ES2929369T3 - Indolinas espirocíclicas como moduladores de IL-17 - Google Patents
Indolinas espirocíclicas como moduladores de IL-17 Download PDFInfo
- Publication number
- ES2929369T3 ES2929369T3 ES18732703T ES18732703T ES2929369T3 ES 2929369 T3 ES2929369 T3 ES 2929369T3 ES 18732703 T ES18732703 T ES 18732703T ES 18732703 T ES18732703 T ES 18732703T ES 2929369 T3 ES2929369 T3 ES 2929369T3
- Authority
- ES
- Spain
- Prior art keywords
- alkyl
- mmol
- formula
- optionally substituted
- amino
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 108050003558 Interleukin-17 Proteins 0.000 title description 40
- 102000013691 Interleukin-17 Human genes 0.000 title description 40
- 150000002476 indolines Chemical class 0.000 title description 2
- 238000011282 treatment Methods 0.000 claims abstract description 36
- 208000027866 inflammatory disease Diseases 0.000 claims abstract description 14
- 208000023275 Autoimmune disease Diseases 0.000 claims abstract description 9
- 230000002757 inflammatory effect Effects 0.000 claims abstract description 7
- 230000002265 prevention Effects 0.000 claims abstract description 6
- -1 amino, imino Chemical group 0.000 claims description 437
- 150000001875 compounds Chemical class 0.000 claims description 304
- 125000000217 alkyl group Chemical group 0.000 claims description 235
- 125000001424 substituent group Chemical group 0.000 claims description 111
- 239000001257 hydrogen Substances 0.000 claims description 54
- 229910052739 hydrogen Inorganic materials 0.000 claims description 54
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 claims description 51
- 125000000592 heterocycloalkyl group Chemical group 0.000 claims description 47
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 45
- 150000003839 salts Chemical class 0.000 claims description 42
- 125000001072 heteroaryl group Chemical group 0.000 claims description 40
- 229910052736 halogen Inorganic materials 0.000 claims description 39
- 150000002367 halogens Chemical class 0.000 claims description 39
- 125000003545 alkoxy group Chemical group 0.000 claims description 33
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 claims description 32
- 125000004093 cyano group Chemical group *C#N 0.000 claims description 31
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims description 31
- 125000003118 aryl group Chemical group 0.000 claims description 29
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 28
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims description 25
- 125000001412 tetrahydropyranyl group Chemical group 0.000 claims description 24
- 150000001602 bicycloalkyls Chemical group 0.000 claims description 23
- 125000000753 cycloalkyl group Chemical group 0.000 claims description 23
- 125000003003 spiro group Chemical group 0.000 claims description 23
- 125000004738 (C1-C6) alkyl sulfinyl group Chemical group 0.000 claims description 22
- 125000004739 (C1-C6) alkylsulfonyl group Chemical group 0.000 claims description 22
- 125000003917 carbamoyl group Chemical group [H]N([H])C(*)=O 0.000 claims description 22
- 125000001028 difluoromethyl group Chemical group [H]C(F)(F)* 0.000 claims description 21
- 125000004043 oxo group Chemical group O=* 0.000 claims description 21
- 125000004786 difluoromethoxy group Chemical group [H]C(F)(F)O* 0.000 claims description 20
- 125000000876 trifluoromethoxy group Chemical group FC(F)(F)O* 0.000 claims description 20
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 claims description 19
- 125000002757 morpholinyl group Chemical group 0.000 claims description 19
- 125000004205 trifluoroethyl group Chemical group [H]C([H])(*)C(F)(F)F 0.000 claims description 19
- 125000004432 carbon atom Chemical group C* 0.000 claims description 18
- 125000004193 piperazinyl group Chemical group 0.000 claims description 18
- 125000004890 (C1-C6) alkylamino group Chemical group 0.000 claims description 17
- 125000004457 alkyl amino carbonyl group Chemical group 0.000 claims description 17
- 125000000719 pyrrolidinyl group Chemical group 0.000 claims description 17
- 125000006272 (C3-C7) cycloalkyl group Chemical group 0.000 claims description 16
- 125000004397 aminosulfonyl group Chemical group NS(=O)(=O)* 0.000 claims description 16
- 125000001207 fluorophenyl group Chemical group 0.000 claims description 16
- 125000004750 (C1-C6) alkylaminosulfonyl group Chemical group 0.000 claims description 15
- 229910020008 S(O) Inorganic materials 0.000 claims description 15
- 125000000475 sulfinyl group Chemical group [*:2]S([*:1])=O 0.000 claims description 15
- 125000006700 (C1-C6) alkylthio group Chemical group 0.000 claims description 14
- 125000003282 alkyl amino group Chemical group 0.000 claims description 14
- 125000004448 alkyl carbonyl group Chemical group 0.000 claims description 14
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 claims description 13
- 125000003710 aryl alkyl group Chemical group 0.000 claims description 11
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 claims description 11
- 125000002485 formyl group Chemical group [H]C(*)=O 0.000 claims description 11
- 125000004845 (C1-C6) alkylsulfonylamino group Chemical group 0.000 claims description 10
- 125000004466 alkoxycarbonylamino group Chemical group 0.000 claims description 10
- 125000004216 fluoromethyl group Chemical group [H]C([H])(F)* 0.000 claims description 10
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 claims description 10
- 125000004453 alkoxycarbonyl group Chemical group 0.000 claims description 9
- 125000003806 alkyl carbonyl amino group Chemical group 0.000 claims description 9
- 229910052799 carbon Inorganic materials 0.000 claims description 8
- 125000004186 cyclopropylmethyl group Chemical group [H]C([H])(*)C1([H])C([H])([H])C1([H])[H] 0.000 claims description 8
- 229910052760 oxygen Inorganic materials 0.000 claims description 8
- 229910052717 sulfur Inorganic materials 0.000 claims description 8
- 125000006001 difluoroethyl group Chemical group 0.000 claims description 7
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 claims description 6
- 239000004480 active ingredient Substances 0.000 claims description 6
- 125000000725 trifluoropropyl group Chemical group [H]C([H])(*)C([H])([H])C(F)(F)F 0.000 claims description 6
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 claims description 5
- 239000008194 pharmaceutical composition Substances 0.000 claims description 5
- 125000000472 sulfonyl group Chemical group *S(*)(=O)=O 0.000 claims description 5
- 125000006545 (C1-C9) alkyl group Chemical group 0.000 claims description 4
- 125000004103 aminoalkyl group Chemical group 0.000 claims description 4
- 239000003937 drug carrier Substances 0.000 claims description 4
- 125000004471 alkyl aminosulfonyl group Chemical group 0.000 claims description 3
- 238000002560 therapeutic procedure Methods 0.000 claims description 3
- 125000006297 carbonyl amino group Chemical group [H]N([*:2])C([*:1])=O 0.000 claims description 2
- 125000000446 sulfanediyl group Chemical group *S* 0.000 claims description 2
- 150000002431 hydrogen Chemical class 0.000 claims 3
- 230000000694 effects Effects 0.000 abstract description 6
- 230000009286 beneficial effect Effects 0.000 abstract description 5
- 101000998146 Homo sapiens Interleukin-17A Proteins 0.000 abstract description 2
- 230000003389 potentiating effect Effects 0.000 abstract description 2
- JYGFTBXVXVMTGB-UHFFFAOYSA-N indolin-2-one Chemical class C1=CC=C2NC(=O)CC2=C1 JYGFTBXVXVMTGB-UHFFFAOYSA-N 0.000 abstract 1
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 459
- 239000000543 intermediate Substances 0.000 description 281
- 238000000034 method Methods 0.000 description 220
- 239000000243 solution Substances 0.000 description 210
- 235000019439 ethyl acetate Nutrition 0.000 description 168
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 166
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 165
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical group N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 128
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 126
- 239000011541 reaction mixture Substances 0.000 description 125
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 122
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 description 110
- 238000000589 high-performance liquid chromatography-mass spectrometry Methods 0.000 description 108
- 239000007787 solid Substances 0.000 description 103
- 239000000203 mixture Substances 0.000 description 97
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 74
- 239000012267 brine Substances 0.000 description 65
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 65
- 229910052757 nitrogen Chemical group 0.000 description 63
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 58
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 56
- 239000002904 solvent Substances 0.000 description 55
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 54
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 54
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 50
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 48
- 238000003818 flash chromatography Methods 0.000 description 48
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 46
- 239000012044 organic layer Substances 0.000 description 45
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 44
- 229920006395 saturated elastomer Polymers 0.000 description 44
- 239000003643 water by type Substances 0.000 description 44
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 42
- 239000000725 suspension Substances 0.000 description 42
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 39
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 38
- 239000003921 oil Substances 0.000 description 38
- 235000019198 oils Nutrition 0.000 description 38
- 239000000284 extract Substances 0.000 description 37
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical compound [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 35
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 35
- 238000006243 chemical reaction Methods 0.000 description 35
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 34
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 34
- 239000000843 powder Substances 0.000 description 34
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 33
- 229910052938 sodium sulfate Inorganic materials 0.000 description 33
- 235000011152 sodium sulphate Nutrition 0.000 description 33
- 239000000706 filtrate Substances 0.000 description 32
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 31
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 30
- 238000003756 stirring Methods 0.000 description 30
- BZLVMXJERCGZMT-UHFFFAOYSA-N Methyl tert-butyl ether Chemical compound COC(C)(C)C BZLVMXJERCGZMT-UHFFFAOYSA-N 0.000 description 29
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 27
- 230000008569 process Effects 0.000 description 25
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 24
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 23
- 239000002585 base Substances 0.000 description 23
- 238000004440 column chromatography Methods 0.000 description 23
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 23
- 235000019341 magnesium sulphate Nutrition 0.000 description 23
- 239000000463 material Substances 0.000 description 22
- 235000017557 sodium bicarbonate Nutrition 0.000 description 21
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 21
- 239000007832 Na2SO4 Substances 0.000 description 20
- DKGAVHZHDRPRBM-UHFFFAOYSA-N Tert-Butanol Chemical compound CC(C)(C)O DKGAVHZHDRPRBM-UHFFFAOYSA-N 0.000 description 20
- 239000005909 Kieselgur Substances 0.000 description 19
- 238000003556 assay Methods 0.000 description 19
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 19
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 18
- 235000019270 ammonium chloride Nutrition 0.000 description 18
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 description 18
- 238000001914 filtration Methods 0.000 description 18
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 17
- 239000008346 aqueous phase Substances 0.000 description 17
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 17
- 239000007821 HATU Substances 0.000 description 16
- 239000010410 layer Substances 0.000 description 16
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 16
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 15
- WMFOQBRAJBCJND-UHFFFAOYSA-M Lithium hydroxide Chemical compound [Li+].[OH-] WMFOQBRAJBCJND-UHFFFAOYSA-M 0.000 description 15
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 15
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 15
- 125000001153 fluoro group Chemical group F* 0.000 description 15
- 239000012074 organic phase Substances 0.000 description 15
- 238000005406 washing Methods 0.000 description 15
- DYHSDKLCOJIUFX-UHFFFAOYSA-N tert-butoxycarbonyl anhydride Chemical compound CC(C)(C)OC(=O)OC(=O)OC(C)(C)C DYHSDKLCOJIUFX-UHFFFAOYSA-N 0.000 description 14
- 235000011054 acetic acid Nutrition 0.000 description 13
- 229960000583 acetic acid Drugs 0.000 description 13
- 229910000024 caesium carbonate Inorganic materials 0.000 description 13
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 13
- 125000001309 chloro group Chemical group Cl* 0.000 description 13
- 238000004587 chromatography analysis Methods 0.000 description 13
- 239000012299 nitrogen atmosphere Substances 0.000 description 13
- 238000000746 purification Methods 0.000 description 13
- 239000003039 volatile agent Substances 0.000 description 13
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 12
- IAYPIBMASNFSPL-UHFFFAOYSA-N Ethylene oxide Chemical compound C1CO1 IAYPIBMASNFSPL-UHFFFAOYSA-N 0.000 description 12
- 239000002253 acid Substances 0.000 description 12
- 239000000460 chlorine Substances 0.000 description 12
- 229910052801 chlorine Inorganic materials 0.000 description 12
- 238000001816 cooling Methods 0.000 description 12
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 12
- 230000002209 hydrophobic effect Effects 0.000 description 12
- 125000004170 methylsulfonyl group Chemical group [H]C([H])([H])S(*)(=O)=O 0.000 description 12
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 12
- 238000002360 preparation method Methods 0.000 description 12
- 229910000029 sodium carbonate Inorganic materials 0.000 description 12
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 12
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 11
- 102000004127 Cytokines Human genes 0.000 description 11
- 108090000695 Cytokines Proteins 0.000 description 11
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 11
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 11
- 150000004292 cyclic ethers Chemical class 0.000 description 11
- PQVSTLUFSYVLTO-UHFFFAOYSA-N ethyl n-ethoxycarbonylcarbamate Chemical compound CCOC(=O)NC(=O)OCC PQVSTLUFSYVLTO-UHFFFAOYSA-N 0.000 description 11
- GLXDVVHUTZTUQK-UHFFFAOYSA-M lithium hydroxide monohydrate Substances [Li+].O.[OH-] GLXDVVHUTZTUQK-UHFFFAOYSA-M 0.000 description 11
- 229940040692 lithium hydroxide monohydrate Drugs 0.000 description 11
- 239000010502 orange oil Substances 0.000 description 11
- 125000003386 piperidinyl group Chemical group 0.000 description 11
- XJDNKRIXUMDJCW-UHFFFAOYSA-J titanium tetrachloride Chemical compound Cl[Ti](Cl)(Cl)Cl XJDNKRIXUMDJCW-UHFFFAOYSA-J 0.000 description 11
- 206010013774 Dry eye Diseases 0.000 description 10
- 235000019502 Orange oil Nutrition 0.000 description 10
- 239000003054 catalyst Substances 0.000 description 10
- 210000004027 cell Anatomy 0.000 description 10
- 125000000640 cyclooctyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C([H])([H])C1([H])[H] 0.000 description 10
- 125000004076 pyridyl group Chemical group 0.000 description 10
- 208000003556 Dry Eye Syndromes Diseases 0.000 description 9
- SIKJAQJRHWYJAI-UHFFFAOYSA-N Indole Chemical compound C1=CC=C2NC=CC2=C1 SIKJAQJRHWYJAI-UHFFFAOYSA-N 0.000 description 9
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 description 9
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 9
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 9
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 9
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 9
- 229910000104 sodium hydride Inorganic materials 0.000 description 9
- 125000003821 2-(trimethylsilyl)ethoxymethyl group Chemical group [H]C([H])([H])[Si](C([H])([H])[H])(C([H])([H])[H])C([H])([H])C(OC([H])([H])[*])([H])[H] 0.000 description 8
- 102100035018 Interleukin-17 receptor A Human genes 0.000 description 8
- 101710186083 Interleukin-17 receptor A Proteins 0.000 description 8
- 150000001412 amines Chemical class 0.000 description 8
- 239000001569 carbon dioxide Substances 0.000 description 8
- 229910002092 carbon dioxide Inorganic materials 0.000 description 8
- 239000003153 chemical reaction reagent Substances 0.000 description 8
- 239000013058 crude material Substances 0.000 description 8
- 239000011737 fluorine Substances 0.000 description 8
- 229910052731 fluorine Inorganic materials 0.000 description 8
- 125000006513 pyridinyl methyl group Chemical group 0.000 description 8
- CXNIUSPIQKWYAI-UHFFFAOYSA-N xantphos Chemical compound C=12OC3=C(P(C=4C=CC=CC=4)C=4C=CC=CC=4)C=CC=C3C(C)(C)C2=CC=CC=1P(C=1C=CC=CC=1)C1=CC=CC=C1 CXNIUSPIQKWYAI-UHFFFAOYSA-N 0.000 description 8
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 7
- 108090001005 Interleukin-6 Proteins 0.000 description 7
- BVCRERJDOOBZOH-UHFFFAOYSA-N bicyclo[2.2.1]heptanyl Chemical group C1C[C+]2CC[C-]1C2 BVCRERJDOOBZOH-UHFFFAOYSA-N 0.000 description 7
- 125000004113 cyclononanyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C([H])([H])C1([H])[H] 0.000 description 7
- 238000002866 fluorescence resonance energy transfer Methods 0.000 description 7
- 125000003392 indanyl group Chemical group C1(CCC2=CC=CC=C12)* 0.000 description 7
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 7
- 125000000286 phenylethyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])C([H])([H])* 0.000 description 7
- 125000006239 protecting group Chemical group 0.000 description 7
- 125000002098 pyridazinyl group Chemical group 0.000 description 7
- 125000004632 tetrahydrothiopyranyl group Chemical group S1C(CCCC1)* 0.000 description 7
- RAXXELZNTBOGNW-UHFFFAOYSA-N 1H-imidazole Chemical compound C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 description 6
- BPXKZEMBEZGUAH-UHFFFAOYSA-N 2-(chloromethoxy)ethyl-trimethylsilane Chemical compound C[Si](C)(C)CCOCCl BPXKZEMBEZGUAH-UHFFFAOYSA-N 0.000 description 6
- FPQQSJJWHUJYPU-UHFFFAOYSA-N 3-(dimethylamino)propyliminomethylidene-ethylazanium;chloride Chemical compound Cl.CCN=C=NCCCN(C)C FPQQSJJWHUJYPU-UHFFFAOYSA-N 0.000 description 6
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 6
- 239000000010 aprotic solvent Substances 0.000 description 6
- 239000012298 atmosphere Substances 0.000 description 6
- 229940090047 auto-injector Drugs 0.000 description 6
- 239000012230 colorless oil Substances 0.000 description 6
- 125000000582 cycloheptyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 6
- 125000004210 cyclohexylmethyl group Chemical group [H]C([H])(*)C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C1([H])[H] 0.000 description 6
- 238000010790 dilution Methods 0.000 description 6
- 239000012895 dilution Substances 0.000 description 6
- 208000035475 disorder Diseases 0.000 description 6
- 125000005843 halogen group Chemical group 0.000 description 6
- IXCSERBJSXMMFS-UHFFFAOYSA-N hydrogen chloride Substances Cl.Cl IXCSERBJSXMMFS-UHFFFAOYSA-N 0.000 description 6
- 229910000041 hydrogen chloride Inorganic materials 0.000 description 6
- 229910003002 lithium salt Inorganic materials 0.000 description 6
- 159000000002 lithium salts Chemical class 0.000 description 6
- 239000012071 phase Substances 0.000 description 6
- 229910000027 potassium carbonate Inorganic materials 0.000 description 6
- 239000000047 product Substances 0.000 description 6
- 238000004007 reversed phase HPLC Methods 0.000 description 6
- 125000003107 substituted aryl group Chemical group 0.000 description 6
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 6
- 125000003718 tetrahydrofuranyl group Chemical group 0.000 description 6
- FYSNRJHAOHDILO-UHFFFAOYSA-N thionyl chloride Chemical compound ClS(Cl)=O FYSNRJHAOHDILO-UHFFFAOYSA-N 0.000 description 6
- UGOMMVLRQDMAQQ-UHFFFAOYSA-N xphos Chemical group CC(C)C1=CC(C(C)C)=CC(C(C)C)=C1C1=CC=CC=C1P(C1CCCCC1)C1CCCCC1 UGOMMVLRQDMAQQ-UHFFFAOYSA-N 0.000 description 6
- DYLIWHYUXAJDOJ-OWOJBTEDSA-N (e)-4-(6-aminopurin-9-yl)but-2-en-1-ol Chemical compound NC1=NC=NC2=C1N=CN2C\C=C\CO DYLIWHYUXAJDOJ-OWOJBTEDSA-N 0.000 description 5
- 125000000022 2-aminoethyl group Chemical group [H]C([*])([H])C([H])([H])N([H])[H] 0.000 description 5
- 125000000339 4-pyridyl group Chemical group N1=C([H])C([H])=C([*])C([H])=C1[H] 0.000 description 5
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 5
- DLFVBJFMPXGRIB-UHFFFAOYSA-N Acetamide Chemical compound CC(N)=O DLFVBJFMPXGRIB-UHFFFAOYSA-N 0.000 description 5
- PCBZRNYXXCIELG-WYFCWLEVSA-N COC1=CC=C(C[C@H](NC(=O)OC2CCCC3(C2)OOC2(O3)C3CC4CC(C3)CC2C4)C(=O)N[C@@H]2[C@@H](CO)O[C@H]([C@@H]2O)N2C=NC3=C2N=CN=C3N(C)C)C=C1 Chemical compound COC1=CC=C(C[C@H](NC(=O)OC2CCCC3(C2)OOC2(O3)C3CC4CC(C3)CC2C4)C(=O)N[C@@H]2[C@@H](CO)O[C@H]([C@@H]2O)N2C=NC3=C2N=CN=C3N(C)C)C=C1 PCBZRNYXXCIELG-WYFCWLEVSA-N 0.000 description 5
- 239000004215 Carbon black (E152) Substances 0.000 description 5
- 208000006545 Chronic Obstructive Pulmonary Disease Diseases 0.000 description 5
- PCLIMKBDDGJMGD-UHFFFAOYSA-N N-bromosuccinimide Chemical compound BrN1C(=O)CCC1=O PCLIMKBDDGJMGD-UHFFFAOYSA-N 0.000 description 5
- 206010028980 Neoplasm Diseases 0.000 description 5
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 5
- 239000007822 coupling agent Substances 0.000 description 5
- 125000006263 dimethyl aminosulfonyl group Chemical group [H]C([H])([H])N(C([H])([H])[H])S(*)(=O)=O 0.000 description 5
- 238000001035 drying Methods 0.000 description 5
- 125000001301 ethoxy group Chemical group [H]C([H])([H])C([H])([H])O* 0.000 description 5
- 125000003754 ethoxycarbonyl group Chemical group C(=O)(OCC)* 0.000 description 5
- 229930195733 hydrocarbon Natural products 0.000 description 5
- 239000005457 ice water Substances 0.000 description 5
- 125000003387 indolinyl group Chemical group N1(CCC2=CC=CC=C12)* 0.000 description 5
- 125000000842 isoxazolyl group Chemical group 0.000 description 5
- 239000007788 liquid Substances 0.000 description 5
- 239000002480 mineral oil Substances 0.000 description 5
- 235000010446 mineral oil Nutrition 0.000 description 5
- 125000001971 neopentyl group Chemical group [H]C([*])([H])C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H] 0.000 description 5
- 125000002971 oxazolyl group Chemical group 0.000 description 5
- 125000004344 phenylpropyl group Chemical group 0.000 description 5
- 102000004169 proteins and genes Human genes 0.000 description 5
- 108090000623 proteins and genes Proteins 0.000 description 5
- 125000000714 pyrimidinyl group Chemical group 0.000 description 5
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 5
- 239000011734 sodium Substances 0.000 description 5
- 239000012312 sodium hydride Substances 0.000 description 5
- 229910052723 transition metal Inorganic materials 0.000 description 5
- 150000003624 transition metals Chemical class 0.000 description 5
- 238000004704 ultra performance liquid chromatography Methods 0.000 description 5
- CYPYTURSJDMMMP-WVCUSYJESA-N (1e,4e)-1,5-diphenylpenta-1,4-dien-3-one;palladium Chemical compound [Pd].[Pd].C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1.C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1.C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1 CYPYTURSJDMMMP-WVCUSYJESA-N 0.000 description 4
- JREJQAWGQCMSIY-UHFFFAOYSA-N 1-methyl-pyrazole-5-carboxylic acid Chemical compound CN1N=CC=C1C(O)=O JREJQAWGQCMSIY-UHFFFAOYSA-N 0.000 description 4
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 4
- OKKJLVBELUTLKV-MZCSYVLQSA-N Deuterated methanol Chemical compound [2H]OC([2H])([2H])[2H] OKKJLVBELUTLKV-MZCSYVLQSA-N 0.000 description 4
- 206010059176 Juvenile idiopathic arthritis Diseases 0.000 description 4
- 208000009319 Keratoconjunctivitis Sicca Diseases 0.000 description 4
- 238000000023 Kugelrohr distillation Methods 0.000 description 4
- 208000002193 Pain Diseases 0.000 description 4
- 208000021386 Sjogren Syndrome Diseases 0.000 description 4
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical group [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 4
- 125000004429 atom Chemical group 0.000 description 4
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical group [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 4
- 125000002393 azetidinyl group Chemical group 0.000 description 4
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 4
- 229910052794 bromium Inorganic materials 0.000 description 4
- 125000004803 chlorobenzyl group Chemical group 0.000 description 4
- 239000010779 crude oil Substances 0.000 description 4
- 238000001514 detection method Methods 0.000 description 4
- 125000002147 dimethylamino group Chemical group [H]C([H])([H])N(*)C([H])([H])[H] 0.000 description 4
- REWLCYPYZCHYSS-UHFFFAOYSA-N ditert-butyl-[3,6-dimethoxy-2-[2,4,6-tri(propan-2-yl)phenyl]phenyl]phosphane Chemical compound COC1=CC=C(OC)C(C=2C(=CC(=CC=2C(C)C)C(C)C)C(C)C)=C1P(C(C)(C)C)C(C)(C)C REWLCYPYZCHYSS-UHFFFAOYSA-N 0.000 description 4
- 230000002500 effect on skin Effects 0.000 description 4
- 210000002950 fibroblast Anatomy 0.000 description 4
- 239000012065 filter cake Substances 0.000 description 4
- 239000011521 glass Substances 0.000 description 4
- 125000005842 heteroatom Chemical group 0.000 description 4
- PZOUSPYUWWUPPK-UHFFFAOYSA-N indole Natural products CC1=CC=CC2=C1C=CN2 PZOUSPYUWWUPPK-UHFFFAOYSA-N 0.000 description 4
- RKJUIXBNRJVNHR-UHFFFAOYSA-N indolenine Natural products C1=CC=C2CC=NC2=C1 RKJUIXBNRJVNHR-UHFFFAOYSA-N 0.000 description 4
- 238000002347 injection Methods 0.000 description 4
- 239000007924 injection Substances 0.000 description 4
- 229910052500 inorganic mineral Inorganic materials 0.000 description 4
- 230000001404 mediated effect Effects 0.000 description 4
- 239000002609 medium Substances 0.000 description 4
- 125000006261 methyl amino sulfonyl group Chemical group [H]N(C([H])([H])[H])S(*)(=O)=O 0.000 description 4
- 125000004458 methylaminocarbonyl group Chemical group [H]N(C(*)=O)C([H])([H])[H] 0.000 description 4
- 235000010755 mineral Nutrition 0.000 description 4
- 239000011707 mineral Substances 0.000 description 4
- XBXCNNQPRYLIDE-UHFFFAOYSA-M n-tert-butylcarbamate Chemical compound CC(C)(C)NC([O-])=O XBXCNNQPRYLIDE-UHFFFAOYSA-M 0.000 description 4
- 150000007524 organic acids Chemical class 0.000 description 4
- 125000003566 oxetanyl group Chemical group 0.000 description 4
- 239000001301 oxygen Substances 0.000 description 4
- MUJIDPITZJWBSW-UHFFFAOYSA-N palladium(2+) Chemical compound [Pd+2] MUJIDPITZJWBSW-UHFFFAOYSA-N 0.000 description 4
- 235000011181 potassium carbonates Nutrition 0.000 description 4
- 238000011321 prophylaxis Methods 0.000 description 4
- 125000003226 pyrazolyl group Chemical group 0.000 description 4
- 125000000168 pyrrolyl group Chemical group 0.000 description 4
- 230000002829 reductive effect Effects 0.000 description 4
- 238000007127 saponification reaction Methods 0.000 description 4
- 238000000926 separation method Methods 0.000 description 4
- 239000011593 sulfur Chemical group 0.000 description 4
- 125000000335 thiazolyl group Chemical group 0.000 description 4
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 4
- LWIHDJKSTIGBAC-UHFFFAOYSA-K tripotassium phosphate Chemical compound [K+].[K+].[K+].[O-]P([O-])([O-])=O LWIHDJKSTIGBAC-UHFFFAOYSA-K 0.000 description 4
- 239000003981 vehicle Substances 0.000 description 4
- 239000011701 zinc Substances 0.000 description 4
- FOZVXADQAHVUSV-UHFFFAOYSA-N 1-bromo-2-(2-bromoethoxy)ethane Chemical compound BrCCOCCBr FOZVXADQAHVUSV-UHFFFAOYSA-N 0.000 description 3
- XWYDQETVMJZUOJ-UHFFFAOYSA-N 1-iodo-2-(2-iodoethoxy)ethane Chemical compound ICCOCCI XWYDQETVMJZUOJ-UHFFFAOYSA-N 0.000 description 3
- PAQZWJGSJMLPMG-UHFFFAOYSA-N 2,4,6-tripropyl-1,3,5,2$l^{5},4$l^{5},6$l^{5}-trioxatriphosphinane 2,4,6-trioxide Chemical compound CCCP1(=O)OP(=O)(CCC)OP(=O)(CCC)O1 PAQZWJGSJMLPMG-UHFFFAOYSA-N 0.000 description 3
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical group [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 3
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 3
- 206010002556 Ankylosing Spondylitis Diseases 0.000 description 3
- KXDHJXZQYSOELW-UHFFFAOYSA-M Carbamate Chemical compound NC([O-])=O KXDHJXZQYSOELW-UHFFFAOYSA-M 0.000 description 3
- XEEYBQQBJWHFJM-UHFFFAOYSA-N Iron Chemical compound [Fe] XEEYBQQBJWHFJM-UHFFFAOYSA-N 0.000 description 3
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 3
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 3
- 201000004681 Psoriasis Diseases 0.000 description 3
- 201000001263 Psoriatic Arthritis Diseases 0.000 description 3
- 208000036824 Psoriatic arthropathy Diseases 0.000 description 3
- 108700012920 TNF Proteins 0.000 description 3
- 125000003342 alkenyl group Chemical group 0.000 description 3
- 239000007900 aqueous suspension Substances 0.000 description 3
- 229910052786 argon Inorganic materials 0.000 description 3
- 125000003785 benzimidazolyl group Chemical group N1=C(NC2=C1C=CC=C2)* 0.000 description 3
- 125000001164 benzothiazolyl group Chemical group S1C(=NC2=C1C=CC=C2)* 0.000 description 3
- 125000004541 benzoxazolyl group Chemical group O1C(=NC2=C1C=CC=C2)* 0.000 description 3
- 239000012455 biphasic mixture Substances 0.000 description 3
- 125000001246 bromo group Chemical group Br* 0.000 description 3
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 3
- 201000011510 cancer Diseases 0.000 description 3
- 229910002091 carbon monoxide Inorganic materials 0.000 description 3
- 238000009903 catalytic hydrogenation reaction Methods 0.000 description 3
- 125000000068 chlorophenyl group Chemical group 0.000 description 3
- 125000000259 cinnolinyl group Chemical group N1=NC(=CC2=CC=CC=C12)* 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- 239000013078 crystal Substances 0.000 description 3
- 125000004850 cyclobutylmethyl group Chemical group C1(CCC1)C* 0.000 description 3
- 125000000723 dihydrobenzofuranyl group Chemical group O1C(CC2=C1C=CC=C2)* 0.000 description 3
- 201000010099 disease Diseases 0.000 description 3
- 239000006185 dispersion Substances 0.000 description 3
- 239000012530 fluid Substances 0.000 description 3
- 125000004175 fluorobenzyl group Chemical group 0.000 description 3
- 238000009472 formulation Methods 0.000 description 3
- 239000007789 gas Substances 0.000 description 3
- 150000002391 heterocyclic compounds Chemical class 0.000 description 3
- COQRGFWWJBEXRC-UHFFFAOYSA-N hydron;methyl 2-aminoacetate;chloride Chemical compound Cl.COC(=O)CN COQRGFWWJBEXRC-UHFFFAOYSA-N 0.000 description 3
- 125000002883 imidazolyl group Chemical group 0.000 description 3
- QNLOWBMKUIXCOW-UHFFFAOYSA-N indol-2-one Chemical compound C1=CC=CC2=NC(=O)C=C21 QNLOWBMKUIXCOW-UHFFFAOYSA-N 0.000 description 3
- 150000007529 inorganic bases Chemical class 0.000 description 3
- 125000003253 isopropoxy group Chemical group [H]C([H])([H])C([H])(O*)C([H])([H])[H] 0.000 description 3
- 125000001786 isothiazolyl group Chemical group 0.000 description 3
- 229910052744 lithium Inorganic materials 0.000 description 3
- 239000012280 lithium aluminium hydride Substances 0.000 description 3
- 125000000250 methylamino group Chemical group [H]N(*)C([H])([H])[H] 0.000 description 3
- 125000006216 methylsulfinyl group Chemical group [H]C([H])([H])S(*)=O 0.000 description 3
- 125000004923 naphthylmethyl group Chemical group C1(=CC=CC2=CC=CC=C12)C* 0.000 description 3
- 125000001715 oxadiazolyl group Chemical group 0.000 description 3
- 230000001575 pathological effect Effects 0.000 description 3
- 238000002953 preparative HPLC Methods 0.000 description 3
- 239000003755 preservative agent Substances 0.000 description 3
- 230000000770 proinflammatory effect Effects 0.000 description 3
- 125000000561 purinyl group Chemical group N1=C(N=C2N=CNC2=C1)* 0.000 description 3
- 125000003373 pyrazinyl group Chemical group 0.000 description 3
- JQRYUMGHOUYJFW-UHFFFAOYSA-N pyridine;trihydrobromide Chemical compound [Br-].[Br-].[Br-].C1=CC=[NH+]C=C1.C1=CC=[NH+]C=C1.C1=CC=[NH+]C=C1 JQRYUMGHOUYJFW-UHFFFAOYSA-N 0.000 description 3
- 239000000741 silica gel Substances 0.000 description 3
- 229910002027 silica gel Inorganic materials 0.000 description 3
- 239000002002 slurry Substances 0.000 description 3
- 239000003826 tablet Substances 0.000 description 3
- 125000005931 tert-butyloxycarbonyl group Chemical group [H]C([H])([H])C(OC(*)=O)(C([H])([H])[H])C([H])([H])[H] 0.000 description 3
- 238000012360 testing method Methods 0.000 description 3
- 125000003831 tetrazolyl group Chemical group 0.000 description 3
- 125000001113 thiadiazolyl group Chemical group 0.000 description 3
- 125000004568 thiomorpholinyl group Chemical group 0.000 description 3
- 238000002054 transplantation Methods 0.000 description 3
- 125000005270 trialkylamine group Chemical group 0.000 description 3
- 125000001425 triazolyl group Chemical group 0.000 description 3
- 125000004191 (C1-C6) alkoxy group Chemical group 0.000 description 2
- FSMHDKRQCDWWGZ-UHFFFAOYSA-N 2-ethylpyrazole-3-carboxylic acid Chemical compound CCN1N=CC=C1C(O)=O FSMHDKRQCDWWGZ-UHFFFAOYSA-N 0.000 description 2
- WKJRYVOTVRPAFN-UHFFFAOYSA-N 2-pyridin-1-ium-4-ylacetic acid;chloride Chemical compound Cl.OC(=O)CC1=CC=NC=C1 WKJRYVOTVRPAFN-UHFFFAOYSA-N 0.000 description 2
- NHQDETIJWKXCTC-UHFFFAOYSA-N 3-chloroperbenzoic acid Chemical compound OOC(=O)C1=CC=CC(Cl)=C1 NHQDETIJWKXCTC-UHFFFAOYSA-N 0.000 description 2
- MCSXGCZMEPXKIW-UHFFFAOYSA-N 3-hydroxy-4-[(4-methyl-2-nitrophenyl)diazenyl]-N-(3-nitrophenyl)naphthalene-2-carboxamide Chemical compound Cc1ccc(N=Nc2c(O)c(cc3ccccc23)C(=O)Nc2cccc(c2)[N+]([O-])=O)c(c1)[N+]([O-])=O MCSXGCZMEPXKIW-UHFFFAOYSA-N 0.000 description 2
- YBLSBWHFPXDRHC-UHFFFAOYSA-N 3-methyl-1,2-oxazole-4-carboxylic acid Chemical compound CC1=NOC=C1C(O)=O YBLSBWHFPXDRHC-UHFFFAOYSA-N 0.000 description 2
- VGVHNLRUAMRIEW-UHFFFAOYSA-N 4-methylcyclohexan-1-one Chemical compound CC1CCC(=O)CC1 VGVHNLRUAMRIEW-UHFFFAOYSA-N 0.000 description 2
- QTDXSEZXAPHVBI-UHFFFAOYSA-N 4-methylcyclohexane-1-carboxylic acid Chemical compound CC1CCC(C(O)=O)CC1 QTDXSEZXAPHVBI-UHFFFAOYSA-N 0.000 description 2
- JARRYVQFBQVOBE-UHFFFAOYSA-N 6-bromo-1,3-dihydroindol-2-one Chemical compound BrC1=CC=C2CC(=O)NC2=C1 JARRYVQFBQVOBE-UHFFFAOYSA-N 0.000 description 2
- WGDOZEMHDIATQE-UHFFFAOYSA-N 6-bromospiro[1h-indole-3,4'-oxane]-2-one Chemical compound C=1C(Br)=CC=C2C=1NC(=O)C21CCOCC1 WGDOZEMHDIATQE-UHFFFAOYSA-N 0.000 description 2
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 2
- 101100096578 Arabidopsis thaliana SQD2 gene Proteins 0.000 description 2
- 206010009900 Colitis ulcerative Diseases 0.000 description 2
- 241001480079 Corymbia calophylla Species 0.000 description 2
- 208000011231 Crohn disease Diseases 0.000 description 2
- 206010012438 Dermatitis atopic Diseases 0.000 description 2
- 206010061218 Inflammation Diseases 0.000 description 2
- 208000022559 Inflammatory bowel disease Diseases 0.000 description 2
- 208000003456 Juvenile Arthritis Diseases 0.000 description 2
- 235000006552 Liquidambar styraciflua Nutrition 0.000 description 2
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 2
- MZRVEZGGRBJDDB-UHFFFAOYSA-N N-Butyllithium Chemical compound [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 description 2
- OFBQJSOFQDEBGM-UHFFFAOYSA-N Pentane Chemical compound CCCCC OFBQJSOFQDEBGM-UHFFFAOYSA-N 0.000 description 2
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 2
- 206010039710 Scleroderma Diseases 0.000 description 2
- 208000006045 Spondylarthropathies Diseases 0.000 description 2
- 201000009594 Systemic Scleroderma Diseases 0.000 description 2
- 206010042953 Systemic sclerosis Diseases 0.000 description 2
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 2
- 201000006704 Ulcerative Colitis Diseases 0.000 description 2
- WETWJCDKMRHUPV-UHFFFAOYSA-N acetyl chloride Chemical compound CC(Cl)=O WETWJCDKMRHUPV-UHFFFAOYSA-N 0.000 description 2
- 239000012346 acetyl chloride Substances 0.000 description 2
- 150000008044 alkali metal hydroxides Chemical class 0.000 description 2
- 125000004390 alkyl sulfonyl group Chemical group 0.000 description 2
- 150000001408 amides Chemical class 0.000 description 2
- 206010003246 arthritis Diseases 0.000 description 2
- 208000006673 asthma Diseases 0.000 description 2
- 201000008937 atopic dermatitis Diseases 0.000 description 2
- 230000001363 autoimmune Effects 0.000 description 2
- 230000037396 body weight Effects 0.000 description 2
- 125000006278 bromobenzyl group Chemical group 0.000 description 2
- 238000004296 chiral HPLC Methods 0.000 description 2
- WGXZDYPGLJYBJW-UHFFFAOYSA-N chloroform;propan-2-ol Chemical compound CC(C)O.ClC(Cl)Cl WGXZDYPGLJYBJW-UHFFFAOYSA-N 0.000 description 2
- 238000007796 conventional method Methods 0.000 description 2
- 239000011258 core-shell material Substances 0.000 description 2
- 238000002425 crystallisation Methods 0.000 description 2
- 125000002946 cyanobenzyl group Chemical group 0.000 description 2
- 125000006286 dichlorobenzyl group Chemical group 0.000 description 2
- 125000004177 diethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 2
- 229910001873 dinitrogen Inorganic materials 0.000 description 2
- DEQYTNZJHKPYEZ-UHFFFAOYSA-N ethyl acetate;heptane Chemical compound CCOC(C)=O.CCCCCCC DEQYTNZJHKPYEZ-UHFFFAOYSA-N 0.000 description 2
- PQJJJMRNHATNKG-UHFFFAOYSA-N ethyl bromoacetate Chemical compound CCOC(=O)CBr PQJJJMRNHATNKG-UHFFFAOYSA-N 0.000 description 2
- 230000005284 excitation Effects 0.000 description 2
- 230000007717 exclusion Effects 0.000 description 2
- 238000004108 freeze drying Methods 0.000 description 2
- 238000010438 heat treatment Methods 0.000 description 2
- 238000005984 hydrogenation reaction Methods 0.000 description 2
- NPZTUJOABDZTLV-UHFFFAOYSA-N hydroxybenzotriazole Substances O=C1C=CC=C2NNN=C12 NPZTUJOABDZTLV-UHFFFAOYSA-N 0.000 description 2
- 125000006289 hydroxybenzyl group Chemical group 0.000 description 2
- 125000002632 imidazolidinyl group Chemical group 0.000 description 2
- 125000001841 imino group Chemical group [H]N=* 0.000 description 2
- 125000001041 indolyl group Chemical group 0.000 description 2
- 125000006301 indolyl methyl group Chemical group 0.000 description 2
- 208000015181 infectious disease Diseases 0.000 description 2
- 230000004054 inflammatory process Effects 0.000 description 2
- 229910001853 inorganic hydroxide Inorganic materials 0.000 description 2
- UEXQBEVWFZKHNB-UHFFFAOYSA-N intermediate 29 Natural products C1=CC(N)=CC=C1NC1=NC=CC=N1 UEXQBEVWFZKHNB-UHFFFAOYSA-N 0.000 description 2
- 229910052740 iodine Inorganic materials 0.000 description 2
- 239000011630 iodine Substances 0.000 description 2
- INQOMBQAUSQDDS-UHFFFAOYSA-N iodomethane Chemical compound IC INQOMBQAUSQDDS-UHFFFAOYSA-N 0.000 description 2
- 125000004628 isothiazolidinyl group Chemical group S1N(CCC1)* 0.000 description 2
- 201000002215 juvenile rheumatoid arthritis Diseases 0.000 description 2
- KWGKDLIKAYFUFQ-UHFFFAOYSA-M lithium chloride Chemical compound [Li+].[Cl-] KWGKDLIKAYFUFQ-UHFFFAOYSA-M 0.000 description 2
- 206010025135 lupus erythematosus Diseases 0.000 description 2
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 2
- 125000001160 methoxycarbonyl group Chemical group [H]C([H])([H])OC(*)=O 0.000 description 2
- 125000004184 methoxymethyl group Chemical group [H]C([H])([H])OC([H])([H])* 0.000 description 2
- 125000006178 methyl benzyl group Chemical group 0.000 description 2
- 238000002156 mixing Methods 0.000 description 2
- 125000002950 monocyclic group Chemical group 0.000 description 2
- 239000002674 ointment Substances 0.000 description 2
- 239000003960 organic solvent Substances 0.000 description 2
- 125000005961 oxazepanyl group Chemical group 0.000 description 2
- 125000000160 oxazolidinyl group Chemical group 0.000 description 2
- YJVFFLUZDVXJQI-UHFFFAOYSA-L palladium(ii) acetate Chemical compound [Pd+2].CC([O-])=O.CC([O-])=O YJVFFLUZDVXJQI-UHFFFAOYSA-L 0.000 description 2
- 238000007911 parenteral administration Methods 0.000 description 2
- 235000019271 petrolatum Nutrition 0.000 description 2
- 239000000546 pharmaceutical excipient Substances 0.000 description 2
- 125000000951 phenoxy group Chemical group [H]C1=C([H])C([H])=C(O*)C([H])=C1[H] 0.000 description 2
- 125000006187 phenyl benzyl group Chemical group 0.000 description 2
- 229910052700 potassium Inorganic materials 0.000 description 2
- 239000011591 potassium Substances 0.000 description 2
- 235000015320 potassium carbonate Nutrition 0.000 description 2
- 239000002244 precipitate Substances 0.000 description 2
- 230000002335 preservative effect Effects 0.000 description 2
- BDERNNFJNOPAEC-UHFFFAOYSA-N propan-1-ol Chemical compound CCCO BDERNNFJNOPAEC-UHFFFAOYSA-N 0.000 description 2
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 2
- 208000005069 pulmonary fibrosis Diseases 0.000 description 2
- 102000005962 receptors Human genes 0.000 description 2
- 108020003175 receptors Proteins 0.000 description 2
- 206010039073 rheumatoid arthritis Diseases 0.000 description 2
- 239000000377 silicon dioxide Substances 0.000 description 2
- MFRIHAYPQRLWNB-UHFFFAOYSA-N sodium tert-butoxide Chemical compound [Na+].CC(C)(C)[O-] MFRIHAYPQRLWNB-UHFFFAOYSA-N 0.000 description 2
- 201000005671 spondyloarthropathy Diseases 0.000 description 2
- 150000003462 sulfoxides Chemical class 0.000 description 2
- 238000001356 surgical procedure Methods 0.000 description 2
- 208000011580 syndromic disease Diseases 0.000 description 2
- 208000020408 systemic-onset juvenile idiopathic arthritis Diseases 0.000 description 2
- FPGGTKZVZWFYPV-UHFFFAOYSA-M tetrabutylammonium fluoride Chemical compound [F-].CCCC[N+](CCCC)(CCCC)CCCC FPGGTKZVZWFYPV-UHFFFAOYSA-M 0.000 description 2
- 125000006173 tetrahydropyranylmethyl group Chemical group 0.000 description 2
- 125000001984 thiazolidinyl group Chemical group 0.000 description 2
- 125000001544 thienyl group Chemical group 0.000 description 2
- 125000005301 thienylmethyl group Chemical group [H]C1=C([H])C([H])=C(S1)C([H])([H])* 0.000 description 2
- 210000001519 tissue Anatomy 0.000 description 2
- JMXKSZRRTHPKDL-UHFFFAOYSA-N titanium ethoxide Chemical compound [Ti+4].CC[O-].CC[O-].CC[O-].CC[O-] JMXKSZRRTHPKDL-UHFFFAOYSA-N 0.000 description 2
- 125000006493 trifluoromethyl benzyl group Chemical group 0.000 description 2
- 229910000404 tripotassium phosphate Inorganic materials 0.000 description 2
- 235000019798 tripotassium phosphate Nutrition 0.000 description 2
- 229920002554 vinyl polymer Polymers 0.000 description 2
- XSQBSVRSFKNYOI-JRSKDTKFSA-N (2S)-2-amino-2-(4-methylcyclohexyl)-N-(2-oxospiro[1H-indole-3,4'-oxane]-6-yl)acetamide Chemical compound N[C@H](C(=O)NC1=CC=C2C(=C1)NC(C21CCOCC1)=O)C1CCC(CC1)C XSQBSVRSFKNYOI-JRSKDTKFSA-N 0.000 description 1
- QXZRCEDDTLDTCN-AWEZNQCLSA-N (2S)-3-(2-chlorophenyl)-2-[(2-pyridin-4-ylacetyl)amino]propanoic acid Chemical compound ClC1=C(C=CC=C1)C[C@@H](C(=O)O)NC(CC1=CC=NC=C1)=O QXZRCEDDTLDTCN-AWEZNQCLSA-N 0.000 description 1
- FMWGYNCAGXLKEQ-YFKPBYRVSA-N (2s)-1-iodo-2-(2-iodoethoxy)propane Chemical compound IC[C@H](C)OCCI FMWGYNCAGXLKEQ-YFKPBYRVSA-N 0.000 description 1
- IBKKDSPQRUFRBM-YFKPBYRVSA-N (2s)-2-(2-hydroxyethoxy)propan-1-ol Chemical compound OC[C@H](C)OCCO IBKKDSPQRUFRBM-YFKPBYRVSA-N 0.000 description 1
- WAMWSIDTKSNDCU-ZETCQYMHSA-N (2s)-2-azaniumyl-2-cyclohexylacetate Chemical compound OC(=O)[C@@H](N)C1CCCCC1 WAMWSIDTKSNDCU-ZETCQYMHSA-N 0.000 description 1
- MZOFCQQQCNRIBI-VMXHOPILSA-N (3s)-4-[[(2s)-1-[[(2s)-1-[[(1s)-1-carboxy-2-hydroxyethyl]amino]-4-methyl-1-oxopentan-2-yl]amino]-5-(diaminomethylideneamino)-1-oxopentan-2-yl]amino]-3-[[2-[[(2s)-2,6-diaminohexanoyl]amino]acetyl]amino]-4-oxobutanoic acid Chemical compound OC[C@@H](C(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCCN=C(N)N)NC(=O)[C@H](CC(O)=O)NC(=O)CNC(=O)[C@@H](N)CCCCN MZOFCQQQCNRIBI-VMXHOPILSA-N 0.000 description 1
- 125000004178 (C1-C4) alkyl group Chemical group 0.000 description 1
- 125000004454 (C1-C6) alkoxycarbonyl group Chemical group 0.000 description 1
- LBUJPTNKIBCYBY-UHFFFAOYSA-N 1,2,3,4-tetrahydroquinoline Chemical compound C1=CC=C2CCCNC2=C1 LBUJPTNKIBCYBY-UHFFFAOYSA-N 0.000 description 1
- DDMOUSALMHHKOS-UHFFFAOYSA-N 1,2-dichloro-1,1,2,2-tetrafluoroethane Chemical compound FC(F)(Cl)C(F)(F)Cl DDMOUSALMHHKOS-UHFFFAOYSA-N 0.000 description 1
- VYCIHDBIKGRENI-UHFFFAOYSA-N 1,3-bis[2,6-di(propan-2-yl)phenyl]-2h-imidazol-1-ium-2-ide Chemical group CC(C)C1=CC=CC(C(C)C)=C1N1C=CN(C=2C(=CC=CC=2C(C)C)C(C)C)[C]1 VYCIHDBIKGRENI-UHFFFAOYSA-N 0.000 description 1
- DOKIJRPCPVSRQF-UHFFFAOYSA-N 1-(3-bicyclo[1.1.1]pentanyl)ethanone Chemical compound C1C2CC1(C(=O)C)C2 DOKIJRPCPVSRQF-UHFFFAOYSA-N 0.000 description 1
- ASOKPJOREAFHNY-UHFFFAOYSA-N 1-Hydroxybenzotriazole Chemical compound C1=CC=C2N(O)N=NC2=C1 ASOKPJOREAFHNY-UHFFFAOYSA-N 0.000 description 1
- HCUARRIEZVDMPT-UHFFFAOYSA-M 1h-indole-2-carboxylate Chemical compound C1=CC=C2NC(C(=O)[O-])=CC2=C1 HCUARRIEZVDMPT-UHFFFAOYSA-M 0.000 description 1
- YNXOKEVENIBAKN-UHFFFAOYSA-N 2'-chloro-7'-(2-trimethylsilylethoxymethyl)spiro[oxane-4,5'-pyrrolo[2,3-d]pyrimidine]-6'-one Chemical compound ClC=1N=CC2=C(N=1)N(C(C21CCOCC1)=O)COCC[Si](C)(C)C YNXOKEVENIBAKN-UHFFFAOYSA-N 0.000 description 1
- LTMRRSWNXVJMBA-UHFFFAOYSA-L 2,2-diethylpropanedioate Chemical compound CCC(CC)(C([O-])=O)C([O-])=O LTMRRSWNXVJMBA-UHFFFAOYSA-L 0.000 description 1
- GLVGZXHESDAIMP-UHFFFAOYSA-N 2,3-dimethylcyclobutan-1-one Chemical compound CC1CC(=O)C1C GLVGZXHESDAIMP-UHFFFAOYSA-N 0.000 description 1
- GHXBPCSSQOKKGB-UHFFFAOYSA-N 2,4-dichloro-7h-pyrrolo[2,3-d]pyrimidine Chemical compound ClC1=NC(Cl)=C2C=CNC2=N1 GHXBPCSSQOKKGB-UHFFFAOYSA-N 0.000 description 1
- LIHDYIAAKPQZSN-UHFFFAOYSA-N 2,5-dihydroxy-5-methyl-3-(morpholin-4-ylamino)cyclopent-2-en-1-one Chemical compound O=C1C(C)(O)CC(NN2CCOCC2)=C1O LIHDYIAAKPQZSN-UHFFFAOYSA-N 0.000 description 1
- MAPLKZRJKHIYAZ-UHFFFAOYSA-N 2-(2-aminopyridin-1-ium-4-yl)acetate Chemical compound NC1=CC(CC(O)=O)=CC=N1 MAPLKZRJKHIYAZ-UHFFFAOYSA-N 0.000 description 1
- LQTIZFMDLWJRNY-UHFFFAOYSA-N 2-(3,3,3-trifluoropropyl)pyrazole-3-carboxylic acid Chemical compound OC(=O)C1=CC=NN1CCC(F)(F)F LQTIZFMDLWJRNY-UHFFFAOYSA-N 0.000 description 1
- MUOXYNIOJBUOCL-UHFFFAOYSA-N 2-(4-methylcyclohexyl)-2-[(2-methylpropan-2-yl)oxycarbonylamino]acetic acid Chemical compound CC1CCC(C(NC(=O)OC(C)(C)C)C(O)=O)CC1 MUOXYNIOJBUOCL-UHFFFAOYSA-N 0.000 description 1
- CVZZNRXMDCOHBG-QMMMGPOBSA-N 2-Chloro-L-phenylalanine Chemical compound OC(=O)[C@@H](N)CC1=CC=CC=C1Cl CVZZNRXMDCOHBG-QMMMGPOBSA-N 0.000 description 1
- HSSJZWWCSQLAQN-UHFFFAOYSA-N 2-[(2,4-dichloropyrrolo[2,3-d]pyrimidin-7-yl)methoxy]ethyl-trimethylsilane Chemical compound N1=C(Cl)N=C2N(COCC[Si](C)(C)C)C=CC2=C1Cl HSSJZWWCSQLAQN-UHFFFAOYSA-N 0.000 description 1
- VNUOLDPKSYKFKV-UHFFFAOYSA-N 2-[(2-chloropyrrolo[2,3-d]pyrimidin-7-yl)methoxy]ethyl-trimethylsilane Chemical compound N1=C(Cl)N=C2N(COCC[Si](C)(C)C)C=CC2=C1 VNUOLDPKSYKFKV-UHFFFAOYSA-N 0.000 description 1
- DYFGBQLPPJPISC-UHFFFAOYSA-N 2-[(2-ethylpyrazole-3-carbonyl)amino]acetic acid Chemical compound C(C)N1N=CC=C1C(=O)NCC(=O)O DYFGBQLPPJPISC-UHFFFAOYSA-N 0.000 description 1
- XIGMVYDCHUYWPA-UHFFFAOYSA-N 2-[(2-methylpyrazole-3-carbonyl)amino]acetic acid Chemical compound CN1N=CC=C1C(=O)NCC(O)=O XIGMVYDCHUYWPA-UHFFFAOYSA-N 0.000 description 1
- XKBLAFHYRFJFIU-UHFFFAOYSA-N 2-[(3-methyl-1,2-oxazole-4-carbonyl)amino]acetic acid Chemical compound CC1=NOC=C1C(=O)NCC(O)=O XKBLAFHYRFJFIU-UHFFFAOYSA-N 0.000 description 1
- HFUKVDSHNKXALI-UHFFFAOYSA-N 2-[(6-chloropyrrolo[2,3-b]pyridin-1-yl)methoxy]ethyl-trimethylsilane Chemical compound ClC1=CC=C2C(=N1)N(C=C2)COCC[Si](C)(C)C HFUKVDSHNKXALI-UHFFFAOYSA-N 0.000 description 1
- NLEZSQHAFMZAGU-UHFFFAOYSA-N 2-bromo-5-chloroaniline Chemical compound NC1=CC(Cl)=CC=C1Br NLEZSQHAFMZAGU-UHFFFAOYSA-N 0.000 description 1
- PJDLGUHPWFCBRD-UHFFFAOYSA-N 2-cyclooctyl-2-[(2-methylpyrazole-3-carbonyl)amino]acetic acid Chemical compound C1(CCCCCCC1)C(C(=O)O)NC(=O)C=1N(N=CC=1)C PJDLGUHPWFCBRD-UHFFFAOYSA-N 0.000 description 1
- SDTMFDGELKWGFT-UHFFFAOYSA-N 2-methylpropan-2-olate Chemical compound CC(C)(C)[O-] SDTMFDGELKWGFT-UHFFFAOYSA-N 0.000 description 1
- JWUJQDFVADABEY-UHFFFAOYSA-N 2-methyltetrahydrofuran Chemical compound CC1CCCO1 JWUJQDFVADABEY-UHFFFAOYSA-N 0.000 description 1
- LEACJMVNYZDSKR-UHFFFAOYSA-N 2-octyldodecan-1-ol Chemical compound CCCCCCCCCCC(CO)CCCCCCCC LEACJMVNYZDSKR-UHFFFAOYSA-N 0.000 description 1
- TWBPWBPGNQWFSJ-UHFFFAOYSA-N 2-phenylaniline Chemical group NC1=CC=CC=C1C1=CC=CC=C1 TWBPWBPGNQWFSJ-UHFFFAOYSA-N 0.000 description 1
- ZIHRJZBCLMMKTC-UHFFFAOYSA-N 2-propan-2-ylpyrazole-3-carboxylic acid Chemical compound CC(C)N1N=CC=C1C(O)=O ZIHRJZBCLMMKTC-UHFFFAOYSA-N 0.000 description 1
- 125000003903 2-propenyl group Chemical group [H]C([*])([H])C([H])=C([H])[H] 0.000 description 1
- CDSCASXRVKUXCN-UHFFFAOYSA-N 3,3-dibromo-1h-indol-2-one Chemical class C1=CC=C2C(Br)(Br)C(=O)NC2=C1 CDSCASXRVKUXCN-UHFFFAOYSA-N 0.000 description 1
- PUAFZMBKKUSJFF-UHFFFAOYSA-N 3,3-dibromo-6-chloro-1-(2-trimethylsilylethoxymethyl)pyrrolo[2,3-b]pyridin-2-one Chemical compound BrC1(C(N(C2=NC(=CC=C21)Cl)COCC[Si](C)(C)C)=O)Br PUAFZMBKKUSJFF-UHFFFAOYSA-N 0.000 description 1
- YXWAHLKOWQLVBJ-UHFFFAOYSA-N 3-chloro-6-(difluoromethyl)pyridazine Chemical compound FC(F)C1=CC=C(Cl)N=N1 YXWAHLKOWQLVBJ-UHFFFAOYSA-N 0.000 description 1
- OKFIXAGANPHTLB-UHFFFAOYSA-N 3-cyclobutyl-1,2-oxazole-4-carboxylic acid Chemical compound OC(=O)C1=CON=C1C1CCC1 OKFIXAGANPHTLB-UHFFFAOYSA-N 0.000 description 1
- NJEPDWZYBJIKMD-UHFFFAOYSA-N 3-cyclopropyl-1,2-oxazole-4-carboxylic acid Chemical compound OC(=O)C1=CON=C1C1CC1 NJEPDWZYBJIKMD-UHFFFAOYSA-N 0.000 description 1
- GWXJFENAVVNMTA-UHFFFAOYSA-N 3-ethyl-1,2-oxazole-4-carboxylic acid Chemical compound CCC1=NOC=C1C(O)=O GWXJFENAVVNMTA-UHFFFAOYSA-N 0.000 description 1
- OJHJUZWZIKNBPB-UHFFFAOYSA-N 4-(1-adamantylmethylidene)-2-(2-methylpyrazol-3-yl)-1,3-oxazol-5-one Chemical compound C12(CC3CC(CC(C1)C3)C2)C=C1N=C(OC1=O)C=1N(N=CC=1)C OJHJUZWZIKNBPB-UHFFFAOYSA-N 0.000 description 1
- NCTMQFGXGALEQA-UHFFFAOYSA-N 4-(2,3-dimethylcyclobutylidene)-2-(2-methylpyrazol-3-yl)-1,3-oxazol-5-one Chemical compound CC1CC(C1C)=C1N=C(OC1=O)C1=CC=NN1C NCTMQFGXGALEQA-UHFFFAOYSA-N 0.000 description 1
- IRZONAKJQNFKND-UHFFFAOYSA-N 4-(3-bicyclo[3.2.1]octanylidene)-2-(2-methylpyrazol-3-yl)-1,3-oxazol-5-one Chemical compound C12CC(CC(CC1)C2)=C1N=C(OC1=O)C=1N(N=CC=1)C IRZONAKJQNFKND-UHFFFAOYSA-N 0.000 description 1
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 1
- YPMWXKBOXZHOLW-UHFFFAOYSA-N 4-[1-(1-bicyclo[1.1.1]pentanyl)ethylidene]-2-(2-methylpyrazol-3-yl)-1,3-oxazol-5-one Chemical compound C12(CC(C1)C2)C(C)=C1N=C(OC1=O)C=1N(N=CC=1)C YPMWXKBOXZHOLW-UHFFFAOYSA-N 0.000 description 1
- YNJDSRPIGAUCEE-JTQLQIEISA-N 4-methylbenzenesulfinamide Chemical compound CC1=CC=C([S@@](N)=O)C=C1 YNJDSRPIGAUCEE-JTQLQIEISA-N 0.000 description 1
- WWQQPSDIIVXFOX-UHFFFAOYSA-N 5-bromo-2-chloro-3-nitropyridine Chemical compound [O-][N+](=O)C1=CC(Br)=CN=C1Cl WWQQPSDIIVXFOX-UHFFFAOYSA-N 0.000 description 1
- MRSHZFYWLHRVTR-UHFFFAOYSA-N 5-chlorobicyclo[4.2.0]octa-1(6),2,4-trien-7-one Chemical compound ClC1=CC=CC2=C1C(=O)C2 MRSHZFYWLHRVTR-UHFFFAOYSA-N 0.000 description 1
- LIVZPRQUTUGNSW-UHFFFAOYSA-N 5-chlorobicyclo[4.2.0]octa-1(6),2,4-triene Chemical compound ClC1=CC=CC2=C1CC2 LIVZPRQUTUGNSW-UHFFFAOYSA-N 0.000 description 1
- RBEPUKMEVUHMAR-UHFFFAOYSA-N 5-methoxybicyclo[4.2.0]octa-1(6),2,4-trien-7-one Chemical compound COC1=CC=CC2=C1C(=O)C2 RBEPUKMEVUHMAR-UHFFFAOYSA-N 0.000 description 1
- OCOCBVKMMMIDLI-UHFFFAOYSA-N 6-amino-1,3-dihydroindol-2-one Chemical compound NC1=CC=C2CC(=O)NC2=C1 OCOCBVKMMMIDLI-UHFFFAOYSA-N 0.000 description 1
- DYAAFUZBRLRTLU-UHFFFAOYSA-N 6-amino-5-fluorospiro[1H-indole-3,4'-oxane]-2-one Chemical compound NC1=C(C=C2C(=C1)NC(C21CCOCC1)=O)F DYAAFUZBRLRTLU-UHFFFAOYSA-N 0.000 description 1
- FYNMKZWPUIKXBT-UHFFFAOYSA-N 6-aminospiro[1H-indole-3,4'-oxane]-2-one Chemical compound NC1=CC=C2C(=C1)NC(C21CCOCC1)=O FYNMKZWPUIKXBT-UHFFFAOYSA-N 0.000 description 1
- CAMLXMKLAOCRGP-UHFFFAOYSA-N 6-aminospiro[1H-indole-3,4'-thiane]-2-one Chemical compound NC1=CC=C2C(=C1)NC(C21CCSCC1)=O CAMLXMKLAOCRGP-UHFFFAOYSA-N 0.000 description 1
- UNVVSMMNEZBZDQ-UHFFFAOYSA-N 6-bromo-4-fluoro-1,3-dihydroindol-2-one Chemical compound FC1=CC(Br)=CC2=C1CC(=O)N2 UNVVSMMNEZBZDQ-UHFFFAOYSA-N 0.000 description 1
- DZBCOTMGAYKEGD-UHFFFAOYSA-N 6-bromospiro[1h-pyrrolo[3,2-b]pyridine-3,4'-oxane]-2-one Chemical compound C=1C(Br)=CN=C2C=1NC(=O)C21CCOCC1 DZBCOTMGAYKEGD-UHFFFAOYSA-N 0.000 description 1
- QSUDHVNTTGSJBO-UHFFFAOYSA-N 6-chloro-1-(2-trimethylsilylethoxymethyl)-3H-pyrrolo[2,3-b]pyridin-2-one Chemical compound ClC1=CC=C2C(=N1)N(C(C2)=O)COCC[Si](C)(C)C QSUDHVNTTGSJBO-UHFFFAOYSA-N 0.000 description 1
- OEXHVQKURTYKTO-UHFFFAOYSA-N 6-chloro-1-(2-trimethylsilylethoxymethyl)spiro[indole-3,4'-thiane]-2-one Chemical compound C[Si](C)(C)CCOCN1C2=C(C=CC(=C2)Cl)C3(C1=O)CCSCC3 OEXHVQKURTYKTO-UHFFFAOYSA-N 0.000 description 1
- CBHXTZKXDLDMJZ-UHFFFAOYSA-N 6-chloro-1h-pyrrolo[3,2-c]pyridine Chemical compound C1=NC(Cl)=CC2=C1C=CN2 CBHXTZKXDLDMJZ-UHFFFAOYSA-N 0.000 description 1
- CJYNVZARKDRURZ-UHFFFAOYSA-N 7-nitro-2,3,4,9-tetrahydro-1h-pyrido[3,4-b]indole Chemical compound C1CNCC2=C1C1=CC=C([N+](=O)[O-])C=C1N2 CJYNVZARKDRURZ-UHFFFAOYSA-N 0.000 description 1
- RZVAJINKPMORJF-UHFFFAOYSA-N Acetaminophen Chemical compound CC(=O)NC1=CC=C(O)C=C1 RZVAJINKPMORJF-UHFFFAOYSA-N 0.000 description 1
- 208000031261 Acute myeloid leukaemia Diseases 0.000 description 1
- 239000012114 Alexa Fluor 647 Substances 0.000 description 1
- 208000024827 Alzheimer disease Diseases 0.000 description 1
- 201000001320 Atherosclerosis Diseases 0.000 description 1
- 206010003645 Atopy Diseases 0.000 description 1
- 208000010839 B-cell chronic lymphocytic leukemia Diseases 0.000 description 1
- 208000032791 BCR-ABL1 positive chronic myelogenous leukemia Diseases 0.000 description 1
- 102000001805 Bromodomains Human genes 0.000 description 1
- 108050009021 Bromodomains Proteins 0.000 description 1
- AQZRCFLGIFZLQS-UHFFFAOYSA-N C(C)N1N=CC=C1C=1OC(CN=1)=O Chemical compound C(C)N1N=CC=C1C=1OC(CN=1)=O AQZRCFLGIFZLQS-UHFFFAOYSA-N 0.000 description 1
- FAZLDATVPDAOBZ-UHFFFAOYSA-N C(C1=CC=CC=C1)N1CCC2(CC1)C(NC1=CC(=CC=C12)Br)=O Chemical compound C(C1=CC=CC=C1)N1CCC2(CC1)C(NC1=CC(=CC=C12)Br)=O FAZLDATVPDAOBZ-UHFFFAOYSA-N 0.000 description 1
- GLSJODTZIJKYAH-UHFFFAOYSA-N C1(CCCCCC1)C1N=C(OC1=O)C=1N(N=CC=1)C Chemical compound C1(CCCCCC1)C1N=C(OC1=O)C=1N(N=CC=1)C GLSJODTZIJKYAH-UHFFFAOYSA-N 0.000 description 1
- LPBGVVVZBHAPBA-UHFFFAOYSA-M C1(CCCCCCC1)C(C(=O)[O-])NC(=O)C=1C(=NOC=1)C.[Li+] Chemical compound C1(CCCCCCC1)C(C(=O)[O-])NC(=O)C=1C(=NOC=1)C.[Li+] LPBGVVVZBHAPBA-UHFFFAOYSA-M 0.000 description 1
- XYJMIPHWFZLBOD-UHFFFAOYSA-N CC1=NOC=C1C(=O)NCC(=O)OC Chemical compound CC1=NOC=C1C(=O)NCC(=O)OC XYJMIPHWFZLBOD-UHFFFAOYSA-N 0.000 description 1
- GBCHLQNTMBDONN-UHFFFAOYSA-N CC1=NOC=C1C=1OC(CN=1)=O Chemical compound CC1=NOC=C1C=1OC(CN=1)=O GBCHLQNTMBDONN-UHFFFAOYSA-N 0.000 description 1
- BKAYPBIMXHWWSA-UHFFFAOYSA-N CC1CCC(CC1)=C1N=C(OC1=O)C=1N(N=CC=1)C Chemical compound CC1CCC(CC1)=C1N=C(OC1=O)C=1N(N=CC=1)C BKAYPBIMXHWWSA-UHFFFAOYSA-N 0.000 description 1
- QNXYFBWVAAYCPW-UHFFFAOYSA-N CCN1N=CC=C1C(=O)NCC(=O)OC Chemical compound CCN1N=CC=C1C(=O)NCC(=O)OC QNXYFBWVAAYCPW-UHFFFAOYSA-N 0.000 description 1
- QYZMLJTTXDPWSL-UHFFFAOYSA-N CN1N=CC=C1C1=NC(C(=O)O1)=C1CCCCCC1 Chemical compound CN1N=CC=C1C1=NC(C(=O)O1)=C1CCCCCC1 QYZMLJTTXDPWSL-UHFFFAOYSA-N 0.000 description 1
- JWOCURAYWHJDFG-UHFFFAOYSA-N CN1N=CC=C1C=1OC(C(N=1)=C1CC2(C1)CCC2)=O Chemical compound CN1N=CC=C1C=1OC(C(N=1)=C1CC2(C1)CCC2)=O JWOCURAYWHJDFG-UHFFFAOYSA-N 0.000 description 1
- NWHKANPDDDIQKF-UHFFFAOYSA-N CN1N=CC=C1C=1OC(CN=1)=O Chemical compound CN1N=CC=C1C=1OC(CN=1)=O NWHKANPDDDIQKF-UHFFFAOYSA-N 0.000 description 1
- PAXVJOJUXBQBDI-UHFFFAOYSA-N COC(=O)C(NC(=O)C1=CON=C1C)C1CCCCCCC1 Chemical compound COC(=O)C(NC(=O)C1=CON=C1C)C1CCCCCCC1 PAXVJOJUXBQBDI-UHFFFAOYSA-N 0.000 description 1
- FQGFWIPCQCIWPR-UHFFFAOYSA-N COC(=O)CNC(=O)C1=CC=NN1C Chemical compound COC(=O)CNC(=O)C1=CC=NN1C FQGFWIPCQCIWPR-UHFFFAOYSA-N 0.000 description 1
- MUOXYNIOJBUOCL-DCAQKATOSA-N C[C@H]1CC[C@@H](CC1)[C@H](NC(=O)OC(C)(C)C)C(O)=O Chemical compound C[C@H]1CC[C@@H](CC1)[C@H](NC(=O)OC(C)(C)C)C(O)=O MUOXYNIOJBUOCL-DCAQKATOSA-N 0.000 description 1
- GAWIXWVDTYZWAW-UHFFFAOYSA-N C[CH]O Chemical group C[CH]O GAWIXWVDTYZWAW-UHFFFAOYSA-N 0.000 description 1
- AXIFAOZVPXWHNE-UHFFFAOYSA-N C[Si](C)(C)CCOCC(C1)OCCC1(C(C(N1)=C2)=CC=C2C(O)=O)C1=O Chemical compound C[Si](C)(C)CCOCC(C1)OCCC1(C(C(N1)=C2)=CC=C2C(O)=O)C1=O AXIFAOZVPXWHNE-UHFFFAOYSA-N 0.000 description 1
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 1
- 208000005024 Castleman disease Diseases 0.000 description 1
- 208000010833 Chronic myeloid leukaemia Diseases 0.000 description 1
- LZVOUGFHWKJAHE-UHFFFAOYSA-N Cl.COC(=O)C(N)C1CCCCCCC1 Chemical compound Cl.COC(=O)C(N)C1CCCCCCC1 LZVOUGFHWKJAHE-UHFFFAOYSA-N 0.000 description 1
- HJYSFADVBYWMNO-AQLQUXDBSA-N Cl.C[C@H]1CC[C@H]([C@H](N)C#N)CC1 Chemical compound Cl.C[C@H]1CC[C@H]([C@H](N)C#N)CC1 HJYSFADVBYWMNO-AQLQUXDBSA-N 0.000 description 1
- NIBZCPLHVAVTCV-WQYNNSOESA-N Cl.C[C@H]1CC[C@H]([C@H](N)C(O)=O)CC1 Chemical compound Cl.C[C@H]1CC[C@H]([C@H](N)C(O)=O)CC1 NIBZCPLHVAVTCV-WQYNNSOESA-N 0.000 description 1
- YKHGDSPNAGYRNC-UHFFFAOYSA-N Cn1nccc1C1=NC(C2CC3(CCC3)C2)C(=O)O1 Chemical compound Cn1nccc1C1=NC(C2CC3(CCC3)C2)C(=O)O1 YKHGDSPNAGYRNC-UHFFFAOYSA-N 0.000 description 1
- NROHXWGROMFYJJ-UHFFFAOYSA-N Cn1nccc1C1=NC(C2CC3CCC(C3)C2)C(=O)O1 Chemical compound Cn1nccc1C1=NC(C2CC3CCC(C3)C2)C(=O)O1 NROHXWGROMFYJJ-UHFFFAOYSA-N 0.000 description 1
- 208000015943 Coeliac disease Diseases 0.000 description 1
- 206010009944 Colon cancer Diseases 0.000 description 1
- 206010010741 Conjunctivitis Diseases 0.000 description 1
- 229920002261 Corn starch Polymers 0.000 description 1
- 206010055665 Corneal neovascularisation Diseases 0.000 description 1
- YZCKVEUIGOORGS-OUBTZVSYSA-N Deuterium Chemical compound [2H] YZCKVEUIGOORGS-OUBTZVSYSA-N 0.000 description 1
- 239000004338 Dichlorodifluoromethane Substances 0.000 description 1
- 239000006144 Dulbecco’s modified Eagle's medium Substances 0.000 description 1
- 206010014824 Endotoxic shock Diseases 0.000 description 1
- 108090000371 Esterases Proteins 0.000 description 1
- 208000030836 Hashimoto thyroiditis Diseases 0.000 description 1
- 208000002250 Hematologic Neoplasms Diseases 0.000 description 1
- 206010020751 Hypersensitivity Diseases 0.000 description 1
- 102000039989 IL-17 family Human genes 0.000 description 1
- 108091069193 IL-17 family Proteins 0.000 description 1
- 102100035012 Interleukin-17 receptor C Human genes 0.000 description 1
- 101710186068 Interleukin-17 receptor C Proteins 0.000 description 1
- 229930194542 Keto Natural products 0.000 description 1
- ZDXPYRJPNDTMRX-VKHMYHEASA-N L-glutamine Chemical compound OC(=O)[C@@H](N)CCC(N)=O ZDXPYRJPNDTMRX-VKHMYHEASA-N 0.000 description 1
- 229930182816 L-glutamine Natural products 0.000 description 1
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 1
- 208000016604 Lyme disease Diseases 0.000 description 1
- 208000031422 Lymphocytic Chronic B-Cell Leukemia Diseases 0.000 description 1
- 206010065062 Meibomian gland dysfunction Diseases 0.000 description 1
- 229920000168 Microcrystalline cellulose Polymers 0.000 description 1
- 241001139947 Mida Species 0.000 description 1
- 208000033761 Myelogenous Chronic BCR-ABL Positive Leukemia Diseases 0.000 description 1
- 208000033776 Myeloid Acute Leukemia Diseases 0.000 description 1
- 208000009525 Myocarditis Diseases 0.000 description 1
- HSHXDCVZWHOWCS-UHFFFAOYSA-N N'-hexadecylthiophene-2-carbohydrazide Chemical compound CCCCCCCCCCCCCCCCNNC(=O)c1cccs1 HSHXDCVZWHOWCS-UHFFFAOYSA-N 0.000 description 1
- JRNVZBWKYDBUCA-UHFFFAOYSA-N N-Chlorosuccinimide Substances ClN1C(=O)CCC1=O JRNVZBWKYDBUCA-UHFFFAOYSA-N 0.000 description 1
- 208000001132 Osteoporosis Diseases 0.000 description 1
- 206010033645 Pancreatitis Diseases 0.000 description 1
- 208000029082 Pelvic Inflammatory Disease Diseases 0.000 description 1
- 206010034277 Pemphigoid Diseases 0.000 description 1
- 208000004362 Penile Induration Diseases 0.000 description 1
- 208000020758 Peyronie disease Diseases 0.000 description 1
- 229920003171 Poly (ethylene oxide) Polymers 0.000 description 1
- 229920001214 Polysorbate 60 Polymers 0.000 description 1
- XBDQKXXYIPTUBI-UHFFFAOYSA-M Propionate Chemical compound CCC([O-])=O XBDQKXXYIPTUBI-UHFFFAOYSA-M 0.000 description 1
- PQUGCKBLVKJMNT-UHFFFAOYSA-N SC560 Chemical compound C1=CC(OC)=CC=C1N1C(C=2C=CC(Cl)=CC=2)=CC(C(F)(F)F)=N1 PQUGCKBLVKJMNT-UHFFFAOYSA-N 0.000 description 1
- 206010039491 Sarcoma Diseases 0.000 description 1
- 206010040070 Septic Shock Diseases 0.000 description 1
- 241000287219 Serinus canaria Species 0.000 description 1
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 1
- DBMJMQXJHONAFJ-UHFFFAOYSA-M Sodium laurylsulphate Chemical compound [Na+].CCCCCCCCCCCCOS([O-])(=O)=O DBMJMQXJHONAFJ-UHFFFAOYSA-M 0.000 description 1
- HVUMOYIDDBPOLL-XWVZOOPGSA-N Sorbitan monostearate Chemical compound CCCCCCCCCCCCCCCCCC(=O)OC[C@@H](O)[C@H]1OC[C@H](O)[C@H]1O HVUMOYIDDBPOLL-XWVZOOPGSA-N 0.000 description 1
- 206010042033 Stevens-Johnson syndrome Diseases 0.000 description 1
- 231100000168 Stevens-Johnson syndrome Toxicity 0.000 description 1
- 208000005718 Stomach Neoplasms Diseases 0.000 description 1
- 206010042618 Surgical procedure repeated Diseases 0.000 description 1
- 229910052771 Terbium Inorganic materials 0.000 description 1
- 206010052779 Transplant rejections Diseases 0.000 description 1
- OKJPEAGHQZHRQV-UHFFFAOYSA-N Triiodomethane Natural products IC(I)I OKJPEAGHQZHRQV-UHFFFAOYSA-N 0.000 description 1
- YZCKVEUIGOORGS-NJFSPNSNSA-N Tritium Chemical compound [3H] YZCKVEUIGOORGS-NJFSPNSNSA-N 0.000 description 1
- 108060008682 Tumor Necrosis Factor Proteins 0.000 description 1
- 102000000852 Tumor Necrosis Factor-alpha Human genes 0.000 description 1
- 206010067584 Type 1 diabetes mellitus Diseases 0.000 description 1
- 206010046851 Uveitis Diseases 0.000 description 1
- 206010047115 Vasculitis Diseases 0.000 description 1
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 1
- AKZWRTCWNXHHFR-PDIZUQLASA-N [(3S)-oxolan-3-yl] N-[(2S,3S)-4-[(5S)-5-benzyl-3-[(2R)-2-carbamoyloxy-2,3-dihydro-1H-inden-1-yl]-4-oxo-3H-pyrrol-5-yl]-3-hydroxy-1-phenylbutan-2-yl]carbamate Chemical compound NC(=O)O[C@@H]1Cc2ccccc2C1C1C=N[C@](C[C@H](O)[C@H](Cc2ccccc2)NC(=O)O[C@H]2CCOC2)(Cc2ccccc2)C1=O AKZWRTCWNXHHFR-PDIZUQLASA-N 0.000 description 1
- OBTDGGBJIYEWMN-UHFFFAOYSA-N [N+](=O)([O-])C1=C(C=CC(=C1)[N+](=O)[O-])C1(CCOCC1)C(=O)OC Chemical compound [N+](=O)([O-])C1=C(C=CC(=C1)[N+](=O)[O-])C1(CCOCC1)C(=O)OC OBTDGGBJIYEWMN-UHFFFAOYSA-N 0.000 description 1
- ZBIKORITPGTTGI-UHFFFAOYSA-N [acetyloxy(phenyl)-$l^{3}-iodanyl] acetate Chemical compound CC(=O)OI(OC(C)=O)C1=CC=CC=C1 ZBIKORITPGTTGI-UHFFFAOYSA-N 0.000 description 1
- QQIRAVWVGBTHMJ-UHFFFAOYSA-N [dimethyl-(trimethylsilylamino)silyl]methane;lithium Chemical compound [Li].C[Si](C)(C)N[Si](C)(C)C QQIRAVWVGBTHMJ-UHFFFAOYSA-N 0.000 description 1
- WJGAPUXHSQQWQF-UHFFFAOYSA-N acetic acid;hydrochloride Chemical compound Cl.CC(O)=O WJGAPUXHSQQWQF-UHFFFAOYSA-N 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 239000013543 active substance Substances 0.000 description 1
- 206010069351 acute lung injury Diseases 0.000 description 1
- 150000001266 acyl halides Chemical class 0.000 description 1
- DZULQZKFBAHSRX-UHFFFAOYSA-N adamantane-1-carbaldehyde Chemical compound C1C(C2)CC3CC2CC1(C=O)C3 DZULQZKFBAHSRX-UHFFFAOYSA-N 0.000 description 1
- 239000000654 additive Substances 0.000 description 1
- 230000002411 adverse Effects 0.000 description 1
- 239000000443 aerosol Substances 0.000 description 1
- 150000001350 alkyl halides Chemical class 0.000 description 1
- 125000004644 alkyl sulfinyl group Chemical group 0.000 description 1
- 125000004414 alkyl thio group Chemical group 0.000 description 1
- 208000026935 allergic disease Diseases 0.000 description 1
- 230000000172 allergic effect Effects 0.000 description 1
- 230000007815 allergy Effects 0.000 description 1
- 229910052782 aluminium Inorganic materials 0.000 description 1
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 1
- PNEYBMLMFCGWSK-UHFFFAOYSA-N aluminium oxide Inorganic materials [O-2].[O-2].[O-2].[Al+3].[Al+3] PNEYBMLMFCGWSK-UHFFFAOYSA-N 0.000 description 1
- 150000001413 amino acids Chemical group 0.000 description 1
- 125000004202 aminomethyl group Chemical group [H]N([H])C([H])([H])* 0.000 description 1
- 229910021529 ammonia Inorganic materials 0.000 description 1
- BVCZEBOGSOYJJT-UHFFFAOYSA-N ammonium carbamate Chemical compound [NH4+].NC([O-])=O BVCZEBOGSOYJJT-UHFFFAOYSA-N 0.000 description 1
- 230000000844 anti-bacterial effect Effects 0.000 description 1
- 230000000919 anti-host Effects 0.000 description 1
- 230000003110 anti-inflammatory effect Effects 0.000 description 1
- 239000006286 aqueous extract Substances 0.000 description 1
- 239000008135 aqueous vehicle Substances 0.000 description 1
- 239000012131 assay buffer Substances 0.000 description 1
- 208000010668 atopic eczema Diseases 0.000 description 1
- 125000003725 azepanyl group Chemical group 0.000 description 1
- 230000001580 bacterial effect Effects 0.000 description 1
- 239000003899 bactericide agent Substances 0.000 description 1
- 235000013871 bee wax Nutrition 0.000 description 1
- 239000012166 beeswax Substances 0.000 description 1
- 229960000686 benzalkonium chloride Drugs 0.000 description 1
- QJCPEOFEBREKQB-UHFFFAOYSA-N benzenesulfinamide Chemical compound NS(=O)C1=CC=CC=C1 QJCPEOFEBREKQB-UHFFFAOYSA-N 0.000 description 1
- 125000004618 benzofuryl group Chemical group O1C(=CC2=C1C=CC=C2)* 0.000 description 1
- 125000004196 benzothienyl group Chemical group S1C(=CC2=C1C=CC=C2)* 0.000 description 1
- 125000003354 benzotriazolyl group Chemical group N1N=NC2=C1C=CC=C2* 0.000 description 1
- 125000004622 benzoxazinyl group Chemical group O1NC(=CC2=C1C=CC=C2)* 0.000 description 1
- 125000003236 benzoyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C(*)=O 0.000 description 1
- CADWTSSKOVRVJC-UHFFFAOYSA-N benzyl(dimethyl)azanium;chloride Chemical compound [Cl-].C[NH+](C)CC1=CC=CC=C1 CADWTSSKOVRVJC-UHFFFAOYSA-N 0.000 description 1
- HIMMOVPGAJKVOS-UHFFFAOYSA-N bicyclo[3.2.1]octan-3-one Chemical compound C1C(=O)CC2CCC1C2 HIMMOVPGAJKVOS-UHFFFAOYSA-N 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- 230000004071 biological effect Effects 0.000 description 1
- 230000036983 biotransformation Effects 0.000 description 1
- 239000000872 buffer Substances 0.000 description 1
- 230000003139 buffering effect Effects 0.000 description 1
- FUFJGUQYACFECW-UHFFFAOYSA-L calcium hydrogenphosphate Chemical compound [Ca+2].OP([O-])([O-])=O FUFJGUQYACFECW-UHFFFAOYSA-L 0.000 description 1
- 239000002775 capsule Substances 0.000 description 1
- 235000011089 carbon dioxide Nutrition 0.000 description 1
- 150000005323 carbonate salts Chemical class 0.000 description 1
- KXDHJXZQYSOELW-UHFFFAOYSA-N carbonic acid monoamide Natural products NC(O)=O KXDHJXZQYSOELW-UHFFFAOYSA-N 0.000 description 1
- 210000003169 central nervous system Anatomy 0.000 description 1
- 229940081733 cetearyl alcohol Drugs 0.000 description 1
- 239000003795 chemical substances by application Substances 0.000 description 1
- 229960002152 chlorhexidine acetate Drugs 0.000 description 1
- 125000006402 chloropyridazinyl group Chemical group 0.000 description 1
- 125000003016 chromanyl group Chemical group O1C(CCC2=CC=CC=C12)* 0.000 description 1
- 125000004230 chromenyl group Chemical group O1C(C=CC2=CC=CC=C12)* 0.000 description 1
- 208000032852 chronic lymphocytic leukemia Diseases 0.000 description 1
- 208000019425 cirrhosis of liver Diseases 0.000 description 1
- 238000005345 coagulation Methods 0.000 description 1
- 230000015271 coagulation Effects 0.000 description 1
- 229940110456 cocoa butter Drugs 0.000 description 1
- 235000019868 cocoa butter Nutrition 0.000 description 1
- 208000029742 colonic neoplasm Diseases 0.000 description 1
- 238000004040 coloring Methods 0.000 description 1
- 238000013270 controlled release Methods 0.000 description 1
- 239000008120 corn starch Substances 0.000 description 1
- 210000004087 cornea Anatomy 0.000 description 1
- 201000000159 corneal neovascularization Diseases 0.000 description 1
- 230000008025 crystallization Effects 0.000 description 1
- 125000004802 cyanophenyl group Chemical group 0.000 description 1
- QPOSYIIMLMZMAZ-UHFFFAOYSA-N cyclobutyl carbonochloridate Chemical compound ClC(=O)OC1CCC1 QPOSYIIMLMZMAZ-UHFFFAOYSA-N 0.000 description 1
- CGZZMOTZOONQIA-UHFFFAOYSA-N cycloheptanone Chemical compound O=C1CCCCCC1 CGZZMOTZOONQIA-UHFFFAOYSA-N 0.000 description 1
- IIRFCWANHMSDCG-UHFFFAOYSA-N cyclooctanone Chemical compound O=C1CCCCCCC1 IIRFCWANHMSDCG-UHFFFAOYSA-N 0.000 description 1
- 230000003247 decreasing effect Effects 0.000 description 1
- 230000007812 deficiency Effects 0.000 description 1
- 201000001981 dermatomyositis Diseases 0.000 description 1
- 229910052805 deuterium Inorganic materials 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- 125000005959 diazepanyl group Chemical group 0.000 description 1
- 125000004987 dibenzofuryl group Chemical group C1(=CC=CC=2OC3=C(C21)C=CC=C3)* 0.000 description 1
- 125000004988 dibenzothienyl group Chemical group C1(=CC=CC=2SC3=C(C21)C=CC=C3)* 0.000 description 1
- 235000019700 dicalcium phosphate Nutrition 0.000 description 1
- PXBRQCKWGAHEHS-UHFFFAOYSA-N dichlorodifluoromethane Chemical compound FC(F)(Cl)Cl PXBRQCKWGAHEHS-UHFFFAOYSA-N 0.000 description 1
- 235000019404 dichlorodifluoromethane Nutrition 0.000 description 1
- 229940087091 dichlorotetrafluoroethane Drugs 0.000 description 1
- LCSNDSFWVKMJCT-UHFFFAOYSA-N dicyclohexyl-(2-phenylphenyl)phosphane Chemical compound C1CCCCC1P(C=1C(=CC=CC=1)C=1C=CC=CC=1)C1CCCCC1 LCSNDSFWVKMJCT-UHFFFAOYSA-N 0.000 description 1
- YWDRRHINJWKDTJ-UHFFFAOYSA-N diethyl 2-(5-bromo-3-nitropyridin-2-yl)propanedioate Chemical compound CCOC(=O)C(C(=O)OCC)C1=NC=C(Br)C=C1[N+]([O-])=O YWDRRHINJWKDTJ-UHFFFAOYSA-N 0.000 description 1
- ZOGHDTBRWUEJDP-UHFFFAOYSA-N diethylalumanylium;cyanide Chemical compound N#[C-].CC[Al+]CC ZOGHDTBRWUEJDP-UHFFFAOYSA-N 0.000 description 1
- 125000006287 difluorobenzyl group Chemical group 0.000 description 1
- 125000004582 dihydrobenzothienyl group Chemical group S1C(CC2=C1C=CC=C2)* 0.000 description 1
- 125000006182 dimethyl benzyl group Chemical group 0.000 description 1
- 125000000597 dioxinyl group Chemical group 0.000 description 1
- 230000006806 disease prevention Effects 0.000 description 1
- 239000007884 disintegrant Substances 0.000 description 1
- 239000002270 dispersing agent Substances 0.000 description 1
- 238000004090 dissolution Methods 0.000 description 1
- 239000002552 dosage form Substances 0.000 description 1
- 239000003814 drug Substances 0.000 description 1
- 230000002526 effect on cardiovascular system Effects 0.000 description 1
- 239000003480 eluent Substances 0.000 description 1
- 239000003995 emulsifying agent Substances 0.000 description 1
- 239000008387 emulsifying waxe Substances 0.000 description 1
- 239000000839 emulsion Substances 0.000 description 1
- 206010014599 encephalitis Diseases 0.000 description 1
- 230000003511 endothelial effect Effects 0.000 description 1
- 150000002085 enols Chemical class 0.000 description 1
- 230000002255 enzymatic effect Effects 0.000 description 1
- 238000010931 ester hydrolysis Methods 0.000 description 1
- 150000002148 esters Chemical class 0.000 description 1
- 125000005745 ethoxymethyl group Chemical group [H]C([H])([H])C([H])([H])OC([H])([H])* 0.000 description 1
- LZCLXQDLBQLTDK-SCSAIBSYSA-N ethyl (2R)-lactate Chemical compound CCOC(=O)[C@@H](C)O LZCLXQDLBQLTDK-SCSAIBSYSA-N 0.000 description 1
- LZCLXQDLBQLTDK-BYPYZUCNSA-N ethyl (2S)-lactate Chemical compound CCOC(=O)[C@H](C)O LZCLXQDLBQLTDK-BYPYZUCNSA-N 0.000 description 1
- SFEWOHUGHVHUCA-ZETCQYMHSA-N ethyl (2s)-2-(2-ethoxy-2-oxoethoxy)propanoate Chemical compound CCOC(=O)CO[C@@H](C)C(=O)OCC SFEWOHUGHVHUCA-ZETCQYMHSA-N 0.000 description 1
- 125000006437 ethyl cyclopropyl group Chemical group 0.000 description 1
- GWQVMPWSEVRGPY-UHFFFAOYSA-N europium cryptate Chemical compound [Eu+3].N=1C2=CC=CC=1CN(CC=1N=C(C=CC=1)C=1N=C(C3)C=CC=1)CC(N=1)=CC(C(=O)NCCN)=CC=1C(N=1)=CC(C(=O)NCCN)=CC=1CN3CC1=CC=CC2=N1 GWQVMPWSEVRGPY-UHFFFAOYSA-N 0.000 description 1
- 208000001936 exophthalmos Diseases 0.000 description 1
- 208000030533 eye disease Diseases 0.000 description 1
- 239000000945 filler Substances 0.000 description 1
- 150000004673 fluoride salts Chemical class 0.000 description 1
- 239000006260 foam Substances 0.000 description 1
- 239000011888 foil Substances 0.000 description 1
- 230000002538 fungal effect Effects 0.000 description 1
- 230000000855 fungicidal effect Effects 0.000 description 1
- 239000000417 fungicide Substances 0.000 description 1
- 125000002541 furyl group Chemical group 0.000 description 1
- 208000020694 gallbladder disease Diseases 0.000 description 1
- 206010017758 gastric cancer Diseases 0.000 description 1
- 239000003102 growth factor Substances 0.000 description 1
- 208000019622 heart disease Diseases 0.000 description 1
- 210000002443 helper t lymphocyte Anatomy 0.000 description 1
- 208000006359 hepatoblastoma Diseases 0.000 description 1
- 125000004446 heteroarylalkyl group Chemical group 0.000 description 1
- 239000003906 humectant Substances 0.000 description 1
- 150000002430 hydrocarbons Chemical class 0.000 description 1
- 125000004464 hydroxyphenyl group Chemical group 0.000 description 1
- 239000001866 hydroxypropyl methyl cellulose Substances 0.000 description 1
- 235000010979 hydroxypropyl methyl cellulose Nutrition 0.000 description 1
- 229920003088 hydroxypropyl methyl cellulose Polymers 0.000 description 1
- 208000026278 immune system disease Diseases 0.000 description 1
- 238000002513 implantation Methods 0.000 description 1
- 238000011534 incubation Methods 0.000 description 1
- 125000003453 indazolyl group Chemical group N1N=C(C2=C1C=CC=C2)* 0.000 description 1
- 230000004968 inflammatory condition Effects 0.000 description 1
- 238000001802 infusion Methods 0.000 description 1
- 230000005764 inhibitory process Effects 0.000 description 1
- 208000014674 injury Diseases 0.000 description 1
- 230000003993 interaction Effects 0.000 description 1
- 102000053460 interleukin-17 receptor activity proteins Human genes 0.000 description 1
- 108040001304 interleukin-17 receptor activity proteins Proteins 0.000 description 1
- 238000010255 intramuscular injection Methods 0.000 description 1
- 239000007927 intramuscular injection Substances 0.000 description 1
- 238000005342 ion exchange Methods 0.000 description 1
- 150000002500 ions Chemical class 0.000 description 1
- 229910052742 iron Inorganic materials 0.000 description 1
- 208000002551 irritable bowel syndrome Diseases 0.000 description 1
- 208000023589 ischemic disease Diseases 0.000 description 1
- 125000004594 isoindolinyl group Chemical group C1(NCC2=CC=CC=C12)* 0.000 description 1
- 125000001972 isopentyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000002183 isoquinolinyl group Chemical group C1(=NC=CC2=CC=CC=C12)* 0.000 description 1
- 206010023332 keratitis Diseases 0.000 description 1
- 125000000468 ketone group Chemical group 0.000 description 1
- 239000008101 lactose Substances 0.000 description 1
- CPNRUDPCGCKPTG-UQKRIMTDSA-M lithium (2S)-2-cyclohexyl-2-[(2-pyridin-4-ylacetyl)amino]acetate Chemical compound C1(CCCCC1)[C@@H](C(=O)[O-])NC(CC1=CC=NC=C1)=O.[Li+] CPNRUDPCGCKPTG-UQKRIMTDSA-M 0.000 description 1
- UKNJXWYBDAAEOB-UHFFFAOYSA-M lithium 2-[(2-methylpyrazole-3-carbonyl)amino]acetate Chemical compound CN1N=CC=C1C(=O)NCC(=O)[O-].[Li+] UKNJXWYBDAAEOB-UHFFFAOYSA-M 0.000 description 1
- YHAZCHSQFVGMMF-UHFFFAOYSA-M lithium 2-cyclooctyl-2-[(2-pyridin-4-ylacetyl)amino]acetate Chemical compound C1(CCCCCCC1)C(C(=O)[O-])NC(CC1=CC=NC=C1)=O.[Li+] YHAZCHSQFVGMMF-UHFFFAOYSA-M 0.000 description 1
- FZRNJOXQNWVMIH-UHFFFAOYSA-N lithium;hydrate Chemical compound [Li].O FZRNJOXQNWVMIH-UHFFFAOYSA-N 0.000 description 1
- 239000006210 lotion Substances 0.000 description 1
- 239000007937 lozenge Substances 0.000 description 1
- 239000000314 lubricant Substances 0.000 description 1
- 235000019359 magnesium stearate Nutrition 0.000 description 1
- 238000004519 manufacturing process Methods 0.000 description 1
- 238000004949 mass spectrometry Methods 0.000 description 1
- 201000001441 melanoma Diseases 0.000 description 1
- 201000011475 meningoencephalitis Diseases 0.000 description 1
- YPQNVBMOAIWJEB-HNNXBMFYSA-N methyl (2S)-2-cyclohexyl-2-[(2-pyridin-4-ylacetyl)amino]acetate Chemical compound C1(CCCCC1)[C@@H](C(=O)OC)NC(CC1=CC=NC=C1)=O YPQNVBMOAIWJEB-HNNXBMFYSA-N 0.000 description 1
- RQVUXUPDMWDERI-HNNXBMFYSA-N methyl (2S)-3-(2-chlorophenyl)-2-[(2-pyridin-4-ylacetyl)amino]propanoate Chemical compound ClC1=C(C=CC=C1)C[C@@H](C(=O)OC)NC(CC1=CC=NC=C1)=O RQVUXUPDMWDERI-HNNXBMFYSA-N 0.000 description 1
- ORJOGDRCFDWIIJ-QRPNPIFTSA-N methyl (2s)-2-amino-2-cyclohexylacetate;hydrochloride Chemical compound [Cl-].COC(=O)[C@@H]([NH3+])C1CCCCC1 ORJOGDRCFDWIIJ-QRPNPIFTSA-N 0.000 description 1
- FXIXAZXWVBFVDS-FVGYRXGTSA-N methyl (2s)-2-amino-3-(2-chlorophenyl)propanoate;hydrochloride Chemical compound Cl.COC(=O)[C@@H](N)CC1=CC=CC=C1Cl FXIXAZXWVBFVDS-FVGYRXGTSA-N 0.000 description 1
- HTANURMJHZJJSO-UHFFFAOYSA-N methyl 2-(2,4-dinitrophenyl)acetate Chemical compound COC(=O)CC1=CC=C([N+]([O-])=O)C=C1[N+]([O-])=O HTANURMJHZJJSO-UHFFFAOYSA-N 0.000 description 1
- DGVZIQRNXKAVHS-UHFFFAOYSA-N methyl 2-(2-aminopyridin-4-yl)acetate Chemical compound COC(=O)Cc1ccnc(N)c1 DGVZIQRNXKAVHS-UHFFFAOYSA-N 0.000 description 1
- FIZOWVAGAKPTML-UHFFFAOYSA-N methyl 2-cyclooctyl-2-[(2-methylpyrazole-3-carbonyl)amino]acetate Chemical compound C1(CCCCCCC1)C(C(=O)OC)NC(=O)C=1N(N=CC=1)C FIZOWVAGAKPTML-UHFFFAOYSA-N 0.000 description 1
- NXICKYVTFMBUNI-UHFFFAOYSA-N methyl 2-cyclooctyl-2-[(2-pyridin-4-ylacetyl)amino]acetate Chemical compound C1(CCCCCCC1)C(C(=O)OC)NC(CC1=CC=NC=C1)=O NXICKYVTFMBUNI-UHFFFAOYSA-N 0.000 description 1
- VAXBTUCCEIMXRL-UHFFFAOYSA-N methyl 2-cyclooctyl-2-formamidoacetate Chemical compound COC(=O)C(NC=O)C1CCCCCCC1 VAXBTUCCEIMXRL-UHFFFAOYSA-N 0.000 description 1
- HKCIIIMEGGDFKR-UHFFFAOYSA-N methyl 2-cyclooctylidene-2-formamidoacetate Chemical compound COC(=O)C(NC=O)=C1CCCCCCC1 HKCIIIMEGGDFKR-UHFFFAOYSA-N 0.000 description 1
- CRXFROMHHBMNAB-UHFFFAOYSA-N methyl 2-isocyanoacetate Chemical compound COC(=O)C[N+]#[C-] CRXFROMHHBMNAB-UHFFFAOYSA-N 0.000 description 1
- 125000006533 methyl amino methyl group Chemical group [H]N(C([H])([H])[H])C([H])([H])* 0.000 description 1
- MQWCXKGKQLNYQG-UHFFFAOYSA-N methyl cyclohexan-4-ol Natural products CC1CCC(O)CC1 MQWCXKGKQLNYQG-UHFFFAOYSA-N 0.000 description 1
- 125000006431 methyl cyclopropyl group Chemical group 0.000 description 1
- 125000000325 methylidene group Chemical group [H]C([H])=* 0.000 description 1
- 125000006405 methylpyridazinyl group Chemical group 0.000 description 1
- 125000006393 methylpyrimidinyl group Chemical group 0.000 description 1
- 230000000813 microbial effect Effects 0.000 description 1
- 235000019813 microcrystalline cellulose Nutrition 0.000 description 1
- 239000008108 microcrystalline cellulose Substances 0.000 description 1
- 229940016286 microcrystalline cellulose Drugs 0.000 description 1
- 229940042472 mineral oil Drugs 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 201000006417 multiple sclerosis Diseases 0.000 description 1
- 208000010125 myocardial infarction Diseases 0.000 description 1
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000001624 naphthyl group Chemical group 0.000 description 1
- 125000004593 naphthyridinyl group Chemical group N1=C(C=CC2=CC=CN=C12)* 0.000 description 1
- 230000007935 neutral effect Effects 0.000 description 1
- 238000006386 neutralization reaction Methods 0.000 description 1
- 150000002825 nitriles Chemical class 0.000 description 1
- 231100000344 non-irritating Toxicity 0.000 description 1
- 239000002687 nonaqueous vehicle Substances 0.000 description 1
- 238000000655 nuclear magnetic resonance spectrum Methods 0.000 description 1
- GLDOVTGHNKAZLK-UHFFFAOYSA-N octadecan-1-ol Chemical compound CCCCCCCCCCCCCCCCCCO GLDOVTGHNKAZLK-UHFFFAOYSA-N 0.000 description 1
- 230000003287 optical effect Effects 0.000 description 1
- 150000007530 organic bases Chemical class 0.000 description 1
- 150000002895 organic esters Chemical class 0.000 description 1
- 230000002611 ovarian Effects 0.000 description 1
- BSCHIACBONPEOB-UHFFFAOYSA-N oxolane;hydrate Chemical compound O.C1CCOC1 BSCHIACBONPEOB-UHFFFAOYSA-N 0.000 description 1
- LXNAVEXFUKBNMK-UHFFFAOYSA-N palladium(II) acetate Substances [Pd].CC(O)=O.CC(O)=O LXNAVEXFUKBNMK-UHFFFAOYSA-N 0.000 description 1
- PIBWKRNGBLPSSY-UHFFFAOYSA-L palladium(II) chloride Chemical compound Cl[Pd]Cl PIBWKRNGBLPSSY-UHFFFAOYSA-L 0.000 description 1
- 230000003071 parasitic effect Effects 0.000 description 1
- 239000008188 pellet Substances 0.000 description 1
- 230000000149 penetrating effect Effects 0.000 description 1
- 125000001147 pentyl group Chemical group C(CCCC)* 0.000 description 1
- 201000001245 periodontitis Diseases 0.000 description 1
- 210000001428 peripheral nervous system Anatomy 0.000 description 1
- 206010034674 peritonitis Diseases 0.000 description 1
- 239000008177 pharmaceutical agent Substances 0.000 description 1
- 239000002831 pharmacologic agent Substances 0.000 description 1
- 230000000144 pharmacologic effect Effects 0.000 description 1
- WVDDGKGOMKODPV-ZQBYOMGUSA-N phenyl(114C)methanol Chemical compound O[14CH2]C1=CC=CC=C1 WVDDGKGOMKODPV-ZQBYOMGUSA-N 0.000 description 1
- PDTFCHSETJBPTR-UHFFFAOYSA-N phenylmercuric nitrate Chemical compound [O-][N+](=O)O[Hg]C1=CC=CC=C1 PDTFCHSETJBPTR-UHFFFAOYSA-N 0.000 description 1
- 125000004592 phthalazinyl group Chemical group C1(=NN=CC2=CC=CC=C12)* 0.000 description 1
- 239000006187 pill Substances 0.000 description 1
- 229960005235 piperonyl butoxide Drugs 0.000 description 1
- 229920001223 polyethylene glycol Polymers 0.000 description 1
- 239000001818 polyoxyethylene sorbitan monostearate Substances 0.000 description 1
- 235000010989 polyoxyethylene sorbitan monostearate Nutrition 0.000 description 1
- 229920001451 polypropylene glycol Polymers 0.000 description 1
- 229940113124 polysorbate 60 Drugs 0.000 description 1
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 1
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 1
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 1
- 229920001592 potato starch Polymers 0.000 description 1
- 238000004237 preparative chromatography Methods 0.000 description 1
- 239000003380 propellant Substances 0.000 description 1
- MCSINKKTEDDPNK-UHFFFAOYSA-N propyl propionate Chemical compound CCCOC(=O)CC MCSINKKTEDDPNK-UHFFFAOYSA-N 0.000 description 1
- 230000004853 protein function Effects 0.000 description 1
- 125000001042 pteridinyl group Chemical group N1=C(N=CC2=NC=CN=C12)* 0.000 description 1
- UBQKCCHYAOITMY-UHFFFAOYSA-N pyridin-2-ol Chemical compound OC1=CC=CC=N1 UBQKCCHYAOITMY-UHFFFAOYSA-N 0.000 description 1
- 125000002294 quinazolinyl group Chemical group N1=C(N=CC2=CC=CC=C12)* 0.000 description 1
- 125000002943 quinolinyl group Chemical group N1=C(C=CC2=CC=CC=C12)* 0.000 description 1
- 125000001567 quinoxalinyl group Chemical group N1=C(C=NC2=CC=CC=C12)* 0.000 description 1
- 125000004621 quinuclidinyl group Chemical group N12C(CC(CC1)CC2)* 0.000 description 1
- 239000002287 radioligand Substances 0.000 description 1
- 239000000985 reactive dye Substances 0.000 description 1
- 210000000664 rectum Anatomy 0.000 description 1
- 238000010992 reflux Methods 0.000 description 1
- 201000002793 renal fibrosis Diseases 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 230000004044 response Effects 0.000 description 1
- 230000037390 scarring Effects 0.000 description 1
- 239000012363 selectfluor Substances 0.000 description 1
- WRIKHQLVHPKCJU-UHFFFAOYSA-N sodium bis(trimethylsilyl)amide Chemical compound C[Si](C)(C)N([Na])[Si](C)(C)C WRIKHQLVHPKCJU-UHFFFAOYSA-N 0.000 description 1
- 229940023144 sodium glycolate Drugs 0.000 description 1
- 235000019333 sodium laurylsulphate Nutrition 0.000 description 1
- XZPVPNZTYPUODG-UHFFFAOYSA-M sodium;chloride;dihydrate Chemical compound O.O.[Na+].[Cl-] XZPVPNZTYPUODG-UHFFFAOYSA-M 0.000 description 1
- 239000012453 solvate Substances 0.000 description 1
- 239000011877 solvent mixture Substances 0.000 description 1
- 239000001587 sorbitan monostearate Substances 0.000 description 1
- 235000011076 sorbitan monostearate Nutrition 0.000 description 1
- 229940035048 sorbitan monostearate Drugs 0.000 description 1
- AOAIWGRQECAIKB-UHFFFAOYSA-N spiro[3.3]heptan-2-one Chemical compound C1C(=O)CC11CCC1 AOAIWGRQECAIKB-UHFFFAOYSA-N 0.000 description 1
- 239000007921 spray Substances 0.000 description 1
- 206010041823 squamous cell carcinoma Diseases 0.000 description 1
- 230000000087 stabilizing effect Effects 0.000 description 1
- 238000010561 standard procedure Methods 0.000 description 1
- 239000007858 starting material Substances 0.000 description 1
- 230000000638 stimulation Effects 0.000 description 1
- 201000011549 stomach cancer Diseases 0.000 description 1
- UDYFLDICVHJSOY-UHFFFAOYSA-N sulfur trioxide-pyridine complex Substances O=S(=O)=O.C1=CC=NC=C1 UDYFLDICVHJSOY-UHFFFAOYSA-N 0.000 description 1
- 239000006228 supernatant Substances 0.000 description 1
- 239000000829 suppository Substances 0.000 description 1
- 239000000375 suspending agent Substances 0.000 description 1
- 230000002195 synergetic effect Effects 0.000 description 1
- 238000003786 synthesis reaction Methods 0.000 description 1
- 239000006188 syrup Substances 0.000 description 1
- 235000020357 syrup Nutrition 0.000 description 1
- 230000009885 systemic effect Effects 0.000 description 1
- 239000000454 talc Substances 0.000 description 1
- 229910052623 talc Inorganic materials 0.000 description 1
- 230000004489 tear production Effects 0.000 description 1
- GZCRRIHWUXGPOV-UHFFFAOYSA-N terbium atom Chemical compound [Tb] GZCRRIHWUXGPOV-UHFFFAOYSA-N 0.000 description 1
- RCIHWAPEJZSCKM-PSLXWICFSA-N tert-butyl (2'S,3S)-2'-methyl-6-[(2-methylpropan-2-yl)oxycarbonylamino]-2-oxospiro[indole-3,4'-oxane]-1-carboxylate Chemical compound C(C)(C)(C)OC(=O)NC1=CC=C2C(=C1)N(C([C@@]21C[C@@H](OCC1)C)=O)C(=O)OC(C)(C)C RCIHWAPEJZSCKM-PSLXWICFSA-N 0.000 description 1
- MHXBHWLGRWOABW-UHFFFAOYSA-N tetradecyl octadecanoate Chemical compound CCCCCCCCCCCCCCCCCC(=O)OCCCCCCCCCCCCCC MHXBHWLGRWOABW-UHFFFAOYSA-N 0.000 description 1
- RWRDLPDLKQPQOW-UHFFFAOYSA-N tetrahydropyrrole Substances C1CCNC1 RWRDLPDLKQPQOW-UHFFFAOYSA-N 0.000 description 1
- 125000003944 tolyl group Chemical group 0.000 description 1
- 238000011200 topical administration Methods 0.000 description 1
- 230000000699 topical effect Effects 0.000 description 1
- 206010044412 transitional cell carcinoma Diseases 0.000 description 1
- 230000008733 trauma Effects 0.000 description 1
- 125000004306 triazinyl group Chemical group 0.000 description 1
- CYRMSUTZVYGINF-UHFFFAOYSA-N trichlorofluoromethane Chemical compound FC(Cl)(Cl)Cl CYRMSUTZVYGINF-UHFFFAOYSA-N 0.000 description 1
- JEJAMASKDTUEBZ-UHFFFAOYSA-N tris(1,1,3-tribromo-2,2-dimethylpropyl) phosphate Chemical compound BrCC(C)(C)C(Br)(Br)OP(=O)(OC(Br)(Br)C(C)(C)CBr)OC(Br)(Br)C(C)(C)CBr JEJAMASKDTUEBZ-UHFFFAOYSA-N 0.000 description 1
- 229910052722 tritium Inorganic materials 0.000 description 1
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 1
- 230000003612 virological effect Effects 0.000 description 1
- 238000010792 warming Methods 0.000 description 1
- 208000005494 xerophthalmia Diseases 0.000 description 1
- 229910052725 zinc Inorganic materials 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4427—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems
- A61K31/4439—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems containing a five-membered ring with nitrogen as a ring hetero atom, e.g. omeprazole
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/40—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil
- A61K31/407—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil condensed with other heterocyclic ring systems, e.g. ketorolac, physostigmine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/415—1,2-Diazoles
- A61K31/4155—1,2-Diazoles non condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/4192—1,2,3-Triazoles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/4196—1,2,4-Triazoles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/42—Oxazoles
- A61K31/422—Oxazoles not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/425—Thiazoles
- A61K31/427—Thiazoles not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/433—Thidiazoles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/4353—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems
- A61K31/437—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems the heterocyclic ring system containing a five-membered ring having nitrogen as a ring hetero atom, e.g. indolizine, beta-carboline
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/50—Pyridazines; Hydrogenated pyridazines
- A61K31/501—Pyridazines; Hydrogenated pyridazines not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/50—Pyridazines; Hydrogenated pyridazines
- A61K31/502—Pyridazines; Hydrogenated pyridazines ortho- or peri-condensed with carbocyclic ring systems, e.g. cinnoline, phthalazine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/506—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim not condensed and containing further heterocyclic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/10—Spiro-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/10—Spiro-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D491/00—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00
- C07D491/02—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00 in which the condensed system contains two hetero rings
- C07D491/10—Spiro-condensed systems
- C07D491/107—Spiro-condensed systems with only one oxygen atom as ring hetero atom in the oxygen-containing ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D491/00—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00
- C07D491/12—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00 in which the condensed system contains three hetero rings
- C07D491/20—Spiro-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D495/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having sulfur atoms as the only ring hetero atoms
- C07D495/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having sulfur atoms as the only ring hetero atoms in which the condensed system contains two hetero rings
- C07D495/10—Spiro-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D519/00—Heterocyclic compounds containing more than one system of two or more relevant hetero rings condensed among themselves or condensed with a common carbocyclic ring system not provided for in groups C07D453/00 or C07D455/00
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Public Health (AREA)
- Epidemiology (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Veterinary Medicine (AREA)
- Medicinal Chemistry (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Oxygen Or Sulfur (AREA)
- Nitrogen And Oxygen Or Sulfur-Condensed Heterocyclic Ring Systems (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
Abstract
Una serie de derivados de 2-oxoindolina espirocíclicos sustituidos y análogos de los mismos, que son potentes moduladores de la actividad de IL-17 humana, son por consiguiente beneficiosos en el tratamiento y/o prevención de diversas dolencias humanas, incluyendo trastornos inflamatorios y autoinmunitarios. (Traducción automática con Google Translate, sin valor legal)
Description
DESCRIPCIÓN
Indolinas espirocíclicas como moduladores de IL-17
La presente invención se refiere a compuestos heterocíclicos y a su uso en terapia. Más particularmente, esta invención se refiere a los derivados de oxoindolinas espirocíclicas farmacológicamente activos y a los análogos de los mismos. Estos compuestos actúan como moduladores de la actividad de la IL-17 y, en consecuencia, son beneficiosos como agentes farmacéuticos para el tratamiento y/o la prevención de estados patológicos, que incluyen trastornos inflamatorios y autoinmunitarios adversos.
La IL-17A (originalmente denominada CTLA-8 y también conocida como IL-17) es una citocina proinflamatoria y el miembro fundador de la familia de las IL-17 (Rouvier et al., J. Immunol., 1993, 150, 5445-5456). Posteriormente, se identificaron otros cinco miembros de la familia (IL-17B a IL-17F), incluido el más estrechamente relacionado, la IL-17F (ML-1), que comparte aproximadamente un 55 % de homología en la secuencia de aminoácidos con la IL-17A (Moseley et al., Cytokine Growth Factor Rev., 2003, 14, 155-174). La IL-17A y la IL-17F las expresan el recientemente definido subconjunto de linfocitos T cooperadores relacionados con la autoinmunidad, Th17, que también expresan las citocinas distintivas IL-21 e IL-22 (Korn et al., Ann. Rev. Immunol., 2009, 27, 485-517). La IL-17A y la IL-17F se expresan como homodímeros, pero también se pueden expresar como heterodímeros con las IL-17A/F (Wright et al., J. Immunol., 2008, 181,2799-2805). La IL-17A y la F transmiten la señal a través de los receptores IL-17R, IL-17RC o un complejo receptor IL-17RA/RC (Gaffen, Cytokine, 2008, 43, 402-407). Tanto la IL-17A como la IL-17F se han relacionado con varias enfermedades autoinmunitarias.
Los compuestos de acuerdo con la presente invención, al ser potentes moduladores de la actividad de la IL-17 humana, son por lo tanto beneficiosos para el tratamiento y/o la prevención de diversas dolencias humanas, que incluyen los trastornos inflamatorios y autoinmunitarios.
Además, los compuestos de acuerdo con la presente invención pueden ser beneficiosos como estándares farmacológicos para el uso en el desarrollo de nuevas pruebas biológicas y en la búsqueda de nuevos agentes farmacológicos. Por tanto, los compuestos de esta invención pueden ser útiles como radioligandos en los ensayos para detectar compuestos farmacológicamente activos.
Las solicitudes de patente internacional WO 2013/116682 y WO 2014/066726 se refieren a clases distintas de compuestos químicos que se afirma que modulan la actividad de la IL-17 y que son útiles para el tratamiento de afecciones médicas, incluidas las enfermedades inflamatorias.
En las solicitudes de patente internacional WO 2015/015378, WO 2016/120849 y WO 2016/120850 se describen varias clases de compuestos heterocíclicos que se afirma que inhiben la actividad de RORy y/o reducen la cantidad de IL-17 en un sujeto, y que son útiles para el tratamiento de diversos trastornos médicos, incluidos los trastornos inmunitarios o inflamatorios.
En la solicitud de patente internacional WO 2015/004533 se da a conocer una clase de compuestos heterocíclicos que se afirma que inhiben la función de la proteína BET al unirse a bromodominios y que son útiles para el tratamiento de afecciones que incluyen cáncer, enfermedades autoinmunitarias y cardiovasculares.
Sin embargo, nada de la técnica anterior disponible hasta la fecha describe o sugiere la clase estructural precisa de los derivados de oxoindolinas espirocíclicas y análogos de los mismos, tal como se da a conocer en la presente invención.
La presente invención da a conocer un compuesto de fórmula (I) o una de sus sales farmacéuticamente aceptables:
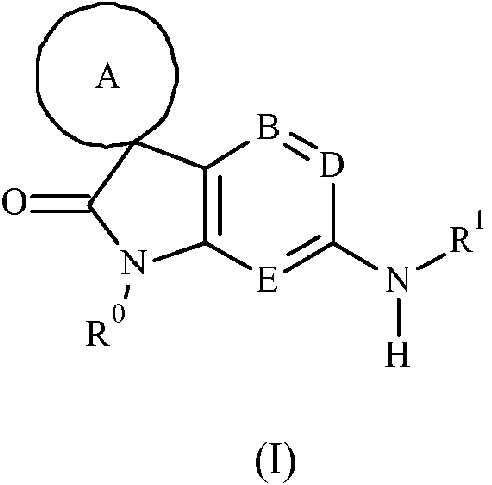
en donde
el anillo A representa cicloalquilo C3-9, heterocicloalquilo C3-7 o heterobicicloalquilo C4-9, cualquiera de cuyos grupos puede estar opcionalmente sustituido con uno, dos o tres sustituyentes seleccionados independientemente de alquilo C1-6, halógeno, ciano, trifluorometilo, hidroxi, oxo, alcoxi C1-6, alquil(C1-6)tío, alquil(C1-e)sulfinilo, alquil(C1-e)sulfonilo, alquil(C2-6)carbonilo, amino, imino, alquil(C1-6)amino y dialquil(C1-6)amino;
B representa C-R2 o N;
D representa C-R3 o N;
E representa C-R4 o N;
R0 representa hidrógeno o alquilo C1-6;
R1 representa -CORa ;
Ra representa -CH(R5)N(H)C(O)R6 , -CH(R5)N(H)S(O)2R6 , -C(=CR5aR5b)N(H)C(O)R6 , -CH(R5)R7 , -CH(R5)N(H)R7 o -CH(R5)C(O)N(H)R7 ;
R2 representa hidrógeno, halógeno, ciano, alquilo C1-6, fluorometilo, difluorometilo, trifluorometilo, hidroxi, alcoxi C1-6, difluorometoxi, trifluorometoxi, alquil(C1-6)sulfinilo o alquil(C1-6)sulfonilo;
R3 representa hidrógeno, halógeno, ciano, alquilo C1-6, fluorometilo, difluorometilo, trifluorometilo, hidroxi, alcoxi C1-6, difluorometoxi, trifluorometoxi, alquil(C1-6)sulfinilo o alquil(C1-6)sulfonilo;
R4 representa hidrógeno, halógeno, ciano, alquilo C1-6, fluorometilo, difluorometilo, trifluorometilo, hidroxi, alcoxi C1-6, difluorometoxi, trifluorometoxi, alquil(C1-6)sulfinilo o alquil(C1-6)sulfonilo;
R5 representa hidrógeno; o R5 representa alquilo C1-5, cicloalquilo C3-9, cicloalquil(C3-9)alquilo C1-5, bicicloalquilo C4-9, bicicloalquil(C4-9)alquilo C1-5, espirocicloalquilo C5-9, espirocicloalquil(C5-9)alquilo C1-5, tricicloalquilo C9-11, tricicloalquil(C9- n )alquilo C1-5, arilo, arilalquilo C1-5, heterocicloalquilo C3-7, heterocicloalquil(C3-7)alquilo C1-5, heteroarilo o heteroarilalquilo C1-5, cualquiera de cuyos grupos puede estar opcionalmente sustituido con uno, dos o tres sustituyentes seleccionados independientemente de halógeno, ciano, nitro, alquilo C1-6, trifluorometilo, trifluoroetilo, fenilo, hidroxi, oxo, alcoxi C1-6, difluorometoxi, trifluorometoxi, alquil(C1-6)tío, alquil(C1-6)sulfinilo, alquil(C1-6)sulfonilo, amino, alquil(C1-6)amino, dialquil(C1-6)amino, alquil(C2-6)carbonilamino, alcoxi(C2-6)carbonilamino, alquil(C1-6)sulfonilamino, formilo, alquil(C2-6)carbonilo, carboxi, alcoxi(C2-6)carbonilo, aminocarbonilo, alquil(C1-6)aminocarbonilo, dialquil(C1-6)aminocarbonilo, aminosulfonilo, alquil(C1-6)aminosulfonilo y dialquil(C1-6)aminosulfonilo;
R5a representa cicloalquilo C3-7, bicicloalquilo C4-9, arilo, heterocicloalquilo C3-7 o heteroarilo, cualquiera de cuyos grupos puede estar opcionalmente sustituido con uno o más sustituyentes seleccionados de alquilo C1-6, halógeno, ciano, trifluorometilo, trifluoroetilo, fenilo, hidroxi, alcoxi C1-6, alquil(C1-6)tío, alquil(C1-6)sulfinilo, alquil(C1-6)sulfonilo, alquil(C2-6)carbonilo, amino, alquil(C1-6)amino y dialquil(C1-6)amino; y
R5b representa hidrógeno o alquilo C1-6; o
R5a y R5b, cuando se toman junto con el átomo de carbono al que ambos están unidos, representan cicloalquilo C3-7, bicicloalquilo C4-9 o heterocicloalquilo C3-7, cualquiera de cuyos grupos puede estar opcionalmente sustituido con uno o dos sustituyentes seleccionados de alquilo C1-6, halógeno, ciano, trifluorometilo, trifluoroetilo, fenilo, hidroxi, alcoxi C1-6, alquil(C1-6)tío, alquil(C1-6)sulfinilo, alquil(C1-6)sulfonilo, alquil(C2-6)carbonilo, amino, alquil(C1-6)amino y dialquil(C1-6)amino;
R6 representa -NR6aR6b u -OR6 c; o R6 representa alquilo C1-9, cicloalquilo C3-9, cicloalquil(C3-9)alquilo C1-6, arilo, arilalquilo C1-6, heterocicloalquilo C3-7, heterocicloalquil(C3-7)alquilo C1-6, heteroarilo, heteroarilalquilo C1-6 o espiro[heterocicloalquil(C3-7)][heteroarilo], cualquiera de cuyos grupos puede estar opcionalmente sustituido con uno, dos o tres sustituyentes seleccionados independientemente entre halógeno, ciano, nitro, alquilo C1-6, difluorometilo, trifluorometilo, difluoroetilo, trifluoroetilo, trifluoropropilo, ciclopropilo, ciclobutilo, ciclopropilmetilo, fenilo, fluorofenilo, hidroxi, hidroxialquilo C1-6, oxo, alcoxi C1-6, alcoxi(C1-6)alquilo C1-6, difluorometoxi, trifluorometoxi, alquil(C1-6)tío, alquil(C1-6)sulfinilo, alquil(C1-6)sulfonilo, amino, aminoalquilo C1-6, alquil(C1-6)amino, dialquil(C1-6)amino, dialquil(C1-6)aminoalquilo C1-6, pirrolidinilo, tetrahidropiranilo, morfolinilo, piperazinilo, alquil(C2-6)carbonilamino, alquil(C2-6)carbonilaminoalquilo C1-6, alcoxi(C2-6)carbonilamino, alquil(C1-6)sulfonilamino, formilo, alquil(C2-6)carbonilo, carboxi, alcoxi(C2-6)carbonilo, aminocarbonilo, alquil(C1-6)aminocarbonilo, dialquil(C1-6)aminocarbonilo, aminosulfonilo, alquil(C1-6)aminosulfonilo y dialquil(C1-6)aminosulfonilo;
R6a representa hidrógeno; o R6a representa alquilo C1-6, cicloalquilo C3-7, cicloalquil(C3-7)alquilo C1-6, arilo, arilalquilo C1-6, heterocicloalquilo C3-7, heterocicloalquil(C3-7)alquilo C1-6, heteroarilo, heteroarilalquilo C1-6 o espiro[heterocicloalquil(C3-7)][heteroarilo], cualquiera de cuyos grupos puede estar opcionalmente sustituido con uno, dos o tres sustituyentes seleccionados independientemente entre halógeno, ciano, nitro, alquilo C1-6, trifluorometilo, fenilo, fluorofenilo, hidroxi, hidroxialquilo C1-6, oxo, alcoxi C1-6, difluorometoxi, trifluorometoxi, alquil(C1-6)tío, alquil(C1-6)sulfinilo, alquil(C1-6)sulfonilo, amino, aminoalquilo C1-6, alquil(C1-6)amino, dialquil(C1-6)amino, pirrolidinilo, morfolinilo, piperazinilo, alquil(C2-6)carbonilamino, alquil(C2-6)carbonilaminoalquilo C1-6, alcoxi(C2-6)carbonilamino, alquil(C1-6)sulfonilamino, formilo, alquil(C2-6)carbonilo, carboxi, alcoxi(C2-6)carbonilo, aminocarbonilo, alquil(C1-6)aminocarbonilo, dialquil(C1-6)aminocarbonilo, aminosulfonilo, alquil(C1-6)aminosulfonilo y dialquil(C1-6)aminosulfonilo;
R6b representa hidrógeno o alquilo C1-6;
R6c representa alquilo C i -6, cicloalquilo C3-7, c¡cloalqu¡l(C3-7)alqu¡lo C1-6, arilo, arilalquilo C1-6, heterocicloalquilo C3-7, heteroc¡cloalqu¡l(C3-7)alqu¡lo C1-6, heteroarilo o heteroarilalquilo C1-6, cualquiera de cuyos grupos puede estar opcionalmente sustituido con uno, dos o tres sustituyentes seleccionados independientemente entre halógeno, ciano, nitro, alquilo C1-6, trifluorometilo, fenilo, fluorofenilo, hidroxi, hidroxialquilo C1-6, oxo, alcoxi C1-6, difluorometoxi, trifluorometoxi, alquil(C1-6)tío, alquil(C1-6)sulfinilo, alquil(C1-6)sulfonilo, amino, aminoalquilo C1-6, alquil(C1-6)amino, dialquil(C1-6)amino, pirrolidinilo, morfolinilo, piperazinilo, alquil(C2-6)carbonilamino, alquil(C2-6)carbonilaminoalquilo C 1-6 , alcoxi(C2-6)carbonilamino, alquil(C1-6)sulfonilamino, formilo, alquil(C2-6)carbonilo, carboxi, alcoxi(C2-6)carbonilo, aminocarbonilo, alquil(C1-6)aminocarbonilo, dialquil(C1-6)aminocarbonilo, aminosulfonilo, alquil(C1-6)aminosulfonilo y dialquil(C1-6)aminosulfonilo; y
R7 representa arilo, heteroarilo o espiro[heterocicloalquil(C3-7)][heteroarilo], cualquiera de cuyos grupos puede estar opcionalmente sustituido con uno, dos o tres sustituyentes seleccionados independientemente entre halógeno, ciano, nitro, alquilo C1-6, difluorometilo, trifluorometilo, fenilo, fluorofenilo, hidroxi, hidroxialquilo C1-6, oxo, alcoxi C1-6, difluorometoxi, trifluorometoxi, alquil(C1-6)tío, alquil(C1-6)sulfinilo, alquil(C1-6)sulfonilo, amino, aminoalquilo C1-6, alquil(C1-6)amino, dialquil(C1-6)amino, pirrolidinilo, morfolinilo, piperazinilo, alquil(C2-6)carbonilamino, alquil(C2-6)carbonilaminoalquilo C1-6, alcoxi(C2-6)carbonilamino, alquil(C1-6)sulfonilamino, formilo, alquil(C2-6)carbonilo, carboxi, alcoxi(C2-6)carbonilo, aminocarbonilo, alquil(C1-6)aminocarbonilo, dialquil(C1-6)aminocarbonilo, aminosulfonilo, alquil(C1-6)aminosulfonilo y dialquil(C1-6)aminosulfonilo.
La presente invención también da a conocer un compuesto de fórmula (I) como se describe anteriormente, o una de sus sales farmacéuticamente aceptables, para uso en terapia.
La presente invención también da a conocer un compuesto de fórmula (I) como se describe anteriormente, o una sal farmacéuticamente aceptable del mismo, para usar en el tratamiento y/o prevención de un trastorno inflamatorio o autoinmunitario.
Cuando se indica que cualquiera de los grupos en los compuestos de fórmula (I) anterior está opcionalmente sustituido, este grupo puede no estar sustituido o estar sustituido con uno o más sustituyentes. Típicamente, dichos grupos no estarán sustituidos o estarán sustituidos por uno, dos o tres sustituyentes. Idóneamente, dichos grupos no estarán sustituidos o estarán sustituidos por uno o dos sustituyentes.
Para su uso en la medicina, las sales de los compuestos de fórmula (I) serán sales farmacéuticamente aceptables. Sin embargo, otras sales pueden ser útiles en la preparación de los compuestos de fórmula (I) o de sus sales farmacéuticamente aceptables. Los principios estándares que subyacen a la selección y la preparación de las sales farmacéuticamente aceptables se describen, por ejemplo, en Handbook o f Pharmaceutical Salts: Properties, Selection and Use, ed. P. H. Stahl y C. G. Wermuth, Wiley-VCH, 2002. Las sales farmacéuticamente aceptables adecuadas de los compuestos de fórmula (I) incluyen las sales de adición de ácido que pueden, por ejemplo, formarse mezclando una solución de un compuesto de fórmula (I) con una solución de un ácido farmacéuticamente aceptable.
Con fines de referencia, la presente descripción también incluye dentro de su alcance los cocristales de los compuestos de la fórmula (I) anterior. El término técnico "cocristal" se utiliza para describir la situación en la que los componentes moleculares neutros están presentes dentro de un compuesto cristalino en una relación estequiométrica definida. La preparación de los cocristales farmacéuticos permite realizar modificaciones en la forma cristalina de un principio activo farmacéutico, que a su vez puede alterar sus propiedades fisicoquímicas sin comprometer su actividad biológica prevista (véase Pharmaceutical Salts and Co-crystals, ed. J. Wouters y L. Quere, RSC Publishing, 2012).
Los grupos alquilo adecuados que pueden estar presentes en los compuestos de uso en la invención incluyen grupos alquilo C1-6 de cadena lineal y ramificada, por ejemplo grupos alquilo C1-4. Los ejemplos típicos incluyen grupos metilo y etilo, y grupos propilo, butilo y pentilo de cadena lineal o ramificada. Los grupos alquilo concretos incluyen metilo, etilo, n-propilo, isopropilo, n-butilo, sec-butilo, isobutilo, tert-butilo, 2,2-dimetilpropilo y 3-metilbutilo. Las expresiones derivadas como "alcoxi C1-6", "alquil(C1-6)tío", "alquil(C1-6)sulfonilo" y "alquil(C1-6)amino" deben interpretarse en consecuencia.
Los grupos alquenilo adecuados que pueden estar presentes en los compuestos de uso en la invención incluyen grupos alquenilo C2-7 de cadena lineal y ramificada, por ejemplo grupos alquenilo C2-4. Los ejemplos típicos incluyen vinilo, alilo y butén-1-ilo.
El término "cicloalquilo C3-9", como se usa en la presente memoria, se refiere a grupos monovalentes de 3 a 9 átomos de carbono derivados de un hidrocarburo monocíclico saturado, y puede comprender análogos benzofusionados del mismo. Los grupos cicloalquilo C3-9 adecuados incluyen ciclopropilo, ciclobutilo, benzociclobutenilo, ciclopentilo, indanilo, ciclohexilo, tetrahidronaftalenilo, cicloheptilo, benzocicloheptenilo, ciclooctilo y ciclononanilo.
El término "cicloalquilidenilo C3-9", como se usa en la presente memoria, se refiere a grupos monovalentes de 3 a 9 átomos de carbono derivados de un hidrocarburo monocíclico saturado, que comprende opcionalmente análogos del mismo benzofusionados, unidos al resto de la molécula a través de un doble enlace C=C. Típicamente, tales grupos incluyen ciclobutilidenilo, ciclopentilidenilo, ciclohexilidenilo, cicloheptilidenilo, ciclooctilidenilo y ciclononanilidenilo.
El término "bicicloalquilo C4-9", tal como se usa en la presente memoria, se refiere a grupos monovalentes de 4 a 9
átomos de carbono derivados de un hidrocarburo bicíclico saturado. Los grupos bicicloalquilo típicos incluyen biciclo[1.1.1]pentanilo, biciclo[3.1.0]hexanilo, biciclo[4.1.0]heptanilo, biciclo[2.2.1]heptanilo, biciclo[2.2.2]octanilo, biciclo[3.3.0]octanilo y biciclo[3.2.1]octanilo.
El término "bicicloalquilidenilo C4-9", como se usa en la presente memoria, se refiere a grupos monovalentes de 4 a 9 átomos de carbono derivados de un hidrocarburo bicíclico saturado, unidos al resto de la molécula a través de un doble enlace C=C. Típicamente, tales grupos incluyen biciclo[3.1.0]hexanilidenilo, biciclo[2.2.1]heptanilidenilo y biciclo[3.2.1]octanilidenilo.
El término "espirocicloalquilo C5-9", como se usa en la presente memoria, se refiere a sistemas de anillos bicíclicos saturados que contienen de 5 a 9 átomos de carbono, en los que los dos anillos están unidos por un átomo común. Los grupos espirocicloalquilo adecuados incluyen espiro[2.3]hexanilo, espiro[2.4]heptanilo, espiro[3.3]heptanilo, espiro[3.4]octanilo, espiro[3.5]nonanilo y espiro[4.4]nonanilo.
El término tricicloalquilo C9-11”, como se usa en la presente memoria, se refiere a grupos monovalentes de 9 a 11 átomos de carbono derivados de un hidrocarburo tricíclico saturado. Los grupos tricicloalquilo típicos incluyen adamantanilo.
El término "arilo", como se usa en la presente memoria, se refiere a grupos aromáticos carbocíclicos monovalentes derivados de un solo anillo aromático o múltiples anillos aromáticos condensados. Los grupos arilo adecuados incluyen fenilo y naftilo, preferiblemente fenilo.
Los grupos de arilalquilo C1-6 adecuados incluyen bencilo, feniletilo, fenilpropilo y naftilmetilo.
El término "heterocicloalquilo C3-7", como se usa en la presente memoria, se refiere a anillos monocíclicos saturados que contienen de 3 a 7 átomos de carbono y al menos un heteroátomo seleccionado entre oxígeno, azufre y nitrógeno, y puede comprender análogos de los mismos benzofusionados. Los grupos heterocicloalquilo adecuados incluyen oxetanilo, azetidinilo, tetrahidrofuranilo, dihidrobenzofuranilo, dihidrobenzotienilo, pirrolidinilo, indolinilo, isoindolinilo, oxazolidinilo, tiazolidinilo, isotiazolidinilo, imidazolidinilo, tetrahidropiranilo, cromanilo, tetrahidrotiopiranilo, piperidinilo, 1,2,3,4-tetrahidroquinolinilo, 1,2,3,4-tetrahidroisoquinolinilo, piperazinilo, 1,2,3,4-tetrahidroquinoxalinilo, hexahidro-[1,2,5]tiadiazolo[2,3-a]pirazinilo, homopiperazinilo, morfolinilo, benzoxazinilo, tiomorfolinilo, azepanilo, oxazepanilo, diazepanilo, tiadiazepanilo y azocanilo.
El término "heterocicloalquilidenilo C3-7", como se usa en la presente memoria, se refiere a anillos monocíclicos saturados que contienen de 3 a 7 átomos de carbono y al menos un heteroátomo seleccionado de oxígeno, azufre y nitrógeno, unido al resto de la molécula a través de un doble enlace C=C. Típicamente, tales grupos incluyen tetrahidropiranilidenilo y piperidinilidenilo.
El término "heterobicicloalquilo C4-9", como se usa en la presente memoria, corresponde al bicicloalquilo C4-9 en el que uno o más de los átomos de carbono han sido reemplazados por uno o más heteroátomos seleccionados de oxígeno, azufre y nitrógeno. Los grupos heterobicicloalquilo típicos incluyen 6-oxabiciclo[3.1.0]hexanilo, 3-azabiciclo[3.1.0]hexanilo, 2-oxa-5-azabiciclo[2.2.1]heptanilo, 6-azabiciclo[3.2.0]heptanilo, 6-oxabiciclo[3.1.1]heptanilo, 3-azabiciclo[3.1.1]heptanilo, 3-azabiciclo[4.1.0]heptanilo, 2-oxabiciclo[2.2.2]octanilo, quinuclidinilo, 2-oxa-5-azabiciclo-[2.2.2]octanilo, 8-oxabiciclo[3.2.1]octanilo, 3-azabiciclo[3.2.1]octanilo, 8-azabiciclo-[3.2.1]octanilo, 3-oxa-8-azabiciclo[3.2.1]octanilo, 3,8-diazabiciclo[3.2.1]octanilo, 3,6-diazabiciclo[3.2.2]nonanilo, 3-oxa-7-azabiciclo[3.3.1]nonanilo, 3,7-dioxa-9-azabiciclo-[3.3.1]nonanilo y 3,9-diazabiciclo[4.2.1]nonanilo.
El término "heteroarilo", como se usa en la presente memoria, se refiere a grupos aromáticos monovalentes que contienen al menos 5 átomos derivados de un solo anillo o múltiples anillos condensados, en donde uno o más átomos de carbono han sido reemplazados por uno o más heteroátomos seleccionados de oxígeno, azufre y nitrógeno. Los grupos heteroarilo adecuados incluyen grupos furilo, benzofurilo, dibenzofurilo, tienilo, benzotienilo, tieno[2,3-c]pirazolilo, tieno[3,4-¿>][1,4]dioxinilo, dibenzotienilo, pirrolilo, indolilo, pirrolo[2,3-¿>]piridinilo, pirrolo[3,2-c]piridinilo, pirrolo[3,4-¿>]piridinilo, pirazolilo, pirazolo[1,5-a]piridinilo, pirazolo[3,4-d]pirimidinilo, pirazolo[1,5-a]pirazinilo, indazolilo, 4,5,6,7-tetrahidroindazolilo, oxazolilo, benzoxazolilo, isoxazolilo, tiazolilo, benzotiazolilo, isotiazolilo, imidazolilo, bencimidazolilo, imidazo-[2,1-¿>]tiazolilo, imidazo[1,2-a]piridinilo, imidazo[4,5-¿>]piridinilo, imidazo[1,2-¿>]-piridazinilo, purinilo, imidazo[1,2-a]pirimidinilo, imidazo[1,2-a]pirazinilo, oxadiazolilo, tiadiazolilo, triazolilo, [1,2,4]triazolo[1,5-a]pirimidinilo, benzotriazolilo, tetrazolilo, piridinilo, quinolinilo, isoquinolinilo, naftiridinilo, piridazinilo, cinnolinilo, ftalazinilo, pirimidinilo, quinazolinilo, pirazinilo, quinoxalinilo, pteridinilo, triazinilo y cromenilo.
El término "halógeno", como se usa en la presente memoria, pretende incluir átomos de flúor, cloro, bromo y yodo, típicamente flúor, cloro o bromo.
Cuando los compuestos de fórmula (I) tienen uno o más centros asimétricos, pueden existir como enantiómeros. Cuando los compuestos según la invención poseen dos o más centros asimétricos, pueden existir adicionalmente como diastereoisómeros. Debe entenderse que la invención se extiende al uso de todos estos enantiómeros y diastereoisómeros, y a sus mezclas en cualquier proporción, incluidos los racematos. La fórmula (I) y las fórmulas representadas a continuación pretenden representar todos los estereoisómeros por separado y todas las mezclas posibles de los mismos, a menos que se indique o muestre lo contrario. Además, los compuestos de fórmula (I) pueden
existir como tautómeros, por ejemplo, tautómeros ceto (CH2C=O) m enol (CH=CHOH) o tautómeros amida (NHC=O) m hidroxiimina (N=COH). La fórmula (I) y las fórmulas representadas a continuación pretenden representar todos los tautómeros por separado y todas las mezclas posibles de los mismos, a menos que se indique o muestre lo contrario. Debe entenderse que cada uno de los átomos presente en la fórmula (I), o en las fórmulas representadas a continuación, puede estar presente en forma de cualquiera de sus isótopos naturales, prefiriéndose los isótopos más abundantes. Así, a modo de ejemplo, cada uno de los átomos de hidrógeno presentes en la fórmula (I), o en las fórmulas que se describen a continuación, puede estar presente como un átomo de 1H, 2H (deuterio) o 3H (tritio), preferiblemente 1H. De manera similar, a modo de ejemplo, cada uno de los átomos de carbono presentes en la fórmula (I), o en las fórmulas que se describen a continuación, puede estar presente como un átomo de 12C, 13C o 14C, preferiblemente 12C.
En una primera realización, el anillo A representa cicloalquilo C3-9 opcionalmente sustituido. En un aspecto de esa realización, el anillo A representa cicloalquilo C4-7 opcionalmente sustituido.
En una segunda realización, el anillo A representa heterocicloalquilo C3-7 opcionalmente sustituido. En un aspecto de esa realización, el anillo A representa heterocicloalquilo C4-6 opcionalmente sustituido.
En una tercera realización, el anillo A representa heterobicicloalquilo C4-9 opcionalmente sustituido. En un aspecto de esa realización, el anillo A representa heterobicicloalquilo C5-7 opcionalmente sustituido.
Típicamente, el anillo A representa ciclopropilo, ciclobutilo, ciclopentilo, ciclohexilo, cicloheptilo, ciclooctilo, ciclononanilo, oxetanilo, azetidinilo, tetrahidrofuranilo, pirrolidinilo, oxazolidinilo, tiazolidinilo, isotiazolidinilo, imidazolidinilo, tetrahidropiranilo, tetrahidrotiopiranilo, piperidinilo, piperazinilo, homopiperazinilo, morfolinilo, tiomorfolinilo, azepanilo, oxazepanilo, diazepanilo, tiadiazepanilo, azocanilo, 6-oxabiciclo[3.1.0]hexanilo, 6-oxabiciclo[3.1.1]heptanilo u 8-oxabiciclo[3.2.1]octanilo, cualquiera de cuyos grupos puede estar opcionalmente sustituido por uno, dos o tres sustituyentes como se ha definido anteriormente.
Oportunamente, el anillo A representa pirrolidinilo, tetrahidropiranilo, tetrahidrotiopiranilo o piperidinilo, cualquiera de cuyos grupos puede estar opcionalmente sustituido con uno, dos o tres sustituyentes como se ha definido anteriormente.
Idóneamente, el anillo A representa tetrahidropiranilo, tetrahidrotiopiranilo o piperidinilo, cualquiera de cuyos grupos puede estar opcionalmente sustituido con uno, dos o tres sustituyentes como se ha definido anteriormente.
En una realización en concreto, el anillo A representa tetrahidropiranilo, cuyo grupo puede estar opcionalmente sustituido con uno, dos o tres sustituyentes como se ha definido anteriormente.
Los ejemplos adecuados de sustituyentes opcionales en el anillo A incluyen uno, dos o tres sustituyentes seleccionados independientemente de alquilo C1-6, oxo e imino.
Los ejemplos típicos de sustituyentes concretos en el anillo A incluyen uno, dos o tres sustituyentes seleccionados independientemente de metilo, fluoro, cloro, bromo, ciano, trifluorometilo, hidroxi, oxo, metoxi, metiltío, metilsulfinilo, metilsulfonilo, acetilo, amino, imino, metilamino y dimetilamino.
Los ejemplos adecuados de sustituyentes concretos en el anillo A incluyen uno, dos o tres sustituyentes seleccionados independientemente de metilo, oxo e imino.
Los valores seleccionados del anillo A incluyen pirrolidinilo, tetrahidropiranilo, (metil)-tetrahidropiranilo, tetrahidrotiopiranilo, (oxo)tetrahidrotiopiranilo, (dioxo)tetrahidrotiopiranilo, (imino)(oxo)tetrahidrotiopiranilo y piperidinilo.
Los valores típicos del anillo A incluyen tetrahidropiranilo, tetrahidrotiopiranilo y piperidinilo.
Un valor concreto del anillo A es tetrahidropiranilo.
En una realización, B representa C-R2. En otra realización, B representa N.
En una realización, D representa C-R3. En otra realización, D representa N.
En una realización, E representa C-R4. En otra realización, E representa N.
En una primera realización, B representa C-R2 , D representa C-R3 y E representa C-R4.
En una segunda realización, B representa C-R2 , D representa C-R3 y E representa N.
En una tercera realización, B representa C-R2 , D representa N y E representa C-R4.
En una cuarta realización, B representa C-R2 , D representa N y E representa N.
En una quinta realización, B representa N, D representa C-R3 y E representa C-R4.
En una sexta realización, B representa N, D representa C-R3 y E representa N.
En una séptima realización, B representa N, D representa N y E representa C-R4. En una octava realización, B representa N, D representa N y E representa N.
Idóneamente, la presente invención da a conocer un compuesto de fórmula (I-1), (I-2), (I-3), (I-4) o (I-5), o una sal farmacéuticamente aceptable del mismo:

(1-5)
en donde A, R0, R1, R2, R3 y R4 son como se definen más arriba.
En una primera realización, R0 representa hidrógeno. En una segunda realización, R0 representa alquilo C1-6 , especialmente metilo.
Idóneamente, R0 representa hidrógeno o metilo.
Típicamente, R2 representa hidrógeno o halógeno.
En una primera realización, R2 representa hidrógeno. En una segunda realización, R2 representa halógeno. En un primer aspecto de esa realización, R2 representa flúor. En un segundo aspecto de esa realización, R2 representa cloro. En una tercera realización, R2 representa ciano. En una cuarta realización, R2 representa alquilo C1-6 , especialmente metilo. En una quinta realización, R2 representa fluorometilo. En una sexta realización, R2 representa difluorometilo. En una séptima realización, R2 representa trifluorometilo. En una octava realización, R2 representa hidroxi. En una novena realización, R2 representa alcoxi C1-6 , especialmente metoxi. En una décima realización, R2 representa difluorometoxi. En una undécima realización, R2 representa trifluorometoxi. En una duodécima realización, R2 representa alquil(C1-6)sulfinilo, especialmente metilsulfinilo. En una decimotercera realización, R2 representa alquil(C1-6)sulfonilo, especialmente metilsulfonilo.
Idóneamente, R2 representa hidrógeno o flúor.
Típicamente, R3 representa hidrógeno o halógeno.
En una primera realización, R3 representa hidrógeno. En una segunda realización, R3 representa halógeno. En un primer aspecto de esta realización, R3 representa flúor. En un segundo aspecto de esta realización, R3 representa cloro. En una tercera realización, R3 representa ciano. En una cuarta realización, R3 representa alquilo C1-6, especialmente metilo. En una quinta realización, R3 representa fluorometilo. En una sexta realización, R3 representa difluorometilo. En una séptima realización, R3 representa trifluorometilo. En una octava realización, R3 representa hidroxi. En una novena realización, R3 representa alcoxi C1-6, especialmente metoxi. En una décima realización, R3 representa difluorometoxi. En una undécima realización, R3 representa trifluorometoxi. En una duodécima realización, R3 representa alquil(C1-6)sulfinilo, especialmente metilsulfinilo. En una decimotercera realización, R3 representa alquil(C1-6)sulfonilo, especialmente metilsulfonilo.
Oportunamente, R3 representa hidrógeno, flúor o cloro.
Idóneamente, R3 representa hidrógeno o flúor.
En una primera realización, R4 representa hidrógeno. En una segunda realización, R4 representa halógeno. En un primer aspecto de esta realización, R4 representa flúor. En un segundo aspecto de esta realización, R4 representa cloro. En una tercera realización, R4 representa ciano. En una cuarta realización, R4 representa alquilo C1-6, especialmente metilo. En una quinta realización, R4 representa fluorometilo. En una sexta realización, R4 representa difluorometilo. En una séptima realización, R4 representa trifluorometilo. En una octava realización, R4 representa hidroxi. En una novena realización, R4 representa alcoxi C1-6, especialmente metoxi. En una décima realización, R4 representa difluorometoxi. En una undécima realización, R4 representa trifluorometoxi. En una duodécima realización, R4 representa alquil(C1-6)sulfinilo, especialmente metilsulfinilo. En una decimotercera realización, R4 representa alquil(C1-6)sulfonilo, especialmente metilsulfonilo.
Una subclase concreta de compuestos según la invención está representada por los compuestos de fórmula (IA) y sus sales farmacéuticamente aceptables:

en donde
A, B, D, E, R0, R5 y R6 son como se definen más arriba.
Una segunda subclase de compuestos según la invención está representada por los compuestos de fórmula (IB) y sus sales farmacéuticamente aceptables:
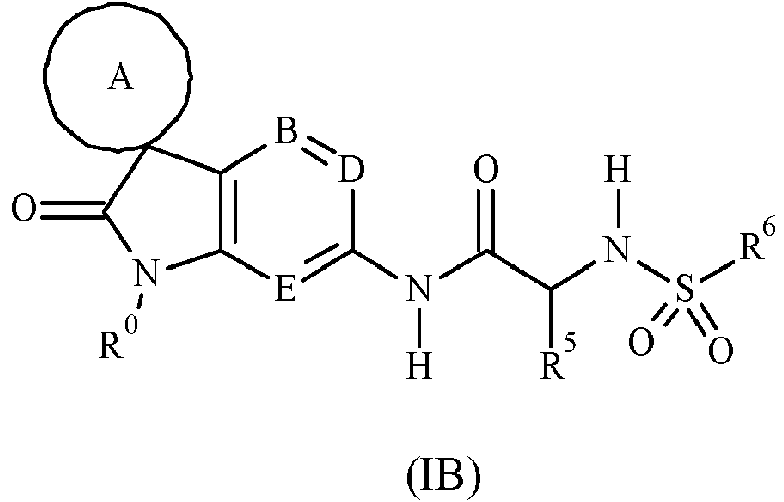
en donde
A, B, D, E, R0, R5 y R6 son como se definen más arriba.
Una tercera subclase de compuestos según la invención está representada por los compuestos de fórmula (IC) y sus sales farmacéuticamente aceptables:

en donde
A, B, D, E, R0, R5 y R7 son como se definen más arriba.
Una cuarta subclase de compuestos según la invención está representada por los compuestos de fórmula (ID) y sus sales farmacéuticamente aceptables:

en donde
A, B, D, E, R0, R5 y R7 son como se definen más arriba.
Una quinta subclase de compuestos según la invención está representada por los compuestos de fórmula (IE) y sus sales farmacéuticamente aceptables:

en donde
A, B, D, E, R0, R5 y R7 son como se definen más arriba.
Una sexta subclase de compuestos según la invención está representada por los compuestos de fórmula (IF) y sus sales farmacéuticamente aceptables:

en donde
A, B, D, E, R0 , R5a, R5b y R6 son como se definen más arriba.
Generalmente, R5 representa hidrógeno; o R5 representa alquilo C1-5, cicloalquilo C3-9, cicloalquil(C3-9)alquilo C1-5, bicicloalquilo C4-9, espirocicloalquilo C5-9, espirocicloalquil(C5-9)alquilo C1-5, tricicloalquilo C9-11, tricicloalquil(C9-11)alquilo C1-5, arilo, arilalquilo C1-5, heterocicloalquilo C3-7, heterocicloalquil(C3-7)alquilo C1-5, heteroarilo o heteroarilalquilo C1-5, cualquiera de cuyos grupos puede estar opcionalmente sustituido con uno, dos o tres sustituyentes como se ha definido anteriormente.
Ventajosamente, R5 representa hidrógeno; o R5 representa alquilo C1-5, cicloalquilo C3-9, cicloalquil(C3-9)alquilo C1-5, bicicloalquilo C4-9, bicicloalquil(C4-9)alquilo C1-5, espirocicloalquilo C5-9, tricicloalquilo C9-11, tricicloalquil(C9- n )alquilo C1-5 , arilo, arilalquilo C1-5, heterocicloalquilo C3-7, heterocicloalquil(C3-7)alquilo C1-5 o heteroarilalquilo C1-5, cualquiera de cuyos grupos puede estar opcionalmente sustituido con uno, dos o tres sustituyentes como se ha definido anteriormente.
Típicamente, R5 representa alquilo C1-5, cicloalquilo C3-9, cicloalquil(C3-9)alquilo C1-5, bicicloalquilo C4-9, espirocicloalquilo C5-9, espirocicloalquil(C5-9)alquilo C1-5, tricicloalquilo C9-11, tricicloalquil(C9- n )alquilo C1-5, arilo, arilalquilo C1-5, heterocicloalquilo C3-7, heterocicloalquil(C3-7)alquilo C1-5, heteroarilo o heteroarilalquilo C1-5, cualquiera de cuyos grupos puede estar opcionalmente sustituido con uno, dos o tres sustituyentes como se ha definido anteriormente. Además, R5 puede representar bicicloalquil(C4-9)alquilo C1-5, cuyo grupo puede estar opcionalmente sustituido con uno, dos o tres sustituyentes como se ha definido anteriormente.
Idóneamente, R5 representa hidrógeno; o R5 representa alquilo C1-5, cicloalquilo C3-9, cicloalquil(C3-9)alquilo C1-5, bicicloalquilo C4-9, espirocicloalquilo C5-9, espirocicloalquil(C5-9)alquilo C1-5, tricicloalquilo C9-11, tricicloalquil(C9-n)alquilo C1-5, arilo, arilalquilo C1-5 o heteroarilalquilo C1-5, cualquiera de cuyos grupos puede estar opcionalmente sustituido con uno, dos o tres sustituyentes como se ha definido anteriormente.
Oportunamente, R5 representa hidrógeno; o R5 representa cicloalquilo C3-9, espirocicloalquilo C5-9 o arilalquilo C1-5, cualquiera de cuyos grupos puede estar opcionalmente sustituido con uno, dos o tres sustituyentes como se ha definido anteriormente.
En una primera realización, R5 representa hidrógeno. En una segunda realización, R5 representa alquilo C1-5 opcionalmente sustituido. En una tercera realización, R5 representa cicloalquilo C3-9 opcionalmente sustituido. En una cuarta realización, R5 representa cicloalquil(C3-9)alquilo C1-5 opcionalmente sustituido. En una quinta realización, R5 representa bicicloalquilo C4-9 opcionalmente sustituido. En una sexta realización, R5 representa bicicloalquil(C4-9)alquilo C1-5 opcionalmente sustituido. En una séptima realización, R5 representa espirocicloalquilo C5-9 opcionalmente sustituido. En una octava realización, R5 representa espirocicloalquil(C5-9)alquilo C1-5 opcionalmente sustituido. En una novena realización, R5 representa tricicloalquilo C9-11 opcionalmente sustituido. En una décima realización, R5 representa tricicloalquil(C9- n )alquilo C1-5 opcionalmente sustituido. En una undécima realización, R5 representa arilo opcionalmente sustituido. En una duodécima realización, R5 representa arilalquilo C1-5 opcionalmente sustituido. En una decimotercera realización, R5 representa heterocicloalquilo C3-7 opcionalmente sustituido. En una decimocuarta realización, R5 representa heterocicloalquil(C3-7)alquilo C1-5 opcionalmente sustituido. En una decimoquinta realización, R5 representa heteroarilo opcionalmente sustituido. En una decimosexta realización, R5 representa heteroarilalquilo C1-5 opcionalmente sustituido.
En una realización concreta, R5 es diferente al hidrógeno.
Los valores típicos de R5 incluyen metilo, isopropilo, 1 -metilpropilo, 2-metilpropilo, ciclopropilo, ciclopentilo, indanilo, ciclohexilo, ciclooctilo, ciclohexilmetilo, espiro[3.3]-heptanilo, espiro[3.3]heptanilmetilo, adamantanilo, adamantanilmetilo, fenilo, bencilo, feniletilo, naftilmetilo, tienilo, indolilo, piridinilo, tienilmetilo, indolilmetilo y piridinilmetilo, cualquiera de cuyos grupos puede estar opcionalmente sustituido con uno, dos o tres sustituyentes como se ha definido anteriormente. Los valores adicionales incluyen ciclobutilo, benzociclobutenilo, tetrahidronaftalenilo, cicloheptilo, benzocicloheptenilo, ciclononanilo, ciclobutilmetilo, ciclobutiletilo, biciclo[3.1.0]hexanilo, biciclo[2.2.1]heptanilo, biciclo[3.3.0]-octanilo, biciclo[3.2.1]octanilo, biciclo[1.1.1]pentanilmetilo,
fenilpropilo, tetrahidropiranilo, azocanilo, dihidrobenzofuranilmetilo y pirroliletilo, cualquiera de cuyos grupos puede estar opcionalmente sustituido con uno, dos o tres sustituyentes como se ha definido anteriormente.
Los valores seleccionados de R5 incluyen metilo, ciclobutilo, benzociclobutenilo, ciclopentilo, indanilo, ciclohexilo, tetrahidronaftalenilo, cicloheptilo, benzocicloheptenilo, ciclooctilo, ciclononanilo, ciclobutilmetilo, ciclobutiletilo, biciclo[3.1.0]hexanilo, biciclo[2.2.1]heptanilo, biciclo[3.3.0]octanilo, biciclo[3.2.1]octanilo, biciclo[1.1.1]pentanilmetilo, espiro[3.3]heptanilo, adamantanilo, adamantanilmetilo, fenilo, bencilo, feniletilo, fenilpropilo, tetrahidropiranilo, azocanilo, dihidrobenzofuranilmetilo y pirroliletilo, cualquiera de los cuales puede ser opcionalmente sustituido con uno, dos o tres sustituyentes como se define anteriormente.
Los valores adecuados de R5 incluyen ciclohexilo, ciclooctilo, espiro[3.3]heptanilo y bencilo, cualquiera de cuyos grupos puede estar opcionalmente sustituido con uno, dos o tres sustituyentes como se ha definido anteriormente.
Los ejemplos adecuados de sustituyentes opcionales en R5 incluyen uno, dos o tres sustituyentes seleccionados independientemente de halógeno, ciano, alquilo C 1-6, trifluorometilo, fenilo, hidroxi, alcoxi C1-6 y aminocarbonilo, especialmente halógeno.
Los ejemplos favoritos de sustituyentes opcionales en R5 incluyen uno, dos o tres sustituyentes seleccionados independientemente de halógeno, alquilo C1-6, trifluorometilo, fenilo y alcoxi C1-6, especialmente halógeno.
Los ejemplos típicos de sustituyentes específicos en R5 incluyen uno, dos o tres sustituyentes seleccionados independientemente de flúor, cloro, bromo, ciano, nitro, metilo, etilo, isopropilo, ferf-butilo, trifluorometilo, fenilo, hidroxi, oxo, metoxi, ferf-butoxi, difluorometoxi, trifluorometoxi, metiltío, metilsulfinilo, metilsulfonilo, amino, metilamino, ferfbutilamino, dimetilamino, acetilamino, metoxicarbonilamino, metilsulfonilamino, formilo, acetilo, carboxi, metoxicarbonilo, etoxicarbonilo, ferf-butoxicarbonilo, aminocarbonilo, metilaminocarbonilo, dimetilaminocarbonilo, aminosulfonilo, metilaminosulfonilo y dimetilaminosulfonilo. Los ejemplos adicionales incluyen trifluoroetilo e isopropoxi.
Los ejemplos adecuados de sustituyentes específicos en R5 incluyen uno, dos o tres sustituyentes seleccionados independientemente de flúor, cloro, bromo, ciano, metilo, trifluorometilo, fenilo, hidroxi, metoxi, ferf-butoxi y aminocarbonilo, especialmente cloro. Los ejemplos adicionales incluyen isopropoxi.
Los ejemplos favoritos de sustituyentes específicos en R5 incluyen uno, dos o tres sustituyentes seleccionados independientemente de fluoro, cloro, bromo, metilo, trifluorometilo, fenilo, isopropoxi y ferf-butoxi, especialmente cloro.
Los valores ilustrativos de R5 incluyen hidrógeno, metilo, isopropilo, 1 -metilpropilo, 2-metilpropilo, ciclopropilo, ciclopentilo, indanilo, ciclohexilo, ciclooctilo, ciclohexilmetilo, espiro[3.3]heptanilo, fenilo, clorofenilo, bencilo, fluorobencilo, clorobencilo, (cloro)(fluoro)bencilo, diclorobencilo, bromobencilo, cianobencilo, metilbencilo, trifluorometilbencilo, fenilbencilo, hidroxibencilo, metoxibencilo, ferf-butoxibencilo, aminocarbonilbencilo, feniletilo, clorofeniletilo, naftilmetilo, tienilmetilo, indolilmetilo y piridinilmetilo. Los valores adicionales incluyen ferf-butoximetilo, ciclobutilo, metilciclobutilo, dimetilciclobutilo, fenilciclobutilo, benzociclobutenilo, metilciclopentilo, difluorociclohexilo, metilciclohexilo, dimetilciclohexilo, trifluorometilciclohexilo, tetrahidronaftalenilo, cicloheptilo, benzocicloheptenilo, ciclononanilo, ciclobutilmetilo, difluorociclobutilmetilo, dimetilciclobutilmetilo, ciclobutiletilo, biciclo[3.1.0]hexanilo, biciclo[2.2.1]heptanilo, biciclo[3.3.0]octanilo, biciclo[3.2.1]octanilo, biciclo[1.1.1 ]pentanilmetilo, adamantanilo, adamantanilmetilo, (cloro)(fluoro)fenilo, (fluoro)(metil)fenilo, (bromo)(cloro)bencilo, (cloro)(isopropoxi)bencilo, fenilpropilo, tetrahidropiranilo, tetrametiltetrahidropiranilo, azocanilo, dihidrobenzofuranilmetilo y metilpirroliletilo.
Los valores seleccionados de R5 incluyen hidrógeno, ferf-butoximetilo, ciclobutilo, metilciclobutilo, dimetilciclobutilo, fenilciclobutilo, benzociclobutenilo, ciclopentilo, metilciclopentilo, indanilo, ciclohexilo, difluorociclohexilo, metilciclohexilo, dimetilciclohexilo, trifluorometilciclohexilo, tetrahidronaftalenilo, cicloheptilo, benzocicloheptenilo, ciclooctilo, ciclononanilo, ciclobutilmetilo, difluorociclobutilmetilo, dimetilciclobutilmetilo, ciclobutiletilo, biciclo[3.1.0]hexanilo, biciclo[2.2.1]heptanilo, biciclo[3.3.0]octanilo, biciclo[3.2.1]octanilo, biciclo[1.1.1]pentanilmetilo, espiro[3.3]heptanilo, adamantanilo, adamantanilmetilo, (cloro)(fluoro)fenilo, (fluoro)(metil)fenilo, fluorobencilo, clorobencilo, (cloro)(fluoro)bencilo, (bromo)(cloro)bencilo, (cloro)(isopropoxi)bencilo, feniletilo, clorofeniletilo, fenilpropilo, tetrahidropiranilo, tetrametiltetrahidropiranilo, azocanilo, dihidrobenzofuranilmetilo y metilpirroliletilo.
Los valores representativos de R5 incluyen hidrógeno, ciclohexilo, ciclooctilo, espiro[3.3]heptanilo y clorobencilo.
Los valores favoritos de R5 incluyen ciclohexilo, 4-metilciclohexilo y ciclooctilo. En una primera realización, R5 representa ciclohexilo. En una segunda realización, R5 representa 4-metilciclohexilo. En una tercera realización, R5 representa ciclooctilo.
En una primera realización, R5a representa cicloalquilo C3-7 opcionalmente sustituido. En una segunda realización, R5a representa bicicloalquilo C4-9 opcionalmente sustituido. En una tercera realización, R5a representa arilo opcionalmente sustituido. En una cuarta realización, R5a representa heterocicloalquilo C3-7 opcionalmente sustituido. En una quinta realización, R5a representa heteroarilo opcionalmente sustituido.
Los valores típicos de R5a incluyen ciclobutilo, ciclopentilo, biciclo[1.1.1]pentanilo, fenilo, dihidrobenzofuranilo y pirrolilo,
cualquiera de cuyos grupos puede estar opcionalmente sustituido con uno o más sustituyentes como se define anteriormente.
Los ejemplos seleccionados de sustituyentes opcionales en R5a incluyen alquilo C1-6 y halógeno.
Los ejemplos típicos de sustituyentes concretos en R5a incluyen metilo, fluoro, cloro, bromo, ciano, trifluorometilo, trifluoroetilo, fenilo, hidroxi, metoxi, metiltío, metilsulfinilo, metilsulfonilo, acetilo, amino, metilamino y dimetilamino. Los ejemplos seleccionados de sustituyentes concretos en R5a incluyen metilo y cloro.
Los valores seleccionados de R5a incluyen ciclobutilo, ciclopentilo, biciclo[1.1.1]pentanilo, fenilo, clorofenilo, dihidrobenzofuranilo y metilpirrolilo.
Idóneamente, R5b representa hidrógeno, metilo o etilo.
En una primera realización, R5b representa hidrógeno. En una segunda realización, R5b representa alquilo C1-6, especialmente metilo o etilo.
Como alternativa, R5a y R5b, cuando se toman junto con el átomo de carbono al que ambos están unidos, pueden representar cicloalquilo C3-7, bicicloalquilo C4-9 o heterocicloalquilo C3-7, cualquiera de cuyos grupos puede estar sin sustituir o sustituido con uno o dos sustituyentes como se ha definido anteriormente.
En una primera realización, R5a y R5b, cuando se toman junto con el átomo de carbono al que ambos están unidos, pueden representar idóneamente cicloalquilo C3-7 opcionalmente sustituido. Los ejemplos incluyen ciclobutilo, benzociclobutenilo, ciclopentilo, indanilo, ciclohexilo, tetrahidronaftalenilo, cicloheptanilo, benzocicloheptenilo, ciclooctanilo y ciclononanilo, cualquiera de cuyos grupos puede estar opcionalmente sustituido con uno o dos sustituyentes como se define anteriormente.
En una segunda realización, R5a y R5b, cuando se toman junto con el átomo de carbono al que ambos están unidos, pueden representar idóneamente bicicloalquilo C4-9 opcionalmente sustituido. Los ejemplos incluyen biciclo[3.1.0]hexanilo, biciclo[2.2.1]heptanilo y biciclo[3.2.1]octanilo, cualquiera de cuyos grupos puede estar opcionalmente sustituido con uno o dos sustituyentes como se definió anteriormente.
En una tercera realización, R5a y R5b, cuando se toman junto con el átomo de carbono al que ambos están unidos, pueden representar idóneamente heterocicloalquilo C3-7 opcionalmente sustituido. Los ejemplos incluyen tetrahidropiranilo y piperidinilo, cualquiera de los cuales puede estar opcionalmente sustituido con uno o dos sustituyentes como se definió anteriormente.
Los ejemplos seleccionados de sustituyentes opcionales en tales grupos incluyen alquilo C1-6, halógeno, trifluorometilo, trifluoroetilo, fenilo y alcoxi C1-6.
Los ejemplos típicos de sustituyentes concretos en dichos grupos incluyen metilo, fluoro, cloro, bromo, ciano, trifluorometilo, trifluoroetilo, fenilo, hidroxi, metoxi, metiltío, metilsulfinilo, metilsulfonilo, acetilo, amino, metilamino y dimetilamino.
Los ejemplos seleccionados de sustituyentes concretos en dichos grupos incluyen metilo, cloro, trifluorometilo, trifluoroetilo, fenilo y metoxi.
Los valores seleccionados de R5a y R5b, cuando se toman junto con el átomo de carbono al que ambos están unidos, incluyen metilciclobutilo, dimetilciclobutilo, fenilciclobutilo, benzociclobutenilo, metilbenzociclobutenilo, clorobenzociclobutenilo, metoxibenzociclobutenilo, ciclopentilo, metilciclopentilo, indanilo, cloroindanilo, ciclohexilo, metilciclohexilo, dimetilciclohexilo, trifluorometilciclohexilo, tetrahidronaftalenilo, cicloheptanilo, benzocicloheptenilo, ciclooctanilo, ciclononanilo, biciclo[3.1.0]hexanilo, biciclo[2.2.1]heptanilo, biciclo[3.2.1]octanilo, tetrametiltetrahidropiranilo y trifluoroetilpiperidinilo.
Generalmente, R6 representa -NR6aR6b; o R6 representa alquilo C1-6, cicloalquilo C3-9, cicloalquil(C3-9)alquilo C1-6, arilo, arilalquilo C1-6, heterocicloalquilo C3-7, heterocicloalquil(C3-7)alquilo C1-6, heteroarilo o heteroarilalquilo C1-6, cualquiera de cuyos grupos puede estar opcionalmente sustituido con uno, dos o tres sustituyentes como se ha definido anteriormente.
Ventajosamente, R6 representa -NR6aR6b u -OR6c; o R6 representa alquilo C1-9, arilo, heterocicloalquilo C3-7, heteroarilo, heteroarilalquilo C1-6 o espiro[heterocicloalquil(C3-7)][heteroarilo], cualquiera de cuyos grupos puede estar opcionalmente sustituido con uno, dos o tres sustituyentes como se ha definido anteriormente.
Típicamente, R6 representa -NR6aR6b; o R6 representa alquilo C1-6, cicloalquilo C3-9, cicloalquil(C3-9)alquilo C1-6, arilo, arilalquilo C1-6, heteroarilo o heteroarilalquilo C1-6, cualquiera de cuyos grupos puede estar opcionalmente sustituido con uno, dos o tres sustituyentes como se ha definido anteriormente. Además, R6 puede representar -OR6c; o R6 puede representar arilo, heterocicloalquilo C3-7 o espiro[heterocicloalquil(C3-7)][heteroarilo], cualquiera de cuyos grupos puede estar opcionalmente sustituido con uno, dos o tres sustituyentes como se ha definido anteriormente.
Idóneamente, R6 representa alquilo C i -6, heteroarilo o heteroarilalquilo C i -6, cualquiera de cuyos grupos puede estar opcionalmente sustituido con uno, dos o tres sustituyentes como se ha definido anteriormente.
En una primera realización, R6 representa alquilo C1-6 opcionalmente sustituido En una segunda realización, R6 representa cicloalquilo C3-9 opcionalmente sustituido. En una tercera realización, R6 representa cicloalquil(C3-9)alquilo C1-6 opcionalmente sustituido. En una cuarta realización, R6 representa arilo opcionalmente sustituido. En una quinta realización, R6 representa arilalquilo C1-6 opcionalmente sustituido. En una sexta realización, R6 representa heterocicloalquilo C3-7 opcionalmente sustituido. En una séptima realización, R6 representa heterocicloalquil(C3-7)alquilo C1-6 opcionalmente sustituido. En una octava realización, R6 representa heteroarilo opcionalmente sustituido. En una novena realización, R6 representa heteroarilalquilo C1-6 opcionalmente sustituido. En una décima realización, R6 representa espiro[heterocicloalquil(C3-7)][heteroarilo] opcionalmente sustituido. En una undécima realización, R6 representa -NR6aR6b. En una duodécima realización, R6 representa -OR6c.
Los valores típicos de R6 incluyen -NR6aR6b; y metilo, etilo, propilo, 2-metilpropilo, butilo, ciclopropilo, ciclobutilo, ciclohexilo, ciclohexilmetilo, fenilo, bencilo, feniletilo, pirazolilo, piridinilo, triazolilmetilo, benzotriazolilmetilo o piridinilmetilo, cualquiera de cuyos grupos puede estar opcionalmente sustituido por uno, dos o tres sustituyentes como se definió anteriormente. Los valores adicionales incluyen -OR6c; y ferf-butilo, heptanilo, pirrolidinilo, indolinilo, piperidinilo, morfolinilo, tiomorfolinilo, piperazinilo, pirrolilo, pirazolo[1,5-a]piridinilo, 4,5,6,7-tetrahidropirazolilo, oxazolilo, isoxazolilo, tiazolilo, isotiazolilo, imidazolilo, oxadiazolilo, tiadiazolilo, triazolilo, tetrazolilo, piridazinilo, pirimidinilo o espiro[tetrahidrofurán][indol], cualquiera de los cuales puede estar opcionalmente sustituido por uno, dos o tres sustituyentes como se define anteriormente.
Los valores seleccionados de R6 incluyen -NR6aR6b y -OR6c; y metilo, ferf-butilo, heptanilo, fenilo, pirrolidinilo, indolinilo, piperidinilo, morfolinilo, tiomorfolinilo, piperazinilo, pirrolilo, pirazolilo, pirazolo[1,5-a]piridinilo, 4,5,6,7-tetrahidropirazolilo, oxazolilo, isoxazolilo, tiazolilo, isotiazolilo, imidazolilo, oxadiazolilo, tiadiazolilo, triazolilo, tetrazolilo, piridinilo, piridazinilo, pirimidinilo, piridinilmetilo o espiro[tetrahidrofurán][indol], cualquiera de cuyos grupos pueden estar opcionalmente sustituidos con uno, dos o tres sustituyentes como se definió anteriormente.
Los valores adecuados de R6 incluyen metilo, pirazolilo y piridinilmetilo, cualquiera de cuyos grupos puede estar opcionalmente sustituido con uno, dos o tres sustituyentes como se definió anteriormente.
Los ejemplos seleccionados de sustituyentes opcionales en R6 incluyen uno, dos o tres sustituyentes seleccionados independientemente de halógeno, alquilo C1-6, difluorometilo, trifluorometilo, difluoroetilo, trifluoroetilo, trifluoropropilo, ciclopropilo, ciclobutilo, ciclopropilmetilo, hidroxi, hidroxialquilo C1-6, oxo, alcoxi(C1-6)alquilo C1-6, alquil(C1-6)sulfonilo, amino, aminoalquilo C1-6, dialquil(C1-6)aminoalquilo C1-6 y tetrahidropiranilo.
Los ejemplos adecuados de los sustituyentes opcionales en R6 incluyen uno, dos o tres sustituyentes seleccionados independientemente de halógeno, ciano, alquilo C1-6, trifluorometilo, fenilo, fluorofenilo, hidroxi, hidroxialquilo C1-6, oxo, alcoxi C1-6, trifluorometoxi, alquil(C1-6)sulfonilo, amino, aminoalquilo C1-6, alquil(C1-6)amino, dialquil(C1-6)amino, pirrolidinilo, morfolinilo, piperazinilo, alquil(C2-6)carbonilaminoalquilo C1-6 y carboxi.
Los ejemplos ilustrativos de sustituyentes opcionales en R6 incluyen uno, dos o tres sustituyentes seleccionados independientemente de alquilo C1-6 y amino.
Los ejemplos típicos de sustituyentes específicos en R6 incluyen uno, dos o tres sustituyentes seleccionados independientemente de flúor, cloro, bromo, ciano, nitro, metilo, etilo, isopropilo, ferf-butilo, trifluorometilo, fenilo, fluorofenilo, hidroxi, hidroximetilo, oxo, metoxi, ferf-butoxi, difluorometoxi, trifluorometoxi, metiltío, metilsulfinilo, metilsulfonilo, amino, aminometilo, aminoetilo, metilamino, ferf-butilamino, dimetilamino, pirrolidinilo, morfolinilo, piperazinilo, acetilamino, acetilaminoetilo, metoxicarbonilamino, metilsulfonilamino, formilo, acetilo, carboxi, metoxicarbonilo, etoxicarbonilo, ferf-butoxicarbonilo, aminocarbonilo, metilaminocarbonilo, dimetilaminocarbonilo, aminosulfonilo, metilaminosulfonilo y dimetilaminosulfonilo. Los ejemplos adicionales incluyen n-propilo, 2-metilpropilo, bután-2-ilo, difluorometilo, difluoroetilo, trifluoroetilo, trifluoropropilo, ciclopropilo, ciclobutilo, ciclopropilmetilo, hidroxietilo, metoximetilo, metoxietilo, aminoisopropilo, dimetilaminoetilo, tetrahidropiranilo.
Los ejemplos seleccionados de sustituyentes específicos en R6 incluyen uno, dos o tres sustituyentes seleccionados independientemente de flúor, metilo, etilo, n-propilo, isopropilo, 2-metilpropilo, bután-2-ilo, ferf-butilo, difluorometilo, trifluorometilo, difluoroetilo, trifluoroetilo, trifluoropropilo, ciclopropilo, ciclobutilo, ciclopropilmetilo, hidroxi, hidroxietilo, oxo, metoximetilo, metoxietilo, metilsulfonilo, amino, aminometilo, aminoisopropilo, dimetilaminoetilo y tetrahidropiranilo.
Los ejemplos adecuados de sustituyentes específicos en R6 incluyen uno, dos o tres sustituyentes seleccionados independientemente de fluoro, cloro, ciano, metilo, etilo, trifluorometilo, fenilo, fluorofenilo, hidroxi, hidroximetilo, oxo, metoxi, trifluorometoxi, metilsulfonilo, amino, aminometilo, aminoetilo, metilamino, dimetilamino, pirrolidinilo, morfolinilo, piperazinilo, acetilaminoetilo y carboxi.
Los ejemplos ilustrativos de sustituyentes específicos en R6 incluyen uno, dos o tres sustituyentes seleccionados independientemente de metilo y amino.
Los valores ilustrativos de R6 incluyen metilo, difluorometilo, metilsulfonilmetilo, aminometilo, metilaminometilo, difluoroetilo, carboxietilo, difluoropropilo, 2-metilpropilo, butilo, cianociclopropilo, metilciclopropilo, etilciclopropilo, dimetilciclopropilo, trifluorometilciclopropilo, fenilciclopropilo, fluorofenilciclopropilo, hidroxiciclopropilo, aminociclopropilo, ciclobutilo, trifluorometilciclobutilo, ciclohexilo, ciclohexilmetilo, fenilo, fluorofenilo, clorofenilo, cianofenilo, metilfenilo, hidroxifenilo, metilsulfonilfenilo, bencilo, fluorobencilo, difluorobencilo, clorobencilo, (cloro)(fluoro)bencilo, diclorobencilo, (cloro)(difluoro)bencilo, bromobencilo, cianobencilo, metilbencilo, dimetilbencilo, trifluorometilbencilo, fenilbencilo, hidroxibencilo, hidroximetilbencilo, benzoílo, metoxibencilo, dimetoxibencilo, trifluorometoxibencilo, metilsulfonilbencilo, aminometilbencilo, aminoetilbencilo, dimetilaminobencilo, pirrolidinilbencilo, (dimetil)(pirrolidinil)bencilo, morfolinilbencilo, (dimetil)(morfolinil)bencilo, piperazinilbencilo, acetilaminoetilbencilo, feniletilo, clorofeniletilo, metilpirazolilo, piridinilo, triazolilmetilo, benzotriazolilmetilo, piridinilmetilo y aminopiridinilmetilo. Otros valores más incluyen -NR6aR6b, -OR6c, ferf-butilo, hidroxiheptanilo, pirrolidinilo, metilpirrolidinilo, indolinilo, piperidinilo, morfolinilo, dioxotiomorfolinilo, metilpiperazinilo, metilpirrolilo, dimetilpirazolilo, etilpirazolilo, (etil)(fluoro)pirazolilo, (etil)(metil)pirazolilo, n-propilpirazolilo, isopropilpirazolilo, 2-metilpropilpirazolilo, bután-2-ilpirazolilo, difluorometilpirazolilo, (difluorometil)(metil)pirazolilo, difluoroetilpirazolilo, trifluoroetilpirazolilo, trifluoropropilpirazolilo, ciclopropilpirazolilo, ciclobutilpirazolilo, ciclopropilmetilpirazolilo, hidroxietilpirazolilo, metoxietilpirazolilo, dimetilaminoetilpirazolilo, tetrahidropiranilpirazolilo, (metil)(tetrahidropiranil)pirazolilo, pirazolo[1,5-a]piridinilo, metil-4,5,6,7-tetrahidropirazolilo, oxazolilo, metiloxazolilo, etiloxazolilo, isoxazolilo, metilisoxazolilo, dimetilisoxazolilo, etilisoxazolilo, isopropilisoxazolilo, ferf-butilisozolilo, trifluorometillisoxazolilo, ciclopropilisoxazolilo, ciclobutilisoxazolilo, trifluorometilisoxazolilo, ciclopropilisoxazolilo, ciclobutilisoxazolilo, metoximetilisoxazolilo, aminometilisoxazolilo, aminoisopropilisoxazolilo, tiazolilo, metiltiazolilo, dimetiltiazolilo, isotiazolilo, metilisotiazolilo, metilimidazolilo, metiloxadiazolilo, metiltiadiazolilo, metiltriazolilo, dimetiltriazolilo, etiltriazolilo, metiltetrazolilo, metilpiridinilo, piridazinilo, pirimidinilo, metilpirimidinilo y espiro[tetrahidrofurán][oxoindol].
Los valores seleccionados de R6 incluyen -NR6aR6b, -OR6c, metilo, ferf-butilo, hidroxiheptanilo, fenilo, fluorofenilo, metilsulfonilfenilo, pirrolidinilo, metilpirrolidinilo, indolinilo, piperidinilo, morfolinilo, dioxotiomorfolinilo, metilpiperazinilo, metilpirrolilo, metilpirazolilo, dimetilpirazolilo, etilpirazolilo, (etil)-(fluoro)pirazolilo, (etil)(metil)pirazolilo, npropilpirazolilo, isopropilpirazolilo, 2-metilpropilpirazolilo, bután-2-ilpirazolilo, difluorometilpirazolilo, (difluorometil)-(metil)pirazolilo, difluoroetilpirazolilo, trifluoroetilpirazolilo, trifluoropropilpirazolilo, ciclopropilpirazolilo, ciclobutilpirazolilo, ciclopropilmetilpirazolilo, hidroxietilpirazolilo, metoxietilpirazolilo, dimetilaminoetilpirazolilo, tetrahidropiranilpirazolilo, (metil)(tetrahidropiranil)pirazolilo, pirazolo[1,5-a]-piridinilo, metil-4,5,6,7-tetrahidropirazolilo, oxazolilo, metiloxazolilo, etiloxazolilo, isoxazolilo, metilisoxazolilo, dimetilisoxazolilo, etilisoxazolilo, isopropilisoxazolilo, ferf-butilisoxazolilo, trifluorometilisoxazolilo, ciclopropilisoxazolilo, ciclobutilisoxazolilo, metoximetilisoxazolilo, aminometilisoxazolilo, aminoisopropilisoxazolilo, tiazolilo, metiltiazolilo, dimetiltiazolilo, isotiazolilo, metilisotiazolilo, metilimidazolilo, metiloxadiazolilo, metiltiadiazolilo, metiltriazolilo, dimetiltriazolilo, etiltriazolilo, metiltetrazolilo, piridinilo, metilpiridinilo, piridazinilo, pirimidinilo, metilpirimidinilo, piridinilmetilo, aminopiridinilmetilo y espiro[tetrahidrofurán]-[oxoindol].
Los valores representativos de R6 incluyen metilo, metilpirazolilo, piridinilmetilo y aminopiridinilmetilo.
Generalmente, R6a representa hidrógeno o alquilo C1-6.
Ventajosamente, R6a representa alquilo C1-6, cicloalquilo C3-7, arilalquilo C1-6, heterocicloalquilo C3-7 o espiro[heterocicloalquil(C3-7)][heteroarilo], cualquiera de cuyos grupos puede estar opcionalmente sustituido con uno, dos o tres sustituyentes como se ha definido anteriormente.
Típicamente, R6a representa hidrógeno o metilo.
En una primera realización, R6a representa hidrógeno. En una segunda realización, R6a representa alquilo C1-6 opcionalmente sustituido En un primer aspecto de esta realización, R6a representa alquilo C1-6 no sustituido, especialmente metilo. En un segundo aspecto de esta realización, R6a representa alquilo C1-6 monosustituido, disustituido o trisustituido. En una tercera realización, R6a representa cicloalquilo C3-7 opcionalmente sustituido. En una cuarta realización, R6a representa cicloalquil(C3-7)alquilo C1-6 opcionalmente sustituido. En una quinta realización, R6a representa arilo opcionalmente sustituido. En una sexta realización, R6a representa aril(C1-6)alquilo opcionalmente sustituido. En una séptima realización, R6a representa heterocicloalquilo C3-7 opcionalmente sustituido. En una octava realización, R6a representa heterocicloalquil(C3-7)alquilo C1-6 opcionalmente sustituido. En una novena realización, R6a representa heteroarilo opcionalmente sustituido. En una décima realización, R6a representa heteroarilalquilo C1-6 opcionalmente sustituido. En una undécima realización, R6a representa espiro[heterocicloalquil(C3-7)][heteroarilo].
Los valores típicos de R6a incluyen metilo, etilo, n-propilo, isopropilo, 2,2-dimetilpropilo, ciclohexilo, bencilo, tetrahidrofuranilo, tetrahidropiranilo, tetrahidrotiopiranilo y espiro[tetrahidrofurán][indol], cualquiera de cuyos grupos puede estar opcionalmente sustituido con uno, dos o tres sustituyentes como se ha definido anteriormente.
Los ejemplos seleccionados de sustituyentes opcionales en R6a incluyen uno, dos o tres sustituyentes seleccionados independientemente de trifluorometilo, oxo y alcoxi C1-6.
Los ejemplos típicos de sustituyentes específicos en R6a incluyen uno, dos o tres sustituyentes seleccionados independientemente de flúor, cloro, bromo, ciano, nitro, metilo, etilo, isopropilo, ferf-butilo, trifluorometilo, fenilo,
fluorofenilo, hidroxi, hidroximetilo, oxo, metoxi, ferf-butoxi, difluorometoxi, trifluorometoxi, metiltío, metilsulfinilo, metilsulfonilo, amino, aminometilo, aminoetilo, metilamino, ferf-butilamino, dimetilamino, pirrolidinilo, morfolinilo, piperazinilo, acetilamino, acetilaminoetilo, metoxicarbonilamino, metilsulfonilamino, formilo, acetilo, carboxi, metoxicarbonilo, etoxicarbonilo, ferf-butoxicarbonilo, aminocarbonilo, metilaminocarbonilo, dimetilaminocarbonilo, aminosulfonilo, metilaminosulfonilo y dimetilaminosulfonilo.
Los ejemplos seleccionados de sustituyentes específicos en R6a incluyen uno, dos o tres sustituyentes seleccionados independientemente de trifluorometilo, oxo y metoxi.
Los valores seleccionados de R6a incluyen metilo, etilo, trifluoroetilo, metoxietilo, n-propilo, isopropilo, 2,2-dimetilpropilo, ciclohexilo, bencilo, tetrahidrofuranilo, tetrahidropiranilo, oxotetrahidrotiopiranilo y espiro[tetrahidrofurán][oxoindol].
Idóneamente, R6b representa hidrógeno, metilo, etilo, n-propilo o isopropilo.
Típicamente, R6b representa hidrógeno o metilo.
En una primera realización, R6b representa hidrógeno. En una segunda realización, R6b representa alquilo C1-6. En un aspecto en concreto de esta realización, R6b representa metilo, etilo, n-propilo o isopropilo, especialmente metilo.
Ventajosamente, R6c representa alquilo C1-6, cicloalquilo C3-7, cicloalquil(C3-7)alquilo C1-6, heterocicloalquilo C3-7, heterocicloalquil(C3-7)alquilo C1-6 o heteroarilalquilo C1-6, cualquiera de cuyos grupos puede estar opcionalmente sustituido con uno, dos o tres sustituyentes como se ha definido anteriormente.
En una primera realización, R6c representa alquilo C1-6 opcionalmente sustituido En una segunda realización, R6c representa cicloalquilo C3-7 opcionalmente sustituido. En una tercera realización, R6c representa cicloalquil(C3-7)alquilo C1-6 opcionalmente sustituido. En una cuarta realización, R6c representa arilo opcionalmente sustituido. En una quinta realización, R6c representa arilalquilo C1-6 opcionalmente sustituido. En una sexta realización, R6c representa heterocicloalquilo C3-7 opcionalmente sustituido. En una séptima realización, R6c representa heterocicloalquil(C3-7)alquilo C1-6 opcionalmente sustituido. En una octava realización, R6c representa heteroarilo opcionalmente sustituido. En una novena realización, R6c representa heteroarilalquilo C1-6 opcionalmente sustituido.
Los valores típicos de R6c incluyen metilo, etilo, isopropilo, 2-metilpropilo, ferf-butilo, 2,2-dimetilpropilo, ciclobutilo, ciclopentilo, ciclohexilo, ciclopropilmetilo, ciclohexilmetilo, oxetanilo, azetidinilo, tetrahidrofuranilo, tetrahidropiranilo, tetrahidropiranilmetilo, pirazolilmetilo, oxazolilmetilo, isoxazolilmetilo, imidazolilmetilo y pirazinilmetilo, cualquiera de cuyos grupos puede estar opcionalmente sustituido con uno, dos o tres sustituyentes como se define más arriba.
Los ejemplos seleccionados de sustituyentes opcionales en R6c incluyen uno, dos o tres sustituyentes seleccionados independientemente de alquilo C1-6, trifluorometilo, alcoxi C1-6 y alcoxi(C2-6)carbonilo.
Los ejemplos típicos de sustituyentes específicos en R6c incluyen uno, dos o tres sustituyentes seleccionados independientemente de flúor, cloro, bromo, ciano, nitro, metilo, etilo, isopropilo, ferf-butilo, trifluorometilo, fenilo, fluorofenilo, hidroxi, hidroximetilo, oxo, metoxi, ferf-butoxi, difluorometoxi, trifluorometoxi, metiltío, metilsulfinilo, metilsulfonilo, amino, aminometilo, aminoetilo, metilamino, ferf-butilamino, dimetilamino, pirrolidinilo, morfolinilo, piperazinilo, acetilamino, acetilaminoetilo, metoxicarbonilamino, metilsulfonilamino, formilo, acetilo, carboxi, metoxicarbonilo, etoxicarbonilo, ferf-butoxicarbonilo, aminocarbonilo, metilaminocarbonilo, dimetilaminocarbonilo, aminosulfonilo, metilaminosulfonilo y dimetilaminosulfonilo.
Los ejemplos seleccionados de sustituyentes específicos en R6c incluyen uno, dos o tres sustituyentes seleccionados independientemente de metilo, trifluorometilo, metoxi y ferf-butoxicarbonilo.
Los valores seleccionados de R6c incluyen metilo, trifluoroetilo, metoxietilo, isopropilo, 2-metilpropilo, ferf-butilo, 2,2-dimetilpropilo, ciclobutilo, ciclopentilo, ciclohexilo, ciclopropilmetilo, ciclohexilmetilo, oxetanilo, metiloxetanilo, azetidinilo, ferf-butoxicarbonilazetidinilo, tetrahidrofuranilo, tetrahidropiranilo, tetrahidropiranilmetilo, metilpirazolilmetilo, oxazolilmetilo, isoxazolilmetilo, metilimidazolilmetilo y pirazinilmetilo.
En una primera realización, R7 representa arilo, cuyo grupo puede estar opcionalmente sustituido con uno, dos o tres sustituyentes como se definió anteriormente. En una segunda realización, R7 representa heteroarilo, cuyo grupo puede estar opcionalmente sustituido con uno, dos o tres sustituyentes como se definió anteriormente. En una tercera realización, R7 representa espiro[heterocicloalquil(C3-7)][heteroarilo], cuyo grupo puede estar opcionalmente sustituido con uno, dos o tres sustituyentes como se definió anteriormente.
Los valores típicos de R7 incluyen fenilo, pirazolo[1,5-a]pirazinilo, benzoxazolilo, benzotiazolilo, bencimidazolilo, imidazo[1,2-¿>]piridazinilo, purinilo, piridinilo, piridazinilo, cinolinilo, pirimidinilo, pirazinilo y espiro[tetrahidropiranil][indol], cualquiera de cuyos grupos puede estar opcionalmente sustituido con uno, dos o tres sustituyentes como se ha definido anteriormente.
Los ejemplos seleccionados de sustituyentes opcionales en R7 incluyen uno, dos o tres sustituyentes seleccionados independientemente de halógeno, ciano, alquilo C1-6, difluorometilo, trifluorometilo, oxo, alcoxi C1-6, difluorometoxi y
dialquil(Ci-6)amino.
Los ejemplos típicos de sustituyentes específicos en R7 incluyen uno, dos o tres sustituyentes seleccionados independientemente de fluoro, cloro, bromo, ciano, nitro, metilo, etilo, isopropilo, tert-butilo, difluorometilo, trifluorometilo, fenilo, fluorofenilo, hidroxi, hidroximetilo, oxo, metoxi, isopropoxi, tert-butoxi, difluorometoxi, trifluorometoxi, metiltío, metilsulfinilo, metilsulfonilo, amino, aminometilo, aminoetilo, metilamino, tert-butilamino, dimetilamino, pirrolidinilo, morfolinilo, piperazinilo, acetilamino, acetilaminoetilo, metoxicarbonilamino, metilsulfonilamino, formilo, acetilo, carboxi, metoxicarbonilo, etoxicarbonilo, tert-butoxicarbonilo, aminocarbonilo, metilaminocarbonilo, dimetilaminocarbonilo, aminosulfonilo, metilaminosulfonilo y dimetilaminosulfonilo.
Los ejemplos seleccionados de sustituyentes específicos en R7 incluyen uno, dos o tres sustituyentes seleccionados independientemente de flúor, cloro, ciano, metilo, etilo, isopropilo, difluorometilo, trifluorometilo, oxo, metoxi, isopropoxi, difluorometoxi y dimetilamino.
Los valores seleccionados de R7 incluyen fenilo, pirazolo[1,5-a]pirazinilo, benzoxazolilo, fluorobenzoxazolilo, metilbenzoxazolilo, benzotiazolilo, bencimidazolilo, fluorobencimidazolilo, imidazo[1,2-b]piridazinilo, purinilo, piridinilo, cianopiridinilo, metilpiridinilo, metoxipiridinilo, piridazinilo, cloropiridazinilo, cianopiridazinilo, metilpiridazinilo, etilpiridazinilo, isopropilpiridazinilo, difluorometilpiridazinilo, trifluorometilpiridazinilo, metoxipiridazinilo, isopropoxipiridazinilo, difluorometoxipiridazinilo, dimetilaminopiridazinilo, cinolinilo, pirimidinilo, pirazinilo, metilpirazinilo y espiro[tetrahidropiranil][oxoindol].
Una subclase de los compuestos de fórmula (IA) anterior está representada por los compuestos de fórmula (IIA) y sus sales farmacéuticamente aceptables:

en donde
W representa O, S, S(O), S(O)2 , S(O)(NH) o N-R17;
R17 representa hidrógeno o alquilo C1-6 ; y
R0, R3, R5 y R6 son como se definen más arriba.
Típicamente, W representa O, S, S(O), S(O)2 o N-R17.
Idóneamente, W representa O, S o N-R17.
En una primera realización, W representa O. En una segunda realización, W representa S. En una tercera realización, W representa S(O). En una cuarta realización, W representa S(O)2. En una quinta realización, W representa S(O)(NH). En una sexta realización, W representa N-R17.
Idóneamente, R17 representa hidrógeno o metilo.
En una primera realización, R17 representa hidrógeno. En una segunda realización, R17 representa alquilo C1-6. En un primer aspecto de esa realización, R17 representa metilo.
Otra subclase de los compuestos de fórmula (IA) de más arriba está representada por los compuestos de fórmula (IIB) y sus sales farmacéuticamente aceptables:

en donde
W, R0, R5 y R6 son como se definen más arriba.
Los nuevos compuestos específicos de acuerdo con la presente invención incluyen cada uno de los compuestos cuya preparación se describe en los ejemplos adjuntos, y sus sales y solvatos farmacéuticamente aceptables.
Los compuestos de acuerdo con la presente invención son beneficiosos para el tratamiento y/o la prevención de diversas dolencias humanas, incluidos los trastornos inflamatorios y autoinmunitarios.
Los compuestos según la presente invención son útiles para el tratamiento y/o la profilaxis de un trastorno patológico que está mediado por una citocina de tipo IL-17 proinflamatoria o está asociado a una mayor cantidad de una citocina de tipo IL-17 proinflamatoria. Generalmente, la afección patológica se selecciona del grupo que consiste en infecciones (víricas, bacterianas, micóticas y parasitarias), choque endotóxico asociado a una infección, artritis, artritis reumatoide, artritis psoriásica, artritis idiopática juvenil (AIJ) de inicio sistémico, lupus eritematoso sistémico (LES), asma, enfermedad obstructiva crónica de las vías respiratorias (COAD), enfermedad pulmonar obstructiva crónica (EPOC), lesión pulmonar aguda, enfermedad inflamatoria pélvica, enfermedad de Alzheimer, enfermedad de Crohn, enteropatía inflamatoria, síndrome del colon irritable, colitis ulcerosa, enfermedad de Castleman, espondilitis anquilosante y otras espondiloartropatías, dermatomiositis, miocarditis, uveítis, exoftalmos, tiroiditis autoinmunitaria, enfermedad de Peyronie, enfermedad celíaca, enfermedad de la vesícula biliar, enfermedad pilonidal, peritonitis, psoriasis, dermatitis atópica, vasculitis, adherencias quirúrgicas, accidente cerebrovascular, diabetes autoinmunitaria, diabetes de tipo I, artritis de Lyme, meningoencefalitis, trastornos inflamatorios inmunomediados del sistema nervioso central y periférico, tal como la esclerosis múltiple y el síndrome de Guillain-Barr, otros trastornos autoinmunitarios, pancreatitis, traumatismo (cirugía), enfermedad de injerto contra huésped, rechazo de trasplantes, trastornos fibrosantes que incluyen fibrosis pulmonar, fibrosis hepática, fibrosis renal, esclerodermia o esclerosis sistémica, cáncer (tanto tumores sólidos tales como melanomas, hepatoblastomas, sarcomas, carcinomas de células escamosas, cánceres de células de transición, cánceres de ovario y neoplasias malignas hematológicas y, en concreto, leucemia mielógena aguda, leucemia mielógena crónica, leucemia linfática crónica, cáncer gástrico y cáncer de colon), enfermedades cardíacas que incluyen enfermedades isquémicas tales como infarto de miocardio así como aterosclerosis, coagulación intravascular, osteoclasia, osteoporosis, periodontitis, hipoclorhidria y dolor (particularmente dolor asociado con la inflamación).
En la solicitud de patente internacional WO 2009/089036 se da a conocer que los moduladores de la actividad de IL-17 pueden administrarse para inhibir o reducir la gravedad de los trastornos inflamatorios oculares, en concreto los trastornos inflamatorios de la superficie ocular, incluido el síndrome del ojo seco (DES). En consecuencia, los compuestos de acuerdo con la presente invención son útiles para el tratamiento y/o la prevención de un trastorno inflamatorio ocular mediado por la IL-17, en concreto un trastorno inflamatorio de la superficie ocular mediado por la IL-17 que incluye el síndrome del ojo seco. Los trastornos inflamatorios de la superficie ocular incluyen síndrome de ojo seco, queratoplastia penetrante, trasplante de córnea, trasplante lamelar o de espesor parcial, trasplante endotelial selectivo, neovascularización corneal, cirugía de queratoprotésica, afecciones inflamatorias de la superficie ocular de la córnea, trastornos de cicatrización conjuntival, afecciones autoinmunitarias oculares, síndrome de penfigoide, síndrome de Stevens-Johnson, alergia ocular, enfermedad ocular alérgica (atópica) grave, conjuntivitis y queratitis microbiana. Las categorías concretas del síndrome del ojo seco incluyen queratoconjuntivitis seca (KCS), síndrome de Sjogren, queratoconjuntivitis seca asociada al síndrome de Sjogren, queratoconjuntivitis seca no asociada al síndrome de Sjogren, queratitis seca, síndrome seco, xeroftalmia, trastorno de la película lagrimal, disminución de la producción de lágrimas, deficiencia de lagrimeo acuoso (ATD), disfunción de las glándulas de Meibomio y pérdida por evaporación.
Ilustrativamente, los compuestos de la presente invención pueden ser útiles para el tratamiento y/o la profilaxis de un trastorno patológico seleccionado del grupo que consiste en artritis, artritis reumatoide, psoriasis, artritis psoriásica, artritis idiopática juvenil (AIJ) de inicio sistémico, lupus eritematoso sistémico (LES), asma, enfermedad obstructiva crónica de las vías respiratorias, enfermedad pulmonar obstructiva crónica, dermatitis atópica, esclerodermia, esclerosis sistémica, fibrosis pulmonar, enteropaías inflamatorias (que incluyen enfermedad de Crohn y colitis ulcerosa), espondilitis anquilosante y otras espondiloartropatías, cáncer y dolor (en concreto dolor asociado a la inflamación).
Idóneamente, los compuestos de la presente invención son útiles para el tratamiento y/o la profilaxis de la psoriasis, la artritis psoriásica o la espondilitis anquilosante.
En la presente invención también se da a conocer una composición farmacéutica que comprende un compuesto de acuerdo con la invención como se describe anteriormente, o una sal farmacéuticamente aceptable del mismo, en asociación con uno o más vehículos farmacéuticamente aceptables.
Las composiciones farmacéuticas según la invención pueden adoptar una forma adecuada para la administración oral, bucal, parenteral, nasal, tópica, oftálmica o rectal, o una forma adecuada para la administración por inhalación o insuflación.
Para la administración oral, las composiciones farmacéuticas pueden tomar la forma de, por ejemplo, comprimidos, pastillas o cápsulas preparadas por medios convencionales con excipientes farmacéuticamente aceptables tales como aglutinantes (por ejemplo, almidón de maíz pregelatinizado, polivinilpirrolidona o hidroxipropilmetilcelulosa); rellenos (por ejemplo, lactosa, celulosa microcristalina o hidrogenofosfato de calcio); lubricantes (por ejemplo, estearato de magnesio, talco o sílice); disgregantes (por ejemplo, almidón de patata o glicolato de sodio); o humectantes (por ejemplo, laurilsulfato de sodio). Los comprimidos pueden recubrir mediante métodos bien conocidos en la técnica. Los preparados líquidos para la administración oral pueden adoptar la forma de, por ejemplo, soluciones, jarabes o suspensiones, o pueden presentarse como un producto seco para su reconstitución con agua u otro vehículo adecuado antes de su uso. Tales preparaciones líquidas se pueden preparar por medios convencionales con aditivos farmacéuticamente aceptables, tales como agentes de suspensión, emulsionantes, vehículos no acuosos o conservantes. Los preparados también pueden contener sales tamponantes, saborizantes, colorantes o edulcorantes, según convenga.
Las preparaciones para administración oral pueden formularse idóneamente para ofrecer una liberación controlada del compuesto activo.
Para la administración bucal, las composiciones pueden tomar la forma de comprimidos o pastillas formuladas de manera convencional.
Los compuestos según la presente invención pueden formularse para la administración parenteral mediante inyección, p. ej., por inyección en bolo o infusión. Las formulaciones para inyección pueden presentarse en forma de dosis unitaria, p. ej., en ampollas de vidrio o envases multidosis, p. ej., viales de vidrio. Las composiciones para inyección pueden adoptar formas tales como suspensiones, soluciones o emulsiones en vehículos oleosos o acuosos, y pueden contener agentes de formulación tales como agentes de suspensión, estabilizantes, conservantes y/o dispersantes. Como alternativa, el principio activo puede estar en forma de polvo para la reconstitución con un vehículo adecuado, p. ej., agua estéril libre de pirógenos, antes de su uso.
Además de las formulaciones descritas anteriormente, los compuestos según la presente invención también se pueden formular como una preparación de efecto prolongado. Dichas formulaciones de acción prolongada pueden administrarse por implantación o por inyección intramuscular.
Para la administración nasal o la administración por inhalación, los compuestos de acuerdo con la presente invención pueden administrarse cómodamente en forma de una presentación de rociador de aerosol para envases presurizados o un nebulizador, con el uso de un propulsor adecuado, p. ej., diclorodifluorometano, fluorotriclorometano, diclorotetrafluoroetano, dióxido de carbono u otro gas o mezcla de gases adecuados.
Las composiciones pueden, si se desea, presentarse en un paquete o dispositivo dispensador que puede contener una o más formas de dosificación unitaria que contienen el principio activo. El envase o dispositivo dispensador puede ir acompañado de instrucciones de administración.
Para la administración tópica, los compuestos según la presente invención se pueden formular cómodamente en un ungüento adecuado que contiene el componente activo suspendido o disuelto en uno o más vehículos farmacéuticamente aceptables. Los vehículos concretos incluyen, por ejemplo, aceite mineral, vaselina líquida, propilenglicol, polioxietileno, polioxipropileno, cera emulsionante y agua. Como alternativa, los compuestos según la presente invención se pueden formular en una loción adecuada que contiene el componente activo suspendido o disuelto en uno o más vehículos farmacéuticamente aceptables. Los vehículos concretos incluyen, por ejemplo, aceite mineral, monoestearato de sorbitano, polisorbato 60, cera de ésteres cetílicos, alcohol cetearílico, alcohol bencílico, 2-octildodecanol y agua.
Para la administración oftálmica, los compuestos según la presente invención pueden formularse cómodamente como suspensiones micronizadas en solución salina isotónica y estéril con el pH ajustado, con o sin un conservante tal como un bactericida o un fungicida, por ejemplo, nitrato fenilmercúrico, cloruro de benzalconio o acetato de clorhexidina. Como alternativa, para la administración oftálmica, los compuestos según la presente invención se pueden formular en un ungüento tal como la vaselina.
Para la administración rectal, los compuestos según la presente invención pueden formularse cómodamente como supositorios. Estos se pueden preparar mezclando el componente activo con un excipiente no irritante adecuado que
sea sólido a temperatura ambiente pero líquido a temperatura rectal y, por lo tanto, se derretirá en el recto para liberar el componente activo. Dichos materiales incluyen, por ejemplo, manteca de cacao, cera de abejas y polietilenglicoles. La cantidad de un compuesto según la presente invención requerida para la profilaxis o el tratamiento de una afección concreta variará dependiendo del compuesto elegido y la afección del paciente a tratar. En general, sin embargo, las dosis diarias pueden oscilar entre alrededor de 10 ng/kg y 1000 mg/kg, normalmente de 100 ng/kg a 100 mg/kg, p. ej., de alrededor de 0,01 mg/kg a 40 mg/kg de peso corporal, para la administración oral o bucal, de alrededor de 10 ng/kg a 50 mg/kg de peso corporal para la administración parenteral, y de alrededor de 0,05 mg a alrededor de 1000 mg, p. ej., de alrededor de 0,5 mg a alrededor de 1000 mg, para la administración nasal o la administración por inhalación o insuflación.
Si se desea, se puede coadministrar un compuesto de acuerdo con la presente invención con otro agente farmacéuticamente activo, p. ej., una molécula antiinflamatoria.
Los compuestos de la fórmula (I) anterior pueden prepararse mediante un procedimiento que comprende hacer reaccionar un ácido carboxílico de fórmula RaCO2 H, o una de sus sales, p. ej., una sal de litio del mismo, con un compuesto de la fórmula (III):

en donde A, B, D, E y Ra son como se definen más arriba, y Rp corresponde al grupo R0 como se define más arriba, o Rp representa un grupo protector de N ; seguido, según sea necesario, por la retirada del grupo Rp protector de N. Los grupos Rp protectores de N serán idóneamente íert-butoxicarbonilo (BOC), bencilo o 2-(trimetilsilil)etoximetilo (SEM).
La reacción se lleva a cabo cómodamente en presencia de un agente de acoplamiento. Los agentes de acoplamiento adecuados incluyen hexafluorofosfato de 2-(7-aza-1 H-benzotriazol-1-il)-1,1,3,3-tetrametil-uronio (HATU).
Cuando el compuesto (III) se hace reaccionar con un ácido carboxílico de fórmula RaCO2H, la reacción se lleva a cabo generalmente en presencia de una base. Las bases adecuadas incluyen aminas orgánicas, p. ej., una trialquilamina tal como N,N-diisopropiletilamina. La reacción se realiza cómodamente a temperatura ambiente en un solvente adecuado, p. ej., un éter cíclico tal como tetrahidrofurano, o un solvente aprótico dipolar tal como N,N-dimetilformamida, o un solvente clorado tal como diclorometano.
Cuando el compuesto (III) se hace reaccionar con la sal de litio de un ácido carboxílico de fórmula RaCO2H, la reacción se lleva a cabo generalmente a temperatura ambiente en un solvente adecuado, p.ej., un solvente aprótico dipolar tal como N,N-dimetilformamida.
Cuando el grupo Rp protector de N es BOC, la subsiguiente retirada del mismo puede efectuarse cómodamente mediante el tratamiento con un ácido, p. ej., un ácido mineral tal como el ácido clorhídrico, o un ácido orgánico tal como el ácido trifluoroacético.
Cuando el grupo Rp protector de N es bencilo, la subsiguiente retirada del mismo puede efectuarse cómodamente mediante la hidrogenación catalítica, normalmente mediante el tratamiento con hidrógeno gaseoso en presencia de un catalizador de hidrogenación, p. ej., paladio sobre carbón vegetal.
Cuando el grupo Rp protector de N es SEM, la subsiguiente retirada del mismo puede efectuarse cómodamente mediante el tratamiento con una sal de fluoruro, p. ej., fluoruro de tetra-n-butilamonio; o por el tratamiento con un ácido, p. ej., un ácido mineral tal como el ácido clorhídrico, o un ácido orgánico tal como el ácido trifluoroacético.
Cuando Ra representa -CH(R5)N(H)C(O)R6 , los intermedios de fórmula RaCO2H puede prepararse mediante un procedimiento de dos etapas que comprende: (i) hacer reaccionar un ácido carboxílico de fórmula R6-CO2H con un compuesto de fórmula (IV):

en donde Alk1 representa alquilo C i -4, p. ej., metilo, y R5 y R6 son como se definen anteriormente; en las condiciones análogas a las descritas anteriormente para la reacción entre el compuesto (III) y un ácido carboxílico de fórmula RaCO2H; y (ii) saponificación del material resultante mediante el tratamiento con una base.
En cuanto a la reacción entre el compuesto (III) y un ácido carboxílico de fórmula RaCO2H, el agente de acoplamiento empleado en la etapa (i) puede ser idóneamente HATU.
Como alternativa, el agente de acoplamiento puede ser 2,4,6-trióxido de 2,4,6-tripropil-1,3,5,2,4,6-trioxatrifosforinano, en cuyo caso la reacción generalmente puede llevarse a cabo en presencia de una base que puede incluir idóneamente aminas orgánicas, p. ej., una trialquilamina tal como N,N-diisopropiletilamina, o una base aromática tal como piridina. La reacción se realiza cómodamente a temperatura ambiente en un solvente adecuado, p. ej., un éster orgánico tal como acetato de etilo y/o un éter cíclico tal como tetrahidrofurano.
Como alternativa, el agente de acoplamiento puede ser una mezcla de hidrocloruro de N-(3-dimetilaminopropil)-N'-etilcarbodiimida y 1-hidroxibenzotriazol, en cuyo caso la reacción puede llevarse a cabo generalmente en presencia de una base, p. ej., una amina orgánica tal como N,N-diisopropiletilamina. La reacción se realiza cómodamente a temperatura ambiente en un solvente adecuado, p. ej., un solvente aprótico dipolar tal como N,N-dimetilformamida.
La reacción de saponificación en la etapa (ii) generalmente se efectuará mediante el tratamiento con una base. Las bases adecuadas incluyen hidróxidos inorgánicos, p. ej., un hidróxido de metal alcalino tal como hidróxido de litio. Cuando se emplea el hidróxido de litio en la etapa (ii) del procedimiento anterior, el producto puede ser la sal de litio del ácido carboxílico de la fórmula RaCO2H.
La etapa (ii) se efectúa cómodamente a temperatura ambiente en agua y un solvente orgánico adecuado, p. ej., un éter cíclico tal como tetrahidrofurano, opcionalmente mezclado con un alcanol C1-4 tal como el metanol.
En otro procedimiento, los compuestos de la fórmula (I) anterior pueden prepararse mediante un procedimiento que comprende hacer reaccionar una amida de la fórmula RaCONH2 con un compuesto de fórmula (V):

en donde A, B, D, E, Ra y Rp son como se definen más arriba, y L1 representa un grupo saliente adecuado; en presencia de un catalizador de metal de transición; seguido, según sea necesario, por la retirada del grupo Rp protector de N.
El grupo saliente L1 es idóneamente un átomo de halógeno, p. ej., cloro o bromo.
El catalizador de tipo metal de transición es idóneamente metanosulfonato de [(2-di-te/t-butilfosfino-3,6-dimetoxi-2',4',6'-triisopropil-1,1'-bifenil)-2-(2'-amino-1,1'-bifenil)]paladio(II) (tBuBrettPhos Pd G3), en cuyo caso la reacción se realizará generalmente en presencia de 2-(di-te/t-butilfosfino)-2',4',6'-triisopropil-3,6-dimetoxi-1,1'-bifenilo (tBuBrettPhos). La reacción se lleva a cabo cómodamente a una temperatura elevada en presencia de una base, p. ej., una base inorgánica tal como carbonato de potasio, en un solvente adecuado, p. ej., un alcanol inferior tal como te/t-butanol.
Como alternativa, el catalizador de metal de transición puede ser idóneamente tris(dibencilidenacetona)dipaladio(0), en cuyo caso la reacción generalmente se realizará en presencia de 2-diciclohexilfosfino-2',4',6'-triisopropilbifenilo (XPhos) o 4,5-bis(difenilfosfino)-9,9-dimetilxanteno (Xantphos). La reacción se lleva a cabo cómodamente a una temperatura elevada en presencia de una base, p. ej., una sal de carbonato tal como carbonato de potasio o carbonato de cesio, en un solvente adecuado, p. ej., un éter cíclico tal como 1,4-dioxano, o un alcanol C1-6 tal como te/t-butanol.
Los intermedios de la fórmula (V) anterior pueden prepararse haciendo reaccionar el a,w-dihaloalcano apropiado con
un compuesto de fórmula (VI):

en donde B, D, E, L1 y Rp son como se definen más arriba.
La reacción se realiza generalmente en presencia de una base, p. ej., una base inorgánica tal como el carbonato de cesio. La reacción se llevará a cabo cómodamente a temperatura ambiente o temperatura elevada, según corresponda, en un solvente adecuado, p. ej., un solvente aprótico dipolar tal como N,N-dimetilformamida, o un solvente carbonílico tal como acetona, o un solvente de tipo sulfóxido tal como dimetilsulfóxido.
Los intermedios de la fórmula (VI) anteriores se pueden preparar mediante un procedimiento de dos etapas a partir de un compuesto de fórmula (VII):

en donde B, D, E, L1 y Rp son como se definen anteriormente; cuyo procedimiento comprende los siguientes etapas: (i) tratamiento del compuesto (VII) con tribromuro de piridinio o N-bromosuccinimida; y
(ii) tratamiento del derivado 3,3-dibromo-2-oxoindolina así obtenido con zinc metálico.
La etapa (i) se lleva a cabo cómodamente a temperatura ambiente en un solvente adecuado, p. ej., un éter cíclico tal como 1,4-dioxano, o un alcanol C1-4 tal como fert-butanol, típicamente mezclado con agua.
La etapa (ii) se lleva a cabo cómodamente en presencia de cloruro de amonio a una temperatura elevada en un solvente adecuado, p. ej., un solvente aprótico dipolar tal como N,N-dimetilformamida, o un éter cíclico tal como tetrahidrofurano. Como alternativa, la etapa (ii) se puede realizar en ácido acético a temperatura ambiente en un solvente adecuado, p. ej., un éter cíclico tal como el tetrahidrofurano.
De manera similar, los intermedios de la fórmula (III) anterior pueden prepararse haciendo reaccionar el a,wdihaloalcano apropiado con un compuesto de fórmula (VI-A):

en donde B, D, E y Rp son como se definen más arriba, y Rq representa hidrógeno o un grupo protector de N ; en las condiciones análogas a las descritas anteriormente para la reacción de un a,w-dihaloalcano con un compuesto de la fórmula (VI); seguido, en su caso, de la retirada del grupo o grupos protectores de N Rp y/o Rq.
El grupo Rq protector de N será idóneamente fe/t-butoxicarbonilo (BOC).
Cuando el grupo Rq protector de N es BOC, la subsiguiente retirada del mismo puede efectuarse cómodamente mediante el tratamiento con un ácido, p. ej., un ácido mineral tal como el ácido clorhídrico, o un ácido orgánico como el ácido trifluoroacético.
En otro procedimiento, los compuestos de la fórmula (IA) anterior pueden prepararse mediante un procedimiento que
comprende hacer reaccionar un compuesto de la fórmula (III) como se define anteriormente con un compuesto de fórmula (VIII):

en donde R5 y R6 son como se definen anteriormente; seguido, según sea necesario, por la retirada del grupo protector de N Rp.
La reacción entre los compuestos (III) y (VIII) se realizará generalmente en presencia de ácido acético. La reacción se lleva a cabo cómodamente a una temperatura elevada en un solvente adecuado, p. ej., un éter cíclico tal como el tetrahidrofurano.
De manera similar, los compuestos de la fórmula (IF) anterior pueden prepararse mediante un procedimiento que comprende hacer reaccionar un compuesto de la fórmula (III) como se definió anteriormente con un compuesto de fórmula (IX):

en donde R5a, R5b y R6 son como se definen anteriormente; en las condiciones análogas a las descritas anteriormente para la reacción entre los compuestos (III) y (VIII); seguido, según sea necesario, por la retirada del grupo Rp protector de N.
Cuando los correspondientes valores de R5, R5a y R5b lo permiten, se puede obtener un intermedio de fórmula (VIII) a partir del correspondiente intermedio de fórmula (IX) mediante la hidrogenación catalítica convencional.
Los intermedios de la fórmula (IX) anterior se pueden preparar al hacer reaccionar un compuesto de la fórmula R5aC(O)R5b con un compuesto de la fórmula (VIII) como se define anteriormente en la que R5 representa un hidrógeno. La reacción se ejecuta cómodamente tratando los reactivos con tetracloruro de titanio; seguido por el tratamiento del material resultante con piridina.
En otro procedimiento, los compuestos de la fórmula (IA) anterior pueden prepararse mediante un procedimiento que comprende hacer reaccionar un ácido carboxílico de fórmula R6-CO2H con un compuesto de fórmula (X):

en donde A, B, D, E, Rp, R5 y R6 son como se definen anteriormente; en las condiciones análogas a las descritas
anteriormente para la reacción entre el compuesto (III) y un ácido carboxílico de la fórmula RaCÜ2H; seguido, según sea necesario, por la retirada del grupo Rp protector de N.
De manera similar, los compuestos de la fórmula (IA) anterior en la que R6 representa -NR6aR6b pueden prepararse mediante un procedimiento que comprende hacer reaccionar un derivado de carbamato de la fórmula L2-C(Ü)NR6aR6b, en donde L2 representa un grupo saliente adecuado, con un compuesto de la fórmula (X) como se define anteriormente; seguido, según sea necesario, por la retirada del grupo Rp protector de N.
El grupo saliente L2 es idóneamente un átomo de halógeno, p. ej., cloro; o L2 es idóneamente fenoxi.
Cuando L2 es un átomo de halógeno, la reacción se lleva a cabo cómodamente a temperatura ambiente en presencia de una base, p. ej., una amina orgánica tal como trietilamina, en un solvente adecuado, p. ej., un solvente clorado como el diclorometano.
Cuando L2 es fenoxi, la reacción se lleva a cabo cómodamente a una temperatura elevada en presencia de 4-(dimetilamino)piridina, en un solvente adecuado, p. ej., un solvente de nitrilo tal como acetonitrilo.
De manera similar, los compuestos de la fórmula (IA) anterior en la que R6 representa -OR6c pueden prepararse mediante un procedimiento que comprende hacer reaccionar un compuesto de la fórmula L3-C(O)OR6c, en donde L3 representa un grupo saliente adecuado, con un compuesto de la fórmula (X) como se define anteriormente; seguido, según sea necesario, por la retirada del grupo Rp protector de N.
El grupo saliente L3 es idóneamente un átomo de halógeno, p. ej., cloro.
La reacción se lleva a cabo cómodamente a temperatura ambiente en presencia de una base, p. ej., una amina orgánica tal como trietilamina, normalmente mezclada con piridina, en un solvente adecuado, p. ej., un éter cíclico tal como tetrahidrofurano.
En otro procedimiento, los compuestos de la fórmula (IB) anterior pueden prepararse mediante un procedimiento que comprende hacer reaccionar un compuesto de la fórmula (X) como se define anteriormente con un compuesto de la fórmula L4-SO)2R6, en donde R6 es como se define más arriba, y L4 representa un grupo saliente adecuado; seguido, según sea necesario, por la retirada del grupo Rp protector de N.
El grupo saliente L4 es idóneamente un átomo de halógeno, p. ej., cloro.
La reacción se lleva a cabo cómodamente a temperatura ambiente en presencia de una base, p. ej., una amina orgánica tal como N,N-diisopropiletilamina, en un solvente adecuado, p. ej., un solvente clorado como el diclorometano.
En otro procedimiento, los compuestos de la fórmula (IC) anterior pueden prepararse mediante un procedimiento que comprende hacer reaccionar un compuesto de la fórmula (X) como se define anteriormente con un compuesto de la fórmula L5-R7, en donde R7 es como se define más arriba, y L5 representa un grupo saliente adecuado; seguido, según sea necesario, por la retirada del grupo Rp protector de N.
El grupo saliente L5 es idóneamente un átomo de halógeno, p. ej., cloro o bromo.
La reacción se lleva a cabo cómodamente en presencia de una base. Las bases adecuadas incluyen aminas orgánicas, p. ej., una trialquilamina tal como N,N-diisopropiletilamina. La reacción se realiza normalmente a una temperatura elevada en un solvente adecuado, p. ej., un éter cíclico tal como 1,4-dioxano.
Como alternativa, la reacción se puede realizar en presencia de un catalizador de tipo metal de transición. Los catalizadores de tipo metal de transición adecuados para usar en este procedimiento incluyen el metanosulfonato de [(2-di-fe/t-butilfosfino-3,6-dimetoxi-2\4',6'-triisopropil-1,1'-bifenil)-2-(2'-amino-1,1'-bifenil)]paladio(II) (tBuBrettPhos PdG3). La reacción se lleva a cabo cómodamente a una temperatura elevada en presencia de una base, p. ej., una base inorgánica tal como el te/t-butóxido de potasio, en un solvente o mezcla de solventes adecuados. El solvente o solventes pueden seleccionarse idóneamente de un éter cíclico tal como 1,4-dioxano y un solvente de tipo sulfóxido tal como dimetilsulfóxido.
Los intermedios de la fórmula (X) anterior pueden prepararse haciendo reaccionar un compuesto de la fórmula (III) como se define anteriormente con un compuesto de fórmula (XI), o una de sus sales, p. ej., una sal de litio del mismo:

en donde R5 y Rq son como se definen anteriormente; en las condiciones análogas a las descritas anteriormente para la reacción entre el compuesto (III) y un ácido carboxílico de la fórmula RaCO2 H; seguido, según sea necesario, por la retirada del grupo Rq protector de N.
En otro procedimiento, los compuestos de la fórmula (ID) anterior pueden prepararse mediante un procedimiento que comprende hacer reaccionar un compuesto de la fórmula R7-NH2 con un compuesto de fórmula (XII):

en donde A, B, D, E, Rp, R5 y R7 son como se definen anteriormente; en las condiciones análogas a las descritas anteriormente para la reacción entre el compuesto (III) y un ácido carboxílico de fórmula RaCO2H; seguido, según sea necesario, por la retirada del grupo Rp protector de N.
Los intermedios de la fórmula (XII) anterior pueden prepararse mediante un procedimiento de dos etapas que comprende: (i) hacer reaccionar un compuesto de la fórmula (III) como se define anteriormente con un compuesto de la fórmula (XIII), o una de sus sales, p. ej., una sal de litio del mismo:
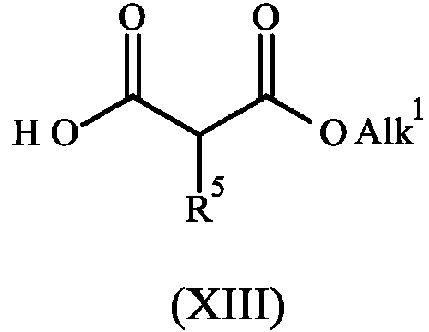
en donde R5 y Alk1 son como se definen anteriormente; en las condiciones análogas a las descritas anteriormente para la reacción entre el compuesto (III) y un ácido carboxílico de fórmula RaCO2H; y (ii) saponificación del material resultante mediante el tratamiento con una base.
La reacción de saponificación en la etapa (ii) generalmente se efectuará mediante el tratamiento con una base. Las bases adecuadas incluyen hidróxidos inorgánicos, p. ej., un hidróxido de metal alcalino tal como hidróxido de litio. Cuando se emplea hidróxido de litio en la etapa (ii) del procedimiento anterior, el producto puede ser la sal de litio del ácido carboxílico de la fórmula (XII).
La etapa (ii) se ejecuta cómodamente a temperatura ambiente en agua y un solvente orgánico adecuado, p. ej., un alcanol C1-4 tal como el etanol.
Cuando no estén disponibles comercialmente, los materiales de partida de las fórmulas (IV), (VI-A), (VII), (XI) y (XIII) pueden prepararse por métodos análogos a los descritos en los ejemplos adjuntos, o por métodos estándares bien conocidos en la técnica.
Se entenderá que cualquier compuesto de la fórmula (I) obtenido inicialmente a partir de cualquiera de los procedimientos anteriores puede, cuando sea apropiado, elaborarse posteriormente en un compuesto adicional de la fórmula (I) mediante las técnicas conocidas en la técnica. A modo de ejemplo, un compuesto de la fórmula (I) que comprende un resto N-BOC (en donde BOC es una abreviatura de tert-butoxicarbonilo) puede convertirse en el compuesto correspondiente que comprende un resto N-H por tratamiento con un ácido, p. ej., un ácido mineral tal como el ácido clorhídrico, o un ácido orgánico tal como el ácido trifluoroacético.
Un compuesto de la fórmula (I) que comprende un resto amino (-NH2) puede acilarse, p. ej., acetilarse, por tratamiento con un haluro de acilo adecuado, p. ej., cloruro de acetilo, normalmente en presencia de una base, p. ej., una base orgánica tal como N,N-diisopropiletilamina.
Un compuesto que contiene un resto N-H puede alquilarse, p. ej., metilarse, por tratamiento con el haluro de alquilo apropiado, p. ej., yodometano, normalmente a temperatura ambiente en presencia de una base, p. ej., hidruro de sodio, en un solvente adecuado, p. ej., un solvente aprótico dipolar tal como N,N-dimetilformamida.
Un compuesto de la fórmula (I) en donde R3 es hidrógeno se puede convertir en el compuesto correspondiente en el
que R3 es fluoro por tratamiento con Selectfluor™ .
Un compuesto de la fórmula (I) en donde R3 es hidrógeno se puede convertir en el compuesto correspondiente en el que R3 es cloro por tratamiento con la W-clorosuccinimida, normalmente en presencia de ácido acético.
Cuando los correspondientes valores de R5 , R5a y R5b lo permiten, se puede obtener un compuesto de la fórmula (IA) a partir del correspondiente compuesto de la fórmula (IF) mediante la hidrogenación catalítica convencional, p. ej., por tratamiento con hidrógeno gaseoso en presencia de un catalizador de hidrogenación tal como paladio sobre carbón vegetal.
Un compuesto que contiene el resto -S- puede convertirse en el correspondiente compuesto que contiene el resto -S(O)- mediante el tratamiento con el ácido 3-cloroperoxibenzoico. Asimismo, un compuesto que contiene el resto -S o -S(O)- puede convertirse en el compuesto correspondiente que contiene el resto -S(O)2- por tratamiento con el ácido 3-cloroperoxibenzoico.
Un compuesto que contiene el resto -S- puede convertirse en el correspondiente compuesto que contiene el resto -S(O)(NH)- mediante tratamiento con carbamato de amonio y (diacetoxiyodo)benceno.
Cuando se obtiene una mezcla de productos a partir de cualquiera de los procedimientos descritos anteriormente para la preparación de compuestos según la invención, el producto deseado puede separarse de la misma en una etapa apropiada mediante los métodos convencionales tales como HPLC preparativa; o cromatografía en columna utilizando, por ejemplo, sílice y/o alúmina junto con un sistema solvente apropiado.
Cuando los procedimientos descritos anteriormente para la preparación de los compuestos según la invención dan lugar a mezclas de estereoisómeros, estos isómeros pueden separarse mediante técnicas convencionales. En concreto, cuando se desea obtener un enantiómero concreto de un compuesto de la fórmula (I), éste puede producirse a partir de una mezcla correspondiente de enantiómeros utilizando cualquier procedimiento convencional adecuado para la resolución de enantiómeros. Así, por ejemplo, los diastereoisómeros derivados, p. ej. sales, pueden producirse por la reacción de una mezcla de enantiómeros de la fórmula (I), p. ej. un racemato, y un compuesto quiral apropiado, p. ej., una base quiral. Los diastereoisómeros pueden luego separarse por cualquier medio cómodo, por ejemplo, por cristalización, y recuperarse el enantiómero deseado, p. ej., por tratamiento con un ácido en el caso de que el diastereoisómero sea una sal. En otro procedimiento de resolución, un racemato de la fórmula (I) puede separarse usando la HPLC quiral. Además, si se desea, se puede obtener un enantiómero concreto usando un intermedio quiral apropiado en uno de los procedimientos descritos anteriormente. Como alternativa, se puede obtener un enantiómero concreto realizando una biotransformación enzimática específica del enantiómero, p. ej., una hidrólisis de éster usando una esterasa, y luego purificando solo el ácido hidrolizado enantioméricamente puro y dejar el éster opuesto sin reaccionar. La cromatografía, la retrocristalización y otros procedimientos de separación convencionales también se pueden usar con productos intermedios o finales cuando se desea obtener un isómero geométrico concreto de la invención.
Durante cualquiera de las secuencias sintéticas anteriores, puede ser necesario y/o deseable proteger los grupos sensibles o reactivos en cualquiera de las moléculas involucradas. Esto se puede lograr por medio de grupos protectores convencionales, tales como los descritos en Greene's Protective Groups in Organic Synthesis , ed. PGM Wuts, John Wiley & Sons, 5.a edición, 2014. Los grupos protectores se pueden retirar con comodidad en cualquier etapa posterior utilizando los métodos conocidos en la técnica.
Los compuestos de acuerdo con esta invención inhiben potentemente la capacidad de la IL-17A para unirse al IL-17RA. Cuando se comprueban en el ensayo FRET de IL-17 descrito a continuación, los compuestos de la presente invención exhiben un valor de CI50 de 10 pM o menos, generalmente de 5 pM o menos, normalmente de 1 pM o menos, típicamente de 500 nM o menos, idóneamente de 100 nM o menos, idealmente de 50 nM o menos, y preferiblemente de 25 nM o menos (el experto apreciará que una cifra más baja de CI50 denota un compuesto más activo).
Además, ciertos compuestos de acuerdo con esta invención inhiben potentemente la liberación de la IL-6 inducida por la IL-17 desde los fibroblastos dérmicos humanos. De hecho, cuando se comprueban en el ensayo con la línea celular HDF que se describe a continuación, los compuestos de la presente invención exhiben un valor de CI50 de 10 pM o menos, generalmente de 5 pM o menos, normalmente de 1 pM o menos, típicamente de 500 nM o menos, idóneamente de 100 nM o menos, idealmente de 50 nM o menos, y preferiblemente de 25 nM o menos (como antes, el experto apreciará que una cifra más baja de CI50 denota un compuesto más activo).
Ensayo de FRET de la IL-17
El propósito de este ensayo es comprobar la capacidad que tienen los compuestos para interrumpir la interacción entre la IL-17A y el receptor A de la IL-17 soluble (IL-17RA). En este ensayo se mide la capacidad de un compuesto para inhibir la unión de la IL-17A al IL-17RA.
Se expresó una construcción IL-17AA-TEV-Fc humana en un sistema de células CHO SXE y se purificó mediante cromatografía con proteína A y exclusión por tamaños. La proteína se marcó con un colorante AlexaFluor 647 reactivo
con las aminas (Thermo Fisher #A20006), según las Instrucciones del fabricante.
Se expresó el IL-17RA (33-317)-HKH-TEV-Fc soluble en un sistema de células Expi HEK293 y se purificó mediante cromatografía con proteína A y exclusión por tamaños. La etiqueta Fc se escindió mediante t Ev , produciendo el IL-17RA (33-317)-HKH, y la proteína se marcó con terbio reactivo con las aminas (Thermo Fisher #PV3581).
En el tampón de ensayo [PBS de Dulbecco (Sigma n.° 14190-094), P20 al 0,05 % (Thermo Scientific n.° 28320), 1 mg/ml de BSA (Sigma n.° A2153-500G)] se prepararon las siguientes soluciones:
Para el ensayo de la IL-17A
• IL-17A-Fc-AF647 a 5 nM
• IL-17RA-HKH-Tb a 5 nM
Los compuestos se diluyeron en serie en DMSO antes de recibir una dilución acuosa en una placa de dilución de 384 pocillos (Greiner #781281), para dar una solución de DMSO al 25 %.
Se añadió la IL-17A (10 gl) a una placa de ensayo negra de bajo volumen (Costar #4511) y se transfirió el compuesto diluido (5 gl) desde la placa de dilución acuosa. Se permitió que la citocina y el compuesto se incubaran durante 1 h, luego se le añadió el IL-17RA (10 gl). Las placas se envolvieron en papel de aluminio y se incubaron a temperatura ambiente durante 18-20 h con agitación suave (<400 rpm) antes de leerlas en un lector de placas Perkin Elmer Envision (excitación: 330 nm; emisión 615/645 nm).
Las concentraciones finales del ensayo fueron IL-17A-AF647 2 nM e IL-17RA-Tb 2 nM, DMSO al 5 %.
Cuando se comprobaron en el ensayo de FRET de IL-17, se encontró que todos los compuestos de los ejemplos adjuntos exhibían unos valores de CI50 de 10 gM o mejores.
Cuando se comprueban en el ensayo de FRET de IL-17, los compuestos de los ejemplos adjuntos exhiben valores de CI50 generalmente en el margen de aproximadamente 0,01 nM a aproximadamente 10 gM, normalmente en el margen de aproximadamente 0,01 nM a aproximadamente 5 gM, típicamente en el margen de aproximadamente 0,01 nM a aproximadamente 1 gM, idóneamente en el margen de aproximadamente 0,01 nM a aproximadamente 500 nM, apropiadamente en el margen de aproximadamente 0,01 nM a aproximadamente 100 nM, idealmente en el margen de aproximadamente 0,01 nM a aproximadamente 50 nM, y preferiblemente en el margen de aproximadamente 0,01 nM a aproximadamente 25 nM.
Inh ib ic ión de la liberación de IL-6 inducida p o r la IL-17A desde la línea ce lu la r de fib rob lastos dérm icos
El propósito de este ensayo es comprobar la capacidad de neutralización que tienen las proteínas de tipo IL-17 en un sistema de células primarias humanas. La estimulación de los fibroblastos dérmicos humanos (HDF) normales con la IL-17 sola produce solo una señal muy débil, pero en combinación con otras citocinas, tales como el TNFa, se puede observar un efecto sinérgico en la producción de las citocinas inflamatorias, es decir, la IL-6.
Los HDF se estimularon con la IL-17A (50 pM) en combinación con el TNF-a (25 pM). A continuación, se midió la respuesta de la IL-6 resultante utilizando un kit de FRET homogéneo de resolución temporal de Cisbio. El kit utiliza dos anticuerpos monoclonales, uno marcado con Eu-Cryptate (donante) y el segundo con d2 o XL665 (aceptor). La intensidad de la señal es proporcional a la concentración de IL-6 presente en la muestra (la relación se calcula con 665/620 x 104).
En este ensayo se mide la capacidad que tiene un compuesto para inhibir la liberación de la IL-6 inducida por la IL-17 desde los fibroblastos dérmicos humanos.
Las células de HDF (Sigma #106-05n) se cultivaron en medios completos (DMEM FCS al 10% L-glutamina a 2 mM) y se mantuvieron en un matraz de cultivo de tejidos usando las técnicas estándares. Las células se recogieron del matraz de cultivo de tejidos en la mañana del ensayo usando TrypLE (Invitrogen #12605036). El TrypLE se neutralizó utilizando el medio completo (45 ml) y las células se centrifugaron a 300 x g durante 3 min. Las células se resuspendieron en el medio completo (5 ml) contadas y ajustadas a una concentración de 3,125 x 104 células/ml antes de agregarse a la placa de ensayo de 384 pocillos (Corning #3701) a 40 gl por pocillo. Las células se dejaron durante un mínimo de 3 h, a 37 °C/5 % de CO2, para adherirse a la placa.
Los compuestos se diluyeron en serie en DMSO antes de recibir una dilución acuosa en una placa de dilución de 384 pocillos (Greiner #781281), donde se transfirieron 5 gl desde la placa de titulación a 45 gl del medio completo y se mezclaron para dar una solución que contenía el DMSO al 10 %.
Se prepararon mezclas de las citocinas TNFa e IL-17 en medio completo hasta la concentración final de TNFa a 25 pM/IL-17A a 50 pM, luego se agregaron 30 gl de la solución a una placa de reactivo de 384 pocillos (Greiner #781281).
Se transfirieron 10 gl desde la placa de dilución acuosa a la placa de reactivos que contenía 30 gl de las citocinas
diluidas, para dar una solución de DMSO al 2,5 %. Los compuestos se incubaron con las mezclas de citocinas durante 1 h a 37 °C. Después de la incubación, se transfirieron 10 pl a la placa de ensayo para obtener una solución de DMSO al 0,5 %, luego se incubó durante 18-20 h a 37 °C/5 % de CO2.
A partir del kit Cisbio de FRET de IL-6 (Cisbio #62IL6PEB) se diluyeron criptato de europio y Alexa 665 en el tampón de reconstitución y se mezclaron a 1:1, según el prospecto del kit. A una placa blanca de 384 pocillos de bajo volumen (Greiner #784075) se le añadieron reactivos de FRET (10 pl), luego se transfirió el sobrenadante (10 pl) desde la placa de ensayo a la placa de reactivos de Greiner. La mezcla se incubó a temperatura ambiente durante 3 h con agitación suave (<400 rpm) antes de leerla en un lector de placas Synergy Neo 2 (excitación: 330 nm; emisión: 615/645 nm).
Cuando se comprobaron en el ensayo anterior, se encontró que los compuestos de los ejemplos adjuntos exhibían valores de CI50 de 10 pM o mejores.
Cuando se comprueban en el ensayo anterior, los compuestos de los ejemplos adjuntos exhiben valores de CI50 generalmente en el margen de aproximadamente 0,01 nM a aproximadamente 10 pM, normalmente en el margen de aproximadamente 0,01 nM a aproximadamente 5 pM, típicamente en el margen de aproximadamente 0,01 nM a aproximadamente 1 pM, idóneamente en el margen de aproximadamente 0,01 nM a aproximadamente 500 nM, apropiadamente en el margen de aproximadamente 0,01 nM a aproximadamente 100 nM, idealmente en el margen de aproximadamente 0,01 nM a aproximadamente 50 nM, y preferiblemente en el margen de aproximadamente 0,01 nM a aproximadamente 25 nM.
Los siguientes ejemplos ilustran la preparación de compuestos según la invención.
Ejemplos
Abreviaturas



Condiciones analíticas
Los compuestos se nombraron con la ayuda de ACD/Name Batch (NetWork), versión 11.01 y/o Accelrys Draw 4.2 y/o Elemental, Dotmatics y/o Chemaxon.
Todas las reacciones en las que intervinieron reactivos sensibles al aire o a la humedad se realizaron en una atmósfera de nitrógeno con solventes secos y material de vidrio.
Los espectros de RMN se registraron en un espectrómetro Bruker Avance III HD a 500 MHz, 400 MHz, 300 MHz o 250 MHz.
Las rotaciones ópticas específicas se midieron usando un polarímetro Rudolph Research Analytical Autopol 1, S2 Serial 32026.
La uPLC-MS se realizó en un sistema Waters Acquity UPLC acoplado a un detector Waters Acquity PDA, un detector ELS y un MSD (Barrido positivo: 150-850).
M étodo 1
Columna de Phenomenex Kinetex-XB, C18 2,1 x 100 mm, 1,7 gm

M étodo 2
Columna de Waters UPLC® CSH™ C18, 2,1 mm x 100 mm, 1,7 gm

HPLC-MS
1. Realizado en un sistema Shimadzu LCMS-2010EV acoplado a los detectores SPD-M20A PDA y PL 2100.
M étodo 3
Columna de Phenomenex Kinetex Core-Shell C8 50 x 2,1 mm, 5 gm, protegida por la columna de Phenomenex 'Security Guard'

M étodo 4
Columna de Waters Atlantis dC18 (2,1 x 100 mm, 3 |um)

2. Realizado en un sistema LCMS de Agilent 1200-6120 acoplado a un espectrómetro de masas (ES) Detection (de 230 a 400 nm y 215 nm) y Mass Spect Detection de Agilent 6120 de 120 a 800 mlz.
M étodo 5
Columna X-Bridge C18 de Waters 2,1 x 20 mm, 2,5 |uM

M étodo 6
Columna X-Bridge C18 de Waters 2,1 x 20 mm, 2,5 |um

M étodo 7
Columna X-Bridge C18 de Waters 2,1 x 20 mm, 2,5 |um


Purificación automatizada por HPLC de fase inversa preparativa
1. Realizada utilizando un sistema de Gilson con una bomba Gilson 306, un autoinyector Gilson 215, un recolector de fracciones Gilson 215 y un detector de UV Gilson 156.
M étodo 8
Columna X-Bridge C18 de Waters 30 x 100 mm, 5 gm

2. Realizado utilizando un sistema de Gilson con una bomba Gilson 331 y 332, un autoinyector Gilson GX281, un recolector de fracciones Gilson GX281 y un detector UV Gilson 159.
M étodo 9
Columna X-Bridge C18 de Waters 30 x 100 mm, 10 gm

M étodo 10
Columna X-Bridge C18 de Waters 30 x 100 mm, 10 gm

M étodo 11
Columna Sunfire C18 de Waters 30 x 100 mm, 10 |um

Las separaciones por cromatografía en columna se realizaron con un sistema Biotage® Isolera 4 con columnas de gel de sílice preempaquetadas Biotage® SNAP KP-Sil.
Análisis de SFC quiral
M étodo 12
Sistema de Waters Thar 3100 SFC conectado a un detector Waters 2998 PDA
M étodo 13
Sistema de Waters UPC2-SQD2
Separación de SFC quiral
M étodo 14
Sistema de Waters Thar SFC con una bomba Waters Thar FDM, un autoinyector Waters Thar Alias, un recolector de fracciones Waters Thar y un detector Waters 2998 PDA
M étodo 15
Waters Prep 100-SQD2
La HPLC-MS se realizó en un sistema ZQ de Waters acoplado a los detectores Waters 2996 PDA y Waters 2420. M étodo 16
Columna de Phenomenex Gemini-NX C182,0 mm x 50 mm, 3 |um

M étodo 17
Columna de Waters Atlantis dC184,6 x 50 mm, 3 gm

M étodo 18
Columna de Waters Atlantis dC184,6 x 50 mm, 3 gm

La HPLC-MS se realizó en un sistema LCMS de Agilent 1200-6120 acoplado a un espectrómetro de masas (ES) Detection (230 a 400 nm y 215 nm) y Mass Spec Detection 6120 de Agilent de 15120 a 800 m/z.
M étodo 19
Columna X-Bridge C18 de Waters 2,1 x 20 mm, 2,5 gm


La purificación automatizada por HPLC de fase inversa preparativa se realizó utilizando un sistema de Gilson con una bomba Gilson 331 y 332, un autoinyector Gilson GX281, un recolector de fracciones Gilson GX281 y un detector UV Gilson 159.
M étodo 20
Columna de Sunfire C18 Waters 30 x 100 mm, 10 pm

La HPLC-MS se realizó en un sistema de LCMS Agilent 1200RR-6140 acoplado a un detector DAD SL.
M étodo 21
Columna XBridge C18 de 2,1 x 20 mm, 2,5 pm

La purificación automatizada por HPLC de fase inversa preparativa se realizó utilizando un sistema Waters FractionLynx Prep acoplado a un espectrómetro de masas SQD2 y un detector Waters 2998 PDA.
M étodo 22
Columna de Waters XBridge Prep C18 19 x 100 mm, 5 pm


La uPLC-MS se realizó en un sistema Waters Classic Acquity-QDa LCMS acoplado a un detector Waters Acquity PDA.
M étodo 23
Columna de Waters Acquity UPLC BEH, C18 2,1 x 50 mm, 1,7 pm

La purificación automatizada por HPLC de fase inversa preparativa se realizó utilizando un sistema Waters FractionLynx Prep acoplado a un espectrómetro de masas SQD2 y un detector Waters 2998 PDA.
M étodo 24
Columna de Waters XBridge Prep Phenyl 19 x 150 mm, 5 pm

M étodo 25
Columna de Waters XBridge Prep Phenyl 19 x 150 mm, 5 pm


M étodo 26
Columna de Waters XBridge Prep C18 19 x 100 mm, 5 gm

La uPLC-MS se realizó en un sistema de LCMS Waters Classic Acquity-QDa acoplado a un detector Waters Acquity PDA.
M étodo 27
Columna de Waters Acquity UPLC BEH, C18 2,1 x 50 mm, 2,5 gm

M étodo 28
Columna de Waters Acquity UPLC BEH, C18 2,1 x 30 mm, 1,7 gm

2,5 95,00  5,00
5,00
La HPLC-MS se realizó en un sistema Shimadzu LCMS-2010EV acoplado a los detectores SPD-M20A PDA y PL 2100.
M étodo 29
Columna de Phenomenex Kinetex Core-Shell C8 50 x 2,1 mm, 2,6 pm protegida por la columna de Phenomenex 'Security Guard'

La purificación automatizada por HPLC de fase inversa preparativa se realizó utilizando un sistema de Gilson con una bomba Gilson 306, un autoinyector Gilson 215, un recolector de fracciones Gilson 215 y un detector UV Gilson 156.
M étodo 30
Columna Sunfire C18 de Waters 19 x 100 mm, 5 pm

M étodo 31
Columna Sunfire C18 de Waters 19 x 100 mm, 5 pm


Separación por HPLC quiral
M étodo 32
Sistema de Gilson con una bomba Gilson 321/322, un autoinyector Gilson GX241, un recolector de fracciones Gilson PREP FC y un detector UV Gilson 171/172.
HPLC-MS
1. Realizada en un Shimadzu LC-20AB & MS 2010.
M étodo 33
Columna Luna-C18(2) 2,0 x 30 mm, 3 pm

2. Realizado en un Shimadzu 20D y MS2020.
M étodo 34
Columna de Zorbax Eclipse XDB-C18 2,1 x 30 mm, 3,5 pm

La SFC quiral se realizó en un sistema de Agilent 1100, DAD.
M étodo 35
Columna de Lux Cellulose-1 4,6 x 150 mm, 3 pm

Intermedio 1
4-(2,4-Dinitrofenil)tetrahidropiran-4-carboxilato de metilo
Se añadió hidruro de sodio (dispersión al 60 % en aceite mineral, 33 mg, 0,84 mmol) a una solución de (2,4-dinitrofenil)acetato de metilo (77 mg, 0,32 mmol) y 1-bromo-2-(2-bromoetoxi)etano (0,046 ml, 0,321 mmol) en DMF (3 ml) a 0 °C. Se retiró el baño de enfriamiento y la mezcla de reacción se agitó durante 10 min a 20 °C, luego se calentó a 90 °C durante 4 h. La mezcla de reacción se enfrió a temperatura ambiente y se le añadió una solución acuosa saturada de cloruro de amonio (5 ml). La mezcla resultante se extrajo con acetato de etilo (2 x 30 ml). La fase orgánica se lavó con salmuera, se secó sobre sulfato de sodio y se concentró al vacío. El residuo se purificó mediante cromatografía en columna ultrarrápida, usando un gradiente de acetato de etilo en heptano (0-100 %), para proporcionar el compuesto del título (49 mg, 49 %) como una goma de color naranja pálido. 5h (250 MHz, CDCh) 8,62 (d, J 2,5 Hz, 1H), 8,48 (dd, J 8,8, 2,5 Hz, 1H), 7,89 (d, J 8,9 Hz, 1H), 3,97 (ddd, J 12,0, 9,5, 2,7 Hz, 2H), 3,80 (td, J 7,8, 4,1 Hz, 2H), 3,73 (s, 3H), 2,40 (d, J 13,8 Hz, 2H), 2,10 (ddd, J 13,8, 9,5, 4,3 Hz, 2H). HPLC-MS (método 3): MH+ mlz no se observó el ion original, TR 1,08 min.
Intermedio 2
6-Aminospiro[indolina-3,4'-tetrahidropiran]-2-ona
Se añadió polvo de hierro (3,79 g, 67,8 mmol) a una solución agitada del intermedio 1 (2,5 g, 4,24 mmol) en una mezcla de etanol/agua/solución acuosa saturada de cloruro amónico (8:1:1; 81 ml) y la suspensión se agitó a 60 °C en una atmósfera de nitrógeno durante 6,5 h. Después de enfriarla a temperatura ambiente, la mezcla se diluyó con agua (100 ml) y etanol (100 ml). La suspensión se filtró a través de un lecho de tierra de diatomeas, lavando secuencialmente con etanol (3 x 50 ml), metanol (3 x 100 ml), agua (100 ml) y acetato de etilo (3 x 100 ml). El filtrado se concentró al vacío. El residuo se diluyó con agua (100 ml) y se extrajo con acetato de etilo (3 x 100 ml), luego con isopropanol/cloroformo 1:1 (3 x 50 ml). Los extractos orgánicos combinados se lavaron con salmuera (100 ml) y se secaron sobre sulfato de sodio, luego se filtraron y concentraron al vacío. El residuo se purificó mediante cromatografía en columna ultrarrápida (columna KP-NH), utilizando un gradiente de acetato de etilo en heptano (50-100 %) seguido de un gradiente de metanol en acetato de etilo (0-10 %), para proporcionar el compuesto del título (571 mg, 61 %) como un polvo tostado. 5h (500 MHz, DMSO-ds) 10,09 (s, 1H), 7,11 (d, J 8,6 Hz, 1H), 6,19-6,07 (m, 2H), 5,07 (s, 2H), 3,97 (ddd, J 11,0, 6,9, 3,8 Hz, 2H), 3,76 (ddd, J 11,3, 7,5, 3,6 Hz, 2H), 1,70 (ddd, J 11,4, 7,5, 3,7 Hz, 2H), 1,52 (ddd, J 13,3, 6,8, 3,5 Hz, 2H). HPLC-MS (método 7): MH+ mlz 219, TR 1,00 min.
Intermedio 3
Hidrocloruro de (2S)-2-amino-3-(2-clorofenil)propanoato de metilo
Se añadió gota a gota cloruro de tionilo (2 ml, 27,42 mmol) a metanol anhidro (7,5 ml) enfriado a 0 °C en nitrógeno. Después de agitar durante 5 min, se le añadió en porciones 2-cloro-L-fenilalanina (5,0 g, 25,05 mmol), seguido de metanol anhidro (7,5 ml). La suspensión se calentó a 50 °C durante 5 h en nitrógeno, luego se eliminó lo volátil al vacío. El residuo se suspendió en éter dietílico (20 ml) y se concentró nuevamente para producir el compuesto del título (5,89 g, 94 %) como un polvo tostado. 5h (500 MHz, DMSO-d6) 8,91 (br s, 3H), 7,49-7,39 (m, 2H), 7,35-7,30 (m, 2H), 4,12 (dd, J 9,0, 6,2 Hz, 1H), 3,59 (s, 3H), 3,39-3,32 (m obs., 1H), 3,23 (dd, J 13,8, 9,0 Hz, 1H). HPLC-MS (método 7): MH+ mlz 214, TR 1,58 min.
Intermedio 4
(2 S )-3-(2-Clorofenil)-2-{[2-(piridin-4-il)acetil]amino}propanoato de metilo
Se añadió lentamente DIPEA (12,4 ml, 75,03 mmol) a una suspensión agitada del intermedio 3 (5,89 g, 23,53 mmol), hidrocloruro de ácido 2-( -4-il)acético (4,29 g, 24,71 mmol) y HATU (9,84 g, 25,88 mmol) en DMF anhidro (47 ml). La mezcla se agitó a 20 °C en nitrógeno durante 16 h. Lo volátil se eliminó al vacío y el residuo se repartió entre acetato de etilo (100 ml) y solución acuosa saturada de carbonato de sodio (100 ml). Se separó la capa acuosa y se extrajo con acetato de etilo (2 x 100 ml). Los extractos orgánicos combinados se lavaron con agua (50 ml) y salmuera (2 x 50
ml), luego se secaron sobre sulfato de magnesio, se filtraron y se concentraron al vacío. La goma de color rojo oscuro en bruto resultante se separó mediante cromatografía en columna ultrarrápida, utilizando un gradiente de metanol en DCM (0-6,5 %), para proporcionar el compuesto del título (5,95 g, 76 %) como un aceite viscoso de color naranja. 5h (500 MHz, DMSO-ds) 8,74 (d, J 8,2 Hz, 1H), 8,47-8,37 (m, 2H), 7,41 (dd, J 7,8, 1,3 Hz, 1H), 7,31-7,19 (m, 3H), 7,16 7,04 (m, 2H), 4,61 (ddd, J 9,9, 8,2, 5,4 Hz, 1H), 3,62 (s, 3H), 3,45 (s, 2H), 3,25 (dd, J 13,8, 5,4 Hz, 1H), 2,97 (dd, J 13,8, 9,9 Hz, 1H). HPLC-MS (método 5): MH+ mlz 333, TR 1,44 min. SFC quiral (método 12, Chiralpak AD-H 25 cm, etanol al 35 %-dióxido de carbono al 65 %, 4 ml/min): TR 2,42 min (100 %).
Intermedio 5
Ácido (2S)-3-(2-clorofenil)-2-{[2-(piridin-4-il)acetil]amino}propanoico
Se añadió una solución de monohidrato de hidróxido de litio (1,20 g, 28,53 mmol) en agua (18 ml) a una solución agitada del intermedio 4 (5,93 g, 17,83 mmol) en THF/MeOH (1:1; 36 ml) a 0 °C. La mezcla se dejó calentar lentamente a 20 °C y se agitó durante un total de 16 h, formando una suspensión espesa. Se eliminó lo volátil al vacío. La suspensión pastosa acuosa se diluyó con agua (150 ml) y una solución acuosa de hidróxido de sodio 1 M (50 ml). La capa acuosa se lavó con tert-butilmetiléter (50 ml). La fase orgánica se separó y se extrajo con una solución acuosa de hidróxido de sodio a 1 M (50 ml). Las capas acuosas combinadas se lavaron con tert-butilmetiléter (50 ml). El pH de la fase acuosa se ajustó a 5-6 usando ácido clorhídrico concentrado. El material se extrajo secuencialmente con 1:1 DCM/isopropanol (8 x 100 ml) y 2-metiltetrahidrofurano (12 x 100 ml). Los extractos orgánicos se lavaron con salmuera (50 ml), se secaron sobre sulfato de magnesio y se filtraron, luego se combinaron y se redujeron al vacío. El polvo bruto amarillo resultante se trituró en DCM (100 ml), luego los sólidos se recogieron por filtración, se lavaron con DCM (2 x 50 ml) y se secaron al vacío a 50 °C durante 4 h para obtener el compuesto del título (5,70 g, cuantitativo) como un polvo blanquecino. 5h (500 MHz, DMSO-d6) 12,87 (br s, 1H), 8,64 (d, J 8,5 Hz, 1H), 8,41 (d, J 5,4 Hz, 2H), 7,39 (dd, J 7,9, 1,2 Hz, 1H), 7,29 (dd, J 7,4, 1,7 Hz, 1H), 7,24 (td, J 7,6, 1,8 Hz, 1H), 7,19 (td, J 7,4, 1,3 Hz, 1H), 7,10 (d, J 5,9 Hz, 2H), 4,56 (ddd, J 10,4, 8,6, 4,7 Hz, 1H), 3,43 (s, 2H), 3,28 (dd, J 13,8, 4,7 Hz, 1H), 2,91 (dd, J 13,9, 10,5 Hz, 1H). HPLC-MS (método 7): MH+ m/z 319, TR 1,26 min. SfC quiral (método 12, Chiralpak AD-H 25 cm, metanol al 25 %, dióxido de carbono al 75 %, 4 ml/min): TR 2,22 min (100 %).
Intermedio 6
Cloruro de [(1S)-1-ciclohexil-2-metoxi-2-oxoetil]amonio
Se añadió gota a gota cloruro de tionilo (8 ml, 110,15 mmol) a metanol anhidro (60 ml) enfriado a 0 °C en nitrógeno. Después de agitar durante 5 min, se le añadió en porciones ácido (2S)-amino-(ciclohexil)etanoico (15,72 g, 100,00 mmol), luego la mezcla de reacción se calentó a 50 °C durante 5 h en nitrógeno. Se eliminó el solvente al vacío y el residuo se trituró en éter dietílico (250 ml). Los sólidos se recogieron por filtración para obtener el compuesto del título (16,9 g, 81 %) como un sólido blanco. 5h (500 MHz, DMSO-ds) 8,52 (s, 3H), 3,81 (d, J 5,1 Hz, 1H), 3,75 (s, 3H), 1,86 1,77 (m, 1H), 1,74-1,67 (m, 3H), 1,65-1,58 (m, 2H), 1,22-1,12 (m, 3H), 1,12-1,03 (m, 1H), 0,97 (qd, J 12,9, 12,2, 3,8 Hz, 1H). HPLC-MS (método 6): m H+ m/z 172, TR 2,32 min.
Intermedio 7
(2 S )-2-Ciclohexil-2-{[2-(piridin-4-il)acetil]amino}acetato de metilo
Se añadió 2,4,6-tripropil-1,3,5,2,4,6-trioxatrifosforinano-2,4,6-trióxido (solución al 50 % en peso en acetato de etilo, 39,2 ml, 66,44 mmol) durante 30 min a una solución agitada del intermedio 6 (6,90 g, 33,22 mmol), hidrocloruro de ácido 2-(piridin-4-il)acético (6,92 g, 39,87 mmol) y DIPEA (19,2 ml, 116,27 mmol) en THF anhidro (210 ml). La mezcla se agitó a 20 °C en nitrógeno durante 3,5 h, luego se diluyó con agua (250 ml) y se lavó con acetato de etilo (2 x 200 ml). El pH de la fase acuosa se ajustó a pH 10 mediante la adición lenta de una solución acuosa de hidróxido de sodio a 4 M (21 ml), luego la fase acuosa se extrajo con acetato de etilo (2 x 300 ml). Los extractos orgánicos se combinaron, se lavaron con agua (250 ml) y salmuera (250 ml), luego se secaron sobre sulfato de magnesio anhidro y se filtraron. Se eliminó el solvente al vacío. El residuo se separó mediante cromatografía en columna ultrarrápida, utilizando un gradiente de metanol en DCM (1 -8 %), para obtener el compuesto del título (4,53 g, 47 %) como un sólido amarillo. 5h (500 MHz, DMSO-de) 8,53-8,44 (m, 3H), 7,30-7,24 (m, 2H), 4,16 (dd, J 8,7, 6,5 Hz, 1H), 3,62 (s, 3H), 3,57 (d, J 14,2 Hz, 1H), 3,54 (d, J 14,4 Hz, 1H), 1,73-1,50 (m, 6H), 1,25-1,03 (m, 4H), 0,98 (qd, J 12,5, 3,5 Hz, 1H). HPLC-MS (método 5): MH+ mlz 291, TR 1,44 min. SFC quiral (método 12, Chiralpak a D-H 25 cm, etanol al 35 %, dióxido de carbono al 65 %, 4 ml/min): TR3,07 min (100 %).
Intermedio 8
(2 S )-2-Ciclohexil-2-{[2-(piridin-4-il)acetil]amino}acetato de litio
Se añadió una solución de monohidrato de hidróxido de litio (0,99 g, 23,70 mmol) en agua (16 ml) a una solución agitada del intermedio 7 (4,53 g, 15,80 mmol) en THF/MeOH (1:1; 32 ml) a 0 °C. La mezcla se dejó calentar lentamente a 20 °C y se agitó durante un total de 16 h, formando una suspensión espesa. Se eliminó lo volátil al vacío, luego el residuo se diluyó con agua (20 ml). Los sólidos se recogieron por filtración, luego se lavaron con agua (20 ml) y tertbutilmetiléter (20 ml), antes del secado al vacío a 50 °C durante 16 h, para obtener el compuesto del título (3,53 g,
79 %) como un polvo blanquecino. 5h (500 MHz, D2O) 8,56-8,45 (m, 2H), 7,42 (d, J 6,1 Hz, 2H), 4,13 (d, J 5,8 Hz, 1H), 3,81 (d, J 14,9 Hz, 1H), 3,72 (d, J 14,9 Hz, 1H), 1,85-1,69 (m, 3H), 1,68-1,54 (m, 3H), 1,24 (dtt, J 16,0, 12,7, 3,3 Hz, 2H), 1,16-0,91 (m, 3H). h Pl C-MS (método 5): Mh mlz 277, TR 1,15 min. SfC quiral (método 12, Chiralpak iC 25 cm, isopropanol al 40 % dietilamina al 0,2 %-dióxido de carbono al 60 %, 4 ml/min): TR 4,40 min (100 %).
Intermedio 9
2-Ciclooctiliden-2-formamidoacetato de metilo
Una solución de ferf-butóxido de potasio en THF (1 M, 48 ml, 48 mmol) se añadió gota a gota a una solución roja de isocianoacetato de metilo (4,0 ml, 41,8 mmol) en THF anhidro (40 ml) a aproximadamente -65 °C en nitrógeno. Después de agitar durante 5 min, se le añadió lentamente a -70°C una solución de ciclooctanona (5 g, 39,62 mmol) en THF anhidro (20 ml). La mezcla de reacción se agitó a -70°C durante 30 min, luego se retiró el baño de enfriamiento y la mezcla se dejó calentar a 20 °C con agitación en nitrógeno durante 60 h. La solución de color rojo intenso resultante se inactivó con agua (100 ml) y se agitó a 20 °C durante 1 h. El residuo se extrajo con acetato de etilo (3 x 100 ml). Los extractos orgánicos combinados se lavaron con salmuera (50 ml) y se secaron sobre sulfato de magnesio, luego se filtraron y se concentraron al vacío. El aceite anaranjado viscoso bruto resultante se separó mediante cromatografía en columna ultrarrápida utilizando un gradiente de acetato de etilo en heptano (0-90 %) para proporcionar el compuesfo del fífulo (5,37 g, 58 %) como un aceite viscoso de color naranja, que se solidificó al reposar. Rotámero principal: 5h (500 MHz, DMSO-ds ) 9,31 (s, 1H), 8,01 (d, J 1,5 Hz, 1H), 3,60 (s, 3H), 2,52-2,47 (m, 2H), 2,31 -2,23 (m, 2H), 1,74-1,60 (m, 4H), 1,50-1,31 (m, 6H). HPLC-MS (método 5): MNa+ m/z 248, TR 1,63 min.
Intermedio 10
2-Ciclooctil-2-formamidoacetato de metilo
Se añadieron cuidadosamente virutas de magnesio (3,15 g, 129,60 mmol) a una solución agitada del infermedio 9 (2,91 g, 12,95 mmol) en metanol anhidro (65 ml) a 0 °C en nitrógeno. La suspensión se agitó a 0 °C durante 1 h, luego se dejó calentar a 20 °C durante 2 h. Se continuó agitando la suspensión turbia a 20 °C durante 16 h. Se le añadió una porción adicional de virutas de magnesio (1 g, 41,14 mmol) y la suspensión se agitó a 20 °C durante 3,5 h en nitrógeno. La mezcla se concentró cuidadosamente al vacío. El residuo se suspendió en acetato de etilo (100 ml) y agua (200 ml), luego se enfrió a 0 °C. Se le añadió con precaución ácido clorhídrico acuoso (1 M, 100 ml), luego se le añadió con precaución ácido clorhídrico concentrado (pH 5) para ayudar a la disolución de los sólidos. La fase orgánica se separó, luego la suspensión acuosa se trató con ácido clorhídrico concentrado (pH 4) y el material se extrajo con acetato de etilo (100 ml). La suspensión acuosa se trató con ácido clorhídrico concentrado (pH 2) y el material se extrajo con acetato de etilo (100 ml). La suspensión acuosa se trató adicionalmente con ácido clorhídrico concentrado (pH 1) y el material se extrajo con acetato de etilo (100 ml). Los extractos orgánicos combinados se lavaron con salmuera (50 ml) y se secaron sobre sulfato de magnesio, luego se filtraron y se concentraron al vacío. El aceite viscoso bruto de color naranja resultante se separó mediante cromatografía en columna ultrarrápida, usando un gradiente de acetato de etilo en heptano (0-80 %), para proporcionar el compuesfo del fífulo (1,53 g, 52 %) como un aceite viscoso de color naranja. Rotámero principal: 5h (500 MHz, DMSO-d6) 8,46 (d, J 8,5 Hz, 1H), 8,06 (s, 1H), 4,29 (dd, J 8,6, 6,1 Hz, 1H), 3,64 (s, 3H), 2,04-1,93 (m, 1H), 1,73-1,19 (m, 14H). HPLC-MS (método 4): MH+ m/z 228, TR 3,94 min.
Intermedio 11
Hidrocloruro de 2-amino-2-ciclooctilacetato de metilo
Se añadió cuidadosamente cloruro de acetilo (1,9 ml, 26,72 mmol) a 0 °C a una solución agitada del infermedio 10 (1,54 g, 6,77 mmol) en metanol (68 ml) en nitrógeno. Después de agitar durante 5 min, la solución se calentó a 50 °C durante 2 h, luego lo volátil se concentró al vacío. El polvo naranja bruto resultante se trituró en éter dietílico (40 ml) y los sólidos se recogieron por filtración, lavando con éter dietílico (2 x 20 ml). Los sólidos se secaron al vacío a 50 °C durante 6 h para obtener el compuesfo del fífulo (1,43 g, 90 %) como un polvo tostado. 5h (500 MHz, DMSO-d6) 8,61 (br s, 3H), 3,86 (d, J 4,4 Hz, 1H), 3,73 (s, 3H), 2,19-2,09 (m, 1H), 1,68-1,37 (m, 13H), 1,32-1,20 (m, 1H). HPLC-MS (método 3): MH+ m/z 200, TR 0,75 y 0,86 min.
Intermedio 12
2-Ciclooctil-2-[(2-metilpirazol-3-carbonil)amino]acetato de metilo
Se añadió DIPEA (1,05 ml, 6,35 mmol) a una solución agitada del infermedio 11 (500 mg, 2,12 mmol), ácido 1-metil-1 H-pirazol-5-carboxílico (269 mg, 2,12 mmol) y HATU (969 mg, 2,55 mmol) en DMF anhidro (10 ml) en una atmósfera de nitrógeno. La mezcla se agitó a 20 °C durante 18 h, luego se inactivó con una solución acuosa saturada de hidrogenocarbonato de sodio (50 ml) y agua (50 ml). El material se extrajo con acetato de etilo (3 x 50 ml). Las capas orgánicas combinadas se lavaron con salmuera (2 x 50 ml) y se secaron sobre sulfato de sodio, luego se filtraron y se concentraron al vacío. El residuo se purificó mediante cromatografía en columna ultrarrápida, usando un gradiente de acetato de etilo en heptano (0-100 %), para proporcionar el compuesfo del fífulo (618 mg, 99 %) como un aceite de color amarillo anaranjado. 5h (500 MHz, DMSO-d6) 8,60 (d, J 8,3 Hz, 1H), 7,46 (d, J 2,1 Hz, 1H), 7,01 (d, J 2,1 Hz, 1H),
4,37 (t, J 8,1 Hz, 1H), 4,01 (s, 3H), 3,66 (s, 3H), 2,22-2,08 (m, 1H), 1,80-1,38 (m, 13H), 1,37-1,29 (m, 1H). HPLC-MS (método 5): MH+ m/z 308, T r 1,87 min.
Intermedio 13
2-Ciclooctil-2-f(2-metilpirazol-3-carbonil)amino]acetato de litio
A una solución agitada del intermedio 12 (519 mg, 1,69 mmol) en THF (9 ml) y agua (4,5 ml) se le añadió monohidrato de hidróxido de litio (0,106 g, 2,53 mmol). La mezcla de reacción se agitó a 20 °C durante 22 h, luego se concentró y se secó al vacío durante 4 h, para proporcionar el compuesto del título (505 mg, cuantitativo) como un sólido amarillo.
5h (500 MHz, DMSO-d6) 7,60 (d, J 7,6 Hz, 1 H), 7,42 (d, J 2,0 Hz, 1H), 6,74 (d, J 2,0 Hz, 1H), 4,01 (s, 3H), 3,88 (dd, J 7,6, 4,3 Hz, 1H), 2,11-2,01 (m, 1H), 1,74-1,22 (m, 14H). HPLC-MS (método 5): MH+ mlz 294, TR 1,71 min.
Intermedio 14
2-(2-Aminopiridin-4-il)acetato de metilo
Se añadió gota a gota cloruro de tionilo (0,3 ml, 4,14 mmol) al ácido (2-aminopiridin-4-il)acético (594 mg, 3,90 mmol) en metanol (12 ml) a 0 °C. La mezcla de reacción se dejó calentar a 20 °C y se agitó durante 18 h, luego se evaporó hasta la sequedad. El residuo se repartió entre isopropanol/cloroformo (1:1; 60 ml) y solución acuosa saturada de bicarbonato de sodio (60 ml). Se separaron las capas, luego la fase acuosa se extrajo con isopropanol/cloroformo (1:1; 2 x 60 ml). Las capas orgánicas combinadas se secaron sobre sulfato de sodio, se filtraron y se concentraron al vacío, para proporcionar el compuesto del título (605 mg, 93 %) como un sólido rosa-rojizo. 5h (500 MHz, DMSO-d6) 7,81 (d, J 5,2 Hz, 1H), 6,37 (dd, J 5,2, 1,4 Hz, 1H), 6,32 (s, 1H), 5,86 (s, 2H), 3,61 (s, 3H), 3,51 (s, 2H). HPLC-MS (método 6): MH+ m/z 167, TR 0,70 min.
Intermedio 15
2-[2-(ferf-Butoxicarbonilamino)piridin-4-il]acetato de metilo
Una solución de dicarbonato de di-terf-butilo (433 mg, 1,98 mmol) en tert-butanol (6 ml) se añadió lentamente a una solución agitada del intermedio 14 (300 mg, 1,81 mmol) en tert-butanol (12 ml). La mezcla se agitó a 20 °C en nitrógeno durante 16 h, luego se eliminó lo volátil al vacío. El residuo se separó mediante cromatografía en columna ultrarrápida, usando un gradiente de acetato de etilo en heptano (0-100 %), para proporcionar el compuesto del título (325 mg, 67 %) como un polvo blanquecino. 5h (500 MHz, DMSO-d6) 9,74 (br s, 1H), 8,16 (d, J 5,1 Hz, 1H), 7,74 (s, 1H), 6,93 (dd, J 5,1, 1,4 Hz, 1H), 3,72 (s, 2H), 3,63 (s, 3H), 1,47 (s, 9H). HPLC-MS (método 5): MH+ mlz 211, TR 1,67 min.
Intermedio 16
Ácido 2-[2-(ferf-butoxicarbonilam ino)piridin-4-il]acético
Se añadió una solución de monohidrato de hidróxido de litio (74 mg, 1,76 mmol) en agua (1,2 ml) a una solución agitada del intermedio 15 (311 mg, 1,17 mmol) en MeOH/THF (1:1; 2,4 ml). La solución se agitó a 20 °C al aire durante 16 h, luego se eliminó lo volátil al vacío. El residuo se diluyó con agua (10 ml) y se lavó con tert-butilmetiléter (2 x 5 ml). Los lavados orgánicos combinados se extrajeron con una solución acuosa de hidróxido de sodio (0,1 M, 5 ml). El pH de la fase acuosa se ajustó a pH 5 con ácido clorhídrico acuoso a 3 M y el material se extrajo con acetato de etilo (3 x 20 ml). Los extractos orgánicos combinados se lavaron con salmuera (20 ml) y se secaron sobre sulfato de magnesio, luego se filtraron y se concentraron al vacío, para proporcionar el compuesto del título (285 mg, 97 %) como un polvo blanco. 5h (500 MHz, DMSO-ds) 12,49 (br s, 1H), 9,70 (br s, 1H), 8,15 (d, J 5,1 Hz, 1H), 7,73 (s, 1H), 6,92 (dd, J 5,1, 1,4 Hz, 1H), 3,59 (s, 2H), 1,47 (s, 9H). HPLC-MS (método 5): [M+2H-tBu]+ mlz 197, TR 1,36 min.
Intermedio 17
2-({2-[2-(fert-butoxicarbonilamino)piridin-4-il]acetil}amino)-2-ciclooctilacetato de metilo
Se añadió DIPEA (380 pl, 2,30 mmol) a una solución agitada del intermedio 11 (180 mg, 0,76 mmol), intermedio 16 (197 mg, 0,77 mmol) y HATU (350 mg, 0,92 mmol) en DMF anhidra (3 ml) en una atmósfera de nitrógeno. La mezcla se agitó a 20 °C durante 18 h, luego se inactivó con una solución acuosa saturada de bicarbonato de sodio (20 ml) y agua (20 ml). El material se extrajo con acetato de etilo (3 x 20 ml). Las capas orgánicas combinadas se lavaron con salmuera (2 x 20 ml) y se secaron sobre sulfato de sodio, luego se filtraron y se concentraron al vacío. El residuo se purificó mediante cromatografía en columna ultrarrápida, utilizando un gradiente de acetato de etilo en heptano (0-100 %), para proporcionar el compuesto del título (282 mg, 85 %) como un aceite de color amarillo anaranjado. 5h (500 MHz, DMSO-ds) 9,65 (s, 1H), 8,48 (d, J 8,5 Hz, 1H), 8,12 (d, J 5,1 Hz, 1H), 7,75 (s, 1H), 6,91 (dd, J 5,1, 1,3 Hz, 1H), 4,22 (dd, J 8,4, 6,5 Hz, 1H), 3,62 (s, 3H), 3,57 (d, J 13,9 Hz, 1H), 3,46 (d, J 13,9 Hz, 1H), 2,03-1,93 (m, 1H), 1,69-1,22 (m, 14h ), 1,45 (s, 9H). HPLC-MS (método 5): MH+ mlz 434, TR 1,97 min.
Intermedio 18
2-({2-[2-(ferf-Butoxicarbonilamino)piridin-4-il]acetil}amino)-2-ciclooctilacetato de litio
Se añadió monohidrato de hidróxido de litio (38 mg, 0,90 mmol) a una solución agitada del intermedio 17 (260 mg, 0,60 mmol) en THF (6 ml) y agua (3 ml). La mezcla de reacción se agitó a 20 °C durante 22 h, luego se concentró y se secó al vacío durante 4 h, para dar el compuesto del título (255 mg, cuantitativo) como un polvo blanquecino. HPLC-MS (método 5): MH+ mlz 420, TR 1,82 min.
Intermedio 19
2-({2-[2-(ferf-Butoxicarbonilamino)piridin-4-il]acetil]amino)-2-ciclooctil-W-(2-oxospiro[indolina-3,4'-tetrahidropiran-6-il)acetamida
Se añadió HATU (210 mg, 0,55 mmol) a una solución agitada del intermedio 2 (100 mg, 0,46 mmol) e intermedio 18 (213 mg, 0,5 mmol) en DMF anhidra (4 ml) en una atmósfera de nitrógeno. La mezcla se agitó a 20 °C durante 15 h, luego se inactivó con una solución acuosa saturada de bicarbonato de sodio (20 ml). Se le añadieron agua (20 ml) y acetato de etilo (20 ml), luego se filtró el precipitado blanco resultante y se lavó con DCM (2 x 10 ml) e isopropanol/cloroformo (1:1; 2 x 10 ml). Las capas acuosa y orgánica se separaron, luego la fase acuosa se extrajo con acetato de etilo (2 x 20 ml). Los extractos y lavados orgánicos combinados se secaron sobre sulfato de sodio, luego se filtraron y se concentraron al vacío. El residuo se trituró con diclorometano. El precipitado de color amarillo pálido resultante se filtró, luego se lavó con diclorometano y se secó al vacío, para proporcionar el compuesto del título (160 mg, 56 %) como un polvo de color amarillo pálido. HpLC-MS (método 5): MH+ m/z 620, TR 1,89 min.
Intermedio 20
2-Ciclooctil-2-{[2-(piridin-4-il)acetil]amino}acetato de metilo
Se añadió HATU (497 mg, 1,31 mmol) a una solución agitada del intermedio 11 (257 mg, 1,09 mmol), hidrocloruro de ácido 2-(piridin-4-il)acético (190 mg, 1,09 mmol) y DIPEA (0,90 ml, 5,45 mmol) en DMF (5 ml) en nitrógeno. La mezcla se agitó a 20 °C en nitrógeno durante 16 h, luego se inactivó con una solución acuosa saturada de bicarbonato de sodio (10 ml). El material se extrajo con acetato de etilo (3 x 10 ml). Los extractos orgánicos combinados se lavaron con salmuera (2 x 10 ml) y se secaron sobre sulfato de sodio, luego se filtraron y se concentraron al vacío. El residuo se separó mediante cromatografía en columna ultrarrápida, utilizando un gradiente de acetato de etilo en heptano (20 100 %) seguido de un gradiente de metanol en acetato de etilo (0-20 %), para proporcionar el compuesto del título (201 mg, 58 %) como un aceite de color naranja pálido. 5h (250 m Hz , CDCb) 8,63-8,58 (m, 2H), 7,26 (d, J 6,0 Hz, 2H), 5,97 (d, J 8,5 Hz, 1H), 4,56 (dt, J 9,1,4,6 Hz, 1H), 3,75 (s, 3H), 3,61 (s, 2H), 2,07 (s, 1H), 1,66-1,39 (m, 14H). HPLC-MS (método 7): Mh m/z 319, TR 1,77 min.
Intermedio 21
2-Ciclooctil-2-{[2-(piridin-4-il)acetil]amino}acetato de litio
Se añadió lentamente una solución de monohidrato de hidróxido de litio (56 mg, 1,33 mmol) en agua (1 ml) a una solución agitada del intermedio 20 (281 mg, 0,88 mmol) en MeOH/THF (1:1; 2 ml) a 20 °C al aire. La mezcla se agitó a 20 °C durante 16 h, luego se diluyó con una porción adicional de MeOH/agua (1:1; 2 ml), y se agitó a 20 °C durante 24 h más. Los sólidos se recogieron por filtración y se lavaron con agua (2 ml) y éter dietílico/MeOH/THF (10:1:1; 12 ml), luego se secaron al vacío a 50 °C durante 5 h, para obtener el compuesto del título (172 mg, 63 %) como un polvo blanco. 5h (500 MHz, DMSO-ds) 8,48-8,40 (m, 2H), 7,57 (d, J 8,4 Hz, 1H), 7,31-7,26 (m, 2H), 3,77 (dd, J 8,4, 4,5 Hz, 1H), 3,56 (d, J 13,9 Hz, 1H), 3,46 (d, J 13,9 Hz, 1H), 1,99-1,89 (m, 1H), 1,62-1,24 (m, 13H), 1,20-1,09 (m, 1H). HPLC-MS (método 5): MH+ m/z 305, TR 1,49 min.
Intermedio 22
2-[(2-Metilpirazol-3-carbonil)amino]acetato de metilo
Se añadió DIPEA (370 pl, 2,24 mmol) a una solución agitada de hidrocloruro de éster metílico de glicina (70 mg, 0,56 mmol), ácido 1 -metil-1 H-pirazol-5-carboxílico (71 mg, 0,57 mmol) y HATU (255 mg, 0,67 mmol) en DMF anhidro (1 ml) en una atmósfera de nitrógeno. La mezcla se agitó a 20 °C durante 15 h, luego se inactivó mediante la adición de una solución acuosa saturada de bicarbonato de sodio (7 ml) y agua (7 ml). El material se extrajo secuencialmente con acetato de etilo (3 x 7 ml) e isopropanol/cloroformo 1:1 (2 x 7 ml). Las capas orgánicas combinadas se lavaron con salmuera (2 x 7 ml) y se secaron sobre sulfato de sodio, luego se filtraron y se concentraron al vacío. El residuo se purificó mediante cromatografía en columna ultrarrápida, usando un gradiente de acetato de etilo en heptano (0-100 %), para proporcionar el compuesto del título (110 mg, cuantitativo) como un aceite marrón anaranjado. 5h (250 MHz, DMSO-ds) 8,92 (t, J 5,6 Hz, 1H), 7,48 (d, J 2,1 Hz, 1H), 6,89 (d, J 2,1 Hz, 1H), 4,04 (s, 3H), 3,98 (d, J 5,9 Hz, 2H), 3,66 (s, 3H). HPLC-MS (método 6): Mh m/z 198, TR 0,52 min.
Intermedio 23
2-[(2-Metilpirazol-3-carbonil)amino]acetato de litio
Se añadió hidróxido de litio monohidrato (36 mg, 0,86 mmol) a una solución agitada del intermedio 22 (140 mg, 0,71 mmol) en THF (3 ml) y agua (1,5 ml). La mezcla de reacción se agitó a 20 °C durante 64 h, luego se concentró y se
secó al vacío durante 4 h, para proporcionar el compuesto del título (134 mg, cuantitativo) como una espuma color canela. HPLC-MS (método 5): MH+ mlz 184, TR 0,12-0,16 min.
Intermedio 24
W-(2-Bromo-5-clorofenil)tetrah¡drot¡op¡rán-4-carboxam¡da
Se añadió lentamente 2,4,6-trióxido de 2,4,6-tripropil-1,3,5,2,4,6-trioxatrifosforinano (solución al 50 % en peso en acetato de etilo, 8,1 ml, 13,6 mmol) a una solución agitada de 2-bromo- 5-cloroanilina (1,5 g, 7,27 mmol), ácido tiano-4-carboxílico (1,0 g, 6,84 mmol) y piridina (2,7 ml, 33,4 mmol) en acetato de etilo (14 ml) a 0 °C en nitrógeno. La mezcla de reacción se dejó calentar lentamente a 20 °C y se agitó durante 64 h. La suspensión resultante se diluyó con acetato de etilo (20 ml) y se inactivó con agua (30 ml). Los sólidos se recogieron por filtración, lavando secuencialmente con acetato de etilo (2 x 20 ml), agua (20 ml) y éter dietílico (2 x 20 ml). Los sólidos se secaron a 50 °C al vacío durante 16 h, para proporcionar el compuesto del título (2,08 g, 86 %) como un polvo de color rosa. 5h (500 MHz, DMSO-d6) 9,51 (br s, 1H), 7,71-7,65 (m, 2H), 7,21 (dd, J 8,6, 2,6 Hz, 1H), 2,71-2,62 (m, 4H), 2,57 (tt, J 11,5, 3,1 Hz, 1H), 2,19 2,08 (m, 2H), 1,78-1,63 (m, 2H). HPLC-MS (método 5): MH+ m/z 334, TR 1,90 min.
Intermedio 25
W-(2-Bromo-5-clorofen¡l)-W-[2-(tr¡met¡ls¡l¡l)etox¡met¡ltetrah¡drot¡onp¡rán-4-carboxam¡da
Se añadió hidruro de sodio (dispersión al 60 % en aceite mineral, 39 mg, 0,98 mmol) a una suspensión agitada del intermedio 24 (250 mg, 0,75 mmol) en THF anhidro (4 ml) a 0 °C en nitrógeno. La mezcla se agitó a 0 °C durante 30 min, luego se le añadió [2-(cloro-metoxi)etil](trimetil)silano (0,2 ml, 1,13 mmol) a 0 °C. La mezcla se dejó calentar gradualmente hasta 20 °C y se agitó durante un total de 16 h, luego se inactivó con agua (20 ml). El material se extrajo con acetato de etilo (3 x 30 ml). Los extractos orgánicos combinados se lavaron con salmuera (20 ml) y se secaron sobre sulfato de magnesio, luego se filtraron y se concentraron al vacío. El aceite bruto viscoso de color rojo resultante se separó mediante cromatografía en columna ultrarrápida, usando un gradiente de terf-butilmetiléter en heptano (0 30 %), para proporcionar el compuesto del título (299 mg, 84 %) como un aceite viscoso incoloro. Rotámero principal: 5h (500 MHz, DMSO-ds) 7,85 (d, J 8,6 Hz, 1H), 7,58 (d, J 2,5 Hz, 1H), 7,49 (dd, J 8,6, 2,5 Hz, 1H), 5,30 (d, J 10,4 Hz, 1H), 4,51 (d, J 10,4 Hz, 1H), 3,64-3,51 (m, 2H), 2,59-2,47 (m, 2H), 2,46-2,38 (m, 1H), 2,34-2,26 (m, 1H), 2,09-1,88 (m, 3H), 1,84-1,65 (m, 1H), 1,62-1,49 (m, 1H), 0,93-0,79 (m, 2H), -0,02 (s, 9H). HPLC-MS (método 5): MH+ m/z 464, TR 2,26 min.
Intermed¡o 26
6-Cloro-1-[2-(trimetilsilil)etoximetil]espiro[indolina-3,4'-tetrahidrotiopirán]-2-ona
El intermedio 25 (290 mg, 0,62 mmol) y el tert-butóxido de sodio (180 mg, 1,87 mmol) se disolvieron en tolueno anhidro (3,1 ml). La mezcla se purgó con nitrógeno y se sonicó durante 5 min, luego se cargó con dicloruro de [1,3-bis(2,6-diisopropilfenil)-imidazol-2-iliden](3-cloropiridil)paladio(II) (42,6 mg, 0,06 mmol). La mezcla se purgó con nitrógeno y se sonicó durante 5 min, luego se selló en nitrógeno y se calentó a 110 °C durante 16 h. Después de enfriar a temperatura ambiente, la mezcla de reacción se repartió con agua (30 ml) y acetato de etilo (30 ml). La mezcla bifásica se filtró a través de un lecho de tierra de diatomeas y se lavó con agua (10 ml) y acetato de etilo (2 x 10 ml). Se separó la fase orgánica y luego se extrajo la fase acuosa con acetato de etilo (2 x 30 ml). Los extractos orgánicos combinados se lavaron con salmuera (20 ml) y se secaron sobre sulfato de magnesio, luego se filtraron y se concentraron al vacío. El aceite bruto viscoso de color rojo resultante se separó mediante cromatografía en columna ultrarrápida, usando un gradiente de terf-butilmetiléter en heptano (0-20 %), para proporcionar el compuesto del título (73 mg, 31 %) como un aceite viscoso de color tostado claro. 5h (500 MHz, DMSO-d6) 7,52 (d, J 8,0 Hz, 1H), 7,18 (d, J 1,9 Hz, 1H), 7,13 (dd, J 8,0, 1,9 Hz, 1H), 5,11 (s, 2H), 3,52-3,45 (m, 2H), 3,13 (ddd, J 13,1,8,6, 4,1 Hz, 2H), 2,72-2,62 (m, 2H), 1,98-1,86 (m, 4H), 0,87-0,81 (m, 2H), -0,08 (s, 9H). HPLC-MS (método 5): MNa+ m /z406, TR 2,28 min.
Intermed¡o 27
W-{2-Oxo-1-[2-(tr¡met¡ls¡l¡l)etox¡met¡l]esp¡ro[¡ndol¡na-3,4'-tetrah¡drot¡op¡rán-6-¡l}carbamato de fert-but¡lo
Se cargó un tubo sellado con el intermedio 26 (73 mg, 0,19 mmol), carbamato de tert-butilo (45 mg, 0,38 mmol) y fosfato tripotásico (57 mg, 0,27 mmol). Los reactivos se suspendieron en tert-butanol (1 ml), luego la mezcla se purgó con nitrógeno y se sonicó durante 5 min. La mezcla de reacción se cargó con metanosulfonato de [(2-di-terf-butilfosfino-3,6-dimetoxi-2',4',6'-triisopropil-1,1'-bifenil)-2-(2'-amino-1,1'-bifenil)]paladio(II) (4,1 mg, 0,005 mmol) y 2-(di-terfbutilfosfino)-2',4',6'-triisopropil-3,6-dimetoxi-1, 1 '-bifenilo (2,4 mg, 0,005 mmol), luego se purgó con nitrógeno y se sonicó durante 5 min. La mezcla se selló en nitrógeno y se calentó a 110 °C durante 16 h. Después de enfriar a temperatura ambiente, la mezcla se diluyó con acetato de etilo (10 ml) y se filtró a través de una capa de tierra de diatomeas, lavando con acetato de etilo (2 x 10 ml). El filtrado se concentró al vacío. El semisólido rojo bruto resultante se separó mediante cromatografía en columna ultrarrápida, usando un gradiente de terf-butilmetiléter en heptano (0 50 %), para proporcionar el compuesto del título (14 mg, 16 %) como un polvo blanquecino. 5h (500 MHz, DMSO-d6) 9,41 (s, 1H), 7,43 (s, 1H), 7,34 (d, J 8,1 Hz, 1H), 7,02 (dd, J 8,1, 1,5 Hz, 1H), 5,03 (s, 2H), 3,52-3,44 (m, 2H), 3,17-3,01 (m, 2H), 2,74-2,65 (m, 2H), 1,97-1,81 (m, 4H), 1,47 (s, 9H), 0,88-0,79 (m, 2H), -0,07 (s, 9H). HPLC-MS (método 5):
MNa+ mlz 487, TR 2,23 min.
Intermedio 28
6-Aminospiro[indolina-3,4'-tetrahidrotiopirán]-2-ona
Se añadió ácido trifluoroacético (46 pl, 0,6 mmol) a una solución agitada del intermedio 27 (14 mg, 0,03 mmol) en DCM (0,5 ml). La mezcla se agitó en nitrógeno a 20 °C durante 16 h, luego se diluyó con DCM (2 ml). Se le añadió una porción adicional de ácido trifluoroacético (46 pl, 0,6 mmol) y se continuó agitando a 20 °C durante 5 días en nitrógeno. La mezcla se diluyó con DCM (2 ml) y se le añadió una porción adicional de ácido trifluoroacético (92 pl, 1,19 mmol). Se continuó agitando durante 24 h a 20 °C en nitrógeno, luego se eliminó lo volátil al vacío. El residuo se destiló azeotrópicamente tres veces con diclorometano, luego se disolvió en metanol (3 ml). Se le añadió DIPEA (50 pl, 0,3 mmol), y luego la mezcla se calentó a 100 °C en nitrógeno durante 1,5 h. Después de enfriar a temperatura ambiente, la mezcla se diluyó con DCM (20 ml) y se repartió con ácido clorhídrico acuoso a 0,5 M (20 ml). Se agitó la mezcla bifásica y las fases se separaron utilizando una frita hidrófoba. La fase acuosa se lavó con DCM/isopropanol (4:1; 20 ml), y luego se ajustó a pH 5-6 con bicarbonato de sodio sólido. El material se extrajo con DCM-isopropanol 4:1 (6 x 20 ml). Los filtrados orgánicos combinados se agitaron con una solución acuosa saturada de hidrogenocarbonato de sodio (10 ml) y la fase orgánica se separó usando una frita hidrófoba. El filtrado orgánico se concentró al vacío para proporcionar el compuesto del título (3,2 mg, 45 %) como un polvo tostado. HPLC-MS (método 5): MH+ m/z 235, T r 1,29 min.
Intermedio 29
1'-Bencil-6-bromospiro[indolina-3,4'-piperidín]-2-ona
Una suspensión de 6-bromo-1,3-dihidro-2H-indol-2-ona (424 mg, 2,00 mmol) en THF anhidro (20 ml) se purgó con nitrógeno y se sonicó durante 10 min. La mezcla se enfrió a -78 °C en nitrógeno y se le añadió lentamente bis(trimetilsilil)amida de sodio a 1 M (10 ml, 10,0 mmol). Después de agitar durante 60 min, se le añadió hidrocloruro de W-bencilbis(2-cloroetil)amina (590 mg, 2,20 mmol) en una porción y la mezcla se agitó a -78 °C durante 1 h. Se retiró el baño de enfriamiento y se dejó calentar la mezcla a 20 °C, luego se calentó a 70 °C durante 16 h. Después de enfriar a temperatura ambiente, la mezcla de reacción se inactivó con una solución acuosa saturada de cloruro de amonio (20 ml) y se diluyó con agua (20 ml). El material se extrajo con acetato de etilo (3 x 100 ml). Los extractos orgánicos combinados se lavaron con salmuera (50 ml) y se secaron sobre sulfato de magnesio, luego se filtraron y se concentraron al vacío. El semisólido bruto de color rojo oscuro resultante se disolvió en DCM caliente (~20 ml) y se dejó enfriar a temperatura ambiente, luego se trituró con heptano (~60 ml). Los sólidos se recogieron por filtración, lavando con heptano (2 x 20 ml). El filtrado se concentró al vacío, y se repitió el procedimiento, para proporcionar el compuesto del título (236 mg, 32 %) como un polvo de color naranja claro después del secado al vacío a 50 °C durante 16 h. 5h (500 MHz, DMSO-ds) 10,50 (br s, 1H), 7,42 (d, J 7,8 Hz, 1H), 7,39-7,30 (m, 4H), 7,29-7,22 (m, 1H), 7,12 (dd, J 8,0, 1,8 Hz, 1H), 6,98 (d, J 1,8 Hz, 1H), 3,61 (s, 2H), 2,91 -2,70 (m, 2H), 2,62-2,48 (m, 2H), 1,87-1,74 (m, 2H), 1,72 1,56 (m, 2H). HPLC-MS (método 5): MH+ mlz 371, TR 1,56 min.
Intermedio 30
6-Amino-1'-bencilespiro[indolina-3,4'-piperidín]-2-ona
Se cargó un tubo sellado con el intermedio 29 (326 mg, 0,88 mmol), carbamato de tert-butilo (206 mg, 1,76 mmol) y fosfato tripotásico (262 mg, 1,23 mmol). Los reactivos se suspendieron en tert-butanol (4,5 ml), luego la mezcla se purgó con nitrógeno y se sonicó durante 5 min. La mezcla de reacción se cargó con metanosulfonato de [(2-di-terfbutilfosfino-3,6-dimetoxi-2',4',6'-triisopropil-1,1'-bifenil)-2-(2'-amino-1,1'-bifenil)]paladio(II) (18,7 mg, 0,02 mmol) y 2-(diterf-butilfosfino)-2',4',6'-triisopropil-3,6-dimetoxi-1,1'-bifenilo (10,8 mg, 0,02 mmol), luego se purgó con nitrógeno y se sonicó durante 5 min. La mezcla se selló en nitrógeno y se calentó a 110 °C durante 16 h. Después de enfriar, la mezcla se repartió entre acetato de etilo (30 ml) y agua (30 ml), y luego se sonicó. Los sólidos se eliminaron por filtración a través de una capa de tierra de diatomeas, lavando con agua (20 ml) y acetato de etilo (20 ml). Se separó la fase orgánica, y luego la capa acuosa se extrajo con acetato de etilo (2 x 30 ml) después de ajustar el pH a 11 con carbonato de sodio. Los extractos orgánicos combinados se lavaron con salmuera (20 ml) y se secaron sobre sulfato de magnesio, luego se filtraron y se redujeron al vacío. El residuo se disolvió en diclorometano (8,3 ml), y luego se le añadió ácido trifluoroacético (0,65 ml, 8,44 mmol). La solución se agitó a 20 °C al aire durante 18 h. Lo volátil se eliminó al vacío y el residuo se adsorbió en un cartucho SCX-2. La columna se lavó con DCM (50 ml) y MeOH (50 ml). El material se eluyó con amoníaco a 1 M en MeOH (50 ml), luego se concentró al vacío. El polvo púrpura resultante se separó mediante cromatografía en columna ultrarrápida (columna KP-NH), usando un gradiente de acetato de etilo en heptano (0-100 %) seguido de un gradiente de MeOH en acetato de etilo (0-10 %), para proporcionar el compuesto del título (24 mg, 9 %) como un aceite viscoso de color canela. HPLC-MS (método 5): MH+ mlz 308, TR 1,07 min.
Intermedio 31
W-{1-Ciclooctil-2-[(1'-bencil-2-oxospiro[indolina-3,4'-piperidín]-6-il)amino]-2-oxoetil}-2-metilpirazol-3-carboxamida
Se añadió HATU (68 mg, 0,18 mmol) a una suspensión agitada del intermedio 30 (46 mg, 0,15 mmol) y del intermedio 13 (50 mg, 0,17 mmol) en DMF anhidra (1 ml). La mezcla se agitó a 20 °C en nitrógeno durante 64 h, luego se inactivó con una solución acuosa saturada de carbonato de sodio (10 ml) y se agitó durante 30 min a 20 °C. La mezcla se diluyó con agua (10 ml) y se repartió con DCM/isopropanol (4:1; 20 ml). La fase orgánica se separó usando una frita hidrófoba, y luego la capa acuosa se extrajo con DCM/isopropanol 4:1 (2 x 20 ml). El filtrado se concentró al vacío. La goma de naranja bruta resultante se separó mediante cromatografía en columna ultrarrápida, usando un gradiente de acetato de etilo en heptano (0-100 %) seguido de un gradiente de MeOH en acetato de etilo (0-20 %), para proporcionar el compuesto del título (24 mg, 28 %) como un polvo blanquecino. 5h (500 MHz CD3OD) 7,46 (d, J 2,1 Hz, 1H), 7,44 7,38 (m, 3H), 7,37-7,31 (m, 3H), 7,31-7,26 (m, 1H), 7,08 (dd, J 8,1, 1,9 Hz, 1H), 6,87 (d, J 2,1 Hz, 1H), 4,50 (d, J 8,7 Hz, 1H), 4,08 (s, 3H), 3,72 (s, 2H), 3,03-2,93 (m, 2H), 2,79-2,70 (m, 2H), 2,28-2,18 (m, 1H), 2,00-1,89 (m, 2H), 1,87 1,43 (m, 16H). HPLC-MS (método 5): MH+ mlz 583, TR 1,75 min.
Intermedio 32
2-(fert-Butoxicarbonilamino)-2-(ciclooctil)acetato de metilo
Se añadió trietilamina (0,71 ml, 5,09 mmol) a una mezcla del intermedio 11 (410 mg, 1,88 mmol) y dicarbonato de diterf-butilo (444 mg, 1,89 mmol) en DCM (16 ml), y la mezcla de reacción se agitó a 20 °C durante 64 h. La mezcla de reacción se lavó con agua (30 ml), una solución acuosa saturada de hidrogenocarbonato de sodio (30 ml) y salmuera (30 ml), luego se secó sobre sulfato de magnesio, se filtró y se concentró al vacío. El residuo se separó mediante cromatografía en columna ultrarrápida usando un gradiente de acetato de etilo en heptano (0-100 %) para proporcionar el compuesto del título (539 mg, 96 %) como un aceite incoloro fluido. 5h (250 MHz, DMsO-d6) 7,14 (d, J 8,6 Hz, 1H), 4,03-3,78 (m, 1H), 3,61 (s, 3H), 2,02-1,84 (m, 1H), 1,75-1,19 (m, 23H).
Intermedio 33
2-(ferf-Butoxicarbonilamino)-2-(ciclooctil)acetato de litio
Se añadió monohidrato de hidróxido de litio (75 mg, 1,78 mmol) a una solución agitada del intermedio 32 (485 mg, 1,62 mmol) en THF-agua 2:1 (12 ml). La mezcla de reacción se agitó a 20 °C durante 15 h, luego se concentró y se secó al vacío durante 2 h, para proporcionar el compuesto del título (471 mg, cuantitativo) como un sólido blanco. 5h (500 MHz, DMSO-ds ) 5,78 (d, J 7,1 Hz, 1H), 3,42-3,38 (m, 1H), 1,96-1,86 (m, 1H), 1,65-1,16 (m, 23H).
Intermedio 34
W-{1-Ciclooctil-2-oxo-2-[(2-oxospiro[indolina-3,4'-tetrahidropiran]-6-il)-amino]etil}carbamato de ferf-butilo
Se añadió HATU (596 mg, 1,57 mmol) a una solución agitada del intermedio 2 (285 mg, 1,31 mmol) y del intermedio 33 (403 mg, 1,39 mmol) en DMF anhidra (6 ml) en una atmósfera de nitrógeno. La mezcla se agitó a 20 °C durante 18 h, luego se inactivó mediante la adición de una solución acuosa saturada de hidrogenocarbonato de sodio (50 ml) y agua (50 ml). La fase acuosa se extrajo con acetato de etilo (2 x 50 ml) e isopropanol-cloroformo 1:1 (2 x 50 ml). Las capas orgánicas combinadas se lavaron con salmuera (2 x 50 ml) y se secaron sobre sulfato de magnesio, luego se filtraron y se concentraron al vacío. El residuo se separó mediante cromatografía en columna ultrarrápida utilizando un gradiente de acetato de etilo en heptano (0-100 %), seguido de un gradiente de metanol en diclorometano (0-100 %), para proporcionar el compuesto del título (502 mg, 79 %) como un sólido amarillo pálido. 5h (500 MHz, DMSO-d6) 10,38 (s, 1H), 9,98 (s, 1H), 7,43 (d, J 8,1 Hz, 1H), 7,37 (d, J 1,8 Hz, 1H), 7,07 (d, J 8,0 Hz, 1H), 6,87 (d, J 8,7 Hz, 1H), 4,00 (ddd, J 11,1, 7,1, 3,5 Hz, 2H), 3,93 (t, J 8,2 Hz, 1H), 3,80 (ddd, J 11,0, 7,0, 3,6 Hz, 2H), 1,93 (s, 1H), 1,79-1,69 (m, 2H), 1,68-1,25 (m, 25H). HPLC-MS (método 6): [M+2H-tBu]+ m/z 430, TR 3,12 min.
Intermedio 35
2-Amino-2-ciclooctil-W-(2-oxospiro[indolina-3,4'-tetrahidropiran]-6-il)acetamida
Se añadió ácido trifluoroacético (0,8 ml, 10,38 mmol) a una solución agitada del intermedio 34 (489 mg, 1,01 mmol) en DCM (20 ml) a 20 °C en una atmósfera de nitrógeno. La mezcla de reacción se agitó a 20 °C durante la noche. Se le añadió más ácido trifluoroacético (0,4 ml, 5,19 mmol) a la mezcla de reacción y se continuó agitando a 20 °C durante 4 h. La mezcla de reacción se diluyó con DCM (30 ml) y se inactivó con solución acuosa saturada de hidrogenocarbonato de sodio (30 ml), y luego se extrajo con isopropanol-cloroformo 1:1 (3 x 30 ml). Las capas orgánicas combinadas se secaron sobre sulfato de magnesio, luego se filtraron y se concentraron al vacío, para proporcionar el compuesto del título (284 mg, 73 %) como un sólido tostado. 5h (500 MHz, DMSO-d6) 10,38 (s, 1H), 9,83 (br s, 1H), 7,47-7,37 (m, 2H), 7,06 (dd, J 8,1, 1,9 Hz, 1H), 4,01 (ddd, J 11,0, 7,1,3,6 Hz, 2H), 3,80 (ddd, J 11,1, 7,1,3,6 Hz, 2H), 3,09 (d, J 5,8 Hz, 1H), 2,26-1,26 (m, 21H). uPLC-MS (método 2): MH+ m/z 386, TR 3,04 min.
Intermedio 36
Ácido 2-[(2-metilpirazol-3-carbonil)amino]acético
Se añadió una solución de monohidrato de hidróxido de litio (2,02 g, 48,10 mmol) en agua (33 ml) a una solución agitada del intermedio 22 (7.3 g, 37,00 mmol) en THF (67 ml). La mezcla de reacción se agitó a 50 °C durante 2 h,
luego se dejó enfriar a temperatura ambiente. Lo volátil se eliminó al vacío. El residuo acuoso se lavó con acetato de etilo (2 x 100 ml), y luego se acidificó a pH 1-2 con ácido clorhídrico a 3 M. La capa acuosa se extrajo con una mezcla de diclorometano:isopropanol (2:1; 3 x 120 ml). Se combinaron los extractos orgánicos, se secaron sobre sulfato de sodio anhidro y se filtraron. El solvente se concentró al vacío para proporcionar el compuesto del título (6,6 g, 97 %) como un sólido de color blanco. 5h (250 MHz, DMSO-d6) 12,66 (s, 1H), 8,81 (t, J 5,9 Hz, 1H), 7,48 (d, J 2,1 Hz, 1H), 6,89 (d, J 2,1 Hz, 1H), 4,05 (s, 3H), 3,90 (d, J 6,0 Hz, 2H). HPLC-MS: MH+ mlz 184, TR 0,174 min.
Intermedio 37
2-(2-Metilpirazol-3-il)-4H-oxazol-5-ona
A la solución agitada del intermedio 36 (7,77 g, 36,06 mmol) en DCM anhidro (72 ml) se le añadió en porciones EDCIHCl (8,99 g, 46,87 mmol). La mezcla de reacción se agitó a temperatura ambiente durante 1,5 h, luego se diluyó con DCM (70 ml) y se inactivó con agua (70 ml). Se separó la capa orgánica, se lavó con agua (3 x 70 ml) y salmuera (50 ml), luego se secó sobre sulfato de sodio anhidro y se filtró. El solvente se concentró al vacío para proporcionar el compuesto del título (7,65 g, 77 %) como un sólido de color rosa, que se utilizó sin purificación adicional. 5h (250 MHz, CDCla) 7,53 (d, J 2,0 Hz, 1H), 6,82 (d, J 2,0 Hz, 1H), 4,42 (s, 2H), 4,22 (s, 3H). HPLC-MS (método 5): [2MNa]+ m/z 353 (débil), TR 0,72 min.
Intermedio 38
2-(2-Metilpirazol-3-il)-4-(espiro[3,3]heptán-2-iliden)oxazol-5-ona
Se añadió tetracloruro de titanio en DCM (1 M, 4,8 ml, 4,80 mmol) a THF anhidro (9 ml) a -10 °C. Una solución del intermedio 37 (200 mg, 1,21 mmol) en THF anhidro (1,5 ml) y una solución de espiro[3,3]heptán-2-ona (267 mg, 2,42 mmol) en THF anhidro (1,5 ml) se le añadieron secuencialmente en porciones y la mezcla de reacción se agitó a 0 °C durante otros 20 min. Se le añadió piridina anhidra (0,784 ml, 9,69 mmol) a 0 °C durante 30 min. La mezcla de reacción se agitó a 0 °C durante 2 h más, y luego a temperatura ambiente durante 16 h. La mezcla de reacción se inactivó mediante la adición de una solución acuosa saturada de cloruro de amonio (20 ml) y se continuó agitando durante 10 min más. La solución se extrajo con acetato de etilo (2 x 40 ml). Los extractos orgánicos se combinaron y se lavaron con salmuera (20 ml), luego se secaron sobre sulfato de sodio anhidro y se filtraron. Se retiró el solvente al vacío. El residuo se purificó mediante cromatografía automatizada, usando un gradiente de acetato de etilo en heptano (5-40 %), para proporcionar el compuesto del título (224 mg, 72 %) como un sólido de color blanco. 5h (250 MHz, CDCl3) 7,53 (d, J 2,1 Hz, 1H), 6,86 (d, J 2,1 Hz, 1H), 4,25 (s, 3H), 3,29-3,24 (m, 2H), 3,19-3,15 (m, 2H), 2,19-2,11 (m, 4H), 1,98-1,84 (m, 2H). HpLC-MS (método 5): MH+ mlz 258, TR 1,93 min.
Intermedio 39
2-(2-Metilpirazol-3-il)-4-(espiro[3.3]heptán-2-il)-4H-oxazol-5-ona
A una solución agitada del intermedio 38 (224 mg, 0,87 mmol) en acetonitrilo anhidro (10 ml) se le añadió paladio al 10 % sobre carbón vegetal (humedad al 50 %, 44 mg, 20 % en peso) en una sola porción. La mezcla de reacción se colocó en una atmósfera de hidrógeno gaseoso y se continuó agitando a temperatura ambiente durante 20 h. Se le añadió una segunda alícuota de paladio al 10 % sobre carbón vegetal (humedad al 50 %, 22 mg, 10 % en peso) y la mezcla de reacción se agitó en hidrógeno durante 43 h más. El catalizador se retiró por filtración sobre tierra de diatomeas, y luego la torta del filtro se enjuagó con acetonitrilo (2 x 5 ml). El solvente se concentró al vacío para proporcionar el compuesto del título (182 mg, 81 %) como un aceite gris. 5h (250 MHz, CDCl3) 7,53 (d, J 2,0 Hz, 1H), 6,81 (d, J 2,0 Hz, 1H), 4,33 (d, J 6,1 Hz, 1H), 4,24 (s, 3H), 2,74 (pd, J 8,4, 6,4 Hz, 1H), 2,23-1,99 (m, 6H), 1,91-1,76 (m, 4H). HPLC-MS (método 6): (M-H)- mlz 258, TR 3,08 min.
Intermedio 40
2-Ciclooctil-2-(3-metilisoxazol-4-carboxamido)acetato de metilo
A una solución de ácido 3-metilisoxazol-4-carboxílico (12,9 g, 66,1 mmol) en DMF seco (100 ml) a 0 °C se le añadieron DIPEA (54,9 g, 424,6 mmol), EDCI.HCl (19,5 g, 101,9 mmol) y HOBt (13,8 g, 101,9 mmol). La mezcla de reacción se agitó durante 15 min a 0 °C, luego se le añadió el intermedio 11 (20.0 g, 84,9 mmol) y la mezcla de reacción se agitó a t.a. durante 48 h. La mezcla de reacción se vertió en agua helada (500 ml) y se extrajo con acetato de etilo (2 x 400 ml). Se separó la capa orgánica, y luego se lavó con agua helada (2 x 100 ml) y HCl 1 N (2 x 50 ml). La capa orgánica se secó sobre Na2SO4 anhidro, luego se filtró y se evaporó al vacío. El residuo bruto se purificó mediante cromatografía en columna ultrarrápida sobre gel de sílice, usando EtOAc al 15 % en hexano como solvente de elución, para proporcionar el compuesto del título (7,9 g, 41,3 %) como un aceite viscoso de color amarillo pálido. LCMS (método 17): MH+ mlz 309, TR 5,5 min.
Intermedio 41
2-Ciclooctil-2-(3-metilisoxazol-4-carboxamido)acetato de litio
A una solución del intermedio 40 (11,01 g, 35,7 mmol) en THF (90 ml) a t.a. se añadieron agua (30 ml) e hidróxido de
litio monohidrato (2,248 g, 53,6 mmol). La mezcla de reacción se agitó durante 16 h y luego se evaporó al vacío. Al residuo se le añadió éter dietílico (50 ml). La mezcla se agitó durante 10 min y luego se filtró. El sólido resultante se lavó con éter dietílico (50 ml) y pentano (50 ml), y luego se secó al vacío para proporcionar el compuesto del título (9,51 g, 91%) como un sólido blanquecino. 5h (400 MHz, DMSO-d6) 9,69 (s, 1H), 8,21 (s, 1H), 4,11 (dd, J 8,0, 4,0 Hz, 1H), 2,35 (s, 3H), 2,05 (br s, 1H), 1,65-1,35 (m, 14H). LCMS (método 18): MH+ mlz 295, TR 5,4 min.
Intermedio 42
frans-(4-Metilciclohexil)metanol
A una solución fría (de -5 °C a -20 °C) de ácido trans-4-metilciclohexanocarboxílico (68,5 g, 0,481 mol) en THF (550 ml) se le añadió una solución de hidruro de litio y aluminio (2,4 M en THF, 200 ml, 0,48 mol) lentamente durante aproximadamente 1 h. La mezcla se agitó a -20 °C durante 1,5 h, luego se dejó calentar a temperatura ambiente. La mezcla se volvió a enfriar en un baño de hielo y sal antes de agregarle lentamente y con precaución agua (16 ml), solución acuosa de hidróxido de sodio (15 % en peso, 16 ml) y agua (40 ml). La mezcla viscosa resultante se agitó durante 10 min y luego se le añadió éter dietílico (500 ml). La suspensión resultante se filtró a través de una capa de tierra de diatomeas. Los solventes se evaporaron a presión reducida para obtener el compuesto del título (63,5 g, 100 %) como un aceite móvil transparente e incoloro. 5h (500 MHz, CDCh) 3,44 (d, J 6,3 Hz, 2h ), 1,79-1,69 (m, 4H), 1,47 1,23 (m, 3H), 1,04-0,89 (m, 4H), 0,88 (d, J 6,6 Hz, 3H).
Intermedio 43
frans-4-Metilciclohexanocarbaldehído
A una solución fría (de -10 °C a -5 °C) del intermedio 42 (30,31 g, 0,229 mol) en DCM (250 ml), DIPEA (122 ml, 1,15 mol) y DMSO (81,4 ml, 0,688 mol) se le añadió un complejo de trióxido de azufre-piridina sólido (73 g, 0,458 mol) en porciones, manteniendo la temperatura interna por debajo de 20 °C. La mezcla de reacción se agitó a temperatura ambiente durante 16 h, y luego se lavó sucesivamente con ácido cítrico acuoso (1 M, 200 ml) y salmuera (200 ml). La capa orgánica se filtró a través de papel de filtro separador de fases. El solvente se retiró a presión reducida para obtener el compuesto del título (34,9 g, 100 %) como un aceite de color amarillo pálido. 5h (250 MHz, CDCh) 9,61 (d, J 1,6 Hz, 1H), 2,28-2,03 (m, 1H), 1,95 (m, 2H), 1,80 (m, 2H), 1,56-1,14 (m, 3H), 1,07-0,80 (m, 5H, incluida la señal Me en d 0,90 (d, J 6,5 Hz)).
Intermedio 44
(S)-4-Metil-W-[(1£)-(frans-4-metilciclohexil)metiliden]bencenosulfinamida
A una solución del intermedio 43 (34,9 g, 229 mmol) y (S)-4-metilbencenosulfinamida (35,6 g, 229 mmol) en DCM (1,2 l) se le añadió etóxido de titanio (IV) (85-90 % de pureza, 174,5 g, 160 ml). La solución resultante se calentó a reflujo durante 2 h. La mezcla de reacción se enfrió a temperatura ambiente, y luego se le añadió lentamente agua (300 ml). La pasta espesa resultante se filtró a través de una capa de tierra de diatomeas, luego se enjuagó con DCM (300 ml) y agua (300 ml). Se separaron las dos fases. La fase de DCM se secó sobre sulfato de sodio anhidro y se filtró, luego se evaporó el solvente para dar el compuesto del título (55,7 g, 78 %) como un aceite de color amarillo, que se solidificó parcialmente al reposar. 5h (250 MHz, CDCla) 8,11 (d, J 4,9 Hz, 1H), 7,70-7,49 (m, 2H), 7,29 (m, 2H), 2,40 (s, 2H), 2,38-2,24 (m, 1H), 2,06-1,66 (m, 4H), 1,53-1,16 (m, 4H), 1,07-0,91 (m, 2H), 0,89 (d, J 6,5 Hz, 3H).
Intermedio 45
W-[(S)-Ciano(frans-4-metilciclohexil)metil]-(S)-4-metilbencenosulfinamida
A una solución de cianuro de dietilaluminio (1 M en tolueno, 103 ml, 103 mmol) en THF (400 ml) a -78 °C se le añadió alcohol isopropílico anhidro (5,3 ml, 69 mmol). La mezcla se agitó a -78 °C durante 30-60 min, luego se canuló en una solución del intermedio 44 (90 % pureza, 20,2 g, 69 mmol) en THF (800 ml) a -78 °C durante unos 45 min. La mezcla se dejó calentar a temperatura ambiente y luego se agitó durante una noche. La mezcla se enfrió en un baño de hielo y agua, y luego se le añadió una solución acuosa saturada de cloruro de amonio (300 ml); se desprendió algo de gas y la temperatura interna aumentó hasta unos 30 °C. Al cabo de 1 h, la mezcla se filtró a través de una capa de tierra de diatomeas, y luego la capa se lavó con agua (300 ml) y acetato de etilo (300 ml). Las capas orgánicas estaban divididas y las capas acuosas se lavaron con más acetato de etilo. Las capas orgánicas combinadas se lavaron con salmuera, se secaron sobre sulfato de sodio anhidro y se filtraron, luego se evaporó el solvente. El aceite de color amarillo pálido resultante, que se solidificó al reposar, se recogió en heptano-acetato de etilo caliente y luego se dejó cristalizar para obtener el compuesto del título (7,78 g, 38 %) como un sólido blanco. Los residuos se evaporaron y se purificaron mediante cromatografía en columna automatizada para dar una mezcla limpia de los dos diastereoisómeros. La retrocristalización de esta mezcla en acetato de etilo-heptano, sembrada con parte de la primera recogida, dio un lote adicional del compuesto del título (4,05 g, 20 %). 5h (250 MHz, CDCl3) 7,61 (d, J 8,3 Hz, 2H), 7,36 (d, J 8,2 Hz, 3H), 4,50 (d, J 7,8 Hz, 1H), 3,95 (dd, J 7,9, 5,8 Hz, 1H), 2,43 (s, 3H), 2,25-1,78 (m, 3H), 1,44-0,91 (m, 5H), 0,89 (d, J 6,5 Hz, 3H).
Intermedio 46
Cloruro de [(S)-ciano(trans-4-metilciclohexil)metil]amonio
A una solución agitada del intermedio 45 (6,6 g, 22,73 mmol) en metanol seco (130 ml) se le añadió gota a gota aproximadamente la mitad del volumen de cloruro de hidrógeno a 4 M en 1,4-dioxano (120 ml) durante 2 min, después de lo cual se produjo una exotermia a 26 °C. La mezcla de reacción se enfrió externamente y se le añadió la mitad del volumen restante de cloruro de hidrógeno a 4 M en 1,4-dioxano durante 3 min. Al cabo de 5 min, se tapó el matraz y la mezcla de reacción se agitó a temperatura ambiente durante 2 h. Lo volátil se concentró al vacío. Se le añadió éter dietílico (100 ml), luego se sonicó la mezcla y se agitó durante 15 min. Se filtraron los sólidos y se lavaron con éter dietílico (3 x 100 ml), y luego se secaron en una corriente de nitrógeno gaseoso para proporcionar el compuesto del título (4,10 g, 96 %) como un sólido blanco. 5h (500 MHz, DMSO-d6) 9,20 (s, 3H), 4,50 (d, J 5,5 Hz, 1H), 1,92-1,77 (m, 3H), 1,77-1,67 (m, 2H), 1,29 (ddp, J 11,4, 6,8, 3,4 Hz, 1H), 1,18-1,01 (m, 2H), 0,95-0,83 (m, 5H). HPLC-MS (método 1): MH+ m/z 153, TR 0,46 min (100 %). Le quiral (método 12, Amylose-225 cm, 80 % de heptano-20 % de 2-propanol, 1 ml/min): TR 8,84 min (S, 93 %).
Intermedio 47
Cloruro de [(S)-carboxi(trans-4-metilciclohexil)metil]amonio
Una solución agitada del intermedio 46 (4,05 g, 21,46 mmol) en una mezcla de ácido acético (17 ml) y ácido clorhídrico concentrado (85 ml) se calentó a una temperatura externa de 130 °C (temperatura interna de 105 °C). Al cabo de 3 h, se le añadió otra porción de ácido clorhídrico concentrado (25 ml), seguido de otra porción (25 ml) después de 2 h más. La mezcla de reacción se calentó durante 1 h y luego se enfrió. Se filtró el sólido precipitado y se enjuagó con tert-butilmetiléteréter, luego se secó al vacío, para proporcionar el compuesto del título (3,04 g, 68 %) como un sólido blanco. 5h (500 MHz, DMSO-ds ) 8,35 (s, 3H), 3,69 (d, J 4,2 Hz, 1H), 1,82-1,65 (m, 4H), 1,64-1,54 (m, 1H), 1,32-1,18 (m, 2H), 1,15-1,02 (m, 1H), 0,93-0,80 (m, 5H). HPLC-MS (método 3): MH+ m/z 172, TR 0,63 min.
Intermedio 48
Ácido (2S)-2-(tert-butoxicarbonilamino)-2-(trans-4-metilciclohexil)acético
A una suspensión agitada del intermedio 47 (25,1 g, 120,8 mmol) en agua (350 ml) se le añadió carbonato de sodio (55 g, 0,52 mol), seguido de dicarbonato de di-terf-butilo (39,6 g, 181 mmol) en 1,4-dioxano (500 ml). La mezcla de reacción se agitó mecánicamente durante 4 h. Lo volátil se eliminó al vacío, luego se enfrió la suspensión y se añadió cuidadosamente ácido clorhídrico a 1 N hasta alcanzar un pH de 1. La mezcla se extrajo con acetato de etilo (3 x 250 ml). Se combinaron las capas orgánicas, se lavaron sucesivamente con agua (200 ml) y salmuera (200 ml), y luego se filtraron a través de papel separador de fases. Se evaporó lo volátil. El sólido resultante se trituró en heptano (500 ml) y se filtró, luego se lavó con heptano (2 x 100 ml) y se secó en estufa para dar el compuesto del título (28,8 g, 87 %) como un sólido blanco. 5h (500 MHz, DMSO-ds ) 12,40 (s, 1H), 6,89 (d, J 8,5 Hz, 1H), 3,81 -3,74 (m, 1H), 1,69 1,53 (m, 5H), 1,37 (s, 9H), 1,28-1,19 (m, 1H), 1,09 (dp, J 22,9, 12,6, 11,6 Hz, 2H), 0,91 -0,76 (m, 5H). HPLC-MS (método 1 ): MH+ m/z 271, TR 3,34 min. SFC quiral (método 12, Chiralpak AS-H 25 cm, 10 % de metanol-90 % de CO2, 4 ml/min): TR2,61 min (100 %). [a]20D = 28,3°(c 3,202, cloroformo).
Intermedio 49
W-[(1S)-1-(4-Metilciclohexil)-2-oxo-2-[(2-oxospirofindolina-3,4'-tetrahidropiran]-6-il)amino]etil]carbamato de tert-butilo (isómero tra n s )
A una solución agitada del intermedio 48 (503 mg, 1,85 mmol) en DMF seca (10 ml) se le añadieron el intermedio 2 (405 mg, 1,86 mmol), HATU (850 mg, 2,24 mmol) y DIPEA (0,92 ml, 5,57 mmol) a t.a. La mezcla de reacción se agitó durante 2,5 días. Con enfriamiento externo (15 °C), se le añadió agua (40 ml). Se filtró el sólido que precipitó y se lavó con agua (2 x 10 ml). La torta del filtro se disolvió en acetato de etilo (10 ml) y se hizo pasar a través de un embudo sinterizado, y luego se enjuagó con acetato de etilo (2 x 5 ml). El exceso de agua se separó del filtrado. La capa orgánica se lavó con una mezcla 1:1 de agua y salmuera (20 ml), y salmuera (10 ml), luego se secó sobre sulfato de magnesio, se filtró y se concentró al vacío. El residuo bruto se adsorbió sobre gel de sílice (4,4 g) usando diclorometano y se purificó por cromatografía automatizada, usando un gradiente de acetato de etilo en heptano (0-100 %), para proporcionar el compuesto del título (648 mg, 70 %) como un sólido blanco. 5h (500 MHz, DMSO-d6) 10,39 (s, 1H), 9,99-9,81 (m, 1H), 7,42 (d, J 8,1 Hz, 1H), 7,38 (d, J 1,8 Hz, 1H), 7,09-7,02 (m, 1H), 6,89-6,48 (m, 1H), 4,06-3,95 (m, 2H), 3,89 (t, J 8,2 Hz, 1H), 3,84-3,66 (m, 2H), 1,79-1,69 (m, 3H), 1,69-1,44 (m, 6H), 1,40-1,28 (m, 9H), 1,28-1,20 (m, 1H), 1,21-1,06 (m, 1H), 1,06-0,93 (m, 1H), 0,90-0,75 (m, 5H). uPLC-MS (método 1): MH+ m/z 472, TR 3,52 min.
Intermedio 50
(2S)-2-Amino-2-(4-metilciclohexil)-N-(2-oxospiro[indolina-3,4'-tetrahidropiran]-6-il)acetamida (isómero tra n s )
A una solución agitada del intermedio 49 (606 mg, 1,29 mmol) en DCM (19 ml) se le añadió ácido trifluoroacético (0,9 ml, 11,68 mmol). La mezcla se agitó durante 18 h, luego se calentó a reflujo durante aproximadamente 1,5 h antes de
agregar ácido trifluoroacético adicional (0,5 ml). Al cabo de 1 h, la mezcla de reacción se enfrió. Con enfriamiento externo, se le añadió NaHCO3 saturado (30 ml), seguido de acetato de etilo (50 ml). La mezcla de dos fases se calentó a t.a., después de lo cual se disolvió el sólido. Se separó la capa orgánica, luego la capa acuosa se volvió a extraer con acetato de etilo (3 x 10 ml). Las capas orgánicas se combinaron y se lavaron con una mezcla 1:1 de agua y salmuera (10 ml), y salmuera (10 ml), luego se secaron sobre sulfato de magnesio, se filtraron y se concentraron al vacío. Se le añadió más acetato de etilo (~3 ml), seguido de heptano (20 ml). El sólido se filtró, luego se lavó con más heptano (4 x 10 ml) y se secó para obtener el compuesto del título (387 mg, 81 %) como un sólido blanco. 5h (500 MHz, DMSO-ds ) 10,37 (s, 1H), 9,79 (s, 1H), 7,45-7,38 (m, 2H), 7,06 (dd, J 8,1, 1,9 Hz, 1H), 4,04-3,97 (m, 2H), 3,86 3,73 (m, 2H), 3,08 (d, J 5,8 Hz, 1H), 1,99 (s, 2H), 1,92-1,56 (m, 7H), 1,56-1,41 (m, 2H), 1,31-1,13 (m, 2H), 1,07-0,94 (m, 1H), 0,92- 0,77 (m, 5H). uPLC-MS (método 3): MH+ m /z372, TR 0,86 min.
Intermedio 51
W-{(1S)-2-[(4-Fluoro-2-oxosp¡ro[¡ndol¡na-3,4'-tetrah¡drop¡ran]-6-¡l)am¡no]-1-(4-met¡lc¡clohex¡l)-2-oxoetil}carbamato de te r t -b u t i lo (¡somero tra n s )
Se añadió DIPEA (0,13 ml, 0,8 mmol) a una suspensión agitada del intermedio 74 (100 mg, 0,38 mmol), intermedio 48 (125 mg, 0,46 mmol) y HATU (188 mg, 0,5 mmol) en DCM anhidro (3,8 ml). La mezcla se agitó a 20 °C en nitrógeno durante 60 h, luego se inactivó con una solución acuosa saturada de carbonato de sodio (10 ml) y se diluyó con agua (10 ml). El material se extrajo con DCM (3 x 25 ml) usando una frita hidrófoba. El filtrado orgánico se concentró al vacío. El aceite anaranjado viscoso resultante se separó mediante cromatografía en columna ultrarrápida, usando un gradiente de acetato de etilo en heptano (0-100 %), para proporcionar el compuesto del título (102 mg, 52 %) como un polvo blanquecino. 5h (500 MHz, DMSO-ds ) 10,58 (s, 1H), 10,14 (s, 1H), 7,09 (d, J 1,5 Hz, 1H), 7,05 (d, 2Jhf 12,4 Hz, 1H), 6,91 (d, J 8,2 Hz, 1H), 4,11-3,98 (m, 2H), 3,86 (t, J 8,2 Hz, 1H), 3,81-3,68 (m, 2H), 2,06-1,95 (m, 2H), 1,80 1,60 (m, 5H), 1,60-1,43 (m, 2H), 1,37 (s, 9H), 1,28-1,20 (m, 1H), 1,19-0,93 (m, 2H), 0,90-0,75 (m, 5H). HPLC-MS (método 5): Mh m/z 490, T r 2,00 min.
Intermed¡o 52
(2S)-2-Am¡no-W-(4-fluoro-2-oxosp¡ro[¡ndol¡na-3,4'-tetrah¡drop¡ran]-6-¡l)-2-(4-met¡lc¡clohex¡l)acetam¡da (Isómero tra n s )
Se le añadió ácido trifluoroacético (161 pl, 2,09 mmol) a una solución agitada del intermedio 51 (102 mg, 0,21 mmol) en DCM (1 ml). La mezcla se agitó a 20 °C en nitrógeno durante 16 h, luego se diluyó con DCM-isopropanol 4:1 (20 ml) y se inactivó con una solución acuosa saturada de carbonato de sodio (10 ml) y agua (10 ml). La mezcla bifásica se agitó a 20 °C durante 30 min, luego la fase orgánica se separó utilizando una frita hidrófoba. La fase acuosa se extrajo con DCM-isopropanol 4:1 (3 x 10 ml). El filtrado orgánico se concentró al vacío para proporcionar el compuesto del título (105 mg, cuantitativo) como un polvo tostado. 5h (500 MHz, DMSO-d6) 10,58 (s, 1H), 7,14 (d, J 1,7 Hz, 1H), 7,08 (dd, 2Jhf 12,5, J 1,6 Hz, 1H), 4,06 (t, J 10,1 Hz, 2H), 3,75 (dt, J 11,2, 3,9 Hz, 2H), 3,06 (d, J 5,8 Hz, 1H), 2,00 (ddd, J 14,3, 10,4, 4,5 Hz, 2H), 1,75-1,61 (m, 5H), 1,54-1,40 (m, 2H), 1,31-1,12 (m, 2H), 1,07-0,94 (m, 1H), 0,93-0,76 (m, 5H). HPLC-MS (método 5): MH+ m /z390, t R 1,60 min.
Intermed¡o 53
2-[(3-Metilisoxazol-4-carbonil)amino]acetato de met¡lo
A una solución agitada de ácido 3-metilisoxazol-4-carboxílico (20 g, 157 mmol) en DMF anhidro (140 ml) en una atmósfera de nitrógeno se le añadieron sucesivamente DIPEA (63,1 g, 488 mmol), EDCI.HCl (36,2 g, 189 mmol), HOBt (25,5 g, 189 mmol) e hidrocloruro de éster metílico de glicina (25,5 g, 157 mmol). La mezcla se agitó a 20 °C durante 12 h, luego se inactivó mediante la adición de una solución acuosa saturada de bicarbonato de sodio (500 ml). El material se extrajo secuencialmente con acetato de etilo (4 x 1,5 l). Las capas orgánicas combinadas se lavaron con salmuera (2 l) y se secaron sobre sulfato de sodio, luego se filtraron y se concentraron al vacío, para proporcionar el compuesto del título (20 g, 64 %) como un aceite bruto de color marrón anaranjado. 5h (400 MHz, CDCh) 8,77 (s, 1H), 6,52 (s, 1H), 4,18 (d, J 4,0 Hz, 2H), 3,81 (s, 3H), 2,51 (s, 3H). HPLC-MS (método 33): MH+ m/z 199, TR 0,86 min.
Intermed¡o 54
Ác¡do 2-[(3-metilisoxazol-4-carbonil)amino]acético
Se añadió una solución de monohidrato de hidróxido de litio (43,4 g, 1,03 mol) en metanol (930 ml) a una solución agitada del intermedio 53 (186 g, 0,94 mol) en THF (1,7 l). La mezcla de reacción se agitó a t.a. durante 3 h, luego se concentró al vacío. El residuo acuoso se acidificó a pH 1 con una solución acuosa de ácido clorhídrico a 6 M (279 ml), y luego se dejó a t.a. Al cabo de 1 h, se filtró el sólido cristalino resultante y luego se secó al vacío para producir el compuesto del título (19 g, 78 %) como un sólido blanquecino. 5h (400 MHz, DMSO-d6) 9,32 (s, 1H), 8,77 (t, J 6,0 Hz, 1H), 3,88 (s, J 8,0 Hz, 2H), 2,37 (s, 3H). LCMS (método 34): MH+ m/z 185, TR 0,29 min.
Intermedio 55
2-(3-Metilisoxazol-4-il)-4H-oxazol-5-ona
A una solución agitada del in term edio 54 (44,2 g, 240 mmol) en DCM anhidro (440 ml) se le añadió en porciones EDCIHCl (59,8 g, 312 mmol). La mezcla de reacción se agitó a temperatura ambiente durante 1,5 h, luego se diluyó con DCM (200 ml) y se inactivó con agua (500 ml). Se separó la capa orgánica y se lavó con salmuera (2 x 500 ml), luego se secó sobre sulfato de sodio anhidro y se filtró. El solvente se concentró al vacío para proporcionar el com puesto de l título (34 g, 86 %) como un sólido amarillo, que se utilizó sin purificación adicional. 5h (400 MHz, CDCh) 8,83 (s, 1H), 4,37 (s, 2H), 2,56 (s, 3H). LCMS (método 34): [M-H]- m lz 167, TR 0,76 min.
Intermedio 56
4-{5-Metoxibiciclo[4.2.0]octa-1(6),2,4-trién-7-iliden}-2-(3-metil-1,2-oxazol-4-il)-4,5-dihidro-1,3-oxazol-5-ona
Se añadió tetracloruro de titanio en DCM (1 M, 2,7 ml, 2,7 mmol) a THF anhidro (4 ml) a -10 °C. Se le agregaron una solución del in term edio 55 (0,146 g, 0,88 mmol) en THF anhidro (1 ml) y una solución de 5-metoxibiciclo[4.2.0]octa-1,3,5-trién-7-ona (0,1 g, 0,67 mmol) en THF anhidro (1 ml) gota a gota secuencialmente. La mezcla de reacción se agitó a 0 °C durante 20 min, luego se le añadió gota a gota piridina anhidra (0,44 ml, 14,5 mmol) a 0 °C durante 30 min. La mezcla de reacción se agitó a 0 °C durante 2 h más, y luego a temperatura ambiente durante 16 h. La mezcla de reacción se inactivó mediante la adición de una solución acuosa saturada de cloruro de amonio (12 ml) y se continuó agitando durante 10 min más. La solución se extrajo con acetato de etilo (2 x 20 ml). Los extractos orgánicos combinados se lavaron con salmuera (20 ml) y se secaron sobre sulfato de magnesio, luego se filtraron y se concentraron al vacío. El residuo se purificó mediante cromatografía en columna ultrarrápida, usando un gradiente de acetato de etilo en heptano (0-100 %), para proporcionar el com puesto de l título (188 mg, 94 %) como un sólido de color amarillo. 5h (500 MHz, DMSO-ds ) 9,66 (s, 1H), 7,53 (dd, J 8,5, 7,2 Hz, 1H), 7,09-6,89 (m, 2H), 3,98 (s, 2H), 3,94 (s, 3H), 2,62 (s, 3H). HPLC-MS (método 5): MH+ m lz 297, TR 1,87 min.
Intermedio 57
6'-Amino-1'-{[2-(trimetilsilil)etoxi]metil}-1',2'-dihidrospiro[oxano-4,3'-pirrolo-[3,2-c]piridin]-2'-ona
Se añadió hexametildisilazano de litio en THF (1 M, 8,13 ml, 8,13 mmol) a una mezcla del in term edio 84 (2,5 g, 6,78 mmol), tris(dibencilidenacetona)dipaladio(0) (0,31 g, 0,34 mmol) y (2-bifenil)diciclohexilfosfina (0,285 g, 0,81 mmol) en THF anhidro (50 ml) en nitrógeno. La mezcla de reacción se agitó a 65 °C en nitrógeno durante 4 h. La mezcla de reacción se enfrió a 0 °C y se le añadió HCl a 1 N (21 ml). La mezcla de reacción se agitó a 0 °C durante 10 min, luego se inactivó con carbonato de sodio (pH 11) y se extrajo con acetato de etilo (3 x 100 ml). Las capas orgánicas combinadas se lavaron con salmuera (100 ml) y se secaron sobre sulfato de magnesio, luego se filtraron y se concentraron al vacío. El residuo se purificó mediante cromatografía en columna ultrarrápida (cartucho KP-NH), utilizando un gradiente de heptano en acetato de etilo (0-100 %), para proporcionar el com puesto de l título (1,65 g, 69 %) como un sólido tostado. 5h (500 MHz, DMSO-ds ) 8,11 (s, 1H), 6,23 (s, 1H), 6,03 (s, 2H), 5,06 (s, 2H), 4,03 (ddd, J 10,2, 5,9, 4,0 Hz, 2H), 3,84 (ddd, J 11,7, 8,4, 3,3 Hz, 2H), 3,59-3,47 (m, 2H), 1,86 (ddd, J 12,7, 8,3, 3,9 Hz, 2H), 1,72 1,57 (m, 2H), 1,01 -0,78 (m, 2H), 0,00 (s, 9H). HPLC-MS (método 3): MH+ m/z 350, TR 0,97 min.
Intermedio 58
2-[(2-Etilpirazol-3-carbonil)amino]acetato de metilo
Se añadió DIPEA (35,4 ml, 214 mmol) a una solución agitada de hidrocloruro de 2-aminoacetato de metilo (8,96 ml, 71,4 mmol), ácido 2-etilpirazol-3-carboxílico (10 g, 71,4 mmol) y HATU (32,56 g, 85,6 mmol) en DMF anhidra (90 ml) en una atmósfera de nitrógeno. La mezcla de reacción se agitó a temperatura ambiente durante 16 h, luego se diluyó con agua (50 ml) y una solución acuosa saturada de bicarbonato de sodio (50 ml). La capa acuosa se extrajo con tert- butilmetiléter (3 x 200 ml), seguido de DCM/MeOH 9:1 (2 x 150 ml), y luego DCM/MeOH 4:1 (2 x 150 ml). Se combinaron los extractos orgánicos y se concentraron al vacío. El material resultante se purificó mediante cromatografía en columna ultrarrápida, utilizando un gradiente de acetato de etilo en heptano (0-80 %), para proporcionar el com puesto de l título (20,9 g, 78 %) como un aceite de color amarillo. 5h (250 MHz, CDCh) 7,47 (d, J 2,0 Hz, 1H), 6,58 (d, J 2,1 Hz, 1H), 6,53 (br s, 1H), 4,59 (q, J 7,2 Hz, 2H), 4,18 (d, J 5,2 Hz, 2H), 3,80 (s, 3H), 1,43 (t, J 7,2 Hz, 3H). HPLC-MS (método 5): MH+ m /z 212, T r 0,86 min.
Intermedio 59
Ácido 2-[(2-etilpirazol-3-carbonil)amino]acético
Se añadió una solución de monohidrato de hidróxido de litio (3,02 g, 72,0 mmol) en agua (60 ml) a una solución agitada del in term edio 58 (pureza del 56 %, 20,88 g, 55,36 mmol) en THF (120 ml). La mezcla de reacción se agitó a 50 °C durante 3 h. Lo volátil se eliminó al vacío y el residuo acuoso se extrajo con acetato de etilo (2 x 100 ml). La fase acuosa se trató con ácido clorhídrico acuoso a 3 M (pH 1 -2) y se extrajo con DCM/MeOH 9:1 (2 x 100 ml), seguido de DCM/MeOH 4:1 (2 x 200 ml). Se combinaron los extractos orgánicos y se concentraron al vacío para dar el com puesto
del título (7,85 g, 37 %) como un aceite amarillo. La fase acuosa se extrajo adicionalmente con isopropanol/DCM 1:1 (4 x 150 ml) para dar un segundo lote del compuesto del título (6,27 g, 40 %) como un sólido blanco. 5h (500 MHz, CDCl3) 7,53 (d, J 2,0 Hz, 1H), 6,61 (d, J 2,0 Hz, 1 H), 6,59-6,51 (m, 1H), 4,63 (q, J 7,2 Hz, 2H), 4,26 (d, J 5,2 Hz, 2H), 1,47 (t, J 7,2 Hz, 3H). HPLC-MS (método 5): m H+ m/z 198, TR 0,33 min.
Intermedio 60
2-(2-Etilpirazol-3-il)-4H-oxazol-5-ona
A la solución agitada del intermedio 59 (pureza del 51 %, 7,85 g, 20,3 mmol) en DCM seco (50 ml) se le añadió en porciones EDCI.HCl (1:1) (5,06 g, 26,39 mmol). La mezcla de reacción se agitó a temperatura ambiente durante 2 h, luego se concentró al vacío. El aceite de naranja resultante se diluyó con agua (50 ml) y se extrajo con tert-butilmetiléter (3 x 70 ml). Los extractos orgánicos se combinaron, se lavaron con agua (3 x 50 ml) y salmuera (50 ml), y se secaron sobre sulfato de sodio, luego se filtraron y se concentraron al vacío, para proporcionar el compuesto del título (2,8 g, 66 %) como un aceite de color naranja. 5h (500 MHz, CDCh) 7,56 (d, J 2,0 Hz, 1H), 6,82 (d, J 2,0 Hz, 1H), 4,66 (q, J 7,2 Hz, 2H), 4,43 (s, 2H), 1,46 (t, J 7,2 Hz, 3H). HPLC-MS (método 3): MH+ m/z 180, TR 0,59 min.
Intermedio 61 (procedimiento F)
4-(5-Clorobiciclo[4.2.0]octa-1,3,5-trién-7-iliden)-2-(1-etil-1H-pirazol-5-il]-4,5-dihidro-1,3-oxazol-5-ona
Se añadió tetracloruro de titanio en DCM (1 M, 2,62 ml, 2,62 mmol) a THF anhidro (3,5 ml) a -10 °C. Una solución del intermedio 60 (178 mg, 0,854 mmol) en THF anhidro (1,5 ml) y una solución de 5-clorobiciclo[4.2.0]octa-1,3,5-trién-7-ona (100 mg, 0,66 mmol) en THF anhidro (1,5 ml) se le añadieron en porciones secuencialmente. La mezcla de reacción se agitó a 0 °C durante 20 min, y luego se le añadió gota a gota piridina anhidra (0,46 ml, 5,69 mmol) a 0 °C durante 30 min. La mezcla de reacción se agitó a 0 °C durante 2 h y a temperatura ambiente durante 16 h más, luego se inactivó mediante la adición de una solución acuosa saturada de cloruro de amonio (7 ml). Se continuó agitando durante 10 min más, y luego la solución se extrajo con acetato de etilo (2 x 15 ml). Se combinaron los extractos orgánicos, se lavaron con salmuera (15 ml) y se secaron sobre sulfato de magnesio, luego se filtraron y se concentraron al vacío. El residuo se purificó mediante cromatografía en columna ultrarrápida, usando un gradiente de acetato de etilo en heptano (0-100 %), para proporcionar el compuesto del título (191 mg, 70 %) como un sólido de color amarillo anaranjado. 5h (500 m Hz , DMSü-da) 7,70 (d, J 2,0 Hz, 1H), 7,61-7,52 (m, 1H), 7,48 (d, J 8,1 Hz, 1H), 7,40 (d, J 7,1 Hz, 1H), 7,01 (d, J 2,0 Hz, 1H), 4,76 (q, J 7,1 Hz, 2H), 4,07 (s, 2H), 1,40 (t, J 7,1 Hz, 3H). HPLC-MS (método 5): MH+ m/z 314 y 316, t R 2,07 min.
Intermedio 62
4-(1-Adamantilmetilén)-2-(2-metilpirazol-3-il)oxazol-5-ona
Se añadió tetracloruro de titanio en DCM (1 M, 3,6 ml, 3,63 mmol) a THF anhidro (6 ml) a -10 °C. Se le añadieron secuencialmente una solución del intermedio 37 (150 mg, 0,908 mmol) en THF anhidro (1,5 ml) y una solución de adamantano-1 -carbaldehído (298 mg, 1,82 mmol) en DCM anhidro (2 ml) en porciones. La mezcla de reacción se agitó a 0 °C durante 20 min, y luego se le añadió piridina anhidra (0,60 ml, 7,42 mmol) a 0 °C durante 30 min. La mezcla de reacción se agitó a 0 °C durante 2 h y a temperatura ambiente durante 16 h más, y luego se inactivó mediante la adición de una solución acuosa saturada de cloruro de amonio (15 ml). Se continuó agitando durante 10 min más, y luego la solución se extrajo con acetato de etilo (2 x 30 ml). Los extractos orgánicos se combinaron y lavaron con salmuera (10 ml), luego se secaron sobre sulfato de sodio anhidro y se filtraron. Se eliminó el solvente al vacío. El residuo se purificó mediante cromatografía automatizada, usando un gradiente de acetato de etilo en heptano (5-40 %), para proporcionar el compuesto del título (24 mg, 8,5 %) como un sólido beige. 5h (250 MHz, CDCh) 7,55 (d, J 2,1 Hz, 1H), 6,90 (d, J 2,1 Hz, 1H), 6,49 (s, 1H), 4,30 (s, 3H), 2,10-2,03 (m, 3H), 2,03-1,97 (m, 6H), 1,80-1,73 (m, 6H). HPLC-MS (método 4 ): MH+ m/z 312, TR 3,20 min.
Intermedio 63 (procedimiento G)
W-{2-(1-Adamantil)-1-[(2-oxospiro[indolín-3,4'-tetrahidropiran]-6-il)carbamoil]-vinil}-2-metilpirazol-3-carboxamida
A una solución agitada del intermedio 2 (17 mg, 0,078 mmol) y del intermedio 62 (24 mg, 0,078 mmol) en acetonitrilo anhidro (3 ml) se le añadió ácido acético (0,045 ml, 0,78 mmol). La mezcla de reacción se agitó a 60 °C durante 19 h. Se eliminó el solvente al vacío y el residuo se trituró en DCM (2 ml). El sólido se recogió por filtración y se secó más al vacío para proporcionar el compuesto del título (20 mg, 42 %) como un sólido de color naranja. HPLC-MS (método 5): MH+ m/z 530, TR 1,82 min (87 %) y TR 2,20 min (13 %).
Intermedio 64
4-[1 -(B iciclo[1.1.1 ]pentán-1 -il)etiliden]-2-(2-metilpirazol-3-il)oxazol-5-ona
Se añadió tetracloruro de titanio en DCM (1 M, 4,8 ml, 4,80 mmol) a THF anhidro (9 ml) a -10 °C. Una solución del intermedio 37 (200 mg, 1,21 mmol) en THF anhidro (1,5 ml) y 1-(biciclo[1.1.1 ]pentán-1 -il)etán-1 -ona (90 %, 297 mg,
2,42 mmol) en THF anhidro (1,5 ml) se añadieron en porciones secuencialmente. La mezcla de reacción se agitó a 0 °C durante 20 min, luego se le añadió piridina anhidra (0,784 ml, 9,69 mmol) a 0 °C durante 30 min. La mezcla de reacción se agitó a 0 °C durante 2 h y a temperatura ambiente durante 16 h más, y luego se inactivó mediante la adición de una solución acuosa saturada de cloruro de amonio (20 ml). Se continuó agitando durante 10 min más, y luego la solución se extrajo con acetato de etilo (2 x 40 ml). Se combinaron los extractos orgánicos y se lavaron con salmuera (20 ml), luego se secaron sobre sulfato de sodio anhidro y se filtraron. Se eliminó el solvente al vacío. El residuo se purificó por cromatografía automatizada, utilizando un gradiente de acetato de etilo en heptano (5-40 %), para proporcionar el compuesto del título (235 mg, 73 %) como un sólido amarillo. 5h (250 MHz, CDCL) 7,53 (d, J 2,1 Hz, 1H), 6,85 (d, J 2,1 Hz, 1H), 4,29 (s, 3H), 2,60 (s, 1H), 2,30 (s, 3H), 2,22 (s, 6H). HPLC-MS (método 5): MH+ m/z 258, TR 2,13 min.
Intermedio 65
4-Cicloheptiliden-2-(2-metilpirazol-3-il)oxazol-5-ona
Se añadió tetracloruro de titanio en DCM (1 M, 4,8 ml, 4,80 mmol) a THF anhidro (9 ml) a -10 °C. Se le añadieron secuencialmente una solución del intermedio 37 (200 mg, 1,21 mmol) en THF anhidro (1,5 ml) y una solución de cicloheptanona (272 mg, 2,42 mmol) en THF anhidro (1,5 ml) en porciones. La mezcla de reacción se agitó a 0 °C durante 20 min, y luego se le añadió piridina anhidra (0,784 ml, 9,69 mmol) a 0 °C durante 30 min. La mezcla de reacción se agitó a 0 °C durante 2 h y a temperatura ambiente durante 16 h más, y luego se inactivó mediante la adición de una solución acuosa saturada de cloruro de amonio (20 ml). Se continuó agitando durante 10 min más, y luego la solución se extrajo con acetato de etilo (2 x 40 ml). Se combinaron los extractos orgánicos y se lavaron con salmuera (20 ml), luego se secaron sobre sulfato de sodio anhidro y se filtraron. Se eliminó el solvente al vacío. El residuo se purificó por cromatografía, utilizando un gradiente de acetato de etilo en heptano (5-40 %), para proporcionar el compuesto del título (201 mg, 64 %) como un sólido beige. 5h (250 MHz, CDCta) 7,53 (d, J 1,9 Hz, 1H), 6,86 (d, J 1,9 Hz, 1H), 4,28 (s, 3H), 3,12-3,05 (m, 2H), 3,02-2,94 (m, 2H), 1,83-1,73 (m, 4H), 1,65-1,57 (m, 4H). HPLC-MS (método 4): MH+ m/z 260, TR 3,09 min.
Intermedio 66 (procedimiento H)
4-Cicloheptil-2-(2-metilpirazol-3-il)-4H-oxazol-5-ona
A una solución agitada del intermedio 65 (120 mg, 0,52 mmol) en THF anhidro (6 ml) se le añadió paladio al 10 % sobre carbón vegetal (humedad al 50 %, 12 mg, 20 % en peso) como una sola porción. La mezcla de reacción se colocó en una atmósfera de hidrógeno gaseoso (3 ciclos de vacío/nitrógeno gaseoso seguidos de 3 ciclos de vacío/hidrógeno gaseoso). Se continuó agitando a temperatura ambiente durante 5 h. Se le añadió acetonitrilo anhidro (6 ml) y se continuó agitando en una atmósfera de hidrógeno gaseoso durante 16 h más. El catalizador se eliminó por filtración sobre tierra de diatomeas, enjuagando la torta del filtro con THF seco (2 x 5 ml). El solvente se concentró al vacío para proporcionar el compuesto del título (66 mg, 65 %) como un aceite gris pálido. HPLC-MS (método 4): (M-H)- m/z 260, TR 3,13 min.
Intermedio 67
4-(Biciclo[3.2.1]octán-3-iliden)-2-(2-metilpirazol-3-il)oxazol-5-ona
Se añadió tetracloruro de titanio en DCM (1 M, 4,8 ml, 4,80 mmol) a THF anhidro (9 ml) a -10 °C. Se le añadieron secuencialmente una solución del intermedio 37 (200 mg, 1,21 mmol) en THF anhidro (1,5 ml) y una solución de biciclo[3.2.1]octán-3-ona (301 mg, 2,42 mmol) en THF anhidro (1,5 ml) en porciones. La mezcla de reacción se agitó a 0 °C durante 20 min, y luego se le añadió piridina anhidra (0,784 ml, 9,69 mmol) a 0 °C durante 30 min. La mezcla de reacción se agitó a 0 °C durante 2 h y a temperatura ambiente durante 16 h más, y luego se inactivó mediante la adición de una solución acuosa saturada de cloruro de amonio (20 ml). Se continuó agitando durante 10 min más, luego la solución se extrajo con acetato de etilo (2 x 40 ml). Se combinaron los extractos orgánicos y lavaron con salmuera (20 ml), luego se secaron sobre sulfato de sodio anhidro y se filtraron. Se eliminó el solvente al vacío. El residuo se purificó por cromatografía, utilizando un gradiente de acetato de etilo en heptano (5-40 %), para proporcionar el compuesto del título (261 mg, 79 %) como un sólido de color amarillo. 5h (250 MHz, CDCh) 7,53 (d, J 2,1 Hz, 1H), 6,86 (d, J 2,1 Hz, 1H), 4,27 (s, 3H), 3,75-3,64 (m, 1H), 3,35-3,24 (m, 1H), 2,56-2,46 (m, 2H), 2,38 (t, J 16,2 Hz, 2H), 1,77 (t, J 11,4 Hz, 2H), 1,71-1,62 (m, 2H), 1,49-1,36 (m, 2H). HPLC-MS (método 4): MH+ m/z 272, TR 3,15 min.
Intermedio 68
4-(Biciclo[3.2.1]octán-3-il)-2-(2-metilpirazol-3-il)-4H-oxazol-5-ona
A una solución agitada del intermedio 67 (200 mg, 0,74 mmol) en acetonitrilo anhidro (10 ml) se le añadió paladio al 10 % sobre carbón vegetal (humedad del 50 %, 40 mg, 20 % en peso) como una sola porción. La mezcla de reacción se colocó en una atmósfera de hidrógeno gaseoso. Se continuó agitando a temperatura ambiente durante 6 h. El catalizador se eliminó por filtración sobre tierra de diatomeas, enjuagando la torta del filtro con acetonitrilo seco (2 x 5 ml). El solvente se concentró al vacío para proporcionar el compuesto del título (200 mg, 84 %) como un aceite gris
pálido. 5h (250 MHz, CDCla) 7,53 (d, J 2,1 Hz, 1H), 6,79 (d, J 2,0 Hz, 1H), 4,33 (d, J 6,6 Hz, 1H), 4,24 (s, 3H), 2,34 2,24 (m, 2H), 2,23-2,02 (m, 3H), 1,95-1,83 (m, 1H), 1,77-1,64 (m, 3H), 1,47-1,35 (m, 3H), 1,21-1,13 (m, 1H). HPLC-MS (método 4): (M-H)- m/z 272, TR 3,13 min.
Intermedio 69
4-(5-Cloro-7-biciclo[4.2.0]octa-1(6),2,4-trieniliden)-2-(2-metilpirazol-3-il)oxazol-5-ona
Se añadió tetracloruro de titanio en DCM (1 M, 4,8 ml, 4,80 mmol) a THF anhidro (9 ml) a -10 °C. Se le añadieron en porciones secuencialmente una solución del intermedio 37 (200 mg, 1,21 mmol) en THF anhidro (1,5 ml) y una solución de 5-clorobiciclo[4.2.0]octa-1,3,5-trién-7-ona (369 mg, 2,42 mmol) en THF anhidro (1,5 ml). La mezcla de reacción se agitó a 0 °C durante 20 min, y luego se le añadió piridina anhidra (0,784 ml, 9,69 mmol) a 0 °C durante 30 min. La mezcla de reacción se agitó a 0 °C durante 2 h y a temperatura ambiente durante 16 h más, y luego se inactivó mediante la adición de una solución acuosa saturada de cloruro de amonio (20 ml). Se continuó agitando durante 10 min más, luego la solución se extrajo con acetato de etilo (2 x 40 ml). Se combinaron los extractos orgánicos y se lavaron con salmuera (20 ml), luego se secaron sobre sulfato de sodio anhidro y se filtraron. Se eliminó el solvente al vacío. El residuo se purificó por cromatografía automatizada, utilizando un gradiente de acetato de etilo en heptano (5-50 %), para obtener el compuesto del título (159 mg, 44 %) como un sólido de color amarillo. 5h (250 MHz, CDCl3) 7,57 (d, J 2,1 Hz, 1H), 7,39 (dd, J 8,2, 6,8 Hz, 1H), 7,33 (d, J 7,5 Hz, 1H), 7,20 (d, J 6,8 Hz, 1H), 6,92 (d, J 2,1 Hz, 1H), 4,38 (s, 3H), 4,09 (s, 2H). HPLC-MS (método 4): MH+ m/z 300, TR 3,18 min.
Intermedio 70
4-(2,3-Dimetilciclobutiliden)-2-(2-metilpirazol-3-il)oxazol-5-ona
Se añadió tetracloruro de titanio en DCM (1 M, 4,8 ml, 4,80 mmol) a THF anhidro (9 ml) a -10 °C. Se le añadieron secuencialmente una solución del intermedio 37 (200 mg, 1,21 mmol) en THF anhidro (1,5 ml) y una solución de 2,3-dimetilciclobután-1-ona (238 mg, 2,42 mmol) en THF anhidro (1,5 ml) en porciones. La mezcla de reacción se agitó a 0 °C durante 20 min, y luego se le añadió piridina anhidra (0,784 ml, 9,69 mmol) a 0 °C durante 30 min. La mezcla de reacción se agitó a 0 °C durante 2 h y a temperatura ambiente durante 16 h más, y luego se inactivó mediante la adición de una solución acuosa saturada de cloruro de amonio (20 ml). Se continuó agitando durante 10 min más, y luego la solución se extrajo con acetato de etilo (2 x 40 ml). Se combinaron los extractos orgánicos y se lavaron con salmuera (20 ml), luego se secaron sobre sulfato de sodio anhidro y se filtraron. Se eliminó el solvente al vacío. El residuo se purificó por cromatografía automatizada, utilizando un gradiente de acetato de etilo en heptano (5-40 %) para obtener el compuesto del título (mezcla de estereoisómeros; 173 mg, 58 %) como un sólido de color amarillo. HPLC-MS (método 4): MH+ m/z 246, TR 2,90 min (49 %) y TR 2,95 min (51 %).
Intermedio 71
6-Bromo-4-fluoro-2-oxoindolín-1-carboxilato de ferf-butilo
Se añadió gota a gota dicarbonato de di-terf-butilo (853,88 mg, 3,91 mmol) en THF (8 ml) a una suspensión agitada de 6-bromo-4-fluoroindolín-2-ona (900 mg, 3,91 mmol) e hidrogenocarbonato de sodio (1,15 g, 13,69 mmol) en THF (10 ml). La mezcla de reacción se calentó, con agitación, a 50 °C durante 4,5 h, luego se eliminó el sólido por filtración y se eliminó el solvente al vacío. El residuo se purificó por cromatografía en columna ultrarrápida, utilizando un gradiente de terf-butilmetiléter en heptano (0-20 %), para proporcionar el compuesto del título (1,04 g, 80 %) como un sólido amarillo. 5h (500 MHz, CDCla) 7,86 (s, 1H), 7,07 (dd, J 7,9, 1,5 Hz, 1H), 3,60 (s, 2H), 1,64 (s, 9H). HPLC-MS (método 5): MH+ m/z 328,2, 330,0, T r 2,05 min.
Intermedio 72
6-Bromo-4-fluoro-2-oxospiro[indolín-3,4'-tetrahidropiran]-1-carboxilato de ferf-butilo
Una solución agitada del intermedio 71 (0,8 g, 2,42 mmol) y 1-yodo-2-(2-yodoetoxi)etano (0,38 ml, 2,67 mmol) en DMF anhidro (16 ml) se enfrió a -15 °C y se purgó con nitrógeno durante 5 min, y luego se le añadió carbonato de cesio (3,16 g, 9,69 mmol). La mezcla de reacción se agitó durante 2 h, con calentamiento a 20 °C. Se le añadió agua (30 ml) y la fase acuosa se extrajo con tert-butilmetiléter (3 x 30 ml). Las capas orgánicas combinadas se lavaron con agua (2 x 30 ml) y salmuera (30 ml), luego se secaron sobre sulfato de sodio, se filtraron y se concentraron. El material bruto resultante se purificó por cromatografía en columna ultrarrápida, usando un gradiente de terf-butilmetiléter en heptano (0-15 %), para proporcionar el compuesto del título (927,9 mg, 86 %) como un sólido amarillo. 5h (500 MHz, CDCls ) 7,90 (d, J 1,3 Hz, 1H), 7,06 (dd, J 9,1, 1,6 Hz, 1H), 4,26 (t, J 11,8 Hz, 2H), 3,89 (dd, J 11,9, 3,6 Hz, 2H), 2,45 2,33 (m, 2H), 1,75-1,69 (m, 2H), 1,65 (s, 9H). HPLC-MS (método 5): [M+H-BOC]+ m/z 300,0, 302,0, TR 2,11 min.
Intermedio 73
6-(ferf-Butoxicarbonilam ino)-4-fluoro-2-oxospiro[indolín-3,4'-tetrahidropiran]-1-carboxilato de ferf-butilo
Se añadió acetato de paladio (II) (17 mg, 0,08 mmol) a una mezcla desgasificada con nitrógeno del intermedio 72 (476 mg, 1,19 mmol), carbamato de tert-butilo (170 mg, 1,45 mmol), XPhos (30 mg, 0,06 mmol) y carbonato de cesio (776
mg, 2,38 mmol) en tolueno anhidro (5 ml). La mezcla de reacción se calentó a 90 °C durante 3 h. La mezcla de reacción enfriada se filtró a través de tierra de diatomeas y se enjuagó con tolueno (2 x 5 ml). El filtrado se concentró al vacío. El residuo bruto se purificó por cromatografía en columna ultrarrápida, utilizando un gradiente de ferf-butilmetiléter en heptano (0-20 %), para producir el compuesto del fífulo (533 mg, 87 %) como un aceite de color marrón pálido que, al reposar, se cristalizó en un sólido blanquecino. 5h (250 MHz, CDCL) 7,63 (d, J 1,6 Hz, 1H), 7,11 (dd, J 11,8, 1,5 Hz, 1H), 6,56 (s, 1H), 4,25 (t, J 11,5 Hz, 2H), 3,95-3,80 (m, 2H), 2,46-2,26 (m, 2H), 1,72 (d, J 14,6 Hz, 2H), 1,64 (s, 9H), 1,51 (s, 9H). HPLC-MS (método 5): [M-H]- m/z 435,0, TR 2,09 min.
Intermedio 74
6-Amino-4-fluoroesp¡ro[¡ndolín-3,4'-tetrah¡drop¡ran]-2-ona
Se añadió ácido trifluoroacético (2,72 ml, 33,4 mmol) a una solución del infermedio 73 (86%, 339 mg, 0,67 mmol) en DCM (4,75 ml) a 20 °C. La mezcla de reacción se agitó a 20 °C durante 3 h, luego se inactivó con una solución acuosa saturada de hidrogenocarbonato de sodio (20 ml) y se extrajo con DCM (3 x 15 ml). Los extractos orgánicos se combinaron, se filtraron a través de una frita hidrófoba y se concentraron al vacío. El material bruto resultante se purificó por cromatografía en columna ultrarrápida, utilizando un gradiente de ferf-butilmetiléter en heptano (0-100 %) seguido de un gradiente de MeOH en ferf-butilmetiléter (0-10 %). Se combinaron las fracciones relevantes y se concentraron al vacío para proporcionar el compuesto del fífulo (53,9 mg, 41 %) como un sólido blanquecino. 5h (500 MHz, DMSO-ds ) 10,27 (s, 1H), 5,95 (d, J 1,6 Hz, 1H), 5,88 (dd, J 12,8, 1,6 Hz, 1H), 5,42 (s, 2H), 4,04 (t, J 10,1 Hz, 2H), 3,79-3,69 (m, 2H), 1,93 (ddd, J 14,2, 10,3, 4,4 Hz, 2H), 1,69-1,59 (m, 2H). uPLC-MS (método 1): MH+ m/z 237,1, TR 1,49 min.
Intermedio 75
4-(4-Metilciclohexiliden)-2-(2-metilpirazol-3-il)oxazol-5-ona
Se añadió tetracloruro de titanio en DCM (1 M, 48 ml, 48,0 mmol) a THF anhidro (90 ml) a -10 °C. Se le añadieron secuencialmente una solución del infermedio 37 (2,00 g, 12,10 mmol) en THF anhidro (15 ml) y una solución de 4-metilciclohexanona (2,72 g, 24,20 mmol) en THF anhidro (15 ml) en porciones. La mezcla de reacción se agitó a 0 °C durante 20 min, y luego se le añadió piridina anhidra (7,84 ml, 96,9 mmol) a 0 °C durante 30 min. La mezcla de reacción se agitó a 0 °C durante 2 h y a 20 °C durante 16 h más, y luego se inactivó mediante la adición de una solución acuosa saturada de cloruro de amonio (200 ml). Se continuó agitando durante 10 min más, y luego la solución se extrajo con acetato de etilo (2 x 400 ml). Se combinaron los extractos orgánicos y se lavaron con salmuera (200 ml), luego se secaron sobre sulfato de sodio anhidro y se filtraron. Se retiró el solvente al vacío. El residuo se purificó mediante cromatografía en columna ultrarrápida, utilizando un gradiente de acetato de etilo en heptano (5-40 %), para proporcionar el compuesto del fífulo (2,90 g, 92 %) como un sólido amarillo pálido. 5h (500 MHz, CDCh) 7,53 (d, J 2,1 Hz, 1H), 6,86 (d, J 2,1 Hz, 1H), 4,28 (s, 3H), 3,89-3,81 (m, 1H), 3,44-3,36 (m, 1H), 2,30-2,18 (m, 2H), 2,04-1,96 (m, 2H), 1,81-1,71 (m, 1H), 1,28-1,18 (m, 2H), 0,97 (d, J 6,6 Hz, 3H). HPLC-MS (método 19): MH+ m/z 260,2, TR 3,21 min.
Intermedio 76
W-{2-[(4-Fluoro-2-oxospiro[indolín-3,4'-tetrahidropiran]-6-il)amino]-1-(4-metil-ciclohexiliden)-2-oxoetil}-2-metilpirazol-3-carboxamida
Se añadió ácido acético (0,09 ml, 1,63 mmol) a una solución agitada del infermedio 74 (73,3 mg, 0,11 mmol) y del infermedio 75 (39 pl, 0,15 mmol) en THF anhidro (1 ml). La mezcla de reacción se agitó a 60°C durante 18 h en nitrógeno en un tubo sellado, y luego se eliminó el solvente al vacío. El residuo se purificó por cromatografía en columna ultrarrápida, utilizando un gradiente de ferf-butilmetiléter en heptano (0-100 %) seguido de un gradiente de MeOH en ferf-butilmetiléter (0-20 %), para proporcionar el compuesfo del fífulo (73 mg, 91 %) como un sólido blanquecino. 5h (500 MHz, CDCla) 9,33 (s, 1H), 8,56 (s, 1H), 8,18 (s, 1H), 7,72 (s, 1H), 7,44 (d, J 2,1 Hz, 1H), 6,82 (d, J 2,1 Hz, 1H), 6,54 (d, J 9,3 Hz, 1H), 4,26 (t, J 10,2 Hz, 2H), 4,16 (s, 3H), 3,94-3,87 (m, 2H), 2,83 (d, J 12,6 Hz, 1H), 2,73-2,67 (m, 1H), 2,30-2,19 (m, 2H), 2,16-2,05 (m, 1H), 2,04-1,94 (m, 1H), 1,90-1,82 (m, 2H), 1,79-1,71 (m, 2H), 1,67 1,61 (m, 1H), 1,16-1,10 (m, 2H), 0,92 (d, J 6,5 Hz, 3H). uPLC-MS (método 1): MH+ m /z496,2, TR 1,78 min.
Intermedio 77
W-{(1S)-1-Ciclohexil-2-oxo-2-[(2-oxospiro[indolín-3,4'-tetrahidropiran]-6-il)amino]etil}carbamato de fert-butilo
Se añadió DIPEA (0,15 ml, 0,92 mmol) a una solución agitada del infermedio 2 (100 mg, 0,46 mmol), ácido (2S)-[(ferfbutoxicarbonil)amino](ciclohexil)etanoico (118 mg, 0,46 mmol) y HATU (209 mg, 0,55 mmol) en THF anhidro (2,5 ml) en una atmósfera de nitrógeno. La mezcla de reacción se agitó a 20 °C durante 72 h, y luego se le añadieron agua (5 ml) y una solución acuosa saturada de bicarbonato de sodio (2,5 ml). Se continuó agitando durante 10 min más. La mezcla de reacción lechosa se extrajo con acetato de etilo (2 x 10 ml). Se combinaron los extractos orgánicos y se lavaron con salmuera (10 ml), luego se secaron sobre sulfato de sodio anhidro y se filtraron. Se eliminó el solvente al vacío. El residuo se purificó mediante cromatografía en columna ultrarrápida, utilizando un gradiente de acetato de etilo en heptano (5-100 %), para proporcionar el compuesfo del fífulo (166 mg, 77 %) como un vidrio incoloro. 5h (250
MHz, DMSO-ds) 10,38 (s, 1H), 9,95 (s, 1H), 7,42 (d, J 8,1 Hz, 1H), 7,38 (d, J 1,8 Hz, 1H), 7,07 (dd, J 8,2, 1,7 Hz, 1H), 6,84 (d, J 8,7 Hz, 1H), 4,08-3,96 (m, 2H), 3,95-3,86 (m, 1H), 3,85-3,72 (m, 2H), 1,81-1,50 (m, 10H), 1,38 (s, 9H), 1,18 0,96 (m, 5H). HPLC-Ms (método 5): m H+ m/z 458,2, TR 1,84 min.
Intermedio 78
(2S)-2-Am¡no-2-c¡clohex¡l-W-(2-oxosp¡ro[¡ndolín-3,4'-tetrah¡drop¡ran]-6-¡l)acetam¡da
Se añadió gota a gota ácido trifluoroacético (0,40 ml, 5,25 mmol) a una suspensión agitada del intermedio 77 (165 mg, 0,35 mmol) en DCM (1,4 ml). La mezcla de reacción se agitó a 20 °C durante 2,5 h. Se le añadieron DCM (10 ml) y agua (5 ml), seguidos de solución acuosa de hidróxido de sodio a 4 M (1,4 ml) y se continuó agitando durante 5 min. Se recogió la fase orgánica y la fase acuosa se extrajo más con una mezcla de DCM-isopropanol (2:1,2 x 10 ml). Se combinaron las fases orgánicas y se agitaron con salmuera (5 ml). La fase orgánica se separó usando una frita hidrófoba, y luego el solvente se concentró al vacío para proporcionar el compuesto del título (111 mg, 79 %) como un sólido vitreo transparente. 5h (250 MHz, DMSO-d6) 10,38 (s, 1H), 9,81 (br s, 1H), 7,48-7,36 (m, 2H), 7,06 (dd, J 8,2, 1,9 Hz, 1H), 4,07-3,93 (m, 2H), 3,87-3,71 (m, 2H), 3,08 (d, J 5,1 Hz, 1H), 1,84-1,42 (m, 10H), 1,25-1,07 (m, 5H). HPLC-MS (método 5): m H+ m/z 358,2, TR 1,45 min.
Intermed¡o 79
2-Isoprop¡l-W-[(1 S)-1-(4-met¡lc¡clohex¡l)-2-oxo-2-({2-oxo-1-[2-(tr¡met¡ls¡l¡l)-etox¡met¡l]esp¡ro[p¡rrolo[3,2-c]p¡r¡d¡n-3,4'- tetrahidropiran]-6-il}amino)etil]-pirazol-3-carboxamida (¡somero tra n s )
Se preparó a partir del intermedio 107 (150 mg, 0,26 mmol) y el ácido 2-isopropilpirazol-3-carboxílico (61,1 mg, 0,4 mmol) de acuerdo con el procedimiento A, y se purificó por cromatografía en columna ultrarrápida, usando un gradiente de tert-butilmetiléter en heptano (0-100 %), para dar el compuesto del título (115,4 mg, 57 %) como un polvo blanco.
5h (500 MHz, DMSO-ds) 10,74 (s, 1H), 8,60 (s, 1H), 8,53 (d, J 8,0 Hz, 1H), 8,05 (s, 1H), 7,61 (d, J 1,9 Hz, 1H), 7,04 (d, J 2,0 Hz, 1H), 5,54-5,44 (m, 1H), 5,19 (s, 2H), 4,60 (t, J 8,2 Hz, 1H), 4,15-4,08 (m, 2H), 3,96 (t, J 8,0 Hz, 2H), 3,64 3,57 (m, 2H), 1,99-1,86 (m, 4H), 1,86-1,74 (m, 4H), 1,71-1,61 (m, 1H), 1,50-1,29 (m, 9H), 1,21-1,10 (m, 1H), 1,02-0,90 (m, 6H), 0,00 (s, 9H). HpLC-MS (método 3 ): MH+ m/z 639,2, Tr 1,45 min.
Intermed¡o 80
(2S)-2-{[6-(D¡fluoromet¡l)p¡r¡daz¡n-3-¡l]am¡no}-2-(4-met¡lc¡clohex¡l)-W-{2-oxo-1-[2-(trim etilsilil)etoxim etil]espiro[pirrolo[3,2-c]piridin-3,4'-tetrahidropiran]-6-il}-acetamida (¡somero tra n s )
El intermedio 107 (100 mg, 0,18 mmol) se disolvió en 1,4-dioxano anhidro (0,5 ml) y DIPEA (0,09 ml, 0,53 mmol) y se trató con 3-cloro-6-(difluorometil)-piridazina (50 mg, 0,3 mmol). La mezcla de reacción se agitó a 140 °C durante 18 h, luego se enfrió a 20 °C y se inactivó con una solución acuosa saturada de hidrogenocarbonato de sodio (10 ml). La capa acuosa se extrajo con DCM (3 x 20 ml). Se combinaron los extractos orgánicos y se lavaron con salmuera (10 ml), luego se secaron sobre sulfato de sodio, se filtraron y se concentraron al vacío. El residuo se purificó por cromatografía en columna ultrarrápida, utilizando un gradiente de terf-butilmetiléter en heptano (0-100 %) seguido de un gradiente de MeOH en terf-butilmetiléter (0-20 %), para proporcionar el compuesto del título (113,3 mg, 33 %) como un aceite de color naranja. HPLC-MS (método 5): Mh m/z 631,0, TR 2,16 min.
Intermed¡o 81
6-Cloro-1-{[2-(tr¡met¡ls¡l¡l)etox¡)met¡l}-1 H-pirrolo[3,2-c]piridina
A una solución de 6-cloro-1 H-pirrolo[3,2-c]piridina (5,00 g, 32,8 mmol) en DMF (20 ml) se le añadió NaH (1,57 g, 39,3 mmol) a 0 °C. La mezcla de reacción se agitó durante 1 h, luego se le añadió SEM-Cl (6,56 g, 39,3 mmol) a 0 °C. La mezcla de reacción se agitó a temperatura ambiente durante 1 h, luego se inactivó con H2O (100 ml) y se extrajo con DCM (3 x 100 ml). Se separó la capa orgánica y se lavó con salmuera (3 x 100 ml), luego se secó sobre Na2SO4 anhidro y se concentró al vacío. El material bruto se purificó por cromatografía en columna (EtOAc al 10-15 % en hexanos) para proporcionar el compuesto del título (7,00 g, 75 %) como un aceite de color amarillo. 5h (400 MHz CD3OD) -0,07 (s, 9H), 0,87 (t, J 7,83 Hz, 2H), 3,52 (t, J 7,83 Hz, 2H), 5,57 (s, 2H), 6,71 (d, J 2,93 Hz, 1H), 7,50 (d, J 3,42 Hz, 1H), 7,64 (s, 1H), 8,61 (s, 1H). HPLC-MS (método 6): MH+ m/z 283,3, TR 2,12 min.
Intermed¡o 82
3,3-D¡bromo-6-cloro-1-[2-(tr¡met¡ls¡l¡l)etox¡met¡l]p¡rrolo[3,2-c]p¡r¡d¡n-2-ona
A una solución del intermedio 81 (3,50 g, 12,4 mmol) en 1,4-dioxano (50 ml) se le añadió tribromuro de piridinio (19,8 g, 61,9 mmol) en porciones a 0 °C. La mezcla de reacción se agitó a temperatura ambiente durante 1 h, luego se inactivó con H2O (100 ml), se agitó durante 10 min y se extrajo con EtOAc (2 x 100 ml). Se separó la capa orgánica y se lavó con salmuera (2 x 100 ml), luego se secó sobre Na2SO4 anhidro y se concentró al vacío, para proporcionar el compuesto del título (5,00 g, 78 %) como un aceite rojo, que se utilizó sin más purificación. 5h (400 MHz, CDCh) 0,00 (s, 9H), 0,92-1,00 (m, 2H), 3,59-3,65 (m, 2H), 5,31 (s, 2H), 7,33-7,37 (m, 1H), 8,75 (s, 1H)). HPLC-MS (método 6): MH+ m/z 457,0, TR 2,40 min.
Intermedio 83
6-Cloro-1-{[2-(tr¡met¡ls¡l¡l)etox¡]met¡l}-1,3-d¡h¡dro-2H-p¡rrolo[3,2-c]p¡r¡d¡n-2-ona
A una solución del intermedio 82 (5,00 g, 10,9 mmol) en THF (70 ml) se le añadió Zn (7,16 g, 109 mmol) a 0 °C, seguido de la adición gota a gota de una solución saturada de NH4Cl (20 ml). La mezcla de reacción se agitó a temperatura ambiente durante 3 h, luego se diluyó con EtOAc (300 ml) y se filtró a través de una capa de Celite. El filtrado se lavó con agua (2 x 100 ml), luego se secó sobre Na2SO4 anhidro y se concentró al vacío. El material bruto se purificó por cromatografía en columna (EtOAc al 25-30 % en hexanos) para producir el compuesto del título (2,00 g, 45 %) como un aceite de color amarillo pálido. 5h (400 MHz CD3OD) 0,01 (s, 9H), 0,90-0,98 (m, 2H), 3,57-3,67 (m, 2H), 3,72 (s, 2H), 5,17 (s, 2H), 7,18 (s, 1H), 8,14 (s, 1H). HPLC-MS (método 6 ): MH+ m/z 299,2, TR 2,08 min.
Intermed¡o 84
6'-Cloro-1'-{[2-(tr¡met¡ls¡l¡l)etox¡]met¡l}-2,3,5,6-tetrah¡drosp¡ro[p¡rán-4,3'-p¡rrolo[3,2-c]p¡r¡d¡n]-2'(1'H)-ona
A una solución del intermedio 83 (2,00 g, 6,69 mmol) en acetona (30 ml) se le añadió Cs2CO3 (6,54 g, 20,1 mmol) a 0 °C. La mezcla de reacción se agitó a 0 °C durante 10 min, y luego se le añadió 1-yodo-2-(2-yodoetoxi)etano (4,36 g, 13,4 mmol) a 0 °C. La mezcla de reacción se agitó a temperatura ambiente durante 6 h, y luego se concentró al vacío. El residuo se diluyó con EtOAc (500 ml) y se lavó con agua (2 x 200 ml). Se separó la capa orgánica y se lavó con salmuera (200 ml), luego se secó sobre Na2SO4 anhidro y se concentró al vacío. El material bruto se purificó por cromatografía en columna (EtOAc al 15-20 % en hexanos) para producir el compuesto del título (1,20 g, 48 %) como un sólido blanquecino. 5h (400 MHz, DMSO-ds) -0,08 (s, 9H), 0,84 (t, J 7,83 Hz, 2H), 1,74-1,85 (m, 4H), 3,50 (t, J 8,07 Hz, 2H), 3,80-3,86 (m, 2H), 3,96-4,06 (m, 2H), 5,13 (s, 2H), 7,31 (s, 1H), 8,58 (s, 1H). HPLC-MS (método 6 ): MH+ m/z 369,0, TR 3,18 min.
Intermed¡o 85
W-(2-Am¡no-1-c¡clooct¡l-2-oxoet¡l)-2-met¡lp¡razol-3-carboxam¡da
A una solución del intermedio 13 (0,30 g, 1,02 mmol) en DCM (15 ml) se le añadieron HATU (0,58 g, 1,53 mmol) y NH4Cl (0 , 22 g, 4,09 mmol). La mezcla de reacción se agitó a 0 °C durante 10 min, seguido de la adición de DIPEA (0,55 ml, 3,07 mmol). La mezcla de reacción se agitó a temperatura ambiente durante 16 h, luego se diluyó con DCM (100 ml) y se lavó con H2O (2 x 50 ml) y salmuera (2 x 25 ml). Se separó la capa orgánica, luego se secó sobre Na2SO4 anhidro y se concentró al vacío. El material bruto se purificó por cromatografía en columna (60-70 % de EtOAc en hexanos) para producir el compuesto del título (0,23 g, 71 %) como un sólido blanquecino. 5h (400 MHz, DMSO-d6) 1,28-1,37 (m, 3H), 1,40-1,49 (m, 4H), 1,51-1,67 (m, 7H), 2,02-2,10 (m, 1H), 4,02 (s, 3H), 4,27 (t, J 8,80 Hz, 1H), 6,99 (d, J 1,96 Hz, 1H), 7,06 (br s, 1H), 7,43 (d, J 1,96 Hz, 1H), 7,50 (br s, 1H), 8,19 (d, J 9,29 Hz, 1H). HPLC-MS (método 6 ): MH+ m/z 293,1, TR 1,66 min.
Intermed¡o 86
W-[1-C¡clooct¡l-2-oxo-2-({2-oxo-1-[2-(tr¡met¡ls¡l¡l)etox¡met¡l]esp¡ro[p¡rrolo[3,2-c]p¡r¡d¡n-3,4'-tetrah¡drop¡ran]-6-¡l}am¡no)et¡l]-2-met¡lp¡razol-3-carboxam¡da
A una solución del intermedio 85 (0,16 g, 0,54 mmol) en 1,4-dioxano (5 ml) se le añadió el intermedio 84 (0,20 g, 0,54 mmol) y Cs2CO3 (0,53 g, 1,63 mmol). La mezcla de reacción se purgó con argón durante 10 min, y luego se purgó con Pd2(dba)3 (0,05 g, 0,05 mmol) y Xantphos (0,03 g, 0,05 mmol). La mezcla de reacción se calentó a 100 °C durante 16 h, luego se diluyó con EtOAc (30 ml), se filtró a través de una capa de Celite y se lavó con agua (2 x 20 ml). Se separó la capa orgánica, se secó sobre Na2SO4 anhidro y se concentró al vacío. El material bruto se purificó por cromatografía en columna (60-70 % de EtOAc en hexanos) para producir el compuesto del título (0,08 g, 23 %) como un sólido amarillo claro. 5h (400 MHz, DMSO-ds) -0,11 (s, 9H), 0,80-0,90 (m, 2H), 1,38-1,59 (m, 10H), 1,62-1,77 (m, 5H), 1,78 1,87 (m, 2H), 2,16-2,19 (m, 1H), 3,49 (t, J 7,58 Hz, 2H), 3,80-3,91 (m, 3H), 3,97-4,00 (m, 1H), 4,02 (s, 4H), 4,59 (t, J 8,31 Hz, 1H), 5,07 (s, 2H), 7,02 (s, 1H), 7,46 (s, 1H), 7,93 (s, 1H), 8,42 (d, J 8,31 Hz, 1H), 8,50 (s, 1H), 10,71 (s, 1H).
HPLC-MS (método 6 ): m H+ m/z 625,3, TR 2,37 min.
Intermed¡o 87
2-(5-Bromo-3-nitropiridin-2-il)malonato de d¡et¡lo
A una solución de malonato de dietilo (25,3 g, 158 mmol) en DMF (45 ml) se le añadió K2CO3 (43,6 g, 316 mmol) a 0 °C. La mezcla se agitó durante 10 min y luego se le añadió 5-bromo-2-cloro-3-nitropiridina (25,0 g, 105 mmol). La mezcla de reacción se agitó a temperatura ambiente durante 16 h, luego se diluyó con EtOAc (250 ml) y se lavó con agua (3 x 250 ml). Se separó la capa orgánica, se secó sobre Na2SO4 anhidro y se concentró al vacío. El material bruto se purificó por cromatografía en columna (EtOAc al 20 % en hexanos) para proporcionar el compuesto del título (26,0 g, 68 %) como un sólido blanquecino. 5h (400 MHz, DMSO-d6) 1,19 (t, J 6,85 Hz, 6H), 4,16-4,27 (m, 4H), 5,58 (s, 1H), 8,89 (s, 1H), 9,09 (s, 1H). HPLC-MS (método 6): MH+ m/z 360,9, TR 2,04 min.
Intermedio 88
2-(5-Bromo-3-n¡trop¡rid¡n-2-¡l)acetato de etilo
A una solución del intermedio 87 (26,0 g, 72,0 mmol) en DMSO:agua (1:1,90 ml) se le añadió cloruro de litio (4,58 g, 108 mmol). La mezcla de reacción se calentó a 100 °C durante 16 h, luego se diluyó con H2O (110 ml) y se extrajo con EtOAc (3 x 110 ml). Se separó la capa orgánica, se secó sobre Na2SO4 anhidro y se concentró al vacío. El residuo bruto se purificó por cromatografía en columna (EtOAc al 10 % en hexanos) para proporcionar el compuesto del título (14,0 g, 67 %) como un sólido blanco. 5h (400 MHz, DMSO-d6) 1,16-1,21 (m, 3H), 4,07-4,11 (m, 2H), 4,21 (s, 2H), 8,82 (d, J 2,45 Hz, 1H), 9,04 (d, J 1,96 Hz, 1H). HPLC-MS (método 6): MH+ m/z 290,8, TR 1,90 min.
Intermedio 89
6-Bromo-1,3-dihidro-2H-pirrolo[3,2-fc]piridin-2-ona
A una solución del intermedio 88 (10,0 g, 34,6 mmol) en ácido acético (200 ml) se le añadió hierro (9,51 g, 173 mmol). La mezcla de reacción se agitó a 60 °C durante 4 h, luego se diluyó con EtOAc (200 ml), se agitó durante 15 min, se filtró a través de una capa de Celite y se lavó con EtOAc (3 x 200 ml). Se separó la capa orgánica, se secó sobre Na2SO4 anhidro y se concentró al vacío. El residuo bruto se disolvió en MeOH al 5 % en EtOAc (30 ml) y se adsorbió sobre Fluorosil. La suspensión resultante se filtró a través de una capa de Celite y se lavó con MeOH al 5 % en EtOAc (3 x 200 ml). La capa orgánica se secó sobre Na2SO4 anhidro y se concentró al vacío, para proporcionar el compuesto del título (5,00 g, 68 %) como un sólido marrón. 5h (400 MHz, DMSO-ds) 3,57 (s, 2H), 7,30 (s, 1H), 8,17 (s, 1H), 10,67 (br s, 1H). HPLC-MS (método 6): MH+ m/z 213,0, Tr 1,31 min.
Intermedio 90
6'-Bromo-2,3,5,6-tetrahidrospiro[pirán-4,3'-pirrolo[3,2-b]piridin]-2'(1'H)-ona
A una solución del intermedio 89 (2,00 g, 9,39 mmol) en DMSO (20 ml) se le añadió Cs2CO3 (3,05 g, 9,39 mmol) a 0 °C. La mezcla de reacción se agitó durante 30 min y luego se le añadió 1-yodo-2-(2-yodoetoxi)etano (3,05 g, 9,39 mmol). La mezcla de reacción se agitó a temperatura ambiente durante 5 h, luego se inactivó con agua (50 ml) y se extrajo con EtOAc (3 x 50 ml). Se separó la capa orgánica, se secó sobre Na2SO4 anhidro y se concentró al vacío. El residuo bruto se purificó por cromatografía en columna (EtOAc al 20 % en hexanos) para proporcionar el compuesto del título (0,45 g, 17 %) como un líquido marrón claro. 5h (400 MHz, DMSO-d6) 1,58-1,67 (m, 2H), 1,77-1,83 (m, 2H), 3,85-3,96 (m, 2H), 3,98-4,08 (m, 2H), 7,39 (d, J 1,96 Hz, 1H), 8,25 (d, J 1,96 Hz, 1H), 10,78 (s, 1H). HPLC-MS (método 6): MH+ m/z 283,1, TR 1,54 min.
Intermedio 91
6'-Bromo-1'-{[2-(trimetilsilil]etoxi]metil}-2,3,5,6-tetrahidrospiro[pirán-4,3'-pirrolo[3,2-fc]piridin]-2'(1'H)-ona
A una solución del intermedio 90 (0,40 g, 1,41 mmol) en THF (10 ml) se le añadió NaH (0,05 g, 2,12 mmol) a 0 °C. La mezcla de reacción se agitó durante 30 min y luego se le añadió SEM-Cl (0,35 g, 2,12 mmol). La mezcla de reacción se agitó a temperatura ambiente durante 3 h, luego se diluyó con agua (50 ml) y se extrajo con DCM (3 x 50 ml). Se separó la capa orgánica, se lavó con H2O (100 ml) y salmuera (100 ml), luego se secó sobre Na2SO4 anhidro y se concentró al vacío. El residuo bruto se purificó por cromatografía en columna (EtOAc al 20 % en hexanos) para proporcionar el compuesto del título (0,40 g, 43 %) como un sólido blanco. HPLC-MS (método 6): MH+ m/z 415.1, TR 2,34 min.
Intermedio 92
W-[1-Ciclooctil-2-oxo-2-({2-oxo-1-[2-(trimetilsilil)etoximetil]espiro[pirrolo[3,2-fc]piridin-3,4'-tetrahidropiran]-6-il}amino)etil]-2-metilpirazol-3-carboxamida
A una solución del intermedio 91 (0,30 g, 0,73 mmol) en tert-butanol (10 ml) se le añadieron el intermedio 85 (0,21 g, 0,73 mmol) y K2CO3 (0,20 g, 1,45 mmol). La mezcla de reacción se purgó con argón durante 10 min, y luego se purgó con Pd2(dba)3 (0,07 g, 0,07 mmol) y XPhos (0,03 g, 0,07 mmol). La mezcla de reacción se calentó a 100 °C durante 16 h, luego se diluyó con EtOAc (100 ml) y se filtró a través de una capa de Celite. El filtrado se lavó con agua (2 x 50 ml). Se separó la capa orgánica, se secó sobre Na2SO4 anhidro y se concentró al vacío. El residuo bruto se purificó por cromatografía en columna (EtOAc al 50-60 % en hexanos) para producir el compuesto del título (0,16 g, 29 %) como un sólido de color amarillo claro. 5h (400 MHz, DMSO-d6) -0,10 (s, 6H), 0,81-0,88 (m, 2H), 1,38-1,48 (m, 4H), 1,50-1,61 (m, 4H), 1,63-1,73 (m, 3H), 1,78-1,86 (m, 2H), 2,69 (s, 3H), 3,15-3,17 (m, 6H), 3,49 (t, J 7,78 Hz, 2H), 3,90 3,98 (m, 2H), 4,03 (s, 2H), 4,07-4,12 (m, 3H), 4,47 (t, J 8,66 Hz, 1H), 5,11 (s, 2H), 7,06 (d, J 2,01 Hz, 1H), 7,46 (d, J 2,01 Hz, 1H), 7,93 (d, J 1,76 Hz, 1H), 8,42 (d, J 1,76 Hz, 1H), 8,57 (d, J 8,28 Hz, 1H), 10,56 (s, 1H). HPLC-MS (método 6): MH+ m/z 625,3, TR 2,34 min.
Intermedio 93
2,4-Dicloro-7-{[2-(trimetilsilil)etoxi]metil}-7H-pirrolo[2,3-d]pirim idina
A una solución de 2,4-dicloropirrolo[2,3-d]pirimidina (10,0 g, 53,2 mmol) en DMF (50 ml) se le añadió NaH (1,91 g, 79.8 mmol) a 0 °C. La mezcla de reacción se agitó a 0 °C durante 30 min, luego se le añadió SEM-Cl (9,30 ml, 79,8 mmol) a 0 °C. La mezcla de reacción se agitó a temperatura ambiente durante 2 h, luego se inactivó con agua helada (200 g) y se extrajo con éter dietílico (3 x 150 ml). Se separó la capa orgánica, se lavó con agua (100 ml) y salmuera (100 ml), luego se secó sobre Na2SÜ4 anhidro y se concentró al vacío. El residuo bruto se purificó por cromatografía en columna (0-20 % de EtOAc en hexanos) para producir el compuesto del título (12,0 g, 71 %) como un aceite de color amarillo. HPLC-MS (método 6): MH+ m/z 317,9, TR 2,47 min.
Intermedio 94
2-[(2-Cloropirrolo[2,3-d]pirim idín-7-il)metoxi]etil(trimetil)silano
A una solución del intermedio 93 (1,50 g, 4,71 mmol) en EtOH (15 ml) se le añadió paladio al 10 % sobre carbón vegetal (0,15 g, 1,41 mmol), seguido de trietilamina (3,28 ml, 23,6 mmol). La mezcla de reacción se agitó a temperatura ambiente durante 1 h con presión de hidrógeno, y luego se filtró a través de una capa de Celite. El filtrado se concentró al vacío. El residuo bruto se purificó por cromatografía en columna (0-20 % de EtOAc en hexanos) para producir el compuesto del título (0,36 g, 27 %) como un aceite amarillo. 5h (400 MHz, DMSO-d6) -0,10 (s, 9H), 0,80-0,88 (m, 2H), 3,52 (t, J 8,07 Hz, 2H), 5,58 (s, 2H), 6,74 (d, J 3,42 Hz, 1H), 7,78 (d, J 3,91 Hz, 1H), 8,97 (s, 1H). HPLC-MS (método 6): MH+ m/z 283,9, TR 2,14 min.
Intermedio 95
5,5-Dibromo-2-cloro-7-{[2-(trimetilsilil]etoxi]metil}-5,7-dihidro-6H-pirrolo[2,3-d]pirim idín-6-ona
A una solución del intermedio 94 (1,00 g, 3,52 mmol) en tert-butanol (10 ml) y agua (10 ml) se le añadió NBS (1,88 g, 10,6 mmol) a temperatura ambiente. La mezcla de reacción se agitó a temperatura ambiente durante 3 h, luego se diluyó con agua (50 ml), se neutralizó con una solución acuosa saturada de NaHCO3 y se extrajo con EtOAc (3 x 50 ml). Se separó la capa orgánica, se lavó con agua (50 ml) y salmuera (50 ml), luego se secó sobre Na2SO4 anhidro y se concentró al vacío, para proporcionar el compuesto del título (2,00 g en bruto) como un sólido blanquecino, que se utilizó sin purificación adicional. 5h (400 MHz, DMSO-d6) -0,06 (s, 9H), 0,88 (t, J 7,83 Hz, 2H), 3,59-3,67 (m, 2H), 5,13 (s, 2H) 8,97 (s, 1H).
Intermedio 96
2-Cloro-7-{[2-(trimetilsilil]etoxi]metil}-5,7-dihidro-6H-pirrolo[2,3-d]pirim idín-6-ona
A una solución del intermedio 95 (2,00 g, 4,37 mmol) en THF (20 ml) y ácido acético (5 ml) se le añadió Zn (1,43 g, 21.9 mmol) a 0 °C. La mezcla de reacción se agitó a temperatura ambiente durante 2 h, y luego se filtró a través de una capa de Celite. El filtrado se diluyó con agua (30 ml) y se extrajo con EtOAc (3 x 50 ml). Se separó la capa orgánica, se lavó con agua (30 ml) y salmuera (30 ml), luego se secó sobre Na2SO4 anhidro y se concentró al vacío. El residuo bruto se purificó por cromatografía en columna (0-20 % de EtOAc en hexanos) para producir el compuesto del título (0,93 g, 71 %) como un aceite rojo. 5h (400 MHz, DMSO-d6) -0,04 (s, 9H), 0,85-0,92 (m, 2H), 3,55-3,64 (m, 2H), 3,77 (s, 2H), 5,03 (s, 2H), 8,35 (s, 1H).
Intermedio 97
2'-Cloro-7'-{[2-(trimetilsilil)etoxi]metil}-2,3,5,6-tetrahidrospiro[pirán-4,5'-pirrolo[2,3-d]pirim idín]-6'(7'H)-ona
A una solución del intermedio 96 (0,90 g, 3,00 mmol) en DMF (10 ml) se le añadió Cs2CO3 (2,93 g, 9,01 mmol), seguido de 1-bromo-2-(2-bromoetoxi)etano (1,04 g, 4,50 mmol). La mezcla de reacción se agitó a temperatura ambiente durante 4 h, luego se vertió en hielo y se extrajo con EtOAc (3 x 100 ml). Se separó la capa orgánica, se lavó con agua (100 ml) y salmuera (100 ml), luego se secó sobre Na2SO4 anhidro y se concentró al vacío. El residuo bruto se purificó por cromatografía en columna (0-20 % de EtOAc en hexanos) para producir el compuesto del título (0,43 g, 38 %) como un sólido de color naranja. 5h (400 MHz, DMSO-d6) -0,06 (s, 9H), 0,87 (t, J 8,07 Hz, 2H), 1,70-1,78 (m, 2H), 1,82-1,95 (m, 2H), 3,55-3,63 (m, 2H), 3,77-3,86 (m, 2H), 3,93-4,00 (m, 2H), 5,05 (s, 2H), 8,85 (s, 1H). HPLC-MS (método 6): MH+ m/z 370,0, TR 2,21 min.
Intermedio 98
W-[1-Ciclooctil-2-oxo-2-({6-oxo-7-[2-(trimetilsilil)etoximetil]espiro[pirrolo[2,3-d]-pirim idín-5,4'-tetrahidropiran]-2-il}amino)etil]-2-metilpirazol-3-carboxamida
A una solución del intermedio 97 (0,25 g, 0,68 mmol) y del intermedio 85 (0,20 g, 0,68 mmol) en 1,4-dioxano (5 ml) se le añadieron Cs2CO3 (0,44 g, 1,35 mmol), Pd2(dba)3 (0,06 g, 0,10 mmol) y Xantphos (0,04 g, 0,07 mmol). La mezcla de reacción se calentó a 80 °C durante 16 h, y luego se filtró a través de una capa de Celite. El filtrado se extrajo con EtOAc (3 x 20 ml). Se separó la capa orgánica, se lavó con agua (10 ml) y salmuera (10 ml), luego se secó sobre Na2SO4 anhidro y se concentró al vacío. El residuo bruto se purificó mediante cromatografía en columna (EtOAc al 0 30 % en hexanos) para proporcionar el compuesto del título (0,26 g, 31 %) como un sólido de color naranja. 5h (400 MHz, DMSO-ds) -0,10 (s, 9H), 0,85-0,88 (m, 2H). 1,43-1,47 (m, 8H), 1,56 (d, J 12,31 Hz, 2H), 1,60-1,74 (m, 6H), 1,85
1,94 (m, 2H), 2,15-2,23 (m, 1H), 3,61 (t, J 7,88 Hz, 2H), 3,83 (t, J 9,35 Hz, 2H), 3,95-3,97 (m, 2H), 4,03 (s, 3H), 4,69 4,74 (m, 1H), 5,07 (s, 2H), 7,03 (d, J 1,97 Hz, 1H), 7,47 (d, J 1,97 Hz, 1H), 8,41 (d, J 8,37 Hz, 1H), 8,77 (s, 1H), 10,81 (s, 1 H). HPLC-MS (método 6): MH+ m/z 626,4, TR 2,26 min.
Intermedio 99
6-Bromo-1-met¡lesp¡ro[¡ndol¡na-3,4'-tetrah¡drop¡ran]-2-ona
Una suspensión de 6-bromo-1,2-dihidrospiro[indol-3,4'-oxan]-2-ona (555 mg, 1,95 mmol) en DMF (8 ml) se trató con yoduro de metilo (135 pl, 2,2 mmol), luego se enfrió a 0 °C en una atmósfera de nitrógeno y se trató con hidruro de sodio (dispersión al 60 % en aceite mineral, 80 mg, 2,0 mmol). La mezcla resultante se puso a calentar a 20 °C durante 2 h, luego se diluyó con agua (50 ml) y se extrajo con EtOAc (2 x 25 ml). Los extractos orgánicos combinados se lavaron con agua (2 x 25 ml) y salmuera (30 ml), luego se secaron sobre MgSO4, se filtraron y se concentraron al vacío, para proporcionar el compuesto del título (550 mg, 94 %) como un sólido blanco. 5h (400 MHz, DMSO-d6) 7,50 (d, J 7,9 Hz, 1H), 7,28 (d, J 1,8 Hz, 1H), 7,22 (dd, J 7,9, 1,8 Hz, 1H), 4,04 (ddd, J 11,6, 6,9, 4,8 Hz, 2H), 3,85-3,75 (m, 2H), 3,13 (s, 3H), 1,72 (ddd, J 5,8, 4,5, 1,9 Hz, 4H).
Intermed¡o 100
W-(1-Met¡l-2-oxosp¡ro[¡ndol¡na-3,4'-tetrah¡drop¡ran]-6-¡l)carbamato de ferf-butMo
Una suspensión del intermedio 99 (550 mg, 1,9 mmol), carbonato de cesio (1,3 g, 4 mmol), carbamato de tert-butilo (250 mg, 2,1 mmol), XPhos (45 mg, 0,09 mmol) y acetato de paladio (25 mg, 0,11 mmol) en tolueno (10 ml) se calentó a 90 °C en atmósfera de nitrógeno durante 18 h. Se enfrió la mezcla de reacción, y luego se diluyó con EtOAc (50 ml) y agua (50 ml). Se separaron las capas y la capa acuosa se extrajo con EtOAc (50 ml, luego 20 ml). Las fases orgánicas combinadas se lavaron con salmuera (40 ml) y se secaron sobre MgSO4, luego se filtraron y se concentraron al vacío. El residuo se purificó mediante cromatografía en columna ultrarrápida, utilizando un gradiente de acetato de etilo en heptano (0-60 %), para proporcionar el compuesto del título (570 mg, 92 %) como un polvo blanco. 5h (400 MHz, DMSO-ds ) 9,42 (s, 1H), 7,43 (d, J 8,1 Hz, 1H), 7,29 (d, J 1,9 Hz, 1H), 6,99 (dd, J 8,1, 1,9 Hz, 1H), 4,04 (m, 2H), 3,81 (ddd, J 11,2, 6,7, 3,9 Hz, 2H), 3,09 (s, 3H), 1,77-1,58 (m, 4H), 1,49 (s, 9H). HPLC-MS (método 6): MH+ m/z 333, TR 1,93 min.
Intermed¡o 101
6-Am¡no-1-met¡lesp¡ro[¡ndol¡na-3,4'-tetrah¡drop¡ran]-2-ona
Una solución del intermedio 100 (740 mg, 2,22 mmol) en DCM (10 ml) se enfrió a 0 °C y luego se trató con TFA (5 ml). La mezcla de reacción se agitó en una atmósfera de nitrógeno durante 18 h, y luego se concentró al vacío. El residuo se aplicó a un cartucho de intercambio iónico SCX2 y se eluyó con metanol, seguido de NH3 en metanol (3 M), para dar el compuesto del título (350 mg, 68 %) como un sólido ceroso beige. 5h (400 MHz, DMSO-d6) 7,21-7,12 (m, 1H), 6,26-6,18 (m, 2H), 5,16 (s, 2H), 4,00 (ddd, J 11,2, 7,2, 3,7 Hz, 2H), 3,78 (ddd, J 11,2, 7,2, 3,7 Hz, 2H), 3,04 (s, 3H), 1,70 (ddd, J 13,4, 7,2, 3,7 Hz, 2H), 1,56 (ddd, J 13,5, 7,3, 3,7 Hz, 2H). HPLC-MS (método 21): MH+ m/z 233, TR 0,57 min.
Intermed¡o 102
W-{1-C¡clooct¡l-2-[(1-met¡l-2-oxosp¡ro[¡ndol¡na-3,4'-tetrah¡drop¡ran]-6-¡l)am¡no]-2-oxoet¡l}carbamato de te r t- but¡lo
Se preparó a partir del intermedio 101 y del intermedio 33 por un método análogo al utilizado para preparar el intermedio 34 para dar el compuesto del título (cuantitativo) como un sólido blanco. 5h (400 MHz, DMSO-d6) 10,09 (s, 1H), 7,49 (d, J 8,1 Hz, 1H), 7,41 (d, J 1,9 Hz, 1H), 7,22 (dd, J 8,1, 1,9 Hz, 1H), 6,90 (d, J 8,8 Hz, 1H), 4,04 (ddd, J 11,2, 7,3, 3,7 Hz, 2H), 3,96 (t, J 8,4 Hz, 1H), 3,82 (ddd, J 11,1, 6,7, 4,0 Hz, 2H), 3,10 (s, 3H), 1,95 (br s, 1H), 1,77-1,25 (m, 27H). HPLC-m S (método 6): MH+ m/z 500, TR 2,58 min.
Intermed¡o 103
2-Am¡no-2-c¡clooct¡l-W-(1-met¡l-2-oxosp¡ro[¡ndol¡na-3,4'-tetrah¡drop¡ran]-6-¡l)-acetam¡da; h¡drocloruro
El intermedio 102 (50 mg, 0,10 mmol) se disolvió en metanol (1 ml) y se le añadió HCl (4 M en 1,4-dioxano, 0,25 ml, 1,00 mmol). La mezcla de reacción se agitó a 20 °C durante 48 h y luego se concentró al vacío para proporcionar el compuesto del título (43 mg, cuantitativo) como un aceite rosa, que se utilizó sin más purificación. HPLC-MS (método 7): MH+ m/z 400, TR 1,21 min.
Intermed¡o 104
W-{1-(4-Met¡lc¡clohex¡l)-2-oxo-2-[(2-oxosp¡ro[¡ndol¡na-3,4'-tetrah¡drop¡ran]-6-¡l)am¡no]et¡l}carbamato de te r t- but¡lo
Se preparó a partir del in term edio 2 (2,4 g, 11,1 mmol) y ácido 2-(terf-butoxicarbonilamino)-2-(4-metilciclohexil)acético (3,6 g, 13,26 mmol) por un método análogo al utilizado para preparar el in term edio 49 para dar el com puesto de l título (4,3 g, 69 %) como un sólido blanco. 5h (400 MHz, DMSO-d6) 10,39 (s, 1H), 10,06-9,84 (m, 1H), 7,48-7,21 (m, 2H), 7,08 (td, J 8,8, 8,2, 1,9 Hz, 1H), 6,88 (m, 1H), 4,15-3,95 (m, 2H), 3,95-3,66 (m, 3H), 1,98-1,43 (m, 9H), 1,38 (s, 14H), 1,01 -0,65 (m, 3H). HPLC-MS (método 7): [M+2H-tBu]+ m/z 416, TR 2,40 min.
Intermedio 105
2- Am¡no-2-(4-met¡lc¡clohex¡l)-W-(2-oxosp¡ro[¡ndolina-3,4'-tetrah¡drop¡ran]-6-¡l)-acetam¡da
Se preparó a partir del in term edio 104 (2,11 g, 4,5 mmol) por un método análogo al utilizado para preparar el in term edio 50 para dar el com puesto de l título (1,53 g, 92 %) como un sólido blanco. HPLC-MS (método 7): MH+ m/z 372, TR 1,11 min.
Intermed¡o 106
W-[(1S)-1-(4-Met¡lc¡clohex¡l)-2-oxo-2-({2-oxo-1-[2-(tr¡met¡ls¡l¡l)etox¡met¡l]esp¡ro[p¡rrolo[3,2-c]p¡r¡d¡n-3,4'-tetrahidropiran]-6-il}am ino)etil]carbamato de tert-butMo (isómero tra n s )
Se preparó a partir del in term edio 57 (2,7 g, 7,74 mmol) y del in term edio 48 (2 g, 7,37 mmol) por un método análogo al utilizado para preparar el in term edio 49 para dar el com puesto de l título (4,2 g, 95 %) como un sólido blanco. 5h (300 MHz, DMSO-ds ) 10,41 (s, 1H), 8,51 (s, 1H), 7,93 (s, 1H), 6,96 (d, J 9,2 Hz, 1H), 5,08 (s, 2H), 3,85 (t, J 8,5 Hz, 2H), 3,50 (t, J 7,8 Hz, 2H), 2,00 (s, 1H), 1,90-1,43 (m, 9H), 1,38 (m, 9H), 1,18 (m, 4H), 0,85 (m, 8H), -0,09 (s, 9H). HPLC-MS (método 6): MH+ m/z 603, t R 1,55 min.
Intermed¡o 107
(2S)-2-Am¡no-2-(4-met¡lc¡clohex¡l)-W-{2-oxo-1-[2-(tr¡met¡ls¡l¡l)etox¡met¡l]-esp¡ro[p¡rrolo[3,2-c]p¡r¡d¡n-3,4'-tetrahidropiran]-6-il}acetamida; h¡drocloruro (Isómero tra n s )
A una solución del in term edio 106 (4,2 g, 7,0 mmol) disuelto en metanol (50 ml) se le añadió HCl (4 M en 1,4-dioxano, 17 ml, 68 mmol) a 20 °C. La mezcla de reacción se agitó durante 18 h y luego se concentró al vacío para proporcionar el com puesto de l título (3,74 g, 100 %) como un sólido de color rosa claro, que se utilizó sin purificación adicional. 5h (300 MHz, DMSO-ds ) 11,19 (s, 1H), 8,55 (s, 1H), 8,40 (s, 2H), 7,87 (s, 1H), 5,19-5,02 (m, 2H), 4,08-3,97 (m, 2H), 3,96 3,79 (m, 3H), 3,61-3,41 (m, 2H), 1,90-1,55 (m, 8H), 1,32-0,97 (m, 3H), 0,96-0,75 (m, 8H), -0,08 (s, 9H). HPLC-MS (método 6): Mh m/z 503, T r 1,32 min.
Intermed¡o 108
3- Et¡l-W-[(1 S)-1-(4-met¡lc¡clohex¡l)-2-oxo-2-({2-oxo-1-[2-(tr¡met¡ls¡l¡l)etox¡met¡l]esp¡ro[p¡rrolo[3,2-c]p¡r¡d¡n-3,4'-tetrahidropiran]-6-il}amino)etil]isoxazol-4-carboxamida (isómero tra n s )
Se preparó a partir del in term edio 107 (2,57 g, 4,77 mmol) y ácido 3-etilisoxazol-4-carboxílico (850 mg, 5,72 mmol) de acuerdo con el procedim iento A , y se purificó por cromatografía en columna ultrarrápida usando un gradiente de 0 100 % de acetato de etilo en hexanos, para dar el com puesto de l título (2,70 g, 91 %) como un sólido blanquecino. 5h (300 MHz, DMSO-de) 10,68 (s, 1H), 9,39 (s, 1H), 8,50 (d, J 0,7 Hz, 1H), 8,40 (d, J 7,9 Hz, 1H), 7,95 (s, 1H), 5,11-5,04 (m, 2H), 4,54 (m, 1H), 4,15-3,99 (m, 2H), 3,90-3,77 (m, 2H), 3,55-3,43 (m, 2H), 3,20-3,12 (m, 2H), 2,80-2,74 (m, 2H), 1,90-1,49 (m, 9H), 1,27-1,06 (m, 6H), 0,95-0,73 (m, 5H), -0,11 (s, 9H). HPLC-MS (método 7): MH+ m/z 626, TR 1,65 min.
Intermed¡o 109
2- Et¡l-W-[(1S)-1-(4-met¡lc¡clohex¡l)-2-oxo-2-({2-oxo-1-[2-(tr¡met¡ls¡l¡l)etox¡met¡l]esp¡ro[p¡rrolo[3,2-c]p¡r¡d¡n-3,4'-tetrahidropiran]-6-il}amino)etil]pirazol-3-carboxamida (Isómero tra n s )
Se preparó a partir del in term edio 107 (2,56 g, 4,75 mmol) y ácido 1 -etil-1 H-pirazol-5-carboxílico (840 mg, 5,69 mmol) de acuerdo con el procedim iento A , y se purificó por cromatografía en columna ultrarrápida usando un gradiente de 0 100 % de acetato de etilo en hexanos, para dar el com puesto de l título (2,73 g, 92 %) como un sólido blanquecino. 5h (300 MHz, DMSO-de) 10,66 (s, 1H), 8,54-8,40 (m, 2H), 7,94 (s, 1H), 7,49 (d, J 2,0 Hz, 1H), 7,00 (d, J 2,0 Hz, 1H), 5,08 (s, 2H), 4,50-4,38 (m, 2H), 4,06-3,94 (m, 1H), 3,90-3,79 (m, 2H), 3,49 (t, J 7,8 Hz, 2H), 1,90-1,51 (m, 10H), 1,35-1,24 (m, 5H), 0,92-0,78 (m, 9H), -0,11 (s, 9H). HPLC-MS (método 6): MH+ m/z 625, TR 1,37 min.
Intermed¡o 110
3- C¡cloprop¡l-W-[(1S)-1-(4-met¡lc¡clohex¡l)-2-oxo-2-({2-oxo-1-[2-(tr¡met¡ls¡l¡l)-etox¡met¡l]esp¡ro[p¡rrolo[3,2-c]piridin-3,4'-tetrahidropiran]-6-il}amino)etil]-isoxazol-4-carboxamida (isómero tra n s )
Se preparó a partir del in term edio 107 (100 mg, 0,19 mmol) y ácido 3-ciclopropilisoxazol-4-carboxílico (36 mg, 0,22 mmol) de acuerdo con el procedim iento A , y se purificó por cromatografía en columna ultrarrápida usando un gradiente
de 0-100 % de acetato de etilo en hexanos, para dar el compuesto del título (100 mg, 85 %) como un aceite claro, que se utilizó sin purificación adicional. HPLC-MS (método 7): MH+ m/z 638, TR 1,57 min.
Intermedio 111 (procedimiento E)
W-[(1S)-1-(4-Metilciclohexil)-2-oxo-2-({2-oxo-1-[2-(trimetilsilil)etoximetil]espiro[pirrolo[3,2-c]piridin-3,4'-tetrahidropiran]-6-il}am ino)etil]carbamato de ciclobutilo (isómero tra n s )
Una solución del intermedio 107 (100 mg, 0,19 mmol) en THF (5 ml) se trató con trietilamina (38 mg, 0,37 mmol) y piridina (22 mg, 0,28 mmol), seguido de la adición lenta de cloroformiato de ciclobutilo (25 mg, 0,19 mmol). La mezcla de reacción se agitó a 20 °C durante 18 h, luego se diluyó con agua y se extrajo con DCM. Las capas orgánicas combinadas se pasaron a través de un cartucho separador de fases de frita hidrófoba y se concentraron al vacío. El aceite amarillo bruto resultante se purificó mediante cromatografía en columna ultrarrápida, utilizando un gradiente de 0-100 % de acetato de etilo en hexanos, para proporcionar el compuesto del título (120 mg, sobrecuant.) como un aceite transparente, que se utilizó sin purificación adicional. HPLC-MS (método 7): MH+ m/z 601, TR 1,61 min. Intermedio 112
W-[(1S)-1-(4-Metilciclohexil)-2-oxo-2-({2-oxo-1-[2-(trimetilsilil)etoximetil]-espiro[pirrolo[3,2-c]piridin-3,4'-tetrahidropiran]-6-il}amino)etil]-2-(3,3,3-trifluoropropil)pirazol-3-carboxamida (isómero tra n s )
Se preparó a partir del intermedio 107 (40 mg, 0,074 mmol) y ácido 1-(3,3,3-trifluoropropil)-1H-pirazol-5-carboxílico (19,5 mg, 0,089 mmol) de acuerdo con el procedimiento A, y se purificó por cromatografía en columna ultrarrápida usando un gradiente de 0-100 % de acetato de etilo en hexanos, para dar el compuesto del título (50 mg, 97 %) como un aceite transparente. HPLC-MS (método 6): MH+ m/z 693, TR 1,44 min.
Intermedio 113
3-Ciclobutil-W-[(1S)-1-(4-metilciclohexil)-2-oxo-2-({2-oxo-1-[2-(trimetilsilil)-etoximetil]espiro[pirrolo[3,2-c]piridin-3,4'-tetrahidropiran]-6-il}amino)etil]-isoxazol-4-carboxamida (isómero tra n s )
Se preparó a partir del intermedio 107 (40 mg, 0,074 mmol) y ácido 3-ciclobutilisoxazol-4-carboxílico (15 mg, 0,089 mmol) de acuerdo con el procedimiento A, y se purificó por cromatografía en columna ultrarrápida usando un gradiente de 0-100 % de acetato de etilo en hexanos, para dar el compuesto del título (45 mg, 93 %) como un aceite transparente. HPLC-MS (método 7): MH+ m/z 652, TR 1,59 min.
Intermedio 114
2-[(6-Cloropirrolo[2,3-b]piridin-1-il)metoxi]etil(trimetil)silano
A una solución de 6-cloro-1 H-pirrolo[2,3-¿>]piridina (10,0 g, 65,5 mmol) en DMF (100 ml) se le añadió NaH (1,89 g, 78,6 mmol) a 0 °C. Se le añadió gota a gota SEM-Cl (13,9 ml, 78,6 mmol) a 0 °C. La mezcla de reacción se agitó a temperatura ambiente durante 3 h, luego se vertió en hielo y se extrajo con EtOAc (3 x 100 ml). Se separaron las capas orgánicas combinadas, se lavaron con agua (100 ml) y salmuera (50 ml), luego se secaron sobre Na2SO4 anhidro y se concentraron al vacío. El residuo bruto se purificó por cromatografía en columna (EtOAc al 0-5 % en hexanos) para proporcionar el compuesto del título (16,0 g, 86 %) como un aceite de color amarillo pálido. 5h (400 MHz, DMSO-ds) -0,12 (s, 9H), 0,79-0,85 (m, 2H), 3,47-3,52 (m, 2H), 5,57 (s, 2H), 6,58 (d, J 3,42 Hz, 1H), 7,18 (d, J 7,83 Hz, 1H), 7,66 (d, J 3,42 Hz, 1H), 8,04 (d, J 8,31 Hz, 1H). HPLC-MS (método 6): MH+ m/z 683,9, TR 2,44 min. Intermedio 115
3,3-Dibromo-6-cloro-1-[2-(trimetilsilil)etoximetil]pirrolo[2,3-b]piridin-2-ona
A una solución de tribromuro de piridinio (3,39 g, 10,6 mmol) en 1,4-dioxano (10 ml) se le añadió la solución del intermedio 114 (1,00 g, 3,54 mmol) en 1,4-dioxano (5 ml) a temperatura ambiente. La mezcla de reacción se agitó durante 15 min, luego se diluyó con agua (500 ml) y se extrajo con EtOAc (3 x 250 ml). Se separaron las capas orgánicas combinadas, se lavaron con agua (250 ml) y salmuera (250 ml), luego se secaron sobre Na2SO4 anhidro y se concentraron al vacío. El residuo bruto se purificó por cromatografía en columna (EtOAc al 0-5 % en hexanos) para proporcionar el compuesto del título (1,2 g, 74 %) como un aceite rojo. 5h (400 m Hz , DMSO-d6) -0,08 (s, 9H), 0,83 0,91 (m, 2H), 3,62 (t, J 8,07 Hz, 2H), 5,14 (s, 2H), 7,40 (d, J 7,83 Hz, 1H), 8,20 (d, J 8,31 Hz, 1H).
Intermedio 116
6-Cloro-1-[2-(trimetilsilil)etoximetil]-3H-pirrolo[2,3-b]piridin-2-ona
A una solución del intermedio 115 (15,0 g, 32,8 mmol) en THF (150 ml) y agua (50 ml) se le añadió Zn (10,7 g, 164 mmol), seguido de la adición de NH4Cl (8,79 g, 164 mmol) a 0 °C. La mezcla de reacción se agitó a temperatura ambiente durante 2 h y luego se filtró a través de una capa de Celite. El filtrado se diluyó con agua (100 ml) y se extrajo con EtOAc (3 x 100 ml). Se separaron las capas orgánicas combinadas, se lavaron con agua (100 ml), salmuera (100
ml), luego se secaron sobre Na2SÜ4 anhidro y se concentraron al vacío. El residuo bruto se purificó por cromatografía en columna (0-20 % de EtOAc en hexanos) para producir el compuesto del título (6,00 g, 61 %) como un aceite incoloro. 5h (400 MHz, DMSO-ds ) -0,05 (s, 9H), 0,83-0,91 (m, 2H), 3,57-3,81 (m, 2H), 3,70 (s, 2H), 5,04 (s, 2H), 7,15 (d, J 7,34 Hz, 1H), 7,69 (d, J 7,82 Hz, 1H). HPLC-MS (método 6): MH+ m/z 296,9, TR 2,23 min.
Intermedio 117
6-Cloro-1-[2-(tr¡met¡ls¡l¡l)etox¡met¡l]esp¡ro[p¡rrolo[2,3-fc]p¡r¡d¡n-3,4'-tetrah¡drop¡ran]-2-ona
A una solución del intermedio 116 (6,00 g, 20,1 mmol) en DMF (120 ml) se le añadió Cs2CO3 (26,2 g, 80,3 mmol), seguido de la adición de 1-bromo-2-(2-bromoetoxi)-etano (6,17 ml, 40,2 mmol). La mezcla de reacción se agitó a temperatura ambiente durante 16 h, luego se diluyó con agua (50 ml) y se extrajo con EtOAc (3 x 100 ml). Se separaron las capas orgánicas combinadas, se lavaron con agua (100 ml) y salmuera (100 ml), luego se secaron sobre Na2SO4 anhidro y se concentraron al vacío. El residuo bruto se purificó por cromatografía en columna (0-20 % de EtOAc en hexanos) para producir el compuesto del título (4,00 g, 45 %) como un sólido rojo. 5h (400 MHz, DMSO-d6) -0,02 (s, 9H), 0,83-0,92 (m, 2H), 1,75-1,85 (m, 4H) 3,58 (t, J 7,91 Hz, 2H) 3,75-3,87 (m, 2H), 3,96-4,06 (m, 2H), 5,06 (s, 2H), 7,19 (d, J 7,53 Hz, 1H), 8,09 (d, J 7,78 Hz, 1H). HPLC-MS (método 6): [(M-100)+H]+ m/z 269,0, TR 3,28 min.
Intermed¡o 118
W-[1-C¡clooct¡l-2-oxo-2-({2-oxo-1-[2-(tr¡met¡ls¡l¡l)etox¡met¡l]esp¡ro[p¡rrolo[2,3-b]-p¡r¡d¡n-3,4'-tetrah¡drop¡ran]-6-il}amino)etil]-2-metilpirazol-3-carboxamida
A una solución del intermedio 117 (0,10 g, 0,27 mmol) en terf-butanol (5 ml) se le añadieron el intermedio 85 (0,08 g, 0,27 mmol) y K2CO3 (0,075 g, 0,54 mmol). La mezcla de reacción se purgó con argón durante 10 min, luego se purgó con Pd2(dba)3 (0,02 g, 0,03 mmol) y XPhos (0,01 g, 0,03 mmol). La mezcla de reacción se calentó a 100 °C durante 16 h, luego se diluyó con EtOAc (100 ml) y se filtró a través de una capa de Celite. La capa orgánica se lavó con agua (2 x 50 ml). Se separó la capa orgánica, se secó sobre Na2SO4 anhidro y se concentró al vacío. El residuo bruto se purificó por cromatografía en columna (40-50 % de acetato de etilo en hexano) para proporcionar el compuesto del título (0,11 g, 50 %) como un sólido amarillo claro. 5h (400 MHz, DMSO-d6) -0,10 (s, 9H), 0,83-0,89 (m, 2H), 1,21-1,26 (m, 2H), 1,37-1,56 (m, 9H), 1,62-1,72 (m, 5H), 1,77-1,86 (m, 2H), 2,15-2,18 (m, 1H), 3,57 (t, J 8,07 Hz, 2H), 3,79-3,88 (m, 2H), 3,96-4,00 (m, 2H), 4,02 (s, 3H), 4,61 -4,70 (m, 1H), 5,10 (s, 2H), 7,02 (d, J 1,96 Hz, 1H), 7,46 (d, J 2,45 Hz, 1H), 7,82 (d, J 8,31 Hz, 1H), 8,03 (d, J 7,82 Hz, 1H), 8,46 (d, J 8,80 Hz, 1H), 10,68 (s, 1H). HPLC-MS (método 6): MH+ m/z 625,8, TR 2,44 min.
Intermed¡o 119
6-Bromo-1-{[2-(tr¡met¡ls¡l¡l)etox¡]met¡l}-1,2-d¡h¡droesp¡ro[¡ndol-3,4'-oxan]-2-ona
A una solución agitada de 6-bromo-1,2-dihidroespiro[indol-3,4'-oxan]-2-ona (10 g, 35,4 mmol) en una mezcla de THF (150 ml) y DMF (150 ml), previamente enfriada a -5 °C, se le añadió en porciones hidruro de sodio (60 %, 1,7 g, 42,5 mmol). Se continuó agitando a 0 °C durante 30 min más, luego se le añadió gota a gota SEM-Cl (3,9 ml, 22,4 mmol). La mezcla de reacción se dejó calentar a temperatura ambiente y se agitó durante una noche, luego se vertió en agua helada (600 ml) y se extrajo con acetato de etilo (3 x 300 ml). Se combinaron los extractos orgánicos, se lavaron con solución acuosa saturada de bicarbonato de sodio (300 ml) y salmuera (2 x 200 ml), luego se secaron (Na2SO4), se filtraron y se concentraron al vacío. El residuo se purificó mediante cromatografía en columna ultrarrápida, usando EtOAc al 0-100 % en heptano como eluyente, para producir el compuesto del título (11,84 g, 81 %) como un sólido blanco. 5h (500 MHz, DMSO-ds ) 7,62 (d, J 8,0 Hz, 1H), 7,39 (d, J 1,8 Hz, 1H), 7,35 (dd, J 8,0, 1,8 Hz, 1H), 5,20 (s, 2H), 4,12 (ddd, J 11,7, 8,3, 3,5 Hz, 2H), 3,99-3,81 (m, 2H), 3,61-3,54 (m, 2H), 1,87 (ddd, J 12,5, 8,3, 4,0 Hz, 2H), 1,83-1,75 (m, 2H), 0,92 (t, J 7,9 Hz, 2H), 0,00 (s, 9H). HPLC-MS (método 5): MH+ m/z 294, TR 2,17 min.
Intermed¡o 120
Ác¡do 2-oxo-[2-(trimetilsilil)etoximetil]espiro[indolina-3,4'-tetrahidropiran]-6-carboxílico
A una solución agitada del intermedio 119 (1,00 g, 2,425 mmol) en THF anhidro (20 ml), previamente enfriada a -78 °C en nitrógeno, se le añadió gota a gota n-butillitio a 2,5 M (0,98 ml, 2,45 mmol). La temperatura se mantuvo a -78 °C durante 30 min, y luego se le añadió en porciones dióxido de carbono (~4,0 g, como gránulos de hielo seco). Se continuó agitando a -78 °C durante 20 min más. La mezcla de reacción se inactivó con solución acuosa saturada de cloruro de amonio (2 ml) y agua (1 ml), luego se dejó calentar a temperatura ambiente y se diluyó con agua (20 ml) y salmuera (20 ml). La mezcla de reacción (pH 8) se extrajo con acetato de etilo (30 ml) y se descartó la fase acuosa. La fase orgánica se extrajo con solución acuosa de hidróxido de sodio a 1 M (2 x 20 ml). Los extractos acuosos básicos se combinaron (pH 12) y el pH se ajustó a pH 4 con ácido clorhídrico a 12 M, y luego a pH 1-2 con ácido clorhídrico a 1 M. La fase acuosa ácida se extrajo con DCM (3 x 20 ml). Los extractos orgánicos se combinaron, se secaron sobre sulfato de sodio anhidro y se filtraron. Se retiró el solvente al vacío para proporcionar el compuesto del título (230 mg, 24 %) como un sólido de color naranja pálido. 5h (250 MHz, DMSO-d6) 7,71 (s, 2H), 7,59 (s, 1H), 5,16 (s, 2H), 4,06 (ddd, J 11,6, 8,1, 3,7 Hz, 2H), 3,89-3,79 (m, 2H), 3,50 (t, J 7,7 Hz, 2H), 1,87-1,67 (m, 4H), 0,84 (t, J 8,1 Hz, 2H), -0,09 (s, 9H). HPLC-MS (método 5): [M-H]- m /z376, Tr 1,90 min.
Intermedio 121
W-{1-C¡clooct¡l-2-oxo-2-[(2-oxosp¡ro[¡ndol¡na-3,4'-tetrah¡drop¡ran]-6-¡l)am¡no]et¡l}-2-oxo-1-[2-(trimetilsilil)etoximetil]espiro[indolina-3,4'-tetrahidropiran]-6-carboxamida
Se preparó a partir del intermedio 35 (50 mg, 0,13 mmol) y del intermedio 120 (54 mg, 0,14 mmol) de acuerdo con el procedimiento A, y se purificó por cromatografía en columna ultrarrápida usando un gradiente de metanol en diclorometano (1-10 %), para dar el compuesto del título (73 mg, 75 %) como un vidrio transparente. 5h (250 MHz, DMSO-d6) 10,38 (s, 1H), 10,22 (s, 1H), 8,52 (d, J 8,6 Hz, 1H), 7,68 (s, 2H), 7,64 (s, 1H), 7,43 (d, J 8,2 Hz, 1H), 7,38 (d, J 1,8 Hz, 1H), 7,13 (dd, J 8,2, 1,8 Hz, 1H), 5,16 (s, 2H), 4,51 (t, J 9,0 Hz, 1H), 4,11-3,96 (m, 4H), 3,85 (d, J 12,0 Hz, 4H), 3,51 (t, J 7,9 Hz, 2H), 2,27-2,12 (m, 1H), 1,82-1,36 (m, 22H), 0,84 (dd, J 8,5, 7,3 Hz, 2H), -0,11 (s, 9H). HPLC-MS (método 5): [M-H]- m/z 743, TR 2,16 min.
Intermedio 122
2- Metil-W-{1-(4-metilciclohexiliden)-2-oxo-2-[(2-oxospirorindolina-3,4'-tetrahidropiran]-6-il)amino]etilo}pirazol-3- carboxamida
Se preparó a partir del intermedio 2 (70 mg, 0,3 mmol) y del intermedio 75 (78 mg, 0,3 mmol) de acuerdo con el procedimiento G, y se purificó por cromatografía preparativa (método 8), para dar el compuesto del título (21,5 mg, 15%) como un sólido blanco. uPLC-MS (método 28): [M-H]- m/z 478, TR 1,28 min.
Intermedio 123
6-Amino-5-fluorospiro[indolina-3,4'-tetrahidropiran]-2-ona
Una suspensión del intermedio 2 (400 mg, 1,83 mmol) en THF (10 ml) se trató con 1-clorometil-4-fluoro-1,4-diazoniabiciclo[2.2.2]octanobis(tetrafluoroborato) (700 mg, 1,98 mmol). La mezcla de reacción se agitó a 20 °C en una atmósfera de nitrógeno durante 24 h, luego se diluyó con una solución acuosa saturada de bicarbonato de sodio (40 ml). Se le añadió más NaHCO3 sólido hasta alcanzar un pH de 8,5. La mezcla se extrajo con EtOAc (2 x 70 ml). Los extractos orgánicos combinados se lavaron con salmuera (50 ml) y se secaron sobre MgSO4 , luego se filtraron y se concentraron al vacío. El residuo bruto se purificó por HPLC de fase inversa (método 8) para proporcionar, después de la liofilización, el compuesto del título (22 mg, 5 %) como un sólido blanco. 5h (250 MHz, DMSO-d6) 10,13 (s, 1H), 7,19 (d, J 11,2 Hz, 1H), 6,32 (d, J 7,8 Hz, 1H), 5,12 (s, 2H), 3,98 (ddd, J 11,3, 7,3, 3,7 Hz, 2H), 3,76 (ddd, J 11,2, 7,1, 3,8 Hz, 2H), 1,69 (ddd, J 13,5, 7,1, 3,7 Hz, 2H), 1,58 (ddd, J 13,5, 7,3, 3,8 Hz, 2H). HPLC-MS (método 6): MH+ m/z 237, TR 0,81 min.
Intermedio 124
-Nitro-1,3,4,9-tetrahidropirido[3,4-b]indol-2-carboxilato de ferf-butilo
El dicarbonato de di-terf-butilo (1,11 g, 5,09 mmol) se añadió a una suspensión agitada de 7-nitro-2,3,4,9-tetrahidro-1 H-beta-carbolina (1 g, 4,6 mmol) en terf-butanol (46 ml) a 20 °C. La mezcla se agitó a 20 °C durante 16 h. Lo volátil se eliminó al vacío, luego el residuo se suspendió en tert-butilmetiléter (20 ml) y se sonicó. Los sólidos se recogieron por filtración y se lavaron con terf-butilmetiléter (3 x 10 ml). Se concentró el filtrado, luego se repitió el procedimiento anterior para proporcionar una segunda cosecha del sólido. Los sólidos combinados se secaron al vacío para proporcionar el compuesto del título (1,37 g, 93 %) como un polvo de color amarillo canario. 5h (500 MHz, DMSO-d6) 11,69 (br s, 1H), 8,27 (d, J 2,0 Hz, 1H), 7,88 (dd, J 8,8, 2,1 Hz, 1H), 7,57 (d, J 8,7 Hz, 1H), 4,65 (s, 2H), 3,68 (t, J 5,7 Hz, 2H), 2,74 (t, J 5,5 Hz, 2H), 1,44 (s, 9H). HPLC-MS (método 7): MH+ m/z 318, TR 2,01 min.
Intermedio 125
-Nitro-2-oxospiro[indolina-3,3'-pirrolidín]-1'-carboxilato de ferf-butilo
La 1-bromopirrolidín-2,5-diona (400 mg, 2,25 mmol) se añadió en porciones durante 10 min a una suspensión agitada del intermedio 124 (680 mg, 2,12 mmol) en THF-ácido acético-agua 1:1:1 (36 ml) a 0 °C en nitrógeno. La mezcla se dejó calentar hasta aproximadamente 20 °C durante 2,5 h, y luego se inactivó con una solución acuosa saturada de carbonato de sodio (pH 10). El material resultante se extrajo con acetato de etilo (3 x 50 ml). Los extractos orgánicos combinados se lavaron con una solución acuosa saturada de carbonato de sodio (30 ml) y salmuera (30 ml), luego se secaron sobre sulfato de magnesio, se filtraron y se concentraron al vacío. La goma de color naranja resultante se separó mediante cromatografía en columna ultrarrápida, utilizando un gradiente de acetato de etilo en heptano (0-80 %), para proporcionar el compuesto del título (679 mg, 93 %) como un polvo de color amarillo pálido. 5h (250 MHz, DMSO-ds , 353K) 10,65 (br s, 1H), 7,88 (dd, J 8,2, 2,2 Hz, 1H), 7,60 (d, J 2,1 Hz, 1H), 7,48 (d, J 8,2 Hz, 1H), 3,78-3,59 (m, 2H), 3,59 (d, J 11,3 Hz, 1H), 3,53 (d, J 11,1 Hz, 1H), 2,36-2,08 (m, 2H), 1,45 (s, 9H). HPLC-MS (método 7): MNa+ m/z 356, TR 1,83 min.
Intermedio 126
6-Amino-2-oxospiro[indolina-3,3'-pirrolidín]-1 '-carboxilato de ferf-butilo
Una suspensión del intermedio 125 (679 mg, 2,04 mmol) y paladio al 10 % sobre carbono (humedad acuosa al 50 %, 200 mg, 0,09 mmol) en etanol (16 ml) se agitó en una atmósfera de hidrógeno durante 4 h. Los sólidos se eliminaron por filtración a través de un lecho de tierra de diatomeas, se lavaron con etanol (4 x 20 ml) y el filtrado se concentró al vacío. El polvo rosa rojizo resultante se separó mediante cromatografía en columna ultrarrápida, usando un gradiente de acetato de etilo en heptano (20-100 %), para producir el compuesto del título (633 mg, 97 %) como un polvo de color canela claro. 5h (250 MHz, DMSO-d6, 353K) 9,93 (br s, 1H), 6,87-6,76 (m, 1H), 6,23-6,14 (m, 2H), 4,88 (br s, 2H), 3,71-3,49 (m, 2H), 3,45 (d, J 10,8 Hz, 1H), 3,33 (d, J 10,8 Hz, 1H), 2,12 (ddd, J 12,5, 7,9, 6,5 Hz, 1H), 2,03-1,88 (m, 1H), 1,43 (s, 9H). HPLC-MS (método 7): MlNa+ m/z 326, TR 1,64 min.
Intermedio 127
6-({2-Ciclooctil-2-[(3-metilisoxazol-4-carbonil)amino]acetil}amino)-2-oxospiro[indolina-3,3'-pirrolidín]-1'-carboxilato de ferf-butilo
Se añadió HATU (143 mg, 0,38 mmol) a una mezcla del intermedio 126 (100 mg, 0,31 mmol) y del intermedio 41 (104 mg, 0,35 mmol) en DMF anhidra (1,6 ml). La mezcla se agitó a 20 °C en nitrógeno durante 16 h, luego se inactivó con una solución acuosa saturada de carbonato de sodio (20 ml). El material resultante se extrajo con acetato de etilo (3 x 30 ml). Los extractos orgánicos combinados se lavaron con agua (20 ml) y salmuera (2 x 20 ml), luego se secaron sobre sulfato de magnesio, se filtraron y se concentraron al vacío. El aceite anaranjado viscoso resultante se separó mediante cromatografía en columna ultrarrápida, utilizando un gradiente de acetato de etilo en heptano (0-100 %), para proporcionar, después de la liofilización, el compuesto del título (89,8 mg, 47 %) como un polvo blanquecino. 5h (250 MHz, DMSO-ds, 353K) 10,23 (br s, 1H), 9,91 (br s, 1H), 9,34 (s, 1H), 8,07 (d, J 8,7 Hz, 1H), 7,35 (s, 1H), 7,17 7,07 (m, 2H), 4,50 (t, J 8,3 Hz, 1H), 3,73-3,54 (m, 2H), 3,50 (d, J 10,9 Hz, 1H), 3,41 (d, J 10,8 Hz, 1H), 2,39 (s, 3H), 2,20-1,96 (m, 3H), 1,79-1,35 (m, 23H). uPLC-MS (método 1): MH+ m/z 580, TR 3,74 min.
Intermedio 128
Ácido 2-ciclooctil-2-[(2-metilpirazol-3-carbonil)amino]acético
Se añadió la solución acuosa de hidróxido de litio (2 M, 2,6 ml, 5,2 mmol) a una solución agitada del intermedio 12 (1,45 g, 4,29 mmol) en THF/MeOH 1:1 (17,2 ml). La mezcla se agitó a 20 °C al aire durante 16 h. Lo volátil se eliminó al vacío y el residuo se diluyó con agua (20 ml), y luego se lavó con terf-butilmetiléter (2 x 20 ml). Los lavados orgánicos combinados se extrajeron con una solución acuosa de hidróxido de sodio (20 ml). Las fases acuosas alcalinas combinadas se trataron con ácido clorhídrico acuoso a 3 M (pH 1) y se extrajeron con acetato de etilo (3 x 30 ml). Los extractos orgánicos combinados se lavaron con salmuera (20 ml) y se secaron sobre sulfato de magnesio, luego se filtraron y se concentraron al vacío, para proporcionar el compuesto del título (1,48 g, 91 %) como un polvo blanco. 5h (500 MHz, DMSO-ds) 12,69 (br s, 1H), 8,42 (d, J 8,6 Hz, 1H), 7,45 (d, J 2,0 Hz, 1H), 7,01 (d, J 2,1 Hz, 1H), 4,33 (dd, J 8,5, 7,3 Hz, 1H), 4,02 (s, 3H), 2,19-2,06 (m, 1H), 1,75-1,27 (m, 14H). HPLC-MS (método 5): MH+ m/z 294, TR 1,71 min.
Intermedio 129
6-(ferf-Butoxicarbonilamino)-2-oxoindolina-1-carboxilato de ferf-butilo
El dicarbonato de di-terf-butilo (33,5 g, 153 mmol) se añadió en porciones a una suspensión agitada de 6-amino-1,3-dihidro-2H-indol-2-ona (9,1 g, 61,4 mmol) e hidrogenocarbonato de sodio (18,1 g, 215 mmol) en THF (120 ml) a 20 °C en nitrógeno. La mezcla se calentó a 70 °C durante 3,5 h. Se le añadió una porción adicional de dicarbonato de ditert-butilo (6,8 g, 31,2 mmol) y se continuó calentando a 70 °C durante 1,5 h. Se le añadió una porción adicional de dicarbonato de di-tertbutilo (6,8 g, 31,2 mmol) y se continuó calentando a 70 °C durante 5 h. Después de enfriar a t.a., la mezcla se diluyó con acetato de etilo (100 ml) y los sólidos se eliminaron por filtración a través de una capa de tierra de diatomeas, lavando con acetato de etilo (2 x 100 ml). El filtrado se concentró al vacío. La goma de color rojo oscuro resultante se separó mediante cromatografía en columna ultrarrápida, usando un gradiente de acetato de etilo en heptano (0-35 %), luego se trituró con acetato de etilo/heptano 4:1, para proporcionar el compuesto del título (16,23 g, 76 %) como un polvo tostado. 5h (500 MHz, DMSO-d6) 9,42 (s, 1H), 8,03 (s, 1H), 7,17-7,08 (m, 2H), 3,64 (s, 2H), 1,57 (s, 9H), 1,47 (s, 9H). HPLC-MS (método 5): MNa+ m/z 371, TR 1,94 min.
Intermedio 130
(2S)-2-(2-Etoxi-2-oxoetoxi)propanoato de etilo
Se añadió (2S)-2-hidroxipropanoato de etilo (4,8 ml, 42,0 mmol) a una suspensión agitada de carbonato de potasio (5,81 g, 42,0 mmol) en DMF anhidra (42 ml). La suspensión se agitó a 20 °C en nitrógeno durante 20 min y luego se le añadió bromoacetato de etilo (4,7 ml, 42,4 mmol). La mezcla de reacción se agitó a 20 °C en nitrógeno durante 64 h, y luego se diluyó con terf-butilmetiléter (50 ml). Los sólidos se eliminaron por filtración y se lavaron con tertbutilmetiléter (2 x 50 ml). El filtrado se concentró al vacío. El aceite de naranja resultante se separó por destilación kugelrohr (160-210 °C, 18 mbar) para producir el compuesto del título (4,57 g, 27 %) como un aceite incoloro. 5h (500 MHz, CDCla) 4,27 (d, J 16,5 Hz, 1H), 4,25-4,17 (m, 4H), 4,14 (q, J 6,9 Hz, 1H), 4,07 (d, J 16,5 Hz, 1H), 1,48 (d, J 6,9 Hz, 3H), 1,31-1,25 (m, 6H).
Intermedio 131
(2S)-2-(2-Hidroxietoxi)propán-1-ol
Se añadió gota a gota una solución de hidruro de litio y aluminio en éter dietílico (4 M, 7,3 ml, 29,2 mmol) a una solución agitada del in term edio 130 (4,57 g, 22,4 mmol) en THF anhidro (45 ml) a 0 °C en nitrógeno. La mezcla se agitó a 0 °C durante 1 h, luego a 20 °C durante 16 h. La mezcla de reacción se inactivó cuidadosamente mediante la adición gota a gota de agua (0,55 ml), seguida de una solución acuosa de hidróxido de sodio a 2 M (1,32 ml) y luego agua (1,64 ml). La suspensión se agitó a temperatura ambiente durante 30 min, luego se diluyó con acetona (50 ml) y se filtró a través de una capa de tierra de diatomeas. Los residuos se lavaron con acetona (2 x 50 ml) y el filtrado se concentró al vacío. El aceite tostado se separó por destilación kuge lrohr (90-120 °C, <1 mbar) para producir el com puesto de l título (818 mg, 22 %) como un aceite incoloro fluido. 5h (500 MHz, CDCh) 3,77-3,69 (m, 3H), 3,64-3,57 (m, 2H), 3,55 3,45 (m, 2H), 3,25 (br s, 2H), 1,12 (d, J 6,2 Hz, 3H).
Intermedio 132
(2S)-1-Yodo-2-(2-yodoetoxi)propano
Se añadió yodo (8,64 g, 34,0 mmol) en porciones a una solución agitada de trifenilfosfina (7,14 g, 27,2 mmol) y 1H-imidazol (1,85 g, 27,2 mmol) en éter dietílico/acetonitrilo 3:1 (54 ml) a 20 °C en nitrógeno. Después de agitar a 20 °C durante 30 min, se le añadió lentamente una solución del in term edio 131 (0,82 g, 6,81 mmol) en éter dietílico (3 ml), seguido de un enjuague con éter dietílico (2 ml). La mezcla de reacción se agitó a 20 °C en la oscuridad durante 22 h. La suspensión se diluyó con terf-butilmetiléter (50 ml) y se filtró a través de un lecho de tierra de diatomeas, lavando bien con tert-butilmetiléter (4 x 30 ml). El filtrado se filtró a través de una segunda almohadilla de tierra de diatomeas, lavando bien con tert-butilmetiléter (2 x 30 ml), y luego se concentró al vacío. El aceite viscoso de color rojo oscuro resultante se separó mediante cromatografía en columna ultrarrápida, usando un gradiente de acetato de etilo en heptano (0-5 %), para proporcionar el com puesto d e l título (1,44 g, 59 %) como un aceite tostado fluido. 5h (500 MHz, CDCla) 3,81-3,69 (m, 2H), 3,54-3,44 (m, 1H), 3,30-3,22 (m, 3H), 3,20 (dd, J 10,3, 5,7 Hz, 1H), 1,29 (d, J 6,1 Hz, 3H).
Intermedio 133
(2'S,3S)-6-(tert-Butoxicarbonilamino)-2'-metil-2-oxospiro[indolina-3,4'-tetrahidropiran]-1-carboxilato de tertbutilo
Una solución agitada del in term edio 129 (600 mg, 1,72 mmol) y del in term edio 132 (640 mg, 1,26 mmol) en DMF anhidro (11,5 ml) se purgó con una corriente de nitrógeno mientras se sonicaba durante 5 min. La solución se enfrió a -10 °C en nitrógeno, luego se le añadió carbonato de cesio (2,24 g, 6,89 mmol) en porciones durante 10 min. La suspensión se dejó calentar lentamente a 20 °C y se agitó durante un total de 16 h. La mezcla se inactivó mediante la adición lenta de ácido acético (0,31 ml) y se agitó a 20 °C durante 5 min. La suspensión se diluyó con agua (50 ml) y se agitó a 20 °C durante 5 min, y luego los sólidos se recogieron por filtración. Los sólidos se lavaron con agua (2 x 25 ml), y luego se disolvieron en acetato de etilo (30 ml). Se recogió el filtrado orgánico y los residuos se lavaron con acetato de etilo (2 x 20 ml). El filtrado orgánico combinado se filtró a través de papel de filtro hidrófobo y se concentró al vacío. La goma resultante de color naranja se separó mediante cromatografía en columna ultrarrápida secuencial, usando un gradiente de acetato de etilo en heptano (0-30 %), y luego HPLC preparativa (método 31), para obtener el com puesto de l título (86,5 mg, 16 %) como un polvo blanco. 5h (500 MHz, DMSO-d6) 9,49 (s, 1H), 8,14 (d, J 1,6 Hz, 1H), 7,74 (d, J 8,4 Hz, 1H), 7,16 (dd, J 8,3, 1,8 Hz, 1H), 4,04-3,83 (m, 3H), 1,90 (td, J 13,1,5,8 Hz, 1H), 1,62-1,54 (m obs., 1H), 1,58 (s, 9H), 1,50-1,44 (m obs., 1H), 1,47 (s, 9H), 1,40 (d, J 12,7 Hz, 1H), 1,10 (d, J 6,0 Hz, 3H). uPLC-MS (método 1): [M+2H-BOC]+ m/z 333, TR 4,01 min. s Fc quiral (método 13, Lux C4 25 cm, MeOH al 20 %-dióxido de carbono al 80 %, 4 ml/min): TR 1,61 min (98 %).
Intermedio 134
(2'S,3fí)-6-(tert-Butoxicarbonilamino)-2'-metil-2-oxospiro[indolina-3,4'-tetrahidropiran]-1-carboxilato de tertbutilo
Una solución agitada del in term edio 129 (600 mg, 1,72 mmol) y del in term edio 132 (640 mg, 1,26 mmol) en DMF anhidro (11,5 ml) se purgó con una corriente de nitrógeno mientras se sonicaba durante 5 min. La solución se enfrió a -10 °C en nitrógeno, luego se le añadió carbonato de cesio (2,24 g, 6,89 mmol) en porciones durante 10 min. La suspensión se dejó calentar lentamente a 20 °C y se agitó durante un total de 16 h. La mezcla se inactivó mediante la adición lenta de ácido acético (0,31 ml) y se agitó a 20 °C durante 5 min. La suspensión se diluyó con agua (50 ml) y se agitó a 20 °C durante 5 min, y luego los sólidos se recogieron por filtración. Los sólidos se lavaron con agua (2 x 25 ml), y luego se disolvieron en acetato de etilo (30 ml). El filtrado orgánico se recogió y los residuos se lavaron con acetato de etilo (2 x 20 ml). El filtrado orgánico combinado se filtró a través de papel de filtro hidrófobo y se concentró al vacío. La goma anaranjada resultante se separó mediante cromatografía en columna ultrarrápida secuencial, usando un gradiente de acetato de etilo en heptano (0-30 %), y luego HPLC preparativa (método 31), para obtener el com puesto de l título (166 mg, 30 %) como un polvo blanquecino. 5h (500 MHz, DMSO-d6) 9,46 (s, 1H), 8,11 -7,99 (m, 1H), 7,23 (d, J 8,2 Hz, 1H), 7,15 (dd, J 8,2, 1,8 Hz, 1H), 4,17-4,00 (m, 2H), 3,77 (dd, J 11,4, 4,5 Hz, 1H), 1,86 (td, J 13,5, 5,1 Hz, 1H), 1,71 (d, J 13,6 Hz, 1H), 1,64 (d, J 13,1 Hz, 1H), 1,58 (s, 9H), 1,52 (dd, J 13,7, 11,3 Hz, 1H), 1,47 (s,
9H), 1,08 (d, J 6,2 Hz, 3H). uPLC-MS (método 1): [M+2H-BOC]+ m/z 333, TR 4,06 min. SFC quiral (método 13, Lux C4 25 cm, MeOH al 15 %-dióxido de carbono al 85 %, 4 ml/min): TR 2,18 min (98 %).
Intermedio 135
(2'S,3fí)-6-Am¡no-2'-met¡lesp¡ro[¡ndol¡na-3,4'-tetrah¡drop¡ran]-2-ona
Se añadió ácido trifluoroacético (0,57 ml, 7,40 mmol) a una solución agitada del in term edio 134 (160 mg, 0,37 mmol) en DCM (1,9 ml). La mezcla se agitó al aire durante 4 h, luego se diluyó con DCM (4 ml) y se inactivó con una solución acuosa saturada de carbonato de sodio (3 ml) y agua (3 ml). La fase orgánica se separó usando una frita hidrófoba y la capa acuosa se extrajo con DCM (2 x 6 ml). El filtrado orgánico se concentró al vacío para proporcionar el com puesto de l título (99 mg, 98 %) como un polvo blanquecino. 5h (500 MHz, DMSO-d6) 9,99 (s, 1H), 6,80 (d, J 7,9 Hz, 1H), 6,13 (dd, J 8,0, 2,0 Hz, 1H), 6,09 (d, J 1,9 Hz, 1H), 5,04 (br s, 2H), 4,24-4,09 (m, 2H), 3,70 (dd, J 11,3, 4,3 Hz, 1H), 1,78 (td, J 13,3, 5,1 Hz, 1H), 1,56-1,39 (m, 3H), 1,05 (d, J 6,2 Hz, 3H). uPLC-MS (método 1): MH+ m /z 233, TR 1,00 min.
Intermed¡o 136
(2' S ,3 S )-6-Am¡no-2'-met¡lesp¡ro[¡ndol¡na-3,4'-tetrah¡drop¡ran]-2-ona
Se añadió ácido trifluoroacético (0,29 ml, 3,76 mmol) a una solución agitada del in term edio 133 (81 mg, 0,19 mmol) en DCM (1 ml). La mezcla se agitó a 20 °C al aire durante 4 h, luego se diluyó con DCM (5 ml) y se inactivó con una solución acuosa saturada de carbonato de sodio (3 ml) y agua (3 ml). La fase orgánica se separó usando una frita hidrófoba y la capa acuosa se extrajo con DCM (2 x 6 ml). El filtrado orgánico se concentró al vacío para proporcionar el com puesto de l título (44,9 mg, 97 %) como un polvo blanquecino. 5h (500 MHz, DMSO-d6) 10,17 (s, 1 H), 7,32 (d, J 8,1 Hz, 1H), 6,16 (d, J 2,1 Hz, 1H), 6,12 (dd, J 8,0, 2,1 Hz, 1H), 5,08 (br s, 2H), 3,96-3,81 (m, 3H), 1,84 (ddd, J 13,0, 11,2, 7,4 Hz, 1H), 1,57-1,48 (m, 1H), 1,30-1,24 (m, 1H), 1,21-1,16 (m, 1H), 1,09 (d, J 6,1 Hz, 3H). uPLC-MS (método 1): MH+ m /z 233, TR 1,00 min.
Intermed¡o 137
(2fí)-2-(2-Etox¡-2-oxoetox¡)propanoato de et¡lo
Se añadió (2R)-2-hidroxipropanoato de etilo (4,8 ml, 42,01 mmol) a una suspensión agitada de carbonato de potasio (5,81 g, 42,0 mmol) en DMF anhidra (42 ml). La suspensión se agitó a 20 °C en nitrógeno durante 20 min y luego se le añadió bromoacetato de etilo (4,7 ml, 42,38 mmol). La mezcla de reacción se agitó a 20 °C en nitrógeno durante 64 h, y luego se diluyó con terf-butilmetiléter (50 ml). Los sólidos se eliminaron por filtración, lavando con tert-butilmetiléter (2 x 50 ml). El filtrado se concentró al vacío. El aceite de color naranja resultante se separó por destilación kuge lrohr (130-185 °C, 11 mbar) para producir el com puesto de l título (4,26 g, 30 %) como un aceite incoloro. 5h (500 MHz, CDCla) 4,27 (d, J 16,5 Hz, 1H), 4,25-4,17 (m, 4H), 4,14 (q, J 6,9 Hz, 1H), 4,07 (d, J 16,5 Hz, 1H), 1,47 (d, J 6,9 Hz, 3H), 1,31-1,26 (m, 6 H).
Intermed¡o 138
(2fí)-2-(2-H¡drox¡etox¡)propán-1-ol
Se añadió gota a gota una solución de hidruro de litio y aluminio en éter dietílico (4 M, 6 , 8 ml, 27,2 mmol) a una solución agitada del in term edio 137 (4,26 g, 20,8 mmol) en THF anhidro (45 ml) a 0 °C en nitrógeno. La mezcla se agitó a 0 °C durante 1 h y a 20 °C durante 16 h. La mezcla de reacción se inactivó cuidadosamente mediante la adición gota a gota de agua (0,51 ml), seguido de la solución acuosa de hidróxido de sodio a 2 M (1,22 ml) y luego agua (1,53 ml). La suspensión se agitó a temperatura ambiente durante 30 min, luego se diluyó con acetona (50 ml) y se filtró a través de una capa de tierra de diatomeas. Los residuos se lavaron con acetona (2 x 50 ml) y el filtrado se concentró al vacío.
El aceite tostado resultante se separó por destilación kuge lrohr (100-120 °C, <1 mbar) para producir el com puesto de l título (904 mg, 29 %) como un aceite incoloro fluido. 5h (500 MHz, CDCl3) 3,78-3,67 (m, 3H), 3,67-3,38 (m, 6 H), 1, 10 (d, J 6,2 Hz, 3H).
Intermed¡o 139
(2fí)-1-Yodo-2-(2-yodoetox¡)propano
Se añadió yodo (9,55 g, 37,63 mmol) en porciones a una solución agitada de trifenilfosfina (7,90 g, 30,12 mmol) y 1H-imidazol (2,06 g, 30,26 mmol) en éter dietílico/acetonitrilo 3:1 (60 ml) a 20 °C en nitrógeno. Después de agitar a 20 °C durante 30 min, se le añadió una solución del in term edio 138 (0,90 g, 7,52 mmol) en éter dietílico (3 ml), seguido de un enjuague con éter dietílico (2 ml). La mezcla de reacción se agitó a 20 °C en la oscuridad durante 22 h. La suspensión se diluyó con terf-butilmetiléter (50 ml) y se filtró a través de una capa de tierra de diatomeas, lavando bien con terf-butilmetiléter (4 x 30 ml). El filtrado se filtró a través de una segunda almohadilla de tierra de diatomeas, lavando bien con terf-butilmetiléter (2 x 30 ml), y luego se concentró al vacío. El aceite viscoso de color rojo oscuro resultante se separó mediante cromatografía en columna ultrarrápida, usando un gradiente de acetato de etilo en heptano (0-5 %), para proporcionar el com puesto de l título (1,32 g, 47 %) como un aceite tostado fluido. 5h (500 MHz, CDCla) 3,80-3,69 (m, 2H), 3,55-3,44 (m, 1H), 3,29-3,22 (m, 3H), 3,20 (dd, J 10,3, 5,7 Hz, 1H), 1,30 (d, J 6,1 Hz, 3H).
Intermedio 140
(2'fí,3S)-6-(ferf-Butox¡carbon¡lam¡no)-2'-met¡l-2-oxosp¡ro[¡ndol¡na-3,4'-tetrah¡drop¡ran]-1-carbox¡lato de te rt- butilo
Una solución agitada del in term edio 129 (600 mg, 1,72 mmol) y del in term edio 139 (640 mg, 1,38 mmol) en DMF anhidro (11,5 ml) se purgó con una corriente de nitrógeno mientras se sonicaba durante 5 min. La solución se enfrió a -10 °C en nitrógeno y se le añadió en porciones carbonato de cesio (2,24 g, 6,89 mmol) durante 10 min. La suspensión se dejó calentar lentamente a 20 °C y se agitó durante un total de 16 h. La mezcla se inactivó mediante la adición lenta de ácido acético (0,31 ml) y se agitó a 20 °C durante 5 min. La suspensión se diluyó con agua (50 ml) y se agitó a 20 °C durante 5 min, y luego los sólidos se recogieron por filtración. Los sólidos se lavaron con agua (2 x 25 ml) y luego se disolvieron en acetato de etilo (30 ml). Se recogió el filtrado orgánico y los residuos se lavaron con acetato de etilo (2 x 20 ml). El filtrado orgánico combinado se filtró a través de papel de filtro hidrófobo y se concentró al vacío. La goma naranja resultante se separó mediante cromatografía en columna ultrarrápida secuencial, usando un gradiente de acetato de etilo en heptano (0-30 %), y luego HPLC preparativa (método 31), para obtener el com puesto de l título (180 mg, 30 %) como un polvo blanco. 5h (500 MHz, DMsO-d6) 9,45 (s, 1H), 8,05 (d, J 1,2 Hz, 1H), 7,23 (d, J 8,2 Hz, 1H), 7,15 (dd, J 8,2, 1,8 Hz, 1H), 4,19-4,00 (m, 2H), 3,77 (dd, J 11,5, 4,4 Hz, 1H), 1,86 (td, J 13,4, 5,0 Hz, 1H), 1,71 (d, J 13,6 Hz, 1H), 1,64 (d, J 13,2 Hz, 1H), 1,58 (s, 9H), 1,52 (dd, J 13,7, 11,3 Hz, 1H), 1,47 (s, 9H), 1,08 (d, J 6,2 Hz, 3H). uPLC-MS (método 1): [M+2H-BOC]+ m/z 333, TR 4,06 min. s Fc quiral (método 13, Lux c 4 25 cm, MeOH al 15 %-dióxido de carbono al 85 %, 4 ml/min): TR 1,96 min (99 %).
Intermed¡o 141
(2'fí,3S)-6-Am¡no-2'-met¡lesp¡ro[¡ndol¡na-3,4'-tetrah¡drop¡ran]-2-ona
Se añadió ácido trifluoroacético (0,62 ml, 8,05 mmol) a una solución agitada del in term edio 140 (175 mg, 0,41 mmol) en DCM (2 ml). La mezcla se agitó a 20 °C al aire durante 4 h, luego se diluyó con DCM (4 ml) y se inactivó con una solución acuosa saturada de carbonato de sodio (3 ml) y agua (3 ml). La fase orgánica se separó usando una frita hidrófoba y la capa acuosa se extrajo con DCM (2 x 6 ml). El filtrado orgánico se concentró al vacío para proporcionar el com puesto de l título (104 mg, cuantitativo) como un polvo blanquecino. 5h (500 MHz, DMSO-d6) 9,99 (s, 1H), 6,80 (d, J 7,9 Hz, 1H), 6,13 (dd, J 7,9, 2,0 Hz, 1H), 6,09 (d, J 1,9 Hz, 1H), 5,04 (br s, 2H), 4,26-4,07 (m, 2H), 3,70 (dd, J 11,3, 4,3 Hz, 1H), 1,78 (td, J 13,3, 5,1 Hz, 1H), 1,56-1,40 (m, 3H), 1,05 (d, J 6,2 Hz, 3H). uPLC-MS (método 1): MH+ m/z 233, TR 1,00 min.
Intermed¡o 142
2-C¡clohex¡l-3-oxo-3-[(2-oxosp¡ro[¡ndol¡na-3,4'-tetrah¡drop¡ran]-6-¡l)am¡no]-propanoato de et¡lo
A una solución del in term edio 2 (390 mg, 1,79 mmol) y sal de litio del ácido 2-ciclohexil-3-etoxi-3-oxopropanoico (398 mg, 1,86) en DMF (7 ml, 90,50 mmol) se le añadió HATU (840 mg, 2,14 mmol). La mezcla de reacción se agitó a 20 °C durante 18 h, luego se repartió entre EtOAc y agua. Se combinaron las capas orgánicas, se secaron y se concentraron al vacío. El residuo se separó mediante cromatografía en columna ultrarrápida, usando un gradiente de acetato de etilo en hexano (0-100 %), para proporcionar el com puesto de l título (623 mg, 84 %). HPLC-MS (método 7): MH+ m/z 415, TR 1,24 min.
Intermed¡o 143
Ác¡do 2-c¡clohex¡l-3-oxo-3-[(2-oxosp¡ro[¡ndol¡na-3,4'-tetrah¡drop¡ran]-6-¡l)am¡no]propano¡co
Se añadió gota a gota una solución de LiOH (36 mg, 1,50 mmol) en agua (5 ml, 277,55 mmol) a una solución del in term edio 142 (623 mg, 1,50 mmol) en etanol (10 ml, 172,00 mmol). La mezcla de reacción se agitó a 20 °C durante 72 h, y luego se concentró al vacío para proporcionar el com puesto de l título (580 mg, 100 %) como la sal de litio. HPLC-MS (método 7): MH+ m/z 387, TR 0,85 min.
Intermed¡o 144
W-(2-Am¡no-3-fluorofen¡l)-2-c¡clohex¡l-W-(2-oxosp¡ro[¡ndol¡na-3,4'-tetrah¡drop¡ran]-6-¡l)propanod¡am¡da
A una solución de 3-fluorobenceno-1,2-diamina (35 mg, 0,26 mmol) y del in term edio 143 (116 mg, 0,30 mmol) en DMF (2 ml, 25,90 mmol) se le añadió HATU (124 mg, 0,32 mmol). La mezcla de reacción se agitó a 20 °C durante 40 h, luego se le añadió agua a 0 °C. El precipitado resultante se recogió por filtración para obtener el com puesto de l título (97 mg, 75 %) como un sólido tostado. Hp LC-MS (método 7): MH+ m/z 495, TR 1,25 min.
Intermed¡o 145
W-(2-Am¡nofen¡l)-2-c¡clohex¡l-W-(2-oxosp¡ro[¡ndol¡na-3,4'-tetrah¡drop¡ran]-6-¡l)-propanod¡am¡da
A una solución de o-fenilendiamina (10 mg, 0,09 mmol) y del in term edio 165 (39 mg, 0,10 mmol) en DMF (1 ml, 12,90 mmol) se le añadió HATU (45 mg, 0,12 mmol). La mezcla de reacción se agitó a 20 °C durante 18 h y luego se repartió
entre EtOAc y agua. Se separaron las capas orgánicas y se lavaron con salmuera, luego se secaron y se concentraron al vacío para producir el com puesto de l título (40 mg, 91 %). HPLC-MS (método 7): MH+ m /z 477, TR 1,18 min.
Intermedio 146
2-(Azocan-1-il)propanodioato de dietilo
A una solución de heptametilenimina (2,25 ml, 17,70 mmol) en acetonitrilo (40 ml, 763,00 mmol) se le añadió K2CO3 (4,90 g, 35,00 mmol), seguido de una solución de 2-bromopropanodioato de dietilo (4,25 g, 17,80 mmol) en acetonitrilo (40 ml, 763,00 mmol). La mezcla de reacción se agitó a 20 °C durante 3 h y luego se concentró al vacío. El residuo se separó mediante cromatografía en columna ultrarrápida, utilizando un gradiente de acetato de etilo en hexano (0-50 %), para proporcionar el com puesto de l título (3,78 g, 78 %) como un aceite incoloro. 5h (400 MHz, CDCta) 4,23 (q, J 7,1 Hz, 4H), 4,12 (s, 1H), 2,83 (t, J 5,4 Hz, 4H), 1,68-1,53 (m, 10H), 1,29 (t, J 7,1 Hz, 6H). LCMS (método 7): MH+ m /z 272, TR 1,59 min.
Intermedio 147
Ácido 2-(azocan-1 -il)-3-etoxi-3-oxo-propanoico
Se añadió gota a gota una solución de LiOH (22 mg, 0,92 mmol) en agua (1 ml, 55,51 mmol) a una solución enfriada (0 °C) del in term edio 146 (250 mg, 0,92 mmol) en EtOH (4 ml, 68,70 mmol). La mezcla de reacción se calentó lentamente a 20 °C y se agitó durante 4,5 h, y luego se concentró al vacío para proporcionar el com puesto de l título (225 mg, 100 %) como la sal de litio. Lc Ms (método 7): MH+ m /z 244, TR 0,85 min.
Intermedio 148
2-(Azocan-1-il)-3-oxo-3-[(2-oxospiro[indolina-3,4'-tetrahidropiran]-6-il)amino]-propanoato de etilo
A una solución del in term edio 2 (190 mg, 0,87 mmol) y del in term edio 147 (224 mg, 0,92 mmol) en DMF (4 ml, 51,70 mmol) se le añadió HATU (410 mg, 1,05 mmol). La mezcla de reacción se agitó a 20 °C durante 18 h, luego se repartió entre EtOAc y agua. Las capas orgánicas se lavaron con salmuera, se secaron y se concentraron al vacío. El residuo se separó mediante cromatografía en columna ultrarrápida, usando un gradiente de acetato de etilo en hexano (0-100 %), para proporcionar el com puesto de l título (125 mg, 32 %) como un sólido blanco. 5h (400 MHz, CDCb) 8,97 (s, 1H), 7,63 (d, J 1,9 Hz, 1H), 7,44 (s, 1H), 7,30 (d, J 8,1 Hz, 1H), 6,87 (dd, J 8,1,2,0 Hz, 1H), 4,38-4,17 (m, 4H), 4,10 (s, 1H), 3,92 (ddd, J 11,8, 5,5, 4,3 Hz, 2H), 2,93-2,72 (m, 4H), 2,08-1,59 (m, 14H), 1,33 (t, J 7,1 Hz, 3H). LCMS (método 6): MH+ m /z 444, TR 2,31 min.
Intermedio 149
Ácido 2-(azocan-1-il)-3-oxo-3-[(2-oxospiro[indolina-3,4'-tetrahidropiran]-6-il)amino]-propanoico
Se añadió gota a gota una solución de LiOH (12 mg, 0,50 mmol) en agua (2 ml, 111,02 mmol) a una solución del in term edio 148 (106 mg, 0,24 mmol) en EtOH (5 ml, 85,90 mmol). La mezcla de reacción se agitó a 20 °C durante 18 h y luego se concentró al vacío para proporcionar el com puesto de l título (99 mg, 100 %) como la sal de litio. LCMS (método 7): MH+ m /z 416, TR 0,91 min.
Intermedio 150
W-[(1S)-1-Ciclohexil-2-oxo-2-({2-oxo-1-[2-(trimetilsilil]etoximetil]-espiro[pirrolo[3,2-c]piridin-3,4'-tetrahidropiran]-6-il}am ino)etilo]carbamato de ferf-butilo
A una solución de ácido (2S)-2-(tert-butoxicarbonilamino)-2-ciclohexilacético (1,47 g, 5,72 mmol) y del in term edio 57 en DCM anhidro (40 ml) se le añadieron HATU (2,69 g, 6,87 mmol) y DIPeA (1,99 ml, 11,44 mmol). La mezcla de reacción se agitó a 20 °C durante 18 h. Se le añadieron además ácido (2S)-2-(terf-butoxicarbonilamino)-2-ciclohexilacético (1,47 g, 5,72 mmol), HATU (2,69 g, 6,87 mmol) y DIPEA (1,99 ml, 11,44 mmol), y la mezcla se agitó durante otras 28 h. La mezcla de reacción se repartió entre DCM y agua, y la fase acuosa se extrajo con DCM (2 x 25 ml). Las capas orgánicas combinadas se separaron a través de un separador de fases de frita hidrófoba y se concentraron al vacío. El residuo bruto se purificó mediante cromatografía en columna ultrarrápida, utilizando un gradiente de 0-100 % de acetato de etilo en hexanos, para proporcionar el com puesto de l título (3,00 g, 89 %) como un sólido blanco. 5h (300 MHz, DMSO-ds ) 10,41 (s, 1H), 8,60-8,34 (m, 1H), 7,93 (s, 1H), 6,94 (d, J 8,6 Hz, 1H), 5,08 (s, 2H), 4,25-3,67 (m, 2H), 3,50 (t, J 7,8 Hz, 2H), 1,94-1,45 (m, 10H), 1,38 (s, 10H), 1,24-0,93 (m, 7H), 0,93-0,74 (m, 2H), -0,09 (s, 9H). HPLC-MS (método 7): MH+ m /z 589, TR 1,69 min. [a]20D = -15,53° (c 7,5, metanol).
Intermedio 151
(2S)-2-Amino-2-ciclohexil-W-(2-oxospiro[1 H-pirrolo[3,2-c]piridin-3,4'-tetrahidropiran]-6-il)acetamida
Se añadió ácido trifluoroacético (12 ml, 150 mmol) a una solución agitada del in term edio 150 (3 g, 5,10 mmol) en DCM (10 ml) a 0 °C. La mezcla se agitó a 20 °C durante 18 h y luego se concentró al vacío. El residuo bruto se disolvió en
acetonitrilo (5 ml), seguido de la adición de una solución acuosa de hidróxido de amonio (5 ml) a 0 °C. La mezcla de reacción se agitó a 20 °C durante 20 min y luego se concentró al vacío. El residuo bruto se repartió entre EtOAc y una solución acuosa de NaOH a 2 M, y la fase acuosa se extrajo con DCM:isopropanol (10 %) (3 x 25 ml). Las capas orgánicas combinadas se concentraron al vacío para proporcionar el com puesto de l título (1,8 g, 74 %) como un sólido.
5h (300 MHz, CDCl3) 9,38 (s, 1H), 8,28 (s, 1H), 8,10 (s, 1H), 4,22 (ddd, J 11,2, 6,9, 3,8 Hz, 2H), 3,93 (ddd, J 11,6, 7,6, 3,6 Hz, 2H), 3,51 (d, J 3,9 Hz, 1H), 2,05 (ddd, J 13,9, 7,7, 3,8 Hz, 4H), 1,86-1,57 (m, 8H), 1,49-1,04 (m, 6H). HPLC-MS (método 7): MH+ m /z 359, TR 0,60 min. [a]20D = -77,80° (c 7,5, metanol).
Intermedio 152
W-(2-Oxospiro[indolina-3,4'-tetrahidropiran]-6-il)carbamato de fenilo
A una solución agitada del in term edio 2 (500 mg, 2,3 mmol) y piridina (0,222 ml, 2,74 mmol) en THF (10 ml) se le añadió gota a gota una solución de cloroformiato de fenilo (0,4 ml, 2,7 mmol) en THF (10 ml). La mezcla se agitó durante 18 h a 20 °C, luego se diluyó con EtOAc (30 ml) y se lavó con una solución acuosa saturada de NaHCO3 (30 ml) seguido de una solución acuosa de HCl a 1 N (30 ml). La capa orgánica se secó sobre Na2SO4 , se filtró y se evaporó hasta la sequedad. El residuo se purificó mediante cromatografía ultrarrápida sobre gel de sílice, eluyendo con un gradiente de EtOAc en hexano (1:9 a 1:0), para producir el com puesto de l título (595 mg, 75 %) como un sólido blanco. 5h (400 MHz, DMSO-ds ) 10,59-10,27 (m, 1H), 9,61-9,18 (m, 1H), 7,47-7,39 (m, 2H), 7,28-7,11 (m, 4H), 6,79 6,71 (m, 2H), 4,13-3,96 (m, 2H), 3,93-3,72 (m, 2H), 1,97-1,82 (m, 2H), 1,80-1,54 (m, 2H). HPLC-MS (método 7): MH+ m /z 339, TR 1,12 min.
Intermedio 153
Ácido 2-(fert-butoxicarbonilamino)-2-(4-cloro-3-fluorofenil)acético
Se añadió dicarbonato de di-tert-butilo (1,29 g, 5,89 mmol) a una suspensión agitada de ácido 2-amino-2-(4-cloro-3-fluorofenil)acético (1,0 g, 4,91 mmol) en THF (3,2 ml) y una solución acuosa de carbonato de sodio a 1 M (9,8 ml, 9,8 mmol). La suspensión se agitó a 20 °C en nitrógeno durante 16 h. La mezcla se diluyó con agua (30 ml) y una solución acuosa de hidróxido de sodio a 1 M (10 ml), y luego se lavó con terf-butilmetiléter (2 x 50 ml). Las capas orgánicas combinadas se extrajeron con una solución acuosa de hidróxido de sodio a 1 M (40 ml). Las capas acuosas combinadas se acidificaron con ácido clorhídrico concentrado (pH 2) y la fase acuosa se extrajo con acetato de etilo (3 x 60 ml). Las capas orgánicas combinadas se lavaron con salmuera (50 ml) y se secaron sobre sulfato de magnesio anhidro, y luego se filtraron y se concentraron al vacío para proporcionar el com puesto de l título (1,52 g, 96 %) como un sólido blanco. 5h (250 MHz, DMSO-ds ) 13,06 (br s, 1H), 7,70 (d, J 8,3 Hz, 1H), 7,58 (t, J 8,1 Hz, 1H), 7,47 (dd, J 10,5, 1,9 Hz, 1H), 7,29 (dd, J 8,2, 1,9 Hz, 1H), 5,18 (d, J 8,6 Hz, 1H), 1,39 (s, 9H). HPLC-MS (método 3): MNa+ m /z 326, TR 1,77 min.
Intermedio 154
W-[1-(4-Cloro-3-fluorofenil)-2-oxo-2-(2-oxo-1-[2-(trimetilsilil)etoximetil]espiro[pirrolo[3,2-c]piridin-3,4'-tetrahidropiran]-6-il}am ino)etil]carbamato de ferf-butilo
Se añadió HATU (999 mg, 2,63 mmol) a una suspensión agitada del in term edio 57 (765 mg, 2,19 mmol), in term edio 153 (700 mg, 2,19 mmol) y DIPEA (0,9 ml, 5,47 mmol) en DCM (25 ml). La mezcla se agitó a 20 °C en nitrógeno durante 16 h, luego se repartió entre DCM (50 ml) y agua (50 ml). Se separó la capa acuosa y se lavó con DCM (2 x 50 ml). Las capas orgánicas combinadas se secaron sobre sulfato de magnesio anhidro, se filtraron y se concentraron al vacío. El residuo se purificó por cromatografía en columna ultrarrápida, usando un gradiente de acetato de etilo en heptano (0-100 %), para dar el com puesto de l título (937 mg, 67 %) como un sólido naranja. 5h (500 MHz, DMSO-d6) 11,01 (s, 1H), 8,61 (s, 1H), 7,95 (s, 1H), 7,84 (d, J 7,8 Hz, 1H), 7,77-7,65 (m, 2H), 7,55 (d, J 8,3 Hz, 1H), 5,67 (d, J 7,5 Hz, 1H), 5,21 (d, J 11,2 Hz, 1H), 5,19 (d, J 11,3 Hz, 1H), 4,16-4,08 (m, 2H), 4,02-3,91 (m, 2H), 3,69-3,53 (m, 2H), 2,03 1,89 (m, 2H), 1,88-1,76 (m, 2H), 1,53 (s, 9H), 1,07-0,86 (m, 2H), 0,00 (s, 9H). HPLC-MS (método 3): MH+ m /z 635, TR 2,19 min.
Intermedio 155
Hidrocloruro de 2-amino-2-(4-cloro-3-fluorofenil)-W-{2-oxo-1-[2-(trimetilsilil)etoximetil]espiro-[pirrolo[3,2-c]piridin-3,4'-tetrahidropiran]-6-il}acetamida
Se añadió una solución de cloruro de hidrógeno en 1,4-dioxano (4 M, 3,66 ml, 14,64 mmol) a una solución agitada del in term edio 154 (929 mg, 1,46 mmol) en MeOH (16 ml) en nitrógeno a t.a. La mezcla de reacción se agitó a t.a. toda la noche y luego se concentró al vacío para proporcionar el com puesto de l título (759 mg, 78 %) como un sólido de color amarillo anaranjado. 5h (500 MHz, DMSO-d6) 11,53 (br s, 1H), 9,17 (br s, 2H), 8,64 (s, 1H), 8,01-7,84 (m, 2H), 7,81 (dd, J 10,2, 1,8 Hz, 1H), 7,64 (d, J 8,3 Hz, 1H), 5,54-5,39 (m, 1H), 5,29-5,12 (m, 2H), 4,21 -4,08 (m, 2H), 4,02-3,93 (m, 2H, obs), 3,66-3,55 (m, 2H), 2,03-1,74 (m, 4H), 1,05-0,88 (m, 2H), 0,00 (s, 9H). HPLC-MS (método 3): MH+ m/z 535, TR 1,85 min.
Intermedio 156
W-[1-(4-Cloro-3-fluorofenil)-2-oxo-2-({2-oxo-1-[2-(trimetilsilil)etoximetil]-espiro[pirrolo[3,2-c]piridin-3,4'-tetrahidropiran]-6-il}amino)etil]-2-etilpirazol-3-carboxamida
Se añadió HATU (206 mg, 0,54 mmol) a una suspensión agitada del in term edio 155 (300 mg, 0,45 mmol), ácido 1-etil-1 H-pirazol-5-carboxílico (63 mg, 0,45 mmol) y DIPEA (0,28 ml, 1,58 mmol) en DCM (6 ml). La mezcla se agitó a 20 °C en nitrógeno durante 16 h, luego se repartió entre DCM (25 ml) y agua (25 ml). Se separó la capa acuosa y se lavó con DCM (2 x 25 ml). Las capas orgánicas combinadas se secaron sobre sulfato de magnesio anhidro, se filtraron y se concentraron al vacío. El residuo se purificó mediante cromatografía en columna ultrarrápida, usando un gradiente de acetato de etilo en heptano (0-100 %), para proporcionar el com puesto de l título (311 mg, 89 %) como una goma naranja. 5h (500 MHz, DMSO-d6) 11,16 (s, 1H), 9,12 (d, J 7,0 Hz, 1H), 8,62 (s, 1H), 8,00 (s, 1H), 7,84-7,71 (m, 2H), 7,63 (d, J 2,0 Hz, 1H), 7,60 (dd, J 8,4, 1,8 Hz, 1H), 7,20 (d, J 2,1 Hz, 1H), 6,05 (d, J 7,0 Hz, 1H), 5,21 (d, J 11,2 Hz, 1H), 5,18 (d, J 11,2 Hz, 1H), 4,59 (q, J 7,2 Hz, 2H), 4,16-4,09 (m, 2H), 4,04-3,90 (m, 2H), 3,66-3,55 (m, 2H), 2,02-1,89 (m, 2H), 1,89-1,78 (m, 2H), 1,41 (t, J 7,1 Hz, 3H), 1,04-0,90 (m, 2H), 0,00 (s, 9H). HPLC-MS (método 3): MH+ m /z 657, TR 2,11 min.
Intermedio 157
Ácido 2-(fert-butoxicarbonilamino)-2-(4-fluoro-3-metilfenil)acético
Se añadió dicarbonato de di-terf-butilo (1,43 g, 6,55 mmol) a una suspensión agitada de ácido 2-amino-2-(4-fluoro-3-metilfenil)acético (1,0 g, 5,46 mmol) en THF (3,6 ml) y una solución acuosa de carbonato de sodio a 1 M (10,9 ml, 10,9 mmol). La suspensión se agitó a 20 °C en nitrógeno durante 16 h. La mezcla se diluyó con agua (30 ml) y una solución acuosa de hidróxido de sodio a 1 M (10 ml), luego se lavó con tert-butilmetiléter (2 x 50 ml). Las capas orgánicas combinadas se extrajeron con una solución acuosa de hidróxido de sodio a 1 M (40 ml). Las capas acuosas combinadas se acidificaron con ácido clorhídrico concentrado (pH 2) y se extrajeron con acetato de etilo (3 x 60 ml). Las capas orgánicas combinadas se lavaron con salmuera (50 ml) y se secaron sobre sulfato de magnesio, luego se filtraron y se concentraron al vacío, para proporcionar el com puesto de l título (1,26 g, 77 %) como un aceite de color naranja transparente. 5h (250 MHz, DMSO-ds ) 12,77 (br s, 1H), 7,53 (d, J 8,0 Hz, 1H), 7,32 (dd, J 7,4, 2,1 Hz, 1H), 7,29-7,19 (m, 1H), 7,17-7,03 (m, 1H), 5,06 (d, J 8,2 Hz, 1H), 2,22 (d, J 1,7 Hz, 3H), 1,39 (s, 9H). HPLC-MS (método 3 ): MNa+ m /z 306 , TR 1,77 min.
Intermedio 158
W-[1-(4-Fluoro-3-metilfenil)-2-oxo-2-({2-oxo-1-[2-(trimetilsilil)etoximetil]espiro[pirrolo[3,2-c]piridin-3,4'-tetrahidropiran]-6-il}am ino)etil]carbamato de ferf-butilo
Se añadió HATU (1,06 g, 2,79 mmol) a una suspensión agitada del in term edio 57 (812 mg, 2,32 mmol), del in term edio 157 (700 mg, 2,32 mmol) y DIPEA (0,96 ml, 5,81 mmol) en DCM (27 ml). La mezcla se agitó a 20 °C en nitrógeno durante 16 h, y luego se repartió entre DCM (50 ml) y agua (50 ml). Se separó la capa acuosa y se lavó con DCM (2 x 50 ml). Las capas orgánicas combinadas se secaron sobre sulfato de magnesio anhidro, se filtraron y se concentraron al vacío. El residuo se purificó por cromatografía en columna ultrarrápida, usando un gradiente de acetato de etilo en heptano (0-100 %), para dar el com puesto de l título (1,09 g, 76 %) como un sólido amarillo. 5h (500 MHz, DMSO-ds ) 10,86 (s, 1H), 8,58 (s, 1H), 7,96 (s, 1H), 7,60 (d, J 7,7 Hz, 1H), 7,55 (dd, J 7,4, 1,8 Hz, 1H), 7,52-7,45 (m, 1H), 7,29-7,17 (m, 1H), 5,55 (d, J 7,4 Hz, 1H), 5,18 (s, 2H), 4,14-4,07 (m, 2H), 4,00-3,86 (m, 2H), 3,65-3,54 (m, 2H), 2,40-2,28 (m, 3H), 1,99-1,87 (m, 2H), 1,87-1,74 (m, 2H), 1,51 (s, 9H), 1,08-0,83 (m, 2H), 0,00 (s, 9H). HPLC-MS (método 3): MH+ m /z 615, TR 2,17 min.
Intermedio 159
Hidrocloruro de 2-amino-2-(4-fluoro-3-metilfenil)-W-{2-oxo-1-[2-(trimetilsilil)etoximetil]espiro-[pirrolo[3,2-c]piridin-3,4'-tetrahidropiran]-6-il}acetamida
Se añadió una solución de cloruro de hidrógeno en 1,4-dioxano (4 M, 4,44 ml, 17,73 mmol) a una solución agitada del in term edio 158 (1,09 g, 1,73 mmol) en MeOH (19 ml) en nitrógeno a t.a. La reacción se agitó a t.a. toda la noche y luego se concentró al vacío, para proporcionar el com puesto de l título (1,1 g, cuantitativo) como un sólido de color amarillo anaranjado. 5h (500 MHz, DMSO-d6) 11,40 (br s, 1H), 8,99 (br d, J 4,2 Hz, 2H), 8,61 (s, 1H), 7,91 (s, 1H), 7,67-7,62 (m, 1H), 7,61-7,56 (m, 1H), 7,37 (t, J 9,1 Hz, 1H), 5,38-5,27 (m, 1H), 5,26-5,13 (m, 2H), 4,17-4,05 (m, 2H, obs.), 3,98-3,88 (m, 2H), 3,64-3,54 (m, 2H), 2,36 (s, 3H), 1,99-1,72 (m, 4H), 1,01-0,88 (m, 2H), 0,00 (s, 9H). HPLC-MS (método 3): Mh m /z 515, TR 1,80 min.
Intermedio 160
2-Etil-W-[1-(4-fluoro-3-metilfenil)-2-oxo-2-({2-oxo-1-[2-(trimetilsilil)etoximetil]espiro[pirrolo[3,2-c]piridin-3,4'-tetrahidropiran]-6-il}amino)etil]pirazol-3-carboxamida
Se añadió HATU (226 mg, 0,59 mmol) a una suspensión agitada del in term edio 159 (300 mg, 0,50 mmol), ácido 1 -etil
1 H-pirazol-5-carboxílico (69 mg, 0,50 mmol) y DIPEA (0,30 ml, 1,73 mmol) en DCM (6 ml). La mezcla se agitó a 20 °C en nitrógeno durante 16 h, y luego se repartió entre DCM (25 ml) y agua (25 ml). Se separó la capa acuosa y se lavó con DCM (2 x 25 ml). Las capas orgánicas combinadas se secaron sobre sulfato de magnesio anhidro, se filtraron y se concentraron al vacío. El residuo se purificó por cromatografía en columna ultrarrápida, usando un gradiente de acetato de etilo en heptano (0-100 %), para dar el com puesto de l títu lo (425 mg, 78 %) como una goma naranja. 5h (500 MHz, DMSO-d6) 10,90 (s, 1H), 8,87 (d, J 6,9 Hz, 1 H), 8,47 (s, 1H), 7,89 (s, 1H), 7,51 -7,46 (m, 2H), 7,45-7,39 (m, 1H), 7,19-7,12 (m, 1H), 7,06 (d, J 2,1 Hz, 1H), 5,83 (d, J 6,8 Hz, 1H), 5,11-5,03 (m, 2H), 4,46 (q, J 7,1 Hz, 2H), 4,07 3,95 (m, 2H), 3,89-3,78 (m, 2H), 3,51 -3,43 (m, 2H), 2,24 (d, J 1,4 Hz, 3H), 1,88-1,76 (m, 2H), 1,75-1,63 (m, 2H), 1,28 (t, J 7,1 Hz, 3H), 0,85-0,78 (m, 2H), -0,12 (s, 9H). HPLC-MS (método 3): MH+ m /z 637, TR 2,08 min.
Ejemplos
Ejemplo 1

(2S)-3-(2-Clorofenil)-W-(2-oxospiro[indolina-3,4'-tetrahidropiran]-6-il)-2-{[2-(piridin-4-amino}propenamida y (2fi)-3-(2-clorofenil)-WL(2-oxospiro-[indolina-3,4'-tetrahidropiran]-6-il)-2-{[2-(piridin-4-il)acetil]amino}propanamida
Se añadió DIPEA (40 pl, 0,24 mmol) a una suspensión agitada del in term edio 2 (40 mg, 0,18 mmol), del in term edio 5 (61 mg, 0,19 mmol) y HATU (77 mg, 0,20 mmol) en THF anhidro (1 ml). La mezcla se agitó a 20 °C en nitrógeno durante 16 h, luego se inactivó con una solución acuosa saturada de carbonato de sodio (6 ml) y se agitó durante 30 min a 20 °C. La mezcla se diluyó con agua (6 ml) y se repartió con DCM/isopropanol (4:1; 30 ml). La fase orgánica se separó usando una frita hidrófoba y luego la capa acuosa se extrajo con DCM/isopropanol 4:1 (2 x 20 ml). El filtrado se concentró al vacío. La goma naranja en bruto resultante se separó por HPLC preparativa (método 10) para producir el com puesto de l títu lo (54 mg, 54 % de rendimiento, 46 % de ee) como un polvo blanquecino después de la liofilización. dH (500 MHz, DMSO-de) 10,39 (s, 1H), 10,09 (s, 1 H), 8,63 (d, J 8,3 Hz, 1H), 8,45-8,38 (m, 2H), 7,42 (d, J 8,1 Hz, 1H), 7,39 (dd, J 7,8, 1,3 Hz, 1H), 7,33-7,27 (m, 2H), 7,23 (td, J 7,6, 1,9 Hz, 1H), 7,19 (td, J 7,4, 1,4 Hz, 1H), 7,16-7,12 (m, 2H), 7,06 (dd, J 8,1, 1,9 Hz, 1H), 4,79 (td, J 8,5, 6,1 Hz, 1 H), 4,01 (ddd, J 11,1,7,2, 3,6 Hz, 2H), 3,79 (ddd, J 11,1,7,0, 3,7 Hz, 2H), 3,52 (d, J 14,3 Hz, 1H), 3,48 (d, J 14,3 Hz, 1H), 3,17 (dd, J 14,1,5,9 Hz, 1H), 3,04 (dd, J 14,1, 8,7 Hz, 1H), 1,77-1,68 (m, 2H), 1,67-1,56 (m, 2H). uPLC-MS (método 2): MH+ m/z 519, TR 2,60 min. Método 12 de SFC quiral, Chiralcel OD-H 25 cm, metanol al 25 %-dióxido de carbono al 75 %, 4 ml/min: TR 5,29 min (27 %, R); TR 6,12 min (73 %, S).
El racemato (29 mg) se separó mediante el método preparativo 15 de SFC quiral, columna Lux Cellulose-1 de 21,2 x 250 mm, 5 pm, MeOH isocrático al 25 % (+0,1 % de NH4OH), para proporcionar el enantiómero (S) (eutómero) (3 mg) y el enantiómero (R) (5 mg) como polvos blanquecinos. Método 13 de SFC quiral, columna Lux Cellulose-1 de 4,6 x 150 mm, 3 pm, velocidad de flujo de 3,5 ml/min, 25 % de MeOH isocrático 0,1 % de NH4OH: TR 5,62 min (100 %, R); TR 6,35 min (100%, S).
Ejemplo 2

(2S)-2-Ciclohexil-W-(2-oxospiro[indolina-3,4'-tetrahidropiran]-6-il)-2-{[2-(piridin-4-il)acetil]amino}acetamida y (2R)-2-ciclohexil-W-(2-oxospiro[indolina-3,4'-tetrahidropiran]-6-il)-2-{[2-(piridin-4-il)acetil]amino}acetamida
Se añadió HATU (90 mg, 0,24 mmol) a una suspensión agitada del in term edio 8 (62 mg, 0,22 mmol) y del interm edio 2 (50 mg, 0,23 mmol) en DMF anhidra (1,1 ml). La suspensión se agitó a 20 °C durante 16 h, luego se inactivó con una solución acuosa saturada de carbonato de sodio (3 ml) y agua (3 ml) y se agitó a 20 °C durante 1 h. La mezcla se
diluyó con agua (6 ml) y luego el material se extrajo con DCM (3 x 10 ml) y DCM/isopropanol 4:1 (3 x 15 ml) usando una frita hidrófoba para separar las fases. Se combinaron los filtrados orgánicos y se concentraron al vacío. La goma naranja en bruto resultante se separó por HPLC preparativa (método 10) para producir el com puesto de l título (77 mg, 70 % de rendimiento, 64 % de ee) como un polvo blanquecino después de la liofilización. 5h (500 MHz, DMSO-d6) 10,39 (br s, 1H), 10,12 (s, 1H), 8,50-8,45 (m, 2H), 8,42 (d, J 8,5 Hz, 1 H), 7,43 (d, J 8,1 Hz, 1H), 7,36 (d, J 1,9 Hz, 1H), 7,32-7,25 (m, 2H), 7,09 (dd, J 8,1, 1,9 Hz, 1H), 4,29 (t, J 8,1 Hz, 1H), 4,01 (ddd, J 11,1, 7,2, 3,6 Hz, 2H), 3,80 (ddd, J 11,0, 7,0, 3,7 Hz, 2H), 3,61 (d, J 14,1 Hz, 1H), 3,56 (d, J 14,1 Hz, 1H), 1,79-1,49 (m, 10H), 1,23-0,92 (m, 5H). uPLC-MS (método 2): MH+ m /z 477, TR 2,61 min. SFC quiral, método 12, Chiralcel OD-H 25 cm, metanol al 30 %-dióxido de carbono al 70 %, 4 ml/min: TR 2,30 min (18 %, R); TR 2,62 min (82 %, S).
Ejemplo 3

W-{1-Ciclooctil-2-oxo-2-[(2-oxospiro[indolina-3,4'-tetrahidropiran]-6-il)amino]etil}-2-metilpirazol-3-carboxamida
Se añadió HATU (210 mg, 0,55 mmol) a una solución agitada del in term edio 2 (100 mg, 0,46 mmol) y del interm edio 13 (150 mg, 0,5 mmol) en DMF anhidra (4 ml) en una atmósfera de nitrógeno. La mezcla se agitó a 20 °C durante 15 h y luego se inactivó con una solución acuosa saturada de carbonato de hidrógeno y sodio (20 ml) y agua (20 ml). El material se extrajo con acetato de etilo (3 x 20 ml). Las capas orgánicas combinadas se lavaron con salmuera (2 x 20 ml) y se secaron sobre sulfato de sodio, luego se filtraron y se concentraron al vacío. El residuo se purificó por HPLC preparativa (método 10) para obtener el com puesto de l título (116 mg, 51 %) como un sólido blanco. 5h (500 MHz, DMSO-de) 10,39 (s, 1H), 10,22 (s, 1H), 8,52 (d, J 8,6 Hz, 1H), 7,45 (d, J 2,1 Hz, 1H), 7,43 (d, J 8,2 Hz, 1H), 7,38 (d, J 1,9 Hz, 1 H), 7,12 (dd, J 8,1, 1,9 Hz, 1H), 7,05 (d, J 2,1 Hz, 1H), 4,44 (t, J 8,9 Hz, 1H), 4,05-3,96 (m, 2H), 4,03 (s, 3H), 3,80 (ddd, J 11,0, 6,9, 3,7 Hz, 2H), 2,22-2,09 (m, 1H), 1,77-1,34 (m, 18H). uPLC-MS (método 2): MH+ m /z 494, TR 3,29 min.
Ejemplo 4

N-{(1 S o 1 R)-1 -Ciclooctil-2-oxo-2-[(2-oxospiro[indolina-3,4'-tetrahidropiran]-6-il)-amino]etil}-2-metilpirazol-3-carboxamida
El ejem plo 3 (100 mg) se separó mediante el método 15 de SFC preparativa quiral, columna ChiralPak IC de 20 x 250 mm, 5 pm, velocidad de flujo de 100 ml/min, gradiente del 25-40 % de MeOH (+0,1 % de NH4OH), para producir el com puesto de l título (eutómero) (25 mg) como un polvo blanquecino. Método 13 de SFC quiral, columna ChiralPak IC de 4,6 x 150 mm, 3 pm, velocidad de flujo de 3,5 ml/min, gradiente del 25-40 % de MeOH (+0,1 % de NH4OH): TR 3,76 min (100% de ee, eutómero).
Ejemplo 5

2-{[2-(2-Aminopiridin-4-il)acetil]amino}-2-ciclooctil-W-(2-oxospiro[indolina-3,4'-tetrahidropiran]-6-il)acetamida Se añadió ácido trifluoroacético (0,18 ml, 2,34 mmol) a una solución agitada del in term edio 19 (142 mg, 0,23 mmol) en DCM (5 ml) a 20 °C en una atmósfera de nitrógeno. La mezcla de reacción se agitó a 20 °C durante la noche y se diluyó con diclorometano (25 ml), luego se inactivó cuidadosamente con una solución acuosa saturada de bicarbonato de sodio (30 ml) y se extrajo con isopropanol/cloroformo 1:1 (3 x 30 ml). Las capas orgánicas combinadas se secaron sobre sulfato de sodio, luego se filtraron y se concentraron al vacío. El residuo se separó por HPLC preparativa (método 10) para producir el com puesto de l título (42,9 mg, 36 %) como un sólido blanco. 5h (500 MHz, DMSO-d6) 10,38 (s, 1H), 10,13 (s, 1 H), 8,32 (d, J 8,9 Hz, 1 H), 7,78 (d, J 5,4 Hz, 1 H), 7,42 (d, J 8,1 Hz, 1H), 7,35 (d, J 1,8 Hz, 1H), 7,09 (dd, J 8,1, 1,8 Hz, 1H), 6,45-6,42 (m, 1 H), 6,37 (s, 1 H), 6,03 (br s, 2H), 4,30 (t, J 8,4 Hz, 1H), 4,00 (ddd, J 11,0, 7,1, 3,6 Hz, 2H), 3,79 (dd, J 11,0, 6,9, 3,7 Hz, 2H), 3,43 (d, J 13,8 Hz, 1H), 3,37-3,24 (m, 1H, enmascarado por el pico del agua), 2,01 -1,92 (m, 1H), 1,77-1,70 (m, 2H), 1,68-1,26 (m, 16H). uPLC-MS (método 1): MH+ m lz 520, TR 2,04 min. Ejemplo 6

2-Ciclooctil-W-(2-oxospiro[indolina-3,4'-tetrahidropiran]-6-il)-2-{[2-(piridin-4-il)-acetil]amino}acetamida Se añadió HATU (228 mg, 0,6 mmol) a una suspensión agitada del in term edio 21 (171 mg, 0,55 mmol) y del interm edio 2 (126 mg, 0,58 mmol) en DMF anhidra (2,9 ml). La mezcla se agitó a 20 °C en nitrógeno durante 16 h, luego se inactivó con una solución acuosa saturada de carbonato de sodio (6 ml) y agua (6 ml) y se agitó a 20 °C durante 30 min. La mezcla se diluyó con agua (12 ml) y luego se extrajo el material con DCM/isopropanol 4:1 (3 x 15 ml), usando una frita hidrófoba para separar las fases. El filtrado orgánico se concentró al vacío. La goma naranja en bruto resultante se separó por HPLC preparativa (método 9) para producir el com puesto de l título (268 mg, 96 %) como un polvo blanquecino después de la liofilización. 5h (500 MHz, DMSO-d6) 10,38 (br s, 1H), 10,14 (br s, 1H), 8,52-8,39 (m, 3H), 7,42 (d, J 8,1 Hz, 1 H), 7,35 (d, J 1,8 Hz, 1H), 7,30-7,25 (m, 2H), 7,08 (dd, J 8,1, 1,8 Hz, 1H), 4,31 (t, J 8,3 Hz, 1H), 4,00 (ddd, J 11,0, 7,1,3,6 Hz, 2H), 3,79 (dd, J 11,0, 7,0, 3,7 Hz, 2H), 3,60 (d, J 14,0 Hz, 1H), 3,53 (d, J 14,0 Hz, 1H), 2,02-1,91 (m, 1H), 1,78-1,68 (m, 2H), 1,68-1,26 (m, 16H). uPLC-MS (método 2): MH+ m /z 505, TR 2,97 min. Ejemplo 7

(2S o 2fi)-2-Ciclooctil-W-(2-oxospiro[indolina-3,4'-tetrahidropiran]-6-il)-2-{[2-(piridin-4-il)acetil]amino}acetamida
El ejem plo 6 (268 mg) se separó mediante el método 14 de SFC preparativa quiral (18:82 metanol-dióxido de carbono, columna Chiralcel OD-H de 25 cm a 15 ml/min) para producir el com puesto de l título (eutómero) (65 mg) como un
polvo blanquecino después de la liofilización. Método 12 de SFC quiral, Chiralcel OD-H 25 cm, metanol al 20 %-dióxido de carbono al 80 %, 4 ml/min: TR 7,97 min (100 %).
Ejemplo 8

2-Metil-W-12-oxo-2-[(2-oxospiro[indolina-3,4'-tetrahidropiran]-6-il)amino]etil}-pirazol-3-carboxamida
Se añadió HATU (67 mg, 0,18 mmol) a una solución agitada del in term edio 2 (32 mg, 0,15 mmol) y del in term edio 23 (30 mg, 0,16 mmol) en DMF anhidra (1 ml) en una atmósfera de nitrógeno. La mezcla se agitó a 20 °C durante 18 h y luego se inactivó mediante la adición de una solución acuosa saturada de bicarbonato de sodio (7 ml) y agua (7 ml). El material se extrajo con acetato de etilo (3 x 7 ml). Las capas orgánicas combinadas se lavaron con salmuera (2 x 7 ml) y se secaron sobre sulfato de sodio, luego se filtraron y se concentraron al vacío. El residuo se separó por HPLC preparativa (método 10) para producir el com puesto de l título (16 mg, 27 %) como un sólido blanco. 5h (500 MHz, DMSO-de) 10,41 (s, 1H), 10,07 (s, 1H), 8,81 (t, J 5,9 Hz, 1H), 7,48 (d, J 2,0 Hz, 1H), 7,44 (d, J 8,1 Hz, 1H), 7,36 (d, J 1.7 Hz, 1 H), 7,08 (dd, J 8,1, 1,8 Hz, 1 H), 6,92 (d, J 2,0 Hz, 1H), 4,05 (s, 3H), 4,03-3,97 (m, 4H), 3,80 (ddd, J 11,1,7,0, 3.7 Hz, 2H), 1,77-1,69 (m, 2H), 1,67-1,58 (m, 2H). uPLC-MS (método 1): MH+ m/z 384, TR 1,60 min.
Ejemplo 9

W-{1-Ciclooctil-2-oxo-2-[(2-oxospiro[indolina-3,4'-tetrahidrotiopirán]-6-il)amino]-etil}-2-metilpirazol-3-carboxamida
Se añadió HATU (10 mg, 0,03 mmol) a una solución agitada del in term edio 28 (4,8 mg, 0,014 mmol) y del interm edio 13 (6 mg, 0,02 mmol) en DMF anhidra (0,2 ml). La mezcla se agitó a 20 °C en nitrógeno durante 16 h, luego se inactivó con una solución acuosa saturada de carbonato de sodio (10 ml) y se agitó durante 30 min a 20 °C. La mezcla se repartió con DCM/isopropanol 4:1 (10 ml). La fase orgánica se separó usando una frita hidrófoba y luego la capa acuosa se extrajo con 4:1 DCM/isopropanol (2 x 10 ml). El filtrado se concentró al vacío. La película de color canela en bruto resultante se separó mediante HPLC preparativa (método 8) para proporcionar el com puesto de l título (0,5 mg, 6 %) como un polvo blanco. 5h (500 MHz, CDCh) 10,32 (br s, 1 H), 8,36 (s, 1H), 8,17 (s, 1 H), 7,91 (d, J 6,9 Hz, 1H), 7,40 (d, J 2,1 Hz, 1 H), 7,22 (d, J 8,0 Hz, 1 H), 6,87 (d, J 2,1 Hz, 1 H), 6,67 (dd, J 8,0, 1,9 Hz, 1H), 4,41 -4,30 (m, 1H), 4,13 (s, 3H), 3,32-3,16 (m, 2H), 2,78-2,65 (m, 2H), 2,35-2,24 (m, 1 H), 2,13-1,99 (m, 2H), 1,98-1,82 (m, 3H), 1,77 1,11 (m, 13H). uPLC-MS (método 1): MH+ m/z 510, TR 3,54 min.
Ejemplo 10

M-{1-Ciclooctil-2-oxo-2-[(2-oxospiro[indolina-3,4'-piperidín]-6-il)amino]etil}-2-metilpirazol-3-carboxamida
Una suspensión del in term edio 31 (34,5 mg, 0,06 mmol) y paladio al 10 % sobre carbono (humedad acuosa del 50 %, 25 mg, 0,012 mmol) en etanol (4 ml) se agitó en una atmósfera de hidrógeno a 20 °C durante 18 h. Los sólidos se retiraron por filtración a través de un lecho de tierra de diatomeas, lavando con etanol (8 x 25 ml). El filtrado se concentró al vacío. El polvo de color canela en bruto resultante se separó mediante HPLC preparativa (método 11), luego las fracciones que contenían el producto se trataron con una solución acuosa saturada de carbonato de sodio (pH 11) y se extrajeron con DCM/isopropanol 4:1 (3 x 20 ml) usando una frita hidrófoba. El filtrado orgánico se concentró al vacío para proporcionar el com puesto de l títu lo (12 mg, 39 %) como un polvo blanco después de la liofilización. 5h (500 MHz, DMSO-de) 10,31 (s, 1H), 10,21 (s, 1H), 8,53 (d, J 8,5 Hz, 1H), 7,45 (d, J 2,0 Hz, 1H), 7,39 (d, J 8,1 Hz, 1 H), 7,37 (d, J 1,7 Hz, 1H), 7,11 (dd, J 8,2, 1,9 Hz, 1 H), 7,05 (d, J 2,1 Hz, 1 H), 4,44 (t, J 8,9 Hz, 1H), 4,03 (s, 3H), 3,10-2,99 (m, 2H), 2,94-2,83 (m, 2H), 2,21-2,11 (m, 1H), 1,73-1,34 (m, 18H). uPLC-MS (método 1): MH+ m /z
493, TR 2,09 min.
Ejemplo 11

2-Acetamido-2-ciclooctil-ftL(2-oxospiro[indolina-3,4'-tetrahidropiran]-6-il)acetamida
Se añadió cloruro de acetilo (15 pl, 0,21 mmol) a una solución del in term edio 35 (70 mg, 0,18 mmol) y DIPEA (63 pl, 0,36 mmol) en DCM (2 ml) y la solución se agitó a 20 °C durante 18 h. Se le añadió más cloruro de acetilo (7,5 pl, 0,11 mmol) y la mezcla de reacción se agitó a 20 °C durante 2 h más. A la mezcla de reacción se le añadieron una solución acuosa saturada de hidrogenocarbonato de sodio (20 ml) e isopropanol-cloroformo 1:1 (20 ml). Se separaron las capas y la capa acuosa se extrajo con isopropanol-cloroformo 1:1 (2 x 20 ml). Las capas orgánicas combinadas se secaron sobre sulfato de magnesio, luego se filtraron y se concentraron al vacío. El residuo se separó por HPLC preparativa (método 9) para obtener el com puesto de l títu lo (34,1 mg, 44 %) como un sólido blanco. 5h (500 MHz, DMSO-d6) 10,38 (s, 1 H), 10,09 (s, 1H), 8,06 (d, J 8,8 Hz, 1H), 7,42 (d, J 8,1 Hz, 1H), 7,36 (d, J 1,8 Hz, 1H), 7,09 (dd, J 8,1, 1,9 Hz, 1H), 4,29 (t, J 8,5 Hz, 1H), 4,00 (ddd, J 11,1,7,2, 3,6 Hz, 2H), 3,80 (ddd, J 11,1,6,9, 3,7 Hz, 2H), 1,94 (s, 1H), 1,87 (s, 3H), 1,77-1,68 (m, 2H), 1,69-1,27 (m, 16H). uPLC-MS (método 2): MH+ m/z 428, TR 2,94 min.
Ejemplo 12

M-{1-Ciclooctil-2-[(5-fluoro-2-oxospiro[indolina-3,4'-tetrahidropiran]-6-il)amino]-2-oxoetil}-2-metilpirazol-3-carboxamida
Se añadió Selectfluor™ (35 mg, 0,101 mmol) a una solución del ejem plo 3 (50 mg, 0,101 mmol) en DMF anhidra (1 ml). La mezcla de reacción se agitó a temperatura ambiente durante 16 h, y luego se le añadieron una solución acuosa saturada de bicarbonato de sodio (5 ml) e isopropanol-cloroformo 1:1 (20 ml). Se separaron las capas y la capa acuosa se extrajo con isopropanol-cloroformo 1:1 (2 x 20 ml). Las capas orgánicas combinadas se secaron sobre sulfato de magnesio, se filtraron y se concentraron al vacío. El residuo se separó por HPLC preparativa (método 9) para obtener el com puesto de l títu lo (13,1 mg, 25 %) como un sólido blanco. 5h (500 MHz, DMSO-d6) 10,34 (s, 1H), 9,94 (s, 1H), 8,48 (s, 1H), 7,50 (d, J 10,4 Hz, 1 H), 7,45 (d, J 2,0 Hz, 1H), 7,39 (d, J 6,5 Hz, 1 H), 7,02 (d, J 2,0 Hz, 1 H), 4,59 (d, J 8,6 Hz, 1H), 4,03 (s, 3H), 4,02-3,96 (m, 2H), 3,84-3,72 (m, 2H), 2,22-2,14 (m, 1H), 1,75-1,61 (m, 7H), 1,59-1,38 (m, 11 H).
19F-RMN (250 MHz, DMSO-de) 5 -132,6. uPLC-MS (método 1) MH+ m/z 512, TR 3,21 min.
Ejemplo 13

-Metil-M-{2-oxo-2-r(2-oxospiro[indolina-3,4'-tetrahidropiran]-6-il)amino]-1-(espiro-[3,3]heptán-2-il)etil}pirazol-3-carboxamida
A una solución agitada del in term edio 2 (25 mg, 0,115 mmol) y del in term edio 39 (30 mg, 0,11 mmol) en THF anhidro (1,1 ml) se le añadió ácido acético (0,066 ml, 11,50 mmol). La mezcla de reacción se agitó a 60 °C durante 3 h, luego se le eliminó el solvente al vacío. El residuo se purificó por HPLC preparativa (método 11) para producir el com puesto de l título (24,6 mg, 44 %) como un sólido blanco. 5h (500 MHz, DMSO-d6) 10,40 (s, 1H), 10,10 (s, 1H), 8,46 (d, J 7,6 Hz, 1 H), 7,46 (d, J 2,0 Hz, 1H), 7,43 (d, J 8,1 Hz, 1H), 7,36 (d, J 1,8 Hz, 1H), 7,10 (dd, J 8,2, 1,9 Hz, 1H), 7,01 (d, J 2,1 Hz, 1 H), 4,43 (dd, J 9,9, 7,6 Hz, 1 H), 4,02 (s, 3H), 4,02-3,96 (m, 2H), 3,80 (ddd, J 11,0, 7,0, 3,7 Hz, 2H), 2,60-2,53 (m, 1H), 2,12 (ddd, J 11,2, 7,8, 3,7 Hz, 1H), 2,02-1,92 (m, 4H), 1,91-1,83 (m, 2H), 1,79-1,74 (m, 3H), 1,74-1,70 (m, 2H),
1,65-1,58 (m, 2H). uPLC-MS (método 1): MH+ m iz 478, TR 2,87 min.
Ejemplo 14

2-Etil-N-{(1 S)-2-[(4-fluoro-2-oxospiro[indolina-3,4'-tetrahidropiran]-6-il)amino]-1-(4-metilciclohexil)-2-oxoetil}pirazol-3-carboxamida (isómero trans)
Se añadió DIPEA (70 pl, 0,42 mmol) a una suspensión de ácido 1 -etil-1 H-pirazol-5-carboxílico (40 mg, 0,29 mmol) y HATU (105 mg, 0,28 mmol) en DCM (1 ml) a 20 °C en nitrógeno. La suspensión se agitó a 20 °C en nitrógeno durante 1 h. Se le añadió una solución del in term edio 52 (81,14 mg, 0,21 mmol) en DCM (1 ml). La mezcla se agitó a 20 °C en nitrógeno durante 16 h, luego se diluyó con DCM (20 ml) y se inactivó con una solución acuosa saturada de carbonato de sodio (10 ml) y agua (10 ml). La mezcla bifásica se agitó a 20 °C durante 30 min y luego la fase orgánica se separó utilizando una frita hidrófoba. La capa acuosa se extrajo con DCM (3 x 20 ml) y el filtrado orgánico se concentró al vacío. El polvo de color canela resultante se separó mediante HPLC preparativa secuencial (método 10), y luego cromatografía en columna ultrarrápida, utilizando un gradiente de acetato de etilo en heptano (25-100 %), para proporcionar, después de la liofilización, el com puesto de l título (26,8 mg, 25 %) como un polvo blanco. 5h (500 MHz, DMSO-d6) 10,59 (s, 1H), 10,35 (s, 1H), 8,52 (d, J 7,9 Hz, 1H), 7,47 (d, J 2,0 Hz, 1H), 7,12-7,06 (m, 2H), 7,01 (d, J 2,0 Hz, 1H), 4,45 (q, J 7,2 Hz, 2H), 4,30 (t, J 8,5 Hz, 1 H), 4,06 (t, J 10,2 Hz, 2H), 3,80-3,69 (m, 2H), 2,01 (ddd, J 14,3, 10,5, 4,4 Hz, 2H), 1,90-1,73 (m, 2H), 1,73-1,64 (m, 4H), 1,60-1,48 (m, 1H), 1,35-1,22 (m, 4H), 1,22-1,11 (m, 1 H), 1,08-0,97 (m, 1H), 0,93-0,80 (m, 5H). uPLC-MS (método 1): MH+ m iz 512, TR 3,36 min.
Ejemplo 15

2-(5-Metoxibiciclo[4.2.0]octa-1,3,5-trién-7-iliden)-2-[(3-metilisoxazol-4-il)-formamido]-N-(2-oxo-1,2-dihidroespiro[indol-3,4'-oxanel-6-il)acetamida (isómero 1)
Se añadió ácido acético (0,1 ml, 1,75 mmol) a una solución del in term edio 56 (50 mg, 0,17 mmol) y del in term edio 57 (89 %, 42 mg, 0,17 mmol) en THF anhidro (1,1 ml). La mezcla de reacción se agitó a 60 °C durante 18 h, luego se enfrió a temperatura ambiente y se concentró al vacío. El residuo se purificó por HPLC preparativa (método 11) para proporcionar, después de la liofilización, el com puesto de l título (20,3 mg, 22 %) como un sólido blanco. 3h (500 MHz, DMSO-de) 10,41 (br s, 1 H), 9,80 (br s, 1H), 9,59 (br s, 1 H), 9,46 (s, 1 H), 7,50 (d, J 1,9 Hz, 1 H), 7,45 (d, J 8,2 Hz, 1H), 7,36 (dd, J 8,4, 7,2 Hz, 1H), 7,24 (dd, J 8,2, 1,9 Hz, 1H), 6,91 (d, J 7,1 Hz, 1H), 6,87 (d, J 8,6 Hz, 1H), 4,03 (ddd, J 11,0, 7,1,3,6 Hz, 2H), 3,91 (s, 2H), 3,82 (ddd, J 11,0, 6,9, 3,6 Hz, 2H), 3,57 (s, 3H), 2,42 (s, 3H), 1,87-1,72 (m, 2H), 1,70-1,56 (m, 2H). HPLC-MS (método 1): MH+ m iz 515, TR 2,66 min.
Ejemplo 16

(2S)-2-[(1 -Metil-1 H-pirazol-5-il)formamido]-N-(2-oxo-1,2-dihidrospiro[indol-3,4'-oxan]-6-il)-2-[(1 r,4S)-4-metilciclohexil]acetamida (isómero trans)
A una solución agitada del in term edio 50 (370 mg, 1 mmol) en DMF seco (8 ml) se le añadió ácido 1 -metil-1 H-pirazol-5-carboxílico (127 mg, 1,01 mmol), y luego HATU (455 mg, 1,2 mmol) y DIPeA (0,5 ml, 3,03 mmol), a t.a. La mezcla de reacción se agitó durante al menos 18 h. Con enfriamiento externo, se le añadió agua (40 ml) a la mezcla de reacción. Se filtró el sólido precipitado y se lavó con agua (2 x 10 ml). La torta del filtro se disolvió en acetato de etilo
(20 ml) y se retiró el exceso de agua. La capa orgánica se lavó con una mezcla 1:1 de agua y salmuera (10 ml), y salmuera (10 ml), luego se secó sobre sulfato de magnesio, se filtró y se concentró al vacío. El residuo bruto se adsorbió sobre gel de sílice (3,1 g), usando DCM, y se purificó por cromatografía automatizada, usando un gradiente de acetato de etilo en heptano (0-100 %), y luego un gradiente de metanol en acetato de etilo (0-7 %). El material aislado se trituró en una mezcla de acetato de etilo (2 ml) y heptano (20 ml), luego se filtró, se lavó con heptano (4 x 10 ml) y se secó para obtener el com puesto de l título (279 mg, 58 %) como un sólido blanco. 5h (500 MHz, DMSO-d6) 10,38 (s, 1H), 10,17 (s, 1H), 8,47 (d, J 8,1 Hz, 1 H), 7,45 (d, J 2,0 Hz, 1 H), 7,43 (d, J 8,1 Hz, 1H), 7,39 (d, J 1,8 Hz, 1 H), 7,11 (dd, J 8,2, 1,9 Hz, 1 H), 7,05 (d, J 2,1 Hz, 1H), 4,34 (t, J 8,5 Hz, 1H), 4,04-3,96 (m, 5H), 3,84-3,75 (m, 2H), 1,85 (d, J 12,6 Hz, 1H), 1,81-1,65 (m, 5H), 1,65-1,52 (m, 3H), 1,34-1,12 (m, 2H), 1,09-0,97 (m, 1H), 0,93-0,79 (m, 5H). uPLC-MS (método 1): MH+ m/z 480, TR 3,01 min. SfC quiral (método 12, Chiralcel OD-H 25 cm, metanol al 10 %, dióxido de carbono al 90 %, 4 ml/min): TR 15,31 min (100 %).
Ejemplo 17

W-{1-(1-Adamantilmetil)-2-oxo-2-[(2-oxospiro[indolina-3,4'-tetrahidropiran]-6-il)-amino]etil}-2-metilpirazol-3-carboxamida
A una suspensión agitada del in term edio 63 (20 mg, 0,038 mmol) en una mezcla de THF (1 ml) y etanol (1 ml) se le añadió paladio al 10 % sobre carbón vegetal (humedad del 50 %, 4 mg, 20 % en peso) como una sola porción. La mezcla de reacción se transfirió a un recipiente a presión y se colocó en una atmósfera de hidrógeno gaseoso. Se continuó agitando a temperatura ambiente durante 24 h en una presión de hidrógeno de 7 bar. Se le añadió una segunda alícuota de paladio al 10 % sobre carbón vegetal (humedad del 50 %, 4 mg, 20 % en peso) como una sola porción y se continuó agitando a temperatura ambiente durante 30 h en una presión de hidrógeno de 7 bar. Se le añadió una tercera alícuota de paladio al 10 % sobre carbón vegetal (50 % húmedo, 4 mg, 20 % en peso) como una sola porción y se continuó agitando a temperatura ambiente durante 70 h en una presión de hidrógeno de 7 bares (tiempo total de reacción: 124 h). El catalizador se eliminó por filtración sobre tierra de diatomeas, enjuagando la torta del filtro con THF (2 x 5 ml). El solvente se concentró al vacío para proporcionar el com puesto de l títu lo (9,5 mg, 42 %) como un sólido beige. 5h (500 MHz, DMSO-d6) 10,40 (s, 1H), 10,07 (s, 1H), 8,58 (d, J 7,9 Hz, 1H), 7,46 (d, J 2,0 Hz, 1 H), 7,43 (d, J 8,2 Hz, 1H), 7,36 (d, J 1,8 Hz, 1H), 7,13 (dd, J 8,2, 1,9 Hz, 1H), 7,00 (d, J 2,1 Hz, 1H), 4,65 (td, J 8,4, 3,8 Hz, 1 H), 4,03 (s, 3H), 4,04-3,97 (m, 2H), 3,80 (ddd, J 11,3, 7,2, 3,8 Hz, 2H), 1,91 (s, 3H), 1,76-1,69 (m, 3H), 1,68-1,62 (m, 6H), 1,57-1,50 (m, 7H), 1,23 (s, 2H). uPLC-MS (método 2): MH+ m /z 532, TR 3,54 min.
Ejemplo 18

M-{2-(Biciclo[1.1.1]pentán-1 -il)-1 -[(2-oxospiro[indolina-3,4'-tetrahidropiran]-6-il)-carbamoil]prop-1 -enil}-2-metilpirazol-3-carboxamida
A una solución agitada del in term edio 2 (50 mg, 0,229 mmol) y del in term edio 64 (59 mg, 0,229 mmol) en THF anhidro (2,3 ml) se le añadió ácido acético (0,131 ml, 2,29 mmol). La mezcla de reacción se agitó a 60 °C durante 16 h. Se eliminó el solvente al vacío, y el residuo se trituró en una mezcla 1:1 de DCM y éter dietílico (1 ml). El sólido se recogió por filtración, se enjuagó la torta con éter dietílico (2 x 0,5 ml) y luego se secó al vacío para proporcionar el com puesto de l títu lo (79,3 mg, 71 %) como un sólido beige. 5h (500 MHz, DMSO-d6) 10,37 (s, 1H), 10,08 (s, 1H), 9,55 (s, 1H), 7,51 (d, J 2,1 Hz, 1H), 7,49 (d, J 1,6 Hz, 1H), 7,43 (d, J 8,2 Hz, 1 H), 7,16 (dd, J 8,2, 1,8 Hz, 1H), 7,07 (d, J 2,1 Hz, 1 H), 4,02 (s, 3H), 4,02-3,97 (m, 2H), 3,80 (ddd, J 11,0, 7,0, 3,7 Hz, 2H), 2,45 (s, 1 H), 2,02 (s, 6H), 1,81 (s, 3H), 1,74 (ddd, J 12,9, 6,8, 3,5 Hz, 2H), 1,62 (ddd, J 13,1,7,0, 3,7 Hz, 2H). uPLC-MS (método 1): MH+ m /z 476, TR 2,69 min.
Ejemplo 19

W-{1-Cicloheptiliden-2-oxo-2-(2-oxospiro[indolina-3,4'-tetrahidropiran]-6-il)amino]-etil}-2-metilpirazol-3-carboxamida
A una solución agitada del in term edio 2 (50 mg, 0,229 mmol) y del in term edio 65 (60 mg, 0,229 mmol) en THF anhidro (2,3 ml) se le añadió ácido acético (0,131 ml, 2,29 mmol). La mezcla de reacción se agitó a 60 °C durante 6 h. Se le añadió ácido acético (0,131 ml, 2,29 mmol) y se continuó calentando a 60 °C durante 21 h más. Se eliminó el solvente al vacío y el residuo se purificó por HPLC preparativa de pH bajo (método 11) para proporcionar, después de la liofilización, el com puesto de l títu lo (35,4 mg, 32 %) como un sólido blanco. 5h (500 MHz, DMSO-d6) 10,36 (s, 1H), 9,88 (s, 1 H), 9,43 (s, 1H), 7,49 (d, J 2,0 Hz, 1H), 7,46 (s, 1H), 7,41 (d, J 8,2 Hz, 1H), 7,14 (dd, J 8,2, 1,7 Hz, 1H), 7,07 (d, J 1,9 Hz, 1 H), 4,01 (s, 3H), 4,04-3,98 (m, 2H), 3,80 (ddd, J 11,1,7,0, 3,7 Hz, 2H), 2,56-2,53 (m, 2H), 2,43-2,39 (m, 2H), 1,73 (ddd, J 12,9, 6,9, 3,6 Hz, 2H), 1,66-1,58 (m, 6H), 1,52 (s, 4H). uPLC-MS (método 1): MH+ m /z 478, TR 2,74 min.
Ejemplo 20 (procedimiento I)

W-{1-Cicloheptil-2-oxo-2-[(2-oxospiro[indolina-3,4'-tetrahidropiran]-6-il)amino]-etil}-2-metilpirazol-3-carboxamida
A una solución agitada del in term edio 2 (30 mg, 0,137 mmol) y del in term edio 66 (60 %, 60 mg, 0,137 mmol) en THF anhidro (1,4 ml) se le añadió ácido acético (0,131 ml, 2,29 mmol). La mezcla de reacción se agitó a 60 °C durante 16 h. Se eliminó el solvente al vacío, y el residuo se purificó por HPLC preparativa de pH bajo (método 11) para proporcionar, después de la liofilización, el com puesto de l título (23,8 mg, 36 %) como un sólido blanco. 3h (500 MHz, DMSO-de) 10,39 (s, 1H), 10,19 (s, 1H), 8,48 (d, J 8,5 Hz, 1H), 7,45 (d, J 2,1 Hz, 1H), 7,43 (d, J 8,1 Hz, 1H), 7,38 (d, J 1,9 Hz, 1 H), 7,12 (dd, J 8,1,2,0 Hz, 1H), 7,05 (d, J 2,1 Hz, 1H), 4,45 (t, J 8,6 Hz, 1H), 4,02 (s, 3H), 4,00 (ddd, J 11,0, 7,1,3,8 Hz, 2H), 3,80 (ddd, J 11,0, 7,0, 3,7 Hz, 2H), 2,11-2,03 (m, 1H), 1,73 (ddd, J 13,0, 6,8, 3,5 Hz, 3H), 1,67-1,59 (m, 5H), 1,57-1,29 (m, 8H). uPLC-MS (método 1): MH+ m lz 480, TR 2,95 min.
Ejemplo 21

W-{1-(Biciclo[3.2.1]octán-3-il)-2-oxo-2-[(2-oxospiro[indolina-3,4'-tetrahidropiran]-6-il)amino]etil}-2-metilpirazol-3-carboxamida (isómero único)
A una solución agitada del in term edio 2 (80 mg, 0,367 mmol) y del in term edio 68 (90 %, 111 mg, 0,367 mmol) en THF anhidro (3,7 ml) se le añadió ácido acético (0,210 ml, 3,67 mmol). La mezcla de reacción se agitó a 60 °C durante 16 h. Se eliminó el solvente al vacío, y el residuo se purificó por HPLC preparativa de pH bajo (método 11). Después de la liofilización, el sólido blanco resultante (91,3 mg) se separó aún más utilizando SFC quiral (método 14, Chiralcel OD-H 25 cm, metanol al 20 %, dióxido de carbono al 80 %, 15 ml/min) para producir el com puesto de l títu lo (30 mg, 36 %) como un sólido blanco. 5h (500 MHz, DMSO-d6) 10,39 (s, 1H), 10,21 (s, 1H), 8,48 (d, J 8,6 Hz, 1H), 7,45 (d, J 2,0 Hz, 1H), 7,43 (d, J 8,2 Hz, 1H), 7,39 (d, J 1,7 Hz, 1H), 7,15 (dd, J 8,2, 1,8 Hz, 1H), 7,03 (d, J 2,0 Hz, 1H), 4,52 (dd, J 10,8, 8,8 Hz, 1H), 4,04 (s, 3H), 4,00 (ddd, J 11,0, 7,2, 3,5 Hz, 2H), 3,80 (ddd, J 11,0, 6,9, 3,7 Hz, 2H), 2,21-2,11 (m, 3H), 1,90 (dt, J 14,1, 7,2 Hz, 1H), 1,81 -1,75 (m, 1H), 1,73 (ddd, J 12,6, 6,8, 3,5 Hz, 2H), 1,67-1,63 (m, 3H), 1,63-1,58 (m, 2H), 1,57-1,51 (m, 1H), 1,46 (d, J 10,9 Hz, 1H), 1,23 (dd, J 13,7, 5,8 Hz, 2H), 1,20-1,14 (m, 1H). uPLC-MS (método 1): MH+ m /z 492, TR 3,03 min. SFC quiral (método 12, Chiralcel OD-H 25 cm, metanol al 20 %-dióxido de carbono al 80 %, 4 ml/min): TR 9,05 min (99 %).
Ejemplo 22

WL{1-(5-Cloro-7-biciclo[4.2.0]octa-1(6),2,4-trieniliden)-2-oxo-2-[(2-oxospiro-[indolina-3,4'-tetrahidropiran]-6-il)amino]etil}-2-metilpirazol-3-carboxamida (isómero 1)
A una solución agitada del in term edio 2 (60 mg, 0,275 mmol) y del in term edio 69 (80 mg, 0,275 mmol) en THF anhidro (1,8 ml) se le añadió ácido acético (0,158 ml, 2,75 mmol). La mezcla de reacción se agitó a 60 °C durante 16 h. Se eliminó el solvente al vacío, y el residuo se trituró en DCm (1 ml), para proporcionar el com puesto de l títu lo (100 mg, 70 %) como un sólido blanquecino. 3h (500 MHz, DMSO-d6) 10,42 (s, 1H), 10,16 (s, 2H), 7,53 (d, J 2,1 Hz, 1H), 7,50 (d, J 1,9 Hz, 1 H), 7,46 (d, J 8,2 Hz, 1 H), 7,38 (dd, J 8,2, 7,1 Hz, 1 H), 7,33-7,27 (m, 2H), 7,22 (dd, J 8,2, 1,9 Hz, 1H), 7,14 (d, J 2,1 Hz, 1 H), 4,06 (s, 3H), 4,02 (ddd, J 11,0, 7,2, 3,7 Hz, 2H), 3,94 (s, 2H), 3,82 (ddd, J 11,1,7,0, 3,8 Hz, 2H), 1,78-1,72 (m, 2H), 1,64 (ddd, J 13,1,7,0, 3,6 Hz, 2H). uPLC-MS (método 1): MH+ m lz 518, TR 2,71 min.
Ejemplo 23

M-{1-(2,3-Dimetilciclobutiliden)-2-oxo-2-[(2-oxospiro[indolina-3,4'-tetrahidropiran]-6-il)amino]etil}-2-metilpirazol-3-carboxamida (isómero 5)
A una solución agitada del in term edio 2 (50 mg, 0,229 mmol) y del in term edio 70 (56 mg, 0,229 mmol) en THF anhidro (2,3 ml) se le añadió ácido acético (0,131 ml, 2,29 mmol). La mezcla de reacción se agitó a 60 °C durante 16 h. Se eliminó el solvente al vacío y el residuo se purificó por HPLC preparativa de pH bajo (método 11). Después de la liofilización, el sólido blanco resultante (72,5 mg) se separó aún más utilizando SFC quiral (método 14, Chiralpak IC 25 cm, metanol al 25 %, dióxido de carbono al 75 %, 15 ml/min) para obtener el com puesto de l títu lo (4,8 mg, 4,5 %) como un sólido blanco. 3h (500 MHz CD3OD) 7,51 (d, J 2,1 Hz, 1H), 7,41 (d, J 8,2 Hz, 1H), 7,39 (d, J 1,9 Hz, 1H), 7,10 (dd, J 8,1,2,0 Hz, 1 H), 6,92 (d, J 2,1 Hz, 1H), 4,16 (ddd, J 11,5, 7,6, 3,7 Hz, 2H), 4,13 (s, 3H), 3,92 (ddd, J 11,2, 6,8, 3,8 Hz, 2H), 3,33 (dd, J 3,7, 2,0 Hz, 1H), 3,22 (ddd, J 17,5, 8,7, 1,5 Hz, 1H), 2,84 (d, J 17,2 Hz, 1 H), 2,66-2,57 (m, 1H), 1,87 (ddd, J 13,4, 6,7, 3,8 Hz, 2H), 1,76 (ddd, J 13,7, 7,6, 3,8 Hz, 2H), 1,11 (d, J 2,9 Hz, 3H), 1,10 (d, J 2,5 Hz, 3H). uPLC-MS (método 1): MH+ m lz 464 , TR 2,52 min. SFC quiral (método 12, Chiralpak IC 25 cm, 25 % metanol-75 % dióxido de carbono, 4 ml/min): TR 17,86 min (90 %, 100 % ee).
Ejemplo 24 (procedimiento A)

W-{1-Ciclooctil-2-oxo-2-[(2-oxospiro[indolina-3,4'-tetrahidropiran]-6-il)amino]etil}-2-etilpirazol-3-carboxamida
Se añadió HATU (148 mg, 0,39 mmol) a una suspensión agitada del in term edio 35 (100 mg, 0,26 mmol) y ácido 1-etil-1 H-pirazol-5-carboxílico (43,6 mg, 0,31 mmol) en DMF anhidro (1 ml) y DIPEA (107 gl, 0,65 mmol). La suspensión se agitó a 20 °C durante 17 h. La mezcla de reacción se inactivó con una solución acuosa saturada de carbonato de sodio (5 ml) y agua (5 ml), se agitó a 20 °C durante 5 min y luego se extrajo con tert-butilmetiléter (2 x 20 ml). Los extractos orgánicos combinados se lavaron con agua (2 x 10 ml) y salmuera (10 ml), luego se secaron sobre sulfato de sodio, se filtraron y se concentraron al vacío. El material en bruto resultante se purificó por cromatografía en columna ultrarrápida, utilizando un gradiente de tert-butilmetiléter en heptano (0-100 %) seguido de un gradiente de MeOH en tert-butilmetiléter (0-10 %), para proporcionar, después de la liofilización, el com puesto de l títu lo (85,7 mg, 59 %) como un sólido incoloro. 5h (500 MHz, CDCh) 10,22 (br s, 1H), 8,36 (s, 1H), 8,15 (s, 1H), 7,90 (d, J 6,6 Hz, 1H), 7,40 (d, J 2,1 Hz, 1H), 7,29-7,26 (m, 1H), 6,83 (d, J 2,0 Hz, 1H), 6,67 (dd, J 8,1, 1,9 Hz, 1H), 4,62-4,47 (m, 2H), 4,40-4,32 (m,
1H), 4,27-4,19 (m, 2H), 3,96-3,87 (m, 2H), 2,36-2,26 (m, 1H), 1,92-1,67 (m, 8H), 1,57-1,41 (m, 8H), 1,38 (t, J 7,2 Hz, 3H), 1,30-1,09 (m, 2H). uPLC-MS (método 1): MH+ m /z 508,4, TR 3,36 min.
Ejemplo 25

N-{(1 S)-1-Ciclooctil-2-oxo-2-[(2-oxospiro[indolina-3,4'-tetrahidropiran]-6-il)amino]etil}-2-etilpirazol-3-carboxamida
El ejem plo 24 (75 mg) se separó mediante SFC preparativa quiral (método 14, columna Chiralpak IC de 25 cm, 70:30 de dióxido de carbono:MeOH, 15 ml/min) para producir el com puesto de l títu lo (eutómero) (5 mg) como un sólido blanco, junto con el racemato (43 mg). El racemato se separó mediante SFC preparativa quiral (método 14, columna Chiralpak IC de 25 cm, dióxido de carbono:MeOH isocrático 70:30, 15 ml/min) para producir más del com puesto del títu lo (eutómero) (13,4 mg) como un sólido blanco. 3h (500 MHz, CDCh) 10,28 (s, 1H), 8,41 (s, 1H), 8,19 (s, 1H), 7,97 (d, J 7,7 Hz, 1 H), 7,40 (d, J 2,0 Hz, 1H), 7,32-7,18 (m, 1H), 6,83 (d, J 2,0 Hz, 1H), 6,66 (dd, J 8,0, 1,8 Hz, 1 H), 4,59 4,44 (m, 2H), 4,42-4,33 (m, 1 H), 4,28-4,16 (m, 2H), 3,97-3,81 (m, 2H), 2,36 (d, J 8,7 Hz, 1 H), 1,98-1,86 (m, 2H), 1,85 1,68 (m, 6H), 1,65-1,60 (m, 2H), 1,57-1,41 (m, 8H), 1,36 (t, J 7,2 Hz, 3H). uPLC-MS (método 1): MH+ m /z 508,3, TR 3,28 min. SFC quiral (método 12, columna Chiralpak IC de 25 cm, dióxido de carbono:MeOH isocrático 70:30, 4 ml/min) TR 3,91 min (100 %).
Ejemplo 26

N-{-Ciclooctil-2-oxo-2-[(2-oxospiro[indolina-3,4'-tetrahidropiran]-6-il)amino]etil}-2-(2,2-difluoroetil)pirazol-3-carboxamida
Se preparó a partir del in term edio 35 (46 mg, 0,12 mmol) y ácido 2-(2,2-difluoroetil)-pirazol-3-carboxílico (25 mg, 0,14 mmol) de acuerdo con el proced im iento A, y se purificó por cromatografía en columna ultrarrápida usando un gradiente de tert-butilmetiléter en heptano (0-100 %) seguido de un gradiente de MeOH en tert-butilmetiléter (0-20 %), para dar el com puesto de l título (20,6 mg, 29 %) como un sólido incoloro. 3h (500 MHz, CDCh) 10,30 (br s, 1H), 8,14-8,07 (m, 2H), 8,01 (d, J 7,2 Hz, 1H), 7,41 (d, J 2,0 Hz, 1H), 7,22 (d, J 8,0 Hz, 1 H), 6,88 (d, J 2,0 Hz, 1H), 6,60 (dd, J 8,0, 1,8 Hz, 1H), 6,02 (tt, J 56,2, 4,5 Hz, 1H), 4,97-4,72 (m, 2H), 4,27 (t, J 9,0 Hz, 1 H), 4,18-4,11 (m, 2H), 3,90-3,80 (m, 2H), 2,23 (d, J 9,0 Hz, 1 H), 1,88-1,52 (m, 10H), 1,46-1,27 (m, 8H). uPLC-MS (método 1): MH+ m /z 544,3, TR 3,38 min.
Ejemplo 27

N-{1-Ciclooctil-2-oxo-2-[(2-oxospiro[indolina-3,4'-tetrahidropiran]-6-il)amino]etil}-3-etilisoxazol-4-carboxamida Se preparó a partir del in term edio 35 (46 mg, 0,12 mmol) y ácido 3-etilisoxazol-4-carboxílico (20,2 mg, 0,14 mmol) de acuerdo con el proced im iento A, y se purificó por cromatografía en columna ultrarrápida usando un gradiente de tert-butilmetiléter en heptano (0-100 %) seguido de un gradiente de MeOH en tert-butilmetiléter (0-20 %), para dar el com puesto de l título (14,1 mg, 21 %) como un sólido incoloro. 3h (500 MHz, DMSO-d6) 10,31 (s, 1H), 10,14 (s, 1H), 9,32 (s, 1H), 8,41 (d, J 8,7 Hz, 1 H), 7,37 (d, J 8,2 Hz, 1H), 7,31 (d, J 1,8 Hz, 1 H), 7,05 (dd, J 8,1, 1,8 Hz, 1H), 4,39 (t, J 8,7 Hz, 1 H), 3,98-3,89 (m, 2H), 3,79-3,67 (m, 2H), 2,77 (q, J 8,0, 7,5 Hz, 2H), 2,02 (m, 1 H), 1,72-1,31 (m, 18H), 1,10 (t, J 7,5 Hz, 3h ). uPLC-MS (método 1): MH+ m /z 509,3, TR 3,40 min.
Ejemplo 28

WL{2-[(4-Fluoro-2-oxospiro[indolina-3,4'-tetrahidropiran]-6-il)amino]-1-(4-metilciclohexil)-2-oxoetil}-2-metilpirazol-3-carboxamida
Se añadió paladio al 10 % sobre carbón (humedad del 50%, 5,3 mg, 2,47 pl) como una sola porción a una suspensión agitada del in term edio 76 (73 mg, 0,1 mmol) en una mezcla de THF (2,5 ml) y etanol (2,5 ml). La mezcla de reacción se colocó en una atmósfera de hidrógeno gaseoso y se agitó a 20 °C durante 18 h. El catalizador se eliminó por filtración sobre tierra de diatomeas, enjuagando la torta del filtro con MeOH (2 x 15 ml). El solvente se concentró al vacío y el residuo se disolvió en una mezcla de THF (2,5 ml) y etanol (2,5 ml). La solución se trató con paladio al 10 % sobre carbón vegetal (húmedo al 50%, 52,52 mg, 0,02 mmol) como una sola porción. La mezcla de reacción se colocó en una atmósfera de hidrógeno gaseoso y se agitó a 20 °C durante 4 h. El catalizador se eliminó por filtración sobre tierra de diatomeas, enjuagando la torta del filtro con MeOH (2 x 15 ml). El solvente se concentró al vacío. El material en bruto resultante se purificó por HPLC preparativa (método 20) y luego se liofilizó para producir el com puesto de l título (7,2 mg, 14 %) como un sólido blanco. Bh (500 MHz, CDCh) 10,38-10,10 (m, 1H), 8,70-8,41 (m, 1H), 7,94 7,74 (m, 2H), 7,43-7,37 (m, 1H), 6,91 -6,81 (m, 1H), 6,48-6,39 (m, 1H), 4,63-4,31 (m, 1H), 4,28-4,22 (m, 2H), 4,15-4,09 (m, 3H), 3,97-3,88 (m, 2H), 2,29-2,16 (m, 2H), 2,11-1,96 (m, 1H), 1,95-1,83 (m, 1H), 1,80-1,68 (m, 4H), 1,37-1,25 (m, 2H), 1,21-1,06 (m, 2H), 0,99-0,84 (m, 5H). uPLC-MS (método 1): MH+ m /z 498,1, TR 3,18 min.
Ejemplo 29

N-{(1 S)-1-Ciclohexil-2-oxo-2-[(2-oxospirorindolina-3,4'-tetrahidropiran]-6-il)amino]-etil}-2-propilpirazol-3-carboxamida
Se preparó a partir del in term edio 78 (50 mg, 0,14 mmol) y ácido 2-propilpirazol-3-carboxílico (25,9 mg, 0,17 mmol) de acuerdo con el proced im iento A, y se purificó por HPLC preparativa (método 20) para dar el com puesto de l títu lo (33,5 mg, 44 %) como un sólido incoloro. Bh (500 MHz, DMsO-d6) 10,39 (s, 1H), 10,17 (s, 1H), 8,48 (d, J 8,1 Hz, 1H), 7,48 (d, J 2,0 Hz, 1H), 7,44 (d, J 8,1 Hz, 1H), 7,39 (d, J 1,9 Hz, 1 H), 7,12 (dd, J 8,2, 1,9 Hz, 1H), 7,01 (d, J 2,0 Hz, 1H), 4,41 (t, J 7,1 Hz, 2H), 4,38 (t, J 8,6 Hz, 1H), 4,06-3,97 (m, 2H), 3,89-3,75 (m, 2H), 1,92-1,78 (m, 2H), 1,78-1,66 (m, 6H), 1,66-1,53 (m, 4H), 1,26-1,09 (m, 4H), 1,08-0,97 (m, 1 H), 0,77 (t, J 7,4 Hz, 3H). uPLC-MS (método 1): MH+ m /z 494,3, TR 3,04 min.
Ejemplo 30

N-{(1 S)-1-Ciclohexil-2-oxo-2-[(2-oxospiro[indolina-3,4'-tetrahidropiran]-6-il)amino]-etil}-2-isopropilpirazol-3-carboxamida
Se preparó a partir del in term edio 78 (50 mg, 0,14 mmol) y ácido 2-isopropilpirazol-3-carboxílico (25,9 mg, 0,17 mmol) de acuerdo con el proced im iento A, y se purificó por HPLC preparativa (método 20), para dar el com puesto de l título (35,3 mg, 49 %) como un sólido incoloro. Bh (500 MHz, DMSO-de) 10,39 (s, 1H), 10,17 (s, 1 H), 8,45 (d, J 8,1 Hz, 1 H), 7,50 (d, J 1,9 Hz, 1H), 7,44 (d, J 8,1 Hz, 1H), 7,40 (d, J 1,8 Hz, 1 H), 7,12 (dd, J 8,2, 1,9 Hz, 1H), 6,95 (d, J 2,0 Hz, 1 H), 5,39 (hept, J 6,5 Hz, 1 H), 4,36 (t, J 8,5 Hz, 1H), 4,05-3,98 (m, 2H), 3,83-3,74 (m, 2H), 1,89-1,79 (m, 2H), 1,77-1,67 (m, 4H), 1,68-1,54 (m, 4H), 1,37 (d, J 6,6 Hz, 3H), 1,35 (d, J 6,6 Hz, 3H), 1,25-1,09 (m, 4H), 1,09-0,94 (m, 1H). uPLC-MS (método 1): MH+ m /z 494,2, TR 3,05 min.
Ejemplo 31

2-Isopropil-W-{(1S)-1-(4-metilciclohexil)-2-oxo-2-[(2-oxospiro[1HLpirrolo[3,2-c]-piridin-3,4'-tetrahidropiran]-6-il)amino]etil}pirazol-3-carboxamida (isómero tra n s )
Se añadió gota a gota ácido trifluoroacético (0,42 ml, 5,42 mmol) a una solución del in term edio 79 (115 mg, 0,18 mmol) en DCM (1 ml) en una atmósfera de nitrógeno y se enfrió a 0 °C. La mezcla de reacción se agitó a 20 °C durante 18 h. Lo volátil se eliminó al vacío, y el residuo se disolvió en acetonitrilo (1 ml) y una solución acuosa de hidróxido de amonio (1 ml). La mezcla de reacción se agitó a 20 °C durante 15 min, luego se eliminó lo volátil al vacío. El residuo se diluyó con agua (3 ml) y la fase acuosa se extrajo con DCM (3 x 10 ml). Los extractos orgánicos combinados se pasaron a través de una frita hidrófoba y se secaron al vacío. El material bruto resultante se purificó mediante HPLC preparativa (método 20) para proporcionar el com puesto de l títu lo (50,8 mg, 54 %) como un sólido blanco. 5h (500 MHz, DMSO-de) 10,78 (s, 1H), 10,47 (s, 1H), 8,39-8,33 (m, 2H), 7,65 (s, 1H), 7,43 (d, J 1,9 Hz, 1H), 6,85 (d, J 1,9 Hz, 1H), 5,28 (h, J 6,5 Hz, 1H), 4,40 (t, J 8,3 Hz, 1H), 3,97-3,89 (m, 2H), 3,82-3,64 (m, 2H), 1,80-1,67 (m, 4H), 1,64-1,45 (m, 5H), 1,28 (dd, J 14,3, 6,6 Hz, 6H), 1,24-1,10 (m, 2H), 1,00 (q, J 12,1, 11,6 Hz, 1H), 0,88-0,70 (m, 5H). uPLC-MS (método 1 ): MH+ m /z 509,3, TR 2,72 min.
Ejemplo 32

(2S)-2-{[6-(Difluorometil)piridazin-3-il]amino}-2-(4-metilciclohexil)-W-(2-oxospiro[1H-pirrolo[3,2-c]piridin-3,4'-tetrahidropiran]-6-il)acetamida (isómero tra n s )
Se añadió gota a gota ácido trifluoroacético (0,27 ml, 3,45 mmol) a una solución del in term edio 80 (113 mg, 0,06 mmol) en DCM (0,5 ml) en una atmósfera de nitrógeno y se enfrió a 0 °C. La mezcla de reacción se agitó a 20 °C durante 18 h. Lo volátil se eliminó al vacío y el residuo se disolvió en acetonitrilo (1 ml) y una solución acuosa de hidróxido de amonio (1 ml). La mezcla se agitó a 20 °C durante 15 min, luego se eliminó lo volátil al vacío. El residuo se diluyó con agua (3 ml) y la fase acuosa se extrajo con DCM (3 x 10 ml). Los extractos orgánicos combinados se pasaron a través de una frita hidrófoba y se concentraron al vacío. El material en bruto resultante se purificó mediante HPLC preparativa (método 10) para proporcionar el com puesto de l títu lo (0,6 mg, 2 %) como una goma blanquecina. 5h (500 MHz, CDCta) 10,84 (s, 1 H), 9,09-8,77 (m, 1H), 8,32-8,22 (m, 1H), 8,07 (s, 1 H), 7,36-7,29 (m, 1 H), 6,92-6,83 (m, 1H), 6,80-6,48 (m, 2H), 4,81 -4,70 (m, 1 H), 4,16-4,09 (m, 2H), 3,91 -3,75 (m, 2H), 2,02-1,93 (m, 2H), 1,91 -1,84 (m, 1H), 1,81 -1,59 (m, 6H), 1,28-1,20 (m, 2H), 1,16-1,09 (m, 1H), 0,83-0,76 (m, 5H). uPLC-MS (método 1): MH+ m /z 501,2, TR 2,54 min.
Ejemplo 33

M-{1-Ciclooctil-2-oxo-2-[(2-oxospiro[1H-pirrolo[3,2-c]piridin-3,4'-tetrahidropiran]-6-il)amino]etil}-2-metilpirazol-3-carboxamida
A una solución del in term edio 86 (0,20 g, 0,29 mmol) en DCM (5 ml) se le añadió TFA (0,44 ml, 5,76 mmol) a 0 °C. La mezcla de reacción se agitó a temperatura ambiente durante 16 h, luego se concentró al vacío. El residuo bruto se disolvió en acetonitrilo (3 ml) y se le añadió una solución acuosa de hidróxido de amonio (al 25 % en agua, 3 ml) a 0 °C. La mezcla de reacción se agitó a temperatura ambiente durante 3 h, luego se concentró al vacío. El residuo bruto se purificó por cromatografía en columna (amoníaco metanólico al 5 % en DCM), seguido de HPLC preparativa (método 8), para producir el com puesto de l títu lo (0,023 g, 16 %) como un sólido blanco. 5h (400 MHz, DMSO-d6) 1,34
1,73 (m, 16H), 1,77-1,89 (m, 2H), 2,15-2,18 (m, 1H), 3,78-3,84 (m, 3H), 3,95-3,98 (m, 1H), 4,01 (s, 3H), 4,57 (t, J 8,31 Hz, 1 H), 7,01 (s, 1 H), 7,47 (s, 1 H), 7,71 (s, 1H), 8,41 (s, 1 H), 8,45 (d, J 8,31 Hz, 1H), 10,63 (br s, 1H), 10,86 (br s, 1 H). HPLC-MS (método 6): MH+ m /z 495,0, Tr 2,44 min.
Ejemplo 34

WL{1-Ciclooctil-2-oxo-2-[(2-oxospiro[1H-pirrolo[3.2-d]piridin-3,4'-tetrahidropiran]-6-il)amino]etil}-2-metilpirazol-3-carboxamida
A una solución del in term edio 92 (0,16 g, 0,26 mmol) en DCM (5 ml) se le añadió TFA (0,78 ml, 10,2 mmol) a 0 °C. La mezcla de reacción se agitó a temperatura ambiente durante 16 h, y luego se concentró al vacío. Se le añadieron acetonitrilo (4 ml) y una solución acuosa de hidróxido de amonio (25 % en agua, 4 ml) a 0 °C. La mezcla de reacción se agitó a temperatura ambiente durante 3 h y luego se concentró al vacío. El residuo bruto se purificó mediante cromatografía en columna (MeOH al 6-8 % en DCM) y HPLC preparativa (método 8) para proporcionar el com puesto de l títu lo (0,021 g, 16 %) como un sólido blanquecino. 3h (400 m Hz , DMSO-d6) 1,36-1,53 (m, 8H), 1,54-1,69 (m, 7H), 1,75-1,79 (m, 3H), 2,15-2,17 (m, 1H), 3,84-3,94 (m, 2H), 4,02 (s, 3H), 4,05-4,10 (m, 2H), 4,45 (t, J 8,56 Hz, 1H), 7,05 (s, 1H), 7,45 (s, 1H), 7,74 (s, 1H), 8,25 (s, 1 H), 8,60 (d, J 8,31 Hz, 1H), 10,49 (br s, 1H), 10,58 (br s, 1H). HPLC-MS (método 6): MH+ m /z 495,0, TR 2,40 min.
Ejemplo 35
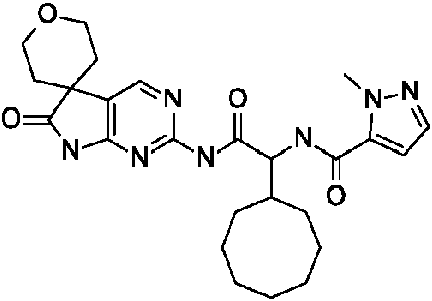
M-{1-Ciclooctil-2-oxo-2-[(6-oxospiro[7H-pirrolo[2,3-d]pirimidín-5,4'-tetrahidropiran]-2-il)amino]etil}-2-metilpirazol-3-carboxamida
A una solución del in term edio 98 (0,25 g, 0,40 mmol) en DCM (5 ml) se le añadió TFA (1,19 ml, 16,0 mmol) a 0 °C. La mezcla de reacción se agitó a temperatura ambiente durante 16 h, luego se concentró al vacío. El residuo se diluyó con acetonitrilo (10 ml) y una solución acuosa de hidróxido de amonio (10 ml) a 0 °C. La mezcla de reacción se agitó a temperatura ambiente durante 3 h y luego se concentró al vacío. El residuo bruto se purificó mediante cromatografía en columna (0-100 % de EtOAc en hexanos) y HPLC preparativa (método 8) para producir el com puesto de l título (0,03 g, 8 %) como un sólido blanco. 5h (400 MHz, DMSO-de) 1,36-1,51 (m, 8H), 1,55 (d, J 10,27 Hz, 2H), 1,61 -1,64 (m, 6H), 1,80-1,93 (m, 2H), 2,15-2,17 (m, 1H), 3,79 (t, J 9,78 Hz, 2H), 3,92-3,99 (m, 2H), 4,02 (s, 3H), 4,61-4,64 (m, 1H), 7,01 (d, J 1,47 Hz, 1 H), 7,46 (d, J 1,47 Hz, 1H), 8,42 (d, J 8,80 Hz, 1H), 8,66 (s, 1H), 10,66 (s, 1H), 11,52 (br s, 1H). HPLC-MS (método 6): MH+ m /z 496,0, TR 3,49 min.
Ejemplo 36

M-{1-Ciclooctil-2-oxo-2-[(2-oxospiro[indolina-3,4'-tetrahidropiran]-6-il)amino]etil}-3-metilisoxazol-4-carboxamida
Se preparó a partir del in term edio 35 y ácido 3-metil-4-isoxazolcarboxílico de acuerdo con el proced im iento A para dar el com puesto de l títu lo (18 %) como un sólido blanco. 5h (400 MHz, DMSO-d6) 10,40 (s, 1H), 10,25 (s, 1H), 9,44 (s, 1H), 8,51 (d, J 8,5 Hz, 1H), 7,44 (d, J 8,1 Hz, 1H), 7,39 (d, J 1,9 Hz, 1H), 7,13 (dd, J 8,1,2,0 Hz, 1H), 4,46 (t, J 8,7 Hz, 1H), 4,01 (ddd, J 11,1,7,1,3,7 Hz, 2H), 3,80 (ddd, J 11,1,7,1,3,8 Hz, 2H), 2,38 (s, 3H), 2,09 (br s, 1H), 1,79-1,36 (m, 18H). HPLC-MS (método 21): MH+ m lz 495, TR 2,17 min.
Ejemplo 37

N-{(1 S)-1-Ciclooctil-2-oxo-2-[(2-oxospiro[indolina-3,4'-tetrahidropiran]-6-il)amino]-etil}-3-metilisoxazol-4-carboxamida
El ejem plo 36 se separó mediante HPLC preparativa quiral utilizando un sistema DAD Agilent 1100 en una columna Lux Cellulose-1 de 21,2 x 250 mm, 5 pm, eluyendo con MeOH al 100 % (+0,1 % de NH4OH) y un caudal de 10 ml/min, para dar el com puesto de l título (2 mg, 2 %) como un sólido blanco. HPLC-MS (método 21): MH+ m /z 495, TR 2,11 min. HPLC quiral en una columna Lux Cellulose-1 de 4,6 x 150 mm, 3 pm, eluyendo con MeOH al 100 % NH4OH al 0,1 %, 1 ml/min, 40 °C, TR 2,20 min (100 %).
Ejemplo 38

N-{(1 S)-1-Ciclohexil-2-oxo-2-[(2-oxospiro[indolina-3,4'-tetrahidropiran]-6-il)amino]etil}-3-metilisoxazol-4-carboxamida
Se preparó a partir del in term edio 78 y ácido 3-metil-4-isoxazolcarboxílico de acuerdo con el proced im iento A para dar el com puesto de l títu lo (216 mg, 79 %) como un sólido blanco. 3h (300 MHz, DMSO-d6) 10,40 (s, 1H), 10,20 (s, 1H), 9,44 (s, 1 H), 8,45 (d, J 8,2 Hz, 1 H), 7,44 (d, J 8,1 Hz, 1H), 7,39 (d, J 1,9 Hz, 1 H), 7,12 (dd, J 8,1,2,0 Hz, 1 H), 4,40 (t, J 8,4 Hz, 1 H), 4,01 (ddd, J 10,9, 7,0, 3,6 Hz, 2H), 3,80 (ddd, J 11,1,6,9, 3,8 Hz, 2H), 2,37 (s, 3H), 1,87-1,51 (m, 10H), 1,17 (m, 5H). HPLC-MS (método 21): MH+ m lz 467, TR 1,81 min.
Ejemplo 39

2-Metil-N-{1-(4-metilciclohexiliden)-2-oxo-2-[(2-oxospiro[indolina-3,4'-tetrahidropiran]-6-il)amino]etil}pirazol-3-carboxamida (isómero 1)
El in term edio 122 se separó mediante HPLC preparativa quiral utilizando un sistema DAD Agilent 1100 en una columna Lux Cellulose-1 de 250 mm x 21,2 mm, 3 pm, eluyendo con MeOH al 12 % (+ 0,1 % de NH4OH) y un caudal de 100 ml/min, para dar el com puesto de l título como un sólido blanco. 5h (400 MHz, DMSO-d6) 10,37 (s, 1H), 10,07 (s, 1H), 9,71 (s, 1 H), 7,48 (m, 2H), 7,42 (d, J 8,2 Hz, 1H), 7,16 (d, J 8,0 Hz, 1H), 7,08 (s, 1H), 4,13-3,93 (m, 5H), 3,81 (ddd, J 11,1,7,2, 3,8 Hz, 2H), 2,80 (d, J 13,9 Hz, 1H), 2,62 (d, J 13,6 Hz, 1H), 2,06 (m, 1H), 1,89 (m, 1H), 1,86-1,54 (m, 2H), 1,28-1,05 (m, 5H), 0,93-0,81 (m 5H). Hp Lc -Ms (método 21): MH+ m /z 478, Tr 1,79 min. SfC quiral (método 35): TR 4,5 min.
Ejemplo 40

W-{1-Ciclooctil-2-[(1-metil-2-oxospiro[indolina-3,4'-tetrahidropiran]-6-il)amino]-2-oxoetil}-3-etilisoxazol-4-carboxamida
Se preparó a partir del in term edio 103 (50 mg, 0,14 mmol) y ácido 3-etilisoxazol-4-carboxílico (14,2 mg, 0,10 mmol) de acuerdo con el proced im iento A, y se purificó por HPLC preparativa (método 22), para dar el com puesto de l título (7 mg, 13 %) como un sólido blanquecino. 5h (400 MHz, DMSO-d6) 10,32 (s, 1H), 9,42 (s, 1H), 8,50 (d, J 8,6 Hz, 1H), 7,53-7,39 (m, 2H), 7,26 (dd, J 8,1, 1,9 Hz, 1H), 4,49 (t, J 8,6 Hz, 1H), 4,04 (ddd, J 11,3, 7,3, 3,8 Hz, 2H), 3,82 (ddd, J 11,1, 6,5, 4,0 Hz, 2H), 3,10 (s, 3H), 2,92-2,77 (m, 2H), 2,15-2,03 (m, 1H), 1,84-1,26 (m, 18H), 1,18 (t, J 7,5 Hz, 3H). HPLC-MS (método 6): MH+ m /z 523, TR 2,33 min.
Ejemplo 41

ferf-Butil-W-{(1S)-1-ciclooctil-2-oxo-2-[(2-oxospiro[indolina-3,4'-tetrahidropiran]-6-il)amino]etil}carbamato
El in term edio 34 (500 mg) se purificó mediante SFC preparativa quiral (método 15). El segundo pico de elución dio el com puesto de l títu lo (133 mg, 27 %) como un sólido blanquecino. 5h (400 MHz, DMSO-d6) 10,38 (s, 1H), 9,99 (s, 1H), 7,43 (d, J 8,1 Hz, 1H), 7,37 (d, J 1,8 Hz, 1 H), 7,08 (dd, J 8,1,2,0 Hz, 1H), 6,89 (d, J 8,8 Hz, 1H), 4,01 (ddd, J 11,1,7,1, 3,7 Hz, 2H), 3,94 (t, J 8,3 Hz, 1H), 3,81 (ddd, J 11,1,7,1,3,8 Hz, 2H), 1,94 (s, 1H), 1,79-1,69 (m, 2H), 1,68-1,25 (m, 25H). HPLC-MS (método 6): [M+2H-tBu]+ m /z 430, TR 2,40 min. uPLC-MS (método 23): [M+2H-tBu]+ m /z 430, TR 2,49 min.
Ejemplo 42 (procedimiento B)

-Ciclooctil-W-(2-oxospiro[indolina-3,4'-tetrahidropiran]-6-il)-2-(piridazin-3-il-amino)acetamida
El in term edio 35 (20 mg, 0,052 mmol), 3-bromopiridazina (8,25 mg, 0,052 mmol), tBuBrettPhos Pd-G3 (4,52 mg, 0,0052 mmol) y tert-butóxido de sodio (10 mg, 0,10 mmol) se añadieron a un vial sellado y se disolvieron en 1,4-dioxano (0,7 ml)/DMSO (0,07 ml). La mezcla de reacción se agitó a 105 °C durante 18 h, luego se filtró a través de Celite y se concentró al vacío. El aceite pardo resultante se purificó por HPLC preparativa (método 24) para proporcionar, después de la liofilización, el com puesto de l título (2 mg, 8 %) como un sólido blanquecino. 5h (400 MHz, DMSO-de) 10,37 (s, 1H), 10,23 (s, 1H), 8,42 (dd, J 4,4, 1,4 Hz, 1H), 7,46-7,36 (m, 2H), 7,23 (dd, J 9,0, 4,4 Hz, 1H), 7,11 (dd, J 8,2, 2,0 Hz, 1H), 6,99 (dt, J 8,7, 1,6 Hz, 2H), 4,66 (t, J 8,1 Hz, 1 H), 4,01 (ddd, J 11,1,7,0, 3,8 Hz, 2H), 3,80 (ddd, J 11,2, 7,1,3,8 Hz, 2H), 1,81-1,34 (m, 19H). HPLC-MS (método 21): [M-H]+ m /z 463, TR 1,77 min.
Ejemplo 43

(2S)-2-Ciclohexil-2-[(6-etilpiridazin-3-il)amino]-W-(2-oxospiro[indolina-3,4'-tetrahidropiran]-6-il)acetamida
Se preparó a partir del in term edio 78 (50 mg, 0,14 mmol) e hidrobromuro de 3-bromo-6-etilpiridazina (31,2 mg, 0,11 mmol) de acuerdo con el procedim iento B, y se purificó por HPLC preparativa (método 22), para dar el com puesto de l títu lo (6 mg, 12 %) como un sólido blanquecino. 5h (300 MHz, DMSO-d6) 10,38 (s, 1H), 10,16 (s, 1H), 7,46-7,36 (m, 2H), 7,21-7,07 (m, 2H), 6,95 (d, J 9,1 Hz, 1H), 6,79 (d, J 8,3 Hz, 1H), 4,55 (t, J 7,8 Hz, 1H), 4,01 (ddd, J 11,1,7,0, 3,7 Hz, 2H), 3,80 (td, J 7,1,3,5 Hz, 2H), 2,67 (q, J 7,6 Hz, 2H), 1,87-1,51 (m, 9H), 1,30-1,01 (m, 9H). HPLC-MS (método 6): MH+ m /z 464, TR 1,80 min.
Ejemplo 44

(2S)-2-Ciclohexil-2-[(6-isopropilpiridazin-3-il)amino]-M-(2-oxospiro[indolina-3,4'-tetrahidropiran]-6-il)acetamida
Se preparó a partir del in term edio 78 (30 mg, 0,084 mmol) y 3-bromo-6-isopropil-piridazina (16,9 mg, 0,084 mmol) de acuerdo con el procedim iento B, y se purificó por HPLC preparativa (método 25), para dar el com puesto de l título (2 mg, 5 %) como un sólido blanquecino. 3h (600 MHz, DMSO-d6) 10,38 (s, 1H), 10,14 (s, 1H), 7,45-7,38 (m, 2H), 7,20 (d, J 9,2 Hz, 1H), 7,12 (dd, J 8,2, 2,0 Hz, 1 H), 6,96 (d, J 9,2 Hz, 1 H), 6,78 (d, J 8,3 Hz, 1 H), 4,54 (t, J 7,9 Hz, 1H), 4,01 (ddd, J 11,2, 7,2, 3,7 Hz, 2H), 3,80 (ddd, J 11,3, 7,2, 3,7 Hz, 2H), 2,98 (m, 1 H), 1,87 (m, 2H), 1,77-1,69 (m, 3H), 1,68 1,55 (s, 3H), 1,37 (m, 1H), 1,23-1,10 (m, 12H). HPLC-MS (método 6): MH+ m /z 478, TR 1,94 min.
Ejemplo 45

(2S)-2-(Cinnolín-3-ilamino)-2-ciclohexil-M-(2-oxospiro[indolina-3,4'-tetrahidropiran]-6-il)acetamida
Se preparó a partir del in term edio 78 (30 mg, 0,084 mmol) y 3-bromocinnolina (14,62 mg, 0,070 mmol) de acuerdo con el proced im iento B, y se purificó por HPLC preparativa (método 25) para dar el com puesto de l título (1,5 mg, 4 %) como un sólido blanquecino. 5h (300 MHz, DMSO-de) 10,38 (s, 1 H), 10,25 (s, 1 H), 8,39 (s, 2H), 8,07 (d, J 8,6 Hz, 1H), 7,67 (d, J 8,3 Hz, 1 H), 7,54 (ddd, J 8,3, 6,6, 1,3 Hz, 1H), 7,49-7,35 (m, 2H), 7,21 -7,06 (m, 2H), 4,66 (t, J 8,0 Hz, 1H), 4,01 (m, 2H), 3,78 (m, 2H), 1,94 (m, 2H), 1,73-1,55 (m, 7H), 1,30-1,10 (m, 6H). HPLC-MS (método 6): MH+ m /z 486, TR 1,97 min.
Ejemplo 46

2-[(6-Etilpiridazin-3-il)amino]-2-(4-metilciclohexil)-M-(2-oxospiro[indolina-3,4'-tetrahidropiran]-6-il)acetamida
Se preparó a partir del in term edio 105 (30 mg, 0,084 mmol) e hidrobromuro de 3-bromo-6-etil-piridazina (38 mg, 0,135 mmol) de acuerdo con el procedim iento B, y se purificó por HPLC preparativa (método 25) para dar el com puesto del títu lo (3 mg, 5 %) como un sólido blanquecino. 3h (400 MHz, DMSO-d6) 10,38 (s, 1H), 10,21 (m, 1H), 7,46-7,30 (m, 2H), 7,24-7,02 (m, 2H), 6,94 (m, 1 H), 6,81 (m, 1H), 4,84-4,42 (m, 1H), 4,01 (ddd, J 11,1,7,1,3,7 Hz, 2H), 3,79 (td, J 7,4, 3,7 Hz, 2H), 2,67 (m, 3H), 1,89 (m, 1H), 1,83-1,34 (m, 7H), 1,17 (m, 6H), 1,03-0,73 (m, 5H). HPLC-MS (método 6): MH+ m /z 478, TR 1,97 min.
Ejemplo 47

2-Etil-W-{(1S)-1-(4-metilciclohexil)-2-oxo-2-[(2-oxospiro[indolina-3,4'-tetrahidropiran-6-il)amino]etil}pirazol-3-carboxamida (isómero trans)
Se preparó a partir del in term edio 50 (30 mg, 0,084 mmol) y ácido 1 -etil-1 H-pirazol-5-carboxílico (49 mg, 0,35 mmol) de acuerdo con el proced im iento A, y se purificó por cromatografía en columna ultrarrápida usando un gradiente de 0 100 % de acetato de etilo en hexanos, para dar el com puesto de l título (81 mg, 47 %) como un sólido blanquecino. 5h (400 MHz, DMSO-d6) 10,40 (s, 1H), 10,18 (s, 1H), 8,50 (d, J 8,1 Hz, 1H), 7,59-7,20 (m, 3H), 7,12 (dd, J 8,2, 2,0 Hz, 1H), 7,02 (d, J 2,1 Hz, 1H), 4,46 (q, J 7,2 Hz, 2H), 4,34 (t, J 8,5 Hz, 1H), 4,13-3,89 (m, 2H), 3,89-3,66 (m, 2H), 1,98 1,42 (m, 9H), 1,42-1,12 (m, 5H), 0,86 (d, J 6,4 Hz, 6H). HPLC-MS (método 6): MH+ m /z 494, TR 2,05 min. uPLC-MS (método 23): MH+ m /z 494, TR 1,62 min. [a]20D = -4,30° (c 10,0, metanol).
Ejemplo 48

3- Etil-W-{(1S)-1-(4-metilciclohexil)-2-oxo-2-[(2-oxospiro[indolina-3,4'-tetrahidropiran]-6-il)amino]etil}isoxazol-4- carboxamida (isómero trans)
Se preparó a partir del in term edio 50 (52 mg, 0,14 mmol) y ácido 3-etilisoxazol-4-carboxílico (19,8 mg, 0,14 mmol) de acuerdo con el proced im iento A, y se purificó por cromatografía en columna ultrarrápida usando un gradiente de 0 100 % de acetato de etilo en hexanos, para dar el com puesto de l título (51 mg, 74 %) como un sólido blanquecino. 3h (400 MHz, DMSO-d6) 10,40 (s, 1H), 10,18 (s, 1H), 9,41 (s, 1H), 8,47 (d, J 8,1 Hz, 1H), 7,56-7,25 (m, 2H), 7,11 (dd, J 8,2, 2,0 Hz, 1 H), 4,37 (t, J 8,3 Hz, 1H), 4,01 (ddd, J 10,9, 6,9, 3,6 Hz, 2H), 3,89-3,70 (m, 2H), 2,84 (q, J 7,5 Hz, 2H), 1,96-1,38 (m, 9H), 1,17 (m, 5H), 0,86 (m, 6H). HPLC-MS (método 6): MH+ m /z 495, TR 2,10 min. uPLC-MS (método 23): MH+ m /z 495, TR 1,67 min. [a]20D = -0,60° (c 10,0, metanol).
Ejemplo 49
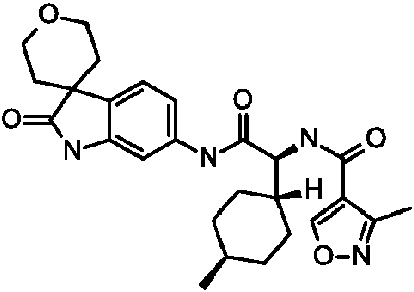
3- Metil-W-{(1S)-1-(4-metilciclohexil)-2-oxo-2-[(2-oxospiro[indolina-3,4'-tetrahidropiran]-6-il)amino]etil}isoxazol-4- carboxamida (isómero trans)
Se preparó a partir del in term edio 50 (52 mg, 0,14 mmol) y ácido 3-metilisoxazol-4-carboxílico (15,4 mg, 0,121 mmol) de acuerdo con el proced im iento A, y se purificó por cromatografía en columna ultrarrápida usando un gradiente de 0 100 % de acetato de etilo en hexanos, para dar el com puesto de l título (51 mg, 88 %) como un sólido blanquecino. 5h (400 MHz, DMSO-de) 10,40 (s, 1H), 10,19 (s, 1H), 9,44 (d, J 0,7 Hz, 1H), 8,46 (d, J 8,2 Hz, 1H), 7,54-7,25 (m, 2H), 7,12 (dd, J 8,2, 2,0 Hz, 1H), 4,38 (t, J 8,4 Hz, 1 H), 4,01 (ddd, J 11,1,7,1,3,7 Hz, 2H), 3,80 (td, J 7,3, 3,6 Hz, 2H), 2,37 (s, 3H), 1,94-1,42 (m, 9H), 1,42-0,93 (m, 3H), 0,86 (d, J 6,5 Hz, 5H). HPLC-MS (método 6): MH+ m /z 481, TR 1,98 min. uPLC-MS (método 23): MH+ m /z 481, TR 1,58 min. [a]20D = 5,65° (c 10,0, metanol).
Ejemplo 50 (procedimiento C)

3-Etil-W-{(1S)-1-(4-metilciclohexil)-2-oxo-2-[(2-oxospiro[1H-pirrolo[3,2-c]-piridin-3,4'-tetrahidropiran]-6-il)amino]etil}isoxazol-4-carboxamida (isómero trans)
A una solución del in term edio 108 (2,70 g, 4,32 mmol) en DCM (20 ml) a 0 °C se le añadió gota a gota ácido trifluoroacético (5 ml, 66,13 mmol). La mezcla se agitó a 20 °C durante 1,5 h. Se le añadió otra porción de ácido
trifluoroacético (5 ml, 66,13 mmol) a 0 °C. La mezcla de reacción se agitó a 20 °C durante 18 h más y luego se concentró al vacío. El residuo se disolvió en una mezcla de acetonitrilo (2 ml) y una solución acuosa de hidróxido de amonio (28 % v/v, 2 ml). La mezcla se agitó a 20 °C durante 1 h. El residuo se concentró al vacío y luego se repartió entre d Cm y agua. La fase acuosa se alcalinizó con una solución acuosa de NaOH a 2 M y luego se extrajo con DCM:MeOH (10:1). La capa orgánica se concentró al vacío. El residuo bruto se purificó mediante cromatografía en columna ultrarrápida, utilizando un gradiente de 0-100 % de DCM en acetato de etilo, para proporcionar, después de la liofilización, el com puesto de l títu lo (1,36 g, 64 %) como un sólido blanco. 5h (300 MHz, DMSO-d6) 10,86 (s, 1H), 10,57 (s, 1H), 9,40 (s, 1H), 8,45-8,36 (m, 2H), 7,71 (s, 1H), 4,52 (t, J 8,0 Hz, 1 H), 4,05-3,94 (m, 2H), 3,84-3,75 (m, 2H), 2,83 (q, J 7,5 Hz, 2H), 1,80-1,57 (m, 10H), 1,32-1,06 (m, 4H), 0,94-0,77 (m, 6H). HPLC-MS (método 6): MH+ m lz 496, TR 2,17 min. uPLC-Ms (método 23): MH+ m /z 496, TR 1,56 min.
Ejemplo 51

2-Etil-N-{(1 S)-1-(4-metilciclohexil)-2-oxo-2-[(2-oxospiro[1H-pirrolo[3,2-c]-piridin-3,4'-tetrahidropiran]-6-il)amino]etil}pirazol-3-carboxamida (isómero tra n s )
Se preparó a partir del in term edio 109 (2,73 g, 4,36 mmol) de acuerdo con el procedim iento C, y se purificó por cromatografía en columna ultrarrápida usando un gradiente de 0-100 % de acetato de etilo en hexanos, para dar el com puesto de l título (1,6 g, 74 %) como un sólido blanco. 5h (400 MHz, DMSO-d6) 10,87 (s, 1H), 10,57 (s, 1H), 8,49 8,39 (m, 2H), 7,72 (s, 1H), 7,49 (d, J 2,0 Hz, 1H), 6,99 (d, J 2,1 Hz, 1H), 4,53-4,40 (m, 3H), 4,06-3,94 (m, 2H), 3,88 3,76 (m, 2H), 1,88-1,50 (m, 7H), 1,33-1,17 (m, 6H), 1,14-0,99 (m, 1H), 0,95-0,78 (m, 6H). HPLC-MS (método 6): MH+ m /z 495, TR 1,92 min. uPLC-MS (método A): MH+ m /z 495, t R 1,51 min. [a]20D = -13,50° (c 10,0, metanol).
Ejemplo 52 (procedimiento D)

(2S)-2-Ciclohexil-2-{metil(tetrahidropiran-4-il)carbamoil]amino}-W-(2-oxospiro-[indolina-3,4'-tetrahidropiran]-6-il)acetamida
El in term edio 78 (30 mg, 0,084 mmol) y el cloruro de W-metil-W-(oxan-4-il)carbamoilo (18 mg, 0,097 mmol) se disolvieron en DCM (1 ml) y se le añadió trietilamina (14 pl, 10,24 mg, 0,1007 mmol). La mezcla de reacción se agitó a 20 °C durante 18 h. Se le añadieron cloruro de W-metil-W-(oxan-4-il)carbamoilo (18 mg, 0,097 mmol) y trietilamina (14 pl, 10,24 mg, 0,1007 mmol). La mezcla de reacción se agitó a 20 °C durante 5 h más y luego se concentró al vacío. El aceite transparente bruto resultante se purificó mediante cromatografía en columna ultrarrápida, utilizando un gradiente de 0-100 % de acetato de etilo en hexanos, para proporcionar, después de la liofilización, el com puesto de l títu lo (15 mg, 36 %) como un sólido blanco. 3h (400 MHz, DMSO-d6) 10,38 (s, 1H), 10,01 (s, 1H), 7,46-7,36 (m, 2H), 7,09 (dd, J 8,1, 1,9 Hz, 1H), 6,05 (d, J 8,3 Hz, 1 H), 4,22-3,96 (m, 4H), 3,88 (dd, J 10,6, 5,3 Hz, 2H), 3,80 (ddd, J 11,1, 7,0, 3,7 Hz, 2H), 3,41-3,31 (m, 2H, ocultado por el pico de agua a 3,31 ppm), 2,72 (s, 3H), 1,86-1,49 (m, 12H), 1,46 1,34 (m, 2H), 1,24-1,06 (m, 4H), 1,02-0,86 (m, 1H). HPLC-MS (método 6): MH+ m /z 499, TR 1,83 min.
Ejemplo 53

3-Ciclopropil-N-{(1S)-1-(4-metilciclohexil)-2-oxo-2-[(2-oxospiro[1H-pirrolo[3,2-c]-piridin-3,4'-tetrahidropiran]-6-il)amino]etil}isoxazol-4-carboxamida (isómero tra n s )
Se preparó a partir del in term edio 110 (100 mg, 0,16 mmol) de acuerdo con el proced im iento C, y se purificó por
cromatografía en columna ultrarrápida usando un gradiente de 0-100 % de acetato de etilo en hexanos, para dar el com puesto de l títu lo (56 mg, 76 % de rendimiento) como un sólido blanco. 5h (400 MHz, DMSO-d6) 10,87 (s, 1H), 10,59 (s, 1 H), 9,37 (s, 1H), 8,44-8,35 (m, 2H), 7,72 (s, 1 H), 4,54 (t, J 8,0 Hz, 1H), 3,99 (dt, J 11,7, 4,8 Hz, 2H), 3,88 3,76 (m, 2H), 2,43 (tt, J 8,4, 5,1 Hz, 1H), 1,88-1,52 (m, 9H), 1,36-1,14 (m, 2H), 1,14-0,9 (m, 10H). HPLC-MS (método 6): MH+ m /z 508, TR 2,29 min.
Ejemplo 54

N-{(1 S)-1-(4-Metilciclohexil)-2-oxo-2-[(2-oxospiro[1H-pirrolo[3,2-c]-piridin-3,4'-tetrahidropiran]-6-il)amino]etil}carbamato de ciclobutilo (isómero trans)
Se preparó a partir del in term edio 111 (60 mg, 0,10 mmol) de acuerdo con el procedim iento C, y se purificó por cromatografía en columna ultrarrápida usando un gradiente de 0-100 % de acetato de etilo en hexanos seguido de HPLC preparativa (método 22), para dar el com puesto de l títu lo (11 mg, 23 %) como un sólido blanco. 3h (400 MHz, DMSO-d6) 10,87 (s, 1 H), 10,35 (s, 1H), 8,39 (s, 1H), 7,69 (s, 1H), 7,27 (d, J 8,4 Hz, 1H), 4,80 (p, J 7,5 Hz, 1H), 4,12 3,94 (m, 3H), 3,81 (dd, J 11,5, 8,6 Hz, 2H), 2,26-2,16 (m, 2H), 2,07-1,86 (m, 2H), 1,85-1,72 (m, 2H), 1,66-1,38 (m, 7H), 1,30-0,93 (m, 4H), 0,93-0,58 (m, 6H). HPLC-MS (método 6): MH+ m /z 471, TR 2,33 min.
Ejemplo 55

N-{(1 S)-1-(4-Metilciclohexil)-2-oxo-2-[(2-oxospiro[1H-pirrolo[3,2-c]piridin-3,4'-tetrahidropiran]-6-il)amino]etil}-2-(3,3,3-trifluoropropil)pirazol-3-carboxamida (isómero trans)
Se preparó a partir del in term edio 112 (50 mg, 0,072 mmol) de acuerdo con el procedim iento C, y se purificó por cromatografía en columna ultrarrápida usando un gradiente de 0-100 % de acetato de etilo en hexanos, para dar el com puesto de l títu lo (21 mg, 52 %) como un sólido blanco. 5h (400 MHz, DMSO-d6) 10,87 (s, 1H), 10,59 (s, 1H), 8,54 (d, J 8,1 Hz, 1H), 8,44-8,39 (m, 1 H), 7,72 (d, J 0,7 Hz, 1H), 7,56 (d, J 2,0 Hz, 1H), 7,10 (d, J 2,1 Hz, 1 H), 4,80-4,63 (m, 2H), 4,52 (t, J 8,3 Hz, 1H), 3,99 (dt, J 10,2, 4,6 Hz, 2H), 3,87-3,77 (m, 2H), 2,87-2,70 (m, 2H), 1,88-1,74 (m, 3H), 1,74 1,40 (m, 4H), 1,36-1,06 (m, 3H), 1,04-0,91 (m, 1H), 0,90-0,75 (m, 6H). HPLC-MS (método 6): MH+ m /z 563, TR 2,17 min.
Ejemplo 56

3-Ciclobutil-N-{(1 S)-1-(4-metilciclohexil)-2-oxo-2-[(2-oxospiro[1H-pirrolo[3,2-c]-piridin-3,4'-tetrahidropiran]-6-il)amino]etil}isoxazol-4-carboxamida (isómero trans)
Se preparó a partir del in term edio 113 (45 mg, 0,069 mmol) de acuerdo con el proced im iento C, y se purificó por cromatografía en columna ultrarrápida usando un gradiente de 0-100 % de acetato de etilo en hexanos seguido de 0 20 % de metanol en acetato de etilo, para dar el com puesto de l título (16 mg, 44 %) como un sólido blanco. 5h (400 MHz, DMSO-de) 10,86 (s, 1H), 10,57 (s, 1H), 9,39 (s, 1H), 8,41 (s, 1H), 8,35 (d, J 8,0 Hz, 1H), 7,71 (s, 1H), 4,51 (t, J 8,1 Hz, 1 H), 4,03-3,94 (m, 2H), 3,91-3,77 (m, 3H), 2,34-2,14 (m, 4H), 1,98-1,91 (m, 1H), 1,89-1,76 (m, 4H), 1,74-1,48 (m, 5H), 1,33-1,23 (m, 2H), 1,11 -0,98 (m, 1H), 0,93-0,76 (m, 6H). HPLC-MS (método 6): MH+ m /z 522, TR 2,19 min.
Ejemplo 57
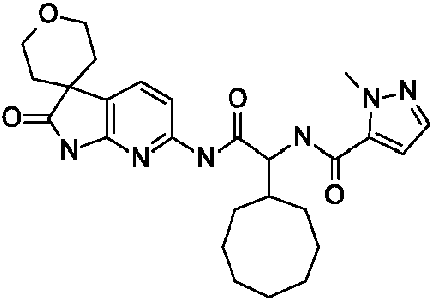
W-{1-Ciclooctil-2-oxo-2-[(2-oxospiro[1H-pirrolo[2,3-b]piridin-3,4'-tetrahidropiran]-6-il)amino]etil}-2-metilpirazol-3-carboxamida
A una solución del in term edio 118 (0,23 g, 0,36 mmol) en DCM (5 ml) se le añadió TFA (0,55 ml, 7,20 mmol) a 0 °C. La mezcla de reacción se agitó a temperatura ambiente durante 16 h y luego se concentró al vacío. Se le añadieron acetonitrilo (3 ml) y NH4OH (solución al 25 % en H2O, 3 ml) a 0 °C. La mezcla de reacción se agitó a temperatura ambiente durante 3 h, y luego se concentró al vacío. El residuo bruto se purificó por cromatografía en columna (MeOH al 3-4 % en DCM) para proporcionar el com puesto de l títu lo (0,042 g, 22 %) como un sólido blanquecino. 5h (400 MHz, DMSO-d6) 1,34-1,70 (m, 16H), 1,77-1,87 (m, 2H), 2,14-2,17 (m, 1H), 3,80 (t, J 8,41 Hz, 2H), 3,93-4,00 (m, 2H), 4,02 (s, 3H), 4,59 (t, J 8,41 Hz, 1H), 7,02 (s, 1H), 7,46 (s, 1H), 7,69 (d, J 8,03 Hz, 1H), 7,94 (d, J 8,03 Hz, 1H), 8,45 (d, J 8,53 Hz, 1 H), 10,49 (s, 1H), 11,01 (br s, 1H). HPLC-MS (método 6): MH+ m /z 495,0, TR 2,65 min.
Ejemplo 58

M-{2-[(5-Cloro-2-oxospirorindolina-3,4'-tetrahidropiran]-6-il)amino]-1-ciclooctil-2-oxoetil}-2-metilpirazol-3-carboxamida
Se añadió NCS (36 mg, 0,27 mmol) en porciones a una solución del ejem plo 3 (45 mg, 0,09 mmol) en ácido acético (2 ml) a 20 °C. La mezcla de reacción se agitó a 20 °C durante 10 min, luego se calentó a 70 °C durante 10 min y luego se concentró al vacío . El residuo se diluyó en acetato de etilo (50 ml), se lavó con solución acuosa saturada de bicarbonato de sodio y salmuera, y luego se secó sobre sulfato de sodio. Las capas orgánicas se concentraron al vacío. El residuo se purificó por HPLC preparativa (método 9) para producir el com puesto de l título (4,5 mg, 9 %) como un polvo blanquecino. 5h (500 MHz, CDCb) 10,44 (s, 1H), 8,41 (s, 1H), 8,40 (s, 1H), 7,93 (d, J 7,4 Hz, 1H), 7,38 (d, J 2,1 Hz, 1 H), 7,37 (s, 1 H), 6,86 (d, J 2,1 Hz, 1H), 4,45-4,37 (m, 1H), 4,27-4,18 (m, 2H), 4,13 (s, 3H), 3,95-3,85 (m, 2H), 2,35-2,23 (m, 1H), 1,92-1,82 (m, 3H), 1,82-1,49 (m, 15H). uPLC-MS (método 2) MH+ m /z 528 y 530, TR 3,56 min.
Ejemplo 59

M-{1-Ciclooctil-2-oxo-2-[(2-oxospiro[indolina-3,4'-tetrahidropiran]-6-il)amino]etil}-2-oxospiro[indolina-3,4'-tetrahidropiran]-6-carboxamida
Se preparó a partir del in term edio 121 (70 mg, 0,094 mmol) de acuerdo con el procedimiento C, y se purificó por HPLC preparativa (método 11), para dar el com puesto de l título (10,5 mg, 18 %) como un sólido blanco. 5h (500 MHz, DMSO-d6) 10,57 (s, 1H), 10,39 (s, 1H), 10,19 (s, 1H), 8,50 (d, J 8,6 Hz, 1 H), 7,60 (d, J 7,8 Hz, 1H), 7,54 (dd, J 7,8, 1,5 Hz, 1H), 7,43 (d, J 8,2 Hz, 1H), 7,39 (d, J 1,9 Hz, 1H), 7,33 (d, J 1,4 Hz, 1H), 7,12 (dd, J 8,2, 1,9 Hz, 1H), 4,46 (t, J 8,8 Hz, 1H), 4,06-3,98 (m, 4H), 3,85-3,77 (m, 4H), 2,22-2,13 (m, 1H), 1,77-1,58 (m, 12H), 1,57-1,38 (m, 10H). HPLC-MS (método 1): MH+ m lz 615, TR 3,13 min.
Ejemplo 60

2-Ciclooctil-2-(metanosulfonamido)-W-(2-oxospiro[indolina-3,4'-tetrahidropiran]-6-il)acetamida
Se añadió cloruro de metanosulfonilo (12 pl, 0,15 mmol) a una solución agitada del in term edio 35 (50 mg, 0,12 mmol) y DIPEA (43 pl, 0,25 mmol) en DCM anhidro (2 ml). La mezcla de reacción se agitó a 20 °C durante 16 h. Se le añadió más cloruro de metanosulfonilo (6 pl, 0,08 mmol). La mezcla de reacción se agitó a 20 °C durante 6 h más y luego se diluyó con DCM (7 ml). El material resultante se lavó con agua (7 ml) y salmuera (7 ml), luego se secó sobre sulfato de magnesio, se filtró y se concentró al vacío. El residuo se purificó por HPLC preparativa (método 10) para producir el com puesto de l título (26,7 mg, 47 %) como un polvo tostado. 5h (500 MHz, DMSü-de) 10,40 (s, 1H), 10,18 (s, 1H), 7,53-7,46 (m, 1H), 7,44 (d, J 8,1 Hz, 1 H), 7,37 (d, J 1,8 Hz, 1H), 7,09 (dd, J 8,1, 1,9 Hz, 1H), 4,01 (ddd, J 11,1,7,1,3,6 Hz, 2H), 3,80 (ddd, J 11,1,7,0, 3,7 Hz, 2H), 3,77-3,69 (m, 1H), 2,81 (s, 3H), 1,98-1,87 (m, 1H), 1,80-1,30 (m, 18H). HPLC-MS (método 1): MH+ m /z 464, TR 2,90 min.
Ejemplo 61

-Ciclooctil-2-[(2-metilpirazol-3-il)sulfonilamino]-W-(2-oxospiro[indolina-3,4'-tetrahidropiran]-6-il)acetamida
Se añadió cloruro de 1 -metil-1 H-pirazol-5-sulfonilo (41 mg, 0,23 mmol) a una solución agitada del in term edio 35 (100 mg, 0,15 mmol) y DIPEA (54 pl, 0,31 mmol) en DCM anhidro (3 ml). La mezcla de reacción se agitó a 20 °C durante 16 h, luego se diluyó con DCM (10 ml). El material resultante se lavó con agua (10 ml) y salmuera (10 ml), luego se secó sobre sulfato de magnesio, se filtró y se concentró al vacío. El residuo se purificó por HPLC preparativa (método 10) para producir el com puesto de l título (20 mg, 24 %) como un polvo de color naranja. 5h (500 MHz, DMSO-d6) 10,36 (s, 1 H), 9,93 (s, 1H), 8,65 (br s, 1H), 7,39 (d, J 8,1 Hz, 1H), 7,34 (d, J 1,7 Hz, 1H), 7,12 (s, 1H), 6,87 (dd, J 8,2, 1,9 Hz, 1H), 6,64 (s, 1 H), 4,05-3,95 (m, 2H), 3,98 (s, 3H), 3,79 (ddd, J 10,7, 6,8, 3,6 Hz, 2H), 3,71 -3,62 (m, 1H), 1,94-1,84 (m, 1H), 1,77-1,68 (m, 2H), 1,68-1,26 (m, 16H). HPLC-MS (método 1): MH+ m /z 530, TR 3,13 min.
Ejemplo 62

W-{1-Ciclooctil-2-oxo-2-[(2-oxospiro[indolina-3,3'-pirrolidín]-6-il)amino]etil}-3-metilisoxazol-4-carboxamida (isómero 1)
Se añadió ácido trifluoroacético (0,23 ml, 2,99 mmol) a una suspensión agitada del in term edio 127 (89,8 mg, 0,15 mmol) en DCM (1,2 ml). La mezcla se agitó a 20 °C durante 2 h, luego se inactivó con una solución acuosa saturada de carbonato de sodio (10 ml). El material resultante se extrajo con DCM/isopropanol 4:1 (4 x 20 ml) usando una frita hidrófoba. El filtrado orgánico se concentró al vacío. La goma de color naranja resultante se separó mediante HPLC preparativa secuencial (método 10), HPLC preparativa quiral (método 32, MeüH dietilamina al 0,2 %, columna Chiralcel OD-H de 25 cm a 9 ml/min) y SFC preparativa quiral (método 14, MeüH 25:75 dietilamina al 0,2 %-dióxido de carbono, columna Chiralpak IC de 25 cm a 15 ml/min), para proporcionar el com puesto de l título (1,4 mg, 2 %) como un polvo blanquecino. 3h (500 MHz CD3OD) 9,12 (s, 1H), 7,43 (s, 1H), 7,27 (d, J 8,1 Hz, 1H), 7,14 (d, J 8,1 Hz, 1H), 4,49 (d, J 8,4 Hz, 1H), 3,47-3,24 (m obs., 3H), 3,11 (d, J 10,8 Hz, 1H), 2,43 (s, 3H), 2,34-2,07 (m, 3H), 1,84-1,44 (m, 14H). uPLC-MS (método 1): MH+ m /z 480, t R 2,21 min. SfC quiral (método 12, Chiralpak IC 25 cm, MeÜH al 35 % dietilamina al 0,2 %-dióxido de carbono al 65 %, 4 ml/min): TR 3,89 min (100 %).
Ejemplo 63

W-{1-Ciclooctil-2-oxo-2-[(2-oxospiro[indolina-3,3'-pirrolidina]-6-il)amino]etil}-3-metilisoxazol-4-carboxamida (isómero 2)
Se añadió ácido trifluoroacético (0,23 ml, 2,99 mmol) a una suspensión agitada del in term edio 127 (89,8 mg, 0,15 mmol) en DCM (1,2 ml). La mezcla se agitó a 20 °C durante 2 h, luego se inactivó con una solución acuosa saturada de carbonato de sodio (10 ml). El material resultante se extrajo con DCM/isopropanol 4:1 (4 x 20 ml) usando una frita hidrófoba. El filtrado orgánico se concentró al vacío. La goma de color naranja resultante se separó mediante HPLC preparativa secuencial (método 10), HPLC preparativa quiral (método 32, MeOH dietilamina al 0,2 %, columna Chiralcel OD-H de 25 cm a 9 ml/min) y SFC preparativa quiral (método 14, MeOH 25:75 dietilamina al 0,2 %-dióxido de carbono, columna Chiralpak IC de 25 cm a 15 ml/min), para proporcionar el com puesto de l título (2,4 mg, 3 %) como un polvo blanquecino. 3h (500 MHz CD3OD) 9,12 (s, 1h), 7,45-7,37 (m, 1H), 7,25 (d, J 8,0 Hz, 1H), 7,18-7,08 (m, 1H), 4,49 (d, J 8,3 Hz, 1H), 3,41 -3,17 (m obs., 3H), 2,99 (d, J 11,8 Hz, 1 H), 2,43 (s, 3H), 2,32-2,12 (m, 2H), 2,05 (dt, J 13,5, 7,1 Hz, 1H), 1,86-1,42 (m, 14H). uPLC-MS (método 1): MH+ m /z 480, TR 2,20 min. SFC quiral (método 12, Chiralpak IC 25 cm, MeOH al 35 % dietilamina al 0,2 %-dióxido de carbono al 65 %, 4 ml/min): TR 3,98 min (93 %).
Ejemplo 64

W-{1-Ciclooctil-2-[(1',2-dioxospiro[indolina-3,4'-tianel-6-il)amino]-2-oxoetil}-2-metilpirazol-3-carboxamida
Se añadió ácido 3-cloroperbenzoico (70 %, 98 mg, 0,4 mmol) a una suspensión agitada del ejem plo 9 (176 mg, 0,34 mmol) en DCM (3,4 ml) a 0 °C en nitrógeno. La mezcla de reacción se agitó a 0 °C durante 3,5 h, luego se inactivó con una solución acuosa de sulfito de sodio al 10%/solución acuosa saturada de carbonato de sodio 1:1 (40 ml). El material resultante se extrajo con acetato de etilo/2-metiltetrahidrofurano 1:1 (3 x 30 ml). Los extractos orgánicos combinados se lavaron con una solución acuosa saturada de carbonato de sodio (20 ml) y salmuera (20 ml), luego se secaron sobre sulfato de magnesio, se filtraron y se concentraron al vacío. El polvo blanquecino bruto resultante se separó mediante HPLC preparativa (método 10) para proporcionar, después de la liofilización, el com puesto de l título (23,3 mg, 13 %) como un polvo blanco. 3h (500 MHz, DMSO-d6) 10,47 (br s, 1H), 10,25 (s, 1 H), 8,53 (d, J 8,5 Hz, 1 H), 7,53 (d, J 8,1 Hz, 1H, menor), 7,46 (d, J 2,0 Hz, 1H), 7,41 (d, J 1,8 Hz, 1H, menor), 7,39 (d, J 1,7 Hz, 1H, mayor), 7,21 (d, J 8,1 Hz, 1H, mayor), 7,16 (dd, J 8,1, 1,8 Hz, 1H, mayor), 7,12 (dd, J 8,2, 1,9 Hz, 1H, menor), 7,05 (d, J 2,1 Hz, 1H), 4,44 (t, J 8,9 Hz, 1H), 4,03 (s, 3H), 3,43-3,23 (m obs., 2H, mayor), 3,14-3,07 (m, 4H, menor), 2,93-2,79 (m, 2H, mayor), 2,56-2,43 (m obs., 2H), 2,41-2,29 (m, 1H, menor), 2,21-2,10 (m, 1H, mayor), 1,75-1,33 (m, 16H). uPLC-MS (método 1): m H+ m /z 526, TR 2,75 min.
Ejemplo 65

ft-{1-Ciclooctil-2-oxo-2-[(1',1',2-trioxospiro[indolina-3,4'-tián]-6-il)amino]etil}-2-metilpirazol-3-carboxamida
Se añadió ácido 3-cloroperbenzoico (70 %, 98 mg, 0,4 mmol) a una suspensión agitada del ejem plo 9 (176 mg, 0,34 mmol) en DCM (3,4 ml) a 0 °C en nitrógeno. La mezcla de reacción se agitó a 0 °C durante 3,5 h, luego se inactivó con una solución acuosa de sulfito de sodio al 10%/solución acuosa saturada de carbonato de sodio 1:1 (40 ml). El
material resultante se extrajo con acetato de etilo/2-metiltetrahidrofurano 1:1 (3 x 30 ml). Los extractos orgánicos combinados se lavaron con una solución acuosa saturada de carbonato de sodio (20 ml) y salmuera (20 ml), luego se secaron sobre sulfato de magnesio, se filtraron y se concentraron al vacío. El polvo blanquecino bruto resultante se separó mediante HPLC preparativa (método 10) para proporcionar, después de la liofilización, el com puesto de l título (71,7 mg, 38 %) como un polvo blanco. 3h (500 MHz, DMSO-d6) 10,62 (br s, 1H), 10,27 (s, 1 h), 8,53 (d, J 8,5 Hz, 1H), 7,45 (d, J 2,0 Hz, 1H), 7,42 (d, J 1,8 Hz, 1H), 7,31 (d, J 8,1 Hz, 1 H), 7,15 (dd, J 8,2, 1,8 Hz, 1H), 7,05 (d, J 2,1 Hz, 1 H), 4,43 (t, J 8,8 Hz, 1 H), 4,02 (s, 3H), 3,74-3,60 (m, 2H), 3,17-3,05 (m, 2H), 2,35-2,25 (m, 2H), 2,22-2,03 (m, 3H), 1,73 1,33 (m, 14H). uPLC-MS (método 1): MH+ m /z 542, TR 3,00 min.
Ejemplo 66

WL(1-Ciclooctil-2-{[(2'S,3fi)-2'-metil-2-oxospiro[indolina-3,4'-tetrahidropiran]-6-il]-amino}-2-oxoetil)-2-metilpirazol-3-carboxamida (isómero 1)
Se añadió DIPEA (76 pl, 0,46 mmol) a una suspensión agitada del in term edio 135 (96,7 mg, 0,35 mmol), in term edio 128 (127 mg, 0,39 mmol) y HATU (161 mg, 0,42 mmol) en THF anhidro (1,75 ml). La mezcla se agitó a 20 °C en nitrógeno durante 16 h, luego se inactivó con una solución acuosa saturada de carbonato de sodio (3 ml) y agua (3 ml), y se agitó a 20 °C durante 1 h. La mezcla se extrajo con DCM/isopropanol 4:1 (4 x 5 ml) usando una frita hidrófoba, y luego el filtrado orgánico se concentró al vacío. El polvo de color canela resultante se separó mediante SFC preparativa quiral (método 14, 15:85 de etanol-dióxido de carbono, columna Chiralpak IC de 25 cm a 15 ml/min) para proporcionar, después de la liofilización, el com puesto de l títu lo (52,2 mg, 28 %) como un polvo blanquecino. 3h (500 MHz, DMSO-d6) 10,30 (s, 1H), 10,20 (s, 1H), 8,52 (d, J 8,6 Hz, 1H), 7,45 (d, J 2,0 Hz, 1H), 7,34 (d, J 1,6 Hz, 1H), 7,15 (d, J 8,1 Hz, 1 H), 7,11 (dd, J 8,1, 1,7 Hz, 1H), 7,05 (d, J 2,0 Hz, 1H), 4,43 (t, J 8,9 Hz, 1H), 4,27-4,10 (m, 2H), 4,03 (s, 3H), 3,73 (dd, J 11,1,4,6 Hz, 1H), 2,22-2,09 (m, 1H), 1,87 (td, J 13,4, 5,0 Hz, 1H), 1,74-1,33 (m, 17H), 1,07 (d, J 6,2 Hz, 3H). uPLC-MS (método 1): Mh m /z 508, TR 3,30 min. SFC quiral (método 12, Chiralpak iC 25 cm, etanol al 20 %-dióxido de carbono al 80 %, 4 ml/min): TR 5,65 min (98 %, isómero 1).
Ejemplo 67

W-(1-Ciclooctil-2-{[(2'S,3fi)-2'-metil-2-oxospiro[indolina-3,4'-tetrahidropiran]-6-il]-amino}-2-oxoetil)-2-metilpirazol-3-carboxamida (isómero 2)
Se añadió DIPEA (76 pl, 0,46 mmol) a una suspensión agitada del in term edio 135 (96,7 mg, 0,35 mmol), in term edio 128 (127 mg, 0,39 mmol) y HATU (161 mg, 0,42 mmol) en THF anhidro (1,75 ml). La mezcla se agitó a 20 °C en nitrógeno durante 16 h, luego se inactivó con una solución acuosa saturada de carbonato de sodio (3 ml) y agua (3 ml), y se agitó a 20 °C durante 1 h. La mezcla se extrajo con DCM/isopropanol 4:1 (4 x 5 ml) usando una frita hidrófoba, y luego el filtrado orgánico se concentró al vacío. El polvo de color canela resultante se separó mediante SFC preparativa quiral (método 14, etanol-dióxido de carbono 15:85, columna Chiralpak IC de 25 cm a 15 ml/min) para proporcionar, después de la liofilización, el com puesto de l título (31,1 mg, 16 %) como un polvo blanco. 3h (500 MHz, DMSO-de) 10,30 (s, 1H), 10,20 (s, 1H), 8,52 (d, J 8,6 Hz, 1H), 7,45 (d, J 2,0 Hz, 1H), 7,35 (d, J 1,6 Hz, 1H), 7,15 (d, J 8,1 Hz, 1H), 7,10 (dd, J 8,1, 1,7 Hz, 1H), 7,05 (d, J 2,0 Hz, 1H), 4,43 (t, J 8,9 Hz, 1H), 4,27-4,10 (m, 2H), 4,03 (s, 3H), 3,73 (dd, J 11,0, 4,7 Hz, 1H), 2,22-2,09 (m, 1H), 1,86 (td, J 13,3, 5,0 Hz, 1H), 1,74-1,32 (m, 17H), 1,07 (d, J 6,2 Hz, 3H). uPLC-MS (método 1): Mh m /z 508, Tr 3,29 min. SfC quiral (método 12, Chiralpak iC 25 cm, etanol al 20 %-dióxido de carbono al 80 %, 4 ml/min: TR 7,23 min (98 %, isómero 2).
Ejemplo 68

WL(1-Ciclooctil-2-{[(2'S,3S)-2'-metil-2-oxospiro[indolina-3,4'-tetrahidropiran]-6-il]-amino}-2-oxoetil)-2-metilpirazol-3-carboxamida (isómero 3)
Se añadió DIPEA (39 pl, 0,24 mmol) a una suspensión agitada del in term edio 136 (44,9 mg, 0,22 mmol), in term edio 128 (65 mg, 0,2 mmol) y HATU (83 mg, 0,22 mmol) en THF anhidro (1 ml). La mezcla se agitó a 20 °C en nitrógeno durante 16 h, luego se inactivó con una solución acuosa saturada de carbonato de sodio (3 ml) y agua (3 ml), y se agitó a 20 °C durante 1 h. La mezcla se extrajo con DCM/isopropanol 4:1 (4 x 5 ml) usando una frita hidrófoba, y luego el filtrado orgánico se concentró al vacío. El polvo tostado resultante se separó mediante HPLC preparativa quiral (método 34, heptano-isopropanol 70:30, columna Chiralpak AD-H de 25 cm a 18 ml/min) para proporcionar, después de la liofilización, el com puesto de l título (30,6 mg, 33 %) como un polvo blanco. 5h (500 MHz, DMSO-d6) 10,48 (s, 1H), 10,24 (s, 1 H), 8,52 (d, J 8,6 Hz, 1H), 7,67 (d, J 8,3 Hz, 1H), 7,46 (d, J 2,0 Hz, 1H), 7,42 (d, J 1,8 Hz, 1H), 7,12 (dd, J 8,2, 1,9 Hz, 1 H), 7,05 (d, J 2,0 Hz, 1 H), 4,44 (t, J 8,9 Hz, 1H), 4,03 (s, 3H), 4,01 -3,81 (m, 3H), 2,23-2,04 (m, 1H), 1,87 (td, J 13,0, 6,1 Hz, 1H), 1,74-1,29 (m, 16H), 1,25 (d, J 13,4 Hz, 1H), 1,10 (d, J 6,0 Hz, 3H). uPLC-MS (método 1): MH+ m /z 508, TR 3,22 min. SFC quiral (método 12, Chiralcel OD-H 25 cm, MeOH al 15 %-dióxido de carbono al 85 %, 4 ml/min): TR 10,70 min (100 %).
Ejemplo 69

M-(1-Ciclooctil-2-{[(2'fí,3S)-2'-metil-2-oxospiro[indolina-3,4'-tetrahidropiran]-6-il]-amino}-2-oxoetil)-2-metilpirazol-3-carboxamida (isómero 4)
Se añadió DIPEA (85 pl, 0,51 mmol) a una suspensión agitada del in term edio 141 (102 mg, 0,4 mmol), in term edio 128 (142 mg, 0,44 mmol) y HATU (180 mg, 0,47 mmol) en THF anhidro (2 ml). La mezcla se agitó a 20 °C en nitrógeno durante 16 h, luego se inactivó con una solución acuosa saturada de carbonato de sodio (3 ml) y agua (3 ml), y se agitó a 20 °C durante 1 h. La mezcla se extrajo con DCM/isopropanol 4:1 (4 x 5 ml) usando una frita hidrófoba, luego el filtrado orgánico se concentró al vacío. El polvo tostado resultante se separó mediante HPLC preparativa quiral (método 32, heptano-isopropanol 50:50, columna Chiralpak AD-H de 25 cm a 18 ml/min) para proporcionar, después de la liofilización, el com puesto de l títu lo (73,4 mg, 36 %) como un polvo blanco. 5h (500 MHz, DMSO-d6) 10,30 (s, 1H), 10,20 (s, 1 H), 8,52 (d, J 8,6 Hz, 1 H), 7,45 (d, J 2,0 Hz, 1H), 7,35 (d, J 1,6 Hz, 1 H), 7,15 (d, J 8,1 Hz, 1H), 7,10 (dd, J 8,1, 1,7 Hz, 1H), 7,05 (d, J 2,0 Hz, 1H), 4,43 (t, J 8,9 Hz, 1H), 4,27-4,11 (m, 2H), 4,03 (s, 3H), 3,73 (dd, J 11,0, 4,6 Hz, 1H), 2,23-2,10 (m, 1H), 1,86 (td, J 13,4, 5,0 Hz, 1H), 1,75-1,32 (m, 17H), 1,07 (d, J 6,2 Hz, 3H). uPLC-MS (método 1): MH+ m /z 508, TR 3,29 min. SFC quiral (método 12, Chiralcel OD-H 25 cm, MeOH al 20 %-dióxido de carbono al 80 %, 4 ml/min): TR 5,29 min (99 %).
Ejemplo 70

M-{1-Ciclooctil-2-[(1'-imino-1',2-dioxospiro[indolina-3,4'-tián]-6-il)amino]-2-oxoetil}-2-metilpirazol-3-carboxamida (mezcla de isómeros 1)
Se disolvieron en MeOH (1 ml) el ejem plo 9 (39 mg, 0,08 mmol), carbamato de amonio (12 mg, 0,15 mmol) y
(diacetoxiyodo)benceno (62 mg, 0,19 mmol). La mezcla de reacción se agitó a 20 °C en nitrógeno durante 16 h, luego se eliminó lo volátil al vacío. El polvo tostado resultante se separó mediante HPLC preparativa (método 10) para proporcionar, después de la liofilización, el com puesto de l títu lo (6,2 mg, 14 %) como un polvo blanco. 5h (500 MHz, DMSO-d6) 10,56 (br s, 1H), 10,26 (s, 1 H), 8,54 (d, J 8,6 Hz, 1H), 7,45 (d, J 2,1 Hz, 1H), 7,40 (d, J 1,8 Hz, 1H), 7,21 (d, J 8,1 Hz, 1H), 7,15 (dd, J 8,2, 1,8 Hz, 1H), 7,05 (d, J 2,1 Hz, 1 H), 4,43 (t, J 8,8 Hz, 1H), 4,02 (s, 3H), 3,68 (s, 1H), 3,67 3,59 (m, 2H), 3,09-2,94 (m, 2H), 2,33-2,24 (m, 2H), 2,21-2,11 (m, 1H), 2,02-1,94 (m, 2H), 1,74-1,32 (m, 14H). uPLC-MS (método 1): MH+ m /z 541, TR 2,54 min.
Ejemplo 71

WL{1-Ciclooctil-2-[(1'-im ino-1',2-dioxospiro[indolina-3,4'-tián]-6-il)amino]-2-oxoetil}-2-metilpirazol-3-carboxamida (mezcla de isómeros 1)
Se disolvieron en MeOH (1 ml) el ejem plo 9 (39 mg, 0,08 mmol), carbamato de amonio (12 mg, 0,15 mmol) y (diacetoxiyodo)benceno (62 mg, 0,19 mmol). La mezcla de reacción se agitó a 20 °C en nitrógeno durante 16 h, luego se eliminó lo volátil al vacío. El polvo tostado resultante se separó mediante HPLC preparativa (método 10) para proporcionar, después de la liofilización, el com puesto de l título (23,5 mg, 53 %) como un polvo blanco. 5h (500 MHz, DMSO-de) 10,56 (br s, 1H), 10,27 (s, 1 H), 8,55 (d, J 8,4 Hz, 1H), 7,45 (d, J 2,1 Hz, 1H), 7,42 (d, J 1,8 Hz, 1H), 7,28 (d, J 8,1 Hz, 1H), 7,15 (dd, J 8,2, 1,9 Hz, 1H), 7,05 (d, J 2,0 Hz, 1 H), 4,43 (t, J 8,7 Hz, 1H), 4,02 (s, 3H), 3,83 (s, 1H), 3,58 3,49 (m, 2H), 3,08-2,94 (m, 2H), 2,23-2,11 (m, 3H), 2,10-2,02 (m, 2H), 1,74-1,33 (m, 14H). uPLC-MS (método 1): MH+ m /z 541, TR 2,62 min.
Ejemplo 72

2-Ciclohexil-2-(4-fluoro-1H-bencimidazol-2-il)-ft-(2-oxospiro[indolina-3,4'-tetrahidropiran]-6-il)acetamida
El in term edio 144 (97 mg, 0,20 mmol) se disolvió en ácido acético (2 ml, 34,90 mmol). La mezcla de reacción se agitó a 65 °C durante 18 h y luego se concentró al vacío. El residuo en bruto se purificó por HPLC preparativa (método 26) para producir el com puesto de l título (2 mg, 2 %) como un sólido blanco. 5h (300 MHz, DMSO-d6) 12,59 (s, 1H), 10,44 (d, J 9,6 Hz, 2H), 7,47-7,27 (m, 3H), 7,19-7,08 (m, 2H), 6,93 (dd, J 11,0, 7,9 Hz, 1 H), 4,07-3,94 (m, 2H), 3,89 (d, J 10,6 Hz, 1 H), 3,85-3,72 (m, 2H), 2,35-2,20 (m, 1H), 1,79-1,57 (m, 8H), 1,31-1,12 (m, 5H), 0,99-0,87 (m, 1H). LCMS (método 6): MH+ m /z 477, TR 2,04 min.
Ejemplo 73

2-(1ft-Benzimidazol-2-il)-2-ciclohexil-N-(2-oxospiro[indolina-3,4'-tetrahidropiran]-6-il)acetamida
El in term edio 145 (40 mg, 0,08 mmol) se disolvió en ácido acético (1 ml, 17,45 mmol). La mezcla de reacción se agitó a 20 °C durante 2 h, luego se calentó a 65 °C durante 18 h. La mezcla de reacción se concentró al vacío y el residuo se separó mediante cromatografía ultrarrápida en columna usando un gradiente de acetato de etilo en hexano (0 100 %). El sólido en bruto resultante se purificó mediante HPLC preparativa (método 22) para proporcionar el com puesto de l título (3 mg, 8 %) como un sólido blanco. 5h (300 MHz, DMSO-d6) 12,28 (s, 1H), 10,48 (s, 1H), 10,42 (s, 1H), 7,63-7,45 (m, 2H), 7,43 (d, J 8,2 Hz, 1 H), 7,37 (d, J 1,9 Hz, 1H), 7,21 -7,05 (m, 3H), 4,00 (ddd, J 10,7, 6,7, 3,3 Hz, 2H), 3,87-3,74 (m, 3H), 2,31-2,23 (m, 1H), 1,81-1,55 (m, 8H), 1,33-1,12 (m, 5H), 0,98-0,85 (m, 1H). HPLC-MS
(método 6): MH+ m lz 459, TR 2,26 min.
Ejemplo 74

2-Ciclohexil-N,M'-bis(2-oxospiro[indolina-3,4'-tetrahidropiran]-6-il)propanodiamida
A una solución del in term edio 2 (63 mg, 0,29 mmol) y del in term edio 143 (116 mg, 0,30 mmol) en DMF (2 ml, 25,90 mmol) se le añadió HATU (136 mg, 0,35 mmol). La mezcla de reacción se agitó a 20 °C durante 2 h. Se le añadió agua a la mezcla de reacción a 0 °C y el sólido resultante se recogió por filtración. El sólido bruto se purificó por HPLC preparativa (método 24) para proporcionar el com puesto de l títu lo (60 mg, 35 %) como un sólido blanco. 5h (300 MHz, DMSO-d6) 10,41 (s, 2H), 9,99 (s, 2H), 7,44 (d, J 8,2 Hz, 2H), 7,38 (d, J 1,9 Hz, 2H), 7,09 (dd, J 8,2, 2,0 Hz, 2H), 4,01 (ddd, J 11,0, 7,1,3,7 Hz, 4H), 3,87-3,72 (m, 4H), 3,21 (d, J 10,5 Hz, 1H), 2,29-2,14 (m, 1H), 1,81-1,55 (m, 13H), 1,28 0,99 (m, 5H). HPLC-MS (método 23): MH+ m lz 587, TR 2,30 min.
Ejemplo 75

2-Ciclohexil-M '-(2-oxospiro[indolina-3,4'-tetrahidropiran]-6-il)-N-fenilpropanodiamida
A una solución de anilina (0,02 ml, 0,2 mmol) y del in term edio 143 (50 mg, 0,13 mmol) en DMF (0,5 ml) se le añadieron HATU (65 mg, 0,17 mmol) y DIPEA (0,09 ml, 0,5 mmol). La mezcla de reacción se agitó a 20 °C durante 1 h, luego se diluyó en EtOAc (10 ml), se lavó con una mezcla 1:1 de agua y salmuera (10 ml) y se secó sobre Na2SO4. El residuo bruto se purificó por cromatografía en columna (de hexano a EtOAc), seguido de HPLC preparativa (método 24), para producir el com puesto de l título (13 mg, 22 %) como un sólido blanco. 3h (400 MHz CD3OD) 7,61-7,55 (m, 2H), 7,48 7,43 (m, 2H), 7,38-7,31 (m, 2H), 7,17-7,10 (m, 2H), 4,18 (m, 2H), 3,94 (m, 2H), 3,12 (d, J 10,1 Hz, 1H), 2,19 (q, J 10,9 Hz, 1H), 1,94-1,68 (m, 9H), 1,27 (m, 6H). HPLC-MS (método 6): MH+ m lz 462, TR 2,27 min.
Ejemplo 76

2-(Azocan-1-il)-W,W-bis(2-oxospiro[indolina-3,4'-tetrahidropiran]-6-il)propanodiamida
A una solución del in term edio 2 (25 mg, 0,12 mmol) y del in term edio 149 (50 mg, 0,12 mmol) en DMF (1 ml, 12,90 mmol) se le añadió HATU (55 mg, 0,15 mmol). La mezcla de reacción se agitó a 20 °C durante 18 h. Se le añadió agua a la mezcla de reacción, y el precipitado blanco resultante se recogió por filtración y se lavó con agua adicional. El residuo en bruto se purificó por HPLC preparativa (método 22) para producir el com puesto de l títu lo (4 mg, 6 %) como un sólido blanco. 3h (400 MHz, DMSO-de) 10,43 (s, 2H), 9,91 (s, 2H), 7,46 (d, J 8,2 Hz, 2H), 7,40 (d, J 2,0 Hz, 2H), 7,06 (dd, J 8,2, 2,0 Hz, 2H), 4,14 (s, 1 H), 4,06-3,98 (m, 4H), 3,81 (ddd, J 11,0, 6,7, 3,5 Hz, 4H), 2,82 (t, J 5,4 Hz, 4H), 1,78-1,52 (m, 18H). LCMS (método 21): MH- m /z 614, TR 1,94 min.
Ejemplo 77

W-{1-Ciclooctil-2-oxo-2-[(2-oxospiro[indolina-3,4'-tetrahidropiran]-6-il)amino]etil}-8-hidroxioctanamida Se preparó a partir del in term edio 35 (50 mg, 0,13 mmol) y ácido 8-hidroxioctanoico (23 mg, 0,14 mmol) de acuerdo con el proced im iento A, y se purificó por HPLC preparativa (método 20), para dar el com puesto de l títu lo (45,2 mg, 66 %) como un polvo blanco. 5h (500 MHz, DMSO-d6) 10,38 (s, 1H), 10,08 (s, 1H), 7,99 (d, J 8,8 Hz, 1H), 7,42 (d, J 8,1 Hz, 1 H), 7,36 (d, J 1,8 Hz, 1H), 7,09 (dd, J 8,2, 1,8 Hz, 1H), 4,35-4,26 (m, 2H), 4,00 (ddd, J 11,0, 7,1,3,6 Hz, 2H), 3,79 (ddd, J 11,0, 6,9, 3,6 Hz, 2H), 3,39-3,28 (m obs., 2H), 2,23-2,06 (m, 2H), 2,00-1,89 (m, 1H), 1,79-1,69 (m, 2H), 1,69-1,28 (m, 20H), 1,27-1,18 (m, 6H). uPLC-MS (método 1): MH+ m /z 528, TR 3,06 min.
Ejemplo 78

2-Ciclooctil-W-(2-oxospiro[indolina-3,4'-tetrahidropiran]-6-il)-2-[(2-oxospiro[indolina-3,4'-tetrahidropiran]-6-il)carbamoilamino]acetamida
Una suspensión del in term edio 152 (65 mg, 0,19 mmol), in term edio 35 (99 mg, 0,26 mmol) y 4-(dimetilamino)piridina (60 mg, 0,49 mmol) en acetonitrilo (3 ml) se calentó a 60 °C en una atmósfera de nitrógeno durante 18 h. La mezcla de reacción se concentró al vacío y se trituró con agua (5 ml). Se filtró el material resultante y se lavó con una pequeña cantidad de agua. El sólido blanco resultante se purificó por HPLC de fase inversa (método 25, con metanol como solvente A) para dar, después de la liofilización, el com puesto de l título (85 mg, 74 %) como un sólido blanco. 5h (400 MHz, DMSO-de) 10,40 (s, 1H), 10,30 (s, 1H), 10,18 (s, 1H), 8,73 (s, 1 H), 7,44 (d, J 8,1 Hz, 1H), 7,37 (d, J 1,9 Hz, 1 H), 7,35 (d, J 8,1 Hz, 1 H), 7,22 (d, J 1,9 Hz, 1H), 7,11 (dd, J 8,2, 1,9 Hz, 1H), 6,75 (dd, J 8,1,2,0 Hz, 1H), 6,45 (d, J 8,8 Hz, 1H), 4,27 (dd, J 8,9, 6,4 Hz, 1 H), 4,01 (ddt, J 11, 1, 7,4, 3,7 Hz, 4H), 3,86-3,74 (m, 4H), 1,96 (d, J 8,4 Hz, 1 H), 1,80-1,21 (m, 22H). HPLC-MS (método 21): MH+ m /z 628, TR 2,16 min.
Ejemplos 79 a 143
Los com puestos de l título se prepararon por una secuencia de tres etapas:
• Etapa 1: reacción del in term edio 37 con el aldehído o cetona comercialmente disponible de acuerdo con el proced im iento F .
• Etapa 2: reacción del material así obtenido de acuerdo con el procedim iento H
• Etapa 3: reacción del material así obtenido con el in term edio 2 de acuerdo con el procedim iento I.
Análisis de SFC quiral (método 12) con las siguientes condiciones:
A: Chiralpak IC 25 cm, metanol al 15 %, dióxido de carbono al 85 %, 4 ml/min
B: Lux C-425 cm, metanol al 35 %-dióxido de carbono al 65 % (0,2 % v/v de NH3), 4 ml/min
C: Chiralcel OD-H 25 cm, metanol al 25 %, dióxido de carbono al 75 %, 4 ml/min
D: Chiralpak AS-H 25 cm, metanol al 30 %-dióxido de carbono al 70 %, 4 ml/min
E: Chiralpak IC 25 cm, metanol al 20 %, dióxido de carbono al 80 %, 15 ml/min
F: Chiralpak IC 25 cm, isopropanol al 25 %, dióxido de carbono al 75 %, 4 ml/min
G: Lux C-325 cm, metanol al 25 %-dióxido de carbono al 75 %, 4 ml/min
H: Chiralcel OD-H 25 cm, metanol al 20 %, dióxido de carbono al 80 %, 4 ml/min I: Chiralpak AS-H 25 cm, etanol al 15 %, dióxido de carbono al 85 %, 4 ml/min







Datos de R M N 1H seleccionados:



Ejemplos 144 y 145
Los com puestos de l título se prepararon por una secuencia de tres etapas:
• Etapa 1: reacción del intermedio 37con el aldehido o cetona comercialmente disponible de acuerdo con el procedimiento F.
• Etapa 2: reacción del material asi obtenido de acuerdo con el procedim iento H.
• Etapa 3: reacción del material asi obtenido con el in term edio 123 de acuerdo con el proced im iento I.

Ejemplos 146 a 183
Los com puestos de l título fueron preparados por una secuencia de dos etapas:
• Etapa 1: reacción de in term edio 37 con el aldehido o cetona comercialmente disponible de acuerdo con el proced im iento F.
• Etapa 2: reacción del material asi obtenido con in term edio 2 de acuerdo con el procedim iento G.




D a to s d e 1H R M N s e le c c io n a d o s :

Ejemplos 184 y 185
Los com puestos de l título se prepararon por una secuencia de dos etapas:
• Etapa 1: reacción del intermedio 55 con el aldehido o cetona comercialmente disponible de acuerdo con el procedimiento F.
• Etapa 2: reacción del material así obtenido con el in term edio 2 de acuerdo con el procedim iento G.


D a to s d e 1H R M N s e le c c io n a d o s :

Ejemplos 186 a 189
Los com puestos de l título se prepararon por una secuencia de tres etapas:
• Etapa 1: reacción del in term edio 55 o del in term edio 60, según corresponda, con el aldehido o la cetona disponible comercialmente de acuerdo con el procedim iento F.
• Etapa 2: reacción del material así obtenido con el in term edio 57 de acuerdo con el procedim iento G.
• Etapa 3: reacción del material así obtenido de acuerdo con el procedim iento C.

Ejemplos 190 a 204
Los com puestos de l título se prepararon de acuerdo con el procedim iento A a partir del in term edio 78 y el ácido
carboxílico apropiado.


Ejemplos 205 a 208
Los com puestos de l título se prepararon de acuerdo con el procedim iento D a partir del in term edio 78 y el cloruro de carbamoilo apropiado.

Ejemplos 209 a 232
Los com puestos de l título se prepararon de acuerdo con el procedim iento E a partir del in term edio 78y el cloroformiato apropiado.



Ejemplos 233 a 246
Los compuestos del título se sintetizaron de acuerdo con el procedimiento B a partir del intermedio 78 y el heterociclo de halógeno apropiado.


D a to s d e 1H R M N s e le c c io n a d o s :

Ejemplos 247 y 248
Los compuestos del título se prepararon de acuerdo con el procedimiento B a partir del intermedio 105 y el heterociclo de halógeno apropiado.

Ejemplos 249 a 263
Los compuestos del título se prepararon de acuerdo con el procedimiento A a partir del intermedio 35 y el ácido carboxílico apropiado.



Ejemplos 264 y 265
Los com puestos de l título se prepararon de acuerdo con el procedim iento D a partir del in term edio 35 y el cloruro de carbamoilo apropiado.

Ejemplos 266 y 267
Los com puestos de l título se prepararon de acuerdo con el procedim iento E a partir del in term edio 35y el cloroformiato apropiado.


Ejemplos 268 a 282
Los compuestos del título se prepararon de acuerdo con el procedimiento B a partir del intermedio 35 y el heterociclo de halógeno apropiado.


D a to s d e 1H R M N s e le c c io n a d o s :

Ejemplo 283
El compuesto del título se preparó a partir del ejemplo 203 de acuerdo con un procedimiento análogo al descrito para la preparación del ejemplo 12.

Ejemplos 284 a 286
Los com puestos de l títu lo se prepararon de acuerdo con el proced im iento A a partir del in term edio 151 y el ácido carboxílico apropiado.

Ejemplo 287
El com puesto de l títu lo se preparó de acuerdo con el procedim iento D a partir del in term edio 151 y el cloruro de carbamoilo apropiado.

Ejemplos 288 a 336
Los com puestos de l títu lo se prepararon por una secuencia de dos etapas:
• Etapa 1: reacción del in term edio 107 con el ácido carboxílico apropiado de acuerdo con el proced im iento A. • Etapa 2: reacción del material así obtenido de acuerdo con el procedim iento C.

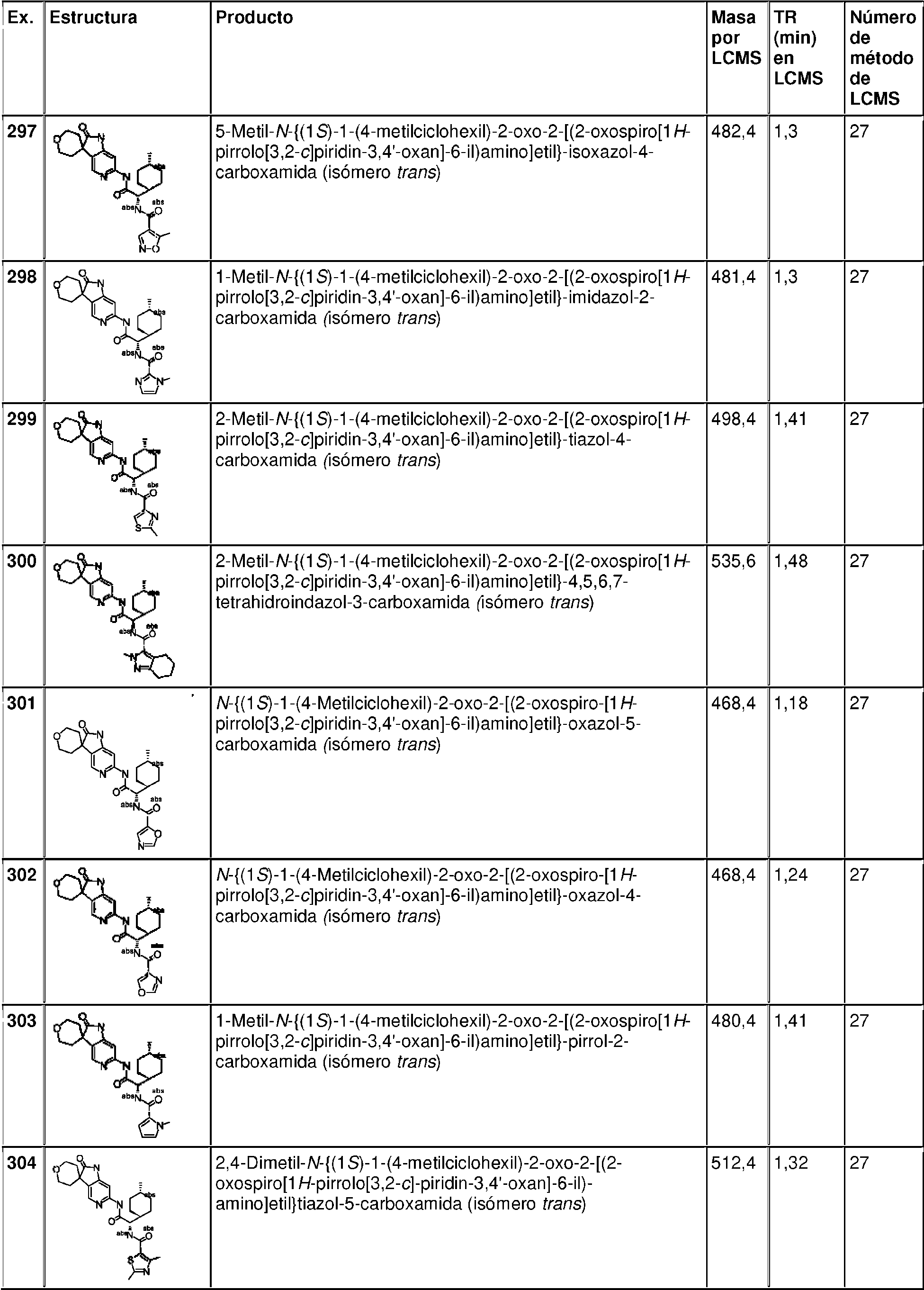




D a to s d e 1H R M N s e le c c io n a d o s :

Ejemplos 337 a 351
Los com puestos de l título se prepararon por una secuencia de dos etapas:
• Etapa 1: reacción del in term edio 107con el cloruro de carbamoilo apropiado de acuerdo con el procedim iento D.
• Etapa 2: reacción del material así obtenido de acuerdo con el procedim iento C.


Ejemplo 352
El c o m p u es to d e l título se preparó por una secuencia de dos etapas:
• Etapa 1: reacción del in te rm e d io 107 con cloroformiato de tert-butilo de acuerdo con el p ro c e d im ie n to E.
• Etapa 2: reacción del material así obtenido de acuerdo con el p ro c e d im ie n to C.

Ejemplos 353 a 355
Los c o m p u e s to s d e l títu lo se prepararon por una secuencia de dos etapas:
• Etapa 1: reacción del in te rm e d io 107 con el heterociclo de halógeno apropiado de acuerdo con el p ro ced im ien to B.
• Etapa 2: reacción del material así obtenido de acuerdo con el p ro c e d im ie n to C.

Ejemplo 356

WL{1-(4-Cloro-3-fluorofenil)-2-oxo-2-[(2-oxospiro[1 H-pirrolo[3,2-c]piridin-3,4'-tetrahidropiran]-6-il)amino]etil}-2-etilpirazol-3-carboxamida
Se añadió gota a gota ácido trifluoroacético (0,93 ml, 12,07 mmol) a una solución enfriada del in te rm e d io 156 (115 mg, 0,18 mmol) en DCM (15 ml) en nitrógeno a 0 °C. La mezcla de reacción se dejó calentar a temperatura ambiente y luego se agitó durante 18 h. Lo volátil se concentró al vacío, y el residuo se disolvió en acetonitrilo (7,5 ml) y una solución acuosa de hidróxido de amonio (7,5 ml). La mezcla de reacción se agitó a 20 °C durante 15 min, luego lo volátil se concentró al vacío. El residuo se diluyó con agua (20 ml) y la fase acuosa se extrajo con DCM (3 x 20 ml). Las capas orgánicas combinadas se secaron sobre sulfato de magnesio anhidro, se filtraron y se concentraron al vacío. El residuo se purificó mediante cromatografía en columna ultrarrápida, usando un gradiente de acetato de etilo en heptano (0-100 %) seguido de un gradiente de MeOH-DCM (0-100 %), para proporcionar, después de la liofilización, el co m p u e s to d e l títu lo (170 mg, 80 %) como un sólido blanco. 5h (500 MHz, DMSO-d6) 10,92 (br s, 1H), 10,88 (br s, 1H), 8,99 (d, J 7,0 Hz, 1H), 8,40 (s, 1 H), 7,72-7,62 (m, 2H), 7,60 (dd, J 10,4, 1,9 Hz, 1 H), 7,49 (d, J 2,0 Hz, 1 H), 7,45 (dd, J 8,4, 1,8 Hz, 1H), 7,05 (d, J 2,0 Hz, 1H), 5,91 (d, J 6,8 Hz, 1H), 4,52-4,39 (m, 2H), 4,04-3,91 (m, 2H), 3,87-3,72 (m, 2H), 1,89-1,73 (m, 2H), 1,69-1,54 (m, 2H), 1,28 (t, J 7,1 Hz, 3H). uPLC-MS (método 1): MH+ m /z 527, TR 2,52 min.
Ejemplo 357

2-Etil-W-{1-(4-fluoro-3-metilfenil)-2-oxo-2-[(2-oxospiro[1 H-pirrolo[3,2-c]-piridin-3,4'-tetrahidropiran]-6-il)amino]etil}pirazol-3-carboxamida
Se añadió gota a gota acido trifluoroacetico (0,93 ml, 12,07 mmol) a una solución del intermedio 160 (469 mg, 0,37 mmol) en DCM (20 ml) en una atmósfera de nitrógeno y se enfrió a 0 °C. La mezcla de reacción se agitó a 20 °C durante 18 h. Lo volátil se concentró al vacío, luego el residuo se disolvió en acetonitrilo (9,5 ml) y una solución acuosa de hidróxido de amonio (9,5 ml). La mezcla de reacción se agitó a 20 °C durante 15 min y lo volátil se concentró al vacío. El residuo se diluyó con agua (20 ml) y la fase acuosa se extrajo con DCM (3 x 20 ml). Las capas orgánicas combinadas se secaron sobre sulfato de magnesio anhidro, se filtraron y se concentraron al vacío. El residuo se purificó por cromatografía en columna ultrarrápida, usando un gradiente de acetato de etilo en heptano (0-100 %) seguido de un gradiente de MeOH-DCM (0-100 %), para dar, después de la liofilización, el compuesto del título (160 mg, 84 %) como un sólido blanco. 5h (500 MHz, DMSO-d6) 10,86 (br s, 1H), 10,79 (br s, 1H), 8,87 (d, J 6,7 Hz, 1H), 8,38 (s, 1 H), 7,67 (s, 1H), 7,55-7,45 (m, 2H), 7,43-7,36 (m, 1H), 7,19-7,11 (m, 1H), 7,04 (d, J 2,0 Hz, 1H), 5,82 (d, J 5,6 Hz, 1H), 4,52-4,39 (m, 2H), 4,03-3,93 (m, 2H), 3,86-3,72 (m, 2H), 2,24 (s, 3H), 1,88-1,75 (m, 2H), 1,68-1,54 (m, 2H), 1,28 (t, J 7,1 Hz, 3H). uPLC-MS (método 1): MH+ m/z 507, TR 2,34 min.
Claims (16)
1. Un compuesto de fórmula (I), o una sal farmacéuticamente aceptable del mismo:

en donde
el anillo A representa cicloalquilo C3-9, heterocicloalquilo C3-7 o heterobicicloalquilo C4-9, cualquiera de cuyos grupos puede estar opcionalmente sustituido con uno, dos o tres sustituyentes seleccionados independientemente de alquilo C1-6, halógeno, ciano, trifluorometilo, hidroxi, oxo, alcoxi C1-6, alquil(C1-6)tío, alquil(C1-6)sulfinilo, alquil(C1-6)sulfonilo, alquil(C2-6)carbonilo, amino, imino, alquil(C1-6)amino y dialquil(C1-6)amino;
B representa C-R2 o N;
D representa C-R3 o N;
E representa C-R4 o N;
R0 representa hidrógeno o alquilo C1-6;
R1 representa -CORa ;
Ra representa -CH(R5)N(H)C(O)R6 , -CH(R5)N(H)S(O)2R6 , -C(=CR5aR5b)N(H)C(O)R6 , -CH(R5)R7 , -CH(R5)N(H)R7 o -CH(R5)C(O)N(H)R7 ;
R2 representa hidrógeno, halógeno, ciano, alquilo C1-6, fluorometilo, difluorometilo, trifluorometilo, hidroxi, alcoxi C1-6 , difluorometoxi, trifluorometoxi, alquil(C1-6)sulfinilo o alquil(C1-6)sulfonilo;
R3 representa hidrógeno, halógeno, ciano, alquilo C1-6, fluorometilo, difluorometilo, trifluorometilo, hidroxi, alcoxi C1-6 , difluorometoxi, trifluorometoxi, alquil(C1-6)sulfinilo o alquil(C1-6)sulfonilo;
R4 representa hidrógeno, halógeno, ciano, alquilo C1-6, fluorometilo, difluorometilo, trifluorometilo, hidroxi, alcoxi C1-6 , difluorometoxi, trifluorometoxi, alquil(C1-6)sulfinilo o alquil(C1-6)sulfonilo;
R5 representa hidrógeno; o R5 representa alquilo C1-5, cicloalquilo C3-9, cicloalquil(C3-9)alquilo C1-5, bicicloalquilo C4-9, bicicloalquil(C4-9)alquilo C1-5, espirocicloalquilo C5-9, espirocicloalquil(C5-9)alquilo C1-5, tricicloalquilo C9-11, tricicloalquil(C9-11)alquilo C1-5, arilo, arilalquilo C1-5, heterocicloalquilo C3-7, heterocicloalquil(C3-7)alquilo C1-5, heteroarilo o heteroarilalquilo C1-5, cualquiera de cuyos grupos puede estar opcionalmente sustituido con uno, dos o tres sustituyentes seleccionados independientemente entre halógeno, ciano, nitro, alquilo C1-6, trifluorometilo, trifluoroetilo, fenilo, hidroxi, oxo, alcoxi C1-6, difluorometoxi, trifluorometoxi, alquil(C1-6)tío, alquil(C1-6)sulfinilo, alquil(C1-6)sulfonilo, amino, alquil(C1-6)amino, dialquil(C1-6)amino, alquil(C2-6)carbonilamino, alcoxi(C2-6)carbonilamino, alquil(C1-6)sulfonilamino, formilo, alquil(C2-6)carbonilo, carboxi, alcoxi(C2-6)carbonilo, aminocarbonilo, alquil(C1-6)aminocarbonilo, dialquil(C1-6)aminocarbonilo, aminosulfonilo, alquil(C1-6)aminosulfonilo y dialquil(C1-6)aminosulfonilo;
R5a representa cicloalquilo C3-7, bicicloalquilo C4-9, arilo, heterocicloalquilo C3-7 o heteroarilo, cualquiera de cuyos grupos puede estar opcionalmente sustituido con uno o más sustituyentes seleccionados de alquilo C1-6, halógeno, ciano, trifluorometilo, trifluoroetilo, fenilo, hidroxi, alcoxi C1-6, alquil(C1-6)tío, alquil(C1-6)sulfinilo, alquil(C1-6)sulfonilo, alquil(C2-6)carbonilo, amino, alquil(C1-6)amino y dialquil(C1-6)amino; y
R5b representa hidrógeno o alquilo C1-6; o
R5a y R5b, cuando se toman junto con el átomo de carbono al que ambos están unidos, representan cicloalquilo C3-7 , bicicloalquilo C4-9 o heterocicloalquilo C3-7, cualquiera de cuyos grupos puede estar opcionalmente sustituido con uno o dos sustituyentes seleccionados de alquilo C1-6, halógeno, ciano, trifluorometilo, trifluoroetilo, fenilo, hidroxi, alcoxi C1-6, alquil(C1-6)tío, alquil(C1-6)sulfinilo, alquil(C1-6)sulfonilo, alquil(C2-6)carbonilo, amino, alquil(C1-6)amino y dialquil(C1-6)amino;
R6 representa -NR6aR6b u -OR6c; o R6 representa alquilo C1-9, cicloalquilo C3-9, cicloalquil(C3-9)alquilo C1-6, arilo, arilalquilo C1-6, heterocicloalquilo C3-7, heterocicloalquil(C3-7)alquilo C1-6, heteroarilo, heteroarilalquilo C1-6 o espiro[heterocicloalquil(C3-7)][heteroarilo], cualquiera de cuyos grupos puede estar opcionalmente sustituido con uno, dos o tres sustituyentes seleccionados independientemente entre halógeno, ciano, nitro, alquilo C1-6, difluorometilo, trifluorometilo, difluoroetilo, trifluoroetilo, trifluoropropilo, ciclopropilo, ciclobutilo, ciclopropilmetilo, fenilo, fluorofenilo, hidroxi, hidroxialquilo C1-6, oxo, alcoxi C1-6, alcoxi(C1-6)alquilo C1-6, difluorometoxi, trifluorometoxi, alquil(C1-6)tío , alquil(C1-6)sulfinilo, alquil(C1-6)sulfonilo, amino, aminoalquilo C1-6, alquil(C1-6)amino, dialquil(C1-6)amino, dialquil(C1-6)aminoalquilo C1-6, pirrolidinilo, tetrahidropiranilo, morfolinilo, piperazinilo, alquil(C2-6)carbonilamino, alquil(C2-6)carbonilaminoalquilo C1-6, alcoxi(C2-6)carbonilamino, alquil(C1-6)sulfonilamino, formilo, alquil(C2-6)carbonilo, carboxi, alcoxi(C2-6)carbonilo, aminocarbonilo, alquil(C1-6)aminocarbonilo, dialquil(C1-6)aminocarbonilo, aminosulfonilo, alquil(C1-6)aminosulfonilo y dialquil(C1-6)aminosulfonilo;
R6a representa hidrógeno; o R6a representa alquilo C1-6, cicloalquilo C3-7, cicloalquil(C3-7)alquilo C1-6, arilo, arilalquilo C1-6, heterocicloalquilo C3-7, heterocicloalquil(C3-7)alquilo C1-6, heteroarilo, heteroarilalquilo C1-6 o espiro[heterocicloalquil(C3-7)][heteroarilo], cualquiera de cuyos grupos puede estar opcionalmente sustituido con uno, dos o tres sustituyentes seleccionados independientemente entre halógeno, ciano, nitro, alquilo C1-6, trifluorometilo, fenilo, fluorofenilo, hidroxi, hidroxialquilo C1-6, oxo, alcoxi C1-6, difluorometoxi, trifluorometoxi, alquil(C1-6)tío, alquil(C1-6)sulfinilo, alquil(C1-6)sulfonilo, amino, aminoalquilo C1-6, alquil(C1-6)amino, dialquil(C1-6)amino, pirrolidinilo, morfolinilo, piperazinilo, alquil(C2-6)carbonilamino, alquil(C2-6)carbonilaminoalquilo C1-6, alcoxi(C2-6)carbonilamino, alquil(C1-6)sulfonilamino, formilo, alquil(C2-6)carbonilo, carboxi, alcoxi(C2-6)carbonilo, aminocarbonilo, alquil(C1-6)aminocarbonilo, dialquil(C1-6)aminocarbonilo, aminosulfonilo, alquil(C1-6)aminosulfonilo y dialquil(C1-6)aminosulfonilo;
R6b representa hidrógeno o alquilo C1-6;
R6c representa alquilo C1-6, cicloalquilo C3-7, cicloalquil(C3-7)alquilo C1-6, arilo, arilalquilo C1-6, heterocicloalquilo C3-7, heterocicloalquil(C3-7)alquilo C1-6, heteroarilo o heteroarilalquilo C1-6, cualquiera de cuyos grupos puede estar opcionalmente sustituido con uno, dos o tres sustituyentes seleccionados independientemente entre halógeno, ciano, nitro, alquilo C1-6, trifluorometilo, fenilo, fluorofenilo, hidroxi, hidroxialquilo C1-6, oxo, alcoxi C1-6, difluorometoxi, trifluorometoxi, alquil(C1-6)tío, alquil(C1-6)sulfinilo, alquil(C1-6)sulfonilo, amino, aminoalquilo C1-6, alquil(C1-6)amino, dialquil(C1-6)amino, pirrolidinilo, morfolinilo, piperazinilo, alquil(C2-6)carbonilamino, alquil(C2-6)carbonilaminoalquilo C1-6, alcoxi(C2-6)carbonilamino, alquil(C1-6)sulfonilamino, formilo, alquil(C2-6)carbonilo, carboxi, alcoxi(C2-6)carbonilo, aminocarbonilo, alquil(C1-6)aminocarbonilo, dialquil(C1-6)aminocarbonilo, aminosulfonilo, alquil(C1-6)aminosulfonilo y dialquil(C1-6)aminosulfonilo; y
R7 representa arilo, heteroarilo o espiro[heterocicloalquil(C3-7)][heteroarilo], cualquiera de cuyos grupos puede estar opcionalmente sustituido con uno, dos o tres sustituyentes seleccionados independientemente entre halógeno, ciano, nitro, alquilo C1-6, difluorometilo, trifluorometilo, fenilo, fluorofenilo, hidroxi, hidroxialquilo C1-6, oxo, alcoxi C1-6, difluorometoxi, trifluorometoxi, alquil(C1-6)tío, alquil(C1-6)sulfinilo, alquil(C1-6)sulfonilo, amino, aminoalquilo C1-6, alquil(C1-6)amino, dialquil(C1-6)amino, pirrolidinilo, morfolinilo, piperazinilo, alquil(C2-6)carbonilamino, alquil(C2-6)carbonilaminoalquilo C1-6, alcoxi(C2-6)carbonilamino, alquil(C1-6)sulfonilamino, formilo, alquil(C2-6)carbonilo, carboxi, alcoxi(C2-6)carbonilo, aminocarbonilo, alquil(C1-6)aminocarbonilo, dialquil(C1-6)aminocarbonilo, aminosulfonilo, alquil(C1-6)aminosulfonilo y dialquil(C1-6)aminosulfonilo.
2. Un compuesto según la reivindicación 1, representado por la fórmula (I-1), (I-2), (I-3), (I-4) o (I-5), o una sal farmacéuticamente aceptable del mismo:


3. Un compuesto según la reivindicación 1, representado por la fórmula (IA) o una sal farmacéuticamente aceptable del mismo:

en donde A, B, D, E, R0, R5 y R6 son como se definen en la reivindicación 1.
4. Un compuesto según la reivindicación 1, representado por la fórmula (IB) o una sal farmacéuticamente aceptable del mismo:
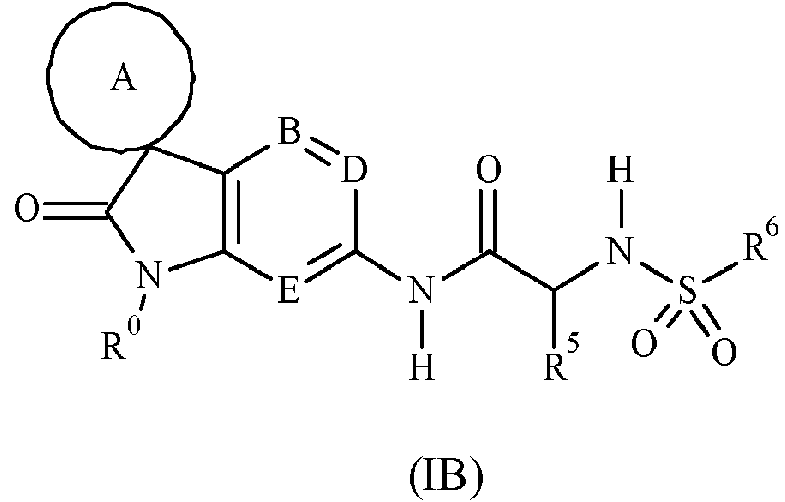
en donde A, B, D, E, R0, R5 y R6 son como se definen en la reivindicación 1.
5. Un compuesto según la reivindicación 1, representado por la fórmula (IC) o una sal farmacéuticamente aceptable del mismo:

en donde A, B, D, E, R0, R5 y R7 son como se definen en la reivindicación 1.
6. Un compuesto según la reivindicación 1, representado por la fórmula (ID) o una sal farmacéuticamente aceptable del mismo:

en donde A, B, D, E, R0, R5 y R7 son como se definen en la reivindicación 1.
7. Un compuesto según la reivindicación 1, representado por la fórmula (IE) o una sal farmacéuticamente aceptable del mismo:
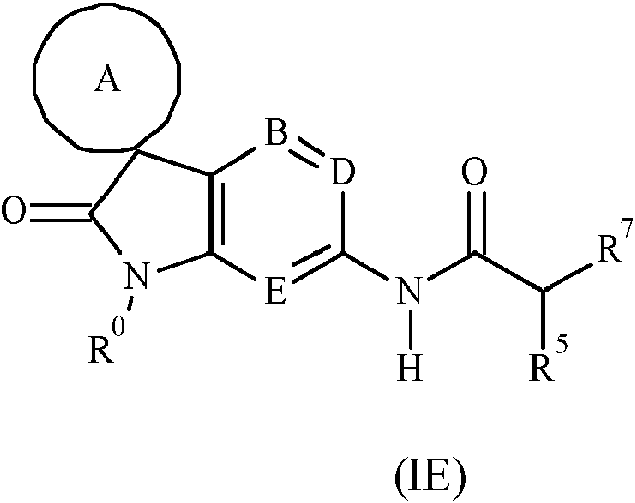
en donde A, B, D, E, R0, R5 y R7 son como se definen en la reivindicación 1.
8. Un compuesto según la reivindicación 1, representado por la fórmula (IF) o una sal farmacéuticamente aceptable del mismo:

en donde A, B, D, E, R0, R5a, R5b y R6 son como se definen en la reivindicación 1.
9. Un compuesto según la reivindicación 3, representado por la fórmula (IIA) o una sal farmacéuticamente aceptable del mismo:

en donde
W representa O, S, S(O), S(O)2, S(O)(NH) o N-R167;
R17 representa hidrógeno o alquilo C1-6; y
R0 , R3 , R5 y R6 son como se definen en la reivindicación 1.
10. Un compuesto según la reivindicación 3, representado por la fórmula (IIB) o una sal farmacéuticamente aceptable del mismo:

en donde
R0 , R5 y R6 son como se definen en la reivindicación 1; y
W es como se define en la reivindicación 9.
11. Un compuesto según una cualquiera de las reivindicaciones 1 a 7, 9 y 10, en el que R5 representa alquilo C1-5, cicloalquilo C3-9, cicloalquil(C3-9)alquilo C1-5, bicicloalquilo C4-9, bicicloalquil(C4-9)alquilo C1-5, espirocicloalquilo C5-9, tricicloalquilo C9-11, tricicloalquil(C9- n )alquilo C1-5, arilo, arilalquilo C1-5, heterocicloalquilo C3-7, heterocicloalquil(C3-7)alquilo C1-5 o heteroarilalquilo C1-5, cualquiera de cuyos grupos puede estar opcionalmente sustituido con uno, dos o tres sustituyentes seleccionados independientemente de halógeno, alquilo C1-6, trifluorometilo, fenilo y alcoxi C1-6.
12. Un compuesto según una cualquiera de las reivindicaciones 1 a 4 y 8 a 11, en el que R6 representa -NR6aR6b u -OR6c; o R6 representa alquilo C1-9, arilo, heterocicloalquilo C3-7, heteroarilo, heteroarilalquilo C1-6 o espiro[heterocicloalquil(C3-7)][heteroarilo], cualquiera de cuyos grupos puede estar opcionalmente sustituido con uno, dos o tres sustituyentes seleccionados independientemente entre halógeno, alquilo C1-6, difluorometilo, trifluorometilo, difluoroetilo, trifluoroetilo, trifluoropropilo, ciclopropilo, ciclobutilo, ciclopropilmetilo, hidroxi, hidroxialquilo C1-6, oxo, alcoxi(C1-6)alquilo C1-6, alquil(C1-6)sulfonilo, amino, aminoalquilo C1-6, dialquil(C1-6)aminoalquilo C1-6 y tetrahidropiranilo.
13. Un compuesto de fórmula (I) según se define en la reivindicación 1, o una sal farmacéuticamente aceptable del mismo, para uso en terapia.
14. Un compuesto de fórmula (I) según se define en la reivindicación 1, o una sal farmacéuticamente aceptable del mismo, para uso en el tratamiento y/o prevención de un trastorno inflamatorio o autoinmunitario.
15. Una composición farmacéutica que comprende un compuesto de fórmula (I) según se define en la reivindicación 1, o una sal farmacéuticamente aceptable del mismo, en asociación con un vehículo farmacéuticamente aceptable.
16. Una composición farmacéutica según la reivindicación 15 que comprende además un principio farmacéuticamente activo adicional.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GBGB1709456.6A GB201709456D0 (en) | 2017-06-14 | 2017-06-14 | Therapeutic agents |
| PCT/EP2018/065558 WO2018229079A1 (en) | 2017-06-14 | 2018-06-12 | Spirocyclic indolines as il-17 modulators |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2929369T3 true ES2929369T3 (es) | 2022-11-28 |
Family
ID=59358375
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES18732703T Active ES2929369T3 (es) | 2017-06-14 | 2018-06-12 | Indolinas espirocíclicas como moduladores de IL-17 |
Country Status (10)
| Country | Link |
|---|---|
| US (1) | US11052076B2 (es) |
| EP (1) | EP3638227B1 (es) |
| JP (1) | JP7163379B2 (es) |
| CN (1) | CN110769821B (es) |
| BR (1) | BR112019026678A2 (es) |
| CA (1) | CA3064156A1 (es) |
| EA (1) | EA202090019A1 (es) |
| ES (1) | ES2929369T3 (es) |
| GB (1) | GB201709456D0 (es) |
| WO (1) | WO2018229079A1 (es) |
Families Citing this family (42)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2018119142A1 (en) | 2016-12-21 | 2018-06-28 | Amgen Inc. | Anti-tnf alpha antibody formulations |
| WO2019138017A1 (en) | 2018-01-15 | 2019-07-18 | Ucb Biopharma Sprl | Fused imidazole derivatives as il-17 modulators |
| JP7349491B2 (ja) | 2018-07-12 | 2023-09-22 | ユーシービー バイオファルマ エスアールエル | Il-17モジュレーターとしてのスピロ環状インダン類似体 |
| GB201820166D0 (en) | 2018-12-11 | 2019-01-23 | Ucb Biopharma Sprl | Therapeutic agents |
| GB201820165D0 (en) * | 2018-12-11 | 2019-01-23 | Ucb Biopharma Sprl | Therapeutic agents |
| BR112021010453A2 (pt) | 2018-12-19 | 2021-08-24 | Leo Pharma A/S | Composto, e, composição farmacêutica |
| TWI752400B (zh) | 2019-01-07 | 2022-01-11 | 美商美國禮來大藥廠 | Il-17a抑制劑 |
| US20220162191A1 (en) | 2019-03-08 | 2022-05-26 | Leo Pharma A/S | Small molecule modulators of il-17 |
| GB201909190D0 (en) | 2019-06-26 | 2019-08-07 | Ucb Biopharma Sprl | Therapeutic agents |
| GB201909194D0 (en) | 2019-06-26 | 2019-08-07 | Ucb Biopharma Sprl | Therapeutic agents |
| GB201909191D0 (en) | 2019-06-26 | 2019-08-07 | Ucb Biopharma Sprl | Therapeutic agents |
| WO2021170631A1 (en) | 2020-02-25 | 2021-09-02 | UCB Biopharma SRL | Difluorocyclohexyl derivatives as il-17 modulators |
| WO2021170627A1 (en) | 2020-02-25 | 2021-09-02 | UCB Biopharma SRL | Difluorocyclohexyl derivatives as il-17 modulators |
| WO2021204800A1 (en) | 2020-04-07 | 2021-10-14 | UCB Biopharma SRL | Difluorocyclohexyl derivatives as il-17 modulators |
| CA3179686A1 (en) | 2020-04-07 | 2021-10-14 | UCB Biopharma SRL | Difluorocyclohexyl derivatives as il-17 modulators |
| WO2021220183A1 (en) * | 2020-04-30 | 2021-11-04 | Janssen Pharmaceutica Nv | Imidazopyrimidines as modulators of il-17 |
| KR20230019507A (ko) | 2020-04-30 | 2023-02-08 | 얀센 파마슈티카 엔.브이. | Il-17의 조절제로서의 이미다조피리다진 |
| US20230286943A1 (en) | 2020-05-27 | 2023-09-14 | Sanofi | Il-17a modulators |
| MX2022014925A (es) | 2020-05-27 | 2023-01-04 | Sanofi Sa | Moduladores de il-17a. |
| GB202103640D0 (en) * | 2021-03-16 | 2021-04-28 | C4X Discovery Ltd | Therapeutic compounds |
| MX2022015554A (es) | 2020-06-12 | 2023-01-30 | Leo Pharma As | Moduladores de molecula peque?a de interleucina 17 (il-17). |
| WO2021255174A1 (en) | 2020-06-18 | 2021-12-23 | Leo Pharma A/S | Small molecule modulators of il-17 |
| WO2021255085A1 (en) | 2020-06-18 | 2021-12-23 | Leo Pharma A/S | Small molecule modulators of il-17 |
| WO2021255086A1 (en) | 2020-06-18 | 2021-12-23 | Leo Pharma A/S | Small molecule modulators of il-17 |
| KR102817731B1 (ko) * | 2020-07-04 | 2025-06-10 | 히트젠 주식회사 | 면역 조절제 |
| EP3943495A1 (en) | 2020-07-24 | 2022-01-26 | Leo Pharma A/S | Small molecule modulators of il-17 |
| UY39484A (es) | 2020-11-02 | 2022-06-30 | Novartis Ag | Inhibidores de interleucina-17 |
| CN116406267A (zh) | 2020-11-09 | 2023-07-07 | Ucb生物制药有限责任公司 | 作为il-17调节剂的二环丙基甲基衍生物 |
| WO2022096411A1 (en) | 2020-11-09 | 2022-05-12 | UCB Biopharma SRL | Dicyclopropylmethyl derivatives as il-17 modulators |
| CA3200594A1 (en) | 2020-12-14 | 2022-06-23 | UCB Biopharma SRL | Imidazopyridazine derivatives as il-17 modulators |
| AR126255A1 (es) | 2021-07-01 | 2023-10-04 | UCB Biopharma SRL | Derivados de imidazotriazina como moduladores de il-17 |
| MX2024000504A (es) * | 2021-07-09 | 2024-04-05 | Dice Alpha Inc | Moduladores de il-17a basados en fenilacetamida y usos de los mismos. |
| WO2023025783A1 (en) | 2021-08-23 | 2023-03-02 | Leo Pharma A/S | Small molecule modulators of il-17 |
| WO2023111181A1 (en) | 2021-12-16 | 2023-06-22 | Leo Pharma A/S | Small molecule modulators of il-17 |
| WO2023166172A1 (en) | 2022-03-04 | 2023-09-07 | Leo Pharma A/S | Small molecule modulators of il-17 |
| CN119053589A (zh) * | 2022-04-21 | 2024-11-29 | 百济神州有限公司 | 小分子il-17a调节剂 |
| GB202210731D0 (en) | 2022-07-22 | 2022-09-07 | UCB Biopharma SRL | Therapeutic agents |
| TW202430165A (zh) | 2022-12-02 | 2024-08-01 | 丹麥商理奧藥品公司 | Il-17之小分子調節劑 |
| KR20250117693A (ko) | 2022-12-09 | 2025-08-05 | 사노피 | 치료용 화합물 |
| WO2025104230A1 (en) | 2023-11-17 | 2025-05-22 | Sanofi | Benzoxazoles as modulators of il-17a |
| WO2025248073A1 (en) | 2024-05-30 | 2025-12-04 | Sanofi | Therapeutic compounds |
| CN121342695A (zh) * | 2025-12-18 | 2026-01-16 | 苏州凯瑞医药科技有限公司 | 一种手性氨基酸的制备方法 |
Family Cites Families (20)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS57102863A (en) * | 1980-12-16 | 1982-06-26 | Takeda Chem Ind Ltd | Spiroindolinone and its preparation |
| PL147842B1 (en) * | 1984-05-12 | 1989-08-31 | Method of obtaining novel pyroisobenzimidazoles | |
| DE3445669A1 (de) * | 1984-12-14 | 1986-06-19 | Boehringer Mannheim Gmbh, 6800 Mannheim | Neue pyrrolo-benzimidazole, verfahren zu ihrer herstellung und diese verbindungen enthaltende arzneimittel |
| US4835280A (en) * | 1985-01-18 | 1989-05-30 | Boehringer Mannheim Gmbh | Indoline compounds for synthesis of pharmaceutically active pyrrolobenzimidazoles |
| DE3803775A1 (de) | 1988-02-09 | 1989-08-17 | Boehringer Mannheim Gmbh | Neue substituierte lactame, verfahren zu ihrer herstellung und arzneimittel, die diese verbindungen enthalten |
| CN1281590C (zh) | 2002-11-27 | 2006-10-25 | 南京凯衡科贸有限公司 | 具有抑制血管生成活性的六员氨基酰胺类衍生物 |
| US7129252B2 (en) | 2003-06-16 | 2006-10-31 | Guoqing P Chen | Six membered amino-amide derivatives an angiogenisis inhibitors |
| WO2008057280A1 (en) | 2006-10-27 | 2008-05-15 | Amgen Inc. | Multi-cyclic compounds and methods of use |
| US9309313B2 (en) | 2008-01-09 | 2016-04-12 | The Schepens Eye Research Institute, Inc. | Therapeutic compositions for treatment of ocular inflammatory disorders |
| US9284283B2 (en) | 2012-02-02 | 2016-03-15 | Ensemble Therapeutics Corporation | Macrocyclic compounds for modulating IL-17 |
| CA2863723C (en) | 2012-02-21 | 2020-09-22 | Carl Deutsch | 8-substituted 2-amino-[1,2,4]triazolo[1,5-a]pyrazines as syk tryrosine kinase inhibitors and gcn2 serin kinase inhibitors |
| US8969361B2 (en) | 2012-04-20 | 2015-03-03 | Annji Pharmaceutical Co., Ltd. | Cyclopropanecarboxylate esters of purine analogues |
| CN104619701B (zh) | 2012-09-13 | 2016-10-19 | 霍夫曼-拉罗奇有限公司 | 用于治疗cns疾病的2-氧代-2,3-二氢-吲哚类化合物 |
| WO2014066726A2 (en) | 2012-10-26 | 2014-05-01 | Ensemble Therapeutics Corporation | Compounds for modulating il-17 |
| EP3010918B1 (en) * | 2013-06-21 | 2018-08-15 | Zenith Epigenetics Ltd. | Novel substituted bicyclic compounds as bromodomain inhibitors |
| LT3027603T (lt) * | 2013-08-02 | 2018-08-10 | Pfizer Inc. | Heterobicikloarilo rorc2 inhibitoriai ir jų panaudojimo būdai |
| US9428515B2 (en) | 2014-05-09 | 2016-08-30 | Boehringer Ingelheim International Gmbh | Benzimidazole derivatives |
| US10385036B2 (en) * | 2015-01-30 | 2019-08-20 | Pfizer Inc. | Sulfonamide-substituted indole modulators of RORC2 and methods of use thereof |
| US10336748B2 (en) * | 2015-01-30 | 2019-07-02 | Pfizer Inc. | Methyoxy-substituted pyrrolopyridine modulators of RORC2 and methods of use thereof |
| WO2017069224A1 (ja) * | 2015-10-22 | 2017-04-27 | 塩野義製薬株式会社 | Mgat2阻害活性を有するスピロ環誘導体 |
-
2017
- 2017-06-14 GB GBGB1709456.6A patent/GB201709456D0/en not_active Ceased
-
2018
- 2018-06-12 US US16/619,573 patent/US11052076B2/en active Active
- 2018-06-12 BR BR112019026678-0A patent/BR112019026678A2/pt not_active Application Discontinuation
- 2018-06-12 ES ES18732703T patent/ES2929369T3/es active Active
- 2018-06-12 EA EA202090019A patent/EA202090019A1/ru unknown
- 2018-06-12 JP JP2020519841A patent/JP7163379B2/ja active Active
- 2018-06-12 CN CN201880039358.1A patent/CN110769821B/zh not_active Expired - Fee Related
- 2018-06-12 CA CA3064156A patent/CA3064156A1/en active Pending
- 2018-06-12 EP EP18732703.6A patent/EP3638227B1/en active Active
- 2018-06-12 WO PCT/EP2018/065558 patent/WO2018229079A1/en not_active Ceased
Also Published As
| Publication number | Publication date |
|---|---|
| JP7163379B2 (ja) | 2022-10-31 |
| EP3638227A1 (en) | 2020-04-22 |
| JP2020523418A (ja) | 2020-08-06 |
| US20200138797A1 (en) | 2020-05-07 |
| GB201709456D0 (en) | 2017-07-26 |
| CN110769821A (zh) | 2020-02-07 |
| WO2018229079A1 (en) | 2018-12-20 |
| US11052076B2 (en) | 2021-07-06 |
| CN110769821B (zh) | 2023-07-07 |
| EP3638227B1 (en) | 2022-08-10 |
| CA3064156A1 (en) | 2018-12-20 |
| EA202090019A1 (ru) | 2020-05-20 |
| BR112019026678A2 (pt) | 2020-06-30 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2929369T3 (es) | Indolinas espirocíclicas como moduladores de IL-17 | |
| ES2977683T3 (es) | Derivados de bencimidazolona y análogos de los mismos, como moduladores de IL-17 | |
| JP7349491B2 (ja) | Il-17モジュレーターとしてのスピロ環状インダン類似体 | |
| ES2963417T3 (es) | Derivados de imidazol condensados como moduladores de IL-17 | |
| EP3894003A1 (en) | Functionalised amine derivatives as il-17 modulators | |
| ES2735099T3 (es) | Inhibidores de dihidropirrolopiridina de ROR-gamma | |
| ES2983724T3 (es) | Derivados de difluorociclohexilo como moduladores de IL-17 | |
| ES2755335T3 (es) | Derivados de imidazoles tricíclicos fusionados como moduladores de la actividad del TNF | |
| WO2023275301A1 (en) | Imidazotriazine derivatives as il-17 modulators | |
| BR112021001044A2 (pt) | Compostos de sulfonimidamida como inibidores da atividade de interleucina-1 | |
| JP2023552864A (ja) | Il-17モジュレータとしてのイミダゾピリダジン誘導体 | |
| WO2021170631A1 (en) | Difluorocyclohexyl derivatives as il-17 modulators | |
| WO2021170627A1 (en) | Difluorocyclohexyl derivatives as il-17 modulators | |
| WO2022096411A1 (en) | Dicyclopropylmethyl derivatives as il-17 modulators | |
| EA039976B1 (ru) | Спироциклические индолины в качестве ингибиторов il-17 |