ES2897496T3 - Procedimiento para la preparación de un compuesto usado como antagonista del receptor de mineralocorticoides - Google Patents
Procedimiento para la preparación de un compuesto usado como antagonista del receptor de mineralocorticoides Download PDFInfo
- Publication number
- ES2897496T3 ES2897496T3 ES13864994T ES13864994T ES2897496T3 ES 2897496 T3 ES2897496 T3 ES 2897496T3 ES 13864994 T ES13864994 T ES 13864994T ES 13864994 T ES13864994 T ES 13864994T ES 2897496 T3 ES2897496 T3 ES 2897496T3
- Authority
- ES
- Spain
- Prior art keywords
- chloro
- pyrazolo
- tetrahydro
- formula
- cyclopentyl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 150000001875 compounds Chemical class 0.000 title claims abstract description 108
- 238000000034 method Methods 0.000 title claims description 19
- 238000002360 preparation method Methods 0.000 title description 26
- 229940083712 aldosterone antagonist Drugs 0.000 title description 3
- UXHQLGLGLZKHTC-CUNXSJBXSA-N 4-[(3s,3ar)-3-cyclopentyl-7-(4-hydroxypiperidine-1-carbonyl)-3,3a,4,5-tetrahydropyrazolo[3,4-f]quinolin-2-yl]-2-chlorobenzonitrile Chemical compound C1CC(O)CCN1C(=O)C1=CC=C(C=2[C@@H]([C@H](C3CCCC3)N(N=2)C=2C=C(Cl)C(C#N)=CC=2)CC2)C2=N1 UXHQLGLGLZKHTC-CUNXSJBXSA-N 0.000 claims abstract description 35
- 229940125269 ocedurenone Drugs 0.000 claims abstract description 21
- 238000004519 manufacturing process Methods 0.000 claims abstract description 12
- 238000006482 condensation reaction Methods 0.000 claims abstract description 10
- 239000002253 acid Substances 0.000 claims abstract description 6
- PGQNOGKEWFKCSM-JTSKRJEESA-N (3s,3ar)-2-(3-chloro-4-cyanophenyl)-3-cyclopentyl-3,3a,4,5-tetrahydropyrazolo[3,4-f]quinoline-7-carboxylic acid Chemical compound C1([C@@H]2N(N=C3C4=CC=C(N=C4CC[C@@H]32)C(=O)O)C=2C=C(Cl)C(C#N)=CC=2)CCCC1 PGQNOGKEWFKCSM-JTSKRJEESA-N 0.000 claims abstract description 5
- HDOWRFHMPULYOA-UHFFFAOYSA-N piperidin-4-ol Chemical compound OC1CCNCC1 HDOWRFHMPULYOA-UHFFFAOYSA-N 0.000 claims abstract description 5
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 claims description 56
- 239000000203 mixture Substances 0.000 claims description 52
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 claims description 45
- 239000000243 solution Substances 0.000 claims description 38
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 claims description 36
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 claims description 22
- 239000007787 solid Substances 0.000 claims description 21
- 238000006243 chemical reaction Methods 0.000 claims description 20
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 claims description 16
- 238000001914 filtration Methods 0.000 claims description 13
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 claims description 12
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 claims description 12
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 claims description 12
- YHHBKPWMEXGLKE-UHFFFAOYSA-N 7,8-dihydro-6h-quinolin-5-one Chemical compound C1=CC=C2C(=O)CCCC2=N1 YHHBKPWMEXGLKE-UHFFFAOYSA-N 0.000 claims description 9
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 claims description 9
- DRUKTSLDCFAWHF-UHFFFAOYSA-N 5-oxo-7,8-dihydro-6h-quinoline-2-carbonitrile Chemical compound N#CC1=CC=C2C(=O)CCCC2=N1 DRUKTSLDCFAWHF-UHFFFAOYSA-N 0.000 claims description 8
- NMTCXHNZUPGKSZ-UHFFFAOYSA-N ethyl 2-(3-chloro-4-cyanophenyl)-3-cyclopentyl-3,3a,4,5-tetrahydropyrazolo[3,4-f]quinoline-7-carboxylate Chemical compound C12CCC3=NC(C(=O)OCC)=CC=C3C2=NN(C=2C=C(Cl)C(C#N)=CC=2)C1C1CCCC1 NMTCXHNZUPGKSZ-UHFFFAOYSA-N 0.000 claims description 8
- JNTBIBBVCBPPLT-UHFFFAOYSA-N ethyl 5-oxo-7,8-dihydro-6h-quinoline-2-carboxylate Chemical compound O=C1CCCC2=NC(C(=O)OCC)=CC=C21 JNTBIBBVCBPPLT-UHFFFAOYSA-N 0.000 claims description 8
- 239000012046 mixed solvent Substances 0.000 claims description 8
- HGFAAUDSQLSSNC-UHFFFAOYSA-N 2-chloro-4-hydrazinylbenzonitrile;hydrochloride Chemical compound Cl.NNC1=CC=C(C#N)C(Cl)=C1 HGFAAUDSQLSSNC-UHFFFAOYSA-N 0.000 claims description 7
- GRQGNMNARDBVOT-UHFFFAOYSA-N [O-][n+]1cccc2C(=O)CCCc12 Chemical compound [O-][n+]1cccc2C(=O)CCCc12 GRQGNMNARDBVOT-UHFFFAOYSA-N 0.000 claims description 7
- 238000001035 drying Methods 0.000 claims description 7
- HGINCPLSRVDWNT-UHFFFAOYSA-N Acrolein Chemical compound C=CC=O HGINCPLSRVDWNT-UHFFFAOYSA-N 0.000 claims description 6
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 claims description 6
- BDERNNFJNOPAEC-UHFFFAOYSA-N propan-1-ol Chemical compound CCCO BDERNNFJNOPAEC-UHFFFAOYSA-N 0.000 claims description 6
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 claims description 6
- NRWOMMXRCHVLFK-UHFFFAOYSA-N 2-chloro-4-hydrazinylbenzonitrile Chemical compound NNC1=CC=C(C#N)C(Cl)=C1 NRWOMMXRCHVLFK-UHFFFAOYSA-N 0.000 claims description 5
- 239000000706 filtrate Substances 0.000 claims description 5
- NMTCXHNZUPGKSZ-CYFREDJKSA-N ethyl (3s,3ar)-2-(3-chloro-4-cyanophenyl)-3-cyclopentyl-3,3a,4,5-tetrahydropyrazolo[3,4-f]quinoline-7-carboxylate Chemical compound C1([C@@H]2N(N=C3C4=CC=C(N=C4CC[C@@H]32)C(=O)OCC)C=2C=C(Cl)C(C#N)=CC=2)CCCC1 NMTCXHNZUPGKSZ-CYFREDJKSA-N 0.000 claims description 4
- ZMYRAZWYGXBBED-ACCUITESSA-N ethyl (6e)-6-(cyclopentylmethylidene)-5-oxo-7,8-dihydroquinoline-2-carboxylate Chemical compound C1CC2=NC(C(=O)OCC)=CC=C2C(=O)\C1=C\C1CCCC1 ZMYRAZWYGXBBED-ACCUITESSA-N 0.000 claims description 4
- 238000010438 heat treatment Methods 0.000 claims description 4
- 238000006467 substitution reaction Methods 0.000 claims description 4
- 238000005406 washing Methods 0.000 claims description 4
- NWZSZGALRFJKBT-KNIFDHDWSA-N (2s)-2,6-diaminohexanoic acid;(2s)-2-hydroxybutanedioic acid Chemical compound OC(=O)[C@@H](O)CC(O)=O.NCCCC[C@H](N)C(O)=O NWZSZGALRFJKBT-KNIFDHDWSA-N 0.000 claims description 3
- PGKPNNMOFHNZJX-UHFFFAOYSA-N 2-chloro-4-fluorobenzonitrile Chemical compound FC1=CC=C(C#N)C(Cl)=C1 PGKPNNMOFHNZJX-UHFFFAOYSA-N 0.000 claims description 3
- USFZMSVCRYTOJT-UHFFFAOYSA-N Ammonium acetate Chemical compound N.CC(O)=O USFZMSVCRYTOJT-UHFFFAOYSA-N 0.000 claims description 3
- 239000005695 Ammonium acetate Substances 0.000 claims description 3
- YIIMEMSDCNDGTB-UHFFFAOYSA-N Dimethylcarbamoyl chloride Chemical compound CN(C)C(Cl)=O YIIMEMSDCNDGTB-UHFFFAOYSA-N 0.000 claims description 3
- 229940043376 ammonium acetate Drugs 0.000 claims description 3
- 235000019257 ammonium acetate Nutrition 0.000 claims description 3
- 238000001816 cooling Methods 0.000 claims description 3
- HJSLFCCWAKVHIW-UHFFFAOYSA-N cyclohexane-1,3-dione Chemical compound O=C1CCCC(=O)C1 HJSLFCCWAKVHIW-UHFFFAOYSA-N 0.000 claims description 3
- VELDYOPRLMJFIK-UHFFFAOYSA-N cyclopentanecarbaldehyde Chemical compound O=CC1CCCC1 VELDYOPRLMJFIK-UHFFFAOYSA-N 0.000 claims description 3
- IKDUDTNKRLTJSI-UHFFFAOYSA-N hydrazine monohydrate Substances O.NN IKDUDTNKRLTJSI-UHFFFAOYSA-N 0.000 claims description 3
- 239000011259 mixed solution Substances 0.000 claims description 3
- LEIMLDGFXIOXMT-UHFFFAOYSA-N trimethylsilyl cyanide Chemical compound C[Si](C)(C)C#N LEIMLDGFXIOXMT-UHFFFAOYSA-N 0.000 claims description 3
- 238000007259 addition reaction Methods 0.000 claims description 2
- 230000005494 condensation Effects 0.000 claims description 2
- 238000005886 esterification reaction Methods 0.000 claims description 2
- 230000007062 hydrolysis Effects 0.000 claims description 2
- 238000006460 hydrolysis reaction Methods 0.000 claims description 2
- 238000007254 oxidation reaction Methods 0.000 claims description 2
- -1 2-(3-chloro-4 ethyl -cyanophenyl)-3-cyclopentyl-3,3a,4,5-tetrahydro-2H-pyrazolo[3,4-f]quinoline-7-carboxylate Chemical compound 0.000 abstract description 4
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 33
- 239000000523 sample Substances 0.000 description 25
- 241000700159 Rattus Species 0.000 description 16
- 239000000126 substance Substances 0.000 description 16
- 239000013078 crystal Substances 0.000 description 14
- 238000012360 testing method Methods 0.000 description 13
- 206010061481 Renal injury Diseases 0.000 description 10
- 208000024172 Cardiovascular disease Diseases 0.000 description 9
- 210000002216 heart Anatomy 0.000 description 9
- 208000037806 kidney injury Diseases 0.000 description 9
- 206010020772 Hypertension Diseases 0.000 description 8
- 230000036772 blood pressure Effects 0.000 description 8
- 230000004224 protection Effects 0.000 description 8
- 238000010992 reflux Methods 0.000 description 8
- 208000013875 Heart injury Diseases 0.000 description 7
- 239000003814 drug Substances 0.000 description 7
- 239000012071 phase Substances 0.000 description 7
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 6
- 206010016654 Fibrosis Diseases 0.000 description 6
- 108090000375 Mineralocorticoid Receptors Proteins 0.000 description 6
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 6
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 6
- 238000002441 X-ray diffraction Methods 0.000 description 6
- 210000004204 blood vessel Anatomy 0.000 description 6
- 238000002474 experimental method Methods 0.000 description 6
- 230000004761 fibrosis Effects 0.000 description 6
- 239000011521 glass Substances 0.000 description 6
- 239000008194 pharmaceutical composition Substances 0.000 description 6
- 238000000634 powder X-ray diffraction Methods 0.000 description 6
- 238000001228 spectrum Methods 0.000 description 6
- 206010002383 Angina Pectoris Diseases 0.000 description 5
- 206010007572 Cardiac hypertrophy Diseases 0.000 description 5
- 208000006029 Cardiomegaly Diseases 0.000 description 5
- 206010019280 Heart failures Diseases 0.000 description 5
- 102100021316 Mineralocorticoid receptor Human genes 0.000 description 5
- 208000009525 Myocarditis Diseases 0.000 description 5
- 206010003119 arrhythmia Diseases 0.000 description 5
- 230000006793 arrhythmia Effects 0.000 description 5
- 108091008698 baroreceptors Proteins 0.000 description 5
- 230000006378 damage Effects 0.000 description 5
- 229940079593 drug Drugs 0.000 description 5
- 230000004064 dysfunction Effects 0.000 description 5
- 238000011835 investigation Methods 0.000 description 5
- 210000003734 kidney Anatomy 0.000 description 5
- 208000010125 myocardial infarction Diseases 0.000 description 5
- 210000001774 pressoreceptor Anatomy 0.000 description 5
- 239000011541 reaction mixture Substances 0.000 description 5
- PQSUYGKTWSAVDQ-ZVIOFETBSA-N Aldosterone Chemical compound C([C@@]1([C@@H](C(=O)CO)CC[C@H]1[C@@H]1CC2)C=O)[C@H](O)[C@@H]1[C@]1(C)C2=CC(=O)CC1 PQSUYGKTWSAVDQ-ZVIOFETBSA-N 0.000 description 4
- PQSUYGKTWSAVDQ-UHFFFAOYSA-N Aldosterone Natural products C1CC2C3CCC(C(=O)CO)C3(C=O)CC(O)C2C2(C)C1=CC(=O)CC2 PQSUYGKTWSAVDQ-UHFFFAOYSA-N 0.000 description 4
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 4
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 4
- 208000027418 Wounds and injury Diseases 0.000 description 4
- 229960002478 aldosterone Drugs 0.000 description 4
- 230000000694 effects Effects 0.000 description 4
- 238000004128 high performance liquid chromatography Methods 0.000 description 4
- 208000014674 injury Diseases 0.000 description 4
- 239000010410 layer Substances 0.000 description 4
- 238000005259 measurement Methods 0.000 description 4
- 239000012074 organic phase Substances 0.000 description 4
- 229910016523 CuKa Inorganic materials 0.000 description 3
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 3
- RWRDLPDLKQPQOW-UHFFFAOYSA-N Pyrrolidine Chemical compound C1CCNC1 RWRDLPDLKQPQOW-UHFFFAOYSA-N 0.000 description 3
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 3
- 239000008346 aqueous phase Substances 0.000 description 3
- 239000007788 liquid Substances 0.000 description 3
- NLKNQRATVPKPDG-UHFFFAOYSA-M potassium iodide Chemical compound [K+].[I-] NLKNQRATVPKPDG-UHFFFAOYSA-M 0.000 description 3
- 230000005855 radiation Effects 0.000 description 3
- 239000002904 solvent Substances 0.000 description 3
- 238000003756 stirring Methods 0.000 description 3
- 230000002861 ventricular Effects 0.000 description 3
- 0 *c(cc1)nc(CCC2C3C4CCCC4)c1C2=NN3c(cc1)cc(Cl)c1C#N Chemical compound *c(cc1)nc(CCC2C3C4CCCC4)c1C2=NN3c(cc1)cc(Cl)c1C#N 0.000 description 2
- 239000004698 Polyethylene Substances 0.000 description 2
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 2
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 2
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 2
- 229910052782 aluminium Inorganic materials 0.000 description 2
- 238000010171 animal model Methods 0.000 description 2
- 238000009530 blood pressure measurement Methods 0.000 description 2
- 238000007796 conventional method Methods 0.000 description 2
- 239000006185 dispersion Substances 0.000 description 2
- 239000003937 drug carrier Substances 0.000 description 2
- 230000002349 favourable effect Effects 0.000 description 2
- 239000012530 fluid Substances 0.000 description 2
- 229920001684 low density polyethylene Polymers 0.000 description 2
- 239000004702 low-density polyethylene Substances 0.000 description 2
- 229910052757 nitrogen Inorganic materials 0.000 description 2
- 238000010606 normalization Methods 0.000 description 2
- 210000000056 organ Anatomy 0.000 description 2
- 238000009512 pharmaceutical packaging Methods 0.000 description 2
- 229920000728 polyester Polymers 0.000 description 2
- 229920000573 polyethylene Polymers 0.000 description 2
- 238000011552 rat model Methods 0.000 description 2
- 150000003839 salts Chemical class 0.000 description 2
- 239000012488 sample solution Substances 0.000 description 2
- 229910052708 sodium Inorganic materials 0.000 description 2
- 239000011734 sodium Substances 0.000 description 2
- 239000011780 sodium chloride Substances 0.000 description 2
- 239000008227 sterile water for injection Substances 0.000 description 2
- 238000003860 storage Methods 0.000 description 2
- 239000000725 suspension Substances 0.000 description 2
- FYSNRJHAOHDILO-UHFFFAOYSA-N thionyl chloride Chemical compound ClS(Cl)=O FYSNRJHAOHDILO-UHFFFAOYSA-N 0.000 description 2
- 238000005160 1H NMR spectroscopy Methods 0.000 description 1
- PGQNOGKEWFKCSM-UHFFFAOYSA-N 2-(3-chloro-4-cyanophenyl)-3-cyclopentyl-3,3a,4,5-tetrahydropyrazolo[3,4-f]quinoline-7-carboxylic acid Chemical compound C12CCC3=NC(C(=O)O)=CC=C3C2=NN(C=2C=C(Cl)C(C#N)=CC=2)C1C1CCCC1 PGQNOGKEWFKCSM-UHFFFAOYSA-N 0.000 description 1
- NHQDETIJWKXCTC-UHFFFAOYSA-N 3-chloroperbenzoic acid Chemical compound OOC(=O)C1=CC=CC(Cl)=C1 NHQDETIJWKXCTC-UHFFFAOYSA-N 0.000 description 1
- 102100032187 Androgen receptor Human genes 0.000 description 1
- WBLLRCSCYCHIST-UHFFFAOYSA-N CC(C)=NNc(cc1Cl)ccc1C#N Chemical compound CC(C)=NNc(cc1Cl)ccc1C#N WBLLRCSCYCHIST-UHFFFAOYSA-N 0.000 description 1
- CJZHFFFIEYMWQJ-UHFFFAOYSA-N CCOC(=O)C1=NC2=CCC3CNNC3=C2C=C1 Chemical compound CCOC(=O)C1=NC2=CCC3CNNC3=C2C=C1 CJZHFFFIEYMWQJ-UHFFFAOYSA-N 0.000 description 1
- 206010014418 Electrolyte imbalance Diseases 0.000 description 1
- 108090000079 Glucocorticoid Receptors Proteins 0.000 description 1
- 102100033417 Glucocorticoid receptor Human genes 0.000 description 1
- 239000007821 HATU Substances 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- 241001465754 Metazoa Species 0.000 description 1
- 102000003979 Mineralocorticoid Receptors Human genes 0.000 description 1
- 206010029164 Nephrotic syndrome Diseases 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- 230000002159 abnormal effect Effects 0.000 description 1
- 230000004913 activation Effects 0.000 description 1
- 210000004404 adrenal cortex Anatomy 0.000 description 1
- 108010080146 androgen receptors Proteins 0.000 description 1
- 238000003556 assay Methods 0.000 description 1
- 230000002146 bilateral effect Effects 0.000 description 1
- 230000027455 binding Effects 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- 210000004556 brain Anatomy 0.000 description 1
- 238000004587 chromatography analysis Methods 0.000 description 1
- 230000009137 competitive binding Effects 0.000 description 1
- 239000002131 composite material Substances 0.000 description 1
- 229940125904 compound 1 Drugs 0.000 description 1
- 229940125773 compound 10 Drugs 0.000 description 1
- 229940126214 compound 3 Drugs 0.000 description 1
- 210000002808 connective tissue Anatomy 0.000 description 1
- 230000003247 decreasing effect Effects 0.000 description 1
- 238000001514 detection method Methods 0.000 description 1
- ZBCBWPMODOFKDW-UHFFFAOYSA-N diethanolamine Chemical compound OCCNCCO ZBCBWPMODOFKDW-UHFFFAOYSA-N 0.000 description 1
- 201000010099 disease Diseases 0.000 description 1
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 1
- 239000007884 disintegrant Substances 0.000 description 1
- 231100000673 dose–response relationship Toxicity 0.000 description 1
- 238000009509 drug development Methods 0.000 description 1
- 239000003792 electrolyte Substances 0.000 description 1
- 230000002124 endocrine Effects 0.000 description 1
- 238000005516 engineering process Methods 0.000 description 1
- 230000029142 excretion Effects 0.000 description 1
- 210000002950 fibroblast Anatomy 0.000 description 1
- 239000012065 filter cake Substances 0.000 description 1
- 230000002496 gastric effect Effects 0.000 description 1
- 238000007489 histopathology method Methods 0.000 description 1
- 239000003906 humectant Substances 0.000 description 1
- 238000001727 in vivo Methods 0.000 description 1
- ZLVXBBHTMQJRSX-VMGNSXQWSA-N jdtic Chemical compound C1([C@]2(C)CCN(C[C@@H]2C)C[C@H](C(C)C)NC(=O)[C@@H]2NCC3=CC(O)=CC=C3C2)=CC=CC(O)=C1 ZLVXBBHTMQJRSX-VMGNSXQWSA-N 0.000 description 1
- 239000011159 matrix material Substances 0.000 description 1
- 230000001404 mediated effect Effects 0.000 description 1
- LULAYUGMBFYYEX-UHFFFAOYSA-N metachloroperbenzoic acid Natural products OC(=O)C1=CC=CC(Cl)=C1 LULAYUGMBFYYEX-UHFFFAOYSA-N 0.000 description 1
- 239000002395 mineralocorticoid Substances 0.000 description 1
- 238000012544 monitoring process Methods 0.000 description 1
- 208000009928 nephrosis Diseases 0.000 description 1
- 231100001027 nephrosis Toxicity 0.000 description 1
- 239000012044 organic layer Substances 0.000 description 1
- 244000052769 pathogen Species 0.000 description 1
- 230000001717 pathogenic effect Effects 0.000 description 1
- 230000001575 pathological effect Effects 0.000 description 1
- 239000000546 pharmaceutical excipient Substances 0.000 description 1
- 230000003285 pharmacodynamic effect Effects 0.000 description 1
- 229910052700 potassium Inorganic materials 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- 230000002265 prevention Effects 0.000 description 1
- WXXVQWSDMOAHHV-UHFFFAOYSA-N quinoline-7-carboxylic acid Chemical compound C1=CC=NC2=CC(C(=O)O)=CC=C21 WXXVQWSDMOAHHV-UHFFFAOYSA-N 0.000 description 1
- 229940044551 receptor antagonist Drugs 0.000 description 1
- 239000002464 receptor antagonist Substances 0.000 description 1
- 102000005962 receptors Human genes 0.000 description 1
- 108020003175 receptors Proteins 0.000 description 1
- 238000002390 rotary evaporation Methods 0.000 description 1
- 229920006395 saturated elastomer Polymers 0.000 description 1
- 238000000926 separation method Methods 0.000 description 1
- 210000000329 smooth muscle myocyte Anatomy 0.000 description 1
- 235000017557 sodium bicarbonate Nutrition 0.000 description 1
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 1
- AKHNMLFCWUSKQB-UHFFFAOYSA-L sodium thiosulfate Chemical class [Na+].[Na+].[O-]S([O-])(=O)=S AKHNMLFCWUSKQB-UHFFFAOYSA-L 0.000 description 1
- 238000010186 staining Methods 0.000 description 1
- 230000035488 systolic blood pressure Effects 0.000 description 1
- 239000002562 thickening agent Substances 0.000 description 1
- 210000003556 vascular endothelial cell Anatomy 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/47—Quinolines; Isoquinolines
- A61K31/4738—Quinolines; Isoquinolines ortho- or peri-condensed with heterocyclic ring systems
- A61K31/4745—Quinolines; Isoquinolines ortho- or peri-condensed with heterocyclic ring systems condensed with ring systems having nitrogen as a ring hetero atom, e.g. phenantrolines
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
- A61P5/38—Drugs for disorders of the endocrine system of the suprarenal hormones
- A61P5/40—Mineralocorticosteroids, e.g. aldosterone; Drugs increasing or potentiating the activity of mineralocorticosteroids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/04—Inotropic agents, i.e. stimulants of cardiac contraction; Drugs for heart failure
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/06—Antiarrhythmics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/12—Antihypertensives
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C255/00—Carboxylic acid nitriles
- C07C255/49—Carboxylic acid nitriles having cyano groups bound to carbon atoms of six-membered aromatic rings of a carbon skeleton
- C07C255/50—Carboxylic acid nitriles having cyano groups bound to carbon atoms of six-membered aromatic rings of a carbon skeleton to carbon atoms of non-condensed six-membered aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D215/00—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems
- C07D215/02—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen atoms or carbon atoms directly attached to the ring nitrogen atom
- C07D215/16—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen atoms or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D215/20—Oxygen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D215/00—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems
- C07D215/02—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen atoms or carbon atoms directly attached to the ring nitrogen atom
- C07D215/16—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen atoms or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D215/48—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D215/00—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems
- C07D215/58—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems with hetero atoms directly attached to the ring nitrogen atom
- C07D215/60—N-oxides
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B2200/00—Indexing scheme relating to specific properties of organic compounds
- C07B2200/13—Crystalline forms, e.g. polymorphs
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Life Sciences & Earth Sciences (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Cardiology (AREA)
- Heart & Thoracic Surgery (AREA)
- Urology & Nephrology (AREA)
- Diabetes (AREA)
- Epidemiology (AREA)
- Rheumatology (AREA)
- Hematology (AREA)
- Vascular Medicine (AREA)
- Pain & Pain Management (AREA)
- Hospice & Palliative Care (AREA)
- Biomedical Technology (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Endocrinology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
Abstract
Procedimiento para preparar un compuesto representado por la fórmula (1), 2-cloro-4-[(3S,3aR)-3-ciclopentil- 7-(4-hidroxipiperidin-1-carbonil)-3,3a,4,5-tetrahidro-2H-pirazolo[3,4-f]quinolin-2-il]benzonitrilo, **(Ver fórmula)** que se caracteriza por comprender las etapas de: (9) Resolver quiralmente 2-(3-cloro-4-cianofenil)-3-ciclopentil-3,3a,4,5-tetrahidro-2H-pirazolo[3,4-f]quinolin- 7-carboxilato de etilo para producir (3S,3aR)-2-(3-cloro-4-cianofenil)-3-ciclopentil-3,3a,4,5-tetrahidro-2H- pirazolo[3,4-f]quinolin-7-carboxilato de etilo; (10) Hidrolizar (3S,3aR)-2-(3-cloro-4-cianofenil)-3-ciclopentil-3,3a,4,5-tetrahidro-2H-pirazolo[3,4-f]quinolin-7- carboxilato de etilo para producir ácido (3S,3aR)-2-(3-cloro-4-cianofenil)-3-ciclopentil-3,3a,4,5-tetrahidro-2H- pirazolo[3,4-f]quinolin-7-carboxílico; (11) Someter ácido (3S,3aR)-2-(3-cloro-4-cianofenil)-3-ciclopentil-3,3a,4,5-tetrahidro-2H-pirazolo[3,4- f]quinolin-7-carboxílico y 4-hidroxipiperidina a una reacción de condensación para producir el compuesto representado por la fórmula (1).
Description
DESCRIPCIÓN
Procedimiento para la preparación de un compuesto usado como antagonista del receptor de mineralocorticoides Campo técnico
La presente invención pertenece al campo de la tecnología farmacéutica. Más específicamente, la presente invención se refiere a un método para preparar un compuesto como antagonista del receptor de mineralocorticoides.
Antecedentes
La aldosterona es una hormona mineralocorticoide sintetizada en la corteza suprarrenal, y puede unirse al receptor de mineralocorticoides y activar al receptor para fomentar la conservación de sodio y la excreción de potasio. Puede tener un papel importante en el mantenimiento del equilibrio hidroelectrolítico y en el cambio de la estructura y función de células endoteliales, células musculares lisas vasculares y fibroblastos en la pared arterial así como en la adventicia arterial y la matriz en sus medios. El alto nivel de aldosterona puede dar como resultado la activación anómala del receptor de mineralocorticoides, que puede provocar un desequilibrio hidroelectrolítico, una lesión de los vasos sanguíneos, fibrosis y similares y dar como resultado una enfermedad cardiovascular tal como hipertensión, lesión a un órgano tal como el riñón, el corazón y el cerebro, alteración endocrina y similares. Por tanto, un fármaco que bloquea la unión de aldosterona y el receptor de mineralocorticoides mediante la unión competitiva al receptor de mineralocorticoides puede inhibir la lesión mediada por aldosterona y reducir la aparición de la enfermedad anteriormente mencionada.
El compuesto representado por la fórmula (1), 2-cloro-4-[(3S,3aR)-3-ciclopentil-7-(4-hidroxipiperidin-1-carbonil)-3,3a,4,5-tetrahidro-2H-pirazolo[3,4-f]quinolin-2-il]benzonitrilo, tal como se da a conocer en la solicitud WO2012022121, es un antagonista del receptor de aldosterona que puede unirse selectivamente al receptor de mineralocorticoides, y tiene una afinidad más baja por el receptor de glucocorticoides y el de andrógenos.
El documento WO2012022121 da a conocer un procedimiento para preparar el compuesto representado por la fórmula (1), que se obtiene resolviendo quiralmente la mezcla racémica que contiene el compuesto representado por la fórmula (1) seguido de la evaporación rotativa hasta sequedad. El compuesto obtenido está en la forma amorfa.
El compuesto de fórmula (1) tiene dos centros quirales. Con el fin de obtener un único isómero, los expertos en la técnica resolverán la mezcla racémica. Según el documento WO2012022121, en primer lugar se obtiene la mezcla racémica que contiene el compuesto representado por la fórmula (1) y después se resuelve para obtener el compuesto representado por la fórmula (1), lo que dificulta la producción del compuesto representado por la fórmula (1) en una planta convencional de BPF, dando como resultado dificultad en la industrialización y un mayor coste de producción.
Sumario de la invención
El estudio de la forma cristalina es muy importante en el desarrollo de fármacos. Las formas cristalinas diferentes de un compuesto darán como resultado la diferencia en la propiedad tal como la estabilidad y la solubilidad. Por tanto, los presentes inventores han llevado a cabo muchas investigaciones de la forma cristalina del compuesto representado por la fórmula (1), e identificaron y hallaron algunas las formas cristalinas útiles del compuesto de fórmula (1).
En la preparación del compuesto de fórmula (1), los presentes inventores han realizado la etapa de resolución de antemano, de modo que puede facilitarse la producción del compuesto representado por la fórmula (1) en un taller convencional de BPF y puede lograrse la industrialización sin problemas.
En el presente documento se describen las formas cristalinas del compuesto de fórmula (1).
El objeto de la presente invención es proporcionar un procedimiento para preparar el compuesto de fórmula (1).
En el presente documento se describen un procedimiento para preparar las formas cristalinas del compuesto de fórmula (1) y un método para convertir una cualquiera de las formas cristalinas en otra forma cristalina.
En el presente documento se describen el uso de las formas cristalinas del compuesto de fórmula (1) para tratar y/o prevenir lesión renal o enfermedad cardiovascular (incluyendo lesión cardíaca, hipertensión, insuficiencia cardíaca, infarto de miocardio, angina de pecho, hipertrofia cardíaca, miocarditis, fibrosis del corazón y los vasos sanguíneos, disfunción del barorreceptor o arritmia) y el uso de las formas cristalinas del compuesto de fórmula (1) en la fabricación de un medicamento para tratar y/o prevenir lesión renal o enfermedad cardiovascular (incluyendo lesión cardíaca, hipertensión, insuficiencia cardíaca, infarto de miocardio, angina de pecho, hipertrofia cardíaca, miocarditis, fibrosis del corazón y los vasos sanguíneos, disfunción del barorreceptor o arritmia).
En el presente documento se da a conocer una forma cristalina de un compuesto representado por la fórmula (1), 2-cloro-4-[(3S,3aR)-3-ciclopentil-7-(4-hidroxipiperidin-1-carbonil)-3,3a,4,5-tetrahidro-2H-pirazolo[3,4-f]quinolin-2-iljbenzonitrilo

que se caracteriza por tener un patrón de difracción de rayos X de polvo que comprende los siguientes picos característicos expresados mediante grados 20, cuando se mide usando radiación CuKa:
Forma cristalina I: 14,8°±0,2°, 17,4°±0,2°, 19,4°±0,2°, 19,8°±0,2°;
Forma cristalina II: 14,6°±0,2°, 19,9°±0,2°, 21,2°±0,2°, 24,6°±0,2°;
Forma cristalina III: 15,3°±0,2°, 19,5°±0,2°, 20,5°±0,2°, 25,0°±0,2° (no forma parte de la presente invención). En el presente documento se da a conocer una forma cristalina del compuesto representado por la fórmula (1), que se caracteriza por tener un patrón de difracción de rayos X de polvo que comprende los siguientes picos característicos expresados mediante grados 20, cuando se mide usando radiación CuKa:
Forma cristalina I: 14,8°±0,2°, 16,9°±0,2°, 17,4°±0,2°, 19,4°±0,2°, 19,8°±0,2°, 26,2°±0,2°;
Forma cristalina II: 14,6°±0,2°, 18,0°±0,2°, 18,7°±0,2°, 19,9°±0,2°, 21,2°±0,2°, 24,6°±0,2°;
Forma cristalina III: 10,0°±0,2°, 15,3°±0,2°, 15,8°±0,2°, 19,5°±0,2°, 20,5°±0,2°, 25,0°±0,2° (no forma parte de la presente invención).
En el presente documento se da a conocer una forma cristalina del compuesto representado por la fórmula (1), que se caracteriza por tener un patrón de difracción de rayos X de polvo que comprende los siguientes picos característicos expresados mediante grados 20, cuando se mide usando radiación CuKa:
Forma cristalina I: 9,8°±0,2°, 12,9°±0,2°, 14,8°±0,2°, 15,4°±0,2°, 16,9°±0,2°, 17,4°±0,2°, 19,4°±0,2°, 19,8°±0,2°, 22,6°±0,2°, 26,2°±0,2°;
Forma cristalina II: 4,5°±0,2°, 9,0°±0,2°, 12,2°±0,2°, 14,0°±0,2°, 14,6°±0,2°, 18,0°±0,2°, 18,7°±0,2°, 19,9°±0,2°, 21,2°±0,2°, 24,6°±0,2°;
Forma cristalina III: 3,8°±0,2°, 10,0°±0,2°, 15,3°±0,2°, 15,8°±0,2°, 17,9°±0,2°, 19,5°±0,2°, 20,5°±0,2°, 25,0°±0,2°, 26,0°±0,2°, 27,2°±0,2° (no forma parte de la presente invención).
Las soluciones técnicas según la presente invención son las siguientes:
1. Un procedimiento para preparar un compuesto representado por la fórmula (1), 2-cloro-4-[(3S,3aR)-3-ciclopentil-7-(4-hidroxipiperidin-1-carbonil)-3,3a,4,5-tetrahidro-2H-pirazolo[3,4-f]quinolin-2-il]benzonitrilo,

que se caracteriza por comprender las etapas de:
(9) Resolver quiralmente 2-(3-cloro-4-cianofenil)-3-ciclopentil-3,3a,4,5-tetrahidro-2H-pirazolo[3,4-f]quinolin-7-carboxilato de etilo para producir (3S,3aR)-2-(3-cloro-4-cianofenil)-3-ciclopentil-3,3a,4,5-tetrahidro-2H-pirazolo[3,4-f]quinolin-7-carboxilato de etilo;
(10) Hidrolizar (3S,3aR)-2-(3-cloro-4-cianofenil)-3-ciclopentil-3,3a,4,5-tetrahidro-2H-pirazolo[3,4-f]quinolin-7-carboxilato de etilo para producir ácido (3S,3aR)-2-(3-cloro-4-cianofenil)-3-ciclopentil-3,3a,4,5-tetrahidro-2H-pirazolo[3,4-f]quinolin-7-carboxílico;
(11) Someter ácido (3S,3aR)-2-(3-cloro-4-cianofenil)-3-ciclopentil-3,3a,4,5-tetrahidro-2H-pirazolo[3,4-f]quinolin-7-carboxílico y 4-hidroxipiperidina a una reacción de condensación para producir el compuesto representado por la fórmula (1).
2. El procedimiento según la solución 1, que comprende además una de las siguientes etapas:
colocar el compuesto representado por la fórmula (1) obtenido en la etapa (11) en un alcohol inferior anhidro, acetonitrilo, un disolvente mixto de acetato de etilo y etanol, un disolvente mixto de metanol y tetrahidrofurano o un disolvente mixto de acetonitrilo y acetona, calentar la disolución resultante hasta que se vuelva transparente, después enfriar la disolución resultante para separar un sólido y filtrar y secar el sólido separado; o
colocar el compuesto representado por la fórmula (1) obtenido en la etapa (11) en un alcohol inferior para disolverlo, después añadir la disolución resultante gota a gota a agua, filtrar la mezcla resultante y opcionalmente secar la sustancia filtrada a vacío; o
lavar el compuesto representado por la fórmula (1) obtenido en la etapa (11) con una disolución mixta de agua y acetonitrilo, filtrar la mezcla resultante y opcionalmente secar la sustancia filtrada a vacío; o disolver el compuesto representado por la fórmula (1) obtenido en la etapa (11) en acetona, añadir la disolución resultante gota a gota a n-heptano y filtrar la mezcla resultante.
2-1. El procedimiento según la solución 1 ó 2, que comprende además la etapa (8) inmediatamente antes de la etapa (9):
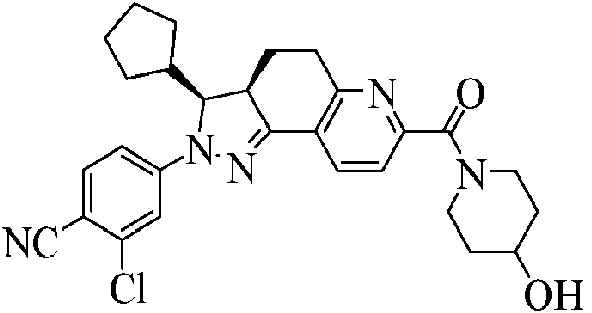
(8) Someter (E)-6-ciclopentilmetilen-5-oxo-5,6,7,8-tetrahidroquinolin-2-carboxilato de etilo y clorhidrato de 2-cloro-4-hidrazinobenzonitrilo a una reacción de condensación para producir 2-(3-cloro-4-cianofenil)-3-ciclopentil-3,3a,4,5-tetrahidro-2H-pirazolo[3,4-f]quinolin-7-carboxilato de etilo.
2-2. El procedimiento según la solución 2-1, que comprende además la etapa (7) inmediatamente antes de la etapa (8):
(7) Someter 2-cloro-4-hidrazinobenzonitrilo y ácido clorhídrico a una reacción de formación de sal para producir clorhidrato de 2-cloro-4-hidrazinobenzonitrilo.
2-3. El procedimiento según la solución 2-2, que comprende además la etapa (6) inmediatamente antes de la etapa (7):
(6) Someter 2-cloro-4-fluorobenzonitrilo e hidrazina hidratada a una reacción de sustitución para producir 2-cloro-4-hidrazinobenzonitrilo.
2-4. El procedimiento según la solución 2-3, que comprende además la etapa (5) inmediatamente antes de la etapa (6):
(5) Someter 5-oxo-5,6,7,8-tetrahidroquinolin-2-carboxilato de etilo y ciclopentanocarbaldehído a una reacción de condensación para producir (E)-6-ciclopentilmetilen-5-oxo-5,6,7,8-tetrahidroquinolin-2-carboxilato de etilo.
2-5. El procedimiento según la solución 2-4, que comprende además la etapa (4) inmediatamente antes de la etapa (5):
(4) Someter 5-oxo-5,6,7,8-tetrahidroquinolin-2-carbonitrilo a reacciones de hidrólisis y esterificación para producir 5-oxo-5,6,7,8-tetrahidroquinolin-2-carboxilato de etilo.
2-6. El procedimiento según la solución 2-5, que comprende además la etapa (3) inmediatamente antes de la etapa (4):
(3) Someter N-óxido de 5-oxo-5,6,7,8-tetrahidroquinolina, cloruro N,N-dimetilcarbámico y cianuro de trimetilsililo a una reacción de sustitución para producir 5-oxo-5,6,7,8-tetrahidroquinolin-2-carbonitrilo.
2-7. El procedimiento según la solución 2-6, que comprende además la etapa (2) inmediatamente antes de la etapa (3):
(2) Someter 5-oxo-5,6,7,8-tetrahidroquinolina a una reacción de oxidación para producir N-óxido de 5-oxo-5,6,7,8-tetrahidroquinolina.
2-8. El procedimiento según la solución 2-7, que comprende además la etapa (1) inmediatamente antes de la etapa (2):
(1) Someter 1,3-ciclohexanodiona, acetato de amonio y acroleína a reacciones de condensación y adición para producir 5-oxo-5,6,7,8-tetrahidroquinolina.
La forma cristalina I del compuesto representado por la fórmula (1), tal como se describió anteriormente, puede prepararse colocando el compuesto 2-cloro-4-[(3S,3aR)-3-ciclopentil-7-(4-hidroxipiperidin-1-carbonil)-3,3a,4,5-tetrahidro-2H-pirazolo[3,4-f]quinolin-2-il]benzonitrilo en un alcohol inferior anhidro, acetonitrilo, una mezcla de acetato de etilo y etanol, una mezcla de metanol y tetrahidrofurano o una mezcla de acetonitrilo y acetona, calentando la disolución resultante hasta que se vuelva transparente, después enfriando la disolución resultante para separar un sólido y filtrando y secando el sólido separado para producir la forma cristalina I; o disolviendo el compuesto 2-cloro-4-[(3S,3aR)-3-ciclopentil-7-(4-hidroxipiperidin-1-carbonil)-3,3a,4,5-tetrahidro-2H-pirazolo[3,4-f]quinolin-2-il]benzonitrilo en acetona, añadiendo la disolución resultante gota a gota a n-heptano y filtrando la mezcla resultante para producir la forma cristalina I.
La forma cristalina III del compuesto representado por la fórmula (1), tal como se describió anteriormente, puede prepararse colocando el compuesto 2-cloro-4-[(3S,3aR)-3-ciclopentil-7-(4-hidroxipiperidin-1-carbonil)-3,3a,4,5-tetrahidro-2H-pirazolo[3,4-f]quinolin-2-il]benzonitrilo en un alcohol inferior para disolverlo, después añadiendo la disolución resultante gota a gota a agua y filtrando la mezcla resultante para producir la forma cristalina III resultante; o lavando el compuesto 2-cloro-4-[(3S,3aR)-3-ciclopentil-7-(4-hidroxipiperidin-1-carbonil)-3,3a,4,5-tetrahidro-2H-pirazolo[3,4-f]quinolin-2-il]benzonitrilo con una mezcla de agua y acetonitrilo y filtrando la mezcla resultante para producir la forma cristalina III resultante.
La forma cristalina II del compuesto representado por la fórmula (1), tal como se describió anteriormente, puede prepararse colocando el compuesto 2-cloro-4-[(3S,3aR)-3-ciclopentil-7-(4-hidroxipiperidin-1-carbonil)-3,3a,4,5-tetrahidro-2H-pirazolo[3,4-f]quinolin-2-il]benzonitrilo en un alcohol inferior para disolverlo, después añadiendo la disolución resultante gota a gota a agua y filtrando la mezcla resultante para producir la forma cristalina III resultante; o lavando el compuesto 2-cloro-4-[(3S,3aR)-3-ciclopentil-7-(4-hidroxipiperidin-1-carbonil)-3,3a,4,5-tetrahidro-2H-pirazolo[3,4-f]quinolin-2-il]benzonitrilo con una mezcla de agua y acetonitrilo y filtrando la mezcla resultante para producir la forma cristalina III resultante; después secando la forma cristalina III resultante a vacío para producir la forma cristalina II.
Según la presente invención, el término "alcohol inferior" se refiere a metanol, etanol o n-propanol.
En el presente documento se da a conocer una composición farmacéutica, que se caracteriza porque dicha composición farmacéutica contiene la forma cristalina anteriormente descrita del compuesto representado por la fórmula (1) y un portador farmacéuticamente aceptable, en la que dicha forma cristalina comprende las formas cristalinas I, II y III o una combinación de las mismas. Esta composición farmacéutica no forma parte de la presente invención.
En el presente documento también se da a conocer una composición farmacéutica que contiene las formas cristalinas I, II y III del compuesto representado por la fórmula (1) o una combinación de las mismas. Dicha composición farmacéutica también puede contener un portador farmacéuticamente aceptable, tal como excipiente, aglutinante, humectante, disgregante, espesante y similares. Esta composición farmacéutica no forma parte de la presente invención.
En el presente documento se da a conocer el uso de la forma cristalina anteriormente descrita del compuesto
representado por la fórmula (1) en la fabricación de un medicamento para tratar y/o prevenir lesión renal o enfermedad cardiovascular, en el que dicha forma cristalina comprende las formas cristalinas I, II, III o una combinación de las mismas. Este uso no forma parte de la presente invención.
Dicha enfermedad cardiovascular puede comprender lesión cardíaca, hipertensión, insuficiencia cardíaca, infarto de miocardio, angina de pecho, hipertrofia cardíaca, miocarditis, fibrosis del corazón y los vasos sanguíneos, disfunción del barorreceptor o arritmia.
En el presente documento se da a conocer un método para tratar y/o prevenir lesión renal o enfermedad cardiovascular, en el que dicho método comprende administrar a un sujeto que lo necesita una cantidad terapéuticamente eficaz de la forma cristalina anteriormente descrita del compuesto representado por la fórmula (1), en el que dicha forma cristalina comprende las formas cristalinas I, II, III o una combinación de las mismas. Este método no forma parte de la presente invención.
Dicha enfermedad cardiovascular puede comprender lesión cardíaca, hipertensión, insuficiencia cardíaca, infarto de miocardio, angina de pecho, hipertrofia cardíaca, miocarditis, fibrosis del corazón y los vasos sanguíneos, disfunción del barorreceptor o arritmia.
En el presente documento se da a conocer la forma cristalina anteriormente descrita del compuesto representado por la fórmula (1) para tratar y/o prevenir lesión renal o enfermedad cardiovascular, en la que dicha forma cristalina comprende las formas cristalinas I, II, III o una combinación de las mismas. Esta no forma parte de la presente invención.
Dicha enfermedad cardiovascular puede comprender lesión cardíaca, hipertensión, insuficiencia cardíaca, infarto de miocardio, angina de pecho, hipertrofia cardíaca, miocarditis, fibrosis del corazón y los vasos sanguíneos, disfunción del barorreceptor o arritmia.
Las formas cristalinas I, II y III y la forma amorfa del compuesto representado por la fórmula (1) pueden convertirse unas en otras en determinadas condiciones. La presente divulgación también proporciona la conversión entre la forma cristalina I, la forma cristalina II, la forma cristalina III y la forma amorfa.
La forma amorfa puede recristalizarse en etanol anhidro para producir la forma cristalina I; las formas cristalinas I, II y III o una combinación de las mismas pueden disolverse en un alcohol inferior como disolvente y después someterse a evaporación rotativa hasta sequedad para producir la forma amorfa;
La forma cristalina II puede recristalizarse en etanol anhidro para producir la forma cristalina I; la forma amorfa puede disolverse en metanol y después se añade la disolución resultante gota a gota a agua para producir la forma cristalina III;
La forma cristalina III puede secarse a temperatura ambiente para producir la forma cristalina II;
La forma cristalina I puede lavarse con un sistema de acetonitrilo y agua para producir la forma cristalina III; y
La forma cristalina III puede recristalizarse en etanol anhidro para producir la forma cristalina I.
Descripción de los dibujos
Figura 1: el espectro de XRD para la forma cristalina I del compuesto de fórmula (1);
Figura 2: el espectro de XRD para la forma cristalina II del compuesto de fórmula (1);
Figura 3: el espectro de XRD para la forma cristalina III del compuesto de fórmula (1);
Figura 4: la relación de conversión entre la forma cristalina I, la forma cristalina II, la forma cristalina III y la forma amorfa del compuesto de fórmula (1), en la que:
1. Se recristaliza con etanol;
2. Se disuelve en un alcohol inferior y después se somete a evaporación rotativa hasta sequedad;
3. Se disuelve en metanol y después se separa con la adición de agua;
4. Se disuelve en un alcohol inferior y después se somete a evaporación rotativa hasta sequedad;
5. Se disuelve en un alcohol inferior y después se somete a evaporación rotativa hasta sequedad;
6. Se recristaliza con etanol;
7. Se lava con acetonitrilo/agua;
8. Se recristaliza con etanol;
9. Se seca a temperatura ambiente.
Realizaciones
La presente invención se ilustrará con detalle mediante las siguientes realizaciones en la forma de ejemplos.
Ejemplo 1: Preparación de 2-cloro-4-[(3S,3aR)-3-ciclopentil-7-(4-hidroxipiperidin-1-carbonil)-3,3a,4,5-tetrahidro-2H-pirazolo[3,4-f]quinolin-2-il]benzonitrilo

1. Preparación de 5-oxo-5,6,7,8-tetrahidroquinolina (01)
Ecuación de reacción:

Se llevaron a cabo dos reacciones en paralelo:
A un recipiente de reacción de 100 l se le añadió tolueno (45 l) y después se le añadió 1,3-ciclohexanodiona (15 kg) con agitación. Se calentó la mezcla resultante hasta que se disolvió el sólido. A la disolución resultante se le añadió acetato de amonio (24 kg). Se calentó la mezcla resultante hasta reflujo durante 12 horas y se enfrió hasta 0°C. A la mezcla resultante se le añadió lentamente un total de 15 kg de acroleína en lotes. Se calentó lentamente la mezcla hasta reflujo, se hizo reaccionar durante 12 horas, se enfrió y se separó en fases. Se lavó la fase inferior con tolueno dos veces (5 l*2). Se combinaron las fases orgánicas y se concentraron hasta sequedad para dar un total de 7,4 kg de 5-oxo-5,6,7,8-tetrahidroquinolina en bruto como un líquido negro,
rendimiento: 18,8%.
2. Preparación de N-óxido de 5-oxo-5,6,7,8-tetrahidroquinolina (02)

Se cargó la 5-oxo-5,6,7,8-tetrahidroquinolina (7,4 kg) en bruto en un recipiente de reacción de 100 l. Se añadió diclorometano al recipiente de reacción hasta el volumen total de 50 l. Se enfrió la mezcla resultante hasta -10°C. se añadió ácido metacloroperbenzoico (13 kg) en lotes. Después se agitó la mezcla durante 20 horas a temperatura ambiente. Después se filtró la mezcla de reacción mediante succión. Se lavó la torta de filtración con diclorometano dos veces y se combinó con el filtrado. Se lavó la disolución orgánica con una disolución saturada de tiosulfato de sodio hasta un nivel tal que el papel indicador de yoduro de potasio ya no mostró azul y se secó con sulfato de sodio anhidro para producir una disolución (50 l), que no se trató posteriormente y se usó directamente en la siguiente etapa.
3. Preparación de 5-oxo-5,6,7,8-tetrahidroquinolin-2-carbonitrilo (03)

A un recipiente de reacción de 100 l se le añadió la disolución anterior de N-óxido de 5-oxo-5,6,7,8-tetrahidroquinolina (50 l), después se le añadió cianuro de trimetilsililo (10 kg) y después se le añadió lentamente cloruro N,N-dimetilcarbámico (11 kg). Se agitó la mezcla de reacción a temperatura ambiente durante 48 horas. Se añadió lentamente una disolución acuosa saturada de hidróxido de sodio en lote para ajustar el pH a 8-9. Se separó la mezcla resultante en fases y se extrajo. Se extrajo la fase acuosa con diclorometano tres veces (8 l*3). Se combinaron las fases orgánicas y se lavaron con agua una vez (20 l). Se secó la fase orgánica resultante sobre sulfato de sodio anhidro, se filtró y se concentró a presión reducida para dar un líquido negro rojizo (aproximadamente 8 l). Se enfrió el líquido y se cristalizó con etanol para producir 5-oxo-5,6,7,8-tetrahidroquinolin-2-carbonitrilo (930 g), rendimiento: 10,7% (calculado basándose en el material de partida de 5-oxo-5,6,7,8-tetrahidroquinolina).
4. Preparación de 5-oxo-5,6,7,8-tetrahidroquinolin-2-carboxilato de etilo (05)

A un matraz de fondo redondo de 2 l se le añadieron etanol anhidro (800 ml) y 5-oxo-5,6,7,8-tetrahidroquinolin-2-carbonitrilo (280 g). Luego, se añadió ácido clorhídrico concentrado (400 ml) en un baño de hielo. Se calentó la mezcla y se agitó durante 16 horas bajo reflujo. Después se enfrió la mezcla de reacción y se concentró. Después de añadir etanol anhidro (1 l), se enfrió la mezcla resultante hasta 0°C. Después de añadir gota a gota cloruro de tionilo (200 ml), se calentó la mezcla resultante y se agitó durante 10 horas bajo reflujo. Se concentró la mezcla de reacción y se disolvió el residuo en diclorometano. Se ajustó la disolución resultante con una disolución de bicarbonato de sodio a pH >7 y se separó en fases. Se extrajo la fase acuosa con diclorometano tres veces. Se combinaron las fases orgánicas, se secaron, se concentraron para dar 5-oxo-5,6,7,8-tetrahidroquinolin-2-carboxilato de etilo (875 g) en total, rendimiento: 8,18%.
5. Preparación de (E)-6-ciclopentilmetilen-5-oxo-5,6,7,8-tetrahidroquinolin-2-carboxilato de etilo (07)

Se llevaron a cabo tres reacciones en paralelo:
A un frasco de una sola boca de 2 l se le añadieron 5-oxo-5,6,7,8-tetrahidroquinolin-2-carboxilato de etilo (291 g) y etanol (450 ml). A -20°C, se añadió adicionalmente ciclopentanocarbaldehído (213 ml) y se agitó la mezcla resultante durante 10 minutos, después se añadió lentamente pirrolidina (110 ml). Bajo protección con nitrógeno y protección de la luz, se agitó la reacción durante 8 horas a temperatura ambiente. Se dejó reposar la disolución a -20°C durante 2 horas y se filtró. Se lavó el sólido obtenido con etanol frío y se secó para dar (E)-6-ciclopentilmetilen-5-oxo-5,6,7,8-tetrahidroquinolin-2-carboxilato de etilo (862 g), rendimiento: 72,3%.
6. Preparación de clorhidrato de 2-cloro-4-hidrazinobenzonitrilo (08)

A un recipiente de reacción de 100 l se le añadieron etanol (40 l) y 2-cloro-4-fluorobenzonitrilo (7 kg). Después se añadió hidrazina hidratada (4 l). Se calentó la mezcla resultante hasta reflujo durante 5 horas, después se enfrió y se sometió a filtración centrífuga. Se introdujo el sólido resultante en un recipiente de reacción de 100 l. Se añadió etanol anhidro (40 l) y después se añadió lentamente ácido clorhídrico concentrado (7,5 l). Se calentó la mezcla resultante hasta reflujo durante 2 horas, se sometió a filtración centrífuga y se secó para producir clorhidrato de 2-cloro-4-hidrazinobenzonitrilo (7 kg), rendimiento: 76,2%.
7. Preparación de 2-(3-cloro-4-cianofenil)-3-ciclopentil-3,3a,4,5-tetrahidro-2H-pirazolo[3,4-f]quinolin-7-carboxilato de etilo (09)
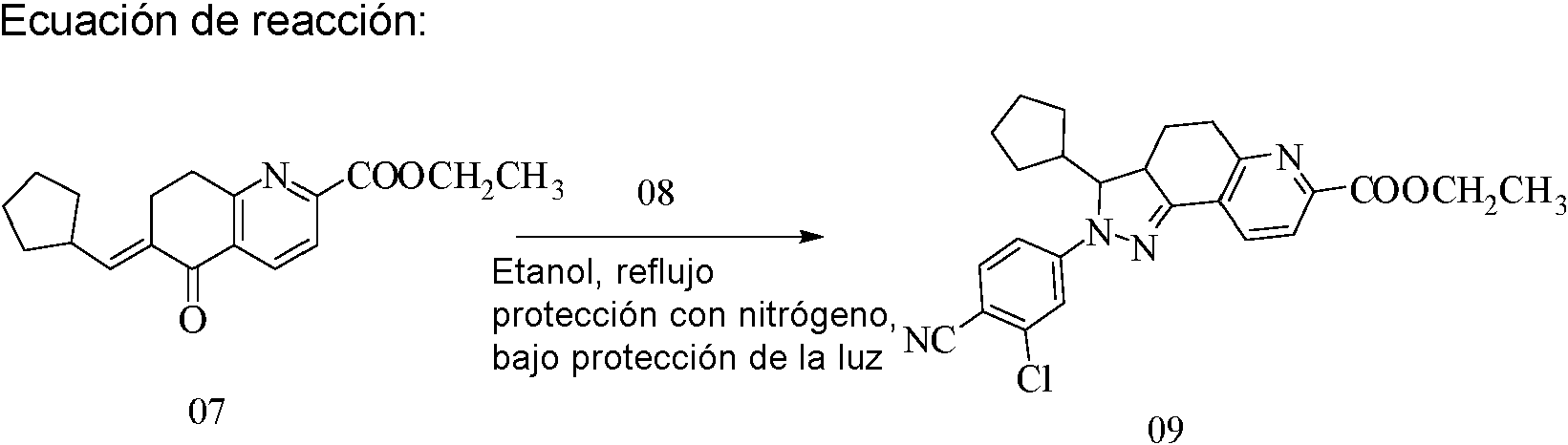
Se llevaron a cabo cuatro reacciones en paralelo:
A un frasco de una sola boca de 2 l se le añadieron (E)-6-ciclopentilmetilen-5-oxo-5,6,7,8-tetrahidroquinolin-2-carboxilato de etilo (215,5 g), clorhidrato de 2-cloro-4-hidrazinobenzonitrilo (191 g) y etanol (900 ml). Bajo protección con nitrógeno y protección de la luz, se calentó la reacción hasta reflujo durante 9 horas a 80°C, se enfrió hasta temperatura ambiente, se dejó reposar a -20°C durante 2 horas y se filtró. Se lavó el sólido resultante con etanol frío y dietil éter, respectivamente, y se secó para producir 2-(3-cloro-4-cianofenil)-3-ciclopentil-3,3a,4,5-tetrahidro-2H-pirazolo[3,4-f]quinolin-7-carboxilato de etilo (1026 g), rendimiento: 79,3%.
8. Preparación de (3S,3aR)-2-(3-cloro-4-cianofenil)-3-ciclopentil-3,3a,4,5-tetrahidro-2H-pirazolo[3,4-f]quinolin-7-carboxilato de etilo (12)

Se resolvió 2-(3-cloro-4-cianofenil)-3-ciclopentil-3,3a,4,5-tetrahidro-2H-pirazolo[3,4-f]quinolin-7-carboxilato de etilo con SFC (cromatografía de fluidos supercríticos) para producir dos isómeros. El primer componente obtenido mediante separación y recogida fue (3S,3aR)-2-(3-cloro-4-cianofenil)-3-ciclopentil-3,3a,4,5-tetrahidro-2H-pirazolo[3,4-f]quinolin-7-carboxilato de etilo;
Las condiciones de resolución:
Instrumento: SFC (Novasep 30-50)
Columna de preparación: Chiralpak IA, 20 |im, 5*25 cm
Fase móvil: la fase A era CO2 supercrítico, la fase B era diclorometano:tetrahidrofurano:dietanolamina = 50:50:0,1 (vol:vol:vol), A:B=50:50 (vol:vol)
Velocidad de flujo: 150 g/min
Longitud de onda de detección: 465 nm
Preparación de muestra: se disolvió con ultrasonidos 2-(3-cloro-4-cianofenil)-3-ciclopentil-3,3a,4,5-tetrahidro-2H-pirazolo[3,4-f]quinolin-7-carboxilato de etilo (1025 g) en diclorometano. Se filtró la mezcla resultante para producir una disolución de muestra (aproximadamente 50 mg/ml).
Se resolvió la disolución de muestra con SFC y se recogió el primer isómero con aparición de pico, es decir (3S,3aR)-2-(3-cloro-4-cianofenil)-3-ciclopentil-3,3a,4,5-tetrahidro-2H-pirazolo[3,4-f]quinolin-7-carboxilato de etilo (601,58 g).
9. Preparación de ácido (3S,3aR)-2-(3-cloro-4-cianofenil)-3-ciclopentil-3,3a,4,5-tetrahidro-2H-pirazolo[3,4-f]quinolin-7-carboxílico (13)

A un recipiente de reacción de 20 l se le añadieron (3S,3aR)-2-(3-cloro-4-cianofenil)-3-ciclopentil-3,3a,4,5-tetrahidro-2H-pirazolo[3,4-f]quinolin-7-carboxilato de etilo (1066 g), tetrahidrofurano (4 l) y metanol (2 l). Se agitó la mezcla a -10°C durante 10 minutos y se le añadió lentamente a la misma una disolución (1,2 l) de hidróxido de sodio (192 g) en agua. Se agitó la mezcla resultante a temperatura ambiente durante 4 horas y se retiró a vacío la mitad del disolvente. Se ajustó la mezcla de reacción con ácido clorhídrico diluido a pH 3-4 y se filtró mediante succión. Se lavó el sólido con metanol frío y dietil éter, respectivamente, y se secó para producir ácido (3S,3aR)-2-(3-cloro-4-cianofenil)-3-ciclopentil-3,3a,4,5-tetrahidro-2H-pirazolo[3,4-f]quinolin-7-carboxílico (867 g), rendimiento: 86,7%.
10. Preparación de 2-cloro-4-[(3S,3aR)-3-ciclopentil-7-(4-hidroxipiperidin-1-carbonil)-3,3a,4,5-tetrahidro-2H-pirazolo[3,4-f]quinolin-2-il]benzonitrilo (compuesto de fórmula (1))
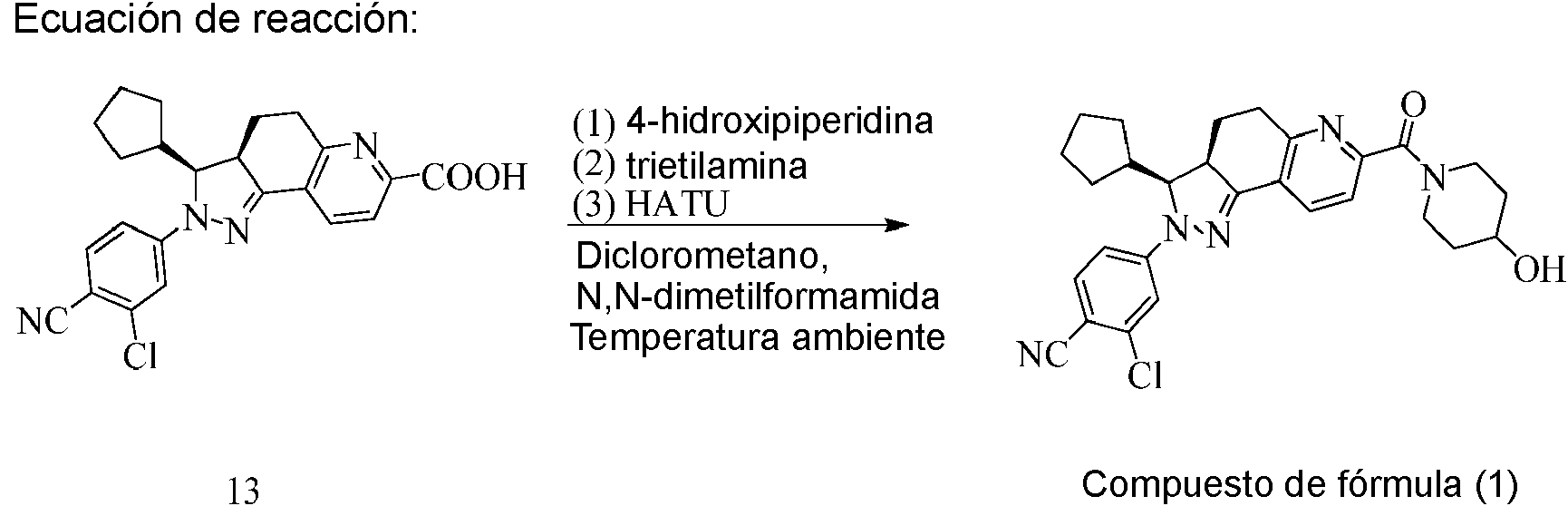
A un recipiente de reacción de 5 l se le añadieron ácido (3S,3aR)-2-(3-cloro-4-cianofenil)-3-ciclopentil-3,3a,4,5-tetrahidro-2H-pirazolo[3,4-f]quinolin-7-carboxílico (380 g), diclorometano (900 ml) y N,N-dimetilformamida (360 ml). Se añadió adicionalmente trietilamina (380 ml) con agitación. Se enfrió la mezcla hasta -10°C y se agitó adicionalmente durante 10 minutos. Se añadió una disolución (700 ml) de 4-hidroxipiperidina (137 g) en diclorometano. Se agitó la mezcla resultante durante 5 minutos. Se añadió hexafluorofosfato de 2-(7-aza-1H-benzotriazol-1-il)-1,1,3,3-tetrametiluronio (HATU) (380 g) y se llevó a cabo la reacción a temperatura ambiente durante 3 horas. Luego, se añadió adicionalmente hexafluorofosfato de 2-(7-aza-1H-benzotriazol-1-il)-1,1,3,3-tetrametiluronio (50 g) y se llevó a cabo la reacción durante 1 hora. Se retiró a vacío el disolvente. Se añadió el residuo gota a gota a una cantidad de 10 veces de agua y se separó un sólido. Se disolvió el sólido en diclorometano (2 l) y se lavó con agua (2 l) una vez. Se extrajo la fase acuosa con diclorometano (500 ml) una vez. Se combinaron las fases orgánicas, se secaron y se concentraron hasta sequedad para producir un sólido de fórmula (1) (290 g), rendimiento: 63,7%.
Fórmula molecular: C2sH30ClN5O2; EM (M+H): 504
1H-RMN (CDCla, 400 MHz): 88,375-8,395 (1H, d), 7,423-7,474 (2H, m), 7,264-7,276 (1H, d), 6,968-6,995 (1H, dd), 4,641-4,678 (1H, dd), 4,17-4,21 (1H, m), 3,99 (1H, s), 3,76-3,79 (1H, m), 3,515 (1H, m), 3,41-3,44 (1H, m), 3,230-3,322 (2H, m), 2,995 (1H, m), 2,321-2,352 (1H, m), 2,10-2,15 (2H, m), 1,978-2,089 (2H, m), 1,863-1,895 (1H, m), 1,758-1,777 (1H, m), 1,433-1,663 (7H, m), 1,221-1,352 (2H, m).
Ejemplo 2: Preparación de la forma cristalina I de 2-cloro-4-[(3S,3aR)-3-ciclopentil-7-(4-hidroxipiperidin-1-carbonil)-3,3a,4,5-tetrahidro-2H-pirazolo[3,4-f]quinolin-2-il]benzonitrilo (1)
Se añadió el compuesto representado por la fórmula (1) (1 g) tal como se preparó en el ejemplo 1 en etanol anhidro (3 ml). Se calentó la mezcla hasta 80°C hasta que la disolución se volvió transparente. Después se enfrió
lentamente la mezcla hasta temperatura ambiente. Se filtró el sólido y se lavó con etanol anhidro tres veces. Se secó el sólido resultante a 60°C a vacío durante 12 horas para producir la forma cristalina I. El espectro de difracción de rayos X de polvo (XRD) de la forma cristalina I se mostró en la figura 1 y sus principales parámetros fueron los siguientes:
Ángulo 29 Valor de d Intensidad
Unidad: grado (°) Unidad: ángstrom Unidad: %
3,423 25,79197 2,2
4,571 19,31406 2,9
5,664 15,58973 3,1
8,343 10,58992 4,1
9,703 910809 47,6
10,764 8,21218 18,1
11,137 7,93840 16,5
12,122 7,29549 30,8
12,785 6,91833 48,9
14,690 6,02527 77,8
15,331 5,77476 39,2
16,834 5,26232 56,3
17,310 5,11893 100,0
17,987 4,92761 19,5
19,342 4,58548 71,7
19,639 4,51659 97,6
20,769 4,27346 11,5
21,191 4,18935 12,5
22,439 3,95900 40,0
23,159 3,83756 9,0
24,498 3,63081 21,9
26,103 3,41101 524
27,586 3,23093 20,4
29,564 3,01911 20,6
30,956 2,88642 12,0
31,954 2,79856 4,2
33,085 2,70537 4,9
33,516 2,67160 8,5
35,260 2,54333 4,0
35,946 2,49634 8,2
36,464 2,46209 9,4
37,729 2,38237 4,2
39,509 2,27904 6,2
40,615 2,21951 6,2
41,018 2,19865 6,8
42,503 2,12517 5,0
42,828 2,10982 6,0
44,139 2,05012 4,3
Ejemplo 3: Preparación de la forma cristalina I de 2-cloro-4-[(3S,3aR)-3-ciclopentil-7-(4-hidroxipiperidin-1-carbonil)-3,3a,4,5-tetrahidro-2H-pirazolo[3,4-f]quinolin-2-il]benzonitrilo (2)
Se disolvió el compuesto representado por la fórmula (1) (146 mg) tal como se preparó en el ejemplo 1 en acetonitrilo (50 ml) a 80°C. Después se enfrió lentamente la mezcla resultante hasta temperatura ambiente, se agitó durante la noche y se filtró para producir la forma cristalina I.
Ejemplo 4: Preparación de la forma cristalina I de 2-cloro-4-[(3S,3aR)-3-ciclopentil-7-(4-hidroxipiperidin-1-carbonil)-3,3a,4,5-tetrahidro-2H-pirazolo[3,4-f]quinolin-2-il]benzonitrilo (3)
Se añadió el compuesto representado por la fórmula (1) (200 mg) tal como se preparó en el ejemplo 1 a un matraz de fondo redondo de 100 ml. Se añadió acetato de etilo (10 ml). Se calentó la mezcla hasta 78°C bajo reflujo. Después se añadió etanol (0,5 ml) y se agitó la mezcla a 80°C. Se enfrió lentamente la disolución resultante hasta temperatura ambiente. Después de 2 días, se filtró la mezcla resultante para producir la forma cristalina I.
Ejemplo 5: Preparación de la forma cristalina I de 2-cloro-4-[(3S,3aR)-3-ciclopentil-7-(4-hidroxipiperidin-1-carbonil)-3,3a,4,5-tetrahidro-2H-pirazolo[3,4-f]quinolin-2-il]benzonitrilo (4)
Se colocó el compuesto representado por la fórmula (1) (100 mg) tal como se preparó en el ejemplo 1 en un matraz de fondo redondo de 100 ml. Se añadió acetona (3 ml) y se disolvió el compuesto. A la mezcla resultante se le añadió gota a gota n-heptano (20 ml) y se separó un sólido. Se filtró la mezcla resultante para producir la forma cristalina I.
Ejemplo 6: Preparación de la forma cristalina I de 2-cloro-4-[(3S,3aR)-3-ciclopentil-7-(4-hidroxipiperidin-1-carbonil)-3,3a,4,5-tetrahidro-2H-pirazolo[3,4-f]quinolin-2-il]benzonitrilo (5)
Se añadió el compuesto representado por la fórmula (1) (100 mg) tal como se preparó en el ejemplo 1 a un matraz de fondo redondo de 100 ml. Se añadió un disolvente mixto (0,5 ml, metanol:tetrahidrofurano=1:1). Se calentó la mezcla resultante hasta 60°C. Después se enfrió lentamente la mezcla hasta temperatura ambiente y se separó un sólido. Se filtró la mezcla resultante para producir la forma cristalina I.
Ejemplo 7: Preparación de la forma cristalina I de 2-cloro-4-[(3S,3aR)-3-ciclopentil-7-(4-hidroxipiperidin-1-carbonil)-3,3a,4,5-tetrahidro-2H-pirazolo[3,4-f]quinolin-2-il]benzonitrilo (6)
Se añadió el compuesto representado por la fórmula (1) (100 mg) tal como se preparó en el ejemplo 1 a un matraz de fondo redondo de 100 ml. Se añadió un disolvente mixto (3,5 ml, acetonitrilo:acetona=1:1) al matraz de fondo redondo. Se disolvió la mezcla bajo calentamiento a 60°C y con agitación, después se enfrió lentamente hasta temperatura ambiente para separar un sólido. Se filtró la mezcla resultante para producir la forma cristalina I.
Ejemplo 8: Preparación de la forma cristalina III de 2-cloro-4-[(3S,3aR)-3-ciclopentil-7-(4-hidroxipiperidin-1-carbonil)-3,3a,4,5-tetrahidro-2H-pirazolo[3,4-f]quinolin-2-il]benzonitrilo (1)
Se añadió el compuesto representado por la fórmula (1) (200 mg) tal como se preparó en el ejemplo 1 a metanol (4 ml). Se disolvió el compuesto a 80°C. Se añadió la disolución resultante gota a gota a agua (40 ml). Se filtró la mezcla resultante para producir la forma cristalina III. El espectro de difracción de rayos X de polvo (XRD) de la forma cristalina III se mostró en la figura 3 y sus principales parámetros fueron los siguientes:
Ángulo 29 Valor de d Intensidad
Unidad: grado (°) Unidad: ángstrom Unidad: %
3,845 22,96100 44,0
6,712 13,15849 5,8
7,697 11,47721 9,4
9,023 9,79324 8,1
9,970 8,86513 56,2
11,600 7,62276 17,7
12,610 7,01426 14,0
14,179 6,24137 23,1
15,266 5,79922 855
15,804 5,60313 49,9
16,834 5,26242 37,2
17,664 5,01696 39,5
19,455 455904 100,0
20,552 4,31804 85,1
21,124 4,20247 14,3
22,123 4,01479 274
22,972 3,86842 31,8
24,109 3,68836 33,2
25,055 3,55133 77,9
26,033 3,42005 46,2
27,241 3,27104 47,3
28,408 3,13930 17,9
29,946 2,98146 19,7
30,949 2,88708 17,5
32,306 2,76887 21,4
32,997 2,71244 125
33,935 2,63952 14,0
34,882 2,57004 18,4
35,381 2,53492 11,6
36,952 2,43069 13,1
38,563 2,33273 10,5
39,280 2,29184 12,5
40,396 2,23102 132
41,806 2,15901 12,4
Ejemplo 9: Preparación de la forma cristalina III de 2-cloro-4-[(3S,3aR)-3-ciclopentil-7-(4-hidroxipiperidin-1-carbonil)-3,3a,4,5-tetrahidro-2H-pirazolo[3,4-f]quinolin-2-il]benzonitrilo (2)
Se añadió el compuesto representado por la fórmula (1) (100 mg) tal como se preparó en el ejemplo 1 a un tubo de centrífuga de 10 ml. Se añadió una disolución mixta (8 ml, agua:acetonitrilo=10:1) al tubo de centrífuga y se agitó. Se filtró la mezcla resultante para producir la forma cristalina III.
Ejemplo 10: Preparación de la forma cristalina II de 2-cloro-4-[(3S,3aR)-3-ciclopentil-7-(4-hidroxipiperidin-1-carbonil)-3,3a,4,5-tetrahidro-2H-pirazolo[3,4-f]quinolin-2-il]benzonitrilo
Se secó la forma cristalina III tal como se preparó en el ejemplo 8 durante 12 horas a vacío a temperatura ambiente para producir la forma cristalina II. El espectro de difracción de rayos X de polvo (XRD) de la forma cristalina II se mostró en la figura 2 y sus principales parámetros fueron los siguientes:
Ángulo 29 Valor de d Intensidad
Unidad: grado (°) Unidad: ángstrom Unidad: %
3,574 24,70236 4,9
4,451 19,83656 56,2
7,515 11,75417 5,7
8,478 10,42104 28,0
9,002 9,81527 44,9
10,638 8,30970 23,2
12,216 7,23974 32,2
13,542 6,53344 20,0
14,049 6,29880 38,1
14,607 6,05934 90,9
16,422 5,39356 31,3
17,123 5,17422 26,2
17,992 4,92615 56,8
18,695 4,74270 57,7
19,873 4,46396 100,0
21,230 4,18168 68,6
22,819 3,89399 28,9
24,589 3,61751 60,0
25,769 3,45445 26,8
26,520 3 35833 31,3
27,023 3,29690 28,0
28,447 3,13509 21,9
30,037 2,97262 10,2
30,853 2,89580 6,8
32,143 2,78249 7,76,2
34,206 2,61925 5,7
35,439 2,53088 6,6
36,144 248317 6,6
39,188 2,29700 4,9
40,726 2,21373 6,5
42,424 2,12895 6,2
43,393 2,08364
Los siguientes ensayos no forman parte de la invención y se presentan únicamente con fines de ilustración.
Ensayo 1: Estabilidad para la forma cristalina I del presente compuesto
Muestra:
Forma cristalina I del compuesto representado por la fórmula (1): se preparó la forma cristalina I según el ejemplo 2.
Condiciones de prueba para investigar los factores influyentes:
Pruebas a alta temperatura:
(1) Se colocó la forma cristalina I del compuesto representado por la fórmula (1) sobre un vidrio de reloj seco y limpio y se mantuvo a 60°C durante 10 días. Se tomaron muestras respectivamente el día 5 y el día 10. Se midieron los contenidos de la sustancia relevante y el compuesto representado por la fórmula (1) en la muestra y se compararon con los contenidos de los mismos en la muestra tomada el día 0;
(2) Se envasó y selló la forma cristalina I del compuesto representado por la fórmula (1) con una bolsa de polietileno de baja densidad para uso farmacéutico en la capa interior y con un película compuesta de poliéster/aluminio/polietileno para envase farmacéutico en la capa exterior, y se mantuvo a 60°C durante 10 días. Se tomaron muestras respectivamente el día 5 y el día 10. Se midieron los contenidos de la sustancia relevante y el compuesto representado por la fórmula (1) en la muestra y se compararon con los contenidos de los mismos en la muestra tomada el día 0.
Prueba a alto contenido en humedad: se colocó la forma cristalina I del compuesto representado por la fórmula (1) sobre un vidrio de reloj seco y limpio y se mantuvo a 25°C bajo una humedad relativa del 90%±5% durante 10 días. Se tomaron muestras respectivamente el día 5 y el día 10. Se midieron los contenidos de la sustancia relevante y el compuesto representado por la fórmula (1) en la muestra y se compararon con los contenidos de los mismos en la muestra tomada el día 0.
Prueba de fotoestabilidad: se colocó la forma cristalina I del compuesto representado por la fórmula (1) sobre un vidrio de reloj seco y limpio y se mantuvo a una iluminancia de 4500 Lx±500 Lx en una caja de iluminación
durante 10 días. Se tomaron muestras respectivamente el día 5 y el día 10. Se midieron los contenidos de la sustancia relevante y el compuesto representado por la fórmula (1) en la muestra y se compararon con los contenidos de los mismos en la muestra tomada el día 0.
Medición del contenido del compuesto representado por la fórmula (1)
Se midió el contenido del compuesto representado por la fórmula (1) usando un método convencional externo según la cromatografía de líquidos de alta resolución en la Farmacopea de China, anexo V D, edición de 2010. Medición del contenido de la sustancia relevante
Se midió el contenido de la sustancia relevante usando un método de normalización de áreas según la cromatografía de líquidos de alta resolución en la Farmacopea de China, anexo V D, edición de 2010.
Los resultados de prueba se mostraron en la tabla 1.
Tabla 1: Resultados de investigación de las pruebas de factores influyentes para la forma cristalina I del compuesto representado por la fórmula (1)
Contenido del compuesto Contenido de la Condiciones Día representado por la fórmula (1) sustancia relevante
(%) (%)
0 99,5 0,54
5 98,9 0,82
60°C-(1)
10 98,1 0,97
5 99,7 0,58
60°C-(2)
10 99,0 0,60
5 99,1 0,56
HR del 90%±5%
10 99,0 0,56
5 98,1 1,2
4500 Lx±500 Lx
10 97,0 2,0
Los presentes inventores investigaron la estabilidad de la forma cristalina I del compuesto representado por la fórmula (1). A partir de los resultados de investigación, pudo aclararse que los contenidos de la sustancia relevante y el compuesto representado por la fórmula (1) en la forma cristalina I del compuesto representado por la fórmula (1) no cambiaron sustancialmente a alta temperatura, a alto contenido en humedad y en la condición de iluminación. La forma cristalina I fue superior a la forma amorfa en cuanto a estabilidad, lo que mostró que la forma cristalina I del compuesto representado por la fórmula (1) tenía una estabilidad relativamente alta que era adecuada para la fabricación, el almacenamiento y el transporte del fármaco y era favorable para garantizar la validez y la seguridad en el uso del fármaco.
Ensayo 2
Estabilidad de la forma cristalina II del presente compuesto
Muestra:
Forma cristalina II del compuesto representado por la fórmula (1): se preparó la forma cristalina II según el ejemplo 4.
Condiciones de prueba para investigar los factores influyentes:
Pruebas a alta temperatura:
(1) Se colocó la forma cristalina II del compuesto representado por la fórmula (1) sobre un vidrio de reloj seco y limpio y se mantuvo a 60°C durante 10 días. Se tomó una muestra el día 10. Se midieron los contenidos de la sustancia relevante y el compuesto representado por la fórmula (1) en la muestra y se compararon con los contenidos de los mismos en la muestra tomada el día 0;
(2) Se envasó y selló la forma cristalina II del compuesto representado por la fórmula (1) con una bolsa de polietileno de baja densidad para uso farmacéutico en la capa interior y con un película compuesta de
poliéster/aluminio/polietileno para envase farmacéutico en la capa exterior, y se mantuvo a 60°C durante 10 días. Se tomó una muestra el día 10. Se midieron los contenidos de la sustancia relevante y el compuesto representado por la fórmula (1) en la muestra y se compararon con los contenidos de los mismos en la muestra tomada el día 0.
Prueba a alto contenido en humedad: se colocó la forma cristalina II del compuesto representado por la fórmula (1) sobre un vidrio de reloj seco y limpio y se mantuvo a 25°C bajo una humedad relativa del 90%±5% durante 10 días. Se tomó una muestra respectivamente el día 10. Se midieron los contenidos de la sustancia relevante y el compuesto representado por la fórmula (1) en la muestra y se compararon con los contenidos de los mismos en la muestra tomada el día 0.
Prueba de fotoestabilidad: se colocó la forma cristalina II del compuesto representado por la fórmula (1) sobre un vidrio de reloj seco y limpio y se mantuvo a una iluminancia de 5000 Lx±500 Lx en una caja de iluminación durante 10 días. Se tomó una muestra el día 10. Se midieron los contenidos de la sustancia relevante y el compuesto representado por la fórmula (1) en la muestra y se compararon con los contenidos de los mismos en la muestra tomada el día 0.
Medición del contenido del compuesto representado por la fórmula (1)
Se midió el contenido del compuesto representado por la fórmula (1) usando un método convencional externo según la cromatografía de líquidos de alta resolución en la Farmacopea de China, anexo V D, edición de 2010. Medición del contenido de la sustancia relevante
Se midió el contenido de la sustancia relevante usando un método de normalización de áreas según la cromatografía de líquidos de alta resolución en la Farmacopea de China, anexo V D, edición de 2010.
Los resultados de prueba se mostraron en la tabla 2.
Tabla 2: Resultados de investigación de las pruebas de factores influyentes para la forma cristalina II del compuesto representado por la fórmula (1)
Contenido del compuesto Contenido de la Condición Día representado por la fórmula (1) sustancia relevante
(%) (%)
0 96,2 2,6
60°C-(1) 10 96,2 2,8
60°C-(2) 10 97,1 2,7
HR del 90%±5% 10 96,9 2,6
5000 Lx±500 Lx 10 95,5 2,8
Los presentes inventores investigaron la estabilidad de la forma cristalina II del compuesto representado por la fórmula (1). A partir de los resultados de investigación, pudo aclararse que los contenidos de la sustancia relevante y el compuesto representado por la fórmula (1) en la forma cristalina II del compuesto representado por la fórmula (1) no cambiaron sustancialmente a alta temperatura, a alto contenido en humedad y en la condición de iluminación. La forma cristalina II fue superior a la forma amorfa en cuanto a estabilidad, lo que mostró que la forma cristalina II del compuesto representado por la fórmula (1) tenía una estabilidad relativamente alta que era adecuada para la fabricación, el almacenamiento y el transporte del fármaco y era favorable para garantizar la validez y la seguridad en el uso del fármaco.
Ensayo 3
Protección de órganos y efecto de despresurización de la forma cristalina I sobre ratas Dahl sensibles a sales (Dahl/SS) inducidas por alto contenido en sales
Muestra y preparación de muestra:
Dispersión de sólidos de la forma cristalina I (forma cristalina I:PVPK30=1:8 (p/p)): se formuló la dispersión de sólidos de la forma cristalina I con una cantidad adecuada de agua estéril para inyección para dar suspensiones que tenían concentraciones de 0,03, 0,10, 0,30 y 1,00 mg/ml. Se preparó la suspensión antes de su uso cada día.
Grupo y modelo animal:
Animales de experimentación: ratas Dahl/ss macho de calidad SPF (libre de patógenos específicos), de 8 9 semanas, adquiridas a través de Vital River Laboratories de HARLAN LABORATORIES, INC. Después de una semana de cuarentena normal, en el experimento se usaron las ratas en buen estado y condición.
Las ratas Dahl/ss se dividieron aleatoriamente en seis grupos según la medición de la tensión arterial antes de la administración:
Control normal (n=10),
Grupo modelo (NaCl al 4%, n=12),
Grupos de tratamiento con la forma cristalina I, cuatro grupos:
Grupo de tratamiento con 0,3 mg/kg/día (n=11),
Grupo de tratamiento con 1 mg/kg/día (n=11),
Grupo de tratamiento con 3 mg/kg/día (n=11),
Grupo de tratamiento con 10 mg/kg/día (n=11),
n es el número de ratas.
Método del experimento:
Se evaluó la actividad farmacodinámica in vivo de la forma cristalina I mediante el modelo de rata Dahl/ss con hipertensión y lesión renal.
Una semana antes del experimento, se monitorizaron las tensiones arteriales de las ratas dos veces mediante medición de la tensión arterial en el manguito de la cola, de modo que las ratas pudieran adaptarse a la operación de monitorización de la tensión arterial. Se monitorizaron las tensiones arteriales de las ratas una vez antes de comenzar el experimento y se usaron como las tensiones arteriales básicas antes de la administración. Se dividieron aleatoriamente las ratas en grupos según las tensiones arteriales medidas antes de la administración. Se estableció el modelo alimentando a las ratas con un pienso con alto contenido en sales (pienso para animales de experimentación AIN-93G que contiene NaCl al 4%) durante 42 días, en los que las ratas tenían acceso libre a alimento y agua. Y las ratas en el grupo de control se alimentaron con un pienso con bajo contenido en sodio.
A las ratas en los grupos de tratamiento con la forma cristalina I se les administraron, respectivamente, la forma cristalina I en una dosificación de 0,3, 1, 3 y 10 mg/kg/día. A las ratas en los grupos de tratamiento se les dosificó por vía oral a través de una sonda gástrica dos veces al día con 5 ml/kg. A las ratas en el grupo modelo y en el control normal se les administraron el mismo volumen de agua estéril para inyección.
Medición de la tensión arterial (tensión arterial sistólica, abreviada como TAS): se midió la tensión arterial una vez a la semana durante seis semanas. Se analizaron los cambios en la tensión arterial para cada grupo.
Exámenes anatomopatológicos del riñón y del corazón: después del experimento, se sacrificaron las ratas de manera indolora. Se recogieron el corazón y los riñones bilaterales para el análisis histopatológico. Se puntuó la lesión renal y se analizó de manera semicuantitativa basándose en la tinción con hematoxilina-eosina (HE). Se analizó la lesión cardíaca mediante la medición del grosor de la pared del ventrículo izquierdo.
Resultado del experimento:
Efecto de despresurización: pudo observarse que la forma cristalina I mostró un efecto de despresurización significativo en el modelo y presentó cierta relación dependiente de la dosis (tabla 3).
Tabla 3: TAS (mmHg, media±DE)
Grupo Antes de la administración Después de la administración (día 41) Control normal 139,3±13,4 149,4±9,6
Grupo modelo 139,9±10,7 183,0±12,8*
Forma cristalina I del
compuesto 0,3 mg/kg/día presente 140,8±9,6 166,5±10,2*#
Forma cristalina I del
compuesto 1 mg/kg/día presente 140,9±9,5 148,7±9,6#
Forma cristalina I del presente 141,Q±9,4
compuesto 3 mg/kg/día 141,6±8,0#
Forma cristalina I del presente 1412±92
compuesto 10 mg/kg/día 141,2±9,6#
*p<0,05, en comparación con el control normal; #p<0,05, en comparación con el grupo modelo.
Protecciones del riñón y del corazón: según las puntuaciones de la lesión renal, los grupos de tratamiento con forma cristalina I mostraron una notable prevención del aumento en la puntuación de la lesión renal (tabla 4). En comparación con el grupo modelo, la forma cristalina I disminuyó significativamente el grosor de la pared del ventrículo izquierdo (tabla 4).
Tabla 4: Protección contra lesión renal y lesión cardíaca
Grupo Puntuaciones de la lesión renal Grosor de la pared del ventrículo izquierdo (media±DE) (cm, media±DE)
Control normal 0,93±0,079 0,401±0,014
Grupo modelo 2,05±0,091* 0,410±0,026
Forma cristalina I 2,00,3 mg/kg/día 2±0,268* 0,392±0,024
Forma cristalina
1 mg/kg/día I 1,25±0,428# 0,383±0,017#
Forma cristalina
3 mg/kg/día I 1,84±0,596* 0,381±0,026#
Forma cristalina
10 mg/kg/día I 1,71 ±0,719* 0,383±0,019#
*p<0,05, en comparación con el control normal; #p<0,05, en comparación con el grupo modelo.
En los modelos de hipertensión y nefrosis de las ratas Dahl/ss inducidas por alto contenido en sales, la forma cristalina I mostró un efecto de despresurización y una protección contra lesión renal notables.
Claims (11)
1. Procedimiento para preparar un compuesto representado por la fórmula (1), 2-cloro-4-[(3S,3aR)-3-ciclopentil-7-(4-hidroxipiperidin-1-carbonil)-3,3a,4,5-tetrahidro-2H-pirazolo[3,4-f]quinolin-2-il]benzonitrilo,

Fórmula (1)
que se caracteriza por comprender las etapas de:
(9) Resolver quiralmente 2-(3-cloro-4-cianofenil)-3-ciclopentil-3,3a,4,5-tetrahidro-2H-pirazolo[3,4-f]quinolin-7-carboxilato de etilo para producir (3S,3aR)-2-(3-cloro-4-cianofenil)-3-ciclopentil-3,3a,4,5-tetrahidro-2H-pirazolo[3,4-f]quinolin-7-carboxilato de etilo;
(10) Hidrolizar (3S,3aR)-2-(3-doro-4-danofenil)-3-ddopentil-3,3a,4,5-tetrahidro-2H-pirazolo[3,4-f]quinolin-7-carboxilato de etilo para producir ácido (3S,3aR)-2-(3-cloro-4-cianofenil)-3-ciclopentil-3,3a,4,5-tetrahidro-2H-pirazolo[3,4-f]quinolin-7-carboxílico;
(11) Someter ácido (3S,3aR)-2-(3-cloro-4-cianofenil)-3-ciclopentil-3,3a,4,5-tetrahidro-2H-pirazolo[3,4-f]quinolin-7-carboxílico y 4-hidroxipiperidina a una reacción de condensación para producir el compuesto representado por la fórmula (1).
2. Procedimiento según la reivindicación 1, que se caracteriza por comprender las etapas de:
(1) Someter 1,3-ciclohexanodiona, acetato de amonio y acroleína a reacciones de condensación y adición para producir 5-oxo-5,6,7,8-tetrahidroquinolina;
(2) Someter 5-oxo-5,6,7,8-tetrahidroquinolina a una reacción de oxidación para producir N-óxido de 5-oxo-5,6,7,8-tetrahidroquinolina;
(3) Someter N-óxido de 5-oxo-5,6,7,8-tetrahidroquinolina, cloruro N,N-dimetilcarbámico y cianuro de trimetilsililo a una reacción de sustitución para producir 5-oxo-5,6,7,8-tetrahidroquinolin-2-carbonitrilo;
(4) Someter 5-oxo-5,6,7,8-tetrahidroquinolin-2-carbonitrilo a reacciones de hidrólisis y esterificación para producir 5-oxo-5,6,7,8-tetrahidroquinolin-2-carboxilato de etilo;
(5) Someter 5-oxo-5,6,7,8-tetrahidroquinolin-2-carboxilato de etilo y ciclopentanocarbaldehído a una reacción de condensación para producir (E)-6-ciclopentilmetilen-5-oxo-5,6,7,8-tetrahidroquinolin-2-carboxilato de etilo;
(6) Someter 2-cloro-4-fluorobenzonitrilo e hidrazina hidratada a una reacción de sustitución para producir 2-cloro-4-hidrazinobenzonitrilo;
(7) Someter 2-cloro-4-hidrazinobenzonitrilo y ácido clorhídrico a una reacción de formación de sal para producir clorhidrato de 2-cloro-4-hidrazinobenzonitrilo;
(8) Someter (E)-6-ciclopentilmetilen-5-oxo-5,6,7,8-tetrahidroquinolin-2-carboxilato de etilo y clorhidrato de 2-cloro-4-hidrazinobenzonitrilo a una reacción de condensación para producir 2-(3-cloro-4-cianofenil)-3-ciclopentil-3,3a,4,5-tetrahidro-2H-pirazolo[3,4-f]quinolin-7-carboxilato de etilo;
(9) Resolver quiralmente 2-(3-cloro-4-cianofenil)-3-ciclopentil-3,3a,4,5-tetrahidro-2H-pirazolo[3,4-f]quinolin-7-carboxilato de etilo para producir (3S,3aR)-2-(3-cloro-4-cianofenil)-3-ciclopentil-3,3a,4,5-tetrahidro-2H-pirazolo[3,4-f]quinolin-7-carboxilato de etilo;
(10) Hidrolizar (3S,3aR)-2-(3-cloro-4-cianofenil)-3-ciclopentil-3,3a,4,5-tetrahidro-2H-pirazolo[3,4-f]quinolin-7-carboxilato de etilo para producir ácido (3S,3aR)-2-(3-cloro-4-cianofenil)-3-ciclopentil-3,3a,4,5-tetrahidro-2H-pirazolo[3,4-f]quinolin-7-carboxílico;
(11) Someter ácido (3S,3aR)-2-(3-cloro-4-cianofenil)-3-ciclopentil-3,3a,4,5-tetrahidro-2H-pirazolo[3,4-f]quinolin-7-carboxílico y 4-hidroxipiperidina a una reacción de condensación para producir el compuesto
representado por la fórmula (1).
3. Procedimiento según la reivindicación 1 ó 2, que comprende una de las siguientes etapas:
colocar el compuesto representado por la fórmula (1) obtenido en la etapa (11) en un alcohol inferior anhidro seleccionado de metanol, etanol y n-propanol, acetonitrilo, un disolvente mixto de acetato de etilo y etanol, un disolvente mixto de metanol y tetrahidrofurano o un disolvente mixto de acetonitrilo y acetona, calentar la disolución resultante hasta que se vuelva transparente, después enfriar la disolución resultante para separar un sólido y filtrar y secar el sólido separado; o
colocar el compuesto representado por la fórmula (1) obtenido en la etapa (11) en un alcohol inferior seleccionado de metanol, etanol y n-propanol para disolverlo, después añadir la disolución resultante gota a gota a agua, filtrar la mezcla resultante y opcionalmente secar la sustancia filtrada a vacío; o
lavar el compuesto representado por la fórmula (1) obtenido en la etapa (11) con una disolución mixta de agua y acetonitrilo, filtrar la mezcla resultante y opcionalmente secar la sustancia filtrada a vacío; o disolver el compuesto representado por la fórmula (1) obtenido en la etapa (11) en acetona, añadir la disolución resultante gota a gota a n-heptano y filtrar la mezcla resultante.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201210563636 | 2012-12-22 | ||
| PCT/CN2013/090252 WO2014094664A1 (zh) | 2012-12-22 | 2013-12-23 | 作为盐皮质激素受体拮抗剂的化合物的晶型及其制备方法 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2897496T3 true ES2897496T3 (es) | 2022-03-01 |
Family
ID=50977609
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES13864994T Active ES2897496T3 (es) | 2012-12-22 | 2013-12-23 | Procedimiento para la preparación de un compuesto usado como antagonista del receptor de mineralocorticoides |
Country Status (16)
| Country | Link |
|---|---|
| US (1) | US9809589B2 (es) |
| EP (2) | EP2937348B1 (es) |
| JP (6) | JP2016503041A (es) |
| KR (1) | KR101737883B1 (es) |
| CN (1) | CN105026391B (es) |
| AU (1) | AU2013362400B2 (es) |
| BR (1) | BR112015014839B1 (es) |
| CA (1) | CA2895968C (es) |
| ES (1) | ES2897496T3 (es) |
| IL (1) | IL239581B (es) |
| MX (1) | MX360889B (es) |
| NZ (1) | NZ709361A (es) |
| RU (1) | RU2622105C2 (es) |
| SG (1) | SG11201504928XA (es) |
| WO (1) | WO2014094664A1 (es) |
| ZA (1) | ZA201504485B (es) |
Families Citing this family (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2014094664A1 (zh) * | 2012-12-22 | 2014-06-26 | 山东亨利医药科技有限责任公司 | 作为盐皮质激素受体拮抗剂的化合物的晶型及其制备方法 |
| CR20240015A (es) * | 2016-09-24 | 2024-04-05 | Novo Nordisk As | COMPOSICIÓN FARMACÉUTICA CON ANTAGONISTA DEL RECEPTOR DE MINERALOCORTICOIDES Y SU USO (Divisional Expediente 2019-203) |
| PE20212179A1 (es) * | 2019-03-01 | 2021-11-09 | Kbp Biosciences Co Ltd | Procedimiento para preparar un compuesto triciclico fusionado, e intermedio del mismo |
Family Cites Families (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS5245716B1 (es) * | 1970-07-10 | 1977-11-17 | ||
| CN85101404A (zh) * | 1985-05-16 | 1987-01-17 | 帝人株式会社 | 头孢菌素衍生物的生产过程 |
| JPH04235188A (ja) * | 1990-06-19 | 1992-08-24 | Takeda Chem Ind Ltd | ペネム化合物の結晶、その製造方法および抗菌剤 |
| DE4235133A1 (de) * | 1992-10-19 | 1994-04-21 | Bayer Ag | Kristallines (R)-(-)-2-Cycloheptyl-N-methylsulfonyl-[4-(2-chinolinyl-methoxy)-phenyl]-acetamid |
| JP3623531B2 (ja) * | 1993-06-07 | 2005-02-23 | ビーエーエスエフ アクチェンゲゼルシャフト | 結晶質l−アスコルビン酸−2−燐酸エステルマグネシウム塩の製造法 |
| WO1998000969A1 (en) * | 1996-06-28 | 1998-01-08 | Philips Electronics N.V. | Method and arrangement for transmitting teletext pages |
| CA2263586A1 (en) * | 1996-09-05 | 1998-03-12 | Eifion Phillips | Novel aryl-pyridazines |
| WO2004048378A1 (en) | 2002-11-22 | 2004-06-10 | Active Biotech Ab | Pyrazoloquinolines with immunomodulating activity |
| CA2596737A1 (en) * | 2005-03-21 | 2006-09-28 | Eisai R & D Management Co., Ltd. | Pyrimidine compound crystal or amorphous form and process for producing the same |
| DE102007054497B3 (de) * | 2007-11-13 | 2009-07-23 | Sanofi-Aventis Deutschland Gmbh | Neue kristalline Diphenylazetidinonhydrate und Verfahren zu deren Herstellung |
| CN102372710A (zh) * | 2010-08-18 | 2012-03-14 | 山东轩竹医药科技有限公司 | 作为盐皮质激素受体拮抗剂的并环类化合物 |
| MY161429A (en) * | 2010-09-30 | 2017-04-14 | Toyama Chemical Co Ltd | Sodium salt of 6-fluoro-3-hydroxy-2-pyrazine carboxamide |
| WO2014094664A1 (zh) * | 2012-12-22 | 2014-06-26 | 山东亨利医药科技有限责任公司 | 作为盐皮质激素受体拮抗剂的化合物的晶型及其制备方法 |
-
2013
- 2013-12-23 WO PCT/CN2013/090252 patent/WO2014094664A1/zh not_active Ceased
- 2013-12-23 EP EP13864994.2A patent/EP2937348B1/en active Active
- 2013-12-23 NZ NZ709361A patent/NZ709361A/en unknown
- 2013-12-23 AU AU2013362400A patent/AU2013362400B2/en active Active
- 2013-12-23 JP JP2015548180A patent/JP2016503041A/ja active Pending
- 2013-12-23 SG SG11201504928XA patent/SG11201504928XA/en unknown
- 2013-12-23 ES ES13864994T patent/ES2897496T3/es active Active
- 2013-12-23 CA CA2895968A patent/CA2895968C/en active Active
- 2013-12-23 BR BR112015014839-5A patent/BR112015014839B1/pt active IP Right Grant
- 2013-12-23 MX MX2015008193A patent/MX360889B/es active IP Right Grant
- 2013-12-23 RU RU2015130222A patent/RU2622105C2/ru active
- 2013-12-23 KR KR1020157019924A patent/KR101737883B1/ko not_active Expired - Fee Related
- 2013-12-23 CN CN201380066871.7A patent/CN105026391B/zh active Active
- 2013-12-23 US US14/653,933 patent/US9809589B2/en not_active Expired - Fee Related
- 2013-12-23 EP EP21194531.6A patent/EP3954688A1/en not_active Withdrawn
-
2015
- 2015-06-22 ZA ZA2015/04485A patent/ZA201504485B/en unknown
- 2015-06-22 IL IL239581A patent/IL239581B/en active IP Right Grant
-
2017
- 2017-07-04 JP JP2017131078A patent/JP2017165787A/ja active Pending
-
2019
- 2019-02-05 JP JP2019018876A patent/JP2019065057A/ja active Pending
-
2020
- 2020-04-16 JP JP2020073415A patent/JP6986197B6/ja active Active
-
2021
- 2021-11-02 JP JP2021179425A patent/JP2022009922A/ja not_active Withdrawn
-
2023
- 2023-11-17 JP JP2023195879A patent/JP2024009114A/ja not_active Withdrawn
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2557318T3 (es) | Hidratos de cloruro de metiltioninio (azul de metileno) cristalinos | |
| WO2022258060A1 (zh) | 一种lanifibranor的晶型及其制备方法 | |
| JP2024009114A (ja) | ミネラルコルチコイド受容体拮抗剤としての化合物の結晶形及びその調製方法 | |
| ES2212759T3 (es) | Nuevas formas polimorfas cristalinas de hidrocloruro de lercanidipino y procedimientos para su preparacion. | |
| WO2019033969A1 (zh) | 替米沙坦与氢氯噻嗪的共晶 | |
| CN102351784B (zh) | 一种盐酸贝尼地平晶型及其应用 | |
| CN119613419A (zh) | 氘代的二芳基甘脲四聚体化合物及其用途 | |
| HK40062778A (en) | Crystal forms of compound used as mineralocorticoid receptor antagonist and methods for their preparation | |
| CN111670191B (zh) | 吡啶酮衍生物的晶型及制备方法和应用 | |
| CN105111195A (zh) | 他克林-联苯双酯杂合物、其制备方法及应用 | |
| HK1212676B (en) | Process for the preparation of a compound used as mineralocorticoid receptor antagonist | |
| CN102020602B (zh) | 盐酸乐卡地平晶型及其制备方法和含有该晶型的药物组合物 | |
| CN100390172C (zh) | 一种甲磺酸多拉司琼晶型及其制备方法 | |
| ES2596293T3 (es) | Sales de solifenacina | |
| CN117105857B (zh) | 一种米力农-肉桂酸晶体以及制备方法 | |
| JP2020528067A (ja) | ステロイド系誘導体fxrアゴニストの結晶又は非晶質、その製造方法及び使用 | |
| CN106831713A (zh) | 一种多靶点Aβ寡聚化抑制剂的合成方法及应用 | |
| CN108929269A (zh) | 苄异喹啉类非去极化肌松剂及其制备方法和用途 | |
| HK1217019B (en) | Crystal form of compound used as mineralocorticoid receptor antagonist and preparation method therefor | |
| CN105330659A (zh) | 一种利伐沙班化合物 | |
| CS219948B2 (en) | Method of preparation of the stereoisomere 1,4,3,6-dianhydro-2,5-diazido-2,5-dideoxyhexite | |
| CN105399727A (zh) | 盐酸瑞伐拉赞新结晶形态 | |
| CN105646387A (zh) | 一种塞利司他化合物 |