ES2905359T3 - Moléculas de suministro de fármacos citotóxicos dirigidas al VIH (CDM-H), actividad citotóxica contra el virus de la inmunodeficiencia humana y métodos de uso - Google Patents
Moléculas de suministro de fármacos citotóxicos dirigidas al VIH (CDM-H), actividad citotóxica contra el virus de la inmunodeficiencia humana y métodos de uso Download PDFInfo
- Publication number
- ES2905359T3 ES2905359T3 ES13780662T ES13780662T ES2905359T3 ES 2905359 T3 ES2905359 T3 ES 2905359T3 ES 13780662 T ES13780662 T ES 13780662T ES 13780662 T ES13780662 T ES 13780662T ES 2905359 T3 ES2905359 T3 ES 2905359T3
- Authority
- ES
- Spain
- Prior art keywords
- hiv
- cells
- cdm
- cytotoxic
- cell
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H15/00—Compounds containing hydrocarbon or substituted hydrocarbon radicals directly attached to hetero atoms of saccharide radicals
- C07H15/26—Acyclic or carbocyclic radicals, substituted by hetero rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/66—Phosphorus compounds
- A61K31/67—Phosphorus compounds having sulfur as a ring hetero atom
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7042—Compounds having saccharide radicals and heterocyclic rings
- A61K31/7052—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides
- A61K31/706—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/54—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic compound
- A61K47/55—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic compound the modifying agent being also a pharmacologically or therapeutically active agent, i.e. the entire conjugate being a codrug
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/56—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic macromolecular compound, e.g. an oligomeric, polymeric or dendrimeric molecule
- A61K47/59—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic macromolecular compound, e.g. an oligomeric, polymeric or dendrimeric molecule obtained otherwise than by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyureas or polyurethanes
- A61K47/60—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic macromolecular compound, e.g. an oligomeric, polymeric or dendrimeric molecule obtained otherwise than by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyureas or polyurethanes the organic macromolecular compound being a polyoxyalkylene oligomer, polymer or dendrimer, e.g. PEG, PPG, PEO or polyglycerol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
- A61P31/18—Antivirals for RNA viruses for HIV
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/14—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing three or more hetero rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7028—Compounds having saccharide radicals attached to non-saccharide compounds by glycosidic linkages
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7028—Compounds having saccharide radicals attached to non-saccharide compounds by glycosidic linkages
- A61K31/7034—Compounds having saccharide radicals attached to non-saccharide compounds by glycosidic linkages attached to a carbocyclic compound, e.g. phloridzin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7028—Compounds having saccharide radicals attached to non-saccharide compounds by glycosidic linkages
- A61K31/7034—Compounds having saccharide radicals attached to non-saccharide compounds by glycosidic linkages attached to a carbocyclic compound, e.g. phloridzin
- A61K31/704—Compounds having saccharide radicals attached to non-saccharide compounds by glycosidic linkages attached to a carbocyclic compound, e.g. phloridzin attached to a condensed carbocyclic ring system, e.g. sennosides, thiocolchicosides, escin, daunorubicin
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Epidemiology (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Molecular Biology (AREA)
- Genetics & Genomics (AREA)
- Biotechnology (AREA)
- Biochemistry (AREA)
- Virology (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Communicable Diseases (AREA)
- Oncology (AREA)
- Tropical Medicine & Parasitology (AREA)
- AIDS & HIV (AREA)
- Biomedical Technology (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Medicinal Preparation (AREA)
- Peptides Or Proteins (AREA)
Abstract
Un compuesto de acuerdo con la estructura química: **(Ver fórmula)** o **(Ver fórmula)** o la amina libre o sal farmacéutica alternativa de la misma.
Description
DESCRIPCIÓN
Moléculas de suministro de fármacos citotóxicos dirigidas al VIH (CDM-H), actividad citotóxica contra el virus de la inmunodeficiencia humana y métodos de uso
Sector de la técnica
La presente invención se refiere a novedosas moléculas pequeñas, como se definen en las reivindicaciones, que tienen la capacidad de suministrar agentes terapéuticos citotóxicos a las células infectadas por el VIH. Estas moléculas, denominadas moléculas de suministro de fármacos citotóxicos dirigidas al VIH (CDM-H), contienen un extremo de unión a la gp 120 del VIH denominado extremo de unión al VIH (VICB, por sus siglas en inglés) que se une al bolsillo de CD4 en la gp120. El VICB está unido covalentemente, a través de un enlazador específicamente lábil (en realizaciones preferidas, a través de un enlazador primario), a un agente citotóxico (CYT).
Estas moléculas bifuncionales de suministro de fármacos citotóxicos de la presente invención se dirigen mecánicamente e inhiben la infección por el virus de la inmunodeficiencia humana (VIH) mediante la unión a la glucoproteína gp 120 del VIH, mientras que también suministran un agente citotóxico para mejorar la terapia del VIH.
Solicitud de prioridad y apoyo de subvenciones
Esta solicitud reivindica la prioridad de la solicitud provisional número de serie US61/638.569 titulada "Cytotoxic-drug Delivering Molecules Targeting HIV", presentada el 26 de abril de 2012.
Estado de la técnica
El uso de terapia antirretroviral de gran actividad (HAART, por sus siglas en inglés) puede reducir la viremia del VIH a niveles casi indetectables en individuos infectados a través de la supresión del ciclo de vida viral, sin embargo, existen problemas crecientes asociados con la toxicidad a largo plazo, el cumplimiento terapéutico, el alto coste y la aparición de cepas resistentes.1, 2 Además, las estrategias de tratamiento supresor conducen a la formación de reservorios latentes de replicación del VIH-1 de bajo nivel. Tras la interrupción del tratamiento, estas fuentes persistentes conducen a un rápido rebote del VIH-1;3-5 por lo tanto, se requiere una estricta adherencia a un tratamiento riguroso de por vida. Dadas estas importantes barreras, las nuevas estrategias terapéuticas que sean capaces de eliminar estos reservorios persistentes son críticas para la erradicación de la infección por VIH.
En la actualidad, una de las estrategias más investigadas destinadas a curar la infección por VIH es el desarrollo de una vacuna contra el VIH, sin embargo, las principales barreras han frustrado estos esfuerzos en el último cuarto de siglo. Estas barreras incluyen la enorme diversidad genética del VIH y la propensión a la recombinación genética, su efecto perjudicial sobre el sistema inmunitario a través de la destrucción de los linfocitos T, las estrategias de evasión inmunitaria altamente evolucionadas del VIH y, por último, el establecimiento de reservorios latentes en los que el VIH es inmunológicamente silencioso.6 La principal característica de la HAART es su capacidad para bloquear la replicación del VIH y prevenir una nueva infección, sin embargo, no logra destruir las células que ya están infectadas. Por lo tanto, las estrategias que complementan la supresión inducida por HAART al destruir directamente las células infectadas (incluida la eliminación de los reservorios latentes) representan un enorme potencial para los esfuerzos por curar el VIH.7
Direccionamiento de las células infectadas por el VIH con conjugados citotóxicos
Debido a que Env reside en las superficies de viriones libres y células infectadas, y también media en la entrada del virus en las células huésped, se han investigado numerosas estrategias para el direccionamiento citotóxico de reservorios celulares que expresan Env. Como se ha analizado a lo largo de la totalidad de esta divulgación, las ARM-H tienen la promesa de ser una estrategia de este tipo, ya que se ha demostrado su éxito no solo en la inhibición de la fusión viral y, por lo tanto, en la supresión de la replicación, sino también en el direccionamiento de las células que expresan gp120 del VIH-1 para la toxicidad inmunomediada. Además, el trabajo pionero se centró en el desarrollo de "inmunotoxinas", construcciones de proteínas dirigidas a Env conjugadas con potentes toxinas celulares. Berger y sus colegas publicaron el primer ejemplo de esta clase con su quimera de exotoxina A de CD4-Pseudomonas (PE) dirigida a gp120, CD4-PE40.8 La toxicidad celular significativa específica de gp120 (tanto de células transfectadas con Env como de líneas celulares constitutivamente infectadas por VIH) y la inhibición de la propagación de la infección por CD4-PE40 se demostraron in vitro.9-12, 96 Este éxito temprano la convirtió en la única inmunotoxina que entró en ensayos clínicos de Fase I, en los que fracasó debido a una falta total de actividad antiviral a la dosis máxima, que estaba limitada por una lesión hepatocelular grave. 13-15 Más recientemente, Pastan y colaboradores conjugaron PE con el fragmento Fv monocatenario del anticuerpo 3B3 anti gp120 ampliamente neutralizante, produciendo una quimera de anticuerpo-toxina, PE38, que demostró una potente toxicidad específica para las células transfectadas con Env, así como una línea celular linfocítica crónicamente infectada por el v Ih .11, 16 Es importante destacar que PE38 demostró una destrucción de células infectadas por el VIH 20-30 veces más eficaz que CD4-PE40 y se supone que posee una hepatotoxicidad significativamente menor.11, 17 Más recientemente, se ha demostrado que estas quimeras aumentan drásticamente la actividad antiviral de HAART en ratones thy/liv-SCID-Hu,17 proporcionando un evidencia
prometedora de que dichas estrategias clínicas sinérgicas pueden conducir a una estrategia de erradicación de la infección por VIH.7 Otros ejemplos notables incluyen el trabajo de Root y col., que recientemente desarrollaron una quimera de proteína-PE de 5 hélices que se une a la gp41 del VIH-1, demostrando una potente citotoxicidad hacia las células infectadas por el VIH-1. En el trabajo de Johansson y col., el potente fármaco de molécula pequeña de intercalación del ADN, doxorrubicina, se conjugó con un mAb anti gp120 y a continuación se administró a ratones que poseían esplenocitos infectados con VIH-1/MuLV, lo que dio como resultado la eliminación de la infección.18
En conjunto, este trabajo demuestra la abrumadora promesa de las estrategias para dirigir la citotoxicidad dirigida contra las células infectadas por VIH. Sin embargo, todas las estrategias informadas previamente utilizan construcciones proteicas, que han mostrado toxicidad aguda e inmunogenicidad en los sistemas celulares7, 19 y están inherentemente limitadas por las características de todos los agentes terapéuticos de proteínas.20 Estas limitaciones incluyen la posibilidad de reacciones alérgicas potencialmente mortales, penetración deficiente en los tejidos, inmunogenicidad (incluso en el caso de proteínas "humanizadas"),21 falta de biodisponibilidad oral, requisito de almacenamiento a baja temperatura y alto coste.22 Dada la promesa de dichos enfoques, se busca superar sus limitaciones utilizando este armazón de molécula pequeña dirigido a gp120 para suministrar compuestos citotóxicos a las células infectadas por el VIH. Además, dichos agentes podrían resultar particularmente útiles si la destrucción mediada por anticuerpos (a través de ARM-H) resulta ineficaz in vivo o en pacientes con sistemas inmunitarios muy comprometidos.
Objeto de la invención
La presente invención se refiere a compuestos bifuncionales de moléculas de suministro de fármacos citotóxicos dirigidos al VIH (CDM-H) de acuerdo con la fórmula general:

o la amina libre o sal farmacéutica alternativa de la misma.
En un aspecto adicional de la invención, una composición farmacéutica comprende una cantidad eficaz de un compuesto bifuncional como se define en las reivindicaciones opcional y preferentemente en combinación con un portador, aditivo o excipiente farmacéuticamente aceptable. En aspectos alternativos, las composiciones de combinación farmacéutica comprenden una cantidad eficaz de un compuesto bifuncional como se define en las reivindicaciones, en combinación con al menos un agente adicional que se usa para tratar el VIH (un agente contra el VIH adicional).
En un aspecto adicional de la invención, los compuestos de acuerdo con la presente invención se usan para tratar y/o reducir la probabilidad de una infección por VIH o un efecto secundario del VIH (tal como SIDA, ARC y patologías o afecciones relacionadas que se producen secundariamente a una infección por VIH) en un paciente. El método para tratar y/o reducir la probabilidad de una infección por VIH o el efecto secundario de un cáncer por VIH no forma parte de la invención y comprende administrar a un paciente que lo necesita una cantidad eficaz de un compuesto bifuncional como se describe en el presente documento en combinación con un portador, aditivo o excipiente farmacéuticamente aceptable, opcionalmente en combinación adicional con al menos un agente adicional que sea eficaz para tratar y/o reducir la probabilidad de una infección por VIH, o una o más de sus afecciones o efectos secundarios.
La presente invención también se refiere a casos en los que la destrucción de células CD4 que están infectadas con el VIH (células CD4 VIH+) puede ser útil para inhibir que las infecciones latentes por VIH se vuelvan activas. En este aspecto de la invención, la destrucción de células CD4 VIH+ en un paciente VIH positivo puede usarse para inhibir o erradicar más completamente una infección por VIH y/o reducir la probabilidad de aparición o recurrencia del VIH en un paciente VIH positivo.
Se desvela un método para unir y eliminar el VIH en un paciente que comprende administrar a un paciente infectado con el VIH una cantidad eficaz de un compuesto bifuncional como se describe de otro modo en el presente documento. Este método no forma parte de la invención.
Se desvela un método para unir y eliminar el VIH en un paciente que tiene una infección latente por VIH que comprende administrar a un paciente infectado con VIH latente una cantidad eficaz de un compuesto bifuncional como se describe en el presente documento en combinación con una molécula activadora de latencia del VIH, incluyendo, por ejemplo, prostratina, bradistatina 1 y análogos relacionados como se expone en la figura 4 del presente documento (véase De Christopher y col., Nature Chemistry, publicado en línea el 15 de julio de 2012, páginas 1-6), briostatina 1, briostatina 2, IL-7, inhibidores de histona desacetilasa, incluyendo zolinza (vorinostat), inhibidores de la metilación del ADN, incluyendo decogen (decitabina) y mezclas de los mismos. Este método no forma parte de la invención.
Por lo tanto, la presente invención presenta moléculas bifuncionales únicas como se define en las reivindicaciones que pueden operar a través de los mecanismos bifuncionales especificados anteriormente en el tratamiento del VIH, incluido el tratamiento de infecciones latentes por VIH.
La comprensión de que los virus pueden ejercer tropismo celular y tisular al unirse a sitios altamente específicos en los receptores de la membrana celular ha alentado a los investigadores en el pasado a buscar agentes que se unan a los sitios del receptor viral de las membranas celulares y, por lo tanto, prevengan la unión de un virus específico a estas células.
Específicamente, se ha demostrado que el VIH se une a una molécula de superficie conocida como receptor CD4 o T4 que está presente en diversas células susceptibles a la infección por VIH, incluyendo los linfocitos T y los macrófagos. La unión se produce a través de la proteína de envoltura del VIH, gp120.
Por lo tanto, un objeto principal de la presente invención es proporcionar compuestos bifuncionales que actuarían para aliviar los síntomas del SIDA uniéndose a una molécula bifuncional que tiene un primer extremo para la unión a la proteína de envoltura gp120, teniendo la molécula bifuncional un segundo extremo citotóxico que provoca la muerte celular tras la introducción en la célula a la que se une el compuesto. Sin quedar limitados a la teoría, se cree que los compuestos de acuerdo con la presente invención se unen a las células a las que se une la gp120 del VIH y/o que expresan células gp120 y al unirse a las células que contienen gp120, se introducen en la célula tras lo cual el resto citotóxico puede causar la muerte celular. Los compuestos bifuncionales de acuerdo con la presente invención se introducen en las células mediante una diversidad de posibles mecanismos que incluyen difusión pasiva, endocitosis (incluida la fagocitosis mediada por clatnina, mediada por caveolas) y pinocitosis. Estas moléculas bifuncionales (cuyo término también incluye multifuncionales) se denominan así genéricamente en el presente documento "moléculas de suministro de fármacos citotóxicos dirigidas al VIH" o (CDM-H) y se definen en las reivindicaciones.
Las moléculas CDM-H de la invención, como se definen en las reivindicaciones, son "bifuncionales" porque poseen al menos un extremo de unión a células de invasión de virus (VICB) y al menos un extremo de citotoxicidad (CYT, por sus siglas en inglés) conectados por al menos un enlazador (preferentemente al menos un enlazador lábil) y una molécula conectora opcional CON. El VICB está diseñado para unirse a la glucoproteína gp120 del VIH (gp120 en la membrana viral, así como la gp120 que se muestra en las células infectadas). El CYT contiene un resto de citotoxicidad
que está diseñado para causar la muerte de cualquier célula a la que se une la molécula bifuncional, inhibiendo así y, en determinados casos, erradicando el VIH de un paciente o sujeto que lo necesite.
En una realización de la invención, la molécula CDM-H bifuncional, como se define en las reivindicaciones, es capaz de suministrar una población de agentes citotóxicos al producto del gen gp120 Env del VIH, en particular, que se expresan en células CD4. La glucoproteína Env, un complejo entre la gp120 y la gp 41 unida a la membrana, se expresa tanto en la superficie del virus del VIH como en las células infectadas por el virus, especialmente las células c D4. (Miranda. L. R.; Schaefer, B. C.; Kupfer. A.; Hu, Z. X.; Franzusoff, A. Proc. Natl. Acad. Sci. U.S.A, 2002, 99, 8031-8036). El componente gp120 de Env media en la primera etapa en la entrada viral en las células humanas al unirse a la proteína CD4.
De acuerdo con la presente invención, la CDM-H, como se define en las reivindicaciones, se une a las células que expresan gp120 Env, se introduce en la célula infectada y provoca la destrucción de estas células mediante la acción del resto citotóxico. Además, dado que las CDM-H se unen a gp120 de manera competitiva con CD4, estos compuestos también inhiben la entrada del VIH vivo en los linfocitos T humanos. Por lo tanto, las CDM-H tienen el potencial de interferir con la supervivencia del VIH a través de múltiples mecanismos complementarios y también pueden funcionar como profilácticos, reduciendo la probabilidad de una infección por VIH o una recaída de una infección por VIH. En determinadas realizaciones de acuerdo con la presente invención, las CDM-H de acuerdo con la presente invención pueden combinarse con al menos uno y preferentemente una mezcla de compuestos activadores de la latencia del VIH para mejorar la terapia contra la infección por VIH. Este enfoque puede ser particularmente útil para erradicar (es decir, curar) las infecciones por VIH en pacientes que lo necesiten.
Los compuestos de CDM-H de la invención representan una estrategia contra el VIH basada en moléculas en lugar de en péptidos o proteínas (aunque determinados enlazadores lábiles pueden depender de péptidos para la degradación de proteasas) para dirigirse al ciclo de vida del virus a través de mecanismos moleculares que se refuerzan mutuamente, que inhiben la entrada del virus mientras se dirigen a las células que expresan Env para el reconocimiento inmunitario y la eliminación (muerte celular). En general, las moléculas CDM-H tienen determinadas ventajas sobre las proteínas desde un punto de vista terapéutico debido a su baja propensión a la inmunogenicidad, alta estabilidad metabólica, capacidad específica para dirigirse a células infectadas por el VIH, producción a gran escala y un coste relativamente bajo. De forma crítica, no se produce una citotoxicidad no específica apreciable en respuesta a los compuestos de CDM-H de acuerdo con la presente invención, limitando así la posibilidad de encontrar efectos secundarios graves del tratamiento con los mismos.
La elucidación de los detalles moleculares que gobiernan las interacciones entre las CDM-H, gp120 y moléculas citotóxicas facilita los esfuerzos de optimización, así como la evaluación de esta estrategia en modelos biológicos más complejos de infección por VIH.
Como se ha indicado anteriormente, la invención está dirigida a moléculas "bifuncionales", como se define en las reivindicaciones, siendo las moléculas de la invención "bifuncionales" en el sentido de que poseen un extremo de unión a células de invasión de virus (VICB) y un resto citotóxico (CYT) conectado por uno o más enlazadores (uno de los cuales es un enlazador lábil que se degrada al introducirse en una célula para liberar el resto de citotoxicidad de la porción restante de la molécula) y un grupo conector opcional (que es principalmente difuncional, pero puede ser multifuncional), como se describe de otro modo en el presente documento. El grupo VICB está diseñado para unirse a la glucoproteína gp120 del VIH (gp120 en la membrana viral, así como la gp120 que se muestra en las células infectadas, generalmente linfocitos T que expresan linfocitos T CD4 o CD4+). El grupo CYT es un resto citotóxico capaz de provocar la muerte celular tras la introducción en una célula, como se describe en detalle en el presente documento.
La presente invención está dirigida a composiciones farmacéuticas que comprenden las moléculas bifuncionales, como se definen en las reivindicaciones, que pueden inhibir la entrada del VIH en una célula diana y/o unirse a las células CD4+ mientras que también suministran uno o más agentes citotóxicos en las células infectadas por el VIH, induciendo así la muerte celular, en un portador, aditivo o excipiente farmacéuticamente aceptable. Como realizaciones adicionales de la invención, por lo tanto, los inventores proporcionan una composición farmacéutica que comprende un compuesto de molécula bifuncional de la invención en asociación con un portador o excipiente farmacéuticamente aceptable, adaptado para su uso en medicina humana o veterinaria. Dichas composiciones se pueden presentar para su uso de manera convencional mezcladas con uno o más portadores o excipientes fisiológicamente aceptables. Las composiciones pueden contener además opcionalmente uno o más agentes terapéuticos que pueden ser, si se desea, un agente antiviral diferente y, preferentemente, un agente contra el VIH adicional y/o un agente anticanceroso.
Los compuestos de molécula bifuncional de acuerdo con la invención se pueden formular para administración oral, bucal, nasal, parenteral, tópica o rectal, entre otras, como se describe de otro modo en el presente documento.
En particular, los compuestos bifuncionales de acuerdo con la invención pueden formularse para inyección o para infusión y pueden presentarse en forma de dosis unitarias en ampollas o en envases multidosis con un conservante añadido. Las composiciones pueden adoptar formas tales como suspensiones, soluciones o emulsiones en vehículos
oleosos o acuosos, y pueden comprender agentes de formulación, tales como agentes de suspensión, estabilización y/o dispersión. Como alternativa, el principio activo puede estar en forma de polvo para constitución con un vehículo adecuado, por ejemplo, agua estéril apirógena, antes de su uso.
Las composiciones farmacéuticas de acuerdo con la invención también pueden contener otros principios activos tales como otros agentes antivirales, incluyendo agentes contra el VIH adicionales, activadores del VIH latente, agentes antineoplásicos, agentes antimicrobianos o conservantes, entre otros.
Las composiciones pueden contener del 0,001-99 % en peso del material activo, a menudo de aproximadamente el 1 % a aproximadamente el 75 % en peso, de aproximadamente el 5 % a aproximadamente el 50 % en peso, de aproximadamente el 10 % a aproximadamente el 50 % en peso.
Un proceso para preparar una composición farmacéutica comprende asociar un compuesto de molécula bifuncional de la invención con un excipiente o portador farmacéuticamente aceptable.
Para la administración por inyección o infusión, las dosis y las concentraciones de fármaco deseadas de las composiciones farmacéuticas desveladas pueden variar según el uso particular, el estado del paciente, la edad, la tolerancia al fármaco, etc., como comprenderá un experto en la materia. En consecuencia, la determinación de una dosificación y/o vía de administración apropiada está bien dentro de la experiencia de un médico común, y los compuestos ciertamente pueden formularse sin demasiada experimentación para la administración en el tratamiento de seres humanos, por ejemplo, usando protocolos de respuesta a la dosis estándar y bien conocidos.
La cantidad de compuesto en una composición farmacéutica de la presente invención que puede combinarse con los materiales portadores para producir una única forma farmacéutica variará según el sujeto o paciente, la enfermedad específica tratada o el uso, así como el modo particular de administración. Preferentemente, las composiciones deben formularse para contener entre aproximadamente 0,05 miligramos y aproximadamente 750 miligramos o más, más preferentemente de aproximadamente 1 miligramo a aproximadamente 600 miligramos, e incluso más preferentemente de aproximadamente 10 miligramos a aproximadamente 500 miligramos de principio activo, en solitario o en combinación con al menos otra CDM-H que puede usarse para tratar la infección por VIH o un efecto secundario o afección de la misma u otro agente tal como un agente contra el VIH adicional, un activador del VIH latente o un agente anticanceroso como se desvela en el presente documento.
Descripción de las figuras
La figura 1 ilustra un esquema de "activación-eliminación" para erradicar la infección por VIH. La infección por VIH latente está caracterizada por células huésped que poseen un genoma del VIH integrado, sin embargo, no expresan activamente las proteínas del VIH. Se sabe que diversas moléculas "activadoras", tales como prostratina, bryostain 1 y análogos de las mismas, activan estos reservorios latentes, induciendo a las células huésped a expresar el genoma del VIH. Una vez activadas, las CDM-H pueden dirigir una respuesta citotóxica que conduce a la muerte de las células infectadas por el VIH.
La figura 2 ilustra dos compuestos de acuerdo con la presente invención. Estos se representan como compuestos 4.19 y 4.20, respectivamente. Estos compuestos se presentan como sus sales farmacéuticas (4.19 como sal de formiato y 4.20 como sal de acetato de amonio). También se pueden presentar fácilmente como la amina libre (como la forma no salina) o como una forma de sal farmacéuticamente aceptable alternativa.
La figura 3 muestra una serie de activadores del VIH latente incluyendo prostratina, briostatina 1 y análogos de briostratina 1 (compuestos 1-7) según se indica.
La figura 4 muestra el esquema químico 1 que está dirigido a la síntesis de amina (S4.11), un precursor de CDM-H 4.19.
La figura 5 muestra el esquema químico 2 que se dirige a las etapas finales para producir CDM-H 4.19 a partir de S4.11 y el NHS-éster S4.12 conocido como se indica.
La figura 6 muestra el esquema químico 3 que se dirige a las etapas finales para producir CDM-H 4.20 a partir de tio S4.14 y la maleimida S4.15 conocida.
La figura 7A muestra el esquema químico 4 que está dirigido a la funcionalización del anillo de indol 3.15 para construir la porción derecha de la molécula para proporcionar un anillo de piperazina que puede funcionalizarse fácilmente para proporcionar grupos Areno 2 que pueden sustituirse opcionalmente con uno o más de un enlazador no lábil, un enlazador lábil, uno o más grupos conectores y un grupo citotóxico. La figura 7B muestra la síntesis química de un resto citotóxico, en este caso doxorrubicina, que contiene un enlazador lábil que se condensa en el resto VICB para proporcionar una CDM-H de acuerdo con la presente invención.
La figura 8 muestra el esquema químico 5 que ilustra un esquema químico genérico para funcionalizar el Areno 1
para poner un grupo citotóxico al final del brazo que se extiende desde el Areno 1.
La figura 9 muestra el esquema químico 6 que ilustra un esquema químico genérico para introducir un resto citotóxico en la parte superior izquierda del anillo de indol.
La figura 10 muestra que la molécula de suministro de fármacos citotóxicos dirigida a la gp120 del VIH (CDM-H) es capaz de proporcionar una respuesta citotóxica a las células que expresan gp120 en ausencia de cualquier otro componente inmunitario. (A) Estructura química del ADN que intercala antraciclina doxorrubicina (4.18), que se usa como carga útil tóxica de las CDM-H y se puede conjugar con una molécula de direccionamiento a través de diversas conjugaciones: hidrazona, éster y amida. (B) Estructura de CDM-H 4.19 en la que la carga útil tóxica se conjuga con TBT a través de un enlace éster. (C) Ensayo de citotoxicidad en el que se incuban 4.18 y 4.19 con células CHO-env (gp120+) o CHO-pSV (gp120-) durante un período de 14 h o 24 h (D). La CDM-H 4.19 demuestra una citotoxicidad selectiva de gp120 a las 14 h de incubación, sin embargo, es citotóxica no específicamente (independiente de gp120) a las 24 h de incubación. (E) Se usa CD4-PE como control positivo. Los datos se representan como la media con barras de error de desviación estándar.
La figura 11 muestra la hidrólisis del éster de CDM-H 4.19, como se detecta por UPLC/HR-MS, con una semivida hidrolítica estimada de 5,7 horas.
La figura 12 muestra la hidrólisis de CDM-H 4.20, como se detecta por UPLC/HR-MS, con una semivida hidrolítica estimada de 56 horas.
La figura 13 muestra el ELISA de inhibición de CD4 de CDM-H 4.19 y 4.20, demostrando que ambos análogos inhiben la interacción CD4-gp120.
La figura 14 muestra la toxicidad del análogo de CDM-H 4.20 frente a células que expresan gp120. (A) Estructura química de 4.20, que se conjuga con doxorrubicina a través de acil hidrazona, medida para tener una semivida de hidrólisis de aproximadamente 55 h. (B) Viabilidad de CHO-env (gp120+) y CHO-pSV (gp120-) en presencia de 4.18 (10 pM), 4.20 (10 pM) o CD4-Pe (1 pg/ml) medida con el instrumento xCELLigence. Viabilidad celular examinada durante un lapso de ~120 h después de una sola adición de todos los compuestos o después de una adición inicial, seguida de un "reciclado" de compuesto como se describe en el texto y la Información de apoyo.
La figura 15 muestra imágenes de inmunofluorescencia de CHO-env (gp120+; paneles A-D) y CHO-pSv (gp120-; paneles E-H) cuando se tiñeron con colorante lisosomal LysoTracker y se incubaron CDM-H 4.20 después de 10 min de incubación. Las imágenes combinadas (D para gp120+ y H para gp120-) muestran que CDM-H coincide con el colorante LysTracker, lo que sugiere que se localizan niveles significativos de 4.20 en el lisosoma, independientemente de la expresión de gp120.
La figura 16 muestra micrografías de fluorescencia cuando se incuba CDM-H 4.20 o doxorrubicina (4.18) con células CHO-env después de 10 min (A-B) o 20 h (C-D) y células CHO-pSv (EH). La formación de partículas fluorescentes, específicamente después de largos períodos de incubación, sugiere la formación de micelas.
La figura 17 muestra los datos de xCelligence sin procesar de CDM-Hs 4.19 y 4.20, doxorrubicina (4.18) y CD4-PE cuando se incubaron con células CHO-env (gp120+) y CHO-pSv (gp120-).
La figura 18 muestra el experimento preliminar de dispersión de luz dinámica (DLS, por sus siglas en inglés) para detectar la formación de agregados.
Descripción detallada de la invención
Los siguientes términos se usan para describir la presente invención. En los casos en que un término no se define específicamente en el presente documento, a ese término se le da un significado reconocido en la técnica por los expertos en la materia aplicando ese término en el contexto para su uso en la descripción de la presente invención.
Cuando se proporciona un intervalo de valores, se entiende que cada valor intermedio, a la décima parte de la unidad del límite inferior a menos que el contexto indique claramente lo contrario (tal como en el caso de un grupo que contiene un número de átomos de carbono, en cuyo caso se proporciona cada número de átomo de carbono que está dentro del intervalo), entre el límite superior e inferior de ese intervalo y cualquier otro valor afirmado o intermedio en este intervalo se incluye dentro de la invención. Los límites superior e inferior de estos intervalos más pequeños pueden incluirse independientemente en los intervalos más pequeños que también se incluyen dentro de la invención, sujetos a cualquier límite específicamente excluido en el intervalo establecido. Cuando el intervalo establecido incluye uno o ambos límites, los intervalos que excluyen cualquiera de los dos límites incluidos también se incluyen en la invención.
El término "compuesto", como se usa en el presente documento, a menos que se indique de otro modo, se refiere a cualquier compuesto químico específico desvelado en el presente documento, e incluye tautómeros, regioisómeros, isómeros geométricos y, cuando sea aplicable, isómeros ópticos (enantiómeros) de los mismos, así como sales y
derivados farmacéuticamente aceptables (incluyendo las formas de profármacos) de los mismos. Dentro de su uso en contexto, el término compuesto se refiere generalmente a un solo compuesto, pero también puede incluir otros compuestos tales como estereoisómeros, regioisómeros y/o isómeros ópticos (incluyendo mezclas racémicas), así como enantiómeros específicos o mezclas enantioméricamente enriquecidas de compuestos desvelados. El término también se refiere, en el contexto, a formas de profármacos de compuestos que han sido modificadas para facilitar la administración y el suministro de compuestos en un sitio de actividad. Se observa que al describir los presentes compuestos, se describen y se eligen numerosos sustituyentes, enlazadores (incluidos enlazadores lábiles) y moléculas conectoras y variables asociadas con los mismos, entre otros, para proporcionar compuestos estables que pueden aislarse y procesarse adicionalmente en una composición farmacéutica.
El término "paciente" o "sujeto" se usa en toda la memoria descriptiva dentro del contexto para describir un animal, generalmente un mamífero, y preferentemente un ser humano, a quien se trata, incluyendo el tratamiento profiláctico (profilaxis), con las composiciones de acuerdo con la presente invención que se proporciona. Para el tratamiento de estas infecciones, afecciones o patologías que son específicas de un animal específico, tal como un paciente humano o un paciente de un género particular, tal como un paciente humano masculino, el término paciente se refiere a ese animal específico. Los compuestos de acuerdo con la presente invención son útiles para tratar y/o reducir la probabilidad de infecciones por VIH o los efectos secundarios de las infecciones por VIH, incluyendo especialmente SIDA y/o ARC.
El término "eficaz" se usa en el presente documento, a menos que se indique de otro modo, para describir una cantidad de un compuesto o de una composición que, en el contexto, se usa para producir o efectuar un resultado deseado, ya sea que ese resultado se relacione con la inhibición de los efectos de un tóxico en un sujeto o el tratamiento de un sujeto por afecciones secundarias, patologías o manifestaciones de exposición a tóxicos como se describe de otro modo en el presente documento. Este término subsume a todas las demás expresiones de cantidad eficaz o concentración eficaz (incluyendo la expresión "terapéuticamente eficaz") que se describen de otro modo en la presente solicitud.
Los términos "tratar", "que trata" y "tratamiento", etc., como se usan en el presente documento, se refieren a cualquier acción que proporcione un beneficio a un paciente en riesgo de infección por VIH o que tenga una infección por VIH, incluyendo una infección por VIH latente, incluida la mejora de la afección a través de la disminución o la supresión de los títulos de VIH o al menos un síntoma de VIH, prevención o retraso en la progresión de la enfermedad, prevención o retraso en la aparición de patologías o afecciones que se producen secundariamente al VIH, incluyendo SIDA o ARC, entre otros, incluyendo la erradicación del VIH. El tratamiento, como se usa en el presente documento, abarca tanto el tratamiento profiláctico como el terapéutico. El término "profiláctico", cuando se usa, significa reducir la probabilidad de una aparición o la gravedad de una aparición dentro del contexto del tratamiento del VIH, como se ha descrito anteriormente de otro modo en el presente documento.
La expresión "virus de la inmunodeficiencia humana" o "VIH" se usará para describir los virus de la inmunodeficiencia humana 1 y 2 (VIH-1 y VIH-2), cuyo crecimiento o replicación puede inhibirse o cuyas patologías pueden tratarse usando uno o más métodos desvelados en el presente documento. Los virus que pueden tratarse incluyen, por ejemplo, virus de la inmunodeficiencia humana 1 y 2 (VIH-1 y VIH-2), entre otros. El término VIH incluye cepas mutantes del VIH que incluyen cepas del virus del VIH "resistentes a fármacos" o "resistentes a múltiples fármacos" que han mutado para volverse resistentes a uno o más agentes contra el VIH aprobados clínicamente, incluyendo, en particular, las cepas del VIH que son resistente a uno o más compuestos NRTI y/o compuestos NNRTI. Las cepas de ejemplo resistentes a fármacos del VIH que pueden tratarse eficazmente usando compuestos de acuerdo con la presente invención incluyen las siguientes, entre otras: (definidas por su mutación de la transcriptasa inversa o mutación RT): XXBRU, K65R, Y115F, F116Y, Q151M, M184V, L74V, V75T, 4XZT, T215Y, K103N, T215Y/M184V, 5705-72, 488-101, C910-6, LA1M184V, G910-6 L100I, K101E, K103N, V106A, D110E, V179D, Y181C, D185E, D186E, Y188H, G190E, E138K, M41L, D67N, K70R, T215Y/F, K219Q/E, Y181C, K103N, L100I, Y188C/H, entre otros, incluyendo aislados del VIH-1 JR-FL, ADA, HXBc2, SF162 y BaL, entre otras.
La expresión "latencia del VIH" se usa para describir la capacidad del VIH para permanecer latente dentro de las células de un paciente, en particular, los linfocitos T positivos para CD4 y formar uno o más reservorios virales. En el VIH, la latencia proviral en tipos específicos de células longevas es la base de los reservorios virales, que se caracterizan por la persistencia o longevidad del virus latente en las células infectadas. En la latencia del VIH, la presencia del VIH competente para la replicación en linfocitos T positivos para CD4 en reposo permite que el virus persista durante años sin evolucionar a pesar de la exposición prolongada a los fármacos antirretrovirales. Este reservorio latente del VIH (VIH latente) puede explicar la incapacidad del tratamiento antirretroviral tradicional para erradicar o curar la infección por VIH en un paciente. La presente invención sirve para destruir células (células positivas para CD4) que albergan reservorios virales y reducir la probabilidad de que se produzca un brote activo de VIH, así como proporcionar una cura real para el VIH a través de la erradicación del VIH en determinados casos. La presente invención sirve para reducir, inhibir y/o eliminar los reservorios virales del VIH y los provirus del VIH en las células de un paciente, especialmente las células positivas para CD4, y reducir la probabilidad de que se produzca una infección o brote activo del VIH en el futuro. Si quedar limitados por la teoría, se cree que los compuestos de acuerdo con la presente invención reducen, inhiben y/o eliminan/erradican el VIH latente (reservorios de VIH latente) y/o provirus del VIH a través de la muerte celular de células positivas para CD4, reduciendo así la probabilidad de que el VIH latente
se convierta en una infección activa por el VIH. Los compuestos actúan liberando agentes citotóxicos después de su introducción en una célula, lo que da como resultado la muerte celular y la inhibición de la capacidad de replicación del VIH. Esta inhibición da como resultado que el VIH latente o el VIH proviral que reside en las células se destruya y/o actúe sobre él mediante un agente contra el VIH administrado conjuntamente que sirve para inhibir el VIH. El resultado del método de tratamiento es que el VIH latente se reduce, se inhibe y/o se elimina y, en determinados casos, puede efectuarse una cura real del VIH porque el VIH latente, así como el VIH activo, se elimina del paciente, lo que da como resultado que no haya más infección.
El VIH se une a la superficie exterior de las células CD4+, entra en las células y a continuación permanece oculto y protegido de las otras células del sistema inmunitario. A salvo dentro de la célula, el virus duplica su ARN. El nuevo ADN viral se integra en el ADN de la célula huésped donde gobierna la producción de nuevos viriones del VIH. Los nuevos viriones abandonan la célula huésped para infectar otras células y la célula huésped CD4+ muere. El cuerpo produce aproximadamente 10 billones de nuevos viriones diariamente, y el sistema inmunitario los destruye y elimina todos menos aproximadamente 100 millones de ellos, que son infecciosos. Los viriones producen y destruyen un número equivalente de células CD4+, creando un equilibrio de lucha de poder entre el virus y las células CD4+.
El ciclo de vida del VIH en su forma activa requiere enzimas específicas, que sirven como dianas para la terapia farmacológica tradicional contra el VIH:
• La transcriptasa inversa ayuda a crear copias de ADN del ARN del VIH. Los fármacos antirretrovirales nucleosídicos y no nucleosídicos bloquean la transcriptasa inversa, evitando que el VIH copie su ARN en el ADN.
• La integrasa ayuda a integrar el ADN viral en el ADN de la célula huésped. La integrasa es una diana potencial para la terapia farmacológica, y los científicos esperan encontrar una manera de bloquearla para evitar que el ADN viral se integre en el ADN de la célula huésped.
• La proteasa ayuda a ensamblar los nuevos viriones. Los inhibidores de proteasa evitan que la proteasa realice esta función.
En algunos casos, el VIH no empieza a replicarse inmediatamente después de entrar en una nueva célula huésped. Una vez que el ADN entra en el genoma de la célula huésped, el VIH puede persistir durante años dentro del cuerpo sin causar los síntomas que definen al SIDA. Pero incluso en esta etapa (que se llama latencia), el virus aún puede transmitirse a otros. La latencia es quizás uno de los mayores desafíos para encontrar una cura o una vacuna para el SIDA y es la razón principal por la cual las personas con SIDA deben tomar fármacos antirretovirales de por vida. La presente invención sirve para reducir los reservorios virales del VIH y los provirus del VIH en las células de un paciente, especialmente las células positivas para CD4, y reducir la probabilidad de que se produzca un brote o una infección activa del VIH. Los compuestos de acuerdo con la presente invención reducen, inhiben y/o eliminan los reservorios de VIH latente y/o los provirus de VIH en solitario o en combinación con al menos un agente contra el VIH adicional y/o uno o más activadores de VIH latente, reduciendo, inhibiendo y/o eliminando así el VIH latente y, por lo tanto, reduciendo la probabilidad de que el VIH latente se convierta en una infección por VIH activa.
La expresión "activador del VIH latente" se usa para describir uno o más compuestos que activan el VIH latente en un estado más activo. Los activadores del VIH que se pueden usar en combinación con CDM-H de acuerdo con la presente invención incluyen prostratina, bradistatina 1 y análogos relacionados como se expone en la figura 4 del presente documento (véase De Christopher y col., Nature Chemistry, publicado en línea el 15 de julio de 2012, páginas 1-6), briostatina 1, briostatina 2, IL-7, inhibidores de histona desacetilasa, incluyendo zolinza (vorinostat), inhibidores de la metilación del ADN, incluyendo decogen (decitabina) y mezclas de los mismos. Los activadores del VIH se usan para regular positivamente el VIH desde un estado de latencia a un estado más activo, de modo que el VIH activo exprese gp120 sobre la superficie de una célula CD4 infectada con el VIH (VIH positivo para el CD4). Los activadores del VIH, cuando se administran a un paciente o sujeto, se administran preferentemente como una mezcla de activadores del VIH, como se ha descrito anteriormente.
Los términos "ARC" y "SIDA" se refieren a síndromes del sistema inmunitario provocados por el virus de la inmunodeficiencia humana, que se caracterizan por la susceptibilidad a determinadas enfermedades y recuentos de linfocitos T reducidos en comparación con los recuentos normales. El VIH progresa de la Categoría 1 (enfermedad asintomática por VIH) a la Categoría 2 (ARC), a la Categoría 3 (SIDA), con la gravedad de la enfermedad.
Una infección por VIH de Categoría 1 se caracteriza por que el paciente o sujeto es VIH positivo, asintomático (sin síntomas) y nunca ha tenido menos de 500 células CD4. Si el paciente ha tenido alguna de las enfermedades definitorias del SIDA enumeradas para las categorías 2 (ARC) o 3 (SIDA), entonces el paciente no está en esta categoría. Si el recuento de linfocitos T del paciente alguna vez ha caído por debajo de 500, ese paciente se considera de Categoría 2 (ARC) o Categoría 3 (SIDA).
Una infección de Categoría 2 (ARC) se caracteriza por los siguientes criterios: Los linfocitos T del paciente han caído por debajo de 500 pero nunca por debajo de 200, y ese paciente nunca ha tenido ninguna enfermedad de Categoría 3 (como se expone a continuación) pero ha tenido al menos una de las siguientes enfermedades definitorias:
Angiomatosis bacilar
Candidiasis, orofaríngea (aftas)
Candidiasis, vulvovaginal; persistente, frecuente o con poca respuesta a la terapia
Displasia cervical (moderada o severa)/carcinoma cervical in situ
Síntomas constitucionales, tales como fiebre (38,5 C) o diarrea que dura más de 1 mes
Leucoplasia vellosa, oral
Herpes zoster (culebrilla), que implica al menos dos episodios distintos o más de un dermatoma
Púrpura trombocitopénica idiopática
Listeriosis
enfermedad pélvica inflamatoria, particularmente si se complica con un absceso tuboovárico
Neuropatía periférica
De acuerdo con el gobierno de los EE.UU., en la ARC de Categoría 2, el sistema inmunitario muestra algunos signos de daño pero no pone en peligro la vida.
Una infección de Categoría 3 (SIDA) se caracteriza por los siguientes criterios:
Los linfocitos T han caído por debajo de 200 o el paciente ha tenido al menos una de las siguientes enfermedades definitorias:
Toxoplasmosis cerebral
Candidiasis de bronquios, tráquea o pulmones
Candidiasis, esofágica
Cáncer del cuello del útero, invasivo**
Coccidioidomicosis, diseminada o extrapulmonar
Criptococosis, extrapulmonar
Criptosporidiosis, intestinal crónica (más de 1 mes de duración)
Enfermedad por citomegalovirus (que no sea del hígado, bazo o ganglios)
Retinitis por citomegalovirus (con pérdida de la visión)
Encefalopatía, relacionada con el VIH
Herpes simple: úlcera(s) crónica(s) (más de 1 mes de duración); o bronquitis, neumonitis o esofagitis
Histoplasmosis, diseminada o extrapulmonar
Isosporiasis, intestinal crónica (más de 1 mes de duración)
Sarcoma de Kaposi
Linfoma, de Burkitt (o término equivalente)
Linfoma, inmunoblástico (o término equivalente)
Linfoma, primario, de cerebro
Complejo Mycobacterium avium o M. kansasii, diseminado o extrapulmonar
Tuberculosis por Mycobacterium, cualquier sitio (pulmonar** o extrapulmonar)
Mycobacterium, otras especies o especies no identificadas, diseminada o extrapulmonar
Neumonía por Pneumocystis carinii
Neumonía, recurrente
Leucoencefalopatía multifocal progresiva
Septicemia por Salmonella, recurrente
Síndrome de desgaste debido al VIH
El término "introducir" se usa para describir la captación por una célula CD4 infectada por el VIH de CDM-H que se han unido a la superficie de la célula CD4 infectada por el VIH en la proteína de envoltura gp120 en la superficie celular. Las CDM-H pueden internalizarse en la célula, después de lo cual el resto citotóxico c Yt puede liberarse de las CDM-H, lo que provoca la muerte de las células CD4 infectadas por el VIH. Las CDM-H se internalizan en una célula CD4 infectada por el VIH después de unirse a través de uno o más de una diversidad de mecanismos que incluyen, pero sin limitación, difusión pasiva, endocitosis (incluyendo fagocitosis mediada por clatnina, mediada por caveolas) y pinocitosis
La expresión "administración conjunta" o "terapia de combinación" significará que al menos dos compuestos o composiciones se administran al paciente al mismo tiempo, de modo que las cantidades o concentraciones eficaces de cada uno de los dos o más compuestos puedan encontrarse en el paciente en un momento dado en el tiempo. Aunque los compuestos de acuerdo con la presente invención pueden administrarse conjuntamente a un paciente al mismo tiempo, el término abarca tanto la administración de dos o más agentes al mismo tiempo como en diferentes momentos, con la condición de que las concentraciones eficaces de todos los compuestos o las composiciones administradas conjuntamente se encuentren en el sujeto en un momento dado. Uno o más de los compuestos de CDM-H bifuncionales descritos anteriormente se administran conjuntamente en combinación con al menos un agente contra el VIH adicional como se describe de otro modo en el presente documento en un cóctel para el tratamiento de infecciones por VIH y/o un compuesto/composición activadora del VIH latente como se describe de otro modo en el presente documento. En particular, la administración conjunta de compuestos da como resultado una actividad contra el VIH sinérgica de la terapia, incluyendo especialmente la inhibición y/o la erradicación del VIH latente.
La expresión "agente contra el VIH adicional" significará un agente contra el VIH tradicional (es decir, un compuesto
de CDM-H no bifuncional como se describe de otro modo en el presente documento) que puede administrarse conjuntamente a un paciente junto con compuestos ARM-HI de acuerdo con la presente invención en el tratamiento de un paciente con VIH. Dichos compuestos incluyen, por ejemplo, agentes tales como inhibidores de transcriptasa inversa nucleosídicos (NRTI, por sus siglas en inglés), inhibidores de transcriptasa inversa no nucleosídicos, inhibidores de proteasa e inhibidores de fusión. Los compuestos de ejemplo incluyen, por ejemplo, amprenivir, abacavir, acemannan, aciclovir, AD-439, AD-519, adefovir dipivoxilo, interferón alfa, ansamicina, 097, AR 177, betafluoro-ddA, BMS-232623 (CGP-73547), BMS-234475 (CGP-61755), CI-1012, cidofovir, sulfato de curdlano, inmunoglobina de citomegalovirus, ganciclovir, didesoxiinosina, DMP-450, efavirenz (DMP-266), EL10, famciclovir, FTC, GS 840, HBY097, hipericina, interferón beta humano recombinante, interferón alfa-n3, indinavir, ISIS-2922, KNI-272, lamivudina (3TC), lobucavir, nelfinavir, nevirapina, novapreno, secuencia octapeptídica de péptido T, fosfonoformiato de trisodio, PNU-140690, probucol, RBC-CD4, ritonavir, saquinavir, valaciclovir, virazol ribavirina, VX-478, zalcitabina, zidovudina (AZT), sal fumarato de tenofovir diisoproxilo, combivir, succinato de abacavir, T-20), AS-101, bropirimina, CL246, EL10, FP-21399, interferón gamma, factor estimulante de colonias de granulocitos y macrófagos (GM-CSF, por sus siglas en inglés), inmunoestimulante de partículas centrales del VIH, interleucina 2 (IL-2), inmunoglobulina intravenosa, IMREG-1, IMREG-2, ditio carbamato de imutio dietilo, interferón alfa-2, metioninaencefalina, MTP-PE (muramilo-tripéptido), factor estimulante de colonias de granulocitos (GCSF, por sus siglas en inglés), remune, rCD4 (CD4-IgG humano soluble recombinante), híbridos de rCD4-IgG, CD4 humano soluble recombinante, interferón alfa 2a, SK&F1-6528, T4 soluble, timopentina, factor de necrosis tumoral (TNF), AK602, alovudina, amdoxovir, AMD070, atazanavir (Reyataz), AVX754 (apricitabina), bevirimat, BI-201, BMS-378806, BMS-488043, BMS-707035, C31G, Carbopol 974P, calanolida A, carragenano, sulfato de celulosa, cianovirina-N, darunavir, delavirdina, didanosina (Videx), efavirenz, elvucitabina, emtricitabina, fosamprenavir (Lexiva), fozivudina tidoxilo, GS 9137, GSK-873,140 (aplaviroc), GSK-364735, GW640385 (brecanavir), HG0004, HGTV43, INCB9471, KP-1461, lopinavir, mifepristona (VGX410), MK-0518, PPL-100, PRO 140, PRO 542, PRO 2000, racivir, SCH-D (vicriviroc), SP01A, SPL7013, TAK-652, tipranavir (aptivus), TNX-355, TMC125 (etravirina), UC-781, UK-427,857 (maraviroc), ácido valproico, VRX496, zalcitabina, valganciclovir, clindamicina con primaquina, pastilla de fluconazol, pastilla de nistatina, eflornitina, pentamidina, isetionato, trimetoprima, trimetoprima/sulfa, piritrexim, isetionato de pentamidina, espiramicina, intraconazol-R51211, trimetrexato, daunorrubicina, eritropoyetina humana recombinante, hormona de crecimiento humano recombinante, acetato de megestrol, testosterona, aldesleucina (proleucina), anfotericina B, azitromicina (zitromax), hidroxiapatita de calcio, doxorrubicina, dronabinol, entecavir, epoyetina alfa, etopósido, fluconazol, isoniazid, itraconazol (Sporanox), megestrol, paclitaxel (Taxol), peginterferón alfa-2, ácido poli-L-láctico (Sculptra), rifabutina (micobutina), rifampina, somatropina y sulfametoxazol/trimetoprima. Los compuestos contra el VIH preferidos para su uso en la presente invención incluyen, por ejemplo, 3TC (lamivudina), AZT (zidovudina), (-)-FTC, ddI (didanosina), ddC (zalcitabina), abacavir (ABC), tenofovir (PMPA), D-D4FC (reverset), D4T (etavudina), racivir, L-FddC, L-FD4C, NVP (nevirapina), DLV (delavirdina), EFV (efavirenz), SQVM (mesilato de saquinavir), Rt V (ritonavir), IDV (indinavir), SQV (saquinavir), NFV (nelfinavir), APV (amprenavir), LPV (lopinavir), inhibidores de la fusión tales como T20, entre otros, fuseón y mezclas de los mismos
La expresión "sal farmacéuticamente aceptable" se usa a lo largo de la memoria descriptiva para describir una forma de sal de uno o más de los compuestos del presente documento que se presentan para aumentar la solubilidad del compuesto en solución salina para suministro parenteral o en los jugos gástricos del tracto gastrointestinal del paciente para promover la disolución y la biodisponibilidad de los compuestos. Las sales farmacéuticamente aceptables incluyen las derivadas de bases y ácidos inorgánicos y orgánicos farmacéuticamente aceptables. Las sales adecuadas incluyen las derivadas de metales alcalinos tales como potasio y sodio, metales alcalinotérreos tales como sales de calcio, magnesio y amonio, entre otros muchos ácidos conocidos en la técnica farmacéutica. Las sales de sodio y potasio pueden ser particularmente preferidas como sales de neutralización de composiciones que contienen ácido carboxílico de acuerdo con la presente invención. El término "sal" significará cualquier sal compatible con el uso de los compuestos de acuerdo con la presente invención. En caso de que los compuestos se usen en indicaciones farmacéuticas, incluyendo el tratamiento de infecciones por VIH, el término "sal" significará una sal farmacéuticamente aceptable, coherente con el uso de los compuestos como agentes farmacéuticos.
La expresión "extremo de citotoxicidad" o "resto de agente citotóxico" (CYT dentro de la fórmula general de los compuestos de acuerdo con la presente invención) se usa para describir la porción de un compuesto de CDM-H de acuerdo con la presente invención que comprende al menos un agente citotóxico que presenta citotoxicidad en el paciente. Las expresiones "resto citotóxico" o "resto citotóxico del VIH" o "citotoxicidad de células infectadas por el VIH" se usan como sinónimos para describir un resto de molécula pequeña que exhibe actividad citotóxica contra una célula que ha sido infectada con el VIH de manera que cuando el resto de unión de la célula infectada por el virus

citotóxico entrarán en la célula infectada y promoverán la muerte celular, dando como resultado la inhibición de la infección por VIH.
La expresión "terminal de unión a células de invasión de virus" ("VICB") (también denominado extremo de unión a la diana o TBT) se usa para describir la porción de un compuesto de acuerdo con la presente invención que comprende al menos una molécula pequeña o resto que puede unirse específicamente o es capaz de unirse a la proteína de la envoltura gp120 en el virus del VIH o en una superficie celular de células CD4 que están infectadas con el VIH (VIH+)
en dicho paciente.
Composiciones farmacéuticas
Las composiciones farmacéuticas comprenden combinaciones de una cantidad eficaz de al menos un compuesto bifuncional de acuerdo con la presente invención, y uno o más de los compuestos descritos de otro modo en el presente documento, todos en cantidades eficaces, en combinación con una cantidad terapéuticamente eficaz de un portador, aditivo o excipiente, representan un aspecto adicional de la presente invención.
Las composiciones de la presente invención pueden formularse de una manera convencional usando uno o más portadores farmacéuticamente aceptables y también se pueden administrar en formulaciones de liberación controlada. Los portadores farmacéuticamente aceptables que se pueden usar en estas composiciones farmacéuticas incluyen, pero sin limitación, intercambiadores iónicos, alúmina, estearato de aluminio, lecitina, proteínas séricas, tales como albúmina sérica humana, sustancias tamponantes tales como fosfatos, glicina, ácido sórbico, sorbato de potasio, mezclas de glicéridos parciales de ácidos grasos vegetales saturados, agua, sales o electrolitos, tales como sulfato de prolamina, hidrogenofosfato de disodio, hidrogenofosfato de potasio, cloruro de sodio, sales de cinc, sílice coloidal, trisilicato de magnesio, polivinilpirrolidona, sustancias a base de celulosa, polietilenglicol, carboximetilcelulosa de sodio, poliacrilatos, ceras, polímeros de bloque de polietileno-polioxipropileno, polietilenglicol y lanolina.
Las composiciones de la presente invención pueden administrarse por vía oral, parenteral, por pulverización por inhalación, tópica, rectal, nasal, bucal, por vía vaginal o a través de un depósito implantado. El término "parenteral", como se usa en el presente documento, incluye inyección subcutánea, intravenosa, intramuscular, intraarticular, intrasinovial, intraesternal, intratecal, intrahepática, intralesional e intracraneal o técnicas de infusión. Preferentemente, las composiciones se administran por vía oral, intraperitoneal o intravenosa.
Las formas inyectables estériles de las composiciones de la presente invención pueden ser una suspensión acuosa u oleosa. Estas suspensiones pueden formularse de acuerdo con técnicas conocidas en la materia usando agentes de dispersión o humectación y agentes de suspensión adecuados. La preparación inyectable estéril también puede ser una solución o suspensión inyectable estéril en un diluyente o disolvente no tóxico parenteralmente aceptable, por ejemplo, en forma de una solución en 1,3-butanodiol. Entre los vehículos y disolventes aceptables que pueden emplearse están agua, solución de Ringer y solución isotónica de cloruro de sodio. Además, se emplean convencionalmente aceites fijos estériles en forma de un disolvente o un medio de suspensión. Para este fin, puede emplearse cualquier aceite suave no volátil, incluyendo monoglicéridos o diglicéridos sintéticos. Los ácidos grasos, tales como ácido oleico y sus derivados de glicéridos, son útiles en la preparación de inyectables, como lo son los aceites naturales farmacéuticamente aceptables, tales como aceite de oliva o aceite de ricino, especialmente en sus versiones polioxietiladas. Estas soluciones o suspensiones oleosas también pueden contener un diluyente o dispersante alcohólico de cadena larga, tal como Ph. Helv o alcohol similar.
Las composiciones farmacéuticas de la presente invención pueden administrarse por vía oral en cualquier forma farmacéutica oralmente aceptable, incluyendo, pero sin limitación, cápsulas, comprimidos, suspensiones o soluciones acuosas. En el caso de los comprimidos para uso oral, los portadores que se usan comúnmente incluyen lactosa y almidón de maíz. Típicamente, también se añadan agentes lubricantes, tal como estearato de magnesio. Para la administración oral en forma de cápsula, los diluyentes útiles incluyen lactosa y almidón de maíz deshidratado. Cuando se necesitan suspensiones acuosas para su uso oral, el principio activo se combina con agentes emulsionantes y de suspensión. Si se desea, también se pueden añadir determinados agentes edulcorantes, saporíferos o colorantes.
Como alternativa, las composiciones farmacéuticas de la presente invención pueden administrarse en forma de supositorios para administración rectal. Estos pueden prepararse mezclando el agente con un excipiente no irritante adecuado que es sólido a temperatura ambiente, pero líquido a temperatura rectal y, por lo tanto, se derretirá en el recto para liberar el fármaco. Dichos materiales incluyen manteca de cacao, cera de abeja y polietilenglicoles.
Las composiciones farmacéuticas de esta invención también se pueden administrar por vía tópica. Las formulaciones tópicas adecuadas se preparan fácilmente para cada una de estas áreas u órganos. La aplicación tópica para el tracto intestinal inferior puede lograrse en una formulación de supositorio rectal (véase anteriormente) o en una formulación de enema adecuada. También pueden usarse parches transdérmicos aceptables para la vía tópica.
Para aplicaciones tópicas, las composiciones farmacéuticas pueden formularse en un ungüento adecuado que contenga el principio activo suspendido o disuelto en uno o más portadores. Los portadores para administración tópica de los compuestos de esta invención incluyen, pero sin limitación, aceite mineral, vaselina líquida, vaselina blanca, propilenglicol, polioxietileno, compuesto de polioxipropileno, cera emulsionante y agua. La crema o loción tópica se puede usar de manera profiláctica para prevenir infecciones cuando se aplica tópicamente en áreas propensas a la infección por virus. En aspectos adicionales, los compuestos de acuerdo con la presente invención se pueden recubrir sobre la superficie interior de un preservativo y utilizarse para reducir la probabilidad de infección durante la actividad sexual.
Como alternativa, las composiciones farmacéuticas pueden formularse en una loción o crema adecuada que contenga
los componentes activos suspendidos o disueltos en uno o más portadores farmacéuticamente aceptables. Los portadores adecuados incluyen, pero sin limitación, aceite mineral, monoestearato de sorbitán, polisorbato 60, cera de ésteres cetílicos, alcohol cetearílico, 2-octildodecanol, alcohol bencílico y agua.
Para uso oftálmico, las composiciones farmacéuticas pueden formularse como suspensiones micronizadas en solución salina estéril isotónica con pH ajustado o, preferentemente, como soluciones en solución salina estéril isotónica de pH ajustado, ya sea con o sin un conservante tales como cloruro de benzalconio. Como alternativa, para sus usos oftálmicos, las composiciones farmacéuticas pueden formularse en un ungüento tal como vaselina.
Las composiciones farmacéuticas de la presente invención también pueden administrarse mediante aerosol nasal o inhalación. Dichas composiciones se preparan de acuerdo con técnicas bien conocidas en la técnica de la formulación farmacéutica y pueden prepararse como soluciones en solución salina, empleando alcohol bencílico u otros conservantes adecuados, promotores de la absorción para potenciar la biodisponibilidad, fluorocarbonos y/u otros agentes solubilizantes o dispersantes convencionales.
La cantidad de compuesto en la composición farmacéutica de la presente invención que puede combinarse con los materiales portadores para producir una forma farmacéutica unitaria variará dependiendo del huésped y de la enfermedad tratada, del modo particular de administración. Preferentemente, las composiciones deben formularse para contener entre aproximadamente 0,05 miligramos y aproximadamente 750 miligramos o más, más preferentemente de aproximadamente 1 miligramo a aproximadamente 600 miligramos, e incluso más preferentemente de aproximadamente 10 miligramos a aproximadamente 500 miligramos de principio activo, en solitario o en combinación con al menos otro compuesto bifuncional de acuerdo con la presente invención u otro agente contra el VIH que puede usarse para tratar la infección por VIH o un efecto secundario o afección de la misma.
Debe entenderse también que un régimen de dosificación y tratamiento específico para cualquier paciente particular dependerán de una diversidad de factores, incluyendo la actividad del compuesto específico empleado, la edad, el peso corporal, el estado de salud general, el sexo, la dieta, el tiempo de administración, la tasa de excreción, la combinación de fármaco y el criterio del médico a cargo del tratamiento y la gravedad de la enfermedad o afección concreta que se está tratando.
Un paciente o sujeto (por ejemplo, un hombre humano) que padece infección por VIH puede tratarse administrando al paciente (sujeto) una cantidad eficaz del compuesto de c DM-H de acuerdo con la presente invención que incluye sales, solvatos o polimorfos farmacéuticamente aceptables del mismo opcionalmente en un portador o diluyente farmacéuticamente aceptable, ya sea en solitario o en combinación con otros agentes antivirales o farmacéuticos conocidos, preferentemente agentes que pueden ayudar en el tratamiento de la infección por VIH, incluido el SIDA o mejorar los efectos secundarios y las afecciones asociadas con la infección por VIH. Este tratamiento también puede administrarse junto con otras terapias convencionales contra el VIH. La administración conjunta de compuestos activadores del VIH latente tales como prostratina, bradistatina 1 y análogos relacionados como se expone en la figura 4 del presente documento (véase De Christopher y col., Nature Chemistry, publicado en línea el 15 de julio de 2012, páginas 1-6), briostatina 1, briostatina 2, IL-7, inhibidores de histona desacetilasa, incluyendo zolinza (vorinostat), los inhibidores de la metilación del ADN, incluido el decogen (decitabina) y mezclas de los mismos, representan un enfoque alternativo para el tratamiento del VIH que puede dar como resultado una terapia eficaz que incluye la erradicación de VIH (cura) de un paciente.
Estos compuestos pueden administrarse por cualquier vía apropiada, por ejemplo, por vía oral, parenteral, intravenosa, intradérmica, subcutánea o tópica, en forma líquida, crema, gel o sólido, o en forma de aerosol.
El compuesto activo se incluye en el portador o diluyente farmacéuticamente aceptable en una cantidad suficiente para suministrar a un paciente una cantidad terapéuticamente eficaz para la indicación deseada, sin provocar efectos tóxicos graves en el paciente tratado. Una dosis preferida del compuesto activo para todas las afecciones mencionadas en el presente documento está en el intervalo de aproximadamente 10 ng/kg a 300 mg/kg, preferentemente, de 0,1 a 100 mg/kg al día, más generalmente, de 0,5 a aproximadamente 25 mg por kilogramo de peso corporal del receptor/paciente al día. Una dosis tópica típica variará entre el 0,01-5 % p/p en un portador adecuado.
El compuesto se administra convenientemente en cualquier forma farmacéutica unitaria adecuada, incluyendo, pero sin limitación, una que contiene menos de 1 mg, de 1 mg a 3000 mg, preferentemente, de 5 a 500 mg de principio activo por forma farmacéutica unitaria. Suele ser conveniente una dosis oral de aproximadamente 25-250 mg.
El principio activo se administra preferentemente para alcanzar concentraciones máximas en plasma del compuesto activo de aproximadamente 0,00001-30 mM, preferentemente, aproximadamente 0,1-30 pM. Esto puede conseguirse, por ejemplo, mediante la inyección intravenosa de una solución o formulación del principio activo, opcionalmente en solución salina, o un medio acuoso o administrado como un bolo del principio activo. La administración oral también es apropiada para generar concentraciones eficaces en plasma de principio activo.
La concentración de compuesto activo en la composición del fármaco dependerá de las tasas de absorción, distribución, inactivación y excreción del fármaco, así como otros factores conocidos por los expertos en la materia.
Cabe destacar que los valores de dosificación también variarán dependiendo de la gravedad de la afección a aliviar. Cabe entender además que para cualquier sujeto particular, los regímenes de dosificación específicos deben ajustarse con el paso del tiempo de acuerdo con las necesidades del individuo y el criterio profesional de la persona que administra o supervisa la administración de las composiciones y que los intervalos de concentración expuestos en el presente documento son solo ilustrativos y no pretenden limitar el alcance o la práctica de la composición reivindicada. El principio activo puede administrarse de una vez o puede dividirse en varias dosis menores para su administración a intervalos de tiempo variables.
Las composiciones orales incluirán generalmente un diluyente inerte o un portador comestible. Pueden encerrarse en cápsulas de gelatina o comprimirse en comprimidos. Con el fin de la administración terapéutica oral, el compuesto activo o su derivado profármaco puede incorporarse con excipientes y usarse en forma de comprimidos, trociscos o cápsulas. Pueden incluirse como parte de la composición agentes aglutinantes y/o materiales adyuvantes farmacéuticamente compatibles.
Los comprimidos, píldoras, cápsulas, trociscos y similares pueden contener cualquiera de los siguientes ingredientes o compuestos de una naturaleza similar: un aglutinante tal como celulosa microcristalina, goma de tragacanto o gelatina; un excipiente tal como almidón o lactosa, un agente dispersante tal como ácido algínico, Primogel o almidón de maíz; un lubricante tal como estearato de magnesio o Sterotes; en deslizante tal como dióxido de silicio coloidal; un agente edulcorante tal como sacarosa o sacarina; o un agente saporífero tal como menta, salicilato de metilo o aroma de naranja. Cuando la forma farmacéutica unitaria es una cápsula, puede contener, además del material del tipo anterior, un portador líquido tal como un aceite graso. Además, las formas farmacéuticas unitarias pueden contener diversos materiales distintos que modifiquen la forma física de la unidad de dosificación, por ejemplo, recubrimientos de azúcar, goma laca o agentes entéricos.
El compuesto activo o la sal farmacéuticamente aceptable del mismo pueden administrarse como un componente de un elixir, suspensión, jarabe, oblea, goma de mascar o similares. Un jarabe puede contener, además de los compuestos activos, sacarosa como agente edulcorante y determinados conservantes, tintes y colorantes y aromas.
El compuesto activo o las sales farmacéuticamente aceptables del mismo también pueden mezclarse con otros materiales activos que no dificultan la acción deseada o con materiales que complementan la acción deseada, tales como otros agentes contra el VIH, antibióticos, antifúngicos, antiinflamatorios o compuestos antivirales. Uno o más compuestos de CDM-H de acuerdo con la presente invención se pueden administrar conjuntamente con otro agente contra el VIH y/u otro agente bioactivo, incluyendo especialmente un activador de VIH latente como se describe de otro modo en el presente documento.
Las soluciones o suspensiones usadas para la aplicación parenteral, intradérmica, subcutánea o tópica pueden incluir los siguientes componentes: un diluyente estéril tal como agua para inyección, solución salina, aceites no volátiles, polietilenglicoles, glicerina, propilenglicol u otros disolventes sintéticos; agentes antibacterianos tales como alcohol bencílico o metilparabenos; antioxidantes tales como ácido ascórbico o bisulfito de sodio; agentes quelantes tales como ácido etilendiaminotetraacético; tampones tales como acetatos, citratos o fosfatos y agentes para el ajuste de la tonicidad tales como cloruro sódico o dextrosa. El preparado parenteral puede encerrarse en ampollas, jeringas desechables o viales multidosis hechos de vidrio o plástico.
Si se administran por vía intravenosa, los portadores preferidos son solución salina fisiológica o solución salina tamponada con fosfato (PBS).
En una realización, los compuestos activos se preparan con portadores que protegerán al compuesto contra su rápida eliminación del organismo, tal como una formulación de liberación controlada, incluyendo implantes y sistemas de administración microencapsulados. Se pueden usar polímeros biocompatibles biodegradables, tales como acetato de etilenvinilo, polianhídridos, ácido poliglicólico, colágeno, poliortoésteres y ácido poliláctico. Los métodos para preparar dichas formulaciones serán evidentes para aquellos expertos en la materia.
Las suspensiones liposómicas también pueden ser portadores farmacéuticamente aceptables. Estos pueden prepararse de acuerdo con métodos conocidos por los expertos en la materia, por ejemplo, como se describe en la Pat. de EE.UU. N.° 4.522.811. Por ejemplo, las formulaciones de liposomas pueden prepararse disolviendo uno o más lípidos apropiados (tales como estearoil fosfatidil etanolamina, estearoil fosfatidilcolina, aracadoilfosfatidilcolina y colesterol) en un disolvente inorgánico que a continuación se evapora, dejando una película fina de lípido seco en la superficie del recipiente. Después se introduce una solución acuosa del compuesto activo en el recipiente. A continuación, el recipiente se hace girar a mano para liberar el material lipídico de los lados del recipiente y dispersar los agregados lipídicos, formándose de esta manera la suspensión liposómica.
Desarrollo de CDM-H
Mientras se desarrollaban moléculas pequeñas de suministro de fármacos citotóxicos dirigidas al VIH (CDM-H), la primera preocupación era cómo el agente citotóxico conjugado entraría en la célula diana para inducir una respuesta citotóxica. Aunque existe abundante bibliografía relacionada con el desarrollo de toxinas quiméricas dirigidas a células
infectadas por VIH, no ha habido estudios que informen cómo estos conjugados pueden conseguir entrar en la célula infectada para suministrar la carga útil citotóxica. Se sabe que el complejo Env posee una cola citoplásmica altamente conservada que contiene una secuencia señal endocítica de tirosina, y se ha demostrado que Env experimenta endocitosis durante el período de infección, posiblemente para evadir el reconocimiento inmunitario.23-25 La endocitosis celular se ha revisado ampliamente,26 así como el desarrollo de estrategias de suministro de fármacos que se dirigen a la ruta endocítica.27, 28 Dado que la mayoría de las rutas endocíticas activas conducen a la fusión de lisosomas,29 la mayoría de las estrategias de suministro de fármacos que se dirigen a las rutas endocíticas utilizan enlazadores (que conectan un resto de direccionamiento a la carga útil) que pueden hidrolizarse a pH bajo, o enlazadores que contienen sustratos peptídicos para proteasas lisosomales (por ejemplo, catepsina B), liberando así el fármaco citotóxico activo u otra carga útil.27 Como alternativa, la carga útil se puede unir a la molécula de suministro/direccionamiento de tal manera que no interrumpa su función. Teniendo en cuenta estos factores, inicialmente se centraron en estrategias de enlazadores químicos que liberarían la carga útil citotóxica elegida en entornos acuosos ácidos; sin embargo, en futuros estudios, también se deben considerar estrategias de conjugación alternativas que no forman parte de la invención (por ejemplo, enlace disulfuro, sustratos de proteasa).
La siguiente consideración para el diseño de CDM-H fue la elección de la molécula citotóxica. De acuerdo con el objetivo general de desarrollar alternativas basadas en moléculas pequeñas para terapias a base de proteínas/péptidos, se optó por no utilizar proteínas citotóxicas, tales como la exotoxina A de Pseudomonas, sino agentes citotóxicos no peptídicos de bajo peso molecular. El uso de moléculas pequeñas citotóxicas para destruir células infectadas por el VIH se limita a un solo estudio.18 En este trabajo, la antraciclina doxorrubicina citotóxica se conjugó con un anticuerpo anti gp120 a través de una acil hidrazona lábil en medio ácido (véase el análisis anterior). Además, dado que la doxorrubicina es un pilar de la terapia contra el cáncer, existe una cantidad significativa de investigaciones que conjugan este agente quimioterapéutico con diversos restos de direccionamiento (por ejemplo, anticuerpos monoclonales,30 péptidos,31 y diversas moléculas pequeñas32), así como péptidos portadores33 y con moléculas de mejora de la semivida.34 Por lo tanto, se sabe mucho no solo sobre su actividad, sino también sobre la química de conjugación.35-37 Dados estos factores, los inventores utilizaron la doxorrubicina como nuestra carga útil citotóxica.
El diseño inicial de CDM-H se basó en estudios realizados por Kiessling y colaboradores, en los que conjugaron doxorrubicina con una molécula dirigida a integrina avp3 no peptídica a través de un enlace éster en el grupo alfa hidroxi.38 CDM-H 4.19 (figura 2) se sintetizó como se muestra en los Esquemas S4.3 (figura 4) y S4.4 (figura 5) y se basa en el armazón de ARM-H (como se describe en el documento W o 2011/046946, 21 de abril de 2011 y el documento WO 2012/068366 del 24 de mayo de 2012) con el enlazador conectado al grupo hidroxilo primario de doxorrubicina en la posición meta del anillo de benzamida. Los inventores primero confirmaron que la conjugación con doxorrubicina no perjudicó la capacidad de 4.19 para unirse a gp120 e inhibir la interacción con CD4. Como se muestra en la figura 10, 4.19 inhibió la interacción CD4-gp120 a una potencia similar observada para el análogo de ARM-H 3.11. A continuación, los inventores intentaron determinar si 4.19 podría destruir selectivamente las células que expresan gp120 del VIH-1 frente a las células de control. Para ello, los inventores desarrollaron un ensayo de viabilidad celular en el que sembraron células CHO-env (gp120+) o las células de control isogénicas CHO-pSV (gp120-) en placas de 96 pocillos, a continuación añadieron concentraciones crecientes de 4.19 y monitorizaron la viabilidad celular durante un tiempo establecido de 14 h. Como puede verse en la figura 10, 4.19 exhibió una citotoxicidad específica de gp120 significativa (~12 %) a concentraciones tan bajas como 309 nM. Más importante aún, no se observó citotoxicidad no específica, ni siquiera a concentraciones tan altas como 67 pM, mientras que la doxorrubicina libre no fue específicamente tóxica. Como control positivo, realizaron un ensayo paralelo con CD4-PE (figura 10). Curiosamente, observaron una toxicidad específica de gp120 de 4.19 a concentraciones en las que no se observó toxicidad para la doxorrubicina libre (a 310 nM y 1,85 pM). Estas observaciones sugieren que la modificación de la doxorrubicina (conjugación con TBT/VICB) reduce su toxicidad general a concentraciones más altas. Sin embargo, aumenta la toxicidad específica de gp120 a concentraciones más bajas, lo que sugiere que la adición del grupo de dirigido a gp120 (TBT/VICB) mejora la concentración eficaz de doxorrubicina en células que expresan gp120 sobre células que no expresan gp120. Sin embargo, cuando el compuesto de CDM-H 4.19 (véase la figura 2) se incubó con células durante más tiempo (24 h), apreciaron una pérdida completa de selectividad entre las células gp120+ y gp120-, así como un aumento en la toxicidad general (4.19 fue más tóxicos en tiempos de incubación más largos que en tiempos más cortos).
Con esta pérdida de selectividad y aumento de la toxicidad, los inventores plantearon la hipótesis de que el enlace éster puede haber sufrido hidrólisis, produciendo la forma activa libre de doxorrubicina. Para investigar esta posibilidad, se desarrolló un método de LC/MS para monitorizar la semivida de 4.19 cuando se incubó en las condiciones de ensayo usadas en el ensayo de citotoxicidad. Curiosamente, se observó que la semivida del enlace éster en el tampón PBS a 37 °C fue de aproximadamente 5,6 h (figura 11), lo que sugiere que la pérdida de selectividad durante tiempos de incubación más prolongados puede ser el resultado de la hidrólisis. Para compensar la labilidad de este enlace éster, se exploran estrategias de conjugación alternativas.
En estudios realizados por Vyas y colaboradores, la doxorrubicina se conjugó con un enlazador a través de un enlace de acil hidrazona, que resultó ser completamente estable a pH 7,4, sin embargo, a pH 5,0 (el pH aproximado del lisosoma es 4-539), poseía una semivida de hidrólisis de aproximadamente 5 h.35 Teniendo en cuenta estas observaciones, se sintetizó un nuevo análogo de CDM-H (4.20, figura 2) que tiene doxorrubicina conjugada con el
resto TBT/VICB a través de acil hidrazona. Además, en esta CDM-H, el enlazador PEG se unió al furano de TBT/VICB, lo que da como resultado una mayor afinidad por gp120. Los inventores demostraron primero que la semivida en hidrólisis de 4.20 a pH 7,4 se extendió a aproximadamente 55 horas (figura 12) y que mantuvo la capacidad de inhibir la interacción CD4-gp120 (figura 13). A continuación, usando el sistema de monitorización de viabilidad celular xCELLigence, se examinó la toxicidad de 4.20 (10 |jM) contra células CHO-env (gp120+) y células CHO-pSv (gp120-) durante un período de 150 h cuando se añadió 4.20 solo una vez y cuando se añadió varias veces (se eliminaron los medios de crecimiento y se repusieron con medios nuevos que contenían 4.20). Como se muestra en la figura 14, en condiciones en las que se añadió 4.20 solo una vez, los inventores observaron una toxicidad selectiva de gp120 constante de ~10 %, sin embargo, cuando se añadió 4.20 varias veces, la toxicidad selectiva de gp120 aumenta a ~35 %o (en ~80 horas). Los inventores plantean la hipótesis de que el reciclaje de los medios elimina cualquier doxorrubicina no específica (a través de 4.20 hidrolizado) y la repone por 4.20 no hidrolizado que aún puede dirigirse a las células que expresan gp120. A pesar de estos resultados, en ambas condiciones se observa un aumento significativo de la toxicidad de 4.20 frente a las células CHO-pSV (gp120-).
Los inventores plantearon la hipótesis de que esta toxicidad no específica es el resultado de la absorción no específica de 4.20. Dado que la propia doxorrubicina es fluorescente,40 los inventores realizaron microscopía de inmunofluorescencia para rastrear la ubicación subcelular de 4.20 en el tiempo. En este experimento, los inventores tiñeron conjuntamente células CHO-env (gp120+) y CHO-pSv (gp120-) con colorante LysoTracker, que tiñe organelos ácidos de las células (es decir, lisosomas) y se incubaron con 4.20. Como se puede ver en la figura 15, existe una ubicación conjunta significativa de (4.20 - doxorrubicina) y fluorescencia (LysoTracker) para las células CHO-env y CHO-pSv, lo que sugiere que 4.20 se localiza en los lisosomas de ambos tipos de células independientemente de la expresión de gp120. Durante el mismo tiempo de incubación, la doxorrubicina libre entra rápidamente en el núcleo de la célula (figura 16). Durante esta incubación, después de aproximadamente 20 h, se apreció que 4.20 comenzó a formar agregados, posiblemente micelas, lo cual no es irrazonable considerando la polaridad de la doxorrubicina (es decir, una amina primaria, 5 grupos hidroxilo) en comparación con la del resto TBT/VICB, que es relativamente hidrófoba. La formación de agregados dependiente de la concentración también se confirmó mediante análisis de dispersión de luz dinámica (figura 16). La endocitosis y pinocitosis no específica de micelas, agregados y nanopartículas es una característica bien conocida de las células de mamífero,41'44 y, por lo tanto, puede explicar los niveles relativamente altos de citotoxicidad no específica de 4.20.
Los inventores plantean la hipótesis de que la longitud del enlazador largo de 4.20 puede favorecer la solvatación separada de la doxorrubicina, así como del VICB, promoviendo así la formación de micelas. En estudios alternativos, la longitud del enlazador se reduce (similar a la de 4.19). La síntesis de análogos con enlazadores más cortos y su evaluación biológica está actualmente en curso en el laboratorio. Además, se investigan estrategias de conjugación alternativas y cargas útiles citotóxicas. Además, los inventores están usando CDM-H de acuerdo con la presente invención en una estrategia de "activación-eliminación"7 para dirigirse a la infección por VIH latente (figura 1). En estos estudios, los inventores examinan la capacidad de las CDM-H para destruir selectivamente las células infectadas de forma latente que se han activado (inducidas a expresar proteínas del VIH)45, 46 con moléculas tales como prostratina47, 48 y otros activadores del VIH latente en diversas células y modelos animales.49-51
La siguiente descripción detallada describe el diseño y la síntesis de varias moléculas pequeñas bifuncionales capaces de suministrar restos citotóxicos en células (células CD4) que están infectadas con el VIH.
La siguiente síntesis química que se presenta en el Esquema 1 (figura 4) y el Esquema 2 (figura 5) puede usarse para sintetizar el compuesto marcado como 4.19 en la figura 2 que muestra una actividad excepcional como agente contra el VIH. La síntesis química del Esquema 1 proporciona el intermedio S4.11 a partir de materiales de partida básicos que representa un resto VICP básico al que se ha unido un enlazador en el grupo benzoílo para proporcionar la amina intermedia S4.11. La amina intermedia S4.11 se puede usar para condensar un agente citotóxico adecuadamente sustituido, tal como doxorrubicina u otros grupos citotóxicos, como se describe en el presente documento, para producir el compuesto final 4.19 de acuerdo con el Esquema 2. Las modificaciones secundarias de la química del Esquema 1 pueden producir enlazadores en diversas posiciones del resto VICB que pueden usarse para unir restos citotóxicos como se describe de otro modo en el presente documento. Las síntesis alternativas que se exponen en los esquemas químicos 4.5 (figura 6), Esquema 4 (figura 7), Esquema 5 (figura 8) y Esquema 6 (figura 9) y como se describe en las solicitudes internacionales relacionadas WO 2011/046946 y WO 2012/068366 proporcionan síntesis químicas estándar de todos los restos VICB relacionados a los que se pueden unir restos citotóxicos en diversas posiciones del resto (usando enlazadores no lábiles y/o enlazadores lábiles y/o restos enlazadores como se describe generalmente en el presente documento). La unión de uno o más de los enlazadores lábiles que se describen en el presente documento se puede realizar fácilmente usando técnicas químicas sintéticas estándar mediante la condensación de un grupo nucleófilo en un grupo electrófilo para producir un resto citotóxico (CYT) que se une a un resto VICB a través de un enlazador lábil y, opcionalmente, un enlazador no lábil y/o un resto enlazador. Esta química estándar se conoce bien en la materia.
Por ejemplo, el Esquema 1 (figura 4) proporciona una síntesis química sencilla para proporcionar una química de grupos funcionales que puede modificarse fácilmente para introducir un enlazador lábil y un resto citotóxico en el resto amina del grupo fenilo como se representa para el compuesto S4.11. En este esquema, el grupo furano permanece sin sustituir (pero puede sustituirse fácilmente usando un precursor de areno 1 sustituido) al igual que el grupo metoxi
en la posición principal del grupo indol. El Esquema 4.4 (figura 5) muestra la condensación de un grupo citotóxico apropiadamente modificado (en este caso, doxorrubicina) que se condensa en el extremo amino del compuesto S4.11 para formar el compuesto 4.19 como la sal formiato.
El Esquema 4.5 (figura 6) muestra la formación del compuesto final 4.20 a partir del compuesto de amina S3.37 (que contiene un grupo Areno 1 sustituido al que se une una construcción de resto citotóxico enlazador, cuya síntesis se representa en el Esquema 5, figura 8 del presente documento) haciendo reaccionar la amina S3.37 con ácido 2-mercaptopropanoico para formar el intermedio S4.14. El intermedio S4.14 se condensa en el resto citotóxico S4.15 que contiene un grupo enlazador lábil en medio ácido y un grupo maleimida en el que se puede introducir el grupo tiol de S4.14 para proporcionar el compuesto 4.20 como la sal acetato.
El Esquema 4 (figura 7A) proporciona una síntesis sencilla del intermedio S3.19 que proporciona un grupo piperazina en un grupo indol sustituido por bromo y metoxi que puede funcionalizarse fácilmente con un grupo areno 2 opcionalmente sustituido, cuyo grupo puede estar sustituido o sin sustituir y puede estar unido covalentemente a un enlazador lábil y un resto citotóxico. Además, el intermedio S3.29 puede modificarse para introducir un grupo Areno 1 opcionalmente sustituido (que puede funcionalizarse aún más para contener un grupo citotóxico) en la posición sustituida por bromo y el grupo indol puede modificarse aún más para contener un resto citotóxico en la posición metoxi del grupo indol. Por lo tanto, el esquema 5, figura 7 muestra la síntesis básica de un intermedio fácilmente funcionalizado a partir del material de partida disponible S3.15 para proporcionar, a través de varias etapas, el intermedio S3.19 que contiene un grupo piperazina que puede funcionalizarse adicionalmente de acuerdo con la presente invención. Además, el grupo bromo en el indol se puede sustituir fácilmente por un grupo Areno 1 funcionalizado como se expone en el Esquema 5, figura 8 del presente documento (por ejemplo, un grupo areno protegido con amina/NHBoc, tal como un grupo furano u otro grupo arilo) para desplazar el grupo bromo, seguido de la unión del grupo amina desprotegido en el grupo Areno I con, por ejemplo, un enlazador no lábil, una molécula enlazadora (por ejemplo, un grupo 1,2,3-triazol), un enlazador lábil y un grupo citotóxico. Estas reacciones se generalizan a partir de la química que se desvela de otro modo en el presente documento. Este enfoque químico para funcionalizar la química del grupo Areno I como se muestra en el Esquema 5, figura 8, también se describe en general en la publicación de patente internacional WO2012/068366.
La figura 7B muestra la modificación del compuesto citotóxico doxorrubicina para proporcionar un enlazador lábil que se une además a un grupo que se puede condensar con el resto VICB según el esquema de síntesis química 4.5 (figura 6). Del mismo modo, cada uno de los compuestos citotóxicos que se pueden usar de acuerdo con la presente invención se puede obtener y funcionalizar fácilmente haciendo reaccionar un grupo funcional (como se indica en la definición de extremo de citotoxicidad o resto de agente citotóxico) en la molécula citotóxica para proporcionar un enlazador lábil, que se une además al resto VICB, a través de un enlazador no lábil y una molécula conectora opcional, para proporcionar compuestos de acuerdo con la presente invención.
El Esquema 6, figura 9, muestra la introducción de un resto citotóxico en la posición principal del resto heteroarilo de pirrolopiridina como se representa. Por consiguiente, se puede permitir la síntesis química de restos de pirrolopiridina que contienen un resto citotóxico en la posición principal. Este enfoque puede adaptarse a otros compuestos que se describen en el presente documento. En el esquema 6, figura 9, la introducción de un enlazador de etilenglicol en la posición principal que se sustituye por una azida puede hacerse reaccionar con el intermedio acetilénico funcionalizado de manera que se forme una molécula conectora de triazina que contenga opcionalmente un segundo enlazador de etilenglicol como se representa. Se pueden proporcionar fácilmente alternativas a este enfoque modificando fácilmente la química representada en el Esquema 6 del presente documento.
Otras síntesis químicas de todos los compuestos descritos en el presente documento se adaptan fácilmente a partir de los esquemas sintéticos descritos o, como alternativa, a partir de esquemas químicos sintéticos que se presentan en los documentos WO 2010/052344 y WO 2012/068366 para proporcionar una diversidad de restos VICB modificados. A estos restos VICB se les puede unir un resto enlazador y un resto conector opcional que se condensan adicionalmente a través de un enlazador lábil como se describe de otro modo en el presente documento en un resto citotóxico para proporcionar todos los compuestos de CDM-H.
Los esquemas químicos descritos anteriormente proporcionan síntesis de ejemplo de compuestos de acuerdo con la presente invención con diversas iteraciones de los mismos proporcionadas por analogía usando métodos bien conocidos como se describe en el presente documento y como entienden los expertos en la materia. Se observa que los esquemas que se proporcionan no se deben considerar limitantes al exponer las enseñanzas que proporcionan compuestos de acuerdo con la presente invención.
Estos compuestos se pueden formular en composiciones farmacéuticas como se describe de otro modo en el presente documento, y pueden usarse en los métodos que se desvelan.
Información sintética detallada y sección experimental biológica
Abreviaturas usadas
AcOH = ácido acético
Boc = ferc-butoxicarbonilo
BSA = albúmina sérica bovina
DCM = diclorometano
DEPBT = 3-(dietoxifosforiloxi)-1,2,3-benzotriazin-4-(3H)-ona
DIPEA = W,W-diisopropiletilamina
DMF = W,W-dimetilformamida
DMSO = dimetilsulfóxido
DNP = 2,4-dinitrofenilo
DPBS = Solución salina tamponada con fosfato de Dulbecco
EDC = 1-etil-3-(3-dimetilaminopropil)carbodiimida
EtOAc = acetato de etilo
Fmoc = 9-fluorenilmetiloxicarbonilo
HI-FBS = suero fetal bovino inactivado por calor
HOBt = hidroxibenzotriazol
MeCN = acetonitrilo
MeOH = metanol
MW = reactor para microondas
NaAsc = ascorbato de sodio
pyr = piridina
fBuOH = t-butanol
TEA = trietilamina
TFA = ácido trifluoroacético
THF = tetrahidrofurano
Materiales e información general
Los materiales de partida adquiridos se usaron tal como se recibieron a menos que se indique lo contrario. Todas las reacciones sensibles a la humedad se realizaron en una atmósfera seca e inerte de nitrógeno en cristalería secada a la llama. Se usaron disolventes de calidad reactiva para las extracciones y la cromatografía ultrarrápida. El progreso de la reacción se comprobó mediante cromatografía analítica de capa fina (TLC, placas de gel de sílice 60 F-254 de Merck). Las placas se controlaron con iluminación ultravioleta o carbonización con tinciones de anisaldehído (panisaldehído al 2,5 %, AcOH al 1 %, H2SO4 (conc.) al 3,5 % en EtOH al 95 %) o ninhidrina (ninhidrina al 0,3 % (p/v), 97:3 de EtOH-AcOH). La cromatografía en columna ultrarrápida se realizó usando gel de sílice (malla de 230-400). Las composiciones de disolventes notificadas para todas las separaciones cromatográficas se basan en volumen/volumen (v/v). Los gráficos y análisis moleculares se realizaron con el paquete UCSF Chimera. Chimera se desarrolla por Resource for Biocomputing, Visualization, and Informatics de la Universidad de California, San Francisco, financiado por subvenciones de los National Institutes of Health National Center for Research Resources (2P41RR001081) y el National Institute of General Medical Sciences (9P41GM103311).
Instrumentación
Los espectros de 1H RMN se registraron a 400 o 500 MHz y se expresan en partes por millón (ppm) en la escala 6 en relación con el disolvente como patrón interno (CDCl3 = 7,26 ppm, DMSO-d6 = 2,49 ppm, Cd 3Od = 3,30). Los datos se informan de la siguiente manera: desplazamiento químico, multiplicidad (s = singlete, d = doblete, t = triplete, c = cuadruplete, a = ancho, m = multiplete), constantes de acoplamiento (Hz) e integración. Los espectros de 13C RMN se registraron a 100 o 125 MHz y se expresan en partes por millón (ppm) en la escala 6 en relación con el disolvente como patrón interno (CDCl3 = 77,0 ppm, DMSO-d6 = 39,5 ppm, CD3OD = 49,0 ppm). La cromatografía líquida de ultra alto rendimiento/espectrometría de masas (UPLC/MS) analítica se realizó en un instrumento Waters UPLC/MS equipado con una columna C18 de fase inversa (tamaño de partícula de 1,7 pm, 2,1 Á~50 mm), ionización química a presión atmosférica dual (API)/detector de espectrometría de masas por electronebulización (ESI) y detector de matriz de fotodiodos. Las muestras se eluyeron con un gradiente lineal de acetonitrilo al 20 %-agua que contenía ácido fórmico al 0,1 % ^ acetonitrilo al 100 % que contenía ácido fórmico al 0,1 % durante 3 min, seguido de acetonitrilo al 100 % que contenía ácido fórmico al 0,1 % durante 1 min, a un caudal de 800 pl/min. La cromatografía líquida de alta resolución-espectrometría de masas (HR-LC/MS) se realizó en un instrumento Waters UPLC/HRMS equipado con un espectrómetro dual de masas de alta resolución API/ESI y un detector de matriz de fotodiodos. Cromatografía líquida de alto rendimiento (HPLC) usando un sistema de suministro de disolvente Dynamax Rainin equipado con un detector Varian Prostar (sistema de datos de cromatografía Galaxie versión 1.8.505.5), y se realizaron mediciones de absorbancia a 214 y 254 nm simultáneamente. Se usó una columna Waters Xterra Prep MS C18 de 7,8 x 150 mm para purificaciones semipreparativas usando un gradiente de agua:acetonitrilo (A:B) que contenía TFA al 0,1 % a 5,0 ml/min, como se especifica a continuación para los compuestos individuales. La cromatografía en columna ultrarrápida (a menos que se indique lo contrario) se realizó usando gel de sílice (malla de 230-400) usando Teledyne Isco CombiFlash Rf 200 equipado con un detector UV y un colector de fracciones. Los espectros infrarrojos de transformada de Fourier (ATR-FTIR) de reflectancia total atenuada se obtuvieron usando un espectrómetro de FTIR Nicolet 6700 de Thermo Electron Corporation. Los datos se representan de la siguiente manera: frecuencia de absorción (cm-1), intensidad de absorción (f = fuerte, m = media, d = débil, a = amplia).
Procedimientos sintéticos químicos
Siguiendo el Esquema 1, figura 4. Síntesis de la amina (S4.11) precursora de CDM-H 4.18

(S4.7) En un matraz equipado con un condensador de reflujo que contenía S4.652 (500 mg, 1,39 mmol) in MeCN (25 ml) se añadieron 3-hidroxibenzoato de metilo (243 mg, 1,6 mmol, 1,15 equiv.) y K2CO3 (221 mg, 1,6 mmol, 1,15 equiv.). Se calentó a 75 °C hasta que la TLC (5:1 de hexanos/EtOAc) indicó la finalización de la reacción (18 h). La reacción se dejó enfriar a TA, a continuación se interrumpió con NH4Cl sat. (30 ml), se extrajo con DCM (3 x 40 ml), a continuación se secó sobre MgSO4 anh. y se filtró. Los volátiles se eliminaron a través de evaporación rotatoria y S4.7 en bruto se purificó por cromatografía ultrarrápida (cromatógrafo automático CombiFlash, columna de 24 g, se cargó en seco con 4 g de columna de carga en seco cargada previamente. Se realizó usando del 100 % de hexanos al 50 % de EtOAc en gradiente de hexanos sobre 20 volúmenes de columna) para producir S4.7 en forma de un sólido pegajoso transparente (391 mg, 83 %). 1H RMN (400 MHz, CDCla) 67,66-7,60 (m, 1H), 7,58 (d, J = 2,5, 1H), 7,33 (t, J = 8,0, 1H), 7,17-7,09 (m, 1H), 4,98 (s a, 1H), 4,19-4,14 (m, 2H), 3,85-3,78 (m, 2H), 3,60 (t, J = 5,1, 2H), 3,34 (m, 2H), 1,43 (s, 9H).

(S4.8) Una solución de S4.7 (310 mg, 0,91 mmol) en THF (12 ml) y NaOH ac. (2 M, 5,7 ml) se calentó a reflujo durante 18 h cuando el análisis por TLC indicó la finalización de la reacción (20:1 de CH2Cl2/CH3OH). La solución se acidificó a un pH de 1 usando HCl ac. 6 M y a continuación se extrajo con CH2Cl2 (3 x 30 ml). Las capas orgánicas combinadas se secaron sobre MgSO4 anh., se filtraron, y todos los disolventes se evaporaron, dando como resultado S4.8 en forma de un aceite viscoso transparente (293 mg, 99 %), que se usó sin purificación adicional. 1H RMN (400 MHz, CDCb) a 12,34-10,19 (s a, 1H), 7,77-7,54 (m, 2H), 7,36 (m, 1H), 7,17 (m, 1H), 5,03 (s, 1H), 4,17 (m, 2H), 3,84 (s a, 2H), 3,62 (m, 2H), 3,35 (m, 2H), 1,43 (s, 9H).

(S4.9) En un matraz secado a la llama que contenía una solución de S4.8 (100 mg, 0,307 mmol, 1,1 equiv.) en DCM (10 ml) se añadieron S3.5a (100 mg, 0,27 mmol), EDC-HCl (48 mg, 0,307 mmol, 1,1 equiv.), HOBT (39 mg, 0,307 mmol, 1,1 equiv.) y DIPEA (120 ul, 0,81 mmol, 3 equiv.). La mezcla resultante se agitó a TA durante 14 h cuando el análisis por TLC (9:1 de DCM/CH3OH) indicó la finalización de la reacción. La mezcla se diluyó con CH2Cl2 (10 ml) y se lavó con NaHCO3 sat. (15 ml), NH4Cl sat. (15 ml) y salmuera (15 ml). Las capas orgánicas combinadas se secaron sobre MgSO4 anh., se filtraron, y todos los disolventes se evaporaron, dando como resultado S4.9 en bruto en forma de un sólido pegajoso. S4.9 en bruto se purificó por cromatografía ultrarrápida (cromatógrafo automático CombiFlash, columna de 12 g, se cargó en seco con 24 g de columna de carga en seco cargada previamente. Se realizó usando del 100 % de DCM al 10 % de MeOH en gradiente de DCM sobre 30 volúmenes de columna) para producir S4.9 en forma de un sólido en polvo incoloro mullido (132 mg, 73 %). 1H RMN (400 MHz, CDCla) 69,77 (s, 1H), 7,93 (s, 1H), 7,36-7,20 (m, 2H), 6,98 (s a, 3H), 6,56 (d, J = 8,5, 1H), 5,10-4,90 (s a, 1H), 4,12 (s, 2H), 3,90 (s, 3H), 3,85-3,20 (m, 14H), 1,43 (s, 9H).

(S4.10) A S4.9 (50 mg, 0,074 mmol) en DCM (500 pl) se le añadió TFA (333 pl), dando como resultado un cambio de color de transparente a transparente-amarillo. Después de 1 h, el análisis por TLC indicó la finalización de la reacción (9:1 de DCM/MeOH). Los volátiles se eliminaron cuidadosamente a través de evaporación rotatoria, con lavado varias
veces con DCM, dando como resultado S4.10 en bruto (42 mg, 0,073 mmol, 99 %) en forma de un sólido pegajoso, que se usó sin purificación adicional. 1H RMN (400 MHz, MeOD-d4) 58,14 (s, 1H), 7,40 (s a, 1H), 7,38 (d, J = 8,4, 1H), 6,72 (d, J = 8,5, 1H), 4,22 (s a, 2H), 3,99-3,41 (m, 15H), 3,16 (s a, 2H).

(S4.11) A una solución de S4.10 (74 mg, 0,11 mmol) en DMF (3,0 ml)/H2O (1,8 ml) en un vial para microondas se le añadieron NaHCO3 (12,8 mg, 0,153 mmol, 1,4 equiv.) y ácido 2-furanilborónico (17,1 mg, 0,153 mmol, 1,4 equiv.). Se eliminó el O2 de la solución mediante burbujeo con N2 durante al menos 10 min. Se añadió cuidadosamente Pd(PPh3)4 (6,3 mg, 5,46 pmol, 5 % en moles), el vial se tapó y se calentó en un reactor para microondas durante 15 min a 150 °C cuando el análisis por UPLC/MS indicó el consumo del material de partida y la formación de producto. Todos los disolventes se evaporaron y el S4.11 en bruto se purificó por cromatografía ultrarrápida (cromatógrafo automático CombiFlash, columna de 12 g, se cargó en seco con 4 g de columna de carga en seco cargada previamente. Se realizó usando del 100 % de DCM al 10 % de MeOH en gradiente de DCM sobre 15 volúmenes de columna, a continuación MeOH al 10-30 % en DCM sobre 5 volúmenes de columna) para producir S4.11 en forma de un polvo de color amarillo (58 mg, 94 %). 1H RMN (400 MHz, CDCI3) 58,03 (s, 1H), 7,51 (s, 1H), 7,41 (d, J = 8,1, 1H), 6,94 (s a, 3H), 6,68 (d, J = 8,0, 1H), 6,63-6,57 (m, 1H), 6,54-6,38 (m, 1H), 4,11 (s a, 2H), 3,89 (s, 3H), 3,85-3,20 (m, 12H), 3,06 (s, 2H). UPLC/HRMS (ESI+) calc. para [M+H]+ C30H33N4O7+ 561,2344, observado 561,2398.
Esquema 2, figura 5-Ensamblaje final de CDM-H 4.19 a partir de la amina S4.11 y el NHS-éster S4.12 conocido.
(S4.13) En un matraz secado a la llama que contenía S4.1238 (27 mg, 0,028 mmol) disuelto en

DMF anhidro (2,2 ml) se le añadió S4.11 (38 mg, 0,068 mmol, 2,4 equiv.), dando como resultado una solución homogénea de color rojo claro que se agitó a TA en una atmósfera de nitrógeno. Después de aproximadamente 4 horas, se añadió DIPEa (8 pl), dando como resultado un cambio de color a rojo oscuro. En 1 hora, el análisis por UPLC/MS indicó el consumo del material de partida y la formación de la masa de producto. Los volátiles se eliminaron cuidadosamente a través de evaporación rotatoria y el S4.13 en bruto purificó por cromatografía ultrarrápida (cromatógrafo automático CombiFlash, columna de 12 g, se cargó en seco con 4 g de columna de carga en seco cargada previamente. Se realizó usando del 100 % de DCM al 7 % de MeOH en gradiente de DCM sobre 40 volúmenes de columna, a continuación lavado de MeOH al 20 %) para producir S4.13 en forma de un residuo de color rojo (19 mg, 48 %). 1H RMN (500 MHz, CDCh) 514,15-13,64 (m, 1H), 13,31-13,00 (m, 1H), 10,34-9,61 (m, 2H), 8,14-7,95 (m, 2H), 7,91-7,28 (m, 11H), 7,18-6,81 (m, 4H), 6,77-6,29 (m, 4H), 5,51-5,33 (m, 2H), 5,25-4,98 (m, 3H), 4,84-4,53 (m, 2H), 4,41-4,26 (s a, 2H), 4,26-4,00 (m, 6H), 3,94-3,18 (m, 18H), 3,05-2,80 (m, 1H), 2,71-1,71 (m, 10H), 1,43-1,10 (m, 5H). UPLC/HRMS (ESI+) calc. para [M+H]+ C77H76NsO22+ 1422,4976, observado 1422,4956, tn 1,86 min.

(4.19) En un matraz secado a la llama que contenía una solución de S4.13 (19 mg, 0,013 mmol) en DMF anh. (1 ml) se le añadió piperadina (100 pl), dando como resultado un cambio de color de rojo a púrpura/azul. Después de 2,5 h, El análisis por UPLC/MS indicó la formación del producto desprotegido con Fmoc y el consumo del material de partida. La reacción se interrumpió con ácido fórmico al 1 % en DMF gota a gota hasta que la solución de reacción cambio de color de azul a rojo (~500 pl) y los volátiles se eliminaron a través de evaporación rotatoria. El 4.19 en bruto resultante se purificó por HPLC de fase inversa (0-80 % de B durante 60 min; Nota: El ácido fórmico al 0,1 % se sustituyó por TFA al 0,1 % para ambos disolventes A y B) y las fracciones puras se combinaron y se secaron a través de un liofilizaron, dando como resultado 4.19 puro (5,6 mg, 36 %) en forma de un polvo de color rojo. 1H RMN (500 MHz, CDCI3) ó 14,06-13,69 (m, 1H), 13,25-12,99 (m, 1H) 10,64-10,47 (m, 1H), 8,02 (s a, 2H), 7,94-7,86 (m, 1H), 7,74-7,65 (m, 1H), 7,55-7,46 (m, 1H), 7,27 (s a, 2H), 7,12-6,89 (m, 2H), 6,60 (s a, 2H), 6,50-6,43 (m, 1H), 5,50-5,42 (m, 1H), 5,35-5,03 (m, 3H), 4,15 (s, 3H), 3,99 (s, 3H), 3,91 (s, 3H), 3,86-3,32 (m, 18H), 3,25-3,14 (m, 2H), 2,92-2,89 (m, 2H), 2,55-2,19(m, 6H), 2,10-1,89 (m, 3H), 1,36-1,11 (m, 5H). Nota: muestra contaminada con sal formiato. UPLC/HRMS (ESI+) calc. para [M+H]+ C62H66N5Ü20+ 1200,4296, observado 1200,4299, tR 1,41 min.
Siguiendo el Esquema 3, Figura 6. Ensamblaje final de CDM-H 4.20 a partir de tiol S4.14 y la maleimida S4.15 conocida.

(S4.14) En un matraz secado a la llama que contenía una solución de S3.37 (55 mg, 0,053 mmol; en DCM (1 ml) se añadieron ácido 3-mercaptanoico (11,5 mg, 7,9 pl 0,11 mmol, 2 equiv.), EDC-HCl (18,5 mg, 0,1 mmol, 1,8 equiv.), HOBT (16 mg, 0,1 mmol, 1,8 equiv.) y DIPEA (44 ul, 3 equiv.). La mezcla resultante se agitó a TA durante 11 h cuando el análisis por UPLC/MS indicó la finalización de la reacción. Todos los disolventes se evaporaron, dando como resultado S4.14 en bruto en forma de un sólido pegajoso, que se purificó por cromatografía ultrarrápida (cromatógrafo automático CombiFlash, columna de 4 g, se cargó en seco con 4 g de columna de carga en seco cargada previamente. Se realizó usando del 100 % de DCM al 20 % de MeOH en gradiente de DCM sobre 80 volúmenes de columna) para producir S4.13 en forma de un sólido pegajoso (19 mg, 30 %). 1H RMN (500 MHz, CDCh) ó 11,34 (s, 1H), 8,14 (d, J = 3,3, 1H), 8,08 (s a, 1H), 7,69 (s, 1H), 7,49-7,36 (m, 6H), 6,69 (d, J = 8,4, 1H), 6,53 (d, J = 3,3, 1H), 6,31 (d, J = 3,3, 1H), 4,65 (s a, 2H), 4,55 (d aparente, J = 6,2, 2H), 4,47-4,43 (m, 2H), 4,04 (s a, 2H), 3,95 (s, 3H), 3,79-3,76 (m, 2H), 3,72-3,34 (m, 44H), 2,78 (dd, J = 6,8, 15,1, 2H), 2,48 (t, J = 6,8, 2H), 1,62 (t, J = 8,3, 1H). UPLC/HRMS (ESI+) calc. para [M+H]+ C55H77N8O17S+ 1153,5122, observado 1153,5189.
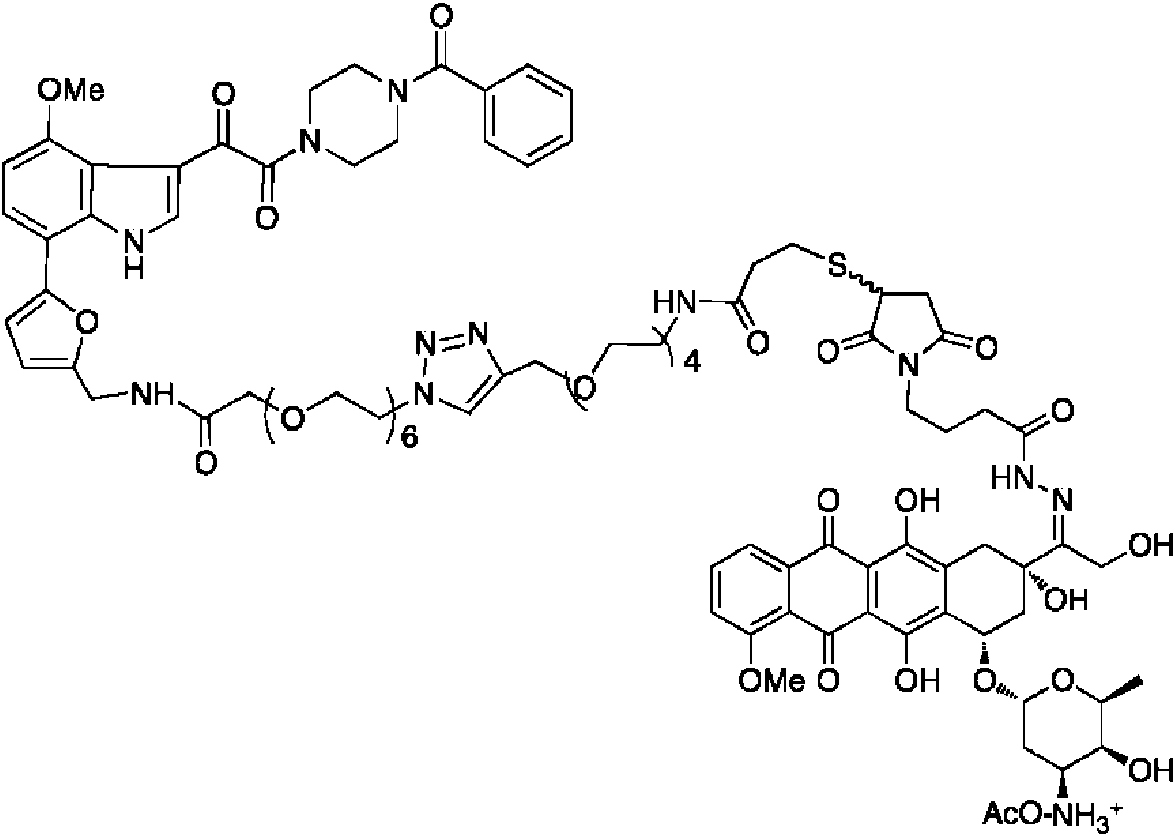
(4.20) En un matraz secado a la llama que contenía S4.14 (19 mg, 0,016 mmol) en MeOH anh. (600 pl) se añadió S4.155354 (15,2 mg, 0,018 mmol, 1,12 equiv.) seguido de DlPEA (0,5 pl), dando como resultado la formación de un
precipitado. La reacción se monitorizó cuidadosamente por UPLC/MS y después de 22 h, el material de partida se consumió y se detectó la masa deseada. Los volátiles se eliminaron y el 4.20 en bruto se purificó por HPLC de fase inversa, en la que el disolvente A = Et3N 30 mM/AcOH en agua desionizada, ajustado a un pH de 6,8; B = MeCN (15 50 % de B durante 46 min, 5 ml/min). Cabe apreciar que si se usa un eluyente no tamponado (por ejemplo, TFA o ácido fórmico), el enlace acil hidrazona se hidroliza. Las fracciones puras se combinaron inmediatamente y se liofilizaron, dando como resultado 4.20 puro (~90 %) (4 mg, 12 %) en forma de un sólido pegajoso de color rojo. 1H RMN (500 MHz, CDCI3) ó 11,50-11,27 (m, 1H), 10,48-10,16 (m, 1H), 8,25-8,17 (m, 1H), 8,17-8,09 (m, 1H), 8,06-7,94 (m, 1H), 7,80-7,55 (m, 3H), 7,51-7,31 (m, 5H), 6,73-6,61 (m, 1H), 6,56-6,42 (m, 1H), 6,34-6,23 (m, 1H), 5,57-5,42 (m, 1H), 5,31-5,15 (m, 1H), 4,77-4,37 (m, 7H), 4,20-3,07 (m, 41H), 2,64-2,33 (m, 6H), 1,78-1,53 (m, 3H), 1,36-1,25 (m, 2H). Nota: muestra contaminada con sal acetato de trietilamonio. UPLC/HRMS (ESI+) calc. para [M+H]+ C90H115N12O30S+ 1875,7563 y 1876,7596, observado 1875,8802 y 1876,8636.
Análisis de la semivida hidrolítica por UPLC/HRMS
Para examinar la estabilidad hidrolítica de diversas conjugaciones citotóxicas, se usó UPLC/MS para calcular la semivida de CDM-H en tampón DPBS. Se preparó una solución de la molécula de prueba (10 pM) en DPBS (Gibco) y se incubó a 37 °C, CO2 al 5 % en viales para LC/MS durante un tiempo específico. La relación relativa de doxorrubicina libre: CDM-H se determinó integrando la señal de absorbancia a una longitud de onda de 490 nm (absorbancia máxima de doxorrubicina) para la doxorrubicina y la CDM-H, que eluyen en tiempos separados, usando el software Waters. Método UPLC/MS descrito en la sección Información general. Datos ajustados usando un modelo de decaimiento de una fase con GraphPad Prism.
% de CDM-H = [(Abs. de CDM-H)/(Abs. de Dox Abs. de CDM-H)] * 100

Hidrólisis del éster de CDM-H 4.19, según se detecta por UPLC/HR-MS.

Hidrólisis de CDM-H acil hidrazona 4.20, según se detecta por UPLC/HR-MS.
Biología
Información general
Todos los reactivos y proteínas usados están disponibles comercialmente y se usan tal como se reciben a menos que se indique lo contrario. A menos que se indique lo contrario, todos los ensayos basados en microplacas se cuantificaron con un lector de microplacas BioTek Synergy 3 y los datos se ajustan y se representan con GraphPad Prism versión 5.00 para Windows (GraphPad Software, San Diego California USA, graphpad.com).
ELISA de inhibición de CD4
Se realizaron análisis ELISA de inhibición de CD4 como se describe a continuación. Los resultados de ELISA para CDM-H 4.19 y 4.20 aparecen en la figura 13 adjunta.
Este procedimiento se adaptó a partir del protocolo notificado previamente.1 Placas de 96 pocillos (Nunc; Immuno) se recubrieron durante una noche (12 h) a 4 °C con VIH-1 recombinante soluble gp120JRFL (Immune Technology; Yonkers, NY) a 1 pg/ml en Tampón C. Las placas se lavaron con DPBS (Gibco, 1 x 100 pl) y a continuación se bloquearon con Tampón A durante 1 h a temperatura ambiente. Después del lavado con Tampón B (3 x 100 pl), se añadieron simultáneamente concentraciones variables del inhibidor (incluido un control "sin molécula") con CD4 de linfocitos T humanos recombinantes (ImmunoDiagnostics, Inc; Woburn, MA) en el Tampón A por triplicado para que la concentración final/pocillo de CD4 fuera de 0,1 pg/ml y las placas se incubaron durante 1 h a temperatura ambiente. Las placas se lavaron con Tampón B (3 x 100 pl) y a continuación se incubaron con anticuerpo IgG anti CD4 OKT4 de ratón (Biolegend; San Diego, CA) a 0,36 pg/ml en Tampón A a TA durante 1 h. Después de los lavados con Tampón B, las placas se incubaron con anticuerpo anti ratón de cabra conjugado con peroxidasa de rábano picante (HRP) (1:2500; Biolegend; San Diego, CA). Después de los lavados con Tampón B (3 x 100 pl), se detectó el anticuerpo unido con 3,3,5,5-tetrametilbencidina (TMB, Pierce Protein Research Products), el sustrato cromogénico para HPR y la absorbancia se leyó a 450 nm después de detener reacción con H2SO42 N (100 pl). A continuación, se representó gráficamente la media (±D.E.) de estas muestras por triplicado frente a la concentración de inhibidor y se generó una curva de ajuste no lineal usando GraphPad Prism. La concentración inhibitoria del 50 % (CI50) se definió como la concentración de inhibidor para reducir la cantidad de CD4 unido a sgp120 en un 50 % del unión máxima. El ensayo inhibitorio se realizó por triplicado al menos dos veces para cada molécula.
(1) Parker, C. G.; Domaoal, R. A.; Anderson, K. S.; Spiegel, D. A., An antibody-recruiting small molecule that targets HIV gp120. JAm Chem Soc 2009, 131, 16392-4.
Cultivo celular
Las células CHO-env que expresan env de VIH-1 de tipo silvestre y CHO-pSv (control isogénico negativo para env) fueron un obsequio del Dr. Edward Berger (NIH), sin embargo, se desarrollaron por Nicholas y colaboradores.55 Las células CHO-pSv se cultivaron en el medio base DMEM descrito a continuación, mientras que las células CHO-env se cultivaron en un medio de selección, que consiste en un medio base que contiene metotrexato 0,25 pM (MTX, Aldrich).
*Nota: Se observó una disminución en la expresión de la envoltura en células CHO-env en varios pases y se recomienda mantener soluciones madre de pase bajas de células en nitrógeno líquido.
Medios base para el cultivo de células CHO
• Glucosa con alto contenido de DMEM, glutamato, piruvato (Gibco, cat. N.° 11995) • MEM al 10 %-NEAA (Gibco) • Tampón HEPES - concentración final 10 mM • FBS inactivado por calor al 10 %
Todas las células se cultivaron en un entorno húmedo a 37 °C, CO2 al 5 %. Las células se cultivaron en matraces de cultivo tisular T-75 y se separaron con EDTA 2,5 mM/EGTA 0,5 mM en DPBS (Gibco) para su pase. Las células se contaron diluyendo 10 pl de la mezcla celular en 90 pl de colorante azul de tripano y a continuación se contaron con un hemocitómetro. Toda la centrifugación se realizó a 1000 rpm.
Ensayo de citotoxicidad celular (CellTiter Glo)
***Nota: todas las incubaciones y diluciones en medios de cultivo de CHO (+MTX para CHO-env)
***T odas las incubaciones se realizaron en placas de 96 pocillos (fondo transparente, lados de color negro; Costar N.° 3603) por triplicado a 37 °C, CO2 al 5 % en un ambiente húmedo.
*** CD4-PE fue un generoso obsequio de Edward Berger (NIH).
Placas de siembra
Los matraces T-75 confluentes (~80 %) de CHO-env y/o CHO-pSv se lavaron una vez con DPBS (3 ml), se separaron, se contaron y a continuación se centrifugaron. Los sedimentos celulares se aspiraron y a continuación se resuspendieron en medio de crecimiento completo a una densidad celular de 1,5E5 células/ml, a continuación se dividieron en alícuotas en una placa de 96 pocillos (50 pl, 7,5E3 células/pocillo), se cubrieron y se incubaron durante 14 h.
Adición de compuestos de prueba
Se prepararon diluciones en serie (1/6) de moléculas en placas vacías de 96 pocillos usando una pipeta multicanal y a continuación se transfirieron a placas que contenían células sembradas (50 pl). También se preparó el control negativo (sin molécula), así como el control de destrucción máxima (Triton X al 2,5 %). Las placas se cubrieron y se incubaron durante 14 h o 24 h. Después de la incubación, se añadieron 100 ul del reactivo CellTiter Glo preparado (N.° de producto Promega G7571) como se describe en el protocolo de ensayo proporcionado. Luminiscencia monitorizada en el lector de microplacas Biotek Synergy 2.
% de citotoxicidad por encima del fondo calculado mediante las siguientes fórmulas:
% de CDC = [100 -((muestra - destrucción máx.)/(sin molécula - destrucción máx.)) * 100]
Las medias de muestra se representaron gráficamente usando el software GraphPad Prism ± desviación estándar (DE.).
Ensayo de citotoxicidad celular (xCELLigence)56
***Nota: todas las incubaciones y diluciones en medios de cultivo de CHO (+MTX para CHO-env)
***Todas las incubaciones se realizaron por duplicado en una placa E-plate 16 (Roche) a 37 °C, CO2 al 5 % en un ambiente húmedo.
*** CD4-PE fue un generoso obsequio de Edward Berger (NIH).
El sistema xCELLigence (modelo RTCA-DP Roche; software RTCA v1.2) es una herramienta diseñada para medir la densidad, viabilidad y morfología celular en tiempo real a través de mediciones de impedancia. Los pocillos de cultivo tisular están recubiertos con electrodos de oro, que forman un circuito cuando el pocillos está lleno de medio. Cuando una celda descansa sobre los electrodos, la impedancia aumenta y se refiere a un "índice celular", y los índices celulares aumentan a medida que las células se adhieren a una fracción mayor del pocillo (placa E-plate). Por consiguiente, un valor de índice celular más alto indica una mayor viabilidad, número de células o una morfología más dispersa. Cuando estas células mueren, se desprenden y, como resultado, los índices celulares disminuyen. A continuación, los cambios en los índices celulares se pueden convertir en cambios en la viabilidad celular:
% de destrucción específica = 100 -[(índice celular)/(índice de crecimiento normal)] x 100
Preparación de las células
Los matraces T-75 confluentes (~80 %) de CHO-env y/o CHO-pSv se lavaron una vez con DPBS (3 ml), se separaron, se contaron y a continuación se centrifugaron. Los sedimentos celulares se aspiraron y a continuación se resuspendieron en medio de crecimiento completo a una densidad celular de 1,25E5 células/ml.
Configuración de xCelligence
Para establecer una impedancia de fondo del medio de cultivo, se añadieron 200 pl del medio de crecimiento respectivo a todos los pocillos de la placa E-plate y se guardó la lectura de fondo. A continuación, las células se dividieron en alícuotas en la placa E-plate (100 pl, 12,5E3 células/pocillo), se cubrieron y se incubaron durante 22 horas para establecer una curva de crecimiento exponencial, escaneando una vez cada 15 minutos. Con el fin de examinar la citotoxicidad de los compuestos de prueba, se eliminaron cuidadosamente 100 pl de medio de las placas E-plate que contenían células adheridas y se reemplazaron por 100 pl de medio fresco que contenía vehículo (es decir, DMSO) o 100 pl de medio que contenía 20 pM de compuesto de prueba (o 2 pg/ml de CD4-PE). A continuación, los índices celulares se controlaron durante 120 h, escaneando una vez cada intervalo de 15 minutos. Se eliminaron 100 pl de solución de los pocillos que se sometieron a un "reciclado" o que recibieron múltiples adiciones y se repusieron con medio fresco que contenía compuesto o vehículo. Los datos de xCelligence sin procesar de CDM-H 4.19 y 4.20, doxorrubicina (4.18) y CD4-PE cuando se incubaron con células CHO-env (gp120+) y CHO-pSv (gp120-) aparecen en la figura 17 de la presente.
Microscopía de inmunofluorescencia
***Nota: todas las incubaciones y diluciones en medios de cultivo de CHO (+MTX para CHO-env)
***Todas las incubaciones se realizaron por duplicado a 37 °C, CO2 al 5 % en un ambiente húmedo. Los matraces T-75 confluentes (~80 %) de CHO-env y/o CHO-pSv se lavaron una vez con DPBS (3 ml), se separaron, se contaron y a continuación se centrifugaron. Los sedimentos celulares se aspiraron y a continuación se resuspendieron en medio de crecimiento completo a una densidad celular de 1,5E5 células/ml. A continuación, la suspensión celular se dividió en alícuotas (300 pl, 3E5 células/pocillo) en pocillos de la cámara portaobjetos de 8 pocillos (LabTek, N.° 12565470, 15411). A continuación, se permitió que las células se adhirieran y crecieran hasta la confluencia (~80 %), lo que tarda aproximadamente 40 horas. El medio se aspiró cuidadosamente y las células se lavaron suavemente con DPBS (300 pl) para eliminar las células muertas. Se añadió a las células medio que contenía la molécula de ensayo (CDM-H o doxorrubicina, 10 pM) y se incubaron durante 1 h. El medio se aspiró cuidadosamente y las células se lavaron suavemente con DPBS (300 pl). Se añadió medio que contenía colorante fluorescente LysoTracker Blue (Invitrogen, cat. N.° L7525), o nada, a las células y se incubó de acuerdo con el protocolo proporcionado. El medio se aspiró cuidadosamente y las células se lavaron suavemente con DPBS (300 pl), a continuación se resuspendieron en medio y se tomaron micrografías de fluorescencia con un microscopio de fluorescencia Zeiss Axiovert 200M equipado con conjuntos de filtros Cy3 y DAPI. Nota: para experimentos de curso temporal, el medio que contenía moléculas de prueba no se eliminó y se tomaron imágenes durante un período de tiempo. La figura 16 muestra micrografías de fluorescencia cuando se incuba CDM-H 4.20 o doxorrubicina (4.18) con células CHO-env después de 10 min (A-B) o 20 h (C-D) y células CHO-pSv (E-H). La formación de partículas fluorescentes, específicamente después de largos períodos de incubación, sugiere la formación de micelas.
Dispersión de luz dinámica (DLS)
La dispersión de luz dinámica es una herramienta analítica que se puede usar para determinar el perfil de distribución de tamaño de partículas pequeñas en solución. Cuando la luz golpea partículas en solución, se dispersa en todas las direcciones, lo que da como resultado una fluctuación dependiente del tiempo en la intensidad de dispersión. Esta fluctuación se debe al hecho de que las moléculas pequeñas en solución experimentan un movimiento browniano, por lo que la distancia entre las dispersiones en la solución cambia constantemente con el tiempo. Es a partir de esta fluctuación de intensidad de donde se puede obtener información sobre la escala temporal dinámica de movimiento y tamaño. La rapidez con la que la intensidad fluctúa con el tiempo se representa mediante una función de autocorrelación. En intervalos de tiempo cortos, la correlación es alta porque las partículas no se mueven significativamente desde el estado inicial en el que se encontraban. Por lo tanto, las dos señales no cambian esencialmente cuando se comparan después de un intervalo de tiempo muy corto. A medida que los intervalos de tiempo de medición se hacen más largos, la correlación decae exponencialmente, lo que significa que, después de que ha transcurrido un largo período de tiempo, no existe correlación entre la intensidad de dispersión de los estados inicial y final. Este decaimiento exponencial está relacionado con el movimiento de las partículas, específicamente con el coeficiente de difusión. Por lo tanto, las partículas que son más grandes tendrán movimientos más lentos, por lo que a tiempos más largos, habrá una mayor correlación entre la intensidad de dispersión de los estados inicial y final, en comparación con las partículas más pequeñas.57
La instalación Keck de la Escuela de Medicina de Yale posee un instrumento DLS en su Biophysical Resource
Laboratory. Como confirmación preliminar de que CDM-H 4.20 está formando agregados de manera dependiente de la concentración y en las concentraciones usadas en los ensayos de citotoxicidad descritos, se examinó su perfil de dispersión en colaboración con Ewa Folta-Stogniew en el centro Keck. Por lo tanto, siguiendo los protocolos proporcionados por el centro de recursos, se realizaron experimentos DLS de 4.20 en DPBS en diversas concentraciones. Como se muestra en la figura S4.7, hay un aumento significativo en la autocorrelación en función de la concentración. Además, a intervalos de tiempo más largos, hay autocorrelaciones significativamente más altas a concentraciones más altas de 4.20 que a concentraciones más bajas, lo que sugiere que, a concentraciones más altas, se está produciendo agregación. Estas observaciones son consistentes con la hipótesis de que 4.20 de hecho está formando agregados y, por lo tanto, se garantiza una mayor investigación y/u optimización de 4.20 (para circunnavegar la agregación). La figura 18 muestra el experimento preliminar de dispersión de luz dinámica (DLS) para detectar la formación de agregados.
Sumario
La presente invención satisface la necesidad estratégica de un nuevo tratamiento para la infección por VIH al proporcionar moléculas pequeñas bifuncionales, como se define en las reivindicaciones, generalmente denominadas ARM-HI que funcionan a través de rutas ortogonales, tanto por inhibición de la interacción gp120-CD4, como mediante el reclutamiento de anticuerpos anti DNP para las células que expresan gp120, en la prevención de la infección celular y la propagación del VIH. Se muestra que: Las ARM-HI de acuerdo con la presente invención exhiben una actividad sustancialmente mayor que los compuestos ARM-H publicados previamente.
El presente enfoque antiviral tiene claras ventajas sobre otras estrategias contra el VIH basadas en vacunas, proteínas y moléculas pequeñas.
Aunque se ha demostrado que la respuesta inmunitaria humana genera anticuerpos neutralizantes anti gp120 en torno a los cuales el virus no muta de manera eficaz, los enfoques basados en vacunas para inducir dichos anticuerpos en huéspedes humanos aún no han demostrado ser exitosos. En teoría, aunque el virus del VIH muta con extrema rapidez en los huéspedes humanos, dado que debe conservar la actividad de unión a CD4 para seguir siendo infeccioso, las moléculas pequeñas reclutadoras de anticuerpos que imitan el motivo de reconocimiento de CD4, tales como las ARM-HI de la invención, tienen la esperanza de cumplir el mismo papel funcional que neutralizar los anticuerpos anti gp120. Además, como moléculas pequeñas, estos materiales probablemente posean ventajas sustanciales sobre los agentes terapéuticos basados en proteínas, incluida una baja propensión a la inmunogenicidad, alta estabilidad metabólica, producción a gran escala y un coste relativamente bajo.
La evidencia sugiere que una respuesta inmunitaria celular es necesaria para la inactivación viral in vivo, y se ha demostrado que las moléculas pequeñas bifuncionales de la invención se dirigen directamente a partículas que expresan gp120 a macrófagos y neutrófilos.
Este enfoque de la terapia antiviral también es ideal como profiláctico, ya que no se espera que el compuesto bifuncional tenga efectos secundarios adversos significativos, ya que solo está activo cuando el virus está presente.
La divulgación completa de todas las patentes, solicitudes de patente y publicaciones y el material disponible electrónicamente (incluyendo, por ejemplo, presentaciones de secuencias de nucleótidos en, por ejemplo, GenBank y RefSeq, y presentaciones de aminoácidos en, por ejemplo, SwissProt, PIR, PRF, PDB y traducciones de regiones codificantes anotadas en GenBank y RefSeq) citadas. La descripción detallada y los ejemplos anteriores se han proporcionado solo para claridad de comprensión. No deben entenderse limitaciones innecesarias de los mismos. La invención se define por las siguientes reivindicaciones.
Referencias
1. Volberding, P. A.; Deeks, S. G., Antiretroviral therapy and management of HIV infection. Lancet 2010, 376, 49 62.
2. Le Douce, V.; Janossy, A.; Hallay, H.; Ali, S.; Riclet, R.; Rohr, O.; Schwartz, C., Achieving a cure for HIV infection: do we have reasons to be optimistic? J Antimicrob Chemother 2012, 67, 1063-74.
3. Finzi, D.; Hermankova, M.; Pierson, T.; Carruth, L. M.; Buck, C.; Chaisson, R. E.; Quinn, T. C.; Chadwick, K.; Margolick, J.; Brookmeyer, R.; Gallant, J.; Markowitz, M.; Ho, D. D.; Richman, D. D.; Siliciano, R. F., Identification of a reservoir for HIV-1 in patients on highly active antiretroviral therapy. Science 1997, 278, 1295-300.
4. Wong, J. K.; Hezareh, M.; Gunthard, H. F.; Havlir, D. V.; Ignacio, C. C.; Spina, C. A.; Richman, D. D., Recovery of replication-competent HIV despite prolonged suppression of plasma viremia. Science 1997, 278, 1291-5. 5. Carter, C. C.; Onafuwa-Nuga, A.; McNamara, L. A.; Riddell, J. t.; Bixby, D.; Savona, M. R.; Collins, K. L., HIV-1 infects multipotent progenitor cells causing cell death and establishing latent cellular reservoirs. Nat Med 2010, 16, 446-51.
6. Walker, B. D.; Burton, D. R., Toward an AIDS vaccine. Science 2008, 320, 760-4.
7. Berger, E. A.; Pastan, I., Immunotoxin complementation of HAART to deplete persisting HIV-infected cell reservoirs. PLoS Pathog 2010, 6, e1000803.
8. Chaudhary, V. K.; Mizukami, T.; Fuerst, T. R.; FitzGerald, D. J.; Moss, B.; Pastan, I.; Berger, E. A., Selective
killing of HlV-infected cells by recombinant human CD4-Pseudomonas exotoxin hybrid protein. Nature 1988, 335, 369-72.
9. Berger, E. A.; Chaudhary, V. K.; Clouse, K. A.; Jaraquemada, D.; Nicholas, J. A.; Rubino, K. L.; Fitzgerald, D. J.; Pastan, I.; Moss, B., Recombinant CD4-Pseudomonas exotoxin hybrid protein displays HIV-specific cytotoxicity without affecting MHC class II-dependent functions. AIDS Res Hum Retroviruses 1990, 6, 795-804.
10. Ashorn, P.; Moss, B.; Berger, E. A., Activity of CD4-Pseudomonas exotoxin against cells expressing diverse forms of the HIV and SIV envelope glycoproteins. J Acquir Immune Defic Syndr 1992, 5, 70-7.
11. Bera, T. K.; Kennedy, P. E.; Berger, E. A.; Barbas, C. F., 3rd; Pastan, I., Specific killing of HIV-infected lymphocytes by a recombinant immunotoxin directed against the HIV-1 envelope glycoprotein. Mol Med 1998, 4, 384-91.
12. Kennedy, P. E.; Moss, B.; Berger, E. A., Primary HIV-1 isolates refractory to neutralization by soluble CD4 are potently inhibited by CD4-Pseudomonas exotoxin. Virology 1993, 192, 375-9.
13. Berger, E. A.; Moss, B.; Pastan, I., Reconsidering targeted toxins to eliminate HIV infection: you gotta have HAART. Proceedings of the National Academy of Sciences of the United States of America 1998, 95, 11511-3. 14. Berger, E. A.; Pastan, I., Immunotoxin complementation of HAART to deplete persisting HIV-infected cell reservoirs. PLoS Pathog 2010, 6, e1000803.
15. Davey, R. T.; Boenning, C. M.; Herpin, B. R.; Batts, D. H.; Metcalf, J. A.; Wathen, L.; Cox, S. R.; Polis, M. A.; Kovacs, J. A.; Falloon, J., Use of recombinant soluble CD4 Pseudomonas exotoxin, a novel immunotoxin, for treatment of persons infected with human immunodeficiency virus. Journal of Infectious Diseases 1994, 170, 1180 8.
16. Lueders, K. K.; De Rosa, S. C.; Valentin, A.; Pavlakis, G. N.; Roederer, M.; Hamer, D. H., A potent anti-HIV immunotoxin blocks spreading infection by primary HIV type 1 isolates in multiple cell types. AIDS Res Hum Retroviruses 2004, 20, 145-50.
17. Goldstein, H.; Pettoello-Mantovani, M.; Bera, T. K.; Pastan, I. H.; Berger, E. A., Chimeric toxins targeted to the human immunodeficiency virus type 1 envelope glycoprotein augment the in vivo activity of combination antiretroviral therapy in thy/liv-SCID-Hu mice. J Infect Dis 2000, 181, 921-6.
18. Johansson, S.; Goldenberg, D. M.; Griffiths, G. L.; Wahren, B.; Hinkula, J., Elimination of HIV-1 infection by treatment with a doxorubicin-conjugated anti-envelope antibody. Aids 2006, 20, 1911-5.
19. Davey, R. T., Jr.; Boenning, C. M.; Herpin, B. R.; Batts, D. H.; Metcalf, J. A.; Wathen, L.; Cox, S. R.; Polis, M. A.; Kovacs, J. A.; Falloon, J.; et al., Use of recombinant soluble CD4 Pseudomonas exotoxin, a novel immunotoxin, for treatment of persons infected with human immunodeficiency virus. JInfect Dis 1994, 170, 1180-8.
20. Allen, T. M., Ligand-targeted therapeutics in anticancer therapy. Nature Reviews Cancer 2002, 2, 750-763. 21. Weber, C. A.; Mehta, P. J.; Ardito, M.; Moise, L.; Martin, B.; De Groot, A. S., T cell epitope: friend or foe? Immunogenicity of biologics in context. In Advanced Drug Delivery Reviews, 2009; Vol. 61, pp 965-76.
22. Hansel, T. T.; Kropshofer, H.; Singer, T.; Mitchell, J. A.; George, A. J. T., The safety and side effects of monoclonal antibodies. Nature Reviews Drug Discovery 2010, 9, 325-38.
23. Egan, M. A.; Carruth, L. M.; Rowell, J. F.; Yu, X.; Siliciano, R. F., Human immunodeficiency virus type 1 envelope protein endocytosis mediated by a highly conserved intrinsic internalization signal in the cytoplasmic domain of gp41 is suppressed in the presence of the Pr55gag precursor protein. J Virol 1996, 70, 6547-56.
24. Cervantes-Acosta, G.; Lodge, R.; Lemay, G.; Cohen, E. A., Influence of human immunodeficiency virus type 1 envelope glycoprotein YXXL endocytosis/polarization signal on viral accessory protein functions. J Hum Virol 2001, 4, 249-59.
25. Fultz, P. N.; Vance, P. J.; Endres, M. J.; Tao, B.; Dvorin, J. D.; Davis, I. C.; Lifson, J. D.; Montefiori, D. C.; Marsh, M.; Malim, M. H.; Hoxie, J. A., In vivo attenuation of simian immunodeficiency virus by disruption of a tyrosinedependent sorting signal in the envelope glycoprotein cytoplasmic tail. J Virol 2001, 75, 278-91.
26. Doherty, G. J.; McMahon, H. T., Mechanisms of endocytosis. Annu Rev Biochem 2009, 78, 857-902.
27. Rajendran, L.; Knolker, H. J.; Simons, K., Subcellular targeting strategies for drug design and delivery. Nat Rev Drug Discov 2010, 9, 29-42.
28. Kovtun, Y. V.; Goldmacher, V. S., Cell killing by antibody-drug conjugates. Cancer Lett 2007, 255, 232-40. 29. Luzio, J. P.; Pryor, P. R.; Bright, N. A., Lysosomes: fusion and function. Nat Rev Mol Cell Biol 2007, 8, 622-32.
30. King, H. D.; Yurgaitis, D.; Willner, D.; Firestone, R. A.; Yang, M. B.; Lasch, S. J.; Hellstrom, K. E.; Trail, P. A., Monoclonal antibody conjugates of doxorubicin prepared with branched linkers: A novel method for increasing the potency of doxorubicin immunoconjugates. Bioconjug Chem 1999, 10, 279-88.
31. Che, C.; Yang, G.; Thiot, C.; Lacoste, M. C.; Currie, J. C.; Demeule, M.; Regina, A.; Beliveau, R.; Castaigne, J. P., New Angiopep-modified doxorubicin (ANG1007) and etoposide (ANG1009) chemotherapeutics with increased brain penetration. J Med Chem 2010, 53, 2814-24.
32. Kasiotis, K. M.; Magiatis, P.; Pratsinis, H.; Skaltsounis, A.; Abadji, V.; Charalambous, A.; Moutsatsou, P.; Haroutounian, S. A., Synthesis and biological evaluation of novel daunorubicin-estrogen conjugates. Steroids 2001, 66, 785-91.
33. Meyer-Losic, F.; Quinonero, J.; Dubois, V.; Alluis, B.; Dechambre, M.; Michel, M.; Cailler, F.; Fernandez, A. M.; Trouet, A.; Kearsey, J., Improved therapeutic efficacy of doxorubicin through conjugation with a novel peptide drug delivery technology (Vectocell). J Med Chem 2006, 49, 6908-16.
34. Kratz, F.; Warnecke, A.; Scheuermann, K.; Stockmar, C.; Schwab, J.; Lazar, P.; Druckes, P.; Esser, N.; Drevs, J.; Rognan, D.; Bissantz, C.; Hinderling, C.; Folkers, G.; Fichtner, I.; Unger, C., Probing the cysteine-34 position of endogenous serum albumin with thiol-binding doxorubicin derivatives. Improved efficacy of an acid-sensitive doxorubicin derivative with specific albumin-binding properties compared to that of the parent compound. J Med
Claims (8)
1. Un compuesto de acuerdo con la estructura química:

o la amina libre o sal farmacéutica alternativa de la misma.
2. Un compuesto de acuerdo con la reivindicación 1 que tiene la estructura química:

3. Un compuesto de acuerdo con la reivindicación 1 que tiene la estructura química:

4. Una composición farmacéutica que comprende una cantidad eficaz de un compuesto de acuerdo con cualquiera de las reivindicaciones 1-3 en combinación con un portador, aditivo o excipiente farmacéuticamente aceptable.
5. La composición de acuerdo con la reivindicación 4 que incluye además un activador del VIH latente.
6. La composición de acuerdo con cualquiera de las reivindicaciones 4 o 5, en la que dicha composición comprende además una cantidad eficaz de un agente contra el VIH adicional.
7. Composición de acuerdo con cualquiera de las reivindicaciones 4-6 para su uso en el tratamiento de una infección por VIH en un paciente.
8. Composición de acuerdo con cualquiera de las reivindicaciones 4-6 para su uso en la reducción de la probabilidad de que un paciente con riesgo de infección por VIH contraiga una infección por VIH o que un paciente infectado por VIH contraiga el SIDA o ARC.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201261638569P | 2012-04-26 | 2012-04-26 | |
| PCT/US2013/032044 WO2013162757A1 (en) | 2012-04-26 | 2013-03-15 | Cytotoxic-drug delivering molecules targeting hiv (cdm-hs), cytotoxic activity against the human immunodeficiency virus and methods of use |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2905359T3 true ES2905359T3 (es) | 2022-04-08 |
Family
ID=49483736
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES13780662T Active ES2905359T3 (es) | 2012-04-26 | 2013-03-15 | Moléculas de suministro de fármacos citotóxicos dirigidas al VIH (CDM-H), actividad citotóxica contra el virus de la inmunodeficiencia humana y métodos de uso |
Country Status (4)
| Country | Link |
|---|---|
| US (1) | US9745334B2 (es) |
| EP (2) | EP4005600A1 (es) |
| ES (1) | ES2905359T3 (es) |
| WO (1) | WO2013162757A1 (es) |
Families Citing this family (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2004060403A2 (en) | 2003-01-06 | 2004-07-22 | Angiochem Inc. | Aprotinin and anglos as carriers across the blood-brain barrier |
| US9365634B2 (en) | 2007-05-29 | 2016-06-14 | Angiochem Inc. | Aprotinin-like polypeptides for delivering agents conjugated thereto to tissues |
| CA2740316A1 (en) | 2008-10-15 | 2010-04-22 | Angiochem Inc. | Conjugates of glp-1 agonists and uses thereof |
| WO2010043049A1 (en) | 2008-10-15 | 2010-04-22 | Angiochem Inc. | Etoposide and doxorubicin conjugates for drug delivery |
| CA2745524C (en) | 2008-12-05 | 2020-06-09 | Angiochem Inc. | Conjugates of neurotensin or neurotensin analogs and uses thereof |
| US8853353B2 (en) | 2008-12-17 | 2014-10-07 | Angiochem, Inc. | Membrane type-1 matrix metalloprotein inhibitors and uses thereof |
| MX2011011023A (es) | 2009-04-20 | 2012-01-20 | Angiochem Inc | Tratamiento de cancer de ovarios usando un agente anti-cancer conjugado con un analogo de angiopep-2. |
| RU2012103240A (ru) | 2009-07-02 | 2013-08-10 | Ангиокем Инк. | Мультимерные пептидные конъюгаты и их применение |
| WO2013070688A1 (en) | 2011-11-11 | 2013-05-16 | Yale University | Reprogramming urokinase into an antibody-recruiting anticancer agent |
| WO2013166110A1 (en) | 2012-05-02 | 2013-11-07 | Yale University | Tlr-agonist-conjugated antibody recruiting molecules (tlr_arms) |
| EP3307326B9 (en) | 2015-06-15 | 2021-02-24 | Angiochem Inc. | Methods for the treatment of leptomeningeal carcinomatosis |
| WO2017023994A1 (en) | 2015-08-06 | 2017-02-09 | Yale University | Small molecule based antibody-recruiting compounds for cancer treatment |
| US10780084B2 (en) | 2016-08-03 | 2020-09-22 | Yale University | Antibody-recruiting molecules for the treatment of cancer |
| CA3038532A1 (en) | 2016-10-05 | 2018-04-12 | The Board Of Trustees Of The Leland Stanford Junior University | Bryostatin compounds and methods of preparing the same |
| CA3069986A1 (en) * | 2017-07-26 | 2019-01-31 | Kleo Pharmaceuticals, Inc. | Universal abt compounds and uses thereof |
| CN111995622B (zh) * | 2020-08-20 | 2022-04-15 | 复旦大学 | 化合物及其用途 |
Family Cites Families (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4522811A (en) | 1982-07-08 | 1985-06-11 | Syntex (U.S.A.) Inc. | Serial injection of muramyldipeptides and liposomes enhances the anti-infective activity of muramyldipeptides |
| US7115262B1 (en) | 1999-03-16 | 2006-10-03 | The United States Of America As Represented By The Department Of Health And Human Services | Chimeric protein for prevention and treatment of HIV infection |
| JP2004537515A (ja) | 2001-05-15 | 2004-12-16 | フォーク ファーマシューティカルス,インク. | ウィルス感染症治療用薬物のターゲッティング送達 |
| HRP20151367T1 (hr) | 2008-05-13 | 2016-01-29 | Yale University | Kimerne male molekule za pojaäśanje odgovora protutijela na stanice raka |
| US8852630B2 (en) | 2008-05-13 | 2014-10-07 | Yale University | Chimeric small molecules for the recruitment of antibodies to cancer cells |
| EA201100741A1 (ru) | 2008-11-10 | 2012-01-30 | Новартис Аг | Антитела к модифицированным пептидам ифр-1/е человека |
| US9181224B2 (en) | 2009-10-13 | 2015-11-10 | Yale University | Bifunctional molecules with antibody-recruiting and entry inhibitory activity against the human immunodeficiency virus |
| SG10201510402WA (en) | 2010-11-18 | 2016-01-28 | Univ Yale | Bifunctional molecules with antibody-recruiting and entry inhibitory activity against the human immunodeficiency virus |
-
2013
- 2013-03-15 EP EP21205664.2A patent/EP4005600A1/en not_active Withdrawn
- 2013-03-15 EP EP13780662.6A patent/EP2841107B1/en active Active
- 2013-03-15 ES ES13780662T patent/ES2905359T3/es active Active
- 2013-03-15 US US14/396,956 patent/US9745334B2/en active Active
- 2013-03-15 WO PCT/US2013/032044 patent/WO2013162757A1/en not_active Ceased
Also Published As
| Publication number | Publication date |
|---|---|
| US20150087609A1 (en) | 2015-03-26 |
| EP2841107B1 (en) | 2021-11-03 |
| EP2841107A4 (en) | 2016-04-27 |
| EP2841107A1 (en) | 2015-03-04 |
| EP4005600A1 (en) | 2022-06-01 |
| WO2013162757A1 (en) | 2013-10-31 |
| US9745334B2 (en) | 2017-08-29 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2905359T3 (es) | Moléculas de suministro de fármacos citotóxicos dirigidas al VIH (CDM-H), actividad citotóxica contra el virus de la inmunodeficiencia humana y métodos de uso | |
| US11858917B2 (en) | Bifunctional molecules with antibody-recruiting and entry inhibitory activity against the human immunodeficiency virus | |
| JP6934846B2 (ja) | 多環式カルバモイルピリドン化合物およびそれらの薬学的使用 | |
| US10188727B2 (en) | Bifunctional molecules with antibody-recruiting and entry inhibitory activity against the human immunodeficiency virus | |
| AU2008314668B2 (en) | Cholesterol derivatives of inhibitors of viral fusion | |
| US20110144069A1 (en) | Compounds for treating viral infections | |
| US20100166806A1 (en) | Combination therapy comprising the use of protein kinase C modulators and Histone Deacetylase inhibitors for treating HIV-1 latency | |
| HK1189219B (en) | Bifunctional molecules with antibody-recruiting and entry inhibitory activity against the human immunodeficiency virus | |
| HK1189219A (en) | Bifunctional molecules with antibody-recruiting and entry inhibitory activity against the human immunodeficiency virus | |
| HK1240932B (zh) | 具有募集抗体并对人体免疫缺陷病毒进入有抑制活性的双功能分子 | |
| HK1240932A1 (en) | Bifunctional molecules with antibody-recruiting and entry inhibitory activity against the human immunodeficiency virus |