ES2912941T3 - Inhibidores de URAT1 para promover la excreción de ácido úrico - Google Patents
Inhibidores de URAT1 para promover la excreción de ácido úrico Download PDFInfo
- Publication number
- ES2912941T3 ES2912941T3 ES18806684T ES18806684T ES2912941T3 ES 2912941 T3 ES2912941 T3 ES 2912941T3 ES 18806684 T ES18806684 T ES 18806684T ES 18806684 T ES18806684 T ES 18806684T ES 2912941 T3 ES2912941 T3 ES 2912941T3
- Authority
- ES
- Spain
- Prior art keywords
- compound
- ring
- substituted
- group
- alkyl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/335—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin
- A61K31/34—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin having five-membered rings with one oxygen as the only ring hetero atom, e.g. isosorbide
- A61K31/343—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin having five-membered rings with one oxygen as the only ring hetero atom, e.g. isosorbide condensed with a carbocyclic ring, e.g. coumaran, bufuralol, befunolol, clobenfurol, amiodarone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/415—1,2-Diazoles
- A61K31/416—1,2-Diazoles condensed with carbocyclic ring systems, e.g. indazole
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/415—1,2-Diazoles
- A61K31/4162—1,2-Diazoles condensed with heterocyclic ring systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/4353—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems
- A61K31/437—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems the heterocyclic ring system containing a five-membered ring having nitrogen as a ring hetero atom, e.g. indolizine, beta-carboline
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/519—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/06—Antigout agents, e.g. antihyperuricemic or uricosuric agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B59/00—Introduction of isotopes of elements into organic compounds ; Labelled organic compounds per se
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B59/00—Introduction of isotopes of elements into organic compounds ; Labelled organic compounds per se
- C07B59/002—Heterocyclic compounds
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D231/00—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings
- C07D231/54—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings condensed with carbocyclic rings or ring systems
- C07D231/56—Benzopyrazoles; Hydrogenated benzopyrazoles
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D307/00—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom
- C07D307/77—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom ortho- or peri-condensed with carbocyclic rings or ring systems
- C07D307/78—Benzo [b] furans; Hydrogenated benzo [b] furans
- C07D307/79—Benzo [b] furans; Hydrogenated benzo [b] furans with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to carbon atoms of the hetero ring
- C07D307/80—Radicals substituted by oxygen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D491/00—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00
- C07D491/02—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00 in which the condensed system contains two hetero rings
- C07D491/04—Ortho-condensed systems
- C07D491/044—Ortho-condensed systems with only one oxygen atom as ring hetero atom in the oxygen-containing ring
- C07D491/052—Ortho-condensed systems with only one oxygen atom as ring hetero atom in the oxygen-containing ring the oxygen-containing ring being six-membered
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B2200/00—Indexing scheme relating to specific properties of organic compounds
- C07B2200/05—Isotopically modified compounds, e.g. labelled
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Animal Behavior & Ethology (AREA)
- Life Sciences & Earth Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Medicinal Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Epidemiology (AREA)
- Physical Education & Sports Medicine (AREA)
- Rheumatology (AREA)
- Pain & Pain Management (AREA)
- Urology & Nephrology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
Description
DESCRIPCIÓN
Inhibidores de URAT1 para promover la excreción de ácido úrico
Campo técnico
La invención pertenece al campo de la química médica y se refiere particularmente a una clase de compuestos inhibidores de URAT1, para aumentar la excreción de ácido úrico, y aplicaciones en medicina.
Antecedentes de la invención
La gota es uno de los tipos más comunes de artritis causada por hiperuricemia. En la actualidad, hay casi 100 millones de pacientes con gota en todo el mundo y el tamaño de su mercado es enorme. De acuerdo con las estadísticas, la incidencia de gota en Europa es de aproximadamente 1-2 %, principalmente en hombres de mediana edad (Michael Doherty, Tim L. Jansen, George Nuki, et al. Gout: why is this curable disease so seldom cured?. Annals of the Rheumatic Diseases. 2012, 71(11): 1765-1770). El número de pacientes con gota en los EE. UU. también ha alcanzado los 8.3 millones (Zhu Y, Pandya BJ, Choi HK. Prevalence of gout y hyperuricemia in the US general population: the National Health y Nutrition Examination Survey 2007-2008. Arthritis Rheumatol 2011, 63(10): 3136-3141). El número de pacientes con hiperuricemia en China es de aproximadamente 120 millones, de los cuales más de 50 millones son pacientes con gota, y el número de pacientes masculinos con gota es mucho mayor que el de mujeres.
Por lo general, cuando el ácido úrico sérico de los pacientes es superior a 6.8 mg/dl, los llamamos pacientes con hiperuricemia. Cuando la concentración de ácido úrico supera la máxima solvencia en el suero, el urato formará depósitos de cristales en el líquido sinovial de los tejidos, el cartílago de las articulaciones periféricas, las aurículas de las orejas y el olécranon de los codos (Richette P, Bardin T Gout. Lancet. 2010, 375 (9711): 318-328), causando artritis inflamatoria repetida, produciendo brotes de gota y, finalmente, provocando una enfermedad articular crónica grave e incluso erosión ósea (Schlesinger N. Difficult- to-treat gouty arthritis: a disease warranting better management. Drugs, 2011, 71(11): 1413-1439). Cuando se forman cristales de urato y se depositan en el tejido subcutáneo, se formarán tofos, que pueden romper el tejido epidérmico humano, causando dolor e incluso infección. En la actualidad, el tratamiento estandarizado para la gota incluye un tratamiento para disminuir la concentración de ácido úrico sérico (terapia reductora de urato, ULT), que puede disminuir la concentración sérica de ácido úrico por debajo de la concentración de saturación sin formar cristales de urato, y puede hacer que los cristales de urato se disuelvan en las lesiones. La gota ya no se formará después de la desaparición de los cristales de urato en el cuerpo. El American College of Rheumatology (ACR) y la European League Against Rheumatism (EULAR) recomiendan que la concentración sérica de ácido úrico de los pacientes con gota general debería ser tratada para que sea inferior a 6 mg/dl, mientras que la concentración sérica de ácido úrico de los pacientes con tofos debería ser tratada para que sea inferior a 5 mg/dl. Múltiples estudios han demostrado que la reducción continua de la concentración sérica de ácido úrico puede reducir la gravedad de la gota clínica, la incidencia de brotes agudos de gota (Shoji A, Yamanaka H, Kamatani N. A retrospective study of the relationship between serum urate level y recurrent attacks of gouty arthritis: evidence for reduction of recurrent gouty arthritis with antihyperuricemic therapy. Artritis Rheum. 2004, 51(3):321-325), ay el tamaño y número de tofos (Perez-Ruiz F, Calabozo M, Pijoan JI, et al. Effect of uratelowering therapy on the velocity of size reduction of tophi in chronic gout. Arthritis Rheumatology, 2002, 47(4): 356-360).
ULT incluye principalmente la terapia de disminución de la producción de ácido úrico (como los inhibidores de la xantina oxidasa) y la terapia de aumento de la excreción de ácido úrico (como los inhibidores de URAT1) de acuerdo con el mecanismo de acción. Los inhibidores de la xantina oxidasa utilizados con mayor frecuencia incluyen principalmente alopurinol y febuxostat, que son los tratamientos de primera línea para los pacientes con gota en Europa y los Estados Unidos, pero una gran cantidad de estudios han demostrado que aproximadamente el 80-85 % de los pacientes con hiperuricemia padecen la enfermedad, debido a la excreción inadecuada de ácido úrico por los riñones. (Cheeseman C. Solute carrier family 2, member 9 y uric acid homeostasis. Current Opinion in Nephrology y Hypertension, 2009, 18(5): 428-432). Por lo tanto, el efecto del tratamiento clínico del inhibidor de la xantina oxidasa no es satisfactorio, aproximadamente el 40-80 % de los pacientes no logran el objetivo de controlar el nivel de ácido úrico sérico mediante el tratamiento con inhibidores de la producción de ácido úrico (Edwards NL. Febuxostat: a new treatment for hyperuricaemia in gout. Rheumatology (Oxford). 2009, 48(2): 15-19). Dado que el alopurinol tiene una eficacia clínica débil (Rashid N, Coburn BW, Wu YL, et al. Modifiable factors associated with allopurinol adherence y outcomes among patients with gout in an integrated healthcare system. Journal of Rheumatology. 2015, 42(3): 504 512), la mayoría de los pacientes que recibieron una dosis de 300 mg/día de alopurinol todavía tienen una concentración sérica de ácido úrico por encima del valor objetivo, y se puede causar una erupción fatal y una variedad de reacciones adversas; aunque 80 mg/día de febuxostat tiene mejor eficacia que el tratamiento de alopurinol 300 mg/día (Schumacher HR, Jr., Becker MA, Wortmann RL, et al. Effects of febuxostat versus allopurinol y placebo in reducing serum urate in subjects with hyperuricemia y gout: a 28-week, phase III, randomized, doubleblind, parallel-group trial. Arthritis Rheumatology. 2008, 59(11): 1540-1548), todavía hay 40 %-52 % de pacientes cuya concentración sérica de ácido úrico no ha descendido al valor objetivo, y febuxostat también tiene efectos secundarios graves de molestias cardiovasculares, gastrointestinales y toxicidad hepática. Por lo tanto, las guías
ACR para el manejo de la gota recomiendan la adición de un fármaco que promueva la excreción de ácido úrico (Khanna D, Fitzgerald JD, Khanna PP, et al. 2012 American College of Rheumatology guidelines for management of gout, part 1: systematic nonpharmacologic y pharmacologic therapeutic Approaches to hyperuricemia. Arthritis Care & Research. 2012, 64(10): 1431-1446).
El aumento de los fármacos de excreción de ácido úrico juega un papel importante en el tratamiento de la hiperuricemia y la gota. El mecanismo de acción es inhibir la reabsorción de ácido úrico en los túbulos contorneados proximales del riñón y aumentar la excreción renal de ácido úrico, disminuyendo así la concentración de ácido úrico sérico. El transportador 1 de aniones de urato humano (hURAT1) es un miembro de la superfamilia de transportadores de aniones orgánicos (OAT). Está codificado por el gen SLC22A12, y su ADNc tiene muchas mutaciones que provocan un metabolismo anormal del ácido úrico. La proteína hURAT1, que se expresa específicamente en la membrana del borde en cepillo de las células epiteliales del riñón proximal contorneado humano, es la proteína de reabsorción de ácido úrico más importante en el cuerpo humano y controla la reabsorción de más del 90 % del ácido úrico después de la filtración glomerular (Michael FW, Jutabha P, Quada B. Developing potent human uric acid transporter 1 (hURAT1) inhibitors. Journal of Medicinal Chemistry. 2011, 54: 2701-2713). La inhibición del transporte de hURAT1 puede reducir eficazmente la reabsorción de ácido úrico, promover la excreción de ácido úrico en el riñón y disminuir los niveles de ácido úrico sérico en el cuerpo (Michael FW, Jutabha P, Quada B. Developing potent human uric acid transporter 1 (hURAT1) inhibitors. Journal of Medicinal Chemistry. 2011, 54:2701-2713).
Los inhibidores de URAT1 que se usan actualmente para el tratamiento de la gota incluyen benzobromarona, Zurampic, probenecid y sulfinpirazona. La benzobromarona es actualmente el fármaco más eficaz del mercado para aumentar la excreción de ácido úrico. Sin embargo, la benzbromarona tiene toxicidad hepática grave y no ha sido aprobada en el mercado estadounidense. Y fue retirado de la mayoría de los países europeos en 2003 (Jansen TL, Reinders MK, van Roon EN, et al. Benzbromarone withdrawn from the European market: another Case of "absence of evidence is evidence of absence". Clinical Experimental Rheumatology, 2004, 22(5): 651). Otra desventaja es que tiene un fuerte efecto inhibidor sobre las enzimas CYP2C9 de P450s del hígado. Sin embargo, debido a la falta de buenos fármacos para la gota en el mercado, todavía se usa en más de 20 países, incluidos China, Alemania, Japón, Brasil y Nueva Zelanda. El probenecid y la sulfinpirazona tienen una eficacia muy baja y efectos secundarios graves. Lesinurad (RDEA-594), nombre comercial como Zurampic, es un nuevo inhibidor de URAT1 desarrollado por Ardea Biosciences. AstraZeneca adquirió el fármaco gastando 1260 millones de dólares en la adquisición de Ardea en 2012. Zurampic fue aprobado en Estados Unidos y Europa en diciembre de 2015 y febrero de 2016 a una dosis de 200 mg/día en combinación con alopurinol, que tiene una eficacia mucho peor que la benzbromarona (50-80 mg/día). Un ensayo clínico de fase III de Zurampic en combinación con febuxostat en el tratamiento de la gota mostró que después de 12 meses de tratamiento de control de ácido úrico, usando un grupo de combinación de 200 mg de Zurampic, 80 mg de febuxostat y un grupo de placebo (80 mg de febuxostat solo), no hubo diferencia significativa en el porcentaje de pacientes con ácido úrico sérico que alcanzaron sUA<5 mg/dl. Zurampic también tiene una variedad de efectos secundarios tóxicos: (1 ) el fármaco puede causar un evento adverso cardiovascular mayor en pacientes con enfermedad cardiovascular fatal, infarto de miocardio no fatal o parálisis cerebral. (2) Hay una reacción adversa relacionada con la función renal inmediatamente después del inicio del tratamiento con Zurampic. Cuando se toman 400 mg de Zurampic solo, existe la mayor incidencia de eventos adversos graves. Por lo tanto, el uso individual de dosis altas de Zurampic está clínicamente prohibido y la función renal debería ser detectada regularmente antes y después del tratamiento. Por lo tanto, la FDA requiere que se indique su toxicidad renal severa con un recuadro negro en la etiqueta del fármaco. (3) El fármaco puede causar daño hepático de leve a moderado. Aunque la FDA aprobó la inclusión de Zurampic en la lista, la falta de eficacia significativa y toxicidad han hecho que la perspectiva del producto sea sombría.
Los documentos CN106432229, CN106065010, US4400387 y WO2018/090921 describen inhibidores de URAT1.
Resumen de la invención
El objetivo de la presente invención es proporcionar una serie de nuevos compuestos basados en las tecnologías actuales, con el objetivo de obtener un inhibidor de URAT1 de baja toxicidad y buena eficacia para el tratamiento de la hiperuricemia o gota.
El objetivo de la invención puede ser logrado mediante las siguientes medidas:
Se proporciona un compuesto de la Fórmula (I)
en la que:
A es un anillo de seis miembros no aromático con o sin un heteroátomo O, N o S;
El anillo B es un anillo aromático de cinco miembros que contiene dos átomos de N o un anillo de furano; Z, E o X son cada uno independientemente un átomo de C o N;
G es un átomo de N o de O; y cuando G es un átomo de O, Z, E y X son todos átomos de C; cuando G es un átomo de N, sólo uno de Z, E y X es un átomo de N;
Y es carbonilo, azufre, sulfona, sulfóxido, metileno o imino opcionalmente sustituidos;
R1 es uno o más seleccionados del grupo que consiste en hidrógeno, deuterio, hidroxi, halógeno, nitro, amino, ciano, alquilo C1-5, alquilo C1-5 sustituido, amino C1-3 sustituido, alcoxi C1-3, alcoxi C1-3 sustituido y alquiltio C1-5;
R2 es uno o más seleccionados del grupo que consiste en hidrógeno, deuterio, hidroxi, halógeno, nitro, amino, ciano, ciano, alquilo C1-4, alquilo C1-3 sustituido, alquenilo C2-3, alquinilo C2-3, amino C1-3 sustituido, alcoxi C1-5, alcoxi C1-5 sustituido y alquiltio C1-5;
R3 es alquilo C1-4, alquilo C1-4 sustituido o cicloalquilo C3-4 ;
m es un número entero de 0 a 3;
n es un número entero de 1 a 3.
El sustituyente en el grupo Y es seleccionado del grupo que consiste en hidroxilo, ciano, nitro, amino, carboxilo o alcoxi C1-3, el sustituyente en el grupo R1, R2 o R3 es seleccionado del grupo que consiste en hidroxilo, halógeno, nitro, amino y ciano. Cuando m en la presente invención es 2 o 3, significa que el compuesto contiene dos grupos R1, y los dos grupos R1 pueden ser iguales, y los grupos definidos por R1 en la presente solicitud también pueden usarse, respectivamente.
Cuando n en la presente invención es 2 o 3, significa que el compuesto contiene dos grupos R2 , y los dos grupos R2 pueden ser iguales, y los grupos definidos por R2 en la presente solicitud también pueden usarse, respectivamente.
En una realización, el Anillo A es un anillo de ciclohexeno o un anillo de seis miembros no aromático que contiene al menos un átomo de O y/o N.
En una realización preferida, el Anillo A es un anillo de ciclohexeno, un anillo de 3,4-dihidro-2H-pirano, un anillo de tetrahidropirano, un anillo de 2,3,4,5-tetrahidropiridina, un anillo de 5,6-dihidro-2H-1,3-oxazina o un anillo de 1,2,5,6-tetrahidropirimidina.
En una realización, R1 es seleccionado de uno o más del grupo que consiste en hidrógeno, deuterio, flúor, cloro, bromo, hidroxi, ciano, alquilo C1-3, haloalquilo C1-3 y alcoxi C1 - 3 ; m es 0, 1 o 2.
En una realización preferida, R1 es seleccionado de uno o más del grupo que consiste en hidrógeno, deuterio, flúor, cloro, bromo, ciano, metilo, etilo, metoxi, etoxi; m es 0, 1 o 2.
En una realización, R2 es seleccionado de uno o más del grupo que consiste en hidrógeno, deuterio, halógeno, ciano, vinilo, etinilo, alquilo C1-2, alquilo C1-2 sustituido, alcoxi C1-2, alcoxi C1-2 sustituido, alquiltio C1-2, alquiltio C1-2 sustituido; el sustituyente es seleccionado del grupo que consiste en deuterio, halógeno, alquilo C1-2, cicloalquilo C3-4 y alcoxi C1-3 ; n es 0, 1 o 2.
En una realización, R2 es seleccionado de uno o más del grupo que consiste en hidrógeno, deuterio, halógeno, ciano, vinilo, etinilo, alquilo C1-2 sustituido o no sustituido, alcoxi C1-2 sustituido o no sustituido, alquiltio C1-2 sustituido o no sustituido ; el sustituyente es seleccionado del grupo que consiste en deuterio, halógeno, alquilo C1-2, cicloalquilo C3-4 o alcoxi C1 - 3 ; n es 0, 1 o 2.
En una realización preferida, R2 es seleccionado de uno o más de hidrógeno, deuterio, halógeno, ciano, alquilo C1-2, haloalquilo C1-2, alcoxi C1-2 o alquiltio C1 - 2 ; n es 1 o 2.
En una realización más preferida, R2 es seleccionado de uno o más del grupo que consiste en bromo, cloro y ciano, y n es 1 o 2.
En una realización, R3 es seleccionado del grupo que consiste en metilo, etilo, n-propilo, isopropilo, ciclopropilo y ciclobutilo.
En una realización más preferida del compuesto de la presente invención o una sal farmacéuticamente aceptable del mismo, el compuesto es seleccionado del grupo que consiste en:

El método de preparación del compuesto representado por la estructura de la Fórmula (I) de la presente invención incluye el método I y el método II.
Método I:

El compuesto de imidazopiridina (o pirazolopiridina) es sometido a una reacción de desmetilación y una reacción de hidrogenación (o deuteración) para dar un compuesto hidroxi correspondiente, que puede ser un producto final o
puede ser el producto objetivo después de una reacción de halogenación, una reacción de reducción u otra. reacción. R1, R2 y R3 son como se definen en las reivindicaciones y la fórmula general del Resumen de la invención.
Método II:

Se hace reaccionar el compuesto de aldehído sustituido con un reactivo de Grignard para obtener un metanol disustituido. El metanol disustituido es sometido a una reacción de oxidación y una reacción de desmetilación para obtener un compuesto hidroxi correspondiente, que puede ser un producto final o puede ser un producto objetivo después de una reacción de halogenación, una reacción de reducción u otra reacción. Z, E, G, R1, R2 y R3 son como se definen en las reivindicaciones y la fórmula general en el Resumen de la invención.
A menos que se indique de otro modo, los siguientes términos utilizados en las reivindicaciones y la descripción tienen los siguientes significados:
"Anillo de seis miembros no aromático" se refiere a un grupo cíclico compuesto por seis átomos en el anillo, que no tiene aromaticidad y no pertenece a un anillo aromático de seis miembros, y el grupo puede contener un enlace simple C-C saturado, también puede contener enlaces dobles insaturados como C=C y C=N, en los que los átomos del anillo pueden ser un heteroátomo distinto del átomo de carbono, como N, S u O, etc., y el número de heteroátomos no está limitado a uno y puede ser dos, tres y más. El anillo de seis miembros no aromático de la presente invención incluye, pero no está limitado a, un anillo de ciclohexeno, un anillo de 3,4-dihidro-2H-pirano, un anillo de 2,3,4,5-tetrahidropiridina y un anillo de 5,6-dihidro-2H-1,3-oxazina, anillo de 1,2,5,6-tetrahidropirimidina y similares.
El "anillo aromático de cinco miembros" se refiere a un grupo anular condensado estructurado en anillo planar conjugado compuesto por cinco átomos anulares, que es aromático y el átomo anular puede ser un átomo distinto de un átomo de carbono, es decir, un heteroátomo. Cuando el anillo aromático de cinco miembros contiene un heteroátomo, el heteroátomo puede ser N, S u O, y el número de heteroátomos no está limitado a uno, y puede ser dos, tres o similares. El anillo aromático de cinco miembros que contiene heteroátomo(s) en la presente invención incluye, pero no está limitado a, un anillo de triazol, un anillo de imidazol, un anillo de tiazol, un anillo de oxazol, un anillo de oxadiazol o un anillo de tiadiazol, etc.
"Hidrógeno" significa protio (1H), que es el principal isótopo estable de hidrógeno.
"Deuterio" significa un isótopo morfológico estable de hidrógeno, también conocido como hidrógeno pesado, y su símbolo elemental es D.
Halógeno significa átomo de flúor, átomo de cloro, átomo de bromo o átomo de yodo.
"Alquilo" significa un grupo hidrocarburo C1-20 alifático saturado, incluidos los grupos de cadena lineal y de cadena ramificada (los rangos numéricos registrados en esta solicitud, como "1 -20", significan que el grupo, cuando es un grupo alquilo, puede contener 1 átomo de carbono, 2 átomos de carbono, 3 átomos de carbono, etc. hasta 20 átomos de carbono). Un grupo alquilo que tiene de 1 a 4 átomos de carbono se denomina grupo alquilo inferior. Cuando el grupo alquilo inferior no tiene sustituyentes, se denomina grupo alquilo inferior no sustituido. Más preferiblemente, el grupo alquilo es un grupo alquilo de tamaño medio que tiene de 2 a 5 átomos de carbono. El grupo alquilo en la presente invención es, por ejemplo, metilo, etilo, propilo, 2-propilo, n-butilo, isobutilo, t-butilo o pentilo. Preferiblemente, el grupo alquilo es un grupo alquilo inferior que tiene de 1 a 4 átomos de carbono, como metilo, etilo, propilo, 2-propilo, n-butilo, isobutilo o t-butilo. El grupo alquilo puede estar sustituido o no sustituido.
"Alcoxi" significa un grupo -O-(alquilo no sustituido) y un grupo -O-(cicloalquilo no sustituido), que denota además -O-(alquilo no sustituido). Los ejemplos representativos incluyen, pero no se limitan a, metoxi, etoxi, propoxi, butoxi, ciclopropoxi, ciclobutoxi, ciclopentiloxi, ciclohexiloxi y similares.
"Carbonilo" significa C=O.
"Grupo sulfona" significa -S (O)2-.
“Grupo tionileno” significa -S(O)-.
"Metileno" significa -CH2-.
"Imino" significa -NH-.
"Hidroxi" significa -OH.
"Nitro" significa -NO2.
"Amino" significa -NH2.
"Carboxi" significa -COOH.
"Ciano" significa -CN.
Una "sal farmacéuticamente aceptable" es una sal que comprende un compuesto de la Fórmula (I) con un ácido orgánico o inorgánico, es decir, aquellas sales que retienen la eficacia biológica y las propiedades del compuesto original. Tales sales incluyen:
(1 ) una sal de adición de ácido obtenida por reacción de una base libre de un compuesto original con un ácido inorgánico u orgánico como, pero no limitado a, ácido clorhídrico, ácido bromhídrico, ácido nítrico, ácido fosfórico, ácido metafosfórico, ácido sulfúrico, ácido sulfuroso y ácido perclórico, etc., ácido orgánico como, pero no limitado a, ácido acético, ácido propiónico, ácido acrílico, ácido oxálico, ácido (D) o (L) mélico, ácido fumárico, ácido maleico, ácido hidroxibenzoico, ácido Y-hidroxibutírico, ácido metoxibenzoico, ácido ftálico, ácido metanosulfónico, ácido etanosulfónico, ácido naftaleno-1-sulfónico, ácido naftaleno-2-sulfónico, ácido p-toluenosulfónico, ácido salicílico, ácido tartárico, ácido cítrico, ácido láctico, ácido mandélico, ácido succínico o ácido malónico.
(2 ) una sal obtenida mediante la sustitución de un protón ácido en un compuesto original con un ion metálico o mediante la formación de complejos con una base orgánica, el ion metálico es, como un ion de metal alcalino, un ion de metal alcalinotérreo o un ion de aluminio, la base orgánica es, tal como etanolamina, dietanolamina, trietanolamina, trometamina, N-metilglucamina y similares.
Una "composición farmacéutica" significa una mezcla de uno o más de los compuestos descritos en el presente documento o sus sales y profármacos farmacéuticamente aceptables con otros ingredientes químicos, tales como vehículos y excipientes farmacéuticamente aceptables. El propósito de la composición farmacéutica es facilitar la administración del compuesto al organismo.
En lo sucesivo, a menos que se especifique de otro modo, los compuestos de la Fórmula (I) como ingredientes activos de los agentes terapéuticos, incluidas todas sus sales farmacéuticamente aceptables, deben entenderse como incluidos dentro del alcance de la invención. En la presente especificación, son denominados simplemente como "compuestos de la Fórmula (I)" solo por conveniencia.
La presente invención incluye una composición farmacéutica que comprende un compuesto de cualquiera de la presente invención como un ingrediente activo, una sal farmacéuticamente aceptable del mismo, o un profármaco fácilmente hidrolizable del mismo, así como un excipiente farmacéuticamente aceptable.
Cada compuesto de la presente invención o una sal farmacéuticamente aceptable del mismo puede usarse para la fabricación de un fármaco para promover la excreción de ácido úrico, en particular, para el tratamiento o prevención de hiperuricemia, enfermedad renal o gota. Los experimentos muestran que los compuestos proporcionados por la presente invención tienen un muy buen efecto inhibidor sobre el transporte de ácido úrico por hURAT1 en células HEK293 transfectadas, lo que indica que los compuestos tienen una buena perspectiva de aplicación en el tratamiento de hiperuricemia o gota.
Realizaciones
La presente invención se describirá adicionalmente a continuación mediante ejemplos, pero el alcance de la presente invención no se limita aquí.
Ejemplo 1: Síntesis de (3,5-dibromo-4-hidroxifenil)(2-etil-5,6,7,8-tetrahidroimidazo[1,2-a]piridin-3-il)metanona (5)

Paso A: A una mezcla de 2-aminopiridina (2.0 g, 21.3 mmol) y trietilamina (2.58 g, 25.5 mmol) en diclorometano (20 ml) se le añadió gota a gota cloruro de propionilo (2.07 g, 22.4 mmol) en un baño de hielo y agua. Una vez completada la adición, se agitó durante la noche a temperatura ambiente la mezcla obtenida. A la mezcla se añadió agua (40 ml) y se le realizó extracción con diclorometano (40 ml x 3), y se lavó la fase orgánica combinada con salmuera (30 ml) y se secó sobre sulfato de sodio anhidro. Se evaporó el disolvente bajo presión reducida y el producto fue purificado mediante cromatografía instantánea (gel de sílice de malla 200-300, eluido con acetato de etilo:éter de petróleo = 1:15 a 1:10) para obtener N-(piridina-2-il)-propionamida (1) (2.74 g). El rendimiento fue del 85.6 %.
Paso B: Se agitó a reflujo durante 48 horas una mezcla que contenía el compuesto 1 (300 mg, 2.0 mmol), 2-bromo-1-(4-metoxifenil) etanona (460 mg, 2.0 mmol) y tolueno (10 ml) . Después de enfriar a temperatura ambiente, se añadió agua (30 ml) y se ajustó el pH a 8-9 con una solución acuosa saturada de carbonato de potasio. A la mezcla se le realizó extracción con diclorometano (40 ml x 3) y se secó sobre sulfato de sodio anhidro. El disolvente es evaporado bajo presión reducida y el producto es purificado mediante cromatografía en columna (gel de sílice de malla 200-300. acetato de etilo:éter de petróleo = 1:30 a 1:1) para dar (2-etilimidazo[1.2-a] piridin-3-il)(4-metoxifenil)metanona (2) (254 mg). El rendimiento fue del 45.3 %. 1H RMN (DMSO-d6. 500 MHz) ó 9.18 (d, J = 7.0 Hz, 1H), 7.74-7.69 (m, 3H), 7.58-7.55 (m, 1H), 7.17-7.14 (m, 1H), 7.09 (d, J = 8.5 Hz, 2H), 3.87 (s, 3H), 2.45 (q, J = 7.5 Hz, 2H), 1.11 (t, J = 7.5 Hz, 3H).MS (EI, m/z): 281.1 [M+H]+.
Paso C: Se agitó durante la noche a 30 °C bajo hidrógeno una mezcla que contenía el compuesto 2 (250 mg, 0.89 mmol), paladio al 10 % sobre carbono (25 mg) y DMF (7 ml) . Después se añadió acetato de etilo (30 ml) y la mezcla se filtró a través de una capa de celite. Se lavó con agua el filtrado (30 ml x 3) y se secó sobre sulfato de sodio anhidro. Se evaporó el solvente bajo presión reducida para dar (2-etil-5.6.7.8-tetrahidroimidazo[1.2-a]piridin-3-il) (4-metoxifenil)metanona (3) (230 mg). El rendimiento fue del 90.7 %.
Paso D: A una solución del compuesto 3 (220 mg, 0.77 mmol) en diclorometano anhidro (10 ml) se le añadió gota a gota una solución 1.0 M de tribromuro de boro en tolueno (2.3 ml) en un baño de agua con hielo. Después de la adición, se agitó la mezcla resultante a temperatura ambiente durante la noche. Se vertió la mezcla de reacción en agua con hielo (30 ml) y se ajustó el pH a 7-8 con bicarbonato de sodio saturado. A la mezcla se le realizó extracción con acetato de etilo (40 ml x 2) y se lavaron las fases orgánicas combinadas, con salmuera saturada (20 ml) y se secaron sobre sulfato de sodio anhidro. Se evaporó el solvente bajo presión reducida para dar (2-etil-5.6.7.8-tetrahidroimidazo[1.2-a]piridin-3-il)(4-hidroxifenil)metanona (4) (205 mg). El rendimiento fue del 98.0 %.
Paso E: Se añadió gota a gota una solución de bromo (260 mg, 1.63 mmol) en ácido acético (1 ml) a una mezcla del compuesto 4 (200 mg, 0.74 mmol) y acetato de sodio anhidro (182 mg, 2.2 mmol) en ácido acético (8 ml). Una vez completada la adición, se agitó la mezcla resultante a temperatura ambiente durante 1 hora. Se añadió gota a gota una solución acuosa diluida de bisulfito de sodio a la mezcla de reacción hasta que se desvaneció el color. Se evaporó el solvente bajo presión reducida, se agregó agua (15 ml) y después se ajustó el valor de pH a 7 - 8 con solución saturada de bicarbonato de sodio. A la mezcla se le realizó extracción con acetato de etilo (40 ml x 2) y se secó sobre sulfato de sodio anhidro. Se evaporó el solvente bajo presión reducida y el producto obtenido se recristalizó en éter de petróleo/acetato de etilo para dar (3.5-dibromo-4-hidroxifenil)(2-etil-5.6.7.8-tetrahidroimidazo[1.2 -a]piridin-3-il) metanona (5) (175 mg). El rendimiento fue del 55.3 %. 1H RMN (DMSO-d6. 400 MHz) ó 7.77 (s, 2H), 4.02-4.00 (m, 2H), 2.86-2.83 (m, 2H), 2.28 (q, J = 7.6 Hz, 2H), 1.92-1.86 (m, 4H), 1.08 (t, J = 7.6 Hz, 3H). MS (EI, m/z): 426.9 [M-H]-
Ejemplo 2: Síntesis de (3.5-dibromo-4-hidroxifenil)(5.6.6.7.8-pentadeuterio-2-etil-5.6.7.8-tetrahidroimidazo[1.2-a]piridin-3- il)metanona (10)

Paso A: Se añadió hidruro de sodio al 60 % (1.68 g, 42 mmol) en porciones a una solución de p-metoxiacetofenona (3.0 g, 20.0 mmol) en DMF (15 ml). Una vez completada la adición, se continuó agitando a esta temperatura durante 40 minutos y después se añadió gota a gota propionato de etilo (2.04 g, 20 mmol). Una vez completada la adición, se agitó la mezcla resultante a temperatura ambiente durante la noche. Después de agregar agua (60 ml), a la mezcla se le realizó extracción con acetato de etilo (30 ml x 3), se lavaron las fases orgánicas combinadas, con salmuera saturada (20 ml x 2) y se secaron sobre sulfato de sodio anhidro. Se evaporó el solvente bajo presión reducida y se purificó el producto mediante cromatografía en columna (gel de sílice de malla 200-300. acetato de etilo:éter de petróleo = 1:30) para dar 1-(4-metoxifenil)pentano-1.3-diona (6) (3.16 g) . El rendimiento fue del 76.6 %.
Paso B: Se disolvieron 2-amino-5-bromopiridina (2.60 g, 15.0 mmol) y el compuesto 6 (3.72 g, 18.0 mmol) en THF (40 ml) y después, en baño de agua con hielo, se añadieron secuencialmente ácido yodoftálico (5.80 g, 18.0 mmol) ) y eterato de trifluoruro de boro (430 mg, 3.03 mmol)) . Una vez completada la adición, se continuó agitando a temperatura ambiente durante la noche. Se añadió agua (40 ml), y se ajustó el valor de pH a 7 - 8 con una solución saturada de bicarbonato de sodio y después se añadió acetato de etilo (50 ml x 3) como agente de extracción. Las capas orgánicas combinadas fueron lavadas con salmuera saturada (20 ml) y se secaron sobre sulfato de sodio anhidro. Se evaporó el solvente bajo presión reducida y se purificó el producto mediante cromatografía en columna (gel de sílice de malla 200-300. acetato de etilo:éter de petróleo = 1:20) para dar (6-bromo-2-etilimidazo[1.2-a ]piridin-3-il)(4-metoxifenil)metanona (7) (1.15 g). El rendimiento fue del 21.3 %.
Paso C: El compuesto 7 (200 mg, 0.557 mmol) fue suspendido en DMF (10 ml). Se añadieron agua pesada (0.5 ml) y paladio al 5 % sobre carbono (20 mg), y se agitó la mezcla resultante a presión atmosférica durante 48 horas bajo deuterio. Después de filtrar a través de una capa de celite, se añadió agua (40 ml) al filtrado, seguido de extracción con acetato de etilo (30 ml x 3). Se lavaron con agua (20 ml x 3) las fases orgánicas combinadas y se secaron sobre sulfato de sodio anhidro. Se evaporó el solvente bajo presión reducida para dar (4-metoxifenil) (5.6.6.7.8-pentadeuterio-2-etil-5.6.7.8-tetrahidroimidazo[1.2-a]-piridin- 3-il)metanona (8) (164 mg). El rendimiento fue del 100 %. 1H RMN (DMSO-d6.400 MHz) ó 7.67 (dd, J = 2.0. 6.8 Hz, 2H), 7.06 (dd, J = 2.0. 6.8 Hz, 2H), 4.03-4.01 (m, 1H), 3.86 (s, 3H), 2.80 -2.78 (m, 1H), 2.18 (q, J = 7.6 Hz, 2H), 1.81-1.79 (m, 1H), 0.99 (t, J = 7.6 Hz, 3H).
Los procedimientos experimentales de los Pasos D y E fueron llevados a cabo de acuerdo con la preparación de los Pasos D y E en el Ejemplo 1 para dar (3.5-dibromo-4-hidroxifenil) (5.6.6.7.8-pentadeuterio-2- etil-5.6.7.8-tetrahidroimidazo[1.2-a]piridin-3-il)-metanona (10). 1H RMN (DMSO-d6.400 MHz) ó 7.80 (s, 2H), 4.05-4.01 (m, 1H), 2.85-2.83 (m, 1H), 2.27 (q, J = 7.2 Hz, 2H), 1.83-1.81 (m, 1H), 1.08 (t, J = 7.2 Hz, 3H).EM (EI, m/z): 434.0 [M+H]+.
Ejemplo 3: Síntesis de 5-(2-etil-5.6.7.8-tetrahidroimidazo[1.2-a]-piridin-3-carbonil)-2-hidroxibenzonitrilo (16)

Paso A: Se añadió 4-metoxiacetofenona (44.0 g, 293 mmol) a una mezcla de bis(tetrafluoroborato) de 1-clorometil-4-fluoro-1.4-diazabiciclo[2.2.2]octano (104 g, 294 mmol), yodo (38.6 g, 152 mmol) y acetonitrilo (440 ml) en un baño de agua con hielo. Una vez completada la adición, se agitó durante la noche a temperatura ambiente la mezcla obtenida. A la mezcla de reacción se añadió agua (1350 ml) y se separó una gran cantidad de sólido. Se filtró y se secó la mezcla para obtener 3-yodo-4-metoxiacetofenona (11) (70.0 g). El rendimiento fue del 86.5 %.
Paso B: Se agitó durante la noche a 130 °C una mezcla del compuesto 11 (70.0 g, 254 mmol), cianuro cuproso (34.0 g, 380 mmol) y DMF (400 ml). Se enfrió la mezcla a temperatura ambiente, se filtró a través de una capa de celite, se le añadió agua (1600 ml) y se realizó extracción con acetato de etilo (800 ml x 3). La fase orgánica combinada fue lavada sucesivamente con agua (400 ml x 2) y salmuera (400 ml) y después se secó sobre sulfato de sodio anhidro. Se evaporó el solvente bajo presión reducida para obtener un compuesto crudo de 5-acetil-2-metoxibenzonitrilo (12) (50.0 g). El compuesto se usó directamente en la siguiente reacción sin tratamiento adicional.
Paso C: Se añadió gota a gota una solución de bromo (49.0 g, 307 mmol) en metanol (50 ml) a una solución del compuesto crudo 12 (45.0 g) en metanol (250 ml). Una vez completada la adición, se agitó durante la noche a temperatura ambiente la mezcla obtenida. A la mezcla se añadió agua (900 ml), se filtró y se secó para dar 5-(2-bromo-acetil)-2-hidroxi-3-metilbenzonitrilo(13) (41.0 g). El rendimiento total de las reacciones en los pasos B y C fue del 70.6 %.
Paso D: Se calentó a reflujo y se agitó durante 48 horas una mezcla del compuesto 13 (41.0 g, 161 mmol), el compuesto 1 (24.0 g, 161 mmol) y metilbenceno (600 ml) . Se enfrió la mezcla a temperatura ambiente, se le añadió agua (400 ml) y se ajustó con solución saturada de bicarbonato de sodio hasta que el valor de pH fue de 7 - 8. A la mezcla se realizó extracción con acetato de etilo (600 ml x 3) y se secó sobre sulfato de sodio anhidro. Se evaporó el solvente bajo presión reducida y se purificó el producto mediante cromatografía instantánea ( gel de sílice de 200 300 mallas, se eluyó con acetato de etilo:éter de petróleo = 1:30 a 2:1) para obtener 5-(2-etilimidazo[ 1.2-a]-piridin-3-carbonil)-2-metoxibenzonitrilo (14) (25.7 g). El rendimiento fue del 52.3 %.
Paso E: Se agitó durante la noche a 30 °C en atmósfera de hidrógeno una mezcla que contenía el compuesto 14 (1.0 g, 3.28 mmol), paladio al 10% sobre carbono(100 mg) y ácido acético (10 ml) . Después de filtrar a través de una capa de celite, se evaporó el solvente bajo presión reducida, se añadió acetato de etilo (70 ml) y se lavó la mezcla con agua (20 ml) y se secó sobre sulfato de sodio. Se evaporó el solvente bajo presión reducida y se purificó el producto por cromatografía en columna (gel de sílice de malla 200-300. acetato de etilo:éter de petróleo = 1:3 a 4:1) para dar 5-(2-etil-5.6. 7.8-tetrahidroimidazo[1.2-a]piridin-3-carbonil)-2-metoxibenzonitrilo (15) (400 mg). El rendimiento fue del 39.5 %.
Paso F: Se añadió hidruro de sodio al 60 % (65 mg, 1.63 mmol) en porciones a una solución de etanotiol (0.12 ml) en THF (10 ml), se agitó la mezcla durante aproximadamente 5 minutos y después se filtró y se recogió la torta del filtro . Posteriormente, se añadió la torta del filtro a la solución del compuesto 15 (100 mg, 0.323 mmol) en DMF (6 ml) y la mezcla obtenida fue agitada durante 1 h a 60 °C. Se enfrió la mezcla a temperatura ambiente, se filtró con celite, se le añadió agua (40 ml) y se ajustó con solución acuosa de ácido cítrico 2 M hasta que el valor de pH fue de 5 - 6. A la mezcla se le realizó extracción con acetato de etilo (40 ml x 3) y se secó sobre sulfato de sodio anhidro. Se evaporó el solvente bajo presión reducida y se purificó el producto mediante cromatografía en columna (gel de sílice de malla 200-300. acetato de etilo:éter de petróleo = 1:2 a 5:1) para dar 5-(2-etil-5.6. 7.8-tetrahidroimidazo[1.2-a]piridin-3-carbonil)-2-hidroxibenzonitrilo (16) (52 mg). El rendimiento fue del 54.5 %.1H RMN (DMSO-d6.400 MHz) 5 7.89 (s, 1H), 7.79 (dd, J = 2.0. 8.8 Hz, 1H), 7.05 (d, J = 8.4 Hz, 1H), 4.04 (t, J = 5.6 Hz, 2H), 2.83-2.81 (m, 2H), 2.18 (q, J = 7.2 Hz, 2H), 1.89-1.83 (m, 4H), 1.01 (t, J = 7.2 Hz, 3H).MS (EI, m/z) : 296.2 [M+H]+.
Ejemplo 4: Síntesis de 3-bromo-5-(2-etil-5.6.7.8-tetrahidroimidazo[1.2-a]piridin-3-carbonil)-2-hidroxibenzonitrilo (17)

Paso A: Se añadió gota a gota una solución de bromo (27 mg, 1.63 mmol) en ácido acético (1 ml) a una solución del compuesto 16 (50 mg, 0.74 mmol) y acetato de sodio anhidro (28 mg, 2.2 mmol) en ácido acético (8 ml). Después de completar la adición, se agitó la mezcla resultante a temperatura ambiente durante 1 hora. Se añadió gota a gota una solución diluida de bisulfito de sodio a la mezcla de reacción hasta que se desvaneció el color. Se evaporó el solvente bajo presión reducida, después se agregó agua (15 ml) y se ajustó el valor de pH a 7 - 8 con bicarbonato de sodio saturado. A la mezcla se le realizó extracción con acetato de etilo (40 ml x 2) y se secó sobre sulfato de sodio anhidro. Se evaporó el solvente bajo presión reducida y el producto obtenido fue recristalizado en éter de petróleo/acetato de etilo para dar 3-bromo-5-(2-etil-5.6.7.8-tetrahidroimidazo[1.2-a]piridina- 3-carbonil) 2-hidroxibenzonitrilo (17) (30 mg). El rendimiento fue del 55.3 %. 1H RMN (DMSO-d6.400 MHz) 57.93 (d, J = 2.4 Hz, 1H), 7.71 (d, J = 2.4 Hz, 1H), 4.00 (t, J = 5.6 Hz, 2H), 2.99-2.97 (m, 2H), 2.37-2.35 (m, 2H), 1.95-1.89 (m, 4H), 1.15 (t, J = 7.6. 3H).MS (EI, m/z): 376.1 [M+H]+.
Ejemplo 5: Síntesis de (3.5-dibromo-4-hidroxifenil)(2-etil-7-hidroxi-5.6.7.8-tetra-hidroimidazo[1.2-a]piridin-3-il)metanona (23) y (3.5-dibromo-4-hidroxifenil)-(2-etil-7-metoxi-5.6.7.8-tetrahidro-imidazo[1.2-a]piridin-3-il)metanona (25)
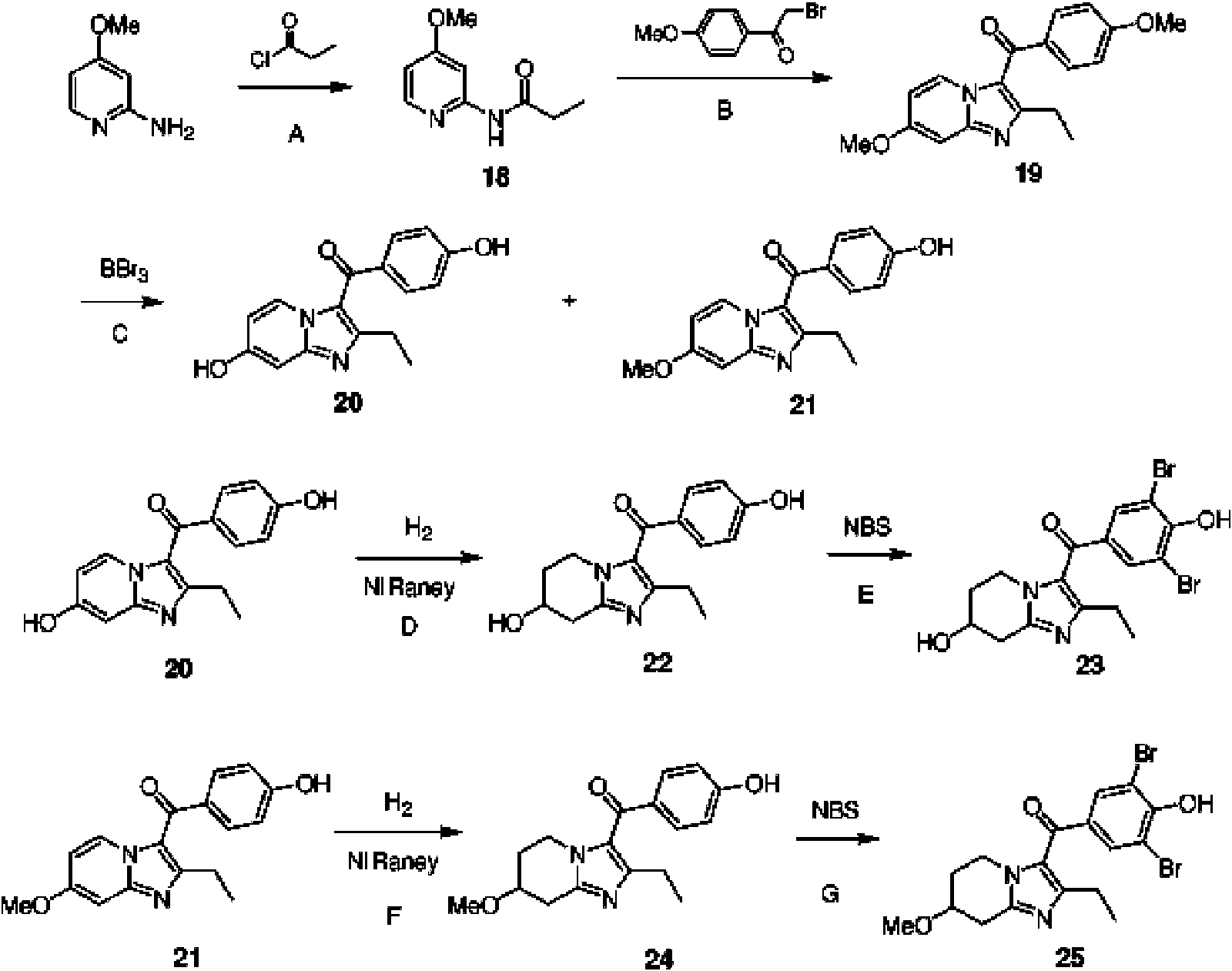
Paso A: Se disolvieron 2-amino-4-metoxipiridina (4.9 g, 39.5 mmol) y trietilamina (4.4 g, 43.5 mmol) en tetrahidrofurano (30 ml), después se añadió gota a gota cloruro de propionilo (4.0 g, 43.5 mmol) en un baño de agua con hielo. y se agitó la mezcla resultante a temperatura ambiente durante la noche. Se añadió agua (100 ml) y a la mezcla se le realizó extracción con acetato de etilo (60 ml x 3). Las capas orgánicas combinadas fueron lavadas con salmuera saturada (30 ml) y se evaporó el solvente bajo presión reducida. Se añadieron al producto carbonato de potasio (4.1 g, 29.7 mmol), metanol (50 ml) y agua (12 ml), y se agitó la mezcla resultante a temperatura ambiente durante 1 hora. Se evaporó el solvente bajo presión reducida, se añadió agua (20 ml), a la mezcla se le realizó extracción con acetato de etilo (30 ml x 3). Las capas orgánicas combinadas fueron lavadas con salmuera saturada (15 ml) y se secaron sobre sulfato de sodio anhidro. Se evaporó el solvente bajo presión reducida para dar N-(4-metoxipiridin-2-il)propanamida (18) (4.85 g). El rendimiento fue del 68.2 %.
Paso B: Se agitó a reflujo durante la noche una mezcla que contenía el compuesto 18 (4.85 g, 26.9 mmol), 2-bromo-1-(4-metoxifenil)etanona (6.14 g, 26.9 mmol) y tolueno (50 ml) . Después de enfriar a temperatura ambiente, se añadió agua (50 ml) y se ajustó el valor de pH a 8 - 9 con una solución de carbonato de potasio 2 M. A la mezcla se le realizó extracción con diclorometano (70 ml x 3) y se secó sobre sulfato de sodio anhidro. Se evaporó el solvente bajo presión reducida y se purificó el producto mediante cromatografía en columna (gel de sílice de malla 200-300. acetato de etilo:éter de petróleo = 1:5 a 2:3) para dar (2-etil-7-metoxiimidazo[1. 2-a]piridin-3-il)(4-metoxifenil)metanona (19)
(900 miligramos). El rendimiento fue del 10.8 %. 1H RMN (DMSO-d6. 400 MHz) 69.08 (d, J = 7.6 Hz, 1H), 7.67 (d, J = 8.8 Hz, 2H), 7.17 (d, J = 2.4 Hz, 1H), 7.08 (d, J = 8.4 Hz, 2H), 6.88-6.86 (m, 1H), 3.91(s, 3H), 3.87 (s, 3H), 2.38 (q, J = 7.2 Hz, 2H), 1.10 (t, J = 7.2 Hz, 3H).
Paso C: Se añadió gota a gota una solución 1.0 M de tribromuro de boro en tolueno (9 ml) a una solución del compuesto 19 (900 mg, 2.9 mmol) en diclorometano anhidro (25 ml). Una vez completada la adición, se agitó la mezcla resultante a temperatura ambiente durante la noche. Se vertió la mezcla de reacción en agua con hielo (50 ml) y se ajustó el valor de pH a 7-8 con bicarbonato de sodio saturado. A la mezcla se le realizó extracción con acetato de etilo (40 ml x 3) y se secó sobre sulfato de sodio anhidro. Se evaporó el solvente bajo presión reducida y se purificó el producto por cromatografía en columna (gel de sílice de malla 200-300. metanol:diclorometano = 1:50 a 1:20) para dar (2-etil-7-hidroxiimidazo[1.2- a]piridin-3-il)(4-hidroxifenil)metanona (20) (477 mg) y (2-etil-7-metoxiimidazo[1.2-a]piridin-3-il) (4-hidroxifenil)metanona ( 21) (277 mg). Los rendimientos fueron 58.3 % y 32.2 % respectivamente. Compuesto 20:1H RMN (DMSO-d6. 400 MHz) 610.83 (s, 1H), 10.22 (s, 1H), 9.06 (d, J = 7.6 Hz, 1H), 7.54 (d, J = 8.4 Hz, 2H), 6.89-6.84 (m, 3H), 6.77-6.75 (m, 1H), 2.37 (q, J = 7.6 Hz, 2H), 1.08 (t, J = 7.6 Hz, 3H).
Compuesto 21:1H NMR (DMSO-d6.400 MHz) 510.25 (s, 1H), 9.03 (d, J = 7.6 Hz, 1H), 7.57 (dd, J = 2.0. 6.8 Hz, 2H), 7.15 (d, J = 2.4 Hz, 1H), 6.91-6.83 (m, 3H), 3.91 (s, 3H), 2.45 (q, J = 7.6 Hz, 2H), 1.11 (t, J = 7.6 Hz, 3H).
Paso D: se agitó bajo hidrógeno a 60 °C durante 6 horas una mezcla que contenía el compuesto 20 (185 mg, 0.66 mmol), Ni Raney (40 mg) y etanol (15 ml), después se añadió Ni Raney a la mezcla de reacción (40 mg) y después se continuó agitando durante 3 horas a 60 °C bajo hidrógeno. Después de enfriar a temperatura ambiente, se filtró la mezcla y se lavó la torta del filtro por goteo con una pequeña cantidad de acetato de etilo. Se evaporó el solvente bajo presión reducida y se purificó el producto mediante cromatografía en columna (gel de sílice de malla 200-300. metanol:diclorometano = 1:50 - 1:30) para dar (2-etil-7-hidroxi-5.6, 7.8-tetrahidroimidazo[1.2-a]piridin-3-il) (4-hidroxifenil)metanona (22) (106 mg). El rendimiento fue del 62.7 %. 1H RMN (DMSO-d6. 400 MHz) 510.33 (s, 1H), 7.58 (d, J = 8.8 Hz, 2H), 6.86 (d, J = 8.8 Hz, 2H), 5.14 (d, J = 3.2 Hz, 1H), 4.17 (s, 1H), 4.12-4.02 (m, 2H), 3.02-2.96 (m, 1H), 2.74-2.68 (m, 1H), 2.20 (q, J = 7.6 Hz, 2H), 1.99-1.88 (m, 2H), 1.00 (t, J = 7.6 Hz, 3H).
Paso E: Se disolvió el compuesto 22 (56 mg, 0.22 mmol) en DMF (3 ml), se añadió NBS (77 mg, 0.44 mmol) y se agitó la mezcla en un baño de agua con hielo durante 0.5 horas. Después de agregar agua (15 ml), a la mezcla se le realizó extracción con acetato de etilo (30 ml x 3) y las capas orgánicas combinadas fueron lavadas con agua (15 ml x 2) y salmuera saturada (15 ml) y se secaron sobre sulfato de sodio anhidro. Se evaporó el solvente bajo presión reducida y se purificó el producto mediante cromatografía en columna (gel de sílice de malla 200 ~ 300. metanol:diclorometano = 1:50) para dar (3.5-dibromo-4-hidroxifenil)(2-etil-7). -hidroxi-5.6.7. 8-tetrahidroimidazo [1.2-a]piridin-3-il)metanona (23) (13 mg). El rendimiento fue del 13.5 %. 1H RMN (DMSO-d6. 400 MHz) 57.81 (s, 2H), 5.22 (s, 1H), 4.21-4.19 (m, 1H), 4.12-4.05 (m, 2H), 3.08-3.03 (m, 1H ), 2.80-2.76 (m, 1H), 2.26 (q, J = 7.6 Hz, 2H), 2.03-1.19 (m, 2H), 1.07 (t, J = 7.6 Hz, 3H). EM (IE, m/z) 442.9 [M-H]-.
Usando el compuesto 21 como materia prima, los procedimientos experimentales de los pasos F y G fueron llevados a cabo de acuerdo con los métodos de preparación de los pasos D y E de este ejemplo para dar (3.5-dibromo-4-hidroxifenil)(2-etil- 7-metoxi-5.6.7.8-tetrahidroimidazo[1.2-a]piridin-3-il)metanona (25). 1 H r Mn (DMSO-d6. 400 MHz) 5 7.81 (s, 2H), 4.09-4.03 (m, 2H), 3.88-3.87 (m, 1H), 3.33 (s, 3H), 3.07-3.06 (m, 1H), 2.94-2.93 (m, 1H), 2.24 (q, J = 7.6 Hz, 2H), 2.17-2.04 (m, 2H), 1.06 (t, J = 7.6 Hz, 3H).MS (EI, m/z): 457.0 [M-H]- .
Ejemplo 6: Síntesis de 3.5-dibromo-4-hidroxifenil)(2-etil-4.5.6.7-tetrahidropirazolo[1.5-a]piridin-3-il)metanona (32)

Paso A: Se agitó una mezcla de yoduro de 1-aminopiridinio (15.5 g, 70.0 mmol), 2-pentinoato de etilo (9.72 g, 77.1 mmol), carbonato de potasio (21.26 g, 154 mmol) y DMF (150 ml) durante 4.5 horas a temperatura ambiente. A la mezcla se añadió agua (450 ml) y se filtró, y se lavó la torta del filtro con agua (100 ml) para dar un compuesto húmedo de 2-etilpirazolo[1.5-a]-piridin-3-formiato de etilo (26 ) (12.25 g). El compuesto se usó directamente en la siguiente reacción sin secado.
Paso B: Se agitó durante la noche a 60 °C una mezcla del compuesto 26 húmedo (12.25 g), etanol (30 ml), THF (30 ml) y solución acuosa de hidróxido de sodio 2 M (70 ml) . Se evaporó aproximadamente la mitad del solvente bajo presión reducida y a la mezcla se agregó agua (150 ml) y se ajustó con ácido clorhídrico 2 M hasta que el valor de pH fue de 5 a 6. La mezcla se filtró para dar un compuesto húmedo de ácido 2-etilpirazolo[1.5-a]piridin-3-fórmico (27) (10.0 g). El compuesto se usó directamente en la siguiente reacción sin secado.
Paso C: El compuesto 27 húmedo (5.60 g) fue suspendido en agua (100 ml) y se le añadió ácido sulfúrico concentrado (4 ml) y la mezcla obtenida se agitó durante 3 h a 80 °C. Se enfrió la mezcla a temperatura ambiente y se ajustó con solución acuosa de hidróxido de sodio 2 M hasta que el valor de pH fue de 8 - 9. A la mezcla se le realizó extracción con acetato de etilo (40 ml x 3) y se lavó la fase orgánica combinada, sucesivamente con agua (30 ml) y salmuera (20 ml) y se secó sobre sulfato de sodio anhidro. Se evaporó el solvente bajo presión reducida para obtener 2-etilpirazolo[1.5-a]piridina (28) (3.18 g). El rendimiento total de las reacciones en los pasos A, B y C fue del 47.7 %.
Paso D: Se agitó durante la noche a 100 °C una mezcla del compuesto 28 (584 mg, 3.99 mmol), cloruro de 4-metoxibenzoilo (680 mg, 3.99 mmol) y tricloruro de aluminio (800 mg, 6.0 mmol) . Se enfrió la mezcla ligeramente, se le añadió acetato de etilo (30 ml) y agua (30 ml) y se ajustó con solución acuosa de hidróxido de sodio 2 M hasta que el valor del pH fue de 9 - 10. Se estratificó la mezcla y se recogió la fase orgánica . A la fase acuosa se le realizó extracción con acetato de etilo (30 ml x 2) y se lavó con salmuera (20 ml) la fase orgánica combinada y se secó sobre sulfato de sodio anhidro. Se evaporó el solvente bajo presión reducida y se purificó el producto por cromatografía instantánea (gel de sílice de malla 200-300, eluido con acetato de etilo:éter de petróleo = 1:30 a 1:10) para obtener (2-etilpirazol[1.5-a]piridin-3-il)(4-metoxifenil)metanona (29) (305 mg). El rendimiento fue del 27.3 %. 1H RMN (DMSO-d6. 300 MHz) 58.79 (d, J = 6.9 Hz, 1H), 7.66 (d, J = 8.7 Hz, 2H), 7.44-7.39 (m, 1H), 7.33-7.30 (m, 1H), 7.08-7.03 (m, 3H), 3.86 (s, 3H), 2.84 (q, J = 7.5 Hz, 2H), 1.20 (t, J = 7.5 Hz, 3H).
Paso E: Se añadió hidruro de sodio al 60 % (218 mg, 5.45 mmol) en porciones a una solución de etanotiol (338 mg, 5.44 mmol) en DMF (3 ml), se agitó la mezcla de reacción durante aproximadamente 5 minutos y después se añadió una solución de el compuesto 29 (305 mg, 1.09 mmol) en DMF (3 ml), y la mezcla obtenida fue agitada durante 2 horas a 120 °C. Se enfrió la mezcla a temperatura ambiente, se le añadió agua (30 ml) y se ajustó con ácido clorhídrico diluido hasta que el valor de pH fue de 7 - 8. Posteriormente, a la mezcla se le realizó extracción con acetato de etilo (30 ml x 3) y la fase orgánica combinada fue lavada sucesivamente con agua (20 ml x 3) y salmuera (20 ml) y se secó sobre sulfato de sodio anhidro. Se evaporó el solvente bajo presión reducida para obtener (2-etilpirazolo[1.5-a]piridin-3-il)(4-hidroxifenil)metanona (30) (420 mg). El compuesto fue usado directamente en la siguiente reacción sin purificación. 1H RMN (DMSO- d6. 300 MHz) 510.27 (s, 1H), 8.76 (d, J = 6.6 Hz, 1H), 7.56 (d, J = 8.4 Hz, 2H), 7.42-7.31 (m, 2H), 7.05-7.01 (m, 1H), 6.87 (d, J = 8.4 Hz, 2H), 2.84 (q, J = 7.5 Hz, 2H), 1.20 (t, J = 7.5 Hz, 3H).
Paso F fue llevado a cabo de acuerdo con el método de preparación del Paso D en el Ejemplo 4 para dar (2-etil-4.5.6.7- tetrahidropirazolo[1.5-a]piridin-3-il)(4-hidroxifenil)-metanona (31).
Paso G: Se añadió NBS (86 mg, 0.483 mmol) a una solución del compuesto 31 (65 mg, 0.240 mmol) en DMF (5 ml) y se agitó durante 1 hora. Después de agregar agua (20 ml), a la mezcla se le realizó extracción con acetato de etilo (20 ml x 3) y se lavaron las fases orgánicas combinadas, con agua (10 ml x 3) y salmuera saturada (10 ml) y se secaron sobre sulfato de sodio. Se evaporó el solvente bajo presión reducida y se purificó el producto mediante cromatografía en columna (gel de sílice de malla 200~300. acetato de etilo:diclorometano = 1:10) para dar 2-etil-4.5.6.7- tetrahidropirazolo[1,5-a]piridin-3-il)metanona (32). 1H RMN (DMSO-d6. 400 MHz) 57.74 (s, 2H), 4.05 (t, J = 6.0 Hz, 2H), 3.36-3.34 (m, 2H), 2.57-2.51 (m, 2H), 1.96-1.95 (m, 2H), 1.72-1.70 (m, 2H), 1.08 (t, J = 7.6 Hz, 3H).MS (EI, m/z): 429.0 [M+H]+.
Ejemplo 7: Síntesis de (3.5-dibromo-4-hidroxifenil) (2-etil-4.5.6.7-tetrahidro-2H-indazol-3-il)metanona (41)
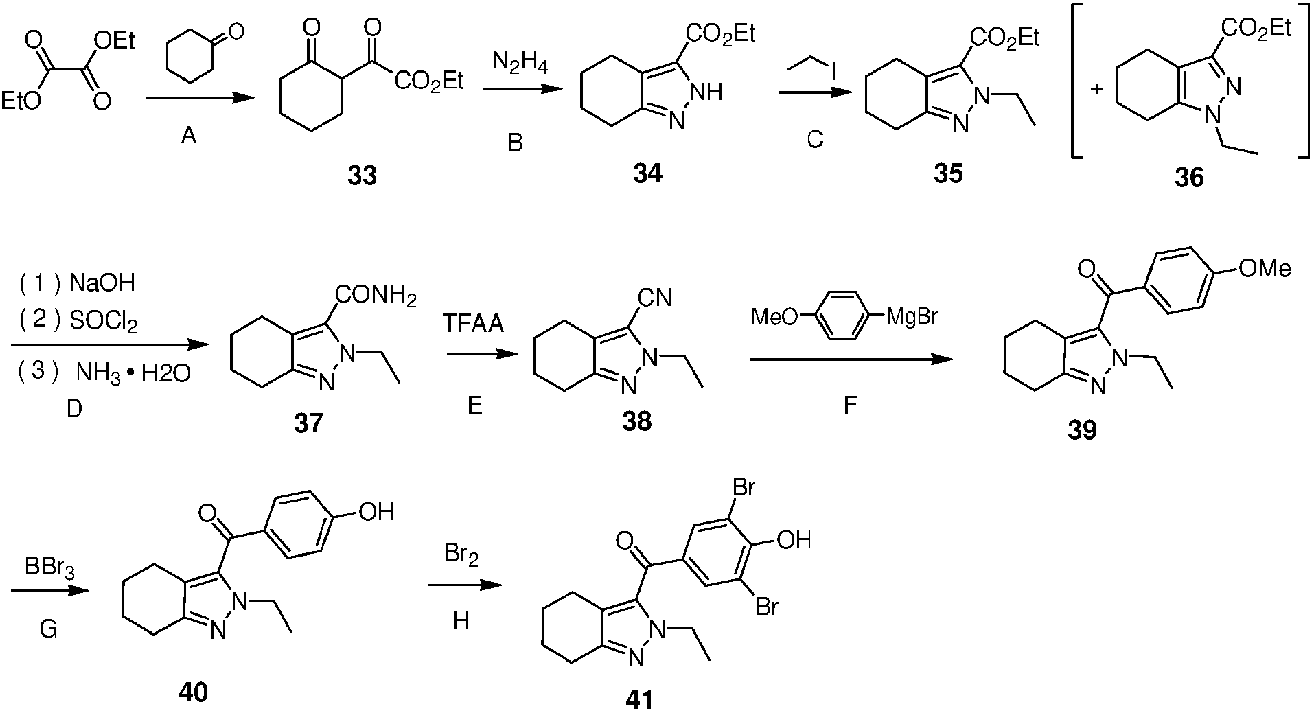
Paso A: A una solución de ciclohexanona (9.81 g, 100 mmol) y oxalato de dietilo (14.6 g, 100 mmol) en THF (100 ml) se añadió en porciones hidruro de sodio al 60 % (4.8 g, 120 mmol). Una vez completada la adición, se calentó la mezcla a 40 °C y se agitó durante 0.5 horas y después se elevó a 50 °C y se agitó durante 1.5 horas. Después de enfriar a temperatura ambiente, se vertió la solución de reacción en una solución de ácido acético (8 ml) en agua (200 ml). A la mezcla se le realizó extracción con metil terc-butil éter (100 ml x 2) y se lavaron las fases orgánicas
combinadas, con salmuera saturada (40 ml). Se evaporó el solvente bajo presión reducida y se purificó el producto mediante cromatografía en columna (gel de sílice de malla 200-300. eluido con éter de petróleo) para dar 2-oxi-2-(2-oxociclohexil)acetato de etilo (33) (10.5 g) . El rendimiento fue del 53.0 %.
Paso B: Se agitó a 60 °C durante 2 horas una solución del compuesto 33 (10.1 g, 51.0 mmol) e hidrato de hidracina al 85 % (1.84 g, 48.8 mmol) en etanol (40 ml) . Se evaporó el solvente bajo presión reducida, se agregó agua (40 ml), a la mezcla se le realizó extracción con acetato de etilo (40 ml x 3) y las capas orgánicas combinadas fueron lavadas con salmuera (30 ml). Se evaporó el solvente bajo presión reducida y se purificó el producto mediante cromatografía en columna (gel de sílice de malla 200~300. éter de petróleo:acetato de etilo = 1:100 a 1:3) para dar etiléster del ácido 4.5.6.7-tetrahidro-2H-indazol-3-carboxílico (34) (5.0 g). El rendimiento fue del 50.5 %.
Paso C: Se agitó a temperatura ambiente durante la noche una mezcla que contenía el compuesto 34 (2.3 g, 11.8 mmol), yoduro de etilo (3.69 g, 23.7 mmol), carbonato de cesio (5.79 g, 17.8 mmol) y DMF (25 ml) . Después de agregar agua (75 ml), a la mezcla se le realizó extracción con acetato de etilo (50 ml * 3) y las capas orgánicas combinadas fueron lavadas con agua (20 ml * 2) y salmuera saturada (15 ml) y se secaron sobre sulfato de sodio anhidro. Se evaporó el solvente bajo presión reducida y se purificó el producto mediante cromatografía en columna (gel de sílice de malla 200~300. éter de petróleo:acetato de etilo = 1:20 a 1:10) para dar éster etílico del ácido 2-etil-4.5.6.7 - tetrahidro-2H-indazol-3-carboxílico (35) (1.58 g, éter de petróleo:acetato de etilo = 1:1. Rf = 0.8) y éster etílico del ácido 1-etil-4.5.6.7-tetrahidro-2H-indazol-3-carboxílico (36) (1.01 g, éter de petróleo:acetato de etilo = 1:1. Rf = 0.5). Los rendimientos fueron 60.2 % y 38.5 % respectivamente.
Paso D: Se agitó a 40 °C durante 1 hora una mezcla que contenía el compuesto 35 (1.58 g, 7.24 mmol), hidróxido de sodio (580 mg, 14.5 mmol), metanol (5 ml) y agua (15 ml) . Se evaporó el agua bajo presión reducida y después se llevó el agua dos veces con tolueno. Se añadieron al residuo cloruro de tionilo (6 ml) y DMF (1 gota), y se agitó la mezcla resultante a reflujo durante 1 hora. Se evaporó el solvente bajo presión reducida, después se añadió THF (15 ml) y la solución de THF anterior se añadió en porciones a amoníaco acuoso concentrado (15 ml) en un baño de agua con hielo. Una vez completada la adición, se continuó agitando durante 20 minutos. Después de agregar agua (30 ml), a la mezcla se le realizó extracción con acetato de etilo (30 ml x 3) y las capas orgánicas combinadas fueron lavadas con salmuera (20 ml) y se secaron sobre sulfato de sodio anhidro. Se evaporó el solvente bajo presión reducida para dar 2-etil-4.5.6.7-tetrahidro-2H-indazol-3-carboxamida (37) (1.18 g). El rendimiento fue del 84.3 %. Paso E: Se agitó a temperatura ambiente durante 3 horas una solución de anhídrido trifluoroacético (1.92 g, 9.14 mmol) y compuesto 37 (1.1 g, 5.69 mmol) en THF (20 ml) . Se evaporó el solvente bajo presión reducida, se agregó agua (20 ml) y se ajustó el valor de pH a 8 - 9 con una solución acuosa de hidróxido de sodio 2 M. A la mezcla se le realizó extracción con acetato de etilo (30 ml x 2) y las capas orgánicas combinadas fueron lavadas con salmuera (15 ml) y se secaron sobre sulfato de sodio anhidro. Se evaporó el solvente bajo presión reducida y se purificó el producto mediante cromatografía en columna (gel de sílice de malla 200-300. acetato de etilo:éter de petróleo = 1:20) para dar 2-etil-4.5.6.7-tetrahidro- 2H-indazol-3-carbonitrilo (38) (640 mg). El rendimiento fue del 64.2 %. 1H RMN (DMSO-d6.400 MHz) ó 4.22 (q, J = 7.2 Hz, 2H), 2.59-2.50 (m, 4H), 1.76-1.68 (m, 4H), 1.37 (t, J = 7.2 Hz, 3H). Paso F: Se añadió gota a gota 1.0 M de bromuro de 4-metoxifenilmagnesio en THF (5.7 ml) a una solución del compuesto 38 (500 mg, 2.85 mmol) en THF (10 ml). Una vez completada la adición, se agitó la mezcla resultante a temperatura ambiente durante la noche. Se añadió solución de ácido clorhídrico 6 M (5 ml) y se ajustó el valor de pH a 8 - 9 con una solución acuosa de hidróxido de sodio 2 M después de agitar durante aproximadamente 1 hora. A la mezcla se le realizó extracción con acetato de etilo (40 ml x 2) y las capas orgánicas combinadas fueron lavadas con salmuera (20 ml) y se secaron sobre sulfato de sodio anhidro. Se evaporó el solvente bajo presión reducida y se purificó el producto por cromatografía en columna (gel de sílice de malla 200-300. acetato de etilo:éter de petróleo = 1:100 a 1:1) para dar (2-etil-4.5.6,7-tetrahidro-2H-indazol-3-il)-(4-metoxifenil)-metanona (39) (300 mg). El rendimiento fue del 37.0 %.
Paso G: Se añadió gota a gota una solución 1.0 M de tribromuro de boro en tolueno (3.2 ml) a una solución del compuesto 39 (300 mg, 1.05 mmol) en diclorometano anhidro (6 ml). Después de la adición, se agitó la mezcla resultante a temperatura ambiente durante la noche. Se vertió la mezcla de reacción en agua con hielo (30 ml) y se ajustó el valor de pH a 7-8 con bicarbonato de sodio saturado. A la mezcla se le realizó extracción con acetato de etilo (30 ml x 3) y las capas orgánicas combinadas fueron lavadas con salmuera (20 ml) y se secaron sobre sulfato de sodio anhidro. Se evaporó el solvente bajo presión reducida para dar (2-etil-4.5.6.7-tetrahidro-2H-indazol-3-il)(4-hidroxifenil)metanona (40) (280 mg). El rendimiento fue del 98.6 %. 1H RMN (DMSO-d6. 400 MHz) ó 10.22 (s, 1H), 7.38 (d, J = 8.4 Hz, 2H ), 6.81 (d, J = 8.4 Hz, 2H ), 3.99-3.96 (m, 2H), 2.58-2.55 (m, 2H), 2.10-2.08 (m, 2H), 1.72-1.70 (m, 2H), 1.58-1.57 (m, 2H), 1.21 (t, J = 7.2 Hz, 3H).
Paso H: Se añadió gota a gota una solución de bromo (124 mg, 0.776 mmol) en ácido acético (3 ml) a una mezcla del compuesto 40 (100 mg, 0.370 mmol) y acetato de sodio anhidro (89 mg, 1.11 mmol) en ácido acético (15 ml). Una vez completada la adición, se agitó la mezcla resultante a temperatura ambiente durante 1 hora. Se añadió gota a gota una solución acuosa diluida de bisulfito de sodio a la mezcla de reacción hasta que se desvaneció el color. Se evaporó el solvente bajo presión reducida, se agregó agua (15 ml) y se ajustó el valor de pH a 7 - 8 con bicarbonato de sodio saturado. A la mezcla se le realizó extracción con acetato de etilo (40 ml x 2) y se secó sobre sulfato de sodio anhidro. Se evaporó el solvente bajo presión reducida y se purificó el producto por cromatografía en columna
(gel de sílice de malla 200-300. acetato de etilo:éter de petróleo = 1:100 a 1:1) para dar (3.5-dibromo-4-hidroxifenil)(2-etil-4.5.6.7-tetrahidro-2H-indazol-3-il)metanona (41). 1H RMN (DMSO-de. 400 MHz) 5 7.83 (s, 2H), 4.20-4.18 (m, 2H), 2.60-2.58 (m, 2H), 2.14-2.11 (m, 2H), 1.73-1.72 (m, 2H), 1.57-1.56 (m, 2H), 1.31 (t, J = 7.2 Hz, 3H).MS (EI, m/z): 426.9 [M-H]-.
Ejemplo 8: Síntesis de 2.6-dibromo-4-[(2-etil-5.6.7.8-tetrahidroimidazo[1.2-a]-piridin-3-il)hidroximetil]fenol (42)

Se añadió hidruro de litio y aluminio (18 mg, 0.474 mmol) a una solución del compuesto 5 (135 mg, 0.315 mmol) en THF (15 ml) y se agitó la mezcla a esta temperatura durante 0.5 horas. Después de agregar agua (15 ml), se ajustó el valor del pH a 5 ~ 6 con una solución de ácido cítrico 2 M y a la mezcla se le realizó extracción con una mezcla de acetato de etilo/THF (20 ml x 3). Se lavaron las fases orgánicas combinadas, con salmuera (15 ml) y se secaron sobre sulfato de sodio anhidro. Se evaporó el solvente bajo presión reducida y se purificó el producto mediante cromatografía en columna (gel de sílice de malla 200-300. diclorometano:metanol = 1:100 a 1:30) para dar 2.6-dibromo-4-[(2-etil-5.6.7.8-tetrahidroimidazo[1.2-a]piridin-3-il)-hidroximetil]fenol (42). 1H RMN (DMSO-d6. 400 MHz) 5 7.36 (s, 2H), 5.95 (s, 1H), 5.78 (s, 1H), 3.88-3.85 (m, 2H), 2.67-2.65 (m, 2H), 2.32 (q, J = 7.6 Hz, 2H), 1.76-1.69 (m, 4H), 1.04 (t, J = 7.6 Hz, 3H).MS (EI, m/z): 431.0 [M+H]+.
Ejemplo 9: Síntesis de (3.5-dibromo-4-hidroxifenil)(2-etil-6-metil-5.6.7.8-tetrahidroimidazo[1.2-a]piridin-3-il)metanona (43)

La síntesis del Compuesto 43 se llevó a cabo de acuerdo con los procedimientos del Ejemplo 1. en el que la 2-aminopiridina en el Paso A del Ejemplo 1 se reemplazó con 2-amino-5-metilpiridina. 1H RMN (DMSO-d6. 400 MHz) 5 7.79 (s, 2H), 4.16-4.11 (m, 1H), 3.57-3.51 (m, 1H), 2.96-2.79 (m, 2H ), 2.27 (q, J = 7.6 Hz, 2H), 2.03-1.91 (m, 2H), 1.56-1.47 (m, 1H), 1.09-1.03 (m, 6H).MS (EI, m/z): 441.0 [M-H]- .
Ejemplo 10: Síntesis de (3-bromo-4-hidroxi-5-metilfenil)(2-etil-5.6.7.8-tetrahidroimidazo[1.2-a]piridin-3-il)metanona (48)
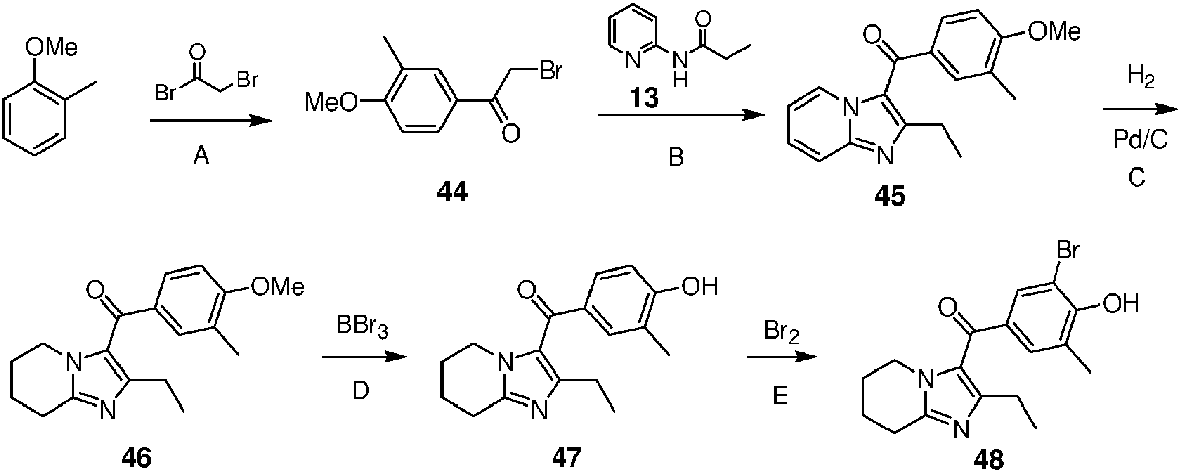
Paso A: Se añadió gota a gota una solución de bromuro de bromoacetilo (9.9 g, 49.0 mmol) en diclorometano (10 ml) a una solución de 2-metilanisol (5.0 g, 40.9 mmol) y tricloruro de aluminio (6.0 g, 45.0 mmol) en diclorometano (40 ml) durante aproximadamente 20 minutos a 0 - 5°C. Una vez completada la adición, se agitó continuamente durante 2 horas a esta temperatura la mezcla obtenida . Se vertió la solución de reacción en una cantidad adecuada de agua con hielo por lotes y se le realizó extracción con diclorometano (60 ml x 3). La fase orgánica combinada fue lavada sucesivamente con agua (30 ml), solución acuosa saturada de bicarbonato de sodio (30 ml x 2), agua (30 ml) y salmuera (30 ml) y después se secó sobre sulfato de sodio anhidro. Se filtró la fase orgánica mediante una columna corta de gel de sílice. Se evaporó el solvente bajo presión reducida y se purificó el producto por cromatografía instantánea (gel de sílice de malla 200-300. acetato de etilo:éter de petróleo = 1:100 a 1:30) para obtener 2-bromo-1-(3- metil-4-metoxifenil)etanona (44) (3.0 g). El rendimiento fue del 30.2 %. Se llevaron a cabo los Pasos B, C, D y E de acuerdo con la preparación de los Pasos B, C, D y E en el Ejemplo 1 para dar (3-bromo-4-hidroxi-5-metilfenil)(2-etil-5.6.7.8-tetrahidroimidazo[1.2-a]piridin-3-il)metanona (48). 1H RMN (DMSO-d6. 400 MHz) 5 7.57 (s, 1H), 7.28 (s, 1H), 3.89-3.87 (m, 2H), 2.76-2.73 (m, 2H), 2.29 (q, J = 7.6 Hz, 2H), 1.96 (s, 3H), 1.87-1.80 (m, 4H), 1.05 (q, J = 7.6 Hz, 3H).MS (EI, m/z): 363.1 [M+H]+.
Ejemplo 11: Síntesis de (3.5-dibromo-4-hidroxifenil)(2-etil-2.4.6.7-tetrahidropirano[4.3-c]pirazol-3-il metanona (58)
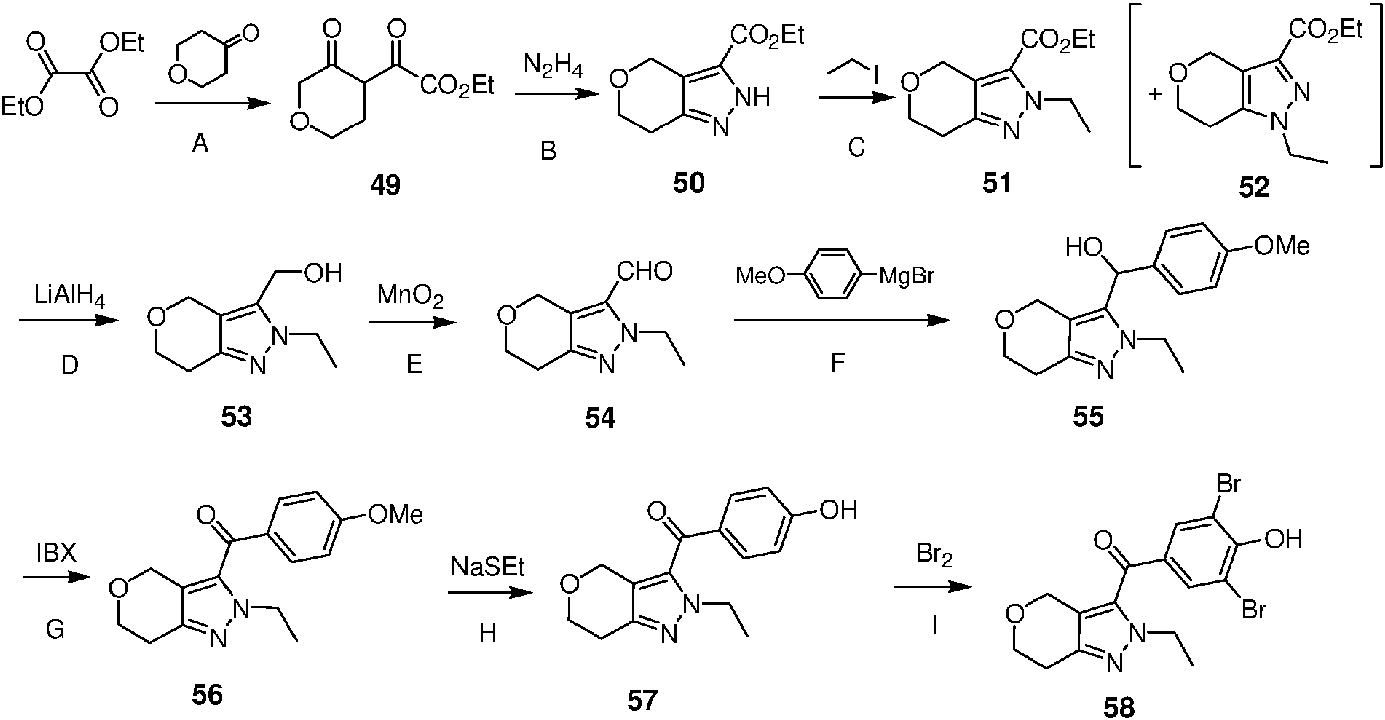
Los procedimientos experimentales de los Pasos A, B y C fueron llevados a cabo de acuerdo con la preparación de los Pasos A, B y C del Ejemplo 7, en los que la ciclohexanona del Paso A del Ejemplo 1 fue reemplazada con tetrahidropirona.
Paso D: Se añadió gota a gota una solución del compuesto 51 (2.5 g, 11.1 mmol) en THF (10 ml) a una mezcla que contenía hidruro de litio y aluminio (846 mg, 22.3 mmol) y THF (15 ml). Una vez completada la adición, se agitó la mezcla resultante adicionalmente a esta temperatura durante 1 hora. A la mezcla de reacción se le añadieron gota a gota agua (1 ml), solución de hidróxido de sodio al 10 % (2 ml) y agua (3 ml). Después de filtrar, la torta del filtro se sometió a un lavado por goteo con THF (15 ml) y el filtrado se secó sobre sulfato de sodio anhidro. Se evaporó el solvente bajo presión reducida para dar (2-etil-2.4.6.7-tetrahidropirano[4.3-c]pirazol-3-il)metanol (53) (2.1 g). El rendimiento fue del 100 %.
Paso E: Se agitó a 45 °C durante la noche una mezcla que contenía el compuesto 53 (2.0 g, 11.0 mmol), dióxido de manganeso (4.78 g, 55.0 mmol) y cloroformo (15 ml) . Los insolubles se eliminaron por filtración y se evaporó el solvente bajo presión reducida. Se purificó el producto por cromatografía en columna (gel de sílice de malla 200-300. acetato de etilo:éter de petróleo = 1:20 a 1:6) para dar (2-etil-2.4.6.7-tetrahidropirano[4.3- c]pirazol-3-il)carbaldehído (54) (928 mg). El rendimiento fue del 46.8 %.
Paso F: Se añadió gota a gota una solución 1.0 M de bromuro de 4-metoxifenilmagnesio en THF (5.5 ml) a una solución del compuesto 54 (900 mg, 4.99 mmol) en THF (15 ml) a -70 °C. Una vez completada la adición, se agitó la mezcla resultante a esta temperatura durante 20 minutos más. Se añadió gota a gota lentamente La mezcla de reacción a agua helada (20 ml) y se le realizó extracción con acetato de etilo (30 ml x 3). Se secaron las fases orgánicas combinadas sobre salmuera (30 ml) y se secaron sobre sulfato de sodio anhidro. Se evaporó el solvente bajo presión reducida y se purificó el producto mediante cromatografía en columna (gel de sílice de malla 200-300. acetato de etilo:éter de petróleo = 1: 10 a 1:1) para dar (2-etil-2.4.6.7-tetrahidropirano[4.3-c]pirazol-3-il) (4-metoxifenil)metanol (55) (1.4 g). El rendimiento fue del 97.3 %.
Paso G: Se agitó a temperatura ambiente durante 1.5 horas una mezcla que contenía el compuesto 55 (1.38 g, 4.79 mmol), ácido 2-yodobenzoico (1.74 g, 55.0 mmol) y DMSO (15 ml) . Después de la adición de agua (45 ml), a la mezcla se le realizó extracción con acetato de etilo (30 ml x 3) y se lavaron las fases orgánicas combinadas sucesivamente con agua (20 ml x 2) y salmuera (20 ml) y se secaron sobre sulfato de sodio anhidro. Se evaporó el solvente bajo presión reducida para dar (2-etil-2.4.6.7-tetrahidropirano[4.3-c]pirazol-3-il)(4-metoxifenil)-metanona (56) (1.32 g) . El rendimiento fue del 96.3 %.
Paso H Los procedimientos experimentales fueron llevados a cabo de acuerdo con el método de preparación del Paso F en el Ejemplo 3 para dar (2-etil-2.4.6.7-tetrahidropirano[4.3-c]pirazol-3-il) (4- hidroxifenil)metanona (57).
Paso I Se llevaron a cabo procedimientos experimentales de acuerdo con el método de preparación del Paso E en el Ejemplo 1 para dar (3.5-dibromo-4-hidroxifenil)(2-etil-2.4.6.7-tetrahidropirano)-[4. 3-c]pirazol-3-il) metanona (58). 1H RMN (DMSO-d6. 400 MHz) 57.84 (s, 2H), 4.26 (q, J = 7.2 Hz, 2H), 4.22 (s, 2H), 3.86 (t, J = 6.4 Hz, 2H ), 2.71 (t, J = 6.4 Hz, 2H), 1.35 (t, J = 7.2 Hz, 3H).MS (EI, m/z): 431.0 [M+H]+.
Ejemplo 12: Síntesis de (3.5-dibromo-4-hidroxifenil)(2-etil-6-fluoro-5.6.7.8-tetrahidroimidazo[1.2-a]piridin-3-il)metanona (62)

Paso A: Se disolvieron 2-amino-5-fluoropiridina (750 mg, 6.69 mmol) y el compuesto 6 (1.65 g, 8.00 mmol) en THF
(15 ml), después se añadieron secuencialmente ácido yodoftálico (2.59 g, 8.05 mmol) y eterato de trifluoruro de boro
(192 mg, 1.35 mmol) en el baño de agua con hielo. Una vez completada la adición, se continuó agitando a temperatura ambiente durante la noche. Se añadió agua (30 ml), y se ajustó el valor de pH a 7-8 con una solución saturada de bicarbonato de sodio y después a la mezcla se le realizó extracción con acetato de etilo (30 ml x 3). Las capas orgánicas combinadas fueron lavadas con salmuera (20 ml) y se secaron sobre sulfato de sodio anhidro. Se evaporó el solvente bajo presión reducida y se purificó el producto por cromatografía en columna (gel de sílice de malla 200 ~ 300. acetato de etilo:éter de petróleo:diclorometano = 1:30:1 a 1:6:1) para dar (6-fluoro -2 etilimidazo[1.2-a]piridin-3-il)-(4-metoxifenil)metanona (59) (390 mg). El rendimiento fue del 19.5 %.
Los procedimientos experimentales de los Pasos B, C y D fueron llevados a cabo de acuerdo con los métodos de preparación de los Pasos C, D y E del Ejemplo 1 para dar (3.5-dibromo-4-hidroxifenil)(2-etil-6-fluoro-5,6.7.8-tetrahidroimidazo[1.2-a]piridin-3-il)metanona (62). 1H RMN (DMSO-d6. 400 MHz) 5 7.80 (s, 2H), 4.98-4.96 (m, 1H), 4.56-4.51 (m, 1H), 4.42-4.37 (m, 1H ), 3.01 (t, J = 6.4 Hz, 2H), 2.33-2.21 (m, 4H), 1.06 (t, J = 7.2 Hz, 3H).MS (EI, m/z): 433.0 [M+H]+ .
Ejemplo 13: Síntesis de 3-bromo-5-((2-etil-5.6.7.8-tetrahidroimidazo[1.2-a]piridin-3-il)hidroximetil)-2-hidroxil benzonitrilo (63)

Se añadió borohidruro de sodio (90 mg, 2.4 mmol) a una solución del compuesto 17 (90 mg, 0.24 mmol) en THF anhidro (7 ml), se agitó a temperatura ambiente durante 1 hora y se ajustó el valor de pH a 5 ~ 6 con solución de ácido cítrico 2 M después de agregar agua (10 ml). A la mezcla se le realizó extracción con acetato de etilo (15 ml x
2) y se secó sobre sulfato de sodio anhidro. Se evaporó el solvente bajo presión reducida y se purificó el producto mediante cromatografía en columna (gel de sílice de malla 200-300. acetato de etilo:éter de petróleo = 1:2 a 5:1) para dar 3-bromo-5-((2-etil)-5.6.7.8-tetrahidroimidazo[1.2-a]piridin-3-il)hidroximetil)-2-hidroxibenzonitrilo (63) (8 mg).
El rendimiento fue del 8.89 %. EM (IE, m/z) 376.10 [M+H]+.
Ejemplo 14 Ensayo de inhibición del transporte de ácido úrico para compuestos en la línea celular de transfección HEK293-hURATl
I. Materiales
Zurampic fue adquirido de Chengdu Yichao Pharmaceutical Technology Co., Ltd. El plásmido pCMV6-hURAT1 fue adquirido de Origene Technologies, Inc. Geneticin (G418) fue adquirida de Sangon Biotech Co., Ltd. La línea celular HEK293 fue adquirida del Cell Resource Center of Shanghai Institutes for Biological Sciences of the Chinese Academy of Sciences. La polilisina fue adquirida de Sigma-Aldrich Co. LLC. El ácido úrico 14C fue adquirido de American Radiolabeled Chemicals, Inc. Gluconato de sodio, gluconato de potasio, gluconato de calcio, KH2PO4.
MgSO4 , glucosa y HEPES fueron adquiridos de Sinopharm Chemical Reagent Co., Ltd. El medio de cultivo DMEM y el suero bovino fetal fueron adquiridos de Thermo Fisher Scientific Inc.
II. Métodos experimentales y resultados.
1. Construcción de una línea celular HEK293 estable con alta expresión de hURAT1. El plásmido pCMV6-hURAT1 fue transfectado en células HEK293. después se obtuvo la cepa estable mediante el cribado de resistencia G418 (concentración final de 500 pg/ml), que es la alta expresión de Proteína hURAT1 de membrana transportadora. Puede ser usado para ensayo de inhibición in vitro del transportador de ácido úrico hURAT1 (Weaver YM, Ehresman DJ, Butenhoff JL, et al. Roles of rat renal organic anion transporters in transporting perfluorinated carboxylates with different chain lengths. Toxicological Sciences, 2009. 113(2):305-314).
2. Revestimiento de placa de 24 pocilios: a una placa de 24 pocilios revestidos se añadieron 200 pl de polilisina 0.1 mg/ml por pocillo y se dejó la placa durante la noche. Se eliminó la polilisina de los pocillos. Los pocillos se limpiaron minuciosamente con agua esterilizada y se secaron para su uso.
3. A la placa de 24 pocillos revestida anterior se añadieron células estables HEK293-hURAT1 (2 x 105 células por pocillo). Las células fueron cultivadas a 37 °C bajo 5 % de CO2 durante 3 días.
4. Preparación del amortiguador HBSS: se pesaron los siguientes reactivos de acuerdo con la concentración final de gluconato de sodio 125 mM, gluconato de potasio 4.8 mM, gluconato de calcio 1.3 mM, 1.2 mM de KH2PO4, 1.2 mM de MgSO4, glucosa 5.6 mM y 25 mM de HEPES . Se añadió agua desionizada para alcanzar el volumen correspondiente y la solución se mezcló por completo para dar HBSS (valor de pH: 7.4). El amortiguador fue almacenado a -20 °C en un refrigerador.
5. El día del experimento, se retiró el amortiguador HBSS del refrigerador y se calentó a 37 °C en un baño de agua. Se retiró la placa de 24 pocillos con células estables HEK293-hURAT1. se retiró el medio de cultivo y lavaron las células con HBSS, después se agregaron 160 pl de HBSS y 20 pl de compuesto de prueba por pocillo. La concentración final del compuesto ensayado por pocillo fue de 500 nM. El pocillo de control en blanco contenía solo 180 pL de HBSS sin el compuesto probado. Se colocó la placa a temperatura ambiente durante 10 minutos.
6. A cada pocillo se le añadieron 20 pL de ácido 14C- 50 pM. La placa de 24 pocillos fue colocada a temperatura ambiente durante 20 minutos.
7. Se eliminó la solución de cada pocillo y se lavaron las células de cada pocillo con el amortiguador HBSS enfriado previamente. A cada pocillo se añadió NaOH 0.2 M para disolver las células. Se recogió la solución que contenía fragmentos de células y se añadió la cantidad adecuada de líquido de centelleo. Después se detectó la intensidad del radioisótopo 14Cdel ácido úrico (valor CPM) utilizando el analizador de centelleo líquido PerkinElmer MicroBeta Trilux 1450.
8. En las líneas celulares HEK293 transfectadas, la fórmula para calcular la tasa de inhibición del transporte de ácido úrico para los compuestos se muestra a continuación (Tabla 1), el valor de CPM de los compuestos probados se representó por CPM(compuesto probado) y el valor de CPM del control en blanco fue representado por CPM(control en blanco). Todas las pruebas se repitieron tres veces, se promediaron los resultados y se calculó la desviación estándar (DE):
Tasa de inhibición (%) (Concentración de compuesto 500 nM) _ (CPM (Control de blanco) — CPM(Compuesto de prueba) / CPM (Control de blanco) X 100%
III. Resultados experimentales
Los resultados mostraron que, en comparación con el fármaco de control Zurampic a la concentración de 500 nM, los compuestos de la invención (en particular, 5, 10, 17, 23, 25, 41, 42, 43 y 48) tienen muy buenos efectos inhibidores del transporte de ácido úrico en la línea celular de transfección HEK293-hURAT 1.
Tabla 1: Tasas de inhibición del transporte de ácido úrico para compuestos de prueba y Zurampic en la línea celular de transfección HEK293-hURAT1

continuación

Claims (10)
1. Un compuesto representado por la estructura de la Fórmula (I) o una de sus sales farmacéuticamente aceptables.

en la que:
El anillo A es un anillo de seis miembros no aromático con o sin un heteroátomo O, N o S;
el anillo B es un anillo aromático de cinco miembros que contiene dos átomos de N o un anillo furano; Z, E o X son cada uno independientemente un átomo de C o N;
G es un átomo de N o de O; y cuando G es un átomo de O, Z, E y X son todos átomos de C; cuando G es un átomo de N, sólo uno de Z, E y X es un átomo de N;
Y es carbonilo, azufre, sulfona, sulfóxido, metileno o imino opcionalmente sustituidos;
R1 es uno o más seleccionados del grupo que consiste en hidrógeno, deuterio, hidroxi, halógeno, nitro, amino, ciano, alquilo C1-5, alquilo C1-5 sustituido, amino C1-3 sustituido, alcoxi C1-3, alcoxi C1-3 sustituido y alquiltio C1-5; R2 es uno o más seleccionados del grupo que consiste en hidrógeno, deuterio, hidroxi, halógeno, nitro, amino, ciano, ciano, alquilo C1-4, alquilo C1-3 sustituido, alquenilo C2-3, alquinilo C2-3, amino C1-3 sustituido, alcoxi C1-5, alcoxi C1-5 sustituido y alquiltio C1-5;
R3 es alquilo C1-4, alquilo C1-4 sustituido o cicloalquilo C3-4 ;
m es un número entero de 0 a 3;
n es un número entero de 1 a 3;
el sustituyente en el grupo Y es seleccionado del grupo que consiste en hidroxilo, ciano, nitro, amino, carboxilo y alcoxi C1-3, el sustituyente en el grupo R1 R2 o R3 es seleccionado del grupo que consiste en hidroxilo, halógeno, nitro, amino y ciano.
2. El compuesto o sal farmacéuticamente aceptable del mismo de acuerdo con la reivindicación 1, en el que el anillo A es un anillo de ciclohexeno o un anillo no aromático de seis miembros que contiene al menos un átomo de O y/o N.
3. El compuesto o sal farmacéuticamente aceptable del mismo de acuerdo con la reivindicación 1, en el que el anillo A es un anillo ciclohexeno, anillo 3,4-dihidro-2H-pirano, anillo de tetrahidropirano, anillo 2,3,4,5-tetrahidropiridina, anillo 5,6-dihidro-2H-1,3-oxazina o anillo 1,2,5,6-tetrahidropirimidina.
4. El compuesto o sal farmacéuticamente aceptable del mismo de acuerdo con la reivindicación 1, en el que R1 es seleccionado de uno o más del grupo que consiste en hidrógeno, deuterio, flúor, cloro, bromo, hidroxi, ciano, alquilo C1-3, haloalquilo C1-3 y alcoxi C1-3 ; m es 0, 1 o 2.
5. El compuesto o sal farmacéuticamente aceptable del mismo de acuerdo con la reivindicación 1, en el que R2 es seleccionado de uno o más del grupo que consiste en hidrógeno, deuterio, halógeno, ciano, vinilo, etinilo, alquilo C1-2 , alquilo C1-2 sustituido, alcoxi C1-2, alcoxi C1-2 sustituido, alquiltio C1-2, alquiltio C1-2 sustituido; el sustituyente es seleccionado del grupo que consiste en deuterio, halógeno, alquilo C1-2, cicloalquilo C3-4 y alcoxi C1-3 ; n es 0, 1 o 2.
6. El compuesto o sal farmacéuticamente aceptable del mismo de acuerdo con la reivindicación 1, en el que R3 es seleccionado del grupo que consiste en metilo, etilo, n-propilo, isopropilo, ciclopropilo y ciclobutilo.
7. El compuesto o sal farmacéuticamente aceptable del mismo de acuerdo con la reivindicación 1, en el que el compuesto es seleccionado de los siguientes:


8. Una composición farmacéutica que comprende el compuesto o una sal farmacéuticamente aceptable del mismo de acuerdo con una cualquiera de las reivindicaciones 1 a 7, como un ingrediente activo o ingrediente activo principal, y excipientes farmacéuticamente aceptables.
9. Compuesto o sal farmacéuticamente aceptable del mismo de acuerdo con una cualquiera de las reivindicaciones 1 a 7 para uso en la promoción de la excreción de ácido úrico.
10. Compuesto o sal farmacéuticamente aceptable del mismo de acuerdo con una cualquiera de las reivindicaciones 1 a 7 para uso en el tratamiento o prevención de hiperuricemia, enfermedad renal o gota.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201710386922 | 2017-05-26 | ||
| PCT/CN2018/088400 WO2018214961A1 (zh) | 2017-05-26 | 2018-05-25 | 促尿酸排泄的urat1抑制剂 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2912941T3 true ES2912941T3 (es) | 2022-05-30 |
Family
ID=63352281
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES18806684T Active ES2912941T3 (es) | 2017-05-26 | 2018-05-25 | Inhibidores de URAT1 para promover la excreción de ácido úrico |
Country Status (11)
| Country | Link |
|---|---|
| US (1) | US11597725B2 (es) |
| EP (1) | EP3632904B1 (es) |
| JP (1) | JP6915907B2 (es) |
| KR (1) | KR102420892B1 (es) |
| CN (1) | CN108484600B (es) |
| CA (1) | CA3066680C (es) |
| DK (1) | DK3632904T3 (es) |
| ES (1) | ES2912941T3 (es) |
| MX (1) | MX394399B (es) |
| PL (1) | PL3632904T3 (es) |
| WO (1) | WO2018214961A1 (es) |
Families Citing this family (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN111410654B (zh) * | 2019-01-19 | 2022-05-17 | 江苏新元素医药科技有限公司 | 3-溴-5-(2-乙基咪唑并[1,2-a]吡啶-3-羰基)-2-羟基苯甲腈的合成 |
| CN111943957B (zh) * | 2019-05-17 | 2023-01-06 | 中国医学科学院药物研究所 | 喹啉甲酰胺类化合物及其制备方法和用途 |
| WO2020253659A1 (en) * | 2019-06-17 | 2020-12-24 | Fochon Pharmaceuticals, Ltd. | Heterocyclic derivatives and use thereof |
| CN119059989A (zh) * | 2023-05-31 | 2024-12-03 | 中国药科大学 | Urat1抑制剂及其制备方法和用途 |
| WO2025237171A1 (zh) * | 2024-05-11 | 2025-11-20 | 广州市联瑞制药有限公司 | 嘧啶并五元氮杂环酚类化合物及其应用 |
| CN119241538A (zh) * | 2024-09-30 | 2025-01-03 | 陕西科技大学 | 一类三唑并吡啶结构的化合物、合成方法及其应用 |
Family Cites Families (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB1565734A (en) * | 1977-05-03 | 1980-04-23 | Laroche Navarron Sa | Fused triazoles |
| NZ193926A (en) * | 1979-07-06 | 1984-05-31 | Labaz Sanofi Nv | 2-(alkyl or phenyl)-3(4-hydroxybenzoyl)indolizines |
| AR042067A1 (es) * | 2002-11-27 | 2005-06-08 | Bayer Pharmaceuticals Corp | Derivados de anilinopirazol utiles en el tratamiento de la diabetes |
| KR20080080584A (ko) * | 2005-11-30 | 2008-09-04 | 버텍스 파마슈티칼스 인코포레이티드 | c-Met의 억제제 및 이의 용도 |
| US8735386B2 (en) * | 2010-07-23 | 2014-05-27 | Boehringer Ingelheim International Gmbh | Aminopyrazoloquinazolines |
| US10005750B2 (en) * | 2010-10-06 | 2018-06-26 | J-Pharma Co., Ltd. | Developing potent urate transporter inhibitors: compounds designed for their uricosuric action |
| GB201321743D0 (en) * | 2013-12-09 | 2014-01-22 | Ucb Pharma Sa | Therapeutic agents |
| CN106065010B (zh) * | 2015-04-23 | 2019-01-01 | 镇江新元素医药科技有限公司 | 用于治疗或预防高尿酸血症或痛风的化合物 |
| KR102109061B1 (ko) | 2015-09-10 | 2020-05-11 | 쟝쑤 애텀 바이오사이언스 앤드 파머수티컬 컴퍼니 리미티드 | 고요산혈증 또는 통풍을 치료 또는 예방하는 화합물 |
| CN106433229A (zh) * | 2016-08-29 | 2017-02-22 | 无锡双诚炭黑有限公司 | 油炉法炭黑反应装置及其反应方法 |
| WO2018090921A1 (zh) * | 2016-11-16 | 2018-05-24 | 江苏新元素医药科技有限公司 | Urat1抑制剂及其应用 |
| CN108084186B (zh) * | 2016-11-16 | 2021-06-25 | 江苏新元素医药科技有限公司 | Urat1抑制剂及其应用 |
-
2018
- 2018-05-25 EP EP18806684.9A patent/EP3632904B1/en active Active
- 2018-05-25 JP JP2019565209A patent/JP6915907B2/ja not_active Expired - Fee Related
- 2018-05-25 MX MX2019014078A patent/MX394399B/es unknown
- 2018-05-25 CN CN201810517139.1A patent/CN108484600B/zh active Active
- 2018-05-25 WO PCT/CN2018/088400 patent/WO2018214961A1/zh not_active Ceased
- 2018-05-25 PL PL18806684.9T patent/PL3632904T3/pl unknown
- 2018-05-25 CA CA3066680A patent/CA3066680C/en active Active
- 2018-05-25 KR KR1020197038336A patent/KR102420892B1/ko active Active
- 2018-05-25 US US16/617,426 patent/US11597725B2/en active Active
- 2018-05-25 ES ES18806684T patent/ES2912941T3/es active Active
- 2018-05-25 DK DK18806684.9T patent/DK3632904T3/da active
Also Published As
| Publication number | Publication date |
|---|---|
| EP3632904A4 (en) | 2021-03-03 |
| KR102420892B1 (ko) | 2022-07-14 |
| JP6915907B2 (ja) | 2021-08-04 |
| US20200262832A1 (en) | 2020-08-20 |
| WO2018214961A1 (zh) | 2018-11-29 |
| CA3066680A1 (en) | 2018-11-29 |
| JP2020521757A (ja) | 2020-07-27 |
| KR20200011974A (ko) | 2020-02-04 |
| EP3632904A1 (en) | 2020-04-08 |
| CA3066680C (en) | 2022-04-12 |
| MX2019014078A (es) | 2020-08-03 |
| CN108484600A (zh) | 2018-09-04 |
| CN108484600B (zh) | 2022-12-13 |
| US11597725B2 (en) | 2023-03-07 |
| DK3632904T3 (da) | 2022-07-25 |
| MX394399B (es) | 2025-03-24 |
| EP3632904B1 (en) | 2022-04-20 |
| PL3632904T3 (pl) | 2022-08-22 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2912941T3 (es) | Inhibidores de URAT1 para promover la excreción de ácido úrico | |
| ES2835553T3 (es) | Inhibidores de indazol de la vía de señalización de Wnt y sus usos terapéuticos | |
| JP2021152064A (ja) | 乳酸脱水素酵素の小分子阻害剤及びその使用方法 | |
| CN106478500B (zh) | 羧酸取代的(杂)芳环类衍生物及其制备方法和用途 | |
| ES2923177T3 (es) | Inhibidor de URAT1 y uso del mismo | |
| WO2013036912A2 (en) | Amido compounds as rorϒtmodulators and uses thereof | |
| PT2877468T (pt) | Compostos e métodos para o tratamento de vih | |
| CN102482276A (zh) | 杂环化合物及其用途 | |
| US9221838B2 (en) | Inhibitors of AKT activity | |
| EP3814331B9 (en) | Novel lxr modulators with bicyclic core moiety | |
| CN107683282A (zh) | 新型urat1抑制剂及其在医药上的应用 | |
| BR112021005086A2 (pt) | tratamento para doença hepática gordurosa não alcoólica | |
| US20200131144A1 (en) | Amine or (thio)amide containing lxr modulators | |
| WO2018157801A1 (zh) | 氰基取代的稠合双环衍生物及其制备方法和用途 | |
| CN118184658A (zh) | Usp1抑制剂 | |
| WO2018202039A1 (zh) | 杂环化合物及其制备方法 | |
| US20260083704A1 (en) | Lxr modulators with bicyclic core moiety for treating dyslipidemias | |
| CN107987006B (zh) | 吲哚类或氮杂吲哚类衍生物、其制备方法和应用 | |
| CN103889989B (zh) | 稠合杂环衍生物及其制药用途 | |
| WO2023208108A1 (zh) | 可用于降尿酸的化合物 | |
| CN115160314A (zh) | 杂环芳酰胺类化合物及其制备方法和应用 | |
| EP4221753A1 (en) | Compositions and methods of treating kidney disease and fibrosis | |
| EP4592302A1 (en) | Small molecule compound having phosphorylated aryl structure, and use thereof | |
| WO2015191640A2 (en) | Bicyclic xanthine oxidase inhibitors and methods of use | |
| WO2015191636A2 (en) | Xanthine oxidase inhibitors and methods of use |