ES2925365T3 - Método para producir derivados de diarilpiridina - Google Patents
Método para producir derivados de diarilpiridina Download PDFInfo
- Publication number
- ES2925365T3 ES2925365T3 ES19831009T ES19831009T ES2925365T3 ES 2925365 T3 ES2925365 T3 ES 2925365T3 ES 19831009 T ES19831009 T ES 19831009T ES 19831009 T ES19831009 T ES 19831009T ES 2925365 T3 ES2925365 T3 ES 2925365T3
- Authority
- ES
- Spain
- Prior art keywords
- formula
- contents
- added
- compound represented
- salt
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Classifications
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/38—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of noble metals
- B01J23/40—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of noble metals of the platinum group metals
- B01J23/44—Palladium
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B61/00—Other general methods
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D319/00—Heterocyclic compounds containing six-membered rings having two oxygen atoms as the only ring hetero atoms
- C07D319/10—1,4-Dioxanes; Hydrogenated 1,4-dioxanes
- C07D319/12—1,4-Dioxanes; Hydrogenated 1,4-dioxanes not condensed with other rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/12—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/14—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing three or more hetero rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4427—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems
- A61K31/444—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems containing a six-membered ring with nitrogen as a ring heteroatom, e.g. amrinone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Engineering & Computer Science (AREA)
- Materials Engineering (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Life Sciences & Earth Sciences (AREA)
- Medicinal Chemistry (AREA)
- Epidemiology (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Plural Heterocyclic Compounds (AREA)
- Pyridine Compounds (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
La presente invención se refiere a un nuevo método para producir derivados de diarilpiridina, y el objeto de la presente invención es proporcionar un nuevo método industrialmente útil. Los presentes inventores desarrollaron un nuevo método para sintetizar anillos de piridina sin usar paladio, una base fuerte o una reacción a alta temperatura. En particular, desarrollaron con éxito un método que se puede utilizar para sintetizar sales de iminio como productos intermedios, sintetizar cianocompuestos a partir de estas sales de iminio y ciclar los cianocompuestos en piridinas en condiciones de reacción muy suaves. (Traducción automática con Google Translate, sin valor legal)
Description
DESCRIPCIÓN
Método para producir derivados de diarilpiridina
Campo técnico
La presente invención se refiere a un método novedoso para producir derivados de diarilpiridina y, más específicamente, a un método de producción que incluye un método novedoso para sintetizar anillos de piridina sin usar paladio.
Técnica anterior
Se sabe que los derivados de diarilpiridina son útiles como productos farmacéuticos o materiales para producir productos farmacéuticos, y útiles para tratar tumores (Documento de patente 1).
El Documento de patente 1 da a conocer varios derivados de diarilpiridina y un método de producción para estos derivados de diarilpiridina. En el método de producción de derivados de diarilpiridina dado a conocer en este documento se usan derivados de piridina sustituidos con átomos de halógeno como compuestos de partida y se introduce un grupo arilo en el anillo de piridina realizando una reacción de acoplamiento que usa paladio (véase, por ejemplo, el producto intermedio 9a y el producto intermedio 10a en los ejemplos). Sin embargo, los derivados de piridina sustituidos con átomos de halógeno son caros, y el uso repetido de paladio significa que hay que prestar atención al paladio residual en el producto diana.
En los métodos conocidos para sintetizar anillos de piridina sin el uso de paladio, se usa el compuesto A a continuación como compuesto de partida,

El compuesto A se somete a cianatación para sintetizar el compuesto B a continuación,
[Fórmula 2]

y el compuesto B se cicliza haciéndolo reaccionar con un compuesto que tiene un grupo amino tal como amoniaco para sintetizar un anillo de piridina (Documentos no de patente 1,2 y 3).
Sin embargo, se requiere una reacción que usa una base fuerte a temperaturas ultrabajas (Documento no de patente 1) o una reacción a altas temperaturas (Documentos no de patente 3, 4) para sintetizar el compuesto A. Además, la reacción de conversión del compuesto A al compuesto B tiene que realizarse en presencia de una base fuerte (Documentos no de patente 2, 3), la ciclización del compuesto B tiene que realizarse en condiciones de alta temperatura (Documentos no de patente 1, 2), es necesario que esté presente una gran cantidad de base fuerte (Documento no de patente 2) o ambos (Documento no de patente 3). Todo esto es difícil de llevar a cabo.
Documentos de la técnica anterior
Documentos de patente
Documento de patente 1: WO 2016006706 A1
Documento de patente 2: EP 2810937 A1
Documentos no de patente
Documento no de patente 1: Synthesis Issue 05, 1979, 376
Documento no de patente 2: J. Prakt. Chem., 5, 1990, 332
Documento no de patente 3: J. Org. Chem. 60, 1995, 3750
Documento no de patente 4: Org. Proc. Res. Dev., vol. 9, n.° 2, 2005, 141
Sumario de la invención
Problema a resolver mediante la invención
La presente invención se refiere a un método novedoso para producir derivados de diarilpiridina, y el objeto de la presente invención es proporcionar un método novedoso, industrialmente útil, para sintetizar anillos de piridina sin usar paladio, una base fuerte o una reacción a alta temperatura.
Medios para solucionar el problema
La presente invención se define mediante las reivindicaciones independientes. Las reivindicaciones dependientes ilustran realizaciones adicionales de la invención.
(1) Un método de producción que comprende la etapa de hacer reaccionar un compuesto representado mediante la fórmula (I):

con un agente de cloración y dimetilformamida para obtener un compuesto representado mediante la fórmula (II):

o una sal del mismo.
(2) Un método de producción según (1), en el que el agente de cloración es cloruro de oxalilo.
(3) Un método de producción que comprende las etapas de producir un compuesto representado mediante la fórmula (II) usando un método de producción según (1) o (2), y hacer reaccionar el compuesto con un compuesto representado mediante la fórmula (III):

en presencia de una base para obtener un compuesto representado mediante la fórmula (IV):

o una sal del mismo.
(4) Un método de producción según (3), en el que la base es 2,6-lutidina.
(5) Un método de producción que comprende las etapas de producir un compuesto representado mediante la fórmula (IV) o una sal del mismo usando un método de producción según (3) o (4), y hacer reaccionar el compuesto con bencilamina para obtener un compuesto representado mediante la fórmula (V):

o una sal del mismo.
(6) Un método de producción que comprende las etapas de: (i) hacer reaccionar un compuesto representado mediante la fórmula (I’) con un reactivo de Wittig, siendo el reactivo de Wittig capaz de obtener un compuesto en el que un doble enlace está sustituido por un grupo alcoxi;

(ii) hacer reaccionar el compuesto obtenido en la etapa (i) con un agente de cloración y dimetilformamida; (iii) hacer reaccionar el compuesto obtenido en la etapa (ii) con un compuesto representado mediante la fórmula (III) anterior en presencia de una base; y
(iv) hacer reaccionar el compuesto obtenido en la etapa (iii) con bencilamina para obtener un compuesto representado mediante la fórmula (V) anterior o una sal del mismo.
(7) Un método según (6), en el que el reactivo de Wittig en la etapa (i) es Ph3P(Cl)CH2OMe.
(8) Un método de producción según (6) o (7), en el que el agente de cloración en la etapa (ii) es cloruro de oxalilo.
(9) Un método de producción según uno cualquiera de (6) a (8), en el que la base en la etapa (iii) es 2,6-lutidina.
(10) Un método de producción que comprende las etapas de producir un compuesto representado mediante la fórmula (V) o una sal del m ismo usando un método de producción según uno cualquiera de (5) a (9), y hacer reaccionar el compuesto con hidrógeno en disolvente y en presencia de un catalizador de paladio sobre carbono para obtener un compuesto representado mediante la fórmula (VI):

o una sal del mismo.
(11) Un método de producción según (10), en el que el compuesto representado mediante la fórmula (V) una o sal del mismo es una sal de ácido clorhídrico de un compuesto representado mediante la fórmula (V), y el disolvente es 1-propanol o N-metilpirrolidona.
(12) Un método de producción que comprende las etapas de producir un compuesto representado mediante la fórmula (VI) o una sal del mismo usando un método de producción según (10) o (11), y condensar el compuesto con un compuesto representado mediante la fórmula (VII):

o una sal del mismo para obtener un compuesto representado mediante la fórmula (VIII):

o una sal del mismo.
(13) Un método de producción según (12), en el que el compuesto representado mediante la fórmula (VIII) o una sal del mismo es una sal de ácido sulfúrico de un compuesto representado mediante la fórmula (VIII). Otro aspecto del método de producción de la presente invención comprende las etapas de producir una sal de ácido sulfúrico de un compuesto representado mediante la fórmula (VIII) usando un método de producción según (13), y cristalizar la sal para obtener cristales de la sal de ácido sulfúrico del compuesto representado mediante la fórmula (VIII).
En la presente invención, los cristales de la sal de ácido sulfúrico del compuesto representado mediante la fórmula (VIII) tienen al menos cinco picos a un ángulo de difracción (20) seleccionado de 3,71 ± 0,2, 6,48 ± 0,2, 7,37 ± 0,2, 9,80 ± 0,2, 10,29 ± 0,2, 11,01 ± 0,2, 18,44 ± 0,2, 20,53 ± 0,2, 22,91 ± 0,2 y 24,15 ± 0,2 en difracción de rayos X de polvo usando radiación CuKa.
Efectos de la invención
La presente invención proporciona un método novedoso para sintetizar anillos de piridina sin usar paladio, una base fuerte o una reacción a alta temperatura. En particular, los presentes inventores descubrieron un método que puede usarse para sintetizar sales de iminio, que solía requerir una reacción que usaba una base fuerte a temperaturas ultrabajas o una reacción a altas temperaturas para sintetizar, en condiciones de reacción muy suaves, y fueron capaces de proporcionar un método de síntesis para la síntesis de sales de iminio en compuestos de ciano y la ciclización a piridinas que no requiere una reacción que use una base fuerte o una reacción en condiciones de alta temperatura como en la técnica anterior.
Breve descripción de los dibujos
[Fig. 1] La Fig. 1 es un diagrama que muestra el gráfico de XRD del compuesto (26) descrito en el ejemplo 7.
Realización de la invención
En la presente invención, el “agente de cloración” puede ser cualquier agente que reaccione con dimetilformamida para producir un reactivo de Vilsmeier. Los ejemplos incluyen cloro, cloruro de oxalilo, cloruro de tionilo y oxicloruro de fósforo. Se prefiere el cloruro de oxalilo.
En la presente invención, la “base” usada para producir un compuesto representado mediante la fórmula (IV) o una sal del mismo puede ser cualquier base capaz de extraer el protón en la posición bencilo en un compuesto representado mediante la fórmula (III). Los ejemplos incluyen 1,8-diazabiciclo[5.4.0]undeca-7-eno (DBU), N,N-diisopropiletilamina (DIPEA), 1,4-diazabiciclo[2.2.2]octano (DABCO), trietilamina, piridina, 2,6-lutidina, N-metilmorfolina y tetrametiletilendiamina (TMEDA). Se prefieren 2,6-lutidina y TMEDA, y se prefiere especialmente 2,6-lutidina.
El “catalizador de paladio sobre carbono” que puede usarse en la presente invención puede ser cualquier catalizador de paladio sobre carbono que pueda usarse en una reacción desbencilación y en una reacción de reducción de un grupo nitro a un grupo amino. Los ejemplos incluyen M (Kawaken Fine Chemicals), PH (Kawaken Fine Chemicals), tipo PE (NE Chemcat) y tipo AER (NE Chemcat). Se prefiere el tipo PE (NE Chemcat).
En la presente invención, el “disolvente” que puede usarse en la reacción que usa el paladio sobre carbono puede ser, por ejemplo, metanol, etanol, 1-propanol, 1,3-dimetil-2-imidazolidinona o N-metilpirrolidona. Se prefiere 1-propanol o N-metilpirrolidona.
En la presente invención, “reactivo de Wittig” significa un reactivo que puede reaccionar con aldehídos y cetonas para formar dobles enlaces carbono-carbono. Se usa un reactivo de Wittig capaz de obtener un compuesto en el que un doble enlace está sustituido por un grupo alcoxi. Los ejemplos incluyen Ph3P(Cl)CH2OMe, PhmP(Br)CH2OMe y Ph3 P(I)CH2OMe.
En la presente invención, los compuestos representados mediante la fórmula (I), los compuestos representados mediante la fórmula (II) o las sales de los mismos, y los compuestos representados mediante la fórmula (IV) o las sales de los mismos incluyen isómeros geométricos.
En la presente invención, los compuestos representados mediante la fórmula (II), los compuestos representados mediante la fórmula (IV), los compuestos representados mediante la fórmula (V), los compuestos representados mediante la fórmula (VI), los compuestos representados mediante la fórmula (VII) y los compuestos representados mediante la fórmula (VIII) pueden convertirse en sales haciéndolos reaccionar con un ácido. Los ejemplos incluyen halohidratos tales como fluorhidrato, clorhidratos, bromhidratos y yodohidratos; sales de ácido inorgánico tales como nitratos, percloratos, sulfatos y fosfatos; sulfonatos de alquilo C1-C6 tales como metanosulfonato, trifluorometanosulfonato y etanosulfonato; sulfonatos de alilo tales como bencenosulfonato y p-toluenosulfonato; sales de ácido orgánico tales como acetatos, malatos, fumaratos, succinatos, citratos, ascorbatos, tartratos, oxalatos y adipatos; y sales de aminoácido tales como sales de glicina, sales de lisina, sales de arginina, sales de ornitina, glutamatos y aspartatos.
Las “sales” en la presente invención pueden ser moléculas formadas a través de enlaces iónicos así como moléculas formadas a través de enlaces hidrógeno y/o enlaces de van der Waals.
En la presente invención, los compuestos representados mediante la fórmula (I), los compuestos representados mediante la fórmula (I’), los compuestos representados mediante la fórmula (II), los compuestos representados mediante la fórmula (III), los compuestos representados mediante la fórmula (IV) o las sales de los mismos, los compuestos representados mediante la fórmula (V) o las sales de los mismos, los compuestos representados mediante la fórmula (VI) o las sales de los mismos, los compuestos representados mediante la fórmula (VII) o las sales de los mismos, y los compuestos representados mediante la fórmula (VIII) o los sales de los mismos pueden dejarse en el aire o recristalizarse para absorber moléculas de agua y convertirse en hidratos. Estos hidratos también se incluyen en la presente invención.
En la presente invención, los compuestos representados mediante la fórmula (I), los compuestos representados mediante la fórmula (I’), los compuestos representados mediante la fórmula (II), los compuestos representados mediante la fórmula (III), los compuestos representados mediante la fórmula (IV) las o sales de los mismos, los compuestos representados mediante la fórmula (V) o las sales de los mismos, los compuestos representados mediante la fórmula (VI) o las sales de los mismos, los compuestos representados mediante la fórmula (VII) o las sales de los mismos, y los compuestos representados mediante la fórmula (VIII) o las sales de los mismos pueden dejarse en un disolvente o recristalizarse para absorber un disolvente y convertirse en solvatos. Estos solvatos también se incluyen en la presente invención.
En la presente invención, “cristal” se refiere a un sólido cuya estructura interna está formada tridimensionalmente por una repetición regular de átomos y moléculas constituyentes, y que se distingue de un sólido amorfo o un cuerpo amorfo que no tiene una estructura interna regular de este tipo.
En la presente invención, la forma cristalina de un compuesto representado mediante la fórmula (VIII) o una sal del mismo puede verificarse mediante observación bajo un microscopio polarizante o realizando un análisis de cristal de rayos X de polvo o una única medición de difracción de rayos X de cristal. El tipo de cristal puede identificarse comparando las características del cristal con datos basados en índices medidos por adelantado. En un aspecto preferido de la presente invención, puede confirmarse que un cristal de la presente invención es un cristal usando tales medios de medición.
En la presente invención, los cristales que tienen ángulos de difracción completamente coincidentes en difracción de rayos X de polvo y los cristales que tienen ángulos de difracción coincidentes dentro de un intervalo de ± 0,2 se incluyen en la presente invención. Esta es una práctica común ya que hay variaciones en los valores pico debido a diferencias en instrumentos, muestras y la preparación de muestras. Dado que los ángulos de difracción (20) en la difracción de rayos X de polvo pueden tener un error dentro del intervalo de ± 0,2, es necesario entender los valores de ángulo de difracción como que incluyen valores numéricos dentro del intervalo de aproximadamente ± 0,2.
Lo siguiente es una descripción de la presente invención. Debe entenderse que las condiciones de reacción de la presente invención no están limitadas a aquellas en los siguientes ejemplos. En la presente invención, los grupos funcionales en los compuestos pueden estar protegidos mediante un grupo protector adecuado. Los ejemplos de grupos funcionales incluyen grupos hidroxilo, grupos carboxi y grupos amino. Para los tipos de grupos protectores y las condiciones para introducir y eliminar estos grupos protectores, véase Protective Groups in Organic Synthesis (T. W. Green y P.G.M. Wuts, John Wiley & Sons, Inc., Nueva York, 2006).
Ejemplos
Lo siguiente es una descripción más detallada de la presente invención con referencia a ejemplos, pero el alcance de la presente invención no está limitado a estos ejemplos.
Las abreviaturas usadas en los ejemplos tienen los siguientes significados. mg: miligramo, g: gramo, kg: kilogramo, ml: mililitro, l: litro, mol: mol, MHz: megahercio. En los ejemplos a continuación, el valor de desplazamiento químico en el espectro de resonancia magnética nuclear (a continuación en el presente documento, 1H RMN: 500 MHz) se describe como un valor 5 (ppm) usando tetrametilsilano como sustancia de referencia. En el patrón de división, s indica una línea simple, d indica una línea doble, t indica una línea triple, q indica un línea cuádruple, m indica una línea múltiple y a indica una línea ancha.
Las condiciones de medición para el difractómetro de rayos X de polvo usados en los ejemplos son tal como sigue. Intervalo de medición: 3-40 grados
Escalón: 0,020 grados
Velocidad: 10 grados/min
Diana: Cu (Ka)
Voltaje del tubo: 40 kV
Corriente del tubo: 15 mA
Temperatura de medición: Temperatura ambiente (25°C)
(Ejemplo de referencia 1)
Producción de (5-metilpiridin-2-il)acetato de potasio (2)

Bajo una atmósfera de nitrógeno, se añadieron agua (25,7 l) e hidrocloruro de acetato de (5-metilpiridin-2-ilo) (1) (19,0 kg, 101 mol) a un recipiente de reacción y se agitó a 0°C. Tras añadir una disolución acuosa de hidróxido de potasio al 48% (23,24 kg) mientras se mantenía la temperatura entre -5°C y 10°C, se ajustó el pH a 12,6 con ácido clorhídrico
concentrado (0,15 kg). Tras confirmar la precipitación de cloruro de potasio, se agitaron los contenidos a 0°C durante 20 minutos y se añadió gota a gota 1-propanol (143 l). Tras completarse la adición gota a gota, se elevó la temperatura hasta 25°C, y los contenidos se agitaron durante 15 minutos y se concentraron a presión reducida hasta que el volumen de líquido alcanzó 66,5 l. Se añadió gota a gota 1-propanol (143 l) a 20°C y se concentraron de nuevo los contenidos a presión reducida hasta que el volumen de líquido alcanzó 66,5 l. Entonces, se añadió gota a gota 1-propanol (143 l) a 25°C, se realizó una filtración caliente a 50°C y se lavaron los contenidos con 1 -propanol (57 l) a 50°C para eliminar la materia insoluble. El filtrado resultante se concentró a presión reducida hasta que el volumen de líquido alcanzó 95 l y se añadió gota a gota una disolución acuosa de 1-propanol (1-propanol 19 l, agua 1,9 l) a 40°C. Entonces, se concentraron los contenidos a presión reducida hasta que el volumen de líquido alcanzó 38 l y se añadió gota a gota acetato de propilo (181 l) a 40°C. Tras agitar a 25°C durante 18 horas, el sólido precipitado se recogió mediante filtración, se lavó con una disolución de acetato de propilo/1-propanol (acetato de propilo 51,3 l, 1-propanol 5,7 l) y se secó a presión reducida a 40°C para obtener el compuesto diana (2) (18,15 kg, rendimiento del 94,7%) como un sólido.
1H RMN (500 MHz, DMSO-d6): 5 = 2,21 (s, 3H), 3,25 (d, J = 3,5 Hz, 2H), 7,16 (d, J = 8,0 Hz, 1H), 7,38 (d, J = 8,0 Hz, 1H), 8,16 (s, 1H).
(Ejemplo de referencia 2)
Producción de (5-metilpiridin-2-il)acetato de potasio (2)

Se añadieron tolueno (315 ml), 2-fluoro-4-metilpiridina (3) (45 g, 405 mmol) y acetonitrilo (20,0 g, 487 mmol) a un recipiente de reacción bajo una atmósfera de nitrógeno, y se agitaron los contenidos a 0°C. Se añadió lentamente gota a gota un disolución en tolueno de hexametildisilazida de potasio (0,5 mol/l, 1,78 l, 890 mmol) mientras se mantenía la temperatura entre 0°C y 10°C. T ras agitar a 25°C durante una hora, se agitaron los contenidos a 50°C durante otras seis horas. Tras enfriar hasta 5°C, se añadió agua (450 ml) y se agitaron los contenidos durante 30 minutos. Se separó el líquido a 5°C y se desechó la fase acuosa. Se añadió una disolución de ácido clorhídrico acuosa 2 N (522 ml) a la fase orgánica para ajustar el pH a 2,4. Tras elevar la temperatura hasta 25°C y agitar durante 15 minutos, se ajustó el pH a 8,7 usando una disolución de hidróxido de sodio acuosa 4 N. Tras agitar a 25°C durante 15 minutos, se desechó la fase acuosa y se añadieron 135 ml de ácido clorhídrico concentrado a 5°C. Se agitaron los contenidos a 25°C durante 15 minutos y se desechó la fase orgánica. La fase acuosa se calentó hasta 80°C y se agitó durante cuatro horas. Tras añadir disolución acuosa de hidróxido de potasio al 48% (204 g) mientras se mantenía la temperatura entre -5°C y 25°C, se agitaron los contenidos a 25°C durante 45 minutos. A continuación, se añadió 1-propanol (450 ml) y se concentró a presión reducida hasta que el volumen de líquido alcanzó 225 ml. Se añadió una disolución de hidróxido de potasio acuosa al 48% (11,8 g) para ajustar el pH a 11,8, 1-propanol (450 ml), y se concentraron los contenidos a presión reducida hasta que el volumen de líquido alcanzó 225 ml. Entonces, se añadió 1-propanol (450 ml), se concentraron los contenidos a presión reducida hasta que el volumen de líquido alcanzó 338 ml y se añadió 1-propanol (338 ml). Se elevó la temperatura hasta 50°C, se realizó una filtración caliente a la misma temperatura y se lavaron los contenidos con 1-propanol (135 ml) a 50°C para eliminar la materia insoluble. Tras enfriar hasta 25°C y confirmar la cristalización, se concentraron los contenidos hasta que el volumen de líquido alcanzó 135 ml. Entonces, se añadieron gota a gota 585 ml de acetato de propilo a 50°C. Tras agitar a 25°C durante ocho horas, el sólido precipitado se recogió mediante filtración, se lavó con disolución de acetato de propilo/1-propanol (acetato de propilo 122 ml, 1-propanol 13,5 ml) y se secó a presión reducida a 40°C para obtener el compuesto diana (2) (68,9 g, rendimiento del 89,9%) como un sólido.
(Ejemplo de referencia 3)
Producción de (5-metilpiridin-2-il)acetato de potasio (2)

(Etapa 1) Producción de (5-metilpiridin-2-il)propanodinitrilo (6)
Bajo una atmósfera de nitrógeno, se añadieron N-metilpirrolidona (7 l) y malononitrilo (797 g, 12,1 mol) a un recipiente de reacción, y se añadió terc-butóxido de sodio (2,64 kg, 27,4 mol) en cuatro porciones. A continuación, se añadió 2-cloro-4-metilpiridina (5) (1,40 kg, 11,0 mol) y se redujo la concentración de oxígeno en la disolución mediante aeración con nitrógeno. Se añadió dicloruro de bis(trifenilfosfina)paladio (77,0 g, 0,11 mol), y se agitaron los contenidos a 55°C durante 1,5 horas y entonces a 80°C durante una hora. T ras enfriar hasta 50°C, se añadió gota a gota una disolución de ácido acético acuosa (1,0 kg de ácido acético, 2,1 kg de agua) a la misma temperatura y se añadió más agua (18,9 kg) mientras se agitaba. Tras enfriar hasta 25°C, se ajustó el pH a 5,6 con una disolución de ácido clorhídrico acuosa 6 N (0,95 kg). Tras agitar durante otra hora, el sólido precipitado se recogió mediante filtración, se lavó con disolución acuosa de N-metilpirrolidona (1,4 l de N-metilpirrolidona, 4,2 l de agua) y entonces con agua (5,6 l), y se secó a presión reducida a 40°C para obtener el compuesto diana (6) (1,67 kg, rendimiento del 96,7%) como un sólido.
1H RMN (500 MHz, DMSO-ds): 5 = 2,15 (s, 3H), 7,04 (d, J = 9,0 Hz, 1H), 7,63-7,66 (m, 2H), 12,85 (s. a., 1H).
(Etapa 2) Producción de (5-metilpiridin-2-il)acetato de potasio (2)
Bajo una atmósfera de nitrógeno, se agitó ácido clorhídrico concentrado (150 ml) a 40°C y se añadió (5-metilpiridin-2-il)propanodinitrilo (6) (50 g, 318 mmol) en cinco porciones cada hora. Tras completarse la adición, se agitaron los contenidos a 40°C durante una hora y entonces a 80°C durante 1,5 horas. Se añadió una disolución acuosa de hidróxido de potasio al 48% (241,7 g) mientras se mantenía la temperatura entre -5°C y 10°C, se elevó la temperatura hasta 25°C y entonces se añadió agua (50 ml). Tras elevar la temperatura hasta 50°C, se realizó una filtración en caliente y se lavaron los contenidos con agua (75 ml) a 50°C para eliminar la materia insoluble. La disolución resultante se concentró a presión reducida hasta que el volumen de líquido alcanzó 250 ml y se añadió gota a gota 1-propanol (500 ml) a de 45 a 50°C. Tras ajustar el pH a 12,6 con ácido clorhídrico concentrado (12,4 ml), se concentraron los contenidos a presión reducida hasta que el volumen de líquido alcanzó 250 ml y se añadió gota a gota 1-propanol (500 ml) a de 45 a 50°C. Se concentraron de nuevo los contenidos a presión reducida hasta que el volumen de líquido alcanzó 250 ml y se añadió gota a gota 1 -propanol (500 ml) a de 45 a 50°C. Se realizó una filtración caliente a 50°C y se lavaron los contenidos con 1-propanol (150 ml) a 50°C para eliminar la materia insoluble. Se añadió agua (50 ml) al filtrado y se concentraron los contenidos a presión reducida hasta que el volumen alcanzó 200 ml. Se añadió acetato de propilo (400 ml) a 40°C y se concentraron los contenidos a presión reducida hasta que el volumen de líquido alcanzó 200 ml. Una vez más, se añadió acetato de propilo (400 ml) a 40°C y se concentraron los contenidos a presión reducida hasta que el volumen de líquido alcanzó 200 ml. El concentrado se enfrío hasta 25°C y se agitó durante otra hora, y el sólido precipitado se recogió mediante filtración, se lavó con una disolución de acetato de propilo/1-propanol (135 ml de acetato de propilo, 15 ml de 1 -propanol) y se secó a 40°C a presión reducida para obtener el compuesto diana (2) (57,05 g, rendimiento del 94,8%) como un sólido.
(Ejemplo de referencia 4)
Producción de carboxilato de 5-metil-4’-oxo-1’-(tetrahidro-2H-piran-4-ilmetil)-1’,4’-dihidro-2,3’-bipiridin-5’-etilo (9)
[Fórmula 6]
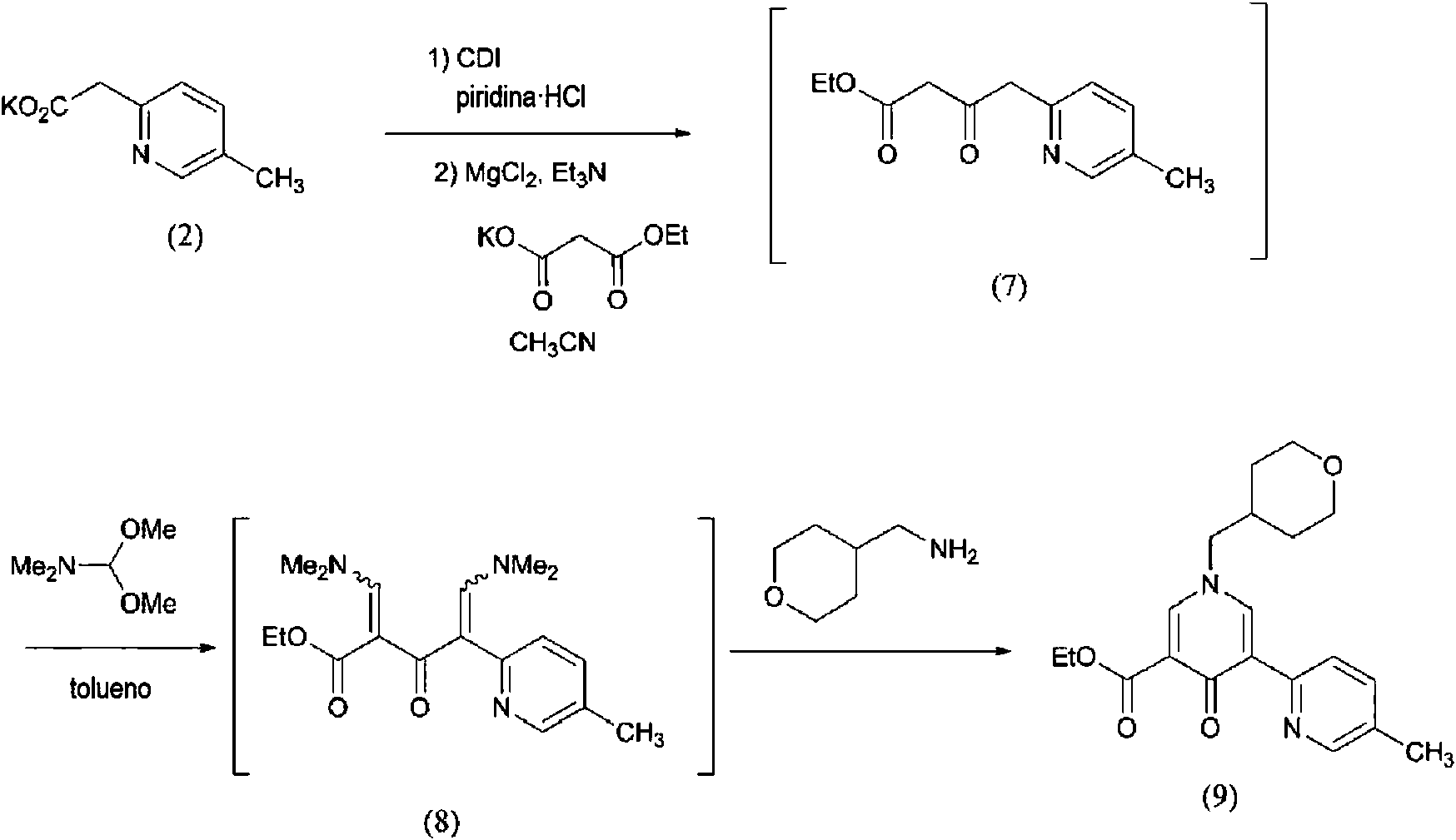
Bajo una atmósfera de nitrógeno, se añadieron acetonitrilo (257 l) y (5-metilpiridin-2-il)potasio-acetato de potasio (2) (17,15 kg, 90,6 mol) a un recipiente de reacción y se calentaron los contenidos a reflujo a 82°C durante dos horas. Se concentraron los contenidos a presión atmosférica hasta que el volumen de líquido alcanzó 86 l y se añadió acetonitrilo (171,5 l) a 25°C. Se añadió hidrocloruro de piridina (12,57 kg, 109 mol) a 0°C y se agitó durante 30 minutos, y se añadió carbonildiimidazol (16,16 kg, 100 mol) a 0°C y se agitó durante 40 minutos. A continuación, se añadieron etilmalonato de potasio (23,14 kg, 136 mol) y trietilamina (18,34 kg, 181 mol) a 0°C, y se añadió cloruro de magnesio (12,08 kg, 127 mol) en diez porciones. T ras agitar a 0°C durante una hora, se agitaron los contenidos a 55°C durante otra hora. Se añadieron tolueno (137,2 l) y agua (51,5 l), y se ajustó el pH a 5,01 con una disolución acuosa de ácido clorhídrico 6 N. Tras desechar la fase acuosa, se lavaron dos veces los contenidos con una disolución de cloruro de sodio acuosa al 10% (51,5 l). Se concentró la fase orgánica a presión reducida hasta que el volumen de líquido alcanzó 69 l, se añadió tolueno (85,8 l) y se concentraron de nuevo los contenidos a presión reducida hasta que el volumen de líquido alcanzó 69 l. Esto se usó como disolución de 4-(5-metilpiridin-2-il)-3-oxobutanoato de etilo (7) en la siguiente reacción. Bajo una atmósfera de nitrógeno, se añadió dimetilacetal de N,N-dimetilformamida (76,8 kg, 645 mol) a otro recipiente de reacción y se ajustó la temperatura a 60°C. La disolución de 4-(5-metilpiridin-2-il)-3-oxobutanoato de etilo (7) se desaló, se filtró y se añadió al recipiente de reacción a 60°C. Los contenidos se lavaron con tolueno (17,2 l) y se agitaron a 60°C durante dos horas. A continuación, se concentraron los contenidos a presión reducida hasta que el volumen de líquido alcanzó 86 l y se repitió cuatro veces la operación de adición de tolueno (85,8 l). Se concentraron de nuevo los contenidos a presión reducida hasta que el volumen de líquido alcanzó 86 l y se añadió 1 -(tetrahidro-2H-piran-4-il)metanamina (10,44 kg, 90,6 mol) a 25°C. Tras agitar a 25°C durante cuatro horas, se añadieron cristales de siembra (9) (2 g). Tras confirmar la cristalización, los contenidos se enfriaron hasta -5°C y se agitaron durante 16 horas. El sólido precipitado se recogió mediante filtración, se lavó con tolueno (51,5 l), se enfrío hasta -5°C y se secó a presión reducida a 40°C para obtener el compuesto diana (9) (17,84 kg, rendimiento del 55,2%) como un sólido.
•Los cristales de siembra (9) se obtuvieron recogiendo algo de la disolución de reacción y concentrándola a presión reducida.
1H RMN (500 MHz, DMSO-ds): 5 = 1,24-1,29 (m, 5H), 1,42-1,45 (m, 2H), 1,99-2,02 (m, 1H), 2,32 (s, 3H), 3,26 (dd, J = 10,0, 10,0 Hz, 2H), 3,85 (dd, J = 11,5, 3,0 Hz, 2H), 4,01 (d, J = 7,5 Hz, 2H), 4,22 (q, J = 7,0 Hz, 2H), 7,63 (dd, J = 7,5, 2,5 Hz, 1H), 8,29 (d, J = 2,5 Hz, 1H), 8,43 (s, 1H), 8,44 (s, 1H), 8,49 (d, J = 2,5 Hz, 1H).
(Ejemplo de referencia 5)
Producción de carboxilato de 5-metil-4’-oxo-1’-(tetrahidro-2H-piran-4-ilmetil)-1’,4’-dihidro-2,3’-bipiridin-5’-etilo (9)
[Fórmula 7]

Bajo una atmósfera de nitrógeno, se añadieron acetonitrilo (97,5 ml) y monohidrocloruro de acetato de (5-metilpiridin-2-ilo) (1) (6,5 g, 0,035 mol) a un recipiente de reacción, se añadió trietilamina (2,74 g, 0,027 mol) a 0°C y se agitaron los contenidos durante 30 minutos, y entonces se añadió carbonildiimidazol (6,18 g, 0,038 mol) a 0°C y se agitaron los contenidos durante una hora. A continuación, se añadieron etilmalonato de potasio (8,84 g, 0,052 mol) y trietilamina (7,01 g, 0,069 mol) a 0°C, y se añadió cloruro de magnesio (4,62 g, 0,049 mol) en diez porciones. Tras agitar a 0°C durante una hora, se agitaron los contenidos a 57°C durante dos horas. Se añadieron tolueno (52 ml) y agua (20 ml), y se ajustó el pH a aproximadamente 5 con una disolución acuosa de ácido clorhídrico 5 N. El líquido se separó a aproximadamente 50°C, se desechó la fase acuosa y se lavaron los contenidos con una disolución de cloruro de sodio acuosa al 10% (20 ml). Entonces, se separó el líquido a 50°C, se desechó la fase acuosa, se enfriaron los contenidos hasta temperatura ambiente, se añadieron 32,5 ml de una disolución acuosa de ácido clorhídrico 2 N y se agitaron los contenidos durante aproximadamente 5 minutos. Tras desechar la fase orgánica separada, se añadieron 32,5 ml de tolueno, se ajustó el pH a aproximadamente 5 con una disolución de cloruro de sodio acuosa al 25% y se agitaron los contenidos durante aproximadamente cinco minutos. Se añadió tolueno (20 ml) a la fase acuosa separada y se agitaron los contenidos durante aproximadamente 5 minutos. Tras desechar la fase acuosa, las fases orgánicas se combinaron y se concentraron a presión reducida hasta que el volumen de líquido alcanzó 26 ml, se añadió tolueno (32,5 ml) y se concentraron de nuevo los contenidos a presión reducida hasta que el volumen de líquido alcanzó 26 ml. Esto se usó como disolución de 4-(5-metilpiridin-2-il)-3-oxobutanoato de etilo (7) en la siguiente reacción.
Bajo una atmósfera de nitrógeno, se añadió dimetilacetal de N,N-dimetilformamida (29,12 g, 0,244 mol) a otro recipiente de reacción y se ajustó la temperatura a 60°C. La disolución de 4-(5-metilpiridin-2-il)-3-oxobutanoato de etilo (7) se desaló, se filtró y se añadió gota a gota al recipiente de reacción a 60°C durante aproximadamente una hora. Los contenidos se lavaron con tolueno (6,5 ml) y se agitaron a 60°C durante dos horas. A continuación, se concentraron los contenidos a presión reducida hasta que el volumen de líquido alcanzó 32,5 ml y se repitió cuatro veces la operación de adición de tolueno (32,5 l). Se concentraron de nuevo los contenidos a presión reducida hasta que el volumen de líquido alcanzó 32,5 ml y se añadió 1-(tetrahidro-2H-piran-4-il)metanamina (3,96 g, 0,034 mol) a 25°C. Tras agitar a 25°C durante cuatro horas, se añadieron cristales de siembra (9) (7 mg). Tras confirmar la cristalización, los contenidos se enfriaron hasta -5°C y se agitaron durante la noche. El sólido precipitado se recogió mediante filtración, se lavó con tolueno (20 ml), se enfrió hasta -5°C y se secó a presión reducida a 40°C para obtener el compuesto diana (9) (5,87 kg, rendimiento del 47,5%) como un sólido.
•Los cristales de siembra (9) se obtuvieron recogiendo algo de la disolución de reacción y concentrándola a presión reducida.
(Ejemplo de referencia 6)
Producción de ácido 5-metil-4’-oxo-1’-(tetrahidro-2H-piran-4-ilmetil)-1’,4’-dihidro-2,3’-bipiridin-5’-carboxílico (10)

Bajo una atmósfera de nitrógeno, se añadieron etanol (50 ml), agua (225 ml), carboxilato de 5-metil-4’-oxo-1’-(tetrahidro-2H-piran-4-ilmetil)-1’,4’-dihidro-2,3’-bipiridin-5’-etilo (9) (50 g, 14,0 mmol) y una disolución acuosa de hidróxido de sodio al 25% (26,9 g, 16,8 mmol) a un recipiente de reacción, y se agitaron los contenidos a 25°C durante dos horas. Se añadieron tolueno (150 ml) y agua (50 ml) y se desechó la fase orgánica. Se añadió etanol (185 ml) a la fase acuosa y se ajustó el pH a 4,8 con una disolución de ácido clorhídrico acuosa 6 N (28 ml). Tras agitar a 25°C durante otros 30 minutos, el sólido precipitado se recogió mediante filtración, se lavó con una disolución de etanol acuosa (75 ml de etanol, 75 ml de agua) y entonces con agua (150 ml), y se secó a presión reducida a 40°C para obtener la diana (10) (42,93 g, rendimiento del 93,1%) como un sólido.
1H RMN (500 MHz, DMSO-d6): 5 = 1,29-1,33 (m, 2H), 1,43-1,45 (m, 2H), 2,06-2,07 (m, 1H), 2,36 (s, 3H), 3,27 (dd, J = 10,0, 9,5 Hz, 2H), 3,84 (dd, J = 11,5, 2,5 Hz, 2H), 4,23 (d, J = 7,5 Hz, 2H), 7,72 (dd, J = 8,0, 2,5 Hz, 1H), 8,38 (d, J = 8,0 Hz, 1H), 8,53 (d, J = 2,5 Hz, 1H), 8,78 (s, 1H), 8,84 (s, 1H).
(Ejemplo de referencia 7)
Producción de (2-fluoro-4-nitrofenil)acetonitrilo (13)
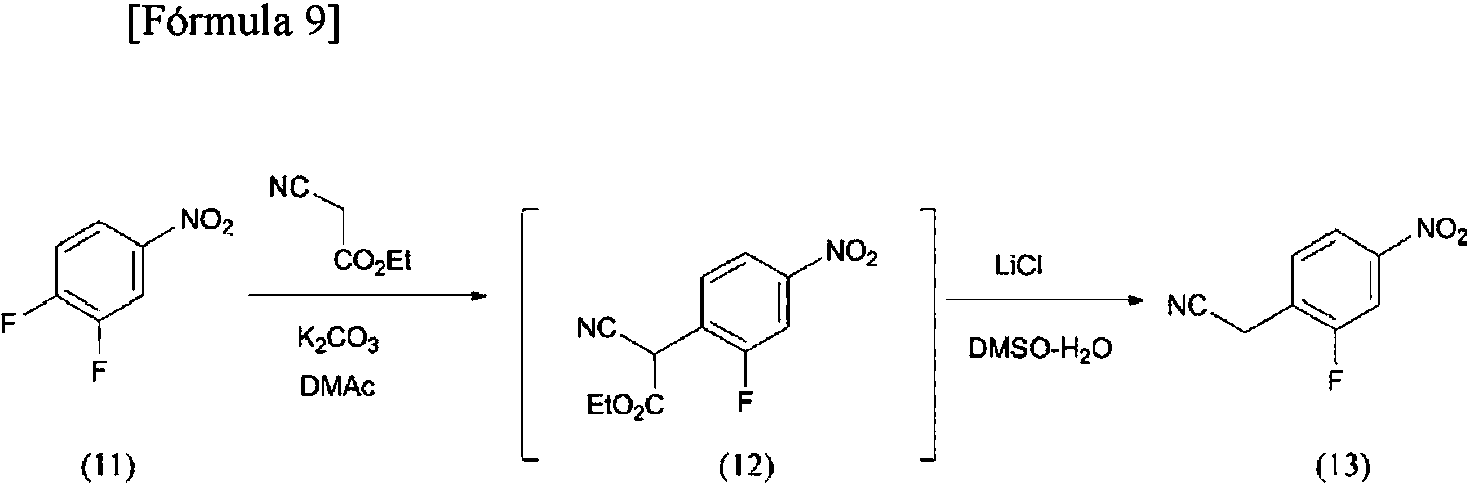
Bajo una atmósfera de nitrógeno, se agitó N,N-dimetilacetamida (5,00 l) a 25°C, se añadieron carbonato de potasio (0,956 kg, 6,92 mol), 1,2-difluoro-4-nitrobenceno (11) (1,00 kg, 6,29 mol) y cianoacetato de etilo (0,782 kg, 6,91 mol), y se elevó la temperatura hasta 90°C. Tras agitar a la misma temperatura durante cuatro horas, se enfriaron los contenidos hasta 25°C. Se añadió gota a gota agua (3,50 l) a la disolución a lo largo de 30 minutos, se añadió gota a gota una disolución acuosa de ácido clorhídrico 2 N (6,91 l) a lo largo de 15 minutos y entonces se añadió acetato de etilo (10,0 l). Tras agitar los contenidos a 25°C durante cinco minutos y permitiéndoles entonces reposar, se desechó la fase acuosa. Se añadieron agua (8,50 l) y cloruro de sodio (1,50 kg) a la fase orgánica, se agitaron los contenidos a 25°C durante 5 minutos y se permitió que reposaran, y entonces se desechó la fase acuosa. La fase orgánica resultante se concentró a presión reducida hasta un volumen de líquido de 3,0 l y se añadió dimetilsulfóxido (2,50 l). Tras concentrar la disolución a presión reducida hasta un volumen de líquido de 4,0 l, se añadió una mezcla de agua (1,50 l) y cloruro de litio (0,400 kg). Tras elevar la temperatura hasta 100°C, se agitaron los contenidos a la misma temperatura durante cinco horas y se enfriaron hasta 50°C. T ras añadir metanol (5,00 l) a la disolución, se añadió gota a gota agua (2,00 l) a lo largo de 10 minutos y entonces se añadieron cristales de siembra (13) (1,00 g) para hacer precipitar cristales. Tras confirmar la precipitación de cristales, se agitaron los contenidos a 50°C durante 30 minutos y se añadió gota a gota agua (2,50 l) a lo largo de una hora. Tras agitar a la misma temperatura durante una hora, se enfriaron los contenidos hasta 25°C a lo largo de otra hora. Entonces, se elevó la temperatura hasta 50°C y se añadió gota a gota agua (4,50 l) a lo largo de 45 minutos. Tras agitar a la misma temperatura durante 30 minutos, se enfriaron los contenidos hasta 25°C a lo largo de una hora. Tras agitar a la misma temperatura durante una hora, el sólido precipitado se recogió mediante filtración, se lavó con una disolución de agua/metanol (3,00 l de agua, 1,00 l de metanol) y se secó a presión reducida a 50°C para obtener el compuesto diana (1,05 kg, rendimiento del 92,8%) como un sólido.
•Los cristales de siembra (13) se obtuvieron recogiendo algo de la disolución de reacción y concentrándola a presión reducida.
1H RMN (500 MHz, CDCla): 5 = 3,89 (s, 2H), 7,71 (dd, J = 8,5, 8,0 Hz 1H), 8,01 (dd, J = 2,0, 8,5 Hz, 1H), 8,12 (dd, J = 8,0, 2,0 Hz, 1H).
(Ejemplo de referencia 8)
Producción de (2R)-1,4-dioxan-2-ilmetil-metanosulfonato (17)
[Fórmula 10]

(Etapa 1) Producción de (2S)-1,4-dioxan-2-ilmetanol (16)
En una atmósfera de nitrógeno, se añadieron 2-cloroetanol (210,0 kg) y trifluoruro de boro-tetrahidrofurano (0,26 kg, 1,86 mol) a un recipiente de reacción, y los contenidos se agitaron y se calentaron hasta 75°C. Se añadió gota a gota (R)-epiclorhidrina (14) (35,00 kg, 378,3 mol) a lo largo de una hora y se añadió xileno (17,5 l). Tras agitar a 75°C durante 1,5 horas, se concentraron los contenidos a presión reducida hasta un volumen de líquido de 70 l. Se añadió xileno (105,0 l) a la disolución y se concentraron de nuevo los contenidos a presión reducida hasta un volumen de líquido de 70 l. La operación de añadir xileno y concentrar a presión reducida se repitió una total de 3 veces. El concentrado resultante se enfrió hasta 15°C y se añadió una disolución acuosa de hidróxido de sodio al 25% (p/p) (302,64 kg, 1,89 kmol). Se elevó la temperatura hasta 65°C, y se agitaron los contenidos a la misma temperatura durante 5,5 horas y entonces se enfriaron hasta 25°C. Se añadió tolueno (105,0 l) a la disolución, se agitaron los contenidos a 25°C durante cinco minutos y se permitió que reposaran, y entonces se desechó la fase orgánica. De nuevo, se añadió tolueno (105,0 l) a la fase acuosa, se agitaron los contenidos a 25°C durante cinco minutos y se permitió que reposaran, y se desechó la fase orgánica. Se añadió ácido clorhídrico concentrado (127,89 kg) a la fase acuosa resultante y se ajustó el pH a 7,2. Se añadió 1-propanol (175,0 l) a la disolución y se concentraron los contenidos a presión reducida hasta un volumen de líquido de 350 l. Se añadió 1-propanol (210,0 l) a la disolución y se concentraron los contenidos a presión reducida hasta un volumen de líquido de 290 l. Se añadió 1-propanol (297,5 l) a la disolución y se concentraron los contenidos a presión reducida hasta un volumen de líquido de 210 l. Se añadió 1-propanol (367,5 l) a la disolución y se concentraron los contenidos a presión reducida hasta un volumen de líquido de 175 l. Se añadió 1-propanol (70,0 l) a la disolución y se concentraron los contenidos a presión reducida hasta un volumen de líquido de 175 l. La suspensión obtenida mediante concentración se filtró usando 1 -propanol (175,0 l) para eliminar las sales inorgánicas precipitadas. El filtrado resultante se concentró a presión reducida hasta un volumen de líquido de 30 l, y se añadió tolueno (52,5 l). Se filtró la suspensión usando tolueno (17,5 l) para eliminar las sales inorgánicas precipitadas. El filtrado resultante se concentró a presión reducida hasta un volumen de líquido de 30 l y se añadió tolueno (70,0 l). Se concentraron adicionalmente los contenidos a presión reducida hasta un volumen de líquido de aproximadamente 20 l para obtener el compuesto diana (16) (22,47 kg, rendimiento del 45,3%).
1H RMN (500 MHz, DMSO-ds): 5 = 3,26 (dd, J = 10,0, 11,5 Hz, 1H), 3,25-3,49 (m, 4H), 3,54 (ddd, J = 2,5, 11,0, 11,5 Hz, 1H), 3,62 (dd, J = 2,5, 11,5 Hz, 1H), 3,68 (dd, J = 3,0, 11,5 Hz, 1H), 3,73 (dd, J = 2,5, 11,0 Hz, 1H), 4,68 (t, J = 5,5Hz, 1H).
(Etapa 2) Producción de (2R)-1,4-dioxan-2-ilmetil-metanosulfonato (17)
El (2S)-1,4-dioxan-2-ilmetanol resultante (16) (22,00 kg, 186,2 mol) se añadió a otro recipiente de reacción. Entonces, se añadieron acetato de etilo (440,0 l), trietilamina (26,57 kg, 262,6 mol) y cloruro de metanosulfonilo (32,64 kg, 284,9 mol) y se agitaron los contenidos a 30°C durante una hora. Se añadió agua (112,2 l) a la disolución, se agitaron los contenidos a 25°C durante 15 minutos y se permitió que reposaran, y entonces se desechó la fase acuosa. La fase orgánica resultante se concentró a presión reducida hasta un volumen de líquido de 40 l. Se añadió metanol (68,2 l) a la disolución y se concentró la disolución a presión reducida hasta un volumen de líquido de 40 l. De nuevo, se añadió metanol (68,2 l) a la disolución y se concentró la disolución a presión reducida hasta un volumen de líquido de 40 l. Se añadió metanol (220,0 l) a la disolución, se enfrió la disolución hasta 5°C y se añadieron cristales de siembra (17) (1 g) para hacer precipitar cristales. Tras la precipitación de cristales, se agitaron los contenidos a 5°C durante una hora.
T ras enfriar hasta -15°C a lo largo de 2 horas, se agitaron los contenidos a la misma temperatura durante 24 horas. El sólido precipitado se recogió mediante filtración, se lavó con metanol (88,0 l), se enfrió hasta -15°C y se secó a presión reducida a 25°C para obtener el compuesto diana (17) (21,19 kg, rendimiento del 63,6%) como un sólido.
•Los cristales de siembra (17) se obtuvieron recogiendo algo de la disolución de reacción y concentrándola a presión reducida.
1H RMN (500 MHz, DMSO-ds): 5 = 3,07 (s, 3H), 3,46 (dd, J = 10,0, 11,5 Hz, 1H), 3,62 (dt, J = 3,0, 11,0 Hz, 1H), 3,70 3,92 (m, 5H), 4,16-4,25 (m, 2H).
(Ejemplo de referencia 9)
Producción de 4-[(2R)-1,4-dioxan-2-ilmetoxi]-3-metoxibenzaldehído (18)

Bajo una atmósfera de nitrógeno, se añadieron N,N-dimetilacetamida (45,5 l), carbonato de sodio (9,96 kg, 93,97 mol) y vainillina (13,00 kg, 85,44 mol), y se agitaron los contenidos a 20°C durante 5 minutos. Se añadió (2R)-1,4-dioxan-2-ilmetil-metanosulfonato (17) (17,60 kg, 89,70 mol) a la suspensión, se elevó la temperatura hasta 120°C y se agitaron los contenidos a la misma temperatura durante 6,5 horas. Tras enfriar hasta 70°C, se añadió gota a gota agua (97,5 l) a lo largo de una hora mientras se mantenía la misma temperatura. Se añadieron cristales de siembra (18) (13 g) a la disolución para hacer precipitar cristales. T ras la precipitación de cristales, se agitaron los contenidos a 70°C durante una hora. Se añadió gota a gota agua (84,5 l) a la suspensión a lo largo de 1,5 horas mientras se mantenía la temperatura a 60°C y se agitaron los contenidos a la misma temperatura durante una hora. Tras enfriar hasta 30°C a lo largo de una hora, se agitaron los contenidos a la misma temperatura durante 30 minutos. Tras enfriar hasta 0°C a lo largo de otras 1,5 horas, se agitaron los contenidos a la misma temperatura durante 15 horas. El sólido precipitado se recogió mediante filtración, se lavó con agua (104,0 l) y se secó a presión reducida a 40°C para obtener el compuesto diana (18) (20,51 kg, rendimiento del 95,2%) como un sólido.
•Los cristales de siembra (18) se obtuvieron recogiendo algo de la disolución de reacción y concentrándola a presión reducida.
1H RMN (500 MHz, DMSO-ds ): 5 = 3,40 (dd, J = 10,0, 11,0 Hz, 1H), 3,47-3,53 (m, 1H), 3,60-3,70 (m, 2H), 3,74-3,79 (m, 1H), 3,80-3,86 (m, 4H), 3,87-3,93 (m, 1H), 4,03-4,10 (m, 2H), 7,19 (d, J = 8,5 Hz, 1H), 7,40 (d, J = 2,0 Hz, 1H), 7,53 (dd, J = 2,0, 8,5 Hz, 1H), 9,84 (s, 1H).
(Ejemplo 1)
Producción de monohidrocloruro de N-bencil-5-{4-[(2R)-1,4-dioxan-2-ilmetoxi]-3-metoxifenil}-3-(2-fluoro-4-nitrofenil)piridin-2-amina (22)
[Fórmula 12]

Bajo una atmósfera de nitrógeno, se añadieron tolueno (475 ml), 4-[(2R)-1,4-dioxan-2-ilmetoxi]-3-metoxibenzaldehído (18) (39,0 g, 0,155 mol) y cloruro de (metoximetil)trifenilfosfonio (58,3 g, 0,17 mol), y se concentraron los contenidos a presión reducida hasta un volumen de líquido de 220 g. La operación de añadir tolueno (136 g) y concentrar los contenidos a presión reducida hasta 135 g se llevó a cabo dos veces. Se añadió tetrahidrofurano (243 g) y se enfriaron los contenidos hasta 0°C. Se añadió gota a gota una disolución de metóxido de sodio/metanol al 28% (13,31 g, 0,185 mol) a la disolución a lo largo de 2 horas y se agitaron los contenidos a la misma temperatura durante 36 horas. Se añadió agua (156,0 ml) a la disolución de reacción, y se agitaron los contenidos a 25°C durante un minuto y se permitió que reposaran. Entonces se desechó la fase acuosa. Se añadieron agua (156,0 ml) y sal de mesa (15,6 g) a la fase orgánica, y se agitaron los contenidos a 25°C durante 5 minutos y se permitió que reposaran. Entonces se desechó la fase acuosa. La fase orgánica resultante se concentró a presión reducida hasta que el efluente alcanzó 477 g. A continuación, se añadió tolueno (169,7 g) y se concentraron los contenidos a presión reducida de nuevo hasta que el efluente alcanzó 166 g. Se añadieron dimetilformamida (33,9 g, 0,464 mol) y tolueno (18 g) a la disolución, y se enfriaron los contenidos hasta aproximadamente 0°C. Entonces, se añadió gota a gota cloruro de oxalilo (27,5 g, 0,217 mol) a lo largo de 20 minutos mientras se mantenía la temperatura a o por debajo de 10°C, se elevó la temperatura hasta 30°C y se agitaron los contenidos a la misma temperatura durante 14 horas. Tras completarse la reacción, se enfrió la temperatura hasta 25°C y se realizó una operación de desaireación cuatro veces. Entonces, se enfrió la temperatura hasta 15°C, se añadió una disolución mixta de N,N-dimetilacetamida (110 g) y (2-fluoro-4-nitrofenil)acetonitrilo (13) (30,6 g, 0,17 mol) a lo largo de 20 minutos, se añadió gota a gota N,N-dimetilacetamida (147 g) a lo largo de 20 minutos y finalmente se añadió gota a gota 2,6-lutidina (49,7 g, 0,464, mol) a lo largo de 15 minutos mientras se mantenía la temperatura a 15°C. Se elevó la temperatura hasta 25°C y se agitaron los contenidos a la misma temperatura durante 17 horas. Se enfrió la disolución de reacción hasta 10°C y se añadió gota a gota bencilamina (49,7 g, 0,464 mol) a lo largo de 15 minutos mientras se mantenía la temperatura a 20°C o menos. Se elevó la temperatura hasta 25°C y se agitaron los contenidos a la misma temperatura durante 9 horas. Tras completarse la reacción, se añadieron agua (546 ml), metilisobutilcetona (468 g) y disolución acuosa de hidróxido de sodio 4 N (39 g), y se agitaron los contenidos a 25°C durante 5 minutos y se permitió que reposaran. Entonces se desechó la fase acuosa. Se añadió agua (585 l) a la fase orgánica, y se agitaron los contenidos a 25°C durante 5 minutos y se permitió que reposaran. Entonces se desechó la fase acuosa. Se añadieron metilisobutilcetona (156 g) y agua (156 g) a la fase orgánica obtenida, se elevó la temperatura hasta 40°C y se añadió gota a gota una disolución acuosa de ácido clorhídrico al 7% (58,5 g) a lo largo de 20 minutos mientras se mantenía la temperatura a 40°C. Se añadieron cristales de siembra (22) (39 mg) a la disolución para hacer precipitar cristales. Tras confirmar la precipitación de cristales, se agitó la suspensión a 40°C durante 30 minutos y se ajustó el pH de la suspensión a 6,67 usando una disolución acuosa de ácido clorhídrico 2 N (39 g). Tras enfriar hasta 25°C, se ajustó el pH de la suspensión
a 6,78 usando una disolución acuosa de ácido clorhídrico 2 N (19,5 g). Tras agitar durante 15 minutos o más, el sólido precipitado se recogió mediante filtración, se lavó con metilisobutilcetona (374 g) y se secó a presión reducida a 40°C para obtener el compuesto diana (22) (62,56 g, rendimiento del 69,5%) como un sólido.
*Los cristales de siembra (22) se obtuvieron recogiendo algo de la disolución de reacción y concentrándola a presión reducida.
1H RMN (500 MHz, DMSO-d6): 5 = 3,39 (dd, J = 11,5, 10 Hz, 1H), 3,49 (dt, J = 3, 11 Hz, 1H), 3,58-3,70 (m, 2H), 3,72 3,78 (m, 1H), 3,79-3,90 (m, 5H), 3,93-4,04 (m, 1H), 5,80 (s, 2H), 7,09 (d, J = 8,5 Hz, 1H), 7,28-7,52 (m, 7H), 7,91 (dd, J = 7,0, 8,0 Hz, 1H), 8,26-8,38 (m, 4H), 8,48 (d, J = 2,5 Hz, 1H), 8,87 (d, J = 1,5 Hz, 1H).
(Ejemplo 2)
Examen de las condiciones de producción para monohidrocloruro de N-bencil-5-{4-[(2R)-1,4-dioxan-2-ilmetoxi]-3-metoxifenil}-3-(2-fluoro-4-nitrofenil)piridin-2-amina (22)

La reacción de ciclización de piridina se examinó usando diversas bases de la misma manera que en el ejemplo 1.
T l 1

(Ejemplo 3)
Producción de monohidrocloruro de 3-(4-amino-2-fluorofenil)-5-{4-[(2R)-1,4-dioxan-2-ilmetoxi]-3-metoxifenil}piridin-2-amina (23)

Bajo una atmósfera de nitrógeno, se añadieron agua (41,7 l), monohidrocloruro de N-bencil-5-{4-[(2R)-1,4-dioxan-2-ilmetoxi]-3-metoxifenil}-3-(2-fluoro-4)-nitrofenil)piridin-2-amina (22) (6,95 kg, 119 mol), 1-propanol (97,3 l) y Pd/C al 5% (TIPO PE, 0,76 kg), y se realizó una sustitución de nitrógeno tres veces a una presión de 0,3 MPaG. Se elevó la temperatura hasta 50°C y se realizó una sustitución de hidrógeno tres veces a una presión de 0,3 MPaG. Entonces, se agitaron los contenidos durante dos horas a una temperatura de 50°C y bajo 0,3 MPaG de presión de hidrógeno. Se elevó la temperatura hasta 70°C, se agitaron los contenidos durante siete horas y se purgó el sistema con nitrógeno. Tras filtrar a la misma temperatura para eliminar el catalizador, se lavaron los contenidos con una disolución de agua/1-propanol (agua 6,3 l, 1-propanol 14,6 l) a 70°C y se enfrió el filtrado resultante hasta 40°C. El filtrado se concentró a presión reducida hasta 52 l mientras se mantenía la temperatura a 40°C y se añadió 1-propanol (69,5 l). El filtrado se concentró a presión reducida hasta que el volumen de líquido alcanzó 52 l una vez más mientras se mantenía la temperatura a 40°C y se añadió 1-propanol (52,1 l). La disolución se elevó hasta 55°C y se agitó durante dos horas. Entonces, los contenidos se enfriaron hasta 25°C y se agitaron durante 18 horas, y el sólido precipitado se recogió mediante filtración, se lavó con 1-propanol (34,8 l) y se secó a presión reducida a 40°C para obtener el compuesto diana (23) (4,97 kg, rendimiento del 90,1%) como un sólido.
1H RMN (500 MHz, DMSO-ds): 5 = 3,43 (dd, J = 10, 11 Hz, 1H), 3,53 (dt, J = 3,0, 11 Hz, 1H), 3,64-3,72 (m, 2H), 3,80 (dd, J = 2,0, 11,5 Hz, 1H), 3,85-3,92 (m, 5H), 3,97-4,04 (m, 2H), 5,86 (a, 1H), 6,51 (dd, J = 2,0, 12,5 Hz, 1H), 6,57 (dd, J = 2,0, 8,5 Hz, 1H), 7,08 (d, J = 8,5 Hz, 1H), 7,17 (t, J = 8,5 Hz, 1H), 7,26 (dd, J = 2,0, 8,5 Hz, 1H), 7,33 (d, J = 2,0 Hz, 1H), 7,65 (a, 2H), 8,17 (d, J = 2,0 Hz, 1H), 8,37 (d, J = 2,0 Hz, 1H).
(Ejemplo 4)
Producción de monohidrocloruro de 3-(4-amino-2-fluorofenil)-5-{4-[(2R)-1,4-dioxan-2-ilmetoxi]-3-metoxifenil}piridin-2-amina (23)
Bajo una atmósfera de nitrógeno, se añadieron monohidrocloruro de N-bencil-5-{4-[(2R)-1,4-dioxan-2-ilmetoxi]-3-metoxifenil}-3-(2-fluoro-4-nitrofenil)piridin-2-amina (22) (40 g, 0,069 mol), N-metilpirrolidona (200 ml) y Pd/C al 5% (TIPO PE, 4,5 g), y se realizaron una sustitución de nitrógeno y una sustitución de hidrógeno tres veces cada una a una presión de 0,3 MPaG. Entonces, se elevó la temperatura hasta 50°C y se agitaron los contenidos durante una hora bajo una presión de hidrógeno de 0,3 MPaG. Se elevó la temperatura hasta 70°C, se agitaron los contenidos durante dos horas y entonces se purgó el sistema con nitrógeno. Se enfriaron los contenidos hasta temperatura ambiente, se añadió una disolución acuosa de hidróxido de sodio al 50% (8,08 g, 0,069 mol) y se agitaron los contenidos durante la noche. Entonces, tras filtrar y eliminar el catalizador, se lavaron los contenidos con N-metilpirrolidona (40 ml), se añadió 2-propanol (240 ml) a la misma temperatura, entonces se añadió disolución acuosa de ácido clorhídrico 6 N (4 g) y se añadieron cristales de siembra (23) (40 mg). T ras la confirmación de la cristalización, se agitaron los contenidos durante dos horas, se añadió 2-propanol (240 ml), se ajustó el pH a 3,5 con una disolución acuosa de ácido clorhídrico 6 N y se agitó la mezcla durante tres horas. El sólido precipitado se recogió mediante filtración, se lavó una primera vez con una mezcla de N-metilpirrolidona (28 ml) y 2-propanol (56 ml), y entonces se lavó una segunda vez con 2-propanol (80 ml). Entonces, se secaron los contenidos a presión reducida a 40°C para obtener el compuesto diana (23) (29,90 kg, rendimiento del 94,2%) como un sólido.
•Los cristales de siembra (23) se obtuvieron recogiendo algo de la disolución de reacción y concentrándola a presión reducida.
(Ejemplo 5)
Producción de N-[4-(2-amino-5-{4-[(2R)-1,4-dioxan-2-ilmetoxi]-3-metoxifenil}piridin-3-il)-3-fluorofenil]-5-metil-4’-oxo-1 (tetrahidro-2H-piran-4-ilmetil)-1 ’,4’-dihidro-2,3’-bipiridin-5’-carboxamida (25)
[Fórmula 15]

Bajo una atmósfera de nitrógeno, se añadieron N,N-dimetilacetamida (35,6 l), ácido 5-metil-4’-oxo-1’-(tetrahidro-2H-piran-4-ilmetil)-1 ’,4’-dihidro-2,3’-bipiridin-5’-carboxílico (10) (3,56 kg, 10,8 mol) y agua purificada (152 g), y se enfriaron los contenidos hasta -8°C. Entonces, se añadió gota a gota cloruro de tionilo (2,46 kg, 20,7 mol) a lo largo de 50 minutos y se agitaron los contenidos a la misma temperatura durante 1,5 horas. A continuación, se añadieron monohidrocloruro de 3-(4-amino-2-fluorofenil)-5-{4-[(2R)-1,4-dioxan-2-ilmetoxi]-3-metoxifenil}piridin-2-amina (23) (4,77 kg, 10,3 mol) y N,N-dimetilacetamida (3,6 l), y se agitaron los contenidos a -8°C durante 40 horas. Entonces, se añadió una disolución mixta de N,N-dimetilacetamida (14,3 l) y agua (3,6 l), y se agitaron los contenidos a -8°C durante 1,5 horas y entonces se calentaron hasta 60°C. Tras añadir agua (7,1 l) a la disolución, se ajustó el pH de la disolución a 5,5 con trietilamina. Entonces, se añadieron cristales de siembra (25) (0,4 g) para hacer precipitar cristales. Tras confirmar la precipitación de cristales, se agitaron los contenidos a 60°C durante 3,5 horas y se añadió gota a gota agua (16,0 l) a lo largo de una hora mientras se mantenía la misma temperatura. Se añadió trietilamina a la suspensión para ajustar el pH a 5,7 y entonces se enfrió la suspensión hasta 25°C a lo largo de una hora. Tras agitar a la misma temperatura durante 14 horas, el sólido precipitado se recogió mediante filtración y se lavó con disolución acuosa de N,N-dimetilacetamida al 33% (N,N-dimetilacetamida 11,7 l, agua 23,8 l) y entonces agua normal (35,6 l), y se secaron los contenidos a presión reducida a 40°C para obtener el compuesto diana (25) (7,17 kg, rendimiento del 94,4%). *Los cristales de siembra (25) se obtuvieron recogiendo algo de la disolución de reacción y concentrándola a presión reducida.
1H RMN (500 MHz, CDCh): 5 = 1,42-1,62 (m, 4H), 2,16 (m, 1H), 2,40 (s, 3H), 3,39 (dt, J = 2,0, 12 Hz, 2H), 3,55 (dd, J = 12, 10 Hz, 1H), 3,65-3,89 (m, 4H), 3,91 (s, 3H), 3,93-4,10 (m, 8H), 4,55 (a, 2H), 6,96 (d, J = 8,0 Hz, 1 H), 7,03 (d, J = 2,0 Hz, 1H), 7,05 (dd, J = 2,0, 8,5 Hz, 1H), 7,38 (t, J = 8,0 Hz, 1 H), 7,50 (dd, J = 2,0, 8,5 Hz, 1H), 7,59 (d, J = 2,5 Hz, 1H), 7,62 (dd, J = 2,5, 8,0 Hz, 1H), 7,92 (dd, J = 4,0, 12 Hz, 1H), 8,31 (d, J = 2,0 Hz, 1H), 8,41 (d, J = 2,5 Hz, 1H), 8,47-8,48 (m, 2H), 8,56 (d, J = 2,5 Hz, 1 H), 13,01 (s, 1H).
(Ejemplo 6)
Producción de N-[4-(2-amino-5-{4-[(2R)-1,4-dioxan-2-ilmetoxi]-3-metoxifenil}piridin-3-il)-3-fluorofenil]-5-metil-4’-oxo-1 (tetrahidro-2H-piran-4-ilmetil)-1 ’,4’-dihidro-2,3’-bipiridin-5’-carboxamida (25)
[Fórmula 16]

Se añadieron N,N-dimetilacetamida (67,5 ml) y ácido 5-metil-4’-oxo-1’-(tetrahidro-2H-piran-4-ilmetil)-1’,4’-dihidro-2,3’-bipiridin-5’-carboxílico (10) (6,72 g, 0,020 mol) al recipiente de reacción 1 bajo una atmósfera de nitrógeno y se enfriaron los contenidos hasta -102C. Entonces, se añadió gota a gota cloruro de tionilo (2,55 g, 0,021 mol) a lo largo de 50 minutos y se agitaron los contenidos a la misma temperatura durante cuatro horas.
Se añadieron N,N-dimetilacetamida (49,5 ml), monohidrocloruro de 3-(4-amino-2-fluorofenil)-5-{4-[(2R)-1,4-dioxan-2-ilmetoxi]-3-metoxifenil}piridin-2-amina (23) (9,00 g, 0,019 mol) y 1,8-diazabiciclo [5.4.0]undeca-7-eno (DBU) (3,11 g, 0,020 mol) al recipiente de reacción 2 bajo una atmósfera de nitrógeno. Tras agitar a temperatura ambiente durante 0,5 horas hasta disolver completamente los contenidos, se enfriaron los contenidos hasta -10°C.
A continuación, se añadió gota a gota la disolución en el recipiente de reacción 2 al recipiente de reacción 1 a lo largo de una hora mientras se mantenía la temperatura a -10°C. Tras agitar a la misma temperatura durante dos horas, se añadió agua (18 ml) y se elevó la temperatura hasta 60°C. Se añadió trietilamina a la disolución para ajustar el pH de la disolución a 7,5 y entonces se añadieron cristales de siembra (25) (1,0 mg) para hacer precipitar cristales. Tras confirmar la precipitación de cristales, se agitaron los contenidos a 60°C durante tres horas y se añadió gota a gota agua (45,0 ml) a lo largo de una hora mientras se mantenía la misma temperatura. Entonces, se enfriaron los contenidos hasta 25°C a lo largo de una hora. Tras agitar a la misma temperatura durante 16 horas, el sólido precipitado se recogió mediante filtración y se mezcló con disolución acuosa de N,N-dimetilacetamida al 33% (N,N
dimetilacetamida 45,2 ml, agua 22,3 ml) y entonces agua normal (67,5 ml), y los contenidos se lavaron y se secaron a presión reducida a 40°C para obtener el compuesto diana (25) (13,55 g, rendimiento del 94,5%) como un sólido.
•Los cristales de siembra (25) se obtuvieron recogiendo algo de la disolución de reacción y concentrándola a presión reducida.
(Ejemplo 7)
Producción de sulfato de N-[4-(2-amino-5-{4-[(2R)-1,4-dioxan-2-ilmetoxi]-3-metoxifenil}piridin-3-il)-3-fluorofenil]-5-metil-4’-oxo-1 ’-(tetrahidro-2H-piran-4-ilmetil)-1 ’,4’-dihidro-2,3’-bipiridin-5’-carboxamida hidratado (26)
Bajo una atmósfera de nitrógeno, se añadieron acetona (49 ml), N-[4-(2-amino-5-{4-[(2R)-1,4-dioxan-2-ilmetoxi]-3-metoxifenil}piridin-3-il)-3-fluorofenil]-5-metil-4’-oxo-1 ’-(tetrahidro-2H-piran-4-ilmetil)-1 ’,4’-dihidro-2,3’-bipiridin-5’-carboxamida (25) (10,00 g, 0,014 mol) y agua purificada (1,7 ml), se elevó la temperatura hasta 45°C y entonces se añadió una disolución acuosa de ácido sulfúrico al 25% (5,87 g) mientras se mantenía la temperatura a 45°C. Tras confirmar la disolución de los cristales, se realizó una filtración a la misma temperatura para eliminar la materia insoluble y se lavaron los contenidos con una disolución mixta de agua purificada y acetona (9 ml de agua purificada, 21 ml de acetona). Después, se añadió una disolución acuosa de ácido sulfúrico al 25% (4,27 g) y entonces se añadieron cristales de siembra (26) (10 mg) para hacer precipitar cristales. Tras confirmar la precipitación de cristales y entonces agitar durante la noche, se añadió agua purificada (3 ml), se elevó la temperatura hasta 55°C y se agitaron los contenidos durante una hora. Tras enfriar hasta 45°C, se añadió gota a gota acetona (40 ml) a lo largo de 30 minutos. Tras agitar a 45°C durante 30 minutos, se añadió gota a gota acetona (40 ml) a lo largo de 30 minutos. Tras agitar a 45°C durante 30 minutos, se añadió gota a gota de nuevo acetona (40 ml) a lo largo de 30 minutos. Tras agitar a 45°C durante 30 minutos, se añadió gota a gota acetona (80 ml) a lo largo de una hora. Entonces, los contenidos se enfriaron hasta 25°C a lo largo de 60 minutos y se agitaron a la misma temperatura durante la noche. El sólido precipitado se recogió mediante filtración, los cristales se lavaron una primera vez con una disolución mixta de acetona y agua purificada (44 ml de acetona, 6 ml de agua purificada) y una segunda vez con acetona (50 ml), y se secaron los cristales a 35°C a presión reducida de 3 kPa para obtener el compuesto diana (26) (11,95 g) como un sólido.
•Los cristales de siembra (26) se obtuvieron recogiendo algo de la disolución de reacción y concentrándola a presión reducida. Se realizó un análisis de estructura cristalina de rayos X de polvo en los cristales resultantes. Los resultados se muestran en la figura 1 y la tabla 2.
1H RMN (500 MHz, DMSO-ds): 5 = 1,37 (dq, J = 4,0, 12 Hz, 2H), 1,52 (d, J = 11,5 Hz, 2H), 2,15 (m, 1H), 2,49 (s, 1H), 3,31 (dt, J = 1,5, 11,5 Hz, 2H), 3,44 (dd, J = 10, 11,5 Hz, 1H), 3,53 (dt, J = 1,5, 11 Hz, 1H), 3,64-3,73 (m, 2H), 3,80 (dd, J = 2,0, 12 Hz, 1H), 3,85-3,92 (m, 5H), 3,98-4,05 (m, 2H), 4,26 (d, J = 7,5 Hz, 2H), 7,10 (d, J = 8,5 Hz, 1H), 7,30 (dd, J = 2,5, 8,5 Hz, 1H), 7,36 (d, J = 2,5 Hz, 1H), 7,57-7,62 (m, 2H), 7,77 (a, 2H), 8,04 (dd, J = 2,0, 13 Hz, 1H), 8,11 (a, 1H), 8,38 (dd, J = 2,5, 9,0 Hz, 2H), 8,48 (d, J = 8,5 Hz, 1H), 8,69 (s, 1H), 8,88 (dd, 2,0, 7,0 Hz, 2H), 13,02 (a, 1H).

Claims (13)
- REIVINDICACIONES1 Un método de producción que comprende la etapa de hacer reaccionar un compuesto representado mediante la fórmula (I):
 con un agente de cloración y dimetilformamida para obtener un compuesto representado mediante la fórmula (II):
con un agente de cloración y dimetilformamida para obtener un compuesto representado mediante la fórmula (II): o una sal del mismo.
o una sal del mismo. - 2. - El método según la reivindicación 1, en el que el agente de cloración es cloruro de oxalilo.
- 3. - El método según la reivindicación 1 o 2, que comprende además hacer reaccionar el compuesto representado mediante la fórmula (II) o una sal del mismo con un compuesto representado mediante la fórmula (III):
 en presencia de una base para obtener un compuesto representado mediante la fórmula (IV):
en presencia de una base para obtener un compuesto representado mediante la fórmula (IV): o una sal del mismo.
o una sal del mismo. - 4.- El método según la reivindicación 3, en el que la base es 2,6-lutidina.
- 5.- El método según la reivindicación 3 o 4, que comprende además hacer reaccionar el compuesto representado mediante la fórmula (IV) o una sal del mismo con bencilamina para obtener un compuesto representado mediante la fórmula (V):
 o una sal del mismo.
o una sal del mismo. - 6.- Un método de producción que comprende las etapas de:(i) hacer reaccionar un compuesto representado mediante la fórmula (I’) con un reactivo de Wittig,siendo el reactivo de Wittig capaz de obtener un compuesto en el que un doble enlace está sustituido por un grupo alcoxi;
 (ii) hacer reaccionar el compuesto obtenido en la etapa (i) con un agente de cloración y dimetilformamida;(iii) hacer reaccionar el compuesto obtenido en la etapa (ii) con un compuesto representado mediante la fórmula (III):
(ii) hacer reaccionar el compuesto obtenido en la etapa (i) con un agente de cloración y dimetilformamida;(iii) hacer reaccionar el compuesto obtenido en la etapa (ii) con un compuesto representado mediante la fórmula (III): en presencia de una base; y(iv) hacer reaccionar el compuesto obtenido en la etapa (iii) con bencilamina para obtener un compuesto representado mediante la fórmula (V):
en presencia de una base; y(iv) hacer reaccionar el compuesto obtenido en la etapa (iii) con bencilamina para obtener un compuesto representado mediante la fórmula (V): o una sal del mismo.
o una sal del mismo. - 7. - El método según la reivindicación 6, en el que el reactivo de Wittig en la etapa (i) es Ph3P(Cl)CH2OMe, Ph3 P(Br)CH2ÜMe o Ph3P(I)CH2OMe.
- 8. - El método según la reivindicación 6 o 7, en el que el agente de cloración en la etapa (ii) es cloruro de oxalilo.
- 9. - El método según una cualquiera de las reivindicaciones 6 a 8, en el que la base en la etapa (iii) es 2,6-lutidina.
- 10. - El método según una cualquiera de las reivindicaciones 5 a 9, que comprende además hacer reaccionar el compuesto representado mediante la fórmula (V) o una sal del mismo con hidrógeno en un disolvente y en presencia de un catalizador de paladio sobre carbono para obtener un compuesto representado mediante la fórmula (VI):
 o una sal del mismo.
o una sal del mismo. - 11. - El método según la reivindicación 10, en el que el compuesto representado mediante la fórmula (V) o una sal del mismo es una sal de ácido clorhídrico de un compuesto representado mediante la fórmula (V), y el disolvente es 1-propanol o N-metilpirrolidona.
- 12. - El método según la reivindicación 10 o la reivindicación 11, que comprende además condensar el compuesto representado mediante la fórmula (VI) o una sal del mismo con un compuesto representado mediante la fórmula (VII):
 o una sal del mismo para obtener un compuesto representado mediante la fórmula (VIII):
o una sal del mismo para obtener un compuesto representado mediante la fórmula (VIII): o una sal del mismo.
o una sal del mismo. - 13.- El método según la reivindicación 12, en el que el compuesto representado mediante la fórmula (VIII) o una sal del mismo es una sal de ácido sulfúrico del compuesto representado mediante la fórmula (VIII).
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2018127197 | 2018-07-04 | ||
| PCT/JP2019/026395 WO2020009132A1 (ja) | 2018-07-04 | 2019-07-03 | ジアリールピリジン誘導体の製造方法 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2925365T3 true ES2925365T3 (es) | 2022-10-17 |
Family
ID=69059540
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES19831009T Active ES2925365T3 (es) | 2018-07-04 | 2019-07-03 | Método para producir derivados de diarilpiridina |
Country Status (10)
| Country | Link |
|---|---|
| US (1) | US11999724B2 (es) |
| EP (1) | EP3819296B1 (es) |
| JP (2) | JP7337058B2 (es) |
| CN (1) | CN112368271A (es) |
| AU (1) | AU2019299152A1 (es) |
| CA (1) | CA3105328A1 (es) |
| ES (1) | ES2925365T3 (es) |
| IL (1) | IL279070B2 (es) |
| TW (1) | TWI840378B (es) |
| WO (1) | WO2020009132A1 (es) |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20250019362A1 (en) | 2021-11-19 | 2025-01-16 | Nippon Soda Co., Ltd. | Method for producing 2-heteroarylpyridine compound |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DK2810937T3 (en) | 2012-01-31 | 2017-03-13 | Daiichi Sankyo Co Ltd | PYRIDONE DERIVATIVES |
| TWI723572B (zh) | 2014-07-07 | 2021-04-01 | 日商第一三共股份有限公司 | 具有四氫吡喃基甲基之吡啶酮衍生物及其用途 |
| TWI796326B (zh) | 2017-03-24 | 2023-03-21 | 日商第一三共股份有限公司 | 含有axl抑制劑與egfr酪胺酸激酶抑制藥的醫藥品及其用途 |
-
2019
- 2019-07-01 TW TW108123125A patent/TWI840378B/zh not_active IP Right Cessation
- 2019-07-03 JP JP2020529019A patent/JP7337058B2/ja active Active
- 2019-07-03 CA CA3105328A patent/CA3105328A1/en active Pending
- 2019-07-03 AU AU2019299152A patent/AU2019299152A1/en not_active Abandoned
- 2019-07-03 US US17/252,469 patent/US11999724B2/en active Active
- 2019-07-03 EP EP19831009.6A patent/EP3819296B1/en active Active
- 2019-07-03 ES ES19831009T patent/ES2925365T3/es active Active
- 2019-07-03 WO PCT/JP2019/026395 patent/WO2020009132A1/ja not_active Ceased
- 2019-07-03 IL IL279070A patent/IL279070B2/en unknown
- 2019-07-03 CN CN201980043226.0A patent/CN112368271A/zh active Pending
-
2023
- 2023-08-22 JP JP2023134392A patent/JP2023159297A/ja active Pending
Also Published As
| Publication number | Publication date |
|---|---|
| JP2023159297A (ja) | 2023-10-31 |
| WO2020009132A1 (ja) | 2020-01-09 |
| AU2019299152A2 (en) | 2021-03-11 |
| TWI840378B (zh) | 2024-05-01 |
| TW202005962A (zh) | 2020-02-01 |
| AU2019299152A1 (en) | 2021-03-04 |
| CA3105328A1 (en) | 2020-01-09 |
| EP3819296B1 (en) | 2022-06-29 |
| EP3819296A4 (en) | 2021-06-09 |
| IL279070B1 (en) | 2024-01-01 |
| IL279070B2 (en) | 2024-05-01 |
| US11999724B2 (en) | 2024-06-04 |
| JP7337058B2 (ja) | 2023-09-01 |
| CN112368271A (zh) | 2021-02-12 |
| IL279070A (en) | 2021-01-31 |
| EP3819296A1 (en) | 2021-05-12 |
| US20210188823A1 (en) | 2021-06-24 |
| JPWO2020009132A1 (ja) | 2021-07-08 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP2026500533A (ja) | Kras g12d阻害剤の結晶形及び調製方法 | |
| US11629153B2 (en) | Forms and compositions of a MK2 inhibitor | |
| CN109153687B (zh) | 一类含有三环杂芳基的化合物 | |
| CN112300153B (zh) | 一种杂环化合物、药物组合物和用途 | |
| BR112015017963A2 (pt) | composto de fenil amino pirimidina deuterado, método para preparar a composição farmacêutica, composição farmacêutica e uso do composto | |
| ES2451140T3 (es) | Cristal de derivado de fenilalanina y método para su producción | |
| KR20160073413A (ko) | 퀴나졸리논 및 이소퀴놀리논 유도체 | |
| CA3160478A1 (en) | Wdr5 inhibitors and modulators | |
| JP2023134749A (ja) | マクロライド誘導体、その製造方法および用途 | |
| ES2925365T3 (es) | Método para producir derivados de diarilpiridina | |
| ES2974328T3 (es) | Formas sólidas de 3-(5-fluorobenzofuran-3-il)-4-(5-metil-5h[1,3]dioxolo[4,5-f]indol-7-il)pirrol-2,5-diona | |
| KR20180033514A (ko) | Fgfr 억제제를 제조하기 위한 방법 | |
| KR102891811B1 (ko) | 3-(5-플루오로벤조푸란-3-일)-4-(5-메틸-5H-[1,3]디옥솔로[4,5-f]인돌-7-일)피롤-2,5-디온의 고체 형태 | |
| JP2020193209A (ja) | チアゾール誘導体の製造方法 | |
| CN120917029A (zh) | 结晶形式 | |
| JP6165335B2 (ja) | ゲフィチニブの新規な結晶形およびその製造方法 | |
| AU2019218150B2 (en) | Crystal form of 3,4-dihydrothieno[3,2-d]pyrimidine compound and preparation method therefor | |
| KR20240089380A (ko) | 마보릭사포의 합성 및 그 중간체 | |
| KR20250174948A (ko) | 질소 함유 테트라시클릭 화합물의 결정 형태 및 이의 제조 방법 | |
| BR122025016219A2 (pt) | Formas de sal de um composto inibidor de cyp11a1 estruturado com 4h-piran-4-ona, métodos para preparar as mesmas, composição farmacêutica e uso das referidas formas de sal para o tratamento de cânceres hormonalmente regulados | |
| BR112020007538B1 (pt) | Forma sólida, processo para preparar a forma sólida, composição farmacêutica, processo para preparar a composição farmacêutica e uso da forma sólida | |
| BR122025019554A2 (pt) | Forma sólida de 3-(5-flúor-benzofuran-3-il)-4-(5-metil-5h-[1,3]dioxolo[4,5- f]indol-7-il)pirrol-2,5-diona, seus usos, composição farmacêutica e processo para preparar a mesma |