ES2912306T3 - Tetratiomolibdato de bis-colina para el tratamiento de enfermedad de Wilson - Google Patents
Tetratiomolibdato de bis-colina para el tratamiento de enfermedad de Wilson Download PDFInfo
- Publication number
- ES2912306T3 ES2912306T3 ES18815624T ES18815624T ES2912306T3 ES 2912306 T3 ES2912306 T3 ES 2912306T3 ES 18815624 T ES18815624 T ES 18815624T ES 18815624 T ES18815624 T ES 18815624T ES 2912306 T3 ES2912306 T3 ES 2912306T3
- Authority
- ES
- Spain
- Prior art keywords
- bis
- choline tetrathiomolybdate
- patient
- dose
- level
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/28—Compounds containing heavy metals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/13—Amines
- A61K31/14—Quaternary ammonium compounds, e.g. edrophonium, choline
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K33/00—Medicinal preparations containing inorganic active ingredients
- A61K33/24—Heavy metals; Compounds thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/0053—Mouth and digestive tract, i.e. intraoral and peroral administration
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/28—Dragees; Coated pills or tablets, e.g. with film or compression coating
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
Landscapes
- Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Epidemiology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Diabetes (AREA)
- Hematology (AREA)
- Obesity (AREA)
- Organic Chemistry (AREA)
- Inorganic Chemistry (AREA)
- Nutrition Science (AREA)
- Physiology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Medicinal Preparation (AREA)
- Investigating Or Analysing Biological Materials (AREA)
- Measuring Or Testing Involving Enzymes Or Micro-Organisms (AREA)
Abstract
Tetratiomolibdato de bis-colina para su uso en el tratamiento de la enfermedad de Wilson en un paciente, en donde el tetratiomolibdato de bis-colina se administra al paciente en un estado en ayunas.
Description
DESCRIPCIÓN
Tetratiomolibdato de bis-colina para el tratamiento de enfermedad de Wilson
REFERENCIA CRUZADA A SOLICITUDES RELACIONADAS
La presente solicitud reivindica prioridad a la solicitud provisional de EE. UU. N.° 62/594.184, presentada el 4 de diciembre de 2017; la solicitud provisional de EE. UU. N.° 62/646.553, presentada el 22 de marzo de 2018; la solicitud provisional de EE. UU. N.° 62/655.568, presentada el 10 de abril de 2018; la solicitud provisional de EE. UU. N.° 62/669.095, presentada el 9 de mayo de 2018; la solicitud provisional de EE. UU. N.° 62/741.313, presentada el 4 de octubre de 2018; y la solicitud provisional de EE. UU. N.° 62/750.595, presentada el 25 de octubre de 2018
ANTECEDENTES
La enfermedad de Wilson (EW) es un trastorno autosómico recesivo de metabolismo alterado del cobre (Cu). Mutaciones en el gen ATP7B producen la producción deficiente de la ATPasa2 del transportador de Cu, que conduce a incorporación alterada del Cu en la ceruloplasmina, eliminación biliar alterada del Cu, elevado Cu libre y unido a albúmina, y acumulación de Cu en el hígado, el cerebro y otros tejidos, con daño del órgano y disfunción resultante. Se estima que la prevalencia de la EW es de 1 cada 30.000 personas, que corresponde a aproximadamente 10.000 individuos en los Estados Unidos y a aproximadamente 15.000 individuos en la Unión Europea.
La presentación clínica típica de la EW es de la adolescencia hasta la adultez temprana. El cribado genético y la correlación genotipo-fenotipo se complica por una multitud de mutaciones de ATP7B asociadas; la mayoría de los individuos con EW son heterocigotos compuestos. Los signos y los síntomas iniciales de la EW son predominantemente hepáticos (~40 %), neurológicos (~40 %) o psiquiátricos (~20 %), pero los pacientes desarrollan frecuentemente enfermedad hepática y neuropsiquiátrica combinada. Los pacientes sin tratar o insuficientemente tratados tienen morbilidad progresiva, y la mortalidad es normalmente secundaria a la cirrosis hepática descompensada e insuficiencia hepática. El trasplante de hígado es la única terapia eficaz para la insuficiencia hepática aguda asociada a la EW; otras causas de muerte asociadas a la EW incluyen tumor maligno hepático y deterioro neurológico con inanición intensa.
El hígado representa uno de los principales órganos de almacenamiento del Cu en los seres humanos. En personas sanas, la homeostasis intracelular del Cu está estrechamente regulada. El cobre es incorporado en las células por el transportador 1 del Cu (CTR1), y luego es transportado desde el CTR1 hasta las chaperonas del Cu, tales como las chaperonas del Cu para el antioxidante 1, la citocromo c oxidasa y la superóxido dismutasa. El cobre que acompaña a la chaperona se suministra a un enzima específica que requiere Cu. Si aparece una cantidad en exceso de Cu, el exceso de Cu se une a la metalotioneína (MT) como Cu monovalente (Cu+) por puentes de tiolato de Cu por restos de cisteína abundantes en MT, por lo que conducen a una desintoxicación del Cu mediante una reducción de su potencial rédox.
En pacientes con EW, la eliminación del Cu está alterada debido a la deficiencia de ATPasa2. Esto produce una acumulación de Cu, principalmente en el hígado y el cerebro, pero también en otros órganos. Dentro de la capacidad amortiguadora de la MT, no existe toxicidad evidente del Cu debido a que la MT se une estrechamente al Cu. Sin embargo, más allá de la capacidad amortiguadora del Cu de la MT, aparecen iones Cu libres y esta excesiva cantidad de Cu intracelular libre desencadena propiedades pro-oxidantes, que conducen a un riesgo elevado de daños a tejidos/órganos con manifestaciones clínicas como resultado. Se supone que la toxicidad del Cu en la EW está mediada por el Cu libre o unido de forma suelta que no está unido fuertemente a la MT debido a la sobrecarga de Cu.
Los objetivos del tratamiento de la EW se centran en compensar la eliminación alterada del Cu causada por la deficiencia de ATPasa2. Los tratamientos actuales para la EW son terapias con quelantes generales, D-penicilamina (CUPRIMINE®, Valeant Pharmaceuticals, DEPEN®, Meda Pharmaceuticals) y trientina (SYPRINE®, Aton Pharma, Inc.), que no quelan específicamente el Cu y promueven la eliminación urinaria de Cu. Además, el cinc (Zn), que bloquea la captación dietética del Cu, se usa principalmente para el tratamiento de mantenimiento. El cinc altera la absorción del Cu por la inducción de la MT en el tubo gastrointestinal (GI).
El control de la enfermedad en pacientes con síntomas neurológicos en el diagnóstico de la EW es un área de preocupación particular. Más de un tercio de los pacientes que presentan síntomas neurológicos no muestran mejoría después de 4 años de tratamiento con quelantes. Este fracaso en responder a la terapia de quelación con presentación neurológica puede reflejar un daño irreversible al sistema nervioso. Por lo tanto, en un estudio reciente, aproximadamente el 50 % de los pacientes tuvieron síntomas neurológicos residuales, a pesar de años de terapia con un agente modulador del Cu. Se ha informado del empeoramiento de los síntomas neurológicos tras el inicio del tratamiento en aproximadamente el 25 % de pacientes que empezaron con penicilamina y trientina, y hasta el 50 % de los pacientes no se recuperaron nunca. Se cree que el mecanismo tras este empeoramiento neurológico "paradójico" es una movilización de Cu del hígado que conduce a elevaciones del Cu en la sangre y el sistema nervioso central, causando deterioro neurológico. Esta teoría está soportada por datos no clínicos.
Los fármacos actualmente disponibles para tratar la EW tienen elevadas tasas de interrupción del tratamiento debido a acontecimientos adversos y a fracaso terapéutico. Sus perfiles de acontecimientos adversos y pautas posológicas complicadas conducen a un mal cumplimiento del tratamiento y a elevadas tasas de fracaso terapéutico, una preocupación importante en una enfermedad tal como la EW que requiere tratamiento de por vida.
Se ha mostrado que el tetratiomolibdato en forma de una sal de amonio proporciona control del Cu y mejora la función hepática después de incluso una dosis única. La mayoría de las pruebas de seguridad y toxicidad clínicas y no clínicas para los tratamientos basados en tetratiomolibdato se han llevado a cabo usando amonio como contraión catiónico.
Weiss Karl Heinz et al., The Lancet. Gastroenterology & Hepatology 122017, vol. 2, no. 12, 5 de octubre de 2017 (05 10-2017), páginas 869-876, desvela el uso de tetratiomolibdato de bis-colina en el tratamiento de la enfermedad de Wilson.
La absorción del fármaco de un agente activo administrado por vía oral puede ser influida por alimento ingerido antes o después de que se administre el agente activo. El alimento puede influir en la absorción del fármaco mediante varios mecanismos. El alimento puede afectar la absorción interactuando con el agente activo o con la formulación farmacéutica en el tubo gastrointestinal, estimulando el flujo de bilis, cambiando el pH gastrointestinal, aumentando la circulación sanguínea esplácnica, o retrasando el vaciamiento gástrico. Por lo tanto, la biodisponibilidad de un agente activo puede verse afectada por la ingestión de alimento en aproximadamente 2 horas antes o aproximadamente 1 hora después de la administración de un agente activo.
Sin embargo, es difícil predecir, de novo, si un agente activo o formulación farmacéutica particular presentará o no un efecto sobre el alimento. Además, aunque esté presente, el efecto sobre el alimento para un agente activo o formulación farmacéutica puede producir un aumento o una disminución en la biodisponibilidad en las condiciones posprandiales en comparación con la administración de una dosis equivalente en condiciones en ayunas. En casos en los que existe un efecto sustancial sobre el alimento (es decir, la ingestión del alimento antes o después de la administración de la dosis provoca un aumento o una disminución sustancial en la absorción del fármaco con respecto a la administración en el estado en ayunas), las formulaciones farmacéuticas administras en las condiciones posprandiales no son bioequivalentes a las mismas formulaciones farmacéuticas administradas en condiciones en ayunas.
Esta ausencia de bioequivalencia puede tener profundas consecuencias clínicas. Por ejemplo, la administración de una composición farmacéutica con alimento puede proporcionar niveles en plasma sanguíneo del fármaco peligrosamente elevados del agente activo que producen efectos adversos clínicos. Alternativamente, la administración de una composición farmacéutica en condiciones en ayunas puede proporcionar una dosis eficaz, mientras que la administración de la composición con alimento puede proporcionar niveles en plasma sanguíneo del fármaco subterapéuticos de forma que la dosis alimentada no sea eficaz.
Por lo tanto, existe una necesidad de desarrollar métodos de tratamiento de la enfermedad de Wilson que sean eficaces en mejorar el metabolismo del Cu, reducir el Cu libre tóxico y mantener los niveles normales de Cu para mejorar los síntomas de los pacientes, sin los efectos secundarios asociados a los tratamientos actualmente disponibles.
Obsérvese que las referencias a los métodos de tratamiento en el Sumario y la Descripción detallada de la invención en esta descripción se deben interpretar como referencias a los compuestos, las composiciones farmacéuticas y los medicamentos de la presente invención para su uso en un método para el tratamiento del cuerpo humano (o animal) por terapia.
SUMARIO DE LA INVENCIÓN
La presente invención se refiere a tetratiomolibdato de bis-colina para su uso en el tratamiento de la enfermedad de Wilson en un paciente, en donde el tetratiomolibdato de bis-colina se administra al paciente en un estado en ayunas. En algunas realizaciones, el tetratiomolibdato de bis-colina tiene la estructura:

En algunas realizaciones, la divulgación se refiere a métodos de tratamiento de la enfermedad de Wilson en un paciente administrando 15 mg de tetratiomolibdato de bis-colina una vez al día o una vez cada día. En algunas realizaciones, la divulgación se refiere a métodos de tratamiento de la enfermedad de Wilson en un paciente administrando desde 30 hasta 90 mg de tetratiomolibdato de bis-colina diariamente. En realizaciones adicionales, la divulgación se refiere a métodos de tratamiento de la enfermedad de Wilson en un paciente administrando desde 30 hasta 90 mg de tetratiomolibdato de bis-colina diariamente, donde el paciente tiene uno o más de los siguientes: un
NCCcorregido superior a 2,3 pm/l, nivel de alanina aminotransferasa (ALT) inferior a 80 Ul/ml, hemoglobina superior a 8 g/dl, plaquetas superior a 30.000/pl, o neutrófilos superior a 1.000/pl o 1 x 103/pl.
En algunas realizaciones, la divulgación se refiere a métodos de modificación de la administración de tetratiomolibdato de bis-colina a un paciente con enfermedad de Wilson que recibe tratamiento con tetratiomolibdato de bis-colina disminuyendo la dosis diaria de tetratiomolibdato de bis-colina o aumentando la dosis diaria de tetratiomolibdato de bis-colina.
En un aspecto, la presente divulgación proporciona además métodos de disminución o aumento de la dosis diaria de tetratiomolibdato de bis-colina en un paciente que presenta un resultado de prueba anormal, que incluye opcionalmente la interrupción del tratamiento durante un periodo de tiempo.
En otro aspecto, la presente divulgación proporciona composiciones farmacéuticas para tratar enfermedad de Wilson en un paciente que comprenden tetratiomolibdato de bis-colina. En otro aspecto, la presente divulgación proporciona kits para tratar enfermedad de Wilson, que comprenden al menos tres conjuntos de unidades de administración farmacéutica; e instrucciones para su uso.
Según la presente invención, el método de tratamiento de la enfermedad de Wilson en un paciente en necesidad del mismo comprende administrar tetratiomolibdato de bis-colina en un estado en ayunas. En ciertos aspectos, el tetratiomolibdato de bis-colina se administra as una formulación con cubierta entérica.
En otro aspecto más, la presente divulgación se refiere a métodos de tratamiento de la enfermedad de Wilson en un paciente en necesidad del mismo que comprenden administrar tetratiomolibdato de bis-colina durante aproximadamente 24 semanas o más, aproximadamente 36 semanas o más, aproximadamente 48 semanas o más, aproximadamente 60 semanas o más, aproximadamente 72 semanas o más, aproximadamente 84 semanas o más, aproximadamente 92 semanas o más, aproximadamente 120 semanas o más, aproximadamente 132 semanas o más, o aproximadamente 144 semanas o más.
En ciertas realizaciones, la divulgación se refiere a una composición farmacéutica que comprende 15 mg de tetratiomolibdato de bis-colina para su uso en el tratamiento de la enfermedad de Wilson en un paciente en estado en ayunas. En realizaciones particulares, la composición farmacéutica que comprende 15 mg de tetratiomolibdato de biscolina es adecuada para administración una vez al día. En realizaciones adicionales, la composición farmacéutica es adecuada para administración una vez cada dos días. En todavía otras realizaciones, la composición farmacéutica es una composición farmacéutica de liberación retardada. En aún otras realizaciones, la composición farmacéutica está en forma de un comprimido o cápsula. En incluso otras realizaciones, la composición farmacéutica está en forma de un comprimido. En realizaciones particulares, la composición farmacéutica está en forma de un comprimido con cubierta entérica.
En ciertas realizaciones de la divulgación, el paciente presenta una reducción en NCCcorregido como se mide después de 24 semanas de administración en comparación con el NCCcorregido del paciente como se mide antes de la administración, tal como una reducción del 20 %, 35 %, 50 % o 75 %, en NCCcorregido como se mide después de 24 semanas de administración en comparación con el NCCcorregido del paciente como se mide antes de la administración.
En realizaciones particulares de la divulgación, el paciente presenta una reducción en NCCcorregido como se mide después de 48 semanas de administración en comparación con el NCCcorregido del paciente como se mide antes de la administración, tal como una reducción del 20 %, 35 %, 50 % o 75 %, en NCCcorregido como se mide después de 48 semanas de administración en comparación con el NCCcorregido del paciente como se mide antes de la administración.
En realizaciones particulares de la divulgación, el paciente presenta una reducción en NCCcorregido como se mide después de 72 semanas de administración en comparación con el NCCcorregido del paciente como se mide antes de la administración, tal como una reducción del 20 %, 35 %, 50 % o 75 %, en NCCcorregido como se mide después de 72 semanas de administración en comparación con el NCCcorregido del paciente como se mide antes de la administración.
En ciertas realizaciones, la divulgación se refiere a una composición farmacéutica que comprende tetratiomolibdato de bis-colina para su uso en el tratamiento de la enfermedad de Wilson en un paciente en necesidad del mismo, en donde se administran 30, 45, 60, 75 o 90 mg de tetratiomolibdato de bis-colina diariamente y el paciente tiene uno o más de los siguientes:
a) un NCCcorregido superior a 2,3 pm/l;
b) nivel de alanina aminotransferasa (ALT) inferior a 80 Ul/ml;
c) hemoglobina superior a 8 g/dl;
d) plaquetas superior a 30.000/pl; o
e) neutrófilos superior a 103/pl.
En realizaciones particulares, la divulgación se refiere a una composición farmacéutica de tetratiomolibdato de biscolina para su uso en el tratamiento de la enfermedad de Wilson en un paciente que recibe tratamiento con tetratiomolibdato de bis-colina, en donde la dosis diaria de tetratiomolibdato de bis-colina se reduce cuando dicho paciente presenta un nivel de alanina aminotransferasa (ALT) de al menos dos veces el nivel de ALT presentado cuando se empieza el tratamiento con tetratiomolibdato de bis-colina. En ciertas realizaciones, si el paciente tomaba una dosis de 15 mg una vez al día de tetratiomolibdato de bis-colina, la dosis se reduce hasta 15 mg de tetratiomolibdato de bis-colina cada dos días; si el paciente tomaba una dosis de 30 mg una vez al día de tetratiomolibdato de bis-colina, la dosis se reduce hasta 15 mg de tetratiomolibdato de bis-colina una vez al día; si el paciente tomaba una dosis de 45 mg una vez al día de tetratiomolibdato de bis-colina, la dosis se reduce hasta 30 mg de tetratiomolibdato de bis-colina una vez al día; si el paciente tomaba una dosis de 60 mg una vez al día de tetratiomolibdato de bis-colina, la dosis se reduce hasta 45 mg de tetratiomolibdato de bis-colina una vez al día; si el paciente tomaba una dosis de 75 mg una vez al día de tetratiomolibdato de bis-colina, la dosis se reduce hasta 60 mg de tetratiomolibdato de bis-colina una vez al día; o si el paciente tomaba una dosis de 90 mg una vez al día de tetratiomolibdato de bis-colina, la dosis se reduce hasta 75 mg de tetratiomolibdato de bis-colina una vez al día.
En realizaciones adicionales, la divulgación se refiere a una composición farmacéutica de tetratiomolibdato de biscolina para su uso en el tratamiento de la enfermedad de Wilson en un paciente que recibe tratamiento con tetratiomolibdato de bis-colina, en donde la dosis diaria de tetratiomolibdato de bis-colina se reduce cuando dicho paciente presenta un nivel de alanina aminotransferasa (ALT) al menos dos veces menos dos veces el límite superior de la normalidad (ULN). En todavía otras realizaciones, si el paciente tomaba una dosis de 15 mg una vez al día de tetratiomolibdato de bis-colina, la dosis se reduce hasta 15 mg de tetratiomolibdato de bis-colina cada dos días; si el paciente tomaba una dosis de 30 mg una vez al día de tetratiomolibdato de bis-colina, la dosis se reduce hasta 15 mg de tetratiomolibdato de bis-colina una vez al día; si el paciente tomaba una dosis de 45 mg una vez al día de tetratiomolibdato de bis-colina, la dosis se reduce hasta 30 mg de tetratiomolibdato de bis-colina una vez al día; si el paciente tomaba una dosis de 60 mg una vez al día de tetratiomolibdato de bis-colina, la dosis se reduce hasta 45 mg de tetratiomolibdato de bis-colina una vez al día; si el paciente tomaba una dosis de 75 mg una vez al día de tetratiomolibdato de bis-colina, la dosis se reduce hasta 60 mg de tetratiomolibdato de bis-colina una vez al día; o si el paciente tomaba una dosis de 90 mg una vez al día de tetratiomolibdato de bis-colina, la dosis se reduce hasta 75 mg de tetratiomolibdato de bis-colina una vez al día. En aún otras realizaciones, el ULN es 30-45 UI/ml. En realizaciones más particulares, el ULN es 34 UI/ml. En todavía realizaciones más particulares, el ULN es 40 UI/ml.
En realizaciones particulares, la divulgación se refiere a una composición farmacéutica de tetratiomolibdato de biscolina para su uso en el tratamiento de la enfermedad de Wilson en un paciente que recibe tratamiento con tetratiomolibdato de bis-colina, en donde la dosis diaria de tetratiomolibdato de bis-colina se reduce cuando dicho paciente presenta un nivel de hemoglobina del 70 % o inferior al nivel de hemoglobina presentado cuando se empieza el tratamiento con tetratiomolibdato de bis-colina. En ciertas realizaciones, si el paciente tomaba una dosis de 15 mg una vez al día de tetratiomolibdato de bis-colina, la dosis se reduce hasta 15 mg de tetratiomolibdato de bis-colina cada dos días; si el paciente tomaba una dosis de 30 mg una vez al día de tetratiomolibdato de bis-colina, la dosis se reduce hasta 15 mg de tetratiomolibdato de bis-colina una vez al día; si el paciente tomaba una dosis de 45 mg una vez al día de tetratiomolibdato de bis-colina, la dosis se reduce hasta 30 mg de tetratiomolibdato de bis-colina una vez al día; si el paciente tomaba una dosis de 60 mg una vez al día de tetratiomolibdato de bis-colina, la dosis se reduce hasta 45 mg de tetratiomolibdato de bis-colina una vez al día; si el paciente tomaba una dosis de 75 mg una vez al día de tetratiomolibdato de bis-colina, la dosis se reduce hasta 60 mg de tetratiomolibdato de bis-colina una vez al día; o si el paciente tomaba una dosis de 90 mg una vez al día de tetratiomolibdato de bis-colina, la dosis se reduce hasta 75 mg de tetratiomolibdato de bis-colina una vez al día.
En realizaciones particulares, la divulgación se refiere a una composición farmacéutica de tetratiomolibdato de biscolina para su uso en el tratamiento de la enfermedad de Wilson en un paciente que recibe tratamiento con tetratiomolibdato de bis-colina, en donde la dosis diaria de tetratiomolibdato de bis-colina se reduce cuando dicho paciente presenta un nivel de plaquetas del 70 % o inferior al nivel de plaquetas presentado cuando se empieza el tratamiento con tetratiomolibdato de bis-colina. En ciertas realizaciones, si el paciente tomaba una dosis de 15 mg una vez al día de tetratiomolibdato de bis-colina, la dosis se reduce hasta 15 mg de tetratiomolibdato de bis-colina cada dos días; si el paciente tomaba una dosis de 30 mg una vez al día de tetratiomolibdato de bis-colina, la dosis se reduce hasta 15 mg de tetratiomolibdato de bis-colina una vez al día; si el paciente tomaba una dosis de 45 mg una vez al día de tetratiomolibdato de bis-colina, la dosis se reduce hasta 30 mg de tetratiomolibdato de bis-colina una vez al día; si el paciente tomaba una dosis de 60 mg una vez al día de tetratiomolibdato de bis-colina, la dosis se reduce hasta 45 mg de tetratiomolibdato de bis-colina una vez al día; si el paciente tomaba una dosis de 75 mg una vez al día de tetratiomolibdato de bis-colina, la dosis se reduce hasta 60 mg de tetratiomolibdato de bis-colina una vez al día; o si el paciente tomaba una dosis de 90 mg una vez al día de tetratiomolibdato de bis-colina, la dosis se reduce hasta 75 mg de tetratiomolibdato de bis-colina una vez al día.
En realizaciones particulares, la divulgación se refiere a una composición farmacéutica de tetratiomolibdato de biscolina para su uso en el tratamiento de la enfermedad de Wilson en un paciente que recibe tratamiento con tetratiomolibdato de bis-colina, en donde la dosis diaria de tetratiomolibdato de bis-colina se reduce cuando dicho paciente presenta un nivel de neutrófilos del 70 % o inferior al nivel de neutrófilos presentado cuando se empieza el
tratamiento con tetratiomolibdato de bis-colina. En ciertas realizaciones, si el paciente tomaba una dosis de 15 mg una vez al día de tetratiomolibdato de bis-colina, la dosis se reduce hasta 15 mg de tetratiomolibdato de bis-colina cada dos días; si el paciente tomaba una dosis de 30 mg una vez al día de tetratiomolibdato de bis-colina, la dosis se reduce hasta 15 mg de tetratiomolibdato de bis-colina una vez al día; si el paciente tomaba una dosis de 45 mg una vez al día de tetratiomolibdato de bis-colina, la dosis se reduce hasta 30 mg de tetratiomolibdato de bis-colina una vez al día; si el paciente tomaba una dosis de 60 mg una vez al día de tetratiomolibdato de bis-colina, la dosis se reduce hasta 45 mg de tetratiomolibdato de bis-colina una vez al día; si el paciente tomaba una dosis de 75 mg una vez al día de tetratiomolibdato de bis-colina, la dosis se reduce hasta 60 mg de tetratiomolibdato de bis-colina una vez al día; o si el paciente tomaba una dosis de 90 mg una vez al día de tetratiomolibdato de bis-colina, la dosis se reduce hasta 75 mg de tetratiomolibdato de bis-colina una vez al día.
En ciertas realizaciones, la divulgación se refiere a una composición farmacéutica de tetratiomolibdato de bis-colina para su uso en el tratamiento de la enfermedad de Wilson en un paciente que recibe tratamiento con tetratiomolibdato de bis-colina, en donde la dosis diaria de tetratiomolibdato de bis-colina se interrumpe cuando dicho paciente presenta un nivel de alanina aminotransferasa (ALT) superior a cinco veces el nivel de ALT presentado cuando se empieza el tratamiento con tetratiomolibdato de bis-colina, y una vez dicho paciente presenta un nivel de ALT inferior a dos veces el nivel presentado cuando se empieza el tratamiento con tetratiomolibdato de bis-colina, el tratamiento se reanuda con 15 mg de tetratiomolibdato de bis-colina cada dos días si el paciente estaba tomando una dosis de 15 mg cada dos días de tetratiomolibdato de bis-colina antes de la interrupción del tetratiomolibdato de bis-colina, o 15 mg de tetratiomolibdato de bis-colina diariamente si el paciente estaba tomando una dosis de 15 a 90 mg una vez al día de tetratiomolibdato de bis-colina antes de la interrupción del tetratiomolibdato de bis-colina.
En ciertas realizaciones, la divulgación se refiere a una composición farmacéutica de tetratiomolibdato de bis-colina para su uso en el tratamiento de la enfermedad de Wilson en un paciente que recibe tratamiento con tetratiomolibdato de bis-colina, en donde la dosis diaria de tetratiomolibdato de bis-colina se interrumpe cuando dicho paciente presenta un nivel de alanina aminotransferasa (ALT) superior a 200 UI/ml, y una vez dicho paciente presenta un nivel de ALT inferior a dos veces el nivel presentado cuando se empieza el tratamiento con tetratiomolibdato de bis-colina, el tratamiento se reanuda con 15 mg de tetratiomolibdato de bis-colina cada dos días si el paciente estaba tomando una dosis de 15 mg cada dos días de tetratiomolibdato de bis-colina antes de la interrupción del tetratiomolibdato de biscolina, o 15 mg de tetratiomolibdato de bis-colina diariamente si el paciente estaba tomando una dosis de 15 a 90 mg una vez al día de tetratiomolibdato de bis-colina antes de la interrupción del tetratiomolibdato de bis-colina.
En ciertas realizaciones, la divulgación se refiere a una composición farmacéutica de tetratiomolibdato de bis-colina para su uso en el tratamiento de la enfermedad de Wilson en un paciente que recibe tratamiento con tetratiomolibdato de bis-colina, en donde la dosis diaria de tetratiomolibdato de bis-colina se interrumpe cuando dicho paciente presenta un nivel de hemoglobina inferior a 8 g/dl, y una vez dicho paciente presenta un nivel de hemoglobina equivalente al presentado cuando se empieza el tratamiento con tetratiomolibdato de bis-colina, el tratamiento se reanuda con 15 mg de tetratiomolibdato de bis-colina cada dos días si el paciente estaba tomando una dosis de 15 mg cada dos días de tetratiomolibdato de bis-colina antes de la interrupción del tetratiomolibdato de bis-colina, o 15 mg de tetratiomolibdato de bis-colina diariamente si el paciente estaba tomando una dosis de 15 a 90 mg una vez al día de tetratiomolibdato de bis-colina antes de la interrupción del tetratiomolibdato de bis-colina.
En ciertas realizaciones, la divulgación se refiere a una composición farmacéutica de tetratiomolibdato de bis-colina para su uso en el tratamiento de la enfermedad de Wilson en un paciente que recibe tratamiento con tetratiomolibdato de bis-colina, en donde la dosis diaria de tetratiomolibdato de bis-colina se interrumpe cuando dicho paciente presenta un nivel de plaquetas inferior a 30.000 gl, y una vez dicho paciente presenta un nivel de plaquetas equivalente al presentado cuando se empieza el tratamiento con tetratiomolibdato de bis-colina, el tratamiento se reanuda con 15 mg de tetratiomolibdato de bis-colina cada dos días si el paciente estaba tomando una dosis de 15 mg cada dos días de tetratiomolibdato de bis-colina antes de la interrupción del tetratiomolibdato de bis-colina, o 15 mg de tetratiomolibdato de bis-colina diariamente si el paciente estaba tomando una dosis de 15 a 90 mg una vez al día de tetratiomolibdato de bis-colina antes de la interrupción del tetratiomolibdato de bis-colina.
En ciertas realizaciones, la divulgación se refiere a una composición farmacéutica de tetratiomolibdato de bis-colina para su uso en el tratamiento de la enfermedad de Wilson en un paciente que recibe tratamiento con tetratiomolibdato de bis-colina, en donde la dosis diaria de tetratiomolibdato de bis-colina se interrumpe cuando dicho paciente presenta niveles de neutrófilos inferiores a 1,0 x 103/gl, y una vez dicho paciente presenta un nivel de neutrófilos equivalente al presentado cuando se empieza el tratamiento con tetratiomolibdato de bis-colina, el tratamiento se reanuda con 15 mg de tetratiomolibdato de bis-colina cada dos días si el paciente estaba tomando una dosis de 15 mg cada dos días de tetratiomolibdato de bis-colina antes de la interrupción del tetratiomolibdato de bis-colina, o 15 mg de tetratiomolibdato de bis-colina diariamente si el paciente estaba tomando una dosis de 15 a 90 mg una vez al día de tetratiomolibdato de bis-colina antes de la interrupción del tetratiomolibdato de bis-colina.
En ciertas realizaciones, la divulgación se refiere a una composición farmacéutica de tetratiomolibdato de bis-colina para su uso en el tratamiento de la enfermedad de Wilson en un paciente que recibe tratamiento con tetratiomolibdato de bis-colina, en donde la dosis diaria de tetratiomolibdato de bis-colina se interrumpe cuando dicho paciente presenta un nivel de bilirrubina superior a 2,4 mg/dl y niveles de alanina aminotransferasa (ALT) superiores a 120 UI/ml, y una
vez dicho paciente presenta un nivel de bilirrubina por debajo del límite superior de la normalidad, el tratamiento se reanuda con 15 mg de tetratiomolibdato de bis-colina cada dos días si el paciente estaba tomando una dosis de 15 mg cada dos días de tetratiomolibdato de bis-colina antes de la interrupción del tetratiomolibdato de bis-colina, o 15 mg de tetratiomolibdato de bis-colina diariamente si el paciente estaba tomando una dosis de 15 a 90 mg una vez al día de tetratiomolibdato de bis-colina antes de la interrupción del tetratiomolibdato de bis-colina.
En ciertas realizaciones, la divulgación se refiere a una composición farmacéutica de tetratiomolibdato de bis-colina para su uso en el tratamiento de la enfermedad de Wilson en un paciente que recibe tratamiento con tetratiomolibdato de bis-colina, en donde la dosis diaria de tetratiomolibdato de bis-colina se interrumpe cuando dicho paciente presenta un nivel de bilirrubina superior a dos veces el límite superior de la normalidad para bilirrubina y niveles de alanina aminotransferasa (ALT) superiores a tres veces el ULN para ALT, y una vez dicho paciente presenta un nivel de bilirrubina por debajo del límite superior de la normalidad, el tratamiento se reanuda con 15 mg de tetratiomolibdato de bis-colina cada dos días si el paciente estaba tomando una dosis de 15 mg cada dos días de tetratiomolibdato de bis-colina antes de la interrupción del tetratiomolibdato de bis-colina, o 15 mg de tetratiomolibdato de bis-colina diariamente si el paciente estaba tomando una dosis de 15 a 90 mg una vez al día de tetratiomolibdato de bis-colina antes de la interrupción del tetratiomolibdato de bis-colina.
En ciertas realizaciones, la divulgación proporciona una composición que comprende tetratiomolibdato de bis-colina para su uso en un método de tratamiento de la enfermedad de Wilson en un paciente. En algunas realizaciones, la composición se define según cualquiera de las composiciones desveladas en el presente documento. En algunas realizaciones, la composición es para su uso en cualquiera de los métodos desvelados en el presente documento. Según la presente invención , el paciente está en un estado en ayunas.
En ciertas realizaciones, la divulgación se refiere a una composición farmacéutica de tetratiomolibdato de bis-colina para su uso en el tratamiento de la enfermedad de Wilson en donde la composición se administra en el estado en ayunas. En realizaciones adicionales, la composición farmacéutica comprende 15 mg de tetratiomolibdato de biscolina. En todavía otras realizaciones la composición farmacéutica es un comprimido con cubierta entérica.
En ciertas realizaciones, la divulgación se refiere a los métodos anteriores para tratar enfermedad de Wilson que comprende administrar tetratiomolibdato de bis-colina a pacientes con enfermedad de Wilson y modificar el tetratiomolibdato de bis-colina para pacientes con enfermedad de Wilson en donde el paciente tiene cirrosis.
En ciertas realizaciones, la divulgación se refiere a los métodos anteriores para tratar enfermedad de Wilson que comprende administrar tetratiomolibdato de bis-colina a pacientes con enfermedad de Wilson y modificar el tetratiomolibdato de bis-colina para pacientes con enfermedad de Wilson en donde el paciente no tiene cirrosis.
En realizaciones particulares, la divulgación se refiere a los métodos anteriores para tratar enfermedad de Wilson que comprende administrar tetratiomolibdato de bis-colina a pacientes con enfermedad de Wilson y modificar el tetratiomolibdato de bis-colina para pacientes con enfermedad de Wilson, en donde el paciente presenta uno o más fenotipos de la enfermedad de Wilson seleccionados de temblor total; marcha total; distonía; agilidad y coordinación de las extremidades; y rigidez; preferentemente en donde el paciente presenta temblor total o agilidad y coordinación de las extremidades, o ambos. En realizaciones adicionales, a) el fenotipo de temblor total comprende una o más manifestaciones neurológicas de la enfermedad de Wilson según la parte III de la Escala unificada de valoración de la enfermedad de Wilson (UWDRS) seleccionada de: temblor de reposo; temblor de la cabeza; brazos - temblor postural y temblor de aleteo; temblor postural - piernas; y temblor de la mandíbula; b) el fenotipo de marcha total comprende una o más manifestaciones neurológicas de la enfermedad de Wilson según la parte III de la Escala unificada de valoración de la enfermedad de Wilson (UWDRS) seleccionadas de: levantarse de la silla; postura - distonía del tronco, ataxia de la postura y parkinsonismo; marcha - distonía de las piernas, ataxia y parkinsonismo; c) el fenotipo de distonía comprende una o más manifestaciones neurológicas de la enfermedad de Wilson según la parte III de la Escala unificada de valoración de la enfermedad de Wilson (UWDRS) seleccionadas de: distonía bucomandibular; distonía cervical; distonía de brazos y manos; distonía del tronco; y marcha - distonía de las piernas; d) el fenotipo de agilidad y coordinación de las extremidades comprende una o más manifestaciones neurológicas de la enfermedad de Wilson según la parte III de la Escala unificada de valoración de la enfermedad de Wilson (UWDRS) seleccionadas de: golpeteo con los dedos; movimientos rápidos y alternantes de las manos; caligrafía; prueba de dedo-nariz; y agilidad de las piernas; e) el fenotipo de rigidez comprende una o más manifestaciones neurológicas de la enfermedad de Wilson según la parte III de la Escala unificada de valoración de la enfermedad de Wilson (UWDRS) seleccionadas de brazos, piernas y cuello.
En todavía otras realizaciones, a) el fenotipo de temblor total se caracteriza por una puntuación de la parte III de UWDRS de 30-45; b) el fenotipo de marcha total se caracteriza por una puntuación de la parte III de UWDRS de 20 32; c) el fenotipo de distonía se caracteriza por una puntuación de la parte III de UWDRS de 15-28; d) el fenotipo de agilidad y coordinación de las extremidades se caracteriza por una puntuación de la parte III de UWDRS de 20-36; y e) el fenotipo de rigidez se caracteriza por una puntuación de la parte III de UWDRS de 10-20.
En realizaciones particulares, la divulgación se refiere a los métodos anteriores para tratar la enfermedad de Wilson que comprende administrar tetratiomolibdato de bis-colina a pacientes con enfermedad de Wilson y modificar el
tetratiomolibdato de bis-colina para pacientes con enfermedad de Wilson, en donde el paciente presenta una manifestación neurológica de la enfermedad de Wilson según la parte III de la Escala unificada de valoración de la enfermedad de Wilson (UWDRS) seleccionada de caligrafía, agilidad de las piernas, y una combinación de las mismas. En realizaciones adicionales, el paciente presenta una puntuación para caligrafía según la parte III de UWDRS de 2 4; una puntuación para agilidad de las piernas según la parte III de UWDRS de 2-8; o una puntuación para caligrafía y agilidad de las piernas según la parte III de UWDRS de 4-12. En todavía otras realizaciones, el paciente presenta una mejoría en una o más manifestaciones neurológicas de la enfermedad de Wilson según la parte III de UWDRS tras la administración de la composición. En aún otras realizaciones, el paciente presenta una reducción en la puntuación de la parte III de UWDRS de uno o más de: a) 5-25 para fenotipo de temblor total; b) 5-20 para el fenotipo de marcha total; c) 5-15 para el fenotipo de distonía; d) 5-20 para el fenotipo de agilidad y coordinación de las extremidades; y e) 5-15 para el fenotipo de rigidez.
En aún otras realizaciones, el paciente presenta uno o más de una reducción en la puntuación de la parte III de UWDRS para caligrafía de 1-3; una reducción en la puntuación de la parte III de UWDRS para agilidad de las piernas de 1-6; y una reducción en la puntuación de la parte III de UWDRS para caligrafía y agilidad de las piernas de 2-9.
En realizaciones particulares, la divulgación proporciona una composición como se describe en el presente documento para su uso en el tratamiento de la enfermedad de Wilson, en donde el paciente presenta uno o más fenotipos de la enfermedad de Wilson seleccionados de temblor total; marcha total; distonía; agilidad y coordinación de las extremidades; y rigidez; preferentemente en donde el paciente presenta temblor total o agilidad y coordinación de las extremidades o ambos. En realizaciones adicionales, a) el fenotipo de temblor total comprende una o más manifestaciones neurológicas de la enfermedad de Wilson según la parte III de la Escala unificada de valoración de la enfermedad de Wilson (UWDRS) seleccionadas de: temblor de reposo; temblor de la cabeza; brazos - temblor postural y temblor de aleteo; temblor postural - piernas; y temblor de la mandíbula; b) el fenotipo de marcha total comprende una o más manifestaciones neurológicas de la enfermedad de Wilson según la parte III de la Escala unificada de valoración de la enfermedad de Wilson (UWDRS) seleccionadas de: levantarse de la silla; postura - distonía del tronco, ataxia de la postura y parkinsonismo; marcha -distonía de las piernas, ataxia y parkinsonismo; c) el fenotipo de distonía comprende una o más manifestaciones neurológicas de la enfermedad de Wilson según la parte III de la Escala unificada de valoración de la enfermedad de Wilson (UWDRS) seleccionadas de: distonía bucomandibular; distonía cervical; distonía de brazos y manos; distonía del tronco; y marcha - distonía de las piernas; d) el fenotipo de agilidad y coordinación de las extremidades comprende una o más manifestaciones neurológicas de la enfermedad de Wilson según la parte III de la Escala unificada de valoración de la enfermedad de Wilson (UWDRS) seleccionadas de: golpeteo con los dedos; movimientos rápidos y alternantes de las manos; caligrafía; prueba de dedo-nariz; y agilidad de las piernas; e) el fenotipo de rigidez comprende una o más manifestaciones neurológicas de la enfermedad de Wilson según la parte III de la Escala unificada de valoración de la enfermedad de Wilson (UWDRS) seleccionadas de brazos, piernas y cuello.
En todavía otras realizaciones, a) el fenotipo de temblor total se caracteriza por una puntuación de la parte III de UWDRS de 30-45; b) el fenotipo de marcha total se caracteriza por una puntuación de la parte III de UWDRS de 20 32; c) el fenotipo de distonía se caracteriza por una puntuación de la parte III de UWDRS de 15-28; d) el fenotipo de agilidad y coordinación de las extremidades se caracteriza por una puntuación de la parte III de UWDRS de 20-36; y e) el fenotipo de rigidez se caracteriza por una puntuación de la parte III de UWDRS de 10-20.
En realizaciones particulares, la divulgación proporciona una composición como se describe en el presente documento para su uso en el tratamiento de la enfermedad de Wilson, en donde el paciente presenta una manifestación neurológica de la enfermedad de Wilson según la parte III de la Escala unificada de valoración de la enfermedad de Wilson (UWDRS) seleccionada de caligrafía, agilidad de las piernas, y una combinación de las mismas. En realizaciones adicionales, el paciente presenta una puntuación para caligrafía según la parte III de UWDRS de 2-4; una puntuación para agilidad de las piernas según la parte III de UWDRS de 2-8; o una puntuación para caligrafía y agilidad de las piernas según la parte III de UWDRS de 4-12. En todavía otras realizaciones, el paciente presenta una mejoría en una o más manifestaciones neurológicas de la enfermedad de Wilson según la parte III de UWDRS tras la administración de la composición. En aún otras realizaciones el paciente presenta una reducción en la puntuación de la parte III de UWDRS de uno o más de: a) 5-25 para fenotipo de temblor total; b) 5-20 para el fenotipo de marcha total; c) 5-15 para el fenotipo de distonía; d) 5-20 para el fenotipo de agilidad y coordinación de las extremidades; y e) 5-15 para el fenotipo de rigidez.
En aún otras realizaciones, el paciente presenta uno o más de una reducción en la puntuación de la parte III de UWDRS para caligrafía de 1-3; una reducción en la puntuación de la parte III de UWDRS para agilidad de las piernas de 1-6; y una reducción en la puntuación de la parte III de UWDRS para caligrafía y agilidad de las piernas de 2-9.
En realizaciones particulares, la divulgación proporciona una composición como se describe en el presente documento para su uso en el tratamiento de la enfermedad de Wilson en donde el paciente tiene cirrosis
En realizaciones particulares, la divulgación proporciona una composición como se describe en el presente documento para su uso en el tratamiento de la enfermedad de Wilson en donde el paciente no tiene cirrosis.
Aspectos y realizaciones adicionales serán evidentes de la descripción detallada que sigue.
BREVE DESCRIPCIÓN DE LOS DIBUJOS
La FIG. 1 representa el perfil del ensayo.
La FIG. 2 representa cambios en las concentraciones de NCCcorregido con el tiempo. Los cambios desde el nivel basal son la media de los mínimos cuadrados (EE) para entre 19 y 25 pacientes en cada momento de tiempo. Un paciente interrumpió el tratamiento en la semana 23, pero dentro de la ventana especificada para la inclusión de la medición de NCCcorregido como un valor para la semana 24. Los valores de p son frente al nivel basal.
La FIG. 3 representa cambios en el estado de incapacidad y neurológico con el tiempo. Los cambios desde el nivel basal de las puntuaciones de la Escala unificada de valoración de la enfermedad de Wilson (parte II de UWDRS) (incapacidad; A) y las puntuaciones de la parte III (signos neurológicos; B) son la media de los mínimos cuadrados (EE) para entre 21 y 28 pacientes.
La FIG. 4 representa los niveles de NCCcorregido con un tratamiento con WTX101 una vez al día.
La FIG. 5 representa cambios en las medidas de la estabilidad hepática con un tratamiento con WTX101 una vez al día.
La FIG. 6 representa cambios en los signos de incapacidad y neurológicos con un tratamiento con WTX101 una vez al día.
La FIG.7 representa el número de acontecimientos adversos (AE) notificados durante los periodos principales y de prolongación.
Las FIG.8A-8B representa la media ± error estándar de las concentraciones plasmáticas de Mo total después de la administración de una dosis única de 60 mg (2 x 30 mg) de comprimidos EC de WTX101 en condiciones en ayunas (Tratamiento A) y posprandiales (Tratamiento B) y UC PPI en condiciones en ayunas (Tratamiento C). Los datos se representa en ejes lineales (FIG. 8A) y semilogarítmicos (FIG. 8B).
La FIG.9 representa los síntomas del estado neurológico de UWDRS (parte III) sufridos por al menos el 50 % de los pacientes en el nivel basal. Los datos se expresan como un porcentaje de pacientes en la población reclutada (N = 28).
Las FIG. 10A-10B representan puntuaciones de ítems del estado neurológico de UWDRS (parte III) para la población del estudio. La FIG. 10A muestra ítems con una puntuación máxima de 4. La FIG. 10A muestra ítems con una puntuación máxima de 8.
Las FIG. 11A-11B representa números de pacientes con cambios en las puntuaciones de ítems del estado neurológico de UWDRS (parte III) entre el nivel basal y la semana 24. Los datos son de todos los pacientes con datos para el ítem dado, que incluyen aquellos con puntuaciones de cero en el nivel basal (n = 22 en cada caso). La FIG. 11A muestra ítems con una puntuación máxima de 4. La FIG. 11A muestra ítems con una puntuación máxima de 8.
La FIG. 12 representa el flujo de pacientes para el estudio de prolongación.
La FIG. 13 representa los niveles de NCCcorr para pacientes con y sin cirrosis. Los niveles de NCC no se corrigieron en el nivel basal, ya que no se había recibido WTX101. BL, nivel basal; LLN, intervalo de referencia del límite inferior de la normalidad (0,8 gmol/l); ULN, intervalo de referencia del límite superior de la normalidad (2,3 gmol/l); NCCcorr, cobre no unido a ceruloplasmina corregido para la cantidad de cobre unido en el complejo de tetratiomolibdato-cobre-albúmina; EEM, error estándar de la media.
La FIG. 14 representa los niveles de ALT para pacientes con y sin cirrosis. ALT, alanina aminotransferasa; BL, nivel basal; EEM, error estándar de la media
La FIG. 15 representa la puntuación de MELD (intensidad de enfermedad hepática; intervalo de puntuación, 6-40) para pacientes con y sin cirrosis. BL, nivel basal; MELD, modelo para enfermedad hepática terminal; EEM, error estándar de la media
La FIG. 16 representa la puntuación de Nazer modificada (índice pronóstico; intervalo de puntuación, 0-20) para pacientes con y sin cirrosis.
La FIG. 17A representa los niveles de albúmina para pacientes con y sin cirrosis. La FIG. 17B representa la relación normalizada internacional para pacientes con y sin cirrosis. BL, nivel basal; INR, relación normalizada internacional; EEM, error estándar de la media
La FIG. 18 representa el tiempo de coagulación de la sangre para pacientes con y sin cirrosis. BL, nivel basal; INR, relación normalizada internacional; EEM, error estándar de la media
Las FIG. 19 representan UWDRS para la parte II (mide la incapacidad basada en las actividades de la vida diaria notificadas por el paciente; intervalo de puntuación, 0-40) (FIG. 19A) y la parte III (mide el estado neurológico como se evalúa por un profesional clínico (intervalo de puntuación; 0-143) (FIG. 19B). Mayores puntuaciones indican peor afección.
DESCRIPCIÓN DETALLADA
Ciertos aspectos de la presente divulgación se basan en el sorprendente descubrimiento de que se pueden reducir los acontecimientos adversos asociados al tratamiento de la enfermedad de Wilson administrando una forma farmacéutica de 15 mg de tetratiomolibdato de bis-colina o modificando una dosis diaria de tetratiomolibdato de biscolina para controlar la función hepática anormal en el paciente. La sal de amonio de tetratiomolibdato se administra, en general, en dosis más grandes, por ejemplo, 90 a 220 mg por día. Brewer et al. Initial Therapy of Patients with Wilson's Disease with Tetrathiomolybdate. Arch. Neurol. 48:42-47 (1991). Según la presente divulgación, la administración de tetratiomolibdato de bis-colina a la misma dosis, o similar, a la dosis de tetratiomolibdato de amonio, conocida en general en la técnica, está asociada con acontecimientos adversos que reducen los métodos de la presente divulgación.
La divulgación proporciona métodos de administración de una dosis terapéuticamente eficaz de tetratiomolibdato de bis-colina a un paciente que ha presentado resultados de pruebas anormales después de que el paciente haya sido tratado con tetratiomolibdato de bis-colina. Debido a que las anomalías en la función hepática pueden ser indicativas de una lesión hepática inducida por el fármaco (hepatotoxicidad), es importante determinar si las anomalías reflejan la lesión del hígado o simplemente indican una toxicidad limitada que se resolverá con el tiempo mientras se sigue tomando el fármaco. Según la presente divulgación, incluso pacientes que presentan una función hepática anormal pueden seguir tomando el tetratiomolibdato de bis-colina a la misma dosis, o seguir tomando el tetratiomolibdato de bis-colina a una dosis reducida, opcionalmente después de la interrupción del tetratiomolibdato de bis-colina durante un periodo de tiempo. Esta pauta de administración tiene la ventaja de maximizar el tiempo con la dosis de fármaco objetivo completa y, por lo tanto, las posibilidades de un efecto terapéutico beneficioso.
Los métodos de la divulgación incluyen opcionalmente identificar la función hepática anormal en un paciente que recibe tetratiomolibdato de bis-colina, y monitorizar los resultados de las pruebas del hígado en un paciente que recibe una dosis reducida de tetratiomolibdato de bis-colina. En cualquiera de los métodos descritos en el presente documento, las ALT pueden estar elevadas, por ejemplo hasta un nivel superior a 34 UI/ml o 64 UI/ml o 170 UI/ml antes de la reducción de dosis. Alternativamente, la hemoglobina puede estar reducida, o las plaquetas pueden estar reducidas, o los neutrófilos pueden estar reducidos antes de la reducción de dosis.
Los métodos de la divulgación incluyen opcionalmente la medición de los niveles de cobre en el cuerpo. Se conocen en la técnica diversos medios de medición de los niveles de cobre en el cuerpo. En una realización, las concentraciones de cobre libre en ultrafiltrado de suero o plasma se miden por espectrometría de masas inductivamente acoplada para ayudar con el diagnóstico y la monitorización de la enfermedad de Wilson. En otra realización, se determina la concentración libre en orina. En otra realización, se determina la secreción biliar de cobre por medición de la concentración de cobre en las heces. En otra realización, se determina el contenido de cobre del pelo. En una realización adicional, se determina la cantidad de cobre libre en suero como la cantidad de cobre no unido que circula en la sangre, que es cobre no unido por ceruloplasmina. Se entenderá que éste es el cobre que está libre para ser acumulado en el hígado y otros órganos. En una realización preferida, el cobre no unido por ceruloplasmina o NCC se determina por espectrometría de masas inductivamente acoplada u otros métodos conocidos en la técnica. En cualquiera de los métodos descritos en el presente documento, el NCCcorregido puede ser elevado, por ejemplo superior a 2,3 pm/l.
Aunque los intervalos para los resultados de las pruebas referenciadas anteriormente y los niveles en individuos sanos pueden variar dependiendo de las condiciones de las pruebas y la metodología de laboratorio, en general, se sabe que los individuos sanos presentan los siguientes: niveles de ALT en el intervalo de 6-34 U/l, 9-34 U/l o 6-41 U/l; hemoglobina en el intervalo de 11,6-16,4 g/dl, 13,6-18,0 g/dl o 12,0-16,0 g/dl; plaquetas en el intervalo de 140 400 x 103/pl; neutrófilos en el intervalo de 1,96-7,23 x 103/pl; y bilirrubina en el intervalo de 0,2-1,2 mg/dl o 0,10-1,10 mg/dl. Información adicional sobre los métodos de laboratorio clínico para el diagnóstico y el tratamiento de la enfermedad de Wilson se proporciona en la European Association for the Study of the Liver (EASL) Clinical Practice Guidelines: Wilson's Disease; J. Hepatology 56:671-685 (2012).
En algunas realizaciones, la divulgación se refiere a métodos de tratamiento de la enfermedad de Wilson en un paciente administrando 15 mg de tetratiomolibdato de bis-colina una vez al día o una vez cada día. La dosis de una
vez al día pueden ser administrada en forma de una dosis única, o dos dosis, opcionalmente dos dosis divididas igualmente, o tres o cuatro o cinco dosis. La dosis se puede administrar por vía oral, por vía intravenosa, por vía intramuscular o en cualquier otro modo conocido en la técnica.
En algunas realizaciones, la divulgación se refiere a métodos de tratamiento de la enfermedad de Wilson en un paciente administrando desde 30 hasta 90 mg de tetratiomolibdato de bis-colina diariamente, donde el paciente tiene uno o más de los siguientes: un NCCcorregido superior a 2,3 gm/l, nivel de alanina aminotransferasa (ALT) inferior a 80 Ul/ml, hemoglobina superior a 8 g/dl, plaquetas superiores a 30.000/gl, o neutrófilos superiores a 1.000/gl o 1 x 103/gl. El paciente puede tener NCCcorregido superior a 2,3 gm/l y nivel de alanina aminotransferasa (ALT) inferior a 80 Ul/ml, o NCCcorregido superior a 2,3 gm/l y hemoglobina superior a 8 g/dl, o cualquier otra combinación de dos o más de estos parámetros. En algunas realizaciones, se medirá uno o más de los parámetros. En una realización, se mide NCCcorregido. En otra realización, se miden dos o más de los parámetros. En otra realización, se miden todos los parámetros.
En algunas realizaciones, la divulgación se refiere a métodos de tratamiento de la enfermedad de Wilson en un paciente administrando desde 30 hasta 90 mg de tetratiomolibdato de bis-colina diariamente, donde el paciente tiene uno o más de los siguientes: un NCCcorregido superior a 2,0, 2,1, 2,2, 2,3, 2,4, 2,5 gm/l o el límite superior de la normalidad (ULN) de NCCcorregido, nivel de alanina aminotransferasa (ALT) inferior a 50, 55, 60, 65, 70, 75, 80, 85, 90, 95, 100 Ul/ml o dos veces el límite superior de la normalidad (ULN) de ALT, hemoglobina superior a 6, 7, 8, 9, o 10 g/dl, plaquetas superior a 20.000, 25.000, 30.000, 35.000 o 40.000/gl, o neutrófilos superior a 0,5 x 103, 1 x 103, 1,5 x 103, 2 x 103 o 2,5 x 103/gl. En ciertas realizaciones, el paciente puede combinar dos o más de estos parámetros. En algunas realizaciones, se medirá uno o más de los parámetros. En otra realización, se mide NCCcorregido. En realizaciones adicionales, se miden dos o más de los parámetros. En aún otras realizaciones, se miden todos.
En algunas realizaciones, la divulgación se refiere a métodos de modificación de la administración de tetratiomolibdato de bis-colina para un paciente con enfermedad de Wilson que recibe tratamiento con tetratiomolibdato de bis-colina aumentando la dosis diaria de tetratiomolibdato de bis-colina. En algunas realizaciones de la divulgación, la dosis de tetratiomolibdato de bis-colina se aumenta en un paciente que presenta ciertos resultados de las pruebas. En algunas realizaciones, la dosis diaria de tetratiomolibdato de bis-colina se aumenta en incrementos de 15 mg. En algunas realizaciones, la divulgación la dosis diaria se aumenta cuando el paciente tiene uno o más de los siguientes: un NCCcorregido superior a 2,3 gm/l, nivel de alanina aminotransferasa (ALT) inferior a 80 Ul/ml, hemoglobina superior a 8 g/dl, plaquetas superior a 30.000/gl, o neutrófilos superior a 1.000/gl o 1 x 103/gl. En realizaciones adicionales, la dosis se aumenta cuando el paciente tiene uno o más de los siguientes: un NCCcorregido superior a 2,0, 2,1,2,2, 2,3, 2,4, 2,5 gm/l o el límite superior de la normalidad (ULN) de NCCcorregido, nivel de alanina aminotransferasa (ALT) inferior a 50, 55, 60, 65, 70, 75, 80, 85, 90, 95, 100 Ul/ml o dos veces el límite superior de la normalidad (ULN) de ALT, hemoglobina superior a 6, 7, 8, 9 o 10 g/dl, plaquetas superior a 20.000, 25.000, 30.000, 35.000 o 40.000/gl, o neutrófilos superior a 0,5 x 103, 1 x 103, 1,5 x 103, 2 x 103 o 2,5 x 103/gl. El paciente puede tener NCCcorregido superior a 2,3 gm/l y nivel de alanina aminotransferasa (ALT) inferior a 80 Ul/ml, o NCCcorregido superior a 2,3 gm/l y hemoglobina superior a 8 g/dl, o cualquier otra combinación de dos o más de estos parámetros. En algunas realizaciones, se medirá uno o más de los parámetros. En una realización, se mide NCCcorregido. En otra realización, se miden dos o más de los parámetros. En otra realización, se miden todos los parámetros. En ciertas realizaciones, la dosis diaria se aumenta por incrementos de 15 mg de tetratiomolibdato de bis-colina. En realizaciones particulares, la dosis diaria se aumenta durante el inicio del tratamiento, normalmente los primeros 3, 4, 5 o 6 meses de tratamiento con tetratiomolibdato de bis-colina. En ciertos aspectos de la divulgación, la dosis de un paciente se aumenta una vez. En aspectos adicionales de la divulgación, la dosis de un paciente se aumenta dos veces. En todavía aspectos adicionales de la divulgación, la dosis de un paciente se aumenta tres o más veces.
En algunas realizaciones, la divulgación se refiere a métodos de modificación de la administración de tetratiomolibdato de bis-colina a un paciente con enfermedad de Wilson que recibe tratamiento con tetratiomolibdato de bis-colina disminuyendo la dosis diaria de tetratiomolibdato de bis-colina. En algunas realizaciones, la dosis diaria de tetratiomolibdato de bis-colina se disminuye en incrementos de 15 mg. En una realización, la dosis diaria se disminuye administrando la dosis cada dos días. En otra realización, la dosis diaria se disminuye administrando 15 mg cada dos días de manera que el paciente reciba en promedio 7,5 mg de tetratiomolibdato de bis-colina por día.
En un aspecto, la presente divulgación proporciona además métodos de disminución de la dosis diaria de tetratiomolibdato de bis-colina en un paciente que presenta un resultado de prueba anormal. En ciertos aspectos de la divulgación, la administración de tetratiomolibdato de bis-colina se interrumpe temporalmente cuando un paciente presenta ciertos resultados de pruebas anormales, y se reanuda, opcionalmente con una dosis menor, cuando el paciente presenta resultados de pruebas mejorados. En una realización, la prueba es una prueba de la función hepática. Se puede emplear cualquiera de las pruebas de la función hepática conocidas en la técnica. En una realización, se usan los niveles de alanina aminotransferasa (ALT) o de bilirrubina. En una realización, la prueba es una prueba de supresión de la médula ósea causada por una eliminación de cobre excesiva a largo plazo que conduce a citopenia. En una realización, se puede usar como prueba el nivel de hemoglobina, el nivel de plaquetas o el nivel de neutrófilos. En una realización, se usan dos o más pruebas test resultados. En una realización, varios pruebas se usan. En realizaciones particulares, la dosis se reducen cuando dos consecutive test resultados are anormal.
En ciertas realizaciones, la dosis diaria de tetratiomolibdato de bis-colina en un paciente que presenta un resultado de prueba anormal se reduce por 15 mg, de forma que la dosis de un paciente que toma 15 mg de tetratiomolibdato de bis-colina diariamente antes del resultado de prueba anormal disminuiría hasta 15 mg de tetratiomolibdato de biscolina cada dos días, la dosis de un paciente que toma 30 mg de tetratiomolibdato de bis-colina diariamente antes del resultado de prueba anormal disminuiría hasta 15 mg diariamente, la dosis de un paciente que toma 45 mg de tetratiomolibdato de bis-colina diariamente antes del resultado de prueba anormal disminuiría hasta 30 mg diariamente, la dosis de un paciente que toma 60 mg de tetratiomolibdato de bis-colina diariamente antes del resultado de prueba anormal disminuiría hasta 45 mg diariamente, la dosis de un paciente que toma 75 mg de tetratiomolibdato de biscolina diariamente antes del resultado de prueba anormal disminuiría hasta 60 mg diariamente, y la dosis de un paciente que toma 90 mg de tetratiomolibdato de bis-colina diariamente antes del resultado de prueba anormal disminuiría hasta 75 mg diariamente. En otras realizaciones, la dosis diaria de tetratiomolibdato de bis-colina en un paciente que presenta un resultado de prueba anormal se reduce la mitad. En otras realizaciones más, la dosis de tetratiomolibdato de bis-colina en un paciente que presenta un resultado de prueba anormal que toma 30-90 mg de tetratiomolibdato de bis-colina diariamente antes del resultado de prueba anormal se reducen hasta una dosis diaria de 15 mg de tetratiomolibdato de bis-colina, y la dosis de tetratiomolibdato de bis-colina en un paciente que presenta un resultado de prueba anormal que toma 15 mg de tetratiomolibdato de bis-colina diariamente antes del resultado de prueba anormal se reducen hasta una dosis de 15 mg de tetratiomolibdato de bis-colina cada dos días.
En realizaciones particulares, la administración de tetratiomolibdato de bis-colina se interrumpe temporalmente cuando un paciente presenta ciertos resultados de pruebas anormales y se reanuda cuando los resultados de pruebas cumplen un umbral particular. En ciertas realizaciones, la administración se interrumpe cuando dos resultados de pruebas consecutivos son anormales. En realizaciones adicionales, la administración se reanuda cuando dos resultados de pruebas consecutivos cumplen un umbral particular. En ciertas realizaciones, la dosis diaria de tetratiomolibdato de bis-colina se reanuda al nivel antes de la interrupción. En realizaciones adicionales, la dosis diaria de tetratiomolibdato de bis-colina se reanuda a un nivel de administración reducido como se ha descrito anteriormente. En aún otras realizaciones de la divulgación, la dosis diaria se reanuda a 15 mg de tetratiomolibdato de bis-colina. En todavía otras realizaciones de la divulgación, la dosis diaria se reanuda a 15 mg de tetratiomolibdato de bis-colina si el paciente estaba tomando 30-90 mg de tetratiomolibdato de bis-colina diariamente antes del resultado de prueba anormal. En incluso otras realizaciones de la divulgación, la dosis se reanuda a 15 mg de tetratiomolibdato de biscolina cada dos días si el paciente estaba tomando 15 mg de tetratiomolibdato de bis-colina diariamente antes del resultado de prueba anormal.
Un resultado de prueba anormal se puede definir en términos de un umbral establecido por encima y por debajo del cual se dice que la función hepática o la función de la médula ósea es normal. En un aspecto opcional, se define un límite superior de la normalidad (ULN) para un resultado de prueba. En un aspecto opcional, la dosis se modifica cuando un paciente presenta dos resultados de pruebas anormales consecutivos. La administración de tetratiomolibdato de bis-colina se puede modificar debido a una resultado de prueba superior al ULN, o dos veces el ULN, o tres veces el ULN, o cuatro veces el ULN, o cinco veces el ULN, o cualquier múltiplo del ULN, o debido a una prueba de la función hepática superior a cualquier múltiplo fraccionario de ULN entre uno, dos, tres, cuatro o cinco veces el ULN. En una realización, la administración diaria de tetratiomolibdato de bis-colina se reduce cuando el resultado de prueba es dos a cinco veces el ULN. En una realización, la administración diaria de tetratiomolibdato de bis-colina se interrumpe cuando el resultado de la prueba es mayor que cinco veces el ULN. En otra realización, la prueba de la función hepática es ALT. Opcionalmente, la administración de tetratiomolibdato de bis-colina se reanuda cuando la ALT es inferior a dos veces el ULN. Opcionalmente, la dosis diaria de tetratiomolibdato de bis-colina se reanuda a una dosis menor cuando la ALT es inferior a dos veces ULN. En una realización, el ULN y límite inferior de la normalidad (LLN) de ALT dependen del ensayo particular usado. En una realización, el ULN de ALT es 30-45 UI/ml o 30-33 UI/ml o 33-36 UI/ml o 36-39 UI/ml o 39-42 UI/ml o 42-45 UI/ml. En una realización, el ULN de ALT es 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44 o 45 UI/ml, o cualquier número fraccionario intermedio. En otra realización, el ULN de ALT es 34 UI/ml. En otra realización, el ULN de Alt es 40 UI/ml.
En otro aspecto opcional, la dosis diaria de tetratiomolibdato de bis-colina se modifica cuando un resultado del hígado disminuye por debajo de una medición basal de la prueba hecha antes de que empiece la administración de tetratiomolibdato de bis-colina. Opcionalmente, este nivel basal es específico del paciente. Opcionalmente, este nivel basal se determina por criterio médico. Opcionalmente, este nivel basal se determina por resultados de ensayo clínico. Opcionalmente, la dosis se modifica cuando un paciente presenta dos resultados de pruebas anormales consecutivos. En una realización, el umbral para modificar la dosis de tetratiomolibdato de bis-colina es el 50 % o 60 % o 70 % u 80 % o 90 % del nivel basal o cualquier porcentaje intermedio. En otra realización, el umbral es el 65 % o 70 % o 75 % del nivel basal. En una realización, el umbral es el 70 % del nivel basal. En un aspecto opcional, la dosis de tetratiomolibdato de bis-colina se modifica cuando el nivel de hemoglobina del paciente es inferior al 70 % de la hemoglobina basal para ese paciente. En otro aspecto opcional, el umbral es del 70 % de las plaquetas basales o 70 % de los neutrófilos basales.
En un aspecto adicional, la dosis de tetratiomolibdato de bis-colina se reduce o interrumpe temporalmente cuando el nivel de hemoglobina del paciente es inferior a 6, 7, 8, 9 o 10 g/dl. En aún otro aspecto, la dosis de tetratiomolibdato de bis-colina se reduce o interrumpe temporalmente cuando el nivel de hemoglobina del paciente es inferior a 8 g/dl. En otro aspecto, la dosis de tetratiomolibdato de bis-colina se reduce o interrumpe temporalmente cuando las
plaquetas son inferiores a 20.000, 25.000, 30.000, 35.000 o 40.000/pl. En otro aspecto más, la dosis de tetratiomolibdato de bis-colina se reduce o interrumpe temporalmente cuando las plaquetas son inferiores a 30.000/pl. En otro aspecto más, la dosis de tetratiomolibdato de bis-colina se reduce o interrumpe temporalmente cuando los neutrófilos son inferiores a 0,5 x 103, 1 x 103, 1,5 x 103, 2 x 103 o 2,5 x 103/pl. En otro aspecto más, la dosis de tetratiomolibdato de bis-colina se reduce o interrumpe temporalmente cuando los neutrófilos son inferiores a 1 x 103/pl. Opcionalmente, la dosis se modifica cuando un paciente presenta dos resultados de pruebas anormales consecutivos.
En realizaciones particulares, cuando la dosis de tetratiomolibdato de bis-colina se interrumpe temporalmente, la administración se reanuda cuando el paciente presenta uno o más de los siguientes: un nivel de hemoglobina igual o superior a 6, 7, 8, 9 o 10 g/dl; plaquetas superior o igual a 20.000, 25.000, 30.000, 35.000 o 40.000/pl; y/o neutrófilos superior o igual a 0,5 x 103, 1 x 103, 1,5 x 103, 2 x 103 o 2,5 x 103/pl. En realizaciones adicionales, cuando la dosis de tetratiomolibdato de bis-colina se interrumpe temporalmente, la administración se reanuda cuando el paciente presenta uno o más de los siguientes: un nivel de hemoglobina igual o superior a 8 g/dl; plaquetas superior o igual a 30.000/pl; y/o neutrófilos superior o igual a 1 x 103/pl. En realizaciones adicionales, cuando la dosis de tetratiomolibdato de biscolina se interrumpe temporalmente, la administración se reanuda cuando el paciente presenta niveles basales de uno o más de hemoglobina, plaquetas o neutrófilos.
Un experto entenderá que este recital de pruebas de la función hepática o de supresión de médula ósea pretende no ser limitante. Se puede realizar otra prueba de la función hepática o supresión de la médula ósea. Se pueden desarrollar y usar nuevas pruebas de la función hepática o supresión de la médula ósea en lugar de las pruebas de la función hepática desveladas en el presente documento.
En otro aspecto, la presente divulgación proporciona además métodos de disminución de la dosis diaria de tetratiomolibdato de bis-colina en un paciente que presenta empeoramiento neurológico. En una realización, el empeoramiento neurológico se evalúa usando la puntuación de la parte III de UWDRS. En una realización, se determina una parte III de UWDRS basal antes de la administración de tetratiomolibdato de bis-colina. En una realización, el empeoramiento neurológico se define como un aumento en la puntuación de la parte III de UWDRS con respecto al nivel basal de 1, 2, 3, 4, 5, 6, 7 u 8 puntos. En otra realización, el empeoramiento neurológico se define como un aumento en la puntuación de la parte III de UWDRS con respecto al nivel basal de 4, 5 o 6. En otra realización, empeoramiento neurológico se define como un aumento en la puntuación de la parte III de UWDRS con respecto al nivel basal de 4 cuando la parte III de UWDRS basal era inferior a 20. En otra realización, el empeoramiento neurológico se define como un aumento en la puntuación de la parte III de UWDRS con respecto al nivel basal de 6 cuando el nivel basal UWDRS Parte III era 20 o mayor. En un aspecto opcional, la modificación comprende la interrupción del tetratiomolibdato de bis-colina. En un aspecto opcional, la modificación comprende después de que dicho paciente no presente más empeoramiento neurológico, administrar una dosis modificada de tetratiomolibdato de bis-colina. En un cierto aspecto, un paciente no presenta más empeoramiento neurológico cuando se determina que se ha estabilizado la puntuación de dicho paciente de la parte III de UWDRS. En una realización, la dosis modificada es una dosis reducida. En algunas realizaciones, la dosis modificada es la mitad de la dosis diaria administrada antes de que el paciente presente empeoramiento neurológico. En otras realizaciones, la dosis modificada es inferior a la dosis diaria administrada antes de que el paciente presente empeoramiento neurológico, tal como 15 mg inferior a la dosis diaria administrada antes de que el paciente presente empeoramiento neurológico. En algunas realizaciones, si el paciente tomaba una dosis de 15 mg una vez al día de tetratiomolibdato de bis-colina, 15 mg de tetratiomolibdato de bis-colina cada dos días; si el paciente tomaba una dosis de 30 mg una vez al día de tetratiomolibdato de bis-colina, 15 mg de tetratiomolibdato de bis-colina una vez al día; si el paciente tomaba una dosis de 45 mg una vez al día de tetratiomolibdato de bis-colina, 15 a 30 mg de tetratiomolibdato de bis-colina una vez al día; si el paciente tomaba una dosis de 60 mg una vez al día de tetratiomolibdato de bis-colina, 30 mg de tetratiomolibdato de bis-colina una vez al día; si el paciente tomaba una dosis de 75 mg una vez al día de tetratiomolibdato de bis-colina, 30 a 45 mg de tetratiomolibdato de bis-colina una vez al día; o si el paciente tomaba una dosis de 90 mg una vez al día de tetratiomolibdato de bis-colina, 45 mg de tetratiomolibdato de bis-colina una vez al día.
En algunas realizaciones, la divulgación se refiere a aumentar o disminuir la dosis de tetratiomolibdato de bis-colina por un incremento fijo. En una realización, el método comprende administrar a un paciente un primer nivel de dosis que comprende aproximadamente 15 a aproximadamente 90 mg por día de tetratiomolibdato de bis-colina durante un periodo de tiempo, seguido de la administración de un segundo nivel de dosis que comprende al menos aproximadamente 15 mg por día menos de tetratiomolibdato de bis-colina que la cantidad de tetratiomolibdato de biscolina en el primer nivel de dosis durante un segundo periodo de tiempo. En una realización, el segundo nivel de dosis es 15 mg cada dos días. Nivel de dosis y dosis diaria se pueden usar indistintamente. Un nivel de dosis puede comprender una, dos, tres, cuatro, cinco o más dosis, administradas en diferentes momentos o a la misma hora del día. Una dosis puede ser opcionalmente un único comprimido o dos comprimidos. Opcionalmente, una dosis se puede proporcionar como un comprimido, cápsula, u otra píldora. Opcionalmente, una dosis puede estar en forma líquida.
En algunas realizaciones, el primer nivel de dosis es 15 mg o 30 mg o 45 mg o 60 mg o 75 mg o 90 mg. En un aspecto opcional, el primer nivel de dosis puede ser superior a 90 mg. En algunas realizaciones, el segundo nivel de dosis es 15 mg o 30 mg o 45 mg o 60 mg o 75 mg o 90 mg. En un aspecto opcional, el segundo nivel de dosis puede ser superior a 90 mg. Un experto entenderá que esta lista de niveles de dosis no es limitante. Opcionalmente, la dosis se
puede ajustar basándose en el peso del sujeto. Opcionalmente, la dosis se puede ajustar midiendo la biodisponibilidad del fármaco, tal como midiendo la concentración en suero de tetratiomolibdato después de la administración de tetratiomolibdato de bis-colina. Opcionalmente, la dosis se puede ajustar midiendo cobre en el suero del paciente. Opcionalmente, la dosis se puede ajustar midiendo NCCcorregido.
En una realización, el primer nivel de dosis es 90 mg y el segundo nivel de dosis es 15 mg. En una realización, el primer nivel de dosis es 90 mg y el segundo nivel de dosis es 75 mg. En otras realizaciones, el primer nivel de dosis es 90 mg o 75 mg o 60 mg o 45 mg y el segundo nivel de dosis es 15 mg inferior al primer nivel de dosis. En otra realización, el segundo nivel de dosis es 30 mg inferior al primer nivel de dosis. En otras realizaciones, el tratamiento con tetratiomolibdato de bis-colina se interrumpe entre el primer nivel de dosis y el segundo nivel de dosis. En una realización, el tratamiento se interrumpe después de resultados de pruebas anormales. En una realización, el tratamiento con el segundo nivel de dosis ocurre después de que el paciente no presente resultado de prueba anormal.
En otro aspecto, la presente divulgación proporciona composiciones farmacéuticas para tratar enfermedad de Wilson en un paciente que comprende tetratiomolibdato de bis-colina. Ciertas composiciones farmacéuticas de tetratiomolibdato de bis-colina se proporcionan por la patente de EE. UU. N.° 7.189.865. Las composiciones farmacéuticas se describen, en general, por Remington: The Science and Practice of Pharmacy, 22a edición (2012).
En algunas realizaciones, la composición farmacéutica comprende tetratiomolibdato de bis-colina y un segundo principio farmacéuticamente activo. En una realización, el segundo principio farmacéuticamente activo es cinc. El cinc se puede proporcionar como acetato de cinc o sulfito de cinc. En una realización, el segundo principio farmacéuticamente activo es una sal de tetratiomolibdato distinta de la sal de bis-colina. Opcionalmente, el segundo principio farmacéuticamente activo es tetratiomolibdato de amonio. En otra realización, el segundo principio farmacéuticamente activo es quelante de cobre. En una realización opcional, el segundo principio farmacéuticamente activo es 2,3,2-tetramina o D-penicilamina.
En algunas realizaciones, las composiciones y métodos de la divulgación se refieren a sales de tetratiomolibdato distintas de una sal de tetratiomolibdato de bis-colina. En algunas realizaciones, la sal de tetratiomolibdato es una sal de tetratiomolibdato y cualquier contraión farmacéuticamente aceptable. Los contraiones a modo de ejemplo incluyen, sin limitación, amonio, colina y acetilcolina. El contraión puede ser, por ejemplo, un ácido orgánico positivamente cargado. La dosis se ajusta según el peso molecular de la sal.
En otro aspecto, la presente divulgación proporciona kits para tratar enfermedad de Wilson, que comprenden al menos tres conjuntos de unidades de administración farmacéutica; e instrucciones para su uso. En una realización, el kit que comprende comprimidos suficientes para un ciclo de tratamiento de 7 días o 30 días o 90 días con instrucciones para su uso. En una realización, las instrucciones para su uso indican la prueba de funciones hepáticas y umbrales para aumentar o disminuir el número de unidades de administración farmacéutica o unidades fraccionarias de la unidad de administración farmacéutica a administrar cada día. En una realización, el kit comprende además comprimidos de cinc. En una realización, el kit comprende además un quelante de cobre distinto de tetratiomolibdato de bis-colina. En otro aspecto, la presente divulgación proporciona composiciones para su uso en cualquiera de los métodos de la divulgación. En otro aspecto, la presente divulgación proporciona composiciones para su uso en la fabricación de un medicamento para su uso en cualquiera de los métodos de la divulgación.
Para administración por vía oral, las composiciones farmacéuticas de la presente divulgación pueden tomar la forma de formas de dosis sólida, por ejemplo, comprimidos (tanto formas tragables como masticables), cápsulas o cápsulas de gel, preparadas mediante medios convencionales con excipientes y vehículos farmacéuticamente aceptables, tales como aglutinantes (por ejemplo almidón de maíz pregelatinizado, polivinilpirrolidona, hidroxipropilmetilcelulosa y similares), cargas (por ejemplo, lactosa, celulosa microcristalina, fosfato de calcio y similares), lubricantes (por ejemplo, estearato de magnesio, talco, sílice y similares), disgregantes (por ejemplo, almidón de patata, glicolato sódico de almidón y similares), humectantes (por ejemplo, laurilsulfato de sodio) y similares. Dichos comprimidos también se pueden recubrir por métodos bien conocidos en la técnica.
La dosis administrada se puede ajustar según la edad, el peso y la afección del paciente, así como la vía de administración, forma farmacéutica y pauta y el resultado deseado. En algunas realizaciones, las dosis administradas al sujeto se valoran hasta que se alcanza un criterio de valoración deseado.
Las composiciones descritas anteriormente se pueden administrar en las formas farmacéuticas como se ha descrito anteriormente en dosis únicas o divididas de una a cuatro veces al día.
Se pueden preparar unidades de administración que incluyen comprimidos, cápsulas y comprimidos oblongos, de diversos tamaños, por ejemplo, de aproximadamente 2 a 10000 mg de peso total, que contienen una o ambas de las sustancias activas en los intervalos descritos anteriormente, siendo el resto un vehículo fisiológicamente aceptable de otros materiales según la práctica farmacéutica aceptada. Estos comprimidos pueden ser, por supuesto, ranurados para proporcionar dosis fraccionarias. Las cápsulas de gelatina se pueden formular similarmente.
En algunas realizaciones, se proporciona tetratiomolibdato de bis-colina en la misma unidad de administración en forma de una unidad de administración divisible. Por ejemplo, en algunas realizaciones, un comprimido ranurado puede proporcionar la unidad de administración. Bajo la dirección de un médico u otro profesional médico, al sujeto se le puede indicar que tome una porción de la unidad de administración, en donde la porción proporcionará el nivel de administración deseado para el intervalo dado. En el siguiente intervalo, se le puede indicar al paciente que tome dos o más porciones de la unidad de administración, en donde las dos o más porciones proporcionarán el nivel de administración deseado para ese intervalo.
También se pueden preparar formulaciones líquidas disolviendo o suspendiendo una o la combinación de sustancias activas en un vehículo líquido convencional aceptable para administración farmacéutica de manera que se proporcione la administración deseada en una a cuatro cucharillas de té.
Dichas formas farmacéuticas se pueden administrar al paciente en una pauta de una a cuatro dosis por día.
Ciertos aspectos de la divulgación se basan en el sorprendente descubrimiento de que la administración de tetratiomolibdato de bis-colina en condiciones posprandiales produce una disminución del 60 % al 75 % en la absorción en comparación con la administración de tetratiomolibdato de bis-colina en condiciones en ayunas. En un aspecto de la divulgación, el tetratiomolibdato de bis-colina se administra en un estado en ayunas a un paciente que padece enfermedad de Wilson. En otras realizaciones, el tetratiomolibdato de bis-colina se administra tras un ayudo durante la noche. En aún otras realizaciones, el tetratiomolibdato de bis-colina se administra en un estómago vacío, después de un ayuno de aproximadamente 1, 2, 3, 4, 5, 6, 7 u 8 horas. En ciertas realizaciones, el tetratiomolibdato de biscolina se administra como una formulación con cubierta entérica.
Todas las características descritas en el presente documento (incluyendo las reivindicaciones, resumen y dibujos adjuntos), y/o todas las etapas de cualquier método así desvelado, se pueden combinar con cualquiera de los aspectos anteriores en cualquier combinación, excepto combinaciones donde al menos algunas de dichas características y/o etapas sean mutuamente excluyentes. En concreto, se puede usar cualquiera de los agentes activos y composiciones descritos en el presente documento en cualquiera de los métodos de tratamiento descritos. Se prevé explícitamente que todas y cada una de dichas combinaciones formen parte de la invención.
Como se usa en el presente documento, los siguientes términos deben tener los siguientes significados:
"Tetratiomolibdato de bis-colina" se refiere a la sal de tetratiomolibdato de bis-colina o a composiciones farmacéuticas de la misma. El tetratiomolibdato de bis-colina también se conoce como tetratiomolibdato de colina o ATN-224 o WTX101 o WTX-101 o DECUPRATE™. "EW" se refiere a enfermedad de Wilson.
"QOD" o "quaque altera die" se refiere a la administración de un tratamiento cada dos días, por ejemplo 15 mg QOD se entiende en la técnica que indica la administración de una dosis de 15 mg cada dos días. "QD" o "quaque die" se refiere a la administración de tratamiento una vez al día, por ejemplo 15 mg QD se entiende en la técnica que indica la administración de una dosis de 15 mg una vez al día. "SoC" se refiere a tratamiento de referencia.
"Resultados de pruebas consecutivos" o "resultados de pruebas anormales consecutivos" se refiere a los resultados de dos o más mediciones del mismo parámetro tomadas en dos momentos diferentes. En ciertas realizaciones, dichas mediciones se toman separadas una semana. En otras realizaciones, dichas mediciones se toman separadas dos semanas.
"Cobre no unido a ceruloplasmina" o "NCC" se refiere a la concentración de cobre libre en suero. En el plasma, el cobre está o unido a la ceruloplasmina, o unido de forma más suelta a otras proteínas del plasma, tales como la albúmina o péptidos circulantes más pequeños. El cobre unido de forma suelta no unido a la ceruloplasmina (es decir, cobre no unido a la ceruloplasmina, o NCC) se denomina algunas veces cobre "libre". En la situación sana (es decir, aquellos sin enfermedad de Wilson), en general, más del 70 % del cobre total en plasma está unido a la ceruloplasmina. Debido a la enfermedad de Wilson, los niveles de cobre unido a la ceruloplasmina son normalmente bajos en la enfermedad de Wilson y puede explicar los bajos niveles de cobre en plasma total observados en los pacientes con enfermedad de Wilson. Sin embargo, cuando el cobre libre (es decir, no unido a la ceruloplasmina) se calcula restando el cobre de ceruloplasmina del nivel de cobre en suero total, normalmente se encuentra que éste está elevado por encima del límite superior de la normalidad (>15 mg/dl). En pacientes con enfermedad de Wilson sin tratar, los niveles de NCC son normalmente superiores a 25 pg/dl. Para calcular el nivel de NCC (en pg/dl), la ceruloplasmina (en mg/dl) se multiplica por 3; este valor se resta entonces del nivel de cobre en suero total (en pg/dl).
"NCCcorregido" o "NCCcorr" se refiere al NCC corregido para el cobre contenido en complejos de tetratiomolibdatocobre-albúmina. Los métodos a modo de ejemplo para determinar NCCcorr se proporcionan en Weiss et al. Lancet Gastroenterol Hepatol. 2:869-876 (2017), que se incorpora en la presente divulgación en su totalidad. Los mecanismos de incorporación de cobre en la ceruloplasmina se describen en Hellman et al. J. Bio. Chem. 48:46632-38 (2002).
"Modelo para enfermedad hepática terminal" o "MELD" se refiere a un sistema de puntuación para evaluar la intensidad de la enfermedad hepática crónica. El MELD usa los valores del sujeto para bilirrubina en suero, creatinina sérica y la
relación normalizada internacional (INR) para el tiempo de protrombina para predecir la supervivencia. Se calcula según la siguiente fórmula: MELD = 3,78xln[bilirrubina en suero (mg/dl)] 11,2xln[INR] 9,57xln[creatinina sérica (mg/dl)] 6,43
"Puntuación de Nazer modificada" se refiere a una evaluación del estado del hígado y consiste en una combinación de 5 parámetros de laboratorio: aspartato aminotransferasa, relación normalizada internacional, bilirrubina, albúmina y número de leucocitos. La puntuación tiene un intervalo total de 0 a 20, y valores más bajos indican mejoría.
"Índice de fibrosis-4/elastografía transitoria" o "índice FIB-4" se refiere a una fórmula usada para predecir la fibrosis hepática basada en valores bioquímicos estándar (ALT, aspartato aminotransferasa y número de plaquetas) y la edad.
"Elastografía transitoria" se refiere a un método no invasivo de obtención de imágenes que evalúa el grado de fibrosis hepática o depósitos grasos en el hígado, por determinación de la velocidad de las ondas del sonido a través del hígado utilizando un sonograma.
"Ceruloplasmina" se refiere a una enzima ferroxidasa que en los seres humanos está codificada por el gen CP. La ceruloplasmina es la principal proteína transportadora del cobre en la sangre, y además, desempeña una función en el metabolismo del hierro.
"Cobre total y molibdeno total" o "análisis de Cu total y Mo total" se refiere a la medición de la concentración total de cobre y la concentración total de molibdeno (Mo) en el suero de un paciente.
"Perfil de especiación" se refiere al perfil de complejos de Mo, Cu y proteína con cromatografía de exclusión por tamaño.
"Escala unificada de valoración de la enfermedad de Wilson" o "UWDRS" se refiere a una escala de valoración clínica diseñada para evaluar las manifestaciones neurológicas de la EW que, en general, se pueden dividir en 3 síndromes del trastorno del movimiento: a. síndrome distónico, b. atáxico y c. parkinsoniano. La UWDRS comprende tres partes: Parte I de UWDRS (consciencia, ítem 1), parte II de UWDRS (una revisión histórica de ítems de la actividad cotidiana [incapacidad], ítems 2 a 11) y parte III de UWDRS (un examen neurológico, ítems 12 a 34). La parte I y la parte III de UWDRS son evaluadas normalmente por un neurólogo. La parte II de UWDRS es informada por el sujeto o la familia. La UWDRS se describe en Czlonkowska A et al. Neurol Neurochir Pol 41:1 -12 (2007), que se incorpora en la presente divulgación en su totalidad.
"Escala de impresión clínica global-gravedad" o "CGI-S" se refiere a una escala de 7 puntos que requiere que el profesional clínico valore la intensidad de las enfermedades del sujeto en el momento de la evaluación, con respecto a la experiencia previa del profesional clínico con sujetos que tienen el mismo diagnóstico. Considerando la experiencia clínica total, un sujeto es evaluado sobre la intensidad de las enfermedades en el momento de la valoración como: 1, normal, no está en absoluto enfermo; 2, marginalmente enfermo; 3, levemente enfermo; 4, moderadamente enfermo; 5, notablemente enfermo; 6, gravemente enfermo; o 7, extremadamente enfermo.
"Escala de impresión clínica global-mejoría" o "CGI-I" se refiere a una escala de 7 puntos que requiere que el profesional clínico evalúe cuánto han mejorado o empeorado las enfermedades del sujeto con respecto a un estado basal al principio de la intervención y valorada como: 1, marcada mejoría; 2, moderada mejoría; 3, mínima mejoría; 4, ningún cambio; 5, mínimo empeoramiento; 6, mucho peor; o 7, marcado empeoramiento.
"Escala breve de evaluación psiquiátrica-24" o "BPRS-24" se refiere a un instrumento de 24 ítems que permite al evaluador medir la gravedad de las manifestaciones psiquiátricas. La BPRS-24 evalúa 24 síntomas psiquiátricos. La presencia y la gravedad de los síntomas psiquiátricos se evalúan en una escala de Likert que varía desde 1 (no presente) hasta 7 (extremadamente grave). La BPRS-24 puede ser realizada por un médico formado.
"5 Dimensiones de EuroQoL" o "EQ-5D" se refiere a una prueba que consiste en el sistema descriptivo EQ-5D-5L y la escala analógica visual EQ. El sistema descriptivo comprende 5 dimensiones (movilidad, cuidado de sí mismo, actividades usuales, dolor/molestia y ansiedad/depresión), cada una de las cuales tiene 5 niveles de intensidad (sin problemas/ligeros problemas/moderados problemas/graves problemas/problemas extremos). Para la puntuación en el sistema descriptivo EQ-5D-5L, se le pregunta a la persona que rellena el cuestionario que indique su estado de salud haciendo tic (o poniendo una cruz) en la casilla con la afirmación más apropiada en cada una de las 5 dimensiones. Esta decisión produce un número de 1 dígito que expresa el nivel seleccionado para esa dimensión. Los dígitos para 5 dimensiones se pueden combinar en un número de 5 dígitos que describe el estado de salud de la persona que rellena el cuestionario.
"Cuestionario de satisfacción con el tratamiento con la medicación" o "TSQM-9" se refiere a una puntuación usada para evaluar el nivel global de satisfacción o descontento con la medicación que están tomando los sujetos. Esta escala compuesta comprende 2 ítems en el cuestionario TSQM-9: ¿En qué medida está satisfecho de que las cosas buenas de esta medicación superen a las malas? Teniendo en cuenta todos los aspectos, ¿cuál es su grado de satisfacción o descontento con esta medicación?
"Tres síntomas más molestos" se refieren a los 3 síntomas más molestos de un sujeto. Cada sujeto, o el sujeto y su cuidador, identifican sus 3 síntomas más molestos, y éstos se documentan en un formulario por escrito, así como el impacto que estos síntomas tienen en sus actividades de la vida diaria. Los 3 síntomas más molestos se graban en una cinta de vídeo de los sujetos que dan su consentimiento, siempre que sea posible y apropiado.
"Prueba de marcha cronometrada de 25 pies" se refiere a una prueba de rendimiento cuantitativo de movilidad y funcionamiento de las piernas basada en una marcha cronometrada de 25 pies. Se dirige al sujeto a un extremo de un recorrido de 25 pies claramente marcado y se le indica que camine 25 pies lo más rápido posible, pero con seguridad. El tiempo se calcula desde el inicio de la instrucción para comenzar y termina cuando el sujeto ha llegado a la marca de 25 pies. La tarea se repite inmediatamente haciendo que el sujeto vuelva a caminar la misma distancia. La puntuación de la prueba de marcha cronometrada de 25F es la media de los 2 ensayos. Los sujetos pueden usar dispositivos de ayuda al realizar esta tarea
"Prueba de clavijas con 9 agujeros" o "9-HPT" es una prueba cuantitativa breve y normalizada de la función de las extremidades superiores. Tanto la mano dominante como la no dominante se prueban dos veces. El sujeto se sienta en una mesa con un pequeño recipiente poco profundo que contiene 9 clavijas y un bloque de madera o plástico que contiene 9 agujeros vacíos. A la orden de inicio, cuando se pone en marcha un cronómetro, el sujeto coge las 9 clavijas de 1 en 1 lo más rápidamente posible, las coloca en los 9 agujeros y, una vez que están en los agujeros, las vuelve a sacar lo más rápidamente posible de 1 en 1, volviéndolas a colocar en el recipiente poco profundo. Dos ensayos consecutivos con la mano dominante son seguidos inmediatamente por 2 ensayos consecutivos con la mano no dominante. La puntuación para 9-HPT es el promedio de los 4 ensayos.
"Prueba de interferencia no verbal de Stroop" se refiere a una medida eficaz del funcionamiento ejecutivo, la capacidad de planificar, aplicar conocimientos y tomar decisiones. En psicología, el efecto Stroop es una demostración de la interferencia en el tiempo de reacción de una tarea. No hay comunicación verbal durante esta prueba. La prueba se imparte con indicaciones no verbales, usando gestos y demostraciones.
"Prueba de amplitud de dígitos" es una prueba que mide el recuerdo de secuencias de dígitos hacia delante y en orden inverso (hacia atrás) y la secuenciación de la amplitud de dígitos. Las secuencias de dígitos se presentan comenzando con una longitud de 2 dígitos y se presentan 2 ensayos con una longitud de lista creciente. La prueba finaliza cuando el sujeto no consigue informar con precisión ninguno de los ensayos con 1 longitud de secuencia o cuando se alcanza la longitud máxima de la lista (9 dígitos, 8 hacia atrás).
"Acontecimiento adverso" se refiere a cualquier manifestación médica no deseada en un sujeto en investigación clínica administrado con un producto farmacéutico, que no tiene necesariamente una relación causal con ese tratamiento. Por lo tanto, un acontecimiento adverso puede ser cualquier signo desfavorable y/o involuntario (incluyendo una resultado de laboratorio anormal), síntoma, o enfermedad temporalmente asociada al uso de un medicamento en investigación, tanto si está como si no relacionado con el medicamento en investigación. Resultados de laboratorio anormales u otros resultados de exámenes clínicamente significativos, que incluyen resultados de exámenes neurológicos, se informan como acontecimientos adversos. El experto ejercerá su criterio médico y científico para decidir si un resultado de laboratorio anormal u otra evaluación anormal es clínicamente significativa. Cualquier prueba anormal que se determine que es un error no requiere su notificación como un acontecimiento adverso.
La intensidad de todos los acontecimientos adversos se clasifica según los Criterios de Terminología Común para Acontecimientos Adversos (CTCAE). Estos criterios se pueden encontrar en http://ctep.cancer.gov/reporting/ctc.html. Para los acontecimientos adversos no listados en CTCAE, se usa el siguiente sistema de clasificación: Leves (grado 1 de CTCAE): síntomas transitorios, conocimiento de signo/síntoma, pero fácilmente tolerados y sin interferencia con las actividades cotidianas del sujeto. Moderados (grado 2 de CTCAE): signos/síntomas significativos que interfieren con las actividades usuales del sujeto, pero todavía aceptables. Intensos (grado 3 de CTCAE): signos/síntomas incapacitantes que causan una interferencia considerable con las actividades cotidianas del sujeto, inaceptables. Potencialmente mortales (grado 4 de CTCAE): acontecimiento adverso que supone una amenaza para la vida o incapacitante. Muerte (grado 5 de CTCAE): acontecimiento adverso relacionado con la muerte.
"Reacción adversa (a un fármaco)" se refiere a todas las respuestas perjudiciales e involuntarias a un medicamento relacionadas con cualquier dosis que debe considerarse una reacción adversa al fármaco. "Respuestas" a un medicamento significa que una relación causal entre un medicamento y un acontecimiento adverso es al menos una posibilidad razonable, es decir, la relación no puede ser descartada.
"Reacción adversa inesperada al fármaco" se refiere a una reacción adversa, cuya naturaleza o gravedad no está de acuerdo con la información sobre el producto.
"Propiedades farmacológicas" se refiere a la absorción, la distribución, el metabolismo y la eliminación de un fármaco que deben ser considerados.
"Acontecimientos adversos de interés especial" se refiere a cualquier síntoma neurológico nuevo o empeoramiento clínicamente significativo de un síntoma neurológico en curso después del inicio de la farmacoterapia del estudio se designará que es un AESI, tanto grave como no grave.
"Acontecimientos adversos graves" se refiere a un acontecimiento adverso o reacción adversa que produce cualquiera de los siguientes desenlaces: muerte o un acontecimiento adverso potencialmente mortal, requiere hospitalización o prolongación de las hospitalizaciones existentes, una discapacidad/incapacidad persistente o significativa, o alteración sustancial de la capacidad para realizar funciones de la vida normal, una anomalía congénita/defecto congénito, o un acontecimiento médico importante.
Un acontecimiento adverso o reacción adversa se considera "potencialmente mortal" si, en vista de cualquiera del investigador o patrocinador, su aparición pone al sujeto en un riesgo inminente de muerte. No incluye un acontecimiento que, en caso de que hubiera ocurrido en una forma más intensa, pudiera haber causado la muerte.
Cualquier ingreso hospitalario de al menos 1 noche se considerará una hospitalización ambulatoria. Una visita al servicio de urgencias sin ingreso en el hospital no se registrará como un SAE bajo este criterio, ni tampoco la hospitalización para un procedimiento programado o planificado antes de firmar el consentimiento informado. Sin embargo, las complicaciones inesperadas y/o la prolongación de la hospitalización que ocurren durante la cirugía electiva se deben registrar como acontecimientos adversos y evaluar para su gravedad. El ingreso en el hospital por motivos sociales o circunstanciales (es decir, está completo, se vive demasiado lejos para las ir a las visitas al hospital) no se considera hospitalizaciones ambulatorias.
Los acontecimientos médicos importantes que pueden no producir la muerte, ser potencialmente mortales, o requieren hospitalización, se pueden considerar un SAE cuando, basándose en el criterio médico apropiado, pueden poner en peligro al sujeto y pueden requerir intervención médica o quirúrgica para prevenir uno de los desenlaces enumerados anteriormente. Los ejemplos de dichos acontecimientos médicos incluyen broncoespasmo alérgica que requiere un tratamiento intenso en un servicio de urgencias o en casa, discrasias sanguíneas o convulsiones que no producen hospitalizaciones ambulatorias, o el desarrollo de dependencia de fármacos.
"Antecedentes médicos" se refiere a la información sobre medicación previa y concomitante, diagnósticos previos y actuales, afecciones y cirugías que se consideran significativas, consumo de tabaco, alcohol y drogas.
"Evaluaciones de laboratorio clínico" o "medidas de laboratorio clínico" incluyen bioquímica, hematología, coagulación y análisis de orina (con microscopía).
"Parámetros de electrocardiograma" se refieren a frecuencia cardíaca, intervalo RR, intervalo PR, anchura de QRS e intervalo QT.
"Constantes vitales" se refiere a frecuencia cardíaca, tensión arterial, frecuencia de respiración, temperatura y peso.
"Exploración física" se refiere a una evaluación de lo siguiente: aspecto general, respiratorio, cardiovascular, abdomen, piel, cabeza y cuello (incluyendo oídos, ojos, nariz y garganta), ganglios linfáticos, tiroides y sistemas musculoesqueléticos (incluyendo columna vertebral y extremidades). La parte III de la Escala unificada de valoración de la enfermedad de Wilson es el examen neurológico usado en este estudio.
El término "puntuación", como se usa en el presente documento, se refiere a un valor relativo, nivel, concentración o grado de un resultado de un ensayo. Puede ser creada artificialmente por un experto en la técnica o usando un algoritmo, algunas veces usando muestras con analitos conocidos, opcionalmente usando muestras con concentraciones o títulos conocidos de los analitos conocidos. Puede ser un número asignado manualmente por un experto en la técnica o generado con una fórmula o algoritmo. También puede ser un símbolo, por ejemplo, "-","+", o "++". Una puntuación se puede generar del cálculo con una fórmula o algoritmo, o puede ser asignada por inspección visual, medición, o estimación del resultado de un ensayo. Cuando se usan muestras con concentraciones o títulos conocidos de analitos conocidos, dichas muestras se pueden ensayar en condiciones diluidas y sin diluir, y se puede generar un intervalo de puntuaciones o una curva patrón de puntuaciones, que se pueden usar para asignar o estimar las puntuaciones de muestras desconocidas ensayadas para los mismos analitos, en algunas realizaciones usando los mismos ensayos.
EJEMPLOS
Ejemplo 1: Tetratiomolibdato de bis-colina en pacientes con enfermedad de Wilson: un estudio de fase 2, multicéntrico y de etiqueta abierta.
Antecedentes
La enfermedad de Wilson es un trastorno genético en el que el cobre se acumula en el hígado, el cerebro y otros tejidos. Las terapias habían estado limitadas por la eficacia, problemas de seguridad y dosis múltiples diarias. El
tetratiomolibdato de bis-colina (WTX101) es la primera molécula oral agregante de cobre-proteína, que se dirige al cobre intracelular hepático y reduce el cobre no unido a la ceruloplasmina (NCC) plasmático formando complejos tripartitos con albúmina y aumentando la eliminación biliar del cobre. La eficacia y la seguridad de WTX101 se evaluó en el tratamiento inicial o primer tratamiento de pacientes con enfermedad de Wilson.
Métodos
Este estudio de fase 2 y etiqueta abierta se realizó en 11 hospitales en los EE. UU. y en Europa. Se reclutó a pacientes (>18 años) con enfermedad de Wilson que no habían sido tratados o que habían recibido no más de 24 meses de tratamiento con quelantes o cinc, tenían una puntuación de Leipzig de 4 o más, y tenían concentraciones de NCC por encima del intervalo de referencia del límite inferior de la normalidad (>0,8 pmol/l). Los pacientes que cumplieron los requisitos recibieron monoterapia con WTX101 con una dosis inicial de 15-60 mg/día basándose en las concentraciones de NCC basal durante las primeras 4-8 semanas, con administración individualizada guiada por respuesta para las semanas restantes hasta la semana 24. Los investigadores, cualquier otro personal del hospital y los pacientes eran conocedores de la identidad del tratamiento. El criterio principal de valoración fue el cambio en las concentraciones de NCC corregido basal para el cobre en los complejos de tetratiomolibdato-cobre-albúmina (NCCcorregido) a las 24 semanas, definiéndose el éxito del tratamiento como el logro o el mantenimiento del NCCcorregido normalizado (<2,3 pmol/l [límite superior de la normalidad]) o el logro de al menos una reducción del 25 % en NCCcorregido desde el nivel basal hasta las 24 semanas. Este estudio se registró en ClinicalTrials.gov, número NCT02273596.
Resultados
Se reclutó a veintiocho pacientes y recibieron WTX101; 22 (79 %) pacientes finalizaron el estudio hasta la semana 24. A las 24 semanas, 20 (71 %, IC del 95 % 51,3-86,8; p<0,0001) de los 28 pacientes cumplieron los criterios para el éxito del tratamiento: 16 (57 %) tratados con WTX101 o lograron o mantuvieron concentraciones normalizadas de NCCcorregido y 4 (14 %) tuvieron al menos una reducción del 25 % desde NCCcorregido basal. NCCcorregido medio se redujo en 72 % desde el nivel basal hasta la semana 24 (diferencia media de los mínimos cuadrados -2,4 pmol/l [EE 0-4], IC del 95 % -3,2 a -1,6; p<0,0001). Sorprendentemente, no se registraron casos de empeoramiento neurológico paradójico relacionado con el fármaco. La función hepática fue estable en todos los pacientes, aunque hubo concentraciones elevadas reversibles de alanina o aspartato aminotransferasa asintomático, o yglutamiltransferasa, sin aumento en la bilirrubina, en 11 (39 %) de los 28 pacientes que recibieron al menos 30 mg/día de WTX101. Se notificaron 11 acontecimientos adversos graves en siete (25 %) de los pacientes, e incluyeron trastornos psiquiátricos (seis acontecimientos en cuatro pacientes), alteración de la marcha (un acontecimiento), aminotransferasas hepáticas elevadas (dos acontecimientos en dos pacientes, uno con agranulocitosis) y deterioro en el funcionamiento neurológico (un acontecimiento, probablemente debido al empeoramiento natural de la enfermedad, aunque no se pudo descartar la causalidad). Se evaluó que era poco probable que los siete acontecimientos adversos graves clasificados como trastornos psiquiátricos y como alteración de la marcha se relacionaran con el fármaco del estudio, mientras que los cuatro acontecimientos restantes estuvieron posiblemente o probablemente relacionados.
Interpretación
Los resultados indicaron que WTX101 podría ser un nuevo enfoque terapéutico prometedor para la enfermedad de Wilson, con un modo de acción único. En vista de su dosis de una vez al día y un perfil de seguridad favorable, WTX101 podría mejorar el tratamiento de pacientes con esta afección debilitante.
Introducción
La enfermedad de Wilson es un trastorno autosómico recesivo de transporte alteado del cobre que conduce a la acumulación de cobre en el hígado, el cerebro y otros tejidos. La enfermedad es causada por mutaciones en el gen ATP7B, que codifica una ATPasa del transportador de Cu. La disminución de la función de ATP7B conduce a la incorporación reducida de cobre en la ceruloplasmina y a una alteración de la eliminación biliar del cobre. La enfermedad de Wilson afecta a aproximadamente una de cada 30000 personas, pero la prevalencia varía entre las poblaciones, y el infradiagnóstico podría ser significativo. La presentación clínica se diferencia ampliamente, e incluye formas de la enfermedad hepática, manifestaciones neurológicas y psiquiátricas, y anillos de Kayser-Fleischer de la córnea. Los datos de laboratorio anormales incluyen elevadas concentraciones de cobre no unido a la ceruloplasmina (NCC) libre en plasma y bajas concentraciones de ceruloplasmina circulante.
Si queda sin diagnosticar y sin tatar, la enfermedad de Wilson es universalmente mortal. Los tratamientos por vía oral autorizados hace varias décadas para reducir las concentraciones de cobre incluyen quelantes (penicilamina y trientina), que aumentan la eliminación urinaria del cobre, o cinc, que inhibe la absorción gastrointestinal del cobre.
Se han hecho algunos estudios prospectivos, si hay alguno, con estos tratamientos, y hay una cantidad considerable de necesidades sin satisfacer con respecto a la eficacia, la seguridad y la simplicidad de las pautas posológicas. Además, los pacientes con presentación neurológica que inician el tratamiento con penicilamina o trientina pueden tener un empeoramiento paradójico prematuro de la enfermedad neurológica, con una rápida aparición de nuevos
signos neurológicos o empeoramiento de los signos neurológicos existentes, que conduce a una marcada incapacidad. En los estudios clínicos, la proporción de pacientes con enfermedad de Wilson neurológica afectados por el empeoramiento prematuro después de empezar con el quelante varía desde el 19 % hasta el 35 %. El empeoramiento neurológico prematuro puede ser irreversible y podría ser debido a una rápida movilización del cobre libre.
El tetratiomolibdato de bis-colina (WTX101) es la primera molécula oral de unión a cobre-proteína en investigación como una monoterapia de una vez al día para la enfermedad de Wilson. Una forma previa del fármaco, el tetratiomolibdato de amonio, controló rápidamente las concentraciones de cobre en estudios clínicos; sin embargo, es demasiado inestable para su uso cotidiano. El resto de bis-colina es un avance importante, puesto que tiene estabilidad mejorada y, a diferencia de otros tratamientos disponibles, parece que WTX101 tiene actividad intracelular directa en hepatocitos, en los que se une al exceso de cobre y promueve la eliminación biliar del cobre. WTX101 también se une rápidamente al cobre plasmático libre, creando un complejo tripartito estable de tetratiomolibdato con cobre y albúmina.
Métodos
El estudio de fase 2 y etiqueta abierta se realizó en 11 hospitales. Los pacientes que cumplieron los requisitos tenían edades de 18 años o más, con un diagnóstico de enfermedad de Wilson establecido por una puntuación de Leipzig de 4 o más. En el momento del reclutamiento, los pacientes no habían recibido tratamiento previo para la enfermedad de Wilson ni habían sido tratados con quelación o cinc durante más de 24 meses, ni tenían concentraciones de NCC por encima del intervalo de referencia del límite inferior de la normalidad (>0,8 pmol/l). Se excluyeron los pacientes con cirrosis hepática descompensada, una puntuación de MELD superior a 11, o una puntuación de Nazer modificada (puntuación de King revisada) superior a 6.
El protocolo y todas las modificaciones fueron autorizadas por los comités institucionales de revisión y comités de ética locales. La realización del estudio fue monitorizada por un comité de monitorización de datos y de la seguridad independiente. Todos los participantes proporcionaron el consentimiento informado por escrito según la Declaración de Helsinki.
Los investigadores, cualquier otro personal del hospital y los pacientes, y el patrocinador del estudio, eran conocedores de la identidad del tratamiento. Los pacientes previamente tratados tuvieron un periodo de lavado de 48 h antes del inicio de WTX101. Los pacientes recibieron una dosis inicial de WTX101 de 15-60 mg/día basándose en las concentraciones de NCC basal durante las primeras 4-8 semanas, con posterior administración individualizada guiada por respuesta durante las 24 semanas restantes. Aunque la administración de WTX101 fue inicialmente dos veces al día, una modificación del protocolo anterior implementó una administración de una vez al día (si se consideró apropiado por el investigador).
Después de una elevada concentración de alanina aminotransferasa (ALT) en un paciente que recibió 120 mg/día, se modificó el esquema posológico para reducir la dosis máxima desde 300 mg/día hasta 60 mg/día.
A criterio del investigador principal, la dosis de WTX101 se pudo ajustar por incrementos predefinidos basándose en diversos factores, que incluían la bioquímica clínica y la hematología, la evaluación clínica, la seguridad y las concentraciones de NCC. El ajuste ascendente de la dosis fue escalonado, con cada aumento restringido a duplicar la dosis previa, y no se permitió si NCC estuvo dentro de o por debajo del intervalo de la normalidad. La dosis se redujo o interrumpió temporalmente después de dos notificaciones consecutivas de concentraciones de ALT o aspartato aminotransferasa (AST) que estuvieron al menos 2-5 veces por encima del intervalo de la normalidad, una reducción del 30 % o más en la hemoglobina basal, o un aumento de 4 puntos o más en los signos neurológicos basándose en la parte III de la Escala unificada de valoración de la enfermedad de Wilson (UWDRS) (un sistema de puntuación neurológica cuantitativa aceptada y validada desarrollada específicamente para la enfermedad de Wilson).
Usando valores para concentraciones de cobre y de ceruloplasmina total en plasma, se calculó el NCC restando la cantidad de cobre unida a la ceruloplasmina de las concentraciones de cobre total (determinadas con espectrometría de masas con plasma acoplado inductivamente). Entonces se corrigieron las mediciones de NCC restando la cantidad de cobre unida en el complejo de tetratiomolibdato-cobre-albúmina después del tratamiento con WTX101, puesto que este no es parte del conjunto de cobre tóxico reactivo. El método de corrección usó la relación molar promedio entre cobre y molibdeno en el complejo tripartito, que se determinó usando la relación entre las concentraciones de NCC y de molibdeno en plasma y se confirmó por dos métodos independientes. El método de corrección de NCC se validó usando selecciones aleatorias de muestras de prueba y de validación.
El criterio principal de valoración fue el cambio en NCCcorregido desde el nivel basal hasta las 24 semanas, que se midió como concentraciones de NCC corregido para cobre contenido en los complejos de tetratiomolibdato-cobre-albúmina (NCCcorregido). El éxito del tratamiento se definió como el logro o el mantenimiento de las concentraciones normalizadas de NCCcorregido (á2,3 pmol/l [límite superior de la normalidad]) o el logro de al menos una reducción del 25 % en NCCcorregido desde el nivel basal hasta las 24 semanas.
Los criterios secundarios de valoración fueron la seguridad y la tolerabilidad, el cambio en y el tiempo hasta la normalización de los niveles de NCCcorregido, la enfermedad neurológica clínica, la función hepática, los síntomas
clínicos, la calidad de vida relacionada con la salud (HRQoL), el estado psiquiátrico y la farmacocinética y el cobre intercambiable, el perfil de especiación y el cobre urinario. También se midieron el estado psiquiátrico, los datos farmacocinéticos y los criterios de valoración del cobre. Se evaluó la enfermedad neurológica como la incapacidad notificada por el paciente, medida con la parte II de UWDRS, y también como el estado neurológico evaluado por el evaluador formado, medido por la parte III de UWDRS. Se evaluó la función sintética del hígado monitorizando la relación normalizada internacional (INR) y las concentraciones de albúmina. Además, se evaluaron cambios en la función hepática por la puntuación de Nazer modificada (basada en bilirrubina, INR, AST, albúmina y número de leucocitos) y, en un análisis a posteriori, por la puntuación de MELD (basada en bilirrubina, creatinina, INR y causa de la enfermedad hepática). HRQoL se midió con la escala analógica visual de 5 dimensiones EuroQoL (EQ VAS).
Se recogió los datos de los acontecimientos adversos (AE) para su aparición, duración, gravedad e intensidad, con relación a la medicación del estudio determinada por el investigador.
El reclutamiento planeado fue 30 pacientes, esperando que al menos 15 hubieran recibido un tratamiento previo limitado (<90 días) con quelantes o cinc. Puesto que el objetivo del estudio era presentar una estadística principalmente descriptiva, no se realizaron cálculos formales de potencia. Los cambios en las concentraciones de cobre y las puntuaciones se resumieron con el tiempo con estadística descriptiva. Se aplicaron análisis de medidas repetidas con modelos mixtos con un término de efectos fijos para la visita clínica, y se aplicó una estructura de covarianza de potencia espacial para modelar los errores intraindividuales. Se seleccionó una estructura de covarianza de potencia espacial debido a que supone que la correlación intraindividual disminuye a medida que aumenta la distancia de tiempo entre las medidas repetidas. Se utilizó SAS (versión 9.3) para proporcionar cambio en la media de los mínimos cuadrados con el tiempo, y los IC del 95 % asociados, EE y valores de p bilaterales.
Se reclutó a veintiocho pacientes y se trataron con WTX101; 22 (79 %) pacientes finalizaron el estudio hasta la semana 24 (FIG. 1). En el nivel basal, 15 (54 %) pacientes fueron mujeres y la edad media fue 34,1 años (DE 11,86) y oscilaron desde 18 hasta 64 años. Los nueve (32 %) pacientes no habían recibido tratamiento previo para la enfermedad de Wilson. Nueve (32 %) pacientes habían sido tratados durante menos de 28 días, y diez (36 %) pacientes habían sido tratados durante entre 28 días y 2 años (mediana de 100 días [intervalo 7-714]). La mayoría de los pacientes tuvieron grados variables de signos neurológicos en el momento del reclutamiento, lo más comúnmente disartria (19 [68 %]), temblor postural (18 [64 %]), movimientos rápidos y alternantes de las manos (18 [64 %]) y marcha anormal (17 [61 %]), con marcha anormal principalmente supeditada por la ataxia (12 [43 %]). La puntuación media de la parte III de UWDRS en el nivel basal fue 22,8 (DE 21,0; intervalo 0-83) con solo tres (11 %) pacientes puntuando 0 (sin anomalías neurológicas). En el nivel basal, 13 (46 %) pacientes tuvieron cirrosis hepática, basada en antecedentes médicos (siete pacientes) o por estimaciones del índice de relación entre AST y plaquetas (seis pacientes). 14 (50 %) pacientes tuvieron 26 casos de una anomalía en la prueba del hígado en el momento de entrar en el estudio (12 ALT, nueve AST, una bilirrubina y cuatro INR). De estas anomalías, 24 estuvieron dentro de 1-2 veces y dos estuvieron dentro de 3-5 veces el límite superior de la normalidad.
En la semana 24, o en la última dosis recibida para pacientes con interrupción prematura, las dosis diarias fueron 15 mg para seis (21 %) pacientes, 30 mg para 13 (46 %) pacientes y 60 mg (32 %) para nueve pacientes. Más del 80 % de la dosis total de WTX101 en el estudio fue una vez al día.
El tratamiento con WTX101 se asoció a mejoras rápidas en NCCcorregido, de forma que las concentraciones medias de NCCcorregido estuvieron por debajo del límite superior de la normalidad en la semana 12 (FIG. 2). A las 24 semanas, 20 (71 %, IC del 95 % 51,3-86,8; p<0,0001) de los 28 pacientes habían logrado el éxito del tratamiento: 16 (57 %) lograron o mantuvieron concentraciones normalizadas de NCCcorregido y 4 (14 %) tuvieron una reducción de al menos el 25 % en NCCcorregido desde el nivel basal. En general, NCCcorregido medio se redujo en el 72 % desde el nivel basal hasta la semana 24 (diferencia de la media de los mínimos cuadrados -2,4 pmol/l [Ee 0-4], IC del 95 % -3,2 a -1,6; p<0,0001; Tabla 1, FIG. 2).
Tabla 1: Cambios desde el nivel basal hasta la semana 24
en los criterios primarios y secundarios de valoración


Los datos en la Tabla 1 son valores medios absolutos basados en casos observados que no incluyen datos faltantes o datos de pacientes que se retiraron. NCCcorregido se refiere a los niveles de cobre no unido a la ceruloplasmina corregido para cobre en los complejos de tetratiomolibdato-cobre-albúmina. UWDRS se refiere a la Escala unificada de valoración de la enfermedad de Wilson. INR se refiere a la relación normalizada internacional. ALT se refiere a alanina aminotransferasa. AST se refiere a aspartato aminotransferasa. GGT se refiere a Y -glutamiltransferasa. EQVAS se refiere a la Escala analógica visual EuroQoL.
La incapacidad relacionada con la enfermedad mejoró significativamente después del tratamiento con WTX101 (Tabla 1, FIG. 3). Las puntuaciones medias de la parte II de UWDRS mejoraron desde 6,6 (DE 10,0) en el nivel basal hasta 4,1 (8,2) en la semana 24 (Tabla 1, FIG. 3A). La puntuación de la parte II de UWDRS mejoró en al menos un punto en 12 (57 %) pacientes y no cambió en nueve (43 %) pacientes; ningún paciente notificó deterioro, excepto un paciente que interrumpió el tratamiento a las 21 semanas debido a un mayor deterioro de la enfermedad neurológica existente.
Las puntuaciones medias para la parte III de UWDRS mejoraron significativamente desde 22,8 (21,0) en el nivel basal hasta 16,6 (17,7) en la semana 24 (Tabla 1, FIG. 3B). La puntuación de la parte III de UWDRS mejoró en 4 o más puntos en 14 (67 %) pacientes y se estabilizó (en 3 puntos del nivel basal) en cinco (24 %) pacientes en la semana 24. Hubo deterioro de cinco puntos en dos (10 %) pacientes: un paciente puntuó 19 en el nivel basal con fluctuaciones entre 16 y 27 durante el estudio, y el segundo paciente puntuó 3 en el nivel basal y 8 en las semanas 18 y 24; la incapacidad se valoró 0 en estos pacientes. Sorprendentemente, no se registraron casos de empeoramiento neurológico paradójico atribuido al fármaco del estudio en las 12 semanas después del inicio del tratamiento.
Aunque las mejoras en INR y albúmina desde el nivel basal hasta la semana 24 fueron estadísticamente significativas, fueron numéricamente pequeñas, que indica función hepática estable (Tabla 1). El estado del hígado, como se estima por MELD y la puntuación de Nazer modificada, tampoco varió en gran medida durante todo el estudio (Tabla 1).
Las mejoras clínicas se reflejaron por puntuaciones de EQ VAS medias significativamente elevadas (Tabla 1).
El tratamiento con WTX101 fue, en general, bien tolerado y la mayoría de los acontecimientos adversos fueron de intensidad leve o moderada (resumido en la Tabla 2). Los presentes inventores registraron aumentos en las concentraciones de enzima, principalmente ALT, AST o Y -glutamiltransferasa, en las pruebas de la función hepática de 11 (39 %) de los 28 pacientes que recibieron WTX101 a 30 mg/día o más, sin elevada bilirrubina. Estos aumentos ocurrieron normalmente después de 4-10 semanas, y fueron principalmente leves o moderados, todos asintomáticos, y se normalizaron en 1-2 semanas después de los ajustes de dosis o la interrupción del tratamiento de hasta 6 semanas. La mediana de la ALT máxima en estos 11 pacientes fue 197 U/l (intervalo 101-1341), un aumento de 7,2 veces desde el nivel basal. Tres (11 %) pacientes con aumentos de ALT de entre 14,3 veces y 29,3 veces desde el nivel basal interrumpieron el tratamiento. La segunda señal de ALT más alta (615 U/l) ocurrió en uno de los primeros pacientes reclutados, que recibió WTX101 a 120 mg/día. La pauta posológica por protocolo fue modificada posteriormente para reducir la dosis máxima a 60 mg/día. El paciente con la señal de ALT más pronunciada (1341 U/l) recibió 30 mg/día de WTX101 y tuvo elevaciones de ALT (máximo 400 U/l) con tratamiento previo con penicilamina antes del reclutamiento. En los tres pacientes en los que el fármaco se interrumpió debido al aumento de las
concentraciones de ALT, los resultados anormales de las pruebas del hígado fueron reversibles y no se asociaron con un aumento notable en la bilirrubina.
Tabla 2: Acontecimientos adversos y acontecimientos adversos graves durante el tratamiento

Se enumeran los acontecimientos adversos surgidos durante el tratamiento que fueron notificados por el investigador en al menos dos pacientes y todos los acontecimientos adversos graves surgidos durante el tratamiento. Un paciente podría haber tenido más de un acontecimiento adverso o acontecimiento adverso grave. Se evaluó que era poco probable que los siete acontecimientos adversos graves clasificados como trastornos psiquiátricos y como alteración de la marcha se relacionaran con el fármaco del estudio, mientras que los cuatro acontecimientos restantes estuvieron posiblemente o probablemente relacionados. El trastorno de adaptación fue notificado en un paciente, que entonces tuvo una exacerbación registrada de la alteración de la situación aguda 6 semanas después (enumerado aquí como un acontecimiento adverso grave). En un paciente diferente, se notificó manía en dos ocasiones separadas en un periodo de 3 semanas durante el estudio de 24 semanas (enumerado aquí como un acontecimiento adverso grave). ALT se refiere a alanina aminotransferasa. GGT se refiere a Y -glutamiltransferasa. AST = aspartato aminotransferasa.
Dos (7 %) pacientes tuvieron leucopenia y uno (4 %) tuvo trombocitopenia notificados como probablemente, posiblemente o definitivamente relacionados con el fármaco del estudio; sin embargo, todos se recuperaron después de los ajustes de la dosis. Algunos pacientes notificaron acontecimientos adversos gastrointestinales o relacionados con la piel con el tratamiento del estudio (Tabla 2).
Se notificaron once acontecimientos adversos graves en siete (25 %) pacientes: trastornos psiquiátricos (seis acontecimientos en cuatro pacientes), aminotransferasas hepáticas elevadas (dos acontecimientos en dos pacientes,
uno con agranulocitosis), alteración de la marcha (un acontecimiento) y deterioro en el funcionamiento neurológico (un acontecimiento; Tabla 2). Se evaluó que los acontecimientos adversos graves psiquiátricos y la alteración de la marcha eran remotos o poco probables que se relacionaran con el fármaco del estudio debido a las manifestaciones preexistentes de enfermedad neurológica o psiquiátrica, mientras que los otros cuatro acontecimientos adversos graves estuvieron posiblemente o probablemente relacionados con el tratamiento.
Un paciente previamente tratado que tuvo empeoramiento neurológico antes del reclutamiento tuvo un mayor deterioro neurológico después de la semana 12, a pesar del tratamiento del estudio, y se retiró en la semana 21, con un aumento de tres puntos en la parte II de UWDRS y un aumento de 11 puntos en la parte III de UWDRS desde el nivel basal. Se evaluó que el deterioro neurológico era probablemente debido al empeoramiento natural de la enfermedad, aunque no se pudo descartar la causalidad. Los investigadores interrumpieron el tratamiento para dos (7 %) pacientes debido a que los síntomas psiquiátricos o conductuales condujeron a su incapacidad para seguir el protocolo. Estos pacientes tuvieron puntuaciones de la parte III de UWDRS mejoradas o inalteradas.
Discusión
Los resultados muestran que WTX101 indujo un control rápido del cobre con reducciones significativas de NCCcorregido después de aproximadamente 3 meses, acompañado de mejoras tempranas significativas en los síntomas neurológicos y la función en la mayoría de pacientes. Este estudio fue el primer ensayo prospectivo multinacional realizado en pacientes con enfermedad de Wilson y tuvo el objetivo de evaluar el tratamiento con una nueva medicación oral, WTX101, con ventajas significativas en la administración y la dosis.
Los tratamientos previamente disponibles podían necesitar varios años para mostrar la mejoría clínica en pacientes con enfermedad de Wilson. La función hepática podría normalizarse durante 1-2 años de las pautas de tratamiento previas en la mayoría de los pacientes con enfermedad hepática o cirrosis compensada en la presentación, mientras que la mejoría de los síntomas en aquellos con enfermedad neurológica era más lenta y podía no mejorar o resolverse tan frecuentemente como la función hepática. Sin desear quedar ligado a teoría particular alguna, es posible que las rápidas mejorías bioquímicas y clínicas observadas con WTX101 estén relacionadas posiblemente con su novedoso, específico por el cobre y directo mecanismo de acción hepático de reducir las concentraciones de cobre libre tóxico en plasma. El tratamiento de pacientes que presentaron síntomas neurológicos fue particularmente exigente debido a que aproximadamente la mitad de aquellos con enfermedad neurológica en la presentación todavía tenían signos residuales, incluso años después de terapia con quelación. Además, se pudo observar empeoramiento neurológico prematuro paradójico en pacientes con enfermedad de Wilson neurológica incluso después del inicio del tratamiento con quelantes habituales, y las deficiencias neurológicas pueden ser irreversibles en de un tercio a la mitad de los pacientes. En un estudio por Litwin y colaboradores, se observó empeoramiento neurológico prematuro en 12 (29 %) de los 42 pacientes con enfermedad de Wilson neurológica tratados con penicilamina, con un tiempo medio hasta el empeoramiento neurológico desde el inicio de 2,3 meses. Litwin et al. Early neurological worsening in patients with Wilson's disease. J. Neurol Sci 355:162-67 (2015). Posiblemente, la unión de WTX101 al cobre en un complejo de proteína inerte y grande que no se puede redistribuir al SNC podría ser la responsable de la evidente ausencia de empeoramiento neurológico prematuro en las primeras 12 semanas de tratamiento. Una posibilidad es que los pacientes tratados previamente hayan recibido la terapia de quelación durante un periodo lo suficientemente largo como para que no se haya observado un empeoramiento neurológico con el tratamiento posterior con WTX101; sin embargo, no se registraron casos de empeoramiento neurológico prematuro en pacientes sin tratamiento previo. Un paciente previamente tratado (<28 días de cinc) con empeoramiento neurológico antes del reclutamiento tuvo un mayor deterioro neurológico, a pesar del aumento gradual de dosis, debido a una respuesta clínica no adecuada a WTX101, y el tratamiento del estudio se interrumpió en la semana 21. Aunque el deterioro neurológico se evaluó como probablemente debido al empeoramiento natural de la enfermedad, no se pudo descartar la causalidad. Además del paciente que se retiró en la semana 21, otros dos participantes interrumpieron el tratamiento con WTX101 por motivos neurológicos o psiquiátricos. Las manifestaciones neurológicas, psiquiátricas, o una combinación de ambas, neurológicas y psiquiátricas, ocurren frecuentemente en pacientes con enfermedad de Wilson y fueron responsables de que estos dos participantes fueran incapaces de seguir los procedimientos del estudio. Las afecciones psiquiátricas preexistentes también requirieron varios ingresos hospitalarios durante el estudio, que por definición se documentaron como acontecimientos adversos graves. De hecho, ocho de los 11 acontecimientos adversos graves notificados fueron de naturaleza neurológica o psiquiátrica y ocurrieron en cinco pacientes.
La función sintética del hígado pareció ser estable con WTX101 durante el estudio de 24 semanas, como se muestra por los resultados para INR y concentraciones de albúmina. Este resultado no se observó en todos los pacientes, independientemente de si tenían evidencia de cirrosis. Cabe señalar que los aumentos reversibles en las pruebas de la función hepática se observaron en el 39 % de los pacientes en el estudio de los presentes inventores, independientemente de la fase de la enfermedad hepática. Los aumentos ocurrieron 4-10 semanas después del inicio, con 30 mg/día o más, y fueron principalmente leves o moderados. Los pacientes que tuvieron elevaciones fueron todos asintomáticos con respecto a su enfermedad hepática. Los resultados de las pruebas de la función hepática se normalizaron en 1-2 semanas después de los ajustes de la dosis o la interrupción del tratamiento. No hubo aumentos notables en la bilirrubina en pacientes con elevados resultados de pruebas de la función hepática, incluyendo a aquellos que dejaron de tomar el fármaco, que indica la ausencia de una grave lesión en el hígado inducida por el fármaco. Ocurrieron aumentos similares reversibles tempranos de la aminotransferasa, dependiente de la dosis, en
pacientes con enfermedad de Wilson tratados con tetratiomolibdato de amonio, que también respondieron a la reducción de dosis o a la interrupción del tratamiento. Sin embargo, no se notificaron anomalías de pruebas hepáticas similares con el uso de tetratiomolibdato en pacientes sin enfermedad hepática o cirrosis biliar primaria, lo que indica que los efectos observados son específicos de la enfermedad de Wilson. Un mecanismo para el aumento de los niveles de enzimas hepáticas en respuesta al tratamiento con WTX101 se podría relacionar con su actividad moduladora del cobre, de forma que la retirada de cobre de los conjuntos hepáticos, que incluyen la metalotioneína, condujo a un aumento transitorio posterior en las aminotransferasas hepáticas. Sin embargo, queda por elucidar el mecanismo exacto.
Los tratamientos previamente disponibles para la enfermedad de Wilson fueron objeto de otros acontecimientos adversos potencialmente graves que frecuentemente condujeron a la interrupción y a cambios en el tratamiento. En un estudio retrospectivo, el 32 % de los pacientes con quelantes y el 11 % de los pacientes con cinc interrumpieron el tratamiento debido a acontecimientos adversos. La penicilamina está asociada con reacciones de sensibilidad prematura, tales como fiebre y urticaria, y diversas reacciones de la piel, y reacciones posteriores que incluyen síndrome de tipo lupus y nefrotoxicidad. Los acontecimientos adversos gastrointestinales se notifican frecuentemente con el tratamiento con cinc; el 40 % de los niños tratados con cinc tuvieron acontecimientos adversos gastrointestinales en una cohorte polaca. Wiernicka et al., Gastrointestinal side effects in children with Wilson's disease treated with zinc sulphate. World J Gastroenterol 19:4356-62 (2013). En el estudio de los presentes inventores, dos pacientes notificaron cada uno náuseas o piel seca, que se consideraron relacionados con el tratamiento del estudio, pero estos fueron, en general, leves o moderados, y no condujeron a interrupción. Seis pacientes interrumpieron el tratamiento del estudio. Estas interrupciones reflejan la naturaleza crónica limitante de la vida de los pacientes sintomáticos con enfermedad de Wilson, teniendo la mayoría de los pacientes reclutados diversos grados de manifestaciones neurológicas o psiquiátricas, y el enfoque exploratorio en el que tuvo que modificarse la pauta posológica inicial.
Los tratamientos previamente disponibles para la enfermedad de Wilson fueron recetados como dosis diarias múltiples, hasta cuatro veces al día, y deben ser tomados sin comida. Los estudios sugieren que hasta el 45 % de los pacientes tratados con las actuales terapias tienen poco cumplimiento terapéutico a largo plazo o es problemático. El fracaso de seguir una terapia de por vida podría conducir a una reaparición de los síntomas y a la progresión de la enfermedad hepática, o síntomas neurológicos o psiquiátricos, aunque hay variabilidad individual en el marco temporal. La administración oral una vez al día es posible con WTX101, y podría conducir a un mejor cumplimiento terapéutico del tratamiento y a mejores resultados de los pacientes.
Aunque el tamaño de muestra es suficiente para un ensayo de fase 2 en una enfermedad huérfana, es relativamente pequeño cuando se evalúan los resultados en una enfermedad clínicamente heterogénea. Sin embargo, todos los resultados fueron coherentes, independientemente del parámetro de evaluación aplicado, lo que soporta el efecto beneficioso global de WTX101 en la población estudiada. Se excluyeron los pacientes con enfermedad hepática descompensada con elevadas puntuaciones de MELD. Fue necesaria una población mixta de pacientes sin tratamiento previo y previamente tratados debido a las dificultades de reclutamiento asociadas a una enfermedad rara, y puesto que la enfermedad de Wilson se trata frecuentemente muy rápidamente después del diagnóstico. La inclusión de esta población en el estudio de los presentes inventores también permitió la evaluación de los efectos de WTX101 en pacientes que previamente recibieron terapia de quelación o cinc. El ensayo fue no controlado y de etiqueta abierta. Aunque se desea un control interno, esto no es siempre factible en las etapas iniciales del desarrollo de un nuevo fármaco para enfermedades raras. Sin embargo, hubo mejoras en los resultados no susceptibles a sesgo, que incluyen control del cobre y función hepática. Aunque el ensayo fue de duración relativamente corta, están en curso estudios de prolongación para investigar más la seguridad y la eficacia a largo plazo de WTX101.
En conclusión, el tratamiento con WTX101 redujo rápidamente el cobre libre en pacientes con enfermedad de Wilson, y este control del cobre se asoció a incapacidad reducida, estado neurológico mejorado y función hepática estable durante 24 semanas. Con los ajustes de dosis, WTX101 mostró un perfil de seguridad favorable con una simple pauta posológica oral de una vez al día sin efectos del alimento. Por lo tanto, WTX101 tiene el potencial de tratar varias necesidad clínicas sin cumplir.
Ejemplo 2: Eficacia y seguridad a largo plazo de WTX101 en la enfermedad de Wilson: Datos de una prolongación en curso de un estudio de fase 2.
En el Ejemplo 1, la monoterapia oral con WTX101 una vez al día redujo y controló rápidamente el NCC, mejoró la incapacidad y el estado neurológico, sin empeoramiento neurológico prematuro inducido por el fármaco, y estabilizó la función hepática en pacientes con EW después de 24 semanas. Los datos de eficacia y seguridad de 72 semanas del periodo de prolongación en curso del estudio de fase 2 representan el primer informe prospectivo sobre el control de la enfermedad a largo plazo con WTX101 en EW.
El Ejemplo 1 fue un ensayo de fase 2 de etiqueta abierta, multicéntrico y de un solo grupo realizado en 28 adultos con un diagnóstico de EW establecido por una puntuación de Leipzig de >4. Para la inclusión, los niveles de NCC tuvieron que estar por encima del intervalo de referencia del límite inferior de la normalidad (>0,8 pM). Los participantes no recibieron ni tratamiento anterior para EW (n=9) ni tratamiento anterior de <24 meses con quelación o cinc (<28 días, n=9; 28 días a 2 años, n=10). Los participantes recibieron WTX101 durante 24 semanas usando una pauta posológica
guiada por respuesta con dosis individualizadas entre 15 y 120 mg/día basadas en los niveles de NCC, evaluaciones clínicas y criterios de seguridad.
Los datos se recogieron desde las primeras 72 semanas del periodo de prolongación para los siguientes parámetros: (a) niveles de NCC, corregidos para cobre unido contenido en complejos de tetratiomolibdato-cobre-albúmina (NCCcorregido); (b) estado del hígado, medido usando medidas de laboratorio habituales y también por la puntuación del modelo para enfermedad hepática terminal (MELD) (basada en bilirrubina, creatinina y la relación normalizada internacional [INR]); (c) incapacidad notificada por el paciente usando la parte II de la Escala unificada de valoración de la enfermedad de Wilson (UWDRS) y el estado neurológico usando la parte III de UWDRS; y (d) la seguridad se muestra a continuación.
En el nivel basal del estudio principal, el 46 % de los pacientes tuvieron cirrosis del hígado basada en los antecedentes médicos (n=7) o por estimaciones del índice de la relación entre AST y plaquetas (n=6). En la semana 24, o en la última dosis recibida para pacientes con interrupción prematura, las dosis diarias fueron 15 mg para 6 pacientes, 30 mg para 13 pacientes y 60 mg para 9 pacientes. Los 22 pacientes que completaron el estudio principal inicial de 24 semanas participaron en el periodo de prolongación descrito en este ejemplo.
Veinte pacientes finalizaron el tratamiento hasta la semana 72. Un paciente interrumpió el tratamiento debido a su deseo de concebir. Un paciente fue incapaz de cumplir los procedimientos del estudio debido a un tratamiento para enfermedad progresiva a pesar del tratamiento en curso.
Tabla 3. Características demográficas y de participantes
en el nivel basal de los periodos principales y de prolongación.

Niveles de cobre libre en plasma
Se redujo el elevado NCCcorregido medio (EEM) en el nivel basal (3,6 [0,4] pM) y se controló en la semana 24 (0,9 [0,2] pM) y siguió controlado en la semana 72 (0,5 [0,2] pM) (FIG. 4).
Función hepática
Pareció que mejoraron la INR media, la albúmina, los niveles de ALT y la puntuación de MELD o no variaron entre la semana 24 y la semana 72, que indica estabilidad de la función hepática (FIG. 5).
No se observaron elevaciones de ALT reversibles que requirieran ajustes de dosis, observadas en el 39 % de los pacientes (con >30 mg/día) hasta la semana 24 (Ejemplo 1), en la prolongación.
Los pacientes mostraron mejorías continuadas en la puntuación media de incapacidad de UWDRS y la puntuación de signos neurológicos desde la semana 24 a 72 (FIG. 6).
WTX101 fue, en general, bien tolerado durante 72 semanas de tratamiento. En general, el número de acontecimientos adversos (AE) notificados y AE graves (SAE) disminuyó en aproximadamente el 50 % desde las semanas 1-24 hasta las semanas 25-72 (FIG. 7 , Tabla 4). Entre la semana 24 y 72, el 89 % de los AE fueron leves o moderados, considerándose el 89 % no relacionados o que probablemente estuvieran poco relacionados con la terapia.
En el nivel basal, bajas cifras de plaquetas (56 %) y neutrófilos (32 %) fueron comunes, con notificación similar durante todo el seguimiento. La mayoría de las apariciones no estuvieron acompañadas por bajos niveles de NCC y fue poco probable que reflejaran deficiencia de cobre. Durante el periodo de seguimiento, la baja hemoglobina fue poco frecuente: el 5,2 % de las mediciones se encontraron por debajo del intervalo de la normalidad y 7 pacientes tuvieron baja hemoglobina en algún momento, pero no hubo mediciones por debajo de 100 g/l. Dos sujetos tuvieron evidencia de neutropenia acompañada de anemia leve y bajos niveles de NCC, posiblemente de acuerdo con la deficiencia de cobre en la semana 36 y 72, respectivamente; ambos respondieron rápidamente a la reducción de dosis.
Tabla 4. SAE durante los periodos principales y de prolongación

Se conservaron las mejorías iniciales en los niveles de cobre libre, y el estado hepático y neurológico, o mejoraron más con WTX101 entre las semanas 24 y 72, lo que demuestra que WTX101 una vez al día proporciona un control de la enfermada a largo plazo en la EW. WTX101 es bien tolerado en pacientes con EW más allá de las 24 semanas de tratamiento. Estos resultados, junto con su pauta posológica simple, indican que WTX101 trata varias necesidades sin cumplir en el tratamiento de EW.
Ejemplo 3: Un estudio de fase 3, aleatorizado, cegado por el evaluador y multicéntrico para evaluar la eficacia y la seguridad de WTX101 administrado durante 48 semanas frente al tratamiento de referencia en sujetos con la enfermedad de Wilson de 18 años y mayores con una fase de prolongación de hasta 60 meses.
Sumario del diseño del estudio
Se realizará un estudio aleatorizado, cegado por el evaluador y multicéntrico que evaluará la eficacia y la seguridad de una pauta posológica individualizada con WTX101 administrado durante 48 semanas, en comparación con SoC, en sujetos con EW de 18 años y mayores.
Se reclutará a aproximadamente 102 sujetos en aproximadamente 5 a 10 sitios en América del Norte y 15 a 25 sitios en el resto del mundo.
Sujetos que cumplen los requisitos con EW, que han recibido terapia SoC (es decir, terapia de quelación con penicilamina o trientina, terapia con Zn, o una combinación de ambas, terapia de quelación y con Zn) durante >28 días (cohorte 1), o que no han recibido tratamiento previo o que han recibido terapia SoC (es decir, terapia de quelación con penicilamina o trientina, terapia con Zn, o una combinación de ambas, terapia de quelación y con Zn) durante <28 días (cohorte 2), serán aleatorizados en una relación 2:1 con respecto al tratamiento con WTX101 o SoC (ya sea como terapia continuada en la cohorte 1 o como terapia continuada o inicial en la cohorte 2). La aleatorización se estratificará por terapia SoC previa.
Los sujetos que cumplen todos los criterios de inclusión y ninguno de los de exclusión serán reclutados en el estudio y estudiados como pacientes ambulatorios. Se requerirá que los sujetos previamente tratados que son aleatorizados para recibir WTX101 se sometan a un periodo de lavado de >48 horas antes de la terapia SoC inmediatamente antes del inicio del tratamiento con WTX101. Los sujetos que cumplen los requisitos aleatorizados a WTX101 recibirán WTX101 como comprimidos de administración retardada para administración por vía oral con dosis que varían desde 15 mg cada dos días (QOD) hasta 60 mg QD. Se deberá considerar y acordar una dosis máxima de hasta 90 mg QD con el monitor médico y solo si se cumplen los siguientes criterios: NCCcorregido es > límite superior de la normalidad (ULN); alanina aminotransferasa (ALT) es <2 x ULN; y parámetros hematológicos (hemoglobina, plaquetas y neutrófilos) siguen por encima de los umbrales que requieren modificación de la dosis según la Tabla 1. Las evaluaciones de eficacia y de seguridad se realizarán en las visitas programadas, mientras que los acontecimientos adversos y las medicaciones concomitantes se monitorizarán continuamente durante todo el estudio.
El esquema de visitas consistirá en la visita de selección, la visita de reclutamiento, la fase de tratamiento y una visita de finalización del estudio (EOS) (o finalización anticipada [ET]). El programa de visitas se resume a continuación.
La visita de selección ocurrirá 28 días antes de la visita de reclutamiento (día 1). Después de la visita del día 1, se producirá una evaluación telefónica en la semana 1, semana 2, semana 30 y semana 42 (día 8, día 15, día 211 y día 295, respectivamente), seguido por visitas del estudio en la semana 4, semana 6, semana 8, semana 12, semana 18, semana 24, semana 36 y semana 48 (día 29, día 43, día 57, día 85, día 127, día 169, día 253 y día 337, respectivamente).
A los sujetos que hayan finalizado el periodo de tratamiento de 48 semanas se les ofrecerá la oportunidad de participar en una fase de prolongación del estudio para evaluar la seguridad y la durabilidad a largo plazo del efecto del tratamiento de WTX101. Si el sujeto elige no participar en esta fase de prolongación, se le ayudará en su transición a SoC para EW con la orientación de su médico local.
La visita EOS se realizará en la semana 52 (día 365) solo para sujetos que no entran en la fase de prolongación. Para cada sujeto que no entra en la fase de prolongación, la fase del estudio principal terminará aproximadamente 52 semanas (~365 días) después del inicio del tratamiento en el día 1 (48 semanas de tratamiento con una visita EOS 4 semanas después de la fecha de la última dosis). Para cada sujeto que entra en la fase de prolongación, la fase del estudio principal terminará aproximadamente 48 semanas (día 337) después del inicio del tratamiento en el día 1.
Modificación de dosis para sujetos individuales
Se detallan criterios específicos para la interrupción temporal de la dosis o la restricción de los aumentos de dosis de WTX101 en la Tabla 5.
Tabla 5: Modificación de dosis individuales



Se deben obtener dos resultados consecutivos (es decir, obtenidos en 2 visitas consecutivas del estudio) sobre parámetros previamente especificados de acuerdo con la toxicidad o el empeoramiento de parámetros específicos de laboratorio o la parte III de UWDRS, para que apliquen los criterios de modificación de la dosis. La repetición semanal de la prueba continuará hasta que los resultados o vuelvan al nivel basal o dentro de los límites del intervalo de la normalidad. Las pruebas se repetirán entonces 2 semanas después. Si los resultados siguen estando en el nivel basal o los límites del intervalo de la normalidad, entonces se reiniciará la coordinación del programa por protocolo.
Duración del estudio
Para cada sujeto, la fase del estudio principal terminará aproximadamente a las 52 semanas (~365 días) después del inicio de tratamiento en el día 1 del estudio (48 semanas de tratamiento con una visita EOS que ocurre 4 semanas después de la fecha de la última dosis, si el sujeto no entra en la fase de prolongación). A los sujetos que han finalizado el periodo de tratamiento de 48 semanas se les ofrecerá la oportunidad de participar en una fase de prolongación para evaluar la seguridad y la eficacia a largo plazo de WTX101. Si el sujeto elige no participar en la fase de prolongación, se le ayudará en su transición a SoC para EW con la orientación de su médico local.
Si el investigador considera necesaria una evaluación clínica adicional fuera del programa de visitas, o si el sujeto cumple los criterios de modificación de la dosis, entonces pueden realizarse visitas sin programar. Además, si existe un claro deterioro neurológico, como se demuestra por signos o síntomas de empeoramiento neurológico, entonces se realizarán evaluaciones neurológicas adicionales a criterio del investigador.
A todos los sujetos que finalicen el periodo de 48 semanas del tratamiento aleatorizado en WTX101-301 se les ofrecerá la oportunidad de participar en una fase de prolongación de etiqueta abierta de hasta 60 meses en la que recibirán terapia con WTX101. El fin de la fase de prolongación es evaluar la durabilidad y establecer la seguridad y la eficacia a largo plazo de WTX101. Los sujetos solo cumplirán los requisitos para seguir en la fase de prolongación hasta que WTX101 esté comercialmente disponible en su país respectivo.
Grupos de tratamiento
Los sujetos se tratarán con 15 mg QOD hasta 60 mg QD de WTX101 o terapia SoC. Los sujetos serán aleatorizados a 1 de 2 cohortes: cohorte 1 - sujetos tratados durante >28 días con terapia de quelación o con Zn o una combinación de ambas, terapia de quelación y con Zn, o cohorte 2 - sujetos que no han recibido tratamiento previo o han sido previamente tratados con terapia de quelación o con Zn o una combinación de ambas, terapia de quelación y con Zn durante <28 días.
Fundamento para la dosis
La dosis de WTX101 se ajustará en sujetos individuales, según convenga, basándose en las pautas especificadas en el protocolo. Una guía de dosis detallada para las modificaciones de la dosis de WTX101 se expone brevemente en la Tabla 5.
La dosis inicial de WTX101 es 15 mg QD para todos los sujetos. Después de 4 semanas, la dosis de WTX101 posterior será individualizada. La orientación de la dosis se basa en una variedad de factores, que incluyen la bioquímica clínica y la hematología, la evaluación clínica, la seguridad y NCCcorregido.
Son posibles los aumentos de dosis a criterio del investigador principal en incrementos de 15 mg separados al menos 4 semanas si la enfermedad no está controlada suficientemente, teniendo en cuenta el estado clínico del sujeto y los niveles de Cu libre en sangre, como se mide por NCC/NCCcorregido, y no aplica ninguno de los criterios
de modificación de dosis. Después del aumento escalonado de la dosis, los sujetos serán monitorizados para acontecimientos adversos y evaluaciones de laboratorio cada 2 semanas durante un periodo de 4 semanas. Si una visita rutinaria del estudio no está programada durante este periodo de tiempo, una enfermera de atención a domicilio puede completar estas evaluaciones.
Cuando los niveles de NCCcorregido han caído dentro del intervalo de la normalidad (<2,3 pmol/l), y el estado clínico del sujeto es estable o mejoró durante 2 evaluaciones consecutivas, se puede mantener o reducir la dosis de WTX101 a criterio del investigador principal. Para evitar el sobretratamiento, la dosis se puede reducir en cualquier momento, a criterio del investigador principal, guiado por lo siguiente: si el estado clínico del sujeto indica un posible sobretratamiento y/o los valores de NCC/NCCcorregido están por debajo del intervalo de la normalidad. Sin embargo, el uso de NCCcorregido como criterio para el ajuste de la dosis es opcional, lo que refleja la diferente práctica clínica en los sitios en el estudio global. La dosis debe ser reducida o interrumpida si se cumple cualquiera de los criterios de modificación de la dosis.
Administración del fármaco del estudio
WTX101 se proporcionará como comprimidos que contienen 15 mg de tetratiomolibdato de bis-colina para administración por vía oral. WTX101 se administrará a dosis que varían desde 15 mg QOD hasta 60 mg QD. Una dosis máxima de hasta 90 mg QD debe ser analizada y acordada con el monitor médico y solo si se cumplen los siguientes criterios: NCCcorregido es >ULN; ALT es <2 x ULN; y los parámetros hematológicos (hemoglobina, plaquetas y neutrófilos) siguen por encima de los umbrales que requieren modificación de la dosis según la Tabla 1.
Se administrará WTX101 durante el periodo de tratamiento de 48 semanas QD o QOD (administrado por la mañana), en el estado en ayunas (1 hora antes o 2 horas después de las comidas). Además, a continuación se incluyen los detalles sobre la administración de WTX101.
Se utilizará la administración individualizada de WTX101 durante todo el estudio basándose en los siguientes parámetros: Criterios clínicos: ajuste de la dosis basado en el estado hepático y neurológico; NCCcorregido: ajuste de la dosis aconsejado basándose en los niveles de NCC ajustados para la cantidad de Cu unido al TPC WTX101; y vigilancia de la seguridad: los criterios de modificación de la dosis se basan en evaluaciones programadas regularmente para efectos hematológicos reconocidos de la reducción de Cu, pruebas hepáticas y pruebas neurológicas.
La dosis de WTX101 también puede ser reducida en caso de que ocurra un acontecimiento adverso que requiera una reducción de dosis. La orientación de la dosis asociada a acontecimientos específicos se proporciona en los Criterios de modificación de la dosis (Tabla 5); las reducciones de dosis para otros acontecimientos serán tratadas caso por caso por el investigador principal del sitio (o subinvestigador, si es apropiado) en colaboración con el monitor médico.
En todos los sujetos, WTX101 se administrará a una dosis inicial de 15 mg QD en el día 1, que continúa durante las primeras 4 semanas. Después de las 4 semanas, se puede realizar el ajuste ascendente de la dosis hasta 30 mg QD a criterio del investigador principal, si la enfermedad no está controlada suficientemente, teniendo en cuenta el estado clínico del sujeto y los niveles de Cu libre en sangre, como se mide por NCC/NCCcorregido, y no aplica ninguno de los criterios de modificación de la dosis. Son posibles aumentos adicionales de dosis a criterio del investigador principal en incrementos de 15 mg separados al menos 4 semanas siguiendo los mismos criterios anteriormente mencionados.
Cuando los niveles de NCCcorregido han caído dentro del intervalo de la normalidad (<2,3 gmol/l), y/o el estado clínico del sujeto es estable o mejoró durante 2 evaluaciones consecutivas, se pueden mantener o reducir la dosis de WTX101 a criterio del investigador principal. Para evitar el sobretratamiento, la dosis se puede reducir en cualquier momento, a criterio del investigador principal, guiado por los siguientes: si el estado clínico del sujeto indica un posible sobretratamiento y/o los valores de NCC/NCCcorregido están por debajo del intervalo de la normalidad. Sin embargo, el uso de NCCcorregido como criterio para el ajuste de la dosis es opcional, lo que refleja la diferente práctica clínica en los sitios en el estudio global. La dosis debe ser reducida o interrumpida si se cumple cualquiera de los criterios de modificación de la dosis.
La dosis máxima esperada es 60 mg QD, pero se pueden considerar caso a caso dosis más altas en colaboración con el monitor médico. Una dosis máxima de hasta 90 mg QD debe ser analizada y acordada con el monitor médico y solo si se cumplen los siguientes criterios: NCCcorregido es >ULN; ALT es <2 x ULN; y los parámetros hematológicos (hemoglobina, plaquetas y neutrófilos) siguen por encima de los umbrales que requieren modificación de la dosis según la Tabla 5.
Para la exactitud en las mediciones, se siguen los siguientes parámetros. Los estrógenos puede interferir con la eliminación biliar de Cu. Se ha usado vitamina E como una terapia complementaria en las pautas de tratamiento de EW. Los sujetos no deben usar vitaminas y/o minerales que contengan Cu, Zn o Mo. Se sabe que los medios
de contraste que contienen gadolinio y yodo interfieren con las pruebas de Mo. Se pide no usar medios de contraste que contengan gadolinio y yodo en las 96 horas antes de la prueba de Mo. Se sabe que los medios de contraste que contienen bario interfieren con las pruebas de Cu. Se pide no usar medios de contraste que contengan bario en las 96 horas antes de las pruebas de Cu. Los sujetos deben evitar tomar alimentos y bebidas con altos contenidos de Cu durante toda la duración del estudio.
Tras la aleatorización y el periodo de lavado (si procede), los sujetos volverán para las visitas y los procedimientos: las visitas de la semana 1 a semana 30 y la visita de la semana 42 ocurrirán en ±3 días desde el momento programado; la visita de la semana 36 y la semana 48 ocurrirán en ±7 días desde el momento programado.
Evaluaciones de eficacia
La evaluación de eficacia primaria será el control de Cu libre, medido como el porcentaje de cambio desde el nivel basal (día 1) hasta 48 semanas en los niveles de NCC. Para sujetos tratados con WTX101, el nivel de NCC se corregirá para la cantidad de Cu unido al complejo tripartito de WTX101 (TPC).
Las evaluaciones de eficacia secundarias incluyen las siguientes: estado hepático usando la puntuación de MELD; incapacidad usando la parte II de UWDRS; estado neurológico usando la parte III de UWDRS; estado clínico usando la Escala de las impresiones globales clínicas (ítems 1 y 2 de la escala); y tasa de respondedores a NCC.
Las evaluaciones de eficacia terciarias incluyen las siguientes: evaluación individualizada de cada uno de los 3 síntomas más molestos de la mayoría de los sujetos; fibrosis hepática usando el índice de FIB-4 y elastografía transitoria; estado hepático usando la puntuación de Nazer modificada; síntomas psiquiátricos usando BPRS-24; y medidas del criterio de valoración de QoL/PRO usando EQ-5D y TSQM-9.
Las evaluaciones de eficacia exploratorias incluyen las sigu ientes:
Evaluaciones del control de Cu usando medidas exploratorias de Cu total, Cu libre, PUF-Cu y especiación del Cu de plasma; evaluación de los niveles de Mo en plasma; evaluación de Cu urinario y Mo urinario durante 24 horas; evaluación de la prueba de marcha cronometrada de 25 pies; evaluación de 9-HPT; evaluación de la prueba de interferencia no verbal de Stroop; y evaluación de la prueba de amplitud de dígitos.
Estadística
Todos los análisis estadísticos se realizarán según los procedimientos reconocidos en la técnica. Se expone brevemente a continuación una descripción general de los métodos estadísticos que se van a usar para analizar los datos de eficacia y seguridad. Los análisis estadísticos se llevarán a cabo usando SAS®, Versión 9.3 o posterior, SAS Institute, Cary, North Carolina, EE. UU.
El criterio principal de valoración es el control del Cu evaluado como el porcentaje de cambio desde el nivel basal hasta las 48 semanas en los niveles de NCC; para sujetos tratados con WTX101, el nivel de NCC se corregirá para la cantidad de Cu unido al TPC de WTX101.
El porcentaje de cambio desde el nivel basal en el nivel de NCC se analizará usando el análisis de medidas repetidas con modelos mixtos (MMRM) estratificado por cohorte y terapia SoC previa. Se incluirá el cambio desde el nivel basal en la semana 4, semana 8, semana 12, semana 24, semana 36 y semana 48. Se usará la estimación de la probabilidad máxima restringida. El modelo se estratificará por cohorte y terapia SoC previa. Para normalizar mejor los datos, el NCC puede ser transformado logarítmicamente antes del análisis. Se incluirán términos de efectos fijos para el tratamiento aleatorizado (WTX101 o SoC), visita e interacción por tratamiento aleatorizado por visita, y el nivel de NCC basal como covariable. La interacción de tratamiento por visita seguirá en el modelo independientemente de la significancia. Se usará una matriz de covarianza no estructurada para modelar el error intraindividual y se usará la aproximación de Kenward-Roger para estimar los grados de libertad. Si el ajuste de la estructura de varianza no estructurada deja de converger, se probarán las siguientes estructuras de covarianza con el fin de llegar hasta la convergencia: Toeplitz con heterogeneidad, autogresiva con heterogeneidad, Toeplitz y autogresiva. El principal contraste de interés será entre los sujetos tratados con WTX101 frente a con SoC en la semana 48. Se proporcionarán estimaciones basadas en modelo de la diferencia entre los tratamientos aleatorizados en el porcentaje medio de cambio en el nivel de NCC en la semana 48, junto con un intervalo de confianza (IC) del 95 % bilateral y el valor de p. Si el menor IC del 95 % bilateral excluye una diferencia de -15 %, entonces se concluirá con no inferioridad en la población general del estudio para WTX101 en relación con SoC; y si el menor IC del 95 % bilateral excluye una diferencia del 0 %, entonces se concluirá con superioridad en la población general del estudio para WTX101 con respecto a SoC. Se extraerán los cambios en la media de los mínimos cuadrados (LS) desde el nivel basal y los errores estándar (EE) asociados en los primeros momentos de tiempo (es decir, semana 4, semana 8, semana 12, semana 24 y semana 36) y se presentarán gráficamente por grupo con el tiempo.
El análisis complementario del criterio principal de valoración en la cohorte 1 reproducirá el descrito para el análisis de la población global, excepto que el análisis ya no se estratificará por cohorte.
El porcentaje de cambio desde el nivel basal en el nivel de NCC se analizará descriptivamente usando análisis de MMRM; no se hará comparación estadística formal entre los grupos de tratamiento aleatorizados. El porcentaje de cambio desde el nivel basal en la semana 4, semana 8, semana 12, semana 24, semana 36 y semana 48 se estimará usando los mismos términos del modelo que se describen para el análisis de los sujetos de la cohorte 1. El principal resultado de interés será la media LS, EE y el valor de p para el porcentaje de cambio desde el nivel basal en el nivel de NCC en la semana 48 dentro de cada uno. Dentro del grupo, se extraerán los cambios de la media LS y los EE desde los primeros momentos de tiempo basales (es decir, semana 4, semana 8, semana 12, semana 24 y semana 36) y se presentarán gráficamente por grupo con el tiempo.
Los criterios de valoración secundarios de la eficacia incluyen los siguientes: cambio desde el nivel basal en el estado hepático a las 48 semanas evaluado por la puntuación de MELD; cambio desde el nivel basal hasta las 48 semanas en la puntuación de la parte II de UWDRS; cambio desde el nivel basal hasta las 48 semanas en la puntuación de la parte III de UWDRS; cambio desde el nivel basal hasta 48 semanas en CGI-I y CGI-S; y tasa de respondedores de NCC a las 48 semanas.
Los criterios de valoración secundarios de la eficacia se analizarán del mismo modo que el criterio principal de valoración, por un análisis de MMRM estratificado por cohortes, siendo el principal contraste entre sujetos tratados con SoC frente a con WTX101 en la semana 48. También se realizará un análisis comparativo complementario en los sujetos de la cohorte 1 y se realizará uno descriptivo complementario en el análisis de grupos en los sujetos de la cohorte 2. Para los criterios de valoración b, c, y d, se analizarán puntuaciones totales.
La respuesta a NCC se define como la proporción de sujetos que logran o mantienen niveles de NCC normalizado o NCCcorregido (0,8 pM a 2,3 pM) o alcanzan una reducción de al menos el 25 % en NCC o NCCcorregido a las 48 semanas. Para sujetos tratados con WTX101, el nivel de NCC se corregirá para la cantidad de Cu unido al TPC WTX101.
Los sujetos sin valores a las 48 semanas se considerarán como no respondedores. Estos datos se analizarán por regresión logística estratificada por cohorte con términos para tratamiento aleatorizado y el nivel de NCC basal. Otra vez, se realizará un análisis comparativo complementario en los sujetos de la cohorte 1 y se realizará uno descriptivo complementario en el análisis de grupos en los sujetos de la cohorte 2.
Ejemplo 4: Absorción de te tratiom olibdato de b is-colina después de una adm in istración de dosis única de una form ulación con cubierta entérica con y sin a lim ento y un form ulación no recubierta coadm inistrada con un inh ib idor de la bomba de protones sin alimento
Se realizó un estudio cruzado, en un solo centro, de etiqueta abierta, aleatorizado, de 3 periodos, 3 tratamientos y 6 secuencias que evaluaba la FC (farmacocinética) de dosis únicas de tetratiomolibdato de bis-colina en sujetos sanos basándose en la medición de la concentración de Mo total en plasma. Dieciocho (18) hombres y mujeres adultos sanos no fumadores recibieron el Tratamiento A, B o C durante el transcurso de 3 periodos como se describe en el diagrama del estudio mostrado en las FIG. 8A y 8B. Los sujetos recibieron cada uno tratamiento en una ocasión. Los sujetos fueron aleatorizados a una de las seis siguientes secuencias de tratamiento: ABC, ACB, BAC, BCA, CAB y CBA. Todas las medicaciones del estudio fueron tomadas por vía oral con aproximadamente 240 ml de agua. Se les pidió a los sujetos que no macharan, dividieran ni masticaran la medicación del estudio.
Tratamiento A: 60 mg de tetratiomolibdato de bis-colina (2 x comprimidos con cubierta entérica (EC) de tetratiomolibdato de bis-colina, 30 mg, véase la Tabla 6) en la hora 0 en el día 1, después de un ayuno durante la noche.
Tratamiento B: 60 mg de tetratiomolibdato de bis-colina (2 x comprimidos EC de tetratiomolibdato de bis-colina 1, 30 mg) en la hora 0 en el día 1, 30 minutos después del comienzo de un desayuno hiperlipídico, precedido por un ayuno durante la noche.
Tratamiento C: 20 mg de omeprazol (1 x 20 mg de cápsula de liberación retardada de un PPI (inhibidor de la bomba de protones)) QD por la mañana de los días -5 a -1 tras un ayuno durante la noche, 20 mg de cápsula de liberación retardada de omeprazol en la hora -1 en el día 1 tras un ayuno durante la noche, y 60 mg de tetratiomolibdato de bis-colina (2 x cápsulas no recubiertas (UC) de tetratiomolibdato de bis-colina, 30 mg. Véase la Tabla 7) en la hora 0 en el día 1.
Tabla 6

Tabla 7

Los 18 sujetos que fueron reclutados y finalizaron el estudio se incluyeron en el análisis de seguridad y de FC. Sin embargo, se excluyó a un sujeto de la estadística descriptiva de FC y los análisis estadísticos para el Tratamiento B debido a una concentración de Mo total en plasma medible previa a la dosis > 40 % de la Cmáx correspondiente (concentración plasmática máxima medida). Por lo tanto, la población de análisis de FC comprendió 18 sujetos para el comprimido EC en ayunas (Tratamiento A) y UC PPI en ayunas (tratamiento C) y 17 sujetos para el comprimido EC posprandial (Tratamiento B).
Resultados farmacocinéticos
Los parámetros FC para Mo total en plasma se calcularon del siguiente modo:
ABCü-t: El área bajo la curva de la concentración plasmática frente al tiempo, desde tiempo 0 hasta la última concentración medible, como se calcula por la regla del trapecio lineal.
ABCü-inf: El área bajo la curva de la concentración plasmática frente al tiempo desde tiempo 0 hasta el infinito. El ABC0-inf se calcula como la suma de ABC0-t más la relación de la última concentración plasmática medible hasta la constante de la velocidad de eliminación.
Cmáx: Máxima concentración plasmática medida durante el periodo de tiempo especificado.
tmáx: Tiempo de la máxima concentración plasmática medida. Si el máximo valor ocurrió en más de un momento de tiempo, tmáx se definió como el primer momento de tiempo con este valor.
Az: Constante de velocidad de la eliminación terminal de primer orden aparente calculada a partir de un gráfico semilogarítmico de la curva de concentración plasmática frente al tiempo. El parámetro se calculó por análisis de regresión de mínimos cuadrados lineal usando el máximo número de puntos en la fase lineal logarítmica terminal (por ejemplo, 3 o más concentraciones plasmáticas distintas de cero), empezando con la última concentración distinta de cero.
t1/ 2 : Se calculó la semivida de eliminación terminal de primer orden aparente como 0,693/Az.
Tlat Tiempo de latencia de la absorción
CL/F: Depuración oral aparente
Vz/F: Volumen de distribución oral aparente
Como se muestra en las FIG. 8A y 8B, la media ± error estándar de las concentraciones plasmáticas de Mo total fue ligeramente menor después de la administración del comprimido EC en ayunas (Tratamiento A) en comparación con las cápsulas no recubiertas (UC) PPI en ayunas (Tratamiento C). Hubo, sin embargo, variabilidad entre los sujetos, mostrando 3 de los 18 concentraciones mucho menores para el comprimido EC, mostrando 5 el mismo patrón que los datos medios, y mostrando 10 concentraciones más comparables o superponibles para ambos tratamientos.
De acuerdo con las concentraciones plasmáticas medias, los valores medios aritméticos (Tabla 8) y geométricos (Tabla 7) para Cmáx, ABCü-t y ABCinf fueron menores para el comprimido EC en ayunas (Tratamiento A) que para la cápsula UC PPI en ayunas (Tratamiento C). Las GMR oscilaron del 75,81 % al 87,16 %, con límites inferiores de los IC al 90 % asociados < 80,00 % (Tabla 9), que indica una disminución en la exposición en promedio. La mediana de Tmáx fue comparable para ambos tratamientos, 4,54 y 4,50 horas, respectivamente, con intervalos comparables (Tabla 8).
Cuatro (4) de los 18 sujetos tuvieron un tiempo de latencia de la absorción después de la administración del comprimido EC en ayunas, con una mediana (intervalo) de 2,00 horas (2,00 a 3,00 horas) (Tabla 8).
La administración del comprimido EC con alimento (Tratamiento B) produjo una gran disminución en las concentraciones medias de Mo total en plasma (Figura 5). Esto también se observó en todos excepto 2 de los 17 sujetos evaluables para este tratamiento. Cmáx, ABC0-t y ABCinf fueron menores después de la administración del comprimido EC con alimento (Tablas 4 y 5), con GMR que variaban desde el 25,20 % hasta el 40,49 %, que indica una disminución sustancial en la absorción. La mediana de Tmáx fue comparable con y sin alimento, 4,55 y 4,54 horas, respectivamente, con intervalos comparables (Tabla 8). En comparación con el comprimido EC en ayunas (Tratamiento A), más sujetos (6 sujetos) tuvieron un tiempo de latencia de la absorción y aumento en la mediana (intervalo) hasta 3,00 horas (2,00 a 5,00 horas) (Tabla 8).
El t / medio fue esencialmente el mismo para los 3 tratamientos (Tabla 8), con una media global de ~48 horas o 2 días. CL/F y Vz/F también fueron comparables para los 2 tratamientos en ayunas pero, debido a la menor biodisponibilidad después de la administración del comprimido EC posprandial, son mayores para ese tratamiento.
Los menores coeficientes de variación interindividuales (BSCV) para Cmáx, ABC0-t y ABCinf se observaron para la cápsula UC PPI en ayunas (Tratamiento C), con valores que variaban desde el 15,8 % hasta el 19,1 % (Tabla 8). La administración del comprimido EC en ayunas (Tratamiento A) produjo mayores BSCV (26,1 % al 35,2 %; Tabla 8). Los BSCV fueron mucho más altos, particularmente para ABC0-t y ABCinf - 81,5 % y 72,6 %, respectivamente, (Tabla 8) -cuando el comprimido EC se administró después de la comida hipercalórica/hiperlipídica. En comparación con el comprimido EC en ayunas, los BSCV para las ABC fueron ~2,2 veces más altos cuando el comprimido EC se administró en condiciones posprandiales.
Como se ilustra en las FIG. 8A y 8B, hubo una ligera disminución en la media ± error estándar de las concentraciones plasmáticas de Mo total después de la administración del comprimido EC en ayunas (Tratamiento A) en comparación con UC PPI en ayunas (Tratamiento C). Se observaron tendencias similares con respecto a Cmáx, ABC(0-t) y ABC(inf) (Tablas 6 y 7). Sin embargo, el examen de datos de sujetos individuales indica que mientras que se observó un patrón similar con algunos de los sujetos individuales, la mayoría tuvo un perfil de concentración de Mo total-tiempo que era comparable para el comprimido EC de tetratiomolibdato de bis-colina y UC de tetratiomolibdato de bis-colina PPI cuando ambos se administraron en condiciones en ayunas. Sin embargo, la administración del comprimido EC de tetratiomolibdato de bis-colina posprandial (Tratamiento B) produjo una disminución del 60 % al 75 % en la absorción que fue coherente entre la mayoría de los sujetos.
Tabla 8: Resumen de parámetros FC de Mo total después de la administración de una única dosis 60 mg (2 x 30 mg) de comprimidos EC de WTX101 en ayunas (Tratamiento A) y condiciones posprandiales (Tratamiento B) y UC PPI en condiciones en ayunas (Tratamiento C)

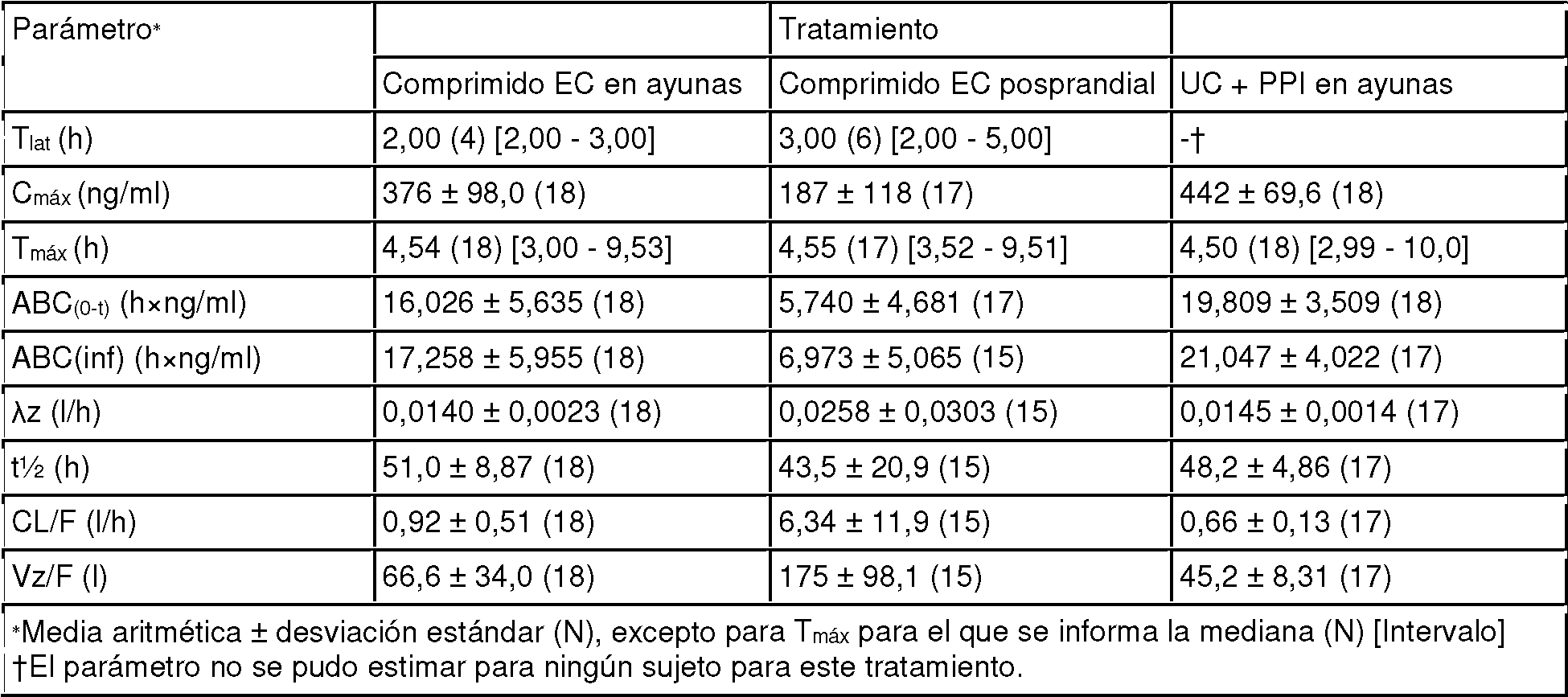
Tabla 9: Análisis estadístico de parámetros FC de Mo total después de la administración de una única dosis 60 mg (2 x 30 mg) de comprimidos EC de WTX101 en ayunas (Tratamiento A) y condiciones posprandiales (Tratamiento B) y UC PPI en condiciones en ayunas (Tratamiento C)

Tabla 10: Resumen de coeficientes de variación interindividual de Mo total, Cmáx, ABC0-t y ABCinf después de la administración de una dosis única de 60 mg (2 x 30 mg) de comprimidos EC de WTX101 en ayunas (Tratamiento A) y condiciones posprandiales (Tratamiento B) y UC PPI en condiciones en ayunas (Tratamiento C)

Ejemplo 5: Mejoría neurológica con el tratamiento con WTX101 en un estudio de fase 2, multicéntrico y de etiqueta abierta en enfermedad de Wilson
Se analizaron más los datos recogidos durante el estudio de fase 2 descrito en el ejemplo 1 para caracterizar los cambios neurológicos específicos después de un tratamiento de 24 semanas con WTX101.
Métodos
Pacientes adultos con EW (sin tratamiento previo o <2 años con terapia de quelación o con cinc) recibieron una dosis de WTX101 individual guiada por la respuesta (15-120 mg una vez al día) durante 24 semanas. Los cambios en el estado neurológico se caracterizaron usando la Escala unificada de valoración de la enfermedad de Wilson (UWDRS).
Resultados
De los 28 pacientes reclutados, 25 tuvieron manifestaciones neurológicas. Las puntuaciones medias basales de la parte II (incapacidad) y III (estado neurológico) de UWDRS fueron 6,6 (DE 10,0; intervalo 0-35) y 22,8 (DE 21,0; intervalo 0-83), respectivamente. En la semana 24, mejoraron tanto la puntuación media [DE] de la parte II de UWDRS (4,1 [8,2]; p<0,001) como la puntuación de la parte III (16,6 [17,7]; p<0,0001). Hubo una relación predictiva altamente significativa entre las puntuaciones totales de las partes II y III tomadas con el tiempo (p<0,0001). Las anomalías más comunes de la parte III de UWDRS en el nivel basal fueron el temblor postural de los brazos (71 %), la disartria (68 %), la marcha (61 %) y los ítems de la escala de destreza de las articulaciones y de coordinación, por ejemplo, movimientos alternantes de las manos (71 %), golpeteo con los dedos (57 %), caligrafía (54 %) y agilidad de las piernas (54 %). La mayoría de los ítems gravemente afectados fueron la caligrafía y la disartria (puntuaciones medias [DE] 2,0 [0,8] y 1,8 [0,9], respectivamente). Las mayores mejorías medias (% de cambio) durante las 24 semanas se observaron para caligrafía (51,4 %), agilidad de las piernas (40,8 %), temblor postural de los brazos (39,5 %) y movimientos alternantes de las manos (35,0 %). La agrupación de los ítems de temblor total o destreza de las articulaciones y coordinación mostraron mejorías similares (34,2 % y 29,2 %, respectivamente). WTX101 fue, en general, bien tolerado y no se observó empeoramiento neurológico prematuro inducido por el fármaco.
Conclusiones
Las manifestaciones neurológicas fueron comunes en esta cohorte de pacientes con EW. El tratamiento con WTX101 mejoró rápidamente la incapacidad y el estado neurológico en un ensayo prospectivo de fase 2 de 24 semanas en EW. La mejoría del estado neurológico después del tratamiento con WTX101 se correlacionó con una reducción de la incapacidad notificada por el paciente.
Ejemplo 6: Mejoría neurológica con el tratamiento con WTX101 en un estudio de fase 2, multicéntrico y de etiqueta abierta en enfermedad de Wilson
El estudio de fase 2 del Ejemplo 1 se analizó más para caracterizar los detalles de las manifestaciones neurológicas en pacientes con enfermedad de Wilson (EW) y los cambios neurológicos específicos después del tratamiento de 24 con WTX101. El análisis de estos datos se realizó, en general, como se describe en el Ejemplo 5.
Aunque las manifestaciones neurológicas pueden llegar a ser rápidamente muy incapacitantes, los tratamientos previamente existentes no siempre aliviaron los síntomas, algunas veces causaron el bien conocido empeoramiento prematuro paradójico de la enfermedad neurológica, y/o fueron poco tolerados por los pacientes. El tetratiomolibdato de bis-colina (WTX101) en un estudio de fase 2 de 24 semanas (NCT02273596; EudraCT 2014-001703-41) mostró un rápido control de los niveles de cobre no unido a la ceruloplasmina (NCC) y un perfil de seguridad favorable donde el nivel de NCC medio cayó por debajo del límite superior de la normalidad en la semana 12.
El análisis de las manifestaciones neurológicas, evaluado usando la Escala unificada de valoración de la enfermedad de Wilson (UWDRS), mostró una mejoría de los síntomas sin empeoramiento paradójico prematuro. Los cambios en las manifestaciones neurológicas en el estudio fueron examinados minuciosamente usando datos de la parte III (estado neurológico) del UWDRS. Además, este ejemplo explora la relación cuantitativa entre las manifestaciones neurológicas notificadas por los pacientes (parte II) y las clínicamente evaluadas (parte III) de la enfermedad de Wilson.
Métodos
Se incluyeron adultos (edad > 18 años) diagnosticados con enfermedad de Wilson (puntuación de Leipzig > 4) si no habían recibido tratamiento previo para enfermedad de Wilson, o habían recibido terapia de quelación o de cinc durante < 24 meses; y tenían una concentración de NCC por encima del intervalo de referencia del límite inferior de la normalidad (es decir, > 0,8 gmol/l). Se excluyeron los pacientes si tenían cirrosis hepática descompensada, una puntuación del modelo para enfermedad hepática terminal (MELD) > 11, o una puntuación de Nazer modificada (puntuación de King revisada) > 6.
El estudio de fase 2, de 24 semanas y de etiqueta abierta se realizó en Europa y los EE. UU. Durante las primeras 4 8 semanas, los pacientes recibieron WTX101 15-60 mg una vez al día, dependiendo de las concentraciones de NCC basal ajustadas para molibdeno en plasma (excepto en el nivel basal). A partir de aquí, la dosis se adaptó según las evaluaciones de laboratorio (incluyendo concentraciones de NCC ajustadas para molibdeno en plasma) y clínicas (máxima dosis diaria administrada: 120 mg).
Se evaluaron la incapacidad y el estado neurológico usando las partes II y III, respectivamente, de UWDRS en el nivel basal y las semanas 4, 8, 12, 18 y 24: Incapacidad (parte II): cuestionario de 10 ítems notificados por el paciente (intervalo para puntuaciones totales: 0-40; puntuaciones más altas indican mayor incapacidad). Estado neurológico (parte III): puntuación de 23 ítems evaluada por el profesional clínico (intervalo para puntuaciones totales: 0-143; puntuaciones más altas indican peor estado neurológico). Los datos (puntuaciones totales para las partes II y III de UWDRS, y AE) se resumen brevemente para el contexto en Resultados.
Se examinó la relación entre las puntuaciones totales de incapacidad y el estado neurológico para entender mejor y cuantificar el impacto del tratamiento sobre las manifestaciones neurológicas para proporcionar pautas de tratamiento óptimas mientras se minimizan los AE. Se calcularon las puntuaciones del estado neurológico de UWDRS en el nivel basal, semana 24, y el cambio desde el nivel basal hasta la semana 24 para: cualquier ítem individual experimentado por al menos el 50 % de los pacientes; y para grupos de ítems (es decir, conjuntos de ítems individuales que se podría considerar que representan fenotipos particulares del paciente [Tabla 11]).
Tabla 11. Composición de grupos de ítems neurológicos de UWDRS
Grupo de ítems Ítems del estado neurológico individual asignados al grupo (número Intervalo para de ítems de UWDRS) puntuación totalb Temblor total Temblor de reposo (15); temblor de la cabeza (16); brazos - temblor postural 0-45
(21A) y temblor de aleteo (21B); temblor postural - piernas (24); temblor de
la mandíbula (31)
Marcha total Levantarse de la silla (27); postura - distonía del tronco (28Aa), ataxia de la 0-32
postura (28B) y parkinsonismo (28C);
Marcha - distonía de las piernas (29Aa), ataxia (29B) y parkinsonismo (29C)
Distonía Distonía bucomandibular (13A); distonía cervical (25); distonía de brazos y 0-28
manos (26); distonía del tronco (28Aa); marcha - distonía de las piernas
(29Aa)
Agilidad y Golpeteo con los dedos (18); movimientos rápidos y alternantes de las 0-36 coordinación de las manos (19); caligrafía (20); prueba de dedo-nariz (22); agilidad de las extremidades piernas (23)
Rigidez Brazos, piernas y cuello (17) 0-20 aEl sub-ítem individual no es único para un grupo; bLas puntuaciones para ítems individuales se suman para dar una puntuación total para el grupo de ítems.
Se calcularon los valores de la media de los mínimos cuadrados (error estándar [EE], intervalos de confianza del 95 % [IC]) para cambios desde el nivel basal para las puntuaciones totales de UWDRS usando un análisis de medidas repetidas con modelos mixtos; se evaluó la significancia usando valores de la p bilaterales. Para investigar y cuantificar la relación entre las puntuaciones totales de UWDRS para las partes II y III, se realizó un análisis de coeficientes
aleatorios usando las puntuaciones de cada paciente de todas las visitas del estudio. Se calculó la estadística resumen para otros resultados.
Resultados
En total, 28 pacientes recibieron WTX101 (Tabla 12). Seis pacientes interrumpieron el tratamiento. Tres sufrieron AE de una naturaleza no neurológica, dos tuvieron dificultades psiquiátricas y no pudieron seguir el protocolo, y uno tuvo empeoramiento neurológico debido a la progresión de la enfermedad.
Tabla 12. Características basales
Pacientes (N = 28)
Mujeres, n (%) 15 (54)
Edad, años, media (DE) [intervalo] 34,1 (11,86) [18-64] Puntuaciones totales de UWDRS, media (DE)
Parte II (incapacidad) 6,6 (10,0)
Parte III (estado neurológico) 22,8 (21,0)
Anomalías neurológicasa, n (%) 25 (89)
Tratamiento previo para enfermedad de Wilson, n (%)
Ninguno 9 (32)
Menos de 28 días 9 (32)
Entre 28 días y 2 años 10 (36)
aPacientes con una puntuación de la parte III de UWDRS de > 1 en el nivel basal.
Incapacidad y estado neurológico de UWDRS: puntuaciones totales
Mejoraron las puntuaciones totales medias (DE) en la semana 24: 4,1 (8,2) para incapacidad y 16,6 (17,7) para estado neurológico. Las mejorías fueron significativas en ambos casos (media de los mínimos cuadrados [EE, IC del 95 %] cambio desde el nivel basal): -3,7 (0,9; -5,5 a -1,8; p = 0,0003) para incapacidad y -8,7 (1,9; -12,5 a -5,0; p < 0,0001) para estado neurológico. Hubo una relación lineal positiva significativa entre las puntuaciones totales de la parte II y la parte III de UWDRS (p < 0,0001).
Estado neurológico de UWDRS: ítems individuales y grupos de ítems
Nivel basal
Ítems individuales: De los seis ítems que afectan a al menos el 50 % de los pacientes, el temblor postural de los brazos, los movimientos rápidos y alternantes de las manos y la disartria (habla) fueron los más comunes (FIG.9). Permitiendo diferencias en las puntuaciones máximas posibles entre los ítems (es decir, 4 u 8), la caligrafía y el habla fueron las más gravemente afectadas (FIG. 10A y 10B).
Grupos de ítems: Se notificó temblor para la mayor proporción de pacientes (23 pacientes, 82 %), seguido por marcha (17, 61 %), distonía (15, 54 %), agilidad y coordinación de las extremidades (15, 54 %) y rigidez (12, 43 %). Permitiendo diferencias en las puntuaciones máximas posibles entre grupos de ítems (Tabla 11), la rigidez fue la más gravemente afectada (puntuación media [desviación estándar, DE]: 3,5 [2,9]), seguido por temblor (7,4 [7,0]), distonía (3,9 [4,4]), marcha (4,4 [4,1]) y agilidad y coordinación de las extremidades (1,3 [0,5]).
Cambios desde el nivel basal hasta la semana 24
Ítems individuales: Basándose en la estadística descriptiva, el porcentaje medio de mejorías fue el mayor para la caligrafía y la agilidad de las piernas (FIG. 11A y 11B) y algunos pacientes mostraron deterioro en las puntuaciones (FIG. 12). Dos pacientes tuvieron un deterioro de 2 puntos (golpeteo con los dedos); todos los otros deterioros fueron por 1 punto.
Grupos de ítems: Las mejoras para temblor (media [DE], 34,2 % [61,7]) y agilidad y coordinación de las extremidades (29,2 % [72,2]) fueron, en general, similares a aquellas para los ítems individuales. De los 22 pacientes en los análisis, cuatro y tres, respectivamente, mostraron deterioro en las puntuaciones (deterioro de 1 punto en cinco casos, 3 puntos en un caso [temblor] y 4 puntos en un caso [agilidad y coordinación] de las extremidades). En general, los deterioros para rigidez (26,9 % [169,6]), distonía (15,0 % [135,2]) y marcha (4,2 % [77,8]) fueron atribuibles a cuatro, tres y seis
individuos, respectivamente (los deterioros entre 1 y 8 puntos para rigidez, 2 y 4 puntos para distonía, y 1 y 6 puntos para marcha total).
Conclusiones
El estudio de fase 2 mostró que el tratamiento con WTX101 tiene el potencial de tratar importantes necesidades sin satisfacer en la enfermedad de Wilson. La mejoría del estado neurológico después del tratamiento con WTX101 se correlacionó con una reducción de la incapacidad notificada por el paciente. WTX101 se asoció con una reducida incapacidad y mejoró el estado neurológico, y fue bien tolerado.
Ejemplo 7: Análisis de pacientes con o sin cirrosis
La mortalidad en la enfermedad de Wilson es normalmente secundaria a la cirrosis hepática descompensada e insuficiencia hepática; por lo tanto, la función hepática es un factor importante en la determinación de las pautas de tratamiento apropiadas. Se analizó el desenlace de los pacientes con y sin cirrosis usando el conjunto de pacientes del Ejemplo 1, complementado con datos de la semana 48 generados en la fase de prolongación de 3 años del ensayo inicial. El Ejemplo 1 fue un estudio de 24 semanas de duración, de etiqueta abierta y de un solo grupo que usó inicialmente 15-60 mg/día de WTX101, luego dosis individualizada guiada por respuesta (máxima administrada, 120 mg/día). La fase de prolongación es un estudio de 3 años de duración, de etiqueta abierta y de un solo grupo que usa dosis individualizada guiada por respuesta continuada de WTX101 una vez al día.
Para los análisis descritos en este ejemplo, los pacientes se caracterizaron por tener cirrosis basada en los antecedentes médicos (biopsia u obtención de imágenes) o basada en estimaciones del índice de relación entre AST y plaquetas (APRI) (AST se refiere a aspartato aminotransferasa). Se calculó la estadística resumen para los datos demográficos basales, niveles de NCCcorr, niveles de ALT, MELD y puntuaciones de Nazer modificadas, concentración de albúmina, relación normalizada internacional (INR), incapacidad y estado neurológico usando la Escala unificada de valoración de la enfermedad de Wilson, y acontecimientos adversos (AE). De los 15 pacientes con cirrosis, 13 completaron el estudio principal, todos los cuales siguen en estudio de prolongación. De los 13 pacientes sin cirrosis, nueve completaron el estudio principal, todos los cuales siguen en el estudio de prolongación. El flujo de pacientes para el estudio de prolongación se representa en la FIG. 12.
NCCcorr en el nivel basal fue 3,6 gmol/l para pacientes con cirrosis y 3,7 gmol/l para pacientes sin cirrosis. WTX101 redujo los niveles de NCCcorr en grados similares en pacientes con y sin cirrosis, desde el nivel basal hasta la semana 24, y las mejoras observadas en la semana 24 se mantuvieron hasta la semana 48. Los niveles de NCCcorr se muestran en la FIG. 13.
En el estudio principal, las elevaciones de ALT que requieren ajustes de la dosis ocurrieron en cinco pacientes con cirrosis y siete pacientes sin cirrosis. Las elevaciones de ALT se asociaron con la retirada del estudio de tres pacientes sin cirrosis. Las elevaciones de ALT durante el estudio principal no estuvieron acompañadas por elevados niveles de bilirrubina. Desde la semana 24 hasta la semana 48, no ocurrieron elevaciones de ALT que requirieran ajustes de la dosis y los niveles de ALT fueron estables independientemente del estado de la cirrosis. Los niveles de ALT para pacientes con y sin cirrosis se muestran en la FIG. 14. La puntuación de MELD (gravedad de la enfermedad hepática; intervalo de puntuación, 6-40) (FIG. 15) y la puntuación de Nazer modificada (índice pronóstico; intervalo de puntuaciones, 0-20) (FIG. 16) fueron bajas en el nivel basal y siguieron siendo estables en el estudio principal y hasta la semana 48.
La concentración media de albúmina (FIG. 17A) y la relación normalizada internacional (INR) (FIG. 17B) fueron estables en el estudio principal y hasta la semana 48 en pacientes tanto con como sin cirrosis. La concentración media de albúmina (FIG. 17A) y el tiempo de coagulación de la sangre (FIG. 18) estuvieron dentro de los intervalos de la normalidad en el nivel basal. Estos siguieron siendo estables en el estudio principal y hasta la semana 48 en pacientes con y sin cirrosis. Se usó UWDRS para evaluar la incapacidad notificada por el paciente y el estado neurológico valorado por el profesional clínico. Las mejorías ocurrieron durante el estudio principal y hasta la semana 48, independientemente del estado de cirrosis (FIG. 19A y 19B). El perfil de tolerabilidad de WTX101 fue favorable y similar para pacientes con y sin cirrosis. Durante el estudio principal, los acontecimientos adversos (AE) ocurrieron en 10 pacientes con cirrosis y 12 sin cirrosis. Los AE más comunes aumentaron los niveles de ALT y GGT (cada uno en tres pacientes con cirrosis y cinco sin cirrosis), aumentaron los niveles de AST (cuatro pacientes en cada grupo) y temblor (cuatro pacientes sin cirrosis). Durante la fase de prolongación, menos pacientes sufrieron AE en comparación con el estudio principal (nueve pacientes con cirrosis y seis pacientes sin cirrosis). El AE más común fue la infección urinaria (cuatro pacientes con cirrosis). Ocurrieron acontecimientos adversos graves (SAE) en cuatro pacientes con cirrosis y seis pacientes sin cirrosis durante las semanas 0-48. Durante la fase de prolongación, dos pacientes en el grupo de cirrosis sufrieron SAE posiblemente relacionados con WTX101. Un paciente tuvo neutropenia y un paciente tuvo degeneración hepatolenticular.
En este análisis, las mejorías en el control de los niveles de cobre con tratamiento con WTX101 en comparación con el nivel basal se mantuvieron desde la semana 24 hasta las 48 semanas de tratamiento con WTX101 en ambos grupos de pacientes. Con WTX101, la función hepática fue estable hasta las 48 semanas en pacientes con y sin cirrosis. El
perfil de tolerabilidad de WTX101 fue favorable y no estuvo influido por la presencia de cirrosis. Por lo tanto, el tratamiento con WTX101 sigue siendo eficaz y bien tolerado, independientemente de si los pacientes tratados tenían o no cirrosis hepática antes del tratamiento.
Un experto en la técnica entendería que los umbrales particulares desvelados en el presente documento pueden variar algo dependiendo de las condiciones particulares de los métodos de prueba y ensayo. Además, un experto en la técnica entendería que los umbrales que se describen como "superior a" o "inferior a" podrían ser, en ciertas realizaciones, "iguales a o superiores a" o "iguales a o inferiores a," respectivamente. Similarmente, un experto en la técnica entendería que los umbrales que se describen como "iguales a o superiores a" o " iguales a o inferiores a" podrían ser, en ciertas realizaciones, "superiores a" o "inferiores a," respectivamente
La anterior descripción detallada se ha realizado para claridad de entendimiento solo y no se deben entender limitaciones innecesarias de la misma, ya que las modificaciones serán obvias para los expertos en la técnica.
Aunque la presente invención se ha descrito a propósito de realizaciones específicas de la misma, se entenderá que es susceptible de otras modificaciones y la presente solicitud pretende cubrir cualquier variación, uso o adaptación de la invención siguiendo, en general, los principios de la divulgación, e incluyendo dichas desviaciones de la presente divulgación que estén dentro de la práctica conocida o habitual dentro de la técnica a la que se refiere la invención y que puedan aplicarse a las características esenciales anteriormente expuestas en el presente documento y como sigue en el alcance de las reivindicaciones adjuntas
Claims (16)
1. Tetratiomolibdato de bis-colina para su uso en el tratamiento de la enfermedad de Wilson en un paciente, en donde el tetratiomolibdato de bis-colina se administra al paciente en un estado en ayunas.
2. Tetratiomolibdato de bis-colina para su uso según la reivindicación 1, en donde se administra 15 mg de tetratiomolibdato de bis-colina al paciente una vez al día.
3. Tetratiomolibdato de bis-colina para su uso según la reivindicación 1 o 2, en donde el tetratiomolibdato de bis-colina se administra al paciente como una forma farmacéutica de liberación retardada, preferentemente en donde la forma farmacéutica es un comprimido o cápsula o la forma farmacéutica es un comprimido con cubierta entérica.
4. Tetratiomolibdato de bis-colina para su uso según una cualquiera de las reivindicaciones 1 a 3, en donde el paciente presenta una reducción en NCCcorregido como se mide después de 24 semanas de la administración en comparación con el NCCcorregido del paciente como se mide antes de la administración, tal como al menos una reducción del 20 %, al menos una reducción del 35 %, al menos una reducción del 50 %, o al menos una reducción del 75 % en NCCcorregido como se mide después de 24 semanas de la administración en comparación con el NCCcorregido del paciente como se mide antes de la administración.
5. Tetratiomolibdato de bis-colina para su uso según una cualquiera de las reivindicaciones 1 a 4, en donde el paciente presenta una reducción en NCCcorregido como se mide después de 48 semanas de la administración en comparación con el NCCcorregido del paciente como se mide antes de la administración, tal como al menos una reducción del 20 %, al menos una reducción del 35 %, al menos una reducción del 50 %, o al menos una reducción del 75 % en NCCcorregido como se mide después de 48 semanas de la administración en comparación con el NCCcorregido del paciente como se mide antes de la administración.
6. Tetratiomolibdato de bis-colina para su uso según la reivindicación 1, en donde la administración de tetratiomolibdato de bis-colina se modifica si el paciente presenta:
(1) un nivel de alanina aminotransferasa (ALT) al menos dos veces el nivel de ALT presentado cuando se empieza el tratamiento con tetratiomolibdato de bis-colina,
(2) un nivel de alanina aminotransferasa (ALT) de al menos dos veces el límite superior de la normalidad (ULN), (3) un nivel de hemoglobina del 70 % o inferior al nivel de hemoglobina presentado cuando se empieza el tratamiento con tetratiomolibdato de bis-colina,
(4) un nivel de plaquetas del 70 % o inferior al nivel de plaquetas presentado cuando se empieza el tratamiento con tetratiomolibdato de bis-colina, o
(5) un nivel de neutrófilos del 70 % o inferior al nivel de neutrófilos presentado cuando se empieza el tratamiento con tetratiomolibdato de bis-colina,
en donde el tetratiomolibdato de bis-colina dosis se reduce y en donde:
si el paciente tomaba una dosis de 15 mg una vez al día de tetratiomolibdato de bis-colina, reducir la dosis hasta 15 mg de tetratiomolibdato de bis-colina cada dos días;
si el paciente tomaba una dosis de 30 mg una vez al día de tetratiomolibdato de bis-colina, reducir la dosis hasta 15 mg de tetratiomolibdato de bis-colina una vez al día;
si el paciente tomaba una dosis de 45 mg una vez al día de tetratiomolibdato de bis-colina, reducir la dosis hasta 30 mg de tetratiomolibdato de bis-colina una vez al día;
si el paciente tomaba una dosis de 60 mg una vez al día de tetratiomolibdato de bis-colina, reducir la dosis hasta 45 mg de tetratiomolibdato de bis-colina una vez al día;
si el paciente tomaba una dosis de 75 mg una vez al día de tetratiomolibdato de bis-colina, reducir la dosis hasta 60 mg de tetratiomolibdato de bis-colina una vez al día; o
si el paciente tomaba una dosis de 90 mg una vez al día de tetratiomolibdato de bis-colina, reducir la dosis hasta 75 mg de tetratiomolibdato de bis-colina una vez al día.
7. Tetratiomolibdato de bis-colina para su uso según la reivindicación 6, en donde la administración de tetratiomolibdato de bis-colina se modifica si el paciente presenta un nivel de alanina aminotransferasa (ALT) de al menos dos veces el límite superior de la normalidad (ULN), en donde el ULN es 30-45 UI/ml, o en donde el ULN es 34 UI/ml, o en donde el ULN es 40 UI/ml.
8. Tetratiomolibdato de bis-colina para su uso según la reivindicación 1, en donde si el paciente presenta un resultado de prueba anormal, el tetratiomolibdato de bis-colina se administra al paciente:
(a) con un primer nivel de dosis que comprende desde aproximadamente 15 hasta aproximadamente 90 mg por día de tetratiomolibdato de bis-colina durante un primer periodo de tiempo, seguido por
(b) un segundo nivel de dosis que comprende al menos aproximadamente 15 mg por día menos de tetratiomolibdato de bis-colina que el primer nivel de dosis durante un segundo periodo de tiempo.
9. Tetratiomolibdato de bis-colina para su uso según la reivindicación 8, en donde:
(i) el primer nivel de dosis comprende aproximadamente 15 mg por día de tetratiomolibdato de bis-colina y el segundo nivel de dosis comprende menos de aproximadamente 15 mg cada dos días de tetratiomolibdato de bis-colina; o
(ii) el primer nivel de dosis comprende aproximadamente 30 mg por día de tetratiomolibdato de bis-colina y el segundo nivel de dosis comprende menos de aproximadamente 15 mg por día de tetratiomolibdato de bis-colina; o
(iii) el primer nivel de dosis comprende aproximadamente 45 mg por día de tetratiomolibdato de bis-colina y el segundo nivel de dosis comprende menos de aproximadamente 30 mg por día de tetratiomolibdato de bis-colina; o
(iv) el primer nivel de dosis comprende aproximadamente 60 mg por día de tetratiomolibdato de bis-colina y el segundo nivel de dosis comprende menos de aproximadamente 45 mg por día de tetratiomolibdato de bis-colina; o
(v) el primer nivel de dosis comprende aproximadamente 75 mg por día de tetratiomolibdato de bis-colina y el segundo nivel de dosis comprende menos de aproximadamente 60 mg por día de tetratiomolibdato de bis-colina; o
(vi) el primer nivel de dosis comprende aproximadamente 90 mg por día de tetratiomolibdato de bis-colina y el segundo nivel de dosis comprende menos de aproximadamente 75 mg por día de tetratiomolibdato de bis-colina.
10. Tetratiomolibdato de bis-colina para su uso según la reivindicación 8 o 9, en donde el resultado de prueba anormal comprende:
(i) un nivel de alanina aminotransferasa (ALT) de (1) al menos dos veces el de una ALT basal medida antes de la administración de tetratiomolibdato de bis-colina o (2) superior a 68 UI/ml; o
(ii) un nivel de hemoglobina de (1) 70 % o inferior a un nivel de hemoglobina basal medido antes de la administración de tetratiomolibdato de bis-colina o (2) 70 % o inferior a un nivel de hemoglobina normal; o
(iii) un nivel de plaquetas de (1) 70 % o inferior a un nivel de plaquetas basal antes de la administración de tetratiomolibdato de bis-colina o (2) 70 % o inferior a un nivel de plaquetas normal; o
(iv) un nivel de neutrófilos de (1) 70 % o inferior a un nivel de neutrófilos basal medido antes de la administración de tetratiomolibdato de bis-colina o (2) 70 % o inferior a un nivel de neutrófilos normal.
11. Tetratiomolibdato de bis-colina para su uso según la reivindicación 8, en donde el resultado de prueba anormal comprende un nivel de alanina aminotransferasa (ALT) de al menos cinco veces el de una ALT basal medida antes de la administración de tetratiomolibdato de bis-colina, y en donde:
(c) el tratamiento se interrumpe durante un tercer periodo de tiempo entre la etapa (a) y la etapa (b) hasta que el paciente presente un nivel de alanina aminotransferasa (ALT) inferior a dos veces la ALT basal medida antes de la administración de tetratiomolibdato de bis-colina; y opcionalmente
en donde la segunda dosis en la etapa (b) es 15 mg cada dos días.
12. Tetratiomolibdato de bis-colina para su uso según la reivindicación 8, en donde el resultado de prueba anormal comprende un nivel de alanina aminotransferasa (ALT) de al menos 200 UI/ml, y en donde:
(c) el tratamiento se interrumpe durante un tercer periodo de tiempo entre la etapa (a) y la etapa (b) hasta que el paciente presente un nivel de alanina aminotransferasa (ALT) inferior a 68 UI/ml; y opcionalmente
en donde la segunda dosis en la etapa (b) es 15 mg cada dos días.
13. Tetratiomolibdato de bis-colina para su uso según la reivindicación 8, en donde el resultado de prueba anormal comprende un nivel de hemoglobina inferior a 8 g/dl en ausencia de sangrado, y en donde:
(c) el tratamiento se interrumpe durante un tercer periodo de tiempo entre la etapa (a) y la etapa (b) hasta que el paciente presente un nivel de hemoglobina equivalente al nivel de hemoglobina medido antes de la administración de tetratiomolibdato de bis-colina; y opcionalmente
en donde la segunda dosis en la etapa (b) es 15 mg cada dos días.
14. Tetratiomolibdato de bis-colina para su uso según la reivindicación 8, en donde el resultado de prueba anormal comprende un nivel de plaquetas inferior a 30.000 pl, y en donde:
(c) el tratamiento se interrumpe durante un tercer periodo de tiempo entre la etapa (a) y la etapa (b) hasta que el paciente presente un nivel de plaquetas equivalente al nivel de plaquetas medido antes de la administración de tetratiomolibdato de bis-colina; y opcionalmente
en donde la segunda dosis en la etapa (b) es 15 mg cada dos días.
15. Tetratiomolibdato de bis-colina para su uso según la reivindicación 8, en donde el resultado de prueba anormal comprende un nivel de neutrófilos inferior a 1,0 x 103/pl, y en donde:
(c) el tratamiento se interrumpe durante un tercer periodo de tiempo entre la etapa (a) y la etapa (b) hasta que el paciente presente un nivel de neutrófilos equivalente al nivel de neutrófilos medido antes de la administración de tetratiomolibdato de bis-colina; y opcionalmente
en donde la segunda dosis en la etapa (b) es 15 mg cada dos días.
16. Tetratiomolibdato de bis-colina para su uso según una cualquiera de las reivindicaciones 4 a 15, que comprende además medir al menos uno de dicho:
a) nivel de NCCcorregido;
b) nivel de alanina aminotransferasa (ALT);
c) nivel de hemoglobina;
d) nivel de plaquetas; y
e) nivel de neutrófilos de dicho paciente.
Applications Claiming Priority (7)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201762594184P | 2017-12-04 | 2017-12-04 | |
| US201862646553P | 2018-03-22 | 2018-03-22 | |
| US201862655568P | 2018-04-10 | 2018-04-10 | |
| US201862669095P | 2018-05-09 | 2018-05-09 | |
| US201862741313P | 2018-10-04 | 2018-10-04 | |
| US201862750595P | 2018-10-25 | 2018-10-25 | |
| PCT/EP2018/083551 WO2019110619A1 (en) | 2017-12-04 | 2018-12-04 | Bis-choline tetrathiomolybdate for treating wilson disease |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2912306T3 true ES2912306T3 (es) | 2022-05-25 |
Family
ID=64661349
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES18815624T Active ES2912306T3 (es) | 2017-12-04 | 2018-12-04 | Tetratiomolibdato de bis-colina para el tratamiento de enfermedad de Wilson |
| ES21215264T Active ES3026756T3 (en) | 2017-12-04 | 2018-12-04 | Bis-choline tetrathiomolybdate for treating wilson disease |
Family Applications After (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES21215264T Active ES3026756T3 (en) | 2017-12-04 | 2018-12-04 | Bis-choline tetrathiomolybdate for treating wilson disease |
Country Status (16)
| Country | Link |
|---|---|
| US (3) | US11510944B2 (es) |
| EP (3) | EP4523745A3 (es) |
| JP (2) | JP2023075247A (es) |
| KR (1) | KR102803161B1 (es) |
| AU (1) | AU2018379255B2 (es) |
| BR (1) | BR112020011055A2 (es) |
| DK (2) | DK4029498T3 (es) |
| ES (2) | ES2912306T3 (es) |
| FI (1) | FI4029498T3 (es) |
| HU (2) | HUE071155T2 (es) |
| IL (1) | IL275115B2 (es) |
| MX (1) | MX2020006284A (es) |
| PL (2) | PL4029498T3 (es) |
| PT (2) | PT3720433T (es) |
| SI (2) | SI4029498T1 (es) |
| WO (1) | WO2019110619A1 (es) |
Families Citing this family (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US11510944B2 (en) * | 2017-12-04 | 2022-11-29 | Alexion Pharmaceuticals, Inc. | Bis-choline tetrathiomolybdate for treating Wilson disease |
| US11419832B2 (en) * | 2017-12-04 | 2022-08-23 | Alexion Pharmaceuticals, Inc. | Bis-choline tetrathiomolybdate for treating Wilson Disease |
| EP3749417A1 (en) | 2018-02-06 | 2020-12-16 | Alexion Pharma International Operations Unlimited Company | Crystalline particles of bis-choline tetrathiomolybdate |
| EP4061353A1 (en) | 2019-11-21 | 2022-09-28 | Alexion Pharmaceuticals, Inc. | Methods of reducing neurological damage in wilson disease patients |
| US12590977B2 (en) | 2020-09-11 | 2026-03-31 | Alexion Pharmaceuticals, Inc. | Anti-ceruloplasmin antibodies and uses thereof |
| JP2023549814A (ja) * | 2020-11-13 | 2023-11-29 | アレクシオン ファーマシューティカルズ, インコーポレイテッド | 銅代謝関連疾患又は障害を治療する方法 |
| US20240100088A1 (en) * | 2021-01-31 | 2024-03-28 | Alexion Pharmaceuticals, Inc. | Novel formulation for treating copper metabolism-associated diseases or disorders |
| US20250082674A1 (en) * | 2021-08-17 | 2025-03-13 | Alexion Pharmaceuticals, Inc. | Methods of treating copper metabolism-associated diseases or disorders |
| JP2026503721A (ja) | 2023-01-30 | 2026-01-29 | アレクシオン ファーマシューティカルズ, インコーポレイテッド | 銅代謝関連疾患又は障害(ウィルソン病)の治療における使用のためのテトラチオモリブデン酸 |
Family Cites Families (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2004009072A2 (en) * | 2002-07-23 | 2004-01-29 | The Regents Of The University Of Michigan | Tetrapropylammonium tetrathiomolybdate and related compounds for anti-angiogenic therapies |
| US7189865B2 (en) * | 2002-07-23 | 2007-03-13 | Attenuon, Llc | Thiomolybdate analogues and uses thereof |
| CA2675230A1 (en) | 2006-01-10 | 2008-07-26 | Pipex, Inc. | Pharmaceutical compositions and methods to achieve and maintain a targeted and stable copper status and prevent and treat copper-related central nervous system diseases |
| US9623021B2 (en) | 2007-01-22 | 2017-04-18 | Gtx, Inc. | Nuclear receptor binding agents |
| US11510944B2 (en) | 2017-12-04 | 2022-11-29 | Alexion Pharmaceuticals, Inc. | Bis-choline tetrathiomolybdate for treating Wilson disease |
| US11419832B2 (en) * | 2017-12-04 | 2022-08-23 | Alexion Pharmaceuticals, Inc. | Bis-choline tetrathiomolybdate for treating Wilson Disease |
| EP3749417A1 (en) | 2018-02-06 | 2020-12-16 | Alexion Pharma International Operations Unlimited Company | Crystalline particles of bis-choline tetrathiomolybdate |
-
2018
- 2018-12-04 US US16/770,022 patent/US11510944B2/en active Active
- 2018-12-04 KR KR1020207019115A patent/KR102803161B1/ko active Active
- 2018-12-04 ES ES18815624T patent/ES2912306T3/es active Active
- 2018-12-04 ES ES21215264T patent/ES3026756T3/es active Active
- 2018-12-04 PT PT188156244T patent/PT3720433T/pt unknown
- 2018-12-04 IL IL275115A patent/IL275115B2/en unknown
- 2018-12-04 BR BR112020011055-8A patent/BR112020011055A2/pt unknown
- 2018-12-04 SI SI201831229T patent/SI4029498T1/sl unknown
- 2018-12-04 FI FIEP21215264.9T patent/FI4029498T3/fi active
- 2018-12-04 PL PL21215264.9T patent/PL4029498T3/pl unknown
- 2018-12-04 PT PT212152649T patent/PT4029498T/pt unknown
- 2018-12-04 DK DK21215264.9T patent/DK4029498T3/da active
- 2018-12-04 MX MX2020006284A patent/MX2020006284A/es unknown
- 2018-12-04 EP EP25154707.1A patent/EP4523745A3/en active Pending
- 2018-12-04 EP EP21215264.9A patent/EP4029498B1/en active Active
- 2018-12-04 SI SI201830650T patent/SI3720433T1/sl unknown
- 2018-12-04 PL PL18815624.4T patent/PL3720433T3/pl unknown
- 2018-12-04 DK DK18815624.4T patent/DK3720433T3/da active
- 2018-12-04 HU HUE21215264A patent/HUE071155T2/hu unknown
- 2018-12-04 EP EP18815624.4A patent/EP3720433B1/en active Active
- 2018-12-04 AU AU2018379255A patent/AU2018379255B2/en active Active
- 2018-12-04 WO PCT/EP2018/083551 patent/WO2019110619A1/en not_active Ceased
- 2018-12-04 HU HUE18815624A patent/HUE058921T2/hu unknown
-
2021
- 2021-02-22 US US17/182,227 patent/US20210177894A1/en not_active Abandoned
-
2022
- 2022-08-23 US US17/821,786 patent/US12233088B2/en active Active
-
2023
- 2023-03-13 JP JP2023038205A patent/JP2023075247A/ja active Pending
-
2025
- 2025-01-24 JP JP2025010250A patent/JP2025066802A/ja active Pending
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2912306T3 (es) | Tetratiomolibdato de bis-colina para el tratamiento de enfermedad de Wilson | |
| JP6612370B2 (ja) | ポンペ病の処置のための投与計画 | |
| CN103974619B (zh) | 治疗法布里病的给药方案 | |
| US11419832B2 (en) | Bis-choline tetrathiomolybdate for treating Wilson Disease | |
| CN114929209A (zh) | 肌萎缩侧索硬化的治疗 | |
| US11793776B1 (en) | Methods of administering solriamfetol to lactating women | |
| JP2016540827A (ja) | ミトコンドリア疾患を治療するためのシステアミン及びその誘導体の使用 | |
| JP2025513844A (ja) | アルギナーゼ1欠損症の処置 | |
| US20160128954A1 (en) | Methods of Treating Huntington's Disease Using Cysteamine Compositions | |
| US12064411B1 (en) | Methods of administering solriamfetol to lactating women | |
| JP7852200B2 (ja) | ウィルソン病を治療するためのビスコリンテトラチオモリブデート | |
| CA3084100C (en) | Bis-choline tetrathiomolybdate for treating wilson disease | |
| JP2022538210A (ja) | 腎機能障害患者における高アンモニア血症の治療 | |
| US12263145B2 (en) | Methods of administering solriamfetol to lactating women | |
| US12036194B1 (en) | Methods of administering solriamfetol to lactating women | |
| WO2025235366A1 (en) | Pyrrolopyrimidine compositions for treatment of atopic dermatitis | |
| WO2025162430A1 (zh) | 一种药物联用及其在预防或治疗神经退行性疾病中的用途 |