ES2909301T3 - Nuevo inhibidor de la quinasa CDK9 dependiente de ciclina - Google Patents
Nuevo inhibidor de la quinasa CDK9 dependiente de ciclina Download PDFInfo
- Publication number
- ES2909301T3 ES2909301T3 ES18787869T ES18787869T ES2909301T3 ES 2909301 T3 ES2909301 T3 ES 2909301T3 ES 18787869 T ES18787869 T ES 18787869T ES 18787869 T ES18787869 T ES 18787869T ES 2909301 T3 ES2909301 T3 ES 2909301T3
- Authority
- ES
- Spain
- Prior art keywords
- amino
- thiazol
- methyl
- pyran
- chloro
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing three or more hetero rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/425—Thiazoles
- A61K31/427—Thiazoles not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4427—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems
- A61K31/4439—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems containing a five-membered ring with nitrogen as a ring hetero atom, e.g. omeprazole
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings directly linked by a ring-member-to-ring-member bond
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Medicinal Chemistry (AREA)
- Public Health (AREA)
- Epidemiology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Hematology (AREA)
- Oncology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
Abstract
Compuesto de fórmula (I): **(Ver fórmula)** o una sal o solvato farmacéuticamente aceptable del mismo, en el que Y se selecciona del grupo que consiste en p-fluorobenzoílo, trans-4-aminociclohexilo en el que N se sustituye opcionalmente con R3 y trans-4-aminociclohexilmetilo en el que N se sustituye opcionalmente con R3, Z se selecciona del grupo que consiste en NH, S y O, R1 se selecciona del grupo que consiste en hidrógeno y halógeno, R2 se selecciona del grupo que consiste en hidrógeno, alquilo C1-C3, cicloalquilo C3-C6, heterocicloalquilo C3-C6 sustituido opcionalmente con R4, y fenilo sustituido opcionalmente con R4, R3 se selecciona de entre el grupo que consiste en alcanoilo C2-C6 y alcoxi C1-C3-alquilo C1-C3, R4 se selecciona del grupo que consiste en ciano y halógeno.
Description
DESCRIPCIÓN
Nuevo inhibidor de la quinasa CDK9 dependiente de ciclina
Campo técnico
La presente solicitud se refiere a compuestos que actúan como inhibidores de la quinasa CDK9 dependiente de ciclina, a composiciones farmacéuticas que comprenden dichos compuestos y a dichos compuestos o composiciones para la utilización en métodos para inhibir la actividad de serina quinasa.
Antecedentes
La proliferación y división de las células eucarióticas es un proceso de regulación preciso y complejo. El proceso de proliferación se lleva a cabo mediante el ciclo celular, y el avance ordenado del ciclo celular ocurre a través de sus estrictos mecanismos moleculares de regulación. Se ha encontrado que existen tres clases principales de moléculas que participan en la regulación del ciclo celular: las quinasas dependientes de ciclina (CDK), las ciclinas y los inhibidores de las quinasas dependientes de ciclina (CKI); entre ellos, CDK está en el centro. Se han encontrado 13 miembros (CDK1 a CDK13) de la familia CDK, que se clasifican en dos categorías según sus funciones intracelulares: CDK que controla el ciclo celular y CDK que controla la transcripción celular. CDK9 pertenece a las serina-quinasas y su complejo formado con la ciclina correspondiente se denomina factor b positivo de elongación de la transcripción (P-TEFb, por sus siglas en inglés). El complejo puede fosforilar la ARN polimerasa II y algunos factores de elongación transcripcional negativos (NELF y N-TEF), permitiendo extender la transcripción desde el sitio de inicio y su molécula nuclear para la elongación transcripcional (Sims RJ 3rd et al., Genes Dev., 2004, 18: 2437-68; Yamaguchi Y et al., Mol. Cell Biol., 2002, 22: 2918-27). Los estudios han encontrado que los niveles de expresión anormales de CDK9 o (y) la actividad de quinasa anormal causará una expresión anormal de diversas proteínas o (y) niveles de ARNm de las mismas anormales en la célula. Entre ellas, las proteínas antiapoptóticas, tales como BcI-2, las proteínas reguladoras asociadas al ciclo celular, tales como ciclina DI, proteínas relacionadas con la ruta de p53, determinadas proteínas de la ruta de NF-kB y proteínas relacionadas con el microambiente tumoral, tales como VEGF y similares, se ha confirmado que están estrechamente relacionadas con los tumores. Puede afirmarse que CDK9 es una de las moléculas más críticas en el desarrollo de los tumores (Shapiro GI, J. Clin. Oncol., 2006, 24: 1770-83).
El documento n° WO 2012/101062 divulga compuestos biheteroarilo sustituidos, tales como inhibidores de CDK9, para el tratamiento de condiciones tales como el cáncer. El documento n° WO 2009/118567 divulga pirimidinas, triazinas y la utilización de las mismas como inhibidores de quinasas. El documento n° WO 2013/156780 divulga compuestos inhibidores de proteína quinasas, especialmente quinasas dependientes de ciclina tales como CDK9.
Descripción resumida de la invención
La invención se refiere a inhibidores de las quinasas dependientes de ciclina. En particular, en la presente invención se proporciona un compuesto de fórmula (I), o una sal o solvato farmacéuticamente aceptable del mismo:

en la que Y se selecciona del grupo que consiste en p-fluorobenzoilo, trans-4-aminociclohexilo en el que N se sustituye opcionalmente con R3 y trans-4-aminociclohexilmetilo en el que N se sustituye opcionalmente con R3,
Z se selecciona del grupo que consiste en NH, S y O,
Ri se selecciona del grupo que consiste en hidrógeno y halógeno,
R2 se selecciona del grupo que consiste en hidrógeno, alquilo C1-C3, cicloalquilo C3-C6, heterocicloalquilo C3-C6 sustituido opcionalmente con R4, y fenilo sustituido opcionalmente con R4,
R3 se selecciona de entre el grupo que consiste en alcanoilo C2-C6 y alcoxi C1-C3-alquilo C1-C3,
R4 se selecciona del grupo que consiste en ciano y halógeno.
En la presente invención se proporciona además una composición farmacéutica que comprende un compuesto de fórmula (I), o una sal o solvato farmacéuticamente aceptable del mismo y un portador o excipiente farmacéuticamente aceptable y opcionalmente otros agentes terapéuticos.
La presente invención se refiere además a la utilización de un compuesto de fórmula (I), o una sal o solvato farmacéuticamente aceptable del mismo en la preparación de un fármaco para el tratamiento, prevención o mejora de
una enfermedad, trastorno o condición regulada o producida por actividad de serina quinasa o relacionada con actividad de quinasa dependiente de ciclina. Entre ellas, la enfermedad, trastorno o condición es cáncer.
Figuras
Las figuras 1a-1d muestran los efectos del compuesto 1 sobre las rutas de señalización celular en las líneas celulares MV4-11 (figura 1a), OCI-AML-3 (figura 1b), HL-60 (figura 1c) y NB4 (figura 1d).
Las figuras 2a-2d muestran los efectos del compuesto 1 sobre las proteínas relacionadas con la apoptosis en las líneas celulares MV4-11 (figura 2a), OCI-AML-3 (figura 2b), HL-60 (figura 2c) y NB4 (figura 2d).
Las figuras 3a-3c muestran los efectos del compuesto 1 sobre el ciclo celular en las líneas celulares MV4-11 (figura 3a), HL-60 (figura 3b) y NB4 (figura 3c).
Las figuras 4a-4c muestran los resultados de un experimento en el que el compuesto 1 inhibe el crecimiento tumoral en un modelo de ratón de tumor, en el que la figura 4a muestra un cambio en el peso corporal relativo de los ratones en los que se han inyectado por vía subcutánea células de leucemia (calculadas basándose en el peso corporal el primer día de administración) durante el tiempo; la figura 4b muestra el cambio en la carga de tamaño tumoral en los ratones durante el tiempo; la figura 4c muestra la tasa finalmente calculada de inhibición del crecimiento tumoral (ICT) para cada grupo y los valores de cada punto de datos mostrados en las figuras reflejan la media de cada grupo experimental.
Descripción detallada de la invención
Términos
A menos que se indique lo contrario, todos los términos técnicos y científicos utilizados en la presente memoria presentan los mismos significados entendidos comúnmente por el experto ordinario en la materia a la que se refiere el objeto reivindicado.
A menos que se indique lo contrario, en la presente invención se utilizan métodos convencionales, tales como espectrometría de masas, RMN, HPLC, química de proteínas, bioquímica, técnicas de ADN recombinante y farmacología comprendidos dentro de los conocimientos del experto en la materia. A menos que se proporcione una definición específica, la nomenclatura y las operaciones y técnicas de laboratorio químicamente relacionadas con química analítica, química orgánica sintética y química médica y farmacéutica indicadas en la presente memoria son conocidas por el experto en la materia. En general, las técnicas y procedimientos anteriormente indicados pueden llevarse a cabo mediante métodos convencionales bien conocidos de la técnica y descritos en diversos documentos generales y más específicos, que se citan y se comentan en la presente especificación.
El término "alquilo" se refiere a un grupo hidrocarburo alifático que puede ser un alquilo ramificado o lineal. Según la estructura, un grupo alquilo puede ser un grupo monovalente o un grupo divalente (es decir, un grupo alquileno). En la presente invención, el grupo alquilo es preferentemente un "grupo alquilo inferior" con 1 a 6 átomos de carbono, y todavía más preferentemente, un "grupo de alquilo inferior" que presenta 1 a 3 átomos de carbono. Entre los grupos alquilo típicos se incluyen, aunque sin limitarse a ellos, metilo, etilo, propilo, isopropilo, butilo, isobutilo, terc-butilo, pentilo, hexilo y similares.
El término "alcoxi" se refiere a un grupo -O-alquilo en el que el alquilo es tal como se define en la presente memoria. Entre los grupos alcoxi típicos se incluyen, aunque sin limitarse a ellos, metoxi, etoxi, propoxi, butoxi, pentiloxi, hexiloxi y similares.
El término "arilo" se refiere a que el anillo plano presenta un sistema de electrones n deslocalizados y contiene 4n+2 n electrones, en donde 'n' es un número entero. El anillo arilo puede estar compuesto de cinco, seis, siete, ocho, nueve o más de nueve átomos. El grupo arilo puede estar opcionalmente sustituido. El término "arilo" incluye grupos arilo carbocíclicos (tales como fenilo) y grupos arilo heterocíclicos (o "heteroarilo" o "heteroaromáticos") (tales como piridina). El término incluye grupos monocíclicos o policíclicos fusionados (es decir, anillos que comparten pares contiguos de átomos de carbono).
El término "arilo" tal como se utiliza en la presente memoria se refiere a que cada uno de los átomos que constituye el anillo en el anillo arilo es un átomo de carbono. El anillo arilo puede estar compuesto de cinco, seis, siete, ocho, nueve o más de nueve átomos. El grupo arilo puede estar opcionalmente sustituido. Entre los grupos arilo se incluyen, aunque sin limitarse a ellos, fenilo, naftilo, fenantrilo, antrilo, fluorenilo y fluorenilo. Según la estructura, un grupo alquilo puede ser un grupo monovalente o un grupo divalente (es decir, un grupo arileno).
El término "alquilo (arilo)" se refiere a un grupo alquilo, tal como se define en la presente memoria, sustituido con un grupo arilo, tal como se define en la presente memoria. Entre los grupos alquilo (arilo) no limitantes se incluyen bencilo, fenetilo y similares.
El término "cicloalquilo" se refiere a un grupo monocíclico o policíclico que contiene únicamente carbono e hidrógeno. El grupo cicloalquilo incluye un grupo con 3 a 10 átomos anulares. Según la estructura, un grupo cicloalquilo puede
ser un grupo monovalente o un grupo divalente (es decir, un grupo cicloalquileno). En la presente invención, el grupo cicloalquilo es preferentemente un grupo cicloalquilo con 1 a 8 átomos de carbono, y todavía más preferentemente, un "grupo de cicloalquilo inferior" con 3 a 6 átomos de carbono.
El término "alquilo (cicloalquilo)" se refiere a un grupo alquilo, tal como se define en la presente memoria, sustituido con un grupo cicloalquilo, tal como se define en la presente memoria. Entre los grupos de alquilo (cicloalquilo) no limitantes se incluyen ciclopropilmetilo, ciclobutilmetilo, ciclopentilmetilo, ciclohexilmetilo y similares.
El término "halo" o "halógeno" se refiere a flúor, cloro, bromo y yodo.
Los términos "haloalquilo" y "haloalcoxi" incluyen estructuras de alquilo o alcoxi, y entre ellas, por lo menos un hidrógeno ha sido sustituido con un átomo de halógeno. En determinadas realizaciones, en el caso de que dos o más átomos de hidrógeno se sustituyan por átomos de halógeno, los átomos de halógeno son iguales o diferentes entre sí.
El término "ciano" tal como se utiliza en la presente memoria se refiere a un radical de fórmula -CN.
El término "carbonilo" es un grupo funcional orgánico (C=O) formado mediante la unión de dos átomos de carbono y oxígeno mediante un doble enlace.
El término "alcanoilo" o "alquilcarbonilo" se refiere a un grupo carbonilo sustituido adicionalmente con un grupo alquilo. Entre los grupos alcanoilo típicos se incluyen, aunque sin limitarse a ellos, acetilo, propionilo, butirilo, pentanoilo, hexanoilo y similares.
El término "amino" se refiere al grupo -NH2. El término "alquilamino" se refiere a un sustituyente amino sustituido adicionalmente con uno o dos grupos alquilo, en particular un grupo -NRR', en el que R y R' se seleccionan, cada uno independientemente, de hidrógeno o alquilo inferior, con la condición de que -NRR' no sea -NH2. El término "aminolaquilo" se refiere a un sustituyente alquilo sustituido adicionalmente con uno o más grupos amino. El término "cianoalquilo" se refiere a un sustituyente alquilo sustituido adicionalmente con uno o más grupos ciano. El término "heteroarilo" tal como se utiliza en la presente memoria se refiere a que uno o más átomos de las cadenas de esqueleto de los grupos alquilo definidos en la presente memoria son heteroátomos, tales como oxígeno, nitrógeno, azufre, silicio, fósforo o combinaciones de los mismos. El heteroátomo o heteroátomos pueden situarse en cualquier sitio dentro del grupo heteroalquilo o en una posición en que el grupo heteroarilo se encuentre unido al resto de la molécula.
El término "heteroarilo" se refiere a un grupo arilo que comprende uno o más heteroátomos anulares seleccionados del grupo que consiste en nitrógeno, oxígeno y azufre. La fracción "heteroarilo" que contiene N se refiere a que por lo menos uno de los átomos de esqueleto en el anillo del grupo arilo es nitrógeno. Según la estructura, un grupo heteroarilo puede ser un grupo monovalente o un grupo divalente (es decir, un grupo heteroarileno). Entre los ejemplos de grupos heteroarilo se incluyen, aunque sin limitarse a ellos, piridilo, imidazolilo, pirimidinilo, pirazolilo, triazolilo, pirazinilo, tetrazolilo, furilo, tienilo, isoxazolilo, tiazolilo, oxazol, isotiazolilo, pirrolilo, quinolilo, isoquinolilo, indolilo, bencimidazolilo, benzofuranilo, indazolilo, indolizinilo, ftalazinilo, piridazinilo, isoindolilo, pteridinilo, purinilo, oxadiazolilo, tiadiazolilo, furilo, benzofurilo, benzotienilo, benzotiazolilo, benzoxazolilo, quinazolinilo, naftiridilo y furopiridinilo, y similares.
El término "heterocicloalquilo" tal como se utiliza en la presente memoria se refiere a que uno o más de los átomos que constituyen el anillo en el anillo no arilo es un heteroátomo seleccionado del grupo que consiste en nitrógeno, oxígeno y azufre. El anillo heterocicloalquilo puede estar compuesto de tres, cuatro, cinco, seis, siete, ocho, nueve o más de nueve átomos. El grupo heterocicloalquilo puede estar opcionalmente sustituido. Entre los ejemplos de grupos heterocicloalquilo se incluyen, aunque sin limitarse a ellos, lactamo, lactona, imina cíclica, tioimina cíclica, carbamato cíclico, tetrahidrotiopirano, 4H-pirano, tetrahidropirano, piperidina, 1,3-dioxina, 1,3-dioxano, 1,4-dioxina, 1,4-dioxano, piperazina, 1,3-oxatiano, 1,4-oxetano, 1,4-oxatiano, tetrahidro-1,4-tiazina, 2H-1,2-oxazina, maleimida, succinimida, ácido barbitúrico, ácido tiobarbitúrico, dioxopiperazina, hidantoína, dihidrouracilo, morfolina, trioxano, hexahidro-1,3,5-triazina, tetrahidrotiofeno, tetrahidrofurano, pirrolina, pirrolidina, imidazolidina, pirrolidona, pirazolina, pirazolidina, imidazolina, imidazolidina, 1,3-dioxol, 1,3-dioxolano, 1,3-ditiolelen, 1,3-dihiolano, isoxazolina, isoxazolidina, oxazolina, oxazolidina, oxazolidinona, tiazolina, tiazolidina y 1,3-oxatiolano. Según la estructura, un grupo heterocicloalquilo puede ser un grupo monovalente o un grupo divalente (es decir, un grupo heterocicloalquileno).
El término "alquil(heteroarilo)" se refiere a un grupo alquilo, tal como se define en la presente memoria, sustituido con un grupo heteroarilo, tal como se define en la presente memoria.
El término "alquil(heterocicloalquilo)" se refiere a un grupo alquilo, tal como se define en la presente memoria, sustituido con un grupo heterocicloalquilo, tal como se define en la presente memoria.
La expresión "sustituido opcionalmente" o "sustituido" se refiere a que el grupo mencionado puede sustituirse con uno o más grupos adicionales, cada uno de los cuales se selecciona individual e independientemente de entre alquilo,
cicloalquilo, arilo, heteroarilo, grupo heterocíclico, hidroxi, alcoxi, ciano, halógeno, amida, nitro, haloalquilo, amino, metilsulfonilo y similares.
Tal como se utiliza en la presente memoria, GI5o se refiere a la concentración de un fármaco necesaria para inhibir al 50% el crecimiento celular; es decir, la concentración de un fármaco en el momento en que el crecimiento de 50% de las células (tal como células de cáncer) ha sido inhibido o controlado.
Tal como se utiliza en la presente memoria, IC50 se refiere a la cantidad, concentración o dosis de un compuesto de ensayo particular a la que se obtiene una inhibición de 50% del efecto máximo en un ensayo que mide un efecto. Inhibidor de quinasa CDK9 de la invención
La invención se refiere a inhibidores de la quinasa dependiente de ciclina CDK9. En particular, en la presente invención se proporciona un compuesto de fórmula (I), o una sal o solvato farmacéuticamente aceptable del mismo:
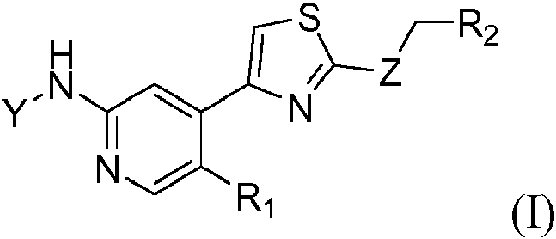
en la que Y se selecciona del grupo que consiste en p-fluorobenzoilo, trans-4-aminociclohexilo en el que N se sustituye opcionalmente con R3 y trans-4-aminociclohexilmetilo en el que N se sustituye opcionalmente con R3 ,
Z se selecciona del grupo que consiste en NH, S y O,
R1 se selecciona del grupo que consiste en hidrógeno y halógeno,
R2 se selecciona del grupo que consiste en hidrógeno, alquilo C1-C3, cicloalquilo C3-C6, heterocicloalquilo C3-C6 sustituido opcionalmente con R4, y fenilo sustituido opcionalmente con R4,
R3 se selecciona de entre el grupo que consiste en alcanoilo C2-C6 y alcoxi C1-C3-alquilo C1-C3,
R4 se selecciona del grupo que consiste en ciano y halógeno.
En determinadas realizaciones preferentes, Y se selecciona de entre las estructuras siguientes:

En una realización preferente, R1 es cloro.
En ora realización preferente, R2 se selecciona del grupo que consiste en hidrógeno, metilo, ciclopropilo, ciclohexilo, 4-tetrahidropiranilo sustituido opcionalmente con ciano y fenilo sustituido opcionalmente con flúor.
En otra realización preferente, R3 se selecciona del grupo que consiste en acetilo, 2-metoxoetilo, (R)-1-metil-2-metoxietilo, y (S)-1-metil-2-metoxietilo.
En la presente invención, entre los compuestos particularmente preferentes se incluyen:
4-(((4-(5-cloro-2-(((1R,4r)-4-(((R)-1-metoxipropil-2-il)amino)ciclohexil)amino)piridín-4-il)tiazol-2-il)amino)metil)tetrahidro-2H-pirán-4-carbonitrilo,
(1r,4r)-N1-(5-cloro-4-(2-(((tetrahidro-2H-pirán-4-il)metil)amino)tiazol-4-il)piridín-2-il)cidohexán-1,4-diamina, N-((1r,4r)-4-((5-cloro-4-(2-(((tetrahidro-2H-pirán-4-il)metil)amino)tiazol-4-il)piridín-2-il)amino)cidohexil)acetamida, (1r,4r)-N1-(5-cloro-4-(2-(((tetrahidro-2H-pirán-4-il)metil)amino)tiazol-4-il)piridín-2-il)-N4-(2-metoxietil)cidohexil-1,4-diamina,
(1S,4r)-N1-(5-cloro-4-(2-(((tetrahidro-2H-pirán-4-il)metil)amino)tiazol-4-il)piridín-2-il)-N4-((S)-1-metoxipropán-2-il)ciclohexil-1,4-diamina,
(1R,4r)-N1-(5-doro-4-(2-(((tetrahidro-2H-pirán-4-il)metil)amino)tiazol-4-il)piridín-2-il)-N4-((R)-1-metoxipropán-2-il)ciclohexán-1,4-diamina,
4-(2-((((1r,4r)-4-aminoddohexil)metil)amino)-5-doropiridm-4-il)-N-((tetrahidro-2H-piran-4-il)metil)tiazol-2-amina, N-(5-cloro-4-(2-(((tetrahidro-2H-pirán-4-il)metil)amino)tiazol-4-il)piridín-2-il)-4-fluorobenzamida,
(1r,4r)-N1-(5-cloro-4-(2-(metilamino)tiazol-4-il)piridín-2-il)-N4-(2-metoxietil)cidohexán-1,4-diamina,
(1r,4r)-N1-(5-cloro-4-(2-((ciclohexilmetil)amino)tiazol-4-il)piridín-2-il)-N4-( 2-metoxietil)ciclohexán-1,4-diamina, (1r,4r)-N1-(4-(2-(bendlamino)tiazol-4-il)-5-doropiridm-2-il)-N4-(2-metoxietil)cidohexán-1,4-diamina,
(1r,4r)-N1-(5-doro-4-(2-((4-fluorobencil)amino)tiazol-4-il)piridín-2-il)-N4-(2-metoxietil)cidohexán-1,4-diamina, (1r,4r)-N1-(5-doro-4-(2-((cidopropilmetil)amino)tiazol-4-il)piridín-2-il)-N4-(2-metoxietil)cidohexán-1,4-diamina, 4-((4-(5-doro-2-(((1R,4R)-4-((2-metoxietil)amino)cidohexil)amino)piridín-4-il)tiazol-2-ilamino)metil)tetrahidro-2H-pirán-4-carbonitrilo,
4-(((4-(5-doro-2-(((1S,4R)-4-(((S)-1-metoxipropN-2-N)amino)ddohexN)amino)pindm-4-N)tiazol-2-il)amino)metil)tetrahidro-2H-pirán-4-carbonitrilo,
(1r,4r)-N1-(5-doro-4-(2-((tetrahidro-2H-pirán-4-il)metoxi)tiazol-4-il)piridín-2-il)-N4-(2-metoxietil)cidohexán-1,4-diamina,
(1r,4r)-N1-(5-doro-4-(2-(((tetrahidro-2H-pirán-4-il)metil)mercapto)tiazol-4-il )piridín-2-il)-N4-(2-metoxietN)ddohexán-1,4-diamina,
(1r,4r)-N1-(2-metoxietil)-N4-(4-(2-(((tetrahidro-2H-pirán-4-il)metil)amino)thi azol-4-il)piridín-2-il)ciclohexán-1,4-diamina.
Las estructuras de los compuestos preferentes de la invención se muestran a continuación.

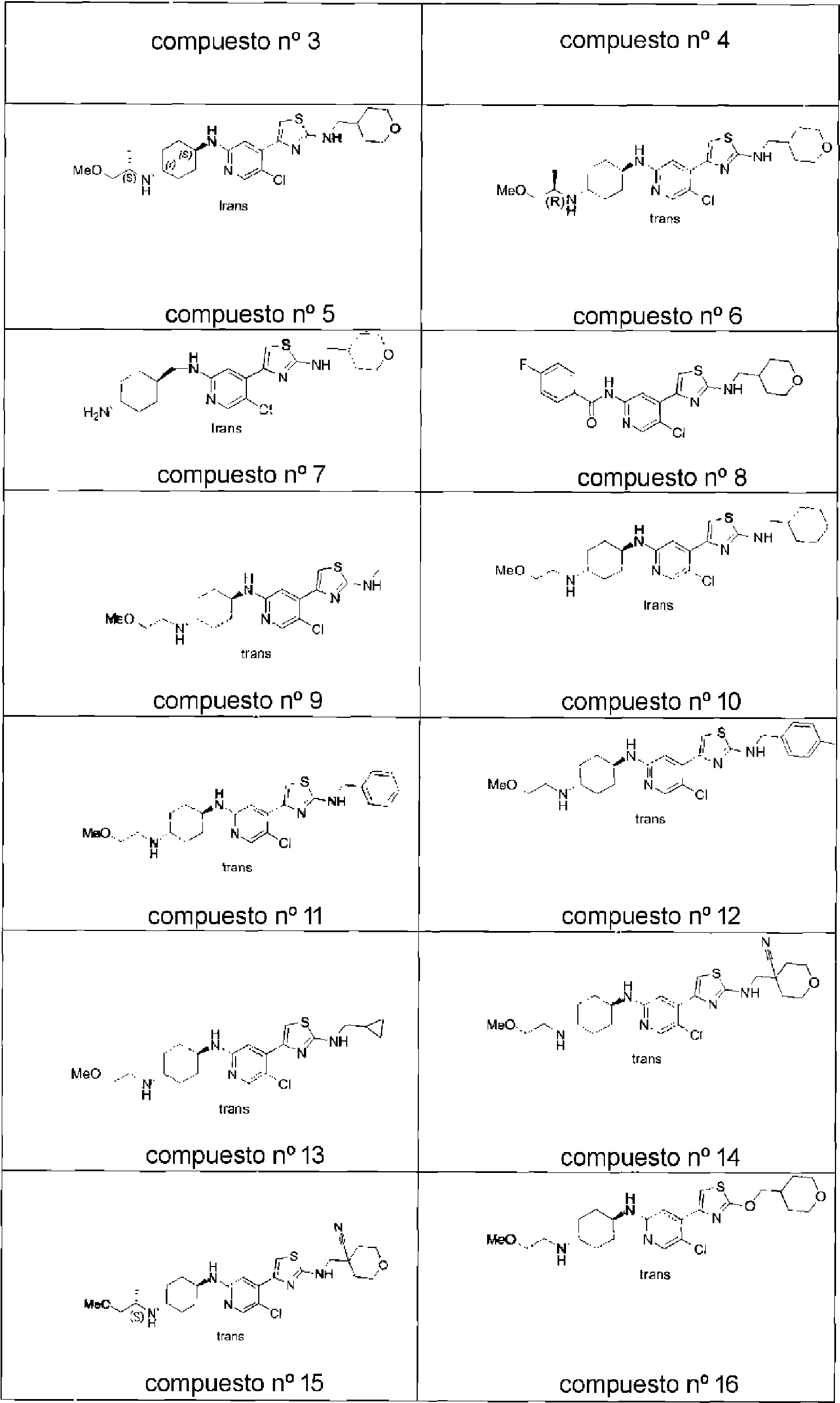

Aunque la tabla, anteriormente, muestra las estructuras de compuestos preferentes de la presente invención, debe entenderse que los dos átomos de carbono, unidos respectivamente al grupo para-amino en el grupo ciclohexilo, no son centros quirales; la representación de enlace químico de
/
o
es meramente indicativa de que el enlace de los dos enlaces químicos al grupo para-amino están orientados en trans con respecto al grupo ciclohexilo y, de esta manera, los compuestos representados mediante intercambio de estos dos enlaces químicos
/
y
también se encuentran comprendidos dentro del alcance de la presente invención.
En la presente memoria se describen nuevos inhibidores de quinasa. En la presente memoria también se describen sales, solvatos, ésteres y ácidos farmacéuticamente aceptables, metabolitos farmacéuticamente activos y profármacos de dicho compuesto; sin embargo, los ésteres, ácidos, metabolitos o profármacos no son partes de la invención.
En realizaciones adicionales de la exposición (que no son parte de la invención), los compuestos indicados en la presente memoria se administran en el sujeto que lo necesita para la metabolización en el cuerpo del mismo, produciendo metabolitos que se utilizan a continuación para producir el efecto deseado, incluyendo el efecto terapéutico deseado.
Los compuestos indicados en la presente memoria pueden convertirse y/o utilizarse en forma de sales farmacéuticamente aceptables. Entre los tipos de sales farmacéuticamente aceptables se incluyen, aunque sin limitarse a ellas, las siguientes: (1) una sal de adición de ácido formada mediante reacción de una forma de base libre del compuesto con un ácido inorgánico farmacéuticamente aceptable, tal como ácido clorhídrico, ácido bromhídrico, ácido sulfúrico, ácido nítrico, ácido fosfórico, ácido metafosfórico o similares, o con un ácido orgánico, tal como ácido acético, ácido propiónico, ácido caproico, ácido ciclopentanopropiónico, ácido glicólico, ácido pirúvico, ácido láctico, ácido malónico, ácido málico, ácido cítrico, ácido succínico, ácido maleico, ácido tartárico, ácido fumárico, ácido trifluoroacético, ácido benzoico, ácido 3-(4-hidroxibenzoil)benzoico, ácido cinámico, ácido mandélico, ácido metanosulfónico, ácido etanosulfónico, ácido 1,2-etanodisulfónico, ácido 2-hdiroxietanosulfónico, ácido bencenosulfónico, ácido toluenosulfónico, ácido 4-metilbiciclo-[2.2.2]oct-2-en-1-carboxílico, ácido 2-naftalenosulfónico, ácido terc-butilacético, ácido glucoheptónico, ácido 4,4'-metilén-bis-( 3-hidroxi-2-en-1-carboxílico, ácido 3-fenilpropiónico, ácido trimetilacético, sulfato de dodecilo, ácido glucónico, ácido glutámico, ácido salicílico, ácido hidroxinaftoico, ácido esteárico, ácido mucónico; (2) una sal de adición de base formada al sustituir un protón ácido de un compuesto parental por un ion metálico, tal como un ion de metal alcalino (p.ej., litio, sodio o potasio), un ion de metal alcalinotérreo (p.ej., magnesio o calcio), o un ion de aluminio, o coordinado con bases orgánicas. Entre las bases orgánicas aceptables se incluyen etanolamina, dietanolamina, trietanolamina, trimetilamina, N-metilglucamina y similares. Entre las bases inorgánicas aceptables se incluyen hidróxido de aluminio, hidróxido de calcio, hidróxido potásico, carbonato sódico, hidróxido sódico y similares.
Los contraiones correspondientes de sales farmacéuticamente aceptables pueden analizarse y caracterizarse utilizando una diversidad de métodos, entre ellos, aunque sin limitación, la cromatografía de intercambio iónico, la cromatografía iónica, la electroforesis capilar, el plasma acoplado por inducción, la espectroscopía de absorción atómica, la espectrometría de masas o cualquiera combinación de las mismas.
La sal se recupera utilizando por lo menos una de las técnicas siguientes: filtración, precipitación con un no solvente seguido de filtración, evaporación del solvente, o liofilización en el caso de una solución acuosa.
El cribado y caracterización de sales farmacéuticamente aceptables, polimorfos y/o solvatos pueden llevarse a cabo utilizando una diversidad de técnicas, entre ellas, aunque sin limitación, el análisis térmico, la difracción de rayos X, la
espectroscopia, la microscopía y el análisis elemental. Entre las diversas técnicas espectrales utilizadas se incluyen, aunque sin limitarse a ellas, Raman, FTIR, UVIS y RMN (de estado líquido y sólido). Entre las diversas técnicas de microscopía se incluyen, aunque sin limitarse a ellas, la microscopía de IR y la microscopía Raman.
Composición farmacéutica de la invención
En la presente invención se proporciona además una composición farmacéutica que comprende por lo menos un compuesto de fórmula (I), o una sal o solvato farmacéuticamente aceptable del mismo y un portador o excipiente farmacéuticamente aceptable y opcionalmente otros agentes terapéuticos.
Durante el tratamiento, puede utilizarse sola o en combinación con otro u otros agentes terapéuticos, según resulte apropiado. El fármaco que comprende un compuesto de la invención puede administrarse en el paciente mediante por lo menos uno de entre inyección, administración oral, inhalación, administración rectal y administración transdérmica.
En una realización de la invención, al tratar un paciente según la presente invención, la cantidad de un fármaco administrado depende de varios factores, tales como el régimen de dosis particular, el tipo de enfermedad o trastorno y su gravedad, y el sujeto en necesidad de tratamiento o la singularidad del huésped (p.ej., el peso corporal); sin embargo, dependiendo de las circunstancias particulares, entre ellas, por ejemplo, el fármaco particular que se ha utilizado, la vía de administración, la condición bajo tratamiento y el sujeto o huésped bajo tratamiento, la dosis administrada puede decidirse mediante métodos rutinariamente conocidos de la técnica. Generalmente, para la utilización en el tratamiento de un adulto, la dosis administrada típicamente estará comprendida entre 0,02 y 5000 mg/día, por ejemplo entre aproximadamente 1 y 1500 mg/día. La dosis deseada puede presentarse convenientemente como una dosis única, o concurrentemente (o en un periodo de tiempo corto) o en dosis divididas a intervalos apropiados, tales como dos, tres, cuatro o más dosis divididas al día. El experto en la materia entenderá que, aunque se proporcionan los intervalos de dosis anteriores, la cantidad eficaz específica puede ajustarse apropiadamente según la condición del paciente y en relación al diagnóstico médico.
Utilización del fármaco de la presente invención
Puede utilizarse un compuesto de fórmula (I), o una sal o solvato farmacéuticamente aceptable del mismo, o una composición farmacéutica que lo comprende, para inhibir la actividad de las quinasas dependientes de ciclina (CDK) y las ciclinas, especialmente la actividad de CDK9. El compuesto de fórmula (I) o una sal o solvato farmacéuticamente aceptable del mismo puede utilizarse para el tratamiento o prevención de una o más enfermedades seleccionadas del grupo que consiste en cáncer pulmonar no microcítico, cáncer pulmonar microcítico, adenocarcinoma pulmonar, carcinoma pulmonar de células escamosas, cáncer pancreático, cáncer prostático, cáncer de vejiga, cáncer hepático, cáncer de piel, glioma, cáncer de mama, melanoma, glioma maligno, rabdomiosarcoma, cáncer ovárico, astrocitoma, sarcoma de Ewing, retinoblastoma, carcinoma de células epiteliales, cáncer de colon, cáncer renal, tumor estromal gastrointestinal, leucemia, linfoma histiocítico y carcinoma nasofaríngeo.
Más preferentemente, el compuesto de fórmula (I) indicado en la presente memoria, o una sal o solvato farmacéuticamente aceptable del mismo, o una composición farmacéutica que lo comprende puede utilizarse como inhibidor de CDK9, que puede utilizarse para el tratamiento de cáncer pulmonar no microcítico, cáncer pulmonar microcítico, adenocarcinoma pulmonar, carcinoma pulmonar de células escamosas, cáncer pancreático, cáncer prostático, cáncer de vejiga, cáncer hepático, cáncer de piel, glioma, cáncer de mama, melanoma, glioma maligno, rabdomiosarcoma, cáncer ovárico, astrocitoma, sarcoma de Ewing, retinoblastoma, carcinoma de células epiteliales, cáncer de colon, cáncer renal, tumor estromal gastrointestinal, leucemia, linfoma histiocítico y carcinoma nasofaríngeo, mediante la utilización del mismo por sí solo o en combinación con otros agentes terapéuticos.
Preparación del compuesto
Los compuestos de fórmula (I) pueden sintetizarse utilizando técnicas sintéticas estándares conocidas por el experto en la materia o utilizando métodos conocidos de la técnica en combinación con los métodos indicados en la presente memoria. Además, los solventes, temperaturas y otras condiciones de reacción presentadas en la presente memoria pueden variar dependiendo del estado de la técnica. Como guía adicional, también pueden utilizarse los métodos sintéticos siguientes.
Las reacciones pueden utilizarse secuencialmente para proporcionar los compuestos indicados en la presente memoria, o pueden utilizarse para sintetizar fragmentos que seguidamente se añaden mediante los métodos indicados en la presente memoria y/o mediante métodos conocidos de la técnica.
En determinadas realizaciones, en la presente memoria se proporcionan métodos de preparación de un compuesto inhibidor de serina quinasa indicado en la presente memoria y dicho compuesto para la utilización en métodos de tratamiento. En determinadas realizaciones, los compuestos indicados en la presente memoria pueden sintetizarse utilizando los esquemas sintéticos siguientes. Los compuestos pueden sintetizarse mediante métodos análogos a los indicados posteriormente, utilizando los materiales de partida apropiados.
Los materiales de partida para la síntesis de los compuestos indicados en la presente memoria pueden sintetizarse o pueden obtenerse de fuentes comerciales. Los compuestos indicados en la presente memoria y otros compuestos relacionados con diferentes sustituyentes pueden sintetizarse utilizando técnicas y materiales de partida conocidos por el experto en la materia. Los métodos generales de preparación de los compuestos divulgados en la presente memoria pueden obtenerse a partir de reacciones conocidas de la técnica, y las reacciones pueden modificarse para introducir diversas fracciones en las moléculas proporcionadas en la presente memoria con reactivos y bajo condiciones consideradas apropiadas por el experto en la materia.
Si se desea, el producto de reacción puede aislarse y purificarse utilizando técnicas convencionales, entre ellas, aunque sin limitación, la filtración, la destilación, la cristalización la cromatografía y similares. Dichos productos pueden caracterizarse utilizando métodos convencionales, entre ellos constantes físicas y datos espectrales.
Se describen a continuación ejemplos no limitativos de esquemas sintéticos para la preparación de compuestos de fórmula (I).
Ejemplos
Los ejemplos no limitativos específicos siguientes sólo deben considerarse ilustrativos y en modo alguno limitativos de la divulgación. Aunque no se indican datos adicionales, se cree que el experto en la materia podrá utilizar por completo la presente divulgación basándose en la descripción en la presente memoria.
Ejemplo 1: síntesis de 4-(((4-(5-cloro-2-(((1R,4r)-4-(((R)-1-metoxi propil-2-il)amino)ciclohexil)amino)piridín-4-il)tiazol-2-il)amino)metil)tetrahidro-2H-pirán-4-carbonitrilo


Etapa 1: síntesis de 5-cloro-2-fluoro-4-(4,4,5,5-tetrametil-1,3,2-dioxaborolán-2-il)piridina
Se añadió ácido 5-cloro-2-fluoropiridina-4-borónico (0,7 g, 4,46 mmoles) y pinacol (0,63 g, 5,35 mmoles) a 50 ml de tolueno, la mezcla se calentó a 120°C y se sometió a reflujo durante la noche, y la cromatografía de capa fina (CCF) mostró que quedaba una pequeña cantidad de material. La solución de reacción se enfrió hasta la temperatura ambiente, se concentró y se secó con una bomba de aceite, proporcionando 0,92 g de 5-cloro-2-fluoro-4-(4,4,5,5
tetrametil-1,3,2-dioxaborolán-2-il)piridina en forma de un sólido blanco, rendimiento: 80% EM (IEP): m/z 258,1 (M H)+.
Etapa 2: síntesis de 4-metilbencenosulfonato de (S)-1-metoxipropán-2-ilo
Se añadió hidruro sódico (NaH) al 60% (6,52 g, 283 mmoles) a tetrahidrofurano (THF) seco (200 ml), que se enfrió a 0°C con un baño de hielo y se protegió bajo nitrógeno, y después se añadió gota a gota (S)-(+)-1-metoxi-2-propanol (21 g, 233 mmoles). Tras completar la adición gota a gota, la mezcla se sometió a agitación a temperatura ambiente durante 1,5 horas. La solución de reacción se enfrió nuevamente a 0°C y a continuación se añadió gota a gota una solución de cloruro de p-toluenosulfonilo (45,3 g, 283 mmoles) en tetrahidrofurano (THF) (200 ml). Tras la adición, la mezcla se sometió a agitación a temperatura ambiente durante la noche. La CCF mostró que el material de partida se había consumido por completo. La mezcla de reacción se diluyó con acetato de etilo (500 ml), se desactivó mediante la adición gota a gota de agua (500 ml) bajo enfriamiento con hielo y se separó. Se extrajo la capa acuosa una vez con acetato de etilo (200 ml). Se agruparon las fases orgánicas, se lavaron con agua (200 ml) y solución hipersalina saturada (200 ml), se secaron sobre sulfato sódico anhidro, se filtraron y se concentraron, proporcionando 43 g de un producto en bruto aceitoso amarillo pálido, que se aisló mediante columna (éter de petróleo/acetato de etilo=5/1), proporcionando 37 g de 4-metilbencenosulfonato de (S)-1-metoxipropán-2-ilo, rendimiento: 65,1%, EM (IEP): m/z 245,1 (M+H)+.
Etapa 3: síntesis de (1R,4R)-N1-((R)-1-metoxipropán-2-il)ciclohexán-1,4-diamina
Se añadió 4-metilbencenosulfonato de (S)-1-metoxipropán-2-ilo (5 g, 20,5 mmoles) y trans-1,4-ciclohexanediamina (5,84 g, 51,2 mmoles) a 50 ml de acetonitrilo, que se calentaron a 90°C y se hicieron reaccionar durante la noche. Tras la reacción se llevó a cabo CCF hasta completarla. La solución de reacción se enfrió y después se filtró, y el filtrado se concentró. El residuo se disolvió en diclorometano, se mezcló con gel de sílice y se aisló mediante columna (diclorometano/metanol=10/1), proporcionando 2,5 g de compuesto de (1R,4R)-N1-((R)-1-metoxipropán-2-il)ciclohexán-1,4-diamina en forma de un líquido amarillo pálido, rendimiento: 65%, EM (IEP): m/z 187,3 (M+H)+.
Etapa 4: síntesis de 5-bromotiazol-2-ilcarbamato de terc-butilo
Se suspendió hidrobromuro de 5-bromotiazol-2-amina (105 g, 403 mmoles) en 500 ml de tetrahidrofurano, y se añadió dimetilaminopiridina (2,41 g, 20 mmoles), formando una solución turbia blanca. Una solución de dicarbonato de diterc-butilo (105,6 g, 484,6 mmoles) en tetrahidrofurano se añadió lentamente gota a gota. La mezcla se hizo reaccionar durante dos días. A continuación, la solución de reacción se concentró, se disolvió en diclorometano (300 ml), se mezcló con gel de sílice y se aisló mediante columna (eluyendo con éter de petróleo/acetato de etilo, gradiente 10/1 a 6/1), proporcionando 45 g de 5-bromotiazol-2-ilcarbamato de terc-butilo en forma de un sólido blanquecino, rendimiento: 40%, EM (IEP): m/z 278,98 (M+H)+.
Etapa 5: síntesis de 4-bromotiazol-2-ilcarbamato de terc-butilo
Una solución de diisopropilamina (64 ml, 446 mmoles) en 200 ml de tetrahidrofurano se añadió a una botella de tres cuellos seca, que se protegió bajo nitrógeno y se enfrió a 0°C y después se añadió n-butil-litio (2,5 M, 173 ml, 431,7 mmoles). La reacción se llevó a cabo durante 1 hora después de completar la adición. Una solución de 5-bromotiazol-2-ilcarbamato de terc-butilo en 400 ml de tetrahidrofurano se añadió gota a gota a 0°C y la reacción se llevó a cabo durante 2 hora después de completar la adición. La CCF mostró que la reacción se había completado. A 0°C, la reacción se desactivó mediante adición lenta de hielo-agua (5 ml), se sometió a agitación durante 30 min y después se añadió solución acuosa saturada de cloruro amónico (500 ml) y se separó. Se extrajo la capa acuosa con diclorometano (2 x 300 ml). Se agruparon las fases orgánicas, se lavaron con solución hipersalina, se secaron sobre sulfato sódico anhidro, se filtraron y se concentraron. El residuo se recristalizó con éter de petróleo:acetato de etilo=30:1, proporcionando 31 g de 4-bromotiazol-2-ilcarbamto de terc-butilo en forma de un sólido blanco, rendimiento: 77,5%, EM (IEP): m/z 278,98 (M+H)+.
Etapa 6: síntesis de carbonato de 4-ciano-tetrahidro-2H-pirán-4-metilo
Se añadió cianoacetato de metilo (39,1 g, 395,3 mmoles) y éter 2,2-dibromoetílico (100 g, 434,8 mmoles) a 600 ml de dimetilformamida y se añadió DBU (90 g, 593 mmoles). La mezcla se calentó a 85°C durante 3 horas. La CCF mostró que el material de partida se había consumido por completo. Se filtró la mezcla para eliminar el sólido, que se lavó con acetato de etilo (2x300 ml). Se concentró el filtrado, proporcionando un aceite marrón, que se destiló bajo presión reducida. La fracción se obtuvo al alcanzar la temperatura interna 65°C a 70°C; era un líquido incoloro, y se sometió a cristalización, proporcionando 42 g de carbonato de 4-ciano-tetrahidro-2H-pirán-4-metilo en forma de un sólido blanco, rendimiento: 62,8%, EM (IEP): m/z 178,3 (M+H)+.
Etapa 7: síntesis de 4-(hidroximetil)-tetrahidro-2H-pirán-4-carbonitrilo
Se disolvió carbonato de 4-ciano-tetrahidro-2H-pirán-4-metilo (42 g, 248,4 mmoles) en 400 ml de dimetil-éter de etilenglicol y 40 ml de metanol, que se enfriaron a 0°C en un baño de hielo y se añadió borohidruro sódico (11,1 g, 149
mimóles) en partes. Tras completar la adición, la mezcla se dejó que se calentase naturalmente hasta la temperatura ambiente y se sometió a agitación durante 16 horas. La CCF mostró que la reacción se había completado. A continuación, se concentró la solución de reacción; después, se concentró nuevamente después de la adición de metanol para neutralizar el exceso de borohidruro sódico, y después se concentró. El residuo se aisló mediante columna (éter de petróleo/acetato de etilo=5:1), proporcionando 28 g de 4-(hidroximetil)-tetrahidro-2H-pirán-4-carbonitrilo en forma de un aceite amarillo pálido, rendimiento: 79,5%, EM (IEP): m/z 142,1 (M+H)+.
Etapa 8: síntesis de (4-bromotiazol-2-il)((4-cianotetrahidro-2H-pirán-4-il)metil)carbamato
Se añadió 4-(hidroximetil)-tetrahidro-2H-pirán-4-carbonitrilo, 4-bromo tiazol-2-ilcarbamato de terc-butilo y trifenilfosfina a tetrahidrofurano anhidro-THF, que se había enfriado a 0°C, y después se añadió gota a gota azodicarboxilato de diisopropilo (DIAD). La mezcla se sometió a agitación a temperatura ambiente durante 10 minutos y después se calentó a 40°C y se sometió a agitación durante la noche. Después, se concentró la solución de reacción. El residuo se disolvió en diclorometano, se mezcló con gel de sílice y se aisló mediante columna (éter de petróleo/acetato de etilo=50/1, 30/1, 20/1), proporcionando 365 mg de (4-bromotiazol-2-il)(4-cianotetrahidro-2H-pirán-4-il)metil)carbamato en forma de un sólido blanco, rendimiento: 50%, EM (IEP): m/z 402,1 (M+H)+.
Etapa 9: síntesis de (4-(5-cloro-2-fluoropiridín-4-il)tiazol-2-il) ((4-ciano-tetrahidro-2H-pirán-4-il)metil)carbamato de tercbutilo
Se añadió 5-cloro-2-fluoro-4-(4,4,5,5-tetrametil-1,3,2-dioxaborolán-2-il)piridina y carbonato sódico en una mezcla de éter dimetílico/H2O/dioxano, que se sustituyó por nitrógeno dos veces, y después se añadió (4-bromotiazol-2-il) ((4-cianotetrahidro-2H-pirán-4-il)metil)carbamato de terc-butilo y tetratrifenilfosfina-paladio Pd(pph3)4. El sistema se sustituyó por nitrógeno tres veces; después, se calentó a 70°C y se hizo reaccionar durante 6 horas. Al mostrar la CCF que solo quedaba la mitad del material de partida, se detuvo el calentamiento y se trató la reacción. La solución de reacción se enfrió hasta la temperatura ambiente y después se añadió acetato de etilo y metanol. Se filtró la mezcla, se lavó la torta con acetato de etilo y se concentró el filtrado. A continuación, el residuo se disolvió en diclorometano, se lavó con solución hipersalina saturada y se separó. La fase orgánica se secó sobre sulfato sódico anhidro y se filtró. El filtrado se mezcló con gel de sílice y se aisló mediante columna (éter de petróleo/acetato de etilo=30/1), proporcionando 3,2 g de (4-(5-cloro-2-fluoropiridín-4-il)tiazol-2-il)((4-ciano-tetrahidro-2H-pirán-4-il)metil)carbamato de terc-butilo en forma de un sólido espumoso blanco, rendimiento: 55%, EM (IEP): m/z 453,1 (M+H)+.
Etapa 10: síntesis de 4-(((4-(5-cloro-2-(((1R,4r)-4-(((R)-1-metoxipropil-2-il)amino)ciclohexil)amino)piridín-4-il)tiazol-2-il)amino)metil)tetrahidro-2H-pirán-4-carbonitrilo
Se añadió (4-(5-cloro-2-fluoropiridín-4-il)tiazol-2-il)((4-ciano-tetrahidro-2H-pirán-4-il)metil)carbamato de terc-butilo (3,2 g, 7,1 mmoles), (1r,4R)-N1-((R)-1-metoxipropán-2-il)ciclohexán-1,4-diamina (3,9 g, 21,2 mmoles) y diisopropiletilamina (DIPEA) a 30 ml de dimetilsulfóxido, que se protegieron bajo nitrógeno y después se calentaron a 100-110°C y se hicieron reaccionar durante dos días. La reacción se monitorizó mediante CCF y CL-EM. Una vez se había consumido por completo el material de partida de (4-(5-cloro-2-fluoropiridín-4-il)tiazol-2-il)((4-ciano-tetrahidro-2H-pirán-4-il) metil)carbamato de terc-butilo y quedaba parte de los intermediarios con la eliminación de BOc, se detuvo la reacción. La mezcla de reacción se enfrió y después se diluyó con acetato de etilo (60 ml), se añadió agua (150 ml) bajo enfriamiento con hielo y se separó. A continuación, se extrajo la fase acuosa con acetato de etilo (2*50 ml). Se agruparon las fases orgánicas, se lavaron con solución hipersalina saturada (100 ml), se secaron sobre sulfato sódico anhidro, se filtraron y el filtrado se concentró, proporcionando un producto en bruto en forma de un aceite marrón amarillento. El producto en bruto se aisló mediante columna (acetonitrilo/agua/ácido trifluoroacético=80/20/0,001), proporcionando 700 mg de 4-(((4-(5-cloro-2-(((1R,4r)-4-(((R)-1-metoxipropil-2-il)amino)ciclohexil)amino)piridín-4-il)tiazol-2-il)amino)metil)tetrahidro-2H-pirán-4-carbonitrilo en forma de un sólido amarillo pálido, rendimiento: 19,1%. RMN 1H (400MHz, CDCla) 88,06 (s, 1H), 7,38 (s, 1H), 6,97 (s, 1H), 5,92 (brs, 1H), 4,45 (d, J=8,0Hz, 1H), 4,02 (dd, J1=2,8Hz, J2=12Hz, 2H), 3,71-3,74 (m, 4H), 3,54-3,56 (m, 1H), 3,35 (s, 3H), 3,21-3,25 (m, 2H), 3,00-3,05 (m, 1H), 2,50-2,60 (m, 1H), 2,15 (d, J=9,6Hz, 2H), 2,04-2,07 (m, 1H), 1,95 (d, J=12,8Hz, 3H), 1,74-1,82 (m, 3H), 1,10-1,30 (m, 4H), 1,00 (d, J=8,4 Hz, 3H), EM (IEP): m/z 519,3 (M+H)+.
Ejemplo 2: síntesis de (1r,4r)-N1-(5-cloro-4-(2-(((tetrqhidro-2H-pirán -4-il)metil)amino)tiazol-4-il)piridín-2-il)ciclohexán-1,4-diamina

Etapa 1: síntesis de (4-bromotiazol-2-il)((tetrahidro-2H-pirán-4-il)metil)carbamato de terc-butilo
Se añadió 4-bromotiazol-2-carbamato de terc-butilo (12,53 g, 107,91 mimóles), (tetrahidro-2H-pirán-4-il)metanol (20 g, 71,94 mimóles) y trifenilfosfina a 360 ml de THF anhidra (redestilada), que se enfrió a -10°C y después se añadió azodicarboxilato de diisopropilo (DIAD) (21,82 g, 107,91 imimoles). La mezcla se sometió a agitación a temperatura ambiente durante 10 minutos y después se calentó a 50°C y se hizo reaccionar durante 3 horas. La CCF mostró la desaparición de los materiales de partida. Después, se concentró la solución de reacción. El residuo se disolvió en diclorometano, se mezcló con gel de sílice y se aisló mediante columna (éter de petróleo/acetato de etilo=30/1, 20/1), proporcionando 21 g de (4-bromotiazol-2-il)((tetrahidro-2H-pirán-4-il)metil)carbamato de terc-butilo en forma de un sólido blanco, rendimiento: 87,5%, EM (IEP): m/z 519,3 (M+H)+.
Etapa 2: síntesis de (4-(5-cloro-2-fluoropiridín-4-il)tiazol-2-il) ((tetrahidro-2H-pirán-4-il)metil)carbamato de terc-butilo
Se añadió (4-bromotiazol-2-il)((tetrahidro-2H-pirán-4-il)metil)carbamato de terc-butilo (21 g, 1,51 mmoles), 5-cloro-2-fluoro-4-(4,4,5,5-tetrametil-1,3,2-dioxaborolán-2-il)piridina (30 g, 3,0 mmoles), Pd(dppf)Ch (2,04 g, 0,151) y Na2CO3(15 g, 3,78 mmoles) a 500 ml de dioxano y 100 ml de agua, que se protegió bajo nitrógeno; después, se calentó a 90°C y se hizo reaccionar durante la noche. La reacción se monitorizó mediante CCF y CL-EM. Una vez se había consumido por completo el (4-bromotiazol-2-il)((tetrahidro-2H-pirán-4-il)metil)carbamato, se detuvo la reacción. Se enfrió la solución de reacción y a continuación se añadió agua (100 ml). La mezcla se extrajo con acetato de etilo (3x100 ml). Se agruparon las fases orgánicas, se secaron sobre sulfato sódico anhidro, se filtraron y se concentraron, proporcionando un producto en bruto en forma de un aceite marrón amarillento. El residuo se aisló mediante columna (éter de petróleo/acetato de etilo=30/1, 25:1), proporcionando 19,4 g de (4-(5-cloro-2-fluoropiridín-4-il)tiazol-2-il)((tetrahidro-2H-pirán-4-il)metil)carbamato de terc-butilo en forma de un sólido blanco, rendimiento: 81,5%, EM (IEP): m/z 428,1 (M+H)+.
Etapa 3: síntesis de ((1r,4r)-4-((5-cloro-4-(2-(((tetrahidro-2H-pirán-4-il)metil)amino)tiazol-4-il) piridín-2-il)amino)ciclohexil)carbamato de terc-butilo
Se añadió (4-(5-cloro-2-fluoropiridín-4-il)tiazol-2-il)((tetrahidro-2H-pirán-4-il)metil)carbamato de terc-butilo y (1r,4r)-(4-aminociclohexil)carbamato de terc-butilo a DMSO y se añadió diisopropiletilamina (DIEA). La mezcla se calentó a 100°C y se hizo reaccionar durante 2 días. Al mostrar la CCF que habían desparecido los materiales de partida, se detuvo el calentamiento y se trató la reacción. La solución de reacción se enfrió hasta la temperatura ambiente y se vertió en hielo-agua. La mezcla se extrajo con diclorometano (3x200 ml). Se lavó la fase orgánica con solución hipersalina saturada, se secó sobre sulfato sódico anhidro y se filtró. El filtrado se mezcló con gel de sílice y se aisló mediante columna (éter de petróleo/acetato de etilo=3/1, 2:1, 1:1), proporcionando 3,6 g de ((1r,4r)-4-((5-cloro-4-(2-(((tetrahidro-2H-pirán-4-il)metil)amino)tiazol-4-il)piridín-2-il)amino)ciclohexil)carbamato de terc-butlo en forma de un sólido amarillo pálido, rendimiento: 40%, EM (lEp): m/z 522,2 (M+H)+.
Etapa 4: síntesis de (1r,4r)-N1-(5-cloro-4-(2-(((tetrahidro-2H-pirán-4-il)metil)amino)tiazol-4-il)piridín-2-il)ciclohexán-1,4-diamina
Se añadió ((1r,4r)-4-((5-cloro-4-(2-(((tetrahidro-2H-pirán-4-il)metil) amino)tiazol-4-il)piridín-2-il)amino)ciclohexil)carbamato de terc-butilo (2,9 g, 5,56 mmoles) a tetrahidrofurano/diclorometano (20 ml/20 ml), que se protegieron bajo nitrógeno y se enfriaron a 0°C; después, se añadieron 20 ml de ácido trifluoroacético gota a gota. La mezcla se hizo reaccionar durante 2 h a temperatura ambiente. La reacción se monitorizó mediante CCF. Se concentró la solución de reacción y después se vertió en hielo-agua lentamente. La mezcla se extrajo con diclorometano (3x30 ml). La fase orgánica se lavó con solución hipersalina saturada, se secó sobre sulfato sódico, se filtró y se concentró, proporcionando un producto en bruto. El producto en bruto se sometió a agitación con diclorometano:acetato de etilo=2:1, se filtró y se secó, proporcionando 1,6 g de (1r,4r)-N1-(5-cloro-4-(2-(((tetrahidro-2H-pirán-4-il)metil)amino)tiazol-4-il)piridín-2-il)ciclohexán-1,4-diamina en forma de un sólido blanco, rendimiento: 68%; RMN 1H (400MHz, CDCla) 88,06 (s, 1H), 7,33 (s, 1H), 6,96 (s, 1H), 5,21-5,30 (m, 1H), 4,32 (d, J=8,0Hz, 1H)), 3,99
4,03 (m, 2H), 3,53-3,61 (m, 1H), 3,38-3,44 (m, 2H), 3,23(t, J=6,4Hz, 2H), 2,68-2,74 (m, 1H), 2,11-2,13 (m, 2H), 1,85 2,13 (m, 3H), 1,70-1,73 (m, 2H), 1,10-1,45 (m, 7H). EM (IEP): m/z 422,2 (M+H)+.
Ejemplo 3: síntesis de N-((1r,4r)-4-((5-cloro-4-(2-(((tetrahidro-2H-piran-4-il)metil)amino)tiazol-4-il)piridín-2-il)amino)cidohexil)acetamida

Se disolvió (1r,4r)-N1-(5-cloro-4-(2-(((tetrahidro-2H-piran-4-il)metil)amino)tiazol-4-il)piridm-2-il)ciclohexán-1,4-diamina (0,422 g, 1 mmol) en 10 ml de diclorometano, que se protegió bajo nitrógeno, y se añadió cloruro de acetilo. Precipitó una gran cantidad de sólidos y la CCF mostró que el material de partida se había consumido por completo. Se filtró la mezcla, se sometió a agitación con éter metil-terc-butílico, y se secó, proporcionando 187 mg de N-((1r,4r)-4-((5-cloro-4-(2-(((tetrahidro-2H-pirán-4 -il)metil)amino)tiazol-4-il)piridín-2-il)amino)ciclohexil)acetamida en forma de un sólido blanco; rendimiento: 41%; RMN 1H (400 MHz, CDCls) 5 8,06 (s, 1H), 7,33 (s, 1H), 6,96 (s, 1H), 5,30-5,34 (m, 1H), 5,20-5,30 (m, 1H), 4,32 (d, J=8,0Hz, 1H), 3,99-4,03 (m, 2H), 3,78-3,83 (m, 1H), 3,62-3,64 (m, 1H), 3,41 (t, J=12Hz, 2H), 3,24 (t, J=6,4Hz, 1H), 2,13-2,15 (m, 2H), 2,00-2,09 (m, 2H), 1,95 (s, 3H), 1,70-1,73 (m, 2H), 1,20-1,49 (m, 7H). EM (IEP): m/z 464,1 (M+H)+.
Ejemplo 4: síntesis de (1r,4r)-N1-(5-cloro-4-(2-(((tetrahidro-2H-piran-4-il)metil)amino)tiazol-4-il)piridín-2-il)-N4-(2-metoxietil)ciclohexán-1,4-diamina

Se añadió (1r,4r)-N1-(5-cloro-4-(2-(((tetrahidro-2H-pirán-4-il)metil)amino)tiazol-4-il)piridín-2-il)ciclohexán-1,4-diamina (0,357 g, 0,846 mmoles), éter 2-bromoetil-metílico (0,118 g, 0,846 mmoles) y carbonato potásico (0,116 g, 0,846 mmoles) a 10 ml de DFM, que se protegieron bajo nitrógeno y después se calentaron a 100°C y se hicieron reaccionar durante dos días. La reacción se monitorizó mediante CCF y CL-Em , y se trató después de detenerse la reacción. La solución de reacción se enfrió y después se vertió en hielo-agua (20 ml). La mezcla se extrajo con acetato de etilo (3x20 ml). La fase orgánica se lavó con solución hipersalina saturada, se secó sobre sulfato sódico anhidro, se filtró y se concentró, proporcionando un producto en bruto en forma de un aceite marrón amarillento. El residuo se aisló mediante cromatografía de columna (diclorometano/metanol=20:1, 15:1, 10:1), proporcionando 0,070 g de (1r,4r)-N1-(5-cloro-4-(2-(((tetrahidro-2H-pirán-4-il)metil)amino)tiazol-4-il)piridín-2-il)-N4-(2-metoxietil)ciclohexán-1,4-diamina en forma de un sólido amarillo pálido; rendimiento: 17%; RMN 1H (400 MHz, c D c l3) 58,06 (s, 1H), 7,33 (s, 1H), 6,96 (s, 1H), 5,65 (brs, 1H), 4,40 (d, J=8,0Hz, 1H), 3,95-4,06 (m, 2H), 3,49-3,70 (m, 3H), 3,28-3,45 (m, 5H), 3,18 (t, J=6,4Hz, 1H), 2,96-3,05 (m, 2H), 2,76-2081 (m, 1H), 2,14-2,28 (m, 6H), 1,85-1,95 (m, 3H), 1,70-1,73 (m, 2H), 1,41-1,60 (m, 2H), 1,13-1,40 (m, 5H). EM (IEP): m/z 480,3 (M+H)+.
Ejemplo 5: síntesis de (1S,4r)-N1-(5-cloro-4-(2-(((tetrahidro-2H-pirán-4-il)metil)amino)tiazol-4-il)piridín-2-il)-N4-((S)-1-metoxipropán-2-il)ciclohexán-1,4-diamina

Etapa 1: síntesis de 4-metilbencenosulfonato de (R)-1-metoxipropán-2-ol
Se añadió hidruro sódico (NaH) (1,46 g, 0,037 mmoles) a tetrahidrofurano (THF) seco (1 l), que se enfrió a 0°C bajo enfriamiento con hielo y se protegió bajo nitrógeno, y después se añadió gota a gota (R)-(-)-1-metoxipropán-2-ol (3 g, 0,033 mmoles). Tras completar la adición gota a gota, la mezcla se calentó hasta la temperatura ambiente y se sometió a agitación durante 1,5 horas. La solución de reacción se enfrió nuevamente a 0°C y a continuación se añadió gota a gota una solución de cloruro de p-toluenosulfonilo (TosCl) (6,47 g, 0,034 mmoles) en tetrahidrofurano (THF) (80 ml). La temperatura era inferior a 10°C durante la adición. Tras la adición, la mezcla se sometió a agitación a temperatura ambiente (32°C) durante la noche. La CCF mostró que el material de partida se había consumido por completo. La
reacción se desactivó mediante la adición gota a gota de solución acuosa saturada de cloruro amónico (20 ml) bajo enfriamiento con hielo, y se separó. Se extrajo la fase acuosa dos veces con acetato de etilo (30 ml). Se agruparon las fases orgánicas, se lavaron con solución hipersalina saturada (50 ml), se secaron sobre sulfato sódico anhidro, se filtraron y se concentraron, proporcionando un producto en bruto en forma de un aceite amarillo pálido. El producto en bruto se aisló mediante columna (éter de petróleo/acetato de etilo=5:1), proporcionando 4,2 g de 4-metilbencenosulfonato de (R)-1-metoxipropán-2-ol en forma de un aceite amarillo pálido, rendimiento: 52%, EM (IEP): m/z 245,1 (M+H)+.
Etapa 2: síntesis de (1S,4r)-N1-(5-cloro-4-(2-(((tetrahidro-2H-pirán-4-il)metil)amino)tiazol-4-il)piridín-2-il)-N4-((S)-1-metoxipropán-2-il)ciclohexán-1,4 -diamina
Se añadió (1 r,4r)-N1-(5-cloro-4-(2-(((tetrahidro-2H-pirán-4-il)metil)amino)tiazol-4-il)py ridin-2-il)ciclohexán-1,4-diamina (600 mg, 1,2 mmoles), 4-metilbencenosulfonato de (R)-1-metoxi propán-2-ol (293 mg, 1,42 mmoles) y carbonato potásico (327 mg, 2,4 mmoles) a 20 ml de acetonitrilo, que se protegieron bajo nitrógeno y se calentaron a 90°C y se sometieron a agitación durante la noche. La reacción se monitorizó mediante CL-EM. La solución de reacción se enfrió hasta la temperatura ambiente, se filtró y se concentró, proporcionando un producto en bruto en forma de un aceite amarillo pálido. El producto en bruto se aisló mediante una placa de capa preparativa (diclorometano/metanol=8/1), proporcionando 30 mg de (1S,4r)-N1-(5-cloro-4-(2-(((tetrahidro-2H-pirán-4-il)metil)amino)tiazol-4-il)piridín-2-il)-N4-((S)-1-metoxipropán-2-il)ciclohexán-1,4-diamina en forma de un sólido blanco; rendimiento: 4,3%; RMN 1H (600 MHz, CDCls) 58,06 (s, 1H), 7,29 (s, 1H), 6,96 (s, 1H), 5,59 (brs, 1H), 4,36 (d, J=8,0Hz, 1H), 3,95-4,06 (m, 2H), 3,49-3,65 (m, 2H), 3,40-3,49 (m, 1H), 3,22-3,39 (m, 6H), 3,11-3,20 (m, 2H), 2,95-3,10 (m, 1H), 2,08-2,30 (m, 4H), 1,79-1,96 (m, 2H), 1,62-1,71 (m, 2H), 1,09-1,40 (m, 12H), 0,72-0,98 (m, 2H). EM (IEP): m/z 494,3 (M+H)+.
Ejemplo 6: síntesis de (1R,4r)-N1-(5-cloro-4-(2-(((tetrahidro-2H-pirán-4-il)metil)amino)tiazol-4-il)piridín-2-il)-N4-((R)-1-metoxipropán-2-il)ciclohexán-1 ,4-diamina

Etapa 1: síntesis de 4-metilbencenosulfonato de (S)-1-metoxipropán-2-ol
Se añadió hidruro sódico (NaH) (al 60%, 1,46 g, 0,037 moles) a tetrahidrofurano (THF) seco (1 l), que se enfrió a 0°C bajo enfriamiento con hielo y se protegió bajo nitrógeno, y después se añadió gota a gota (S)-(+)-1-metoxipropán-2-ol (3 g, 0,033 mmoles). Tras completar la adición gota a gota, la mezcla se calentó hasta la temperatura ambiente y se sometió a agitación durante 1,5 horas. La solución de reacción se enfrió nuevamente a 0°C y a continuación se añadió gota a gota una solución de cloruro de p-toluenosulfonilo (TosCl) en tetrahidrofurano (THF). La temperatura era inferior a 10°C durante la adición. Tras la adición, la mezcla se sometió a agitación a temperatura ambiente (32°C) durante la noche. La CCF mostró que el material de partida se había consumido por completo. La reacción se desactivó mediante la adición gota a gota de solución acuosa saturada de cloruro amónico (20 ml) bajo enfriamiento con hielo, y se separó. Se extrajo la fase acuosa dos veces con acetato de etilo (30 ml). Se agruparon las fases orgánicas, se lavaron con solución hipersalina saturada (50 ml), se secaron sobre sulfato sódico anhidro, se filtraron y se concentraron, proporcionando un producto en bruto en forma de un aceite amarillo pálido. El producto en bruto se aisló mediante columna (éter de petróleo/acetato de etilo=5:1), proporcionando 4,5 g de 4-metilbencenosulfonato de (S)-1-metoxipropán-2-ol en forma de un aceite amarillo pálido, rendimiento: 55%, EM (IEP): m/z 245,1 (M+H)+.
Etapa 2: síntesis de (1R,4r)-N1-(5-cloro-4-(2-(((tetrahidro-2H-pirán-4-il)metil)amino)tiazol-4-il)piridín-2-il)-N4-((R)-1-metoxipropán-2-il)ciclohexán-1,4-diamina
Se añadió (1r,4r)-N1-(5-cloro-4-(2-(((tetrahidro-2H-pirán-4-il)metil)amino)tiazol-4-il)piridín-2-il)ciclohexán-1,4-diamina (422 mg, 1 mmol), 4-metilbencenosulfonato de (S)-1-metoxipropán-2-ol (122 mg, 0,5 mmoles) y carbonato potásico (276 mg, 2 mmoles) a 15 ml de acetonitrilo, que se protegieron bajo nitrógeno y se calentaron a 90°C y se sometieron a agitación durante la noche. La reacción se monitorizó mediante CL-EM hasta completarse al 25%. La solución de reacción se enfrió hasta la temperatura ambiente, se filtró y se concentró, proporcionando un producto en bruto en forma de un aceite amarillo pálido. El producto en bruto se aisló mediante placa de capa preparativa (diclorometano/metanol=8/1), proporcionando 83 mg de (1R,4r)-N1-(5-cloro-4-(2-(((tetrahidro-2H-pirán-4-il)metil)amino)tiazol-4-il)piridín-2-il)-N4-((R)-1-metoxipropán-2-il)ciclohexán-1,4-diamina en forma de un sólido blanco; rendimiento: 17%; RMN 1H (400 MHz, CDCls) 58,06 (s, 1H), 7,33 (s, 1H), 6,96 (s, 1H), 5,30 (brs, 1H), 4,37 (d, J=8,0Hz, 1H), 3,99-4,03 (m, 2H), 3,52-3,59 (m, 1H), 3,25-3,49 (m, 4H), 3,36 (s, 3H), 3,16-3,25 (m, 2H), 3,06-3,10 (m, 1H), 2,60 2,65 (m, 1H), 2,16 (d, J=10,8Hz, 2H), 2,00-2,08 (m, 2H), 1,89-1,95 (m, 2H), 1,33-1,45 (m, 4H), 1,12-1,29 (m, 4H), 1,07 (d, J=6,4Hz, 3H). EM (IEP): m/z 494,2 (M+H)+.
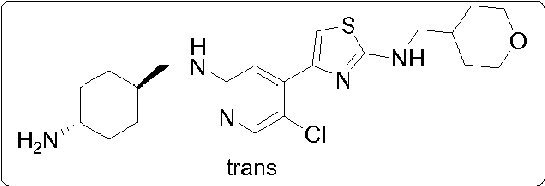
Ejemplo 7 : síntesis de 4-(2-((((1r,4r)-4-aminociclohexil)metil)amino)-5-cloropiridm-4-il)-N-((tetrahidro-2H-piran-4-il)metil)tiazol-2-amina

Etapa 1: síntesis de (1r,4r)-4-(((5-cloro-4-(2-(((tetrahidro-2H-piran-4-il)metil)amino)tiazol-4-il)piridm-2-il)amino)metil)ciclohexil)carbamato de terc-butilo
Se añadió (4-(5-cloro-2-fluoropiridm-4-il)tiazol-2-il)((tetrahidro-2H-piran-4-il)metil)carbamato de terc-butilo (0,7 g, 1,6 mmoles), (1r,4r)-4-(aminometil)ciclohexilcarbamato de terc-butilo (0,748 g, 3,2 mmoles) y trietilamina (0,458 g, 4,8 mmoles) a 10 ml de dimetilsulfóxido. La mezcla se calentó a 110°C y se sometió a agitación durante 48 horas. La CCF mostró que el material de partida se había consumido por completo. Tras enfriar hasta la temperatura ambiente, la solución de reacción se vertió en hielo-agua. La mezcla se extrajo con acetato de etilo (3x20 ml). Se agruparon las fases orgánicas, se lavaron con solución hipersalina, se secaron sobre sulfato sódico anhidro, se filtraron y se concentraron. El residuo se aisló mediante cromatografía de columna (éter de petróleo/acetato de etilo=10:2, 2:1), proporcionando ((1r,4r)-4-(((5-cloro-4-(2-(((tetrahidro-2H-piran-4-il)metil)amino)tiazol-4-il)piridm-2-il)amino)metil)ciclohexil)carbamato de terc-butilo en forma de un sólido amarillo; rendimiento: 26%; EM (IEP): m/z 536,2 (M+H)+.
Etapa 2: síntesis de 4-(2-((((1r,4r)-4-aminociclohexil)metil)amino)-5-cloropiridín-4-il)-N-((tetrahidro-2H-pirán-4-il)metil)tiazol-2-amina
Se añadió ((1r,4r)-4-(((5-cloro-4-(2-(((tetrahidro-2H-piran-4-il)metil)amino)tiazol-4-il)piridm-2-il)amino)metil)ciclohexil)carbamato de terc-butilo (230 mg, 0,43 mmoles) a diclorometano (10 ml), que se protegió bajo nitrógeno y se enfrió a 0°C, y después se añadió gota a gota ácido trifluoroacético. La mezcla se hizo reaccionar durante 1 h a temperatura ambiente. La reacción se monitorizó mediante CCF. Se concentró la solución de reacción y después se vertió en hielo-agua lentamente. La mezcla se extrajo con diclorometano (3x30 ml). La fase orgánica se lavó con solución hipersalina saturada, se secó sobre sulfato sódico, se filtró y se concentró, proporcionando un producto en bruto. El producto en bruto se aisló mediante placa de capa preparativa (diclorometano/metanol=5/1), proporcionando 0,065 g de 4-(2-((((1r,4r)-4-aminociclohexil)metil)amino)-5-cloropiridm-4-il)-N-((tetrahidro-2H-piran-4-il)metil)tiazol-2-amina en forma de un aceite amarillo pálido; rendimiento: 34,8%; RMN 1H (400 Mh z , MeOD) 57,82 (s, 1H), 7,11 (s, 1H), 6,95 (s, 1H), 3,84-3,88 (m, 2H), 3,32 (t, J=11,2Hz, 2H), 3,16-3,17 (m, 2H), 3,16 (d, J=6,8Hz, 2H), 3,04 (d, J=6,8Hz, 2H), 2,75-2,80 (m, 1H), 1,81-1,92 (m, 5H), 1,61-1,64 (m, 2H), 1,49-1,51 (m, 1H), 1,12-1,29 (m, 5H), 0,92-1,05 (m, 2H). EM (IEP): m/z 436,3 (M+H)+.
Ejemplo 8: síntesis de N-(5-cloro-4-(2-(((tetrahidro-2H-pirán-4-il)metil)amino)tiazol-4-il)piridín-2-il)-4-fluorobenzamida
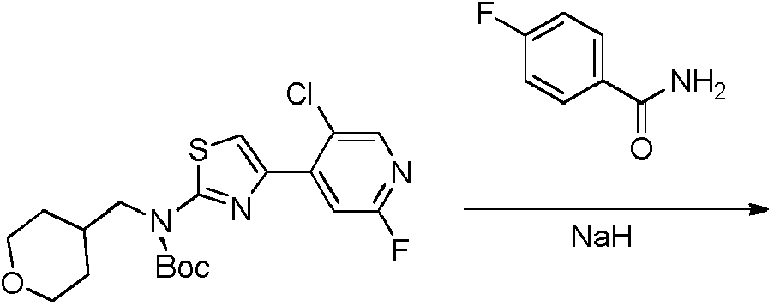

Se disolvió 4-fluorobenzamida (0,65 g, 4,68 mmoles) en N,N-dimetilformamida (DMF) (15 ml) y se añadió NaH (0,19 g, 4,68 mmoles) a temperatura ambiente. La solución de reacción se sometió a agitación a temperatura ambiente durante 10 min y después se añadió (4-(5-cloro-2-fluoropiridín-4-il)tiazol-2-il((tetrahidro-2H-pirán-4-il)metil)carbamato de terc-butilo (1 g, 2,34 mmoles). La solución de reacción se calentó a 55°C y se hizo reaccionar durante 4 h. La reacción se monitorizó mediante CCF. Se detuvo la reacción y seguidamente la solución de reacción se vertió en agua y se extrajo con EA (3x20 ml). La fase orgánica se lavó con solución hipersalina saturada, se secó sobre sulfato sódico, se filtró y se concentró, proporcionando un producto en bruto. El producto en bruto se aisló mediante placa de capa preparativa (PE:EA=1:1), proporcionando 0,032 g de N-(5-cloro-4-(2-(((tetrahidro-2H-pirán-4-il)metil)amino)tiazol-4-il)piridín-2-il)-4-fluorobenzamida en forma de un sólido blanco; rendimiento: 3,1%; RMN 1H (400 MHz, CDCh) 58,96 (s, 1H), 8,52 (s, 1H), 8,30 (s, 1H), 7,93-7,96 (m, 2H), 7,41 (s, 1H), 7,19 (t, J=8,4Hz, 2H), 5,35-5,38 (m, 1H), 4,00-4,04
(m, 2H), 3,40-3,50 (m, 2H), 3,24 (t, J=6,4Hz, 2H), 1,95-2,01 (m, 1H), 1,72-1,76 (m, 2H), 1,36-1,45 (m, 2H). (IEP+): m/z 447,1 [M+H]+.
Ejemplo 9: síntesis de (1r,4r)-N1-(5-cloro-4-(2-(metilamino)tiazol-4-il)piridín-2-il)-N4-(2-metoxietil)ciclohexán-1,4-diamina

Etapa 1: síntesis de ((1r,4r)-4-((2-metoxietil)amino)ciclohexil)carbamato de terc-butilo
Se añadió (1r,4r)-(4-aminociclohexil)carbamato de terc-butilo (10,0 g, 46,7 mmoles), éter 2-bromoetil-metílico (5,2 g, 37,4 mmoles) y carbonato potásico (12,9 g, 93,4 mmoles) a acetonitrilo (150 ml). La reacción se sometió a agitación a 80°C durante 16 h. La reacción se monitorizó mediante CCF. Una vez quedaba una parte menor del material de partida, se detuvo la reacción. La solución de reacción se enfrió hasta la temperatura ambiente y se filtró. El filtrado se secó mediante evaporación rotatoria, se mezcló con gel de sílice y se aisló mediante cromatografía en columna de gel de sílice (diclorometano/metanol=20:1), proporcionando 6,3 g de ((1r,4r)-4-((2-metoxietil)amino)ciclohexil)carbamato de terc-butilo en forma de un sólido blanco amarillento; rendimiento: 50%, e M (IEP): m/z 273,2 (M+H)+.
Etapa 2: síntesis de (1r,4r)-N1-(2-metoxietil)ciclohexán-1,4-diamina
Se disolvió ((1r,4r)-4-((2-metoxietil)amino)ciclohexil)carbamato de terc-butilo (6 g, 22,0 mmoles) en ácido clorhídrico diluido-tetrahidrofurano (80 ml). La reacción se sometió a agitación a temperatura ambiente durante 2 h y precipitó una gran cantidad de sólido. Se filtró la solución de reacción. Se secó la torta, obteniendo 5,1 g de (1r,4r)-N1-(2-metoxietil)ciclohexán-1,4-diamina (dihidrocloruro) en forma de un sólido blanco; rendimiento: 94,8%; e M (IEP): m/z 173,2 (M+H)+.
Etapa 3: síntesis de (4-(5-cloro-2-fluoropiridín-4-il)tiazol-2-il)carbamato de terc-butilo
Se disolvieron 4-bromotiazol-2-il carbamato de terc-butilo (20,0 g, 71,7 mmoles), 5-cloro-2-fluoro-4-(4,4,5,5-tetrametil-1,3,2-dioxaborolán-2-il)piridina (37,0 g, 143,4 mmoles), Pd(dppf)Cl2 (2,6 g, 0,151) y Na2CO3 (22,8 g, 245 mmoles) en 1,4-dioxano/H2O (350 ml/40ml), que se sustituyó por nitrógeno tres veces y después se sometieron a agitación a 90°C durante 16 h. La reacción se monitorizó mediante CL-EM. Quedó una cantidad reducida de materiales de partida y se añadió adicionalmente 5-cloro-2-fluoro-4-(4,4,5,5-tetrametil-1,3,2-dioxaborolán-2-il)piridina (18,5 g, 71,7 mmoles). Se sustituyó el nitrógeno tres veces en la reacción y se sometió a agitación a 85°C durante 18 h adicionales. La reacción se monitorizó mediante CL-EM. Aproximadamente 95% de los materiales de partida se habían convertido en productos. La solución de reacción se enfrió hasta la temperatura ambiente y se filtró. El filtrado se secó mediante evaporación rotatoria, se mezcló con gel de sílice y se aisló mediante cromatografía en columna de gel de sílice (éter de petróleo/acetato de etilo=10: 1), proporcionando 11,0 g de (4-(5-cloro-2-fluoropiridín-4-il)tiazol-2-il)carbamato de terc-butilo en forma de un sólido blanco; rendimiento: 47% y otros 10 g de producto en bruto. EM (lEP): m/z 330,0 (M+H)+.
Etapa 4: síntesis de (4-(5-cloro-2-fluoropiridín-4-il)tiazol-2-il) (metil)carbamato de terc-butilo
Se disolvió (4-(5-cloro-2-fluoropiridín-4-il)tiazol-2-il)carbamato de terc-butilo (200 mg, 0,61 mmoles) y trifenilfosfina (239 mg, 0,91 mmoles) en THF (4 ml), que se sustituyó por nitrógeno tres veces y se añadió metanol (MeOH) (78 mg, 2,43 mmoles). La mezcla se sometió a agitación a temperatura ambiente durante 1 minuto y después se añadió azodicarboxilato de diisopropilo (DIAD) (184 mg, 0,91 mmoles). La reacción se sometió a agitación a temperatura ambiente durante 2 h. La CCF mostró que el material de partida se había consumido por completo. La solución de reacción se aisló mediante cromatografía CCF preparativa con un solvente de revelado de éter de petróleo/acetato de etilo=10: 1, proporcionando 205 mg de (4-(5-cloro-2-fluoropiridín-4-il)tiazol-2-il)(metil)carbamato de terc-butilo en forma de un sólido blanco; rendimiento: 98%; e M (IEP): m/z 344,1 (M+H)+.
Etapa 5: síntesis de (1r, 4r)-N1-(5-cloro-4-(2-(metilamino)tiazol-4-il)piridín-2-il)-N4-(2-metoxietil)ciclohexán-1,4-diamina Se disolvió (4-(5-cloro-2-fluoropiridín-4-il)tiazol-2-il)(metil)carbamato de terc-butilo (200 mg, 0,58 mmoles), (1r, 4r)-N1-(2-metoxietil)ciclohexán-1,4-diamina (150 mg, 0,64 mmoles), diisopropiletilamina (DIEA) (375 mg, 2,9 moles) y fluoruro de cesio (265 mg, 1,74 mmoles) en dimetilsulfóxido (3 ml). La reacción se sometió a agitación a 120°C durante 2 días. La reacción se monitorizó mediante CL-EM. Una vez generado el producto, se añadió agua (40 ml) a la solución de reacción. La mezcla se extrajo con acetato de etilo (2x30 ml). El extracto se secó sobre sulfato sódico anhidro, se
concentró mediante evaporación rotatoria y después se aisló mediante cromatografía CCF preparativa con un solvente de revelado de diclorometano/metanol=6:1, proporcionando 80 mg de (1r,4r)-N1-(5-cloro-4-(2-(metilamino)tiazol-4-il)piridín-2-il)-N4-(2-metoxietil)cido hexán-1,4-diamina en forma de un sólido amarillo pálido; rendimiento: 35%; RMN 1H (400 MHz, DMSO) 57,97 (s, 1H), 7,61-7,62 (m, 1H), 7,29 (s, 1H), 7,04 (s, 1H), 6,70 (d, J=7,6Hz, 1H), 3,59 3,61 (m, 2H), 3,37-3,42 (m, 3H), 3,25 (s, 3H), 2,87 (d, J=4,8Hz, 2H), 2,74-2,77 (m, 2H), 1,90-1,96 (m, 4H), 1,12-1,23 (m, 4H). (IEP+): m/z 396,2 [M+H]+.
Ejemplo 10: síntesis de (1r,4r)-N1-(5-cloro-4-(2-((ciclohexilmetil)amino)tiazol-4-il)piridín-2-il)-N4-(2-metoxietil)ciclohexán-1,4-diamina

Etapa 1: síntesis de (4-(5-cloro-2-fluoropiridín-4-il)tiazol-2-il)(ciclohexilmetil)carbamato de terc-butilo
Se disolvió (4-(5-cloro-2-fluoropiridín-4-il)tiazol-2-il)carbamato de terc-butilo (200 mg, 0,61 mmoles) y trifenilfosfina (239 mg, 0,91 mmoles) en Th F (5 ml), que se sustituyó por nitrógeno tres veces y se añadió ciclohexilmetanol (207 mg, 1,82 mmoles). La mezcla se sometió a agitación a temperatura ambiente durante 5 minuto y después se añadió azodicarboxilato de diisopropilo (DIAD) (184 mg, 0,91 mmoles). La reacción se sometió a agitación a temperatura ambiente durante 2 h. La CCF mostró que el material de partida se había consumido por completo. La solución de reacción se aisló mediante cromatografía CCF preparativa con un solvente de revelado de éter de petróleo/acetato de etilo=10:1, proporcionando 255 mg de (4-(5-cloro-2-fluoropiridín-4-il)tiazol-2-il)(ciclohexilmetil) carbamato de terc-butilo en forma de un sólido blanco, rendimiento: 99% (IEP+): m/z 426,1 [m H]+.
Etapa 2: síntesis de (1r,4r)-N1-(5-cloro-4-(2-((ciclohexilmetil)amino)tiazol-4-il)piridín-2-il)-N4-(2-metoxietil)ciclohexán-1,4-diamina
Se disolvió(4-(5-cloro-2-fluoropiridín-4-il)tiazol-2-il)(ciclohexilmetil)carbamato de terc-butilo (250 mg, 0,59 mmoles), (1R,4R)-N1-(2-metoxietil) ciclohexán-1,4-diamina (288 mg, 1,17 mmoles), diisopropiletilamina (DIEA) (379 mg, 2,93 moles) y fluoruro de cesio (268 mg, 1,76 mmoles) en dimetilsulfóxido (8 ml). La reacción se sometió a agitación a 120°C durante 2 días. La reacción se monitorizó mediante CL-EM. Una vez generado el producto, se añadió agua (30 ml) a la solución de reacción. La mezcla se extrajo con diclorometano/isopropanol=3:1 (2x30 ml). El extracto se lavó con solución hipersalina, se secó sobre sulfato sódico anhidro, se concentró mediante evaporación rotatoria; después, se mezcló con gel de sílice y se aisló mediante cromatografía en columna de gel de sílice (diclorometano/metanol=50:1^20:1), proporcionando un producto en bruto en forma de aceite amarillo. El producto en bruto se aisló mediante cromatografía CCF preparativa con un solvente de revelado de diclorometano/metanol=8:1, proporcionando 100 mg de (1r,4r)-N1-(5-cloro-4-(2-(ciclohexilmetil)amino)tiazol-4-il)piridín-2-il)-N4-(2-metoxietil)ciclohexán-1,4-diamina en forma de un sólido amarillo pálido; rendimiento: 30%; RMN 1H (400 MHz, DMSO) 5 7,97 (s, 1H), 7,67-7,69 (m, 1H), 7,25 (s, 1H), 7,01 (s, 1H), 6,71 (d, J=7,6Hz, 1H), 3,50-3,53 (m, 1H), 3,36-3,47 (m, 2H), 3,13 (t, J=6,0Hz, 2H), 2,94-2,97 (m, 2H), 2,72-2,81 (m, 1H), 1,99-2,02 (m, 4H), 1,61-1,77 (m, 5H), 1,19-1,33 (m, 7H), 0,91-1,01 (m, 2H). (IEP+): m/z 478,3 [M+H]+.
Ejemplo 11: síntesis de (1r,4r)-N1-(4-(2-(bencilamino)tiazol-4-il)-5-cloropiridín-2-il)-N4-(2-metoxietil)ciclohexán-1,4-diamina

Etapa 1: síntesis de bencil-(4-(5-cloro-2-fluoropiridín-4-il)tiazol-2-il)carbamato de terc-butilo
Se disolvió (4-(5-cloro-2-fluoropiridín-4-il)tiazol-2-il)carbamato de terc-butilo (200 mg, 0,61 mmoles) y trifenilfosfina (239 mg, 0,91 mmoles) en THF (5 ml), que se sustituyó por nitrógeno tres veces y después se añadió alcohol bencílico (131 mg, 1,21 mmoles). La mezcla se sometió a agitación a temperatura ambiente durante 5 minutos y después se añadió diisopropiletilamina (DIEA) (184 mg, 0,91 mmoles). La reacción se sometió a agitación a temperatura ambiente durante 2 h. La CCF mostró que el material de partida se había consumido por completo. La solución de reacción se aisló mediante cromatografía CCF preparativa con un solvente de revelado de éter de petróleo/acetato de etilo=8: 1, proporcionando 246 mg de bencil-(4-(5-cloro-2-fluoropiridín-4-il)tiazol-2-il)carbamato de terc-butilo en forma de un sólido blanco; rendimiento: 97%; (IEP+): m/z 420,1 [M+H]+.
Etapa 2: síntesis de (1r,4r)-N1-(4-(2-(bencilamino)tiazol-4-il)-5-cloropiridín-2-il)-N4-(2-metoxietil)ciclohexán-1,4-diamina
Se disolvió bencil-(4-(5-cloro-2-fluoropiridín-4-il)tiazol-2-il)carbamato de terc-butilo (240 mg, 0,57 mmoles), (1r,4r)-N1-(2-metoxietil)cidohexán-1,4-diamina (280 mg, 1,14 mmoles), diisopropiletilamina (DIEA) (369 mg, 2,86 moles) y fluoruro de cesio (268 mg, 1,71 mmoles) en dimetilsulfóxido (8 ml). La reacción se sometió a agitación a 120°C durante 3 días. La reacción se monitorizó mediante CL-EM. Una vez generado el producto, se añadió agua (30 ml) a la solución de reacción. La mezcla se extrajo con diclorometano/isopropanol=3:1 (2x35 ml). El extracto se lavó con solución hipersalina, se secó sobre sulfato sódico anhidro, se concentró mediante evaporación rotatoria; después, se mezcló con gel de sílice y se aisló mediante cromatografía en columna de gel de sílice (diclorometano/metanol=20:1), proporcionando un producto en bruto en forma de aceite amarillo. El producto en bruto se aisló mediante cromatografía CCF preparativa con un solvente de revelado de diclorometano/metanol=6:1, proporcionando 100 mg de (1r,4r)-N1-(4-(2-(bencilamino) tiazol-4-il)-5-cloropiridín-2-il)-N4-(2-metoxietil)ciclohexán-1,4-diamina en forma de un sólido amarillo pálido; rendimiento: 30%; RMN 1H (400 MHz, DMSO) 58,21 (t, J=6,0Hz, 1H), 7,98 (s, 1H), 7,26-7,40 (m, 6H), 7,05 (s, 1H), 6,72 (d, J=7,6Hz, 1H), 4,52 (d, J=5,6Hz, 2H), 3,46-3,53 (m, 4H), 2,97 (brs, 2H), 2,81 (brs, 1H), 1,99-2,01 (m, 4H), 1,18-1,34 (m, 4H). (IEP+): m/z 472,1 [M+H]+.
Ejemplo 12: síntesis de (1r,4r)-N1-(5-cloro-4-(2-((4-fluorobencil)amino)tiazol-4-il)piridín-2-il-N4-(2-metoxietil)ciclohexán-1,4-diamina

Etapa 1: síntesis de (4-(5-cloro-2-fluoropiridín-4-il)tiazol-2-il) (4-fluorobencil)carbamato de terc-butilo
Se disolvió (4-(5-cloro-2-fluoropiridín-4-il)tiazol-2-il)carbamato de terc-butilo (200 mg, 0,61 mmoles) y trifenilfosfina (239 mg, 0,91 mmoles) en THF (5 ml), que se sustituyó por nitrógeno tres veces y después se añadió alcohol 4-fluorobencílico (153 mg, 1,21 mmoles). La mezcla se sometió a agitación a temperatura ambiente durante 5 minutos y después se añadió DIAD (184 mg, 0,91 mmoles). La reacción se sometió a agitación a temperatura ambiente durante 2 h. La CCF mostró que el material de partida se había consumido por completo. La solución de reacción se aisló mediante cromatografía CCF preparativa con una solución de revelado de p E/EA=10:1, proporcionando 248 mg de (4-(5-cloro-2-fluoropiridín-4-il)tiazol-2-il)(4-fluorobencil)carbamato de terc-butilo en forma de un sólido amarillo pálido; rendimiento: 93% (IEP+): m/z 438,1 [M+H]+.
Etapa 2: síntesis de (1r,4r)-N1-(5-cloro-4-(2-((4-fluorobencil)amino)tiazol-4-il)piridín-2-il)-N4-(2-metoxietil)ciclohexán-1,4-diamina
Se disolvió (4-(5-cloro-2-fluoropiridín-4-il)tiazol-2-il)(4-fluorobencil)carbamato de terc-butilo (240 mg, 0,55 mmoles), (1 r, 4r)-N1-(2-metoxietil)ciclohexán-1,4-diamina (268 mg, 1,10 mmoles), diisopropiletilamina (DIEA) (353 mg, 2,74 moles) y fluoruro de cesio (251 mg, 1,65 mmoles) en dimetilsulfóxido/N,N-dimetilacetamida (3 ml/3 ml). La reacción se sometió a agitación a 120°C durante 3 días. La reacción se monitorizó mediante CL-EM. Una vez generado el producto, se añadió agua (35 ml) a la solución de reacción. La mezcla se extrajo con diclorometano/isopropanol=3:1 (2x30 ml). El extracto se lavó con solución hipersalina, se secó sobre sulfato sódico anhidro, se concentró mediante evaporación rotatoria; después, se mezcló con gel de sílice y se aisló mediante cromatografía en columna de gel de sílice (diclorometano/metanol=20:1), proporcionando un producto en bruto en forma de aceite amarillo. El producto en bruto se aisló mediante cromatografía CCF preparativa con un solvente de revelado de diclorometano/metanol=8:1, proporcionando 100 mg de (1r,4r)-N1-(5-cloro-4-(2-((4-fluorobencil))amino)tiazol-4-il)piridín-2-il)-N4-(2-metoxi etil)ciclohexán-1,4-diamina en forma de un sólido amarillo pálido; rendimiento: 30%; RMN 1H (400 MHz, DMSO) 5 8,22-8,24 (m, 1H), 7,98 (s, 1H), 7,41-7,44 (m, 2H), 7,32 (s, 1H), 7,18 (t, J=8,8Hz, 2H), 7,04 (s, 1H), 6,73 (d, J=7,6Hz, 1H), 4,50 (d, J=5,6Hz, 2H), 3,52-3,55 (m, 3H), 3,29 (s, 3H), 2,96 (brs, 2H), 2,97 (brs, 1H), 1,98-2,01 (m, 4H), 1,21-1,23 (m, 4H). (IEP+): m/z 490,2 [M+H]+.
Ejemplo 13: síntesis de (1r,4r)-N1-(5-cloro-4-(2-((ciclopropilmetil)amino)tiazol-4-il)piridín-2-il)-N4-(2-metoxietil)ciclohexán-1,4-diamina

Etapa 1: síntesis de (4-(5-cloro-2-fluoropiridín-4-il)tiazol-2-il)(ciclohexilmetil)carbamato de terc-butilo
Se disolvió (4-(5-cloro-2-fluoropiridín-4-il)tiazol-2-il)carbamato de terc-butilo (200 mg, 0,61 mmoles) y trifenilfosfina (239 mg, 0,91 mmoles) en tetrahidrofurano (5 ml), que se sustituyó por nitrógeno tres veces y después se añadió ciclopropilmetanol (131 mg, 1,82 mmoles). La mezcla se sometió a agitación a temperatura ambiente durante 5 minuto y después se añadió azodicarboxilato de diisopropilo (DIAD) (184 mg, 0,91 mmoles). La reacción se sometió a agitación a temperatura ambiente durante 2 h. La CCF mostró que el material de partida se había consumido por completo. La solución de reacción se aisló mediante cromatografía CCF preparativa con un solvente de revelado de éter de petróleo/acetato de etilo=10: 1, proporcionando 230 mg de (4-(5-cloro-2-fluoropiridín-4-il)tiazol-2-il)(ciclopropilmetil)carbamato de terc-butilo en forma de un sólido blanco amarillento; rendimiento: 98%; (IEP+): m/z 384,1 [M+H]+.
Etapa 2: síntesis de (1r,4r)-N1-(5-cloro-4-(2-((ciclopropilmetil)amino)tiazol-4-il)piridín-2-il)-N4-(2-metoxietil)ciclohexán-1,4-diamina
Se disolvió (4-(5-cloro-2-fluoropiridín-4-il)tiazol-2-il)(ciclopropilmetil)carbamato de terc-butilo (220 mg, 0,57 mmoles), (1r,4r)-N1-(2-metoxietil) ciclo hexán-1,4-diamina (280 mg, 1,15 mmoles), diisopropiletilamina (DIEA) (370 mg, 2,86 moles) y fluoruro de cesio (262 mg, 1,72 mmoles) en dimetilsulfóxido/N,N-dimetilacetamida (3 ml/3 ml). La reacción se sometió a agitación a 120°C durante 2 días. La reacción se monitorizó mediante CL-EM. Una vez generado el producto, se añadió agua (35 ml) a la solución de reacción. La mezcla se extrajo con diclorometano/isopropanol=3:1 (2x30 ml). El extracto se lavó con solución hipersalina, se secó sobre sulfato sódico anhidro, se concentró mediante evaporación rotatoria; después, se mezcló con gel de sílice y se aisló mediante cromatografía en columna de gel de sílice (diclorometano/metanol=20:1), proporcionando un producto en bruto en forma de aceite amarillo. El producto en bruto se aisló mediante cromatografía CCF preparativa con un solvente de revelado de diclorometano/metanol=7:1, proporcionando 100 mg de (1r,4r)-N1-(5-cloro-4-(2-((ciclopropilmetil)amino)tiazol-4-il)piridín-2-il)-N4-(2-metoxietil)ciclohexán-1,4-diamina en forma de un sólido amarillo pálido; rendimiento: 35%; RMN 1H (400 MHz, DMSO) 5 7,97 (s, 1H), 7,84 (t, J=5,6Hz, 1H), 7,28 (s, 1H), 7,04 (s, 1H), 7,74 (d, J=8,0Hz, 2H), 3,52-3,55 (m, 3H), 3,29 (s, 3H), 3,17 (t, J=6,4Hz, 2H), 2,94 (brs, 1H), 2,70-2,85 (m, 1H), 1,97-2,01 (m, 4H), 1,18-1,23 (m, 5H), 0,46-0,49 (m, 2H), 0,23 0,24 (m, 2H). (IEP+): m/z 436,3 [M+H]+.
Ejemplo 14: síntesis de 4-((4-(5-cloro-2-(((1r,4r)-4-((2-metoxietil)amino)ciclohexil)amino)piridín-4-il)tiazol-2-ilamino)metil)tetrahidro-2H-pirán-4-carbonitrilo

Se disolvió (4-(5-cloro-2-fluoropiridín-4-il)tiazol-2-il)((4-ciano-tetrahidro-2H-pirán-4-il)metil)carbamato de terc-butilo (250 mg, 0,55 mmoles), (1r,4r)-N1-(2-metoxietil) ciclohexán-1,4-diamina (270 mg, 1,10 mmoles), diisopropiletilamina (DIEA) (355 mg, 2,75 moles) y fluoruro de cesio (251 mg, 1,65 mmoles) en dimetilsulfóxido/N,N-dimetilacetamida (3 ml/3 ml). La reacción se sometió a agitación a 120°C durante 3 días. La reacción se monitorizó mediante CL-EM. Una vez generado el producto, se añadió agua (30 ml) a la solución de reacción. La mezcla se extrajo con diclorometano/isopropanol=3:1 (3x30 ml). El extracto se lavó con sulfato sódico anhidro, se concentró mediante evaporación rotatoria; después, se mezcló con gel de sílice y se aisló mediante cromatografía en columna de gel de sílice (diclorometano/metanol=10:1), proporcionando un producto en bruto en forma de sólido amarillo. El producto en bruto se aisló mediante cromatografía CCF preparativa con un solvente de revelado de diclorometano/metanol 5:1, proporcionando 80 mg de 4-((4-(5-cloro-2-(((1r,4r)-4-((2-metoxietil)amino)ciclohexil)amino)piridín-4-il) tiazol-2-ilamino)metil)tetrahidro-2H-pirán-4-carbonitrilo en forma de un sólido amarillo; rendimiento: 28%; RMN 1H (400 MHz, DMSO) 58,13 (t, J=6,0Hz, 1H), 7,99 (s, 1H), 7,35 (s, 1H), 7,03 (s, 1H), 6,71 (d, J=7,6Hz, 1H), 3,91-3,95 (m, 2H), 3,67 (d, J=6,4Hz, 2H), 3,45-3,54 (m, 6H), 3,30 (s, 3H), 2,98 (brs, 2H), 2,81 (brs, 1H), 2,00-2,02 (m, 4H), 1,86-1,89 (m, 2H), 1,69-1,72 (m, 2H), 1,19-1,32 (m, 5H). (IEP+): m/z 505,3 [M+H]+.
Ejemplo 15: síntesis de 4-(((4-(5-cloro-2-(((1S,4r)-4-(((S)-1-metoxipropil-2-il)amino)ciclohexil)amino)piridín-4-il)tiazol-2-il)amino)metil)tetrahidro-2H-pirán-4-carbonitrilo
Etapa 1: síntesis de (1r,4S)-N1-((S)-1-metoxipropán-2-il)ciclohexán-1,4-diamina
Se disolvió 4-metilbencenosulfonato de (R)-1-metoxipropán-2-ol (2,0 g, 8,2 mmoles) en acetonitrilo (20 ml) y se añadió frans-1,4-ciclohexanediamina (2,34 g, 20,5 mmoles). La reacción se sometió a agitación y a reflujo a 85°C durante 16 h. La CCF mostró que el material de partida se había consumido por completo. La solución de reacción se enfrió hasta la temperatura ambiente y se filtró. El filtrado se secó mediante evaporación rotatoria; después, se mezcló con gel de sílice y se aisló mediante cromatografía en columna de gel de sílice (diclorometano/metanol (que contenía 0,1% de una solución acuosa al 28% de amoníaco)=10:1), proporcionando 600 mg de (1R,4S)-N1-((S)-1-metoxi propán-2-il)ciclohexán-1,4-diamina en forma de aceite amarillo; rendimiento: 40%; (IEP+): m/z 187,2 [M+H]+.
Etapa 2: síntesis de 4-(((4-(5-cloro-2-(((1S,4r)-4-(((S)-1-metoxipropil-2-il)amino)ciclohexil)amino)piridín-4-il)tiazol-2-il)amino)metil)tetrahidro-2H-pirán-4-carbonitrilo
Se disolvieron (4-(5-cloro-2-fluoropiridín-4-il)tiazol-2-il)((4-ciano-tetra hidro-2H-pirán-4-il)metil)carbamato de terc-butilo (200 mg, 0,44 mmoles), (1r,4S)-N1-((S) -1-metoxipropán-2-il)ciclohexán-1,4-diamina (200 mg, 1,08 mmoles) y diisopropiletilamina (DIp Ea ) (284 mg, 2,9 moles) en dimetilsulfóxido (2 ml). La reacción se sometió a agitación a 130°C durante 2,5 días. La reacción se monitorizó mediante CL-EM. Una vez generado el producto, se añadió agua (30 ml) a la solución de reacción. La mezcla se extrajo con diclorometano/isopropanol=3:1 (3x30 ml). El extracto se lavó con sulfato sódico anhidro, se concentró mediante evaporación rotatoria; después, se mezcló con gel de sílice y se aisló mediante cromatografía en columna de gel de sílice (diclorometano/metanol=10:1), proporcionando un producto en bruto en forma de aceite marrón. El producto en bruto se aisló mediante cromatografía CCF preparativa con un solvente de revelado de diclorometano/metanol=8:1, proporcionando 50 mg de 4-(((4-(5-cloro-2-(((1S,4r)-4-(((S)-1-metoxipropil-2-il)amino)ciclohexil)amino)piridín-4-il)tiazol-2-il)amino)metil)tetrahidro-2H-pirán-4-carbonitrilo en forma de un sólido amarillo; rendimiento: 22%; RMN 1H (400 MHz, DMSO) 58,12 (t, J=6,0Hz, 1H), 7,98 (s, 1H), 7,35 (s, 1H), 7,03 (s, 1H), 6,69 (d, J=8,0Hz, 1H), 3,91-3,95 (m, 2H), 3,66 (d, J=6,4Hz, 2H), 3,55-3,65 (m, 1H), 3,47-3,51 (m, 3H), 3,29 (s, 3H), 3,17 (d, J=4,8Hz, 1H), 1,86-1,99 (m, 6H), 1,66-1,74 (m, 2H), 0,99-1,26 (m, 8H). (IEP+): m/z 519,3 [M+H]+. Ejemplo 16: síntesis de (1r,4r)-N1-(5-cloro-4-(2-((tetrahidro-2H-piran-4-il)metoxi)tiazol-4-il)piridín-2-il)-N4-(2-metoxietil)ciclohexán-1,4-diamina

Etapa 1: síntesis de carbonato de 4-bromo-2-((tetrahidro-2H-pirán-4-il)metoxi)tiazol
Se disolvió (tetrahidro-2H-pirán-4-il)metanol (5,0 g, 20,8 mmoles) en 50 ml de tetrahidrofurano (50 ml) y después se añadió NaH (996 mg, 24,9 mmoles). La mezcla se sometió a agitación durante 10 minutos a temperatura ambiente y después se añadió 2,4-dibromotiazol (5,0 g, 20,8 mmoles). A continuación, la mezcla se sometió a agitación a temperatura ambiente durante la noche. Tras añadir 100 ml de una solución saturada de cloruro amónico a la solución de reacción, la mezcla se extrajo con acetato de etilo dos veces, 50 ml cada vez. A continuación, se agruparon las fases orgánicas, se secaron sobre sulfato sódico anhidro y después se concentraron mediante evaporación rotatoria. El residuo se aisló mediante cromatografía de columna (éter de petróleo:acetato de etilo=100.1), proporcionando 4,2 g de 4-bromo-2-((tetrahidro-2H-pirán-4-il)metoxi)tiazol en forma de un sólido blanco; rendimiento: 73%. (IEP+): m/z 278,0 [M+H]+.
Etapa 2: síntesis de 4-(5-cloro-2-fluoropiridín-4-il)-2-((tetrahidro-2H-pirán-4-il)metoxi)tiazol
Se añadió 4-bromo-2-((tetrahidro-2H-pirán-4-il)metoxi)tiazol (2,0 g, 7,22 mmoles) y 5-cloro-2-fluoro-4-(4,4,5,5-tetrametil-1,3,2-dioxaborolán-2-il)piridina (3,71 g, 14,44 mmoles) a un solvente mixto de 20 ml de dioxano y 4 ml de agua, y se añadieron además Pd(dppf)Cl2 (161 mg, 0,22 mmoles) y Na2CO3 (2,3 g, 21,66 mmoles). Bajo protección con nitrógeno, la mezcla se calentó a 80°C y se sometió a agitación durante la noche. Tras añadir 50 ml de agua a la solución de reacción, la mezcla se extrajo con acetato de etilo dos veces, 50 ml cada vez. A continuación, se agruparon las fases orgánicas, se secaron sobre sulfato sódico anhidro y después se concentraron mediante evaporación rotatoria. El residuo se aisló mediante cromatografía de columna (éter de petróleo:acetato de etilo=30:1), proporcionando 1,45 g de 4-(5-cloro-2-fluoropiridín-4-il)-2-((tetrahidro-2H-pirán-4-il)metoxi)tiazol en forma de un sólido blanco; rendimiento: 61,2%. (IEP+): m/z 329,1 [M+H]+.
Etapa 3: síntesis de (1r,4r)-N1-(5-cloro-4-(2-((tetrahidro-2H-pirán-4-il)metoxi)tiazol-4-il)piridín-2-il)-N4-(2-metoxietil)ciclohexán-1,4-diamina
Se añadió 4-(5-cloro-2-fluoropiridín-4-il)-2-((tetrahidro-2H-pirán-4-il)metoxi)tiazol (300 mg, 0,915 mmoles), (1R,4R-N1-(2-metoxietil)ciclohexán-1,4-diamina (245 mg, 1,006 mmoles) y K2CO3 (104 mg, 2,745 mmoles) a 5 ml de DMSO. A continuación, la mezcla se calentó a 100°C bajo agitación y se hizo reaccionar durante 48 horas. La reacción se monitorizó mediante CL-EM y la mayor parte de los materiales de partida había reaccionado por completo. La solución
de reacción se enfrió hasta la temperatura ambiente y después se añadieron 50 ml de agua. A continuación, la mezcla se extrajo con acetato de etilo dos veces, 10 ml cada vez. A continuación, se agruparon las fases orgánicas, se secaron sobre sulfato sódico anhidro y después se concentraron mediante evaporación rotatoria. El producto en bruto obtenido se aisló mediante cromatografía de columna (diclorometano:metanol=20:1). Finalmente, se obtuvieron 111,0 mg de (1r,4r)-N1-(5-cloro-4-(2-((tetrahidro-2H-pirán-4-il)metoxi)tiazol-4-il)piridín-2-il)-N4-(2-metoxietil)ciclohexán-1,4-diamina en forma de un sólido marrón pálido; rendimiento: 25,3%. RMN 1H (400 MHz, CDCh) 88,07 (s, 1H), 7,50 (s, 1H), 6,95 (s, 1H), 4,39 (d, J=8,0Hz, 1H), 4,31 (d, J=6,4Hz, 2H), 4,03 (dd, J1=3,2Hz, J2=11,2Hz, 2H), 3,59-3,61 (m, 1H), 3,54 (t, J=4,8Hz, 2H), 3,44-3,47 (m, 2H), 3,37 (s, 3H), 2,85 (t, J=5,2Hz, 2H), 2,53-2,54 (m, 1H), 2,15-2,18 (m, 3H), 2,00-2,03 (m, 2H), 1,73-1,76 (m, 2H), 1,46-1,52 (m, 2H), 1,15-1,37 (m, 5H). (IEP+): m/z 481,2 [M+H]+.
Ejemplo 17: síntesis de (1r,4r)-N1-(5-cloro-4-(2-(((tetrahidro-2H-pirán-4-il)metil)mercapto)tiazol-4-il)piridín-2-il)-N4-(2-metoxietil)ciclohexán-1,4-diamina

Etapa 1: síntesis de 4-(bromometil)-tetrahidro-2H-pirano
Se añadió tetrahidro-2H-pirán-4-il)metanol (8,12 g, 10 mmoles) y N-bromosuccinimida (NBS) (13,71 g, 2448 mmoles) a 400 ml de diclorometano, que se enfriaron a 0°C y después se añadió lentamente, en partes, trifenilfósforo. La reacción se sometió a agitación a temperatura ambiente durante 1-2 horas y la CCF mostró que los materiales de partida habían desaparecido. La solución de reacción se vertió en agua (100 ml). La mezcla se extrajo con diclorometano. La fase orgánica se lavó con salmuera saturada, se secó sobre sulfato sódico y se aisló mediante cromatografía de columna (éter de petróleo/acetato de etilo=20/1), proporcionando 6,2 g de 4-(bromometil)-tetrahidro-2H-pirano en forma de un líquido incoloro; rendimiento: 49%; (IEP+): m/z 179,0 [M+H]+.
Etapa 2: síntesis de S-(tetrahidro-2H-pirán-4-il)tioacetato de metilo
Se añadió 4-(bromometil)-tetrahidro-2H-pirano y tioacetato potásico a 60 ml de DMF. La mezcla se calentó a 90°C y se hizo reaccionar durante 2 horas. Al mostrar la CCF que habían desparecido los materiales de partida, se detuvo el calentamiento y se trató la reacción. La solución de reacción se enfrió hasta la temperatura ambiente y se vertió en hielo-agua. La mezcla se extrajo con acetato de etilo (3x30 ml). Se lavó la fase orgánica con solución hipersalina saturada, se secó sobre sulfato sódico anhidro y se filtró. El filtrado se mezcló con gel de sílice y se aisló mediante columna (éter de petróleo/acetato de etilo=30/1, 20:1, 10:1), proporcionando 1,8 g de S-(tetrahidro-2H-pirán-4-il)tioacetato de metilo en forma de aceite amarillo; rendimiento: 69%. (IEP+): m/z 175,1 [M+H]+.
Etapa 3: síntesis de (tetrahidro-2H-pirán-4-il)metil-mercaptano
Se añadió S-(tetrahidro-2H-pirán-4-il)tioacetato de metilo a THF, que se protegió bajo nitrógeno y se enfrió a 0°C. Se añadió lentamente, en lotes, hidruro de litio-aluminio, y se hizo reaccionar durante la noche. La reacción se monitorizó mediante CCF. La solución de reacción se diluyó con tetrahidrofurano (50 ml) y se añadió lentamente, en lotes, una cantidad apropiada de decahidrato de sulfato sódico. La mezcla se sometió a agitación durante 10 min, se filtró y se concentró, proporcionando 0,68 g de un producto en bruto (tetrahidro-2H-pirán-4-il)metil-mercaptano en forma de aceite amarillo; rendimiento: 100%. (IEP+): m/z 133,1 [M+H]+.
Etapa 4: síntesis de 4-bromo-2-(((tetrahidro-2H-pirán-4-il)metil)sulfhidril)tiazol
Se disolvió (tetrahidro-2H-pirán-4-il)metil-mercaptano (0,632 g, 4,8 mmoles) en tetrahidrofurano (THF), que se protegió bajo nitrógeno y se enfrió a 0°C, y seguidamente se añadió lentamente en lotes hidruro sódico (NaH) (0,2 g, 4,8 mmoles), y se hizo reaccionar a temperatura ambiente durante 10 min. Se añadió gota a gota una solución de 2,4-dibromotiazol en 30 ml de tetrahidrofurano (THF) y se hizo reaccionar durante la noche. Una vez la CCF había mostrado que se había completado prácticamente la reacción, ésta se detuvo. La solución de reacción se vertió en cloruro amónico saturado y se desactivó. La mezcla se extrajo con acetato de etilo (3x30 ml). Se agruparon las fases orgánicas, se secaron sobre sulfato sódico anhidro, se filtraron y se concentraron, proporcionando un producto en bruto en forma de un aceite marrón amarillento. El producto en bruto se aisló mediante cromatografía de columna (éter
de petróleo/acetato de etMo=25:1, 20:1), proporcionando 0,7 g de 4-bromo-2-(((tetrahidro-2H-pirán-4-il)metil)sulfhidril)tiazol en forma de un sólido blanquecino; rendimiento: 60%. (IEP+): m/z 294,0 [M+H]+.
Etapa 5: síntesis de 4-(5-cloro-2-fluoropiridín-4-il)-2-(((tetrahidro-2H-pirán-4-il)metil)sulfhidril)tiazol
Se añadió 4-bromo-2-(((tetrahidro-2H-pirán-4-il)metil)sulfhidril)tiazol (0,45 g, 1,512 mmoles), 5-cloro-2-fluoro-4-(4,4,5,5-tetrametil-1,3,2-dioxaborolán-2-il) piridina (0,788 g, 3,0 mmoles), tetrakistrifenilfosfina-paladio, Pd(PPh3)4 (0,18 g, 0,151 mmoles) y carbonato sódico (0,405 g, 3,78 mmoles) a 20 ml de dioxano y 4 ml de agua, que se protegieron bajo nitrógeno; después se calentaron a 90°C y se hicieron reaccionar durante la noche. La reacción se monitorizó mediante CCF y CL-EM. Una vez los materiales de partida habían desaparecido por completo, se detuvo la reacción. Se enfrió la solución de reacción y a continuación se añadió agua (80 ml). La mezcla se extrajo con acetato de etilo (3x30 ml). Se agruparon las fases orgánicas, se secaron sobre sulfato sódico anhidro, se filtraron y se concentraron, proporcionando un producto en bruto en forma de un aceite marrón amarillento. Se aisló el producto en bruto mediante cromatografía (éter de petróleo/acetato de etilo=30:1, 25:1), proporcionando 0,27 g de 4-(5-cloro-2-fluoropiridín-4-il)-2-(((tetrahidro-2H-pirán-4-il)metil)sulfhidril)tiazol en forma de aceite amarillo; rendimiento: 42%. (IEP+): m/z 345,0 [M+H]+.
Etapa 6 : síntesis de (1r,4r)-N1-(5-cloro-4-(2-(((tetrahidro-2H-pirán-4-il)metil)mercapto)tiazol-4-il)piridín-2-il)-N4-(2-metoxietil)ciclohexán-1,4-diamina
Se añadió 4-(5-cloro-2-fluoropiridín-4-il)-2-(((tetrahidro-2H-pirán-4-il)metil)sulfhidril)tiazol (0,27 g, 0,756 mmoles), (1R,4R-N1-(2-metoxietil)ciclohexán-1,4-diamina (0,203 g, 0,831 mmoles) y K2CO3 (0,313 g, 2,268 mmoles) a DMSO, que se protegió bajo nitrógeno y después se calentó a 100°C y se hizo reaccionar durante dos días. La reacción se monitorizó mediante CCF y CL-EM. Se retuvo el material de partida, 4-(5-cloro-2-fluoropiridín-4-il)-2-(((tetrahidro-2H-pirán-4-il)metil)sulfhidril)tiazol y se detuvo la reacción. Se enfrió la mezcla de reacción y se diluyó con acetato de etilo (20 ml), se añadió agua (800 ml) en un baño de hielo, y se separó. A continuación, se extrajo la fase acuosa con acetato de etilo (2*20 ml). Se agruparon las fases orgánicas, se secaron sobre sulfato sódico anhidro, se filtraron y se concentraron, proporcionando un producto en bruto en forma de un aceite marrón amarillento. El producto en bruto se aisló mediante cromatografía (diclorometano/metanol=15:1), proporcionando 0,135 g de (1r,4r)-N1-(5-cloro-4-(2-(((tetrahidro-2H-pirán-4-il)metil)sulfhidril)tiazol-4-il)piridín-2-il)-N4-(2-metoxietil)ciclohexán-1,4-diamina en forma de un sólido amarillo; rendimiento: 34,5%. RMN 1H (400 MHz, CDCla) 8 8,07 (s, 1H), 7,98 (s, 1H), 6,99 (s, 1H), 4,42 (brs, 1H), 3,91-4,10 (m, 2H), 3,55-3,71 (m, 3H), 2,83-3,52 (m, 12H), 2,13-2,17 (m, 4H), 1,95-2,05 (m, 1H), 1,69-1,87 (m, 2H), 1,31-1,56 (m, 5H), 1,02-1,35 (m, 4H), 0,79-0,95 (m, 1H). (IEP+): m/z 497,2 [M+H]+.
Ejemplo 18: síntesis de (1r,4r)-N1-(2-metoxietil)-N4-(4-(2-(((tetrahidro-2H-pirán-4-il)metil)amino)tiazol-4-il)piridín-2-il)ciclohexán-1,4-diamina

Etapa 1: síntesis de (4-(2-cloropiridín-4-il)tiazol-2-il)((tetrahidro-2H-pirán-4-il)metil)carbamato de terc-butilo
Se añadió (4-bromotiazol-2-il)((tetrahidro-2H-pirán-4-il)metil)carbamato de terc-butilo (1 g, 1,512 mmoles), 2-cloro-4-(4,4,5,5-tetrametil-1,3,2-dioxaborolán-2-il)piridina (0,95 g, 3,0 mmoles), Pd(dppf)Ch (0,22 g, 0,151 mmoles) y Na2CO3 (0,703 g, 3,78 mmoles) a 15 ml de dioxano y 30 ml de agua, que se protegieron bajo nitrógeno; después, se calentaron a 80°C y se hicieron reaccionar durante la noche. La reacción se monitorizó mediante CCF y CL-EM. Una vez los materiales de partida habían desaparecido por completo, se detuvo la reacción. Se enfrió la solución de reacción y a continuación se añadió agua (50 ml). La mezcla se extrajo con acetato de etilo (3x30 ml). Se agruparon las fases orgánicas, se secaron sobre sulfato sódico anhidro, se filtraron y se concentraron, proporcionando un producto en bruto en forma de un aceite marrón amarillento. Se aisló el producto en bruto mediante cromatografía (éter de petróleo/acetato de etilo=30:1), proporcionando 0,27 g de (4-(2-cloropiridín-4-il)tiazol-2-il) ((tetrahidro-2H-pirán-4-il)metil)carbamato de terc-butilo en forma de aceite amarillo; rendimiento: 42%. (IEP+): m/z 410,1 [M+H]+.
Etapa 2: síntesis de ((1r,4r)-4-((4-(2-(((tetrahidro-2H-pirán-4-il)metil)amino)tiazol-4-il)piridín-2-il)amino)ciclohexil)carbamato
Se añadió (4-(2-doropiridín-4-il)tiazol-2-il)((tetrahidro-2H-pirán-4-il)metil)carbamato de terc-butilo (0,388 g, 0,95 mimóles), ((1R,4R)-4-aiminocidohexN)carbaimato de terc-butilo (0,244 g, 1,14 mmoles), Pd2(dba)3 (0,026 g, 3,8 mmoles), terc-butóxico sódico, (±)-2,2'-bis-(difenilfosfino)-1,1'-bina1tilo (BINAP) (0,035 g, 0,0285 mmoles) a tolueno, que se protegió bajo nitrógeno y después se calentó a 120°C y se hizo reaccionar durante la noche. La reacción se monitorizó mediante CCF. Una vez los materiales de partida habían desaparecido por completo, se detuvo la reacción. Se enfrió la solución de reacción y se vertió en una solución acuosa saturada de cloruro amónico (20 ml) y se separó. A continuación, se extrajo la fase acuosa con acetato de etilo (2*20 ml). Se agruparon las fases orgánicas, se secaron sobre sulfato sódico anhidro, se filtraron y se concentraron, proporcionando 600 mg de ((1R,4R)-4-((4-(2-(((tetrahidro-2H-pirán-4-il)metil)amino) tiazol-4-il)piridín-2-il)amino)ciclohexil)carbamato en forma de aceite marrón amarillento. El producto en bruto se utilizó directamente en la etapa siguiente, y se calculó el rendimiento en la etapa siguiente. (IEP+): m/z 488,3 [M+H]+.
Etapa 3: síntesis de (1r,4r)-N1-(4-(2-(((tetrahidro-2H-pirán-4-il)metil)amino)tiazol-4-il)piridín-2-il)ciclohexán-1,4-diamina
Se añadió ((1R,4R)-4-((4-(2-(((tetrahidro-2H-pirán-4-il)metil)amino)tiazol-4-il)piridín-2-il)amino)ciclohexil)carbamato de terc-butilo (0,6 g, 0,95 mmoles) a metanol y se añadieron 5 ml de ácido clorhídrico (6 N) y se hicieron reaccionar durante la noche. A continuación, se concentró la solución de reacción y se añadió solución saturada de bicarbonato sódico para ajustar el pH a 7. A continuación, se extrajo la fase acuosa con acetato de etilo (2*20 ml) y se concentró. El residuo se sumergió durante la noche en diclorometano:metanol=10:1 y se filtró. El filtrado se concentró, proporcionando 0,12 g de (1r,4r)-N1-(4-(2-(((tetrahidro-2H-pirán-4-il)metil)amino)tiazol-4-il)piridín-2-il)ciclohexán-1,4-diamina en forma de aceite amarillo pálido; rendimiento: 31% (2 etapas). (IEP+): m/z 388,2 [M+H]+.
Etapa 4: síntesis de (1r,4r)-N1-(2-metoxietil)-N4-(4-(2-(((tetrahidro-2H-pirán-4-il)metil)amino)tiazol-4-il)piridín-2-il)ciclohexán-1,4-diamina
Se añadió (1r,4r)-N1-(4-(2-(((tetrahidro-2H-pirán-4-il)metil)amino)tiazol-4-il)piridín-2-il)ciclohexán-1,4-diamina (0,66 g, 1,7 mmoles), éter 2-bromoetil-metílico (0,240 g, 1,7 mmoles) y carbonato potásico (0,235 g, 1,7 mmoles) a N,N dimetilformamida, que se protegió con nitrógeno y después se calentó a 100°C y se hizo reaccionar durante dos días. La reacción se monitorizó mediante CCF y CL-EM. Aunque quedaba una pequeña cantidad de material de partida, se detuvo la reacción. Se enfrió la solución de reacción y a continuación se añadió agua (30 ml). La mezcla se extrajo con acetato de etilo (3x20 ml). Se agruparon las fases orgánicas, se secaron sobre sulfato sódico anhidro, se filtraron y se concentraron, proporcionando un producto en bruto en forma de un aceite marrón amarillento. El producto en bruto se aisló mediante placa de capa preparativa (diclorometano/metanol=8:1), proporcionando 0,057 g de (1r,4r)-N1-(2-metoxietil)-N4-(4-(2-(((tetrahidro-2H-pirán-4-il)metil)amino)tiazol-4-il)piridín-2-il)ciclohexán-1,4-diamina en forma de aceite amarillo; rendimiento: 15%. RMN 1H (400 MHz, CDCla) 87,95-8,02 (m, 1H), 6,79-6,89 (m, 3H), 5,48 (brs, 1H), 4,65-4,85 (m, 1H), 3,98-4,01 (m, 2H), 3,57-3,61 (m, 3H), 3,35-3,42 (m, 5H), 3,18-3,20 (m, 2H), 2,89-2,92 (m, 2H), 2,61 2,68 (m, 1H), 2,17-2,27 (m, 3H), 2,04-2,08 (m, 3H), 1,87-1,96 (m, 1H), 1,69-1,73 (m, 2H), 1,20-1,50 (m, 10H), 0,79 0,95 (m, 2H). (IEP+): m/z 446,3 [M+H]+.
Ejemplo 19: efecto de los inhibidores de CDK9 sobre el crecimiento de las células de cáncer
Mediante el ensayo del efecto de inhibidores de CK9 sobre el crecimiento de las células de cáncer, los presentes inventores evaluaron la selectividad de los compuestos en la inhibición de la proliferación de las células de cáncer.
En los ejemplos, los presentes inventores utilizando línea celular de leucemia mielocítica aguda (LMA) OCI-AML-3, línea celular de leucemia promielocítica aguda NB-4, línea celular SKM-1 MDS-RAEB (síndrome mielodisplásico de tipo exceso de blastos), células de leucemia humana Nomo-1, línea celular de leucemia mieloide aguda MOLM14, línea celular de leucemia mieloide aguda MOLM13, línea celular de leucemia mieloide aguda MV4-11, línea celular de leucemia mieloide aguda HL-60, línea celular de leucemia mieloide aguda OCI-AML-2, linfoma histiocítico U-937, línea celular de leucemia de células B aguda MEC-1, línea celular de leucemia de células B aguda MEC-2, leucemia megacarioblástica aguda CMK, células pulmonares de hámster CHL, células ováricas de hámster CHO, células de cáncer pulmonar no microcítico humano H1975, células de cáncer pulmonar no microcítico H358, células de cáncer pulmonar microcítico humano H209, células de adenocarcinoma pulmonar humano H1395, células de cáncer pulmonar no microcítico humano PC-9, células de cáncer pulmonar humano H3122, células de cáncer pulmonar no microcítico humano H2122, células de cáncer pulmonar no microcítico humano H1915, células de adenocarcinoma pulmonar humano H1355, células de cáncer pulmonar no microcítico humano HCC827, células de cáncer de mama humano MDA-MB-231, células de cáncer de mama humano MDA-MB-468, células de cáncer de mama humano MCF-7, células de cáncer de mama humano T47D, células de cáncer de mama humano SK-Br-3; las células anteriormente indicadas se adquirieron de la ATCC. Además, como compuestos de control se utilizaron Palbociclib (un inhibidor selectivo de CDK4/6 adquirido de Shanghai Haoyuan Chemical), HY-16462 (CDK9-IN-2, adquirido de Shanghai Haoyuan) y Dinaciclib (un inhibidor de CDK1/2/5/9, adquirido de Shanghai Haoyuan Chemical).
En los ejemplos, los compuestos de la presente invención con diferentes concentraciones (0,000508 pM, 0,00152 pM, 0,00457 pM, 0,0137 pM, 0,0411 pM, 0,123 pM, 0,370 pM, 1,11 pM, 3,33 pM y 10 pM) y los compuestos de control se añadieron por separado a las células anteriormente indicadas, que se incubaron durante 72 horas. Se utilizó el kit de ensayo de viabilidad celular de autoluminiscencia química Cell Titer-Glo® (Promega, EE.UU.) para detectar el número
de células viables mediante la medición cuantitativa de ATP en las células vivas, y a partir de estas mediciones se calculó GI50 e IC50. Se muestran los resultados en las Tablas 1 a 3: las Tablas 1 y 2 muestran la GI50 de los compuestos de la invención frente a las líneas celulares de enfermedad hemática; la Tabla 3 muestra la IC50 del compuesto n° 1 frente a células de tipo canceroso de cáncer no hemático.
Basándose en los resultados en las Tablas 1 y 2, se encontró que los compuestos de la presente invención sometidos a ensayos presentaban fuertes efectos de inhibición de las células de cáncer sometidas a ensayo, tales como células de leucemia y células de linfoma, y los compuestos n° 1 y n° 14 también mostraron una buena selectividad: no presentaban efectos de inhibición de las células normales CHL y de las células CHO, mientras que el fármaco de referencia Dinaciclib y HY-16462 presentaron ciertos efectos de inhibición de CHL Y CHO. Los resultados en la Tabla 3 también mostraron que el compuesto n° 1 de la presente invención también mostraba efectos inhibitorios significativos de las células de cáncer pulmonar no microcítico humano, de las células de cáncer pulmonar microcítico humano, de las células de adenocarcinoma pulmonar y de las células de cáncer de mama, mientras que Palbociclib no mostró ninguna inhibición evidente de las células de cáncer sometidas a ensayo. Dichos resultados proporcionan una importante base teórica para la utilización del compuesto n° 1 como inhibidor de quinasa CDK9 selectivo menos tóxico para el tratamiento de dichos cánceres.
Tabla 1

Tabla 2

Tabla 3


Ejemplo 20: ensayo enzimático de inhibición de la proteína CDK in vitro
Los compuestos n° 1 y n° 14 diluidos en DMSO se mezclaron con proteína CDK detectada (Invitrogen, EE.UU.), respectivamente, y se incubaron a temperatura ambiente durante 30 minutos y a continuación se mezclaron con mezcla de quinasas/sustratos peptídicos Z'-LYTE™ y 4xATP. El sistema mixto se transfirió una placa opaca blanca de 384 pocillos para la reacción durante 1 hora a temperatura ambiente; se añadieron 5 j l de solución de revelado (Invitrogen, EE.UU.) para la reacción a temperatura ambiente durante 1 hora y finalmente se añadió reactivo de parada (Invitrogen, EE.UU.) para terminar la reacción y se utilizó el lector de microplacas MD SpectraMax I3X (Molecular Devices, EE.UU.) para leer los valores de fluorescencia. Se calcularon los valores de IC50 de los compuestos n° 1 y n° 14 frente a las proteínas CDK sometidas a ensayo, basándose en los valores de fluorescencia leídos utilizando Prism 5.0 (software GraphPad, San Diego, CA) y se muestran en la Tabla 4, a continuación.
Tabla 4

Ejemplo 21: efecto de los inhibidores de CDK9 sobre la ruta de señalización
Se evaluó el efecto del compuesto n° 1 sobre CDK9 en las células y otras proteína quinasas relacionadas con su ruta de señalización, tal como RNAPII, XIAP, MCL-1, c-MYC, BCL-2, etc., frente a cuatro líneas celulares de leucemia mielocítica aguda (LMA): OCI-AML-3, línea celular de leucemia promielocítica aguda NB-4, línea celular de leucemia mielocítica aguda (LMA) HL-60 y línea celular de leucemia mielocítica aguda (LMA) MV4-11 (todas adquiridas de la ATCC), mediante la medición de criterios de evaluación bioquímicos y funcionales de múltiples células. Se utilizó el compuesto n° 1 (en DMSO) a diferentes concentraciones: 0 jM , 0,03 jM , 0,1 jM , 0,3 jM , 1 jM y 3 jM , y los fármacos de referencia Dinaciclib y Hy -16462 (CDK9-IN-2) (adquiridos de Shanghai Haoyuan) (en DMSO) a 1 jM , para tratar dichas líneas celulares durante 2 horas y después se recogieron muestras. Se determinó el efecto del compuesto n° 1 sobre la fosforilación de CDK9, RNAPII, XIAP, MCL-1, c-MYC, BCL-2 en dichas líneas celulares (fig. 1a-d).
En las cuatro líneas celulares: línea celular de leucemia mielocítica aguda (LMA) OCI-AML-3, línea celular de leucemia promielocítica aguda NB-4, línea celular de leucemia mielocítica aguda (LMA) HL-60 y línea celular de leucemia mielocítica aguda (LMA) MV4-11, se encontró que el compuesto n° 1 presentaba un efecto inhibitorio significativo de la fosforilación de RNAPII, MCL-1 y c-MYC directamente corriente abajo de la proteína CDK9.
Ejemplo 22: efecto de los nuevos inhibidores de quinasa sobre la apoptosis
Con el fin de probar si la muerte celular se producía mediante apoptosis o necrosis, en las cuatro líneas celulares: línea celular de leucemia mielocítica aguda (LMA) OCI-AML-3, línea celular de leucemia promielocítica aguda NB-4, línea celular de leucemia mielocítica aguda (LMA) HL-60 y línea celular de leucemia mielocítica aguda (LMA) MV4-11 (todas adquiridas de la ATCC), se detectó en las células el efecto del compuesto n° 1 sobre el enzima de reparación del ADN poliadenosina-difosfato-ribosa polimerasa (PARP), estrechamente relacionado con la apoptosis, y el corte de proteínas por el enzima aspartato proteolítico que contiene cisteína Caspasa 3. Se utilizó el compuesto n° 1 (en DMSO) a diferentes concentraciones: 0 jM , 0,01 jM , 0,03 jM y 0,1 jM , Dinaciclib (en DMSO) a 0,01 jM y HY-16462 (en DMSO) a 0,1 jM para tratar diferentes células y posteriormente se recogieron las células tras 24 horas. Se utilizó la transferencia Western para detectar los efectos de los fármacos a diferentes concentraciones sobre el enzima de
reparación del ADN poliadenosina-difosfato-ribosa polimerasa (PARP) y el corte de proteínas del enzima aspartato proteolítico que contiene cisteína Caspasa 3 en diferentes intervalos de tiempo.
Se muestran los resultados experimentales en las figuras 2a-d; en las cuatro líneas celulares: línea celular de leucemia mielocítica aguda (LMA) OCI-AML-3, línea celular de leucemia promielocítica aguda NB-4, línea celular de leucemia mielocítica aguda (LMA) HL-60 y línea celular de leucemia mielocítica aguda (LMA) MV4-11, resultaba evidente que se producía corte parcial del enzima de reparación del ADN poliadenosina-difosfato-ribosa polimerasa (PARP) o de la Caspasa 3 cadena abajo de PARP. Lo anterior demuestra que el compuesto n° 1 puede causar apoptosis en las cuatro células: línea celular de leucemia mielocítica aguda (LMA) OCI-AML-3, línea celular de leucemia promielocítica aguda NB-4, línea celular de leucemia mielocítica aguda (LMA) HL-60 y línea celular de leucemia mielocítica aguda (LMA) MV4-11.
Ejemplo 23: efecto de los nuevos inhibidores de quinasa sobre el ciclo celular
Con el fin de estudiar en qué ciclo se paraban las células tras la administración, en las tres líneas celulares: línea celular de leucemia promielocítica aguda NB-4, línea celular de leucemia mielocítica aguda (LMA) HL-60 y línea celular de leucemia mielocítica aguda (LMA) MV4-11, se sometieron a ensayo los efectos del compuesto n° 2 sobre la distribución del ciclo celular de dichas líneas celulares. Se utilizó compuesto n° 1 a diferentes concentraciones: 0 j M, 0,01 j M, 0,03 j M y 0,1 j M (en DMSO), inhibidor de quinasa CDK9 Dinaciclib a 0,01 j M (en DMSO) y HY-16462 a 0,1 j M (en DMSO) para tratar las líneas celulares HL-60, MV4-11 o NB-4 durante 12 horas, 24 horas o 48 horas, y después se recogieron las células, se lavaron dos veces con 1x tampón de PBS, se fijaron con etanol al 75% a -20°C durante 24 horas y después se lavaron dos veces con 1x tampón de PBS. Se añadieron 0,5 ml de 1x tampón de PBS y 0,5 ml de solución de tinción PI (adquirida de BD Bioscience, EE.UU.) a las células y éstas se dejaron en la oscuridad a 37°C durante 15 minutos para la tinción. Se midió la distribución del ciclo celular mediante citometría de flujo (BD FACS Calibur). Se muestran los resultados en las figuras 3a-c.
Los resultados se muestran en las figuras 3a-c: tras tratar las tres líneas celulares: línea celular de leucemia mielocítica aguda (LMA) HL-60, línea celular de leucemia mielocítica aguda (LMA) MV4-11 y línea celular de leucemia promielocítica aguda NB-4, durante 12 horas, 24 horas o 48 horas, respectivamente, se encontró que el compuesto n° 1 presentaba un efecto sobre el ciclo celular de dichas tres líneas celulares, es decir, el compuesto n° 1 bloqueaba las células en la fase G0-G1.
Ejemplo 24: resultados experimentales del compuesto n° 1 en el modelo de ratón de leucemia granulocítica aguda humana MV4-11
Se adquirieron ratones Balb/c hembra, de 4 a 6 semanas, de Shanghai Slack Laboratory Animals Co., Ltd., y se guardaron en el laboratorio libre de patógenos específicos (LPE). El agua de consumo y las almohadillas se trataron asépticamente en un autoclave. Todas las operaciones se llevaron a cabo bajo condiciones asépticas. El día 0, se inyectaron por vía subcutánea 5x106 células de leucemia granulocítica aguda MV4-11 (adquiridas de la ATCC) en el flanco izquierdo del lomo de todos los ratones. A partir del día 15, todos los ratones fueron divididos en cuatro grupos (6 ratones en cada grupo); se administró por vía oral solvente de metilcelulosa (HKI) en el primer grupo de ratones cada día; se administró por vía oral compuesto n° 1 a una dosis de 10 mg/kg de peso corporal en el segundo grupo de ratones cada día; se administró por vía oral compuesto n° 1 a una dosis de 20 mg/kg de peso corporal en el tercer grupo de ratones cada día, y se administró compuesto n° 1 a una dosis de 30 mg/kg de peso corporal de ratón en el cuarto grupo de ratones cada día. Desde el inicio de la administración se midió la longitud/anchura del tumor subcutáneo con un calibrador de Vernier cada día y se registró el peso corporal de cada ratón cada día y se observó el efecto del compuesto n° 1 sobre el peso corporal de los ratones. El día 43, se sacrificaron los ratones, se extrajeron los tumores subcutáneos y se pesaron y compararon los tumores, y después se preparó una muestra de lisado de proteínas a partir del tejido de muestra tumoral para su utilización. La tendencia de crecimiento de tumor subcutáneo se estimó entre los días 16 y 43 y se calculó el volumen tumoral como longitud x anchura x anchura / 2 mm3.
Se muestran los resultados experimentales en la figura. Los resultados muestran que para el compuesto inhibidor n° 1 divulgado en la presente invención, el grupo de dosis alta (20, 30 mg/kg) afectó al peso corporal de los ratones Balb/c, mientras que en el grupo de dosis baja (10 mg/kg), el peso del tumor subcutáneo se había reducido significativamente y no se observó ningún efecto significativo sobre el peso corporal de los ratones; la inhibición del crecimiento tumoral (ICT) del grupo de dosis alta (20, 30 mg/kg) podía alcanzar el 98,7%. Lo anterior indica que el compuesto n° 1 resultaba eficaz en la inhibición del crecimiento de los tumores subcutáneos (fig. 4a-c).
Aplicabilidad industrial
La presente invención proporciona un inhibidor de la quinasa dependiente de ciclina CDK9 que puede utilizarse en el tratamiento, prevención o mejora de una enfermedad, trastorno o condición regulada o afectada por la actividad de serina quinasa, o relacionada con la actividad de quinasa dependiente de ciclina. De esta manera, puede convertirse en un fármaco correspondiente adecuado para aplicaciones industriales.
Aunque la presente invención ha sido descrita en detalle en la presente memoria, la presente invención no se encuentra limitada a la misma, y el experto en la materia podrá realizar modificaciones de acuerdo con los principios
de la presente invención. Por lo tanto, diversas modificaciones de acuerdo con los principios de la presente invención deben considerarse que se encuentran comprendidas dentro del alcance de la presente invención.
Claims (5)
- REIVINDICACIONESi . Compuesto de fórmula (I):
 o una sal o solvato farmacéuticamente aceptable del mismo,en el que Y se selecciona del grupo que consiste en p-fluorobenzoílo, trans-4-aminocidohexilo en el que N se sustituye opcionalmente con R3 y trans-4-aminociclohexilmetilo en el que N se sustituye opcionalmente con R3,Z se selecciona del grupo que consiste en NH, S y O,R1 se selecciona del grupo que consiste en hidrógeno y halógeno,R2 se selecciona del grupo que consiste en hidrógeno, alquilo C1-C3 , cicloalquilo C3-C6, heterocicloalquilo C3-C6 sustituido opcionalmente con R4 , y fenilo sustituido opcionalmente con R4,R3 se selecciona de entre el grupo que consiste en alcanoilo C2-C6 y alcoxi C1-C3-alquilo C1-C3 , R4 se selecciona del grupo que consiste en ciano y halógeno.
o una sal o solvato farmacéuticamente aceptable del mismo,en el que Y se selecciona del grupo que consiste en p-fluorobenzoílo, trans-4-aminocidohexilo en el que N se sustituye opcionalmente con R3 y trans-4-aminociclohexilmetilo en el que N se sustituye opcionalmente con R3,Z se selecciona del grupo que consiste en NH, S y O,R1 se selecciona del grupo que consiste en hidrógeno y halógeno,R2 se selecciona del grupo que consiste en hidrógeno, alquilo C1-C3 , cicloalquilo C3-C6, heterocicloalquilo C3-C6 sustituido opcionalmente con R4 , y fenilo sustituido opcionalmente con R4,R3 se selecciona de entre el grupo que consiste en alcanoilo C2-C6 y alcoxi C1-C3-alquilo C1-C3 , R4 se selecciona del grupo que consiste en ciano y halógeno. - 2. Compuesto o una sal o solvato farmacéuticamente aceptable del mismo según la reivindicación 1, en el que R1 es cloro.
- 3. Compuesto o una sal o solvato farmacéuticamente aceptable del mismo según la reivindicación 1, en el que R2 se selecciona del grupo que consiste en hidrógeno, metilo, ciclopropilo, ciclohexilo, 4-tetrahidropiranilo sustituido opcionalmente con ciano, y fenilo sustituido opcionalmente con flúor.
- 4. Compuesto o una sal o solvato farmacéuticamente aceptable del mismo según la reivindicación 1, en el que R3 se selecciona del grupo que consiste en acetilo, 2-metoxietilo, (R)-1-metil-2-metoxietilo y (S)-1-metil-2-metoxietilo.
- 5. Compuesto o una sal o solvato farmacéuticamente aceptable del mismo según cualquiera de las reivindicaciones 1 a 4, en el que el compuesto se selecciona de entre:4-(((4-(5-cloro-2-(((1R,4r)-4-(((R)-1-metoxipropil-2-il)amino)ciclohexil)amino)piridín-4-il)tiazol-2-il)amino)metil)tetrahidro-2H-pirán-4-carbonitrilo,(1r,4r)-N1-(5-cloro-4-(2-(((tetrahidro-2H-pirán-4-il)metil)amino)tiazol-4-il)piridín-2-il)ciclohexán-1,4-diamina,N-((1r,4r)-4-((5-cloro-4-(2-(((tetrahidro-2H-pirán-4-il)metil)amino)tiazol-4-il)piridín-2-il)amino)ciclohexil)acetamida,(1r,4r)-N1-(5-cloro-4-(2-(((tetrahidro-2H-pirán-4-il)metil)amino)tiazol-4-il)piridín-2-il)-N4-(2-metoxietil)ciclohexil-1,4-diamina,(1S,4r)-N1-(5-cloro-4-(2-(((tetrahidro-2H-pirán-4-il)metil)amino)tiazol-4-il)piridín-2-il)-N4-((S)-1-metoxipropán-2-il)ciclohexil-1,4-diamina,(1R,4r)-N1-(5-cloro-4-(2-(((tetrahidro-2H-pirán-4-il)metil)amino)tiazol-4-il)piridín-2-il)-N4-((R)-1-metoxipropán-2-il)ciclohexán-1,4-diamina,4-(2-((((1r,4r)-4-aminociclohexil)metil)amino)-5-cloropiridín-4-il)-N-((tetrahidro-2H-pirán-4-il)metil)tiazol-2-amina,N-(5-cloro-4-(2-(((tetrahidro-2H-pirán-4-il)metil)amino)tiazol-4-il)piridín-2-il)-4-fluorobenzamida, (1r,4r)-N1-(5-cloro-4-(2-(metilamino)tiazol-4-il)piridín-2-il)-N4-(2-metoxietil)ciclohexán-1,4-diamina, (1r,4r)-N1-(5-cloro-4-(2-((ciclohexilmetil)amino)tiazol-4-il)piridín-2-il)-N4-(2-metoxietil)ciclohexán-1,4-diamina,(1r,4r)-N1-(4-(2-(bencilamino)tiazol-4-il)-5-cloropiridín-2-il)-N4-(2-metoxietil)ciclohexán-1,4-diamina, (1r,4r)-N1-(5-cloro-4-(2-((4-fluorobencil)amino)tiazol-4-il)piridín-2-il)-N4-(2-metoxietil)ciclohexán-1,4-diamina,(1r,4r)-N1-(5-cloro-4-(2-((ciclopropilmetil)amino)tiazol-4-il)piridín-2-il)-N4-(2-metoxietil)ciclohexán-1,4-diamina,4-((4-(5-cloro-2-(((1r,4r)-4-((2-metoxietil)amino)ciclohexil)amino)piridín-4-il)tiazol-2-ilamino)metil)tetrahidro-2H-pirán-4-carbonitrilo,4-(((4-(5-cloro-2-(((1S,4r)-4-(((S)-1-metoxipropil-2-il)amino)ciclohexil)amino)piridín-4-il)tiazol-2-il)amino)metil)tetrahidro-2H-pirán-4-carbonitrilo,(1r,4r)-N1-(5-cloro-4-(2-((tetrahidro-2H-pirán-4-il)metoxi)tiazol-4-il)piridín-2-il)-N4-(2-metoxietil)ciclohexán-1,4-diamina,(1r,4r)-N1-(5-doro-4-(2-(((tetrahidro-2H-pirán-4-il)metil)mercapto)tiazol-4-il)piridín-2-il)-N4-(2-metoxietil)cidohexán- 1,4-diamina y(1r,4r)-N1-(2-metoxietil)-N4-(4-(2-(((tetrahidro-2H-pirán-4-il)metil)amino)tiazol-4-il)piridm-2-il)ciclohexán-1,4-diamina.Composición farmacéutica que comprende el compuesto o solvato farmacéuticamente aceptable del mismo según una cualquiera de las reivindicaciones 1 a 5 y un portador o excipiente farmacéuticamente aceptable, y opcionalmente otros agentes terapéuticos.Compuesto o una sal o solvato farmacéuticamente aceptable del mismo según una cualquiera de las reivindicaciones 1 a 5, para la utilización en el tratamiento, prevención o mejora de una enfermedad, trastorno o condición regulada o producida por la actividad de serina quinasa o relacionada con la actividad de quinasa dependiente de ciclina, en el que la enfermedad, trastorno o condición es cáncer.Compuesto o una sal o solvato farmacéuticamente aceptable del mismo para la utilización según la reivindicación 7, en el que la enfermedad, trastorno o condición se selecciona del grupo que consiste en cáncer pulmonar no microcítico, cáncer pulmonar microcítico, adenocarcinoma pulmonar, carcinoma pulmonar de células escamosas, cáncer pancreático, cáncer prostático, cáncer de vejiga, cáncer hepático, cáncer de piel, glioma, cáncer de mama, melanoma, glioma maligno, rabdomiosarcoma, cáncer ovárico, astrocitoma, sarcoma de Ewing, retinoblastoma, carcinoma de células epiteliales, cáncer de colon, cáncer renal, tumor estromal gastrointestinal, leucemia, linfoma histiocítico y carcinoma nasofaríngeo.Compuesto o una sal o solvato farmacéuticamente aceptable del mismo para la utilización según la reivindicación 7, en el que la enfermedad, trastorno o condición se selecciona del grupo que consiste en MDS-RAEB (síndrome mielodisplásico de tipo exceso de blastos), linfoma histiocítico, leucemia de células B aguda, leucemia megacarioblástica aguda, leucemia mieloide agua y leucemia promielocítica aguda.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201710257652.7A CN108727363B (zh) | 2017-04-19 | 2017-04-19 | 一种新型细胞周期蛋白依赖性激酶cdk9抑制剂 |
| PCT/CN2018/070108 WO2018192273A1 (zh) | 2017-04-19 | 2018-01-03 | 一种新型细胞周期蛋白依赖性激酶cdk9抑制剂 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2909301T3 true ES2909301T3 (es) | 2022-05-06 |
Family
ID=63856449
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES18787869T Active ES2909301T3 (es) | 2017-04-19 | 2018-01-03 | Nuevo inhibidor de la quinasa CDK9 dependiente de ciclina |
Country Status (12)
| Country | Link |
|---|---|
| US (1) | US10952999B2 (es) |
| EP (1) | EP3613737B1 (es) |
| JP (1) | JP6866967B2 (es) |
| KR (1) | KR102309986B1 (es) |
| CN (1) | CN108727363B (es) |
| AU (1) | AU2018253655B2 (es) |
| CA (1) | CA3059622C (es) |
| DK (1) | DK3613737T3 (es) |
| ES (1) | ES2909301T3 (es) |
| PT (1) | PT3613737T (es) |
| RU (1) | RU2738654C1 (es) |
| WO (1) | WO2018192273A1 (es) |
Families Citing this family (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN108727363B (zh) | 2017-04-19 | 2020-06-19 | 劲方医药科技(上海)有限公司 | 一种新型细胞周期蛋白依赖性激酶cdk9抑制剂 |
| US12384774B2 (en) | 2019-06-06 | 2025-08-12 | Genfleet Therapeutics (Shanghai) Inc. | Polymorph of CDK9 inhibitor and preparation method for polymorph and use thereof |
| US20220267321A1 (en) * | 2019-06-27 | 2022-08-25 | Medshine Discovery Inc. | Azaindole pyrazole compounds as cdk9 inhibitors |
| EP4140997A4 (en) | 2020-05-12 | 2023-09-06 | Suzhou Alphama Biotechnology Co., Ltd. | PYRIDINE ACETAMIDE DERIVATIVE AS CDK INHIBITOR, PREPARATION METHOD AND USE THEREOF |
| EP4141004A4 (en) * | 2020-05-12 | 2023-08-23 | Suzhou Alphama Biotechnology Co., Ltd. | POLYCYCLIC AMIDE DERIVATIVE SERVING AS A CDK9 INHIBITOR, METHOD FOR PREPARING IT AND ITS USE |
| US20230416221A1 (en) * | 2020-11-20 | 2023-12-28 | Hefei Institutes Of Physical Science, Chinese Academy Of Sciences | Dihydroisoquinolinone and isoindolinone derivatives and uses thereof |
| TWI809330B (zh) * | 2020-11-20 | 2023-07-21 | 大陸商勁方醫藥科技(上海)有限公司 | Cdk9抑制劑的多晶型物及其製法和用途 |
| TWI846516B (zh) * | 2020-11-20 | 2024-06-21 | 大陸商勁方醫藥科技(上海)有限公司 | Cdk9抑制劑的多晶型物及其製法和用途 |
| CN116270658B (zh) * | 2023-05-24 | 2023-10-27 | 劲方医药科技(上海)有限公司 | Cdk9抑制剂与btk抑制剂联用的药物组合及其用途 |
| CN116270644A (zh) * | 2023-05-22 | 2023-06-23 | 劲方医药科技(上海)有限公司 | Cdk9抑制剂与bcl-2抑制剂联用的药物组合及其用途 |
| WO2024239512A1 (zh) * | 2023-05-22 | 2024-11-28 | 劲方医药科技(上海)有限公司 | 药物组合及其在治疗癌症中的用途 |
| CN116496267B (zh) * | 2023-06-26 | 2023-09-22 | 劲方医药科技(上海)有限公司 | Cdk9抑制剂及其制备方法和用途 |
| WO2025217597A1 (en) | 2024-04-12 | 2025-10-16 | Sellas Life Sciences Group, Inc. | A cdk9 inhibitor for use in the treatment of cancer in a subject having an asxl1 mutation |
Family Cites Families (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN1333994A (zh) * | 1998-11-16 | 2002-01-30 | 伊利诺伊大学评议会 | 双路立体声信号处理技术 |
| CN100355751C (zh) * | 2000-03-29 | 2007-12-19 | 西克拉塞尔有限公司 | 2-取代的4-杂芳基-嘧啶、其组合物及其用途 |
| EP1389616B1 (en) * | 2001-04-27 | 2011-07-27 | Mitsubishi Tanabe Pharma Corporation | 3,4-Dihalobenzylpiperidine derivatives and their medical use |
| RU2008148962A (ru) * | 2006-05-12 | 2010-06-20 | Вертекс Фармасьютикалз Инкорпорейтед (Us) | Избирательно действующие ингибиторы rоск протеинкиназы и их применение |
| GB0805477D0 (en) | 2008-03-26 | 2008-04-30 | Univ Nottingham | Pyrimidines triazines and their use as pharmaceutical agents |
| CN103339110A (zh) * | 2011-01-28 | 2013-10-02 | 诺瓦提斯公司 | 作为cdk9抑制剂的取代的杂-联芳基化合物及其用途 |
| GB201206908D0 (en) * | 2012-04-19 | 2012-06-06 | Cancer Rec Tech Ltd | Therapeutic compounds |
| CN108727363B (zh) | 2017-04-19 | 2020-06-19 | 劲方医药科技(上海)有限公司 | 一种新型细胞周期蛋白依赖性激酶cdk9抑制剂 |
-
2017
- 2017-04-19 CN CN201710257652.7A patent/CN108727363B/zh active Active
-
2018
- 2018-01-03 RU RU2019133203A patent/RU2738654C1/ru active
- 2018-01-03 CA CA3059622A patent/CA3059622C/en active Active
- 2018-01-03 WO PCT/CN2018/070108 patent/WO2018192273A1/zh not_active Ceased
- 2018-01-03 US US16/606,136 patent/US10952999B2/en active Active
- 2018-01-03 AU AU2018253655A patent/AU2018253655B2/en active Active
- 2018-01-03 EP EP18787869.9A patent/EP3613737B1/en active Active
- 2018-01-03 DK DK18787869.9T patent/DK3613737T3/da active
- 2018-01-03 KR KR1020197033935A patent/KR102309986B1/ko active Active
- 2018-01-03 JP JP2019555490A patent/JP6866967B2/ja active Active
- 2018-01-03 ES ES18787869T patent/ES2909301T3/es active Active
- 2018-01-03 PT PT187878699T patent/PT3613737T/pt unknown
Also Published As
| Publication number | Publication date |
|---|---|
| CA3059622C (en) | 2021-09-07 |
| WO2018192273A1 (zh) | 2018-10-25 |
| RU2738654C1 (ru) | 2020-12-15 |
| PT3613737T (pt) | 2022-03-11 |
| AU2018253655A1 (en) | 2019-10-17 |
| EP3613737A4 (en) | 2020-08-26 |
| EP3613737A1 (en) | 2020-02-26 |
| DK3613737T3 (da) | 2022-03-07 |
| EP3613737B1 (en) | 2021-12-29 |
| JP2020517595A (ja) | 2020-06-18 |
| KR20200039616A (ko) | 2020-04-16 |
| CA3059622A1 (en) | 2018-10-25 |
| AU2018253655B2 (en) | 2020-12-17 |
| US20200078343A1 (en) | 2020-03-12 |
| CN108727363B (zh) | 2020-06-19 |
| BR112019021794A2 (pt) | 2020-05-05 |
| KR102309986B1 (ko) | 2021-10-12 |
| US10952999B2 (en) | 2021-03-23 |
| CN108727363A (zh) | 2018-11-02 |
| JP6866967B2 (ja) | 2021-04-28 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2909301T3 (es) | Nuevo inhibidor de la quinasa CDK9 dependiente de ciclina | |
| ES3011855T3 (en) | Tyk2 inhibitors and uses thereof | |
| AU2016245864B2 (en) | Substituted quinazoline compounds and methods of use thereof | |
| JP6954567B2 (ja) | 2−置換芳香族環−ピリミジン系誘導体及びその調製と医学的用途 | |
| AU2021203051A1 (en) | Inhibitors of cyclin-dependent kinase 7 (CDK7) | |
| KR20250070059A (ko) | 신규한 테트라헤테로사이클 화합물 | |
| BR112020026748A2 (pt) | Inibidores de quinases dependentes de ciclina | |
| AU2018286221B2 (en) | Aminopyrimidine compound, preparation method therefor and use thereof | |
| CA2926328A1 (en) | Substituted quinazolinyl and quinolinyl derivatives and pharmaceutical compositions thereof useful as inhibitors of kras g12c | |
| KR20140021350A (ko) | N2,n4-비스(4-(피페라진-1-일)페닐)피리미딘-2,4-디아민 유도체 또는 이의 약학적으로 허용가능한 염 및 이를 유효성분으로 함유하는 암의 예방 또는 치료용 약학적 조성물 | |
| BR112018014536B1 (pt) | Derivados de amônio, processo para sua preparação, composição farmacêutica contendo os mesmos, combinação e seu uso | |
| ES3033417T3 (en) | Cyclin-dependent kinase 7 (cdk7) non-covalent inhibitors | |
| WO2023224998A1 (en) | Inhibitors of parg | |
| WO2021074251A1 (en) | Pyrrolo[2,3-d]pyrimidine derivatives and their use in the treatment of cancer | |
| JP2025522274A (ja) | Pargの阻害剤 | |
| CA3054459A1 (en) | Azetidine derivative | |
| BR112019021794B1 (pt) | Inibidores de quinase dependente de ciclina cdk9, seus usos e composição farmacêutica | |
| HK40026502A (en) | Novel inhibitor of cyclin-dependent kinase cdk9 | |
| HK40026502B (en) | Novel inhibitor of cyclin-dependent kinase cdk9 | |
| ES2994690T3 (en) | N-linked macrocyclic 4-(pyrazol-5-yl)-indole derivatives as inhibitors of mcl-1 | |
| Chen et al. | Synthesis of One Novel Thieno [2, 3-D] Pyrimidine Derivative Bearing a Sulfonylurea Moiety | |
| HK40001081A (en) | Substituted 2, 4-diamino-quinoline derivatives for use in the treatment of proliferative diseases | |
| NZ712576B2 (en) | Dna-pk inhibitors |