ES2925198T3 - Amidas de pirrolidinas sustituidas I - Google Patents
Amidas de pirrolidinas sustituidas I Download PDFInfo
- Publication number
- ES2925198T3 ES2925198T3 ES18816101T ES18816101T ES2925198T3 ES 2925198 T3 ES2925198 T3 ES 2925198T3 ES 18816101 T ES18816101 T ES 18816101T ES 18816101 T ES18816101 T ES 18816101T ES 2925198 T3 ES2925198 T3 ES 2925198T3
- Authority
- ES
- Spain
- Prior art keywords
- alkyl
- oxo
- methyl
- mmol
- pyridyl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- -1 pyrrolidine amides Chemical class 0.000 title claims description 117
- RWRDLPDLKQPQOW-UHFFFAOYSA-N tetrahydropyrrole Natural products C1CCNC1 RWRDLPDLKQPQOW-UHFFFAOYSA-N 0.000 title description 6
- 150000001875 compounds Chemical class 0.000 claims abstract description 103
- 238000011282 treatment Methods 0.000 claims abstract description 13
- 238000011321 prophylaxis Methods 0.000 claims abstract description 9
- 125000000592 heterocycloalkyl group Chemical group 0.000 claims description 84
- 125000001424 substituent group Chemical group 0.000 claims description 50
- QLNJFJADRCOGBJ-UHFFFAOYSA-N propionamide Chemical compound CCC(N)=O QLNJFJADRCOGBJ-UHFFFAOYSA-N 0.000 claims description 45
- 125000006570 (C5-C6) heteroaryl group Chemical group 0.000 claims description 44
- 125000003118 aryl group Chemical group 0.000 claims description 26
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 26
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims description 23
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 21
- 125000004537 indazol-5-yl group Chemical group N1N=CC2=CC(=CC=C12)* 0.000 claims description 19
- 125000001072 heteroaryl group Chemical group 0.000 claims description 18
- 229920006395 saturated elastomer Polymers 0.000 claims description 17
- 201000010099 disease Diseases 0.000 claims description 15
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 claims description 14
- 150000003839 salts Chemical class 0.000 claims description 10
- AIMMVWOEOZMVMS-UHFFFAOYSA-N cyclopropanecarboxamide Chemical compound NC(=O)C1CC1 AIMMVWOEOZMVMS-UHFFFAOYSA-N 0.000 claims description 9
- 201000008482 osteoarthritis Diseases 0.000 claims description 8
- 208000006673 asthma Diseases 0.000 claims description 7
- 125000003226 pyrazolyl group Chemical group 0.000 claims description 7
- 239000002552 dosage form Substances 0.000 claims description 6
- 125000004076 pyridyl group Chemical group 0.000 claims description 6
- 206010039073 rheumatoid arthritis Diseases 0.000 claims description 6
- 206010009900 Colitis ulcerative Diseases 0.000 claims description 5
- 208000011231 Crohn disease Diseases 0.000 claims description 5
- 201000006704 Ulcerative Colitis Diseases 0.000 claims description 5
- 125000001786 isothiazolyl group Chemical group 0.000 claims description 5
- 125000000842 isoxazolyl group Chemical group 0.000 claims description 5
- 125000002971 oxazolyl group Chemical group 0.000 claims description 5
- 125000000335 thiazolyl group Chemical group 0.000 claims description 5
- 125000004506 1,2,5-oxadiazolyl group Chemical group 0.000 claims description 4
- 125000004105 2-pyridyl group Chemical group N1=C([*])C([H])=C([H])C([H])=C1[H] 0.000 claims description 4
- 206010001052 Acute respiratory distress syndrome Diseases 0.000 claims description 4
- 208000006545 Chronic Obstructive Pulmonary Disease Diseases 0.000 claims description 4
- 201000003883 Cystic fibrosis Diseases 0.000 claims description 4
- 208000007465 Giant cell arteritis Diseases 0.000 claims description 4
- 206010061218 Inflammation Diseases 0.000 claims description 4
- 208000007048 Polymyalgia Rheumatica Diseases 0.000 claims description 4
- 208000013616 Respiratory Distress Syndrome Diseases 0.000 claims description 4
- 201000000028 adult respiratory distress syndrome Diseases 0.000 claims description 4
- 125000001028 difluoromethyl group Chemical group [H]C(F)(F)* 0.000 claims description 4
- 230000004054 inflammatory process Effects 0.000 claims description 4
- 125000003386 piperidinyl group Chemical group 0.000 claims description 4
- 206010043207 temporal arteritis Diseases 0.000 claims description 4
- VSPGWMCXTHEHDO-BKMJKUGQSA-N CC1(CC1)C(=O)N[C@@H]1[C@H](N(C(C1)=O)C=1C=C2C=NN(C2=CC=1)C1=CN(C(C=C1)=O)C)C1=CC=CC=C1 Chemical compound CC1(CC1)C(=O)N[C@@H]1[C@H](N(C(C1)=O)C=1C=C2C=NN(C2=CC=1)C1=CN(C(C=C1)=O)C)C1=CC=CC=C1 VSPGWMCXTHEHDO-BKMJKUGQSA-N 0.000 claims description 3
- WVDSTTFBHSJDPC-HFZDXXHNSA-N CN1C=C(C=CC1=O)N1N=CC2=CC(=CC=C12)N1[C@@H]([C@H](CC1=O)NC(=O)C=1N=CSC=1)C1=CC=CC=C1 Chemical compound CN1C=C(C=CC1=O)N1N=CC2=CC(=CC=C12)N1[C@@H]([C@H](CC1=O)NC(=O)C=1N=CSC=1)C1=CC=CC=C1 WVDSTTFBHSJDPC-HFZDXXHNSA-N 0.000 claims description 3
- HEYUMVVDUHJXME-SQJMNOBHSA-N CN1C=C(C=CC1=O)N1N=CC2=CC(=CC=C12)N1[C@@H]([C@H](CC1=O)NC(=O)C=1OC=CN=1)C1=CC=CC=C1 Chemical compound CN1C=C(C=CC1=O)N1N=CC2=CC(=CC=C12)N1[C@@H]([C@H](CC1=O)NC(=O)C=1OC=CN=1)C1=CC=CC=C1 HEYUMVVDUHJXME-SQJMNOBHSA-N 0.000 claims description 3
- 206010013801 Duchenne Muscular Dystrophy Diseases 0.000 claims description 3
- LEDGCBVULGYNJK-NBGIEHNGSA-N FC(C(=O)N[C@@H]1[C@H](N(C(C1)=O)C=1C=C2C=NN(C2=CC=1)C1=CN(C(C=C1)=O)C)C1=CC(=C(C=C1)F)OC)(C)F Chemical compound FC(C(=O)N[C@@H]1[C@H](N(C(C1)=O)C=1C=C2C=NN(C2=CC=1)C1=CN(C(C=C1)=O)C)C1=CC(=C(C=C1)F)OC)(C)F LEDGCBVULGYNJK-NBGIEHNGSA-N 0.000 claims description 3
- 208000021386 Sjogren Syndrome Diseases 0.000 claims description 3
- 206010047115 Vasculitis Diseases 0.000 claims description 3
- 208000002551 irritable bowel syndrome Diseases 0.000 claims description 3
- 125000000714 pyrimidinyl group Chemical group 0.000 claims description 3
- WNXJIVFYUVYPPR-UHFFFAOYSA-N 1,3-dioxolane Chemical group C1COCO1 WNXJIVFYUVYPPR-UHFFFAOYSA-N 0.000 claims description 2
- QWEWLLNSJDTOKH-UHFFFAOYSA-N 1,3-thiazole-2-carboxamide Chemical compound NC(=O)C1=NC=CS1 QWEWLLNSJDTOKH-UHFFFAOYSA-N 0.000 claims description 2
- 125000004575 3-pyrrolidinyl group Chemical group [H]N1C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 claims description 2
- SLRWZGQWRLEQEF-SQJMNOBHSA-N CC1=NC(=NO1)C(=O)N[C@@H]1[C@H](N(C(C1)=O)C=1C=C2C=NN(C2=CC=1)C1=CN(C(C=C1)=O)C)C1=CC=CC=C1 Chemical compound CC1=NC(=NO1)C(=O)N[C@@H]1[C@H](N(C(C1)=O)C=1C=C2C=NN(C2=CC=1)C1=CN(C(C=C1)=O)C)C1=CC=CC=C1 SLRWZGQWRLEQEF-SQJMNOBHSA-N 0.000 claims description 2
- QQBMNBRRJZNSHA-WNCULLNHSA-N CN1C=C(C=CC1=O)N1N=CC2=CC(=CC=C12)N1[C@@H]([C@H](CC1=O)NC(=O)C1CC1)C1=C(C=CC=C1)C Chemical compound CN1C=C(C=CC1=O)N1N=CC2=CC(=CC=C12)N1[C@@H]([C@H](CC1=O)NC(=O)C1CC1)C1=C(C=CC=C1)C QQBMNBRRJZNSHA-WNCULLNHSA-N 0.000 claims description 2
- ORSUIFYTTXOECT-CAKYRVLISA-N CN1C=C(C=CC1=O)N1N=CC2=CC(=CC=C12)N1[C@@H]([C@H](CC1=O)NC(=O)[C@@H]1OCCC1)C1=CC=CC=C1 Chemical compound CN1C=C(C=CC1=O)N1N=CC2=CC(=CC=C12)N1[C@@H]([C@H](CC1=O)NC(=O)[C@@H]1OCCC1)C1=CC=CC=C1 ORSUIFYTTXOECT-CAKYRVLISA-N 0.000 claims description 2
- GFDFKVXLXQRVKG-WNCULLNHSA-N CN1N=C(C=C1)C(=O)N[C@@H]1[C@H](N(C(C1)=O)C=1C=C2C=NN(C2=CC=1)C1=CN(C(C=C1)=O)C)C1=CC=CC=C1 Chemical compound CN1N=C(C=C1)C(=O)N[C@@H]1[C@H](N(C(C1)=O)C=1C=C2C=NN(C2=CC=1)C1=CN(C(C=C1)=O)C)C1=CC=CC=C1 GFDFKVXLXQRVKG-WNCULLNHSA-N 0.000 claims description 2
- YYDWEUANGAYJOG-UHFFFAOYSA-N COC1=NC=CC(=C1)CN1N=CC2=CC(=CC=C12)N1C(C(CC1=O)NC(=O)C1CC1)C1=CC=CC=C1 Chemical compound COC1=NC=CC(=C1)CN1N=CC2=CC(=CC=C12)N1C(C(CC1=O)NC(=O)C1CC1)C1=CC=CC=C1 YYDWEUANGAYJOG-UHFFFAOYSA-N 0.000 claims description 2
- PXPGCPMFBSDGSJ-UHFFFAOYSA-N FC(C(=O)NC1C(N(C(C1)=O)C=1C=C2C=NN(C2=CC=1)C1=CN(C(C=C1)=O)C)C1=C(C=C(C=C1)F)F)(C)F Chemical compound FC(C(=O)NC1C(N(C(C1)=O)C=1C=C2C=NN(C2=CC=1)C1=CN(C(C=C1)=O)C)C1=C(C=C(C=C1)F)F)(C)F PXPGCPMFBSDGSJ-UHFFFAOYSA-N 0.000 claims description 2
- UHFDKSBHOKTXHA-UHFFFAOYSA-N FC(C(=O)NC1C(N(C(C1)=O)C=1C=C2C=NN(C2=CC=1)C1=CN(C(C=C1)=O)C)C1=CC2=C(OCCO2)C=C1)(C)F Chemical compound FC(C(=O)NC1C(N(C(C1)=O)C=1C=C2C=NN(C2=CC=1)C1=CN(C(C=C1)=O)C)C1=CC2=C(OCCO2)C=C1)(C)F UHFDKSBHOKTXHA-UHFFFAOYSA-N 0.000 claims description 2
- OMSBLLDIQHSVNE-UHFFFAOYSA-N FC(C(=O)NC1C(N(C(C1)=O)C=1C=C2C=NN(C2=CC=1)C1=NC=C(C=N1)F)C1=CC=CC=C1)(C)F Chemical compound FC(C(=O)NC1C(N(C(C1)=O)C=1C=C2C=NN(C2=CC=1)C1=NC=C(C=N1)F)C1=CC=CC=C1)(C)F OMSBLLDIQHSVNE-UHFFFAOYSA-N 0.000 claims description 2
- LFSWZZGELQFBCG-UHFFFAOYSA-N FC(C(=O)NC1C(N(C(C1)=O)C=1C=C2C=NN(C2=CC=1)C1CCN(CC1)C(=O)C1CC1)C1=CC=CC=C1)(C)F Chemical compound FC(C(=O)NC1C(N(C(C1)=O)C=1C=C2C=NN(C2=CC=1)C1CCN(CC1)C(=O)C1CC1)C1=CC=CC=C1)(C)F LFSWZZGELQFBCG-UHFFFAOYSA-N 0.000 claims description 2
- OHVFFFTUSFPCNO-UHFFFAOYSA-N FC(C(=O)NC1C(N(C(C1)=O)C=1C=C2C=NN(C2=CC=1)C=1C=NC=CC=1)C1=CC=CC=C1)(C)F Chemical compound FC(C(=O)NC1C(N(C(C1)=O)C=1C=C2C=NN(C2=CC=1)C=1C=NC=CC=1)C1=CC=CC=C1)(C)F OHVFFFTUSFPCNO-UHFFFAOYSA-N 0.000 claims description 2
- BGNLBDZJKPOYMM-UHFFFAOYSA-N FC(C(=O)NC1C(N(C(C1)=O)C=1C=C2C=NN(C2=CC=1)C=1C=NN(C=1)C)C1=CC=CC=C1)(C)F Chemical compound FC(C(=O)NC1C(N(C(C1)=O)C=1C=C2C=NN(C2=CC=1)C=1C=NN(C=1)C)C1=CC=CC=C1)(C)F BGNLBDZJKPOYMM-UHFFFAOYSA-N 0.000 claims description 2
- VRZXBJKFLIUSDV-UHFFFAOYSA-N FC(C(=O)NC1C(N(C(C1)=O)C=1C=C2C=NN(C2=CC=1)CC1=CC(=NC=C1)OC)C1=CC=CC=C1)(C)F Chemical compound FC(C(=O)NC1C(N(C(C1)=O)C=1C=C2C=NN(C2=CC=1)CC1=CC(=NC=C1)OC)C1=CC=CC=C1)(C)F VRZXBJKFLIUSDV-UHFFFAOYSA-N 0.000 claims description 2
- FFQWOJBOTRTRNS-UHFFFAOYSA-N FC(C(=O)NC1C(N(C(C1)=O)C=1C=C2C=NN(C2=CC=1)CC1=CN(C(C=C1)=O)C)C1=CC=CC=C1)(C)F Chemical compound FC(C(=O)NC1C(N(C(C1)=O)C=1C=C2C=NN(C2=CC=1)CC1=CN(C(C=C1)=O)C)C1=CC=CC=C1)(C)F FFQWOJBOTRTRNS-UHFFFAOYSA-N 0.000 claims description 2
- UFURWCQWCSDHMC-UHFFFAOYSA-N FC(C(=O)NC1C(N(C(C1)=O)C=1C=C2C=NN(C2=CC=1)CC1=NN(C=C1)C)C1=CC=CC=C1)(C)F Chemical compound FC(C(=O)NC1C(N(C(C1)=O)C=1C=C2C=NN(C2=CC=1)CC1=NN(C=C1)C)C1=CC=CC=C1)(C)F UFURWCQWCSDHMC-UHFFFAOYSA-N 0.000 claims description 2
- RAJFIIMAKASHNG-UHFFFAOYSA-N FC(C(=O)NC1C(N(C(C1)=O)C=1C=C2C=NN(C2=CC=1)CC=1C=NC(=CC=1)OC)C1=CC=CC=C1)(C)F Chemical compound FC(C(=O)NC1C(N(C(C1)=O)C=1C=C2C=NN(C2=CC=1)CC=1C=NC(=CC=1)OC)C1=CC=CC=C1)(C)F RAJFIIMAKASHNG-UHFFFAOYSA-N 0.000 claims description 2
- JEWRKENUIQWVRM-GBXCKJPGSA-N FC(C(=O)N[C@@H]1[C@H](N(C(C1)=O)C=1C=C2C=NN(C2=CC=1)C1=CN(C(C=C1)=O)C)C1=C(C=CC=C1)F)(C)F Chemical compound FC(C(=O)N[C@@H]1[C@H](N(C(C1)=O)C=1C=C2C=NN(C2=CC=1)C1=CN(C(C=C1)=O)C)C1=C(C=CC=C1)F)(C)F JEWRKENUIQWVRM-GBXCKJPGSA-N 0.000 claims description 2
- TXYSVTRFJRHWSG-GBXCKJPGSA-N FC(C(=O)N[C@@H]1[C@H](N(C(C1)=O)C=1C=C2C=NN(C2=CC=1)C1=CN(C(C=C1)=O)C)C1=CC(=CC=C1)F)(C)F Chemical compound FC(C(=O)N[C@@H]1[C@H](N(C(C1)=O)C=1C=C2C=NN(C2=CC=1)C1=CN(C(C=C1)=O)C)C1=CC(=CC=C1)F)(C)F TXYSVTRFJRHWSG-GBXCKJPGSA-N 0.000 claims description 2
- BUIDZJAALKLQRD-YADARESESA-N FC(C(=O)N[C@@H]1[C@H](N(C(C1)=O)C=1C=C2C=NN(C2=CC=1)C1=CN(C(C=C1)=O)C)C1=CC(=NC=C1)OC)(C)F Chemical compound FC(C(=O)N[C@@H]1[C@H](N(C(C1)=O)C=1C=C2C=NN(C2=CC=1)C1=CN(C(C=C1)=O)C)C1=CC(=NC=C1)OC)(C)F BUIDZJAALKLQRD-YADARESESA-N 0.000 claims description 2
- NMTZIOYVEAJZNE-GBXCKJPGSA-N FC(C(=O)N[C@@H]1[C@H](N(C(C1)=O)C=1C=C2C=NN(C2=CC=1)C1=CN(C(C=C1)=O)C)C1=CC=C(C=C1)F)(C)F Chemical compound FC(C(=O)N[C@@H]1[C@H](N(C(C1)=O)C=1C=C2C=NN(C2=CC=1)C1=CN(C(C=C1)=O)C)C1=CC=C(C=C1)F)(C)F NMTZIOYVEAJZNE-GBXCKJPGSA-N 0.000 claims description 2
- NGLDFORLOPWDSV-GBXCKJPGSA-N FC(C(=O)N[C@@H]1[C@H](N(C(C1)=O)C=1C=C2C=NN(C2=CC=1)C1=CN(C(C=C1)=O)C)C1=CC=CC=C1)(C)F Chemical compound FC(C(=O)N[C@@H]1[C@H](N(C(C1)=O)C=1C=C2C=NN(C2=CC=1)C1=CN(C(C=C1)=O)C)C1=CC=CC=C1)(C)F NGLDFORLOPWDSV-GBXCKJPGSA-N 0.000 claims description 2
- UEEUNUVWIGFNRA-SQJMNOBHSA-N FC1(CC1)C(=O)N[C@@H]1[C@H](N(C(C1)=O)C=1C=C2C=NN(C2=CC=1)C1=CN(C(C=C1)=O)C)C1=CC=CC=C1 Chemical compound FC1(CC1)C(=O)N[C@@H]1[C@H](N(C(C1)=O)C=1C=C2C=NN(C2=CC=1)C1=CN(C(C=C1)=O)C)C1=CC=CC=C1 UEEUNUVWIGFNRA-SQJMNOBHSA-N 0.000 claims description 2
- 208000002193 Pain Diseases 0.000 claims description 2
- 125000000246 pyrimidin-2-yl group Chemical group [H]C1=NC(*)=NC([H])=C1[H] 0.000 claims description 2
- ZMECITWNTVCEJH-UHFFFAOYSA-N FC(C(=O)NC1C(N(C(C1)=O)C=1C=C2C=NN(C2=CC=1)C1=NC=C(C=C1)F)C1=CC=CC=C1)(C)F Chemical compound FC(C(=O)NC1C(N(C(C1)=O)C=1C=C2C=NN(C2=CC=1)C1=NC=C(C=C1)F)C1=CC=CC=C1)(C)F ZMECITWNTVCEJH-UHFFFAOYSA-N 0.000 claims 1
- HUFOUUNZWCYQHO-SQJMNOBHSA-N FC(C(=O)N[C@@H]1[C@H](N(C(C1)=O)C=1C=C2C=NN(C2=CC=1)C1=CN(C(C=C1)=O)C)C1=C(C=CC=C1)C)(C)F Chemical compound FC(C(=O)N[C@@H]1[C@H](N(C(C1)=O)C=1C=C2C=NN(C2=CC=1)C1=CN(C(C=C1)=O)C)C1=C(C=CC=C1)C)(C)F HUFOUUNZWCYQHO-SQJMNOBHSA-N 0.000 claims 1
- 102000003676 Glucocorticoid Receptors Human genes 0.000 abstract description 30
- 108090000079 Glucocorticoid Receptors Proteins 0.000 abstract description 30
- 230000001404 mediated effect Effects 0.000 abstract description 8
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 300
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 242
- 239000011541 reaction mixture Substances 0.000 description 196
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 183
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical group ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 171
- 238000006243 chemical reaction Methods 0.000 description 164
- 239000000243 solution Substances 0.000 description 164
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 140
- 235000019439 ethyl acetate Nutrition 0.000 description 120
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 93
- 239000012043 crude product Substances 0.000 description 88
- 238000004440 column chromatography Methods 0.000 description 78
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 76
- 239000000741 silica gel Substances 0.000 description 76
- 229910002027 silica gel Inorganic materials 0.000 description 76
- 239000012044 organic layer Substances 0.000 description 70
- 230000002829 reductive effect Effects 0.000 description 67
- 239000007832 Na2SO4 Substances 0.000 description 63
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 63
- 229910052938 sodium sulfate Inorganic materials 0.000 description 63
- 235000011152 sodium sulphate Nutrition 0.000 description 63
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 58
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 57
- 239000007787 solid Substances 0.000 description 50
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 43
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 39
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 38
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 34
- 238000005481 NMR spectroscopy Methods 0.000 description 34
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 33
- 238000003786 synthesis reaction Methods 0.000 description 32
- 230000015572 biosynthetic process Effects 0.000 description 31
- 239000012267 brine Substances 0.000 description 31
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 31
- WVDDGKGOMKODPV-UHFFFAOYSA-N Benzyl alcohol Chemical compound OCC1=CC=CC=C1 WVDDGKGOMKODPV-UHFFFAOYSA-N 0.000 description 30
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 29
- 239000012071 phase Substances 0.000 description 28
- 239000002904 solvent Substances 0.000 description 28
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 27
- WGLUMOCWFMKWIL-UHFFFAOYSA-N dichloromethane;methanol Chemical compound OC.ClCCl WGLUMOCWFMKWIL-UHFFFAOYSA-N 0.000 description 27
- 239000005457 ice water Substances 0.000 description 23
- 239000000203 mixture Substances 0.000 description 23
- HPNMFZURTQLUMO-UHFFFAOYSA-N diethylamine Chemical compound CCNCC HPNMFZURTQLUMO-UHFFFAOYSA-N 0.000 description 22
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 22
- 229910052786 argon Inorganic materials 0.000 description 19
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 18
- 239000007821 HATU Substances 0.000 description 18
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 18
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical class [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 17
- 239000010410 layer Substances 0.000 description 17
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 15
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 15
- 229910052739 hydrogen Inorganic materials 0.000 description 15
- 239000001257 hydrogen Substances 0.000 description 15
- 238000000926 separation method Methods 0.000 description 14
- 241000521257 Hydrops Species 0.000 description 13
- 206010030113 Oedema Diseases 0.000 description 13
- 239000002253 acid Substances 0.000 description 13
- 210000004027 cell Anatomy 0.000 description 13
- 238000004296 chiral HPLC Methods 0.000 description 12
- 239000000706 filtrate Substances 0.000 description 12
- INQOMBQAUSQDDS-UHFFFAOYSA-N iodomethane Chemical compound IC INQOMBQAUSQDDS-UHFFFAOYSA-N 0.000 description 12
- 238000000034 method Methods 0.000 description 12
- 238000006467 substitution reaction Methods 0.000 description 12
- WLHCBQAPPJAULW-UHFFFAOYSA-N 4-methylbenzenethiol Chemical compound CC1=CC=C(S)C=C1 WLHCBQAPPJAULW-UHFFFAOYSA-N 0.000 description 11
- 239000007868 Raney catalyst Substances 0.000 description 11
- NPXOKRUENSOPAO-UHFFFAOYSA-N Raney nickel Chemical compound [Al].[Ni] NPXOKRUENSOPAO-UHFFFAOYSA-N 0.000 description 11
- 229910000564 Raney nickel Inorganic materials 0.000 description 11
- FPYJFEHAWHCUMM-UHFFFAOYSA-N maleic anhydride Chemical compound O=C1OC(=O)C=C1 FPYJFEHAWHCUMM-UHFFFAOYSA-N 0.000 description 11
- 229910000027 potassium carbonate Inorganic materials 0.000 description 11
- 238000000746 purification Methods 0.000 description 11
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 10
- 235000019445 benzyl alcohol Nutrition 0.000 description 10
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 10
- 229910052757 nitrogen Inorganic materials 0.000 description 10
- 241000400611 Eucalyptus deanei Species 0.000 description 9
- 238000003556 assay Methods 0.000 description 9
- 238000001816 cooling Methods 0.000 description 9
- 230000000694 effects Effects 0.000 description 9
- 239000003862 glucocorticoid Substances 0.000 description 9
- 239000007788 liquid Substances 0.000 description 9
- 238000010992 reflux Methods 0.000 description 9
- 230000001154 acute effect Effects 0.000 description 8
- 125000004432 carbon atom Chemical group C* 0.000 description 8
- 125000004122 cyclic group Chemical group 0.000 description 8
- 229940037128 systemic glucocorticoids Drugs 0.000 description 8
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 8
- DTQVDTLACAAQTR-UHFFFAOYSA-N trifluoroacetic acid Substances OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 8
- JRHPOFJADXHYBR-HTQZYQBOSA-N (1r,2r)-1-n,2-n-dimethylcyclohexane-1,2-diamine Chemical compound CN[C@@H]1CCCC[C@H]1NC JRHPOFJADXHYBR-HTQZYQBOSA-N 0.000 description 7
- QOWBXWFYRXSBAS-UHFFFAOYSA-N (2,4-dimethoxyphenyl)methanamine Chemical compound COC1=CC=C(CN)C(OC)=C1 QOWBXWFYRXSBAS-UHFFFAOYSA-N 0.000 description 7
- 229910052799 carbon Inorganic materials 0.000 description 7
- 238000005859 coupling reaction Methods 0.000 description 7
- 239000013067 intermediate product Substances 0.000 description 7
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 7
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 7
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 7
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 7
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 7
- 238000003756 stirring Methods 0.000 description 7
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 7
- SNGOPWAUPAJDBW-SCZZXKLOSA-N (4R,5S)-4-amino-5-phenylpyrrolidin-2-one Chemical compound N[C@@H]1CC(=O)N[C@H]1c1ccccc1 SNGOPWAUPAJDBW-SCZZXKLOSA-N 0.000 description 6
- 125000004973 1-butenyl group Chemical group C(=CCC)* 0.000 description 6
- 125000004972 1-butynyl group Chemical group [H]C([H])([H])C([H])([H])C#C* 0.000 description 6
- 125000000530 1-propynyl group Chemical group [H]C([H])([H])C#C* 0.000 description 6
- 125000004974 2-butenyl group Chemical group C(C=CC)* 0.000 description 6
- 125000000069 2-butynyl group Chemical group [H]C([H])([H])C#CC([H])([H])* 0.000 description 6
- 125000004493 2-methylbut-1-yl group Chemical group CC(C*)CC 0.000 description 6
- 125000001494 2-propynyl group Chemical group [H]C#CC([H])([H])* 0.000 description 6
- CGCHCLICSHIAAM-UHFFFAOYSA-N 5-iodo-1h-indazole Chemical compound IC1=CC=C2NN=CC2=C1 CGCHCLICSHIAAM-UHFFFAOYSA-N 0.000 description 6
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical class [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 6
- 229940117965 Glucocorticoid receptor modulator Drugs 0.000 description 6
- 239000000556 agonist Substances 0.000 description 6
- 239000005557 antagonist Substances 0.000 description 6
- 125000001972 isopentyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])C([H])([H])* 0.000 description 6
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 6
- 125000001280 n-hexyl group Chemical group C(CCCCC)* 0.000 description 6
- 125000000740 n-pentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 6
- 125000001971 neopentyl group Chemical group [H]C([*])([H])C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H] 0.000 description 6
- 239000000047 product Substances 0.000 description 6
- 125000004368 propenyl group Chemical group C(=CC)* 0.000 description 6
- HNJBEVLQSNELDL-UHFFFAOYSA-N pyrrolidin-2-one Chemical compound O=C1CCCN1 HNJBEVLQSNELDL-UHFFFAOYSA-N 0.000 description 6
- 239000000725 suspension Substances 0.000 description 6
- 125000006023 1-pentenyl group Chemical group 0.000 description 5
- 125000006024 2-pentenyl group Chemical group 0.000 description 5
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 5
- 101000926939 Homo sapiens Glucocorticoid receptor Proteins 0.000 description 5
- BLEBFDYUDVZRFG-UHFFFAOYSA-N dichloromethane;propan-2-ol Chemical compound ClCCl.CC(C)O BLEBFDYUDVZRFG-UHFFFAOYSA-N 0.000 description 5
- 208000035475 disorder Diseases 0.000 description 5
- 125000005842 heteroatom Chemical group 0.000 description 5
- 125000003538 pentan-3-yl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])C([H])([H])[H] 0.000 description 5
- JAEIBKXSIXOLOL-UHFFFAOYSA-N pyrrolidin-1-ium-3-carboxylate Chemical compound OC(=O)C1CCNC1 JAEIBKXSIXOLOL-UHFFFAOYSA-N 0.000 description 5
- 230000009467 reduction Effects 0.000 description 5
- 235000017557 sodium bicarbonate Nutrition 0.000 description 5
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 5
- 239000000126 substance Substances 0.000 description 5
- PAQZWJGSJMLPMG-UHFFFAOYSA-N 2,4,6-tripropyl-1,3,5,2$l^{5},4$l^{5},6$l^{5}-trioxatriphosphinane 2,4,6-trioxide Chemical compound CCCP1(=O)OP(=O)(CCC)OP(=O)(CCC)O1 PAQZWJGSJMLPMG-UHFFFAOYSA-N 0.000 description 4
- USFZMSVCRYTOJT-UHFFFAOYSA-N Ammonium acetate Chemical compound N.CC(O)=O USFZMSVCRYTOJT-UHFFFAOYSA-N 0.000 description 4
- 208000002691 Choroiditis Diseases 0.000 description 4
- 208000003971 Posterior uveitis Diseases 0.000 description 4
- 125000000753 cycloalkyl group Chemical group 0.000 description 4
- YMGUBTXCNDTFJI-UHFFFAOYSA-N cyclopropanecarboxylic acid Chemical compound OC(=O)C1CC1 YMGUBTXCNDTFJI-UHFFFAOYSA-N 0.000 description 4
- 229960003957 dexamethasone Drugs 0.000 description 4
- 239000000499 gel Substances 0.000 description 4
- 125000000623 heterocyclic group Chemical group 0.000 description 4
- 230000005764 inhibitory process Effects 0.000 description 4
- 150000003951 lactams Chemical class 0.000 description 4
- ZCSHNCUQKCANBX-UHFFFAOYSA-N lithium diisopropylamide Chemical compound [Li+].CC(C)[N-]C(C)C ZCSHNCUQKCANBX-UHFFFAOYSA-N 0.000 description 4
- 125000001570 methylene group Chemical group [H]C([H])([*:1])[*:2] 0.000 description 4
- 125000000325 methylidene group Chemical group [H]C([H])=* 0.000 description 4
- 238000002953 preparative HPLC Methods 0.000 description 4
- PMWGIVRHUIAIII-UHFFFAOYSA-N 2,2-difluoropropanoic acid Chemical compound CC(F)(F)C(O)=O PMWGIVRHUIAIII-UHFFFAOYSA-N 0.000 description 3
- 125000005916 2-methylpentyl group Chemical group 0.000 description 3
- 125000005917 3-methylpentyl group Chemical group 0.000 description 3
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 3
- 239000005695 Ammonium acetate Substances 0.000 description 3
- PIMHBWDLYGXVQA-UHFFFAOYSA-N ClC=1C=C(C=CC=1)C1NC(CC1(C(=O)O)SC1=CC=C(C=C1)C)=O Chemical compound ClC=1C=C(C=CC=1)C1NC(CC1(C(=O)O)SC1=CC=C(C=C1)C)=O PIMHBWDLYGXVQA-UHFFFAOYSA-N 0.000 description 3
- JQIAOQWLFWCWBU-KOLCDFICSA-N FC(C(=O)N[C@H]1[C@@H](NC(C1)=O)C1=CC=CC=C1)(C)F Chemical compound FC(C(=O)N[C@H]1[C@@H](NC(C1)=O)C1=CC=CC=C1)(C)F JQIAOQWLFWCWBU-KOLCDFICSA-N 0.000 description 3
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 3
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 3
- 229910020667 PBr3 Inorganic materials 0.000 description 3
- 229940043376 ammonium acetate Drugs 0.000 description 3
- 235000019257 ammonium acetate Nutrition 0.000 description 3
- 125000004429 atom Chemical group 0.000 description 3
- 125000002619 bicyclic group Chemical group 0.000 description 3
- 230000027455 binding Effects 0.000 description 3
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 3
- 125000000596 cyclohexenyl group Chemical group C1(=CCCCC1)* 0.000 description 3
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 3
- 125000002433 cyclopentenyl group Chemical group C1(=CCCC1)* 0.000 description 3
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 3
- 210000000172 cytosol Anatomy 0.000 description 3
- 125000004216 fluoromethyl group Chemical group [H]C([H])(F)* 0.000 description 3
- 208000027866 inflammatory disease Diseases 0.000 description 3
- 230000002757 inflammatory effect Effects 0.000 description 3
- 125000004491 isohexyl group Chemical group C(CCC(C)C)* 0.000 description 3
- FEWJPZIEWOKRBE-LWMBPPNESA-N levotartaric acid Chemical compound OC(=O)[C@@H](O)[C@H](O)C(O)=O FEWJPZIEWOKRBE-LWMBPPNESA-N 0.000 description 3
- 239000003446 ligand Substances 0.000 description 3
- MZRVEZGGRBJDDB-UHFFFAOYSA-N n-Butyllithium Substances [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 description 3
- IPNPIHIZVLFAFP-UHFFFAOYSA-N phosphorus tribromide Chemical compound BrP(Br)Br IPNPIHIZVLFAFP-UHFFFAOYSA-N 0.000 description 3
- 230000023603 positive regulation of transcription initiation, DNA-dependent Effects 0.000 description 3
- 108090000623 proteins and genes Proteins 0.000 description 3
- UBQKCCHYAOITMY-UHFFFAOYSA-N pyridin-2-ol Chemical compound OC1=CC=CC=N1 UBQKCCHYAOITMY-UHFFFAOYSA-N 0.000 description 3
- 239000002287 radioligand Substances 0.000 description 3
- 239000000377 silicon dioxide Substances 0.000 description 3
- FVAUCKIRQBBSSJ-UHFFFAOYSA-M sodium iodide Chemical compound [Na+].[I-] FVAUCKIRQBBSSJ-UHFFFAOYSA-M 0.000 description 3
- 229910052717 sulfur Inorganic materials 0.000 description 3
- 230000009885 systemic effect Effects 0.000 description 3
- 238000012360 testing method Methods 0.000 description 3
- 125000003718 tetrahydrofuranyl group Chemical group 0.000 description 3
- 125000001412 tetrahydropyranyl group Chemical group 0.000 description 3
- 230000035903 transrepression Effects 0.000 description 3
- 238000001291 vacuum drying Methods 0.000 description 3
- 229920002554 vinyl polymer Polymers 0.000 description 3
- AIDKSONIWROXFQ-UHFFFAOYSA-N (1-methyl-2-oxopyridine-4-carbonyl) 1-methyl-2-oxopyridine-4-carboxylate Chemical compound CN1C(C=C(C=C1)C(=O)OC(=O)C1=CC(N(C=C1)C)=O)=O AIDKSONIWROXFQ-UHFFFAOYSA-N 0.000 description 2
- GHGADOKTLYRRQO-XCBNKYQSSA-N (2R,3R)-2-(2-fluorophenyl)-5-oxopyrrolidine-3-carboxylic acid Chemical compound FC1=C(C=CC=C1)[C@@H]1NC(C[C@H]1C(=O)O)=O GHGADOKTLYRRQO-XCBNKYQSSA-N 0.000 description 2
- YYFAMTRJHLEGJC-XCBNKYQSSA-N (2R,3R)-2-(2-methoxypyridin-4-yl)-5-oxopyrrolidine-3-carboxylic acid Chemical compound COC1=NC=CC(=C1)[C@@H]1NC(C[C@H]1C(=O)O)=O YYFAMTRJHLEGJC-XCBNKYQSSA-N 0.000 description 2
- OTWLZMJISLAKFE-KOLCDFICSA-N (2R,3R)-2-(2-methylphenyl)-5-oxopyrrolidine-3-carboxylic acid Chemical compound O=C1C[C@H]([C@@H](N1)C1=C(C=CC=C1)C)C(=O)O OTWLZMJISLAKFE-KOLCDFICSA-N 0.000 description 2
- SNTOSHSJOVPZLA-SCZZXKLOSA-N (2R,3R)-2-(3-chlorophenyl)-5-oxopyrrolidine-3-carboxylic acid Chemical compound OC(=O)[C@@H]1CC(=O)N[C@H]1c1cccc(Cl)c1 SNTOSHSJOVPZLA-SCZZXKLOSA-N 0.000 description 2
- OKGNNJPTYBTWAC-SCZZXKLOSA-N (2R,3R)-2-(3-fluorophenyl)-5-oxopyrrolidine-3-carboxylic acid Chemical compound FC=1C=C(C=CC=1)[C@@H]1NC(C[C@H]1C(=O)O)=O OKGNNJPTYBTWAC-SCZZXKLOSA-N 0.000 description 2
- ATYFORDPFVYPIJ-SCZZXKLOSA-N (2R,3R)-2-(4-fluorophenyl)-5-oxopyrrolidine-3-carboxylic acid Chemical compound FC1=CC=C(C=C1)[C@@H]1NC(C[C@H]1C(=O)O)=O ATYFORDPFVYPIJ-SCZZXKLOSA-N 0.000 description 2
- YBNCUBKXPBVKKB-SCZZXKLOSA-N (2R,3R)-5-oxo-2-phenylpyrrolidine-3-carboxylic acid Chemical compound O=C1C[C@H]([C@@H](N1)C1=CC=CC=C1)C(=O)O YBNCUBKXPBVKKB-SCZZXKLOSA-N 0.000 description 2
- BIOKXHSODVPINM-PELKAZGASA-N (4R,5S)-4-amino-5-(2,3-dihydro-1,4-benzodioxin-6-yl)pyrrolidin-2-one Chemical compound N[C@@H]1CC(N[C@H]1C1=CC2=C(OCCO2)C=C1)=O BIOKXHSODVPINM-PELKAZGASA-N 0.000 description 2
- JJHZZWWJRSJLTK-SCZZXKLOSA-N (4R,5S)-4-amino-5-(2-fluorophenyl)pyrrolidin-2-one Chemical compound N[C@@H]1CC(N[C@H]1C1=C(C=CC=C1)F)=O JJHZZWWJRSJLTK-SCZZXKLOSA-N 0.000 description 2
- LECOQDLYGICXTJ-SCZZXKLOSA-N (4R,5S)-4-amino-5-(3-fluorophenyl)pyrrolidin-2-one Chemical compound N[C@@H]1CC(N[C@H]1C1=CC(=CC=C1)F)=O LECOQDLYGICXTJ-SCZZXKLOSA-N 0.000 description 2
- GISCHAJISJSNAE-KCJUWKMLSA-N (4R,5S)-4-amino-5-(4-fluoro-3-methoxyphenyl)pyrrolidin-2-one Chemical compound N[C@@H]1CC(N[C@H]1C1=CC(=C(C=C1)F)OC)=O GISCHAJISJSNAE-KCJUWKMLSA-N 0.000 description 2
- FCBZLYWLHIPKTK-SCZZXKLOSA-N (4R,5S)-4-amino-5-(4-fluorophenyl)pyrrolidin-2-one Chemical compound N[C@@H]1CC(N[C@H]1C1=CC=C(C=C1)F)=O FCBZLYWLHIPKTK-SCZZXKLOSA-N 0.000 description 2
- SNGOPWAUPAJDBW-WCBMZHEXSA-N (4s,5r)-4-amino-5-phenylpyrrolidin-2-one Chemical compound N[C@H]1CC(=O)N[C@@H]1C1=CC=CC=C1 SNGOPWAUPAJDBW-WCBMZHEXSA-N 0.000 description 2
- 125000004178 (C1-C4) alkyl group Chemical group 0.000 description 2
- ZWDVQMVZZYIAHO-UHFFFAOYSA-N 2-fluorobenzaldehyde Chemical compound FC1=CC=CC=C1C=O ZWDVQMVZZYIAHO-UHFFFAOYSA-N 0.000 description 2
- MYHUFJLABUEBLV-UHFFFAOYSA-N 3-(bromomethyl)-1-methylpyrazole Chemical compound CN1C=CC(CBr)=N1 MYHUFJLABUEBLV-UHFFFAOYSA-N 0.000 description 2
- 125000003349 3-pyridyl group Chemical group N1=C([H])C([*])=C([H])C([H])=C1[H] 0.000 description 2
- TXLBLTVJLCDDJQ-UHFFFAOYSA-N 4-(bromomethyl)-1-methylpyridin-2-one Chemical compound CN1C=CC(CBr)=CC1=O TXLBLTVJLCDDJQ-UHFFFAOYSA-N 0.000 description 2
- ROPDTLJSDLQQGZ-UHFFFAOYSA-N 4-(bromomethyl)-2-methoxypyridine Chemical compound COC1=CC(CBr)=CC=N1 ROPDTLJSDLQQGZ-UHFFFAOYSA-N 0.000 description 2
- ZBAXPNQGKAJZAZ-UHFFFAOYSA-N 4-(hydroxymethyl)-1-methylpyridin-2-one Chemical compound CN1C=CC(CO)=CC1=O ZBAXPNQGKAJZAZ-UHFFFAOYSA-N 0.000 description 2
- DWOZNANUEDYIOF-UHFFFAOYSA-L 4-ditert-butylphosphanyl-n,n-dimethylaniline;dichloropalladium Chemical compound Cl[Pd]Cl.CN(C)C1=CC=C(P(C(C)(C)C)C(C)(C)C)C=C1.CN(C)C1=CC=C(P(C(C)(C)C)C(C)(C)C)C=C1 DWOZNANUEDYIOF-UHFFFAOYSA-L 0.000 description 2
- JFUAWXPBHXKZGA-IBGZPJMESA-N 4-fluoro-2-[(4r)-5,5,5-trifluoro-4-hydroxy-2-methyl-4-(1h-pyrrolo[2,3-c]pyridin-2-ylmethyl)pentan-2-yl]phenol Chemical compound C([C@@](O)(CC=1NC2=CN=CC=C2C=1)C(F)(F)F)C(C)(C)C1=CC(F)=CC=C1O JFUAWXPBHXKZGA-IBGZPJMESA-N 0.000 description 2
- 125000000339 4-pyridyl group Chemical group N1=C([H])C([H])=C([*])C([H])=C1[H] 0.000 description 2
- HGJZMANIFHWVII-UHFFFAOYSA-N 5-(bromomethyl)-1h-pyridin-2-one;hydrobromide Chemical compound Br.BrCC=1C=CC(=O)NC=1 HGJZMANIFHWVII-UHFFFAOYSA-N 0.000 description 2
- LCLDNQYXVDKJMX-UHFFFAOYSA-N 5-(bromomethyl)-2-methoxypyridine Chemical compound COC1=CC=C(CBr)C=N1 LCLDNQYXVDKJMX-UHFFFAOYSA-N 0.000 description 2
- 208000026872 Addison Disease Diseases 0.000 description 2
- 206010001382 Adrenal suppression Diseases 0.000 description 2
- 208000005676 Adrenogenital syndrome Diseases 0.000 description 2
- 208000035285 Allergic Seasonal Rhinitis Diseases 0.000 description 2
- 206010027654 Allergic conditions Diseases 0.000 description 2
- 206010002556 Ankylosing Spondylitis Diseases 0.000 description 2
- 206010002965 Aplasia pure red cell Diseases 0.000 description 2
- 206010004485 Berylliosis Diseases 0.000 description 2
- CGRYNDUAVGUVCG-UHFFFAOYSA-N C1(CC1)C(=O)N1CCC(CC1)N1N=CC2=CC(=CC=C12)I Chemical compound C1(CC1)C(=O)N1CCC(CC1)N1N=CC2=CC(=CC=C12)I CGRYNDUAVGUVCG-UHFFFAOYSA-N 0.000 description 2
- GJPGWNWGUHHNCJ-HFZDXXHNSA-N CN1C=C(C=CC1=O)N1N=CC2=CC(=CC=C12)N1[C@@H]([C@H](CC1=O)NC(=O)C=1N=COC=1)C1=CC=CC=C1 Chemical compound CN1C=C(C=CC1=O)N1N=CC2=CC(=CC=C12)N1[C@@H]([C@H](CC1=O)NC(=O)C=1N=COC=1)C1=CC=CC=C1 GJPGWNWGUHHNCJ-HFZDXXHNSA-N 0.000 description 2
- HUYSKORLHZASHI-VIZCGCQYSA-N CN1C=C(C=CC1=O)N1N=CC2=CC(=CC=C12)N1[C@H]([C@@H](CC1=O)NC(OCC1=CC=CC=C1)=O)C1=CC=CC=C1 Chemical compound CN1C=C(C=CC1=O)N1N=CC2=CC(=CC=C12)N1[C@H]([C@@H](CC1=O)NC(OCC1=CC=CC=C1)=O)C1=CC=CC=C1 HUYSKORLHZASHI-VIZCGCQYSA-N 0.000 description 2
- ALFJUOSCIJAEGW-UHFFFAOYSA-N COC1=NC=CC(=C1)C1NC(CC1C(=O)OC)=O Chemical compound COC1=NC=CC(=C1)C1NC(CC1C(=O)OC)=O ALFJUOSCIJAEGW-UHFFFAOYSA-N 0.000 description 2
- KXDHJXZQYSOELW-UHFFFAOYSA-M Carbamate Chemical compound NC([O-])=O KXDHJXZQYSOELW-UHFFFAOYSA-M 0.000 description 2
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 2
- 208000023355 Chronic beryllium disease Diseases 0.000 description 2
- LGYWNDJHJHMUAS-KOLCDFICSA-N ClC=1C=C(C=CC=1)[C@@H]1NC(C[C@H]1NC(C(C)(F)F)=O)=O Chemical compound ClC=1C=C(C=CC=1)[C@@H]1NC(C[C@H]1NC(C(C)(F)F)=O)=O LGYWNDJHJHMUAS-KOLCDFICSA-N 0.000 description 2
- 208000027932 Collagen disease Diseases 0.000 description 2
- 208000008448 Congenital adrenal hyperplasia Diseases 0.000 description 2
- 206010010744 Conjunctivitis allergic Diseases 0.000 description 2
- 238000006969 Curtius rearrangement reaction Methods 0.000 description 2
- 206010012438 Dermatitis atopic Diseases 0.000 description 2
- 206010012441 Dermatitis bullous Diseases 0.000 description 2
- 206010012442 Dermatitis contact Diseases 0.000 description 2
- 206010012455 Dermatitis exfoliative Diseases 0.000 description 2
- 229920002307 Dextran Polymers 0.000 description 2
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical class OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 2
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 2
- 206010013700 Drug hypersensitivity Diseases 0.000 description 2
- 208000017701 Endocrine disease Diseases 0.000 description 2
- 201000011275 Epicondylitis Diseases 0.000 description 2
- 206010015218 Erythema multiforme Diseases 0.000 description 2
- VGGSQFUCUMXWEO-UHFFFAOYSA-N Ethene Chemical compound C=C VGGSQFUCUMXWEO-UHFFFAOYSA-N 0.000 description 2
- 239000005977 Ethylene Substances 0.000 description 2
- AXRLRGMFZZYPSN-UHFFFAOYSA-N FC1=C(C=C(C=C1)OC)C1NC(CC1C(=O)OC)=O Chemical compound FC1=C(C=C(C=C1)OC)C1NC(CC1C(=O)OC)=O AXRLRGMFZZYPSN-UHFFFAOYSA-N 0.000 description 2
- NBRAJKAZNZYFAG-KCJUWKMLSA-N FC1=C(C=C(C=C1)OC)[C@@H]1NC(C[C@H]1C(=O)O)=O Chemical compound FC1=C(C=C(C=C1)OC)[C@@H]1NC(C[C@H]1C(=O)O)=O NBRAJKAZNZYFAG-KCJUWKMLSA-N 0.000 description 2
- YCFCJTKDDUYBSZ-HQJQHLMTSA-N FC1=C(C=C(C=C1)[C@@H]1NC(C[C@H]1C(=O)O)=O)OC Chemical compound FC1=C(C=C(C=C1)[C@@H]1NC(C[C@H]1C(=O)O)=O)OC YCFCJTKDDUYBSZ-HQJQHLMTSA-N 0.000 description 2
- NGSUFZWDGVUWKN-UHFFFAOYSA-N FC1=C(C=CC(=C1)F)SC1(C(NC(C1)=O)C1=CC=CC=C1)C(=O)O Chemical compound FC1=C(C=CC(=C1)F)SC1(C(NC(C1)=O)C1=CC=CC=C1)C(=O)O NGSUFZWDGVUWKN-UHFFFAOYSA-N 0.000 description 2
- ANIXSRAKWFXNFR-XCBNKYQSSA-N FC1=C(C=CC(=C1)F)[C@@H]1NC(C[C@H]1C(=O)O)=O Chemical compound FC1=C(C=CC(=C1)F)[C@@H]1NC(C[C@H]1C(=O)O)=O ANIXSRAKWFXNFR-XCBNKYQSSA-N 0.000 description 2
- UIBKUAXQSSSXQV-UHFFFAOYSA-N FC1=C(C=CC=C1)C1NC(CC1C(=O)OC)=O Chemical compound FC1=C(C=CC=C1)C1NC(CC1C(=O)OC)=O UIBKUAXQSSSXQV-UHFFFAOYSA-N 0.000 description 2
- VDGZNJVCWFIIIL-YPMHNXCESA-N FC1=C(C=CC=C1)[C@@H]1NC(C[C@H]1NC(=O)C1CC1)=O Chemical compound FC1=C(C=CC=C1)[C@@H]1NC(C[C@H]1NC(=O)C1CC1)=O VDGZNJVCWFIIIL-YPMHNXCESA-N 0.000 description 2
- FKHCVBKMVWXSTO-UHFFFAOYSA-N FC1=CC=C(C=C1)C1NC(CC1C(=O)OC)=O Chemical compound FC1=CC=C(C=C1)C1NC(CC1C(=O)OC)=O FKHCVBKMVWXSTO-UHFFFAOYSA-N 0.000 description 2
- ZYXCKDYRSCAAFY-UHFFFAOYSA-N FC=1C=C(C=CC=1)C1NC(CC1C(=O)OC)=O Chemical compound FC=1C=C(C=CC=1)C1NC(CC1C(=O)OC)=O ZYXCKDYRSCAAFY-UHFFFAOYSA-N 0.000 description 2
- 208000018522 Gastrointestinal disease Diseases 0.000 description 2
- 201000005569 Gout Diseases 0.000 description 2
- 206010018634 Gouty Arthritis Diseases 0.000 description 2
- 208000003809 Herpes Zoster Ophthalmicus Diseases 0.000 description 2
- 208000037147 Hypercalcaemia Diseases 0.000 description 2
- 208000034961 Hypoplastic Congenital Anemia Diseases 0.000 description 2
- XAWVMHOHMIRBJM-UHFFFAOYSA-N IC=1C=C2C=NN(C2=CC=1)C1CCNCC1 Chemical compound IC=1C=C2C=NN(C2=CC=1)C1CCNCC1 XAWVMHOHMIRBJM-UHFFFAOYSA-N 0.000 description 2
- ODELEVMCRNQMSU-UHFFFAOYSA-N IC=1C=C2C=NN(C2=CC=1)C=1C=CC(N(C=1)C)=O Chemical compound IC=1C=C2C=NN(C2=CC=1)C=1C=CC(N(C=1)C)=O ODELEVMCRNQMSU-UHFFFAOYSA-N 0.000 description 2
- TWUQLTPBIKEOKS-UHFFFAOYSA-N IC=1C=C2C=NN(C2=CC=1)CC1=CC(=NC=C1)OC Chemical compound IC=1C=C2C=NN(C2=CC=1)CC1=CC(=NC=C1)OC TWUQLTPBIKEOKS-UHFFFAOYSA-N 0.000 description 2
- KYVWXAMNWOXBMI-UHFFFAOYSA-N IC=1C=C2C=NN(C2=CC=1)CC1=CC(N(C=C1)C)=O Chemical compound IC=1C=C2C=NN(C2=CC=1)CC1=CC(N(C=C1)C)=O KYVWXAMNWOXBMI-UHFFFAOYSA-N 0.000 description 2
- KBEPKBUKRZZICY-UHFFFAOYSA-N IC=1C=C2C=NN(C2=CC=1)CC1=NN(C=C1)C Chemical compound IC=1C=C2C=NN(C2=CC=1)CC1=NN(C=C1)C KBEPKBUKRZZICY-UHFFFAOYSA-N 0.000 description 2
- VPCQJYZUSOJFDE-UHFFFAOYSA-N IC=1C=C2C=NN(C2=CC=1)CC=1C=CC(N(C=1)C)=O Chemical compound IC=1C=C2C=NN(C2=CC=1)CC=1C=CC(N(C=1)C)=O VPCQJYZUSOJFDE-UHFFFAOYSA-N 0.000 description 2
- QYUMFHAVVIESIY-UHFFFAOYSA-N IC=1C=C2C=NN(C2=CC=1)CC=1C=CC(NC=1)=O Chemical compound IC=1C=C2C=NN(C2=CC=1)CC=1C=CC(NC=1)=O QYUMFHAVVIESIY-UHFFFAOYSA-N 0.000 description 2
- AOPOGQFDKQKYLO-UHFFFAOYSA-N IC=1C=C2C=NN(C2=CC=1)CC=1C=NC(=CC=1)OC Chemical compound IC=1C=C2C=NN(C2=CC=1)CC=1C=NC(=CC=1)OC AOPOGQFDKQKYLO-UHFFFAOYSA-N 0.000 description 2
- 206010021245 Idiopathic thrombocytopenic purpura Diseases 0.000 description 2
- 206010022941 Iridocyclitis Diseases 0.000 description 2
- 208000003456 Juvenile Arthritis Diseases 0.000 description 2
- 206010059176 Juvenile idiopathic arthritis Diseases 0.000 description 2
- 201000009324 Loeffler syndrome Diseases 0.000 description 2
- 206010025323 Lymphomas Diseases 0.000 description 2
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 2
- DRKUBOCNAACHGK-AEFFLSMTSA-N N1N=CC2=CC(=CC=C12)N1[C@H]([C@@H](CC1=O)NC(C(C)(F)F)=O)C1=CC=CC=C1 Chemical compound N1N=CC2=CC(=CC=C12)N1[C@H]([C@@H](CC1=O)NC(C(C)(F)F)=O)C1=CC=CC=C1 DRKUBOCNAACHGK-AEFFLSMTSA-N 0.000 description 2
- UUCQGBMEIYDZBF-SCZZXKLOSA-N N[C@@H]1CC(N[C@H]1C1=C(C=C(C=C1)F)F)=O Chemical compound N[C@@H]1CC(N[C@H]1C1=C(C=C(C=C1)F)F)=O UUCQGBMEIYDZBF-SCZZXKLOSA-N 0.000 description 2
- NGNHSYRFRRZYPV-KOLCDFICSA-N N[C@@H]1CC(N[C@H]1C1=C(C=CC(=C1)OC)F)=O Chemical compound N[C@@H]1CC(N[C@H]1C1=C(C=CC(=C1)OC)F)=O NGNHSYRFRRZYPV-KOLCDFICSA-N 0.000 description 2
- RQFYLCLNSRVAAI-XCBNKYQSSA-N N[C@@H]1CC(N[C@H]1C1=CC(=NC=C1)OC)=O Chemical compound N[C@@H]1CC(N[C@H]1C1=CC(=NC=C1)OC)=O RQFYLCLNSRVAAI-XCBNKYQSSA-N 0.000 description 2
- 206010028980 Neoplasm Diseases 0.000 description 2
- YGVWQTBWQXSIHM-PELKAZGASA-N O1C2=C(OCC1)C=C(C=C2)[C@@H]1NC(C[C@H]1C(=O)O)=O Chemical compound O1C2=C(OCC1)C=C(C=C2)[C@@H]1NC(C[C@H]1C(=O)O)=O YGVWQTBWQXSIHM-PELKAZGASA-N 0.000 description 2
- QCONPPLSJMDYGA-UHFFFAOYSA-N O=C1CC(C(N1)C1=CC=CC=C1)(C(=O)O)SC1=CC=C(C=C1)C Chemical compound O=C1CC(C(N1)C1=CC=CC=C1)(C(=O)O)SC1=CC=C(C=C1)C QCONPPLSJMDYGA-UHFFFAOYSA-N 0.000 description 2
- WLNXQGODHUUHFG-YPMHNXCESA-N O=C1C[C@H]([C@@H](N1)C1=CC=CC=C1)NC(=O)C1CC1 Chemical compound O=C1C[C@H]([C@@H](N1)C1=CC=CC=C1)NC(=O)C1CC1 WLNXQGODHUUHFG-YPMHNXCESA-N 0.000 description 2
- YDTMGIURMLOWEJ-UHFFFAOYSA-N O=C1NC(C2=CC=CC=C2)C(C(=O)OC)C1 Chemical compound O=C1NC(C2=CC=CC=C2)C(C(=O)OC)C1 YDTMGIURMLOWEJ-UHFFFAOYSA-N 0.000 description 2
- 206010030865 Ophthalmic herpes zoster Diseases 0.000 description 2
- 208000003435 Optic Neuritis Diseases 0.000 description 2
- 208000001132 Osteoporosis Diseases 0.000 description 2
- 241000721454 Pemphigus Species 0.000 description 2
- 208000008469 Peptic Ulcer Diseases 0.000 description 2
- 206010035669 Pneumonia aspiration Diseases 0.000 description 2
- XBDQKXXYIPTUBI-UHFFFAOYSA-N Propionic acid Substances CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 2
- 201000004681 Psoriasis Diseases 0.000 description 2
- 201000001263 Psoriatic Arthritis Diseases 0.000 description 2
- 208000036824 Psoriatic arthropathy Diseases 0.000 description 2
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 2
- 208000025747 Rheumatic disease Diseases 0.000 description 2
- 206010039094 Rhinitis perennial Diseases 0.000 description 2
- 208000036284 Rhinitis seasonal Diseases 0.000 description 2
- 206010039793 Seborrhoeic dermatitis Diseases 0.000 description 2
- 206010039807 Secondary adrenocortical insufficiency Diseases 0.000 description 2
- 206010042033 Stevens-Johnson syndrome Diseases 0.000 description 2
- 231100000168 Stevens-Johnson syndrome Toxicity 0.000 description 2
- 208000007271 Substance Withdrawal Syndrome Diseases 0.000 description 2
- 206010042742 Sympathetic ophthalmia Diseases 0.000 description 2
- 208000004760 Tenosynovitis Diseases 0.000 description 2
- 208000031981 Thrombocytopenic Idiopathic Purpura Diseases 0.000 description 2
- 238000002441 X-ray diffraction Methods 0.000 description 2
- 201000004208 acquired thrombocytopenia Diseases 0.000 description 2
- 125000005073 adamantyl group Chemical group C12(CC3CC(CC(C1)C3)C2)* 0.000 description 2
- 239000000654 additive Substances 0.000 description 2
- 230000002411 adverse Effects 0.000 description 2
- 239000000443 aerosol Substances 0.000 description 2
- 125000001931 aliphatic group Chemical group 0.000 description 2
- 208000002205 allergic conjunctivitis Diseases 0.000 description 2
- 230000000172 allergic effect Effects 0.000 description 2
- 201000010105 allergic rhinitis Diseases 0.000 description 2
- 150000001408 amides Chemical class 0.000 description 2
- 150000001412 amines Chemical class 0.000 description 2
- 235000019270 ammonium chloride Nutrition 0.000 description 2
- 208000007502 anemia Diseases 0.000 description 2
- 201000004612 anterior uveitis Diseases 0.000 description 2
- 230000003110 anti-inflammatory effect Effects 0.000 description 2
- 230000002365 anti-tubercular Effects 0.000 description 2
- 239000007864 aqueous solution Substances 0.000 description 2
- 201000009408 aspiration pneumonitis Diseases 0.000 description 2
- 208000024998 atopic conjunctivitis Diseases 0.000 description 2
- 201000008937 atopic dermatitis Diseases 0.000 description 2
- 208000010668 atopic eczema Diseases 0.000 description 2
- 230000001363 autoimmune Effects 0.000 description 2
- 201000003710 autoimmune thrombocytopenic purpura Diseases 0.000 description 2
- 125000002393 azetidinyl group Chemical group 0.000 description 2
- 125000004069 aziridinyl group Chemical group 0.000 description 2
- 230000008901 benefit Effects 0.000 description 2
- HUMNYLRZRPPJDN-UHFFFAOYSA-N benzaldehyde Chemical compound O=CC1=CC=CC=C1 HUMNYLRZRPPJDN-UHFFFAOYSA-N 0.000 description 2
- YDNWYCYRPLBZKQ-WBVHZDCISA-N benzyl N-[(2S,3R)-2-(2,4-difluorophenyl)-5-oxopyrrolidin-3-yl]carbamate Chemical compound FC1=C(C=CC(=C1)F)[C@@H]1NC(C[C@H]1NC(OCC1=CC=CC=C1)=O)=O YDNWYCYRPLBZKQ-WBVHZDCISA-N 0.000 description 2
- DGXORDUAEGXMCC-AEFFLSMTSA-N benzyl N-[(2S,3R)-2-(2-fluoro-5-methoxyphenyl)-5-oxopyrrolidin-3-yl]carbamate Chemical compound FC1=C(C=C(C=C1)OC)[C@@H]1NC(C[C@H]1NC(OCC1=CC=CC=C1)=O)=O DGXORDUAEGXMCC-AEFFLSMTSA-N 0.000 description 2
- QZWHVZCPNGHALA-WBVHZDCISA-N benzyl N-[(2S,3R)-2-(2-fluorophenyl)-5-oxopyrrolidin-3-yl]carbamate Chemical compound FC1=C(C=CC=C1)[C@@H]1NC(C[C@H]1NC(OCC1=CC=CC=C1)=O)=O QZWHVZCPNGHALA-WBVHZDCISA-N 0.000 description 2
- WDWBVVUAWQIQSV-PBHICJAKSA-N benzyl N-[(2S,3R)-2-(2-methoxypyridin-4-yl)-5-oxopyrrolidin-3-yl]carbamate Chemical compound COC1=NC=CC(=C1)[C@@H]1NC(C[C@H]1NC(OCC1=CC=CC=C1)=O)=O WDWBVVUAWQIQSV-PBHICJAKSA-N 0.000 description 2
- FWEJEAWFAOUPSD-AEFFLSMTSA-N benzyl N-[(2S,3R)-2-(2-methylphenyl)-5-oxopyrrolidin-3-yl]carbamate Chemical compound O=C1C[C@H]([C@@H](N1)C1=C(C=CC=C1)C)NC(OCC1=CC=CC=C1)=O FWEJEAWFAOUPSD-AEFFLSMTSA-N 0.000 description 2
- SQKYQYPREDJGBN-WBVHZDCISA-N benzyl N-[(2S,3R)-2-(3-fluorophenyl)-5-oxopyrrolidin-3-yl]carbamate Chemical compound FC=1C=C(C=CC=1)[C@@H]1NC(C[C@H]1NC(OCC1=CC=CC=C1)=O)=O SQKYQYPREDJGBN-WBVHZDCISA-N 0.000 description 2
- ITNFKCVXLZMTEY-QAPCUYQASA-N benzyl N-[(2S,3R)-2-(4-fluoro-3-methoxyphenyl)-5-oxopyrrolidin-3-yl]carbamate Chemical compound FC1=C(C=C(C=C1)[C@@H]1NC(C[C@H]1NC(OCC1=CC=CC=C1)=O)=O)OC ITNFKCVXLZMTEY-QAPCUYQASA-N 0.000 description 2
- KHNGQPONWQQJLK-WBVHZDCISA-N benzyl N-[(2S,3R)-2-(4-fluorophenyl)-5-oxopyrrolidin-3-yl]carbamate Chemical compound FC1=CC=C(C=C1)[C@@H]1NC(C[C@H]1NC(OCC1=CC=CC=C1)=O)=O KHNGQPONWQQJLK-WBVHZDCISA-N 0.000 description 2
- 201000011510 cancer Diseases 0.000 description 2
- 239000002775 capsule Substances 0.000 description 2
- 239000000969 carrier Substances 0.000 description 2
- 238000002512 chemotherapy Methods 0.000 description 2
- 201000002797 childhood leukemia Diseases 0.000 description 2
- IJOOHPMOJXWVHK-UHFFFAOYSA-N chlorotrimethylsilane Chemical compound C[Si](C)(C)Cl IJOOHPMOJXWVHK-UHFFFAOYSA-N 0.000 description 2
- 201000004709 chorioretinitis Diseases 0.000 description 2
- 208000019069 chronic childhood arthritis Diseases 0.000 description 2
- 208000025302 chronic primary adrenal insufficiency Diseases 0.000 description 2
- 201000004425 congenital hypoplastic anemia Diseases 0.000 description 2
- 208000010247 contact dermatitis Diseases 0.000 description 2
- 230000008878 coupling Effects 0.000 description 2
- 238000010168 coupling process Methods 0.000 description 2
- 238000010511 deprotection reaction Methods 0.000 description 2
- 201000001981 dermatomyositis Diseases 0.000 description 2
- UREBDLICKHMUKA-CXSFZGCWSA-N dexamethasone Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1C[C@@H](C)[C@@](C(=O)CO)(O)[C@@]1(C)C[C@@H]2O UREBDLICKHMUKA-CXSFZGCWSA-N 0.000 description 2
- 206010012601 diabetes mellitus Diseases 0.000 description 2
- 125000004852 dihydrofuranyl group Chemical group O1C(CC=C1)* 0.000 description 2
- 125000005045 dihydroisoquinolinyl group Chemical group C1(NC=CC2=CC=CC=C12)* 0.000 description 2
- 125000005048 dihydroisoxazolyl group Chemical group O1N(CC=C1)* 0.000 description 2
- 125000005050 dihydrooxazolyl group Chemical group O1C(NC=C1)* 0.000 description 2
- 125000004655 dihydropyridinyl group Chemical group N1(CC=CC=C1)* 0.000 description 2
- 125000005054 dihydropyrrolyl group Chemical group [H]C1=C([H])C([H])([H])C([H])([H])N1* 0.000 description 2
- 125000005044 dihydroquinolinyl group Chemical group N1(CC=CC2=CC=CC=C12)* 0.000 description 2
- 125000005411 dithiolanyl group Chemical group S1SC(CC1)* 0.000 description 2
- 239000003814 drug Substances 0.000 description 2
- 230000000925 erythroid effect Effects 0.000 description 2
- 208000004526 exfoliative dermatitis Diseases 0.000 description 2
- 238000001914 filtration Methods 0.000 description 2
- 238000003818 flash chromatography Methods 0.000 description 2
- 125000000524 functional group Chemical group 0.000 description 2
- 229940124750 glucocorticoid receptor agonist Drugs 0.000 description 2
- 229940126013 glucocorticoid receptor antagonist Drugs 0.000 description 2
- 239000008187 granular material Substances 0.000 description 2
- 208000014951 hematologic disease Diseases 0.000 description 2
- 208000007475 hemolytic anemia Diseases 0.000 description 2
- 238000004128 high performance liquid chromatography Methods 0.000 description 2
- 239000011539 homogenization buffer Substances 0.000 description 2
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 2
- 230000002209 hydrophobic effect Effects 0.000 description 2
- 230000000148 hypercalcaemia Effects 0.000 description 2
- 208000030915 hypercalcemia disease Diseases 0.000 description 2
- 125000002632 imidazolidinyl group Chemical group 0.000 description 2
- 238000011534 incubation Methods 0.000 description 2
- 208000030603 inherited susceptibility to asthma Diseases 0.000 description 2
- 201000004614 iritis Diseases 0.000 description 2
- HEBMCVBCEDMUOF-UHFFFAOYSA-N isochromane Chemical compound C1=CC=C2COCCC2=C1 HEBMCVBCEDMUOF-UHFFFAOYSA-N 0.000 description 2
- 230000000155 isotopic effect Effects 0.000 description 2
- 125000003965 isoxazolidinyl group Chemical group 0.000 description 2
- 201000002215 juvenile rheumatoid arthritis Diseases 0.000 description 2
- 206010023332 keratitis Diseases 0.000 description 2
- 208000032839 leukemia Diseases 0.000 description 2
- 238000000670 ligand binding assay Methods 0.000 description 2
- YNESATAKKCNGOF-UHFFFAOYSA-N lithium bis(trimethylsilyl)amide Chemical compound [Li+].C[Si](C)(C)[N-][Si](C)(C)C YNESATAKKCNGOF-UHFFFAOYSA-N 0.000 description 2
- DWLILKFCTWFGMA-UHFFFAOYSA-N methyl 3-(4-methylphenyl)sulfanyl-5-oxo-2-phenylpyrrolidine-3-carboxylate Chemical compound C=1C=C(C)C=CC=1SC1(C(=O)OC)CC(=O)NC1C1=CC=CC=C1 DWLILKFCTWFGMA-UHFFFAOYSA-N 0.000 description 2
- VVWWZOKQKXPVIV-UHFFFAOYSA-N methyl pyrrolidine-3-carboxylate Chemical compound COC(=O)C1CCNC1 VVWWZOKQKXPVIV-UHFFFAOYSA-N 0.000 description 2
- 125000002757 morpholinyl group Chemical group 0.000 description 2
- 201000005962 mycosis fungoides Diseases 0.000 description 2
- 125000003136 n-heptyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 2
- 230000001613 neoplastic effect Effects 0.000 description 2
- 125000000160 oxazolidinyl group Chemical group 0.000 description 2
- 230000037361 pathway Effects 0.000 description 2
- 208000022719 perennial allergic rhinitis Diseases 0.000 description 2
- 125000004193 piperazinyl group Chemical group 0.000 description 2
- 125000003367 polycyclic group Chemical group 0.000 description 2
- 208000005987 polymyositis Diseases 0.000 description 2
- 230000003389 potentiating effect Effects 0.000 description 2
- 238000002360 preparation method Methods 0.000 description 2
- 150000003141 primary amines Chemical class 0.000 description 2
- 125000006238 prop-1-en-1-yl group Chemical group [H]\C(*)=C(/[H])C([H])([H])[H] 0.000 description 2
- 125000006239 protecting group Chemical group 0.000 description 2
- 208000008128 pulmonary tuberculosis Diseases 0.000 description 2
- 125000004309 pyranyl group Chemical group O1C(C=CC=C1)* 0.000 description 2
- 125000003072 pyrazolidinyl group Chemical group 0.000 description 2
- 150000003222 pyridines Chemical class 0.000 description 2
- 125000000719 pyrrolidinyl group Chemical group 0.000 description 2
- 102000005962 receptors Human genes 0.000 description 2
- 108020003175 receptors Proteins 0.000 description 2
- 208000023504 respiratory system disease Diseases 0.000 description 2
- 201000007529 rheumatic myocarditis Diseases 0.000 description 2
- 201000000306 sarcoidosis Diseases 0.000 description 2
- 208000017022 seasonal allergic rhinitis Diseases 0.000 description 2
- 230000001932 seasonal effect Effects 0.000 description 2
- 208000008742 seborrheic dermatitis Diseases 0.000 description 2
- 206010040400 serum sickness Diseases 0.000 description 2
- 239000012453 solvate Substances 0.000 description 2
- UCSJYZPVAKXKNQ-HZYVHMACSA-N streptomycin Chemical compound CN[C@H]1[C@H](O)[C@@H](O)[C@H](CO)O[C@H]1O[C@@H]1[C@](C=O)(O)[C@H](C)O[C@H]1O[C@@H]1[C@@H](NC(N)=N)[C@H](O)[C@@H](NC(N)=N)[C@H](O)[C@H]1O UCSJYZPVAKXKNQ-HZYVHMACSA-N 0.000 description 2
- 229940124530 sulfonamide Drugs 0.000 description 2
- 150000003456 sulfonamides Chemical class 0.000 description 2
- 239000006228 supernatant Substances 0.000 description 2
- 201000004595 synovitis Diseases 0.000 description 2
- 201000000596 systemic lupus erythematosus Diseases 0.000 description 2
- 239000003826 tablet Substances 0.000 description 2
- WOEQSXAIPTXOPY-UHFFFAOYSA-N tert-butyl 4-methylsulfonyloxypiperidine-1-carboxylate Chemical compound CC(C)(C)OC(=O)N1CCC(OS(C)(=O)=O)CC1 WOEQSXAIPTXOPY-UHFFFAOYSA-N 0.000 description 2
- HJUGFYREWKUQJT-UHFFFAOYSA-N tetrabromomethane Chemical compound BrC(Br)(Br)Br HJUGFYREWKUQJT-UHFFFAOYSA-N 0.000 description 2
- 125000004853 tetrahydropyridinyl group Chemical group N1(CCCC=C1)* 0.000 description 2
- 125000003554 tetrahydropyrrolyl group Chemical group 0.000 description 2
- 125000003507 tetrahydrothiofenyl group Chemical group 0.000 description 2
- 230000001225 therapeutic effect Effects 0.000 description 2
- 125000001984 thiazolidinyl group Chemical group 0.000 description 2
- 125000001544 thienyl group Chemical group 0.000 description 2
- 125000004568 thiomorpholinyl group Chemical group 0.000 description 2
- 206010043554 thrombocytopenia Diseases 0.000 description 2
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 2
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 2
- LWIHDJKSTIGBAC-UHFFFAOYSA-K tripotassium phosphate Chemical compound [K+].[K+].[K+].[O-]P([O-])([O-])=O LWIHDJKSTIGBAC-UHFFFAOYSA-K 0.000 description 2
- DGVVWUTYPXICAM-UHFFFAOYSA-N β‐Mercaptoethanol Chemical compound OCCS DGVVWUTYPXICAM-UHFFFAOYSA-N 0.000 description 2
- WCKJRVKJTUIAFW-UHFFFAOYSA-N (1-methylpyrazol-3-yl)methanol Chemical compound CN1C=CC(CO)=N1 WCKJRVKJTUIAFW-UHFFFAOYSA-N 0.000 description 1
- RNKOUSCCPHSCFE-UHFFFAOYSA-N (2,4-dimethoxyphenyl)methanol Chemical compound COC1=CC=C(CO)C(OC)=C1 RNKOUSCCPHSCFE-UHFFFAOYSA-N 0.000 description 1
- YENBVKZRNXXJSF-UHFFFAOYSA-N (2-methoxypyridin-4-yl)methanol Chemical compound COC1=CC(CO)=CC=N1 YENBVKZRNXXJSF-UHFFFAOYSA-N 0.000 description 1
- UZSOQCCLZOUTAN-KOLCDFICSA-N (4R,5S)-4-amino-5-(2-methylphenyl)pyrrolidin-2-one Chemical compound N[C@@H]1CC(N[C@H]1C1=C(C=CC=C1)C)=O UZSOQCCLZOUTAN-KOLCDFICSA-N 0.000 description 1
- FXIWUPZJZLQOPQ-SCZZXKLOSA-N (4R,5S)-4-amino-5-(3-chlorophenyl)pyrrolidin-2-one Chemical compound N[C@@H]1CC(N[C@H]1C1=CC(=CC=C1)Cl)=O FXIWUPZJZLQOPQ-SCZZXKLOSA-N 0.000 description 1
- DHADXDMPEUWEAS-UHFFFAOYSA-N (6-methoxypyridin-3-yl)boronic acid Chemical compound COC1=CC=C(B(O)O)C=N1 DHADXDMPEUWEAS-UHFFFAOYSA-N 0.000 description 1
- YKTYUHMWNUVAAM-UHFFFAOYSA-N (6-methoxypyridin-3-yl)methanol Chemical compound COC1=CC=C(CO)C=N1 YKTYUHMWNUVAAM-UHFFFAOYSA-N 0.000 description 1
- KZPYGQFFRCFCPP-UHFFFAOYSA-N 1,1'-bis(diphenylphosphino)ferrocene Chemical compound [Fe+2].C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1 KZPYGQFFRCFCPP-UHFFFAOYSA-N 0.000 description 1
- NDCPERCVXDYEFU-UHFFFAOYSA-N 1-fluorocyclopropane-1-carboxylic acid Chemical compound OC(=O)C1(F)CC1 NDCPERCVXDYEFU-UHFFFAOYSA-N 0.000 description 1
- XUKWOJNDLIOXSC-UHFFFAOYSA-N 1-methyl-2-oxopyridine-4-carboxylic acid Chemical compound CN1C=CC(C(O)=O)=CC1=O XUKWOJNDLIOXSC-UHFFFAOYSA-N 0.000 description 1
- DIZKLZKLNKQFGB-UHFFFAOYSA-N 1-methylcyclopropane-1-carboxylic acid Chemical compound OC(=O)C1(C)CC1 DIZKLZKLNKQFGB-UHFFFAOYSA-N 0.000 description 1
- 125000001637 1-naphthyl group Chemical group [H]C1=C([H])C([H])=C2C(*)=C([H])C([H])=C([H])C2=C1[H] 0.000 description 1
- 238000004229 14N NMR spectroscopy Methods 0.000 description 1
- 238000005451 15N solid-state nuclear magnetic resonance spectroscopy Methods 0.000 description 1
- CWKXDPPQCVWXAG-UHFFFAOYSA-N 2,3-dihydro-1,4-benzodioxine-6-carbaldehyde Chemical compound O1CCOC2=CC(C=O)=CC=C21 CWKXDPPQCVWXAG-UHFFFAOYSA-N 0.000 description 1
- WCGPCBACLBHDCI-UHFFFAOYSA-N 2,4-difluorobenzaldehyde Chemical compound FC1=CC=C(C=O)C(F)=C1 WCGPCBACLBHDCI-UHFFFAOYSA-N 0.000 description 1
- XNCSCQSQSGDGES-UHFFFAOYSA-N 2-[2-[bis(carboxymethyl)amino]propyl-(carboxymethyl)amino]acetic acid Chemical compound OC(=O)CN(CC(O)=O)C(C)CN(CC(O)=O)CC(O)=O XNCSCQSQSGDGES-UHFFFAOYSA-N 0.000 description 1
- UODINHBLNPPDPD-UHFFFAOYSA-N 2-bromo-5-fluoropyridine Chemical compound FC1=CC=C(Br)N=C1 UODINHBLNPPDPD-UHFFFAOYSA-N 0.000 description 1
- ANSMRNCOBLTNBO-UHFFFAOYSA-N 2-bromo-5-fluoropyrimidine Chemical group FC1=CN=C(Br)N=C1 ANSMRNCOBLTNBO-UHFFFAOYSA-N 0.000 description 1
- DKIQXHIAEMGZGO-UHFFFAOYSA-N 2-fluoro-5-methoxybenzaldehyde Chemical compound COC1=CC=C(F)C(C=O)=C1 DKIQXHIAEMGZGO-UHFFFAOYSA-N 0.000 description 1
- VOCKNCWQVHJMAE-UHFFFAOYSA-N 2-methoxypyridine-4-carbaldehyde Chemical compound COC1=CC(C=O)=CC=N1 VOCKNCWQVHJMAE-UHFFFAOYSA-N 0.000 description 1
- 125000001622 2-naphthyl group Chemical group [H]C1=C([H])C([H])=C2C([H])=C(*)C([H])=C([H])C2=C1[H] 0.000 description 1
- JHUUPUMBZGWODW-UHFFFAOYSA-N 3,6-dihydro-1,2-dioxine Chemical compound C1OOCC=C1 JHUUPUMBZGWODW-UHFFFAOYSA-N 0.000 description 1
- VSGOSHZXPRWOJS-UHFFFAOYSA-N 3-(4-methylphenyl)sulfanylpyrrolidine-3-carboxylic acid Chemical compound C1(=CC=C(C=C1)SC1(CNCC1)C(=O)O)C VSGOSHZXPRWOJS-UHFFFAOYSA-N 0.000 description 1
- SRWILAKSARHZPR-UHFFFAOYSA-N 3-chlorobenzaldehyde Chemical compound ClC1=CC=CC(C=O)=C1 SRWILAKSARHZPR-UHFFFAOYSA-N 0.000 description 1
- PIKNVEVCWAAOMJ-UHFFFAOYSA-N 3-fluorobenzaldehyde Chemical compound FC1=CC=CC(C=O)=C1 PIKNVEVCWAAOMJ-UHFFFAOYSA-N 0.000 description 1
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 1
- IXJSDKIJPVSPKF-UHFFFAOYSA-N 4-bromo-1-methylpyrazole Chemical compound CN1C=C(Br)C=N1 IXJSDKIJPVSPKF-UHFFFAOYSA-N 0.000 description 1
- YFTGMMXMLPTTAY-UHFFFAOYSA-N 4-bromo-2-methoxypyridine Chemical compound COC1=CC(Br)=CC=N1 YFTGMMXMLPTTAY-UHFFFAOYSA-N 0.000 description 1
- JFBMFWHEXBLFCR-UHFFFAOYSA-N 4-bromo-2-methylpyridine Chemical compound CC1=CC(Br)=CC=N1 JFBMFWHEXBLFCR-UHFFFAOYSA-N 0.000 description 1
- BSDGZUDFPKIYQG-UHFFFAOYSA-N 4-bromopyridine Chemical compound BrC1=CC=NC=C1 BSDGZUDFPKIYQG-UHFFFAOYSA-N 0.000 description 1
- NALVGTOMKSKFFV-UHFFFAOYSA-N 4-fluoro-3-methoxybenzaldehyde Chemical compound COC1=CC(C=O)=CC=C1F NALVGTOMKSKFFV-UHFFFAOYSA-N 0.000 description 1
- UOQXIWFBQSVDPP-UHFFFAOYSA-N 4-fluorobenzaldehyde Chemical compound FC1=CC=C(C=O)C=C1 UOQXIWFBQSVDPP-UHFFFAOYSA-N 0.000 description 1
- VVGRROXUVXNCIY-UHFFFAOYSA-N 4-piperazin-1-ylpyrrolidin-2-one Chemical class C1NC(=O)CC1N1CCNCC1 VVGRROXUVXNCIY-UHFFFAOYSA-N 0.000 description 1
- JLPOBAADYFDVAV-UHFFFAOYSA-N 5-(hydroxymethyl)-1h-pyridin-2-one Chemical compound OCC1=CC=C(O)N=C1 JLPOBAADYFDVAV-UHFFFAOYSA-N 0.000 description 1
- OFKWIQJLYCKDNY-UHFFFAOYSA-N 5-bromo-2-methylpyridine Chemical compound CC1=CC=C(Br)C=N1 OFKWIQJLYCKDNY-UHFFFAOYSA-N 0.000 description 1
- UXGKQBPGDONEDV-UHFFFAOYSA-N 5-iodo-1-(oxan-2-yl)indazole Chemical compound N1=CC2=CC(I)=CC=C2N1C1CCCCO1 UXGKQBPGDONEDV-UHFFFAOYSA-N 0.000 description 1
- UYDGNRLMYZMDOG-UHFFFAOYSA-N 5-methyl-1,3-thiazole-2-carboxylic acid Chemical compound CC1=CN=C(C(O)=O)S1 UYDGNRLMYZMDOG-UHFFFAOYSA-N 0.000 description 1
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 1
- OUKGEYIRXXUHTJ-UHFFFAOYSA-N CC(C(C1(C(O)=O)SC2=CC=C(C)C=C2)=O)N(CC(C=CC(OC)=C2)=C2OC)C1C(C=C1)=CC(OC)=C1F Chemical compound CC(C(C1(C(O)=O)SC2=CC=C(C)C=C2)=O)N(CC(C=CC(OC)=C2)=C2OC)C1C(C=C1)=CC(OC)=C1F OUKGEYIRXXUHTJ-UHFFFAOYSA-N 0.000 description 1
- XAQSXNMGPPOUGD-UHFFFAOYSA-N CC(C(N(CC1)CC1C(O)=O)=CC(C1)=O)=C1SC(C=CC(F)=C1)=C1F Chemical compound CC(C(N(CC1)CC1C(O)=O)=CC(C1)=O)=C1SC(C=CC(F)=C1)=C1F XAQSXNMGPPOUGD-UHFFFAOYSA-N 0.000 description 1
- VSPGWMCXTHEHDO-GJZUVCINSA-N CC1(CC1)C(=O)N[C@H]1[C@@H](N(C(C1)=O)C=1C=C2C=NN(C2=CC=1)C1=CN(C(C=C1)=O)C)C1=CC=CC=C1 Chemical compound CC1(CC1)C(=O)N[C@H]1[C@@H](N(C(C1)=O)C=1C=C2C=NN(C2=CC=1)C1=CN(C(C=C1)=O)C)C1=CC=CC=C1 VSPGWMCXTHEHDO-GJZUVCINSA-N 0.000 description 1
- JQLNYDHQOPZZFK-YPMHNXCESA-N CC1(CC1)C(=O)N[C@H]1[C@@H](NC(C1)=O)C1=CC=CC=C1 Chemical compound CC1(CC1)C(=O)N[C@H]1[C@@H](NC(C1)=O)C1=CC=CC=C1 JQLNYDHQOPZZFK-YPMHNXCESA-N 0.000 description 1
- IJSZMKIJAFOQAP-UHFFFAOYSA-N COC1=C(CN2C(C(CC2=O)(C(=O)O)SC2=CC=C(C=C2)C)C2=C(C=CC=C2)C)C=CC(=C1)OC Chemical compound COC1=C(CN2C(C(CC2=O)(C(=O)O)SC2=CC=C(C=C2)C)C2=C(C=CC=C2)C)C=CC(=C1)OC IJSZMKIJAFOQAP-UHFFFAOYSA-N 0.000 description 1
- LRQFSDHTCNEIOH-UHFFFAOYSA-N COC1=C(CN2C(C(CC2=O)(C(=O)O)SC2=CC=C(C=C2)C)C2=C(C=CC=C2)F)C=CC(=C1)OC Chemical compound COC1=C(CN2C(C(CC2=O)(C(=O)O)SC2=CC=C(C=C2)C)C2=C(C=CC=C2)F)C=CC(=C1)OC LRQFSDHTCNEIOH-UHFFFAOYSA-N 0.000 description 1
- RXBHCVNQAREXQQ-UHFFFAOYSA-N COC1=C(CN2C(C(CC2=O)(C(=O)O)SC2=CC=C(C=C2)C)C2=CC(=C(C=C2)F)OC)C=CC(=C1)OC Chemical compound COC1=C(CN2C(C(CC2=O)(C(=O)O)SC2=CC=C(C=C2)C)C2=CC(=C(C=C2)F)OC)C=CC(=C1)OC RXBHCVNQAREXQQ-UHFFFAOYSA-N 0.000 description 1
- AZOUAABQJRHODO-UHFFFAOYSA-N COC1=C(CN2C(C(CC2=O)(C(=O)O)SC2=CC=C(C=C2)C)C2=CC(=CC=C2)F)C=CC(=C1)OC Chemical compound COC1=C(CN2C(C(CC2=O)(C(=O)O)SC2=CC=C(C=C2)C)C2=CC(=CC=C2)F)C=CC(=C1)OC AZOUAABQJRHODO-UHFFFAOYSA-N 0.000 description 1
- SJUQDQGVRDAICN-UHFFFAOYSA-N COC1=C(CN2C(C(CC2=O)(C(=O)O)SC2=CC=C(C=C2)C)C2=CC(=NC=C2)OC)C=CC(=C1)OC Chemical compound COC1=C(CN2C(C(CC2=O)(C(=O)O)SC2=CC=C(C=C2)C)C2=CC(=NC=C2)OC)C=CC(=C1)OC SJUQDQGVRDAICN-UHFFFAOYSA-N 0.000 description 1
- QVUDQOAWJGHYFV-UHFFFAOYSA-N COC1=C(CN2C(C(CC2=O)(C(=O)O)SC2=CC=C(C=C2)C)C2=CC=C(C=C2)F)C=CC(=C1)OC Chemical compound COC1=C(CN2C(C(CC2=O)(C(=O)O)SC2=CC=C(C=C2)C)C2=CC=C(C=C2)F)C=CC(=C1)OC QVUDQOAWJGHYFV-UHFFFAOYSA-N 0.000 description 1
- KMDYXTJKEYBRFP-UHFFFAOYSA-N COC1=C(CN2C(C(CC2=O)(C(=O)OC)SC2=CC=C(C=C2)C)C2=C(C=CC(=C2)OC)F)C=CC(=C1)OC Chemical compound COC1=C(CN2C(C(CC2=O)(C(=O)OC)SC2=CC=C(C=C2)C)C2=C(C=CC(=C2)OC)F)C=CC(=C1)OC KMDYXTJKEYBRFP-UHFFFAOYSA-N 0.000 description 1
- RVBAWEIDAYQOTH-UHFFFAOYSA-N COC1=C(CN2C(C(CC2=O)(C(=O)OC)SC2=CC=C(C=C2)C)C2=C(C=CC=C2)C)C=CC(=C1)OC Chemical compound COC1=C(CN2C(C(CC2=O)(C(=O)OC)SC2=CC=C(C=C2)C)C2=C(C=CC=C2)C)C=CC(=C1)OC RVBAWEIDAYQOTH-UHFFFAOYSA-N 0.000 description 1
- KFMVYOMEFYLWHD-UHFFFAOYSA-N COC1=C(CN2C(C(CC2=O)(C(=O)OC)SC2=CC=C(C=C2)C)C2=C(C=CC=C2)F)C=CC(=C1)OC Chemical compound COC1=C(CN2C(C(CC2=O)(C(=O)OC)SC2=CC=C(C=C2)C)C2=C(C=CC=C2)F)C=CC(=C1)OC KFMVYOMEFYLWHD-UHFFFAOYSA-N 0.000 description 1
- XWOVXGBDXMKOEE-UHFFFAOYSA-N COC1=C(CN2C(C(CC2=O)(C(=O)OC)SC2=CC=C(C=C2)C)C2=CC(=C(C=C2)F)OC)C=CC(=C1)OC Chemical compound COC1=C(CN2C(C(CC2=O)(C(=O)OC)SC2=CC=C(C=C2)C)C2=CC(=C(C=C2)F)OC)C=CC(=C1)OC XWOVXGBDXMKOEE-UHFFFAOYSA-N 0.000 description 1
- DTKLNJLBCVEKFJ-UHFFFAOYSA-N COC1=C(CN2C(C(CC2=O)(C(=O)OC)SC2=CC=C(C=C2)C)C2=CC(=CC=C2)F)C=CC(=C1)OC Chemical compound COC1=C(CN2C(C(CC2=O)(C(=O)OC)SC2=CC=C(C=C2)C)C2=CC(=CC=C2)F)C=CC(=C1)OC DTKLNJLBCVEKFJ-UHFFFAOYSA-N 0.000 description 1
- YDIOHRBMCUZINY-UHFFFAOYSA-N COC1=C(CN2C(C(CC2=O)(C(=O)OC)SC2=CC=C(C=C2)C)C2=CC(=NC=C2)OC)C=CC(=C1)OC Chemical compound COC1=C(CN2C(C(CC2=O)(C(=O)OC)SC2=CC=C(C=C2)C)C2=CC(=NC=C2)OC)C=CC(=C1)OC YDIOHRBMCUZINY-UHFFFAOYSA-N 0.000 description 1
- NIUQQUCJPTZASJ-UHFFFAOYSA-N COC1=C(CN2C(C(CC2=O)(C(=O)OC)SC2=CC=C(C=C2)C)C2=CC=C(C=C2)F)C=CC(=C1)OC Chemical compound COC1=C(CN2C(C(CC2=O)(C(=O)OC)SC2=CC=C(C=C2)C)C2=CC=C(C=C2)F)C=CC(=C1)OC NIUQQUCJPTZASJ-UHFFFAOYSA-N 0.000 description 1
- JRBOHPRZEDVGTP-UHFFFAOYSA-N COC1=NC=CC(=C1)C1NC(CC1(C(=O)OC)SC1=CC=C(C=C1)C)=O Chemical compound COC1=NC=CC(=C1)C1NC(CC1(C(=O)OC)SC1=CC=C(C=C1)C)=O JRBOHPRZEDVGTP-UHFFFAOYSA-N 0.000 description 1
- LZRBJGWOWIQBEZ-UHFFFAOYSA-N ClC=1C=C(C=CC=1)C1NC(CC1(C(=O)OC)SC1=CC=C(C=C1)C)=O Chemical compound ClC=1C=C(C=CC=1)C1NC(CC1(C(=O)OC)SC1=CC=C(C=C1)C)=O LZRBJGWOWIQBEZ-UHFFFAOYSA-N 0.000 description 1
- DRSMAUNXCUWHEN-UHFFFAOYSA-N ClC=1C=C(C=CC=1)C1NC(CC1C(=O)OC)=O Chemical compound ClC=1C=C(C=CC=1)C1NC(CC1C(=O)OC)=O DRSMAUNXCUWHEN-UHFFFAOYSA-N 0.000 description 1
- FNCZNVMOIARYJX-YKSBVNFPSA-N ClC=1C=C(C=CC=1)[C@@H]1N(C(C[C@H]1NC(C(C)(F)F)=O)=O)C=1C=C2C=NN(C2=CC=1)C1=CN(C(C=C1)=O)C Chemical compound ClC=1C=C(C=CC=1)[C@@H]1N(C(C[C@H]1NC(C(C)(F)F)=O)=O)C=1C=C2C=NN(C2=CC=1)C1=CN(C(C=C1)=O)C FNCZNVMOIARYJX-YKSBVNFPSA-N 0.000 description 1
- AGOFDKUTRYUMSA-APWZRJJASA-N ClC=1C=C(C=CC=1)[C@@H]1NC(C[C@H]1NC(OCC1=C(C=C(C=C1)OC)OC)=O)=O Chemical compound ClC=1C=C(C=CC=1)[C@@H]1NC(C[C@H]1NC(OCC1=C(C=C(C=C1)OC)OC)=O)=O AGOFDKUTRYUMSA-APWZRJJASA-N 0.000 description 1
- FNCZNVMOIARYJX-GBXCKJPGSA-N ClC=1C=C(C=CC=1)[C@H]1N(C(C[C@@H]1NC(C(C)(F)F)=O)=O)C=1C=C2C=NN(C2=CC=1)C1=CN(C(C=C1)=O)C Chemical compound ClC=1C=C(C=CC=1)[C@H]1N(C(C[C@@H]1NC(C(C)(F)F)=O)=O)C=1C=C2C=NN(C2=CC=1)C1=CN(C(C=C1)=O)C FNCZNVMOIARYJX-GBXCKJPGSA-N 0.000 description 1
- YZCKVEUIGOORGS-OUBTZVSYSA-N Deuterium Chemical compound [2H] YZCKVEUIGOORGS-OUBTZVSYSA-N 0.000 description 1
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 1
- UDRZGZQEYVWOII-UHFFFAOYSA-N FC(C(=O)NC1C(N(C(C1)=O)C=1C=C2C=NN(C2=CC=1)CC1=CC(N(C=C1)C)=O)C1=CC=CC=C1)(C)F Chemical compound FC(C(=O)NC1C(N(C(C1)=O)C=1C=C2C=NN(C2=CC=1)CC1=CC(N(C=C1)C)=O)C1=CC=CC=C1)(C)F UDRZGZQEYVWOII-UHFFFAOYSA-N 0.000 description 1
- BMNOWNFLYRGRMF-QREQTRJTSA-N FC(C(=O)N[C@H]1[C@@H](N(C(C1)=O)C=1C=C2C=NN(C2=CC=1)C1OCCCC1)C1=CC=CC=C1)(C)F Chemical compound FC(C(=O)N[C@H]1[C@@H](N(C(C1)=O)C=1C=C2C=NN(C2=CC=1)C1OCCCC1)C1=CC=CC=C1)(C)F BMNOWNFLYRGRMF-QREQTRJTSA-N 0.000 description 1
- PXWHBSFZDBPOGM-YKSBVNFPSA-N FC(C(=O)N[C@H]1[C@@H](N(C(C1)=O)C=1C=C2C=NN(C2=CC=1)C=1C=NC(=CC=1)OC)C1=CC=CC=C1)(C)F Chemical compound FC(C(=O)N[C@H]1[C@@H](N(C(C1)=O)C=1C=C2C=NN(C2=CC=1)C=1C=NC(=CC=1)OC)C1=CC=CC=C1)(C)F PXWHBSFZDBPOGM-YKSBVNFPSA-N 0.000 description 1
- BMDYCKKVCBGQOM-UHFFFAOYSA-N FC1=C(C=C(C=C1)C1NC(CC1(C(=O)OC)SC1=CC=C(C=C1)C)=O)OC Chemical compound FC1=C(C=C(C=C1)C1NC(CC1(C(=O)OC)SC1=CC=C(C=C1)C)=O)OC BMDYCKKVCBGQOM-UHFFFAOYSA-N 0.000 description 1
- QMHDEPJVSMFACZ-UHFFFAOYSA-N FC1=C(C=C(C=C1)C1NC(CC1C(=O)OC)=O)OC Chemical compound FC1=C(C=C(C=C1)C1NC(CC1C(=O)OC)=O)OC QMHDEPJVSMFACZ-UHFFFAOYSA-N 0.000 description 1
- NHKOAXZIIVQDAK-UHFFFAOYSA-N FC1=C(C=C(C=C1)OC)C1NC(CC1(C(=O)OC)SC1=CC=C(C=C1)C)=O Chemical compound FC1=C(C=C(C=C1)OC)C1NC(CC1(C(=O)OC)SC1=CC=C(C=C1)C)=O NHKOAXZIIVQDAK-UHFFFAOYSA-N 0.000 description 1
- FQCZMUCEYOLDLN-UHFFFAOYSA-N FC1=C(C=CC(=C1)F)C1NC(CC1C(=O)OC)=O Chemical compound FC1=C(C=CC(=C1)F)C1NC(CC1C(=O)OC)=O FQCZMUCEYOLDLN-UHFFFAOYSA-N 0.000 description 1
- CCYAROILGIKCAV-UHFFFAOYSA-N FC1=C(C=CC(=C1)F)SC1(C(NC(C1)=O)C1=CC=CC=C1)C(=O)OC Chemical compound FC1=C(C=CC(=C1)F)SC1(C(NC(C1)=O)C1=CC=CC=C1)C(=O)OC CCYAROILGIKCAV-UHFFFAOYSA-N 0.000 description 1
- JTHBRKPDAZQMMN-UHFFFAOYSA-N FC1=C(C=CC=C1)C1NC(CC1(C(=O)OC)SC1=CC=C(C=C1)C)=O Chemical compound FC1=C(C=CC=C1)C1NC(CC1(C(=O)OC)SC1=CC=C(C=C1)C)=O JTHBRKPDAZQMMN-UHFFFAOYSA-N 0.000 description 1
- PBRWWZKNRUGEGH-BKMJKUGQSA-N FC1=C(C=CC=C1)[C@H]1N(C(C[C@@H]1NC(=O)C1CC1)=O)C=1C=C2C=NN(C2=CC=1)C1=CN(C(C=C1)=O)C Chemical compound FC1=C(C=CC=C1)[C@H]1N(C(C[C@@H]1NC(=O)C1CC1)=O)C=1C=C2C=NN(C2=CC=1)C1=CN(C(C=C1)=O)C PBRWWZKNRUGEGH-BKMJKUGQSA-N 0.000 description 1
- LNRODCMNTLSLBY-UHFFFAOYSA-N FC1=CC=C(C=C1)C1NC(CC1(C(=O)OC)SC1=CC=C(C=C1)C)=O Chemical compound FC1=CC=C(C=C1)C1NC(CC1(C(=O)OC)SC1=CC=C(C=C1)C)=O LNRODCMNTLSLBY-UHFFFAOYSA-N 0.000 description 1
- PRTUJBYBFFMLQS-UHFFFAOYSA-N FC=1C=C(C=CC=1)C1NC(CC1(C(=O)OC)SC1=CC=C(C=C1)C)=O Chemical compound FC=1C=C(C=CC=1)C1NC(CC1(C(=O)OC)SC1=CC=C(C=C1)C)=O PRTUJBYBFFMLQS-UHFFFAOYSA-N 0.000 description 1
- 208000010412 Glaucoma Diseases 0.000 description 1
- 241000282412 Homo Species 0.000 description 1
- 206010020751 Hypersensitivity Diseases 0.000 description 1
- 206010020772 Hypertension Diseases 0.000 description 1
- IIQUHLNMQQKHQD-UHFFFAOYSA-N I.c1n[nH]c2ccccc12 Chemical class I.c1n[nH]c2ccccc12 IIQUHLNMQQKHQD-UHFFFAOYSA-N 0.000 description 1
- WBYKAQFELFTKTA-UHFFFAOYSA-N IC=1C=C2C=NN(C2=CC=1)C1CCN(CC1)C(=O)OC(C)(C)C Chemical compound IC=1C=C2C=NN(C2=CC=1)C1CCN(CC1)C(=O)OC(C)(C)C WBYKAQFELFTKTA-UHFFFAOYSA-N 0.000 description 1
- 208000022559 Inflammatory bowel disease Diseases 0.000 description 1
- 206010065390 Inflammatory pain Diseases 0.000 description 1
- 206010022489 Insulin Resistance Diseases 0.000 description 1
- ZDXPYRJPNDTMRX-VKHMYHEASA-N L-glutamine Chemical compound OC(=O)[C@@H](N)CCC(N)=O ZDXPYRJPNDTMRX-VKHMYHEASA-N 0.000 description 1
- 241000124008 Mammalia Species 0.000 description 1
- 239000012359 Methanesulfonyl chloride Substances 0.000 description 1
- 206010028289 Muscle atrophy Diseases 0.000 description 1
- DEBIUXTUEMHXKO-WMZHIEFXSA-N N[C@@H]1[C@H](N(C(C1)=O)C=1C=C2C=NN(C2=CC=1)C=1C=CC(N(C=1)C)=O)C1=CC=CC=C1 Chemical compound N[C@@H]1[C@H](N(C(C1)=O)C=1C=C2C=NN(C2=CC=1)C=1C=CC(N(C=1)C)=O)C1=CC=CC=C1 DEBIUXTUEMHXKO-WMZHIEFXSA-N 0.000 description 1
- CPZYMQAZDGEHJL-UHFFFAOYSA-N O1C2=C(OCC1)C=C(C=C2)C1NC(CC1(C(=O)O)SC1=CC=C(C=C1)C)=O Chemical compound O1C2=C(OCC1)C=C(C=C2)C1NC(CC1(C(=O)O)SC1=CC=C(C=C1)C)=O CPZYMQAZDGEHJL-UHFFFAOYSA-N 0.000 description 1
- SVLOVJSPMDWAKB-UHFFFAOYSA-N O1C2=C(OCC1)C=C(C=C2)C1NC(CC1(C(=O)OC)SC1=CC=C(C=C1)C)=O Chemical compound O1C2=C(OCC1)C=C(C=C2)C1NC(CC1(C(=O)OC)SC1=CC=C(C=C1)C)=O SVLOVJSPMDWAKB-UHFFFAOYSA-N 0.000 description 1
- CRUWGWBSCYGTPY-UHFFFAOYSA-N O1C2=C(OCC1)C=C(C=C2)C1NC(CC1C(=O)OC)=O Chemical compound O1C2=C(OCC1)C=C(C=C2)C1NC(CC1C(=O)OC)=O CRUWGWBSCYGTPY-UHFFFAOYSA-N 0.000 description 1
- GKENHEXFJZQTFN-UHFFFAOYSA-N O=C1CC(C(N1)C1=C(C=CC=C1)C)(C(=O)OC)SC1=CC=C(C=C1)C Chemical compound O=C1CC(C(N1)C1=C(C=CC=C1)C)(C(=O)OC)SC1=CC=C(C=C1)C GKENHEXFJZQTFN-UHFFFAOYSA-N 0.000 description 1
- PEMAIDFXQSVOIL-UHFFFAOYSA-N O=C1CC(C(N1)C1=C(C=CC=C1)C)C(=O)OC Chemical compound O=C1CC(C(N1)C1=C(C=CC=C1)C)C(=O)OC PEMAIDFXQSVOIL-UHFFFAOYSA-N 0.000 description 1
- SZUZCNUTOZNOAB-UHFFFAOYSA-N O=C1CC(CN1)(C(=O)O)SC1=CC=C(C=C1)C Chemical compound O=C1CC(CN1)(C(=O)O)SC1=CC=C(C=C1)C SZUZCNUTOZNOAB-UHFFFAOYSA-N 0.000 description 1
- RKFLUPNAWVPATL-WBVHZDCISA-N O=C1C[C@H]([C@@H](N1)C1=CC=CC=C1)NC(OCC1=CC=CC=C1)=O Chemical compound O=C1C[C@H]([C@@H](N1)C1=CC=CC=C1)NC(OCC1=CC=CC=C1)=O RKFLUPNAWVPATL-WBVHZDCISA-N 0.000 description 1
- RWHIBAXNFAIJQB-LVVITFJHSA-N O[C@@H](C(=O)O)[C@H](C(=O)O)O.N[C@@H]1CC(N[C@H]1C1=CC=CC=C1)=O Chemical compound O[C@@H](C(=O)O)[C@H](C(=O)O)O.N[C@@H]1CC(N[C@H]1C1=CC=CC=C1)=O RWHIBAXNFAIJQB-LVVITFJHSA-N 0.000 description 1
- 229930182555 Penicillin Natural products 0.000 description 1
- JGSARLDLIJGVTE-MBNYWOFBSA-N Penicillin G Chemical compound N([C@H]1[C@H]2SC([C@@H](N2C1=O)C(O)=O)(C)C)C(=O)CC1=CC=CC=C1 JGSARLDLIJGVTE-MBNYWOFBSA-N 0.000 description 1
- 239000012980 RPMI-1640 medium Substances 0.000 description 1
- 238000001069 Raman spectroscopy Methods 0.000 description 1
- XUIMIQQOPSSXEZ-UHFFFAOYSA-N Silicon Chemical compound [Si] XUIMIQQOPSSXEZ-UHFFFAOYSA-N 0.000 description 1
- 239000007983 Tris buffer Substances 0.000 description 1
- YZCKVEUIGOORGS-NJFSPNSNSA-N Tritium Chemical compound [3H] YZCKVEUIGOORGS-NJFSPNSNSA-N 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 150000007513 acids Chemical class 0.000 description 1
- 230000004913 activation Effects 0.000 description 1
- 125000002015 acyclic group Chemical group 0.000 description 1
- 230000010933 acylation Effects 0.000 description 1
- 238000005917 acylation reaction Methods 0.000 description 1
- 230000001270 agonistic effect Effects 0.000 description 1
- 125000000217 alkyl group Chemical group 0.000 description 1
- 208000026935 allergic disease Diseases 0.000 description 1
- 230000007815 allergy Effects 0.000 description 1
- 230000003042 antagnostic effect Effects 0.000 description 1
- 125000002178 anthracenyl group Chemical group C1(=CC=CC2=CC3=CC=CC=C3C=C12)* 0.000 description 1
- 230000000947 anti-immunosuppressive effect Effects 0.000 description 1
- 229940124599 anti-inflammatory drug Drugs 0.000 description 1
- 239000008346 aqueous phase Substances 0.000 description 1
- 150000004945 aromatic hydrocarbons Chemical class 0.000 description 1
- 150000001502 aryl halides Chemical class 0.000 description 1
- 238000003149 assay kit Methods 0.000 description 1
- WAJGGTDMTRBQFE-BEFAXECRSA-N benzyl N-[(2S,3R)-2-(2,3-dihydro-1,4-benzodioxin-6-yl)-5-oxopyrrolidin-3-yl]carbamate Chemical compound O1C2=C(OCC1)C=C(C=C2)[C@@H]1NC(C[C@H]1NC(OCC1=CC=CC=C1)=O)=O WAJGGTDMTRBQFE-BEFAXECRSA-N 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- 230000037396 body weight Effects 0.000 description 1
- RBHJBMIOOPYDBQ-UHFFFAOYSA-N carbon dioxide;propan-2-one Chemical compound O=C=O.CC(C)=O RBHJBMIOOPYDBQ-UHFFFAOYSA-N 0.000 description 1
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 1
- 150000001805 chlorine compounds Chemical class 0.000 description 1
- 238000004587 chromatography analysis Methods 0.000 description 1
- 230000001684 chronic effect Effects 0.000 description 1
- 239000003086 colorant Substances 0.000 description 1
- 238000012875 competitive assay Methods 0.000 description 1
- 230000002860 competitive effect Effects 0.000 description 1
- 239000010949 copper Substances 0.000 description 1
- GBRBMTNGQBKBQE-UHFFFAOYSA-L copper;diiodide Chemical compound I[Cu]I GBRBMTNGQBKBQE-UHFFFAOYSA-L 0.000 description 1
- 239000006071 cream Substances 0.000 description 1
- 238000006880 cross-coupling reaction Methods 0.000 description 1
- 239000013078 crystal Substances 0.000 description 1
- 125000001162 cycloheptenyl group Chemical group C1(=CCCCCC1)* 0.000 description 1
- 125000000582 cycloheptyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000006547 cyclononyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C([H])([H])C1([H])[H] 0.000 description 1
- 125000000522 cyclooctenyl group Chemical group C1(=CCCCCCC1)* 0.000 description 1
- 125000000640 cyclooctyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C([H])([H])C1([H])[H] 0.000 description 1
- 229960001270 d- tartaric acid Drugs 0.000 description 1
- 230000001419 dependent effect Effects 0.000 description 1
- 229910052805 deuterium Inorganic materials 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- 125000004611 dihydroisoindolyl group Chemical group C1(NCC2=CC=CC=C12)* 0.000 description 1
- 239000003085 diluting agent Substances 0.000 description 1
- MKRTXPORKIRPDG-UHFFFAOYSA-N diphenylphosphoryl azide Chemical compound C=1C=CC=CC=1P(=O)(N=[N+]=[N-])C1=CC=CC=C1 MKRTXPORKIRPDG-UHFFFAOYSA-N 0.000 description 1
- 208000037765 diseases and disorders Diseases 0.000 description 1
- 229940079593 drug Drugs 0.000 description 1
- 238000001035 drying Methods 0.000 description 1
- 239000000839 emulsion Substances 0.000 description 1
- 125000005610 enamide group Chemical group 0.000 description 1
- RIFGWPKJUGCATF-UHFFFAOYSA-N ethyl chloroformate Chemical compound CCOC(Cl)=O RIFGWPKJUGCATF-UHFFFAOYSA-N 0.000 description 1
- 238000002474 experimental method Methods 0.000 description 1
- 239000000284 extract Substances 0.000 description 1
- 230000002349 favourable effect Effects 0.000 description 1
- KTWOOEGAPBSYNW-UHFFFAOYSA-N ferrocene Chemical compound [Fe+2].C=1C=C[CH-]C=1.C=1C=C[CH-]C=1 KTWOOEGAPBSYNW-UHFFFAOYSA-N 0.000 description 1
- 239000012091 fetal bovine serum Substances 0.000 description 1
- 239000000945 filler Substances 0.000 description 1
- 125000003983 fluorenyl group Chemical group C1(=CC=CC=2C3=CC=CC=C3CC12)* 0.000 description 1
- SAVROPQJUYSBDD-UHFFFAOYSA-N formyl(dimethyl)azanium;chloride Chemical compound [Cl-].C[N+](C)=CO SAVROPQJUYSBDD-UHFFFAOYSA-N 0.000 description 1
- 235000011389 fruit/vegetable juice Nutrition 0.000 description 1
- 125000002541 furyl group Chemical group 0.000 description 1
- 230000008297 genomic mechanism Effects 0.000 description 1
- 125000004836 hexamethylene group Chemical group [H]C([H])([*:2])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[*:1] 0.000 description 1
- 229930195733 hydrocarbon Natural products 0.000 description 1
- 150000002430 hydrocarbons Chemical class 0.000 description 1
- FUKUFMFMCZIRNT-UHFFFAOYSA-N hydron;methanol;chloride Chemical compound Cl.OC FUKUFMFMCZIRNT-UHFFFAOYSA-N 0.000 description 1
- 125000002883 imidazolyl group Chemical group 0.000 description 1
- 150000002466 imines Chemical class 0.000 description 1
- 238000001727 in vivo Methods 0.000 description 1
- 150000002473 indoazoles Chemical group 0.000 description 1
- 230000000670 limiting effect Effects 0.000 description 1
- DLEDOFVPSDKWEF-UHFFFAOYSA-N lithium butane Chemical compound [Li+].CCC[CH2-] DLEDOFVPSDKWEF-UHFFFAOYSA-N 0.000 description 1
- 239000006210 lotion Substances 0.000 description 1
- 230000014759 maintenance of location Effects 0.000 description 1
- 229910052751 metal Inorganic materials 0.000 description 1
- 239000002184 metal Substances 0.000 description 1
- LVWZTYCIRDMTEY-UHFFFAOYSA-N metamizole Chemical compound O=C1C(N(CS(O)(=O)=O)C)=C(C)N(C)N1C1=CC=CC=C1 LVWZTYCIRDMTEY-UHFFFAOYSA-N 0.000 description 1
- QARBMVPHQWIHKH-UHFFFAOYSA-N methanesulfonyl chloride Chemical compound CS(Cl)(=O)=O QARBMVPHQWIHKH-UHFFFAOYSA-N 0.000 description 1
- 230000020763 muscle atrophy Effects 0.000 description 1
- 201000000585 muscular atrophy Diseases 0.000 description 1
- VOVZXURTCKPRDQ-CQSZACIVSA-N n-[4-[chloro(difluoro)methoxy]phenyl]-6-[(3r)-3-hydroxypyrrolidin-1-yl]-5-(1h-pyrazol-5-yl)pyridine-3-carboxamide Chemical compound C1[C@H](O)CCN1C1=NC=C(C(=O)NC=2C=CC(OC(F)(F)Cl)=CC=2)C=C1C1=CC=NN1 VOVZXURTCKPRDQ-CQSZACIVSA-N 0.000 description 1
- 125000001624 naphthyl group Chemical group 0.000 description 1
- 210000003739 neck Anatomy 0.000 description 1
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 description 1
- 125000002347 octyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 239000002674 ointment Substances 0.000 description 1
- 239000012074 organic phase Substances 0.000 description 1
- 125000003566 oxetanyl group Chemical group 0.000 description 1
- 229910052760 oxygen Inorganic materials 0.000 description 1
- QNGNSVIICDLXHT-UHFFFAOYSA-N para-ethylbenzaldehyde Natural products CCC1=CC=C(C=O)C=C1 QNGNSVIICDLXHT-UHFFFAOYSA-N 0.000 description 1
- 239000004031 partial agonist Substances 0.000 description 1
- 229940049954 penicillin Drugs 0.000 description 1
- 125000004817 pentamethylene group Chemical group [H]C([H])([*:2])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[*:1] 0.000 description 1
- 239000000546 pharmaceutical excipient Substances 0.000 description 1
- 229940124531 pharmaceutical excipient Drugs 0.000 description 1
- 239000002831 pharmacologic agent Substances 0.000 description 1
- 125000001791 phenazinyl group Chemical group C1(=CC=CC2=NC3=CC=CC=C3N=C12)* 0.000 description 1
- 229910000160 potassium phosphate Inorganic materials 0.000 description 1
- 235000011009 potassium phosphates Nutrition 0.000 description 1
- 239000000843 powder Substances 0.000 description 1
- 229960005205 prednisolone Drugs 0.000 description 1
- OIGNJSKKLXVSLS-VWUMJDOOSA-N prednisolone Chemical compound O=C1C=C[C@]2(C)[C@H]3[C@@H](O)C[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 OIGNJSKKLXVSLS-VWUMJDOOSA-N 0.000 description 1
- 229960004618 prednisone Drugs 0.000 description 1
- XOFYZVNMUHMLCC-ZPOLXVRWSA-N prednisone Chemical compound O=C1C=C[C@]2(C)[C@H]3C(=O)C[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 XOFYZVNMUHMLCC-ZPOLXVRWSA-N 0.000 description 1
- 230000002265 prevention Effects 0.000 description 1
- QQONPFPTGQHPMA-UHFFFAOYSA-N propylene Natural products CC=C QQONPFPTGQHPMA-UHFFFAOYSA-N 0.000 description 1
- 125000004805 propylene group Chemical group [H]C([H])([H])C([H])([*:1])C([H])([H])[*:2] 0.000 description 1
- 238000000159 protein binding assay Methods 0.000 description 1
- 102000004169 proteins and genes Human genes 0.000 description 1
- 230000005588 protonation Effects 0.000 description 1
- 125000000561 purinyl group Chemical group N1=C(N=C2N=CNC2=C1)* 0.000 description 1
- 125000003373 pyrazinyl group Chemical group 0.000 description 1
- 125000002098 pyridazinyl group Chemical group 0.000 description 1
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 1
- JUJWROOIHBZHMG-UHFFFAOYSA-O pyridinium Chemical compound C1=CC=[NH+]C=C1 JUJWROOIHBZHMG-UHFFFAOYSA-O 0.000 description 1
- 125000000168 pyrrolyl group Chemical group 0.000 description 1
- 238000003653 radioligand binding assay Methods 0.000 description 1
- 230000035484 reaction time Effects 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 229910052710 silicon Inorganic materials 0.000 description 1
- 239000010703 silicon Substances 0.000 description 1
- 239000012279 sodium borohydride Substances 0.000 description 1
- 229910000033 sodium borohydride Inorganic materials 0.000 description 1
- 235000009518 sodium iodide Nutrition 0.000 description 1
- 239000011684 sodium molybdate Substances 0.000 description 1
- 235000015393 sodium molybdate Nutrition 0.000 description 1
- TVXXNOYZHKPKGW-UHFFFAOYSA-N sodium molybdate (anhydrous) Chemical compound [Na+].[Na+].[O-][Mo]([O-])(=O)=O TVXXNOYZHKPKGW-UHFFFAOYSA-N 0.000 description 1
- 239000011343 solid material Substances 0.000 description 1
- 230000009870 specific binding Effects 0.000 description 1
- 238000004611 spectroscopical analysis Methods 0.000 description 1
- 239000012258 stirred mixture Substances 0.000 description 1
- 229960005322 streptomycin Drugs 0.000 description 1
- 239000012134 supernatant fraction Substances 0.000 description 1
- 239000000829 suppository Substances 0.000 description 1
- 201000006489 suppurative thyroiditis Diseases 0.000 description 1
- 235000020357 syrup Nutrition 0.000 description 1
- 239000006188 syrup Substances 0.000 description 1
- 230000008685 targeting Effects 0.000 description 1
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 1
- 125000003039 tetrahydroisoquinolinyl group Chemical group C1(NCCC2=CC=CC=C12)* 0.000 description 1
- 125000000147 tetrahydroquinolinyl group Chemical group N1(CCCC2=CC=CC=C12)* 0.000 description 1
- 125000000383 tetramethylene group Chemical group [H]C([H])([*:1])C([H])([H])C([H])([H])C([H])([H])[*:2] 0.000 description 1
- 125000003831 tetrazolyl group Chemical group 0.000 description 1
- 238000002560 therapeutic procedure Methods 0.000 description 1
- 125000001113 thiadiazolyl group Chemical group 0.000 description 1
- 206010043778 thyroiditis Diseases 0.000 description 1
- 230000000699 topical effect Effects 0.000 description 1
- 125000004306 triazinyl group Chemical group 0.000 description 1
- 125000001425 triazolyl group Chemical group 0.000 description 1
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 1
- 229910052722 tritium Inorganic materials 0.000 description 1
- 208000001072 type 2 diabetes mellitus Diseases 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/06—Antiasthmatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/14—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing three or more hetero rings
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Rheumatology (AREA)
- Pulmonology (AREA)
- Neurosurgery (AREA)
- Neurology (AREA)
- Pain & Pain Management (AREA)
- Biomedical Technology (AREA)
- Immunology (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Physical Education & Sports Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
Abstract
La invención se refiere a compuestos según la fórmula general (I), que actúan como moduladores del receptor de glucocorticoides y pueden usarse en el tratamiento y/o profilaxis de trastornos que están mediados al menos parcialmente por el receptor de glucocorticoides. (Traducción automática con Google Translate, sin valor legal)
Description
DESCRIPCIÓN
Amidas de pirrolidinas sustituidas I
La invención se refiere a compuestos según la fórmula general (I),

que actúa como un modulador del receptor de glucocorticoides y se puede usar en el tratamiento y/o la profilaxis de trastornos que están al menos parcialmente mediados por el receptor de glucocorticoides.
Los glucocorticoides (GC) tienen fuertes efectos antiinflamatorios, inmunosupresores y terapéuticos de modificación de enfermedad, mediados por el receptor de glucocorticoides (RG) Se han utilizado ampliamente para tratar enfermedades inflamatorias e inmunitarias durante décadas y aún representan la terapia más efectiva en esas condiciones. Sin embargo, el tratamiento crónico con GC de enfermedades inflamatorias como asma, artritis reumatoide, enfermedad inflamatoria intestinal, enfermedad pulmonar obstructiva crónica, síndrome de dificultad respiratoria aguda, fibrosis quística, osteoartritis, polimialgia reumática y arteritis de células gigantes se ve obstaculizado por los efectos adversos asociados con los GC. Estos efectos secundarios no deseados incluyen resistencia a la insulina, diabetes, hipertensión, glaucoma, depresión, osteoporosis, supresión suprarrenal y atrofia muscular, la osteoporosis y la diabetes siendo las más graves desde el punto de vista del médico (Hapgood JP. y col, Pharmacol Ther. Septiembre de 2016, 165: 93-113; Buttgereit F. y col., Clin Exp Rheumatol. 2015 Jul-Aug;33(Sup. 492):S29-33; Hartmann K. y col., Physiol Rev. 2016 Apr;96(2):409-47).
Un ejemplo de un glucocorticoide oral es la prednisona, que se prescribe con frecuencia para el tratamiento de varios trastornos inflamatorios (De Bosscher K. y col., Trends Pharmacol Sci. Enero de 2016;37(1):4-16 ; Buttgereit F. y col., JAMA. 2016;315(22):2442-2458 ). Como los GC causan supresión suprarrenal, los síntomas de abstinencia de prednisolona pueden ser severos si el medicamento se suspende abruptamente cuando todos los signos de la enfermedad han desaparecido. Por lo tanto, la reducción gradual de GC a dosis fisiológicas es con frecuencia parte de los protocolos de tratamiento para reducir el riesgo de recaída y otros síntomas de abstinencia (Liu D. y col., Allergy Asthma Clin Immunol. Agosto de 2013 15;9(1):30). Por lo tanto, existe una gran necesidad médica de nuevos fármacos antiinflamatorios potentes con menos efectos adversos.
La investigación reciente se ha centrado en el desarrollo de agonistas parciales o moduladores selectivos de los receptores de glucocorticoides que activan las vías de inhibición de la inflamación pero evitan dirigirse a las vías que conducen a los efectos adversos asociados con los GC. Se ha demostrado que la mayoría de estos efectos están mediados por diferentes mecanismos genómicos dependientes del RG, denominados transactivación y transrepresión. Las acciones antiinflamatorias de g C se atribuyen principalmente a la transrepresión de genes inflamatorios, mientras que ciertos efectos secundarios están mediados predominantemente por la transactivación de varios genes. Según la naturaleza de un ligando, el RG puede modularse selectivamente en una conformación específica que favorece la transrepresión sobre la transactivación, lo que resulta en un beneficio terapéutico mejorado (De Bosscher K y col., Trends Pharmacol Sci. Enero de 2016;37(1):4-16). El concepto de tales ligandos de disociación ya se definió hace alrededor de dos décadas y se han identificado y evaluado varios compuestos en pruebas preclínicas y clínicas, pero ninguno de ellos ha sido aprobado todavía para el uso clínico.
Los compuestos que están activos como moduladores del receptor de glucocorticoides también se conocen, por ejemplo, de los documentos WO 2007/122165, WO 2008/076048 y WO 2008/043789, WO 2009/035067, WO 2009/142571, WO 2016/046260, WO 2017/034006. WO 2006/108699, WO 2008/063116 y WO 2009/142569.
Los compuestos de 4-(piperazin-1-il)-pirrolidin-2-ona se conocen a partir del documento WO 2015/099196.
Era un objetivo de la invención proporcionar nuevos compuestos que son moduladores del receptor de
glucocorticoides y que preferiblemente tienen ventajas sobre los compuestos de la técnica anterior. En particular, los nuevos compuestos deben ser adecuados para su uso en el tratamiento y/o profilaxis de trastornos o enfermedades que están mediados al menos parcialmente por el receptor de glucocorticoides. Este objetivo se ha conseguido por la materia objeto de las reivindicaciones de patente.
Sorprendentemente, se descubrió que los compuestos según la invención son moduladores muy potentes del receptor de glucocorticoides.
La invención se refiere a un compuesto según la fórmula general (I)

donde
Ri representa un -C1-10-alquilo; un -C3-10-cicloalquilo; un -C1-6-alquileno-C3-10-cicloalquilo; un heterocicloalquilo de 3 a 7 miembros; un -C1-6-alquileno-(heterocicloalquilo de 3 a 7 miembros); un arilo; un -C1-6-alquileno-arilo; un heteroarilo de 5 o 6 miembros; o un -C1-6-alquileno-(heteroarilo de 5 o 6 miembros); R2 representa un -C(=O)-C1-10-alquilo; un -C(=O)-C3-10-cicloalquilo; un -C(=O)-C1-6-alquileno-C3-10-cicloalquilo; un -C(=O)-(heterocicloalquilo de 3 a 7 miembros); un -C(=O)-C1-6-alquileno-(heterocicloalquilo de 3 a 7 miembros); un -C(=O)-arilo; un -C(=O)-C1-6-alquileno-arilo; un -C(=O)-(heteroarilo de 5 o 6 miembros); un -C(=O)-C1-6-alquileno-(heteroarilo de 5 o 6 miembros); un -S(=O)1-2-C1-10-alquilo; un -S(=O)1-2-C3-10-cicloalquilo; un -S(=O)1-2-C1-6-alquileno-C3-10-cicloalquilo; un -S(=O)1-2-(heterocicloalquilo de 3 a 7 miembros); un -S(=O)1-2-C1-6-alquileno-(heterocicloalquilo de 3 a 7 miembros); un -S(=O)1-2-arilo; un -S(=O)1-2-C1-6-alquileno-arilo; un -S(=O)1-2-(heteroarilo de 5 o 6 miembros); o un -S(=O)1-2-C1-6-alquileno-(heteroarilo de 5 o 6 miembros);
R3 representa un heterocicloalquilo de 3 a 7 miembros; un -C1-6-alquileno -(heterocicloalquilo de 3 a 7 miembros); heteroarilo de 5 o 6 miembros; -C1-6-alquileno-(heteroarilo de 5 o 6 miembros); -C(=O)-(heterocicloalquilo de 3 a 7 miembros); -C(=O)-C1-6-alquileno-(heterocicloalquilo de 3 a 7 miembros); -C(=o )-(heteroarilo de 5 o 6 miembros); -C(=O)-C1-6-alquileno-(heteroarilo de 5 o 6 miembros); -S(=O)1-2-(heterocicloalquilo de 3 a 7 miembros); -S(=O) 1-2-C1-6-alquileno-(heterocicloalquilo de 3 a 7 miembros); -S(=O)1-2-(heteroarilo de 5 o 6 miembros); o -S(=O) 1-2-C1-6-alquileno-(heteroarilo de 5 o 6 miembros);
R4 representa un -H; -F; -Cl; -Br; -I; -CN; -CF3 ; -CF2H; -CFH2 o un ciclopropilo;
X representa un CR5 ; donde R5 representa un -H; -F; -Cl; -Br; -I; -CN; -C1-10-alquilo o -C3-10-cicloalquilo;
Y representa un CR6; donde R6 representa un -H; -F; -Cl; -Br; -I; -CN; -C1-10-alquilo o -C3-10-cicloalquilo;
Z representa un CR7 ; donde R7 representa un -H; -F; -Cl; -Br; -I; -CN; -C1-10-alquilo o -C3-10-cicloalquilo;
donde -C1-10-alquilo, -C1-4-alquilo y -C1-6-alquileno- en cada caso independientemente entre sí son lineales o ramificados, saturados o insaturados;
donde un -C1-10-alquilo, -C1-4-alquilo, -C1-6-alquileno-, -C3-10-cicloalquilo y un heterocicloalquilo de 3 a 7 miembros, en cada caso independientemente entre sí, no están sustituidos, o están mono o polisustituidos con uno o más sustituyentes seleccionados de entre -F; -Cl; -Br; -I; -CN; -C1-6-alquilo; -CF3 ; -CF2H; -CFH2 ; -CF2Cl; -CFCl2 ; -C(=O)-C1-6-alquilo; -C(=O)-OH; -C(=O)-OC1-6-alquilo; -C(=O)-NH2 ; -C(=O)-NH(C1-6-alquilo); -C(=O)-N(C1-6-alquilol)2 ; -OH; =O; -OCF3 ; -OCF2 H; -OCFH2 ; - OCF2Cl; -OCFCh; -O-C1-6-alquilo; -O-C(=O)-C1-6-alquilo; -O-C(=O)-O-C1-6-alquilo; -O-(CO)-NH(C1-6-alquilo); -O-C(=O)-N(C1-6-alquilo)2 ; -O-S(=O)2-NH2 ; -O-S(=O)2-NH(C1-6-alquilo); -O-S(=O)2-N(C1-6-alquilo)2 ; -NH2 ; -NH(C1-6-alquilo); -N(C1-6-alquilo)2 ; -NH-C(=O)-C1-6-alquilo; -NH-C(=O)-O-C1-6-alquilo; -NH-C(=O)-NH2 ; -NH-C(=O)-NH(C1-6-alquilo); -NH-C(=O)-N(C1-6-alquilo)2 ; -N(C1-6alquilo)-C(=O)-Ci-6-alquilo; -N(Ci-6-alquilo)-C(=O)-O-Ci-6-alquilo; -N(Ci-6-alquilo)-C(=O)-NH2 ; -N(Ci-6-alquilo)-C(=O)-NH(Ci-6-alquilo); -N(Ci-6-alquilo)-C(=O)-N(Ci-6-alquilo)2 ; -NH-S(=O)2OH; NH-S(=O)2-C i-6-alquilo; -NH-S(=O)2-O-Ci-6-alquilo; -NH-S(=O)2-NH2 ; -NH-S(=O)2-NH(Ci-6-alquilo); -NH-S(=O)2N(Ci-6-alquilo)2 ; -N(Ci-6-alquilo)-S(=O)2-OH; -N(Ci-6-alquilo)-S(=O)2-C i-6-alquilo; -N(Ci-6-alquilo)-S(=O)2-O-Ci-6-alquilo; -N(Ci-6-alquilo)-S(=O)2-NH2 ; -N(Ci-6-alquilo)-S(=O)2-NH(Ci-6-alquilo); -N(Ci-6-alquilo)-S(=O)2-N(Ci-6-alquilo)2 ; -SCFa; -SCF2H; -SCFH2 ; -S-Ci-e-alquilo; -S(=O)-Ci-6-alquilo; -S(=O)2-C i-6-alquilo; -S(=O)2-OH; -S(=O)2-O-Ci-6-alquilo; -S(=O)2-NH2 ; -S(=O)2-NH(Ci-e-alquilo); - S(=O)2-N(Ci-6-alquilo)2 ; -C3-e-cicloalquilo; un heterocicloalquilo de 3 a 6 miembros; un fenilo; un heteroarilo de 5 o 6 miembros; -O-C3-6-cicloalquilo; -O-(un heterocicloalquilo de 3 a 6 miembros); -O-fenilo; -O-(heteroarilo de 5 a 6 miembros); -C(=O)-C3-6-cicloalquilo; -C(=O)-(heterocicloalquilo de 3 a 6 miembros); -C(=O)-fenilo; -C(=O)-(heteroarilo de 5 a 6 miembros); - S(=O)2-(C3-6-cicloalquilo; -S(=O)2-(heterocicloalquilo de 3 a 6 miembros); -S(=O)2-fenilo o -S(=O)2-(heteroarilo de 5 a 6 miembros);
donde un arilo y un heteroarilo de 5 o 6 miembros en cada caso, independientemente entre sí, no están sustituidos o están mono o polisustituidos con uno o más sustituyentes seleccionados de entre -F; -Cl; -Br; -I; -CN; -Ci-6-alquilo; -CF3 ; -CF2H; -CFH2 ; -CF2Cl; -CFCh; -Ci-4-alquileno-CF3; -Ci-4-alquileno-CF2H; -C1-4-alquileno-CFH2 ; -C(=O)-Ci-6-alquilo; -C(=O)-OH; - C(=O)-OCi-6-alquilo; -C(=O)-NH(OH); -C(=O)-NH2 ; -C(=O)-NH(Ci-6-alquilo); -C(=O)-N(Ci-6-alquilo)2 ; -OH; =O; -OCF3 ; -OCF2H; -OCFH2 ; -OCF2Cl; -OCFCl2 ; -O-C1-6-alquilo; -O-C3-6-cicloalquilo; -O-(heterocicloalquilo de 3 a 6 miembros); -NH2 ; -NH(Ci-6-alquilo); -N(Ci-6-alquilo)2 ; -NH-C(=O)-Ci-6-alquilo; -N(Ci-6-alquilo)-C(=O)-Ci-6-alquilo; -NH-C(=O)-NH2 ; -NH-C(=O)-NH(Ci-6-alquilo); -NH-C(=O)-N(Ci-6-alquilo)2 ; -N(Ci-6-alquilo)-C(=O)-NH(Ci-6-alquilo); -N(Ci-6-alquilo)-C(=O)-N(Ci-6-alquilo)2 ; -NH-S(=O)2-C i-6-alquilo; -SCF3 ; -S-Ci-6-alquilo; -S(=O)-Ci-6-alquilo; -S(=O)2-C i-6-alquilo; -S(=O)2-NH2 ; -S(=O)2-NH(Ci-6-alquilo); -S(=O)2-N(Ci-6-alquilo)2 ; -C3-6-cicloalquilo; -Ci-4-alquileno-C3-6-cicloalquilo; heterocicloalquilo de 3 a 6 miembros; -Ci-4-alquileno-(heterocicloalquilo de 3 a 6 miembros); fenilo o heteroarilo de 5 o 6 miembros;
en forma del compuesto libre o una sal fisiológicamente aceptable del mismo.
En una realización preferida, el compuesto según la invención está presente en la forma de un compuesto libre. A los efectos de la memoria descriptiva, "compuesto libre" significa preferiblemente que el compuesto según la invención no está presente en forma de sal. Los procedimientos para determinar si una sustancia química está presente como el compuesto libre o como una sal son conocidos para el experto en la materia, por ejemplo, la RMN de estado sólido 14N o 15N, difracción de rayos X, la difracción de rayos X de polvo, la RI, las imágenes Raman y la EFRX. La RMN H 1 registrada en una solución también se puede usar para considerar la presencia de la protonación.
En otra realización preferida, el compuesto según la invención está presente en forma de una sal fisiológicamente aceptable. A los efectos de esta memoria descriptiva, el término "sal fisiológicamente aceptable" se refiere preferiblemente a una sal obtenida a partir de un compuesto según la invención y un ácido o base fisiológicamente aceptable.
Según la invención, el compuesto según la invención puede estar presente en cualquier forma posible, incluidos solvatos, cocristales y polimorfos. A los efectos de esta memoria descriptiva, el término "solvato" se refiere preferiblemente a un aducto de (i) un compuesto según la invención y/o a una sal fisiológicamente aceptable del mismo con (ii) distintos equivalentes moleculares de uno o más solventes.
Además, el compuesto según la invención puede estar presente en forma de racemato, enantiómeros, diastereómeros, tautómeros o cualquier mezcla de los mismos.
La invención también incluye isómeros isotópicos de un compuesto según la invención, donde al menos un átomo de los compuestos se remplaza con un isótopo del átomo respectivo que es diferente del isótopo que se produce predominantemente de manera natural, así como también cualquier mezcla de isómeros isotópicos de dicho compuesto. Los isótopos preferidos son 2H (deuterio), 3H (tritio), 13C y 14C. Los isómeros isotópicos de un compuesto de la invención generalmente se pueden preparar mediante procedimientos convencionales conocidos por los expertos en la materia.
Según la invención, los términos "-Ci-10-alquilo", "-Ci-8-alquilo", "-Ci-6-alquilo" y "-Ci-4-alquilo" preferiblemente significan residuos de hidrocarburo alifáticos (es decir, no aromáticos) saturados o insaturados acíclicos, que pueden ser lineales (es decir, no ramificados) o ramificados, y que pueden no estar sustituidos o estar mono o polisustituidos, y que contienen de 1 a 10 (es decir, 1, 2, 3, 4, 5, 6, 7, 8, 9 o 10), de 1 a 8 (es decir, 1, 2, 3, 4, 5, 6, 7 u 8), de 1 a 6 (es decir, 1, 2, 3, 4, 5 o 6) y de 1 a 4 (es decir, 1, 2, 3 o 4) átomos de carbono, respectivamente. En una realización preferida, -C i-10-alquilo, -Ci-8-alquilo, -Ci-6-alquilo y -Ci-4-alquilo están saturados. En otra realización preferida, -C i-10-alquilo, -Ci-8-alquilo, -Ci-6-alquilo y -Ci-4-alquilo no están saturados. Según esta realización, -C i-10-alquilo, -Ci-8-alquilo, -Ci-6-alquilo y -Ci-4-alquilo comprenden al menos un doble enlace C-C (un enlace C=C) o al menos un triple enlace C-C (un enlace C=C). En incluso otra realización preferida, -C i-10-alquilo, -Ci-8-alquilo, -Ci-6-alquilo y -Ci-4-alquilo están (i) saturados o (ii) insaturados, donde -C i-10-alquilo, -Ci-8-alquilo, -Ci-6-alquilo y -Ci-4-alquilo comprenden al menos,
preferiblemente, un triple enlace C-C (un enlace C=C).
Los grupos -C i-10-alquilo preferidos se seleccionan de entre metilo, etilo, etenilo (vinilo), n-propilo, 2-propilo, 1-propinilo, 2-propinilo, propenilo (-CH2CH=CH2 , -CH=CH-CH3, -C(=CH2)-CH3), n-butilo, 1-butinilo, 2-butinilo, 1-butenilo, 2-butenilo, isobutilo, sec-butilo, terc-butilo, n-pentilo, 2-pentilo, 3-pentilo, 1-pentenilo, 2-pentenilo, 1-pentinilo, 2-pentinilo, 2-metilbutilo, 3-metilbutilo, 3-metilbut-2-ilo, 2-metilbut-2-ilo, 3-metilbut-1-inilo, 2,2-dimetilpropilo, n-hexilo, 2-hexilo, 3-hexilo, 2-metilpentilo, 4-metilpentilo, 4-metilpent-2-ilo, 2-metilpent-2-ilo, 3,3-dimetilbutilo, 3,3-dimetilbut-2-ilo, 3-metilpentilo, 3-metilpent-2-ilo y 3-metilpent-3-ilo; más preferiblemente metilo, etilo, n-propilo, 2-propilo, 1-propinilo, 2-propinilo, propenilo (-CH2CH=CH2 , -CH=CH-CH3, -C(=CH2)-CH3), n-butilo, 1-butinilo, 2-butinilo, 1-butenilo, 2-butenilo, isobutilo, sec-butilo, terc-butilo, n-pentilo, 2-pentilo, 3-pentilo, 1-pentenilo, 2-pentenilo, 1-pentinilo, 2-pentinilo, 2-metilbutilo, 3-metilbutilo, 3-metilbut-2-ilo, 2-metilbut-2-ilo, 3-metilbut-1-inilo, 2,2-dimetilpropilo, n-hexilo, n-heptilo, n-octilo, n-nonilo y n-decilo.
Los grupos -C1-8-alquilo preferidos se seleccionan de entre metilo, etilo, etenilo (vinilo), n-propilo, 2-propilo, 1-propinilo, 2-propinilo, propenilo (-CH2CH=CH2 , -CH=CH-CH3, -C(=CH2)-CH3), n-butilo, 1-butinilo, 2-butinilo, 1-butenilo, 2-butenilo, isobutilo, sec-butilo, terc-butilo, n-pentilo, 2-pentilo, 3-pentilo, 1-pentenilo, 2-pentenilo, 1-pentinilo, 2-pentinilo, 2-metilbutilo, 3-metilbutilo, 3-metilbut-2-ilo, 2-metilbut-2-ilo, 3-metilbut-1-inilo, 2,2-dimetilpropilo, n-hexilo, 2-hexilo, 3-hexilo, 2-metilpentilo, 4-metilpentilo, 4-metilpent-2-ilo, 2-metilpent-2-ilo, 3,3-dimetilbutilo, 3,3-dimetilbut-2-ilo, 3-metilpentilo, 3-metilpent-2-ilo y 3-metilpent-3-ilo; más preferiblemente metilo, etilo, n-propilo, 2-propilo, 1-propinilo, 2-propinilo, propenilo (-CH2CH=CH2 , -CH=CH-CH3, -C(=CH2)-CH3), n-butilo, 1-butinilo, 2-butinilo, 1-butenilo, 2-butenilo, isobutilo, sec-butilo, terc-butilo, n-pentilo, 2-pentilo, 3-pentilo, 1-pentenilo, 2-pentenilo, 1-pentinilo, 2-pentinilo, 2-metilbutilo, 3-metilbutilo, 3-metilbut-2-ilo, 2-metilbut-2-ilo, 3-metilbut-1-inilo, 2,2-dimetilpropilo, n-hexilo, n-heptilo y n-octilo.
Los grupos -C1-6-alquilo preferidos se seleccionan de entre metilo, etilo, etenilo (vinio), n-propilo, 2-propilo, nbutilo, isobutilo, sec-butilo, terc-butilo, n-pentilo, 2-pentilo, 3-pentilo, 2-metilbutilo, 3-metilbutilo, 3-metilbut-2-ilo, 2-metilbut-2-ilo, 2,2-dimetilpropilo, n-hexilo, 2-hexilo, 3-hexilo, 2-metilpentilo, 4-metilpentilo, 4-metilpent-2-ilo, 2-metilpent-2-ilo, 3,3-dimetilbutilo, 3,3-dimetilbut-2-ilo, 3-metilpentilo, 3-metilpent-2-ilo y 3-metilpent-3-ilo; más preferiblemente metilo, etilo, n-propilo, 2-propilo, 1-propinilo, 2-propinilo, propenilo (-CH2-CH=CH2 , -CH=CH-CH3 , -C(=CH2)-CH3), n-butilo, 1-butinilo, 2-butinilo, 1-butenilo, 2-butenilo, isobutilo, sec-butilo, terc-butilo, npentilo, 2-pentilo, 3,-pentilo, 1-pentenilo, 2-pentenilo, 1-pentinilo, 2-pentinilo, 2-metilbutilo, 3-metilbutilo, 3-metilbut-2-ilo, 2-metilbut-2-ilo, 3-metilbut-1-inilo, 2,2-dimetilpropilo, n-hexilo. Los grupos -C1-6-alquilo particularmente preferidos se seleccionaron de entre los grupos -C1-4-alquilo.
Los grupos -C1-4-alquilo preferidos se seleccionan de entre metilo, etilo, etenilo (vinilo), n-propilo, 2-propilo, 1-propinilo, 2-propinilo, propenilo (-CH2CH=CH2 , -CH=CH-CH3, -C(=CH2)-CH3), n-butilol, 1-butinilo, 2-butinilo, 1-butenilo, 2-butenilo, isobutilo, sec-butilo, terc-butilo y 3-metilbut-1-inilo.
Además, según la invención, los términos "-C1-6-alquileno-"; "-C1-4-alquileno-" y "-C1-2-alquileno-" se refieren a residuos lineales o ramificados, preferiblemente lineales y preferiblemente residuos alifáticos saturados que preferiblemente se seleccionan de entre el grupo que consiste en metileno (-CH2-), etileno (-CH2CH2-), propileno (-CH2CH2CH2- o -C(CH3)2-), butileno (-CH2CH2CH2CH2-), pentileno (-CH2CH2CH2CH2CH2-) y hexileno (-CH2CH2CH2CH2CH2CH2-); más preferiblemente metileno (-CH2-) y etileno (-CH2CH2-) y más preferiblemente metileno (-CH2-). Preferiblemente, el -C1-6-alquileno- se selecciona del -C1-4-alquileno-, más preferiblemente del -C1-2-alquileno-.
También, según la invención, los términos "-C3-10-cicloalquilo" y "-C3-6-cicloalquilo" preferiblemente significan hidrocarburos alifáticos cíclicos que contienen 3, 4, 5, 6, 7, 8, 9 o 10 átomos de carbono y 3, 4, 5 o 6 átomos de carbono, respectivamente, donde los hidrocarburos en cada caso pueden estar saturados o insaturados (sin ser aromáticos), no sustituidos, mono o polisustituidos.
Preferiblemente, -C3-10-cicloalquilo y -C3-6-cicloalquilo están saturados. El -C3-10-cicloalquilo y el -C3-6-cicloalquilo pueden estar unidos a la estructura general superordinada respectiva mediante cualquier miembro de anillo posible o deseado del grupo cicloalquilo. Los grupos -C3-10-cicloalquilo y -C3-6-cicloalquilo también se pueden condensar con sistemas de anillo saturados, (parcialmente) insaturados, (hetero)cíclicos, aromáticos o heteroaromáticos adicionales, es decir, con residuos cicloalquilo, heterociclilo, arilo o heteroarilo, los cuales, en cada caso, a su vez, pueden no estar sustituidos o estar mono o polisustituidos. Además, el -C3-10-cicloalquilo y el -C3-6-cicloalquilo se pueden puentear de manera simple o múltiple, como, por ejemplo, en el caso del adamantilo, biciclo[2.2.1]heptil o biciclo[2.2.2]octilo. Sin embargo, preferiblemente, ni el -C3-10-cicloalquilo ni el -C3-6-cicloalquilo se puentean ni condensan con sistemas de anillo adicionales. Más preferiblemente, ni el -C3-10-cicloalquilo ni el -C3-6-cicloalquilo están condensados con otros sistemas de anillo ni presentan puentes ni están saturados. Los grupos de -C3-10-cicloalquilo se seleccionan de entre el grupo que consiste en ciclopropilo, ciclobutilo, ciclopentilo, ciclohexilo, ciclopentenilo, ciclohexenilo, cicloheptilo, ciclooctilo, ciclononilo, ciclodecilo, adamantilo, ciclopentenilo, ciclohexenilo, cicloheptenilo, ciclooctenilo, biciclo[2.2.1]heptilo y biciclo[2.2.2]octilo. Los grupos -C3-10-cicloalquilo particularmente preferidos se seleccionaron de entre los grupos -C3-6-cicloalquilo.
Los grupos -C3-6-cicloalquilo preferidos se seleccionaron de entre el grupo que consiste en ciclopropilo, ciclobutilo, ciclopentilo, ciclohexilo, ciclopentenilo y ciclohexenilo. Los grupos -C3-6-cicloalquilo particularmente preferidos se seleccionaron de entre el grupo que consiste en ciclopropilo, ciclobutilo, ciclopentilo y ciclohexilo, más preferiblemente ciclopropilo.
Según la invención, los términos "heterocicloalquilo de 3 a 7 miembros" y "heterocicloalquilo de 3 a 6 miembros" preferiblemente significan residuos heterocicloalifáticos saturados o insaturados (pero no aromáticos) que tienen de 3 a 7, es decir, 3, 4, 5, 6 o 7 miembros de anillo y de 3 a 6, es decir, 3, 4, 5 o 6 miembros de anillo, respectivamente, donde, en cada caso, al menos uno, si corresponde, también dos o tres átomos de carbono son reemplazados con un heteroátomo o un grupo de heteroátomos, cada uno seleccionado independientemente entre sí de entre el grupo que consiste en O, S, S(=O), S(=O)2 , N, NH y N(C-M-alquilo), tal como N(CH3), donde los átomos de carbono del anillo pueden no estar sustituidos o estar mono o polisustituidos.
Preferiblemente, el heterocicloalquilo de 3 a 7 miembros y el heterocicloalquilo de 3 a 6 miembros están saturados. Los grupos heterocicloalquilo de 3 a 7 miembros y heterocicloalquilo de 3 a 6 miembros también se pueden condensar con otros sistemas de anillos cicloalquilo o heterociclilo, aromáticos o heteroaromáticos, saturados o (parcialmente) insaturados. Sin embargo, más preferiblemente, el heterocicloalquilo de 3 a 7 miembros y el heterocicloalquilo de 3 a 6 miembros no se condensan con otros sistemas de anillo. Aún más preferiblemente, el heterocicloalquilo de 3 a 7 miembros y el heterocicloalquilo de 3 a 6 miembros no se condensan con otros sistemas de anillo y están saturados. El heterocicloalquilo de 3 a 7 miembros y el grupo heterocicloalquilo de 3 a 6 miembros se pueden unir a la estructura general superordinada a través de cualquier miembro del anillo deseado y posible del residuo heterocicloalifático si no se indica lo contrario. En una realización preferida, el heterocicloalquilo de 3 a 7 miembros y el heterocicloalquilo de 3 a 6 miembros están unidos a la estructura general superordinada a través de un átomo de carbono.
Los grupos heterocicloalquilo preferidos de 3 a 7 miembros se seleccionan de entre el grupo que consiste en azepanilo, dioxepanilo, oxazepanilo, diazepanilo, tiazolidinilo, tetrahidrotiofenilo, tetrahidropiridinilo, tiomorfolinilo, tetrahidropiranilo, oxetanilo, oxiranilo, tetrahidrofuranilo, morfolinilo, pirrolidinilo, 4-metilpiperazinilo, morfolinonilo, azetidinilo, aziridinilo, ditiolanilo, dihidropirrolilo, dioxanilo, dioxolanilo, dihidropiridinilo, dihidrofuranilo, dihidroisoxazolilo, dihidrooxazolilo, imidazolidinilo, isoxazolidinilo, oxazolidinilo, piperazinilo, piperidinilo, pirazolidinilo, piranilo; tetrahidropirrolilo, dihidroquinolinilo, dihidroisoquinolinilo, dihidroindolinilo, dihidroisoindolilo, tetrahidroquinolinilo, tetrahidroisoquinolinilo y tetrahidroindolinilo. Los grupos heterocicloalquilo de 3 a 7 miembros particularmente preferidos se seleccionan de entre grupos heterocicloalquilo de 3 a 6 miembros.
Los grupos heterocicloalquilo preferidos de 3 a 6 miembros se seleccionan de entre el grupo que consiste en tetrahidropiranilo, oxetanilo, oxiranilo, tetrahidrofuranilo, tiazolidinilo, tetrahidrotiofenilo, tetrahidropiridinilo, tiomorfolinilo, morfolinilo, pirrolidinilo, 4-metilpiperazinilo, morfolinonilo, azetidinilo, aziridinilo, ditiolanilo, dihidropirrolilo, dioxanilo, dioxolanilo, dihidropiridinilo, dihidrofuranilo, dihidroisoxazolilo, dihidrooxazolilo, imidazolidinilo, isoxazolidinilo, oxazolidinilo, piperazinilo, piperidinilo, pirazolidinilo, piranilo; tetrahidropirrolilo, dihidroquinolinilo, dihidroisoquinolinilo, dihidroindolinilo, dihidroisoindolilo y tetrahidroindolinilo. Preferiblemente, los grupos de heterocicloalquilo de 3 a 6 miembros se seleccionan de entre el grupo que consiste en tetrahidropiranilo, oxetanilo, oxiranilo y tetrahidrofuranilo.
Según la invención, el término "arilo" significa preferiblemente hidrocarburos aromáticos que tienen de 6 a 14, es decir, 6, 7, 8, 9, 10, 11, 12, 13 o 14 miembros en el anillo, preferiblemente que tienen de 6 a 10, es decir, 6, 7, 8, 9 o 10 miembros del anillo, incluidos fenilos y naftilos. Cada residuo arilo puede estar sin sustituir, mono o polisustituido. El arilo se puede unir a la estructura general superordinada a través de cualquier miembro de anillo deseado y posible del residuo de arilo. Los residuos arilo también pueden estar condensados con otros sistemas de anillo cicloalquilo o heterocicloalquilo, aromáticos o heteroaromáticos, saturados o (parcialmente) insaturados, que, a su vez, pueden no estar sustituidos o estar mono o polisustituidos. En una realización preferida, el arilo se condensa con un sistema de anillo adicional. Los ejemplos de residuos de arilo condensados son el 2H-benzo[b][1,4]oxacin-3(4H)-onilo, 1H-benzo[d]imidazolilo, 2,3-dihidro-1H-indenilo, tetrahidronaftalenilo, isocromán, 1,3-dihidroisobenzofuranilo, benzodioxolanilo y benzodioxanilo.
Preferiblemente, el arilo se selecciona de entre el grupo que consiste en fenilo, 1H-benzo[d]imidazolilo, 2H-benzo[b][1,4]oxazin-3(4H)-onilo, 2,3-dihidro-1H-indenilo, tetrahidronaftalenilo, isocromán, 1,3-dihidroisobenzofuranilo, 1-naftilo, 2-naftilo, fluorenilo y antracenilo, cada uno de los cuales puede no estar sustituido o estar mono o polisustituido. En otra realización preferida, el arilo no se condensa con ningún sistema de anillo adicional. Un arilo particularmente preferido es fenilo, no sustituido o mono o polisustituido.
Según la invención, el término "heteroarilo de 5 a 6 miembros" significa preferiblemente un residuo aromático cíclico de 5 o 6 miembros que contiene al menos 1, dado el caso también 2, 3, 4 o 5 heteroátomos, donde cada uno de heteroátomos se selecciona independientemente entre sí del grupo que consiste en S, N y O y el residuo
heteroarilo puede no estar sustituido o estar mono o polisustituido, si no se indica lo contrario. En el caso de sustitución en el heteroarilo, los sustituyentes pueden ser iguales o diferentes y estar en cualquier posición deseada y posible del heteroarilo. La unión a la estructura general superordinada se puede llevar a cabo a través de cualquier miembro del anillo deseado y posible del residuo heteroarilo, si no se indica lo contrario. Preferiblemente, el heteroarilo de 5 a 6 miembros está unido a la estructura general supraordinada a través de un átomo de carbono del heterociclo. El heteroarilo también puede ser parte de un sistema bi- o policíclico que tiene hasta 14 miembros de anillo, donde el sistema de anillo puede estar formado con más sistemas de anillo cicloalquilo o heterocicloalquilo saturados o (parcialmente) insaturados, aromáticos o heteroaromáticos, que, a su vez, pueden no estar sustituidos o estar mono o polisustituidos, si no se indica lo contrario. En una realización preferida, el heteroarilo de 5 a 6 miembros es parte de un sistema bicíclico o policíclico, preferiblemente bicíclico. En otra realización preferida, el heteroarilo de 5 a 6 miembros no forma parte de un sistema bicíclico o policíclico bicíclico.
Preferiblemente, el heteroarilo de 5 a 6 miembros se selecciona de entre el grupo que consiste en piridilo (es decir, 2-piridilo, 3-piridilo, 4-piridilo), piridona (piridinona), pirimidinilo, piridazinilo, pirazinilo, pirrolilo, imidazolilo, pirazolilo, oxazolilo, isoxazolilo, tiazolilo, isotiazolilo, furanilo, tienilo (tiofenilo), triazolilo, tiadiazolilo, 4,5,6,7-tetrahidro-2H-indazolilo, 2,4,5,6-tetrahidrociclopenta[c]pirazolilo, benzofuranilo, benzoimidazolilo, benzotienilo, benzotiadiazolilo, benzotiazolilo, benzotriazolilo, benzooxazolilo, benzooxadiazolilo, quinazolinilo, quinoxalinilo, carbazolilo, quinolinilo, dibenzofuranilo, dibenzotienilo, imidazotiazolilo, indazolilo, indolizinilo, indolilo, isoquinolinilo, napthyridinilo, oxazolilo, oxadiazolilo, fenazinilo, fenotiazinilo, ftalazinilo, purinilo, fenazinilo, tetrazolilo y triazinilo. Los heteroarilo de 5 a 6 miembros particularmente preferidos se seleccionan de entre el grupo que consiste en piridilo (es decir, 2-piridilo, 3-piridilo, 4-piridilo). Como piridonas pueden considerarse piridinas que están sustituidas con =O, a los efectos de la memoria descriptiva, la definición de piridinas que pueden sustituirse opcionalmente por =O cubre las piridonas.
Los compuestos según la invención se definen mediante los sustituyentes, por ejemplo, mediante Ri, R2 , R3 y R4 (sustituyentes de 1era generación), que, a su vez, opcionalmente pueden estar sustituidos (sustituyentes de 2da generación). Dependiendo de la definición, estos sustituyentes de los sustituyentes pueden estar opcionalmente resustituidos en parte (sustituyentes de3ra generación). Si, por ejemplo, Ri = -C-M0-alquilo (sustituyente de 1era generación), a continuación, el -C-M0-alquilo puede, por su parte, estar sustituido, por ejemplo, con un -NH(C1-6-alquilo) (sustituyente de 2da generación). Esto produce el grupo funcional Ri = (-C1-10-alquilo-NH-C1-6-alquilo). El -NH-C1-6-alquilo, a continuación, puede, por su parte, volver a sustituirse, por ejemplo, con -Cl (sustituyente de 3era generación). En general, esto produce el grupo funcional Ri = -C1-10-alquilo-NH-C1-6-alquilo, donde el -C1-6-alquilo del -NH-C1-6-alquilo se sustituye con -Cl.
Sin embargo, en una realización preferida de la invención, los sustituyentes de 3era generación pueden no volver a sustituirse, es decir que, a continuación, puede no haber sustituyentes de 4ta generación. Más preferiblemente, los sustituyentes de 2da generación pueden no estar sustituidos nuevamente, es decir, es posible que no haya sustituyentes de 3era generación.
Si ocurriese que un residuo se multiplicase dentro de una molécula, a continuación, este residuo puede tener diferentes significados respectivamente para varios sustituyentes: si, por ejemplo, tanto R2 como R3 denotan -C1-6-alquilo, a continuación, -C1-6-alquilo puede representar, por ejemplo, un etilo para R2 y puede representar un metilo para R3.
En conexión con los términos "-C1-10-alquilo", "-C1-6-alquilo", "-C1-4-alquilo","-C3-10-cicloalquilo", "-C3-6-cicloalquilo", "heterocicloalquilo de 3 a 7 miembros", "heterocicloalquilo de 3 a 6 miembros", "-C1-6-alquileno-", "-C1-4-alquileno-" y "-C1-2-alquileno-", el término "sustituido" se refiere, en el sentido de la invención, con respecto a los residuos o grupos correspondientes, a la sustitución simple (monosustitución) o a la sustitución múltiple (polisustitución), por ejemplo, a la disustitución o trisustitución; más preferiblemente a la monosustitución o disustitución; de uno o más átomos de hidrógenos, cada uno independientemente entre sí, con al menos un sustituyente. En el caso de una sustitución múltiple, es decir, en el caso de residuos polisustituidos, tales como residuos disustituidos o trisustituidos, estos residuos pueden estar polisustituidos en diferentes o en los mismos átomos, por ejemplo, trisustituidos en el mismo átomo de carbono, como en el caso de -CF3 , -CH2CF3 o disustituido como en el caso del 1,1-difluorociclohexilo, o en varios puntos, como en el caso del -CH(OH)-CH=CH-CHCb o 1-cloro-3-fluorociclohexilo. La sustitución múltiple se puede realizar utilizando los mismos o diferentes sustituyentes.
En relación con los términos "arilo", "fenilo", "heteroarilo" y "heteroarilo de 5 a 6 miembros", el término "sustituido" se refiere, en el sentido de esta invención, a la sustitución simple (monosustitución) o a la sustitución múltiple (polisustitución), por ejemplo, a la disustitución o trisustitución, de uno o más átomos de hidrógeno, cada uno independientemente entre sí, con al menos un sustituyente. La sustitución múltiple se puede realizar utilizando los mismos o diferentes sustituyentes.
Según la invención, preferiblemente, el -C1-10-alquilo-, -C1-6-alquilo, -C1-4-alquilo, -C3-10-cicloalquilo, -C3-6-cicloalquilo, heterocicloalquilo de 3 a 7 miembros, heterocicloalquilo de 3 a 6 miembros, -C1-6-alquileno-, -C1-4alquileno- y -C1-2-alquileno-, en cada caso, independientemente entre sí, no están sustituidos o están mono o polisustituidos con uno o más sustituyentes seleccionados de entre-F; -Cl; -Br; -I; -CN; -C1-6-alquilo; -CF3 ; -CF2H; -CFH2 ; -CF2Cl; -CFCb; -C(=O)-C1-6-alquilo; -C(=O)-OH; -C(=O)-OC1-6-alquilo; -C(=O)-NH2 ; -C(=O)-NH(C1-6-alquilo); -C(=O)-N(C1-6-alquilo)2 ; -OH; =O; - OCF3 ; -OCF2H; -OCFH2 ; -OCF2Cl; -OCFCb; -O-C1-6-alquilo; -O-C(=O)-C1-6-alquilo; -O-C(=O)-O-C1-6-alquilo; -O-(CO)-NH(C1-6-alquilo); -O-C(=O)-N(C1-6-alquilo)2 ; -O-S(=O)2-NH2 ; -O-S(=O)2-NH(C1-6-alquilo); -O-S(=O)2-N(C1-6-alquilo)2 ; - NH2 ; -NH(C1-6-alquilo); -N(C1-6-alquilo)2 ; -NH-C(=O)-C1-6-alquilo; -NH-C(=O)-O-C1-6-alquilo; -NH-C(=O)-NH2 ; -NH-C(=O)-NH(C1-6-alquilo); -NH-C(=O)-N(C1-6-alquilo)2 ; -N(C1-6-alquilo)-C(=O)-C1-6-alquilo; -N(C1-6-alquilo)-C(=O)-O-C1-6-alquilo; -N(C1-6-alquilo)-C(=O)-NH2 ; -N(C1-6-alquilo-C(=O)-NH(C1-6-alquilo); -N(C1-6-alquilo)-C(=O)-N(C1-6-alquilo)2 ; -NH-S(=O)2OH; -NH-S(=O)2-C1-6-alquilo; -NH-S(=O)2-O-C1-6-alquilo; -NH-S(=O)2-NH2 ; -NH-S(=O)2-NH(C1-6-alquilo); -NH-S(=O)2N(C1-6-alquilo)2 ; -N(C1-6-alquilo)-S(=O)2-OH; -N(C1-6-alquilo)-S(=O)2-C1-6-alquilo; -N(C1-6-alquilo)-S(=O)2-O-C1-6-alquilo; -N(C1-6-alquilo)-S(=O)2-NH2 ; -N(C1-6-alquilo)-S(=O)2-NH(C1-6-alquilo); -N(C1-6-alquilo)-S(=O)2-N(C1-6-alquilo)2 ; - SCF3 ; -SCF2H; -SCFH2 ; -S-C1-6-alquilo; -S(=O)-C1-6-alquilo; -S(=O)2-C1-6-alquilo; -S(=O)2-OH; -S(=O)2-O-C1-6-alquilo; - S(=O)2-NH2 ; -S(=O)2-NH(C1-6-alquilo); -S(=O)2-N(C1-6-alquilo)2 ; -C3-6-cicloalquilo; heterocicloalquilo de 3 a 6 miembros; fenilo; heteroarilo de 5 a 6 miembros; -O-C3-6-cicloalquilo; -O-(heterocicloalquilo de 3 a 6 miembros); -O-fenilo; -O-(heteroarilo de 5 a 6 miembros); -C(=O)-C3-6-cicloalquilo; C(=O)-(heterocicloalquilo de 3 a 6 miembros); -C(=O)-fenilo; - C(=O)-(heteroarilo de 5 a 6 miembros); -S(=O)2-(C3-6-cicloalquilo); -S(=O)2-(heterocicloalquilo de 3 a 6 miembros); - S(=O)2-fenilo y -S(=O)2-(heteroarilo de 5 o 6 miembros).
Los sustituyentes preferidos de -C1-10-alquilo, -C1-6-alquilo, -C1-4-alquilo, -C3-10-cicloalquilo, -C3-6-cicloalquilo, heterocicloalquilo de 3 a 7 miembros, heterocicloalquilo de 3 a 6 miembros, -C1-6-alquileno- y -C1-4-alquilenose seleccionan de entre el grupo que consiste en -F; -Cl; -Br; -I; -CN; -C1-6-alquilo; -CF3 ; -CF2H; -CFH2 ; -C(=O)-NH2 ; -C(=O)-NH(C1-6-alquilo); -C(=O)-N(C1-6-alquilo)2 ; -OH; -OCF3 ; -OCF2H; -OCFH2 ; -O-C1-6-alquilo; -NH2 ; -NH(C1-6-alquilo); -N(C1-6-alquilo)2 ; -SCF3 ; -SCF2H; -SCFH2 ; -S-C1-6-alquilo; -S(=O)-C1-6-alquilo; -S(=O)2-C1-6-alquilo; -C3-6-cicloalquilo; heterocicloalquilo de 3 a 6 miembros; fenilo y heteroarilo de 5 a 6 miembros; y preferiblemente y en particular -F, -CN, -CH3 , -CH2CH3 , -CF3 ; - CF2H; -CFH2 ; -C(=O)-NH2 ; -C(=O)-NH(CH3); -C(=O)-N(CH3)2; -OH, -NH2 , -OCH3 , -SCH3, -S(=O)2(CH3), - S(=O)(CH3), -N(CH3)2, ciclopropilo y oxetanilo. Según esta realización, -C1-10-alquilo, -C1-6-alquilo, -C1-4-alquilo, -C3-10-cicloalquilo, -C3-6-cicloalquilo, heterocicloalquilo de 3 a 7 miembros, heterocicloalquilo de 3 a 6 miembros están preferiblemente, cada uno, independientemente entre sí, sin sustituir, mono, di o trisustituidos, más preferiblemente no sustituidos o mono o disustituidos con un sustituyente seleccionado de entre el grupo que consiste en -F; -Cl; - Br; -I; -CN; -C1-6-alquilo; -CF3 ; -CF2H; -CFH2; -C(=O)-NH2 ; -C(=O)-NH(C1-6-alquilo); -C(=O)-N(C1-6-alquilo)2 ; -OH; - OCF3 ; -OCF2H; -OCFH2 ; -O-C1-6-alquilo; -NH2 ; -NH(C1-6-alquilo); -N(C1-6-alquilo)2 ; -SCF3 ; -SCF2H; -SCFH2; -S-C1-6-alquilo; -S(=O)-C1-6-alquilo; -S(=O)2-C1-6-alquilo; -C3-6-cicloalquilo; heterocicloalquilo de 3 a 6 miembros; fenilo y heteroarilo de 5 o 6 miembros. Preferiblemente, los grupos -C1-6-alquileno y los grupos -C1-4-alquileno no se sustituyen.
Según la invención, preferiblemente el arilo, el fenilo y el heteroarilo de 5 o 6 miembros en cada caso, independientemente entre sí, no están sustituidos o están mono o polisustituidos con uno o más sustituyentes seleccionados de entre -F; -Cl; -Br; -I; -CN; -C1-6-alquilo; -CF3 ; -CF2H; -CFH2 ; -CF2Cl; -CFCb; -C1-4-alquileno-CF3 ; -C1-4-alquileno-CF2H; -C1-4-alquileno-CFH2; -C(=O)-C1-6-alquilo; -C(=O)-OH; - C(=O)-OC1-6-alquilo; -C(=O)-NH(OH); -C(=O)-NH2 ; -C(=O)-NH(C1-6-alquilo); -C(=O)-N(C1-6-alquilo)2 ; =O; -OH; -OCF3 ; -OCF2H; -OCFH2 ; -OCF2Cl; -OCFCl2 ; -O-C1-6-alquilo; -O-C3-6-cicloalquilo; -O-(heterocicloalquilo de 3 a 6 miembros); -NH2 ; -NH(C1-6-alquilo); -N(C1-6-alquilo)2 ; -NH-C(=O)-C1-6-alquilo; -N(C1-6-alquilo)-C(=O)-C1-6-alquilo; -NH-C(=O)-NH2 ; -NH-C(=O)-NH(C1-6-alquilo); -NH-C(=O)-N(C1-6-alquilo)2 ; -N(C1-6-alquilo)-C(=O)-NH(C1-6-alquilo); -N(C1-6-alquilo)-C(=O)-N(C1-6-alquilo)2 ; -NH-S(=O)2-C1-6-alquilo; -SCF3 ; -S-C1-6-alquilo; -S(=O)-C1-6-alquilo; -S(=O)2-C1-6-alquilo; -S(=O)2-n H2 ; -S(=O)2-NH(C1-6-alquilo); -S(=O)2-N(C1-6-alquilo)2 ; -C3-6-cicloalquilo; -C1-4-alquileno-C3-6-cicloalquilo; heterocicloalquilo de 3 a 6 miembros; -C1-4-alquileno-(heterocicloalquilo de 3 a 6 miembros); fenilo o heteroarilo de 5 o 6 miembros.
Los sustituyentes preferidos del arilo, el fenilo y el heteroarilo de 5 o 6 miembros se seleccionan de entre el grupo que consiste en -F; -Cl; -Br; -I; -CN; -C1-6-alquilo; -CF3 ; -CF2H; -CFH2 ; -C1-4-alquileno-CF3; -C1-4-alquileno-CF2H; -C1-4-alquileno-CFH2; -OH; -OCF3; -OCF2H; -OCFH2 ; -O-C1-6-alquilo; -O-C3-6-cicloalquilo y -C3-6-cicloalquilo; y preferiblemente y en particular de entre -F; -Cl; -Br; -CN; -CH3 ; -CH2CH3 ; -CF3 ; -CF2H; -CFH2 ; -CH2-CF3 ; =O; -OH; -OCF3 ; -OCF2H; -OCFH2 ; -O-CH3 ; -O-ciclopropilo y ciclopropilo. Según esta realización, cada uno del arilo, el fenilo y el heteroarilo de 5 o 6 miembros, preferible e independientemente entre sí, no están sustituidos o están mono, di o trisustituidos, más preferiblemente no sustituidos o mono o disustituidos con un sustituyente seleccionado de entre el grupo que consiste en -F; -Cl; -Br; -I; -CN; -C1-6-alquilo; -CF3 ; -CF2H; -CFH2 ; -C1-4-alquileno-CF3; -C1-4-alquileno-CF2H; -C1-4-alquileno-CFH2; =O; -OH; -OCF3 ; -OCF2H; -OCFH2 ; -O-C1-6-alquilo; -O-C3-6-cicloalquilo y -C3-6-cicloalquilo. Un heteroarilo sustituido de 5 ó 6 miembros particularmente preferido es N-metil-2-oxo-piridilo.
En una realización preferida, el compuesto según la invención tiene una estereoquímica según la fórmula general (II), (III), (IV) o (V)

En una realización preferida, el compuesto según la invención tiene una estereoquímica según las fórmulas generales (II) o (III), de modo tal que los residuos -R1 y -NH-R2 en el anillo de pirrolidina tienen una orientación cis. Preferiblemente, el compuesto según la invención tiene una estereoquímica según la fórmula general (II). Preferiblemente, el compuesto según la invención tiene una estereoquímica según la fórmula general (III). Se prefiere particularmente la estereoquímica según la fórmula general (II).
En otra realización preferida, el compuesto según la invención tiene una estereoquímica según la fórmula general (IV) o (V), de modo tal que los residuos -R1 y -NH-R2 en el anillo de pirrolidina tienen una orientación cis. Preferiblemente, el compuesto según la invención tiene una estereoquímica según la fórmula general (IV). Preferiblemente, el compuesto según la invención tiene una estereoquímica según la fórmula general (V).
En el compuesto de la invención según cualquiera de las fórmulas generales (I), (II), (III), (IV) o (V), Ri representa un -C1-10-alquilo; -C3-10-cicloalquilo; -C1-6-alquileno-C3-10-cicloalquilo; heterocicloalquilo de 3 a 7 miembros; -C1-6-alquileno-(heterocicloalquilo de 3 a 7 miembros); arilo; -C1-6-alquileno-arilo; heteroarilo de 5 o 6 miembros; o -C1-6-alquileno-(heteroarilo de 5 o 6 miembros).
En una realización preferida, Ri representa -C3-10-cicloalquilo; -C1-6-alquileno-C3-10-cicloalquilo; arilo; o heteroarilo de 5 o 6 miembros.
En realizaciones particularmente preferidas, Ri representa
(i) ciclopropilo, sin sustituir;
(ii) -CH2-ciclopropilo, sin sustituir;
(iii) fenilo, no sustituido o mono o disustituido con sustituyentes independientemente entre sí seleccionados de entre el grupo que consiste en -F, -Cl, -Br, -CH3 , -CF3 , -CN, ciclopropilo y -OCH3 , donde el fenilo está opcionalmente recocido a un anillo de dioxolano mediante un -O-CH2CH2-O- sustituyente; o
(iv) piridilo, no sustituido o mono o disustituido con sustituyentes, independientemente entre sí, seleccionados de entre el grupo que consiste en -F, -Cl, -Br, -CH3 , -CF3 , -CN y -OCH3.
En el compuesto de la invención según cualquiera de las fórmulas generales (I), (II), (III), (IV) o (V), R2 representa un -C(=O)-Ci-io-alquilo; un -C(=O)-C3-10-cicloalquilo; un -C(=O)-C1-6-alquileno-C3-10-cicloalquilo; un -C(=O)-(heterocicloalquilo de 3 a 7 miembros); un -C(=O)-C1-6-alquileno-(heterocicloalquilo de 3 a 7 miembros); un -C(=O)-arilo; un -C(=O)-C1-6-alquileno-arilo; un -C(=O)-(heteroarilo de 5 o 6 miembros); un -C(=O)-C1-6-alquileno-(heteroarilo de 5 o 6 miembros); un -S(=O)1-2-C1-10-alquilo; un -S(=O)1-2-C3-10-cicloalquilo; un -S(=O)1-2-C1-6-alquileno-C3-10-cicloalquilo; un -S(=O)1-2-(heterocicloalquilo de 3 a 7 miembros); un -S(=O)1-2-C1-6-alquileno-(heterocicloalquilo de 3 a 7 miembros); un -S(=O)1-2-arilo; un -S(=O)1-2-C1-6-alquileno-arilo; un -S(=O)1-2-(heteroarilo de 5 o 6 miembros); o un -S(=O)1-2-C1-6-alquileno-(heteroarilo de 5 o 6 miembros);
En una realización preferida, R2 representa -C(=O)-C1-10-alquilo; -C(=O)-C3-10-cicloalquilo; -C(=O)-C1-6-alquileno-C3-10-cicloalquilo; -C(=O)-(heterocicloalquilo de 3 a 7 miembros); -C(=O)-(heteroarilo de 5 o 6 miembros); -S(=O)2-C1-10-alquilo; -S(=O)2-C3-10-cicloalquilo; -S(=O)2-C1-6-alquileno-C3-10-cicloalquilo; o -S(=O)2-(heteroarilo de 5 o 6 miembros).
En realizaciones particularmente preferidas, R2 representa
(i) -C(=O)-Ci-io-alquilo, no sustituido o mono o disustituido con sustituyentes, independientemente entre sí, seleccionados de entre el grupo que consiste en -F, -Cl y -Br;
(ii) -C(=O)-ciclopropilo, no sustituido o mono o disustituido con sustituyentes, independientemente entre sí, seleccionados de entre el grupo que consiste en -F, -Cl, -Br, -CH3 , -CF3 , -CN y -OCH3 ;
(iii) -C(=O)-ciclobutilo, no sustituido o mono o disustituido con sustituyentes, independientemente entre sí, seleccionados de entre el grupo que consiste en -F, -Cl, -Br, -CH3 , -CF3 , -CN y -OCH3 ;
(iv) -C(=O)-2-tetrahidrofuranilo, no sustituido;
(v) -C(=O)-(heteroarilo de 5 a 6 miembros), donde dicho heteroarilo de 5 a 6 miembros se selecciona de entre el grupo que consiste en tiazolilo, pirazolilo, oxazolilo y 1-oxa-2,4-diazolilo, 1,2,5-oxadiazolilo, isoxazolilo, isotiazolilo, donde, en cada caso, dicho heteroarilo de 5 a 6 miembros no está sustituido o está mono o disustituido con sustituyentes, independientemente entre sí, seleccionados de entre el grupo que consiste en -F, -Cl, -Br, -CH3 , -CF3 , -CN, =O y -OCH3 ;
(vi) -S(=O)2-C1-10-alquilo, no sustituido;
(vii) -S(=O)2-ciclopropilo, no sustituido;
(viii) -S(=O)2-CH2-ciclopropilo, no sustituido; o
(ix) -S(=O)2-(heteroarilo de 5 a 6 miembros), donde dicho heteroarilo de 5 a 6 miembros se selecciona de entre el grupo que consiste en tiazolilo, pirazolilo, oxazolilo y 1-oxa-2,4-diazolilo, 1,2,5-oxadiazolilo, isoxazolilo, isotiazolilo, donde, en cada caso, dicho heteroarilo de 5 a 6 miembros no está sustituido o está mono o disustituido con sustituyentes, independientemente entre sí, seleccionados de entre el grupo que consiste en -F, -Cl, -Br, -CH3 , -CF3 , -CN, =O y -OCH3.
En el compuesto de la invención según cualquiera de las fórmulas generales (I), (II), (III), (IV) o (V), R3 representa un
heterocicloalquilo de 3 a 7 miembros; un -C1-6-alquileno -(heterocicloalquilo de 3 a 7 miembros); heteroarilo de 5 o 6 miembros; -C1-6-alquileno-(heteroarilo de 5 o 6 miembros); -C(=O)-(heterocicloalquilo de 3 a 7 miembros); -C(=O)-C1-6-alquileno-(heterocicloalquilo de 3 a 7 miembros); -C(=O)-(heteroarilo de 5 o 6 miembros); -C(=O)-C1-6-alquileno-(heteroarilo de 5 o 6 miembros); -S(=O)1-2-(heterocicloalquilo de 3 a 7 miembros); -S(=O) 1-2-C1-6-alquileno-(heterocicloalquilo de 3 a 7 miembros); -S(=O)1-2-(heteroarilo de 5 o 6 miembros); o -S(=O) 1-2-C1-6-alquileno-(heteroarilo de 5 o 6 miembros);
En una realización preferida, R3 un heterocicloalquilo de 3 a 7 miembros; heteroarilo de 5 o 6 miembros; o - C1-6-alquileno-(heteroarilo de 5 o 6 miembros).
En realizaciones particularmente preferidas, R3 representa
(i) piperidinilo, no sustituido o sustituido con -C(=O)-ciclopropilo;
(ii) heteroarilo de 5 a 6 miembros seleccionado de entre el grupo que consiste en pirazolilo, piridilo y pirimidinilo, en el que en cada caso dicho heteroarilo de 5 a 6 miembros no está sustituido o es mono o disustituido con sustituyentes independientemente entre sí seleccionados de entre el grupo que consiste en -F, -Cl, -Br, -CH 3 , -CF3 , -CN, =O y -OCH3; o
(iii) -CH2-(heteroarilo de 5 a 6 miembros) seleccionado de entre el grupo que consiste en -CH2-pirazolilo, -CH2-piridilo y -CH2-pirimidinilo, en el que en cada caso dicho heteroarilo de 5 a 6 miembros no está sustituido o es mono o disustituido con sustituyentes independientemente entre sí seleccionados de entre el grupo que consiste en -F, -Cl, -Br, -CH3 , -CF3 , -CN, =O y -OCH3.
En el compuesto de la invención según cualquiera de las fórmulas generales (I), (II), (III), (IV) o (V), R4 representa -H; -F; -Cl; -Br; -I; -CN; -CH3 ; -CF3 ; -CF2H; -CFH2 o ciclopropilo.
En una realización preferida, R4 representa -H.
En el compuesto de la invención según cualquiera de las fórmulas generales (I), (II), (III), (IV) o (V), X representa CR5 ; donde R5 representa -H; -F; -Cl; -Br; -I; -CN; -C1-10-alquilo o -C3-10-cicloalquilo.
En una realización preferida, X representa CH.
En los compuestos de la invención según cualquier fórmula general (I), (II), (III), (IV) o (V), Y representa CR6; donde R6 representa -H; -F; -Cl; -Br; -I; -CN; -C1-10-alquilo o -C3-10-cicloalquilo.
En una realización preferida, Y representa CH.
En los compuestos de la invención según cualquier fórmula general (I), (II), (III), (IV) o (V), Z representa CR7 ; donde R7 representa -H; -F; -Cl; -Br; -I; -CN; -C1-10-alquilo o -C3-10-cicloalquilo.
En una realización preferida, Z representa CH.
En una realización particularmente preferida,
(i) X representa CH; Y representa CH; y Z representa CH.
En realizaciones particularmente preferidas de la invención según cualquiera de las fórmulas generales (I), (II), (III), (IV) o (V),
Ri representa al fenilo, no sustituido, mono o disustituido con sustituyentes, independientemente entre sí, seleccionados de entre el grupo que consiste en -F, -Cl, -Br, -CH3 , -CF3 , -CN y -OCH3 ; y/o
R2 representa un -C(=O)-C1-6-alquilo; -C(=O)-ciclopropilo; o -C(=O)-ciclobutilo, no sustituido, mono o disustituido con sustituyentes, independientemente entre sí, seleccionados de entre el grupo que consiste en -F,-Cl y -Br; y/o R3 representa un N-metil-2-oxo-piridilo.
En una realización preferida, el compuesto según la invención se selecciona de entre el grupo que consiste en
1 N-(2R,3S)-2-(3-clorofenil)-1-[1-(1-metil-6-oxo-3-piridil)indazol-5-il]-5-oxo-pirrolidin-3-il]-2,2-difluoropropanamida
2 2,2-difluoro-N-[rac-(2R,3S)-2-(2,4-difluorofenil)-1-[1-(1-metil-6-oxo-3-piridil)indazol-5-il]-5-oxopirrolidin-3 -il]propanamida
3 2,2-difluoro-N-[rac-(2R,3S)-2-(2,3-dihidro-1,4-benzodioxin-6-il)-1-[1-(1-metil-6-oxo-3-piridil)indazol-5-il]-5-oxo-pirrolidin-3-il]propanamida
4 2,2-difluoro-N-[(2R,3S)-1-[1-(1-metil-6-oxo-3-piridil)indazol-5-il]-5-oxo-2-fenil-pirrolidin-3-il]propanamida 5 2,2-difluoro-N-[(2R,3S)-2-(3-fluorofenil)-1-[1-(1-metil-6-oxo-3-piridil)indazol-5-il]-5-oxo-pirrolidin-3-il]propanamida
6 2,2-difluoro-N-[(2R,3S)-2-(2-fluorofenil)-1-[1-(1-metil-6-oxo-3-piridil)indazol-5-il]-5-oxo-pirrolidin-3-il]propanamida
7 2,2-difluoro-N-[rac-(2R,3S)-5-oxo-2-fenil-1-[1-(3-piridil)indazol-5-il]pirrolidin-3-il]propanamida
9 2,2-difluoro-N-[rac-(2R,3S)-1-[1-5-fluoro-2-piridil)indazol-5-il]-5-oxo-2-fenil-pirrolidin-3-il]propanamida 13 5-metil-N-[(2R,3S)-1-[1-(1-metil-6-oxo-3-piridil)indazol-5-il]-5-oxo-2-fenil-pirrolidin-3- i l]tiazol-2-carboxamida
15 2,2-difluoro-N-[(2R,3S)-2-(4-fluoro-3-metoxi-fenil)-1-[1-(1-metil-6-oxo-3-piridil)indazol-5-il]-5-oxopirrolidin-3-il]propanamida
17 2,2-difluoro-N-[(2R,3S)-2-(4-fluorofenil)-1-[1-(1-metil-6-oxo-3-piridil)indazol-5-il]-5-oxo-pirrolidin-3-il]propanamida
18 1-fluoro-N-[(2R,3S)-1-[1-(1-metil-6-oxo-3-piridil)indazol-5-il]-5-oxo-2-fenil-pirrolidin-3-il]ciclopropanocarboxamida
222,2-difluoro-N-[rac-(2R,3S)-1-[1-(6-metoxi-3-piridil)indazol-5-il]-5-oxo-2-fenil-pirrolidin-3-il]propanamida 232,2-difluoro-N-[rac-(2R,3S)-5-oxo-2-fenil-1-[1-(4-piridil)indazol-5-il]pirrolidin-3-il]propanamida
242,2-difluoro-N-[-(2R,3S)-1-[1-(6-metil-3-piridil)indazol-5-il]-5-oxo-2-fenil-pirrolidin-3-il]propanamida 252,2-difluoro-N-[rac-(2R,3S)-1-[1-(2-metil-4-piridil)indazol-5-il]-5-oxo-2-fenil-pirrolidin-3-il]propanamida 26 1-metil-N-[(2R,3S)-1-[1-(1-metil-6-oxo-3-piridil)indazol-5-il]-5-oxo-2-fenil-pirrolidin-3-il]ciclopropanocarboxamida
272,2-difluoro-N-[rac-(2R,3S)-1-[1-(2-metoxi-4-piridil)indazol-5-il]-5-oxo-2-fenil-pirrolidin-3-il]propanamida 31 2,2-difluoro-N-[rac-(2R,3S)-1-[1-[(2-metoxi-4-piridil)metil]indazol-5-il]-5-oxo-2-fenil-pirrolidin-3-il]propanamida
32 2,2-difluoro-N-[rac-(2R,3S)-1-[1-[(1-metilpirazol-3-il)metil]indazol-5-il]-5-oxo-2-fenil-pirrolidin-3-il]propanamida
33 2,2-difluoro-N-[rac-(2R,3S)-1-[1-[(6-metoxi-3-piridil)metil]indazol-5-il]-5-oxo-2-fenil-pirrolidin-3-il]propanamida
34 2,2-difluoro-N-[rac-(2R,3S)-1-[1-[(1-metil-6-oxo-3-piridil)metil]indazol-5-il]-5-oxo-2-fenil-pirrolidin-3-il]propanamida
35 2,2-difluoro-N-[rac-(2R,3S)-1-[1-[1-(ciclopropanocarbonil)-4-piperidil]indazol-5-il]-5-oxo-2-fenil-pirrolidin-3-il]propanamida
38 2,2-difluoro-N-[rac-(2R,3S)-1-[1-[(1-metil-2-oxo-4-piridil)metil]indazol-5-il]-5-oxo-2-fenil-pirrolidin-3-il]propanamida
39 N-[(2R,3S)-2-(2-fluorofenil)-1-[1-(1-metil-6-oxo-3-piridil)indazol-5-il]-5-oxo-pirrolidin-3-yil]ciclopropanocarboxamida
40 N-[rac-(2R,3S)-1-[1-[(2-metoxi-4-piridil)metil]indazol-5-il]-5-oxo-2-fenil-pirrolidin-3-il]ciclopropanocarboxamida
41 N-[(2R,3S)-1-[1-(1-metil-6-oxo-3-piridil)indazol-5-il]-2-(o-tolil)-5-oxo-pirrolidin-3-il]ciclopropanocarboxamida
42 2,2-difluoro-N-[(2R,3S)-2-(2-metoxi-4-piridil)-1-[1-(1-metil-6-oxo-3-piridil)indazol-5-il]-5-oxo-pirrolidin-3-il]propanamida
43 2,2-difluoro-N-[(2R, 3S)-1-[1-(1-metil-6-oxo-3-piridilo)indazol-5-M]-2-(o-toMlo)-5-oxo-pirrolidin-3-ilo]propanamida
44 N-[(2R,3S)-1-[1-(1-metil-6-oxo-3-piridil)indazol-5-il]-5-oxo-2-fenil-pirrolidin-3-ilo]tiazol-4-carboxamida 45 1-metilo-N-[(2R,3S)-1-[1-(1-metil-6-oxo-3-piridilo)indazol-5-il]-5-oxo-2-fenil-pirrolidin-3-ilo]pirazol-3-carboxamida
46 2,2-difluoro-N-[rac-(2R,3S)-1-[1-(1-metilpirazol-4-il)indazol-5-il]-5-oxo-2-fenil-pirrolidin-3-ilo]propanamida
47 2,2-difluoro-N-[rac-(2R,3S)-1-[1-(5-fluoropirimidin-2-il)indazol-5-il]-5-oxo-2-fenil-pirrolidin-3-ilo]propanamida
48 (R)-N-[(2R,3S)-1-[1-(1-metil-6-oxo-3-piridil)indazol-5-il]-5-oxo-2-fenil-pirrolidin-3-ilo]-tetrahidrofurano-2-carboxamida
49 N-[(2R,3S)-1-[1-(1-metil-6-oxo-3-piridil)indazol-5-yl]-5-oxo-2-fenilo-pirrolidin-3-ilo]oxazol-2-carboxamida 50 N-[(2R,3S)-1-[1-(1-metil-6-oxo-3-piridil)indazol-5-il]-5-oxo-2-fenil-pirrolidin-3-ilo]oxazol-4-carboxamida 51 5-metil-N-[(2R,3S)-1-[1-(1-metil-6-oxo-3-piridilo)indazol-5-il]-5-oxo-2-fenil-pirrolidin-3-il]-1,2,4-oxadiazol-3-carboxamida
En cada caso, en forma del compuesto libre o una sal fisiológicamente aceptable del mismo.
Los compuestos según la invención se pueden sintetizar mediante reacciones estándares en el campo de la química orgánica conocidas por el experto en la materia o de la manera que se describe en esta invención (véanse los esquemas de reacción a continuación) o de manera análoga. Las condiciones de reacción en las rutas de síntesis descritas en esta invención son conocidas por los expertos y, en algunos casos, también se ejemplifican en los Ejemplos descritos en esta invención.
Esquema de reacción 1:

Las fracciones de indazol sustituidas en los compuestos de la fórmula (D) y la fórmula (F) se introducen sometiendo el lactámico (B) o el lactámico (E) en una reacción de acoplamiento C-N catalizadas por metal regioselectivo con los haluros de indazol correspondientes (C), preferiblemente con los yoduros de indazol correspondientes. Las reacciones de acoplamiento de C-N catalizadas por metal son generalmente conocidas en la técnica (Current Organic Synthesis, 2011,8, 53). Las reacciones de acoplamiento C-N favorables son las reacciones de acoplamiento cruzado catalizadas por paladio y cobre (Chem. Rev., 2016, 116, 12564; Chem. Soc. Rev., 2014, 43, 3525; Chem. Sci., 2010, 1, 13). Los acoplamientos C-N regioselectivos con arilhaluros son conocidos en la técnica (Chem. Sci., 2011, 2, 27; J. Am. química Soc., 2001, 123, 7727).
Las aminas primarias (A) y (G) se convierten a las amidas y sulfonamidas correspondientes (acilación y formación de sulfonamidas) (B) y (D) usando ácidos comercialmente disponibles (activación de ácidos usando, por ejemplo, HATU) o cloruros de ácido en condiciones de reacción de acoplamiento de amidas estándares (March's Advanced Organic Chemistry, 2007, 6ta edición, página 1427-1474).
La introducción de diferentes grupos ortogonales de protección GP (por ejemplo, Boc, Cbz) para convertir (A) a (E), así como también la desprotección de los compuestos de la fórmula (E) a (A) se describe claramente en
la bibliografía (T. W. Green, P. G. M. Wuts, Protective Groups in Organic Synthesis, Wiley-Interscience, Nueva York, 1999).
Esquema de reacción 1.1:
Los compuestos (A) y (E) se pueden sintetizar según los procedimientos que se describen en la bibliografía.

Ruta 1: Los compuestos de las fórmulas (A) y (E) se pueden sintetizar a partir de los compuestos de la fórmula (H) (J. Org. Chem., 2010, 76, 948).
Ruta 2: La síntesis de los compuestos de las fórmulas (M) y (L) se describe en la bibliografía (J. Org. Chem., 2007, 72, 5016; Org. Lett., 2007, 9, 4077; J. Org. Chem., 2012, 77, 160). Los compuestos de la fórmula (A) y (E) se pueden sintetizar utilizando el reordenamiento de Curtius como etapa clave para convertir el ácido carboxílico (L) en la amina primaria correspondiente (A) o (E). El reordenamiento de Curtius es bien conocido en la técnica (Tetrahedron Letters, 2010, 385).
Ruta 3: La síntesis de los compuestos de la fórmula (J) se describe en la bibliografía (Org. Lett., 2009, 11, 4512;ACS Sustainable Chem. Eng., 2015, 3, 1873). La reducción de lactámicos altamente funcionalizados (J) proporciona una ruta alternativa para la síntesis de los compuestos de las fórmulas (A) y (E). La reducción de grupos nitro es bien conocida en la técnica (Advanced Organic Chemistry de March, 2007, 6a edición, página 1815f).
Ruta 4: La síntesis de los compuestos de la fórmula (K) se describe en la bibliografía (J. Heterocyclic Chem., 2014, 51, E25). La reducción de lactámicos altamente funcionalizados (K) proporciona una ruta alternativa para la síntesis de los compuestos de las fórmulas (A) y (E). La reducción de enamidas/iminas es bien conocida en la técnica ( Advanced Organic Chemistry de March, 2007, 6 a edición, página 1053f y página 1811f).
Esquema de reacción 2:

Los compuestos de la fórmula (D) se pueden sintetizar mediante el acoplamiento de C-N regioselectivo del compuesto (O). Las reacciones de acoplamiento de C-N adecuadas para N-H que contienen heterociclos son conocidas en la técnica (Synthesis, 2011, 829; Chem. Sci., 2011, 2, 27; Beilstein J. Org. Chem., 2011, 7, 59;
J. Org. Chem., 2004, 69, 5578). El compuesto de la fórmula (O) se sintetiza mediante la desprotección del compuesto (N) en condiciones ácidas.
Los compuestos según la invención se pueden producir en la manera que se describe en esta invención o de una manera análoga.
En una realización preferida, los compuestos según la invención son moduladores del receptor de glucocorticoides. En el sentido de la invención, el término "modulador selectivo del receptor de glucocorticoides (modulador del receptor de glucocorticoides)" significa preferiblemente que el compuesto respectivo exhibe, en un ensayo de compromiso de diana celular que evalúa la potencia agonista o antagonista en el receptor de glucocorticoides, un valor EC50 o IC50 en el receptor de glucocorticoides de como máximo 15 pM (1010-6 mol/L) o como máximo 10 pM; más preferiblemente como máximo 1 pM; aún más preferiblemente como máximo 500 nM(10-9 mol/L); aún más preferiblemente como máximo 300 nM; incluso más preferiblemente como máximo 100 nM; lo más preferiblemente como máximo 10 nM; y en particular como máximo 1 nM. En una realización preferida, el compuesto según la invención exhibe, en un ensayo de compromiso de diana celular que evalúa la potencia agonista o antagonista del receptor de glucocorticoides, un valor EC50 o IC50 en el receptor de glucocorticoides en el intervalo de 1 pM a 15 pM, más preferiblemente de 100 nM a 1 pM, más preferiblemente por debajo de 100 nM.
El experto en la materia sabe cómo probar compuestos para la modulación (agonista o antagonista) de la actividad del receptor de glucocorticoides. Los ensayos de participación de diana preferidos para probar los compuestos en cuanto a su potencia agonista o antagonista (EC50, IC50) en el receptor de glucocorticoides se describen a continuación en esta invención:
Ensayo de unión al ligando del receptor de glucocorticoides humano (hGR)
Los posibles moduladores selectivos de los receptores de glucocorticoides de esta intervención se pueden probar para determinar su afinidad de unión en el receptor de glucocorticoides mediante el ensayo de unión que se describe a continuación.
Preferiblemente, el receptor de glucocorticoides extraído del citosol de las células IM9 se usa para ensayos competitivos de unión de radioligandos para calcular los valores IC50 y la afinidad de unión (valor Ki) de los compuestos según la presente invención. Preferiblemente, una concentración fija del radioligando 3H-dexametasona y un intervalo de concentraciones de compuestos según la presente invención (como competidores no marcados de dexametasona) se mezclan con el receptor de glucocorticoides extraído para medir la potencia/afinidad con la que compiten por la unión del radioligando. Preferiblemente, mediante el uso de curvas de competición se determina la IC50, que es la concentración de ligando competidor que desplaza el 50% de la unión específica del radioligando. Finalmente, este valor IC50 se convierte en un valor Ki.
En una realización preferida, el compuesto según la invención exhibe, en un ensayo de compromiso de diana celular que evalúa la potencia agonista o antagonista en el receptor de glucocorticoides, un valor EC50 o IC50 en el receptor de glucocorticoides de como máximo 1 pM (10-6 mol/l); incluso más preferiblemente como máximo 500 nM(10-9 mol/l); aún más preferiblemente como máximo 300 nM; incluso más preferiblemente como máximo 100 nM; aún más preferiblemente como máximo 50 nM; y, en particular, como máximo 10 nMo como máximo 1 nM.
En una realización preferida, el compuesto según la invención exhibe, en un ensayo de compromiso de diana celular que evalúa la potencia agonista o antagonista del receptor de glucocorticoides, un valor EC50 o IC50 en el receptor de glucocorticoides en el intervalo de 1 pM a 15 pM, más preferiblemente de 100 nM a 1 pM, más preferiblemente por debajo de 100 nM.
En una realización preferida, el compuesto según la invención exhibe, en un ensayo de compromiso de diana celular que evalúa la potencia agonista o antagonista en el receptor de glucocorticoides, un valor EC50 o IC50 en el receptor de glucocorticoides en el intervalo de desde 0,1 nM(10-9 mol/l) hasta 1000 nM; incluso más preferiblemente de 1 nM a 800 nM; aún más preferiblemente de 1 nM a 500 nM; incluso más preferiblemente de 1 nM a 300 nM; más preferiblemente de 1 nM a 100 nM; y, en particular, de 1 nM a 80 nM.
En una realización preferida, el compuesto según la invención exhibe, en un ensayo de compromiso de diana celular para potencia agonista o antagonista en el receptor de glucocorticoides, una inhibición a 1 pM de al menos el 40 %, más preferiblemente al menos el 60 %, lo más preferiblemente al menos el 85 %. En una realización preferida, el compuesto según la invención exhibe, en un ensayo de compromiso de diana celular para potencia agonista o antagonista en el receptor de glucocorticoides, una inhibición a 1 pM en el intervalo del 40 % al 60 %, más preferiblemente de más del 60 % al 85 %, lo más preferiblemente mayor que el 85 %.
Preferiblemente, los compuestos según la invención son útiles como moduladores selectivos del receptor de glucocorticoides.
Por lo tanto, los compuestos según la invención son preferiblemente útiles para el tratamiento o la prevención in vivo de enfermedades en las que está implicada la participación del receptor de glucocorticoides.
Por lo tanto, la invención además se refiere a un compuesto según la invención para su uso en la modulación de la actividad del receptor de glucocorticoides.
Por lo tanto, otro aspecto de la invención se refiere a un compuesto según la invención para su uso en el tratamiento y/o la profilaxis de un trastorno que está mediado al menos en parte por el receptor de glucocorticoides.
Un aspecto adicional de la invención se refiere al uso de un compuesto según la invención como un medicamento.
Otro aspecto de la invención se refiere a una forma de dosificación farmacéutica que comprende un compuesto según la invención. Preferiblemente, la forma de dosificación farmacéutica comprende un compuesto según la invención y uno o más excipientes farmacéuticos, tales como vehículos fisiológicamente aceptables, aditivos y/o sustancias auxiliares; y opcionalmente uno o más ingredientes farmacológicamente activos adicionales. Los ejemplos de vehículos, aditivos y/o sustancias auxiliares fisiológicamente aceptables son rellenos, solventes, diluyentes, colorantes y/o aglutinantes. Estas sustancias son conocidas por el experto en la materia (véase H. P. Fiedler, Lexikon der Hilfsstoffe fur Pharmazie, Kosmetik und angrenzende Gebiete, Editio Cantor Aulendoff).
La forma de dosificación farmacéutica según la invención está destinada preferiblemente a un uso sistémico, tópico o local y preferiblemente a una administración por vía oral. Por lo tanto, la forma de dosificación farmacéutica puede ser líquida, semisólida o sólida, por ejemplo, en forma de soluciones inyectables, gotas, jugos, jarabes, aerosoles, suspensiones, comprimidos, parches, películas, cápsulas, emplastos, supositorios, ungüentos, cremas, lociones, geles, emulsiones, aerosoles o en forma multiparticulada, por ejemplo en forma de gránulos o gránulos, dado el caso, prensados en comprimidos, decantados en cápsulas o suspendidos en un líquido, pudiendo también administrarse como tales.
La forma de dosificación farmacéutica según la invención se prepara preferiblemente con la ayuda de medios, dispositivos y procedimientos convencionales conocidos en la técnica. La cantidad del compuesto según la invención a administrar al paciente puede variar y depende, por ejemplo, del peso o la edad del paciente y también del tipo de administración, la indicación y la gravedad del trastorno. Preferiblemente se administra de 0,001 a 100 mg/kg, más preferiblemente de 0,05 a 75 mg/kg e incluso más preferiblemente de 0,05 a 50 mg de un compuesto según la invención por kg de peso corporal del paciente.
Se cree que el receptor de glucocorticoides tiene el potencial para modificar una variedad de enfermedades o trastornos en mamíferos como los humanos. Estos incluyen, en particular, enfermedades inflamatorias, asma, la artritis reumatoidea, la enfermedad del intestino irritable, la enfermedad pulmonar obstructiva crónica, el síndrome de insuficiencia respiratoria aguda, la fibrosis quística, la osteoartritis, la polimialgia reumática, la arteritis de células gigantes, el síndrome de Sjogren, la distrofia muscular de Duchenne, la vasculitis, la enfermedad de Behget, la colitis ulcerativa y la enfermedad de Crohn.
Otras enfermedades y trastornos que se cree que están modulados por el receptor de glucocorticoides incluyen trastornos endocrinos, preferiblemente seleccionados de entre insuficiencia adrenocortical primaria o secundaria, hiperplasia suprarrenal congénita, hipercalcemia asociada con cáncer y tiroiditis no supurativa; trastornos reumáticos; preferiblemente seleccionados de entre artritis psoriásica, artritis reumatoide, artritis reumatoide juvenil, espondilitis anquilosante, bursistis aguda y subaguda, tenosinovitis aguda no específica, artritis gotosa aguda, osteoartritis postraumática, sinovitis de osteoartritis y epicondilitis; enfermedades del colágeno, preferiblemente seleccionadas de entre lupus eritematoso sistémico, dermatomiositis sistémica (polimiositis) y carditis reumática aguda; enfermedades dermatológicas, preferiblemente seleccionadas de entre pénfigo, dermatitis ampollosa herpetiforme, eritema multiforme grave (síndrome de Stevens-Johnson), dermatitis exfoliativa, micosis fungoide, psoriasis y dermatitis seborreica; estados alérgicos, preferiblemente seleccionados de entre rinitis alérgica estacional o perenne, asma bronquial, dermatitis de contacto, dermatitis atópica, enfermedad del suero y reacciones de hipersensibilidad a fármacos; enfermedades oftálmicas, preferiblemente seleccionadas de entre úlceras marginales corneales alérgicas, herpes zoster oftálmico, inflamación del segmento anterior, uveítis posterior difusa y coroiditis, oftalmía simpática, conjuntivitis alérgica, queratitis, coriorretinitis, neuritis óptica, iritis e iridociclitis; enfermedades respiratorias, preferiblemente seleccionadas de entre sarcoidosis sintomática, síndrome de Loeffler, beriliosis, tuberculosis pulmonar fulminante o diseminada cuando se usa junto con quimioterapia antituberculosa, neumonitis por aspiración; trastornos hematológicos, preferiblemente seleccionados de entre púrpura trombocitopénica idiopática, trombocitopenia secundaria, anemia hemolítica adquirida (autoinmune), eritroblastopenia (anemia RBC), anemia hipoplásica congénita (eritroide); enfermedades neoplásicas, preferiblemente seleccionadas de entre leucemias y linfomas, leucemia aguda de la infancia; enfermedades gastrointestinales, preferiblemente
seleccionadas de entre colitis ulcerosa y enteritis regional.
Otro aspecto de la invención se refiere a un compuesto según la invención para su uso en el tratamiento y/o profilaxis del dolor y/o inflamación; más preferiblemente para el dolor inflamatorio.
Otro aspecto de la invención se refiere a un compuesto según la invención para su uso en el tratamiento y/o la profilaxis del asma, la artritis reumatoidea, la enfermedad del intestino irritable, la enfermedad pulmonar obstructiva crónica, el síndrome de insuficiencia respiratoria aguda, la fibrosis quística, la osteoartritis, la polimialgia reumática, la arteritis de células gigantes, el síndrome de Sjogren, la distrofia muscular de Duchenne, la vasculitis, la enfermedad de Behget, la colitis ulcerativa y/o la enfermedad de Crohn.
Incluso otro aspecto de la invención se refiere a un compuesto según la invención para su uso en el tratamiento y/o la profilaxis de trastornos endocrinos, preferiblemente seleccionados de entre insuficiencia adrenocortical primaria o secundaria, hiperplasia suprarrenal congénita, hipercalcemia asociada con cáncer y tiroiditis no supurativa; trastornos reumáticos; preferiblemente seleccionados de entre artritis psoriásica, artritis reumatoide, artritis reumatoide juvenil, espondilitis anquilosante, bursistis aguda y subaguda, tenosinovitis aguda no específica, artritis gotosa aguda, osteoartritis postraumática, sinovitis de osteoartritis y epicondilitis; enfermedades del colágeno, preferiblemente seleccionadas de entre lupus eritematoso sistémico, dermatomiositis sistémica (polimiositis) y carditis reumática aguda; enfermedades dermatológicas, preferiblemente seleccionadas de entre pénfigo, dermatitis ampollosa herpetiforme, eritema multiforme grave (síndrome de Stevens-Johnson), dermatitis exfoliativa, micosis fungoide, psoriasis y dermatitis seborreica; estados alérgicos, preferiblemente seleccionados de entre rinitis alérgica estacional o perenne, asma bronquial, dermatitis de contacto, dermatitis atópica, enfermedad del suero y reacciones de hipersensibilidad a fármacos; enfermedades oftálmicas, preferiblemente seleccionadas de entre úlceras marginales corneales alérgicas, herpes zoster oftálmico, inflamación del segmento anterior, uveítis posterior difusa y coroiditis, oftalmía simpática, conjuntivitis alérgica, queratitis, coriorretinitis, neuritis óptica, iritis e iridociclitis; enfermedades respiratorias, preferiblemente seleccionadas de entre sarcoidosis sintomática, síndrome de Loeffler, beriliosis, tuberculosis pulmonar fulminante o diseminada cuando se usa junto con quimioterapia antituberculosa, neumonitis por aspiración; trastornos hematológicos, preferiblemente seleccionados de entre púrpura trombocitopénica idiopática, trombocitopenia secundaria, anemia hemolítica adquirida (autoinmune), eritroblastopenia (anemia RBC), anemia hipoplásica congénita (eritroide); enfermedades neoplásicas, preferiblemente seleccionadas de entre leucemias y linfomas, leucemia aguda de la infancia; enfermedades gastrointestinales, preferiblemente seleccionadas de entre colitis ulcerosa y enteritis regional.
Los siguientes ejemplos ilustran detalladamente la invención, pero no han de considerarse como limitantes de su alcance.
Las siguientes abreviaturas se usan en las descripciones de los experimentos: AcOH = ácido acético; Atafos = bis(di-terc-butil(4 dimetilaminofenil)fosfina)dicloropaladio(II); Cbz = carboxibencilo; DCM = diclorometano; DEA = dietilamina; DIPEA = N,N-diisopropiletilamina; DMa P = 4-(dimetilamino)-piridina; DMF = N,N-dimetilformamida; DMSO = dimetilsulfóxido; DPPA = difenil fosforil azida; dppf = 1,1'; bis(difenilfosfanil)ferroceno; EA = acetato de etilo; EtOAc = acetato de etilo; EtOH = etanol; HATU = hexafluorofosfato de 3-óxido de 1-[bis(dimetilamino)metileno]-1H-1,2,3-triazol[4,5-6]piridinio; h = hora; LDA = litiodiisopropilamida; LiHMDS = bis(trimetilsilil)amida de litio; MeOH = metanol; min = minuto; n-BuLi = nbutillitio; sat. = saturado(a); TA = temperatura ambiente; TR = tiempo de retención; terc = terciario(a); TEA = trietilamina; TFA = ácido trifluoro acético; THF = tetrahidrofurano; p-TSA = ácido paratoluenosulfónico; TMSCl = cloruro de trimetilsililo.
Síntesis de trans-4-amino-5-(3-dorofenil)pirrolidin-2-ona (producto intermedio A1)

P roducto in te rm ed io A1
Etapa 1: En un tubo sellado, se colocó anhídrico maleico (9,8 g, 100 mmol, 1,0 eq), p-tiocresol (12,4 g, 100 mmol, 1,0 eq), acetato de amonio (7,8 g, 100 mmol, 1,0 eq), 3-clorobenzaldehído (11,5 ml, 100 mmol, 1,0 eq) y tolueno (100 ml). La mezcla de reacción se agitó a TA durante 1 hora y después se agitó a 150 °C durante 16 h. Después de enfriar a TA, el solvente se evaporó bajo presión reducida y el residuo se basificó con solución una solución de NaHCO3 sat. y se extrajo con DCM. La capa acuosa se acidificó con HCl al 2 N bajo enfriamiento con hielo y el producto crudo se extrajo dos veces con EtOAc. Las capas orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4, se filtraron y se concentraron para obtener el ácido 2-(3-clorofenil)-5-oxo-3-(p-toliltio)pirrolidin-3-carboxílico crudo (10,0 g).
Etapa 2: A una solución de ácido 2-(3-clorofenil)-5-oxo-3-(p-toliltio)pirrolidin-3-carboxílico crudo (10,0 g, 27,7 mmol, 1,0 eq) en acetona (100 ml), se le añadió carbonato de potasio (15,3 g, 110,8 mmol, 4,0 eq) y yoduro de metilo (7,0 ml, 110,8 mmol, 4,0 eq) a 0 °C y la mezcla de reacción se agitó durante 16 h a TA. El solvente se eliminó bajo presión reducida y el residuo se repartió entre DCM y agua. La capa acuosa se extrajo dos veces con d Cm . Las fases orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4, se filtraron y se concentraron. El producto crudo se purificó mediante cromatografía en columna (gel de sílice 100 200, 50 % de EtOAc:hexano) para dar 2-(3-clorofenil) -5-oxo-3-(p-toliltio)pirrolidin-3-carboxilato de metilo como un sólido blanquecino (4,0 g, 38 %).
Etapa 3: A una solución agitada de 2-(3-clorofenil)-5-oxo-3-(p-toliltio)pirrolidin-3-carboxilato de metilo (10,0 g, 26,66 mmol, 1,0 eq) en EtOH:THF (100 ml, 2:1), se le añadió níquel Raney (2,5 g) y la mezcla de reacción se agitó durante 2 h a TA. Después de completar la reacción, la mezcla de reacción se filtró a través de un lecho de celite y, a continuación, este último se lavó de 2 a 3 veces con EtOAc. Las capas orgánicas combinadas se concentraron y el producto crudo se purificó mediante cromatografía en columna (gel de sílice 100-200, con un 50 % de EtOAc:hexanos) para dar 2-(3-clorofenil)-5-oxopirrolidin-3-carboxilato de metilo como un sólido blanquecino (6,0 g, 89 %) (sin:anti, mezcla de 1:1).
Etapa 4: A una solución agitada de 2-(3-clorofenil)-5-oxopirrolidin-3-carboxilato de metilo (3,0 g, 11,85 mmol, 1.0 eq) en MeOH (50 ml), se le añadió una solución de NaOH al 2 N (10 ml) y la mezcla de reacción se agitó a 80 °C durante 2 h. Después de completar la reacción (monitorizada mediante CLEM), la mezcla de reacción se concentró y se acidificó con una solución de HCl al 2 N y, a continuación, el producto crudo se extrajo con un 30 % de isopropanol-DCM. Las capas orgánicas combinadas se secaron sobre Na2SO4 y se concentraron bajo presión reducida para obtener ácido trans-2-(3-clorofenil)-5-oxopirrolidin-3-carboxílico (2,5 g, 88 %).
Etapa 5: A una solución agitada de ácido trans-2-(3-clorofenil)-5-oxopirrolidin-3-carboxílico (2,0 g, 8,36 mmol, 1.0 eq) en benceno:THF (100 ml, 4:1), se le añadió TEA (2,35 ml, 16,73 mmol, 2,0 eq) y DPPA (2,35 ml, 10,8 mmol, 1,3 eq) y la mezcla de reacción se agitó a TA durante 2 h. A continuación, se añadió alcohol 2,4-dimetoxi bencílico (1,8 g, 10,87 mmol, 1,3 eq) a la mezcla de reacción y esta última se calentó a reglujo durante 16 h. Una vez completa, la mezcla de reacción se concentró bajo presión reducida para obtener el producto crudo que se extrajo con agua y EtOAc. Las capas orgánicas combinadas se secaron sobre Na2SO4 y se concentraron bajo presión reducida para obtener el producto crudo que se purificó mediante cromatografía en columan (gel de sílice de malla 100-200; con MeOH al 2 %-DCM; Rf-valor-0,5) para obtener trans-2,4
dimetoxibencil (2-(3-dorofenil)-5-oxopirrolidin-3-il)carbamato (1,5 g, 44 %).
Etapa 6: A una solución agitada de trans-2,4-dimetoxibencil (2-(3-clorofenil)-5-oxopirrolidin-3-il)carbamato (0,5 g, 1,23 mmol, 1,0 eq) en DCM (10 ml) se le añadió TFA (2 ml) a 0 °C, y la reacción se agitó durante 3 h a TA. Después de completar la reacción, la mezcla de reacción se diluyó con EtOAc y se lavó con una solución de NaHCO3 saturada. Las capas orgánicas combinadas se secaron sobre Na2SO4 y se concentraron para obtener la trans-4-amino-5-(3-clorofenil)pirrolidin-2-ona deseada como un sólido blanco (0,25 g, 96 %).
Síntesis de trans-4-amino-5-fenilpirrolidin-2-ona (producto intermedio A2)

Etapa 1: En un tubo sellado, se colocó anhídrido maleico (9,8 g, 100 mmol, 1,0 eq), p-tiocresol (12,4 g, 100 mmol, 1,0 eq), acetato de amonio (7,8 g, 100 mmol, 1,0 eq) y benzaldehído (10 ml, 100 mmol, 1,0 eq) y se añadieron 100 ml de tolueno. La mezcla de reacción se agitó a TA durante 1 hora y después se agitó a 150 °C durante 16 h. Después de enfriar a TA, el solvente se evaporó bajo presión reducida y el residuo se basificó con solución una solución de NaHCO3 saturada y se extrajo con DCM. La capa acuosa se acidificó con HCl al 2 N bajo enfriamiento con hielo y el producto crudo se extrajo dos veces con EtOAc. Las capas orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4, se filtraron y se concentraron para obtener un ácido 5-oxo-2-fenil-3-(p-toliltio)pirrolidin-3-carboxílico crudo (10,0 g, crudo).
Etapa 2: A una solución agitada de ácido 5-oxo-2-fenil-3-(p-toliltio)pirrolidin-3-carboxílico crudo (10,0 g, 30,58 mmol, 1,0 eq) en acetona (100 ml), se le añadió carbonato de potasio (16,8 g, 122,32 mmol, 4,0 eq) y yoduro de metilo (7,6 ml, 122,32 mmol, 4,0 eq) a 0 °C, y la reacción se agitó durante 16 h a TA. El solvente se eliminó bajo presión reducida y el residuo se repartió entre DCM y agua. La capa acuosa se extrajo dos veces con DCM. Las fases orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4, se filtraron y se concentraron. El producto crudo se purificó mediante cromatografía en columna (gel de sílice 100-200, 50 % de EtOAc:hexanos) para dar 5-oxo-2-fenil-3-(p-toliltio)pirrolidin-3-carboxilato de metilo (4,0 g, 38 %) como un sólido blanquecino.
Etapa 3: A una solución agitada de 5-oxo-2-fenil-3-(p-toliltio)pirrolidin-3-carboxilato de metilo (4,0 g, 11,73 mmol, 1,0 eq) en EtOH:THF (100 ml, 2:1), se le añadió níquel Raney (1 g) y la mezcla de reacción se agitó durante 2 h a TA. Después de completar la reacción, la mezcla de reacción se filtró a través de un lecho de celite y este último se lavó de 2 a 3 veces con EtOAc.. Las capas orgánicas combinadas se concentraron y el producto crudo se purificó mediante cromatografía en columna (gel de sílice 100-200, 50 % de EtOAc:hexanos) para dar 5-oxo-2-fenilpirrolidin-3-carboxilato de metilo (2,2 g, 88 %, sin:anti, mezcla de 1:1) como un sólido blanquecino.
Etapa 4: A una solución agitada de 5-oxo-2-fenilpirrolidin-3-carboxilato de metilo (1,0 g, 4,56 mmol, 1,0 eq) en MeOH (25 ml), se le añadió una solución de NaOH al 2 N (5 ml) y la mezcla de reacción se agitó a 80 °C durante 2 h. Después de completar la reacción (monitorizada mediante CLEM), la mezcla de reacción se concentró, se acidificó con una solución de HCl al 2 N y se extrajo con un 30 % de isopropanol-DCM. Las capas orgánicas combinadas se secaron sobre Na2SO4 y se concentraron bajo presión reducida para obtener el ácido trans-5-oxo-2-fenilpirrolidin-3-carboxílico deseado (0,8 g, 85 %).
Etapa 5: A una solución agitada de ácido trans-5-oxo-2-fenilpirrolidin-3-carboxílico (0,5 g, 2,43 mmol, 1,0 eq) en benceno:THF (25 ml, 4:1), se le añadió TEA (0,68 ml, 4,87 mmol, 2,0 eq) y DPpA (0,68 ml, 3,17 mmol, 1,3 eq) y la mezcla de reacción se agitó a TA durante 2 h. Se añadió alcohol bencílico (0,33 ml, 3,17 mmol, 1,3 eq) y la mezcla de reacción se calentó a reflujo durante 16 h. Una vez completa, la mezcla de reacción se concentró bajo presión reducida para obtener el compuesto crudo que se extrajo con agua y EtOAc. Las capas orgánicas
combinadas se secaron sobre Na2SO4 y se concentraron bajo presión reducida para obtener el producto crudo que se purificó mediante cromatografía en columna (gel de sílice de malla 100-200; con MeOH al 2 %-DCM; Rf-valor-0,5) para obtener (5-oxo-2-fenilpirrolidin-3-il)carbamato de terc-bencilo (0,38 g, 50 %).
Etapa 6: A una solución agitada de (5-oxo-2-fenilpirrolidin-3-il)carbamato de terc-bencilo (1,7 g, 5,48 mmol, 1,0 eq) en MeOH (20 ml, 2:1), se le añadió Pd/C (0,058 g, 0,548 mmol, 0,1 eq) y la reacción se agitó con un globo de hidrógeno durante 2 h a TA. Después de completar la reacción, la mezcla de reacción se filtró a través de un lecho de celite y se lavó de 2 a 3 veces con EtOAc. Las capas orgánicas combinadas se concentraron para obtener la trans-4-amino-5-fenilpirrolidin-2-ona deseada como una goma marrón (0,9 g, 93 %).
Síntesis de (4S,5R)-4-amino-5-fenilpirrolidin-2-ona (producto intermedio A2-ent2)
resolución
quiral


producto intermedio A2 producto intermedio A2-ent2
Racémico 
A una solución agitada de trans-4-amino-5-fenil-pirrolidin-2-ona (producto intermedio A2) (10,0 g, 0,056 mol) en EtOH (180 ml) y acetonitrilo (200 ml), se le añadió ácido L-tartárico (8,5 g, 0,056 mol) a TA. La suspensión resultante se agitó a 90 °C durante 1 h. A esta suspensión en reflujo se le añadió lentamente agua (110 ml). La mezcla de reacción resultante se mantuvo a 90 °C y se agitó durante 4 h. La solución clara resultante se enfrió lentamente a TA y se la dejó reposar a TA durante 24 h. Por consiguiente, el sólido precipitado se recogió mediante filtración y se lavó con EtOH (100 ml) para obtener 7,5 g de quiral (ent-2) como la sal de L-tartrato correspondiente. Este material sólido se trató con una solución acuosa de NaOH al 1N a TA. A continuación, la solución acuosa básica resultante se extrajo con MeOH al 10 % en DCM (100 ml * 5 a 6 veces) para obtener (4S,5R)-4-amino-5-fenil-pirrolidin-2-ona (3 g, 60 %) como un sólido blanco (producto intermedio A2-ent2).
Exceso enantiomérico (ee) determinado por HPLC quiral (nombre de la columna: Chiralpak IA (4,6 * 250 mm), 5 pM; fase móvil: hexano/EtOH/IP amina: 80/20/0,1; caudal: 1,0 ml/min; TR = 25,0 min): ee = 99,7 %
Rotación específica: [+29,9°] a 25 °C, C = 1 % en EtOH.
Síntesis de (4R,5S)-4-amino-5-fenilpirrolidin-2-ona (producto intermedio A2-ent1)

A una solución agitada de trans-4-amino-5-fenil-pirrolidin-2-ona (producto intermedio A2) (7,0 g, 39,77 mmol) en EtOH (126 ml) y acetonitrilo (140 ml), se le añadió ácido D-tartárico (5,96 g, 39,77 mmol) a TA. La suspensión resultante se agitó a 90 °C durante 1 h. A esta suspensión en reflujo se le añadió lentamente agua (77 ml). La mezcla de reacción resultante se mantuvo a 90 °C durante 4 h. La solución clara resultante se enfrió lentamente a TA y se la dejó reposar a TA durante 24 h. Por consiguiente, el sólido precipitado se recogió mediante filtración y se lavó con EtOH (70 ml) para obtener 5,2 g de quiral (ent-1) como la sal de D-tartrato correspondiente, como un sólido blanquecino. El (4R,5S)-4-amino-5-fenilpirrolidin-2-ona (2R,3R)-2,3-dihidroxisuccinato (5,2 g) se trató con una solución de NaOH al 1 N a TA. A continuación, la solución acuosa básica resultante se extrajo con MeOH al 10 % en DCM (4 * 50 ml) para obtener (4R,5S)-4-amino-5-fenilpirrolidin-2-ona (2,4 g, 34 %) como un sólido blanco.
Exceso enantiomérico (ee) determinado por HPLC quiral (nombre de la columna: Chiralpak IA (4,6 * 250 mm), 5 pM; fase móvil: hexano/EtOH/IP amina: 80/20/0,1; caudal: 1,0 ml/min; TR = 17,65 min): ee = 99,1 %
Rotación específica: [-34,5°] a 25 °C, C = 1,0 % en EtOH.
Síntesis de trans-4-amino-5-(2,4-difluorofenil)pirrolidin-2-ona (producto intermedio A3)

Etapa 1: En un tubo sellado, se colocó anhídrido maleico (28,9 g, 295,7 mmol, 1,0 eq), p-tiocresol (36,6 g, 295.7 mmol, 1,0 eq), acetato de amonio (22,7 g, 295,7 mmol, 1,0 eq) y 2,4-difluorobenzaldehído (42,0 g, 295,7 mmol, 1,0 eq) y se agregaron 100 ml de tolueno. La mezcla de reacción se agitó a TA durante 1 hora y después se agitó a 150 °C durante 16 h. Después de enfriar a TA, el solvente se evaporó bajo presión reducida y el residuo se basificó con solución una solución de NaHCO3 sat. y se extrajo con DCM. La capa acuosa se acidificó con HCl al 2 N, bajo enfriamiento con hielo y, a continuación, se extrajo dos veces con EtOAc. Las capas orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4, se filtraron y se concentraron para obtener el ácido 3-((2,4-difluorofenil)tio)-5-oxo-2-fenilpirrolidin-3-carboxílico crudo (120,0 g).
Etapa 2: A una solución agitada de ácido 3-((2,4-difluorofenil)tio)-5-oxo-2-fenilpirrolidin-3-carboxílico crudo (107,0 g, crudo) en acetona (600 ml), se le añadió carbonato de potasio (162,7 g, 1170 mmol, 4,0 eq) y yoduro de metilo (73,3 ml, 1170 mmol, 4,0 eq) a 0 °C, y la mezcla de reacción se agitó durante 16 h a TA. El solvente se eliminó bajo presión reducida y el residuo se repartió entre DCM y agua. La capa acuosa se extrajo dos veces con d Cm . Las fases orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4, se filtraron y se concentraron. El producto crudo se purificó mediante cromatografía en columna (gel de sílice de 100-200, con un 50 % de EtOAc:hexanos), lo que dio el 3-((2,4-difluorofenil)tio)-5-oxo-2-fenilpirrolidin-3-carboxilato de metilo como un sólido blanquecino (6,0 g, 5 %).
Etapa 3: A una solución agitada de 3-((2,4-difluorofenil)tio)-5-oxo-2-fenilpirrolidin-3-carboxilato de metilo (6,0 g, 15,9 mmol, 1,0 eq) en EtOH:THF (225 ml, 2:1), se le añadió níquel Raney (60,0 g) y la reacción se agitó durante 2 h a TA. Después de completar la reacción, la mezcla de reacción se filtró a través de un lecho de celite y se lavó de 2 a 3 veces con EtOAc. Las capas orgánicas combinadas se concentraron y el producto crudo se purificó mediante cromatografía en columna (gel de sílice 100-200, 50 % de EtOAc:hexanos) que dio 2-(2,4-difluorofenil)-5-oxopirrolidin-3-carboxilato de metilo (2,8 g, 69 %, sin:anti, 1:1) como un sólido blanquecino.
Etapa 4: A una solución agitada de 2-(2,4-difluorofenil)-5-oxopirrolidin-3-carboxilato de metilo (2,0 g, 7,84 mmol, 1,0 eq) en MeOH (47 ml), se le añadió una solución de NaOH al 2 N (12 ml) y la mezcla de reacción se agitó a 70 °C durante 3 h. Después de completar la reacción (monitorizada mediante CLEM), la mezcla de reacción se concentró y se acidificó con una solución de HCl al 2 N y, a continuación, se extrajo con un 30 % de isopropanol-DCM. Las capas orgánicas combinadas se secaron sobre Na2SO4 y se concentraron bajo presión reducida para obtener el ácido trans-2-(-2,4-difluorofenil)-5-oxopirrolidin-3-carboxílico deseado (1,8 g, 95 %).
Etapa 5: A una solución agitada de ácido trans-2-(2,4-difluorofenil)-5-oxopirrolidin-3-carboxílico (1,8 g, 7,46 mmol, 1,0 eq) en benceno:THF (60 ml, 4:1), se le añadió TEA (2,07 ml, 14,93 mmol, 2,0 eq) y DPPA (2,1 ml, 9.7 mmol, 1,3 eq) y la mezcla de reacción se agitó a TA durante 2 h. Se añadió alcohol bencílico (1,0 ml, 9,7 mmol, 1,3 eq) y la mezcla de reacción se calentó a reflujo durante 16 h. Una vez completa, la mezcla de reacción se concentró bajo presión reducida para obtener el producto crudo que se extrajo con agua y EtOAc. Las capas orgánicas combinadas se secaron sobre Na2SO4 y se concentraron bajo presión reducida para obtener el producto crudo que se purificó mediante cromatografía en columan (gel de sílice de malla 100-200; con MeOH al 2 %-DCM; Rf-valor-0,5) para obtener (2-(2,4-difluorofenil)-5-oxopirrolidin-3-il)carbamato de transbencilo (1,2 g, 46 %) como un sólido blanquecino.
Step 6: A una solución agitada de (2-(2,4-difluorofenil)-5-oxopirrolidin-3-il)carbamato de trans-bencilo (1,2 g, 3,46 mmol, 1,0 eq) en MeOH (15 ml), se le añadió Pd/C (0,12 g, 10 % en peso) y la reacción se agitó con un globo de hidrógeno durante 2 h a TA. Después de completar la reacción, la mezcla de reacción se filtró a través
de un lecho de celite y se lavó de 2 a 3 veces con EtOAc. Las capas orgánicas combinadas se concentraron para obtener la trans-4-amino-5-(2,4-difluorofenil)pirrolidin-2-ona deseada (0,85 g) como un sólido blanquecino.
Síntesis de trans-4-amino-5-(2,3-dihidrobenzo[b][1,4]dioxin-6-il)pirrolidin-2-ona (producto intermedio A4)

Etapa 1: En un tubo sellado, se colocó anhídrido maleico (5,97 g, 60,9 mmol, 1,0 eq), p-tiocresol (7,55 g, 60,9 mmol, 1,0 eq), acetato de amonio (4,68 g, 60,9 mmol, 1,0 eq) y 2,3-dihidro-1,4-benzodioxin-6-carbaldehído (10,0 g, 60,9 mmol, 1,0 eq), seguido de la adición de 80 ml de tolueno. La mezcla de reacción se agitó a TA durante 1 hora y, a continuación, se calentó a 150 °C durante 16 h. Después de enfriar a TA, el solvente se evaporó bajo presión reducida y el residuo se basificó con solución una solución de NaHCO3 sat. y se extrajo con DCM. La capa acuosa se acidificó con HCl al 2 N, bajo enfriamiento con hielo, y se extrajo dos veces con EtOAc. Las capas orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4, se filtraron y se concentraron para obtener el ácido 2-(2,3-dihidrobenzo[b][1,4]dioxin-6-il)-5-oxo-3-(p-toliltio)pirrolidin-3-carboxílico crudo (2 , 2 0 g).
Etapa 2: A una solución agitada de ácido 2-(2,3-dihidrobenzo[b][1,4]dioxin-6-il)-5-oxo-3-(p-toliltio) pirrolidin-3-carboxílico crudo (2,2 g, 5,707 mmol, 1,0 eq) en acetona (100 ml), se le añadió carbonato de potasio (3,2 g, 22,831 mmol, 4,0 eq) y yoduro de metilo (1,42 ml, 22,831 mmol, 4,0 eq) a 0 °C, y la reacción se agitó durante 16 h a TA. El solvente se eliminó bajo presión reducida y el residuo se repartió entre DCM y agua. La capa acuosa se extrajo dos veces con DCM. Las fases orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4, se filtraron y se concentraron. El producto crudo se purificó mediante cromatografía en columna (gel de sílice 100-200, 50 % de EtOAc:hexanos) que dio 2-(2,3-dihidrobenzo[b][1,4]dioxin-6-il)-5-oxo-3-(ptoliltio-pirrolidin-3-carboxilato de metilo como un sólido blanquecino (0,9 g, 41 %).
Etapa 3: A una solución agitada de 2-(2,3-dihidrobenzo[b][1,4]dioxin-6-il)-5-oxo-3-(p-toliltio)-pirrolidin-3-carboxilato de metilo (0,9 g, 2,253 mmol, 1,0 eq) en EtOH:THF ( 60 ml, 2:1), se le añadió níquel Raney (1,0 g) y la reacción se agitó durante 2 h a TA. Después de completar la reacción, la mezcla de reacción se filtró a través de un lecho de celite y se lavó de 2 a 3 veces con EtOAc. Las capas orgánicas combinadas se concentraron y los restos crudos se purificaron mediante cromatografía en columna (gel de sílice 100-200, 50 % de EtOAc:hexanos) que dio 2-(2,3-dihidrobenzo[b][1,4]dioxin-6-il)-5-oxopirrolidin-3-carboxilato de metilo (0,6 g, 96 %, sin:anti, 1:1) como un sólido blanquecino.
Etapa 4: A una solución agitada de 2-(2,3-dihidrobenzo[b][1,4]dioxin-6-il)-5-oxopirrolidin-3-carboxilato de metilo (0,7 g, 2,524 mmol, 1,0 eq) en MeOH (15 ml), se le añadió una solución de NaOH al 2 N (3,7 ml) y la mezcla de reacción se agitó a 80 °C durante 2 h. Después de completar la reacción (monitorizada mediante CLEM), la mezcla de reacción se concentró y se acidificó con una solución de HCl al 2 N y, a continuación, se extrajo con un 30 % de isopropanol-DCM. La capa orgánica se secó sobre Na2SO4 anhidro y se concentró bajo presión reducida para obtener el ácido trans-2-(2,3-dihidrobenzo[b][1,4]dioxin-6-il)-5-oxopirrolidin-3-carboxílico deseado (0,5 g, 75 %).
Etapa 5: A una solución agitada de ácido trans-2-(2,3-dihidrobenzo[b][1,4]dioxin-6-il)-5-oxopirrolidin-3-carboxílico (0,3 g, 1,139 mmol, 1,0 eq) en benceno:THF (15 ml, 4:1), se le añadió TEA (0,31 ml, 4,87 mmol, 2,0 eq) y DPPA (0,32 ml, 1,48 mmol, 1,3 eq) y la mezcla de reacción se agitó a TA durante 2 h. Se añadió alcohol bencílico (3 ml) y la mezcla de reacción se calentó a reflujo durante 16 h. Después de completada, la mezcla de reacción se concentró bajo presión reducida para dar el producto crudo que se extrajo con agua y EtOAc. La capa orgánica se secó sobre Na2SO4 anhidro y se concentró bajo presión reducida para obtener le producto crudo que se purificó mediante cromatografía en columna (gel de sílice de malla 100-200; con MeOH al 2 %-DCM; Rf-valor-0,5) para obtener (-2-(2,3-dihidrobenzo[b][1,4]dioxin-6-il)-5-oxopirrolidin-3-il)carbamato de trans-bencilo (0,2 g, 47 %).
Etapa 6: A una solución agitada de (-2-(2,3-dihidrobenzo[b][1,4]dioxin-6-il)-5-oxopirrolidin-3-il)carbamato de trans-bencilo (0,32 g, 0,869 mmol, 1,0 eq) en MeOH:THF (20 ml, 2:1), se le añadió Pd/C (50,0 mg) y la reacción se agitó con un globo de hidrógeno durante 2 h a TA. Después de completar la reacción, la mezcla de reacción se filtró a través de un lecho de celite y se lavó de 2 a 3 veces con EtOAc. La capa orgánica combinada se concentró para obtener la trans-4-amino-5-(2,3-dihidrobenzo[b][1,4]dioxin-6-il)pirrolidin-2-ona deseada (0,2 g, 98 %) como una goma marrón.
Síntesis de trans-4-amino-5-(3-fluorofenil)pirrolidin-2-ona (producto intermedio A5)

Etapa 1: En un matraz de base redonda, se colocó anhídrido maleico (19,7 g, 201,61 mmol, 1,0 eq), p-tiocresol (25,0 g, 201,61 mmol, 1,0 eq), 2,4-dimetoxi bencilamina (33,6 g, 201,61 mmol, 1,0 eq), y 3-fluorobenzaldehído (25,0 g, 201,61 mmol, 1,0 eq), seguido de la adición de 250 ml de tolueno. La mezcla de reacción se calentó a reflujo durante 16 h con agitación vigorosa. Después de completar la reacción (monitorizada por TLC), la mezcla de reacción se enfrió a la TA y el solvente se evaporó bajo presión reducida para obtener un ácido 1-(2,4-dimetoxibencil)-2-(3-fluorofenil)-5-oxo-3-(p-toliltio)pirrolidin-3-carboxílico crudo (89,0 g, 89 %) como un líquido gomoso, el cual se usó en la etapa siguiente sin purificación adicional.
Etapa 2: A una solución agitada de ácido 1-(2,4-dimetoxibencil)-2-(3-fluorofenil)-5-oxo-3-(p-toliltio)-pirrolidin-3-carboxílico (99,7 g, 201,4 mmol, 1,0 eq) en acetona (1 l), se le añadió carbonato de potasio (111,3 g, 805,6 mmol, 4,0 eq) y yoduro de metilo (51,0 ml, 805,6 mmol, 4,0 eq) a 0 °C y la reacción se agitó durante 16 h a TA. Después de completar la reacción (monitorizada mediante TLC), el solvente se eliminó bajo presión reducida y el residuo se repartió entre EtOAc y agua. La capa acuosa se extrajo dos veces con EtOAc. Las fases orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4, se filtraron y se concentraron. El producto crudo se purificó mediante cromatografía en columna (gel de sílice 100-200, EtOAc al 40 % en hexano) para proporcionar 1-(2,4-dimetoxibencil)-2-(3-fluorofenil)-5-oxo-3-(p -toliltio)pirrolidin-3-carboxilato de metilo (79,0 g, 77 %) como un sólido blanquecino.
Etapa 3: A una solución agitada de 1-(2,4-dimetoxibencil)-2-(3-fluorofenil)-5-oxo-3-(p-toliltio)-pirrolidin-3-carboxilato de metilo (78,0 g, 153,2 mmol, 1,0 eq) en acetonitrilo (500 ml), se le añadió Ca n (251.9 g, 459.6 mmol, 3,0 eq) disuelto en agua gota a gota a 0 °C a través de un embudo de adición. A continuación, la mezcla de reacción se agitó a TA durante 16 h. Después de completar la reacción (monitorizada mediante TLC), la mezcla de reacción se diluyó con agua y se extrajo dos veces con EtOAc. Las fases orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4, se filtraron y se concentraron. El producto crudo se purificó mediante cromatografía en columna (gel de sílice 230-400, EtOAc al 40-50 % EtOAc:hexano), para dar 2-(3-fluorofenil)-5-oxo-3-(p -toliltio)pirrolidin-3-carboxilato de metilo (47,0 g, 85 %) como un sólido blanquecino.
Etapa 4: A una solución agitada de 2-(3-fluorofenil)-5-oxo-3-(p-toliltio)pirrolidin-3-carboxilato de metilo (30,0 g,
83,5 mmol, 1,0 eq) en EtOH:THF (500 ml:500 ml, 1:1), se le añadió níquel Raney (20,0 g) y la reacción se agitó bajo una atmósfera de hidrógeno durante 16 h a TA. Después de completar la reacción (monitorizada mediante TLC), la mezcla de reacción se filtró a través de un lecho de celite, el cual se lavó de 4 a 5 veces con THF. El filtrado se concentró para obtener 2-(3-fluorofenil)-5-oxopirrolidin-3-carboxilato de metilo (15,2 g, 77 %, mezcla de sin:anti) como un sólido blanquecino.
Etapa 5: A una solución agitada de 2-(3-fluorofenil)-5-oxopirrolidin-3-carboxilato de metilo (16,0 g, 67,4 mmol, 1.0 eq) en MeOH (320 ml), se le añadió una solución de NaOH al 2 N (75 ml) y la mezcla de reacción se agitó a 80 °C durante 16 h. Después de completar la reacción (monitorizada mediante TLC), la mezcla de reacción se concentró y se acidificó con una solución de HCl al 2 N para obtener un sólido, el cual se filtró y se lavó con éter dietílico y, a continuación, se secó al vacío, para obtener ácido trans-2-(3-fluorofenil)-5-oxopirrolidin-3-carboxílico (9,3 g, 62 %).
Etapa 6: A una solución agitada de ácido trans-2-(3-fluorofenil)-5-oxopirrolidin-3-carboxílico (13,0 g, 58,3 mmol, 1.0 eq) en tolueno (130 ml), se le añadió TEA (8.5 ml, 61,2 mmol, 1,05 eq) y DPPA (19.3g, 70,0 mmol, 1,2 eq) y la mezcla de reacción se agitó a 90 °C durante 30 min. El alcohol bencílico (12.6g, 116,6 mmol, 2,0 eq) se añadió a la mezcla de reacción y se calentó a reflujo durante 16 h. Después de completar la reacción (monitorizada mediante TLC), la mezcla de reacción se concentró bajo presión reducida. A continuación, el residuo se diluyó con EtOAc (100 ml), se lavó con agua (2 x 100 ml), se secó sobre Na2SO4 anhidro y se concentró bajo presión reducida para obtener el producto crudo que se purificó mediante cromatografía en columna (gel de sílice de malla 230-400; MeOH del 0 al 2 % en DCM) para obtener (2-(3-fluorofenil)-5-oxopirrolidin-3-il)carbamato de trans-bencilo (7,0 g, 37 %).
Etapa 7: A una solución agitada de (2-(3-fluorofenil)-5-oxopirrolidin-3-il)carbamato de trans-bencilo (7,0 g, 21,3 mmol, 1,0 eq) en MeOH (50 ml) y Th F (20 ml), se le añadió Pd-C (1,5 g, 14,9 mmol, 0,7 eq) y la mezcla de reacción se agitó con un globo de hidrógeno durante 2 h a TA. Después de completar la reacción (monitorizada mediante TLC), la mezcla de reacción se filtró a través de un lecho de celite, el cual se lavó de 2 a 3 veces con THF. El filtrado se concentró para obtener la trans-4-amino-5-(3-fluorofenil)pirrolidin-2-ona (3,8 g, 92 %) deseada como una goma marrón.
Síntesis de trans-4-amino-5-(2-fluorofenil)pirrolidin-2-ona (producto intermedio A6)

Etapa 1: Se recogió anhídrido maleico (19,7 g, 201,4 mmol, 1,0 eq), p-tiocresol (25,0 g, 201,4 mmol, 1,0 eq), 2,4-dimetoxi bencilamina (33,6 g, 201,4 mmol, 1,0 eq) y 2-fluorobenzaldehído (25,0 g, 201,4 mmol, 1,0 eq) en 300 ml de tolueno. La mezcla de reacción se calentó a reflujo durante 16 h con agitación vigorosa. Después de completar la reacción (monitorizada por TLC, el sistema de TLC con MeOH al 5 % en DCM, Rf-0,1), la mezcla de reacción se enfrió a la TA y el solvente se evaporó bajo presión reducida para dar ácido 1-(2,4-dimetoxibencil)-2-(2-fluorofenil)-5-oxo-3-(p-toliltio)pirrolidin-3-carboxílico crudo (95,0 g, 95 %) como un líquido gomoso, el cual se usó en la etapa siguiente sin purificación adicional.
Etapa 2: A una solución agitada de ácido 1-(2,4-dimetoxibencil)-2-(2-fluorofenil)-5-oxo-3-(p-toliltio)-pirrolidin-3carboxílico crudo (95,0 g, 191,7 mmol, 1,0 eq) en acetona (1 l), se le añadió carbonato de potasio (111,3 g, 805.0 mmol, 4,2 eq) y yoduro de metilo (50,0 ml, 805,0 mmol, 4,2 eq) a 0 °C y la mezcla de reacción se agitó a TA durante 16 h. Después de completar la reacción (monitorizada mediante TLC, sistema de TLC con 30 % de EtOAc en hexano, Rf-0,3), el solvente se eliminó bajo presión reducida y el residuo se repartió entre EtOAc y agua. La capa acuosa se extrajo dos veces con EtOAc. Las fases orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4, se filtraron y se concentraron. El producto crudo se purificó mediante cromatografía en columna (gel de sílice 100-200, 40 % de EtOAc en hexano) para obtener 1-(2,4-dimetoxibencil)-2-(2-fluorofenil)-5-oxo-3-(p-toliltio)pirrolidin-3-carboxilato de metilo como un sólido blanquecino (55,0 g, 56 %).
Etapa 3: A una solución agitada de 1-(2,4-dimetoxibencil)-2-(2-fluorofenil)-5-oxo-3-(p-toliltio)-pirrolidin-3-carboxilato de metilo (55,0 g, 108,0 mmol, 1,0 eq) en acetonitrilo (300 ml), se le añadió Ca n (178,0 g, 324,0 mmol, 3,0 eq) en agua (300 ml) gota a gota a 0 °C a través de un embudo de adición. A continuación, la mezcla de reacción se agitó a TA durante 16 h. Después de completar la reacción (monitorizada mediante TLC, sistema de TLC con un 50 % de EtOAc en hexano, Rf-0,3), la mezcla de reacción se diluyó con agua y se extrajo dos veces con EtOAc. Las fases orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4, se filtraron y se concentraron. El producto crudo se purificó mediante cromatografía en columna (gel de sílice 230 400, 40-50 % de EtOAc:hexano), lo que dio 2-(2-fluorofenil)-5-oxo-3-(p-toliltio)pirrolidin-3-carboxilato de metilo como un sólido blanquecino (15,0 g, 39 %).
Etapa 4: A una solución agitada de 2-(2-fluorofenil)-5-oxo-3-(p-toliltio)pirrolidin-3-carboxilato de metilo (15,0 g, 41.7 mmol, 1,0 eq) en EtOH:THF (300:300 ml, 1:1), se le añadió níquel Raney (15 g) y la reacción se agitó bajo una atmósfera de hidrógeno durante 16 h a TA. Después de completar la reacción (monitorizada mediante TLC, sistema de TLC con un 70 % de EtOAc en hexano, Rf-0,4), la mezcla de reacción se filtró a través de un lecho de celite, el cual se lavó de 4 a 5 veces con THF. El filtrado se concentró para obtener 2-(2-fluorofenil)-5-oxopirrolidin-3-carboxilato de metilo como un sólido blanco (9,0 g, 91 %; mezcla de sin:anti).
Etapa 5: A una solución agitada de 2-(2-fluorofenil)-5-oxopirrolidin-3-carboxilato de metilo (9,0 g, 37,9 mmol, 1.0 eq) en MeOH (180 ml), se le añadió una solución de NaOH al 2 N (40 ml) y la mezcla de reacción se agitó a 80 °C durante 16 h. Después de completar la reacción (monitorizada mediante TLC, sistema de TLC, MeOH al 5 % en DCM, Rf-0,1), la mezcla de reacción se concentró y se acidificó con una solución de HCl de 2 N para obtener un sólido, el cual se filtró, a continuación, se lavó con éter dietílico y se secó al vacío, para obtener ácido trans -2-(2-fluorofenil)-5-oxopirrolidin-3-carboxílico (7,0 g, 83 %).
Etapa 6: A una solución agitada de ácido trans-2-(2-fluorofenil)-5-oxopirrolidin-3-carboxílico (7,0 g, 31,4 mmol, 1.00 eq) en tolueno (80 ml), se le añadió TEA (4,6 ml, 33,0 mmol, 1,05 eq) y DPPA (10,4 g, 37,7 mmol, 1,2 eq) y la mezcla de reacción se agitó a 90 °C durante 30 min. A continuación, se añadió alcohol bencílico (6,8 g, 62.8 mmol, 2,0 eq) y la mezcla de reacción se calentó a reflujo durante 16 h. Después de completar la reacción (monitorizada mediante TLC, sistema de TLC con MeOH al 5 % en DCM, Rf-0,3, la mezcla de reacción se concentró bajo presión reducida y después se diluyó con EtOAc (100 ml), se lavó con agua (2 x 100 ml), se secó sobre Na2SO4 y se concentró bajo presión reducida para obtener el producto crudo que se purificó mediante cromatografía en columna (gel de sílice de malla 230-400; MeOH del 0 al 2 % en DCM) para obtener (2-(2-fluorofenil)-5-oxopirrolidin-3-il)carbamato de trans-bencilo (4,7 g, 46 %).
Etapa 7:A una solución agitada de (2-(2-fluorofenil)-5-oxopirrolidin-3-il)carbamato de trans-bencilo (4,7 g, 14,3 mmol, 1,0 eq) en MeOH:THF (20 ml, 2:1), se le añadió Pd/C (2,0 g, 10 % húmedo) y la reacción se agitó con un globo de hidrógeno durante 2 h a TA. Después de completar la reacción (monitorizada mediante TLC, sistema de TLC con MeOH al 5 % en DCM, Rf-0,2), la mezcla de reacción se filtró a través de un lecho de celite, el cual se lavó de 2 a 3 veces con THF. El filtrado se concentró para obtener la trans-4-amino-5-(2-fluorofenil)pirrolidin-2-ona deseada como una goma marrón (2,5 g, 90 %).
Síntesis de trans-4-amino-5-(4-fluoro-3-metoxifenil)pirrolidin-2-ona (producto intermedio A7)

Etapa 1: Se disolvió anhídrido maleico (14,6 g, 149,7 mmol, 1,0 eq), p-tiocresol (18,5 g, 149,7 mmol, 1,0 eq), 2,4-dimetoxi bencil amina (25,0 g, 149,7 mmol, 1,0 eq) y 4-fluoro-3-metoxi benzaldehído (23,0 g, 149,7 mmol, 1,0 eq) en 500 ml de tolueno en un matraz de fondo redondo y dos cuellos provisto de una trampa de Dean Stark y un condensador. A continuación, la mezcla de reacción se calentó a 150 °C durante 16 h. Después de enfriar a TA, el solvente se evaporó bajo presión reducida para obtener el ácido 1-(2,4-dimetoxibencil)-2-(4-fluoro-3-metoxifenil)-5-oxo-3-(p-toliltio)-pirrolidin-3-carboxílico crudo, el cual se llevó a la etapa siguiente sin purificación adicional.
Etapa 2: A una solución agitada de ácido 1-(2,4-dimetoxibencil) -2-(4-fluoro-3-metoxifenil)-5-oxo-3-(p-toliltio) pirrolidin-3-carboxílico crudo (máx. 149,7 mmol, 1,0 eq) en acetona (500 ml), se le añadió carbonato de potasio (82,0 g, 598,0 mmol, 4,0 eq) y yoduro de metilo (37,5 ml, 598,0 mmol, 4,0 eq) a 0 °C, y la reacción se agitó durante 16 h a TA. El solvente se eliminó bajo presión reducida y el residuo se repartió entre DCM y agua. La capa acuosa se extrajo dos veces con DCM. Las fases orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4, se filtraron y se concentraron. El producto crudo se purificó mediante cromatografía en columna (gel de sílice 100-200, 50 % EtOAc:hexanos), lo que dio 1-(2,4-dimetoxibencil)-2-(4-fluoro-3-metoxifenil)-5-oxo-3-(p-toliltio)pirrolidin-3-carboxilato de metilo (72,0 g, 88 %) como un sólido blanquecino.
Etapa 3: A una solución agitada de 1-(2,4-dimetoxibencil)-2-(4-fluoro-3-metoxifenil)-5-oxo-3-(p-toliltio)pirrolidin-3-carboxilato de metilo (70,0 g, 129,0 mmol, 1,0 eq) en acetonitrilo:agua (500 ml, en 1:1), se le añadió CAN a 0 °C y la reacción se agitó durante 16 h a TA. El solvente se eliminó bajo presión reducida y el residuo se repartió entre EtOAc y agua. La capa acuosa se extrajo dos veces con EtOAc. Las fases orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4, se filtraron y se concentraron. El producto curdo se purificó mediante cromatografía en columna (gel de sílice 100-200, con un 50 % de EtOAc:hexano), lo que dio 2-(4-fluoro-3-metoxifenil)-5-oxo-3-(p-toliltio)pirrolidin-3-carboxilato de metilo (25,0 g, 50 %) como un sólido blanquecino.
Etapa 4: A una solución agitada de 2-(4-fluoro-3-metoxifenil)-5-oxo-3-(p-toliltio)pirrolidin-3-carboxilato de metilo (15,0 g, 64,3 mmol, 1,0 eq) en EtOH:THF (300 ml, 2:1), se le añadió níquel Raney Nickel (5,0 g) y la mezcla de reacción se agitó durante 2 h a TA. Después de completar la reacción, la mezcla de reacción se filtró a través de un lecho de celite y se lavó de 2 a 3 veces con EtOAc. Las capas orgánicas combinadas se concentraron y el producto crudo se purificó mediante cromatografía en columna (gel de sílice 100-200, 50 % de EtOAc:hexanos) que dio 2-(4-fluoro-3-metoxifenil)-5-oxopirrolidin-3-carboxilato de metilo (10,0 g, 98 %, sin:anti, mezcla de 1:1) como un sólido blanquecino.
Etapa 5: A una solución agitada de 2-(4-fluoro-3-metoxifenil)-5-oxopirrolidin-3-carboxilato de metilo (10,0 g, 37,5 mmol, 1,0 eq) en MeOH (250 ml), se le añadió una solución de NaOH al 2 N (50 ml) y la mezcla de reacción se agitó a 80 °C durante 2 h. Después de completar la reacción (monitorizada mediante CLEM), la mezcla de reacción se concentró, se acidificó con una solución de HCl al 2 N y, a continuación, se extrajo con un 30 % de isopropanol-DCM. Las capas orgánicas combinadas se secaron sobre Na2SO4 anhidro y se
concentraron bajo presión reducida para obtener el ácido trans-2-(4-fluoro-3-metoxifenil)-5-oxopirrolidin-3-carboxílico deseado (8,0 g, 84 %).
Etapa 6: A una solución agitada de ácido trans-2-(4-fluoro-3-metoxifenil)-5-oxopirrolidin-3-carboxílico (2,0 g, 7,90 mmol, 1,0 eq) en benceno:THF (100 ml, 4:1), se le añadió TEA (2,2 ml, 15,81 mmol, 2,0 eq) y DPPA (2,2 ml, 10,27 mmol, 1,3 eq) y la mezcla de reacción se agitó a TA durante 2 h. A continuación, se añadió alcohol bencílico (1,0 ml, 10,27 mmol, 1,3 eq) a la mezcla de reacción y esta última se calentó a reflujo durante 16 h. Una vez completa, la mezcla de reacción se concentró bajo presión reducida para obtener el producto crudo que se extrajo con agua y EtOAc. La capa orgánica se secó sobre Na2SO4 anhidro y se concentró bajo presión reducida para obtener el producto crudo que se purificó mediante cromatografía en columna (gel de sílice de malla 100-200; MeOH al 2 %-DCM; Rf-valor-0,5) para obtener (2-(4-fluoro-3-metoxifenil)-5-oxopirrolidin-3-il)carbamato de trans-bencilo (1,4 g, 50 %).
Etapa 7: A una solución agitada de (2-(4-fluoro-3-metoxifenil)-5-oxopirrolidin-3-il)carbamato de trans-bencilo (7 g, 19,55 mmol, 1 eq) en MeOH:THF (20 ml, 2:1), se le añadió Pd-C (0,7 g) y la reacción se agitó durante 2 h a TA. Después de completar la reacción, la mezcla de reacción se filtró través del lecho de celite y se lavó de 2 a 3 veces con EtOAc. La capa orgánica combinada se concentró para obtener trans-4-amino-5-(4-fluoro-3-metoxifenil)pirrolidin-2-ona (4 g, 91 %) como una goma marrón.
Síntesis de trans-4-amino-5-(4-fluorofenil)pirrolidin-2-ona (producto intermedio A8)



Etapa 1: Se recogió anhídrido maleico (19,7 g, 201,6 mmol, 1,0 eq), p-tiocresol (25,0 g, 201,6 mmol, 1,0 eq), 2,4 dimetoxi bencilamina (33,6 g, 201,6 mmol, 1,0 eq) y 4-fluorobenzaldehído (25,0 g, 201,6 mmol, 1,0 eq) en 250 ml de tolueno. La mezcla de reacción se calentó a reflujo durante 16 h con agitación vigorosa. Después de completar la reacción (monitorizada por TLC, el sistema de TLC con MeOH al 5 % en DCM, Rf-0,1), la mezcla de reacción se enfrió a la TA y el solvente se evaporó bajo presión reducida para dar ácido 1-(2,4-dimetoxibencil)-2-(4-fluorofenil)-5-oxo-3-(p-toliltio)pirrolidin-3-carboxílico crudo (92,0 g, 92 %) como un líquido gomoso, el cual se usó en la etapa siguiente sin purificación adicional.
Etapa 2: A una solución agitada de ácido 1-(2,4-dimetoxibencil)-2-(4-fluorofenil)-5-oxo-3-(p-toliltio)-pirrolidin-3-carboxílico crudo (92,0 g, 201,4 mmol, 1,0 eq) en acetona (1 l), se le añadió carbonato de potasio (111,3 g, 805,6 mmol, 4,0 eq) y yoduro de metilo (50,0 ml, 805,6 mmol, 4,0 eq) a 0 °C y la reacción se agitó durante 16 h a TA. Después de completar la reacción (monitorizada mediante TLC), el solvente se eliminó bajo presión reducida y el residuo se repartió entre EtOAc y agua. La capa acuosa se extrajo dos veces con EtOAc. Las
fases orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4, se filtraron y se concentraron. El producto crudo se purificó mediante cromatografía en columna (gel de sílice 100-200, EtOAc al 40 % en hexano) para proporcionar 1-(2,4-dimetoxibencil)-2-(4-fluorofenil)-5-oxo-3-(p -toliltio)pirrolidin-3-carboxilato de metilo (79,0 g, 84 %) como un sólido blanquecino.
Etapa 3: A una solución agitada de 1-(2,4-dimetoxibencil)-2-(4-fluorofenil)-5-oxo-3-(p-toliltio)-pirrolidin-3-carboxilato de metilo (92,0 g, 180,7 mmol, 1,0 eq) en acetonitrilo, de añadió CAN (297,0 g, 542,1 mmol, 3,0 eq) en agua gota a gota a la mezcla de reacción a 0 °C a través de un embudo de adición. A continuación, la reacción se agitó a TA durante 16 h. Después de completar la reacción (monitorizada mediante TLC, sistema de TLC con un 50 % de EtOAc en hexano, Rf-0,3), la mezcla de reacción se diluyó con agua y se extrajo dos veces con EtOAc. Las fases orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4, se filtraron y se concentraron. El producto crudo se purificó mediante cromatografía en columna (gel de sílice 230 400, 40-50 % EtOAc:hexano), lo que dio 2-(4-fluorofenil)-5-oxo-3-(p -toliltio)pirrolidin-3-carboxilato de metilo (41,0 g, 63 %) como un sólido blanquecino.
Etapa 4: A una solución agitada de 2-(4-fluorofenil)-5-oxo-3-(p-toliltio)pirrolidin-3-carboxilato de metilo (13,0 g, 36,2 mmol, 1,0 eq) en EtOH:THF (260:130 ml, 2:1), se le añadió níquel Raney (13,0 g) y la mezcla de reacción se agitó bajo una atmósfera de hidrógeno durante 16 h a TA. Después de completar la reacción (monitorizada mediante TLC), la mezcla de reacción se filtró a través de un lecho de celite, el cual se lavó de 4 a 5 veces con THF. El filtrado se concentró para dar 2-(4-fluorofenil)-5-oxopirrolidin-3-carboxilato de metilo (6,7 g, 78 %, mezcla de sin:anti) como un sólido blanco.
Etapa 5: A una solución agitada de 2-(4-fluorofenil)-5-oxopirrolidin-3-carboxilato de metilo (10,0 g, 42,2 mmol, 1.0 eq) en MeOH (200 ml), se le añadió una solución de NaOH al 2 N (48 ml) y la mezcla de reacción se agitó a 80 °C durante 16 h. Después de completar la reacción (monitorizada mediante TLC, sistema de TLC, 5 % en DCM, Rf-0,1), la mezcla de reacción se concentró y se acidificó con una solución de HCl de 2 N para obtener un sólido, el cual se filtró y se lavó con éter dietílico, seguido de un secado al vacío, para obtener ácido trans 2-(4-fluorofenil)-5-oxopirrolidin-3-carboxílico (6,4 g, 68 %).
Etapa 6: A una solución agitada de ácido trans2-(4-fluorofenil)-5-oxopirrolidin-3-carboxílico (5,0 g, 22,4 mmol, 1.00 eq) en tolueno (50 ml), se le añadió TEA (3,3 ml, 23,5 mmol, 1,05 eq) y DPPA (7,4 g, 26,9 mmol, 1,20 eq) y la mezcla de reacción se calentó a 90 °C durante 30 min. A continuación, se añadió alcohol bencílico (4,8 g, 44,8 mmol, 2,00 eq) y la mezcla de reacción se calentó a reflujo durante 16 h. Después de completar la reacción (monitorizada mediante TLC), la mezcla de reacción se concentró bajo presión reducida. A continuación, el residuo se diluyó con EtOAc (100 ml), se lavó con agua (2 x 100 ml), se secó sobre Na2SO4 y se concentró, finalmente, bajo presión reducida para obtener el producto crudo que se purificó mediante cromatografía en columna (gel de sílice de malla 230-400; 0-2 % de MeOH en DCM) para obtener (2-(4-fluorofenil)-5-oxopirrolidin-3-il)carbamato de trans-bencilo (4,1 g, 56 %).
Etapa 7: A una solución agitada de (2-(4-fluorofenil)-5-oxopirrolidin-3-il)carbamato de trans-bencilo (2,0 g, 6,1 mmol, 1,0 eq) en MeOH (50 ml) y THF (20 ml), Pd/C (0,3 g, 3,0 mmol, 0,5 eq) y la reacción se agitó con un globo de hidrógeno durante 2 h a TA. Después de completar la reacción (monitorizada mediante TLC), la mezcla de reacción se filtró a través de un lecho de celite, el cual se lavó de 2 a 3 veces con THF. El filtrado se concentró para obtener la trans4-amino-5-(4-fluorofenil)pirrolidin-2-ona deseada (1,1 g, 93 %) como una goma marrón.
Síntesis de trans-4-amino-5-(2-metoxipiridin-4-il)pirrolidin-2-ona (producto intermedio A9)

Etapa 1: Se recogió anhídrido maleico (17,2 g, 175,0 mmol, 1,0 eq), p-tiocresol (21,7 g, 175,0 mmol, 1,0 eq), 2,4-dimetoxi bencilamina (29,2 g, 175,0 mmol, 1,0 eq), y 2-metoxipiridin-4-carbaldehído (24,0 g, 175,0 mmol, 1,0 eq) en 300 ml de tolueno. La mezcla de reacción se calentó a reflujo durante 16 h con agitación vigorosa. Después de completar la reacción (monitorizada por TLC, el sistema de TLC con MeOH al 5 % en DCM, Rf-0,1), la mezcla de reacción se enfrió a la TA y el solvente se evaporó bajo presión reducida para dar ácido 1-(2,4-dimetoxibencil)-2-(2-metoxipiridin-4-il)-5-oxo-3-(p-toliltio)pirrolidin-3-carboxílico crudo (80,0 g) como un líquido gomoso, el cual se usó en la etapa siguiente sin purificación adicional.
Etapa 2: A una solución agitada de ácido 1-(2,4-dimetoxibencil)-2-(2-metoxipiridin-4-il)-5-oxo-3-(p-toliltio)-pirrolidin-3-carboxílico (57,0 g, 112,1 mmol, 1,0 eq) en acetona (300 ml), se le añadió carbonato de potasio (61,9 g, 448,3 mmol, 4,0 eq) y yoduro de metilo (28,0 ml, 448,3 mmol, 4,0 eq) a 0 °C, y la reacción se agitó a TA durante 16 h. Después de completar la reacción (monitorizada mediante TLC, sistema de TLC con 30 % de EtOAc en hexano, Rf-0,3), el solvente se eliminó bajo presión reducida y el residuo se repartió entre EtOAc y agua. La capa acuosa se extrajo dos veces con EtOAc. Las fases orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4, se filtraron y se concentraron. El producto crudo se purificó mediante cromatografía en columna (gel de sílice 100-200, 40 % de EtOAc en hexano) para obtener 1-(2,4-dimetoxibencil)-2-(2-metoxipiridin-4-il)-5-oxo-3-(p-toliltio)pirrolidin-3-carboxilato de metilo como un sólido blanquecino (35,0 g, 60 %).
Etapa 3: A una solución agitada de 1-(2,4-dimetoxibencil)-2-(2-metoxipiridin-4-il)-5-oxo-3-(p-toliltio)pirrolidin-3-carboxilato de metilo (60,0 g, 114,8 mmol, 1,0 eq) en acetonitrilo (300 ml), se le añadió Ca n (188,8 g, 344,4 mmol, 3,0 eq) en agua (300 ml) gota a gota a través de un embudo de adición y, a continuación, la mezcla de reacción se agitó a TA durante 16 h. Después de completar la reacción (monitorizada mediante TLC, sistema de TLC con un 70 % de EtOAc en hexano, Rf-0,3), la mezcla de reacción se diluyó con agua y se extrajo dos veces con EtOAc. Las fases orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4, se filtraron y se concentraron. El producto crudo se purificó mediante cromatografía en columna (gel de sílice 230 400, 40-50 % de EtOAc:hexano) para dar 2-(2-metoxipiridin-4-il)-5-oxo-3-(p-toliltio)pirrolidin-3-carboxilato de metilo como un sólido blanquecino (12,0 g, 28 %).
Etapa 4: A una solución agitada de 2-(2-metoxipiridin-4-il)-5-oxo-3-(p-toliltio)pirrolidin-3-carboxilato de metilo (11,4 g, 30,6 mmol, 1,0 eq) en EtOH:THF (50:100 ml, 1:2), se le añadió níquel Raney (18 g) y la reacción se agitó bajo una atmósfera de hidrógeno durante 16 h a TA. Después de completar la reacción (monitorizada mediante TLC, sistema de TLC con un 70 % de EtOAc en hexano, Rf-0,4), la mezcla de reacción se filtró a través de un lecho de celite, el cual se lavó de 4 a 5 veces con THF. El filtrado se concentró para obtener 2-(2-metoxipiridin-4-il)-5-oxopirrolidin-3-carboxilato de metilo como un sólido blanco (7,1 g, 93 %, mezcla de sin:anti).
Etapa 5: A una solución agitada de 2-(2-metoxipiridin-4-il)-5-oxopirrolidin-3-carboxilato de metilo (0,7 g, 2,8 mmol, 1 eq) en MeOH (10 ml), se le añadió una solución de NaOH al 2 N (6 ml) y la mezcla de reacción se agitó a 80 °C durante 16 h. Después de completar la reacción (monitorizada mediante TLC, sistema de TLC con MeOH al 5 % en DCM, Rf-0,1), la mezcla de reacción se concentró y se acidificó con una solución de HCl al 2 N para obtener un sólido que se filtró y se lavó con éter dietílico. Después de secar al vacío se obtuvo
ácido trans-2-(2-metoxipiridin-4-il)-5-oxopirrolidin-3-carboxílico (0,4 g, 61 %).
Etapa 6: A una solución agitada de ácido trans-2-(2-metoxipiridin-4-il)-5-oxopirrolidin-3-carboxílico (0,37 g, 1,58 mmol, 1,00 eq) en tolueno (20 ml), se le añadió t Ea (0,30 ml, 1,66 mmol, 1,05 eq) y DPPA (0,40 ml, 1,89 mmol, 1,20 eq) y la mezcla de reacción se agitó a 90 °C durante 30 min. El alcohol bencílico (0,40 ml, 3,16 mmol, 2.00 eq) se añadió a la mezcla de reacción y se calentó a reflujo durante 16 h. Después de completar la reacción (monitorizada mediante TLC, sistema de TLC con MeOH al 5 % en DCM, Rf-0,3), la mezcla de reacción se concentró bajo presión reducida. A continuación, el residuo se diluyó con EtOAc (100 ml), se lavó con agua (2 x 100 ml), se secó sobre Na2SO4 y se concentró bajo presión reducida para obtener el producto crudo que se purificó mediante cromatografía en columna (gel de sílice de malla 230-400; 0-2 % en DCM) para obtener (2-(2-metoxipiridin-4-il)-5-oxopirrolidin-3-il)carbamato de trans-bencilo (0,20 g, 37 %).
Etapa 7: A una solución agitada de (2-(2-metoxipiridin-4-il)-5-oxopirrolidin-3-il)carbamato trans-bencilo (0,2 g, 24.0 mmol, 1,0 eq) en MeOH:THF (20 ml, 2:1), se le añadió Pd/C (0,2 g, 10 % húmedo) y la reacción se agitó con un globo de hidrógeno durante 2 h a TA. Después de completar la reacción (monitorizada mediante TLC, sistema de TLC con un 5 % de MeOH en DCM, Rf-0,2), la mezcla de reacción se filtró a través de un lecho de celite, el cual se lavó de 2 a 3 veces con THF. El filtrado se concentró para obtener trans-4-amino-5-(2-metoxipiridin-4-il)pirrolidin-2-ona como una goma marrón (0,1 g, 82 %).
Síntesis de trans-4-amino-5-(o-tolil)pirrolidin-2-ona (producto intermedio A10)

Etapa 1: Se recogió anhídrido maleico (20,3 g, 208,2 mmol, 1,0 eq), p-tiocresol (25,8 g, 208,2 mmol, 1,0 eq), 2,4-dimetoxi bencilamina (34,7 g, 208,2 mmol, 1,0 eq) y 2-fluorobenzaldehído (25,0 g, 208,2 mmol, 1,0 eq) en 300 ml de tolueno. La mezcla de reacción se calentó a reflujo durante 16 h con agitación vigorosa. Después de completar la reacción (monitorizada por TLC, el sistema de TLC MeOH al 5 % en DCM, Rf-0,1), la mezcla de reacción se enfrió a la TA y el solvente se evaporó bajo presión reducida para dar el ácido 1-(2,4-dimetoxibencil)-5-oxo-2-(o-toliltio) -3-(p-toliltio)pirrolidin-3-carboxílico crudo (101,0 g) como un líquido gomoso, el cual se usó en la etapa siguiente sin purificación adicional.
Etapa 2: A una solución agitada de ácido 1-(2,4-dimetoxibencil)-5-oxo-2-(o-tolil)-3-(p-toliltio)pirrolidin-3-carboxílico crudo (101,0 g, 208,2 mmol, 1,0 eq) en acetona (1 l), se le añadió carbonato de potasio (115,0 g, 832,8 mmol, 4,0 eq) y yoduro de metilo (52,0 ml, 832,8 mmol, 4,0 eq) a 0 °C y la reacción se agitó a TA durante 16 h. Después de completar la reacción (monitorizada mediante TLC, sistema de TLC con 30 % de EtOAc en hexano, Rf-0,3), el solvente se eliminó bajo presión reducida y el residuo se repartió entre EtOAc y agua. La capa acuosa se extrajo dos veces con EtOAc. Las fases orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4, se filtraron y se concentraron. El producto crudo se purificó mediante cromatografía en columna (gel de sílice 100-200, 40 % de EtOAc en hexano) para obtener 1-(2,4-dimetoxibencil)-5-oxo-2-(otolil)-3-(p-toliltio)pirrolidin-3-carboxilato de metilo como sólido blanquecino (80,0 g, 76 %).
Etapa 3: A una solución agitada de 1-(2,4-dimetoxibencil)-5-oxo-2-(o-tolil)-3-(p-toliltio)pirrolidin-3-carboxilato de metilo (80,0 g, 158,0 mmol, 1,0 eq) en acetonitrilo (300 ml), se le añadió Ca N (260,0 g, 475,0 mmol, 3,0 eq) en agua (300 ml) gota a gota a la mezcla de reacción, a 0 °C, a través de un embudo de adición, y la mezcla de
reacción se agitó a TA durante 16 h. Después de completar la reacción (monitorizada mediante TLC, sistema de TLC con un 50 % de EtOAc en hexano, Rf-0,3), la mezcla de reacción se diluyó con agua y se extrajo dos veces con EtOAc. Las fases orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4, se filtraron y se concentraron. El producto crudo se purificó mediante cromatografía en columna (gel de sílice 230 400, con un 40-50 % de EtOAc:hexano), lo que dio 5-oxo-2-(o-tolil)-3-)p-toliltio)pirrolidin-3-carboxilato de metilo como un sólido blanquecino (21,5 g, 38 %).
Etapa 4: A una solución agitada de 5-oxo-2-(o-tolil)-3-(p-toliltio)pirrolidin-3-carboxilato de metilo (21,5 g, 60,5 mmol, 1,0 eq) en EtOH:THF (300:300 ml, 1:1), se le añadió níquel Raney (~18 g) y la reacción se agitó bajo una atmósfera de hidrógeno durante 16 h a TA. Después de completar la reacción (monitorizada mediante TLC, sistema de TLC con un 70 % de EtOAc en hexano, Rf-0,4), la mezcla de reacción se filtró a través de un lecho de celite, el cual se lavó de 4 a 5 veces con THF. El filtrado se concentró para obtener 5-oxo-2-(otolil)pirrolidin-3-carboxilato de metilo como un sólido blanco (11,5 g, 82 %, mezcla de sin:anti).
Etapa 5: A una solución agitada de 5-oxo-2-(o-tolil)pirrolidin-3-carboxilato de metilo (11,5 g, 49,3 mmol, 1,0 eq) en MeOH (400 ml), se le añadió una solución de NaOH al 2 N (80 ml) y la mezcla de reacción se agitó a 80 °C durante 16 h. Después de completar la reacción (monitorizada mediante TLC, sistema de TLC con MeOH al 5 % en DCM, Rf-0,1), la mezcla de reacción se concentró y se acidificó con una solución de HCl al 2 N para obtener un sólido que se filtró y se lavó con éter dietílico. Después de secar al vacío, se obtuvo ácido trans-5-oxo-2-(o-tolil)pirrolidin-3-carboxílico (8,5 g, 79 %).
Etapa 6: A una solución agitada de ácido trans-5-oxo-2-(o-tolil)pirrolidin-3-carboxílico (8,5 g, 38,0 mmol, 1,00 eq) en tolueno (110 ml), se le añadió TEA (5,5 ml, 39,9 mmol, 1,05 eq) y DPPA (10,5 g, 45,0 mmol, 1,20 eq) y la mezcla de reacción se agitó a 90 °C durante 30 min. Después de 30 minutos, se añadió alcohol bencílico (8,4 g, 77,0 mmol, 2,00 eq) y la mezcla de reacción se calentó a reflujo durante 16 h. Después de completar la reacción (monitorizada mediante TLC, sistema de TLC con MeOH al 5 % en DCM, Rf-0,3), la mezcla de reacción se concentró bajo presión reducida. A continuación, el residuo se diluyó con EtOAc (100 ml), se lavó con agua (2 x 100 ml), se secó sobre Na2SO4 anhidro y, a continuación, se concentró bajo presión reducida para obtener el producto crudo que se purificó mediante cromatografía en columna (gel de sílice de malla 230 400; MeOH al 0-2 % en DCM) para obtener (5-oxo-2-(o-tolil)pirrolidin-3-il)carbamato de trans-bencilo (8,0 g, 65 %).
Etapa 7: A una solución agitada de (5-oxo-2-(o-tolil)pirrolidin-3-il)carbamato de trans-bencilo (8,0 g, 24,0 mmol, 1,0 eq) en MeOH:THF (20 ml, 2:1), se le añadió Pd/C (2,0 g, 10 %, húmedo) y la mezcla de reacción se agitó con un globo de hidrógeno durante 2 h a TA. Después de completar la reacción (monitorizada mediante TLC, sistema de TLC con MeOH al 5 % en DCM, Rf-0,2), la mezcla de reacción se filtró a través de un lecho de celite, el cual se lavó de 2 a 3 veces con THF. El filtrado se concentró para obtener la trans-4-amino-5-(ptolil)pirrolidin-2-ona deseada como una goma marrón (4,5 g, 99 %).
Síntesis de trans-4-amino-5-(2-fluoro-5-metoxifenil)pirrolidin-2-ona (producto intermedio A11)

Step 1: Se recogió anhídrido maleico (14,6 g, 149.7 mmol, 1,0 eq), p-tiocresol (18,5 g, 149,7 mmol, 1,0 eq), 2,4-dimetoxi bencilamina (25,0 g, 149,7 mmol, 1,0 eq) y 2-fluoro-5-metoxibenzaldehído (23,0 g, 149,7 mmol, 1,0 eq) en 300 ml de tolueno. La mezcla de reacción se calentó a reflujo durante 16 h con agitación vigorosa. Después de completar la reacción (monitorizada mediante TLC, sistema de TLC con MeOH al 5 % en DCM, Rf-0,1), la mezcla de reacción se enfrió a TA y, a continuación, se evaporó el solvente bajo presión reducida para obtener el producto crudo como un líquido gomoso (75,0 g, 96 %), que se usó en la etapa siguiente sin purificación adicional.
Etapa 2: A una solución agitada de ácido 1-(2,4-dimetoxibencil)-2-(2-fluoro-5-metoxifenil)-5-oxo-3-(ptoliltio)pirrolidin-3-carboxílico crudo (75,0 g, 142,9 mmol, 1,0 eq) en acetona (1 l), se le añadió carbonato de potasio (78,9 g, 571,4 mmol, 4,0 eq) y yoduro de metilo (35,0 ml, 571,4 mmol, 4,0 eq) a 0 °C y la mezcla de reacción se agitó a TA durante 16 h. Después de completar la reacción (monitorizada mediante TLC, sistema de TLC con 30 % de EtOAc en hexano, Rf-0,3), el solvente se eliminó bajo presión reducida y el residuo se repartió entre EtOAc y agua. La capa acuosa se extrajo dos veces con EtOAc. Las fases orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4, se filtraron y se concentraron. El producto crudo se purificó mediante cromatografía en columna (gel de sílice 100-200, 40 % de EtOAc en hexano) para obtener el 1-(2,4-dimetoxibencil)-2-(2-fluoro-5-metoxifenil)-5-oxo-3-(p-toliltio)pirrolidin-3-carboxilato de metilo deseado (45,0 g, 58 %) como un sólido blanquecino.
Etapa 3: A una solución agitada de 1-(2,4-dimetoxibencil)-2-(2-fluoro-5-metoxifenil)-5-oxo-3-(p-toliltio)pirrolidin-3-carboxilato de metilo (45,0 g, 83,5 mmol, 1,0 eq) en acetonitrilo, se le añadió CAN (137,3 g, 250,4 mmol, 3,0 eq) en agua gota a gota, usando un embudo de adición, a la mezcla de reacción, a 0 °C, y la mezcla de reacción se agitó a TA durante 16 h. Después de completar la reacción (monitorizada mediante TLC, sistema de TLC con un 50 % de EtOAc en hexano, Rf-0,3), la mezcla de reacción se diluyó con agua y se extrajo dos veces con EtOAc. Las fases orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4, se filtraron y se concentraron. El producto curdo se purificó mediante cromatografía en columna (gel de sílice 230-400, 40-50 % de EtOAc: hexano) para dar 2-(2-fluoro-5-metoxifenil)-5-oxo-3-(p-toliltio)pirrolidin-3-carboxilato de metilo (17,0 g, 52 %) como un sólido blanquecino.
Etapa 4: A una solución agitada de 2-(2-fluoro-5-metoxifenil)-5-oxo-3-(p-toliltio)pirrolidin-3-carboxilato de metilo (17,0 g, 43,7 mmol, 1,0 eq) en EtOH:THF (300:300 ml, 1:1), se le añadió níquel Raney (17 g) y la mezcla de reacción se agitó bajo una atmósfera de hidrógeno durante 16 h a TA. Después de completar la reacción (monitorizada mediante TLC, sistema de TLC con un 70 % de EtOAc en hexano, Rf-0,4), la mezcla de reacción se filtró a través de un lecho de celite, el cual se lavó de 4 a 5 veces con THF. El filtrado se concentró para obtener el 2-(2-fluoro-5-metoxifenil)-5-oxopirrolidin-3-carboxilato de metilo deseado (9,0 g, 77 %, mezcla de sin:anti) como un sólido blanco.
Etapa 5: A una solución agitada de 2-(2-fluoro-5-metoxifenil)-5-oxopirrolidin-3-carboxilato de metilo (9,0 g, 33,7 mmol, 1 eq) en (180 ml), se le añadió una solución de NaOH al 2 N (36 ml) y la mezcla de reacción se agitó a
80 °C durante 16 h. Después de completar la reacción (monitorizada mediante TLC, sistema de TLC con MeOH al 5 % en DCM, Rf-0,1), la mezcla de reacción se concentró y se acidificó con una solución de HCl al 2 N para obtener un sólido que se filtró y, a continuación, se lavó con éter dietílico. El secado al vacío proporcionó ácido trans-2-(2-fluoro-5-metoxifenil)-5-oxopirrolidin-3-carboxílico (7,9 g, 93 %).
Etapa 6: A una solución agitada de ácido trans-2-(2-fluoro-5-metoxifenil)-5-oxopirrolidin-3-carboxílico (7,9 g, 31,2 mmol, 1,00 eq) en tolueno (80 ml), se le añadió TEA (4,6 ml, 32,8 mmol, 1,05 eq) y DPPA (10,3 g, 37,46 mmol, 1,20 eq) y la mezcla de reacción se agitó a 90 °C durante 30 min. Después de 30 minutos, se añadió alcohol bencílico (6,7 g, 62,4 mmol, 2,00 eq) y la mezcla de reacción se calentó a reflujo durante 16 h. Después de completar la reacción (monitorizada mediante TLC, sistema de TLC con MeOH al 5 % en DCM, Rf-0,3), la mezcla de reacción se concentró bajo presión reducida. A continuación, el residuo se diluyó con EtOAc (100 ml), se lavó con agua (2 x 100 ml), se secó sobre Na2SO4 y se concentró bajo presión reducida para obtener el producto crudo que se purificó mediante cromatografía en columna (gel de sílice de malla 230-400; 0-2 % en DCM) para obtener bencil (trans-2-(2-fluoro-5-metoxifenil)-5-oxopirrolidin-3-il)carbamato (1,5 g, 13 %).
Etapa 7: A una solución agitada de (trans-2-(2-fluoro-5-metoxifenil)-5-oxopirrolidin-3-il)carbamato de bencilo (1,5 g, 4,2 mmol, 1,0 eq) en MeOH:THF (20 ml, en 2:1), se le añadió Pd/C (0,3 g, 0,548 mmol, 0,1 eq) y la mezcla de reacción se agitó con un globo de hidrógeno durante 2 h a TA. Después de completar la reacción (monitorizada mediante TLC, sistema de TLC con MeOH al 5 % en DCM, Rf-0,2), la mezcla de reacción se filtró a través de un lecho de celite, el cual se lavó de 2 a 3 veces con THF. El filtrado se concentró para obtener la trans-4-amino-5-(2-fluoro-5-metoxifenil)pirrolidin-2-ona (0,9 g, 96 %) deseada como una goma marrón.
Síntesis de trans-N-(1-(1H-indazol-5-il)-5-oxo-2-fenilpirrolidin-3-il)-2,2-difluoropropanamida (producto intermedio B1)

Etapa 1: Una solución agitada del producto intermedio A2 (1,2 g, 4,477 mmol, 1,0 eq), 5-yodo-1-(tetrahidro-2H-piran-2-il)-1H-indazol (1,8 g, 5,373 mmol, 1,2 eq) y K3PO4 (1,9 g, 8,955 mmol, 2,0 eq) en 1,4-dioxano (20 ml) se desgasificó con argón durante 30 min. A continuación, se añadió trans-N,N'-dimetilciclohexano-1,2-diamina (0,3 g, 1,791 mmol, 0,4 eq) y CuI (0,2 g, 0,985 mmol, 0,2 eq) y la mezcla de reacción se agitó durante 16 h a 90 °C en un tubo sellado. Después de completar la reacción (monitorizada mediante TLC, sistema de TLC con MeOH al 5 % en DCM, Rf-0,5), la mezcla de reacción se filtró a través de un lecho de celite, el cual se lavó de 2 a 3 veces con 1,4-dioxano. Las capas orgánicas combinadas se concentraron para obtener el producto crudo que se purificó mediante cromatografía en columna (gel de sílice de malla 230-400; MeOH al 0 al 2 % en DCM) para obtener la trans-2,2-difluoro-N-(5-oxo-2-fenil-1-(1-(tetrahidro-2H-piran-2-il)-1H-indazol-5-il)pirrolidin-3-il)propanamida deseada (1,5 g, 72 %).
Etapa 2: A una solución agitada de trans-2,2-difluoro-N-(5-oxo-2-fenil-1-(1-(tetrahidro-2H-piran-2-il)-1H-indazol-5-il)pirrolidin-3-il)propanamida (1,5 g, 3,20 mmol, 1,0 eq) en DCM (20 ml), se le añadió TFA (15 ml) a 0 °C y la reacción se agitó durante 16 h a TA. Después de completar la reacción (monitorizada por TLC, sistema de TLC con MeOH al 5 % en DCM, Rf-0,3), la mezcla de reacción se concentró y se basificó con una solución de NaHCO3. La fase acuosa se extrajo con DCM (3 x 100 ml). Las capas orgánicas combinadas se secaron sobre Na2SO4, se filtraron y concentraron para dar trans-N-(1-(1H-indazol-5-il)-5-oxo-2-fenilpirrolidin-3-il)-2,2-difluoropropanamida (1,1 g, 89 %) como un sólido.
Síntesis de 5-yodo-1-((2-metoxipiridin-4-il)metil)-1H-indazol (producto intermedio C1)

Etapa 1: A una solución agitada de (2-metoxipiridin-4-il)metanol (2,0 g, 14,372 mmol, 1,0 eq) en DCM (20 ml), se le añadió PBr3 (1,63 ml, 17,247 mmol, 1,2 eq) a 0 °C y la reacción se agitó a TA durante 2 h. Después de completar la reacción (monitorizada mediante TLC, sistema de TLC con MeOH al 5 % en DCM, Rf-0,3), la reacción se templó con una solución de NaHCO3 (150 ml), y el producto se extrajo con DCM (3 x 150 ml), se secó sobre Na2SO4 y se concentró para obtener 4-(bromometil)-2-metoxipiridina (2,8 g, 96 %).
Etapa 2: A una solución agitada de 5-yodo-1H-indazol (2,2 g, 9,018 mmol, 1,0 eq) en DMF (20 ml), se le añadió NaH (50 %) (0.432 g, 9,018 mmol, 1,0 eq) a 0 °C, seguido de la adición de 4-(bromometil)-2-metoxipiridina (2,7 g, 13,527 mmol, 1,5 eq) y la mezcla de reacción se dejó calentar a TA durante 16 h. Después de completar la reacción (monitorizada mediante TLC, sistema de TLC con MeOH al 5 %/DCM, Rf-0,4), la mezcla de reacción se templó con agua helada (100 ml), se extrajo con EtOAc (3 x 100 ml), se lavó con salmuera (50 ml), se secó sobre Na2SO4 y se concentró para obtener el producto crudo que se purificó mediante cromatografía en columna (gel de sílice de malla 230-400; MeOH del 0 al 4 %-DCM) para producir 5-yodo-1-((2-metoxipiridin-4-il)metilo)-1H-indazol (0,7 g, 21 %) como regioisómero puro.
RMN H1 (DMSO-d6) 8: 8,21 (s, 1H), 8,11 (s, 1H), 8,05 (d, 1H), 7,62-7,67 (m, 1H), 7,55-7,57 (m, 1H), 6,67 ( d, 1H), 6,45 (s, 1H), 5,67 (s, 2H), 3,78 (s, 3H).
Síntesis de 5-yodo-1-((1-metil-1H-pirazol-3-il)metil)-1H-indazol (producto intermedio C2)

Etapa 1: A una solución agitada de (1-metil-1H-pirazol-3-il)metanol (1,2 g, 10,708 mmol, 1,0 eq) en DCM (15 ml), se le añadió CBr4 (7,2 g, 21,417 mmol, 2,0 eq) y trifenilfosfina (5,7 g, 21,417 mmol, 2,0 eq) a TA y, a continuación, la reacción se agitó durante 16 h. Una vez completada la reacción (monitorizada mediante TLC, sistema de TLC con MeOH al 5 % en DCM, Rf:0,3), la mezcla de reacción se diluyó con agua (150 ml), se extrajo con DCM (3 x 150 ml), se lavó con salmuera (50 ml), se secó sobre Na2SO4 y se concentró para obtener el producto crudo que se purificó mediante cromatografía en columna (gel de sílice de malla 230-400; MeOH del 0 al 2 %-DCM) para obtener 3-(bromometil)-1-metil-1H-pirazol (1,25 g, 67 %).
Etapa 2: A una solución agitada de 5-yodo-1H-indazol (1,34 g, 5,517 mmol, 0,8 eq) en DMF (20 ml) se le añadió NaH (50 %) (0,40 g, 8,276 mmol, 1,2 eq) a 0 °C, seguido de la adición de 3-(bromometil)-1-metil-1H-pirazol (1,20 g, 6,897 mmol, 1,0 eq). La mezcla de reacción se agitó a TA durante 16 h. Una vez completada la reacción (monitorizada mediante TLC, sistema de TLC con MeOH al 5 %/DCM, Rf:0,4), la mezcla de reacción se templó con agua helada (100 ml), se extrajo con EtOAc (3 x 100 ml), se lavó con salmuera (50 ml), se secó sobre Na2SO4 y se concentró para obtener el producto bruto que se purificó mediante cromatografía en columna (gel de sílice de malla 230-400; MeOH del 0 al 4 %-DCM) para obtener 5-yodo-1-((1-metil-1H-pirazol-3-il)metilo)-1 H-indazol (0,7 g, 30 %).
Síntesis de 5-yodo-1-((6-metoxipiridin-3-il)metil)-1H-indazol (producto intermedio C3)

Etapa 1: A una solución agitada de (6-metoxipiridin-3-il)metanol (0,60 g, 4,316 mmol, 1,0 eq) en DCM (20 ml), se le añadió PBr3 (0,50 ml, 5,179 mmol, 1,2 eq) a 0 °C y, a continuación, la reacción se agitó a TA durante 2 h. Una vez completada la reacción (monitorizada mediante TLC, sistema de TLC con MeOH al 5 % en DCM, Rf:0,3), la reacción se templó con una solución de NaHCO3 saturada (50 ml), se extrajo con DCM (3x50 ml), se secó sobre Na2SO4 y se concentró para obtener 5-(bromometil)-2-metoxipiridina (0,55 g, 63 %).
Etapa 2: A una solución agitada de 5-yodo-1H-indazol (0,49 g, 1,990 mmol, 0,8 eq) en DMF (20 ml), se le añadió NaH (50 %, 0,14 g, 2,985 mmol, 1,2 eq) a 0 °C, seguido de la adición de 5-(bromometil)-2-metoxipiridina (0,50 g, 2,488 mmol, 1,0 eq). La mezcla de reacción se agitó a TA durante 16 h. Una vez completada la reacción (monitorizada mediante TLC, sistema de TLC con MeOH al 5 %/DCM, Rf:0,4), la mezcla de reacción se templó con agua helada (100 ml), se extrajo con EtOAc (3 x 100 ml), se lavó con salmuera (50 ml), se secó sobre Na2SO4 y se concentró para obtener el producto bruto que se purificó mediante cromatografía en columna (gel de sílice de malla 230-400; MeOH del 0 al 4 %-DCM) para obtener 5-yodo-1-((6-metoxipiridin-3-il)metil)-1 H-indazol (0,40 g, 44 %).
RMN H1 (DMSO-d6) 8: 8,14 (s, 1H), 8,08 (m, 1H), 7,99 (s, 1H), 7,62 (mc, 1H), 7,52 (mc, 1H), 7,43 (mc, 1H), 6,70 (d, 1H), 5,54 (s, 2H), 3,85 (s, 3H).
Síntesis de 5-((5-yodo-1H-indazol-1-il)metil)-1-metilpiridin-2(1H)-ona (producto intermedio C4)

Etapa 1: Se agitó 5-(hidroximetil)-1,2-dihidropiridin-2-ona (1,0 g, 7,991 mmol, 1,0 eq) en HBr acuoso (48 %) a 110 °C durante 3 h. Una vez completada la reacción (monitorizada mediante TLC, sistema de TLC con MeOH al 5 % en DCM, Rf:0,1), se eliminó el solvente bajo presión reducida para obtener el producto bruto. El producto bruto se destiló azeotrópicamente con tolueno para obtener bromhidrato de 5-(bromometil)piridin-2(1H)-ona (2,0 g, 93 %).
Etapa 2: A una solución agitada de 5-yodo-1H-indazol (1,46 g, 5,99 mmol, 0,75 eq) en DMF (20 ml), se le añadió NaH (50 %, 1,15 g, 23,97 mmol, 3,0 eq) a 0 °C, seguido de la adición de bromhidrato de 5-(bromometil)piridin-2(1H)-ona (2,15 g, 7,99 mmol, 1,0 eq). A continuación, la mezcla de reacción se agitó a TA durante 16 h. Una vez completada la reacción (monitorizada mediante TLC, sistema de TLC con metanol al 5 %/DCM, Rf:0,3), la mezcla de reacción se templó con agua helada (150 ml), se extrajo con EtOAc (3 x 150 ml), se lavó con salmuera (100 ml), se secó sobre Na2SO4 y se concentró para obtener el producto crudo que se purificó mediante cromatografía en columna (gel de sílice de malla 230-400; MeOH del 0 al 4 %-DCM) para obtener 5-((5-yodo-1H-indazol-1-il)metil)piridin-2(1H)-ona (0,19 g, 7 %).
Etapa 3: A una solución agitada de 5-((5-yodo-1H-indazol-1-il)metil)piridin-2(1H)-ona (0,09 g, 0,256 mmol, 1,0 eq) en DMF (10 ml) se le añadió NaH (50 %, 24,6 mg, 0,512 mmol, 2,0 eq) a 0 °C seguido de la adición de yoduro de metilo (0,04 ml, 0,512 mmol, 2,0 eq). La reacción se agitó a TA durante 16 h. Una vez completada la reacción (monitorizada mediante TLC, sistema de TLC con MeOH al 5 %/DCM, Rf:0,4), la mezcla de reacción se templó con agua helada (50 ml), se extrajo con EtOAc (3 x 50 ml), se lavó con salmuera (20 ml), se secó sobre Na2SO4 y se concentró para obtener el producto crudo que se purificó mediante cromatografía en columna (gel de sílice de malla 230-400; MeOH del 0 a 3 %-d Cm ) para obtener 5-((5-yodo-1H-indazol-1-il)metil)-1 -metilpiridin-2(1H)-ona (0,09 g, 96 %).
RMN H1 (DMSO-d6) 8: 8,18 (s, 1H), 8,04 (s, 1H), 7,85 (s, 1H), 7,65 (mc, 2H), 7,29 (dd, 1H), 6,30 (d, 1H), 5,35 (s, 2H), 3,34 (s, 3H).
Síntesis de ciclopropil(4-(5-yodo-1H-indazol-1-il)piperidin-1-il)metanona (producto intermedio C5)

Etapa 1: A una solución agitada de 4-hidroxipipendin-1-carboxilato de terc-butilo (1,19 g, 4,88 mmol, 1,0 eq) en DCM (20 ml) a 0 °C se le añadió TEA (1,04 ml, 7,45 mmol, 1,5 eq) y la reacción se agitó durante 5 minutos. Se añadió gota a gota cloruro de metanosulfonilo (0,46 ml, 5,96 mmol, 1,2 eq) a 0 °C. A continuación, la mezcla de reacción se agitó a 0 °C durante 1 h. Una vez completada, la mezcla de reacción se diluyó con DCM y se lavó con agua y una solución de NH4Cl saturada. La capa orgánica combinada se concentró para obtener el producto bruto que se purificó mediante cromatografía en columna (gel de sílice de malla 100-200; EtOAc al 50 %/hexano; valor Rr: 0,5) para obtener terc-butil 4-((metilsulfonil)oxi)piperidin-1-carboxilato (1,35 g, 98 %).
Etapa 2: A una solución agitada de 5-yodo-1H-indazol (1,19 g, 4,88 mmol, 1,0 eq) en DMF (20 ml) a 0 °C se le añadió NaH (0,26 g, 5,37 mmol, 1,1 eq) y la mezcla de reacción se agitó durante 15 min. A continuación, se añadió 4-((metilsulfonil)oxi)piperidin-1-carboxilato de terc-butilo (1,5 g, 5,37 mmol, 1,1 eq) gota a gota a 0 °C disuelto en DMF (10 ml). A continuación, la mezcla de reacción se calentó a 100 °C durante 16 h. Una vez completada, la mezcla de reacción se diluyó con EtOAc y se lavó con agua helada. Las capas orgánicas combinadas se concentraron para obtener el producto crudo que se purificó mediante cromatografía en columna (gel de sílice de malla 100-200; EtOAc al 50 %/hexano; valor de Rf:0,5, separación de isómeros) para obtener 4-(5-yodo-1H-indazol-1-il)piperidin-1-carboxilato de terc-butilo (0,82 g, 41 %) como regioisómero puro.
Etapa 3: A una solución de 4-(5-yodo-1H-indazol-1-il)piperidin-1-carboxilato de terc-butilo (0,82 g, 1,92 mmol, 1,0 eq) en DCM (20 ml) a 0 °C se le añadió TFA (5 ml) gota a gota, y la mezcla de reacción se agitó durante 1 hora a TA. Una vez completada, la mezcla de reacción se concetró para obtener el 5-yodo-1-(piperidin-4-il)-1H-indazol crudo como la sal de TFA (0,1 g, crudo).
Etapa 4: A una solución agitada de 5-yodo-1 -(piperidin-4-il)-1 H-indazol (sal de TFA, 0,6 g, 1,83 mmol, 1,0 eq) en DMF (20 ml), se le añadió HATU (1,0 g, 2,75 mmol, 1,5 eq), DIPeA (1,6 ml, 9,17 mmol, 5,0 eq) y ácido ciclopropanocarboxílico (0,23 g, 2,75 mmol, 1,5 eq) y la mezcla de reacción se agitó durante 16 h a TA. Una vez completada, la mezcla de reacción se diluyó con EtOAc y se lavó con agua helada, NaHCO3 saturado y una solución de NH4Cl saturada. Las capas orgánicas combinadas se concentraron para obtener el producto bruto que se purificó mediante cromatografía en columna (gel de sílice de malla 100-200; MeOH al 2 %-DCM; valor de Rf:0,5) para obtener ciclopropil(4-(5-yodo-1H-indazol-1-il)piperidin-1-il)metanona (0,3 g).
Síntesis de 4-((5-yodo-1H-indazol-1-il)metil)-1-metilpiridin-2(1H)-ona (producto intermedio C6)

Etapa 1: A una solución agitada de ácido 1-metil-2-oxo-1,2-dihidropiridin-4-carboxílico (3,0 g, 19,60 mmol, 1,0 eq) en DCM (30 ml), se le añadió TEA (4,1 ml, 29,40 mmol, 1,5 eq)) y cloroformiato de etilo (2,24 ml, 23,52 mmol, 1,2 eq) y la mezcla de reacción se agitó a TA durante 2 h. Una vez completada de la reacción
(monitorizada mediante TLC, sistema de TLC con MeOH al 5 % en DCM, Rf-0,7), la mezcla de reacción se concentró para obtener el anhídrido 1-metil-2-oxo-1,2-dihidropiridin-4-carboxílico (etil carbónico) bruto que se usó en la siguiente etapa sin más purificación (3,0 g, 68 %).
Etapa 2: A una solución agitada de anhídrido 1-metil-2-oxo-1,2-dihidropiridin-4-carboxílico crudo (etil carbónico) (3.0 g, 13.329 mmol, 1,0 eq) en THF:EtOH (80 ml, 3:1), se le añadió NaBH4 (2,5 g, 66,648 mmol, 5,0 eq) y la mezcla de reacción se agitó a TA durante 2 h. Una vez completada la reacción (monitorizada mediante TLC, sistema de TLC con EtOAc al 50 % en hexano, Rf:0,1), la mezcla de reacción se templó con agua helada (75 ml), se extrajo con MeOH al 5 % en DCM (3 x 150 ml), se secó sobre Na2SO4 y se concentró bajo presión reducida para obtener la 4-(hidroximetil)-1-metilpiridin-2(1H)-ona cruda que se usó en la siguiente etapa sin más purificación (1,7 g, 92%).
Etapa 3: A una solución agitada de 4-(hidroximetil)-1-metilpiridin-2(1H)-ona cruda (1,2 g, 8,63 mmol, 1,0 eq) en DCM (15 ml), se le añadió PBr3 (1,0 ml, 10,36 mmol, 1,2 eq) a 0 °C y, a continuación, la mezcla de reacción se agitó a TA durante 2 h. Una vez completada la reacción (monitorizada mediante TLC, sistema de TLC con EtOAc al 50 % en hexano, Rf:0,4), la mezcla de reacción se templó con una solución de NaHCO3 (50 ml), se extrajo con DCM (3x 50 ml), se secó sobre Na2SO4 y se concentró bajo presión reducida para obtener 4-(bromometil)-1-metilpiridin-2(1H)-ona (1,0 g, 57 %).
Etapa 4: A una solución agitada de 5-yodo-1H-indazol (0,970 g, 3,980 mmol, 0,8 eq) en DMF (20 ml) se le añadió NaH (50 %, 0,238 g, 4,975 mmol, 1,0 eq) a 0 °C, seguido de la adición de 4-(bromometil)-1-metilpiridin-2(1H)-ona (1,0 g, 4,975 mmol, 1,0 eq). A continuación, la mezcla de reacción se agitó a TA durante 16 h. Una vez completada la reacción (monitorizada mediante TLC, sistema de TLC con MeOH al 5 %/DCM, Rf:0,4), la mezcla de reacción se templó con agua helada (50 ml), se extrajo con EtOAc (3 x 50 ml), se lavó con salmuera (50 ml), se secó sobre Na2SO4 y se concentró para obtener el producto crudo que se purificó mediante cromatografía en columna (gel de sílice de malla 230-400; MeOH del 0 a 3 %-DCM) para obtener 4-((5-yodo-1H-indazol-1-il)metil)-1-metilpiridin-2(1H)-ona (0,360 g, 20 %) como regioisómero puro.
RMN H1 (DMSO-d6) 8: 8,22 (s, 1H), 8,10 (s, 1H), 7,54-7,66 (m, 3H), 5,94 (s, 1H), 5,90 (d, 1H), 5,51 (s, 2H), 3,33 (s, 3H).
Síntesis de 5-(5-((2R,3S)-3-amino-5-oxo-2-fenilpirrolidin-1-il)-1H-indazol-1-il)-1-metilpiridin-2(1H)-ona (producto intermedio D1-ent2)

Etapa 1: A una solución agitada de N-[(trans)-2-fenil-5-oxo-pirrolidin-3-il]carbamato de bencilo (producto intermedio A2-Cbz,1,0 g, 3,22 mmol, 1,0 eq) y 5-(5-yodo-1H-indazol-1-il)-1-metilpiridin-2(1H)-ona (1,1 g, 3,22 mmol, 1,0 eq) en 1,4-dioxano (80 ml) se le añadió fosfato de potasio (1,4 g, 6,44 mmol, 2,0 eq), seguido de trans-N,N'-dimetilciclohexano-1,2-diamina (1,02 ml, 0,65 mmol, 0,2 eq) y la mezcla de reacción se desgasificó bajo una atmósfera de argón durante 30 minutos,, a continuación se añadio Cul (61,3 mg, 0,32 mmol, 0,1 eq) y la reacción se calentó en un tubo sellado a 90 °C durante 16 h (monitorizado mediante LCMS). La mezcla de reacción se filtró a través de un lecho de celite y el lecho de celite se lavó con EtOAc (500 ml), y las capas orgánicas combinadas se concentraron bajo presión reducida. El residuo crudo se purificó mediante cromatografía en columna (gel de sílice 100-200, MeOH del 3 al 5 %-DCM como eluyente) para obtener bencilo N-[(trans)-1-(1-(1 -metil-6-oxo-1,6-dihidropiridin-3-il)-1 H-indazol-5-il)-2-fenilo-5-oxo-pirrolidin-3-ilo]-carbamato (750 mg, 44 %).
Etapa 2: Una suspensión agitada de bencilo N-[(trans)-1-(1-(1-metil-6-oxo-1,6-dihidropiridin-3-il)-1H-indazol-5-il)-2-fenilo-5-oxo-pirrolidin-3-ilo]carbamato (22,0 g, 41,2 mmol) en TFA (80 ml) se calentó a 80 °C durante 2 h. Una vez completada la reacción (monitorizada mediante LCMS), la mezcla de reacción se enfrió a TA y se eliminó el TFA bajo presión reducida como un azeótropo con tolueno. El residuo se alcalinizó (pH~8) con una
solución saturada de NaHCO3 y se extrajo con MeOH al 10 %/DCM (5 x 150 ml). Las capas orgánicas combinadas se secaron sobre Na2SO4, se filtraron y se concentraron bajo presión reducida. El residuo bruto se purificó mediante cromatografía en columna (gel de sílice 100-200, MeOH al 5-10 %/DCM como eluyente) para obtener (trans)-4-amino-5-(fenil)-1 -(1-(1 -metil-6-oxo-1,6-dihidropiridin-3-M)-1 H-indazol-5-il)pirrolidin-2-ona (11,0 g, 67 %) como un sólido gris.
Etapa 3: Separación quiral
El compuesto racémico se separó mediante una HPLC quiral preparativa (ID de columna: CHIRALPAK IB (4,6 x 250 nm), 5 pm; fase móvil: MeOH/DEA (100/0,1); caudal: 1 ml/min; temperatura: 25 °C) para obtener el producto intermedio D1-ent1 (pico 1; 4,915 g; 100 % ee) y el producto intermedio D1-ent2 (pico 2; 2,763 g; 99,60 % ee).
Ejemplo______1______N-((2R,3S)-2-(3-clorofenil)-1-(1-(1-metil-6-oxo-1,6-dihidropiridin-3-il)-1H-indazol-5-il)-5-oxop¡rrolid¡n-3-¡l)-2.2-d¡fluoropropanam¡da

Etapa 1: A una solución agitada del producto intermedio A1 (0,25 g, 1,19 mmol, 1,0 eq) en DMF (10 ml) se le añadió HATU (0,68 g, 1,78 mmol, 1,5 eq), DIPEA (1,0 ml, 5,95 mmol, 5,0 eq) y ácido 2,2-difluoropropanoico (0,17 g, 1,54 mmol, 1,3 eq) y, a continuación, la mezcla de reacción se agitó durante 16 h a t A. Una vez completa, la mezcla de reacción se diluyó con EtOAc y se lavó con agua helada y una solución de NaHCO3 y NH4Cl saturada. Las capas orgánicas combinadas se concentraron para obtener el producto crudo, que se purificó mediante cromatografía en columna (gel de sílice de malla 100-200; MeOH al 2 %-DCM; Rf-valor-0,5) para proporcionar trans-N-(2-(3-clorofenil)-5-oxopirrolidin-3-il)-2,2-difluoropropanamida (0,19 g, 53 %).
Etapa 2: Una solución agitada de trans-N-(2-(3-clorofenil)-5-oxopirrolidin-3-il)-2,2-difluoropropanamida (0,30 g, 0,99 mmol, 1,0 eq), 5-5-yodo-1H-indazol-1-il)-1-metilpiridin-2(1H)-ona (0,41 g, 1,19 mmol, 1,2 eq) y K3PO4 (0,42 g, 1,98 mmol, 2,0 eq) en 1,4-dioxano (20 ml) se desgasificó con argón durante 15 min. A continuación, se añadieron trans-N,N'-dimetilciclohexano-1,2-diamina (0,056 g, 0,40 mmol, 0,4 eq) y Cul (0,038 g, 0,20 mmol, 0,2 eq) y la mezcla de reacción se agitó durante 16 h a 90 °C. Una vez completada, la mezcla de reacción se filtró a través de un lecho de celite y el lecho de celite se lavó 2-3 veces con EtOAc. Las capas orgánicas combinadas se concentraron para obtener el producto bruto que se purificó mediante cromatografía en columna (gel de sílice de malla 100-200; MeOH al 5 %-DCM; valor de Rf:0,5) para obtener el producto racémico y la posterior separación de enantiómeros se realizó mediante una HPLc quiral preparativa para obtener N-((2R,3S)-2-(3-clorofenil)-1-(1-(1-metil-6-oxo-1,6-dihidropiridin-3-il)-1H-indazol-5-il)-5-oxo-pirrolidin-3-il)-2,2-difluoropropanamida (0,07g, TR=15,9 min, nombre de la columna: CHIRALPAK Al (250x4,6 mm) 5 pm, fase móvil: hexano/EA/EtOH/DEA : 70/15/15/0,1, caudal : 1,0 ml/min) [y N-((2S,3R)-2-(3-clorofenil)-1-(1-(1-metil-6-oxo-1,6-dihidropiridin-3-il)-1H-indazol-5-il)-5-oxopirrolidin-3-il)-2,2-difluoropropanamida (0,06 g; TR=10,6 min, nombre de la columna : CHIRALpA k AI (250x4,6 mm) 5 pm, fase móvil: hexano/EA/EtOH/DEA : 70/15/15/0.1, caudal: 1,0 ml/min)].
RMN H1 (DMSO-d6) 8: 9,46 (d, 1H), 8,26 (s, 1H), 8,19 (d, 1H), 7,86 (s, 1H), 7,71 (dd, 1H), 7,62-7,58 (m, 2H), 7,45 (s, 1H), 7,34-7,28 (m, 3H), 6,53 (d, 1H), 5,34 (d, 1H), 4,30 (bs, 1H), 3,49 (s, 3H), 3,14-3,08 (m, 1H), 2,67 2,62 (m, 1H), 1,78 (t, 3H).
Ejemplo 2: trans-2.2-d¡fluoro-N-(5-oxo-2-(2.4-d¡fluorofen¡l)-1-(1-(1-met¡l-6-oxo-1.6-d¡h¡drop¡r¡d¡n-3-¡l)-1H-

Etapa 1: Una soluc¡ón del producto intermedio A3 (0.85 g. 4.09 mmol. 1.0 eq) en DMF (12 ml) se trató con ác¡do 2.2-d¡fluoropropano¡co (0.57 g. 5.21 mmol. 1.3 eq) en presenc¡a de HATU (3.04 g. 8.01 mmol. 2.0 eq) y DIPEA (3.5 ml. 20.04 mmol. 2.0 eq) y la mezcla se ag¡tó a TA durante 16 h. A cont¡nuac¡ón. la mezcla de reacc¡ón se repart¡ó entre EtOAc y agua. los extractos orgán¡cos se lavaron con salmuera. se secaron y se concentraron para obtener el producto bruto que se purificó med¡ante cromatografía en columna ultrarrápida (gel de síl¡ce de malla 230-400; 5 % de MeOH/EtOAc; valor de Rf:0.4) para obtener la trans-N-(-2-(2.4-d¡fluorofen¡l)-5-oxop¡rrol¡d¡n-3-¡l)-2.2-d¡fluoropropanam¡da (0.7 g. 58 %) como un sól¡do blanco
Etapa 2: A una soluc¡ón ag¡tada de trans-N-(-2-(2.4-d¡fluorofen¡l)-5-oxop¡rrol¡d¡n-3-¡l)-2.2-d¡fluoropropanam¡da (0.30 g. 0.98 mmol. 1.0 eq) y 5-(5-yodo-1H-¡ndazol-1-¡l)-1-met¡lp¡r¡d¡n-2(1H)-ona (0.35 g. 0.98 mmol. 1.0 eq) en 1.4-d¡oxano (5 ml) se le añad¡ó K3PO4 (0.41 g. 1.97 mmol. 2.0 eq). Cul (0.038 g. 0.19 mmol. 0.2 eq) y la trans-N.N'-d¡met¡l-c¡clohexano-1.2-d¡am¡na (0.056 g. 0.39 mmol. 0.4 eq) a TA bajo una atmósfera de n¡trógeno y la mezcla se desgas¡f¡có con una comente de n¡trógeno durante 5 m¡n. La mezcla resultante se calentó a 90 °C durante 16 h. La mezcla de reacc¡ón se dejó enfr¡ar a TA. a cont¡nuac¡ón. se f¡ltró y se concentró para obtener el producto bruto que se pur¡f¡có med¡ante cromatografía en columna ultrarráp¡da (gel de síl¡ce de malla 230 400; 5 % de MeOH/EtOAc; valor de Rf: 0.4) para obtener la trans-N-(-2-(2.4-d¡fluorofen¡l)-1-(1-(1-met¡l-6-oxo-1.6-d¡h¡drop¡r¡d¡n-3-¡l)-1H-¡ndazol-5-¡l)-5-oxop¡rrol¡d¡n-3-¡l)-2.2-d¡fluoropropanam¡da (0.16 g. 31 %) como un sól¡do blanquec¡no.
RMN H1 (DMSO-d6) 6: 9.4 (d. 1H). 8.27 (s. 1H). 8.20 (d. 1H). 7.77 (s. 1H). 7.71 (dd. 1H). 7.61 (d. 1H). 7.52 7.43 (m. 2H). 7.20 (t. 1H). 7.00 (t. 1H). 6.53 (d. 1H). 5.50 (d. 1H). 4.49 (t. 1H). 3.49 (s. 3H). 3.14-3.07 (m. 1H).
2.7 (dd. 1H). 1.75 (t. 3H).
Ejemplo 3: trans-2.2-d¡fluoro-N-(5-oxo-2-(2.3-d¡h¡drobenzo[b1[1.41d¡ox¡n-6-¡l)-1-(1-(1-met¡l-6-oxo-1.6-d¡h¡drpp¡r¡d¡n-3-¡l)-1H-¡ndazpl-5-¡l)p¡rrplid¡n-3-¡l)prppanam¡da

le añad¡ó HATU (0.49 g. 1.30 mmol. 1.5 eq). DIPEA (0.75 ml. 4.30 mmol. 5.0 eq) y 2.2-d¡fluoro-prop¡ón¡co (0.12 g. 1.12 mmol. 1.3 eq) y la reacc¡ón se ag¡tó durante 16 h a TA. Una vez completada. la mezcla de reacc¡ón se d¡luyó con EtOAc y se lavó con agua helada. NaHCO3 saturado y una soluc¡ón de NH4Cl saturada. Las capas orgán¡cas comb¡nadas se concentraron para obtener el producto bruto que se pur¡f¡có med¡ante cromatografía en columna (gel de síl¡ce de malla 100-200; MeOH al 2%-DCM; valor de Rf:0.5) para obtener la trans-N-(2-(2.3-d¡h¡drobenzo[b1[1.41d¡ox¡n-6-¡l)-5-oxop¡rrol¡d¡n-3-¡l)-2.2-d¡fluoropropanam¡da (0.20 g. 71 %).
Etapa 2: Una soluc¡ón ag¡tada de trans-N-(2-(2.3-d¡h¡drobenzo[b1[1.41d¡ox¡n-6-¡l)-5-oxop¡rrol¡d¡n-3-¡l)-2.2-d¡fluoropropanam¡da (0.20 g. 0.613 mmol. 1.0 eq). 5-(5-yodo-1H-¡ndazol-1-¡l)-1-met¡lp¡r¡d¡n-2(1H)-ona (0.26 g.
0.736 mmol. 1.2 eq) y K3PO4 (0.26 g. 1.22 mmol. 2.0 eq) en 1.4-d¡oxano (20 ml) se desgas¡f¡có con argón durante 15 m¡n. A cont¡nuac¡ón. se añad¡eron trans-N.N'-d¡met¡lc¡clohexano-1.2-d¡am¡na (0.035 g. 0.245 mmol.
0.4 eq) y Cul (0.025 g. 0.122 mmol. 0.2 eq) y la reacc¡ón se ag¡tó durante 16 h a 90 °C. Una vez completada. la mezcla de reacc¡ón se f¡ltró a través de un lecho de cel¡te y el lecho de cel¡te se lavó 2-3 veces con EtOAc. Las capas orgán¡cas comb¡nadas se concentraron para obtener el producto bruto que se pur¡f¡có med¡ante cromatografía en columna (gel de síl¡ce de malla 100-200; MeOH al 5 %-DCM; valor de Rf:0.3) para obtener la trans-N-(2-(2.3-d¡h¡drobenzo[b1[1.41d¡ox¡n-6-¡l)-1-(1-(1-met¡l-6-oxo-1.6-d¡h¡drop¡r¡d¡n-3-¡l)-1H-¡ndazol-5-¡l)-5-oxop¡rrol¡d¡n-3-¡l)-2.2-d¡fluoropropanam¡da (0.12 g. 36 %).
RMN H1 (DMSO-da) 8: 9,42 (d, 1H), 8,26 (s, 1H), 8,19 (d, 1H), 7,85 (s, 1H), 7,72 (dd, 1H), 7,62-7,56 (m, 2H), 6,82 (s,1H), 6,77 (s, 2H), 6,54 (d, 1H), 5,20 (d, 1H), 4,24-4,20 (m, 1H), 4,16 (s, 4H), 3,50 (s, 3H), 3,10-3,04 (m, 1H) 2,60-2,56 (m, 1H), 1,78 (t, 3H).
Ejemplo 4: 2,2-d¡fluoro-N-((2R,3S)-1-(1-(1-met¡l-6-oxo-1,6-d¡h¡drop¡r¡d¡n-3-¡l)-1H-¡ndazol-5-¡l)-5-oxo-2-fenilp¡rrol¡d¡n-3-¡l)propanamida

Etapa 1: A una soluc¡ón ag¡tada del producto intermedio A2 (1,0 g, 5,68 mmol, 1,0 eq) en DMF (20 ml), se le añad¡ó HATU (3,2 g, 8,52 mmol, 1,5 eq), DIPEA (4,9 ml, 28,40 mmol, 5,0 eq) y 2,2-d¡fluoro-prop¡ón¡co (0,8 g, 7,38 mmol, 1,3 eq). La mezcla de reacc¡ón se ag¡tó a TA durante 16 h. Una vez completada, la mezcla de reacc¡ón se diluyó con EtOAc y se lavó con agua helada, NaHCO3 saturado y una soluc¡ón de NH4Cl saturada. Las capas orgán¡cas comb¡nadas se concentraron para obtener el producto bruto que se pur¡f¡có med¡ante cromatografía en columna (gel de síl¡ce de malla 100-200; MeOH al 2%-DCM; valor de Rf:0,5) para obtener la trans-2,2-d¡fluoro-N-(5-oxo-2-fen¡lp¡rrol¡d¡n-3-¡l)propanam¡da (1,4 g, 93 %).
Etapa 2: Una soluc¡ón ag¡tada de trans-2,2-d¡fluoro-N-(5-oxo-2-fen¡lp¡rrol¡d¡n-3-¡l)propanam¡da (0,3 g, 1,11 mmol, 1,0 eq), 5-(5-yodo-1H-¡ndazol-1-¡l)-1-met¡lp¡r¡d¡n-2(1H)-ona (0,47 g, 1,34 mmol, 1,2 eq) y K3 PO4 (0,47 g, 2,23 mmol, 2,0 eq) en 1,4-d¡oxano (20 ml) se desgas¡f¡có con argón durante 15 m¡n. A cont¡nuac¡ón, se añad¡eron trans-N,N'-d¡met¡lc¡clohexano-1,2-d¡am¡na (0,06 g, 0,45 mmol, 0,4 eq) y Cul (0,04 g, 0,22 mmol, 0,2 eq) y la reacc¡ón se ag¡tó durante 16 h a 90 °C. Una vez completada, la mezcla de reacc¡ón se f¡ltró a través de un lecho de cel¡te y el lecho de cel¡te se lavó 2-3 veces con EtOAc.. Las capas orgán¡cas comb¡nadas se concentraron para obtener el producto bruto que se pur¡f¡có med¡ante cromatografía en columna (gel de síl¡ce de malla 100-200; MeOH-DCM al 5 %; valor de Rf:0,5) para obtener el producto racém¡co y se real¡zó una separac¡ón de enant¡ómeros ad¡c¡onal med¡ante una HPLC qu¡ral preparat¡va para obtener 2,2-difluoro-N-((2R,3S)-1-(1-(1-metN-6-oxo-1,6-dihidropiridm-3-N)-1H-mdazol-5-N)-5-oxo-2-femlpirroNdm-3-il)propanamida (0,10 g; TR=8,06 m¡n; nombre de la columna: Ch¡ralpak IA (250x4,6 mm) 5 pm, fase móv¡l: hexano/EA/EtOH/DEA: 50/25/25/ 0,1, caudal: 1,0 ml/m¡n) [y 2,2-d¡fluoro-N-((2S,3R)-1-(1-(1-met¡l-6-oxo-1,6-d¡h¡drop¡r¡d¡n-3-¡l)-1H-¡ndazol-5-¡l)-5-oxo-2-fen¡lp¡rrol¡d¡n-3-¡l)propanam¡da (0,14 g; TR=5,88 m¡n; nombre de la columna: Ch¡ralpak IA (250x4,6 mm) 5 pm, fase móv¡l: hexano/EA/EtOH/DEA: 50/25/25/0,1, caudal: 1,0 ml/m¡n)].
RMN H1 (DMSO-d6) 8: 9,47 (d, 1H), 8,25 (s, 1H), 8,18 (d, 1H), 7,85 (s, 1H), 7,70 (dd, 1H), 7,58 (s, 2H), 7,35 7,29 (m, 4H), 7,24-7,22 (m, 1H), 6,53 (d, 1H), 5,30 (d, 1H), 4,24 (bs, 1H), 3,49 (s, 3H), 3,08-3,06 (m, 1H) 2,64 2,63 (m, 1H), 1,78 (t, 3H).
Ejemplo 5: 2.2-d¡fluoro-N-((2R.3S)-2-(3-fluorofen¡l)-1-(1-(1-met¡l-6-oxo-1.6-d¡h¡drop¡r¡d¡n-3-¡l)-1H-¡ndazol-5-¡l)-5-oxop¡rrolid¡n-3-¡l)propanam¡da

Etapa 1: A una soluc¡ón ag¡tada de ác¡do 2.2-d¡fluoro-prop¡ón¡co (0.68 g. 6.185 mmol. 1.2 eq) en DMF (10 ml), HATU (3.9 g. 10.309 mmol. 2.0 eq). se le añad¡ó DlpEA (4.5 ml. 25.773 mmol. 5.0 eq) y el producto intermedio A5 (1.00 g. 5.1545 mmol. 1.0 eq) a 0 °C y. a cont¡nuac¡ón. la reacc¡ón se ag¡tó a TA durante 16 h. Una vez completada la reacc¡ón (mon¡tor¡zada med¡ante TLC). la mezcla de reacc¡ón se diluyó con EtOAc (25 ml). se lavó con agua helada (3x25 ml). se secó sobre Na2SO4 y se concentró para obtener el producto bruto. que se pur¡f¡có med¡ante cromatografía en columna (gel de síl¡ce de malla 230-400; MeOH del 0 al 2 %-DCM) para obtener la trans-2.2-d¡fluoro-N-(2-(3-fluorofen¡l)-5-oxop¡rrol¡d¡n-3-¡l)propanam¡da (0.56 g. 38 %).
Etapa 2: Una soluc¡ón ag¡tada de trans-2.2-d¡fluoro-N-(2-(3-fluorofen¡l)-5-oxop¡rrol¡d¡n-3-¡l)propanam¡da (0.55 g.
1.923 mmol. 1 eq). 5-(5-yodo-1H-¡ndazol-1-¡l)-1-met¡lp¡r¡d¡n-2(1H)-ona (0.81 g. 2.307 mmol. 1.2 eq) y K3PO4 (0.82 g. 3.846 mmol. 2.0 eq) en 1.4-d¡oxano (25 ml) se desgas¡f¡có con argón durante 30 m¡n. A cont¡nuac¡ón. se añad¡eron trans-N.N'-d¡met¡lc¡clohexano-1.2-d¡am¡na (0.11 grs. 0.769 mmol. 0.4 eq) y Cul (0.07 g.
0.384 mmol. 0.2 eq) y la reacc¡ón se agitó durante 16 h a 90 °C en un tubo sellado. Una vez completada la reacc¡ón (mon¡tor¡zada med¡ante TLC). la mezcla de reacc¡ón se f¡ltró a través de un lecho de cel¡te y el lecho de cel¡te se lavó 2-3 veces con 1.4-d¡oxano. Las capas orgán¡cas comb¡nadas se concentraron para obtener el producto bruto que se pur¡f¡có med¡ante cromatografía en columna (gel de síl¡ce de malla 230-400; MeOH del 0 al 2 % en Dc M) para obtener la trans-2.2-d¡fluoro-N-((2R.3S)-2-(3-fluorofen¡l)-1-(1-(1-met¡l-6-oxo-1.6-d¡h¡drop¡r¡d¡n-3-¡l)-1H-¡ndazol-5-¡l)-5-oxop¡rrol¡d¡n-3-¡l)propanam¡da. Se real¡zó una separac¡ón ad¡c¡onal de los enant¡ómeros med¡ante una HPLC qu¡ral preparat¡va para obtener 2.2-d¡fluoro-N-((2S.3R)-2-(3-fluorofen¡l)-1-(1-(1-met¡l-6-oxo-1.6-d¡h¡drop¡r¡d¡n-3-¡l)-1H-¡ndazol-5-¡l)-5-oxop¡rrol¡d¡n-3-¡l)propanam¡da (0.13 g; TR=5.40 m¡n; nombre de la columna: Ch¡ralpak IA (250x4.6 mm) 5 pm. fase móv¡l: hexano/EA/EtOH/DEA: 50/25/25/0.1. caudal: 1.0 ml/m¡n) y 2,2-difluoro-N-((2R,3S)-2-(3-fluorofeml)-1-(1-(1-metN-6-oxo-1,6-dihidropiridm-3-M)-1H-indazol-5-il)-5-oxopirrolidin-3-il)propanamida (0.13 g; TA=7.14 m¡n. nombre de la columna: Ch¡ralpak IA (250 x 4.6 mm) 5 pm. fase móv¡l: hexano/EA/EtOH/DEA: 50/25/25/0.1. caudal: 1.0 ml/m¡n).
RMN H1 (DMSO-d6) 8: 9.48-9.46 (m. 1H). 8.26-8.19 (m. 2H). 7.86 (s. 1H). 7.72-7.70 (m. 1H). 7.60-7.59 (m. 2H).
7.35-7.33 (m. 1H). 7.23-7.18 (m. 2H). 7.06 (s. 1H). 6.54 (d. 1H). 5.35-5.33 (m. 1H). 4.27-4.33 (s. 1H). 3.49 (s.
3H). 3.14-3.08 (m. 1H). 2.66-2.61 (m. 1H). 1.83-1.73 (m. 3H).
Ejemplo 6: 2.2-d¡fluoro-N-((2R.3S)-2-(2-fluorofen¡l)-1-(1-(1-met¡l-6-oxo-1.6-d¡h¡drop¡r¡d¡n-3-¡l)-1H-¡ndazol-5-¡l)-5-oxop¡rrol¡d¡n-3-¡l)propanam¡da

Etapa 1: A una soluc¡ón ag¡tada de ác¡do 2,2-d¡fluoropropano¡co (0.68 g. 6.18 mmol. 1.2 eq) en DMF (8 ml), HATU (4.00 g. 10.30 mmol. 2.0 eq). se le añad¡ó DIPeA (4.5 ml. 25.75 mmol. 5.0 eq) y el producto intermedio A6 (1.00 g. 5.15 mmol. 1.0 eq) a 0 °C y. a cont¡nuac¡ón. la reacc¡ón se agitó a TA durante 16 h. Una vez completada la reacc¡ón (mon¡tor¡zada med¡ante TLC. s¡stema de TLC con MeOH al 5 % en DCM. Rf:0.3). la mezcla de reacc¡ón se d¡luyó con EtOAc (25 ml) y se lavó con agua helada (3 x 25 ml). se secó sobre Na2SO4 y se concentró para obtener el producto bruto que se pur¡f¡có med¡ante cromatografía en columna (gel de síl¡ce de malla 230-400; MeOH del 0 al 2 %-d Cm ;) para obtener la trans-2.2-d¡fluoro-N-(2-(2-fluorofen¡l)-5-oxop¡rrol¡d¡n-3-¡l)propanam¡da (0.51 g. 35 %).
Etapa 2: Una soluc¡ón ag¡tada de trans-2.2-d¡fluoro-N-(2-(2-fluorofen¡l)-5-oxop¡rrol¡d¡n-3-¡l)propanam¡da (0.25 g.
0.873 mmol. 1.0 eq). 5-(5-yodo-1H-¡ndazol-1-¡l)-1-met¡lp¡r¡d¡n-2(1H)-ona (0.37 g. 1.047 mmol. 1.2 eq) y K3PO4 (0.37 g. 1.746 mmol. 2.0 eq) en 1.4-d¡oxano (10 ml) se desgas¡f¡có con argón durante 30 m¡n. A cont¡nuac¡ón. se añad¡eron trans-N.N'-d¡met¡lc¡clohexano-1.2-d¡am¡na (0.05 g. 0.349 mmol. 0.4 eq) y Cul (0.03 g. 0.175 mmol.
0.2 eq) y la mezcla de reacc¡ón se ag¡tó durante 16 h a 90 °C en un tubo sellado. Una vez completada la reacc¡ón (mon¡tor¡zada med¡ante TLC. s¡stema de TLC con MeOH al 5 % en DCM. Rf:0.4). la mezcla de reacc¡ón se f¡ltró a través de un lecho de cel¡te y el lecho de cel¡te se lavó 2-3 veces con 1.4-d¡oxano. Las capas orgán¡cas comb¡nadas se concentraron para obtener el producto bruto que se pur¡f¡có med¡ante cromatografía en columna (gel de síl¡ce de malla 230-400; MeOH del 0 al 2 % en Dc M) para obtener la trans-2.2-d¡fluoro-N-(2-(2-fluorofen¡l)-1-(1-(1-met¡l-6-oxo-1.6-d¡h¡drop¡r¡d¡n-3-¡l)-1H-¡ndazol-5-¡l)-5-oxop¡rrol¡d¡n-3-¡l)propanam¡da y se real¡zó una separac¡ón de enant¡ómeros ad¡c¡onal med¡ante una HPLC qu¡ral preparat¡va para obtener 2.2-d¡fluoro-N-((2S.3R)-2-(2-fluorofen¡l)-1-(1-(1-met¡l-6-oxo-1.6-d¡h¡drop¡r¡d¡n-3-¡l)-1H-¡ndazol-5-¡l)-5-oxop¡rrol¡d¡n-3-¡l)propanam¡da (0.06 g. 13 %; TR=5.90 m¡n; nombre de la columna: Ch¡ralpak IA (250x4.6 mm) 5 pm. fase móv¡l: hexano/EA/EtOH/DEA: 50/25/25/0.1. caudal: 1.0 ml/m¡n) y 2,2-difluoro-N-((2R,3S)-2-(2-fluorofeml)-1-(1-(1-metil-6-oxo-1,6-dihidropiridin-3-il)-1H-indazol-5-il)-5-oxopirrolidin-3-il)propanamida (0.07 g. 16 %; TR=9,56 min; nombre de la columna: Ch¡ralpak IA (250 x 4.6 mm) 5 pm. fase móv¡l: hexano/EA/EtOH/DEA: 50/25/25/0.1. caudal: 1.0 ml/m¡n).
RMN H1 (DMSO-d6) 8: 9.45-9.43 (m. 1H). 8.27 (s. 1H). 8.19-8.18 (m. 1H). 7.78 (s. 1H). 7.72-7.68 (m. 1H). 7.62 7.59 (m. 1H). 7.48-7.45 (m. 1H). 7.42-7.38 (m. 1H). 7.27-7.25 (m. 1H). 7.16-7.08 (m. 2H). 6.54-6.51 (m. 1H).
5.53-5.52 (m. 1H). 4.48-4.46 (m. 1H). 3.49 (s. 3H). 3.16-3.09 (m. 1H). 2.70-2.64 (m. 1H). 1.80-1.71 (m. 3H).

A part¡r del producto intermedio B1. el ejemplo 7 se s¡ntet¡zó de manera análoga al proced¡m¡ento de síntes¡s descr¡to para el ejemplo 9. Rend¡m¡ento: 34 %
RMN H1 (DMSO-da) 8: 9,51-9,50 (m, 1H), 8,99 (s, 1H), 8,58-8,57 (m, 1H), 8,39 (s, 1H), 8,18-8,16 (m, 1H), 7,91 (s, 1H), 7,85-7,82 (m, 1H), 7,69-7,67 (m, 1H), 7,61-7,58 (m, 1H), 7,37-7,23 (m, 5H), 5,34-5,32 (m, 1H), 4,31 4,23 (m, 1H), 3,14-3,08 (m, 1H), 2,65-2,60 (m, 1H), 1,83-1,74 (m, 3H).
Ejemplo______9:______trans-2.2-d¡fluoro-N-(1-(1-(5-fluoropv¡¡d¡n-2-¡l)-1H-¡ndazol-5-il)-5-oxo-2-fen¡lp¡rrol¡d¡n-3-il)propanamida

Una soluc¡ón ag¡tada del producto intermedio B1 (0,200 g, 0,5208 mmol, 1,0 eq), 2-bromo-5-fluorop¡r¡d¡na (0,109 g, 0,624 mmol, 1,2 eq) y K3PO4 (0,220 g, 1,0416 mmol, 2,0 eq) en 1,4-d¡oxano (10 ml) se desgas¡f¡có con argón durante 30 m¡n. A cont¡nuac¡ón, se añad¡eron trans-N,N'-d¡met¡lc¡dohexano-1,2-d¡am¡na (0,030 g, 0,2083 mmol, 0,4 eq) y Cul (0,020 g, 0,1041 mmol, 0,2 eq) y la reacc¡ón se ag¡tó durante 16 h a 90 °C en un tubo sellado. Una vez completada la reacc¡ón (mon¡tor¡zada med¡ante TLC, s¡stema de TLC con MeOH al 5 % en DCM, Rf:0,4), la mezcla de reacc¡ón se f¡ltró a través de un lecho de cel¡te y el lecho de cel¡te se lavó 2-3 veces con 1,4-d¡oxano. Las capas orgán¡cas comb¡nadas se concentraron para obtener el producto bruto que se purificó med¡ante cromatografía en columna (gel de síl¡ce de malla 230-400; MeOH del 0 al 2 % en DCM) para obtener el compuesto y se real¡zó una pur¡f¡cac¡ón ad¡c¡onal med¡ante una HPLC preparat¡va para obtener la trans-2,2-d¡fluoro-N-(1-(1-(5-fluorop¡r¡d¡n-2-¡l)-1H-¡ndazol-5-¡l)-5-oxo-2-fen¡lp¡rrol¡d¡n-3-¡l)propanam¡da (0,057 g, 23 %).
RMN H1 (DMSO-d6) 8: 9,51-9,49 (m, 1H), 8,53-8,50 (m, 2H), 8,39 (s, 1H), 7,99-7,92 (m, 3H), 7,72-7,69 (m, 1H), 7,36-7,29 (m, 4H), 7,23-7,20 (m, 1H), 5,35-5,33 (m, 1H), 4,34-4,26 (m, 1H), 3,14-3,08 (m, 1H), 2,66-2,61 (m, 1H), 1,83-1,73 (m, 3H).
Ejemplo 13: 5-met¡l-N-(trans-1-(1-(1-met¡l-6-oxo-1.6-d¡h¡drop¡r¡d¡n-3-¡l)-1H-¡ndazol-5-¡l)-5-oxo-2-fen¡lp¡rrol¡d¡n-3-¡l)t¡azol-2-carboxam¡da

A una soluc¡ón de 5-met¡lt¡azol-2-ác¡do carboxíl¡co (20 mg, 0,138 mmol, 1,1 eq) en DCM (1,3 ml) se le añad¡ó tr¡et¡lam¡na (0,09 ml, 0,626 mmol, 5,0 eq) y soluc¡ón de anhídr¡do prop¡lfosfón¡co (>50 % en peso en EtOAc, T3P, 0,15 ml, 0,250 mmol, 2,0 eq) y la mezcla de reacc¡ón se ag¡tó a TA durante 30 m¡n. A esta mezcla ag¡tada, se le añad¡ó una soluc¡ón de 5-[5-(3-am¡no-5-oxo-2-fen¡l-p¡rrol¡d¡n-1-¡l)¡ndazol-1-¡l]-1-met¡l-p¡r¡d¡n-2-ona (50 mg, 0,125 mmol, 1,0 eq) en DCM (1,3 ml) lentamente gota a gota y la mezcla resultante se ag¡tó a TA durante la noche.. A cont¡nuac¡ón, la mezcla de reacc¡ón se d¡luyó con DCM, se añad¡ó una soluc¡ón de NaHCO3 saturada, y las fases se separaron med¡ante una fr¡ta h¡drofób¡ca.Después de la el¡m¡nac¡ón del solvente bajo pres¡ón reduc¡da, el res¡duo crudo se pur¡f¡có med¡ante HPLC para obtener 5-(5-((2R,3S)-3-am¡no-5-oxo-2-fen¡lp¡rrol¡d¡n-1-¡l)-1H-¡ndazol-1-¡l)-1-met¡lp¡r¡d¡n-2(1H)-ona (39 mg, 59 %).
RMN H1 (DMSO-d6) 8: 9,53 (d, 1H), 8,24 (s, 1H), 8,17 (d, 1H), 7,85 - 7,81 (m, 1H), 7,75 (d, 1H), 7,71 (dd, 1H), 7,63 - 7,53 (m, 2H), 7,40 - 7,33 (m, 2H), 7,30 (t, 2H), 7,25 - 7,18 (m, 1H), 6,54 (d, 1H), 5,45 (d, 1H), 4,51-4,43 (m, 1H), 3,50 (s, 3H), 3,09 (dd, 1H), 2,81 - 2,72 (m, 1H), 2,52 (d, 3H).
Ejemplo 15: 2.2-d¡fluoro-N-((2R.3S)-2-(4-fluoro-3-)-1-(1-(1-met¡l-6-oxo-1.6-d¡h¡dro-p¡r¡d¡n-3-¡l)-1H-¡ndazol-5-¡l)-5-oxop¡rrol¡d¡n-3-¡l)propanam¡da


ejemplo 15
Etapa 1: A una soluc¡ón ag¡tada del producto intermedio A7 (3.12 g. 13.92 mmol. 1.0 eq) en DMF (30 ml), se le añad¡ó HATU (7.90 g. 20.89 mmol. 1.5 eq). DIPEA (12.0 ml. 69.64 mmol. 5.0 eq) y 2.2-d¡fluoropropano¡co ác¡do (2.00 g. 18.10 mmol. 1.3 eq) y la mezcla de reacc¡ón se ag¡tó durante 16 h a TA. Una vez completada. la mezcla de reacc¡ón se d¡luyó con EtOAc y se lavó con agua helada. NaHCO3 saturado y una soluc¡ón de NH4Cl saturada. Las capas orgán¡cas comb¡nadas se concentraron para obtener el producto bruto que se pur¡f¡có med¡ante cromatografía en columna (gel de síl¡ce de malla 100-200; MeOH-DCM al 2 %; valor de Rf:0.5) para obtener la trans-2.2-d¡fluoro-N-(2-(4-fluoro-3-metox¡fen¡l)-5-oxop¡rrol¡d¡n-3-¡l)propanam¡da (3.50 g. 80 %).
Etapa 2: Una soluc¡ón ag¡tada de trans-2.2-d¡fluoro-N-(2-(4-fluoro-3-metox¡fen¡l)-5-oxop¡rrol¡d¡n-3-¡l)propanam¡da (0.30 g. 0.95 mmol. 1:0 eq). 5-(5-yodo-1H-¡ndazol-1-¡l)-1-met¡lp¡r¡d¡n-2(1H)-ona (0.40 g.
1.13 mmol. 1.2 eq) y K3PO4 (0.40 g. 1.89 mmol. 2.0 eq) en 1.4-d¡oxano (20 ml) se desgas¡f¡có con argón durante 15 m¡n. A cont¡nuac¡ón. se añad¡eron trans-N.N'-d¡met¡lc¡clohexano-1.2-d¡am¡na (0.06 g. 0.38 mmol.
0.4 eq) y Cul (0.04 g. 0.19 mmol. 0.2 eq) y la reacc¡ón se ag¡tó durante 16 h a 90 °C. Una vez completada. la mezcla de reacc¡ón se f¡ltró a través de un lecho de cel¡te y el lecho de cel¡te se lavó 2-3 veces con EtOAc. Las capas orgán¡cas comb¡nadas se concentraron para obtener el producto bruto que se pur¡f¡có med¡ante cromatografía en columna (gel de síl¡ce de malla 100-200; MeOH al 5 %-DCM; valor de Rf:0.5) para obtener la trans-2.2-d¡fluoro-N-(2-(4-fluoro-3-metox¡fen¡l)-1-(1-(1-met¡l-6-oxo-1.6-d¡h¡drop¡r¡d¡n-3-¡l)-1H-¡ndazol-5-¡l)-5-oxop¡rrol¡d¡n-3-¡l)propanam¡da racém¡ca. Se real¡zó una separac¡ón de enant¡ómeros ad¡c¡onal med¡ante una Hp Lc qu¡ral preparat¡va para obtener 2.2-d¡fluoro-N-((2S.3R)-2-(4-fluoro-3-metox¡fen¡l)-1-(1-(1-met¡l-6-oxo-1.6-d¡h¡drop¡r¡d¡n-3-¡l)-1H-¡ndazol-5-¡l)-5-oxop¡rrol¡d¡n-3-¡l)propanam¡da (0.10 g; TR=10.31 m¡n. Ch¡ralpak ID (250x4.6mm) 5 pm. fase móv¡l: EtOH. caudal: 0.5 ml/m¡n) y 2,2-difluoro-N-((2R,3S)-2-(4-fluoro-3-metoxifenilo)-1-(1-(1-metil-6-oxo-1,6-dihidropiridin-3-il)-1H-indazol-5-il)-5-oxopirrolidin-3-il)propanamida (0.10 g; TR=13.00 m¡n. Ch¡ralpak ID (250x4.6 mm) 5 pm; fase móv¡l: EtOH. caudal: 0.5 ml/m¡n) como un sól¡do blanco.
RMN H1 (DMSO-d6) 8: 9.43-9.41 (m. 1H). 8.26 (s. 1H). 8.20 (d. 1H). 7.84 (s. 1H). 7.73-7.69 (m. 1H). 7.62-7.54 (m. 2H). 7.19-7.17 (m. 1H). 7.12-7.07 (m. 1H). 6.87-6.84 (m. 1H). 6.54-6.52 (m. 1H). 5.29-5.27 (m. 1H). 4.32 4.30 (m. 1H). 3.78 (s. 3H). 3.49 (s. 3H). 3.12-3.06 (m. 1H). 2.66-2.61 (m. 1H). 1.83-1.73 (m. 3H).
empo : . - ¡ uoro- - . - - - uoro en¡ - - - -me¡- -oxo- . - ¡ ¡ rop¡r¡ ¡n- -¡ - -¡n azo- -¡ --oxop¡rrol¡d¡n-3-¡l)propanam¡da

Etapa-2

ejemplo 17
Etapa 1: A una soluc¡ón ag¡tada de ác¡do 2.2-d¡fluoro-prop¡ón¡co (0.44 g. 4.020 imimol, 1.2 eq) en DMF (6 ml), se le añad¡ó HATU (2.55 g. 6.701 mmol. 2.0 eq). DIPEA (2.95 ml. 6.701 mmol. 5.0 eq) y el producto intermedio 8 (0.65 g. 3.350 mmol. 1.0 eq) a 0 °C y la reacc¡ón se ag¡tó a TA durante 16 h. Una vez completada la reacc¡ón (mon¡tor¡zada med¡ante TLC). la mezcla de reacc¡ón se d¡luyó con EtOAc (25 ml). se lavó con agua helada (3x25 ml). se secó sobre Na2SO4 y se concentró bajo pres¡ón reduc¡da para obtener el producto bruto. que se pur¡f¡có med¡ante cromatografía en columna (gel de síl¡ce de malla 230-400; MeOH al 2 %-DCM) para obtener la trans-2.2-d¡fluoro-N-(2-(4-fluorofen¡l)-5-oxop¡rrol¡d¡n-3-¡l)propanam¡da (0.60 g. 63 %).
Etapa 2: Una soluc¡ón ag¡tada de trans-2.2-d¡fluoro-N-(2-(4-fluorofen¡l)-5-oxop¡rrol¡d¡n-3-¡l)propanam¡da (0.30 g.
1.048 mmol. 1.0 eq). 5-(5-yodo-1H-¡ndazol-1-¡l)-1-met¡lp¡r¡d¡n-2(1H)-ona (0.44 g. 1.258 mmol. 1.2 eq) y K3PO4 (0.44 g. 2.097 mmol. 2.0 eq) en 1.4-d¡oxano (10 ml) se desgas¡f¡có con argón durante 30 m¡n. A cont¡nuac¡ón. se añad¡eron trans-N.N'-d¡met¡lc¡clohexano-1.2-d¡am¡na (0.06 g. 0.419 mmol. 0.4 eq) y Cul (0.04 g. 0.209 mmol.
0.2 eq) y la reacc¡ón se ag¡tó durante 16 h a 90 °C en un tubo sellado. Una vez completada la reacc¡ón (mon¡tor¡zada med¡ante TLC). la mezcla de reacc¡ón se f¡ltró a través de un lecho de cel¡te y el lecho de cel¡te se lavó 2-3 veces con 1.4-d¡oxano. Las capas orgán¡cas comb¡nadas se concentraron para obtener el producto bruto que se pur¡f¡có med¡ante cromatografía en columna (gel de síl¡ce de malla 230-400; MeOH al 2 % en DCM) para obtener la trans-2.2-d¡fluoro-N-(2-(4-fluorofen¡l)-1-(1-(1-met¡l-6-oxo-1.6-d¡h¡drop¡r¡d¡n-3-¡l)-1H-¡ndazol-5-¡l)-5-oxop¡rrol¡d¡n-3-¡l)propanam¡da. Se real¡zó una separac¡ón de enant¡ómeros ad¡c¡onal med¡ante una HPLC qu¡ral preparat¡va para obtener 2.2-d¡fluoro-N-((2S.3R)-2-(4-fluorofen¡l)-1-(1-(1-met¡l-6-oxo-1.6-d¡h¡drop¡r¡d¡n-3-¡l)-1H-¡ndazol-5-¡l)-5-oxop¡rrol¡d¡n-3-¡l)propanam¡da (0.12 g. 23 %; TR=6.17 m¡n; nombre de la columna: Ch¡ralpak IA (250 * 4.6 mm) 5 pm. fase móv¡l: hexano/EA/EtOH/DEA: 50/25/25/0.1. caudal: 1.0 ml/m¡n) y 2,2-difluoro-N-((2R,3S)-2-(4-fluorofenM)-1-(1-(1-metN-6-oxo-1,6-dihidropiridm-3-M)-1H-indazol-5-il)-5-oxopirrolidin-3-il)propanamida (0.12 g.22 %; TR=8,46 min; nombre de la columna: Ch¡ralpak IA (250 * 4.6 mm) 5 pm. fase móv¡l: hexano/EA/EtOH/DEA: 50/25/25/0.1. caudal: 1.0 ml/m¡n).
RMN H1 (DMSO-d6) 8: 9.46-9.44 (m. 1H). 8.25 (s. 1H). 8.19-8.18 (m. 1H). 7.83 (s. 1H). 7.72-7.69 (m. 1H). 7.61 7.53 (m. 2H). 7.42-7.38 (m. 2H). 7.15-7.11 (m. 2H). 6.54 (d. 1H). 5.33-5.31 (m. 1H). 4.32-4.26 (m. 1H). 3.49 (s.
3H). 3.12-3.05 (m. 1H). 2.66-2.61 (m. 1H). 2.49 (s. 1H). 1.82-1.72 (m. 3H).
Ejemplo 18; 1-fluoro-N-((2R.3S)-1-(1-(1-met¡l-6-oxo-1.6-d¡h¡drop¡nd¡n-3-¡l)-1H-¡ndazol-5-¡l)-5-oxo-2-

Etapa 1: A una soluc¡ón ag¡tada de ác¡do 1-fluoroc¡clopropano-1-carboxíl¡co (0.71 g. 6.818 mmol. 1.2 eq) en DMF (10 ml). se le añad¡ó HATU (4.32 g. 11.364 mmol. 2.0 eq). DIPEA (5.0 ml. 28.409 mmol. 5.0 eq) y el producto intermedio A2 (1.00 g. 5.682 mmol. 1.0 eq) a 0 °C y. a cont¡nuac¡ón. la reacc¡ón se ag¡tó a TA durante 16 h. Una vez completada la reacc¡ón (mon¡tor¡zada med¡ante TLC. s¡stema de TLC con MeOH al 5 % en DCM. Rf:0.3). la mezcla de reacc¡ón se diluyó con EtOAc (35 ml). se lavó con agua helada (3x25 ml). se secó sobre Na2SO4 y se concentró para obtener el producto crudo que se pur¡f¡có med¡ante cromatografía en columna (gel de síl¡ce de malla 230-400; MeOH del 0 a 3 %-DCM;) para obtener la trans-1-fluoro-N-(5-oxo-2-fen¡lp¡rrol¡d¡n-3-¡l) c¡clopropanocarboxam¡da (0.64 g. 43 %).
Etapa 2: Una soluc¡ón ag¡tada de trans-1-fluoro-N-(5-oxo-2-fen¡lp¡rrol¡d¡n-3-¡l)c¡clopropanocarboxam¡da (0.32 g.
1.220 mmol. 1.0 eq). 5-(5-yodo-1H-¡ndazol-1-¡l)-1-met¡lp¡r¡d¡n-2(1H)-ona (0.51 g. 1.465 mmol. 1.2 eq) y K3PO4 (0.52 g. 2.441 mmol. 2.0 eq) en 1.4 d¡oxano (20 ml) se desgas¡f¡có con argón durante 30 m¡n. A cont¡nuac¡ón. se añad¡eron trans-N.N'-d¡met¡lc¡clohexano-1.2-d¡am¡na (0.07 g. 0.480 mmol. 0.4 eq) y Cul (0.05 g. 0.244 mmol.
0.2 eq) y la reacc¡ón se ag¡tó durante 16 h a 90 °C en un tubo sellado. Una vez completada la reacc¡ón (mon¡tor¡zada med¡ante TLC. s¡stema de TLC con MeOH al 5 % en DCM. Rf:0.4). la mezcla de reacc¡ón se f¡ltró a través de un lecho de cel¡te y el lecho de cel¡te se lavó 2-3 veces con 1.4-d¡oxano. Las capas orgán¡cas comb¡nadas se concentraron para obtener el producto bruto que se pur¡f¡có med¡ante cromatografía en columna (gel de síl¡ce de malla 230-400; MeOH del 0 al 2 % en DCM) para obtener la trans-1-fluoro-N-(1-(1-(1-met¡l-6-oxo-1.6-d¡h¡drop¡r¡d¡n-3-¡l)-1 H-¡ndazol-5-¡l)-5-oxo-2-fen¡lp¡rrol¡-d¡n-3-¡l)c¡clopropanocarboxam¡da. Se real¡zó una separac¡ón de enant¡ómeros ad¡c¡onal med¡ante una HPLC qu¡ral preparat¡va para obtener 1-fluoro-N-((2S.3R)-1-(1-(1-met¡l-6-oxo-1.6-d¡h¡drop¡r¡d¡n-3-¡l)-1H-¡ndazol-5-¡l)-5-oxo-2-fen¡lp¡rrol¡d¡n-3-¡l)c¡clopropanocarboxam¡da (0.13 g. 22 %; TR=6.68 m¡n. nombre de la columna: Ch¡ralpak IA (250x4.6 mm) 5 pm. fase móv¡l: hexano/EA/EtOH/DEA: 50/25/25/0.1. caudal: 1.0 ml/m¡n) y 1-fluoro-N-((2R,3S)-1-(1-(1-metil-6-oxo-1,6-dihidropiridm-3-M)-1H-mdazol-5-M)-5-oxo-2-femlpirroMdm-3-M)cidopropanocarboxamida (0.11 g. 18 %; TR=8,97 min. nombre de la columna: Ch¡ralpak lA (250 x 4 .6 mm) 5 pm. fase móv¡l: hexano/EA/EtOH/DEA: 50/25/25/0.1. caudal: 1.0 ml/m¡n).
RMN H1 (DMSO-d6) 8: 9.18-9.17 (m. 1H). 8.24 (s. 1H). 8.18 (s. 1H). 7.84 (s. 1H). 7.72-7.69 (m. 1H). 7.59 (s.
2H). 7.36-7.34 (m. 2H). 7.31-7.27 (m. 2H). 7.23-7.21 (m. 1H). 6.54-6.51 (m. 1H). 5.35-5.34 (m. 1H). 4.36-4.28 (m. 1H). 3.49 (s. 3H). 3.09-3.03 (m. 1H). 2.67-2.62 (m. 1H). 1.35-1.29 (m. 2H). 1.22-1.19 (m. 2H).
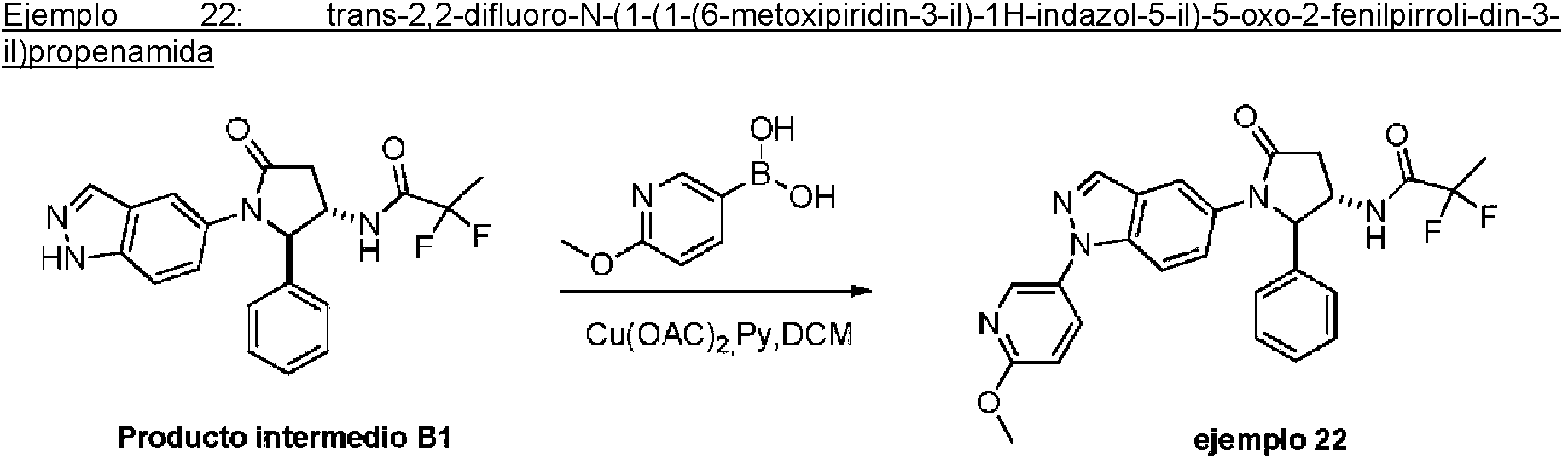
A una solución agitada del producto intermedio B1 (0,250 g, 0,651 mmol, 1,0 eq), ácido (6-metoxipiridin-3-il)borónico (0,200 g, 1,302 mmol, 2,0 eq) y piridina (0,1 ml, 1.302 mmol, 2,0 eq) en DCM (20 ml) se le añadió Cu(OAc)2 (0,177 g, 0,976 mmol, 1,5 eq) y la reacción se agitó durante 16 h a TA. Una vez completada la reacción (monitorizada mediante TLC, sistema de TLC con MeOH al 5 % en DCM, Rf:0,4), el solvente se eliminó bajo presión reducida y el residuo se repartió entre DCM y agua. La capa acuosa se extrajo dos veces con DCM (2x50 ml). Las capas orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4, se filtraron y se concentraron. bajo presión reducida. El producto bruto se purificó mediante cromatografía en columna de HPLC preparativa para obtener la trans-2,2-difluoro-N-(1-(1-(6-metoxipiridin-3-il)-1 H-indazol-5-il)-5-oxo-2-fenilpirrolidin-3-il)propanamida (0,105 g, 33 %).
RMN H1 (DMSO-d6) 8: 9,50-9,49 (m, 1H), 8,51-8,50 (m, 1H), 8,31 (s, 1H),8,05-8,02 (m, 1H), 7,86 (s, 1H), 7,68 7,62 (m, 2H), 7,36-7,29 (m, 4H), 7,24-7,21 (m, 1H), 7,02 (d, 1H), 5,32-5,31 (m, 1H), 4,28-4,26 (m, 1H), 3,92 (s, 3H), 3,13-3,07 (m, 1H), 2,66-2,59 (m, 1H), 1,83-1,73 (m, 3H).
Ejemplo 23: trans-2.2-d¡fluoro-N-(5-oxo-2-fen¡l-1-(1-(p¡r¡d¡n-4-¡l)-1H-¡ndazol-5-¡l)p¡rrolid¡n-3-¡l)propanam¡da

Una solución agitada del producto intermedio B1 (0,200 g, 0,521 mmol, 1,0 eq), 4-bromo-piridina (0,120 g, 0,624 mmol, 1,2 eq) y K3PO4 (0,276 g, 1,302 mmol, 2,5 eq) en 1,4-dioxano (10 ml) se desgasificó con argón durante 30 min. A continuación, se añadieron trans-N,N'-dimetilciclohexano-1,2-diamina (0,030 g, 0,208 mmol, 0,4 eq) y Cul (0,020 g, 0,104 mmol, 0,2 eq) y la reacción se agitó durante 16 h a 90 °C en un tubo sellado. Una vez completada la reacción (monitorizada mediante TLC, sistema de TLC con metanol al 5 % en DCM, Rf:0,4), la mezcla de reacción se filtró a través de un lecho de celite y el lecho de celite se lavó 2-3 veces con 1,4-dioxano. Las capas orgánicas combinadas se concentraron para obtener el producto bruto que primero se purificó mediante cromatografía en columna (gel de sílice de malla 230-400; MeOH del 0 al 2 % en DCM) para obtener el producto deseado que se purificó adicionalmente mediante una HPLC preparativa para obtener la trans-2,2-difluoro-N-(5-oxo-2-fenil-1-(1-(piridin-4-il)-1H-indazol-5-il)pirrolidin-3-il)propanamida (0,052 g, 22 %).
RMN H1 (DMSO-d6) 8: 9,52-9,50 (m, 1H), 8,68-8,66 (m, 2H), 8,45 (s, 1H), 8,06-8,03 (m, 1H), 7,95 (s, 1H), 7,85 7,84 (m, 2H), 7,75-7,72 (m, 1H), 7,37-7,30 (m, 4H), 7,25-7,23 (m, 1H), 5,36-5,34 (m, 1H), 4,32-4,26 (m, 1H), 3,15-3,08 (m, 1H), 2,66-2,60 (m, 1H), 1,83-1,74 (m, 3H).
Ejemplo______24______trans-2,2-difluoro-N-(1-(1-(6-metilpiridin-3-il)-1H-indazol-5-il)-5-oxo-2-fenilpirrolidin-3-il)propanamida

Una solución agitada del producto intermedio B1 (0,200 g, 0,521 mmol, 1,0 eq), 5-bromo-2-metil-piridina (0,106 g, 0,624 mmol, 1,2 eq) y K3PO4 (0,220 g, 1,042 mmol, 2,0 eq)) en 1,4-dioxano (10 ml) se desgasificó con argón durante 30 min. A continuación, se añadieron trans-N,N'-dimetilciclohexano-1,2-diamina (0,030 g, 0,208 mmol, 0,4 eq) y Cul (0,020 g, 0,104 mmol, 0,2 eq) y la reacción se agitó durante 16 h a 90 °C en un tubo sellado. Una vez finalizada la reacción (monitorizada mediante TLC, sistema de TLC con MeOH al 5 % in DCM, Rf-0.4), la mezcla de reacción se filtró a través de un lecho de celite y el lecho de celite se lavó 2-3 veces con
1,4-dioxano. La capa orgánica combinada se concentró para obtener el producto bruto que se purificó mediante cromatografía en columna (gel de sílice de malla 230-400; MeOH del 0 al 2 % en DCM) para obtener el compuesto bruto y se realizó una purificación adicional mediante una HPLC preparativa para obtener la trans-2,2-difluoro-N-(1 -(1-(6-metMpiridin-3-il)-1 H-indazol-5-il)-5-oxo-2-fenilpirrolidin-3-il)propanamida (0,041 g, 17 %).
RMN H1 (DMSO-d6) 8: 9,51-9,50 (m, 1H), 8,81 (s, 1H), 8,35 (s, 1H), 8,04-8,02 (m, 1H), 7,89 (s, 1H), 7,78-7,75 (m, 1H), 7,67-7,65 (m, 1H), 7,45-7,43 (m, 1H), 7,36-7,29 (m, 3H), 7,24-7,23 (m, 1H), 5,33-5,31 (m, 1H), 4,30 4,24 (m, 1H), 3,14-3,09 (m, 1H), 2,64-2,59 (m, 1H), 2,54 (s, 3H), 1,83-1,73 (m, 3H).
Ejemplo______25______trans-2,2-difluoro-N-(1-(1-(2-metilpiridin-4-il)-1H-indazol-5-il)-5-oxo-2-fenilpirrolidin-3-il)propanamida

A partir del producto intermedio B1 y 4-bromo-2-metilpiridina, el ejemplo 25 se sintetizó de manera análoga al procedimiento sintético descrito para el ejemplo 24.
RMN H1 (DMSO-d6) 8: 9,52-9,50 (m, 1H), 8,53-8,52 (m, 1H), 8,43 (s, 1H), 8,05 (d, 1H), 7,95 (s, 1H), 7,72-7,63 (m, 3H), 7,37-7,23 (m, 5H), 5,35-5,34 (m, 1H), 4,31-4,26 (m, 1H), 3,15-3,08 (m, 1H), 2,66-2,60 (m, 1H), 2,55 (s, 3H), 1,83-1,74 (m, 3H).
Ejemplo_____ 26_____ 1-metil-N-((2R,3S)-1-(1-(1-metil-6-oxo-1,6-dil'iidropiridin-3-il)-1H-indazol-5-il)-5-oxo-2-fen¡lp¡rrolid¡n-3-¡l)c¡clopropano-1-carboxam¡da

Etapa 1: A una solución agitada de ácido 1-metilciclopropano-1-carboxílico (0,68 g, 6,818 mmol, 1,2 eq) en DMF (10 ml), se le añadió HATU (4,32 g, 11,363 mmol, 2,0 eq), DIPEA (5,0 ml, 28,409 mmol, 5,0 eq) y el producto intermedio A2 (1,00 g, 5,682 mmol, 1,0 eq) a 0 °C y, a continuación, la reacción se agitó a TA durante 16 h. Una vez completada la reacción (monitorizada mediante TLC, sistema de TLC con MeOH al 5 % en DCM, Rf:0,3), la mezcla de reacción se diluyó con EtOAc (35 ml), se lavó con agua helada (3 x 25 ml), se secó sobre Na2SO4 y se concentró bajo presión reducida para obtener el producto bruto que se purificó mediante cromatografía en columna (gel de sílice de malla 230-400; MeOH del 0 a 3 %-DCM) para obtener la trans-1-metil-N-(5-oxo-2-fenilpirrolidin-3-il)ciclopropanocarboxamida (0,78 g, 53 %).
Etapa 2: Una solución agitada de trans-1-metil-N-(5-oxo-2-fenilpirrolidin-3-il)cidopropanocarboxamida (0,31 g, 1,20 mmol, 1,0 eq), 5-(5-yodo-1H-mdazol-1-il)-1-metilpindm-2(1H)-ona (0,51 g, 1,44 mmol, 1,2 eq) y K3PO4 (0,51 g, 2,40 mmol, 2,0 eq) en 1,4-dioxano (30 ml) se desgasificó con argón durante 30 minutos. A continuación, se añadieron trans-N,N'-dimetilciclohexano-1,2-diamina (0,07 g, 0,48 mmol, 0,4 eq) y CuI (0,05 g, 0,24 mmol, 0,2 eq) y la reacción se agitó durante 16 h a 90 °C en un tubo sellado. Una vez finalizada la reacción (monitorizada mediante TLC, sistema de TLC con MeOH al 5 % in DCM, Rf-0.4), la mezcla de reacción se filtró a través de un lecho de celite y el lecho de celite se lavó 2-3 veces con 1,4-dioxano. Las capas orgánicas combinadas se concentraron para obtener el producto bruto que se purificó mediante cromatografía en columna (gel de sílice de malla 230-400; MeOH del 0 al 2 % en Dc M) para obtener la trans-1-metil-N-(1-(1-(1-metil-6-oxo-1,6-dihidropiridin-3-il)-1 H-indazol-5-il)-5-oxo-2-fenilpirrolidin-3-il)ciclopropanocarboxamida racémica. Se realizó una separación de enantiómeros adicional mediante una HPLC quiral preparativa para obtener 1-metil-N-((2S,3R)-1-(l-(1-metil-6-oxo-1,6-dihidropiridin-3-il)-1H-indazol-5-il)-5-oxo-2-fenilpirrolidin-3-il)ciclopropanocarboxamida (0,12 g, 21 %; TR=6,47 min, nombre de la columna: Chiralpak IA (250 * 4,6 mm) 5 pm, fase móvil: hexano/EA/EtOH/DEA: 50/25/25/0,1, caudal: 1,0 ml/min) y 1-metil-N-((2R,3S)-1-(1-(1-metil-6-oxo-1,6-dihidropiridin-3-il)-1H-indazol-5-il)-5-oxo-2-fenilpirrolidin-3-il)ciclopropanocarboxamida (0,09 g, 16 %; TR=8,22 min, nombre de la columna: Chiralpak IA (250 * 4,6 mm) 5 pm, fase móvil: hexano/EA/EtOH/DEA: 50/25/25/0,1, caudal: 1,0 ml/min).
RMN H1 (DMSO-d6) 8: 8,24 (s, 1H), 8,18-8,15 (m, 2H), 7,84 (s, 1H), 7,71-7,70 (m, 1H), 7,59 (s, 2H), 7,34-7,27 (m, 4H), 7,22-7,20 (m, 1H), 6,54 (d, 1H), 5,25-5,23 (m, 1H), 4,24-4,16 (m, 1H), 3,49 (s, 3H), 3,04-2,97 (m, 1H), 2,61-2,56 (m, 1H), 1,30 (s, 3H), 1,01-0,99 (m, 2H), 0,55-0,54 (m, 2H).
Ejemplo 27:_____trans-2,2-difluoro-N-(1-(1-(2-metoxipindin-4-il)-1H-indazol-5-il)-5-oxo-2-fenilpirroli-din-3-il)propanamida

A partir del producto intermedio B1 y 4-bromo-2-metoxipiridina, el ejemplo 27 se sintetizó de manera análoga al procedimiento sintético descrito para el ejemplo 24.
RMN H1 (DMSO-d6) 8: 9,51-9,50 (m, 1H), 8,42 (s, 1H), 8,28-8,26 (m, 1H), 8,01-7,99 (m, 1H), 7,93 (s, 1H), 7,74 7,72 (m, 1H), 7,48-7,47 (m, 1H), 7,36-7,30 (m, 4H), 7,25-7,23 (m, 1H), 7,16 (s, 1H), 5,35-5,33 (m, 1H), 4,30 4,23 (m, 1H), 3,91 (s, 3H), 3,15-3,08 (m, 1H), 2,65-2,60 (m, 1H), 1,83-1,74 (m, 3H).
Ejemplo 31: trans-2.2-d¡fluoro-N-(1-(1-((1-met¡l-2-oxo-1.2-d¡h¡drop¡r¡d¡n-4-¡l)met¡l)-1H-¡ndazol-5-¡l)-5-oxo-2-fenilp¡rrol¡d¡n-3-¡l)propanamida

Una solución agitada de trans-2,2-difluoro-N-(5-oxo-2-fenilpirrolidin-3-il)propanamida (para la síntesis, véase el ejemplo 4) (0,200 g, 0,746 mmol, 1,0 eq), el producto intermedio C1 (0,326 g, 0,985 mmol, 1,2 eq) y K3PO4 (0,316 g, 1,492 mmol, 2,0 eq) en 1,4-dioxano (15 ml) se desgasificó con argón durante 30 min. A continuación, se añadieron trans-2,2-difluoro-N-(5-oxo-2-fenilpirrolidin-3-il)propanamida (0,042 g, 0,298 mmol, 0,4 eq) y CuI (0,028 g, 0,149 mmol, 0,2 eq) y la reacción se agitó durante 16 h a 90 °C en un tubo sellado. Una vez completada la reacción (monitorizada mediante TLC, sistema de TLC con metanol al 5 % en DCM, Rf:0,4), la mezcla de reacción se filtró a través de un lecho de celite y el lecho de celite se lavó 2-3 veces con 1,4-dioxano. Las capas orgánicas combinadas se concentraron para obtener el producto bruto que se purificó mediante una
HPLC preparativa para obtener la trans-2,2-difluoro-N-(1-(1-((1-metil-2-oxo-1,2-dihidropiridin-4-M)metil)-1H-indazol-5-il)-5-oxo-2-fenilpirrolidin-3-il)propanamida (0,130 g, 34 %).
RMN H1 (DMSO-da) 8: 9,47-9,45 (m, 1H), 8,07 (s, 1H), 8,03-8,02 (m, 1H), 7,76 (s, 1H), 7,59-7,57 (m, 1H), 7,51 7,49 (m, 1H), 7,34-7,28 (m, 4H), 7,23-7,20 (m, 1H), 6,66 (d, 1H), 6,45 (s, 1H), 5,59 (s, 2H), 5,27-5,26 (m, 1H), 4,27-4,24 (m, 1H), 3,76 (s, 3H), 3,10-3,04 (m, 1H), 2,63-2,57 (m, 1H), 1,82-1,73 (m, 3H).

A partir del producto intermedio C4 y la trans-2,2-difluoro-N-(5-oxo-2-fenilpirrolidin-3-il)propanamida, el ejemplo 34 se sintetizó de manera análoga al procedimiento sintético descrito para el ejemplo 31.
RMN H1 (DMSO-d6) 8: 9,45-9,47 (m, 1H), 8,00 (s, 1H), 7,83 (s, 1H), 7,73 (s, 1H), 7,68-7,71 (m, 1H), 7,49-7,51 (m, 1H), 7,21-7,34 (m, 6H), 6,29 (d, 1H), 5,25-5,28 (m, 3H), 4,28-4,22 (m, 1H), 3,37 (s, 3H), 3,03-3,10 (m, 1H), 2,61-2,62 (m, 1H), 1,73-1,82 (m, 3H).
Ejemplo 35: trans-2.2-d¡fluoro-N-(1-(1-(1-(c¡clopropanocarbon¡l)p¡per¡d¡n-4-¡l)-1H-¡ndazol-5-¡l)-5-oxo-2-fen¡lp¡rrol¡d¡n-3-¡l)propanam¡da

A part¡r del producto intermedio C5 y la trans-2.2-d¡fluoro-N-(5-oxo-2-fen¡lp¡rrol¡d¡n-3-¡l)propanam¡da. el ejemplo 35 se s¡ntet¡zó de manera análoga al proced¡m¡ento s¡ntét¡co descr¡to para el ejemplo 31.
RMN H1 (DMSO-da) 8: 9.47-9.45 (m. 1H). 8.00 (s. 1H). 7.72-7.71 (m, 1H). 7.66-7.64 (m, 1H). 7.52-7.50 (m, 1H).
7.35-7.28 (m. 4H). 7.23-7.21 (m. 1H). 5.28-5.26 (m. 1H). 4.88-4.83 (m. 1H). 4.61-4.56 (m. 2H). 4.27-4.25 (m.
1H). 3.11-3.05 (m. 1H). 2.95-2.93 (m. 1H). 2.63-2.58 (m. 1H). 2.02-1.97 (m. 5H). 1.83-1.73 (m. 4H). 0.73-0.69 (m. 4H).
Ejemplo 38: trans-2.2-d¡fluoro-N-(1-(1-((1-met¡l-2-oxo-1.2-d¡h¡drop¡r¡d¡n-4-¡l)met¡l)-1H-¡ndazol-5-¡l)-5-oxo-2-fen¡lp¡rrol¡d¡n-3-¡l)propanam¡da

A part¡r del producto intermedio C6 y la trans-2.2-d¡fluoro-N-(5-oxo-2-fen¡lp¡rrol¡d¡n-3-¡l)propanam¡da. el ejemplo 38 se s¡ntet¡zó de manera análoga al proced¡m¡ento s¡ntét¡co descr¡to para el ejemplo 31.
RMN H1 (DMSO-d6) 8: 9.47-9.45 (m. 1H). 8.07 (s. 1H). 7.76 (s. 1H). 7.58-7.49 (m. 3H). 7.34-7.28 (m. 4H). 7.23 7.21 (m. 1H). 5.96 (s. 1H). 5.90-5.88 (m. 1H). 5.43 (s. 2H). 5.28-5.26 (m. 1H). 4.26-4.24 (m. 1H). 3.10-3.04 (m.
1H). 2.62-2.57 (m. 1H). 1.82-1.73 (m. 3H).
Ejemplo 39:_____N-((2R.3S)-2-(2-fluorofen¡l)-1-(1-(1-met¡l-6-oxo-1.6-d¡h¡drop¡r¡d¡n-3-¡l)-1H-¡ndazol-5-¡l)-5-oxop¡rrol¡d¡n-3-¡l)c¡clopropanocarboxam¡da

Etapa 1: A una soluc¡ón ag¡tada de ác¡do c¡clopropanocarboxíl¡co (0.53 g. 6.18 mmol. 1.2 eq) en DMF (8ml) se le añad¡ó HATU (4.00 g. 10.30 mmol. 2.0 eq). DIPEA (4.5 ml. 25.75 mmol. 5.0 eq) y el producto intermedio A6 (1.00 g. 5.15 mmol. 1.0 eq) a 0 °C y la reacc¡ón. a cont¡nuac¡ón. se ag¡tó a t A durante 16 h. Después de completar la reacc¡ón (mon¡tor¡zada med¡ante TLC. s¡stema de TLC con metanol al 5 % en DCM. Rf-0.3). la
mezcla de reacción se diluyó con EtOAc (25 ml) y se lavó con agua helada (3x25 ml), se secó sobre Na2SO4 y se concentró bajo presión reducida para obtener el producto crudo que se purificó mediante cromatografía en columna (gel de sílice de malla 230-400; MeOH del 0 al 2 %-DCM) para obtener una trans-N-(2-(2-fluorofenil)-5-oxopirrolidin-3-il)ciclopropanocarboxamida (0,56 g, 41 %).
Etapa 2: Una solución agitada de trans-N-(2-(2-fluorofenil)-5-oxopirrolidin-3-il)ciclopropanocarboxamida (0,250 g, 0,953 mmol, 1 eq), 5-(5-yodo-1H-indazol-1-il)-1-metilpiridin-2(1H)-ona (0,401 g, 1,144 mmol, 1,2 eq) y K3PO4 (0,404 g, 1,906 mmol, 2,0 eq) en 1,4-dioxano (10 ml) se desgasificó con argón durante 30 min. A continuación, se añadieron trans-N,N'-dimetilciclohexano-1,2-diamina (0,054 g, 0,381 mmol, 0,4 eq) y CuI (0,036 g, 0,191 mmol, 0,2 eq) y la reacción se agitó durante 16 h a 90 °C en un tubo sellado. Una vez completada la reacción (monitorizada mediante TLC, sistema de TLC con metanol al 5 % en DCM, Rf:0,4), la mezcla de reacción se filtró a través de un lecho de celite y el lecho de celite se lavó 2-3 veces con 1,4-dioxano. Las capas orgánicas combinadas se concentraron bajo presión reducida para obtener el producto bruto que se purificó mediante cromatografía en columna (gel de sílice de malla 230-400; MeOH del 0 al 2 % en DCM) para obtener el producto racémico. Se realizó una separación de enantiómeros adicional mediante una HPLC quiral preparativa para obtener N-((2S,3R)-2-(2-fluorofenM)-1-(1-(1-metN-6-oxo-1,6-dihidropiridm-3-N)-1H-indazol-5-il)-5-oxopirrolidin-3-il)ciclopropanocarboxamida (0,042 g, 9%; TR=6,96 min, nombre de la columna: Chiralpak IA (250X4,6 mm) 5 pm, fase móvil: hexano/EA/EtOH/DEA: 50/25/25/0,1, caudal: 1,0 ml/min) y N-((2R,3s)-2-(2-fluorofenil)-1 -(1-(1-metil-6-oxo-1,6-dihidropiridin-3-il)-1H-indazol-5-il)-5-oxopirrolidin-3-il)ciclopropanocarboxamida (0,042 g, 9%; TR=9,77 min, nombre de la columna: Chiralpak IA (250 * 4,6 mm) 5 pm, fase móvil: hexano/EA/EtOH/DEA: 50/25/25/0,1, caudal: 1,0 ml/min).
RMN H1 (DMSO-d6) 8: 8,86-8,85 (m, 1H), 8,27 (s, 1H), 8,19 (s, 1H), 7,81 (s, 1H), 7,71-7,69 (m, 1H), 7,62-7,60 (m, 1H), 7,52-7,49 (m, 1H), 7,39-7,35 (m, 1H), 7,25 (s, 1H), 7,15-7,09 (m, 2H), 6,54-6,52 (m, 1H), 5,44-5,43 (m, 1H), 4,36-4,31 (m, 1H), 3,49 (s, 3H), 3,13-3,06 (m, 1H), 1,58-1,55 (m, 1H), 0,68 (s, 4H).
Ejemplo_______ 40:_______ trans-N-(1-(1-((2-metoxipiridin-4-il)metil)-1H-indazol-5-il)-5-oxo-2-fenilpirrolidin-3-¡l)c¡clopropanocarboxamida

Etapa 1: A una solución agitada de ácido ciclopropanocarboxílico (2,35 g, 27,27 mmol, 1,2 eq) en DMF (40 ml), se le añadió HATU (17,22 g, 45,45 mmol, 2,0 eq), DIPEA (19,75 ml, 113,64 mmol, 5,0 eq) y el producto intermedio A2 (4,00 g, 22,73 mmol, 1,0 eq) a 0 °C y, a continuación, la mezcla de reacción se agitó a t A durante 16 h. Una vez completada la reacción (monitorizada mediante TLC, sistema de TLC con metanol al 5 % en DCM, Rf:0,3), la mezcla de reacción se diluyó con EtOAc (250 ml), se lavó con agua helada (3 x 150 ml), se secó sobre Na2SO4 y se concentró en presión reducida para obtener el producto bruto que se purificó mediante cromatografía en columna (gel de sílice de malla 230-400; MeOH del 0 a 3 %-DCM) para obtener la trans-N-(5-oxo-2-fenilpirrolidin-3-il)ciclopropanocarboxamida (2,1 g, 39 %).
Etapa 2: A partir del producto intermedio C7 y la trans-N-(5-oxo-2-fenilpirrolidin-3-il)ciclopropanocarboxamida, el ejemplo 40 se sintetizó de manera análoga al procedimiento sintético descrito para el ejemplo 31.
RMN H1 (DMSO-d6) 8: 8,87-8,88 (m, 1H), 8,02-8,08 (m, 2H), 7,79 (s, 1H), 7,57-7,60 (m, 2H), 7,22-7,32 (m, 5H), 6,65-6,66 (m, 1H), 6,45 (s, 1H), 5,60 (s, 2H), 5,21-5,19 (m, 1H), 4,15-4,08 (m, 1H), 3,77 (s, 3H), 3,00-3,06 (m, 1H), 2,32-2,44 (m, 1H), 1,59 (s, 1H), 0,69-0,72 (m, 4H).
Ejemplo 41: N-((2R.3S)-1-(1-(1-met¡l-6-oxo-1.6-d¡h¡drop¡r¡d¡n-3-¡l)-1H-¡ndazol-5-¡l)-5-oxo-2-(o-tol¡l)p¡rrol¡d¡n-3-¡l)c¡clopropanocarboxam¡da

Etapa 1: A una soluc¡ón ag¡tada de ác¡do c¡clopropanocarboxíl¡co (0.54 g. 6.31 mmol. 1.2 eq) en DMF (8.0 ml), HATU (3.90 g. 10.52 mmol. 2.0 eq). se le añad¡ó DlPEA (4.7 ml. 26.32 mmol. 5.0 eq) y el producto intermedio A10 (1.00 g. 5.26 mmol. 1.0 eq) a 0 °C y. a cont¡nuac¡ón. la mezcla de reacc¡ón se ag¡tó a TA durante 16 h. Una vez completada la reacc¡ón (mon¡tor¡zada med¡ante TLC. s¡stema de TLC con MeOH al 5 % en DCM. Rf:0.3). la mezcla de reacc¡ón se diluyó con EtOAc (25 ml). se lavó con agua helada (3 x 25 ml). se secó sobre Na2SO4 y se concentró bajo pres¡ón reduc¡da para obtener el producto bruto que se pur¡f¡có med¡ante cromatografía en columna (gel de síl¡ce de malla 230-400; MeOH del 0 al 2 %-DCM) para obtener la trans-N-(5-oxo-2-(o-tol¡l)p¡rrol¡d¡n-3-¡l)c¡clopropanocarboxam¡da (0.60 g. 43 %).
Etapa 2: Una soluc¡ón ag¡tada de trans-N-(5-oxo-2-(o-tol¡l)p¡rrol¡d¡n-3-¡l)c¡clopropanocarboxam¡da (0.40 g.
1.55 mmol. 1.0 eq). 5-(5-yodo-1H-¡ndazol-1-¡l)-1-met¡lp¡r¡d¡n-2(1H)-ona (0.65 g. 1.86 mmol. 1.2 eq) y K3PO4 (0.66 g. 3.10 mmol. 2.0 eq) en 1.4-d¡oxano (10 ml) se desgas¡f¡có con argón durante 30 m¡n. A cont¡nuac¡ón. se añad¡eron trans-N.N'-d¡met¡lc¡clohexano-1.2-d¡am¡na (0.09 g. 0.62 mmol. 0.4 eq) y Cul (0.06 g. 0.31 mmol.
0.2 eq) y la mezcla de reacc¡ón se ag¡tó durante 16 h a 90 °C en un tubo sellado. Una vez f¡nal¡zada la reacc¡ón (mon¡tor¡zada med¡ante TLC. s¡stema de TLC con MeOH al 5 % ¡n DCM. Rf-0.4). la mezcla de reacc¡ón se f¡ltró a través de un lecho de cel¡te y el lecho de cel¡te se lavó 2-3 veces con 1.4-d¡oxano. Las capas orgán¡cas comb¡nadas se concentraron para obtener el producto bruto que se pur¡f¡có med¡ante cromatografía en columna (gel de síl¡ce de malla 230-400; MeOH del 0 al 2 % en DCM) para obtener el producto racém¡co. A cont¡nuac¡ón. se real¡zó una separac¡ón de enant¡ómeros ad¡c¡onal med¡ante una HPLC qu¡ral preparat¡va para obtener N-((2S.3R)-1-(1-(1-met¡l-6-oxo-1.6-d¡h¡drop¡r¡d¡n-3-¡l)-1H-¡ndazol-5-¡l)-5-oxo-2-(o-tol¡l)p¡rrol¡d¡n-3-¡l)c¡clopropanocarboxam¡da (0.14 g. 19 %; TR=6.72 m¡n. nombre de la columna Ch¡ralpak IA (250 * 4.6 mm) 5 pm. fase móv¡l: hexano/EA/EtOH/DEA: 50/25/25/0.1. caudal: 1.0 ml/m¡n) y N-((2R,3S)-1-(1-(1-metil-6-oxo-1,6-dihidropiridm-3-M)-1H-mdazol-5-M)-5-oxo-2-(o-toMl)pirroMdm-3-M)cidopropanocarboxamida (0.10 g.
14 %; TR=8.13 m¡n. nombre de la columna: Ch¡ralpak IA (250 * 4.6 mm) 5 pm. fase móv¡l: hexano/EA/EtOH/DEA: 50/25/25/0.1. caudal: 1.0 ml/m¡n).
RMN H1 (DMSO-d6) 8: 9.00-8.99 (m. 1H). 8.26 (s. 1H). 8.18 (s. 1H). 7.91 (s. 1H). 7.72-7.70 (m. 1H). 7.61 (s.
2H). 7.16-7.11 (m. 4H). 6.54 (d. 1H). 5.41-5.40 (m. 1H). 4.18-4.16 (m. 1H). 3.49 (s. 3H). 3.10-3.03 (m. 1H).
2.43-2.41 (m. 4H). 1.61-1.60 (m. 1H). 0.71-0.69 (m. 4H).
Ejemplo 42: 2.2-d¡fluoro-N-((2R.3S)-2-(2-metox¡p¡r¡d¡n-4-¡l)-1-(1-(1-met¡l-6-oxo-1.6-d¡h¡drop¡r¡d¡n-3-¡l)-1H-¡ndazol-5-¡l)-5-oxop¡rrol¡d¡n-3-¡l)propanam¡da
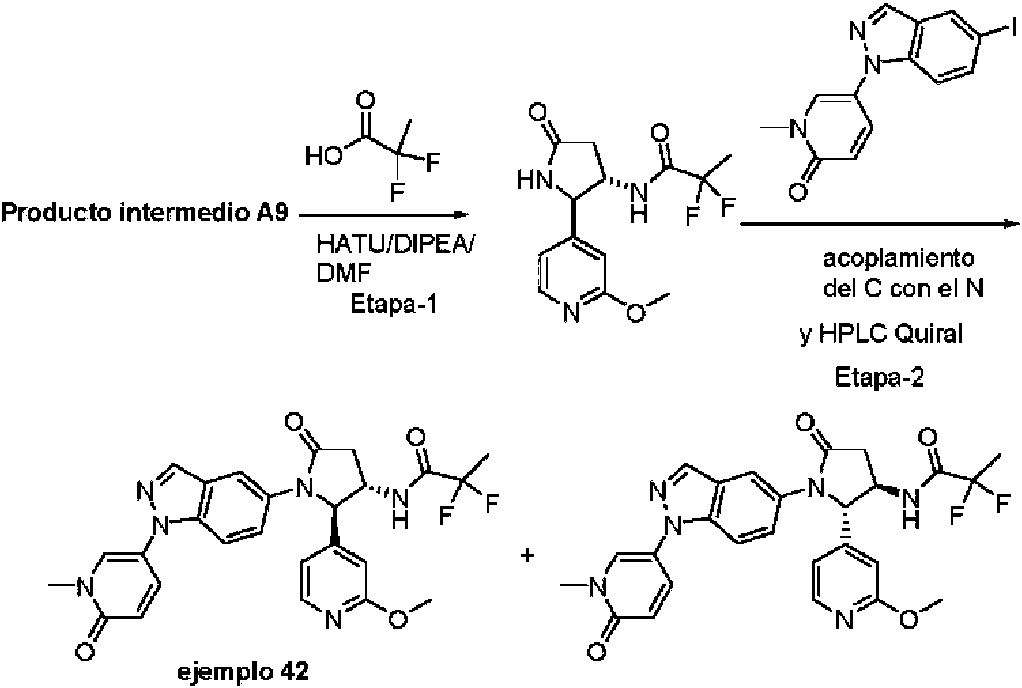
Etapa 1: A una soluc¡ón ag¡tada de 2,2-d¡fluoropropano¡co ác¡do (0.64 g. 5.79 mmol. 1.2 eq) en DMF (10 ml), se le añad¡ó HATU (3.60 g. 9.65 mmol. 2.0 eq). DlpEA (4.2 ml. 24.13 mmol. 5.0 eq) y el producto intermedio A9 (1.00 g. 4.83 mmol. 1.0 eq) a 0 °C y la mezcla de reacc¡ón se ag¡tó a TA durante 16 h. Una vez completada la reacc¡ón. (mon¡tor¡zada med¡ante TLC. s¡stema de TLC con MeOH al 5 % en DCM. Rf:0.3). la mezcla de reacc¡ón se diluyó con EtOAc (25 ml). se lavó con agua helada (3x25 ml). se secó sobre Na2SO4 y se concentró bajo pres¡ón reduc¡da para obtener el producto crudo que se pur¡f¡có med¡ante cromatografía en columna (gel de síl¡ce de malla 230-400; MeOH del 0 al 2 %-DCM;) para obtener la trans-2.2-d¡fluoro-N-(2-(2-metox¡p¡r¡d¡n-4-¡l)-5-oxop¡rrol¡d¡n-3-¡l)propanam¡da (0.76 g. 52 %).
Etapa 2: Una soluc¡ón ag¡tada de trans-2.2-d¡fluoro-N-(5-oxo-2-(2-metox¡p¡r¡d¡n-4-¡l)p¡rrol¡d¡n-3-¡l)-propanam¡da (0.378 g. 1.26 mmol. 1.0 eq). 5-5-yodo-1H-¡ndazol-1-¡l)-1-met¡lp¡r¡d¡n-2(1H)-ona (0.531 g. 1.52 mmol. 1.2 eq) y K3PO4 (0.370 g. 2.53 mmol. 2.0 eq) en 1.4-d¡oxano (10 ml) se desgas¡f¡có con argón durante 30 m¡n. A cont¡nuac¡ón. se añad¡eron trans-N.N'-d¡met¡lc¡clohexano-1.2-d¡am¡na (0.072 g. 0.51 mmol. 0.4 eq) y Cul (0.048 g. 0.25 mmol. 0.2 eq) y la reacc¡ón se ag¡tó durante 16 h a 90 °C en un tubo sellado. Una vez completada la reacc¡ón (mon¡tor¡zada med¡ante TLC. s¡stema de TLC con metanol al 5 % en DCM. Rf:0.4). la mezcla de reacc¡ón se f¡ltró a través de un lecho de cel¡te y el lecho de cel¡te se lavó 2-3 veces con 1.4-d¡oxano. Las capas orgán¡cas comb¡nadas se concentraron para obtener el producto crudo que se pur¡f¡có med¡ante cromatografía en columna (gel de síl¡ce de malla 230-400; MeOH del 0 a 7 % en DCM) para obtener la trans-2.2-d¡fluoro-N-(2-(2-metox¡p¡r¡d¡n-4-¡l)-1-(1-(1-met¡l-6-oxo-1.6-d¡h¡drop¡r¡d¡n-3-¡l)-1H-¡ndazol-5-¡l)-5-oxop¡rrol¡d¡n-3-¡l)propanam¡da racém¡ca. Se real¡zó una separac¡ón ad¡c¡onal de enant¡ómeros med¡ante una HPLC qu¡ral preparat¡va para obtener 2.2-d¡fluoro-N-((2S.3R)-2-(2-metox¡p¡r¡d¡n-4-¡l)-1-(1-(1-met¡l-6-oxo-1.6-d¡h¡drop¡r¡d¡n-3¡l)-1H-¡ndazol-5¡l)-5-oxop¡rrol¡d¡n-3-¡l)propanam¡da pura (0.035 g. 5 %; TR=11.93 m¡n. nombre de la columna: Ch¡ralpak IA (250 * 4.6 mm) 5 pm. fase móv¡l: hexano/EA/EtOH/DEA: 70/15/15/0.1. caudal: 1.0 ml/m¡n) y 2,2-difluoro-N-((2R,3S)-2-(2-metoxipiNdm-4-il)-1-(1-(1-metil-6-oxo-1,6-dihidropiNdm-3-il)-1H-mdazol-5-il)-5-oxopirrolidin-3-il)propanamida (0.027 g. 4 %; TR=14,72 min. nombre de la columna: Ch¡ralpak IA (250 * 4.6 mm) 5 pm. fase móv¡l: hexano/EA/EtOH/DEA: 70/15/15/0.1. caudal: 1.0 ml/m¡n).
RMN H1 (DMSO-d6) 8: 9.48-9.46 (m. 1H). 8.26 (s. 1H). 8.20 (d. 1H). 8.09 (d. 1H). 7.87 (s. 1H). 7.73-7.70 (m.
1H). 7.63-7.60 (m. 2H). 7.00-6.98 (m. 1H). 6.75 (s. 1H). 6.54-6.52 (m. 1H). 5.32-5.31 (m. 1H). 4.31-4.29 (m.
1H). 3.77 (s. 3H). 3.49 (s. 3H). 3.13-3.06 (m. 1H). 2.68-2.62 (m. 1H). 1.83-1.74 (m. 3H).
Ejemplo 43: 2.2-d¡fluoro-N-((2R.3S)-1-(1-(1-met¡l-6-oxo-1.6-d¡h¡drop¡r¡d¡n-3-¡l)-1H-¡ndazol-5-¡l)-5-oxo-2-(otol¡l)p¡rrol¡d¡n-3-¡l)propanam¡da

Etapa 1: A una soluc¡ón ag¡tada de ác¡do 2.2-d¡fluoropropano¡co (0.70 g. 6.32 mmol. 1.2 eq) en DMF (8 ml), se le añad¡ó HATU (3.90 g. 10.52 mmol. 2.0 eq). DIPEA (4.7 ml. 26.32 mmol. 5.0 eq)) y el producto intermedio A10 (1.00 g. 5.26 mmol. 1.0 eq) a 0 °C y. a cont¡nuac¡ón. la reacc¡ón se ag¡tó a Ta durante 16 h. Una vez completada la reacc¡ón (mon¡tor¡zada med¡ante TLC. s¡stema de TLC con MeOH al 5 % en DCM. Rf:0.3). la mezcla de reacc¡ón se diluyó con EtOAc (25 ml). se lavó con agua helada (3 x 25 ml). se secó sobre Na2SO4 y se concentró bajo pres¡ón reduc¡da para obtener el producto bruto que se pur¡f¡có med¡ante cromatografía en columna (gel de síl¡ce de malla 230-400; MeOH del 0 al 2 %-DCM) para obtener la trans-2.2-d¡fluoro-N-(5-oxo-2-(o-tol¡l)p¡rrol¡d¡n-3-¡l)propanam¡da (0.60 g. 40 %).
Etapa 2: Una soluc¡ón ag¡tada de trans-2.2-d¡fluoro-N-(5-oxo-2-(o-tol¡l)p¡rrol¡d¡n-3-¡l)propanam¡da (0.25 g.
0.873 mmol. 1 eq). 5-(5-yodo-1H-¡ndazol-1-¡l)-1-met¡lp¡r¡d¡n-2(1H)-ona (0.37 g. 1.04 mmol. 1.2 eq) y K3PO4 (0.37 g. 1.746 mmol. 2.0 eq) en 1.4-d¡oxano (10 ml) se desgas¡f¡có con argón durante 30 m¡n. A cont¡nuac¡ón. se añad¡eron trans-N.N'-d¡met¡lc¡clohexano-1.2-d¡am¡na (0.050 g. 0.35 mmol. 0.4 eq) y Cul (0.033 g.
0.175 mmol. 0.2 eq) y la reacc¡ón se ag¡tó durante 16 h a 90 °C en un tubo sellado. Una vez f¡nal¡zada la reacc¡ón (mon¡tor¡zada med¡ante TLC. s¡stema de TLC con MeOH al 5 % ¡n DCM. Rf-0.4). la mezcla de reacc¡ón se f¡ltró a través de un lecho de cel¡te y el lecho de cel¡te se lavó 2-3 veces con 1.4-d¡oxano. Las capas orgán¡cas comb¡nadas se concentraron para obtener el producto bruto que se pur¡f¡có med¡ante cromatografía en columna (gel de síl¡ce de malla 230-400; MeOH del 0 al 2 % en DCM) para obtener la trans-2.2-d¡fluoro-N-(1-(1-(1-met¡l-6-oxo-1.6-d¡h¡drop¡r¡d¡n-3-¡l)-1 H-¡ndazol-5-¡l)-5-oxo-2-(o-tol¡l)p¡rrol¡d¡n-3-¡l)propanam¡da racém¡ca. Se real¡zó una separac¡ón de enant¡ómeros ad¡c¡onal med¡ante una HpLC qu¡ral preparat¡va para obtener 2.2-d¡fluoro-N-((2S.3R)-1-(1-(1-met¡l-6-oxo-1.6-d¡h¡drop¡r¡d¡n-3-¡l)-1H--¡ndazol-5-¡l)-5-oxo-2-(o-tol¡l)p¡rrol¡d¡n-3-¡l)propanam¡da (0.08 g. 17 %; TR=5.57 m¡n. nombre de la columna: Ch¡ralpak IA (250 * 4 .6 mm) 5 pm. fase móv¡l: hexano/EA/EtOH/DEA: 50/25/25/0.1. caudal: 1,0 ml/m¡n) y 2,2-difluoro-N-((2R,3S)-1-(1-(1-metN-6-oxo-1,6-dihidropiridin-3-il)-1H-indazol-5-il)-5-oxo-2-(o-tolil)pirrolidin-3-il)propanamida (0.07 g. 15 %; TR=7,91 min. nombre de la columna: Ch¡ralpak IA (250x4.6 mm) 5 pm. fase móv¡l: hexano/EA/EtOH/DEA: 50/25/25/0.1. caudal: 1.0 ml/m¡n).
RMN H1 (DMSO-d6) 8: 9.61-9.60 (m. 1H). 8.26 (s. 1H). 8.18 (s. 1H). 7.86 (s. 1H). 7.71-7.69 (m. 1H). 7.62-7.55 (m. 2H). 7.15-7.12 (m. 4H). 6.54-6.51 (m. 1H). 5.54-5.52 (m. 1H). 4.30-4.24 (m. 1H). 3.49 (s. 3H). 3.16-3.09 (m.
1H). 2.58-2.54 (m. 1H). 2.37 (s. 3H). 1.83-1.73 (m. 3H).
Ejemplo______ 46______ N-(trans-1-(1-(1-met¡l-1H-p¡razol-4-¡l)-1H-¡ndazol-5-il)-5-oxo-2-fen¡lp¡rrol¡d¡n-3-¡l)-2,2-difluoropropanamida

El producto intermedio B1 (80,0 mg, 0,208 mmol, 1,0 eq), el yoduro de cobre (7,9 mg, 0,042 mmol, 0,2 eq), el yoduro de sod¡o (93,6 mg, 0,624 mmol, 3,0 eq), el 4-bromo-1-met¡lp¡razol (67,0 mg, 0,416 mmol, 2,0 eq) y el K3PO4 (132,5 mg, 0,624 mmol, 3,0 eq) se pesaron en un v¡al, se añad¡ó una barra de ag¡tac¡ón, se selló el v¡al y se purgó con n¡trógeno. Se añad¡ó 1,4-d¡oxano (1,5 ml), segu¡do de trans-N,N-d¡met¡lddohexano-1,2-d¡am¡na (0,012 g, 0,083 mmol, 0,4 eq). La mezcla se calentó a 110 °C durante 16 horas. La mezcla se enfrió a TA y, a cont¡nuac¡ón, se d¡luyó con DCM y agua. La mezcla se f¡ltró a través de una frita h¡drófoba y, a cont¡nuac¡ón, se purificó med¡ante cromatografía en columna para obtener N-(trans-1-(1-(1-metil-1H-p¡razol-4-¡l)-1H-indazol-5¡l)-5-oxo-2-fen¡lp¡rrol¡d¡n-3-¡l)-2,2-d¡fluoropropanam¡da (73,4 mg, 76 %).
RMN H1 (DMSO-d6) 5: 9,48 (d, 1H), 8,24 (s, 1H), 8,20 (d, 1H), 7,84 (d, 1H), 7,82 (d, 1H), 7,64 - 7,56 (m, 2H), 7,35 (d, 2H), 7,31 (t, 2H), 7,24 - 7,20 (m, 1H), 5,30 (d, 1H), 4,34 - 4,24 (m, 1H), 3,90 (s, 3H), 3,12 - 3,07 (m, 1H), 2,63 (dd, 1H), 1,78 (t, 3H).
Ejemplo_______47_______ N-(trans-1-(1-(5-fluorop¡r¡m¡d¡n-2-¡l)-1H-¡ndazol-5-¡l)-5-oxo-2-fen¡lp¡rrol¡d¡n-3-¡l)-2,2-

A partir del producto intermedio B1, el ejemplo 47 se s¡ntet¡zó de manera análoga al proced¡m¡ento de síntesis descrito para el ejemplo 46, sustituyendo 2-bromo-5-fluoropirim¡d¡na por 4-bromo-1-metilpirazol y prolongando el tiempo de reacción a 40 horas. El Ejemplo 47 se obtuvo con un rendimiento del 57 % (57,1 mg).
RMN H1 (DMSO-d6) 5: 9,49 (d, 1H), 8,97 (s, 2H), 8,45 (dt, 1H), 8,42 (d, 1H), 7,95 (d, 1H), 7,72 (dd, 1H), 7,36 (dd, 2H), 7,31 (t, 2H), 7,25 - 7,20 (m, 1H), 5,36 (d, 1H), 4,40 - 4,24 (m, 1H), 3,12 (dd, 1H), 2,66 (dd, 1H), 1,77 (t, 3H).
Los ejemplos de la Tabla 1 se sintetizaron de manera análoga al Ejemplo 13.


Ensayo de unión al ligando del receptor de glucocorticoides humano (hGR)
La línea celular de linfoblastos humanos IM9 (ATCC, Bethesda, MD) se cultivó en un medio RPMI 1640 que contenía suero fetal bovino al 10 %, penicilina (100 U/ml), estreptomicina (100 |jg/ml) y 2 mM L-glutamina a 37° y 7 % de CO2 en una incubadora humidificada. Las células se centrifugaron durante 10 minutos a 1500 g y se lavaron en PBS y se repelieron. A continuación, las células se resuspendieron en un tampón de homogeneización que consistía en: TES 10 mM, molibdato de sodio 10 mM, MEDTA 1 mM, pH 7,4, 2-mercaptoetanol 20 mM y glicerol al 10 %. La ruptura de las células se realizó mediante cavitación con nitrógeno utilizando 2 * 15 minutos a 600 a 750 psi de nitrógeno en un cavitador de N2 a 0 °C. A continuación, la preparación celular se centrifugó a 27.000 g durante 15 minutos y el sobrenadante resultante (= citosol de células IM9) se centrifugó a 103.000 g durante 60 minutos a 4°C. La cantidad de proteína en la fracción sobrenadante se determinó utilizando un kit de ensayo BCA y las partes alícuotas se congelaron instantáneamente en un baño de hielo seco y acetona y se almacenaron a -70 °C. Los ensayos de unión competitiva se realizaron por duplicado en un tampón de homogeneización con un volumen total de 200 jl. Con esta finalidad, se mezclaron 1 mg de citosol IM9, 0,05 jC i (1,5 nM) de 3H-dexametasona (1 jM ) y compuestos según la presente invención (= competidores no marcados de dexametasona; intervalo de concentraciones). La reacción se detuvo después de la incubación a 0°C durante 16 a 18 h mediante la adición de 100 j l de una mezcla de carbón vegetal-dextrano (carbón activado del 2 %, dextrano del 0,5 % en 10 mM de Tris, 1 mM de EDTA, pH 7,4). Siguió otra etapa de incubación a 0°C durante 10 minutos antes de centrifugar las muestras durante 5 minutos a 8200 g. 100 j l del sobrenadante) finalmente se realizó un ensayo de radioactividad mediante espectrometría de centelleo líquido, y los valores IC50 se determinaron gráficamente y se convirtieron en valores Ki.
Los resultados se resumen en la Tabla 2 a continuación (% de inhibición hGR a 1 pM; 40 % < A < 60 %, 60 % < B < 85 %, 85 % < C).
Tabla 2:

Claims (14)
1. Un compuesto según la fórmula general (I)

donde
Ri representa un -C1-10-alquilo; un -C3-10-cidoalquilo; un -C1-6-alquileno-C3-10-cidoalquilo; un heterocicloalquilo de 3 a 7 miembros; un -C1-6-alquileno-(heteroddoalquilo de 3 a 7 miembros); un arilo; un -C1-6-alquileno-arilo; un heteroarilo de 5 o 6 miembros; o un -C1-6-alquileno-(heteroarilo de 5 o 6 miembros); R2 representa un -C(=O)-C1-10-alquilo; un -C(=O)-C3-10-cicloalquilo; un -C(=O)-C1-6-alquileno-C3-10-cicloalquilo; un -C(=O)-(heterocicloalquilo de 3 a 7 miembros); un -C(=O)-C1-6-alquileno-(heterocicloalquilo de 3 a 7 miembros); un -C(=O)-arilo; un -C(=O)-C1-6-alquileno-arilo; un -C(=O)-(heteroarilo de 5 o 6 miembros); un -C(=O)-C1-6-alquileno-(heteroarilo de 5 o 6 miembros); un -S(=O)1-2-C1-10-alquilo; un -S(=O)1-2-C3-10-cicloalquilo; un -S(=O)1-2-C1-6-alquileno-C3-10-cicloalquilo; un -S(=O)1-2-(heterocicloalquilo de 3 a 7 miembros); un -S(=O)1-2-C1-6-alquileno-(heterocicloalquilo de 3 a 7 miembros); un -S(=O)1-2-arilo; un -S(=O)1-2-C1-6-alquileno-arilo; un -S(=O)1-2-(heteroarilo de 5 o 6 miembros); o un -S(=O)1-2-C1-6-alquileno-(heteroarilo de 5 o 6 miembros);
R3 representa un heterocicloalquilo de 3 a 7 miembros; un -C1-6-alquileno-(heterocicloalquilo de 3 a 7 miembros); un heteroarilo de 5 o 6 miembros; un -C1-6-alquileno-(heteroarilo de 5 o 6 miembros); un -C(=O)-(heterocicloalquilo de 3 a 7 miembros); un -C(=O)-C1-6-alquileno-(heterocicloalquilo de 3 a 7 miembros); un -C(=O)-(heteroarilo de 5 o 6 miembros); un -C(=O)-C1-6-alquileno(heteroarilo de 5 o 6 miembros); un -S(=O) 1-2-(heterocicloalquilo de 3 a 7 miembros); un -S(=O) 1-2-C1-6-alquileno-(heterocicloalquilo de 3 a 7 miembros); un -S(=O) 1-2-(heteroarilo de 5 o 6 miembros); o un -S(=O) 1-2-C1-6-alquileno-(heteroarilo de 5 o 6 miembros);
R4 representa un -H; -F; -Cl; -Br; -I; -CN; -CH3 ; -CF3 ; -CF2H; -CFH2 o ciclopropilo;
X representa un CR5 ; donde R5 representa un -H; -F; -Cl; -Br; -I; -CN; -C1-10-alquilo o -C3-10-cicloalquilo; Y representa un CR6; donde R6 representa un -H; -F; -Cl; -Br; -I; -CN; -C1-10-alquilo o -C3-10-cicloalquilo; Z representa un CR7 ; donde R7 representa un -H; -F; -Cl; -Br; -I; -CN; -C1-10-alquilo o -C3-10-cicloalquilo; donde -C1-10-alquilo, -C1-4-alquilo y -C1-6-alquileno- en cada caso independientemente entre sí son lineales o ramificados, saturados o insaturados;
donde un -C1-10-alquilo, -C1-4-alquilo, -C1-6-alquileno-, -C3-10-cicloalquilo y un heterocicloalquilo de 3 a 7 miembros, en cada caso independientemente entre sí, no están sustituidos, o están mono o polisustituidos con uno o más sustituyentes seleccionados de entre -F; -Cl; -Br; -I; -CN; -C1-6-alquilo; -CF3 ; -CF2H; -CFH2 ; -CF2Cl; -CFCh; -C(=O)-C1-6-alquilo; -C(=O)-OH; -C(=O)-OC1-6-alquilo; -C(=O)-NH2 ; -C(=O)-NH(C1-6-alquilo); -C(=O)-N(C1-6-alquilol)2 ; -OH; =O; -OCF3 ; -OCF2H; -OCFH2 ; - OCF2Cl; -OCFCl2 ; -O-C1-6-alquilo; -O-C(=O)-C1-6-alquilo; -O-C(=O)-O-C1-6-alquilo; -O-(CO)-NH(C1-6-alquilo); -O-C(=O)-N(C1-6-alquilo)2 ; -OS(=O) 2-NH2 ; -O-S(=O) 2-NH(C1-6-alquilo); -O-S(=O) 2-N(C1-6-alquilo) 2 ; -NH2 ; -NH(C1-6-alquilo); -N(C1-6-alquilo)2 ; -NH-C(=O)-C1-6-alquilo; -NH-C(=O)-O-C1-6-alquilo; -NH-C(=O)-NH2 ; -NH-C(=O)-NH(C1-6-alquilo); -NH-C(=O)-N(C1-6-alquilo) 2 ; -N(C1-6-alquilo)-C(=O)-C1-6-alquilo; -N(C1-6-alquilo)-C(=O)-O-C1-6-alquilo; -N(C1-6-alquilo)-C(=O)-NH2 ; -N(C1-6-alquilo)-C(=O)-NH(C1-6-alquilo); -N(C1-6-alquilo)-C(=O)-N(C1-6-alquilo) 2 ; -NH-S(=O) 2OH; NH-S(=O) 2-C1-6-alquilo; -NH-S(=O) 2-O-C1-6-alquilo; -NH-S(=O) 2-NH2; -NH-S(=O) 2-NH(C1-6-alquilo); -NH-S(=O) 2 N(C1-6-alquilo) 2 ; -N(C1-6-alquilo)-S(=O) 2-Oh ; -N(C1-6-alquilo)-S(=O) 2-C1-6-alquilo; -N(C1-6-alquilo)-S(=O) 2-O-C1-6-alquilo; -N(C1-6-alquilo)-S(=O) 2-NH2 ; -N(C1-6-alquilo)-S(=O) 2-NH(C1-6-alquilo); -N(C1-6-alquilo)-S(=O)2-N(C1-6-alquilo)2 ; -SCF3 ; -SCF2H; -SCFH2 ; -S-C1-6-alquilo; -S(=O)-C1-6-alquilo; -S(=O)2-C1-6-alquilo; -S(=O)2-OH; -S(=O)2-O-C1-6-alquilo; -S(=O)2-NH2 ; -S(=O)2-NH(C1-6-alquilo); - S(=O)2-N(C1-6-alquilo)2 ; -C3-6-cicloalquilo; un heterocicloalquilo de 3 a 6 miembros; un fenilo; un heteroarilo de 5 o 6 miembros; -O-C3-6-cicloalquilo; -O-(un heterocicloalquilo de 3 a 6 miembros); -O-fenilo; -O-(heteroarilo de 5 a 6 miembros); -C(=O)-C3-6-cicloalquilo; -C(=O)-(heterocicloalquilo de 3 a 6 miembros); -C(=O)-fenilo; -C(=O)-(heteroarilo de 5 a 6 miembros); - S(=O)2-(C3-6-cicloalquilo; -S(=O)2-(heterocicloalquilo de 3 a 6 miembros); -S(=O)2-fenilo o -S(=O)2-(heteroarilo de 5 a 6 miembros);
donde un arilo y un heteroarilo de 5 o 6 miembros en cada caso, independientemente entre sí, no están sustituidos o están mono o polisustituidos con uno o más sustituyentes seleccionados de entre -F; -Cl; -Br; -I; -CN; -C1-6-alquilo; -CF3 ; -CF2 H; -CFH2 ; -CF2Cl; -CFCh; -C1-4-alquileno-CF3; -C1-4-alquileno-CF2H; -C1-4-alquileno-CFH2 ; -C(=O)-C1-6-alquilo; -C(=O)-OH; - C(=O)-OC1-6-alquilo; -C(=O)-NH(OH); -C(=O)-NH2 ;
C(=O)-NH(Ci-4-alquilo); -C(=O)-N(Ci-6-alquilo)2 ; -OH; =O; -OCFa; -OCF2H; -OCFH2 ; -OCF2CI; -OCFCI2 ; -O-Ci-6-alquilo; -O-C3-6-cicloalquilo; -O-(heterocicloalquilo de 3 a 6 miembros); -NH2 ; -NH(Ci-6-alquilo); -N(Ci-6-alquilo)2 ; -NH-C(=O)-Ci-6-alquilo; -N(Ci-6-alquilo)-C(=O)-Ci-6-alquilo; -NH-C(=O)-NH2 ; -NH-C(=O)-NH(Ci-6-alquilo); -NH-C(=O)-N(Ci-6-alquilo)2 ; -N(Ci-6-alquilo)-C(=O)-NH(Ci-6-alquilo); -N(Ci-6-alquilo)-C(=O)-N(Ci-6-alquilo)2 ; -NH-S(=O)2-Ci-6-alquilo; -SCF3 ; -S-Ci-6-alquilo; -S(=O)-Ci-6-alquilo; -S(=O)2-Ci-e-alquilo; -S(=O)2-NH2 ; -S(=O)2-NH(Ci-6-alquilo); -S(=O)2-N(Ci-6-alquilo)2 ; -C3-6-cicloalquilo; -Ci-4-alquileno-C3-6-cicloalquilo; heterocicloalquilo de 3 a 6 miembros; -Ci-4-alquileno-(heterocicloalquilo de 3 a 6 miembros); fenilo o heteroarilo de 5 o 6 miembros;
en forma del compuesto libre o una sal fisiológicamente aceptable del mismo.
2. El compuesto según la reivindicación 1, donde R5 representa un-H; R6 representa un -H; y/o R7 representa un -H.
3. El compuesto según la reivindicación 1 o 2, donde el Ri representa un -C3-i0-cicloalquilo; un -Ci-6-alquileno-C3-i0-cicloalquilo; un arilo; o un heteroarilo de 5 o 6 miembros.
4. El compuesto según cualquiera de las reivindicaciones anteriores, donde:
R2 representa un -C(=O) Ci-10-alquilo; -C(=O)-C3-i0-cicloalquilo; -C(=O)-Ci-6-alquileno-C3-i0-cicloalquilo; -C(=O)-(heterocicloalquilo de 3 a 7 miembros); -C(=O)-(heteroarilo de 5 o 6 miembros); -S(=O) 2-Ci-i0-alquilo; -S(=O) 2-C3-i0-cicloalquilo; -S(=O) 2-Ci-6-alquileno-C3-i0-cicloalquilo; o -S(=O)2-(heteroarilo de 5 o 6 miembros).
5. El compuesto según cualquiera de las reivindicaciones anteriores, donde:
R3 representa un heterocicloalquilo de 3 a 7 miembros; un heteroarilo de 5 o 6 miembros; o un -C1-6-alquileno-(heteroarilo de 5 o 6 miembros).
6. El compuesto según cualquiera de las reivindicaciones anteriores, donde:
R4 representa un -H.
7. El compuesto según cualquiera de las reivindicaciones anteriores, donde:
Ri representa ciclopropilo, sin sustituir;
-CH2-ciclopropilo, sin sustituir;
fenilo, no sustituido o mono o disustituido con sustituyentes independientemente entre sí, seleccionados de entre el grupo que consiste en -F, -Cl, -Br, -CH3 , -CF3 , -CN, ciclopropilo y -OCH3 , donde el fenilo está opcionalmente recocido a un anillo de dioxolano mediante un -O-CH2CH2-O- sustituyente; o
piridilo, no sustituido o mono o disustituido con sustituyentes, independientemente entre sí, seleccionados de entre el grupo que consiste en -F, -Cl, -Br, -CH3, -CF3 , -CN y -OCH3.
8. El compuesto según cualquiera de las reivindicaciones anteriores, donde:
R2 representa -C(=O)-Ci-i0-alquilo, no sustituido o mono o disustituido con sustituyentes, independientemente entre sí, seleccionados de entre el grupo que consiste en -F, -Cl y -Br;
-C(=O)-ciclopropilo, no sustituido o mono o disustituido con sustituyentes, independientemente entre sí, seleccionados de entre el grupo que consiste en -F, -Cl, -Br, -CH3 , -CF3 , -CN y -OCH3 ;
-C(=O)-ciclopropilo, no sustituido o mono o disustituido con sustituyentes, independientemente entre sí, seleccionados de entre el grupo que consiste en -F, -Cl, -Br, -CH3 , -CF3 , -CN y -OCH3 ;
-C(=O)-2-tetrahidrofuranilo, sin sustituir;
-C(=O)-(heteroarilo de 5 a 6 miembros), donde dicho heteroarilo de 5 a 6 miembros se selecciona de entre el grupo que consiste en tiazolilo, pirazolilo, oxazolilo y 1-oxa-2,4-diazolilo, 1,2,5-oxadiazolilo, isoxazolilo, isotiazolilo, donde, en cada caso, dicho heteroarilo de 5 a 6 miembros no está sustituido o está mono o disustituido con sustituyentes, independientemente entre sí, seleccionados de entre el grupo que consiste en -F, -Cl, -Br, -CH3 , -CF3 , -CN, =O y -OCH3 ;
-S(=O)2-Ci-i0-alquilo, sin sustituir;
-S(=O)2-ciclopropilo, sin sustituir;
-S(=O)2-CH2-ciclopropilo, sin sustituir; o
-S(=O)2-(heteroarilo de 5 a 6 miembros), donde dicho heteroarilo de 5 a 6 miembros se selecciona de entre el grupo que consiste en tiazolilo, pirazolilo, oxazolilo y 1-oxa-2,4-diazolilo, 1,2,5-oxadiazolilo, isoxazolilo, isotiazolilo, donde, en cada caso, dicho heteroarilo de 5 a 6 miembros no está sustituido o está mono o disustituido con sustituyentes, independientemente entre sí, seleccionados de entre el grupo que consiste en -F, -Cl, -Br, -CH3 , -CF3 , -CN, =O y -OCH3.
9. El compuesto según cualquiera de las reivindicaciones anteriores, donde:
R3 representa piperidinilo, no sustituido o sustituido con -C(=O)-ciclopropilo;
heteroarilo de 5 a 6 miembros seleccionado de entre el grupo que consiste en pirazolilo, piridilo y pirimidinilo, donde en cada caso dicho heteroarilo de 5 a 6 miembros no está sustituido o está mono o disustituido con sustituyentes independientemente entre sí seleccionados de entre el grupo que consiste en -F, -Cl, -Br, -CHa, -CFa, -CN, =O y -OCH3 ; o
-CH2-(heteroarilo de 5 a 6 miembros) seleccionado de entre el grupo que consiste en -CH2-pirazolilo,-CH2-piridilo y -CH2-pirimidinilo, donde en cada caso dicho heteroarilo de 5 a 6 miembros no está sustituido o está mono o disustituido con sustituyentes independientemente entre sí seleccionados de entre el grupo que consiste en -F, -Cl, -Br, -CH3 , -CF3 , -CN, =O y -OCH3.
10. El compuesto según cualquiera de las reivindicaciones anteriores, donde:
Ri representa fenilo, no sustituido o mono o disustituido con sustituyentes, independientemente entre sí, seleccionados de entre el grupo que consiste en -F, -Cl, -Br, -CH3 , -CF3 , -CN, ciclopropilo y -OCH3 ; y/o R2 representa -C(=O)-Ci-6-alquilo; -C(=O)-ciclopropilo; o -C(=O)-ciclobutilo, no sustituido o mono-o disustituido con sustituyentes independientemente entre sí seleccionados de entre el grupo que consiste en -F, -Cl y -Br; y/o
R3 representa N-metil-2-oxo-piridilo.
11. El compuesto según cualquiera de las reivindicaciones anteriores seleccionado de entre el grupo que consiste en
1 N-(2R,3S)-2-(3-clorofenil)-1-[1-(1-metil-6-oxo-3-piridil)indazol-5-il]-5-oxo-pirrolidin-3-il]-2,2-difluoropropanamida
2 2,2-difluoro-N-[rac-(2R,3S)-2-(2,4-difluorofenil)-1-[1-(1-metil-6-oxo-3-piridil)indazol-5-il]-5-oxopirrolidin-3 -il]propanamida
3 2,2-difluoro-N-[rac-(2R,3S)-2-(2,3-dihidro-1,4-benzodioxin-6-il)-1-[1-(1-metil-6-oxo-3-piridil)indazol-5-il]-5-oxo-pirrolidin-3-il]propanamida
4 2,2-difluoro-N-[(2R,3S)-1-[1-(1-metil-6-oxo-3-piridil)indazol-5-il]-5-oxo-2-fenil-pirrolidin-3-il]propanamida 5 2,2-difluoro-N-[(2R,3S)-2-(3-fluorofenil)-1-[1-(1-metil-6-oxo-3-piridil)indazol-5-il]-5-oxo-pirrolidin-3-il]propanamida
6 2,2-difluoro-N-[(2R,3S)-2-(2-fluorofenil)-1-[1-(1-metil-6-oxo-3-piridil)indazol-5-il]-5-oxo-pirrolidin-3-il]propanamida
7 2,2-difluoro-N-[rac-(2R,3S)-5-oxo-2-fenil-1-[1-(3-piridil)indazol-5-il]pirrolidin-3-il]propanamida
9 2,2-difluoro-N-[rac-(2R,3S)-1-[1-(5-fluoro-2-piridil)indazol-5-il]-5-oxo-2-fenil-pirrolidin-3-il]propanamida 13 5-metil-N-[(2R,3S)-1-[1-(1-metil-6-oxo-3-piridil)indazol-5-il]-5-oxo-2-fenil-pirrolidin-3- i l]tiazol-2-carboxamida
15 2,2-difluoro-N-[(2R,3S)-2-(4-fluoro-3-metoxi-fenil)-1-[1-(1-metil-6-oxo-3-piridil)indazol-5-il]-5-oxopirrolidin-3-il]propanamida
17 2,2-difluoro-N-[(2R,3S)-2-(4-fluorofenil)-1-[1-(1-metil-6-oxo-3-piridil)indazol-5-il]-5-oxo-pirrolidin-3-il]propanamida
18 1-fluoro-N-[(2R,3S)-1-[1-(1-metil-6-oxo-3-piridil)indazol-5-il]-5-oxo-2-fenil-pirrolidin-3-il]ciclopropanocarboxamida
222,2-difluoro-N-[rac-(2R,3S)-1-[1-(6-metoxi-3-piridil)indazol-5-il]-5-oxo-2-fenil-pirrolidin-3-il]propanamida 232,2-difluoro-N-[rac-(2R,3s)-5-oxo-2-fenil-1-[1-(4-piridil)indazol-5-il]pirrolidin-3-il]propanamida
242,2-difluoro-N-[-(2R,3S)-1-[1-(6-metil-3-piridil)indazol-5-il]-5-oxo-2-fenil-pirrolidin-3-il]propanamida 252,2-difluoro-N-[rac-(2R,3S)-1-[1-(2-metil-4-piridil)indazol-5-il]-5-oxo-2-fenil-pirrolidin-3-il]propanamida 26 1-metil-N-[(2R,3S)-1-[1-(1-metil-6-oxo-3-piridil)indazol-5-il]-5-oxo-2-fenil-pirrolidin-3-il]ciclopropanocarboxamida
272,2-difluoro-N-[rac-(2R,3S)-1-[1-(2-metoxi-4-piridil)indazol-5-il]-5-oxo-2-fenil-pirrolidin-3-il]propanamida 31 2,2-difluoro-N-[rac-(2R,3S)-1-[1-[(2-metoxi-4-piridil)metil]indazol-5-il]-5-oxo-2-fenil-pirrolidin-3-il]propanamida
32 2,2-difluoro-N-[rac-(2R,3S)-1-[1-[(1-metilpirazol-3-il)metil]indazol-5-il]-5-oxo-2-fenil-pirrolidin-3-il]propanamida
33 2,2-difluoro-N-[rac-(2R,3S)-1-[1-[(6-metoxi-3-piridil)metil]indazol-5-il]-5-oxo-2-fenil-pirrolidin-3-il]propanamida
34 2,2-difluoro-N-[rac-(2R,3S)-1-[1-[(1-metil-6-oxo-3-piridil)metil]indazol-5-il]-5-oxo-2-fenil-pirrolidin-3-il]propanamida
35 2,2-difluoro-N-[rac-(2R,3S)-1-[1-[1-(ciclopropanocarbonil)-4-piperidil]indazol-5-il]-5-oxo-2-fenil-pirrolidin-3-il]propanamida
38 2,2-difluoro-N-[rac-(2R,3S)-1-[1-[(1-metil-2-oxo-4-piridil)metil]indazol-5-il]-5-oxo-2-fenil-pirrolidin-3-il]propanamida
39 N-[(2R,3S)-2-(2-fluorofenil)-1-[1-(1-metil-6-oxo-3-piridil)indazol-5-il]-5-oxo-pirrolidin-3-yil]ciclopropanocarboxamida
40 N-[rac-(2R,3S)-1-[1-[(2-metoxi-4-piridil)metil]indazol-5-il]-5-oxo-2-fenil-pirrolidin-3-il]ciclopropanocarboxamida
41 N-[(2R,3S)-1-[1-(1-metil-6-oxo-3-piridil)indazol-5-il]-2-(o-tolil)-5-oxo-pirrolidin-3-il]ciclopropanocarboxamida
42 2,2-difluoro-N-[(2R,3S)-2-(2-metoxi-4-piridil)-1-[1-(1-metil-6-oxo-3-piridil)indazol-5-il]-5-oxo-pirrolidin-3-il]propanamida
43 2,2-difluoro-N-[(2R, 3S)-1-[1-(1-metil-6-oxo-3-piridilo)indazol-5-il]-2-(o-tolilo)-5-oxo-pirrolidin-3-ilo]propanamida
44 N-[(2R,3S)-1-[1-(1-metil-6-oxo-3-piridil)indazol-5-il]-5-oxo-2-fenil-pirrolidin-3-ilo]tiazol-4-carboxamida 45 1-metilo-N-[(2R,3S)-1-[1-(1-metil-6-oxo-3-piridilo)indazol-5-il]-5-oxo-2-fenil-pirrolidin-3-ilo]pirazol-3-carboxamida
46 2,2-difluoro-N-[rac-(2R,3S)-1-[1-(1-metilpirazol-4-il)indazol-5-il]-5-oxo-2-fenil-pirrolidin-3-ilo]propanamida
47 2,2-difluoro-N-[rac-(2R,3S)-1-[1-(5-fluoropirimidin-2-il)indazol-5-il]-5-oxo-2-fenil-pirrolidin-3-ilo]propanamida
48 (R)-N-[(2R,3S)-1-[1-(1-metil-6-oxo-3-piridil)indazol-5-il]-5-oxo-2-fenil-pirrolidin-3-ilo]-tetrahidrofurano-2-carboxamida
49 N-[(2R,3S)-1-[1-(1-metil-6-oxo-3-piridil)indazol-5-yl]-5-oxo-2-fenilo-pirrolidin-3-ilo]oxazol-2-carboxamida 50 N-[(2R,3S)-1-[l-(l-metil-6-oxo-3-piridil)indazol-5-il]-5-oxo-2-fenil-pirrolidin-3-il]oxazol-4-carboxamida 51 5-metil-N-[(2R,3S)-1-[1-(1-metil-6-oxo-3-piridilo)indazol-5-il]-5-oxo-2-fenil-pirrolidin-3-il]-1,2,4-oxadiazol-3-carboxamida
en la forma del compuesto libre o una sal fisiológicamente aceptable del mismo.
12. Una forma de dosificación farmacéutica que comprende un compuesto según cualquiera de las reivindicaciones 1 a 11.
13. El compuesto según cualquiera de las reivindicaciones 1 a 11 para su uso en el tratamiento y/o profilaxis del dolor y/o la inflamación.
14. El compuesto según la reivindicación 13, para su uso en el tratamiento y/o la profilaxis del asma, la artritis reumatoidea, la enfermedad del intestino irritable, la enfermedad pulmonar obstructiva crónica, el síndrome de insuficiencia respiratoria aguda, la fibrosis quística, la osteoartritis, la polimialgia reumática, la arteritis de células gigantes, el síndrome de Sjogren, la distrofia muscular de Duchenne, la vasculitis, la enfermedad de Behget, la colitis ulcerativa y/o la enfermedad de Crohn.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP17208175 | 2017-12-18 | ||
| PCT/EP2018/085383 WO2019121606A1 (en) | 2017-12-18 | 2018-12-18 | Substituted pyrrolidine amides i |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2925198T3 true ES2925198T3 (es) | 2022-10-14 |
Family
ID=60673845
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES18816101T Active ES2925198T3 (es) | 2017-12-18 | 2018-12-18 | Amidas de pirrolidinas sustituidas I |
Country Status (14)
| Country | Link |
|---|---|
| US (1) | US10626106B2 (es) |
| EP (1) | EP3728219B1 (es) |
| JP (1) | JP2021506964A (es) |
| CN (1) | CN111556867A (es) |
| AR (1) | AR113967A1 (es) |
| AU (1) | AU2018387793A1 (es) |
| BR (1) | BR112020012217A2 (es) |
| CA (1) | CA3085874A1 (es) |
| EA (1) | EA202091429A1 (es) |
| ES (1) | ES2925198T3 (es) |
| IL (1) | IL275374A (es) |
| MX (1) | MX2020006348A (es) |
| TW (1) | TW201927770A (es) |
| WO (1) | WO2019121606A1 (es) |
Families Citing this family (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| TW201927769A (zh) * | 2017-12-18 | 2019-07-16 | 德商歌林達有限公司 | 經取代之吡咯啶醯胺ii |
| BR112021013557A2 (pt) | 2019-01-11 | 2021-09-21 | Grünenthal GmbH | Amidas de pirrolidina substituídas iii |
| WO2020254551A1 (en) * | 2019-06-19 | 2020-12-24 | Grünenthal GmbH | Substituted pyrrolidine amides iv |
| WO2020254552A2 (en) * | 2019-06-19 | 2020-12-24 | Grünenthal GmbH | Substituted pyrrolidine amides v |
| WO2022008705A1 (en) | 2020-07-09 | 2022-01-13 | Grünenthal GmbH | Substituted pyrrolidine amines and amides as mediator of the glucocortoid receptor |
| WO2022081725A1 (en) * | 2020-10-16 | 2022-04-21 | The Scripps Research Institute | Therapeutic uses of glucocorticoids with anabolic effects in skeletal muscle |
Family Cites Families (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7388953B2 (en) | 1999-09-24 | 2008-06-17 | Verizon Business Global Llc | Method and system for providing intelligent network control services in IP telephony |
| JP2008535884A (ja) | 2005-04-14 | 2008-09-04 | グラクソ グループ リミテッド | グルココルチコイド受容体リガンドとしてのインダゾール |
| MX2008013411A (es) | 2006-04-20 | 2008-11-04 | Glaxo Group Ltd | Nuevos compuestos. |
| GB0620385D0 (en) | 2006-10-13 | 2006-11-22 | Glaxo Group Ltd | Novel compounds |
| TW200829578A (en) * | 2006-11-23 | 2008-07-16 | Astrazeneca Ab | Chemical compounds 537 |
| JO2754B1 (en) | 2006-12-21 | 2014-03-15 | استرازينكا ايه بي | Amylendazoleil derivatives for the treatment of glucocorticoid-mediated disorders |
| JP2009084273A (ja) | 2007-09-13 | 2009-04-23 | Santen Pharmaceut Co Ltd | 1,3,3−トリメチル−7−フェニル−3,4−ジヒドロ−1h−キノキサリン−2−オン誘導体からなるグルココルチコイド受容体アゴニスト |
| UY31832A (es) | 2008-05-20 | 2010-01-05 | Astrazeneca Ab | Derivados de indazol sustituidos con fenilo y benzodioxinilo |
| EP3087067B1 (en) | 2013-12-26 | 2018-10-24 | Takeda Pharmaceutical Company Limited | 4-(piperrazin-1-yl)-pyrrolidin-2-one compounds as monoacylglycerol lipase (magl) inhibitors |
| ES2751640T3 (es) | 2014-09-26 | 2020-04-01 | Astrazeneca Ab | Compuestos de 1-alquil-6-oxo-1,6-dihidropiridin-3-ilo y uso como moduladores de SGRM |
| CN107922356A (zh) | 2015-08-25 | 2018-04-17 | 参天制药株式会社 | [4‑(1,3,3‑三甲基‑2‑氧代‑3,4‑二氢‑1h‑喹喔啉‑7‑基)苯氧基]乙基氧基化合物或其盐 |
| TW201927769A (zh) | 2017-12-18 | 2019-07-16 | 德商歌林達有限公司 | 經取代之吡咯啶醯胺ii |
-
2018
- 2018-12-18 TW TW107145626A patent/TW201927770A/zh unknown
- 2018-12-18 EA EA202091429A patent/EA202091429A1/ru unknown
- 2018-12-18 EP EP18816101.2A patent/EP3728219B1/en active Active
- 2018-12-18 AR ARP180103705A patent/AR113967A1/es unknown
- 2018-12-18 BR BR112020012217-3A patent/BR112020012217A2/pt not_active Application Discontinuation
- 2018-12-18 US US16/223,845 patent/US10626106B2/en active Active
- 2018-12-18 CN CN201880081532.9A patent/CN111556867A/zh active Pending
- 2018-12-18 JP JP2020552140A patent/JP2021506964A/ja not_active Withdrawn
- 2018-12-18 MX MX2020006348A patent/MX2020006348A/es unknown
- 2018-12-18 CA CA3085874A patent/CA3085874A1/en active Pending
- 2018-12-18 ES ES18816101T patent/ES2925198T3/es active Active
- 2018-12-18 WO PCT/EP2018/085383 patent/WO2019121606A1/en not_active Ceased
- 2018-12-18 AU AU2018387793A patent/AU2018387793A1/en not_active Abandoned
-
2020
- 2020-06-15 IL IL275374A patent/IL275374A/en unknown
Also Published As
| Publication number | Publication date |
|---|---|
| CN111556867A (zh) | 2020-08-18 |
| EP3728219B1 (en) | 2022-04-13 |
| TW201927770A (zh) | 2019-07-16 |
| AU2018387793A1 (en) | 2020-07-30 |
| EA202091429A1 (ru) | 2020-10-27 |
| MX2020006348A (es) | 2020-09-03 |
| BR112020012217A2 (pt) | 2020-11-24 |
| IL275374A (en) | 2020-07-30 |
| US10626106B2 (en) | 2020-04-21 |
| AR113967A1 (es) | 2020-07-01 |
| US20190185455A1 (en) | 2019-06-20 |
| JP2021506964A (ja) | 2021-02-22 |
| WO2019121606A1 (en) | 2019-06-27 |
| EP3728219A1 (en) | 2020-10-28 |
| CA3085874A1 (en) | 2019-06-27 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2925198T3 (es) | Amidas de pirrolidinas sustituidas I | |
| ES2920359T3 (es) | Amidas de pirrolidinas sustituidas II | |
| KR101919680B1 (ko) | 신규한 피롤리딘 화합물 및 멜라노코르틴 수용체 아고니스트로서의 용도 | |
| BR112021013557A2 (pt) | Amidas de pirrolidina substituídas iii | |
| TWI911262B (zh) | 化合物、組合物及方法 | |
| WO2010058846A1 (ja) | 4,6-ジアミノニコチンアミド化合物 | |
| CA2975196A1 (en) | 1-(het)arylsulfonyl-(pyrrolidine or piperidine)-2-carboxamide derivatives and their use as trpa1 antagonists | |
| CA2917193A1 (en) | Substituted heterocyclic sulfonamide compounds useful as trpa1 modulators | |
| CN102177136A (zh) | 吡咯烷n-苄基衍生物 | |
| US12331034B2 (en) | Substituted pyrrolidine amides V | |
| CN110072859A (zh) | 作为RORγ调节剂的化合物 | |
| JP2019531347A (ja) | ウレア及びフェニルで置換された6員環式アミンまたはラクタム | |
| ES2946888T3 (es) | Pirrolidina-amidas sustituidas IV | |
| CN114269736A (zh) | 一种酰胺化合物、药物组合物及其应用 | |
| ES3057363T3 (en) | Substituted pyrrolidine amines and amides as mediator of the glucocortoid receptor | |
| EA044347B1 (ru) | Замещенные амиды пирролидина iii | |
| CN117545752A (zh) | Cereblon结合化合物、其组合物及其用于治疗的方法 |