ES2926580T3 - Proceso para preparar 5-[[4-[2-[5-(1-hidroxietil)-2-piridinil]etoxi]fenil]metil]-2,4-tiazolidindiona y sales de la misma - Google Patents
Proceso para preparar 5-[[4-[2-[5-(1-hidroxietil)-2-piridinil]etoxi]fenil]metil]-2,4-tiazolidindiona y sales de la misma Download PDFInfo
- Publication number
- ES2926580T3 ES2926580T3 ES17829042T ES17829042T ES2926580T3 ES 2926580 T3 ES2926580 T3 ES 2926580T3 ES 17829042 T ES17829042 T ES 17829042T ES 17829042 T ES17829042 T ES 17829042T ES 2926580 T3 ES2926580 T3 ES 2926580T3
- Authority
- ES
- Spain
- Prior art keywords
- compound
- formula
- solvent
- reaction mixture
- group
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 150000003839 salts Chemical class 0.000 title claims abstract description 39
- 238000004519 manufacturing process Methods 0.000 title abstract description 18
- OXVFDZYQLGRLCD-UHFFFAOYSA-N hydroxypioglitazone Chemical compound N1=CC(C(O)C)=CC=C1CCOC(C=C1)=CC=C1CC1C(=O)NC(=O)S1 OXVFDZYQLGRLCD-UHFFFAOYSA-N 0.000 title description 22
- 150000001875 compounds Chemical class 0.000 claims abstract description 196
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 claims description 165
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 claims description 150
- 239000011541 reaction mixture Substances 0.000 claims description 130
- 239000002904 solvent Substances 0.000 claims description 118
- 239000000203 mixture Substances 0.000 claims description 96
- 238000000034 method Methods 0.000 claims description 88
- 230000008569 process Effects 0.000 claims description 78
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 claims description 74
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 claims description 69
- 238000006243 chemical reaction Methods 0.000 claims description 68
- 239000000243 solution Substances 0.000 claims description 65
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical group CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 claims description 54
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 claims description 51
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical group CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 claims description 42
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 claims description 41
- 125000006239 protecting group Chemical group 0.000 claims description 41
- -1 alkyl lithium Chemical compound 0.000 claims description 36
- IAYPIBMASNFSPL-UHFFFAOYSA-N Ethylene oxide Chemical compound C1CO1 IAYPIBMASNFSPL-UHFFFAOYSA-N 0.000 claims description 34
- 229910021595 Copper(I) iodide Inorganic materials 0.000 claims description 30
- LSXDOTMGLUJQCM-UHFFFAOYSA-M copper(i) iodide Chemical compound I[Cu] LSXDOTMGLUJQCM-UHFFFAOYSA-M 0.000 claims description 30
- 230000015572 biosynthetic process Effects 0.000 claims description 26
- 239000002841 Lewis acid Substances 0.000 claims description 25
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-dimethylformamide Substances CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 claims description 25
- 150000007517 lewis acids Chemical class 0.000 claims description 25
- 125000001181 organosilyl group Chemical group [SiH3]* 0.000 claims description 25
- VMQMZMRVKUZKQL-UHFFFAOYSA-N Cu+ Chemical class [Cu+] VMQMZMRVKUZKQL-UHFFFAOYSA-N 0.000 claims description 24
- 229910052744 lithium Inorganic materials 0.000 claims description 24
- MZRVEZGGRBJDDB-UHFFFAOYSA-N N-Butyllithium Chemical group [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 claims description 22
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 claims description 21
- 239000002585 base Substances 0.000 claims description 21
- 125000001412 tetrahydropyranyl group Chemical group 0.000 claims description 20
- 125000004184 methoxymethyl group Chemical group [H]C([H])([H])OC([H])([H])* 0.000 claims description 19
- BZLVMXJERCGZMT-UHFFFAOYSA-N Methyl tert-butyl ether Chemical compound COC(C)(C)C BZLVMXJERCGZMT-UHFFFAOYSA-N 0.000 claims description 17
- LMBFAGIMSUYTBN-MPZNNTNKSA-N teixobactin Chemical compound C([C@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CO)C(=O)N[C@H](CCC(N)=O)C(=O)N[C@H]([C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CO)C(=O)N[C@H]1C(N[C@@H](C)C(=O)N[C@@H](C[C@@H]2NC(=N)NC2)C(=O)N[C@H](C(=O)O[C@H]1C)[C@@H](C)CC)=O)NC)C1=CC=CC=C1 LMBFAGIMSUYTBN-MPZNNTNKSA-N 0.000 claims description 17
- 238000001914 filtration Methods 0.000 claims description 16
- 239000012279 sodium borohydride Substances 0.000 claims description 16
- 229910000033 sodium borohydride Inorganic materials 0.000 claims description 16
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Natural products C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 claims description 15
- 239000003960 organic solvent Substances 0.000 claims description 15
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 claims description 14
- RGHHSNMVTDWUBI-UHFFFAOYSA-N 4-hydroxybenzaldehyde Chemical compound OC1=CC=C(C=O)C=C1 RGHHSNMVTDWUBI-UHFFFAOYSA-N 0.000 claims description 13
- 150000007524 organic acids Chemical class 0.000 claims description 12
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical group [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 claims description 11
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims description 10
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 claims description 10
- 239000003880 polar aprotic solvent Substances 0.000 claims description 10
- 239000002244 precipitate Substances 0.000 claims description 10
- 239000000376 reactant Substances 0.000 claims description 10
- 125000000217 alkyl group Chemical group 0.000 claims description 9
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 claims description 9
- 238000000746 purification Methods 0.000 claims description 9
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 claims description 8
- 150000008044 alkali metal hydroxides Chemical class 0.000 claims description 8
- 239000012299 nitrogen atmosphere Substances 0.000 claims description 8
- ZOBPZXTWZATXDG-UHFFFAOYSA-N 1,3-thiazolidine-2,4-dione Chemical compound O=C1CSC(=O)N1 ZOBPZXTWZATXDG-UHFFFAOYSA-N 0.000 claims description 7
- IKHGUXGNUITLKF-UHFFFAOYSA-N Acetaldehyde Chemical compound CC=O IKHGUXGNUITLKF-UHFFFAOYSA-N 0.000 claims description 7
- JGUQDUKBUKFFRO-CIIODKQPSA-N dimethylglyoxime Chemical compound O/N=C(/C)\C(\C)=N\O JGUQDUKBUKFFRO-CIIODKQPSA-N 0.000 claims description 7
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 claims description 7
- 229910021645 metal ion Inorganic materials 0.000 claims description 7
- JRMUNVKIHCOMHV-UHFFFAOYSA-M tetrabutylammonium bromide Chemical group [Br-].CCCC[N+](CCCC)(CCCC)CCCC JRMUNVKIHCOMHV-UHFFFAOYSA-M 0.000 claims description 7
- 239000002253 acid Substances 0.000 claims description 6
- HSZCZNFXUDYRKD-UHFFFAOYSA-M lithium iodide Inorganic materials [Li+].[I-] HSZCZNFXUDYRKD-UHFFFAOYSA-M 0.000 claims description 6
- 230000009467 reduction Effects 0.000 claims description 6
- LSNNMFCWUKXFEE-UHFFFAOYSA-L sulfite Chemical group [O-]S([O-])=O LSNNMFCWUKXFEE-UHFFFAOYSA-L 0.000 claims description 6
- VDZOOKBUILJEDG-UHFFFAOYSA-M tetrabutylammonium hydroxide Chemical compound [OH-].CCCC[N+](CCCC)(CCCC)CCCC VDZOOKBUILJEDG-UHFFFAOYSA-M 0.000 claims description 6
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 claims description 5
- LSNNMFCWUKXFEE-UHFFFAOYSA-M Bisulfite Chemical compound OS([O-])=O LSNNMFCWUKXFEE-UHFFFAOYSA-M 0.000 claims description 5
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 claims description 5
- 229910052783 alkali metal Inorganic materials 0.000 claims description 5
- 229910000288 alkali metal carbonate Inorganic materials 0.000 claims description 5
- 150000008041 alkali metal carbonates Chemical class 0.000 claims description 5
- 150000001412 amines Chemical class 0.000 claims description 5
- 239000003638 chemical reducing agent Substances 0.000 claims description 5
- 238000010511 deprotection reaction Methods 0.000 claims description 5
- 229910000027 potassium carbonate Inorganic materials 0.000 claims description 5
- 125000003944 tolyl group Chemical group 0.000 claims description 5
- XLJKHNWPARRRJB-UHFFFAOYSA-N cobalt(2+) Chemical compound [Co+2] XLJKHNWPARRRJB-UHFFFAOYSA-N 0.000 claims description 4
- 125000001301 ethoxy group Chemical group [H]C([H])([H])C([H])([H])O* 0.000 claims description 4
- 235000019253 formic acid Nutrition 0.000 claims description 4
- 239000003446 ligand Substances 0.000 claims description 4
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 4
- 150000003242 quaternary ammonium salts Chemical class 0.000 claims description 4
- ILMRJRBKQSSXGY-UHFFFAOYSA-N tert-butyl(dimethyl)silicon Chemical compound C[Si](C)C(C)(C)C ILMRJRBKQSSXGY-UHFFFAOYSA-N 0.000 claims description 4
- BCNZYOJHNLTNEZ-UHFFFAOYSA-N tert-butyldimethylsilyl chloride Chemical compound CC(C)(C)[Si](C)(C)Cl BCNZYOJHNLTNEZ-UHFFFAOYSA-N 0.000 claims description 4
- 125000001981 tert-butyldimethylsilyl group Chemical group [H]C([H])([H])[Si]([H])(C([H])([H])[H])[*]C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H] 0.000 claims description 4
- 150000004791 alkyl magnesium halides Chemical class 0.000 claims description 3
- 239000007864 aqueous solution Substances 0.000 claims description 3
- KOPOQZFJUQMUML-UHFFFAOYSA-N chlorosilane Chemical compound Cl[SiH3] KOPOQZFJUQMUML-UHFFFAOYSA-N 0.000 claims description 3
- 238000001556 precipitation Methods 0.000 claims description 3
- 125000000037 tert-butyldiphenylsilyl group Chemical group [H]C1=C([H])C([H])=C([H])C([H])=C1[Si]([H])([*]C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H])C1=C([H])C([H])=C([H])C([H])=C1[H] 0.000 claims description 3
- 125000002088 tosyl group Chemical group [H]C1=C([H])C(=C([H])C([H])=C1C([H])([H])[H])S(*)(=O)=O 0.000 claims description 3
- 125000005270 trialkylamine group Chemical group 0.000 claims description 3
- 125000000025 triisopropylsilyl group Chemical group C(C)(C)[Si](C(C)C)(C(C)C)* 0.000 claims description 3
- JWUJQDFVADABEY-UHFFFAOYSA-N 2-methyltetrahydrofuran Chemical compound CC1CCCO1 JWUJQDFVADABEY-UHFFFAOYSA-N 0.000 claims description 2
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 claims description 2
- XDTMQSROBMDMFD-UHFFFAOYSA-N Cyclohexane Chemical compound C1CCCCC1 XDTMQSROBMDMFD-UHFFFAOYSA-N 0.000 claims description 2
- KRHYYFGTRYWZRS-UHFFFAOYSA-M Fluoride anion Chemical compound [F-] KRHYYFGTRYWZRS-UHFFFAOYSA-M 0.000 claims description 2
- 125000005104 aryl silyl group Chemical group 0.000 claims description 2
- 239000012298 atmosphere Substances 0.000 claims description 2
- KQIADDMXRMTWHZ-UHFFFAOYSA-N chloro-tri(propan-2-yl)silane Chemical compound CC(C)[Si](Cl)(C(C)C)C(C)C KQIADDMXRMTWHZ-UHFFFAOYSA-N 0.000 claims description 2
- IJOOHPMOJXWVHK-UHFFFAOYSA-N chlorotrimethylsilane Chemical compound C[Si](C)(C)Cl IJOOHPMOJXWVHK-UHFFFAOYSA-N 0.000 claims description 2
- 239000008139 complexing agent Substances 0.000 claims description 2
- 125000004170 methylsulfonyl group Chemical group [H]C([H])([H])S(*)(=O)=O 0.000 claims description 2
- 230000001376 precipitating effect Effects 0.000 claims description 2
- MHYGQXWCZAYSLJ-UHFFFAOYSA-N tert-butyl-chloro-diphenylsilane Chemical compound C=1C=CC=CC=1[Si](Cl)(C(C)(C)C)C1=CC=CC=C1 MHYGQXWCZAYSLJ-UHFFFAOYSA-N 0.000 claims description 2
- 125000004665 trialkylsilyl group Chemical group 0.000 claims description 2
- JOCBASBOOFNAJA-UHFFFAOYSA-N N-tris(hydroxymethyl)methyl-2-aminoethanesulfonic acid Chemical compound OCC(CO)(CO)NCCS(O)(=O)=O JOCBASBOOFNAJA-UHFFFAOYSA-N 0.000 claims 1
- 235000015320 potassium carbonate Nutrition 0.000 claims 1
- CZDYPVPMEAXLPK-UHFFFAOYSA-N tetramethylsilane Chemical compound C[Si](C)(C)C CZDYPVPMEAXLPK-UHFFFAOYSA-N 0.000 claims 1
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 57
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 52
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 32
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 description 30
- 239000010410 layer Substances 0.000 description 30
- 239000003921 oil Substances 0.000 description 29
- 235000019198 oils Nutrition 0.000 description 29
- 239000000725 suspension Substances 0.000 description 28
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 26
- 239000007787 solid Substances 0.000 description 25
- 235000019439 ethyl acetate Nutrition 0.000 description 23
- 238000003786 synthesis reaction Methods 0.000 description 23
- 239000012044 organic layer Substances 0.000 description 19
- ZWEHNKRNPOVVGH-UHFFFAOYSA-N 2-Butanone Chemical compound CCC(C)=O ZWEHNKRNPOVVGH-UHFFFAOYSA-N 0.000 description 18
- OMSAAQUHZXHUGS-JWGURIENSA-N (5Z)-5-[[4-[2-[5-[2-[tert-butyl(dimethyl)silyl]oxyethyl]pyridin-2-yl]ethoxy]phenyl]methylidene]-1,3-thiazolidine-2,4-dione Chemical compound CC(C)(C)[Si](C)(C)OCCC1=CN=C(CCOC2=CC=C(\C=C3/SC(=O)NC3=O)C=C2)C=C1 OMSAAQUHZXHUGS-JWGURIENSA-N 0.000 description 16
- OUTOIAUNYUCUGG-UHFFFAOYSA-N 4-[2-[5-[2-[tert-butyl(dimethyl)silyl]oxyethyl]pyridin-2-yl]ethoxy]benzaldehyde Chemical compound CC(C)(C)[Si](C)(C)OCCC1=CN=C(CCOC2=CC=C(C=O)C=C2)C=C1 OUTOIAUNYUCUGG-UHFFFAOYSA-N 0.000 description 16
- SBHOQYCDAHAMDW-UHFFFAOYSA-N 5-[[4-[2-[5-(1-hydroxyethyl)pyridin-2-yl]ethoxy]phenyl]methyl]-1,3-thiazolidine-2,4-dione;hydrochloride Chemical compound Cl.N1=CC(C(O)C)=CC=C1CCOC(C=C1)=CC=C1CC1C(=O)NC(=O)S1 SBHOQYCDAHAMDW-UHFFFAOYSA-N 0.000 description 16
- UORVGPXVDQYIDP-UHFFFAOYSA-N borane Chemical compound B UORVGPXVDQYIDP-UHFFFAOYSA-N 0.000 description 16
- 239000000284 extract Substances 0.000 description 16
- HYAFETHFCAUJAY-UHFFFAOYSA-N pioglitazone Chemical class N1=CC(CC)=CC=C1CCOC(C=C1)=CC=C1CC1C(=O)NC(=O)S1 HYAFETHFCAUJAY-UHFFFAOYSA-N 0.000 description 15
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 14
- ILAHWRKJUDSMFH-UHFFFAOYSA-N boron tribromide Chemical compound BrB(Br)Br ILAHWRKJUDSMFH-UHFFFAOYSA-N 0.000 description 14
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 13
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical compound [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 12
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 12
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 12
- LDGYVEPFXQIYTH-UHFFFAOYSA-N 5-[[4-[2-[5-[2-[tert-butyl(dimethyl)silyl]oxyethyl]pyridin-2-yl]ethoxy]phenyl]methyl]-1,3-thiazolidine-2,4-dione Chemical compound CC(C)(C)[Si](OCCC=1C=CC(=NC=1)CCOC1=CC=C(C=C1)CC1C(NC(S1)=O)=O)(C)C LDGYVEPFXQIYTH-UHFFFAOYSA-N 0.000 description 11
- YZCKVEUIGOORGS-OUBTZVSYSA-N Deuterium Chemical compound [2H] YZCKVEUIGOORGS-OUBTZVSYSA-N 0.000 description 11
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 11
- 229910052805 deuterium Inorganic materials 0.000 description 11
- 239000000047 product Substances 0.000 description 11
- 238000003756 stirring Methods 0.000 description 11
- 238000005160 1H NMR spectroscopy Methods 0.000 description 10
- DHNIYCWDTAPKJI-UHFFFAOYSA-N 2-[5-[1-[tert-butyl(dimethyl)silyl]oxyethyl]pyridin-2-yl]ethanol Chemical compound [Si](C)(C)(C(C)(C)C)OC(C)C=1C=CC(=NC=1)CCO DHNIYCWDTAPKJI-UHFFFAOYSA-N 0.000 description 10
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 10
- 239000003153 chemical reaction reagent Substances 0.000 description 10
- 238000002330 electrospray ionisation mass spectrometry Methods 0.000 description 10
- 238000005481 NMR spectroscopy Methods 0.000 description 9
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 9
- 229910000085 borane Inorganic materials 0.000 description 8
- 239000000706 filtrate Substances 0.000 description 8
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 8
- 238000002360 preparation method Methods 0.000 description 8
- QOQYGKBTOFYPKJ-UHFFFAOYSA-N 1-(6-bromopyridin-3-yl)ethoxy-tert-butyl-dimethylsilane Chemical compound CC(C)(C)[Si](C)(C)OC(C)C1=CC=C(Br)N=C1 QOQYGKBTOFYPKJ-UHFFFAOYSA-N 0.000 description 7
- 229910015845 BBr3 Inorganic materials 0.000 description 7
- 229940125898 compound 5 Drugs 0.000 description 7
- 229960005095 pioglitazone Drugs 0.000 description 7
- ZHXUWDPHUQHFOV-UHFFFAOYSA-N 2,5-dibromopyridine Chemical compound BrC1=CC=C(Br)N=C1 ZHXUWDPHUQHFOV-UHFFFAOYSA-N 0.000 description 6
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 6
- DWAQJAXMDSEUJJ-UHFFFAOYSA-M Sodium bisulfite Chemical compound [Na+].OS([O-])=O DWAQJAXMDSEUJJ-UHFFFAOYSA-M 0.000 description 6
- 235000019270 ammonium chloride Nutrition 0.000 description 6
- 239000000010 aprotic solvent Substances 0.000 description 6
- GVPFVAHMJGGAJG-UHFFFAOYSA-L cobalt dichloride Chemical compound [Cl-].[Cl-].[Co+2] GVPFVAHMJGGAJG-UHFFFAOYSA-L 0.000 description 6
- 238000004440 column chromatography Methods 0.000 description 6
- 229910052757 nitrogen Inorganic materials 0.000 description 6
- 238000010992 reflux Methods 0.000 description 6
- 235000010267 sodium hydrogen sulphite Nutrition 0.000 description 6
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 5
- 238000001816 cooling Methods 0.000 description 5
- 238000003818 flash chromatography Methods 0.000 description 5
- 238000010438 heat treatment Methods 0.000 description 5
- 230000035484 reaction time Effects 0.000 description 5
- 229910052938 sodium sulfate Inorganic materials 0.000 description 5
- 235000011152 sodium sulphate Nutrition 0.000 description 5
- 238000005406 washing Methods 0.000 description 5
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 4
- YYROPELSRYBVMQ-UHFFFAOYSA-N 4-toluenesulfonyl chloride Chemical compound CC1=CC=C(S(Cl)(=O)=O)C=C1 YYROPELSRYBVMQ-UHFFFAOYSA-N 0.000 description 4
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 4
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 4
- FGUUSXIOTUKUDN-IBGZPJMESA-N C1(=CC=CC=C1)N1C2=C(NC([C@H](C1)NC=1OC(=NN=1)C1=CC=CC=C1)=O)C=CC=C2 Chemical compound C1(=CC=CC=C1)N1C2=C(NC([C@H](C1)NC=1OC(=NN=1)C1=CC=CC=C1)=O)C=CC=C2 FGUUSXIOTUKUDN-IBGZPJMESA-N 0.000 description 4
- WSFSSNUMVMOOMR-UHFFFAOYSA-N Formaldehyde Chemical compound O=C WSFSSNUMVMOOMR-UHFFFAOYSA-N 0.000 description 4
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 4
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 4
- 229940126214 compound 3 Drugs 0.000 description 4
- 239000010949 copper Substances 0.000 description 4
- 239000012043 crude product Substances 0.000 description 4
- 238000004821 distillation Methods 0.000 description 4
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 4
- IUYHWZFSGMZEOG-UHFFFAOYSA-M magnesium;propane;chloride Chemical compound [Mg+2].[Cl-].C[CH-]C IUYHWZFSGMZEOG-UHFFFAOYSA-M 0.000 description 4
- 239000002207 metabolite Substances 0.000 description 4
- 239000003495 polar organic solvent Substances 0.000 description 4
- 238000010926 purge Methods 0.000 description 4
- 238000006722 reduction reaction Methods 0.000 description 4
- 239000013557 residual solvent Substances 0.000 description 4
- 229920006395 saturated elastomer Polymers 0.000 description 4
- 239000007858 starting material Substances 0.000 description 4
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 4
- FPGGTKZVZWFYPV-UHFFFAOYSA-M tetrabutylammonium fluoride Chemical compound [F-].CCCC[N+](CCCC)(CCCC)CCCC FPGGTKZVZWFYPV-UHFFFAOYSA-M 0.000 description 4
- 239000003039 volatile agent Substances 0.000 description 4
- QDOJZKXYIAXLDB-UHFFFAOYSA-N 1-(6-bromopyridin-3-yl)ethanol Chemical compound CC(O)C1=CC=C(Br)N=C1 QDOJZKXYIAXLDB-UHFFFAOYSA-N 0.000 description 3
- DCYLRSYWXFRFOX-UHFFFAOYSA-N 2-[5-[1-(methoxymethoxy)ethyl]pyridin-2-yl]ethanol Chemical compound COCOC(C)C1=CC=C(CCO)N=C1 DCYLRSYWXFRFOX-UHFFFAOYSA-N 0.000 description 3
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 3
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 3
- 150000001340 alkali metals Chemical class 0.000 description 3
- 229940125782 compound 2 Drugs 0.000 description 3
- 229910052802 copper Inorganic materials 0.000 description 3
- 229940079593 drug Drugs 0.000 description 3
- 239000003814 drug Substances 0.000 description 3
- 238000004128 high performance liquid chromatography Methods 0.000 description 3
- 229910052739 hydrogen Inorganic materials 0.000 description 3
- 239000001257 hydrogen Substances 0.000 description 3
- 239000007788 liquid Substances 0.000 description 3
- 238000005259 measurement Methods 0.000 description 3
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 3
- 239000000377 silicon dioxide Substances 0.000 description 3
- 239000011734 sodium Substances 0.000 description 3
- 239000011550 stock solution Substances 0.000 description 3
- 238000012360 testing method Methods 0.000 description 3
- FANCTJAFZSYTIS-IQUVVAJASA-N (1r,3s,5z)-5-[(2e)-2-[(1r,3as,7ar)-7a-methyl-1-[(2r)-4-(phenylsulfonimidoyl)butan-2-yl]-2,3,3a,5,6,7-hexahydro-1h-inden-4-ylidene]ethylidene]-4-methylidenecyclohexane-1,3-diol Chemical compound C([C@@H](C)[C@@H]1[C@]2(CCCC(/[C@@H]2CC1)=C\C=C\1C([C@@H](O)C[C@H](O)C/1)=C)C)CS(=N)(=O)C1=CC=CC=C1 FANCTJAFZSYTIS-IQUVVAJASA-N 0.000 description 2
- SZUVGFMDDVSKSI-WIFOCOSTSA-N (1s,2s,3s,5r)-1-(carboxymethyl)-3,5-bis[(4-phenoxyphenyl)methyl-propylcarbamoyl]cyclopentane-1,2-dicarboxylic acid Chemical compound O=C([C@@H]1[C@@H]([C@](CC(O)=O)([C@H](C(=O)N(CCC)CC=2C=CC(OC=3C=CC=CC=3)=CC=2)C1)C(O)=O)C(O)=O)N(CCC)CC(C=C1)=CC=C1OC1=CC=CC=C1 SZUVGFMDDVSKSI-WIFOCOSTSA-N 0.000 description 2
- SHAHPWSYJFYMRX-GDLCADMTSA-N (2S)-2-(4-{[(1R,2S)-2-hydroxycyclopentyl]methyl}phenyl)propanoic acid Chemical compound C1=CC([C@@H](C(O)=O)C)=CC=C1C[C@@H]1[C@@H](O)CCC1 SHAHPWSYJFYMRX-GDLCADMTSA-N 0.000 description 2
- LJIOTBMDLVHTBO-CUYJMHBOSA-N (2s)-2-amino-n-[(1r,2r)-1-cyano-2-[4-[4-(4-methylpiperazin-1-yl)sulfonylphenyl]phenyl]cyclopropyl]butanamide Chemical compound CC[C@H](N)C(=O)N[C@]1(C#N)C[C@@H]1C1=CC=C(C=2C=CC(=CC=2)S(=O)(=O)N2CCN(C)CC2)C=C1 LJIOTBMDLVHTBO-CUYJMHBOSA-N 0.000 description 2
- VIMMECPCYZXUCI-MIMFYIINSA-N (4s,6r)-6-[(1e)-4,4-bis(4-fluorophenyl)-3-(1-methyltetrazol-5-yl)buta-1,3-dienyl]-4-hydroxyoxan-2-one Chemical compound CN1N=NN=C1C(\C=C\[C@@H]1OC(=O)C[C@@H](O)C1)=C(C=1C=CC(F)=CC=1)C1=CC=C(F)C=C1 VIMMECPCYZXUCI-MIMFYIINSA-N 0.000 description 2
- IGVKWAAPMVVTFX-BUHFOSPRSA-N (e)-octadec-5-en-7,9-diynoic acid Chemical compound CCCCCCCCC#CC#C\C=C\CCCC(O)=O IGVKWAAPMVVTFX-BUHFOSPRSA-N 0.000 description 2
- PVRYOKQFLBSILA-UHFFFAOYSA-N 1-(6-methylpyridin-3-yl)ethanone Chemical compound CC(=O)C1=CC=C(C)N=C1 PVRYOKQFLBSILA-UHFFFAOYSA-N 0.000 description 2
- VBIBXVPTBBPIOC-UHFFFAOYSA-N 2-[5-[2-[tert-butyl(dimethyl)silyl]oxyethyl]pyridin-2-yl]ethanol Chemical compound CC(C)(C)[Si](OCCC=1C=CC(=NC=1)CCO)(C)C VBIBXVPTBBPIOC-UHFFFAOYSA-N 0.000 description 2
- WFIFHOWQEJADPH-UHFFFAOYSA-N 2-chloro-4-(2-oxo-4-phenylpyrrolidin-1-yl)benzenesulfonamide Chemical compound C1=C(Cl)C(S(=O)(=O)N)=CC=C1N1C(=O)CC(C=2C=CC=CC=2)C1 WFIFHOWQEJADPH-UHFFFAOYSA-N 0.000 description 2
- GNFTZDOKVXKIBK-UHFFFAOYSA-N 3-(2-methoxyethoxy)benzohydrazide Chemical compound COCCOC1=CC=CC(C(=O)NN)=C1 GNFTZDOKVXKIBK-UHFFFAOYSA-N 0.000 description 2
- PPCLBBXZDXYYQR-UHFFFAOYSA-N 4-[2-[5-[1-(methoxymethoxy)ethyl]pyridin-2-yl]ethoxy]benzaldehyde Chemical compound N1=CC(C(C)OCOC)=CC=C1CCOC1=CC=C(C=O)C=C1 PPCLBBXZDXYYQR-UHFFFAOYSA-N 0.000 description 2
- AOHSDRCYABCFTI-UHFFFAOYSA-N 5-[1-(methoxymethoxy)ethyl]-2-methylpyridine Chemical compound COCOC(C)C1=CC=C(C)N=C1 AOHSDRCYABCFTI-UHFFFAOYSA-N 0.000 description 2
- NPAXFQZLUHJHMA-UHFFFAOYSA-N 5-[[4-[2-[5-[1-(methoxymethoxy)ethyl]pyridin-2-yl]ethoxy]phenyl]methylidene]-1,3-thiazolidine-2,4-dione Chemical compound N1=CC(C(C)OCOC)=CC=C1CCOC(C=C1)=CC=C1C=C1C(=O)NC(=O)S1 NPAXFQZLUHJHMA-UHFFFAOYSA-N 0.000 description 2
- 229910015844 BCl3 Inorganic materials 0.000 description 2
- 239000005711 Benzoic acid Substances 0.000 description 2
- BVKZGUZCCUSVTD-UHFFFAOYSA-L Carbonate Chemical compound [O-]C([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-L 0.000 description 2
- XJUZRXYOEPSWMB-UHFFFAOYSA-N Chloromethyl methyl ether Chemical compound COCCl XJUZRXYOEPSWMB-UHFFFAOYSA-N 0.000 description 2
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical compound [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 description 2
- 229910021589 Copper(I) bromide Inorganic materials 0.000 description 2
- 229910021591 Copper(I) chloride Inorganic materials 0.000 description 2
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 2
- 102000000536 PPAR gamma Human genes 0.000 description 2
- 108010016731 PPAR gamma Proteins 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 2
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 2
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 2
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 2
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 2
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 2
- 150000001338 aliphatic hydrocarbons Chemical class 0.000 description 2
- 125000003545 alkoxy group Chemical group 0.000 description 2
- 229910021529 ammonia Inorganic materials 0.000 description 2
- 239000006286 aqueous extract Substances 0.000 description 2
- 239000008346 aqueous phase Substances 0.000 description 2
- 238000010936 aqueous wash Methods 0.000 description 2
- 125000004429 atom Chemical group 0.000 description 2
- 235000010233 benzoic acid Nutrition 0.000 description 2
- 229910052799 carbon Inorganic materials 0.000 description 2
- 210000003169 central nervous system Anatomy 0.000 description 2
- PBAYDYUZOSNJGU-UHFFFAOYSA-N chelidonic acid Natural products OC(=O)C1=CC(=O)C=C(C(O)=O)O1 PBAYDYUZOSNJGU-UHFFFAOYSA-N 0.000 description 2
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Natural products OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 2
- 229910017052 cobalt Inorganic materials 0.000 description 2
- 239000010941 cobalt Substances 0.000 description 2
- GUTLYIVDDKVIGB-UHFFFAOYSA-N cobalt atom Chemical compound [Co] GUTLYIVDDKVIGB-UHFFFAOYSA-N 0.000 description 2
- 230000000295 complement effect Effects 0.000 description 2
- 229940126543 compound 14 Drugs 0.000 description 2
- OXBLHERUFWYNTN-UHFFFAOYSA-M copper(I) chloride Chemical compound [Cu]Cl OXBLHERUFWYNTN-UHFFFAOYSA-M 0.000 description 2
- NKNDPYCGAZPOFS-UHFFFAOYSA-M copper(i) bromide Chemical compound Br[Cu] NKNDPYCGAZPOFS-UHFFFAOYSA-M 0.000 description 2
- XBDQKXXYIPTUBI-UHFFFAOYSA-N dimethylselenoniopropionate Natural products CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 2
- 229910001873 dinitrogen Inorganic materials 0.000 description 2
- UZUODNWWWUQRIR-UHFFFAOYSA-L disodium;3-aminonaphthalene-1,5-disulfonate Chemical compound [Na+].[Na+].C1=CC=C(S([O-])(=O)=O)C2=CC(N)=CC(S([O-])(=O)=O)=C21 UZUODNWWWUQRIR-UHFFFAOYSA-L 0.000 description 2
- 230000000694 effects Effects 0.000 description 2
- 238000001704 evaporation Methods 0.000 description 2
- 230000008020 evaporation Effects 0.000 description 2
- 238000000605 extraction Methods 0.000 description 2
- 238000000589 high-performance liquid chromatography-mass spectrometry Methods 0.000 description 2
- RWSOTUBLDIXVET-UHFFFAOYSA-M hydrosulfide Chemical compound [SH-] RWSOTUBLDIXVET-UHFFFAOYSA-M 0.000 description 2
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 2
- WGOPGODQLGJZGL-UHFFFAOYSA-N lithium;butane Chemical compound [Li+].CC[CH-]C WGOPGODQLGJZGL-UHFFFAOYSA-N 0.000 description 2
- CETVQRFGPOGIQJ-UHFFFAOYSA-N lithium;hexane Chemical compound [Li+].CCCCC[CH2-] CETVQRFGPOGIQJ-UHFFFAOYSA-N 0.000 description 2
- 238000002156 mixing Methods 0.000 description 2
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 2
- 235000005985 organic acids Nutrition 0.000 description 2
- 239000012074 organic phase Substances 0.000 description 2
- 229910052700 potassium Inorganic materials 0.000 description 2
- 239000011591 potassium Substances 0.000 description 2
- LPNYRYFBWFDTMA-UHFFFAOYSA-N potassium tert-butoxide Chemical compound [K+].CC(C)(C)[O-] LPNYRYFBWFDTMA-UHFFFAOYSA-N 0.000 description 2
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 2
- 238000000425 proton nuclear magnetic resonance spectrum Methods 0.000 description 2
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 2
- 238000000926 separation method Methods 0.000 description 2
- 239000000741 silica gel Substances 0.000 description 2
- 229910002027 silica gel Inorganic materials 0.000 description 2
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 2
- 235000017557 sodium bicarbonate Nutrition 0.000 description 2
- 239000000126 substance Substances 0.000 description 2
- FAQYAMRNWDIXMY-UHFFFAOYSA-N trichloroborane Chemical compound ClB(Cl)Cl FAQYAMRNWDIXMY-UHFFFAOYSA-N 0.000 description 2
- JQSHBVHOMNKWFT-DTORHVGOSA-N varenicline Chemical compound C12=CC3=NC=CN=C3C=C2[C@H]2C[C@@H]1CNC2 JQSHBVHOMNKWFT-DTORHVGOSA-N 0.000 description 2
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 description 1
- UNILWMWFPHPYOR-KXEYIPSPSA-M 1-[6-[2-[3-[3-[3-[2-[2-[3-[[2-[2-[[(2r)-1-[[2-[[(2r)-1-[3-[2-[2-[3-[[2-(2-amino-2-oxoethoxy)acetyl]amino]propoxy]ethoxy]ethoxy]propylamino]-3-hydroxy-1-oxopropan-2-yl]amino]-2-oxoethyl]amino]-3-[(2r)-2,3-di(hexadecanoyloxy)propyl]sulfanyl-1-oxopropan-2-yl Chemical compound O=C1C(SCCC(=O)NCCCOCCOCCOCCCNC(=O)COCC(=O)N[C@@H](CSC[C@@H](COC(=O)CCCCCCCCCCCCCCC)OC(=O)CCCCCCCCCCCCCCC)C(=O)NCC(=O)N[C@H](CO)C(=O)NCCCOCCOCCOCCCNC(=O)COCC(N)=O)CC(=O)N1CCNC(=O)CCCCCN\1C2=CC=C(S([O-])(=O)=O)C=C2CC/1=C/C=C/C=C/C1=[N+](CC)C2=CC=C(S([O-])(=O)=O)C=C2C1 UNILWMWFPHPYOR-KXEYIPSPSA-M 0.000 description 1
- YQTCQNIPQMJNTI-UHFFFAOYSA-N 2,2-dimethylpropan-1-one Chemical group CC(C)(C)[C]=O YQTCQNIPQMJNTI-UHFFFAOYSA-N 0.000 description 1
- 125000003903 2-propenyl group Chemical group [H]C([*])([H])C([H])=C([H])[H] 0.000 description 1
- 125000004105 2-pyridyl group Chemical group N1=C([*])C([H])=C([H])C([H])=C1[H] 0.000 description 1
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 description 1
- UDDCGCGNWXOFEI-UHFFFAOYSA-N 5-[[4-[2-[5-[1-(methoxymethoxy)ethyl]pyridin-2-yl]ethoxy]phenyl]methyl]-1,3-thiazolidine-2,4-dione Chemical compound N1=CC(C(C)OCOC)=CC=C1CCOC(C=C1)=CC=C1CC1C(=O)NC(=O)S1 UDDCGCGNWXOFEI-UHFFFAOYSA-N 0.000 description 1
- PXRKCOCTEMYUEG-UHFFFAOYSA-N 5-aminoisoindole-1,3-dione Chemical compound NC1=CC=C2C(=O)NC(=O)C2=C1 PXRKCOCTEMYUEG-UHFFFAOYSA-N 0.000 description 1
- IDCLTMRSSAXUNY-UHFFFAOYSA-N 5-hydroxylansoprazole Chemical compound CC1=C(OCC(F)(F)F)C=CN=C1CS(=O)C1=NC2=CC(O)=CC=C2N1 IDCLTMRSSAXUNY-UHFFFAOYSA-N 0.000 description 1
- 208000024827 Alzheimer disease Diseases 0.000 description 1
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical compound [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 description 1
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 1
- ROFVEXUMMXZLPA-UHFFFAOYSA-N Bipyridyl Chemical group N1=CC=CC=C1C1=CC=CC=N1 ROFVEXUMMXZLPA-UHFFFAOYSA-N 0.000 description 1
- 206010005003 Bladder cancer Diseases 0.000 description 1
- VKYXHVFPMYEGSY-UHFFFAOYSA-N CS(=O)(=O)OCCC1=NC=C(C=C1)C(C)OCOC Chemical compound CS(=O)(=O)OCCC1=NC=C(C=C1)C(C)OCOC VKYXHVFPMYEGSY-UHFFFAOYSA-N 0.000 description 1
- DCERHCFNWRGHLK-UHFFFAOYSA-N C[Si](C)C Chemical compound C[Si](C)C DCERHCFNWRGHLK-UHFFFAOYSA-N 0.000 description 1
- 208000024412 Friedreich ataxia Diseases 0.000 description 1
- 241000871495 Heeria argentea Species 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- 206010022998 Irritability Diseases 0.000 description 1
- 239000012448 Lithium borohydride Substances 0.000 description 1
- 241001465754 Metazoa Species 0.000 description 1
- 239000012359 Methanesulfonyl chloride Substances 0.000 description 1
- 235000019502 Orange oil Nutrition 0.000 description 1
- 208000018737 Parkinson disease Diseases 0.000 description 1
- 235000002492 Rungia klossii Nutrition 0.000 description 1
- 244000117054 Rungia klossii Species 0.000 description 1
- XUIMIQQOPSSXEZ-UHFFFAOYSA-N Silicon Chemical group [Si] XUIMIQQOPSSXEZ-UHFFFAOYSA-N 0.000 description 1
- 229910008433 SnCU Inorganic materials 0.000 description 1
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 1
- 229910010165 TiCu Inorganic materials 0.000 description 1
- 208000007097 Urinary Bladder Neoplasms Diseases 0.000 description 1
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 1
- DGEZNRSVGBDHLK-UHFFFAOYSA-N [1,10]phenanthroline Chemical compound C1=CN=C2C3=NC=CC=C3C=CC2=C1 DGEZNRSVGBDHLK-UHFFFAOYSA-N 0.000 description 1
- YLEIFZAVNWDOBM-ZTNXSLBXSA-N ac1l9hc7 Chemical compound C([C@H]12)C[C@@H](C([C@@H](O)CC3)(C)C)[C@@]43C[C@@]14CC[C@@]1(C)[C@@]2(C)C[C@@H]2O[C@]3(O)[C@H](O)C(C)(C)O[C@@H]3[C@@H](C)[C@H]12 YLEIFZAVNWDOBM-ZTNXSLBXSA-N 0.000 description 1
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 description 1
- 150000007513 acids Chemical class 0.000 description 1
- YBCVMFKXIKNREZ-UHFFFAOYSA-N acoh acetic acid Chemical compound CC(O)=O.CC(O)=O YBCVMFKXIKNREZ-UHFFFAOYSA-N 0.000 description 1
- 239000000556 agonist Substances 0.000 description 1
- 150000001298 alcohols Chemical class 0.000 description 1
- 229910001854 alkali hydroxide Inorganic materials 0.000 description 1
- 239000000908 ammonium hydroxide Substances 0.000 description 1
- 206010002026 amyotrophic lateral sclerosis Diseases 0.000 description 1
- 238000004458 analytical method Methods 0.000 description 1
- 125000003236 benzoyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C(*)=O 0.000 description 1
- 244000309464 bull Species 0.000 description 1
- 229910052792 caesium Inorganic materials 0.000 description 1
- TVFDJXOCXUVLDH-UHFFFAOYSA-N caesium atom Chemical compound [Cs] TVFDJXOCXUVLDH-UHFFFAOYSA-N 0.000 description 1
- 230000000711 cancerogenic effect Effects 0.000 description 1
- 150000001721 carbon Chemical group 0.000 description 1
- 125000004432 carbon atom Chemical group C* 0.000 description 1
- 150000005323 carbonate salts Chemical class 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-N carbonic acid Chemical compound OC(O)=O BVKZGUZCCUSVTD-UHFFFAOYSA-N 0.000 description 1
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 1
- 150000001735 carboxylic acids Chemical class 0.000 description 1
- 231100000357 carcinogen Toxicity 0.000 description 1
- 239000003183 carcinogenic agent Substances 0.000 description 1
- 230000008859 change Effects 0.000 description 1
- 239000003795 chemical substances by application Substances 0.000 description 1
- 229940061627 chloromethyl methyl ether Drugs 0.000 description 1
- 238000003776 cleavage reaction Methods 0.000 description 1
- 229910001429 cobalt ion Inorganic materials 0.000 description 1
- QAHREYKOYSIQPH-UHFFFAOYSA-L cobalt(II) acetate Chemical compound [Co+2].CC([O-])=O.CC([O-])=O QAHREYKOYSIQPH-UHFFFAOYSA-L 0.000 description 1
- 230000000052 comparative effect Effects 0.000 description 1
- 229940125904 compound 1 Drugs 0.000 description 1
- 150000001879 copper Chemical class 0.000 description 1
- 239000013058 crude material Substances 0.000 description 1
- 125000004122 cyclic group Chemical group 0.000 description 1
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 1
- 230000001419 dependent effect Effects 0.000 description 1
- 206010012601 diabetes mellitus Diseases 0.000 description 1
- 125000000532 dioxanyl group Chemical group 0.000 description 1
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 1
- KPUWHANPEXNPJT-UHFFFAOYSA-N disiloxane Chemical compound [SiH3]O[SiH3] KPUWHANPEXNPJT-UHFFFAOYSA-N 0.000 description 1
- 208000035475 disorder Diseases 0.000 description 1
- 230000002526 effect on cardiovascular system Effects 0.000 description 1
- 239000003480 eluent Substances 0.000 description 1
- 238000005516 engineering process Methods 0.000 description 1
- DZGCGKFAPXFTNM-UHFFFAOYSA-N ethanol;hydron;chloride Chemical compound Cl.CCO DZGCGKFAPXFTNM-UHFFFAOYSA-N 0.000 description 1
- 239000004210 ether based solvent Substances 0.000 description 1
- 150000002170 ethers Chemical class 0.000 description 1
- 238000002474 experimental method Methods 0.000 description 1
- 150000002222 fluorine compounds Chemical class 0.000 description 1
- 239000008098 formaldehyde solution Substances 0.000 description 1
- 125000000524 functional group Chemical group 0.000 description 1
- 239000011521 glass Substances 0.000 description 1
- 150000004820 halides Chemical class 0.000 description 1
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 150000003840 hydrochlorides Chemical class 0.000 description 1
- 239000005457 ice water Substances 0.000 description 1
- 238000001727 in vivo Methods 0.000 description 1
- 238000010348 incorporation Methods 0.000 description 1
- 239000013067 intermediate product Substances 0.000 description 1
- 125000003253 isopropoxy group Chemical group [H]C([H])([H])C([H])(O*)C([H])([H])[H] 0.000 description 1
- 230000000155 isotopic effect Effects 0.000 description 1
- 125000005647 linker group Chemical group 0.000 description 1
- YNESATAKKCNGOF-UHFFFAOYSA-N lithium bis(trimethylsilyl)amide Chemical compound [Li+].C[Si](C)(C)[N-][Si](C)(C)C YNESATAKKCNGOF-UHFFFAOYSA-N 0.000 description 1
- UKZCGMDMXDLAGZ-UHFFFAOYSA-M magnesium;2-methylpropane;bromide Chemical compound [Mg+2].[Br-].C[C-](C)C UKZCGMDMXDLAGZ-UHFFFAOYSA-M 0.000 description 1
- CQRPUKWAZPZXTO-UHFFFAOYSA-M magnesium;2-methylpropane;chloride Chemical compound [Mg+2].[Cl-].C[C-](C)C CQRPUKWAZPZXTO-UHFFFAOYSA-M 0.000 description 1
- 238000004949 mass spectrometry Methods 0.000 description 1
- 238000002844 melting Methods 0.000 description 1
- 230000008018 melting Effects 0.000 description 1
- 230000037353 metabolic pathway Effects 0.000 description 1
- QARBMVPHQWIHKH-UHFFFAOYSA-N methanesulfonyl chloride Chemical compound CS(Cl)(=O)=O QARBMVPHQWIHKH-UHFFFAOYSA-N 0.000 description 1
- 125000000325 methylidene group Chemical group [H]C([H])=* 0.000 description 1
- 150000007522 mineralic acids Chemical class 0.000 description 1
- GVOISEJVFFIGQE-YCZSINBZSA-N n-[(1r,2s,5r)-5-[methyl(propan-2-yl)amino]-2-[(3s)-2-oxo-3-[[6-(trifluoromethyl)quinazolin-4-yl]amino]pyrrolidin-1-yl]cyclohexyl]acetamide Chemical compound CC(=O)N[C@@H]1C[C@H](N(C)C(C)C)CC[C@@H]1N1C(=O)[C@@H](NC=2C3=CC(=CC=C3N=CN=2)C(F)(F)F)CC1 GVOISEJVFFIGQE-YCZSINBZSA-N 0.000 description 1
- 230000004770 neurodegeneration Effects 0.000 description 1
- 208000015122 neurodegenerative disease Diseases 0.000 description 1
- 239000010502 orange oil Substances 0.000 description 1
- 150000002891 organic anions Chemical class 0.000 description 1
- 125000000962 organic group Chemical group 0.000 description 1
- 125000004430 oxygen atom Chemical group O* 0.000 description 1
- 125000001147 pentyl group Chemical group C(CCCC)* 0.000 description 1
- 230000000144 pharmacologic effect Effects 0.000 description 1
- 230000003389 potentiating effect Effects 0.000 description 1
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 1
- IUVKMZGDUIUOCP-BTNSXGMBSA-N quinbolone Chemical compound O([C@H]1CC[C@H]2[C@H]3[C@@H]([C@]4(C=CC(=O)C=C4CC3)C)CC[C@@]21C)C1=CCCC1 IUVKMZGDUIUOCP-BTNSXGMBSA-N 0.000 description 1
- 239000012429 reaction media Substances 0.000 description 1
- 238000001953 recrystallisation Methods 0.000 description 1
- 238000013341 scale-up Methods 0.000 description 1
- 230000007017 scission Effects 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- 239000011780 sodium chloride Substances 0.000 description 1
- 239000012312 sodium hydride Substances 0.000 description 1
- 229910000104 sodium hydride Inorganic materials 0.000 description 1
- 230000002194 synthesizing effect Effects 0.000 description 1
- 230000009885 systemic effect Effects 0.000 description 1
- 125000005207 tetraalkylammonium group Chemical group 0.000 description 1
- 125000000026 trimethylsilyl group Chemical group [H]C([H])([H])[Si]([*])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 208000001072 type 2 diabetes mellitus Diseases 0.000 description 1
- 201000005112 urinary bladder cancer Diseases 0.000 description 1
- 239000003643 water by type Substances 0.000 description 1
- 230000004584 weight gain Effects 0.000 description 1
- 235000019786 weight gain Nutrition 0.000 description 1
- 239000011701 zinc Substances 0.000 description 1
- 229910052725 zinc Inorganic materials 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4427—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/06—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom containing only hydrogen and carbon atoms in addition to the ring nitrogen atom
- C07D213/127—Preparation from compounds containing pyridine rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F7/00—Compounds containing elements of Groups 4 or 14 of the Periodic Table
- C07F7/02—Silicon compounds
- C07F7/08—Compounds having one or more C—Si linkages
- C07F7/10—Compounds having one or more C—Si linkages containing nitrogen having a Si-N linkage
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F7/00—Compounds containing elements of Groups 4 or 14 of the Periodic Table
- C07F7/02—Silicon compounds
- C07F7/08—Compounds having one or more C—Si linkages
- C07F7/18—Compounds having one or more C—Si linkages as well as one or more C—O—Si linkages
- C07F7/1804—Compounds having Si-O-C linkages
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Epidemiology (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
- Plural Heterocyclic Compounds (AREA)
- Pyridine Compounds (AREA)
Abstract
La descripción proporciona un proceso para preparar el compuesto de fórmula I y sus sales farmacéuticamente aceptables: y el proceso para preparar el intermedio de fórmula III: en el que PG se define como se establece en la memoria descriptiva. (Traducción automática con Google Translate, sin valor legal)
Description
DESCRIPCIÓN
Proceso para preparar 5-[[4-[2-[5-(1-hidroxietil)-2-piridinil]etoxi]fenil]metil]-2,4-tiazolidindiona y sales de la misma Campo de divulgación
La presente solicitud se refiere a un proceso novedoso para preparar 5-[[4-[2-[5-(1-hidroxietil)-2-piridinil]etoxi]fenil]metil]-2,4-tiazolidindiona, también conocido como el metabolito M-IV de pioglitazona, y sales farmacéuticamente aceptables de la misma.
Antecedentes de la técnica relacionada
La pioglitazona es un fármaco comercializado para su uso en el tratamiento de la diabetes mellitus tipo 2. La pioglitazona es un potente agonista para el receptor gamma activado por proliferador de peroxisomas (PPARy) y se han propuesto para el tratamiento de algunas enfermedades neurodegenerativas incluyendo enfermedad de Alzheimer, enfermedad de Parkinson, ALS y ataxia de Friedreich. La pioglitazona se ha asociado con efectos secundarios no deseados que incluyen efectos cardiovasculares, retención de líquidos, aumento de peso y cáncer de vejiga. Por lo tanto, altas dosis de pioglitazona no son deseables, ya que es probable que una alta exposición sistémica dé como resultado efectos secundarios graves.
La pioglitazona es un fármaco “inespecífico” que se convierte en muchos metabolitos in vivo. La ruta metabólica de pioglitazona después de la administración oral se ha estudiado en varias especies animales y en seres humanos y los metabolitos se han descrito en la bibliografía (véase, por ejemplo, los documentos de Sohda et al., Chem. Pharm. Bull., 1995, 43(12), 2168-2172) y de Maeshiba et al., Arzneim.-Forsch/Drug Res, 1997, 47 (I), 29-35). Se han identificado al menos seis metabolitos, denominados M-I a M-VI. Entre estos metabolitos, M-II, M-III y M-IV muestran cierta actividad farmacológica pero son menos activos que pioglitazona en modelos preclínicos diabéticos.
La 5-[[4-[2-[5-(1-hidroxietil)-2-piridinil]etoxi]fenil]metil]-2,4-tiazolidindiona tiene la siguiente estructura:

El documento de Tanis et al. (J. Med. Chem. 39(26):5053-5063 (1996)) describe la síntesis de 5-[[4-[2-[5-(1-hidroxietil)-2-piridinil]etoxi]fenil]metil]-2,4-tiazolidindiona de la siguiente manera:
Esquema 1

Tanis et al. describen que el producto intermedio 14 se obtuvo con un rendimiento del 27 % haciendo reaccionar el
compuesto 13 en un formaldehído acuoso al 37 % a 170 °C durante 6 horas. En este proceso, se obtuvo 5-[[4-[2-[5-(1-hidroxietil)-2-piridinil]etoxi]fenil]metil]-2,4-tiazolidindiona (compuesto 6 en el esquema 1) con un rendimiento global del 2,47 %.
El documento WO 2015/150476 A1 describe el uso de 5-[[4-[2-[5-(1-hidroxietil)-2-piridinil]etoxi]fenil]metil]-2,4-tiazolidindiona, y sus sales farmacéuticamente aceptables, en el tratamiento de trastornos del sistema nervioso central (SNC). El documento WO 2015/150476 A1 describe que se preparó 5-[[4-[2-[5-(1-hidroxietil)-2-piridinil]etoxi]fenil]metil]-2, 4-tiazolidindiona según el proceso de Tanis et al. (mencionado anteriormente) donde el producto intermedio correspondiente al compuesto 14 de Tanis et al. se preparó de manera similar a 160 °C durante 5 horas proporcionando un rendimiento del 17%. El rendimiento global de 5-[[4-[2-[5-(1-hidroxietil)-2-piridinil]etoxi]fenil]metil]-2,4-tiazolidindiona fue de aproximadamente el 1,5 %.
Debido al bajo rendimiento del producto intermedio 2-[5-(1-metoximetoxi-etil)piridin-2-il]etanol, la etapa de proceso para preparar este producto intermedio es crítica para el rendimiento global del producto, 5-[[4-[2-[5-(1-hidroxietil)-2-piridinil]etoxi]fenil]metil]-2,4-tiazolidindiona. Además, el proceso de la técnica anterior para obtener el compuesto 14 es difícil de escalar porque la reacción se lleva a cabo en un recipiente a presión a una temperatura muy alta y es una reacción muy inespecífica.
Por consiguiente, los procesos descritos en la técnica proporcionan el producto 5-[[4-[2-[5-(1-hidroxietil)-2-piridinil]etoxi]fenil]metil]-2,4-tiazolidindiona solo con un rendimiento global muy bajo y, por lo tanto, no son adecuados para la síntesis a gran escala. Además, el proceso de la técnica anterior emplea CH3OCH2Cl, un carcinógeno conocido, para proteger el grupo hidroxilo en el producto intermedio clave. Existe la necesidad de un proceso mejorado para sintetizar 5-[[4-[2-[5-(1-hidroxietil)-2-piridinil]etoxi]fenil]metil]-2,4-tiazolidindiona, y sus sales farmacéuticamente aceptables.
Breve sumario de la invención
La presente divulgación proporciona un proceso mejorado para preparar un compuesto de fórmula I:

5-[[4-[2-[5-(1-hidroxietil)-2-piridinil]etoxi]fenil]metil]-2,4-tiazolidindiona, y sales farmacéuticamente aceptables de la misma, denominados colectivamente en el presente documento “compuestos de la divulgación” (cada uno se denomina individualmente en lo sucesivo “compuesto de la divulgación”).
Un aspecto de la presente divulgación se refiere a un proceso para preparar un producto intermedio sintético que tiene la fórmula III:

en donde PG es un grupo protector. En una realización, PG es un grupo protector de sililo, tetrahidropiranilo, metoximetilo, o bencilo. En otra realización, PG es un grupo protector de sililo, tetrahidropiranilo, o metoximetilo. Un compuesto que tiene la fórmula III se usa en procesos de preparación de compuestos de la divulgación.
El proceso de preparación de un compuesto que tiene la fórmula III comprende hacer reaccionar un compuesto de fórmula II:

en donde PG es un grupo protector y LG1 es un grupo saliente,
con óxido de etileno en presencia de
(a) un alquil-litio y una sal de cobre (I), o
(b) un alquil-litio y un ácido de Lewis, y
un disolvente,
para dar un compuesto de fórmula III:

Otro aspecto de la divulgación se refiere a un proceso para preparar un compuesto de fórmula IV:

que comprende hacer reaccionar el compuesto de fórmula III:

en donde PG es como se definió anteriormente, con R1-Cl, en donde R1 es un organosulfonato, en presencia de una primera base.
En otro aspecto, la divulgación se refiere a un proceso de preparación de un compuesto de fórmula V:

en donde PG es como se definió anteriormente, que comprende hacer reaccionar el compuesto de fórmula IV con 4-hidroxibenzaldehído que tiene la estructura

en presencia de una segunda base, tal como un carbonato de metal alcalino, y opcionalmente un disolvente.
En otro aspecto, la divulgación se refiere a un proceso de preparación de un compuesto de fórmula VI:

en donde PG es como se definió anteriormente, que comprende hacer reaccionar el compuesto de fórmula V con 2,4
tiazolidindiona de fórmula

en presencia de piperidina y opcionalmente un disolvente, tal como un alcohol, y opcionalmente un ácido orgánico. Preferiblemente, esta reacción se lleva a cabo en presencia de piperidina y un disolvente, y opcionalmente un ácido orgánico.
En otro aspecto, la divulgación se refiere a un proceso de preparación de un compuesto de fórmula VII:

en donde PG es como se definió anteriormente, que comprende reducir el compuesto de fórmula VI.
En otro aspecto, la divulgación se refiere a un proceso para preparar un compuesto de fórmula I:

o una sal farmacéuticamente aceptable del mismo, que comprende desproteger el compuesto de fórmula VII y opcionalmente tratar adicionalmente con un ácido para formar una sal.
En otro aspecto, la divulgación se refiere a un compuesto de fórmula III en donde PG es un grupo protector seleccionado del grupo que consiste en un grupo protector de sililo, tetrahidropiranilo y bencilo, obtenido mediante un proceso descrito en el presente documento haciendo reaccionar el compuesto de fórmula II.
Se expondrán realizaciones y ventajas adicionales de la divulgación, en parte, en la descripción que aparece a continuación, y se desprenderán de la descripción, o pueden aprenderse mediante la puesta en práctica de la divulgación. Las realizaciones y ventajas de la divulgación se realizarán y alcanzarán por medio de los elementos y combinaciones parTiCUlarmente indicados en las reivindicaciones adjuntas.
Debe entenderse que tanto el sumario anterior como la siguiente descripción detallada son solo a modo de ejemplo y explicativos, y no son restrictivos de la invención como se reivindica.
Descripción detallada de la invención
Los inventores han realizado una serie de intentos para encontrar una ruta para sintetizar 5-[[4-[2-[5-(1-hidroxietil)-2-piridinil]etoxi]fenil]metil]-2,4-tiazolidindiona, así como el producto intermedio de fórmula III como se define a continuación, a escala comercial, obtener un rendimiento global mejorado de 5-[[4-[2-[5-(1-hidroxietil)-2-piridinil]etoxi]fenil]metil]-2,4-tiazolidindiona. Ahora se ha descubierto que los compuestos de la divulgación pueden sintetizarse en un proceso simple aplicable a gran escala y proporcionando el producto con una alta pureza y un rendimiento global mejorado.
La presente divulgación proporciona un proceso para preparar el producto intermedio sintético de fórmula III que tiene la estructura:

en donde PG es un grupo protector, dicho proceso comprende hacer reaccionar un compuesto de fórmula II:

en donde PG es un grupo protector y LG1 es un grupo saliente,
con óxido de etileno en presencia de
(a) un alquil-litio y una sal de cobre (I), o
(b) un alquil-litio y un ácido de Lewis, y
un disolvente,
para dar un compuesto de fórmula III:

En una realización, PG es un grupo protector de sililo, tetrahidropiranilo, metoximetilo, o bencilo. En otra realización, PG es un grupo protector de sililo, tetrahidropiranilo o metoximetilo. En otra realización, PG es un grupo protector de sililo.
En una realización, la sal de cobre (I) se selecciona del grupo que consiste en bromuro de cobre (I), cloruro de cobre (I), y yoduro de cobre (I). En otra realización, la sal de cobre (I) es yoduro de cobre (I).
En otra realización, el ácido de Lewis se selecciona del grupo que consiste en AlBr3, AlCh , FeBr3, FeCh , SnCU, Ti(OiPr)4, BF3, BF3 • O(Et)2, BBr3, BCh , TiCl4, y ZnCh . En otra realización, el ácido de Lewis se selecciona del grupo que consiste en BF3 • O(Et)2 , BBr3, BCh , TiCU, y ZnCl2. En otra realización, el ácido de Lewis se selecciona del grupo que consiste en BF3 • o(Et)2 , BBr3 y BCh .
En otra realización, el grupo saliente (LG1) es -Br.
En otra realización, el proceso comprende un alquil-litio y yoduro de cobre (I).
La presente divulgación proporciona un proceso para preparar el producto intermedio sintético de fórmula III que tiene la estructura:

en donde PG es un grupo protector, dicho proceso comprende hacer reaccionar un compuesto de fórmula II:

en donde PG es un grupo protector y LG1 es un grupo saliente,
con óxido de etileno en presencia de
(a) un alquil-litio y una sal de cobre (I), por ejemplo, yoduro de cobre (I); o
(b) un alquil-litio y un ácido de Lewis, por ejemplo, BF3 • O(Et)2, BBr3 o BCh, y
un disolvente,
para dar un compuesto de fórmula III:

En otra realización, PG es un grupo protector seleccionado del grupo que consiste en un grupo protector de sililo, tetrahidropiranilo, metoximetilo y bencilo. En otra realización, PG es un grupo protector seleccionado del grupo que consiste en un grupo protector de sililo, tetrahidropiranilo y metoximetilo.
En una realización, los reactivos, es decir, el óxido de etileno, alquil-litio y sal de cobre (I), por ejemplo, yoduro de cobre (I), o el ácido de Lewis, se añaden a la mezcla de reacción que comprende el compuesto de fórmula II a baja temperatura, por ejemplo, a una temperatura de aproximadamente -20 °C, aproximadamente -25 °C, aproximadamente -3o °C, aproximadamente -35 °C, aproximadamente -40 °C, aproximadamente -45 °C, aproximadamente -50 °C, aproximadamente -55 °C, aproximadamente -60 °C, aproximadamente -65 °C, aproximadamente -70 °C o aproximadamente -75 °C. En otra realización, los reactivos se añaden mientras se mantiene la temperatura de la mezcla de reacción por debajo de -20 °C. En otra realización, los reactivos se añaden mientras se mantiene la temperatura de la mezcla de reacción a aproximadamente < -55 °C. En otra realización, los reactivos se añaden a la mezcla de reacción a aproximadamente < -55 °C.
En otra realización, el óxido de etileno se añade a la mezcla de reacción después de añadir primero el alquil-litio. En otra realización, el óxido de etileno se añade a la mezcla de reacción después de añadir el alquil-litio y la sal de cobre (I), por ejemplo, yoduro de cobre (I), o el ácido de Lewis. En otra realización, se permite que la mezcla de reacción se caliente a temperatura ambiente, por ejemplo, aproximadamente 20-25 °C, después de la adición de los reactivos. En otra realización, la mezcla de reacción se mantiene a 20 °C-25 °C durante al menos 4 horas, durante al menos 6 horas o durante al menos 8 horas. En otra realización, la mezcla de reacción se mantiene a 20 °C-25 °C durante al menos 10 horas.
En una realización de este aspecto de la divulgación, la reacción se lleva a cabo en presencia de un alquil-litio y una sal de cobre (I). En otra realización, la reacción se lleva a cabo en presencia de un alquil-litio y yoduro de cobre (I). En otra realización, la reacción se lleva a cabo en presencia de un alquil-litio y un ácido de Lewis. En otra realización, la reacción se realiza en presencia de un alquil-litio y un ácido de Lewis seleccionado del grupo que consiste en BF3 • O(Et)2, BBr3 y BCh.
En otra realización, el proceso de la descripción comprende hacer reaccionar un compuesto de fórmula II:

en donde PG es un grupo protector seleccionado del grupo que consiste en un grupo protector de sililo, tetrahidropiranilo y metoximetilo y
LG1 es un grupo saliente,
con óxido de etileno en presencia de un alquil-litio y yoduro de cobre (I) y
un disolvente, en donde
la temperatura de reacción se mantiene por debajo de -20 °C cuando se añade el alquil-litio y el yoduro de cobre (I) a la mezcla de reacción,
para dar un compuesto de fórmula III:

En una realización, el óxido de etileno se añade a la mezcla de reacción después de añadir primero el alquil-litio. En otra realización, el óxido de etileno se añade a la mezcla de reacción mientras se mantiene la temperatura de reacción por debajo de -20 °C. En otra realización, el alquil-litio y la sal de cobre (I), por ejemplo, yoduro de cobre (I), se añaden mientras se mantiene la temperatura de la mezcla de reacción por debajo de -55 °C. En otra realización, el óxido de etileno se añade a la mezcla de reacción mientras se mantiene la temperatura de reacción por debajo de -55 °C. En otra realización, se permite que la mezcla de reacción se caliente a temperatura ambiente, por ejemplo, aproximadamente 20-25 °C, después de la adición de los reactivos. En otra realización, la mezcla de reacción se mantiene a 20 °C-25 °C durante al menos 4 horas, durante al menos 6 horas o durante al menos 8 horas. En otra realización, la mezcla de reacción se mantiene a 20 °C-25 °C durante al menos 10 horas.
En una realización, el disolvente se selecciona del grupo que consiste en un disolvente orgánico no polar y un disolvente orgánico polar, o una mezcla de los mismos. En otra realización, el disolvente se selecciona del grupo que consiste en un disolvente orgánico aprótico no polar y un disolvente orgánico aprótico polar, o una mezcla de los mismos. En otra realización, el disolvente es un disolvente orgánico no polar. En otra realización, el disolvente es un disolvente orgánico aprótico no polar. En otra realización, el disolvente es dietiléter o metil terc-butiléter. En otra realización, el disolvente es un disolvente orgánico polar. En otra realización, el disolvente es un disolvente orgánico aprótico polar. En otra realización, el disolvente se selecciona del grupo que consiste en tetrahidrofurano, dioxano y DMSO, o una mezcla de los mismos.
En otra realización, el proceso de la divulgación proporciona un proceso para preparar un compuesto que tiene la fórmula III:

comprendiendo el proceso:
(a) hacer reaccionar un compuesto que tiene la fórmula II:

con un alquil-litio en un primer disolvente a una temperatura para dar una primera mezcla de reacción;
en donde:
la temperatura está por debajo de -20 °C;
PG es un grupo protector, por ejemplo, un grupo protector seleccionado del grupo que consiste en un grupo protector de sililo, tetrahidropiranilo, metoximetilo y bencilo; y
LG1 es un grupo saliente, por ejemplo, Br,
(b) añadir una disolución de óxido de etileno en un segundo disolvente a dicha temperatura a dicha primera mezcla de reacción a dicha temperatura para dar una segunda mezcla de reacción; y
(c) añadir una sal de cobre (I), por ejemplo, yoduro de cobre (I), o un ácido de Lewis, por ejemplo, BF3 • O(Et)2 , BBr3 o BCl3, a dicha segunda mezcla de reacción a dicha temperatura para dar una tercera mezcla de reacción que comprende un compuesto que tiene la fórmula III.
En otra realización, el proceso de la divulgación proporciona un proceso para preparar un compuesto que tiene la fórmula III:

comprendiendo el proceso:
(a) hacer reaccionar un compuesto que tiene la fórmula II:

con un alquil-litio en un primer disolvente a una temperatura para dar una primera mezcla de reacción;
en donde:
la temperatura está por debajo de -20 °C;
PG es un grupo protector, por ejemplo, un grupo protector seleccionado del grupo que consiste en un grupo protector de sililo, tetrahidropiranilo y metoximetilo; y
LG1 es un grupo saliente,
(b) añadir una sal de cobre (I), por ejemplo, yoduro de cobre (I), o un ácido de Lewis, por ejemplo, BF3 • O(Et)2, BBr3 o BCl3, a la primera mezcla de reacción a dicha temperatura para dar una segunda mezcla de reacción; y
(c) añadir una disolución de óxido de etileno en un segundo disolvente a dicha temperatura a la segunda mezcla de reacción a dicha temperatura para dar una tercera mezcla de reacción que comprende un compuesto que tiene la fórmula III.
En una realización de estos aspectos de la divulgación, la temperatura está por debajo de -55 °C. En otra realización, se permite que la tercera mezcla de reacción se caliente a una temperatura de 20 °C-25 °C. En otra realización, la tercera mezcla de reacción se mantiene a 20 °C-25 °C durante al menos 4 horas. En otra realización, la tercera mezcla de reacción se mantiene a 20 °C-25 °C durante al menos 6 horas. En otra realización, la mezcla de reacción se mantiene a 20 °C-25 °C durante al menos 8 horas. En otra realización, la tercera mezcla de reacción se mantiene a 20 °C-25 °C durante al menos 10 horas. En una realización, el primer disolvente y el segundo disolvente son diferentes. En otra realización, el primer disolvente y el segundo disolvente son iguales, tales como, por ejemplo, dietiléter. Los disolventes adecuados son los descritos anteriormente. En otra realización, LG1 es bromuro, es decir, -Br.
En otra realización, el primer disolvente se selecciona del grupo que consiste en un disolvente aprótico no polar y un disolvente aprótico polar, o una mezcla de los mismos. En una realización, el primer disolvente es un disolvente aprótico no polar. En otra realización, el primer disolvente se selecciona del grupo que consiste en dietiléter y metil terc-butiléter, o una mezcla de los mismos. En otra realización, el primer disolvente es un disolvente aprótico polar. En otra realización, el primer disolvente se selecciona del grupo que consiste en THF, dioxano y DMSO, o una mezcla de los mismos.
En otra realización, el segundo disolvente se selecciona del grupo que consiste en un disolvente aprótico no polar y un disolvente aprótico polar, o una mezcla de los mismos. En una realización, el segundo disolvente es un disolvente aprótico no polar. En otra realización, el segundo disolvente se selecciona del grupo que consiste en dietiléter y metil terc-butiléter, o una mezcla de los mismos. En otra realización, el segundo disolvente es un disolvente aprótico polar. En otra realización, el segundo disolvente se selecciona del grupo que consiste en THF, dioxano, DMSO y una mezcla de los mismos.
En otra realización, el primer disolvente y el segundo disolvente se seleccionan cada uno independientemente del grupo que consiste en un disolvente aprótico no polar y un disolvente aprótico polar, o una mezcla de los mismos. En otra realización, el primer disolvente y el segundo disolvente son diferentes. En otra realización, el primer disolvente y el segundo disolvente son los mismos disolventes. En otra realización, tanto el primer disolvente como el segundo disolvente son dietiléter. En otra realización, el proceso comprende además aislar el compuesto que tiene la fórmula III.
En otra realización, el proceso de la divulgación proporciona un proceso, que comprende
combinar los reactantes:
(a) óxido de etileno;
(b) un compuesto que tiene la fórmula II:

(c) un alquil-litio; y
(d) una sal de cobre (I), por ejemplo, yoduro de cobre (I), o un ácido de Lewis, por ejemplo, BF3 • O(Et)2, BBr3 o BCh,
en un disolvente a una temperatura inferior a -20 °C,
en donde:
PG es un grupo protector, por ejemplo, un grupo protector seleccionado del grupo que consiste en un grupo protector de sililo, tetrahidropiranilo, metoximetilo y bencilo; y
LG1 es un grupo saliente,
para dar un compuesto que tiene la fórmula III:

El término “reactante” o “reactivo”, como se usa en el presente documento, se refiere a una sustancia que participa y/o experimenta un cambio durante una reacción química.
En los procesos descritos en el presente documento, los reactantes pueden combinarse en cualquier orden que proporcione la fórmula III con un rendimiento y pureza aceptables. Los reactantes también pueden combinarse tal cual o como una disolución. Por ejemplo, puede añadirse un disolvente al óxido de etileno para formar una disolución y esta disolución puede añadirse a otro reactante.
En una realización de este aspecto de la invención, los reactantes se combinan en un disolvente por debajo de -55 °C. En una realización, el alquil-litio se añade a una disolución del compuesto que tiene la fórmula II en el disolvente a la temperatura para formar una primera mezcla de reacción. En otra realización, una disolución del óxido de etileno en el disolvente se enfría a la temperatura y se añade a la primera mezcla de reacción a la temperatura para dar una segunda mezcla de reacción. En una realización, el disolvente se selecciona del grupo que consiste en un disolvente orgánico aprótico no polar y un disolvente orgánico aprótico polar, o una mezcla de los mismos. En otra realización, el disolvente es un disolvente orgánico aprótico no polar. En otra realización, el disolvente se selecciona del grupo que consiste en dietiléter y metil terc-butiléter, o una mezcla de los mismos. En otra realización, el disolvente es un disolvente orgánico aprótico polar. En otra realización, el disolvente se selecciona del grupo que consiste en tetrahidrofurano, dioxano y DMSO, o una mezcla de los mismos.
En una realización, la sal de cobre (I), por ejemplo, yoduro de cobre (I), o ácido de Lewis, por ejemplo, BF3 • O(Et)2, BBr3 o BCl3, se añade a la segunda mezcla de reacción a la temperatura para dar una tercera mezcla de reacción. En otra realización, se añade yoduro de cobre (I) a la segunda mezcla de reacción a la temperatura para dar una tercera mezcla de reacción. En otra realización, se permite que la tercera mezcla de reacción se caliente a una temperatura de 20 °C-25 °C. En otra realización, la tercera mezcla de reacción se mantiene a 20 °C-25 °C durante al menos 4 horas. En otra realización, la tercera mezcla de reacción se mantiene a 20 °C-25 °C durante al menos 6 horas. En otra realización, la tercera mezcla de reacción se mantiene a 20 °C-25 °C durante al menos 8 horas. En otra realización, la tercera mezcla de reacción se mantiene a 20 °C-25 °C durante al menos 10 horas.
En otra realización, la sal de cobre (I), por ejemplo, yoduro de cobre (I), o el ácido de Lewis, por ejemplo, BF3 • O(Et)2, BBr3 o BCh se añade a la primera mezcla de reacción a la temperatura para dar una cuarta mezcla de reacción. En otra realización, se añade yoduro de cobre (I) a la primera mezcla de reacción a la temperatura para dar una cuarta mezcla de reacción. En otra realización, una disolución del óxido de etileno en el disolvente se enfría a la temperatura y se añade a la cuarta mezcla de reacción a la temperatura para dar una quinta mezcla de reacción. En otra realización, se permite que la quinta mezcla de reacción se caliente a una temperatura de 20 °C-25 °C. En otra realización, la quinta mezcla de reacción se mantiene a 20 °C-25 °C durante al menos 4 horas. En otra realización, la quinta mezcla de reacción se mantiene a 20 °C-25 °C durante al menos 6 horas. En otra realización, la quinta mezcla de reacción se mantiene a 20 °C-25 °C durante al menos 8 horas. En otra realización, la quinta mezcla de reacción se mantiene a 20 °C-25 °C durante al menos 10 horas. En otra realización, LG1 es -Br.
En otra realización, la presente divulgación proporciona un proceso para preparar el producto intermedio clave usado en el proceso de preparación de compuestos de la divulgación, que tiene la fórmula III:

en donde PG es un grupo protector seleccionado de grupos protectores de sililo, tetrahidropiranilo o metoximetilo, comprendiendo dicho proceso hacer reaccionar un compuesto de fórmula II que tiene la estructura:
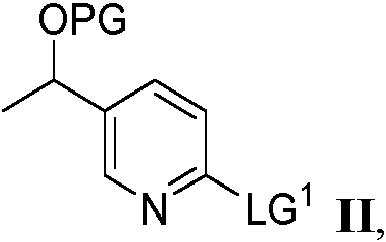
en donde PG es tal como se definió anteriormente y LG1 es un grupo saliente, con óxido de etileno en presencia de (a) un alquil-litio y yoduro de cobre (I) o (b) un alquil-litio y un ácido de Lewis seleccionado del grupo que consiste en BF3 • O(Et)2, BBr3 y BCl3 y un disolvente, en donde la temperatura de reacción se mantiene por debajo de -20 °C para dar el compuesto de fórmula III.
En una realización, PG se selecciona de grupos protectores de sililo. En otra realización, PG es un grupo alquil o aril sililo o una combinación de los mismos. En otra realización, PG es un grupo trialquilsililo. En otra realización, PG se selecciona del grupo que consiste en terc-butildimetilsililo (TBDMS), trimetilsililo (TMS), trietilsililo (TES), tercbutildifenilsililo (Tb Dp S) y triisopropilsililo (TIPS). En otra realización, p G es terc-butildimetilsililo (TBDMS).
En otra realización, PG es tetrahidropiranilo. En otra realización, PG es metoximetilo. En otra realización, PG es bencilo. Preferiblemente, PG es un grupo protector de sililo.
Grupos salientes adecuados para LG1 incluyen cloruro (-Cl), bromuro (-Br), yoduro (-I) y fluoruro (-F). Preferiblemente, LG1 es bromuro.
El disolvente usado en esta reacción puede ser un disolvente orgánico polar o no polar. En una realización, el disolvente es un disolvente orgánico aprótico polar o no polar. En una realización, el disolvente es un disolvente aprótico no polar seleccionado de éteres, tales como, por ejemplo, dietiléter y metil terc-butiléter. En otra realización, el disolvente es un disolvente aprótico polar, tal como, por ejemplo, tetrahidrofurano (THF). En otra realización, el disolvente aprótico polar es dioxano o dimetilsulfóxido (DMSO).
En una realización, la reacción se lleva a cabo en presencia de un alquil-litio y una sal de cobre (I), por ejemplo, yoduro de cobre (I), bromuro de cobre (I) o cloruro de cobre (I). En otra realización, la reacción se lleva a cabo en presencia de un alquil-litio y yoduro de cobre (I). En otra realización, el alquil-litio es alquil-litio C1-6. En otra realización, el alquillitio se selecciona del grupo que consiste en n-butil-litio (n-BuLi), sec-butil-litio (sec-BuLi) y hexil-litio (HxLi). Una relación molar adecuada del alquil-litio y el yoduro de cobre (I) usados en la reacción es de aproximadamente 8:1 a aproximadamente 2:1. En una realización, la relación molar del alquil-litio y yoduro de cobre (I) es de aproximadamente 2:1. En una realización, el cobre complejado se retira después de la reacción lavando con amoniaco al 10 %.
En otra realización, la reacción se lleva a cabo en presencia de un alquil-litio y un ácido de Lewis. En otra realización, la reacción se lleva a cabo en presencia de un alquil-litio y un ácido de Lewis seleccionado del grupo que consiste en BF3 • O(Et)2, BBr3 y BCh.En otra realización, el ácido de Lewis es BF3 • O(Et)2. Compuestos de alquil-litio adecuados se han descrito anteriormente.
En una realización, la temperatura de reacción, es decir, la temperatura de la mezcla de reacción, para formar el compuesto de fórmula III se mantiene a bajas temperaturas, tal como de aproximadamente -20 °C a aproximadamente -85 °C, cuando se añaden los reactivos a la mezcla de reacción que comprende el compuesto de fórmula II, seguido de agitación a temperatura ambiente, por ejemplo, aproximadamente 20-25 °C. En otra realización, la adición a la mezcla de reacción se realiza a una temperatura de aproximadamente -40 °C a aproximadamente -70 °C. En otra realización, la adición a la mezcla de reacción se realiza a una temperatura de aproximadamente -50 °C a aproximadamente -65 °C. En otra realización, la temperatura de reacción se mantiene por debajo de aproximadamente -55 °C cuando se añaden los reactivos.
En una realización, el tiempo de reacción para formar el compuesto de fórmula III a partir de un compuesto de fórmula II es de al menos 24 horas. En otra realización, los tiempos de reacción para formar el compuesto de fórmula III a partir de un compuesto de fórmula II son de aproximadamente 24 a aproximadamente 40 horas o aproximadamente 27 horas o aproximadamente 30 horas o aproximadamente 36 horas.
Después de que se complete la reacción para formar el compuesto de fórmula III a partir del compuesto de fórmula II, el compuesto de fórmula III se aísla de la mezcla de reacción y preferiblemente se purifica, por ejemplo, por cromatografía en columna o recristalización y preferiblemente por cromatografía en columna, tal como HPLC. El compuesto de fórmula III puede obtenerse con un buen rendimiento con alta pureza. En determinados aspectos de la divulgación, los inventores han obtenido el compuesto de fórmula III usando el proceso descrito anteriormente con aproximadamente un rendimiento del 95 % y aproximadamente una pureza del 80 %. En determinados aspectos de la divulgación, los inventores han obtenido el compuesto de fórmula III usando el proceso descrito anteriormente con aproximadamente un rendimiento del 50 % y aproximadamente una pureza del 99 %.
El proceso de la divulgación comprende además hacer reaccionar el compuesto de fórmula III con R1-Cl, en donde R1 es un organosulfonato, tal como un grupo tosilo (Ts) o un grupo mesilo (Ms), en presencia de una primera base para dar un compuesto de fórmula IV:

en donde PG y R1 son como se han definido anteriormente. La primera base puede ser, por ejemplo, uno o más de una amina, una sal de amonio cuaternario en combinación con una disolución acuosa de un hidróxido de metal alcalino, hidróxido de tetrabutilamonio o un hidróxido de metal alcalino. En una realización, la primera base es una amina. Aminas adecuadas incluyen, por ejemplo, trialquilamina, tales como trietilamina y piridina. En otra realización, la primera base es una sal de amonio cuaternario, tal como bromuro de tetra-n-butilamonio o fluoruro de tetra-nbutilamonio en combinación con una disolución acuosa de un hidróxido de metal alcalino. Hidróxidos metal alcalino adecuados incluyen NaOH y KOH. En otra realización, la primera base es hidróxido de tetrabutilamonio. En otra realización, la primera base es bromuro de tetra-n-butilamonio en disolución acuosa de hidróxido de alcalino. En otra realización, la primera base es bromuro de tetra-n-butilamonio en NaOH al 30 %.
En otra realización, R1-Cl se usa en una cantidad de desde aproximadamente 1 equivalente hasta aproximadamente 1,50 equivalentes. En otra realización, R1-Cl es p-Ts-Cl en una cantidad de desde 1 equivalente hasta aproximadamente 1,1 equivalentes.
Ventajosamente, la reacción para preparar el compuesto de fórmula IV se lleva a cabo en presencia de un disolvente, tal como, por ejemplo, tolueno o DCM. En una realización, el disolvente es tolueno.
En otra realización, la reacción para preparar el compuesto de fórmula IV se obtiene haciendo reaccionar p-Ts-Cl (de aproximadamente 1 eq a aproximadamente 1,1 eq) con el compuesto de fórmula III en presencia de tolueno, NaOH acuoso al 30 % y bromuro de tetra-n-butilamonio.
En una realización, la reacción para preparar el compuesto de fórmula IV se lleva a cabo a una temperatura de desde aproximadamente 15 °C hasta aproximadamente 30 °C y preferiblemente a una temperatura de desde aproximadamente 20 °C hasta aproximadamente 25 °C y preferiblemente a aproximadamente 20 °C.
En una realización, los tiempos de reacción para formar el compuesto de fórmula IV a partir del compuesto de fórmula III son de desde aproximadamente 5 hasta aproximadamente 26 horas o desde aproximadamente 10 hasta aproximadamente 26 o desde aproximadamente 15 hasta aproximadamente 25 horas o aproximadamente 22 horas.
El proceso de la divulgación comprende además hacer reaccionar el compuesto de fórmula IV con 4-hidroxibenzaldehído:

en presencia de una segunda base y un disolvente para dar un compuesto de fórmula V

en donde PG es como se definió anteriormente. Segundas bases adecuadas incluyen, por ejemplo, carbonatos de metal alcalino (tales como carbonato de potasio (K2CO3)), trialquilaminas (tal como trietilamina (Et3N)) y alcóxidos de metales alcalinos (tales como terc-butóxido de potasio (KOtBu)). En una realización, la segunda base es K2CO3. Ventajosamente, la cantidad de la segunda base es de desde aproximadamente 1 eq hasta aproximadamente 2 eq y preferiblemente aproximadamente 1 eq.
En una realización, el disolvente se selecciona del grupo que consiste en tolueno, etanol, 2-propanol, THF, 2-MeTHF y agua o combinaciones de los mismos. En otra realización, el disolvente es una mezcla de tolueno y etanol. En otra realización, el disolvente es una mezcla de tolueno y etanol en una relación de aproximadamente 6:4.
En una realización, la reacción para preparar el compuesto de fórmula V se realiza a una temperatura de desde aproximadamente 50 °C hasta aproximadamente 80 °C y preferiblemente a aproximadamente 80 °C.
En una realización, se añade agua adicional a la mezcla de reacción. En una realización, la cantidad de agua añadida es de desde aproximadamente 2 % hasta aproximadamente 7 % en v/v (es decir, el volumen de agua en relación con el volumen de disolventes utilizados). En otra realización, se añade agua en una cantidad de aproximadamente el 4 % en v/v, aproximadamente el 5 % en v/v o aproximadamente el 6 % en v/v. En otra realización, se añade agua en una cantidad del 5 % en v/v. En otra realización, la temperatura de la reacción es la mezcla es de desde aproximadamente 20 °C hasta aproximadamente 30 °C. En otra realización, se añade agua a aproximadamente 20 °C.
En una realización, los tiempos de reacción para formar el compuesto de fórmula V a partir del compuesto de fórmula IV son de desde aproximadamente 6 hasta aproximadamente 24 horas o de desde aproximadamente 10 hasta aproximadamente 22 horas o aproximadamente 16 horas o aproximadamente 18 horas.
En otra realización, el proceso comprende aislar y purificar el compuesto de fórmula V. En una realización, la purificación se realiza mediante extracción. En otra realización, la purificación comprende extraer con una disolución acuosa de bisulfito. En otra realización, la purificación comprende extraer una disolución que comprende el compuesto de fórmula V y etanol con una disolución acuosa de bisulfito que comprende etanol; separar la fase acuosa; extraer la fase acuosa con una mezcla de tolueno y heptano en donde el pH de la mezcla se ajusta a pH 12; separar la capa orgánica; y recuperar dicho compuesto purificado de fórmula V. En una realización, el pH se ajusta a pH 12 mediante la adición de NaOH acuoso.
El proceso de la presente divulgación comprende además hacer reaccionar el compuesto de fórmula V con 2,4-tiazolidindiona:

en presencia de piperidina y opcionalmente un disolvente, y opcionalmente un ácido orgánico, para dar un compuesto de fórmula VI:

en donde PG es como se definió anteriormente.
En una realización, la reacción para formar un compuesto de fórmula VI se lleva a cabo en presencia de piperidina y un disolvente y opcionalmente un ácido orgánico. Disolventes adecuados en esta reacción incluyen tolueno, alcoholes inferiores (tales como metanol y etanol), hexano, ciclohexano y mezclas de los mismos. En un aspecto, el disolvente es tolueno o metanol, o una mezcla de los mismos. Ventajosamente, el disolvente es metanol.
En una realización, la reacción se lleva a cabo en presencia de un ácido orgánico. Ácidos orgánicos adecuados incluyen ácidos carboxílicos, tales como, por ejemplo, ácidos carboxílicos de alquilo (C1-12) (por ejemplo, ácido acético), ácido fórmico y ácido benzoico. En otra realización, la reacción se lleva a cabo sin un ácido orgánico.
Temperaturas de reacción adecuadas para obtener el compuesto de fórmula VI dependen del disolvente usado en la reacción. Por ejemplo, la temperatura de reacción adecuada puede variar dentro del intervalo de desde aproximadamente 45 °C hasta aproximadamente 80 °C. Preferiblemente, la temperatura de reacción es de aproximadamente 47 °C cuando el disolvente es metanol. Preferiblemente, la temperatura de reacción es de aproximadamente 78 °C cuando el disolvente es tolueno. El tiempo de reacción varía dependiendo del disolvente y puede ser de desde aproximadamente 5 horas hasta aproximadamente 37 horas o desde aproximadamente 9 hasta aproximadamente 25 horas.
En otra realización, la reacción para formar el compuesto de fórmula VI se lleva a cabo en ausencia de un disolvente, a una temperatura que es suficientemente alta para causar la fusión al menos parcial de la mezcla de reacción. Una temperatura preferida de este tipo está en el intervalo de desde 100 °C hasta 250 °C y especialmente de desde 140 °C hasta 200 °C.
Preferiblemente, la reacción para formar el compuesto de fórmula VI se lleva a cabo en presencia de un disolvente, y opcionalmente un ácido orgánico.
El proceso de la presente divulgación comprende además reducir el compuesto de fórmula VI para dar un compuesto de fórmula VII:

en donde PG es como se definió anteriormente. La reducción se realiza típicamente permitiendo que el compuesto de fórmula VI reaccione con un agente reductor en presencia de un ion metálico y un agente complejante para el ion metálico (un ligando).
La temperatura de reacción de esta reacción de reducción puede variar entre -20 °C y 45 °C y está preferiblemente entre 20 °C y 35 °C. Ventajosamente, la temperatura de reacción es de aproximadamente 30 °C.
Disolventes adecuados para usar en esta etapa de reducción incluyen metanol, etanol, i-propanol, dimetilformamida (DMF) y tetrahidrofurano (THF). Si se usan DMF o THF como disolventes, deben estar presentes agua o un alcohol (tal como metanol, etanol o i-propanol) (típicamente en una cantidad de > 1 eq). Ventajosamente, la combinación de disolventes contiene agua y THF. En una realización, la combinación de disolventes contiene una disolución acuosa de NaOH, un ácido carboxílico (tal como ácido acético) y THF.
En una realización, el pH de la mezcla de reacción se mantiene a de aproximadamente pH 9,5 a aproximadamente pH 10,5.
Agentes reductores adecuados incluyen borohidruro de sodio (NaBH4), borohidruro de litio, borohidruro de potasio, borohidruro de tetraalquilamonio y borohidruro de zinc. En una realización, el agente reductor es NaBH4.
Preferiblemente, el ion metálico es cobalto (Co+2 o Co+3). Las fuentes de cobalto incluyen dicloruro de cobalto (CoCh), diacetato de cobalto (Co(OAc)2 y CoCh. Preferiblemente, el ion metálico es Co2+.
Ligandos adecuados incluyen dimetilglioxima, 2,2'-bipiridilo y 1,10-fenantrolina y preferiblemente dimetilglioxima. El ligando debe usarse al menos a una relación molar de 2:1 con el ion cobalto y preferiblemente a una relación de 50:1.
En otra realización, la reducción se realiza en una atmósfera inerte, tales como, por ejemplo, en una atmósfera de nitrógeno.
El proceso de la presente divulgación comprende además desproteger el compuesto de fórmula VII, y opcionalmente tratar de manera adicional con un ácido, para dar un compuesto de fórmula I:

o una sal farmacéuticamente aceptable del mismo. La desprotección (es decir, la retirada del grupo protector de éter de sililo) puede llevarse a cabo en un disolvente adecuado mediante tratamiento con ácidos o fluoruros (por ejemplo, fluoruro de tetra-n-butilamonio) como se describe, por ejemplo, en el documento de Greene, T. W. y Wuts, P. G. M., Protective Groups in Organic Synthesis, pág. 114 (J. Wiley & Sons, 1999). Posteriormente, el compuesto de fórmula I desprotegido puede tratarse con un ácido adecuado (tal como ácido clorhídrico) para formar la sal del compuesto de fórmula I.
En una realización, la desprotección del compuesto de fórmula VII y la formación de sal se realizan en dos etapas separadas como se describió anteriormente.
En otra realización, la desprotección del compuesto de fórmula VII y la formación de sal son simultáneas en las condiciones de reacción. El término “simultáneas” en el presente documento significa que la desprotección y la formación de sal se producen al mismo tiempo o secuencialmente. Por ejemplo, el compuesto de la sal de ácido clorhídrico de fórmula I puede obtenerse simultáneamente tratando el compuesto de fórmula VII con ácido clorhídrico al 30 % en metanol a una temperatura elevada de desde aproximadamente 35 °C hasta aproximadamente 45 °C y preferiblemente a aproximadamente 40 °C.
En una realización, el proceso comprende además aislar el compuesto de fórmula I, o una sal farmacéuticamente aceptable del mismo, y opcionalmente purificar el compuesto aislado de fórmula I, o una sal farmacéuticamente aceptable del mismo.
En otra realización, el proceso comprende además precipitar el compuesto de fórmula I, o una sal farmacéuticamente aceptable del mismo. En una realización, la precipitación se realiza tratando la mezcla de reacción con un disolvente aprótico polar, tal como acetonitrilo, a una temperatura elevada, tal como a temperatura de reflujo y luego permitiendo que la mezcla de reacción se enfríe a temperatura ambiente, por ejemplo, 20-25 °C. En otra realización, el proceso comprende además aislar el precipitado que comprende el compuesto de fórmula I, o una sal farmacéuticamente aceptable del mismo, a partir de la mezcla de reacción. En una realización, el precipitado se aísla por filtración. En otra realización, el precipitado aislado se purifica adicionalmente, por ejemplo, lavando con el disolvente usado en la precipitación y/o una mezcla del disolvente y agua. En otra realización, el precipitado aislado se lava con acetonitrilo y/o una mezcla de acetonitrilo y agua, para obtener el compuesto aislado de fórmula I, o una sal farmacéuticamente aceptable del mismo.
En otra realización, el compuesto aislado de fórmula I, o una sal farmacéuticamente aceptable del mismo, se purifica adicionalmente. En una realización, el compuesto aislado de fórmula I, o una sal farmacéuticamente aceptable del mismo, se purifica disolviendo el compuesto aislado en un disolvente adecuado (por ejemplo, en metanol acuoso) y tratando la disolución con carbón activado (por ejemplo, una suspensión en metanol) a una temperatura elevada (por ejemplo, a aproximadamente 45 °C) durante un tiempo suficiente (por ejemplo, 1 hora), filtrando el carbón activado y luego recuperando el compuesto purificado de fórmula I, o una sal farmacéuticamente aceptable del mismo, a partir del filtrado.
El compuesto de fórmula I y sales farmacéuticamente aceptables del mismo, puede obtenerse con un buen rendimiento con alta pureza. En determinados aspectos de la divulgación, los inventores han obtenido el compuesto de fórmula I usando el proceso descrito anteriormente con aproximadamente un rendimiento global del 13%. En determinados aspectos de la divulgación, los inventores han obtenido el compuesto de fórmula I usando el proceso descrito anteriormente con aproximadamente un rendimiento global del 6 %. Como se describió anteriormente, el rendimiento del compuesto para la fórmula I depende del rendimiento y la pureza del producto intermedio de fórmula III.
En otra realización, el proceso comprende además deuterar el compuesto de fórmula I, o una sal farmacéuticamente aceptable del mismo. En otra realización, se deutera el compuesto aislado de fórmula I, o una sal farmacéuticamente aceptable del mismo. Métodos conocidos en la técnica pueden usarse para preparar los compuestos deuterados de fórmula I y las sales farmacéuticamente aceptables de los mismos. Por ejemplo, tales métodos se describen en el documento WO 2014/152843 A1.
En una realización de la presente divulgación, el compuesto de fórmula II se prepara protegiendo el grupo hidroxilo de un compuesto de fórmula VIII:

en donde LG1 es un grupo saliente, como se definió anteriormente, con PG, en donde PG es un grupo protector, por ejemplo, un grupo protector seleccionado del grupo que consiste en grupos protectores de sililo, tetrahidropiranilo, metoximetilo y bencilo. La protección del grupo hidroxilo por un grupo protector de sililo, tetrahidropiranilo, metoximetilo o bencilo puede realizarse por métodos conocidos en la técnica. Por ejemplo, si PG es un grupo protector de sililo, el compuesto de fórmula VIII puede hacerse reaccionar con un cloruro de sililo (tal como TBDMS-Cl, TMS-Cl, TBDPS-Cl y TIPS-Cl) usando una base de amina (tal como imidazol). Un posible procedimiento es el protocolo de Corey en el que el compuesto de fórmula VIII se hace reaccionar con un cloruro de sililo e imidazol a alta concentración en DMF
En una realización de la presente divulgación, el compuesto de fórmula VIII se prepara haciendo reaccionar un compuesto de fórmula IX:

en donde LG1 es un grupo saliente, como se definió anteriormente, con CH3CHO en presencia de un haluro de alquilmagnesio en presencia de un disolvente para dar el compuesto de fórmula VIII. Haluros de alquil-magnesio adecuados incluyen, por ejemplo, i-PrMgCl, t-BuMgCl y t-BuMgBr. Compuestos de fórmula IX donde LG1 es un halogenuro también están disponibles comercialmente de, por ejemplo, OxChem Corporation. Disolventes adecuados incluyen disolventes etéreos anhidros, tales como THF y dietilétery preferiblemente THF.
El término “sal farmacéuticamente aceptable” se refiere a una sal preparada a partir de ácidos inorgánicos y orgánicos farmacéuticamente aceptables. Sales de adición de ácido farmacéuticamente aceptables a modo de ejemplo de los compuestos de la divulgación incluyen, sin limitación, ácido clorhídrico, bromhídrico, yodhídrico, sulfúrico, fosfórico, fórmico, acético, trifluoroacético, propiónico, cítrico y benzoico.
El término “grupo protector de hidroxilo” o un “grupo protector” como se usa en el presente documento se refiere al grupo que bloquea (es decir, protege) la funcionalidad de hidroxilo mientras que las reacciones se llevan a cabo en otros grupos funcionales o partes de la molécula. Los expertos en la técnica estarán familiarizados con la selección, fijación y escisión de grupos protectores y apreciarán que se conocen muchos grupos protectores diferentes en la técnica, la idoneidad de un grupo protector u otro depende del esquema de síntesis particular planificado. Grupos protectores de hidroxilo adecuados generalmente pueden introducirse y retirarse selectivamente usando condiciones de reacción suaves que no interfieren con otras partes de los compuestos en cuestión. Estos grupos protectores pueden introducirse o retirarse en una etapa conveniente usando métodos conocidos en la técnica. Las propiedades químicas de tales grupos, métodos para su introducción y retirada son conocidos en la técnica y pueden encontrarse, por ejemplo, en el documento de Wuts, P. G. M. & Greene, T. W., Greene’s Preotective Groups in Organic Synthesis, 4a Ed., págs. 16-430 (J. Wiley & Sons, 2007), se incorporan en el presente documento como referencia en su totalidad. A menos que se especifique específicamente, grupos protectores de hidroxilo adecuados se dan a conocer en el documento de Wuts, P. G. M. & Greene, T. W., mencionado anteriormente. Pueden encontrarse grupos protectores de hidroxilo adicionales, por ejemplo, en la patente estadounidense n.° 5.952.495, la publicación de solicitud de patente estadounidense n.° 2008/0312411, los documentos WO 2006/035195 y WO 98/02033, incorporados en el presente documento en su totalidad. Grupos protectores de hidroxilo adecuados incluyen el grupo metoximetilo, tetrahidropiranilo, terc-butilo, alilo, terc-butildimetilsililo, terc-butildifenilsililo, acetilo, pivaloílo, benzoílo, bencilo (Bn) y p-metoxibencilo.
El término “aproximadamente”, como se usa en el presente documento en relación con una cantidad medida, se refiere a las variaciones normales en esa cantidad medida, según lo esperado por el experto en la técnica, haciendo que la medición y el ejercicio de un nivel de cuidado sean proporcionales al objetivo de medición y la precisión del equipo de medición.
Algunos de los compuestos dados a conocer en el presente documento pueden contener uno o más centros asimétricos y, por lo tanto, pueden dar lugar a enantiómeros, diastereómeros y otras formas estereoisoméricas, tales como epímeros. La presente invención se pretende que abarque los usos de todas estas formas posibles, así como sus formas racémicas y resueltas y mezclas de las mismas. Los enantiómeros individuales pueden separarse según métodos conocidos por los expertos en la técnica en vista de la presente divulgación. Cuando los compuestos descritos en el presente documento contienen dobles enlaces olefínicos u otros centros de asimetría geométrica y a menos que se especifique de otro modo, se pretende que incluyan ambos isómeros geométricos E y Z. Todos los tautómeros también se pretende que se abarquen por la presente invención.
Como se usa en el presente documento, el término “estereoisómeros” es un término general para todos los isómeros de moléculas individuales que difieren solo en la orientación de sus átomos en el espacio. Este incluye enantiómeros e isómeros de compuestos con más de un centro quiral que no son imágenes especulares entre sí (diastereómeros).
El término “centro quiral” se refiere a un átomo de carbono al que están unidos cuatro grupos diferentes.
El término “epímero” se refiere a diastereómeros que tienen una configuración opuesta en solo uno de dos o más centros estereogénicos tetraédricos presentes en las entidades moleculares respectivas.
El término “centro estereogénico” es un átomo, que porta grupos de manera que un intercambio de dos grupos cualquiera conduce a un estereoisómero.
Los términos “enantiómero” y “enantiomérico” se refieren a una molécula que no puede superponerse a su imagen especular y, por lo tanto, es ópticamente activa en donde el enantiómero rota el plano de luz polarizada en un sentido y su compuesto de imagen especular rota el plano de luz polarizada en el sentido opuesto.
El término “racémico” se refiere a una mezcla de partes iguales de enantiómeros y mezcla que es ópticamente inactiva.
El término “resolución” se refiere a la separación o concentración o agotamiento de una de las dos formas enantioméricas de una molécula.
Los términos “un” y “una” se refieren a uno/una o más.
Términos abiertos tales como “incluir”, “que incluye”, “contener”, “que contiene” y similares significan “que comprende”.
El término “metal alcalino”, como se usa en el presente documento, se refiere a sodio (Na), potasio (K) y litio (Li). En una realización, el término “metal alcalino” también puede referirse a cesio (Cs).
El término “carbonato”, como se usa en el presente documento, se refiere a una sal de ácido carbónico caracterizada por la presencia del ion carbonato CO32-.
El término “carbonato de metal alcalino” como se usa en el presente documento se refiere a una sal de carbonato de cualquiera de los metales alcalinos mencionados anteriormente. Carbonatos de metal alcalino incluyen K2CO3, Na2CO3, LisCO3, KHCO3 y NaHCO3.
El término “alquilo” como se usa en el presente documento por sí mismo o como parte de otro grupo se refiere a hidrocarburo alifático ramificado, cíclico o de cadena lineal no sustituido que contiene de uno a doce átomos de carbono, es decir, alquilo C1 -12. En una realización, el grupo alquilo es alquilo C1-6. Grupos alquilo C1-6 a modo de ejemplo incluyen metilo, etilo, propilo, iso-propilo, n-butilo, terc-butilo, sec-butilo, pentilo, hexilo, ciclopropilo, ciclobutilo, ciclopentilo y ciclohexilo.
El término “alquilo inferior” se refiere a alquilo C1-4 que es un hidrocarburo alifático de cadena lineal o ramificada. Grupos alquilo inferior incluyen metilo, etilo, propilo, iso-propilo, n-butilo, sec-butilo y terc-butilo.
El término “organosulfonato”, como se usa en el presente documento, se refiere a un anión orgánico R-SO3', en donde R puede ser un grupo orgánico, tales como metilo, etilo, benceno y p-tolueno.
El término “grupos protectores de sililo” como se usa en el presente documento se refiere a un grupo de compuestos químicos que contienen átomos de silicio que pueden unirse covalentemente a un grupo alcoxilo. Por lo tanto, se forma un éter de sililo.
El término “alcoxilo”, como se usa en el presente documento, se refiere a un radical de la fórmula -O-R2, donde R2 es un grupo que tiene un grupo alquilo unido al átomo de oxígeno.
Los términos “deuteración” o “deuterar” y similares, como se usan en el presente documento, se refieren a la incorporación de deuterio en una o más posiciones de la fórmula I en lugar de hidrógeno en esa(s) posición(es). En una realización, el enriquecimiento de deuterio es aproximadamente el 15% o más, es decir, al menos aproximadamente 1000 veces mayor que la abundancia natural de deuterio. En otra realización, el enriquecimiento de deuterio es aproximadamente el 20 % o más, aproximadamente el 25 % o más, aproximadamente el 30 % o más, aproximadamente el 35 % o más, aproximadamente el 40 % o más, aproximadamente el 45 % o más, aproximadamente el 50 % o más, aproximadamente el 55 % o más, aproximadamente el 60 % o más, aproximadamente el 65 % o más, aproximadamente el 70 % o más, aproximadamente el 75 % o más, aproximadamente el 80 % o más, aproximadamente el 85 % o más, aproximadamente el 90 % o más, aproximadamente el 95 % o más, aproximadamente el 98 % o más o aproximadamente el 99 % o más. En otra realización, el enriquecimiento de deuterio es de aproximadamente el 100%. El enriquecimiento de deuterio puede
determinarse usando métodos analíticos convencionales conocidos por un experto en la técnica, incluyendo espectrometría de masas (LC-MS) y espectroscopia de resonancia magnética nuclear (RMN).
Cuando una posición de las fórmulas I se designa específicamente como “H” o “hidrógeno”, se entiende que la posición tiene hidrógeno en su composición isotópica de abundancia natural.
Cuando una posición de las fórmulas I se designa específicamente como “D” o “deuterio”, se entiende que la posición tiene deuterio en una abundancia que es al menos aproximadamente 1000 veces mayor que la abundancia natural de deuterio, que es aproximadamente un 0,015 %.
En una realización, un compuesto deuterado de fórmula I tiene la siguiente estructura:

Véase, la preparación en el ejemplo 8.
En otra realización, el deuterio se incorpora en más de una posición disponible de la fórmula I, por ejemplo, 1,2, 3, 4, 5 o 6 posiciones disponibles. En otra realización, se incorpora deuterio en todas las posiciones disponibles de la fórmula I. Compuestos de partida deuterados pueden usarse para preparar un compuesto deuterado de fórmula I, o una sal farmacéuticamente aceptable del mismo. Tales compuestos de partida pueden ser, por ejemplo, acetaldehído deuterado, óxido de etileno deuterado o 4-hidroxibenzaldehído deuterado.
En una realización, la divulgación proporciona un proceso para preparar el compuesto de fórmula I ilustrado por el esquema 2:
Esquema 2
I <-20°C o ácido de Lewis


des protección
formación de 
sal opcional
HCI
I (sal de HCI)
En otra realización, la divulgación proporciona un proceso para preparar el compuesto de fórmula I ilustrado por el esquema 3:

En otra realización en el esquema 3, etapa c, el orden de mezclado de los reactivos puede ser el siguiente: 1. n-BuLi, 2. óxido de etileno y 3. Cu I. Este orden de mezclado se describe en el ejemplo 2.
En la etapa a, 2,5-dibromopiridina (1) se hace reaccionar con i-PrMgCl en THF y luego adicionalmente con acetaldehído para obtener el compuesto 2. La mezcla de reacción se filtra preferiblemente sobre Celite® después de la reacción para retirar la mayoría de las sales. En una realización, la adición de acetaldehído se realiza a una temperatura entre -15 °C y -10 °C para controlar la reacción exotérmica.
En la etapa b, el compuesto 2 se hace reaccionar con TBDMS-Cl en presencia de imidazol que tiene DMF como disolvente. El producto en bruto 3 se purifica ventajosamente mediante una filtración de conector corto.
En la etapa c, el compuesto 3 protegido con hidroxilo se hace reaccionar con óxido de etileno en presencia de n-BuLi y yoduro de Cu (I) mientras se mantiene la temperatura de reacción, es decir, la temperatura de mezcla de reacción, por debajo de -20 °C. En una realización, la temperatura de reacción se mantiene por debajo de -55 °C mientras se añade n-BuLi y yoduro de Cu (I) a la mezcla de reacción. En otra realización, la temperatura de la mezcla de reacción se mantiene por debajo de -55 °C mientras se añade n-BuLi, seguido de óxido de etileno y luego yoduro de Cu (I) a la mezcla de reacción. En otra realización, la temperatura de la mezcla de reacción se mantiene por debajo de -55 °C mientras se añade n-BuLi a la mezcla de reacción, seguido de óxido de etileno. En esta realización, después se añade yoduro de Cu (I) a la mezcla de reacción mientras la temperatura de la mezcla de reacción se mantiene por debajo de -20 °C y preferiblemente por debajo de -55 °C. Se permite entonces que la mezcla de reacción se caliente lentamente a temperatura ambiente después de la adición de los reactivos y se agita a temperatura ambiente, por ejemplo, 20 25 °C, durante la noche. Este proceso se describe en detalle en el ejemplo 2. Después de la reacción, el cobre complejado se retira ventajosamente lavando con amoniaco al 10 %. El compuesto en bruto 4 puede purificarse por cromatografía en columna para dar >99 % de producto puro con un rendimiento de aproximadamente el 52 %.
Los siguientes ejemplos son ilustrativos, pero no limitantes, de los métodos de la presente invención.
Ejemplos
Ejemplo comparativo 1
Síntesis de 5-[[4-[2-[5-(1-hidroxietil)-2-piridinil]etoxi]fenil]metil]-2,4-tiazolidindiona (9a) según el proceso descrito en el documento WO 2015/150476 A1
Esquema 4

Se añadió gota a gota LiHMDS (1,0 M en tetrahidrofurano, 463 ml, 0,463 mol) a una disolución enfriada de 6-metilnicotinato de metilo (1a) (20 g, 0,132 mol) y acetato de etilo (82 g, 0,927 mol) en dimetilformamida a -50 °C; se aumentó gradualmente la temperatura a temperatura ambiente y se agitó a la misma temperatura. Después de 1 h, la mezcla de reacción se enfrió a 0 °C; se diluyó lentamente con ácido sulfúrico al 20 % y se calentó a reflujo. Después de 4 h, la mezcla de reacción se enfrió a temperatura ambiente, y además a 0 °C y se basificó con carbonato de potasio. El medio de reacción se diluyó con agua y se extrajo en acetato de etilo (3x50 ml). El extracto orgánico combinado se secó sobre sulfato de sodio y se concentró para proporcionar 1-(6-metilpiridin-3-il)etan-1-ona en bruto (2a) (20,0 g) que se llevó a la siguiente etapa sin ninguna purificación. ES-MS [M+1]+: 136,1.
Se añadió borohidruro de sodio (2,3 g, 0.06 mol) en pequeñas porciones durante 30 min, a una disolución del compuesto 2a (16,4 g, 0,121 mol) en etanol (160 ml) a 0 °C y la mezcla de reacción se agitó a la misma temperatura. Después de 1 h, la mezcla de reacción se diluyó con disolución de bicarbonato de sodio (saturada) (2x200 ml) y se extrajo con diclorometano (2x500 ml). El extracto orgánico combinado se secó sobre sulfato de sodio anhidro y se concentró para proporcionar un aceite amarillo pálido, que se purificó por cromatografía en columna ultrarrápida (5 % de metanol/diclorometano) para proporcionar el compuesto 3a (17,0 g; 93 % de rendimiento en 2 etapas) en forma de un aceite amarillo pálido. ES-MS [M+1]+ : 138,1. 1H RMN (400 MHz, CDCla): 8 8,35 (d, J = 2,0 Hz, 1H), 7,63 (dd, J = 8,0, 2,4 Hz, 1H), 7,12 (d, J = 8,0 Hz, 1H), 4,89 (q, J = 6,5 Hz, 1H), 3,30 (s. a., 1H), 2,50 (s, 3H), 1,48 (d, J = 6,5 Hz, 3H).
(b) Síntesis de 5-(1-metoximetoxi-etil)-2-metil-piridina (4a):
El compuesto 3a (15 g, 0,109 mol) se añadió, gota a gota, a una suspensión enfriada de hidruro de sodio (6,56 g, 0,164 mol) en tetrahidrofurano (150 ml) y se agitó a 0 °C. Después de 30 min, se añadió gota a gota clorometil metil éter (13,2 g, 0,164 mol) mientras se agitaba y se mantenía la temperatura interna alrededor de 0 °C. Después de que se terminó la adición, la mezcla de reacción se agitó a la misma temperatura durante 1 h. La reacción se desactivó con agua enfriada con hielo (80 ml) y se extrajo con acetato de etilo (3x50 ml). El extracto orgánico combinado se secó sobre sulfato de sodio anhidro y se concentró para proporcionar un aceite de color naranja, que se purificó por cromatografía en columna ultrarrápida (1 % de metanol/diclorometano) para proporcionar el compuesto 4a (10,0 g; 51 % de rendimiento) en forma de un aceite amarillo pálido. ES-MS [M+1]+: 182,2. 1H RMN (400 MHz, CDCla): 8 8,45 (d, J = 2,0 Hz, 1H), 7,56 (dd, J = 8,0, 2,0 Hz, 1H), 7,14 (d, J = 8,0 Hz, 1H), 4,75 (q, J = 6,4 Hz, 1H), 4,57 (ABq, 2H), 3,36 (s, 3H), 2,53 (s, 3H), 1,48 (d, J = 6,6 Hz, 3H).
(c) Síntesis de 2-[5-(1-metoximetoxi-etil)-piridin-2-il]-etanol (5a):
Una mezcla del compuesto 4a (7,0 g, 0,0386 mol) y disolución de formaldehído al 37 % (5,8 g, 0,077 mol) se calentó a 160 °C en un tubo de vidrio sellado durante 5 h. La mezcla de reacción se enfrió a temperatura ambiente y se concentró a presión reducida para proporcionar un compuesto en bruto que se purificó por cromatografía en columna ultrarrápida (1% de metanol/diclorometano) para proporcionar el compuesto 5 (1,2 g; 17 % de rendimiento) en forma de un aceite amarillo pálido. ES-MS [M+1]+: 212,1. 1H RMN (400 MHz, CDCla): 8 8,42 (d, J = 2,0 Hz, 1H), 7,65 (dd, J = 8,0, 2,4 Hz, 1H), 7,25 (d, J = 8,0 Hz, 1H), 4,72 (q, J = 6,6 Hz, 1H), 4,65 (t, J = 5,6 Hz, 1H), 4,52 (ABq, 2H), 3,73 (m, 2H), 3,24 (s, 3H), 2,86 (t, J = 7,2 Hz, 2H), 1,49 (d, J = 6,4 Hz, 3H).
El rendimiento total para el compuesto 5a a partir del compuesto 1a fue del 8 % molar.
(d) Síntesis de 4-{2-[5-(1-metoximetoxi-etil)-piridin-2-il]-etoxi}-benzaldehído (6a):
Se añadió, gota a gota, cloruro de metanosulfonilo (1,19 g, 0,01 mol) a una suspensión enfriada de compuesto 5a (1,7 g, 0,008 mol) y trietilamina (1,79 ml, 0,013 mol) en diclorometano (20 ml) a 0°C y se agitó a la misma temperatura durante 1 h. La mezcla de reacción se diluyó con agua (50 ml) y se extrajo con diclorometano (3x50 ml). El extracto orgánico combinado se secó sobre sulfato de sodio anhidro y se concentró para proporcionar metanosulfonato de 2-(5-(1-(metoximetoxi)etil)piridin-2-il)etilo (2,04 g; 88 % de rendimiento) en forma de un aceite de amarillo, que se llevó a la siguiente etapa sin purificación. ES-MS [M+1]+: 290.
Se añadió metanosulfonato de 2-(5-(1-(metoximetoxi)etil)piridin-2-il)etilo (2,3 g, 0,008 mol) a una suspensión agitada de 4-hidroxibenzaldehído (1,65 g, 0,0137 mol) y carbonato de potasio (1,86 g, 0,0137 mol) en mezcla de tolueno (25 ml) y etanol (25 ml); se agitó a 85 °C durante 5 h. Después del consumo de los materiales de partida, la mezcla de reacción se diluyó con agua (30 ml) y se extrajo con acetato de etilo (2x100 ml). El extracto orgánico combinado se lavó con agua; se secó sobre sulfato de sodio anhidro y se concentró para proporcionar un líquido amarillo oscuro en bruto. El producto en bruto se purificó por cromatografía en columna ultrarrápida (1 % de metanol/diclorometano) para proporcionar el compuesto 6a (1,5 g; 60 % de rendimiento) en forma de un líquido amarillo pálido. ES-MS [M+1]+: 316,1.
(e) Síntesis de 5-(4-{2-[5-(1-metoximetoxi-etil)-piridin-2-il]-etoxi}-bencilideno)-tiazolidin-2,4-diona (7a):
Se añadió piperidina (80 mg, 0,95 mmol) a una disolución del compuesto 6a (0,6 g, 1,9 mmol) y tiazolidin-2,4-diona (0,22 g, 1,9 mmol) en etanol (15 ml) y la mezcla se calentó a reflujo durante la noche. Después de 15 h, la mezcla de reacción se enfrió a temperatura ambiente y se concentró a presión reducida para proporcionar la mezcla en bruto, que se purificó por cromatografía en columna ultrarrápida (2 % de metanol/diclorometano) para proporcionar el compuesto 7 (500 mg; 64 % de rendimiento) en forma de un sólido amarillo. ES-MS [M+1]+ : 415,1. 1H Rm N (400 MHz, DMSO-d6): 8 12,25 (s. a., 1H), 8,47 (d, J = 2,0 Hz, 1H), 7,70 (dd, J = 8,0, 2,0 Hz, 1H), 7,54 (d, J = 8,8 Hz, 2H), 7,36 (d, J = 8,0 Hz, 1H), 7,21 (d, J = 8,8 Hz, 2H), 4,73 (m, 1H), 4,60-4,40 (m, 4H), 4,22 (t, J = 6,2 Hz, 1H), 3,24 (s, 3H), 3,20 (t, J = 6,8 Hz, 2H), 1,41 (d, J = 6,0 Hz, 3H).
(f) Síntesis de 5-(4-{2-[5-(1-hidroxi-etil)-piridin-2-il]-etoxi}-bencil)-tiazolidin-2,4-diona (9a):
Una disolución de borohidruro de sodio (115 mg, 3,017 mmol) en hidróxido de sodio 0,2 N (1,2 ml) se añadió lentamente a una disolución agitada del compuesto 7 (0,5 g, 1,207 mmol), dimetilglioxima (42 mg, 0,36 mmol) y CoCl2.6H2O (23 mg, 0,096 mmol) en una mezcla de agua (6 ml): tetrahidrofurano (6 ml) y disolución de hidróxido de sodio 1 M (1 ml) a 10 °C y después de la adición, la mezcla de reacción se agitó a temperatura ambiente. Después de 1 h, el color de la reacción se aclaró y se añadieron cantidades adicionales de borohidruro de sodio (46 mg, 1,207 mmol) y CoCl2.6H2O (22 mg, 0,096 mmol) y se continuó agitando a temperatura ambiente. Después de 12 h, la reacción se neutralizó con ácido acético (pH~7); se diluyó con agua (10 ml) y se extrajo en acetato de etilo (3x50 ml). El extracto orgánico combinado se secó sobre sulfato de sodio anhidro y se concentró para proporcionar el compuesto en bruto 8a, 5-(4-(2-(5-(1-(metoximetoxi)etil)piridin-2-il)etoxi)bencil)tiazolidin-2,4-diona, (0,4 g) como semisólido amarillo pálido, que se llevó a la siguiente etapa sin purificación. ES-MS [M+1]+ : 417,5.
Se añadió HCl 2 N (2 ml) a una disolución del compuesto 8a (0,4 g, 0,96 mmol) en metanol (20 ml) y la mezcla se calentó a reflujo. Después de 4 h, la mezcla de reacción se enfrió a temperatura ambiente y después se concentró a presión reducida para proporcionar un residuo que se disolvió en agua y la disolución se neutralizó usando una disolución de bicarbonato de sodio (sat.). El precipitado blanco resultante se recogió por filtración para proporcionar el compuesto 9a (250 mg; 56 % de rendimiento en 2 etapas) en forma de un sólido de color blanquecino. ES-MS [M+1]+: 373,4. 1H RMN (400 MHz, DMSO-d6): 8 12,00 (s. a., -NH), 8,46 (d, J = 2,0 Hz, 1H), 7,66 (dd, J = 8,0, 2,4 Hz, 1H), 7,30 (d, J = 8,0 Hz, 1H), 7,13 (d, J = 8,4 Hz, 2H), 6,86 (d, J = 8,4 Hz, 2H), 5,25 (d, J = 4,4 Hz, 1H), 4,86 (m, 1H), 4,75 (m, 1H), 4,30 (t, J = 6,8 Hz, 2H), 3,30 (m, 1H), 3,14 (t, J = 6,4 Hz, 2H), 3,04 (m, 1H), 1,34 (d, J = 6,4 Hz, 3H).
El rendimiento global del compuesto 9a fue del 1,5 % molar.
Ejemplo 2
Síntesis de 2-(5-(1-((terc-butildimetilsilil)oxi)etil)piridin-2-il)etan-1-ol
La síntesis de 2-(5-(1-((terc-butildimetilsilil)oxi)etM)piridin-2-il)etan-1-ol se realizó según el esquema 5 usando los reactivos y disolventes enumerados en la tabla 1 a continuación:
Esquema 5

Los datos de LC-MS se obtuvieron en un aparato Agilent serie 1290 con detector UV y detector de masa HP 6130 MSD usando como columna Waters XBridge BEH XP (2,1 x 50 mm; 2,5 |im) y como eluyente acetato de amonio (10 mM); Agua/Metanol/Acetonitrilo.
(a) 1-(6-bromopiridin-3-il)etan-1-ol (2)
Se colocó un recipiente de 20 l en atmósfera de nitrógeno y se cargó con tetrahidrofurano (5,5 l) y se añadió 2,5-dibromopiridina (1) (2000 g, 8,44 mol, 1,0 eq) (OxChem Corporation). La mezcla se enfrió a -10 °C y se añadió lentamente cloruro de isopropil magnesio (20 % en THF, 6,02 l, 11,82 mol, 1,4 eq) (Rockwood Lithium) durante 1 h, manteniendo la temperatura de reacción por debajo de 5 °C. Después de la adición, el baño de enfriamiento se retiró y la temperatura se mantuvo por debajo de 30 °C (se necesita un enfriamiento adicional para lograr esto) y la mezcla de reacción se agitó durante la noche. Después de 16 h, se tomó una muestra; se desactivó con cloruro de amonio acuoso saturado y se extrajo con metil terc-butiléter (TBME). El TBME se evaporó al vacío. 1H-RMN en cloroformo deuterado mostró conversión completa.
La mezcla de reacción se enfrió a -15 °C y una disolución de acetaldehído (472 g, 10,72 mol, 1,27 eq) (Acros) en tetrahidrofurano (200 ml) se añadió gota a gota, mientras se mantuvo la temperatura por debajo de -10 °C. Una vez completada la adición, se retiró el baño de enfriamiento y se dejó que la temperatura aumentara hasta un máximo de 5-8 °C. Después de 1,5 h, se tomó una muestra y la reacción se desactivó con cloruro de amonio acuoso como se describió anteriormente. 1H-RMN mostró que la reacción se había completado.
Se combinaron dos lotes para pruebas complementarias.
La mezcla de reacción se desactivó vertiendo la mezcla en una disolución de cloruro de amonio acuoso (1 kg en 5 l de agua) y se agitó durante 15 min, se filtró sobre Celite® y se enjuagó a fondo con tolueno. El filtrado se transfirió a un embudo de separación y el sistema de dos capas obtenido se separó. La capa acuosa se extrajo con tolueno (2 l). Las capas orgánicas combinadas se secaron sobre sulfato de sodio y se filtraron. La evaporación del filtrado a sequedad al vacío produjo 3,49 kg (99 %) del material en bruto deseado. 1H RMN (300 MHz, CDCh): 88,30 (d, J = 2,5 Hz, 1H), 7,59 (dd, J = 8,0, 2,5 Hz, 1H), 7,44 (d, J = 8,0 Hz, 1H), 4,91 (q, J = 6,5 Hz, 1H), 1,49 (d, J = 6,5 Hz, 3H). (b) 2-bromo-5-(1-((terc-butildimetilsilil)oxi)etil)piridina (3)
Un reactor de 50 l en atmósfera de nitrógeno se cargó con el compuesto 2 (10,0 kg, alrededor de 49,5 mol) y DMF (16 l). La mezcla se enfrió a 10 °C e imidazol (6,74 kg, 99 mol, 2,0 eq) (Apollo Scientific Ltd.) se añadió en porciones en 30 min. La mezcla se enfrió a 0 °C y TBDMS-Cl (7,46 kg, 49,5 mol, 1,0 eq) (Fluorochem) se añadió en porciones en 5 h, manteniendo la temperatura por debajo de 3 °C. La temperatura de reacción de mezcla se dejó alcanzar la temperatura ambiente y se agitó durante la noche. 1H RMN de una muestra mostró una conversión completa.
La mezcla de reacción se transfirió a un recipiente de extracción de 100 l y el producto se extrajo con heptano (2x7,5 L, 10 l). Las capas de heptano combinadas se lavaron con agua (2x6 l, 3 l) para retirar pequeñas cantidades de DMF, se secaron sobre sulfato de sodio y se evaporaron al vacío para dar el compuesto en bruto 3 (15,5 kg, 49,0 mol) en un 99,0 % de rendimiento. Este producto en bruto se purificó mediante una filtración de conector corto, usando 10 kg de sílice/heptano y eluyendo con heptano (aprox. 50 l). Las fracciones de producto se combinaron y se evaporaron al vacío para dar 12,0 kg de compuesto purificado 3 (38 mol) como un aceite marrón en un 76,8 % de rendimiento molar. (Rendimiento promedio para 3 experimentos fue del 78 %). HPLC-MS: T. a. = 2,6 min, M+1=316,1 y 318,1; 1H RMN (300 MHz, CDCla): 88,55 (d, J = 2,2 Hz, 1H), 7,54 (dd, J = 8,2, 2,2 Hz, 1H), 7,42 (d, J = 8,2 Hz, 1H), 4,86 (q, J = 6,5
Hz, 1H), 1,40 (d, J = 6,5 Hz, 3H), 0,88 (s, 9H), 0,02 (d, J = 26 Hz, 2x3H).
(c) 2-(5-(1-((terc-butildimetilsilil)oxi)etil)piridin-2-il)etan-1-ol (4)
La disolución de óxido de etileno en dietiléter se preparó de antemano. Dietiléter (1,2 l) en un matraz de tres bocas de 3 l se enfrió a -65 °C y se añadió óxido de etileno (462,3 g, 10,5 mol, 1,06 eq) (Linde) y se agitó a -70 °C. Alternativamente, la disolución de óxido de etileno puede prepararse a aproximadamente -20 °C y luego añadirse gradualmente a la mezcla de reacción que tiene una temperatura a aproximadamente -60 °C.
A una disolución de 2-bromo-5-(1-((terc-butildimetilsilil)oxi)etil)piridina (3) (3,13 kg, 9,90 mol, 1,0 eq) en dietiléter (7,5 l) enfriada a -59 °C, se añadió n-butil-litio (4 l, 10,0 mol, 2,5 M en hexanos, 1,01 eq) (Aldrich Chemistry) mientras se mantenía la temperatura entre -58 °C y -62 °C. Después de la adición, la mezcla se agitó durante 1 h mientras se mantenía la temperatura entre -60 °C y -68 °C. La disolución de óxido de etileno preparada por adelantado se añadió de una vez a la mezcla de reacción, mientras que la temperatura era de aproximadamente -62 °C. Posteriormente, yoduro de cobre (I) (962,3 g, 5,05 mol, 0,51 eq) (Acros Organics) se añadió en porciones de 120 g, cada 10 min, manteniendo la temperatura entre -61 °C y -63 °C. La agitación continuó durante 1 h después de la adición manteniendo la temperatura entre -61 °C y -63 °C. El baño de enfriamiento se retiró y se permitió que la temperatura aumentara a aproximadamente 15 °C y adicionalmente a 25 °C con un baño de agua durante la noche.
Prueba complementaria: La mezcla de reacción se vertió en una disolución de 1 kg de cloruro de amonio en 5 l de agua y se agitó durante 30 min, después se separaron las capas. La capa orgánica se lavó con hidróxido de amonio acuoso (10 %, 2,5 l, 4x) para retirar el complejo de Cu (el color azul desapareció). Las capas orgánicas combinadas se secaron sobre sulfato de sodio y se evaporaron para dar 3,12 kg (máx. 9,90 mol) de compuesto 4 en bruto como un aceite marrón. El compuesto en bruto se purificó sobre 20 kg de sílice (heptano/EtOAc) eluyendo con 80 l de heptano/EtOAc, 20 l de EtOAc, 25 l de EtOAc/MeOH 95/5, 25 l de EtOAc/MeOH 9/1 y 10 l de EtOAc/MeOH 8/2, para dar 1,47 kg de compuesto 4 purificado (5,22 mol) como un aceite marrón (con tendencia a solidificarse) en un rendimiento molar promedio del 52,7% (pureza por HPLC del 99,5%). (Rendimiento promedio durante 12 experimentos del 52 %). HPLC-MS: t. a. = 2,3 min, M+1=282,1; 1H RMN (300 MHz, CDCla): 88,42 (d, J = 2,1 Hz, 1H), 7,61 (dd, J = 8,3, 2,1 Hz, 1H), 7,11 (d, J = 8,3 Hz, 1H), 4,88 (q, J = 7,0 Hz, 1H), 4,01 (t, J=6,0 Hz, 2 H), 3,00 (t, J=6,0 Hz, 2 H), 1,41 (d, J = 7,0 Hz, 3H), 0,90 (s, 9H), 0,02 (d, J = 26 Hz, 2x3H).
Otro 2,5 % del producto se aisló volviendo a purificar la fracción de producto impuro. El rendimiento total del compuesto 4 a partir del compuesto 1 fue del 39,6 % molar.
Síntesis de clorhidrato de 5-[[4-[2-[5-(1-hidroxietil)-2-piridinil]etoxi]fenil]metil]-2,4-tiazolidindiona (9)


etapa f etapa g

Los espectros de 1H RMN se registraron con un espectrómetro de RMN Avance Bruker de 400 MHz. Los datos de LC-MS se obtuvieron en un Agilent Technologies 6130 Quadrapole LC/MS usando como columna Agilent XDB-C18 y como eluyente 0,1 % de ácido fórmico (ac.) y 0,05 % de ácido fórmico en acetonitrilo.
Etapas d y e: Síntesis de 4-[2-[5-[[[(1,1 -dimetiletil)dimetilsilil]oxi]etil]-2-piridinil]etoxi]-benzaldehído (6)
A una disolución bien agitada de 5-[[[(1,1-dimetiletil)dimetilsilil]-oxi]etil]-2-piridinaetanol (4) (obtenido como se describe en el ejemplo 2) (1,91 kg) en tolueno (8,6 l) a 5 °C se añadieron hidróxido de sodio (acuoso al 30 %, 2,79 l) y bromuro de tetrabutilamonio (7,2 g). A continuación se añadió cloruro de p-toluenosulfonilo (1,62 kg) en porciones durante 5 min. Después de la adición, se permitió que la mezcla de reacción alcanzara la temperatura ambiente en 0,5 h y se agitó a esta temperatura durante 18 h. A continuación se añadió agua (7,3 l) y la mezcla se mezcló bien. Una vez que se disolvieron los sólidos, las capas se dejaron sedimentar y la capa orgánica se separó. Esta fase orgánica se lavó con agua (5,7 l, 2x), seguido de lavado con una disolución de cloruro de sodio (57 g) en agua (5,7 l). Los disolventes se concentraron a presión reducida hasta una cantidad de 2,5 kg de un aceite marrón (compuesto 5).
A este aceite marrón bien agitado se añadieron posteriormente etanol (7,8 l), agua (0,86 L), 4-hidroxibenzaldehído (0,88 kg) y carbonato de potasio (1,17 kg) y después la mezcla se calentó a 75 °C durante 18 h. A continuación, el disolvente se evaporó mientras se añadía tolueno (7,7 l) durante 6 h y después la mezcla de reacción se dejó enfriar. A 30 °C, se añadió agua (7,6 l), se agitó hasta que todos los sólidos se disolvieron y la mezcla se enfrió a temperatura ambiente. Las capas se dejaron sedimentar y separarse. La capa orgánica se lavó con agua (7,6 l). El primer extracto acuoso se extrajo con tolueno (2,8 l) y este extracto orgánico se usó para extraer también el lavado acuoso. Los extractos orgánicos se combinaron y se concentraron al vacío para dar 3,49 kg de un aceite negro (compuesto del título en bruto 6).
Se disolvieron 1,73 kg de este aceite negro en etanol (0,74 L) y se añadieron a una disolución bien agitada de bisulfito de sodio (1,36 kg) en una mezcla de agua (3,27 l) y etanol (0,74 l). El recipiente del aceite negro se enjuagó con etanol (0,37 l) dos veces y estos dos enjuagues también se añadieron a la mezcla de reacción de bisulfito. Después de 75 min, se añadió heptano (5,3 l), se mezclaron bien durante 5 min y las capas se dejaron sedimentar y separarse. A la capa orgánica se le añadió una disolución de bisulfito de sodio (0,55 kg) en agua (2,65 l) y etanol (1,06 l). Después de agitar durante 30 min, las capas se dejaron sedimentar y separarse. Los dos extractos acuosos de bisulfuro se combinaron y los matraces se enjuagaron con agua (2,12 l). A continuación, se añadieron tolueno (4,5 l) y heptano (4,5 l), la mezcla se agitó bien y el pH se ajustó a 12 usando hidróxido de sodio (ac. al 10 %) (la temperatura pasó a ser 32 °C). Después de agitar durante 5 min adicionales, las capas se dejaron sedimentar y se separaron a 30 °C. La capa acuosa se extrajo con una mezcla de tolueno (1,5 l) y heptano (3,0 l). Las capas se separaron y se combinaron las capas orgánicas. Las capas orgánicas combinadas se lavaron con agua (5 l, 2x) y se concentraron al vacío para dar el compuesto del título purificado 6. Este proceso se repitió con otros 1,73 kg del aceite negro (compuesto del título
en bruto 6) para dar en total 2,77 kg de 4-[2-[5-[[[(1,1-dimetiletil)dimetilsilil]oxi]etil]-2-piridinil]etoxi]-benzaldehído (6) como aceite marrón que contenía un 24 % en m/m de tolueno según 1H RMN (rendimiento = 80 %, calculado a partir del compuesto 4 y corregido en cuanto a tolueno residual).
1H-RMN (CDCla) 8 : 0,00 (s, 3H), 0,09 (s, 3H), 0,91 (s, 9H), 1,44 (d, J = 6 Hz, 3H), 3,30 (t, J = 7 Hz, 2H), 4,47 (t, J = 7 Hz, 2H), 4,92 (q, J = 6 Hz, 1H), 6,99 -7,30 (m, 3H), 7,62-7,67 (m, 1H), 7,80 -7,85 (m, 2H), 8,5- 8,54 (m, 1H) y 9,88 (s, 1H).
LC-MS; t. a. 7,5 min: ES: M+ 387, 386.
Etapa f: Síntesis de (5Z)-5-[[4-[2-[5-[[(1,1-dimetiletil)dimetilsilil]oxi]etil]-2-piridinil]etoxi]fenil]metilen]-2,4-tiazolidindiona (7)
Una disolución de 4-[2-[5-[[[(1,1-dimetiletil)dimetilsilil]oxi]etil]-2-piridinil]-etoxi]-benzaldehído (6) (2,75 kg, que contiene un 24 % en m/m de tolueno) y piperidina (6,0 g) en metanol (3,16 l) se concentró a 40 °C a presión reducida. El residuo se disolvió en metanol (10,4 l) y se añadieron 2,4-tiazolidindiona (759 g) y piperidina (230 g). La mezcla se calentó a 47 °C. Después de 25 h, la mezcla de reacción se dejó enfriar a temperatura ambiente. La mezcla se mantuvo a pH 5-6 ajustándola con ácido acético, si es necesario. Después de una noche a temperatura ambiente, se añadió agua (1,56 L) y la suspensión se agitó a temperatura ambiente durante 2 h adicionales. Los sólidos se aislaron por filtración, se lavaron con metanol (1 l, 2x) y se secaron al vacío para dar el compuesto en bruto 7 (1,65 kg). El compuesto en bruto se mezcló con metanol (10 l) y diclorometano (8,6 l) y se calentó a 32 °C hasta que se disolvieron todos los sólidos. A continuación, los disolventes se retiraron por destilación hasta que la temperatura de la mezcla alcanzó 34 °C a una presión de 333 mbar. A continuación, se dejó enfriar a temperatura ambiente durante la noche y se agitó a 2 °C durante 2 h adicionales. Los sólidos se aislaron por filtración, se lavaron con metanol (0,5 l, 2x) y se secaron al vacío para dar el compuesto del título 7 (1,50 kg) (rendimiento = 61 %).
1H RMN (CDCla) 8 0,00 (s, 3H), 0,08 (s, 3H), 0,90 (s, 9H), 1,43 (d, J = 6 Hz, 3H), 3,32 (t, J = 7 Hz, 2H), 4,48 (t, J = 7 Hz, 2H), 4,92 (q, J = 6 Hz, 1H), 6,95 - 7,00 (m, 2H), 7,24 - 7,28 (m, 1H), 7,38 - 7,42 (m, 2H), 7,67 (s, 1H), 7,69 - 7,73 (m, 1H) y 8,48 (d, J = 3 Hz, 1H).
LC-MS; t. a. 7,5 min: ES: M+ 487, 486, 485.
Etapa g: Síntesis de 5-[[4-[2-[5-[[[(1,1-dimetiletil)dimetilsilil]oxi]etil]-2-piridinil]etoxi]fenil]metil]-2,4-tiazolidindiona (8)
Se añadió a una suspensión agitada de (5Z)-5-[[4-[2-[5-[[[(1,1-dimetiletil)dimetilsilil]oxi]-etil]-2-piridinil]etoxi]fenil]metilen]-2,4-tiazolidindiona (7) (10 g) en THF (10 ml) e hidróxido de sodio (ac. 1 N, 21 ml) de una disolución de cloruro de cobalto (26 mg) y de dimetilglioxima (930 mg) en THF (2,3 ml) y agua (1,0 ml). Después, la suspensión se puso en una atmósfera de nitrógeno aplicando la secuencia de vacío y purgando con nitrógeno (4x). Posteriormente, la suspensión se calentó a 30 °C. A continuación, se preparó una disolución madre de borohidruro de sodio disolviendo borohidruro de sodio (2,7 g) en una mezcla de agua (15,8 ml) y una disolución de hidróxido de sodio (ac. 1 N, 3,5 ml), que se puso en una atmósfera de nitrógeno aplicando una secuencia de vacío y purgando con nitrógeno (3x). Esto se añadió a la suspensión del compuesto 7 a una velocidad de 4,5 ml/h. Simultáneamente, se añadió ácido acético saturado con gas nitrógeno a la suspensión a una velocidad de 0,7 ml/h para mantener un pH de 10,0-10,5. Después de 1 h 30 min, la velocidad de adición de la disolución de borohidruro de sodio y ácido acético se redujo a la mitad. A continuación, 3 h 45 min después del inicio de la adición, se detuvo la adición de borohidruro de sodio y ácido acético. La mezcla se dejó enfriar a temperatura ambiente y se añadió acetona (2,5 ml) durante un período de 1 minuto. Después de agitar la mezcla de reacción durante 15 min se añadió ácido acético hasta que el pH fue 5,5-6,0 (se requieren aproximadamente 3 ml). A continuación, se añadió una mezcla de acetato de etilo/tolueno (1/3 en v/v, 30 ml), se mezcló bien y las capas se dejaron sedimentar. La capa acuosa se separó y lavando con acetato de etilo/tolueno (1/3 en v/v, 10 ml). Ambos extractos orgánicos se juntaron y se añadió agua (40 ml), se mezcló bien y las capas se dejaron sedimentar. El pH de la capa acuosa se ajustó a 5,5-6 usando disolución saturada de hidrogenocarbonato de sodio (ac.) y nuevamente se mezcló con la capa orgánica. Las capas se dejaron sedimentar y la capa orgánica se separó y se concentró al vacío para dar 11,09 g de aceite amarillo (mezcla en bruto que contiene el compuesto del título 8 y su complejo de borano). Se combinaron varios lotes para pruebas complementarias.
Se disolvieron 33,1 g de la mezcla en bruto que contenía el compuesto del título 8 y su complejo de borano (sin corregir en cuanto a disolventes residuales) en tolueno (30 ml) y se filtraron. El filtrado se sometió a cromatografía en columna (gel de sílice, gradiente de tolueno a tolueno/acetato de etilo 1/1) para dar 30,0 g de mezcla de 5-[[4-[2-[5-[[[(1,1-dimetiletil)dimetilsilil]oxi]etil]-2-piridinil]etoxi]fenil]metil]-2,4-tiazolidindiona (8) y su complejo de borano como un aceite ligeramente amarillo (rendimiento = 100 % a partir del compuesto 4, sin corregir en cuanto a disolventes residuales).
1H-RMN (CDCls ) 8 : -0,03 -0,10 (m, 6H), 0,87 -0,93 (m, 9H), 1,42 (d, J = 6 Hz, 3H), 3,05-3,71 (m, 4H), 4,30 -4,51 (m, 3H), 4,87 -4,94 (m, 1H), 6,82 -6,88 (m, 2H), 7,10-7,92 (m, 5H), 8,49 (d, J = 3 Hz, 0,6H) y 8,72 (s. a., 0,4 H).
LC-MS; t. a. 6,8 min: ES: M+ 489, 488, 487, M'487, 486, 485; t. a. 8,1 min: ES M' 501, 500, 499, 498, 485.
Etapa h: Síntesis de clorhidrato de 5-[[4-[2-[5-(1 -hidroxietil)-2-piridinil]etoxi]fenil]-metM]-2,4-tiazolidindiona (9)
A una disolución agitada de la mezcla de (5-[[4-[2-[5-[[[(1,1 -dimetiMetil)-dimetilsilil]oxi]etil]-2-piridinil]etoxi]fenil]metil]-2,4-tiazolidindiona y su complejo de borano (8) (5,17 g) en metanol (25,2 ml) a 22 °C se añadió ácido clorhídrico (30 %, 2,75 ml) en aproximadamente 5 min para dar un aumento de temperatura a 28 °C. Esta disolución se calentó a 40 °C. Tres horas después de la adición, los 11 g de sustancias volátiles se retiraron a presión reducida. A continuación, se añadió acetonitrilo (40,3 ml) y la mezcla se calentó a reflujo durante 0,5 h. A continuación, la suspensión se dejó enfriar a temperatura ambiente y se agitó durante 1 h a temperatura ambiente. Se aislaron por filtración sólidos, se lavaron con una mezcla de acetonitrilo/agua (20/1 en v/v, 10 ml) y con acetonitrilo (10 ml) y se secaron al vacío a 40 °C para dar 4,00 g de sólidos blancos (compuesto 9 en bruto) (rendimiento = 77 %, sin corregir en cuanto a disolventes residuales).
Purificación de clorhidrato de 5-[[4-[2-[5-(1-hidroxietil)-2-piridinil]etoxi]fenil]metil]-2,4-tiazolidindiona (9):
La mezcla en bruto de clorhidrato de 5-[[4-[2-[5-(1-hidroxietil)-2-piridinil]etoxi]fenil]-metil]-2,4-tiazolidindiona (3,95 g, compuesto 9 en bruto) se disolvió en metanol/agua (7/2 en v/v, 80 ml) calentándola a 49 °C. A esta disolución se añadió Norit lavado (obtenido calentando una suspensión de Norit (6 g) en metanol/agua (7/2 en v/v, 90 ml) a 45 °C durante 1 h, después se aisló el Norit por filtración y lavando dos veces con metanol/agua (7/2 en v/v, 30 ml) y se secó al vacío a 40 °C). El equipo se enjuagó con metanol/agua (7/2 en v/v, 18 ml). Después de 0,5 h de agitación a 46 °C, la suspensión caliente se filtró para retirar el Norit y el filtro se lavó dos veces con metanol/agua (7/2 en v/v, 18 ml). El filtrado se concentró al vacío a una temperatura del baño de 60 °C hasta una masa de 11,8 g (1 v de compuesto y 2 v de agua). A la suspensión se añadió butanona (19,7 ml, 5 v) y la mezcla se calentó a una temperatura de baño de 95 °C. En destilación a un volumen constante, se añadió butanona (95 ml). A continuación, se detuvo el calentamiento y se permitió que la suspensión alcanzara la temperatura ambiente en aproximadamente 0,5 h. Posteriormente se agitó durante 0,75 h a temperatura ambiente. Se aislaron por filtración sólidos, se lavaron con una mezcla de butanona/agua (95/5 en v/v, 18 ml) y butanona (18 ml) y se secaron al vacío a 40 °C para dar 3,57 g del compuesto 9 en forma de sólidos blancos (rendimiento = 91 %).
1H RMN (DMSO-da): 812,00 (s. a., -NH), 8,71 (d, J = 2,0 Hz, 1H), 8,45 (dd, J = 8,3, 1,7 Hz, 1H), 7,98 (d, J = 8,3 Hz, 1H), 7,15 (d, J = 8,7 Hz, 2H), 6,88 (d, J = 8,7 Hz, 2H), 5,57 (s, OH), 4,95 (q, J = 6,5 Hz, 1H), 4,86 (dd, J = 8,9, 4,4 Hz, 1H), 4,40 (t, J = 6,3 Hz, 2H), 3,49 (t, J = 6,2 Hz, 2H), 3,29 (dd, J = 14,2, 4,4 Hz, 1H), 3,06 (dd, J = 14,2, 9,0 Hz, 1H), 1,41 (d, J = 6,5 Hz, 3H).
LC-MS; t. a. 3,5 min: ES: M+ 374, 373, M- 372, 371.
Ejemplo 4
Condiciones sometidas a prueba en la preparación del compuesto 5 en la etapa d
Las condiciones descritas en la tabla 2 a continuación se sometieron a prueba en la etapa d en la preparación del compuesto 5 a partir del compuesto 4 proporcionando un buen rendimiento del compuesto 5:
Tabla 2

Ejemplo 5
Condiciones sometidas a prueba en la preparación del compuesto 6 en la etapa e
Las condiciones descritas en la tabla 3 a continuación se sometieron a prueba en la etapa e en la preparación del compuesto 6 a partir del compuesto 5 proporcionando un buen rendimiento del compuesto 6:
Tabla 3

Ejemplo 6
Condiciones sometidas a prueba en la preparación del compuesto 7 en la etapa f
Las condiciones descritas en la tabla 4 a continuación se sometieron a prueba en la etapa f en la preparación del compuesto 7 a partir del compuesto 6 proporcionando un buen rendimiento del compuesto 7:
Tabla 4

Ejemplo 7
Síntesis a gran escala de clorhidrato de 5-[[4-[2-[5-(1-hidroxietil)-2-piridinil]etoxi]fenil]metil]-2,4-tiazolidindiona (9)
Etapas d, e, f, g y h se refieren a las etapas descritas en el esquema del ejemplo 3 anterior. Los espectros de 1H-RMN y los datos de lC-MS de los compuestos preparados se confirmaron como se describe en el ejemplo 3.
Etapas d y e: Síntesis de 4-[2-[5-[[[(1,1 -dimetiletil)dimetilsilil]oxi]etil]-2-piridinil]etoxi]-benzaldehído (6)
A una disolución bien agitada de 5-[[[(1,1-dimetiletil)dimetilsilil]-oxi]etil]-2-piridinaetanol (4) (obtenido como se describe en el ejemplo 2) (8,14 kg) en tolueno (33 l) a 5 °C se añadieron hidróxido de sodio (acuoso al 30 %, 11,65 l) y bromuro de tetrabutilamonio (30,13 g). Cloruro de p-toluenosulfonilo (6,76 kg) se añadió a continuación, el equipo de adición se enjuagó con tolueno (2 l) y el enjuague se añadió al reactor. Después de la adición, la mezcla de reacción se dejó alcanzar la temperatura ambiente y se agitó a esta temperatura durante la noche. Luego se añadió agua (32 l) y la mezcla se mezcló bien. Una vez que se disolvieron los sólidos, las capas se dejaron sedimentar y la capa orgánica se separó. Esta fase orgánica se lavó con agua dos veces (24 l, 2x). Los disolventes se evaporaron parcialmente a presión reducida para producir un residuo de 29,5 kg de un aceite marrón (compuesto 5).
A este aceite marrón bien agitado se añadieron etanol (32 l), agua (3,6 l), 4-hidroxibenzaldehído (3,69 kg) y carbonato de potasio (4,87 kg) y después la mezcla se calentó a 75 °C durante la noche. A continuación, se añadió tolueno (32 l) y las sustancias volátiles (32 l) se evaporaron. Posteriormente, la mezcla de reacción se dejó enfriar. A 20 °C, se añadió agua (32 l), se agitó hasta que todos los sólidos se disolvieron y la mezcla se enfrió a temperatura ambiente. Las capas se dejaron sedimentar y separarse. La capa orgánica se lavó con agua (32 l). El primer extracto acuoso se extrajo con tolueno (12 l) y este extracto orgánico se usó para extraer también el lavado acuoso. Los extractos orgánicos se combinaron y se concentraron al vacío para dar 15,6 kg de un aceite negro (compuesto del título en bruto 6 que contiene tolueno residual). Este procedimiento se repitió usando 8,14 kg de compuesto 4 para dar otros 13,3 kg de un aceite negro (compuesto del título en bruto 6 que contiene tolueno residual).
Ambos lotes del compuesto en bruto 6 se juntaron. Los 4,76 kg de este aceite negro juntado se concentraron al vacío, se disolvieron en etanol (0,9 l) y se añadieron a una disolución bien agitada de bisulfito de sodio (7,3 l de una disolución
madre preparada disolviendo 2,91 kg de bisulfito de sodio en 10 l de agua) y etanol (1,1 l). El recipiente del aceite negro se enjuagó con etanol (0,55 l 2x) dos veces y estos dos enjuagues también se añadieron a la mezcla de reacción de bisulfito. Después de 75 min, se añadió heptano (8 l), se mezcló bien durante 15 min, las capas se dejaron sedimentar y separarse. A la capa orgánica se añadió una disolución de bisulfito de sodio (2,8 l de una disolución madre preparada disolviendo 2,91 kg de bisulfito de sodio en 10 l de agua), agua (2,4 l) y etanol (1,6 l). Después de agitar durante 30 min, las capas se dejaron sedimentar y separarse. Los dos extractos acuosos de bisulfuro se combinaron y los matraces se enjuagaron con agua (3,2 l). A continuación, se añadieron tolueno (7 l) y heptano (7 l), la mezcla se agitó bien y el pH se ajustó a 12 usando hidróxido de sodio (ac. al 10 %) (la temperatura pasó a ser 28 °C). Después de agitar durante 20 min adicionales, las capas se dejaron sedimentar y se separaron a 30 °C. La capa acuosa se extrajo con una mezcla de tolueno (2,3 l) y heptano (4,6 l). Las capas se separaron y se combinaron las capas orgánicas. Las capas orgánicas combinadas se lavaron con agua (7,7 l) y se concentraron al vacío para dar el compuesto del título purificado 6 (4,17 kg que contienen 1,06 kg de tolueno y 3,11 kg de compuesto 6). Este procedimiento se repitió con los 24 kg restantes del aceite negro (compuesto del título en bruto 6) y en dos ejecuciones dio en total 14,23 kg adicionales de 4-[2-[5-[[[(1,1-dimetiletil)dimetilsilil]oxi]etil]-2-piridinil]etoxi]-benzaldehído (6) como aceite marrón corregido en cuanto a residuo de tolueno (basado en 1H RMN) y 0,13 kg de compuesto 6 como enjuague del equipo en metanol (rendimiento total = 79 %, calculado a partir del compuesto 4 y corregido en cuanto a disolventes).
Etapa f: Síntesis de (5Z)-5-[[4-[2-[5-[[[(1,1-dimetiletil)dimetilsilil]oxi]etil]-2-piridinil]etoxi]fenil]metilen]-2,4-tiazolidindiona (7)
Una disolución de 4-[2-[5-[[[(1,1-dimetiletil)dimetilsilil]oxi]etil]-2-piridinil]-etoxi]-benzaldehído (6) (14,36 kg de compuesto 6 en tolueno/metanol), tolueno (22,2 l), piperidina (44 g) y metanol (14.5 l) se concentró a 45 °C (temperatura de camisa) a presión reducida. El residuo se disolvió en metanol (61,6 l) y se añadieron 2,4-tiazolidindiona (5257 g) y piperidina (1544 g). El equipo de adición se enjuagó con metanol (en total 10,8 l), el enjuague también se añadió a la mezcla de reacción y la mezcla se calentó a 47 °C. Después de 28 h, la mezcla de reacción se dejó enfriar a temperatura ambiente. La mezcla de pH se ajustó a pH 5-6 mediante la adición de ácido acético (se requirieron 0,73 kg). Después de una noche a temperatura ambiente, se añadió agua (10,9 l) y la suspensión se agitó a temperatura ambiente durante 2 h adicionales. Los sólidos se aislaron por filtración, se lavaron con metanol (7,3 l) y se secaron al vacío para dar el compuesto en bruto 7 (13,6 kg). El compuesto en bruto se mezcló con metanol (16 l) y diclorometano (54 l) y se calentó a 32 °C hasta que se disolvieron todos los sólidos. A continuación, el disolvente se retiró por destilación a presión reducida a una temperatura de camisa de 45 °C manteniendo un volumen constante mediante la adición de metanol hasta que la temperatura de la mezcla alcanzó 34 °C a una presión de 333 mbar (se retiraron 84 l de destilado y se añadieron 84 l de metanol). A continuación, se agitó a una temperatura de camisa establecida de 2 °C durante una hora. Los sólidos se aislaron por filtración, se lavaron con metanol (8 l 2x) y se secaron al vacío a 30 °C para dar el compuesto del título 7 (12,38 kg) (rendimiento = 68 %).
Etapa g: Síntesis de 5-[[4-[2-[5-[[[(1,1-dimetiletil)dimetilsilil]oxi]etil]-2-piridinil]etoxi]fenil]metil]-2,4-tiazolidindiona (8)
A una suspensión agitada de (5Z)-5-[[4-[2-[5-[[[(1,1-dimetiletil)dimetilsilil]oxi]-etil]-2-piridinil]etoxi]fenil]metilen]-2,4-tiazolidindiona (7) (12,38 kg) en THF (25 l) e hidróxido de sodio (ac. 1 N, 26 l) se añadió una disolución de cloruro de cobalto (8,25 g) y de dimetilglioxima (289 g) en THF (2,65 L) y agua (0,66 L). El matraz que contenía el cloruro de cobalto/dimetilglioxima se enjuagó con una mezcla de THF y agua (1/1 en v/v, 1 l) y el enjuague se añadió al contenido del reactor. Posteriormente, la suspensión se calentó a 30 °C y se puso en una atmósfera de nitrógeno aplicando la secuencia de vacío y purgando con nitrógeno (4x). Se preparó una disolución madre de borohidruro de sodio disolviendo borohidruro de sodio (5,0 kg) en una mezcla de agua (23,3 l) y una disolución de hidróxido de sodio (ac. 1 N, 6,2 l), que se puso en una atmósfera de nitrógeno aplicando una secuencia de vacío y purgando con nitrógeno (3x). Esto se añadió a la suspensión del compuesto 7 a una velocidad de 6,0 l/h. Simultáneamente, se añadió ácido acético saturado con gas nitrógeno a la suspensión a una velocidad tal que se mantenía un pH de 9,5-10,5. Después de una hora, la velocidad de adición de la disolución de borohidruro de sodio se redujo a 2,6 l/h. Después de la adición de 23 l de disolución de borohidruro de sodio, se detuvo la adición de borohidruro de sodio y ácido acético. La mezcla se dejó enfriar a 20 °C y se agitó a esta temperatura durante la noche. A continuación, se añadió acetona (3,1 l) lentamente. Después de agitar la mezcla de reacción durante 15 minutos, se añadió ácido acético hasta que el pH fue 5,5-6,0 (se requirieron 5,2 l). A continuación, se añadió una mezcla de acetato de etilo/tolueno (1/3 en v/v, 37,1 l), se mezcló bien y las capas se dejaron sedimentar. La capa acuosa se separó y se lavó con acetato de etilo/tolueno (1/3 en v/v, 12,4 l). Ambos extractos orgánicos se juntaron y se añadió agua (12,4 l), se mezcló bien y las capas se dejaron sedimentar. El pH de la capa acuosa se ajustó a 5,5-6 usando una disolución de hidróxido de sodio 1 M (ac.) y se mezcló nuevamente con la capa orgánica. Las capas se dejaron sedimentar y la capa orgánica se separó y se concentró al vacío para dar 23,15 kg de aceite amarillo (mezcla en bruto que contiene el compuesto del título 8 y su complejo de borano y tolueno). El equipo se enjuagó con tolueno (4 l) para dar una disolución (4,7 kg) que contenía producto en bruto en tolueno.
Se juntaron los 23,15 kg de la mezcla en bruto que contenían el compuesto del título 8 y su complejo de borano y tolueno y el enjuague de los 4,7 kg que contienen producto en tolueno, se añadió tolueno adicional (30 l) y se agitó durante 30 minutos. A continuación, la suspensión se filtró y los sólidos se lavaron con tolueno (10 l). El filtrado se sometió a cromatografía en columna (gel de sílice (70 kg), gradiente de tolueno con respecto a tolueno/acetato de etilo
1/1) para dar 18,7 kg de una disolución de tolueno que contenía 10,96 kg de una mezcla de 5-[[4-[2-[5-[[[(1,1-dimetiletil)dimetilsilil]oxi]etil]-2-piridinil]etoxi]fenil]-metil]-2,4-tiazolidindiona (8) y su complejo de borano como un aceite ligeramente amarillo (rendimiento = 85 % a partir del compuesto 7).
Etapa h: Síntesis de clorhidrato de 5-[[4-[2-[5-(1-hidroxietil)-2-piridinil]etoxi]fenil]-metil]-2,4-tiazolidindiona (9)
Una disolución de tolueno de 18,7 kg que contenía 10,96 kg de una mezcla de 5-[[4-[2-[5-[[[(1,1-dimetiletil)dimetilsilil]oxi]etil]-2-piridinil]etoxi]fenil]metil]-2,4-tiazolidindiona (8) y su complejo de borano se transfirieron a un reactor, los recipientes se enjuagaron con tolueno (4 l) y el enjuague también se añadió al reactor. El nivel de tolueno se redujo al 25 % en p/p por destilación. Después se diluyó con metanol (54 l) a 20 °C y ácido clorhídrico (30 %, 6,15 l) en aproximadamente 15 minutos. Esta disolución se calentó a 40 °C. Dos horas después de la adición, se retiraron 40 l de sustancias volátiles a presión reducida. A continuación, se añadió acetonitrilo (76 l) y la mezcla se calentó a reflujo durante 0,5 h. A continuación, la suspensión se dejó enfriar a temperatura ambiente y se agitó durante dos horas a temperatura ambiente. Se aislaron por filtración sólidos, se lavaron con una mezcla de acetonitrilo/agua (20/1 en v/v, 8,4 l), acetonitrilo (14,5 l) y agua (10 l). Los sólidos aislados se transfirieron a un reactor, se añadió agua (33 l), la suspensión se agitó bien y el pH se ajustó a 2 usando ácido clorhídrico 2 M. La suspensión se agitó a 50 °C durante dos horas y después se dejó enfriar a temperatura ambiente. Después de una hora a temperatura ambiente, los sólidos se aislaron por filtración, se lavaron con agua (20 l) y se secó al vacío a 40 °C para dar 7,77 kg de sólidos blancos (compuesto en bruto 9) (rendimiento = 85 %, sin corregir en cuanto a disolventes residuales).
Purificación de clorhidrato de 5-[[4-[2-[5-(1-hidroxietil)-2-piridinil]etoxi]fenil]metil]-2,4-tiazolidindiona (9):
El clorhidrato de 5-[[4-[2-[5-(1-hidroxietil)-2-piridinil]etoxi]fenil]-metil]-2,4-tiazolidindiona en bruto (7,77 kg, compuesto en bruto 9) se disolvió en una mezcla de agua (35 l) y metanol (124 l) por calentamiento. A esta disolución a 45 °C se añadió una suspensión de carbón activado (0,78 kg) en metanol (4 l). El equipo se enjuagó con metanol (38,4 l) y agua (12,6 l) y se añaden enjuagues al reactor. Después de 0,5 h de agitación a 45 °C, la suspensión caliente se filtró para retirar el carbón activado y el filtro se lavó dos veces con metanol/agua (7/2 en v/v, 18 l). El filtrado se concentró al vacío a una temperatura de camisa de 50 °C hasta un volumen de 23,3 l. A la suspensión se añadió butanona (38,9 l). Después, las sustancias volátiles se retiraron por destilación con adición simultánea de butanona a una velocidad tal que el volumen fue constante hasta que se alcanzó una temperatura de proceso de 76-77 °C (se requirió 251 l de butanona). A continuación, se detuvo el calentamiento y se permitió que la suspensión alcanzara la temperatura ambiente. Después de 0,75 h a temperatura ambiente, los sólidos se aislaron por filtración, se lavaron con una mezcla de butanona/agua (95/5 en v/v, 38 l) y butanona (38 l) y se secaron al vacío a 40 °C para dar 7,14 kg de compuesto 9 como un sólido blanco (rendimiento = 92 %).
Ejemplo 8
Deuteración de 5-[[4-[2-[5-(1-hidroxietil)-2-piridinil]etoxi]fenil]metil]-2,4-tiazolidindiona

Deuteración de 5-[[4-[2-[5-(1-hidroxietil)-2-piridinil]etoxi]fenil]-metil]-2,4-tiazolidindiona se realizó de la siguiente manera:
Se añadió N,N-(diisopropil)aminometilpoliestireno (agente inorgánico al 1 %) (PS-DIEA; 3,2 eq) a una disolución de 5-[[4-[2-[5-(1-hidroxietil)-2-piridinil]etoxi]fenil]-metil]-2,4-tiazolidindiona (1,0 eq.) en CD3OD (60 ml) y la mezcla se agitó a temperatura ambiente durante 7-8 días. La mezcla de reacción se filtró y se lavó con CD3OD (10 ml). El filtrado se concentró, el residuo se diluyó con CH2Cl2 y se concentró completamente para proporcionar 5-[[4-[2-[5-(1 -hidroxietil)-2-piridinil]etoxi]fenil]-metil]-2,4-tiazolidindiona deuterada como un sólido blanco (como una mezcla de los 4 isómeros deuterados). Rendimiento: 73 % (440 mg).
ES-MS [M+H]+: 374,1.
1H RMN (400 MHz, DMSO-d6): 811,98 (s. a., 1H), 8,46 (d, J = 1,6 Hz, 1H), 7,67 (dd, J = 8,0, 2,4 Hz, 1H), 7,30 (d, J = 8,0 Hz, 1H), 7,13 (d, J = 8,8 Hz, 2H), 6,86 (d, J = 8,4 Hz, 2H), 5,24 (d, J = 4,4 Hz, 1H), 4,75 (m, 1H), 4,30 (t, J = 6,4 Hz, 2H), 3,31 (m, 1H), 3,14 (t, J = 6,4 Hz, 2H), 3,03 (d, J = 14,4 Hz, 1H), 1,34 (d, J = 6,4 Hz, 3H). Se observaron trazas de material de partida no deuterado.
Ejemplo 9
2-(5-(1-((terc-butildimetilsilil)oxi)etil)piridin-2-il)etan-1-ol (4)
Un matraz de 20 l equipado con agitador mecánico, en atmósfera de nitrógeno, se cargó con 3160 g (10,0 mol, 1,0 eq.) de compuesto 3 y 8 l de dietiléter. La mezcla se enfrió a -60 °C y 4 l de 2,5 M n-BuLi en hexanos (10,0 mol, 1,0 eq. n-BuLi) se añadió gota a gota en 1 hora, mientras se mantenía la temperatura < -55 °C. La mezcla se agitó durante 1 hora a -80 °C. Se añadió yoduro de cobre (I) (953 g, 5,0 mol, 0,5 eq.) se añadió de una vez, mientras que se dejó que la temperatura aumentara a -43 °C. La mezcla se enfrió de nuevo a -55 °C a -60 °C. Una disolución de óxido de etileno (528 g, 12,0 mol, 1,2 eq.) en 900 g de dietiléter a -20 °C (preparado por separado en un recipiente de 2 l) se añadió gota a gota (rápidamente) en 20 minutos a la mezcla de reacción. Se observó una reacción exotérmica y se requirió enfriamiento externo con nitrógeno líquido para mantener la temperatura por debajo de -20 °C y finalmente para enfriar de nuevo a -60 °C. La mezcla se agitó durante la noche, mientras que la mezcla de reacción se dejó alcanzar la temperatura ambiente.
En un recipiente de separación de 40 l, se disolvieron 1,2 kg de cloruro de amonio en 6 l de agua. La mezcla de reacción se vertió en la disolución de cloruro de amonio agitada y se agitó durante 1 hora para destruir la mayoría de los complejos de cobre. Las capas se separaron y la capa de agua se extrajo con heptano (2 x 2 l). Las capas orgánicas combinadas se secaron sobre sulfato de sodio y se filtraron sobre una ruta de Celite® (para retirar más sales de cobre). Después de la evaporación de los disolventes, 3088 g (máx. 10,0 mol) del compuesto en bruto 4 se aislaron en forma de un aceite de color pardo oscuro, que se purificó por cromatografía en columna, usando 22,5 kg de sílice. Se aplicó un gradiente para eluir el producto, comenzando con 80 l de heptano/EtOAc 1/1, seguido de 20 l de EtOAc, 40 l de EtOAc/MeOH 95/5, 20 l de EtOAc/MeOH 9/1, 20 l de EtOAc/MeOH 8/2 y finalmente 40 l de EtOAc/MeOH 7/3. Las fracciones de producto con alta pureza se combinaron y se evaporaron para dar 1390 g (4,94 mol, rendimiento 9,4 %) de compuesto 4 en forma de un aceite marrón con una pureza por HPLC del 99,3 %.
La amplitud y el alcance de la presente invención no deberían estar limitados por ninguna de las realizaciones a modo de ejemplo descritas anteriormente, pero deben definirse solo según las siguientes reivindicaciones.
Claims (32)
1. Proceso, que comprende:
hacer reaccionar un compuesto de fórmula II:
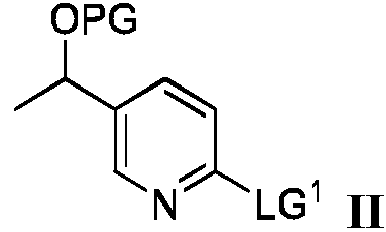
en donde PG es un grupo protector y LG1 es un grupo saliente,
con óxido de etileno en presencia de
(a) un alquil-litio y una sal de cobre (I); o
(b) un alquil-litio y un ácido de Lewis, y
un disolvente,
para dar un compuesto de fórmula III:

2. Proceso según la reivindicación 1, en donde PG es un grupo protector seleccionado del grupo que consiste en un grupo protector de sililo, tetrahidropiranilo, metoximetilo, y bencilo; y preferiblemente se añaden el óxido de etileno, alquil-litio, y sal de cobre (I) o el ácido de Lewis a la mezcla de reacción que comprende el compuesto de fórmula II mientras se mantiene la temperatura de la mezcla de reacción por debajo de -20 °C; y se añaden más preferiblemente el óxido de etileno, alquil-litio, y sal de cobre (I) o el ácido de Lewis mientras se mantiene la temperatura de la mezcla de reacción por debajo de -55 °C; y más preferiblemente el óxido de etileno se añade a la mezcla de reacción después de añadir primero el alquil-litio.
3. Proceso según la reivindicación 1 o 2, en el que se permite que la mezcla de reacción se caliente a temperatura ambiente después de la adición de los reactivos.
4. Proceso según una cualquiera de las reivindicaciones 1-3, en el que la reacción se lleva a cabo en presencia de un alquil-litio y yoduro de cobre (I); y se añaden preferiblemente el alquil-litio y el yoduro de cobre (I) mientras se mantiene la temperatura de reacción por debajo de -55 °C.
5. Proceso según la reivindicación 1, que comprende
hacer reaccionar un compuesto de fórmula II:
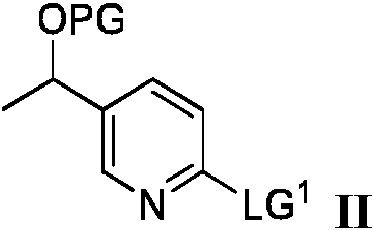
en donde PG es un grupo protector seleccionado del grupo que consiste en un grupo protector de sililo, tetrahidropiranilo, y metoximetilo, y LG1 es un grupo saliente,
con óxido de etileno en presencia de un alquil-litio y yoduro de cobre (I), y
un disolvente, en el que
la temperatura de reacción se mantiene por debajo de -20 °C cuando se añade el alquil-litio y el yoduro de cobre (I) a la mezcla de reacción,
para dar un compuesto de fórmula III:

6. Proceso según la reivindicación 5, en el que el óxido de etileno se añade a la mezcla de reacción después de añadir primero el alquil-litio; y se añaden preferiblemente el alquil-litio y el yoduro de cobre (I) mientras se mantiene la temperatura de reacción por debajo de -55 °C; y más preferiblemente en el que se permite que la mezcla de reacción se caliente a temperatura ambiente después de la adición de los reactivos.
7. Proceso según una cualquiera de las reivindicaciones 1-6, en el que el disolvente se selecciona del grupo que consiste en un disolvente orgánico aprótico no polar y un disolvente orgánico aprótico polar, o una mezcla de los mismos; y preferiblemente el disolvente es un disolvente orgánico aprótico no polar; y más preferiblemente el disolvente es dietiléter o terc-butilmetiléter, o una mezcla de los mismos.
8. Proceso según la reivindicación 1, en el que el proceso comprende:
(a) hacer reaccionar el compuesto que tiene la fórmula II:

con el alquil-litio en un primer disolvente a una temperatura para dar una primera mezcla de reacción; en el que:
la temperatura está por debajo de -20 °C;
PG es un grupo protector seleccionado del grupo que consiste en un grupo protector de sililo, tetrahidropiranilo, metoximetilo, y bencilo; y
LG1 es un grupo saliente, y o bien
(b) añadir una disolución del óxido de etileno en un segundo disolvente a dicha temperatura a dicha primera mezcla de reacción a dicha temperatura para dar una segunda mezcla de reacción; y
(c) añadir la sal de cobre (I) o ácido de Lewis a dicha segunda mezcla de reacción a dicha temperatura para dar una tercera mezcla de reacción que comprende un compuesto que tiene la fórmula III; o bien
(b) añadir la sal de cobre (I) o ácido de Lewis a dicha primera mezcla de reacción a dicha temperatura para dar una segunda mezcla de reacción; y
(c) añadir una disolución del óxido de etileno en un segundo disolvente a dicha temperatura a dicha segunda mezcla de reacción a dicha temperatura para dar una tercera mezcla de reacción que comprende un compuesto que tiene la fórmula III.
9. Proceso según la reivindicación 8, en el que la temperatura está por debajo de -55 °C.
10. Proceso según la reivindicación 8 o 9, en el que se permite que dicha tercera mezcla de reacción se caliente a una temperatura de 20 °C-25 °C; y preferiblemente dicha tercera mezcla de reacción se mantiene a 20 °C-25 °C durante al menos 4 horas, durante al menos 6 horas, durante al menos 8 horas, o durante al menos 10 horas.
11. Proceso según una cualquiera de las reivindicaciones 8-10, en el que dicho primer disolvente y dicho segundo disolvente se seleccionan cada uno independientemente del grupo que consiste en un disolvente orgánico aprótico no polar y un disolvente orgánico aprótico polar, o una mezcla de los mismos; y preferiblemente dicho primer disolvente y dicho segundo disolvente se seleccionan del grupo que consiste en dietilétery metil
terc-butiléter, o una mezcla de los mismos.
12. Proceso según una cualquiera de las reivindicaciones 8-11, en el que el proceso comprende yoduro de cobre (I).
13. Proceso según una cualquiera de las reivindicaciones 1-12, en el que PG se selecciona de grupos protectores de sililo; y preferiblemente PG es un grupo alquil o aril sililo, o una combinación de los mismos; y más preferiblemente PG es un grupo trialquilsililo; y más preferiblemente PG es TBDMS, TMS, TES, TIPS, o TBDPS; y más preferiblemente PG es terc-butildimetilsililo (TBDMS).
14. Proceso según una cualquiera de las reivindicaciones 1-13, en el que LG1 es cloruro, bromuro, yoduro, o fluoruro; y preferiblemente LG1 es bromuro; y en el que el alquil-litio es alquil-litio C-i-a; y preferiblemente el alquil-litio es n-butil-litio.
15. Proceso según una cualquiera de las reivindicaciones 1-14, que comprende además aislar dicho compuesto de fórmula III y opcionalmente purificar el compuesto aislado de fórmula III.
16. Proceso según una cualquiera de las reivindicaciones 1-15, que comprende además hacer reaccionar dicho compuesto de fórmula III con R1-Cl, en donde R1 es un organosulfonato, en presencia de una primera base para dar un compuesto de fórmula IV:

y preferiblemente el organosulfonato es un grupo tosilo (Ts) o un grupo mesilo (Ms); y más preferiblemente el organosulfonato es un grupo tosilo; y más preferiblemente la primera base es una o más de una amina, una sal de amonio cuaternario en combinación con una disolución acuosa de un hidróxido de metal alcalino, hidróxido de tetrabutilamonio, o un hidróxido de metal alcalino; y más preferiblemente la primera base es una sal de amonio cuaternario en combinación con una disolución acuosa de un hidróxido de metal alcalino; y más preferiblemente la primera base es bromuro de tetra-n-butilamonio en NaOH acuoso.
17. Proceso según la reivindicación 16, en el que la reacción se realiza en presencia de un disolvente.
18. Proceso según la reivindicación 16 o 17, que comprende además hacer reaccionar dicho compuesto de fórmula IV con 4-hidroxibenzaldehído:

en presencia de una segunda base y un disolvente para dar un compuesto de fórmula V:

y preferiblemente la segunda base es un carbonato de metal alcalino, una trialquilamina, o un alcóxido de metal alcalino; y más preferiblemente la segunda base es K2CO3; y más preferiblemente el disolvente se selecciona del grupo que consiste en tolueno, etanol, 2-propanol, t Hf , 2-MeTHF, y agua, o una mezcla de los mismos; y más preferiblemente el disolvente es una mezcla de tolueno y etanol.
19. Proceso según la reivindicación 18, que comprende además añadir agua a la mezcla de reacción; y preferiblemente la cantidad de agua añadida es de un 2 % a un 7 % en v/v; y preferiblemente que comprende además aislar y purificar dicho compuesto de fórmula V; y preferiblemente dicha purificación comprende extraer con una disolución acuosa de bisulfito.
20. Proceso según la reivindicación 18 o 19, que comprende además hacer reaccionar dicho compuesto de fórmula V con 2,4-tiazolidindiona:

en presencia de piperidina y opcionalmente un disolvente, y opcionalmente un ácido orgánico, para dar un compuesto de fórmula VI:

21. Proceso según la reivindicación 20, en el que la reacción se realiza en presencia del disolvente; y preferiblemente el disolvente se selecciona del grupo que consiste en tolueno, un alcohol inferior, hexano, y ciclohexano, o una mezcla de los mismos; y más preferiblemente el disolvente es tolueno o metanol, o una mezcla de los mismos.
22. Proceso según la reivindicación 20 o 21, en el que la reacción se realiza en presencia del ácido orgánico; y preferiblemente el ácido orgánico es ácido acético o ácido fórmico.
23. Proceso según una cualquiera de las reivindicaciones 20-22, en el que la reacción se lleva a cabo a una temperatura de desde 45 °C hasta 80 °C; y preferiblemente el disolvente es metanol y el proceso se lleva a cabo a 47 °C.
24. Proceso según una cualquiera de las reivindicaciones 20-23, que comprende además reducir dicho compuesto de fórmula VI para dar un compuesto de fórmula VII:

y preferiblemente la reducción se realiza permitiendo que dicho compuesto de fórmula VI reaccione con un agente reductor en presencia de un ion metálico y un agente complejante para el ion metálico; y más preferiblemente el agente reductor es NaBH4, el ion metálico es Co2+, y el ligando es dimetilglioxima; y más preferiblemente el pH de la mezcla de reacción se mantiene a pH de 9,5 a 10,5; y más preferiblemente la reducción se realiza en una atmósfera inerte; y más preferiblemente la reducción se realiza en una atmósfera de nitrógeno.
25. Proceso según la reivindicación 24, que comprende además desproteger dicho compuesto de fórmula VII, y opcionalmente tratar además con un ácido, para dar un compuesto de fórmula I:

o una sal farmacéuticamente aceptable del mismo; y preferiblemente la desprotección de dicho compuesto de fórmula VII y la formación de sal se realizan simultáneamente; y más preferiblemente que comprende además precipitar dicho compuesto de fórmula I, o una sal farmacéuticamente aceptable del mismo; y más preferiblemente dicha precipitación se realiza tratando la mezcla de reacción con un disolvente aprótico polar a una temperatura elevada y luego permitiendo que la mezcla de reacción se enfríe a temperatura ambiente; y más preferiblemente el disolvente aprótico polar es acetonitrilo.
26. Proceso según la reivindicación 25, que comprende además aislar dicho precipitado que comprende el compuesto de fórmula I, o una sal farmacéuticamente aceptable del mismo; y preferiblemente dicho precipitado se aísla por filtración para obtener un precipitado aislado; y más preferiblemente dicho precipitado
aislado se lava con acetonitrilo y/o una mezcla de acetonitrilo y agua; y más preferiblemente el proceso comprende además purificar dicho precipitado aislado.
27. Proceso según la reivindicación 25 o 26, en el que dicho compuesto de fórmula I se aísla como su sal farmacéuticamente aceptable.
28. Proceso según la reivindicación 25 o 26, en el que dicho compuesto de fórmula I es sal de clorhidrato de 5-[[4-[2-[5-(1-hidroxietil)-2-piridinil]etoxi]fenil]metil]-2,4-tiazolidindiona.
29. Proceso según una cualquiera de las reivindicaciones 25-28, que comprende además deuterar el compuesto de fórmula I, o una sal farmacéuticamente aceptable del mismo.
30. Proceso según la reivindicación 1 u 8, en el que dicho compuesto de fórmula II se prepara protegiendo el grupo hidroxilo de un compuesto de fórmula VIII:

en donde LG1 es un grupo saliente, con un grupo protector PG, en donde PG se selecciona del grupo que consiste en un grupo protector de sililo, tetrahidropiranilo, metoximetilo, y bencilo para dar el compuesto de fórmula II; y preferiblemente dicho compuesto de fórmula VIII se hace reaccionar con un cloruro de sililo seleccionado del grupo que consiste en TBDMS-Cl, TMS-Cl, TBDPS-Cl, y TIPS-Cl e imidazol a alta concentración en DMF.
31. Proceso según la reivindicación 30, en el que dicho compuesto de fórmula VIII se prepara haciendo reaccionar un compuesto de fórmula IX:

en donde LG1 es un grupo saliente, con CH3CHO en presencia de un haluro de alquil-magnesio y un disolvente para dar el compuesto de fórmula VIII.
32. Compuesto de fórmula III:

en donde PG es un grupo protector seleccionado del grupo que consiste en un grupo protector de sililo, y bencilo, preparado por el proceso como se reivindica en una cualquiera de las reivindicaciones 1-15.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP16382648 | 2016-12-23 | ||
| PCT/IB2017/058374 WO2018116281A1 (en) | 2016-12-23 | 2017-12-22 | Process for preparing 5-[[4-[2-[5-(1-hydroxyethyl)-2-pyridinyl]ethoxy]phenyl]methyl]-2,4-thiazolidinedione and salts thereof |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2926580T3 true ES2926580T3 (es) | 2022-10-27 |
Family
ID=57799523
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES17829042T Active ES2926580T3 (es) | 2016-12-23 | 2017-12-22 | Proceso para preparar 5-[[4-[2-[5-(1-hidroxietil)-2-piridinil]etoxi]fenil]metil]-2,4-tiazolidindiona y sales de la misma |
Country Status (19)
| Country | Link |
|---|---|
| US (2) | US11124505B2 (es) |
| EP (1) | EP3559010B1 (es) |
| JP (2) | JP7294661B2 (es) |
| KR (1) | KR102609373B1 (es) |
| CN (1) | CN110198945B (es) |
| BR (1) | BR112019012972B1 (es) |
| CA (1) | CA3046744A1 (es) |
| CY (1) | CY1125531T1 (es) |
| DK (1) | DK3559010T3 (es) |
| ES (1) | ES2926580T3 (es) |
| HR (1) | HRP20221067T1 (es) |
| HU (1) | HUE059604T2 (es) |
| IL (1) | IL267557B (es) |
| LT (1) | LT3559010T (es) |
| MX (1) | MX379709B (es) |
| PL (1) | PL3559010T3 (es) |
| PT (1) | PT3559010T (es) |
| SI (1) | SI3559010T1 (es) |
| WO (1) | WO2018116281A1 (es) |
Families Citing this family (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| ES2926580T3 (es) | 2016-12-23 | 2022-10-27 | Minoryx Therapeutics S L | Proceso para preparar 5-[[4-[2-[5-(1-hidroxietil)-2-piridinil]etoxi]fenil]metil]-2,4-tiazolidindiona y sales de la misma |
| CN112823004A (zh) | 2018-06-06 | 2021-05-18 | 米尼奥尔克斯治疗有限公司 | 5-[[4-[2-[5-乙酰基吡啶-2-基]乙氧基]苄基]-1,3-噻唑烷-2,4-二酮及其盐的用途 |
| EP3801515B1 (en) | 2018-06-06 | 2025-04-09 | Minoryx Therapeutics S.L. | 5-[[4-[2-[5-(1-hydroxyethyl)pyridin-2-yl]ethoxy]phenyl]methyl]-1,3-thiazolidine-2,4-dione and its salts for use in the treatment of mitochondrial diseases |
| CN112512524A (zh) | 2018-06-06 | 2021-03-16 | 米尼奥尔克斯治疗有限公司 | 给予治疗有效量的5-[[4-[2-[5-(1-羟基乙基)吡啶-2-基]乙氧基]苯基]甲基]-1,3-噻唑烷-2,4-二酮的方法 |
| EP4642456A1 (en) | 2022-12-28 | 2025-11-05 | Minoryx Therapeutics S.L. | Optimized dosing of leriglitazone |
| WO2024231881A2 (en) | 2023-05-09 | 2024-11-14 | Minoryx Therapeutics S.L. | Polymorphic forms and formulations of leriglitazone |
Family Cites Families (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE69231886T2 (de) * | 1991-04-11 | 2002-03-28 | Pharmacia & Upjohn Co., Kalamazoo | Thiazolidindionderivate, herstellung und anwendung |
| US5952509A (en) | 1996-06-27 | 1999-09-14 | Takeda Chemical Industries, Ltd. | Production of benzaldehyde compounds |
| US6067749A (en) | 1996-07-11 | 2000-05-30 | Tasmanian Alkaloids Pty. Ltd. | Papaver somniferum strain with high concentration of thebaine and oripavine |
| US5869669A (en) | 1996-07-26 | 1999-02-09 | Penick Corporation | Preparation of 14-hydroxynormorphinones from normorphinone dienol acylates |
| HUP0304096A2 (hu) | 2003-12-19 | 2005-08-29 | Richter Gedeon Vegyészeti Gyár Rt. | Eljárás pioglitazon hidrogénklorid előállítására és intermedierjei |
| GB0421687D0 (en) | 2004-09-30 | 2004-11-03 | Johnson Matthey Plc | Preparation of opiate analgesics |
| CA2594666A1 (en) | 2005-02-16 | 2006-08-24 | Micromet Ag | Use of activated polymers for separation of protein and polypeptide multimers |
| US20090247560A1 (en) * | 2006-09-28 | 2009-10-01 | Banyu Pharmaceutical Co., Ltd. | Diaryl ketimine derivative |
| JPWO2008136428A1 (ja) * | 2007-04-27 | 2010-07-29 | 武田薬品工業株式会社 | 含窒素5員複素環化合物 |
| EP2855489B1 (en) * | 2012-04-26 | 2017-01-04 | Bristol-Myers Squibb Company | Imidazothiadiazole and imidazopyridazine derivatives as protease activated receptor 4 (par4) inhibitors for treating platelet aggregation |
| WO2014152843A1 (en) | 2013-03-14 | 2014-09-25 | Deuterx, Llc | Deuterium-enriched 2,4-thiazolidinediones and methods of treatment |
| EA202091120A3 (ru) | 2014-04-02 | 2020-12-30 | Минорикс Терапьютикс С.Л. | Производные 2,4-тиазолинидона в лечении расстройств центральной нервной системы |
| ES2926580T3 (es) | 2016-12-23 | 2022-10-27 | Minoryx Therapeutics S L | Proceso para preparar 5-[[4-[2-[5-(1-hidroxietil)-2-piridinil]etoxi]fenil]metil]-2,4-tiazolidindiona y sales de la misma |
-
2017
- 2017-12-22 ES ES17829042T patent/ES2926580T3/es active Active
- 2017-12-22 SI SI201731203T patent/SI3559010T1/sl unknown
- 2017-12-22 HR HRP20221067TT patent/HRP20221067T1/hr unknown
- 2017-12-22 BR BR112019012972-3A patent/BR112019012972B1/pt active IP Right Grant
- 2017-12-22 EP EP17829042.5A patent/EP3559010B1/en active Active
- 2017-12-22 HU HUE17829042A patent/HUE059604T2/hu unknown
- 2017-12-22 US US16/470,866 patent/US11124505B2/en active Active
- 2017-12-22 KR KR1020197021529A patent/KR102609373B1/ko active Active
- 2017-12-22 WO PCT/IB2017/058374 patent/WO2018116281A1/en not_active Ceased
- 2017-12-22 PL PL17829042.5T patent/PL3559010T3/pl unknown
- 2017-12-22 JP JP2019534154A patent/JP7294661B2/ja active Active
- 2017-12-22 DK DK17829042.5T patent/DK3559010T3/da active
- 2017-12-22 LT LTEPPCT/IB2017/058374T patent/LT3559010T/lt unknown
- 2017-12-22 MX MX2019007327A patent/MX379709B/es unknown
- 2017-12-22 PT PT178290425T patent/PT3559010T/pt unknown
- 2017-12-22 CN CN201780084302.3A patent/CN110198945B/zh active Active
- 2017-12-22 CA CA3046744A patent/CA3046744A1/en active Pending
-
2019
- 2019-06-20 IL IL267557A patent/IL267557B/en unknown
-
2021
- 2021-09-20 US US17/479,838 patent/US11731963B2/en active Active
-
2022
- 2022-09-06 CY CY20221100593T patent/CY1125531T1/el unknown
-
2023
- 2023-02-06 JP JP2023016009A patent/JP2023052918A/ja not_active Withdrawn
Also Published As
| Publication number | Publication date |
|---|---|
| US20190389852A1 (en) | 2019-12-26 |
| CN110198945A (zh) | 2019-09-03 |
| HRP20221067T1 (hr) | 2022-11-25 |
| IL267557B (en) | 2021-10-31 |
| EP3559010A1 (en) | 2019-10-30 |
| IL267557A (en) | 2019-08-29 |
| PL3559010T3 (pl) | 2022-11-21 |
| BR112019012972B1 (pt) | 2023-04-18 |
| SI3559010T1 (sl) | 2022-10-28 |
| PT3559010T (pt) | 2022-09-01 |
| CY1125531T1 (el) | 2026-02-25 |
| MX379709B (es) | 2025-03-11 |
| HUE059604T2 (hu) | 2022-11-28 |
| KR20190100300A (ko) | 2019-08-28 |
| BR112019012972A2 (pt) | 2019-12-31 |
| US20220204496A1 (en) | 2022-06-30 |
| DK3559010T3 (da) | 2022-08-15 |
| US11731963B2 (en) | 2023-08-22 |
| JP2023052918A (ja) | 2023-04-12 |
| JP2020502228A (ja) | 2020-01-23 |
| CN110198945B (zh) | 2023-06-27 |
| LT3559010T (lt) | 2022-10-10 |
| EP3559010B1 (en) | 2022-06-08 |
| CA3046744A1 (en) | 2018-06-28 |
| JP7294661B2 (ja) | 2023-06-20 |
| KR102609373B1 (ko) | 2023-12-01 |
| MX2019007327A (es) | 2019-09-02 |
| WO2018116281A1 (en) | 2018-06-28 |
| US11124505B2 (en) | 2021-09-21 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2926580T3 (es) | Proceso para preparar 5-[[4-[2-[5-(1-hidroxietil)-2-piridinil]etoxi]fenil]metil]-2,4-tiazolidindiona y sales de la misma | |
| US20070225510A1 (en) | Convergent Process for the Synthesis of Taxane Derivatives | |
| US12202782B2 (en) | Processes and intermediates for preparing MCL1 inhibitors | |
| US6175023B1 (en) | Synthesis of water soluble 9-dihydro-paclitaxel derivatives from 9-dihydro-13-acetylbaccatin III | |
| US9434740B2 (en) | Taxane compounds, compositions and methods | |
| CN109415383A (zh) | 制备艾日布林的方法及其中间体 | |
| HK40014767A (en) | Process for preparing 5-[[4-[2-[5-(1-hydroxyethyl)-2-pyridinyl]ethoxy]phenyl]methyl]-2,4-thiazolidinedione and salts thereof | |
| HK40014767B (en) | Process for preparing 5-[[4-[2-[5-(1-hydroxyethyl)-2-pyridinyl]ethoxy]phenyl]methyl]-2,4-thiazolidinedione and salts thereof | |
| TWI914718B (zh) | 用於製備mcl1抑制劑之程序及中間物 | |
| WO2017106649A1 (en) | Synthetic route to anhydroryanodol, ryanodol, and structural analogues | |
| HK40096379A (zh) | 用於制备大环mcl1抑制剂的方法和中间体 | |
| US20090292131A1 (en) | Processes for preparation of taxanes and intermediates thereof | |
| HU231431B1 (hu) | Remdesivir köztitermékek | |
| HK40013289B (zh) | 用於制备噻唑烷二酮化合物及其盐的方法 | |
| HK40108339A (zh) | 用於合成缬苯那嗪的方法 | |
| HK40013289A (en) | Process for preparing thiazolidinedione compounds and salts thereof | |
| US20140249186A1 (en) | Taxane compounds, compositions and methods | |
| JP2010513459A (ja) | セコタキサンの調製方法 | |
| JP2011020944A (ja) | ポジトロン放出源化合物の製造方法 | |
| CN102643276A (zh) | 一种免疫抑制剂fr901483中间体及其合成方法 |