ó ppm 9,76 (s, 1 H), 9,48 (s, 1 H), 7,45 - 7,53 (m, 5 H), 7,33 - 7,36 (m, 5 H), 4,38 (s, 2 H), 4,30 (s, 2 H), 4,17 (s, 2 H). Esta sustancia se utilizó directamente para la siguiente etapa sin purificación adicional.
Etapas c-e. El compuesto del título se sintetizó a partir del intermedio anterior utilizando un procedimiento similar al descrito para el Ejemplo 89. LCMS: Método A, RT 4,41 min, MS: ES+ 377,06; RMN H1 (400 MHz, DMSO-d6) 5 ppm 7,45 - 7,51 (m, 4 H), 7,26 - 7,40 (m, 6 H), 6,69 (d, J = 6,8 Hz, 1 H), 4,20 (s, 2 H), 4,14 - 4,17 (m, 1 H), 4,13 (s, 4 H), 3,44 - 3,53 (m, 2 H), 3,35 - 3,41 (m, 1 H), 3,13 - 3,16 (m, 1 H), 1,98 - 2,03 (m, 1 H), 1,76 - 1,81 (m, 1 H).
Ejemplo 239 (R)-N-(1-cianopirrolidin-3-il)-1-(4-ciclopropilpirimidin-2-il)indolin-5-carboxamida

Etapa a. A una solución de 2-cloro-4-ciclopropilpirimidina (1,080 g, 7,01 mmoles) en DMF (10 ml) se le añadieron indolin-5-carboxilato de metilo (Número Ca S 339007-88-4) (1,000 g, 5,65 mmoles) y Cs2CO3 (5,510 g, 16,90 mmoles) a rt. La mezcla de reacción se desgasificó durante 30 min antes de la adición de Xantphos (0,320 g, 0,553 mmoles) y Pd2(dba)3 (0,250 g, 0,273 mmoles) a rt. La mezcla de reacción se calentó a 120°C durante 4 h. La mezcla de reacción resultante se enfrió a rt y se vertió en agua helada (100 ml) y se extrajo con EtOAc (3 x 30 ml). La capa orgánica combinada se lavó con agua helada (25 ml), se secó sobre Na2SO4, se filtró y se concentró a presión reducida. El residuo resultante se trituró con éter dietílico (25 ml) y se secó a alto vacío produciendo 2-cloro-4-ciclopropilpirimidina (1,25 g, 4,21 mmoles). LCMS: Método C, RT 2,81 min, MS: ES+ 296,40; RMN H1 (400 MHz, CDCh) 5 ppm 8,31 - 8,35 (m, 2 H), 7,89 (dd, J = 8,8, 8,8 Hz, 1 H), 7,85 (s, 1 H), 6,70 (d, J = 5,2 Hz, 1 H), 4,28 (t, J = 8,8 Hz, 2 H), 3,90 (s, 3 H), 3,21 (t, J = 8,8 Hz, 2 H), 1,94 - 1,98 (m, 1 H), 1,23 - 1,28 (m, 2 H), 1,11 - 1,08 (m, 2 H). Esta sustancia se utilizó directamente para la siguiente etapa sin purificación adicional.
Etapas b-e. El compuesto del título se sintetizó a partir del intermedio anterior utilizando un procedimiento similar al descrito para las etapas b-e del Ejemplo 2. LCMS: Método B, RT 4,13 min, MS: ES+ 375,22; RMN H1 (400 MHz, CDCl3) 5 ppm 8,31 - 8,35 (m, 2 H), 7,60 - 7,64 (m, 2 H), 6,71 (d, J = 5,2 Hz, 1 H), 6,16 (d, J = 6,8 Hz, 1 H), 4,71 - 4,73 (m, 1 H), 4,26 - 4,30 (m, 2 H), 3,75 - 3,80 (m, 1 H), 3,54 - 3,66 (m, 2 H), 3,39 - 3,42 (m, 1 H), 3,19 - 3,24 (m, 2 H),
2,27 - 2,34 (m, 1 H), 1,95 - 2,07 (m, 2 H), 1,22 - 1,27 (m, 2 H), 1,19 - 1,21 (m, 2 H).
Ejemplo 240 (R)-N-(1-cianopirrolidin-3-il)-1-(4-ciclopropilpirimidin-2-il)-N-metilindolin-5-carboxamida
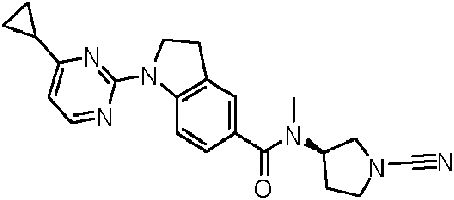
Sintetizada mediante un procedimiento similar al descrito para el Ejemplo 239 utilizando (R)-3-(metilamino)pirrolidin-1-carboxilato de terc-butilo (Número CAS 199336-83-9) en la etapa c. lCm S: Método A, RT 4,48 min, MS: eS+ 389,10; RMN H1 (400 MHz, CDCh) 5 ppm 8,30 - 8,34 (m, 2 H), 7,26 - 7,29 (m, 2 H), 6,67 (d, J = 4,8 Hz, 1 H), 5,01 - 5,03 (m, 1 H), 4,27 (t, J = 8,8 Hz, 2 H), 3,61 - 3,66 (m, 2 H), 3,41 - 3,48 (m, 2 H), 3,21 (t, J = 8,8 Hz, 2 H), 3,01 (s, 3 H), 2,10 -2,19 (m, 2 H), 1,93 - 2,00 (m, 1 H), 1,21 - 1,27 (m, 2 H), 1,08 - 1,11 (m, 2 H).
Ejemplo 241 (3aR,6aR)-5-ciano-N-(3-(2-metilpiridin-4-il)fenil)hexahidropirrolo[3,4-b]pirrol-1(2H)-carboxamida
Etapa a. A una solución de ácido (2-metilpiridin-4-il)borónico (Número CAS 579476-63-4) (0,500 g, 3,65 mmoles) y 3-bromoanilina (0,620 g, 3,60 mmoles) en DMF:agua (8: 2, 10 ml) se le añadió Cs2CO3 (3,570 g, 10,95 mmoles) a rt. La mezcla de reacción se desgasificó durante 30 min antes de la adición de Pd(PPh3)4 (0,420 g, 0,363 mmoles) a rt. La mezcla de reacción se calentó a 90°C durante 16 h. La mezcla de reacción resultante se enfrió a rt y se vertió en agua (100 ml). La mezcla obtenida se extrajo con EtOAc (2 x 100 ml). La fase orgánica combinada se secó sobre Na2SO4, se filtró y se concentró a presión reducida. El residuo obtenido se purificó mediante cromatografía ultrarrápida (EtOAc al 70% en hexano) produciendo 3-(2-metilpiridin-4-il)anilina (0,450 g, 2,445 mmoles). LCMS: Método C, Rt 0,97 min, MS: ES+ 185,09
Etapas b-d. El compuesto del título se sintetizó a partir del intermedio anterior utilizando un procedimiento similar al descrito para el Ejemplo 89, utilizando (3aR,6aR)-5-N-BOC-hexahidro-pirrolo[3,4-b]pirrol (Número CAS 370882-39-6) en la etapa b. LCMS: Método B, RT 2,57 min, m S: ES+ 348,16; RMN H1 (400 MHz, DMSO-d6) 5 ppm 8,49 (d, J = 5,6 Hz, 1 H), 8,43 (s, 1 H), 7,94 (s, 1 H), 7,61 - 7,63 (m, 1 H), 7,49 (s, 1 H), 7,40 - 7,42 (m, 1 H), 7,34 - 7,36 (m, 2 H),
,36 - 4,40 (m, 1 H), 3,52 - 3,60 (m, 4 H), 3,41 - 3,47 (m, 1 H), 3,25 - 3,29 (m, 1 H), 2,92 - 3,97 (m, 1 H), 2,45 (s, 3 H), 1,99 - 2,08 (m, 1 H), 1,82 - 1,86 (m, 1 H).
Los compuestos de la Tabla 11 se sintetizaron utilizando un procedimiento similar al descrito para el Ejemplo 241.
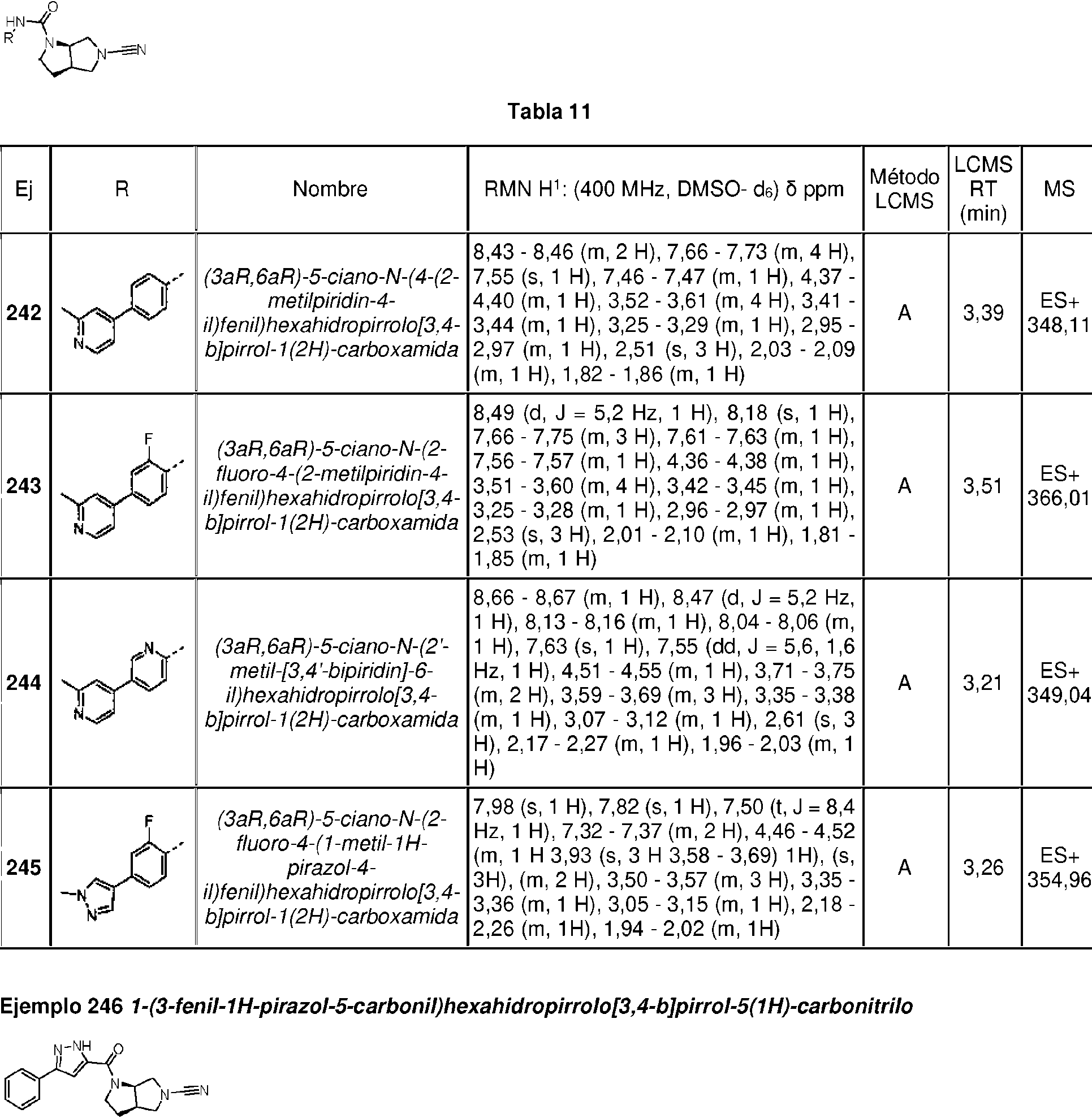
El Ejemplo 52 se sometió adicionalmente a una separación enantiomérica utilizando HPLC preparativa; fase móvil: (A) hexano (B) IPA: MeOH (50:50), columna: CHIRa LpAK IC 250x21,0 mm, 5 pm, caudal: 15 ml/min para proporcionar el compuesto del título. Lc Ms : Método A, RT 3,56 min, MS: ES+ 308,06; HPLC quiral: Método X, r T 14,99 min; RMN H1 (400 MHz, DMSO-de, 80°C) 5 ppm 13,51 (s, 1 H), 7,80 - 7,84 (m, 2 H), 7,44 - 7,48 (m, 2 H), 7,37 - 7,38 (m, 1 H), 7,06 (s, 1 H), 4,61 - 5,10 (m, 1 H), 3,70 - 3,98 (m, 2 H), 3,60 - 3,62 (m, 1 H), 3,57 - 3,59 (m, 1 H), 3,30 - 3,33 (m, 2 H), 3,02 - 3,10 (m, 1 H), 2,06 - 2,08 (m, 1 H), 1,87 - 1,88 (m, 1 H).
Ejemplo 247 (3aR,6aR)-1-(3-fenoxiazetidin-1-carbonil)hexahidropirrolo[3,4-b]pirrol-5(1H)-carbonitrilo
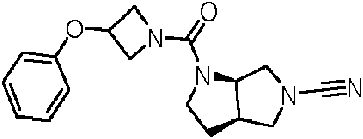
Sintetizado mediante un procedimiento similar al descrito para el Ejemplo 89, utilizando (3aR,6aR)-5-N-BOC-hexahidro-pirrolo[3,4-b]pirrol (Número CAS 370882-39-6) en la etapa a. lCm S: Método B, RT 3,53 min, MS: ES+ 313,34; RMN H1 (400 MHz, DMSO-d6) 5 ppm 7,30 (t, J = 8,4 Hz, 2 H), 6,97 (t, J = 7,6 Hz, 1 H), 6,83 (d, J = 7,6 Hz, 2 H), 4,98 - 5,01 (m, 1 H), 4,40 - 4,44 (m, 1 H), 4,25 - 4,29 (m, 2 H), 3,90 - 3,94 (m, 1 H), 3,78 - 3,82 (m, 1 H), 3,49 - 3,54 (m, 2 H), 3,34 - 3,40 (m, 3 H), 3,18 - 3,22 (m, 1 H), 2,85 - 2,87 (m, 1 H), 1,88 - 1,95 (m, 1 H), 1,75 - 1,76 (m, 1 H). Ejemplo 248 N-(1-cianopiperidin-3-il)-[1,1'-bifenil]-3-carboxamida

Etapa a. A una solución de ácido 3-fenilbenzoico (0,2 mmoles) en DCM (1 ml) se le añadió HATU (0,2 mmoles). La mezcla de reacción se agitó a 0°C durante 20 min. Se añadieron 3-aminopipendin-1-carboxilato de terc-butilo (0,2 mmoles) y DIPEA (0,6 mmoles) a la mezcla de reacción a rt. La mezcla de reacción se agitó a rt durante 16 h. La mezcla resultante se concentró a presión reducida. El residuo resultante se purificó mediante TLC prep. (PE/EtOAc = 1:2) produciendo 3-([1,1'-bifenil]-3-ilcarboxamido)piperidin-1 -carboxilato de terc-butilo. MS: ES+ 381,4.
Etapas b, c. El compuesto del título se sintetizó a partir del intermedio anterior utilizando un procedimiento similar al descrito para las etapas b, c del Ejemplo 1. LCMS: Método D, RT 2,82 min, MS: ES+ 306,2.
Ejemplo 2491-(3-bencilfenil)-3-(1-cianopiperidin-3-il)urea

Etapa a. A una solución de 1-bencil-3-isocianatobenceno (0,2 mmoles) en DCM (1 ml) se le añadió 3-aminopiperidin-1-carboxilato de terc-butilo (0,2 mmoles) y DIPEA (0,6 mmoles). La mezcla de reacción se agitó a rt durante 16 h. La mezcla resultante se concentró a presión reducida. El residuo resultante se purificó mediante TLC prep. (PE/EtOAc = 1: 2) produciendo 3-(3-(3-bencilfenil)ureido)piperidin-1-carboxilato de terc-butilo. MS: ES+ 410,5.
Etapas b, C. El compuesto del título se sintetizó a partir del intermedio anterior utilizando un procedimiento similar al descrito para las etapas b, c del Ejemplo 1, MS: e S+ 335,2.
Ejemplo 2501-(1-cianopiperidin-3-il)-3-(3-fenoxifenil)urea

Sintetizada mediante un procedimiento similar al descrito para el Ejemplo 249. LCMS: Método D, 2,79 min, MS: ES+ 337,2. Los compuestos de la Tabla 12 se sintetizaron utilizando un procedimiento similar al descrito para el Ejemplo 249 utilizando 3-amino-1-pirrolidinacarboxilato de terc-butilo (Número CAS 186550-13-0) en la etapa a.

Tabla 12


Actividad biológica de los compuestos de la invención
Abreviaturas:
TAMRA carboxitetrametilrrodamina
PCR reacción en cadena de la polimerasa
PBS solución salina tamponada con fosfato
EDTA Ácido etilendiaminotetracético
Tris 2-amino-2-(hidroximetil)-1,3-propanodiol
NP-40 Nonidet P-40, octilfenoxipolietoxietanol
BSA albúmina de suero bovino
PNS sistema nervioso periférico
BH3 Dominio 3 de homología de Bcl-2
PTEN homólogo de fosfatasa y tensina
Ensayo de inhibición in vitro de USP30 FP
Ensayo cinético bioquímico de USP30. Las reacciones se realizaron por duplicado en placas de color negro de 384 pocilios (volumen pequeño, Greiner 784076) en un volumen de reacción final de 21 gl. Se diluyó USP30 CD (57-517, núm. 64-0057-050 ubiquigent) en tampón de reacción (Tris 40 mM, pH 7,5, Tween 20 al 0,005%, 0,5 mg/ml de BSA, beta-mercaptoetanol 5 mM) hasta el equivalente de 0, 0,005, 0,01, 0,05, 0,1 y 0,5 gl/pocillo. El tampón se optimizó para temperatura, pH, agente reductor, sales, tiempo de incubación y detergente óptimos. Las reacciones se iniciaron
mediante la adición de 50 nM de péptido marcado con TAMRA ligado a ubiquitina mediante un enlace isopeptídico como sustrato de polarización de fluorescencia. Las reacciones se incubaron a temperatura ambiente y se leyeron cada 2 min durante 120 min. Las lecturas se realizaron en un Pherastar Plus (BMG Labtech). A Excitación 540 nm; A Emisión 590 nm.
Ensayo bioquímico de CI50 de USP30
Se prepararon placas de dilución a 21 veces la concentración final (2100 pM para una concentración final de 100 pM) en DMSO al 50% en una placa de fondo en V de polipropileno de 96 pocillos (Greiner núm. 651201). Una serie de típica dilución de 8 puntos sería 100, 30, 10, 3, 1, 0,3, 0,1, 0,03 pM final. Las reacciones se realizaron por duplicado en placas de color negro de 384 pocillos (volumen pequeño, Greiner 784076) en un volumen de reacción final de 21 pl. Se añadió a la placa 1 pl de DMSO al 50% o compuesto diluido. Se diluyó USP30 en tampón de reacción (Tris 40 mM, pH 7,5, Tween 20 al 0,005%, 0,5 mg/ml de BSA, beta-mercaptoetanol 5 mM) hasta el equivalente de 0,05 pl/pocillo y se añadieron 10 pl de USP30 diluida al compuesto. La enzima y el compuesto se incubaron durante 30 min a temperatura ambiente. Las reacciones se iniciaron mediante la adición de 50 nM de péptido marcado con TAMRA ligado a ubiquitina mediante un enlace isopeptídico como sustrato de polarización de fluorescencia. Las reacciones se leyeron inmediatamente después de la adición del sustrato y después de una incubación de 2 horas a temperatura ambiente. Las lecturas se realizaron en un Pherastar Plus (BMG Labtech). A Excitación 540 nm; A Emisión 590 nm.
Ensayo de inhibición in vitro de USP30 FI
Ensayo cinético de intensidad de fluorescencia bioquímica de USP30. Las reacciones se realizaron por duplicado en placas de color negro de 384 pocillos (volumen pequeño, Greiner 784076) en un volumen de reacción final de 21 pl. Se diluyó USP30 CD (Boston Biochem E-582 o 57-517, núm. 64-0057-050 Ubiquigent) en tampón de reacción (Tris 40 mM, pH 7,5, Tween 20 al 0,005%, 0,5 mg/ml de BSA, beta-mercaptoetanol 5 mM) hasta el equivalente de 0, 0,0005, 0,001, 0,005 y 0,01 pl/pocillo. El tampón se optimizó para temperatura, pH, agente reductor, sales, tiempo de incubación y detergente óptimos. Las reacciones se iniciaron mediante la adición de ubiquitina-rodamina 110 (U-555, Boston Biochem) a una concentración final de 100 nM. Las reacciones se incubaron a temperatura ambiente y se leyeron cada 2 min durante 120 min. Las lecturas se realizaron en un Pherastar Plus (BMG Labtech). A Excitación 487 nm; A Emisión 535 nm.
Ensayo bioquímico de CI 50 de intensidad de fluorescencia de USP30
Se prepararon placas de dilución a 21 veces la concentración final (2100 pM para una concentración final de 100 pM) en DMSO al 50% en una placa de fondo en V de polipropileno de 96 pocillos (Greiner núm. 651201). Una serie típica de dilución de 8 puntos sería 100, 30, 10, 3, 1, 0,3, 0,1,0,03 pM final. Las reacciones se realizaron por duplicado en placas de color negro de 384 pocillos (volumen pequeño, Greiner 784076) en un volumen de reacción final de 21 pl.
Se añadió a la placa 1 pl de DMSO al 50% o compuesto diluido. Se diluyó USP30 en tampón de reacción (Tris 40 mM, pH 7,5, Tween 20 al 0,005%, 0,5 mg/ml de BSA, beta-mercaptoetanol 5 mM) hasta el equivalente de 0,001 pl/pocillo y se añadieron 10 pl de USP30 diluida al compuesto. La enzima y el compuesto se incubaron durante 30 min a temperatura ambiente. Las reacciones se iniciaron mediante la adición de ubiquitina-rodamina 110 (U-555; Boston Biochem) a una concentración final de 100 nM. Las reacciones se leyeron inmediatamente después de la adición del sustrato y después de una incubación de 2 h a temperatura ambiente. Las lecturas se realizaron en un Pherastar Plus (BMG Labtech). A Excitación 487 nm; A Emisión 535 nm.
Actividad de Compuestos ilustrativos en el ensayo bioquímico FP o FI de CI50 de USP30
Rangos
A<0,1 pM;
0,1<B<1 pM;
1pM<C<10 pM;
10pM<D<30 pM





Claims (1)
- REIVINDICACIONES1. Un compuesto de fórmula (II)
 un tautómero del mismo, o una sal farmacéuticamente aceptable de dicho compuesto o tautómero, en donde: m es 0 o 1;cuando m es 1, Z es -C(R6)(R7)-;R2 representa un átomo de hidrógeno, un grupo alquilo C1-C6, o alcoxi C1-C6 opcionalmente sustituidos;R3, R4 y R5 representan cada uno independientemente un átomo de hidrógeno, un grupo alquilo C1-C3, o alcoxi C1-C3 opcionalmente sustituidos;R1, R6, R7 y R8 representan cada uno independientemente un átomo de hidrógeno, un átomo de flúor, ciano, hidroxilo, un grupo alquilo C1-C3, o alcoxi C1-C3 opcionalmente sustituidos;R9 representa un átomo de hidrógeno, un átomo de flúor, ciano, hidroxilo, alquilo C1-C6 , un grupo alcoxi C1-C3, o un anillo de cicloalquilo de 3 a 8 miembros, o forma un anillo heterocíclico con R10;R10 representa un átomo de hidrógeno, alquilo C1-C6 opcionalmente sustituido, o forma un anillo heterocíclico con R9, o forma un anillo monocíclico con R11;Y representa un enlace covalente, -alquileno(C0-C3)-N(R11)-alquileno C0-C3 o alquileno C1-C3 ;R11 representa un átomo de hidrógeno, alquilo C1-C6, o forma un anillo monocíclico con R10;R11 representa un anillo de heteroarilo, heterociclilo o cicloalquilo de 3 a 14 miembros monocíclico sustituido, bicíclico opcionalmente sustituido o tricíclico opcionalmente sustituido;donde R12, cuando está sustituido, está sustituido con uno o más de -Q1-(R13)p, en donde:p es 0 o 1;Q1 representa un átomo de halógeno, ciano, oxo, hidroxilo, un enlace covalente, -alquilen(C0-C3)-NR14-, -alquilen(C0-C3)-NR14R15, -alquilen(C0-C3)-CONR14-, -alquilen(C0-C3)-NR14CO-, -alquilen(C0-C3)-NR14SO2-, un átomo de oxígeno, -alquilen(C0-C3)-CO-, -alquilen(C0-C3)-S(O)q-, -alquilen(C0-C3)-SO2NR14, -alquilen(C0-C3)-SO2NR14R15, -alcoxi C1-C6, haloalcoxi C1-C6, hidroxialquilo C1-C6, -alquilen(C0-C3)-SO2R14, -alquilen(C0-C3)-NR14COR15, -alquilen(C0-C3)-NR14CONR15R16, -alquilen(C0-C3)-NR14SO2NR15R16, -alquilen(C0-C3)-CONR14R15, -alquilen(C0-C3)-CO2R14, -alquilen(C0-C3)-NR14CO2R15, -alquilen(C0-C3)-SO2NR14R15, -alquilen(C0-C3)-C(O)R14 y alquilen(C1-C6)-NR14SO2R15, NO2, alquileno C1-C6, -alquenileno C2-C6 , o un -grupo alquilo C1-C6 opcionalmente sustituido;q es 0, 1 o 2;R14, R15 y R16 representan cada uno independientemente un átomo de hidrógeno, alquilo C1-C6, o un grupo alquileno C1-C6 ;cuando p es 1, R13 representa un anillo de heteroarilo, heterociclilo, arilo de 4 a 10 miembros o cicloalquilo de 3 a 8 miembros; en donde R13 puede estar opcionalmente sustituido con uno o más sustituyentes seleccionados entre halógeno, haloalquilo C1-C6, alcoxi C1-C6, haloalcoxi C1-C6, alquilo C1-C6, alquenilo C2-C6, alquinilo C2-C6, hidroxialquilo C1-C6 , oxo, ciano, -Q2-R17, -Q2-NR17CONR18R19, -Q2-NR17R18, -Q2-COR17, -Q2-NR17COR18, -Q2-NR17CO2R18, -Q2-SO2R17, Q2-CONR17R18, -Q2-CO2R17, -Q2-SO2NR17R18, -Q2-NR17SO2R18, heterociclilo, cicloalquilo, heteroarilo y arilo; en donde dichos anillos de heterociclilo, cicloalquilo, heteroarilo y arilo pueden estar opcionalmente sustituidos con uno o más sustituyentes, que pueden ser iguales o diferentes, seleccionados entre Cl, F, OMe, Me, COCH3, CONH2 , NHC(O)CH(CHs)2 , y CO2CH2CH3 ;Q2 representa un enlace covalente, un átomo de oxígeno, -CO- o un grupo alquileno C1-C6 o alquenileno C2-C6 ;R16, R17, R18 representan cada uno independientemente hidrógeno, alquilo C1-C6, heterociclilo, heteroarilo, arilo o cicloalquilo; yen donde los sustituyentes opcionales de alquilo de R1, R2, R3,R7, R8, R10 y diferentes, se seleccionan entre alcoxi C1-C3, halógeno, hidroxilo, tiol, ciano, amino, amido, nitro y SF5.
un tautómero del mismo, o una sal farmacéuticamente aceptable de dicho compuesto o tautómero, en donde: m es 0 o 1;cuando m es 1, Z es -C(R6)(R7)-;R2 representa un átomo de hidrógeno, un grupo alquilo C1-C6, o alcoxi C1-C6 opcionalmente sustituidos;R3, R4 y R5 representan cada uno independientemente un átomo de hidrógeno, un grupo alquilo C1-C3, o alcoxi C1-C3 opcionalmente sustituidos;R1, R6, R7 y R8 representan cada uno independientemente un átomo de hidrógeno, un átomo de flúor, ciano, hidroxilo, un grupo alquilo C1-C3, o alcoxi C1-C3 opcionalmente sustituidos;R9 representa un átomo de hidrógeno, un átomo de flúor, ciano, hidroxilo, alquilo C1-C6 , un grupo alcoxi C1-C3, o un anillo de cicloalquilo de 3 a 8 miembros, o forma un anillo heterocíclico con R10;R10 representa un átomo de hidrógeno, alquilo C1-C6 opcionalmente sustituido, o forma un anillo heterocíclico con R9, o forma un anillo monocíclico con R11;Y representa un enlace covalente, -alquileno(C0-C3)-N(R11)-alquileno C0-C3 o alquileno C1-C3 ;R11 representa un átomo de hidrógeno, alquilo C1-C6, o forma un anillo monocíclico con R10;R11 representa un anillo de heteroarilo, heterociclilo o cicloalquilo de 3 a 14 miembros monocíclico sustituido, bicíclico opcionalmente sustituido o tricíclico opcionalmente sustituido;donde R12, cuando está sustituido, está sustituido con uno o más de -Q1-(R13)p, en donde:p es 0 o 1;Q1 representa un átomo de halógeno, ciano, oxo, hidroxilo, un enlace covalente, -alquilen(C0-C3)-NR14-, -alquilen(C0-C3)-NR14R15, -alquilen(C0-C3)-CONR14-, -alquilen(C0-C3)-NR14CO-, -alquilen(C0-C3)-NR14SO2-, un átomo de oxígeno, -alquilen(C0-C3)-CO-, -alquilen(C0-C3)-S(O)q-, -alquilen(C0-C3)-SO2NR14, -alquilen(C0-C3)-SO2NR14R15, -alcoxi C1-C6, haloalcoxi C1-C6, hidroxialquilo C1-C6, -alquilen(C0-C3)-SO2R14, -alquilen(C0-C3)-NR14COR15, -alquilen(C0-C3)-NR14CONR15R16, -alquilen(C0-C3)-NR14SO2NR15R16, -alquilen(C0-C3)-CONR14R15, -alquilen(C0-C3)-CO2R14, -alquilen(C0-C3)-NR14CO2R15, -alquilen(C0-C3)-SO2NR14R15, -alquilen(C0-C3)-C(O)R14 y alquilen(C1-C6)-NR14SO2R15, NO2, alquileno C1-C6, -alquenileno C2-C6 , o un -grupo alquilo C1-C6 opcionalmente sustituido;q es 0, 1 o 2;R14, R15 y R16 representan cada uno independientemente un átomo de hidrógeno, alquilo C1-C6, o un grupo alquileno C1-C6 ;cuando p es 1, R13 representa un anillo de heteroarilo, heterociclilo, arilo de 4 a 10 miembros o cicloalquilo de 3 a 8 miembros; en donde R13 puede estar opcionalmente sustituido con uno o más sustituyentes seleccionados entre halógeno, haloalquilo C1-C6, alcoxi C1-C6, haloalcoxi C1-C6, alquilo C1-C6, alquenilo C2-C6, alquinilo C2-C6, hidroxialquilo C1-C6 , oxo, ciano, -Q2-R17, -Q2-NR17CONR18R19, -Q2-NR17R18, -Q2-COR17, -Q2-NR17COR18, -Q2-NR17CO2R18, -Q2-SO2R17, Q2-CONR17R18, -Q2-CO2R17, -Q2-SO2NR17R18, -Q2-NR17SO2R18, heterociclilo, cicloalquilo, heteroarilo y arilo; en donde dichos anillos de heterociclilo, cicloalquilo, heteroarilo y arilo pueden estar opcionalmente sustituidos con uno o más sustituyentes, que pueden ser iguales o diferentes, seleccionados entre Cl, F, OMe, Me, COCH3, CONH2 , NHC(O)CH(CHs)2 , y CO2CH2CH3 ;Q2 representa un enlace covalente, un átomo de oxígeno, -CO- o un grupo alquileno C1-C6 o alquenileno C2-C6 ;R16, R17, R18 representan cada uno independientemente hidrógeno, alquilo C1-C6, heterociclilo, heteroarilo, arilo o cicloalquilo; yen donde los sustituyentes opcionales de alquilo de R1, R2, R3,R7, R8, R10 y diferentes, se seleccionan entre alcoxi C1-C3, halógeno, hidroxilo, tiol, ciano, amino, amido, nitro y SF5. 2. Un compuesto según la reivindicación 1, en donde el anillo de heteroarilo, heterociclilo o cicloalquilo de R12 se selecciona del grupo que consiste en pirrolidinilo, tiazolilo, piridinilo, dihidropiridinilo, isoxazolilo, oxazolilo, imidazolilo, pirazolilo, piridazinilo, pirimidinilo, indolilo, benzimidazolilo, quinolinilo, azetidinilo, indazolilo, pirazolopiridinilo, imidazopiridinilo, indolinilo, piperazinilo, morfolinilo, diazepanilo, tetrahidro-2H-pirido[3,4-b]indolilo, benzomorfolinilo y pirrolopiridinilo.3. Un compuesto según la reivindicación 1 o la reivindicación 2, en donde el anillo de heterociclilo de R12 es un anillo que contiene nitrógeno seleccionado entre azetidinilo y pirrolidinilo, e Y es un enlace covalente que está anclado al nitrógeno del anillo de azetidinilo o pirrolidinilo.4. Un compuesto según una cualquiera de las reivindicaciones 1 a 3, en donde el anillo de heteroarilo, heterociclilo, arilo o cicloalquilo de R13 se selecciona entre fenilo, piridinilo, pirazolilo, imidazolilo, isoxazolilo, morfolinilo, piperidinilo, piperazinilo, quinolinilo, pirrolidinilo, benzopirazolilo, isoindolinilo, tetrahidroquinolinilo, homopiperazinilo, pirimidinilo, imidazopirimidinilo, imidazopiridinilo, indazolilo, pirrolopiridinilo, isoxazolilo, benzimidazolilo, piridazinilo, ciclopropilo, pirazolopirimidinilo, pirrolopirimidinilo, dihidroisoquinolinilo e imidazopirazinilo.5. Un compuesto de fórmula (II)
2. Un compuesto según la reivindicación 1, en donde el anillo de heteroarilo, heterociclilo o cicloalquilo de R12 se selecciona del grupo que consiste en pirrolidinilo, tiazolilo, piridinilo, dihidropiridinilo, isoxazolilo, oxazolilo, imidazolilo, pirazolilo, piridazinilo, pirimidinilo, indolilo, benzimidazolilo, quinolinilo, azetidinilo, indazolilo, pirazolopiridinilo, imidazopiridinilo, indolinilo, piperazinilo, morfolinilo, diazepanilo, tetrahidro-2H-pirido[3,4-b]indolilo, benzomorfolinilo y pirrolopiridinilo.3. Un compuesto según la reivindicación 1 o la reivindicación 2, en donde el anillo de heterociclilo de R12 es un anillo que contiene nitrógeno seleccionado entre azetidinilo y pirrolidinilo, e Y es un enlace covalente que está anclado al nitrógeno del anillo de azetidinilo o pirrolidinilo.4. Un compuesto según una cualquiera de las reivindicaciones 1 a 3, en donde el anillo de heteroarilo, heterociclilo, arilo o cicloalquilo de R13 se selecciona entre fenilo, piridinilo, pirazolilo, imidazolilo, isoxazolilo, morfolinilo, piperidinilo, piperazinilo, quinolinilo, pirrolidinilo, benzopirazolilo, isoindolinilo, tetrahidroquinolinilo, homopiperazinilo, pirimidinilo, imidazopirimidinilo, imidazopiridinilo, indazolilo, pirrolopiridinilo, isoxazolilo, benzimidazolilo, piridazinilo, ciclopropilo, pirazolopirimidinilo, pirrolopirimidinilo, dihidroisoquinolinilo e imidazopirazinilo.5. Un compuesto de fórmula (II) un tautómero del mismo, o una sal farmacéuticamente aceptable de dicho compuesto o tautómero, en donde:m es 0 o 1;cuando m es 1, Z es -C(R6)(R7)-;R2 representa un átomo de hidrógeno, un grupo alquilo C1-C6, o alcoxi C1-C6 opcionalmente sustituidos;R3, R4 y R5 representan cada uno independientemente un átomo de hidrógeno, un grupo alquilo C1-C3, o alcoxi C1-C3 opcionalmente sustituidos;R1, R6, R7 y R8 representan cada uno independientemente un átomo de hidrógeno, un átomo de flúor, ciano, hidroxilo, un grupo alquilo C1-C3, o alcoxi C1-C3 opcionalmente sustituidos;R9 representa un átomo de hidrógeno, un átomo de flúor, ciano, hidroxilo, alquilo C1-C6 , un grupo alcoxi C1-C3, o un anillo de cicloalquilo de 3 a 8 miembros, o forma un anillo heterocíclico con R10;R10 representa un átomo de hidrógeno, alquilo C1-C6 opcionalmente sustituido, o forma un anillo heterocíclico con R9, o forma un anillo monocíclico con R11;Y representa un enlace covalente, -alquileno(C0-C3)-N(R11)-alquileno C0-C3 o alquileno C1-C3 ;R11 representa un átomo de hidrógeno, alquilo C1-C6, o forma un anillo monocíclico con R10;R12 representa un anillo de arilo de 3 a 14 miembros monocíclico sustituido, bicíclico opcionalmente sustituido otricíclico opcionalmente sustituido;en donde R12, cuando está sustituido, está sustituido con uno o más de -Q1-(R13)p, en donde:p es 0 o 1;Q1 representa un átomo de halógeno, ciano, oxo, hidroxilo, un enlace covalente, -alquilen(C0-C3)-NR14-, -alquilen(C0-C3)-NR14R15, -alquilen(C0-C3)-CONR14-, -alquilen(C0-C3)-NR14CO-, -alquilen(C0-C3)-NR14SO2-, un átomo de oxígeno, -alquilen(C0-C3)-CO-, -alquilen(C0-C3)-S(O)q-, -alquilen(C0-C3)-SO2NR14R15, -alcoxi C1-C6 , haloalcoxi C1-C6, hidroxialquilo C1-C6, -alquilen(C0-C3)-SO2R14, -alquilen(C0-C3)-NR14COR15, -alquilen(C0-C3)-NR14CONR15R16, -alquilen(C0-C3)-NR14SO2NR15R16, -alquilen(C0-C3)-CONR14R15, -alquilen(C0-C3)-CO2R14, -alquilen(C0-C3)-NR14CO2R15, -alquilen(C0-C3)-SO2NR14R15, -alquilen(C0-C3)-C(O)R14 y alquilen(CrC6)-NR14SO2R15, NO2, alquileno C1-C6, -alquenileno C2-C6, o un -grupo alquilo C1-C6 opcionalmente sustituido;q es 0, 1 o 2;R14, R15 y R16 representan cada uno independientemente un átomo de hidrógeno, alquilo C1-C6, o un grupo alquileno C1-C6 ; cuando p es 1, R13 representa un anillo de heteroarilo de 4 a 10 miembros, heterociclilo o cicloalquilo de 3 a 8 miembros; en donde R13 puede estar opcionalmente sustituido con uno o más sustituyentes seleccionados entre halógeno, haloalquilo C1-C6, alcoxi C1-C6, haloalcoxi C1-C6, alquilo C1-C6, alquenilo C2-C6, alquinilo C2-C6 , hidroxialquilo C1-C6 , oxo, ciano, -Q2-R17, -Q2-NR17CONR18R19, -Q2-NR17R18, -Q2-COR17, -Q2-NR17COR18, -Q2-NR17CO2R18, -Q2-SO2R17, Q2-CONR17R18, -Q2-CO2R17, -Q2-SO2NR17R18, -Q2-NR17SO2R18, heterociclilo, cicloalquilo, heteroarilo y arilo; en donde dichos anillos de heterociclilo, cicloalquilo, heteroarilo y arilo pueden estar opcionalmente sustituidos con uno o más sustituyentes, que pueden ser iguales o diferentes, seleccionados entre Cl, F, OMe, Me, COCH3 , CONH2, NHC(O)CH(CH3)2, y CO2CH2CH3 ;Q2 representa un enlace covalente, un átomo de oxígeno, -CO- o un grupo alquileno C1-C6 o alquenileno C2-C6 ; R16, R17, R18 representan cada uno independientemente hidrógeno, alquilo C1-C6, heterociclilo, heteroarilo, arilo o cicloalquilo;en donde los sustituyentes opcionales de alquilo de R1, R2, R3, R4, R5, R6, R7, R8, R10 y Q1, que pueden ser iguales o diferentes, se seleccionan entre alcoxi C1-C3 , halógeno, hidroxilo, tiol, ciano, amino, amido, nitro y SF5 ; y donde el compuesto no tiene la fórmula:
un tautómero del mismo, o una sal farmacéuticamente aceptable de dicho compuesto o tautómero, en donde:m es 0 o 1;cuando m es 1, Z es -C(R6)(R7)-;R2 representa un átomo de hidrógeno, un grupo alquilo C1-C6, o alcoxi C1-C6 opcionalmente sustituidos;R3, R4 y R5 representan cada uno independientemente un átomo de hidrógeno, un grupo alquilo C1-C3, o alcoxi C1-C3 opcionalmente sustituidos;R1, R6, R7 y R8 representan cada uno independientemente un átomo de hidrógeno, un átomo de flúor, ciano, hidroxilo, un grupo alquilo C1-C3, o alcoxi C1-C3 opcionalmente sustituidos;R9 representa un átomo de hidrógeno, un átomo de flúor, ciano, hidroxilo, alquilo C1-C6 , un grupo alcoxi C1-C3, o un anillo de cicloalquilo de 3 a 8 miembros, o forma un anillo heterocíclico con R10;R10 representa un átomo de hidrógeno, alquilo C1-C6 opcionalmente sustituido, o forma un anillo heterocíclico con R9, o forma un anillo monocíclico con R11;Y representa un enlace covalente, -alquileno(C0-C3)-N(R11)-alquileno C0-C3 o alquileno C1-C3 ;R11 representa un átomo de hidrógeno, alquilo C1-C6, o forma un anillo monocíclico con R10;R12 representa un anillo de arilo de 3 a 14 miembros monocíclico sustituido, bicíclico opcionalmente sustituido otricíclico opcionalmente sustituido;en donde R12, cuando está sustituido, está sustituido con uno o más de -Q1-(R13)p, en donde:p es 0 o 1;Q1 representa un átomo de halógeno, ciano, oxo, hidroxilo, un enlace covalente, -alquilen(C0-C3)-NR14-, -alquilen(C0-C3)-NR14R15, -alquilen(C0-C3)-CONR14-, -alquilen(C0-C3)-NR14CO-, -alquilen(C0-C3)-NR14SO2-, un átomo de oxígeno, -alquilen(C0-C3)-CO-, -alquilen(C0-C3)-S(O)q-, -alquilen(C0-C3)-SO2NR14R15, -alcoxi C1-C6 , haloalcoxi C1-C6, hidroxialquilo C1-C6, -alquilen(C0-C3)-SO2R14, -alquilen(C0-C3)-NR14COR15, -alquilen(C0-C3)-NR14CONR15R16, -alquilen(C0-C3)-NR14SO2NR15R16, -alquilen(C0-C3)-CONR14R15, -alquilen(C0-C3)-CO2R14, -alquilen(C0-C3)-NR14CO2R15, -alquilen(C0-C3)-SO2NR14R15, -alquilen(C0-C3)-C(O)R14 y alquilen(CrC6)-NR14SO2R15, NO2, alquileno C1-C6, -alquenileno C2-C6, o un -grupo alquilo C1-C6 opcionalmente sustituido;q es 0, 1 o 2;R14, R15 y R16 representan cada uno independientemente un átomo de hidrógeno, alquilo C1-C6, o un grupo alquileno C1-C6 ; cuando p es 1, R13 representa un anillo de heteroarilo de 4 a 10 miembros, heterociclilo o cicloalquilo de 3 a 8 miembros; en donde R13 puede estar opcionalmente sustituido con uno o más sustituyentes seleccionados entre halógeno, haloalquilo C1-C6, alcoxi C1-C6, haloalcoxi C1-C6, alquilo C1-C6, alquenilo C2-C6, alquinilo C2-C6 , hidroxialquilo C1-C6 , oxo, ciano, -Q2-R17, -Q2-NR17CONR18R19, -Q2-NR17R18, -Q2-COR17, -Q2-NR17COR18, -Q2-NR17CO2R18, -Q2-SO2R17, Q2-CONR17R18, -Q2-CO2R17, -Q2-SO2NR17R18, -Q2-NR17SO2R18, heterociclilo, cicloalquilo, heteroarilo y arilo; en donde dichos anillos de heterociclilo, cicloalquilo, heteroarilo y arilo pueden estar opcionalmente sustituidos con uno o más sustituyentes, que pueden ser iguales o diferentes, seleccionados entre Cl, F, OMe, Me, COCH3 , CONH2, NHC(O)CH(CH3)2, y CO2CH2CH3 ;Q2 representa un enlace covalente, un átomo de oxígeno, -CO- o un grupo alquileno C1-C6 o alquenileno C2-C6 ; R16, R17, R18 representan cada uno independientemente hidrógeno, alquilo C1-C6, heterociclilo, heteroarilo, arilo o cicloalquilo;en donde los sustituyentes opcionales de alquilo de R1, R2, R3, R4, R5, R6, R7, R8, R10 y Q1, que pueden ser iguales o diferentes, se seleccionan entre alcoxi C1-C3 , halógeno, hidroxilo, tiol, ciano, amino, amido, nitro y SF5 ; y donde el compuesto no tiene la fórmula: 6. Un compuesto según la reivindicación 5, en donde el anillo de arilo de R12 es fenilo.7. Un compuesto según la reivindicación 5 o la reivindicación 6, en donde el anillo de heteroarilo, heterociclilo o cicloalquilo de R13 se selecciona entre piridinilo, pirazolilo, imidazolilo, isoxazolilo, morfolinilo, piperidinilo, piperazinilo, quinolinilo, pirrolidinilo, benzopirazolilo, isoindolinilo, tetrahidroquinolinilo, homopiperazinilo, pirimidinilo, imidazopirimidinilo, imidazopiridinilo, indazolilo, pirrolopiridinilo, isoxazolilo, benzimidazolilo, piridazinilo, ciclopropilo, pirazolopirimidinilo, pirrolopirimidinilo, dihidroisoquinolinilo e imidazopirazinilo.8. Un compuesto según una cualquiera de las reivindicaciones 1 a 7, en donde Q1 se selecciona entre halógeno, ciano, oxo, un enlace covalente, -NR14R15, -NR14CO-, un átomo de oxígeno, alcoxi C1-C6, -NR14-, -NR14COR15, alquilo C1-C6, alquileno C1-C3 , en donde el alquilo y el alcoxi están opcionalmente sustituidos con flúor; y R14 y R15 representan cada uno independientemente un átomo de hidrógeno o alquilo C1-C3.9. Un compuesto de fórmula (II)
6. Un compuesto según la reivindicación 5, en donde el anillo de arilo de R12 es fenilo.7. Un compuesto según la reivindicación 5 o la reivindicación 6, en donde el anillo de heteroarilo, heterociclilo o cicloalquilo de R13 se selecciona entre piridinilo, pirazolilo, imidazolilo, isoxazolilo, morfolinilo, piperidinilo, piperazinilo, quinolinilo, pirrolidinilo, benzopirazolilo, isoindolinilo, tetrahidroquinolinilo, homopiperazinilo, pirimidinilo, imidazopirimidinilo, imidazopiridinilo, indazolilo, pirrolopiridinilo, isoxazolilo, benzimidazolilo, piridazinilo, ciclopropilo, pirazolopirimidinilo, pirrolopirimidinilo, dihidroisoquinolinilo e imidazopirazinilo.8. Un compuesto según una cualquiera de las reivindicaciones 1 a 7, en donde Q1 se selecciona entre halógeno, ciano, oxo, un enlace covalente, -NR14R15, -NR14CO-, un átomo de oxígeno, alcoxi C1-C6, -NR14-, -NR14COR15, alquilo C1-C6, alquileno C1-C3 , en donde el alquilo y el alcoxi están opcionalmente sustituidos con flúor; y R14 y R15 representan cada uno independientemente un átomo de hidrógeno o alquilo C1-C3.9. Un compuesto de fórmula (II)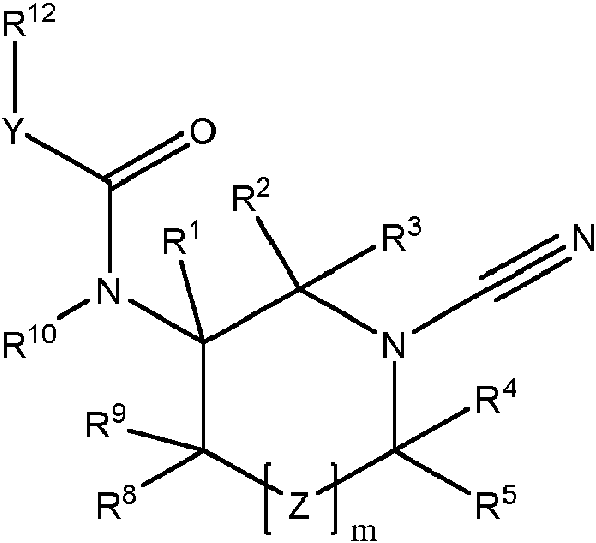 un tautómero del mismo, o una sal farmacéuticamente aceptable de dicho compuesto o tautómero, en donde: m es 0 o 1;cuando m es 1, Z es -C(R6)(R7)-;R2 representa un átomo de hidrógeno, un grupo alquilo C1-C6, o alcoxi C1-C6 opcionalmente sustituidos;R3, R4 y R5 representan cada uno independientemente un átomo de hidrógeno, un grupo alquilo C1-C3 , o alcoxi C1-C3 opcionalmente sustituidos;R1, R6, R7 y R8 representan cada uno independientemente un átomo de hidrógeno, un átomo de flúor, ciano, hidroxilo, un grupo alquilo C1-C3, o alcoxi C1-C3 opcionalmente sustituidos;R9 representa un átomo de hidrógeno, un átomo de flúor, ciano, hidroxilo, alquilo C1-C6 , un grupo alcoxi C1-C3, o un anillo de cicloalquilo de 3 a 8 miembros, o forma un anillo heterocíclico con R10;R10 representa un átomo de hidrógeno, alquilo C1-C6 opcionalmente sustituido, o forma un anillo heterocíclico con R9, o forma un anillo monocíclico con R11;Y representa un enlace covalente, -alquileno(C0-C3)-N(R11)-alquileno C0-C3 o alquileno C1-C3 ;R11 representa un átomo de hidrógeno, alquilo C1-C6, o forma un anillo monocíclico con R10;R12 representa un anillo de arilo monocíclico, bicíclico o tricíclico de 3 a 14 miembros;donde R12 está sustituido con uno o más de -Q1-(R13)p, en donde:p es 1;Q1 representa ciano, oxo, hidroxilo, un enlace covalente, -alquilen(C0-C3)-NR14-, -alquilen(C0-C3)-NR14R15, -alquilen(C0-C3)-CONR14-, -alquilen(C0-C3)-NR14CO-, -alquilen(C0-C3)-NR14SO2-, un átomo de oxígeno, -alquilen(C0-C3)-CO-, -alquilen(C0-C3)-S(O)q-, -alquilen(C0-C3)-SO2NR14, -alquilen(C0-C3)-SO2NR14R15, -alcoxi C1-C6, haloalcoxi C1-C6, hidroxialquilo C1-C6, -alquilen(C0-C3)-SO2R14, -alquilen(C0-C3)-NR14COR15, -alquilen(C0-C3)-NR14CONR15R16, -alquilen(C0-C3)-NR14SO2NR15R16, -alquilen(C0-C3)-CONR14R15, -alquilen(C0-C3)-CO2R14, -alquilen(C0-C3)-NR14CO2R15, -alquilen(C0-C3)-SO2NR14R15, -alquilen(C0-C3)-C(O)R14 y alquilen(C1-C6)-NR14SO2R15, NO2, alquileno C1-C6, -alquenileno C2-C6, o un -grupo alquilo C1-C6 opcionalmente sustituido;q es 0, 1 o 2;R14, R15 y R16 representan cada uno independientemente un átomo de hidrógeno, alquilo C1-C6 , o un grupo alquileno C1-C6 ; cuando p es 1, R13 representa un anillo de arilo de 4 a 10 miembros; en donde R13 está sustituido con uno o más sustituyentes seleccionados entre haloalquilo C1-C6, alcoxi C1-C6, haloalcoxi C1-C6, alquilo C1-C6, alquenilo C2-C6, alquinilo C2-C6 , hidroxialquilo C1-C6 , oxo, ciano, -Q2-R17, -Q2-NR17CONR18R19, -Q2-NR17R18, -Q2-COR17, -Q2-NR17COR18, -Q2-NR17CO2R18, -Q2-SO2R17, Q2-CONR17R18, -Q2-CO2R17, -Q2-SO2NR17R18, -Q2-NR17SO2R18, heterociclilo, cicloalquilo, heteroarilo y arilo; en donde dichos anillos de heterociclilo, cicloalquilo, heteroarilo y arilo pueden estar opcionalmente sustituidos con uno o más sustituyentes, que pueden ser iguales o diferentes, seleccionados entre Cl, F, OMe, Me, COCH3, CONH2, NHC(O)CH(CH3)2, y CO2CH2CH3;Q2 representa un enlace covalente, un átomo de oxígeno, -CO- o un grupo alquileno C1-C6 o alquenileno C2-C6 ; R16, R17, R18 representan cada uno independientemente hidrógeno, alquilo C1-C6, heterociclilo, heteroarilo, arilo o cicloalquilo; yen donde los sustituyentes opcionales de alquilo de R1, R2, R3, R4, R5, R6, R7, R8, R10 y Q1, que pueden ser iguales o diferentes, se seleccionan entre alcoxi C1-C3, halógeno, hidroxilo, tiol, ciano, amino, amido, nitro y SF5.10. Un compuesto según la reivindicación 9, en donde Q1 se selecciona entre ciano, oxo, un enlace covalente, -NR14R15, -NR14CO-, un átomo de oxígeno, alcoxi C1-C6, -NR14-, -NR14COR15, alquilo C1-C6 , alquileno C1-C3, en donde el alquilo y el alcoxi están opcionalmente sustituidos con flúor; y R14 y R15 representan cada uno independientemente un átomo de hidrógeno o alquilo C1-C3.11. Un compuesto según una cualquiera de las reivindicaciones 1 a 10, en donde m es 0.12. Un compuesto según cualquiera de las reivindicaciones 1 a 11, en donde R10 representa un átomo de hidrógeno, alquilo C1-C6 opcionalmente sustituido, o forma un anillo heterocíclico opcionalmente sustituido con R9, o forma un anillo monocíclico de 5 o 6 miembros opcionalmente sustituido con R11-13. Un compuesto según una cualquiera de las reivindicaciones 1 a 12, en donde R1, R3, R4, R5 y R6, R7 y R8 si están presentes, representan cada uno independientemente un átomo de hidrógeno, y en donde R2 representa hidrógeno o metilo.14. Un compuesto según cualquiera de las reivindicaciones 1 a 13, en donde R9 representa hidrógeno, átomo de flúor, hidroxilo, metilo, metoxi, ciclopropilo o forma un anillo heterocíclico de 5 miembros con R1a15. Un compuesto según una cualquiera de las reivindicaciones 1 a 14, en donde R10 forma un anillo heterocíclico de 5 miembros con R916. Un compuesto según una cualquiera de las reivindicaciones 1 a 15, que tiene la estructura de fórmula (III)
un tautómero del mismo, o una sal farmacéuticamente aceptable de dicho compuesto o tautómero, en donde: m es 0 o 1;cuando m es 1, Z es -C(R6)(R7)-;R2 representa un átomo de hidrógeno, un grupo alquilo C1-C6, o alcoxi C1-C6 opcionalmente sustituidos;R3, R4 y R5 representan cada uno independientemente un átomo de hidrógeno, un grupo alquilo C1-C3 , o alcoxi C1-C3 opcionalmente sustituidos;R1, R6, R7 y R8 representan cada uno independientemente un átomo de hidrógeno, un átomo de flúor, ciano, hidroxilo, un grupo alquilo C1-C3, o alcoxi C1-C3 opcionalmente sustituidos;R9 representa un átomo de hidrógeno, un átomo de flúor, ciano, hidroxilo, alquilo C1-C6 , un grupo alcoxi C1-C3, o un anillo de cicloalquilo de 3 a 8 miembros, o forma un anillo heterocíclico con R10;R10 representa un átomo de hidrógeno, alquilo C1-C6 opcionalmente sustituido, o forma un anillo heterocíclico con R9, o forma un anillo monocíclico con R11;Y representa un enlace covalente, -alquileno(C0-C3)-N(R11)-alquileno C0-C3 o alquileno C1-C3 ;R11 representa un átomo de hidrógeno, alquilo C1-C6, o forma un anillo monocíclico con R10;R12 representa un anillo de arilo monocíclico, bicíclico o tricíclico de 3 a 14 miembros;donde R12 está sustituido con uno o más de -Q1-(R13)p, en donde:p es 1;Q1 representa ciano, oxo, hidroxilo, un enlace covalente, -alquilen(C0-C3)-NR14-, -alquilen(C0-C3)-NR14R15, -alquilen(C0-C3)-CONR14-, -alquilen(C0-C3)-NR14CO-, -alquilen(C0-C3)-NR14SO2-, un átomo de oxígeno, -alquilen(C0-C3)-CO-, -alquilen(C0-C3)-S(O)q-, -alquilen(C0-C3)-SO2NR14, -alquilen(C0-C3)-SO2NR14R15, -alcoxi C1-C6, haloalcoxi C1-C6, hidroxialquilo C1-C6, -alquilen(C0-C3)-SO2R14, -alquilen(C0-C3)-NR14COR15, -alquilen(C0-C3)-NR14CONR15R16, -alquilen(C0-C3)-NR14SO2NR15R16, -alquilen(C0-C3)-CONR14R15, -alquilen(C0-C3)-CO2R14, -alquilen(C0-C3)-NR14CO2R15, -alquilen(C0-C3)-SO2NR14R15, -alquilen(C0-C3)-C(O)R14 y alquilen(C1-C6)-NR14SO2R15, NO2, alquileno C1-C6, -alquenileno C2-C6, o un -grupo alquilo C1-C6 opcionalmente sustituido;q es 0, 1 o 2;R14, R15 y R16 representan cada uno independientemente un átomo de hidrógeno, alquilo C1-C6 , o un grupo alquileno C1-C6 ; cuando p es 1, R13 representa un anillo de arilo de 4 a 10 miembros; en donde R13 está sustituido con uno o más sustituyentes seleccionados entre haloalquilo C1-C6, alcoxi C1-C6, haloalcoxi C1-C6, alquilo C1-C6, alquenilo C2-C6, alquinilo C2-C6 , hidroxialquilo C1-C6 , oxo, ciano, -Q2-R17, -Q2-NR17CONR18R19, -Q2-NR17R18, -Q2-COR17, -Q2-NR17COR18, -Q2-NR17CO2R18, -Q2-SO2R17, Q2-CONR17R18, -Q2-CO2R17, -Q2-SO2NR17R18, -Q2-NR17SO2R18, heterociclilo, cicloalquilo, heteroarilo y arilo; en donde dichos anillos de heterociclilo, cicloalquilo, heteroarilo y arilo pueden estar opcionalmente sustituidos con uno o más sustituyentes, que pueden ser iguales o diferentes, seleccionados entre Cl, F, OMe, Me, COCH3, CONH2, NHC(O)CH(CH3)2, y CO2CH2CH3;Q2 representa un enlace covalente, un átomo de oxígeno, -CO- o un grupo alquileno C1-C6 o alquenileno C2-C6 ; R16, R17, R18 representan cada uno independientemente hidrógeno, alquilo C1-C6, heterociclilo, heteroarilo, arilo o cicloalquilo; yen donde los sustituyentes opcionales de alquilo de R1, R2, R3, R4, R5, R6, R7, R8, R10 y Q1, que pueden ser iguales o diferentes, se seleccionan entre alcoxi C1-C3, halógeno, hidroxilo, tiol, ciano, amino, amido, nitro y SF5.10. Un compuesto según la reivindicación 9, en donde Q1 se selecciona entre ciano, oxo, un enlace covalente, -NR14R15, -NR14CO-, un átomo de oxígeno, alcoxi C1-C6, -NR14-, -NR14COR15, alquilo C1-C6 , alquileno C1-C3, en donde el alquilo y el alcoxi están opcionalmente sustituidos con flúor; y R14 y R15 representan cada uno independientemente un átomo de hidrógeno o alquilo C1-C3.11. Un compuesto según una cualquiera de las reivindicaciones 1 a 10, en donde m es 0.12. Un compuesto según cualquiera de las reivindicaciones 1 a 11, en donde R10 representa un átomo de hidrógeno, alquilo C1-C6 opcionalmente sustituido, o forma un anillo heterocíclico opcionalmente sustituido con R9, o forma un anillo monocíclico de 5 o 6 miembros opcionalmente sustituido con R11-13. Un compuesto según una cualquiera de las reivindicaciones 1 a 12, en donde R1, R3, R4, R5 y R6, R7 y R8 si están presentes, representan cada uno independientemente un átomo de hidrógeno, y en donde R2 representa hidrógeno o metilo.14. Un compuesto según cualquiera de las reivindicaciones 1 a 13, en donde R9 representa hidrógeno, átomo de flúor, hidroxilo, metilo, metoxi, ciclopropilo o forma un anillo heterocíclico de 5 miembros con R1a15. Un compuesto según una cualquiera de las reivindicaciones 1 a 14, en donde R10 forma un anillo heterocíclico de 5 miembros con R916. Un compuesto según una cualquiera de las reivindicaciones 1 a 15, que tiene la estructura de fórmula (III) un tautómero del mismo, o una sal farmacéuticamente aceptable de dicho compuesto o tautómero, en donde m, Z, R1, R2, R3, R4, R5, R8, R9, R10, R12 e Y se definen en cualquiera de las reivindicaciones 1 a 15 para los compuestos de fórmula (II).17. Un compuesto de fórmula (II) según la reivindicación 1, que se selecciona del grupo que consiste en:(R)-N-(1-cianopirrolidin-3-il)-5-fenilpicolinamida;6-(bencil(metil)amino)-N-(1-cianopirrolidin-3-il)nicotinamida;(R)-N-(1 -cianopirrolidin-3-il)-3-fenilazetidin-1 -carboxamida;N-(1-cianopirrolidin-3-il)-4-feniltiazol-2-carboxamida;3-(3-clorofenil)-N-(1-cianopirrolidin-3-il)isoxazol-5-carboxamida;N-(1 -cianopirrolidin-3-il)-1 -fenil-1 H-imidazol-4-carboxamida;N-(1-cianopirrolidin-3-il)-1-(2,4-difluorobencil)-5-oxopirrolidin-3-carboxamida;N-(1-cianopirrolidin-3-il)-5-oxo-1-fenilpirrolidin-3-carboxamida;N-(1-cianopirrolidin-3-il)-2-fenilquinolin-4-carboxamida;6-(4-carbamoilpiperidin-1-il)-N-(1-cianopirrolidin-3-il)nicotinamida;N-(1-cianopirrolidin-3-il)-6-(4-(2,4-difluorofenil)piperazin-1-il)nicotinamida;4-(5-((1 -cianopirrolidin-3-il)carbamoil)piridin-2-il)piperazin-1 -carboxilato de etilo;N-(1 -cianopirrolidin-3-il)-6-(2-(piridin-3-il)pirrolidin-1 -il)nicotinam ida;N-(1-cianopirrolidin-3-il)-6-(4-fenoxipiperidin-1-il)nicotinamida;N-(1 -cianopirrolidin-3-il)-6-(4-(piridin-4-il)piperidin-1 -il)nicotinam ida;6-(bencil(metil)amino)-N-(1-cianopirrolidin-3-il)picolinamida;N-(1 -cianopirrolidin-3-il)-6-(3,4-dihidroisoquinolin-2 (1 H)-il)picolinamida;N-(1-cianopirrolidin-3-il)-6-(4-fenoxipiperidin-1-il)picolinamida;N-(1 -cianopirrolidin-3-il)-2-(3,4-dihidroisoquinolin-2 (1 H)-il)isonicotinamida;2-(4-acetil-1,4-diazepan-1 -il)-N-(1 -cianopirrolidin-3-il)isonicotinamida;(R)-N-(1-cianopirrolidin-3-il)-6-fenilpicolinamida;(R)-N-(1-cianopirrolidin-3-il)-4-fenilpicolinamida;(R)-N-(1-cianopirrolidin-3-il)-3-fenilisoxazol-5-carboxamida;(R)-N-(1-cianopirrolidin-3-il)-5-(piridin-4-il)isoxazol-3-carboxamida;(R)-6-(4-clorofenil)-N-(1-cianopirrolidin-3-il)nicotinamida;(R)-2-(2-clorofenil)-N-(1-cianopirrolidin-3-il)tiazol-5-carboxamida;(R) -N-(1 -cianopirrolidin-3-il)-5-(1 -metil-1 H-pirazol-4-il)pirimidin-2-carboxamida;N-((R)-1 -cianopirrolidin-3-il)-3-fenilpirrolidin-1 -carboxamida;(S) -N-(1-cianopirrolidin-3-il)-6-fenilpicolinamida;(R)-1 -(1 -cianopirrolidin-3-il)-3-(imidazo[1,2-a]piridin-2-il)-1 -metilurea;(3aR,6aR)-1-(3-fenil-1 H-pirazol-5-carbonil)hexahidropirrolo[3,4-b]pirrol-5(1 H)-carbonitrilo;(3aR,6aR)-1-(3-fenilisoxazol-5-carbonil)hexahidropirrolo[3,4-b]pirrol-5(1 H)-carbonitrilo;(3aR,6aR)-1 -(1 -fenil-1 H-imidazol-4-carbonil)hexahidropirrolo[3,4-b]pirrol-5(1 H)-carbonitrilo;(3aR,6aR)-1-(3-(4-metoxifenil)-1 H-pirazol-5-carbonil)hexahidropirrolo[3,4-b]pirrol-5(1 H)-carbonitrilo; (3aR,6aR)-1-(3-(4-metoxifenil)isoxazol-5-carbonil)hexahidropirrolo[3,4-b]pirrol-5(1 H)-carbonitrilo; (3aR,6aR)-1-(2-oxo-6-fenil-1,2-dihidropiridin-3-carbonil)hexahidropirrolo[3,4-b]pirrol-5(1 H)-carbonitrilo; (R)-N-(1-cianopirrolidin-3-il)-5-fenilpirimidin-2-carboxamida;(R)-N-(1-cianopirrolidin-3-il)-3-(piridin-4-il)isoxazol-5-carboxamida;(R)-N-(1-cianopirrolidin-3-il)-3-(piridin-3-il)isoxazol-5-carboxamida;(R)-N-(1-cianopirrolidin-3-il)-3-(piridin-2-il)isoxazol-5-carboxamida;N-(1-cianopirrolidin-3-il)-5-fenilpiridazin-3-carboxamida;N-((3S, 4R)-1-ciano-4-metilpirrolidin-3-il)-2-feniltiazol-5-carboxamida;N-((3R,4S)-1-ciano-4-metilpirrolidin-3-il)-2-feniltiazol-5-carboxamida;N-((3S, 4R)-1-ciano-4-metilpirrolidin-3-il)-5-feniltiazol-2-carboxamida;N-((3R,4S)-1-ciano-4-metilpirrolidin-3-il)-5-feniltiazol-2-carboxamida;(R)-N-(1-cianopirrolidin-3-il)-2-(isoindolin-2-il)isonicotinamida;(R)-N-(1 -cianopirrolidin-3-il)-2-(3,4-dihidroisoquinolin-2 (1 H)-il)isonicotinamida;(R)-N-(1 -cianopirrolidin-3-il)-6-(1 -metil-1 H-pirazol-4-il)-1 H-benzo[d]imidazol-2-carboxamida;(R)-N-(1 -cianopirrolidin-3-il)-6-(1 -metil-1 H-pirazol-4-il)-1 H-indol-2-carboxamida;(R)-N-(1 -cianopirrolidin-3-il)-N-metil-6-(1 -metil-1 H-pirazol-4-il)-1 H-benzo[d]imidazol-2-carboxamida; (R)-N-(1-cianopirrolidin-3-il)-N-metil-3-fenoxiazetidin-1-carboxamida;2-(2-clorofenil)-N-((3R,4R)-1-ciano-4-hidroxipirrolidin-3-il)tiazol-5-carboxamida;(R)-N-(1-cianopirrolidin-3-il)-5-(4-metoxifenil)-1 H-pirazol-3-carboxamida;(R)-N-(1-cianopirrolidin-3-il)-5-(piridin-2-il)-1 H-pirazol-3-carboxamida;(R)-N-(1-cianopirrolidin-3-il)-5-(2-metoxifenil)-1 H-pirazol-3-carboxamida;(R)-N-(1-cianopirrolidin-3-il)-5-(2-fluorofenil)-1 H-pirazol-3-carboxamida;(R)-N-(1-cianopirrolidin-3-il)-6-morfolinonicotinamida;(R)-N-(1-cianopirrolidin-3-il)-3-(4-metoxifenil)isoxazol-5-carboxamida;(R)-N-(1 -cianopirrolidin-3-il)-1 H-indazol-3-carboxamida;(R)-N-(1-cianopirrolidin-3-il)-5-fenil-1 H-pirazol-3-carboxamida;(R)-N-(1-cianopirrolidin-3-il)-5-(piridin-3-il)-1 H-pirazol-3-carboxamida;(R)-N-(1 -cianopirrolidin-3-il)-5-(1 -metil-1 H-pirazol-4-il)picolinamida;(R)-N-(1 -cianopirrolidin-3-il)-6-(1 -metil-1 H-pirazol-4-il)nicotinamida;(R)-N-(1 -cianopirrolidin-3-il)-6-(1 -metil-1 H-pirazol-4-il)pirazolo[1,5-a]piridin-3-carboxamida;(R)-6-(3-cianofenil)-N-(1-cianopirrolidin-3-il)imidazo[1,2-a]piridin-2-carboxamida;(R)-6-(4-cianofenil)-N-(1-cianopirrolidin-3-il)imidazo[1,2-a]piridin-2-carboxamida;(R)-N-(1 -cianopirrolidin-3-il)-3-fluoro-5-(1 -metil-1 H-indazol-5-il)picolinamida;(R)-N-(1 -cianopirrolidin-3-il)-3-fluoro-5-(1 -metil-1 H-pirrolo[2,3-b]piridin-5-il)picolinamida;(R)-N-(1 -cianopirrolidin-3-il)-5-(1,3-dimetil-1 H-pirazol-4-il)-3-fluoropicolinamida;(R)-3-cloro-N-(1-cianopirrolidin-3-il)-5-(4-fluorofenil)picolinamida;(R)-N-(1 -cianopirrolidin-3-il)-6-(1 -metil-1 H-pirazol-4-il)imidazo[1,2-a]piridin-2-carboxamida;(R)-N-(1 -cianopirrolidin-3-il)-6-(1,3-dimetil-1 H-pirazol-4-il)imidazo[1,2-a]piridin-2-carboxamida; (R)-N-(1 -cianopirrolidin-3-il)-5-(1 -metil-1 H-pirazol-4-il)-1 H-indazol-3-carboxamida;(R)-N-(1 -cianopirrolidin-3-il)-6-(1 -metil-1 H-pirazol-4-il)-1 H-indazol-3-carboxamida;(R)-N-(1-cianopirrolidin-3-il)-6-(3,5-dimetilisoxazol-4-il)-1 H-indol-2-carboxamida;(R)-N-(1 -cianopirrolidin-3-il)-6-(1,3-dimetil-1 H-pirazol-4-il)-1 H-indol-2-carboxamida;(R)-N-(1 -cianopirrolidin-3-il)-5-(1 -metil-1 H-pirazol-4-il)-1 H-indol-2-carboxamida;(R)-N-(1 -cianopirrolidin-3-il)-5-(1 -etil-1 H-pirazol-4-il)-4-metilpicolinamida;(R)-N-(1-cianopirrolidin-3-il)-3-fenoxiazetidin-1-carboxamida;(R)-3-(1 H-benzo[d]imidazol-2-il)-N-(1 -cianopirrolidin-3-il)azetidin-1 -carboxamida;(R)-N-(1-cianopirrolidin-3-il)-4-fenilpiperazin-1-carboxamida;N-((R)-1-cianopirrolidin-3-il)-2-fenilmorfolin-4-carboxamida;(R)-4-(2-cloro-6-fluorobencil)-N-(1 -cianopirrolidin-3-il)-1,4-diazepano-1 -carboxamida; (R)-4-bencil-N-(1 -cianopirrolidin-3-il)-1,4-diazepano-1 -carboxamida;(R)-N-(1 -cianopirrolidin-3-il)-1,3,4,9-tetrahidro-2H-pirido [3,4-b] indol-2-carboxamida;N-((R)-1-cianopirrolidin-3-il)-2-((2S, 6R)-2,6-dimetilmorfolino)-5-fluoroisonicotinamida;(R)-N-(1-cianopirrolidin-3-il)-5-fluoro-2-(isoindolin-2-il)isonicotinamida;N-((R)-1 -cianopirrolidin-3-il)-5-metil-1 -(1 -feniletil)-1 H-pirazol-3-carboxamida;(R)-N-(1 -cianopirrolidin-3-il)-5-metil-1 -(piridin-2-ilmetil)-1 H-pirazol-3-carboxamida;(R)-N-(1 -cianopirrolidin-3-il)-1 -isobutil-6-(1 -metil-1 H-pirazol-4-il)-1 H-indazol-3-carboxamida;(R)-N-(1 -cianopirrolidin-3-il)-6-(3,5-dimetilisoxazol-4-il)-1 -isobutil-1 H-indazol-3-carboxamida;(R)-N-(1-cianopirrolidin-3-il)-1-(ciclopropilmetil)-6-(3,5-dimetilisoxazol-4-il)-1 H-indazol-3-carboxamida; (R)-N-(1 -cianopirrolidin-3-il)-N-metil-6-(1 -metil-1 H-pirazol-4-il)imidazo[1,2-a]piridin-2-carboxamida; (R)-N-(1 -cianopirrolidin-3-il)-N-metil-5-(1 -metil-1 H-pirazol-4-il)-1 H-indol-2-carboxamida;(R)-N-(1-cianopirrolidin-3-il)-6-(3,5-dimetilisoxazol-4-il)-N-metil-1 H-benzo[d]imidazol-2-carboxamida; (R)-N-(1 -cianopirrolidin-3-il)-7-(1 -metil-1 H-pirazol-4-il)imidazo[1,2-a]piridin-3-carboxamida;(R)-7-(3-cianofenil)-N-(1-cianopirrolidin-3-il)imidazo[1,2-a]piridin-3-carboxamida;(R)-N-(1-cianopirrolidin-3-il)-N-metil-7-(2-metilpiridin-4-il)imidazo[1,2-a]piridin-3-carboxamida;(R)-N-(1-cianopirrolidin-3-il)-N-metil-7-(6-metilpiridin-3-il)imidazo[1,2-a]piridin-3-carboxamida;(R)-N-(1 -cianopirrolidin-3-il)-7-(1,3-dimetil-1 H-pirazol-4-il)-N-metilimidazo[1,2-a]piridin-3-carboxamida; (R)-N-(1-cianopirrolidin-3-il)-7-(2,6-dimetilpiridin-4-il)-N-metilimidazo[1,2-a]piridin-3-carboxamida; (R)-N-(1-cianopirrolidin-3-il)-N-etil-7-(2-metilpiridin-4-il)imidazo[1,2-a]piridin-3-carboxamida;(R)-N-(1-cianopirrolidin-3-il)-7-morfolinoimidazo[1,2-a]piridin-3-carboxamida;(R)-6-(3-cianofenil)-N-(1-cianopirrolidin-3-il)-3-fluoroimidazo[1,2-a]piridin-2-carboxamida;(R)-N-(1 -cianopirrolidin-3-il)-3-fluoro-6-(1 -metil-1 H-pirazol-4-il)imidazo[1,2-a]piridin-2-carboxamida; (R)-N-(1 -cianopirrolidin-3-il)-6-(1 -etil-1 H-pirazol-4-il)-3-fluoroimidazo[1,2-a]piridin-2-carboxamida; (R)-N-(1 -cianopirrolidin-3-il)-6-(1,3-dimetil-1 H-pirazol-4-il)-3-fluoroimidazo[1,2-a]piridin-2-carboxamida; (R)-N-(1-cianopirrolidin-3-il)-6-(3,5-dimetilisoxazol-4-il)-3-fluoroimidazo1,2-a]piridin-2-carboxamida; (R)-N-(1-cianopirrolidin-3-il)-5-(4-fluorofenil)picolinamida;N-((2R, 3R)-1-ciano-2-metilpirrolidin-3-il)-5-(4-fluorofenil)picolinamida;(R)-N-(1 -cianopirrolidin-3-il)-5-(1,3-dimetil-1 H-pirazol-4-il)picolinamida;(R)-N-(1-cianopirrolidin-3-il)-5-(2-metil-6-(trifluorometil)pirimidin-4-il)picolinamida;(R)-N-(1-cianopirrolidin-3-il)-3-fluoro-5-(pirazolo1,5-a]pirimidin-5-il)picolinamida;(R)-N-(1-cianopirrolidin-3-il)-3-fluoro-5-(imidazo[1,2-a]piridin-6-il)picolinamida;(R)-N-(1-cianopirrolidin-3-il)-3-metoxi-3-fenilazetidin-1-carboxamida;(R)-N-(1-cianopirrolidin-3-il)-N-metil-3-fenilazetidin-1-carboxamida;(R)-N-(1-cianopirrolidin-3-il)-3-(4-metoxifenil)azetidin-1-carboxamida;(R)-3-(4-clorofenil)-N-(1-cianopirrolidin-3-il)azetidin-1-carboxamida;(R)-3-(3-clorofenil)-N-(1-cianopirrolidin-3-il)azetidin-1-carboxamida;(3aR,6aR)-1 -(3-fenilazetidin-1 -carbonil)hexahidropirrolo[3,4-b]pirrol-5(1 H)-carbonitrilo; (3aR,6aR)-5-ciano-N-(5-fenilpiridin-2-il)hexahidropirrolo[3,4-b]pirrol-1 (2H)-carboxamida;(R)-N-(1-cianopirrolidin-3-il)-N-etil-3-fenilazetidin-1-carboxamida;(R)-3-(2-oxo-3-(4-feniltiazol-2-il)imidazolidin-1 -il)pirrolidin-1 -carbonitrilo;(R)-3-(2-oxo-3-(4-feniltiazol-2-il)tetrahidropirimidin-1 (2H)-il)pirrolidin-1 -carbonitrilo;(R)-N-(1-cianopirrolidin-3-il)-4-(pirimidin-2-il)-3,4-dihidro-2H-benzo[b][1,4]oxazin-7-carboxamida;(R)-N-(1-cianopirrolidin-3-il)-4-(4-ciclopropilpirimidin-2-il)-3,4-dihidro-2H-benzo[b][1,4]oxazin-7-carboxamida ; (R)-N-(1-cianopirrolidin-3-il)-4-(N-metilisobutiramido)picolinamida;(R)-N-(1-cianopirrolidin-3-il)-[2,3'-bipiridin]-6'-carboxamida;(R)-N-(1-cianopirroldin-3-il)-[2,4'-bipiridin]-2'-carboxamida;(R)-3-(4-clorofenil)-N-(1-cianopirrolidin-3-il)isoxazol-5-carboxamida;(R)-N-(1-cianopirrolidin-3-il)-3-(4-(trifluorometil)fenil)isoxazol-5-carboxamida;(R)-N-(1-cianopirrolidin-3-il)-3-(3,4-dimetoxifenil)isoxazol-5-carboxamida;(R)-N-(1-cianopirrolidin-3-il)-3-(3-metoxifenil)isoxazol-5-carboxamida;N-((R)-1 -cianopirrolidin-3-il)-1 -fenilpirrolidin-3-carboxamida;N-((R)-1 -cianopirrolidin-3-il)-3-(piridin-2-il)pirrolidin-1 -carboxamida;N-((R)-1 -cianopirrolidin-3-il)-3-(1 -metil-1 H-pirazol-4-il)pirrolidin-1 -carboxamida;(R)-N-(1-cianopirrolidin-3-il)-3-(2-metoxipiridin-4-il)-N-metilisoxazol-5-carboxamida;(R)-N-(1 -cianopirrolidin-3-il)-1 -metil-6-(1 -metil-1 H-pirazol-4-il)-1 H-indol-2-carboxamida;(R)-N-(1 -cianopirrolidin-3-il)-1 -metil-5-(1 -metil-1 H-pirazol-4-il)-1 H-indol-2-carboxamida;(R)-1 -(1 -cianopirrolidin-3-il)-3-(2-(isoindolin-2-il)piridin-4-il)-1 -metilurea;(R)-N-(1 -cianopirrolidin-3-il)-3-f luoro-1 -metil-5-(1 -metil-1 H-pirazol-4-il)-1 H-indol-2-carboxamida;(R)-N-(1 -cianopirrolidin-3-il)-7-(1 -metil-1 H-pirazol-4-il)imidazo[1,5-a]piridin-3-carboxamida;(R)-N-(1 -cianopirrolidin-3-il)-3-(1 -fenil-1 H-pirazol-3-il)azetidin-1 -carboxamida;(R)-N-(1 -cianopirrolidin-3-il)-3-(1 -(pirazin-2-il)-1 H-pirazol-3-il)azetidin-1 -carboxamida;(R)-N-(1 -cianopirrolidin-3-il)-3-(2-fenilpirimidin-4-il)azetidin-1 -carboxamida;(R)-3-(2-(4-clorofenil)pirimidin-4-il)-N-(1-cianopirrolidin-3-il)azetidin-1-carboxamida;(R)-3-(benciloxi)-N-(1-cianopirrolidin-3-il)-3-fenilazetidin-1-carboxamida;(R)-N-(1 -cianopirrolidin-3-il)-1 -(4-ciclopropilpirimidin-2-il)indolin-5-carboxam ida;(R)-N-(1-cianopirrolidin-3-il)-1-(4-ciclopropilpirimidin-2-il)-N-metilindolin-5-carboxamida;(3aR,6aR)-5-ciano-N-(2'-metil-[3,4'-bipiridin]-6-il)hexahidropirrolo[3,4-b]pirrol-1 (2H)-carboxamida;1 -(3-fenil-1 H-pirazol-5-carbonil)hexahidropirrolo[3,4-b]pirrol-5(1 H)-carbonitrilo;(3aR,6aR)-1 -(3-fenoxiazetidin-1 -carbonil)hexahidropirrolo[3,4-b]pirrol-5(1 H)-carbonitrilo;(R)-N-(1-cianopirrolidin-3-il)-6-(3,5-dimetilisoxazol-4-il)-N-metil-1 H-indol-2-carboxamida;(R)-N-(1 -cianopirrolidin-3-il)-N-metil-5-(1 -metil-1 H-pirazol-4-il)-1 H-pirrolo[2,3-c]piridin-2- carboxamida;N-((R)-1-cianopirrolidin-3-il)-N-metil-2-fenilmorfolin-4-carboxamida;(R)-N-(1 -cianopirrolidin-3-il)-N-metilindolin-1 -carboxamida;(R)-1 -(1 -cianopirrolidin-3-il)-1 -metil-3-(6-(trifluorometil)piridin-3-il)urea;(R)-3-(5-cloropiridin-2-il)-1 -(1 -cianopirrolidin-3-il)-1 -metilurea;(3aR,6aR)-1 -(indolin-1 -carbonil)hexahidropirrolo[3,4-b]pirrol-5(1 H)-carbonitrilo;(R)-N-(1-cianopirrolidin-3-il)-3-(2-metilpiridin-4-il)isoxazol-5-carboxamida;(R)-N-(1-cianopirrolidin-3-il)-3-(3,4-dimetilfenil)isoxazol-5-carboxamida;(R)-N-(1-cianopirrolidin-3-il)-3-(2,4-difluorofenil)isoxazol-5-carboxamida; y(R)-N-(1-cianopirrolidin-3-il)-N-metil-3-(2-metilpiridin-4-il)isoxazol-5-carboxamida;un tautómero del mismo, o una sal farmacéuticamente aceptable de dicho compuesto o tautómero. 18. Un compuesto según la reivindicación 5, que se selecciona entre:(R)-N-(1 -cianopirrolidin-3-il)-3-metoxi-4-(1 -metil-1 H-pirazol-4-il)benzamida;N-((R)-1-cianopirrolidin-3-il)-4-((2S, 6R)-2,6-dimetilmorfolino)-3-fluorobenzamida;N-(1-cianopirrolidin-3-il)-4-(3,5-dimetilisoxazol-4-il)benzamida;(R)-N-(1-cianopirrolidin-3-il)-4-morfolinobenzamida;(R)-N-(1-cianopirrolidin-3-il)-3-fluoro-4-morfolinobenzamida;(R)-4-(3-cloropiridin-4-il)-N-(1-cianopirrolidin-3-il)benzamida;(R)-4-(3-cloropiridin-4-il)-N-(1-cianopirrolidin-3-il)-3-metoxibenzamida;(R)-N-(1-cianopirrolidin-3-il)-3-metoxi-4-(2-metilpiridin-4-il)benzamida;(R)-N-(1-cianopirrolidin-3-il)-3-metoxi-4-(2-morfolinopiridin-4-il)benzamida;(R)-N-(1 -cianopirrolidin-3-il)-4-fluoro-3-(piridin-4-il)benzam ida;(R)-N-(1 -cianopirrolidin-3-il)-4-fluoro-3-(1 -metil-1 H-pirazol-4-il)benzamida;(R) -N-(1 -cianopirrolidin-3-il)-4-(1 -metil-1 H-pirazol-4-il)benzamida;(S) -N-(1-cianopirrolidin-3-il)-4-(piridin-4-il)benzamida;(R)-4-(3-cloropiridin-4-il)-N-(1-cianopirrolidin-3-il)-N-metilbenzamida;(3aR,6aR)-1-(4-fluoro-3-(piridin-4-il)benzoil)hexahidropirrolo[3,4-b]pirrol-5(1 H)-carbonitrilo;(3aR,6aR)-1 -(4-fluoro-3-(1 -metil-1 H-pirazol-4-il)benzoil)hexahidropirrolo[3,4-b]pirrol-5(1 H)-carbonitrilo; (3aR,6aR)-1-(4-(3-cloropiridin-4-il)-3-metoxibenzoil)hexahidropirrolo[3,4-b]pirrol-5(1 H)-carbonitrilo; (3aR,6aR)-1 -(3-metoxi-4-(1 -metil-1 H-pirazol-4-il)benzoil)hexahidropirrolo[3,4-b]pirrol-5(1 H)-carbonitrilo; (R)-N-(1-cianopirrolidin-3-il)-3-(N-metilisobutiramido)benzamida;(R)-N-(1 -cianopirrolidin-3-il)-2-fluoro-4-(4-metil-1 H-imidazol-1 -il)benzamida;(R)-N-(1 -cianopirrolidin-3-il)-3-fluoro-4-(1 -metil-1 H-pirazol-4-il)benzamida;(R)-N-(1 -cianopirrolidin-3-il)-2-fluoro-4-(1 -metil-1 H-pirazol-4-il)benzamida;(R)-N-(1 -cianopirrolidin-3-il)-2,5-difluoro-4-(1 -metil-1 H-pirazol-4-il)benzamida;(R)-N-(1 -cianopirrolidin-3-il)-4-(1,3-dimetil-1 H-pirazol-4-il)-3-fluorobenzamida;(R)-N-(1 -cianopirrolidin-3-il)-4-(1,3-dimetil-1 H-pirazol-4-il)-2-fluorobenzamida;(R)-N-(1 -cianopirrolidin-3-il)-4-(1 -etil-1 H-pirazol-4-il)-2-fluorobenzamida;(R)-N-(1 -cianopirrolidin-3-il)-2-fluoro-4-(1 -(2-metoxietil)-1 H-pirazol-4-il)benzamida;(R)-N-(1-cianopirrolidin-3-il)-2-fluoro-4-(5-(trifluorometil)-1 H-pirazol-4-il)benzamida;(R)-N-(1 -cianopirrolidin-3-il)-2-fluoro-4-(1 -metil-1 H-indazol-5-il)benzamida;(R)-N-(1 -cianopirroldin-3-il)-3-fluoro-N-metil-4-(1 -metil-1 H-pirazol-4-il)benzamida;(3aR,6aR)-5-ciano-N-(2-fluoro-4-(trifluorometil)fenil)hexahidropirrolo[3,4-b]pirrol-1 (2H)-carboxamida; (R)-N-(1-cianopirrolidin-3-il)-2-fluoro-4-(pirimidin-2-ilamino)benzamida;N-((3R,4S)-1-ciano-4-metilpirrolidin-3-il)-2-fluoro-4-((R)-3-metoxipirrolidin-1-il)benzamida;N-(1 -ciano-3-metilpirrolidin-3-il)-2-fluoro-4-(1 -metil-1 H-pirazol-4-il)benzamida;(R)-N-(1 -cianopirrolidin-3-il)-2-fluoro-3-(1 -metil-1 H-pirazol-4-il)benzamida;(R)-N-(1-cianopirrolidin-3-il)-2-fluoro-4-(2-metilpirimidin-4-il)benzamida;(R)-N-(1 -cianopirrolidin-3-il)-2-fluoro-5-(1 -metil-1 H-pirazol-4-il)benzamida;(R)-N-(1-cianopirrolidin-3-il)-2-fluoro-4-(pirimidin-4-il)benzamida;(R)-N-(1-cianopirroldin-3-il)-2-fluoro-4-(imidazo[1,2-a]pirimidin-6-il)benzamida;(R)-N-(1 -cianopirrolidin-3-il)-2-metoxi-4-(1 -metil-1 H-pirazol-4-il)benzamida(R)-N-(1 -cianopirrolidin-3-il)-3,5-difluoro-4-(1 -metil-1 H-pirazol-4-il)benzamida;(R)-N-(1 -cianopirrolidin-3-il)-2,6-difluoro-4-(1 -metil-1 H-pirazol-4-il)benzamida;(R)-N-(1 -cianopirrolidin-3-il)-2-fluoro-3-metoxi-4-(1 -metil-1 H-pirazol-4-il)benzamida;(R)-N-(1-cianopirrolidin-3-il)-2-fluoro-4-(imidazo[1,2-a]piridin-6-il)benzamida;(R)-N-(1-cianopirrolidin-3-il)-2-fluoro-4-(2-morfolinopiridin-4-il)benzamida;(R)-N-(1 -cianopirrolidin-3-il)-2,3-difluoro-4-(1 -metil-1 H-pirazol-4-il)benzamida;(R)-N-(1-cianopirrolidin-3-il)-3-fluoro-4-(pirimidin-2-ilamino)benzamida;(R)-N-(1-cianopirrolidin-3-il)-2-fluoro-4-(pirrolidin-1-il)benzamida;(R)-N-(1-cianopirrolidin-3-il)-2,5-difluoro-4-morfolinobenzamida;(R)-N-(1-cianopirrolidin-3-il)-2,5-difluoro-4-(pirrolidin-1-il)benzamida;N-((R)-1-cianopirrolidin-3-il)-2-fluoro-4-((R)-3-metoxipirrolidin-1-il)benzamida;(R)-N-(1-cianopirrolidin-3-il)-3-metoxi-4-(pirimidin-2-ilamino)benzamida;(R)-N-(1-cianopirrolidin-3-il)-3-metoxi-4-((4-metilpirimidin-2-il)amino)benzamida;(R)-N-(1-cianopirrolidin-3-il)-2-fluoro-4-((4-metoxipirimidin-2-il)amino)benzamida;(R)-N-(1-cianopirrolidin-3-il)-2-fluoro-4-(piridazin-4-il)benzamida;(R)-N-(1-cianopirrolidin-3-il)-2-fluoro-4-(pirazolo1,5-a]pirimidin-5-il)benzamida;3-cloro-N-((3R,4S)-1-ciano-4-metilpirrolidin-3-il)-4-morfolinobenzamida;N-((3R,4R)-1 -ciano-4-ciclopropilpirrolidin-3-il)-3-fluoro-4-(1 -metil-1 H-pirazol-4-il)benzamida;N-((3S, 4S)-1 -ciano-4-metoxipirrolidin-3-il)-N-metil-4-(1 -metil-1 H-pirazol-4-il)benzamida;(R)-N-(1-cianopirrolidin-3-il)-4-(2,6-dimetilpirimidin-4-il)-2-fluorobenzamida;(R)-N-(1-cianopirrolidin-3-il)-2-fluoro-4-(5-fluoro-2-metilpirimidin-4-il)benzamida;(R)-N-(1 -cianopirrolidin-3-il)-2-fluoro-4-(2-(trifluorometil)pirimidin-4-il)benzam ida;(R)-N-(1-cianopirrolidin-3-il)-2-fluoro-4-(2-metil-3H-pirrolo[2,3-d]pirimidin-4-il)benzamida;(R)-N-(1-cianopirrolidin-3-il)-2-fluoro-4-(imidazo[1,2-a]pirazin-3-il)benzamida;(R)-1 -(1 -cianopirrolidin-3-il)-1 -metil-3-(4-(1 -metil (-1 H-pirazol-4-il)fenil)urea;(R)-1 -(1 -cianopirrolidin-3-il)-1 -metil-3-(4-(trifluorometil)fenil)urea;(3aR,6aR)-N-(4-cloro-2-fluorofenil)-5-cianohexahidropirrolo[3,4-b]pirrol-1 (2H)-carboxamida;(3aR,6aR)-5-ciano-N-(2-fluoro-4-(trifluorometoxi) fenil)hexahidropirrolo[3,4-b]pirrol-1 (2H)-carboxamida; (3aR,6aR)-5-ciano-N-(4-ciano-2-fluorofenil)hexahidropirrolo[3,4-b]pirrol-1 (2H)-carboxamida;(3aR,6aR)-5-ciano-N-(4-ciano-2,5-difluorofenil)hexahidropirrolo[3,4-b]pirrol-1 (2H)-carboxamida;(3aR,6aR)-N-(5-cloro-2-fluorofenil)-5-cianohexahidropirrolo[3,4-b]pirrol-1 (2H)-carboxamida;(3aR,6aR)-5-ciano-N-(2-fluoro-5-(trifluorometil)fenil)hexahidropirrolo[3,4-b]pirrol-1 (2H)-carboxamida;(3aR,6aR)-5-ciano-N-(4-(trifluorometil)fenil)hexahidropirrolo[3,4-b]pirrol-1 (2H)-carboxamida;(R)-1 -(1 -cianopirrolidin-3-il)-1 -etil-3-(4-(trifluorometil)fenil)urea;1 -(1 -cianopirrolidin-3-il)-1 -(2-metoxietil)-3-(4-(trifluorometil)fenil)urea;(R)-N-(1 -cianopirrolidin-3-il)-N-etil-3-fluoro-4-(1 -metil-1 H-pirazol-4-il)benzamida;(R)-3-(3-(3-morfolinofenil)-2-oxoimidazolidin-1 -il)pirrolidin-1 -carbonitrilo;(R)-N-(1-cianopirrolidin-3-il)-4-((4-ciclopropilpirimidin-2-il)amino)-3-fluorobenzamida;(R)-N-(1-cianopirrolidin-3-il)-4-((4-ciclopropilpirimidin-2-il)amino)-2,3-difluorobenzamida;(R)-N-(1 -cianopirrolidin-3-il)-3-fluoro-4-(4-metil-1 H-imidazol-1 -il)benzamida;(R)-N-(1 -cianopirrolidin-3-il)-3-fluoro-N-metil-4-(4-metil-1 H-imidazol-1 -il)benzamida;(3aR,6aR)-5-ciano-N-(3-(2-metilpiridin-4-il)fenil)hexahidropirrolo[3,4-b]pirrol-1 (2H)-carboxamida;(3aR,6aR)-5-ciano-N-(4-(2-metilpiridin-4-il)fenil)hexahidropirrolo[3,4-b]pirrol-1 (2H)-carboxamida;(3aR,6aR)-5-ciano-N-(2-fluoro-4-(2-metilpiridin-4-il)fenil)hexahidropirrolo[3,4-b]pirrol-1 (2H)-carboxamida; (3aR,6aR)-5-ciano-N-(2-fluoro-4-(1-metil-1 H-pirazol-4-il)fenil)hexahidropirrolo[3,4-b]pirrol-1(2H)-carboxamida; 1 -(1 -cianopirrolidin-3-il)-3-(2,4-diclorofenil)urea;1 -(1 -cianopirrolidin-3-il)-3-(4-(trifluorometil)fenil)urea;3-(3-clorofenil)-1 -(1 -cianopirrolidin-3-il)-1 -metilurea;1 -(1 -cianopirrolidin-3-il)-3-(2,4-diclorofenil)-1 -metilurea;1 -(1 -cianopirrolidin-3-il)-1 -metil-3-(4-(trifluorometil)fenil)urea; y(3aR,6aR)-1-(3-cloro-4-morfolinobenzoil)hexahidropirrolo[3,4-b]pirrol-5(1 H)-carbonitrilo;un tautómero del mismo, o una sal farmacéuticamente aceptable de dicho compuesto o tautómero.19. Un compuesto según la reivindicación 9, que se selecciona entre:N-(1 -cianopirrolidin-3-il)-2'-metoxi-[1,1'-bifenil]-4-carboxamida;N-(1-cianopirrolidin-3-il)-4-fenoxibenzamida;2-([1,1'-bifenil]-4-il)-N-(1-cianopirrolidin-3-il)acetamida;(R)-N-(1 -cianopirrolidin-3-il)-[1,1'-bifenil]-4-carboxamida;(3aR,6aR)-1-([1,1'-bifenil]-3-carbonil)hexahidropirrolo[3,4-b]pirrol-5(1 H)-carbonitrilo;N-(1 -cianopirrolidin-3-il)-N-metil-[1,1'-bifenil]-4-carboxamida;N-((3R,4R)-1-ciano-4-fluoropirrolidin-3-il)-[1,1'-bifenil]-4-carboxamida;N-(1-cianopiperidin-3-il)-[1,1'-bifenil]-3-carboxamida;1 -(3-bencilfenil)-3-(1 -cianopiperidin-3-il)urea;1 -(1 -cianopiperidin-3-il)-3-(3-fenoxifenil)urea;1 -(3-bencilfenil)-3-(1 -cianopirrolidin-3-il)urea;1 -([1,1'-bifenil]-4-il)-3-(1 -cianopirrolidin-3-il)urea;1 -(1 -cianopirrolidin-3-il)-3-(3-fenoxifenil)urea;3-(3-bencilfenil)-)- 1 -(1 -cianopirrolidin-3-il)-1 -metilurea;1 -(1 -cianopirrolidin-3-il)-1 -metil-3-(3-fenoxifenil)urea; y3-([1,1'-bifenil]-4-il)-1 -(1 -cianopirrolidin-3-il)-1 -metilurea;un tautómero del mismo, o una sal farmacéuticamente aceptable de dicho compuesto o tautómero.20. Un compuesto seleccionado entre:2'-cloro-N-(1-cianopirrolidin-3-il)-[1,1'-bifenil]-4-carboxamida;3'-cloro-N-(1-cianopirrolidin-3-il)-[1,1'-bifenil]-4-carboxamida; y(R)-N-(1-cianopirrolidin-3-il)-2-fluoro-4-(N-metilfenilsulfonamido)benzamida;un tautómero del mismo, o una sal farmacéuticamente aceptable de dicho compuesto o tautómero.21. Un compuesto según una cualquiera de las reivindicaciones 1 a 20, un tautómero del mismo, o una sal farmacéuticamente aceptable de dicho compuesto o tautómero, para uso como medicamento.22. Un compuesto según una cualquiera de las reivindicaciones 1 a 20, un tautómero del mismo, o una sal farmacéuticamente aceptable de dicho compuesto o tautómero, para su uso en el tratamiento de una afección que implica disfunción mitocondrial o un cáncer.23. Un compuesto para su uso según la reivindicación 22, en donde la afección que implica la disfunción mitocondrial se selecciona entre una enfermedad neurodegenerativa; encefalopatía mitocondrial, acidosis láctica y síndrome de episodios similares a apoplejía; Neuropatía óptica hereditaria de Leber; cáncer; neuropatía, ataxia, retinitis pigmentaria-síndrome de Leigh heredado de la madre; Enfermedad de Danon; diabetes; trastornos metabólicos; cardiopatía isquémica que conduce a infarto de miocardio; enfermedades psiquiátricas, por ejemplo, esquizofrenia; deficiencia múltiple de sulfatasa; mucolipidosis II; mucolipidosis III; mucolipidosis IV; gangliosidosis GM1; lipofuscinosis ceroides neuronales; enfermedad de Alpers; síndrome de Barth; defectos de beta-oxidación; deficiencia de carnitinaacil-carnitina; deficiencia de carnitina; síndromes de deficiencia de creatina; deficiencia de coenzima Q10; deficiencia de complejo I; deficiencia de complejo II; deficiencia de complejo III; deficiencia de complejo IV; deficiencia de complejo V; deficiencia de COX; síndrome de oftalmoplejía externa progresiva crónica; deficiencia de CPT I; deficiencia de CPT II; aciduria glutárica tipo II; síndrome de Kearns-Sayre; acidosis láctica; deficiencia de acil-CoA deshidrogenasa de cadena larga; enfermedad o síndrome de Leigh; miocardiopatía infantil letal; enfermedad de Luft; aciduria glutárica tipo II; deficiencia de acil-CoA deshidrogenasa de cadena media; epilepsia mioclónica y síndrome de fibra roja irregular; citopatía mitocondrial; síndrome de ataxia mitocondrial recesiva; síndrome de depleción del ADN mitocondrial; trastorno mioneurogastrointestinal y encefalopatía; síndrome de Pearson; deficiencia de piruvato deshidrogenasa; deficiencia de piruvato carboxilasa; mutaciones POLG; deficiencia de 3-hidroxiacil-CoA deshidrogenasa de cadena media/corta; y deficiencia de acil-CoA deshidrogenasa de cadena muy larga.24. Un compuesto para su uso según la reivindicación 23, en donde la enfermedad neurodegenerativa se selecciona entre enfermedad de Parkinson, enfermedad de Alzheimer, esclerosis lateral amiotrófica, enfermedad de Huntington, isquemia, accidente cerebrovascular, demencia con cuerpos de Lewy y demencia frontotemporal; y la enfermedad de Parkinson relacionada con mutaciones en a-sinucleína, parkina y PINK1, enfermedad de Parkinson juvenil autosómica recesiva, en donde la parkina está mutada.25. Un compuesto para su uso según la reivindicación 22, en donde el cáncer se selecciona entre cáncer de mama, ovario, próstata, pulmón, riñón, gástrico, colon, testicular, cabeza y cuello, páncreas, cerebro, melanoma, hueso u otros cánceres de tejidos, órganos y cánceres de las células sanguíneas tales como linfomas y leucemias, linfoma, mieloma múltiple, cáncer colorrectal y carcinoma de pulmón de células no pequeñas; cáncer donde las vías apoptóticas están desreguladas; y cáncer en donde las proteínas de la familia BCL-2 están mutadas o expresadas en exceso o insuficientemente.26. Una composición farmacéutica que comprende un compuesto de fórmula (II) como se define en una cualquiera de las reivindicaciones 1 a 20, un tautómero del mismo o una sal farmacéuticamente aceptable de dicho compuesto o tautómero, junto con uno o más excipientes farmacéuticamente aceptables.
un tautómero del mismo, o una sal farmacéuticamente aceptable de dicho compuesto o tautómero, en donde m, Z, R1, R2, R3, R4, R5, R8, R9, R10, R12 e Y se definen en cualquiera de las reivindicaciones 1 a 15 para los compuestos de fórmula (II).17. Un compuesto de fórmula (II) según la reivindicación 1, que se selecciona del grupo que consiste en:(R)-N-(1-cianopirrolidin-3-il)-5-fenilpicolinamida;6-(bencil(metil)amino)-N-(1-cianopirrolidin-3-il)nicotinamida;(R)-N-(1 -cianopirrolidin-3-il)-3-fenilazetidin-1 -carboxamida;N-(1-cianopirrolidin-3-il)-4-feniltiazol-2-carboxamida;3-(3-clorofenil)-N-(1-cianopirrolidin-3-il)isoxazol-5-carboxamida;N-(1 -cianopirrolidin-3-il)-1 -fenil-1 H-imidazol-4-carboxamida;N-(1-cianopirrolidin-3-il)-1-(2,4-difluorobencil)-5-oxopirrolidin-3-carboxamida;N-(1-cianopirrolidin-3-il)-5-oxo-1-fenilpirrolidin-3-carboxamida;N-(1-cianopirrolidin-3-il)-2-fenilquinolin-4-carboxamida;6-(4-carbamoilpiperidin-1-il)-N-(1-cianopirrolidin-3-il)nicotinamida;N-(1-cianopirrolidin-3-il)-6-(4-(2,4-difluorofenil)piperazin-1-il)nicotinamida;4-(5-((1 -cianopirrolidin-3-il)carbamoil)piridin-2-il)piperazin-1 -carboxilato de etilo;N-(1 -cianopirrolidin-3-il)-6-(2-(piridin-3-il)pirrolidin-1 -il)nicotinam ida;N-(1-cianopirrolidin-3-il)-6-(4-fenoxipiperidin-1-il)nicotinamida;N-(1 -cianopirrolidin-3-il)-6-(4-(piridin-4-il)piperidin-1 -il)nicotinam ida;6-(bencil(metil)amino)-N-(1-cianopirrolidin-3-il)picolinamida;N-(1 -cianopirrolidin-3-il)-6-(3,4-dihidroisoquinolin-2 (1 H)-il)picolinamida;N-(1-cianopirrolidin-3-il)-6-(4-fenoxipiperidin-1-il)picolinamida;N-(1 -cianopirrolidin-3-il)-2-(3,4-dihidroisoquinolin-2 (1 H)-il)isonicotinamida;2-(4-acetil-1,4-diazepan-1 -il)-N-(1 -cianopirrolidin-3-il)isonicotinamida;(R)-N-(1-cianopirrolidin-3-il)-6-fenilpicolinamida;(R)-N-(1-cianopirrolidin-3-il)-4-fenilpicolinamida;(R)-N-(1-cianopirrolidin-3-il)-3-fenilisoxazol-5-carboxamida;(R)-N-(1-cianopirrolidin-3-il)-5-(piridin-4-il)isoxazol-3-carboxamida;(R)-6-(4-clorofenil)-N-(1-cianopirrolidin-3-il)nicotinamida;(R)-2-(2-clorofenil)-N-(1-cianopirrolidin-3-il)tiazol-5-carboxamida;(R) -N-(1 -cianopirrolidin-3-il)-5-(1 -metil-1 H-pirazol-4-il)pirimidin-2-carboxamida;N-((R)-1 -cianopirrolidin-3-il)-3-fenilpirrolidin-1 -carboxamida;(S) -N-(1-cianopirrolidin-3-il)-6-fenilpicolinamida;(R)-1 -(1 -cianopirrolidin-3-il)-3-(imidazo[1,2-a]piridin-2-il)-1 -metilurea;(3aR,6aR)-1-(3-fenil-1 H-pirazol-5-carbonil)hexahidropirrolo[3,4-b]pirrol-5(1 H)-carbonitrilo;(3aR,6aR)-1-(3-fenilisoxazol-5-carbonil)hexahidropirrolo[3,4-b]pirrol-5(1 H)-carbonitrilo;(3aR,6aR)-1 -(1 -fenil-1 H-imidazol-4-carbonil)hexahidropirrolo[3,4-b]pirrol-5(1 H)-carbonitrilo;(3aR,6aR)-1-(3-(4-metoxifenil)-1 H-pirazol-5-carbonil)hexahidropirrolo[3,4-b]pirrol-5(1 H)-carbonitrilo; (3aR,6aR)-1-(3-(4-metoxifenil)isoxazol-5-carbonil)hexahidropirrolo[3,4-b]pirrol-5(1 H)-carbonitrilo; (3aR,6aR)-1-(2-oxo-6-fenil-1,2-dihidropiridin-3-carbonil)hexahidropirrolo[3,4-b]pirrol-5(1 H)-carbonitrilo; (R)-N-(1-cianopirrolidin-3-il)-5-fenilpirimidin-2-carboxamida;(R)-N-(1-cianopirrolidin-3-il)-3-(piridin-4-il)isoxazol-5-carboxamida;(R)-N-(1-cianopirrolidin-3-il)-3-(piridin-3-il)isoxazol-5-carboxamida;(R)-N-(1-cianopirrolidin-3-il)-3-(piridin-2-il)isoxazol-5-carboxamida;N-(1-cianopirrolidin-3-il)-5-fenilpiridazin-3-carboxamida;N-((3S, 4R)-1-ciano-4-metilpirrolidin-3-il)-2-feniltiazol-5-carboxamida;N-((3R,4S)-1-ciano-4-metilpirrolidin-3-il)-2-feniltiazol-5-carboxamida;N-((3S, 4R)-1-ciano-4-metilpirrolidin-3-il)-5-feniltiazol-2-carboxamida;N-((3R,4S)-1-ciano-4-metilpirrolidin-3-il)-5-feniltiazol-2-carboxamida;(R)-N-(1-cianopirrolidin-3-il)-2-(isoindolin-2-il)isonicotinamida;(R)-N-(1 -cianopirrolidin-3-il)-2-(3,4-dihidroisoquinolin-2 (1 H)-il)isonicotinamida;(R)-N-(1 -cianopirrolidin-3-il)-6-(1 -metil-1 H-pirazol-4-il)-1 H-benzo[d]imidazol-2-carboxamida;(R)-N-(1 -cianopirrolidin-3-il)-6-(1 -metil-1 H-pirazol-4-il)-1 H-indol-2-carboxamida;(R)-N-(1 -cianopirrolidin-3-il)-N-metil-6-(1 -metil-1 H-pirazol-4-il)-1 H-benzo[d]imidazol-2-carboxamida; (R)-N-(1-cianopirrolidin-3-il)-N-metil-3-fenoxiazetidin-1-carboxamida;2-(2-clorofenil)-N-((3R,4R)-1-ciano-4-hidroxipirrolidin-3-il)tiazol-5-carboxamida;(R)-N-(1-cianopirrolidin-3-il)-5-(4-metoxifenil)-1 H-pirazol-3-carboxamida;(R)-N-(1-cianopirrolidin-3-il)-5-(piridin-2-il)-1 H-pirazol-3-carboxamida;(R)-N-(1-cianopirrolidin-3-il)-5-(2-metoxifenil)-1 H-pirazol-3-carboxamida;(R)-N-(1-cianopirrolidin-3-il)-5-(2-fluorofenil)-1 H-pirazol-3-carboxamida;(R)-N-(1-cianopirrolidin-3-il)-6-morfolinonicotinamida;(R)-N-(1-cianopirrolidin-3-il)-3-(4-metoxifenil)isoxazol-5-carboxamida;(R)-N-(1 -cianopirrolidin-3-il)-1 H-indazol-3-carboxamida;(R)-N-(1-cianopirrolidin-3-il)-5-fenil-1 H-pirazol-3-carboxamida;(R)-N-(1-cianopirrolidin-3-il)-5-(piridin-3-il)-1 H-pirazol-3-carboxamida;(R)-N-(1 -cianopirrolidin-3-il)-5-(1 -metil-1 H-pirazol-4-il)picolinamida;(R)-N-(1 -cianopirrolidin-3-il)-6-(1 -metil-1 H-pirazol-4-il)nicotinamida;(R)-N-(1 -cianopirrolidin-3-il)-6-(1 -metil-1 H-pirazol-4-il)pirazolo[1,5-a]piridin-3-carboxamida;(R)-6-(3-cianofenil)-N-(1-cianopirrolidin-3-il)imidazo[1,2-a]piridin-2-carboxamida;(R)-6-(4-cianofenil)-N-(1-cianopirrolidin-3-il)imidazo[1,2-a]piridin-2-carboxamida;(R)-N-(1 -cianopirrolidin-3-il)-3-fluoro-5-(1 -metil-1 H-indazol-5-il)picolinamida;(R)-N-(1 -cianopirrolidin-3-il)-3-fluoro-5-(1 -metil-1 H-pirrolo[2,3-b]piridin-5-il)picolinamida;(R)-N-(1 -cianopirrolidin-3-il)-5-(1,3-dimetil-1 H-pirazol-4-il)-3-fluoropicolinamida;(R)-3-cloro-N-(1-cianopirrolidin-3-il)-5-(4-fluorofenil)picolinamida;(R)-N-(1 -cianopirrolidin-3-il)-6-(1 -metil-1 H-pirazol-4-il)imidazo[1,2-a]piridin-2-carboxamida;(R)-N-(1 -cianopirrolidin-3-il)-6-(1,3-dimetil-1 H-pirazol-4-il)imidazo[1,2-a]piridin-2-carboxamida; (R)-N-(1 -cianopirrolidin-3-il)-5-(1 -metil-1 H-pirazol-4-il)-1 H-indazol-3-carboxamida;(R)-N-(1 -cianopirrolidin-3-il)-6-(1 -metil-1 H-pirazol-4-il)-1 H-indazol-3-carboxamida;(R)-N-(1-cianopirrolidin-3-il)-6-(3,5-dimetilisoxazol-4-il)-1 H-indol-2-carboxamida;(R)-N-(1 -cianopirrolidin-3-il)-6-(1,3-dimetil-1 H-pirazol-4-il)-1 H-indol-2-carboxamida;(R)-N-(1 -cianopirrolidin-3-il)-5-(1 -metil-1 H-pirazol-4-il)-1 H-indol-2-carboxamida;(R)-N-(1 -cianopirrolidin-3-il)-5-(1 -etil-1 H-pirazol-4-il)-4-metilpicolinamida;(R)-N-(1-cianopirrolidin-3-il)-3-fenoxiazetidin-1-carboxamida;(R)-3-(1 H-benzo[d]imidazol-2-il)-N-(1 -cianopirrolidin-3-il)azetidin-1 -carboxamida;(R)-N-(1-cianopirrolidin-3-il)-4-fenilpiperazin-1-carboxamida;N-((R)-1-cianopirrolidin-3-il)-2-fenilmorfolin-4-carboxamida;(R)-4-(2-cloro-6-fluorobencil)-N-(1 -cianopirrolidin-3-il)-1,4-diazepano-1 -carboxamida; (R)-4-bencil-N-(1 -cianopirrolidin-3-il)-1,4-diazepano-1 -carboxamida;(R)-N-(1 -cianopirrolidin-3-il)-1,3,4,9-tetrahidro-2H-pirido [3,4-b] indol-2-carboxamida;N-((R)-1-cianopirrolidin-3-il)-2-((2S, 6R)-2,6-dimetilmorfolino)-5-fluoroisonicotinamida;(R)-N-(1-cianopirrolidin-3-il)-5-fluoro-2-(isoindolin-2-il)isonicotinamida;N-((R)-1 -cianopirrolidin-3-il)-5-metil-1 -(1 -feniletil)-1 H-pirazol-3-carboxamida;(R)-N-(1 -cianopirrolidin-3-il)-5-metil-1 -(piridin-2-ilmetil)-1 H-pirazol-3-carboxamida;(R)-N-(1 -cianopirrolidin-3-il)-1 -isobutil-6-(1 -metil-1 H-pirazol-4-il)-1 H-indazol-3-carboxamida;(R)-N-(1 -cianopirrolidin-3-il)-6-(3,5-dimetilisoxazol-4-il)-1 -isobutil-1 H-indazol-3-carboxamida;(R)-N-(1-cianopirrolidin-3-il)-1-(ciclopropilmetil)-6-(3,5-dimetilisoxazol-4-il)-1 H-indazol-3-carboxamida; (R)-N-(1 -cianopirrolidin-3-il)-N-metil-6-(1 -metil-1 H-pirazol-4-il)imidazo[1,2-a]piridin-2-carboxamida; (R)-N-(1 -cianopirrolidin-3-il)-N-metil-5-(1 -metil-1 H-pirazol-4-il)-1 H-indol-2-carboxamida;(R)-N-(1-cianopirrolidin-3-il)-6-(3,5-dimetilisoxazol-4-il)-N-metil-1 H-benzo[d]imidazol-2-carboxamida; (R)-N-(1 -cianopirrolidin-3-il)-7-(1 -metil-1 H-pirazol-4-il)imidazo[1,2-a]piridin-3-carboxamida;(R)-7-(3-cianofenil)-N-(1-cianopirrolidin-3-il)imidazo[1,2-a]piridin-3-carboxamida;(R)-N-(1-cianopirrolidin-3-il)-N-metil-7-(2-metilpiridin-4-il)imidazo[1,2-a]piridin-3-carboxamida;(R)-N-(1-cianopirrolidin-3-il)-N-metil-7-(6-metilpiridin-3-il)imidazo[1,2-a]piridin-3-carboxamida;(R)-N-(1 -cianopirrolidin-3-il)-7-(1,3-dimetil-1 H-pirazol-4-il)-N-metilimidazo[1,2-a]piridin-3-carboxamida; (R)-N-(1-cianopirrolidin-3-il)-7-(2,6-dimetilpiridin-4-il)-N-metilimidazo[1,2-a]piridin-3-carboxamida; (R)-N-(1-cianopirrolidin-3-il)-N-etil-7-(2-metilpiridin-4-il)imidazo[1,2-a]piridin-3-carboxamida;(R)-N-(1-cianopirrolidin-3-il)-7-morfolinoimidazo[1,2-a]piridin-3-carboxamida;(R)-6-(3-cianofenil)-N-(1-cianopirrolidin-3-il)-3-fluoroimidazo[1,2-a]piridin-2-carboxamida;(R)-N-(1 -cianopirrolidin-3-il)-3-fluoro-6-(1 -metil-1 H-pirazol-4-il)imidazo[1,2-a]piridin-2-carboxamida; (R)-N-(1 -cianopirrolidin-3-il)-6-(1 -etil-1 H-pirazol-4-il)-3-fluoroimidazo[1,2-a]piridin-2-carboxamida; (R)-N-(1 -cianopirrolidin-3-il)-6-(1,3-dimetil-1 H-pirazol-4-il)-3-fluoroimidazo[1,2-a]piridin-2-carboxamida; (R)-N-(1-cianopirrolidin-3-il)-6-(3,5-dimetilisoxazol-4-il)-3-fluoroimidazo1,2-a]piridin-2-carboxamida; (R)-N-(1-cianopirrolidin-3-il)-5-(4-fluorofenil)picolinamida;N-((2R, 3R)-1-ciano-2-metilpirrolidin-3-il)-5-(4-fluorofenil)picolinamida;(R)-N-(1 -cianopirrolidin-3-il)-5-(1,3-dimetil-1 H-pirazol-4-il)picolinamida;(R)-N-(1-cianopirrolidin-3-il)-5-(2-metil-6-(trifluorometil)pirimidin-4-il)picolinamida;(R)-N-(1-cianopirrolidin-3-il)-3-fluoro-5-(pirazolo1,5-a]pirimidin-5-il)picolinamida;(R)-N-(1-cianopirrolidin-3-il)-3-fluoro-5-(imidazo[1,2-a]piridin-6-il)picolinamida;(R)-N-(1-cianopirrolidin-3-il)-3-metoxi-3-fenilazetidin-1-carboxamida;(R)-N-(1-cianopirrolidin-3-il)-N-metil-3-fenilazetidin-1-carboxamida;(R)-N-(1-cianopirrolidin-3-il)-3-(4-metoxifenil)azetidin-1-carboxamida;(R)-3-(4-clorofenil)-N-(1-cianopirrolidin-3-il)azetidin-1-carboxamida;(R)-3-(3-clorofenil)-N-(1-cianopirrolidin-3-il)azetidin-1-carboxamida;(3aR,6aR)-1 -(3-fenilazetidin-1 -carbonil)hexahidropirrolo[3,4-b]pirrol-5(1 H)-carbonitrilo; (3aR,6aR)-5-ciano-N-(5-fenilpiridin-2-il)hexahidropirrolo[3,4-b]pirrol-1 (2H)-carboxamida;(R)-N-(1-cianopirrolidin-3-il)-N-etil-3-fenilazetidin-1-carboxamida;(R)-3-(2-oxo-3-(4-feniltiazol-2-il)imidazolidin-1 -il)pirrolidin-1 -carbonitrilo;(R)-3-(2-oxo-3-(4-feniltiazol-2-il)tetrahidropirimidin-1 (2H)-il)pirrolidin-1 -carbonitrilo;(R)-N-(1-cianopirrolidin-3-il)-4-(pirimidin-2-il)-3,4-dihidro-2H-benzo[b][1,4]oxazin-7-carboxamida;(R)-N-(1-cianopirrolidin-3-il)-4-(4-ciclopropilpirimidin-2-il)-3,4-dihidro-2H-benzo[b][1,4]oxazin-7-carboxamida ; (R)-N-(1-cianopirrolidin-3-il)-4-(N-metilisobutiramido)picolinamida;(R)-N-(1-cianopirrolidin-3-il)-[2,3'-bipiridin]-6'-carboxamida;(R)-N-(1-cianopirroldin-3-il)-[2,4'-bipiridin]-2'-carboxamida;(R)-3-(4-clorofenil)-N-(1-cianopirrolidin-3-il)isoxazol-5-carboxamida;(R)-N-(1-cianopirrolidin-3-il)-3-(4-(trifluorometil)fenil)isoxazol-5-carboxamida;(R)-N-(1-cianopirrolidin-3-il)-3-(3,4-dimetoxifenil)isoxazol-5-carboxamida;(R)-N-(1-cianopirrolidin-3-il)-3-(3-metoxifenil)isoxazol-5-carboxamida;N-((R)-1 -cianopirrolidin-3-il)-1 -fenilpirrolidin-3-carboxamida;N-((R)-1 -cianopirrolidin-3-il)-3-(piridin-2-il)pirrolidin-1 -carboxamida;N-((R)-1 -cianopirrolidin-3-il)-3-(1 -metil-1 H-pirazol-4-il)pirrolidin-1 -carboxamida;(R)-N-(1-cianopirrolidin-3-il)-3-(2-metoxipiridin-4-il)-N-metilisoxazol-5-carboxamida;(R)-N-(1 -cianopirrolidin-3-il)-1 -metil-6-(1 -metil-1 H-pirazol-4-il)-1 H-indol-2-carboxamida;(R)-N-(1 -cianopirrolidin-3-il)-1 -metil-5-(1 -metil-1 H-pirazol-4-il)-1 H-indol-2-carboxamida;(R)-1 -(1 -cianopirrolidin-3-il)-3-(2-(isoindolin-2-il)piridin-4-il)-1 -metilurea;(R)-N-(1 -cianopirrolidin-3-il)-3-f luoro-1 -metil-5-(1 -metil-1 H-pirazol-4-il)-1 H-indol-2-carboxamida;(R)-N-(1 -cianopirrolidin-3-il)-7-(1 -metil-1 H-pirazol-4-il)imidazo[1,5-a]piridin-3-carboxamida;(R)-N-(1 -cianopirrolidin-3-il)-3-(1 -fenil-1 H-pirazol-3-il)azetidin-1 -carboxamida;(R)-N-(1 -cianopirrolidin-3-il)-3-(1 -(pirazin-2-il)-1 H-pirazol-3-il)azetidin-1 -carboxamida;(R)-N-(1 -cianopirrolidin-3-il)-3-(2-fenilpirimidin-4-il)azetidin-1 -carboxamida;(R)-3-(2-(4-clorofenil)pirimidin-4-il)-N-(1-cianopirrolidin-3-il)azetidin-1-carboxamida;(R)-3-(benciloxi)-N-(1-cianopirrolidin-3-il)-3-fenilazetidin-1-carboxamida;(R)-N-(1 -cianopirrolidin-3-il)-1 -(4-ciclopropilpirimidin-2-il)indolin-5-carboxam ida;(R)-N-(1-cianopirrolidin-3-il)-1-(4-ciclopropilpirimidin-2-il)-N-metilindolin-5-carboxamida;(3aR,6aR)-5-ciano-N-(2'-metil-[3,4'-bipiridin]-6-il)hexahidropirrolo[3,4-b]pirrol-1 (2H)-carboxamida;1 -(3-fenil-1 H-pirazol-5-carbonil)hexahidropirrolo[3,4-b]pirrol-5(1 H)-carbonitrilo;(3aR,6aR)-1 -(3-fenoxiazetidin-1 -carbonil)hexahidropirrolo[3,4-b]pirrol-5(1 H)-carbonitrilo;(R)-N-(1-cianopirrolidin-3-il)-6-(3,5-dimetilisoxazol-4-il)-N-metil-1 H-indol-2-carboxamida;(R)-N-(1 -cianopirrolidin-3-il)-N-metil-5-(1 -metil-1 H-pirazol-4-il)-1 H-pirrolo[2,3-c]piridin-2- carboxamida;N-((R)-1-cianopirrolidin-3-il)-N-metil-2-fenilmorfolin-4-carboxamida;(R)-N-(1 -cianopirrolidin-3-il)-N-metilindolin-1 -carboxamida;(R)-1 -(1 -cianopirrolidin-3-il)-1 -metil-3-(6-(trifluorometil)piridin-3-il)urea;(R)-3-(5-cloropiridin-2-il)-1 -(1 -cianopirrolidin-3-il)-1 -metilurea;(3aR,6aR)-1 -(indolin-1 -carbonil)hexahidropirrolo[3,4-b]pirrol-5(1 H)-carbonitrilo;(R)-N-(1-cianopirrolidin-3-il)-3-(2-metilpiridin-4-il)isoxazol-5-carboxamida;(R)-N-(1-cianopirrolidin-3-il)-3-(3,4-dimetilfenil)isoxazol-5-carboxamida;(R)-N-(1-cianopirrolidin-3-il)-3-(2,4-difluorofenil)isoxazol-5-carboxamida; y(R)-N-(1-cianopirrolidin-3-il)-N-metil-3-(2-metilpiridin-4-il)isoxazol-5-carboxamida;un tautómero del mismo, o una sal farmacéuticamente aceptable de dicho compuesto o tautómero. 18. Un compuesto según la reivindicación 5, que se selecciona entre:(R)-N-(1 -cianopirrolidin-3-il)-3-metoxi-4-(1 -metil-1 H-pirazol-4-il)benzamida;N-((R)-1-cianopirrolidin-3-il)-4-((2S, 6R)-2,6-dimetilmorfolino)-3-fluorobenzamida;N-(1-cianopirrolidin-3-il)-4-(3,5-dimetilisoxazol-4-il)benzamida;(R)-N-(1-cianopirrolidin-3-il)-4-morfolinobenzamida;(R)-N-(1-cianopirrolidin-3-il)-3-fluoro-4-morfolinobenzamida;(R)-4-(3-cloropiridin-4-il)-N-(1-cianopirrolidin-3-il)benzamida;(R)-4-(3-cloropiridin-4-il)-N-(1-cianopirrolidin-3-il)-3-metoxibenzamida;(R)-N-(1-cianopirrolidin-3-il)-3-metoxi-4-(2-metilpiridin-4-il)benzamida;(R)-N-(1-cianopirrolidin-3-il)-3-metoxi-4-(2-morfolinopiridin-4-il)benzamida;(R)-N-(1 -cianopirrolidin-3-il)-4-fluoro-3-(piridin-4-il)benzam ida;(R)-N-(1 -cianopirrolidin-3-il)-4-fluoro-3-(1 -metil-1 H-pirazol-4-il)benzamida;(R) -N-(1 -cianopirrolidin-3-il)-4-(1 -metil-1 H-pirazol-4-il)benzamida;(S) -N-(1-cianopirrolidin-3-il)-4-(piridin-4-il)benzamida;(R)-4-(3-cloropiridin-4-il)-N-(1-cianopirrolidin-3-il)-N-metilbenzamida;(3aR,6aR)-1-(4-fluoro-3-(piridin-4-il)benzoil)hexahidropirrolo[3,4-b]pirrol-5(1 H)-carbonitrilo;(3aR,6aR)-1 -(4-fluoro-3-(1 -metil-1 H-pirazol-4-il)benzoil)hexahidropirrolo[3,4-b]pirrol-5(1 H)-carbonitrilo; (3aR,6aR)-1-(4-(3-cloropiridin-4-il)-3-metoxibenzoil)hexahidropirrolo[3,4-b]pirrol-5(1 H)-carbonitrilo; (3aR,6aR)-1 -(3-metoxi-4-(1 -metil-1 H-pirazol-4-il)benzoil)hexahidropirrolo[3,4-b]pirrol-5(1 H)-carbonitrilo; (R)-N-(1-cianopirrolidin-3-il)-3-(N-metilisobutiramido)benzamida;(R)-N-(1 -cianopirrolidin-3-il)-2-fluoro-4-(4-metil-1 H-imidazol-1 -il)benzamida;(R)-N-(1 -cianopirrolidin-3-il)-3-fluoro-4-(1 -metil-1 H-pirazol-4-il)benzamida;(R)-N-(1 -cianopirrolidin-3-il)-2-fluoro-4-(1 -metil-1 H-pirazol-4-il)benzamida;(R)-N-(1 -cianopirrolidin-3-il)-2,5-difluoro-4-(1 -metil-1 H-pirazol-4-il)benzamida;(R)-N-(1 -cianopirrolidin-3-il)-4-(1,3-dimetil-1 H-pirazol-4-il)-3-fluorobenzamida;(R)-N-(1 -cianopirrolidin-3-il)-4-(1,3-dimetil-1 H-pirazol-4-il)-2-fluorobenzamida;(R)-N-(1 -cianopirrolidin-3-il)-4-(1 -etil-1 H-pirazol-4-il)-2-fluorobenzamida;(R)-N-(1 -cianopirrolidin-3-il)-2-fluoro-4-(1 -(2-metoxietil)-1 H-pirazol-4-il)benzamida;(R)-N-(1-cianopirrolidin-3-il)-2-fluoro-4-(5-(trifluorometil)-1 H-pirazol-4-il)benzamida;(R)-N-(1 -cianopirrolidin-3-il)-2-fluoro-4-(1 -metil-1 H-indazol-5-il)benzamida;(R)-N-(1 -cianopirroldin-3-il)-3-fluoro-N-metil-4-(1 -metil-1 H-pirazol-4-il)benzamida;(3aR,6aR)-5-ciano-N-(2-fluoro-4-(trifluorometil)fenil)hexahidropirrolo[3,4-b]pirrol-1 (2H)-carboxamida; (R)-N-(1-cianopirrolidin-3-il)-2-fluoro-4-(pirimidin-2-ilamino)benzamida;N-((3R,4S)-1-ciano-4-metilpirrolidin-3-il)-2-fluoro-4-((R)-3-metoxipirrolidin-1-il)benzamida;N-(1 -ciano-3-metilpirrolidin-3-il)-2-fluoro-4-(1 -metil-1 H-pirazol-4-il)benzamida;(R)-N-(1 -cianopirrolidin-3-il)-2-fluoro-3-(1 -metil-1 H-pirazol-4-il)benzamida;(R)-N-(1-cianopirrolidin-3-il)-2-fluoro-4-(2-metilpirimidin-4-il)benzamida;(R)-N-(1 -cianopirrolidin-3-il)-2-fluoro-5-(1 -metil-1 H-pirazol-4-il)benzamida;(R)-N-(1-cianopirrolidin-3-il)-2-fluoro-4-(pirimidin-4-il)benzamida;(R)-N-(1-cianopirroldin-3-il)-2-fluoro-4-(imidazo[1,2-a]pirimidin-6-il)benzamida;(R)-N-(1 -cianopirrolidin-3-il)-2-metoxi-4-(1 -metil-1 H-pirazol-4-il)benzamida(R)-N-(1 -cianopirrolidin-3-il)-3,5-difluoro-4-(1 -metil-1 H-pirazol-4-il)benzamida;(R)-N-(1 -cianopirrolidin-3-il)-2,6-difluoro-4-(1 -metil-1 H-pirazol-4-il)benzamida;(R)-N-(1 -cianopirrolidin-3-il)-2-fluoro-3-metoxi-4-(1 -metil-1 H-pirazol-4-il)benzamida;(R)-N-(1-cianopirrolidin-3-il)-2-fluoro-4-(imidazo[1,2-a]piridin-6-il)benzamida;(R)-N-(1-cianopirrolidin-3-il)-2-fluoro-4-(2-morfolinopiridin-4-il)benzamida;(R)-N-(1 -cianopirrolidin-3-il)-2,3-difluoro-4-(1 -metil-1 H-pirazol-4-il)benzamida;(R)-N-(1-cianopirrolidin-3-il)-3-fluoro-4-(pirimidin-2-ilamino)benzamida;(R)-N-(1-cianopirrolidin-3-il)-2-fluoro-4-(pirrolidin-1-il)benzamida;(R)-N-(1-cianopirrolidin-3-il)-2,5-difluoro-4-morfolinobenzamida;(R)-N-(1-cianopirrolidin-3-il)-2,5-difluoro-4-(pirrolidin-1-il)benzamida;N-((R)-1-cianopirrolidin-3-il)-2-fluoro-4-((R)-3-metoxipirrolidin-1-il)benzamida;(R)-N-(1-cianopirrolidin-3-il)-3-metoxi-4-(pirimidin-2-ilamino)benzamida;(R)-N-(1-cianopirrolidin-3-il)-3-metoxi-4-((4-metilpirimidin-2-il)amino)benzamida;(R)-N-(1-cianopirrolidin-3-il)-2-fluoro-4-((4-metoxipirimidin-2-il)amino)benzamida;(R)-N-(1-cianopirrolidin-3-il)-2-fluoro-4-(piridazin-4-il)benzamida;(R)-N-(1-cianopirrolidin-3-il)-2-fluoro-4-(pirazolo1,5-a]pirimidin-5-il)benzamida;3-cloro-N-((3R,4S)-1-ciano-4-metilpirrolidin-3-il)-4-morfolinobenzamida;N-((3R,4R)-1 -ciano-4-ciclopropilpirrolidin-3-il)-3-fluoro-4-(1 -metil-1 H-pirazol-4-il)benzamida;N-((3S, 4S)-1 -ciano-4-metoxipirrolidin-3-il)-N-metil-4-(1 -metil-1 H-pirazol-4-il)benzamida;(R)-N-(1-cianopirrolidin-3-il)-4-(2,6-dimetilpirimidin-4-il)-2-fluorobenzamida;(R)-N-(1-cianopirrolidin-3-il)-2-fluoro-4-(5-fluoro-2-metilpirimidin-4-il)benzamida;(R)-N-(1 -cianopirrolidin-3-il)-2-fluoro-4-(2-(trifluorometil)pirimidin-4-il)benzam ida;(R)-N-(1-cianopirrolidin-3-il)-2-fluoro-4-(2-metil-3H-pirrolo[2,3-d]pirimidin-4-il)benzamida;(R)-N-(1-cianopirrolidin-3-il)-2-fluoro-4-(imidazo[1,2-a]pirazin-3-il)benzamida;(R)-1 -(1 -cianopirrolidin-3-il)-1 -metil-3-(4-(1 -metil (-1 H-pirazol-4-il)fenil)urea;(R)-1 -(1 -cianopirrolidin-3-il)-1 -metil-3-(4-(trifluorometil)fenil)urea;(3aR,6aR)-N-(4-cloro-2-fluorofenil)-5-cianohexahidropirrolo[3,4-b]pirrol-1 (2H)-carboxamida;(3aR,6aR)-5-ciano-N-(2-fluoro-4-(trifluorometoxi) fenil)hexahidropirrolo[3,4-b]pirrol-1 (2H)-carboxamida; (3aR,6aR)-5-ciano-N-(4-ciano-2-fluorofenil)hexahidropirrolo[3,4-b]pirrol-1 (2H)-carboxamida;(3aR,6aR)-5-ciano-N-(4-ciano-2,5-difluorofenil)hexahidropirrolo[3,4-b]pirrol-1 (2H)-carboxamida;(3aR,6aR)-N-(5-cloro-2-fluorofenil)-5-cianohexahidropirrolo[3,4-b]pirrol-1 (2H)-carboxamida;(3aR,6aR)-5-ciano-N-(2-fluoro-5-(trifluorometil)fenil)hexahidropirrolo[3,4-b]pirrol-1 (2H)-carboxamida;(3aR,6aR)-5-ciano-N-(4-(trifluorometil)fenil)hexahidropirrolo[3,4-b]pirrol-1 (2H)-carboxamida;(R)-1 -(1 -cianopirrolidin-3-il)-1 -etil-3-(4-(trifluorometil)fenil)urea;1 -(1 -cianopirrolidin-3-il)-1 -(2-metoxietil)-3-(4-(trifluorometil)fenil)urea;(R)-N-(1 -cianopirrolidin-3-il)-N-etil-3-fluoro-4-(1 -metil-1 H-pirazol-4-il)benzamida;(R)-3-(3-(3-morfolinofenil)-2-oxoimidazolidin-1 -il)pirrolidin-1 -carbonitrilo;(R)-N-(1-cianopirrolidin-3-il)-4-((4-ciclopropilpirimidin-2-il)amino)-3-fluorobenzamida;(R)-N-(1-cianopirrolidin-3-il)-4-((4-ciclopropilpirimidin-2-il)amino)-2,3-difluorobenzamida;(R)-N-(1 -cianopirrolidin-3-il)-3-fluoro-4-(4-metil-1 H-imidazol-1 -il)benzamida;(R)-N-(1 -cianopirrolidin-3-il)-3-fluoro-N-metil-4-(4-metil-1 H-imidazol-1 -il)benzamida;(3aR,6aR)-5-ciano-N-(3-(2-metilpiridin-4-il)fenil)hexahidropirrolo[3,4-b]pirrol-1 (2H)-carboxamida;(3aR,6aR)-5-ciano-N-(4-(2-metilpiridin-4-il)fenil)hexahidropirrolo[3,4-b]pirrol-1 (2H)-carboxamida;(3aR,6aR)-5-ciano-N-(2-fluoro-4-(2-metilpiridin-4-il)fenil)hexahidropirrolo[3,4-b]pirrol-1 (2H)-carboxamida; (3aR,6aR)-5-ciano-N-(2-fluoro-4-(1-metil-1 H-pirazol-4-il)fenil)hexahidropirrolo[3,4-b]pirrol-1(2H)-carboxamida; 1 -(1 -cianopirrolidin-3-il)-3-(2,4-diclorofenil)urea;1 -(1 -cianopirrolidin-3-il)-3-(4-(trifluorometil)fenil)urea;3-(3-clorofenil)-1 -(1 -cianopirrolidin-3-il)-1 -metilurea;1 -(1 -cianopirrolidin-3-il)-3-(2,4-diclorofenil)-1 -metilurea;1 -(1 -cianopirrolidin-3-il)-1 -metil-3-(4-(trifluorometil)fenil)urea; y(3aR,6aR)-1-(3-cloro-4-morfolinobenzoil)hexahidropirrolo[3,4-b]pirrol-5(1 H)-carbonitrilo;un tautómero del mismo, o una sal farmacéuticamente aceptable de dicho compuesto o tautómero.19. Un compuesto según la reivindicación 9, que se selecciona entre:N-(1 -cianopirrolidin-3-il)-2'-metoxi-[1,1'-bifenil]-4-carboxamida;N-(1-cianopirrolidin-3-il)-4-fenoxibenzamida;2-([1,1'-bifenil]-4-il)-N-(1-cianopirrolidin-3-il)acetamida;(R)-N-(1 -cianopirrolidin-3-il)-[1,1'-bifenil]-4-carboxamida;(3aR,6aR)-1-([1,1'-bifenil]-3-carbonil)hexahidropirrolo[3,4-b]pirrol-5(1 H)-carbonitrilo;N-(1 -cianopirrolidin-3-il)-N-metil-[1,1'-bifenil]-4-carboxamida;N-((3R,4R)-1-ciano-4-fluoropirrolidin-3-il)-[1,1'-bifenil]-4-carboxamida;N-(1-cianopiperidin-3-il)-[1,1'-bifenil]-3-carboxamida;1 -(3-bencilfenil)-3-(1 -cianopiperidin-3-il)urea;1 -(1 -cianopiperidin-3-il)-3-(3-fenoxifenil)urea;1 -(3-bencilfenil)-3-(1 -cianopirrolidin-3-il)urea;1 -([1,1'-bifenil]-4-il)-3-(1 -cianopirrolidin-3-il)urea;1 -(1 -cianopirrolidin-3-il)-3-(3-fenoxifenil)urea;3-(3-bencilfenil)-)- 1 -(1 -cianopirrolidin-3-il)-1 -metilurea;1 -(1 -cianopirrolidin-3-il)-1 -metil-3-(3-fenoxifenil)urea; y3-([1,1'-bifenil]-4-il)-1 -(1 -cianopirrolidin-3-il)-1 -metilurea;un tautómero del mismo, o una sal farmacéuticamente aceptable de dicho compuesto o tautómero.20. Un compuesto seleccionado entre:2'-cloro-N-(1-cianopirrolidin-3-il)-[1,1'-bifenil]-4-carboxamida;3'-cloro-N-(1-cianopirrolidin-3-il)-[1,1'-bifenil]-4-carboxamida; y(R)-N-(1-cianopirrolidin-3-il)-2-fluoro-4-(N-metilfenilsulfonamido)benzamida;un tautómero del mismo, o una sal farmacéuticamente aceptable de dicho compuesto o tautómero.21. Un compuesto según una cualquiera de las reivindicaciones 1 a 20, un tautómero del mismo, o una sal farmacéuticamente aceptable de dicho compuesto o tautómero, para uso como medicamento.22. Un compuesto según una cualquiera de las reivindicaciones 1 a 20, un tautómero del mismo, o una sal farmacéuticamente aceptable de dicho compuesto o tautómero, para su uso en el tratamiento de una afección que implica disfunción mitocondrial o un cáncer.23. Un compuesto para su uso según la reivindicación 22, en donde la afección que implica la disfunción mitocondrial se selecciona entre una enfermedad neurodegenerativa; encefalopatía mitocondrial, acidosis láctica y síndrome de episodios similares a apoplejía; Neuropatía óptica hereditaria de Leber; cáncer; neuropatía, ataxia, retinitis pigmentaria-síndrome de Leigh heredado de la madre; Enfermedad de Danon; diabetes; trastornos metabólicos; cardiopatía isquémica que conduce a infarto de miocardio; enfermedades psiquiátricas, por ejemplo, esquizofrenia; deficiencia múltiple de sulfatasa; mucolipidosis II; mucolipidosis III; mucolipidosis IV; gangliosidosis GM1; lipofuscinosis ceroides neuronales; enfermedad de Alpers; síndrome de Barth; defectos de beta-oxidación; deficiencia de carnitinaacil-carnitina; deficiencia de carnitina; síndromes de deficiencia de creatina; deficiencia de coenzima Q10; deficiencia de complejo I; deficiencia de complejo II; deficiencia de complejo III; deficiencia de complejo IV; deficiencia de complejo V; deficiencia de COX; síndrome de oftalmoplejía externa progresiva crónica; deficiencia de CPT I; deficiencia de CPT II; aciduria glutárica tipo II; síndrome de Kearns-Sayre; acidosis láctica; deficiencia de acil-CoA deshidrogenasa de cadena larga; enfermedad o síndrome de Leigh; miocardiopatía infantil letal; enfermedad de Luft; aciduria glutárica tipo II; deficiencia de acil-CoA deshidrogenasa de cadena media; epilepsia mioclónica y síndrome de fibra roja irregular; citopatía mitocondrial; síndrome de ataxia mitocondrial recesiva; síndrome de depleción del ADN mitocondrial; trastorno mioneurogastrointestinal y encefalopatía; síndrome de Pearson; deficiencia de piruvato deshidrogenasa; deficiencia de piruvato carboxilasa; mutaciones POLG; deficiencia de 3-hidroxiacil-CoA deshidrogenasa de cadena media/corta; y deficiencia de acil-CoA deshidrogenasa de cadena muy larga.24. Un compuesto para su uso según la reivindicación 23, en donde la enfermedad neurodegenerativa se selecciona entre enfermedad de Parkinson, enfermedad de Alzheimer, esclerosis lateral amiotrófica, enfermedad de Huntington, isquemia, accidente cerebrovascular, demencia con cuerpos de Lewy y demencia frontotemporal; y la enfermedad de Parkinson relacionada con mutaciones en a-sinucleína, parkina y PINK1, enfermedad de Parkinson juvenil autosómica recesiva, en donde la parkina está mutada.25. Un compuesto para su uso según la reivindicación 22, en donde el cáncer se selecciona entre cáncer de mama, ovario, próstata, pulmón, riñón, gástrico, colon, testicular, cabeza y cuello, páncreas, cerebro, melanoma, hueso u otros cánceres de tejidos, órganos y cánceres de las células sanguíneas tales como linfomas y leucemias, linfoma, mieloma múltiple, cáncer colorrectal y carcinoma de pulmón de células no pequeñas; cáncer donde las vías apoptóticas están desreguladas; y cáncer en donde las proteínas de la familia BCL-2 están mutadas o expresadas en exceso o insuficientemente.26. Una composición farmacéutica que comprende un compuesto de fórmula (II) como se define en una cualquiera de las reivindicaciones 1 a 20, un tautómero del mismo o una sal farmacéuticamente aceptable de dicho compuesto o tautómero, junto con uno o más excipientes farmacéuticamente aceptables.
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP16204257.6A EP3335815B1 (de) | 2016-12-15 | 2016-12-15 | Blindnietsetzgerät und blindnietverarbeitungseinrichtung mit einem blindnietsetzgerät |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2864948T3 true ES2864948T3 (es) | 2021-10-14 |
Family
ID=57614132
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES16204257T Active ES2864948T3 (es) | 2016-12-15 | 2016-12-15 | Aparato de colocación de remaches ciegos y dispositivo de procesamiento de remaches ciegos con un aparato de colocación de remaches ciegos |
Country Status (4)
| Country | Link |
|---|---|
| EP (1) | EP3335815B1 (es) |
| ES (1) | ES2864948T3 (es) |
| HU (1) | HUE054276T2 (es) |
| PL (1) | PL3335815T3 (es) |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE102019107380B4 (de) * | 2019-03-22 | 2023-03-23 | Honsel Distribution Gmbh & Co. Kg | Elektrisch betriebenes Nietgerät mit schaltbarem Getriebe |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN104439022B (zh) * | 2014-10-24 | 2015-09-16 | 洪劲松 | 电动铆钉工具 |
-
2016
- 2016-12-15 ES ES16204257T patent/ES2864948T3/es active Active
- 2016-12-15 EP EP16204257.6A patent/EP3335815B1/de active Active
- 2016-12-15 HU HUE16204257A patent/HUE054276T2/hu unknown
- 2016-12-15 PL PL16204257T patent/PL3335815T3/pl unknown
Also Published As
| Publication number | Publication date |
|---|---|
| EP3335815A1 (de) | 2018-06-20 |
| EP3335815B1 (de) | 2021-01-20 |
| PL3335815T3 (pl) | 2021-09-27 |
| HUE054276T2 (hu) | 2021-08-30 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP7389856B2 (ja) | Usp30阻害剤としての1-シアノ-ピロリジン化合物 | |
| ES2798473T3 (es) | Compuestos nuevos | |
| ES2773046T3 (es) | Cianopirrolidinas como inhibidores de DUB para el tratamiento del cáncer | |
| ES2864948T3 (es) | Aparato de colocación de remaches ciegos y dispositivo de procesamiento de remaches ciegos con un aparato de colocación de remaches ciegos | |
| HK40043421A (en) | 1-cyano-pyrrolidine compounds as usp30 inhibitors | |
| HK40043421B (zh) | 作为usp30抑制剂的1-氰基吡咯烷化合物 | |
| HK1245266B (zh) | 作为usp30抑制剂的1-氰基吡咯烷化合物 | |
| BR112017005887B1 (pt) | Composto, método para produção e uso do mesmo, bem como composição farmacêutica |