ES2907981T3 - Derivado de benzazepina, método de preparación, composición farmacéutica y uso del mismo - Google Patents
Derivado de benzazepina, método de preparación, composición farmacéutica y uso del mismo Download PDFInfo
- Publication number
- ES2907981T3 ES2907981T3 ES17792495T ES17792495T ES2907981T3 ES 2907981 T3 ES2907981 T3 ES 2907981T3 ES 17792495 T ES17792495 T ES 17792495T ES 17792495 T ES17792495 T ES 17792495T ES 2907981 T3 ES2907981 T3 ES 2907981T3
- Authority
- ES
- Spain
- Prior art keywords
- alkyl
- compound
- nrdre
- ord
- alkynyl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 239000008194 pharmaceutical composition Substances 0.000 title claims description 17
- 238000002360 preparation method Methods 0.000 title description 9
- 150000008038 benzoazepines Chemical class 0.000 title description 5
- 150000001875 compounds Chemical class 0.000 claims abstract description 366
- 125000000217 alkyl group Chemical group 0.000 claims abstract description 99
- -1 amino, carboxyl Chemical group 0.000 claims abstract description 66
- 125000000592 heterocycloalkyl group Chemical group 0.000 claims abstract description 66
- 150000003839 salts Chemical class 0.000 claims abstract description 58
- 125000003601 C2-C6 alkynyl group Chemical group 0.000 claims abstract description 44
- 125000001424 substituent group Chemical group 0.000 claims abstract description 41
- 125000000882 C2-C6 alkenyl group Chemical group 0.000 claims abstract description 40
- 229910052739 hydrogen Inorganic materials 0.000 claims abstract description 37
- 229910052736 halogen Inorganic materials 0.000 claims abstract description 36
- 150000002367 halogens Chemical class 0.000 claims abstract description 36
- 239000001257 hydrogen Substances 0.000 claims abstract description 34
- 125000004093 cyano group Chemical group *C#N 0.000 claims abstract description 32
- 125000006552 (C3-C8) cycloalkyl group Chemical group 0.000 claims abstract description 29
- 125000003118 aryl group Chemical group 0.000 claims abstract description 25
- 125000003342 alkenyl group Chemical group 0.000 claims abstract description 23
- 125000000171 (C1-C6) haloalkyl group Chemical group 0.000 claims abstract description 19
- 125000000304 alkynyl group Chemical group 0.000 claims abstract description 19
- 125000006570 (C5-C6) heteroaryl group Chemical group 0.000 claims abstract description 18
- 229910052757 nitrogen Inorganic materials 0.000 claims abstract description 18
- 125000004432 carbon atom Chemical group C* 0.000 claims abstract description 17
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims abstract description 17
- 125000001072 heteroaryl group Chemical group 0.000 claims abstract description 16
- 125000000753 cycloalkyl group Chemical group 0.000 claims abstract description 15
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims abstract description 14
- 125000004438 haloalkoxy group Chemical group 0.000 claims abstract description 13
- 125000001188 haloalkyl group Chemical group 0.000 claims abstract description 13
- 125000004433 nitrogen atom Chemical group N* 0.000 claims abstract description 13
- 229910052760 oxygen Inorganic materials 0.000 claims abstract description 13
- 229910052717 sulfur Inorganic materials 0.000 claims abstract description 13
- 125000004737 (C1-C6) haloalkoxy group Chemical group 0.000 claims abstract description 12
- 101100162205 Aspergillus parasiticus (strain ATCC 56775 / NRRL 5862 / SRRC 143 / SU-1) aflI gene Proteins 0.000 claims abstract description 11
- 229910052805 deuterium Inorganic materials 0.000 claims abstract description 11
- 125000005842 heteroatom Chemical group 0.000 claims abstract description 11
- YZCKVEUIGOORGS-OUBTZVSYSA-N Deuterium Chemical group [2H] YZCKVEUIGOORGS-OUBTZVSYSA-N 0.000 claims abstract description 9
- 125000004103 aminoalkyl group Chemical group 0.000 claims abstract description 9
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 claims abstract description 8
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 claims abstract description 8
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 claims abstract description 7
- 125000002768 hydroxyalkyl group Chemical group 0.000 claims abstract description 6
- 125000004890 (C1-C6) alkylamino group Chemical group 0.000 claims abstract description 4
- 125000003282 alkyl amino group Chemical group 0.000 claims abstract description 4
- 125000006590 (C2-C6) alkenylene group Chemical group 0.000 claims abstract description 3
- 125000006591 (C2-C6) alkynylene group Chemical group 0.000 claims abstract description 3
- 125000006577 C1-C6 hydroxyalkyl group Chemical group 0.000 claims abstract description 3
- 150000002431 hydrogen Chemical class 0.000 claims abstract 19
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 claims abstract 11
- 230000000155 isotopic effect Effects 0.000 claims description 47
- 102000002689 Toll-like receptor Human genes 0.000 claims description 31
- 108020000411 Toll-like receptor Proteins 0.000 claims description 31
- 101000800483 Homo sapiens Toll-like receptor 8 Proteins 0.000 claims description 24
- 206010028980 Neoplasm Diseases 0.000 claims description 24
- 102100033110 Toll-like receptor 8 Human genes 0.000 claims description 24
- 238000000034 method Methods 0.000 claims description 21
- 125000003545 alkoxy group Chemical group 0.000 claims description 19
- 201000010099 disease Diseases 0.000 claims description 18
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims description 18
- 101000669402 Homo sapiens Toll-like receptor 7 Proteins 0.000 claims description 14
- 102100039390 Toll-like receptor 7 Human genes 0.000 claims description 14
- 229910052799 carbon Inorganic materials 0.000 claims description 14
- 239000000460 chlorine Substances 0.000 claims description 13
- 230000001404 mediated effect Effects 0.000 claims description 11
- 229910052794 bromium Inorganic materials 0.000 claims description 10
- 229910052801 chlorine Inorganic materials 0.000 claims description 10
- 239000003814 drug Substances 0.000 claims description 9
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 9
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 claims description 8
- 229910052731 fluorine Inorganic materials 0.000 claims description 8
- 208000023275 Autoimmune disease Diseases 0.000 claims description 7
- 239000000546 pharmaceutical excipient Substances 0.000 claims description 7
- 229940124597 therapeutic agent Drugs 0.000 claims description 7
- 125000006583 (C1-C3) haloalkyl group Chemical group 0.000 claims description 6
- 208000036142 Viral infection Diseases 0.000 claims description 6
- 230000002265 prevention Effects 0.000 claims description 6
- 238000002560 therapeutic procedure Methods 0.000 claims description 6
- 230000009385 viral infection Effects 0.000 claims description 6
- 238000006069 Suzuki reaction reaction Methods 0.000 claims description 5
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 claims description 5
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 claims description 5
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 claims description 5
- 125000006559 (C1-C3) alkylamino group Chemical group 0.000 claims description 4
- 206010061218 Inflammation Diseases 0.000 claims description 4
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 claims description 4
- 230000004054 inflammatory process Effects 0.000 claims description 4
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 claims description 4
- 125000004076 pyridyl group Chemical group 0.000 claims description 4
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 claims description 4
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical group [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 claims description 3
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 3
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 claims description 3
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 claims description 3
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 claims description 3
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 claims description 3
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 claims description 3
- 125000000714 pyrimidinyl group Chemical group 0.000 claims description 3
- 229910052702 rhenium Inorganic materials 0.000 claims description 3
- 101150100019 NRDC gene Proteins 0.000 claims description 2
- 125000001246 bromo group Chemical group Br* 0.000 claims description 2
- PNDPGZBMCMUPRI-UHFFFAOYSA-N iodine Chemical group II PNDPGZBMCMUPRI-UHFFFAOYSA-N 0.000 claims description 2
- 125000001971 neopentyl group Chemical group [H]C([*])([H])C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H] 0.000 claims description 2
- 229910052720 vanadium Inorganic materials 0.000 claims description 2
- 125000006376 (C3-C10) cycloalkyl group Chemical group 0.000 claims 1
- 125000006584 (C3-C10) heterocycloalkyl group Chemical group 0.000 claims 1
- 125000001313 C5-C10 heteroaryl group Chemical group 0.000 claims 1
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 claims 1
- 125000000876 trifluoromethoxy group Chemical group FC(F)(F)O* 0.000 claims 1
- 125000004191 (C1-C6) alkoxy group Chemical group 0.000 abstract 6
- 125000000041 C6-C10 aryl group Chemical group 0.000 abstract 3
- 125000006273 (C1-C3) alkyl group Chemical group 0.000 abstract 1
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 141
- 239000000243 solution Substances 0.000 description 108
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 105
- 238000003786 synthesis reaction Methods 0.000 description 104
- 230000015572 biosynthetic process Effects 0.000 description 103
- 238000006243 chemical reaction Methods 0.000 description 70
- AICOOMRHRUFYCM-ZRRPKQBOSA-N oxazine, 1 Chemical compound C([C@@H]1[C@H](C(C[C@]2(C)[C@@H]([C@H](C)N(C)C)[C@H](O)C[C@]21C)=O)CC1=CC2)C[C@H]1[C@@]1(C)[C@H]2N=C(C(C)C)OC1 AICOOMRHRUFYCM-ZRRPKQBOSA-N 0.000 description 55
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 52
- 239000007787 solid Substances 0.000 description 49
- 239000012074 organic phase Substances 0.000 description 44
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 40
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 40
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 39
- 239000000203 mixture Substances 0.000 description 38
- 239000002904 solvent Substances 0.000 description 37
- 238000010189 synthetic method Methods 0.000 description 37
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 35
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 33
- 210000004027 cell Anatomy 0.000 description 32
- ZMXDDKWLCZADIW-UHFFFAOYSA-N dimethylformamide Substances CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 24
- 239000003208 petroleum Substances 0.000 description 24
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 23
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 22
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 20
- ZDYXSEQHOVSTPA-UHFFFAOYSA-N 6-bromo-2h-isoquinolin-1-one Chemical compound C1=CNC(=O)C=2C1=CC(Br)=CC=2 ZDYXSEQHOVSTPA-UHFFFAOYSA-N 0.000 description 17
- 125000004435 hydrogen atom Chemical class [H]* 0.000 description 17
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 16
- 238000010898 silica gel chromatography Methods 0.000 description 14
- 239000012065 filter cake Substances 0.000 description 13
- 239000005457 ice water Substances 0.000 description 13
- 238000005160 1H NMR spectroscopy Methods 0.000 description 12
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 12
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 12
- 239000007864 aqueous solution Substances 0.000 description 11
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 10
- 230000004913 activation Effects 0.000 description 10
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 9
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical class [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 9
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 9
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 9
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 9
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonium chloride Substances [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 8
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 8
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 8
- 239000000706 filtrate Substances 0.000 description 8
- 239000003921 oil Substances 0.000 description 8
- 235000019198 oils Nutrition 0.000 description 8
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 8
- 238000010992 reflux Methods 0.000 description 8
- 229920006395 saturated elastomer Polymers 0.000 description 8
- 229960003010 sodium sulfate Drugs 0.000 description 8
- 229910052938 sodium sulfate Inorganic materials 0.000 description 8
- 235000011152 sodium sulphate Nutrition 0.000 description 8
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 8
- 229920002472 Starch Polymers 0.000 description 7
- 229940124614 TLR 8 agonist Drugs 0.000 description 7
- 108010060818 Toll-Like Receptor 9 Proteins 0.000 description 7
- 102100033117 Toll-like receptor 9 Human genes 0.000 description 7
- XHXFXVLFKHQFAL-UHFFFAOYSA-N phosphoryl trichloride Chemical compound ClP(Cl)(Cl)=O XHXFXVLFKHQFAL-UHFFFAOYSA-N 0.000 description 7
- 239000000843 powder Substances 0.000 description 7
- 239000008107 starch Substances 0.000 description 7
- 235000019698 starch Nutrition 0.000 description 7
- 239000000725 suspension Substances 0.000 description 7
- 239000003826 tablet Substances 0.000 description 7
- UJORPFQWUKFXIE-UHFFFAOYSA-N 2-[1-[(4-ethoxy-2,6-difluorophenyl)methyl]-5,6-dihydro-4h-cyclopenta[c]pyrazol-3-yl]-5-methoxy-n-pyridin-4-ylpyrimidin-4-amine Chemical compound FC1=CC(OCC)=CC(F)=C1CN1C(CCC2)=C2C(C=2N=C(NC=3C=CN=CC=3)C(OC)=CN=2)=N1 UJORPFQWUKFXIE-UHFFFAOYSA-N 0.000 description 6
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 6
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 6
- MHAJPDPJQMAIIY-UHFFFAOYSA-N Hydrogen peroxide Chemical compound OO MHAJPDPJQMAIIY-UHFFFAOYSA-N 0.000 description 6
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 6
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 6
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 6
- 239000002253 acid Substances 0.000 description 6
- 125000004429 atom Chemical group 0.000 description 6
- 201000011510 cancer Diseases 0.000 description 6
- 239000011203 carbon fibre reinforced carbon Substances 0.000 description 6
- 230000000694 effects Effects 0.000 description 6
- 239000001963 growth medium Substances 0.000 description 6
- 208000015181 infectious disease Diseases 0.000 description 6
- 125000006578 monocyclic heterocycloalkyl group Chemical group 0.000 description 6
- 238000002953 preparative HPLC Methods 0.000 description 6
- 239000011541 reaction mixture Substances 0.000 description 6
- 229910052708 sodium Inorganic materials 0.000 description 6
- 239000011734 sodium Substances 0.000 description 6
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 5
- ABRVLXLNVJHDRQ-UHFFFAOYSA-N [2-pyridin-3-yl-6-(trifluoromethyl)pyridin-4-yl]methanamine Chemical compound FC(C1=CC(=CC(=N1)C=1C=NC=CC=1)CN)(F)F ABRVLXLNVJHDRQ-UHFFFAOYSA-N 0.000 description 5
- 239000000556 agonist Substances 0.000 description 5
- 235000011114 ammonium hydroxide Nutrition 0.000 description 5
- 230000010056 antibody-dependent cellular cytotoxicity Effects 0.000 description 5
- 150000001721 carbon Chemical group 0.000 description 5
- HEDRZPFGACZZDS-UHFFFAOYSA-N chloroform Substances ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 5
- 238000010511 deprotection reaction Methods 0.000 description 5
- 238000001914 filtration Methods 0.000 description 5
- 238000003818 flash chromatography Methods 0.000 description 5
- 210000002865 immune cell Anatomy 0.000 description 5
- 239000007924 injection Substances 0.000 description 5
- 238000002347 injection Methods 0.000 description 5
- 239000000126 substance Substances 0.000 description 5
- 238000004809 thin layer chromatography Methods 0.000 description 5
- KZPYGQFFRCFCPP-UHFFFAOYSA-N 1,1'-bis(diphenylphosphino)ferrocene Chemical compound [Fe+2].C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1 KZPYGQFFRCFCPP-UHFFFAOYSA-N 0.000 description 4
- VTYYLEPIZMXCLO-UHFFFAOYSA-L Calcium carbonate Chemical compound [Ca+2].[O-]C([O-])=O VTYYLEPIZMXCLO-UHFFFAOYSA-L 0.000 description 4
- 102000004127 Cytokines Human genes 0.000 description 4
- 108090000695 Cytokines Proteins 0.000 description 4
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 4
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 4
- 241000711549 Hepacivirus C Species 0.000 description 4
- 206010062016 Immunosuppression Diseases 0.000 description 4
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 4
- BAVYZALUXZFZLV-UHFFFAOYSA-N Methylamine Chemical compound NC BAVYZALUXZFZLV-UHFFFAOYSA-N 0.000 description 4
- PCLIMKBDDGJMGD-UHFFFAOYSA-N N-bromosuccinimide Chemical compound BrN1C(=O)CCC1=O PCLIMKBDDGJMGD-UHFFFAOYSA-N 0.000 description 4
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Chemical compound NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 4
- 241000711975 Vesicular stomatitis virus Species 0.000 description 4
- 241000700605 Viruses Species 0.000 description 4
- 238000003556 assay Methods 0.000 description 4
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 description 4
- 229910000024 caesium carbonate Inorganic materials 0.000 description 4
- 238000000423 cell based assay Methods 0.000 description 4
- 239000006285 cell suspension Substances 0.000 description 4
- 239000012230 colorless oil Substances 0.000 description 4
- 125000004122 cyclic group Chemical group 0.000 description 4
- BGTOWKSIORTVQH-UHFFFAOYSA-N cyclopentanone Chemical compound O=C1CCCC1 BGTOWKSIORTVQH-UHFFFAOYSA-N 0.000 description 4
- 238000010828 elution Methods 0.000 description 4
- 230000001506 immunosuppresive effect Effects 0.000 description 4
- 239000008101 lactose Substances 0.000 description 4
- 239000003446 ligand Substances 0.000 description 4
- 238000001819 mass spectrum Methods 0.000 description 4
- 125000002950 monocyclic group Chemical group 0.000 description 4
- 239000013641 positive control Substances 0.000 description 4
- SCVFZCLFOSHCOH-UHFFFAOYSA-M potassium acetate Chemical compound [K+].CC([O-])=O SCVFZCLFOSHCOH-UHFFFAOYSA-M 0.000 description 4
- 229910000027 potassium carbonate Inorganic materials 0.000 description 4
- 238000012746 preparative thin layer chromatography Methods 0.000 description 4
- 125000006239 protecting group Chemical group 0.000 description 4
- 230000019491 signal transduction Effects 0.000 description 4
- 239000000741 silica gel Substances 0.000 description 4
- 229910002027 silica gel Inorganic materials 0.000 description 4
- 229910000029 sodium carbonate Inorganic materials 0.000 description 4
- 239000000829 suppository Substances 0.000 description 4
- WNWHHMBRJJOGFJ-UHFFFAOYSA-N 16-methylheptadecan-1-ol Chemical class CC(C)CCCCCCCCCCCCCCCO WNWHHMBRJJOGFJ-UHFFFAOYSA-N 0.000 description 3
- OISVCGZHLKNMSJ-UHFFFAOYSA-N 2,6-dimethylpyridine Chemical compound CC1=CC=CC(C)=N1 OISVCGZHLKNMSJ-UHFFFAOYSA-N 0.000 description 3
- NHQDETIJWKXCTC-UHFFFAOYSA-N 3-chloroperbenzoic acid Chemical compound OOC(=O)C1=CC=CC(Cl)=C1 NHQDETIJWKXCTC-UHFFFAOYSA-N 0.000 description 3
- GSXUXSXBEUJRAJ-UHFFFAOYSA-N 4-bromo-2-nitrobenzaldehyde Chemical compound [O-][N+](=O)C1=CC(Br)=CC=C1C=O GSXUXSXBEUJRAJ-UHFFFAOYSA-N 0.000 description 3
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 3
- 239000005995 Aluminium silicate Substances 0.000 description 3
- 241000711404 Avian avulavirus 1 Species 0.000 description 3
- 108010010803 Gelatin Proteins 0.000 description 3
- 101000669447 Homo sapiens Toll-like receptor 4 Proteins 0.000 description 3
- 241000725303 Human immunodeficiency virus Species 0.000 description 3
- WMFOQBRAJBCJND-UHFFFAOYSA-M Lithium hydroxide Chemical compound [Li+].[OH-] WMFOQBRAJBCJND-UHFFFAOYSA-M 0.000 description 3
- BZLVMXJERCGZMT-UHFFFAOYSA-N Methyl tert-butyl ether Chemical compound COC(C)(C)C BZLVMXJERCGZMT-UHFFFAOYSA-N 0.000 description 3
- 102000010168 Myeloid Differentiation Factor 88 Human genes 0.000 description 3
- 108010077432 Myeloid Differentiation Factor 88 Proteins 0.000 description 3
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 3
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 3
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 3
- 102100039360 Toll-like receptor 4 Human genes 0.000 description 3
- 235000012211 aluminium silicate Nutrition 0.000 description 3
- 125000003277 amino group Chemical group 0.000 description 3
- 230000000840 anti-viral effect Effects 0.000 description 3
- 239000002585 base Substances 0.000 description 3
- IPWKHHSGDUIRAH-UHFFFAOYSA-N bis(pinacolato)diboron Chemical compound O1C(C)(C)C(C)(C)OB1B1OC(C)(C)C(C)(C)O1 IPWKHHSGDUIRAH-UHFFFAOYSA-N 0.000 description 3
- 239000003054 catalyst Substances 0.000 description 3
- 210000000182 cd11c+cd123- dc Anatomy 0.000 description 3
- 239000003795 chemical substances by application Substances 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- 235000019864 coconut oil Nutrition 0.000 description 3
- 239000003240 coconut oil Substances 0.000 description 3
- XHFGWHUWQXTGAT-UHFFFAOYSA-N dimethylamine hydrochloride Natural products CNC(C)C XHFGWHUWQXTGAT-UHFFFAOYSA-N 0.000 description 3
- IQDGSYLLQPDQDV-UHFFFAOYSA-N dimethylazanium;chloride Chemical compound Cl.CNC IQDGSYLLQPDQDV-UHFFFAOYSA-N 0.000 description 3
- 239000000839 emulsion Substances 0.000 description 3
- 239000008273 gelatin Substances 0.000 description 3
- 229920000159 gelatin Polymers 0.000 description 3
- 235000019322 gelatine Nutrition 0.000 description 3
- 235000011852 gelatine desserts Nutrition 0.000 description 3
- 239000008103 glucose Substances 0.000 description 3
- 230000028993 immune response Effects 0.000 description 3
- 125000003392 indanyl group Chemical group C1(CCC2=CC=CC=C12)* 0.000 description 3
- NLYAJNPCOHFWQQ-UHFFFAOYSA-N kaolin Chemical compound O.O.O=[Al]O[Si](=O)O[Si](=O)O[Al]=O NLYAJNPCOHFWQQ-UHFFFAOYSA-N 0.000 description 3
- 208000032839 leukemia Diseases 0.000 description 3
- 239000012046 mixed solvent Substances 0.000 description 3
- 239000012044 organic layer Substances 0.000 description 3
- KJIFKLIQANRMOU-UHFFFAOYSA-N oxidanium;4-methylbenzenesulfonate Chemical compound O.CC1=CC=C(S(O)(=O)=O)C=C1 KJIFKLIQANRMOU-UHFFFAOYSA-N 0.000 description 3
- 239000001301 oxygen Chemical group 0.000 description 3
- 239000012071 phase Substances 0.000 description 3
- 239000006187 pill Substances 0.000 description 3
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 3
- 230000004044 response Effects 0.000 description 3
- 125000006413 ring segment Chemical group 0.000 description 3
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 3
- 235000017557 sodium bicarbonate Nutrition 0.000 description 3
- FVAUCKIRQBBSSJ-UHFFFAOYSA-M sodium iodide Chemical compound [Na+].[I-] FVAUCKIRQBBSSJ-UHFFFAOYSA-M 0.000 description 3
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 3
- 230000003612 virological effect Effects 0.000 description 3
- HZAXFHJVJLSVMW-UHFFFAOYSA-N 2-Aminoethan-1-ol Chemical compound NCCO HZAXFHJVJLSVMW-UHFFFAOYSA-N 0.000 description 2
- LDLCZOVUSADOIV-UHFFFAOYSA-N 2-bromoethanol Chemical compound OCCBr LDLCZOVUSADOIV-UHFFFAOYSA-N 0.000 description 2
- HOZLOOPIXHWKCI-UHFFFAOYSA-N 2-chloro-n-methylacetamide Chemical compound CNC(=O)CCl HOZLOOPIXHWKCI-UHFFFAOYSA-N 0.000 description 2
- RHKWIGHJGOEUSM-UHFFFAOYSA-N 3h-imidazo[4,5-h]quinoline Chemical class C1=CN=C2C(N=CN3)=C3C=CC2=C1 RHKWIGHJGOEUSM-UHFFFAOYSA-N 0.000 description 2
- RVCJOGNLYVNRDN-UHFFFAOYSA-N 4-bromo-2-methylbenzoic acid Chemical compound CC1=CC(Br)=CC=C1C(O)=O RVCJOGNLYVNRDN-UHFFFAOYSA-N 0.000 description 2
- KRNSYSYRLQDHDK-UHFFFAOYSA-N 6,7-dihydro-5h-cyclopenta[b]pyridine Chemical compound C1=CN=C2CCCC2=C1 KRNSYSYRLQDHDK-UHFFFAOYSA-N 0.000 description 2
- FQPKKECSRKYXIZ-UHFFFAOYSA-N 6-bromo-3,4-dihydro-2h-isoquinolin-1-one Chemical compound O=C1NCCC2=CC(Br)=CC=C21 FQPKKECSRKYXIZ-UHFFFAOYSA-N 0.000 description 2
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 2
- 229920001817 Agar Polymers 0.000 description 2
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 2
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 2
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 2
- 208000010839 B-cell chronic lymphocytic leukemia Diseases 0.000 description 2
- 241001474374 Blennius Species 0.000 description 2
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 2
- 206010008342 Cervix carcinoma Diseases 0.000 description 2
- 241000701022 Cytomegalovirus Species 0.000 description 2
- 241000725619 Dengue virus Species 0.000 description 2
- OKKJLVBELUTLKV-MZCSYVLQSA-N Deuterated methanol Chemical compound [2H]OC([2H])([2H])[2H] OKKJLVBELUTLKV-MZCSYVLQSA-N 0.000 description 2
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 2
- 239000006144 Dulbecco’s modified Eagle's medium Substances 0.000 description 2
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 2
- 241000701044 Human gammaherpesvirus 4 Species 0.000 description 2
- 102000013462 Interleukin-12 Human genes 0.000 description 2
- 108010065805 Interleukin-12 Proteins 0.000 description 2
- WQZGKKKJIJFFOK-ZZWDRFIYSA-N L-glucose Chemical compound OC[C@@H]1OC(O)[C@@H](O)[C@H](O)[C@H]1O WQZGKKKJIJFFOK-ZZWDRFIYSA-N 0.000 description 2
- 206010058467 Lung neoplasm malignant Diseases 0.000 description 2
- 208000031422 Lymphocytic Chronic B-Cell Leukemia Diseases 0.000 description 2
- 241000124008 Mammalia Species 0.000 description 2
- 208000003250 Mixed connective tissue disease Diseases 0.000 description 2
- 241000711408 Murine respirovirus Species 0.000 description 2
- 101000574441 Mus musculus Alkaline phosphatase, germ cell type Proteins 0.000 description 2
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 2
- MZRVEZGGRBJDDB-UHFFFAOYSA-N N-Butyllithium Chemical compound [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 description 2
- 238000005481 NMR spectroscopy Methods 0.000 description 2
- 239000004698 Polyethylene Substances 0.000 description 2
- 239000002202 Polyethylene glycol Substances 0.000 description 2
- 108700008625 Reporter Genes Proteins 0.000 description 2
- DBMJMQXJHONAFJ-UHFFFAOYSA-M Sodium laurylsulphate Chemical compound [Na+].CCCCCCCCCCCCOS([O-])(=O)=O DBMJMQXJHONAFJ-UHFFFAOYSA-M 0.000 description 2
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 2
- 229930006000 Sucrose Natural products 0.000 description 2
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical group [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 2
- 210000001744 T-lymphocyte Anatomy 0.000 description 2
- DKGAVHZHDRPRBM-UHFFFAOYSA-N Tert-Butanol Chemical compound CC(C)(C)O DKGAVHZHDRPRBM-UHFFFAOYSA-N 0.000 description 2
- 208000006105 Uterine Cervical Neoplasms Diseases 0.000 description 2
- WETWJCDKMRHUPV-UHFFFAOYSA-N acetyl chloride Chemical compound CC(Cl)=O WETWJCDKMRHUPV-UHFFFAOYSA-N 0.000 description 2
- 239000012346 acetyl chloride Substances 0.000 description 2
- 230000033289 adaptive immune response Effects 0.000 description 2
- 239000008272 agar Substances 0.000 description 2
- 235000010419 agar Nutrition 0.000 description 2
- 150000001298 alcohols Chemical class 0.000 description 2
- 150000001413 amino acids Chemical class 0.000 description 2
- 239000000427 antigen Substances 0.000 description 2
- 108091007433 antigens Proteins 0.000 description 2
- 102000036639 antigens Human genes 0.000 description 2
- 239000008346 aqueous phase Substances 0.000 description 2
- 125000003710 aryl alkyl group Chemical group 0.000 description 2
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical group [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 2
- 125000000499 benzofuranyl group Chemical group O1C(=CC2=C1C=CC=C2)* 0.000 description 2
- 125000001164 benzothiazolyl group Chemical group S1C(=NC2=C1C=CC=C2)* 0.000 description 2
- 125000004196 benzothienyl group Chemical group S1C(=CC2=C1C=CC=C2)* 0.000 description 2
- 125000004541 benzoxazolyl group Chemical group O1C(=NC2=C1C=CC=C2)* 0.000 description 2
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 2
- 239000011230 binding agent Substances 0.000 description 2
- 150000001649 bromium compounds Chemical class 0.000 description 2
- 229910000019 calcium carbonate Inorganic materials 0.000 description 2
- 238000002619 cancer immunotherapy Methods 0.000 description 2
- 239000002775 capsule Substances 0.000 description 2
- 239000004202 carbamide Substances 0.000 description 2
- 229910002091 carbon monoxide Inorganic materials 0.000 description 2
- 239000000969 carrier Substances 0.000 description 2
- XMPZTFVPEKAKFH-UHFFFAOYSA-P ceric ammonium nitrate Chemical compound [NH4+].[NH4+].[Ce+4].[O-][N+]([O-])=O.[O-][N+]([O-])=O.[O-][N+]([O-])=O.[O-][N+]([O-])=O.[O-][N+]([O-])=O.[O-][N+]([O-])=O XMPZTFVPEKAKFH-UHFFFAOYSA-P 0.000 description 2
- 201000010881 cervical cancer Diseases 0.000 description 2
- 208000032852 chronic lymphocytic leukemia Diseases 0.000 description 2
- 239000011248 coating agent Substances 0.000 description 2
- 238000000576 coating method Methods 0.000 description 2
- 238000004440 column chromatography Methods 0.000 description 2
- 229940125890 compound Ia Drugs 0.000 description 2
- 125000001316 cycloalkyl alkyl group Chemical group 0.000 description 2
- 125000000596 cyclohexenyl group Chemical group C1(=CCCCC1)* 0.000 description 2
- JCWIWBWXCVGEAN-UHFFFAOYSA-L cyclopentyl(diphenyl)phosphane;dichloropalladium;iron Chemical compound [Fe].Cl[Pd]Cl.[CH]1[CH][CH][CH][C]1P(C=1C=CC=CC=1)C1=CC=CC=C1.[CH]1[CH][CH][CH][C]1P(C=1C=CC=CC=1)C1=CC=CC=C1 JCWIWBWXCVGEAN-UHFFFAOYSA-L 0.000 description 2
- 210000004443 dendritic cell Anatomy 0.000 description 2
- 230000001419 dependent effect Effects 0.000 description 2
- 125000004431 deuterium atom Chemical group 0.000 description 2
- 238000011161 development Methods 0.000 description 2
- ZUOUZKKEUPVFJK-UHFFFAOYSA-N diphenyl Chemical compound C1=CC=CC=C1C1=CC=CC=C1 ZUOUZKKEUPVFJK-UHFFFAOYSA-N 0.000 description 2
- USIUVYZYUHIAEV-UHFFFAOYSA-N diphenyl ether Chemical compound C=1C=CC=CC=1OC1=CC=CC=C1 USIUVYZYUHIAEV-UHFFFAOYSA-N 0.000 description 2
- 239000002552 dosage form Substances 0.000 description 2
- 231100000673 dose–response relationship Toxicity 0.000 description 2
- 238000002474 experimental method Methods 0.000 description 2
- 235000011187 glycerol Nutrition 0.000 description 2
- 239000008187 granular material Substances 0.000 description 2
- 125000003187 heptyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 2
- 125000004446 heteroarylalkyl group Chemical group 0.000 description 2
- 125000005885 heterocycloalkylalkyl group Chemical group 0.000 description 2
- 238000004128 high performance liquid chromatography Methods 0.000 description 2
- IXCSERBJSXMMFS-UHFFFAOYSA-N hydrogen chloride Substances Cl.Cl IXCSERBJSXMMFS-UHFFFAOYSA-N 0.000 description 2
- 229910000041 hydrogen chloride Inorganic materials 0.000 description 2
- 239000003112 inhibitor Substances 0.000 description 2
- 229940117681 interleukin-12 Drugs 0.000 description 2
- 125000000842 isoxazolyl group Chemical group 0.000 description 2
- 210000003292 kidney cell Anatomy 0.000 description 2
- 239000007788 liquid Substances 0.000 description 2
- GLXDVVHUTZTUQK-UHFFFAOYSA-M lithium;hydroxide;hydrate Chemical compound [Li+].O.[OH-] GLXDVVHUTZTUQK-UHFFFAOYSA-M 0.000 description 2
- 201000005202 lung cancer Diseases 0.000 description 2
- 208000020816 lung neoplasm Diseases 0.000 description 2
- NXPHGHWWQRMDIA-UHFFFAOYSA-M magnesium;carbanide;bromide Chemical compound [CH3-].[Mg+2].[Br-] NXPHGHWWQRMDIA-UHFFFAOYSA-M 0.000 description 2
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 2
- 239000012528 membrane Substances 0.000 description 2
- QPJVMBTYPHYUOC-UHFFFAOYSA-N methyl benzoate Chemical compound COC(=O)C1=CC=CC=C1 QPJVMBTYPHYUOC-UHFFFAOYSA-N 0.000 description 2
- 210000001616 monocyte Anatomy 0.000 description 2
- 125000001624 naphthyl group Chemical group 0.000 description 2
- 239000013642 negative control Substances 0.000 description 2
- 230000001613 neoplastic effect Effects 0.000 description 2
- 230000007935 neutral effect Effects 0.000 description 2
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 2
- 108020004707 nucleic acids Proteins 0.000 description 2
- 102000039446 nucleic acids Human genes 0.000 description 2
- 150000007523 nucleic acids Chemical class 0.000 description 2
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 2
- 230000003287 optical effect Effects 0.000 description 2
- CTSLXHKWHWQRSH-UHFFFAOYSA-N oxalyl chloride Chemical compound ClC(=O)C(Cl)=O CTSLXHKWHWQRSH-UHFFFAOYSA-N 0.000 description 2
- 125000002971 oxazolyl group Chemical group 0.000 description 2
- 125000003367 polycyclic group Chemical group 0.000 description 2
- 125000005592 polycycloalkyl group Polymers 0.000 description 2
- 229920000573 polyethylene Polymers 0.000 description 2
- 229920001223 polyethylene glycol Polymers 0.000 description 2
- 235000011056 potassium acetate Nutrition 0.000 description 2
- 238000000746 purification Methods 0.000 description 2
- 125000005493 quinolyl group Chemical group 0.000 description 2
- 230000001105 regulatory effect Effects 0.000 description 2
- 238000000926 separation method Methods 0.000 description 2
- RMAQACBXLXPBSY-UHFFFAOYSA-N silicic acid Chemical compound O[Si](O)(O)O RMAQACBXLXPBSY-UHFFFAOYSA-N 0.000 description 2
- 235000012239 silicon dioxide Nutrition 0.000 description 2
- 239000011780 sodium chloride Substances 0.000 description 2
- 235000019333 sodium laurylsulphate Nutrition 0.000 description 2
- LPXPTNMVRIOKMN-UHFFFAOYSA-M sodium nitrite Chemical compound [Na+].[O-]N=O LPXPTNMVRIOKMN-UHFFFAOYSA-M 0.000 description 2
- JQWHASGSAFIOCM-UHFFFAOYSA-M sodium periodate Chemical compound [Na+].[O-]I(=O)(=O)=O JQWHASGSAFIOCM-UHFFFAOYSA-M 0.000 description 2
- 241000114864 ssRNA viruses Species 0.000 description 2
- 239000007858 starting material Substances 0.000 description 2
- 230000000638 stimulation Effects 0.000 description 2
- 239000000758 substrate Substances 0.000 description 2
- KDYFGRWQOYBRFD-UHFFFAOYSA-L succinate(2-) Chemical compound [O-]C(=O)CCC([O-])=O KDYFGRWQOYBRFD-UHFFFAOYSA-L 0.000 description 2
- 239000011593 sulfur Chemical group 0.000 description 2
- 239000006228 supernatant Substances 0.000 description 2
- 239000000454 talc Substances 0.000 description 2
- 229910052623 talc Inorganic materials 0.000 description 2
- 235000012222 talc Nutrition 0.000 description 2
- DYHSDKLCOJIUFX-UHFFFAOYSA-N tert-butoxycarbonyl anhydride Chemical compound CC(C)(C)OC(=O)OC(=O)OC(C)(C)C DYHSDKLCOJIUFX-UHFFFAOYSA-N 0.000 description 2
- 238000012360 testing method Methods 0.000 description 2
- CZDYPVPMEAXLPK-UHFFFAOYSA-N tetramethylsilane Chemical compound C[Si](C)(C)C CZDYPVPMEAXLPK-UHFFFAOYSA-N 0.000 description 2
- WROMPOXWARCANT-UHFFFAOYSA-N tfa trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F.OC(=O)C(F)(F)F WROMPOXWARCANT-UHFFFAOYSA-N 0.000 description 2
- 230000001225 therapeutic effect Effects 0.000 description 2
- 125000001544 thienyl group Chemical group 0.000 description 2
- FYSNRJHAOHDILO-UHFFFAOYSA-N thionyl chloride Chemical compound ClS(Cl)=O FYSNRJHAOHDILO-UHFFFAOYSA-N 0.000 description 2
- LWIHDJKSTIGBAC-UHFFFAOYSA-K tripotassium phosphate Chemical compound [K+].[K+].[K+].[O-]P([O-])([O-])=O LWIHDJKSTIGBAC-UHFFFAOYSA-K 0.000 description 2
- DCXXMTOCNZCJGO-UHFFFAOYSA-N tristearoylglycerol Chemical compound CCCCCCCCCCCCCCCCCC(=O)OCC(OC(=O)CCCCCCCCCCCCCCCCC)COC(=O)CCCCCCCCCCCCCCCCC DCXXMTOCNZCJGO-UHFFFAOYSA-N 0.000 description 2
- 210000004881 tumor cell Anatomy 0.000 description 2
- 229910052721 tungsten Inorganic materials 0.000 description 2
- 241000712461 unidentified influenza virus Species 0.000 description 2
- DTGKSKDOIYIVQL-WEDXCCLWSA-N (+)-borneol Chemical compound C1C[C@@]2(C)[C@@H](O)C[C@@H]1C2(C)C DTGKSKDOIYIVQL-WEDXCCLWSA-N 0.000 description 1
- REPVLJRCJUVQFA-UHFFFAOYSA-N (-)-isopinocampheol Natural products C1C(O)C(C)C2C(C)(C)C1C2 REPVLJRCJUVQFA-UHFFFAOYSA-N 0.000 description 1
- TXUICONDJPYNPY-UHFFFAOYSA-N (1,10,13-trimethyl-3-oxo-4,5,6,7,8,9,11,12,14,15,16,17-dodecahydrocyclopenta[a]phenanthren-17-yl) heptanoate Chemical compound C1CC2CC(=O)C=C(C)C2(C)C2C1C1CCC(OC(=O)CCCCCC)C1(C)CC2 TXUICONDJPYNPY-UHFFFAOYSA-N 0.000 description 1
- JNYAEWCLZODPBN-JGWLITMVSA-N (2r,3r,4s)-2-[(1r)-1,2-dihydroxyethyl]oxolane-3,4-diol Chemical compound OC[C@@H](O)[C@H]1OC[C@H](O)[C@H]1O JNYAEWCLZODPBN-JGWLITMVSA-N 0.000 description 1
- LNAZSHAWQACDHT-XIYTZBAFSA-N (2r,3r,4s,5r,6s)-4,5-dimethoxy-2-(methoxymethyl)-3-[(2s,3r,4s,5r,6r)-3,4,5-trimethoxy-6-(methoxymethyl)oxan-2-yl]oxy-6-[(2r,3r,4s,5r,6r)-4,5,6-trimethoxy-2-(methoxymethyl)oxan-3-yl]oxyoxane Chemical compound CO[C@@H]1[C@@H](OC)[C@H](OC)[C@@H](COC)O[C@H]1O[C@H]1[C@H](OC)[C@@H](OC)[C@H](O[C@H]2[C@@H]([C@@H](OC)[C@H](OC)O[C@@H]2COC)OC)O[C@@H]1COC LNAZSHAWQACDHT-XIYTZBAFSA-N 0.000 description 1
- 108091032973 (ribonucleotides)n+m Proteins 0.000 description 1
- 102000040650 (ribonucleotides)n+m Human genes 0.000 description 1
- 125000001399 1,2,3-triazolyl group Chemical group N1N=NC(=C1)* 0.000 description 1
- 125000004504 1,2,4-oxadiazolyl group Chemical group 0.000 description 1
- 125000001376 1,2,4-triazolyl group Chemical group N1N=C(N=C1)* 0.000 description 1
- 125000004506 1,2,5-oxadiazolyl group Chemical group 0.000 description 1
- 125000001781 1,3,4-oxadiazolyl group Chemical group 0.000 description 1
- MBIZXFATKUQOOA-UHFFFAOYSA-N 1,3,4-thiadiazole Chemical compound C1=NN=CS1 MBIZXFATKUQOOA-UHFFFAOYSA-N 0.000 description 1
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 1
- ASOKPJOREAFHNY-UHFFFAOYSA-N 1-Hydroxybenzotriazole Chemical compound C1=CC=C2N(O)N=NC2=C1 ASOKPJOREAFHNY-UHFFFAOYSA-N 0.000 description 1
- OUKQTRFCDKSEPL-UHFFFAOYSA-N 1-Methyl-2-pyrrolecarboxaldehyde Chemical compound CN1C=CC=C1C=O OUKQTRFCDKSEPL-UHFFFAOYSA-N 0.000 description 1
- JNOZGFXJZQXOSU-UHFFFAOYSA-N 1-chloro-2-methylpropan-2-ol Chemical compound CC(C)(O)CCl JNOZGFXJZQXOSU-UHFFFAOYSA-N 0.000 description 1
- IXPNQXFRVYWDDI-UHFFFAOYSA-N 1-methyl-2,4-dioxo-1,3-diazinane-5-carboximidamide Chemical compound CN1CC(C(N)=N)C(=O)NC1=O IXPNQXFRVYWDDI-UHFFFAOYSA-N 0.000 description 1
- VBICKXHEKHSIBG-UHFFFAOYSA-N 1-monostearoylglycerol Chemical compound CCCCCCCCCCCCCCCCCC(=O)OCC(O)CO VBICKXHEKHSIBG-UHFFFAOYSA-N 0.000 description 1
- 125000004214 1-pyrrolidinyl group Chemical group [H]C1([H])N(*)C([H])([H])C([H])([H])C1([H])[H] 0.000 description 1
- AAILEWXSEQLMNI-UHFFFAOYSA-N 1h-pyridazin-6-one Chemical compound OC1=CC=CN=N1 AAILEWXSEQLMNI-UHFFFAOYSA-N 0.000 description 1
- NAGGYODWMPFKJQ-UHFFFAOYSA-N 2,4-dibromobenzoic acid Chemical compound OC(=O)C1=CC=C(Br)C=C1Br NAGGYODWMPFKJQ-UHFFFAOYSA-N 0.000 description 1
- OZAIFHULBGXAKX-UHFFFAOYSA-N 2-(2-cyanopropan-2-yldiazenyl)-2-methylpropanenitrile Chemical compound N#CC(C)(C)N=NC(C)(C)C#N OZAIFHULBGXAKX-UHFFFAOYSA-N 0.000 description 1
- BCLSJHWBDUYDTR-UHFFFAOYSA-N 2-(propylamino)ethanol Chemical compound CCCNCCO BCLSJHWBDUYDTR-UHFFFAOYSA-N 0.000 description 1
- REXUYBKPWIPONM-UHFFFAOYSA-N 2-bromoacetonitrile Chemical compound BrCC#N REXUYBKPWIPONM-UHFFFAOYSA-N 0.000 description 1
- XWKFPIODWVPXLX-UHFFFAOYSA-N 2-methyl-5-methylpyridine Natural products CC1=CC=C(C)N=C1 XWKFPIODWVPXLX-UHFFFAOYSA-N 0.000 description 1
- MGACSNHAFXGSIV-UHFFFAOYSA-N 4-bromo-2-chloro-6-nitrobenzaldehyde Chemical compound BrC1=CC(=C(C=O)C(=C1)[N+](=O)[O-])Cl MGACSNHAFXGSIV-UHFFFAOYSA-N 0.000 description 1
- IKIBCVVUGNCJEZ-UHFFFAOYSA-N 4-bromo-2-fluoro-6-nitrobenzaldehyde Chemical compound [O-][N+](=O)c1cc(Br)cc(F)c1C=O IKIBCVVUGNCJEZ-UHFFFAOYSA-N 0.000 description 1
- ZQQSRVPOAHYHEL-UHFFFAOYSA-N 4-bromo-2-fluorobenzoic acid Chemical compound OC(=O)C1=CC=C(Br)C=C1F ZQQSRVPOAHYHEL-UHFFFAOYSA-N 0.000 description 1
- RCBAWHDKFODODY-UHFFFAOYSA-N 4-bromo-2-methoxy-6-nitrobenzaldehyde Chemical compound BrC1=CC(=C(C=O)C(=C1)[N+](=O)[O-])OC RCBAWHDKFODODY-UHFFFAOYSA-N 0.000 description 1
- MOERBJIWBODDFC-UHFFFAOYSA-N 4-bromo-2-methyl-6-nitrobenzaldehyde Chemical compound BrC1=CC(=C(C=O)C(=C1)[N+](=O)[O-])C MOERBJIWBODDFC-UHFFFAOYSA-N 0.000 description 1
- AYSVJBVDPZARKA-UHFFFAOYSA-N 4-bromo-5-fluoro-2-nitrobenzaldehyde Chemical compound [O-][N+](=O)C1=CC(Br)=C(F)C=C1C=O AYSVJBVDPZARKA-UHFFFAOYSA-N 0.000 description 1
- IRPQLEYYDURDBA-UHFFFAOYSA-N 5,6-dihydro-4h-cyclopenta[b]thiophene Chemical compound C1=CSC2=C1CCC2 IRPQLEYYDURDBA-UHFFFAOYSA-N 0.000 description 1
- OOKFCQHMHNKVKJ-UHFFFAOYSA-N 5-bromo-2-methyl-3-nitroaniline Chemical compound CC1=C(N)C=C(Br)C=C1[N+]([O-])=O OOKFCQHMHNKVKJ-UHFFFAOYSA-N 0.000 description 1
- GSOCTKHBFREQCV-UHFFFAOYSA-N 5-bromo-3-methylpyridine-2-carboxylic acid Chemical compound CC1=CC(Br)=CN=C1C(O)=O GSOCTKHBFREQCV-UHFFFAOYSA-N 0.000 description 1
- GFBVUFQNHLUCPX-UHFFFAOYSA-N 5-bromothiophene-2-carbaldehyde Chemical compound BrC1=CC=C(C=O)S1 GFBVUFQNHLUCPX-UHFFFAOYSA-N 0.000 description 1
- QMONLZVJOOMKRW-UHFFFAOYSA-N 6-bromo-2h-phthalazin-1-one Chemical compound C1=NNC(=O)C=2C1=CC(Br)=CC=2 QMONLZVJOOMKRW-UHFFFAOYSA-N 0.000 description 1
- SHSNAKRNZGOTJL-UHFFFAOYSA-N 6h-furo[2,3-c]pyridin-7-one Chemical compound OC1=NC=CC2=C1OC=C2 SHSNAKRNZGOTJL-UHFFFAOYSA-N 0.000 description 1
- FUGKKMYSCAYXJJ-UHFFFAOYSA-N 7-bromo-1h-quinazolin-4-one Chemical compound N1C=NC(=O)C=2C1=CC(Br)=CC=2 FUGKKMYSCAYXJJ-UHFFFAOYSA-N 0.000 description 1
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 1
- 244000215068 Acacia senegal Species 0.000 description 1
- NIXOWILDQLNWCW-UHFFFAOYSA-M Acrylate Chemical compound [O-]C(=O)C=C NIXOWILDQLNWCW-UHFFFAOYSA-M 0.000 description 1
- 208000024893 Acute lymphoblastic leukemia Diseases 0.000 description 1
- 208000014697 Acute lymphocytic leukaemia Diseases 0.000 description 1
- 208000031261 Acute myeloid leukaemia Diseases 0.000 description 1
- 241001251200 Agelas Species 0.000 description 1
- ATRRKUHOCOJYRX-UHFFFAOYSA-N Ammonium bicarbonate Chemical compound [NH4+].OC([O-])=O ATRRKUHOCOJYRX-UHFFFAOYSA-N 0.000 description 1
- 229910000013 Ammonium bicarbonate Inorganic materials 0.000 description 1
- 239000004475 Arginine Substances 0.000 description 1
- 241000416162 Astragalus gummifer Species 0.000 description 1
- 206010003827 Autoimmune hepatitis Diseases 0.000 description 1
- 208000032791 BCR-ABL1 positive chronic myelogenous leukemia Diseases 0.000 description 1
- 241000894006 Bacteria Species 0.000 description 1
- 208000008439 Biliary Liver Cirrhosis Diseases 0.000 description 1
- 208000033222 Biliary cirrhosis primary Diseases 0.000 description 1
- LSNNMFCWUKXFEE-UHFFFAOYSA-M Bisulfite Chemical compound OS([O-])=O LSNNMFCWUKXFEE-UHFFFAOYSA-M 0.000 description 1
- 206010005003 Bladder cancer Diseases 0.000 description 1
- 206010005949 Bone cancer Diseases 0.000 description 1
- 208000018084 Bone neoplasm Diseases 0.000 description 1
- 208000003174 Brain Neoplasms Diseases 0.000 description 1
- 206010006187 Breast cancer Diseases 0.000 description 1
- 208000026310 Breast neoplasm Diseases 0.000 description 1
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 1
- 201000002829 CREST Syndrome Diseases 0.000 description 1
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 1
- 229920002134 Carboxymethyl cellulose Polymers 0.000 description 1
- 206010008609 Cholangitis sclerosing Diseases 0.000 description 1
- 208000010833 Chronic myeloid leukaemia Diseases 0.000 description 1
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 1
- 206010009944 Colon cancer Diseases 0.000 description 1
- 229910021589 Copper(I) bromide Inorganic materials 0.000 description 1
- 241000709687 Coxsackievirus Species 0.000 description 1
- SNRUBQQJIBEYMU-UHFFFAOYSA-N Dodecane Natural products CCCCCCCCCCCC SNRUBQQJIBEYMU-UHFFFAOYSA-N 0.000 description 1
- 241000991587 Enterovirus C Species 0.000 description 1
- 208000000461 Esophageal Neoplasms Diseases 0.000 description 1
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 1
- BDAGIHXWWSANSR-UHFFFAOYSA-M Formate Chemical compound [O-]C=O BDAGIHXWWSANSR-UHFFFAOYSA-M 0.000 description 1
- 241000233866 Fungi Species 0.000 description 1
- 208000022072 Gallbladder Neoplasms Diseases 0.000 description 1
- 206010018338 Glioma Diseases 0.000 description 1
- 206010018366 Glomerulonephritis acute Diseases 0.000 description 1
- 206010018367 Glomerulonephritis chronic Diseases 0.000 description 1
- 208000009329 Graft vs Host Disease Diseases 0.000 description 1
- 229920000084 Gum arabic Polymers 0.000 description 1
- 208000001688 Herpes Genitalis Diseases 0.000 description 1
- 208000007514 Herpes zoster Diseases 0.000 description 1
- 208000017604 Hodgkin disease Diseases 0.000 description 1
- 208000021519 Hodgkin lymphoma Diseases 0.000 description 1
- 208000010747 Hodgkins lymphoma Diseases 0.000 description 1
- 241000282412 Homo Species 0.000 description 1
- 101000763579 Homo sapiens Toll-like receptor 1 Proteins 0.000 description 1
- 101000831567 Homo sapiens Toll-like receptor 2 Proteins 0.000 description 1
- 101000831496 Homo sapiens Toll-like receptor 3 Proteins 0.000 description 1
- 101000669460 Homo sapiens Toll-like receptor 5 Proteins 0.000 description 1
- 101000669406 Homo sapiens Toll-like receptor 6 Proteins 0.000 description 1
- 241000713772 Human immunodeficiency virus 1 Species 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical compound Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 1
- 206010020850 Hyperthyroidism Diseases 0.000 description 1
- 102000006992 Interferon-alpha Human genes 0.000 description 1
- 108010047761 Interferon-alpha Proteins 0.000 description 1
- 102000006940 Interleukin-1 Receptor-Associated Kinases Human genes 0.000 description 1
- 108010072621 Interleukin-1 Receptor-Associated Kinases Proteins 0.000 description 1
- XEEYBQQBJWHFJM-UHFFFAOYSA-N Iron Chemical compound [Fe] XEEYBQQBJWHFJM-UHFFFAOYSA-N 0.000 description 1
- 208000008839 Kidney Neoplasms Diseases 0.000 description 1
- JVTAAEKCZFNVCJ-UHFFFAOYSA-M Lactate Chemical compound CC(O)C([O-])=O JVTAAEKCZFNVCJ-UHFFFAOYSA-M 0.000 description 1
- 206010023825 Laryngeal cancer Diseases 0.000 description 1
- 239000012448 Lithium borohydride Substances 0.000 description 1
- 208000028018 Lymphocytic leukaemia Diseases 0.000 description 1
- 206010025323 Lymphomas Diseases 0.000 description 1
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 1
- 239000004472 Lysine Substances 0.000 description 1
- OFOBLEOULBTSOW-UHFFFAOYSA-L Malonate Chemical compound [O-]C(=O)CC([O-])=O OFOBLEOULBTSOW-UHFFFAOYSA-L 0.000 description 1
- 208000000172 Medulloblastoma Diseases 0.000 description 1
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 1
- 239000012359 Methanesulfonyl chloride Substances 0.000 description 1
- 208000003445 Mouth Neoplasms Diseases 0.000 description 1
- 208000033761 Myelogenous Chronic BCR-ABL Positive Leukemia Diseases 0.000 description 1
- 208000033776 Myeloid Acute Leukemia Diseases 0.000 description 1
- MBBZMMPHUWSWHV-BDVNFPICSA-N N-methylglucamine Chemical compound CNC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO MBBZMMPHUWSWHV-BDVNFPICSA-N 0.000 description 1
- 230000006051 NK cell activation Effects 0.000 description 1
- 229910002651 NO3 Inorganic materials 0.000 description 1
- 208000001894 Nasopharyngeal Neoplasms Diseases 0.000 description 1
- 206010061306 Nasopharyngeal cancer Diseases 0.000 description 1
- 208000010359 Newcastle Disease Diseases 0.000 description 1
- NHNBFGGVMKEFGY-UHFFFAOYSA-N Nitrate Chemical compound [O-][N+]([O-])=O NHNBFGGVMKEFGY-UHFFFAOYSA-N 0.000 description 1
- 208000015914 Non-Hodgkin lymphomas Diseases 0.000 description 1
- 206010030155 Oesophageal carcinoma Diseases 0.000 description 1
- 108091034117 Oligonucleotide Proteins 0.000 description 1
- 235000019502 Orange oil Nutrition 0.000 description 1
- 206010033128 Ovarian cancer Diseases 0.000 description 1
- 206010061535 Ovarian neoplasm Diseases 0.000 description 1
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 1
- 229910019142 PO4 Inorganic materials 0.000 description 1
- 206010061902 Pancreatic neoplasm Diseases 0.000 description 1
- 208000000821 Parathyroid Neoplasms Diseases 0.000 description 1
- 208000031845 Pernicious anaemia Diseases 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-L Phosphate ion(2-) Chemical compound OP([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-L 0.000 description 1
- 206010035226 Plasma cell myeloma Diseases 0.000 description 1
- 208000006664 Precursor Cell Lymphoblastic Leukemia-Lymphoma Diseases 0.000 description 1
- 208000012654 Primary biliary cholangitis Diseases 0.000 description 1
- 208000033759 Prolymphocytic T-Cell Leukemia Diseases 0.000 description 1
- OFOBLEOULBTSOW-UHFFFAOYSA-N Propanedioic acid Natural products OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 1
- XBDQKXXYIPTUBI-UHFFFAOYSA-M Propionate Chemical compound CCC([O-])=O XBDQKXXYIPTUBI-UHFFFAOYSA-M 0.000 description 1
- XBDQKXXYIPTUBI-UHFFFAOYSA-N Propionic acid Chemical compound CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 1
- 206010060862 Prostate cancer Diseases 0.000 description 1
- 208000000236 Prostatic Neoplasms Diseases 0.000 description 1
- 102000001253 Protein Kinase Human genes 0.000 description 1
- LCTONWCANYUPML-UHFFFAOYSA-M Pyruvate Chemical compound CC(=O)C([O-])=O LCTONWCANYUPML-UHFFFAOYSA-M 0.000 description 1
- 208000015634 Rectal Neoplasms Diseases 0.000 description 1
- 206010038389 Renal cancer Diseases 0.000 description 1
- 229920001800 Shellac Polymers 0.000 description 1
- 208000000453 Skin Neoplasms Diseases 0.000 description 1
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 1
- VMHLLURERBWHNL-UHFFFAOYSA-M Sodium acetate Chemical compound [Na+].CC([O-])=O VMHLLURERBWHNL-UHFFFAOYSA-M 0.000 description 1
- PXIPVTKHYLBLMZ-UHFFFAOYSA-N Sodium azide Chemical class [Na+].[N-]=[N+]=[N-] PXIPVTKHYLBLMZ-UHFFFAOYSA-N 0.000 description 1
- UIIMBOGNXHQVGW-DEQYMQKBSA-M Sodium bicarbonate-14C Chemical compound [Na+].O[14C]([O-])=O UIIMBOGNXHQVGW-DEQYMQKBSA-M 0.000 description 1
- 208000005718 Stomach Neoplasms Diseases 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-N Succinic acid Natural products OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 1
- LSNNMFCWUKXFEE-UHFFFAOYSA-N Sulfurous acid Chemical compound OS(O)=O LSNNMFCWUKXFEE-UHFFFAOYSA-N 0.000 description 1
- 201000009594 Systemic Scleroderma Diseases 0.000 description 1
- 206010042953 Systemic sclerosis Diseases 0.000 description 1
- 230000005867 T cell response Effects 0.000 description 1
- 206010042970 T-cell chronic lymphocytic leukaemia Diseases 0.000 description 1
- 208000026651 T-cell prolymphocytic leukemia Diseases 0.000 description 1
- 108700012920 TNF Proteins 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 1
- 208000024313 Testicular Neoplasms Diseases 0.000 description 1
- 206010057644 Testis cancer Diseases 0.000 description 1
- 208000024770 Thyroid neoplasm Diseases 0.000 description 1
- 229910021626 Tin(II) chloride Inorganic materials 0.000 description 1
- 102100027010 Toll-like receptor 1 Human genes 0.000 description 1
- 102100024333 Toll-like receptor 2 Human genes 0.000 description 1
- 102100024324 Toll-like receptor 3 Human genes 0.000 description 1
- 102100039357 Toll-like receptor 5 Human genes 0.000 description 1
- 102100039387 Toll-like receptor 6 Human genes 0.000 description 1
- 206010062129 Tongue neoplasm Diseases 0.000 description 1
- 229920001615 Tragacanth Polymers 0.000 description 1
- 102000040945 Transcription factor Human genes 0.000 description 1
- 108091023040 Transcription factor Proteins 0.000 description 1
- 206010052779 Transplant rejections Diseases 0.000 description 1
- DTQVDTLACAAQTR-UHFFFAOYSA-M Trifluoroacetate Chemical compound [O-]C(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-M 0.000 description 1
- OKJPEAGHQZHRQV-UHFFFAOYSA-N Triiodomethane Natural products IC(I)I OKJPEAGHQZHRQV-UHFFFAOYSA-N 0.000 description 1
- 206010067584 Type 1 diabetes mellitus Diseases 0.000 description 1
- 208000007097 Urinary Bladder Neoplasms Diseases 0.000 description 1
- 206010047115 Vasculitis Diseases 0.000 description 1
- ZEEBGORNQSEQBE-UHFFFAOYSA-N [2-(3-phenylphenoxy)-6-(trifluoromethyl)pyridin-4-yl]methanamine Chemical compound C1(=CC(=CC=C1)OC1=NC(=CC(=C1)CN)C(F)(F)F)C1=CC=CC=C1 ZEEBGORNQSEQBE-UHFFFAOYSA-N 0.000 description 1
- JLCPHMBAVCMARE-UHFFFAOYSA-N [3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-hydroxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methyl [5-(6-aminopurin-9-yl)-2-(hydroxymethyl)oxolan-3-yl] hydrogen phosphate Polymers Cc1cn(C2CC(OP(O)(=O)OCC3OC(CC3OP(O)(=O)OCC3OC(CC3O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c3nc(N)[nH]c4=O)C(COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3CO)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cc(C)c(=O)[nH]c3=O)n3cc(C)c(=O)[nH]c3=O)n3ccc(N)nc3=O)n3cc(C)c(=O)[nH]c3=O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)O2)c(=O)[nH]c1=O JLCPHMBAVCMARE-UHFFFAOYSA-N 0.000 description 1
- NGFFLHMFSINFGB-UHFFFAOYSA-N [chloro(methoxy)phosphoryl]oxymethane Chemical compound COP(Cl)(=O)OC NGFFLHMFSINFGB-UHFFFAOYSA-N 0.000 description 1
- 238000010521 absorption reaction Methods 0.000 description 1
- 235000010489 acacia gum Nutrition 0.000 description 1
- 239000000205 acacia gum Substances 0.000 description 1
- 239000003377 acid catalyst Substances 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 231100000851 acute glomerulonephritis Toxicity 0.000 description 1
- 125000005073 adamantyl group Chemical group C12(CC3CC(CC(C1)C3)C2)* 0.000 description 1
- 230000003044 adaptive effect Effects 0.000 description 1
- 208000009956 adenocarcinoma Diseases 0.000 description 1
- 239000003463 adsorbent Substances 0.000 description 1
- 229910052783 alkali metal Inorganic materials 0.000 description 1
- 229910052784 alkaline earth metal Inorganic materials 0.000 description 1
- 150000003973 alkyl amines Chemical class 0.000 description 1
- HSFWRNGVRCDJHI-UHFFFAOYSA-N alpha-acetylene Natural products C#C HSFWRNGVRCDJHI-UHFFFAOYSA-N 0.000 description 1
- BJEPYKJPYRNKOW-UHFFFAOYSA-N alpha-hydroxysuccinic acid Natural products OC(=O)C(O)CC(O)=O BJEPYKJPYRNKOW-UHFFFAOYSA-N 0.000 description 1
- 150000001412 amines Chemical class 0.000 description 1
- 229910021529 ammonia Inorganic materials 0.000 description 1
- 235000012538 ammonium bicarbonate Nutrition 0.000 description 1
- 239000001099 ammonium carbonate Substances 0.000 description 1
- 235000019270 ammonium chloride Nutrition 0.000 description 1
- 229940035676 analgesics Drugs 0.000 description 1
- 239000000730 antalgic agent Substances 0.000 description 1
- 230000003266 anti-allergic effect Effects 0.000 description 1
- 230000002924 anti-infective effect Effects 0.000 description 1
- 230000003110 anti-inflammatory effect Effects 0.000 description 1
- 230000000259 anti-tumor effect Effects 0.000 description 1
- 239000002246 antineoplastic agent Substances 0.000 description 1
- 229940041181 antineoplastic drug Drugs 0.000 description 1
- 230000001640 apoptogenic effect Effects 0.000 description 1
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 1
- 229940072107 ascorbate Drugs 0.000 description 1
- 235000010323 ascorbic acid Nutrition 0.000 description 1
- 239000011668 ascorbic acid Substances 0.000 description 1
- 230000001363 autoimmune Effects 0.000 description 1
- 208000018093 autoimmune cholangitis Diseases 0.000 description 1
- 230000006472 autoimmune response Effects 0.000 description 1
- 125000004069 aziridinyl group Chemical group 0.000 description 1
- 210000003719 b-lymphocyte Anatomy 0.000 description 1
- 239000000440 bentonite Substances 0.000 description 1
- 229910000278 bentonite Inorganic materials 0.000 description 1
- SVPXDRXYRYOSEX-UHFFFAOYSA-N bentoquatam Chemical compound O.O=[Si]=O.O=[Al]O[Al]=O SVPXDRXYRYOSEX-UHFFFAOYSA-N 0.000 description 1
- 125000003785 benzimidazolyl group Chemical group N1=C(NC2=C1C=CC=C2)* 0.000 description 1
- UMIVXZPTRXBADB-UHFFFAOYSA-N benzocyclobutene Chemical compound C1=CC=C2CCC2=C1 UMIVXZPTRXBADB-UHFFFAOYSA-N 0.000 description 1
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 1
- 125000002619 bicyclic group Chemical group 0.000 description 1
- 238000004166 bioassay Methods 0.000 description 1
- 239000004305 biphenyl Substances 0.000 description 1
- 235000010290 biphenyl Nutrition 0.000 description 1
- 210000004369 blood Anatomy 0.000 description 1
- 239000008280 blood Substances 0.000 description 1
- KGBXLFKZBHKPEV-UHFFFAOYSA-N boric acid Chemical compound OB(O)O KGBXLFKZBHKPEV-UHFFFAOYSA-N 0.000 description 1
- 239000004327 boric acid Substances 0.000 description 1
- 229940116229 borneol Drugs 0.000 description 1
- CKDOCTFBFTVPSN-UHFFFAOYSA-N borneol Natural products C1CC2(C)C(C)CC1C2(C)C CKDOCTFBFTVPSN-UHFFFAOYSA-N 0.000 description 1
- 239000000872 buffer Substances 0.000 description 1
- KDYFGRWQOYBRFD-NUQCWPJISA-N butanedioic acid Chemical compound O[14C](=O)CC[14C](O)=O KDYFGRWQOYBRFD-NUQCWPJISA-N 0.000 description 1
- 125000004369 butenyl group Chemical group C(=CCC)* 0.000 description 1
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000000480 butynyl group Chemical group [*]C#CC([H])([H])C([H])([H])[H] 0.000 description 1
- 239000011575 calcium Substances 0.000 description 1
- 229910052791 calcium Inorganic materials 0.000 description 1
- 239000001768 carboxy methyl cellulose Substances 0.000 description 1
- 235000010948 carboxy methyl cellulose Nutrition 0.000 description 1
- 150000007942 carboxylates Chemical group 0.000 description 1
- 239000008112 carboxymethyl-cellulose Substances 0.000 description 1
- 230000022534 cell killing Effects 0.000 description 1
- 210000000170 cell membrane Anatomy 0.000 description 1
- 239000001913 cellulose Substances 0.000 description 1
- 229920002678 cellulose Polymers 0.000 description 1
- 239000003153 chemical reaction reagent Substances 0.000 description 1
- 238000002512 chemotherapy Methods 0.000 description 1
- 150000001805 chlorine compounds Chemical class 0.000 description 1
- WRJWRGBVPUUDLA-UHFFFAOYSA-N chlorosulfonyl isocyanate Chemical compound ClS(=O)(=O)N=C=O WRJWRGBVPUUDLA-UHFFFAOYSA-N 0.000 description 1
- 208000006990 cholangiocarcinoma Diseases 0.000 description 1
- 238000004587 chromatography analysis Methods 0.000 description 1
- 208000029742 colonic neoplasm Diseases 0.000 description 1
- 238000009833 condensation Methods 0.000 description 1
- 230000005494 condensation Effects 0.000 description 1
- 238000001816 cooling Methods 0.000 description 1
- NKNDPYCGAZPOFS-UHFFFAOYSA-M copper(i) bromide Chemical compound Br[Cu] NKNDPYCGAZPOFS-UHFFFAOYSA-M 0.000 description 1
- 239000012043 crude product Substances 0.000 description 1
- 239000013078 crystal Substances 0.000 description 1
- 125000000000 cycloalkoxy group Chemical group 0.000 description 1
- 125000000582 cycloheptyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000000640 cyclooctyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C([H])([H])C1([H])[H] 0.000 description 1
- NXQGGXCHGDYOHB-UHFFFAOYSA-L cyclopenta-1,4-dien-1-yl(diphenyl)phosphane;dichloropalladium;iron(2+) Chemical compound [Fe+2].Cl[Pd]Cl.[CH-]1C=CC(P(C=2C=CC=CC=2)C=2C=CC=CC=2)=C1.[CH-]1C=CC(P(C=2C=CC=CC=2)C=2C=CC=CC=2)=C1 NXQGGXCHGDYOHB-UHFFFAOYSA-L 0.000 description 1
- 210000004544 dc2 Anatomy 0.000 description 1
- NNBZCPXTIHJBJL-UHFFFAOYSA-N decalin Chemical compound C1CCCC2CCCCC21 NNBZCPXTIHJBJL-UHFFFAOYSA-N 0.000 description 1
- 125000002704 decyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 230000005860 defense response to virus Effects 0.000 description 1
- 201000001981 dermatomyositis Diseases 0.000 description 1
- 238000001514 detection method Methods 0.000 description 1
- 235000014113 dietary fatty acids Nutrition 0.000 description 1
- 125000004586 dihydrobenzopyranyl group Chemical group O1C(CCC2=C1C=CC=C2)* 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-M dihydrogenphosphate Chemical compound OP(O)([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-M 0.000 description 1
- 239000003085 diluting agent Substances 0.000 description 1
- 239000000539 dimer Substances 0.000 description 1
- XPPKVPWEQAFLFU-UHFFFAOYSA-J diphosphate(4-) Chemical compound [O-]P([O-])(=O)OP([O-])([O-])=O XPPKVPWEQAFLFU-UHFFFAOYSA-J 0.000 description 1
- 235000011180 diphosphates Nutrition 0.000 description 1
- 238000009826 distribution Methods 0.000 description 1
- DTGKSKDOIYIVQL-UHFFFAOYSA-N dl-isoborneol Natural products C1CC2(C)C(O)CC1C2(C)C DTGKSKDOIYIVQL-UHFFFAOYSA-N 0.000 description 1
- 125000003438 dodecyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 239000008298 dragée Substances 0.000 description 1
- 229940079593 drug Drugs 0.000 description 1
- 239000003937 drug carrier Substances 0.000 description 1
- 238000009510 drug design Methods 0.000 description 1
- 239000003480 eluent Substances 0.000 description 1
- 210000000750 endocrine system Anatomy 0.000 description 1
- 238000005516 engineering process Methods 0.000 description 1
- 201000004101 esophageal cancer Diseases 0.000 description 1
- 150000002148 esters Chemical class 0.000 description 1
- 125000002534 ethynyl group Chemical group [H]C#C* 0.000 description 1
- 230000029142 excretion Effects 0.000 description 1
- 208000024519 eye neoplasm Diseases 0.000 description 1
- 239000000194 fatty acid Substances 0.000 description 1
- 229930195729 fatty acid Natural products 0.000 description 1
- 239000007941 film coated tablet Substances 0.000 description 1
- 125000003983 fluorenyl group Chemical group C1(=CC=CC=2C3=CC=CC=C3CC12)* 0.000 description 1
- 239000011737 fluorine Substances 0.000 description 1
- 239000006260 foam Substances 0.000 description 1
- 238000009472 formulation Methods 0.000 description 1
- 239000012458 free base Substances 0.000 description 1
- 239000001530 fumaric acid Substances 0.000 description 1
- 125000002541 furyl group Chemical group 0.000 description 1
- 201000010175 gallbladder cancer Diseases 0.000 description 1
- 206010017758 gastric cancer Diseases 0.000 description 1
- 108010025899 gelatin film Proteins 0.000 description 1
- 201000004946 genital herpes Diseases 0.000 description 1
- 125000005456 glyceride group Chemical group 0.000 description 1
- 230000002710 gonadal effect Effects 0.000 description 1
- 208000024908 graft versus host disease Diseases 0.000 description 1
- 238000011554 guinea pig model Methods 0.000 description 1
- 201000010536 head and neck cancer Diseases 0.000 description 1
- 208000014829 head and neck neoplasm Diseases 0.000 description 1
- 201000005787 hematologic cancer Diseases 0.000 description 1
- 230000002008 hemorrhagic effect Effects 0.000 description 1
- MNWFXJYAOYHMED-UHFFFAOYSA-N heptanoic acid Chemical compound CCCCCCC(O)=O MNWFXJYAOYHMED-UHFFFAOYSA-N 0.000 description 1
- FUZZWVXGSFPDMH-UHFFFAOYSA-N hexanoic acid Chemical compound CCCCCC(O)=O FUZZWVXGSFPDMH-UHFFFAOYSA-N 0.000 description 1
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 208000029824 high grade glioma Diseases 0.000 description 1
- 150000002430 hydrocarbons Chemical group 0.000 description 1
- 238000005984 hydrogenation reaction Methods 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-M hydrogensulfate Chemical compound OS([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-M 0.000 description 1
- 125000002883 imidazolyl group Chemical group 0.000 description 1
- 230000001900 immune effect Effects 0.000 description 1
- 230000036039 immunity Effects 0.000 description 1
- 230000008975 immunomodulatory function Effects 0.000 description 1
- 239000012535 impurity Substances 0.000 description 1
- 238000000338 in vitro Methods 0.000 description 1
- 238000001727 in vivo Methods 0.000 description 1
- 125000003453 indazolyl group Chemical group N1N=C(C2=C1C=CC=C2)* 0.000 description 1
- 125000001041 indolyl group Chemical group 0.000 description 1
- 230000002757 inflammatory effect Effects 0.000 description 1
- 230000005764 inhibitory process Effects 0.000 description 1
- 230000015788 innate immune response Effects 0.000 description 1
- 150000007529 inorganic bases Chemical class 0.000 description 1
- 150000004694 iodide salts Chemical class 0.000 description 1
- 229910052740 iodine Inorganic materials 0.000 description 1
- 239000011630 iodine Substances 0.000 description 1
- HVTICUPFWKNHNG-UHFFFAOYSA-N iodoethane Chemical compound CCI HVTICUPFWKNHNG-UHFFFAOYSA-N 0.000 description 1
- INQOMBQAUSQDDS-UHFFFAOYSA-N iodomethane Chemical compound IC INQOMBQAUSQDDS-UHFFFAOYSA-N 0.000 description 1
- KQNPFQTWMSNSAP-UHFFFAOYSA-N isobutyric acid Chemical compound CC(C)C(O)=O KQNPFQTWMSNSAP-UHFFFAOYSA-N 0.000 description 1
- 125000000904 isoindolyl group Chemical group C=1(NC=C2C=CC=CC12)* 0.000 description 1
- VDBNYAPERZTOOF-UHFFFAOYSA-N isoquinolin-1(2H)-one Chemical compound C1=CC=C2C(=O)NC=CC2=C1 VDBNYAPERZTOOF-UHFFFAOYSA-N 0.000 description 1
- 125000002183 isoquinolinyl group Chemical group C1(=NC=CC2=CC=CC=C12)* 0.000 description 1
- 125000005956 isoquinolyl group Chemical group 0.000 description 1
- 201000010982 kidney cancer Diseases 0.000 description 1
- 208000017169 kidney disease Diseases 0.000 description 1
- 206010023841 laryngeal neoplasm Diseases 0.000 description 1
- CFHGBZLNZZVTAY-UHFFFAOYSA-N lawesson's reagent Chemical compound C1=CC(OC)=CC=C1P1(=S)SP(=S)(C=2C=CC(OC)=CC=2)S1 CFHGBZLNZZVTAY-UHFFFAOYSA-N 0.000 description 1
- 239000010410 layer Substances 0.000 description 1
- 125000005647 linker group Chemical group 0.000 description 1
- 208000012987 lip and oral cavity carcinoma Diseases 0.000 description 1
- 201000007270 liver cancer Diseases 0.000 description 1
- 208000019423 liver disease Diseases 0.000 description 1
- 208000014018 liver neoplasm Diseases 0.000 description 1
- 210000004072 lung Anatomy 0.000 description 1
- 201000003866 lung sarcoma Diseases 0.000 description 1
- 208000003747 lymphoid leukemia Diseases 0.000 description 1
- 159000000003 magnesium salts Chemical class 0.000 description 1
- 201000004792 malaria Diseases 0.000 description 1
- 239000011976 maleic acid Substances 0.000 description 1
- 201000011614 malignant glioma Diseases 0.000 description 1
- 208000015486 malignant pancreatic neoplasm Diseases 0.000 description 1
- 208000026045 malignant tumor of parathyroid gland Diseases 0.000 description 1
- IWYDHOAUDWTVEP-UHFFFAOYSA-M mandelate Chemical compound [O-]C(=O)C(O)C1=CC=CC=C1 IWYDHOAUDWTVEP-UHFFFAOYSA-M 0.000 description 1
- 238000004519 manufacturing process Methods 0.000 description 1
- 239000000463 material Substances 0.000 description 1
- 230000007246 mechanism Effects 0.000 description 1
- 230000004060 metabolic process Effects 0.000 description 1
- 125000005341 metaphosphate group Chemical group 0.000 description 1
- 230000001394 metastastic effect Effects 0.000 description 1
- 208000037819 metastatic cancer Diseases 0.000 description 1
- 208000011575 metastatic malignant neoplasm Diseases 0.000 description 1
- 206010061289 metastatic neoplasm Diseases 0.000 description 1
- VNWKTOKETHGBQD-UHFFFAOYSA-N methane Natural products C VNWKTOKETHGBQD-UHFFFAOYSA-N 0.000 description 1
- QARBMVPHQWIHKH-UHFFFAOYSA-N methanesulfonyl chloride Chemical compound CS(Cl)(=O)=O QARBMVPHQWIHKH-UHFFFAOYSA-N 0.000 description 1
- 229940095102 methyl benzoate Drugs 0.000 description 1
- 229920000609 methyl cellulose Polymers 0.000 description 1
- 239000001923 methylcellulose Substances 0.000 description 1
- 235000010981 methylcellulose Nutrition 0.000 description 1
- 244000000010 microbial pathogen Species 0.000 description 1
- 150000007522 mineralic acids Chemical class 0.000 description 1
- 239000003226 mitogen Substances 0.000 description 1
- 230000003020 moisturizing effect Effects 0.000 description 1
- 238000002625 monoclonal antibody therapy Methods 0.000 description 1
- 125000004573 morpholin-4-yl group Chemical group N1(CCOCC1)* 0.000 description 1
- 201000000050 myeloid neoplasm Diseases 0.000 description 1
- ULWOJODHECIZAU-UHFFFAOYSA-N n,n-diethylpropan-2-amine Chemical compound CCN(CC)C(C)C ULWOJODHECIZAU-UHFFFAOYSA-N 0.000 description 1
- DAZSWUUAFHBCGE-KRWDZBQOSA-N n-[(2s)-3-methyl-1-oxo-1-pyrrolidin-1-ylbutan-2-yl]-3-phenylpropanamide Chemical compound N([C@@H](C(C)C)C(=O)N1CCCC1)C(=O)CCC1=CC=CC=C1 DAZSWUUAFHBCGE-KRWDZBQOSA-N 0.000 description 1
- PSZYNBSKGUBXEH-UHFFFAOYSA-N naphthalene-1-sulfonic acid Chemical compound C1=CC=C2C(S(=O)(=O)O)=CC=CC2=C1 PSZYNBSKGUBXEH-UHFFFAOYSA-N 0.000 description 1
- 125000004593 naphthyridinyl group Chemical group N1=C(C=CC2=CC=CN=C12)* 0.000 description 1
- 210000000822 natural killer cell Anatomy 0.000 description 1
- SLCVBVWXLSEKPL-UHFFFAOYSA-N neopentyl glycol Chemical compound OCC(C)(C)CO SLCVBVWXLSEKPL-UHFFFAOYSA-N 0.000 description 1
- 125000006574 non-aromatic ring group Chemical group 0.000 description 1
- 230000000683 nonmetastatic effect Effects 0.000 description 1
- 231100000252 nontoxic Toxicity 0.000 description 1
- 230000003000 nontoxic effect Effects 0.000 description 1
- 125000001400 nonyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 238000000655 nuclear magnetic resonance spectrum Methods 0.000 description 1
- WWZKQHOCKIZLMA-UHFFFAOYSA-M octanoate Chemical compound CCCCCCCC([O-])=O WWZKQHOCKIZLMA-UHFFFAOYSA-M 0.000 description 1
- 125000002347 octyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 201000008106 ocular cancer Diseases 0.000 description 1
- 238000005457 optimization Methods 0.000 description 1
- 239000010502 orange oil Substances 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 150000007530 organic bases Chemical class 0.000 description 1
- 125000004430 oxygen atom Chemical group O* 0.000 description 1
- 235000019629 palatability Nutrition 0.000 description 1
- 229910052763 palladium Inorganic materials 0.000 description 1
- 201000002528 pancreatic cancer Diseases 0.000 description 1
- 208000008443 pancreatic carcinoma Diseases 0.000 description 1
- 239000004031 partial agonist Substances 0.000 description 1
- 244000052769 pathogen Species 0.000 description 1
- 230000001717 pathogenic effect Effects 0.000 description 1
- 230000037361 pathway Effects 0.000 description 1
- 125000001147 pentyl group Chemical group C(CCCC)* 0.000 description 1
- 238000005191 phase separation Methods 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 1
- 239000010452 phosphate Substances 0.000 description 1
- CYQAYERJWZKYML-UHFFFAOYSA-N phosphorus pentasulfide Chemical compound S1P(S2)(=S)SP3(=S)SP1(=S)SP2(=S)S3 CYQAYERJWZKYML-UHFFFAOYSA-N 0.000 description 1
- XNGIFLGASWRNHJ-UHFFFAOYSA-L phthalate(2-) Chemical compound [O-]C(=O)C1=CC=CC=C1C([O-])=O XNGIFLGASWRNHJ-UHFFFAOYSA-L 0.000 description 1
- 125000004194 piperazin-1-yl group Chemical group [H]N1C([H])([H])C([H])([H])N(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000004193 piperazinyl group Chemical group 0.000 description 1
- 125000000587 piperidin-1-yl group Chemical group [H]C1([H])N(*)C([H])([H])C([H])([H])C([H])([H])C1([H])[H] 0.000 description 1
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 1
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 1
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 1
- XAEFZNCEHLXOMS-UHFFFAOYSA-M potassium benzoate Chemical compound [K+].[O-]C(=O)C1=CC=CC=C1 XAEFZNCEHLXOMS-UHFFFAOYSA-M 0.000 description 1
- 229910000160 potassium phosphate Inorganic materials 0.000 description 1
- 235000011009 potassium phosphates Nutrition 0.000 description 1
- 230000003389 potentiating effect Effects 0.000 description 1
- 238000000039 preparative column chromatography Methods 0.000 description 1
- 201000000742 primary sclerosing cholangitis Diseases 0.000 description 1
- 239000000047 product Substances 0.000 description 1
- 230000000770 proinflammatory effect Effects 0.000 description 1
- 230000035755 proliferation Effects 0.000 description 1
- 201000008171 proliferative glomerulonephritis Diseases 0.000 description 1
- BDERNNFJNOPAEC-UHFFFAOYSA-N propan-1-ol Chemical compound CCCO BDERNNFJNOPAEC-UHFFFAOYSA-N 0.000 description 1
- 125000004368 propenyl group Chemical group C(=CC)* 0.000 description 1
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000002568 propynyl group Chemical group [*]C#CC([H])([H])[H] 0.000 description 1
- 108060006633 protein kinase Proteins 0.000 description 1
- 108090000623 proteins and genes Proteins 0.000 description 1
- 102000004169 proteins and genes Human genes 0.000 description 1
- 125000001042 pteridinyl group Chemical group N1=C(N=CC2=NC=CN=C12)* 0.000 description 1
- 230000002685 pulmonary effect Effects 0.000 description 1
- 125000000561 purinyl group Chemical group N1=C(N=C2N=CNC2=C1)* 0.000 description 1
- 125000003226 pyrazolyl group Chemical group 0.000 description 1
- 125000000168 pyrrolyl group Chemical group 0.000 description 1
- 229940076788 pyruvate Drugs 0.000 description 1
- 125000001453 quaternary ammonium group Chemical group 0.000 description 1
- 150000003242 quaternary ammonium salts Chemical class 0.000 description 1
- 125000002294 quinazolinyl group Chemical group N1=C(N=CC2=CC=CC=C12)* 0.000 description 1
- 230000035484 reaction time Effects 0.000 description 1
- 206010038038 rectal cancer Diseases 0.000 description 1
- 210000000664 rectum Anatomy 0.000 description 1
- 201000001275 rectum cancer Diseases 0.000 description 1
- 210000003289 regulatory T cell Anatomy 0.000 description 1
- 206010039073 rheumatoid arthritis Diseases 0.000 description 1
- 229960004641 rituximab Drugs 0.000 description 1
- YGSDEFSMJLZEOE-UHFFFAOYSA-M salicylate Chemical compound OC1=CC=CC=C1C([O-])=O YGSDEFSMJLZEOE-UHFFFAOYSA-M 0.000 description 1
- 229960001860 salicylate Drugs 0.000 description 1
- 229930195734 saturated hydrocarbon Natural products 0.000 description 1
- 239000012047 saturated solution Substances 0.000 description 1
- 208000010157 sclerosing cholangitis Diseases 0.000 description 1
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 230000028327 secretion Effects 0.000 description 1
- ZLGIYFNHBLSMPS-ATJNOEHPSA-N shellac Chemical compound OCCCCCC(O)C(O)CCCCCCCC(O)=O.C1C23[C@H](C(O)=O)CCC2[C@](C)(CO)[C@@H]1C(C(O)=O)=C[C@@H]3O ZLGIYFNHBLSMPS-ATJNOEHPSA-N 0.000 description 1
- 239000004208 shellac Substances 0.000 description 1
- 235000013874 shellac Nutrition 0.000 description 1
- 229940113147 shellac Drugs 0.000 description 1
- 238000007086 side reaction Methods 0.000 description 1
- 230000011664 signaling Effects 0.000 description 1
- 201000000849 skin cancer Diseases 0.000 description 1
- 201000002314 small intestine cancer Diseases 0.000 description 1
- 239000001632 sodium acetate Substances 0.000 description 1
- 235000017281 sodium acetate Nutrition 0.000 description 1
- 235000010413 sodium alginate Nutrition 0.000 description 1
- 239000000661 sodium alginate Substances 0.000 description 1
- 229940005550 sodium alginate Drugs 0.000 description 1
- 239000012312 sodium hydride Substances 0.000 description 1
- 229910000104 sodium hydride Inorganic materials 0.000 description 1
- 235000009518 sodium iodide Nutrition 0.000 description 1
- 235000010288 sodium nitrite Nutrition 0.000 description 1
- 229940056729 sodium sulfate anhydrous Drugs 0.000 description 1
- 239000011877 solvent mixture Substances 0.000 description 1
- 238000001179 sorption measurement Methods 0.000 description 1
- 206010041823 squamous cell carcinoma Diseases 0.000 description 1
- 239000001119 stannous chloride Substances 0.000 description 1
- 235000011150 stannous chloride Nutrition 0.000 description 1
- 229940114926 stearate Drugs 0.000 description 1
- 201000011549 stomach cancer Diseases 0.000 description 1
- TYFQFVWCELRYAO-UHFFFAOYSA-L suberate(2-) Chemical compound [O-]C(=O)CCCCCCC([O-])=O TYFQFVWCELRYAO-UHFFFAOYSA-L 0.000 description 1
- 238000006467 substitution reaction Methods 0.000 description 1
- 239000007940 sugar coated tablet Substances 0.000 description 1
- 125000004434 sulfur atom Chemical group 0.000 description 1
- 230000001629 suppression Effects 0.000 description 1
- 208000024891 symptom Diseases 0.000 description 1
- 208000011580 syndromic disease Diseases 0.000 description 1
- 238000001308 synthesis method Methods 0.000 description 1
- 239000006188 syrup Substances 0.000 description 1
- 235000020357 syrup Nutrition 0.000 description 1
- 201000000596 systemic lupus erythematosus Diseases 0.000 description 1
- 125000005931 tert-butyloxycarbonyl group Chemical group [H]C([H])([H])C(OC(*)=O)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 150000003512 tertiary amines Chemical class 0.000 description 1
- 201000003120 testicular cancer Diseases 0.000 description 1
- FPGGTKZVZWFYPV-UHFFFAOYSA-M tetrabutylammonium fluoride Chemical compound [F-].CCCC[N+](CCCC)(CCCC)CCCC FPGGTKZVZWFYPV-UHFFFAOYSA-M 0.000 description 1
- 125000004192 tetrahydrofuran-2-yl group Chemical group [H]C1([H])OC([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000001712 tetrahydronaphthyl group Chemical group C1(CCCC2=CC=CC=C12)* 0.000 description 1
- 125000003831 tetrazolyl group Chemical group 0.000 description 1
- 125000000335 thiazolyl group Chemical group 0.000 description 1
- 125000004149 thio group Chemical group *S* 0.000 description 1
- 125000004571 thiomorpholin-4-yl group Chemical group N1(CCSCC1)* 0.000 description 1
- 201000002510 thyroid cancer Diseases 0.000 description 1
- 201000006134 tongue cancer Diseases 0.000 description 1
- 235000010487 tragacanth Nutrition 0.000 description 1
- 239000000196 tragacanth Substances 0.000 description 1
- 229940116362 tragacanth Drugs 0.000 description 1
- 229960000575 trastuzumab Drugs 0.000 description 1
- ITMCEJHCFYSIIV-UHFFFAOYSA-M triflate Chemical compound [O-]S(=O)(=O)C(F)(F)F ITMCEJHCFYSIIV-UHFFFAOYSA-M 0.000 description 1
- FTVLMFQEYACZNP-UHFFFAOYSA-N trimethylsilyl trifluoromethanesulfonate Chemical compound C[Si](C)(C)OS(=O)(=O)C(F)(F)F FTVLMFQEYACZNP-UHFFFAOYSA-N 0.000 description 1
- IIHPVYJPDKJYOU-UHFFFAOYSA-N triphenylcarbethoxymethylenephosphorane Chemical compound C=1C=CC=CC=1P(C=1C=CC=CC=1)(=CC(=O)OCC)C1=CC=CC=C1 IIHPVYJPDKJYOU-UHFFFAOYSA-N 0.000 description 1
- 230000005760 tumorsuppression Effects 0.000 description 1
- 125000002948 undecyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 201000005112 urinary bladder cancer Diseases 0.000 description 1
- 238000001291 vacuum drying Methods 0.000 description 1
- 235000015112 vegetable and seed oil Nutrition 0.000 description 1
- 239000008158 vegetable oil Substances 0.000 description 1
- 239000003981 vehicle Substances 0.000 description 1
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 1
- 229920002554 vinyl polymer Polymers 0.000 description 1
- 230000029812 viral genome replication Effects 0.000 description 1
- 239000003643 water by type Substances 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/55—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having seven-membered rings, e.g. azelastine, pentylenetetrazole
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/16—Drugs for disorders of the alimentary tract or the digestive system for liver or gallbladder disorders, e.g. hepatoprotective agents, cholagogues, litholytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/04—Drugs for skeletal disorders for non-specific disorders of the connective tissue
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
- A61P31/16—Antivirals for RNA viruses for influenza or rhinoviruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
- A61P31/18—Antivirals for RNA viruses for HIV
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/20—Antivirals for DNA viruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P33/00—Antiparasitic agents
- A61P33/02—Antiprotozoals, e.g. for leishmaniasis, trichomoniasis, toxoplasmosis
- A61P33/06—Antimalarials
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
- A61P5/14—Drugs for disorders of the endocrine system of the thyroid hormones, e.g. T3, T4
- A61P5/16—Drugs for disorders of the endocrine system of the thyroid hormones, e.g. T3, T4 for decreasing, blocking or antagonising the activity of the thyroid hormones
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/06—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D491/00—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00
- C07D491/02—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00 in which the condensed system contains two hetero rings
- C07D491/04—Ortho-condensed systems
- C07D491/044—Ortho-condensed systems with only one oxygen atom as ring hetero atom in the oxygen-containing ring
- C07D491/048—Ortho-condensed systems with only one oxygen atom as ring hetero atom in the oxygen-containing ring the oxygen-containing ring being five-membered
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D495/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having sulfur atoms as the only ring hetero atoms
- C07D495/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having sulfur atoms as the only ring hetero atoms in which the condensed system contains two hetero rings
- C07D495/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic Table
- C07F9/02—Phosphorus compounds
- C07F9/547—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom
- C07F9/6558—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom containing at least two different or differently substituted hetero rings neither condensed among themselves nor condensed with a common carbocyclic ring or ring system
- C07F9/65583—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom containing at least two different or differently substituted hetero rings neither condensed among themselves nor condensed with a common carbocyclic ring or ring system each of the hetero rings containing nitrogen as ring hetero atom
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Animal Behavior & Ethology (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Public Health (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Virology (AREA)
- Oncology (AREA)
- Communicable Diseases (AREA)
- Molecular Biology (AREA)
- Immunology (AREA)
- Epidemiology (AREA)
- Diabetes (AREA)
- Tropical Medicine & Parasitology (AREA)
- Endocrinology (AREA)
- Biochemistry (AREA)
- Physical Education & Sports Medicine (AREA)
- Rheumatology (AREA)
- Hematology (AREA)
- Pain & Pain Management (AREA)
- Dermatology (AREA)
- AIDS & HIV (AREA)
- Pulmonology (AREA)
- Obesity (AREA)
- Emergency Medicine (AREA)
- Biomedical Technology (AREA)
- Transplantation (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Biotechnology (AREA)
- Reproductive Health (AREA)
- Urology & Nephrology (AREA)
Abstract
Un compuesto de fórmula (I), un estereoisómero, un derivado isotópico estable o una sal farmacéuticamente aceptable del mismo; **(Ver fórmula)** en donde, L es -C(O)-, -C(S)- o -S(O)2-; L1 es un enlace; R se selecciona del grupo que consiste en R-1 a R-19: **(Ver fórmula)** cada uno de R7 y R7a se selecciona independientemente de hidrógeno, halógeno, alquilo, haloalquilo, alquenilo, alquinilo, Cy1, -L2-Cy1, -SRd, -ORd, -C(O)ORd, -C(O)Rd, - C(O)NRdRe, -C(O)NRdS(O)2Re, -C(=NH)Re, -C(=NH)NRdRe, -S(O)2Re y S(O)2NRdRe; en donde el alquilo, alquenilo o alquinilo no está sustituido o está sustituido en cualquier posición por uno o más de un sustituyente seleccionado del grupo que consiste en -CN, -NO2, -SRd, -ORd, -OC(O)Rd, -OC(O)ORd, -OC(O)NRdRe, -C(O)ORd, -C(O)Rd, -C(O)NRdRe, -C(O)NRdS(O)2Re, -NRdRe, -NRdC(O)Re, -N(Rd)C(O)ORe, -N(Rd)C(O)NRdRe, -NRdC(=NH)Re, -NRdC(=NH)NRdRe, -NRdS(O)2Re, -NRdS(O)2NRdRe, -N(Rd)C(O)N(Rd)S(O)2Re, -S(O)1-2Re, -S(O)2NRdRe, -S(O)(=NCN)Re, -S(O)(=NRd)Re, -S(O)(=NSO2Rd)Re, -S(O)2N(Rd)C(O)Re, -S(O)2N(Rd)C(O)NRdRe, -P(O)(ORd)2, -OP(O)(ORd)2 o -B(ORd)2; o R7 es **(Ver fórmula)** cada uno de R8 es independientemente hidrógeno, halógeno, alquilo, haloalquilo, haloalcoxi, alquenilo, alquinilo, Cy1, -L2-Cy1, -CN, -NO2, -SRd, -ORd, -OC(O)Rd, -OC(O)ORd, -OC(O)NRdRe, -C(O)ORd, -C(O)Rd, -C(O)NRdRe, -C(O)NRdS(O)2Re, -C(=NH)Re, -C(=NH)NRdRe, -NRdRe, -NRdC(O)Re, -N(Rd)C(O)ORe, -N(Rd)C(O)NRdRe, -NRdS(O)2Re, -NRdC(=NH)Re, -NRdC(=NH)NRdRe, -S(O)1-2Re, -S(O)2NRdRe y -NRdS(O)2NRdRe; en donde el alquilo, alquenilo o alquinilo no está sustituido o está sustituido en cualquier posición por uno o más de un sustituyente seleccionado del grupo que consiste en -CN, -NO2, -SRd, -ORd, -OC(O)Rd, -OC(O)ORd, -OC(O)NRdRe, -C(O)ORd, -C(O)Rd, -C(O)NRdRe, -C(O)NRdS(O)2Re, -NRdRe, -NRdC(O)Re, -N(Rd)C(O)ORe, -N(Rd)C(O)NRdRe, -NRdC(=NH)Re, -NRdC(=NH)NRdRe, -NRdS(O)2Re, -NRdS(O)2NRdRe, -N(Rd)C(O)N(Rd)S(O)2Re, -S(O)1-2Re, -S(O)2NRdRe, -S(O)2N(Rd)C(O)Re o -S(O) 2N(Rd)C(O)NRdRe; cada uno de n es independientemente 1, 2 ó 3; L2 es un enlace, alquenileno de C2-6, alquinileno de C2-6, o -(CRa1Rb1)m-; cada uno de Cy1 es independientemente cicloalquilo, heterocicloalquilo, arilo o heteroarilo; el Cy1 no está sustituido o está sustituido selectivamente en cualquier posición por uno o más de un sustituyente seleccionado del grupo que consiste en halógeno, alquilo, haloalquilo, haloalcoxi, hidroxialquilo, aminoalquilo, alquenilo, alquinilo, -CN, -NO2, -SRd2, -ORd2, -OC(O)Rd2, - OC(O)ORd2, -OC(O)NRd2Re2, -C(O)ORd2, -C(O)Rd2, -C(O)NRd2Re2, -NRd2Re2, -NRd2C(O)Re2, -N(Rd2)C(O)ORe2, -N(Rd2)C(O)NRd2Re2, -NRd2S(O)2Re2, -NRd2C(=NH)Re2, -NRd2C(=NH)NRd2Re2, -S(O)1-2Re2, -S(O)2NRd2Re2 y - NRd2S(O)2NRd2Re2; R1, R2 y R3 se seleccionan independientemente de hidrógeno, deuterio o alquilo; o, R2 y R3 junto con el átomo de C al que están unidos, forman un cicloalquilo de C3-8 o un heterocicloalquilo de 3 a 8 miembros; R4, R5 y R6 se seleccionan independientemente de hidrógeno, deuterio, halógeno, amino, ciano, nitro, alquilo de C1-6, haloalquilo de C1-6, haloalcoxi de C1-6, alcoxi de C1-6, alquenilo de C2-6, alquinilo de C2-6, hidroxialquilo de C1-6, cicloalquilo de C3-8, heterocicloalquilo de 3 a 8 miembros, fenilo o heteroarilo de 5 a 6 miembros; RA se selecciona de -ORd1 o -NRd1Re1; RB es -NRd1Re1; cada uno de Ra1 y Rb1 se selecciona independientemente de hidrógeno, deuterio, halógeno, alquilo de C1-6, alcoxi de C1-6, alquenilo de C2-6, alquinilo de C2-6, cicloalquilo de C3-8, heterocicloalquilo de 3 a 8 miembros, arilo de C6-10, heteroarilo de 5 a 6 miembros, cicloalquilo de C3-8 alquilo de C1-6, heterocicloalquilo de 3 a 8 miembros alquilo de C1-6, fenilo alquilo de C1-6 o heteroarilo de 5 a 6 miembros alquilo de C1-6; el alquilo de C1-6, alcoxi de C1-6, alquenilo de C2-6, alquinilo de C2-6, cicloalquilo de C3-8, heterocicloalquilo de 3 a 8 miembros, arilo de C6-10 o heteroarilo de 5 a 6 miembros no está sustituido o está sustituido en cualquier posición por 1 a 3 sustituyentes seleccionados del grupo que consiste en halógeno, hidroxilo, amino, carboxilo, alquenilo de C2-6, alquinilo de C2-6, alquilo de C1-6, haloalquilo de C1-6, alcoxi de C1-6 y haloalcoxi de C1-6; o Ra1 y Rb1 junto con el átomo de C al que están unidos, forman un cicloalquilo de C3-8 o un heterocicloalquilo de 3 a 8 miembros; cada uno de Rd, Re, Rd1 y Re1 se selecciona independientemente de hidrógeno, alquilo de C1-6, alquenilo de C2-6, alquinilo de C2-6, cicloalquilo de C3-8, heterocicloalquilo de 3 a 8 miembros, arilo de C6-10, heteroarilo de 5 a 6 miembros, cicloalquilo de C3-8 alquilo de C1-6, heterocicloalquilo de 3 a 8 miembros alquilo de C1-6, fenilo alquilo de C1-6 o heteroarilo de 5 a 6 miembros alquilo de C1-6; Rd, Re, Rd1 o Re1 no están sustituidos o están sustituidos en cualquier posición por 1 a 3 sustituyentes seleccionados del grupo que consiste en halógeno, hidroxilo, amino, carboxilo, alquilo de C1-6, alcoxi de C1-6, alquilamino de C1-6, haloalquilo de C1-6, haloalcoxi de C1-6, alquenilo de C2-6 y alquinilo de C2-6; o, Rd y Re, o Rd1 y Re1 junto con el átomo de N al que están unidos, forman un heterocicloalquilo de 3 a 8 miembros; el heterocicloalquilo puede comprender además de 1 a 3 heteroátomos seleccionados del grupo que consiste en N, O y S; el heterocicloalquilo no está sustituido o está sustituido en cualquier posición por 1 a 3 sustituyentes seleccionados del grupo que consiste en halógeno, amino, hidroxilo, carboxilo, ciano, alquilo de C1-6, haloalquilo de C1-6, alquenilo de C2-6, alquinilo de C2-6, alquilo de C1-3, amino alquilo de C1-3, -ORd2, -OC(O)Rd2, -OC(O)ORd2, -OC(O)NRd2Re2, -C(O)ORd2, -C(O)Rd2, -C(O)NRd2Re2, -NRd2Re2, - NRd2C(O)Re2, -N(Rd2)C(O)ORe2, -N(Rd2)C(O)NRd2Re2, -NRd2S(O)2Re2, -NRd2C(=NH)Re2, -NRd2C(=NH)NRd2Re2, -NRd2S(O)2NRd2Re2, -S(O)1-2Rd2 y -S(O)2NRd2Re2; cada uno de Rd2 y Re2 se selecciona independientemente de hidrógeno, alquilo de C1-6, alquenilo de C2-6, alquinilo de C2-6, cicloalquilo de C3-8, heterocicloalquilo de 3 a 8 miembros, arilo de C6-10, heteroarilo de 5 a 6 miembros, cicloalquilo de C3-8 alquilo de C1-6, heterocicloalquilo de 3 a 8 miembros alquilo de C1-6, fenilo alquilo de C1-6 o heteroarilo de 5 a 0 6 miembros alquilo de C1-6; Rd2 o Re2 no están sustituidos o están sustituidos en cualquier posición por 1 a 3 sustituyentes seleccionados del grupo que consiste en halógeno, hidroxilo, amino, carboxilo, alquilo de C1-6, alcoxi de C1-6, alquilamino de C1-6, haloalquilo de C1-6, haloalcoxi de C1-6, alquenilo de C2-6 y alquinilo de C2-6; o, Rd2 y Re2 junto con el átomo de N al que están unidos, forman un heterocicloalquilo de 3 a 8 miembros; cada uno de m es 1, 2, 3, 4, 5 o 6.
Description
d e s c r ip c ió n
Derivado de benzazepina, método de preparación, composición farmacéutica y uso del mismo
Campo de la invención
La presente invención se refiere a un derivado de benzazepina, un método de preparación, una composición farmacéutica y uso del mismo.
Técnicas anteriores
Los receptores tipo Toll (TLRs) son proteínas importantes que reconocen patrones moleculares asociados a patógenos, detectan e inician respuestas inmunitarias innatas y promueven el desarrollo de respuestas inmunitarias adaptativas. Los TLRs se expresan principalmente en células inmunitarias tales como las células dendríticas mieloides (mDC), las células dendríticas similares al plasma (pDC), los monocitos, las células B y los pulmones. En humanos, se considera que más de 10 TLRs tienen funciones significativas. TLR1, TLR2, TLR4, TLR5 y TLR6 se encuentran en la membrana celular y su función principal es reconocer ligandos macromoleculares extracelulares de bacterias y hongos. Por el contrario, TLR3, TLR7, TLR8 y TLR9 se encuentran en la membrana endosomal de las células, cuya función principal es reconocer los ácidos nucleicos exógenos de las células patógenas. Aunque la mayoría de los t Lr funcionan a través de vías de señalización específicas (principalmente a través de la vía dependiente de MyD88), diferentes TLRs pueden coordinar diferentes moléculas posteriormente. La adición de TLRs específicos conduce a la activación de diferentes poblaciones de células y la producción de diferentes citocinas y otros mediadores inflamatorios, lo que da como resultado diferentes respuestas inmunitarias. Para la realización, cuando se une a un ligando, TLR8 forma un dímero y los cambios conformacionales dan como resultado la participación de la proteína adaptadora MyD88, que recluta una quinasa asociada al receptor de interleucina-1, lo que lleva a la activación posterior de vías de señalización, incluyendo la proteína quinasa asociada a mitógeno y el factor de transcripción NF-k B.
Los TLRs, principalmente TLR7, TLR8 y TLR9, ubicados en connotaciones, se han considerado como nuevas dianas atractivas para la inmunoterapia contra el cáncer (Kanzler, et al, 2007; Kreig 2008; Smits, et al, 2008; Hennessy, et al, 2010; Kaczanowska, et al, 2013; Beesu, et al, 2016). Para la realización, TLR7 activa pDC en respuesta a infecciones virales, induce altos niveles de interferón alfa e induce respuestas de células T adaptativas de las células principales a antígenos virales endógenos (Liu, et al, 2009). En comparación con TLR7 y TLR9, TLR8 se expresa más ampliamente en células inmunitarias de diferentes subtipos. Las células T reguladoras (Treg) tienen una potente inhibición de la respuesta inmunitaria y son obstáculos importantes para la inmunoterapia eficaz contra el cáncer. Se ha demostrado que la vía de señalización de TLR8 es necesaria y suficiente para la reversión de la función supresora de células Treg y la supresión de un tumor fuerte. Los agonistas selectivos de TLR8 activan de manera efectiva una variedad de células inmunitarias, incluyendo mDCs y monocitos (Gorden, et al, 2005), y promueven respuestas inmunitarias adaptativas a las células cancerosas (Krug, et al, 2003; Schnurr, et al, 2005). Las mDCs activadas fagocitan las células tumorales muertas y apoptóticas, y luego presentan los antígenos asociados con el tumor de forma cruzada a CD8+ CTL de manera más eficaz que los pDCs (Berard, et al, 2000; Dalgaard, et al, 2005). Además, la activación de mDCs conduce a la liberación de TNFa e interleucina 12 (IL-12), lo que estimula la activación de células T y células NK. La activación de las células NK es el mecanismo principal de la citotoxicidad mediada por células dependiente de anticuerpos (ADCC). Por lo tanto, la muerte mejorada de células tumorales por parte de ADCC puede presentar una oportunidad terapéutica importante para los inhibidores selectivos de TLR8 (Lu, et al, 2011). Algunas terapias con anticuerpos monoclonales se utilizan ampliamente en pacientes con cáncer, tal como rituximab y trastuzumab, que pueden tratarse con ADCC (Ferris, et al, 2010). De hecho, la adición de agonistas de TLR8 a la terapia con mAb puede mejorar la ADCC y aumentar la eficacia de la terapia con mAb (Ferris, et al, 2015). Además, estudios recientes han demostrado que los agonistas de TLR8 tienen un efecto antitumoral directo, independiente de su función inmunomoduladora (Ignatz-Hoover, et al, 2015). Por lo tanto, los agonistas de TLR8 no solo pueden actuar como un solo fármaco para la terapia, sino que también pueden mejorar la respuesta inmunitaria del huésped para mejorar la eficacia de una variedad de quimioterapia y fármacos anticancerosos dirigidos.
Entre los miembros de la familia de TLRs que reconocen el ácido nucleico de microorganismos patógenos, TLR7 y TLR8 tienen una alta homología y pueden reconocer algunas pequeñas moléculas sintéticas con actividad antiviral, tal como las imidazoquinolinas (ligandos de TLR7 y TLR8). Las imidazoquinolinas se estudiaron en un modelo de herpes genital de cobayas infectadas con HSV y se encontró que tenían un efecto pequeño sobre la replicación viral in vitro, pero tenían un fuerte efecto in vivo, lo que indica que estos compuestos pueden promover las citoquinas proinflamatorias y regular las citoquinas en células inmunitarias y conducir a una respuesta antiviral (Int Immunopharmacol 2002; 2: 443-451). Más importante aún, TLR7 y TLR8 pueden reconocer ssRNA viral. Los estudios han demostrado que los virus ssRNA son ligandos naturales de TLR7 y TLR8, tal como el virus de la inmunodeficiencia humana (HIV) de tipo I, el virus de la gripe, el virus Sendai, el virus del dengue, el virus de la enfermedad de Newcastle (NDV), el virus de la estomatitis vesicular (VSV) y el virus de la hepatitis C (HCV), etc. TLR8 reconoce compuestos antivirales, virus ssRNA, oligonucleótidos sintéticos, etc., induce Th1, inhibe la secreción de citoquinas Th2 y la proliferación de Tregs a través de la vía de señalización dependiente de MyD88, media la inmunidad antiviral y ejerce efectos anti-infecciosos y anti-alérgicos.
La estructura cristalina de rayos X de la proteína TLR8 y sus miembros de la familia de TLR asociados se han estudiado
ampliamente, lo que promoverá aún más el diseño y la optimización de fármacos basados en la estructura.
El documento de Patente WO2011022508 describe un compuesto que tiene una fórmula de

y composiciones y métodos útiles para modular la señalización mediada por TLR7 y/o TLR8.
Por lo tanto, TLR8 es actualmente una diana terapéutica atractiva. Aunque ha habido más estudios sobre los TLRs, todavía hay grandes oportunidades para una mayor expansión de sus aplicaciones y ventajas. Los compuestos y aplicaciones descritos en la presente memoria contribuirán al desarrollo de agonistas de TLR8 para satisfacer necesidades clínicamente insatisfechas.
Contenido de la divulgación
El problema técnico a resolver en la presente divulgación es proporcionar un nuevo derivado de benzazepina, un método de preparación, una composición farmacéutica y el uso de los mismos. El derivado de benzazepina de la presente divulgación tiene un buen efecto regulador sobre los TLRs y puede tratar, aliviar y/o prevenir de forma eficaz varias enfermedades relacionadas provocadas por la inmunosupresión, tal como el cáncer, enfermedades autoinmunes, infección, inflamación, rechazo de trasplantes, injerto contra enfermedad del huésped, etc.
La presente invención proporciona un compuesto de fórmula (I), un estereoisómero, un derivado isotópico estable o una sale farmacéuticamente aceptable;

en donde, L es -C(O)-, -C(S)- o -S(O)2-;
L1 es un enlace;
R se selecciona del grupo que consiste en R-1 a R-19:

cada uno de R7 y R7a se selecciona independientemente de hidrógeno, halógeno, alquilo, haloalquilo, alquenilo, alquinilo, Cy1, -L2-Cy1, -SRd, -ORd, -C(O)ORd, -C(O)Rd, -C(O)NRdRe, -C(O)NRdS(O)2Re, -C(=NH)Re, -C(=NH)NRdRe, -S(O)2Re y S(O)2NRdRe; en donde el alquilo, alquenilo o alquinilo no está sustituido o está sustituido en cualquier posición por uno o más de un sustituyente seleccionado del grupo que consiste en -CN, -NO2 , -SRd, -ORd, -OC(O)Rd, -OC O ORd, -OC O NRdRe, -C O ORd, -C O Rd, -C O NRdRe, -C O NRdS O 2Re, -NRdRe, -NRdC O Re,

cada uno de R8 es independientemente hidrógeno, halógeno, alquilo, haloalquilo, haloalcoxi, alquenilo, alquinilo, Cy1, -L2-Cy1, -CN, -NO2 , -SRd, -ORd, -OC(O)Rd, -OC(O)ORd, -OC(O)NRdRe, -C(O)ORd, -C(O)Rd, -C(O)NRdRe, -C(O)NRdS(O)2Re, -C(=NH)Re, -C(=NH)NRdRe, -NRdRe, -NRdC(O)Re, -N(Rd)C(O)ORe, -N(Rd)C(O)NRdRe, -NRdS(O)2 Re, -NRdC(=NH)Re, -NRdC(=NH)NRdRe, -S(O)1-2Re, -S(O)2NRdRe y -NRdS(O)2NRdRe; en donde el alquilo, alquenilo o alquinilo no está sustituido o está sustituido en cualquier posición por uno o más de un sustituyente seleccionado del grupo que consiste en -CN, -NO2 , -SRd, -ORd, -OC(O)Rd, -OC(O)ORd, -OC(O)NRdRe, -C(O)ORd, -C(O)Rd, -C(O)NRdRe, - C(O)NRdS(O)2Re, -NRdRe, -NRdC(O)Re, -N(Rd)C(O)ORe, -N(Rd)C(O)NRdRe, -NRdC(=NH)Re, -NRdC(=NH)NRdRe, -NRdS(O)2Re, -NRdS(O)2NRdRe, -N(Rd)C(O)N(Rd)S(O)2Re, -S(O)1-2Re, -S(O)2NRdRe, -S(O)2N(Rd)C(O)Re o S(O)2N(Rd)C(O)NRdRe;
cada uno de n es independientemente 1,2 ó 3;
L2 es un enlace, alquenileno de C2-6, alquinileno de C2-6, o -(CRa1Rb1)m-;
cada uno de Cy1 es independientemente cicloalquilo, heterocicloalquilo, arilo o heteroarilo; el Cy1 no está sustituido o está sustituido selectivamente en cualquier posición por uno o más de un sustituyente seleccionado del grupo que consiste en halógeno, alquilo, haloalquilo, haloalcoxi, hidroxialquilo, aminoalquilo, alquenilo, alquinilo, -CN, -NO2 , -SRd2, -ORd2, -OC(O)Rd2, -OC(O)ORd2, -OC(O)NRd2Re2, -C(O)ORd2, -C(O)Rd2, -C(O)NRd2Re2, -NRd2Re2, -NRd2C(O)Re2, -N(Rd2)C(O)ORe2, -N(Rd2)C(O)NRd2Re2, -NRd2S(O)2Re2, -NRd2C(=NH)Re2, -NRd2C(=NH)NRd2Re2, -S(O)1-2Re2, -S(O)2NRd2Re2 y -NRd2S(O)2NRd2Re2;
R1, R2 y R3 se seleccionan independientemente de hidrógeno, deuterio o alquilo;
o, R2 y R3 junto con el átomo de C al que están unidos, forman un cicloalquilo de C3-8 o un heterocicloalquilo de 3 a 8 miembros;
R4 , R5 y R6 se seleccionan independientemente de hidrógeno, deuterio, halógeno, amino, ciano, nitro, alquilo de C1-6, haloalquilo de C1-6, haloalcoxi de C1-6, alcoxi de C1-6, alquenilo de C2-6, alquinilo de C2-6, hidroxialquilo de C1-6, cicloalquilo de C3-8, heterocicloalquilo de 3 a 8 miembros, fenilo o heteroarilo de 5 a 6 miembros;
RA se selecciona de -ORd1 o -NRd1 Re1;
RB es -NRd1Re1;
cada uno de Ra1 y Rb1 se selecciona independientemente de hidrógeno, deuterio, halógeno, alquilo de C1-6, alcoxi de C1-6, alquenilo de C2-6, alquinilo de C2-6, cicloalquilo de C3-8, heterocicloalquilo de 3 a 8 miembros arilo de C6-10,
heteroarilo de 5 a 6 miembros, cicloalquilo de C3-8 alquilo de C1-6, heterocicloalquilo de 3 a 8 miembros alquilo de C1-6, fenil alquilo de C1-6 o heteroarilo de 5 a 6 miembros alquilo de C1-6; el alquilo de C1-6, alcoxi de C1-6, alquenilo de C2-6, alquinilo de C2-6, cicloalquilo de C3-8, heterocicloalquilo de 3 a 8 miembros, arilo de C6-10 o heteroarilo de 5 a 6 miembros no están sustituidos o están sustituidos en cualquier posición por 1 a 3 sustituyentes seleccionados del grupo que consiste en halógeno, hidroxilo, amino, carboxilo, alquenilo de C2-6, alquinilo de C2-6, alquilo de C1-6, haloalquilo de C1-6, alcoxi de C1-6 y haloalcoxi de C1 - 6 ;
o Ra1 y Rb1 junto con el átomo de C al que están unidos, forman un cicloalquilo de C3-8 o un heterocicloalquilo de 3 a
8 miembros;
cada uno de Rd, Re, Rd1 y Re1 se seleccionan independientemente de hidrógeno, alquilo de C1-6, alquenilo de C2-6, alquinilo de C2-6, cicloalquilo de C3-8, heterocicloalquilo de 3 a 8 miembros, arilo de C6-10, heteroarilo de 5 a 6 miembros, cicloalquilo de C3-8, alquilo de C1-6, heterocicloalquilo de 3 a 8 miembros alquilo de C1-6, fenilo alquilo de C1-6 o heteroarilo de 5 a 6 miembros alquilo de C1-6; Rd, Re, Rd1 o Re1 no están sustituidos o están sustituidos en cualquier posición por 1 a 3 sustituyentes seleccionados del grupo que consiste en halógeno, hidroxilo, amino, carboxilo, alquilo de C1-6, alcoxi de C1-6, alquilamino de C1-6, haloalquilo de C1-6, haloalcoxi de C1-6, alquenilo de C2-6 y alquinilo de C2-6 ;
o, Rd y Re, o Rd1 y Re1 junto con el átomo de N al que están unidos, forman un heterocicloalquilo de 3 a 8 miembros;
el heterocicloalquilo puede comprender además de 1 a 3 heteroátomos seleccionados del grupo que consiste en N, O y S; el heterocicloalquilo no está sustituido o está sustituido en cualquier posición por 1 a 3 sustituyentes seleccionados del grupo que consiste en halógeno, amino, hidroxilo, carboxilo, ciano, alquilo de C1-6, haloalquilo de C1-6, alquenilo de
C2-6, alquinilo de C2-6, alquilo de C1-3, amino alquilo de C1-3, -ORd2, -OC(O)Rd2 , -OC(O)ORd2 , -OC(O)NRd2 Re2 , -C(O)ORd2, -C(O)Rd2, -C(O)NRd2Re2, -NRd2Re2, -NRd2C(O)Re2, - N(Rd2)C(O)ORe2, -N(Rd2)C(O)NRd2Re2, -NRd2S(O)2Re2, -NRd2C(=NH)Re2 , -NRd2C(=NH)NRd2 Re2 , -NRd2S(O)2NRd2 Re2 , -S(O)1-2Rd2 y -S(O)2NRd2 Re2 ;
cada uno de Rd2 y Re2 se selecciona independientemente de hidrógeno, alquilo de C1-6, alquenilo de C2-6, alquinilo de
C2-6, cicloalquilo de C3-8, heterocicloalquilo de 3 a 8 miembros, arilo de C6-10, heteroarilo de 5 a 6 miembros, cicloalquilo de C3-8 alquilo de C1-6, heterocicloalquilo de 3 a 8 miembros alquilo de C1-6, fenilo alquilo de C1-6 o heteroarilo de 5 a
6 miembros alquilo de C1-6; Rd2 o Re2 no están sustituidos o están sustituidos en cualquier posición por 1 a 3 sustituyentes seleccionados del grupo que consiste en halógeno, hidroxilo, amino, carboxilo, alquilo de C1-6, alcoxi de
C1-6, alquilamino de C1-6, haloalquilo de C1-6, haloalcoxi de C1-6, alquenilo de C2-6 y alquinilo de C2-6 ; o, Rd2 y Re2 junto con el átomo de N al que están unidos, forman un heterocicloalquilo de 3 a 8 miembros;
cada uno de m es 1, 2, 3, 4, 5 o 6.
En una realización preferida de la presente invención, R1 es preferiblemente H.
En una realización preferida de la presente invención, R2 es preferiblemente H.
En una realización preferida de la presente invención, R3 es preferiblemente H.
En una realización preferida de la presente invención, cada uno de R4 , R5 y R6 se selecciona preferiblemente de H, D, F, Cl, Br, -CH3 , -OCH3 , -CF3 , -CH2 F, -CHF2 , -OCF3, -CN o -NH2 ;
en otra realización preferida de la presente invención, cada uno de R4 , R5 y R6 se selecciona preferiblemente de H, I,
F, Cl, Br, -CH3 , -OCH3 , -CF3 , -CH2 F, -CHF2 , -OCF3, -CN o -NH2 ;
en otra realización preferida de la presente invención, cada uno de R4 , R5 y R6 se selecciona preferiblemente de H, F,
Cl, Br, -CH3 , -OCH3 , -CF3 , -CH2 F, -CHF2 , -OCF3 , -CN o -NH2.
En una realización preferida de la presente invención, en RA, cada uno de Rd1 y Re1 se selecciona preferiblemente de hidrógeno, alquilo de C1-6, haloalquilo de C1-6, cicloalquilo de C3-8 o heterocicloalquilo de 3 a 8 miembros; Rd1 o Re1 no están sustituidos o están sustituidos selectivamente en cualquier posición por un hidroxilo.
o Rd1 y Re1 junto con el átomo de N al que están unidos, forman un heterocicloalquilo de C3-8; el heterocicloalquilo puede contener además 1-3 heteroátomos seleccionados del grupo que consiste en N, O y S; el heterocicloalquilo no está sustituido o está sustituido adicionalmente en cualquier posición por un sustituyente seleccionado del grupo que consiste en alquilo de C1-4, haloalquilo de C1-3, o aminoalquilo de C1-3.
En una realización preferida de la presente invención, RB es preferiblemente -NH2.
En una realización preferida de la presente invención, R se selecciona preferiblemente del grupo que consiste en R-1 a R-11 y R-13 a R-16; cada uno de R7 y R7a se define preferiblemente como antes, en el que el alquilo, alquenilo o alquinilo en la definición de R7 o R7a no está sustituido o está sustituido selectivamente en cualquier posición por uno o más de un sustituyente como se describe anteriormente excepto por -S(O)(=NCN)Re, -S(O)(=NRd)Re, -S(O)(=NSO2 Rd)Re, -P(O)(ORd)2 , -OP(O)(ORd)2 o -B(ORd)2 ; R8 se define preferiblemente como antes.
En una realización preferida de la presente invención, R se selecciona preferiblemente del grupo que consiste en R-1 a R-16; cada uno de R7 y R7a se define preferiblemente como antes, en el que el alquilo, alquenilo o alquinilo en la
definición de R7 o R7a no está sustituido o está sustituido selectivamente en cualquier posición por uno o más de un sustituyente excepto por -P(O)(ORd)2 , -OP(O)(ORd)2 o -B(ORd)2 ; R8 se define preferiblemente como anteriormente.
En una realización preferida de la presente invención, R se selecciona preferiblemente del grupo que consiste en R-1 a R-18; cada uno de R7 y R7a se define preferiblemente como antes.
En una realización preferida de la presente invención, en la definición de R, R7 es preferiblemente hidrógeno, alquilo de C1-6, haloalquilo de C1-6, alquenilo de C2-6, alquinilo de C2-6, Cy1, -L2-Cy1, - SRd, -ORd, -C(O)ORd, -C(O)Rd, -C(O)NRdRe, -C(=NH)Re, -C(=NH)NRdRe, -S(O)2 Re o -S(O)2NRdRe; en donde el alquilo de C1-6, alquenilo de C2-6 o alquinilo de C2-6 no están sustituidos o están sustituidos selectivamente en cualquier posición por 1 a 3 sustituyentes seleccionados del grupo que consiste en -CN, -NO2 , -SRd, -ORd, -OC(O)Rd, -OC(O)ORd, -OC(O)NRdRe, -C(O)ORd, -C(O)Rd, -C(O)NRdRe, -NRdRe, -NRdC(O)Re, -N(Rd)C(O)ORe, -N(Rd)C(O)NRdRe, -NRdS(O)2Re, -NRdC(=NH)Re, -NRdC(=NH)NRdRe, -S(O)1-2Re, -S(O)(=NCN)Re, -S(O)(=NRd)Re, -S(O)(=NSO2Rd)Re, -S(O)2NRdRe, -NRdS(O)2NRdRe, -P(O)(ORd)2 , -OP(O)(ORd)2 o -B(ORd)2 ; L2 , Cy1, Rd y Re se definen como anteriormente.
En una realización preferida de la presente invención, en la definición de R, R7 preferiblemente se define como anteriormente, en donde el alquilo de C1-6, alquenilo de C2-6 o alquinilo de C2-6 no están sustituidos o están sustituidos selectivamente en cualquier posición por 1 a 3 sustituyentes como se define anteriormente distintos de -S(O)(=NCN)Re, -S(O)(=NRd)Re, -S(O)(=NSO2Rd)Re, -P(O)(ORd)2, -OP(O)(ORd)2 o -B(ORd)2.
En otra realización preferida de la invención, en la definición de R, R7 se define preferiblemente como anteriormente, en el que el alquilo de C1-6, alquenilo de C2-6 o alquinilo de C2-6 no están sustituidos o están sustituidos selectivamente en cualquier posición por 1 a 3 sustituyentes distintos de -P(O)(ORd)2 , -OP(O)(ORd)2 o -B(ORd)2.
En una realización preferida de la presente invención, en la definición de R, R7 es preferiblemente hidrógeno, metilo, etilo, propilo, butilo, isopropilo, isobutilo, n-butilo, neopentilo, terc-amilo, ciclopropilo, ciclobutilo, ciclopentilo, ciclohexilo,

En una realización preferida de la presente invención, en la definición de R, R7 es más preferiblemente
En una realización preferida de la presente invención, en la definición de R, R7 es más preferiblemente bencilo,

En otra realización preferida de la presente invención, en la definición de R, R7 es más preferiblemente bencilo, piridilo, pirimidinilo,

En una realización preferida de la invención, en la definición de R, R7 es más preferiblemente

En una realización preferida de la presente invención, en la definición de R, R7a es preferiblemente hidrógeno, alquilo de C1-6, haloalquilo de C1-6, alquenilo de C2-6, alquinilo de C2-6, Cy1 o -L2-Cy1, en donde el alquilo de C1-6, alquenilo de C2-6 o alquinilo de C2-6 no está sustituido o está sustituido selectivamente en cualquier posición por 1 a 3 sustituyentes seleccionados del grupo que consiste en -CN, -NO2 , -SRd, -ORd, -OC(O)Rd, -OC(O)ORd, -OC(O)NRdRe, -C(O)ORd, -C(O)Rd, -C(O)NRdRe, -NRdRe, -NRdC(O)Re, -N(Rd)C(O)ORe, -N(Rd)C(O)NRdRe, -NRdS(O)2Re, -NRdC(=NH)Re, -NRdC(=NH)NRdRe, -S(O)1-2Re, -S(O)2NRdRe y -NRdS(O)2NRdRe; L2 , Cy1, Rd y Rei se definen como anteriormente. En una realización preferida de la presente invención, en la definición de R, R7a es más preferiblemente hidrógeno o alquilo de C1-6 (tal como, metilo o etilo).
En una realización preferida de la presente invención, en la definición de R, R8 es más preferiblemente hidrógeno, metilo, etilo, n-propilo, terc-butilo, isopropilo, isobutilo, n-butilo, -CN, -NO2 , -NH2 , -CF3 , -OCF3 , -CH3 , -OCH3 , F, Cl o Br; o R8 es preferiblemente un sustituyente distinto de -OCH3.
En la definición de R, n es preferentemente 1 ó 2; n es más preferiblemente 1.
En una realización preferida de la presente invención, L2 es preferiblemente un enlace o -(CRa1Rb1)m-.
En otra realización preferida de la presente invención, L2 es preferiblemente un enlace, -CH2-, -CH2CH2-, -CH2-CH2CH2-, -CH2CH2CH2CH2-, -CH2CH(CH2)CH2- o

En otra realización preferida de la presente invención, L2 es preferiblemente un enlace, -CH2-, -CH2CH2-, -CH2CH2CH2-, -CH2CH2CH2CH2- o -CH2CH(CH2)CH2-.
En una realización preferida de la presente invención, Rd, Re, Rd1, Re1, Rd2 o Re2 están preferiblemente no sustituidos o sustituidos selectivamente en cualquier posición por 1 a 3 sustituyentes seleccionados del grupo que consiste en halógeno, hidroxilo, amino, carboxilo, alquilo de C1-4, haloalquilo de C1-3, haloalcoxi de C1-3, alquenilo de C2-6 y alquinilo de C2-6.
En una realización preferida de la presente invención, Rd, Re, Rdi, Reí, Rd2 o Re2 están preferiblemente no sustituidos o sustituidos selectivamente en cualquier posición por 1 a 3 sustituyentes seleccionados del grupo que consiste en halógeno, hidroxilo, amino, carboxilo, alquilo de C1-4, haloalquilo de C1-3, haloalcoxi de C1-3, alcoxi de C1-3, alquilamino de C1-3, alquenilo de C2-6 y alquinilo de C2-6.
En una realización preferida de la presente invención, Rd y Re, o Rd1 y Re1, o Rd2 y Re2 junto con el átomo de N al que están unidos, forman un heterocicloalquilo de 3 a 8 miembros; el heterocicloalquilo contiene además de 1 a 3 heteroátomos seleccionados del grupo que consiste en N, O y S; el heterocicloalquilo está preferiblemente no sustituido o sustituido en cualquier posición por 1 a 3 sustituyentes seleccionados del grupo que consiste en halógeno, amino, carboxilo, ciano, alquilo de C1-4, haloalquilo de C1-3, haloalcoxi de C1-3, alquenilo de C2-6, alquinilo de C2-6, hidroxialquilo, aminoalquilo, -ORd2 , -OC(O)Rd2 , -OC(O)ORd2 , -OC(O)NRd2Re2 , -C(O)ORd2 , -C(O)Rd2 , -C(O)NRd2Re2 , -NRd2Re2, -NRd2C(O)Re2, -N(Rd2)C(O)ORe2, -N(Rd2)C(O)NRd2Re2, -NRd2S(O)2Re2, -NRd2C(=NH)Re2, -NRd2C(=NH)NRd2Re2 , -S(O)1-2Rd2 y -S(O)2NRd2Re2.
El compuesto de fórmula (I), el estereoisómero, el derivado isotópico estable o la sal farmacéuticamente aceptable del mismo es preferiblemente un compuesto de fórmula (IA), un estereoisómero, un derivado isotópico estable o la sal farmacéuticamente aceptable del mismo;

en donde,
es un enlace simple o un enlace doble, U es N, C(Rs) o C(O), V es N, C(Rs) o N(R7a);
— , U y V son cualquier combinación de lo siguiente:
es un doble enlace, U es N, V es C(Rs);
es un doble enlace, U es C(Rs), V es N;
es un doble enlace, U es C(Rs), V es C(Rs);
es un enlace sencillo, U es C(Rs), V es C(Rs);
es un enlace sencillo, U es C(O), V es N(R7a);
Y es O o S;
n es 1;
L1, Rd1, Re1, R4 , R5 , R6, R7 , R7a y R8 se definen como anteriormente.
El compuesto de fórmula (I), el estereoisómero, el derivado isotópico estable o la sal farmacéuticamente aceptable del mismo es preferiblemente un compuesto de fórmula (IB) o (IC), un estereoisómero, un derivado isotópico estable o la sal farmacéuticamente aceptable del mismo;

en donde, es un enlace simple o un enlace doble;
Rd1, Re1, R4 , R5 , R6, R7 , R8 e Y se definen como anteriormente.
En una realización preferida de la presente invención, en el compuesto de fórmula (IB) o (IC), el estereoisómero, el derivado isotópico estable o la sal farmacéuticamente aceptable, Y es preferiblemente O.
El compuesto de fórmula (I), el estereoisómero, el derivado isotópico estable o la sal farmacéuticamente aceptable del mismo es preferiblemente un compuesto de fórmula (ID), (IE), (IF), (IG), (IH) o (II), un estereoisómero, un derivado isotópico estable o una sal farmacéuticamente aceptable del mismo:

en donde, Rd1, Re1, R4 , R5 , R6, R7 , R7a, R8 e Y se definen como anteriormente.
En una realización preferida de la presente invención, en el compuesto de fórmula (ID) o (IF), el estereoisómero, un derivado isotópico estable o la sal farmacéuticamente aceptable del mismo, Y es preferiblemente O.
El compuesto de fórmula (I), el estereoisómero, el derivado isotópico estable o la sal farmacéuticamente aceptable del mismo es preferiblemente un compuesto de fórmula (IJ), un estereoisómero, un derivado isotópico estable o una sal farmacéuticamente aceptable del mismo:

en donde, es un enlace simple o un enlace doble; Z es N(F¡7a) o S;
n es 1 ó 2;
L1, Rd1, Re1, R4 , R5 , R6, R7 , R7a y R8 se definen como anteriormente.
El compuesto de fórmula (I), el estereoisómero, el derivado isotópico estable o la sal farmacéuticamente aceptable del mismo es preferiblemente un compuesto de fórmula (IK) o (IL), un estereoisómero, un derivado isotópico estable o una sal farmacéuticamente aceptable del mismo:
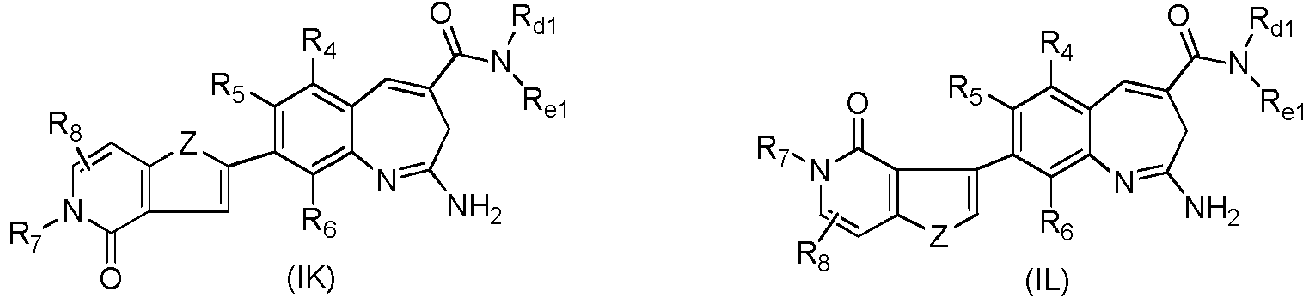
en donde, Z es NH, N(CH3) o S;
Rdi, Reí, R4, Rs, R6, R7 y R8 se definen como anteriormente.
El compuesto de fórmula (I), el estereoisómero, el derivado isotópico estable o la sal farmacéuticamente aceptable del mismo se seleccionan preferiblemente del grupo que consiste en:









La presente invención también proporciona un método de preparación del compuesto de fórmula (I), el estereoisómero, el derivado isotópico estable o la sal farmacéuticamente aceptable del mismo,
en donde, el método de preparación es uno cualquiera de los siguientes métodos:
Método 1 que comprende realizar una reacción de acoplamiento de Suzuki con el compuesto I-a y
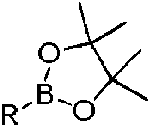
para obtener el compuesto de fórmula (I);

Método 2 que comprende realizar una reacción de acoplamiento de Suzuki con el compuesto I-b y R-M para obtener el compuesto de fórmula (I);

en donde, M es bromo, cloro, yodo o -OS(O)2CF3; R1, R2 , R3 , R4 , R5 , R6, RA, RB, R y L se definen como anteriormente.
En el método 1 y el método 2, las condiciones para la reacción de acoplamiento de Suzuki pueden ser las condiciones convencionales en este tipo de reacción en la técnica, y las siguientes condiciones de reacción son particularmente preferidas en la presente invención: bajo nitrógeno, en una mezcla de disolventes (tal como, tetrahidrofurano/agua, acetonitrilo/agua, o A/,W-dimetilformamida/agua), base (carbonato de potasio, carbonato de cesio o dietilisopropilamina) y catalizador (preferiblemente dicloruro de [1,1'-bis(difenilfosfino)ferroceno] paladio (Pd(dppf)CL) o el complejo de diclorometano cloruro de [1,1'-bis(difenilfosfino)ferroceno] paladio (Pd(dppf)2Cl2), la mezcla de disolventes se usa preferiblemente en una cantidad de 1 a 50 mL/mmol de compuesto Ia o Ib, y el tiempo de reacción es preferiblemente de 0 a 24 horas, y la temperatura es preferiblemente de temperatura ambiente a reflujo, más preferiblemente de 60 a 90°C, y la relación molar del compuesto Ia o Ib al catalizador es preferiblemente de 1:0,01 a 1:0,1.
En la etapa final del método de preparación, puede usarse un sistema ácido tal como ácido p-toluenosulfónico, ácido clorhídrico, cloruro de hidrógeno o ácido trifluoroacético, etc., o en el proceso de purificación, por ejemplo, cuando el sistema ácido mencionado anteriormente está presente en la fase móvil del HPLC preparativo, el compuesto de fórmula (I) será el correspondiente p-toluenosulfonato, clorhidrato o trifluoroacetato, etc.
En el método anterior, cuando un amino, un hidroxilo o un carboxilo están presentes en el compuesto de fórmula I-a, I-b,

o R-M, el amino, hidroxilo o carboxilo se puede proteger mediante un grupo protector para evitar reacciones secundarias. Si está presente el grupo protector de amino o el grupo protector de hidroxi, es necesario someterse a una etapa de desprotección posterior para dar el compuesto I-1. Puede usarse cualquier grupo protector de amino adecuado, por ejemplo, un terc-butoxicarbonilo (Boc), para proteger el amino. Si se utiliza Boc como grupo protector, las reacciones de desprotección posteriores se pueden llevar a cabo en condiciones estándar, por ejemplo, sistema de ácido p-toluenosulfónico/metanol, sistema de diclorometano/ácido trifluoroacético, solución saturada de cloruro de hidrógeno en éter o el sistema de trifluorometanosulfonato de trimetilsililo/2,6- -lutidina/diclorometano; cualquier grupo protector de hidroxilo adecuado, tal como un bencilo, puede usarse para proteger el grupo amino, y las reacciones de desprotección posteriores pueden llevarse a cabo en condiciones estándar, por ejemplo, paladio sobre carbono/hidrógeno; cualquier grupo protector de carboxilo adecuado, por ejemplo, para formar un grupo carboxilato (tal como carboxilato de metilo, carboxilato de etilo), puede usarse para proteger el grupo carboxilo, y la desprotección posterior puede llevarse a cabo en condiciones estándar, tal como usar hidróxido de sodio, hidróxido de potasio, hidróxido de litio para desproteger en tetrahidrofurano, agua y/o metanol. La desprotección anterior se lleva a cabo preferiblemente en la última etapa.
El compuesto de fórmula (I), o la sal farmacéuticamente aceptable del mismo, puede sintetizarse según un método general de síntesis química.
En general, la preparación de la sal puede realizarse haciendo reaccionar la base libre o el ácido con un equivalente químico equivalente o un exceso de un ácido (ácido inorgánico u orgánico) o una base (base inorgánica u orgánica) en un disolvente adecuado o composición de disolvente.
La presente invención también proporciona una composición farmacéutica, que comprende un componente seleccionado del grupo que consiste en la cantidad terapéuticamente eficaz del compuesto de fórmula (I), el estereoisómero, el derivado isotópico estable y la sal farmacéuticamente aceptable del mismo, y un excipiente farmacéuticamente estable.
La composición farmacéutica también puede incluir otros agentes terapéuticos para el tratamiento, alivio y/o prevención del cáncer, infecciones virales o enfermedades autoinmunes.
En la composición farmacéutica, el excipiente farmacéuticamente aceptable puede incluir un vehículo, diluyente y/o excipiente farmacéuticamente aceptable.
Según el propósito del tratamiento, la composición farmacéutica se puede formular en varios tipos de dosis unitarias, tales como comprimidos, píldoras, polvos, líquidos, suspensiones, emulsiones, gránulos, cápsulas, supositorios e inyecciones (soluciones y suspensiones) y similares, y preferiblemente líquidos, suspensiones, emulsiones, supositorios e inyecciones (soluciones y suspensiones), etc.
Para dar forma a la composición farmacéutica en forma de comprimido se puede utilizar cualquier excipiente conocido y ampliamente utilizado en la técnica. Por ejemplo, vehículos tales como lactosa, azúcar blanco, cloruro de sodio, glucosa, urea, almidón, carbonato de calcio, caolín, celulosa cristalina y ácido silícico; aglutinantes tales como agua, etanol, propanol, jarabe común, solución de dextrosa, solución de almidón, solución de gelatina, carboximetilcelulosa, goma laca, metilcelulosa y fosfato de potasio, polivinilpirrolidona, etc.; agentes disgregantes, tales como almidón seco, alginato de sodio, polvo de agar, polvo de algas marinas, bicarbonato de sodio, carbonato de calcio, polietilensorbitán, laurilsulfato de sodio, estearato de monoglicerilo, almidón y lactosa; inhibidores de disgregación tales como azúcar blanco, triestearato de glicerilo, aceite de coco y aceite de hidrogenación; promotores de adsorción tales como bases de amonio cuaternario y laurilsulfato de sodio; agentes humectantes tales como glicerina, almidón, etc.; adsorbentes tales como almidón, lactosa, caolín, bentonita y ácido silícico coloidal; tal como talco puro, estearato, polvo de ácido bórico y polietilenglicol. También es posible usar un material de recubrimiento habitual para formular un comprimido recubierto de azúcar, un comprimido de película de gelatina, un comprimido de cubierta, un comprimido recubierto con película, un comprimido de película de dos capas y un comprimido multicapa.
Para dar forma a la composición farmacéutica en forma de píldora se puede utilizar cualquier excipiente conocido y ampliamente utilizado en la técnica, por ejemplo, vehículos tales como lactosa, almidón, aceite de coco, aceite vegetal endurecido, caolín y talco, etc.; aglutinantes tales como polvo de goma arábiga, polvo de tragacanto, gelatina y etanol, etc.; agentes disgregantes tales como agar y polvo de algas marinas.
Para dar forma a la composición farmacéutica en forma de supositorio se puede utilizar cualquier excipiente conocido y ampliamente utilizado en la técnica, por ejemplo, polietilenglicol, aceite de coco, alcoholes superiores, ésteres de alcoholes superiores, gelatina y glicéridos semisintéticos, etc.
Para la preparación de una composición farmacéutica en forma de inyección, la solución o suspensión puede esterilizarse (preferiblemente añadiendo una cantidad apropiada de cloruro de sodio, glucosa o glicerina, etc.) para preparar una inyección que es isotónica con la sangre. También se puede utilizar cualquiera de los vehículos comúnmente utilizados en la técnica. Por ejemplo, agua, etanol, propilenglicol, alcohol isoestearílico etoxilado, alcohol isoestearílico polioxilado y ésteres de ácidos grasos de polietilensorbitán. Además, se pueden añadir solubilizantes, tampones, analgésicos, etc. habituales.
En la presente invención, el contenido del compuesto de fórmula (I), el estereoisómero, el derivado isotópico estable y la sal farmacéuticamente aceptable del mismo, y/o el otro agente terapéutico comprendido en la composición farmacéutica no tiene limitación específica, que puede seleccionarse dentro de un amplio intervalo, normalmente del 5 % al 95 % en peso, preferiblemente del 30 % al 80 % en peso.
En la presente invención, el método de administración de la composición farmacéutica no está particularmente limitado. Se pueden seleccionar formulaciones de varias formas de dosificación dependiendo de la edad, sexo y otras condiciones y síntomas del paciente. Por ejemplo, los comprimidos, píldoras, soluciones, suspensiones, emulsiones, gránulos o cápsulas se administran por vía oral; las inyecciones se pueden administrar solas o en combinación con soluciones inyectables (por ejemplo, solución de glucosa y solución de aminoácidos); los supositorios se administran en el recto.
La divulgación también proporciona el uso del compuesto de fórmula (I), el estereoisómero, el derivado isotópico estable o la sal farmacéuticamente aceptable del mismo para su uso como modulador de TLRs. El modulador de TLRs es preferiblemente un agonista de TLRs o un agonista parcial de TLRs. Los TLRs se seleccionan preferiblemente del grupo que consiste en TLR7, TLR8 y TLR9.
La presente divulgación también proporciona el compuesto de fórmula (I), el estereoisómero, el derivado isotópico estable o la sal farmacéuticamente aceptable del mismo para su uso en la modulación de células T y otras células inmunitarias.
La presente divulgación también proporciona el compuesto de fórmula (I), el estereoisómero, el derivado isotópico estable o la sal farmacéuticamente aceptable del mismo para el tratamiento, alivio y/o prevención de una enfermedad mediada por TLRs; la presente invención proporciona preferiblemente un uso del compuesto de fórmula (I), el estereoisómero, el derivado isotópico estable o la sal farmacéuticamente aceptable del mismo para su uso en el tratamiento, alivio y/o prevención de enfermedades relacionadas mediadas por TLR8; las enfermedades incluyen tumores y enfermedades no neoplásicas. Tales enfermedades incluyen, pero no se limitan a, cáncer, virus y otras infecciones, enfermedades causadas por inmunosupresión y enfermedades autoinmunes, etc.
La presente divulgación proporciona preferiblemente el compuesto de fórmula (I), el estereoisómero, el derivado isotópico estable o la sal farmacéuticamente aceptable del mismo para su uso en el tratamiento y/o alivio de cánceres. Los cánceres están preferiblemente asociados con la inhibición inmunológica, que se refiere a la inmunosupresión específica del tumor.
La presente divulgación proporciona además el compuesto de fórmula (I), el estereoisómero, el derivado isotópico estable, la sal farmacéuticamente aceptable del mismo o la composición farmacéutica para su uso en un método para tratar cánceres, infecciones virales y de otro tipo, o enfermedades autoinmunes, comprendiendo el método administrar una cantidad terapéuticamente requerida del compuesto de fórmula (I), el estereoisómero, el derivado isotópico estable o la sal farmacéuticamente aceptable del mismo, o la composición farmacéutica, a un mamífero.
El mamífero es preferiblemente un ser humano.
La presente divulgación proporciona además el compuesto de fórmula (I), el estereoisómero, el derivado isotópico estable o la sal farmacéuticamente aceptable del mismo para su uso en combinación con uno o más de otros agentes terapéuticos y/o métodos terapéuticos para el tratamiento, alivio y /o prevención de enfermedades relacionadas mediadas por TLRs, en particular por TLR8. La enfermedad relacionada mediada por TLR8 se refiere a una enfermedad causada por inmunosupresión mediada por TLR8, que puede incluir cánceres, infecciones virales, inflamaciones, enfermedades autoinmunes, rechazos de trasplantes, enfermedades de trasplante contra huésped, etc.
La presente divulgación proporciona preferiblemente el compuesto de fórmula (I), el estereoisómero, el derivado isotópico estable o la sal farmacéuticamente aceptable del mismo para su uso en combinación con uno o más de otros agentes terapéuticos y/o métodos terapéuticos para el tratamiento y/o alivio de enfermedades mediadas por TLR8, preferiblemente cánceres.
El otro agente terapéutico (tal como, otros agentes terapéuticos para el tratamiento de cánceres) puede formularse en una forma de dosificación única con el compuesto de fórmula (I) o administrarse secuencialmente.
Las infecciones virales pueden incluir: la infección causada por el virus de la gripe, el virus Sendai, el virus Coxsackie, el virus del dengue, el virus de la enfermedad de Newcastle (NDV), el virus de la estomatitis vesicular (VSV), el virus de la hepatitis C (HCV), las infecciones papilares humanas causadas por virus tales como el virus neoplásico (HPV), el citomegalovirus (CMV), el virus de Epstein-Barr (EBV), el poliovirus, el virus de la varicela-zóster o el virus de la inmunodeficiencia humana de tipo I (HIV), etc.
Dichos cánceres incluyen cánceres metastásicos y no metastásicos, así como cánceres hereditarios y esporádicos familiares, y también pueden incluir tumores sólidos y tumores no sólidos.
Las realizaciones específicas del tumor sólido pueden incluir, pero no se limitan a, cánceres seleccionados del grupo que consiste en cáncer de ojo, cáncer de hueso, cáncer de pulmón, cáncer de estómago, cáncer de páncreas, cáncer de mama, cáncer de próstata, cáncer de cerebro (incluyendo glioma maligno, meduloblastoma), cáncer de ovario, cáncer de vejiga, cáncer de cuello uterino, cáncer de testículo, cáncer de riñón (incluyendo el adenocarcinoma y el cáncer de nefroblastos), cáncer bucal (incluyendo el carcinoma de células escamosas), cáncer de lengua, cáncer de laringe, cáncer de nasofaringe, cáncer de cabeza y cuello, cáncer de colon, cáncer de intestino delgado, cáncer de recto, cáncer de paratiroides, cáncer de tiroides, cáncer de esófago, cáncer de vesícula biliar, colangiocarcinoma, cáncer de cuello uterino, cáncer de hígado, cáncer de pulmón, sarcoma y cáncer de piel.
Las realizaciones específicas de los tumores no sólidos (incluyendo los tumores hematológicos) pueden incluir, pero no se limitan a, uno o más de leucemia linfocítica (incluyendo la leucemia linfocítica aguda, linfoma, mieloma, leucemia linfocítica crónica, linfoma de Hodgkin, linfoma no Hodgkin, leucemia linfocítica crónica de células T, leucemia linfocítica crónica de células B), leucemia asociada a mieloide (incluyendo leucemia mieloide aguda, leucemia mieloide crónica) y leucemia asociada a AIDs.
Las enfermedades autoinmunes pueden incluir, pero no se limitan a, artritis reumatoide, lupus eritematoso sistémico, enfermedad mixta del tejido conjuntivo (MCTD), esclerodermia sistémica (incluyendo: síndrome CREST), dermatomiositis, vasculitis nodular segmentaria, nefropatía (incluyendo: síndrome de nefritis hemorrágica pulmonar, glomerulonefritis aguda, glomerulonefritis proliferativa de membrana primaria, etc.), enfermedades relacionadas con el sistema endocrino (incluyendo: diabetes de tipo I, insuficiencia gonadal, anemia perniciosa, hipertiroidismo, etc.),
enfermedad hepática (incluyendo: cirrosis biliar primaria, colangitis autoinmune, hepatitis autoinmune, colangitis esclerosante primaria, etc.) y respuesta autoinmune debido a la infección (tal como: uno o más de AIDs, malaria, etc.).
En la presente invención, a menos que se indique lo contrario, el término "sustituido selectivamente en cualquier posición por un sustituyente seleccionado del grupo que consiste en" se refiere a que uno o más de un átomo de hidrógeno unido a uno o más de un átomo indicado se reemplaza con el indicado sustituyente, siempre que el enlace unido al átomo no exceda la valencia del átomo indicado, la posición de sustitución es cualquier posición razonable en la técnica.
En la presente invención, a menos que se indique lo contrario, un enlace químico representado por una línea discontinua indica que el enlace está opcionalmente presente o ausente. Por ejemplo, una línea punteada paralela a un enlace simple sólido indica un enlace simple o un enlace doble.
En la presente invención, cuando un enlace de un sustituyente se cruza con un enlace formado por dos átomos del anillo, dicho sustituyente puede unirse a cualquier átomo del anillo enlazable.
A menos que se indique lo contrario, los siguientes términos cuando se usan en las descripciones y las reivindicaciones de la presente invención tienen los siguientes significados:
El término "alquilo" se refiere a grupos hidrocarbonados saturados de cadena lineal o ramificada que comprenden de 1 a 20 átomos de carbono, preferiblemente de 1 a 10 átomos de carbono, más preferiblemente de 1 a 8, de 1 a 6, de 1 a 4, de 1 a 3 átomos de carbono, tales como metilo, etilo, n-propilo, isopropilo, n-butilo, sec-butilo, t-butilo, isobutilo, pentilo, hexilo, heptilo, octilo, nonilo, decilo, 4,4-dimetilpentilo, 2,2,4-trimetilpentilo, undecilo, dodecilo y varios isómeros de los mismos. Cuando se usa "alquilo" como grupo de enlace para otros grupos, tal como -(CH2)m-, puede ser una cadena lineal o ramificada, incluyendo las realizaciones, pero limitadas a, -CH2-, -CH2CH2-, -CH2CH(CH3)-.
El término "cicloalquilo" se refiere a un grupo monocíclico o policíclico saturado o parcialmente insaturado (que contiene 1 o 2 dobles enlaces) que comprende de 3 a 20 átomos de carbono. "monocicloalquilo" es preferiblemente un monocicloalquilo de 3 a 10 miembros, más preferiblemente un monocicloalquilo de 3 a 8 miembros, tal como ciclopropilo, ciclobutilo, ciclopentilo, ciclohexilo, cicloheptilo, ciclooctilo, ciclodecilo, ciclododecilo, ciclohexenilo. "policicloalquilo" incluye "anillo con puente", "cicloalquilo condensado" y "espirocicloalquilo", y realizaciones representativas de "cicloalquilo con puente" incluyen, pero no se limitan a, borneol, biciclo[2.2.1]heptenilo, biciclo[3.1.1] heptilo, biciclo[2.2.1] heptilo, biciclo[2.2.2]octilo, biciclo[3.2.2]nonilo, biciclo[3.3.1]nonilo, biciclo[4.2.1]nonanilo, adamantilo, etc. "Cicloalquilo fusionado" incluye un cicloalquilo fusionado con un fenilo, un cicloalquilo o un heteroarilo, incluyendo, pero no limitado a, benzociclobuteno, 2,3-dihidro-1-H-oxima, 2,3-ciclopentenopiridina, 5,6-dihidro-4H-ciclopenta[B]tiofeno, naftano y similares. Se pueden unir monocicloalquilo o policicloalquilo al resto molecular principal a través de cualquier átomo de carbono en el anillo.
El término "heterocicloalquilo" se refiere a grupos cíclicos no aromáticos saturados o parcialmente insaturados que comprenden átomos de carbono y heteroátomos seleccionados de nitrógeno, oxígeno o azufre, etc. El grupo cíclico puede ser monocíclico o policíclico. En la presente invención, el número de heteroátomos en el heterocicloalquilo es preferiblemente 1, 2, 3 ó 4, y el átomo de nitrógeno, carbono o azufre en el grupo heterocicloalquilo puede estar opcionalmente oxidado. El átomo de nitrógeno puede estar opcionalmente sustituido adicionalmente con otros grupos para formar una amina terciaria o una sal de amonio cuaternario. El "heterocicloalquilo monocíclico" es preferiblemente un heterocicloalquilo monocíclico de 3 a 10 miembros, más preferiblemente un heterocicloalquilo monocíclico de 3 a 8 miembros. Tales como: aziridinilo, tetrahidrofuran-2-ilo, morfolin-4-ilo, tiomorfolin-4-ilo, tiomorfolina-S-óxido-4-ilo, piperidin-1-ilo, N-alquilpiperidin-4-ilo, pirrolidin-1-ilo, N-alquilpirrolidin-2-ilo, piperazin-1-ilo, 4-alquilpiperazin-1-ilo y similares. "policicloheterocicloalquilo" incluye "heterocicloalquilo fusionado", "espiroheterociclilo" y "heterocicloalquilo puente". “Heterocicloalquilo fusionado” incluye un anillo de heterocicloalquilo monocíclico fusionado a un fenilo, cicloalquilo, heterocicloalquilo o heteroarilo, incluyendo, pero no limitado a: 2,3-dihidrobenzofuranillo, 1,3-dihidroisobenzofuranilo, indanilo, 2,3-dihidrobenzo[b]tienilo, dihidrobenzopiranilo, 1,2,3,4-tetrahidroquinolilo y similares. El heterocicloalquilo monocíclico y el heterocicloalquilo policíclico se pueden unir al resto molecular principal a través de cualquier átomo en el anillo. Los átomos del anillo anteriores se refieren específicamente a átomos de carbono y/o átomos de nitrógeno que constituyen el anillo.
El término "cicloalquilalquilo" se refiere a un enlace alquilo entre un cicloalquilo y un resto molecular principal. Por lo tanto, "cicloalquilalquilo" incluye la definición de alquilo y cicloalquilo como se describe anteriormente.
El término "heterocicloalquilalquilo" se refiere a un enlace alquilo entre un grupo heterocicloalquilo y un resto molecular principal. Por lo tanto, "heterocicloalquilalquilo" incluye las definiciones de alquilo y heterocicloalquilo como se describe anteriormente.
El término "alcoxi" se refiere a un alquilo cíclico o acíclico que tiene los átomos de carbono indicados unidos a través de un puente de oxígeno, e incluye alquiloxi, cicloalquiloxi y heterocicloalquiloxi. Por lo tanto, "alcoxi" incluye las definiciones de alquilo, heterocicloalquilo y cicloalquilo como se describe anteriormente.
El término "hidroxialquilo" se refiere a que cualquiera de los átomos de hidrógeno en el alquilo se reemplaza por un hidroxilo, incluyendo, pero no limitado a, -CH2OH, -CH2CH2OH, -CH2CH2C(CH3)2OH.
El término "alquenilo" se refiere a un hidrocarburo no aromático lineal, ramificado o cíclico que contiene al menos un doble enlace carbono-carbono. Puede haber de 1 a 3 dobles enlaces carbono-carbono, preferiblemente un doble enlace carbono-carbono. El término "alquenilo de C2-4" se refiere a un alquenilo que tiene de 2 a 4 átomos de carbono, y el término "alquenilo de C2-6" se refiere a un grupo alquenilo que tiene de 2 a 6 átomos de carbono, incluyendo vinilo, propenilo y butenilo, 2-metilbutenilo y ciclohexenilo. El alquenilo puede estar sustituido.
El término "alquinilo" se refiere a un hidrocarburo lineal, ramificado o cíclico que contiene al menos un triple enlace carbono-carbono. Puede haber de 1 a 3 triples enlaces carbono-carbono, preferiblemente un triple enlace carbonocarbono. El término "alquinilo de C2-6" se refiere a un alquinilo que tiene de 2 a 6 átomos de carbono e incluye etinilo, propinilo, butinilo y 3-metilbutinilo.
El término "arilo" se refiere a cualquier grupo aromático monocíclico o policíclico estable de 6 a 20 miembros, tal como fenilo, naftilo, tetrahidronaftilo, indanilo o bifenilo.
El término "heteroarilo" se refiere a un anillo aromático en el que al menos un átomo de carbono está reemplazado por un heteroátomo seleccionado de nitrógeno, oxígeno o azufre, que puede ser un anillo monocíclico de 5 a 7 miembros o un anillo fusionado de 7 a 20 miembros, preferiblemente un heteroarilo de 5 a 6 miembros. En la presente invención, el número de heteroátomos es preferiblemente 1,2 ó 3 e incluye: piridilo, pirimidinilo, piperazinilo, piridazin-3(2H)-ona, furilo, tienilo, tiazolilo, pirrolilo, imidazolilo, pirazolilo, oxazolilo, isoxazolilo, 1,2,5-oxadiazolilo, 1,2,4-oxadiazolilo, 1,3,4-oxadiazolilo, 1,3,4-tiadiazol, 1,2,4-triazolilo, 1,2,3-triazolilo, tetrazolilo, oxazolilo, isoxazolilo, fluorenilo, isoindolilo, benzofuranilo, benzotienilo, benzo[d][1,3]dioxolanilo, benzotiazolilo, benzoxazolilo, quinolilo, isoquinolinilo, isoquinolinona, quinazolinilo, 4-hidroxitieno[3,2-c]piridilo, 4,5-dihidro-4-oxofuro[3,2]piridinilo, 4-hidroxil-5-azaindolilo, furo[2,3-c]piridin-7(6H)-ona, tien[2,3-c]piridin-7(6H)-ona, y similares.
El término "anillo condensado" se refiere a una estructura cíclica en la que dos, tres o cuatro anillos comparten dos átomos adyacentes, y cada uno de los cuales puede ser un arilo monocíclico, un heteroarilo monocíclico, un monocicloalquilo o un heterocicloalquilo monocíclico. El anillo condensado al que se hace referencia en la presente invención es una estructura de anillo condensado saturado, insaturado o parcialmente saturado, y preferiblemente al menos un anillo es un anillo aromático. Más preferiblemente, un anillo condensado bicíclico o tricíclico, y al menos un anillo es un anillo aromático. El anillo no aromático en el anillo fusionado puede comprender además de 1 a 2 grupos oxo o tio. En la presente invención, el anillo fusionado es un anillo fusionado de 8 a 20 miembros, preferiblemente un grupo fusionado de 8 a 15 miembros. Las realizaciones específicas del anillo fusionado incluyen, pero no se limitan a, benzociclobutenilo, 2,3-dihidro-1 -H-indenilo, 1,2,3,4-tetrahidronaftilo, 6,7,8,9-tetrahidro-5H-benzo[7]anulenilo, 6,9-dihidro-5H-benzo[7]anulenilo, 5,6,7,8,9,10-hexahidrobenzo[8]anulenilo, 2,3-ciclopentenopiridilo, 5,6-dihidro-4H-ciclopenta[B]tienilo, 5,6-dihidro-4H-ciclopenta[B]furanilo, 2,3-dihidrobenzofuranilo, 1,3-dihidroisobenzofuranilo, indanilo, 2,3-dihidrobenzo[b]tienilo, dihidrobenzopipenilo, 1 ,2,3,4-tetrahidroquinolilo, 2,3-dihidro-1,4-benzodioxilo, 3,4-dihidro-2H-1,4-benzoxazinilo, naftiridinilo, naftilo, benzofuranilo, benzotienilo, benzopirrolilo, benzotiazolilo, benzoxazolilo, indazolilo, benzopiridazinilo, bencimidazolilo, indolilo, quinolilo, isoquinolilo, purinilo, pteridinilo,

el anillo condensado se puede unir al resto molecular principal a través de un átomo de carbono del anillo, preferiblemente a través de un átomo de carbono comprendido en el anillo aromático. El anillo condensado puede no estar sustituido o estar opcionalmente sustituido en cualquier posición por uno o más de un sustituyente.
El término "anillo aromático" incluye "arilo" y "heteroarilo".
El término "arilalquilo" se refiere a un arilo unido al resto molecular principal a través de un alquilo. Por lo tanto, "arilalquilo" incluye la definición de alquilo y arilo como se define anteriormente.
El término "heteroarilalquilo" se refiere a un heterocicloalquilo unido al resto molecular principal a través de un alquilo. Por lo tanto, "heteroarilalquilo" incluye las definiciones de alquilo y heteroarilo como se define anteriormente.
El término "halógeno" se refiere a flúor, cloro, bromo o yodo.
El término "haloalquilo" se refiere a un alquilo opcionalmente sustituido con halógeno. Por lo tanto, "haloalquilo" incluye las definiciones de halógeno y alquilo como se definen anteriormente.
El término "haloalcoxi" se refiere a un grupo alcoxi opcionalmente sustituido por halógeno. Por lo tanto, "haloalcoxi" incluye las definiciones de halógeno y alcoxi como se definen anteriormente.
El término "amino" se refiere a -NH2 , y el término "alquilamino" se refiere a que al menos un átomo de hidrógeno en el grupo amino está sustituido por un alquilo, que incluye, pero no se limita a, -NHCH3 , -N(CH3)2, -NHCH2CH3 , -N(CH2CH3)2. El término "aminoalquilo" significa que uno cualquiera de los átomos de hidrógeno en el alquilo está sustituido por un grupo amino, que incluye, pero no se limita a, -CH2NH2 , -CH2CH2NH2. Por lo tanto, "aminoalquilo" y "alquilamino" incluyen la definición de alquilo y amino como se define anteriormente.
El término "nitro" se refiere a -NO2.
El término "ciano" se refiere a -CN.
El símbolo ” ” se refiere a un doble enlace; el símbolo “ ” se refiere a un doble enlace o un enlace simple. "Temperatura ambiente" utilizada en la presente memoria significa de 15 a 30°C.
El derivado sustituido con isótopo incluye un derivado sustituido con isótopo en el que cualquier átomo de hidrógeno del compuesto de fórmula I se reemplaza por 1 a 5 átomos de deuterio, o cualquier átomo de carbono del compuesto de fórmula I se reemplaza por 1-3 átomos de C14, o cualquier átomo de oxígeno del compuesto de fórmula I se reemplaza por 1 a 3 átomos de O18.
Las "sales farmacéuticamente aceptables" como se describen en la presente memoria, se analizan en Berge, et al., "Pharmaceutically aceptable salts", J. Pharm. Sci., 66, 1-19 (1977), y es evidente para los químicos farmacéuticos que las sales son sustancialmente no tóxicas y pueden proporcionar las propiedades farmacocinéticas deseadas, palatabilidad, absorción, distribución, metabolismo o excreción, y similares. Los compuestos de la presente invención pueden tener un grupo ácido, un grupo básico o un grupo anfótero, y las sales farmacéuticamente aceptables típicas incluyen aquellas preparadas haciendo reaccionar el compuesto de la presente invención con un ácido, por ejemplo, hidrocloruro, hidrobromuro, sulfato, pirosulfato, bisulfato, sulfito, bisulfito, fosfato, monohidrógeno fosfato, dihidrógeno fosfato, metafosfato, pirofosfato, nitrato, acetato, propionato, citrato, octanoato, formiato, acrilato, isobutirato, hexanoato, heptanoato, oxalato, malonato, succinato, suberato, benzoato, metil benzoato, ftalato, maleato, metanosulfonato, p-toluenosulfonato, ácido (D,L)-tartárico, ácido cítrico, ácido maleico, ácido (D,L)-málico, ácido fumárico, ácido succínico, succinato, lactato, triflato, naftalen-1-sulfonato, mandelato, piruvato, estearato, ascorbato, salicilato. Cuando el compuesto de la presente invención contiene un grupo ácido, la sal farmacéuticamente aceptable del mismo puede incluir además una sal de metal alcalino tal como una sal de sodio o potasio; una sal de metal alcalinotérreo tal como una sal de calcio o magnesio; una sal de base orgánica tal como la formada por amoníaco, alquilaminas, hidroxialquilaminas, aminoácidos (lisina, arginina) o N-metilglucamina, etc.
El compuesto de fórmula (I) de la presente invención puede tener centros asimétricos y racematos, mezclas racémicas y diastereómeros individuales, todos los cuales, incluyendo los estereoisómeros, los isómeros geométricos, están todos incluidos en la presente invención. En la presente invención, cuando el compuesto de fórmula I o una sal del mismo está presente en forma estereoisómera (por ejemplo, cuando contiene uno o más átomos de carbono asimétricos), los estereoisómeros individuales (enantiómeros y diastereómeros) y mezclas de los mismos están incluidos dentro del alcance de la invención La presente invención también incluye isómeros individuales de los compuestos de fórmula I o sales, así como mezclas con isómeros en los que uno o más centros quirales están invertidos. El alcance de la invención incluye mezclas de estereoisómeros, así como enantiómeros purificados o
mezclas enantioméricamente/diastereoméricamente enriquecidas. La presente invención incluye mezclas de estereoisómeros de todas las diferentes combinaciones posibles de todos los enantiómeros y diastereómeros. La presente invención incluye todas las combinaciones y subconjuntos de estereoisómeros de todos los grupos específicos definidos anteriormente. La presente invención también incluye isómeros geométricos del compuesto de fórmula I o la sal del mismo, incluyendo los isómeros cis o trans.
Las condiciones preferidas anteriores de la presente invención pueden combinarse arbitrariamente sin apartarse del conocimiento general en la técnica para obtener las realizaciones preferidas de la presente invención.
Los reactivos y las materias primas utilizadas en la presente invención están disponibles comercialmente.
Descripción detallada de las realizaciones
Todas las estructuras de los compuestos en la presente divulgación fueron confirmadas por Resonancia Magnética Nuclear (1H NMR) y/o espectros de masas (MS).
Los desplazamientos químicos de 1H NMR (5) se registraron en ppm (10-6). Los espectros de NMR se registraron en un espectrómetro Bruker AVANCE-400. Los disolventes adecuados son cloroformo-d (CDCh), metanol-D4 (CD3OD), dimetilsulfóxido-D6 (DMSO-ds). Tetrametilsilano como patrón interno (TMS).
Los espectros de masas (MS) analíticos de baja resolución se registraron en Agilent 1200 HPLC/6120 utilizando una XBridge C18, 4,6 x 50 mm, 3,5 pm. El método de elución en gradiente 1: 80-5 % de disolvente A1 y 20-95% de disolvente B1 (1,8 min), y después 95% de disolvente B1 y 5% de disolvente A1 (más de 3 minutos). "%" como se usa en la presente memoria es el porcentaje en volumen del volumen de un disolvente en el volumen total de disolvente. Disolvente A1 : solución acuosa de ácido trifluoroacético (TFA) al 0,01 %; Disolvente B1 : solución en acetonitrilo de ácido trifluoroacético al 0,01%. "%" es el volumen de un disolvente en el volumen total de disolvente. El método de elución en gradiente 2: 80-5 % de disolvente A2 y 20-95% de disolvente B2 (1,5 min), y después 95% de disolvente B2 y 5% de disolvente A2 (más de 2 min), "%" es el volumen de un disolvente en el volumen total de disolvente. Disolvente A2 : solución acuosa de bicarbonato de amonio 10 mM; Disolvente B2 : acetonitrilo.
Todos los compuestos de la presente divulgación se separaron mediante cromatografía líquida de alta resolución preparativa, cromatografía en columna o cromatografía en columna flash.
La purificación por cromatografía líquida de alta resolución preparativa (HPLC preparativa) se realizó en Shimadzu LC-20 HPLC, columna cromatográfica: waters xbridge Pre C18, 10um, 19mm*250mm. Método preparativo: fase móvil A: solución acuosa de ácido trifluoroacético al 0,05% (% es porcentaje en volumen), fase móvil B: acetonitrilo; el método de elución en gradiente: 25-75 % de disolvente A y 75-25 % de disolvente B; longitud de onda de detección: 214 y/o 254 y/o 262 nM. El caudal: 10,0 mL/min;
La cromatografía en columna flash (sistema flash/Cheetah™) se realizó en Agela Technologies MP200, la columna de separación se utilizó Flash column Silica-CS (80 g), Cat No. CS140080-0.
Se utilizó cromatografía de capa fina Yantai xinnuo chemical, el espesor del recubrimiento es de 0,2 ± 0,03 mm. La cromatografía en columna generalmente usaba gel de sílice de 200-300 mesh de Yantai Huanghai como vehículo. Realización 1: Síntesis de los compuestos 1.7 y 1.8

Etapa 1: síntesis del compuesto 1.2
El compuesto 1.1 (20 g, 57,4 mmol) y bromoacetonitrilo (6,9 g, 57,4 mmol) se disolvieron en acetato de etilo (200 mL), el sistema de reacción se calentó a reflujo y se agitó durante 3 h, el sólido se eliminó por filtración. La torta del filtro se lavó dos veces con acetato de etilo. El filtrado se concentró a presión reducida para eliminar el disolvente y proporcionar el compuesto 1.2 (17 g, rendimiento: 76 %) como un sólido de color amarillo claro, que puede usarse directamente para la siguiente etapa.
Etapa 2: síntesis del compuesto 1.3
Una solución de 4-bromo-2-nitrobenzaldehído (10 g, 43,9 mmol), compuesto 1.2 (17 g, 43,9 mmol) y tolueno (170 mL) se agitó a reflujo durante 2 h, la mezcla resultante se enfrió a temperatura ambiente y se filtró a través de una almohadilla corta de gel de sílice, se eluyó con una solución al 25% de acetato de etilo en éter de petróleo hasta que el producto no pudo ser detectado por TLC, la mayor parte del eluyente se eliminó por concentración a presión reducida y la solución residual se colocó a -18°C durante 16h. Después de la filtración, la torta del filtro se secó para dar el compuesto 1.3 (8,2 g, rendimiento: 55%) como un sólido blanquecino.
Etapa 3: síntesis del compuesto 1.4
Una mezcla del compuesto 1.3 (4,2 g, 12,4 mmol) y ácido acético (80 mL) se calentó a 80°C. A la mezcla se le añadió polvo de hierro (4,1 g, 74,3 mmol) en 15 min, la temperatura de reacción se mantuvo a no más de 90°C y se agitó durante 3 h. El sistema de reacción se enfrió a temperatura ambiente, se filtró a través de celita y se enjuagó 3 veces con acetato de etilo. El filtrado se concentró a presión reducida, el residuo se diluyó con agua fría y se ajustó a pH > 8 con una solución acuosa de bicarbonato de sodio saturada. Las fases orgánicas combinadas se lavaron con una disolución saturada de NaCl y la fase orgánica se separó y se secó sobre sulfato de sodio anhidro, se filtró y se concentró. El residuo se trituró con una disolución al 10% de acetato de etilo en éter de petróleo, se filtró y la torta del filtro se secó para proporcionar el compuesto 1.4 (3,0 g, rendimiento: 78 %) como un sólido blanquecino.
Etapa 4: síntesis del compuesto 1.5
A una solución del compuesto 1.4 (3,0 g, 9,7 mmol) y trietilamina (1,47 g, 14,6 mmol) en diclorometano (50 mL) se añadió dicarbonato de di-terc-butilo ((BOC)2O, 3,2 g, 14,6 mmol). El sistema de reacción se agitó a temperatura ambiente durante 2 días y después se diluyó con diclorometano (100 mL). La fase orgánica se lavó sucesivamente con ácido clorhídrico (3,0 M), una solución acuosa de bicarbonato de sodio saturada y una disolución saturada de NaCl, la fase orgánica se separó y se secó sobre sulfato de sodio anhidro, se filtró y se concentró. El residuo se trituró con una disolución al 10% de acetato de etilo en éter de petróleo, se filtró y la torta del filtro se secó para proporcionar el compuesto 1.5 (1,6 g, rendimiento: 40 %) como un sólido blanquecino.
m/z: [M+H]+ 409
Etapa 5: síntesis del compuesto 1.6
A una solución enfriada con hielo del compuesto 1.5 (1,6 g, 3,91 mmol) en tetrahidrofurano (THF) (50 mL) se añadió una solución acuosa de hidróxido de sodio (1,0 M, 5,9 mL, 5,9 mmol). El sistema de reacción se agitó a temperatura ambiente durante 16 h y después se ajustó el pH a 6 con ácido clorhídrico (0,5 M). La mezcla resultante se extrajo dos veces con acetato de etilo, las fases orgánicas combinadas se lavaron con una disolución saturada de NaCl y la fase orgánica se separó y se secó sobre sulfato de sodio anhidro, se filtró y se concentró para proporcionar el compuesto 1.6 (1,1 g, rendimiento: 74 %) como una espuma amarilla clara.
m/z: [M+H]+ 381
Etapa 6: síntesis del compuesto 1.7
La mezcla del compuesto 1.6 (1,1 g, 2,8 mmol), hexafluorfosfato de 2-(7-azabenzotriazol-1-il)-N,W,W',W'-tetrametiluronio (1,6 g, 4,33 mmol), clorhidrato de dimetilamina (470 mg, 5,77 mmol) y N,N-diisopropiletilamina (560 mg, 4,37 mmol) en N,N-dimetilformamida (DMF) (10 mL) se agitó a temperatura ambiente durante 3 h. El sistema de reacción se diluyó con acetato de etilo (100 mL) y se lavó sucesivamente con agua y una disolución saturada de NaCl, la fase orgánica se separó y se secó sobre sulfato de sodio anhidro, se filtró y se concentró, y el residuo se purificó por cromatografía en columna sobre gel de sílice (metanol/diclorometano (DCM) = 1/20) para proporcionar el compuesto 1.7 (400 mg, rendimiento: 34%) como un sólido amarillo claro.
m/z: [M+H]+ 408
Etapa 8: síntesis del compuesto 1.8
La mezcla del compuesto 1.7 (380 mg, 0,93 mmol), acetato de potasio (274 mg, 2,79 mmol), bis(pinacolato)diboro (354 mg, 1,40 mmol) y Pd(dppf)2Cl2 en 1,4-dioxano (10 mL) se agitó a 80°C durante 5 h bajo la protección de N2 . El sistema de reacción se enfrió a temperatura ambiente, se filtró a través de celita, la celita se enjuagó con una solución de metanol al 10 % en DCM, el filtrado se concentró y el residuo se purificó mediante cromatografía en columna sobre gel de sílice (metanol/DCM = 1/20~1/10) para dar el compuesto 1.8 (208 mg, rendimiento: 49%) como un sólido rojo.
m/z: [M+H]+ 456
Realización 2: Síntesis del compuesto 2.4

Etapa 1: síntesis del compuesto 2.1
A una suspensión enfriada con hielo de 5-bromo-2-metil-3-nitroanilina (2,0 g, 8,66 mmol), agua (8 mL) y ácido clorhídrico (6 N, 8 mL) se añadió a la solución acuosa de nitrito de sodio (627 mg, 9,09 mmol, 5 mL), y después se le añadió una solución concentrada de ácido clorhídrico de cloruro estannoso (5,4 g, 25,9 mmol, 5 mL), el sistema de reacción se agitó a 0°C durante 30 min. La solución de reacción se neutralizó con una solución concentrada de amoníaco a pH=8 y después se extrajo con acetato de etilo. Las fases orgánicas combinadas se lavaron con una disolución saturada de NaCl, se secaron sobre sulfato de sodio anhidro, se filtraron y se concentraron. El producto bruto se trituró con terc-butil metil éter y se filtró para dar el compuesto 2.1 (700 mg, rendimiento: 33%) como un sólido amarillo claro.
m/z: [M+H]+ 246
Etapa 2: síntesis del compuesto 2.2
A una solución de nitrato de amonio cérico (3,28 g, 5,97 mmol) en metanol-D4 (7 mL) se añadió el compuesto 2.1 (700 mg, 2,84 mmol) en una porción. El sistema de reacción se agitó a temperatura ambiente durante 10 min y después se filtró a través de celita, el filtrado se concentró y se purificó mediante cromatografía en columna sobre gel de sílice (100 % éter de petróleo) para proporcionar el compuesto 2.2 (300 mg, rendimiento: 49 %) como un aceite amarillo claro. Etapa 3: síntesis del compuesto 2.3
El Compuesto 2.2 (0,85 g, 3,92 mmol) y W,W-dimetilformamida dimetil acetal (5 mL) se disolvieron en DMF (10 mL), el sistema de reacción se agitó a 105°C durante la noche y después se concentró a presión reducida para producir el compuesto 2.3 (1,0 g, rendimiento: 94 %) como un compuesto rojo sólido.
m/z: [M+H]+ 272
Etapa 4: síntesis del compuesto 2.4
A una solución del compuesto 2.3 (1,0 g, 3,93 mmol) en una mezcla de disolventes de THF y H2O (5 mL/5 mL) se añadió peryodato de sodio (2,5 g, 11,8 mmol), el sistema de reacción se agitó a temperatura ambiente durante 16 h, la sustancia no disuelta se filtró, el filtrado se extrajo con acetato de etilo, las fases orgánicas combinadas se lavaron con una disolución saturada de NaCl, se secaron sobre sulfato de sodio anhidro, se concentraron y el residuo se purificó por cromatografía en columna sobre gel de sílice (éter de petróleo/acetato de etilo = 10/1 ~5/1) para producir el compuesto 2.4 (200 mg, rendimiento: 22 %) como un sólido de color amarillo claro.
1H NMR (400 MHz, CDCh): 510,38 (s, 1 H), 8,26 (s, 1 H), 7,93 (s, 1 H).
Realización 3: Síntesis de los compuestos 1.9~1.13
Los compuestos 1.9~1.13 se sintetizaron siguiendo el método sintético utilizado para el compuesto 1.7 de la Realización 1, reemplazando el clorhidrato de dimetilamina con las aminas correspondientes en la etapa 6:



Realización 4: Síntesis de Ios compuestos 1.14~1.17
Los compuestos 1.14~1.17 se sintetizaron siguiendo el método sintético utilizado para el compuesto 1.7 de la Realización 1, reemplazando el 4-bromo-2-nitrobenzaldehído con 4-bromo-2-metoxi-6-nitrobenzaldehído, 4-bromo-2-metil-6-nitrobenzaldehído, 4-bromo-2-cloro-6-nitrobenzaldehído, o compuesto el 2.4 en la etapa 2:


Realización 5: Síntesis del compuesto 1.19
El compuesto 1.19 se sintetizó siguiendo el método sintético utilizado para el compuesto 1.8 de la Realización 1, reemplazando el clorhidrato de dimetilamina con 2-(propilamino) etanol en la etapa 6:

m/z: [M+H]+ 514
Realización 6: Síntesis de compuestos 1.20~1.22
El compuesto 1.20 se sintetizó siguiendo el método sintético utilizado para el compuesto 1.8 de la Realización 1, reemplazando el compuesto 1.7 con el compuesto 1.10 en la etapa 7:
Los compuestos 1.21 y 1.22 se sintetizaron siguiendo el método sintético utilizado para el compuesto 1.8 de la Realización 1, reemplazando el 4-bromo-2-nitrobenzaldehído con 4-bromo-5-fluoro-2-nitrobenzaldehído o 4-bromo-2-fluoro-6-nitrobenzaldehído en la etapa 2:


Realización 7: Síntesis del compuesto 3.8

1, Síntesis del catalizador del ácido p-toluenosulfónico: el monohidrato del ácido p-toluenosulfónico (100 mg) y benceno (20 mL) se calentaron a reflujo durante 2 h en un aparato Dean-stark hasta que la solución se volvió transparente.
2, La solución de 1-metil-1H-pirrol-2-carbaldehído (2,0 g, 18,3 mmol) y 2,2-dimetilpropano-1,3-diol (4,9 g, 47,6 mmol) en benceno (20 mL) se calentó a reflujo durante 15 min en un aparato Dean-stark, el catalizador del ácido ptoluenosulfónico preparado se añadió lentamente gota a gota a la solución anterior, después de la adición, el sistema de reacción se agitó a 85°C durante 2 h y después se enfrió a temperatura ambiente, se filtró, el filtrado se concentró a presión reducida, el residuo se purificó por cromatografía en columna sobre gel de sílice (acetato de etilo/éter de petróleo = 1/100) para proporcionar el compuesto 3.1 (1,0 g, rendimiento: 28%) como un aceite incoloro.
1H NMR (400 MHz, CDCla): 56,59-6,58 (m, 1H), 6,25-6,23 (m, 1H), 6,06-6,05 (m, 1H), 5,45 (s, 1H), 3,80 (s, 3H), 3,78 (d, J = 11,2 Hz, 2H), 3,63 (d, J = 10,8 Hz, 2H), 1,33 (s, 3H), 0,82 (s, 3H).
Etapa 2: síntesis del compuesto 3.2
A una solución del compuesto 3.1 (1,0 g, 5,12 mmol) en THF (10 mL) se añadió W-bromosuccinimida (NBS) (960 mg, 5,38 mmol) en pequeñas porciones, el sistema de reacción se agitó a temperatura ambiente durante 1 h y después se diluyó con acetato de etilo (100 mL), la fase orgánica se lavó con la solución acuosa de hidróxido de sodio (2,0 M) y una disolución saturada de NaCl, separación de la fase orgánica, secado sobre sulfato sódico, filtrado y concentrado, el residuo se purificó mediante cromatografía en columna sobre gel de sílice (acetato de etilo/éter de petróleo = 1/100~ 1/10) para producir el compuesto 3.2 (800 mg, rendimiento: 57%) como un aceite incoloro, que puede usarse directamente para la siguiente etapa.
Etapa 3: síntesis del compuesto 3.3
A una solución del compuesto 3.2 (800 mg, 2,92 mmol) en acetona (4 mL) se le añadió ácido clorhídrico concentrado (1 mL), el sistema de reacción se agitó a 40°C durante 1 hora y después se diluyó con agua (50 mL), la mezcla se extrajo 3 veces con acetato de etilo, las fases orgánicas combinadas se lavaron con una disolución saturada de NaCl, se secaron sobre sulfato de sodio anhidro, se filtraron y concentraron, el residuo se purificó mediante cromatografía en columna sobre gel de sílice (acetato de etilo/éter de petróleo = 1/9) para proporcionar el compuesto 3.3 (145 mg, rendimiento: 26%) como un aceite marrón.
1H NMR (400 MHz, CDCla): 59,45 (s, 1H), 6,90 (d, J = 4,0 Hz, 1H), 6,23 (d, J = 4,4 Hz, 1H), 3,96 (s, 3H).
Etapa 4: síntesis del compuesto 3.4
A una solución del compuesto 3.3 (145 mg, 0,77 mmol) en THF (10 mL) se añadió acetato de etil (trifenilfosforanilideno) (537 g, 1,54 mmol) en pequeñas porciones. El sistema de reacción se agitó a temperatura ambiente durante 2 días hasta que la TLC detectó que la reacción estaba completada, la reacción se detuvo mediante la adición de agua (30 mL), la mezcla se extrajo con acetato de etilo (20 mL x 2). Las fases orgánicas combinadas se lavaron con una disolución saturada de NaCl, la fase orgánica se separó, se secó sobre sulfato de sodio anhidro, se filtró y se concentró, y el residuo se purificó por cromatografía en columna sobre gel de sílice (disolución de acetato de etilo al 25% en éter de petróleo) para producir el compuesto 3.4 (200 mg, rendimiento: 100%) como un sólido blanquecino.
Etapa 5: síntesis del compuesto 3.5
A una solución del compuesto 3.4 (200 mg, 0,77 mmol) en una mezcla de disolventes de metanol (2 mL), THF (0,5 mL) y H2O (0,5 mL) se añadió hidróxido de litio hidratado (162 mg, 3,87 mmol). El sistema de reacción se agitó a temperatura ambiente durante 16 h y después se detuvo la reacción mediante la adición de ácido clorhídrico (3,0 M) y se ajustó el pH a 5. La mezcla se extrajo dos veces con acetato de etilo, las fases orgánicas combinadas se lavaron con una disolución saturada de NaCl, la fase orgánica se separó, se secó sobre sulfato de sodio anhidro, se filtró y se concentró para dar el compuesto 3.5 (170 mg, rendimiento: 95 %) como un sólido amarillo pálido, que se puede usar directamente para la siguiente etapa.
Etapa 6: síntesis del compuesto 3.6
A una solución del compuesto 3.5 (170 mg, 0,74 mmol) en cloroformo (2 mL) se le añadió cloruro de tionilo (1 mL) y una gota de DMF bajo la protección de N2 , después de la adición, el sistema de reacción se calentó a reflujo y se agitó durante 1 hora. El sistema de reacción se enfrió a temperatura ambiente y se concentró a presión reducida para proporcionar el compuesto 3.6 (165 mg, rendimiento: 90 %) como un sólido marrón, que puede usarse directamente para la siguiente etapa.
Etapa 7: síntesis del compuesto 3.7
A una solución enfriada con hielo del compuesto 3.6 (165 mg, 0,66 mmol) en acetona (5,0 mL) se añadió la solución acuosa de azida de sodio saturada (1 ml). El sistema de reacción se agitó vigorosamente en un baño de hielo hasta que la TLC detectó que la reacción se había completado. El sistema de reacción se diluyó con diclorometano y se lavó con agua y una disolución saturada de NaCl, la fase orgánica se separó, se secó sobre sulfato de sodio anhidro, se filtró y se concentró para proporcionar el compuesto 3.7 (188 mg, rendimiento: 100 %) como un sólido amarillo pálido, que puede utilizarse directamente para la siguiente etapa.
Etapa 8: síntesis del compuesto 3.8
Se añadió difenil éter (3 mL) a un matraz de fondo redondo de 10 mL con un tubo de condensación de aire, el matraz se calentó a 240°C, a la solución anterior se le inyectó lentamente una solución del compuesto 3.7 (188 mg, 0,74 mmol) en diclorometano (3 mL) con una jeringa. El sistema de reacción se agitó a 240°C durante 20 min y luego se enfrió a 50°C, el sistema de reacción se vertió en éter de petróleo (50 mL) y se agitó durante 2 h, la mezcla resultante se filtró, la torta del filtro se enjuagó con petróleo éter y se secó para proporcionar el compuesto 3.8 (90 mg, rendimiento: 54%) como un sólido gris oscuro.
m/z: [M+H]+ 227
Realización 8: Síntesis del compuesto 3.9
El compuesto 3.9 se sintetizó siguiendo el método sintético utilizado para el compuesto 3.8 de la Realización 7, reemplazando el compuesto 3.3 con 5-bromotiofen-2-carbaldehído en la etapa 4:

m/z: [M+H]+ 230
Realización 9: Síntesis del compuesto 4.2

A una solución de ácido 2,4-dibromobenzoico (1,0 g, 3,57 mmol) en DMF (10 mL) se le añadió sucesivamente 1-hidroxibenzotriazol (530 mg, 3,93 mmol) y clorhidrato de 1-(3-dimetilaminopropil)-3-etilcarbodiimida. (820 mg, 4,29 mmoles). El sistema de reacción se enfrió a 0°C y se añadió una solución de amoníaco (20 mL, 25 %) después de la adición, el sistema de reacción se calentó a temperatura ambiente y se agitó durante la noche. Se añadió agua (50 mL) al sistema de reacción, se filtró, la torta del filtro se lavó 3 veces con agua, se secó para proporcionar el compuesto 4.1 (660 mg, rendimiento: 66%) como un sólido blanco.
m/z: [M+H]+ 278
Etapa 2: síntesis del compuesto 4.2
Se añadieron bromuro cuproso (15,4 mg, 0,11 mmol), carbonato de cesio (701 mg, 2,15 mmol), el compuesto 4.1 (300 mg, 1,08 mmol), ciclopentanona (136 mg, 1,61 mmol) y dimetilsulfóxido (DMSO) (10 mL) en un tubo estanco (20 mL), el tubo se reemplazó 3 veces con N2. El sistema de reacción se agitó a 80°C durante la noche y después se enfrió a temperatura ambiente, al sistema de reacción se le añadió una disolución saturada de NaCl (50 mL) y acetato de etilo (50 mL), separación de la fase orgánica, la fase acuosa se extrajo con acetato de etilo (20 mL x 2). Las fases orgánicas combinadas se lavaron con una disolución saturada de NaCl, se secaron sobre sulfato de sodio anhidro, se filtraron y concentraron, el residuo se trituró con una mezcla de disolventes de terc-butil metil éter (50 mL) y diclorometano (5 mL), se filtró, la torta del filtro se secó para proporcionar el compuesto 4.2 (150 mg, rendimiento: 42%) como un sólido amarillo.
m/z: [M+H]+ 264
Realización 10: Síntesis del compuesto 4.3
El compuesto 4.3 se sintetizó siguiendo el método sintético utilizado para el compuesto 4.2 de la Realización 9, reemplazando la ciclopentanona con acetona en la etapa 2:

m/z: [M+H]+ 238
Realización 11: Síntesis del compuesto 5.3

Etapa 1: síntesis del compuesto 5.1
A una solución de ácido 4-bromo-2-metilbenzoico (1,0 g, 4,65 mmol) en DMF (10 mL) se le añadió sucesivamente carbonato de potasio (670 mg, 4,89 mmol) y yodometano (900 mg, 6,98 mmol). El sistema de reacción se agitó a temperatura ambiente durante 2 h y después se paró la reacción mediante la adición de agua (60 mL), la mezcla se extrajo con acetato de etilo (40 mL x 2), las fases orgánicas combinadas se lavaron con una disolución saturada de NaCl, la separación de la fase orgánica, se secó sobre sulfato de sodio, se filtró y se concentró para proporcionar el compuesto 5.1 (1,0 g, rendimiento: 93%) como un aceite de color rojo claro, que se puede usar directamente para la siguiente etapa. Etapa 2: síntesis del compuesto 5.2
A una solución del compuesto 5.1 (300 mg, 1,31 mmol) en cloroformo (10 mL) se le añadió NBS (350 mg, 1,96 mmol) y azodiisobutironitrilo (100 mg, 0,65 mmol) en una porción. El sistema de reacción se calentó a 80°C y se agitó durante 2 h y después se enfrió a temperatura ambiente, se filtró, el filtrado se lavó con una disolución saturada de NaCl, la fase orgánica se separó, se secó sobre sulfato de sodio, se filtró y se concentró para producir el compuesto 5.2 (300 mg, rendimiento: 75%) como un aceite rojo, que se puede utilizar directamente para la siguiente etapa.
Etapa 3: síntesis del compuesto 5.3
A una solución del compuesto 5.2 (300 mg, 0,97 mmol) en THF (2 mL) se le añadió una solución de amoníaco (2 mL). El sistema de reacción se agitó a 20°C durante 12 h. El sólido resultante se filtró, después del tratamiento de secado al vacío para proporcionar el compuesto 5.3 (100 mg, rendimiento: 50%) como un sólido blanquecino.
m/z: [M+H]+ 212
Realización 12: Síntesis del compuesto 5.4
El compuesto 5.4 se sintetizó siguiendo el método sintético utilizado para el compuesto 5.3 de la Realización 11, reemplazando el ácido 4-bromo-2-metilbenzoico con ácido 5-bromo-3-metilpiridin-2-carboxílico en la etapa 1:

m/z: [M+H]+ 213
Realización 13: Síntesis del compuesto 6.2

Etapa 1: síntesis del compuesto 6.1
Se disolvió una solución de metilamina en tetrahidrofurano (2,0 M, 11 mL, 27,8 mmol) en THF (10 ml) en un baño de
agua con hielo y se añadió gota a gota al sistema n-butillitio (8,2 mL, 20,5 mmol). Después de la adición, la reacción se agitó a 0°C durante 1 hora y después el sistema de reacción se enfrió hasta -78°C. Se añadió al sistema una solución de ácido 4-bromo-2-fluorobenzoico (1,0 g, 4,57 mmol) en THF (5 mL), se agitó a -78°C durante 0,5 h y se paró la reacción mediante la adición de ácido clorhídrico (1,0 M), se extrajo con acetato de etilo (5 ml x 3). Las fases orgánicas combinadas se lavaron con una disolución saturada de NaCl, se secaron sobre sulfato sódico, se filtraron y concentraron para dar el compuesto 6.1 (700 mg, rendimiento: 68%) como un sólido amarillo claro.
m/z: [M+H]+ 230
Etapa 2: síntesis del compuesto 6.2
El compuesto 6.1 (580 mg, 2,52 mmol) y la urea (3 g) se calentaron hasta fundir y se agitaron a 150°C durante 7 h. El sistema de reacción se enfrió a temperatura ambiente y después se paró la reacción mediante la adición de agua, se extrajo con acetato de etilo. Las fases orgánicas combinadas se lavaron con una disolución saturada de NaCl, se secaron sobre sulfato de sodio, se filtraron y concentraron para dar el compuesto 6.2 (320 mg, rendimiento: 35 %) como un sólido gris.
m/z: [M+H]+ 255
Realización 14: Síntesis de los compuestos 15.1 y 15.2

Etapa 1: síntesis del compuesto 15.1
A una solución enfriada con hielo de 6-bromoisoquinolin-1(2H)-ona (800 mg, 3,57 mmol) en DMF (8 mL) se añadió hidruro de sodio (160 mg, 3,93 mmol, 60%) en pequeñas porciones, después de la adición, el sistema de reacción se calentó a temperatura ambiente y se agitó durante 20 min, se añadió iodoetano (800 mg, 5,36 mmol). Después de la adición, el sistema de reacción se agitó a temperatura ambiente durante 2 h, la reacción se paró mediante la adición de agua (20 mL), se extrajo con acetato de etilo (20 mL x 2), las fases orgánicas combinadas se lavaron con una disolución saturada de NaCl, se secaron sobre sulfato de sodio, la fase orgánica se separó, se secó sobre sulfato de sodio anhidro, se filtró y se concentró, el residuo se purificó por cromatografía en columna sobre gel de sílice (acetato de etilo: éter de petróleo = 1:4) para proporcionar el compuesto 15.1 (800 mg, rendimiento: 91 %) como un sólido blanco.
1H NMR (400 MHz, CDCla): 5 8.30 (d, J = 8,8 Hz, 1 H), 7,67 (s, 1 H), 7,58 (d, J = 8,8 Hz, 1 H), 7,12 (d, J = 7,2 Hz, 1 H), 6,43 (d, J = 7,2 Hz, 1 H), 4,08 - 4,02 (m, 2 H), 1,40 (t, 3 H).
Etapa 2: síntesis del compuesto 15.2
A una solución del compuesto 15.1 (500 mg, 1,98 mmol) en 1,4-dioxano (6 mL) se le añadió sucesivamente acetato de potasio (580 mg, 5,95 mmol), bis(pinacolato)diboro (760 mg, 2,97 mmol) y Pd (dppf)Cl2 (160 mg, 0,19 mmol) bajo protección de N2. El sistema de reacción se agitó a 80°C durante 2 h. La reacción se enfrió a temperatura ambiente y se paró mediante la adición de agua (60 mL), se extrajo con acetato de etilo (40 mL x 2), las fases orgánicas combinadas se lavaron con una disolución saturada de NaCl, la fase orgánica se separó, se secó sobre sulfato de sodio anhidro, se filtró y se concentró, el residuo se purificó por cromatografía en columna sobre gel de sílice (acetato de etilo : éter de petróleo = 1:4) para dar el compuesto 15.2 (500 mg, rendimiento: 83%) como un sólido amarillo.
m/z: [M+H]+ 300
Realización 15: Síntesis de los compuestos 15.3~15.10
Los compuestos 15.3~15.10 se sintetizaron siguiendo el método sintético utilizado para el compuesto 15.1 de la Realización 14, por reacción de 6-bromoisoquinolin-1(2H)-ona, 6-bromo-3,4-dihidroisoquinolin-1(2H)-ona, los compuestos 3.9, 4.2, 4.3 o 5.4 con los ioduros o bromuros correspondientes:

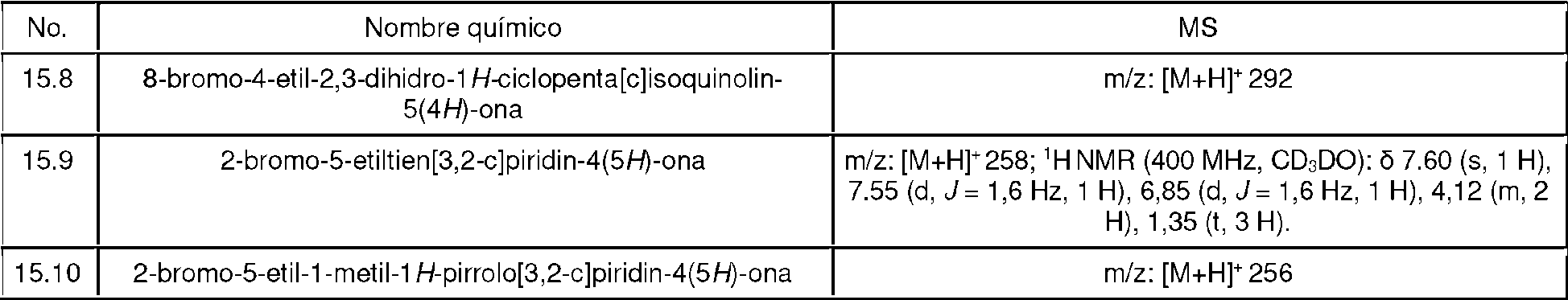
Realización 16: Síntesis del compuesto 16.1

La solución de 6-bromoisoquinolin-1(2H)-ona (150 mg, 0,67 mmol), carbonato de cesio (650 mg, 2,01 mmol) y 2 bromoetanol (170 mg, 1,34 mmol) en DMF (2 mL) se agitó a temperatura ambiente durante 2 días, el sistema de reacción se diluyó con acetato de etilo (100 mL), la mezcla se lavó con agua y una disolución saturada de NaCl, la fase orgánica se separó y se secó sobre sulfato de sodio anhidro, se filtró y se concentró, el residuo se trituró con éter de petróleo, se filtró, la torta del filtro se secó para producir el compuesto 16.1 (120 mg, rendimiento: 60%) como un sólido blanquecino.
m/z: [M+H]+ 268
Realización 17: Síntesis de los compuestos 16.2~16.75
Los compuestos 16.2~16.75 se sintetizaron siguiendo el método sintético utilizado para el compuesto 16.1 de la Realización 16, por reacción de 6-bromoisoquinolin-1(2H)-ona, 6-bromo-3,4-dihidroisoquinolin-1(2H)-ona, 7-bromoquinazolin-4(3H)-ona, 6-bromoftalazin-1(2H)-one, los compuestos 4.3, 5.3, 5.4 o 6.2 con los correspondientes bromuros o cloruros:



Realización 18: Síntesis del compuesto 17.1

El compuesto 16.5 (50,0 mg, 0,154 mmol) se disolvió en THF (5 mL), la disolución se enfrio a 0°C con un baño de hielo y agua. A esta solución se añadió bromuro de metilmagnesio (0,18 mL, 3 M, 0,54 mmol) bajo protección de N2. A continuación, se retiró el baño de agua helada y la mezcla de reacción se agitó a temperatura ambiente durante 1,5 h, se añadió una solución acuosa saturada de cloruro de amonio (20 mL), la mezcla se extrajo con acetato de etilo (20 mL x 2), las fases orgánicas combinadas se lavaron con una disolución saturada de NaCl, se secaron sobre sulfato de sodio anhidro, se filtraron y se concentraron a presión reducida. El residuo se purificó por TLC preparativa (éter de petróleo/acetato de etilo = 1/1) para dar el compuesto 17.1 (13 mg, rendimiento: 27%) como un sólido amarillo. m/z: [M+H]+ 310
Realización 19: Síntesis de los compuestos 17.2~17.5
Los compuestos 17.2~17.5 se sintetizaron siguiendo el método sintético utilizado para el compuesto 17.1 de la Realización 18, reemplazando el compuesto 16.5 con 16.6, 16.23, 16.26 o 16.27:

Realización 20: Síntesis del compuesto 18.1

Se añadieron el compuesto 16.8 (0,2 g, 0,76 mmol), carbonato de potasio (0,21 g, 1,52 mmol), peróxido de hidrógeno (0,5 mL, 30 %) y DMSO (2,0 mL) a un matraz de 25 mL. El sistema de reacción se agitó a temperatura ambiente durante 1 hora. Se le añadió agua y después se filtró, la torta del filtro se lavó para proporcionar el compuesto 18.1 (160 mg, rendimiento: 80%) como un sólido de color amarillo claro.
m/z: [M+H]+ 281
Realización 21: Síntesis de los compuestos 18.2-18.4
Los compuestos 18.2~18.4 se sintetizaron siguiendo el método sintético utilizado para el compuesto 11.1 de la Realización 20, reemplazando el compuesto 16.8 con 16.14, 16.15 o 16.19:

Realización 22: Síntesis del compuesto 19.1

Se añadieron el compuesto 16.10 (55 mg, 0,18 mmol), borohidruro de litio (0,5 mL, solución en THF 2 M) y THF (1,0 mL) a un matraz de 25 mL. El sistema de reacción se agitó a temperatura ambiente durante 2 horas y después se filtró, el filtrado se añadió a agua (10 mL), la mezcla se extrajo con acetato de etilo (10 mL x 2), las fases orgánicas combinadas se lavaron con una disolución saturada de NaCl, se secaron sobre sulfato de sodio anhidro, se filtró, el disolvente se evaporó a presión reducida, el residuo se purificó por cromatografía en columna sobre gel de sílice (metanol/DCM = 1/10) para proporcionar el compuesto 19.1 (23 mg, rendimiento: 50 %) como un blanco sólido. m/z: [M+H]+ 256
Realización 23: Síntesis del compuesto 19.2
El compuesto 19.2 se sintetizó siguiendo el método sintético utilizado para el compuesto 19.1 de la Realización 22, reemplazando el compuesto 16.10 con 16.29:
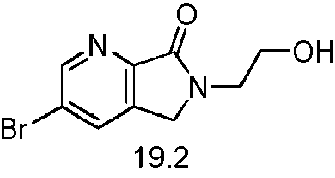
m/z: [M+H]+ 257
Realización 24: Síntesis del compuesto 20.2

Etapa 1: síntesis del compuesto 20.1
El compuesto 16.16 (2,80 g, 7,62 mmol) se disolvió en DCM (30 mL), se le añadió ácido trifluoroacético (TFA) (15 mL) en un baño de hielo y agua. Se retiró el baño de agua helada y la mezcla de reacción se agitó a temperatura ambiente durante 1 h. La mezcla se concentró directamente a sequedad bajo presión reducida. El residuo se ajustó a pH=7~8 con la solución acuosa de carbonato de sodio saturado, se extrajo con acetato de etilo (50 mL x 2), las fases orgánicas combinadas se lavaron con una disolución saturada de NaCl, se secaron sobre sulfato de sodio anhidro y se filtraron. El disolvente se evaporó a presión reducida para dar el compuesto 20.1 (2,00 g, rendimiento: 98%) como un sólido amarillo. m/z: [M+H]+ 267
Etapa 2: síntesis del compuesto 20.2
Se añadieron el compuesto 20.1 (150 mg, 0,56 mmol) y trietilamina (284 mg, 2,81 mmol) a DCM (3 mL), se añadió cloruro de acetilo (66 mg, 0,84 mmol) en un baño de agua con hielo y, después de retirar el baño de hielo-agua, la mezcla de reacción se agitó a temperatura ambiente durante 2 h. Se añadió agua (5 mL) y la solución se extrajo con DCM (10 mL x 2), las fases orgánicas combinadas se lavaron con una disolución saturada de NaCl, se secaron sobre sulfato de sodio anhidro y se filtraron. El disolvente se evaporó a presión reducida para proporcionar el compuesto 20.2 (90 mg, rendimiento: 52 %) como un sólido amarillo.
m/z: [M+H]+ 309
Realización 25: Síntesis de los compuestos 20.3-20.6
Los compuestos 20.3~20.6 se sintetizaron siguiendo el método sintético utilizado para el compuesto 20.2 de la
Realización 24, mediante la reacción del cloruro de metanosulfonilo que se utilizó en lugar del cloruro de acetilo en la etapa 2, con el compuesto 16.16, 16.55, 16.56 o 16.57:

Realización 26: Síntesis del compuesto 21.5

Etapa 1: síntesis del compuesto 21.2
Se añadieron el compuesto 21.1 (10,0 g, 66,2 mmol), ácido m-cloroperbenzoico (m-CPBA) (16,3 g, 94,5 mmol) y DCM (140 mL) a un matraz de 250 mL y se agitó a temperatura ambiente durante la noche. Se añadió una solución acuosa de bicarbonato de sodio saturado para ajustar el pH a 7~8, la mezcla se extrajo con DCM (50 mL x 2), las fases orgánicas combinadas se lavaron con una disolución saturada de NaCl, se secaron sobre sulfato de sodio anhidro y se filtraron, el disolvente se destiló a presión reducida. Al residuo se añadió oxicloruro de fósforo (60 mL) y se calentó a reflujo y se agitó durante 4 h. El oxicloruro de fósforo se evaporó a presión reducida, el residuo se vertió en agua con hielo y el pH se ajustó a neutro con carbonato de sodio. Después de la filtración, el disolvente se evaporó a presión reducida, el residuo se purificó mediante cromatografía en columna Flash (éter de petróleo/acetato de etilo = 0 %~30 %) para proporcionar el compuesto 21.2 (1,94 g, rendimiento: 16 %) como un aceite naranja.
m/z: [M+H]+ 186
Etapa 2: síntesis del compuesto 21.3
Se añadieron el compuesto 21.3 (1,94 g, 40,5 mmol), m-CPBA (2,25 g, 13,1 mmol) y DCM (25 mL) a un matraz de 100 mL y se agitó a temperatura ambiente durante la noche. Se añadió una solución acuosa saturada de bicarbonato de sodio para ajustar el pH a 7~8, la mezcla se extrajo con DCM (50 mL x 2), las fases orgánicas combinadas se lavaron con una disolución saturada de NaCl, se secaron sobre sulfato de sodio anhidro, se filtraron y se concentraron a presión reducida para producir el compuesto 21.3 (1,90 g, rendimiento: 90%) como un sólido blanco.
m/z: [M+H]+ 202
Etapa 3: síntesis del compuesto 21.4
El compuesto 21.3 (1,90 g, 9,42 mmol) se añadió a oxicloruro de fósforo (20 mL) y se agitó a reflujo durante 4 h. El oxicloruro de fósforo se evaporó a presión reducida, el residuo se vertió en agua con hielo y el pH se ajustó a neutro con carbonato de sodio. La mezcla resultante se extrajo con acetato de etilo (50 mL x 3), las fases orgánicas combinadas se lavaron con una disolución saturada de NaCl, se secaron sobre sulfato de sodio anhidro, se filtraron y concentraron a presión reducida, el residuo se purificó mediante cromatografía en columna Flash (elución: acetato de etilo /éter de petróleo = 0%~50%) para proporcionar el compuesto 21.4 (1,40 g, rendimiento: 68%) como un sólido amarillo.
m/z: [M+H]+ 220
Etapa 4: síntesis del compuesto 21.5
Se añadió el compuesto 21.4 (0,40 g, 1,82 mmol), etanolamina (1,8 mL) y metanol (40 mL) a un matraz de 100 mL y se agitó a temperatura ambiente durante la noche, el disolvente se evaporó a presión reducida, el residuo se purificó mediante cromatografía en columna Flash (metanol/DCM = 0%~5%) para proporcionar el compuesto 21.5 (0,37 g, rendimiento: 95%) como un aceite amarillo.
m/z: [M+H]+ 213
Realización 27: Síntesis del compuesto 22.1

El compuesto 16.17 (500 mg, 1,67 mmol) se disolvió en acido acético (2,5 mL), se le añadió gota a gota peróxido de hidrógeno (2,5 mL) en un baño de agua con hielo, el sistema se agitó a temperatura ambiente durante la noche. Parte del disolvente se evaporó a presión reducida usando una bomba de agua, el residuo se vertió en agua (100 mL), se enfrió a 0°C y se agitó durante 15 min y después se filtró usando un embudo Buchner, el sólido obtenido se lavó con agua (10 mL x 2) y éter de petróleo (10 mL x 2) y se secó para proporcionar el compuesto 22.1 (460 mg, rendimiento: 83 %) como un sólido blanco.
m/z: [M+H]+ 331
Realización 28: Síntesis de los compuestos 22.2~22.6
Los Compuestos 22.2~22.6 se sintetizaron siguiendo el método sintético utilizado para el compuesto 22.1 de la Realización 27, reemplazando el compuesto 16.7 con 16.23, 16.25, 16.28, 16.31 o 16.58:

Realización 29: Síntesis de los compuestos 22.7~22.8
Los compuestos 22.7~22.8 se sintetizaron siguiendo el método sintético utilizado para el compuesto 24.1 de la Realización 14 en la etapa 2, por reacción del compuesto 22.4 o 16.64 con bis(pinacolato)diboro:

m/z: [M+H]+379

m/z: [M+H]+365
Realización 30: Síntesis del compuesto 22.11

El compuesto 16.31 (1,22 g, 4,08 mmol) se disolvió en acido acético (10 mL), se añadió peróxido de hidrógeno (10 mL) en un baño de agua con hielo, el sistema se agitó a temperatura ambiente durante 1,5 h. La solución de reacción se vertió en agua (100 mL), la mezcla se enfrió a 0°C y el pH se ajustó a una alcalinidad débil con la solución acuosa saturada de bicarbonato de sodio, se extrajo con una solución al 10% de metanol en DCM (30 mL x 3 ), las fases orgánicas combinadas se lavaron con una disolución saturada de NaCl, se secaron sobre sulfato de sodio anhidro, se filtraron y se concentraron a presión reducida para proporcionar el compuesto 22.11 (1,15 g, rendimiento: 89%) como un sólido amarillo claro.
m/z: [M+H]+ 315
Realización 31: Síntesis del compuesto 23.1

El compuesto 6.2 (150 mg, 0,59 mmol), 2-bromoetanol (81 mg, 0,65 mmol) y carbonato de potasio (122 mg, 0,88 mmol) se añadieron a DMF (8 mL), el sistema se agitó a 90°C durante 1 h. La reacción se paró mediante la adición de agua y se extrajo con acetato de etilo después de enfriar a temperatura ambiente. Las fases orgánicas combinadas se lavaron con una disolución saturada de NaCl, se secaron sobre sulfato de sodio anhidro, se filtraron y concentraron, el residuo se purificó mediante cromatografía en columna sobre gel de sílice (éter de petróleo/acetato de etilo = 1/1) para producir el compuesto 23.1 (70 mg, rendimiento: 27%) como un sólido blanco.
m/z: [M+H]+ 299
Realización 32: Síntesis del compuesto 24.1

El compuesto 6.2 (150 mg, 0,59 mmol), 2-cloro-N-metilacetamida (70 mg, 0,65 mmol) y carbonato de cesio (287 mg, 0,88 mmol) se disolvieron en DMF (5 mL), el sistema se agitó a 75°C durante 2h. La reacción se paró mediante la adición de agua. Los sólidos se precipitaron, filtraron y lavaron con agua, la torta del filtro se secó para proporcionar el compuesto 24.1 (85 mg, rendimiento: 44%) como un sólido gris.
m/z: [M+H]+ 326
Realización 33: Síntesis del compuesto 24.2
El compuesto 24.2 se sintetizó siguiendo el método sintético utilizado para el compuesto 24.1 de la Realización 32, reemplazando 2-cloro-N-metilacetamida con 1 -cloro-2-metil-2-propanol:

m/z: [M+H]+ 327
Realización 34: Síntesis del compuesto 25.1

Se añadió el compuesto 16.30 (0,3 g, 0,8 mmol), una solución de metilamina en tetrahidrofurano (2,0 M, 10,0 mL) a un tubo estanco de 25 mL. El sistema de reacción se agitó a temperatura ambiente durante 12 h y después se filtró. La torta del filtro se lavó con éter de petróleo y se secó para proporcionar el compuesto 25.1 (180 mg, rendimiento: 66%) como un sólido blanco.
m/z: [M+H]+ 312
Realización 35: Síntesis del compuesto 26.1

Se añadieron isocianato de clorosulfonilo (0,2 g, 1,41 mmol), terc-butanol (0,19 g, 2,54 mmol), DCM (3,0 mL) a un matraz de 25 mL. El sistema de reacción se agitó a 0°C durante 1,5 horas. Y después a esta solución se le añadió el compuesto 20.1 (0,38 g, 1,41 mmol) y una solución de trietilamina (0,71 g, 7,0 mmol) en DCM (2 mL) a 0°C. La mezcla resultante se agitó a temperatura ambiente durante 3 horas. La reacción se paró mediante la adición de H2O (20 mL), se extrajo con acetato de etilo (20 mL x 2), las fases orgánicas combinadas se lavaron con una disolución saturada de NaCl, se secaron sobre sulfato de sodio anhidro, se filtraron, el disolvente se evaporó a presión reducida, el residuo se purificó por cromatografía en columna en gel de sílice (DCM/metanol = 10/1) para proporcionar el compuesto 26.1 (0,26 g, rendimiento: 42%) como un sólido blanco.
m/z: [M+H]+ 446
Realización 36: Síntesis del compuesto 27.1

A una solución enfriada con hielo del compuesto 16.1 (100 mg, 0,373 mmol) y fosforocloridato de dimetilo (108 mg, 0,746 mmol) en DCM (5 mL) se añadió piridina (148 mg, 1,86 mmol). El sistema de reacción se agitó a temperatura ambiente durante 1 h y después se concentró a presión reducida, el residuo se purificó por TLC preparativa (éter de petróleo: acetato de etilo = 1:1) para proporcionar el compuesto 27.1 (140 mg, rendimiento: 99%) como un aceite incoloro. m/z: [M+H]+ 376
Realización 37: Síntesis del compuesto 28.3

Se añadieron el compuesto 16.60 (1,55 g, 3,99 mmol) y yoduro de sodio (1,79 g, 8,93 mmol) a piridina (15 mL), el sistema se agitó a 115°C durante la noche. El sistema se enfrió a temperatura ambiente y después se concentró a presión reducida, el residuo se disolvió en agua, se extrajo con terc-butil metil éter para eliminar la impureza. La fase acuosa se ajustó a pH=2 con ácido clorhídrico 2N y después se extrajo con DCM. Las fases orgánicas combinadas se lavaron con una disolución saturada de NaCl, se secaron sobre sulfato sódico anhidro, se filtraron y se concentraron para dar el compuesto 28.1 (1,15 g, rendimiento: 80%) como un sólido blanco.
m/z: [M+H]+ 360
Etapa 2: síntesis del compuesto 28.2
Se añadió cloruro de oxalilo (0,81 g, 6,39 mmol) a DCM (20 mL), se añadieron 2 gotas de DMF y luego se agitó a temperatura ambiente durante 15 min. A esta solución se añadió gota a gota una solución del compuesto 28.1 (1,15 g, 3,19 mmol) en DCM (20 mL) y la mezcla resultante se agitó a temperatura ambiente durante 40 min, después se concentró a presión reducida para producir el compuesto 1.4 (1,21 g, rendimiento : 100%) como un aceite amarillo. Etapa 3: síntesis del compuesto 28.3
A una solución del compuesto 28.2 (1,21 g, 3,19 mmol) en THF (50 mL) se le añadió una solución de bromuro de metilmagnesio en éter etílico (2,1 mL, 6,30 mmol, 3 M) a -30°C. El sistema de reacción se agitó a -30°C durante 1,5 horas y después se paró la reacción mediante la adición de la solución acuosa de cloruro de amonio saturado y se extrajo con acetato de etilo. Las fases orgánicas combinadas se lavaron con una disolución saturada de NaCl, se secaron sobre sulfato de sodio anhidro, se filtraron y se concentraron. El residuo se purificó mediante cromatografía en columna sobre gel de sílice (DCM/metanol = 10/1) para proporcionar el compuesto 28.3 (1,0 g, rendimiento: 87%) como un aceite amarillo.
m/z: [M+H]+ 358
Realización 38: Síntesis del compuesto 28.4
El compuesto 28.4 se sintetizó siguiendo el método sintético utilizado para el compuesto 28.3 de la Realización 37, reemplazando el compuesto 16.60 con el compuesto 16.61:

m/z: [M+H]+ 359
Realización 39: Síntesis del compuesto 29.1

El compuesto 16.45 (100 mg, 0,314 mmol) y el reactivo de Lawesson (380 mg, 0,942 mmol) se disolvieron en tolueno (3 mL), el sistema se agitó a 110°C durante 7 días y después se concentró a presión reducida. El residuo se purificó mediante TLC preparativa (DCM/metanol = 20/1) para proporcionar el compuesto 29.1 (75 mg, rendimiento: 71%) como un sólido marrón claro.
m/z: [M+H]+ 334
Realización 40: Síntesis del compuesto 29.2
El compuesto 29.2 se sintetizó siguiendo el método sintético utilizado para el compuesto 29.1 de la Realización 39, reemplazando el compuesto 16.45 con el compuesto 16.64:

m/z: [M+H]+ 335
Realización 41: Síntesis del compuesto 30.1

A una mezcla del compuesto 22.4 (500 mg, 1,51 mmol) y pentasulfuro de fósforo (1,01 g, 4,53 mmol) se le añadió piridina (20 mL), la mezcla de reacción se agitó a 110°C durante 2 h y después se calentó hasta 130°C y se agitó durante 2 h. La mezcla de reacción se concentró a presión reducida y el residuo se dispersó en una solución de amoníaco (10 mL) y se agitó durante 10 min. Después de la filtración, la torta del filtro se dispersó nuevamente en una solución de amoníaco (10 mL), se agitó durante 10 min. Después de la filtración, la torta del filtro se secó al vacío para proporcionar el compuesto 30.1 (410 mg, rendimiento: 78 %) como un sólido gris.
m/z: [M+H]+ 347
Realización 42: Síntesis de los compuestos 30.2-30.6
Los compuestos 30.2~30.6 se sintetizaron siguiendo el método sintético utilizado para el compuesto 30.1 de la Realización 41, reemplazando el compuesto 22.4 con los compuestos 16.42, 16.62, 16.68, 16.74 o 16.75:

Realización 43: Síntesis del compuesto 1-1-1

Etapa 1: síntesis del compuesto 1-1
A una solución del compuesto 1.20 (101 mg, 0,19 mmol) en THF (5 mL) se le añadió sucesivamente 6-bromoisoquinolin-1(2H)-ona (44 mg, 0,2 mmol), solución acuosa de carbonato de sodio (2,0 M, 2,0 mL) y Pd(dppf)Cl2 (10 mg) bajo protección de N2 , después de la adición, el sistema de reacción se reemplazó 3 veces por N2 , se agitó a 70°C durante de 20 min a 2 h, hasta que la TLC detectó que la reacción se había completado. La reacción se paró mediante la adición de agua (10 mL), la mezcla se extrajo con acetato de etilo (10 mL x 2), las fases orgánicas combinadas se lavaron con una disolución saturada de NaCl, la capa orgánica se separó, se secó sobre sulfato de sodio anhidro, se filtró y se concentró, y el residuo se purificó por cromatografía en columna sobre gel de sílice (éter de petróleo/acetato de etilo = 1/1) para proporcionar el compuesto 1-1 (44 mg, rendimiento: 43%) como un aceite incoloro.
Etapa 2: síntesis del compuesto 1-1-1
A una solución enfriada con hielo del compuesto 1-1-1 (20 mg, 0,04 mmol) en DCM (2 mL) se añadió TFA (2 mL). Después de la adición, el sistema de reacción se agitó a 0°C durante 20 min y después el disolvente se concentró a presión reducida, el residuo se lavó con una solución de acetato de etilo al 50 % en éter de petróleo y se filtró para producir el compuesto 1-1. -1 (9,4 mg, rendimiento: 44%) como un sólido blanco.
H1 NMR (400 MHz, CD3OD): 58.37 (d, J = 8,0 Hz, 1 H), 7,95 (s, 1 H), 7,84 (d, J =8,0 Hz, 1 H), 7,54 (s, 1 H), 7,46 (s, 2 H), 7,21 (d, J = 8,0 Hz, 1 H), 6,91 (s, 1 H), 6,77 (d, J =8,0 Hz, 1 H), 3,43 (t, J = 7,0 Hz, 4 H), 3,30 (s, 2 H), 1,76-1,61 (m, 4 H), 1,08 - 0,79 (m, 6 H); m/z: [M+H]+ 429.
Realización 44: Síntesis de los compuestos 1 -1 -2~1 -1 -4
Los compuestos 1-1 -2~1 -1 -4 se sintetizaron siguiendo el método sintético utilizado para el compuesto 1-1-1 de la Realización 43, mediante la reacción del compuesto 1.8 que se utilizó en lugar del compuesto 1.20 en la etapa 1, con el compuesto 15.1, 15.9 o 15.10:


Realización 45: Síntesis de los compuestos 1-2-1-1-2-51
Los compuestos 1 -2-1 ~1-2-51 se sintetizaron siguiendo el método sintético utilizado para el compuesto 1-1-1 de la Realización 43, por reacción del compuesto correspondiente 15.1, 15.5, 15.6, 15.7, 15.8, 16.1, 16.2, 16.3, 16.9, 16.11, 16.12, 16.13, 16.16, 16.22, 16.25, 17.1, 17.2, 18.1, 18.2, 20.3, 22.3, 26.1, 16.33, 16.34, 16.35, 16.37, 16.38, 16.40, 16.41, 16.42, 16.43, 16.45, 16.46, 16.47, 16.48, 16.49, 16.50, 16.51, 16.52, 16.53, 16.54, 16.59, 16.62, 16.68, 16.69, 16.70, 16.71,16.72, 27.1 o 28.3 que se usó en lugar de 6-bromoisoquinolin-1 (2H)-ona en la etapa 1, con el compuesto 1.20:





1H NMR (400 MHz, CDsOD): 58,68 (s, 1 H), 8,10 (dd, J = 8,0, 4,0 Hz, 1 H), 7,82 - 7,76 (m, 3 H), 7,67 (d, J = 8,0 Hz, 1 H), 7,44 (d, J = 8,0 Hz, 1 H), 7,12 (s, 1 H), 6,78 (d, J = 4,0 Hz, 1 H), 4,22 (t, J = 5,2 Hz, 2 H), 3,93 (t, J = 5,2 Hz, 2 H), 3,50 (ba. s, 4 H), 3,42 - 3,37 (m, 2 H), 1,73 - 1,65 (m, 4 H), 0,95 (ba. s, 6 H); m/z: [M+H]+ 473.
Realización 46: Síntesis del compuesto 1-2-52

El compuesto 2-1 se sintetizó siguiendo el método sintético usado para el compuesto 1-1 de la Realización 43, por reacción del compuesto 16.59 correspondiente que se usó en lugar de 6-bromoisoquinolin-1(2H)-ona en la etapa 1, con el compuesto 1.20.
A una solución del compuesto 2-1 (100 mg, 0,15 mmol) en DCM (3,0 mL) se le añadió TFA (1,5 mL), el sistema de reacción se agitó a temperatura ambiente durante 1,5 h y después se concentró a presión reducida. El residuo se recristalizó de etanol para dar el compuesto 1 -2-52 (50 mg, rendimiento: 53%) como un sólido blanco.
1H NMR (400 MHz, DMSO-afe): 59,88 (s, 1H), 9,12 (s, 1H), 8,35 (d, J = 8.8 Hz, 1H), 8,03 (d, J = 1,6 Hz, 1H), 7,89 -7,80 (m, 3 H), 7,72 (d, J = 8,4 Hz, 1 H), 7,48 (d, J = 7,2 Hz, 1H), 7,06 (s, 1H), 6,74 (d, J = 7.2 Hz, 1H), 4,37 (d, J = 12.4 Hz, 2H), 3,57 (s, 2 H), 3,35 (s, 6 H), 1,61 - 1,54 (m, 4H), 0,91- 0,82 (m, 6H); m/z: [M+H]+ 523.
Realización 47: Síntesis de los compuestos 1-3-1 ~ 1-3-2
Los compuestos 1 -3-1 ~1 -3-2 se sintetizaron siguiendo el método sintético utilizado para el compuesto 1-1-1 de la Realización 43, mediante la reacción del compuesto 15.4 o 19.1 correspondiente que se utilizó en lugar de 6 bromoisoquinolin-1 (2H)-ona en la etapa 1, con el compuesto 1.20:


Realización 48: Síntesis de los compuestos 1-4-1 ~ 1-4-2
Los compuestos 1-4-1 ~ 1-4-2 se sintetizaron siguiendo el método sintético utilizado para el compuesto 1-1-1 de la Realización 43, mediante la reacción del compuesto 15.3 o 17.3 correspondiente que se utilizó en lugar de 6-bromoisoquinolin-1 (2H)-ona en la etapa 1, con el compuesto 1.20:


Realización 49: Síntesis de Io s compuestos 1-6-1 y 1-6-2
Los compuestos 1-5-1 y 1-5-2 se sintetizaron siguiendo el método sintético utilizado para el compuesto 1-1-1 de la Realización 43, mediante la reacción del compuesto 15.9 o 15.10 correspondiente que se utilizó en lugar de 6 bromoisoquinolin-1 (2H)-ona en la etapa 1, con el compuesto 1.20:


Realización 50: Síntesis de los compuestos 1-6-1 y 1-6-2
Los compuestos 1-6-1 y 1-6-2 se sintetizaron siguiendo el método sintético utilizado para el compuesto 1-1-1 de la Realización 43, mediante la reacción del compuesto 19.2 o 21.5 correspondiente que se utilizó en lugar de 6-bromoisoquinolin-1 (2H)-ona en la etapa 1, con el compuesto 1.20:


Realización 51: Síntesis de los compuestos 1-7-1-1-7-6
Los compuestos 1 -7-1 ~1 -7-6 se sintetizaron siguiendo el método sintético utilizado para el compuesto 1-1-1 de la Realización 43, por reacción del compuesto correspondiente 16.18, 16.20, 17.5, 18.3, 22.1 o 20.5 que se utilizó en lugar de 6-bromoisoquinolin-1 (2H)-ona en la etapa 1, con el compuesto 1.20:



Realización 52: Síntesis de Ios compuestos 1-8-1-1-8-22
Los compuestos 1 -8-1 ~1 -8-22 se sintetizaron siguiendo el método sintético utilizado para el compuesto 1-1-1 de la Realización 43, por reacción del compuesto correspondiente 18.4, 16.21, 16.24, 17.4, 22.4, 22.5, 16.32, 20.6, 16.36, 16.39, 22.11, 22.6, 16.44, 16.60, 16.63, 16.64, 28.4, 16.66, 16.67, 16.73 o 16.75 que se usó en lugar de 6-bromoisoquinolin-1 (2H)-ona en la etapa 1, con compuesto 1.20 :



Realización 53: Síntesis de Io s compuestos 1-9-1-1-9-6
Los compuestos 1 -9-1 ~1 -9-6 se sintetizaron siguiendo el método sintético utilizado para el compuesto 1-1-1 de la Realización 43, mediante la reacción del compuesto correspondiente 17.3, 23.1,24.1,24.2, 25.1 o 20.4 que se utilizó en su lugar de 6-bromoisoquinolin-1 (2H)-ona en la etapa 1, con el compuesto 1.20:


Realización 54: Síntesis de los compuestos 1-10-1-1-10-11
Los compuestos 1 -10-1 ~1 -10-11 se sintetizaron siguiendo el método sintético utilizado para el compuesto 1-1-1 de la Realización 43, reemplazando la 6-bromoisoquinolin-1(2H)-ona y el compuesto 1.20 con el correspondiente compuesto 18.1,22.4, 16.22, 22.3, 24.1, 16.21, 16.34, 16.18, 16.33, 22.2 o 22.1 y 1.22 en la etapa 1:


Realización 56: Síntesis del compuesto 1-11-1
El compuesto 1-11-1 se sintetizó siguiendo el método sintético utilizado para el compuesto 1-1-1 de la Realización 43, reemplazando la 6-bromoisoquinolin-1 (2H)-ona y el compuesto 1.20 con los correspondientes compuestos 18.1 y 1.21 en la etapa 1:

1H NMR (400 MHz, CD3OD): 58,42 (d, J = 8,4 Hz, 1 H), 7,91 (s, 1 H), 7,75 (d, J = 8,4 Hz, 1 H), 7,62 (d, J = 6.8 Hz, 1
H), 7,51 (d, J = 11.2 Hz, 1 H), 7,39 (d, J = 7.6 Hz, 1 H), 7,08 (s, 1 H), 6,77 (d, J = 7.2 Hz, 1 H), 4,76 (s, 2 H), 3,55 - 3,41 (m, 4 H), 3,32 (superposición con disolvente, 2 H), 1,77 - 1,65 (m, 4 H), 1,05 - 0,85 (m, 6 H); m/z: [M+H]+ 504.
Realización 57: Síntesis del compuesto 1-12-1
El compuesto 1-12-1 se sintetizó siguiendo el método sintético utilizado para el compuesto 1-1-1 de la Realización 43, reemplazando la 6-bromoisoquinolin-1 (2H)-ona y el compuesto 1.20 con los correspondientes compuestos 18.4 y 1.19 en la etapa 1:

1H NMR (400 MHz, CD3OD): 58,40 (d, J = 8,0 Hz, 1 H), 7,98 (s, 1 H), 7,90 - 7,79 (m, 2 H), 7,78 - 7,65 (m, 2 H), 7,37 (d, J = 8,0 Hz, 1 H), 7,16 (s, 1 H), 6,79 (d, J = 8,0 Hz, 1 H), 4,75 (s, 2 H), 3,71 - 3,44 (m, 6 H), 3,25 - 3,23 (m, 2 H), 1,76 - 1,64 (m, 2 H), 1,03 - 0,80 ( m,3H); m/z: [M+H]+ 488.
Realización 58: Síntesis de los compuestos 1-14-1-1-14-8
Los compuestos 1-14-1~1-14-8 se sintetizaron siguiendo el método sintético utilizado para el compuesto 1-1-1 de la Realización 43, por reacción del compuesto correspondiente 29.1, 29.2, 30.1, 30.2, 30.3, 30.4, 30.5 o 30.6 que se utilizó en lugar de 6-bromoisoquinolin-1 (2H)-ona en la etapa 1, con el compuesto 1.20:



Realización 59: Síntesis del compuesto 1-4-10

Etapa 1: síntesis del compuesto 1-3
Los compuestos 1 -2 se sintetizaron siguiendo el método de síntesis usado para el compuesto 1 -1 de la Realización 43, usando los compuestos 1.20 y 16.4 como materiales de partida.
El compuesto 1-2 (15 mg, 0,022 mmol) se disolvió en THF (5 mL), se le añadió TBAF (8,5 mg, 0,033 mmol) y luego se agitó a temperatura ambiente durante 2 h. La mezcla se añadió a una solución acuosa de bicarbonato de sodio saturado (10 mL) y se extrajo con acetato de etilo (10 mL x 2). Las fases orgánicas combinadas se lavaron con una disolución saturada de NaCl, se secaron sobre sulfato de sodio anhidro, se filtraron, el disolvente se evaporó a presión reducida y el residuo se purificó por TLC preparativa (acetato de etilo) para proporcionar el compuesto 1-3 (12 mg, rendimiento: 96%) como un sólido amarillo.
m/z: [M+H]+ 575
Etapa 2: síntesis del compuesto 1-4-10
El compuesto 1-3 (12 mg, 0,021 mmol) se disolvió en DCM (2 mL), se añadió TFA (1 mL) en un baño de agua con hielo y luego se retiró el baño de agua con hielo y la mezcla se agitó a temperatura ambiente durante 1 hora. La mezcla se concentró directamente a sequedad bajo presión reducida. El residuo se purificó mediante HPLC preparativo para proporcionar el compuesto 1-4-10 (1,55 mg, rendimiento: 16%) como un sólido amarillo.
1H NMR (400 MHz, CD3OD): 58,07 - 8,04 (m, 1 H), 7,78 - 7,74 (m, 1 H), 7,72 - 7,67 (m, 2 H), 7,67 - 7,62 (m, 2 H), 7,09 (s, 1 H ), 3,84 - 3,69 (m, 6 H), 3,55 - 3,40 (m, 4 H), 3,38 - 3,33 (m, 2 H), 3,16 - 3,11 (m, 2 H), 1,76 - 1,65 (m, 4 H), 1,02 - 0,88 (m, 6 H); m/z: [M+H]+ 475.
Realización 60: Síntesis del compuesto 2-1-1

Etapa 1: síntesis del compuesto 2-1
A una solución del compuesto 1.12 (30 mg, 0,07 mmol) en THF (1 mL) se le añadió sucesivamente una solución acuosa de carbonato de sodio (2,0 M, 0,5 mL), Pd(dppf)Cl2 (10 mg, 0,01 mmol) y el compuesto 15.2 (30 mg, 0,10 mmol) bajo protección de N2. Después de la adición, el sistema de reacción se reemplazó 3 veces por N2 y se agitó a 80°C durante 30 min. La reacción se paró mediante la adición de agua (25 mL), la mezcla se extrajo con acetato de etilo (10 mL x 2), las fases orgánicas combinadas se lavaron con una disolución saturada de NaCl y la fase orgánica se separó, se secó sobre sulfato de sodio anhidro, se filtró y se concentró. El residuo se purificó mediante cromatografía en columna sobre gel de sílice (acetato de etilo:éter de petróleo = 1:1) para proporcionar el compuesto 2-1 (35 mg, rendimiento: 97%) como un sólido blanco.
Etapa 2: síntesis del compuesto 2-1-1
A una solución enfriada con hielo del compuesto 2-1 (35 mg, 0,07 mmol) en DCM (1 mL) se añadió TFA (1 mL) y después el sistema de reacción se agitó a 20°C durante 2 h. La reacción se paró mediante la adición de agua (10 mL), la mezcla se extrajo con acetato de etilo (10 mL x 2), las fases orgánicas combinadas se lavaron con una disolución saturada de NaCl, la fase orgánica se separó, se secó sobre sulfato de sodio anhidro, se filtró y se concentró, y el residuo se purificó por HPLC preparativo para proporcionar el compuesto 2-1-1 (15 mg, rendimiento: 55%) como un sólido blanco.
1H NMR (400 MHz, CD3OD): 58.42 (d, J = 8.2 Hz, 1 H), 7,97 (s, 1 H), 7,88 - 7,80 (m, 2 H), 7,75 (s, 1 H), 7,70 (d, J = 8,0 Hz, 1H), 7,45 (d, J = 7,2 Hz, 1 H), 7,16 (s, 1 H), 6,80 (d, J = 7,2 Hz, 1 H), 4,11 (c, J = 7,2 Hz, 2 H), 3,73 (s, 8 H), 3,38 (s, 2 H), 1,38 (t, J = 7,2 Hz, 3 H); m/z: [M+H]+ 443.
Realización 61: Síntesis de los compuestos 2-1-2-2-1-4
Los compuestos 2-1-2~2-1-4 se sintetizaron siguiendo el método sintético utilizado para el compuesto 2-1-1 de la Realización 60, mediante la reacción del compuesto 1.9, 1.11 o 1.13 correspondiente que se utilizó en lugar del compuesto 1.12 en la etapa 1, con el compuesto 15.2:


Realización 62: Síntesis de los compuestos 2-2-1 ~2-2-4
Los compuestos 2-2-1~2-2-4 se sintetizaron siguiendo el método sintético usado para el compuesto 2-1-1 de la Realización 60, por reacción del compuesto 1.14, 1.15, 1.16 o 1.17 correspondiente que se usó en lugar del compuesto 1.12 en la etapa 1, con el compuesto 22.7:



Realización 63: Síntesis de Ios compuestos 2-3-1-2-3-3
Los compuestos 2-3-1~2-3-3 se sintetizaron siguiendo el método sintético utilizado para el compuesto 2-1-1 de la Realización 60, mediante la reacción del compuesto 1.15, 1.16 o 1.17 correspondiente que se utilizó en lugar del compuesto 1.12 en la etapa 1, con el compuesto 22.8:


Realizaciones de bioensayos
Realización 1: ensayo basado en células TLR8
En este ensayo, la bioactividad del compuesto de fórmula I se midió usando un ensayo basado en células. El ensayo se realizó en células de riñón embrionario humano (HEK293) que expresan TLR8 u otros miembros de la familia de TLR como TLR4, TLR7 y TLR9 (para pantallas de selectividad). La actividad de los agonistas de TLR específicos se evaluó utilizando el gen informador de la fosfatasa alcalina embrionaria secretada (SEAP) que está vinculado a la activación de NF-k B en respuesta a la estimulación de TLR. La actividad del SEAP se midió usando el sustrato Quanti-Blue (InvivoGen) después del tratamiento con agonista de TLR.
El método experimental detallado es el siguiente:
La línea celular HEK-BLUE-hTLR8 se adquirió de InvivoGen. Las células se cultivaron en medio de Eagle modificado por Dulbecco que contenía L-glucosa 4,5 g/L (Sigma-Aldrich) y FBS al 10%. Las células se cultivaron a 37°C, 95 % de humedad y 5 % de CO2.
Las concentraciones de prueba de los compuestos oscilaron entre 0,5 nM y 15 pM con 10 puntos. Se añadió un agonista de TLR8 conocido como control positivo y se añadió 1 pl de DMSO como control negativo.
Las células se prepararon como sigue: Las células se sacaron de la incubadora y se descartó el medio de cultivo. Las células se enjuagaron suavemente con 10 mL de PBS precalentado para un matraz T-150. Se añadieron 12 mL de medio de cultivo precalentado y las células se separaron golpeando el matraz. Los grupos de células se disociaron pipeteando suavemente hacia arriba y hacia abajo. Se contaron las células. Se preparó una suspensión de células individuales a 200.000 células por mL en medio de cultivo y se añadieron inmediatamente 200 pl de la suspensión celular (40.000 células por pocillo) a cada pocillo de la placa. La concentración final de DMSO de cada pocillo fue del 0,5 %.
Las células se cultivaron y trataron con los compuestos a 37°C, CO2 al 5% durante 24 h.
Se añadieron 20 pL del sobrenadante de cada pocillo a 180 pL de QUANTI-Blue y las placas se incubaron a 37°C durante 1,5 horas. La densidad óptica se midió usando un espectrofotómetro a 650 nm (datos de OD). La activación
se calculó usando la siguiente ecuación:
% de activación = (valor medio de OD del com puesto -va lo r medio de OD del
DMSO)/(valor medio de OD del control p os itivo - valor medio de OD del DMSO) x 100
Los datos se analizaron utilizando GraphPad para obtener los valores de EC50 y ajustar las curvas de respuesta a la dosis de los compuestos.
Realización 2: ensayo basado en células TLR7
En este ensayo, la bioactividad del compuesto de fórmula (I) se midió utilizando un ensayo basado en células. El ensayo se realizó en células de riñón embrionario humano (HEK293) que expresan TLR7 u otros miembros de la familia de TLR tal como TLR4 y TLR9 (para pantallas de selectividad). La actividad de los agonistas de TLR específicos se evaluó utilizando el gen informador de la fosfatasa alcalina embrionaria secretada (SEAP) que está vinculado a la activación de NF-k B en respuesta a la estimulación de TLR. La actividad de SEAP se midió usando el sustrato Quanti-Blue (InvivoGen) después del tratamiento con agonista de TLR.
El método experimental detallado es el siguiente:
La línea celular HEK-BLUE-hTLR7 se adquirió de InvivoGen. Las células se cultivaron en medio de Eagle modificado por Dulbecco que contenía L-glucosa 4,5 g/L (Sigma-Aldrich) y FBS al 10%. Las células se cultivaron a 37°C, 95 % de humedad y 5 % de CO2.
Las concentraciones de prueba de los compuestos oscilaron entre 0,5 nM y 15 pM con 10 puntos. Se añadió un agonista de TLR8 conocido como control positivo y se añadió 1 pl de DMSO como control negativo.
Las células se prepararon como sigue. Se sacaron las células de la incubadora y se descartó el medio de cultivo. Las células se enjuagaron suavemente con 10 mL de PBS precalentado para un matraz T-150. Se añadieron 12 mL de medio de cultivo precalentado y las células se separaron golpeando el matraz. Los grupos de células se disociaron pipeteando suavemente hacia arriba y hacia abajo. Se contaron las células. Se preparó una suspensión de células individuales a 200.000 células por mL en medio de cultivo y se añadieron inmediatamente 200 pl de la suspensión celular (40.000 células por pocillo) a cada pocillo de la placa. La concentración final de DMSO de cada pocillo fue del 0,5 %.
Las células se cultivaron y trataron con los compuestos a 37°C, CO2 al 5% durante 24 horas.
Se añadieron 20 pL del sobrenadante de cada pocillo a 180 pL de QUANTI-Blue y las placas se incubaron a 37°C durante 1,5 horas. La densidad óptica se midió usando un espectrofotómetro a 650 nm (datos de OD). La activación se calculó usando la siguiente ecuación:
% de activación = (valor medio de OD del com puesto -va lo r medio de OD del
DMSO)/(valor medio de OD del control p o s itivo - valor medio de OD del DMSO) x 100
Los datos se analizaron utilizando GraphPad para obtener los valores de EC50 y ajustar las curvas de respuesta a la dosis de los compuestos.
El intervalo de valores de EC50 de TLR8 y TLR9 es el siguiente: - representa > 50 pM, -- representa 10-50 pM, representa 1-10 pM, + representa 0,1-1 pM, ++ representa < 0,1 pM, /representa No hecho.



Claims (13)
1. Un compuesto de fórmula (I), un estereoisómero, un derivado isotópico estable o una sal farmacéuticamente aceptable del mismo;

en donde, L es -C(O)-, -C(S)- o -S(O)2-;
L1 es un enlace;
R se selecciona del grupo que consiste en R-1 a R-19:

cada uno de R7 y R7a se selecciona independientemente de hidrógeno, halógeno, alquilo, haloalquilo, alquenilo, alquinilo, Cy1, -L2-Cy1, -SRd, -ORd, -C(O)ORd, -C(O)Rd, - C(O)NRdRe, -C(O)NRdS(O)2Re, -C(= -S(O)2 Re y S(O)2NRdRe; en donde el alquilo, alquenilo o alquinilo no está sustituido o está sustituido en cualquier posición por uno o más de un sustituyente seleccionado del grupo que consiste en -CN, -NO2 , -SRd, -ORd, -OC(O)Rd, -OC(O)ORd, -OC(O)NRdRe, -C(O)ORd, -C(O)Rd, -C(O)NRdRe, -C(O)NRdS(O)2Re, -NRdRe, -NRdC(O)Re, -N(Rd)C(O)ORe, -N(Rd)C(O)NRdRe, -NRdC(=NH)Re, -NRdC(=NH)NRdRe, -NRdS(O)2Re, -NRdS(O)2NRdRe, -N(Rd)C(O)N(Rd)S(O)2Re, -S(O)1-2Re, -S(O)2NRdRe, -S(O)(=NCN)Re, -S(O)(=NRd)Re, -S(O)(=NSO2Rd)Re, -S(O)2N(Rd)C(O)Re, -S(O)2N(Rd)C(O)NRdRe, -P(O)(ORd)2 , -OP(O)(ORd)2 o -B(ORd)2 ; o R7 es

cada uno de R8 es independientemente hidrógeno, halógeno, alquilo, haloalquilo, haloalcoxi, alquenilo, alquinilo, Cy1,
-L2-Cy1, -CN, -NO2 , -SRd, -ORd, -OC(O)Rd, -OC(O)ORd, -OC(O)NRdRe, -C(O)ORd, -C(O)Rd, -C(O)NRdRe,
-C(O)NRdS(O)2Re, -C(=NH)Re, -C(=NH)NRdRe, -NRdRe, -NRdC(O)Re, -N(Rd)C(O)ORe, -N(Rd)C(O)NRdRe, -NRdS(O)2 Re, -NRdC(=NH)Re, -NRdC(=NH)NRdRe, -S(O)i-2Re, -S(O)2NRdRe y -NRdS(O)2NRdRe; en donde el alquilo, alquenilo o alquinilo no está sustituido o está sustituido en cualquier posición por uno o más de un sustituyente seleccionado del grupo que consiste en -CN, -NO2 , -SRd, -ORd, -OC(O)Rd, -OC(O)ORd, -OC(O)NRdRe, -C(O)ORd, -C(O)Rd, -C(O)NRdRe, -C(O)NRdS(O)2Re, -NRdRe, -NRdC(O)Re, -N(Rd)C(O)ORe, -N(Rd)C(O)NRdRe, -NRdC(=NH)Re, -NRdC(=NH)NRdRe, -NRdS(O)2Re, -NRdS(O)2NRdRe, -N(Rd)C(O)N(Rd)S(O)2Re, -S(O)1-2Re, -S(O)2NRdRe, -S(O)2N(Rd)C(O)Re o -S(O) 2N(Rd)C(O)NRdRe;
cada uno de n es independientemente 1, 2 ó 3;
L2 es un enlace, alquenileno de C2-6, alquinileno de C2-6, o -(CRa1Rb1)m-;
cada uno de Cy1 es independientemente cicloalquilo, heterocicloalquilo, arilo o heteroarilo; el Cy1 no está sustituido o está sustituido selectivamente en cualquier posición por uno o más de un sustituyente seleccionado del grupo que consiste en halógeno, alquilo, haloalquilo, haloalcoxi, hidroxialquilo, aminoalquilo, alquenilo, alquinilo, -CN, -NO2 , -SRd2, -ORd2, -OC(O)Rd2, - OC(O)ORd2, -OC(O)NRd2Re2, -C(O)ORd2, -C(O)Rd2, -C(O)NRd2Re2, -NRd2Re2, -NRd2C(O)Re2, -N(Rd2)C(O)ORe2, -N(Rd2)C(O)NRd2Re2, -NRd2S(O)2Re2, -NRd2C(=NH)Re2, -NRd2C(=NH)NRd2Re2, -S(O)1-2Re2 , -S(O)2NRd2 Re2 y - NRd2S(O)2NRd2 Re2 ;
R1, R2 y R3 se seleccionan independientemente de hidrógeno, deuterio o alquilo;
o, R2 y R3 junto con el átomo de C al que están unidos, forman un cicloalquilo de C3-8 o un heterocicloalquilo de 3 a 8 miembros;
R4 , R5 y R6 se seleccionan independientemente de hidrógeno, deuterio, halógeno, amino, ciano, nitro, alquilo de C1-6, haloalquilo de C1-6, haloalcoxi de C1-6, alcoxi de C1-6, alquenilo de C2-6, alquinilo de C2-6, hidroxialquilo de C1-6, cicloalquilo de C3-8, heterocicloalquilo de 3 a 8 miembros, fenilo o heteroarilo de 5 a 6 miembros;
RA se selecciona de -ORd1 o -NRd1Re1 ;
RB es -NRd1Re1 ;
cada uno de Ra1 y Rb1 se selecciona independientemente de hidrógeno, deuterio, halógeno, alquilo de C1-6, alcoxi de C1-6, alquenilo de C2-6, alquinilo de C2-6, cicloalquilo de C3-8, heterocicloalquilo de 3 a 8 miembros, arilo de C6-10, heteroarilo de 5 a 6 miembros, cicloalquilo de C3-8 alquilo de C1-6, heterocicloalquilo de 3 a 8 miembros alquilo de C1-6, fenilo alquilo de C1-6 o heteroarilo de 5 a 6 miembros alquilo de C1-6; el alquilo de C1-6, alcoxi de C1-6, alquenilo de C2-6, alquinilo de C2-6, cicloalquilo de C3-8, heterocicloalquilo de 3 a 8 miembros, arilo de C6-10 o heteroarilo de 5 a 6 miembros no está sustituido o está sustituido en cualquier posición por 1 a 3 sustituyentes seleccionados del grupo que consiste en halógeno, hidroxilo, amino, carboxilo, alquenilo de C2-6, alquinilo de C2-6, alquilo de C1-6, haloalquilo de C1-6, alcoxi de C1-6 y haloalcoxi de C1-6;
o Ra1 y Rb1 junto con el átomo de C al que están unidos, forman un cicloalquilo de C3-8 o un heterocicloalquilo de 3 a 8 miembros;
cada uno de Rd, Re, Rd1 y Re1 se selecciona independientemente de hidrógeno, alquilo de C1-6, alquenilo de C2-6, alquinilo de C2-6, cicloalquilo de C3-8, heterocicloalquilo de 3 a 8 miembros, arilo de C6-10, heteroarilo de 5 a 6 miembros, cicloalquilo de C3-8 alquilo de C1-6, heterocicloalquilo de 3 a 8 miembros alquilo de C1-6, fenilo alquilo de C1-6 o heteroarilo de 5 a 6 miembros alquilo de C1-6; Rd, Re, Rd1 o Re1 no están sustituidos o están sustituidos en cualquier posición por 1 a 3 sustituyentes seleccionados del grupo que consiste en halógeno, hidroxilo, amino, carboxilo, alquilo de C1-6, alcoxi de C1-6, alquilamino de C1-6, haloalquilo de C1-6, haloalcoxi de C1-6, alquenilo de C2-6 y alquinilo de C2-6 ;
o, Rd y Re, o Rd1 y Re1 junto con el átomo de N al que están unidos, forman un heterocicloalquilo de 3 a 8 miembros; el heterocicloalquilo puede comprender además de 1 a 3 heteroátomos seleccionados del grupo que consiste en N, O y S; el heterocicloalquilo no está sustituido o está sustituido en cualquier posición por 1 a 3 sustituyentes seleccionados del grupo que consiste en halógeno, amino, hidroxilo, carboxilo, ciano, alquilo de C1-6, haloalquilo de C1-6, alquenilo de C2-6, alquinilo de C2-6, alquilo de C1-3, amino alquilo de C1-3, -ORd2, -OC(O)Rd2 , -OC(O)ORd2 , -OC(O)NRd2 Re2 , -C(O)ORd2, -C(O)Rd2, -C(O)NRd2Re2, -NRd2Re2, - NRd2C(O)Re2, -N(Rd2)C(O)ORe2, -N(Rd2)C(O)NRd2Re2, -NRd2S(O)2Re2, -NRd2C(=NH)Re2 , -NRd2C(=NH)NRd2 Re2 , -NRd2S(O)2NRd2 Re2, -S(O)1-2Rd2 y -S(O)2NRd2Re2 ;
cada uno de Rd2 y Re2 se selecciona independientemente de hidrógeno, alquilo de C1-6, alquenilo de C2-6, alquinilo de C2-6, cicloalquilo de C3-8, heterocicloalquilo de 3 a 8 miembros, arilo de C6-10, heteroarilo de 5 a 6 miembros, cicloalquilo de C3-8 alquilo de C1-6, heterocicloalquilo de 3 a 8 miembros alquilo de C1-6, fenilo alquilo de C1-6 o heteroarilo de 5 a 6 miembros alquilo de C1-6; Rd2 o Re2 no están sustituidos o están sustituidos en cualquier posición por 1 a 3 sustituyentes seleccionados del grupo que consiste en halógeno, hidroxilo, amino, carboxilo, alquilo de C1-6, alcoxi de C1-6, alquilamino de C1-6, haloalquilo de C1-6, haloalcoxi de C1-6, alquenilo de C2-6 y alquinilo de C2-6 ; o, Rd2 y Re2 junto con el átomo de N al que están unidos, forman un heterocicloalquilo de 3 a 8 miembros;
cada uno de m es 1, 2, 3, 4, 5 o 6.
2. El compuesto de fórmula (I), el estereoisómero, el derivado isotópico estable o la sal farmacéuticamente aceptable del mismo según la reivindicación 1, en el que cada uno de R1, R2 y R3 es H; cada uno de R4 , R5 y R6 se selecciona independientemente de H, D, F, Cl, Br, -CN, -NH2, -CH3 , -OCH3 , -OCF3 , -CH2F, -CHF2 o -CF3.
3. El compuesto de fórmula (I), el estereoisómero, el derivado isotópico estable o la sal farmacéuticamente aceptable del mismo según la reivindicación 1, en el que cada uno de Rd1 y Re1 se seleccionan independientemente
de hidrógeno, alquilo de C1-6, haloalquilo de C1-6, cicloalquilo de C3-8 o un heterocicloalquilo de 3 a 8 miembros; Rd1 o
Re1 no están sustituidos o están opcionalmente sustituidos en cualquier posición por un hidroxilo;
o, Rd1 y Re1 junto con el átomo de N al que están unidos, forman un heterocicloalquilo de C3-8; el heterocicloalquilo
puede contener además 1-3 heteroátomos seleccionados de N, O y S; el heterocicloalquilo no está sustituido o está sustituido en cualquier posición por un sustituyente seleccionado del grupo que consiste en alquilo de C1-4, haloalquilo
de C1-3 y aminoalquilo de C1-3.
4. El compuesto de fórmula (I), el estereoisómero, el derivado isotópico estable o la sal farmacéuticamente aceptable del mismo según la reivindicación 1, en el que RB es -NH2.
5. El compuesto de fórmula (I), el estereoisómero, el derivado isotópico estable o la sal farmacéuticamente aceptable del mismo según la reivindicación 1, en el que,
cada uno de R7 es independientemente hidrógeno, alquilo de C1-6, haloalquilo de C1-6, alquenilo de C2-6, alquinilo de
C2-6, Cy1, -L2-Cy1, -SRd, -ORd, -C(O)ORd, -C(O)Rd, -C(O)NRdRe, -C(=NH)Re, -C(=NH)NRdRe, -S(O)2Re o -S(O)2NRaRe;
en donde el alquilo de C1-6, alquenilo de C2-6 o alquinilo de C2-6 no están sustituidos o están sustituidos en cualquier posición por 1 a 3 sustituyentes seleccionados del grupo que consiste en -CN, -NO2 , -CN, -SRd, -ORd, -OC(O)Rd, -OC(O)ORd, - OC(O)NRdRe, -C(O)ORd, -C(O)Rd, -C(O)NRdRe, -NRdRe, -NRdC(O)Re, -N(Rd)C(O)ORe, -N(Rd)C(O)NRdRe, -NRDS(O)2Re, -NRdC(=NH)Re, -NRdC(=NH)NRdRe, -S(O)1-2Re, -S(O)2NRdRe, -S(O)(=NCN)Re, -S(O)(=NRd)Re, -S(O)(=NSO2Rd)Re, -NRdS(O)2NRdRe, -P(O)(ORd)2, -OP(O)(ORd)2, -B(ORd)2,

y/o, cada uno de R7a es independientemente hidrógeno o alquilo de C1-6;
y/o, cada uno de R8 es independientemente hidrógeno, metilo, etilo, n-propilo, terc-butilo, isopropilo, isobutilo, n-butilo,
-CN, -NO2 , -NH2 , -CF3 , -OCF3 , -CH3 , -OCH3 , F, Cl o Br;
y/o L2 es un enlace, -CH2-, -CH2CH2-, -CH2CH2CH2-, -CH2CH2C(CH3)2-, -CH2CH2CH2CH2- o -CH2CH(CH3)CH2-;
y/o, cada uno de Cy1 es independientemente cicloalquilo de C3-10, heterocicloalquilo de 3 a 10 miembros, arilo de C6-10 o heteroarilo de 5 a 10 miembros; el Cy1 no está sustituido o está sustituido en cualquier posición por 1 a 3 sustituyentes seleccionados del grupo que consiste en F, Cl, Br, alquilo de C1-4, haloalquilo de C1-3, haloalcoxi de C1-3, hidroxialquilo de
C1-3, aminoalquilo de C1-3, alquenilo de C2-6, alquinilo de C2-6, -CN, -NO2 , -SRd2 , -ORd2 , -OC( -OC(O)NRd2Re2, -C(O)ORd2, -C(O)Rd2, -C(O)NRd2Re2, -NRd2Re2, -NRd2C(O)Re2, - N(Rd2)C(O)ORe2, -N(Rd2)C(O)NRd2Re2, -NRd2S(O)2Re2, -NRd2C(=NH)Re2, -NRd2C(=NH)NRd2Re2, -S(O)1-2Re2, -S(O)2NRd2Re2 y -N Rd2S(O)2NRd2 Re2;
y/o, cada uno de Rd, Re, Rd2 y Re2 se selecciona independientemente de hidrógeno, alquilo de C1-6, haloalquilo de C1-6, alquenilo de C2-6, alquinilo de C2-6, cicloalquilo de C3-8, heterocicloalquilo de 3 a 8 miembros, arilo de C6-10, heteroarilo
de 5 a 6 miembros, cicloalquilo de C3-8 alquilo de C1-6, heterocicloalquilo de 3 a 8 miembros alquilo de C1-6, fenilo
alquilo de C1-6 o heteroarilo de 5 a 6 miembros alquilo de C1-6; Rd, Re, Rd2 o Re2 no están sustituidos o están sustituidos en cualquier posición por 1 a 3 sustituyentes seleccionados del grupo que consiste en halógeno, hidroxilo, amino, carboxilo, alquilo de C1-4, haloalquilo de C1-3, haloalcoxi de C1-3, alcoxi de C1-3, alquilamino de C1-3, alquenilo de C2-6 y alquinilo de C2-6;
o, Rd y Re junto con el átomo de N al que están unidos, forman un heterocicloalquilo de 3 a 8 miembros; el heterocicloalquilo puede contener además de 1 a 3 heteroátomos seleccionados entre N, O y S;
o, Rd2 y Re2 junto con el átomo de N al que están unidos, forman un heterocicloalquilo de 3 a 8 miembros.
6. El compuesto de fórmula (I), el estereoisómero, el derivado isotópico estable o la sal farmacéuticamente aceptable del mismo según la reivindicación 1 ó 5, en el que cada uno de R7 es independientemente hidrógeno, metilo, etilo, n-propilo, terc-butilo, isopropilo, isobutilo, n-butilo, neopentilo, terc-amilo, ciclopropilo, ciclobutilo, ciclopentilo, ciclohexilo, fenilo, piridilo, pirimidinilo, bencilo,


7. El compuesto de fórmula (I), el estereoisómero, el derivado isotópico estable o la sal farmacéuticamente aceptable del mismo según al menos una de las reivindicaciones 1-6, que se muestra como el compuesto de fórmula (IA), (IB), (IC ), (ID), (IE), (IF), (IG), (IH), (II), (IJ), (IK) o (IL), el estereoisómero, el derivado isotópico estable o la sal farmacéuticamente aceptable del mismo;
el compuesto de fórmula (IA), el estereoisómero, el derivado isotópico estable o la sal farmacéuticamente aceptable del mismo:

en donde, es un enlace simple o un enlace doble;
Y es O S;
U es N, C(R8) o C(O), V es N, C(R8) o N(R7a); y, , U y V son cualquier combinación de los siguientes:
1) es un doble enlace, U es N, V es C(R8);
2) es un doble enlace, U es C(Rs), V es N;
3) es un doble enlace, U es C(Rs), V es C(Rs);
4) es un enlace sencillo, U es C(Rs), V es C(Rs);
5) es un enlace sencillo, U es C(O), V es N(R7a);
n es 1;
Li, Rdi, Reí, R4, R5 y R6 se definen como en una cualquiera de las reivindicaciones 1-6; R7 , Rs y R7a se definen como en una cualquiera de las reivindicaciones 1,5 y 6;
el compuesto de fórmula (IB) o (IC), el estereoisómero, el derivado isotópico estable o la sal farmacéuticamente aceptable del mismo:

en donde, es un enlace simple o un enlace doble; Y es O S;
Rd1, Re1, R4 , R5 y R6 se definen como en una cualquiera de las reivindicaciones 1-6; R7 y Rs se definen como en una cualquiera de las reivindicaciones 1,5 y 6;
el compuesto de fórmula (ID) o (IE), el estereoisómero, el derivado isotópico estable o la sal farmacéuticamente aceptable del mismo:

en donde, Y es O o S; Rd1, Re1, R4 , R5 y R6 se definen como en una cualquiera de las reivindicaciones 1-6; R7 y Rs se definen como en una cualquiera de las reivindicaciones 1, 5 y 6;
el compuesto de fórmula (IF) o (IG), el estereoisómero, el derivado isotópico estable o la sal farmacéuticamente aceptable del mismo:

en donde, Y es O o S; Rdi, Reí, R4, R5 y R6 se definen como en una cualquiera de las reivindicaciones 1-6; R7 y Rs se definen como en una cualquiera de las reivindicaciones 1,5 y 6;
el compuesto de fórmula (IH) o (II), el estereoisómero, el derivado isotópico estable o la sal farmacéuticamente aceptable del mismo:

en donde, Rd1, Re1, R4 , R5 y R6 se definen como en una cualquiera de las reivindicaciones 1-6; R7 y R7a se definen como en una cualquiera de las reivindicaciones 1, 5 y 6;
el compuesto de fórmula (IJ), el estereoisómero, el derivado isotópico estable, la sal farmacéuticamente aceptable del mismo:

en donde, es un enlace simple o un enlace doble; Z es N(R7a) o S;
n es 1 ó 2;
L1, Rd1, Re1, R4 , R5 y R6 se definen como en una cualquiera de las reivindicaciones 1-6; R7 , R7a y Rs se definen como en una cualquiera de las reivindicaciones 1, 5 y 6;
el compuesto de fórmula (IK) o (IL), el estereoisómero, el derivado isotópico estable, la sal farmacéuticamente aceptable del mismo:

en donde, Z es NH, N(CH3) o S;
Rd1, Re1, R4 , R5 y R6 se definen como en una cualquiera de las reivindicaciones 1-6; R7 y Rs se definen como en una cualquiera de las reivindicaciones 1,5 y 6.
8. El compuesto de fórmula (I), el estereoisómero, el derivado isotópico estable o la sal farmacéuticamente aceptable del mismo según la reivindicación 1, en el que el compuesto de fórmula (I) se selecciona del grupo que consiste en










9. Un método de preparación del compuesto de fórmula (I), el estereoisómero, el derivado isotópico estable o la sal farmacéuticamente aceptable del mismo según al menos una de las reivindicaciones 1-8, que es uno cualquiera de los siguientes métodos:
método 1 que comprende realizar una reacción de acoplamiento de Suzuki con el compuesto de fórmula I-a y

para obtener el compuesto de fórmula (I);

método 2 que comprende realizar una reacción de acoplamiento de Suzuki con el compuesto I-b y R-M para obtener el compuesto de fórmula (I);

en donde, M es bromo, cloro, yodo o -OS(O)2FC3; R1, R2 , R3 , R4 , R5 , R6, RA, RB, R y L se definen como en una cualquiera de las reivindicaciones 1-8.
10. Una composición farmacéutica que comprende una cantidad terapéuticamente eficaz del compuesto de fórmula (I), el estereoisómero, el derivado isotópico estable o la sal farmacéuticamente aceptable del mismo según al menos una de las reivindicaciones 1-8, y excipiente(s) farmacéuticamente aceptable(s).
11. El compuesto de fórmula (I), el estereoisómero, el derivado isotópico estable o la sal farmacéuticamente aceptable del mismo según al menos una de las reivindicaciones 1-8 para su uso en el tratamiento, alivio y/o prevención de enfermedades relacionadas mediadas por TLRs, en donde las enfermedades mediadas por TLRs se seleccionan del grupo que consiste en cánceres, infecciones virales, inflamaciones y enfermedades autoinmunes.
12. El compuesto de fórmula (I), el estereoisómero, el derivado isotópico estable o la sal farmacéuticamente aceptable del mismo para su uso según la reivindicación 11, en el que el medicamento se usa además en combinación con uno o más de otros tipos de agentes terapéuticos y/o métodos terapéuticos para el tratamiento, mejora y/o prevención de enfermedades relacionadas mediadas por TLRs, en los que las enfermedades mediadas por TLRs se seleccionan del grupo que consiste en cánceres, infecciones virales, inflamaciones y enfermedades autoinmunes.
13. El compuesto de fórmula (I), el estereoisómero, el derivado isotópico estable o la sal farmacéuticamente aceptable del mismo para su uso según al menos una de las reivindicaciones 11-12, en el que los TLRs son TLR7 y/o TLR8.
Applications Claiming Priority (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201610296748 | 2016-05-06 | ||
| CN201610589219 | 2016-07-25 | ||
| CN201610965360 | 2016-11-04 | ||
| CN201710020298 | 2017-01-12 | ||
| CN201710054038 | 2017-01-24 | ||
| PCT/CN2017/083031 WO2017190669A1 (zh) | 2016-05-06 | 2017-05-04 | 苯并氮杂卓衍生物、其制备方法、药物组合物及应用 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2907981T3 true ES2907981T3 (es) | 2022-04-27 |
Family
ID=60202778
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES17792495T Active ES2907981T3 (es) | 2016-05-06 | 2017-05-04 | Derivado de benzazepina, método de preparación, composición farmacéutica y uso del mismo |
Country Status (22)
| Country | Link |
|---|---|
| US (1) | US10669252B2 (es) |
| EP (1) | EP3453707B1 (es) |
| JP (1) | JP6739548B2 (es) |
| KR (1) | KR102274557B1 (es) |
| CN (1) | CN107344931B (es) |
| AU (1) | AU2017259654B2 (es) |
| CA (1) | CA3023154C (es) |
| CY (1) | CY1126367T1 (es) |
| DK (1) | DK3453707T3 (es) |
| ES (1) | ES2907981T3 (es) |
| HR (1) | HRP20220577T1 (es) |
| HU (1) | HUE057924T2 (es) |
| LT (1) | LT3453707T (es) |
| MX (1) | MX382727B (es) |
| PH (1) | PH12018502336A1 (es) |
| PL (1) | PL3453707T3 (es) |
| PT (1) | PT3453707T (es) |
| RS (1) | RS63116B1 (es) |
| SG (1) | SG11201809859VA (es) |
| SI (1) | SI3453707T1 (es) |
| SM (1) | SMT202200122T1 (es) |
| WO (1) | WO2017190669A1 (es) |
Families Citing this family (68)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| PT3052485T (pt) | 2013-10-04 | 2021-10-22 | Infinity Pharmaceuticals Inc | Compostos heterocíclicos e suas utilizações |
| MX382033B (es) | 2014-03-19 | 2025-03-13 | Infinity Pharmaceuticals Inc | Compuestos heterocíclicos para utilizarlos en el tratamiento de trastornos mediados por pi3k-gamma. |
| WO2017048702A1 (en) | 2015-09-14 | 2017-03-23 | Infinity Pharmaceuticals, Inc. | Solid forms of isoquinolinone derivatives, process of making, compositions comprising, and methods of using the same |
| JP6918838B2 (ja) | 2016-05-23 | 2021-08-11 | エフ・ホフマン−ラ・ロシュ・アクチェンゲゼルシャフト | 第三級アミド基を有するベンズアゼピンジカルボキサミド化合物 |
| CN109311854B (zh) | 2016-05-23 | 2021-08-10 | 豪夫迈·罗氏有限公司 | 具有仲酰胺官能团的苯并氮杂卓二甲酰胺化合物 |
| JP7012668B2 (ja) | 2016-06-12 | 2022-02-14 | エフ・ホフマン-ラ・ロシュ・アクチェンゲゼルシャフト | ジヒドロピリミジニルベンズアゼピンジカルボキサミド化合物 |
| KR20230010826A (ko) | 2016-10-14 | 2023-01-19 | 프리시젼 바이오사이언시스 인코포레이티드 | B형 간염 바이러스 게놈 내의 인식 서열에 대해 특이적인 조작된 메가뉴클레아제 |
| SG10202110182PA (en) | 2017-03-15 | 2021-10-28 | Silverback Therapeutics Inc | Benzazepine compounds, conjugates, and uses thereof |
| US10899733B2 (en) | 2017-08-23 | 2021-01-26 | Oregon Health & Science University | Inhibitors of PARPs that catalyze mono-ADP-ribosylation |
| WO2019084060A1 (en) | 2017-10-24 | 2019-05-02 | Silverback Therapeutics, Inc. | CONJUGATES AND METHODS OF USE FOR THE SELECTIVE DELIVERY OF IMMUNOMODULATORY AGENTS |
| JP2021506827A (ja) | 2017-12-15 | 2021-02-22 | シルバーバック セラピューティックス インコーポレイテッド | 肝炎の治療用の抗体コンストラクト−薬物コンジュゲート |
| EP3728283B1 (en) | 2017-12-20 | 2023-11-22 | Institute of Organic Chemistry and Biochemistry ASCR, V.V.I. | 3'3' cyclic dinucleotides with phosphonate bond activating the sting adaptor protein |
| WO2019123339A1 (en) | 2017-12-20 | 2019-06-27 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 2'3' cyclic dinucleotides with phosphonate bond activating the sting adaptor protein |
| CN111433201B (zh) * | 2017-12-21 | 2022-05-27 | 江苏恒瑞医药股份有限公司 | 苯并氮杂䓬衍生物、其制备方法及其在医药上的应用 |
| CN109956903B (zh) * | 2017-12-22 | 2022-08-23 | 江苏恒瑞医药股份有限公司 | 苯并氮杂䓬衍生物、其制备方法及其在医药上的应用 |
| JP7050165B2 (ja) | 2018-02-26 | 2022-04-07 | ギリアード サイエンシーズ, インコーポレイテッド | Hbv複製阻害剤としての置換ピロリジン化合物 |
| US10870691B2 (en) | 2018-04-05 | 2020-12-22 | Gilead Sciences, Inc. | Antibodies and fragments thereof that bind hepatitis B virus protein X |
| TWI818007B (zh) | 2018-04-06 | 2023-10-11 | 捷克科學院有機化學與生物化學研究所 | 2'3'-環二核苷酸 |
| TW202005654A (zh) | 2018-04-06 | 2020-02-01 | 捷克科學院有機化學與生物化學研究所 | 2,2,─環二核苷酸 |
| TWI833744B (zh) | 2018-04-06 | 2024-03-01 | 捷克科學院有機化學與生物化學研究所 | 3'3'-環二核苷酸 |
| US11142750B2 (en) | 2018-04-12 | 2021-10-12 | Precision Biosciences, Inc. | Optimized engineered meganucleases having specificity for a recognition sequence in the Hepatitis B virus genome |
| US20190359645A1 (en) | 2018-05-03 | 2019-11-28 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 2'3'-cyclic dinucleotides comprising carbocyclic nucleotide |
| WO2020028097A1 (en) | 2018-08-01 | 2020-02-06 | Gilead Sciences, Inc. | Solid forms of (r)-11-(methoxymethyl)-12-(3-methoxypropoxy)-3,3-dimethyl-8-0x0-2,3,8,13b-tetrahydro-1h-pyrido[2,1-a]pyrrolo[1,2-c] phthalazine-7-c arboxylic acid |
| MX2021002764A (es) | 2018-09-12 | 2021-05-12 | Silverback Therapeutics Inc | Composiciones para el tratamiento de enfermedades con conjugados inmunoestimuladores. |
| AU2019337654A1 (en) * | 2018-09-12 | 2021-04-08 | Silverback Therapeutics, Inc. | Substituted benzazepine compounds, conjugates, and uses thereof |
| WO2020092528A1 (en) | 2018-10-31 | 2020-05-07 | Gilead Sciences, Inc. | Substituted 6-azabenzimidazole compounds having hpk1 inhibitory activity |
| US11203591B2 (en) | 2018-10-31 | 2021-12-21 | Gilead Sciences, Inc. | Substituted 6-azabenzimidazole compounds |
| EP3934757B1 (en) | 2019-03-07 | 2023-02-22 | Institute of Organic Chemistry and Biochemistry ASCR, V.V.I. | 2'3'-cyclic dinucleotides and prodrugs thereof |
| WO2020178770A1 (en) | 2019-03-07 | 2020-09-10 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 3'3'-cyclic dinucleotides and prodrugs thereof |
| WO2020178768A1 (en) | 2019-03-07 | 2020-09-10 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 3'3'-cyclic dinucleotide analogue comprising a cyclopentanyl modified nucleotide as sting modulator |
| TWI751516B (zh) | 2019-04-17 | 2022-01-01 | 美商基利科學股份有限公司 | 類鐸受體調節劑之固體形式 |
| TW202212339A (zh) | 2019-04-17 | 2022-04-01 | 美商基利科學股份有限公司 | 類鐸受體調節劑之固體形式 |
| EP3972695A1 (en) | 2019-05-23 | 2022-03-30 | Gilead Sciences, Inc. | Substituted exo-methylene-oxindoles which are hpk1/map4k1 inhibitors |
| KR20220035908A (ko) | 2019-06-19 | 2022-03-22 | 실버백 테라퓨틱스, 인크. | 항-메소텔린 항체 및 이의 면역접합체 |
| CN114667134A (zh) | 2019-08-15 | 2022-06-24 | 希沃尔拜克治疗公司 | 苯并氮杂䓬缀合物的制剂及其用途 |
| WO2021034804A1 (en) | 2019-08-19 | 2021-02-25 | Gilead Sciences, Inc. | Pharmaceutical formulations of tenofovir alafenamide |
| DK4037708T3 (da) | 2019-09-30 | 2024-09-30 | Gilead Sciences Inc | HBV-vacciner og fremgangsmåder til at behandle HBV |
| CA3151322A1 (en) | 2019-10-01 | 2021-04-08 | Silverback Therapeutics, Inc. | Combination therapy with immune stimulatory conjugates |
| DK4069729T3 (da) | 2019-12-06 | 2025-04-07 | Prec Biosciences Inc | Optimerede, modificerede meganukleaser med specificitet for en genkendelsessekvens i hepatitis b-virusgenomet |
| JP7735291B2 (ja) | 2020-02-21 | 2025-09-08 | エーアールエス ファーマシューティカルズ、インコーポレイテッド | ネクチン-4抗体コンジュゲートおよびその使用 |
| WO2021188959A1 (en) | 2020-03-20 | 2021-09-23 | Gilead Sciences, Inc. | Prodrugs of 4'-c-substituted-2-halo-2'-deoxyadenosine nucleosides and methods of making and using the same |
| JP2023520916A (ja) * | 2020-04-08 | 2023-05-22 | リミックス セラピューティクス インコーポレイテッド | スプライシングを調整するための化合物及び方法 |
| JP2023532304A (ja) | 2020-07-01 | 2023-07-27 | エーアールエス ファーマシューティカルズ オペレーションズ,インク. | 抗asgr1抗体コンジュゲートおよびその使用 |
| CN113880866B (zh) * | 2020-08-07 | 2022-09-27 | 上海维申医药有限公司 | 氮杂四氢萘啶类化合物、其制备方法、药物组合物及其用途 |
| CN114075219B (zh) * | 2020-08-14 | 2023-11-14 | 江苏恒瑞医药股份有限公司 | 喹啉稠环类衍生物、其制备方法及其在医药上的应用 |
| CA3200168A1 (en) * | 2020-12-11 | 2022-06-16 | Shelley Erin ACKERMAN | Anti-pd-l1 immunoconjugates, and uses thereof |
| CA3200056A1 (en) * | 2020-12-11 | 2022-06-16 | Shelley Erin ACKERMAN | Anti-her2 immunoconjugates, and uses thereof |
| CN112778201B (zh) * | 2021-01-14 | 2023-07-11 | 合肥工业大学 | 一种苯并[b]氮杂䓬-查尔酮杂合物及其制备方法和用途 |
| CN114874217A (zh) * | 2021-02-05 | 2022-08-09 | 上海迪诺医药科技有限公司 | 苯并氮杂卓衍生物的盐、溶剂合物、多晶型物、制备方法及应用 |
| US20240209080A1 (en) | 2021-04-10 | 2024-06-27 | Profoundbio Us Co. | Folr1 binding agents, conjugates thereof and methods of using the same |
| CA3216459A1 (en) | 2021-04-23 | 2022-10-27 | Profoundbio Us Co. | Anti-cd70 antibodies, conjugates thereof and methods of using the same |
| GB202106871D0 (en) * | 2021-05-13 | 2021-06-30 | Addex Pharmaceuticals Sa | Novel compounds |
| WO2022241134A1 (en) | 2021-05-13 | 2022-11-17 | Gilead Sciences, Inc. | COMBINATION OF A TLR8 MODULATING COMPOUND AND ANTI-HBV siRNA THERAPEUTICS |
| MX2023014762A (es) | 2021-06-23 | 2024-01-15 | Gilead Sciences Inc | Compuestos moduladores de diacilglicerol quinasa. |
| JP7686091B2 (ja) | 2021-06-23 | 2025-05-30 | ギリアード サイエンシーズ, インコーポレイテッド | ジアシルグリセロールキナーゼ調節化合物 |
| AU2022299051B2 (en) | 2021-06-23 | 2025-03-13 | Gilead Sciences, Inc. | Diacylglyercol kinase modulating compounds |
| JP7651018B2 (ja) | 2021-06-23 | 2025-03-25 | ギリアード サイエンシーズ, インコーポレイテッド | ジアシルグリセロールキナーゼ調節化合物 |
| TW202320857A (zh) | 2021-07-06 | 2023-06-01 | 美商普方生物製藥美國公司 | 連接子、藥物連接子及其結合物及其使用方法 |
| WO2023166067A1 (en) | 2022-03-02 | 2023-09-07 | Syngenta Crop Protection Ag | Microbiocidal pyridazinone amide derivatives |
| WO2024132895A1 (en) | 2022-12-19 | 2024-06-27 | Syngenta Crop Protection Ag | Microbiocidal dihydrooxadiazinyl pyridazinone compounds |
| CN116675684B (zh) * | 2023-08-02 | 2023-11-07 | 上海翰森生物医药科技有限公司 | 含炔基稠环类衍生物拮抗剂、其制备方法和应用 |
| AR133955A1 (es) | 2023-09-26 | 2025-11-19 | Profoundbio Us Co | Agentes de unión a ptk7, conjugados de éstos y métodos de uso de los mismos |
| WO2025149661A1 (en) | 2024-01-10 | 2025-07-17 | Genmab A/S | Slitrk6 binding agents, conjugates thereof and methods of using the same |
| US20250381289A1 (en) | 2024-02-29 | 2025-12-18 | Genmab A/S | Egfr and c-met bispecific binding agents, conjugates thereof and methods of using the same |
| WO2025240242A1 (en) | 2024-05-13 | 2025-11-20 | Gilead Sciences, Inc. | Combination therapies with ribavirin |
| WO2025240243A1 (en) | 2024-05-13 | 2025-11-20 | Gilead Sciences, Inc. | Combination therapies with bulevirtide and an inhibitory nucleic acid targeting hepatitis b virus |
| WO2025240244A1 (en) | 2024-05-13 | 2025-11-20 | Gilead Sciences, Inc. | Combination therapies comprising bulevirtide and lonafarnib for use in the treatment of hepatitis d virus infection |
| WO2025240246A1 (en) | 2024-05-13 | 2025-11-20 | Gilead Sciences, Inc. | Combination therapies with ribavirin |
Family Cites Families (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN102766142A (zh) | 2004-10-04 | 2012-11-07 | 千禧药品公司 | 有效作为蛋白激酶抑制剂的内酰胺化合物 |
| WO2010014913A1 (en) * | 2008-08-01 | 2010-02-04 | Ventirx Pharmaceuticals, Inc. | Toll-like receptor agonist formulations and their use |
| RS53347B (sr) * | 2008-12-09 | 2014-10-31 | Gilead Sciences, Inc. | Modulatori toll-sličnih receptora |
| RU2016108987A (ru) | 2009-08-18 | 2018-11-26 | Вентиркс Фармасьютикалз, Инк. | Замещенные бензоазепины в качестве модуляторов toll-подобного рецептора |
| CN105669553A (zh) | 2009-08-18 | 2016-06-15 | 文蒂雷克斯药品公司 | 作为toll样受体调节剂的取代的苯并氮杂* |
| DK2663555T3 (en) * | 2011-01-12 | 2017-03-27 | Ventirx Pharmaceuticals Inc | SUBSTITUTED BENZOAZEPINS AS MODULATORS OF TOLL-LIKE RECEPTORS |
| EP3208263A1 (en) * | 2011-01-12 | 2017-08-23 | VentiRx Pharmaceuticals, Inc. | Substituted benzoazepines as toll-like receptor modulators |
-
2017
- 2017-05-04 CN CN201710308602.7A patent/CN107344931B/zh active Active
- 2017-05-04 DK DK17792495.8T patent/DK3453707T3/da active
- 2017-05-04 EP EP17792495.8A patent/EP3453707B1/en active Active
- 2017-05-04 SM SM20220122T patent/SMT202200122T1/it unknown
- 2017-05-04 PT PT177924958T patent/PT3453707T/pt unknown
- 2017-05-04 SG SG11201809859VA patent/SG11201809859VA/en unknown
- 2017-05-04 US US16/098,972 patent/US10669252B2/en active Active
- 2017-05-04 HR HRP20220577TT patent/HRP20220577T1/hr unknown
- 2017-05-04 MX MX2018013467A patent/MX382727B/es unknown
- 2017-05-04 LT LTEPPCT/CN2017/083031T patent/LT3453707T/lt unknown
- 2017-05-04 SI SI201731103T patent/SI3453707T1/sl unknown
- 2017-05-04 CA CA3023154A patent/CA3023154C/en active Active
- 2017-05-04 AU AU2017259654A patent/AU2017259654B2/en not_active Ceased
- 2017-05-04 HU HUE17792495A patent/HUE057924T2/hu unknown
- 2017-05-04 JP JP2018558270A patent/JP6739548B2/ja not_active Expired - Fee Related
- 2017-05-04 KR KR1020187033064A patent/KR102274557B1/ko not_active Expired - Fee Related
- 2017-05-04 ES ES17792495T patent/ES2907981T3/es active Active
- 2017-05-04 WO PCT/CN2017/083031 patent/WO2017190669A1/zh not_active Ceased
- 2017-05-04 RS RS20220362A patent/RS63116B1/sr unknown
- 2017-05-04 PL PL17792495T patent/PL3453707T3/pl unknown
-
2018
- 2018-11-06 PH PH12018502336A patent/PH12018502336A1/en unknown
-
2022
- 2022-04-07 CY CY20221100261T patent/CY1126367T1/el unknown
Also Published As
| Publication number | Publication date |
|---|---|
| SMT202200122T1 (it) | 2022-05-12 |
| CN107344931A (zh) | 2017-11-14 |
| PT3453707T (pt) | 2022-03-15 |
| CN107344931B (zh) | 2020-09-15 |
| RS63116B1 (sr) | 2022-05-31 |
| JP6739548B2 (ja) | 2020-08-12 |
| JP2019518728A (ja) | 2019-07-04 |
| KR102274557B1 (ko) | 2021-07-07 |
| HUE057924T2 (hu) | 2022-06-28 |
| EP3453707A4 (en) | 2020-01-08 |
| BR112018072591A2 (pt) | 2019-02-19 |
| PH12018502336A1 (en) | 2019-07-29 |
| CA3023154A1 (en) | 2017-11-09 |
| MX2018013467A (es) | 2019-07-08 |
| EP3453707A1 (en) | 2019-03-13 |
| HRP20220577T1 (hr) | 2022-06-24 |
| CY1126367T1 (el) | 2024-12-13 |
| US10669252B2 (en) | 2020-06-02 |
| AU2017259654B2 (en) | 2020-02-27 |
| US20190152941A1 (en) | 2019-05-23 |
| PL3453707T3 (pl) | 2022-06-13 |
| EP3453707B1 (en) | 2022-02-16 |
| DK3453707T3 (da) | 2022-05-09 |
| SI3453707T1 (sl) | 2022-04-29 |
| AU2017259654A1 (en) | 2019-01-03 |
| MX382727B (es) | 2025-03-13 |
| SG11201809859VA (en) | 2018-12-28 |
| WO2017190669A1 (zh) | 2017-11-09 |
| CA3023154C (en) | 2021-01-12 |
| LT3453707T (lt) | 2022-03-10 |
| KR20190003572A (ko) | 2019-01-09 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2907981T3 (es) | Derivado de benzazepina, método de preparación, composición farmacéutica y uso del mismo | |
| ES3014294T3 (en) | Heterocyclic compounds as immunomodulators | |
| ES2906460T3 (es) | Compuestos heterocíclicos como inmunomoduladores | |
| ES2927346T3 (es) | Inhibidores de HPK1, método de preparación y aplicación de los mismos | |
| CN102146081B (zh) | 吲哚乙酸衍生物及其制备方法和应用 | |
| KR20190056380A (ko) | 항인플루엔자 바이러스 피리미딘 유도체 | |
| ES2714100T3 (es) | Compuestos de quinolina fusionados como inhibidores de mTor, pi3k | |
| BRPI1015367B1 (pt) | Derivados imidazo[2,1-b][1,3,4]tiadiazol | |
| CN109721597B (zh) | 吡啶并氮杂卓衍生物、其药物组合物及应用 | |
| CN116375707B (zh) | Menin抑制剂及其用途 | |
| US11932663B2 (en) | Phosphorus imidazoquinoline amine derivatives, pharmaceutical compositions and therapeutic methods thereof | |
| JP2022500476A (ja) | 環状ジヌクレオチド類似体、その医薬組成物及び使用 | |
| CN121263406A (zh) | 抗病毒杂环化合物 | |
| CN116253719B (zh) | 一种雌激素和雄激素受体双重降解剂及其应用 | |
| BR112021004094A2 (pt) | formas cristalinas de um inibidor de fosfoinositídeo 3-quinase (pi3k) | |
| CN105693729B (zh) | 吲哚并 [ 3 , 2 ‑ a ] 咔唑衍生物及其应用 | |
| CN112851679B (zh) | 2,4,7-三取代嘧啶并吲哚化合物抗肿瘤作用 | |
| WO2023237116A1 (en) | Protein degraders and uses thereof | |
| BR112018072591B1 (pt) | Derivado de benzazepina, método de preparação, composição farmacêutica e uso do mesmo | |
| TWI700281B (zh) | 苯並氮雜䓬衍生物、其製備方法及其在醫藥上的應用 | |
| TW202237101A (zh) | Ctla-4小分子降解劑及其應用 | |
| HK1262638B (en) | Benzazepine derivative, preparation method, pharmaceutical composition and use thereof | |
| HK1262638A1 (en) | Benzazepine derivative, preparation method, pharmaceutical composition and use thereof | |
| EA037488B1 (ru) | Производное бензазепина, способ получения, фармацевтическая композиция и его применение | |
| WO2025140656A1 (en) | Heteroaryl compounds as multi-target protein kinase inhibitors |