ES2908498T3 - Análogo de pirido[1,2-a]pirimidona, forma cristalina del mismo, intermedio del mismo y método de preparación del mismo - Google Patents
Análogo de pirido[1,2-a]pirimidona, forma cristalina del mismo, intermedio del mismo y método de preparación del mismo Download PDFInfo
- Publication number
- ES2908498T3 ES2908498T3 ES16874904T ES16874904T ES2908498T3 ES 2908498 T3 ES2908498 T3 ES 2908498T3 ES 16874904 T ES16874904 T ES 16874904T ES 16874904 T ES16874904 T ES 16874904T ES 2908498 T3 ES2908498 T3 ES 2908498T3
- Authority
- ES
- Spain
- Prior art keywords
- compound
- crystalline form
- present
- xrpd pattern
- salts
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 238000002360 preparation method Methods 0.000 title claims description 31
- 239000013078 crystal Substances 0.000 title description 34
- FNKRSSCRFSZXBE-UHFFFAOYSA-N pyrido[1,2-a]pyrimidin-2-one Chemical class C1=CC=CC2=NC(=O)C=CN21 FNKRSSCRFSZXBE-UHFFFAOYSA-N 0.000 title description 4
- 229940125904 compound 1 Drugs 0.000 claims abstract description 54
- 238000002441 X-ray diffraction Methods 0.000 claims abstract description 20
- 229940125782 compound 2 Drugs 0.000 claims description 84
- 150000001875 compounds Chemical class 0.000 claims description 74
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 claims description 59
- 206010028980 Neoplasm Diseases 0.000 claims description 46
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 claims description 33
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 claims description 32
- 239000002904 solvent Substances 0.000 claims description 31
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 claims description 30
- 238000004458 analytical method Methods 0.000 claims description 30
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 claims description 27
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 claims description 24
- 238000000034 method Methods 0.000 claims description 24
- 239000008194 pharmaceutical composition Substances 0.000 claims description 24
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 claims description 22
- 102000038030 PI3Ks Human genes 0.000 claims description 19
- 108091007960 PI3Ks Proteins 0.000 claims description 19
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 claims description 18
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 claims description 18
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 claims description 18
- 229940126214 compound 3 Drugs 0.000 claims description 16
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 claims description 16
- 239000003513 alkali Substances 0.000 claims description 15
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 claims description 14
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 claims description 13
- 150000003839 salts Chemical class 0.000 claims description 12
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical group C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 claims description 11
- WMFOQBRAJBCJND-UHFFFAOYSA-M Lithium hydroxide Chemical compound [Li+].[OH-] WMFOQBRAJBCJND-UHFFFAOYSA-M 0.000 claims description 11
- 229910000027 potassium carbonate Inorganic materials 0.000 claims description 11
- 229910000024 caesium carbonate Inorganic materials 0.000 claims description 10
- 239000000546 pharmaceutical excipient Substances 0.000 claims description 10
- OISVCGZHLKNMSJ-UHFFFAOYSA-N 2,6-dimethylpyridine Chemical compound CC1=CC=CC(C)=N1 OISVCGZHLKNMSJ-UHFFFAOYSA-N 0.000 claims description 9
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 claims description 9
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 claims description 9
- 201000010099 disease Diseases 0.000 claims description 9
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims description 9
- SECXISVLQFMRJM-UHFFFAOYSA-N N-Methylpyrrolidone Chemical compound CN1CCCC1=O SECXISVLQFMRJM-UHFFFAOYSA-N 0.000 claims description 8
- 239000003937 drug carrier Substances 0.000 claims description 8
- 206010009944 Colon cancer Diseases 0.000 claims description 7
- 208000029742 colonic neoplasm Diseases 0.000 claims description 7
- 229910000029 sodium carbonate Inorganic materials 0.000 claims description 7
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 claims description 6
- 208000005718 Stomach Neoplasms Diseases 0.000 claims description 6
- 206010017758 gastric cancer Diseases 0.000 claims description 6
- 229960005235 piperonyl butoxide Drugs 0.000 claims description 6
- SCVFZCLFOSHCOH-UHFFFAOYSA-M potassium acetate Chemical compound [K+].CC([O-])=O SCVFZCLFOSHCOH-UHFFFAOYSA-M 0.000 claims description 6
- NROKBHXJSPEDAR-UHFFFAOYSA-M potassium fluoride Chemical group [F-].[K+] NROKBHXJSPEDAR-UHFFFAOYSA-M 0.000 claims description 6
- 201000011549 stomach cancer Diseases 0.000 claims description 6
- LWIHDJKSTIGBAC-UHFFFAOYSA-K tripotassium phosphate Chemical group [K+].[K+].[K+].[O-]P([O-])([O-])=O LWIHDJKSTIGBAC-UHFFFAOYSA-K 0.000 claims description 6
- KZPYGQFFRCFCPP-UHFFFAOYSA-N 1,1'-bis(diphenylphosphino)ferrocene Chemical compound [Fe+2].C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1 KZPYGQFFRCFCPP-UHFFFAOYSA-N 0.000 claims description 5
- LVCHXPHUKPLVRQ-UHFFFAOYSA-N 2-bromo-n,n-dimethylethanamine Chemical compound CN(C)CCBr LVCHXPHUKPLVRQ-UHFFFAOYSA-N 0.000 claims description 4
- WQMAANNAZKNUDL-UHFFFAOYSA-N 2-dimethylaminoethyl chloride Chemical compound CN(C)CCCl WQMAANNAZKNUDL-UHFFFAOYSA-N 0.000 claims description 4
- GSNUFIFRDBKVIE-UHFFFAOYSA-N DMF Natural products CC1=CC=C(C)O1 GSNUFIFRDBKVIE-UHFFFAOYSA-N 0.000 claims description 4
- NFHFRUOZVGFOOS-UHFFFAOYSA-N Pd(PPh3)4 Substances [Pd].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 NFHFRUOZVGFOOS-UHFFFAOYSA-N 0.000 claims description 4
- XJHCXCQVJFPJIK-UHFFFAOYSA-M caesium fluoride Chemical group [F-].[Cs+] XJHCXCQVJFPJIK-UHFFFAOYSA-M 0.000 claims description 4
- 239000003054 catalyst Substances 0.000 claims description 4
- XWKFPIODWVPXLX-UHFFFAOYSA-N 2-methyl-5-methylpyridine Natural products CC1=CC=C(C)N=C1 XWKFPIODWVPXLX-UHFFFAOYSA-N 0.000 claims description 3
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 claims description 3
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical group [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 claims description 3
- VMHLLURERBWHNL-UHFFFAOYSA-M Sodium acetate Chemical compound [Na+].CC([O-])=O VMHLLURERBWHNL-UHFFFAOYSA-M 0.000 claims description 3
- RQPZNWPYLFFXCP-UHFFFAOYSA-L barium dihydroxide Chemical group [OH-].[OH-].[Ba+2] RQPZNWPYLFFXCP-UHFFFAOYSA-L 0.000 claims description 3
- 229910001863 barium hydroxide Inorganic materials 0.000 claims description 3
- 239000011591 potassium Chemical group 0.000 claims description 3
- 229910052700 potassium Inorganic materials 0.000 claims description 3
- 235000011056 potassium acetate Nutrition 0.000 claims description 3
- 239000011698 potassium fluoride Chemical group 0.000 claims description 3
- 235000003270 potassium fluoride Nutrition 0.000 claims description 3
- 229910000160 potassium phosphate Inorganic materials 0.000 claims description 3
- 235000011009 potassium phosphates Nutrition 0.000 claims description 3
- 229910052708 sodium Inorganic materials 0.000 claims description 3
- 239000011734 sodium Substances 0.000 claims description 3
- 239000001632 sodium acetate Substances 0.000 claims description 3
- 235000017281 sodium acetate Nutrition 0.000 claims description 3
- 238000000634 powder X-ray diffraction Methods 0.000 claims 9
- -1 Et 3 N Chemical compound 0.000 claims 2
- AHVYPIQETPWLSZ-UHFFFAOYSA-N N-methyl-pyrrolidine Natural products CN1CC=CC1 AHVYPIQETPWLSZ-UHFFFAOYSA-N 0.000 claims 1
- 239000000203 mixture Substances 0.000 description 72
- 238000006243 chemical reaction Methods 0.000 description 62
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 51
- 239000000243 solution Substances 0.000 description 49
- 239000012065 filter cake Substances 0.000 description 31
- 239000007787 solid Substances 0.000 description 26
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical group CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 24
- 241001465754 Metazoa Species 0.000 description 24
- 238000012360 testing method Methods 0.000 description 24
- 239000012453 solvate Substances 0.000 description 21
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 18
- 230000002829 reductive effect Effects 0.000 description 18
- 239000003814 drug Substances 0.000 description 16
- 229940079593 drug Drugs 0.000 description 13
- 238000002474 experimental method Methods 0.000 description 13
- 108091000080 Phosphotransferase Proteins 0.000 description 12
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 12
- 210000004027 cell Anatomy 0.000 description 12
- 230000000875 corresponding effect Effects 0.000 description 12
- 102000020233 phosphotransferase Human genes 0.000 description 12
- 238000003756 stirring Methods 0.000 description 12
- 238000005119 centrifugation Methods 0.000 description 11
- 230000000694 effects Effects 0.000 description 11
- 239000000758 substrate Substances 0.000 description 11
- 238000005160 1H NMR spectroscopy Methods 0.000 description 10
- 239000003153 chemical reaction reagent Substances 0.000 description 10
- 239000000047 product Substances 0.000 description 10
- 229910002483 Cu Ka Inorganic materials 0.000 description 9
- 239000003960 organic solvent Substances 0.000 description 9
- 230000005855 radiation Effects 0.000 description 9
- LQLJZSJKRYTKTP-UHFFFAOYSA-N 2-dimethylaminoethyl chloride hydrochloride Chemical compound Cl.CN(C)CCCl LQLJZSJKRYTKTP-UHFFFAOYSA-N 0.000 description 8
- GDINMGXNCCVWQB-UHFFFAOYSA-N 7-(5-amino-4-methoxy-2H-pyrimidin-1-yl)-3-[2-(dimethylamino)ethoxy]pyrido[1,2-a]pyrimidin-4-one Chemical compound NC1=CN(CN=C1OC)C=1C=CC=2N(C(C(=CN=2)OCCN(C)C)=O)C=1 GDINMGXNCCVWQB-UHFFFAOYSA-N 0.000 description 8
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 8
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 8
- 238000003556 assay Methods 0.000 description 8
- 238000001514 detection method Methods 0.000 description 8
- 238000001914 filtration Methods 0.000 description 8
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 8
- FCFPJKHVCHKCMP-UHFFFAOYSA-N 2-chloro-4-fluorobenzenesulfonyl chloride Chemical compound FC1=CC=C(S(Cl)(=O)=O)C(Cl)=C1 FCFPJKHVCHKCMP-UHFFFAOYSA-N 0.000 description 7
- JIEPYPKCNCZCOF-UHFFFAOYSA-N 7-bromo-3-phenylmethoxypyrido[1,2-a]pyrimidin-4-one Chemical compound C(C1=CC=CC=C1)OC1=CN=C2N(C1=O)C=C(C=C2)Br JIEPYPKCNCZCOF-UHFFFAOYSA-N 0.000 description 7
- 239000012043 crude product Substances 0.000 description 7
- 238000000132 electrospray ionisation Methods 0.000 description 7
- 239000000706 filtrate Substances 0.000 description 7
- 239000012074 organic phase Substances 0.000 description 7
- 235000011181 potassium carbonates Nutrition 0.000 description 7
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 description 6
- 229940125898 compound 5 Drugs 0.000 description 6
- 230000009089 cytolysis Effects 0.000 description 6
- 230000005764 inhibitory process Effects 0.000 description 6
- 150000002632 lipids Chemical class 0.000 description 6
- 239000003921 oil Substances 0.000 description 6
- 239000012265 solid product Substances 0.000 description 6
- 239000000725 suspension Substances 0.000 description 6
- 230000004614 tumor growth Effects 0.000 description 6
- CDPFSWUTOYYXOA-UHFFFAOYSA-N 7-bromo-3-hydroxypyrido[1,2-a]pyrimidin-4-one Chemical compound BrC=1C=CC=2N(C(C(=CN=2)O)=O)C=1 CDPFSWUTOYYXOA-UHFFFAOYSA-N 0.000 description 5
- CWHUFRVAEUJCEF-UHFFFAOYSA-N BKM120 Chemical compound C1=NC(N)=CC(C(F)(F)F)=C1C1=CC(N2CCOCC2)=NC(N2CCOCC2)=N1 CWHUFRVAEUJCEF-UHFFFAOYSA-N 0.000 description 5
- 102000004190 Enzymes Human genes 0.000 description 5
- 108090000790 Enzymes Proteins 0.000 description 5
- 238000010171 animal model Methods 0.000 description 5
- 229950003628 buparlisib Drugs 0.000 description 5
- 239000000543 intermediate Substances 0.000 description 5
- QNEIZSSWCVSZOS-UHFFFAOYSA-N methyl 2-phenylmethoxyacetate Chemical compound COC(=O)COCC1=CC=CC=C1 QNEIZSSWCVSZOS-UHFFFAOYSA-N 0.000 description 5
- XSNNLQBNWWVONY-UHFFFAOYSA-N methyl 3-(dimethylamino)-2-phenylmethoxyprop-2-enoate Chemical compound COC(=O)C(=CN(C)C)OCC1=CC=CC=C1 XSNNLQBNWWVONY-UHFFFAOYSA-N 0.000 description 5
- KDLHZDBZIXYQEI-UHFFFAOYSA-N palladium Substances [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 5
- QCQCHGYLTSGIGX-GHXANHINSA-N 4-[[(3ar,5ar,5br,7ar,9s,11ar,11br,13as)-5a,5b,8,8,11a-pentamethyl-3a-[(5-methylpyridine-3-carbonyl)amino]-2-oxo-1-propan-2-yl-4,5,6,7,7a,9,10,11,11b,12,13,13a-dodecahydro-3h-cyclopenta[a]chrysen-9-yl]oxy]-2,2-dimethyl-4-oxobutanoic acid Chemical compound N([C@@]12CC[C@@]3(C)[C@]4(C)CC[C@H]5C(C)(C)[C@@H](OC(=O)CC(C)(C)C(O)=O)CC[C@]5(C)[C@H]4CC[C@@H]3C1=C(C(C2)=O)C(C)C)C(=O)C1=CN=CC(C)=C1 QCQCHGYLTSGIGX-GHXANHINSA-N 0.000 description 4
- AYYSPBOIVICWKT-UHFFFAOYSA-N 7-bromo-3-[2-(dimethylamino)ethoxy]pyrido[1,2-a]pyrimidin-4-one Chemical compound BrC=1C=CC=2N(C(C(=CN=2)OCCN(C)C)=O)C=1 AYYSPBOIVICWKT-UHFFFAOYSA-N 0.000 description 4
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 4
- 239000005909 Kieselgur Substances 0.000 description 4
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 4
- 230000037396 body weight Effects 0.000 description 4
- 239000003651 drinking water Substances 0.000 description 4
- 235000020188 drinking water Nutrition 0.000 description 4
- 238000000338 in vitro Methods 0.000 description 4
- 238000001727 in vivo Methods 0.000 description 4
- NOESYZHRGYRDHS-UHFFFAOYSA-N insulin Chemical compound N1C(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(NC(=O)CN)C(C)CC)CSSCC(C(NC(CO)C(=O)NC(CC(C)C)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CCC(N)=O)C(=O)NC(CC(C)C)C(=O)NC(CCC(O)=O)C(=O)NC(CC(N)=O)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CSSCC(NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2C=CC(O)=CC=2)NC(=O)C(CC(C)C)NC(=O)C(C)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2NC=NC=2)NC(=O)C(CO)NC(=O)CNC2=O)C(=O)NCC(=O)NC(CCC(O)=O)C(=O)NC(CCCNC(N)=N)C(=O)NCC(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC(O)=CC=3)C(=O)NC(C(C)O)C(=O)N3C(CCC3)C(=O)NC(CCCCN)C(=O)NC(C)C(O)=O)C(=O)NC(CC(N)=O)C(O)=O)=O)NC(=O)C(C(C)CC)NC(=O)C(CO)NC(=O)C(C(C)O)NC(=O)C1CSSCC2NC(=O)C(CC(C)C)NC(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CC(N)=O)NC(=O)C(NC(=O)C(N)CC=1C=CC=CC=1)C(C)C)CC1=CN=CN1 NOESYZHRGYRDHS-UHFFFAOYSA-N 0.000 description 4
- 239000013641 positive control Substances 0.000 description 4
- 239000011535 reaction buffer Substances 0.000 description 4
- 238000002390 rotary evaporation Methods 0.000 description 4
- 238000005070 sampling Methods 0.000 description 4
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 4
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 3
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 3
- 241000699670 Mus sp. Species 0.000 description 3
- 102100032543 Phosphatidylinositol 3,4,5-trisphosphate 3-phosphatase and dual-specificity protein phosphatase PTEN Human genes 0.000 description 3
- 108010065917 TOR Serine-Threonine Kinases Proteins 0.000 description 3
- 102000013530 TOR Serine-Threonine Kinases Human genes 0.000 description 3
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 3
- 229910052782 aluminium Inorganic materials 0.000 description 3
- 201000011510 cancer Diseases 0.000 description 3
- HEDRZPFGACZZDS-UHFFFAOYSA-N chloroform Substances ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- 238000007405 data analysis Methods 0.000 description 3
- 229910001873 dinitrogen Inorganic materials 0.000 description 3
- 239000011521 glass Substances 0.000 description 3
- 239000001963 growth medium Substances 0.000 description 3
- 238000010438 heat treatment Methods 0.000 description 3
- 238000004128 high performance liquid chromatography Methods 0.000 description 3
- 230000002401 inhibitory effect Effects 0.000 description 3
- 238000011081 inoculation Methods 0.000 description 3
- 238000004519 manufacturing process Methods 0.000 description 3
- 238000005259 measurement Methods 0.000 description 3
- 239000002609 medium Substances 0.000 description 3
- 230000037361 pathway Effects 0.000 description 3
- 239000012071 phase Substances 0.000 description 3
- 150000003916 phosphatidylinositol 3,4,5-trisphosphates Chemical class 0.000 description 3
- 230000008569 process Effects 0.000 description 3
- 238000013112 stability test Methods 0.000 description 3
- 239000003981 vehicle Substances 0.000 description 3
- 239000011534 wash buffer Substances 0.000 description 3
- KYYKGOURQXPERA-UHFFFAOYSA-N 2-methoxy-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridin-3-amine Chemical compound C1=C(N)C(OC)=NC=C1B1OC(C)(C)C(C)(C)O1 KYYKGOURQXPERA-UHFFFAOYSA-N 0.000 description 2
- 238000011729 BALB/c nude mouse Methods 0.000 description 2
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 2
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 2
- 102000004877 Insulin Human genes 0.000 description 2
- 108090001061 Insulin Proteins 0.000 description 2
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 2
- 241000699666 Mus <mouse, genus> Species 0.000 description 2
- 108010011536 PTEN Phosphohydrolase Proteins 0.000 description 2
- 108090000430 Phosphatidylinositol 3-kinases Proteins 0.000 description 2
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 2
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 2
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 2
- 230000003321 amplification Effects 0.000 description 2
- 230000000259 anti-tumor effect Effects 0.000 description 2
- 229910052799 carbon Inorganic materials 0.000 description 2
- 239000000969 carrier Substances 0.000 description 2
- 230000004663 cell proliferation Effects 0.000 description 2
- 230000001276 controlling effect Effects 0.000 description 2
- 230000034994 death Effects 0.000 description 2
- 231100000517 death Toxicity 0.000 description 2
- 238000013401 experimental design Methods 0.000 description 2
- 239000011888 foil Substances 0.000 description 2
- 239000005457 ice water Substances 0.000 description 2
- 238000002513 implantation Methods 0.000 description 2
- 239000004615 ingredient Substances 0.000 description 2
- 229940125396 insulin Drugs 0.000 description 2
- 238000004255 ion exchange chromatography Methods 0.000 description 2
- 230000033001 locomotion Effects 0.000 description 2
- 239000011259 mixed solution Substances 0.000 description 2
- 239000012046 mixed solvent Substances 0.000 description 2
- 230000007935 neutral effect Effects 0.000 description 2
- 238000003199 nucleic acid amplification method Methods 0.000 description 2
- 238000011580 nude mouse model Methods 0.000 description 2
- AICOOMRHRUFYCM-ZRRPKQBOSA-N oxazine, 1 Chemical compound C([C@@H]1[C@H](C(C[C@]2(C)[C@@H]([C@H](C)N(C)C)[C@H](O)C[C@]21C)=O)CC1=CC2)C[C@H]1[C@@]1(C)[C@H]2N=C(C(C)C)OC1 AICOOMRHRUFYCM-ZRRPKQBOSA-N 0.000 description 2
- 239000003208 petroleum Substances 0.000 description 2
- 230000003285 pharmacodynamic effect Effects 0.000 description 2
- 150000003905 phosphatidylinositols Chemical class 0.000 description 2
- BASFCYQUMIYNBI-UHFFFAOYSA-N platinum Chemical compound [Pt] BASFCYQUMIYNBI-UHFFFAOYSA-N 0.000 description 2
- 235000015320 potassium carbonate Nutrition 0.000 description 2
- 238000002953 preparative HPLC Methods 0.000 description 2
- 125000006239 protecting group Chemical group 0.000 description 2
- 238000000746 purification Methods 0.000 description 2
- 238000000926 separation method Methods 0.000 description 2
- 238000007920 subcutaneous administration Methods 0.000 description 2
- 239000000126 substance Substances 0.000 description 2
- 238000003786 synthesis reaction Methods 0.000 description 2
- 238000004809 thin layer chromatography Methods 0.000 description 2
- 210000001519 tissue Anatomy 0.000 description 2
- 238000002054 transplantation Methods 0.000 description 2
- WZRRRFSJFQTGGB-UHFFFAOYSA-N 1,3,5-triazinane-2,4,6-trithione Chemical compound S=C1NC(=S)NC(=S)N1 WZRRRFSJFQTGGB-UHFFFAOYSA-N 0.000 description 1
- JKMHFZQWWAIEOD-UHFFFAOYSA-N 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid Chemical compound OCC[NH+]1CCN(CCS([O-])(=O)=O)CC1 JKMHFZQWWAIEOD-UHFFFAOYSA-N 0.000 description 1
- GPPJPLJOCJMMLS-UHFFFAOYSA-N 2-bromo-n,n-dimethylethanamine;hydrochloride Chemical compound Cl.CN(C)CCBr GPPJPLJOCJMMLS-UHFFFAOYSA-N 0.000 description 1
- QISAUDWTBBNJIR-UHFFFAOYSA-N 2-phenylmethoxyacetyl chloride Chemical compound ClC(=O)COCC1=CC=CC=C1 QISAUDWTBBNJIR-UHFFFAOYSA-N 0.000 description 1
- QBWKPGNFQQJGFY-QLFBSQMISA-N 3-[(1r)-1-[(2r,6s)-2,6-dimethylmorpholin-4-yl]ethyl]-n-[6-methyl-3-(1h-pyrazol-4-yl)imidazo[1,2-a]pyrazin-8-yl]-1,2-thiazol-5-amine Chemical compound N1([C@H](C)C2=NSC(NC=3C4=NC=C(N4C=C(C)N=3)C3=CNN=C3)=C2)C[C@H](C)O[C@H](C)C1 QBWKPGNFQQJGFY-QLFBSQMISA-N 0.000 description 1
- WGOLHUGPTDEKCF-UHFFFAOYSA-N 5-bromopyridin-2-amine Chemical compound NC1=CC=C(Br)C=N1 WGOLHUGPTDEKCF-UHFFFAOYSA-N 0.000 description 1
- 238000003727 ADP Glo Kinase Assay Methods 0.000 description 1
- 230000007730 Akt signaling Effects 0.000 description 1
- 241000894006 Bacteria Species 0.000 description 1
- 208000005623 Carcinogenesis Diseases 0.000 description 1
- 206010010904 Convulsion Diseases 0.000 description 1
- 206010012735 Diarrhoea Diseases 0.000 description 1
- 101100520033 Dictyostelium discoideum pikC gene Proteins 0.000 description 1
- KRHYYFGTRYWZRS-UHFFFAOYSA-M Fluoride anion Chemical group [F-] KRHYYFGTRYWZRS-UHFFFAOYSA-M 0.000 description 1
- 206010064571 Gene mutation Diseases 0.000 description 1
- 206010053759 Growth retardation Diseases 0.000 description 1
- 241000238631 Hexapoda Species 0.000 description 1
- 241000699660 Mus musculus Species 0.000 description 1
- 238000005481 NMR spectroscopy Methods 0.000 description 1
- 229910020700 Na3VO4 Inorganic materials 0.000 description 1
- 101100030361 Neurospora crassa (strain ATCC 24698 / 74-OR23-1A / CBS 708.71 / DSM 1257 / FGSC 987) pph-3 gene Proteins 0.000 description 1
- 239000012828 PI3K inhibitor Substances 0.000 description 1
- 101710132081 Phosphatidylinositol 3,4,5-trisphosphate 3-phosphatase and dual-specificity protein phosphatase PTEN Proteins 0.000 description 1
- 206010035039 Piloerection Diseases 0.000 description 1
- OFOBLEOULBTSOW-UHFFFAOYSA-N Propanedioic acid Natural products OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 1
- 108700025716 Tumor Suppressor Genes Proteins 0.000 description 1
- 102000044209 Tumor Suppressor Genes Human genes 0.000 description 1
- 230000002159 abnormal effect Effects 0.000 description 1
- 230000004913 activation Effects 0.000 description 1
- 239000004480 active ingredient Substances 0.000 description 1
- 239000000853 adhesive Substances 0.000 description 1
- 230000001070 adhesive effect Effects 0.000 description 1
- 239000012080 ambient air Substances 0.000 description 1
- 150000001413 amino acids Chemical class 0.000 description 1
- 125000003277 amino group Chemical group 0.000 description 1
- 238000000540 analysis of variance Methods 0.000 description 1
- 238000004164 analytical calibration Methods 0.000 description 1
- 230000006907 apoptotic process Effects 0.000 description 1
- 230000033228 biological regulation Effects 0.000 description 1
- 230000005587 bubbling Effects 0.000 description 1
- 229910052792 caesium Inorganic materials 0.000 description 1
- TVFDJXOCXUVLDH-UHFFFAOYSA-N caesium atom Chemical compound [Cs] TVFDJXOCXUVLDH-UHFFFAOYSA-N 0.000 description 1
- 230000036952 cancer formation Effects 0.000 description 1
- 231100000504 carcinogenesis Toxicity 0.000 description 1
- 230000003197 catalytic effect Effects 0.000 description 1
- 230000020411 cell activation Effects 0.000 description 1
- 239000013592 cell lysate Substances 0.000 description 1
- 230000001413 cellular effect Effects 0.000 description 1
- 230000008859 change Effects 0.000 description 1
- 210000000349 chromosome Anatomy 0.000 description 1
- 239000012230 colorless oil Substances 0.000 description 1
- 229940125846 compound 25 Drugs 0.000 description 1
- 239000000356 contaminant Substances 0.000 description 1
- 238000001816 cooling Methods 0.000 description 1
- 230000002596 correlated effect Effects 0.000 description 1
- 238000002425 crystallisation Methods 0.000 description 1
- 230000008025 crystallization Effects 0.000 description 1
- 210000004748 cultured cell Anatomy 0.000 description 1
- NXQGGXCHGDYOHB-UHFFFAOYSA-L cyclopenta-1,4-dien-1-yl(diphenyl)phosphane;dichloropalladium;iron(2+) Chemical compound [Fe+2].Cl[Pd]Cl.[CH-]1C=CC(P(C=2C=CC=CC=2)C=2C=CC=CC=2)=C1.[CH-]1C=CC(P(C=2C=CC=CC=2)C=2C=CC=CC=2)=C1 NXQGGXCHGDYOHB-UHFFFAOYSA-L 0.000 description 1
- 230000007423 decrease Effects 0.000 description 1
- 230000018044 dehydration Effects 0.000 description 1
- 238000006297 dehydration reaction Methods 0.000 description 1
- 230000003111 delayed effect Effects 0.000 description 1
- 238000002050 diffraction method Methods 0.000 description 1
- 238000010790 dilution Methods 0.000 description 1
- 239000012895 dilution Substances 0.000 description 1
- 239000006185 dispersion Substances 0.000 description 1
- 239000012154 double-distilled water Substances 0.000 description 1
- 230000003828 downregulation Effects 0.000 description 1
- 238000005516 engineering process Methods 0.000 description 1
- 239000003623 enhancer Substances 0.000 description 1
- 230000002708 enhancing effect Effects 0.000 description 1
- DEFVIWRASFVYLL-UHFFFAOYSA-N ethylene glycol bis(2-aminoethyl)tetraacetic acid Chemical compound OC(=O)CN(CC(O)=O)CCOCCOCCN(CC(O)=O)CC(O)=O DEFVIWRASFVYLL-UHFFFAOYSA-N 0.000 description 1
- SFNALCNOMXIBKG-UHFFFAOYSA-N ethylene glycol monododecyl ether Chemical compound CCCCCCCCCCCCOCCO SFNALCNOMXIBKG-UHFFFAOYSA-N 0.000 description 1
- 239000000374 eutectic mixture Substances 0.000 description 1
- 238000011156 evaluation Methods 0.000 description 1
- 238000000605 extraction Methods 0.000 description 1
- 235000021050 feed intake Nutrition 0.000 description 1
- 239000012634 fragment Substances 0.000 description 1
- 239000012458 free base Substances 0.000 description 1
- 125000000524 functional group Chemical group 0.000 description 1
- 231100000001 growth retardation Toxicity 0.000 description 1
- 230000036541 health Effects 0.000 description 1
- 230000003862 health status Effects 0.000 description 1
- 231100000086 high toxicity Toxicity 0.000 description 1
- 238000011534 incubation Methods 0.000 description 1
- 239000003112 inhibitor Substances 0.000 description 1
- CDAISMWEOUEBRE-GPIVLXJGSA-N inositol Chemical group O[C@H]1[C@H](O)[C@@H](O)[C@H](O)[C@H](O)[C@@H]1O CDAISMWEOUEBRE-GPIVLXJGSA-N 0.000 description 1
- 238000007689 inspection Methods 0.000 description 1
- 238000000021 kinase assay Methods 0.000 description 1
- 239000004816 latex Substances 0.000 description 1
- 229920000126 latex Polymers 0.000 description 1
- 239000007788 liquid Substances 0.000 description 1
- 230000002934 lysing effect Effects 0.000 description 1
- 239000012139 lysis buffer Substances 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 239000011976 maleic acid Substances 0.000 description 1
- 239000003550 marker Substances 0.000 description 1
- 238000001819 mass spectrum Methods 0.000 description 1
- 239000000463 material Substances 0.000 description 1
- 230000008018 melting Effects 0.000 description 1
- 238000002844 melting Methods 0.000 description 1
- 230000004060 metabolic process Effects 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 230000035772 mutation Effects 0.000 description 1
- HXRAMSFGUAOAJR-UHFFFAOYSA-N n,n,n',n'-tetramethyl-1-[(2-methylpropan-2-yl)oxy]methanediamine Chemical compound CN(C)C(N(C)C)OC(C)(C)C HXRAMSFGUAOAJR-UHFFFAOYSA-N 0.000 description 1
- 230000035407 negative regulation of cell proliferation Effects 0.000 description 1
- 238000001543 one-way ANOVA Methods 0.000 description 1
- 229910052763 palladium Inorganic materials 0.000 description 1
- 239000002245 particle Substances 0.000 description 1
- 230000002085 persistent effect Effects 0.000 description 1
- 229940043441 phosphoinositide 3-kinase inhibitor Drugs 0.000 description 1
- 230000026731 phosphorylation Effects 0.000 description 1
- 238000006366 phosphorylation reaction Methods 0.000 description 1
- 230000005371 pilomotor reflex Effects 0.000 description 1
- 229910052697 platinum Inorganic materials 0.000 description 1
- 239000000843 powder Substances 0.000 description 1
- 239000003755 preservative agent Substances 0.000 description 1
- 230000002335 preservative effect Effects 0.000 description 1
- 230000035755 proliferation Effects 0.000 description 1
- 230000001681 protective effect Effects 0.000 description 1
- 125000005493 quinolyl group Chemical group 0.000 description 1
- 238000003044 randomized block design Methods 0.000 description 1
- 238000010992 reflux Methods 0.000 description 1
- 230000001105 regulatory effect Effects 0.000 description 1
- 230000029058 respiratory gaseous exchange Effects 0.000 description 1
- 229920006395 saturated elastomer Polymers 0.000 description 1
- 238000007789 sealing Methods 0.000 description 1
- 210000002966 serum Anatomy 0.000 description 1
- 239000004017 serum-free culture medium Substances 0.000 description 1
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 1
- 235000017557 sodium bicarbonate Nutrition 0.000 description 1
- 238000001228 spectrum Methods 0.000 description 1
- 239000007858 starting material Substances 0.000 description 1
- 230000001954 sterilising effect Effects 0.000 description 1
- 238000004659 sterilization and disinfection Methods 0.000 description 1
- 239000012089 stop solution Substances 0.000 description 1
- 238000003860 storage Methods 0.000 description 1
- 230000004083 survival effect Effects 0.000 description 1
- 230000002194 synthesizing effect Effects 0.000 description 1
- 238000010189 synthetic method Methods 0.000 description 1
- 238000010998 test method Methods 0.000 description 1
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 1
- CZDYPVPMEAXLPK-UHFFFAOYSA-N tetramethylsilane Chemical compound C[Si](C)(C)C CZDYPVPMEAXLPK-UHFFFAOYSA-N 0.000 description 1
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 1
- IHIXIJGXTJIKRB-UHFFFAOYSA-N trisodium vanadate Chemical compound [Na+].[Na+].[Na+].[O-][V]([O-])([O-])=O IHIXIJGXTJIKRB-UHFFFAOYSA-N 0.000 description 1
- 238000001291 vacuum drying Methods 0.000 description 1
- 238000003260 vortexing Methods 0.000 description 1
- 230000003442 weekly effect Effects 0.000 description 1
- 230000004580 weight loss Effects 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/519—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B2200/00—Indexing scheme relating to specific properties of organic compounds
- C07B2200/13—Crystalline forms, e.g. polymorphs
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Epidemiology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
Abstract
Forma cristalina IX del compuesto 1 según representa la siguiente fórmula: **(Ver fórmula)** caracterizada por que la forma cristalina IX tiene picos de difracción en 2θ = 7,947°, 10,073°, 14,531°, 19,187°, 21,237°, 24,055°, 25,497°, en un patrón de difracción de rayos X.
Description
DESCRIPCIÓN
Análogo de pirido[1,2-a]pirimidona, forma cristalina del mismo, intermedio del mismo y método de preparación del mismo Campo de la invención
La presente invención se refiere a una forma cristalina de un análogo de pirido[1,2-a]pirimidona, un método de preparación del mismo y un intermedio del mismo.
Antecedentes de la invención
La ruta de la PI3K en las células cancerosas humanas es una vía en donde las mutaciones ocurren con mayor frecuencia y pueden conducir a la proliferación y activación celular, así como a la amplificación de las señales celulares. La cinasa PI3K (fosfatidilinositol-3-cinasa, PI3Ks) pertenece a la familia de las cinasas de lípidos y puede fosforilar el extremo 3'-OH del anillo de inositol del fosfatidilinositol. La PI3K es una cinasa de lípidos compuesta por una subunidad reguladora p85 o p101 y una subunidad catalítica p110, y desempeña un papel clave en la proliferación, la supervivencia y el metabolismo celular, etc., al catalizar la fosforilación de 4,5-bisfosfato de fosfatidilinositol (PIP2) para formar 3,4,5-trifosfato de fosfatidilinositol (PIP3) y activar así posteriormente la Akt y similares. Por lo tanto, la inhibición de la fosfatidilinositol-3-cinasa puede afectar a la ruta de la PI3K y, por lo tanto, inhibir la proliferación y activación de las células cancerosas.
El gen supresor de tumores PTEN (homólogo de la fosfatasa y la tensina eliminado del cromosoma diez) desfosforila PIP3 para generar PIP2, con lo que consigue una regulación negativa de la ruta de señalización de PI3K/Akt, con inhibición de la proliferación celular y promoción de la apoptosis. La frecuente aparición de la mutación y amplificación del gen PI3K, así como la pérdida de PTEN en el cáncer y similares, indican que la PI3K está estrechamente relacionada con la tumorigénesis.
El documento WO 2014/022128 describe inhibidores de la PIK3 que tienen una estructura central que es un anillo de quinolilo sustituido y no una piridino[1,2-A]pirimidona como en los compuestos de la presente invención, así como un proceso para su preparación.
El documento EP 3159341 A1 describe un compuesto (compuesto 25) que tiene la misma fórmula estructural general que el compuesto 1 según la presente solicitud, pero no describe ninguna forma cristalina definida del mismo ni da ninguna indicación de cómo obtener tales formas cristalinas definidas.
Compendio de la invención
La presente invención proporciona un método para preparar el compuesto 1,

que comprende los siguientes pasos:

en donde
X se selecciona entre Cl o Br;
el álcali C se selecciona entre piridina, 2,6-lutidina, Et3N, 4-DMAP, LiOH, Cs2CO3 o K2CO3 ;
el disolvente c se selecciona entre piridina, diclorometano, tolueno, acetonitrilo, acetona, DMF o THF;
la relación molar entre el compuesto 7 y el compuesto 8 es de 1:1 ~3;
la relación molar entre el compuesto 7 y el álcali C es de 1:1~3.
En algunas realizaciones de la presente invención, la relación molar entre el compuesto 7 y el compuesto 8 es de 1:1,2~1,6.
En algunas realizaciones de la presente invención, la preparación de dicho compuesto 1 comprende los siguientes pasos:

en donde
el álcali A se selecciona entre carbonato de potasio, carbonato de sodio, carbonato de cesio, hidróxido de potasio o hidróxido de sodio;
el disolvente a se selecciona entre DMF, DMSO o NMP.
Aquí, el cloruro de 2-dimetilaminoetilo o el bromuro de 2-dimetilaminoetilo pueden usarse en forma de una de sus sales, tales como clorhidrato de cloruro de 2-dimetilaminoetilo o clorhidrato de bromuro de 2-dimetilaminoetilo. En algunas realizaciones de la presente invención, la relación molar entre el compuesto 5 y el cloruro de 2-dimetilaminoetilo (o su sal clorhidrato) o el bromuro de 2-dimetilaminoetilo (o su sal clorhidrato) es de 1:1~2.
En algunas realizaciones de la presente invención, la relación molar entre el compuesto 5 y el cloruro de 2-dimetilaminoetilo (o su sal clorhidrato) o el bromuro de 2-dimetilaminoetilo (o su sal clorhidrato) es 1:1,1~1,3.
En algunas realizaciones de la presente invención, la preparación de dicho compuesto 1 comprende los siguientes pasos:

en donde
el álcali B se selecciona entre carbonato de potasio, carbonato de sodio, hidróxido de bario, fosfato de potasio, carbonato de cesio, fluoruro de potasio, fluoruro de cesio, hidróxido de sodio, f-butóxido de potasio, f-butóxido de sodio, acetato de potasio o acetato de sodio;
el disolvente b se selecciona entre 1,4-dioxano, DMSO, THF, 1,4-dioxano/agua o THF/agua;
la relación en volumen entre el 1,4-dioxano o el THF y el agua en el disolvente b es de 3-6:1, preferiblemente de 5:1; el catalizador se selecciona entre Pd(dppf)Cl2 o Pd(PPh3)4.
En algunas realizaciones de la presente invención, la relación en volumen entre el 1,4-dioxano o el THF y el agua en dicho disolvente b es de 5:1.
En algunas realizaciones de la presente invención, la preparación de dicho compuesto 1 comprende los siguientes pasos:

En el esquema de reacción anterior, la reacción del compuesto 5 con clorhidrato de cloruro de 2-dimetilaminoetilo para generar el compuesto 6 se realiza preferiblemente en presencia del álcali A y el disolvente a, en donde el álcali A se selecciona entre carbonato de potasio, carbonato de sodio, carbonato de cesio, hidróxido de potasio o hidróxido de sodio; y el disolvente a se selecciona entre DMF, DMSO o NMP. En algunas realizaciones de la presente invención, la relación molar entre el compuesto 5 y el clorhidrato de cloruro de 2-dimetilaminoetilo es de 1:1~2.
En algunas realizaciones de la presente invención, la relación molar entre el compuesto 5 y el clorhidrato de cloruro de 2-dimetilaminoetilo es de 1:1,1 ~1,3.
En el esquema de reacción anterior, la reacción del compuesto 6 con 2-metoxi-5-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)piridin-3-amina para generar el compuesto 7 se realiza preferiblemente en presencia del álcali B, el disolvente b y un catalizador, en donde el álcali B se selecciona entre carbonato de potasio, carbonato de sodio, hidróxido de bario, fosfato de potasio, carbonato de cesio, fluoruro de potasio, fluoruro de cesio, hidróxido de sodio, f-butóxido de potasio , f-butóxido de sodio, acetato de potasio o acetato de sodio; el disolvente b se selecciona entre 1,4-dioxano, DMSO, THF, 1,4-dioxano/agua o THF/agua, en donde la relación en volumen entre el 1,4-dioxano o el THF y el agua en el disolvente b es de 3~6:1, preferiblemente de 5:1; y el catalizador se selecciona entre Pd(dppf)Cl2 o Pd(PPh3)4.
En el esquema de reacción anterior, la reacción del compuesto 7 con cloruro de 2-cloro-4-fluorobencenosulfonilo para generar el compuesto 1 se realiza preferiblemente en presencia del álcali C y el disolvente c, en donde el álcali C se selecciona entre piridina, 2,6-lutidina , Et3N, 4-DMAP, LiOH, Cs2CO3, o K2CO3 ; el disolvente c se selecciona entre piridina, diclorometano, tolueno, acetonitrilo, acetona, DMF o THF; y la relación molar entre el compuesto 7 y el cloruro de 2-cloro-4-fluorobencenosulfonilo es de 1:1 ~3; y la relación molar entre el compuesto 7 y el álcali C es de 1:1 ~3. En algunas realizaciones de la presente invención, la relación molar entre el compuesto 7 y el cloruro de 2-cloro-4-fluorobencenosulfonilo es de 1:1,2~1,6.
La presente invención también proporciona compuestos representados por la siguiente fórmula como intermedios para la preparación del compuesto 1:

La presente invención proporciona la forma cristalina IX del compuesto 1, caracterizada por tener picos de difracción en 20 = 7,947°, 10,073°, 14,531 °, 19,187°, 21,237°, 24,055°, 25,497°; normalmente en 20 = 7,947°, 10,073°, 11,970°, 13,468°, 14,531°, 15,911°, 19,187°, 21,237°, 24,055°, 25,497°; más normalmente en 20 = 7,947°, 10,073°, 11,970°, 13,468°, 14,531°, 15,911°, 19,187°, 19,561°, 21,237°, 23,446°, 24,055°, 25,497°, 27,074°, en un patrón de difracción de rayos X (XRD).
La presente invención proporciona la forma cristalina IX del compuesto 1, que tiene un patrón de XRPD como se muestra en la figura 25.
Los datos del análisis del patrón de XRPD de dicha forma cristalina IX del compuesto 1 en algunas realizaciones de la presente invención se muestran en la tabla 1.
Tabla 1: datos del análisis del patrón de XRPD de la forma cristalina IX del compuesto 1

En algunas realizaciones de la presente invención, el patrón de DSC de dicha forma cristalina IX del compuesto 1 es como se muestra en la figura 26.
En algunas realizaciones de la presente invención, el patrón de TGA de dicha forma cristalina IX del compuesto 1 es como se muestra en la figura 27.
La forma cristalina IX del compuesto 1 puede presentarse en forma de cristal no solvatado o de cristal solvatado. El solvato en esta memoria se refiere a un solvato formado por un disolvente orgánico y/o agua con el compuesto correspondiente.
La presente invención proporciona una composición cristalina de la forma cristalina IX. En algunas realizaciones de la presente invención, la forma cristalina IX representa el 50 % o más, preferiblemente el 80 % o más, más preferiblemente el 90 % o más, lo más preferiblemente el 95 % o más del peso de la composición cristalina.
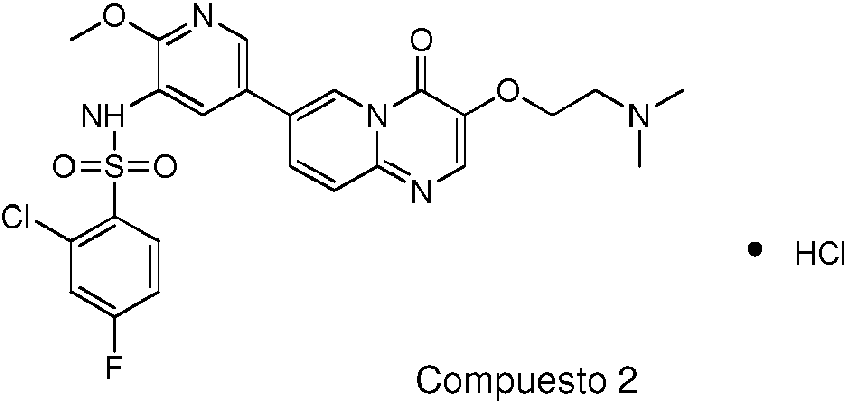
La presente invención proporciona una composición farmacéutica de la forma cristalina IX, que comprende una cantidad terapéuticamente eficaz de la forma cristalina IX o una composición cristalina de la forma cristalina IX. Además, la composición farmacéutica puede contener o no vehículos, excipientes y/o medios farmacéuticamente aceptables. La presente invención proporciona el compuesto 2 representado por la siguiente fórmula:
La presente invención proporciona la forma cristalina I del compuesto 2, caracterizada por tener picos de difracción en 20 = 10,154°, 12,285°, 14,511°, 16,328°, 24,311°, 26,188°; normalmente en 20 = 7,270°, 10,154°, 12,285°, 13,206°, 14,511°, 16,328°, 24,311°, 26,188°, 27,724°; más normalmente en 20 = 7,270°, 10,154°, 12,285°, 13,206°, 14,511°, 16,328°, 19,008°, 20,702°, 21,259°, 24,311°, 26,188°, 27,724°, en un patrón de difracción de rayos X (XRD).
La presente invención proporciona la forma cristalina I del compuesto 2, que tiene un patrón de XRPD como se muestra en la figura 1.
Los datos del análisis del patrón de XRPD de dicha forma cristalina I del compuesto 2 en algunas realizaciones de la presente invención se muestran en la tabla 2.
Tabla 2: datos del análisis del patrón de XRPD de la forma cristalina I del compuesto 2

En algunas realizaciones de la presente invención, el patrón de DSC de la forma cristalina I del compuesto 2 es como se muestra en la figura 2.
En algunas realizaciones de la presente invención, el patrón de TGA de la forma cristalina I del compuesto 2 es como se muestra en la figura 3.
La forma cristalina I del compuesto 2 puede presentarse en forma de cristal no solvatado o de cristal solvatado. El solvato en esta memoria se refiere a un solvato formado por un disolvente orgánico y/o agua con el compuesto correspondiente.
La presente invención proporciona una composición cristalina de la forma cristalina I. En algunas realizaciones de la presente invención, la forma cristalina I representa el 50 % o más, preferiblemente el 80 % o más, más preferiblemente el 90 % o más, lo más preferiblemente el 95 % o más del peso de la composición cristalina.
La presente invención proporciona una composición farmacéutica de la forma cristalina I, que comprende una cantidad terapéuticamente eficaz de la forma cristalina I o una composición cristalina de la forma cristalina I. Además, la composición farmacéutica puede contener o no vehículos, excipientes y/o medios farmacéuticamente aceptables.
La presente invención proporciona la forma cristalina II del compuesto 2, caracterizada por tener picos de difracción en 20 = 6,524°, 7,782°, 13,895°, 15,495°, 17,487°, 19,322°; normalmente en 20 = 6,524°, 7,782°, 11,628°, 13,895°, 15,495°, 17,487°, 19,322°, 20,962°, 23,269°; más normalmente en 20 = 6,524°, 7,782°, 11,628°, 13,895°, 15,495°, 17,487°, 19,322°, 20,962°, 23,269°, 24,257°, 26,009°, 31,533°, en un patrón de difracción de rayos X (XRD).
La presente invención proporciona la forma cristalina II del compuesto 2, que tiene un patrón de XRPD como se muestra en la figura 4.
Los datos del análisis del patrón de XRPD de la forma cristalina II del compuesto 2 en algunas realizaciones de la presente invención se muestran en la tabla 3.
Tabla 3: datos del análisis del patrón de XRPD de la forma cristalina II del compuesto 2

En algunas realizaciones de la presente invención, el patrón de DSC de la forma cristalina II del compuesto 2 es como se muestra en la figura 5.
En algunas realizaciones de la presente invención, el patrón de TGA de la forma cristalina II del compuesto 2 es como se muestra en la figura 6.
La forma cristalina II del compuesto 2 puede presentarse en forma de cristal no solvatado o de cristal solvatado. El solvato en esta memoria se refiere a un solvato formado por un disolvente orgánico y/o agua con el compuesto correspondiente.
La presente invención proporciona una composición cristalina de la forma cristalina II. En algunas realizaciones de la presente invención, la forma cristalina II representa el 50 % o más, preferiblemente el 80 % o más, más preferiblemente el 90 % o más, lo más preferiblemente el 95 % o más del peso de la composición cristalina.
La presente invención proporciona una composición farmacéutica de la forma cristalina II, que comprende una cantidad terapéuticamente eficaz de la forma cristalina II o una composición cristalina de la forma cristalina II. Además, la composición farmacéutica puede contener o no vehículos, excipientes y/o medios farmacéuticamente aceptables. La presente invención proporciona la forma cristalina III del compuesto 2, caracterizada por tener picos de difracción en 20 = 6,979°, 9,939°, 14,392°, 16,107°, 20,982°, 25,990°; normalmente en 20 = 6,187°, 6,979°, 9,939°, 11,910°, 14,392°, 16,107°, 20,982°, 22,755°, 25,990°; más normalmente en 20 = 6,187°, 6,979°, 9,939°, 11,910°, 13,148°, 14,392°, 16,107°, 20,982°, 22,755°, 23,975°, 25,990°, 29,006°, en un patrón de difracción de rayos X (XRD).
La presente invención proporciona la forma cristalina III del compuesto 2, que tiene un patrón de XRPD como se muestra en la figura 7.
Los datos del análisis del patrón de XRPD de dicha forma cristalina III del compuesto 2 en algunas realizaciones de la presente invención se muestran en la tabla 4.
Tabla 4: datos del análisis del patrón de XRPD de la forma cristalina III del compuesto 2

En algunas realizaciones de la presente invención, el patrón de DSC de la forma cristalina III del compuesto 2 es como se muestra en la figura 8.
En algunas realizaciones de la presente invención, el patrón de TGA de la forma cristalina III del compuesto 2 es como se muestra en la figura 9.
La forma cristalina III del compuesto 2 puede presentarse en forma de cristal no solvatado o de cristal solvatado. El solvato en esta memoria se refiere a un solvato formado por un disolvente orgánico y/o agua con el compuesto correspondiente.
La presente invención proporciona una composición cristalina de la forma cristalina III. En algunas realizaciones de la presente invención, la forma cristalina III representa el 50 % o más, preferiblemente el 80 % o más, más preferiblemente el 90 % o más, lo más preferiblemente el 95 % o más del peso de la composición cristalina.
La presente invención proporciona una composición farmacéutica de la forma cristalina III, que comprende una cantidad terapéuticamente eficaz de la forma cristalina III o una composición cristalina de la forma cristalina III. Además, la composición farmacéutica puede contener o no vehículos, excipientes y/o medios farmacéuticamente aceptables. La presente invención proporciona la forma cristalina IV del compuesto 2, caracterizada por tener picos de difracción en 20 = 6,388°, 7,278°, 11,076°, 15,454°, 21,256°; normalmente en 20 = 6,388°, 7,278°, 11,076°, 12,102°, 15,454°,
16,091°, 18,912°, 21,256°; más normalmente en 20 = 6,388°, 7,278°, 11,076°, 12,102°, 15,103°, 15,454°, 16,091°, 18,912°, 21,256°, 21,846°, en un patrón de difracción de rayos X (XRD).
La presente invención proporciona la forma cristalina IV del compuesto 2, que tiene un patrón de XRPD como se muestra en la figura 10.
Los datos del análisis del patrón de XRPD de dicha forma cristalina IV del compuesto 2 en algunas realizaciones de la presente invención se muestran en la tabla 5.
Tabla 5: datos del análisis del patrón de XRPD de la forma cristalina IV del compuesto 2

En algunas realizaciones de la presente invención, el patrón de DSC de la forma cristalina IV del compuesto 2 es como se muestra en la figura 11.
En algunas realizaciones de la presente invención, el patrón de TGA de la forma cristalina IV del compuesto 2 es como se muestra en la figura 12.
La forma cristalina IV del compuesto 2 puede presentarse en forma de cristal no solvatado o de cristal solvatado. El solvato en esta memoria se refiere a un solvato formado por un disolvente orgánico y/o agua con el compuesto correspondiente.
La presente invención proporciona una composición cristalina de la forma cristalina IV. En algunas realizaciones de la presente invención, la forma cristalina IV representa el 50 % o más, preferiblemente el 80 % o más, más preferiblemente el 90 % o más, lo más preferiblemente el 95 % o más del peso de la composición cristalina.
La presente invención proporciona una composición farmacéutica de la forma cristalina IV, que comprende una cantidad terapéuticamente eficaz de la forma cristalina IV o una composición cristalina de la forma cristalina IV. Además, la composición farmacéutica puede contener o no vehículos, excipientes y/o medios farmacéuticamente aceptables. La presente invención proporciona la forma cristalina V del compuesto 2, caracterizada por tener picos de difracción en 20 = 7,116°, 14,137°, 15,911°, 22,223°, 24,610°; normalmente en 20 = 7,116°, 14,137°, 15,911°, 21,691°, 22,223°, 24,213°, 24,610°, 28,987°, en un patrón de difracción de rayos X (XRD).
La presente invención proporciona la forma cristalina V del compuesto 2, que tiene un patrón de XRPD como se muestra en la figura 13.
Los datos del análisis del patrón de XRPD de dicha forma cristalina V del compuesto 2 en algunas realizaciones de la presente invención se muestran en la tabla 6.
Tabla 6: datos del análisis del patrón de XRPD de la forma cristalina V del compuesto 2

En algunas realizaciones de la presente invención, el patrón de DSC de la forma cristalina V del compuesto 2 es como se muestra en la figura 14.
En algunas realizaciones de la presente invención, el patrón de TGA de la forma cristalina V del compuesto 2 es como se muestra en la figura 15.
La forma cristalina V del compuesto 2 puede presentarse en forma de cristal no solvatado o de cristal solvatado. El solvato en esta memoria se refiere a un solvato formado por un disolvente orgánico y/o agua con el compuesto correspondiente.
La presente invención proporciona una composición cristalina de la forma cristalina V. En algunas realizaciones de la presente invención, la forma cristalina V representa el 50 % o más, preferiblemente el 80 % o más, más preferiblemente el 90 % o más, lo más preferiblemente el 95 % o más del peso de la composición cristalina.
La presente invención proporciona una composición farmacéutica de la forma cristalina V, que comprende una cantidad terapéuticamente eficaz de la forma cristalina V o una composición cristalina de la forma cristalina V. Además, la composición farmacéutica puede contener o no vehículos, excipientes y/o medios farmacéuticamente aceptables.
La presente invención proporciona la forma cristalina VI del compuesto 2, caracterizada por tener picos de difracción en 20 = 5,775°, 11,770°, 14,415°, 15,753°, 22,518°, 26,623°; normalmente en 20 = 5,775°, 11,770°, 14,415°, 15,753°, 17,132°, 20,939°, 22,518°, 26,623°; más normalmente en 20 = 5,775°, 11,770°, 14,415°, 15,753°, 17,132°, 20,939°, 22,518°, 23,745°, 26,623°, 31,295°, en un patrón de difracción de rayos X (XRD).
La presente invención proporciona la forma cristalina VI del compuesto 2, que tiene un patrón de XRPD como se muestra en la figura 16.
Los datos del análisis del patrón de XRPD de dicha forma cristalina VI del compuesto 2 en algunas realizaciones de la presente invención se muestran en la tabla 7.
Tabla 7: datos del análisis del patrón de XRPD de la forma cristalina VI del compuesto 2

En algunas realizaciones de la presente invención, el patrón de DSC de la forma cristalina VI del compuesto 2 es como se muestra en la figura 17.
En algunas realizaciones de la presente invención, el patrón de TGA de la forma cristalina VI del compuesto 2 es como se muestra en la figura 18.
La forma cristalina VI del compuesto 2 puede presentarse en forma de cristal no solvatado o de cristal solvatado. El solvato en esta memoria se refiere a un solvato formado por un disolvente orgánico y/o agua con el compuesto correspondiente.
La presente invención proporciona una composición cristalina de la forma cristalina VI. En algunas realizaciones de la presente invención, la forma cristalina VI representa el 50 % o más, preferiblemente el 80 % o más, más preferiblemente el 90 % o más, lo más preferiblemente el 95 % o más del peso de la composición cristalina.
La presente invención proporciona una composición farmacéutica de la forma cristalina VI, que comprende una cantidad terapéuticamente eficaz de la forma cristalina VI o una composición cristalina de la forma cristalina VI. Además, la composición farmacéutica puede contener o no vehículos, excipientes y/o medios farmacéuticamente aceptables. La presente invención proporciona el compuesto 3 representado por la siguiente fórmula:

La presente invención proporciona la forma cristalina VII del compuesto 3, caracterizada por tener picos de difracción en 20 = 6,325°, 12,677°, 15,813°, 21,395°, 22,519°, 27,133°; normalmente en 20 = 6,325°, 12,677°, 13,251°, 15,813°, 18,954°, 21,395°, 22,519°, 25,161°, 27,133°; más normalmente en 20 = 6,325°, 12,677°, 13,251°, 15,813°, 16,565°, 18,954°, 21,395°, 22,519°, 24,117°, 25,161°, 26,405°, 27,133°, en un patrón de difracción de rayos X (XRD).
La presente invención proporciona la forma cristalina VII del compuesto 3, que tiene un patrón de XRPD como se muestra en la figura 19.
Los datos del análisis del patrón de XRPD de dicha forma cristalina VII del compuesto 3 en algunas realizaciones de la presente invención se muestran en la tabla 8.
Tabla 8: datos del análisis del patrón de XRPD de la forma cristalina VII del compuesto 3

En algunas realizaciones de la presente invención, el patrón de DSC de la forma cristalina VII del compuesto 3 es como se muestra en la figura 20.
En algunas realizaciones de la presente invención, el patrón de TGA de la forma cristalina VII del compuesto 3 es como se muestra en la figura 21.
La forma cristalina VII del compuesto 3 puede estar presente en forma de cristal no solvatado o de cristal solvatado. El solvato en esta memoria se refiere a un solvato formado por un disolvente orgánico y/o agua con el compuesto correspondiente.
La presente invención proporciona una composición cristalina de la forma cristalina VII. En algunas realizaciones de la presente invención, la forma cristalina VII representa el 50 % o más, preferiblemente el 80 % o más, más preferiblemente el 90 % o más, lo más preferiblemente el 95 % o más del peso de la composición cristalina.
La presente invención proporciona una composición farmacéutica de la forma cristalina VII, que comprende una cantidad terapéuticamente eficaz de la forma cristalina VII o una composición cristalina de la forma cristalina VII. Además, la composición farmacéutica puede contener o no vehículos, excipientes y/o medios farmacéuticamente aceptables. La presente invención proporciona el compuesto 4 representado por la siguiente fórmula:

La presente invención proporciona la forma cristalina VIII del compuesto 4, caracterizada por tener picos de difracción en 20 = 5,889°, 11,002°, 12,518°, 14,906°, 17,825°, 22,814°, 25,555°; normalmente en 20 = 5,889°, 7,173°, 11,002°, 11,396°, 12,518°, 12,895°, 14,906°, 17,825°, 22,814°, 25,555°; más normalmente en 20 = 5,889°, 7,173°, 11,002°, 11,396°, 12,518°, 12,895°, 14,906°, 16,169°, 17,825°, 19,875°, 21,574°, 22,814°, 25,555°, 27,254° en un patrón de difracción de rayos X (XRD).
En algunas realizaciones de la presente invención, el patrón de XRPD de la forma cristalina VIII del compuesto 4 es como se muestra en la figura 22.
Los datos del análisis del patrón de XRPD de dicha forma cristalina VIII del compuesto 4 en algunas realizaciones de la presente invención se muestran en la tabla 9.
Tabla 9: datos del análisis del patrón de XRPD de la forma cristalina VIII del compuesto 4

En algunas realizaciones de la presente invención, el patrón de DSC de la forma cristalina VIII del compuesto 4 es como se muestra en la figura 23.
En algunas realizaciones de la presente invención, el patrón de TGA de la forma cristalina VIII del compuesto 4 es como se muestra en la figura 24.
La forma cristalina VIII del compuesto 4 puede presentarse en forma de cristal no solvatado o de cristal solvatado. El solvato en esta memoria se refiere a un solvato formado por un disolvente orgánico y/o agua con el compuesto correspondiente.
La presente invención proporciona una composición cristalina de la forma cristalina VIII. En algunas realizaciones de la presente invención, la forma cristalina VIII representa el 50 % o más, preferiblemente el 80 % o más, más preferiblemente el 90 % o más, lo más preferiblemente el 95 % o más del peso de la composición cristalina.
La presente invención proporciona una composición farmacéutica de la forma cristalina VIII, que comprende una cantidad terapéuticamente eficaz de la forma cristalina VIII o una composición cristalina de la forma cristalina VIII. Además, la composición farmacéutica puede contener o no vehículos, excipientes y/o medios farmacéuticamente aceptables. Un objetivo de la presente invención también es proporcionar los compuestos 1, 2, 3 y 4, o una composición farmacéutica de los mismos, para usar en la fabricación de un medicamento para tratar enfermedades relacionadas con receptores de PI3K.
Otro objetivo de la presente invención es proporcionar las formas cristalinas I, II, III, IV, V, VI, VII, VIII y IX, composiciones cristalinas de las mismas y una composición farmacéutica de las mismas para usar en la fabricación de un medicamento para tratar enfermedades relacionadas con la cinasa PI3K.
En algunas realizaciones de la presente invención, las enfermedades relacionadas con la cinasa PI3K se seleccionan entre cánceres, tales como cáncer de colon, cáncer gástrico y similares.
La presente solicitud describe un método para tratar enfermedades relacionadas con la cinasa PI3K, que comprende administrar una cantidad terapéuticamente eficaz de los compuestos 1,2, 3 y 4, o una composición farmacéutica de los mismos, a un paciente que lo necesite.
La presente solicitud describe además un método para tratar enfermedades relacionadas con la cinasa PI3K, que comprende administrar una cantidad terapéuticamente eficaz de las formas cristalinas I, II, III, IV, V, VI, VII, VIII y IX de la presente invención, una composición cristalina de las mismas y una composición farmacéutica de las mismas a un paciente que lo necesite.
En algunas realizaciones, las enfermedades relacionadas con la cinasa PI3K se seleccionan entre cánceres, tales como cáncer de colon, cáncer gástrico y similares.
Definición y descripción
A menos que se indique lo contrario, los términos y frases utilizados en esta memoria pretenden tener los siguientes significados. Una frase o un término específico no debe considerarse incierto o poco claro en ausencia de una definición específica y debe entenderse de acuerdo con el sentido común. Cuando en esta memoria aparece un nombre comercial, se pretende hacer referencia al producto correspondiente o al ingrediente activo del mismo.
Los compuestos intermedios de la presente invención pueden prepararse mediante diversos métodos sintéticos bien conocidos por los expertos en la técnica, incluidas las realizaciones específicas que se ilustran a continuación, las realizaciones formadas junto con otros métodos de síntesis química y equivalentes bien conocidos por los expertos en la técnica. Las realizaciones preferidas incluyen, pero no se limitan a los ejemplos de la presente invención.
Las reacciones químicas en las realizaciones específicas de la presente invención se realizan en disolventes adecuados, que deben ser adecuados para los cambios químicos, así como para los reactivos y materiales requeridos en la presente invención. Para obtener los compuestos de la presente invención, en ocasiones es necesario que los expertos en la técnica modifiquen o seleccionen los pasos de síntesis o los esquemas de reacción sobre la base de estas realizaciones en esta memoria.
Un factor de consideración importante para la planificación de cualquier ruta sintética en la técnica es la selección de un grupo protector adecuado para un grupo funcional reactivo (tal como el grupo amino de la presente invención). La publicación Protective Groups In Organic Synthesis, Wiley y Sons, 1991, de Greene y Wuts es la autoridad en este campo para los expertos en la técnica. Todas las referencias citadas en la presente invención se incorporan a esta memoria en su totalidad como referencia.
La presente invención se describirá específicamente a continuación mediante ejemplos, pero no se pretende que la presente invención quede limitada a estos ejemplos.
Todos los disolventes usados en la presente invención están disponibles comercialmente y se usan sin purificación adicional. Las reacciones generalmente se llevan a cabo bajo gas nitrógeno inerte en el disolvente anhidro. Los datos de resonancia magnética nuclear de protones se registran en un espectrómetro Bruker Avance III 400 (400 MHz), en donde los desplazamientos químicos se indican como (ppm) de tetrametilsilano para un campo de baja intensidad. Los espectros de masas se miden en un aparato Agilent 1200 Series plus 6110 (&1956A). El sistema de CL/EM o EM de Shimadzu contiene un equipo DAD: SPD-M20A (LC) y un detector Shimadzu Micromass 2020. El espectrómetro de masas está equipado con una fuente de ionización por electropulverización (ESI) que funciona en modo positivo o negativo.
Cabe indicar que, en el espectro de difracción de rayos X, el patrón de difracción obtenido a partir de un compuesto cristalino suele ser característico de una forma cristalina específica, en donde la intensidad relativa de las bandas (especialmente para ángulos pequeños) puede variar debido a un efecto de orientación dominante derivado de la diferencia de las condiciones de cristalización, el tamaño de partícula y otras condiciones de medición. Por lo tanto, la intensidad relativa de los picos de difracción no es característica de la forma cristalina objetivo. Al juzgar si una forma cristalina es idéntica a la conocida, lo que debe tenerse en cuenta son las posiciones relativas de los picos más que las intensidades relativas de los mismos. Además, para cualquier forma cristalina dada, puede haber un ligero error en las posiciones de los picos, lo que también es bien conocido en el campo de la cristalografía. Por ejemplo, la posición de un pico puede desplazarse debido a cambios de la temperatura, el movimiento de la muestra o la calibración de los instrumentos durante el análisis de la muestra, y el error de medición del valor de 20 es a veces de aproximadamente ±0,5°, preferiblemente de aproximadamente ±0,3°, más preferiblemente de aproximadamente ±0,2°. Por lo tanto, al determinar la estructura de cada forma cristalina, debe tenerse en cuenta dicho error, y los valores de 20 dentro del error también se encuentran dentro del alcance de la presente invención. En un patrón de XRD, la posición del pico generalmente se representa mediante un ángulo 20 o un espaciamiento interplanar d, y existe una fórmula de conversión simple entre ambos: d = A/2sen0, en donde d representa el espaciamiento interplanar, A representa la longitud de onda de los rayos X incidentes y 0 es el ángulo de difracción. Para la misma forma cristalina del mismo tipo de compuesto, las posiciones de los picos en los espectros de XRD de la misma son similares en su conjunto y la intensidad relativa puede tener un error mayor. También debe señalarse que, en la identificación de una mezcla, pueden perderse parte de las líneas de difracción debido a factores como la disminución del contenido y similares. En este caso, incluso una banda también puede ser también característica del cristal dado y no es necesario contar con todas las bandas que se observan en una muestra de alta pureza.
Debe saberse que, durante la preparación de una forma cristalina de un fármaco, cuando la molécula del fármaco y una molécula del disolvente entran en contacto entre sí, es difícil evitar que la molécula del disolvente forme una mezcla eutéctica con la molécula del compuesto y permanezca en el sólido debido a condiciones externas y factores internos, para formar así un solvato, incluyendo específicamente solvatos estequiométricos y no estequiométricos. Tales solvatos están incluidos dentro del alcance de la presente invención.
La estequiometría del ion cloruro en el compuesto 2 (un clorhidrato) preparado por la presente invención puede determinarse mediante cromatografía iónica. El instrumento utilizado es 883 Basic IC plus 1; la columna seleccionada es Metrosep A Supp 5-150/4,0; caudal de 0,700 ml/min; tiempo de ejecución de 10 minutos.
La presente invención emplea las siguientes abreviaturas: DCM representa diclorometano; PE representa éter de petróleo; EA representa acetato de etilo; DMF representa A/,W-dimetilformamida; DMSO representa sulfóxido de dimetilo; THF representa tetrahidrofurano; MeOH representa metanol; NMP representa W-metilpirrolidona; Et3N representa trietilamina; 4-DMAP representa 4-dimetilaminopiridina; LiOH representa hidróxido de litio; Cs2CO3 representa carbonato de cesio; K2CO3 representa carbonato de potasio; PPh3 representa trifenilfosfina; Pd(PPh3)4 representa tetratrifenilfosfina-paladio; Pd(dppf)Cl2 representa cloruro de 1,1 '-bis(difenilfosfino)ferroceno-paladio.
Método del difractómetro de rayos X en polvo (XRPD) en la presente invención:
Modelo de instrumento: difractómetro de rayos X Bruker D8 Advance
Condiciones de prueba: los parámetros detallados de la XRPD son los siguientes:
generador de rayos X: Cu, Ka, (A = 1,54056 Á)
tensión del tubo: 40 kV, corriente del tubo: 40 mA
rendija de dispersión: 0,60 mm
rendija del detector: 10,50 mm
rendija antidispersión: 7,10 mm
intervalo de barrido: 4-40°
longitud de paso: 0,02°
velocidad: 0,1 s
velocidad de rotación de una placa de muestra: 15 rpm
Método de calorímetro diferencial de barrido (DSC) en la presente invención:
Modelo de instrumento: calorímetro diferencial de barrido TA Q2000
Condiciones de prueba: se toma una muestra (0,5~ 1 mg) y se coloca en una bandeja de aluminio de DSC para la prueba; el método es: desde la temperatura ambiente hasta 300 °C, a una velocidad de calentamiento de 10 °C/min. Método del analizador termogravimétrico (TGA) en la presente invención:
Modelo de instrumento: analizador termogravimétrico TA Q5000IR
Condiciones de prueba: se toma una muestra (2~5 mg) y se coloca en una bandeja de platino para TGA para la prueba; el método es: desde la temperatura ambiente hasta 300 °C, a una velocidad de calentamiento de 10 °C/min. Efecto técnico
Los compuestos 2, 3 y 4, la forma cristalina IX del compuesto 1, las formas cristalinas I, II, III, IV, V y VI del compuesto 2, la forma cristalina VII del compuesto 3 y la forma cristalina VIII del compuesto 4 proporcionados en la presente invención son de naturaleza estable y tienen buena solubilidad y excelente higroscopicidad, por lo que su futuro como fármacos es prometedor.
Los procesos para sintetizar el compuesto 1 y los intermedios del mismo proporcionados por la presente invención, en los que las materias primas son económicas y accesibles, superan las desventajas de la alta toxicidad de los reactivos utilizados, las rigurosas condiciones de reacción, las dificultades en la separación y la purificación y que no sean fáciles de industrializar y similares.
Descripción de los dibujos adjuntos
La figura 1 es un patrón de XRPD de la forma cristalina I del compuesto 2 usando radiación de Cu-Ka.
La figura 2 es un patrón de DSC de la forma cristalina I del compuesto 2.
La figura 3 es un patrón de TGA de la forma cristalina I del compuesto 2.
La figura 4 es un patrón de XRPD de la forma cristalina II del compuesto 2 usando radiación de Cu-Ka.
La figura 5 es un patrón de DSC de la forma cristalina II del compuesto 2.
La figura 6 es un patrón de TGA de la forma cristalina II del compuesto 2.
La figura 7 es un patrón de XRPD de la forma cristalina III del compuesto 2 usando radiación de Cu-Ka.
La figura 8 es un patrón de DSC de la forma cristalina III del compuesto 2.
La figura 9 es un patrón de TGA de la forma cristalina III del compuesto 2.
La figura 10 es un patrón de XRPD de la forma cristalina IV del compuesto 2 usando radiación de Cu-Ka.
La figura 11 es un patrón de DSC de la forma cristalina IV del compuesto 2.
La figura 12 es un patrón de TGA de la forma cristalina IV del compuesto 2.
La figura 13 es un patrón de XRPD de la forma cristalina V del compuesto 2 usando radiación de Cu-Ka.
La figura 14 es un patrón de DSC de la forma cristalina V del compuesto 2.
La figura 15 es un patrón de TGA de la forma cristalina V del compuesto 2.
La figura 16 es un patrón de XRPD de la forma cristalina VI del compuesto 2 usando radiación de Cu-Ka.
La figura 17 es un patrón de DSC de la forma cristalina VI del compuesto 2.
La figura 18 es un patrón de TGA de la forma cristalina VI del compuesto 2.
La figura 19 es un patrón de XRPD de la forma cristalina VII del compuesto 3 usando radiación de Cu-Ka.
La figura 20 es un patrón de DSC de la forma cristalina VII del compuesto 3.
La figura 21 es un patrón de TGA de la forma cristalina VII del compuesto 3.
La figura 22 es un patrón de XRPD de la forma cristalina VIII del compuesto 4 usando radiación de Cu-Ka.
La figura 23 es un patrón de DSC de la forma cristalina VIII del compuesto 4.
La figura 24 es un patrón de TGA de la forma cristalina VIII del compuesto 4.
La figura 25 es un patrón de XRPD de la forma cristalina IX del compuesto 1 usando radiación de Cu-Ka.
La figura 26 es un patrón de DSC de la forma cristalina IX del compuesto 1.
La figura 27 es un patrón de TGA de la forma cristalina IX del compuesto 1.
Descripción detallada
Para que el contenido de la presente invención se comprenda mejor, se describirá más detalladamente combinando los siguientes ejemplos específicos que no constituyen ninguna limitación del contenido de la presente invención. Ejemplo de referencia 1: preparación del compuesto 5
Preparación de 2-(benciloxi)acetato de metilo (2)

En un matraz de fondo redondo de tres bocas de 3,0 l se puso diclorometano (960 ml), se añadieron metanol (197,6 g, 247 ml) y piridina (304,78 ml, 311 mol) y la mezcla se enfrió a 0 °C en un baño de agua helada. Bajo la protección de gas nitrógeno, se añadió gota a gota cloruro de 2-(benciloxi)acetilo (300 g, 1,54 mol) al matraz de fondo redondo, y la temperatura se controló a 0-10 °C durante la adición. Después de la adición, se retiró el baño de hielo y agua y la solución de reacción se agitó a 20 °C durante 1,5 horas. Después del muestreo y la detección, la CCF (éter de petróleo/acetato de etilo = 5/1) muestra que la reacción se había completado. Se añadió agua (1,5 l) al matraz de fondo redondo y se agitó durante 10 minutos, se dejaron separar las fases y se recogió la fase orgánica; luego la fase orgánica se lavó con ácido clorhídrico diluido a 1,0 mol/l (900 ml x 2), se dejaron separar las fases y se recogió la fase orgánica; la fase orgánica se lavó adicionalmente con una solución de carbonato de sodio al 20 % (600 ml), se dejaron separar las fases y se recogió la fase orgánica, que se secó con sulfato de sodio anhidro (150 g) y se filtró, y el filtrado se concentró a presión reducida para dar como producto un aceite incoloro (284 g, 1,53 mol, 97 % de rendimiento, 99 % de pureza). RMN 1H (400 MHz, cloroformo-d) ppm 7,37-7,32 (m, 5H), 4,63 (s, 2H), 4,11 (s, 2H), 3,76 (s, 3H); CLEM (ESI) m/z: 202,8 (M+23).
Preparación de 2-(benciloxi)-3-(dimetilamino)acrilato de metilo (3)

Compuesto 3
Se puso 2-(benciloxi)acetato de metilo (506 g, 2,72 mol) en un matraz de fondo redondo de 3 l, se añadió tbutoxibis(dimetilamino)metano (569 g, 3,26 mol) y la temperatura de reacción se controló a 90-100 °C en una reacción de 14 horas. Después del muestreo y la detección, la CCF (PE/EA = 5/1) muestra que la reacción se había completado. La solución de reacción se enfrió a 60 °C y se concentró mediante una bomba de aceite para dar como producto un aceite amarillo (699 g, producto bruto), que se usó directamente en el siguiente paso.
RMN 1H (400 MHz, cloroformo-d) ppm 7,44-7,2 (m, 2H), 7,37-7,28 (m, 3H), 6,87 (s, 1H), 4,72 (s, 2H), 3,73 (s, 3H), 2,98 (s, 6H).
Preparación de 3-(benciloxi)-7-bromo-4H-pirido[1,2-a]pirimidin-4-ona (4)

Se puso 2-(benciloxi)-3-(dimetilamino)acrilato de metilo (318 g, 1,35 mol) en un matraz de fondo redondo de 5 l y se le añadieron ácido acético (3 l) y 2-amino-5-bromopiridina (246 g, 1,35 mol). La solución de reacción se dejó reaccionar durante 14 horas con agitación, controlando la temperatura a 120-130 °C. Después del muestreo y la detección, la CLEM muestra que la reacción se había completado sustancialmente. La solución de reacción se enfrió a 60 °C, se concentró y se evaporó para eliminar el disolvente, se le añadió acetato de etilo (750 ml) y la mezcla se agitó durante 10 minutos y se filtró. A la torta de filtración obtenida se le añadió acetato de etilo (500 ml), la mezcla se agitó durante 10 minutos y se filtró. La nueva torta de filtración obtenida se lavó con acetato de etilo (150 ml) y se secó por centrifugación para dar un compuesto sólido amarillo (319 g, 95 % de pureza, 67,79 % de rendimiento).
RMN 1H (400 MHz, cloroformo-d) d = 9,13 (d, J = 2,0 Hz, 1H), 8,05 (s, 1H), 7,56 (dd, J = 2,0, 9,6 Hz, 1H), 7,46-7,42 (m, 3H), 7,37-7,33 (m, 3H), 5,30 (s, 2H); CLEM (ESI) m/z: 332,6 (isótopo M+1).
Preparación de 7-bromo-3-hidroxi-4H-pirido[1,2-a]pirimidin-4-ona (5)

Se puso ácido trifluoroacético (1,2 l) en un matraz de fondo redondo de 3 l, se le añadió 3-(benciloxi)-7-bromo-4H-pirido[1,2-a]pirimidin-4-ona (313 g, 897,9 mmol) y la solución de reacción se dejó reaccionar durante 2 horas con agitación, controlando la temperatura a 80-90 °C. Después del muestreo y la detección, la CLEM muestra que la reacción se había completado sustancialmente. La solución de reacción se enfrió a 60 °C, se concentró y se evaporó para eliminar el disolvente, se le añadió acetato de etilo (1,2 l) y la mezcla se agitó durante 60 minutos y se filtró. A la torta de filtración obtenida se le añadió acetato de etilo (400 ml), la mezcla se agitó durante 60 minutos y se filtró. La torta de filtración adicional obtenida se secó a presión reducida y 40 °C durante 70 horas, para dar un compuesto sólido amarillo (191 g, 95,6 % de contenido, 100 % de pureza, 84,59 % de rendimiento).
RMN 1H (400MHz, DMSO-d6) d = 9,92 (sa, 1H), 8,90 (s, 1H), 8,07 (s, 1H), 7,73 (dd, J = 2,0, 9,6 Hz, 1H), 7,53 (d, J = 9,6 Hz, 1H); EM m/z: 240,9 (M+1), 242,8 (isótopo M+1).
Ejemplo 1: preparación del compuesto 1
Preparación de 7-bromo-3-(2-(dimetilamino)etoxi)-4H-pirido[1,2-a]pirimidin-4-ona (6)

Se pusieron 7-bromo-3-hidroxi-4H-pirido[1,2-a]pirimidin-4-ona (300 g, 1,2 mol) y N,N-dimetilformamida (3 l) en un matraz de fondo redondo y la temperatura del reactor se ajustó a 95-100 °C. Se añadió carbonato de potasio (497,4 g, 3,6 mol) al matraz de reacción y se agitó durante 30 minutos. A continuación, el clorhidrato de cloruro de 2-dimetilaminoetilo se dividió en tres porciones y se añadió como sigue: adición de clorhidrato de cloruro de 2-dimetilaminoetilo (70,6 g, 0,49 mol) al matraz de reacción y agitación durante 30 minutos; adición de clorhidrato de cloruro de 2-dimetilaminoetilo (70,6 g, 0,49 mol) al matraz de reacción y agitación durante 30 minutos; y adición de clorhidrato de cloruro de 2-dimetilaminoetilo (70,6 g, 0,49 mol) al matraz de reacción y agitación durante 2-2,5 horas.
Una vez completada la reacción (según control mediante HPLC), la temperatura del reactor se ajustó a 15 ± 5 °C. La solución de reacción se añadió a agua (15 l), a lo que siguió la extracción con diclorometano (4,5 l x 4). La fase orgánica se combinó y se concentró a presión reducida y 35 ± 5 °C hasta peso constante. Al producto obtenido después de la concentración, se le añadió n-heptano (1,8 l) y la mezcla se agitó a 15 ± 5 °C durante 15-16 horas. Después de la filtración, la torta de filtración obtenida se sometió a rotaevaporación a presión reducida y a 35 ± 5 °C para dar un producto sólido verde (280 g, 74,09 % de rendimiento, 98,22 % de pureza).
RMN 1H (400 MHz, CDCh) d = 2,35 (s, 6 H), 2,78 (t, J = 5,6 Hz, 2H), 4,25 (t, J = 6,0 Hz, 2H), 7,45 (d, J = 9,6 Hz, 1H), 7,55 (dd, J = 9,6 Hz, 2 Hz, 1H), 8,13 (s, 1H), 9,09 (d, J = 2,0 Hz, 1H); CLEM (ESI) m/z: 312 (isótopo M+1).
Preparación de 7-(5-amino-6-metoxipirimidin-3-il)-3-(2-(dimetilamino)etoxi)pirido[1,2-a]pirimidin-4-ona (7)

Se pusieron sucesivamente 7-bromo-3-(2-(dimetilamino)etoxi)-4H-pirido[1,2-a]pirimidin-4-ona (275 g, 0,87 mol), 2-metoxi-5-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)piridin-3-amina (249 g, 0,96 mol), 1,4-dioxano (2,75 l), agua (550 ml) y carbonato de potasio (362 g, 2,62 mol) en un matraz de reacción; después de burbujear durante 30-60 minutos, se añadió Pd(dppf)Cl2 (19,2 g, 26 mmol) al matraz de reacción y el volumen de dicho matraz de reacción se reemplazó 5 veces con gas nitrógeno; la temperatura del matraz de reacción se ajustó a 95 ± 5 °C y la mezcla se agitó durante
2-2,5 horas a esta temperatura. Una vez completada la reacción (según control mediante HPLC), la temperatura del reactor se ajustó a 15 ± 5 °C. La solución de reacción se añadió a n-heptano (6,6 l) y, después de ajustar la temperatura a 15 ± 5 °C, la mezcla se agitó durante 2-2,5 horas a esta temperatura. Después de la filtración, la torta de filtración obtenida se secó por centrifugación a presión reducida y 45 ± 5 °C. Al residuo obtenido se le añadió diclorometano/metanol (V/V = 8/1,2,75 l), la mezcla se agitó a 15 ± 5 °C durante 30-60 minutos y se filtró para obtener una torta de filtración; a la torta de filtración se le añadió diclorometano/metanol (V/V = 8/1, 1,375 l), la mezcla se agitó durante 30-60 minutos a 15 ± 5 °C y se filtró nuevamente para obtener otra torta de filtración, que luego se lavó con diclorometano/metanol (V/V = 8/1, 1,375 l). Los dos filtrados obtenidos se combinaron y concentraron a presión reducida y 45 ± 5 °C. Al residuo concentrado se le añadió diclorometano/metanol (V/V = 2/1,4,125 l) y la mezcla se disolvió con agitación. A la solución resultante se le añadieron ácido tiocianúrico (13,93 g) y carbón activado (27,5 g), la mezcla se agitó a 15 ± 5 °C durante 15-16 horas, se filtró con tierra de diatomeas (137,5 g), y la torta de filtración obtenida se lavó con diclorometano/metanol (V/V = 2/1, 1,375 l x 2). El filtrado se concentró a presión reducida y 45 ± 5 °C. Al residuo concentrado se le añadió metanol (1,1 l), la mezcla se agitó a 15 ± 5 °C durante 2-3 horas y se filtró, y la torta de filtración obtenida se lavó con metanol (137,5 ml) y se sometió a rotaevaporación a presión reducida y a 45 ± 5 °C para dar un producto sólido amarillo (270 g, 97,98 % de pureza, 84,24 % de rendimiento).
RMN 1H (400 MHz, DMSO-d6) d = 8,92 (d, J = 1,6 Hz, 1H), 8,24 (s, 1 H), 8,04 (dd, J = 9,6 Hz, 2 Hz, 1H), 7,80 (d, J = 2 Hz, 1H), 7,67 (d, J = 9,6 Hz, 1H), 7,27 (d, J = 2,0 Hz, 1H), 5,24 (s, 2H), 4,19 (t, J = 6,0 Hz, 2H), 3,93 (s, 3H), 2,66 (t, J = 6,0 Hz, 2H), 2,25 (s, 6H); CLEM (ESI) m/z: 356 (M+1).
Preparación del compuesto 1

Se pusieron 7-(5-amino-6-metoxipirimidin-3-il)-3-(2-(dimetilamino)etoxi)pirido[1,2-a]pirimidin-4-ona (265 g, 0,72 mol) y piridina (2,65 l) en un matraz de reacción; después de enfriar a 5 ± 5 °C, se añadió gota a gota al matraz de reacción una solución de cloruro de 2-cloro-4-fluorobencenosulfonilo (252 g, 1,08 mol) en piridina (504 ml); cuando se completó la adición, la solución de reacción se ajustó a la temperatura de 30-35 °C y se agitó durante 2-3 horas a esta temperatura. Una vez completada la reacción (según control mediante HPLC), la solución de reacción se concentró a presión reducida y 45 ± 5 °C hasta un peso de 2,5-3 veces el del compuesto 7. Al producto concentrado se le añadió diclorometano (3,7 l), la mezcla se agitó a 25 ± 5 °C durante 30 minutos y luego se concentró a presión reducida y 45 ± 5 °C hasta un peso de 2,5-3 veces el del compuesto 7. Al producto concentrado se le añadió diclorometano (3,7 l) y la mezcla se suspendió a 25 ± 5 °C durante 2-3 horas. Después de la filtración, la torta de filtración se recogió y se sometió a rotaevaporación a 45 ± 5 °C hasta un peso de 1,3-1,7 veces el del compuesto 7. Al producto de la rotaevaporación se le añadió diclorometano (1,85 l) y la mezcla se suspendió a 25 ± 5 °C durante 2-3 horas. Después de la filtración, la torta de filtración se recogió, se sometió a rotaevaporación a 45 ± 5 °C hasta un peso de 1,2-1,4 veces el del compuesto 7 y luego se secó al vacío a 45 °C durante 3-4 horas hasta un peso de 1,2-1,3 veces el del compuesto 7. Al producto bruto obtenido se le añadió acetonitrilo (2,12 l) y la mezcla se suspendió a 55 ± 5 °C durante 15-16 horas. La solución después de la suspensión se enfrió a 25 ± 5 °C y se filtró, y la torta de filtración obtenida se recogió y se sometió a rotaevaporación a 45 ± 5 °C hasta un peso de 1,1-1,2 veces el del compuesto 7. Al producto de la rotaevaporación se le añadió acetonitrilo (1,9 l) y la mezcla se suspendió a 55 ± 5 °C durante 15-16 horas. La solución después de la suspensión se enfrió a 25 ± 5 °C y se filtró, y la torta de filtración obtenida se sometió a rotaevaporación a 45 ± 5 °C hasta un peso de 1,0-1,1 veces el del compuesto 7. Al producto de la rotaevaporación se le añadieron metanol (5,3 l) y carbón activado (53 g), y la mezcla se agitó a 75 ± 5 °C durante 2-3 horas. La filtración se realizó con tierra de diatomeas (132 g), y la torta de filtración obtenida se recogió, se le añadió un disolvente mixto de diclorometano y metanol (V/V = 4/1,7,95 l) y se agitó a 25 ± 5 °C durante 30-60 minutos. La filtración se repitió y los dos filtrados obtenidos se combinaron y concentraron a 45 ± 5 °C hasta un peso de 1,01-1,03 veces el del compuesto 7. Al producto concentrado se le añadieron agua (4,24 l) y etanol (1,06 l), la mezcla se agitó a 25 ± 5 °C durante 5-10 minutos, se le añadió gota a gota una solución acuosa saturada de hidrogenocarbonato de sodio (1,3 l) y se agitó adicionalmente durante 2-3 horas. Después de la filtración, la torta de filtración se sometió a rotaevaporación a 45 ± 5 °C hasta un peso de 1,1-1,3 veces el del compuesto 7. Al producto de la rotaevaporación se le añadió etanol (1,59 l) y la mezcla se suspendió a 75 ± 5 °C durante 15-16 horas. La solución después de la suspensión se enfrió a 25 ± 5 °C y se filtró, y la torta de filtración se recogió y se sometió a rotaevaporación a 45 ± 5 °C hasta un peso de 0,89-0,92 veces el del compuesto 7. Al producto de la rotaevaporación se le añadió etanol (1,59 l) y la mezcla se suspendió a 75 ± 5 °C durante 15-16 horas. La solución después de la suspensión se enfrió a 25 ± 5 °C y se filtró, y la torta de filtración obtenida se recogió y se sometió a rotaevaporación a 45 ± 5 °C hasta un peso de 0,87-0,9 veces el del compuesto 7. Al producto de la rotaevaporación se le añadió agua (2,35 l) y la mezcla se agitó a 45 ± 5 °C durante 61 ± 1 horas. La solución mezclada se enfrió a 25 ± 5 °C y se filtró. La torta de filtración obtenida se recogió,
se le añadió agua (2,35 l), se agitó a 25 ± 5 °C durante 2-3 horas y se filtró. La torta de filtración obtenida se recogió, se secó al vacío a 60 °C durante 15-16 horas y luego se tamizó con un tamiz de malla 60 para dar un producto sólido de color amarillo claro (190 g, 98,33 % de pureza, 47,36 % de rendimiento).
RMN 1H (400 MHz, DMSO-d6-d) d = 2,96 (t, J = 6,0 Hz, 2H), 3,72 (s, 3H), 4,27 (t, J = 5,2 Hz, 2H), 7,32 (td, J = 8,8, 2,8 Hz, 1H), 7,60 (dd, J = 8,4, 2,4 Hz, 1H), 7,70 (d, J = 9,2 Hz, 1H), 7,75 (d, J = 2,0 Hz, 1H), 7,95-8,05 (m, 2H), 8,15 (d, J = 1,6 Hz, 1H), 8,27 (s, 1H), 8,85 (s, 1H); CLEM (ESI) m/z: 548 (M+1).
Ejemplo 2: preparación de la forma cristalina IX del compuesto 1
Se disolvió 7-(5-amino-6-metoxipirimidin-3-il)-3-(2-(dimetilamino)etoxi)pirido[1,2-a]pirimidin-4-ona (2,5 g, 6,75 mmol, 1,0 eq) en piridina (25 ml), se le añadió gota a gota cloruro de 2-cloro-4-fluorobencenosulfonilo (2,01 g, 8,78 mmol, 1,3 eq) a 0 °C y la mezcla se agitó a 10-20 °C durante 16 horas. Una vez completada la reacción, el disolvente se secó por centrifugación para dar un producto bruto. El producto bruto se purificó mediante una columna (DCM/MeOH: 10/1-4/1). Se obtuvo un producto sólido amarillo (2,4 g, 98,31 % de pureza, 63,79 % de rendimiento). El sólido amarillo anterior (1,3 g, 2,37 mmol) se separó mediante HPLC preparativa (neutra). El líquido de la separación por HPLC preparativa (neutra) se extrajo con DCM (500 ml x 3). La fase orgánica se secó sobre sulfato de sodio anhidro (100 g) y luego se filtró, y el filtrado obtenido se secó por centrifugación para dar un producto sólido blanco, la forma cristalina IX del compuesto 1 (970 mg, 1,75 mmol, 99 % de pureza, 73,94 % de rendimiento).
Ejemplo 3: preparación de la forma cristalina I del compuesto 2
Se pusieron 7-(5-amino-6-metoxipirimidin-3-il)-3-(2-(dimetilamino)etoxi)pirido[1,2-a]pirimidin-4-ona (29,0 g, 81,60 mmol, 1,0 eq) y piridina (290 ml) en un matraz de fondo redondo y tres bocas de 1,0 l R1 equipado con un agitador. R1 se colocó en un baño de hielo y se enfrió a 0-5°C. Se añadió gota a gota a R1 una solución de cloruro de 2-cloro-4-fluorobencenosulfonilo (24,30 g, 106,08 mmol, 1,3 eq) en piridina (60 ml), lo que llevó aproximadamente 30 minutos, y la solución de reacción se calentó de forma natural hasta 20 °C y se dejó reaccionar con agitación durante 16 horas. Una vez completada la reacción, la solución de reacción se concentró a presión reducida para eliminar la piridina y dar 80 g de un producto bruto sólido rojo. Se tomaron 64 g del producto bruto anterior y se pusieron en un matraz de fondo redondo de 1,0 l R2, al que se añadió diclorometano (350 ml) en R2. Luego, R2 se agitó a 15°C durante 2 horas y se filtró, y la torta de filtración obtenida se recogió y se secó para dar un sólido rojo claro (33,4 g, 77 % de rendimiento, 99,4 % de pureza). Se tomaron 30 g del sólido anterior y se pusieron en un matraz de fondo redondo de 1 l R3, al que se añadieron metanol (600 ml) y carbón activado (6 g, 20 %) en R3. La mezcla se colocó en un baño de aceite a 70°C y se agitó durante 12 horas. La mezcla se filtró con tierra de diatomeas (15 g) estando caliente. El filtrado se recogió y se secó por centrifugación para dar un producto sólido amarillo (22,6 g, 97,47 % de pureza). Al sólido anterior se le añadió acetonitrilo (150 ml); la mezcla resultante se agitó en un baño de aceite a 85 °C durante 12 horas, se enfrió a 20°C y se filtró; la torta de filtración se recogió y se secó para dar el producto del título como un sólido blanco, la forma cristalina I del compuesto 2 (21 g, 44,3 % de rendimiento, 100 % de pureza). La relación molar entre el ion cloruro en el compuesto 2 y en el compuesto 1 fue de 1:1 según se determinó por cromatografía iónica.
RMN 1H (400MHz, DMSO-d6-d) d = 2,91 (s, 6H), 3,53 (t, 2H), 3,71 (s, 3H), 4,52 (t, 2H), 7,38 (m, 1H), 7,77 (m, 2H), 7,97 (m, 2H), 8,16 (m, 1H), 8,45 (m, 2H), 8,98 (s, 1H).
Ejemplo 4: preparación de la forma cristalina II del compuesto 2
Se tomaron aproximadamente 50 mg de la forma cristalina I del compuesto 2 y se le añadieron 0,4 ml de acetona para formar una suspensión. La muestra en suspensión se colocó en un mezclador a temperatura constante (40°C) y se agitó durante 2 días (protegida de la luz). El sólido residual se centrifugó y se secó en un horno de secado al vacío a 40°C durante la noche, para dar la forma cristalina II del compuesto 2.
Ejemplo 5: preparación de la forma cristalina III del compuesto 2
El procedimiento de preparación de la forma cristalina III es idéntico al de la forma cristalina II, excepto porque el disolvente de acetona se sustituye por isopropanol. Se obtuvo la forma cristalina III del compuesto 2.
Ejemplo 6: preparación de la forma cristalina IV del compuesto 2
El procedimiento de preparación de la forma cristalina IV es idéntico al de la forma cristalina II, excepto únicamente porque el disolvente de acetona se sustituye por acetato de etilo. Se obtuvo la forma cristalina IV del compuesto 2.
Ejemplo 7: preparación de la forma cristalina V del compuesto 2
La forma cristalina I del compuesto 2 (2,0 g, 3,42 mmol) se puso en un matraz de una boca de 500 ml R1 y el sólido se disolvió por completo añadiendo DCM/MeOH (2/1,200 ml) con agitación. La solución se sometió a presión reducida y 40 °C para eliminar el disolvente y dar 2,0 g de un sólido amarillo; se tomó 1 g del sólido y se puso en un matraz de una boca de 50 ml, a lo que siguió la adición de etanol (6 ml); la mezcla obtenida se colocó en un baño de aceite a 80 °C con agitación durante 12 horas y luego se detuvo el calentamiento. La mezcla se enfrió a 20 °C con agitación y luego se filtró y la torta de filtración se secó para dar la forma cristalina V del compuesto 2.
Ejemplo 7: preparación de la forma cristalina VI del compuesto 2
Se pusieron 7-(5-amino-6-metoxipirimidin-3-il)-3-(2-(dimetilamino)etoxi)pirido[1,2-a]pirimidin-4-ona (70,0 g, 222,90 mmol, 1,0 eq, 99,4 % de pureza) y piridina (700 ml) en un matraz de fondo redondo de tres bocas de 2,0 l R1, equipado con un agitador mecánico, y R1 se colocó en un baño de hielo y se enfrió a 0-5 °C. Se añadió gota a gota a R1 una solución de cloruro de 2-cloro-4-fluorobencenosulfonilo (70,81 g, 293,67 mmol, 1,5 eq, 95 % de pureza) en piridina (140 ml), lo que llevó aproximadamente 30 minutos. R1 se colocó en un baño de aceite a 30 °C y la mezcla se dejó reaccionar con agitación durante 2 horas. Una vez completada la reacción, la solución de reacción se concentró a presión reducida para eliminar el disolvente piridina, y dar un producto bruto sólido rojo (200 g). Al residuo se le añadió diclorometano (1,0 l) y la mezcla se agitó a 20 °C durante 3 horas. Después de la filtración, se recogió la torta de filtración. Se añadió acetonitrilo (1,2 l) a la torta de filtración; la solución de reacción obtenida se calentó a reflujo a 85 °C durante 12 horas, se enfrió a 20 °C y se filtró; se recogió otra torta de filtración y se secó para dar un sólido (92 g). Al sólido de la torta de filtración se le añadieron metanol (2 l) y carbón activado (14 g), la mezcla se calentó a reflujo con agitación durante 3 horas, se filtró con tierra de diatomeas (40 g) cuando aún estaba caliente y se lavó con 500 ml. El filtrado se secó por centrifugación a presión reducida y 40 °C para dar un sólido (83 g). Al sólido obtenido se le añadió acetonitrilo (800 ml) y la mezcla se calentó a reflujo a 85 °C durante la noche, se enfrió a 20 °C y se filtró, y la torta de filtración resultante se secó para dar 77 g de un sólido blanco. Se tomaron 72 g del sólido blanco y se disolvieron por completo en metanol y se secaron por centrifugación para dar la forma cristalina VI del compuesto 2.
Ejemplo 8: preparación de la forma cristalina VII del compuesto 3
El compuesto 1 (997,34 mg, 1,82 mmol, 1,00 eq) se puso en un vial de vidrio de 5 ml, se añadió etanol/agua (7,5 ml/2,5 ml) y se agitó a temperatura ambiente (15 °C) durante 0,1 horas, durante las que gran cantidad de sólido no se disolvió. Se añadió ácido maleico (211,25 mg, 1,82 mmol, 1,00 eq) a la mezcla y esta se agitó a temperatura ambiente (15 °C) durante 18 horas, y se permitió que el sólido se disolviera por completo y formara una solución amarilla. La solución obtenida se secó por centrifugación a presión reducida y 40 °C hasta un volumen de 2 ml, se le añadió EA (20 ml), se agitó durante 0,5 horas y se filtró; la torta de filtración formada se secó por centrifugación a presión reducida y 40 °C para dar la forma cristalina VII del compuesto 3.
RMN 1H (400 MHz, DMSO-d6) d ppm 2,94 (s, 6H) 3,51-3,56 (m, 2H) 3,71 (s, 3H) 4,36-4,59 (m, 2H) 6,03 (s, 2H) 7,18 7,48 (m, 1H) 7,65-7,90 (m, 2H) 7,92-8,09 (m, 2H) 8,17 (dd, J = 9,29, 1,76 Hz, 1H) 8,35-8,55 (m, 2H) 8,99 (s, 1H).
Ejemplo 9: preparación de la forma cristalina VIII del compuesto 4
El compuesto 1 (997,34 mg, 1,82 mmol, 1,00 eq) se puso en un vial de vidrio de 5 ml, se añadió etanol/agua (7,5 ml/2,5 ml) y se agitó a temperatura ambiente (15 °C) durante 0,5 horas, durante las que gran cantidad de sólido no se disolvió. Se añadió ácido cítrico (382,45 mg, 1,82 mmol, 1,00 eq) a la mezcla y esta se agitó a temperatura ambiente (15 °C) durante 18 horas para dar una suspensión blanca lechosa en el vial de reacción. La solución mezclada obtenida se secó por centrifugación a presión reducida y 40 °C hasta un volumen de 2 ml, se le añadió EA (20 ml), se agitó durante 0,5 horas y se filtró; la torta de filtración formada se secó por centrifugación a presión reducida y 40 °C para dar la forma cristalina VIII del compuesto 4.
RMN 1H (400 MHz, DMSO-d6) d ppm 2,56-2,68 (m, 4H) 2,76 (s, 6 H) 3,31 (m, 2H) 3,72 (s, 3H) 4,37-4,40 (m, 2H) 7,35 7,37 (m, 1H) 7,71-7,78 (m, 2H) 7,91-7,95 (m, 1H) 7,95-8,09 (m, 1H) 8,11-8,13 (m, 1H) 8,36-8,37 (m, 2H) 8,96 (d, J = 1,6, 1H).
Ejemplo de prueba 1: prueba de estabilidad de la forma cristalina IX del compuesto 1 en diferentes disolventes
Se tomaron varias muestras de la forma cristalina IX del compuesto 1 en una cantidad adecuada, se añadieron respectivamente a 0,3-0,4 ml de un solo disolvente o un disolvente mixto como se muestra en la tabla siguiente y se agitaron a 40 °C. Después de agitar durante 2 días, las muestras se centrifugaron. Se recogió el sólido en las muestras y se detectó su estado cristalino por XRPD. Los resultados se muestran en la tabla 10.
Tabla 10: experimentos de estabilidad de la forma cristalina IX en forma de base libre en diferentes disolventes


Ejemplo de prueba 2: prueba de estabilidad en estado sólido de la forma cristalina IX del compuesto 1 en condiciones de alta temperatura, alta humedad y luz intensa.
Se pesaron aproximadamente 10 mg de la forma cristalina IX del compuesto 1 y se pusieron en el fondo de una botella de muestras de vidrio para formar una capa delgada. Para las muestras sometidas a las condiciones de 60 °C y temperatura ambiente / 92,5 % HR, la parte superior de las botellas se cubrió con papel de aluminio, en el que se hicieron varios orificios para garantizar que las muestras estuvieran completamente expuestas al aire del ambiente; para las muestras sometidas a luz intensa (5 klx), las botellas se sellaron con un tapón de rosca. Se tomó otra muestra de 15 mg de la forma cristalina IX y se trató de acuerdo con el método descrito anteriormente para detectar la forma cristalina de la muestra. Las muestras sometidas a diferentes condiciones se muestrearon y detectaron el día 5 y el día 10, y los resultados de la detección se compararon con los resultados de la detección inicial el día 0. Los resultados de la prueba se muestran en la tabla 11 a continuación.15*203
Tabla 11: prueba de estabilidad en estado sólido de la forma cristalina IX del compuesto 1

Resultados experimentales: las formas cristalinas de la presente invención tienen buena estabilidad y pueden utilizarse fácilmente para fabricar un medicamento.
Ejemplo de prueba 3: pruebas de actividad enzimática in vitro
Ensayo ADP-Glo I
Dilución de los compuestos:
Los compuestos de prueba se diluyeron en un gradiente de concentración triple y se obtuvieron en total 10 concentraciones (10.000 nM a 0,5 nM).
Método experimental:
Se transfirieron 50 nl de los compuestos de prueba de la presente invención a una placa de reacción (PerkinElmer #6007299) y se les añadieron 3 pl de una mezcla de enzima/sustrato (PI3Ka 0,33 nM, Millipore #14-602-K / PIP2166,5 pM); después de 20 minutos de incubación, se añadieron 2 pl de una solución de ATP (100 pM) para iniciar la reacción; después de 2 horas de reacción a temperatura ambiente, la reacción de la cinasa se detuvo añadiendo 5 pl del reactivo ADP-Glo e incubando a temperatura ambiente durante 60 minutos, lo que permitió que el ATP restante sin reaccionar se digiriera por completo; a la solución resultante se le añadieron 10 pl de un reactivo de detección de cinasas y la solución se incubó a temperatura ambiente durante 40 minutos, y luego se leyó la fluorescencia con un sistema Envision. PIP2, ATP, el reactivo ADP-Glo y los reactivos del ensayo de cinasas provienen todos de un kit de ensayo de cinasas ADP-Glo (Promega n.° V1792).
Análisis de los datos:
La CI50 se calculó utilizando el método estándar de ajuste de 4 parámetros (modelo 205, XL-fit, iDBS).
La actividad de los compuestos de prueba de la presente invención sobre la cinasa mTOR se analizó mediante el método de prueba siguiente.
Tampón de reacción: Hepes 20 mM (pH 7,5), MgCl2 10 mM, MnCl22 mM, EGTA 1 mM, Brij35 al 0,02 %, BSA 0,02 mg/ml, Na3VO40,1 mM, Dt T 2 mM, Dm SO al 2 %.
Enzima para la reacción: fragmento de mTOR recombinante humanizado con una etiqueta de GST N-terminal (aminoácidos 1360-2549, peso molecular = 163,9 kDa), expresado en células de insecto.
Sustrato de reacción: 4EBP1 recombinante humanizada de longitud completa con una etiqueta de His N-terminal (peso molecular = 13,6 kDa), expresada en bacterias.
Condiciones de reacción: 4EBP1 3 pM y ATP 10 pM.
Procedimiento de la reacción:
1) El sustrato de reacción y los otros factores de reacción se añadieron al tampón de reacción recién preparado. 2) La cinasa se añadió al sustrato de reacción y se mezcló suavemente.
3) El compuesto disuelto en DMSO al 100 % se transfirió a la solución de reacción de la cinasa usando un aparato de Acoustic Technology (Echo 550; rango de nanolitros) y luego se incubó a temperatura ambiente durante 20 minutos.
4) Se añadió al sistema de reacción una concentración apropiada de 32P-ATP.
5) Se incubó a temperatura ambiente durante 2 horas.
6) La actividad de la cinasa se detectó mediante el método de unión a filtros P81.
Los resultados experimentales se muestran en la Tabla 12.
Tabla 12: resultados de las pruebas de actividad enzimática in vitro

Nota: A < 1 nM; 200 nM < D.
Conclusión: el compuesto 1 tiene un efecto inhibidor significativo sobre PI3K (p110a), pero su efecto inhibidor sobre mTOR es más débil.
Ensayo ADP-Glo II
Procedimiento experimental:
1) El compuesto se diluyó mediante un aparato Echo de Labcyte Company, con el que se transfirieron 50 nl del compuesto a una placa de ensayo que se centrifugó a una velocidad de 1.000 rpm durante 10 segundos.
2) Se prepararon una solución mixta de cinasa/sustrato lipídico y una solución mixta de tampón de reacción de cinasa/sustrato lipídico; la solución mixta de cinasa/sustrato lipídico se añadió a las columnas 3-24 de la placa de ensayo, 3 pl por pocillo; la solución mixta de tampón de reacción de cinasa/sustrato lipídico se añadió a las columnas 1 -2 de la placa de ensayo, 3 pl por pocillo; y la placa se centrifugó a una velocidad de 1.000 rpm durante 10 segundos.
3) Se preparó una solución de ATP y se añadió a la placa de ensayo, 2 pl por pocillo; la placa se centrifugó a una velocidad de 1.000 rpm durante 10 segundos, se agitó y se mezcló en el modo de la 2.a marcha en un agitador de placas durante 1 minuto, se volvió a centrifugar a una velocidad de 1.000 rpm durante 10 segundos y se incubó durante 120 minutos a 23 °C.
4) Se preparó el reactivo ADP-Glo® y se añadió a la placa de ensayo, 5 pl por pocillo; la placa se centrifugó a una velocidad de 1.000 rpm durante 10 segundos, se agitó y se mezcló en el modo de la 2.a marcha en un agitador de placas durante 1 minuto, se volvió a centrifugar a una velocidad de 1.000 rpm durante 10 segundos y se incubó durante 60 minutos a 23 °C.
5) Se preparó un reactivo de detección de cinasas y se añadió a la placa de ensayo, 10 pl por pocillo; la placa se centrifugó a una velocidad de 1.000 rpm durante 10 segundos, se agitó y se mezcló en el modo de la 2a velocidad en un agitador de placas durante 1 minuto, se volvió a centrifugar a una velocidad de 1.000 rpm durante 10 segundos y se incubó durante 30 minutos a 23 °C; y luego se leyó con el detector de marcadores múltiples Envision.
Análisis de los datos:
Los resultados de la CI50 se analizaron mediante XLfit5 (fórmula 205) de la empresa IDBS.
Los experimentos y análisis correspondientes se realizaron empleando el procedimiento experimental descrito anteriormente, con el compuesto BKM120 como fármaco de control positivo.
Resultados experimentales:
Los valores de la CI50 para la inhibición por el compuesto 1 de la actividad de PI3Ka, PI3Kp, PI3K5 y PI3Ky fueron respectivamente de 0,6 ± 0,2 nM, 9,9 ± 2,7 nM, 0,5 ± 0,1 nM y 7,0 ± 0,9 nM (n = 2). En contraste, los valores de la CI50 para la inhibición por el fármaco de control positivo BKM120 (inhibidor de PI3K, Buparlisib) de la actividad de PI3Ka, PI3Kp, PI3K5 y PI3Ky fueron respectivamente de 24,7 ± 4,7 nM, 241,6 ± 50,6 nM, 68,8 ± 25,0 nM y 111,9 ± 15,2 nM. Conclusión: el compuesto 1 exhibe una actividad inhibidora muy alta sobre los cuatro subtipos de PI3K.
Ejemplo de prueba 4: pruebas de actividad celular in vitro
Pasos y métodos experimentales:
1) Se inocularon células MCF-7 a una densidad de 2,5 x 104 células/pocillo en placas de 96 pocillos (el medio de cultivo utilizado debe ser un medio de cultivo completo con FBS al 10%).
2) Al día siguiente, se extrajo el medio de cada pocillo. Se disolvió una determinada concentración (selección preliminar) o una serie de concentraciones (prueba de la CI50) de los compuestos de prueba en un medio de cultivo sin suero y se añadió a las placas de 96 pocillos para cultivar las células durante 2 horas.
3) Se disolvió insulina en un medio de cultivo sin suero, se añadió a las células cultivadas y se incubó durante 30 minutos, en donde la concentración final de insulina fue de 10 mg/ml.
4) Se preparó una solución de lisis de acuerdo con el siguiente método durante el período de espera para la reacción: a) La solución potenciadora se retiró del refrigerador para derretirla previamente.
b) La solución potenciadora se diluyó 10 veces con el tampón de lisis 5x para dar una solución de lisis concentrada. c) La solución de lisis concentrada se diluyó 5 veces con agua bidestilada para dar la solución de lisis.
5) El medio de cada pocillo se eliminó por completo y los pocillos se lavaron rápidamente una vez con PBS.
6) Se añadieron 150 pl de la solución de lisis recién preparada a cada pocillo y se agitó a temperatura ambiente durante 10 minutos.
7) Después de confirmar el desprendimiento de todas las células, la solución de lisis junto con los restos celulares se transfirió a un tubo de 1,5 ml.
8) El tubo se agitó varias veces en un vórtex, para permitir que la solución de lisis y las células se mezclaran por completo, y luego la mezcla se centrifugó a 4 °C a 12.000 g durante 10 minutos.
9) Se calculó el número de tiras de placa de micropocillos ELISA-One requeridas. Las tiras innecesarias se retiraron del marco, se volvieron a colocar en la bolsa de almacenamiento y se sellaron. Antes de usar las tiras de placa de micropocillos, se usaron 200 pl de agua bidestilada para lavar los pocillos y eliminar el conservante.
10) Se añadieron 50 pl de la mezcla de anticuerpos a cada pocillo. (La solución de la mezcla de anticuerpos se preparó mezclando el reactivo del medio de los anticuerpos y el reactivo de los anticuerpos marcado enzimáticamente en la misma proporción. La preparación de la mezcla de anticuerpos no necesitó vórtex).
11) Se añadieron 25 pl de lisados celulares a cada pocillo de la placa de micropocillos ELISA-One. La placa de micropocillos se cubrió con película sellante adhesiva y se incubó en un instrumento de oscilación de placas de micropocillos a temperatura ambiente durante 1 hora.
12) Los pocillos se lavaron 3 veces con 150 pl de tampón de lavado 1x. Después del último lavado, se eliminó por completo el tampón de lavado de los pocillos. Si es necesario, puede permitirse que el tampón de lavado 1x permanezca en la placa de micropocillos hasta 30 minutos para que la solución mixta de sustratos pueda prepararse durante ese período de tiempo.13
13) La solución mixta de sustratos debe prepararse justo antes de cada uso, se añadieron 100 pl de la solución mixta de sustratos a cada pocillo y la placa de micropocillos se selló con papel de aluminio y se incubó en el instrumento de oscilación de placas de micropocillos a temperatura ambiente durante 10 minutos.
14) Se añadieron 10 pl de la solución de parada a cada pocilio y se mezcló ligeramente (5-10 segundos) en el instrumento de oscilación de placas de micropocillos.
15) Se ensambló el grupo de filtros ELISA-One correspondiente y se usó para leer la intensidad de la señal de fluorescencia.
Los resultados experimentales se muestran en la tabla 13.
Tabla 13: resultados de las pruebas de actividad celular in vitro

Nota: A < 50 nM.
Ejemplo de prueba 5: experimento de eficacia in vivo
Los estudios se realizaron para examinar si los fármacos de prueba tienen eficacia in vivo en el modelo animal de cáncer de colon humano CO-04-0032 y en el modelo animal de cáncer gástrico ST-02-0013. Las descripciones sobre la alimentación de los animales, los ingredientes del pienso, la observación experimental, los índices experimentales, la terminación experimental, así como el análisis de los datos en las pruebas fueron las siguientes:
Alimentación de los animales; a su llegada, los animales deben ser alimentados en el ambiente experimental durante 3 a 7 días antes de comenzar el experimento. Los animales se alojaron en jaulas IVC (con sistema de ventilación independiente) (5 animales por jaula) en alojamientos para animales de grado SPF. Todas las jaulas, el acolchado y el agua potable deben esterilizarse antes de su uso, y los registros de esterilización se muestran en el anexo. Todo el personal de laboratorio en los alojamientos de los animales debe usar ropa protectora y guantes de látex durante el trabajo. Las tarjetas de información de las jaulas deben indicar el número de animales en la jaula, género, raza, fecha de llegada, régimen de dosificación, número de experimento, grupo y fecha de inicio de los experimentos. Las jaulas, el pienso y el agua de bebida se reemplazaron dos veces por semana. El entorno de alimentación y las condiciones de luz son las siguientes:
temperatura: 20-26 °C
humedad: 40~70%
ciclo de luz y oscuridad: 12 h de luz, 12 h sin luz
Ingredientes del pienso: pienso conforme al estándar de identificación de alimentos para animales de laboratorio. El contenido máximo de contaminantes está dentro del rango controlable y el fabricante es responsable de la inspección rutinaria. Como agua de bebida se utilizó agua potable esterilizada en autoclave.
Agrupación de los animales: los animales se pesaron y se midió el volumen tumoral antes de la administración. Los animales se agruparon aleatoriamente según el volumen tumoral (diseño de bloques al azar).
Observación: el protocolo experimental y cualquier modificación del mismo se realizaron con la aprobación del Comité Institucional para el Cuidado y el Uso de Animales (IACUC) de WuXi AppTec, Shanghái, China. El uso y bienestar de los animales de experimentación siguió las normas de la Asociación para la Evaluación y Acreditación del Cuidado de Animales de Laboratorio (AAALAC). La salud y la muerte de los animales se monitorearon diariamente y los exámenes rutinarios incluyeron la observación del crecimiento tumoral y la influencia del tratamiento con los fármacos en el comportamiento y el rendimiento diarios de los animales, tal como el comportamiento, la ingesta de alimento y agua, el cambio de peso corporal (con mediciones del peso corporal dos veces por semana), el aspecto u otra situación anormal. Las muertes de animales y los efectos secundarios se registraron en función del número de animales en cada grupo, y los registros correlacionados pueden verse en el apéndice.
Índices experimentales: los índices experimentales se usaron para examinar si el crecimiento tumoral se inhibe, retrasa o cura. El diámetro tumoral se midió dos veces por semana con un pie de rey. El volumen tumoral se calculó mediante la fórmula: V = 0,5a x b2, en donde a y b representan el diámetro largo y el diámetro corto de un tumor, respectivamente. La inhibición del crecimiento tumoral (ICT) de un compuesto se evaluó con el T-C (día) y el T/C (%). El T-C (día) refleja el índice de retraso del crecimiento tumoral, en donde T representa el número medio de días necesarios para alcanzar un volumen tumoral predeterminado (p. ej., 1.000 mm3) en un grupo de administración, y C representa el número medio de días necesarios para alcanzar el mismo volumen tumoral en un grupo de control. El valor porcentual de T/C (%) refleja la tasa de inhibición del crecimiento tumoral, en donde T y C representan el peso tumoral (volumen tumoral) en el grupo de administración y un grupo de control en un día determinado, respectivamente.
La tasa de inhibición del crecimiento tumoral se calculó mediante la siguiente fórmula: ICT (%) = [1-(Ti-T0)/(Vi-Vü)] x 100, en donde Ti es el volumen tumoral medio del grupo de administración en un día determinado y T0 es el volumen tumoral medio del grupo de administración al comienzo de la administración; Vi es el volumen tumoral medio de un
grupo de control con vehículo en un día determinado (el mismo día que Ti), y V0 es el volumen tumoral medio del grupo de control con vehículo al comienzo de la administración. Al final del experimento, se midió el peso tumoral y se calculó el porcentaje T/C, en donde T y C representan el peso tumoral del grupo de administración y el grupo de control con vehículo, respectivamente.
Terminación del experimento: si el estado de salud de un animal continúa deteriorándose o un animal tiene más de 2.000 mm3 de volumen tumoral o una enfermedad o dolor grave, debe ser sacrificado. Si aparecen las siguientes dolencias, se notificará a un veterinario y los animales serán sacrificados:
obvia delgadez, pérdida de peso superior al 20 %;
imposibilidad de acceder libremente al alimento y el agua;
el volumen tumoral medio de un grupo de control alcanza los 2.000 mm3, en cuyo caso el experimento debe terminarse.
Un animal tiene las siguientes manifestaciones clínicas y continúa deteriorándose:
piloerección
espalda arqueada
oído, nariz, ojo o pie pálidos
respiración apresurada
convulsiones
diarrea persistente
deshidratación
movimiento lento
sonidos
Análisis de los datos: se usó ANOVA unidireccional para la comparación de tres o más grupos. Si hay una diferencia significativa en los valores de F, deben realizarse comparaciones múltiples después del análisis ANOVA. Todos los datos se analizaron con SPSS 17.0. p < 0,05 indicó una diferencia significativa.
Estudios farmacodinámicos in vivo de los fármacos de prueba en un modelo tumoral de xenoinjerto subcutáneo de células de cáncer de colon humano CO-04-0032:
Diseño experimental:
Establecimiento del modelo de trasplante tumoral humano: el modelo de cáncer de colon humano CO-04-0032 se derivó originalmente de muestras de tumores extraídas quirúrgicamente. La adquisición y el uso de las muestras cumplieron estrictamente con las leyes y reglamentos éticos nacionales, así como de los hospitales y empresas, incluido el consentimiento informado del paciente. El proceso de establecimiento del modelo siguió estrictamente el SOP interno de la empresa. Las reglas para nombrar la generación de las transferencias fueron las siguientes: el tumor después de la inoculación de la muestra tumoral en un ratón desnudo se denominó generación P0, la transferencia continuada se denominó generación P1 y así sucesivamente; y la muestra revivida se denominó FP. Los tejidos tumorales usados en este experimento fueron de la generación FP4.
Animales: ratones desnudos BALB/c, hembra, 6-8 semanas de edad, 18-20 g de peso corporal, proporcionados por Shanghai Sippr-BK Laboratory Animal Co., Ltd.
Inoculación tumoral: un volumen de aproximadamente 30 mm3 de masa tumoral CO-04-0032 se inoculó por vía subcutánea en el dorso derecho de cada ratón; los ratones se dividieron en grupos y los fármacos se administraron a cada grupo cuando el volumen tumoral medio había alcanzado aproximadamente de 100 a 200 mm3.
Resultados experimentales: prácticamente no hay aumento del volumen tumoral desde el día 15 hasta el día 30 desde la administración del compuesto 1 (que comprende la forma cristalina IX del mismo) de la presente invención; y, en comparación con el fármaco de control positivo BKM120, el compuesto 1 (que comprende la forma cristalina IX del mismo) de la presente invención tiene una actividad antitumoral más excelente frente al cáncer de colon.
Estudios farmacodinámicos in vivo de los fármacos de prueba en un modelo tumoral de xenoinjerto subcutáneo de cáncer gástrico humano ST-02-0013
Diseño experimental:
Establecimiento del modelo de trasplante tumoral humano: el modelo PDX para ST-02-0013 se derivó originalmente de una muestra clínica extraída quirúrgicamente, que se implantó en ratones desnudos y se denominó generación P0. La siguiente generación de la implantación con tumores P0 se denominó generación P1, y así sucesivamente para los
tumores por implantación continua de generación en generación de ratones. Los tumores FP2 se revivieron para obtener tumores FP3. Los tumores FP3 se transfirieron para obtener tumores FP4, que se usaron para este estudio.
Animales: ratones desnudos BALB/c, hembra, 6-8 semanas de edad, 18-22 g de peso corporal, proporcionados por Shanghai Ling Chang Biotechnology Co., Ltd.
Inoculación tumoral: un volumen de aproximadamente 30 mm3 de tejido tumoral ST-02-0013 FP4 se inoculó por vía subcutánea en el dorso derecho de cada ratón; los ratones se dividieron en grupos y los fármacos se administraron a cada grupo, cuando el volumen tumoral medio había alcanzado aproximadamente de 150 a 200 mm3.
Resultados experimentales: prácticamente no hay aumento del volumen tumoral desde el día 15 hasta el día 30 desde la administración del compuesto 1 (que comprende la forma cristalina IX del mismo) de la presente invención; y, en comparación con el fármaco de control positivo BKM120, el compuesto 1 (que comprende la forma cristalina IX del mismo) de la presente invención tiene una actividad antitumoral más excelente frente al cáncer gástrico.
Claims (15)
1. Forma cristalina IX del compuesto 1 según representa la siguiente fórmula:

Compuesto 1
caracterizada por que la forma cristalina IX tiene picos de difracción en 20 = 7,947°, 10,073°, 14,531°, 19,187°, 21,237°, 24,055°, 25,497°, en un patrón de difracción de rayos X.
2. La forma cristalina IX de la reivindicación 1, caracterizada por que la forma cristalina IX tiene picos de difracción en 20 = 7,947°, 10,073°, 11,970°, 13,468°, 14,531°, 15,911°, 19,187°, 21,237°, 24,055°, 25,497°, en un patrón de difracción de rayos X.
3. La forma cristalina IX de la reivindicación 1, caracterizada por que la forma cristalina IX tiene picos de difracción en 20 = 7,947°, 10,073°, 11,970°, 13,468°, 14,531°, 15,911°, 19,187°, 19,561°, 21,237°, 23,446°, 24,055°, 25,497°, 27,074°, en un patrón de difracción de rayos X.
4. La forma cristalina IX de la reivindicación 1, caracterizada por que los datos del análisis del patrón de XRPD de la misma son los siguientes:

5. Sales del compuesto 1 según representan las siguientes fórmulas:

6. Las sales del compuesto 1 de la reivindicación 5, caracterizadas por que la forma cristalina I del compuesto 2 tiene los siguientes datos de análisis del patrón de XRPD:

7. Las sales del compuesto 1 de la reivindicación 5, caracterizadas por que la forma cristalina II del compuesto 2 tiene los siguientes datos de análisis del patrón de XRPD:

8. Las sales del compuesto 1 de la reivindicación 5, caracterizadas por que la forma cristalina III del compuesto 2 tiene los siguientes datos de análisis del patrón de XRPD:


9. Las sales del compuesto 1 de la reivindicación 5, caracterizadas por que la forma cristalina IV del compuesto 2 tiene los siguientes datos de análisis del patrón de XRPD:

o caracterizadas por que la forma cristalina V del compuesto 2 tiene los siguientes datos de análisis del patrón de XRPD:


o caracterizadas por que la forma cristalina VI del compuesto 2 tiene los siguientes datos de análisis del patrón de XRPD:

10. Las sales del compuesto 1 de la reivindicación 5, caracterizadas por que la forma cristalina VII del compuesto 3 tiene los siguientes datos de análisis del patrón de XRPD:

11. Las sales del compuesto 1 de la reivindicación 5, caracterizadas por que la forma cristalina VIII del compuesto 4 tiene los siguientes datos de análisis del patrón de XRPD:

12. Una composición farmacéutica que comprende la forma cristalina IX del compuesto 1 de una cualquiera de las reivindicaciones 1-4, o las sales del compuesto 1 de cualquiera de las reivindicaciones 5-11, en donde la composición farmacéutica comprende una cantidad terapéuticamente eficaz de la forma cristalina IX del compuesto 1 o las sales del compuesto 1 y, opcionalmente, vehículos, excipientes y/o medios farmacéuticamente aceptables.
13. La forma cristalina IX del compuesto 1 de cualquiera de las reivindicaciones 1-4, o las sales del compuesto 1 de cualquiera de las reivindicaciones 5-11, para usar en el tratamiento de enfermedades relacionadas con la cinasa PI3K, en donde la enfermedad relacionada con la cinasa PI3K se selecciona preferiblemente entre cánceres, más preferiblemente cáncer de colon y cáncer gástrico.
14. Un método para preparar el compuesto 1 según representa la siguiente fórmula,

que comprende los siguientes pasos,

en donde
X se selecciona entre Cl o Br;
el álcali C se selecciona entre piridina, 2,6-lutidina, Et3N, 4-DMAP, LiOH, Cs2CO3, o K2CO3 ;
el disolvente c se selecciona entre piridina, diclorometano, tolueno, acetonitrilo, acetona, DMF o THF;
la relación molar entre el compuesto 7 y el compuesto 8 es de 1:1-3, preferiblemente 1:1,2~1,6;
la relación molar entre el compuesto 7 y el álcali C es de 1:1 ~3;
en donde un método para preparar el compuesto 7 comprende los siguientes pasos,

en donde
el álcali A se selecciona entre carbonato de potasio, carbonato de sodio, carbonato de cesio, hidróxido de potasio o hidróxido de sodio;
el disolvente a se selecciona entre DMF, DMSO o NMP;
el álcali B se selecciona entre carbonato de potasio, carbonato de sodio, hidróxido de bario, fosfato de potasio, carbonato de cesio, fluoruro de potasio, fluoruro de cesio, hidróxido de sodio, f-butóxido de potasio, f-butóxido de sodio, acetato de potasio o acetato de sodio;
el disolvente b se selecciona entre 1,4-dioxano, DMSO, THF, 1,4-dioxano/agua o THF/agua;
la relación en volumen entre el 1,4-dioxano o el THF y el agua en dicho disolvente b es de 3~6:1, preferiblemente de 5:1; el catalizador se selecciona entre Pd(dppf)Cl2 , o Pd(PPh3)4;
y el 2-cloruro de dimetilaminoetilo o el bromuro de 2-dimetilaminoetilo puede estar en forma de su sal.
15. Un compuesto representado por la siguiente fórmula, como intermedio para la preparación del compuesto 1,
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201510951482 | 2015-12-16 | ||
| PCT/CN2016/110284 WO2017101847A1 (zh) | 2015-12-16 | 2016-12-16 | 吡啶并[1,2-a]嘧啶酮类似物、其晶型、其中间体及其制备方法 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2908498T3 true ES2908498T3 (es) | 2022-04-29 |
Family
ID=59055711
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES16874904T Active ES2908498T3 (es) | 2015-12-16 | 2016-12-16 | Análogo de pirido[1,2-a]pirimidona, forma cristalina del mismo, intermedio del mismo y método de preparación del mismo |
Country Status (13)
| Country | Link |
|---|---|
| US (1) | US10479789B2 (es) |
| EP (1) | EP3395817B1 (es) |
| JP (1) | JP7037483B2 (es) |
| KR (1) | KR102731762B1 (es) |
| CN (1) | CN108602815B (es) |
| AU (1) | AU2016369835B2 (es) |
| CA (1) | CA3008689A1 (es) |
| DK (1) | DK3395817T3 (es) |
| ES (1) | ES2908498T3 (es) |
| PL (1) | PL3395817T3 (es) |
| RU (1) | RU2753696C2 (es) |
| WO (1) | WO2017101847A1 (es) |
| ZA (1) | ZA201804007B (es) |
Families Citing this family (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2020228729A1 (zh) * | 2019-05-13 | 2020-11-19 | 罗欣药业(上海)有限公司 | 喹唑啉酮类化合物的晶型及其制备方法 |
| CN115003674A (zh) * | 2020-02-10 | 2022-09-02 | 正大天晴药业集团股份有限公司 | 吡啶并[1,2-a]嘧啶酮化合物的治疗淋巴瘤的用途 |
| WO2021180111A1 (zh) * | 2020-03-10 | 2021-09-16 | 正大天晴药业集团股份有限公司 | 包括吡啶并[1,2-a]嘧啶酮化合物的药物组合 |
| WO2022057812A1 (zh) * | 2020-09-15 | 2022-03-24 | 正大天晴药业集团股份有限公司 | 吡啶并[1,2-a]嘧啶酮化合物的治疗外周T细胞淋巴瘤的用途 |
| WO2022121929A1 (zh) * | 2020-12-08 | 2022-06-16 | 正大天晴药业集团股份有限公司 | 吡啶并[1,2-a]嘧啶酮化合物的治疗妇科肿瘤的用途 |
| CN115671289B (zh) * | 2021-07-27 | 2024-04-26 | 广州嘉越医药科技有限公司 | 药物组合及其应用 |
| WO2023169488A1 (zh) * | 2022-03-09 | 2023-09-14 | 正大天晴药业集团股份有限公司 | 包括吡啶并[1,2-a]嘧啶酮化合物和EGFR抑制剂的药物组合 |
Family Cites Families (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| AP1699A (en) * | 2001-03-21 | 2006-12-26 | Warner Lambert Co | New spirotricyclic derivatives and their use as phosphodiesterase-7 inhibitors |
| EP2211615A4 (en) * | 2007-10-22 | 2010-10-13 | Glaxosmithkline Llc | PYRIDOSULFONAMIDE DERIVATIVES AS PI3 KINASE INHIBITORS |
| CN102206222B (zh) * | 2011-04-14 | 2013-04-10 | 中山大学 | 一种双脂肪氨基取代吲哚喹啉衍生物及其制备方法和制备抗肿瘤药物中的应用 |
| CN103539777B (zh) * | 2012-07-13 | 2016-03-02 | 广东东阳光药业有限公司 | Pi3激酶调节剂及其使用方法和用途 |
| WO2014022128A1 (en) | 2012-07-29 | 2014-02-06 | Calitor Sciences, Llc | Pi3 kinase modulators and methods of use |
| CN103788071A (zh) * | 2012-11-01 | 2014-05-14 | 中国人民解放军第二军医大学 | N-(5-(喹啉-6-基)吡啶-3-基)苯磺酰胺衍生物、制备方法及治疗用途 |
| WO2015192761A1 (zh) * | 2014-06-17 | 2015-12-23 | 辰欣药业股份有限公司 | 作为mTOR/PI3K抑制剂的吡啶并[1,2-a]嘧啶酮类似物 |
| CN105461711B (zh) | 2014-06-17 | 2018-11-06 | 正大天晴药业集团股份有限公司 | 作为PI3K抑制剂的吡啶并[1,2-a]嘧啶酮类似物 |
| WO2015192760A1 (zh) * | 2014-06-17 | 2015-12-23 | 南京明德新药研发股份有限公司 | 作为pi3k抑制剂的吡啶并[1,2-a]嘧啶酮类似物 |
-
2016
- 2016-12-16 PL PL16874904T patent/PL3395817T3/pl unknown
- 2016-12-16 KR KR1020187019466A patent/KR102731762B1/ko active Active
- 2016-12-16 ES ES16874904T patent/ES2908498T3/es active Active
- 2016-12-16 EP EP16874904.2A patent/EP3395817B1/en active Active
- 2016-12-16 WO PCT/CN2016/110284 patent/WO2017101847A1/zh not_active Ceased
- 2016-12-16 CN CN201680072381.1A patent/CN108602815B/zh active Active
- 2016-12-16 CA CA3008689A patent/CA3008689A1/en active Pending
- 2016-12-16 AU AU2016369835A patent/AU2016369835B2/en active Active
- 2016-12-16 US US16/061,279 patent/US10479789B2/en active Active
- 2016-12-16 RU RU2018125475A patent/RU2753696C2/ru active
- 2016-12-16 DK DK16874904.2T patent/DK3395817T3/da active
- 2016-12-16 JP JP2018531358A patent/JP7037483B2/ja active Active
-
2018
- 2018-06-15 ZA ZA2018/04007A patent/ZA201804007B/en unknown
Also Published As
| Publication number | Publication date |
|---|---|
| RU2018125475A3 (es) | 2020-02-28 |
| WO2017101847A1 (zh) | 2017-06-22 |
| DK3395817T3 (da) | 2022-03-21 |
| KR20180095565A (ko) | 2018-08-27 |
| CN108602815A (zh) | 2018-09-28 |
| US20180319799A1 (en) | 2018-11-08 |
| EP3395817B1 (en) | 2022-02-23 |
| CA3008689A1 (en) | 2017-06-22 |
| EP3395817A1 (en) | 2018-10-31 |
| EP3395817A4 (en) | 2019-08-07 |
| HK1256044A1 (zh) | 2019-09-13 |
| AU2016369835B2 (en) | 2020-10-15 |
| JP7037483B2 (ja) | 2022-03-16 |
| RU2018125475A (ru) | 2020-01-16 |
| AU2016369835A1 (en) | 2018-07-19 |
| CN108602815B (zh) | 2020-07-28 |
| RU2753696C2 (ru) | 2021-08-19 |
| JP2018537497A (ja) | 2018-12-20 |
| KR102731762B1 (ko) | 2024-11-18 |
| US10479789B2 (en) | 2019-11-19 |
| ZA201804007B (en) | 2019-05-29 |
| PL3395817T3 (pl) | 2022-05-02 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2908498T3 (es) | Análogo de pirido[1,2-a]pirimidona, forma cristalina del mismo, intermedio del mismo y método de preparación del mismo | |
| DK2663564T3 (en) | IMIDAZO [4,5-C] QUINOLIN-2-ON COMPOUND AND ITS USE AS PI3-KINASE / MTOR DUAL INHIBITOR | |
| CN109689641B (zh) | 一种取代的2-氢-吡唑衍生物的晶型、盐型及其制备方法 | |
| US20150166539A1 (en) | Three-ring pi3k and/or mtor inhibitor | |
| ES2930081T3 (es) | Pirazolopirimidinas sustituidas útiles como inhibidores de quinasas | |
| CN106831812B (zh) | 含联芳基酰胺结构的杂环并嘧啶或吡嗪类化合物及其应用 | |
| AU2019348120A1 (en) | FGFR4 inhibitor and use thereof | |
| ES2958528T3 (es) | Macrociclos sustituidos útiles como inhibidores de quinasas | |
| US20250115609A1 (en) | Aromatic heterocyclic compounds, preparation method therefor and uses thereof | |
| MX2015000309A (es) | Terapia combinada para tratamiento de cancer e inmunosupresion. | |
| CN102731525A (zh) | 苯并吗啉衍生物 | |
| CN105121442B (zh) | PI3K和/或mTOR抑制剂的前药 | |
| HK1256044B (en) | Pyrido[1,2-a]pyrimidone analog, crystal form thereof, intermediate thereof and preparation method therefor | |
| NZ611541B2 (en) | Imidazo[4,5-c]quinolin-2-one compound and its use as pi3 kinase / mtor dual inhibitor | |
| HK1188454B (en) | Imidazo [4, 5 -c]quinolin- 2 -one compound and its use as pi3 kinase / mtor dual inhibitor | |
| HK1213242B (en) | Pi3k and/or mtor inhibitor prodrug |