ES2915502T3 - Procedimiento para preparar microesferas biodegradables con estabilidad mejorada y estabilidad al almacenamiento - Google Patents
Procedimiento para preparar microesferas biodegradables con estabilidad mejorada y estabilidad al almacenamiento Download PDFInfo
- Publication number
- ES2915502T3 ES2915502T3 ES18883786T ES18883786T ES2915502T3 ES 2915502 T3 ES2915502 T3 ES 2915502T3 ES 18883786 T ES18883786 T ES 18883786T ES 18883786 T ES18883786 T ES 18883786T ES 2915502 T3 ES2915502 T3 ES 2915502T3
- Authority
- ES
- Spain
- Prior art keywords
- continuous phase
- microspheres
- phase
- surfactant
- organic solvent
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 239000004005 microsphere Substances 0.000 title claims abstract description 253
- 238000000034 method Methods 0.000 title claims abstract description 50
- 238000003860 storage Methods 0.000 title description 21
- 239000003960 organic solvent Substances 0.000 claims abstract description 97
- 229920002988 biodegradable polymer Polymers 0.000 claims abstract description 74
- 239000004621 biodegradable polymer Substances 0.000 claims abstract description 74
- 239000007864 aqueous solution Substances 0.000 claims abstract description 62
- 239000000243 solution Substances 0.000 claims abstract description 58
- 239000004094 surface-active agent Substances 0.000 claims abstract description 48
- 239000003814 drug Substances 0.000 claims abstract description 39
- 239000000839 emulsion Substances 0.000 claims abstract description 30
- 229940079593 drug Drugs 0.000 claims abstract description 27
- 238000002156 mixing Methods 0.000 claims abstract description 16
- 239000011859 microparticle Substances 0.000 claims abstract description 15
- 238000001704 evaporation Methods 0.000 claims abstract description 12
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 claims description 135
- ADEBPBSSDYVVLD-UHFFFAOYSA-N donepezil Chemical compound O=C1C=2C=C(OC)C(OC)=CC=2CC1CC(CC1)CCN1CC1=CC=CC=C1 ADEBPBSSDYVVLD-UHFFFAOYSA-N 0.000 claims description 56
- 239000004372 Polyvinyl alcohol Substances 0.000 claims description 55
- 229920002451 polyvinyl alcohol Polymers 0.000 claims description 55
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 claims description 39
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 claims description 32
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 claims description 30
- 229960003530 donepezil Drugs 0.000 claims description 28
- 239000002904 solvent Substances 0.000 claims description 28
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 claims description 24
- 235000019441 ethanol Nutrition 0.000 claims description 19
- 239000000203 mixture Substances 0.000 claims description 14
- BDERNNFJNOPAEC-UHFFFAOYSA-N propan-1-ol Chemical compound CCCO BDERNNFJNOPAEC-UHFFFAOYSA-N 0.000 claims description 12
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 claims description 9
- AEMRFAOFKBGASW-UHFFFAOYSA-N Glycolic acid Polymers OCC(O)=O AEMRFAOFKBGASW-UHFFFAOYSA-N 0.000 claims description 8
- ZWEHNKRNPOVVGH-UHFFFAOYSA-N 2-Butanone Chemical compound CCC(C)=O ZWEHNKRNPOVVGH-UHFFFAOYSA-N 0.000 claims description 6
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 claims description 6
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 claims description 6
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 claims description 6
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 claims description 6
- -1 fatty acid ester Chemical class 0.000 claims description 6
- JVTAAEKCZFNVCJ-UHFFFAOYSA-N lactic acid Chemical compound CC(O)C(O)=O JVTAAEKCZFNVCJ-UHFFFAOYSA-N 0.000 claims description 6
- 229920001606 poly(lactic acid-co-glycolic acid) Polymers 0.000 claims description 6
- LEBVLXFERQHONN-UHFFFAOYSA-N 1-butyl-N-(2,6-dimethylphenyl)piperidine-2-carboxamide Chemical compound CCCCN1CCCCC1C(=O)NC1=C(C)C=CC=C1C LEBVLXFERQHONN-UHFFFAOYSA-N 0.000 claims description 4
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 claims description 4
- XSVMFMHYUFZWBK-NSHDSACASA-N Rivastigmine Chemical compound CCN(C)C(=O)OC1=CC=CC([C@H](C)N(C)C)=C1 XSVMFMHYUFZWBK-NSHDSACASA-N 0.000 claims description 4
- MUMGGOZAMZWBJJ-DYKIIFRCSA-N Testostosterone Chemical compound O=C1CC[C@]2(C)[C@H]3CC[C@](C)([C@H](CC4)O)[C@@H]4[C@@H]3CCC2=C1 MUMGGOZAMZWBJJ-DYKIIFRCSA-N 0.000 claims description 4
- 108700025485 deslorelin Proteins 0.000 claims description 4
- 239000012046 mixed solvent Substances 0.000 claims description 4
- 239000004632 polycaprolactone Substances 0.000 claims description 4
- 229920001610 polycaprolactone Polymers 0.000 claims description 4
- 229960004136 rivastigmine Drugs 0.000 claims description 4
- GJKXGJCSJWBJEZ-XRSSZCMZSA-N Deslorelin Chemical compound CCNC(=O)[C@@H]1CCCN1C(=O)[C@H](CCCN=C(N)N)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CO)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H](CC=1NC=NC=1)NC(=O)[C@H]1NC(=O)CC1)CC1=CNC2=CC=CC=C12 GJKXGJCSJWBJEZ-XRSSZCMZSA-N 0.000 claims description 3
- 229960003150 bupivacaine Drugs 0.000 claims description 3
- 229960005408 deslorelin Drugs 0.000 claims description 3
- 239000004310 lactic acid Substances 0.000 claims description 3
- 235000014655 lactic acid Nutrition 0.000 claims description 3
- 229920000747 poly(lactic acid) Polymers 0.000 claims description 3
- 150000003839 salts Chemical class 0.000 claims description 3
- DEQANNDTNATYII-OULOTJBUSA-N (4r,7s,10s,13r,16s,19r)-10-(4-aminobutyl)-19-[[(2r)-2-amino-3-phenylpropanoyl]amino]-16-benzyl-n-[(2r,3r)-1,3-dihydroxybutan-2-yl]-7-[(1r)-1-hydroxyethyl]-13-(1h-indol-3-ylmethyl)-6,9,12,15,18-pentaoxo-1,2-dithia-5,8,11,14,17-pentazacycloicosane-4-carboxa Chemical compound C([C@@H](N)C(=O)N[C@H]1CSSC[C@H](NC(=O)[C@H]([C@@H](C)O)NC(=O)[C@H](CCCCN)NC(=O)[C@@H](CC=2C3=CC=CC=C3NC=2)NC(=O)[C@H](CC=2C=CC=CC=2)NC1=O)C(=O)N[C@H](CO)[C@H](O)C)C1=CC=CC=C1 DEQANNDTNATYII-OULOTJBUSA-N 0.000 claims description 2
- SWXOGPJRIDTIRL-DOUNNPEJSA-N (4r,7s,10s,13r,16s,19r)-10-(4-aminobutyl)-n-[(2s)-1-amino-3-(1h-indol-3-yl)-1-oxopropan-2-yl]-19-[[(2r)-2-amino-3-phenylpropanoyl]amino]-16-[(4-hydroxyphenyl)methyl]-13-(1h-indol-3-ylmethyl)-6,9,12,15,18-pentaoxo-7-propan-2-yl-1,2-dithia-5,8,11,14,17-pent Chemical compound C([C@H]1C(=O)N[C@H](CC=2C3=CC=CC=C3NC=2)C(=O)N[C@@H](CCCCN)C(=O)N[C@H](C(N[C@@H](CSSC[C@@H](C(=O)N1)NC(=O)[C@H](N)CC=1C=CC=CC=1)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(N)=O)=O)C(C)C)C1=CC=C(O)C=C1 SWXOGPJRIDTIRL-DOUNNPEJSA-N 0.000 claims description 2
- PUDHBTGHUJUUFI-SCTWWAJVSA-N (4r,7s,10s,13r,16s,19r)-10-(4-aminobutyl)-n-[(2s,3r)-1-amino-3-hydroxy-1-oxobutan-2-yl]-19-[[(2r)-2-amino-3-naphthalen-2-ylpropanoyl]amino]-16-[(4-hydroxyphenyl)methyl]-13-(1h-indol-3-ylmethyl)-6,9,12,15,18-pentaoxo-7-propan-2-yl-1,2-dithia-5,8,11,14,17-p Chemical compound C([C@H]1C(=O)N[C@H](CC=2C3=CC=CC=C3NC=2)C(=O)N[C@@H](CCCCN)C(=O)N[C@H](C(N[C@@H](CSSC[C@@H](C(=O)N1)NC(=O)[C@H](N)CC=1C=C2C=CC=CC2=CC=1)C(=O)N[C@@H]([C@@H](C)O)C(N)=O)=O)C(C)C)C1=CC=C(O)C=C1 PUDHBTGHUJUUFI-SCTWWAJVSA-N 0.000 claims description 2
- ZKMNUMMKYBVTFN-HNNXBMFYSA-N (S)-ropivacaine Chemical compound CCCN1CCCC[C@H]1C(=O)NC1=C(C)C=CC=C1C ZKMNUMMKYBVTFN-HNNXBMFYSA-N 0.000 claims description 2
- IIZPXYDJLKNOIY-JXPKJXOSSA-N 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCC\C=C/C\C=C/C\C=C/C\C=C/CCCCC IIZPXYDJLKNOIY-JXPKJXOSSA-N 0.000 claims description 2
- VOXZDWNPVJITMN-ZBRFXRBCSA-N 17β-estradiol Chemical compound OC1=CC=C2[C@H]3CC[C@](C)([C@H](CC4)O)[C@@H]4[C@@H]3CCC2=C1 VOXZDWNPVJITMN-ZBRFXRBCSA-N 0.000 claims description 2
- PMXMIIMHBWHSKN-UHFFFAOYSA-N 3-{2-[4-(6-fluoro-1,2-benzoxazol-3-yl)piperidin-1-yl]ethyl}-9-hydroxy-2-methyl-6,7,8,9-tetrahydropyrido[1,2-a]pyrimidin-4-one Chemical compound FC1=CC=C2C(C3CCN(CC3)CCC=3C(=O)N4CCCC(O)C4=NC=3C)=NOC2=C1 PMXMIIMHBWHSKN-UHFFFAOYSA-N 0.000 claims description 2
- IJWPAFMIFNSIGD-UHFFFAOYSA-N 4-[3-(3-fluorophenyl)-5,5-dimethyl-4-oxofuran-2-yl]benzenesulfonamide Chemical compound O=C1C(C)(C)OC(C=2C=CC(=CC=2)S(N)(=O)=O)=C1C1=CC=CC(F)=C1 IJWPAFMIFNSIGD-UHFFFAOYSA-N 0.000 claims description 2
- CEUORZQYGODEFX-UHFFFAOYSA-N Aripirazole Chemical compound ClC1=CC=CC(N2CCN(CCCCOC=3C=C4NC(=O)CCC4=CC=3)CC2)=C1Cl CEUORZQYGODEFX-UHFFFAOYSA-N 0.000 claims description 2
- 108010037003 Buserelin Proteins 0.000 claims description 2
- 229920002134 Carboxymethyl cellulose Polymers 0.000 claims description 2
- DLGOEMSEDOSKAD-UHFFFAOYSA-N Carmustine Chemical compound ClCCNC(=O)N(N=O)CCCl DLGOEMSEDOSKAD-UHFFFAOYSA-N 0.000 claims description 2
- UHDGCWIWMRVCDJ-CCXZUQQUSA-N Cytarabine Chemical compound O=C1N=C(N)C=CN1[C@H]1[C@@H](O)[C@H](O)[C@@H](CO)O1 UHDGCWIWMRVCDJ-CCXZUQQUSA-N 0.000 claims description 2
- HTQBXNHDCUEHJF-XWLPCZSASA-N Exenatide Chemical compound C([C@@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(N)=O)C(=O)NCC(=O)NCC(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CO)C(=O)N[C@@H](CO)C(=O)NCC(=O)N[C@@H](C)C(=O)N1[C@@H](CCC1)C(=O)N1[C@@H](CCC1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CO)C(N)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@@H](NC(=O)[C@H](C)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCSC)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CO)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@@H](NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)CNC(=O)[C@@H](N)CC=1NC=NC=1)[C@@H](C)O)[C@@H](C)O)C(C)C)C1=CC=CC=C1 HTQBXNHDCUEHJF-XWLPCZSASA-N 0.000 claims description 2
- 108010011459 Exenatide Proteins 0.000 claims description 2
- 108010010803 Gelatin Proteins 0.000 claims description 2
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 claims description 2
- BLCLNMBMMGCOAS-URPVMXJPSA-N Goserelin Chemical compound C([C@@H](C(=O)N[C@H](COC(C)(C)C)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCN=C(N)N)C(=O)N1[C@@H](CCC1)C(=O)NNC(N)=O)NC(=O)[C@H](CO)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H](CC=1NC=NC=1)NC(=O)[C@H]1NC(=O)CC1)C1=CC=C(O)C=C1 BLCLNMBMMGCOAS-URPVMXJPSA-N 0.000 claims description 2
- 108010069236 Goserelin Proteins 0.000 claims description 2
- 108010000817 Leuprolide Proteins 0.000 claims description 2
- YSDQQAXHVYUZIW-QCIJIYAXSA-N Liraglutide Chemical compound C([C@@H](C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(O)=O)C(=O)NCC(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](C)C(=O)N[C@@H](C)C(=O)N[C@@H](CCCCNC(=O)CC[C@H](NC(=O)CCCCCCCCCCCCCCC)C(O)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](C)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)NCC(=O)N[C@@H](CCCNC(N)=N)C(=O)NCC(O)=O)NC(=O)[C@H](CO)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@@H](NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](C)NC(=O)[C@@H](N)CC=1NC=NC=1)[C@@H](C)O)[C@@H](C)O)C(C)C)C1=CC=C(O)C=C1 YSDQQAXHVYUZIW-QCIJIYAXSA-N 0.000 claims description 2
- 108010019598 Liraglutide Proteins 0.000 claims description 2
- XVVOERDUTLJJHN-UHFFFAOYSA-N Lixisenatide Chemical compound C=1NC2=CC=CC=C2C=1CC(C(=O)NC(CC(C)C)C(=O)NC(CCCCN)C(=O)NC(CC(N)=O)C(=O)NCC(=O)NCC(=O)N1C(CCC1)C(=O)NC(CO)C(=O)NC(CO)C(=O)NCC(=O)NC(C)C(=O)N1C(CCC1)C(=O)N1C(CCC1)C(=O)NC(CO)C(=O)NC(CCCCN)C(=O)NC(CCCCN)C(=O)NC(CCCCN)C(=O)NC(CCCCN)C(=O)NC(CCCCN)C(=O)NC(CCCCN)C(N)=O)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)CC)NC(=O)C(NC(=O)C(CC(C)C)NC(=O)C(CCCNC(N)=N)NC(=O)C(NC(=O)C(C)NC(=O)C(CCC(O)=O)NC(=O)C(CCC(O)=O)NC(=O)C(CCC(O)=O)NC(=O)C(CCSC)NC(=O)C(CCC(N)=O)NC(=O)C(CCCCN)NC(=O)C(CO)NC(=O)C(CC(C)C)NC(=O)C(CC(O)=O)NC(=O)C(CO)NC(=O)C(NC(=O)C(CC=1C=CC=CC=1)NC(=O)C(NC(=O)CNC(=O)C(CCC(O)=O)NC(=O)CNC(=O)C(N)CC=1NC=NC=1)C(C)O)C(C)O)C(C)C)CC1=CC=CC=C1 XVVOERDUTLJJHN-UHFFFAOYSA-N 0.000 claims description 2
- ZRVUJXDFFKFLMG-UHFFFAOYSA-N Meloxicam Chemical compound OC=1C2=CC=CC=C2S(=O)(=O)N(C)C=1C(=O)NC1=NC=C(C)S1 ZRVUJXDFFKFLMG-UHFFFAOYSA-N 0.000 claims description 2
- 108010021717 Nafarelin Proteins 0.000 claims description 2
- 108010016076 Octreotide Proteins 0.000 claims description 2
- 229920003171 Poly (ethylene oxide) Polymers 0.000 claims description 2
- 229920000954 Polyglycolide Polymers 0.000 claims description 2
- 229920001214 Polysorbate 60 Polymers 0.000 claims description 2
- DLSWIYLPEUIQAV-UHFFFAOYSA-N Semaglutide Chemical compound CCC(C)C(NC(=O)C(Cc1ccccc1)NC(=O)C(CCC(O)=O)NC(=O)C(CCCCNC(=O)COCCOCCNC(=O)COCCOCCNC(=O)CCC(NC(=O)CCCCCCCCCCCCCCCCC(O)=O)C(O)=O)NC(=O)C(C)NC(=O)C(C)NC(=O)C(CCC(N)=O)NC(=O)CNC(=O)C(CCC(O)=O)NC(=O)C(CC(C)C)NC(=O)C(Cc1ccc(O)cc1)NC(=O)C(CO)NC(=O)C(CO)NC(=O)C(NC(=O)C(CC(O)=O)NC(=O)C(CO)NC(=O)C(NC(=O)C(Cc1ccccc1)NC(=O)C(NC(=O)CNC(=O)C(CCC(O)=O)NC(=O)C(C)(C)NC(=O)C(N)Cc1cnc[nH]1)C(C)O)C(C)O)C(C)C)C(=O)NC(C)C(=O)NC(Cc1c[nH]c2ccccc12)C(=O)NC(CC(C)C)C(=O)NC(C(C)C)C(=O)NC(CCCNC(N)=N)C(=O)NCC(=O)NC(CCCNC(N)=N)C(=O)NCC(O)=O DLSWIYLPEUIQAV-UHFFFAOYSA-N 0.000 claims description 2
- DRHKJLXJIQTDTD-OAHLLOKOSA-N Tamsulosine Chemical compound CCOC1=CC=CC=C1OCCN[C@H](C)CC1=CC=C(OC)C(S(N)(=O)=O)=C1 DRHKJLXJIQTDTD-OAHLLOKOSA-N 0.000 claims description 2
- 108010050144 Triptorelin Pamoate Proteins 0.000 claims description 2
- 229960004372 aripiprazole Drugs 0.000 claims description 2
- RMRJXGBAOAMLHD-IHFGGWKQSA-N buprenorphine Chemical compound C([C@]12[C@H]3OC=4C(O)=CC=C(C2=4)C[C@@H]2[C@]11CC[C@]3([C@H](C1)[C@](C)(O)C(C)(C)C)OC)CN2CC1CC1 RMRJXGBAOAMLHD-IHFGGWKQSA-N 0.000 claims description 2
- 229960001736 buprenorphine Drugs 0.000 claims description 2
- CUWODFFVMXJOKD-UVLQAERKSA-N buserelin Chemical compound CCNC(=O)[C@@H]1CCCN1C(=O)[C@H](CCCN=C(N)N)NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](COC(C)(C)C)NC(=O)[C@@H](NC(=O)[C@H](CO)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H](CC=1NC=NC=1)NC(=O)[C@H]1NC(=O)CC1)CC1=CC=C(O)C=C1 CUWODFFVMXJOKD-UVLQAERKSA-N 0.000 claims description 2
- 229960002719 buserelin Drugs 0.000 claims description 2
- 239000001768 carboxy methyl cellulose Substances 0.000 claims description 2
- 235000010948 carboxy methyl cellulose Nutrition 0.000 claims description 2
- 239000008112 carboxymethyl-cellulose Substances 0.000 claims description 2
- 229960005243 carmustine Drugs 0.000 claims description 2
- 239000004359 castor oil Substances 0.000 claims description 2
- 235000019438 castor oil Nutrition 0.000 claims description 2
- 229960000684 cytarabine Drugs 0.000 claims description 2
- 235000014113 dietary fatty acids Nutrition 0.000 claims description 2
- 229960000980 entecavir Drugs 0.000 claims description 2
- YXPVEXCTPGULBZ-WQYNNSOESA-N entecavir hydrate Chemical compound O.C1=NC=2C(=O)NC(N)=NC=2N1[C@H]1C[C@H](O)[C@@H](CO)C1=C YXPVEXCTPGULBZ-WQYNNSOESA-N 0.000 claims description 2
- 229960005309 estradiol Drugs 0.000 claims description 2
- 229930182833 estradiol Natural products 0.000 claims description 2
- 229960001519 exenatide Drugs 0.000 claims description 2
- 229930195729 fatty acid Natural products 0.000 claims description 2
- 239000000194 fatty acid Substances 0.000 claims description 2
- 229960002428 fentanyl Drugs 0.000 claims description 2
- 229920000159 gelatin Polymers 0.000 claims description 2
- 239000008273 gelatin Substances 0.000 claims description 2
- 235000019322 gelatine Nutrition 0.000 claims description 2
- 235000011852 gelatine desserts Nutrition 0.000 claims description 2
- 239000008103 glucose Substances 0.000 claims description 2
- ZEMPKEQAKRGZGQ-XOQCFJPHSA-N glycerol triricinoleate Natural products CCCCCC[C@@H](O)CC=CCCCCCCCC(=O)OC[C@@H](COC(=O)CCCCCCCC=CC[C@@H](O)CCCCCC)OC(=O)CCCCCCCC=CC[C@H](O)CCCCCC ZEMPKEQAKRGZGQ-XOQCFJPHSA-N 0.000 claims description 2
- 229960002913 goserelin Drugs 0.000 claims description 2
- 229960003727 granisetron Drugs 0.000 claims description 2
- MFWNKCLOYSRHCJ-BTTYYORXSA-N granisetron Chemical compound C1=CC=C2C(C(=O)N[C@H]3C[C@H]4CCC[C@@H](C3)N4C)=NN(C)C2=C1 MFWNKCLOYSRHCJ-BTTYYORXSA-N 0.000 claims description 2
- JTEGQNOMFQHVDC-NKWVEPMBSA-N lamivudine Chemical compound O=C1N=C(N)C=CN1[C@H]1O[C@@H](CO)SC1 JTEGQNOMFQHVDC-NKWVEPMBSA-N 0.000 claims description 2
- 229960001627 lamivudine Drugs 0.000 claims description 2
- 229960002437 lanreotide Drugs 0.000 claims description 2
- 108010021336 lanreotide Proteins 0.000 claims description 2
- 239000000787 lecithin Substances 0.000 claims description 2
- 235000010445 lecithin Nutrition 0.000 claims description 2
- 229940067606 lecithin Drugs 0.000 claims description 2
- GFIJNRVAKGFPGQ-LIJARHBVSA-N leuprolide Chemical compound CCNC(=O)[C@@H]1CCCN1C(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](CC(C)C)NC(=O)[C@@H](NC(=O)[C@H](CO)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H](CC=1N=CNC=1)NC(=O)[C@H]1NC(=O)CC1)CC1=CC=C(O)C=C1 GFIJNRVAKGFPGQ-LIJARHBVSA-N 0.000 claims description 2
- 229960004338 leuprorelin Drugs 0.000 claims description 2
- 229960002701 liraglutide Drugs 0.000 claims description 2
- 108010004367 lixisenatide Proteins 0.000 claims description 2
- 229960001093 lixisenatide Drugs 0.000 claims description 2
- 229960001929 meloxicam Drugs 0.000 claims description 2
- 229960004640 memantine Drugs 0.000 claims description 2
- BUGYDGFZZOZRHP-UHFFFAOYSA-N memantine Chemical compound C1C(C2)CC3(C)CC1(C)CC2(N)C3 BUGYDGFZZOZRHP-UHFFFAOYSA-N 0.000 claims description 2
- 229920000609 methyl cellulose Polymers 0.000 claims description 2
- 239000001923 methylcellulose Substances 0.000 claims description 2
- 235000010981 methylcellulose Nutrition 0.000 claims description 2
- RWHUEXWOYVBUCI-ITQXDASVSA-N nafarelin Chemical compound C([C@@H](C(=O)N[C@H](CC=1C=C2C=CC=CC2=CC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCN=C(N)N)C(=O)N1[C@@H](CCC1)C(=O)NCC(N)=O)NC(=O)[C@H](CO)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H](CC=1NC=NC=1)NC(=O)[C@H]1NC(=O)CC1)C1=CC=C(O)C=C1 RWHUEXWOYVBUCI-ITQXDASVSA-N 0.000 claims description 2
- 229960002333 nafarelin Drugs 0.000 claims description 2
- 229960002700 octreotide Drugs 0.000 claims description 2
- 229960005017 olanzapine Drugs 0.000 claims description 2
- KVWDHTXUZHCGIO-UHFFFAOYSA-N olanzapine Chemical compound C1CN(C)CCN1C1=NC2=CC=CC=C2NC2=C1C=C(C)S2 KVWDHTXUZHCGIO-UHFFFAOYSA-N 0.000 claims description 2
- 229960001057 paliperidone Drugs 0.000 claims description 2
- VMZMNAABQBOLAK-DBILLSOUSA-N pasireotide Chemical compound C([C@H]1C(=O)N2C[C@@H](C[C@H]2C(=O)N[C@H](C(=O)N[C@H](CC=2C3=CC=CC=C3NC=2)C(=O)N[C@H](C(N[C@@H](CC=2C=CC(OCC=3C=CC=CC=3)=CC=2)C(=O)N1)=O)CCCCN)C=1C=CC=CC=1)OC(=O)NCCN)C1=CC=CC=C1 VMZMNAABQBOLAK-DBILLSOUSA-N 0.000 claims description 2
- 229960005415 pasireotide Drugs 0.000 claims description 2
- 108700017947 pasireotide Proteins 0.000 claims description 2
- 229950006009 polmacoxib Drugs 0.000 claims description 2
- 235000019422 polyvinyl alcohol Nutrition 0.000 claims description 2
- 239000001267 polyvinylpyrrolidone Substances 0.000 claims description 2
- 229920000036 polyvinylpyrrolidone Polymers 0.000 claims description 2
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 claims description 2
- RAPZEAPATHNIPO-UHFFFAOYSA-N risperidone Chemical compound FC1=CC=C2C(C3CCN(CC3)CCC=3C(=O)N4CCCCC4=NC=3C)=NOC2=C1 RAPZEAPATHNIPO-UHFFFAOYSA-N 0.000 claims description 2
- 229960001534 risperidone Drugs 0.000 claims description 2
- UHSKFQJFRQCDBE-UHFFFAOYSA-N ropinirole Chemical compound CCCN(CCC)CCC1=CC=CC2=C1CC(=O)N2 UHSKFQJFRQCDBE-UHFFFAOYSA-N 0.000 claims description 2
- 229960001879 ropinirole Drugs 0.000 claims description 2
- 229960001549 ropivacaine Drugs 0.000 claims description 2
- KFQYTPMOWPVWEJ-INIZCTEOSA-N rotigotine Chemical compound CCCN([C@@H]1CC2=CC=CC(O)=C2CC1)CCC1=CC=CS1 KFQYTPMOWPVWEJ-INIZCTEOSA-N 0.000 claims description 2
- 229960003179 rotigotine Drugs 0.000 claims description 2
- 229950011186 semaglutide Drugs 0.000 claims description 2
- 108010060325 semaglutide Proteins 0.000 claims description 2
- 229960002613 tamsulosin Drugs 0.000 claims description 2
- 229960003604 testosterone Drugs 0.000 claims description 2
- GFNANZIMVAIWHM-OBYCQNJPSA-N triamcinolone Chemical compound O=C1C=C[C@]2(C)[C@@]3(F)[C@@H](O)C[C@](C)([C@@]([C@H](O)C4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 GFNANZIMVAIWHM-OBYCQNJPSA-N 0.000 claims description 2
- 229960005294 triamcinolone Drugs 0.000 claims description 2
- VXKHXGOKWPXYNA-PGBVPBMZSA-N triptorelin Chemical compound C([C@@H](C(=O)N[C@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N1[C@@H](CCC1)C(=O)NCC(N)=O)NC(=O)[C@H](CO)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H](CC=1N=CNC=1)NC(=O)[C@H]1NC(=O)CC1)C1=CC=C(O)C=C1 VXKHXGOKWPXYNA-PGBVPBMZSA-N 0.000 claims description 2
- 229960004824 triptorelin Drugs 0.000 claims description 2
- 229960002730 vapreotide Drugs 0.000 claims description 2
- 108700029852 vapreotide Proteins 0.000 claims description 2
- PJMPHNIQZUBGLI-UHFFFAOYSA-N fentanyl Chemical compound C=1C=CC=CC=1N(C(=O)CC)C(CC1)CCN1CCC1=CC=CC=C1 PJMPHNIQZUBGLI-UHFFFAOYSA-N 0.000 claims 1
- 239000012071 phase Substances 0.000 description 375
- 239000000725 suspension Substances 0.000 description 92
- 238000002360 preparation method Methods 0.000 description 83
- 238000003756 stirring Methods 0.000 description 47
- 239000012528 membrane Substances 0.000 description 36
- 230000001804 emulsifying effect Effects 0.000 description 35
- 239000008367 deionised water Substances 0.000 description 27
- 229910021641 deionized water Inorganic materials 0.000 description 27
- 229920000249 biocompatible polymer Polymers 0.000 description 25
- 230000000052 comparative effect Effects 0.000 description 22
- 238000004519 manufacturing process Methods 0.000 description 20
- 238000002347 injection Methods 0.000 description 19
- 239000007924 injection Substances 0.000 description 19
- 239000011148 porous material Substances 0.000 description 17
- 239000013557 residual solvent Substances 0.000 description 15
- 239000002245 particle Substances 0.000 description 13
- 229940124597 therapeutic agent Drugs 0.000 description 12
- VZSRBBMJRBPUNF-UHFFFAOYSA-N 2-(2,3-dihydro-1H-inden-2-ylamino)-N-[3-oxo-3-(2,4,6,7-tetrahydrotriazolo[4,5-c]pyridin-5-yl)propyl]pyrimidine-5-carboxamide Chemical compound C1C(CC2=CC=CC=C12)NC1=NC=C(C=N1)C(=O)NCCC(N1CC2=C(CC1)NN=N2)=O VZSRBBMJRBPUNF-UHFFFAOYSA-N 0.000 description 10
- 230000000877 morphologic effect Effects 0.000 description 9
- 102100022258 Disks large homolog 5 Human genes 0.000 description 8
- 101100063489 Homo sapiens DLG5 gene Proteins 0.000 description 8
- 238000004458 analytical method Methods 0.000 description 8
- 229920000642 polymer Polymers 0.000 description 7
- 238000005406 washing Methods 0.000 description 7
- 239000013543 active substance Substances 0.000 description 6
- 238000001914 filtration Methods 0.000 description 6
- 238000012512 characterization method Methods 0.000 description 5
- 238000004090 dissolution Methods 0.000 description 5
- 230000008569 process Effects 0.000 description 5
- 239000001961 anticonvulsive agent Substances 0.000 description 4
- 239000011877 solvent mixture Substances 0.000 description 4
- 231100000331 toxic Toxicity 0.000 description 4
- 230000002588 toxic effect Effects 0.000 description 4
- 230000000202 analgesic effect Effects 0.000 description 3
- 238000000605 extraction Methods 0.000 description 3
- 239000012467 final product Substances 0.000 description 3
- 230000007062 hydrolysis Effects 0.000 description 3
- 238000006460 hydrolysis reaction Methods 0.000 description 3
- 238000000638 solvent extraction Methods 0.000 description 3
- 238000012360 testing method Methods 0.000 description 3
- KZEVSDGEBAJOTK-UHFFFAOYSA-N 1-(2,4,6,7-tetrahydrotriazolo[4,5-c]pyridin-5-yl)-2-[5-[2-[[3-(trifluoromethoxy)phenyl]methylamino]pyrimidin-5-yl]-1,3,4-oxadiazol-2-yl]ethanone Chemical compound N1N=NC=2CN(CCC=21)C(CC=1OC(=NN=1)C=1C=NC(=NC=1)NCC1=CC(=CC=C1)OC(F)(F)F)=O KZEVSDGEBAJOTK-UHFFFAOYSA-N 0.000 description 2
- SXAMGRAIZSSWIH-UHFFFAOYSA-N 2-[3-[2-(2,3-dihydro-1H-inden-2-ylamino)pyrimidin-5-yl]-1,2,4-oxadiazol-5-yl]-1-(2,4,6,7-tetrahydrotriazolo[4,5-c]pyridin-5-yl)ethanone Chemical compound C1C(CC2=CC=CC=C12)NC1=NC=C(C=N1)C1=NOC(=N1)CC(=O)N1CC2=C(CC1)NN=N2 SXAMGRAIZSSWIH-UHFFFAOYSA-N 0.000 description 2
- YJLUBHOZZTYQIP-UHFFFAOYSA-N 2-[5-[2-(2,3-dihydro-1H-inden-2-ylamino)pyrimidin-5-yl]-1,3,4-oxadiazol-2-yl]-1-(2,4,6,7-tetrahydrotriazolo[4,5-c]pyridin-5-yl)ethanone Chemical compound C1C(CC2=CC=CC=C12)NC1=NC=C(C=N1)C1=NN=C(O1)CC(=O)N1CC2=C(CC1)NN=N2 YJLUBHOZZTYQIP-UHFFFAOYSA-N 0.000 description 2
- 206010012289 Dementia Diseases 0.000 description 2
- 208000010228 Erectile Dysfunction Diseases 0.000 description 2
- SECXISVLQFMRJM-UHFFFAOYSA-N N-Methylpyrrolidone Chemical compound CN1CCCC1=O SECXISVLQFMRJM-UHFFFAOYSA-N 0.000 description 2
- 208000001132 Osteoporosis Diseases 0.000 description 2
- 208000018737 Parkinson disease Diseases 0.000 description 2
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 2
- 229910052782 aluminium Inorganic materials 0.000 description 2
- 239000003242 anti bacterial agent Substances 0.000 description 2
- 230000001754 anti-pyretic effect Effects 0.000 description 2
- 230000002365 anti-tubercular Effects 0.000 description 2
- 229940088710 antibiotic agent Drugs 0.000 description 2
- 239000003146 anticoagulant agent Substances 0.000 description 2
- 239000003472 antidiabetic agent Substances 0.000 description 2
- 229940121375 antifungal agent Drugs 0.000 description 2
- 239000003429 antifungal agent Substances 0.000 description 2
- 239000002246 antineoplastic agent Substances 0.000 description 2
- 239000002221 antipyretic Substances 0.000 description 2
- 229960004676 antithrombotic agent Drugs 0.000 description 2
- 239000003443 antiviral agent Substances 0.000 description 2
- 239000008346 aqueous phase Substances 0.000 description 2
- 230000008859 change Effects 0.000 description 2
- 239000003795 chemical substances by application Substances 0.000 description 2
- 150000001875 compounds Chemical class 0.000 description 2
- 238000001035 drying Methods 0.000 description 2
- 238000004945 emulsification Methods 0.000 description 2
- 230000008020 evaporation Effects 0.000 description 2
- 230000002496 gastric effect Effects 0.000 description 2
- 201000001881 impotence Diseases 0.000 description 2
- 239000003158 myorelaxant agent Substances 0.000 description 2
- BASFCYQUMIYNBI-UHFFFAOYSA-N platinum Chemical compound [Pt] BASFCYQUMIYNBI-UHFFFAOYSA-N 0.000 description 2
- 239000000047 product Substances 0.000 description 2
- 108090000623 proteins and genes Proteins 0.000 description 2
- 230000002829 reductive effect Effects 0.000 description 2
- OHVLMTFVQDZYHP-UHFFFAOYSA-N 1-(2,4,6,7-tetrahydrotriazolo[4,5-c]pyridin-5-yl)-2-[4-[2-[[3-(trifluoromethoxy)phenyl]methylamino]pyrimidin-5-yl]piperazin-1-yl]ethanone Chemical compound N1N=NC=2CN(CCC=21)C(CN1CCN(CC1)C=1C=NC(=NC=1)NCC1=CC(=CC=C1)OC(F)(F)F)=O OHVLMTFVQDZYHP-UHFFFAOYSA-N 0.000 description 1
- WZFUQSJFWNHZHM-UHFFFAOYSA-N 2-[4-[2-(2,3-dihydro-1H-inden-2-ylamino)pyrimidin-5-yl]piperazin-1-yl]-1-(2,4,6,7-tetrahydrotriazolo[4,5-c]pyridin-5-yl)ethanone Chemical compound C1C(CC2=CC=CC=C12)NC1=NC=C(C=N1)N1CCN(CC1)CC(=O)N1CC2=C(CC1)NN=N2 WZFUQSJFWNHZHM-UHFFFAOYSA-N 0.000 description 1
- IHCCLXNEEPMSIO-UHFFFAOYSA-N 2-[4-[2-(2,3-dihydro-1H-inden-2-ylamino)pyrimidin-5-yl]piperidin-1-yl]-1-(2,4,6,7-tetrahydrotriazolo[4,5-c]pyridin-5-yl)ethanone Chemical compound C1C(CC2=CC=CC=C12)NC1=NC=C(C=N1)C1CCN(CC1)CC(=O)N1CC2=C(CC1)NN=N2 IHCCLXNEEPMSIO-UHFFFAOYSA-N 0.000 description 1
- XBBVURRQGJPTHH-UHFFFAOYSA-N 2-hydroxyacetic acid;2-hydroxypropanoic acid Chemical compound OCC(O)=O.CC(O)C(O)=O XBBVURRQGJPTHH-UHFFFAOYSA-N 0.000 description 1
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 1
- 208000031226 Hyperlipidaemia Diseases 0.000 description 1
- 206010020772 Hypertension Diseases 0.000 description 1
- 208000001953 Hypotension Diseases 0.000 description 1
- AFCARXCZXQIEQB-UHFFFAOYSA-N N-[3-oxo-3-(2,4,6,7-tetrahydrotriazolo[4,5-c]pyridin-5-yl)propyl]-2-[[3-(trifluoromethoxy)phenyl]methylamino]pyrimidine-5-carboxamide Chemical compound O=C(CCNC(=O)C=1C=NC(=NC=1)NCC1=CC(=CC=C1)OC(F)(F)F)N1CC2=C(CC1)NN=N2 AFCARXCZXQIEQB-UHFFFAOYSA-N 0.000 description 1
- 229920001244 Poly(D,L-lactide) Polymers 0.000 description 1
- 208000025747 Rheumatic disease Diseases 0.000 description 1
- LYCYLGFSIXIXAB-NUZRHMIVSA-N acetic acid;(2s)-n-[(2s)-1-[[(2s)-1-[[(2s)-1-[[(2s)-1-[[(2r)-1-[[(2s)-1-[[(2s)-5-(diaminomethylideneamino)-1-[(2s)-2-(ethylcarbamoyl)pyrrolidin-1-yl]-1-oxopentan-2-yl]amino]-4-methyl-1-oxopentan-2-yl]amino]-3-(1h-indol-3-yl)-1-oxopropan-2-yl]amino]-3-(4-h Chemical compound CC(O)=O.CCNC(=O)[C@@H]1CCCN1C(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CO)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H](CC=1N=CNC=1)NC(=O)[C@H]1NC(=O)CC1)CC1=CNC2=CC=CC=C12 LYCYLGFSIXIXAB-NUZRHMIVSA-N 0.000 description 1
- 230000002411 adverse Effects 0.000 description 1
- 150000001335 aliphatic alkanes Chemical class 0.000 description 1
- 230000003444 anaesthetic effect Effects 0.000 description 1
- 229940035676 analgesics Drugs 0.000 description 1
- 239000000730 antalgic agent Substances 0.000 description 1
- 230000000567 anti-anemic effect Effects 0.000 description 1
- 230000001088 anti-asthma Effects 0.000 description 1
- 230000003178 anti-diabetic effect Effects 0.000 description 1
- 230000003474 anti-emetic effect Effects 0.000 description 1
- 230000003556 anti-epileptic effect Effects 0.000 description 1
- 229940121363 anti-inflammatory agent Drugs 0.000 description 1
- 239000002260 anti-inflammatory agent Substances 0.000 description 1
- 229940124599 anti-inflammatory drug Drugs 0.000 description 1
- 229940035678 anti-parkinson drug Drugs 0.000 description 1
- 230000000561 anti-psychotic effect Effects 0.000 description 1
- 230000002921 anti-spasmodic effect Effects 0.000 description 1
- 229940124344 antianaemic agent Drugs 0.000 description 1
- 239000003416 antiarrhythmic agent Substances 0.000 description 1
- 239000000924 antiasthmatic agent Substances 0.000 description 1
- 229940125681 anticonvulsant agent Drugs 0.000 description 1
- 239000000935 antidepressant agent Substances 0.000 description 1
- 229940005513 antidepressants Drugs 0.000 description 1
- 229940125708 antidiabetic agent Drugs 0.000 description 1
- 239000000729 antidote Substances 0.000 description 1
- 229940125683 antiemetic agent Drugs 0.000 description 1
- 239000002111 antiemetic agent Substances 0.000 description 1
- 229960003965 antiepileptics Drugs 0.000 description 1
- 229940030225 antihemorrhagics Drugs 0.000 description 1
- 229940125715 antihistaminic agent Drugs 0.000 description 1
- 239000000739 antihistaminic agent Substances 0.000 description 1
- 239000002220 antihypertensive agent Substances 0.000 description 1
- 229940030600 antihypertensive agent Drugs 0.000 description 1
- 239000003524 antilipemic agent Substances 0.000 description 1
- 229940125684 antimigraine agent Drugs 0.000 description 1
- 239000002282 antimigraine agent Substances 0.000 description 1
- 229940127218 antiplatelet drug Drugs 0.000 description 1
- 239000003904 antiprotozoal agent Substances 0.000 description 1
- 239000000164 antipsychotic agent Substances 0.000 description 1
- 229940005529 antipsychotics Drugs 0.000 description 1
- 239000003435 antirheumatic agent Substances 0.000 description 1
- 229940124575 antispasmodic agent Drugs 0.000 description 1
- 239000003699 antiulcer agent Substances 0.000 description 1
- 239000002249 anxiolytic agent Substances 0.000 description 1
- 230000000949 anxiolytic effect Effects 0.000 description 1
- 229940005530 anxiolytics Drugs 0.000 description 1
- 230000006793 arrhythmia Effects 0.000 description 1
- 206010003119 arrhythmia Diseases 0.000 description 1
- 230000008901 benefit Effects 0.000 description 1
- 229960002537 betamethasone Drugs 0.000 description 1
- UREBDLICKHMUKA-DVTGEIKXSA-N betamethasone Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1C[C@H](C)[C@@](C(=O)CO)(O)[C@@]1(C)C[C@@H]2O UREBDLICKHMUKA-DVTGEIKXSA-N 0.000 description 1
- 238000009835 boiling Methods 0.000 description 1
- 229940124630 bronchodilator Drugs 0.000 description 1
- 230000000711 cancerogenic effect Effects 0.000 description 1
- 229910052799 carbon Inorganic materials 0.000 description 1
- 125000004432 carbon atom Chemical group C* 0.000 description 1
- 239000000496 cardiotonic agent Substances 0.000 description 1
- 230000003177 cardiotonic effect Effects 0.000 description 1
- 238000005119 centrifugation Methods 0.000 description 1
- 239000000812 cholinergic antagonist Substances 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- 229920001577 copolymer Polymers 0.000 description 1
- 239000003246 corticosteroid Substances 0.000 description 1
- 229960001334 corticosteroids Drugs 0.000 description 1
- 229940091137 cough suppressants and expectorants Drugs 0.000 description 1
- 238000000354 decomposition reaction Methods 0.000 description 1
- 230000003247 decreasing effect Effects 0.000 description 1
- 230000003111 delayed effect Effects 0.000 description 1
- 229960004165 deslorelin acetate Drugs 0.000 description 1
- 230000006866 deterioration Effects 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- 239000006185 dispersion Substances 0.000 description 1
- 239000002934 diuretic Substances 0.000 description 1
- 229940030606 diuretics Drugs 0.000 description 1
- 230000002526 effect on cardiovascular system Effects 0.000 description 1
- 230000000694 effects Effects 0.000 description 1
- 238000002474 experimental method Methods 0.000 description 1
- 230000001815 facial effect Effects 0.000 description 1
- IVLVTNPOHDFFCJ-UHFFFAOYSA-N fentanyl citrate Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O.C=1C=CC=CC=1N(C(=O)CC)C(CC1)CCN1CCC1=CC=CC=C1 IVLVTNPOHDFFCJ-UHFFFAOYSA-N 0.000 description 1
- 239000000945 filler Substances 0.000 description 1
- 238000004108 freeze drying Methods 0.000 description 1
- 239000003163 gonadal steroid hormone Substances 0.000 description 1
- 150000008282 halocarbons Chemical class 0.000 description 1
- 239000002874 hemostatic agent Substances 0.000 description 1
- 229940125697 hormonal agent Drugs 0.000 description 1
- 229940088597 hormone Drugs 0.000 description 1
- 239000005556 hormone Substances 0.000 description 1
- 229920001477 hydrophilic polymer Polymers 0.000 description 1
- 230000002209 hydrophobic effect Effects 0.000 description 1
- 230000036543 hypotension Effects 0.000 description 1
- 239000002955 immunomodulating agent Substances 0.000 description 1
- 229940121354 immunomodulator Drugs 0.000 description 1
- 230000000670 limiting effect Effects 0.000 description 1
- 239000007788 liquid Substances 0.000 description 1
- 239000007791 liquid phase Substances 0.000 description 1
- 238000002483 medication Methods 0.000 description 1
- 230000004660 morphological change Effects 0.000 description 1
- 229940035363 muscle relaxants Drugs 0.000 description 1
- 210000005036 nerve Anatomy 0.000 description 1
- 239000007764 o/w emulsion Substances 0.000 description 1
- 239000003791 organic solvent mixture Substances 0.000 description 1
- 230000002572 peristaltic effect Effects 0.000 description 1
- WVDDGKGOMKODPV-ZQBYOMGUSA-N phenyl(114C)methanol Chemical compound O[14CH2]C1=CC=CC=C1 WVDDGKGOMKODPV-ZQBYOMGUSA-N 0.000 description 1
- 239000000106 platelet aggregation inhibitor Substances 0.000 description 1
- 229910052697 platinum Inorganic materials 0.000 description 1
- 229920000728 polyester Polymers 0.000 description 1
- 238000003825 pressing Methods 0.000 description 1
- 230000001737 promoting effect Effects 0.000 description 1
- 102000004169 proteins and genes Human genes 0.000 description 1
- 239000004089 psychotropic agent Substances 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 230000000717 retained effect Effects 0.000 description 1
- 238000004626 scanning electron microscopy Methods 0.000 description 1
- 239000003381 stabilizer Substances 0.000 description 1
- 239000000126 substance Substances 0.000 description 1
- 238000006467 substitution reaction Methods 0.000 description 1
- 230000002459 sustained effect Effects 0.000 description 1
- 238000013268 sustained release Methods 0.000 description 1
- 239000012730 sustained-release form Substances 0.000 description 1
- 231100000419 toxicity Toxicity 0.000 description 1
- 230000001988 toxicity Effects 0.000 description 1
- 229940124549 vasodilator Drugs 0.000 description 1
- 239000003071 vasodilator agent Substances 0.000 description 1
- 239000013585 weight reducing agent Substances 0.000 description 1
- 230000037303 wrinkles Effects 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
- A61K9/16—Agglomerates; Granulates; Microbeadlets ; Microspheres; Pellets; Solid products obtained by spray drying, spray freeze drying, spray congealing,(multiple) emulsion solvent evaporation or extraction
- A61K9/1682—Processes
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J13/00—Colloid chemistry, e.g. the production of colloidal materials or their solutions, not otherwise provided for; Making microcapsules or microballoons
- B01J13/02—Making microcapsules or microballoons
- B01J13/06—Making microcapsules or microballoons by phase separation
- B01J13/12—Making microcapsules or microballoons by phase separation removing solvent from the wall-forming material solution
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/445—Non condensed piperidines, e.g. piperocaine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/21—Esters, e.g. nitroglycerine, selenocyanates
- A61K31/27—Esters, e.g. nitroglycerine, selenocyanates of carbamic or thiocarbamic acids, meprobamate, carbachol, neostigmine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/04—Peptides having up to 20 amino acids in a fully defined sequence; Derivatives thereof
- A61K38/08—Peptides having 5 to 11 amino acids
- A61K38/09—Luteinising hormone-releasing hormone [LHRH], i.e. Gonadotropin-releasing hormone [GnRH]; Related peptides
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
- A61K9/16—Agglomerates; Granulates; Microbeadlets ; Microspheres; Pellets; Solid products obtained by spray drying, spray freeze drying, spray congealing,(multiple) emulsion solvent evaporation or extraction
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
- A61K9/16—Agglomerates; Granulates; Microbeadlets ; Microspheres; Pellets; Solid products obtained by spray drying, spray freeze drying, spray congealing,(multiple) emulsion solvent evaporation or extraction
- A61K9/1605—Excipients; Inactive ingredients
- A61K9/1629—Organic macromolecular compounds
- A61K9/1635—Organic macromolecular compounds obtained by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyvinyl pyrrolidone, poly(meth)acrylates
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
- A61K9/16—Agglomerates; Granulates; Microbeadlets ; Microspheres; Pellets; Solid products obtained by spray drying, spray freeze drying, spray congealing,(multiple) emulsion solvent evaporation or extraction
- A61K9/1605—Excipients; Inactive ingredients
- A61K9/1629—Organic macromolecular compounds
- A61K9/1641—Organic macromolecular compounds obtained otherwise than by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyethylene glycol, poloxamers
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
- A61K9/16—Agglomerates; Granulates; Microbeadlets ; Microspheres; Pellets; Solid products obtained by spray drying, spray freeze drying, spray congealing,(multiple) emulsion solvent evaporation or extraction
- A61K9/1605—Excipients; Inactive ingredients
- A61K9/1629—Organic macromolecular compounds
- A61K9/1641—Organic macromolecular compounds obtained otherwise than by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyethylene glycol, poloxamers
- A61K9/1647—Polyesters, e.g. poly(lactide-co-glycolide)
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
- A61K9/16—Agglomerates; Granulates; Microbeadlets ; Microspheres; Pellets; Solid products obtained by spray drying, spray freeze drying, spray congealing,(multiple) emulsion solvent evaporation or extraction
- A61K9/1605—Excipients; Inactive ingredients
- A61K9/1629—Organic macromolecular compounds
- A61K9/1652—Polysaccharides, e.g. alginate, cellulose derivatives; Cyclodextrin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
- A61K9/16—Agglomerates; Granulates; Microbeadlets ; Microspheres; Pellets; Solid products obtained by spray drying, spray freeze drying, spray congealing,(multiple) emulsion solvent evaporation or extraction
- A61K9/1682—Processes
- A61K9/1694—Processes resulting in granules or microspheres of the matrix type containing more than 5% of excipient
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
- A61K9/19—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles lyophilised, i.e. freeze-dried, solutions or dispersions
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G63/00—Macromolecular compounds obtained by reactions forming a carboxylic ester link in the main chain of the macromolecule
- C08G63/02—Polyesters derived from hydroxycarboxylic acids or from polycarboxylic acids and polyhydroxy compounds
- C08G63/06—Polyesters derived from hydroxycarboxylic acids or from polycarboxylic acids and polyhydroxy compounds derived from hydroxycarboxylic acids
- C08G63/08—Lactones or lactides
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G63/00—Macromolecular compounds obtained by reactions forming a carboxylic ester link in the main chain of the macromolecule
- C08G63/88—Post-polymerisation treatment
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G63/00—Macromolecular compounds obtained by reactions forming a carboxylic ester link in the main chain of the macromolecule
- C08G63/88—Post-polymerisation treatment
- C08G63/89—Recovery of the polymer
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J3/00—Processes of treating or compounding macromolecular substances
- C08J3/12—Powdering or granulating
- C08J3/16—Powdering or granulating by coagulating dispersions
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L67/00—Compositions of polyesters obtained by reactions forming a carboxylic ester link in the main chain; Compositions of derivatives of such polymers
- C08L67/04—Polyesters derived from hydroxycarboxylic acids, e.g. lactones
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L71/00—Compositions of polyethers obtained by reactions forming an ether link in the main chain; Compositions of derivatives of such polymers
- C08L71/02—Polyalkylene oxides
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2367/00—Characterised by the use of polyesters obtained by reactions forming a carboxylic ester link in the main chain; Derivatives of such polymers
- C08J2367/04—Polyesters derived from hydroxy carboxylic acids, e.g. lactones
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K5/00—Use of organic ingredients
- C08K5/16—Nitrogen-containing compounds
- C08K5/34—Heterocyclic compounds having nitrogen in the ring
- C08K5/3412—Heterocyclic compounds having nitrogen in the ring having one nitrogen atom in the ring
- C08K5/3432—Six-membered rings
- C08K5/3435—Piperidines
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L2201/00—Properties
- C08L2201/06—Biodegradable
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L2203/00—Applications
- C08L2203/02—Applications for biomedical use
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Life Sciences & Earth Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Epidemiology (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Organic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Polymers & Plastics (AREA)
- Dispersion Chemistry (AREA)
- Immunology (AREA)
- Reproductive Health (AREA)
- Gastroenterology & Hepatology (AREA)
- Endocrinology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Emergency Medicine (AREA)
- Medicinal Preparation (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Manufacturing Of Micro-Capsules (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Cosmetics (AREA)
Abstract
Un método para preparar microesferas biodegradables, que comprende: (a) formar una disolución de polímero biodegradable disolviendo un polímero biodegradable solo; o un polímero biodegradable y un fármaco en un disolvente orgánico; (b) mezclar uniformemente la disolución de polímero biodegradable preparada en la etapa (a) con una disolución acuosa que contiene un tensioactivo, para formar una emulsión que contiene la disolución de polímero biodegradable como fase dispersa y una disolución acuosa que contiene el tensioactivo como una fase continua; (c) extraer y evaporar el disolvente orgánico de la fase dispersa en la emulsión de la etapa (b) a la fase continua para producir microesferas, en el que se elimina una parte de la fase continua que contiene el disolvente orgánico extraído, mientras que se suministra una nueva fase continua; y (d) recuperar las microesferas de la fase continua que contiene las microesferas producidas de la etapa (c), en el que la parte de la fase continua en la etapa (c) se elimina de forma discontinua o continua, y (i) cuando la parte de la fase continua en la etapa (c) se elimina de forma discontinua, la fase continua eliminada es una relación de volumen de 40% (v/v) a 99% (v/v), basada en el volumen total de la fase continua, o es una relación en peso de una fase continua restante, excepto la relación en peso puntual de la fase continua de la micropartícula en la etapa (c), o (ii) cuando se elimina continuamente la parte de la fase continua en la etapa (c), la tasa de eliminación de la fase continua está en el intervalo de 2% (v/v) a 200% (v/v)/min, basado en el volumen total de la fase continua.
Description
DESCRIPCIÓN
Procedimiento para preparar microesferas biodegradables con estabilidad mejorada y estabilidad al almacenamiento
CAMPO TÉCNICO
[Referencia cruzada a solicitud(es) relacionada(s)]
Esta solicitud reivindica el beneficio de prioridad basado en la Solicitud de Patente Coreana No. 10-2017-0163105, con fecha de 30 de noviembre, 2017.
La presente invención se refiere a un método para producir microesferas biodegradables, y más particularmente, a un método para producir microesferas biodegradables que tienen alta seguridad al reducir notablemente el disolvente residual, menos deformaciones de las microesferas, y estabilidad de almacenamiento mejorada.
TÉCNICA RELACIONADA
El polímero biodegradable se puede preparar en forma de microesferas (por ejemplo, partículas que tienen un tamaño de partícula promedio en el intervalo de nanometros a milímetros, particularmente 1 a 500 pm, particularmente 10 a 150 pm) mediante diversas técnicas conocidas. Las microesferas poliméricas biodegradables se pueden usar como cargas de partículas para mejorar las arrugas faciales por sí mismas, y son muy usadas con el fin de proporcionar una liberación sostenida o retardada de un fármaco u otro agente activo encapsulando dicho fármaco u otro agente activo. El método más frecuentemente usado para preparar dicha microesfera de polímero biodegradable es disolver el polímero biodegradable, o el polímero biodegradable y la sustancia (fármaco u otro agente activo) a encapsular en dicho polímero, en un disolvente, y entonces dispersar o emulsionar la disolución resultante en una disolución acuosa. A continuación, el disolvente de las microesferas se elimina para obtener el producto final de microesferas. Los disolventes tóxicos tales como el diclorometano o el cloroformo se usan a menudo para disolver polímeros biodegradables y agentes activos en el procedimiento de producción de microesferas según la técnica conocida. Los residuos de estos disolventes tóxicos en el producto final son indeseables debido a su toxicidad general y a sus efectos cancerígenos potenciales. Además, se usa una mezcla de diversos tipos de disolventes orgánicos para disolver homogéneamente el polímero biodegradable y el agente activo simultáneamente. Esta mezcla de disolventes se dispersa en una disolución acuosa para formar una emulsión, y después se extrae y se evapora en una capa de disolución acuosa según la solubilidad de cada disolvente en una disolución acuosa, la afinidad con el polímero biodegradable, etc. Sin embargo, se ha encontrado que la mezcla de tales disolventes orgánicos no se elimina lo suficiente durante la producción de microesferas según la técnica anterior conocida, y el disolvente residual afecta negativamente la estabilidad del producto final, tal como al promover la descomposición del polímero durante el período de almacenamiento de las microesferas finalmente producidas. En consecuencia, existe la necesidad de desarrollar un método de producción de microesferas poliméricas biodegradables capaz de aumentar la vida útil de un producto reduciendo la cantidad residual de mezcla de disolvente tóxico y disolvente orgánico. El documento WO 02/49620 describe un procedimiento para la producción de micropartículas poliméricas, que comprende disolver un polímero en un disolvente, para formar una disolución de polímero; añadir un agente activo no soluble en agua a la disolución de polímero, para formar una fase de fármaco contenida en una vasija; añadir con mezclamiento una cantidad predeterminada de una fase acuosa de tensioactivo a la vasija que contiene la fase de fármaco, siendo suficiente dicha cantidad predeterminada para hacer que la fase de tensioactivo se convierta en la fase continua y el medio de extracción, para extraer una cantidad de dicho disolvente de dicha fase de fármaco de manera que se produce una suspensión de micropartículas tras la adición de la fase de tensioactivo a la fase de fármaco, sin necesidad de eliminar el disolvente de la vasija. En este procedimiento, el disolvente se elimina mediante lavado, filtración, vacío, o evaporación. El documentoEP 2 982 367 describe un método para preparar una microesfera de donepezilo, que comprende las etapas de: disolver en uno o más disolventes donepezilo o una de sus sales farmacéuticamente aceptables y un polímero biocompatible y biodegradable; formar una microesfera añadiendo la disolución de donepezilo y polímero biocompatible biodegradable a una disolución acuosa de polímero hidrófilo, y agitando; y eliminar el disolvente, en el que el método comprende además una etapa de lavar la microesfera. El documentoWO 2014/202214 describe un procedimiento para la preparación de micropartículas biodegradables de polímero de poli(D,L láctida-co-glicólida) (PLGA), que tiene un perfil de liberación sigmoidal de un compuesto básico o hidrófobo, contenido dentro de las micropartículas, que comprende preparar una emulsión de aceite en agua, en la que la fase oleosa comprende PLGA, el compuesto y un disolvente orgánico, y la fase acuosa comprende agua, tensioactivo, opcionalmente, un amortiguador, y el mismo disolvente orgánico que en la fase oleosa, y entonces llevar a cabo una etapa de extracción/evaporación de la emulsión con disolvente, seguido de una única etapa de secado de las micropartículas endurecidas. El documento EP 1 629 833 describe un procedimiento para la producción de micropartículas, en el que se disuelve betametasona y un polímero biodegradable en un disolvente que comprende un hidrocarburo halogenado y alcohol fenilalquílico para formar una fase oleosa, la fase oleosa se mezcla con una fase acuosa para formar una emulsión de tipo o/w, y después los disolventes mencionados anteriormente se eliminan para producir las micropartículas. El documento KR 20160019020 se refiere al desarrollo de un método para producir micropartículas de liberación sostenida, que puede eliminar fácilmente el disolvente de alcano halogenado y minimizar la hidrólisis del polímero biodegradable.
DESCRIPCIÓN
[Problema técnico]
En consecuencia, la presente invención se ha realizado para resolver el problema mencionado anteriormente de la técnica convencional. De este modo, un objeto de la presente invención es proporcionar un método para producir microesferas biodegradables que tienen alta seguridad al reducir notablemente el disolvente residual, menos deformaciones de las microesferas, y estabilidad de almacenamiento mejorada.
[Solución técnica]
Como un aspecto para lograr dicho propósito, la presente invención se refiere a un método para preparar microesferas biodegradables, que comprende:
(a) formar una disolución de polímero biodegradable disolviendo un polímero biodegradable solo; o un polímero biodegradable y un fármaco en un disolvente orgánico;
(b) mezclar uniformemente la disolución de polímero biodegradable preparada en la etapa (a) en una disolución acuosa que contiene un tensioactivo, para formar una emulsión que contiene la disolución de polímero biodegradable como fase dispersa y una disolución acuosa que contiene el tensioactivo como una fase continua; (c) extraer y evaporar el disolvente orgánico de la fase dispersa en la emulsión de la etapa (b) a la fase continua para producir microesferas, en el que se elimina una parte de la fase continua que contiene el disolvente orgánico extraído, mientras que se suministra una nueva fase continua; y
(d) recuperar las microesferas de la fase continua que contiene las microesferas producidas en la etapa (c), en el que la parte de la fase continua en la etapa (c) se elimina de forma discontinua o continua, y (i) cuando la parte de la fase continua en la etapa (c) se elimina de forma discontinua, la fase continua eliminada es una relación de volumen de 40% (v/v) a 99% (v/v), basado en el volumen total de la fase continua, o es una relación en peso de una fase continua restante, excepto la relación en peso puntual de la fase continua de la micropartícula en la etapa (c), o (ii) cuando la parte de la fase continua en la etapa (c) se elimina de forma continua, la tasa de eliminación de la fase continua está en el intervalo de 2% (v/v) a 200% (v/v)/min, basado en el volumen total de la fase continua. EFECTOS VENTAJOSOS
En la producción de una microesfera de polímero biodegradable que contiene un polímero biodegradable en sí mismo, o una microesfera biodegradable que contiene un polímero biodegradable y un fármaco fisiológicamente activo, es muy importante minimizar la cantidad residual de disolvente tóxico o mezcla de disolvente orgánico que queda en la microesfera. La presente invención proporciona un método para preparar de manera eficaz microesferas biodegradables que tienen alta seguridad y excelente estabilidad de almacenamiento formando una emulsión y después eliminando de la fase dispersa una parte de una fase continua que comprende un disolvente orgánico extraído, al tiempo que se suministra una nueva disolución acuosa para reemplazar dicha fase continua. De este modo, es posible eliminar rápida y fácilmente el disolvente orgánico residual en las microesferas, al tiempo que se minimizan los cambios morfológicos de las microesferas y la reducción del peso molecular debido a la hidrólisis del polímero biodegradable en las microesferas.
BREVE DESCRIPCIÓN DE LOS DIBUJOS FIG. 1a es una fotografía de microscopio electrónico de barrido (SEM) para el análisis de la caracterización morfológica de las microesferas preparadas en el Ejemplo 1-1 de la presente invención, que confirma que la mayoría de las microesferas mantienen la morfología esférica completa.
FIG. 1b es una fotografía SEM para el análisis de la caracterización morfológica de las microesferas preparadas en el Ejemplo Comparativo 1 (cuando el tiempo de intercambio del disolvente de fase continua es más rápido que el tiempo de endurecimiento de la superficie de la microesfera), lo que confirma que las microesferas no pueden mantener la forma esférica.
FIG. 1c es una fotografía de las microesferas preparadas en el Ejemplo Comparativo 2 (cuando se eliminó todo el disolvente de la fase continua) para el análisis de caracterización morfológica, y se confirmó mediante el microscopio electrónico de barrido que la forma de las partículas de las microesferas cambiaba.
FIG. 1d es una fotografía de las microesferas preparadas en el Ejemplo 5 (cuando el disolvente de fase continua contiene una cantidad apropiada de alcohol etílico) para el análisis de caracterización morfológica usando un SEM, lo que confirma que la mayoría de las microesferas mantienen la morfología esférica completa.
FIG. 1e es una fotografía de las microesferas preparadas en el Ejemplo Comparativo 3 (cuando el disolvente de fase continua contiene 50% (v/v) o más de alcohol etílico) preparadas en el Ejemplo Comparativo 3), para el análisis de caracterización morfológica usando un SEM, que muestra que la morfología de las microesferas se deformó por la eliminación brusca del diclorometano.
A continuación, la presente invención se describirá con más detalle.
La presente invención se refiere a un método para producir una microesfera mediante la preparación de una disolución de polímero biodegradable que comprende un polímero biodegradable y un disolvente orgánico como fase dispersa, preparando una emulsión que contiene un tensioactivo como fase continua, extrayendo y evaporando el disolvente orgánico de la fase dispersa en una fase continua, en el que una parte de la fase continua que comprende el disolvente orgánico extraído de la fase dispersa se elimina del sistema de emulsión, mientras se suministra una nueva disolución acuosa que contiene un tensioactivo al sistema de emulsión para reemplazar dicha fase continua eliminada, para proporcionar microesferas biodegradables que tienen una deformación reducida de las microesferas y una estabilidad de almacenamiento mejorada.
Como una realización de la presente invención, la presente invención se refiere a un método para preparar microesferas biodegradables, que comprende:
(a) formar una disolución de polímero biodegradable disolviendo un polímero biodegradable solo; o un polímero biodegradable y un fármaco en un disolvente orgánico;
(b) mezclar uniformemente la disolución de polímero biodegradable preparada en la etapa (a) en una disolución acuosa que contiene un tensioactivo, para formar una emulsión que contiene la disolución de polímero biodegradable como fase dispersa y una disolución acuosa que contiene el tensioactivo como una fase continua;
(c) extraer y evaporar el disolvente orgánico de la fase dispersa en la emulsión de la etapa (b) a la fase continua para producir microesferas, en el que se elimina una parte de la fase continua que contiene el disolvente orgánico extraído, mientras que se suministra una nueva fase continua; y
(d) recuperar las microesferas de la fase continua que contiene las microesferas producidas en la etapa (c), en el que la parte de la fase continua en la etapa (c) se elimina de forma discontinua o continua, y (i) cuando la parte de la fase continua en la etapa (c) se elimina de forma discontinua, la fase continua eliminada es una relación de volumen de 40% (v/v) a 99% (v/v), basado en el volumen total de la fase continua, o es una relación en peso de una fase continua restante, excepto la relación en peso puntual de la fase continua de la micropartícula en la etapa (c), o (ii) cuando la parte de la fase continua en la etapa (c) se elimina de forma continua, la tasa de eliminación de la fase continua está en el intervalo de 2% (v/v) a 200% (v/v)/min, basado en el volumen total de la fase continua.
A continuación, se describirá en detalle un método para producir las microesferas de polímero biodegradable de la presente invención.
Como se usa aquí, la expresión “método de extracción y evaporación con disolvente” se refiere a un método en el que una disolución de polímero biodegradable preparada disolviendo en un disolvente orgánico un polímero biodegradable solo, o una mezcla de un polímero biodegradable y un fármaco, se añade a una fase continua en forma de una disolución acuosa que contiene un tensioactivo, extrayendo y evaporando el disolvente orgánico de la fase dispersa de la emulsión en una fase continua para formar microesferas, y recuperando microesferas de la fase continua para producir microesferas poliméricas biodegradables.
El método de preparación de la presente invención comprende (a) disolver en un disolvente orgánico un polímero biodegradable solo, o una mezcla de un polímero biodegradable y un fármaco, para formar una disolución de polímero biodegradable.
La etapa (a) puede comprender un procedimiento de formación de una disolución de polímero biodegradable al disolver un polímero biodegradable en un disolvente orgánico para formar una disolución de polímero biodegradable, o un procedimiento de disolución de un polímero biodegradable y un fármaco simultáneamente en una mezcla de disolvente orgánico para formar una disolución de polímero biodegradable que contiene fármaco.
El peso molecular medio ponderal del polímero biodegradable no está particularmente limitado, pero el límite inferior puede ser 5.000 o más, preferiblemente 10.000 o más, y el límite superior puede ser 500.000 o menos, preferiblemente 200.000 o menos.
El tipo de polímero biodegradable no está particularmente limitado, pero se pueden usar preferiblemente poliésteres. En particular, el polímero biodegradable se puede seleccionar del grupo que consiste en polilactida, poliglicolida, poli(lactida-co-glicolida), poli(lactida-co-glicolida) glucosa, policaprolactona, y mezclas de los mismos. Más preferiblemente, se pueden usar polilactida, poli(lactida-co-glicolida), y policaprolactona.
Cuando se usa poli(lactida-co-glicolida) como polímero biodegradable, la relación molar de ácido láctico a ácido glicólico (ácido láctico:ácido glicólico) en el copolímero puede ser de 99:1 a 50:50, preferiblemente 50:50, 75:25, u 85:15. Los ejemplos de polímeros biodegradables comercialmente disponibles que se pueden usar en la presente invención incluyen la serie Resomer de Evonik, por ejemplo RG502H, RG503H, RG504H, RG502, RG503, RG504, RG653H, RG752H, RG752S, RG755S, RG756S, RG858S, R202H, R203H, R205H, R202S, R203S and R205S, PDL 02A, PDL 02, PDL 04, PDL 05, PDLG 7502A, PDLG 7502, PDLG 7507, PDLG 5002A, PDLG 5002, PDLG 5004A,
PDLG 5004, PDLG 5010, PL 10, PL 18, PL 24, PL 32, PL 38, PDL 20, PDL 45, PC 02, PC 04, PC 12, PC 17 y PC 24 de Corbion.
En una realización específica, para preparar las microesferas según la presente invención se usaron Resomer R203H, RG502H, RG653H, RG752H, RG757S, RG858S, R202H y R205S, y Purasorb PC 04.
El fármaco usado en la etapa (a) no está particularmente limitado, y los ejemplos del mismo incluyen un agente para el tratamiento de la demencia; un agente terapéutico para la enfermedad de Parkinson; agentes anticancerígenos; antipsicóticos, tales como ansiolíticos, antidepresivos, estabilizadores nerviosos, y agentes psicotrópicos; un agente terapéutico cardiovascular, tal como un agente terapéutico para la hiperlipidemia, un agente terapéutico para la hipertensión, un agente terapéutico para la hipotensión, un agente antitrombótico, un vasodilatador, y un remedio para la arritmia; antiepilépticos; agentes terapéuticos gastrointestinales, tales como agentes antiulcerosos; un agente terapéutico para enfermedades reumáticas; antiespamódicos; fármacos antituberculosos; relajantes musculares; un agente terapéutico para la osteoporosis; medicamentos para la disfunción eréctil; agente hematostático; hormonas, tales como hormonas sexuales y similares; agentes antidiabéticos; antibióticos; agentes antifúngicos; agentes antivirales; fármacos antipiréticos, analgésicos y antiinflamatorios; reguladores autonómicos; corticosteroides; diuréticos; analgésico; anestésico; antihistamínicos; agente antiprotozoario; antianémicos; antiasmáticos; anticonvulsivos; antídoto; agente antimigrañoso; antieméticos; fármaco antiparkinsoniano; agentes antiepilépticos; agentes antiplaquetarios; antitusivos y expectorantes; broncodilatador; cardiotónicos; inmunomoduladores; fármacos proteicos; droga genética; y una mezcla de los mismos. Preferiblemente, el fármaco se puede seleccionar del grupo que consiste en el agente terapéutico para la demencia, un fármaco para la enfermedad de Parkinson, un agente anticanceroso, un fármaco antipsicótico, un agente antihiperlipidémico, un agente antihipertensivo, un agente antiepiléptico, un agente terapéutico gastrointestinal, un fármaco antirreumático, un fármaco antiespasmódico, fármacos antituberculosos, relajante muscular, un agente antiarrítmico, un agente terapéutico para la osteoporosis, un agente terapéutico para la disfunción eréctil, un agente hemostático, un agente antiviral, un agente hormonal, antibióticos, antidiabéticos, un agente antifúngico, un agente antitrombótico, un agente antipirético, analgésico y antiinflamatorio, y una mezcla de los mismos.
Los ejemplos no limitativos de los fármacos mencionados anteriormente incluyen donepezilo, memantina, rivastigmina, entecavir, lamivudina, rotigotina, ropinirol, bupivacaína, ropivacaína, meloxicam, buprenorfina, fentanilo, granisetrón, triamcinolona, citarabina, carmustina, tamsulosina, polmacoxib, testosterona, estradiol, risperidona, paliperidona, olanzapina, aripiprazol, goserelina, leuprolida, triptorelina, buserelina, nafarelina, deslorelina, octreotida, pasireotida, lanreotida, vapreotida, exenatida, liraglutida, lixisenatida, semaglutida, y sales o una mezcla de los mismos.
En la etapa (a), el disolvente usado para disolver el polímero biodegradable tiene preferiblemente la propiedad de no ser miscible con agua. Debido a la propiedad de que el disolvente orgánico no es miscible con agua, la emulsión se puede formar mezclando homogéneamente la disolución de polímero biodegradable en la fase continua en la etapa (b) que se describirá más adelante. El ejemplo del disolvente para disolver el polímero biodegradable no está particularmente limitado, pero es preferiblemente uno o más disolventes seleccionados del grupo que consiste en diclorometano, cloroformo, acetato de etilo, acetona, acetonitrilo, dimetilsulfóxido, dimetilformamida, metiletilcetona, ácido acético, alcohol metílico, alcohol etílico, alcohol propílico, alcohol bencílico, y una mezcla de disolventes de los mismos, más preferiblemente diclorometano, acetato de etilo, o una mezcla de disolventes de los mismos.
El método de preparación de la presente invención comprende la etapa (b) mezclar uniformemente la disolución de polímero biodegradable preparada en la etapa (a) en una disolución acuosa que contiene un tensioactivo, para formar una emulsión que contiene la disolución de polímero biodegradable como fase dispersa y una disolución acuosa que contiene el tensioactivo como una fase continua.
Para mezclar homogéneamente una disolución acuosa que contiene una disolución de polímero biodegradable y un tensioactivo en la etapa (b), se pueden usar diversos métodos de mezclamiento, y ejemplos no limitantes de los mismos son un mezclador de alta velocidad, un mezclador en línea, un método de emulsión de membrana o un método de emulsión microfluídica.
Cuando se forma una emulsión que contiene una disolución acuosa que contiene la disolución de polímero biodegradable y el tensioactivo como en la etapa (b), la disolución de polímero biodegradable se dispersa homogéneamente en la disolución acuosa para formar una fase dispersa en forma de gotitas.
Por lo tanto, la disolución acuosa que contiene el tensioactivo como fase continua usada en la etapa (b) no es miscible con el disolvente orgánico en la disolución de polímero biodegradable como fase dispersa.
El tensioactivo usado en la etapa (b) no está particularmente limitado, y se puede usar cualquier tipo de tensioactivo siempre que ayude a la disolución de polímero biodegradable a formar una fase dispersa de gotitas líquidas estables en una fase líquida continua. El tensioactivo se selecciona preferiblemente del grupo que consiste en metilcelulosa, polivinilpirrolidona, carboximetilcelulosa, lecitina, gelatina, alcohol polivinílico, éster de ácido graso con polioxietilensorbitán, derivados de aceite de ricino polioxietilenados, y una mezcla de los mismos. Además, lo más preferible, se puede usar alcohol polivinílico.
En la etapa (b), el contenido del tensioactivo en la disolución acuosa que contiene el tensioactivo es 0,01 % (p/v) a 20% (p/v), basado en el volumen total de la disolución acuosa que contiene el tensioactivo, preferiblemente 0,1% (p/v) a 5% (p/v). Si el contenido del tensioactivo es menor que 0,01% (p/v), es posible que no se forme una fase dispersa en forma de gotitas en la disolución acuosa o una emulsión. Si el contenido de tensioactivo supera el 20% (p/v), es difícil eliminar el tensioactivo después de que se hayan formado las microesferas en la disolución acuosa, debido a la cantidad excesiva del tensioactivo.
Además, en una realización preferida, la fase continua usada en la etapa (b) puede comprender además uno o más disolventes seleccionados de un grupo que consiste en alcohol metílico, alcohol etílico, alcohol propílico, y acetato de etilo, para controlar la tasa de extracción del disolvente orgánico de la emulsión así como el agua y el tensioactivo. Es preferible que dicho disolvente pueda estar contenido en una cantidad de 0,1% (v/v) a 40% (v/v) en base al volumen total de la fase continua.
El método de la presente invención comprende una etapa (c) de extraer y evaporar el disolvente orgánico de la fase dispersa en la emulsión de la etapa (b) a la fase continua para producir microesferas, en el que se elimina una parte de la fase continua que contiene el disolvente orgánico extraído, mientras que se suministra a la emulsión una nueva fase continua.
En la etapa (c), cuando la emulsión que contiene la disolución de polímero biodegradable (fase dispersa) en forma de gotitas y la fase continua se mantiene o se agita a una temperatura menor que el punto de ebullición del disolvente orgánico durante un tiempo predeterminado, por ejemplo 2 a 48 horas, el disolvente orgánico se puede extraer de la disolución de polímero biodegradable en forma de gotitas (una fase dispersa) a la fase continua. Parte del disolvente orgánico extraído a la fase continua se puede evaporar de la superficie de la fase continua que contiene las microesferas producidas. A medida que el disolvente orgánico se extrae de la disolución de polímero biodegradable (fase dispersa) en forma de gotitas y se evapora, la fase dispersa en forma de gotitas se puede solidificar para formar microesferas.
En el método convencional para producir una microesfera usando un método de extracción y evaporación con disolvente, a veces se aplica calor durante mucho tiempo para eliminar suficientemente el disolvente orgánico de la fase dispersa en forma de gotitas, y el calor degrada el polímero biodegradable, reduciendo así el peso molecular del polímero biodegradable.
Sin embargo, en la presente invención, en la etapa (c), se elimina una parte de la fase continua que contiene el disolvente orgánico extraído de la fase dispersa, y se suministra a la emulsión una nueva disolución acuosa que contiene un tensioactivo que reemplaza a la fase continua eliminada, por lo que la cantidad residual del disolvente orgánico se puede minimizar de forma eficiente extrayendo suficientemente el disolvente orgánico en una fase continua y evaporando.
La presente invención se caracteriza por que solo se elimina una parte de la fase continua y se suministra a la emulsión una nueva disolución acuosa que comprende un tensioactivo, de modo que el procedimiento de extracción con disolvente de la fase dispersa se puede llevar a cabo de forma continua. Preferiblemente, el tiempo de intercambio para la fase continua es superior a 5 minutos, más preferiblemente 10 minutos a 60 minutos desde el momento en que la superficie de las microesferas comienza a endurecerse. Cuando la fase continua se intercambia más rápido que el tiempo anterior, las microesferas no pueden mantener la forma esférica y pueden deformarse.
Cuando la fase continua se elimina de forma discontinua, es que se elimina del sistema de emulsión (i) 40% (v/v) a 99% (v/v) de la fase continua, o (ii) el resto de la fase continua excepto una (1) vez del peso de las microesferas. Si la fase continua se elimina más allá del intervalo anterior (por ejemplo, la cantidad eliminada es menor que 99% (v/v), o la mitad (0,5) del peso de la microesfera), la forma de las microesferas producidas puede deformarse. Además, cuando se elimina el 35% (v/v) o menos de la fase continua del 35% (v/v) o menos, la eliminación del disolvente orgánico puede no ser eficaz.
Al eliminar una parte de la fase continua, las microesferas inicialmente formadas como fase dispersa pueden retenerse usando un filtro o similar, y solo una parte de la fase continua puede eliminarse usando una bomba peristáltica o similar.
Como otro método para la etapa (c), puede usarse un método de suministro continuo de una disolución acuosa reciente que comprende un tensioactivo, mientras se elimina simultáneamente una parte de la fase continua.
En la eliminación de la fase continua, se elimina a una tasa del 2% (v/v) al 200% (v/v) con respecto al volumen total de la fase continua por minuto.
La disolución acuosa que contiene el tensioactivo como una nueva fase continua que se suministra adicionalmente en la etapa (c) puede comprender agua o un disolvente mixto que comprende (i) agua y (ii) uno o más seleccionados del grupo que consiste en alcoholes alifáticos que tienen 1 a 4 átomos de carbono (preferiblemente, alcohol metílico, alcohol etílico, o alcohol propílico) y acetato de etilo, en una cantidad predeterminada. Además, es preferible que el disolvente mixto esté contenido en una cantidad de 0,1% (v/v) a 40% (v/v) en base al volumen total de la fase continua que se va a añadir de nuevo.
Mediante la eliminación continua del disolvente orgánico extraído por el método de sustitución de fase continua de la etapa (c) de la presente invención, el disolvente orgánico puede extraerse eficazmente, y el disolvente residual puede minimizarse.
Para eliminar más eficientemente el disolvente orgánico en la etapa (c) de la presente invención, se puede aplicar calor durante un cierto período de tiempo para mantener constante la temperatura de la fase continua.
El método de la presente invención comprende la etapa (d) de recuperar las microesferas de la emulsión preparada en dicha etapa (c). Se pueden usar diversas técnicas conocidas para recuperar las microesferas, tales como filtración o centrifugación.
En el método de la presente invención, el tensioactivo residual puede eliminarse mediante filtración y lavado entre la etapa (c) y la etapa (d), y las microesferas pueden recuperarse mediante filtración adicional.
La etapa de lavado para eliminar el tensioactivo residual normalmente se puede llevar a cabo usando agua, y la etapa de lavado se puede repetir varias veces.
En el método de la presente invención, después de la etapa (d), o después de la etapa de filtración y lavado anterior, las microesferas obtenidas pueden secarse usando un método de secado convencional para obtener finalmente microesferas secas.
Preferiblemente, en el método de la presente invención, las microesferas obtenidas pueden suspenderse en una suspensión después de la etapa (d), o después de la etapa de filtración y lavado entre las etapas (c) y (d), y posteriormente pueden introducirse en un recipiente farmacéuticamente aceptable, por ejemplo jeringa desechable o similar, para obtener un producto final.
El método descrito anteriormente permite eliminar eficazmente el disolvente residual en las microesferas biodegradables, minimizar la reducción del peso molecular debido a la hidrólisis del polímero biodegradable en la microesfera, y proporcionar microesferas de polímero biodegradable que tienen una seguridad y una estabilidad de almacenamiento excelentes.
Preferiblemente, cuando las micropartículas se producen mediante el método según la presente invención, el contenido de disolvente residual en las micropartículas secas después de la producción es 1000 ppm o menos, más preferiblemente 600 ppm o menos.
[MODO PARA LA INVENCIÓN]
Ejemplo 1: Producción de microesferas preparadas mediante diferentes tiempos de intercambio de fase continua
3.5 g de un polímero biocompatible Resomer R203H (fabricante: Evonik, Alemania; poli(D,L-lactida) Mw: 18000 a 28000) y 1,5 g de base de donepezilo (fabricante: Neuland Laboratories, India) se disolvieron en 9,2 g de diclorometano (JT Baker, USA) para preparar una fase dispersa. La fase dispersa se preparó agitando durante 30 minutos o más para una disolución suficiente, y entonces se usó. Se usó una disolución acuosa de alcohol polivinílico al 1% (p/v) (viscosidad: 4,8 a 5,8 mPas) como fase continua. Se conectó un recipiente que incluía 460 ml de la fase continua a un aparato de emulsionamiento equipado con una membrana que tenía poros de 40 gm de diámetro, mientras que la fase dispersa anterior se inyectó simultáneamente en el aparato para producir la suspensión de microesferas. Después, la suspensión de microesferas resultante se colocó en una vasija de preparación, y se agitó a 150 rpm.
La temperatura del aparato de emulsionamiento de membrana y de la vasija de preparación se mantuvo a 25°C, y la agitación se mantuvo durante 15 minutos. Se retiraron 455 ml de la fase continua, al tiempo que quedaban 5 ml de la fase continua (la misma cantidad de peso total del polímero biodegradable y base de donepezilo usada en la preparación de la fase dispersa). Después, se añadió una nueva disolución de fase continua al aparato, en la misma cantidad que la fase continua eliminada. El disolvente orgánico se eliminó mientras que se mantiene la temperatura a 45°C durante 3 horas. Después de eliminar el disolvente orgánico, la temperatura de la suspensión de microesferas se redujo a 25°C. La suspensión de microesferas se lavó tres veces con agua desionizada para eliminar el alcohol polivinílico residual, y las microesferas resultantes se liofilizaron.
Ejemplo 1-1: Producción de microesferas preparadas mediante diferentes tiempos de intercambio de fase continua
3.5 g de un polímero biocompatible Resomer R203H (fabricante: Evonik, Alemania) y 1,5 g de base de donepezilo (fabricante: Neuland Laboratories, India) se disolvieron en 9,2 g de diclorometano (JT Baker, USA) para preparar una fase dispersa. La fase dispersa se preparó agitando durante 30 minutos o más para una disolución suficiente, y entonces se usó. Se usó una disolución acuosa de alcohol polivinílico al 1% (p/v) (viscosidad: 4,8 a 5,8 mPas) como fase continua. Se conectó un recipiente que incluía 460 ml de la fase continua a un aparato de emulsionamiento equipado con una membrana que tenía poros de 40 gm de diámetro, mientras que la fase dispersa anterior se inyectó
simultáneamente en el aparato para producir la suspensión de microesferas. Después, la suspensión de microesferas resultante se colocó en una vasija de preparación, y se agitó a 150 rpm.
La temperatura del aparato de emulsionamiento de membrana y de la vasija de preparación se mantuvo a 25°C, y la agitación se mantuvo durante 30 minutos. Se retiraron 455 ml de la fase continua, al tiempo que quedaban 5 ml de la fase continua (la misma cantidad de peso total del polímero biodegradable y base de donepezilo usada en la preparación de la fase dispersa). Después, se añadió una nueva disolución de fase continua al aparato, en la misma cantidad que la fase continua eliminada. El disolvente orgánico se eliminó mientras que se mantiene la temperatura a 45°C durante 3 horas. Después de eliminar el disolvente orgánico, la temperatura de la suspensión de microesferas se redujo a 25°C. La suspensión de microesferas se lavó tres veces con agua desionizada para eliminar el alcohol polivinílico residual, y las microesferas resultantes se liofilizaron.
Ejemplo 1-2: Producción de microesferas preparadas mediante diferentes tiempos de intercambio de fase continua
3.5 g de un polímero biocompatible Resomer R203H (fabricante: Evonik, Alemania) y 1,5 g de base de donepezilo (fabricante: Neuland Laboratories, India) se mezclaron en 9,2 g de diclorometano (JT Baker, USA) para preparar una fase dispersa. La fase dispersa se preparó agitando durante 30 minutos o más para una disolución suficiente, y entonces se usó. Se usó una disolución acuosa de alcohol polivinílico al 1% (p/v) (viscosidad: 4,8 a 5,8 mPas) como fase continua. Se conectó un recipiente que incluía 460 ml de la fase continua a un aparato de emulsionamiento equipado con una membrana que tenía poros de 40 pm de diámetro, mientras que la fase dispersa anterior se inyectó simultáneamente en el aparato para producir la suspensión de microesferas. Después, la suspensión de microesferas resultante se colocó en una vasija de preparación, y se agitó a 150 rpm.
La temperatura del aparato de emulsionamiento de membrana y de la vasija de preparación se mantuvo a 25°C, y la agitación se mantuvo durante 60 minutos. Se retiraron 455 ml de la fase continua, al tiempo que quedaban 5 ml de la fase continua (la misma cantidad de peso total del polímero biodegradable y base de donepezilo usada en la preparación de la fase dispersa). Después, se añadió una nueva disolución de fase continua al aparato, en la misma cantidad que la fase continua eliminada. El disolvente orgánico se eliminó mientras que se mantiene la temperatura a 45°C durante 3 horas. Después de eliminar el disolvente orgánico, la temperatura de la suspensión de microesferas se redujo a 25°C. La suspensión de microesferas se lavó tres veces con agua desionizada para eliminar el alcohol polivinílico residual, y las microesferas resultantes se liofilizaron.
Ejemplo 2: Producción de microesferas preparadas mediante diferentes cantidades de intercambio de fase continua
3.5 g de un polímero biocompatible Resomer R203H (fabricante: Evonik, Alemania) y 1,5 g de base de donepezilo (fabricante: Neuland Laboratories, India) se disolvieron en 9,2 g de diclorometano (JT Baker, USA). La fase dispersa se preparó agitando durante 30 minutos o más para una disolución suficiente, y entonces se usó. Se usó una disolución acuosa de alcohol polivinílico al 1% (p/v) (viscosidad: 4,8 a 5,8 mPas) como fase continua. Se conectó un recipiente que incluía 460 ml de la fase continua a un aparato de emulsionamiento equipado con una membrana que tenía poros de 40 pm de diámetro, mientras que la fase dispersa anterior se inyectó simultáneamente en el aparato para producir la suspensión de microesferas. Después, la suspensión de microesferas resultante se colocó en una vasija de preparación, y se agitó a 150 rpm.
La temperatura del aparato de emulsionamiento de membrana y de la vasija de preparación se mantuvo a 25°C, y la agitación se mantuvo durante 30 minutos. Se eliminaron 230 ml de la fase continua (50% del volumen total de la fase continua), y se añadió una nueva disolución de fase continua al aparato en la misma cantidad que la fase continua retirada (230 ml). El disolvente orgánico se eliminó mientras que se mantiene la temperatura a 45°C durante 3 horas. Después de eliminar el disolvente orgánico, la temperatura de la suspensión de microesferas se redujo a 25°C. La suspensión de microesferas se lavó tres veces con agua desionizada para eliminar el alcohol polivinílico residual, y las microesferas resultantes se liofilizaron.
Ejemplo 3 Preparación de microesferas usando acetato de etilo
La fase dispersa se preparó mezclando 3,5 g de un polímero biocompatible, Resomer R203H (fabricante: Evonik, Alemania) y 1,5 g de base de donepezilo (fabricante: Neuland Laboratories, India) con 10,5 g de acetato de etilo (Sigma Aldrich, EE. UU.). La fase dispersa se disolvió suficientemente agitando durante 30 minutos o más, y entonces se usó. Se usó una disolución acuosa de alcohol polivinílico al 1% (p/v) (viscosidad: 4,8 a 5,8 mPas) como fase continua. Se conectó un recipiente que incluía 530 ml de la fase continua a un aparato de emulsionamiento equipado con una membrana que tenía poros de 20 pm de diámetro, mientras que la fase dispersa anterior se inyectó simultáneamente en el aparato para producir la suspensión de microesferas. Después, la suspensión de microesferas resultante se colocó en una vasija de preparación, y se agitó a 300 rpm.
La temperatura del aparato de emulsionamiento de membrana y de la vasija de preparación se mantuvo a 25°C, y la agitación se mantuvo durante 30 minutos. Se eliminaron 525 ml de la fase continua, al tiempo que quedaron 5 ml de la fase continua. Después, se añadió una nueva disolución de fase continua al aparato, en la misma cantidad que la fase continua eliminada. El disolvente orgánico se eliminó mientras que se mantiene la temperatura a 45°C durante 3
horas. Después de eliminar el disolvente orgánico, la temperatura de la suspensión de microesferas se redujo a 25°C. La suspensión de microesferas se lavó tres veces con agua desionizada para eliminar el alcohol polivinílico residual, y las microesferas resultantes se liofilizaron.
Ejemplo 4: Producción de microesferas usando RG502H
La fase dispersa se preparó mezclando 3,5 g de un polímero biocompatible, Resomer RG502H (fabricante: Evonik, Alemania; poli(D,L-lactida-co-glicolida) 50:50; Mw: 7.000 a 17.000) y una base de donepezilo (fabricada por Neuland Laboratories, India) con 7,8 g de diclorometano (fabricante: JT Baker, EE. UU.). La fase dispersa se disolvió suficientemente agitando durante 30 minutos o más, y entonces se usó. Se usó una disolución acuosa de alcohol polivinílico al 1% (p/v) (viscosidad: 4,8-5,8 mPa s) como fase continua. Se vertieron 1750 ml de la fase continua en el vasija de preparación, y se agitó con un mezclador de alta velocidad (L4RT, Silverson, UK) a 3000 rpm, mientras se inyectaba la fase dispersa a un caudal de 7 ml por minuto.
Cuando se completó la inyección de la fase dispersa, la suspensión en el vasija de preparación se mantuvo a 25°C durante 30 minutos mientras se agitaba a 200 rpm. Se eliminaron 1745 ml de la fase continua, al tiempo que quedaban 5 ml de fase continua. Después, se añadió la misma cantidad de nueva disolución de fase continua al vasija de preparación. El disolvente orgánico se eliminó mientras que se mantiene la temperatura a 40°C durante 3 horas. Después de eliminar el disolvente orgánico, la temperatura de la suspensión de microesferas se redujo a 25°C. La suspensión que comprende microesferas se lavó tres veces con agua desionizada para eliminar el alcohol polivinílico residual, y las microesferas se liofilizaron.
Ejemplo 4-1: Preparación de microesferas usando RG653H
3.5 g de un polímero biocompatible Resomer RG653H (fabricante: Evonik, Alemania; poli(D,L-lactida-co-glicolida) 65:35; Mw: 24.000 a 38.000) y 1,5 g de una base de donepezilo (fabricado por Neuland Laboratories, India ) se disolvieron en 17,5 g de diclorometano (JT Baker, EE. UU.) para preparar la fase dispersa. La fase dispersa se agitó durante 30 minutos o más para una disolución suficiente, y entonces se usó. Se usó una disolución acuosa de alcohol polivinílico al 2% (p/v) (viscosidad: 4,8 a 5,8 mPas) como fase continua. Se conectó un recipiente que incluía 880 ml de la fase continua a un aparato de emulsionamiento equipado con una membrana que tenía poros de 30 gm de diámetro, mientras que la fase dispersa anterior se inyectó simultáneamente en el aparato para producir la suspensión de microesferas. Después, la suspensión de microesferas resultante se colocó en una vasija de preparación, y se agitó a 180 rpm.
La temperatura del aparato de emulsionamiento de membrana y de la vasija de preparación se mantuvo a 25°C, y la agitación se mantuvo durante 30 minutos. Se eliminaron 875 ml de la fase continua, al tiempo que quedaban 5 ml de fase continua. Después, se añadió la misma cantidad de nueva disolución de fase continua al vasija de preparación. El disolvente orgánico se eliminó mientras que se mantiene la temperatura a 40°C durante 3 horas. Después de eliminar el disolvente orgánico, la temperatura de la suspensión de microesferas se redujo a 25°C. La suspensión de microesferas se lavó tres veces con agua desionizada para eliminar el alcohol polivinílico residual, y las microesferas resultantes se liofilizaron.
Ejemplo 4-2: Preparación de microesferas usando polímero RG752H
3.5 g de un polímero biocompatible Resomer RG752H (fabricante: Evonik, Alemania; poli(D,L-lactida-co-glicolida) 75:25; Mw: 4.000 a 15.000) y 1,5 g de una base de donepezilo (fabricado por Neuland Laboratories, India ) se mezclaron con 10,0 g de diclorometano (fabricante: JT Baker, EE. UU.) para preparar la fase dispersa. La fase dispersa se disolvió suficientemente agitando durante 30 minutos o más, y entonces se usó. Se usó una disolución acuosa de alcohol polivinílico al 1% (p/v) (viscosidad: 4,8-5,8 mPas) como fase continua. Se vertieron 500 ml de la fase continua en el vasija de preparación, y se agitó con un mezclador de alta velocidad (L4RT, Silverson, UK) a 3000 rpm, mientras se inyectaba la fase dispersa a un caudal de 15 ml por minuto.
Cuando se completó la inyección de la fase dispersa, la suspensión en el vasija de preparación se mantuvo a 25°C durante 30 minutos mientras se agitaba a 200 rpm. Se eliminaron 495 ml de la fase continua, al tiempo que quedaban 5 ml de fase continua. Después, se añadió la misma cantidad de nueva disolución de fase continua al vasija de preparación. El disolvente orgánico se eliminó mientras que se mantiene la temperatura a 40°C durante 3 horas. Después de eliminar el disolvente orgánico, la temperatura de la suspensión de microesferas se redujo a 25°C. La suspensión que comprende microesfera se lavó tres veces con agua desionizada para eliminar el alcohol polivinílico residual, y las microesferas se liofilizaron.
Ejemplo 4-3: Preparación de microesferas usando RG757S
3.5 g de un polímero biocompatible Resomer RG757S (fabricante: Evonik, Alemania; poli(D,L-lactida-co-glicolida) 75:25; Mw: 110.000 a 180.000) y 1,5 g de base de donepezilo (fabricado por Neuland Laboratories, India ) se mezclaron con 26,9 g de diclorometano (fabricante: JT Baker, EE. UU.) para preparar la fase dispersa. La fase dispersa se disolvió suficientemente agitando durante 30 minutos o más, y entonces se usó. Se usó una disolución acuosa de alcohol polivinílico al 4% (p/v) (viscosidad: 4,8-5,8 mPas) como fase continua. Se vertieron 1400 ml de la fase continua
en el vasija de preparación equipada con un mezclador de alta velocidad (L4RT, Silverson, UK), y se agitó a 5000 rpm, mientras se inyectaba la fase dispersa a un caudal de 10 ml por minuto.
Cuando se completó la inyección de la fase dispersa, la suspensión en el vasija de preparación se mantuvo a 25°C durante 30 minutos mientras se agitaba a 150 rpm. Se eliminaron 1395 ml de la fase continua, al tiempo que quedaban 5 ml de fase continua. Después, se añadió la misma cantidad de nueva disolución de fase continua al vasija de preparación. El disolvente orgánico se eliminó mientras que se mantiene la temperatura a 40°C durante 3 horas. Después de eliminar el disolvente orgánico, la temperatura de la suspensión de microesferas se redujo a 25°C. La suspensión que comprende microesfera se lavó tres veces con agua desionizada para eliminar el alcohol polivinílico residual, y las microesferas se liofilizaron.
Ejemplo 4-4: Preparación de microesferas usando RG858S
3.5 g de un polímero biocompatible Resomer RG858S (fabricante: Evonik, Alemania; poli(D,L-lactida-co-glicolida) 85:15; Mw: 190.000 a 240.000) y 1,5 g de base de donepezilo (fabricado por Neuland Laboratories, India ) se mezclaron con 38,9 g de diclorometano (fabricante: JT Baker, EE. UU.) para preparar la fase dispersa. La fase dispersa se disolvió suficientemente agitando durante 30 minutos o más, y entonces se usó. Se usó una disolución acuosa de alcohol polivinílico al 2,5% (p/v) (viscosidad: 4,8-5,8 mPas) como fase continua.
Se conectó un recipiente que incluía 2000 ml de la fase continua a un aparato de emulsionamiento equipado con una membrana que tenía poros de 20 gm de diámetro, mientras que la fase dispersa anterior se inyectó simultáneamente en el aparato para producir la suspensión de microesferas. Después, la suspensión de microesferas resultante se colocó en una vasija de preparación, y se agitó a 250 rpm.
La temperatura del aparato de emulsionamiento de membrana y de la vasija de preparación se mantuvo a 25°C, y la agitación se mantuvo durante 30 minutos. Se eliminaron 1995 ml de la fase continua, al tiempo que quedaron 5 ml de la fase continua. Después, se añadió una nueva disolución de fase continua al aparato, en la misma cantidad que la fase continua eliminada. El disolvente orgánico se eliminó mientras que se mantiene la temperatura a 40°C durante 3 horas. Después de eliminar el disolvente orgánico, la temperatura de la suspensión de microesferas se redujo a 25°C. La suspensión de microesferas se lavó tres veces con agua desionizada para eliminar el alcohol polivinílico residual, y las microesferas resultantes se liofilizaron.
Ejemplo 4-5: Preparación de microesferas usando R202H
3.5 g de un polímero biocompatible Resomer R202H (fabricante: Evonik, Alemania; poli(D,L-lactida) Mw: 10.000 a 18.000) y 1,5 g de base de donepezilo (fabricado por Neuland Laboratories, India ) se mezclaron con 7,8 g de diclorometano (fabricante: JT Baker, EE. UU.) para preparar la fase dispersa. La fase dispersa se disolvió suficientemente agitando durante 30 minutos o más, y entonces se usó. Se usó una disolución acuosa de alcohol polivinílico al 2,5% (p/v) (viscosidad: 4,8-5,8 mPas) como fase continua.
Se conectó un recipiente que incluía 390 ml de la fase continua a un aparato de emulsionamiento equipado con una membrana que tenía poros de 50 gm de diámetro, mientras que la fase dispersa anterior se inyectó simultáneamente en el aparato para producir la suspensión de microesferas. Después, la suspensión de microesferas resultante se colocó en una vasija de preparación, y se agitó a 250 rpm.
La temperatura del aparato de emulsionamiento de membrana y de la vasija de preparación se mantuvo a 25°C, y la agitación se mantuvo durante 30 minutos. Se eliminaron 385 ml de la fase continua, al tiempo que quedaron 5 ml de la fase continua. Después, se añadió una nueva disolución de fase continua al aparato, en la misma cantidad que la fase continua eliminada. El disolvente orgánico se eliminó mientras que se mantiene la temperatura a 40°C durante 3 horas. Después de eliminar el disolvente orgánico, la temperatura de la suspensión de microesferas se redujo a 25°C. La suspensión de microesferas se lavó tres veces con agua desionizada para eliminar el alcohol polivinílico residual, y las microesferas resultantes se liofilizaron.
Ejemplo 4-6: Preparación de microesferas usando R205S
3.5 g de un polímero biocompatible Resomer R205S (fabricante: Evonik, Alemania; poli(D,L-lactida) Mw: 58000 a 89000) y 1,5 g de base de donepezilo (fabricante: Neuland Laboratories, India) se disolvieron en 17,5 g de diclorometano (fabricado por JT Baker, USA) para formar la fase dispersa. La fase dispersa se disolvió suficientemente agitando durante 30 minutos o más, y entonces se usó. Como fase continua, se usó una disolución acuosa de alcohol polivinílico al 1% (viscosidad: 4,8-5,8 mPas), y se conectó un recipiente que contenía 880 ml de la fase continua a un aparato de emulsionamiento equipado con una membrana de 40 gm de diámetro de poros, mientras se inyecta la fase dispersa para formar la suspensión de las microesferas. La suspensión de las microesferas se colocó en un vasija de preparación y se agitó a 100 rpm.
La temperatura del aparato de emulsionamiento de membrana y de la vasija de preparación se mantuvo a 25°C, y la agitación se mantuvo durante 30 minutos. Se eliminaron 875 ml de la fase continua, al tiempo que quedaron 5 ml de la fase continua. Después, se añadió una nueva disolución de fase continua al aparato, en la misma cantidad que la fase continua eliminada. El disolvente orgánico se eliminó mientras que se mantiene la temperatura a 40°C durante 3
horas. Después de eliminar el disolvente orgánico, la temperatura de la suspensión de microesferas se redujo a 25°C. La suspensión de microesferas se lavó tres veces con agua desionizada para eliminar el alcohol polivinílico residual, y las microesferas resultantes se liofilizaron.
Ejemplo 4-7: Producción de microesferas usando PC04
La fase dispersa se preparó mezclando 5 g de un polímero biocompatible, Purasorb PC04 (fabricante: Corbion, Países Bajos; policaprolactona; Mw: 28000 a 38000) con 20,0 g de diclorometano (fabricante: J.T Baker, EE. UU.). La fase dispersa se disolvió suficientemente agitando durante 30 minutos o más antes del uso. La fase continua fue una disolución acuosa de alcohol polivinílico al 1% (p/v) (viscosidad: 4,8-5,8 mPas), y se conectó un recipiente que contenía 700 ml de la fase continua a un aparato de emulsionamiento equipado con una membrana de 40 gm de diámetro de poros, mientras se inyecta simultáneamente la fase dispersa preparada para producir las microesferas en suspensión. La suspensión de microesferas resultante se colocó en una vasija de preparación, y se agitó a 150 rpm.
La temperatura del aparato de emulsionamiento de membrana y de la vasija de preparación se mantuvo a 25°C, y la agitación se mantuvo durante 30 minutos. Se eliminaron 455 ml de la fase continua, al tiempo que quedaron 695 ml de la fase continua. Después, se añadió una nueva disolución de fase continua al aparato, en la misma cantidad que la fase continua eliminada. El disolvente orgánico se eliminó mientras que se mantiene la temperatura a 40°C durante 3 horas. Después de eliminar el disolvente orgánico, la temperatura de la suspensión de microesferas se redujo a 25°C. La suspensión de microesferas se lavó tres veces con agua desionizada para eliminar el alcohol polivinílico residual, y las microesferas resultantes se liofilizaron.
Ejemplo 5 Preparación de microesferas usando fase continua que comprende alcohol etílico
La fase dispersa se preparó mezclando 3,5 g de un polímero biocompatible, Resomer R203H (fabricante: Evonik, Alemania) y 1,5 g de base de donepezilo (fabricante: Neuland Laboratories, India) con 9,2 g de diclorometano (fabricante: J.T Baker, EE. UU.). La fase dispersa se disolvió suficientemente agitando durante 30 minutos o más, y entonces se usó. La fase continua fue una disolución acuosa de alcohol polivinílico al 1% (p/v) (viscosidad: 4,8-5,8 mPa s) y alcohol etílico al 30% (v/v). Se conectó un recipiente que incluía la fase continua a un aparato de emulsionamiento equipado con una membrana que tenía poros de 40 gm de diámetro, mientras se inyectaba simultáneamente la fase dispersa preparada para producir las microesferas en suspensión. La suspensión de microesferas resultante se colocó en una vasija de preparación, y se agitó a 200 rpm.
La temperatura del aparato de emulsionamiento de membrana y de la vasija de preparación se mantuvo a 25°C. Cuando se completó la inyección de la fase dispersa, la agitación se mantuvo durante 30 minutos. En esta etapa, se eliminaron 455 ml de la fase continua remanente, al tiempo que se dejaron 5 ml de fase continua, y se añadió la misma cantidad de nueva disolución de fase continua. El disolvente orgánico se eliminó mientras que se mantiene la temperatura a 40°C durante 3 horas. Después de eliminar el disolvente orgánico, la temperatura de la suspensión de microesferas se redujo a 25°C. La suspensión de microesferas se lavó tres veces con agua desionizada para eliminar el alcohol polivinílico residual, y las microesferas se liofilizaron.
Ejemplo 6: Preparación de microesferas con un procedimiento para añadir y eliminar continuamente una fase continua
3.5 g de un polímero biocompatible Resomer R203H (fabricante: Evonik, Alemania) y 1,5 g de base de donepezilo (fabricante: Neuland Laboratories, India) se mezclaron con 9,2 g de diclorometano (fabricante: J.T Baker, EE. UU.) para preparar la fase dispersa. La fase dispersa se disolvió suficientemente agitando durante 30 minutos o más, y entonces se usó. Se usó una disolución acuosa de alcohol polivinílico al 1% (p/v) (viscosidad: 4,8-5,8 mPas) como fase continua. Se vertieron 460 ml de la fase continua en el vasija de preparación equipada con un mezclador de alta velocidad (L4RT, Silverson, UK), y se agitó a 2000 rpm, mientras se inyectaba la fase dispersa a un caudal de 10 ml por minuto. La temperatura del aparato de emulsionamiento de membrana y de la vasija de preparación se mantuvo a 25°C. Cuando se completó la inyección de la fase dispersa, la fase continua se eliminó a un caudal de 13,8 ml por minuto (3% del volumen total de la fase continua), mientras se inyectaba una nueva fase continua a la misma velocidad de eliminación durante 1 hora. Después, la temperatura del aparato se mantuvo a 45°C durante 3 horas. Cuando se completó la eliminación del disolvente orgánico, la temperatura de la suspensión de microesferas se redujo a 25°C. La suspensión que comprende microesfera se lavó tres veces con agua desionizada para eliminar el alcohol polivinílico residual, y las microesferas se liofilizaron.
Ejemplo 6-1: Preparación de microesferas con un procedimiento de añadir y eliminar continuamente una fase continua
3.5 g de un polímero biocompatible Resomer R203H (fabricante: Evonik, Alemania) y 1,5 g de base de donepezilo (fabricante: Neuland Laboratories, India) se mezclaron con 9,2 g de diclorometano (fabricante: J.T Baker, EE. UU.) para preparar la fase dispersa. La fase dispersa se disolvió suficientemente agitando durante 30 minutos o más, y entonces se usó. Se usó una disolución acuosa de alcohol polivinílico al 1% (p/v) (viscosidad: 4,8-5,8 mPas) como fase continua. Se vertieron 460 ml de la fase continua en el vasija de preparación equipada con mezclador de alta velocidad (L4RT, Silverson, UK), y se agitó a 2000 rpm, mientras se inyectaba la fase dispersa a un caudal de 10 ml por minuto y se mantenía la temperatura de la vasija de preparación a 25°C. Cuando se completó la inyección de la
fase dispersa, la fase continua se eliminó a un caudal de 27,6 ml por minuto (6% del volumen total de la fase continua), mientras se inyectaba una nueva fase continua a la misma velocidad de eliminación durante 1 hora. Después, la temperatura de la vasija de preparación se mantuvo a 452C durante 3 horas. Cuando se completó la eliminación del disolvente orgánico, la temperatura de la suspensión de microesferas se redujo a 25°C. La suspensión que comprende microesfera se lavó tres veces con agua desionizada para eliminar el alcohol polivinílico residual, y las microesferas se liofilizaron.
Ejemplo 6-2: Preparación de microesferas que comprende además una etapa de añadir y eliminar continuamente una fase continua
3.5 g de un polímero biocompatible Resomer R203H (fabricante: Evonik, Alemania) y 1,5 g de base de donepezilo (fabricante: Neuland Laboratories, India) se mezclaron con 9,2 g de diclorometano (fabricante: J.T Baker, EE. UU.) para formar la fase dispersa. La fase dispersa se disolvió suficientemente agitando durante 30 minutos o más, y entonces se usó. Se usó una disolución acuosa de alcohol polivinílico al 1% (p/v) (viscosidad: 4,8-5,8 mPas) como fase continua. Se vertieron 460 ml de la fase continua en el vasija de preparación equipada con mezclador de alta velocidad (L4RT, Silverson, UK), y se agitó a 2000 rpm, mientras se inyectaba la fase dispersa a un caudal de 10 ml por minuto y se mantenía la temperatura de la vasija de preparación a 25°C. Cuando se completó la inyección de la fase dispersa, la fase continua se eliminó a un caudal de 26,7 ml por minuto (6% del volumen total de la fase continua), mientras se inyectaba una nueva fase continua a la misma velocidad de eliminación durante 0,5 hora. Después, la temperatura de la vasija de preparación se mantuvo a 45°C durante 3 horas. Cuando se completó la eliminación del disolvente orgánico, la temperatura de la suspensión de microesferas se redujo a 25°C. La suspensión que comprende microesfera se lavó tres veces con agua desionizada para eliminar el alcohol polivinílico residual, y las microesferas se liofilizaron.
Ejemplo 6-3: Preparación de microesferas con una etapa de añadir y elminar continuamente una fase continua
3.5 g de un polímero biocompatible Resomer R203H (fabricante: Evonik, Alemania) y 1,5 g de base de donepezilo (fabricante: Neuland Laboratories, India) se mezclaron con 9,2 g de diclorometano (fabricante: J.T Baker, EE. UU.) para formar la fase dispersa. La fase dispersa se disolvió suficientemente agitando durante 30 minutos o más, y entonces se usó. Se usó una disolución acuosa de alcohol polivinílico al 1% (p/v) (viscosidad: 4,8-5,8 mPas) como fase continua. Se vertieron 460 ml de la fase continua en el vasija de preparación equipada con mezclador de alta velocidad (L4RT, Silverson, UK), y se agitó a 2000 rpm, mientras se inyectaba la fase dispersa a un caudal de 10 ml por minuto y se mantenía la temperatura de la vasija de preparación a 25°C. Cuando se completó la inyección de la fase dispersa, la fase continua se eliminó a un caudal de 920 ml por minuto (200% del volumen total de la fase continua), mientras se inyectaba una nueva fase continua a la misma velocidad de eliminación durante 10 minutos. Después, la temperatura de la vasija de preparación se mantuvo a 45°C durante 3 horas. Cuando se completó la eliminación del disolvente orgánico, la temperatura de la suspensión de microesferas se redujo a 25°C. La suspensión que comprende microesfera se lavó tres veces con agua desionizada para eliminar el alcohol polivinílico residual, y las microesferas se liofilizaron.
Ejemplo 7: Preparación de microesferas que comprenden rivastigmina
La fase dispersa se preparó mezclando 4,0 g de un polímero biocompatible, Resomer R203H (fabricante: Evonik, Alemania) y 1,0 g de base de rivastigmina (fabricante: Hwail Pharmaceutical Co., Ltd., Corea) con 10,0 g de diclorometano (fabricante: JT Baker, EE. UU.). La fase dispersa se disolvió suficientemente agitando durante 30 minutos o más, y entonces se usó. La fase continua fue una disolución acuosa de alcohol polivinílico al 1% (p/v) (viscosidad: 4,8-5,8 mPa s). Se conectó un recipiente que incluía 500 ml de la fase continua a un aparato de emulsionamiento equipado con una membrana que tenía poros de 30 gm de diámetro, mientras se inyectaba simultáneamente la fase dispersa preparada para producir las microesferas en suspensión. La suspensión de microesferas resultante se colocó en una vasija de preparación, y se agitó a 200 rpm.
La temperatura del aparato de emulsionamiento de membrana y de la vasija de preparación se mantuvo a 25°C. Cuando se completó la inyección de la fase dispersa, la agitación se mantuvo durante 30 minutos. En esta etapa, se eliminaron 495 ml de la fase continua, al tiempo que se dejaron 5 ml de fase continua, y se añadió la misma cantidad de nueva disolución de fase continua. El disolvente orgánico se eliminó mientras que se mantiene la temperatura a 45°C durante 3 horas. Después de eliminar el disolvente orgánico, la temperatura de la suspensión de microesferas se redujo a 25°C. La suspensión de microesferas se lavó tres veces con agua desionizada para eliminar el alcohol polivinílico residual, y las microesferas se liofilizaron.
Ejemplo 7-1: Fabricación de microesferas de deslorelina
4,4 g de Resomer R203H (fabricante: Evonik, Alemania) y 0,6 g de acetato de deslorelina (fabricante: Chengdu Kaijie Biopharm, China) se mezclaron con 15,7 g de diclorometano (fabricante: JT Baker, EE. UU.) y 7,5 g de alcohol metílico (fabricado por Sigma Aldrich, EE. UU.) para formar una fase dispersa. La fase dispersa se disolvió suficientemente agitando durante 30 minutos o más, y entonces se usó.
La fase continua fue una disolución acuosa de alcohol polivinílico al 1% (p/v) (viscosidad: 4,8-5,8 mPas). Se conectó un recipiente que incluía 790 ml de la fase continua a un aparato de emulsionamiento equipado con una membrana
que tenía poros de 30 pm de diámetro, mientras se inyectaba simultáneamente la fase dispersa preparada para producir las microesferas en suspensión a un caudal de 5 ml por minuto. La suspensión de microesferas resultante se colocó en una vasija de preparación, y se agitó a 180 rpm.
La temperatura del aparato de emulsionamiento de membrana y de la vasija de preparación se mantuvo a 25°C. Cuando se completó la inyección de la fase dispersa, la agitación se mantuvo durante 30 minutos. En esta etapa, se eliminaron 785 ml de la fase continua, al tiempo que se dejaron 5 ml de fase continua, y se añadió la misma cantidad de nueva disolución de fase continua. El disolvente orgánico se eliminó mientras que se mantiene la temperatura a 40°C durante 3 horas. Después de eliminar el disolvente orgánico, la temperatura de la suspensión de microesferas se redujo a 25°C. La suspensión de microesferas se lavó tres veces con agua desionizada para eliminar el alcohol polivinílico residual, y las microesferas se liofilizaron.
Ejemplo 7-2: Preparación de una microesfera de bupivacaína
La fase dispersa se preparó mezclando 4,5 g de Resomer R203H (fabricante: Evonik, Alemania), un polímero biocompatible, y 0,5 g de una base de bupivacaína (fabricante: Dishman, India) en 12 g de diclorometano (fabricante: J.T Baker, EE. UU.). La fase dispersa se disolvió suficientemente agitando durante 30 minutos o más, y entonces se usó.
Se usó una disolución acuosa de alcohol polivinílico al 1% (p/v) (viscosidad: 4,8-5,8 mPas) como fase continua. Se vertieron 600 ml de la fase continua en el vasija de preparación equipada con mezclador de alta velocidad (L4RT, Silverson, UK), y se agitó a 4000 rpm, mientras se inyectaba la fase dispersa a un caudal de 12 ml por minuto y se mantenía la temperatura de la vasija de preparación a 25°C. Cuando se completó la inyección de la fase dispersa, la suspensión en el vasija de preparación se mantuvo en agitación a 25°C durante 30 minutos.
Se eliminaron 595 ml de la fase continua, al tiempo que se dejaron 5 ml de fase continua, y se añadió la misma cantidad de nueva disolución de fase continua. El disolvente orgánico se eliminó mientras que se mantiene la temperatura a 45°C durante 3 horas. Después de eliminar el disolvente orgánico, la temperatura de la suspensión de microesferas se redujo a 25°C. La suspensión de microesferas se lavó tres veces con agua desionizada para eliminar el alcohol polivinílico residual, y las microesferas se liofilizaron.
Ejemplo Comparativo 1: Producción de microesferas preparadas usando diferentes tiempos de intercambio de fase continua
La fase dispersa se preparó mezclando 3,5 g de un polímero biocompatible, Resomer R203H (fabricante: Evonik, Alemania) y 1,5 g de base de donepezilo (fabricante: Neuland Laboratories, India) con 9,2 g de diclorometano (fabricante: J.T Baker, EE. UU.). La fase dispersa se disolvió suficientemente agitando durante 30 minutos o más, y entonces se usó.
Se usó una disolución acuosa de alcohol polivinílico al 1% (p/v) (viscosidad: 4,8-5,8 mPas) como fase continua. Se conectó un recipiente que incluía 460 ml de la fase continua a un aparato de emulsionamiento equipado con una membrana que tenía poros de 40 pm de diámetro, mientras se inyectaba simultáneamente la fase dispersa preparada para producir las microesferas en suspensión. La suspensión de microesferas resultante se colocó en una vasija de preparación, y se agitó a 150 rpm.
La temperatura del aparato de emulsionamiento de membrana y de la vasija de preparación se mantuvo a 25°C. Cuando se completó la inyección de la fase dispersa, la agitación se mantuvo durante 5 minutos. En esta etapa, se eliminaron 455 ml de la fase continua, al tiempo que se dejaron 5 ml de fase continua, y se añadió la misma cantidad de nueva disolución de fase continua. El disolvente orgánico se eliminó mientras que se mantiene la temperatura a 451 °C durante 3 horas. Después de eliminar el disolvente orgánico, la temperatura de la suspensión de microesferas se redujo a 25°C. La suspensión de microesferas se lavó tres veces con agua desionizada para eliminar el alcohol polivinílico residual, y las microesferas se liofilizaron.
Ejemplo Comparativo 2: Producción de microesferas usando diferentes cantidades de intercambio de fase continua
3,5 g de un polímero biocompatible Resomer R203H (fabricante: Evonik, Alemania) y 1,5 g de base de donepezilo (fabricante: Neuland Laboratories, India) se mezclaron con 9,2 g de diclorometano (fabricante: J.T Baker, EE. UU.) para preparar una fase dispersa. La fase dispersa se disolvió suficientemente agitando durante 30 minutos o más, y entonces se usó.
Se usó una disolución acuosa de alcohol polivinílico al 1% (p/v) (viscosidad: 4,8-5,8 mPas) como fase continua. Se conectó un recipiente que incluía 460 ml de la fase continua a un aparato de emulsionamiento equipado con una membrana que tenía poros de 40 pm de diámetro, mientras se inyectaba simultáneamente la fase dispersa preparada para producir las microesferas en suspensión. La suspensión de microesferas resultante se colocó en una vasija de preparación, y se agitó a 150 rpm.
La temperatura del aparato de emulsionamiento de membrana y de la vasija de preparación se mantuvo a 252C. Cuando se completó la inyección de la fase dispersa, la agitación se mantuvo durante 30 minutos. En esta etapa, se eliminó toda la fase continua, y había 2,5 ml de fase continua (50% (v/v) de la microesfera) en la microesfera (es decir, la microesfera existe en forma hidratada con la fase continua), mientras que se añadieron 460 ml de nueva disolución de fase continua. El disolvente orgánico se eliminó mientras que se mantiene la temperatura a 45°C durante 3 horas. Después de eliminar el disolvente orgánico, la temperatura de la suspensión de microesferas se redujo a 25°C. La suspensión de microesferas se lavó tres veces con agua desionizada para eliminar el alcohol polivinílico residual, y las microesferas se liofilizaron.
Ejemplo Comparativo 2-1: Producción de microesferas usando diferentes cantidades de intercambio de fase continua
3.5 g de un polímero biocompatible Resomer R203H (fabricante: Evonik, Alemania) y 1,5 g de base de donepezilo (fabricante: Neuland Laboratories, India) se mezclaron con 9,2 g de diclorometano (fabricante: J.T Baker, EE. UU.) para formar una fase dispersa. La fase dispersa se disolvió suficientemente agitando durante 30 minutos o más, y entonces se usó. Se usó una disolución acuosa de alcohol polivinílico al 1% (p/v) (viscosidad: 4,8-5,8 mPas) como fase continua. Se conectó un recipiente que incluía 460 ml de la fase continua a un aparato de emulsionamiento equipado con una membrana que tenía poros de 40 pm de diámetro, mientras se inyectaba simultáneamente la fase dispersa preparada para producir las microesferas en suspensión. La suspensión de microesferas resultante se colocó en una vasija de preparación, y se agitó a 150 rpm.
La temperatura del aparato de emulsionamiento de membrana y de la vasija de preparación se mantuvo a 25°C. Cuando se completó la inyección de la fase dispersa, la agitación se mantuvo durante 30 minutos. En esta etapa, se eliminaron 161 ml (35% (v/v) del volumen total de la fase continua) de la fase continua, mientras se añadían 460 ml de nueva disolución de fase continua. El disolvente orgánico se eliminó mientras que se mantiene la temperatura a 45°C durante 3 horas. Después de eliminar el disolvente orgánico, la temperatura de la suspensión de microesferas se redujo a 25°C. La suspensión de microesferas se lavó tres veces con agua desionizada para eliminar el alcohol polivinílico residual, y las microesferas se liofilizaron.
Ejemplo Comparativo 3: Preparación de microesferas usando una fase continua en la que se mezcló alcohol etílico
3.5 g de un polímero biocompatible Resomer R203H (fabricante: Evonik, Alemania) y 1,5 g de base de donepezilo (fabricante: Neuland Laboratories, India) se mezclaron con 9,2 g de diclorometano (fabricante: J.T Baker, EE. UU.) para formar una fase dispersa. La fase dispersa se disolvió suficientemente agitando durante 30 minutos o más, y entonces se usó.
Se usó una disolución acuosa que comprende alcohol polivinílico al 1% (p/v) (viscosidad: 4,8-5,8 mPas) y alcohol etílico al 50% (v/v) como fase continua. Se conectó un recipiente que incluía la fase continua a un aparato de emulsionamiento equipado con una membrana que tenía poros de 40 pm de diámetro, mientras se inyectaba simultáneamente la fase dispersa preparada para producir las microesferas en suspensión. La suspensión de microesferas resultante se colocó en una vasija de preparación, y se agitó a 200 rpm.
La temperatura del aparato de emulsionamiento de membrana y de la vasija de preparación se mantuvo a 25°C. Cuando se completó la inyección de la fase dispersa, la agitación se mantuvo durante 30 minutos. En esta etapa, se eliminaron 455 ml de la fase continua, mientras se añadía la misma cantidad de una nueva disolución de fase continua. El disolvente orgánico se eliminó mientras que se mantiene la temperatura a 40°C durante 3 horas. Después de eliminar el disolvente orgánico, la temperatura de la suspensión de microesferas se redujo a 25°C. La suspensión de microesferas se lavó tres veces con agua desionizada para eliminar el alcohol polivinílico residual, y las microesferas se liofilizaron.
Ejemplo Comparativo 4: Preparación de microesferas con un procedimiento de añadir y eliminar continuamente una fase continua
La fase dispersa se preparó mezclando 3,5 g de un polímero biocompatible, Resomer R203H (fabricante: Evonik, Alemania) y 1,5 g de base de donepezilo (fabricante: Neuland Laboratories, India) con 9,2 g de diclorometano (fabricante: J.T Baker, EE. UU.). La fase dispersa se disolvió suficientemente agitando durante 30 minutos o más, y entonces se usó.
Se usó una disolución acuosa de alcohol polivinílico al 1% (p/v) (viscosidad: 4,8-5,8 mPas) como fase continua. Se vertieron 460 ml de la fase continua en el vasija de preparación equipada con mezclador de alta velocidad (L4RT, Silverson, UK), y se agitó a 2000 rpm, mientras se inyectaba la fase dispersa a un caudal de 10 ml por minuto y se mantenía la temperatura de la vasija de preparación a 25°C. Cuando se completó la inyección de la fase dispersa, la fase continua se eliminó de la emulsión a un caudal de 4,6 ml por minuto (1% (v/v) del volumen total de la fase continua), mientras se añadía una nueva fase continua a la vasija de preparación al mismo caudal de la eliminación de la fase continua durante 1 hora.
El disolvente orgánico se eliminó mientras que se mantiene la temperatura a 45°C durante 3 horas. Después de eliminar el disolvente orgánico, la temperatura de la suspensión de microesferas se redujo a 25°C. La suspensión de microesferas se lavó tres veces con agua desionizada para eliminar el alcohol polivinílico residual, y las microesferas se liofilizaron.
Ejemplo Experimental 1: Análisis morfológico de microesferas por microscopio electrónico
Este ensayo se realizó para analizar las características morfológicas de las microesferas preparadas, y el procedimiento experimental detallado es el siguiente. Se colocaron 5 mg de microesferas en un trozo de aluminio con una cinta de carbón, y se recubrieron con platino usando ION-COATER (COXEM, Corea). El trozo de aluminio se montó en un microscopio electrónico de barrido (COXEM EM-30, Corea), y se observaron las características morfológicas a un voltaje de aceleración de 15 kV.
Como se muestra en la FIG. 1A y FIG. 1B, en el Ejemplo Comparativo 1, el tiempo de intercambio de fase continua fue más rápido que el tiempo de endurecimiento de la superficie de las microesferas, y las microesferas se deformaron sin mantener la forma esférica, mientras que en el Ejemplo 1-1, la mayoría de las microesferas eran completamente esféricas. Por lo tanto, se considera que la fase continua debe reemplazarse después de más de 5 minutos, apropiadamente después de que la superficie esté curada.
Como se puede ver en la FIG. 1 (c), se confirmó que la deformación de la forma de las partículas podría ocurrir durante el procedimiento continuo de intercambio de fase cuando se eliminaron todas las fases continuas. En consecuencia, se confirmó que la forma de las partículas se conservaba eliminando una cierta cantidad de la fase continua, como en el Ejemplo 1-1, en lugar de eliminar toda la fase continua.
Como se muestra en la FIG. 1d y la FIG. 1e, en el caso en el que la fase continua contenía alcohol etílico, en particular, en el Ejemplo 5, se predijo que el alcohol etílico aumentaba la solubilidad del diclorometano para facilitar la eliminación. Por otro lado, en el Ejemplo Comparativo 3, se confirmó que cuando se añadía 50% (v/v) o más de alcohol etílico a la fase continua, la eliminación rápida del diclorometano daba como resultado la deformación de las microesferas.
Ejemplo Experimental 2: Análisis de disolvente orgánico residual
Se llevó a cabo el siguiente ensayo para confirmar la estabilidad y la estabilidad de almacenamiento de las microesferas midiendo la cantidad de disolvente residual en las microesferas preparadas mediante liofilización.
Para la determinación del diclorometano residual y acetato de etilo, se colocan 100 mg de microesferas liofilizadas en un matraz volumétrico, y se añaden 10 ml de dimetilformamida. La disolución se diluye, se transfiere a un vial de 5 ml, y se coloca en un cromatógrafo de gases equipado con un muestreador de espacio de cabeza. Y se midió el disolvente residual. La columna usada fue DB-624 (fabricada por Agilent, EE.UU.) (30 m x 0,53 mm, 3 gm), la cantidad de inyección de muestra fue 1 gl, y la temperatura del detector de ionización de llama se fijó en 250°C.
Para medir el alcohol metílico residual, se colocaron 100 mg de microesferas liofilizadas en un matraz volumétrico, se añadieron 5 ml de N-metilpirrolidona, y la disolución resultante se diluyó y se transfirió a un vial de 5 ml. El disolvente residual se midió con un cromatógrafo de gases equipado con un muestreador de espacio de cabeza. La columna usada fue DB-624 (fabricada por Agilent, EE.UU.) (30 m x 0,53 mm, 3 gm), la cantidad de inyección de muestra fue 1 gl, y la temperatura del detector de ionización de llama se fijó en 250°C.
Para medir el alcohol etílico residual, se colocan 100 mg de microesferas liofilizadas en un matraz volumétrico, se le añaden 5 ml de N-metilpirrolidona, y la disolución resultante se diluye y se transfiere a un vial de 5 ml. El disolvente residual se midió con un cromatógrafo de gases equipado con un muestreador de espacio de cabeza. La columna fue DB-624 (fabricada por Agilent, EE.UU.) (30 m x 0,53 mm, 3 gm), la cantidad de inyección de muestra fue 1 gl, y la temperatura del detector de ionización de llama se fijó en 240°C.
[Tabla 1]


Como se muestra en la Tabla 1, el Ejemplo 1, el Ejemplo 1-1 y el Ejemplo Comparativo 1 muestran el cambio en la cantidad de diclorometano residual dependiendo del tiempo de intercambio de fase continua; y después de 15 minutos del tiempo de intercambio de fase continua, la cantidad de diclorometano residual es 600 ppm o menos.
Como puede verse a partir de los resultados del Ejemplo 1-1, Ejemplo 2 y Ejemplo Comparativo 2, se confirmó que cuando la fase continua se intercambió de forma discontinua, el disolvente residual disminuyó en proporción a la cantidad de la eliminación de la fase continua. Sin embargo, como puede verse a partir del resultado del Ejemplo Comparativo 2, en el que se eliminan todas las fases continuas, las microesferas se deformaron morfológicamente al aplicar presión a las microesferas. Además, como en el Ejemplo Comparativo 2-1, en el que el volumen de fase continua a intercambiar no fue suficiente, el diclorometano no se eliminó de forma eficaz, y quedó una gran cantidad de diclorometano. Es preferible mantener la cantidad residual de diclorometano en las microesferas a 1000 ppm o menos para mejorar la estabilidad durante el almacenamiento. Preferiblemente, el diclorometano residual se mantiene a 600 ppm o menos.
Se ha encontrado que cuando se usa una fase continua que comprende alcohol etílico como el Ejemplo 5 y el Ejemplo Comparativo 3, se facilita la eliminación del diclorometano residual. Como se muestra en la Tabla 1, Ejemplo 6, Ejemplo 6-1, Ejemplo 6-2, Ejemplo 6-3, en los que la fase continua se añadió y eliminó continuamente, el diclorometano residual se redujo durante un período corto de tiempo, y parecía ser ventajoso para la eliminación del diclorometano residual. Sin embargo, se confirmó que cuando la cantidad de la fase continua a intercambiar fue menor que 1% (v/v) de la fase continua total como en el Ejemplo Comparativo 4, no era eficaz para eliminar el diclorometano.
Como se muestra en la Tabla 1, se confirmó que los disolventes residuales se eliminaron de manera eficiente incluso cuando se usaron disolventes orgánicos distintos al diclorometano como Ejemplo 3, Ejemplo 5, Ejemplo 7-1, y Ejemplo Comparativo 3.
Ejemplo Experimental 3: Análisis de estabilidad de almacenamiento acelerado de microesferas de la presente invención
Para confirmar la estabilidad de almacenamiento de las microesferas preparadas comparando el comportamiento de las microesferas almacenadas en las duras condiciones de almacenamiento, se realizaron los siguientes ensayos.
Cada 1 g de las microesferas del Ejemplo 1-1 de acuerdo con la presente invención y las microesferas del Ejemplo Comparativo 1 se colocaron en viales de 5 ml y se almacenaron en una incubadora a 40°C durante 14 días, y entonces se midió el tamaño de partícula promedio con un analizador de tamaño de partículas (Microtrac Bluewave, Japón). Se mezclaron 50 mg de las microesferas sacadas de la incubadora con 1 ml de agua desionizada, se mezcló con un mezclador de vórtice durante 20 segundos, y se dispersó en un generador ultrasónico durante 1 minuto. La dispersión de microesferas resultante se colocó en un analizador de tamaño de partículas, y se midió durante 20 segundos.
El valor de amplitud como índice de la uniformidad del tamaño de partícula se obtuvo mediante la siguiente ecuación (1).
[Ecuación 1]
Valor de amplitud = (Dv> °-9 " Dv> °-1) / Dv> 0.5
[Tabla 2]
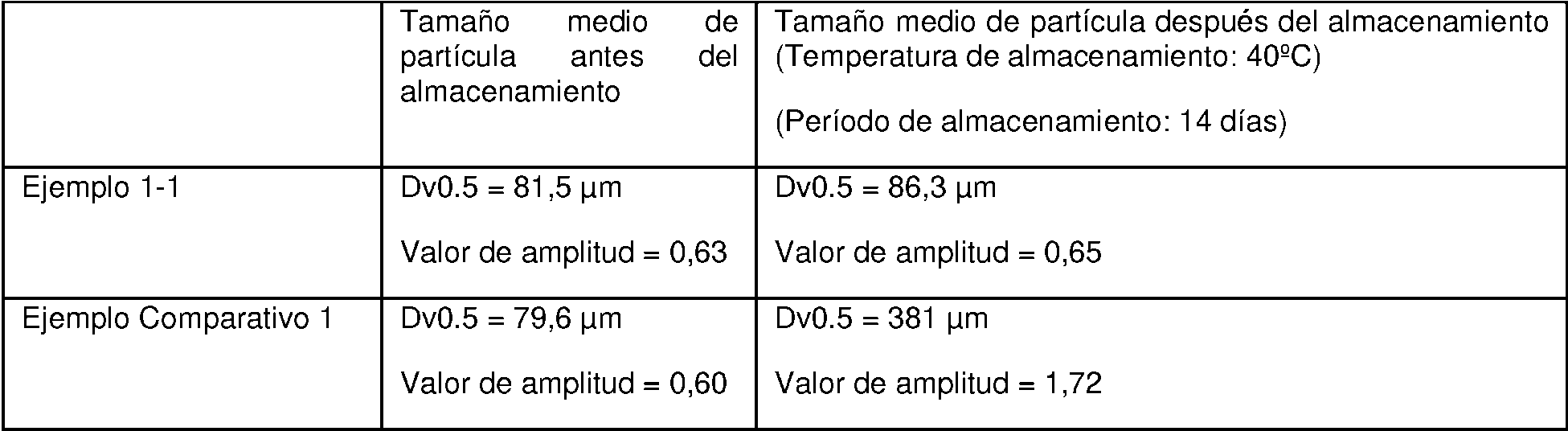
Como se muestra en la Tabla 2, se confirmó que el tamaño de partícula del Ejemplo 1-1 mantenía un nivel similar de tamaño de partícula después del almacenamiento a 40°C.
Por otro lado, las microesferas del Ejemplo Comparativo 1 se curaron a una temperatura de almacenamiento de 40°C, y todas las microesferas apenas se endurecieron en su conjunto. Después de medir el tamaño de partícula de las microesferas suspendidas, se confirma que el tamaño medio de partícula de las microesferas aumentó significativamente. Como resultado, se considera que el diclorometano residual en las microesferas del Ejemplo 1 -1 y el Ejemplo Comparativo 1 es una causa del deterioro de la estabilidad al almacenamiento de las microesferas.
Además, el valor de amplitud del Ejemplo 1-1 después del almacenamiento, en el que el disolvente residual se eliminó de manera eficiente, mostró un aumento de solo 3% en comparación con el anterior al almacenamiento, mientras que en el Ejemplo Comparativo 1, en el que la eliminación del disolvente residual no fue eficiente, el valor de amplitud muestra un aumento del 286% en comparación con los anteriores al almacenamiento. De este modo, se consideró que el disolvente residual había afectado negativamente a las características morfológicas de las microesferas. Por lo tanto, la microesfera con estabilidad de almacenamiento de la presente invención muestra 100% o menos de cambio del valor de intervalo basado en el valor de intervalo inicial, preferiblemente 50%, a una temperatura de almacenamiento de 40°C durante 14 días.
Claims (12)
1. Un método para preparar microesferas biodegradables, que comprende:
(a) formar una disolución de polímero biodegradable disolviendo un polímero biodegradable solo; o un polímero biodegradable y un fármaco en un disolvente orgánico;
(b) mezclar uniformemente la disolución de polímero biodegradable preparada en la etapa (a) con una disolución acuosa que contiene un tensioactivo, para formar una emulsión que contiene la disolución de polímero biodegradable como fase dispersa y una disolución acuosa que contiene el tensioactivo como una fase continua;
(c) extraer y evaporar el disolvente orgánico de la fase dispersa en la emulsión de la etapa (b) a la fase continua para producir microesferas, en el que se elimina una parte de la fase continua que contiene el disolvente orgánico extraído, mientras que se suministra una nueva fase continua; y
(d) recuperar las microesferas de la fase continua que contiene las microesferas producidas de la etapa (c), en el que la parte de la fase continua en la etapa (c) se elimina de forma discontinua o continua, y
(i) cuando la parte de la fase continua en la etapa (c) se elimina de forma discontinua, la fase continua eliminada es una relación de volumen de 40% (v/v) a 99% (v/v), basada en el volumen total de la fase continua, o es una relación en peso de una fase continua restante, excepto la relación en peso puntual de la fase continua de la micropartícula en la etapa (c), o
(ii) cuando se elimina continuamente la parte de la fase continua en la etapa (c), la tasa de eliminación de la fase continua está en el intervalo de 2% (v/v) a 200% (v/v)/min, basado en el volumen total de la fase continua.
2. El método de la reivindicación 1, en el que el polímero biodegradable es uno o más seleccionados del grupo que consiste en polilactida, poliglicolida, poli(lactida-co-glicolida), poli(lactida-co-glicolida) glucosa, y policaprolactona.
3. El método de la reivindicación 2, en el que la relación molar de ácido láctico a ácido glicólico en la poli(lactida-coglicolida) es 99:1 a 50:50.
4. El método de la reivindicación 1, en el que el disolvente orgánico es uno o más seleccionados del grupo que consiste en diclorometano, cloroformo, acetato de etilo, acetona, acetonitrilo, dimetilsulfóxido, dimetilformamida, metiletilcetona, ácido acético, alcohol metílico, alcohol etílico, alcohol propílico, y una mezcla de los mismos.
5. El método según la reivindicación 1, en el que la disolución acuosa que contiene el tensioactivo de la etapa (b) comprende
(i) agua, o un disolvente mixto que comprende agua y uno o más disolventes orgánicos seleccionados del grupo que consiste en alcohol metílico, alcohol etílico, alcohol propílico, y acetato de etilo como disolvente; y (ii) un tensioactivo.
6. El método según la reivindicación 1, en el que el tensioactivo en la etapa (b) es uno o más seleccionados del grupo que consiste en metilcelulosa, polivinilpirrolidona, carboximetilcelulosa, lecitina, gelatina, alcohol polivinílico, éster de ácido graso con polioxietilensorbitán, derivados de aceite de ricino polioxietilenados, y una mezcla de los mismos.
7. El método según la reivindicación 1, en el que el tensioactivo de la etapa (b) está en una cantidad de 0,01% (p/v) a 20% (p/v), basado en el volumen total de la disolución acuosa que contiene el tensioactivo.
8. El método según la reivindicación 1, en el que la nueva disolución acuosa que contiene un tensioactivo en la etapa (c) se suministra de forma discontinua o continua.
9. El método según la reivindicación 1, en el que la etapa de eliminar una parte de la fase continua en la etapa (c) y suministrar una nueva disolución acuosa que contiene un tensioactivo comienza 10 a 60 minutos a partir del inicio del endurecimiento de la superficie de la microesfera.
10. El método de la reivindicación 1, en el que la nueva disolución acuosa que comprende el tensioactivo de la etapa (c) comprende (i)
(i) agua, o un disolvente mixto que comprende agua y uno o más disolventes orgánicos seleccionados de un grupo que consiste en alcohol metílico, alcohol etílico, alcohol propílico, y acetato de etilo como disolvente; y (ii) un tensioactivo.
11. El método de la reivindicación 1, en el que la eliminación de la fase continua y el suministro de una nueva disolución acuosa que contiene tensioactivo en la etapa (c) proceden de forma simultánea o asíncrona.
12. El método de la reivindicación 1, en el que el fármaco se selecciona del grupo que consiste en donepezilo, memantina, rivastigmina, entecavir, lamivudina, rotigotina, ropinirol, bupivacaína, ropivacaína, meloxicam, buprenorfina, fentanilo, granisetrón, triamcinolona, citarabina, carmustina, tamsulosina, polmacoxib, testosterona, estradiol, risperidona, paliperidona, olanzapina, aripiprazol, goserelina, leuprolida, triptorelina, buserelina, nafarelina, deslorelina, octreotida, pasireotida, lanreotida, vapreotida, exenatida, liraglutida, lixisenatida, semaglutida, y sales de los mismos.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| KR20170163105 | 2017-11-30 | ||
| PCT/KR2018/015121 WO2019108030A1 (ko) | 2017-11-30 | 2018-11-30 | 안정성 및 저장 안정성이 향상된 생분해성 미립구의 제조방법 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2915502T3 true ES2915502T3 (es) | 2022-06-22 |
Family
ID=66665152
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES18883786T Active ES2915502T3 (es) | 2017-11-30 | 2018-11-30 | Procedimiento para preparar microesferas biodegradables con estabilidad mejorada y estabilidad al almacenamiento |
Country Status (12)
| Country | Link |
|---|---|
| US (1) | US11311854B2 (es) |
| EP (1) | EP3586828B1 (es) |
| JP (2) | JP2020521805A (es) |
| KR (1) | KR102047983B1 (es) |
| CN (1) | CN110709067B (es) |
| AU (1) | AU2018271381B2 (es) |
| CA (1) | CA3058743C (es) |
| ES (1) | ES2915502T3 (es) |
| MX (1) | MX392548B (es) |
| RU (1) | RU2722358C1 (es) |
| WO (1) | WO2019108030A1 (es) |
| ZA (1) | ZA201808116B (es) |
Families Citing this family (33)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR102181231B1 (ko) * | 2018-11-22 | 2020-11-20 | 주식회사 메디포럼제약 | 로티고틴 함유 고분자 미립자의 제조방법 |
| KR102249104B1 (ko) * | 2019-04-30 | 2021-05-07 | (주)인벤티지랩 | 데슬로렐린을 포함하는 서방성 마이크로 입자 및 이의 제조 방법 |
| MX2022000489A (es) * | 2019-07-12 | 2022-02-03 | G2Gbio Inc | Formulacion de larga duracion que contiene rivastigmina y metodo para prepararla. |
| KR102212717B1 (ko) * | 2019-11-08 | 2021-02-08 | 환인제약 주식회사 | 지속 방출을 위한 마이크로스피어 및 이의 제조 방법 |
| KR102212722B1 (ko) * | 2019-12-23 | 2021-02-08 | 환인제약 주식회사 | 로피니롤을 포함하는 마이크로스피어 및 이를 함유하는 주사제 조성물 |
| EP4104820A4 (en) * | 2020-02-14 | 2024-04-17 | G2GBIO, Inc. | Pharmaceutical composition comprising sustained-release microspheres including glp-1 analogue or pharmaceutically acceptable salt thereof |
| US20240033221A1 (en) * | 2020-04-22 | 2024-02-01 | Scienture, Inc. | Long-acting bupivacaine microsphere formulations |
| KR102583029B1 (ko) * | 2020-05-28 | 2023-09-26 | 주식회사 아울바이오 | 글루카곤 유사 펩타이드 1 작용제 함유 제어방출 미립구 및 이의 제조방법 |
| KR20210158232A (ko) * | 2020-06-23 | 2021-12-30 | 주식회사 아울바이오 | 치매치료를 위한 장기지속형 주사제 |
| CN111973561B (zh) * | 2020-08-31 | 2022-06-07 | 常州药物研究所有限公司 | 去除聚合物微球溶剂中有机溶剂残留方法 |
| CN114225104B (zh) * | 2020-09-09 | 2024-09-27 | 渼颜空间(河北)生物科技有限公司 | 一种可生物降解的注射填充剂微球及其制备方法和其应用 |
| CN112156170B (zh) * | 2020-11-03 | 2022-10-21 | 北京康欣东弘医药科技有限公司 | 可供皮下注射的曲普瑞林缓释微球及其制备方法和用途 |
| KR102266385B1 (ko) * | 2021-01-22 | 2021-06-21 | 주식회사 울트라브이 | 필러용 생분해성 고분자 미세입자, 그 제조방법 및 이를 포함하는 동결건조체 및 필러용 주사제 |
| WO2022270956A1 (ko) * | 2021-06-23 | 2022-12-29 | 주식회사 펩트론 | 세마글루타이드 또는 이의 약학적으로 허용가능한 염을 포함하는 서방형 제제 조성물 |
| US11865213B2 (en) | 2021-07-05 | 2024-01-09 | Mapi Pharma Ltd. | Semaglutide depot systems and use thereof |
| US12514908B2 (en) | 2021-07-05 | 2026-01-06 | Mapi Pharma Ltd. | Semaglutide depot systems and use thereof |
| KR20230036886A (ko) * | 2021-09-08 | 2023-03-15 | 주식회사 아울바이오 | 생분해성 고분자를 이용한 서방형 미립구 및 이의 제조방법 |
| KR102567292B1 (ko) * | 2021-11-05 | 2023-08-17 | 주식회사 지투지바이오 | 덱사메타손을 포함하는 서방형 주사제제 및 그 제조방법 |
| EP4427740A4 (en) * | 2021-11-05 | 2025-10-22 | G2Gbio Inc | PHARMACEUTICAL KIT FOR PARENTERAL CO-ADMINISTRATION |
| CN114081867B (zh) * | 2021-11-08 | 2023-12-19 | 中国药科大学 | 一种提高微球载药量的方法 |
| CA3236685A1 (en) * | 2021-11-18 | 2023-05-25 | Inventage Lab Inc. | Method for preparing microparticles containing poorly soluble drugs |
| KR102707065B1 (ko) * | 2022-03-08 | 2024-09-19 | 동국제약 주식회사 | 코아세르베이션법을 이용하여 현탁성이 개선된 미립구의 제조방법 |
| KR102764486B1 (ko) * | 2022-07-21 | 2025-02-07 | 주식회사 지투지바이오 | 5-알파 환원효소 저해제를 포함하는 서방성 제제 |
| KR102700719B1 (ko) * | 2022-11-07 | 2024-08-29 | 주식회사 지투지바이오 | 덱사메타손 아세테이트를 포함하는 서방형 주사제제 및 그 제조방법 |
| WO2024122991A1 (ko) * | 2022-12-05 | 2024-06-13 | (주)인벤티지랩 | 복합 수상 용액를 이용한 마이크로 입자에 잔류하는 유기 용매의 제거 방법 |
| KR102527600B1 (ko) * | 2022-12-21 | 2023-05-02 | 주식회사 이오바이오 | 의료용 생분해성 고분자를 이용한 필러용 미립구의 제조방법 |
| CN117554505A (zh) * | 2023-03-08 | 2024-02-13 | 西岭(镇江)医疗科技有限公司 | 一种三氯甲烷试剂残留检测方法 |
| AR133300A1 (es) * | 2023-07-20 | 2025-09-17 | Ferring Bv | Micropartículas cargadas con fármaco de liberación prolongada |
| KR102743491B1 (ko) * | 2023-08-01 | 2024-12-17 | (주)인벤티지랩 | 세마글루타이드 또는 이의 약학적으로 허용 가능한 염을 포함하는 마이크로 입자 및 이의 제조 방법 |
| WO2025135778A1 (ko) * | 2023-12-18 | 2025-06-26 | 주식회사 포스테라헬스사이언스 | 약물의 특성에 따른 활성형 미립자의 제조 방법, 이의 제조 방법으로 제조된 활성형 미립자 및 상기 활성형 미립자를 이용한 약물을 포함하는 장기 지속 제형의 제조 방법 |
| KR20250161987A (ko) * | 2024-05-10 | 2025-11-18 | 주식회사 대웅제약 | 로피바케인 및 비스테로이드성 항염증제를 포함하는 서방형 미립구 및 이의 제조방법 |
| KR20260002273A (ko) * | 2024-06-28 | 2026-01-06 | 주식회사 지투지바이오 | 고생산량을 갖는 미립구의 생산방법 |
| CN120267621B (zh) * | 2025-04-16 | 2026-03-10 | 鲁南制药集团股份有限公司 | 一种多奈哌齐缓释制剂及其制备方法 |
Family Cites Families (27)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5945126A (en) | 1997-02-13 | 1999-08-31 | Oakwood Laboratories L.L.C. | Continuous microsphere process |
| EP1277465A1 (en) * | 2000-04-24 | 2003-01-22 | Tanabe Seiyaku Co., Ltd. | Process for producing microsphere |
| CA2405205C (en) | 2000-04-28 | 2007-11-20 | Tanabe Seiyaku Co., Ltd. | Method for preparing microspheres |
| EP1343478B1 (en) | 2000-12-21 | 2007-10-10 | Alrise Biosystems GmbH | Induced phase transition method for the production of microparticles containing hydrophilic active agents |
| EP1629833A4 (en) | 2003-06-03 | 2010-04-21 | Santen Pharmaceutical Co Ltd | METHOD FOR PRODUCING A MICROPARTICLE |
| KR100722607B1 (ko) * | 2006-05-11 | 2007-05-28 | 주식회사 펩트론 | 분산성 및 주사 투여능이 향상된 서방성 미립구의 제조방법 |
| RU2326655C1 (ru) | 2006-11-09 | 2008-06-20 | Федеральное государственное учреждение "48 Центральный научно-исследовательский институт Министерства обороны Российской Федерации" | Способ инкапсулирования белоксодержащих веществ в микросферы из сополимера полилактид-полигликолид |
| NZ580700A (en) * | 2007-04-19 | 2012-01-12 | Dong A Pharm Co Ltd | A biodegradable microsphere composition suitable for the controlled release of glucose controlling peptide and formulation thereof |
| ES2718612T3 (es) * | 2007-12-20 | 2019-07-03 | Evonik Corp | Procedimiento para preparar micropartículas que tienen un bajo volumen de disolvente residual |
| US8034382B2 (en) * | 2008-07-31 | 2011-10-11 | The Board Of Trustees Of The University Of Arkansas | Preparation method for biodegradable micro-particles containing drugs |
| KR101113044B1 (ko) * | 2008-08-29 | 2012-02-27 | 동국제약 주식회사 | 용매교류증발법에 의한 서방출성 미립구의 제조방법 |
| US20100086597A1 (en) * | 2008-10-06 | 2010-04-08 | Oakwood Laboratories LLC | Microspheres for the sustained release of octreotide with a low initial burst |
| CN103386118B (zh) * | 2008-12-23 | 2015-04-08 | 成都地奥九泓制药厂 | 控制胸腺肽α1微球中有机溶剂残留的方法 |
| KR101105292B1 (ko) | 2009-06-05 | 2012-01-17 | 주식회사 리젠 바이오텍 | 생분해성 고분자 미세입자와 그의 제조방법 |
| CN101693179B (zh) * | 2009-11-06 | 2012-02-15 | 上海交通大学 | 可生物降解纳米微球的制备方法 |
| KR101149809B1 (ko) * | 2010-09-09 | 2012-05-24 | 한국과학기술원 | 유아용 온도 패치 |
| CN102670518B (zh) * | 2011-03-14 | 2014-08-20 | 齐鲁制药有限公司 | 一种难溶性药物球形颗粒的制备方法 |
| WO2013005094A1 (en) | 2011-07-05 | 2013-01-10 | Torrent Pharmaceuticals Ltd | Acid addition salt of donepezil and pharmaceutical composition thereof |
| CN102516565B (zh) * | 2011-12-13 | 2013-04-17 | 扬州大学 | 一种制备聚乳酸纳微米球的方法 |
| CN102440964A (zh) * | 2011-12-16 | 2012-05-09 | 深圳市健元医药科技有限公司 | 戈舍瑞林缓释微球制剂及其制备方法 |
| CN103462901B (zh) * | 2012-06-08 | 2016-03-09 | 浙江海正药业股份有限公司 | 一种控制聚合物微球制剂中二氯甲烷残留的方法 |
| KR101486132B1 (ko) * | 2013-03-20 | 2015-01-23 | 씨제이헬스케어 주식회사 | 졸-겔 변환 특성을 갖는 고분자를 이용한 미립구의 제조방법 및 이로부터 제조된 미립구 |
| KR101811797B1 (ko) | 2013-04-03 | 2017-12-22 | 동국제약 주식회사 | 도네페질을 포함하는 비경구투여용 약제학적 조성물 |
| RU2658004C2 (ru) * | 2013-06-20 | 2018-06-19 | Фарматен С.А. | Получение полилактидно-полигликолидных микрочастиц, характеризующихся сигмоидальным профилем высвобождения |
| BR112016002342A8 (pt) | 2013-08-06 | 2018-01-23 | Dong Kook Pharm Co Ltd | microesfera entecavir, método para preparação de uma microesfera entecavir, e, composição farmacêutica |
| KR101738127B1 (ko) * | 2014-08-08 | 2017-05-22 | (주)비씨월드제약 | 약물 함유 서방성 미립자의 제조 방법 |
| KR101583351B1 (ko) | 2014-11-28 | 2016-01-07 | 동국제약 주식회사 | 초기 방출 억제 및 잔류용매 제거율을 향상시킨 서방출성 미립구 및 이의 제조방법 |
-
2018
- 2018-11-29 KR KR1020180151359A patent/KR102047983B1/ko active Active
- 2018-11-30 ZA ZA2018/08116A patent/ZA201808116B/en unknown
- 2018-11-30 US US16/495,825 patent/US11311854B2/en active Active
- 2018-11-30 CN CN201880037391.0A patent/CN110709067B/zh active Active
- 2018-11-30 AU AU2018271381A patent/AU2018271381B2/en active Active
- 2018-11-30 CA CA3058743A patent/CA3058743C/en active Active
- 2018-11-30 JP JP2019566734A patent/JP2020521805A/ja active Pending
- 2018-11-30 EP EP18883786.8A patent/EP3586828B1/en active Active
- 2018-11-30 ES ES18883786T patent/ES2915502T3/es active Active
- 2018-11-30 WO PCT/KR2018/015121 patent/WO2019108030A1/ko not_active Ceased
- 2018-11-30 MX MX2019012021A patent/MX392548B/es unknown
- 2018-11-30 RU RU2019132377A patent/RU2722358C1/ru active
-
2021
- 2021-11-08 JP JP2021181687A patent/JP7468909B2/ja active Active
Also Published As
| Publication number | Publication date |
|---|---|
| EP3586828A1 (en) | 2020-01-01 |
| CA3058743A1 (en) | 2019-06-06 |
| RU2722358C1 (ru) | 2020-05-29 |
| KR20190064509A (ko) | 2019-06-10 |
| US20200298196A1 (en) | 2020-09-24 |
| KR102047983B1 (ko) | 2019-11-22 |
| ZA201808116B (en) | 2019-08-28 |
| CN110709067B (zh) | 2022-10-14 |
| CN110709067A (zh) | 2020-01-17 |
| AU2018271381A1 (en) | 2019-06-13 |
| BR112019021791A2 (pt) | 2020-05-05 |
| US11311854B2 (en) | 2022-04-26 |
| EP3586828A4 (en) | 2021-01-13 |
| WO2019108030A1 (ko) | 2019-06-06 |
| EP3586828B1 (en) | 2022-03-02 |
| JP2022023219A (ja) | 2022-02-07 |
| JP7468909B2 (ja) | 2024-04-16 |
| JP2020521805A (ja) | 2020-07-27 |
| CA3058743C (en) | 2022-11-29 |
| MX392548B (es) | 2025-03-24 |
| AU2018271381B2 (en) | 2020-04-09 |
| MX2019012021A (es) | 2019-11-18 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2915502T3 (es) | Procedimiento para preparar microesferas biodegradables con estabilidad mejorada y estabilidad al almacenamiento | |
| ES2391315T5 (es) | Método para preparar micropartículas que tienen un peso molecular polimérico seleccionado | |
| Lu et al. | Microparticles produced by the hydrogel template method for sustained drug delivery | |
| JP7437074B2 (ja) | リバスチグミンを含む長期持続型製剤およびその製造方法 | |
| ES2284776T3 (es) | Microparticulas. | |
| JP5851518B2 (ja) | 生理活性ペプチドを含む微小粒子及びその製造方法、及びそれらを含む薬剤学的組成物 | |
| Bain et al. | Solvent influence on spray-dried biodegradable microspheres | |
| JP2020524171A (ja) | ドネペジルを含有する徐放性注射製剤およびその製造方法 | |
| CN113473973A (zh) | 含有地洛瑞林的缓释注射剂及其制备方法 | |
| CN106474070B (zh) | 一种克服停滞期、恒速释放疏水性药物的微球及制备方法 | |
| JP6249584B2 (ja) | 薬物含有徐放性微粒子の製造方法 | |
| KR102427305B1 (ko) | 국소 투여용 이중 봉입 생분해성 고분자 미립구의 제조방법 및 이를 포함하는 주사제 | |
| KR101831417B1 (ko) | 자발적 공극 폐쇄 기능성을 갖는 고분자 미립구 및 이의 제조방법 | |
| KR20200127964A (ko) | 난용성 약물 서방출용 미립구의 제조방법 | |
| PT996426E (pt) | Sistema multifásico | |
| Sohier et al. | Release of small water-soluble drugs from multiblock copolymer microspheres: a feasibility study | |
| BR112019021791B1 (pt) | Método para preparar microesferas biodegradáveis tendo melhor estabilidade e estabilidade de armazenamento | |
| Zhang | Biodegradable microparticles and in situ forming implants/microparticles containing drugs in different physical states | |
| JP2025523523A (ja) | 薬物とパモ酸を含有する徐放性微粒球 | |
| BR112019021366B1 (pt) | Preparação de injeção de liberação sustentada que contém donepezil e método de preparação do mesmo |