ES2909793T3 - 4-fenilpiperidinas sustituidas, su preparación y uso - Google Patents
4-fenilpiperidinas sustituidas, su preparación y uso Download PDFInfo
- Publication number
- ES2909793T3 ES2909793T3 ES15785329T ES15785329T ES2909793T3 ES 2909793 T3 ES2909793 T3 ES 2909793T3 ES 15785329 T ES15785329 T ES 15785329T ES 15785329 T ES15785329 T ES 15785329T ES 2909793 T3 ES2909793 T3 ES 2909793T3
- Authority
- ES
- Spain
- Prior art keywords
- alkyl
- trifluoromethyl
- phenyl
- methanone
- mmol
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/4353—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems
- A61K31/437—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems the heterocyclic ring system containing a five-membered ring having nitrogen as a ring hetero atom, e.g. indolizine, beta-carboline
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/445—Non condensed piperidines, e.g. piperocaine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/445—Non condensed piperidines, e.g. piperocaine
- A61K31/451—Non condensed piperidines, e.g. piperocaine having a carbocyclic group directly attached to the heterocyclic ring, e.g. glutethimide, meperidine, loperamide, phencyclidine, piminodine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/445—Non condensed piperidines, e.g. piperocaine
- A61K31/4523—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems
- A61K31/454—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems containing a five-membered ring with nitrogen as a ring hetero atom, e.g. pimozide, domperidone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/445—Non condensed piperidines, e.g. piperocaine
- A61K31/4523—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems
- A61K31/4545—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems containing a six-membered ring with nitrogen as a ring hetero atom, e.g. pipamperone, anabasine
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/06—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Epidemiology (AREA)
- Ophthalmology & Optometry (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Plural Heterocyclic Compounds (AREA)
- Hydrogenated Pyridines (AREA)
Abstract
Un compuesto que tiene la estructura: **(Ver fórmula)** en donde R1, R2, R3, R4 y R5 son cada uno independientemente H, halógeno, CF3 o alquilo C1-C4, en donde dos o más de R1, R2, R3, R4 o R5 son distintos de H; R6 es H o halógeno; y B tiene la estructura: **(Ver fórmula)** en donde n es un número entero de 0-1; α, β, χ, δ, ε y φ están presentes o ausentes cada uno independientemente, y cuando están presentes cada uno es un enlace; Z1 es N; Z2 es N o NR7; en donde R7 es H, alquilo C1-C4 u oxetano; X es C o N; Y1, Y2, Y3 e Y4 son cada uno independientemente CR8, CH2, N o N-R9, en donde R8 es H, halógeno, OCH3, CN o CF3; y R9 es H, CN, oxetano, alquilo C1-C6, cicloalquilo C3-C6, (alquilo C1-C4)(cicloalquilo C3-C6), (alquilo C1-C6)-OCH3, (alquilo C1-C6)-CF3, C(O)-(alquilo C1-C6), C(O)2-(alquilo C1-C6), C(O)-NH2, C(O)NH-(alquilo C1-C6), C(O)-(arilo C6), C(O)- (heteroarilo C6), C(O)-pirrolidina, C(O)-piperidina, C(O)-piperazina, (alquilo C1-C6)-CO2H, (alquilo C1-C6)-CO2 (alquilo C1- C6) o SO2-(alquilo C1-C6), en donde cuando R1 es CF3, R2 es H, R3 es F, R4 es H, y R5 es H, o R1 es H, R2 es CF3, R3 es H, R4 es CF3, y R5 es H, o R1 es Cl, R2 es H, R3 es H, R4 es F, y R5 es H, o R1 es CF3, R2 es H, R3 es F, R4 es H, y R5 es H, o R1 es CF3, R2 es F, R3 es H, R4 es H, y R5 es H, o R1 es Cl, R2 es F, R3 es H, R4 es H, y R5 es H, entonces B es distinto de **(Ver fórmula)** o una sal farmacéuticamente aceptable de los mismos.
Description
DESCRIPCIÓN
4-fenilpiperidinas sustituidas, su preparación y uso
Campo técnico
La presente solicitud reivindica la prioridad de la Solicitud Provisional de EE. UU. No. 61/986.578, presentada el 30 de abril de 2014.
La invención se realizó con el apoyo del gobierno bajo los números de subvención NS067594 y NS074476 otorgados por los Institutos Nacionales de Salud. El gobierno tiene ciertos derechos en la invención.
Antecedentes de la invención
La degeneración macular asociada a la edad (DMAE) es la principal causa de ceguera en los países desarrollados. Se estima que 62,9 millones de personas en todo el mundo tienen la forma atrófica (seca) de DMAE más prevalente; 8 millones de ellos son estadounidenses. Debido al aumento de la esperanza de vida y la demografía actual, se espera que este número se triplique para el año 2020. Actualmente no existe un tratamiento aprobado por la Administración de Alimentos y Medicamentos (“FDA”, por sus siglas en inglés) para la DMAE seca. Dada la falta de tratamiento y la alta prevalencia, el desarrollo de fármacos para la DMAE seca es de suma importancia. Clínicamente, la DMAE atrófica representa un trastorno neurodegenerativo de progresión lenta en el que neuronas especializadas (fotorreceptores de conos y bastones) mueren en la parte central de la retina denominada mácula (1). Los estudios histopatológicos y de imagenología clínica indican que la degeneración de los fotorreceptores en la DMAE seca se desencadena por anomalías en el epitelio pigmentario de la retina (“RPE”, por sus siglas en inglés) que se encuentra debajo de los fotorreceptores y proporciona un soporte metabólico crítico para estas células neuronales sensibles a la luz. Los datos experimentales y clínicos indican que la acumulación excesiva de agregados de lípidos, proteínas y retinoides autofluorescentes citotóxicos (lipofuscina) en el RPE es un factor desencadenante importante de la DMAE seca (2-9). Además de DMAE, la acumulación dramática de lipofuscina es el sello distintivo de la enfermedad de Stargardt (“STGD”, por sus siglas en inglés), una forma hereditaria de degeneración macular de inicio juvenil. El principal componente citotóxico de la lipofuscina de RPE es el bisretinoide A2E de piridinio (Figura 1). Los bisretinoides citotóxicos adicionales son isoA2E, atRAL di-PE y A2-DHP-PE (40, 41). La formación de A2E y otros bisretinoides de lipofuscina, tales como A2-DHP-PE (A2-dihidropiridinafosfatidiletanolamina) y atRALdi-PE (dimerfosfatidiletanolamina todo-frans-retinal), comienza en las células fotorreceptoras de manera no enzimática y puede considerarse como un subproducto del correcto funcionamiento del ciclo visual.
A2E es un producto de la condensación de todo-frans-retinaldehído con fosfatidil-etanolamina que se produce en la retina de forma no enzimática y, como se ilustra en la Figura 4, puede considerarse un subproducto de un ciclo visual que funciona correctamente (10). La isomerización inducida por la luz del 11-c/s-retinaldehído a su forma todo-frans es el primer paso en una cascada de señalización que media la percepción de la luz. El ciclo visual es una cadena de reacciones bioquímicas que regeneran el pigmento visual (11-c/s-retinaldehído conjugado con opsina) después de la exposición a la luz.
Como los bisretinoides citotóxicos se forman durante el curso de un ciclo visual que funciona normalmente, la inhibición farmacológica parcial del ciclo visual puede representar una estrategia de tratamiento para la DMAE seca y otros trastornos caracterizados por una acumulación excesiva de lipofuscina (25-27, 40, 41).
Petrukhin et al. (WO 2013/166037) describe compuestos antagonistas no retinoides para su uso en el tratamiento de una enfermedad caracterizada por una acumulación excesiva de lipofuscina en la retina de un mamífero que la padece.
Breve descripción de la invención
La presente invención proporciona un compuesto que tiene la estructura:

en donde
Ri , R2, R3, R4 y R5 son cada uno independientemente H, halógeno, CF3 o alquilo C1-C4,
en donde dos o más de R1, R2, R3, R4 o R5 son distintos de H;
R6 es H o halógeno; y
B tiene la estructura:

en donde
n es un número entero de 0-1;
a, p, x, 5, £ y 9 están presentes o ausentes cada uno independientemente, y cuando están presentes cada uno es un enlace;
Z1 es N;
Z2 es N o NR7;
en donde R7 es H, alquilo C1-C4 u oxetano;
X es C o N;
Y1, Y2, Y3 e Y4 son cada uno independientemente CRs, CH2, N, o N-Rg;
en donde
R8 es H, halógeno, OCH3, CN o CF3; y
Rg es H, CN, oxetano, alquilo C1-C6, cicloalquilo C3-C6, (alquilo C1-C4) (cicloalquilo C3-C6), (alquilo C1-C6)-OCH3, (alquilo
C1-Ca)-CF3,C(O)-(alquilo C1-Ca), C(O)2-(alquilo C1-Ce), C(O)-NH2, C(O)NH-(alquilo C1-Ce), C(O)-(arilo Ce), C(O)-(heteroarilo Ca), C(O)-pirrolidina, C(O)-piperidina, C(O)-piperazina, (alquilo C1-Ca)-CO2H, (alquilo C1-Ca)-CO2 (alquilo C1-Ca) o SO2-(alquilo C1-Ca),
en donde cuando R1 es CF3, R2 es H, R3 es F, R4 es H, y R5 es H, o R1 es H, R2 es CF3, R3 es H, R4 es CF3, y R5 es H, o
R1 es Cl, R2 es H, R3 es H, R4 es F, y R5 es H, o R1 es CF3, R2 es H, R3 es F, R4 es H, y R5 es H, o R1 es CF3, R2 es F,
R3 es H, R4 es H, y R5 es H, o R1 es Cl, R2 es F, R3 es H, R4 es H, y R5 es H, entonces B es di

o una sal farmacéuticamente aceptable de los mismos.
Breve descripción de los dibujos
Figura 1. Estructura de bisretinoide A2E, un componente citotóxico de la lipofuscina retiniana.
Figura 2. Estructura del bisretinoide atRAL di-PE (todo-frans-retinal dímero-fosfatidil etanolamina), un componente citotóxico de la lipofuscina retiniana. R1 y R2 se refieren a varios constituyentes de ácidos grasos.
Figura 3. Estructura de bisretinoide A2-DHP-PE, un componente citotóxico de la lipofuscina retiniana.
Figura 4. Ciclo visual y biosíntesis de A2E. La biosíntesis de A2E comienza cuando una parte de todo frans-retinal escapa del ciclo visual (recuadro amarillo) y reacciona de forma no enzimática con la fosfatidil-etanolamina formando el precursor de A2E, A2-PE. La captación de retinol sérico en el RPE (recuadro gris) alimenta el ciclo.
Figura 5. Estructura tridimensional del complejo RBP4-TTR-retinol. La TTR tetrameica se muestra en azul, azul claro, verde y amarillo (región encuadrada grande). RBP se muestra en rojo (región sin recuadro) y el retinol se muestra en gris (región con recuadro pequeño) (28).
Figura 6. Estructura de fenretinida, [N-(4-hidroxi-fenil)retinamida, 4HRP], un antagonista del retinoide RBP4.
Figura 7. Representación esquemática del formato de ensayo basado en HTRF para la caracterización de antagonistas de RBP4 que interrumpen la interacción RBP4-TTR inducida por retinol.
Figura 8. Efecto del tratamiento con el compuesto 81 sobre la acumulación de bisretinoides en ojos de ratones Abca4-/-(P = 0,006; prueba t no apareada).
Figura 9. Niveles séricos de RBP4 en ratones Abca4-/- tratados con el compuesto 81 y el vehículo.
Descripción detallada de la invención
En la presente se describen compuestos que tienen la estructura:
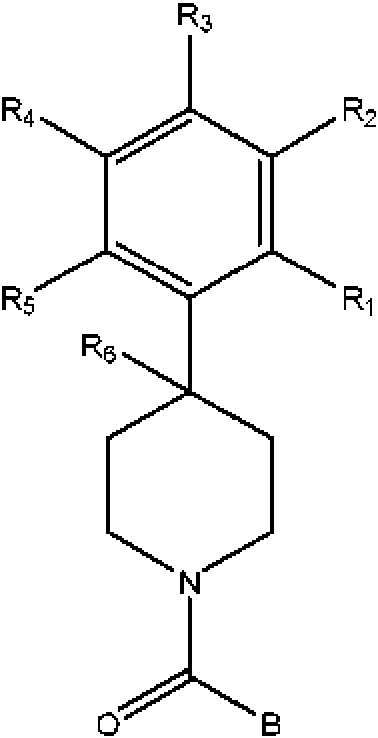
en donde
R1, R2, R3, R4 y R5 son cada uno independientemente H, halógeno, CF3 o alquilo C1-C4,
en donde dos o más de R1, R2, R3, R4 o R5 son distintos de H;
R6 es H, OH o halógeno; y
B es un heterobiciclo sustituido o no sustituido,
en donde cuando R1 es CF3, R2 es H, R3 es F, R4 es H, y R5 es H, o R1 es H, R2 es CF3, R3 es H, R4 es CF3, y R5 es H, o R1 es Cl, R2 es H, R3 es H, R4 es F, y R5 es H, o R1 es CF3, R2 es H, R3 es F, R4 es H, y R5 es H, o R1 es CF3, R2 es F, R3 es H, R4 es H, y R5 es H, o R1 es Cl, R2 es F, R3 es H, R4 es H, y R5 es H, entonces B es distinto de

o una sal farmacéuticamente aceptable de los mismos.
También se describen en la presente compuestos que tienen la estructura:

en donde
R1, R2, R3, R4 y R5 son cada uno independientemente H, halógeno, CF3 o alquilo C1-C4,
en donde dos o más de R1, R2, R3, R4 o R5 son distintos de H;
R6 es H, OH o halógeno; y
B es un heterobiciclo sustituido o no sustituido,
en donde cuando R1 es CF3, R2 es H, R3 es F, R4 es H, y R5 es H, o R1 es H, R2 es CF3, R3 es H, R4 es C R1 es Cl, R2 es H, R3 es H, R4 es F, y R5 es H, o R1 es CF3, R2 es H, R3 es F, R4 es H, y R5 es H, o R1 es CF3, R2 es F, R3 es H, R4 es H, y R5 es H, o R1 es Cl, R2 es F, R3 es H, R4 es H, y R5 es H, entonces B es distinto de

o una sal farmacéuticamente aceptable de los mismos.
En algunas realizaciones, el compuesto
en donde cuando R1 es CF3, R2 es H, R3 es F, R4 es H, y R5 es H, o R1 es H, R2 es CF3, R3 es H, R4 es CF3, y R5 es H, o
R1 es Cl, R2 es H, R3 es H, R4 es F, y R5 es H, o R1 es CF3, R2 es H, R3 es F, R4 es H, y R5 es H, o R1 es CF3, R2 es F, R3 es H, R4 es H, y R5 es H, o R1 es Cl, R2 es F, R3 es H, R4 es H, y R5 es H, entonces B es distinto de

En algunas realizaciones, el compuesto
en donde cuando R1 es CF3, R2 es H, R3 es F, R4 es H, y R5 es H, o R1 es H, R2 es CF3, R3 es H, R4 es CF3, R1 es Cl, R2 es H, R3 es H, R4 es F, y R5 es H, o R1 es CF3, R2 es H, R3 es F, R4 es H, y R5 es H, o R1 es CF3, R2 es F, R3 es H, R4 es H, y R5 es H, o R1 es Cl, R2 es F, R3 es H, R4 es H, y R5 es H, entonces B es distinto de
o una sal farmacéuticamente aceptable de los mismos.
En algunas realizaciones, el compuesto tiene la estructura:

En algunas realizaciones, el compuesto en donde
Ri , R2, R3, R4, R5 y R6 son cada uno independientemente H, Cl, F o CF3.
En algunas realizaciones, el compuesto en donde
R1 es CF3, R2 es F, R3 es F, R4 es H, y R5 es H, o
R1 es CF3, R2 es F, R3 es H, R4 es H, y R5 es H, o
R1 es CF3, R2 es F, R3 es H, R4 es F, y R5 es H, o
R1 es CF3, R2 es H, R3 es F, R4 es F, y R5 es H, o
R1 es CF3, R2 es H, R3 es H, R4 es H, y R5 es F, o
R1 es CF3, R2 es H, R3 es F, R4 es H, y R5 es H, o
R1 es CF3, R2 es H, R3 es H, R4 es Cl, y R5 es H, o
R1 es CF3, R2 es Cl, R3 es H, R4 es H, y R5 es H, o
R1 es H, R2 es CF3, R3 es H, R4 es CF3, y R5 es H, o
R1 es Cl, R2 es H, R3 es H, R4 es F, y R5 es H, o
R1 es Cl, R2 es F, R3 es H, R4 es H, y R5 es H.
También se describen en la presente compuestos en donde B tiene la estructura:

en donde
a, p, x y 6 están presentes o ausentes cada uno independientemente, y cuando están presentes, cada uno es un enlace; X es C o N;
Zi es N;
Z2 es N o NR7,
en donde R7 es H, alquilo C1-C4 u oxetano;
Q es una estructura de anillo de 5, 6 o 7 miembros sustituida o no sustituida.
También se describen en la presente compuestos en donde B tiene la estructura:

en donde
cuando a está presente, entonces Zi y Z2 son N, X es N, p está presente, y x y 6 están ausentes; y
cuando a está ausente, entonces Zi es N, Z2 es N-R7, X es C, p y 6 están presentes, y x está ausente.
En algunas realizaciones, el compuesto en donde B tiene la estructura:

en donde
n es un número entero de 0-2;
a, p, x, 6, £ y 9 están presentes o ausentes cada uno independientemente, y cuando están presentes cada uno es un enlace;
Zi es N;
Z2 es N o N-R7,
en donde R7 es H, alquilo C1-C10 u oxetano;
X es C o N; y
Yi , Y2, Y3, y cada ocurrencia de Y4 son cada uno independientemente CRs, CH2, o N-Rg,
en donde
R8 es H, halógeno, OCH3, CN o CF3; y
Rg es H, CN, oxetano, alquilo C1-C6, cicloalquilo C3-C6, (alquilo C1-C4) (cicloalquilo C3-C6), (alquilo Ci-C6)-OCH3, (alquilo Ci -Ca)-CF3, C(O)-(alquilo Ci -Ca), C(O)2-(alquilo Ci -Ce), C(O)-NH2 C(O)NH-(alquilo Ci-Ce), C(O)-(arilo Ce), C(O)-(Ce heteroarilo), C(O)-pirrolidina, C(O)-piperidina, C(O)-piperazina, (alquilo Ci -Ca)-CO2H, (alquilo Ci -Ca)-CO2(alquilo Ci-Ca) o SO2-(alquilo Ci-Ca).
En algunas realizaciones, el compuesto en donde B tiene la estructura:

en donde
n es 0;
R7 es H, alquilo Ci -C4 u oxetano;
Yi e Y3 son cada uno CH2; y
Y2 es N-Rg,
en donde
Rg es H, CN, oxetano, alquilo Ci-Ca, cicloalquilo C3-Ca, (alquilo Ci -C4) (cicloalquilo C3-Ca), (alquilo Ci-Ca)-OCH3, (alquilo Ci -Ca)-CF3, C(O)-(alquilo Ci -Ca), C(O)2-(alquilo C-Ca), C(O)-NH2 C(O)NH-(alquilo Ci-Ca), C(O)-(arilo Ca), C(O)-(Ca heteroarilo), C(O)-pirrolidina, C(O)-piperidina, C(O)-piperazina, (alquilo Ci -Ca)-CO2H, (alquilo Ci -Ca)-CO2(alquilo Ci-Ca) o SO2-(alquilo Ci-Ca).
En algunas realizaciones, el compuesto en donde B tiene la estructura:

en donde
n es 1;
R7 es H, alquilo C1-C4 u oxetano;
Y1, Y2 e Y4 son cada uno CH2; y
Y3 es N-R9,
en donde
R9 es H, CN, oxetano, alquilo C1-C6, cicloalquilo C3-C6, (alquilo C1-C4) (cicloalquilo C3-C6), (alquilo C1- Ca)-OCH3, (alquilo C1-Ca)-CF3, C(O)-(alquilo C1-C6), C(O)2-(alquilo Cr Ca), C(O)-NH2 C(O)NH-(alquilo C1- Ca), C(O)-(arilo Ca), C(O)-(heteroarilo Ca), C(O)-pirrolidina, C(O)-piperidina, C(O)-piperazina, (alquilo C1-Ca)-CO2H, (alquilo C1-Ca)-CO2(alquilo C1-Ca) o SO2-(alquilo C1-Ca).
En algunas realizaciones, el compuesto en donde B tiene la estructura:

en donde
n es 1;
R7 es H, alquilo C1-C4 u oxetano;
Y1, Y3 e Y4 son cada uno CH2; y
Y2 es N-R9,
en donde
R9 es H, CN, oxetano, alquilo C1-Ca, cicloalquilo C3-Ca, (alquilo C1-C4) (cicloalquilo C3-Ca), (alquilo C1-Ca)-OCH3, (alquilo C1-Ca)-CF3, C(O)-(alquilo C1-Ca), C(O)2-(alquilo C1-Ca), C(O)-NH2 C(O)NH-(alquilo C1- Ca), C(O)-(arilo Ca), C(O)-(heteroarilo Ca), C(O)-pirrolidina, C(O)-piperidina, C(O)-piperazina, (alquilo C1-Ca)-CO2H, (alquilo C1-Ca)-CO2(alquilo C1-Ca) o SO2-(alquilo C1-Ca).
En algunas realizaciones, el compuesto en donde B tiene la estructura:

En algunas realizaciones, el compuesto en donde
R9 es H, CN, CH3, CH2CH3, CH2CH2CH3, CH(CH3)2, CH2CH(CH3)2, t-Bu, CH2CH(CH3)2, CH2C(CH3)3, CH2CF3, CH2CH2CF3, CH2OCH3, CH2CH2OCH3,

En algunas realizaciones, el compuesto en donde
Rg es SO2-CH3, C(O)-CH3, C(O)-CHgCH3, C(O)-CHgCHgCH3, C(O)-CH(CH3)2, C(O)-CH2CH(CH3)2, C(O)-t-Bu, C(O)-OCH3, C(O)-NHCH3,

En algunas realizaciones, el compuesto en donde
R7 es H, CH3, CH2CH3, CH(CH3)2, o

En algunas realizaciones, el compuesto en donde B tiene la estructura:

en donde
Y1, Y2, Y3 e Y4 son cada uno independientemente CRs o N,
en donde cada R8 es independientemente H, halógeno, OCH3, CN o CF3.
En algunas realizaciones, el compuesto en donde B tiene la estructura:

En algunas realizaciones, el compuesto en donde cada R8 es CN u OCH3.
En algunas realizaciones, el compuesto tiene la estructura:





o una sal farmacéuticamente aceptable del compuesto.
En algunas realizaciones, el compuesto en donde la estructura:

o una sal farmacéuticamente aceptable del compuesto.
En algunas realizaciones, el compuesto tiene la estructura:

,
N
NH
0 Y N V NH
o una sal farmacéuticamente aceptable del compuesto.
La presente invención proporciona una composición farmacéutica que comprende cualquiera de los compuestos anteriores y un portador farmacéuticamente aceptable.
La presente invención proporciona compuestos o composiciones para usar en el tratamiento de una enfermedad caracterizada por una acumulación excesiva de lipofuscina en la retina en un sujeto que la padece, que comprende administrar al sujeto una cantidad efectiva de cualquiera de los compuestos anteriores.
La presente invención proporciona una composición farmacéutica que comprende el compuesto de la presente invención y un portador farmacéuticamente aceptable.
La presente invención proporciona compuestos o composiciones para usar en el tratamiento de una enfermedad caracterizada por una acumulación excesiva de lipofuscina en la retina en un sujeto afectado que comprende administrar al sujeto una cantidad efectiva del compuesto de la presente invención o una composición de la presente invención. En algunas realizaciones, la enfermedad se caracteriza además por degeneración macular mediada por bisretinoides.
En algunas realizaciones, el bisretinoide es A2E. En algunas realizaciones, el bisretinoide es isoA2E. En algunas realizaciones, el bisretinoide es A2-DHP-PE. En algunas realizaciones, el bisretinoide es atRAL di-PE.
En algunas realizaciones, la enfermedad caracterizada por una acumulación excesiva de lipofuscina en la retina es la degeneración macular asociada a la edad.
En algunas realizaciones, la enfermedad caracterizada por una acumulación excesiva de lipofuscina en la retina es la degeneración macular asociada a la edad seca (atrófica).
En algunas realizaciones, la enfermedad caracterizada por una acumulación excesiva de lipofuscina en la retina es la enfermedad de Stargardt.
En algunas realizaciones, la enfermedad caracterizada por una acumulación excesiva de lipofuscina en la retina es la enfermedad de Best.
En algunas realizaciones, la enfermedad caracterizada por una acumulación excesiva de lipofuscina en la retina es la maculopatía viteliforme del adulto.
En algunas realizaciones, la enfermedad caracterizada por una acumulación excesiva de lipofuscina en la retina es la distrofia macular tipo Stargardt.
En algunas realizaciones, el sujeto es un mamífero. En algunas realizaciones, el mamífero es un ser humano.
En algunas realizaciones, Rg es H, alquilo C1-C4, cicloalquilo C3-C6, (alquilo C1- C4)-CF3, (alquilo C-i-C4)-OCH3, (alquilo C1-C4)-halógeno, SO2-(alquilo C1-C4), SO2-(alquilo Ci -C4)-CF3, SO2-(alquilo C1-C4HOCH3, S o2-(alquilo Ci -C4)-halógeno, C(O)-(alquilo C1-C4), C(O)-(alquilo C1-C4)-CF3, C(O)-(alquilo C1-C4HOCH3, C(O)-(alquilo C1-C4)-halógeno, C(O)-NH-(alquilo C1-C4), C(o)-N(alquilo C1-C4)2, (alquilo C1-C4)-C(O)OH, C(O)-NH2 u oxetano.
En algunas realizaciones, Rg es H, CH3, CH2CH3, CH2CH2CH3, CH(CH3)2, CH2CH(CH3)2, t-Bu, CH2OCH3, CH2CF3, CH2Cl, CH2F, CH2CH2OCH3, CH2CH2CF3, CH2CH2CI, CH2CH2F, o


o una sal farmacéuticamente aceptable de los mismos.
En algunas realizaciones del compuesto, B tiene la estructura:











La presente invención proporciona una composición farmacéutica que comprende un compuesto de la presente invención y un portador farmacéuticamente aceptable.
La presente invención proporciona compuestos o composiciones para usar en el tratamiento de una enfermedad caracterizada por una acumulación excesiva de lipofuscina en la retina en un mamífero que la padece.
En algunas realizaciones de los compuestos o composiciones para uso, la enfermedad se caracteriza además por degeneración macular mediada por bisretinoides.
En algunas realizaciones de los compuestos o composiciones para uso, el bisretinoide es A2E. En algunas realizaciones de los compuestos o composiciones para uso, el bisretinoide es isoA2E. En algunas realizaciones de los compuestos o composiciones para uso, el bisretinoide es A2-DHP-PE. En algunas realizaciones de los compuestos o composiciones para uso, el bisretinoide es atRAL di-PE.
En algunas realizaciones de los compuestos o composiciones para uso, la enfermedad caracterizada por una acumulación excesiva de lipofuscina en la retina es la degeneración macular asociada a la edad.
En algunas realizaciones de los compuestos o composiciones para el uso, la enfermedad caracterizada por una acumulación excesiva de lipofuscina en la retina es la degeneración macular asociada a la edad seca (atrófica).
En algunas realizaciones de los compuestos o composiciones para uso, la enfermedad caracterizada por una acumulación excesiva de lipofuscina en la retina es la enfermedad de Stargardt.
En algunas realizaciones de los compuestos o composiciones para uso, la enfermedad caracterizada por una acumulación excesiva de lipofuscina en la retina es la enfermedad de Best.
En algunas realizaciones de los compuestos o composiciones para uso, la enfermedad caracterizada por una acumulación excesiva de lipofuscina en la retina es la maculopatía viteliforme del adulto.
En algunas realizaciones de los compuestos o composiciones para uso, la enfermedad caracterizada por una acumulación excesiva de lipofuscina en la retina es la distrofia macular tipo Stargardt.
En algunas realizaciones, la degeneración macular mediada por bisretinoides es degeneración macular asociada a la edad o enfermedad de Stargardt.
En algunas realizaciones, la degeneración macular mediada por bisretinoides es degeneración macular asociada a la edad.
En algunas realizaciones, la degeneración macular mediada por bisretinoides es degeneración macular asociada a la edad seca (atrófica).
En algunas realizaciones, la degeneración macular mediada por bisretinoides es la enfermedad de Stargardt.
En algunas realizaciones, la degeneración macular mediada por bisretinoides es la enfermedad de Best.
En algunas realizaciones, la degeneración macular mediada por bisretinoides es la maculopatía viteliforme del adulto. En algunas realizaciones, la degeneración macular mediada por bisretinoides es la distrofia macular tipo Stargardt. La degeneración macular mediada por bisretinoides puede comprender la acumulación de depósitos de lipofuscina en el epitelio pigmentario de la retina.
Como se usa en la presente, “lipofuscina bisretinoide” es lipofuscina que contiene un bisretinoide citotóxico. Los bisretinoides citotóxicos incluyen, entre otros, A2E, isoA2E, atRAL di-PE y A2-DHP-PE (Figura 1,2 y 3).
La presente invención proporciona compuestos de piperidina sin retinol que comprenden un resto 3,4-difluoro-2-(trifluorometil)fenilo. Esta característica aumenta significativamente la potencia y mejora las características farmacocinéticas de las moléculas.
La presente invención proporciona compuestos de piperidina sin retinol que comprenden un resto 3,5-difluoro-2-(trifluorometil)fenilo. Esta característica aumenta significativamente la potencia y mejora las características farmacocinéticas de las moléculas.
La presente invención proporciona compuestos de piperidina sin retinol que comprenden un resto fenilo di- o tri-sustituido. Esta característica aumenta significativamente la potencia y mejora las características farmacocinéticas de las moléculas. Salvo que se especifique lo contrario, cuando la estructura de un compuesto de esta invención incluye un átomo de carbono asimétrico, se entiende que el compuesto se presenta como un racemato, una mezcla racémica y un enantiómero único aislado. Todas estas formas isoméricas de estos compuestos se incluyen expresamente en esta invención. Salvo que se especifique lo contrario, cada carbono estereogénico puede tener la configuración R o S. En consecuencia, debe entenderse que los isómeros que surgen de tal asimetría (por ejemplo, todos los enantiómeros y diastereómeros) están incluidos dentro del alcance de esta invención, a menos que se indique lo contrario. Dichos isómeros se pueden obtener en forma sustancialmente pura mediante técnicas clásicas de separación y mediante síntesis estereoquímicamente controlada, tales como las descritas en “Enantiomers, Racemates and Resolutions” por J. Jacques, A. Collet & S. Wilen, Pub. John Wiley & Sons, NY, 1981. Por ejemplo, la resolución puede realizarse mediante cromatografía preparativa en una columna quiral.
El objeto de la invención también pretende incluir todos los isótopos de átomos que se encuentran en los compuestos descritos en la presente. Los isótopos incluyen aquellos átomos que tienen el mismo número atómico, pero diferente número de masa. A modo de ejemplo general y sin limitación, los isótopos de hidrógeno incluyen tritio y deuterio. Los isótopos de carbono incluyen C -13 y C -14.
Se observará que cualquier notación de un carbono en estructuras a lo largo de esta solicitud, cuando se usa sin notación adicional, pretende representar todos los isótopos de carbono, tales como 12C, 13C o 14C. Además, cualquier compuesto que contenga 13C o 14C puede tener específicamente la estructura de cualquiera de los compuestos descritos en la presente.
También se observará que cualquier notación de hidrógeno en estructuras a lo largo de esta solicitud, cuando se usa sin notación adicional, pretende representar todos los isótopos de hidrógeno, tales como 1H, 2H o 3H. Además, cualquier compuesto que contenga 2H o 3H puede tener específicamente la estructura de cualquiera de los compuestos descritos en la presente.
Los compuestos marcados con isótopos generalmente se pueden preparar mediante técnicas convencionales conocidas por los expertos en la técnica usando reactivos etiquetados con isótopos apropiados en lugar de los reactivos no etiquetados empleados.
El término “sustitución”, “sustituido” y “sustituyente” se refiere a un grupo funcional como se describió anteriormente en el que uno o más enlaces a un átomo de hidrógeno contenido en él se reemplazan por un enlace a átomos que no son de hidrógeno ni de carbono, siempre que se mantienen las valencias normales y que la sustitución da como resultado un compuesto estable. Los grupos sustituidos también incluyen grupos en los que uno o más enlaces a un átomo de carbono o hidrógeno se reemplazan por uno o más enlaces, incluyendo enlaces dobles o triples, a un heteroátomo. Los ejemplos de grupos sustituyentes incluyen los grupos funcionales descritos anteriormente y halógenos (es decir, F, Cl, Br e I); grupos alquilo, tales como metilo, etilo, n-propilo, isopropilo, n-butilo, terc-butilo, y trifluorometilo; hidroxilo; grupos alcoxi, tales como metoxi, etoxi, n-propoxi e isopropoxi; grupos ariloxi, tales como fenoxi; arilalquiloxi, tales como benciloxi (fenilmetoxi) y p-trifluorometilbenciloxi (4-trifluorometilfenil-metoxi); grupos heteroariloxi; grupos sulfonilo, tales como trifluorometanosulfonilo, metanosulfonilo y p-toluenosulfonilo; nitro, nitrosilo; mercapto; grupos sulfanilo, tales como metilsulfanilo, etilsulfanilo y propilsulfanilo; ciano; grupos amino, tales como amino, metilamino, dimetilamino, etilamino y dietilamino; y carboxilo. Cuando se describen o reivindican múltiples restos sustituyentes, el compuesto sustituido puede sustituirse independientemente por uno o más de los restos sustituyentes descritos o reivindicados, en singular o en plural. Por sustituidos independientemente, se entiende que los (dos o más) sustituyentes pueden ser iguales o diferentes.
En los compuestos usados en la presente invención, los sustituyentes pueden estar sustituidos o no sustituidos, a menos que se defina específicamente lo contrario.
En los compuestos usados en la presente invención, los grupos alquilo, heteroalquilo, monociclo, biciclo, arilo, heteroarilo y heterociclo pueden sustituirse adicionalmente reemplazando uno o más átomos de hidrógeno con grupos alternativos que no son hidrógeno. Estos incluyen, pero no se limitan a, halo, hidroxi, mercapto, amino, carboxi, ciano y carbamoílo.
Se entiende que los sustituyentes y los patrones de sustitución de los compuestos usados en la presente invención pueden ser seleccionados por un experto en la técnica para proporcionar compuestos que sean químicamente estables y que puedan sintetizarse fácilmente mediante técnicas conocidas en el arte a partir de materiales de partida fácilmente disponibles. Si un mismo sustituyente está sustituido con más de un grupo, se entiende que estos múltiples grupos pueden estar en el mismo carbono o en diferentes carbonos, siempre que de como resultado una estructura estable.
Al elegir los compuestos utilizados en la presente invención, un experto normal en la técnica reconocerá que los diversos sustituyentes, es decir, R1, R2, etc., deben elegirse de conformidad con principios bien conocidos de conectividad estructural química.
Como se usa en la presente, “alquilo” pretende incluir grupos hidrocarbonados alifáticos saturados de cadena lineal y ramificada que tienen el número especificado de átomos de carbono. Por lo tanto, C1-Cn como en “alquilo C1-Cn” se define para incluir grupos que tienen 1, 2..., n-1 o n carbonos en una disposición lineal o ramificada, y específicamente incluye metilo, etilo, propilo, butilo, pentilo, hexilo, heptilo, isopropilo, isobutilo, secbutilo, y así sucesivamente. Una realización puede ser alquilo C1-C12, alquilo C2-C12, alquilo C3-C12, alquilo C4-C12, y así sucesivamente. Una realización puede ser alquilo C1-C8, alquilo C2-C8, alquilo C3-C8, alquilo C4-C8, y así sucesivamente. “Alcoxi” representa un grupo alquilo como se describió anteriormente unido a través de un puente de oxígeno.
El término “alquenilo” se refiere a un radical hidrocarburo no aromático, lineal o ramificado, que contiene al menos 1 enlace doble carbono-carbono, y puede estar presente hasta el máximo número posible de dobles enlaces carbono-carbono no aromáticos. Por lo tanto, alquenilo C2-Cn se define para incluir grupos que tienen 1,2...., n-1 o n carbonos. Por ejemplo, “alquenilo C2-C6” significa un radical alquenilo que tiene 2, 3, 4, 5 o 6 átomos de carbono y al menos 1 enlace doble carbono-carbono y hasta, por ejemplo, 3 enlaces dobles carbono-carbono en el caso de un alquenilo C6, respectivamente. Los grupos alquenilo incluyen etenilo, propenilo, butenilo y ciclohexenilo. Como se describió anteriormente con respecto al alquilo, la porción lineal, ramificada o cíclica del grupo alquenilo puede contener enlaces dobles y puede estar sustituido si se indica un grupo alquenilo sustituido. Una realización puede ser alquenilo C2-C12 o alquenilo C2-C8.
El término “alquinilo” se refiere a un radical hidrocarbonado lineal o ramificado, que contiene al menos 1 enlace triple carbono-carbono, y puede estar presente hasta el máximo número posible de enlaces triples carbono-carbono no aromáticos. Por lo tanto, alquinilo C2-Cn se define para incluir grupos que tienen 1, 2...., n-1 o n carbonos. Por ejemplo, “alquinilo C2-C6” significa un radical alquinilo que tiene 2 o 3 átomos de carbono y 1 enlace triple carbono-carbono, o que tiene 4 o 5 átomos de carbono y hasta 2 enlaces triples carbono-carbono, o que tiene 6 átomos de carbono y hasta 3 enlaces triples carbono-carbono. Los grupos alquinilo incluyen etinilo, propinilo y butinilo. Como se describió anteriormente con respecto al alquilo, la porción lineal o ramificada del grupo alquinilo puede contener enlaces triples y puede estar sustituido si se indica un grupo alquinilo sustituido. Una realización puede ser un alquinilo C2-Cn. Una realización puede ser alquinilo C2-C12 o alquinilo C3-C8.
Los grupos alquilo pueden estar no sustituidos o sustituidos con uno o más sustituyentes, incluyendo, entre otros, halógeno, alcoxi, alquiltio, trifluorometilo, difluorometilo, metoxi e hidroxilo.
Como se usa en la presente, “alquilo C1-C4” incluye alquilo C1-C4 tanto de cadena lineal como ramificada.
Como se usa en la presente, “heteroalquilo” incluye grupos hidrocarbonados alifáticos saturados tanto de cadena lineal como ramificada que tienen al menos 1 heteroátomo dentro de la cadena o ramificación.
Como se usa en la presente, “cicloalquilo” incluye anillos cíclicos de alcanos de tres a ocho átomos de carbono totales, o cualquier número dentro de este rango (es decir, ciclopropilo, ciclobutilo, ciclopentilo, ciclohexilo, cicloheptilo o ciclooctilo).
Como se usa en la presente, “heterocicloalquilo” pretende significar un anillo no aromático de 5 a 10 miembros que contiene de 1 a 4 heteroátomos seleccionados del grupo que consiste en O, N y S, e incluye grupos bicíclicos. Por lo tanto, “heterociclilo” incluye, pero no se limita a, lo siguiente: imidazolilo, piperazinilo, piperidinilo, pirrolidinilo, morfolinilo, tiomorfolinilo, tetrahidropiranilo, dihidropiperidinilo, tetrahidrotiofenilo y similares. Si el heterociclo contiene nitrógeno, se entiende que los correspondientes N-óxidos del mismo también están incluidos en esta definición.
Como se usa en la presente, “arilo” significa cualquier anillo de carbono monocíclico, bicíclico o policíclico estable de hasta 10 átomos en cada anillo, en donde al menos un anillo es aromático y puede estar sustituido o no sustituido. Los ejemplos de tales elementos arilo incluyen, pero no se limitan a: fenilo, p-toluenilo (4-metilfenilo), naftilo, tetrahidronaftilo, indanilo, fenantrilo, antrilo o acenaftilo. En los casos en los que el sustituyente arilo es bicíclico y un anillo no es aromático, se entiende que la unión se realiza a través del anillo aromático.
El término “alquilarilo” se refiere a grupos alquilo como se describió anteriormente en donde uno o más enlaces con el hidrógeno contenido en ellos se reemplazan por un enlace con un grupo arilo como se describió anteriormente. Se entiende que un grupo “alquilarilo” está conectado a una molécula central a través de un enlace del grupo alquilo y que el grupo arilo actúa como sustituyente del grupo alquilo. Los ejemplos de restos arilalquilo incluyen, pero no se limitan a, bencilo (fenilmetilo), p-trifluorometilbencilo (4-trifluorometilfenilmetilo), 1 -feniletilo, 2-feniletilo, 3-fenilpropilo, 2-fenilpropilo y similares.
El término “heteroarilo”, como se usa en la presente, representa un anillo monocíclico, bicíclico o policíclico estable de hasta 10 átomos en cada anillo, en donde al menos un anillo es aromático y contiene de 1 a 4 heteroátomos seleccionados del grupo que consiste en O, N y S. Los grupos heteroarilo aromáticos bicíclicos incluyen, entre otros, anillos de fenilo, piridina, pirimidina o piridizina que están (a) fusionados con un anillo heterocíclico aromático (insaturado) de 6 miembros que tiene un átomo de nitrógeno; (b) fusionados con un anillo heterocíclico aromático (insaturado) de 5 o 6 miembros que tiene dos átomos de nitrógeno; (c) fusionados con un anillo heterocíclico aromático (insaturado) de 5 miembros que tiene un átomo de nitrógeno junto con un átomo de oxígeno o uno de azufre; o (d) fusionados con un anillo heterocíclico aromático (insaturado) de 5 miembros que tiene un heteroátomo seleccionado de O, N o S. Los grupos heteroarilo dentro del alcance de esta definición incluyen pero no se limitan a: benzoimidazolilo, benzofuranilo, benzofurazanilo, benzopirazolilo, benzotriazolilo, benzotiofenilo, benzoxazolilo, carbazolilo, carbolinilo, cinolinilo, furanilo, indolinilo, indolilo, indolacinilo, indazolilo, isobenzofuranilo, isoindolilo, isoquinolilo, isotiazolilo, isoxazolilo, naftpiridinilo, oxadiazolilo, oxazolilo, oxazolina, isoxazolina, oxetanilo, piranilo, pirazinilo, pirazolilo, piridazinilo, piridopiridinilo, piridazinilo, piridilo, pirimidilo, pirrolilo, quinazolinilo, quinolilo, quinoxalinilo, tetrazolilo, tetrazolopiridilo, tiadiazolilo, tiazolilo, tienilo, triazolilo, azetidinilo, aziridinilo, 1,4-dioxanilo, hexahidroazepinilo, dihidrobenzoimidazolilo, dihidrobenzofuranilo, dihidrobenzotiofenilo, dihidrobenzoxazolilo, dihidrofuranilo, dihidroimidazolilo, dihidroindolilo, dihidroisooxazolilo, dihidroisotiazolilo, dihidrooxadiazolilo, dihidrooxazolilo, dihidropirazinilo, dihidropirazolilo, dihidropiridinilo, dihidropirimidinilo, dihidropirrolilo, dihidroquinolinilo, dihidrotetrazolilo, dihidrotiadiazolilo, dihidrotiazolilo, dihidrotienilo, dihidrotriazolilo, dihidroazetidinilo, metilendioxibenzoilo, tetrahidrofuranilo, tetrahidrotienilo, acridinilo, carbazolilo, cinnolinilo, quinoxalinilo, pirrazolilo, indolilo, benzotriazolilo, benzotiazolilo, benzoxazolilo, isoxazolilo, isotiazolilo, furanilo, tienilo, benzotienilo, benzofuranilo, quinolinilo, isoquinolinilo, oxazolilo, isoxazolilo, indolilo, pirazinilo, piridazinilo, piridinilo, pirimidinilo, pirrolilo, tetrahidroquinolina. En los casos en los que el sustituyente heteroarilo es bicíclico y un anillo no es aromático o no contiene heteroátomos, se entiende que la unión se realiza mediante el anillo aromático o mediante el anillo que contiene heteroátomos, respectivamente. Si el heteroarilo contiene átomos de nitrógeno, se entiende que los correspondientes N-óxidos del mismo también están incluidos en esta definición.
Como se usa en la presente, “monociclo” incluye cualquier anillo de carbono policíclico estable de hasta 10 átomos y puede estar sustituido o no sustituido. Los ejemplos de tales elementos de monociclo no aromáticos incluyen, pero no se limitan a: ciclobutilo, ciclopentilo, ciclohexilo y cicloheptilo. Los ejemplos de dichos elementos de monociclo aromático incluyen, pero no se limitan a: fenilo. Como se usa en la presente, “heteromonociclo” incluye cualquier monociclo que contenga al menos un heteroátomo.
Como se usa en la presente, “biciclo” incluye cualquier anillo de carbono policíclico estable de hasta 10 átomos que está fusionado con un anillo de carbono policíclico de hasta 10 átomos, estando cada anillo independientemente sustituido o no sustituido. Los ejemplos de tales elementos biciclos no aromáticos incluyen, pero no se limitan a: decahidronaftaleno. Los ejemplos de tales elementos biciclos aromáticos incluyen, pero no se limitan a: naftaleno. Como se usa en la presente, “heterobiciclo” incluye cualquier biciclo que contenga al menos un heteroátomo.
El término “fenilo” pretende significar un anillo aromático de seis miembros que contiene seis carbonos y cualquier derivado sustituido del mismo.
El término “bencilo” pretende significar un metileno unido directamente a un anillo de benceno. Un grupo bencilo es un grupo metilo en donde un hidrógeno se reemplaza con un grupo fenilo y cualquier derivado sustituido del mismo.
El término “piridina” pretende significar un heteroarilo que tiene un anillo de seis miembros que contiene 5 átomos de carbono y 1 átomo de nitrógeno, y cualquier derivado sustituido del mismo.
El término “pirazol” pretende significar un heteroarilo que tiene un anillo de cinco miembros que contiene tres átomos de carbono y dos átomos de nitrógeno en donde los átomos de nitrógeno son adyacentes entre sí, y cualquier derivado sustituido del mismo.
El término “indol” pretende significar un heteroarilo que tiene un anillo de cinco miembros condensado con un anillo de fenilo con el anillo de cinco miembros que contiene 1 átomo de nitrógeno unido directamente al anillo de fenilo.
El término “oxatano” pretende significar un anillo de cuatro miembros no aromático que contiene tres átomos de carbono y un átomo de oxígeno, y cualquier derivado sustituido del mismo.
Los compuestos utilizados en la presente invención se pueden preparar mediante técnicas bien conocidas en la síntesis orgánica y familiares para un experto en la materia. Sin embargo, estos pueden no ser los únicos medios para sintetizar
u obtener los compuestos deseados.
Los compuestos de la presente invención pueden prepararse mediante técnicas descritas en Vogel's Textbook of Practical Organic Chemistry, A.I. Vogel, A.R. Tatchell, B.S. Furnis, A.J. Hannaford, P.W.G. Smith, (Prentice Hall) 5a edición (1996), March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, Michael B. Smith, Jerry March, (Wileylnterscience) 5a edición (2007), y las referencias que contienen. Sin embargo, estos pueden no ser los únicos medios para sintetizar u obtener los compuestos deseados.
Los compuestos de la presente invención pueden prepararse mediante técnicas descritas en la presente. Los métodos sintéticos usados para preparar los Ejemplos 1-103 se usan para preparar compuestos de piperidina adicionales que se describen en las realizaciones de la presente.
Los diversos grupos R unidos a los anillos aromáticos de los compuestos descritos en la presente pueden añadirse a los anillos mediante procedimientos estándar, por ejemplo, los establecidos en Advanced Organic Chemistry: Part B: Reaction and Synthesis, Francis Carey and Richard Sundberg, (Springer) 5a edición. (2007).
Otro aspecto de la invención comprende un compuesto de la presente invención como una composición farmacéutica.
Como se usa en la presente, el término “agente farmacéuticamente activo” significa cualquier sustancia o compuesto adecuado para la administración a un sujeto y proporciona actividad biológica u otro efecto directo en el tratamiento, cura, mitigación, diagnóstico o prevención de enfermedades, o afecta la estructura o cualquier función del sujeto. Los agentes farmacéuticamente activos incluyen, entre otros, sustancias y compuestos descritos en Physicians' Desk Reference (PDR Network, LLC; 64a edición; 15 de noviembre de 2009) y “Approved Drug Products with Therapeutic Equivalence Evaluations” (Departamento de Salud y Servicios Humanos de EE. UU., 30a edición, 2010). Los agentes farmacéuticamente activos que tienen grupos pendientes de ácido carboxílico pueden modificarse de acuerdo con la presente invención usando reacciones de esterificación estándar y métodos fácilmente disponibles y conocidos por los expertos en la técnica de la síntesis química. Cuando un agente farmacéuticamente activo no posee un grupo de ácido carboxílico, el experto en la materia podrá diseñar e incorporar un grupo de ácido carboxílico en el agente farmacéuticamente activo en donde la esterificación puede llevarse a cabo posteriormente siempre que la modificación no interfiera con la actividad o efecto biológico del agente farmacéuticamente activo.
Los compuestos de la presente invención pueden estar en forma de una sal. Como se usa en la presente, una “sal” es una sal de los presentes compuestos que se ha modificado haciendo sales ácidas o básicas de los compuestos. En el caso de compuestos usados para tratar una enfermedad, la sal es farmacéuticamente aceptable. Los ejemplos de sales farmacéuticamente aceptables incluyen, pero no se limitan a, sales de ácidos minerales u orgánicos de residuos básicos, tales como aminas; sales alcalinas u orgánicas de residuos ácidos, tales como fenoles. Las sales se pueden preparar usando un ácido orgánico o inorgánico. Tales sales de ácido son cloruros, bromuros, sulfatos, nitratos, fosfatos, sulfonatos, formiatos, tartratos, maleatos, malatos, citratos, benzoatos, salicilatos, ascorbatos y similares. Las sales de fenolato son las sales de metales alcalinotérreos, sodio, potasio o litio. El término “sal farmacéuticamente aceptable” a este respecto, se refiere a las sales de adición de ácido o base inorgánicas y orgánicas, relativamente no tóxicas, de los compuestos de la presente invención. Estas sales se pueden preparar in situ durante el aislamiento final y la purificación de los compuestos de la invención, o haciendo reaccionar por separado un compuesto purificado de la invención en su forma de base libre o ácido libre con una base o ácido orgánico o inorgánico adecuado, y aislando la sal así formada. Las sales representativas incluyen bromhidrato, clorhidrato, sulfato, bisulfato, fosfato, nitrato, acetato, valerato, oleato, palmitato, estearato, laurato, benzoato, lactato, fosfato, tosilato, citrato, maleato, fumarato, succinato, tartrato, naftilato, mesilato, sales de glucoheptonato, lactobionato y laurilsulfonato, y similares. (Véase, por ejemplo, Berge et al. (1977) “Pharmaceutical Salts”, J. Pharm. Sci. 66:1-19).
Se contempla una sal o una sal farmacéuticamente aceptable para todos los compuestos descritos en la presente. En algunas realizaciones, una sal farmacéuticamente aceptable o una sal de cualquiera de los compuestos anteriores de la presente invención.
Como se usa en la presente, “tratar” significa prevenir, ralentizar, detener o revertir la progresión de una enfermedad o infección. Tratar también puede significar mejorar uno o más síntomas de una enfermedad o infección.
Los compuestos de la presente invención se pueden administrar de diversas formas, incluyendo las que se detallan en la presente. El tratamiento con el compuesto puede ser un componente de una terapia de combinación o una terapia adjunta, es decir, el sujeto o paciente que necesita el fármaco se trata o se le administra otro fármaco para la enfermedad junto con uno o más de los presentes compuestos. Esta terapia de combinación puede ser una terapia secuencial en la que el paciente se trata primero con un fármaco y luego con el otro, o los dos fármacos se administran simultáneamente. Estos pueden administrarse independientemente por la misma vía, o por dos o más vías de administración diferentes dependiendo de las formas de dosificación empleadas.
Como se usa en la presente, un “portador farmacéuticamente aceptable” es un disolvente, agente de suspensión o vehículo farmacéuticamente aceptable, para administrar los presentes compuestos al animal o al ser humano. El portador puede ser líquido o sólido y se selecciona teniendo en cuenta la forma de administración planeada. Los liposomas también
son un portador farmacéuticamente aceptable.
La dosificación de los compuestos administrados en el tratamiento variará dependiendo de factores tales como las características farmacodinámicas de un agente quimioterapéutico específico y su modo y vía de administración; la edad, sexo, tasa metabólica, eficiencia de absorción, salud y peso del receptor; la naturaleza y extensión de los síntomas; el tipo de tratamiento concurrente que se administra; la frecuencia de tratamiento con el mismo; y el efecto terapéutico deseado.
Una unidad de dosificación de los compuestos usados en la presente invención puede comprender un solo compuesto o mezclas de los mismos con agentes adicionales. Los compuestos se pueden administrar en formas de dosificación oral como comprimidos, cápsulas, píldoras, polvos, gránulos, elixires, tinturas, suspensiones, jarabes y emulsiones. Los compuestos también pueden administrarse en forma intravenosa (bolo o infusión), intraperitoneal, subcutánea o intramuscular, o introducirse directamente, por ejemplo, por inyección, aplicación tópica u otros métodos, en o sobre un sitio de infección, todos usando formas de dosificación bien conocidas por los expertos en las técnicas farmacéuticas.
Los compuestos usados en la presente invención pueden administrarse mezclados con diluyentes, extensores, excipientes o portadores farmacéuticos adecuados (denominados colectivamente en la presente como un portador farmacéuticamente aceptable) adecuadamente seleccionados con respecto a la forma de administración prevista y de acuerdo con las practicas farmacéuticas convencionales. La unidad estará en una forma adecuada para administración oral, rectal, tópica, intravenosa, o inyección directa o parenteral. Los compuestos se pueden administrar solos o mezclados con un portador farmacéuticamente aceptable. Este portador puede ser sólido o líquido, y el tipo de portador generalmente se elige en función del tipo de administración que se utilice. El principio activo se puede co-administrar en forma de un comprimido o cápsula, liposoma, como un polvo aglomerado o en forma líquida. Los ejemplos de portadores sólidos adecuados incluyen lactosa, sacarosa, gelatina y agar. Las cápsulas o comprimidos se pueden formular fácilmente y se pueden hacer fáciles de tragar o masticar; otras formas sólidas incluyen gránulos y polvos a granel. Los comprimidos pueden contener aglutinantes, lubricantes, diluyentes, agentes disgregantes, colorantes, saborizantes, agentes inductores de flujo y agentes de fusión adecuados. Los ejemplos de formas de dosificación líquidas adecuadas incluyen soluciones o suspensiones en agua, grasas y aceites farmacéuticamente aceptables, alcoholes u otros disolventes orgánicos, incluyendo ésteres, emulsiones, jarabes o elixires, suspensiones, soluciones y/o suspensiones reconstituidas a partir de gránulos no efervescentes y preparaciones efervescentes reconstituidas a partir de gránulos efervescentes. Tales formas de dosificación líquidas pueden contener, por ejemplo, disolventes adecuados, conservantes, agentes emulsionantes, agentes de suspensión, diluyentes, edulcorantes, espesantes y agentes de fusión. Las formas de dosificación orales contienen opcionalmente saborizantes y colorantes. Las formas parenterales e intravenosas también pueden incluir minerales y otros materiales para hacerlos compatibles con el tipo de inyección o sistema de administración elegido.
Las técnicas y composiciones para hacer formas de dosificación útiles en la presente invención se describen en las siguientes referencias: 7 Modern Pharmaceutics, Capítulos 9 y 10 (Bankers Rhodes, Editores, 1979); Pharmaceutical Dosage Forms: Tablets (Lieberman et al., 1981); Ansel, Introduction to Pharmaceutical Dosage Forms, 2a edición(1976); Remington's Pharmaceutical Sciences, 17a edición. (Mack Publishing Company, Easton, Pa., 1985); Advances in Pharmaceutical Sciences (David Ganderton, Trevor Jones, Eds., 1992); Advances in Pharmaceutical Sciences Vol. 7. (David Ganderton, Trevor Jones, James McGinity, Eds., 1995); Aqueous Polymeric Coatings for Pharmaceutical Dosage Forms (Drugs and the Pharmaceutical Sciences, Series 36 (James McGinity, Ed., 1989); Pharmaceutical Particulate Carriers: Therapeutic Applications: Drugs and the Pharmaceutical Sciences, Vol. 61 (Alain Rolland, Ed., 1993); Drug Delivery to the Gastrointestinal Tract (Ellis Horwood Books in the Biological Sciences. Series in Pharmaceutical Technology; J. G. Hardy, S. S. Davis, Clive G. Wilson, Eds.); Modem Pharmaceutics Drugs and the Pharmaceutical Sciences, Vol. 40 (Gilbert S. Banker, Christopher T. Rhodes, Eds.).
Los comprimidos pueden contener aglutinantes, lubricantes, agentes disgregantes, agentes colorantes, agentes saborizantes, agentes inductores de flujo y agentes de fusión adecuados. Por ejemplo, para la administración oral en la forma de unidad de dosificación de un comprimido o cápsula, el componente de fármaco activo se puede combinar con un portador inerte, no tóxico, farmacéuticamente aceptable, oral, tal como lactosa, gelatina, agar, almidón, sacarosa, glucosa, metilcelulosa, estearato de magnesio, fosfato dicálcico, sulfato de calcio, manitol, sorbitol y similares. Los aglutinantes adecuados incluyen almidón, gelatina, azúcares naturales, tales como glucosa o beta-lactosa, edulcorantes de maíz, gomas naturales y sintéticas, tales como acacia, tragacanto o alginato de sodio, carboximetilcelulosa, polietilenglicol, ceras y similares. Los lubricantes usados en estas formas de dosificación incluyen oleato de sodio, estearato de sodio, estearato de magnesio, benzoato de sodio, acetato de sodio, cloruro de sodio y similares. Los disgregantes incluyen, sin limitación, almidón, metilcelulosa, agar, bentonita, goma xantana y similares.
Los compuestos utilizados en la presente invención también se pueden administrar en forma de sistemas de administración de liposomas, tales como vesículas unilamelares pequeñas, vesículas unilamelares grandes y vesículas multilamelares. Los liposomas se pueden formar a partir de diversos fosfolípidos, tales como colesterol, estearilamina o fosfatidilcolinas. Los compuestos pueden administrarse como componentes de emulsiones dirigidas al tejido.
Los compuestos usados en la presente invención también se pueden acoplar a polímeros solubles como portadores de fármacos dirigidos o como profármacos. Dichos polímeros incluyen polivinilpirrolidona, copolímero de pirano, polihidroxipropilmetacrilamida-fenol, polihidroxietilaspartamidefenol o polietilenóxido-polilisina sustituida con residuos de
palmitoílo. Además, los compuestos pueden acoplarse a una clase de polímeros biodegradables útiles para lograr la liberación controlada de un fármaco, por ejemplo, ácido poliláctico, ácido poliglicólico, copolímeros de ácido poliláctico y poliglicólico, poliepsilon caprolactona, ácido polihidroxibutírico, poliortoésteres, poliacetales, polidihidropiranos, policianoacilatos y copolímeros de bloque reticulados o anfipáticos de hidrogeles.
Las cápsulas de gelatina pueden contener los compuestos del ingrediente activo y portadores en polvo, tales como lactosa, almidón, derivados de celulosa, estearato de magnesio, ácido esteárico y similares. Se pueden usar diluyentes similares para preparar comprimidos. Tanto los comprimidos como las cápsulas se pueden fabricar como productos de liberación inmediata o como productos de liberación sostenida para proporcionar una liberación continua del medicamento durante un período de horas. Los comprimidos se pueden recubrir con azúcar o con película para enmascarar cualquier sabor desagradable y proteger el comprimido de la atmósfera, o con recubrimiento entérico para la desintegración selectiva en el tracto gastrointestinal.
Para la administración oral en forma de dosificación líquida, los componentes del fármaco oral se combinan con cualquier portador inerte oral, no tóxico, farmacéuticamente aceptable, tal como etanol, glicerol, agua y similares. Los ejemplos de formas de dosificación líquidas adecuadas incluyen soluciones o suspensiones en agua, grasas y aceites farmacéuticamente aceptables, alcoholes u otros disolventes orgánicos, incluyendo ésteres, emulsiones, jarabes o elixires, suspensiones, soluciones y/o suspensiones reconstituidas a partir de gránulos no efervescentes y preparaciones efervescentes reconstituidas a partir de gránulos efervescentes. Tales formas de dosificación líquidas pueden contener, por ejemplo, disolventes adecuados, conservantes, agentes emulsionantes, agentes de suspensión, diluyentes, edulcorantes, espesantes y agentes de fusión.
Las formas de dosificación líquidas para administración oral pueden contener colorantes y saborizantes para aumentar la aceptación del paciente. En general, el agua, un aceite adecuado, solución salina, dextrosa acuosa (glucosa) y soluciones de azúcar relacionadas y glicoles, tales como propilenglicol o polietilenglicoles son portadores adecuados para soluciones parenterales. Las soluciones para administración parenteral contienen preferiblemente una sal soluble en agua del ingrediente activo, agentes estabilizantes adecuados y, si es necesario, sustancias amortiguadoras. Los agentes antioxidantes, tales como bisulfito de sodio, sulfito de sodio o ácido ascórbico, solos o combinados, son agentes estabilizadores adecuados. También se utilizan ácido cítrico y sus sales, y ácido etilendiaminotetraacético (“EDTA”, por sus siglas en inglés) sódico. Además, las soluciones parenterales pueden contener conservantes, tales como cloruro de benzalconio, metilparabeno o propilparabeno y clorobutanol. Los portadores farmacéuticos adecuados se describen en Remington's Pharmaceutical Sciences, Mack Publishing Company, un texto de referencia estándar en este campo. Los compuestos utilizados en la presente invención también pueden administrarse en forma intranasal mediante el uso de vehículos intranasales adecuados, o mediante vías transdérmicas, utilizando aquellas formas de parches cutáneos transdérmicos bien conocidos por los expertos en la materia. Para administrarse en forma de un sistema de suministro transdérmico, la administración de la dosis generalmente será continua en lugar de intermitente a lo largo del régimen de dosificación.
Las formas parenterales e intravenosas también pueden incluir minerales y otros materiales para hacerlos compatibles con el tipo de inyección o sistema de administración elegido.
Cada realización descrita en la presente se contempla como aplicable a cada una de las otras realizaciones descritas. Por lo tanto, todas las combinaciones de los diversos elementos descritos en la presente están dentro del alcance de la invención.
Esta invención se entenderá mejor con referencia a los Detalles experimentales a continuación, pero los expertos en la técnica entenderán fácilmente que los experimentos específicos detallados son sólo ilustrativos de la invención tal como se describe con más detalle en las reivindicaciones que siguen a continuación.
Detalles experimentales
Materiales y métodos
Ensayo TR-FRET para la interacción RBP4-TTR inducida por retinol
La unión de un antagonista RBP4 deseado desplaza el retinol e induce un obstáculo para la interacción RBP4-TTR, lo que da como resultado la disminución de la señal FRET (Figura 7). En este ensayo se usaron MBP-RBP4 expresado en bacterias y TTR no etiquetada. Para el uso en el ensayo de transferencia de energía de fluorescencia resuelta en el tiempo (“TR-FRET”, por sus siglas en inglés), el fragmento RBP4 humano marcado con proteína de unión a maltosa (“MBP”, por sus siglas en inglés) (aminoácidos 19-201) se expresó en la cepa Gold(DE3)pLysS de E. co li (Stratagene) usando el vector pMAL-c4x. Después de la lisis celular, la RBP4 recombinante se purificó a partir de la fracción soluble usando el sistema ACTA FPLC (Ge Healthcare) equipado con la columna MBP Trap HP de 5 mL. La TTR humana no etiquetada se adquirió de Calbiochem. La TTR no etiquetada se etiquetó directamente con Eu3+ Cryptate-NHS utilizando el kit de etiquetado de criptado HTRF de CisBio siguiendo las recomendaciones del fabricante. El ensayo HTRF se realizó en placas blancas de 384 pocillos de bajo volumen (Greiner-Bio) en un volumen de ensayo final de 16 julios por pocillo. La
solución amortiguadora de reacción contenía Tris-HCl 10 mM pH 7,5, DTT 1 mM, NP-40 al 0,05%, Prionex al 0,05%, glicerol al 6 % y KF 400 mM. Cada reacción contenía MBP-RBP460 nM y TTR-Eu 2 nM junto con 26,7 nM de anticuerpo anti-MBP conjugado con d2 (Cisbio). La titulación de los compuestos de prueba en este ensayo se realizó en presencia de retinol 1 pM. Todas las reacciones se ensamblaron en la oscuridad bajo luz roja tenue y se incubaron durante la noche a 4°C envueltas en papel de aluminio. La señal TR-FRET se midió en el lector de placas multimodo SpectraMax M5e (Molecular Device). Se excitó la fluorescencia a 337 nm y se tomaron dos lecturas por pocillo: Lectura 1 para transferencia de energía controlada por tiempo de Eu(K) a d2 (excitación de 337 nm, emisión de 668 nm, retardo de recuento de 75 microsegundos, ventana de recuento de 100 microsegundos) y Lectura 2 para fluorescencia controlada por tiempo de Eu(K) (excitación de 337 nm, emisión de 620 nm, retardo de recuento de 400 microsegundos, ventana de recuento de 400 microsegundos). La señal TR-FRET se expresó como la relación de intensidad de fluorescencia: Flu665/Flu620 x 10.000.
Ensayo de unión a RBP4 de proximidad de centelleo
La RBP4 humana no etiquetada purificada a partir de la orina de pacientes con proteinuria tubular se adquirió de Fitzgerald Industries International. Se biotiniló usando el Kit de biotinilación EZ-Link Sulfo-NHS-LC de Pierce siguiendo las recomendaciones del fabricante. Los experimentos de unión se realizaron en placas de 96 pocillos (OptiPlate, PerkinElmer) en un volumen de ensayo final de 100 pL por pocillo en solución amortiguadora de ensayo de proximidad de centelleo (“SPA”, por sus siglas en inglés) (1X PBS, pH 7,4, EDTA 1 mM, BSA al 0 ,1% , CHAPS al 0,5%). La mezcla de reacción contenía 3H-Retinol 10 nM (48,7 Ci/mmol; PerkinElmer), perlas de estreptavidina-PVT 0,3 mg/pocillo, RBP4 biotinilado 50 nM y un compuesto de prueba. La unión no específica se determinó en presencia de 20 pm de retinol no etiquetado. La mezcla de reacción se ensambló en la oscuridad bajo una luz roja tenue. Las placas se sellaron con cinta transparente (TopSeal-A: microplaca de 96 pocillos, PerkinElmer), envuelta en papel de aluminio y se dejó equilibrar durante 6 horas a temperatura ambiente, seguido de una incubación durante la noche a 4°C. Los recuentos de radio se midieron utilizando un contador TopCount NXT (Packard Instrument Company).
Procedimiento general (“GP”, por sus siglas en inglés) para preparar productos intermedios para la síntesis de compuestos de piperidina

Condiciones: A1) ácido carboxílico, HBTU, Et3N, DMF; A2) ácido carboxílico, EDCI, HOBt, /-P^NEt, DMF; A3) cloruro de ácido, EtaN, CH2CI2.
Procedimiento general (GP-A1) para la formación de carboxamida: Una mezcla de amina I (1 equiv), ácido carboxílico deseado (1 equiv), trietilamina (Et3N) (3 equiv) y hexafluorofosfato de 2-(1H-benzotriazol-1-il)-1,1,3,3-tetrametiluronio (HBTU) (1,5 equiv) en DMF (0,25 M) se agitó a temperatura ambiente hasta que la reacción se completó mediante cromatografía líquida con espectrometría de masas (“LC-MS”, por sus siglas en inglés). La mezcla se diluyó con H2O y se extrajo con EtoAc. Los extractos orgánicos combinados se lavaron con H2O y salmuera, se secaron sobre Na2SO4, se filtraron y se concentraron a presión reducida. El residuo resultante se purificó mediante cromatografía en gel de sílice (los eluyentes típicos incluían una mezcla de, o hexanos y EtOAc o una mezcla de CH2CI2 y una mezcla 90:9:1 de CH2CI2/CH3OH/NH4OH concentrado) para proporcionar la carboxamida II deseada. La estructura del producto se verificó mediante resonancia magnética nuclear protónica (“1 H NMR”, por sus siglas en inglés) y mediante análisis de masas.
Procedimiento general (GP-A2) para la formación de carboxamida: Una mezcla de amina I (1 equiv), ácido carboxílico deseado (1 equiv), W,A/-diisopropiletilamina (/-Pr2NEt) (3 equiv), 1-etil-3-(3-dimetilaminopropil)carbodiimida (EDCI) (1,5 equiv) e hidroxibenzotriazol (HOBt) (1,5 equiv) en DMF (0,25 M) se agitó a temperatura ambiente hasta que la reacción se completó mediante LC-MS. La mezcla se diluyó con H2O y se extrajo con EtOAc. Los extractos orgánicos combinados se lavaron con H2O y salmuera, se secaron sobre Na2SO4, se filtraron y se concentraron a presión reducida. El residuo resultante se purificó mediante cromatografía en gel de sílice (los eluyentes típicos incluían una mezcla de, o hexanos y EtOAc o una mezcla de CH2CI2 y una mezcla 90:9:1 de CH2CI2/CH3OH/NH4OH concentrado) para proporcionar la carboxamida II deseada. La estructura del producto se verificó mediante 1 H NMR y mediante análisis de masas.
Procedimiento general (GP-A3) para la formación de carboxamida: Una mezcla de amina I (1 equiv), Et3N (3 equiv) y cloruro de ácido (1 equiv) en CH2Ch (0,25 M) se agitó a temperatura ambiente hasta que la reacción se completó mediante
LC-MS. La mezcla se lavó con H2O, salmuera, se secó sobre Na2SO4, se filtró y se concentró a presión reducida. El residuo resultante se purificó mediante cromatografía en gel de sílice (los eluyentes típicos incluían una mezcla de, o hexanos y EtOAc o una mezcla de CH2Ch y una mezcla 90:9:1 de CH2CI2/CH3OH/NH4OH concentrado) para proporcionar las carboxamidas II deseadas. La estructura del producto se verificó mediante 1 H NMR y mediante análisis de masas.
Procedimientos generales para preparar carboxamidas de (4-fenilpiperidin-1-il)(4,5,6,7-tetrahidro-1H-pirazolo[4,3-c]piridin-3-il)metanona IV

Condiciones: B) cloruro de ácido, Et3N, CH2CI2.
Procedimiento general (GP-B) para la formación de carboxamida: Una mezcla de amina III (1 equiv), cloruro de ácido deseado (1 equiv) y trietilamina (Et3N) (3 equiv) en CH2Ch (0,25 M) se agitó desde 0°C hasta temperatura ambiente hasta que la reacción se completó mediante LC-MS. La mezcla se diluyó con H2O y se extrajo con CH2Ch. Los extractos orgánicos combinados se lavaron con H2O y salmuera, se secaron sobre Na2SO4, se filtraron y se concentraron a presión reducida. El residuo resultante se purificó mediante cromatografía en gel de sílice (los eluyentes típicos incluían una mezcla de, o hexanos y EtOAc o una mezcla de CH2CI2 y una mezcla 90:9:1 de CH2CI2/CH3OH/NH4OH concentrado) para proporcionar las carboxamidas IV deseadas. La estructura del producto se verificó mediante 1 H NMR y mediante análisis de masas.
Procedimientos generales para preparar sulfonamidas de (4-fenilpiperidin-1-il)(4,5,6,7-tetrahidro-1H-pirazolo[4,3-c]piridin-3-il)metanona V

Condiciones: C) cloruro de sulfonilo, /-P^NEt, CH2CI2.
Procedimiento general (GP-C) para la formación de sulfonamidas: Una mezcla de amina III (1 equiv), cloruro de sulfonilo deseado (1 equiv) e /-Pr2NEt (3 equiv) en CH2Ch (0,25 M) se agitó desde 0°C hasta temperatura ambiente hasta que la reacción se completó mediante LC-MS. La mezcla se diluyó con H2O y se extrajo con CH2Ch. Los extractos orgánicos combinados se lavaron con H2O y salmuera, se secaron sobre Na2SO4, se filtraron y se concentraron a presión reducida. El residuo resultante se purificó mediante cromatografía en gel de sílice (los eluyentes típicos incluían una mezcla de, o hexanos y EtOAc o una mezcla de CH2Ch y una mezcla 90:9:1 de CH2CI2/CH3OH/NH4OH concentrado) para proporcionar las sulfonamidas V deseadas. La estructura del producto se verificó mediante 1 H NMR y mediante análisis de masas.
Procedimientos generales para preparar (4-fenilpiperidin-1-il)(4,5,6,7-tetrahidro-1H-pirazolo[4,3-c]piridin-3-il)metanonas alquiladas VI

Condiciones: D) aldehido o cetona, NaBH(OAc)3, CH2CI2.
Procedimiento general (GP-D) para la formación de sulfonamidas: Una mezcla de amina III (1 equiv), aldehído o cetona deseada (1,5 equiv) y HOAc (6 equiv) en CH2Ch (0,25 M) se agitó durante 16 horas a temperatura ambiente. A esto se añadió triacetoxiborohidruro de sodio (NaBH(OAc^) y la mezcla se agitó a temperatura ambiente hasta que la reacción se completó mediante LC-MS. La mezcla se diluyó con solución acuosa saturada de NaHCO3 y se extrajo con CH2Ch. Los extractos orgánicos combinados se lavaron con H2O y salmuera, se secaron sobre Na2SO4, se filtraron y se concentraron a presión reducida. El residuo resultante se purificó mediante cromatografía en gel de sílice (los eluyentes típicos incluían una mezcla de, o hexanos y EtOAc o una mezcla de CH2Ch y una mezcla 90:9:1 de CH2CI2/CH3OH/NH4OH concentrado) para proporcionar las aminas VI deseadas. La estructura del producto se verificó mediante 1 H NMR y mediante análisis de masas.
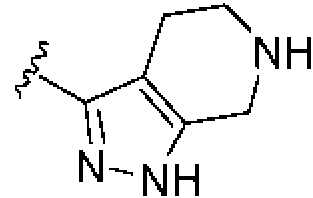
Procedimiento general para preparar carboxamidas de (4-fenilpiperidin-1-il)(4,5,6,7-tetrahidro-1H-pirazolo[3,4-c]piridin-3-il)metanona VIII

Condiciones: E) cloruro de ácido, Et3N, CH2CI2.
Procedimiento general (GP-E) para la formación de carboxamida: Una mezcla de amina VII (1 equiv), cloruro de ácido deseado (1 equiv) y trietilamina (Et3N) (3 equiv) en CH2Ch (0,25 M) se agitó desde 0°C hasta temperatura ambiente hasta que la reacción se completó mediante LC-MS. La mezcla se diluyó con H2O y se extrajo con CH2Ch. Los extractos orgánicos combinados se lavaron con H2O y salmuera, se secaron sobre Na2SO4, se filtraron y se concentraron a presión reducida. El residuo resultante se purificó mediante cromatografía en gel de sílice (los eluyentes típicos incluían una mezcla de, o hexanos y EtOAc o una mezcla de CH2CI2 y una mezcla 90:9:1 de CH2CI2/CH3OH/NH4OH concentrado) para proporcionar las carboxamidas VIII deseadas. La estructura del producto se verificó mediante 1 H NMR y mediante análisis de masas.
P ro c e d im ie n to s g e n e ra le s p a ra p re p a ra r s u lfo n a m id a s d e (4 - fe n ilp ip e r id in -1 - i l) (4 ,5 ,6 ,7 - te t ra h id ro -1 H -p ira z o lo [3 ,4 -c ]p ir id in -3 - i l)m e ta n o n a IX

Condiciones: F) cloruro de sulfonilo, /-P^NEt, CH2CI2.
Procedimiento general (GP-F) para la formación de sulfonamidas: Una mezcla de amina VII (1 equiv), cloruro de sulfonilo deseado (1 equiv) e /-Pr2NEt (3 equiv) en CH2Ch (0,25 M) se agitó desde 0°C hasta temperatura ambiente hasta que la reacción se completó mediante lC-MS. La mezcla se diluyó con H2O y se extrajo con CH2Ch. Los extractos orgánicos combinados se lavaron con H2O y salmuera, se secaron sobre Na2SO4, se filtraron y se concentraron a presión reducida. El residuo resultante se purificó mediante cromatografía en gel de sílice (los eluyentes típicos incluían una mezcla de, o hexanos y EtOAc o una mezcla de CH2Ch y una mezcla 90:9:1 de CH2CI2/CH3OH/NH4OH concentrado) para proporcionar las sulfonamidas IX deseadas La estructura del producto se verificó mediante 1 H NMR y mediante análisis de masas.
Procedimientos generales para preparar (4-fenilpiperidin-1-il)(4,5,6,7-tetrahidro-1H-pirazolo[3,4-c]piridin-3-il)metanonas alquiladas X
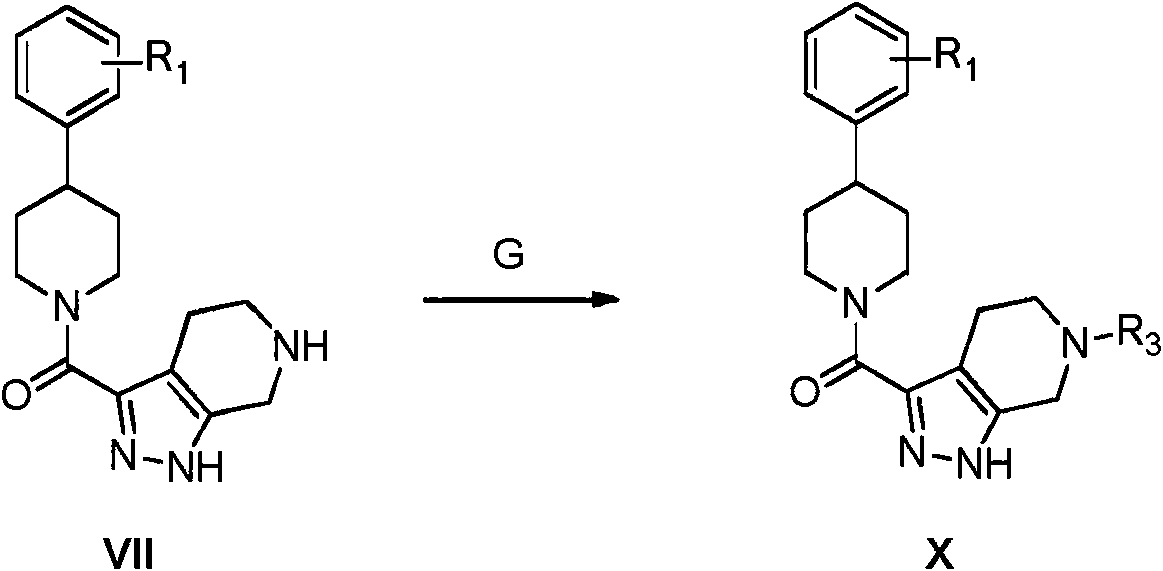
Condiciones: G) aldehído o cetona, NaBH(OAc)3, CH2CI2.
Procedimiento general (GP-G) para la formación de sulfonamidas: Una mezcla de amina VII (1 equiv), aldehído o cetona deseada (1,5 equiv) y HOAc (6 equiv) en CH2Ch (0,25 M) se agitó durante 16 horas a temperatura ambiente. A esto se añadió triacetoxiborohidruro de sodio (NaBH(OAc)3) y la mezcla se agitó a temperatura ambiente hasta que la reacción se completó mediante LC-MS. La mezcla se diluyó con solución acuosa saturada de NaHCO3 y se extrajo con CH2Ch. Los extractos orgánicos combinados se lavaron con H2O y salmuera, se secaron sobre Na2SO4, se filtraron y se concentraron a presión reducida. El residuo resultante se purificó mediante cromatografía en gel de sílice (los eluyentes típicos incluían una mezcla de, o hexanos y EtOAc o una mezcla de CH2Ch y una mezcla 90:9:1 de CH2CI2/CH3OH/NH4OH concentrado) para proporcionar las aminas X deseadas. La estructura del producto se verificó mediante 1 H NMR y mediante análisis de masas.


Condiciones: H) cloruro de ácido, Et3N, CH2CI2.
Procedimiento general (GP-H) para la formación de carboxamida: Una mezcla de amina XI (1 equiv), cloruro de ácido deseado (1 equiv) y trietilamina (Et3N) (3 equiv) en CH2Ch (0,25 M) se agitó desde 0°C hasta temperatura ambiente hasta que la reacción se completó mediante LC-MS. La mezcla se diluyó con H2O y se extrajo con CH2Ch. Los extractos orgánicos combinados se lavaron con H2O y salmuera, se secaron sobre Na2SO4, se filtraron y se concentraron a presión reducida. El residuo resultante se purificó mediante cromatografía en gel de sílice (los eluyentes típicos incluían una mezcla de, o hexanos y EtOAc o una mezcla de CH2CI2 y una mezcla 90:9:1 de CH2CI2/CH3OH/NH4OH concentrado) para proporcionar las carboxamidas XII deseadas. La estructura del producto se verificó mediante 1 H NMR y mediante análisis de masas.
Procedimientos generales para preparar sulfonamidas de (4-fenilpiperidin-1-M)(1,4,5,6-tetrahidropirrolo[3,4-c]pirazol-3-il)metanona XIII

Condiciones: I) cloruro de sulfonilo, /-P^NEt, CH2CI2.
Procedimiento general (GP-I) para la formación de sulfonamidas: Una mezcla de amina XI (1 equiv), cloruro de sulfonilo deseado (1 equiv) e /-Pr2NEt (3 equiv) en CH2Ch (0,25 M) se agitó desde 0°C hasta temperatura ambiente hasta que la reacción se completó mediante LC-MS. La mezcla se diluyó con H2O y se extrajo con CH2Ch. Los extractos orgánicos combinados se lavaron con H2O y salmuera, se secaron sobre Na2SO4, se filtraron y se concentraron a presión reducida. El residuo resultante se purificó mediante cromatografía en gel de sílice (los eluyentes típicos incluían una mezcla de, o hexanos y EtOAc o una mezcla de CH2Ch y una mezcla 90:9:1 de CH2CI2/CH3OH/NH4OH concentrado) para proporcionar las sulfonamidas XIII deseadas. La estructura del producto se verificó mediante 1 H NMR y mediante análisis de masas.
P ro c e d im ie n to s g e n e ra le s p a ra p re p a ra r (4 - fe n ilp ip e r id in -1 - i l) (1 ,4 ,5 ,6 - te t ra h id ro p ir ro lo [3 ,4 -c ]p ira z o l-3 - i l)m e ta n o n a a lq u ila d a X IV

Condiciones: J) aldehido o cetona, NaBH(OAc)3, CH2CI2.
Procedimiento general (GP-J) para la formación de sulfonamidas: Una mezcla de amina XI (1 equiv), aldehido o cetona deseada (1,5 equiv) y HOAc (6 equiv) en CH2Ch (0,25 M) se agitó durante 16 horas a temperatura ambiente. A esto se añadió triacetoxiborohidruro de sodio (NaBH(OAc)3) y la mezcla se agitó a temperatura ambiente hasta que la reacción se completó mediante LC-MS. La mezcla se diluyó con solución acuosa saturada de NaHCO3 y se extrajo con CH2Ch. Los extractos orgánicos combinados se lavaron con H2O y salmuera, se secaron sobre Na2SO4, se filtraron y se concentraron a presión reducida. El residuo resultante se purificó mediante cromatografía en gel de sílice (los eluyentes típicos incluían una mezcla de, o hexanos y EtOAc o una mezcla de CH2Ch y una mezcla 90:9:1 de CH2CI2/CH3OH/NH4OH concentrado) para proporcionar las aminas XIV deseadas. La estructura del producto se verificó mediante 1 H NMR y mediante análisis de masas.
Preparación de clorhidrato de 4-(3-fluoro-2-(trifluorometil)fenil)piperidina (5)

Paso A: Una solución de 4-oxopiperidina-1-carboxilato de terc-butilo (1, 1,0 g, 5,02 mmol) en THF (30 mL) se enfrió a -78°C. Se añadió gota a gota LiHMDS (solución 1,0 M en THF, 6,52 mL) durante 30 min. La reacción se agitó a -78°C durante 1 h, luego se añadió gota a gota una solución de N-fenil-bis(trifluorometanosulfonimida) (2,52 g, 7,05 mmol) en THF (5,0 mL) durante 30 min. La mezcla se agitó a 0°C durante 3 h y luego se concentró a presión reducida. El residuo se cromatografió sobre gel de sílice (unidad Isco CombiFlash Rf, columna Redisep de 24 g, 0% a 100% de EtOAc en hexanos) para proporcionar 4-(((trifluorometil)sulfonil)oxi)-5,6-dihidropiridina-1(2H)-carboxilato de ferc-butilo (2) como un aceite de color amarillo claro (1,50 g, 90%): 1 H NMR (300 MHz, CDCI3) 65,75 (br s, 1H), 4,05-4,02 (m, 2H), 3,64-3,60 (m, 2H), 2,44-2,42 (m, 2H), 1,46 (s, 9H).
Paso B: Una mezcla de ácido 4-(((trifluorometil)sulfonil)oxi)-5,6-dihidropiridina-1(2H)-carboxilato de ferc-butilo (2, 3,50 g, 10,6 mmol), 3-fluoro-(2-trifluorometil)fenil borónico (2,19 g, 10,6 mmol), Pd(PPh3)4 (1,22 g, 1,06 mmol) y Na2cO 32 M (62 mL) en DME (120 mL) se calentó a 80°C durante 6 h. La mezcla se enfrió a temperatura ambiente y se diluyó con una solución acuosa de LiCl al 5% (100 mL). La mezcla se extrajo con EtOAc (3 x 50 mL), los extractos orgánicos combinados se lavaron con salmuera (2 x 50 mL) y se concentraron a presión reducida. El residuo se diluyó en CH2Ch (100 mL) y se envió a través de un tapón de gel de sílice de 300 mL, eluyendo con EtOAc al 10% en hexanos (800 mL). El filtrado resultante se concentró a presión reducida y se cromatografió sobre gel de sílice (unidad Isco CombiFlash Rf, columna
Redisep de 80 g, 0 % a 50% de EtOAc en hexanos) para proporcionar 4-(3-fluoro-2-(trifluorometM)fenil)-5,6-dihidropiridina-1(2H)-carboxilato de ferc-butilo (3) como un aceite amarillo claro (2,39 g, 69%): 1H NMR (300 MHz, DMSO- d6) 8 7,75 7,61 (m, 1H), 7,49-7,36 (m, 1H), 7,17 (d, J = 7,8 Hz, 1H), 5,63-5,54 (m, 1H), 3,97-3,86 (m, 2H), 3,57-3,45 (m, 2H), 2,31 2,18 (m, 2H), 1,42 (s, 9H).
Paso C: Una mezcla de 4-(3-fluoro-2-(trifluorometN)fenil)-5,6-dihidropindina-1(2H)-carboxilato de ferc-butilo (3, 4,7 g, 13,6 mmol) y Pd/C al 10 % (1,0 g) en EtOH (100 mL) bajo una atmósfera de H2 (2,10 kg/cm2 (30 libras por pulgada cuadrada (“psi”, por sus siglas en inglés))) a temperatura ambiente durante 18 h. La mezcla se filtró a través de Celite y el filtrado se concentró a presión reducida para proporcionar 4-(3-fluoro-2-(trifluorometil)fenil)piperidina-1-carboxilato de ferc-butilo (4) como un aceite transparente (4,80 g, >99%): 1H NMR (300 MHz, DMSO-d6) 8 7,72-7,60 (m, 1H), 7,46 (d, J = 8,1 Hz, 1H), 7,30 (dd, J = 12,3, 8,1 Hz, 1H), 4,18-4,00 (m, 2H), 3,11-2,95 (m, 1H), 2,92-2,64 (m, 2H), 1,76-1,51 (m, 4H), 1,42 (s, 9H).
Paso D: A una solución de 4-(3-fluoro-2(trifluorometil)fenil)piperidina-1-carboxilato de terc-butilo (4, 4,70 g, 13,6 mmol) en CH2Cl2 (40 mL) se le añadió HCl 2 N (2,0 M en Et2O, 40 mL). La mezcla se agitó a temperatura ambiente durante 18 h y se diluyó con Et2O (100 mL). El precipitado resultante se recolectó por filtración para proporcionar clorhidrato de 4-(3-fluoro-2-(trifluorometil)fenilpiperidina como un polvo blanco (5, 3,69 g, 96%): 1H NMR (300 MHz, DMSO-d6) 8 9,09-8,80 (m, 2H), 7,83-7,70 (m, 1H), 7,44-7,29 (m, 2H), 3,42-3,31 (m, 2H), 3,29-3,15 (m, 1H), 3,14-2,95 (m, 2H), 2,11-1,91 (m, 2H), 1,89, 1,76 (m, 2H); ESI MS m /z 248 [M H]+.
Preparación de 4-(3,4-difluoro-2-(trifluorometil)fenil)piperidina (8 )

Paso A: Una mezcla de 4-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)-5,6-dihidropiridina-1(2H)-carboxilato de ferc-butilo (6 , 57,4 g, 185 mmol), 3 1-bromo-3,4-difluoro-2-(trifluorometil)benceno (48,5 g, 185 mmol), Pd(PPh3)4 (21,5 g, 18,5 mmol) y Na2CO 32 M (150 mL) en DME (500 mL) se calentó a 80°C durante 16 h. La mezcla se enfrió a temperatura ambiente y se diluyó con una solución acuosa de LiCl al 5 % (100 mL). La mezcla se extrajo con EtOAc (3 x 200 mL), los extractos orgánicos combinados se lavaron con salmuera (2 x 200 mL) y se concentraron a presión reducida. El residuo se diluyó en CH2Cl2 (100 mL) y se envió a través de un tapón de gel de sílice de 300 mL, eluyendo con EtOAc al 10 % en hexanos (800 mL). El filtrado resultante se concentró a presión reducida y se cromatografió sobre gel de sílice (unidad Isco CombiFlash Rf, 3 columnas Redisep de 330 g, 0 % a 50 % de EtOAc en hexanos) para proporcionar 4-(3,4-difluoro-2-(trifluorometil)fenil)-5,6-dihidropiridina-1(2H)-carboxilato de ferc-butilo (7) como un sólido blanco (59,0 g, 92%): 1 H NMR (300 MHz, CDCI3) 8 7,34-7,28 (m, 1H), 6,93 (m, 1H), 5,55 (br, 1H), 4,01 (br, 2H), 3,60 (m, 2H), 2,30 (m, 2H), 1,50 (s, 9H); MS (ESI+) m /z 308 [M+H-C4Hs]+.
Paso B: Una mezcla de 4-(3,4-difluoro-2-(trifluorometil)fenil)-5,6-dihidropiridina-1(2H)-carboxilato de ferc-butilo (7, 59,0 g, 162,3 mmol) y 10 % de Pd/C (5,0 g) en EtOH ( 200 mL) bajo una atmósfera de H2 (2 , 10 kg/cm2 (30 psi)) a temperatura ambiente durante 72 h. La mezcla se filtró a través de Celite y el filtrado se concentró a presión reducida para proporcionar 4-(3,4-difluoro-2-(trifluorometil)fenil)piperidina-1-carboxilato de ferc-butilo (8 ) como un sólido blanco (57,9 g, 97%): 1 H NMR (300 MHz, CDCI3) 8 7,36-7,28 (m, 1H), 7,12 (m, 1H), 4,24 (br, 2H), 3,06 (m, 1H), 2,80 (m, 2H), 1,78-1,52 (m, 4H), 1,48 (s, 9H); MS (ESI+) m /z 310 [M+H-C4Hs]+.
Preparación de clorhidrato de 4-(5-fluoro-2-(trifluorometil)fenil)piperidina (11)

Paso A: Una mezcla de 4-(((tr¡fluoromet¡l)sulfon¡l)ox¡)-5,6-d¡h¡drop¡nd¡na-1(2H)-carbox¡lato de ferc-but¡lo (2, 1,10 g, 3,32 mmol), ác¡do 5-fluoro-(2-tr¡fluoromet¡l)fen¡l borón¡co (0,69 g, 3,32 mmol), pd(PPh3)4 (0,384 g, 0,332 mmol) y Na2CO32 M (20 mL) en DME (50 mL) se calentó a 80°C durante 6 h. La mezcla se enfr¡ó a temperatura amb¡ente y los sól¡dos resultantes se el¡m¡naron por f¡ltrac¡ón a través de una almohad¡lla de Cel¡te. El f¡ltrado se lavó con soluc¡ón de salmuera (4 x 50 mL) y se concentró a pres¡ón reduc¡da. El res¡duo resultante se cromatograf¡ó sobre gel de síl¡ce (un¡dad Isco Comb¡Flash Rf, columna Red¡sep de 40 g, 0% a 80% de EtOAc en hexanos) para proporc¡onar 4-(5-fluoro-2-(tnfluoromet¡l)fen¡l)-5,6-d¡h¡drop¡nd¡na-1(2H)-carboxNato de ferc-but¡lo (9) como un ace¡te transparente (0,542 g, 48%): 1 H NMR (300 MHz, DMSO-da) 87,80 (dd, J = 8,4, 6,0 Hz, 1H), 7,42-7,27 (m, 2H), 5,62 (br s, 1H), 3,97-3,87 (m, 2H), 3,51 (t, J = 5,7 Hz, 2H), 2,34-2,23 (m, 2H), 1,42 (s, 9H).
Paso B: Una mezcla de 4-(5-fluoro-2-(tr¡fluoromet¡l)fen¡l)-5,6-d¡h¡drop¡nd¡na-1(2H)-carbox¡lato de ferc-but¡lo (9, 0,542 g, 1,58 mmol) y HCl (soluc¡ón 2 N en Et2O, 10 mL) en CH2Ch (20 mL) ag¡tado a temperatura amb¡ente durante 18 h. La mezcla de reacc¡ón se d¡luyó con Et2O (30 mL) y el prec¡p¡tado resultante se recolectó por f¡ltrac¡ón para proporc¡onar clorh¡drato de 4-(5-fluoro-2-(tr¡fluoromet¡l)fen¡l)-1,2,3,6-tetrah¡drop¡r¡d¡na (10) como un sól¡do blanco (0,393 g, 88%): 1 H NMR (300 MHz, DMSO-d6) 89,26-9,00 (m, 2H), 7,84 (dd, J = 8,7, 5,4 Hz, 1H), 7,46-7,36 (m, 1H), 7,24 (dd, J = 9,3, 2,4 Hz, 1H), 5,67 (br s, 1H), 3,76-3,64 (m, 2H), 3,27 (t, J = 5,1 Hz, 2H), 2,70-2,40 (m, 2H).
Paso C: Una mezcla de clorh¡drato de 4-(5-fluoro-2-(tr¡fluoromet¡l)fen¡l)-1,2,3,6-tetrah¡drop¡r¡d¡na (10, 0,393 g, 1,41 mmol) y PtO2 (0,095 mg, 0,42 mmol) en EtOAc (14 mL) se ag¡tó a temperatura amb¡ente durante 18 h bajo un globo de H2. La mezcla se f¡ltró sobre Cel¡te y el f¡ltrado se concentró a pres¡ón reduc¡da y se d¡solv¡ó en CH2Ch (4 mL). A esta soluc¡ón se le añad¡ó HCl (2 N en Et2O, 4,0 mL) y la mezcla resultante se ag¡tó a temperatura amb¡ente durante 20 m¡n. La suspens¡ón resultante se d¡luyó con Et2O (20 mL) y los sól¡dos se recolectaron por f¡ltrac¡ón para proporc¡onar clorh¡drato de 4-(5-fluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡na (11) como un sól¡do blanco (309 mg, 78%): 1 H NMR (300 MHz, DMSO-cfó) 8 8,81 (br s, 2H), 7,80 (dd, J = 9,3, 6,0 Hz, 1H), 7,39-7,26 (m, 2H), 3,43-3,30 (m, 1H, se superpone con H2O), 3,24-2,97 (m, 3H), 2,11-1,90 (m, 2H), 1,88-1,75 (m, 2H); ESI MS m/z 248 [M H]+.
Preparac¡ón de clorh¡drato de 4-(2-cloro-3-fluorofen¡l)p¡per¡d¡na (14)

Paso A: Una mezcla de 4-(((tr¡fluoromet¡l)sulfon¡l)ox¡)-5,6-d¡h¡drop¡r¡d¡na-1(2H)-carbox¡lato de ferc-butilo (2, 1,18 g, 3,56 mmol), ácido 2-cloro-3-fluorofenil borónico (0,621 g, 3,56 mmol), Pd(PPh3)4 (0,411 g, 0,356 mmol) y Na2CO32 M (20 mL) en DME (50 mL) se calentó a 80°C durante 6 h. La mezcla se enfrió a temperatura ambiente y los sólidos resultantes se eliminaron por filtración a través de una almohadilla de Celite. El filtrado se lavó con solución de salmuera (4 x 50 mL) y se concentró a presión reducida. El residuo resultante se cromatografió sobre gel de sílice (unidad Isco CombiFlash Rf, columna Redisep de 40 g, 0% a 80% de EtOAc en hexanos) para proporcionar 4-(2-cloro-3-fluorofenil)-5,6-dihidro-pir¡dina-1(2H)-carboxilato de ferc-butilo (12) como un aceite transparente (0,579 g, 52%): 1 H NMR (300 MHz, DMSO-d6) 87,43 7,31 (m, 2H), 7,16-7,10 (m, 1H), 5,81-5,72 (m, 1H), 4,03-3,93 (m, 2H), 3,53 (t, J = 5,7 Hz, 2H), 2,41-2,31 (m, 2H), 1,43 (s, 9H).
Paso B: Una mezcla de 4-(2-cloro-3-fluorofen¡l)-5,6-d¡h¡drop¡r¡d¡na-1(2H)-carbox¡lato de ferc-butilo (12, 0,488 g, 1,41 mmol) y PtO2 (0,109 g, 0,48 mmol) en EtOAc (15 mL) se agitó a temperatura ambiente durante 18 h bajo un globo de H2. La mezcla se filtró sobre Celite y el filtrado se concentró a presión reducida y se disolvió en CH2Ch (4 mL). A esta solución se le añadió HCl (2 N en Et2O, 4,0 mL) y la mezcla resultante se agitó a temperatura ambiente durante 20 min. La suspensión resultante se diluyó con Et2O (20 mL) y los sólidos se recolectaron por filtración para proporcionar 4-(2-cloro-3-fluorofenil)piperidina-1 carboxilato de ferc-butilo (13) como un semisólido transparente (0,471 g, 95%): 1 H NMR (300 MHz, DMSO-d6) 87,43-7,19 (m, 3H), 4,17-4,01 (m, 2H), 3,20-3,03 (m, 1H), 2,95-2,68 (m, 2H), 1,79-1,65 (m, 2H), 1,58 1,45 (m, 2H), 1,41 (s, 9H).
Paso C: A una solución de 4-(2-cloro-3-fluorofenil)piper¡d¡na-1-carbox¡lato de terc-butilo (13, 0,520 g, 1,66 mmol) en CH2CI2 (10 mL) bajo una atmósfera de N2 se le añadió HCl (2 N en Et2O, 10 mL), la solución se agitó a temperatura ambiente durante 18 h. La mezcla de reacción se diluyó con Et2O (20 mL). El precipitado resultante se recolectó por filtración y se lavó con Et2O para proporcionar clorhidrato de 4-(2-cloro-3-fluorofenil)piperidina (14) como un sólido blanco (309 mg, 74%): 1 H NMR (300 MHz, DMSO-d6) 88,81-8,55 (m, 2H), 7,47-7,37 (m, 1H), 7,36-7,27 (m, 1H), 7,21-7,13 (m, 1H), 3,43-3,20, (m, 3H), 3,17-2,97 (m, 2H), 2,00-1,73 (m, 4H).
Preparación de clorhidrato de 4-(2-cloro-5-fluorofenil)piper¡d¡na (17)

Paso A: Una mezcla de 4-(((tr¡fluoromet¡l)sulfon¡l)ox¡)-5,6-d¡h¡drop¡r¡d¡na-1(2H)-carbox¡lato de ferc-butilo (2, 1,10 g, 3,3 mmol), ácido 2-cloro-5-fluorofenil borónico (0,58 g, 3,3 mmol), Pd(PPh3)4 (0,38 g, 0,33 mmol) y Na2CO32 M (20 mL) en DME (50 mL) se calentó a 80°C durante 6 h. La mezcla se enfrió a temperatura ambiente y los sólidos resultantes se eliminaron por filtración a través de una almohadilla de Celite. El filtrado se lavó con solución de salmuera (4 x 50 mL) y se concentró a presión reducida. El residuo resultante se cromatografió sobre gel de sílice (unidad Isco CombiFlash Rf, columna Redisep de 40 g, 0% a 80% de EtOAc en hexanos) para proporcionar 4-(2-cloro-5 fluorofenil)-5,6-dih¡dropiridina-1(2H)-carboxilato de ferc-butilo (15) como un aceite transparente (0,57 g, 55%): 1 H NMR (300 MHz, DMSO-d6) 87,53 7,46 (m, 1H), 7,23-7,14 (m, 2H), 5,79-5,74 (m, 1H), 4,00-3,92 (m, 2H), 3,52 (t, J = 5,7 Hz, 2H), 2,40-2,32 (m, 2H), 1,43 (s, 9H).
Paso B: A una solución de 4-(2-cloro-5-fluorofen¡l)-5,6-d¡h¡drop¡r¡d¡na-1(2H)-carboxilato de terc-butilo (15, 0,573 g, 1,84 mmol) en CH2Ch (11 mL) se le añadió HCl (solución 2,0 N en Et2O, 11,0 mL) y la mezcla se agitó a temperatura ambiente durante 18 h. La mezcla de reacción se diluyó con Et2O (30 mL), y el precipitado resultante se recolectó por filtración para proporcionar clorhidrato de 4-(2-cloro-5-fluorofenil)-1,2,3,6-tetrahidrop¡r¡d¡na (16) como un sólido blanco (0,267 g, 80%): 1 H NMR (300 MHz, DMSO-d6) 89,15 (br s, 2H), 7,54 (dd, J = 9,0, 5,4 Hz, 1H), 7,29-7,17 (m, 1H), 7,14 (dd, J= 9,3, 3,0 Hz, 1H), 5,84-5,79(m, 1H) , 3,76-3,68(m, 2H), 3,28 (t, J = 5,7 Hz, 2H), 2,62-2,53 (m,2H); ESI MS m/z 212 [M H]+.
Paso C: Una mezcla de clorhidrato de 4-(5-fluoro-2-(trifluoromet¡l)fen¡l)-1,2,3,6-tetrah¡drop¡rid¡na (16, 0,310 g, 1,31 mmol) PtO2 (0,085 g, 0,37 mmol) y HOAc (71 pL, 1,31 mmol) en EtOAc (12 mL) se agitó a temperatura ambiente durante 18 bajo una atmósfera de H2 (1 atm). La mezcla de reacción se diluyó con EtOAc (50 mL) y CH3OH (5 mL) y se filtró sobre Celite, y el filtrado se concentró a presión reducida y se disolvió en CH2Ch (5 mL). A esta solución se le añadió HCl (solución 2,0 N en Et2O, 2,0 mL) y la mezcla se agitó a temperatura ambiente durante 5 min. La suspensión resultante se diluyó con Et2O (20 mL) y los sólidos se recolectaron por filtración para proporcionar clorhidrato de 4-(2-cloro-5 fluorofenil)piper¡dina (17) como un sólido blanquecino (215 mg, 48%): 1 H NMR (300 MHz, DMSO-d6) 88,93-8,20 (m, 2H), 7,58-7,48 (m, 1H), 7,22-7,12 (m, 1H), 7,11-7,01 (m, 1H), 3,43-3,30 (m, 2H), 3,29-3,16 (m, 1H), 3,14-2,89 (m, 2H), 2,01-1,68 (m, 4H); ESI MS m/z 214 [M H]+.
Preparación de clorhidrato de 4-(3,5-bis(trifluorometil)fenil)piperidina (20)

Paso A: Una solución de 4-(((trifluoromet¡l)sulfoml)oxi)-5,6-d¡h¡drop¡nd¡na-1(2H)-carbox¡lato de ferc-but¡lo (2, 1,10 g, 3,32 mmol) y ác¡do (3,5-b¡s(tr¡fluoromet¡l)fen¡l)borón¡co (1,42 g, 3,32 mmol), Pd(Pph3)4 (0,38 g, 0,33 mmol) y Na2CO32 M (20 mL) en DME (50 mL) se calentó a 80°C durante 6 h. La mezcla se enfr¡ó a temperatura amb¡ente y los sól¡dos resultantes se el¡m¡naron por f¡ltrac¡ón a través de una almohad¡lla de Celíte. El f¡ltrado se lavó con soluc¡ón de salmuera (4 x 50 mL) y se concentró a pres¡ón reduc¡da. El res¡duo resultante se cromatograf¡ó sobre gel de síl¡ce (un¡dad Isco Comb¡Flash Rf, columna Red¡sep de 40 g, 0% a 80% de EtOAc en hexanos) para proporc¡onar 4-(3,5-b¡s(tr¡fluoromet¡l)fen¡l)-5,6-d¡h¡drop¡r¡d¡na-1(2H)-carbox¡lato de ferc-but¡lo (18) como un ace¡te amar¡llo (0,891 g, 68%): 1 H NMR (300 MHz, d Ms O-da) 88,09-8,04 (m, 2H), 8,00-7,96 (m, 1H), 6,53-6,42 (m, 1H), 4,09-4,00 (m, 2H), 3,55 (t, J = 5,7 Hz, 2H), 2,60-2,52 (m, 2H), 1,43 (s, 9H).
Paso B: A una soluc¡ón de 4-(3,5 b¡s(tr¡fluoromet¡l)fen¡l)-5,a-d¡h¡drop¡r¡d¡na-1(2H)-carbox¡lato de ferc-but¡lo (18, 0,891 g, 2,25 mmol) en CH2Ch (13,5 mL) en CH2Ch (11 mL) se añad¡ó HCl (soluc¡ón 2,0 N en Et2O, 11,0 mL) y la mezcla se ag¡tó a temperatura amb¡ente durante 18 h. La mezcla de reacc¡ón se d¡luyó con Et2O (30 mL), y el prec¡p¡tado resultante se recolectó por f¡ltrac¡ón para proporc¡onar clorh¡drato de 4-(3,5-b¡s(tr¡fluoromet¡l)fen¡l)-1,2,3,6-tetrah¡drop¡r¡d¡na (19) como un sól¡do blanco (0,452 g, 60%): 1 H NMR (300 MHz, DMSO-d6) 89,34 (br s, 2H), 8,14-8,09 (m, 2H), 8,08-8,04 (m, 1H), 6,59-6,53 (m, 1H), 3,83-3,74 (m, 2H), 3,38-3,25 (m, 2H), 2,83-2,71 (m, 2H); ESI MS m/z 296 [M H]+.
Paso C: Una mezcla de clorh¡drato de 4-(3,5-b¡s(tr¡fluoromet¡l)fen¡l)-1,2,3,6-tetrah¡drop¡r¡d¡na (19, 452 mg, 1,37 mmol), form¡ato de amon¡o (0,863 g, 13,7 mmol) y 10% de Pd/C (0,332 g) en CH3OH (10 mL) se calentó a reflujo durante 7 h. La mezcla se enfr¡ó a temperatura amb¡ente y se f¡ltró sobre Cel¡te. El f¡ltrado se concentró y el res¡duo resultante se d¡luyó en CH2CI2 (8 mL) y CH3OH (2 mL). A esta soluc¡ón se le añad¡ó HCl (soluc¡ón 2,0 N en Et2O, 6 mL). Los sól¡dos resultantes se reclectaron por f¡ltrac¡ón para proporc¡onar clorh¡drato de 4-(3,5-b¡s(tr¡fluoromet¡l)fen¡l)p¡per¡d¡na (20) como un sól¡do blanco (376 mg, 82%): 1 H NMR (300 MHz, DMSO-de) 89,05-8,58 (m, 2H), 8,03-7,97 (m, 1H), 7,95-7,87 (m, 2H), 3,44 3,29 (m, 2H, se superpone con H2O), 3,19-2,88 (m, 3H), 2,09-1,80 (m, 4H); ESI MS m/z 298 [M H]+.
Preparac¡ón de clorh¡drato de 4-(2-fluoro-6-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡na (23)

Paso A: Una mezcla de 4-(((tr¡fluoromet¡l)sulfon¡l)ox¡)-5,6-d¡h¡drop¡r¡d¡na-1(2H)-carbox¡lato de terc-but¡lo (2, 1,20 g, 3,62 mmol) y ác¡do 6-fluoro-(2-tr¡fluoromet¡l)fen¡l borón¡co (0,528 g, 2,53 mmol), Pd(PPh3)4 (0,292 g, 0,253 mmol) y Na2CO32 M (20 mL) en DME (30 mL) se calentó a 80°C durante 4 h. La mezcla se enfr¡ó a temperatura amb¡ente, se d¡luyó con EtOAc (50 mL) y se f¡ltró a través de una capa de Cel¡te. El f¡ltrado orgán¡co se lavó con soluc¡ón saturada de b¡carbonato de sod¡o (2 x 30 mL), H2O (30 mL) y se concentró a pres¡ón reduc¡da. El res¡duo resultante se cromatograf¡ó sobre gel de sílice (un¡dad Isco Comb¡Flash Compan¡on, columna Red¡sep de 40 g, 0% a 10% de EtOAc en hexanos) para proporc¡onar 4-(2-fluoro-6-(tr¡fluoromet¡l)fen¡l)-5,6-d¡h¡drop¡r¡d¡na-1(2H)-carbox¡lato de terc-but¡lo (21) como un ace¡te transparente (0,479 g, 39%): 1 H NMR (300 MHz, DMSO-da) 87,66-7,51 (m, 3H), 5,68 (s, 1H), 4,04-3,82 (m, 2H), 3,67-3,39 (m, 2H), 2,39-2,02 (m, 2H), 1,43 (s, 9H).
Paso B: Una mezcla de 4-(2-fIuoro-6-(tr¡fluoromet¡l)fen¡l)-5,6-d¡h¡drop¡r¡d¡na-1(2H)-carbox¡lato de terc-but¡lo (21, 0,479 g, 1,41 mmol) y PtO2 (0,095 g, 0,42 mmol) en EtOAc (15 mL) y HOAc (82 pL, 1,4 mmol) se ag¡tó a temperatura amb¡ente durante 72 h bajo una atmósfera de H2 (1 atm). La mezcla se d¡luyó con EtOAc (50 mL) y se f¡ltró sobre Cel¡te. El f¡ltrado se concentró y el res¡duo se cromatograf¡ó sobre gel de síl¡ce (un¡dad Isco Comb¡Flash Compan¡on, columna Red¡sep de 24 g, 0% a 15% de EtOAc en hexanos) para proporc¡onar 4-(2-fluoro-6-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡na-1-carbox¡Iato de terc-butílo (22) como un sól¡do blanco (0,219 g, 45%): 1 H NMR (300 MHz, DMSO-d6) 87,62-7,48 (m, 3H), 4,15-3,94 (m, 1H), 3,10-2,94 (m, 2H), 2,93-2,67 (m, 2H), 2,00-1,79 (m, 2H), 1,67-1,55 (m, 2H), 1,42 (s, 9H).
Paso C: A una soluc¡ón de 4-(2-fluoro-6-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡na-1-carbox¡lato de ferc-but¡lo (22, 0,219 g, 0,63 mmol) en CH2Cl2 (4 mL) se le añad¡ó HCl 2 N (soluc¡ón 2,0 N en Et2O, 4 mL), y la mezcla se ag¡tó a temperatura amb¡ente durante 4 h. La mezcla de reacc¡ón se d¡luyó con Et2O (50 mL) y los sól¡dos se recolectaron por f¡ltrac¡ón para proporc¡onar clorh¡drato de 4-(2-fluoro-6-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡na (23) como un sól¡do blanquec¡no (158 mg, 88%): 1 H NMR (300 MHz, DMSO-d6) 88,82 (brs, 1H), 8,50 (brs, 1H), 7,66-7,48 (m, 3H), 3,42-3,33 (m, 2H), 3,24-2,95 (m, 3H), 2,35-2,15 (m, 2H), 1,87-1,74 (m, 2H); ESI MS m/z 248 [M H]+.
Preparac¡ón de clorh¡drato de 4-(3,5-d¡fluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡na (28)

Paso A: Una suspensión de 3,5-difluoro-2-(trifluorometil)anilina (24, 1,0 g, 5,07 mmol) en HBr acuoso al 48% (8 mL) y H2O (8 mL) se agitó a -5°C durante 5 min. A la suspensión, se añadió gota a gota NaNO2 (350 mg, 5,07 mmol) en una solución acuosa de 10 mL manteniendo -5°C. La mezcla resultante se agitó a -5°C durante 1 h, luego se añadió en porciones CuBr (1,09 g, 7,63 mmol) y la suspensión resultante se dejó calentar lentamente a temperatura ambiente. Después de 4 horas, la solución resultante se extrajo con hexanos (3 x 75 mL). Los extractos orgánicos combinados se secaron sobre Na2SO4, se filtraron y se concentraron a presión reducida. El residuo resultante se cromatografió sobre gel de sílice (columna Redisep de 40 g, hexanos puros) para producir 1-bromo-3,5-difluoro-2-(trifluorometil)benceno (25) como un líquido amarillo claro (1,01 g, 68%). 1 H NMR (300 MHz, CDCI3) 87,34-7,28 (m, 1H), 6,99-6,85 (m, 1H).
Paso B: Una mezcla de 1-bromo-3,5-difluoro-2-(trifluorometil)benceno (25, 0,200 g, 0,76 mmol) y 4-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)-5,6-dihidropindina-1(2H)-carboxNato de ferc-butilo (0,237 g, 0,76 mmol), Pd(dppf) (0,056 g, 0,077 mmol) y Na2CO32 N (2 mL, 4 mmol) en Dm E (3 mL) se calentó a 85°C durante 5 h. La mezcla se diluyó con H2O (50 mL) y se extrajo con CH2Ch (3 x 75 mL). Los extractos orgánicos combinados se secaron sobre Na2SO4, se concentraron a presión reducida y el residuo resultante se cromatografió sobre gel de sílice (columna Redisep de 24 g, 0-25% de EtOAc en hexanos) para producir 4-(3,5-difluoro-2-(trifluorometil)fenil)-5,6-dihidropindina-1(2H)-carboxilato de ferc-butilo (26) como un aceite amarillo claro (0,325 g, 90%). 1 H NMR (300 MHz, CDCI3) 86,92-6,80 (m, 1H), 6,78-6,68 (m, 1H), 5,58 (s, 1H), 4,06-3,94 (m, 2H), 3,69-3,53 (m, 2H), 2,36-1,24 (m, 2H), 1,50 (s, 9H).
Paso C: Una mezcla de 4-(3,5-difluoro-2-(trifluorometil)fenil)-5,6-dihidropiridina-1(2H)-carboxilato de ferc-butilo (26, 0,750 g, 2,11 mmol) y 10% de Pd/C (1,0 g) en EtOH (50 mL) agitado a temperatura ambiente en atmósfera de H2 durante 24 h. La mezcla se filtró a través de Celite y el filtrado se concentró a presión reducida para proporcionar 4-(3,5-difluoro-2-(trifluorometil)fenil)piperidina-1 -carboxilato de ferc-butilo (27) como un sólido blanco (535 mg, 82%). 1 H NMR (300 MHz, CDCI3) 86,97-6,85 (m, 1H), 6,85-6,69 (m, 1H), 4,37 - 4,16 (m, 2H), 3,23-3,05 (m, 2H), 2,89-2,71 (m, 2H), 1,86-1,51 (m, 4H), 1,48 (s, 9H).
Paso D: Una solución de 4-(3,5-difluoro-2-(trifluorometil)fenil)piperidin-1-carboxilato de terc-butilo (27, 0,590 g, 1,61 mmol) en CH2Cl2 (10 mL) y HCl (solución 2,0 N en Et2O, 10 mL) agitada a temperatura ambiente durante 18 h. Los sólidos resultantes se filtraron para producir clorhidrato de 4-(3,5-difluoro-2-(trifluorometil)fenil)piperidina (28) como un sólido blanco (0,309 g, 63%). 1 H NMR (300 MHz, CDCI3) 87,01-6,94 (m, 1H), 6,94-6,76 (m, 1H), 3,82-3,60 (m, 2H), 3,42-3,02 (m, 3H), 2,22-1,99 (m, 4H).
Preparación de clorhidrato de (4-(3-fluoro-2-(trifluorometil)fenil)piperidin-1-il)(4,5,6,7-tetrahidro-1H-pirazolo[3,4-c]piridin-3-il)metanona (30)

Paso A: A una solución de clorhidrato de 4-(3-fluoro-2-trifluorometil)fenMpiperidina (5, 0,080 g, 0,28 mmol), ácido 6-(tercbutoxicarbonN)-4,5,6,7-tetrahidro-1H-pirazolo[3,4-c]pmdina-3-carboxílico (0,098 g, 0,67 mmol), y diisopropiletilamina (0,15 mL, 0,85 mmol) en DMF (5,3 mL) se le añadió EDCI (0,065 mg, 0,34 mmol) y HOBt (46 mg, 0,34 mmol), y la mezcla se agitó a temperatura ambiente durante 24 h. La mezcla se diluyó con H2O (30 mL) y se extrajo con EtOAc (4 x 30 mL). Los extractos orgánicos combinados se lavaron con una solución de salmuera saturada (4 x 30 mL) y se concentraron a sequedad bajo presión reducida. El residuo obtenido se cromatografió sobre gel de sílice (unidad Isco CombiFlash Rf, columna Redisep de 12 g, 0% a 10% de CH3OH en CH2Ch con 0,1% de NH4OH en CH2Ch) para proporcionar 3-(4-(3-fluoro-2-(trifluorometil)feml)pipendina-1-carboml)-4,5-dihidro-1H-pirazolo[3,4-c]pmdina-6(7H)-carboxilato de terc-butilo (29) como un sólido blanco (66 mg, 47%): 1 H NMR (300 MHz, DMSO-de) 813,20-12,78 (m, 1H), 7,73-7,59 (m, 1H), 7,46 (d, J = 7,2 Hz, 1H), 7,37-7,24 (m, 1H), 4,90-4,60 (m, 2H), 4,53-4,43 (m, 2H), 3,60-3,48 (m, 2H), 3,28-2,98 (m, 2H), 2,85 2,69 (m, 1H), 2,65-2,50 (m, 2H, se superpone con el disolvente), 1,87-1,56 (m, 4H), 1,42 (s, 9H); ESI MS m/z 497 [M H]+.
Paso B: A una solución de 3-(4-(3-fluoro-2-(trifluorometil)fenil)piperidina-1-carbonil)-4,5-dihidro-1H-pirazolo[3,4-c]piridina-6(7H)-carboxilato de terc-butilo (29, 0,066 g, 0,13 mmol) en CH2Ch (2 mL) se añadió HCl (2 mL, solución 2,0 N en Et2O). La mezcla se agitó a temperatura ambiente durante 18 h, se diluyó con Et2O (30 mL) y los sólidos resultantes se recolectaron por filtración para proporcionar clorhidrato de (4-(3-fluoro-2-(trifluorometil)fenil)piperidin-1-il)(4,5,6,7-tetrahidro-1H-pirazolo[3,4-c]piridin-3-il)metanona (30) como un sólido blanco (0,027 g, 47%): 1 H n Mr (300 MHz, DMSO-d6) 89,46-9,20 (m, 2H), 7,74-7,61 (m, 1H), 7,46 (d, J = 8,1 Hz, 1H), 7,37-7,25 (m, 1H), 4,70-4,44 (m, 2H), 4,34-4,22 (m, 2H), 3,50-3,10 (m, 4H), 2,93-2,76 (m, 3H), 1,86-1,60 (m, 4H); ESI MS m/z 468 [M H]+.
Preparación de clorhidrato de ((4-(3-fluoro-2-(trifluorometil)fenil)piperidin-1-il) (4,5,6,7-tetrahidro-1H-pirazolo[4,3-c]piridin-3-il)metanona (32)

Paso A: A una solución de clorhidrato de 4-(3-fluoro-2-trifluorometil)fenMpiperidina (5, 0,080 g, 0,28 mmol), ácido 5-(tercbutoxicarbonN)-4,5,6,7-tetrahidro-1H-pirazolo[3,4-c]pmdina-3-carboxílico (0,098 g, 0,67 mmol), y diisopropiletilamina (0,15 mL, 0,85 mmol) en DMF (5,3 mL) se le añadió EDCl (0,065 mg, 0,34 mmol) y HOBt (46 mg, 0,34 mmol), y la mezcla se agitó a temperatura ambiente durante 24 h. La mezcla se diluyó con H2O (30 mL) y se extrajo con EtOAc (4 x 30 mL). Los extractos orgánicos combinados se lavaron con una solución de salmuera saturada (4 x 30 mL) y se concentraron a sequedad bajo presión reducida. El residuo obtenido se cromatografió sobre gel de sílice (unidad Isco CombiFlash Rf, columna Redisep de 12 g, 0% a 10% de CH3OH en CH2Ch con 0,1% de NH4OH en CH2Ch) para proporcionar 3-(4-(3-fluoro-2-(trifluorometil)feml)pipendin-1-carboml)-6,7-dihidro-1H-pirazolo[4,3-c]pmdina-5(4H)-carboxNato de terc-butilo (31) como un sólido blanco (0,109 g, 77%): 1 H NMR (300 MHz, DMSO-de) 813,20-12,78 (m, 1H), 7,73-7,59 (m, 1H), 7,46 (d, J = 7,2 Hz, 1H), 7,37-7,24 (m, 1H), 4,90-4,60 (m, 2H), 4,53-4,43 (m, 2H), 3,60-3,48 (m, 2H), 3,28-2,98 (m, 2H), 2,85-2,69 (m, 1H), 2,65-2,50 (m, 2H, se superpone con el disolvente), 1,87-1,56 (m, 4H), 1,42 (s, 9H); ESI MS m/z 497 [M H]+.
Paso B: A una solución de 3-(4-(3-fluoro-2-(trifluorometil)fenil)piperidina-1-carbonil)-6,7-dihidro-1H-pirazolo[4,3-c]piridina-5(4H)-carboxilato de terc-butilo (31, 0,148 g, 0,30 mmol) en CH2Ch (2 mL) se le añadió HCl (2 mL, solución 2,0 N en Et2O). La mezcla se agitó a temperatura ambiente durante 18 h, se diluyó con Et2O (30 mL) y los sólidos resultantes se recolectaron por filtración para proporcionar clorhidrato de (4-(3-fluoro-2-(trifluorometil)fenil)piperidin-1-il)(4,5,6,7-tetrahidro-1H-pirazolo[4,3-c]piridin-3-il)metanona (32) como un sólido blanco (0,097 g, 75%): 1 H n Mr (300 MHz, DMSO-d6) 89,46-9,20 (m, 2H), 7,74-7,61 (m, 1H), 7,46 (d, J = 8,1 Hz, 1H), 7,37-7,25 (m, 1H), 4,70-4,44 (m, 2H), 4,34-4,22 (m, 2H), 3,50-3,10 (m, 4H), 2,93-2,76 (m, 3H), 1,86-1,60 (m, 4H); ESI MS m/z 468 [M H]+.
Preparación de sal de ácido trifluoroacético de ((4-(3,4-difluoro-2-(trifluorometil)fenil)piperidin-1-il)(4,5,6,7-tetrahidro-1H-pirazolo[3,4-c]piridin-3-il)metanona (34)

Paso A: A una solución de 4-(3,4-difluoro-2-(trifluorometil)fenil)piperidina-1-carboxilato de terc-butilo (9, 41,1 g, 113 mmol) en CH2Cl2 (50 mL) se le añadió ácido trifluoroacético (“TFA”, por sus siglas en inglés) (50 mL). La mezcla se agitó a temperatura ambiente durante 1 hora y se concentró a presión reducida. El residuo se disolvió en DMF (240 mL) y a esta solución se le añadió W,A/-diisopropiletilamina (72,4 g, 560 mmol), seguido de ácido 6-(terc-butoxicarbonil)-4,5,6,7-tetrahidro-1H-pirazolo[3,4-c]piridina-3-carboxílico (30,1 g, 113 mmol) y HBTU (74,7 g, 169 mmol). La mezcla se agitó a temperatura ambiente durante 16 h, se diluyó con EtOAc (1 L) y se lavó con H2O (1,4 L). La capa orgánica se lavó con salmuera (3 x 600 mL), se secó sobre Na2SO4, se filtró y se concentró a presión reducida. El residuo resultante se cromatografió sobre gel de sílice (30-80% de EtOAc en hexanos) para proporcionar 3-(4-(3,4-difluoro-2-(trifluorometil)fenil)piperidina-1-carbonil)-4,5-dihidro-1H-pirazolo[3,4-c]piridina-6(7H)-carboxilato de ferc-butilo (33) como un sólido blanco (41,2 g, 71%): 1 H NMR (300 MHz, CDCI3) 510,47 (br, 1H), 7,37-7,29 (m, 1H), 7,15 (m, 1H), 4,74 (br, 2H), 4,60 (s, 2H), 3,66 (br, 2H), 3,23 (m, 1H), 3,02 (br, 2H), 2,72 (m, 2H), 1,91-1,65 (m, 4H), 1,89-1,66 (m, 4H), 1,49 (s, 9H); MS (ESI+) m/z 515 [M H]+.
Paso B: A una solución de 3-(4-(3,4-difluoro-2-(trifluorometil)fenil)piperidina-1-carbonil)-4,5-dihidro-1H-pirazolo[3,4-c]piridina-6(7H)-carboxilato ferc-butilo (33, 41,2 g, 80,0 mmol) en CH2Cl2 (150 mL) se añadió TFA (70 mL). La mezcla se agitó a temperatura ambiente durante 16 h y luego se concentró a presión reducida para proporcionar sal TFA de ((4-(3,4-difluoro-2-(trifluorometil)fenil)piperidin-1-il)(4,5,6,7-tetrahidro-1H-pirazolo[3,4-c]piridin-3-il)metanona (34) como un sólido blanquecino (40,0 g, >99%). El material se usó tal cual sin caracterización espectral.
Preparación de sal de ácido trifluoroacético de (4-(3,4-difluoro-2-(trifluorometil)fenil)piperidin-1-il)(4,5,6,7-tetrahidro-1H-pirazolo[4,3-c]piridin-3-il)metanona (36)

Paso A: A una solución de 4-(3,4-difluoro-2-(trifluorometil)fenil)piperidina-1-carboxilato de terc-butilo (9, 41,1 g, 113 mmol) en CH2Cl2 (50 mL) se le añadió TFA (50 mL). La mezcla se agitó a temperatura ambiente durante 1 hora y se concentró a presión reducida. El residuo se disolvió en DMF (240 mL) y a esta solución se le añadió W,W-diisopropiletilamina (72,4 g, 560 mmol), seguido de ácido 5-(terc-butoxicarbonil)-4,5,6,7-tetrahidro-1H-pirazolo[3,4-c]piridina-3-carboxílico (30,1 g, 113 mmol) y HBTU (74,7 g, 169 mmol). La mezcla se agitó a temperatura ambiente durante 16 h, se diluyó con EtOAc (1 L) y se lavó con H2O (1,4 L). La capa orgánica se lavó con salmuera (3 x 600 mL), se secó sobre Na2SO4, se filtró y se concentró a presión reducida. El residuo resultante se cromatografió sobre gel de sílice (30-80% de EtOAc en hexanos) para proporcionar 3-(4-(3,4-difluoro-2-(trifluorometil) fenil)piperidina-1-carbonil)-6,7-dihidro-1H-pirazolo[4,3-c]piridina-5(4H)-carboxilato de ferc-butilo (35) como un sólido blanco (0,068 g, 80%): 1 H NMR (300 MHz, CDCI3) 810,23 (br, 1H), 7,36-7,28 (m, 1H), 7,15 (m, 1H), 4,86 (br, 2H), 4,62 (s, 2H), 3,72 (br, 2H), 3,27-2,74 (m, 5H), 1,90-1,64 (m, 4H), 1,48 (s, 9H); MS (ESI+) m/z 515 [M H]+.
Paso B: A una mezcla de 3-(4-(3,4-difluoro-2-(trifluorometil)fenil)piperidina-1-carbonil)-6,7-dihidro-1H-pirazolo[4,3-c]piridina-5(4H)-carboxilato de ferc-butilo (35, 41,2 g, 80,0 mmol) y C ^ C h (150 mL) se le añadió TFA (70 mL). La mezcla se agitó a temperatura ambiente durante 16 hora y se concentró a presión reducida. El residuo se disolvió en CH2Ch y la solución se lavó con NaHCO3 saturado, se secó sobre Na2SO4, se filtró y se concentró a presión reducida. El residuo resultante se cromatografió sobre gel de sílice (0-20% de CH3OH en CH2Ch) para proporcionar (4-(3,4-difluoro-2-(trifluorometil)fenil)piperidin-1-il) (4,5,6,7-tetrahidro-1H-pirazolo[4,3-c]piridin-3-il)metanona (36) como un sólido blanco (0,052 g, 95%): 1 H NMR (300 MHz, CDCI3) 87,36-7,27 (m, 1H), 7,14 (m, 1H), 4,92 (m, 2H), 4,04 (s, 2H), 3,27-2,69 (m, 7H), 1,89-1,65 (m, 4H); MS (ESI+) m/z 415 [M H]+.
Preparación de clorhidrato de (4-(3,4-difluoro-2-(trifluorometil)fenil)piperidin-1-il) (1,4,5,6-tetrahidropirrolo[3,4-c]pirazol-3-il)metanona (38)

Paso A: A una solución de 4-(3,4-difluoro-2-(trifluorometil)fenil)piperidina-1-carboxilato de terc-butilo (9, 0,100 g, 0,27 mmol) en CH2Cl2 (10 mL) se le añadió TFA (2 mL). La mezcla se agitó a temperatura ambiente durante 1 hora y se concentró a presión reducida. El residuo se disolvió en DMF (2 mL) y a esta solución se le añadió ácido 5-(tercbutoxicarbonil)-1,4,5,6-tetrahidropirrolo[3,4-c]pirazol-3-carboxílico (0,073 g, 0,28 mmol), HBTU (0,191 g, 0,43 mmol) y W,W-diisopropiletilamina (0,11 g, 0,864 mmol). La mezcla se agitó a temperatura ambiente durante 16 h y se diluyó con EtOAc, se lavó con salmuera, se secó sobre Na2SO4, se filtró y se concentró a presión reducida. El residuo resultante se cromatografió sobre gel de sílice (0-90% de EtOAc en hexanos) para dar 3-(4-(3,4-difluoro-2-(trifluorometil)fenil)piperidina-1-carbonil)-4,6-dihidropirrolo[3,4-c]pirazolo-5(1H)-carboxilato de ferc-butilo (37) como un sólido blanco (0,132 g, 91%): 1 H NMR (300 MHz, CDCI3) 810,39 (br, 1H), 7,38-7,30 (m, 1H), 7,15-7,08 (m, 1H), 4,80-4,26 (m, 6H), 3,26-3,20 (m, 2H), 2,92 (br, 1H), 1,96-1,66 (m, 4H), 1,51 (s, 9H); MS (ESI+) m/z 501 [M H]+.
Paso B: A una mezcla de 3-(4-(3,4-difluoro-2-(trifluorometil)fenil)piperidina-1-carbonil)-4,6-dihidropirrolo[3,4-c]pirazolo-5(1H)-carboxilato de ferc-butilo (37, 0,132 g, 0,26 mmol) y C ^ C h (1 mL) se le añadió TfA (1 mL). La mezcla se agitó a temperatura ambiente durante 2 horas y se concentró a presión reducida. El residuo se disolvió en CH2Cl2 y la solución se lavó con NaHCO3 saturado, se secó sobre Na2SO4, se filtró y se concentró a presión reducida. El residuo resultante se cromatografió sobre gel de sílice (0-20% de CH3OH en CH2Ch) para proporcionar (4-(3,4-difluoro-2-(trifluorometil)fenil)piperidin-1-il) (1,4,5,6 -tetrahidropirrolo[3,4-c]pirazol-3-il)metanona (38) como un sólido blanco (0,070 g, 66 %): 1 H NMR (500 MHz, CDCI3) 57,36-7,31 (m, 1H), 7,12 (m, 1H), 4,82 (br, 1H), 4,38 (br, 1H), 4,11 (s, 2H), 4,09 (s, 2H), 3,24 (m, 2H), 2,89 (br, 1H), 1,93-1,68 (m, 4H); MS (ESI+) m/z 401 [M+H]+.
Preparación de clorhidrato de (4-(2-cloro-3-fluorofenil)piperidin-1-il)(4,5,6,7-tetrahidro-1Hpirazolo[3,4-c]piridin-3-il)metanona (40)
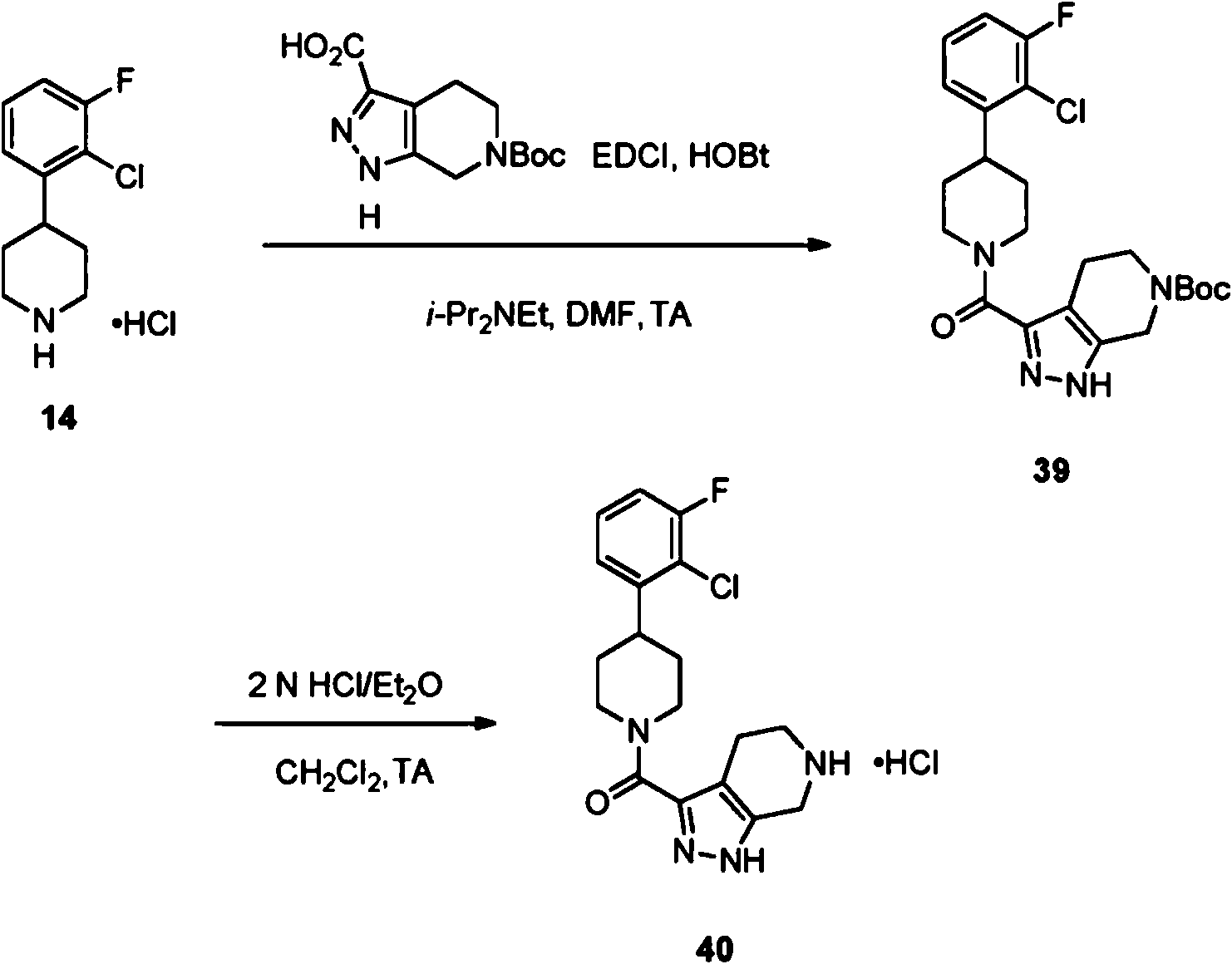
Paso A: A una mezcla de clorhidrato de 4-(2-doro-3-fluorofeml)pipendina (14, 0,796 g, 3,18 mmol), 6-(terc-butoxicarbonil)-4,5,6,7-tetrahidro-1H-pirazolo[3,4-c]ád do piridina-3-carboxílico (0,935 g, 3,50 mmol) y diisopropiletMamina (1,7 mL, 9,76 mmol) en DMF (30 mL) se le añadió EDCI (0,853 g, 4,45 mmol) y HOBt (0,601 g, 4,45 mmol). La mezcla se agitó a temperatura ambiente durante 120 h, se concentró a presión reducida y el residuo obtenido se cromatografió sobre gel de sílice (unidad Isco CombiFlash Rf, columna Redisep de 40 g, 0% a 100% de acetato de etilo en hexanos) para proporcionar 3-(4-(2-doro-3-(fluorofen¡l)p¡per¡d¡na-1-carbon¡l)-4,5-d¡h¡dro-1H-p¡razolo[3,4-c]p¡r¡d¡na-6(7H)-carbox¡lato de ferc-butilo (39) como un sólido blanco (0,694 g, 47%): 1 H NMR (300 MHz, CDCI3) 87,24-7,18 (m, 1H), 7,06-7,00 (m, 2H), 4,93-4,42 (m, 3H), 3,67-3,65 (m, 2H), 3,39-3,01 (m, 3H), 2,73-2,70 (m, 2H), 2,14-1,94 (m, 2H), 1,71-1,68 (m, 2H), 1,49 1,44 (m, 11H); ESI MS m/z 463 [M H]+.
Paso B: A una solución de 3-(4-(2-doro-3-(fluorofen¡l)p¡pe^d¡na-1-carbon¡l)-4,5-d¡h¡dro-1H-p¡razolo[3,4-c]p¡^d¡na-6(7H)-carboxilato de ferc-butilo (39, 0,694 g, 1,50 mmol) en CH2Ch (7 mL) se le añadió HCl 2 M en Et2O (16 mL). La mezcla se agitó a temperatura ambiente durante 6 h, se diluyó con Et2O (30 mL) y los sólidos resultantes se recolectaron por filtración para proporcionar clorhidrato de (4-(2-doro-3-fluorofen¡l)p¡per¡d¡n-1-¡l) (4,5,6,7-tetrah¡dro-1H-p¡razolo[4,3-c]p¡r¡d¡n-3-il)metanona (40) como un sólido blanquecino (0,509 g, 94%): 1 H NMR (300 MHz, DMSO-d6) 813,18 (br s, 1H), 9,31 (brs, 2H), 7,38-7,23 (m, 3H), 4,69-4,65 (m, 2H), 4,49-4,21 (m, 2H), 3,39-3,11 (m, 4H), 2,99-2,84 (m, 3H), 1,94-1,54 (m, 4H); ESI MS m/z 363 [M H]+.
Preparación de clorhidrato de (4-(2-cloro-3-fluorofen¡l)p¡perid¡n-1-¡l)(4,5,6,7-tetrah¡dro-1Hp¡razolo[4,3-c]p¡rid¡n-3-il)metanona (42)

Paso A: A una solución de clorhidrato de 4-(2-cloro-3-fluorofenil)piperidina (14, 90 mg, 0,36 mmol), ácido 5-(terc-butoxicarbonil)-4,5,6,7-tetrahidro-1H-pirazolo[4,3-c]piridina-3-carboxílico (96 mg, 0,36 mmol), y diisopropiletMamina (0,19 mL, 1,08 mmol) en DMF (7,8 mL) se le añadió EDCI (83 mg, 0,43 mmol) y HOBt (58 mg, 0,43 mmol). La solución resultante se agitó a temperatura ambiente durante 24 h. La mezcla de reacción se diluyó con H2O (30 mL) y el precipitado resultante se recolectó mediante filtración. Los sólidos obtenidos se cromatografiaron sobre gel de sílice (unidad Isco CombiFlash Rf, columna Redisep de 12 g, 0% a 5% de CH3OH en CH2Ch con 0,1% de NH4OH en CH2Ch) para proporcionar 3-(4-(2-cloro-3-fluorofenil)piperidina-1-carbonil)-6,7-dihidro-1Hpirazolo[4,3-c]piridina-5(4H)-carboxilato de terc-butilo (41) como una espuma blanca (139 mg, 82%): 1 H NMR (500 MHz, DMSO-cfe) 8 13,11-12,91 (m, 1H), 7,39-7,32 (m, 1H), 7,30-7,22 (m, 2H), 5,28-5,13 (m, 1H), 4,75-4,60 (m, 1H), 4,49-4,36 (m, 2H), 3,65-3,53 (m, 2H), 3,33-3,25 (m, 1H, se superpone con H2O), 3,24-3,08 (m, 1H), 2,91-276 (m, 1H), 2,67 (t, J = 5,5 Hz, 2H), 1,91-1,75 (m, 2H), 1,67-1,50 (m, 2H), 1,41 (s, 9H); ESI MS m/z 463 [M H]+.
Paso B: A una solución de 3-(4-(2-cloro-3-fluorofenil)piperidina-1-carbonil)-6,7-dihidro-1H-pirazolo[4,3-c]piridina-5(4H)-carboxilato de terc-butilo (41, 125 mg, 0,27 mmol) en C ^ C h (2 mL) se añadió HCl (solución 2,0 N en Et2O, 2 mL). La mezcla se agitó durante 18 h a temperatura ambiente. La mezcla de reacción se diluyó con Et2O (20 mL) y los sólidos resultantes se recolectaron por filtración para proporcionar clorhidrato de (4-(2-cloro-3-fluorofenil)piperidin-1-il)(4,5,6,7-tetrahidro-1H-pirazolo[4,3-c]piridin-3-il)metanona como un sólido blanco (42, 93 mg, 86%): 1 H NMR (300 MHz, DMSO-cfe) 8 9,37 (br s, 2H), 7,42-7,21 (m, 3H), 5,31-5,13 (m, 1H), 4,74-4,57 (m, 1H), 4,25-4,14 (m, 2H), 3,44-3,14 (m, 4H, se superpone con H2O), 3,00-2,75 (m, 3H), 1,93-1,77 (m, 2H), 1,70-1,47 (m, 2H) falta N-H pirazol; ESI MS m/z 363 [M H]+.
Preparación de clorhidrato de (4-(5-fluoro-2-(trifluorometil)fenil)piperidin-1-il)(4,5,6,7-tetrahidro-1H-pirazolo[3,4-c]piridin-3-il)metanona (44)

Paso A: Paso A: A una solución de clorhidrato de 4-(5-fluoro-2-(trifluorometil)fenM)piperidina (11, 93 mg, 0,33 mmol), ácido 6-(terc-butoxicarboml)-4,5,6,7-tetrahidro-1H-pirazolo[3,4-c]pindina-3-carboxílico (88 mg, 0,33 mmol), y diisopropiletilamina (0,17 mL, 0,99 mmol) en DMF (6,0 mL) se le añadió EDCI (76 mg, 0,40 mmol) y HOBt (54 mg, 0,40 mmol). La solución resultante se agitó a temperatura ambiente durante 24 h. La mezcla de reacción se diluyó con H2O (10 mL) y se extrajo con EtOAc (3 x 30 mL). Los extractos orgánicos combinados se lavaron con una solución de salmuera saturada (4 x 20 mL) y se concentraron a sequedad bajo presión reducida. El residuo obtenido se cromatografió sobre gel de sílice (unidad Isco CombiFlash Rf, columna Redisep de 12 g, 0% a 5% de MeOH en CH2Ch con 0,1% de NH4OH en CH2Ch) para proporcionar 3-(4-(5-fluoro-2-(trifluorometil)feml)pipendin-1-carboml)-4,5-dihidro-1H-pirazolo[3,4-c]pmdina-6(7H)-carboxilato de terc-butilo (43) como una película blanca (110 mg, 67%): 1 H Nm R (300 MHz, DMSO-cfe) 813,16-12,76 (m, 1H), 7,82-7,71 (m, 1H), 7,62-7,50 (m, 1H), 7,33-7,18 (m, 1H), 4,92-4,76 (m, 1H), 4,74-4,59 (m, 1H), 4,53-4,39 (m, 2H), 3,54 (t, J = 5,7 Hz, 2H), 3,21-3,01 (m, 2H), 2,86-2,69 (m, 1H), 2,66-2,53 (m, 2H), 1,83-1,62 (m, 4H), 1,42 (s, 9H); ESI MS m/z 497 [M H]+.
Paso B: A una solución de 3-(4-(5-fluoro-2-(trifluorometil)fenil)piperidina-1-carbonil)-4,5-dihidro-1H-pirazolo[3,4-c]piridina-6(7H)-carboxilato de terc-butilo (43, 107 mg, 0,21 mmol) en C ^ C h (2 mL) se le añadió HCl (2N en Et2O, 2 mL). La mezcla se agitó durante 18 h a temperatura ambiente. La mezcla de reacción se diluyó con Et2O (20 mL) y los sólidos resultantes se recolectaron por filtración para proporcionar clorhidrato de (4-(5-fluoro-2-(trifluorometil)fenil)piperidin-1-il)(4,5,6,7-tetrahidro-1H-pirazolo[3,4-c]piridin-3-il)metanona (44) como un sólido blanco (66 mg, 71%): 1 H n Mr (500 MHz, DMSO-cfe) 813,52-13,13 (m, 1H), 9,41 (br s, 2H), 7,77 (dd, J = 9,0, 5,7 Hz, 1H), 7,62-7,50 (m, 1H), 7,32-7,21 (m, 1H), 5,00-4,83 (m, 1H), 4,75-4,58 (m, 1H), 4,37-4,19 (m, 2H), 3,41-3,24 (m, 2H, se superpone con H2O), 3,22-3,04 (m, 2H), 2,94-2,73 (m, 3H), 1,86-1,64 (m, 4H); ESI MS m/z 397 [M H]+.
Preparación de clorhidrato de (4-(5-fluoro-2-(trifluorometil)fenil)piperidin-1-il)(4,5,6,7-tetrahidro-1H-pirazolo[4,3-c]piridin-3-il)metanona (46)

Paso A: A una solución de clorhidrato de 4-(5-fluoro-2-trifluorometil)fenMpiperidina (11, 90 mg, 0,32 mmol), ácido 5-(terc-butoxicarbonil)-4,5,6,7-tetrahidro-1H-pirazolo[4,3-c]piridina-3-carboxílico (85 mg, 0,32 mmol), y diisopropiletilamina (0,17 mL, 0,96 mmol) en DMF (5,8 mL) se le añadió EDCI (74 mg, 0,38 mmol) y HOBt (52 mg, 0,38 mmol). La solución resultante se agitó a temperatura ambiente durante 24 h. La mezcla de reacción se diluyó con H2O (10 mL) y se extrajo con EtOAc (3 x 30 mL). Los extractos orgánicos combinados se lavaron con una solución de salmuera saturada (4 x 20 mL) y se concentraron a sequedad bajo presión reducida. El residuo obtenido se cromatografió sobre gel de sílice (unidad Isco CombiFlash Rf, columna Redisep de 12 g, 0% a 5% de MeOH en CH2Ch con 0,1% de NH4OH en CH2Ch) para proporcionar 3-(4-(5-fluoro-2-(trifluorometil)fenil)piperidin-1-carbonil)-6,7-dihidro-1H-pirazolo[4,3-c]piridina-5(4H)-carboxilato de terc-butilo (45) como una película blanca (120 mg, 80%): 1 H Nm R (300 MHz, DMSO-cfe) 612,96 (br s, 1H), 7,76 (dd, J = 9,0, 5,7 Hz, 1H), 7,61-7,51 (m, 1H), 7,30-7,18 (m, 1H), 5,34-5,16 (m, 1H), 4,76-4,58 (m, 1H), 4,53-4,38 (m, 2H), 3,65-3,52 (m, 2H), 3,22-3,01 (m, 2H), 2,60-2,43 (m, 3H, se superpone con el disolvente), 1,83-1,65 (m, 4H), 1,42 (s, 9H); ESI MS m/z 497 [M H]+.
Paso B: A una solución de 3-(4-(5-fluoro-2-(trifluorometil)fenil)piperidina-1-carbonil)-6,7-dihidro-1H-pirazolo[4,3-c]piridina-5(4H)-carboxilato de terc-butilo (45, 120 mg, 0,24 mmol) en CH2Cl2 (2 mL) se añadió HCl (2N en Et2O, 2 mL). La mezcla se agitó durante 18 h a temperatura ambiente. Se añadió HCl adicional (2 N en Et2O, 1 mL) y la mezcla se agitó a temperatura ambiente durante 3 h. La mezcla de reacción se diluyó con Et2O (20 mL) y los sólidos resultantes se recogieron por filtración para proporcionar clorhidrato de (4-(5-fluoro-2-(trifluorometil)fenil)piperidin-1-il) (4,5,6,7-tetrahidro-1H-pirazolo[4,3-c]piridin-3-il)metanona (46) como un sólido blanco (104 mg, 99%): 1 H n MR (300 MHz, DMSO-cfe) 69,54 9,19 (m, 2H), 7,84 (dd, J = 9,0, 5,7 Hz, 1H), 7,60-7,51 (m, 1H), 7,32-7,20 (m, 1H), 5,32-5,12 (m, 1H), 4,78-4,60 (m, 1H), 4,29-4,16 (m, 2H), 3,43-3,30 (m, 2H, se superpone con H2O), 3,26-3,06 (m, 2H), 2,95 (t, J = 5,4 Hz, 2H), 2,89-2,72 (m, 1H), 1,84-1,65 (m, 4H) falta N-H pirazol; ESI MS m/z 397 [M H]+.
Preparación de clorhidrato de (4-(2-cloro-5-fluorofenil)piperidin-1-il)(4,5,6,7-tetrahidro-1H-pirazolo[3,4-c]piridin-3-il)metanona (48)

Paso A: A una solución de clorhidrato de 4-(2-cloro-5-fluorofenil)piperidina (17, 70 mg, 0,28 mmol), ácido 6-(terc-butoxicarbonil)-4,5,6,7-tetrahidro-1H-pirazolo[3,4-c]piridina-3-carboxílico (104 mg, 0,39 mmol), y diisopropiletilamina (0,15 mL, 0,84 mmol) en DMF (5,4 mL) se le añadió EDCI (65 mg, 0,34 mmol) y HOBt (45 mg, 0,34 mmol). La solución resultante se agitó a temperatura ambiente durante 18 h. La mezcla de reacción se diluyó con H2O (20 mL) y se extrajo con EtOAc (4 x 20 mL). Los extractos orgánicos combinados se lavaron con una solución de salmuera saturada (9 x 20 mL), H2O (2 x 20 mL) y se concentraron a sequedad bajo presión reducida. El residuo obtenido se cromatografió sobre gel de sílice (unidad Isco CombiFlash Rf, columna Redisep de 12 g, 0% a 100% de EtOAc en hexanos) para proporcionar 3-(4-(2-cloro-5-fluorofenil)piperidina-1-carbonil)-4,5-dihidro-1H-pirazolo[3,4-c]piridina-6(7H)-carboxilato de terc-butilo (47) como un sólido blanco (57 mg, 44%): 1 H NMR (300 MHz, DMSO-d6) 613,16-12,78 (m, 1H), 7,48 (dd, J = 9,0, 5,7 Hz, 1H), 7,28 (dd, J = 10,5, 3,3 Hz, 1H), 7,17-7,05 (m, 1H), 4,90-4,59 (m, 2H), 4,53-4,40 (m, 2H), 3,62-3,48 (m, 2H), 3,28-3,07 (m, 2H), 2,92-2,73 (m, 1H), 2,64-2,50 (m, 2H, se superpone con el disolvente), 1,93-1,69 (m, 2H), 1,68-1,49 (m, 2H), 1,42 (S, 9H); ESI MS m/z 463 [M H]+.
Paso B: A una solución de 3-(4-(2-cloro-5-fluorofenil) piperidina-1-carbonil)-4,5-dihidro-1H-pirazolo[3,4-c]piridina-6(7H)-carboxilato de terc-butilo (47, 57 mg, 0,12 mmol) en CH2Cl2 (2 mL) se le añadió HCl (2N en Et2O, 2 mL). La mezcla se agitó durante 18 h a temperatura ambiente. La mezcla de reacción se concentró a presión reducida para producir clorhidrato de (4-(2-cloro-5-fluorofenil)piperidin-1-il) (4,5,6,7-tetrahidro-1H-pirazolo[3,4-c]piridina-3-il)metanona (48) como un sólido blanco (43 mg, 87%): 1 H NMR (300 MHz, DMSO-d6) 69,42 (br s, 2H), 7,49 (dd, J = 8,7, 5,4 Hz, 1H), 7,28 (dd, J = 10,2, 3,0 Hz, 1H), 7,16-7,07 (m, 1H), 4,71-4,43 (m, 2H), 4,32-4,23 (m, 2H), 3,38-3,14 (m, 4H,se superpone con H2O), 2,91-2,72 (m, 3H), 1,89-1,73 (m, 2H), 1,69-1,50 (m, 2H), falta N-H pirazol; ESI MS m/z 363 [M H]+.
Preparación de clorhidrato de (4-(2-cloro-5-fluorofenil)piperidin-1-il)(4,5,6,7-tetrahidro-1Hpirazolo[4,3-c]piridin-3-il)metanona (50)

Paso A: A una solución de clorhidrato de 4-(2-cloro-5-fluorofenil)piperidina (17, 70 mg, 0,28 mmol), ácido 5-(terc-butoxicarbonil)-4,5,6,7-tetrahidro-1H-pirazolo[4,3-c]piridina-3-carboxílico (89 mg, 0,34 mmol), y diisopropiletMamina (0,15 mL, 0,84 mmol) en DMF (5,4 mL) se le añadió EDCI (64 mg, 0,34 mmol) y HOBt (45 mg, 0,34 mmol). La solución resultante se agitó a temperatura ambiente durante 24 h. La mezcla de reacción se diluyó con H2O (20 mL) y se extrajo con EtOAc (4 x 20 mL). Los extractos orgánicos combinados se lavaron con una solución de salmuera saturada (8 x 20 mL), H2O (20 mL) y se concentraron a sequedad bajo presión reducida. El residuo obtenido se cromatografió sobre gel de sílice (unidad Isco CombiFlash Rf, columna Redisep de 12 g, 0% a 100% de EtOAc en hexanos) para proporcionar 3-(4-(2-cloro-5-fluorofenil)piperidina-1-carbonil)-6,7-dihidro-1H-pirazolo[4,3-c]piridina-5(4H)-carboxilato de terc-butilo (49) como un sólido blanco (78 mg, 60%): 1 H NMR (300 MHz, DMSO-d6) 612,96 (s, 1H), 7,48 (dd, J = 8,7, 5,4 Hz, 1H), 7,34-7,21 (m, 1H), 7,16-7,06 (m, 1H), 5,28-5,11 (m, 1H), 4,77-4,57 (m, 1H), 4,53-4,38 (m, 2H), 3,66-3,52 (m, 2H), 3,30-3,04 (m, 2H, se superpone con H2O), 2,92-2,61 (m, 3H), 1,92-1,74 (m, 2H), 1,69-1,49 (m, 2H), 1,41 (s, 9H); ESI MS m/z 462 [M H]+.
Paso B: A una solución de 3-(4-(2-cloro-5-fluorofenil)piperidina-1-carbonil)-6,7-dihidro-1H-pirazolo[4,3-c]piridina-5(4H)-carboxilato de terc-butilo (49, 78 mg, 0,17 mmol) en CH2Ch (2 mL) se le añadió HCl (2N en Et2O, 2 mL). La mezcla se agitó durante 18 h a temperatura ambiente. La mezcla de reacción se concentró a presión reducida para producir clorhidrato de (4-(2-cloro-5-fluorofenil)piperidin-1-il)(4,5,6,7-tetrahidro-1H-pirazolo[4,3-c]piridina-3-il)metanona (50) como un sólido blanco (64 mg, 94%): 1 H NMR (300 MHz, DMSO-d6) 69,19 (br s, 2H), 7,49 (dd, J = 8,7, 5,4 Hz, 1H), 7,27 (dd, J = 10,2, 3,0 Hz, 1H), 7,17-7,07 (m, 1H), 5,34-5,05 (m, 1H), 4,79-4,56 (m, 1H), 4,38-4,19 (m, 2H), 3,44-3,07 (m, 4H, se superpone con H2O), 3,01-2,76 (m, 3H), 1,92-1,73 (m, 2H), 1,71-1,50 (m, 2H), falta N-H pirazol; ESI MS m/z 363 [M H]+.
Preparación de clorhidrato de ((4-(3,5-bis(trifluorometil)fenil)piperidin-1-il) (4,5,6,7-tetrahidro-1H-pirazolo[3,4-c]piridin-3-il)metanona (52)

Paso A: Paso A: A una solución de clorhidrato de 4-(3,5-bis(trifluorometM)fenil)piperidina (20, 100 mg, 0,31 mmol), ácido 6-(terc-butoxicarboml)-4,5,6,7-tetrahidro-1H-pirazolo[3,4-c]pindina-3-carboxílico (96 mg, 0,36 mmol), y diisopropiletilamina (0,16 mL, 0,90 mmol) en DMF (5,6 mL) se añadió EDCI (69 mg, 0,36 mmol) y HOBt (49 mg, 0,36 mmol). La solución resultante se agitó a temperatura ambiente durante 18 h. La mezcla de reacción se diluyó con H2O (30 mL) y se extrajo con EtOAc (3 x 30 mL). Los extractos orgánicos combinados se lavaron con una solución de salmuera saturada (4 x 30 mL) y se concentraron a sequedad bajo presión reducida. El residuo obtenido se cromatografió sobre gel de sílice (unidad Isco CombiFlash Rf, columna Redisep de 12 g, 0% a 5% de CH3OH en CH2Ch con 0,1% de NH4OH en CH2Ch) para proporcionar 3-(4-(3,5-bis(trifluorometil)feml)pipendina-1-carboml)-4,5-dihidro-1H-pirazolo[3,4-c]pmdina-6(7H)-carboxilato de terc-butilo (51) como una película blanca (76 mg, 45%): 1 H NMR (300 MHz, DMSO-d6) 8 13,13-12,77 (m, 1H), 8,02 7,98 (m, 2H), 7,96-7,91 (m, 1H), 4,91-4,59 (m, 2H), 4,53-4,41 (m, 2H), 3,60-3,46 (m, 2H), 3,21-3,03 (m, 2H), 2,85-2,69 (m, 1H), 2,63-2,54 (m, 2H, se superpone con el disolvente), 1,97-1,78 (m, 2H), 1,77-1,58 (m, 2H), 1,42 (s, 9H); ESI MS m/z 547 [M H]+.
Paso B: A una solución de 3-(4-(3,5-bis(trifluorometil)fenil)piperidina-1-carbonil)-4,5-dihidro-1Hpirazolo[3,4-c]piridina-6(7H)-carboxilato de terc-butilo (51, 75 mg, 0,14 mmol) en CH2Ch (1 mL) se le añadió HCl (2N en Et2O, 1 mL). La mezcla se agitó durante 18 h a temperatura ambiente. La mezcla de reacción se diluyó con Et2O (50 mL) y se concentró para producir clorhidrato de (4-(3,5-bis(trifluorometil)fenil)piperidin-1-il) (4,5,6,7-tetrahidro-1H-pirazolo[3,4-c]piridin-3-il)metanona (52) como un sólido amarillo (79 mg, >99%): 1 H NMR (300 MHz, DMSO-d6) 89,37 (br s, 2H), 8,05-7,86 (m, 3H), 4,75-4,44 (m, 2H), 4,39-4,18 (m, 2H), 3,42-3,25 (m, 2H, se superpone con H2O), 3,20- 3,03 (m, 2H), 2,95-2,75 (m, 3H), 2,03-1,80 (m, 2H), 1,79-1,62 (m, 2H), falta N-H pirazol; ESI MS m/z 447 [M H]+.
Preparación de clorhidrato de (4-(3,5-bis(trifluorometil)fenil)piperidin-1-il)(4,5,6,7-tetrahidro-1H-pirazolo[4,3-c]piridin-3-il)metanona (54)

Paso A: A una solución de clorhidrato de 4-(3,5-bis(trifluorometil)fenM)piperidina (20, 100 mg, 0,30 mmol), ácido 5-(terc-butoxicarbonil)-4,5,6,7-tetrahidro-1H-pirazolo[4,3-c]piridina-3-carboxílico (81 mg, 0,30 mmol), y diisopropiletilamina (0,18 mL, 0,90 mmol) en DMF (5,6 mL) se añadió EDCI (69 mg, 0,36 mmol) y HOBt (49 mg, 0,36 mmol). La solución resultante se agitó a temperatura ambiente durante 18 h. La mezcla de reacción se diluyó con H2O (30 mL). El precipitado resultante se recolectó por filtración para producir 3-(4-(3,5-bis(trifluorometN)fenN)piperidina-1-carbonN)-6,7-dihidro-1H-pirazolo[4,3-c]piridina-5(4H)-carboxilato de terc-butilo (53) como un sólido blanco (142 mg, 86%): 1 H NMR (300 MHz, DMSO-ak) 8 12,95 (s, 1H), 8,03-7,97 (m, 2H), 7,95-7,90 (m, 1H), 5,31-5,13 (m, 1H), 4,76-4,58 (m, 1H), 4,52-4,39 (m, 2H), 3,64-3,53 (m, 2H), 3,20-3,03 (m, 2H), 2,87-2,61 (m, 3H), 1,97-1,81 (m, 2H), 1,78-1,58 (m, 2H), 1,41(s, 9H); ESI MS m/z 547 [M H]+.
Paso B: A una solución de 3-(4-(3,5-bis(trifluorometil)fenil)piperidina-1-carbonil)-6,7-dihidro-1H-pirazolo[4,3-c]piridina-5(4H)-carboxilato de terc-butilo (53, 142 mg, 0,26 mmol) en 1:1 MeOH/CH2Ch (2 mL) se añadió HCl (2N en Et2O, 2 mL). La mezcla se agitó durante 18 h a temperatura ambiente. La mezcla de reacción se diluyó con Et2O (20 mL) y los sólidos resultantes se recolectaron por filtración para proporcionar clorhidrato de (4-(3,5-bis(trifluorometil)fenil)piperidin-1-il) (4,5,6,7-tetrahidro-1H-pirazolo[4,3-c]piridin-3-il)metanona (54) como un sólido blanquecino (127 mg, >99%): 1 H NMR (500 MHz, DMSO-cfe) 89,28 (br s, 2H), 8,02-7,98 (m, 2H), 7,96-7,92 (m, 1H), 5,30-5,09 (m, 1H), 4,78-4,55 (m, 1H), 4,28-4,14 (m, 2H), 3,43-3,28 (m, 2H, se superpone con H2O), 3,26-3,07 (m, 2H), 3,02-2,90 (m, 2H), 2,89-2,75 (m, 1H), 2,00-1,82 (m, 2H), 1,80-1,61 (m, 2H), falta N-H pirazol; ESI MS m/z 447 [M H]+.
Preparación de clorhidrato de (4-(2-fluoro-6-(trifluorometil)fenil)piperidin-1-il)(4,5,6,7-tetrahidro-1H-pirazolo[3,4-c]piridin-3-il)metanona (56)

Paso A: A una solución de clorhidrato de 4-(2-fluoro-6-(trifluorometil)fenil)piperidina (20, 83 mg, 0,29 mmol), ácido 6-(terc-butoxicarbonil)-4,5,6,7-tetrahidro-1H-pirazolo[3,4-c]piridina-3-carboxílico (78 mg, 0,29 mmol), y diisopropiletilamina (0,15 mL, 0,88 mmol) en DMF (6,3 mL) se le añadió EDCI (67 mg, 0,35 mmol) y HOBt (47 mg, 0,35 mmol). La solución resultante se agitó a temperatura ambiente durante 18 h. La mezcla de reacción se diluyó con H2O (20 mL) y los sólidos resultantes se recolectaron por filtración. Los sólidos obtenidos se cromatografiaron sobre gel de sílice (unidad Isco CombiFlash Rf, columna Redisep de 12 g, 0% a 5% de MeOH en CH2Ch con 0,1% de NH4OH en CH2Ch) para proporcionar 3-(4-(2-fluoro-6-(trifluorometil)fenil)piperidina-1-carbonil)-4,5-dihidro-1H-pirazolo[3,4-c]piridina-6(7H)-carboxilato de terc-butilo (55) como una película transparente (95 mg, 65%): 1 H NMR (300 MHz, DMSO-da) 813,27-12,72 (m, 1H), 7,63-7,45 (m, 3H), 4,86-4,56 (m, 2H), 4,55-4,38 (m, 2H), 3,63-3,43 (m, 2H), 3,27-3,00 (m, 2H), 2,92-2,41 (m, 3H, se superpone con el disolvente), 2,13-1,84 (m, 2H), 1,80-1,61 (m, 2H), 1,42 (s, 9H); ESI MS m/z 496 [M H]+.
Paso B: A una solución de 3-(4-(2-fluoro-6-(trifluorometil)fenil)piperidina-1-carbonil)-4,5-dihidro-1H-pirazolo[3,4-c]piridina-(7H)-carboxilato de terc-butilo (55, 94 mg, 0,19 mmol) en C ^ C h (3 mL) se le añadió HCl (2N en Et2O, 3 mL). La mezcla se agitó durante 18 h a temperatura ambiente. La mezcla de reacción se diluyó con Et2O (20 mL) y la mezcla se concentró a presión reducida para producir clorhidrato de (4-(2-fluoro-6-(trifluorometil)fenil)piperidin-1-il) (4,5,6,7-tetrahidro-1H-pirazolo[3,4-c]piridin-3-il)metanona (56) como un sólido blanco (80 mg, 97%): 1 H n MR (300 MHz, DMSO-cfe) 89,39-9,26 (m, 2H), 7,63-7,47 (m, 3H), 4,76-4,40 (m, 1H), 4,35-4,25 (m, 2H), 3,78-3,39 (m, 4H), 3,25-3,08 (m, 2H), 2,91-2,78 (m, 3H), 2,11-1,88(m, 2H), 1,81-1,66 (m, 2H); ESI MS m/z 397 [M H]+.
Preparación de clorhidrato de (4-(2-fluoro-6-(trifluorometil)fenil)piperidin-1-il)(4,5,6,7-tetrahidro-1H-pirazolo[4,3-c]piridin-3-il)metanona (58)

Paso A: A una solución de clorhidrato de 4-(3,5-bis(trifluorometil)fenM)piperidina (20, 100 mg, 0,30 mmol), ácido 5-(terc-butoxicarbonil)-4,5,6,7-tetrahidro-1H-pirazolo[4,3-c]piridina-3-carboxílico (81 mg, 0,30 mmol), y diisopropiletilamina (0,18 mL, 0,90 mmol) en DMF (5,6 mL) se le añadió EDCI (69 mg, 0,36 mmol) y HOBt (49 mg, 0,36 mmol). La solución resultante se agitó a temperatura ambiente durante 18 h. La mezcla de reacción se diluyó con H2O (30 mL). El precipitado resultante se recolectó por filtración para producir 3-(4-(3,5-bis(trifluorometil)fenil)pipendina-1-carbonil)-6,7-dihidro-1H-pirazolo[4,3-c]piridina-5(4H)-carboxilato de terc-butilo (53) como un sólido blanco (142 mg, 86%): 1 H NMR (300 MHz, DMSO-ak) 8 12,95 (s, 1H), 8,03-7,97 (m, 2H), 7,95-7,90 (m, 1H), 5,31-5,13 (m, 1H), 4,76-4,58 (m, 1H), 4,52-4,39 (m, 2H), 3,64-3,53 (m, 2H), 3,20-3,03 (m, 2H), 2,87-2,61 (m, 3H), 1,97-1,81 (m, 2H), 1,78-1,58 (m,2H), 1,41(s, 9H); ESI MS m/z 547 [M H]+.
Paso B: A una solución de 3-(4-(3,5-bis(trifluorometil)fenil)pipendina-1-carbonil)-6,7-dihidro-1H-pirazolo[4,3-c]pindina-5(4H)-carboxilato de terc-butilo (53, 142 mg, 0,26 mmol) en 1:1 MeOH/CH2Ch (2 mL) se le añadió HCl (2n en Et2O, 2 mL). La mezcla se agitó durante 18 h a temperatura ambiente. La mezcla de reacción se diluyó con Et2O (20 mL) y los sólidos resultantes se recolectaron por filtración para proporcionar clorhidrato de (4-(3,5-bis(trifluorometil)fenil)piperidin-1-il) (4,5,6,7-tetrahidro-1H-pirazolo[4,3-c]piridin-3-il)metanona (54) como un sólido blanquecino (127 mg, >99%): 1 H NMR (500 MHz, DMSO-cfe) 89,28 (br s, 2H), 8,02-7,98 (m, 2H), 7,96-7,92 (m, 1H), 5,30-5,09 (m, 1H), 4,78-4,55 (m, 1H), 4,28 4,14 (m, 2H), 3,43-3,28 (m, 2H, se superpone con H2O), 3,26-3,07 (m, 2H), 3,02-2,90 (m, 2H), 2,89-2,75 (m, 1H), 2,00 1,82 (m, 2H), 1,80-1,61 (m, 2H), falta N-H pirazol; ESI MS m/z 447 [M H]+.
Preparación de clorhidrato de (4-(3,5-difluoro-2-(trifluorometil)fenil)piperidin-1-il)(4,5,6,7-tetrahidro-1H-pirazolo[3,4-c]piridin-3-il)metanona (60)

Paso A: A una suspensión de clorhidrato de 4-(3,5-difluoro-2-(trifluorometil)fenil)piperidina (28, 500 mg, 1,66 mmol), ácido 6-(ferc-butoxicarboml)-4,5,6,7-tetrah¡dro-1Hp¡razolo[3,4-c]p¡nd¡na-3-carboxílico (443 mg, 1,66 mmol), y diisopropiletilamina (36 mL, 2,04 mmol) en DMF (5 mL) se le añadió HBTU (1,10 g, 2,49 mmol). La mezcla resultante se agitó a temperatura ambiente durante 18 h. La reacción se diluyó en H2O (50 mL) y se extrajo con EtOAc (3 x 75 mL). Los extractos orgánicos combinados se secaron sobre Na2SO4, se filtraron y se concentraron a presión reducida. El residuo resultante se cromatografió sobre gel de sílice (columna Redisep de 40 g, 0 -100 % EtOAc en hexanos)
para producir 3-(4-(3,5-d¡fluoro-2-(trifluoromet¡l)fenil)p¡perid¡na-1 -carbonil)-4,5 d¡h¡dro-1H-p¡razolo[3,4-c]p¡^d¡na-6(7H)-carboxilato de terc-butilo (59) como un sólido blanco (200 mg, 23%),1H NMR (300 MHz, cD ch) 8 7,017-6,876 (m, 1H), 6,876-6,741 (m, 1H), 5,306 (s, 1H), 4,632 (s, 2H), 3,776-3,581 (m, 2H), 2,762-2,631 (m, 2H), 1,986-1,653 (m, 4H), 1,500 (s, 9H), 1,399-1,189 (m, 4H).
Paso B: A una solución de 3-(4-(3,5-d¡fluoro-2-(tr¡fluoromet¡l)fen¡l)p¡pe^d¡na-1-carbon¡l)-4,5-d¡h¡dro-1H-p¡razolo[3,4-c]pmdina-6(7H)-carboxNato de terc-butilo (59, 94 mg, 0,19 mmol) en C ^ C h (3 mL) se le añadió HCl (solución 2 , 0 N en Et2O, 3 mL). La mezcla se agitó durante 18 h a temperatura ambiente. La mezcla de reacción se diluyó con Et2O (20 mL) y la mezcla se concentró a presión reducida para producir clorhidrato de (4-(3,5-d¡fluoro-2-(tr¡fluoromet¡l)fenil)piper¡din-1-il) (4,5,6,7-tetrah¡dro-1H-p¡razolo[3,4-c]pmd¡n-3-¡l)metanona (60) como un sólido blanco (80 mg, 97%):
Preparación de (4-(3,5-d¡fluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡n-1-¡l)(4,5,6,7-tetrah¡dro-1H-p¡razolo[4,3-c]p¡r¡d¡n-3-il)metanona (62)

Paso A: A una suspensión de clorhidrato de 4-(3,5-difluoro-2-(trifluorometil)fenii)piperidina (28, 500 mg, 1,66 mmol), ácido 5-(ferc-butoxicarboml)-4,5,6,7-tetrahidro-1Hpirazolo[3,4-c]pindina-3-carboxílico (443 mg, 1,66 mmol), y diisopropiletilamina (36 mL, 2,04 mmol) en DMF (5 mL) se le añadió HBTU (1,10 g, 2,49 mmol). La mezcla resultante se agitó a temperatura ambiente durante 18 h. La reacción se diluyó en H2O (50 mL) y se extrajo con EtOAc (3 x 75 mL). Los extractos orgánicos combinados se secaron sobre Na2SO4, se filtraron y se concentraron a presión reducida. El residuo resultante se cromatografió sobre gel de sílice (columna Redisep de 40 g, 0-100% de EtOAc en hexanos) para producir 3-(4-(3,5-difluoro-2-(trifluorometil)feml)pipendina-1-carboml)-6,7-dihidro-1H-pirazolo[4,3-c]pindina-5(4H)-carboxilato de ferc-butilo (61) como un sólido blanco (200 mg, 23%).
Paso B: A una solución de 3-(4-(3,5-difluoro-2-(trifluorometil)feml)pipendina-1-carbonil)-6,7-dihidro-1H pirazolo[4,3-c]piridina-5(4H)-carboxilato de ferc-butilo (61, 94 mg, 0,19 mmol) en CH2Cl2 (3 mL) se le añadió HCl (solución 2,0 N en Et2O, 3 mL). La mezcla se agitó durante 18 h a temperatura ambiente. La mezcla de reacción se diluyó con Et2O (20 mL), se lavó con una solución saturada de NaHCO3 y se concentró a presión reducida para producir (4-(3,5-difluoro-2-(trifluorometN)fenil)piperidin-1-il) ( 4,5,6,7-tetrahidro-1H-pirazolo[4,3-c]pmdin-3-il)metanona (62) como un sólido blanco (80 mg, 97%): 1H NMR (500 MHz, DMSO-d6) 812,73 (s, 1H), 7,487-7,336 (m, 2H), 5,193-5,008 (m, 1H), 4,76-4,58 (br s, 1H), 3,75 (s, 2H), 3,25-3,04 (m, 2H), 2,94-2,84 (m, 1H), 2,84 - 2,71 (br s, 1H), 2,60-2,53 (m, 2H), 1,89-1,58 (m, 4H); ESI MS m /z 415,1 [M H]+.
Ejemplo 1: Preparación de 1-(3-(4-(3-fluoro-2-(trifluorometil)fenil)piperidina-1-carbonil)-4,5-dihidro-1H-pirazolo[3,4-c]piridin-6(7H) )-il)etanona (63)
Paso A: Siguiendo el procedimiento general G P-E1, clorhidrato de ((4-(3-fluoro-2-(trifluorometil)fenil)piperidin-1-il)(4,5,6,7-tetrahidro-1H-pirazolo[3,4-c] piridin-3-il)metanona (30) y cloruro de acetilo se convirtieron en 1-(3-(4-(3-fluoro-2-(trifluorometN)fenil)piperidina-1-carbonil)-4,5-dihidro-1H-pirazolo[3,4-c]piridin-6(7H)-il)etanona como un sólido blanco (20 mg, 73%): 1H NMR (500 MHz, DMSO-d6) 813,18-12,83 (m, 1H), 7,73-7,60 (m, 1H), 7,46 (d, J = 8,0 Hz, 1H), 7,35-7,26 (m, 1H), 4,91-4,49 (m, 4H), 3,71-3,57 (m, 2H), 3,25-3,06 (m, 2H), 2,85-2,48 (m, 3H, se superpone con el disolvente), 2,12 2,05 (m, 3H), 1,83-1,66 (m, 4H); ESI MS m /z 439 [M H]+.
Ejemplo 2: Preparación de (4-(3-fluoro-2-(trifluorometN)feml)piperidin-1-il)(5-(metilsulfonil)-4,5,6,7-tetrahidro-1H-pirazolo[4,3- c ]piridin-3-il)metanona (64)
Paso A: Siguiendo el procedimiento general GP-C, clorhidrato de (4-(3-fluoro-2-(trifluorometil)fenil)piperidin-1-il)(4,5,6,7-tetrahidro-1H-pirazolo[4,3-c]piridin-3-il)metanona (32) y cloruro de metanosulfonilo se convirtieron en (4-(3-fluoro-2-(trifluorometN)fenil)piperidin-1-il) (5-(metNsulfonil)-4,5,6,7-tetrahidro-1H-pirazolo[4, 3-c]piridin-3-il)metanona como un sólido blanco (23 mg, 41%): punto de fusión (“mp”, por sus siglas en inglés) 243-246°C; 1H NMR (500 MHz, DMSO-d6) 8 13,03 (br s, 1H), 7,69-7,60 (m, 1H), 7,46 (d, J = 7,5 Hz, 1H), 7,33-7,26 (m, 1H), 5,30-5,21 (m, 1H), 4,72-4,62 (m, 1H), 4,41 4,24 (m, 2H), 3,52-3,39 (m, 2H), 3,27-3,09 (m, 2H), 2,95 (s, 3H), 2,85-2,74 (m, 3H), 1,85-1,61 (m, 4H); ESI MS m /z 475 [M H]+.
Ejemplo 3: Preparación de 3-(4-(3-fluoro-2-(trifluorometil)fenil)piperidina-1-carbonil)-[1,2,4]triazolo[4,3-a]piridina-6-carbonitrilo (65)
Paso A: A una solución de 6-bromo-[1,2,4]triazolo[4,3-a]piridina-3-carboxilato de etilo (75 mg, 0,28 mmol) en THF (2,3 mL) se le añadió una solución de L iO H ^ O (23 mg, 0,56 mmol) en H2O (1,5 mL). La mezcla se agitó durante 20 min y se neutralizó con HCl 2 N. La mezcla se concentró a presión reducida. El residuo obtenido se diluyó en DMF (3,0 mL) en atmósfera de N2. A esta mezcla se le añadió clorhidrato de 4-(3-fluoro-2-trifluorometil)fenilpiperidina (5, 78 mg, 0,28 mmol), hexafluorofosfato de benzotriazol-1-il-oxi-tris-(dimetilamino)-fosfonio (245 mg, 0,556 mmol ) y diisopropiletilamina (107 mg, 0,834 mmol). La mezcla se agitó a temperatura ambiente durante 18 h. La mezcla resultante se diluyó con H2O (20 mL). La mezcla se extrajo con EtOAc (4 x 30 mL). Las capas orgánicas combinadas se lavaron con solución de cloruro de litio al 5 % (4 x 20 mL) y se concentraron a presión reducida. El residuo resultante se cromatografió sobre gel de sílice (unidad Isco CombiFlash Rf, columna Redisep de 12 g, 0 % a 50 % de EtOAc en hexanos) para proporcionar (6 -bromo-[1,2,4]triazolo[4,3-a]piridin-3-il)(4-(3-fluoro-2-(trifluorometil)fenil)piperidin-1-il)metanona como una película naranja (87 mg, 6 6 %): 1H NMR (300 MHz, DMSO-cfe) 8 9,13-9,10 (m, 1H), 7,75-7,62 (m, 2H), 7,52-7,46 (m, 1H), 7,38-7,25 (m, 1H), 5,30 5,17 (m, 1H), 4,78-4,64 (m, 1H), 3,42-3,28 (m, 3H, se superpone con H2O), 3,11-2,92 (m, 1H), 1,98-1,70 (m, 4H); ESI MS m /z 471 [M H]+.
Paso B: Una solución de (6-bromo-[1,2,4]triazolo[4,3-a]piridin-3-il)(4-(3-fluoro-2-(trifluorometil)fenil)piperidin-1-il)metanona (87 mg, 0,19 mmol) y cianuro de zinc (43 mg, 037 mmol) en DMF (2,0 mL) se burbujeó con Ar durante 10 min. A la solución se le añadió Pd(PPIi3)4 (21 mg, 0,019 mmol), el recipiente se selló y se calentó a 130°C con microondas durante 30 min. La mezcla se diluyó con solución saturada de bicarbonato de sodio (30 mL) y se extrajo con EtOAc (3 x 30 mL). Los extractos orgánicos combinados se concentraron a sequedad bajo presión reducida. El residuo resultante se cromatografió sobre gel de sílice (unidad Isco CombiFlash Rf, columna Redisep de 12 g, 0 % a 70% de EtOAc en hexanos) y se liofilizó para proporcionar 3-(4-(3-fluoro-2-(trifluorometil)fenil)piperidina-1-carbonil)-[1,2,4]triazolo[4,3-a]piridina-6-carbonitrilo como un sólido blanco (52 mg, 67%): mp 188-190°C; 1H NMR (500 MHz, DMSO-cfe) 8 9,54-9,51 (m, 1H), 8,13 (dd, J = 9,5, 1,0 Hz, 1H), 7,81 (dd, J = 9,5, 1,5 Hz, 1H), 7,71-7,65 (m, 1H), 7,48 (d, J = 8,0 Hz, 1H), 7,32 (dd, J = 12,5, 8,5 Hz, 1H), 5,17-5,09 (m, 1H), 4,78-4,70 (m, 1H), 3,44-3,28 (m, 2H, se superpone con H2O), 3,09-3,00 (m, 1H), 1,97-1,75 (m, 4H); ESI MS m /z 418 [M H]+.
Ejemplo 4: Preparación de 3-(4-(3-fluoro-2-(trifluorometil)fenil)piperidina-1-carbonil)-A/-metil-6,7-dihidro-1H-pirazolo[3,4-c]piridina-5(4H)-carboxamida (6 6 )
Paso A: Siguiendo el procedimiento general GP-E2, clorhidrato de ((4-(3-fluoro-2-(trifluorometil)fenil)piperidin-1-il)(4,5,6,7-tetrahidro-1H-pirazolo[3,4-c]piridin-3-il)metanona (30) e isocianato de metilo se convirtieron en 3-(4-(3-fluoro-2-(trifluorometil)fenil)piperidina-1-carbonil)-N-metil-6,7-dihidro-1H-pirazolo[3,4-c]piridina- 5(4H)-carboxamida como un sólido blanco (32 mg, 41%): mp 165-170°C; 1H NMR (500 MHz, DMSO-cfe) 8 13,03-12,85 (m, 1H), 7,70-7,62 (m, 1H), 7,46 (d, J = 8,0 Hz, 1H), 7,30 (dd, J = 12,0, 8,0 Hz, 1H), 6,58-6,49 (m, 1H), 5,19-5,06 (m, 2H), 4,76-4,62 (m, 2H), 3,63-3,50 (m, 2H), 3,27-3,09 (m, 2H), 2,86-2,72 (m, 1H), 2,64 (t, J = 5,5 Hz, 2H), 2,59-2,54 (m, 3H), 1,84-1,59 (m, 4H); ESI MS m /z 454 [M H]+.
Ejemplo 5: Preparación de (5-etil-4,5,6,7-tetrahidro-1H-pirazolo[4,3-c]piridin-3-il)(4-(3-fluoro-2-(trifluorometil)fenil)piperidin-1-il)metanona (67)
Paso A: Siguiendo el procedimiento general GP-D, clorhidrato de (4-(3-fluoro-2-(trifluorometil)fenil)piperidin-1-il)(4,5,6,7-tetrahidro-1H-pirazolo[4,3-c]piridina-3-il)metanona (32) y acetaldehído se convirtieron en (5-etil-4,5,6,7-tetrahidro-1H-pirazolo[4,3-c]piridin-3-il) (4-(3-fluoro-2-(trifluorometil)fenil)piperidin-1-il)metanona como un sólido blanco (2,5 mg, 2%): 1H NMR (500 MHz, CD3OD) 8 7,64-7,53 (m, 1H), 7,38 (d, J = 8,0 Hz, 1H), 7,13 (dd, J = 12,0, 8,5 Hz, 1H), 4,90-4,72 (m, 2H, se superpone con H2O), 3,62 (br s, 2H), 3,34-3,17 (m, 2H, se superpone con el disolvente), 2,92-2,78 (m, 5H), 2,68 (q, J = 7,0 Hz, 2H), 1,96-1,74 (m, 4H), 1,20 (t, J = 7,5 Hz, 3H) falta NH-pirazol; ESI MS m /z 425 [M H]+.
Ejemplo 6: Preparación de 3-(4-(3-fluoro-2-(trifluorometil)fenil)piperidina-1-carbonil)-6,7-dihidro-1H-pirazolo[4,3-c]piridina-5(4H)-carboxilato (6 8 )
Paso A: Siguiendo el procedimiento general GP-B1, clorhidrato de (4-(3-fluoro-2-(trifluorometil)fenil)piperidin-1-il) (4,5,6,7-tetrahidro-1H-pirazolo[4,3-c]piridin-3-il)metanona (32) y cloroformiato de metilo se convirtieron en 3-(4-(3-fluoro-2-(trifluorometil)fenil)piperidina-1-carbonil)-6,7-dihidro-1H-pirazolo[4,3-c]piridina-5(4H)-carboxilato como un sólido blanco (62 mg, 60%): 1H NMR (500 MHz, DMSO-cfe) 8 13,11-12,94 (m, 1H), 7,68-7,61 (m, 1H), 7,46 (d, J = 8,0 Hz, 1H), 7,29 (dd, J = 12,5, 8,5 Hz, 1H), 5,33-5,16 (m, 1H), 4,74-4,60 (m, 1H), 4,56-4,41 (m, 2H), 3,68-3,58 (m, 5H), 3,26-3,08 (m, 2H), 2,85 2,74 (m, 1H), 2,70 (t, J = 5,5 Hz, 2H), 1,85-1,59 (m, 4H); ESI MS m /z 455 [M H]+.
Ejemplo 7: Preparación de (5-(ciclopropilmetil)-4,5,6,7-tetrahidro-1Hpirazolo[4,3-c]piridin-3-il)(4-(3-fluoro-2-(trifluorometil)fenil)piperidina-1-il)metanona (69)
Paso A: Siguiendo el procedimiento general GP-D1, clorhidrato de (4-(3-fluoro-2-(trifluorometil)fenil)piperidin-1-il)(4,5,6,7-tetrahidro-1H-pirazolo[4,3-c]piridin-3-il)metanona (32) y ciclopropano carboxaldehído se convirtieron (5-(ciclopropilmetil)
4,5,6,7-tetrahidro-1H-p¡razolo[4,3-c]p¡r¡d¡n-3-¡l) (4-(3-fluoro-2-(trifluoro-metil)fenil)piperidin-1-il)metanona como un sólido blanco (33 mg, 81%): 1H NMR (500 MHz, DMSO-da) 8 12,92-12,73 (m, 1H), 7,69-7,61 (m, 1H), 7,45 (d, J = 8,0 Hz, 1H), 7,29 (dd, J = 12,0, 8,5 Hz, 1H), 5,13-5,00 (m, 1H), 4,73-4,60 (m, 1H), 3,60-3,46 (m, 2H), 3,25-3,03 (m, 2H), 2,83-2,64 (m, 5H), 2,41-2,33 (m, 2H), 1,85-1,59 (m, 4H), 0,94-0,83 (m, 1H), 0,52-0,44 (m, 2H), 0,14-0,08 (m, 2H); ESI MS m/z 451 [M H]+.
Ejemplo 8: Preparación de 3-(4-(3-fluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡na-1-carbon¡l)-6,7-d¡hidro-1H-p¡razolo[4,3-c]p¡r¡d¡na-5(4H)-carbon¡tr¡lo (70)
Paso A: S¡gu¡endo el proced¡m¡ento general GP-D2, clorh¡drato de (4-(3-fluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡n-1-¡l) (4,5,6,7-tetrahidro-1H-p¡razolo[4,3-c]p¡r¡d¡n-3-¡l)metanona (32) y bromuro de c¡anógeno se conv¡rt¡eron en 3-(4-(3-fluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡na-1-carbon¡l)-6,7-d¡hidro-1H-p¡razolo[4,3-c]p¡r¡d¡na-5(4H)-carbon¡tr¡lo como un sól¡do blanco (35 mg, 70%): 1H NMR (500 MHz, DMSO-d6) 8 13,26-13,05 (m, 1H), 7,68-7,61 (m, 1H), 7,61 (d, J = 8,0 Hz, 1H), 7,32 (dd, J = 12,5, 8,5 Hz, 1H), 5,32-5,20 (m, 1H), 4,71-4,61 (m, 1H), 4,44-4,29 (m, 2H), 3,46 (t, J = 5,5 Hz, 2H), 3,27-3,09 (m, 2H), 2,87-2,73 (m, 3H), 1,85-1,59 (m, 4H); ESI MS m /z 422 [M H]+.
Ejemplo 9: Preparac¡ón de (4-(3-fluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡n-1-¡l)(5-(2,2,2-tr¡fluoroet¡l)-4,5,6,7-tetrahidro-1H-p¡razolo[4,3-c]p¡r¡d¡n-3-¡l)metanona (71)
Paso A: S¡gu¡endo el proced¡m¡ento general GP-D2, clorhidrato de (4-(3-fluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡n-1-¡l) (4,5,6,7-tetrahidro-1H-p¡razolo[4,3-c]p¡r¡d¡n-3-¡l)metanona (32) y tnfluorometanosulfonato de 2,2,2-tr¡fluoroet¡lo se conv¡rt¡eron en (4-(3-fluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡n-1-¡l) (5-(2,2,2-tr¡fluoroet¡l)-4,5,6,7-tetrahidro-1H-p¡razolo[4,3-c]p¡r¡d¡n-3-¡l)metanona como un sól¡do blanco (71 mg, 64%): mp 144-151°C; 1H NMR (500 MHz, DMS-d6) 8 12,99-12,81 (m, 1H), 7,68-7,61 (m, 1H), 7,45 (d, J = 8,0 Hz, 1H), 7,38-7,26 (m, 1H), 5,20-5,08 (m, 1H), 4,69-4,61 (m, 1H), 3,82-3,69 (m, 2H), 3,50-3,30 (m, 2H, se superpone con H2O), 3,24-3,06 (m, 2H), 2,92 (t, J = 6,5 Hz, 2H), 2,83-2,73 (m, 1H), 2,70 (t, J = 5,5 Hz, 2H), 1,86-1,58 (m, 4H); ESI MS m /z 479 [M H]+.
Ejemplo 10: Preparac¡ón de (4-(3-fluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡n-1-¡l)(5-(3,3,3-tr¡fluoroprop¡l)-4,5,6,7-tetrahidro-1H-p¡razolo[4,3-c]p¡r¡d¡n-3-¡l)metanona (72)
Paso A: S¡gu¡endo el proced¡m¡ento general GP-D2, clorhidrato de (4-(3-fluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡n-1-¡l)(4,5,6,7-tetrahidro-1H-p¡razolo[4,3-c]p¡r¡d¡n-3-¡l)metanona (32) y 3-bromo-1,1,1-tr¡fluoropropano se conv¡rt¡eron en (4-(3-fluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡n-1-¡l)(5-(3,3,3-tr¡fluoroprop¡l)-4,5,6,7-tetrahidro-1H-p¡razolo[4,3-c]p¡r¡d¡n-3-¡l)metanona como un sól¡do blanco (26 mg, 32 %): mp 152-159°C; 1H NMR (500 MHz, DMSO-d6) 8 12,93-12,75 (m, 1H), 7,69-7,61 (m, 1H), 7 ,4 7 - 7 , 4 3 (m, 1H), 7,29 (dd, J = 12,0, 8,5 Hz, 1H), 5,11-5,00 (m, 1H), 4,71-4,62 (m, 1H), 3,58-3,44 (m, 2H), 3,25-3,06 (m, 2H), 2,83-2,61 (m, 7H), 2,58-2,46 (m, 1H, se superpone con el d¡solvente), 1,84-1,59 (m, 4H); ESI MS m /z 493 [M H]+.
Ejemplo 11: Preparac¡ón de (4-(3-fluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡n-1-¡l)(5-(2-metox¡et¡l)-4,5,6,7-tetrahidro-3aH-p¡razolo[4,3-c]p¡r¡d¡n-3-¡l)metanona (73)
Paso A: S¡gu¡endo el proced¡m¡ento general GP-G2, clorhidrato de ((4-(3-fluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡n-1-¡l)(4,5,6,7-tetrah¡dro-1H-p¡razolo[4,3-c]p¡r¡d¡n-3-¡l)metanona (32) y bromoet¡lmet¡léter se conv¡rt¡eron en 4-(3-fluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡na-1-¡l)(5-(2-metox¡et¡l)-4,5,6,7-tetrahidro-3aH-p¡razolo[4 ,3-c]p¡r¡d¡n-3-¡l)metanona como un sól¡do blanquec¡no (28 mg, 56%): mp 147-151°C ; 1H NMR (300 MHz, DMSO-d6) 82,82 (br s, 1H), 7,65-7,27 (m, 3H), 5,18 5,09 (m, 1H), 4,75-4,60 (m, 1H), 3,51-3,45 (m, 2H), 3,27-3,11 (m, 6 H), 2,84-2,70 (m, 8 H), 1,87-1,63 (m, 4H); ESI MS m /z 475 [M H] .
Ejemplo 12: Preparac¡ón de 4-(3-fluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡na-1-¡l)(5-(oxetan-3-¡l)-4,5,6,7-tetrahidro-1H-p¡razolo[4,3-c]p¡r¡d¡n-3-¡l)metanona (74)
Paso A: S¡gu¡endo el proced¡m¡ento general GP-D1, clorhidrato de (4-(3-fluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡n-1-¡l)(4,5,6,7-tetrahidro-1H-p¡razolo[4,3-c]p¡r¡d¡n-3-¡l)metanona (32) y 3-oxetanona se conv¡rt¡eron en 4-(3-fluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡n-1-¡l)(5-(oxetan-3-¡l)-4, 5,6,7-tetrahidro-1Hp¡razolo[4,3-c]p¡r¡d¡n-3-¡l)metanona como una espuma blanca (30 mg, 29%): 1H NMR (300 MHz, CDCla) 8 10,04 (br s, 1H), 7,48-7,42 (m, 1H), 7,20-7,17 (m, 1H), 7,06 7,02 (m, 1H), 5,22-4,81 (m, 2H), 4,74 (d, J = 6 ,6 Hz, 4H), 3,89-3,81 (m, 1H), 3,62 (br s, 2H), 3,31-3,24 (m, 1H), 3,21-2,68 (m, 5H), 1,91-1,72 (m, 5H); ESI MS m /z 453 [M H] .
Ejemplo 13: Preparac¡ón de (6-et¡l-4,5,6,7-tetrahidro-1H-p¡razolo[3,4-c]p¡r¡d¡n-3-¡l)(4-(3-fluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡na-1-¡l)metanona (75)
Paso A: S¡gu¡endo el proced¡m¡ento general GP-G1, clorhidrato de ((4-(3-fluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡n-1-¡l) (4,5,6,7-tetrahidro-1H-p¡razolo[3,4-c]p¡r¡d¡n-3-¡l)metanona (30) y acetaldehído se conv¡rt¡eron en (6-et¡l-4,5,6,7-tetrah¡dro-1H-p¡razolo[3,4-c]p¡r¡d¡n-3-¡l) (4-(3-fluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡n-1-¡l)metanona como un sól¡do blanco (21 mg, 31%): 1H NMR (300 MHz, DMSO-d6) 8 12,74 (br s, 1H), 7,72-7,65 (m, 1H), 7,48-7,27 (m, 2H), 4,92-4,63 (m, 2H), 3,68 3,04 (m, 4H), 2,90-2,39 (m, 7H), 1,74-1,55 (m, 4H), 1,18-1,02 (m, 3H); ESI MS m /z 425 [M H]+.
Ejemplo 14: Preparación de (4-(3-fluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡n-1-¡l)(6-(3,3,3-tr¡fluoroprop¡l)-4,5,6,7-tetrahidro-1H-pirazolo[3,4-c]piridin-3-il)metanona (76)
Paso A: S¡gu¡endo el proced¡m¡ento general GP-G1, clorhidrato de ((4-(3-fluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡n-1-¡l) (4,5,6,7-tetrah¡dro-1H-p¡razolo[3,4-c]p¡r¡d¡n-3-¡l)metanona (30) y bromotr¡fluoromet¡l propano se conv¡rt¡eron en 4-(3-fluoro-2 -(tr¡fluoromet¡l) fen¡l) p¡per¡d¡n-1 -¡l) (6-(3,3,3-tr¡fluoroprop¡l)-4,5,6,7-tetrah¡dro-lH -p¡razolo[3,4-c]p¡r¡d¡n-3-¡l)metanona como un sól¡do blanco (19 mg, 15%): mp 162-166°C; 1H NMR (300 MHz, DMSO-da) 8 12,78 (br s, 1H), 7,72-7,65 (m, 1H), 7,48-7,27 (m, 2H), 4,92-4,63 (m, 2H), 3,57-3,53 (m, 2H), 3,27-3,09 (m, 2H), 2,88-2,39 (m, 9H), 1,77-1,72 (m, 4H); ESI MS m /z 493 [M H]+.
Ejemplo 15: Preparac¡ón de (4-(3-fluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡n-1-¡l) (6-(2-metox¡et¡l)-4,5,6,7-tetrahidro-3aH-p¡razolo[3,4- c ]p¡r¡d¡n-3-¡l)metanona (77)
Paso A: S¡gu¡endo el proced¡m¡ento general GP-G1, clorhidrato de ((4-(3-fluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡n-1-¡l) (4,5,6,7-tetrahidro-1H-p¡razolo[3,4-c]p¡r¡d¡n-3-¡l)metanona (30) y bromoet¡lmet¡léter se conv¡rt¡eron en 4-(3-fluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡n-1-¡l) (6-(2-metox¡et¡l)-4,5,6,7-tetrahidro-3aH-p¡razolo[3,4-c]p¡r¡d¡n-3-¡l)metanona como un sól¡do blanco (23 mg, 30%): 1H NMR (300 MHz, DMSO-da) 8 12,72 (br s, 1H), 7,67-7,62 (m, 1H), 7,46 (d, J= 8,1 Hz, 1H), 7.33- 7,27 (m, 1H), 4,92-4,63 (m, 2H), 3,57-3,48 (m, 4H), 3,27-3,05 (m, 5H), 2,81-2,49 (m, 7H), 1,77-1,72 (m, 4H); ESI MS m /z 455 [M H]+.
Ejemplo 16: Preparac¡ón de 3-(4-(3-fluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡na-1-carbon¡l)-4,5-d¡hidro-1H-p¡razolo[3,4-c]p¡r¡d¡na-6(4H)-carbon¡tr¡lo (78)
Paso A: S¡gu¡endo el proced¡m¡ento general GP-G2, clorhidrato de ((4-(3-fluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡n-1-¡l)(4,5,6,7-tetrah¡dro-1H-p¡razolo [3,4-c]p¡r¡d¡n-3-¡l)metanona (30) y bromuro de c¡anógeno se conv¡rt¡eron en 3-(4-(3-fluoro-2-(tr¡fluormet¡l)fen¡l)p¡per¡d¡na-1-carbon¡l)-4,5-d¡hidro-1H-p¡razolo[3,4-c]p¡r¡d¡na-6(7H)-carbon¡tr¡lo como un sól¡do blanco (38 mg, 53%): mp 194-198°C; 1H NMR (300 MHz, DMSO-d6) 8 12,98 (br s, 1H), 7,68-7,65 (m, 1H), 7,47 (d, J= 7,8 Hz, 1H), 7.34- 7,27 (m, 1H), 4,92-4,63 (m, 2H), 4,48-4,40 (m, 2H), 3,43-3,38 (m, 2H), 3,27-3,05 (m, 2H), 2,88-2,71 (m, 3H), 1,77 1,72 (m, 4H); ESI MS m /z 422 [M H]+.
Ejemplo 17: Preparac¡ón de (4-(3-fluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡n-1-¡l)(6-oxetan-3-¡l)-4,5,6,7-tetrahidro-1H-p¡razolo[3,4- c ]p¡r¡d¡n-3-¡l)metanona (79)
Paso A: S¡gu¡endo el proced¡m¡ento general GP-G1, clorhidrato de ((4-(3-fluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡n-1-¡l)(4,5,6,7-tetrah¡dro-1H-p¡razolo [3,4-c]p¡r¡d¡n-3-¡l)metanona (30) y oxetan-3-ona se conv¡rt¡eron en (4-(3-fluoro-2-(tr¡fluormet¡l)p¡per¡d¡n-1-¡l) (6-oxetan-3-¡l)-4,5,6,7-tetrahidro-1H-p¡razolo[3,4-c]p¡r¡d¡n-3-¡l)metanona como un sól¡do blanco (30 mg, 39%): mp 148-151°C; 1H NMR (300 MHz, DMSO-d6) 8 12,77 (br s, 1H), 7,70-7,63 (m, 1H), 7,47 (d, J= 8,1 Hz, 1H),7,34-7,27 (m, 1H), 4,91-4,47 (m, 6 H), 3,71-3,66 (m, 1H), 3,47-3,34 (m, 2H), 3,28-3,06 (m, 2H), 2,93-2,78 (m, 1H), 2,74-2,53(m, 2H), 1,91-1,78 (m, 2H), 1,89-1,55 (m, 4H); ESI MS m /z 453 [M H]+.
Ejemplo 18: Preparac¡ón de (6-(c¡cloprop¡lmet¡l)-4,5,6,7-tetrahidro-1H-p¡razolo[3,4-c]p¡r¡d¡n-3-¡l)(4-(3-fluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡n-1 -¡l)metanona (80)
Paso A: S¡gu¡endo el proced¡m¡ento general GP-G1, clorhidrato de ((4-(3-fluoro-2-(tr¡fluoro-met¡l)fen¡l)p¡per¡d¡n-1-¡l) (4,5,6,7-tetrahidro-1H-p¡razolo[3,4-c]p¡r¡d¡n-3-¡l)metanona (30) y c¡clopropano carbaldehído se conv¡rt¡eron en (6 -(c¡cloprop¡lmet¡l)-4,5,6,7-tetrahidro-1H-p¡razolo[3,4-c]p¡r¡d¡n-3-¡l)(4-(3-fluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡n-1-¡l)metanona como un sól¡do blanco (23 mg, 34%): 1H NMR (300 MHz, DMSO-d6) 8 12,74 (br s, 1H), 7,72-7,65 (m, 1H), 7,47 (d, J= 7,8 Hz, 1H), 7,34-7,27 (m, 1H), 4,92-4,63 (m, 2H), 3,59-3,53 (m, 2H), 3,32-3,09 (m, 2H), 2,90- 2,39 (m, 7H), 1,81-1,62 (m, 4H), 0,92-0,85 (m, 1H), 0,52-0,48 (m, 2H), 0,14-0,10 (m, 2H); ESI MS m /z 451 [M H]+.
Ejemplo 19: 1-(3-(4-(3,4-d¡fluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡n-1-carbon¡l)-4,5-d¡hidro-1H-p¡razolo[3,4-c]p¡r¡d¡n-6(7H)-¡l)etanona (81)
Paso A: S¡gu¡endo el proced¡m¡ento general GP-G1, sal TFA de (4-(3,4-d¡fluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡n-1-¡l) (4,5,6,7-tetrahidro-1H-p¡razol[3,4-c]p¡r¡d¡n-3-¡l)metanona (34) y cloruro de acet¡lo se conv¡rt¡eron para proporc¡onar 1-(3-(4-(3,4-d¡fluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡na-1-carbon¡l)-4,5-d¡hidro-1H-p¡razolo[3,4-c]p¡r¡d¡n-6(7H)-¡l)etanona como un sól¡do blanco (29,2 g, 80%): 1H NMR (500 MHz, CDCla) 8 10,59 (br, 1H), 7,36-7,29 (m, 1H), 7,15 (m, 1H), 4,81 (br, 2H), 4,77 y 4,65 (s, 2H), 3,85 (br, 1H), 3,68 (m, 1H), 3,26-2,69 (m, 5H), 2,21 y 2,19 (s, 3H), 1,89-1,73 (m, 4H) MS (ESI+) m /z 457 [M+H]+.
Ejemplo 20: 1-(3-(4-(3,4-d¡fluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡n-1-carbon¡l)-6,7-d¡hidro-1H-p¡razolo[3,4-c]p¡r¡d¡n-5(4H)-¡l)etanona (82)
Paso A: S¡gu¡endo el proced¡m¡ento general GP-B1, (4-(3,4-d¡fluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡n-1-¡l) (4,5,6,7-tetrahidro-1H-p¡razolo[4,3-c]p¡r¡d¡n-3-¡l)metanona (36) y cloruro de acet¡lo se conv¡rt¡eron en 1-(3-(4-(3,4-d¡fluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡n-1-carbon¡l)-6,7-d¡hidro-1H-p¡razolo[4,3-c]p¡r¡d¡n-5(4H)-¡l)etanona como un sól¡do blanco
(0 ,046 g, 85 ): 1H N M R (300 M H z , C D C I3) 5 10 ,15 (b r, 1 H ), 7 ,36 -7 ,27 (m , 1 H ), 7 ,15 (m , 1 H ), 5 ,33 -4 ,72 (m , 4 H ), 3 ,90 -3 ,73 (m , 2 H ), 3 ,30 - 2 ,76 (m , 5 H ), 2 ,20 (s, 3 H ), 1 ,89 -1 ,70 (m , 4 H ); M S (E S I+ ) m /z 457 [M H ]+ .
Ejemplo 21: 1-(3-(4-(3,4-d¡fluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡na-1-carbon¡l)p¡rrolo[3,4-c]p¡razol-5(1H,4H,6H)-¡l)etanona (83)
Paso A: S¡gu¡endo el proced¡m¡ento general GP-B1, (4-(3,4-d¡fluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡n-1-¡l)(1,4,5,6-tetrah¡drop¡rrolo [3,4-c]p¡razol-3-¡l)metanona (38) y cloruro de acet¡lo se conv¡rt¡eron en 1-(3-(4-(3,4-d¡fluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡na-1-carbon¡l)p¡rrolo[3,4-c]p¡razol-5(1H,4H,6H)-¡l)etanona como un sól¡do blanco (0,045 g, 72%): 1H NMR (500 MHz, CDCl3) 510,92 (br, 1H), 7,37-7,32 (m, 1H), 7,12 (m, 1H), 4,81-4,21 (m, 6 H), 3,29-2,88 (m, 3H), 2,18 y 2,16 (s, 3H), 1,97-1,70 (m, 4H); MS (ESI+) m /z 443 [M+H]+.
Ejemplo 22: Preparac¡ón de 1-(3-(4-(3,4-d¡fluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡na-1-carbon¡l)p¡rrolo[3,4-c]p¡razol-5(1H,4H,6H)-¡l)propan-1-ona (84)
Paso A: S¡gu¡endo el proced¡m¡ento general GP-H1, (4-(3,4-d¡fluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡n-1-¡l)(1,4,5,6-tetrah¡drop¡rrolo [3,4-c]p¡razol-3-¡l)metanona (38) y cloruro de prop¡on¡lo se conv¡rt¡eron en 1-(3-(4-(3,4-d¡fluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡na-1-carbon¡l)p¡rrol[3,4-c]p¡razol-5(1H,4H, 6H)-¡l)propan-1-ona como un sól¡do blanco (37 mg, 51%): mp >260°C; 1H NMR (500 MHz, CDCla) 57,38-7,30 (m, 1H), 7,13-7,10 (m, 1H), 4,99-4,61 (m, 5,5H), 4,46-4,19 (m, 0,5H), 3,46-2,72 (m, 3H), 2,42-2,35 (m, 2H), 1,99-1,92 (m, 2H), 1,82-1,57 (m, 2H), 1,35-1,16 (m, 3H), falta N-H p¡razol; ESI MS m /z 457 [M H]+.
Ejemplo 23: Preparac¡ón de 1-(3-(4-(3,4-d¡fluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡na-1-carbon¡l)p¡rrolo[3,4-c]p¡razol-5(1H,4H,6H)-¡l)-2-met¡lpropan-1-ona (85)
Paso A: S¡gu¡endo el proced¡m¡ento general GP-H1, (4-(3,4-d¡fluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡n-1-¡l) (1,4,5,6-tetrah¡drop¡rrolo[3,4-c]p¡razol-3-¡l)metanona (38) y cloruro de ¡sobut¡r¡lo se conv¡rt¡eron en 1-(3-(4-(3,4-d¡fluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡na-1-carbon¡l)p¡rrolo[3,4-c]p¡razol-5(1H,4H,6H)-¡l)-2-met¡lpropan-1-ona como un sól¡do blanco (29 mg, 38%): mp 249-253°C; 1H NMR (500 MHz, DMSO-cfe) 5 13,21, (br s, 1H), 7,81-7,68 (m, 1H), 7,57-7,47 (m, 1H), 4,83-3,65 (m, 6 H), 3,29-2,67 (m, 4H), 1,82-1,61 (m, 4H), 1,05 (d, J = 9 Hz, 6 H); ESI MS m /z 471 [M H]+.
Ejemplo 24: Preparac¡ón de 1-(3-(4-(3,4-d¡fluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡na-1-carbon¡l)p¡rrolo[3,4-c]p¡razol-5(1H,4H,6H)-¡l)-3-met¡lbutan-1-ona (86 )
Paso A: S¡gu¡endo el proced¡m¡ento general GP-H1, (4-(3,4-d¡fluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡n-1-¡l)(1,4,5,6-tetrah¡drop¡rrolo[3,4-c]p¡razol-3-¡l)metanona (38) y cloruro de ¡sovaler¡lo se conv¡rt¡eron en 1-(3-(4-(3,4-d¡fluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡na-1-carbon¡l)p¡rrolo[3,4-c]p¡razol-5(1H,4H,6H)-¡l)-3-met¡lbutan-1-ona como un sól¡do blanco (42 mg, 54%): 1H NMR (500 MHz, CDCla) 57,37-7,28 (m, 1H), 7,17-7,08 (m, 1H), 4,95-4,53 (m, 5,5H), 4,43-3,90 (m, 0,5H), 3,34-2,70 (m, 3H), 2,30-2,19 (m, 3H), 1,98-1,89 (m, 2H), 1,80-1,58 (m, 2H), 1,05-0,94 (m, 6 H) falta N-H p¡razol; ESI MS m /z 485 [M H]+.
Ejemplo 25: Preparac¡ón de (4-(3,4-d¡fluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡n-1-¡l) (5-et¡l-4,5,6,7-tetrah¡dro-1H-p¡razolo[4,3-c]p¡r¡d¡n-3-¡l)metanona (87)
Paso A: S¡gu¡endo el proced¡m¡ento general GP-D1, (4-(3,4-d¡fluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡n-1-¡l) (4,5,6,7-tetrah¡dro-1H-p¡razolo[4,3-c]p¡r¡d¡n-3-¡l)metanona (36) y acetaldehído se conv¡rt¡eron en (4-(3,4-d¡fluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡n-1-¡l) (5-et¡l-4,5,6,7-tetrah¡dro-1H-p¡razolo[4,3-c]p¡r¡d¡n-3-¡l)metanona como un sól¡do blanco (35 mg, 36%): mp 185-190°C; 1H NMR (300 MHz, DMSO-d6) 5 12,793 (s, 1H), 7,903-7,609 (m, 1H), 7,60-7,40 (m, 1H), 5,24-4,93 (m, 1H), 4,85-4,49 (m, 1H), 3,45 (s, 2H), 3,13 (s, 2H), 2,96-2,71 (m, 1H), 2,61 (s, 4H), 2,58-2,52 (m, 1H), 1,84 1,57 (br s, 4H), 1,19-0,97 (t, 3H); ESI MS m /z 433,1 [M H]+.
Ejemplo 26: Preparac¡ón de (5-(c¡cloprop¡lmet¡l)-4,5,6,7-tetrah¡dro-1Hp¡razolo[4,3-c]p¡r¡d¡n-3-¡l)(4-(3,4-d¡fluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡n-1 -¡l)metanona (88 )
Paso A: S¡gu¡endo el proced¡m¡ento general GP-D1, (4-(3,4-d¡fluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡n-1-¡l) (4,5,6,7-tetrah¡dro-1H-p¡razolo[4,3-c]p¡r¡d¡n-3-¡l)metanona (36) y c¡clopropano carboxaldehído se conv¡rt¡eron en (5-(c¡cloprop¡lmet¡l)-4,5,6,7-tetrah¡dro-1H-p¡razolo[4,3-c]p¡r¡d¡n-3-¡l) (4-(3,4-d¡fluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡n-1-¡l)metanona como sól¡do blanco (38 mg, 37%): No se observó fus¡ón clara; 1H NMR (500 MHz, DMSO-cfe) 5 12,80 (s, 1H), 7,80-7,66 (m, 1H), 7,54-7,45 (m, 1H), 5,19-5,03 (br s, 1H), 4,73-4,58 (m, 1H), 3,66-3,46 (br s, 1H), 3,22-3,05 (br s, 2H), 2,85-2,63 (m, 4H), 1,83-1,59 (br s, 4H), 0,99-0,82 (br s, 1H), 0,58-0,43 (m, 2H), 0,21-0,07 (br s, 2H); ESI MS m /z 469,2 [M H]+.
Ejemplo 27: Preparac¡ón de (4-(3,4-d¡fluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡n-1-¡l)(5-(oxetan-3-¡l)-4,5,6,7-tetrah¡dro-1H-p¡razolo[4,3-c]p¡r¡d¡n-3-¡l)metanona (89)
Paso A: S¡gu¡endo el proced¡m¡ento general GP-D1, (4-(3,4-d¡fluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡n-1-¡l)(4,5,6,7-tetrah¡dro-1H-p¡razolo[4,3-c]p¡r¡d¡n-3-¡l)metanona (36) y 3-oxetanona se conv¡rt¡eron en (4-(3,4-d¡fluoro-2(trifluorometil)fenil)piperidin-1-il) (5-(oxetan-3-¡l)-4,5,6,7-tetrah¡dro-1Hp¡razolo[4,3-c]p¡r¡d¡n-3-¡l)metanona como un sólido blanco (28 mg, 27%): mp 212-215°C ; 1H NMR (300 MHz, DMSO-da) 8 12,847 (s, 1H), 7,796-7,684 (m, 1H), 7,546-7,458 (m, 1H), 5,151 (d, 1H), 4,737-4,554 (m, 3H), 4,554-4,423 (m, 2H), 3,755-3,600 (m, 1H), 3,379 (s, 2H), 3,222-3,050 (br s, 2H), 2,844-2,643 (m, 3H), 1,898-1,515 (br s, 4H); ESI MS m /z 471,2 [M H]+.
Ejemplo 28: Preparación de (4-(3,4-difluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡n-1-il) (5-neopent¡l-4,5,6,7-tetrah¡dro-1H-pirazolo[4,3-c]pir¡d¡n-3-¡l)metanona metanona (90)
Paso A: S¡gu¡endo el proced¡m¡ento general GP-D1, (4-(3,4-d¡fluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡n-1-¡l)(4,5,6,7-tetrah¡dro-1H-p¡razolo[4,3-c]p¡r¡d¡n-3-¡l)metanona (36) y p¡valdehído se conv¡rt¡eron en (4-(3,4-d¡fluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡n-1-¡l) (5-neopent¡l-4,5,6,7-tetrah¡dro-1H-p¡razolo[4,3-c]p¡r¡d¡n-3-¡l)metanona metanona como un sól¡do blanco (11 mg, 10%): mp 203-210°C; 1H NMR (500 MHz, DMSO-cfe) 8 12,789 (s, 1H), 7,821-7,681 (m, 1H), 7,553-7,424 (m, 1H), 5,216-5,064 (br s, 1H), 4,755-4,579 (br s, 1H), 3,686-3,546 (m, 2H), 3,172-3,070 (m, 2H), 2,845-2,710 (m, 1H), 2,249 (s, 2H), 1,799 1,612 (m, 4H), 0,864 (s, 9H); ESI MS m /z 485,2 [M H]+.
Ejemplo 29: Preparac¡ón de 3-(4-(3,4-d¡fluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡na-1-carbon¡l)-6,7-d¡h¡dro-1H-p¡razolo[4,3-c]p¡r¡d¡na-5(4H))-carbox¡lato de met¡lo (91)
Paso A: S¡gu¡endo el proced¡m¡ento general GP-B1, (4-(3,4-d¡fluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡n-1-¡l)(4,5,6,7-tetrah¡dro-1H-p¡razolo[4,3-c]p¡r¡d¡n-3-¡l)metanona (36) y cloroform¡ato de met¡lo se conv¡rt¡eron en 3-(4-(3,4-d¡fluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡na-1-carbon¡l)-6,7-d¡h¡dro-1H-p¡razolo[4,3-c]p¡r¡d¡na-5(4H)- carbox¡lato de met¡lo como un sól¡do blanco (21 mg, 20%): mp 248-252°C; 1H NMR (500 MHz, DMSO-cfe) 8 12,924 (s, 1H), 7,763-7,692 (m, 1H), 7,540-7,472 (m, 1H), 5,358-5,152 (br s, 1H), 4,754-4,605 (br s, 1H), 4,650-4,418 (m, 2H), 3,705-3,581 (m, 6 H), 3,119-3,100 (m, 3H), 2,860-2,731 (m, 4H), 1,296-1,237 (m, 6 H); ESI MS m /z 473,1 [M H]+.
Ejemplo 30: Preparac¡ón de 3-(4-(3,4-d¡fluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡na-1-carbon¡l)-N-met¡l-6,7-d¡h¡dro-1H-p¡razolo[4,3-c]p¡r¡d¡na-5(4H)-carboxam¡da (92)
Paso A: S¡gu¡endo el proced¡m¡ento general GP-B2, (4-(3,4-d¡fluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡n-1-¡l)(4,5,6,7-tetrah¡dro-1H-p¡razolo[4,3-c]p¡r¡d¡n-3-¡l)metanona (36) e ¡soc¡anato de met¡lo se conv¡rt¡eron en 3-(4-(3,4-d¡fluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡n-1-carbon¡l)-W-met¡l-6,7-d¡h¡dro-1H-p¡razolo[4,3-c]p¡r¡d¡na-5(4H)-carboxam¡da como un sól¡do blanco (28 mg, 36%): No se observó fus¡ón clara; 1H NMR (500 MHz, DMSO-cfe) 8 12,895 (s, 1H), 7,780-7,692 (m, 1H), 7,538-7,472 (m, 1H), 6,579-6,490 (m, 1H), 5,202-5,086 (m, 1H), 4,743-4,622 (m, 1H), 4,465-4,332 (m, 2H), 3,630-3,499 (m, 2H), 3,209-3,095 (m, 2H), 2,849-2,730 (m, 1H), 2,673-2,602 (m, 2H), 2,602-2,542 (m, 3H), 1,912-1,588 (m, 5H); ESI MS m /z 472,2 [M H]+.
Ejemplo 31: Preparac¡ón de (4-(3,4-d¡fluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡n-1-¡l) (5-(3,3,3-tr¡fluoroprop¡l)-4,5,6,7-tetrah¡dro-1 H-p¡razolo[4,3-c]p¡r¡d¡n-3-¡l)metanona (93)
Paso A: S¡gu¡endo el proced¡m¡ento general GP-D2, (4-(3,4-d¡fluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡n-1-¡l)(4,5,6,7-tetrah¡dro-1H-p¡razolo[4,3-c]p¡r¡d¡n-3-¡l)metanona (36) y 1,1,1-tr¡fluoro-3-bromopropano se conv¡rt¡eron en (4-(3,4-d¡fluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡n-1-¡l) (5-(3,3,3-tr¡fluoroprop¡l)-4,5,6,7-tetrah¡dro-1H-p¡razolo[4,3-c]p¡r¡d¡n-3-¡l)metanona como un sól¡do blanco (24 mg, 21% ): mp 190-195°C; 1H NMR (500 MHz, DMSO-cfe) 8 12,939-12,764 (m, 1H), 7,847-7,665 (m, 1H), 7,591-7,417 (m, 1H), 5,213-4,965 (m, 1H), 4,729-4,555 (m, 1H), 3,637-3,456 (m, 2H), 3,223-3,039 (br s, 2H), 2,853 2,698 (m, 5H), 2,698-2,623 (m, 2H), 2,596-2,522 (m, 1H), 1,859-1,589 (m, 4H); ESI MS m /z 511,1 [M H]+.
Ejemplo 32: Preparac¡ón de (4-(3,4-d¡fluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡n-1-¡l) (5-(2-metox¡et¡l)-4,5,6,7-tetrah¡dro-1H-p¡razolo[4,3-c]p¡r¡d¡n-3-¡l)metanona (94)
Paso A: S¡gu¡endo el proced¡m¡ento general GP-D2, (4-(3,4-d¡fluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡n-1-¡l)(4,5,6,7-tetrah¡dro-1H-p¡razolo [4,3-c]p¡r¡d¡n-3-¡l) metanona (36) y 2-metox¡bromoetano se conv¡rt¡eron en (4-(3,4-d¡fluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡n-1-¡l)(5-(2-metox¡et¡l)-4,5,6,7-tetrah¡dro-1H-p¡razolo[4,3-c]p¡r¡d¡n-3-¡l)metanona como un sól¡do blanco (24 mg, 23%): mp 179-182°C; 1H NMR (500 MHz, DMSO-d6) 8 12,805 (s, 1H), 7,824-7,654 (m, 1H), 7,563-7,438 (m, 1H), 5,085 (s, 1H), 4,660 (s, 1H), 3,517 (s, 4H), 3,268 (s, 3H), 3,187-3,063 (m, 2H), 2,906-2,604 (m, 6 H), 1,885-1,550 (m, 4H); ESI MS m /z 473,2 [M H]+.
Ejemplo 33: Preparac¡ón de 3-(4-(3,4-d¡fluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡na-1-carbon¡l)-6,7-d¡h¡dro-1Hp¡razolo[4,3-c]p¡r¡d¡na-5(4H)-carbon¡tr¡lo (95)
Paso A: S¡gu¡endo el proced¡m¡ento general GP-D2, (4-(3,4-d¡fluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡n-1-¡l)(4,5,6,7-tetrah¡dro-1H-p¡razolo[4,3-c]p¡r¡d¡n-3-¡l)metanona (36) y bromuro de c¡anógeno se conv¡rt¡eron en 3-(4-(3,4-d¡fluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡na-1-carbon¡l)-6,7-d¡h¡dro-1H-p¡razolo[4,3-c]p¡r¡d¡na-5(4H)-carbon¡tr¡lo como un sól¡do blanco (95 mg, cant.): No se observó fus¡ón clara; 1H NMR (500 MHz, DMSO-d6) 8 13 ,115 (s, 1H), 7,833-7,627 (m, 1H), 7,627 7,409 (m, 1H), 5,188-5,067 (m, 1H), 4,558-4,469 (m, 1H), 4,374 (s, 2H), 3,538-3,404 (m, 2H), 3,074-2,98 (br s, 2H), 2,877 2,805 (m, 2H), 1,801-1,694 (br s, 4H); ESI MS m /z 440 [M H]+.
Ejemplo 34: Preparación de (4-(3,4-difluoro-2(trifluorometil)fenil)piperidin-1-il)(5-etil-1,4,5,6-tetrahidropirrolo[3,4-c]pirazol-3-il)metanona (96)
Paso A: Siguiendo el procedimiento general GP-J1, (4-(3,4-difluoro-2-(trifluorometil)fenil)piperidin-1-il)(1,4,5,6-tetrahidropirrolo[3,4-c]pirazol-3-il)metanona (38) y acetaldehído se convirtieron en (4-(3,4-difluoro-2-(trifluorometil)fenil)piperidin-1-il) (5-etil-1,4,5,6-tetrahidropirrolo[3,4-c]pirazol-3-il)metanona como un sólido blanco (36 mg, 76%): No se observó fusión clara; 1H NMR (500 MHz, DMSO-cfe) 8 13,140-12,832 (m, 1H), 7,879-7,669 (m, 1H), 7,669 7,408 (m, 1H), 5,271-3,901 (m, 2H), 3,901-3,559 (m, 4H), 3,216-3,013 (m, 2H), 2,960-2,684 (m, 3H), 1,854-1,569 (m, 4H), 1,162-1,005 (m, 3H); ESI MS m/z 429,2 [M H]+.
Ejemplo 35: Preparación de (5-(ciclopropilmetil)-1,4,5,6-tetrahidropirrolo[3,4-c]pirazol-3-il)(4-(3,4-difluoro-2-(trifluorometil)fenil)piperidin-1-il)metanona (97)
Paso A: Siguiendo el procedimiento general GP-J1, (4-(3,4-difluoro-2-(trifluorometil)fenil)piperidin-1-il)(1,4,5,6-tetrahidropirrolo[3,4-c]pirazol-3-il)metanona (38) y ciclopropano carboxaldehído se convirtieron en ((5-(ciclopropilmetil)-1,4,5,6-tetrahidropirrolo[3,4-c]pirazol-3-il) (4-(3,4-difluoro-2-(trifluorometil)fenil)piperidin-1-il)metanona como un sólido blanco (36 mg, 76%): No se observó fusión clara; 1H NMR (500 MHz, DMSO-cfe) 8 13,101-12,818 (m, 1H), 7,887-7,665 (m, 1H), 7,665-7,423 (m, 1H), 5,223-3,923 (m, 2H), 3,923-3,594 (m, 4H), 3,258-3,667 (m, 3H), 2,667-2,533 (m, 2H), 1,827 1,606 (m, 4H), 0,988-0,793 (m, 1H), 0,598-0,417 (m, 2H), 0,235-0,087 (m, 2H); ESI MS m/z 455,1 [M H]+.
Ejemplo 36: Preparación de (5-(ciclopropilmetil)-1,4,5,6-tetrahidropirrolo[3,4-c]pirazol-3-il)(4-(3,4-difluoro-2-(trifluorometil)fenil)piperidin-1-il)metanona (98)
Paso A: Siguiendo el procedimiento general GP-J1, (4-(3,4-difluoro-2-(trifluorometil)fenil)piperidin-1-il)(1,4,5,6-tetrahidropirrolo[3,4-c]pirazol-3-il)metanona (38) y 3-oxetanona se convirtieron en (4-(3,4-difluoro-2-(trifluorometil)fenil) piperidin-1-il) (5-(oxetan-3-il)-1,4,5,6-tetrahidropirrolo[3,4-c]pirazol-3-il)metanona como un sólido blanco (39 mg, 74%): No se observó fusión clara; 1H NMR (500 MHz, DMSO-cfe) 8 13,105-12,925 (m, 1H), 7,874-7,651 (m, 1H), 7,602-7,442 (m, 1H), 5,250-4,509 (m, 1H), 4,245-3,601 (m, 5H), 3,221-3,074 (m, 1H), 3,016-2,723 (m, 3H), 2,596-2,518 (m, 2H), 1,854 1,610 (m, 4H); ESI MS m/z 457,1 [M H]+.
Ejemplo 37: Preparación de (4-(3,4-difluoro-2-(trifluorometil)fenil)piperidin-1-il) (5-neopentil-1,4,5,6-tetrahidropirrolo[3,4-c]pirazol-3-il)metanona (99)
Paso A: Siguiendo el procedimiento general GP-J1, (4-(3,4-difluoro-2-(trifluorometil)fenil)piperidin-1-il)(1,4,5,6-tetrahidropirrolo[3,4-c]pirazol-3-il)metanona (38) y pivaldehído se convirtieron en (4-(3,4-difluoro-2-(trifluorometil)fenil)piperidin-1-il) (5-neopentilo-1,4,5,6-tetrahidropirrolo[3,4-c]pirazol-3-il)metanona como un sólido blanco (25 mg, 46%): No se observó fusión clara; 1H NMR (500 MHz, DMSO-cfe) 813,060-12,840 (m, 1H), 7,840-7,440 (m, 2H), 5,315-4,473 (m, 1H), 4,210-3,642 (m, 5H), 3,272-2,728 (m, 3H), 2,555 (s, 2H), 1,867-1,597 (m, 4H), 0,905 (s, 9H); ESI MS m/z 471,2 [M H]+.
Ejemplo 38: Preparación de 3-(4-(3,4-difluoro-2-(trifluorometil)fenil)piperidina-1-carbonil)-N-metil-4,6-dihidropirrolo[3,4-c]pirazol-5(1 H)-carboxamida (100)
Paso A: Siguiendo el procedimiento general GP-H2, (4-(3,4-difluoro-2-(trifluorometil)fenil)piperidin-1-il) (1,4,5,6-tetrahidropirrolo[3,4-c]pirazol-3-il)metanona (38) e isocianato de metilo se convirtieron en 3-(4-(3,4-difluoro-2-(trifluorometil)fenil)piperidina-1-carbonil)-N-metil-4,6-dihidropirrolo[3,4-c]pirazol-5(1H)carboxamida como un sólido blanco (55 mg, 53%): mp >260°C; 1H NMR (300 MHz, DMSO-cfe) 813,449-12,960 (m, 1H), 7,719-7,620 (m, 1H), 7,620-7,379 (m, 1H), 6,272 (s, 1H), 5,454-3,850 (m, 6H), 3,240-2,737 (m, 3H), 2,623 (s, 3H), 1,979- 1,523 (m, 4H; ESI MS m/z 458,1 [M H]+.
Ejemplo 39: Preparación de 3-(4-(3,4-difluoro-2-(trifluorometil)fenil)piperidina-1-carbonil)-4,6-dihidropirrolo[3,4-c]pirazol-5(1H)-carboxilato de metilo (101)
Paso A: Siguiendo el procedimiento general GP-H1, (4-(3,4-difluoro-2-(trifluorometil)fenil)piperidin-1-il)(1,4,5,6-tetrahidropirrolo[3,4-c]pirazol-3-il)metanona (38) y cloroformiato de metilo se convirtieron en 3-(4-(3,4-difluoro-2-(trifluorometil)fenil)piperidina-1-carbonil)-4,6-dihidropirrolo[3,4-c]pirazol-5(1H)-carboxilato como un sólido blanco (30 mg, 55%): No se observó fusión clara; 1H NMR (500 MHz, DMSO-cfe) 813,503-13,110 (br s, 1H), 7,310-7,721 (m, 1H), 7,587 7,471 (m, 1H), 4,808-4,531 (br s, 1H), 4,531-4,370 (m, 4H), 3,673 (s, 3H), 3,277-3,102 (m, 2H), 3,012-2,722 (br s, 1H), 1,851-1,621 (m, 4H); ESI MS m/z 459,2 [M H]+.
Ejemplo 40: Preparación de (5-benzoil-1,4,5,6-tetrahidropirrolo[3,4-c]pirazol-3-il) (4-(3,4-difluoro-2-(trifluorometil)fenil)piperidin-1-il)metanona (102)
Paso A: Siguiendo el procedimiento general GP-H1, (4-(3,4-difluoro-2-(trifluorometil)fenil)piperidin-1-il)(1,4,5,6-tetrahidropirrolo[3,4-c]pirazol-3-il)metanona (38) y cloruro de benzoílo se convirtieron en (5-benzoil-1,4,5,6-tetrahidropirrolo[3,4-c]pirazol-3-il) (4-(3,4-difluoro-2-(trifluorometil)fenil)piperidin-1-il)metanona como un sólido blanco (30
m g , 55% ): m p > 260 °C ; 1H N M R (500 M H z , D M S O -d a ) 5 13 ,697 -13 ,007 (m , 1H ), 7 ,867 -7 ,668 (m , 1 H ), 7 ,668 -7 ,545 (m , 2 H ), 7 ,545 -7 ,384 (m , 4 H ), 5 ,416 -3 ,891 (m , 6 H ), 3 ,248 -2 ,620 (m , 3 H ), 1 ,923 -1 ,524 (m , 4 H ); E S I M S m /z 505 [M H ]+ .
Ejemplo 41: Preparación de (4-(3,4-difluoro-2(trifluorometil)fenil)piperidin-1-il)(5-picolinoil-1,4,5,6-tetrahidropirrolo[3,4-c]pirazol-3-il)metanona (103)
Paso A: Siguiendo el procedimiento general GP-H1, (4-(3,4-difluoro-2-(trifluorometil)fenil)piperidin-1-il)(1,4,5,6-tetrahidropirrolo[3,4-c]pirazol-3-N)metanona (38) y cloruro de picolinoilo se convirtieron en (4-(3,4-difluoro-2-(trifluorometil)fenil)piperidin-1-il) (5-picolinoil-1,4,5,6-tetrahidropirrolo[3,4-c]pirazol-3-il)metanona como un sólido blanco (13 mg, 22%): No se observó fusión clara; 1 H NMR (300 MHz, DMSO-d6) 513,632-13,061 (m, 1H), 8,722-8,139 (m, 1H), 8,140-7,908 (m, 1H), 7,908-7,684 (m, 2H), 7,684-7,398 (m, 2H), 5,115-4,428 (m, 5H), 3,316-2,598 (m, 3H), 1,927-1,133 (m, 5H); ESI MS m/z 506,1 [M H]+.
Ejemplo 42: Preparación de (4-(3,4-difluoro-2-(trifluorometil)fenil)piperidin-1-il)(5-nicotinoil-1,4,5,6-tetrahidropirrolo[3,4-c]pirazol-3-il)metanona (104)
Paso A: Siguiendo el procedimiento general GP-H1, (4-(3,4-difluoro-2-(trifluorometil)fenil)piperidin-1-il) (1,4,5,6-tetrahidropirrolo[3,4-c]pirazol-3-il)metanona (38) y cloruro de nicotinoílo se convirtieron en (4-(3,4-difluoro-2-(trifluorometil)fenil)piperidin-1-il) (5-nicotinoil-1,4,5,6-tetrahidropirrolo[3,4-c]pirazol-3-il)metanona como un sólido blanco (34 mg, 59%): No se observó fusión clara; 1 H NMR (500 MHz, DMSO-de) 513,589-13,123 (br s, 1H), 8,807 (s, 1H), 8,140 7,991 (m, 1H), 7,843-7,678 (m, 1H), 7,628-7,364 (m, 2H), 5,433-3,721 (m, 6H), 3,257-2,701 (m, 3H), 1,933-1,522 (m, 4H); ESI MS m/z 506,1 [M H]+.
Ejemplo 43: Preparación de (4-(3,4-difluoro-2(trifluorometil)fenil)piperidin-1-il)(5-isonicotinoil-1,4,5,6-tetrahidropirrolo[3,4-c]pirazol-3-il)metanona (105)
Paso A: Siguiendo el procedimiento general GP-H1, (4-(3,4-difluoro-2-(trifluorometil)fenil)piperidin-1-il) (1,4,5,6-tetrahidropirrolo[3,4-c]pirazol-3-il)metanona (38) y cloruro de isonicotinoilo se convirtieron en (4-(3,4-difluoro-2-(trifluorometil)fenil)piperidin-1-il) (5-isonicotinoil-1,4,5,6-tetrahidropirrolo[3,4-c]pirazol-3-il)metanona como un sólido blanco (14 mg, 24%): mp >260°C; 1 H NMR (500 MHz, DMSO-d6) 513,420-13,120 (m, 1H), 8,770-8,620 (m, 2H), 7,833-7,679 (m, 1H), 7,612-7,369 (m, 3H), 5,410-3,820 (m, 6H), 3,240-3,060 (m, 2H), 1,860-1,530 (m, 4H), 1,290-1,190 (m, 1H); ESI MS m/z 506,1 [M H]+.
Ejemplo 44: Preparación de (4-(3,4-difluoro-2-(trifluorometil)fenil)piperidin-1-il) (5-(pirrolidin-1-carbonil)-1,4,5,6-tetrahidropirrolo[3,4-c]pirazol-3-il)metanona (106)
Paso A: Siguiendo el procedimiento general GP-H2, (4-(3,4-difluoro-2-(trifluorometil)fenil)piperidin-1-il) (1,4,5,6-tetrahidropirrolo[3,4-c]pirazol-3-il)metanona (38) y cloruro de 1-pirolocarbamoilo se convirtieron en (4-(3,4-difluoro-2-(trifluorometil)fenil)piperidin-1 -il)(5-(pirrolidin-1 -carbonil)-1,4,5,6-tetrahidropirrolo[3,4-c]pirazol-3-il)metanona como un sólido blanco (22 mg, 38%): mp >260°C; 1 H NMR (500 MHz, DMSO-d6) 5 13,389-13,022 (m, 1H), 7,858-7,407 (m, 2H), 5,439-3,846 (m, 6H), 3,412-3,326 (m, 4H), 3,253-2,742 (m, 3H), 1,868-1,638 (m, 8H); ESI MS m/z 498,2 [M H]+.
Ejemplo 45: Preparación de 4-(3,4-difluoro-2-(trifluorometil)fenil) piperidin-1 -il)(5-(2,2,2-trifluoroetil)-1,4,5,6-tetrahidropirrolo [3,4 -c]pirazol-3-il)metanona (107)
Paso A: Siguiendo el procedimiento general GP-J2, (4-(3,4-difluoro-2-(trifluorometil)fenil)piperidin-1-il) (1,4,5,6-tetrahidropirrolo[3,4-c]pirazol-3-il)metanona (38) y trifluorometanosulfonato de 2,2,2-trifluoroetilo se convirtieron en (4-(3,4-difluoro-2-(trifluorometil)fenil)piperidin-1-il) (5-(2,2,2-trifluoroetil)-1,4,5,6-tetrahidropirrolo[3,4-c]pirazol-3-il)metanona como un sólido blanco (16 mg, 28%): No se observó fusión clara; 1 H NMR (500 MHz, DMSO-de) 513,274-12,902 (m, 1H), 7,343 7,681 (m, 1H), 7,601-7,441 (m, 1H), 5,368-3,811 (m, 6H), 3,695-3,520 (m, 2H), 3,273-2,71 (m, 3H), 1,906-1,571 (m, 4H); ESI MS m/z 483,1 [M H]+.
Ejemplo 46: Preparación de (4-(3,4-difluoro-2(trifluorometil)fenil)piperidin-1-il)(5-(3,3,3-trifluoropropil)-1,4,5,6-tetrahidropirrolo[3,4-c]pirazol-3-il)metanona (108)
Paso A: Siguiendo el procedimiento general GP-J2, (4-(3,4-difluoro-2-(trifluorometil)fenil)piperidin-1-il)(1,4,5,6-tetrahidropirrolo[3,4-c]pirazol-3-il)metanona (38) y 1,1,1-trifluoro-3-bromopropano se convirtieron en (4-(3,4-difluoro-2-(trifluorometil)fenil)piperidin-1-il) (5-(3,3,3-trifluoropropil)-1,4,5,6-tetrahidropirrolo[3,4-c]pirazol-3-il)metanona como un sólido blanco (13 mg, 22%): No se observó fusión clara; 1 H NMR (500 MHz, DMSO-de) 513,091-12,860 (m, 1H), 7,837 7,651 (m, 1H), 7,638-7,430 (m, 1H), 5,271-4,038 (m, 7H), 3,899-3,673 (m, 4H), 3,210-3,059 (m, 2H), 2,955-2,681 (br s, 1H), 1,869-1,558 (m, 4H); ESI MS m/z 497 [M H]+.
Ejemplo 47: Preparación de (4-(3,4-difluoro-2(trifluorometil)fenil)piperidin-1-il)(5-(2-metoxietil)-1,4,5,6-tetrahidropirrolo[3,4-c]pirazol-3-il)metanona(109)
Paso A: Siguiendo el procedimiento general GP-J2, (4-(3,4-difluoro-2-(trifluorometil)fenil)piperidin-1-il)(1,4,5,6tetrahidropirrolo[3,4-c]pirazol-3-il)metanona (38) y 2-metoxi-bromoetano se convirtieron en (4-(3,4-difluoro-2-(trifluorometil)fenil)piperidin-1-il) (5-(2-metoxietil)-1,4,5,6-tetrahidropirrolo[3,4-c]pirazol-3-il)metanona como un sólido blanco (14 mg, 22%): No se observó fusión clara; 1H NMR (300 MHz, DMSO-de) 8 13,070-12,860 (m, 1H), 7,852-7,648 (m, 1H), 7,616-7,449 (m, 1H), 5,365-3,596 (m, 6 H), 3,560-3,413 (m, 2H), 3,298-3,219 (m, 4H), 3,219-3,047 (m, 2H), 2,937 2,832 (m, 3H), 1,843-1,576 (m, 5H); ESI MS m /z 459,2 [M H]+.
Ejemplo 48: Preparación de 3-(4-(3,4-difluoro-2-(trifluorometil)fenil)piperidina-1-carbonil)-4,6-dihidropirrolo[3,4-c]pirazol-5(1H)-carbonitrilo (110)
Paso A: Siguiendo el procedimiento general GP-J2, (4-(3,4-difluoro-2-(trifluorometil)fenil)piperidin-1-il)(1,4,5,6-tetrahidropirrolo[3,4-c]pirazol-3-il)metanona (38) y bromuro de cianógeno se convirtieron en -(4-(3,4-difluoro-2-(trifluorometil)fenil)piperidina-1-carbonil)-4,6-dihidropirrolo[3,4-c]pirazol-5(1H)-carbonitrilo como un sólido blanco (23 mg, 79%): No se observó fusión clara; 1H NMR (500 MHz, DMSO-d6) 8 13,623-13,172 (m, 1H), 7,850-7,664 (m, 1H), 7,664 7,470 (m, 1H), 5,552-3,846 (m, 6 H), 3,271-2,652 (m, 3H), 2,042-1,503 (m, 4H); ESI MS m /z 426 [M H]+.
Ejemplo 49: Preparación de (4-(3,4-difluoro-2-(trifluorometil)fenil)piperidin-1-il)(6-etil-4,5,6,7-tetrahidro-1H-pirazolo[3,4-c]piridin-3-il)metanona (111)
Paso A: Siguiendo el procedimiento general GP-G1, sal TFA de (4-(3,4-difluoro-2-(trifluorometil)fenil)piperidin-1-il) (4,5,6,7-tetrahidro-1H-pirazolo[3,4-c]piridin-3-il)metanona (34) y acetaldehído se convirtieron en (4-(3,4-difluoro-2-(trifluorometil)fenil)piperidin-1-il) (6-etil-4,5,6,7-tetrahidro-1H-pirazolo[3,4-c]piridin-3-il)metanona como un sólido blanco (13 mg, 21% ): mp 207-210°C; 1H NMR (500 MHz, DMSO-d6) 8 12,90 (br s, 0,25H), 12,71 (br s, 0,75H), 7,78-7,70 (m, 1H), 7.50- 7,49 (m, 1H), 4,87-4,85 (m, 1H), 4,68-4,66 (m, 1H), 3,48-3,45 (m, 2H), 3,14-3,13 (m, 2H), 2,78-2,77 (m, 1H), 2,61 2,55 (m, 6 H, partialy merged with DMSO peak), 1,76-1,70 (m, 4H), 1,07 (t, J = 7,0 Hz, 3H); ESI MS m /z 443 [M H]+. Ejemplo 50: Preparación de 3-(4-(3,4-difluoro-2-(trifluorometil)fenil)piperidina-1-carbonil)-W-metil-1,4,5,7-tetrahidro-6Hpirazol[3,4-c]piridina-6-carboxamida (112)
Paso A: Siguiendo el procedimiento general GP-E2, sal TFA de (4-(3,4-difluoro-2-(trifluorometil)fenil)piperidin-1-il)(4,5,6,7-tetrahidro-1H-pirazolo[3,4-c]piridin-3-il)metanona (34) e isocianatometano se convirtieron en 3-(4-(3,4-difluoro-2-(trifluorometil)fenil)piperidina-1-carbonil)-W-metil-1,4,5,7-tetrahidro-6Hpirazolo[3,4-c]piridina-6-carboxamida como un sólido blanquecino (6 mg, 27%): mp 220-225°C; 1H NMR (500 MHz, DMSO-d6) 8 13,05 (br s, 0,25H), 12,85 (br s, 0,75H), 7,79-7,70 (m, 1H), 7,51-7,48 (m, 1H), 6,61-6,60 (m, 0,75H), 6,55-6,54 (m, 0,25H), 4,86-4,84 (m, 1H), 4,68-4,65 (m, 1H), 4,47-4,42 (m, 2H), 3,54-3,50 (m, 2H), 3,14-3,13 (m, 2H), 2,78-2,77 (m, 1H), 2,59-2,56 (m, 5H, partially merged with DMSO peak), 1,76-1,67 (m, 4H); ESI MS m /z 472 [M H]+.
Ejemplo 51: Preparación de (4-(3,4-difluoro-2-(trifluorometil)fenil)piperidin-1-il)(6-metil-4,5,6,7-tetrahidro-1H-pirazolo[3,4-c]piridin-3-il)metanona (113)
Paso A: Siguiendo el procedimiento general GP-G1, sal TFA de (4-(3,4-difluoro-2-(trifluorometil)fenil)piperidin-1-il)(4,5,6,7-tetrahidro-1H-pirazolo[3,4-c]piridin-3-il)metanona (34) y formaldehído se convirtieron en (4-(3,4-difluoro-2-(trifluorometil)fenil)piperidin-1-il) (6-metil-4,5,6,7-tetrahidro-1H-pirazolo[3,4-c]piridin-3-il)metanona como un sólido blanco (18 mg, 55%): mp 210 -211°C ; 1H NMR (500 MHz, DMSO-d6) 8 12,91 (br s, 0,25H), 12,72 (br s, 0,75H), 7,79-7,70 (m, 1H), 7.50- 7,49 (m, 1H), 4,88-4,86 (m, 1H), 4,67-4,65 (m, 1H), 3,43-3,40 (m, 2H), 3,15-3,13 (m, 2H), 2,77-2,76 (m, 1H), 2,65 2,60 (m, 4H), 2,36 (s, 3H), 1,76-1,69 (m, 4H); ESI MS m /z 429 [M H]+.
Ejemplo 52: Preparación de 3-(4-(3-fluoro-2-(trifluorometil)fenil)piperidina-1-carbonil)-A/-metil-6,7-dihidro-1H-pirazolo[4,3-c]piridina-5(4H)-carboxamida (114)
Paso A: Siguiendo el procedimiento general GP-C, clorhidrato de (4-(3-fluoro-2-(trifluorometil)fenil)piperidin-1-il)(4,5,6,7-tetrahidro-1H-pirazolo[4,3-c]piridin-3-il)metanona (32) e isocianato de metilo se convirtieron en 3-(4-(3-fluoro-2-(trifluorometil)fenil)piperidina-1-carbonil)-W-metil-6,7-dihidro-1H-pirazolo[4,3-c]piridina-5(4H)-carboxamida como un sólido blanco (32 mg, 41%): mp 165-170°C; 1H NMR (500 MHz, DMSO-d6) 8 13,03-12,85 (m, 1H), 7,70-7,62 (m, 1H), 7,46 (d, J = 8,0 Hz, 1H), 7,30 (dd, J = 12,0, 8,0 Hz, 1H), 6,58-6,49 (m, 1H), 5,19-5,06 (m, 2H), 4,76-4,62 (m, 2H), 3,63-3,50 (m, 2H), 3,27-3,09 (m, 2H), 2,86-2,72 (m, 1H), 2,64 (t, J = 5,5 Hz, 2H), 2,59-2,54 (m, 3H), 1,84-1,59 (m, 4H); ESI MS m /z 454 [M H]+.
Ejemplo 53: Preparación de ácido 2-(3-(4-(3,4-difluoro-2-(trifluorometil)fenil)piperidina-1-carbonil)-1,4,5,7-tetrahidro-6H-pirazolo[3,4-c]piridin-6-il)acético (115)
Paso A: Siguiendo el procedimiento general GP-G2, sal TFA de (4-(3,4-difluoro-2-(trifluorometil)fenil)piperidin-1-il) (4,5,6,7-tetrahidro-1H-pirazolo[3,4-c]piridin-3-il)metanona (34) y 2-bromoacetato de ferc-butilo se convirtieron para proporcionar 2-(3-(4-(3,4-difluoro-2-(trifluorometil)fenil)piperidina-1-carbonil)-1,4,5,7-tetrahidro-6H-pirazolo[3,4-c]piridin-6-il)acetato de ferc-butilo como un sólido vitreo transparente (55 mg, 69%): 1H NMR (500 MHz, DMSO-d6) 8 12,94 (br s, 0,25H), 12,71 (br s, 0,75H), 7,37-7,33 (m, 1H), 7,50-7,49 (m, 1H), 4,86-4,84 (m, 1H), 4,68-4,66 (m, 1H), 3,67-3,63 (m, 2H), 3,34-3,30 (m, 2H, fusionado parcialmente con el pico de H2O), 3,14-3,13 (m, 2H), 2,59-2,58 (m, 3H), 2,59-2,50 (m, 2H,
fusionado parcialmente con el pico de DMSO), 1,76-1,70 (m, 4H), 1,43 (s, 9H); ESI MS m /z 529 [M H]+.
Paso B: Una solución de 2-(3-(4-(3,4-difluoro-2-(trifluorometil)fenil)piperidina-1-carbonil)-1,4,5,7-tetrahidro-6H-pirazolo[3,4-c]piridin-6-il)acetato de ferc-butilo (53 mg, 0,10 mmol) en CH2Ch anhidro (3 mL) se trató con TFA (3 mL) y se agitó bajo una atmósfera de N2 a temperatura ambiente durante 8 h. Después de este tiempo, la mezcla se concentró a sequedad bajo presión reducida y el disolvente se intercambió con CH2Ch (10 mL). El residuo se diluyó en CH2Ch anhidro (10 mL), se trató con MP-carbonato (0,50 g) y se agitó a temperatura ambiente durante 15 min. Pasado este tiempo, la solución se filtró y la resina se lavó con C^Cfe (2 x 10 mL). El filtrado se concentró a sequedad bajo presión reducida para proporcionar ácido 2-(3-(4-(3,4-difluoro-2-(trifluorometil)fenil)piperidin-1-carbonil)-1,4,5,7-tetrahidro-6H-pirazolo[3,4-c]piridin-6 -il)acético como un sólido blanquecino (40 mg, 85%): mp 151-153°C ; 1H NMR (500 MHz, DMSO-cfe) 513,29 (br s, 0,25H), 12,89 (br s, 0,75H), 7,56-7,55 (m, 1H), 7,51-7,50 (m, 1H), 4,87-4,86 (m, 1H), 4,66-4,64 (m, 1H), 4,04-4,02 (m, 2H), 3,76-3,72 (m, 2H), 3,34-3,32 (m, 2H, fusionado parcialmente con el pico de H2O), 3,15-3,11 (m, 4H), 2,76- 2,74 (m, 2H), 1,76-1,70 (m, 4H); ESI MS m /z 473 [M H]+.
Ejemplo 54: Preparación de 3-(4-(3,4-difluoro-2-(trifluorometil)fenil)piperidina-1-carbonil)-1,4,5,7-tetrahidro-6H-pirazolo[3,4-c]piridina-6-carboxilato de metilo (116)
Paso A: Siguiendo el procedimiento general GP-E1, sal TFA de (4-(3,4-difluoro-2-(trifluorometil)fenil)piperidin-1-il)(4,5,6,7-tetrahidro-1H-pirazolo[3,4-c]piridin-3-il)metanona (34) y el carbonocloridato de metilo se convirtieron en 3-(4-(3,4-difluoro-2-(trifluorometil)fenil)piperidin-1-carbonil)-1,4,5,7-tetrahidro-6Hpirazolo[3,4-c]piridina-6-carboxilato como un sólido naranja claro (30 mg, 60%): mp 238-240°C; 1H NMR (500 MHz, DMSO-cfe) 5 13,13 (br s, 0,25H), 12,86 (br s, 0,75H), 7,78-7,70 (m, 1H), 7,51-7,48 (m, 1H), 4,82-4,80 (m, 1H), 4,67-4,65 (m, 1H), 4,54 (s, 1,5H), 4,50 (s, 0,5H), 3,64 (s, 3H), 3,60-3,57 (m, 2H), 3,15-3,13 (m, 2H), 2,79-2,78 (m, 1H), 2,64-2,59 (m, 2H), 1,76-1,68 (m, 4H); ESI MS m /z 473 [M H]+.
Ejemplo 55: Preparación de (4-(3,4-difluoro-2-(trifluorometil)fenil)piperidin-1-il) (6-(oxetan-3-il)-4,5,6,7-tetrahidro-1H-pirazolo[3,4-c]piridin-3-il)metanona (117)
Paso A: Siguiendo el procedimiento general G P-G1, sal TFA de (4-(3,4-difluoro-2-(trifluorometil)fenil)piperidin-1-il) (4,5,6,7-tetrahidro-1H-pirazolo[3,4-c]piridin-3-il)metanona (34) y oxetan-3-ona se convirtieron en (4-(3,4-difluoro-2-(trifluorometil)fenil)piperidin-1-il)(6-(oxetan-3-il)-4,5,6,7-tetrahidro-1Hpirazolo[3,4-c]piridin-3-il)metanona como un sólido blanquecino (32 mg, 50%): mp 206-207°C; 1H NMR (500 MHz, DMSO-cfe) 512,96 (br s, 0,25H), 12,76 (br s, 0,75H), 7,80 7,70 (m, 1H), 7,51-7,50 (m, 1H), 4,86-4,84 (m, 1H), 4,68-4,65 (m, 1H), 4,60 (t aparente, J = 6,5 Hz, 2H), 4,50 (t aparente, J = 6,0 Hz, 2H), 3,71-3,64 (m, 1H), 3,43-3,38 (m, 2H), 3,15-3,13 (m, 2H), 2,78-2,77 (m, 1H), 2,64-2,60 (m, 2H), 1,79-1,68 (m, 4H), CH2 oscurecido por el pico de disolvente; ESI MS m /z 471 [M H]+.
Ejemplo 56: Preparación de (6-(ciclopropilmetil)-4,5,6,7-tetrahidro-1Hpirazolo[3,4-c]piridin-3-il) (4-(3,4-difluoro-2-(trifluorometil)fenil)piperidin-1 -il)metanona (118)
Paso A: Siguiendo el procedimiento general GP-G1, sal TFA de (4-(3,4-difluoro-2-(trifluorometil)fenil)piperidin-1-il) (4,5,6,7-tetrahidro-1H-pirazolo[3,4-c]piridin-3-il)metanona (34) y ciclopropanocarbaldehído se convirtieron en (6 -(ciclopropilmetil)-4,5,6,7-tetrahidro-1H-pirazolo[3,4-c]piridin-3-il)(4-(3,4-difluoro-2-(trifluorometil)fenil)piperidin-1-il)metanona como un sólido blanco (30 mg, 58%): mp 184-185°C; 1H NMR (500 MHz, CD3OD) 57,53 (dd, J = 17,5, 9,0 Hz, 1H), 7,39 (dd, J = 9,0, 4,0 Hz, 1H), 4,83-4,82 (m, 1H, fusionado parcialmente con el pico de H2O), 4,65-4,63 (m, 1H), 3,94-3,92 (m, 2H) , 3,29-3,26 (m, 2H, fusionado parcialmente con el pico de CH3OH), 3,05-3,03 (m, 2H), 2,90-2,84 (m, 3H), 2,69-2,67 (m, 2H), 1,89-1,81 (m, 4H), 1,04-1,02 (m, 1H), 0,65 (d, J = 7,0 Hz, 2H), 0,29-0,27 (m, 2H), protón NH no observado; ESI MS m /z 469 [M H]+.
Ejemplo 57: Preparación de (4-(3,4-difluoro-2(trifluorometil)fenil)piperidin-1-il)(6-(3,3,3-trifluoropropil)-4,5,6,7-tetrahidro-1 H-pirazolo[3,4-c]piridin-3-il)metanona (119)
Paso A: Siguiendo el procedimiento general GP-G1, sal TFA de (4-(3,4-difluoro-2-(trifluorometil)fenil)piperidin-1-il)(4,5,6,7-tetrahidro-1H-pirazolo[3,4-c]piridin-3-il)metanona (34) y 3,3,3-trifluoropropanal se convirtieron en (4-(3,4-difluoro-2-(trifluorometil)fenil)piperidin-1-il) (6-(3,3,3-trifluoropropil)-4,5,6,7-tetrahidro-1H-pirazolo[3,4-c]piridin-3-il)metanona como un sólido blanquecino (18 mg, 36%): mp 194-195°C; 1H NMR (500 MHz, DMSO-cfe) 5 12,94 (br s, 0,25H), 12,76 (br s, 0,75H), 7,79-7,70 (m, 1H), 7,50-7,49 (m, 1H), 4,87-4,85 (m, 1H), 4,68-4,66 (m, 1H), 3,57-3,53 (m, 2H), 3,15-3,13 (m, 2H), 2,75 (t, J = 7,5 Hz, 2H), 2,69-2,67 (m, 2H), 2,58-2,50 (m, 5H, fusionado parcialmente con el pico de DMSO), 1,76-1,68 (m, 4H); ESI MS m /z 511 [M H]+.
Ejemplo 58: Preparación de 3-(4-(3,4-difluoro-2-(trifluorometil)fenil)piperidina-1-carbonil)-1,4,5,7-tetrahidro-6H-pirazolo[3,4-c]piridina-6-carbonitrilo (120)
Paso A: Siguiendo el procedimiento general GP-G2, sal TFA de (4-(3,4-difluoro-2-(trifluorometil)fenil)piperidin-1-il) (4,5,6,7-tetrahidro-1H-pirazolo[3,4-c]piridin-3-il)metanona (34) y bromuro de cianógeno se convirtieron en 3-(4-(3,4-difluoro-2-(trifluorometil)fenil)piperidina-1-carbonil)-1,4,5,7-tetrahidro-6H-pirazolo[3,4-c]piridina-6-carbonitrilo como un sólido blanco (36 mg, 83%): mp 248-250°C; 1H NMR (500 MHz, DMSO-cfe) 513,26 (br s, 0,25H), 12,97 (br s, 0,75H), 7,78 7,75 (m, 1H), 7,51-7,49 (m, 1H), 4,85 (d aparente, J = 8,0 Hz, 1H), 4,68-4,66 (m, 1H), 4,47-4,40 (m, 2H), 3,43-3,40 (m,
2 H ), 3 ,16 -3 ,14 (m , 2 H ), 2 ,77 -2 ,72 (m , 3 H ), 1 ,77 -1 ,71 (m , 4 H ); E S I M S m /z 440 [M H ]+ .
Ejemplo 59: Preparación de 1-(3-(4-(3,4-d¡fluoro-2-(tr¡fluoromet¡l)fen¡l)p¡perid¡na-1-carbon¡l)-1,4,5,7-tetrah¡dro-6H-pirazolo[3,4-c]piridin-6-il)-2-metilpropan-1-ona (121)
Paso A: Siguiendo el procedimiento general G P-E1, sal TFA de (4-(3,4-difluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡n-1-il) (4,5,6,7-tetrah¡dro-1H-p¡razolo[3,4-c]p¡rid¡n-3-¡l)metanona (34) y cloruro de isobutirilo se convirtieron en 1-(3-(4-(3,4-difluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡na-1-carbon¡l)-1,4,5,7-tetrah¡dro-6Hp¡razolo[3,4-c]p¡rid¡n-6-¡l)-2-met¡lpropan-1-ona como un sólido blanco (29 mg, 62%): mp 228-229°C; 1H NMR (500 MHz, DMSO-da) 8 13,12 (br s, 0,25H), 12,87 (br s, 0,75H), 7,79 7,70 (m, 1H), 7,50-7,48 (m, 1H), 4,88-4,86 (m, 1H), 4,68-4,56 (m, 3H), 3,70 (s, 2H), 3,15-3,13 (m, 2H), 2,99-2,97 (m, 1H), 2.79- 2,77 (m, 1H), 2,70-2,64 (m, 2H), 1,76- 1,68 (m, 4H), 1,04-1,01 (m, 6 H); ESI MS m /z 485 [M H]+.
Ejemplo 60: Preparación de 1-(3-(4-(3,4-d¡fluoro-2-(tr¡fluoromet¡l)fen¡l)p¡perid¡na-1-carbon¡l)-1,4,5,7-tetrah¡dro-6H-pirazolo[3,4-c]pir¡d¡n-6-¡l)propan-1-ona (122)
Paso A: Siguiendo el procedimiento general G P-E1, sal TFA de (4-(3,4-difluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡n-1-il) (4,5,6,7-tetrahidro-1H-pirazolo [3,4-c]pirid¡n-3-¡l)metanona (34) y cloruro de propionilo se convirtieron en 1-(3-(4-(3,4-difluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡na-1-carbon¡l)-1,4,5,7-tetrah¡dro-6Hpirazolo[3,4-c]p¡r¡d¡n-6-¡l)propan-1-ona como un sólido blanco (35 mg, 63%): mp 182-187°C; 1H NMR (500 MHz, DMSO-cfe) 813,15-13,10 (m, 0,25H), 12,87 (br s, 0,75H), 7,76 7,72 (m, 1H), 7,51-7,49 (m, 1H), 4,85-4,83 (m, 1H), 4,68-4,57 (m, 3H), 3,69 (s, 0,5H), 3,63 (s, 1,5H), 3,15-3,13 (m, 2H), 2.79- 2,77 (m, 1H), 2,68-2,55 (m, 2H, fusionado parcialmente con el pico de DMSO), 2,46-2,38 (m, 2H), 1,76-1,71 (m, 4H), 1,02 (t, J = 7,5 Hz, 3H); ESI MS m /z 471 [M H]+.
Ejemplo 61: Preparación de 1-(3-(4-(3,4-d¡fluoro-2-(tr¡fluoromet¡l)fen¡l)p¡perid¡na-1-carbon¡l)-1,4,5,7-tetrah¡dro-6H-pirazolo[3,4-c]p¡r¡din-6-¡l)-3-met¡lbutan-1-ona (123)
Paso A: Siguiendo el procedimiento general G P-E1, sal TFA de (4-(3,4-difluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡n-1-il) (4,5,6,7-tetrah¡dro-1H-p¡razolo[3,4-c]p¡rid¡n-3-¡l)metanona (34) y cloruro de 3-metilbutanoilo se convirtieron en 1-(3-(4-(3,4-difluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡na-1-carbon¡l)-1,4,5,7-tetrah¡dro-6Hp¡razolo[3,4-c]pir¡d¡n-6-¡l)-3-met¡lbutan-1-ona como un sólido blanco(37 mg, 63%): mp 184-188°C; 1H NMR (500 MHz, DMSO-cfe) 8 13 ,13 -13 ,11 (m, 0,25H), 12,87-12,84 (m, 0,75H), 7,79-7,70 (m, 1H), 7,51-7,49 (m, 1H), 4,89-4,86 (m, 1H), 4,67-4,57 (m, 3H), 3,67-3,64 (m, 2H), 3,15-3,13 (m, 2H), 2.79- 2,77 (m, 1H), 2,68-2,64 (m, 2H), 2,55-2,51 (m, 1H, fusionado parcialmente con el pico de DMSO), 2,02 (pent, J = 7,0 Hz, 1H), 1,76-1,70 (m, 4H), 1,26 (t, J = 7,0 Hz, 1H), 0,92-0,90 (m, 6 H); ESI MS m/z 499 [M H]+.
Ejemplo 62: Preparación de (4-(3,4-difluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡n-1-il) (6-(2-metoxietil)-4,5,6,7-tetrah¡dro-1H-pirazolo[3,4-c]pir¡d¡n-3-¡l)metanona (124)
Paso A: Siguiendo el procedimiento general GP-G2, sal TFA de (4-(3,4-difluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡n-1-il) (4,5,6,7-tetrahidro-1H-p¡razolo[3,4-c]p¡r¡d¡n-3-¡l)metanona (34) y 1-bromo-2-metoxietano se convirtieron en (4-(3,4-difluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡n-1-il) (6-(2-metox¡et¡l)-4,5,6,7-tetrahidro-1H-p¡razolo[3,4-c]p¡r¡d¡n-3-¡l)metanona como un sólido blanco (33 mg, 48%): mp 171-173°C ; 1H NMR (500 MHz, DMSO-cfe) 8 12,91 (br s, 0,25H), 12,71 (br s, 0,75H), 7,78-7,70 (m, 1H), 7,51-7,48 (m, 1H), 4,85 (d aparente, J = 11,0 Hz, 1H), 4,66 (d aparente, J = 11,0 Hz, 1H), 3,57 3,49 (m, 4H) , 3,25 (s, 3H), 3,16-3,11 (m, 2H), 2,77- 2,58 (m, 7H), 1,76-1,68 (m, 4H); ESI MS m /z 473 [M H]+.
Ejemplo 63: Preparación de (4-(3,4-difluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡n-1-il) (6-(2,2,2-trifluoroetil)-4,5,6,7-tetrahidro-1H-pirazolo[3,4-c]p¡r¡d¡n-3-¡l)metanona (125)
Paso A: Siguiendo el procedimiento general GP-G2, sal TFA de (4-(3,4-difluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡n-1-il) (4,5,6,7-tetrahidro-1H-p¡razolo[3,4-c]p¡r¡d¡n-3-¡l)metanona (34) y trifluorometanosulfonato de 2,2,2-trifluoroetilo se convirtieron en (4-(3,4-difluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡n-1-il) (6-(2,2,2-trifluoroetilo)-4,5,6,7-tetrahidro-1H-pirazolo[3,4-c]pir¡d¡n-3-¡l)metanona como un sólido blanquecino (39 mg, 72%): mp 192-193°C; 1H NMR (500 MHz, DMSO-cfe) 8 13,00 (br s, 0,25H), 12,76 (br s, 0,75H), 7,80-7,70 (m, 1H), 7,50-7,48 (m, 1H), 4,85 (d aparente, J = 11,0 Hz, 1H), 4,66 (d aparente, J = 11,0 Hz, 1H), 3,79 (s, 1,5H), 3,75 (s, 0,5H), 3,42-3,35 (m, 2H), 3,15-3,13 (m, 2H), 2,87 (t, J = 6,0 Hz, 2H), 2,78-2,76 (m, 1H), 2,64-2,62 (m, 2H), 1,79-1,68 (m, 4H); ESI MS m /z 497 [M H]+.
Ejemplo 64: Preparación de (4-(3,4-d¡fluoro-2(tr¡fluoromet¡l)fen¡l)p¡perid¡n-1-¡l)(6-neopent¡l-4,5,6,7-tetrah¡dro-1H-pirazolo[3,4-c]pir¡d¡n-3-¡l)metanona (126)
Paso A: Siguiendo el procedimiento general GP-G1, sal TFA de (4-(3,4-difluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡n-1-il)(4,5,6,7-tetrahidro-1H-p¡razolo[3,4-c]p¡rid¡n-3-¡l)metanona (34) y pivalaldehído se convirtieron en (4-(3,4-difluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡n-1-¡l)(6-neopent¡l-4,5,6,7-tetrahidro-1H-p¡razolo[3,4-c]p¡r¡d¡n-3-¡l)metanona como un sólido blanco (28 mg, 49%): mp 218-220 °C; 1H NMR (500 MHz, DMSO-cfe) 812,90 (br s, 0,25H), 12,67 (br s, 0,75H), 7,75-7,70 (m, 1H), 7,49 (dd, J = 8,0, 4,0 Hz, 1H), 4,88 (d aparente, J = 12,0 Hz, 1H), 4,67 (d aparente, J = 10,0 Hz, 1H), 3,61 (s, 1,5H), 3,59 (s, 0,5H), 3,15-3,13 (m, 2H), 2,78-2,76 (m, 1H), 2,71 (t, J = 5,5 Hz, 2H), 2,64-2,58 (m, 2H), 2,25 (s, 2H), 1,79 1 ,68 (m, 4H), 0,88 (s, 9H); ESI MS m /z 485 [M H]+.
Ejemplo 65: Preparación de (4-(3,5-difluoro-2-(trifluorometil)fenil)piperidin-1-il) (5-(2-metoxietil)-4,5,6,7-tetrah¡dro-1H-pirazolo[4,3-c]piridin-3-il)metanona (127)
Paso A: Siguiendo el procedimiento general GP-G2, clorhidrato de (4-(3,5-d¡fluoro-2-(tr¡fluoromet¡l)fenil)p¡per¡d¡n-1-¡l)(4,5,6,7-tetrah¡dro-1H-p¡razolo[3,4-c]p¡rid¡n-3-¡l)metanona (60) y bromoetilmetil éter se convirtieron en (4-(3,5-difluoro-2-(trifluoromet¡l)fen¡l)p¡per¡din-1-¡l) (5-(2-metox¡et¡lo)-4,5,6,7-tetrah¡dro-1H-p¡razolo[4,3-c]p¡rid¡n-3-¡l)metanona como un sólido blanco (32 mg, 41%): 1H NMR (500 MHz, DMSO-da) 8 12,85 (m, 1H), 7,48 (m, 2H), 5,12 (m, 1H), 4,67 (m, 1H), 3,50 (m, 4H), 3,01-3,32 (m, 5H), 2,73 (m, 7H), 3,63-3,50 (m, 2H), 1,62 (m, 4H); ESI MS m /z 473 [M H]+.
Ejemplo 66: Preparación de (4-(3,4-difluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡n-1-il) (5-(p¡perid¡na-1-carbon¡l)-1,4,5,6-tetrahidropirrolo[3,4-c]p¡razol-3-¡l)metanona (128)
Paso A: Siguiendo el procedimiento general GP-H2, (4-(3,4-d¡fluoro-2-(tr¡fluoromet¡l)fenil)p¡per¡d¡n-1-¡l)(1,4,5,6-tetrahidropirrolo[3,4-c]p¡razol-3-¡l)metanona (38) y cloruro de piperidina-1-carbonilo se convirtieron en (4-(3,4-difluoro-2-(trifluoromet¡l)fen¡l)p¡per¡din-1-¡l) (5-(piperid¡na-1-carbon¡l)-1,4,5,6-tetrahidropirrolo[3,4-c] pirazol-3-il)metanona como un sólido blanco (45 mg, 77%): mp 236-237°C; 1H NMR (500 MHz, DMSO-cfe) 8 13,26 (br s, 0,6H), 13,06 (br s, 0,4H), 7,79 7,70 (m, 1H), 7,56-7,50 (m, 1H), 5,25-5,23 (m, 0,4H), 4,65-4,62 (m, 0,6H), 4,57 (s, 2H), 4,47 (s, 2H), 4,17-3,91 (m, 1H), 3,26-3,03 (m, 6 H), 3,01-2,73 (m, 1H), 1,80-1,71 (m, 4H), 1,53-1,49 (m, 6 H); ESI MS m /z 512 [M H]+.
Ejemplo 67: Preparación de (4-(3,5-difluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡n-1-il) (5-(3,3,3-trifluoroprop¡l)-4,5,6,7-tetrah¡dro-1H-pirazolo[4,3-c]p¡r¡d¡n-3-¡l)metanona (129)
Paso A: Siguiendo el procedimiento general GP-G2, clorhidrato de (4-(3,5-d¡fluoro-2-(tr¡fluoromet¡l)fen¡l)p¡perid¡n-1-¡l)(4,5,6,7-tetrah¡dro-1H-p¡razolo[3,4-c]p¡rid¡n-3-¡l)metanona (60) y 3-bromo-1,1,1-trifluoropropano se convirtieron en (4-(3,5-difluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡n-1-il) (5-(2-metox¡et¡l)-4,5,6,7-tetrahidro-1H-p¡razolo[4,3-c]p¡r¡d¡n-3-¡l)metanona como un sólido blanco (41 mg, 51%): 1H NMR (500 MHz, DMSO-cfe) 8 12,83 (m, 1H), 7,43 (m, 2H), 5,12 (m, 1H), 4,67 (m, 1H), 3,53 (m, 2H), 2,50-3,12 (m, 11H), 1,68 (m, 4H); ESI MS m /z 511 [M H]+.
Ejemplo 68: Preparación de 3-(4-(3,5-d¡fluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡na-1-carbonil)-1,4,5,7-tetrah¡dro-6H-pirazolo[3,4-c]pir¡d¡na-6-carbox¡lato de metilo (130)
Paso A: Siguiendo el procedimiento general GP-E2, clorhidrato de (4-(3,5-bis(tr¡fluoromet¡l)fen¡l)p¡per¡d¡n-1-il) (4,5,6,7-tetrah¡dro-1H-p¡razolo[3,4-c]p¡rid¡n-3-¡l)metanona (52) y el carbonocloridato de metilo se convirtieron en 3-(4-(3,5-difluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡na-1-carbon¡l)-1,4,5,7-tetrah¡dro-6H-p¡razolo[3,4-c]pir¡d¡na-6-carbox¡lato de metilo como un sólido blanquecino (7 mg, 20%): mp 253-254°C; 1H NMR (500 MHz, DMSO-cfe) 8 13,12 (br s, 0,25H), 12,86 (br s, 0,75H), 7,45-7,38 (m, 2H), 4,84-4,82 (m, 1H), 4,68-4,66 (m, 1H), 4,54-4,51 (m, 2H), 3,64 (s, 3H), 3,60-3,58 (m, 2H), 3,20-3,13 (m, 2H), 2,79-2,77 (m, 1H), 2,65-2,62 (m, 2H), 1,76-1,72 (m, 4H); ESI MS m /z 473 [M H]+.
Ejemplo 69: 1-(3-(4-(3,5-d¡fluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡na-1-carbon¡l)-4,5-d¡h¡dro-1H-pirazolo[3,4-c]p¡r¡d¡n-6(7H)-il)etanona (131)
Paso A: Siguiendo el procedimiento general GP-E1, clorhidrato de (4-(3,5-b¡s(tr¡fluoromet¡l)fen¡l)piper¡d¡n-1-¡l)(4,5,6,7-tetrah¡dro-1H-p¡razolo[3,4-c]p¡rid¡n-3-¡l)metanona (52) y cloruro de acetilo se convirtieron en 1-(3-(4-(3,5-difluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡na-1-carbon¡l)-4,5-d¡h¡dro-1H-p¡razolo[3,4-c]p¡r¡din-6(7H)-¡l)etanona como un sólido blanco (0,087 g, 41%): 1H NMR (300 MHz, CDCfe) 8 11,01 (br, 1H), 6,93 (m, 1H), 6,78 (m, 1H), 4,84-4,66 (m, 4H), 3,85-3,67 (m, 2H), 3,34-2,68 (m, 5H), 2,22 and 2,19 (s, 3H), 1,89-1,66 (m, 4H); MS (ESI+) m /z 457 [M+H]+,77 (m, 1H), 2,65-2,62 (m, 2H), 1,76-1,72 (m, 4H); ESI MS m /z 473 [M H]+.
Ejemplo 70: 3-(4-(3,5-d¡fluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡na-1-carbon¡l)-[1,2,4]tr¡azolo[4,3-a]p¡rid¡na-6-carbon¡tr¡lo (132) Paso A: A una solución de 4-(3,5-difluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡na-1-carboxilato de ferc-butilo (0,246 g, 0,675 mmol) en diclorometano (5 mL) se añadió HCl (2 M en éter, 10 mL). La mezcla se agitó durante 6 h y se evaporó para proporcionar un sólido que se disolvió en DMF (4 mL). En un matraz aparte, a una solución de 6-bromo-[1,2,4]triazolo[4,3-a]p¡r¡d¡na-3-carboxilato de etilo (0,182 g, 0,675 mmol) en THF (5 mL) se le añadió una solución de hidróxido de litio hidratado (0,028 g, 0,675 mmol) en agua (2 mL). La mezcla se agitó durante 20 min, se acidificó con HCl 2 N a pH 6 y se evaporó hasta sequedad. A este residuo se le añadieron hexafluorofosfato de benzotriazol-1-¡l-ox¡tr¡s(d¡met¡lam¡no)fosfonio (0,448 g, 1,01 mmol), W,W-diisoprop¡let¡lam¡na (0,349 g, 2,70 mmol) y la solución de DMF obtenida de la primera reacción. La mezcla se agitó a temperatura ambiente durante 16 horas y se vertió en agua. La mezcla se extrajo con acetato de etilo y la capa orgánica se lavó tres veces con salmuera, se secó (Na2SO4), se filtró y se concentró a presión reducida. El residuo resultante se cromatografió sobre gel de sílice (0-60% de EtOAc en hexanos) para proporcionar (6 -bromo-[1,2,4]triazolo[4,3-a]pir¡d¡n-3-¡l) (4-(3,5-difluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡n-1-il)metanona como un sólido blanquecino (0,115 g, 34%): 1H NMR (300 MHz, CDCfe) 8 9,37 (m, 1H), 7,79 (dd, J = 9,6, 0,9 Hz, 1H), 7,50 (dd, J = 9,6, 1,7 Hz, 1H), 6,96 (d, J = 9,7 Hz, 1H), 6,84-6,77 (m, 1H), 5,77-5,72 (m, 1H), 5,00-4,95 (m, 1H), 3,44-3,29 (m, 2H), 3,01-2,92 (m, 1H), 2,01-1,69 (m, 4H); MS (ESI+) m /z 489 [M H]+.
Paso B: Una mezcla de (6-bromo-[1,2,4]tr¡azolo[4,3-a]p¡r¡din-3-¡l)(4-(3,5-d¡fluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡n-1
il)metanona (0,115 g, 0,235 mmol), cianuro de zinc (0,055 g, 0,470 mmol), tetrakis(trifenilfosfina)paladio (0,027 g, 0,0235 mmol) y DMF (4 mL) se calentó con radiación de microondas a 130°C durante 30 minutos. Después de enfriar a temperatura ambiente, la mezcla se diluyó con agua (80 mL) y se extrajo con EtOAc (80 mL). El extracto se lavó con salmuera (2 x 80 mL), se secó (Na2SO4), se filtró y se concentró a presión reducida. El residuo resultante se cromatografió sobre gel de sílice (EtOAc al 0-50% en hexanos) para proporcionar 3-(4-(3,5-difluoro-2-(trifluorometil)fenil)piperidina-1-carbonil)-[1,2,4]triazolo[4,3-a]piridina-6-carbonitrilo como un sólido blanco (0,050 g, 49%): 1H NMR (300 m Hz , CDCh) 8 9,71 (m, 1H), 7,98 (dd, J = 9,5, 1,0 Hz, 1H), 7,51 (dd, J = 9,5, 1,6 Hz, 1H), 6,95 (d, J = 9,5 Hz, 1H), 6,84-6,77 (m, 1H), 5,76 (m, 1H), 5,01-4,96 (m, 1H), 3,46-3,31 (m, 2H), 3,03-2,94 (m, 1H), 2,07-1,70 (m, 4H); MS (ESI+) m/z 436 [M H]+.
Ejemplo 71: Preparación de (4-(3,5-difluoro-2-(trifluorometil)fenil)piperidin-1-il) (5-etil-4,5,6,7-tetrahidro-1H-pirazolo[4,3-c]piridin-3-il)metanona (133)
Paso A: Siguiendo el procedimiento general GP-G1, clorhidrato de (4-(3,5-bis(trifluorometil)fenil)piperidin-1-il) (4,5,6,7-tetrahidro-1H-pirazolo[3,4-c]piridin-3-il)metanona (52) y acetaldehído se convirtieron en (4-(3,5-difluoro-2-(trifluorometil)fenil)piperidin-1-il) (5-etil-4,5,6,7-tetrahidro-1H-pirazolo[4,3-c]piridin-3-il)metanona como un sólido blanco (29 mg, 67%): mp 149-155°C; 1H NMR (500 MHz, DMSO-d6) 8 12,766 (s, 1H), 7,481-7,353 (m, 1H), 5,196-5,049 (m, 1H), 4,765-4,569 (m, 1H), 3,558-3,378 (m, 2H), 3,263-3,048 (m, 2H), 2,850-2,715 (m, 1H), 2,664 (s, 4H), 2,575-2,515 (m, 2H), 1,858-1,606 (br s, 4H), 1,121-1,018 (m, 4H); ESI MS m /z 443,2 [M H]+.
Ejemplo 72: Preparación de (5-(ciclopropilmetil)-4,5,6,7-tetrahidro-1H-pirazolo[4,3-c]piridin-3-il) (4-(3,5-difluoro-2-(trifluorometil)fenil)piperidin-1-il)metanona (134)
Paso A: Siguiendo el procedimiento general GP-G1, clorhidrato de (4-(3,5-bis(trifluorometil)fenil)piperidin-1-il) (4,5,6,7-tetrahidro-1H-pirazolo[3,4-c]piridin-3-il)metanona (52) y el acetaldehído de ciclopropilo se convirtieron en (5-(ciclopropilmetil)-4,5,6,7-tetrahidro-1H-pirazolo[4,3-c]piridin-3-il) (4-(3,5-difluoro-2-(trifluorometil)fenil)piperidin-1-il)metanona como un sólido blanco (29 mg, 67%): No se observó fusión clara; 1H NMR (500 MHz, DMSO-de) 8 12,764 (s, 1H), 7,522-7,347 (m, 2H), 5,225-5,016 (br s, 1H), 4,766-4,580 (br s, 1H), 3,673-3,444 (m, 2H), 3,258-3,034 (m, 2H), 2,868 2,601 (m, 5H), 2,443-2,334 (m, 2H), 1,858-1,620 (br s, 4H), 0,995- 0,829 (m, 1H), 0,551-0,410 (m, 2H), 0,195-0,075 (m, 2H) ; ESI MS m /z 469,1 [M H]+.
Ejemplo 73 : Preparación de (4-(3,5-difluoro-2-(trifluorometil)fenil)piperidin-1-il) (5-(oxetan-3-il)-4,5,6,7-tetrahidro-1H-pirazolo[4,3-c]piridin-3-il)metanona (135)
Paso A: Siguiendo el procedimiento general GP-G1, clorhidrato de (4-(3,5-bis(trifluorometil)fenil)piperidin-1-il) (4,5,6,7-tetrahidro-1H-pirazolo[3,4-c]piridin-3-il)metanona (52) y 3-oxetanona se convirtieron en (4-(3,5-difluoro-2-(trifluorometil)fenil)piperidin-1-il) (5-(oxetan-3-il)-4,5,6,7-tetrahidro-1H-pirazolo[4,3-c]piridin-3-il)metanona como un sólido blanco (35 mg, 67%): No se observó fusión clara; 1H NMR (300 MHz, DMSO-de) 8 12,843 (s, 1H), 7,483-7,346 (m, 1H), 5,234-5,065 (br s, 1H), 4,731-4,627 (br s, 1H), 4,627-4,562 (m, 2H), 4,562-4,452 (m, 2H), 3,714-3,643 (m, 1H), 3,473 3,337 (br s, 2H), 3,260-3,058 (m, 2H), 2,850-2,724 (m, 1H), 2,724-2,658 (m, 2H), 2,582-2,516 (m, 2H), 1,876-1,586 (br s, 4H); ESI MS m /z 471 [M H]+.
Ejemplo 74: Preparación de 3-(4-(3,5-difluoro-2-(trifluorometil)fenil)piperidina-1-carbonil)-6,7-dihidro-1Hpirazol[4,3-c]piridina-5(4H)-carbonitrilo (136)
Paso A: Siguiendo el procedimiento general GP-G2, clorhidrato de (4-(3,5-bis(trifluorometil)fenil)piperidin-1-il) (4,5,6,7-tetrahidro-1H-pirazolo[3,4-c]piridin-3-il)metanona (52) y bromuro de cianógeno se convirtieron en 3-(4-(3,5-difluoro-2-(trifluorometil)fenil)piperidin-1-carbonil)-6,7-dihidro-1H-pirazolo[4,3-c]piridina-5(4H)-carbonitrilo como un sólido blanco (16 mg, 55%): No se observó fusión clara; 1H NMR (500 MHz, DMSO-de) 8 13,146-13,060 (m, 1H), 7,508-7,356 (m, 2H), 5,402-4,584 (m, 2H), 4,433-4,327 (m, 2H), 3,536-3,412 (m, 2H), 3,261-3,092 (m, 2H), 2,905-2,728 (m, 3H), 1,884-1,608 (m, 4H); ESI MS m /z 440 [M H]+.
Ejemplo 75: Preparación de (4-(3,5-difluoro-2-(trifluorometil)fenil)piperidin-1-il) (5-(2,2,2-trifluoroetil)-4,5,6,7-tetrahidro-1 H-pirazolo[4,3-c]piridin-3-il)metanona (137)
Paso A: Siguiendo el procedimiento general GP-G2, clorhidrato de (4-(3,5-bis(trifluorometil)fenil)piperidin-1-il) (4,5,6,7-tetrahidro-1H-pirazolo[3,4-c]piridin-3-il)metanona (52) y el trifluorometanosulfonato de 2 ,2 ,2 -trifluoroetilo se convirtieron en (4-(3,5-difluoro-2-(trifluorometil)fenil)piperidin-1-il) (5-(2,2,2-trifluoroetil)-4,5,6,7-tetrahidro-1H-pirazolo[4,3-c]piridin-3-il)metanona como un sólido blanco (16 mg, 55%): No se observó fusión clara; 1H NMR (500 MHz, DMSO-de) 8 13,033 12,768 (m, 1H), 7,506-7,333 (m, 2H), 5,290-4,578 (m, 2H), 3,894-3,657 (m, 2H), 3,447-3,320 (m, 2H), 3,256-3,037 (m, 2H), 2,937 (s, 2H),2,709 (s, 3H), 1,906-1,605 (br s, 4H); ESI MS m /z 497 [M H]+.
Ejemplo 76: Preparación de 3-(4-(3,5-difluoro-2-(trifluorometil)fenil)piperidina-1-carbonil)-6,7-dihidro-1H-pirazolo[4,3-c]piridina-5(4H)-carboxilato de metilo (138)
Paso A: Siguiendo el procedimiento general GP-B2, clorhidrato de (4-(3,5-difluoro-2-(trifluorometil)fenil)piperidin-1-il)(4,5,6,7-tetrahidro-1H-pirazolo[3,4-c]piridin-3-il)metanona (60) y cloroformiato de metilo se convirtieron en 3-(4-(3,5d¡fluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡na-1-carbon¡l)-6,7-d¡hidro-1H-p¡razolo[4,3-c]p¡r¡d¡na-5(4H)-carbox¡lato de metilo como un sól¡do blanco (16 mg, 55%): 1H NMR (300 MHz, DMSO-da) 8 13,11-12,93 (m, 1H), 7,56-7,32 (m, 2H), 5,40-5,16 (m, 1H), 4,81-4,59 (m, 1H), 4,59-4,40 (m, 2H), 3,64 (s, 5H), 3,28-3,03 (br s, 2H), 2,89-2,62 (m, 3H), 1,94-1,61 (br s, 4H); ESI MS m /z 472 [M H]+.
Ejemplo 77: Preparac¡ón de 3-(4-(3,5-d¡fluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡na-1-carbon¡l)-A/-met¡l-6,7-d¡hidro-1H-p¡razolo[4,3-c]p¡r¡d¡na-5(4H)-carboxam¡da (139)
Paso A: S¡gu¡endo el proced¡m¡ento general GP-E2, clorhidrato de (4-(3,5-b¡s(tr¡fluoromet¡l)fen¡l)p¡per¡d¡n-1-¡l) (4,5,6,7-tetrahidro-1H-p¡razolo[3,4-c]p¡r¡d¡n-3-¡l)metanona (52) e ¡soc¡anato de met¡lo se conv¡rt¡eron en 3-(4-(3,5-d¡fluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡na-1-carbon¡l)-W-met¡l-6,7-d¡hidro-1Hp¡razolo[4,3-c]p¡r¡d¡na-5(4H)-carboxam¡da como un sól¡do blanco (16 mg, 55%): S¡n fus¡ón clara; 1H NMR (500 MHz, DMSO-cfe) 8 13,043-12,777 (m, 1H), 7,523-7,348 (m, 2H), 6,536 (s, 1H), 5,275-4,595 (m, 2H), 4,536-4,296 (m, 2H), 3,561 (s, 2H), 3,273-3,076 (m, 2H), 2,891-2,706 (br s, 1H), 2,706-2,614 (m, 2H), 2,576 (s, 3H), 1,906-1,585 (m, 4H); ESI MS m /z 472 [M H]+.
Ejemplo 78: Preparac¡ón de 1-(3-(4-(5-fluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡na-1-carbon¡l)-4,5-d¡hidro-1H-p¡razolo[3,4-c]p¡r¡d¡n-6(7H) )-¡l)etanona (140)
Paso A: S¡gu¡endo el proced¡m¡ento general GP-E1, clorhidrato de (4-(5-fluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡n-1-¡l) (4,5,6,7-tetrahidro-1H-p¡razolo[3,4-c]p¡r¡d¡n-3-¡l)metanona (44) y cloruro de acet¡lo se conv¡rt¡eron en 1-(3-(4-(5-fluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡na-1-carbon¡l)-4,5-d¡hidro-1H-p¡razolo[3,4-c]p¡r¡d¡na-6(7H)-¡l)etanona como un sól¡do blanco (27 mg, 41%): mp 190-195°C; 1H NMR (500 MHz, DMSO-cfe) 8 13,17-12,85 (m, 1H), 7,79-7,73 (m, 1H), 7,57-7,52 (m, 1H), 7,30-7,22 (m, 1H), 4,94-4,77 (m, 1H), 4,74-4,63 (m, 1H), 4,62-4,51 (m, 2H), 3,73-3,56 (m, 2H), 3,19-3,06 (m, 2H), 2,83 2,53 (m, 3H), 2,14-2,05 (m, 3H), 1,80-1,64 (m, 4H); ESI MS m /z 439 [M H]+.
Ejemplo 79: Preparac¡ón de (4-(5-fluoro-2-(tr¡fluoromet¡l)fen¡l) p¡per¡d¡n-1-¡l)(5-(met¡lsulfon¡l)-4,5,6,7-tetrahidro-1H-p¡razolo[4,3-c]p¡r¡d¡n-3-¡l)metanona (141)
Paso A: S¡gu¡endo el proced¡m¡ento general GP-C, clorhidrato de (4-(5-fluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡n-1-¡l)(4,5,6,7-tetrahidro-1H-p¡razolo[4,3-c]p¡r¡d¡n-3-¡l)metanona (46) y cloruro de metanosulfon¡lo se conv¡rt¡eron en (4-(5-fluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡n-1-¡l) (5-(met¡lsulfon¡l)-4,5,6,7-tetrahidro-1H-p¡razolo[4,3-c]p¡r¡d¡n-3-¡l)metanona como un sól¡do blanco (105 mg, 60%): mp >260°C; 1H NMR (500 MHz, DMSO-cfe) 8 13,03 (s, 1H), 7,76 (dd, J = 9,0, 6,0 Hz, 1H), 7,55 (dd, J = 10,5, 2,5 Hz, 1H), 7,28-7,21 (m, 1H), 5,34-5,23 (m, 1H), 4,72-4,64 (m, 1H), 4,43-4,26 (m, 2H), 3,54-3,39 (m, 2H), 3,22 3,09 (m, 2H), 2,94 (s, 3H), 2,86-2,72 (m, 3H), 1,83-1,67 (m, 4H); ESI MS m /z 475 [M H]+.
Ejemplo 80: Preparac¡ón de 3-(4-(5-fluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡na-1-carbon¡l)-[1,2,4]tr¡azolo[4,3-a]p¡r¡d¡na-6-carbon¡tr¡lo (142)
Paso A: A una soluc¡ón de 6-bromo-[1,2,4]tr¡azolo[4,3-a]p¡r¡d¡na-3-carbox¡lato de et¡lo (85 mg, 0,32 mmol) en THF (2,6 mL) se le añad¡ó una soluc¡ón de monohidrato de L¡OH (15 mg, 0,35 mmol) en H2O (1,8 mL). La mezcla se ag¡tó durante 20 m¡n y se neutral¡zó con HCl 2 N. La mezcla se concentró a pres¡ón reduc¡da. El res¡duo obten¡do se d¡luyó en DMF (3,0 mL) en atmósfera de N2. A esta mezcla se le añad¡ó clorhidrato de 4-(5-fluoro-2-tr¡fluoromet¡l)fen¡lp¡per¡d¡na (11, 89 mg, 0,32 mmol), hexafluorofosfato de benzotr¡azol-1-¡l-ox¡-tr¡s-(d¡met¡lam¡no)-fosfon¡o (280 mg, 0,63 mmol) y d¡¡soprop¡let¡lam¡na (0,17 mL, 0,95 mmol). La mezcla se ag¡tó a temperatura amb¡ente durante 18 h. La mezcla resultante se d¡luyó con H2O (20 mL). La soluc¡ón se extrajo con EtOAc (3 x 20 mL). Las capas orgán¡cas comb¡nadas se lavaron con una soluc¡ón de salmuera saturada (3 x 20 mL) y se concentraron a pres¡ón reduc¡da. El res¡duo resultante se cromatograf¡ó sobre gel de síl¡ce (un¡dad Isco Comb¡Flash Rf, columna Red¡sep de 12 g, 0 % a 50% de EtOAc en hexanos) para proporc¡onar (6-bromo-[1,2,4]tr¡azolo[4,3-a]p¡r¡d¡n-3-¡l)(4-(5-fluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡n-1-¡l)metanona como un sól¡do blanco (97 mg, 65%): 1H NMR (500 MHz, DMSO-cfe) 8 9,16-9,14 (m, 1H), 7,98 (dd, J = 10,0, 1,0 Hz, 1H), 7,77 (dd, J = 8,5, 5,5 Hz, 1H), 7,72 (dd, J = 9,5, 1,5 Hz, 1H), 7,52 (dd, J = 11,0, 3,5Hz, 1H), 7,29-7,23 (m, 1H), 5,31-5,24 (m, 1H), 4,76-4,70 (m, 1H), 3,42-3,32 (m, 1H), 3,27-3,19 (m, 1H), 3,06-2,96 (m, 1H), 1,98-1,75 (m, 4H).
Paso B: Una soluc¡ón de (6-bromo-[1,2,4]tr¡azolo[4,3-a]p¡r¡d¡n-3-¡l)(4-(5-fluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡n-1-¡l)metanona (97 mg, 0,21 mmol) en DMF (2,2 mL) se burbujeó con Ar durante 20 m¡n. Se añad¡ó c¡anuro de z¡nc (48 mg, 0,41 mmol) y la soluc¡ón se burbujeó con Ar durante 10 m¡n. A la soluc¡ón se le añad¡ó Pd(PPh3)4 (24 mg, 0,021 mmol) y el rec¡p¡ente se selló y calentó a 130°C con m¡croondas durante 30 m¡n. La mezcla se d¡luyó con soluc¡ón saturada de b¡carbonato de sod¡o (10 mL) y se extrajo con EtOAc (4 x 30 mL). Los extractos orgán¡cos comb¡nados se concentraron a sequedad bajo pres¡ón reduc¡da. El res¡duo resultante se cromatograf¡ó sobre gel de síl¡ce (un¡dad Isco Comb¡Flash Rf, columna Red¡sep de 12 g, 0 % a 50 % de EtOAc en hexanos) y se l¡of¡l¡zó para proporc¡onar 3-(4-(5-fluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡na-1-carbon¡l)-[1,2,4]tr¡azolo[4,3-a]p¡r¡d¡na-6-carbon¡tr¡lo como un sól¡do blanco (35 mg, 41%): mp 190-197°C; 1H NMR (500 MHz, DMSO-cfe) 8 9,54-9,53 (m, 1H), 8,14 (dd, J = 9,5, 1,5Hz, 1H), 7,82 (d, J = 9,5, 1,5Hz, 1H), 7,78 (dd, J = 9,0, 6,0 Hz, 1H), 7,56 (dd, J = 10,5, 2,5 Hz, 1H), 7,30-7,24 (m, 1H), 5,20-5,10 (m, 1H), 4,73-4,71 (m, 1H), 3,43-3,34 (m, 1H), 3,29-3,20 (m, 1H), 3,08-3,00 (m, 1H), 1,99-1,77 (m, 4H); ESI MS m /z 418 [M H]+.
Ejemplo 81: Preparac¡ón de 1-(3-(4-(2-cloro-5-fluorofen¡l)p¡per¡d¡na-1-carbon¡l)-4,5-d¡hidro-1H-p¡razolo[3,4-c]p¡r¡d¡n-6(7H)-¡l)etanona (143)
Paso A: Siguiendo el procedimiento general GP-E1, clorhidrato de ((4-(2-cloro-5-fluorofenil)piperidin-1-il) (4,5,6,7-tetrahidro-1H-pirazolo[3,4-c]piridin-3-il)metanona (48) y cloruro de acetilo se convirtieron en 1-(3-(4-(2-cloro-5-fluorofenil)piperidina-1-carbonil)-4,5-dihidro-1H-pirazolo[3,4-c]piridin-6(7H)-il)etanona como un sólido blanco (27 mg, 63%): 1H NMR (500 MHz, DMSO-da) 8 13,99-12,18 (m, 1H), 7,48 (dd, J= 9,0, 5,5 Hz, 1H), 7,72 (dd, J= 10,5, 3,5 Hz, 1H), 7,15-7,08 (m, 1H), 4,90-4,74 (m, 1H), 4,73- 4,53 (m, 3H), 3,71-3,55 (m, 2H), 3,28-3,10 (m, 2H), 2,87-2,50 (m, 3H, se superpone con el disolvente), 2,12-2,04 (m, 3H), 1,91-1,71 (m, 2H), 1,66-1,52 (m, 2H); ESI MS m/z 405 [M H]+.
Ejemplo 82: Preparación de (4-(2-cloro-5-fluorofenil)piperidin-1-il) (5-(metilsulfonil)-4,5,6,7-tetrahidro-1H-pirazolo[4,3- c ]piridin-3-il)metanona (144)
Paso A: Siguiendo el procedimiento general GP-C, clorhidrato de (4-(2-cloro-5-fluorofenil)piperidin-1-il)(4,5,6,7-tetrahidro-1H-pirazolo[4,3-c]piridin-3-il)metanona (50) y cloruro de metanosulfonilo se convirtieron en (4-(2-cloro-5-fluorofenil)piperidin-1-il)(5-(metilsulfonil)-4,5,6,7-tetrahidro-1H-pirazolo[4,3-c]piridin-3-il)metanona como un sólido blanco (36 mg, 51%): mp 260-267°C; 1H NMR (500 MHz, DMSO-cfe) 8 13,03 (s, 1H), 7,48 (dd, J = 8,5, 5,0 Hz, 1H), 7,28 (dd, J = 10,5, 3,5 Hz, 1H), 7,13-7,08 (m, 1H), 5,30-5,20 (m, 1H), 4,72-4,63 (m, 1H), 4,41-4,24 (m, 2H), 3,53-3,40 (m, 2H), 3,28 3,13 (m, 2H), 2,93 (s, 3H), 2,89-2,73 (m, 3H), 1,90-1,74 (m, 2H), 1,70-1,51 (m, 2H); ESI MS m /z 441 [M H]+.
Ejemplo 83: Preparación de 3-(4-(2-cloro-5-fluorofenil)piperidina-1-carbonil)-[1,2,4]triazolo[4,3-a]piridina-6-carbonitrilo (145)
Paso A: A una solución de 6-bromo-[1,2,4]triazolo[4,3-a]piridina-3-carboxilato de etilo (73 mg, 0,27 mmol) en THF (2,2 mL) se le añadió una solución de monohidrato de LiOH (13 mg, 0,30 mmol) en H2O (1,5 mL). La mezcla se agitó durante 20 min y se neutralizó con HCl 2 N. La mezcla se concentró a presión reducida. El residuo obtenido se diluyó en DMF (2,9 mL) en atmósfera de N2. A esta mezcla se le añadió clorhidrato de 4-(2-cloro-5-fluorofenil)piperidina (17, 68 mg, 0,27 mmol), hexafluorofosfato de benzotriazol-1-il-oxi-tris-(dimetilamino)-fosfonio (240 mg, 0,54 mmol) y diisopropiletilamina (0,14 mL, 0,81 mmol). La mezcla se agitó a temperatura ambiente durante 18 h. La mezcla resultante se diluyó con H2O (20 mL) y el precipitado resultante se recolectó mediante filtración. Los sólidos obtenidos se cromatografiaron sobre gel de sílice (unidad Isco CombiFlash Rf, columna Redisep de 12 g, 0 a 50% de EtOAc en hexanos) para proporcionar (6 -bromo-[1,2,4]triazolo[4,3-a]piridin-3-il) (4-(2-cloro-5-fluorofenil)piperidin-1-il)metanona como un sólido blanco (50 mg, 42%): 1H NMR (500 MHz, DMSO-cfe) 8 9,13-9,11 (m, 1H), 7,98 (dd, J = 9,5, 1,0 Hz, 1H), 7,71 (dd, J = 9,5, 1,5 Hz, 1H), 7,50 (dd, J = 9,0, 5,5 Hz, 1H), 7,28 (dd, J = 25, 3,0 Hz, 1H), 7,15-7,10 (m, 1H), 5,28-5,20 (m, 1H), 4,76-4,70 (m, 1H), 3,42 3,29 (m, 2H, se superpone con H2O), 3,07-2,98 (m, 1H), 1,96-1,65 (m, 4H).
Paso B: Una solución de (6-bromo-[1,2,4]triazolo[4,3-a]piridin-3-il)(4-(2-cloro-5-fluorofenil)piperidin-1-il)metanona (50 mg, 0,21 mmol) en DMF (2,0 mL) y cianuro de zinc (48 mg, 0,41 mmol) se burbujeó con Ar durante 20 min. A la solución se le añadió Pd(PPh3)4 (13 mg, 0,011 mmol) y el recipiente se selló y calentó a 130°C con microondas durante 30 min. La mezcla se diluyó con solución saturada de bicarbonato de sodio (10 mL) y se extrajo con EtOAc (4 x 30 mL). Los extractos orgánicos combinados se concentraron a sequedad bajo presión reducida. El residuo resultante se cromatografió sobre gel de sílice (unidad Isco CombiFlash Rf, columna Redisep de 12 g, 0 % a 50 % de EtOAc en hexanos) y se liofilizó para proporcionar 3-(4-(2-cloro-5-fluorofenil)piperidina-1-carbonil)-[1,2,4]triazolo[4,3-a]piridina-6-carbonitrilo como un sólido blanco (21 mg, 54%):mp 211-214 °C ; 1H NMR (500 MHz, DMSO-cfe) 8 9,52-9,50 (m, 1H), 8,13 (dd, J = 9,5, 1,0 Hz, 1H), 7,81 (dd, J = 9,5, 1,5 Hz, 1H), 7,50 (dd, J = 9,0, 5,5 Hz, 1H), 7,28 (dd, J = 10,5, 3,5 Hz, 1H), 7,16-7,11 (m, 1H), 5,16-5,11 (m, 1H), 4,78-4,71 (m, 1H), 3,46-3,30 (m, 2H, se superpone con H2O), 3,11-3,01 (m, 1H), 1,99-1,70 (m, 4H); ESI MS m /z 384 [M H]+.
Ejemplo 84: Preparación de (4-(2-cloro-3-(fluorofenil)piperidin-1-il) (6-ciclopropilmetil)-4,5,6,7-tetrahidro-1H-pirazolo[3,4-c]piridin-3-il)metanona (146)
Paso A: Siguiendo el procedimiento general GP-G2, clorhidrato de (4-(2-cloro-3-fluorofenil)piperidin-1-il) (4,5,6,7-tetrahidro-1H-pirazolo[4,3-c]piridin-3-il)metanona (40) y ciclopropano carbaldehído se convirtieron en (4-(2-cloro-3-(fluorofenil)piperidin-1-il)(6-ciclopropilmetil)-4,5,6,7-tetrahidro-1H-pirazolo[3,4-c]piridin-3-il)metanona como un sólido blanco (21 mg, 36%): mp 187-191°C; 1H NMR (300 MHz, DMSO-de) 8 12,74 (br s, 1H), 7,38-7,24 (m, 3H), 4,96-4,55 (m, 2H), 3,57 (s, 2H), 3,28-3,17 (m, 2H), 2,91-2,38 (m, 7H), 1,91-1,79 (m, 2H), 1,64-1,56 (m, 2H), 0,92-0,85 (m, 1H), 0,52-0,48 (m, 2H), 0,16-0,08 (m, 2H); ESI MS m /z 417 [M H]+.
Ejemplo 85: Preparación de 3-(4-(2-cloro-3-fluorofenil)piperidina-1-carbonil)-4,5-dihidro-1H-pirazolo[3,4-c]piridina-6(7H)-carboxilato de metilo (147)
Paso A: Siguiendo el procedimiento general GP-E1, clorhidrato de (4-(2-cloro-3-fluorofenil)piperidin-1-il)(4,5,6,7-tetrahidro-1H-pirazolo[4,3-c]piridin-3-il)metanona (40) y cloroformiato de metilo se convirtieron en 3-(4-(2-cloro-3-fluorofenil)piperidina-1-carbonil)-4,5-dihidro-1H-pirazolo[3,4-c]piridina-6(7H)-carboxilato de metilo como un sólido blanco(30 mg, 65%): mp 182-185°C; 1H NMR (300 MHz, DMSO-cfe) 8 12,88 (br s, 1H), 7,39-7,24 (m, 3H), 4,85-4,61 (m, 2H), 4,54-4,49 (m, 2H), 3,64-3,56 (m, 5H), 3,28-3,12 (m, 2H), 2,93-2,75 (m, 1H), 2,61-2,55 (m, 2H), 1,91-1,78 (m, 2H), 1,64-1,55 (m, 2H); ESI MS m /z 421 [M H]+.
Ejemplo 86: Preparación de 3-(4-(2-cloro-3-fluorofen¡l)p¡per¡d¡na-1-carbon¡l)-4,5-d¡h¡dro-1H-p¡razolo[3,4-c]p¡r¡d¡na-6(7H)-carbonitrilo (148)
Paso A: Siguiendo el procedimiento general GP-G2, clorhidrato de (4-(2-cloro-3-fluorofen¡l)p¡per¡d¡n-1-¡l)(4,5,6,7-tetrah¡dro-1H-p¡razolo[4,3-c]p¡rid¡n-3-¡l)metanona (40) y bromuro de cianógeno se convirtieron en 3-(4-(2-cloro-3-fluorofen¡l)p¡per¡d¡na-1-carbon¡l)-4,5-d¡h¡dro-1H-p¡razolo[3,4-c]p¡r¡d¡na-6(7H)-carbonitr¡lo como un sólido blanco (19mg, 35%): mp 182-185°C; 1 H NMR (300 MHz, DMSO-de) 812,98 (br s, 1H), 7,37- 7,24 (m, 3H), 4,91-4,65 (m, 2H), 4,47-4,40 (m, 2H), 3,43-3,12 (m, 4H), 2,93-2,69 (m, 3H), 1,91-1,78 (m, 2H), 1,76-1,55 (m, 2H); ESI MS m/z 388 [M H]+.
Ejemplo 87: Preparación de (4-(2-cloro-3-(fluorofen¡l)p¡per¡d¡n-1-¡l)(6-oxetan-3-¡l)-4,5,6,7-tetrah¡dro-1H-p¡razolo[3,4-c]p¡r¡d¡n-3-¡l)metanona (149)
Paso A: Siguiendo el procedimiento general GP-G1, clorhidrato de (4-(2-cloro-3-fluorofenil)piper¡d¡n-1 -il)(4,5,6,7-tetrahidro-1 H-p¡razolo[4,3-c]p¡rid¡n-3-¡l)metanona (40) y oxetan-3-ona se convirtieron en (4-(2-cloro-3-(fluorofen¡l)p¡per¡d¡n-1-¡l)(6-oxetan-3-¡l)-4,5,6,7-tetrah¡dro-1H-pirazolo[3,4-c]p¡r¡d¡n-3-¡l)metanona como un sólido blanco (17 mg, 31%): 1 H NMR (300 MHz, DMSO-de) 812,77 (br s, 1H), 7,37-7,24 (m, 3H), 4,91-4,47 (m, 6H), 3,71-3,61 (m, 1H), 3,47-3,12 (m, 4H), 2,93 2,78 (m, 1H), 2,74-2,53(m, 4H), 1,91-1,78 (m, 2H), 1,76-1,55 (m, 2H); ESI MS m/z 419 [M H]+.
Ejemplo 88: Preparación de 1-(3-(4-(2-cloro-3-fluorofen¡l)p¡per¡d¡na-1-carbon¡l)-4,5-d¡h¡dro-1H-p¡razolo[3,4-c]p¡r¡d¡n-6(7H)-il)etanona (150)
Paso A: Siguiendo el procedimiento general GP-E1, clorhidrato de (4-(2-cloro-3-fluorofen¡l)p¡per¡d¡n-1-¡l)(4,5,6,7-tetrah¡dro-1H-p¡razolo[4,3-c]p¡rid¡n-3-¡l)metanona (40) y cloruro de acetilo se convirtieron en 1-(3-(4-(2-cloro-3-fluorofen¡l)p¡per¡d¡na-1-carbon¡l)-4,5-d¡hidro-1H-p¡razolo[3,4-c]p¡r¡d¡n-6(7H)-¡l)etanona como un sólido blanco (14 mg, 25%): 1 H NMR (500 MHz, DMSO-de) 813,18-12,83 (m, 1H), 7,41-7,31 (m, 1H), 7,30-7,22 (m, 2H), 4,87-4,74 (m, 1H), 4,73 4,63 (m, 1H), 4,62-4,53 (m, 2H), 3,71-3,58 (m, 2H), 3,31-3,24 (m, 1H, se superpone con H2O), 3,20-3,12 (m, 1H), 2,90 2,76 (m, 1H), 2,74-2,52 (m, 2H), 2,11-2,07 (m, 3H), 1,89-1,72 (m, 2H), 1,65-1,52 (m, 2H); ESI MS m/z 405 [M H]+; HPLC >99% de pureza (Método H). (m, 3H), 4,91-4,47 (m, 6H), 3,71-3,61 (m, 1H), 3,47-3,12 (m, 4H), 2,93-2,78 (m, 1H), 2,74 2,53 (m, 4H), 1,91-1,78 (m, 2H), 1,76-1,55 (m, 2H); ESI MS m/z 419 [M H]+.
Ejemplo 89: Preparación de (4-(2-cloro-3-fluorofen¡l)p¡per¡d¡n-1-¡l)(5-(met¡lsulfon¡l)-4,5,6,7-tetrah¡dro-1H-p¡razolo[4,3-c]pirid¡n-3-¡lmetanona (151)
Paso A: Siguiendo el procedimiento general GP-C, clorhidrato de ((4-(2-cloro-3-fluorofen¡l)p¡per¡d¡n-1-¡l)(4,5,6,7-tetrah¡dro-1H-pirazolo[4,3-c]p¡r¡d¡n-3-¡l)metanona (40) y cloruro de metanosulfonilo se convirtieron en 1-(3-(4-(2-cloro-3-fluorofen¡l)p¡per¡d¡na-1-carbon¡l)-4,5-d¡h¡dro-1H-p¡razolo[3,4-c(4-(2-cloro-3-fluorofen¡l)p¡per¡d¡n-1-¡l)(5-(met¡lsulfon¡l)-4,5,6,7-tetrahidro-1H-p¡razolo[4,3-c]p¡r¡d¡n-3-¡lmetanona como un sólido blanco (21 mg, 34%): mp 247-253°C decomp.; 1 H NMR (500 MHz, DMSO-de) 813,03 (br s, 1H), 7,39-7,31 (m, 1H), 7,29-7,22 (m, 2H), 5,28-5,19 (m, 1H), 4,72-4,63 (m, 1H), 4,39-4,28 (m, 2H), 3,50-3,14 (m, 7H, se superpone con H2O), 2,86-2,79 (m, 3H), 1,91-1,76 (m, 2H), 1,69-1,53 (m, 2H); ESI MS m/z 441 [M H]+.
Ejemplo 90: Preparación de (6-bromo-[1,2,4]tr¡azolo[4,3-a]p¡r¡d¡n-3-¡l)(4-(2-cloro-3-fluorofen¡l)p¡perid¡n-1-¡l)metanona (152)
Paso A: A una solución de 6-bromo-[1,2,4]triazolo[4,3-a]pir¡d¡na-3-carbox¡lato de etilo (102 mg, 0,38 mmol) en THF (3,1 mL) se le añadió una solución de monohidrato de LiOH (16 mg, 0,38 mmol) en H2O (2,0 mL). La mezcla se agitó durante 20 min y se neutralizó con HCl 2 N. La mezcla se concentró a presión reducida. El residuo obtenido se diluyó en DMF (4,0 mL) en atmósfera de N2. A esta mezcla se le añadió clorhidrato de 4-(2-cloro-3-fluorofenil)p¡per¡d¡na (14, 94 mg, 0,38 mmol), hexafluorofosfato de benzotriazol-1-¡l-ox¡-tr¡s-(d¡met¡lam¡no)-fosfonio (334 mg, 0,76 mmol) y diisopropiletilamina (0,20 mL, 1,1 mmol). La mezcla se agitó a temperatura ambiente durante 18 h. La mezcla resultante se diluyó con H2O (20 mL) y el precipitado resultante se recolectó mediante filtración. Los sólidos obtenidos se cromatografiaron sobre gel de sílice (unidad Isco CombiFlash Rf, columna Redisep de 12 g, 0 a 50% de EtOAc en hexanos) para proporcionar (6-bromo-[1,2,4]triazolo[4,3-a]pir¡d¡n-3-¡l) (4-(2-cloro-3-fluorofen¡l)p¡perid¡n-1-¡l)metanona como un sólido blanco (80 mg, 48%): 1 H NMR (300 MHz, DMSO-cfe) 89,11 (br s, 1H), 8,03-7,95 (m, 1H), 7,71 (dd, J = 9,6, 1,8 Hz, 1H), 7,43-7,23 (m, 3H), 5,27-5,17 (m, 1H), 4,80-4,69 (m, 1H), 3,48-3,34 (m, 2H), 3,12-2,96 (m, 1H), 2,02-1,59 (m, 4H); ESI MS m/z 438 [M H]+.
Paso B: Una solución de (6-bromo-[1,2,4]tr¡azolo[4,3-a]p¡r¡d¡n-3-¡l)(4-(2-cloro-3-fluorofen¡l)p¡perid¡n-1-¡l)metanona (80 mg, 0,18 mmol) en DMF (2,0 mL) con cianuro de zinc (43 mg, 0,65 mmol) se burbujeó con Ar durante 20 min. A la solución se le añadió Pd(PPh3)4 (21 mg, 0,018 mmol) y el recipiente se selló y calentó a 130°C con microondas durante 30 min. La mezcla se diluyó con H2O (10 mL) y se extrajo con EtOAc (4 x 30 mL). Los extractos orgánicos combinados se concentraron a sequedad bajo presión reducida. El residuo resultante se cromatografió sobre gel de sílice (unidad Isco CombiFlash Rf, columna Redisep de 12 g, 0% a 60% de EtOAc en hexanos) y se liofilizó para proporcionar 3-(4-(2-cloro-3-fluorofenil)piper¡d¡na-1 -carbonil)-[1,2,4]tr¡azolo[4,3-a]p¡r¡d¡na-6-carbonitr¡lo como un sólido blanco (38 mg, 55%): mp 158-163°C; 1 H NMR (500 MHz, DMSO-de) 89,51-9,50 (m, 1H), 8,12 (dd, J = 9,5, 1,0 Hz, 1H), 7,81 (dd, J = 9,5, 1,5 Hz, 1H), 7,41-7,34 (m, 1H), 7,31-7,24 (m, 2H), 5,16-5,09 (m, 1H), 4,78-4,71 (m, 1H), 3,47-3,37 (m, 2H), 3,12-3,03 (m, 1H), 2,00-1,64 (m, 4H); ESI MS m/z 384 [M H]+.
Ejemplo 91: Preparación de 1-(3-(4-(3,5-b¡s(tr¡fluoromet¡l)fen¡l)p¡per¡d¡na-1-carbon¡l)-4,5-dih¡dro-1H-p¡razolo[3,4-c]p¡r¡d¡n-6(7H)-il)etanona (153)
Paso A: Siguiendo el procedimiento general GP-E1, clorhidrato de 4-(3,5-b¡s(tr¡fluoromet¡l)fen¡l)p¡perid¡n-1-¡l)(4,5,6,7-tetrahidro-1H-p¡razolo[3,4-c]p¡rid¡n-3-¡l)metanona (52) y cloruro de acetilo se convirtieron en 1-(3-(4-(3,5-b¡s(tr¡fluoromet¡l)fen¡l)p¡perid¡na-1-carbon¡l)-4,5-d¡h¡dro-1H-p¡razolo[3,4-c]p¡r¡d¡n- 6(7H)-il)etanona como un sólido blanco (33 mg, 50%):mp 204-205°C; 1H NMR (500 MHz, DMSO-de) 8 13,13-12,82 (m, 1H), 8,01-7,97 (m, 2H), 7,96-7,90 (m, 1H),4,92-4,79 (m, 1H), 4,75-4,63 (m, 1H), 4,62-4,53 (m, 2H), 3,72-3,58 (m, 2H), 3,18-3,05 (m, 2H), 2,84-2,48 (m, 3H, se superpone con el disolvente), 2,12-2,05 (m, 3H), 1,97-1,78 (m, 2H), 1,76-1,61 (m, 2H); ESI MS m /z 489 [M H]+.
Ejemplo 92: Preparación de (4-(3,5-b¡s(tr¡fluoromet¡l)fen¡l)p¡per¡din-1-¡l)(5-(met¡lsulfon¡l)-4,5,6,7-tetrah¡dro-1H-p¡razolo [4,3-c]pirid¡n-3-¡l)metanona (154)
Paso A: Siguiendo el procedimiento general GP-C, clorhidrato de (4-(3,5-bis(tr¡fluoromet¡l)fen¡l)p¡per¡d¡n-1-il) (4,5,6,7-tetrah¡dro-1H-p¡razolo[4,3-c]p¡rid¡n-3-¡l)metanona (52) y cloruro de metanosulfonilo se convirtieron en (4-(3,5-b¡s(tr¡fluoromet¡l)fen¡l)p¡per¡d¡n-1-¡l-(5-(met¡lsulfon¡l)-4,5,6,7-tetrahidro-1H-p¡razolo[4,3-c]p¡r¡d¡n-3-¡l)metanona como un sólido blanco (25 mg, 37%): 1H NMR (500 MHz, DMSO-cfe) 8 13,02 (s, 1H), 8,01-7,98 (m, 2H), 7,94-7,91 (m, 1H), 5,31 5,24 (m, 1H), 4,73-4,63 (m, 1H), 4,40-4,26 (m, 2H), 3,51-3,41 (m, 2H), 3,21-3,06 (m, 2H), 2,93(s, 3H), 2,85-2,74 (m, 3H), 1,96-1,80 (m, 2H), 1,79-1,64 (m, 2H); ESI MS m /z 525 [M H]+.
Ejemplo 93: Preparación de (4-(3,5-b¡s(tr¡fluoromet¡l)fen¡l)p¡per¡din-1-¡l)(5-(met¡lsulfon¡l)-4,5,6,7-tetrah¡dro-1H-pirazolo[4,3-c]pir¡d¡n-3-¡l)metanona (155)
Paso A: A una solución de 6-metoxi-[1,2,4]tr¡azolo[4,3-a]p¡rid¡na-3-carbox¡lato de etilo (66 mg, 0,30 mmol) en THF (2,5 mL) se le añadió una solución de monohidrato de LiOH (25 mg, 0,60 mmol) en H2O (1,6 mL). La mezcla se agitó durante 20 min y se neutralizó con HCl 2 N. La mezcla se concentró a presión reducida. El residuo obtenido se diluyó en DMF (3,3 mL) en atmósfera de N2. A esta mezcla se le añadió clorhidrato de 4-(3,5-bis(tr¡fluoromet¡l)fen¡l)p¡perid¡na (20, 100 mg, 0,30 mmol), hexafluorofosfato de benzotriazol-1-¡l-ox¡-tr¡s-(d¡met¡lam¡no)-fosfonio (266 mg, 0,60 mmol) y diisopropiletilamina (0,16 mL, 0,90 mmol). La mezcla se agitó a temperatura ambiente durante 18 h. La mezcla resultante se diluyó con H2O (20 mL) y se extrajo con EtOAc (4 x 30 mL). Los extractos orgánicos combinados se lavaron con una solución de salmuera saturada (3 x 30 mL) y se concentraron a presión reducida. El residuo obtenido se cromatografió sobre gel de sílice (unidad Isco CombiFlash Rf, columna Redisep de 24 g, 0 % a 2 % de MeOH en CH2Ch con 0 ,1% de NH4OH en CH2Cl2). El residuo obtenido se disolvió en CH2Ch (10 mL) y hexanos (100 mL). La solución se concentró parcialmente y los sólidos resultantes se recolectaron por filtración para proporcionar (4-(3,5-b¡s(tr¡fluorometil)fen¡l)p¡per¡d¡n-1-¡l) (6-metoxi-[1,2,4]tr¡azolo[4,3-a]p¡rid¡n-3-¡l)metanona como un sólido blanquecino (80 mg, 56%): mp 146-149 °C; 1H NMR (500 MHz, DMSO-cfe) 8 8,55 (d, J = 2,0 Hz, 1H), 8,03 (s, 2H), 7,95-7,89 (m, 2H), 7,38 (dd, J = 10, 2,5 Hz, 1H), 5,40-5,33 (m, 1H), 4,81-4,73 (m, 1H), 3,85 (s, 3H), 3,39-3,31 (m, 1H, se superpone con H2O), 3,26-3,16 (m, 1H), 3,02-2,93 (m, 1H), 2,03-1,77 (m, 4H); ESI MS m /z 473 [M H]+.
Ejemplo 94: Preparación de 1-(3-(4-(2-fluoro-6-(tr¡fluoromet¡l)fen¡l)p¡perid¡na-1-carbon¡l)-4,5-d¡h¡dro-1H-p¡razolo[3,4-c]piridin-6(7H) )-¡l)etanona (156)
Paso A: Siguiendo el procedimiento general GP-E1, clorhidrato de (4-(2-fluoro-6-(trifluoromet¡l)fen¡l)p¡per¡din-1-¡l) (4,5,6,7-tetrah¡dro-1H-p¡razolo[3,4-c]p¡rid¡n-3-¡l)metanona (56) y cloruro de acetilo se convirtieron en 1-(3-(4-(2-fluoro-6-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡na-1-carbon¡l)-4,5-d¡h¡dro-1H-pirazolo[3,4-c]p¡r¡d¡n-6(7H)-¡l)etanona como un sólido blanco (33 mg, 43%): 1H NMR (500 MHz, DMSO-cfe) 8 13,26-12,80 (m, 1H), 7,61-7,56 (m, 1H), 7,56-7,47 (m, 2H), 4,87-4,49 (m, 4H), 3,72-3,56 (m, 2H), 3,25-3,04 (m, 2H), 2,84-2,53 (m, 3H), 2,07-1,89 (m, 5H), 1,79-1,64 (m, 2H); ESI MS m /z 439 [M H]+.
Ejemplo 95: Preparación de 3-(4-(2-fluoro-6-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡na-1-carbon¡l)-[1,2,4]tr¡azolo[4,3-a]p¡rid¡na-6-carbonitrilo (157)
Paso A: A una solución de 6-bromo-[1,2,4]triazolo[4,3-a]pir¡d¡na-3-carbox¡lato de etilo (72 mg, 0,27 mmol) en THF (2,2 mL) se le añadió una solución de monohidrato de LiOH (12 mg, 0,29 mmol) en H2O (1,5 mL). La mezcla se agitó durante 20 min y se neutralizó con HCl 2 N. La mezcla se concentró a presión reducida. El residuo obtenido se diluyó en DMF (2,8 mL) en atmósfera de N2. A esta mezcla se le añadió clorhidrato de 4-(2-fluoro-6-(trifluoromet¡l)fen¡l)piper¡d¡na (23, 75 mg, 0,27 mmol), hexafluorofosfato de benzotriazol-1-¡l-ox¡-tr¡s-(d¡met¡lam¡no)-fosfonio (236 mg , 0,53 mmol) y diisopropiletilamina (0,14 mL, 0,80 mmol). La mezcla se agitó a temperatura ambiente durante 18 h. La mezcla resultante se diluyó con H2O (20 mL) y se extrajo con EtOAc (3 x 30 mL). Los extractos orgánicos combinados se lavaron con una solución de salmuera saturada (30 mL) y se concentraron a presión reducida. El residuo obtenido se cromatografió sobre gel de sílice (unidad Isco CombiFlash Rf, columna Redisep de 12 g, 0 a 50 % de EtOAc en hexanos) para proporcionar (6 -bromo-[1,2,4]tr¡azolo[4,3-a]p¡r¡dina-3-¡l)(4-(2-fluoro-6-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡n-1-¡l)metanona como una película de color naranja claro (80 mg, 64%): 1H NMR (300 MHz, DMSO-cfe) 8 9,12-9,10 (m, 1H), 8,00-7,95 (m, 1H), 7,74-7,68 (m, 1H), 7,65-7,50 (m, 3H), 5,29-5,15 (m, 1H), 4,82-4,68 (m, 1H), 3,41-3,19 (m, 2H, se superpone con H2O), 3,07-2,97 (m, 1H), 2,34-2,19 (m, 1H), 2,15-2,02 (m, 1H), 1,93-1,75 (m, 2H); ESI MS m /z 472 [M H]+.
Paso B: Una solución de (6-bromo-[1,2,4]triazolo[4,3-a]piridin-3-il)(4-(2-fluoro-6-(trifluorometil)fenil)piperidin-1-il)metanona (80 mg, 0,17 mmol) en DMF (2,0 mL) con cianuro de zinc (40 mg, 0,34 mmol) se burbujeó con Ar durante 15 min. A la solución se le añadió Pd(PPh3)4 (19 mg, 0,017 mmol) y el recipiente se selló y calentó a 130°C con microondas durante 30 min. La mezcla se diluyó con solución saturada de bicarbonato de sodio (20 mL) y se extrajo con EtOAc (3 x 30 mL). Los extractos orgánicos combinados se lavaron con una solución de salmuera saturada (2 x 30 mL) y se concentraron a sequedad bajo presión reducida. El residuo resultante se cromatografió sobre gel de sílice (unidad Isco CombiFlash Rf, columna Redisep de 12 g, 0% a 50% de EtOAc en hexanos) seguido de cromatografía líquida de alta resolución (“HPLC”, por sus siglas en inglés) (Phenomenex Luna C18 (2), 250,0 x 50,0 mm, 15 micras, H2O con 0,05% de TFA y CH3CN con 0,05% de TFA), se lavó con solución saturada de bicarbonato de sodio y se liofilizó para proporcionar 3-(4-(2-fluoro-6-(trifluorometil)fenil)piperidina-1-carbonil)-[1,2,4]triazolo[4,3-a]piridina-6-carbonitrilo como un sólido blanco (23 mg, 33%): 1 H NMR (500 MHz, DMSO-da) 89,54-9,52 (m, 1H), 8,14-8,11 (m, 1H), 7,82-7,78 (m, 1H), 7,64-7,59 (m, 1H), 7,57-7,50 (m, 2H), 5,17-5,10 (m, 1H), 4,79-4,72 (m, 1H), 3,40-3,24 (m, 2H, se superpone con H2O), 3,07-2,98 127 (m, 1H), 2,30-2,19 (m, 1H), 2,14-2,03 (m, 1H), 1,91-1,79 (m, 2H); ESI MS m/z 418 [M H]+.
Ejemplo 96: Preparación de 1-(3-(4-(4-fluoro-2-(trifluorometil)fenil)piperidina-1-carbonil)-4,5-dihidro-1H-pirazolo[3,4-c]piridin-6(7H) )-il)etanona (158)
Paso A: Una solución de 4-(((trifluorometil)sulfonil)oxi)-5,6-dihidropiridina-1(2H)-carboxilato de ferc-butilo (1,50 g, 4,53 mmol) en 1,2-dimetoxietano (25 mL) se burbujeó con N2 durante 30 min. Se añadió ácido 4-fluoro-(2-trifluorometil)fenil borónico (1,13 g, 5,43 mmol) seguido de una solución 2 M de carbonato de sodio (2,9 mL). La mezcla resultante se burbujeó con N2 durante 10 min. Se añadió Pd(PPh3)4 (260 mg, 0,225 mmol) y la mezcla resultante se calentó a 80°C bajo una atmósfera de N2. Después de 72 h, la solución resultante se enfrió a temperatura ambiente y se diluyó con una solución de cloruro de litio al 5% (100 mL). La solución se extrajo con EtOAc (3 x 50 mL). Los extractos orgánicos combinados se lavaron con salmuera saturada (2 x 50 mL) y se concentraron a sequedad bajo presión reducida. El residuo se cromatografió sobre gel de sílice (unidad Isco CombiFlash Rf, columna Redisep de 24 g, 0% a 100% de EtOAc en hexanos) para proporcionar 4-(4-fluoro-2-(trifluorometil)fenil)-5,6-dihidropiridina-1(2H)-carboxilato de ferc-butilo como un aceite marrón (1,29 g, 83%): 1 H NMR (300 MHz, CDCh) 87,37-7,08 (m, 3H), 5,57 (br s, 1H), 4,02-3,99 (m, 2H), 3,62-3,58 (m, 2H), 2,32 (br s, 2H), 1,46 (s, 9H).
Paso B: Una solución de 4-(4-fluoro-2-(trifluorometil)fenil)-5,6-dihidropiridina-1(2H)-carboxilato de ferc-butilo (1,29 g, 3,74 mmol) en acetato de etilo (20 mL) y ácido acético (0,22 mL) se burbujeó con N2. Se añadió dióxido de platino (84 mg) y la suspensión resultante se burbujeó con N2 durante 5 min. La mezcla se colocó bajo una atmósfera de H2 a 1 atm. Después de 18 h, la reacción se burbujeó con N2 durante 15 min, se filtró a través de una capa de tierra de diatomeas, se recargó con dióxido de platino (100 mg) y se agitó bajo una atmósfera de hidrógeno de 1 atm. La filtración y recarga de la reacción se repitió tres veces durante las siguientes 64 h. La reacción se filtró a través de tierra de diatomeas. El filtrado obtenido se lavó con solución de bicarbonato de sodio, se secó (Na2SO4) y se concentró a presión reducida. El residuo se cromatografió sobre gel de sílice (unidad Isco CombiFlash Rf, columna Redisep de 24 g, 0% a 100% de EtOAc en hexanos) para proporcionar 4-(4-fluoro-2-(trifluorometil)fenil)piperidina-1-carboxilato de ferc-butilo como un aceite amarillo (0,813 g, 63%): 1 H NMR (300 MHz, CDCh) 87,37-7,20 (m, 3H), 4,27-4,22 (m, 2H), 3,07-2,99 (m, 1H), 2,85-2,75 (m, 2H), 2,04-1,46 (m, 13H).
Paso C: A una solución de 4-(4-fluoro-2-(trifluorometil)fenil)piperidina-1-carboxilato de ferc-butilo (0,813 g, 2,34 mmol) en éter dietílico (6 mL) se le añadió HCl 2 M en éter dietílico (10 mL). La mezcla se agitó durante 18 h a temperatura ambiente. La mezcla de reacción se concentró a presión reducida y el residuo se trituró con éter dietílico (10 mL). Los sólidos se recolectaron por filtración para proporcionar clorhidrato de 4-(4-fluoro-2-(trifluorometil)fenilpiperidina como un sólido blanco (0,244 g, 37%): 1 H NMR (300 MHz, CDCh) 89,79 (br s, 1H), 7,59-7,54 (m, 1H), 7,37-7,24 (m, 2H), 3,68-3,64 (m, 2H), 3,27-3,03 (m, 4H), 2,39-2,27 (m, 2H), 2,01-1,96 (m, 2H); ESI MS m/z 248 [M H]+.
Paso D: A una solución de clorhidrato de 4-(4-fluoro-2-trifluorometil)fenilpiperidina (75 mg, 0,26 mmol), ácido 6-(tercbutoxicarbonil)-4,5,6,7-tetrahidro-1H-pirazolo[3,4-c]piridina-3-carboxílico (75 mg, 0,29 mmol), y diisopropiletilamina (0,14 mL, 0,80 mmol) en DMF (3 mL) bajo una atmósfera de N2 se le añadió EDC (70 mg, 0,37 mmol) y HOBt (49 mg, 0,36 mmol). La solución resultante se agitó a temperatura ambiente durante 24 h. La mezcla de reacción se diluyó con H2O (30 mL) y se extrajo con EtOAc (3 x 10 mL). Los extractos orgánicos combinados se lavaron con salmuera (1 x 20 mL) y se concentraron a sequedad bajo presión reducida. El residuo obtenido se cromatografió sobre gel de sílice (unidad Isco CombiFlash Rf, columna Redisep de 12 g, 0% a 100% de acetato de etilo en hexanos) para proporcionar 3-(4-(4-fluoro-2-(trifluorometil)fenil)piperidina-1-carbonil)-4,5-dihidro-1H-pirazolo[3,4-c]piridina-6(7H)-carboxilato de ferc-butilo como un sólido blanco (75 mg, 58%): 1 H NMR (300 MHz, DMSO-cfe) 812,04 (br s, 1H), 7,72-7,68 (m, 1H), 7,57-7,49 (m, 2H), 4,84-4,64 (m, 1H), 4,49-4,45 (m, 2H), 3,56-3,53 (m, 2H), 3,08 (br s, 2H), 2,78-2,50 (m, 4H), 1,75 (br s, 4H), 1,42 (s, 9H); ESI MS m/z 497 [M H]+.
Paso E: A una solución de 3-(4-(4-fluoro-2-(trifluorometil)fenil)piperidina-1-carbonil)-4,5-dihidro-1H-pirazolo[3,4-c]piridina-6(7H)-carboxilato de ferc-butilo (74 mg, 0,15 mmol) en CH2Cl2 (3 mL) y metanol (1 mL) se le añadió HCl 2 N (2 mL, 2 M en Et2O). La mezcla se agitó durante 4 h a temperatura ambiente. La mezcla de reacción se diluyó con Et2O (30 mL) y los sólidos resultantes se recolectaron por filtración para proporcionar clorhidrato de (4-(4-fluoro-2-(trifluorometil)fenil)piperidin-1-il)(4,5,6,7-tetrahidro-1H-pirazolo[3,4-c]piridin-3-il)metanona como un sólido blanco (64 mg, 98%): 1 H NMR (300 MHz, DMSO-cfe) 813,14 (s, 1H), 9,16 (br s, 2H), 7,73-7,68 (m, 1H), 7,59-7,50 (m, 2H), 4,84-4,69 (m,
1H), 4,32-4,25 (m, 2H), 3,26-3,03 (m, 2H), 2,89-2,81 (m, 2H), 2,68-2,48 (m, 4H), 1,73 (m, 4H); ESI MS m/z 397 [M H]+.
Paso F: A una solución de clorhidrato de (4-(4-fluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡n-1-¡l)(4,5,6,7-tetrah¡dro-1H-p¡razolo[3,4-c]piridin-3-il)metanona (63 mg, 0,15 mmol) y d¡¡soprop¡let¡lam¡na (70 mL, 0,40 mmol) en DMF (3,0 mL) se le añad¡ó cloruro de acet¡lo (11 mL, 0,15 mmol). La mezcla se ag¡tó durante 16 horas. El d¡solvente se el¡m¡nó a pres¡ón reduc¡da y el res¡duo se d¡luyó con H2O (10 mL) y se extrajo con EtOAc (2 x 10 mL). Los extractos orgán¡cos comb¡nados se lavaron con salmuera saturada (1 x 20 mL), se secaron sobre Na2SO4 y se concentraron a sequedad bajo pres¡ón reduc¡da. El res¡duo resultante se cromatograf¡ó sobre gel de síl¡ce (un¡dad Isco Comb¡Flash Rf, columna Red¡sep de 12 g, 0% a 100% (10% de CH3OH en CH2Ch con 0,01% NH4OH) en CH2Cl2) y se pur¡f¡có ad¡c¡onalmente por cromatografía de fase ¡nversa (un¡dad Isco Comb¡Flash Rf, columna de oro Red¡sep c18 de 12 g, 0% a 100% de aceton¡tr¡lo en agua) para proporc¡onar 1-(3-(4-(4-fluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡na-1-carbon¡l)-4,5-d¡h¡dro-1H-p¡razolo[3,4-c]p¡r¡d¡n-6(7H)-¡l)etanona como un sól¡do blanco (20 mg, 94%): mp 176-180°C; 1 H NMR (500 MHz, DMSO-d6) 812,86 (s, 1H), 7,71-7,68 (m, 1H), 7,56-7,49 (m, 2H), 4,94-4,55 (m, 3H), 3,66-3,62 (m, 2H), 3,10 (br s, 2H), 2,85-2,48 (m, 4H), 2,10-2,08 (m, 3H), 1,73 (m, 4H); ESI MS m/z 439 [M H]+.
Ejemplo 97: Preparac¡ón de (4-(4-fluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡n-1-¡l)(5-(met¡lsulfon¡l)-4,5,6,7-tetrah¡dro-1H-p¡razolo[4,3-c]p¡r¡d¡n-3-¡l)metanona (159)
Paso A: A una soluc¡ón de clorh¡drato de 4-(4-fluoro-2-tr¡fluoromet¡l)fen¡lp¡per¡d¡na (75 mg, 0,26 mmol), ác¡do 5-(tercbutox¡carbon¡l)-4,5,6,7-tetrah¡dro-1H-p¡razolo[4,3-c]p¡r¡d¡na-3-carboxíl¡co (75 mg, 0,29 mmol), y d¡¡soprop¡let¡lam¡na (0,14 mL, 0,80 mmol) en DMF (3,0 mL) bajo una atmósfera de N2 se le añad¡ó EDCI (70 mg, 0,37 mmol) y HOBt (49 mg, 0,36 mmol). La soluc¡ón resultante se ag¡tó a temperatura amb¡ente durante 16 h. La mezcla de reacc¡ón se d¡luyó con H2O (30 mL) y se extrajo con EtOAc (3 x 30 mL). Los extractos orgán¡cos comb¡nados se lavaron con salmuera (1 x 30 mL) y se concentraron a sequedad bajo pres¡ón reduc¡da. El res¡duo obten¡do se cromatograf¡ó sobre gel de síl¡ce (un¡dad Isco Comb¡Flash Rf, columna Red¡sep de 12 g, 0% a 100% de acetato de et¡lo en hexanos) para proporc¡onar 3-(4-(4-fluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡na-1-carbon¡l)-6,7-d¡h¡dro-1H-p¡razolo[4,3-c]p¡r¡d¡na-5(4H)-carbox¡lato de ferc-but¡lo como un sól¡do blanco (109 mg, 77%): 1 H NMR (300 MHz, DMSO-de) 8 12,96 (s, 1H), 7,70-7,68 (m, 1H), 7,57-7,49 (m, 2H), 5,32-5,13 (m, 1H), 4,74-4,64 (m, 1H), 4,47-4,45 (m, 2H), 3,59 (br s, 2H), 3,22-3,10 (m, 2H), 2,82-2,50 (m, 3H), 1,73 (br s, 4H), 1,42 (s, 9H); ESI MS m/z 497 [M H]+.
Paso B: A una soluc¡ón de 3-(4-(4-fluoro-2-(tr¡fluoromet¡lfen¡l)p¡per¡d¡na-1-carbon¡l)-4,5-d¡h¡dro-1H-p¡razolo[3,4-c]-p¡r¡d¡na-5(4H)-carbox¡lato de ferc-but¡lo (85 mg, 0,17 mmol) en C ^ C h (3 mL) y metanol (1 mL) se le añad¡ó HCl 2 N (2 mL, 2 M en Et2O). La mezcla se ag¡tó durante 4 h a temperatura amb¡ente. La mezcla de reacc¡ón se d¡luyó con Et2O (30 mL) y los sól¡dos resultantes se recolectaron por f¡ltrac¡ón para proporc¡onar clorh¡drato de (4-(4-fluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡n-1-¡l) (4,5,6,7-tetrah¡dro-1H-p¡razolo[4,3-c]p¡r¡d¡n-3-¡l)metanona como un sól¡do blanquec¡no (78 mg, >99%): 1 H NMR (300 MHz, DMSO-de) 8 13,14 (br s, 1H), 9,13 (br s, 2H), 7,73-7,67 (m, 1H), 7,58-7,50 (m, 2H), 5,32-4,69 (m, 3H), 4,24 (s, 2H), 3,38 (br s, 2H), 3,21 3,07 (m, 2H), 2,96-2,72 (m, 3H), 1,75 (m, 4H); ESI MS m/z 397 [M H]+.
Paso C: A una soluc¡ón de clorh¡drato de (4-(4-fluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡n-1-¡l) (4,5,6,7-tetrah¡dro-1H-p¡razolo[4,3-c]p¡r¡d¡n-3-¡l)metanona (38 mg, 0,088 mmol) y d¡¡soprop¡let¡lam¡na (35 mL, 0,20 mmol) en DMF (2,0 mL) se le añad¡ó cloruro de metanosulfon¡lo (9 mL, 0,12 mmol). La mezcla se ag¡tó durante 16 h a temperatura amb¡ente. El d¡solvente se el¡m¡nó a pres¡ón reduc¡da y el res¡duo se d¡luyó con H2O (10 mL) y se extrajo con acetato de et¡lo (3 x 10 mL). Los extractos orgán¡cos comb¡nados se secaron (Na2SO4) y se concentraron a pres¡ón reduc¡da. Los sól¡dos obten¡dos se cromatograf¡aron sobre gel de síl¡ce (un¡dad Isco Comb¡Flash Rf, columna Red¡sep de 12 g, 0% a 100% (10% de CH3OH en CH2Ch con 0,01% de NH4OH) en CH2Ch) y se l¡of¡l¡zaron para proporc¡onar (4-(4-fluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡n-1-¡l) (5-(met¡lsulfon¡l)-4,5,6,7-tetrah¡dro-1H-p¡razolo[4,3-c]p¡r¡d¡n-3-¡l)metanona como un sól¡do blanco (4 mg, 10%): 1 H NMR (300 MHz, DMSO-cfe) 813,05 (s, 1H), 7,73-7,69 (m, 1H), 7,57-7,46 (m, 2H), 5,28-5,24 (m, 1H), 4,70-4,65 (m, 1H), 4,38-4,33 (m, 2H), 3,51-3,45 (m, 2H), 3,22-3,10 (m, 2H), 2,94 (s, 3H), 2,84-2,70 (m, 3H), 1,73 (br s, 4H); ESI MS m/z 475 [M H]+.
Ejemplo 98: Preparac¡ón de 3-(4-(4-fluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡na-1-carbon¡l)-[1,2,4]tr¡azolo[4,3-a]p¡r¡d¡na-6-carbon¡tr¡lo (160)
Paso A: A una soluc¡ón de 6-bromo-[1,2,4]tr¡azolo[4,3-a]p¡r¡d¡na-3-carbox¡lato de et¡lo (81 mg, 0,30 mmol) en THF (2,5 mL) se le añad¡ó una soluc¡ón de monoh¡drato de L¡OH (14 mg, 0,30 mmol) en H2O (1,7 mL). La mezcla se ag¡tó durante 20 m¡n y se neutral¡zó con HCl 2 N. La mezcla se concentró a pres¡ón reduc¡da. El res¡duo obten¡do se d¡luyó en DMF (3,2 mL) en atmósfera de N2. A esta mezcla se le añad¡ó clorh¡drato de 4-(4-fluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡na (85 mg, 0,30 mmol), hexafluorofosfato de benzotr¡azol-1-¡l-ox¡-tr¡s-(d¡met¡lam¡no)-fosfon¡o (267 mg , 0,60 mmol) y d¡¡soprop¡let¡lam¡na (0,15 mL, 0,91 mmol). La mezcla se ag¡tó a temperatura amb¡ente durante 18 h. La mezcla resultante se d¡luyó con H2O (20 mL) y el prec¡p¡tado resultante se recolectó med¡ante f¡ltrac¡ón. Los sól¡dos obten¡dos se cromatograf¡aron sobre gel de síl¡ce (un¡dad Isco Comb¡Flash Rf, columna Red¡sep de 12 g, 0 a 50% de EtOAc en hexanos) para proporc¡onar (6-bromo-[1,2,4]tr¡azolo[4,3-a]p¡r¡d¡n-3-¡l) (4-(4-fluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡n-1-¡l)metanona como una película naranja (92 mg, 64%): 1 H NMR (300 MHz, DMSO-cfe) 89,13 (dd, J = 1,7, 0,9 Hz 1H), 7,98 (dd, J = 10, 0,9 Hz, 1H), 7,77-7,69 (m, 2H), 7,60-7,47 (m, 2H), 5,30-5,18 (m, 1H), 4,77-4,68 (m, 1H), 3,43-3,34 (m, 1H), 3,28-3,12 (m, 1H), 3,11-2,90 (m, 1H), 1,97-1,69 (m, 4H); ESI MS m/z 472 [M H]+.
Paso B: Una solución de (6-bromo-[1,2,4]triazolo[4,3-a]piridin-3-il)(4-(4-fluoro-2-(trifluorometil)fenil)piperidin-1-il)metanona (90 mg, 0,19 mmol) en DMF (2,2 mL) y cianuro de zinc (45 mg, 0,38 mmol) se burbujeó con Ar durante 15 min. A la solución se le añadió Pd(PPh3)4 (22 mg, 0,019 mmol) y el recipiente se selló y calentó a 130°C con microondas durante 30 min. La mezcla se diluyó con solución saturada de bicarbonato de sodio (10 mL) y se extrajo con EtOAc (3 x 20 mL). Los extractos orgánicos combinados se lavaron con solución de salmuera saturada (30 mL) y se concentraron a sequedad bajo presión reducida. El residuo resultante se cromatografió sobre gel de sílice (unidad Isco CombiFlash Rf, columna Redisep de 12 g, 0 % a 50 % de EtOAc en hexanos) seguido de HPLC (Phenomenex Luna C18 (2), 250,0 x 50,0 mm, 15 micras, H2O con 0,05% TFA y CH3CN con 0,05% TFA) y se lavó con solución saturada de bicarbonato de sodio (3 x 30 mL), luego se liofilizó para proporcionar 3-(4-(4-fluoro-2-(trifluorometil)fenil)piperidina-1-carbonil)-[1,2,4] triazolo[4,3-a]piridina-6 -carbonitrilo como un sólido blanco (32 mg, 40%): mp 158-162°C; 1H NMR (300 MHz, DMsO-cfe) 8 9,53 (s, 1 H), 8,14 (dd, J = 9,0, 0,9 Hz, 1H), 7,82 (dd, J = 9,6, 1,5 Hz, 1H), 7,77-7,67 (m, 1H), 7,61-7,47 (m, 2H), 5,19-5,04 (m, 1H), 4,80 4,67 (m, 1H), 3,46-3,14 (m, 2H, se superpone con H2O), 3,12-2,94 (m, 1H), 2,02-1,70 (m, 4H); ESI MS m/z 418 [M H]+.
Ejemplo 99: Preparación de 3-(3-(4-(5-cloro-2-(trifluorometil)fenil)piperidina-1-carbonil)-[1,2,4]triazolo[4,3-a]piridina-6-carbonitrilo (161)
Paso A: Una mezcla de ácido (5-cloro-2-(trifluorometil)fenil)borónico (0,453 g, 2,02 mmol), 4-(((trifluorometil)sulfonil)oxi)-5.6- dihidropiridina-1(2H)-carboxilato de ferc-butilo (0,669 g, 2,02 mmol), tetrakis(trifenilfosfina)paladio (0,117 g, 0,1 mmol), carbonato de sodio ( 2 M, 5 mL), y 1,2-dimetoxietano (10 mL) se calentó a 80°C bajo irradiación de microondas durante 1,5 h. Después de enfriar a temperatura ambiente, la mezcla se diluyó con agua (80 mL) y se extrajo con acetato de etilo (80 mL). El extracto se lavó con salmuera (2 x 50 mL), se secó sobre Na2SO4, se filtró y se concentró a presión reducida. El residuo resultante se cromatografió sobre gel de sílice (EtOAc al 0 -25% en hexanos) para proporcionar 4-(5-cloro-2-(trifluorometil)fenil)-5,6-dihidropiridina-1(2H)-carboxilato de ferc-butilo como aceite incoloro (0,614 g, 84%): 1H NMR (300 MHz, CDCla) 8 7,59 (d, J = 8,5 Hz, 1H), 7,37-7,22 (m, 1H), 7,22 (d, J = 1,68 Hz, 1H), 5,60 (br. 1H), 4,02 (br, 2H), 3,61 (br, 2H), 2,34 (br, 2H), 1,50 (s, 9H); MS (ESI+) m /z 306 [M H]+.
Paso B: Una mezcla de 4-(5-cloro-2-(trifluorometil)fenil)-5,6-dihidropiridina-1(2H)-carboxilato de terc-butilo (0,614 g, 1,70 mmol), óxido de platino (0,200 g, 0,881 mmol), ácido acético (1 mL) y acetato de etilo (15 mL) se hidrogenaron usando un globo de H2 durante 16 h y se filtraron. Después de la concentración, el residuo se cromatografió sobre gel de sílice (0-30 % de EtOAc en hexanos) para proporcionar 4-(5-cloro-2-(trifluorometil)fenil)piperidina-1-carboxilato de ferc-butilo como un aceite espeso incoloro (0,302 g, 48%): 1H NMR (300 MHz, CDCla) 8 7,56 (d, J = 8,5 Hz, 1H), 7,38 (s, 1H), 7,29 (d, J = 1.3 Hz, 1H), 4,26 (br, 2H), 3,04 (m, 1H), 2,80 (m, 2H), 1,80-1,55 (m, 4H), 1,49 (s, 9H); MS (ESI+) m /z 308 [M H]+.
Paso C: A una solución de 4-(5-cloro-2-(trifluorometil)fenil)piperidina-1-carboxilato de ferc-butilo (0,302 g, 0,830 mmol) en diclorometano (5 mL) se le añadió solución de HCl (2 M en éter, 5 mL). La mezcla se agitó durante 4 h y se evaporó para proporcionar un sólido que se disolvió en DMF (8 mL). En un matraz aparte, a una solución de 6-bromo-[1,2,4]triazolo[4,3-a]piridina-3-carboxilato de etilo (0,224 g, 0,830 mmol) en THF (5 mL) se le añadió una solución de hidróxido de litio hidratado (0,035 g, 0,830 mmol) en agua (2 mL). La mezcla se agitó durante 20 min, se acidificó con HCl 2 N a pH 6 y se evaporó hasta sequedad. A este residuo se le añadieron hexafluorofosfato de benzotriazol-1-iloxitris(dimetilamino)fosfonio (0,550 g, 1,25 mmol), W,W-diisopropiletilamina (0,646 g, 5,00 mmol) y la solución de DMF obtenida de la primera reacción. La mezcla se agitó a temperatura ambiente durante 16 horas y se vertió en agua. La mezcla se extrajo con acetato de etilo y la capa orgánica se lavó tres veces con salmuera, se secó sobre Na2SO4, se filtró y se concentró a presión reducida. El residuo resultante se cromatografió sobre gel de sílice (EtOAc al 0-60% en hexanos) para proporcionar (6-bromo-[1,2,4]triazolo[4,3-a]piridin-3-il) (4-(5-cloro-2-(trifluorometil)fenil)piperidin-1-il)metanona como un sólido (0,205 g, 50%): 1H NMR (300 MHz, CDCh) 8 9,38 (m, 1H), 7,79 (dd, J = 9,6, 0,9 Hz, 1H), 7,60 (d, J = 8,5 Hz, 1H), 7,50 (dd, J = 9,6, 1,7 Hz, 1H), 7,42 (d, J = 1,4 Hz, 1H), 7,31 (dd, J = 8,5, 1,3 Hz, 1H), 5,76-5,71 (m, 1H), 5,01-4,95 (m, 1H), 3,38-3,26 (m, 2H), 3,02-2,92 (m, 1H), 2,01-1,82 (m, 4H); MS (ESI+) m /z 489 [M H]+.
Paso D: Una mezcla de (6-bromo-[1,2,4]triazolo[4,3-a]piridin-3-il)(4-(5-cloro-2-(trifluorometil)fenil)piperidin-1-il)metanona (0,205 g, 0,42 mmol), cianuro de zinc (0,099 g, 0,840 mmol), tetrakis(trifenilfosfina)paladio (0,048 g, 0,042 mmol) y DMF (4 mL) se calentó con radiación de microondas a 130°C durante 30 minutos. Después de enfriar a temperatura ambiente, la mezcla se diluyó con agua (80 mL) y se extrajo con acetato de etilo (80 mL). El extracto se lavó con salmuera ( 2 x 80 mL), se secó (Na2SO4), se filtró y se concentró a presión reducida. El residuo resultante se cromatografió sobre gel de sílice (EtOAc al 0-60% en hexanos) para proporcionar 3-(4-(5-cloro-2-(trifluorometil)fenil)piperidina-1 -carbonil)-[1,2,4]triazolo[4,3-a]piridina-6-carbonitrilo como un sólido blanco (0,115 g, 63%): 1H NMR (300 MHz, CDCh) 8 9,72 (s, 1H), 7,98 (dd, J = 9,5, 1,1 Hz, 1H), 7,60 (d, J = 8,5 Hz, 1H), 7,51 (dd, J = 9,5, 1,6 Hz, 1H), 7,41 (s, 1H), 7,31 (dd, J = 8,5, 1.3 Hz, 1H), 5,77-5,72 (m, 1H), 5,02-4,96 (m, 1H), 3,40 (m, 2H), 3,04-2,94 (m, 1H), 2,06-1,80 (m, 4H); MS (ESI+) m /z 434 [M H]+.
Ejemplo 100: Preparación de 3-(4-(3-cloro-2-(trifluorometil)fenil)piperidina-1-carbonil)-[1,2,4]triazolo[4,3-a]piridina-6-carbonitrilo (162)
Paso A: Una mezcla de ácido (3-cloro-2-(trifluorometil)fenil)borónico (0,453 g, 2,02 mmol), 4-(((trifluorometil)sulfonil)oxi)-5.6- dihidropiridina-1(2H)-carboxilato de terc-butilo (0,669 g, 2,02 mmol), tetrakis(trifenilfosfina)paladio (0,117 g, 0,1 mmol), carbonato de sodio (2 M, 5 mL), y 1,2-dimetoxietano (10 mL) se calentó a 80°C bajo irradiación de microondas durante 1
h. Después de enfriar a temperatura ambiente, la mezcla se diluyó con agua (80 mL) y se extrajo con acetato de etilo (80 mL). El extracto se lavó con salmuera (2 x 50 mL), se secó (Na2SO4), se filtró y se concentró a presión reducida. El residuo resultante se cromatografió sobre gel de sílice (EtOAc al 0-30% en hexanos) para proporcionar 4-(3-cloro-2-(trifluorometil)fenil)-5,6-dihidropiridin-1(2H)-carboxilato de ferc-butilo como aceite incoloro (0,438 g, 60%): 1H NMR (300 MHz, CDCla) 8 7,44-7,34 (m, 2H), 7,09 (m, 1H), 5,49 (br. 1H), 4,01 (br, 2H), 3,60 (br, 2H), 2,30 (br, 2H), 1,50 (s, 9H); MS (ESI+) m /z 306 [M H]+.
Paso B: Una mezcla de 4-(3-cloro-2-(trifluorometil)fenil)-5,6-dihidropiridin-1(2H)-carboxilato de ferc-butilo (0,438 g, 1,21 mmol), óxido de platino (0,082 g, 0,363 mmol), ácido acético (0,073 g, 1,21 mmol) y acetato de etilo (20 mL) se hidrogenaron usando un globo durante 20 h y se filtraron. El material se volvió a someter a hidrogenación a 80°C durante 16 h y se filtró. Después de la concentración, el residuo se cromatografió sobre gel de sílice (0-30% de EtOAc en hexanos) para proporcionar 4-(3-cloro-2-(trifluorometil)fenil)piperidina-1-carboxilato de ferc-butilo como un aceite espeso incoloro (0,115 g, 26%): 1H NMR (300 MHz, CDCla) 8 7,42-7,30 (m, 3H), 4,25 (br, 2H), 3,21 (m, 1H), 2,81 (m, 2H), 1,80-1,60 (m, 4H), 1,49 (s, 9H); MS (ESI+) m /z 308 [M H]+.
Paso C: A una solución de 4-(3-cloro-2-(trifluorometil)fenil)piperidina-1-carboxilato de ferc-butilo (0,115 g, 0,316 mmol) en diclorometano (3 mL) se le añadió HCl (2 M en éter, 3 mL). La mezcla se agitó durante 3 h y se evaporó para proporcionar un sólido que se disolvió en DMF (3 mL). En un matraz aparte, a una solución de 6-bromo-[1,2,4]triazolo[4,3-a]piridina-3-carboxilato de etilo (0,094 g, 0,348 mmol) en THF (3 mL) se le añadió una solución de hidróxido de litio hidratado (0,015 g, 0,348 mmol) en agua (1 mL). La mezcla se agitó durante 20 min, se acidificó con HCl 2 N a pH 6 y se evaporó. Al residuo se le añadieron hexafluorofosfato de benzotriazol-1-il-oxi-tris(dimetilamino)fosfonio (0,210 g, 0,474 mmol), N ,N- diisopropiletilamina (0,163 g, 1,26 mmol) y la solución de DMF obtenida de la primera reacción. La mezcla se agitó a temperatura ambiente durante 16 horas y se vertió en agua. La mezcla se extrajo con acetato de etilo y la capa orgánica se lavó tres veces con salmuera, se secó (Na2SO4), se filtró y se concentró a presión reducida. El residuo resultante se cromatografió sobre gel de sílice (EtOAc al 0-60% en hexanos) para proporcionar (6-bromo-[1,2,4]triazolo[4,3-a]piridin-3-il) (4-(3-cloro-2-(trifluorometil)fenil)piperidin-1-il)metanona como un sólido (0,076 g, 49%): 1H NMR (300 MHz, CDCh) 8 9,36 (m, 1H), 7,78 (dd, J = 9,6, 0,9 Hz, 1H), 7,50-7,34 (m, 4H), 5,73-5,68 (m, 1H), 5,00-4,94 (m, 1H), 3,52-3,28 (m, 2H), 2,97 (m, 1H), 2,01-1,74 (m, 4H); MS (ESI+) m /z 489 [M H]+.
Paso D: Una mezcla de (6-bromo-[1,2,4]triazolo[4,3-a]piridin-3-il)(4-(3-cloro-2-(trifluorometil)fenil)piperidin-1-il)metanona (0,076 g, 0,156 mmol), cianuro de zinc (0,037 g, 0,312 mmol), paladio tetrakis(trifenilfosfina) (0,018 g, 0,0156 mmol) y DMF (2 mL) se calentó con radiación de microondas a 130°C durante 30 min. Después de enfriar a temperatura ambiente, la mezcla se diluyó con agua (50 mL) y se extrajo con acetato de etilo (50 mL). El extracto se lavó con salmuera (2 x 50 mL), se secó sobre Na2SO4, se filtró y se concentró a presión reducida. El residuo resultante se cromatografió sobre gel de sílice (EtOAc al 0-60% en hexanos) para proporcionar 3-(4-(3-cloro-2-(trifluorometil)fenil)piperidina-1 -carbonil)-[1,2,4]triazolo[4,3-a]piridina-6-carbonitrilo como un sólido blanco (0,026 g, 38%): 1H NMR (300 MHz, CDCh) 8 9,70 (s, 1H), 7,97 (dd, J = 9,5, 1,0 Hz, 1H), 7,52-7,32 (m, 4H), 5,74-5,69 (m, 1H), 5,00-4,95 (m, 1H), 3,52-3,31 (m, 2H), 2,99 (m, 1H), 2,06-1,75 (m, 4H); MS (ESI+) m /z 434 [M H]+.
Ejemplo 101: Preparación de (4-(2-cloro-3-fluorofenil)piperidin-1-il)(5-etil-4,5,6,7-tetrahidro-1H-pirazolo[4,3-c]piridin-3-il)metanona (163)
Paso A: Siguiendo el procedimiento general GP-D1, clorhidrato de (4-(2-cloro-3-fluorofenil)piperidin-1-il) (4,5,6,7-tetrahidro-1H-pirazolo[4,3-c]piridin-3-il)metanona (42) y formaldehído se convirtieron en (4-(2-cloro-3-fluorofenil)piperidin-1-il)(5-etil-4,5,6,7-tetrahidro-1H-pirazolo[4,3-c]piridin-3-il)metanona como un sólido blanco (25 mg, 32%): 1H Nm R (300 MHz, DMSO-cfe) 8 12,93 (m, 1H), 7,56-7,32 (m, 3H), 5,16 (m, 1H), 4,81 (m, 1H), 2,61-3,42 (m, 9H), 1,94-1,61 (m, 4H); ESI MS m /z 391 [M H]+.
Ejemplo 102: Preparación de (4-(2-cloro-3-fluorofenil)piperidin-1-il)(5-(ciclopropilmetil)-4,5,6,7-tetrahidro-1H-pirazolo[4,3-c]piridin-3-il)metanona (164)
Paso A: Siguiendo el procedimiento general GP-D1, clorhidrato de (4-(2-cloro-3-fluorofenil)piperidin-1-il) (4,5,6,7-tetrahidro-1H-pirazolo[4,3-c]piridin-3-il)metanona (42) y ciclopropil-carboxaldehído se convirtieron en (4-(2-cloro-3-fluorofenil)piperidin-1-il) (5-(ciclopropilmetil)-4,5,6,7-tetrahidro-1H-pirazolo[4,3-c]piridin-3-il)metanona como un sólido blanco (41 mg, 65%): 1H NMR (300 MHz, DMSO-cfe) 8 12,91 (m, 1H), 7,56-7,39 (m, 3H), 5,15 (m, 1H), 4,85 (m, 1H), 3,53 (m, 2H), 3,12 (m, 1H), 2,62-2,35 (m, 5H), 2,43 (m, 2H), 1,87 (m, 2H), 1,63 (m, 2H), 0,98 (m, 1H), 0,55 (m, 2H), 0,021 (m, 2H); ESI MS m /z 417 [M H]+.
Ejemplo 103: Preparación de (4-(2-cloro-3-fluorofenil)piperidin-1-il)(5-(oxetan-3-il)-4,5,6,7-tetrahidro-1H-pirazolo[4,3-c]piridin-3-il)metanona (165)
Paso A: Siguiendo el procedimiento general GP-D1, clorhidrato de (4-(2-cloro-3-fluorofenil)piperidin-1-il) (4,5,6,7-tetrahidro-1H-pirazolo[4,3-c]piridin-3-il)metanona (42) y 3-oxetanona se convirtieron en (4-(2-cloro-3-fluorofenil)piperidin-1-il)(5-(oxetan-3-il)-4,5,6,7-tetrahidro-1H-pirazolo[4,3-c]piridin-3-il)metanona como un sólido blanco (53 mg, 62%): 1H NMR (300 MHz, DMSO-cfe) 8 12,91 (m, 1H), 7,56-7,39 (m, 3H), 5,15 (m, 1H), 4,52-4,85 (m, 5H), 3,53 (m, 1H), 3,12-3,40 (m, 3H), 2,82-265 (m, 3H), 1,72 (m, 2H), 1,53 (m, 2H); ESI MS m /z 419 [M H]+.
Ejemplo 104: Preparación de (4-(2-cloro-3-fluorofen¡l)p¡per¡d¡n-1-¡l)(5-(2,2,2-tr¡fluoroet¡l)-4,5,6,7-tetrahidro-1H-pirazolo[4,3-c]piridin-3-il)metanona (166)
Paso A: S¡gu¡endo el proced¡m¡ento general GP-D2, clorhidrato de (4-(2-cloro-3-fluorofen¡l)p¡per¡d¡n-1-¡l) (4,5,6,7-tetrahidro-1H-p¡razolo[4,3-c]p¡r¡d¡n-3-¡l)metanona (42) y tr¡fluorometanosulfonato de 2,2,2-tr¡fluoroet¡lo se conv¡rt¡eron en (4-(2-cloro-3-fluorofen¡l) p¡per¡d¡n-1-¡l) (5-(2,2,2-tr¡fluoroet¡l) 4,5,6,7-tetrahidro-1H-p¡razolo[4,3-c]p¡r¡d¡n-3-¡l)metanona como un sól¡do blanco (56 mg, 62%): 1 H NMR (300 MHz, DMSO-da) 812,91 (m, 1H), 7,56-7,39 (m, 3H), 5,15 (m, 1H), 4,73 (m, 1H), 3,82 (m, 2H), 3,32 (m, 1H), 2,89 (m, 2H),2,65 (m, 2H), 1,89 (m, 2H), 1,56 (m, 2H); ESI MS m/z 445 [M H]+.
Ejemplo 105: Preparac¡ón de (4-(2-cloro-3-fluorofen¡l)p¡per¡d¡n-1-¡l)(5-(3,3,3-tr¡fluoroprop¡l)-4,5,6,7-tetrahidro-1H-p¡razolo[4,3-c]p¡r¡d¡n-3-¡l)metanona (167)
Paso A: S¡gu¡endo el proced¡m¡ento general GP-D2, clorhidrato de (4-(2-cloro-3-fluorofen¡l)p¡per¡d¡n-1-¡l) (4,5,6,7-tetrah¡dro-1H-p¡razolo [4,3-c]p¡r¡d¡n-3-¡l)metanona (42) y 3-bromo-1,1,1-tr¡fluoropropano se conv¡rt¡eron en (4-(2-cloro-3-fluorofen¡l)p¡per¡d¡n-1-¡l) (5-(3,3,3-tr¡fluoroprop¡l)-4,5,6,7-tetrahidro-1H-p¡razolo[4,3-c]p¡r¡d¡n-3-¡l)metanona como un sól¡do blanco (36 mg, 43%): 1 H NMR (300 MHz, DMSO-d6) 813,01 (m, 1H), 7,56-7,39 (m, 3H), 5,15 (m, 1H), 4,52 (m, 2H), 3,41 (m, 2H), 2,82 (m, 3H), 1,89 (m, 2H), 1,56 (m, 2H); ESI MS m/z 459 [M H]+.
Ejemplo 106: Preparac¡ón de (4-(2-cloro-3-fluorofen¡l)p¡per¡d¡n-1-¡l)(5-(2-metox¡et¡l)-4,5,6,7-tetrahidro-1H-p¡razolo[4,3-c]p¡r¡d¡n-3-¡l)metanona (168)
Paso A: S¡gu¡endo el proced¡m¡ento general GP-D2, clorhidrato de (4-(2-cloro-3-fluorofen¡l)p¡per¡d¡n-1-¡l) (4,5,6,7-tetrahidro-1H-p¡razolo[4,3-c]p¡r¡d¡n-3-¡l)metanona (42) y bromoet¡lmet¡l éter se conv¡rt¡eron en (4-(2-cloro-3-fluorofen¡l)p¡per¡d¡n-1-¡l)(5-(2-metox¡et¡l)-4,5,6,7-tetrahidro-1H-p¡razolo[4,3-c]p¡r¡d¡n-3-¡l)metanona como un sól¡do blanco (53 mg, 45%): 1 H NMR (300 MHz, DMSO-d6) 812,89 (m, 1H), 7,56-7,39 (m, 3H), 5,15 (m, 1H), 4,52 (m, 2H), 3,51 (m, 4H), 3,23 (m, 4H), 2,72 (m, 6H), 1,89 (m, 2H), 1,56 (m, 2H); ESI MS m/z 421 [M H]+.
Ejemplo 107: Preparac¡ón de (4-(3,4-d¡fluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡n-1-¡l)(5-(p¡peraz¡na-1-carbon¡l)-1,4,5,6-tetrah¡drop¡rrolo[3,4-c]p¡razol-3-¡l)metanona (169)
Paso A: Una soluc¡ón de p¡peraz¡na-1-carbox¡lato de terc-but¡lo (210 mg, 1,13 mmol) y p¡r¡d¡na (137 mg, 1,73 mmol) en CH2Cl2 anhidro (2 mL) se enfr¡ó a 0°C bajo una atmósfera de N2, se trató con una soluc¡ón de tr¡fosgeno (402 mg, 1,35 mmol) en CH2Ch anhidro (2 mL) y se ag¡tó a 0°C durante 1 h. Luego se ret¡ró el baño de enfr¡am¡ento y la reacc¡ón se ag¡tó a temperatura amb¡ente durante 1 h más. Transcurr¡do este t¡empo, la mezcla se d¡luyó con ác¡do clorhídr¡co 1 M (25 mL) y se extrajo con CH2Ch (3 x 20 mL). Los extractos orgán¡cos comb¡nados se lavaron con salmuera (20 mL), se secaron sobre Na2SO4 y el agente desecante se el¡m¡nó por f¡ltrac¡ón. El f¡ltrado se concentró a sequedad bajo pres¡ón reduc¡da para proporc¡onar 4-(clorocarbon¡l) p¡peraz¡na-1-carbox¡lato de -but¡lo como un sól¡do blanco (280 mg, 100%): 1 H NMR (500 MHz, CDCla) 810,76 (br s, 1H), 7,33 (dd, J = 17,0, 9,0 Hz, 1H) , 7,11 (dd, J = 9,0, 4,0 Hz, 1H), 4,88 4,52 (m, 2H), 4,69 (br s, 2H), 4,62 (s, 2H), 3,49 (t aparente, J = 4,5 Hz, 4H), 3,32 (t aparente, J = 4,5 Hz, 4H), 3,25 (t aparente, J = 12,5 Hz, 1H), 3,14-2,88 (m, 2H), 1,94 (d, J = 12,5 Hz, 2H), 1,72-1,66 (m, 2H), 1,48 (s, 9H).
Paso B: Una soluc¡ón de sal de clorhidrato de (4-(3,4-d¡fluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡n-1-¡l) (1,4,5,6-tetrah¡drop¡rrolo[3,4-c]p¡razol-3-¡l)metanona (50 mg, 0,11 mmol), W,W,W-d¡¡soprop¡let¡lam¡na (0,05 mL, 0,3 mmol) y DMAP (0,5 mg, 0,004 mmol) en CH2Ch anhidro (1 mL) se enfr¡ó a 0°C en una atmósfera de N2, se trató con 4-(clorocarbon¡l)p¡peraz¡na-1-carbox¡lato de terc-but¡lo (30 mg, 0,12 mmol) y se ag¡tó a 0°C durante 1 h. Luego se ret¡ró el baño de enfr¡am¡ento y la reacc¡ón se ag¡tó a temperatura amb¡ente durante 4 h más. Después de este t¡empo, la mezcla se cromatograf¡ó sobre gel de síl¡ce (un¡dad Isco Comb¡Flash Rf, columna de oro Red¡sep de 12 g, 0% a 10% de CH3OH en CH2Cl2) para proporc¡onar 4-(3-(4-(3,4- d¡fluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡na-1-carbon¡l)-1,4,5,6-tetrahidrop¡rrolo[3,4-c]p¡razol-5-carbon¡l)p¡peraz¡na-1-carbox¡lato de terc-but¡lo como un sól¡do blanco (48 mg, 71%): 1 H NMR (500 MHz, CDCla) 8 10,76 (br s, 1H), 7,33 (dd, J = 17,0, 9,0 Hz, 1H), 7,11 (dd, J = 9,0, 4,0 Hz, 1H), 4,88-4,52 (m, 2H), 4,69 (br s, 2H), 4,62 (s, 2H), 3,49 (t aparente, J = 4,5 Hz, 4H), 3,32 (t aparente, J = 4,5 Hz, 4H), 3,25 (t aparente, J = 12,5 Hz, 1H), 3,14-2,88 (m, 2H), 1,94 (d, J = 12,5 Hz, 2H), 1,72-1,66 (m, 2H), 1,48 (s, 9H).
Paso C: Una soluc¡ón de 4-(3-(4-(3,4-d¡fluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡na-1-carbon¡l)-1,4,5,6-tetrah¡dro-p¡rrolo[3,4-c]p¡razolo-5-carbon¡l)p¡peraz¡na-1-carbox¡lato de terc-but¡lo (47 mg, 0,077 mmol) en CH2Ch anhidro (1,5 mL) se enfr¡ó a 0°C bajo una atmósfera de N2 y se trató con TFA (1,5 mL). Cuando se completó la ad¡c¡ón, se ret¡ró el baño de enfr¡am¡ento y la reacc¡ón se ag¡tó a temperatura amb¡ente durante 1 h. Pasado este t¡empo, la mezcla se concentró a sequedad bajo pres¡ón reduc¡da, se d¡luyó en CH2Ch (100 mL) y se lavó con NaOH acuoso 2 M (2 x 50 mL). La capa orgán¡ca se secó sobre Na2SO4 y el agente desecante se el¡m¡nó por f¡ltrac¡ón. El f¡ltrado se concentró a sequedad bajo pres¡ón reduc¡da y el res¡duo resultante se cromatograf¡ó sobre gel de síl¡ce (un¡dad Isco Comb¡Flash Rf, columna Red¡sep de 120 g, 0% a 40% de CH3OH en CH2Ch). Las fracc¡ones de columna comb¡nadas se concentraron a sequedad bajo pres¡ón reduc¡da y se encontró que contenían TFA res¡dual (~17%). El res¡duo resultante (21 mg) se d¡luyó en una mezcla de CH2Ch (5 mL) y CH3OH (1 mL), se trató con MP-carbonato y se ag¡tó a temperatura amb¡ente durante 2 h. Pasado este t¡empo la mezcla se f¡ltró y el f¡ltrado se concentró a sequedad bajo pres¡ón reduc¡da para proporc¡onar (4-(3,4-d¡fluoro-2-(tr¡fluoromet¡l)fen¡l)p¡per¡d¡n-1-¡l) (5-(p¡peraz¡na-1-carbon¡l)-1,4,5,6-tetrah¡drop¡rrolo[3,4-c] p¡razol-3-¡l)metanona como un
sólido blanco (13 mg, 33%): mp 153-155°C; 1H NMR (500 MHz, DMSO-da) 5 13,20 (br s, 1H), 7,75 (dd, J = 18,0, 9,0 Hz, 1H), 7,55-7,52 (m, 1H), 4,65-4,51 (m, 6H), 3,24-3,22 (m, 2H), 3,14 (t, J = 5,0 Hz, 4H), 2,96-2,80 (m, 1H), 2,70-2,68 (m, 3H), 2,32-2,22 (m, 1H), 1,79-1,70 (m, 4H), 1 protón no se observa fácilmente; ESI MS m/z 513 [M H]+.
Ejemplo 108: Unión a RPB4 de compuestos de piperidina sustituidos
Los compuestos enumerados en la Tabla 1 se probaron en dos ensayos in vitro, unión de RBP4 (SPA) e interacción RBP4-TTR dependiente de retinol (HTRF). Los compuestos se unieron a RBP4 y/o antagonizaron la interacción RBP4-TTR dependiente de retinol (Tabla 2). Esta actividad indicó que los compuestos reducen los niveles de RBP4 y retinol en suero.
Ċ
TABLA 1.

continuación

continuación

Ċ
continuación

Ċ
(continuación)

(continuación)

(continuación)

Ċ
(continuación)

( c o n t in u a c ió n )

( c o n t in u a c ió n )

Ċ
continuación

( c o n t in u a c ió n )

Ċ
continuación

(continuación)

Ċ
Ĩcontinuación)

Ċ
continuación

Ċ
Ĩcontinuación)

Ċ
Ĩcontinuación)

c o n t in u a c ió n

(continuación)

(continuación)

TABLA 2

continuación

continuación

continuación

continuación

Ejemplo 109: Unión a RPB4 de compuestos de piperidina sustituidos adicionales
Un aspecto adicional de la invención proporciona análogos de los compuestos de la Tabla 1 que son activos como antagonistas de RBP4. Estos análogos contienen un anillo de fenilo di-sustituido o tri-sustituido ubicado en la posición 4 del núcleo de piperidina. Los análogos de los Compuestos 63-162 descritos en la presente se unen de manera análoga a RBP4 y antagonizan la interacción RBP4-TTR dependiente de retinol.
Se prueban compuestos de piperidina adicionales, que son análogos de los descritos en la Tabla 1, en dos ensayos in vitro, unión de RBP4 (SPA) e interacción RBP4-TTR dependiente de retinol (HTRF). Estos compuestos de piperidina se unen a RBP4 y antagonizan la interacción RBP4-TTR dependiente de retinol. Esta actividad indica que los compuestos reducen el nivel de RBP4 y retinol en suero.
Ejemplo 110: Eficacia en un modelo de mamífero
La eficacia de los compuestos enumerados en la Tabla 1 se prueba en ratones de tipo silvestre y Abca4-/-. El modelo de ratón Abca4-/- manifiesta una acumulación acelerada de lipofuscina en el RPE y se considera un modelo de eficacia preclínica para un fármaco que reduce la acumulación de lipofuscina. Los compuestos se dosifican por vía oral durante 3 semanas a 30 mg/kg. Hay una reducción en el nivel sérico de RBP4 en los animales tratados. Los niveles de A2E/isoA2E y otros bisretinoides se reducen en los ratones tratados. Los niveles de A2-DHP-PE y atRAL di-PE también se reducen.
La eficacia de compuestos de piperidina adicionales, que son análogos de los descritos en la Tabla 1, se prueba en ratones de tipo silvestre y Abca4-/-. El modelo de ratón Abca4-/- manifiesta una acumulación acelerada de lipofuscina en el RPE y se considera un modelo de eficacia preclínica para un fármaco que reduce la acumulación de lipofuscina. Los compuestos se dosifican por vía oral durante 3 semanas a 30 mg/kg. Hay una reducción en el nivel sérico de RBP4 en los animales tratados. Los niveles de A2E/isoA2E y otros bisretinoides se reducen en los ratones tratados. Los niveles de A2-DHP-PE y atRAL di-PE también se reducen.
Ejemplo 111: Eficacia del compuesto 81 en un modelo de mamífero
El compuesto 81 inhibió la acumulación de bisretinoides en el modelo de ratón Abca4-/-. Los ratones Abca4-/- tienen niveles de A2E en el rango de ~15-19 pmoles/ojo a las 17 semanas de edad. Los ratones, a partir de las 17 semanas de edad, se trataron con 25 mg/kg del Compuesto 81 durante 12 semanas. Hubo una reducción del 53% en el contenido de bisretinoides en los ratones tratados con el Compuesto 81 frente a los controles tratados con vehículo (Figura 8). Estos datos fueron consistentes con la detención completa de la síntesis de bisretinoides desde el inicio del régimen de dosificación. La acumulación reducida de bisretinoides dio como resultado una reducción significativa de RBP4 en suero en los ratones tratados con el Compuesto 81 (Figura 9).
Ejemplo 112: Eficacia de compuestos adicionales en un modelo de mamífero
Los Compuestos 34, 36 y 38 inhiben la acumulación de bisretinoides en el modelo de ratón Abca4-/-. Los ratones, a partir de las 17 semanas de edad, se tratan con 25 mg/kg de los Compuestos 34, 36 o 38 durante 12 semanas. Hay una reducción en el contenido de bisretinoides en los ratones tratados frente a los controles tratados con vehículo. Estos datos son consistentes con la detención completa de la síntesis de bisretinoides desde el inicio del régimen de dosificación. La acumulación reducida de bisretinoides da como resultado una reducción significativa de RBP4 en suero en los ratones tratados.
Los Compuestos 30, 40 y 42 inhiben la acumulación de bisretinoides en el modelo de ratón Abca4-/-. Los ratones, a partir de las 17 semanas de edad, se tratan con 25 mg/kg de los Compuestos 30, 40 o 42 durante 12 semanas. Hay una reducción en el contenido de bisretinoides en los ratones tratados frente a los controles tratados con vehículo. Estos datos son consistentes con la detención completa de la síntesis de bisretinoides desde el inicio del régimen de dosificación. La
a c u m u la c ió n re d u c id a d e b is re t in o id e s d a c o m o re s u lta d o u n a re d u c c ió n s ig n if ic a t iv a d e R B P 4 en s u e ro en lo s ra to n e s tra ta d o s .
Cada uno de los Compuestos 63-80 o 82-169 inhibe la acumulación de bisretinoides en el modelo de ratón Abca4-/-. Los ratones, a partir de las 17 semanas de edad, se tratan con 25 mg/kg de cualquiera de los Compuestos 63-80 o 82-169 durante 12 semanas. Hay una reducción en el contenido de bisretinoides en los ratones tratados frente a los controles tratados con vehículo. Estos datos son consistentes con la detención completa de la síntesis de bisretinoides desde el inicio del régimen de dosificación. La acumulación reducida de bisretinoides da como resultado una reducción significativa de RBP4 en suero en los ratones tratados.
Ejemplo 113: Administración a un sujeto
Se administra una cantidad de un compuesto 81 al ojo de un sujeto afectado por DMAE. La cantidad del compuesto es efectiva para tratar al sujeto.
Se administra una cantidad de un Compuesto 81 al ojo de un sujeto afectado por la enfermedad de Stargardt. La cantidad del compuesto es efectiva para tratar al sujeto.
Se administra una cantidad de cualquiera de los compuestos 63-80 o 82-169 al ojo de un sujeto afectado por DMAE. La cantidad del compuesto es efectiva para tratar al sujeto.
Se administra una cantidad de cualquiera de los compuestos 63-80 o 82-169 al ojo de un sujeto afectado por la enfermedad de Stargardt. La cantidad del compuesto es efectiva para tratar al sujeto.
Discusión
La degeneración macular asociada a la edad (DMAE) es la principal causa de ceguera en los países desarrollados. Su prevalencia es superior a la de la enfermedad de Alzheimer. No existe tratamiento para la forma seca más común de DMAE. La DMAE seca se desencadena por anomalías en el epitelio pigmentario de la retina (RPE) que se encuentra debajo de las células fotorreceptoras y proporciona un soporte metabólico crítico para estas células sensibles a la luz. La disfunción del RPE induce una degeneración secundaria de los fotorreceptores en la parte central de la retina denominada mácula. Los datos experimentales indican que los altos niveles de lipofuscina inducen la degeneración del RPE y los fotorreceptores adyacentes en las retinas de DMAE atrófica. Además de DMAE, la acumulación dramática de lipofuscina es el sello distintivo de la enfermedad de Stargardt (STGD), una forma hereditaria de degeneración macular de inicio juvenil. El principal componente citotóxico de la lipofuscina de RPE es un bisretinoide A2E de piridinio. La formación de A2E se produce en la retina de forma no enzimática y puede considerarse un subproducto de un ciclo visual que funcione correctamente. Dados los efectos citotóxicos establecidos de A2E en el RPE y los fotorreceptores, la inhibición de la formación de A2E podría retrasar la pérdida visual en pacientes con DMAE seca y STGD. Se sugirió que los inhibidores del ciclo visual de molécula pequeña pueden reducir la formación de A2E en la retina y prolongar la supervivencia del RPE y de los fotorreceptores en pacientes con DMAE seca y STGD. Las tasas del ciclo visual y la producción de A2E en la retina dependen de la entrada de todo-frans-retinol desde el suero al RPE. La captación de retinol por el RPE depende de las concentraciones séricas de retinol. La regulación a la baja farmacológica del retinol sérico es una estrategia de tratamiento válida para la DMAE seca y las STGD. El retinol sérico se mantiene en circulación como un complejo terciario con la proteína de unión al retinol (RBP4) y la transtiretina (TTR). Sin interactuar con TTR, el complejo RBP4-retinol se elimina rápidamente debido a la filtración glomerular. Se requiere la unión de retinol a RBP4 para la formación del complejo RBP4-TTR; apo-RBP4 no interactúa con TTR. Es importante destacar que el sitio de unión de retinol en RBP4 está estéricamente proximal a la interfaz que media la interacción RBP4-TTR. Sin desear limitarse a ninguna teoría científica en particular, los datos aquí presentados muestran que los antagonistas de RBP4 de molécula pequeña que desplazan el retinol de RBP4 y alteran la interacción RBP4-TTR reducirán la concentración de retinol sérico, inhibirán la absorción de retinol en la retina y actuarán como inhibidores indirectos del ciclo visual reduciendo la formación de A2E citotóxico.
RBP4 en suero como un objetivo farmacológico para la inhibición farmacológica del ciclo visual
Dado que las tasas del ciclo visual y la producción de A2E en la retina dependen de la entrada de todo-frans-retinol del suero al RPE (Figura 4), se ha sugerido que la regulación a la baja farmacológica parcial del retinol sérico puede representar un área objetivo en el tratamiento de la DMAE seca (11). El retinol sérico se une a la proteína fijadora de retinol (RBP4) y se mantiene en circulación como un complejo terciario con RBP4 y transtiretina (TTR) (Figura 5). Sin interactuar con TTR, el complejo RBP4-retinol se elimina rápidamente de la circulación debido a la filtración glomerular. Además, se requiere la formación del complejo RBP4-TTRretinol para la captación de todo-frans-retinol mediada por receptores desde el suero hasta la retina.
Sin desear limitarse a ninguna teoría científica, los inhibidores del ciclo visual pueden reducir la formación de bisretinoides tóxicos y prolongar la supervivencia del RPE y de los fotorreceptores en la DMAE seca. Las tasas del ciclo visual y la producción de A2E dependen de la entrada de todo-frans-retinol desde el suero hasta el RPE. La formación del complejo de proteína 4 de unión a retinol terciario (RBP4)-transtiretina (TTR)-retinol en el suero es necesaria para la captación de retinol de la circulación al RPE. El sitio de unión a retinol en RBP4 está estéricamente proximal a la interfaz que media la
in te ra c c ió n R B P 4 -T T R . L o s a n ta g o n is ta s d e R B P 4 q u e c o m p ite n c o n e l re tin o l s é r ic o p o r u n irs e a R B P 4 m ie n tra s b lo q u e a n la in te ra c c ió n R B P 4 -T T R re d u c ir ía n e l re t in o l s é r ic o , ra le n t iz a r ía n e l c ic lo v is u a l e in h ib ir ía n la fo rm a c ió n d e b is re t in o id e s c ito tó x ic o s .
RBP4 representa un objetivo farmacológico atractivo para la inhibición farmacológica indirecta del ciclo visual y la formación de A2E. El sitio de unión de retinol en RBP4 está estéricamente proximal a la interfaz que media la interacción RBP4-TTR. Los antagonistas del retinol que compiten con el retinol sérico por unirse a RBP4 mientras bloquean la interacción RBP4-TTR reducirían los niveles séricos de RBP4 y retinol, lo que conduciría a una reducción de la absorción de retinol en la retina. El resultado sería la inhibición del ciclo visual con la consiguiente reducción de la síntesis de A2E.
Se descubrió que un retinoide sintético llamado fenretinida [N-(4-hidroxifenil)retinamida, 4HRP] (Figura 6) considerado anteriormente como un tratamiento contra el cáncer (29) se une a RBP4, desplaza el todo-trans-retinol de RBP4 (13) e interrumpe la interacción RBP4-TTR (13,14).
Se demostró que la fenretinida reduce la RBP4 en suero y el retinol (15), inhibe la captación de todo-trans-retinol ocular y ralentiza el ciclo visual (11). Es importante destacar que la administración de fenretinida redujo la producción de A2E en un modelo animal de acumulación excesiva de bisretinoides, ratones Abca4-/- (11). Los experimentos preclínicos con fenretinida validaron a RBP4 como un objetivo farmacológico para la DMAE seca. Sin embargo, la fenretinida no es selectiva y es tóxica. Independientemente de su actividad como antagonista de la unión del retinol a RBP4, la fenretinida es un inductor extremadamente activo de la apoptosis en muchos tipos de células (16-19), incluyendo las células del epitelio pigmentario de la retina (20). Se ha sugerido que los efectos adversos de la fenretinida están mediados por su acción como ligando de un receptor nuclear RAR (21-24). Además, al igual que otros retinoides, se informa que la fenretinida estimula la formación de hemangiosarcomas en ratones. Además, la fenretinida es teratogénica, lo que hace que su uso sea problemático en pacientes con enfermedad de Stargardt en edad fértil.
Dado que el perfil de seguridad de fenretinida puede ser incompatible con la dosificación a largo plazo en personas con condiciones de ceguera, pero que no amenazan la vida, la identificación de nuevas clases de antagonistas de RBP4 es de gran importancia. Los compuestos de la presente invención desplazan el retinol de RBP4, interrumpen la interacción RBP4-TTR inducida por retinol y reducen los niveles de REBP4 en suero. Los compuestos de la presente invención inhiben la acumulación de bisretinoides en el modelo de ratón Abca4-/- de lipofuscinogénesis excesiva, lo que indica la utilidad de un tratamiento para la DMAE seca y la enfermedad de Stargardt.
La presente invención se refiere a moléculas pequeñas para uso en el tratamiento de la degeneración macular y la enfermedad de Stargardt. En la presente se describe el uso oftálmico de las moléculas pequeñas como antagonistas de RBP4 no retinoides. Se ha demostrado que el compuesto enumerado en la Tabla 2 se une a RBP4 in vitro y/o antagoniza la interacción RBP4-TTR in vitro en concentraciones biológicamente significativas. Los compuestos adicionales descritos en la presente, que son análogos del compuesto enumerado en la Tabla 2, se unen de forma análoga a RBP4 in vitro y antagonizan la interacción RBP4-TTR in vitro a concentraciones biológicamente significativas.
Actualmente, no existe un tratamiento aprobado por la FDA para la DMAE seca o la enfermedad de Stargardt, que afecta a millones de pacientes. Se afirma que un cóctel de vitaminas antioxidantes y zinc de venta libre y no aprobado por la FDA (fórmula AREDS) es beneficioso en un subconjunto de pacientes con DMAE seca. No existen tratamientos para la enfermedad de Stargardt. La presente invención identificó antagonistas de RBP4 no retinoides que son útiles para el tratamiento de DMAE seca y otras condiciones caracterizadas por una acumulación excesiva de lipofuscina. Sin desear limitarse a ninguna teoría científica, dado que la acumulación de lipofuscina parece ser una causa directa de la muerte de RPE y fotorreceptores en la retina de DMAE y STGD, los compuestos descritos en la presente son agentes modificadores de la enfermedad, ya que abordan directamente la causa raíz de estas enfermedades. La presente invención proporciona un compuesto novedoso para su uso en el tratamiento que preservará la visión en pacientes con DMAE y enfermedad de Stargardt, y pacientes que padecen afecciones caracterizadas por una acumulación excesiva de lipofuscina.
Referencias
1. Petrukhin K. New therapeutic targets in atrophic age-related macular degeneration. Expert Opin. Ther. Targets. 2007, 11(5): 625-639.
2. C. Delori, D.G. Goger & C.K. Dorey, Age-related accumulation and spatial distribution of lipofuscin in RPE of normal subjects. Investigative Ophthalmology and Visual Science 42 (2001), pp. 1855-1866.
3. F.C. Delori, RPE lipofuscin in ageing and age-related macular degeneration. En: G. Coscas & F.C. Piccolino, Editors, Retinal Pigment Epithelium and Macular Disease (Documenta Ophthalmologica) vol. 62, Kluwer Academic Publishers, Dordrecht, Países Bajos (1995), pp. 37-45.
4. C.K. Dorey, G. Wu, D. Ebenstein, A. Garsd & J.J. Weiter, Cell loss in the aging retina. Relationship to lipofuscin accumulation and macular degeneration. Investigative Ophthalmology and Visual Science 30 (1989), pp. 1691-1699.
5. L. Feeney-Burns, E.S. Hilderbrand & S. Eldridge, Aging human RPE: morphometric analysis of macular, equatorial, and
p e r ip h e ra l c e lls . In v e s t ig a t iv e O p h th a lm o lo g y a n d V is u a l S c ie n c e 25 (1984 ) , pp. 195 -200.
6. F.G. Holz, C. Bellman, S. Staudt, F. Schutt & H.E. Volcker, Fundus autofluorescence and development of geographic atrophy in age-related macular degeneration. Investigative Ophthalmology and Visual Science 42 (2001), pp. 1051-1056.
7. F.G. Holz, C. Bellmann, M. Margaritidis, F. Schutt, T.P. Otto & H.E. Volcker, Patterns of increased in vivo fundus autofluorescence in the junctional zone of geographic atrophy of the retinal pigment epithelium associated with agerelated macular degeneration. Graefe's Archive for Clinical and Experimental Ophthalmology 237 (1999), pp. 145-152.
8. A. von Rückmann, F.W. Fitzke & A.C. Bird, Fundus autofluorescence in age-related macular disease imaged with a laser scanning ophthalmoscope. Investigative Ophthalmology and Visual Science 38 (1997), pp. 478-486.
9. F.G. Holz, C. Bellman, S. Staudt, F. Schutt & H.E. Volcker, Fundus autofluorescence and development of geographic atrophy in age-related macular degeneration. Investigative Ophthalmology and Visual Science 42 (2001), pp. 1051-1056.
10. Sparrow JR, Fishkin N, Zhou J, Cai B, Jang YP, Krane S, Itagaki Y, Nakanishi K. A2E, a byproduct of the visual cycle. Vision Res. Dic 2003;43(28):2983-90.
11. Radu RA, Han Y, Bui TV, Nusinowitz S, Bok D, Lichter J, Widder K, Travis GH, Mata NL. Reductions in serum vitamin A arrest accumulation of toxic retinal fluorophores: a potential therapy for treatment of lipofuscin-based retinal diseases. Invest Ophthalmol Vis Sci. Dic 2005;46(12):4393-401.
12. Motani A, Wang Z, Conn M, Siegler K, Zhang Y, Liu Q, Johnstone S, Xu H, Thibault S, Wang Y, Fan P, Connors R, Le H, Xu G, Walker N, Shan B, Coward P. Identification and characterization of a non-retinoid ligand for retinolbinding protein 4 which lowers serum retinol-binding protein 4 levels in vivo. J Biol Chem. 20 Mar 2009;284(12):7673-80.
13. Berni R, Formelli F. In vitro interaction of fenretinide with plasma retinol-binding protein and its functional consequences. FEBS Lett. 10 Ago 1992;308(1):43-5.
14. Schaffer EM, Ritter SJ, Smith JE. N-(4-hydroxyphenyl)retinamide (fenretinide) induces retinol-binding protein secretion from liver and accumulation in the kidneys in rats. J Nutr. Sep 1993;123(9):1497-503.
15. Adams WR, Smith JE, Green MH. Effects of N-(4-hydroxyphenyl)retinamide on vitamin A metabolism in rats. Proc Soc Exp Biol Med. Feb 1995;208(2):178-85.
16. Puduvalli VK, Saito Y, Xu R, Kouraklis GP, Levin VA, Kyritsis AP. Fenretinide activates caspases and induces apoptosis in gliomas. Clin Cancer Res. Ago 1999;5(8):2230-5.
17. Holmes WF, Soprano DR, Soprano KJ. Synthetic retinoids as inducers of apoptosis in ovarian carcinoma cell lines. J Cell Physiol. Jun 2004;199(3):317-29.
18. Simeone AM, Ekmekcioglu S, Broemeling LD, Grimm EA, Tari AM. A novel mechanism by which N-(4-hydroxyphenyl)retinamide inhibits breast cancer cell growth: the production of nitric oxide. Mol Cancer Ther. Oct 2002;1(12):1009-17.
19. Fontana JA, Rishi AK. Classical and novel retinoids: their targets in cancer therapy. Leukemia. Abr 2002;16(4):463-72.
20. Samuel W, Kutty RK, Nagineni S, Vijayasarathy C, Chandraratna RA, Wiggert B. N-(4-hydroxyphenyl)retinamide induces apoptosis in human retinal pigment epithelial cells: retinoic acid receptors regulate apoptosis, reactive oxygen species generation, and the expression of heme oxygenase-1 and Gadd153. J Cell Physiol. Dic 2006;209(3):854-65.
21. Fontana JA, Rishi AK. Classical and novel retinoids: their targets in cancer therapy. Leukemia. Abr 2002;16(4):463-72.
22. Samuel W, Kutty RK, Nagineni S, Vijayasarathy C, Chandraratna RA, Wiggert B. N-(4-hydroxyphenyl)retinamide induces apoptosis in human retinal pigment epithelial cells: retinoic acid receptors regulate apoptosis, reactive oxygen species generation, and the expression of heme oxygenase-1 and Gadd153. J Cell Physiol. Dic 2006;209(3):854-65.
23. Sabichi AL, Xu H, Fischer S, Zou C, Yang X, Steele VE, Kelloff GJ, Lotan R, Clifford JL. Retinoid receptordependent and independent biological activities of novel fenretinide analogues and metabolites. Clin Cancer Res. 1 Oct 2003;9(12):4606-13.
24. Clifford JL, Menter DG, Wang M, Lotan R, Lippman SM. Retinoid receptor-dependent and -independent effects of N-(4-hydroxyphenyl)retinamide in F9 embryonal carcinoma cells. Cancer Res. 1 Ene 1999;59(1):14-8.
25. Gollapalli DR, Rando RR. The specific binding of retinoic acid to RPE65 and approaches to the treatment of macular degeneration. Proc Natl Acad Sci U S A. 6 Jul 2004;101(27):10030-5.
26. Maiti P, Kong J, Kim SR, Sparrow JR, Allikmets R, Rando RR. Small molecule RPE65 antagonists limit the visual cycle and prevent lipofuscin formation. Biochemistry. 24 Ene 2006;45(3):852-60.
27. Radu RA, Mata NL, Nusinowitz S, Liu X, Sieving PA, Travis GH. Treatment with isotretinoin inhibits lipofuscin accumulation in a mouse model of recessive Stargardt's macular degeneration. Proc Natl Acad Sci U S A,15 Abr 2003;100(8):4742-7.
28. Monaco HL, Rizzi M, Coda A. Structure of a complex of two plasma proteins: transthyretin and retinol-binding protein. Science. 19 May 1995;268(5213):1039-41.
29. Bonanni B, Lazzeroni M, Veronesi U. Synthetic retinoid fenretinide in breast cancer chemoprevention. Expert Rev Anticancer Ther. Abr 2007;7(4):423-32.
30. Sunness JS, et al. The long-term natural history of geographic atrophy from age-related macular degeneration: enlargement of atrophy and implications for interventional clinical trials. Ophthalmology. Feb 2007;114(2):271-7.
31. Glickman JF, et al. A comparison of ALPHAScreen, TR-FRET, and TRF as assay methods for FXR nuclear receptors. J. Biomol. Screening 2002;7:3-10.
32. Fujimura T, et al. Unique properties of coactivator recruitment caused by differential binding of FK614, an antidiabetic agent, to PPARgamma. Biol. Pharm. Bull. 2006;29:423-429.
33. Zhou G, et al. Nuclear receptors have distinct affinities fo coactivators: characterization by FRET. Mol. Endocrinol.
1998;12:1594-1605.
34. Cogan U, Kopelman M, Mokady S, Shinitzky M. Binding affinities of retinol and related compounds to retinol binding proteins. Eur J Biochem. 17 May 1976;65(1):71-8.
35. Decensi A, Torrisi R, Polizzi A, Gesi R, Brezzo V, Rolando M, Rondanina G, Orengo MA, Formelli F, Costa A. Effect of the synthetic retinoid fenretinide on dark adaptation and the ocular surface. J Natl Cancer Inst. 19 Ene 1994;86(2):105-10.
36. Conley B, et al. Pilot trial of the safety, tolerability, and retinoid levels of N-(4-hydroxyphenyl) retinamide in combination with tamoxifen in patients at high risk for developing invasive breast cancer. J Clin Oncol. Ene 2000;18(2):275-83.
37. Fain GL, Lisman JE. Photoreceptor degeneration in vitamin A deprivation and retinitis pigmentosa: the equivalent light hypothesis. Exp Eye Res. Sep 1993;57(3):335-40.
38. Makimura H, Wei J, Dolan-Looby SE, Ricchiuti V, Grinspoon S. Retinol-Binding Protein Levels are Increased in Association with Gonadotropin Levels in Healthy Women. Metabolism. Abril 2009; 58(4): 479-487.
39. Yang Q, et al. Serum retinol binding protein 4 contributes to insulin resistance in obesity and type 2 diabetes. Nature.
21 Jul 2005;436(7049):356-62.
40. Kim SR, et al. The all-trans-retinal dimer series of lipofuscin pigments in retinal pigment epithelial cells in a recessive Stargardt disease model. PNAS. 4 de diciembre de 2007, Vol. 104, No. 49, 19273-8.
41. Wu Y, et al. Novel Lipofuscin Bisretinoids Prominent in Human Retina and in a Model of Recessive Stargardt Disease. Journal of Biological Chemistry. 24 de julio de 2009, Vol. 284, No. 30, 20155-20166.
42. F.G. Holz, et al. Patterns of increased in vivo fundus autofluorescence in the junctional zone of geographic atrophy of the retinal pigment epithelium associated with age-related macular degeneration. Graefe's Archive for Clinical and Experimental Ophthalmology 237 (1999), pp. 145-152.
Claims (15)
1. Un compuesto que tiene la estructura:

en donde
R1, R2, R3, R4 y R5 son cada uno independientemente H, halógeno, CF3 o alquilo C1-C4,
en donde dos o más de R1, R2 , R3, R4 o R5 son distintos de H;
R6 es H o halógeno; y
B tiene la estructura:

en donde
n es un número entero de 0-1;
a, p, x, 5, £ y 9 están presentes o ausentes cada uno independientemente, y cuando están presentes cada uno es un enlace;
Z1 es N;
Z2 es N o NR7;
en donde R7 es H, alquilo C1-C4 u oxetano;
X es C o N;
Y1 , Y2, Y3 e Y4 son cada uno independientemente CR8, CH2 , N o N-Rg,
en donde R8 es H, halógeno, OCH3, CN o CF3; y
Rg es H, CN, oxetano, alquilo C1-C6, cicloalquilo C3-C6, (alquilo C1-C4)(cicloalquilo C3-C6), (alquilo C1 -C6)-OCH3, (alquilo C1-Ca)-CF3, C(O)-(alquilo C1-C6), C(O)2-(alquilo C1-Ca), C(O)-NH2, C(O)NH-(alquilo C1 -Ca), C(O)-(arilo Ca), C(O)-(heteroarilo Ca), C(O)-pirrolidina, C(O)-piperidina, C(O)-piperazina, (alquilo C1-Ca)-CO2H, (alquilo C1-Ca)-CO2 (alquilo C1-Ca) o SO2-(alquilo C1-Ca),
en donde cuando R1 es CF3, R2 es H, R3 es F, R4 es H, y R5 es H, o
R1 es H, R2 es CF3, R3 es H, R4 es CF3, y R5 es H, o
R1 es Cl, R2 es H, R3 es H, R4 es F, y R5 es H, o
R1 es CF3, R2 es H, R3 es F, R4 es H, y R5 es H, o
R1 es CF3, R2 es F, R3 es H, R4 es H, y R5 es H, o
R1 es Cl, R2 es F, R3 es H, R4 es H, y R5 es H,
entonces B es distinto de

o una sal farmacéuticamente aceptable de los mismos.
H a
2. El compuesto de la reivindicación 1, en donde
R1 es CF3, R2 es F, R3 es F, R4 es H, y R5 es H, o
R1 es CF3, R2 es F, R3 es H, R4 es H, y R5 es H, o
R1 es CF3, R2 es F, R3 es H, R4 es F, y R5 es H, o
R1 es CF3, R2 es H, R3 es F, R4 es F, y R5 es H, o
R1 es CF3, R2 es H, R3 es H, R4 es H, y R5 es F, o
R1 es CF3, R2 es H, R3 es F, R4 es H, y R5 es H, o
R1 es CF3, R2 es H, R3 es H, R4 es Cl , y R5 es H, o
R1 es CF3, R2 es Cl , R3 es H, R4 es H, y R5 es H, o
R1 es H, R2 es CF3, R3 es H, R4 es CF3, y R5 es H, o
R1 es Cl, R2 es H, R3 es H, R4 es F, y R5 es H, o
R1 es Cl, R2 es F, R3 es H, R4 es H, y R5 es H.
3. El compuesto de la reivindicación 1, en donde B tiene la estructura:

en donde
n es 0;
R7 es H, alquilo C1 -C4 u oxetano;
Y1 e Y3 son cada uno CH2 ; y
Y2 es N-R9,
en donde
R9 es H, CN, oxetano, alquilo C1-C6, cicloalquilo C3-C6, (alquilo Ci -C4)(cicloalquilo C3-C6), (alquilo Ci -C6)-Ci -Ca)-CF3, C(O)-(alquilo C1-C6), C(O)2-(alquilo C1-6), C(O)NH2 , C(O)NH-(alquilo C1-C6), C(O)-(arilo Ca), C(O)-(heteroarilo
Ce), C(O)-pirrolidina, C(O)-piperidina, C(O)-piperazina, (alquilo C1 -Ca)-CO2H, (alquilo C1-Ca)-CO2(alquilo C1-Ca) o SO2-(alquilo C1-Ca); o
en donde B tiene la estructura:

en donde
n es 1;
R7 es H, alquilo C1 -C4 u oxetano;
Y1, Y2 e Y4 son cada uno CH2 ; y
Y3 es N-R9,
en donde
R9 es H, CN, oxetano, alquilo C1-Ca, cicloalquilo C3-Ca, (alquilo C1-C4)(cicloalquilo C3-Ca), (alquilo C1 -Ca)-OCH3, (alquilo
C1-Ca)-CF3, C(O)-(alquilo C1-Ca), C(O)2-(alquilo C1-Ca), C(O)-NH2, C(O)NH-(alquilo C1 -Ca), C(O)-(arilo Ca), C(O)-(heteroarilo Ca), C(O)-pirrolidina, C(O)-piperidina, C(O)-piperazina, (alquilo C1-Ca)-CO2H, (alquilo C1-Ca)-CO2(alquilo C1-Ca) o SO2-(alquilo C1-Ca); o
en donde B tiene la estructura:

en donde
n es 1;
R7 es H, alquilo C1 -C4 u oxetano;
Y1, Y3 e Y4 son cada uno CH2 ; y
Y2 es N-R9,
en donde
R9 es H, CN, oxetano, alquilo C1-C6, cicloalquilo C3-C6, (alquilo C1-C4)(cicloalquilo C3-C6), (alquilo C1 -C6)-OCH3, (alquilo C1-Ca)-CF3, C(O)-(alquilo C1-C6), C(O)2-(alquilo C^C a), C(O)-NH2, C(O)NH-(alquilo C^C a), C(O)-(arilo Ca), C(O)-(heteroarilo Ca), C(O)-pirrolidina, C(O)-piperidina, C(O)-piperazina, (alquilo C1-Ca)-CO2H, (alquilo C1-Ca)-CO2(alquilo C1-Ca) o SO2-(alquilo C1-Ca).
4. El compuesto de la reivindicación 3, en donde B tiene la estructura:

5. El compuesto de la reivindicación 4,
en donde R9 es H, CN, CH3, CH2CH3, CH2CH2CH3, CH(CH3)2, CH2CH(CH3)2, /-Bu, CH2CH(CH3)2, CH2C(CH3)3, CH2CF3, CH2CH2CF3, CH2OCH3, CH2CH2OCH3,

en donde R9 es SO2-CH3, C(O)-CH3, C(O)-CH2CH3, C(O)-CH2CH2CH3, C(O)-CH(CH3)2, C(O)-CH2CH(CH3)2, C(O)-/-Bu, C(O)-OCH3, C(O)-NHCH3,

a. El compuesto de la reivindicación 1, en donde B tiene la estructura:

en donde
Yi , Y2, Y3 e Y4 son cada uno independientemente CR8 o N,
en donde cada R8 es independientemente, OCH3, CN o CF3; o
en donde B tiene la estructura:

en donde cada R8 es independientemente H, halógeno, OCH3, CN o CF3.
7. El compuesto de la reivindicación 1 que tiene la estructura:





o una sal farmacéuticamente aceptable del compuesto.
8. Una composición farmacéutica que comprende el compuesto de cualquiera de las reivindicaciones 1-7 y un portador farmacéuticamente aceptable.
9. El compuesto de cualquiera de las reivindicaciones 1-7 o la composición de la reivindicación 8 para uso en el tratamiento de una enfermedad caracterizada por una acumulación excesiva de lipofuscina en la retina en un sujeto que padece dicha enfermedad.
10. El compuesto o composición para uso de acuerdo con la reivindicación 9, en donde la enfermedad se caracteriza además por degeneración macular mediada por bisretinoides, en donde el bisretinoide es A2E, isoA2E, A2-DHP-PE o atRAL di-PE.
11. El compuesto o composición para uso de acuerdo con la reivindicación 9 o 10, en donde la enfermedad caracterizada por una acumulación excesiva de lipofuscina en la retina es la degeneración macular asociada a la edad, degeneración macular asociada a la edad seca (atrófica), enfermedad de Stargardt, enfermedad de Best, maculopatía viteliforme del adulto o distrofia macular tipo Stargardt.
12. El compuesto o composición para uso de acuerdo con cualquiera de las reivindicaciones 9-11, en donde el sujeto es un mamífero.
13. El compuesto o composición para uso de acuerdo con la reivindicación 12, en donde el compuesto o composición se administra al mamífero por vía intravenosa, intravítrea u oral.
14. El compuesto o composición para uso de acuerdo con la reivindicación 9, en donde el compuesto o composición se administra por vía oral.
15. El compuesto o composición para uso de acuerdo con la reivindicación 14, en donde el compuesto o composición se administra como una dosificación oral en forma de una cápsula o comprimido.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201461986578P | 2014-04-30 | 2014-04-30 | |
| PCT/US2015/028293 WO2015168286A1 (en) | 2014-04-30 | 2015-04-29 | Substituted 4-phenylpiperidines, their preparaiton and use |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2909793T3 true ES2909793T3 (es) | 2022-05-10 |
Family
ID=54354744
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES15785329T Active ES2909793T3 (es) | 2014-04-30 | 2015-04-29 | 4-fenilpiperidinas sustituidas, su preparación y uso |
Country Status (18)
| Country | Link |
|---|---|
| US (8) | US9434727B2 (es) |
| EP (3) | EP3137168B1 (es) |
| JP (1) | JP6676541B2 (es) |
| KR (1) | KR102429220B1 (es) |
| CN (2) | CN106794364B (es) |
| AU (1) | AU2015253232B2 (es) |
| BR (1) | BR112016025356B1 (es) |
| CA (1) | CA2947174C (es) |
| DK (2) | DK4036094T3 (es) |
| ES (1) | ES2909793T3 (es) |
| FI (1) | FI4036094T3 (es) |
| HR (1) | HRP20260361T1 (es) |
| MX (1) | MX384244B (es) |
| PH (1) | PH12016502147B1 (es) |
| PL (1) | PL4036094T3 (es) |
| PT (1) | PT4036094T (es) |
| SG (1) | SG11201608943VA (es) |
| WO (1) | WO2015168286A1 (es) |
Families Citing this family (19)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9333202B2 (en) | 2012-05-01 | 2016-05-10 | The Trustees Of Columbia University In The City Of New York | Non-retinoid antagonists for treatment of age-related macular degeneration and stargardt disease |
| WO2014152018A1 (en) | 2013-03-14 | 2014-09-25 | The Trustees Of Columbia University In The City Of New York | Octahydrocyclopentapyrroles, their preparation and use |
| US9944644B2 (en) | 2013-03-14 | 2018-04-17 | The Trustees Of Columbia University In The City Of New York | Octahydropyrrolopyrroles their preparation and use |
| EP3495357B1 (en) | 2013-03-14 | 2021-05-05 | The Trustees of Columbia University in the City of New York | 4-phenylpiperidines, their preparation and use |
| US9938291B2 (en) | 2013-03-14 | 2018-04-10 | The Trustess Of Columbia University In The City Of New York | N-alkyl-2-phenoxyethanamines, their preparation and use |
| SG11201608943VA (en) | 2014-04-30 | 2016-11-29 | Univ Columbia | Substituted 4-phenylpiperidines, their preparaiton and use |
| GB201604638D0 (en) * | 2016-03-18 | 2016-05-04 | Mission Therapeutics Ltd | Novel compounds |
| US10245259B2 (en) | 2017-06-15 | 2019-04-02 | Belite Bio, Inc | Methods of treating RBP4 related diseases with triazolopyridines |
| US11389444B2 (en) | 2017-06-15 | 2022-07-19 | Belite Bio, Inc | Methods of treating metabolic diseases with fused bicyclic pyrazoles |
| US12440486B2 (en) * | 2018-08-01 | 2025-10-14 | The Trustees Of Columbia University In The City Of New York | RBP4 antagonists for treatment and prevention of non-alcoholic fatty liver disease and gout |
| AU2020310120B2 (en) * | 2019-07-08 | 2025-05-29 | Belite Bio (HK) Limited | Formulations of RBP4 inhibitors and methods of use |
| KR102399432B1 (ko) | 2020-04-20 | 2022-05-19 | 주식회사 엘파니 | 전도성 고분자 구조체 표면에 코팅된 금속 박막 층을 포함하는, 하이브리드 구조체 및 이의 제조 방법 |
| CA3203127A1 (en) | 2020-12-23 | 2022-06-30 | Soon-Ee Cheah | Transaction data processing systems and methods |
| EP4340701A4 (en) * | 2021-05-21 | 2025-04-09 | Belite Bio, LLC | Biomarkers for age-related macular degeneration |
| CA3238550A1 (en) * | 2021-11-23 | 2023-06-01 | Cheng-Chi Irene Wang | Tablet formulations of rbp4 inhibitors and methods of use |
| WO2025108420A1 (zh) * | 2023-11-24 | 2025-05-30 | 北京双鹤润创科技有限公司 | 一种降低rbp4的血清浓度的化合物及用途 |
| WO2025195508A1 (zh) * | 2024-03-22 | 2025-09-25 | 润尔眼科药物(广州)有限公司 | 一种苯基杂环化合物及其应用 |
| WO2025195018A1 (zh) * | 2024-03-22 | 2025-09-25 | 润尔眼科药物(广州)有限公司 | 一种苯基哌啶化合物及其应用 |
| WO2026018214A1 (en) | 2024-07-19 | 2026-01-22 | Tapi Czech Industries S.R.O. | Solid state forms of tinlarebant and process for preparation thereof |
Family Cites Families (126)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE3926606A1 (de) | 1989-08-11 | 1991-02-14 | Hoechst Ag | Verfahren zur behandlung der cardialen sowie der vasculaeren hypertrophie und hyperplasie |
| DE4130514A1 (de) | 1991-09-13 | 1993-03-18 | Chemie Linz Deutschland | Verfahren zur herstellung reiner n,n'-unsymmetrisch substituierter phenylharnstoffe |
| US5532243A (en) | 1992-02-14 | 1996-07-02 | The Dupont Merck Pharmaceutical Company | Antipsychotic nitrogen-containing bicyclic compounds |
| US5312814A (en) | 1992-12-09 | 1994-05-17 | Bristol-Myers Squibb Co. | α-phosphonocarboxylate squalene synthetase inhibitors |
| DE4341403A1 (de) | 1993-12-04 | 1995-06-08 | Basf Ag | N-substituierte 3-Azabicycloalkan-Derivate, ihre Herstellung und Verwendung |
| US5523430A (en) | 1994-04-14 | 1996-06-04 | Bristol-Myers Squibb Company | Protein farnesyl transferase inhibitors |
| US5837716A (en) | 1995-11-13 | 1998-11-17 | Albany Medical College | Analgesic heterocyclic compounds |
| EP0944388A4 (en) | 1996-04-03 | 2001-08-16 | Merck & Co Inc | INHIBITORS OF FARNESYL PROTEIN TRANSFERASE |
| ES2200313T3 (es) | 1997-03-03 | 2004-03-01 | Eisai Co., Ltd. | Uso de inhibidores de la colinesterasa para tratar trastornos de la atencion. |
| TR200002182T2 (tr) | 1998-01-27 | 2000-12-21 | Aventis Pharmaceuticals Products Inc. | İkame edilmiş aksozaherosayklil faktör xa inhibitörleri |
| CA2333554A1 (en) | 1998-06-17 | 1999-12-23 | Chu-Baio Xue | Cyclic hydroxamic acids as metalloproteinase inhibitors |
| EP1121150A4 (en) | 1998-10-09 | 2003-06-04 | Merck & Co Inc | DELTA-6 FATTY ACID DESATURASE |
| AU3215500A (en) | 1999-01-25 | 2000-08-07 | Smithkline Beecham Corporation | Compounds and methods |
| ATE346152T1 (de) | 1999-04-14 | 2006-12-15 | Merck & Co Inc | Neuartige menschliche kalium-potentialabhängige kanäle |
| AU4612300A (en) | 1999-05-13 | 2001-05-10 | Shionogi & Co., Ltd. | Preventive or therapeutic drugs for diabetes |
| CA2374947A1 (en) | 1999-05-24 | 2000-11-30 | Robert M. Scarborough | Inhibitors of factor xa |
| CN1420882A (zh) | 1999-07-28 | 2003-05-28 | 阿温蒂斯药物公司 | 取代的氧代氮杂杂环基化合物 |
| US6372793B1 (en) | 1999-08-20 | 2002-04-16 | Florida Agricultural & Mechanical University | Method for treatment of a neurological disease characterized by impaired neuromodulator function |
| MY125533A (en) * | 1999-12-06 | 2006-08-30 | Bristol Myers Squibb Co | Heterocyclic dihydropyrimidine compounds |
| EP2133078A1 (en) | 2000-03-03 | 2009-12-16 | Eisai R&D Management Co., Ltd. | Use of a cholinesterase inhibitor for the treatment of dementia and cognitive impairments |
| US20020045613A1 (en) | 2000-04-27 | 2002-04-18 | Heinz Pauls | 1-aroyl-piperidinyl benzamidines |
| EP1283897A4 (en) | 2000-05-16 | 2004-08-04 | Merck & Co Inc | GENE RESPONSIBLE FOR STARGARDT-LIKE, DOMINANT, MAKULA-DYSTROPHY |
| AU2001280599A1 (en) | 2000-07-15 | 2002-01-30 | Smith Kline Beecham Corporation | Compounds and methods |
| CA2445568A1 (en) | 2001-04-27 | 2002-11-07 | Vertex Pharmaceuticals Incorporated | Triazole-derived kinase inhibitors and uses thereof |
| AU2002320958B2 (en) | 2001-07-06 | 2008-02-28 | Neurosearch A/S | Novel compounds, their preparation and use |
| WO2003024456A1 (en) | 2001-09-20 | 2003-03-27 | Eisai Co., Ltd. | Methods for treating and preventing migraines |
| WO2003024450A1 (en) | 2001-09-20 | 2003-03-27 | Eisai Co., Ltd. | Methods for treating prion diseases |
| WO2003032914A2 (en) | 2001-10-17 | 2003-04-24 | Eisai Co., Ltd. | Methods for treating substance abuse with cholinesterase inhibitors |
| DE60327922D1 (de) | 2002-02-05 | 2009-07-23 | Lilly Co Eli | Harnstoff-linker verbindungen und ihre verwendung als ppar regulatoren |
| US20030195192A1 (en) | 2002-04-05 | 2003-10-16 | Fortuna Haviv | Nicotinamides having antiangiogenic activity |
| MXPA04008797A (es) | 2002-03-13 | 2004-11-26 | Janssen Pharmaceutica Nv | Inhibidores de histona desacetilasa. |
| AU2003228796A1 (en) | 2002-05-01 | 2003-11-17 | Eisai Co., Ltd. | Cholinesterase inhibitors to prevent injuries caused by chemicals |
| WO2004034963A2 (en) | 2002-05-17 | 2004-04-29 | Eisai Co., Ltd. | Methods and compositions using cholinesterase inhibitors |
| JPWO2004002531A1 (ja) | 2002-06-26 | 2005-10-27 | 小野薬品工業株式会社 | 血管の収縮または拡張による疾患治療剤 |
| EP1526852A1 (en) | 2002-07-25 | 2005-05-04 | Pharmacia Italia S.p.A. | Bicyclo-pyrazoles active as kinase inhibitors, process for their preparation and pharmaceutical compositions comprising them |
| AU2003256922A1 (en) | 2002-07-31 | 2004-02-16 | Smithkline Beecham Corporation | Substituted heterocyclic compounds as modulators of the ccr5 receptor |
| AU2003255845A1 (en) | 2002-08-22 | 2004-03-11 | Piramed Limited | Phosphadidylinositol 3,5-biphosphate inhibitors as anti-viral agents |
| US7196082B2 (en) | 2002-11-08 | 2007-03-27 | Merck & Co. Inc. | Ophthalmic compositions for treating ocular hypertension |
| WO2004089416A2 (en) | 2003-04-11 | 2004-10-21 | Novo Nordisk A/S | Combination of an 11beta-hydroxysteroid dehydrogenase type 1 inhibitor and an antihypertensive agent |
| US7700583B2 (en) * | 2003-04-11 | 2010-04-20 | High Point Pharmaceuticals, Llc | 11β-hydroxysteroid dehydrogenase type 1 active compounds |
| US7501405B2 (en) * | 2003-04-11 | 2009-03-10 | High Point Pharmaceuticals, Llc | Combination therapy using an 11β-hydroxysteroid dehydrogenase type 1 inhibitor and an antihypertensive agent for the treatment of metabolic syndrome and related diseases and disorders |
| WO2004108135A1 (en) | 2003-05-30 | 2004-12-16 | Merck & Co., Inc. | Composition and method for treating macular disorders |
| BRPI0410630A (pt) | 2003-06-19 | 2006-06-13 | Pfizer Prod Inc | antagonista de nk1 |
| FR2857363B1 (fr) * | 2003-07-10 | 2007-09-07 | Aventis Pharma Sa | 4,5,6,7-tetrahydro-1h-pyrazolo[3,4-c] pyridines substituees compositions les contenant et utilisation |
| AR045037A1 (es) * | 2003-07-10 | 2005-10-12 | Aventis Pharma Sa | Tetrahidro-1h-pirazolo [3,4-c] piridinas sustituidas, composiciones que las contienen y su utilizacion. |
| WO2005074535A2 (en) | 2004-01-30 | 2005-08-18 | Eisai Co., Ltd. | Cholinesterase inhibitors for spinal cord disorders |
| US7435831B2 (en) | 2004-03-03 | 2008-10-14 | Chemocentryx, Inc. | Bicyclic and bridged nitrogen heterocycles |
| EP1729761A4 (en) | 2004-03-05 | 2008-09-03 | Eisai Co Ltd | TREATMENT OF CADASIL WITH CHOLINESTERASE INHIBITORS |
| CA2565599C (en) | 2004-05-18 | 2012-07-31 | Schering Corporation | Substituted 2-quinolyl-oxazoles useful as pde4 inhibitors |
| EP1779867A4 (en) | 2004-07-01 | 2009-12-02 | Eisai R&D Man Co Ltd | MEANS FOR PROMOTING NERVE REVENERATION |
| FR2872416B1 (fr) | 2004-07-01 | 2006-09-22 | Oreal | Utilisation de derives de piperidine pour lutter contre les rides |
| EP1778685B1 (de) | 2004-08-02 | 2008-03-26 | Schwarz Pharma Ag | Carboxamide des indolizins und seiner aza- und diazaderivate |
| JP2006077006A (ja) | 2004-08-13 | 2006-03-23 | Eisai Co Ltd | 加齢に伴う過活動膀胱治療薬 |
| AU2005287343A1 (en) | 2004-08-18 | 2006-03-30 | Sirion Therapeutics, Inc. | Combination compositions comprising 13-cis-retinyl derivatives and uses thereof to treat opthalmic disorders |
| JP4639696B2 (ja) | 2004-08-31 | 2011-02-23 | Jsr株式会社 | 磁性体複合粒子およびその製造方法 |
| MX2007003332A (es) | 2004-09-20 | 2007-06-05 | Xenon Pharmaceuticals Inc | Derivados heterociclicos y su uso como inhibidores de estearoil-coa-desaturasa. |
| EP1827438B2 (en) | 2004-09-20 | 2014-12-10 | Xenon Pharmaceuticals Inc. | Piperazin derivatives for inhibiting human stearoyl-coa-desaturase |
| KR20070074638A (ko) | 2004-10-27 | 2007-07-12 | 얀센 파마슈티카 엔.브이. | 테트라하이드로 피리디닐 피라졸 카나비노이드 모듈레이터 |
| JP2008519054A (ja) | 2004-11-04 | 2008-06-05 | シリオン セラピューティクス, インコーポレイテッド | レチノール−レチノール結合タンパク質(rbp)−トランスチレチン(ttr)複合体の形成のモジュレーター |
| WO2006058088A2 (en) | 2004-11-23 | 2006-06-01 | Ptc Therapeutics, Inc. | Carbazole, carboline and indole derivatives useful in the inhibition of vegf production |
| JP2006176503A (ja) | 2004-11-26 | 2006-07-06 | Tohoku Univ | 脳血管障害を伴うアルツハイマー病治療薬 |
| CA2584845C (en) | 2004-12-08 | 2009-02-17 | Sirion Therapeutics, Inc. | Methods, assays and compositions for treating retinol-related diseases |
| EP1838690A2 (en) * | 2004-12-21 | 2007-10-03 | Devgen N.V. | Compounds with kv4 ion channel activity |
| GB0503056D0 (en) | 2005-02-14 | 2005-03-23 | Smithkline Beecham Corp | Chemical compounds |
| WO2006094235A1 (en) | 2005-03-03 | 2006-09-08 | Sirtris Pharmaceuticals, Inc. | Fused heterocyclic compounds and their use as sirtuin modulators |
| EP2392328A1 (en) | 2005-05-09 | 2011-12-07 | Hydra Biosciences, Inc. | Compounds for modulating TRPV3 Function |
| US7632837B2 (en) | 2005-06-17 | 2009-12-15 | Bristol-Myers Squibb Company | Bicyclic heterocycles as cannabinoid-1 receptor modulators |
| UA81382C2 (en) | 2005-07-11 | 2007-12-25 | Composition for treating retinol-related diseases by modulation of retinol binding | |
| EP2392571A3 (en) * | 2005-07-29 | 2012-03-14 | F. Hoffmann-La Roche AG | Indol-3-yl-carbonyl-piperidin and piperazin derivatives |
| JPWO2007020888A1 (ja) | 2005-08-12 | 2009-02-26 | 武田薬品工業株式会社 | 脳・神経細胞保護剤および睡眠障害治療薬 |
| JP2009507791A (ja) | 2005-08-29 | 2009-02-26 | メルク エンド カムパニー インコーポレーテッド | ナイアシン受容体アゴニスト、このような化合物を含有する組成物、および治療方法 |
| EP2407459A1 (en) | 2005-09-27 | 2012-01-18 | Shionogi & Co., Ltd. | Synthetic intermediate in the production of a sulfonamide derivative having PGD2 receptor antagonistic activity |
| WO2007073432A2 (en) | 2005-10-11 | 2007-06-28 | Chemocentryx, Inc. | Piperidine derivatives and methods of use |
| DE102005049954A1 (de) * | 2005-10-19 | 2007-05-31 | Sanofi-Aventis Deutschland Gmbh | Triazolopyridin-derivate als Inhibitoren von Lipasen und Phospholipasen |
| WO2007058322A1 (ja) | 2005-11-18 | 2007-05-24 | Ono Pharmaceutical Co., Ltd. | 塩基性基を含有する化合物およびその用途 |
| WO2007086584A1 (ja) | 2006-01-30 | 2007-08-02 | Meiji Seika Kaisha, Ltd. | 新規FabKおよびFabI/K阻害剤 |
| US20070254911A1 (en) | 2006-03-27 | 2007-11-01 | Mingde Xia | Tetrahydro-Pyrazolo[3,4-c]Pyridine Cannabinoid Modulators |
| CA2647545C (en) | 2006-04-03 | 2016-02-23 | Istituto Di Ricerche Di Biologia Molecolare P. Angeletti S.P.A. | Amide substituted indazole and benzotriazole derivatives as poly(adp-ribose)polymerase (parp) inhibitors |
| ATE481409T1 (de) | 2006-05-18 | 2010-10-15 | Hoffmann La Roche | Thiazolopyramidin/pyridinharnstoffderivate als antagonisten am adenosin-a2b-rezeptor |
| WO2008045393A2 (en) | 2006-10-11 | 2008-04-17 | Amgen Inc. | Imidazo- and triazolo-pyridine compounds and methods of use therof |
| ATE527249T1 (de) | 2006-12-08 | 2011-10-15 | Hoffmann La Roche | Indole |
| DE102006060598A1 (de) | 2006-12-21 | 2008-06-26 | Merck Patent Gmbh | Tetrahydrobenzoisoxazole |
| PL2170340T3 (pl) | 2007-06-21 | 2016-06-30 | Neuronascent Inc | Sposoby i kompozycje stymulacji neurogenezy i hamowania degeneracji neuronów z wykorzystaniem izotiazolopirymidonów |
| CA2695989A1 (en) | 2007-08-10 | 2009-02-19 | Glaxosmithkline Llc | Certain nitrogen containing bicyclic chemical entities for treating viral infections |
| US7973079B2 (en) | 2007-09-27 | 2011-07-05 | Revision Therapeutics, Inc. | Methods and compounds for treating retinol-related diseases |
| CN101873852A (zh) | 2007-09-27 | 2010-10-27 | 锐视医药公司 | 治疗视黄醇相关性疾病的方法和化合物 |
| WO2009051244A1 (ja) | 2007-10-18 | 2009-04-23 | Takeda Pharmaceutical Company Limited | 複素環化合物 |
| EP2085398A1 (en) | 2008-02-01 | 2009-08-05 | Merz Pharma GmbH & Co. KGaA | Pyrazolopyrimidines, a process for their preparation and their use as medicine |
| JP5566288B2 (ja) | 2008-05-30 | 2014-08-06 | 武田薬品工業株式会社 | 複素環化合物 |
| WO2014097304A1 (en) | 2012-12-22 | 2014-06-26 | Maytronics Ltd. | Autonomous pool cleaning robot |
| CN102186479A (zh) | 2008-09-10 | 2011-09-14 | 凯利普西斯公司 | 用于治疗疾病的组胺受体的氨基嘧啶抑制剂 |
| WO2010077915A1 (en) | 2008-12-17 | 2010-07-08 | Forest Laboratories Holdings Limited | Novel compounds useful as cc chemokine receptor ligands |
| WO2010088050A2 (en) | 2009-01-28 | 2010-08-05 | Cara Therapeutics, Inc. | Bicyclic pyrazolo-heterocycles |
| US20100204265A1 (en) | 2009-02-09 | 2010-08-12 | Genelabs Technologies, Inc. | Certain Nitrogen Containing Bicyclic Chemical Entities for Treating Viral Infections |
| JP5642661B2 (ja) * | 2009-03-05 | 2014-12-17 | 塩野義製薬株式会社 | Npyy5受容体拮抗作用を有するピペリジンおよびピロリジン誘導体 |
| TW201041868A (en) | 2009-03-20 | 2010-12-01 | Intervet Int Bv | Anthelmintic agents and their use |
| US20120010186A1 (en) | 2009-03-23 | 2012-01-12 | Merck Frosst Canada Ltd. | Heterocyclic compounds as inhibitors of stearoyl-coenzyme a delta-9 desaturase |
| AU2010236685A1 (en) | 2009-04-13 | 2011-12-01 | Irm Llc | Compositions and methods for modulating retinol binding to retinol binding protein 4 (RBP4) |
| US8853215B2 (en) | 2009-04-16 | 2014-10-07 | Takeda Pharmaceutical Company Limited | Derivatives of N-acyl-N′-phenylpiperazine useful (inter alia) for the prophylaxis or treatment of diabetes |
| US8102887B2 (en) | 2009-05-26 | 2012-01-24 | Corning Incorporated | Edge bonded optical packages |
| CN102459248A (zh) | 2009-05-26 | 2012-05-16 | 埃克塞里艾克西斯公司 | 作为PI3K/mTOR抑制剂的苯并氧杂环庚三烯以及它们使用与制造方法 |
| WO2010137738A1 (en) | 2009-05-28 | 2010-12-02 | Otsuka Pharmaceutical Co., Ltd. | Heterocyclic compounds for the treatment of stress-related conditions |
| US8642621B2 (en) | 2009-09-16 | 2014-02-04 | The University Of Edinburgh | (4-phenyl-piperidin-1-yl)-[5-(1H-pyrazol-4-yl)-thiophen-3-yl]-methanone compounds and their use |
| US20130189246A1 (en) | 2009-11-12 | 2013-07-25 | The Trustees Of Columbia University In The City Of New York | Treatment of ophthalmic conditions with fluorenone derivatives |
| CA2781888C (en) | 2009-12-11 | 2019-06-18 | Nono Inc. | Agents and methods for treating ischemic and other diseases |
| JP5879273B2 (ja) | 2010-03-01 | 2016-03-08 | ジーティーエックス・インコーポレイテッド | 癌を処置するための化合物 |
| WO2011116123A1 (en) | 2010-03-19 | 2011-09-22 | Irm Llc | Tafamidis for the treatment of ophthalmic diseases |
| WO2011116356A2 (en) | 2010-03-19 | 2011-09-22 | Sanford-Burnham Medical Research Institute | Positive allosteric modulators of group ii mglurs |
| US8796470B2 (en) | 2010-05-25 | 2014-08-05 | Abbvie Inc. | Substituted octahydrocyclopenta[c]pyrroles as calcium channel modulators |
| WO2011156632A2 (en) | 2010-06-09 | 2011-12-15 | Georgetown University | Compositions and methods of treatment for tumors in the nervous system |
| US20130085131A1 (en) * | 2010-07-01 | 2013-04-04 | Amgen Inc. | Heterocyclic compounds and their uses |
| CA2807738A1 (en) | 2010-08-27 | 2012-03-01 | Novartis Ag | Hydroxamate-based inhibitors of deacetylases |
| JP5951625B2 (ja) | 2010-11-24 | 2016-07-13 | ザ・トラスティーズ・オブ・コランビア・ユニバーシティー・イン・ザ・シティー・オブ・ニューヨークThe Trustees Of Columbia University In The City Of New York | 加齢性黄斑変性症およびシュタルガルト病の処置のための非レチノイドrbp4アンタゴニスト |
| US9290453B2 (en) | 2010-12-23 | 2016-03-22 | Merck Sharp & Dohme Corp. | Quinolines and aza-quinolines as CRTH2 receptor modulators |
| WO2012125904A1 (en) | 2011-03-17 | 2012-09-20 | The Trustees Of The University Of Pennsylvania | Mutation mimicking compounds that bind to the kinase domain of egfr |
| JP6130827B2 (ja) | 2011-05-17 | 2017-05-17 | 塩野義製薬株式会社 | ヘテロ環化合物 |
| US9333202B2 (en) | 2012-05-01 | 2016-05-10 | The Trustees Of Columbia University In The City Of New York | Non-retinoid antagonists for treatment of age-related macular degeneration and stargardt disease |
| WO2013166041A1 (en) | 2012-05-01 | 2013-11-07 | The Trustees Of Columbia University In The City Of New York | Transthyretin ligands capable of inhibiting retinol-dependent rbp4-ttr interaction for treatment of age-related macular |
| WO2013166040A1 (en) | 2012-05-01 | 2013-11-07 | The Trustees Of Columbia University In The City Of New York | S-fta and s-fta analogues capable of inhibiting retinol-dependent rbp4-ttr interaction for treatment of age-related macular degeneration, stargardt disease, and other retinal disease characterized by excessive lipofuscin accumulation |
| TW201501713A (zh) * | 2013-03-01 | 2015-01-16 | Kyowa Hakko Kirin Co Ltd | 眼炎症性疾病之預防及/或治療劑 |
| US9944644B2 (en) | 2013-03-14 | 2018-04-17 | The Trustees Of Columbia University In The City Of New York | Octahydropyrrolopyrroles their preparation and use |
| US9938291B2 (en) | 2013-03-14 | 2018-04-10 | The Trustess Of Columbia University In The City Of New York | N-alkyl-2-phenoxyethanamines, their preparation and use |
| US20160030422A1 (en) | 2013-03-14 | 2016-02-04 | Konstantin Petrukhin | Rbp4 antagonists for the treatment of age-related macular degeneration and stargardt disease |
| WO2014152018A1 (en) | 2013-03-14 | 2014-09-25 | The Trustees Of Columbia University In The City Of New York | Octahydrocyclopentapyrroles, their preparation and use |
| EP3495357B1 (en) | 2013-03-14 | 2021-05-05 | The Trustees of Columbia University in the City of New York | 4-phenylpiperidines, their preparation and use |
| SG11201608943VA (en) | 2014-04-30 | 2016-11-29 | Univ Columbia | Substituted 4-phenylpiperidines, their preparaiton and use |
-
2015
- 2015-04-29 SG SG11201608943VA patent/SG11201608943VA/en unknown
- 2015-04-29 PL PL21217254.8T patent/PL4036094T3/pl unknown
- 2015-04-29 JP JP2016565008A patent/JP6676541B2/ja active Active
- 2015-04-29 CA CA2947174A patent/CA2947174C/en active Active
- 2015-04-29 BR BR112016025356-6A patent/BR112016025356B1/pt active IP Right Grant
- 2015-04-29 US US14/699,672 patent/US9434727B2/en active Active
- 2015-04-29 CN CN201580036136.0A patent/CN106794364B/zh active Active
- 2015-04-29 EP EP15785329.2A patent/EP3137168B1/en active Active
- 2015-04-29 DK DK21217254.8T patent/DK4036094T3/da active
- 2015-04-29 EP EP25221420.0A patent/EP4691465A3/en active Pending
- 2015-04-29 PT PT212172548T patent/PT4036094T/pt unknown
- 2015-04-29 KR KR1020167033645A patent/KR102429220B1/ko active Active
- 2015-04-29 CN CN201911252390.0A patent/CN111393434B/zh active Active
- 2015-04-29 HR HRP20260361TT patent/HRP20260361T1/hr unknown
- 2015-04-29 AU AU2015253232A patent/AU2015253232B2/en active Active
- 2015-04-29 MX MX2016014197A patent/MX384244B/es unknown
- 2015-04-29 EP EP21217254.8A patent/EP4036094B1/en active Active
- 2015-04-29 DK DK15785329.2T patent/DK3137168T3/da active
- 2015-04-29 FI FIEP21217254.8T patent/FI4036094T3/fi active
- 2015-04-29 WO PCT/US2015/028293 patent/WO2015168286A1/en not_active Ceased
- 2015-04-29 ES ES15785329T patent/ES2909793T3/es active Active
-
2016
- 2016-09-01 US US15/254,966 patent/US9777010B2/en active Active
- 2016-10-27 PH PH12016502147A patent/PH12016502147B1/en unknown
-
2017
- 2017-10-02 US US15/722,760 patent/US10072016B2/en active Active
-
2018
- 2018-08-08 US US16/058,299 patent/US10407433B2/en active Active
-
2019
- 2019-07-24 US US16/520,886 patent/US10913746B2/en active Active
-
2020
- 2020-11-16 US US17/099,231 patent/US11649240B2/en active Active
-
2022
- 2022-12-19 US US18/068,005 patent/US12404277B2/en active Active
-
2025
- 2025-06-26 US US19/251,371 patent/US20250320218A1/en active Pending
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2909793T3 (es) | 4-fenilpiperidinas sustituidas, su preparación y uso | |
| DK2968304T3 (en) | 4-PHENYLPIPERIDINES, THEIR PREPARATION AND USE. | |
| ES2700541T3 (es) | Octahidrociclopentapirroles, su preparación y uso | |
| US10787453B2 (en) | Octahydropyrrolopyrroles their preparation and use | |
| HK40075816A (en) | Substituted 4-phenylpiperidines, their preparation and use | |
| HK1232169B (en) | Substituted 4-phenylpiperidines, their preparation and use | |
| HK40010051A (en) | 4-phenylpiperidines, their preparation and use | |
| NZ726053B2 (en) | Substituted 4-phenylpiperidines, their preparation and use |