ES2942319T3 - Inhibidores de oxacina monoacilglicerol lipasa (MAGL) - Google Patents
Inhibidores de oxacina monoacilglicerol lipasa (MAGL) Download PDFInfo
- Publication number
- ES2942319T3 ES2942319T3 ES19711920T ES19711920T ES2942319T3 ES 2942319 T3 ES2942319 T3 ES 2942319T3 ES 19711920 T ES19711920 T ES 19711920T ES 19711920 T ES19711920 T ES 19711920T ES 2942319 T3 ES2942319 T3 ES 2942319T3
- Authority
- ES
- Spain
- Prior art keywords
- carbonyl
- oxazin
- pyrido
- hexahydro
- acetidine
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D498/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D498/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms in which the condensed system contains two hetero rings
- C07D498/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/535—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines
- A61K31/5375—1,4-Oxazines, e.g. morpholine
- A61K31/5383—1,4-Oxazines, e.g. morpholine ortho- or peri-condensed with heterocyclic ring systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/2004—Excipients; Inactive ingredients
- A61K9/2009—Inorganic compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/2004—Excipients; Inactive ingredients
- A61K9/2022—Organic macromolecular compounds
- A61K9/205—Polysaccharides, e.g. alginate, gums; Cyclodextrin
- A61K9/2054—Cellulose; Cellulose derivatives, e.g. hydroxypropyl methylcellulose
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/2004—Excipients; Inactive ingredients
- A61K9/2022—Organic macromolecular compounds
- A61K9/205—Polysaccharides, e.g. alginate, gums; Cyclodextrin
- A61K9/2059—Starch, including chemically or physically modified derivatives; Amylose; Amylopectin; Dextrin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/48—Preparations in capsules, e.g. of gelatin, of chocolate
- A61K9/4841—Filling excipients; Inactive ingredients
- A61K9/485—Inorganic compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/48—Preparations in capsules, e.g. of gelatin, of chocolate
- A61K9/4841—Filling excipients; Inactive ingredients
- A61K9/4858—Organic compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/48—Preparations in capsules, e.g. of gelatin, of chocolate
- A61K9/4841—Filling excipients; Inactive ingredients
- A61K9/4866—Organic macromolecular compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/06—Antimigraine agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/08—Antiepileptics; Anticonvulsants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D519/00—Heterocyclic compounds containing more than one system of two or more relevant hetero rings condensed among themselves or condensed with a common carbocyclic ring system not provided for in groups C07D453/00 or C07D455/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B2200/00—Indexing scheme relating to specific properties of organic compounds
- C07B2200/07—Optical isomers
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Veterinary Medicine (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Epidemiology (AREA)
- Biomedical Technology (AREA)
- Neurosurgery (AREA)
- Neurology (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Pain & Pain Management (AREA)
- Inorganic Chemistry (AREA)
- Rheumatology (AREA)
- Hospice & Palliative Care (AREA)
- Psychiatry (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Nitrogen And Oxygen As The Only Ring Hetero Atoms (AREA)
- Nitrogen And Oxygen Or Sulfur-Condensed Heterocyclic Ring Systems (AREA)
- Plural Heterocyclic Compounds (AREA)
Abstract
La invención proporciona nuevos compuestos heterocíclicos que tienen la fórmula general (Ic) en la que A, L, X, m, n y R20 a R23 son como se describen en este documento, composiciones que incluyen los compuestos, procesos de fabricación de los compuestos y métodos de uso de los compuestos. (Traducción automática con Google Translate, sin valor legal)
Description
DESCRIPCIÓN
Inhibidores de oxacina monoacilglicerol lipasa (MAGL)
Campo de la invención
La presente invención se refiere a compuestos orgánicos útiles para tratamiento o profilaxis en un mamífero, y en particular a inhibidores de monoacilglicerol lipasa (MAGL) para el tratamiento o la profilaxis de neuroinflamación, enfermedades neurodegenerativas, dolor, cáncer, trastornos mentales, esclerosis múltiple, enfermedad de Alzheimer, enfermedad de Parkinson, esclerosis lateral amiotrófica, traumatismo craneoencefálico, neurotoxicidad, apoplejía, epilepsia, ansiedad, migraña y/o depresión en un mamífero.
Antecedentes de la invención
Los endocanabinoides (EC) son lípidos de señalización que ejercen sus acciones biológicas al interactuar con los receptores de canabinoides (CBR), CB1 y CB2. Modulan múltiples procesos fisiológicos que incluyen neuroinflamación, neurodegeneración y regeneración de tejidos (Iannotti, F.A., et al., Progress in lipid research 2016, 62, 107-28). En el cerebro, el principal endocanabinoide, 2-araquidonoilglicerol (2-AG), se produce por diacilglicerol lipasas (DAGL) y se hidroliza por la monoacilglicerol lipasa, MAGL. MAGL hidroliza un 85 % de 2-AG; hidrolizándose el 15 % restante por ABHD6 y ABDH12 (Nomura, D.K., et al., Science 2011, 334, 809). MAGL se expresa en todo el cerebro y en la mayoría de los tipos de células cerebrales, incluyendo las neuronas, los astrocitos, los oligodendrocitos y los microgliocitos (Chanda, P.K., et al., Molecular pharmacology 2010, 78, 996; Viader, A., et al., Cell reports 2015, 12, 798). La hidrólisis de 2-AG da como resultado la formación de ácido araquidónico (AA), el precursor de las prostaglandinas (PG) y los leucotrienos (LT). El metabolismo oxidativo del AA se incrementa en los tejidos inflamados. Existen dos vías enzimáticas principales de oxigenación del ácido araquidónico implicadas en procesos inflamatorios, la ciclooxigenasa que produce GP y la 5-lipoxigenasa que produce LT. De los diversos productos de la ciclooxigenasa formados durante la inflamación, PGE2 es una de las más importantes. Estos productos se han detectado en sitios de inflamación, por ejemplo, en el líquido cefalorraquídeo de pacientes que padecen trastornos neurodegenerativos y se cree que contribuyen a la respuesta inflamatoria y a la progresión de la enfermedad. Los ratones que carecen de MAGL (Mgll-/-) presentan una actividad hidrolasa de 2-AG drásticamente reducida y niveles elevados de 2-AG en el sistema nervioso, mientras que otras especies de lípidos neutros y fosfolípidos que contienen araquidonoilo, incluyendo la anandamida (AEA), así como otros ácidos grasos libres, están inalterados. Por el contrario, los niveles de AA y prostaglandinas derivadas de AA y otros icosanoides, incluyendo prostaglandina E2 (PGE2), D2 (PGD2), F2 (Pg F2) y tromboxano B2 (TXB2), están fuertemente disminuidos. Las enzimas fosfolipasa A2 (PLA2) se han visto como la fuente principal de AA, pero los ratones carentes de cPLA2 tienen niveles de Aa inalterados en su cerebro, lo que refuerza el papel clave de MAGL en el cerebro para la producción de AA y la regulación de los procesos inflamatorios del cerebro.
La neuroinflamación es un cambio patológico común característico de las enfermedades del cerebro, incluyendo, pero no restringido a, enfermedades neurodegenerativas (por ejemplo, esclerosis múltiple, enfermedad de Alzheimer, enfermedad de Parkinson, esclerosis lateral amiotrófica, traumatismo craneoencefálico, neurotoxicidad, apoplejía, epilepsia y trastornos mentales tales como ansiedad y migraña). En el cerebro, la producción de icosanoides y prostaglandinas controla el proceso de neuroinflamación. El agente proinflamatorio lipopolisacárido (LPS) produce un incremento consistente y dependiente del tiempo en los icosanoides cerebrales que se atenúa notablemente en ratones Mgll-/-. El tratamiento con LPS también induce una elevación generalizada de las citocinas proinflamatorias, incluyendo la interleucina-1-a (IL-1-a), IL-1 b, IL-6 y el factor de necrosis tumoral-a (TNF-a) que se previene en ratones M gll-/-.
La neuroinflamación se caracteriza por la activación de las células inmunitarias inherentes del sistema nervioso central, la microglia y los astrocitos. Se ha informado de que los fármacos antiinflamatorios pueden suprimir en modelos preclínicos la activación de los neurogliocitos y la progresión de enfermedades que incluyen la enfermedad de Alzheimer y la esclerosis múltiple (Lleo A., Cell Mol Life Sci. 2007, 64, 1403). De forma importante, la alteración genética y/o farmacológica de la actividad MAGL también bloquea la activación inducida por LPS de microgliocitos en el cerebro (Nomura, D.K., et al., Science 2011, 334, 809).
Además, se demostró que la alteración genética y/o farmacológica de la actividad MAGL es protectora en varios modelos animales de neurodegeneración, incluyendo, pero no restringidos a, la enfermedad de Alzheimer, la enfermedad de Parkinson y la esclerosis múltiple. Por ejemplo, un inhibidor de MAGL irreversible se ha usado ampliamente en modelos preclínicos de neuroinflamación y neurodegeneración (Long, J.Z., et al., Nature chemical biology 2009, 5, 37). La inyección sistémica de dicho inhibidor da lugar al fenotipo de ratones Mgll-/- en el cerebro, incluyendo un incremento en los niveles de 2-AG, una reducción en los niveles de AA y la producción de icosanoides relacionados, así como la prevención de la producción de citocinas y la activación de la microglia después de la neuroinflamación inducida por LPS (Nomura, D.K., et al., Science 2011, 334, 809), lo que confirma en conjunto que MAGL es una diana farmacológica.
Como consecuencia de la alteración genética y/o farmacológica de la actividad MAGL, se incrementan los niveles
endógenos del sustrato natural de MAGL en el cerebro, 2-AG. Se ha informado de que 2-AG muestra efectos beneficiosos sobre el dolor con, por ejemplo, efectos antinocisensibles en ratones (Ignatowska-Jankowska B. et al., J. Pharmacol. Exp. Ther. 2015, 353, 424) y en trastornos mentales, tales como la depresión en modelos de estrés crónico (Zhong P. et al., Neuropsychopharmacology 2014, 39, 1763).
Además, los oligodendrocitos (OL), las células mielinizantes del sistema nervioso central y sus precursores (OPC) expresan el receptor canabinoide 2 (CB2) en su membrana. 2-AG es el ligando endógeno de los receptores CB1 y CB2. Se ha informado de que tanto los canabinoides como la inhibición farmacológica de MAGL atenúan la vulnerabilidad de OL y OPC a las agresiones excitotóxicas y, por lo tanto, pueden ser neuroprotectores (Bernal-Chico, A., et al., Glia 2015, 63, 163). Adicionalmente, la inhibición farmacológica de MAGL incrementa el número de OL mielinizantes en el cerebro de ratones, lo que sugiere que la inhibición de MAGL puede promover la diferenciación de OPC en OL mielinizantes in vivo (Alpar, A., et al., Nature comunicaciones 2014, 5, 4421). También se demostró que la inhibición de MAGL promueve la remielinización y la recuperación funcional en un modelo de ratón de esclerosis múltiple progresiva (Feliu A. et al., Journal of Neuroscience 2017, 37 (35), 8385).
Finalmente, en los últimos años se habla de un metabolismo altamente importante en la investigación contra el cáncer, especialmente del metabolismo de los lípidos. Los investigadores creen que la síntesis de ácidos grasos de novo juega un papel importante en la aparición de tumores. Muchos estudios ilustraron que los endocanabinoides tienen acciones antioncógenas, que incluyen efectos antiproliferación, inducción de apoptosis y antimetastásicos. MAGL como una enzima de descomposición importante tanto para el metabolismo de los lípidos como para el sistema de endocanabinoides, adicionalmente como parte de un distintivo de expresión génica, contribuye a diferentes aspectos de la oncogénesis (Qin, H., et al., Cell Biochem. Biophys. 2014, 70, 33, Nomura D.K. et al., Cell 2009, 140(1), 49-61, Nomura D.K. et al., Chem. Biol. 2011, 18(7), 846-856).
En conclusión, suprimir la acción y/o la activación de MAGL es una nueva estrategia terapéutica prometedora para el tratamiento o prevención de la neuroinflamación, enfermedades neurodegenerativas, dolor, cáncer y trastornos mentales. Además, suprimir la acción y/o la activación de MAGL es una nueva estrategia terapéutica prometedora para proporcionar neuroprotección y regeneración de mielina. En consecuencia, existe una gran necesidad médica no cubierta de nuevos inhibidores de MAGL.
Sumario de la invención
En un primer aspecto, la presente invención proporciona compuestos de fórmula (Ic)

en la que A, L, X, m, n y de R20 a R23 son como se define en el presente documento.
En otro aspecto, la presente invención proporciona un procedimiento de fabricación de los compuestos de fórmula (Ic) como se describe en el presente documento, que comprende:
hacer reaccionar 4a,5,6,7,8,8a-hexahidro-4H-pirido[4,3-b][1,4]oxacin-3-ona (1),

con una amina heterocíclica 2a, en la que A, L, X, m, n y de R20 a R23 son como se define en el presente documento

en presencia de una base y un reactivo formador de urea,
para formar dichos compuestos de fórmula (Ic).
En otro aspecto, la presente invención proporciona un compuesto de fórmula (Ic) como se describe en el presente documento, cuando se fabrica de acuerdo con los procedimientos descritos en el presente documento.
En otro aspecto, la presente invención proporciona un compuesto de fórmula (Ic) como se describe en el presente documento, para su uso como sustancia terapéuticamente activa.
En otro aspecto, la presente invención proporciona una composición farmacéutica que comprende un compuesto de fórmula (Ic) como se describe en el presente documento y un vehículo terapéuticamente inerte.
En otro aspecto, la presente invención proporciona el uso de un compuesto de fórmula (Ic) como se describe en el presente documento para inhibir monoacilglicerol lipasa (MAGL) en un mamífero.
En otro aspecto, la presente invención proporciona el uso de un compuesto de fórmula (Ic) como se describe en el presente documento para el tratamiento o la profilaxis de neuroinflamación, enfermedades neurodegenerativas, dolor, cáncer y/o trastornos mentales en un mamífero.
En otro aspecto, la presente invención proporciona el uso de un compuesto de fórmula (Ic) como se describe en el presente documento para el tratamiento o la profilaxis de esclerosis múltiple, enfermedad de Alzheimer, enfermedad de Parkinson, esclerosis lateral amiotrófica, traumatismo craneoencefálico, neurotoxicidad, apoplejía, epilepsia, ansiedad, migraña, depresión, carcinoma hepatocelular, carcinogénesis de colon, cáncer de ovario, dolor neuropático, neuropatía inducida por quimioterapia, dolor agudo, dolor crónico y/o espasticidad asociada con dolor en un mamífero.
En otro aspecto, la presente invención proporciona un compuesto de fórmula (Ic) como se describe en el presente documento para su uso en un procedimiento de inhibición de monoacilglicerol lipasa en un mamífero.
En otro aspecto, la presente invención proporciona un compuesto de fórmula (Ic) como se describe en el presente documento para su uso en el tratamiento o la profilaxis de neuroinflamación, enfermedades neurodegenerativas, dolor, cáncer y/o trastornos mentales en un mamífero.
En otro aspecto, la presente invención proporciona un compuesto de fórmula (Ic) como se describe en el presente documento, para su uso en el tratamiento o la profilaxis de esclerosis múltiple, enfermedad de Alzheimer, enfermedad de Parkinson, esclerosis lateral amiotrófica, traumatismo craneoencefálico, neurotoxicidad, apoplejía, epilepsia, ansiedad, migraña, depresión, carcinoma hepatocelular, carcinogénesis de colon, cáncer de ovario, dolor neuropático, neuropatía inducida por quimioterapia, dolor agudo, dolor crónico y/o espasticidad asociada con dolor en un mamífero.
En otro aspecto, la presente invención proporciona el uso de un compuesto de fórmula (Ic) como se describe en el presente documento para la preparación de un medicamento para inhibir monoacilglicerol lipasa en un mamífero.
En otro aspecto, la presente invención proporciona el uso de un compuesto de fórmula (Ic) como se describe en el presente documento para la preparación de un medicamento para el tratamiento o la profilaxis de neuroinflamación, enfermedades neurodegenerativas, dolor, cáncer y/o trastornos mentales en un mamífero.
En otro aspecto, la presente invención proporciona el uso de un compuesto de fórmula (Ic) como se describe en el presente documento para la preparación de un medicamento para el tratamiento o la profilaxis de esclerosis múltiple, enfermedad de Alzheimer, enfermedad de Parkinson, esclerosis lateral amiotrófica, traumatismo craneoencefálico, neurotoxicidad, apoplejía, epilepsia, ansiedad, migraña, depresión, carcinoma hepatocelular, carcinogénesis de colon, cáncer de ovario, dolor neuropático, neuropatía inducida por quimioterapia, dolor agudo, dolor crónico y/o espasticidad asociada con dolor en un mamífero.
Descripción detallada de la invención
Definiciones
Se ha de entender que los rasgos característicos, números enteros, características, compuestos, restos o grupos químicos descritos junto con un aspecto, modo de realización o ejemplo particular de la invención son aplicables a cualquier otro aspecto, modo de realización o ejemplo descrito en el presente documento, a menos que sea incompatible con el mismo. Todos los rasgos característicos divulgados en esta memoria descriptiva (incluyendo cualquier reivindicación adjunta, el resumen y los dibujos), y/o todas las etapas de cualquier procedimiento o proceso así divulgado, se pueden combinar en cualquier combinación, excepto combinaciones donde al menos algunos de dichos rasgos característicos y/o etapas son mutuamente excluyentes. La invención no se limita a los
detalles de ninguno de los modos de realización anteriores. La invención se extiende a cualquier rasgo novedoso o a cualquier combinación novedosa de los rasgos característicos divulgados en esta memoria descriptiva (incluyendo cualquier reivindicación adjunta, el resumen y los dibujos), o a cualquier etapa novedosa o a cualquier combinación novedosa de las etapas de cualquier procedimiento o proceso así divulgado.
El término "alquilo" se refiere a un grupo hidrocarburo saturado mono- o multivalente, por ejemplo, mono- o bivalente, lineal o ramificado, de 1 a 12 átomos de carbono. En algunos modos de realización preferentes, el grupo alquilo contiene de 1 a 6 átomos de carbono, por ejemplo, 1, 2, 3, 4, 5 o 6 átomos de carbono. En otros modos de realización, el grupo alquilo contiene de 1 a 3 átomos de carbono, por ejemplo, 1,2 o 3 átomos de carbono. Algunos ejemplos no limitantes de alquilo incluyen metilo, etilo, propilo, 2-propilo (isopropilo), n-butilo, /so-butilo, sec-butilo, ferc-butilo y 2,2-dimetilpropilo. Un ejemplo en particular preferente, aunque no limitante, de alquilo es metilo. Un grupo alquilo puede estar sustituido. Por tanto, el término "alquilo sustituido" se refiere a un grupo alquilo en el que al menos uno de los átomos de hidrógeno del grupo alquilo se ha reemplazado por un sustituyente como se describe en el presente documento, preferentemente por un sustituyente seleccionado de halógeno, hidroxi, alcoxi, arilalcoxi, trialquilsililoxi, cicloalquilo sustituido o no sustituido y heterociclilo sustituido o no sustituido. Lo más preferentemente, "alquilo sustituido" se refiere a un grupo alquilo en el que 1,2 o 3 de los átomos de hidrógeno del grupo alquilo se han reemplazado por un sustituyente seleccionado de halógeno, hidroxi, alcoxi, arilalcoxi, trialquilsililoxi, cicloalquilo sustituido o no sustituido y heterociclilo sustituido o no sustituido. Los ejemplos particulares, aunque no limitantes, de alquilo sustituido son 2-hidroxietilo, 2-metoxietilo, hidroximetilo, metoximetilo, trifluorometilo, oxetan-3-il-metilo, (1-ferc-butoxicarbonilacetidin-3-il)metilo, ciclopropilmetilo, 1-(clorometil)-2-hidroxi-etilo, 2-[ferc-butil(dimetil)silil]oxietilo y benciloximetilo.
El término "alcoxi" se refiere a un grupo alquilo, como se define previamente, unido al resto molecular original por medio de un átomo de oxígeno. A menos que se especifique de otro modo, el grupo alcoxi contiene de 1 a 12 átomos de carbono. En algunos modos de realización preferentes, el grupo alcoxi contiene de 1 a 6 átomos de carbono. En otros modos de realización, el grupo alcoxi contiene de 1 a 4 átomos de carbono. Todavía en otros modos de realización, el grupo alcoxi contiene de 1 a 3 átomos de carbono. Algunos ejemplos no limitantes de grupos alcoxi incluyen metoxi, etoxi, n-propoxi, isopropoxi, n-butoxi, isobutoxi y ferc-butoxi. Un ejemplo de alcoxi en particular preferente, aunque no limitante, es metoxi.
El término "halógeno" o "halo" se refiere a fluoro (F), cloro (Cl), bromo (Br) o yodo (I). Preferentemente, el término "halógeno" o "halo" se refiere a fluoro (F), cloro (Cl) o bromo (Br). Los ejemplos en particular preferentes, aunque no limitantes, de "halógeno" o "halo" son fluoro (F) y cloro (Cl).
El término "cicloalquilo", como se usa en el presente documento, se refiere a un grupo hidrocarburo monocíclico o bicíclico saturado o parcialmente insaturado de 3 a 10 átomos de carbono en el anillo. En algunos modos de realización preferentes, el grupo cicloalquilo es un grupo hidrocarburo monocíclico saturado de 3 a 8 átomos de carbono en el anillo. "Cicloalquilo bicíclico" se refiere a restos cicloalquilo que consisten en dos carbociclos saturados que tienen dos átomos de carbono en común, es decir, el puente que separa los dos anillos es un enlace simple o bien una cadena de uno o dos átomos de anillo, y a restos espirocíclicos, es decir, los dos anillos están conectados por medio de un átomo de anillo común. Preferentemente, el grupo cicloalquilo es un grupo hidrocarburo monocíclico saturado de 3 a 6 átomos de carbono en el anillo, por ejemplo, de 3, 4, 5 o 6 átomos de carbono. Algunos ejemplos no limitantes de cicloalquilo incluyen ciclopropilo, ciclobutilo, ciclopentilo, ciclohexilo y cicloheptilo.
El término "cicloalquiloxi" se refiere a un grupo cicloalquil-O-, es decir, un grupo cicloalquilo sustituido con un grupo oxi y unido al resto molecular original por medio de dicho grupo oxi.
El término "heterociclilo", como se usa en el presente documento, se refiere a un sistema de anillos saturado o parcialmente insaturado, mono o bicíclico, preferentemente monocíclico, de 3 a 10 átomos de anillo, preferentemente de 3 a 8 átomos de anillo, en el que 1, 2 o 3 de dichos átomos de anillo son heteroátomos seleccionados de N, O y S, siendo los átomos de anillo restantes, carbono. Preferentemente, de 1 a 2 de dichos átomos de anillo se seleccionan de N y O, siendo los átomos de anillo restantes, carbono. "Heterociclilo bicíclico" se refiere a restos heterocíclicos que consisten en dos ciclos que tienen dos átomos de anillo en común, es decir, el puente que separa los dos anillos es un enlace simple o bien una cadena de uno o dos átomos de anillo, y a restos espirocíclicos, es decir, los dos anillos están conectados por medio de un átomo de anillo común. Algunos ejemplos no limitantes de grupos heterociclilo monocíclicos incluyen acetidin-3-ilo, acetidin-2-ilo, oxetan-3-ilo, oxetan-2-ilo, 2-oxopirrolidin-1-ilo, 2-oxopirrolidin-3-ilo, 5-oxopirrolidin-2-ilo, 5-oxopirrolidin-3-ilo, 2-oxo-1-piperidilo, 2-oxo-3-piperidilo, 2-oxo-4-piperidilo, 6-oxo-2-piperidilo, 6-oxo-3-piperidilo, 1-piperidinilo, 2-piperidinilo, 3-piperidinilo, 4-piperidinilo, morfolino, morfolin-2-ilo y morfolin-3-ilo. Un grupo heterociclilo puede estar sustituido. Por tanto, el término "heterociclilo sustituido" se refiere a un grupo heterociclilo en el que al menos uno de los átomos de hidrógeno del grupo heterociclilo se ha reemplazado por un sustituyente como se describe en el presente documento, preferentemente por un sustituyente seleccionado de alquilo sustituido o no sustituido, halógeno y alcoxi, en el que dicho alquilo sustituido está sustituido con 1-3 sustituyentes seleccionados de hidroxi, halógeno, alcoxi, arilalcoxi y cicloalquilo. Lo más preferentemente, "heterociclilo sustituido" se refiere a un grupo heterociclilo en el que 1-2 de los átomos de hidrógeno del grupo heterociclilo se han reemplazados por un
sustituyente seleccionado de alquilo sustituido o no sustituido, halógeno y alcoxi, en el que dicho alquilo sustituido está sustituido con 1-3 sustituyentes seleccionados de hidroxi, halógeno, alcoxi, arilalcoxi y cicloalquilo. Los ejemplos particulares, aunque no limitantes, de heterociclilo sustituido son 2-metil-5-oxo-pirrolidin-1-ilo, 4,4-difluoro-1-piperidilo, 1-ferc-butoxicarbonilacetidin-3-ilo y 1-ferc-butoxicarbonilacetidin-2-ilo.
El término "heterocicliloxi" se refiere a un grupo heterociclilo-O-, es decir, un grupo heterociclilo sustituido con un grupo oxi y unido al resto molecular original por medio de dicho grupo oxi. Un ejemplo no limitante de un grupo heterocicliloxi es oxetaniloxi, tal como oxetan-3-iloxi.
El término "arilo" se refiere a un sistema de anillo carbocíclico monocíclico, bicíclico o tricíclico que tiene un total de 6 a 14 miembros de anillo, preferentemente, de 6 a 12 miembros de anillo, y más preferentemente de 6 a 10 miembros de anillo, y en el que al menos un anillo en el sistema es aromático. Algunos ejemplos no limitantes de arilo incluyen fenilo y 9H-fluorenilo (por ejemplo, 9H-fluoren-9-ilo). Un ejemplo en particular preferente, aunque no limitante, de arilo es fenilo. Un grupo arilo puede estar sustituido. Por tanto, el término "arilo sustituido" se refiere a un grupo arilo en el que al menos uno de los átomos de hidrógeno del grupo arilo se ha reemplazado por un sustituyente como se describe en el presente documento, por ejemplo, por un sustituyente seleccionado de halógeno, ciano, alcoxi, haloalcoxi, alquilo sustituido o no sustituido, cicloalquilo sustituido o no sustituido, cicloalquilalquilo sustituido o no sustituido, cicloalquiloxi sustituido o no sustituido, cicloalquiloxialquilo sustituido o no sustituido, cicloalquilalcoxi sustituido o no sustituido, heterociclilo sustituido o no sustituido, heterocicliloxi sustituido o no sustituido, arilo sustituido o no sustituido y ariloxi sustituido o no sustituido. Preferentemente, el término "arilo sustituido" se refiere a un grupo arilo en el que al menos uno de los átomos de hidrógeno del grupo arilo se ha reemplazado por un sustituyente seleccionado de halógeno y haloalquilo. Lo más preferentemente, "arilo sustituido" se refiere a un grupo arilo en el que 1-3 de los átomos de hidrógeno del grupo arilo se han reemplazado por un sustituyente seleccionado de halógeno y haloalquilo. Los ejemplos particulares, aunque no limitantes, de arilo sustituido son 4-fluorofenilo, 4-clorofenilo, 2-cloro-4-fluoro-fenilo, 4-(trifluorometil)fenilo y 3,4-difluorofenilo.
El término "heteroarilo" se refiere a un sistema de anillos mono- o multivalente, monocíclico, bicíclico o tricíclico, preferentemente bicíclico, que tiene un total de 5 a 14 miembros de anillo, preferentemente, de 5 a 12 miembros de anillo, y más preferentemente de 5 a 10 miembros de anillo, en el que al menos un anillo del sistema es aromático y al menos un anillo en el sistema contiene uno o más heteroátomos. Preferentemente, "heteroarilo" se refiere a un heteroarilo de 5-10 miembros que comprende 1, 2, 3 o 4 heteroátomos seleccionados independientemente de O, S y N. Lo más preferentemente, "heteroarilo" se refiere a un heteroarilo de 5-10 miembros que comprende de 1 a 2 heteroátomos seleccionados independientemente de O y N. Algunos ejemplos no limitantes de heteroarilo incluyen espiro[ciclopropano-1,3'-indolina] (por ejemplo, espiro[ciclopropano-1,3'-indolina]-1'-ilo), 2-piridilo, 3-piridilo, 4-piridilo, indol-1-ilo, 1 H-indol-2-ilo, 1 H-indol-3-ilo, 1 H-indol-4-ilo, 1 H-indol-5-ilo, 1 H-indol-6-ilo, 1 H-indol-7-ilo, 1,2-benzoxazol-3-ilo, 1,2-benzoxazol-4-ilo, 1,2-benzoxazol-5-ilo, 1,2-benzoxazol-6-ilo, 1,2-benzoxazol-7-ilo, 1H-indazol-3-ilo, 1H-indazol-4-ilo, 1H-indazol-5-ilo, 1H-indazol-6-ilo, 1H-indazol-7-ilo, pirazol-1-ilo, 1 H-pirazol-3-ilo, 1 H-pirazol-4-ilo, 1 H-pirazol-5-ilo, imidazol-1-ilo, 1H-imidazol-2-ilo, 1H-imidazol-4-ilo, 1H-imidazol-5-ilo, oxazol-2-ilo, oxazol-4-ilo y oxazol-5-ilo. Un ejemplo en particular preferente, aunque no limitante, de heteroarilo es indolilo, en particular 1 H-indol-3-ilo. Un grupo heteroarilo puede estar sustituido. Por tanto, el término "heteroarilo sustituido" se refiere a un grupo heteroarilo en el que al menos uno de los átomos de hidrógeno del grupo heteroarilo se ha reemplazado por un sustituyente como se describe en el presente documento, preferentemente por un sustituyente seleccionado de halógeno, alquilo sustituido o no sustituido, cicloalquilo, heterociclilo y heterociclilo sustituido con alcoxicarbonilo, en el que el alquilo sustituido está sustituido con 1-3 sustituyentes seleccionados de halógeno, hidroxi, heterociclilo, trialquilsililoxi, cicloalquilo y heterociclilo sustituido con alcoxicarbonilo. Lo más preferentemente, "heteroarilo sustituido" se refiere a un grupo heteroarilo en el que 1 2 de los átomos de hidrógeno del grupo heteroarilo se han reemplazado por un sustituyente seleccionado de halógeno, alquilo sustituido o no sustituido, cicloalquilo, heterociclilo y heterociclilo sustituido con alcoxicarbonilo, en el que el alquilo sustituido está sustituido con 1-3 sustituyentes seleccionados de halógeno, hidroxi, heterociclilo, trialquilsililoxi, cicloalquilo y heterociclilo sustituido con alcoxicarbonilo. Los ejemplos particulares, aunque no limitantes, de heteroarilo sustituido son 5-metil-1,2,4-oxadiazol-3-ilo, 5-fluoro-1-metil-indol-3-ilo, 5-cloro-1-metilindol-3-ilo, 5-cloro-1-ciclopropil-indol-3-ilo, 5-cloro-1-oxetanil-indol-3-ilo, 5-cloro-1-(oxetan-3-ilmetil)indol-3-ilo, 5-cloro-1 -(2-hidroxietil)indol-3-ilo, 1-(1-ferc-butoxicarbonilacetidin-3-il)-5-cloro-indol-3-ilo, 1-[(1-fercbutoxicarbonilacetidin-3-il)metil]-5-cloro-indol-3-ilo, 5-(trifluorometil)-2-piridilo, 5-(trifluorometil)-3-piridilo, 4-(trifluorometil)imidazol-1-ilo, 4-(trifluorometil)pirazol-1 -ilo, 4-ferc-butilpirazol-1-ilo, 4-ferc-butiloxazol-2-ilo, 5-cloro-1-(ciclopropilmetil)indol-3-ilo, 6-fluoro-1 H-indol-3-ilo, 5-fluoro-1,2-benzoxazol-3-ilo, 5-cloro-1 H-indol-3-ilo, 1-metilindazol-5-ilo, 5-cloro-1-[1-(clorometil)-2-hidroxi-etil]indol-3-ilo y 1-[2-[ferc-butil(dimetil)silil]oxietil]-5-cloro-indol-3-ilo.
El término "hidroxi" se refiere a un grupo -OH.
El término "ciano" se refiere a un grupo -CN (nitrilo).
El término "cicloalquilalquilo" se refiere a un grupo alquilo, en el que al menos uno de los átomos de hidrógeno del grupo alquilo se ha reemplazado por un grupo cicloalquilo. Preferentemente, "cicloalquilalquilo" se refiere a un
grupo alquilo en el que 1, 2 o 3 átomos de hidrógeno, lo más preferentemente 1 átomo de hidrógeno del grupo alquilo, se han reemplazado por un grupo cicloalquilo. Un ejemplo en particular preferente, aunque no limitante, de cicloalquilalquilo es ciclopropilmetilo.
El término "haloalquilo" se refiere a un grupo alquilo, en el que al menos uno de los átomos de hidrógeno del grupo alquilo se ha reemplazado por un átomo de halógeno, preferentemente fluoro. Preferentemente, "haloalquilo" se refiere a un grupo alquilo en el que 1, 2 o 3 átomos de hidrógeno del grupo alquilo se han reemplazado por un átomo de halógeno, lo más preferentemente fluoro. Los ejemplos en particular preferentes, aunque no limitantes, de haloalquilo son trifluorometilo y trifluoroetilo.
El término "haloalcoxi" se refiere a un grupo alcoxi, en el que al menos uno de los átomos de hidrógeno del grupo alcoxi se ha reemplazado por un átomo de halógeno, preferentemente fluoro. Preferentemente, "haloalcoxi" se refiere a un grupo alcoxi en el que 1,2 o 3 átomos de hidrógeno del grupo alcoxi se han reemplazado por un átomo de halógeno, lo más preferentemente fluoro. Un ejemplo en particular preferente, aunque no limitante, de haloalcoxi es trifluorometoxi (-OCF3).
El término "cicloalquilalcoxi" se refiere a un grupo alcoxi, en el que al menos uno de los átomos de hidrógeno del grupo alcoxi se ha reemplazado por un grupo cicloalquilo. Preferentemente, "cicloalquilalcoxi" se refiere a un grupo alcoxi en el que 1, 2 o 3 átomos de hidrógeno, lo más preferentemente 1 átomo de hidrógeno del grupo alcoxi, se han reemplazado por un grupo cicloalquilo. Un ejemplo en particular preferente, aunque no limitante, de cicloalquilalcoxi es ciclopropilmetoxi.
El término "cicloalquiloxialquilo" se refiere a un grupo alquilo, en el que al menos uno de los átomos de hidrógeno del grupo alquilo se ha reemplazado por un grupo cicloalquiloxi como se define en el presente documento. Preferentemente, "cicloalquiloxialquilo" se refiere a un grupo alquilo en el que 1, 2 o 3 átomos de hidrógeno, lo más preferentemente 1 átomo de hidrógeno del grupo alquilo, se han reemplazado por un grupo cicloalquiloxi. Un ejemplo en particular preferente, aunque no limitante, de cicloalquiloxialquilo es ciclopropoximetilo.
El término "hidroxialquilo" se refiere a un grupo alquilo, en el que al menos uno de los átomos de hidrógeno del grupo alquilo se ha reemplazado por un grupo hidroxi. Preferentemente, "hidroxialquilo" se refiere a un grupo alquilo en el que 1, 2 o 3 átomos de hidrógeno, lo más preferentemente 1 átomo de hidrógeno del grupo alquilo, se han reemplazado por un grupo hidroxi. Los ejemplos preferentes, aunque no limitantes, de hidroxialquilo son hidroximetilo e hidroxietilo (por ejemplo, 2-hidroxietilo). Un ejemplo en particular preferente, aunque no limitante, de hidroxialquilo es hidroximetilo.
El término "arilalcoxi" se refiere a un grupo alcoxi, en el que al menos uno de los átomos de hidrógeno del grupo alcoxi, se ha reemplazado por un grupo arilo. Preferentemente, "arilalcoxi" se refiere a un grupo alcoxi en el que 1, 2 o 3 átomos de hidrógeno, lo más preferentemente 1 átomo de hidrógeno del grupo alcoxi, se han reemplazado por un grupo arilo. Un ejemplo en particular preferente, aunque no limitante, de arilalcoxi es benciloxi.
El término "arilalcoxialquilo" se refiere a un grupo alquilo, en el que al menos uno de los átomos de hidrógeno del grupo alquilo se ha reemplazado por un grupo arilalcoxi como se define en el presente documento. Preferentemente, "arilalcoxialquilo" se refiere a un grupo alquilo en el que 1, 2 o 3 átomos de hidrógeno, lo más preferentemente 1 átomo de hidrógeno del grupo alquilo, se han reemplazado por un grupo arilalcoxi. Un ejemplo en particular preferente, aunque no limitante, de arilalcoxialquilo es benciloximetilo.
El término "alcoxialquilo" se refiere a un grupo alquilo, en el que al menos uno de los átomos de hidrógeno del grupo alquilo se ha reemplazado por un grupo alcoxi. Preferentemente, "alcoxialquilo" se refiere a un grupo alquilo en el que 1, 2 o 3 átomos de hidrógeno, lo más preferentemente 1 átomo de hidrógeno del grupo alquilo, se han reemplazado por un grupo alcoxi. Un ejemplo en particular preferente, aunque no limitante, de alcoxialquilo es 2-metoxietilo.
El término "alcoxicarbonilo" se refiere a un grupo alquil-OC(O)-(es decir, un éster alquílico).
El término "trialquilsililoxi" se refiere a un grupo (alquil)3Si-O-. Un ejemplo en particular preferente, aunque no limitante, de trialquilsililoxi es [ferc-butil(dimetil)silil]oxi.
El término "haloarilo" se refiere a un grupo arilo, en el que al menos uno de los átomos de hidrógeno del grupo arilo se ha reemplazado por un átomo de halógeno. Preferentemente, "haloarilo" se refiere a un grupo arilo en el que 1, 2 o 3 átomos de hidrógeno, más preferentemente 1 o 2 átomos de hidrógeno, lo más preferentemente 1 átomo de hidrógeno del grupo arilo, se han reemplazado por un átomo de halógeno. Un ejemplo en particular preferente, aunque no limitante, de haloarilo es fluorofenilo, en particular 4-fluorofenilo.
El término "sal farmacéuticamente aceptable" se refiere a las sales que conservan la eficacia biológica y las propiedades de las bases libres o ácidos libres, que no son biológicamente o de otro modo indeseables. Las sales se forman con ácidos inorgánicos tales como ácido clorhídrico, ácido bromhídrico, ácido sulfúrico, ácido nítrico,
ácido fosfórico y similares, en particular ácido clorhídrico, y ácidos orgánicos tales como ácido acético, ácido propiónico, ácido glicólico, ácido pirúvico, ácido oxálico, ácido maleico, ácido malónico, ácido succínico, ácido fumárico, ácido tartárico, ácido cítrico, ácido benzoico, ácido cinámico, ácido mandélico, ácido metanosulfónico, ácido etanosulfónico, ácido p-toluenosulfónico, ácido salicílico, N-acetilcisteína y similares. Además, estas sales se pueden preparar por adición de una base inorgánica o una base orgánica al ácido libre. Las sales derivadas de una base inorgánica incluyen, pero no se limitan a, sales de sodio, potasio, litio, amonio, calcio, magnesio y similares. Las sales derivadas de bases orgánicas incluyen, pero no se limitan a, sales de aminas primarias, secundarias y terciarias, aminas sustituidas que incluyen aminas sustituidas naturales, aminas cíclicas y resinas de intercambio iónico básicas, tales como resinas de isopropilamina, trimetilamina, dietilamina, trietilamina, tripropilamina, etanolamina, lisina, arginina, N-etilpiperidina, piperidina, poliimina y similares. Las sales farmacéuticamente aceptables particulares de los compuestos de fórmula (I) son las sales clorhidrato.
El término "éster farmacéuticamente aceptable" se refiere a los ésteres que se hidrolizan in vivo e incluyen los que se descomponen fácilmente en el cuerpo humano para dejar el compuesto original o una sal del mismo. Los grupos éster adecuados incluyen, por ejemplo, los derivados de ácidos carboxílicos alifáticos farmacéuticamente aceptables, en particular ácidos alcanoico, alquenoico, cicloalcanoico y alcanodioico, en los que cada resto alquilo o alquenilo, de forma ventajosa, no tiene más de 6 átomos de carbono. Los ejemplos representativos de ésteres particulares incluyen, pero no se limitan a, formiatos, acetatos, propionatos, butiratos, acrilatos y etilsuccinatos. Se describen ejemplos de tipos de profármacos farmacéuticamente aceptables en Higuchi y Stella, Pro-drugs as Novel Delivery Systems, vol. 14 de la A.C.S. Serie de simposios, y en Roche, ed., Bioreversible Carriers in Drug Design, American Pharmaceutical Association and Pergamon Press, 1987.
El término "grupo protector" (GP) indica un grupo que bloquea selectivamente un sitio reactivo en un compuesto multifuncional de modo que se pueda llevar a cabo selectivamente una reacción química en otro sitio reactivo desprotegido en el sentido convencionalmente asociado con él en química de síntesis. Los grupos protectores se pueden retirar en el momento apropiado. Los grupos protectores ejemplares son grupos protectores amino, grupos protectores carboxi o grupos protectores hidroxi. Los grupos protectores particulares son terc-butoxicarbonilo (Boc), benciloxicarbonilo (Cbz), fluorenilmetoxicarbonilo (Fmoc) y bencilo (Bn). Otros grupos protectores particulares son terc-butoxicarbonilo (Boc) y fluorenilmetoxicarbonilo (Fmoc). Un grupo protector más particular es terc-butoxicarbonilo (Boc). Los grupos protectores ejemplares y su aplicación en síntesis orgánica se describen, por ejemplo, en "Protective Groups in Organic Chemistry" de T.W. Greene y P.G.M. Wutts, 5.a ed., 2014, John Wiley & Sons, N.Y.
El término "reactivo formador de urea" se refiere a un compuesto químico que puede convertir una primera amina en una especie que reaccionará con una segunda amina, formando de este modo un derivado de urea. Los ejemplos no limitantes de un reactivo formador de urea incluyen carbonato de bis(triclorometilo), fosgeno, cloroformiato de triclorometilo, carbonato de 4-nitrofenilo y 1,1'-carbonildiimidazol. Los reactivos formadores de urea descritos en G. Sartori et al., Green Chemistry 2000, 2, 140 se incorporan en el presente documento por referencia.
Los compuestos de fórmula (I) pueden contener varios centros asimétricos y pueden estar presentes en forma de enantiómeros ópticamente puros, mezclas de enantiómeros, tales como, por ejemplo, racematos, diastereoisómeros ópticamente puros, mezclas de diastereoisómeros, racematos diastereoisómeros o mezclas de racematos diastereoisómeros.
De acuerdo con la convención de Cahn-Ingold-Prelog, el átomo de carbono asimétrico puede tener la configuración "R" o "S".
La abreviatura "MAGL" se refiere a la enzima monoacilglicerol lipasa. Los términos "MAGL" y "monoacilglicerol lipasa" se usan en el presente documento de manera intercambiable.
El término "tratamiento" como se usa en el presente documento incluye: (1) inhibir el estado, trastorno o afección (por ejemplo, detener, reducir o retrasar la aparición de la enfermedad, o una recidiva de la misma en caso de tratamiento de mantenimiento, de al menos un síntoma clínico o subclínico de la misma); y/o (2) aliviar la afección (es decir, provocar la regresión del estado, trastorno o afección o al menos uno de sus síntomas clínicos o subclínicos). El beneficio para un paciente que se va a tratar es estadísticamente significativo o bien al menos perceptible para el paciente o para el médico. Sin embargo, se apreciará que cuando se administra un medicamento a un paciente para tratar una enfermedad, el resultado puede no ser siempre un tratamiento eficaz.
El término "profilaxis", como se usa en el presente documento, incluye: prevenir o retrasar la aparición de síntomas clínicos del estado, trastorno o afección que padece un mamífero y especialmente un ser humano que puede estar aquejado de, o predispuesto a, el estado, trastorno o afección pero aún no experimenta o presenta síntomas clínicos o subclínicos del estado, trastorno o afección.
El término "neuroinflamación", como se usa en el presente documento, se refiere a la inflamación aguda y crónica del tejido nervioso, que es el principal componente tisular de las dos partes del sistema nervioso; el cerebro y la
médula espinal del sistema nervioso central (SNC), y los nervios periféricos ramificados del sistema nervioso periférico (SNP). La neuroinflamación crónica está asociada con enfermedades neurodegenerativas tales como la enfermedad de Alzheimer, la enfermedad de Parkinson y la esclerosis múltiple. La neuroinflamación aguda normalmente sigue de inmediato a una lesión del sistema nervioso central, por ejemplo, como resultado de un traumatismo craneoencefálico (TCE).
El término "traumatismo craneoencefálico" ("TCE", también conocido como "lesión intracraneal"), se refiere al daño al cerebro que resulta de una fuerza mecánica externa, tal como una aceleración o desaceleración rápida, un impacto, ondas expansivas o penetración de un proyectil.
El término "enfermedades neurodegenerativas" se refiere a enfermedades que están relacionadas con la pérdida progresiva de la estructura o la función de las neuronas, incluyendo la muerte de las neuronas. Los ejemplos de enfermedades neurodegenerativas incluyen, pero no se limitan a, esclerosis múltiple, enfermedad de Alzheimer, enfermedad de Parkinson y esclerosis lateral amiotrófica.
El término "trastornos mentales" (también llamados enfermedades mentales o trastornos psiquiátricos) se refiere a patrones conductuales o mentales que pueden provocar sufrimiento o una capacidad deficiente para funcionar en la vida. Dichos rasgos característicos pueden ser constantes, recidivantes y remisivos, o producirse como un episodio único. Los ejemplos de trastornos mentales incluyen, pero no se limitan a, ansiedad y depresión.
El término "dolor" se relaciona con una experiencia sensorial y emocional desagradable asociada con un daño tisular real o potencial. Los ejemplos de dolor incluyen, pero no se limitan a, dolor nocisensible, dolor crónico (incluyendo el dolor idiopático), dolor neuropático que incluye neuropatía inducida por quimioterapia, dolor fantasma y dolor psicogénico. Un ejemplo particular de dolor es el dolor neuropático, que se provoca por daño o enfermedad que afecta a cualquier parte del sistema nervioso implicada en las sensaciones corporales (es decir, el sistema somatosensitivo). En un modo de realización, "dolor" es dolor neuropático resultante de amputación o toracotomía. En un modo de realización, "dolor" es neuropatía inducida por quimioterapia.
El término "neurotoxicidad" se refiere a la toxicidad en el sistema nervioso. Se produce cuando la exposición a sustancias tóxicas naturales o artificiales (neurotoxinas) alteran la actividad normal del sistema nervioso de tal manera que provocan daño al tejido nervioso. Los ejemplos de neurotoxicidad incluyen, pero no se limitan a, la neurotoxicidad resultante de la exposición a sustancias usadas en quimioterapia, radioterapia, farmacoterapias, drogadicción y trasplantes de órganos, así como la exposición a metales pesados, determinados alimentos y aditivos alimentarios, plaguicidas, disolventes industriales y/o de limpieza, cosméticos y algunas sustancias naturales.
El término "cáncer" se refiere a una enfermedad caracterizada por la presencia de una neoplasia o tumor resultante del crecimiento anómalo descontrolado de las células (siendo dichas células "células cancerosas"). Como se usa en el presente documento, el término cáncer incluye explícitamente, pero no se limita a, carcinoma hepatocelular, carcinogénesis de colon y cáncer de ovario.
El término "mamífero" como se usa en el presente documento incluye tanto seres humanos como no humanos e incluye pero no se limita a seres humanos, primates no humanos, caninos, felinos, murinos, bovinos, equinos y porcinos. En un modo de realización en particular preferente, el término "mamífero" se refiere a seres humanos.
Compuestos de la invención
La presente invención proporciona un compuesto de fórmula (Ic)

o una sal farmacéuticamente aceptable del mismo, en la que:
L se selecciona de -C R 1R2-(CH2)p-, -(CH 2)p-CR1R2-, -OCR3R4-, -CR3R4O - y un enlace covalente;
m es 0, n es 0 o 1 y X es CR24; o
m es 1, n es 1 o 2 y X es CR24 o N;
p es 0, 1 o 2;
R1 se selecciona de halógeno, alcoxi Ci-e, alquilo Ci-e, halo-alquilo Ci-e, hidroxi-alquilo Ci-e, alcoxi Ci-e-alquil Ci-e-

halo-alcoxi Ci-6, halo-alcoxi Ci-6-alquil C1-6--, ciano y un grupo ; y R2 se selecciona de hidrógeno, halógeno e hidroxi; o
R1 y R2, tomados conjuntamente con el átomo de carbono al que están unidos, forman un cicloalquilo C3-12 o un heterociclilo C2-9;
R3 se selecciona de alquilo C1-6, halo-alquilo C1-6, cicloalquilo C3-12 y arilo C6-14; y R4 es hidrógeno; o
R3 y R4, tomados conjuntamente con el átomo de carbono al que están unidos, forman un cicloalquilo C3-12 o un heterociclilo C2-9;
R20 es hidrógeno o alquilo C1-6;
R21, R22 y R23 se seleccionan independientemente de hidrógeno, halógeno, ciano, hidroxi, alcoxi C1-6, halo-alcoxi C1-6, alquilo C1-6, halo-alquilo C1-6, hidroxi-alquilo C1-6, (halo-alquil C1-6)-hidroxi-alquilo C1-6, alcoxicarbonil C1-6-alquil C1-6- , alcoxicarbonil C1-6-NH-alcoxi C1 -6-, alcoxicarbonil C1-6-NH-(alcoxi C1 -6 )2-alquil C1-6-C(O)-NH-alcoxi C1 -6 -, alcoxicarbonil Ci-6-NH-alcox¡ Ci-6-alquil-Ci-6-C(0)-NH-alcox¡ C1-6- , SFs, (alquil Ci-6)3Si-0-alquil C1-6-, un grupo

y un grupo ;
R24 se selecciona de hidrógeno, halógeno, hidroxi, halo-alquilo C1-6 y alquilo C1-6;
R25 y R26 se seleccionan independientemente de hidrógeno, halógeno, alquilo C1-6, alcoxi C1-6, halo-alquilo C1-6, halo-alcoxi C1-6, alquil C1-6-SO2- y cicloalquilo C3-12;
R27 y R28 se seleccionan independientemente de hidrógeno, halógeno, alcoxi C1-6, halo-alcoxi C1-6, alquilo C1-6, halo-alquilo C1-6, hidroxi-alquilo C1-6, halógeno, hidroxi, alquilsulfonilo C1-6, carbamoílo, ciano, cicloalquil-alcoxi C1-6-, alquil C1-6-NH-C(O)- y cicloalquilo;
A y B se seleccionan independientemente de arilo C6-14, heteroarilo C1-13, cicloalquilo C3-12 y heterociclilo C2-9; C1 es cicloalquilo C3-12 o heterociclilo C2 -9 ;
C2 es arilo C6-14;
L2 se selecciona de un enlace covalente, -O -, -CH 2O-, -CH 2OCH2- y -CH 2-;
L3 se selecciona de un enlace covalente, -O -, -CH 2O-, -OCH2-, -CH 2OCH2- y -CH 2-; y
L3a se selecciona de un enlace covalente, -CH 2O-, -OCH2- , -CH 2OCH2- y -CH 2-.
En un modo de realización, la presente invención proporciona un compuesto de fórmula (Ic) como se describe en el presente documento, o una sal farmacéuticamente aceptable del mismo, en la que el compuesto de fórmula (Ic) es un compuesto de fórmula (Id):

en la que A, L, X, m, n y R20-R23 son como se define en la reivindicación 1.
En un modo de realización, la presente invención proporciona un compuesto de fórmula (Ic) como se describe en el presente documento, o una sal farmacéuticamente aceptable del mismo, en la que el compuesto de fórmula (Ic) es un compuesto de fórmula (Ie):

en la que A, L, X, m, n y R20-R23 son como se define en la reivindicación 1.
En un modo de realización, la presente invención proporciona un compuesto de fórmula (Ic) como se describe en el presente documento, o una sal farmacéuticamente aceptable del mismo, en la que el compuesto de fórmula (Ic) es un compuesto de fórmula (If):

en la que A, L, X, m, n y R20-R23 son como se define en la reivindicación 1.
En un modo de realización, la presente invención proporciona un compuesto de fórmula (Ic) como se describe en el presente documento, o una sal farmacéuticamente aceptable del mismo, en la que el compuesto de fórmula (Ic) es un compuesto de fórmula (Ig):

en la que A, L, X, m, n y R20-R23 son como se define en la reivindicación 1.
En un modo de realización, la presente invención proporciona un compuesto de fórmula (Ic) como se describe en el presente documento, o una sal farmacéuticamente aceptable del mismo, en la que:
m es 0, n es 0 o 1 y X es CR24; o
m es 1, n es 1 y X es CR24 o N.
En un modo de realización, la presente invención proporciona un compuesto de fórmula (Ic) como se describe en el presente documento, o una sal farmacéuticamente aceptable del mismo, en la que p es 0 o 1.
En un modo de realización preferente, la presente invención proporciona un compuesto de fórmula (Ic) como se describe en el presente documento, o una sal farmacéuticamente aceptable del mismo, en la que p es 0.
En un modo de realización, la presente invención proporciona un compuesto de fórmula (Ic) como se describe en el presente documento, o una sal farmacéuticamente aceptable del mismo, en la que:
R1 se selecciona de halógeno, alcoxi C1-6 , alquilo C1-6 , halo-alquilo C1-6, hidroxi-alquilo C1-6, alcoxi C1-6-alquil C1-6-,

halo-alcoxi Ci-6, halo-alcoxi Ci-6-alquil C1-6--, ciano y un grupo y R2 se selecciona de hidrógeno, halógeno e hidroxi; o
R1 y R2, tomados conjuntamente con el átomo de carbono al que están unidos, forman un cicloalquilo C3 -12.
En un modo de realización preferente, la presente invención proporciona un compuesto de fórmula (Ic) como se describe en el presente documento, o una sal farmacéuticamente aceptable del mismo, en la que R1 se selecciona de halógeno, alquilo C1-6 , hidroxi-alquilo C1-6 , alcoxi Ci-e-alquil C1-6-, halo-alcoxi C1-6 y un grupo

En un modo de realización en particular preferente, la presente invención proporciona un compuesto de fórmula (le) como se describe en el presente documento, o una sal farmacéuticamente aceptable del mismo, en la que R1

se selecciona de 2-metoxietilo, metilo, 2,2,2-trifluoroetoxi, fluoro, 2-hidroxietilo y un grupo ; y R2 es hidrógeno o fluoro.
En un modo de realización, la presente invención proporciona un compuesto de fórmula (Ic) como se describe en el presente documento, o una sal farmacéuticamente aceptable del mismo, en la que R3 es alquilo C1-6 o haloalquilo C1-6; y R4 es hidrógeno.
En un modo de realización preferente, la presente invención proporciona un compuesto de fórmula (Ic) como se describe en el presente documento, o una sal farmacéuticamente aceptable del mismo, en la que R3 es alquilo C1-6; y R4 es hidrógeno.
En un modo de realización en particular preferente, la presente invención proporciona un compuesto de fórmula (Ic) como se describe en el presente documento, o una sal farmacéuticamente aceptable del mismo, en la que R3 es metilo; y R4 es hidrógeno.
En un modo de realización preferente, la presente invención proporciona un compuesto de fórmula (Ic) como se describe en el presente documento, o una sal farmacéuticamente aceptable del mismo, en la que R20 es hidrógeno.
En un modo de realización, la presente invención proporciona un compuesto de fórmula (Ic) como se describe en el presente documento, o una sal farmacéuticamente aceptable del mismo, en la que R21 se selecciona de hidrógeno, halógeno, hidroxi, alcoxi C1-6, halo-alcoxi C1-6, alquilo C1-6, halo-alquilo C1-6, hidroxi-alquilo C1-6, (haloalquil C1-6)-hidroxi-alquilo C1-6, alcoxicarbonil C1-6-alquil C1 -6- , alcoxicarbonil C1-6-NH-alcoxi C1 -6-, alcoxicarbonil Ci-6-NH-(alcox¡ Ci-e)2-alquil Ci-6-C(0)-NH-alcox¡ C1-6-, alcoxicarbonil Ci-6-NH-alcox¡ Ci-e-alquil Ci-e-C(0)-NH-


alcoxi C1-6-, SFs, (alquil Ci-6)3Si-0-alquil C1-6-, un grupo y un grupo en la que R27, R28, C1, C2, L3 y L3a son como se define en el presente documento.
En un modo de realización preferente, la presente invención proporciona un compuesto de fórmula (Ic) como se describe en el presente documento, o una sal farmacéuticamente aceptable del mismo, en la que R21 se selecciona de halógeno, alcoxi C1-6, halo-alcoxi C1-6, alquilo C1-6, halo-alquilo C1-6, SF5 , arilo C6-14 y un grupo

en la que R27, R28, C1 y L3 son como se define en el presente documento.
En un modo de realización en particular preferente, la presente invención proporciona un compuesto de fórmula (Ic) como se describe en el presente documento, o una sal farmacéuticamente aceptable del mismo, en la que R21 se selecciona de fluoro, cloro, bromo, metilo, metoxi, ferc-butilo, propilo, trifluorometoxi, 2-fluoroetoxi, 2,2,2-
trifluoroetoxi, trifluorometilo, 2,2,2-trifluoroetilo, 1,1 -difluoroetilo, SF5 , fenilo y un grupo 
que R27, R28, C1 y L3 son como se define en el presente documento.
En un modo de realización, la presente invención proporciona un compuesto de fórmula (Ic) como se describe en el presente documento, o una sal farmacéuticamente aceptable del mismo, en la que R22 se selecciona de hidrógeno, halógeno, alcoxi C1-6, halo-alcoxi C1-6, alquilo C1-6 y ciano.
En un modo de realización preferente, la presente invención proporciona un compuesto de fórmula (Ic) como se describe en el presente documento, o una sal farmacéuticamente aceptable del mismo, en la que R22 se selecciona de hidrógeno, halógeno, alcoxi C1-6 y halo-alcoxi C1-6.
En un modo de realización en particular preferente, la presente invención proporciona un compuesto de fórmula (Ic) como se describe en el presente documento, o una sal farmacéuticamente aceptable del mismo, en la que R22 se selecciona de hidrógeno, fluoro, cloro, metoxi, metilo y trifluorometilo.
En un modo de realización preferente, la presente invención proporciona un compuesto de fórmula (Ic) como se describe en el presente documento, o una sal farmacéuticamente aceptable del mismo, en la que R23 es hidrógeno o halógeno.
En un modo de realización en particular preferente, la presente invención proporciona un compuesto de fórmula (Ic) como se describe en el presente documento, o una sal farmacéuticamente aceptable del mismo, en la que R23 es hidrógeno o fluoro.
En un modo de realización, la presente invención proporciona un compuesto de fórmula (Ic) como se describe en el presente documento, o una sal farmacéuticamente aceptable del mismo, en la que R24 es hidrógeno.
En un modo de realización, la presente invención proporciona un compuesto de fórmula (Ic) como se describe en el presente documento, o una sal farmacéuticamente aceptable del mismo, en la que R25 se selecciona de hidrógeno, halógeno, alquilo C1-6, alcoxi C1-6, halo-alquilo C1-6, halo-alcoxi C1-6, alquil C1-6-SO2- y cicloalquilo C3-12.
En un modo de realización preferente, la presente invención proporciona un compuesto de fórmula (Ic) como se describe en el presente documento, o una sal farmacéuticamente aceptable del mismo, en la que R25 se selecciona de hidrógeno, halógeno, alcoxi C1-6 y cicloalquilo C3-12.
En un modo de realización en particular preferente, la presente invención proporciona un compuesto de fórmula (Ic) como se describe en el presente documento, o una sal farmacéuticamente aceptable del mismo, en la que R25 se selecciona de hidrógeno, metoxi, fluoro y ciclopropilo.
En un modo de realización, la presente invención proporciona un compuesto de fórmula (Ic) como se describe en el presente documento, o una sal farmacéuticamente aceptable del mismo, en la que R26 se selecciona de hidrógeno, alquilo C1-6 y alcoxi C1-6.
En un modo de realización preferente, la presente invención proporciona un compuesto de fórmula (Ic) como se describe en el presente documento, o una sal farmacéuticamente aceptable del mismo, en la que R26 es hidrógeno o alcoxi C1-6.
En un modo de realización en particular preferente, la presente invención proporciona un compuesto de fórmula (Ic) como se describe en el presente documento, o una sal farmacéuticamente aceptable del mismo, en la que R26 es hidrógeno o metoxi.
En un modo de realización preferente, la presente invención proporciona un compuesto de fórmula (Ic) como se
describe en el presente documento, o una sal farmacéuticamente aceptable del mismo, en la que R27 se selecciona de hidrógeno, halo-alcoxi C1-6, alquilo C1-6, halo-alquilo C1-6 y halógeno.
En un modo de realización en particular preferente, la presente invención proporciona un compuesto de fórmula (Ic) como se describe en el presente documento, o una sal farmacéuticamente aceptable del mismo, en la que R27 se selecciona de metilo, trifluorometoxi, trifluorometilo, 2,2,2-trifluoro-1-metil-etoxi, 2,2,2-trifluoro-1, 1 -dimetil-etoxi, 2,2,2-trifluoroetoxi, fluoro y cloro.
En un modo de realización preferente, la presente invención proporciona un compuesto de fórmula (Ic) como se describe en el presente documento, o una sal farmacéuticamente aceptable del mismo, en la que R28 se selecciona de hidrógeno, alquilo C1-6 y halógeno.
En un modo de realización en particular preferente, la presente invención proporciona un compuesto de fórmula (Ic) como se describe en el presente documento, o una sal farmacéuticamente aceptable del mismo, en la que R28 se selecciona de hidrógeno, metilo, fluoro y cloro.
En un modo de realización preferente, la presente invención proporciona un compuesto de fórmula (Ic) como se describe en el presente documento, o una sal farmacéuticamente aceptable del mismo, en la que A es arilo C6-14 o heteroarilo C1-13.
En un modo de realización en particular preferente, la presente invención proporciona un compuesto de fórmula (Ic) como se describe en el presente documento, o una sal farmacéuticamente aceptable del mismo, en la que A se selecciona de fenilo, indol-3-ilo, 2-piridilo y 3-piridilo.
En un modo de realización preferente, la presente invención proporciona un compuesto de fórmula (Ic) como se describe en el presente documento, o una sal farmacéuticamente aceptable del mismo, en la que B es arilo C6-14 o heteroarilo C1-13.
En un modo de realización en particular preferente, la presente invención proporciona un compuesto de fórmula (Ic) como se describe en el presente documento, o una sal farmacéuticamente aceptable del mismo, en la que B es fenilo o 1,2,4-oxadiazol-5-ilo.
En un modo de realización preferente, la presente invención proporciona un compuesto de fórmula (Ic) como se describe en el presente documento, o una sal farmacéuticamente aceptable del mismo, en la que L se selecciona de -C R 1R2-(CH2)p-, -OCR3R4-, -CR 3R4O - y un enlace covalente; en la que de R1 a R4 y p son como se define en el presente documento.
En un modo de realización, la presente invención proporciona un compuesto de fórmula (Ic) como se describe en el presente documento, o una sal farmacéuticamente aceptable del mismo, en la que L2 se selecciona de un enlace covalente, -O - y -CH 2-.
En un modo de realización preferente, la presente invención proporciona un compuesto de fórmula (Ic) como se describe en el presente documento, o una sal farmacéuticamente aceptable del mismo, en la que L2 es un enlace covalente.
En un modo de realización, la presente invención proporciona un compuesto de fórmula (Ic) como se describe en el presente documento, o una sal farmacéuticamente aceptable del mismo, en la que:
m es 0, n es 0 o 1 y X es CR24; o
m es 1, n es 1 y X es CR24 o N;
L se selecciona de -CR1R2-(CH2)p- , -OCHR3-, -CHR3O - y un enlace covalente;
p es 0 o 1;
R1 se selecciona de halógeno, alcoxi C1-6 , alquilo C1-6 , halo-alquilo C1-6, hidroxi-alquilo C1-6, alcoxi Ci-e-alquil C1-6-,

halo-alcoxi C1-6 , halo-alcoxi Ci-e-alquil C1-6--, ciano y un grupo ; y R2 se selecciona de hidrógeno, halógeno e hidroxi; o
R1 y R2, tomados conjuntamente con el átomo de carbono al que están unidos, forman un cicloalquilo C3 -12 ;
R3 es alquilo Ci-6 o halo-alquilo Ci-a;
R20 es hidrógeno o alquilo Ci-a;
R21 se selecciona de hidrógeno, halógeno, hidroxi, alcoxi Ci-a, halo-alcoxi Ci-a, alquilo Ci-a, halo-alquilo Ci-a, hidroxialquilo C1-6 , (halo-alquil Ci-6)-hidroxi-alquilo Ci-e, alcoxicarbonil Ci-e-alquil Ci-e-, alcoxicarbonil Ci-6-NH-alcox¡


Ci-a-, SF5 , (alquil Ci-a)3Si-O-alquil Ci-a-, un grupo y un grupo R22 se selecciona de hidrógeno, halógeno, alcoxi Ci-a, halo-alcoxi Ci-a, alquilo Ci-a y ciano;
R23 es hidrógeno o halógeno;
R24 se selecciona de hidrógeno, halógeno, hidroxi y alquilo Ci-a;
R25 se selecciona de hidrógeno, halógeno, alquilo Ci-a, alcoxi Ci-a, halo-alquilo Ci-a, halo-alcoxi Ci-a, alquil Ci-a-SO2- y cicloalquilo C3-i2;
R2a se selecciona de hidrógeno, alquilo Ci-a y alcoxi Ci-a;
R27 se selecciona de hidrógeno, halógeno, alcoxi Ci-a, halo-alcoxi Ci-a, alquilo Ci-a, halo-alquilo Ci-a, hidroxi-alquilo Ci-a, halógeno, hidroxi, alquilsulfonilo Ci-a, carbamoílo, ciano, cicloalquil-alcoxi Ci-a- y cicloalquilo;
R28 se selecciona de hidrógeno, alquilo Ci-a y halógeno;
A es arilo Ca-i4 o heteroarilo Ci-i3;
B es arilo Ca-i4 o heteroarilo Ci-i3;
C1 es cicloalquilo C3-i2 o heterociclilo C2 -9 ;
C2 es arilo Ca-i4;
L2 se selecciona de un enlace covalente, -O - y -CH 2-;
L3 se selecciona de un enlace covalente, -CH 2O - y -CH 2- ; y
L3a es un enlace covalente o -CH 2-.
En un modo de realización preferente, la presente invención proporciona un compuesto de fórmula (Ic) como se describe en el presente documento, o una sal farmacéuticamente aceptable del mismo, en la que:
m es 0, n es 0 o i y X es CH; o
m es i, n es i y X es CH o N;
L se selecciona de -CR i R2-, -OCHR3-, -CHR3O - y un enlace covalente;
R1 se selecciona de halógeno, alquilo C1-6 , hidroxi-alquilo C1-6 , alcoxi Ci-e-alquil C1-6- , halo-alcoxi C1-6 y un grupo

R2 es hidrógeno o halógeno;
R3 es alquilo Ci-a;
R20 es hidrógeno;
i5
R21 se selecciona de halógeno, alcoxi Ci-e, halo-alcoxi Ci-e, alquilo Ci-e, halo-alquilo Ci-e, SFs, arilo Ce-14 y un grupo

R22 se selecciona de hidrógeno, halógeno, alcoxi Ci-6 y halo-alcoxi Ci-a;
R23 es hidrógeno o halógeno;
R25 se selecciona de hidrógeno, halógeno, alcoxi Ci-a y cicloalquilo C3-12;
R26 es hidrógeno o alcoxi Ci-a;
R27 se selecciona de hidrógeno, halo-alcoxi Ci-a, alquilo Ci-a, halo-alquilo Ci-a y halógeno;
R28 se selecciona de hidrógeno, alquilo Ci-a y halógeno;
A es arilo Ca-i4 o heteroarilo Ci-i3;
B es arilo Ca-i4 o heteroarilo Ci-i3;
Ci es cicloalquilo C3-12 o heterociclilo C2 -9 ;
L3 es un enlace covalente o -CH 2-.
En un modo de realización en particular preferente, la presente invención proporciona un compuesto de fórmula (Ic) como se describe en el presente documento, o una sal farmacéuticamente aceptable del mismo, en la que: m es 0, n es 0 o i y X es CH; o
m es i, n es i y X es CH o N;
L se selecciona de -C R i R2-, -OCHR3-, -CHR3O - y un enlace covalente;

R1 se selecciona de 2-metoxietilo, metilo, 2,2,2-trifluoroetoxi, fluoro, 2-hidroxietilo y un grupo ; R2 es hidrógeno o fluoro;
R3 es metilo;
R20 es hidrógeno;
R21 se selecciona de fluoro, cloro, bromo, metilo, metoxi, ferc-butilo, propilo, trifluorometoxi, 2-fluoroetox¡, 2,2,2-
trifluoroetoxi, trifluorometilo, 2,2,2-trifluoroetilo,
R22 se selecciona de hidrógeno, fluoro, cloro, metoxi, metilo y trifluorometilo;
R23 es hidrógeno o fluoro;
R25 se selecciona de hidrógeno, metoxi, fluoro y ciclopropilo;
í 6
R26 es hidrógeno o metoxi;
R27 se selecciona de metilo, trifluorometoxi, trifluorometilo, 2,2,2-trifluoro-1-metil-etoxi, 2,2,2-trifluoro-1, 1-dimetiletoxi, 2,2,2-trifluoroetoxi, fluoro y cloro;
R28 se selecciona de hidrógeno, metilo, fluoro y cloro;
A se selecciona de fenilo, indol-3-ilo, 2-piridilo y 3-piridilo;
B es fenilo o 1,2,4-oxadiazol-5-ilo;
C1 se selecciona de acetidin-1-ilo, pirrolidin-1-ilo, ciclopropilo y oxetan-3-ilo; y
L3 es un enlace covalente o -CH 2-.
En un modo de realización, la presente invención proporciona un compuesto de fórmula (Ic) como se describe en el presente documento, o una sal farmacéuticamente aceptable del mismo, en la que:
m es 0, n es 0 o 1 y X es CR24; o
m es 1, n es 1 y X es CR24 o N; y
R24 se selecciona de hidrógeno, halógeno, hidroxi y alquilo C1-6.
En un modo de realización preferente, la presente invención proporciona un compuesto de fórmula (Ic) como se describe en el presente documento, o una sal farmacéuticamente aceptable del mismo, en la que:
m es 0, n es 0 o 1 y X es CH; o
m es 1, n es 1 y X es CH o N.
En un modo de realización, la presente invención proporciona un compuesto de fórmula (Ic) como se describe en el presente documento, o una sal farmacéuticamente aceptable del mismo, en la que:
L se selecciona de -CR1R2-(CH2)p- , -OCHR3-, -CHR3O - y un enlace covalente;
p es 0 o 1;
R1 se selecciona de halógeno, alcoxi C1-6 , alquilo C1-6 , halo-alquilo C1-6, hidroxi-alquilo C1-6, alcoxi Ci-e-alquil C1-6-,

halo-alcoxi C1-6, halo-alcoxi Ci-e-alquil C1-6--, ciano y un grupo ; y R2 se selecciona de hidrógeno, halógeno e hidroxi; o
R1 y R2, tomados conjuntamente con el átomo de carbono al que están unidos, forman un cicloalquilo C3 -12 ; R3 es alquilo C1-6 o halo-alquilo C1-6;
R25 se selecciona de hidrógeno, halógeno, alquilo C1-6, alcoxi C1-6, halo-alquilo C1-6, halo-alcoxi C1-6, alquil C1-6-SO2- y cicloalquilo C3-12;
R26 se selecciona de hidrógeno, alquilo C1-6 y alcoxi C1-6;
B es arilo C6-14 o heteroarilo C1-13; y
L2 se selecciona de un enlace covalente, -O - y -CH 2-.
En un modo de realización preferente, la presente invención proporciona un compuesto de fórmula (Ic) como se describe en el presente documento, o una sal farmacéuticamente aceptable del mismo, en la que:
L se selecciona de -C R 1R2-, -OCHR3-, -CHR3O - y un enlace covalente;
R1 se selecciona de halógeno, alquilo C1-6, hidroxi-alquilo C1-6, alcoxi C1-6-alquil C1-6-, halo-alcoxi C1-6 y un grupo

R2 es hidrógeno o halógeno;
R3 es alquilo Ci-a;
R25 se selecciona de hidrógeno, halógeno, alcoxi Ci-a y cicloalquilo C3-12;
R2a es hidrógeno o alcoxi Ci-a; y
B es arilo Ca-14 o heteroarilo C1-13.
En un modo de realización en particular preferente, la presente invención proporciona un compuesto de fórmula (Ic) como se describe en el presente documento, o una sal farmacéuticamente aceptable del mismo, en la que: L se selecciona de -C R 1R2-, -OCHR3-, -CHR3O - y un enlace covalente;
R1 se selecciona de 2-metoxietilo, metilo, 2,2,2-trifluoroetoxi, fluoro, 2-hidroxietilo y un grupo
R2 es hidrógeno o fluoro;
R3 es metilo;
R25 se selecciona de hidrógeno, metoxi, fluoro y ciclopropilo;
R2a es hidrógeno o metoxi; y
B es fenilo o 1,2,4-oxadiazol-5-ilo.
En un modo de realización, la presente invención proporciona un compuesto de fórmula (Ic) como se describe en el presente documento, o una sal farmacéuticamente aceptable del mismo, en la que:
R2i se selecciona de hidrógeno, halógeno, hidroxi, alcoxi Ci-a, halo-alcoxi Ci-a, alquilo Ci-a, halo-alquilo Ci-a, hidroxialquilo C1-6 , (halo-alquil Ci-6)-hidrox¡-alquilo C1-6 , alcoxicarbonil Ci-e-alquil C1-6-, alcoxicarbonil Ci-6-NH-alcox¡
Ci-a-, SF5 , (alquil Ci-a)3Si-O-alquil Ci-a-, un grupo
R22 se selecciona de hidrógeno, halógeno, alcoxi Ci-a, halo-alcoxi Ci-a, alquilo Ci-a y ciano;
R23 es hidrógeno o halógeno;
R27 se selecciona de hidrógeno, halógeno, alcoxi Ci-a, halo-alcoxi Ci-a, alquilo Ci-a, halo-alquilo Ci-a, hidroxi-alquilo Ci-a, halógeno, hidroxi, alquilsulfonilo Ci-a, carbamoílo, ciano, cicloalquil-alcoxi Ci-a- y cicloalquilo;
R28 se selecciona de hidrógeno, alquilo Ci-a y halógeno;
A es arilo Ca-i4 o heteroarilo Ci-i3;
Ci es cicloalquilo C3-i2 o heterociclilo C2 -9 ;
C2 es arilo Ca-i4;
i8
L3 se selecciona de un enlace covalente, -CH 2O - y -CH 2- ; y
L3a es un enlace covalente o -CH 2-.
En un modo de realización preferente, la presente invención proporciona un compuesto de fórmula (Ic) como se describe en el presente documento, o una sal farmacéuticamente aceptable del mismo, en la que:
R21 se selecciona de halógeno, alcoxi C1-6 , halo-alcoxi C1-6 , alquilo C1-6 , halo-alquilo C1-6 , SFs, arilo Ce-14 y un grupo

R22 se selecciona de hidrógeno, halógeno, alcoxi C1-6 y halo-alcoxi C1-6;
R23 es hidrógeno o halógeno;
R27 se selecciona de hidrógeno, halo-alcoxi C1-6, alquilo C1-6, halo-alquilo C1-6 y halógeno;
R28 se selecciona de hidrógeno, alquilo C1-6 y halógeno;
A es arilo C6-14 o heteroarilo C1-13;
C1 es cicloalquilo C3-12 o heterociclilo C2 -9 ; y
L3 es un enlace covalente o -CH 2-.
En un modo de realización en particular preferente, la presente invención proporciona un compuesto de fórmula (Ic) como se describe en el presente documento, o una sal farmacéuticamente aceptable del mismo, en la que: R21 se selecciona de fluoro, cloro, bromo, metilo, metoxi, ferc-butilo, propilo, trifluorometoxi, 2-fluoroetox¡, 2,2,2-
trifluoroetoxi, trifluorometilo, 2,2,2-trifluoroetilo, 1, 1 -difluoroetilo, SF 
5 , fenilo y un grupo R22 se selecciona de hidrógeno, fluoro, cloro, metoxi, metilo y trifluorometilo;
R23 es hidrógeno o fluoro;
R27 se selecciona de metilo, trifluorometoxi, trifluorometilo, 2,2,2-trifluoro-1-metil-etoxi, 2,2,2-trifluoro-1,1-dimetiletoxi, 2,2,2-trifluoroetoxi, fluoro y cloro;
R28 se selecciona de hidrógeno, metilo, fluoro y cloro;
A se selecciona de fenilo, indol-3-ilo, 2-piridilo y 3-piridilo;
C1 se selecciona de acetidin-1-ilo, pirrolidin-1-ilo, ciclopropilo y oxetan-3-ilo; y
L3 es un enlace covalente o -CH 2-.
En un modo de realización, la presente invención proporciona un compuesto de fórmula (Ic) como se describe en el presente documento, o una sal farmacéuticamente aceptable del mismo, seleccionado de los compuestos presentados en la tabla 1 y la tabla 3.
En un modo de realización preferente, la presente invención proporciona un compuesto de fórmula (Ic) como se describe en el presente documento, o una sal farmacéuticamente aceptable del mismo, seleccionado de los compuestos presentados en la tabla 1.
En un modo de realización en particular preferente, la presente invención proporciona un compuesto de fórmula (Ic) como se describe en el presente documento, o una sal farmacéuticamente aceptable del mismo, en la que el
compuesto de fórmula (Ic) se selecciona de:
rac-cis-6-(4-(5-doro-1-(cidopropilmetil)-1H-indol-3-il)piperidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
rac-cis-6-(4-(5-doro-1-metil-1H-indol-3-il)piperidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona; rac-cis-6-(4-(1-(4-fluorofenil)-3-metoxipropil)piperidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona; rac-cis-6-(4-((4-clorofenil)(fenil)metil)piperacina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona; (+)-cis-6-(4-(bis(4-fluorofenil)metil)piperacina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(+)- o (-)-ds-6-(4-(5-don>1-ddopropiMH-mdol-3-N)piperidma-1-carbonN)hexahidn>2H-pirido[4,3-b][1,4]oxadn-3(4H)-ona;
(+)- o (-)-cis-6-(4-(5-cloro-1-(oxetan-3-il)-1H-indol-3-il)piperidina-1-carbonil)hexahidro-2H-pirido [4,3-b][1,4]oxacin-3(4H)-ona;
rac-cis-6-(4-(1-(4-fluorofenil)-3-hidroxipropil)piperidina-1-carbonil)hexahidro-2H-pi rido[4,3-b][1,4]oxacin-3(4H)-ona; (4aR,8aS)-6-(4-(difluoro(4-(trifluorometil)fenil)metil)piperidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(+) o (-)-cis-6-(4-((R o S)-1-(2-cloro-4-fluorofenoxi)etil)piperidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(+) o (-)-cis-6-(4-((4-clorofenil)difluorometil)piperidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona; (+) o (-)-cis-6-(4-((2-cloro-4-fluorofenil)difluorometil)piperidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(4-((R o S)-1-(4-(trifluorometil)fenil)etil)piperidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(4-((S o R)-(4-fluorofenil)(2,2,2-trifluoroetoxi)metil)piperidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-[4-[(S o R)-(4-fluorofenil)-(3-metoxifenil)metil]piperidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona;
(4aR,8aS)-6-(4-((R o S)-(4-fluorofenil)(3-metoxifenil)metil)piperidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(4-((S o R)-(3-(2-fluoroetoxi)fenil)(fenil)metil)piperidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(4-((3-ciclopropil-1,2,4-oxadiazol-5-il)(4-fluorofenil)metil)piperidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(4-((S o R)-(4-fluorofenil)(4-metoxifenil)metil)piperidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(4-((S o R)-(3,4-dimetoxifenil)(4-fluorofenil)metil)piperidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(4-((R o S)-(4-fluorofenil)(4-metoxifenil)metil)piperidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(4-((S o R)-(4-(2-fluoroetoxi)fenil)(4-fluorofenil)metil)piperidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-[3-[4-(trifluorometoxi)fenil]acetidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona; (4aR,8aS)-6-(3-(2'-trifluorometoxi)-[1,1'-bifenil]-4-il)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-(2'-(trifluorometil)-[1,1'-bifenil]-4-il)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-(2',4'-difluoro-[1,1'-bifenil]-4-il)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-(4-(3-(trifluorometil)acetidin-1-il)fenil)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-(4-bromo-3-dorofenil)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona; (4aR,8aS)-6-[3-[4-(4-doro-2-fluoro-fenil)fenil]acetidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona;
(4aR,8aS)-6-[3-[4-(2-doro-4-fluoro-fenil)fenil]acetidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona;
(4aR,8aS)-6-(3-(4-(3-((1,1,1-trifluoropropan-2-il)oxi)acetidin-1-il)fenil)acetidina-1-carbonil)hexahidro-2H-pi rido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-(4-(terc-butN)fenN)acetidma-1-carbonN)hexahidn>2H-pirido[4,3-b][1,4]oxadn-3(4H)-ona;
(4aR,8aS)-6-(3-(4-(trifluorometil)fenil)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-(4-(3-((1,1,1-trifluoro-2-metilpropan-2-il)oxi)acetidin-1-il)fenil)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-(4-(1,1-difluoroetil)fenil)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona; (4aR,8aS)-6-(3-(4-(3,3-dimetilpirrolidin-1-il)fenil)acetidina-1-carbonil)hexahidro-2H-pi rido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-(4-(3-(trifluorometoxi)acetidin-1-il)fenil)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-(6-(2,4-didorofenil)piridin-3-il)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-[3-[4-(2,2,2-trifluoroetoxi)fenil]acetidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona;
(4aR,8aS)-6-[3-[4-[3-[(1S o 1R)-2,2,2-trifluoro-1-metil-etoxi]acetidin-1-il]fenil]acetidina-1-carbonil]-4,4a,5,7,8,8ahexahidropirido[4,3-b][l,4]oxacin-3-ona;
(4aR,8aS)-6-[3-[4-[3-[(1R o 1S)-2,2,2-trifluoro-1-metil-etoxi]acetidin-1-il]fenil]acetidina-1-carbonil]-4,4a,5,7,8,8ahexahidropirido[4,3-b][l,4]oxacin-3-ona;
(4aR,8aS)-6-(3-(4-(2-(trifluorometil)pirrolidin-1-il)fenil)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-(4-(3-(trifluorometil)pirrolidin-1-il)fenil)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-(4-(3-(2,2,2-trifluoroetoxi)acetidin-1-il)fenil)acetidin-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-(6-(2-(trifluorometil)pirrolidin-1-il)piridin-3-il)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-(4-(pentafluoro-16-sulfanoil)fenil)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-(2-fluoro-4-(trifluorometoxi)fenil)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-[3-[4-(2,2,2-trifluoroetil)fenil]acetidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona;
(4aR,8aS)-6-[3-[4-[1-(trifluorometil)cidopropil]fenil]acetidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona;
(4aR,8aS)-6-[3-[4-(6,6-difluoro-2-azaespi ro[3.3]heptan-2-il)fenil]acetidina-1-carbonil]-4,4a,5,7,8,8ahexahidropirido[4,3-b][1,4]oxacin-3-ona;
(4aR,8aS)-6-(3-(5-(2,4-didorofenil)piridin-2-il)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-(4-((R o S)-2-(trifluorometN)pirroNdm-1-N)fenN)acetidma-1-carbonN)hexahidn>2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-(4-((S o R)-2-(trifluorometN)pirroNdm-1-N)fenN)acetidma-1-carbonN)hexahidn>2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-(4-((R o S)-3-(trifluorometN)pirroNdm-1-N)fenN)acetidma-1-carbonN)hexahidn>2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-(4-((S o R)-3-(trifluorometN)pirroNdm-1-N)fenN)acetidma-1-carbonN)hexahidn>2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-(3-fluoro-4-(trifluorometoxi)fenil)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-(3-metil-4-(trifluorometoxi)fenil)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-(3,5-difluoro-4-(trifluorometoxi)fenil)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-(3-doro-4-(trifluorometoxi)fenil)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-((R o S)-3-(3-don>5-(2,2,2-trifluoroetoxi)fenN)pirroNdma-1-carbonN)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-((S o R)-3-(3-don>5-(2,2,2-trifluoroetoxi)fenN)pirroNdma-1-carbonN)hexahidn>2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-(4-(terc-butN)-3-metoxifenN)acetidma-1-carbonN)hexahidro-2H-pirido[4,3-b][1,4]oxadn-3(4H)-ona; (4aR,8aS)-6-(3-(4-propilfenil)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-(4-(trifluorometoxi)-3-(trifluorometil)fenil)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-(5-((R o S)-3-(trifluorometN)pirroNdm-1-N)piridm-2-N)acetidma-1-carbonN)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-(5-((S o R)-3-(trifluorometN)pirroNdm-1-N)pmdm-2-N)acetidma-1-carbonN)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-[3-[6-[3-(R o S)-(trifluorometil)pirrolidin-1-il]-3-piridil]acetidina-1-carbonil]-4,4a,5,7,8,8ahexahidropirido[4,3-b][1,4]oxacin-3-ona;
(4aR,8aS)-6-[3-[6-[3-(S o R)-(trifluorometil)pirrolidin-1-il]-3-piridil]acetidina-1-carbonil]-4,4a,5,7,8,8ahexahidropirido[4,3-b][1,4]oxacin-3-ona;
(4aR,8aS)-6-[3-(4-tetrahidropiran-3-ilfenil)acetidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropi rido[4,3-b][1,4]oxacin-3-ona;
(4aR,8aS)-6-[3-[2-metoxi-4-(2,2,2-trifluoroetil)fenil]acetidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropi rido[4,3-b][1,4]oxacin-3-ona; y
(4aR,8aS)-6-(3-(4-(neopentiloxi)fenil)acetidina-1-carbonil)hexahidro-2H-pi rido[4,3-b][1,4]oxacin-3(4H)-ona.
En un modo de realización, la presente invención proporciona sales o ésteres farmacéuticamente aceptables de los compuestos de fórmula (Ic) como se describe en el presente documento. En un modo de realización particular, la presente invención proporciona sales farmacéuticamente aceptables de los compuestos de acuerdo con la fórmula (Ic) como se describe en el presente documento, especialmente sales clorhidrato. En otro modo de realización particular, la presente invención proporciona ésteres farmacéuticamente aceptables de los compuestos de acuerdo con la fórmula (Ic) como se describe en el presente documento. Aún en otro modo de realización particular, la presente invención proporciona compuestos de acuerdo con la fórmula (I) como se describe en el presente documento.
Procedimientos de fabricación
La preparación de compuestos de fórmula (Ic) de la presente invención se puede llevar a cabo en vías sintéticas secuenciales o convergentes. Las síntesis de la invención se muestran en los siguientes esquemas generales. Las habilidades requeridas para llevar a cabo la reacción y purificación de los productos resultantes son conocidas por los expertos en la técnica. Los sustituyentes e índices usados en la siguiente descripción de los procedimientos tienen el significado dado en el presente documento, a menos que se indique lo contrario.
Si uno de los materiales de partida, intermedios o compuestos de fórmula (Ic) contienen uno o más grupos funcionales que no son estables o son reactivos en las condiciones de reacción de una o más etapas de reacción, se pueden introducir los grupos protectores apropiados (como se describe, por ejemplo, en "Protective Groups in Organic Chemistry" de T.W. Greene y P.G.M. Wutts, 5.a ed., 2014, John Wiley & Sons, N.Y.) antes de la etapa crucial aplicando procedimientos bien conocidos en la técnica. Dichos grupos protectores se pueden retirar en una fase posterior de la síntesis usando procedimientos estándar descritos en la literatura.
Si los materiales de partida o los intermedios contienen centros estereogénicos, los compuestos de fórmula (Ic) se pueden obtener como mezclas de diastereómeros o enantiómeros, que se pueden separar por procedimientos bien conocidos en la técnica, por ejemplo, HPLC quiral, SFC quiral o cristalización quiral. Los compuestos racémicos, por ejemplo, se pueden separar en sus antípodas por medio de sales diastereoméricas por cristalización con ácidos ópticamente puros o por separación de las antípodas por procedimientos cromatográficos específicos usando un adsorbente quiral o un eluyente quiral. Es igualmente posible separar materiales de partida e intermedios que contienen centros estereogénicos para dar materiales de partida e intermedios enriquecidos de forma diastereomérica/enantiomérica. El uso de dichos materiales de partida e intermedios enriquecidos de forma diastereomérica/enantiomérica en la síntesis de compuestos de fórmula (Ic) típicamente dará lugar a los respectivos compuestos de fórmula (Ic) enriquecidos de forma diastereomérica/enantiomérica. Un experto en la técnica reconocerá que en la síntesis de compuestos de fórmula (I), en la medida en que no se desee de otro modo, se aplicará una "estrategia de grupo de protección ortogonal", que permite la escisión de varios grupos protectores uno a la vez sin afectar a otros grupos protectores en la molécula. El principio de protección ortogonal es bien conocido en la técnica y también se ha descrito en la literatura (por ejemplo, Barany y R. B. Merrifield, J. Am. Chem. Soc. 1977, 99, 7363; H. Waldmann et al., Angew. Chem. Int. Ed. Engl. 1996, 35, 2056).
Un experto en la técnica reconocerá que la secuencia de reacciones puede variar dependiendo de la reactividad y la naturaleza de los intermedios.
En más detalle, los compuestos de fórmula (Ic) se pueden fabricar por los procedimientos dados a continuación, por los procedimientos dados en los ejemplos o por procedimientos análogos. Las condiciones de reacción apropiadas para las etapas de reacción individuales son conocidas para un experto en la técnica. Además, para las condiciones de reacción descritas en la literatura que afectan a las reacciones descritas, véase, por ejemplo: Comprehensive Organic Transformations: A Guide to Functional Group Preparations, 2.a edición, Richard C. Larock. John Wiley & Sons, New York, NY. 1999). Se encontró práctico llevar a cabo las reacciones en presencia o ausencia de un disolvente. No existe una restricción particular sobre la naturaleza del disolvente que se va a emplear, siempre que no tenga un efecto adverso sobre la reacción o los reactivos implicados y que pueda disolver los reactivos, al menos hasta cierto punto. Las reacciones descritas pueden tener lugar en un amplio intervalo de temperaturas, y la temperatura de reacción precisa no es crucial para la invención. Es práctico llevar a cabo las reacciones descritas en un intervalo de temperaturas entre -78 °C hasta reflujo. El tiempo requerido para la reacción también puede variar ampliamente, dependiendo de muchos factores, notablemente la temperatura de reacción y la naturaleza de los reactivos. Sin embargo, normalmente será suficiente un período de 0,5 horas a varios días para proporcionar los intermedios y compuestos descritos. La secuencia de reacción no se limita a la presentada en los esquemas, sin embargo, dependiendo de los materiales de partida y de su respectiva reactividad, se puede modificar libremente la secuencia de las etapas de reacción.
Si los materiales de partida o los intermedios no están disponibles comercialmente o su síntesis no está descrita en la literatura, se pueden preparar de forma análoga a los procedimientos existentes para análogos cercanos o como se explica en la sección experimental.
Las siguientes abreviaturas se usan en el presente texto:
AcOH = ácido acético, ACN = acetonitrilo, Boc = terc-butiloxicarbonilo, CAS RN = número de registro de compendios químicos, Cbz = benciloxicarbonilo, Cs2CO3 = carbonato de cesio, CO = monóxido de carbono, CuCl = cloruro de cobre(I), CuCN = cianuro de cobre(I), CuI = yoduro de cobre(I), DMAP = 4-dimetilaminopiridina, DME = dimetoxietano, DMEDA = N,N'-dimetiletilendiamina, DMF = N,N-dimetilformamida, DIPEA = N,N-diisopropiletilamina, dppf = 1,1 bis(difenil fosfino)ferroceno, EDC.HCl = clorhidrato de N-(3-dimetilaminopropil)-N'-etilcarbodiimida, EI = impacto de electrones, ESI = ionización por electronebulización, EtOAc = acetato de etilo, EtOH = etanol, h = hora(s), FA = ácido fórmico, H2O = agua, H2SO4= ácido sulfúrico, Hal = halógeno, HATU = hexafluorofosfato de 1-[bis(dimetilamino)metileno]-1H-1,2,3-triazolo[4,5-b]piridinio-3-óxido, HBTU = hexafluorofosfato de O-benzotriazol-N,N,N',N'-tetrametil-uronio, HCl = cloruro de hidrógeno, HOBt = 1-hidroxi-1 H-benzotriazolo; HPLC = cromatografía de líquidos de alto rendimiento, iPrMgCl = cloruro de isopropilmagnesio, I2 = yodo, IPA = 2-propanol, (Ir[dF(CF3)ppy]2(dtbpy))PF6 = hexafluorofosfato de [4,4'-bis(1,1 -dimetiletil)-2,2'-bipiridina-N1,N1 ']bis[3,5-difluoro-2-[5-(trifluorometil)-2-piridinil-N]fenil-C]iridio(IM), ISP = positivo para nebulización de iones (modo), ISN = negativo para nebulización de iones (modo), K2CO3 = carbonato de potasio, KHCO3 = bicarbonato de potasio, KI = yoduro de potasio, KOH = hidróxido de potasio, K3PO4 = fosfato de potasio tribásico, LiAlH4 o LAH = hidruro de litio y aluminio, LiHMDS = bis(trimetilsilil)amida de litio, LiOH = hidróxido de litio, MgSO4 = sulfato de magnesio, min = minuto(s), ml = mililitro, MPLC = cromatografía de líquidos a presión media, EM = espectro de masas, NaH = hidruro de sodio, NaHCO3 = carbonato hidrógeno de sodio, NaNO2 = nitrito de sodio, NaOH = hidróxido de sodio, Na2CO3 = carbonato de sodio, Na2SO4 = sulfato de sodio, Na2S2O3 = tiosulfato de sodio, NBS = N-bromosuccinimida, nBuLi = n-butillitio, NEt3 = trietilamina (TEA), NH4Cl = cloruro de amonio, NiCh glyme = complejo de cloruro de níquel(II)-éter dimetílico de etilenglicol, NMP = N-metil-2-pirrolidona, OAc = acetoxi, T3P = anhídrido propilfosfónico, P2O5 = pentóxido de fósforo, PE = éter de petróleo, GP = grupo protector, Pd-C = paladio sobre carbón activado, PdCh(dppf)-CH2Cl2 = complejo de dicloruro de 1,1'-bis(difenilfosfino)ferrocenopaladio(II)/diclorometano, Pd2(dba)3 = tris(dibencilidenoacetona)dipaladio(0), Pd(OAc)2 = acetato de paladio(II), Pd(OH)2 = hidróxido de paladio, Pd(PPh3)4 = tetraquis(trifenilfosfina)paladio(0), PTSA = ácido p-toluenosulfónico, R = cualquier grupo, SFC = cromatografía de fluidos supercríticos, S-PHOS = 2-diciclohexilfosfino-2',6'-dimetoxibifenil, T3P = anhídrido propilfosfónico, t. a. = temperatura ambiente, TBAI = yoduro de tetrabutilamonio, TEA = trietilamina, TFA = ácido trifluoroacético, THF = tetrahidrofurano, TMEDA = N,N,N',N'-tetrametiletilenodiamina, ZnCl2 = cloruro de cinc, Xantphos = 4,5-bis(difenilfosfino)-9,9-dimetilxanteno.
Los compuestos de fórmula I, en la que A, L, X, n y m son como se describe en el presente documento se pueden sintetizar de forma análoga a los procedimientos de la literatura y/o como se representa, por ejemplo, en el esquema 1.

Esquema 1
En consecuencia, las 4a,5,6,7,8,8a-hexahidro-4H-pirido[4,3-b][1,4]oxacin-3-onas 1 se hacen reaccionar con los intermedios 2 en presencia de un reactivo formador de urea tal como carbonato de bis(triclorometilo) usando una base adecuada y un disolvente tal como, por ejemplo, bicarbonato de sodio en DCM, para dar los compuestos de fórmula I (etapa a). Otros reactivos formadores de urea incluyen pero no se limitan a fosgeno, cloroformiato de triclorometilo, carbonato de 4-nitrofenilo o 1,1'-carbonildiimidazol. Las reacciones de este tipo y el uso de estos reactivos están descritos ampliamente en la literatura (por ejemplo, G. Sartori et al., Green Chemistry 2000, 2, 140). Un experto en la técnica reconocerá que el orden de la adición de los reactivos puede ser importante en este tipo de reacciones debido a la reactividad y estabilidad de los cloruros de carbamoílo intermediarios formados, así como para evitar la formación de subproductos de urea simétricos no deseados.
Los compuestos de fórmula (Ic) descritos en el presente documento se pueden preparar de forma análoga a los compuestos de fórmula (I), como se describe en el presente documento en los esquemas de 1 a 18. Por ejemplo, de forma análoga al procedimiento descrito en el esquema 1 anterior, las 4a,5,6,7,8,8a-hexahidro-4H-pirido[4,3-b][1,4]oxacin-3-onas 1 se hacen reaccionar con los intermedios 2a para proporcionar los compuestos de fórmula (Ic) (esquema 1A, etapa a).

Esquema 1A
Los intermedios 1 se pueden sintetizar como se representa, por ejemplo, en el esquema 2 y/o de forma análoga a los procedimientos descritos en la literatura.

Esquema 2
Por tanto, los derivados de 3-aminopiperidin-4-ol 3 en los que "GP" significa un grupo protector adecuado tal como un grupo protector Cbz o Boc, se pueden acilar, por ejemplo, con cloruro de cloro- o bromoacetilo 4 , en el que "GS" significa un grupo saliente adecuado (por ejemplo, Cl o Br), usando una base adecuada tal como carbonato de sodio o potasio, hidróxido de sodio o acetato de sodio en un disolvente apropiado tal como THF, agua, acetona o mezclas de los mismos, para proporcionar los intermedios 5 (etapa a).
Los intermedios 5 se pueden ciclar a los intermedios 6 usando procedimientos bien conocidos en la técnica, por ejemplo, por tratamiento de 5 con hidruro de sodio en THF o terc-butóxido de potasio en IPA y agua (etapa b). Las reacciones de ese tipo se describen en la literatura (por ejemplo, Z. Rafinski et al., J. Org. Chem. 2015, 80, 7468; S. Dugar et al., Synthesis 2015, 47(5), 712; documento WO2005/066187).
La retirada del grupo protector en los intermedios 6 , aplicando procedimientos conocidos en la técnica (por ejemplo, un grupo Boc usando TFA en DCM a temperaturas entre 0 °C y temperatura ambiente, un grupo Cbz usando hidrógeno en presencia de un catalizador adecuado tal como Pd o Pd(OH)2 sobre carbón vegetal en un disolvente adecuado tal como MeOH, EtOH, EtOAc o mezclas de los mismos y como se describe, por ejemplo, en "Protective Groups in Organic Chemistry" de T.W. Greene y P.G.M. Wuts, 4.a ed., 2006, Wiley N.Y.), proporciona los intermedios 1 (etapa c).
Los intermedios 1 se pueden obtener como mezclas de diastereómeros y enantiómeros, respectivamente, o como estereoisómeros individuales dependiendo de si se emplean en sus síntesis mezclas racémicas o formas enantioméricamente puras de derivados cis- o trans-3-aminopiperidin-4-ol 3. Los intermedios 3 están disponibles comercialmente y su síntesis también se ha descrito en la literatura (por ejemplo, documento WO2005/066187; documento WO2011/0059118; documento WO2016/185279).
Los intermedios 1B y 1C configurados en cis ópticamente puros se pueden obtener, por ejemplo, de acuerdo con el esquema 3 por separación quiral de (cis-rac)-4a,5,6,7,8,8a-hexahidro-4H-pirido[4,3-b][1,4]oxacin-3-ona (1A) disponible comercialmente (opcionalmente en forma de una sal tal como, por ejemplo, una sal clorhidrato) usando procedimientos conocidos en la técnica, por ejemplo, por cristalización de sal diastereomérica o por cromatografía quiral (etapa a).
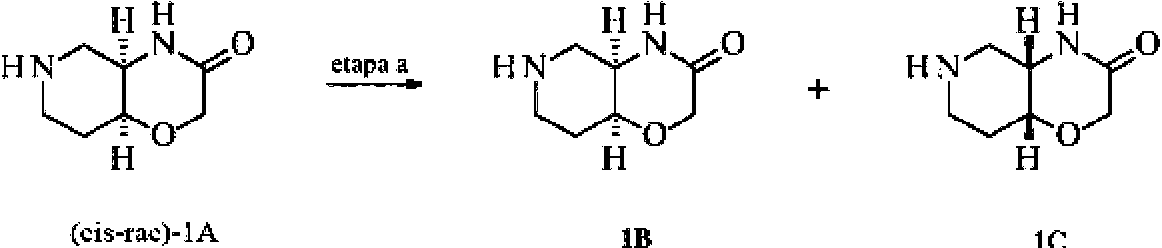
Esquema 3
En algún modo de realización, los intermedios 2 son intermedios de tipo B, C, D o E. Los intermedios de tipo B, C,
D o E se pueden preparar, por ejemplo, por los procedimientos de síntesis explicados en los esquemas 4, 5, 6 y 7.
Los intermedios de tipo B en los que R1 es como se define en el presente documento, se pueden preparar por una variedad de condiciones, que se pueden ejemplificar por el procedimiento de síntesis general explicado en el esquema 4.

Esquema 4
Comenzando a partir de haluros de arilo o heteroarilo 7, en los que X1 se selecciona de Cl, Br o I y A es como se define en el presente documento, se puede realizar una reacción de intercambio de halógeno de litio usando una solución de LiHMDS o n-BuLi, preferentemente n-BuLi en un disolvente como THF, éter dietílico, n-pentano, nhexano o mezclas de los mismos, preferentemente THF y en un intervalo de temperatura entre -20 °C y -78 °C, preferentemente a -78 °C, para generar el correspondiente intermedio arilo o heteroarilo litiado. La adición nucleófila del intermedio de arilo o heteroarilo litiado preparado in situ a las cetonas de tipo 8 , en las que R1, m y n son como se definen en el presente documento, en un disolvente tal como THF y preferentemente a una temperatura de -78 °C da los correspondientes alcoholes terciarios 9 (etapa a).
La eliminación posterior del grupo hidroxi terciario con la retirada simultánea del grupo protector Boc usando condiciones ácidas tales como HCl 4 M en dioxano en un disolvente como MeOH o, preferentemente, TFA en DCM proporciona las correspondientes olefinas 10 (etapa b).
La hidrogenación catalítica heterogénea de olefinas 10 usando un catalizador tal como Pd(OH)2 o Pd/C en un disolvente como THF, MeOH, EtOH, EtOAc o una mezcla de los mismos, preferentemente Pd/C en THF a, por ejemplo, presión atmosférica de hidrógeno, da intermedios de tipo B (etapa c).
Los intermedios 8 están disponibles comercialmente y/o se pueden preparar de forma análoga a los procedimientos descritos en la literatura, por ejemplo, Bioorg. Med. Chem. Lett. 2011, 27(18), 5191, documentos WO2012/155199, WO2016/180536, Bioorg. Med. Chem. Lett. 2008, 18(18), 5087, documento WO2007/117557, J. Am. Chem. Soc.
2017, 139(33), 11353, J. Med. Chem. 2017, 60(13), 5507.
Los intermedios de tipo C, en los que A es como se define en el presente documento y n = 1, 2 o 3, se pueden preparar por una variedad de condiciones, que se pueden ejemplificar por los procedimientos de síntesis generales explicados en el esquema 5.

Esquema 5
El tratamiento de una mezcla de compuestos de arilo o heteroarilo 11, en los que A es como se define en el presente documento, preferentemente en los que A es heteroarilo sustituido como se define en el presente documento, lo más preferentemente indolilo sustituido como se define en el presente documento, y cetonas 12, en las que n es como se define en el presente documento, con una base tal como NaOH o KOH en un disolvente como EtOH o MeOH y en un intervalo de temperatura entre temperatura ambiente y 80 °C, preferentemente alrededor de la temperatura de reflujo de la mezcla, da las olefinas 13 (etapa a).
La hidrogenación catalítica heterogénea posterior usando un catalizador de metal de transición, tal como PtO2 en un disolvente polar como MeOH, EtOH, AcOEt, AcOH o una mezcla de los mismos, preferentemente una mezcla de EtOH/AcOH a temperatura ambiente desde baja a alta presión, preferentemente alrededor de 5 bar de presión
de hidrógeno gaseoso, proporciona los intermedios 14 (etapa b).
La retirada del grupo protector Boc usando condiciones ácidas tales como el tratamiento con TFA en DCM o preferentemente con HCl 4 M en dioxano en un disolvente como MeOH da los correspondientes intermedios de tipo C (etapa c).
En algunos modos de realización, el intermedio 14 es un intermedio de fórmula 14a, en el que el anillo A es heteroarilo que comprende un grupo amino secundario (es decir, "-N H-", tal como en indolilo) y m y n son como se definen en el presente documento. Los intermedios 14a se pueden transformar en intermedios de tipo D, en los que A es heteroarilo que comprende al menos un átomo de nitrógeno, m y n son como se definen en el presente documento y R14 se selecciona de alquilo, cicloalquilo, hidroxialquilo, cicloalquilalquilo, heterociclilo y heterociclilalquilo, preferentemente de metilo, ciclopropilo, ciclopropilmetilo, hidroxietilo, oxetan-3-ilo y oxetan-3-ilmetilo, por ejemplo, como se explica en el esquema 6.

Esquema 6
Por tanto, los intermedios 14a se pueden N-funcionalizar por tratamiento con una base apropiada, tal como NaH, KH, NaHMDS, LiHMDS, LDA, preferentemente con NaH en un disolvente adecuado como DMF, THF, dioxano o una mezcla de los mismos, preferentemente DMF y en un intervalo de temperatura entre -78 °C y temperatura ambiente, preferentemente a 0 °C, seguido de la adición de compuestos R14-GS en los que GS significa un grupo saliente apropiado tal como cloro, bromo, yodo, OSO2 alquilo (por ejemplo, mesilato (metanosulfonato), OSO2 fluoroalquilo (por ejemplo, triflato (trifluorometanosulfonato) u OSO2 arilo (por ejemplo, tosilato (p-toluenosulfonato)) para dar los correspondientes compuestos N-funcionalizados 15, en los que R14 es como se define anteriormente para el intermedio D. Un experto en la técnica reconocerá que, dependiendo de la naturaleza de R14, se pueden aplicar reactivos adicionales o diferentes, tal como usar ácido ciclopropilborónico en presencia de acetato de cobre(II) y una base tal como DMAP con NaHMDS en un disolvente como tolueno a temperaturas hasta el punto de ebullición del disolvente en el caso de que R14 signifique un grupo ciclopropilo (etapa a).
La desprotección de los compuestos 15 usando las mismas condiciones como se describe anteriormente para los compuestos 14 (véase el esquema 5, etapa c) da intermedios de tipo D (etapa b).
En un modo de realización, el intermedio D es un derivado de indazol de tipo E, en el que m, n y R14 son como se define en el presente documento. Además del procedimiento explicado en el esquema 6, los intermedios de tipo E se pueden preparar por una variedad de condiciones, en particular por el procedimiento de síntesis general explicado en el esquema 7.

Esquema 7
En consecuencia, la condensación de los intermedios 16, en los que GP es un grupo protector tal como Boc, Cbz o Bn y m y n son como se describe en el presente documento, con un derivado de hidracina de tipo R14NHNH2 en el que R14 es como se describe en el presente documento en un disolvente como n-BuOH, DMA, DMF, DMSO, o una mezcla de los mismos, preferentemente en n-BuOH, en un recipiente de reacción sellado a temperatura elevada, por ejemplo, 120 °C, proporciona compuestos de indazol 17 (etapa a).
Posteriormente, la retirada del grupo protector, en el caso de un grupo Boc usando, por ejemplo, condiciones ácidas, tales como el tratamiento con HCl en dioxano o TFA en DCM, preferentemente con HCl 4 M en dioxano en
un disolvente como MeOH, preferentemente a alrededor de temperatura ambiente, da intermedios de tipo E (etapa b).
En algunos modos de realización, los intermedios 2 son intermedios de tipo F, G, H, J , K, L y M . Los intermedios de tipo F, G, H, J , K, L y M en los que A, m y n son como se describe en el presente documento, R15 y R16 se seleccionan cada uno independientemente de alquilo, cicloalquilo, hidroxialquilo, cicloalquilalquilo, heterociclilo y heterociclilalquilo, preferentemente de metilo, ciclopropilo, ciclopropilmetilo, hidroxietilo, oxetan-3-ilo y oxetan-3-ilmetilo; y R17 se selecciona de arilo sustituido o no sustituido, heteroarilo sustituido o no sustituido y alquilo sustituido o no sustituido, se pueden preparar por procedimientos bien conocidos por un experto en la técnica y como se ejemplifica por los procedimientos de síntesis generales explicados en el esquema 8.

Esquema 8
El grupo carbonilo en los intermedios 18, en los que A, m y n son como se describe en el presente documento y GP es un grupo protector adecuado tal como un grupo Boc, Cbz o Bn, disponible comercialmente o bien preparado de acuerdo con los procedimientos descritos en la literatura (por ejemplo, J. Am. Chem. Soc. 2017, 139(33), 11353, J. Med. Chem. 20 l7 , 60(13), 5507, RSC Advances 2015, 5(61), 40964), se puede reducir por procedimientos bien conocidos en el técnica, por ejemplo, usando NaBP4 en MeOH, para dar los intermedios 19 (etapa a).
La retirada del grupo protector de los intermedios 19 aplicando procedimientos conocidos en la técnica (por ejemplo, un grupo Boc usando TFA en DCM o HCl 4 M en dioxano a temperaturas entre 0 °C y temperatura ambiente, un grupo Cbz usando hidrógeno en presencia de un catalizador adecuado tal como Pd o Pd(OH)2 sobre carbón vegetal en un disolvente adecuado tal como MeOH, EtOH, EtOAc o mezclas de los mismos y como se describe, por ejemplo, en "Protective Groups in Organic Chemistry" de T.W. Greene y P.G.M. Wuts, 4.a ed., 2006, Wiley N.Y.), proporciona los intermedios G (etapa b).
La alquilación del grupo hidroxi de los intermedios 19 con un agente alquilante de tipo R15GS en el que GS es un grupo saliente adecuado tal como cloro, bromo, yodo, OSO2alquilo (por ejemplo, mesilato (metanosulfonato),
OSO2fluoroalquilo (por ejemplo, triflato (trifluorometanosulfonato) u OSO2arilo (por ejemplo, tosilato (ptoluenosulfonato)) y R15 es como se describe en el presente documento, usando una base adecuada en un disolvente apropiado (por ejemplo, hidruro de sodio en DMF) a temperaturas entre 0 °C y la temperatura de ebullición del disolvente da los intermedios 20 (etapa c).
La retirada del grupo protector de los intermedios 20 aplicando procedimientos bien conocidos en la técnica y como se describe anteriormente (etapa b) proporciona los intermedios F (etapa d).
Los intermedios 18 se pueden convertir en intermedios 21 en los que Ra es alquilo, preferentemente metilo o etilo usando una reacción de olefinación tal como la reacción de Wittig u Horner Emmons ampliamente descrita, por ejemplo, usando 2-(dietoxifosforil)acetato de etilo y LiHMDS en dioxano a temperaturas que van de 0 °C al punto de ebullición del disolvente (etapa e).
El doble enlace en los intermedios 21 se puede reducir, por ejemplo, por hidrogenación en presencia de un catalizador adecuado tal como Pd-C en un disolvente adecuado tal como MeOH, EtOH o EtOAc o mezclas de los mismos para proporcionar los intermedios 22 (etapa f).
La reducción de la funcionalidad éster en los intermedios 22 usando, por ejemplo, LiBH4 en THF a temperaturas que van de 0°C al punto de ebullición del disolvente proporciona los intermedios 23 (etapa g).
La retirada del grupo protector de los intermedios 23 aplicando procedimientos bien conocidos en la técnica y como se describe anteriormente (etapa b) proporciona los intermedios G (etapa h).
La alquilación del grupo hidroxi de los intermedios 23 con un agente alquilante de tipo R16GS en el que GS es un grupo saliente adecuado y R16 como se describe en el presente documento, usando las condiciones como se describe anteriormente (etapa c), da los intermedios 24 (etapa i).
La retirada del grupo protector de los intermedios 24 aplicando procedimientos bien conocidos en la técnica y como se describe anteriormente (etapa b) proporciona los intermedios H (etapa j).
La adición de un compuesto organometálico de tipo R17M en el que M es, por ejemplo, MgCl, MgCl o Li y R17 como se describe en el presente documento a los intermedios 18 proporciona los intermedios 25 (etapa k). Las reacciones de este tipo son bien conocidas en la técnica y se describen en la literatura (J. Med. Chem. 1989, 32(1), 105, J. Med. Chem. 2014, 57(4), 1543, Bioorg. Med. Chem Lett. 2015, 25(13), 2720).
La retirada del grupo protector de los intermedios 25 aplicando procedimientos bien conocidos en la técnica y como se describe anteriormente (etapa b) proporciona los intermedios J (etapa l).
La alquilación del grupo hidroxi de los intermedios 25 con un agente alquilante de tipo R15GS en el que GS es un grupo saliente adecuado y R15 como se describe en el presente documento, usando las condiciones como se describe anteriormente (etapa c), da los intermedios 26 (etapa m).
La retirada del grupo protector de los intermedios 26 aplicando procedimientos bien conocidos en la técnica y como se describe anteriormente (etapa b) proporciona los intermedios K (etapa n).
Los intermedios 18 se pueden convertir en intermedios 27 aplicando procedimientos conocidos en la técnica y descritos en la literatura (por ejemplo, J. Med. Chem. 2003, 46(25), 5445; documento WO2016/205590), por ejemplo por olefinación de Wittig usando, por ejemplo, metil trifenilfosfonio bromuro y terc-butóxido de potasio en tolueno o LiHMDS en THF (etapa o).
La reducción del doble enlace de los intermedios 27 aplicando, por ejemplo, condiciones de hidrogenación (por ejemplo, hidrógeno en presencia de un catalizador adecuado tal como Pd-C o PtO2) o usando 9-borabiciclo(3.3.1)nonano en THF (de forma análoga a los procedimientos de la literatura, por ejemplo, documento W02007/002057) da los intermedios 28 (etapa p).
La retirada del grupo protector de los intermedios 28 aplicando procedimientos bien conocidos en la técnica y como se describe anteriormente (etapa b) proporciona los intermedios L (etapa q).
La conversión de la cetona de los intermedios 18 aplicando, por ejemplo, condiciones de fluoración usando aminosulfuranos tales como DAST, da los intermedios 29 (etapa r).
La retirada del grupo protector de los intermedios 29 aplicando procedimientos bien conocidos en la técnica y como se describe anteriormente (etapa b) proporciona los intermedios M (etapa s).
En algún modo de realización, los intermedios 2 son intermedios de tipo N y O en los que A, m, n y R15 son como se describe en el presente documento, se pueden preparar por procedimientos bien conocidos por un experto en
la técnica y como se ejemplifica por los procedimientos de síntesis generales explicados en el Esquema 9.

Esquema 9
En consecuencia, los intermedios 30a en los que A, m y son como se define en el presente documento, GP significa un grupo protector adecuado tal como Boc, Cbz o Bn e Y es un grupo formilo (disponible comercialmente o preparado por procedimientos de la literatura, por ejemplo, Bioorg. Med. Chem. Lett. 2006, 76(14), 3668; documentos WO2013/179024) se pueden tratar con un agente reductor tal como NaBH4 en MeOH para proporcionar los intermedios 31. Las reacciones de este tipo también se describen en la literatura (por ejemplo, documento WO2013/179024) (etapa a).
De forma alternativa, los intermedios 31 se pueden preparar a partir de los intermedios 30b en los que Y es un grupo carboxilo (disponible comercialmente o preparado de forma análoga a los procedimientos descritos en la literatura, por ejemplo, Bioorg. Med. Chem. 2013, 21(15), 4600; documento WO2016/109501) usando un agente reductor apropiado tal como un complejo de tetrahidrofurano de borano en un disolvente tal como THF. Las reacciones de este tipo también están ampliamente descritas en la literatura, por ejemplo, Bioorg. Med. Chem.
2013, 21(15), 4600) (etapa a).
La retirada del grupo protector de los intermedios 31 aplicando procedimientos bien conocidos en la técnica y como se describe anteriormente proporciona los intermedios N (etapa b).
La alquilación del grupo hidroxi de los intermedios 31 con un agente alquilante de tipo R15GS en el que GS es un grupo saliente adecuado y R15 como se describe en el presente documento, usando las condiciones como se describe anteriormente, da los intermedios 32 (etapa c).
La retirada del grupo protector de los intermedios 32 aplicando procedimientos bien conocidos en la técnica y como se describe en el esquema 8, etapa b, proporciona los intermedios O (etapa d).
En otro modo de realización, los intermedios 2 son intermedios de tipo P, en los que A es como se define en el presente documento, m es 1 o 2, Ar significa un grupo arilo y HET significa un grupo heterociclilo o heteroarilo. Los intermedios de ese tipo se pueden preparar de forma análoga a los procedimientos de la literatura (por ejemplo, Bioorg. Med. Chem. 2013, 21(7) 1756) o como se ejemplifica por el procedimiento de síntesis explicado en el esquema 10.

Esquema 10
El grupo carbonilo en los intermedios 33, en los que Ar es arilo y HET es heteroarilo o heterociclilo, se puede reducir por procedimientos bien conocidos en la técnica, por ejemplo, usando NaBH4 en MeOH, para dar los intermedios 34 (etapa a).
El grupo hidroxilo de los intermedios 34 se puede convertir en un grupo saliente adecuado tal como cloro, bromo, OSO2alquilo (por ejemplo, mesilato (metanosulfonato), OSO2fluoroalquilo (por ejemplo, triflato (trifluorometanosulfonato) u OSO2arilo (por ejemplo, tosilato (p-toluenosulfonato))), por ejemplo, un grupo cloro por reacción con cloruro de tionilo en tolueno, para dar los intermedios 34 (etapa b).
Los intermedios 35 se pueden hacer reaccionar con piperacinas monoprotegidas (m = 1) u homopiperacinas (m = 2) 36 disponibles comercialmente, en las que GP es un grupo protector, tal como Boc, Cbz o Bn, opcionalmente en presencia de KI, usando una base y disolvente adecuados tal como TEA o base de Huenig en AcCN para proporcionar los intermedios 37 (etapa c).
La retirada del grupo protector de los intermedios 37 aplicando procedimientos bien conocidos en la técnica y como se describe anteriormente proporciona los intermedios P (etapa d).
En otro modo de realización, los intermedios 2 son intermedios de tipo Q en los que A es como se define en el presente documento, m es 1 o 2, Ar significa un grupo arilo y HET significa un grupo heteroarilo. Los intermedios de ese tipo se pueden preparar de forma análoga a los procedimientos de la literatura (por ejemplo, Bioorg. Med. Chem. Lett. 2O06, 76(16), 4349; Bioorg. Med. Chem. Lett. 2010, 20(12), 3788) y como se ejemplifica por el procedimiento de síntesis explicado en el esquema 11.

Esquema 11
La condensación de aldehidos de arilo o heteroarilo 38 con piperacinas monoprotegidas (m = 1) u homopiperacinas (m = 2) 36 en las que GP es un grupo protector adecuado tal como un grupo Boc y benzotriazol, por ejemplo, en tolueno a reflujo con retirada azeotrópica de H2O da los intermedios 39 (etapa a).
Los intermedios 39 se hacen reaccionar in situ con reactivos bencílicos de Grignard o cinc 40 del tipo Ar/HETCH2MX, siendo MX un grupo tal como MgCl, MgBr, ZnCl o ZnBr para dar los intermedios 41 (etapa b). La retirada del grupo protector de los intermedios 41 aplicando procedimientos bien conocidos en la técnica y como se describe anteriormente proporciona los intermedios Q (etapa c).
En otro modo de realización, los intermedios 2 en los que X = N son intermedios de tipo R y S en los que m y R14 son como se define en el presente documento y R18 y R19 se seleccionan cada uno independientemente de hidrógeno, alquilo sustituido o no sustituido, ciano, alcoxi y halógeno, en los que el alquilo sustituido es como se define en el presente documento. Los intermedios de ese tipo se pueden preparar de forma análoga a los procedimientos de la literatura (por ejemplo, documento WO2007/098418) y como se ejemplifica por el procedimiento de síntesis explicado en el esquema 12.

Esquema 12
Los intermedios de indol 42 se pueden hacer reaccionar con piperacinas monoprotegidas (m = 1) u homopiperacinas (m = 2) 36 en las que GP es un grupo protector adecuado tal como un grupo Boc, Cbz o Bn, y GS es un grupo saliente apropiado tal como -OAc o yodo, para proporcionar los intermedios 43 (etapa a). Las reacciones de este tipo se describen en la literatura (por ejemplo, documento WO2007/098418).
La retirada simultánea o secuencial de los grupos protectores de los intermedios 43 aplicando procedimientos bien conocidos en la técnica y como se describe anteriormente proporciona los intermedios R (etapa b).
La retirada selectiva del grupo protector indol de los intermedios 43 por procedimientos bien conocidos en la técnica da los intermedios 44 (etapa c). Los intermedios 44 se pueden convertir en intermedios 45 usando compuestos del tipo R14GS por procedimientos conocidos en la técnica y como se describe en el esquema 6, etapa a (etapa d). La retirada del grupo protector de los intermedios 45 aplicando procedimientos bien conocidos en la técnica y como se describe anteriormente proporciona los intermedios S (etapa e).
En otro modo de realización, los intermedios 2 son intermedios de tipo T , en los que A es como se define en el presente documento, m es 1 o 2 y R1 se selecciona de alquilo sustituido o no sustituido, cicloalquilo sustituido o no sustituido, arilo sustituido o no sustituido y heteroarilo sustituido o no sustituido. Los intermedios de ese tipo se pueden preparar de forma análoga a los procedimientos de la literatura (por ejemplo, Tetrahedron Letters 1990, 31(39), 5547) o como se ejemplifica por el procedimiento de síntesis explicado en el esquema 13.
El grupo carbonilo en los intermedios 46 se hace reaccionar por aminación reductora con una amina 36 por procedimientos bien conocidos en la técnica, por ejemplo, catalizada por un ácido, tal como TiCU, lo que da lugar a la imina, que a continuación se reduce directamente in situ al correspondiente intermedio de amina 47 con un agente reductor tal como cianoborohidruro de sodio (etapa a).
La retirada del grupo protector de los intermedios 47 aplicando procedimientos bien conocidos en la técnica y como se describe anteriormente proporciona los intermedios T (etapa b).

Esquema 13
En otro modo de realización, los intermedios 2 son intermedios de tipo U, en los que A es un pirazol sustituido con arilo y m es como se describe en el presente documento. Los intermedios de ese tipo se pueden preparar de forma análoga a los procedimientos de la literatura (por ejemplo, J. Med. Chem. 2018, 61(7), 3008) o como se ejemplifica por el procedimiento de síntesis explicado en el esquema 14.

Esquema 14
La cetona en los intermedios 48 se hace reaccionar con N,N-dimetilformamida-dimetilacetal y diclorhidrato de hidracina para proporcionar el intermedio de pirazol 49 (etapa a).
A continuación, se hace reaccionar un ácido aril-borónico, usando los reactivos acetato de cobre(II) y piridina para facilitar la reacción, para formar el intermedio 50 (etapa b). La retirada del grupo protector de los intermedios 50 aplicando procedimientos bien conocidos en la técnica y como se describe anteriormente proporciona los intermedios U (etapa c).
En algunos modos de realización, los intermedios 2 son intermedios de tipo V en los que m, n son como se describe en el presente documento, A es un anillo de arilo o heteroarilo opcionalmente sustituido adicionalmente y cada uno de R21 a R23 se selecciona independientemente de hidrógeno, (ciclo)alquilo sustituido o no sustituido, (ciclo)alcoxi, arilo sustituido o no sustituido, RbRcN, ciano, heterociclo, metilsulfonilo y halógeno, en los que alquilo, arilo y heteroarilo sustituidos son como se define en el presente documento. Los intermedios de ese tipo se pueden preparar por procedimientos bien conocidos en la técnica y como se ejemplifica por los procedimientos de síntesis generales explicados en el esquema 15.

Esquema 15
Los intermedios 51 disponibles comercialmente en los que GP significa un grupo protector adecuado y X es bromuro o yoduro se pueden someter a reacciones de acoplamiento cruzado tales como las reacciones de acoplamiento de Negishi, Heck, Stille, Suzuki, Sonogashira o Buchwald-Hartwig con los compuestos 52, disponibles comercialmente o bien preparados por procedimientos conocidos en la técnica, en los que GF significa un grupo funcional adecuado tal como, por ejemplo, cloro, bromo, yodo, -OSO 2alquilo (por ejemplo, mesilato (metanosulfonato), -OSO 2 fluoroalquilo (por ejemplo, triflato (trifluorometanosulfonato) u -OSO 2arilo (por ejemplo, tosilato (p-toluenosulfonato) (etapa a).
Las reacciones de este tipo se describen ampliamente en la literatura y son bien conocidas por los expertos en la técnica.
Por ejemplo, los intermedios 51 se pueden hacer reaccionar con ácidos borónicos 52a (GF = B(OH)2) o ésteres borónicos 52b (GF = por ejemplo, éster de 4,4,5,5-tetrametil-2-fenil-1,3,2-dioxaborolano (pinacol)) de arilo o heteroarilo disponibles comercialmente o bien preparados usando procedimientos de la literatura como se describe, por ejemplo, en "Boronic Acids - Preparation and Applications in Organic Synthesis and Medicine" de Dennis G. Hall (ed.) 1.a ed., 2005, John Wiley & Sons, New York) usando un catalizador adecuado (por ejemplo, aducto de diclorometano-dicloro[1,1'-bis(difenilfosfino)-ferroceno]paladio(II), tetraquis(trifenilfosfina)paladio(0) o acetato de paladio(II) con trifenilfosfina) en un disolvente apropiado (por ejemplo, dioxano, dimetoxietano, agua, tolueno, DMF o mezclas de los mismos) y una base adecuada (por ejemplo, Na2CO3, NaHCO3, KF, K2CO3 o TEA) a temperaturas entre la temperatura ambiente y el punto de ebullición del disolvente o mezcla de disolventes, para proporcionar los intermedios 53 (etapa a). Las reacciones de Suzuki de este tipo se describen ampliamente en la literatura (por ejemplo, A. Suzuki, Pure Appl. Chem. 1991, 63, 419-422; A. Suzuki, N. Miyaura, Chem. Rev. 1995, 95, 2457-2483; A. Suzuki, J. Organomet. Chem. 1999, 576, 147-168; V. Polshettiwar et al., Chem. Sus. Chem. 2010, 3, 502-522) y son bien conocidas por los expertos en la técnica. De forma alternativa, se pueden usar trifluoroboratos de arilo o heteroarilo 52c (GF = BF3) en la reacción de acoplamiento cruzado aplicando un catalizador de paladio tal como, por ejemplo, tetraquis(trifenilfosfina)-paladio (0), acetato de paladio(II) o aducto de diclorometano-dicloro[1,1'-bis(difenilfosfino)ferroceno]-paladio(II) en presencia de una base adecuada tal como carbonato de cesio o fosfato de potasio en disolventes tales como tolueno, THF, dioxano, agua o mezclas de los mismos, a temperaturas entre temperatura ambiente y el punto de ebullición del disolvente o mezcla de disolventes.
De forma alternativa, los intermedios 51 se pueden hacer reaccionar con estannanos de arilo o heteroarilo 52d en los que GF es Sn(alquilo)3 y alquilo es preferentemente n-butilo o metilo, usando un catalizador y disolvente adecuados tales como, por ejemplo, tetraquis(trifenilfosfina)-paladio(0) en DMF a temperaturas entre la temperatura ambiente y el punto de ebullición del disolvente o mezcla de disolventes para proporcionar los intermedios 53 (etapa a). Las reacciones de Stille de ese tipo son bien conocidas en la técnica y se describen en la literatura, por ejemplo, Org. React. 1997, 50, 1-652, ACS Catal. 2015, 5, 3040-3053.
Además, los intermedios 51 se pueden hacer reaccionar con haluros de arilo o heteroarilcinc 52e en los que GF
es ZnHal y Hal preferentemente bromuro o yoduro, disponibles comercialmente o bien preparados por procedimientos de la literatura, usando un catalizador y un sistema de disolventes apropiados tales como, por ejemplo, [1,1'-bis(difenilfosfino)ferroceno]dicloropaladio(N) y yoduro de cobre(I) en DMA, o tetraquis(trifenilfosfina)paladio(0) en THF o DMF a temperaturas entre la temperatura ambiente y el punto de ebullición del disolvente para proporcionar los intermedios 53. (etapa a). Las reacciones de Negishi de ese tipo son bien conocidas en la técnica y también se describen en la literatura, por ejemplo, Org. Lett., 2005, 7, 4871, ACS Catal. 2016, 6 (3), 1540-1552. Ace. Chem. Res. 1982, 15 (11), pp. 340-348. De forma alternativa, los intermedios 53 se pueden preparar convirtiendo los intermedios 51 en los que X es, por ejemplo, yoduro en las correspondientes especies de cinc aplicando procedimientos de la literatura (por ejemplo, reacción de 51 con Zn en polvo en presencia de clorotrimetilsilano y 1,2-dibromoetano en un disolvente adecuado tal como DMA) y el acoplamiento de las especies de cinc con bromuros o yoduros de arilo o heteroarilo en las condiciones mencionadas anteriormente.
De forma alternativa, los intermedios 51 en los que X es preferentemente bromuro se pueden someter a un acoplamiento electrofílico cruzado con bromuros de arilo o heteroarilo 52f en los que GF significa bromuro bajo irradiación con una lámpara de luz azul de 420 nm usando un fotocatalizador apropiado tal como [Ir{dF(CF3)ppy}2(dtbpy)]PF6 (hexafluorofosfato de [4,4-bis(1,1-dimetiletil)-2,2-bipiridina-N1,N1']bis[3,5-difluoro-2-[5-(trifluorometil)-2-piridinil-N]fenil-C]/r/d/o(NI)), un catalizador de níquel como NiCh glyme (dicloro(dimetoxietano)níquel), 4,4-di-terc-butil-2,2'-dipiridilo y tris(trimetilsilil)silano, en presencia de una base adecuada tal como carbonato de sodio anhidro en un disolvente como DME. Las reacciones de este tipo se describen en la literatura, por ejemplo, J. Am. Chem. Soc. 2016, 138, 8084. (etapa a).
La retirada del grupo protector de los intermedios 53 aplicando procedimientos bien conocidos en la técnica y como se describe, por ejemplo, en el esquema 2, etapa c, proporciona los intermedios V (etapa b).
Los intermedios 53 se pueden preparar de forma alternativa a partir de los intermedios 51 y bromuros de arilo o heteroarilo 54, disponibles comercialmente o bien preparados por procedimientos conocidos en la técnica, aplicando las transformaciones descritas anteriormente en la etapa a para proporcionar los intermedios 55 (etapa c).
Los intermedios 55 se pueden hacer reaccionar además con los compuestos 56 aplicando las mismas estrategias de síntesis como se describe anteriormente en la etapa a para proporcionar los intermedios 53 (etapa d).
Los intermedios 53 en los que R23 significa un grupo amino de tipo RbRcN en el que Rb es hidrógeno, alquilo o arilo y Rc es alquilo o arilo o en el que Rb y Rc, tomados conjuntamente con el átomo de nitrógeno al que están unidos, forman un anillo heterocíclico mono- o bicíclico, de 4-11 miembros sustituido adicionalmente, se pueden sintetizar, por ejemplo, a partir de la reacción de 55 con aminas primarias o secundarias RbRcNH y usando, por ejemplo, un catalizador (por ejemplo, Pd(OAc)2 , Pd2(dba)3), ligando (por ejemplo, BINAP, Xphos, BrettPhos, RuPhos), base (por ejemplo, Cs2CO3, K2CO3 , KOt-Bu, LiHMDS, K3PO4) y disolvente (por ejemplo, tolueno, THF, dioxano) adecuados. Las reacciones de Buchwald-Hartwig de ese tipo son conocidas en la técnica y se describen en la literatura (por ejemplo, Angew. Chem. Int. Ed. 2008, 47, 6338; Chem. Rev. 2008, 108, 3054, J. Organomet. Chem.
2018, 861, 17) (etapa d).
En algún modo de realización, los compuestos de fórmula (I) son compuestos Id y le en los que m y n son como se describe en el presente documento, R21 y R22 se seleccionan cada uno independientemente de hidrógeno, (ciclo)alquilo sustituido o no sustituido, haloalquilo, (ciclo)alcoxi, arilo sustituido o no sustituido, RbRcN, ciano, heterociclo, metilsulfonilo y halógeno, X es Hal, A es arilo opcionalmente sustituido o heteroarilo opcionalmente sustituido, C es arilo opcionalmente sustituido o un heterociclo de 4-7 miembros que contiene al menos un átomo de nitrógeno al que está unido al anillo A. Los intermedios de ese tipo se pueden preparar por procedimientos bien conocidos en la técnica y como se ejemplifica por los procedimientos de síntesis generales explicados en el esquema 16.

Esquema 16
El grupo protector de los intermedios 55 se puede retirar aplicando procedimientos bien conocidos en la técnica y como se describe, por ejemplo, en el esquema 2, etapa c, para dar los intermedios 57 (etapa a).
Los intermedios 50 se pueden acoplar con los intermedios 1 en las condiciones descritas en el esquema 1, etapa a, para proporcionar los compuestos 1d (etapa b).
El sustituyente bromo o yodo en los compuestos 1d se puede convertir en un ácido borónico o éster borónico (por ejemplo, éster de pinacol) de acuerdo con los procedimientos descritos en la literatura o como se explica en el esquema 15, etapa a, para proporcionar los intermedios 58 (etapa c).
Los intermedios 58 se pueden hacer reaccionar con los compuestos 59, disponibles comercialmente o bien preparados por procedimientos de la literatura y en los que GF es un grupo funcional apropiado tal como cloruro, bromuro, OSO2alquilo (por ejemplo, mesilato (metanosulfonato), -OSO 2fluoroalquilo (por ejemplo, triflato (trifluorometanosulfonato) u -OSO 2 arilo (por ejemplo, tosilato (p-toluenosulfonato) en un acoplamiento de Suzuki usando, por ejemplo, las condiciones de reacción descritas en el esquema 15, etapa a, para proporcionar los compuestos 1e (etapa d).
Los compuestos 1e en los que el anillo C es un heterociclo de 4-7 miembros enlazado por medio de su nitrógeno al anillo A se pueden preparar, por ejemplo, por la reacción de acoplamiento de Buchwald-Hartwig de los compuestos 1d con los compuestos 60 aplicando, por ejemplo, las condiciones descritas en el esquema 15, etapa d (etapa e).
En algún modo de realización, los intermedios 2 son intermedios de tipo W, X e Y en los que m, n son como se describe en el presente documento, L es un enlace covalente y A es un anillo heteroarilo de 5 miembros sustituido adicionalmente por un anillo arilo opcionalmente sustituido adicionalmente. Los compuestos de ese tipo se pueden preparar por procedimientos bien conocidos en la técnica o por los procedimientos ejemplificados en los esquemas 17, 18 y 19 a continuación.
Los intermedios de tipo W , en los que cada Y es independientemente CH o un heteroátomo, por ejemplo, un heteroátomo seleccionado de N, O y S, se pueden preparar por una variedad de condiciones, que se pueden ejemplificar por el procedimiento de síntesis general explicado en el esquema 17. Por ejemplo, si el anillo de heteroarilo de 5 miembros en los compuestos 62 es un 1 H-pirrol, 1H-imidazol, 1H-pirazol o 1 H-triazol sustituido con bromo, se puede someter a una reacción de acoplamiento de Suzuki con ácidos fenilborónicos opcionalmente sustituidos 61a (B(ORa)2 = B(OH)2) o ésteres borónicos 61b (por ejemplo, B(ORa)2 = éster de 4,4,5,5-tetrametil-2-fenil-1,3,2-dioxaborolano (pinacol))) en los que R27 y R28 se seleccionan cada uno independientemente de hidrógeno, (ciclo)alquilo sustituido o no sustituido, haloalquilo, (ciclo)alcoxi, arilo sustituido o no sustituido, RbRcN, ciano, heteroarilo sustituido o no sustituido, heterociclo, metilsulfonilo y halógeno aplicando procedimientos conocidos en la técnica y como se describe en el esquema 15, etapa a, para proporcionar los intermedios 63 (etapa a).
Los intermedios 63 se pueden hacer reaccionar con los compuestos 64, disponibles comercialmente o bien preparados de forma análoga a los procedimientos de la literatura, por ejemplo, aplicando condiciones fotooxidorreducción como se describe en el esquema 15, etapa a, para dar los intermedios 65 (etapa b).
La retirada del grupo protector de los intermedios 65 aplicando procedimientos bien conocidos en la técnica y como se describe, por ejemplo, en el esquema 2, etapa c, proporciona los intermedios W (etapa c).

Los intermedios de tipo X en los que el anillo de heteroarilo de 5 miembros es un 1,2,4-oxadiazol sustituido adicionalmente en la posición 3 con un anillo de fenilo opcionalmente sustituido adicionalmente se pueden preparar, por ejemplo, por el procedimiento de síntesis general explicado en el esquema 18.

Esquema 18
Los benzonitrilos 66, disponibles comercialmente o sintetizados por procedimientos conocidos en la técnica, se pueden hacer reaccionar con hidroxilamina, por ejemplo, usando clorhidrato de hidroxilamina, y K2CO3 en EtOH a temperaturas que van de la t. a. al punto de ebullición del disolvente para proporcionar las amidoximas 67 (etapa a).
El acoplamiento de los intermedios 67 con ácidos carboxílicos activados 68a (X = H) o cloruros de ácidos carboxílicos 68b (X = Cl), en los que GP significa un grupo protector adecuado, con ciclodeshidratación posterior proporciona los intermedios 69 (etapa b). Los acoplamientos de amida de este tipo se describen ampliamente en la literatura y se pueden lograr por el uso de reactivos de acoplamiento tales como CDI, DCC, HATU, HBTU, HOBT, TBTU, T3P o reactivo de Mukaiyama (Mukaiyama T. Angew. Chem., Int. Ed. Engl. 1979, 18, 707) en un disolvente adecuado, por ejemplo, DMF, DMA, DCM o dioxano, opcionalmente en presencia de una base (por ejemplo, NEt3, DIPEA (base de Huenig) o DMAP). De forma alternativa, los ácidos carboxílicos 68a se pueden convertir en sus cloruros de ácido 68b por tratamiento, por ejemplo, con cloruro de tionilo o cloruro de oxalilo, puro u opcionalmente en un disolvente tal como DCM. La reacción del cloruro de ácido con los intermedios 68 en un disolvente apropiado tal como DCM o DMF y una base, por ejemplo, NEt3, base de Huenig, piridina, DMAP o bis(trimetilsilil)amida de litio a temperaturas que van de 0 °C a la temperatura de reflujo del disolvente o mezcla de disolventes proporciona la forma acilada de los intermedios 67 que se pueden deshidratar, por ejemplo, a temperaturas elevadas para proporcionar los intermedios 69 (etapa b).
La retirada del grupo protector de los intermedios 69 aplicando procedimientos bien conocidos en la técnica y como se describe, por ejemplo, en el esquema 2, etapa c, proporciona los intermedios X (etapa c).
Los intermedios de tipo Y en los que el anillo de heteroarilo de 5 miembros es un 1,3,4-oxadiazol sustituido adicionalmente en la posición 5 con un anillo de fenilo opcionalmente sustituido adicionalmente se pueden preparar por procedimientos descritos en la literatura o, por ejemplo, por el procedimiento de síntesis general explicado en el esquema 19.

Esquema 19
La acilación de las acilhidracidas 70, disponibles comercialmente o bien preparadas por procedimientos bien conocidos en la técnica, con ácidos carboxílicos activados 68a (X = H) o cloruros de ácido carboxílico 68b (X = Cl) aplicando las condiciones descritas en el esquema 18, etapa b, proporciona los intermedios 71 (etapa a).
La ciclodeshidratación posterior de los intermedios 71, por ejemplo, por calentamiento, proporciona los intermedios 72 (etapa b).
La retirada del grupo protector de los intermedios 72 aplicando procedimientos bien conocidos en la técnica y como se describe, por ejemplo, en el esquema 2, etapa c, proporciona los intermedios Y (etapa c).
En un aspecto, la presente invención proporciona un procedimiento de fabricación de los compuestos de fórmula (I) como se describe en el presente documento, que comprende:
hacer reaccionar 4a,5,6,7,8,8a-hexahidro-4H-pirido[4,3-b][1,4]oxacin-3-ona (1),

con una amina heterocíclica 2, en la que A, L, X, m y n son como se define en el presente documento
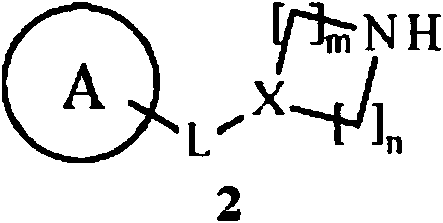
en presencia de una base y un reactivo formador de urea, para formar dichos compuestos de fórmula (I).
En otro aspecto, la presente invención proporciona un procedimiento de fabricación de los compuestos de fórmula (Ic) como se describe en el presente documento, que comprende:
hacer reaccionar 4a,5,6,7,8,8a-hexahidro-4H-pirido[4,3-b][1,4]oxacin-3-ona (1),

con una amina heterocíclica 2a, en la que A, L, X, m, n y de R20 a R23 son como se define en el presente documento

en presencia de una base y un reactivo formador de urea, para formar dichos compuestos de fórmula (Ic).
En un modo de realización, se proporciona un procedimiento de acuerdo con la invención, en el que dicha base es bicarbonato de sodio.
En un modo de realización, se proporciona un procedimiento de la invención, en el que dicho reactivo formador de urea se selecciona de carbonato de bis(triclorometilo), fosgeno, cloroformiato de triclorometilo, carbonato de (4-nitrofenilo) y 1,1'-carbonildiimidazol, preferentemente en el que dicho reactivo formador de urea es carbonato de bis(triclorometilo).
Actividad inhibidora de MAGL
Los compuestos de la presente invención son inhibidores de MAGL. Por tanto, en un aspecto, la presente invención proporciona el uso de compuestos de fórmula (Ic) como se describe en el presente documento para inhibir MAGL en un mamífero.
En otro aspecto, la presente invención proporciona compuestos de fórmula (Ic) como se describe en el presente documento para su uso en un procedimiento de inhibición de MAGL en un mamífero.
En otro aspecto, la presente invención proporciona el uso de compuestos de fórmula (Ic) como se describe en el presente documento para la preparación de un medicamento para inhibir MAGL en un mamífero.
En otro aspecto, la presente invención proporciona un procedimiento para inhibir MAGL en un mamífero, comprendiendo dicho procedimiento administrar al mamífero una cantidad eficaz de un compuesto de fórmula (Ic) como se describe en el presente documento.
Los compuestos se perfilaron para obtener la actividad inhibidora de MAGL midiendo la actividad enzimática de MAGL siguiendo la hidrólisis de acetato de 4-nitrofenilo que da como resultado 4-nitrofenol, que se absorbe a 405 412 nm (G.G. Muccioli, G. Labar, D.M. Lambert, Chem. Bio. Chem. 2008, 9, 2704-2710). Este ensayo se abrevia a continuación en el presente documento como "ensayo 4-NPA".
El ensayo se llevó a cabo en placas de ensayo de 384 pocillos (negras con fondo transparente, superficie de no unión tratada, ref. de Corning 3655) en un volumen total de 40 pl. Las diluciones del compuesto se realizaron en DMSO al 100 % (VWR Chemicals 23500.297) en una placa de polipropileno en etapas de dilución de 3 veces para dar un intervalo de concentración final en el ensayo de 25 pM a 1,7 pM. Se añadieron diluciones de compuesto de 1 pl (DMSO al 100 %) a 19 pl de MAGL (natural recombinante) en tampón de ensayo (TRIS 50 mM (GIBCO, 15567 027), EDTA 1 mM (Fluka, 03690-100 ml)). Se agitó la placa durante 1 min a 2000 rpm (Variomag Teleshake) y a continuación se incubó durante 15 min a t. a. Para iniciar la reacción, se añadieron 20 pl de acetato de 4-nitrofenilo (Sigma N-8130) en tampón de ensayo con EtOH al 6 %. Las concentraciones finales en el ensayo fueron 1 nM de MAGL y 300 pM de acetato de 4-nitrofenilo. Después de agitar (1 min, 2000 rpm) y 5 min de incubación a t. a., se midió la absorbancia a 405 nm por primera vez (Molecular Devices, SpectraMax Paradigm). A continuación se realizó una segunda medición después de la incubación durante 80 min a t. a. A partir de las dos mediciones, se
calculó la pendiente restando la primera de la segunda medición.
De forma alternativa, los compuestos se perfilaron para obtener la actividad inhibidora de MAGL determinando la actividad enzimática siguiendo la hidrólisis del sustrato natural 2-araquidonoilglicerol (2-AG) que da como resultado ácido araquidónico, que se puede seguir por espectrometría de masas. Este ensayo se abrevia a continuación en el presente documento como "ensayo 2-AG".
El ensayo de 2-AG se llevó a cabo en placas de ensayo de 384 pocillos (PP, cat. de Greiner n.° 784201) en un volumen total de 20 pl. Las diluciones del compuesto se realizaron en DMSO al 100 % (VWR Chemicals 23500.297) en una placa de polipropileno en etapas de dilución de 3 veces para dar un intervalo de concentración final en el ensayo de 12,5 pM a 0,8 pM. Se añadieron diluciones de compuesto de 0,25 |jl (DMSO al 100 %) a 9 |jl de MAGL en tampón de ensayo (TRIS 50 mM (GIBCO, 15567-027), Ed Ta 1 mM (Fluka, 03690-100 ml), Tween al 0,01 % (v/v). Después de agitar, se incubó la placa durante 15 min a t. a. Para iniciar la reacción, se añadieron 10 pl de 2-araquidonoilglicerol en tampón de ensayo. Las concentraciones finales en el ensayo fueron 50 pM de MAGL y 8 pM de 2-araquidonoilglierol. Después de agitar y 30 min de incubación a t. a., se detuvo la reacción por la adición de 40 pl de ACN que contenía 4 pM de ácido d8-araquidónico. Se rastreó la cantidad de ácido araquidónico por un sistema SPE en línea (Agilent Rapidfire) acoplado a un espectrómetro de masas de triple cuadrupolo (Agilent 6460). Se usó un cartucho SPE C18 (G9205A) en una configuración de líquido de ACN/agua. El espectrómetro de masas se hizo funcionar en modo de electronebulización negativa siguiendo las transiciones de masas 303,1 ^ 259,1 para ácido araquidónico y 311,1 ^ 267,0 para ácido d8-araquidónico. Se calculó la actividad de los compuestos en base a la proporción de intensidades [ácido araquidónico/ácido d8-araquidónico].
Tabla 1
































[a]: si no se indica de otro modo (véase [b]), se midió la actividad en el ensayo 4-NPA; [b]: medido en el ensayo 2-AG.
En un aspecto, la presente invención proporciona compuestos de fórmula (Ic) y sus sales o ésteres farmacéuticamente aceptables como se describe en el presente documento, en los que dichos compuestos de fórmula (Ic) y sus sales o ésteres farmacéuticamente aceptables tienen CI50 para la inhibición de MAGL por debajo de 25 |jM, preferentemente por debajo de 10 |jM, más preferentemente por debajo de 5 |jM como se mide en el ensayo de MAGL descrito en el presente documento.
En un modo de realización, los compuestos de fórmula (Ic) y sus sales o ésteres farmacéuticamente aceptables como se describe en el presente documento tienen valores de CI50 (inhibición de MAGL) entre 0,000001 |jM y 25 j M, los compuestos particulares tienen valores de CI50 entre 0,000005 j M y 10 j M, otros compuestos particulares tienen valores de CI50 entre 0,00005 j M y 5 j M, como se mide en el ensayo de MAGL descrito en el presente documento.
En un modo de realización, la presente invención proporciona compuestos de fórmula (Ic) y sus sales o ésteres farmacéuticamente aceptables como se describe en el presente documento, en los que dichos compuestos de fórmula (Ic) y sus sales o ésteres farmacéuticamente aceptables tienen un CI50 para MAGL por debajo de 25 j M, preferentemente por debajo de 10 j M, más preferentemente por debajo de 5 j M como se mide en un ensayo que comprende las etapas de:
a) proporcionar una solución de un compuesto de fórmula (Ic), o una sal o éster farmacéuticamente aceptable del mismo, en DMSO;
b) proporcionar una solución de MAGL (natural recombinante) en tampón de ensayo (tris(hidroximetil)aminometano 50 mM; ácido etilendiaminatetraacético 1 mM);
c) añadir 1 jl de solución de compuesto de la etapa a) a 19 jl de solución de MAGL de la etapa b);
d) agitar la mezcla durante 1 min a 2000 rpm;
e) incubar durante 15 min a t. a.;
f) añadir 20 jl de una solución de acetato de 4-nitrofenilo en tampón de ensayo (tris(hidroximetil)aminometano 50 mM; ácido etilendiaminatetraacético 1 mM, EtOH al 6 %);
g) agitar la mezcla durante 1 min a 2000 rpm;
h) incubar durante 5 min a t. a.;
i) medir la absorbancia de la mezcla a 405 nm por primera vez;
j) incubar durante otros 80 min a t. a.;
k) medir la absorbancia de la mezcla a 405 nm por segunda vez;
l) restar la absorbancia medida en i) de la absorbancia medida en k) y calcular la pendiente de absorbancia; en los que:
i) la concentración del compuesto de fórmula (Ic), o la sal o éster farmacéuticamente aceptable del mismo en el ensayo después de la etapa f) está en el intervalo de 25 |jM a 1,7 nM;
ii) la concentración de MAGL en el ensayo después de la etapa f) es de 1 nM;
iii) la concentración de acetato de 4-nitrofenilo en el ensayo después de la etapa f) es de 300 j M; y iv) las etapas de a) a 1) se repiten al menos 3 veces, cada vez con una concentración diferente del compuesto de fórmula (Ic), o la sal o éster farmacéuticamente aceptable del mismo.
Uso de los compuestos de la invención
En un aspecto, la presente invención proporciona compuestos de fórmula (Ic) como se describe en el presente documento para su uso como sustancia terapéuticamente activa.
En otro aspecto, la presente invención proporciona el uso de compuestos de fórmula (Ic) como se describe en el presente documento para el tratamiento o la profilaxis de neuroinflamación, enfermedades neurodegenerativas, dolor, cáncer y/o trastornos mentales en un mamífero.
En un modo de realización, la presente invención proporciona el uso de compuestos de fórmula (Ic) como se describe en el presente documento para el tratamiento o la profilaxis de neuroinflamación y/o enfermedades neurodegenerativas en un mamífero.
En un modo de realización, la presente invención proporciona el uso de compuestos de fórmula (Ic) como se describe en el presente documento para el tratamiento o la profilaxis de enfermedades neurodegenerativas en un mamífero.
En un modo de realización, la presente invención proporciona el uso de compuestos de fórmula (Ic) como se describe en el presente documento para el tratamiento o la profilaxis de cáncer en un mamífero.
En un aspecto, la presente invención proporciona el uso de compuestos de fórmula (Ic) como se describe en el presente documento para el tratamiento o la profilaxis de esclerosis múltiple, enfermedad de Alzheimer, enfermedad de Parkinson, esclerosis lateral amiotrófica, traumatismo craneoencefálico, neurotoxicidad, apoplejía, epilepsia, ansiedad, migraña, depresión, carcinoma hepatocelular, carcinogénesis de colon, cáncer de ovario, dolor neuropático, neuropatía inducida por quimioterapia, dolor agudo, dolor crónico y/o espasticidad asociada con dolor en un mamífero.
En un modo de realización preferente, la presente invención proporciona el uso de compuestos de fórmula (Ic) como se describe en el presente documento para el tratamiento o la profilaxis de esclerosis múltiple, enfermedad de Alzheimer y/o enfermedad de Parkinson en un mamífero.
En un modo de realización en particular preferente, la presente invención proporciona el uso de compuestos de fórmula (Ic) como se describe en el presente documento para el tratamiento o la profilaxis de esclerosis múltiple en un mamífero.
En un aspecto, la presente invención proporciona compuestos de fórmula (Ic) como se describe en el presente documento para su uso en el tratamiento o la profilaxis de neuroinflamación, enfermedades neurodegenerativas, dolor, cáncer y/o trastornos mentales en un mamífero.
En un modo de realización, la presente invención proporciona compuestos de fórmula (Ic) como se describe en el presente documento para su uso en el tratamiento o la profilaxis de neuroinflamación y/o enfermedades neurodegenerativas en un mamífero.
En un modo de realización, la presente invención proporciona compuestos de fórmula (Ic) como se describe en el
presente documento para su uso en el tratamiento o la profilaxis de cáncer en un mamífero.
En un modo de realización, la presente invención proporciona compuestos de fórmula (Ic) como se describe en el presente documento para su uso en el tratamiento o la profilaxis de enfermedades neurodegenerativas en un mamífero.
En un aspecto, la presente invención proporciona compuestos de fórmula (Ic) como se describe en el presente documento para su uso en el tratamiento o la profilaxis de esclerosis múltiple, enfermedad de Alzheimer, enfermedad de Parkinson, esclerosis lateral amiotrófica, traumatismo craneoencefálico, neurotoxicidad, apoplejía, epilepsia, ansiedad, migraña, depresión, carcinoma hepatocelular, carcinogénesis de colon, cáncer de ovario, dolor neuropático, neuropatía inducida por quimioterapia, dolor agudo, dolor crónico y/o espasticidad asociada con dolor en un mamífero.
En un modo de realización preferente, la presente invención proporciona compuestos de fórmula (Ic) como se describe en el presente documento para su uso en el tratamiento o la profilaxis de esclerosis múltiple, enfermedad de Alzheimer y/o enfermedad de Parkinson en un mamífero.
En un modo de realización en particular preferente, la presente invención proporciona compuestos de fórmula (Ic) como se describe en el presente documento para su uso en el tratamiento o la profilaxis de esclerosis múltiple en un mamífero.
En un aspecto, la presente invención proporciona el uso de compuestos de fórmula (Ic) como se describe en el presente documento para la preparación de un medicamento para el tratamiento o la profilaxis de neuroinflamación, enfermedades neurodegenerativas, dolor, cáncer y/o trastornos mentales en un mamífero.
En un modo de realización, la presente invención proporciona el uso de compuestos de fórmula (Ic) como se describe en el presente documento para la preparación de un medicamento para el tratamiento o la profilaxis de neuroinflamación y/o enfermedades neurodegenerativas en un mamífero.
En un modo de realización, la presente invención proporciona el uso de compuestos de fórmula (Ic) como se describe en el presente documento para la preparación de un medicamento para el tratamiento o la profilaxis de enfermedades neurodegenerativas en un mamífero.
En un modo de realización, la presente invención proporciona el uso de compuestos de fórmula (Ic) como se describe en el presente documento para la preparación de un medicamento para el tratamiento o la profilaxis de cáncer en un mamífero.
En otro aspecto, la presente invención proporciona el uso de compuestos de fórmula (Ic) como se describe en el presente documento para la preparación de un medicamento para el tratamiento o la profilaxis de esclerosis múltiple, enfermedad de Alzheimer, enfermedad de Parkinson, esclerosis lateral amiotrófica, traumatismo craneoencefálico, neurotoxicidad, apoplejía, epilepsia, ansiedad, migraña, depresión, carcinoma hepatocelular, carcinogénesis de colon, cáncer de ovario, dolor neuropático, neuropatía inducida por quimioterapia, dolor agudo, dolor crónico y/o espasticidad asociada con dolor en un mamífero.
En un modo de realización preferente, la presente invención proporciona el uso de compuestos de fórmula (Ic) como se describe en el presente documento para la preparación de un medicamento para el tratamiento o la profilaxis de esclerosis múltiple, enfermedad de Alzheimer y/o enfermedad de Parkinson en un mamífero.
En un modo de realización en particular preferente, la presente invención proporciona el uso de compuestos de fórmula (Ic) como se describe en el presente documento para la preparación de un medicamento para el tratamiento o la profilaxis de esclerosis múltiple en un mamífero.
Composiciones farmacéuticas y administración
En un aspecto, la presente invención proporciona una composición farmacéutica que comprende un compuesto de fórmula (Ic) como se describe en el presente documento y un vehículo terapéuticamente inerte.
Los compuestos de fórmula (Ic) y sus sales y ésteres farmacéuticamente aceptables se pueden usar como medicamentos (por ejemplo, en forma de preparaciones farmacéuticas). Las preparaciones farmacéuticas se pueden administrar por vía interna, tal como por vía oral (por ejemplo, en forma de comprimidos, comprimidos recubiertos, grageas, cápsulas de gelatina dura y blanda, soluciones, emulsiones o suspensiones), por vía nasal (por ejemplo, en forma de pulverizadores nasales) o por vía rectal (por ejemplo, en forma de supositorios). Sin embargo, la administración también se puede efectuar por vía parenteral, tal como por vía intramuscular o intravenosa (por ejemplo, en forma de soluciones inyectables).
Los compuestos de fórmula (Ic) y sus sales y ésteres farmacéuticamente aceptables se pueden procesar con
adyuvantes inorgánicos u orgánicos farmacéuticamente inertes para la producción de comprimidos, comprimidos recubiertos, grageas y cápsulas de gelatina dura. Se pueden usar lactosa, almidón de maíz o derivados del mismo, talco, ácido esteárico o sus sales, etc., por ejemplo, como dichos adyuvantes para comprimidos, grageas y cápsulas de gelatina dura.
Los adyuvantes adecuados para cápsulas de gelatina blanda son, por ejemplo, aceites vegetales, ceras, grasas, sustancias semisólidas y polioles líquidos, etc.
Los adyuvantes adecuados para la producción de soluciones y jarabes son, por ejemplo, agua, polioles, sacarosa, azúcar invertido, glucosa, etc.
Los adyuvantes adecuados para soluciones inyectables son, por ejemplo, agua, alcoholes, polioles, glicerol, aceites vegetales, etc.
Los adyuvantes adecuados para supositorios son, por ejemplo, aceites naturales o hidrogenados, ceras, grasas, polioles semisólidos o líquidos, etc.
Además, las preparaciones farmacéuticas pueden contener conservantes, solubilizantes, sustancias que incrementan la viscosidad, estabilizantes, agentes humectantes, emulsionantes, edulcorantes, colorantes, saborizantes, sales para variar la presión osmótica, tampones, agentes de enmascaramiento o antioxidantes. También pueden contener todavía otras sustancias terapéuticamente valiosas. La dosificación puede variar en límites amplios y, por supuesto, se ajustará a los requisitos individuales en cada caso particular. En general, en el caso de administración oral, debería ser apropiada una dosificación diaria de aproximadamente 0,1 mg a 20 mg por kg de peso corporal, preferentemente de aproximadamente 0,5 mg a 4 mg por kg de peso corporal (por ejemplo, aproximadamente 300 mg por persona), dividida en preferentemente 1-3 dosis individuales, que pueden consistir, por ejemplo, en las mismas cantidades. Quedará claro, sin embargo, que el límite superior dado en el presente documento se puede exceder cuando se demuestre que está indicado.
De acuerdo con la invención, los compuestos de fórmula (Ic) o sus sales y ésteres farmacéuticamente aceptables se pueden usar para el tratamiento o la profilaxis de complicaciones microvasculares relacionadas con la diabetes de tipo 2 (tales como, pero sin limitarse a retinopatía diabética, neuropatía diabética y nefropatía diabética), arteriopatía coronaria, obesidad y enfermedades inflamatorias subyacentes, enfermedades inflamatorias crónicas y autoinmunitarias/inflamatorias.
Ejemplos
La invención se entenderá más completamente por referencia a los siguientes ejemplos. Sin embargo, las reivindicaciones no se deben interpretar como limitadas al alcance de los ejemplos.
En caso de que los ejemplos preparativos se obtengan como una mezcla de enantiómeros, los enantiómeros puros se pueden separar por procedimientos descritos en el presente documento o por procedimientos conocidos para el experto en la técnica, tales como, por ejemplo, cromatografía quiral (por ejemplo, SFC quiral) o cristalización.
Todos los ejemplos de reacción y los intermedios se prepararon en atmósfera de argón si no se especifica de otro modo.
Procedimiento A1
Ejemplo 1
mc-(4aR,8aS)-6-(4-benzhidnlpipendina-1-carbonil)-4,4a,5,7,8,8a-hexahidmpirido[4,3-b][1,4]oxadn-3-ona
A una solución de 4-benzhidrilpiperidina-1-carboxilato de 4-nitrofenilo (40 mg, 96 |jmol, BB1) en DMF (1 ml), se le añadió TEA (19,4 mg, 26,8 jl, 192 jmol), rac-cis-hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona (15 mg, 96 jmol, ChemBridge Corporation) y se calentó la mezcla de reacción resultante a 80 °C durante 18 horas dando como resultado la finalización de la reacción. Se diluyó la mezcla de reacción con H2O y NaHCO3 y se extrajo tres veces con EtOAc. Se lavaron las capas orgánicas combinadas con salmuera, se secó sobre Na2SO4, se filtró y evaporó. Se purificó el producto por HPLC preparativa (columna Gemini NX) usando un gradiente de ACN: H2O (que contiene FA al 0,1 %) (de 20: 80 a 98: 2) para proporcionar el compuesto deseado como un sólido incoloro (0,015 g; 36,5 %). EM (ESI): m/z = 434,2 [M+H]+.
Procedimiento A2
Ejemplo 2
rac-(4aR,8aS)-6-(4-(5-doro-1-(ddopropilmetil)-1H-indol-3-il)piperidina-1-carbonil)hexahidro-2H-pirido[4,3- b][1,4]oxacin-3(4H)-ona
A una suspensión helada de carbonato de bis(tridorometilo) (45,3 mg, 153 |jmol, CAS RN 32315-10-9) y NaHCO3 (73,3 mg, 873 jmol) en DCM (1 ml) se le añadió acetato y formiato de 5-cloro-1-(ciclopropilmetil)-3-(piperidin-4-il)-1 H-indol (76,1 mg, 218 jmol, BB2) en una porción y se agitó la mezcla a t. a. durante la noche. Después de enfriar en un baño de hielo se añadieron diclorhidrato de rac-cis-hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona (50 mg, 218 jmol, ChemBridge Corporation) y DIPEA (112 mg, 152 jl, 870 |jmol). Se agitó la suspensión a t. a. durante 6 horas. Se vertió la mezcla de reacción sobre H2O y DCM y se separaron las capas. Se extrajo la capa acuosa dos veces con DCM. Se secaron las capas orgánicas sobre MgSO4, se filtró, se trató con gel de sílice y se evaporó. Se purificó el compuesto por cromatografía en gel de sílice en una columna de 4 g usando un sistema MPLC que se eluyó con un gradiente de DCM: MeOH (de 100: 0 a 90: 10) para obtener el compuesto deseado en forma de goma incolora (0,070 g; 68,1 %). EM (ESI): m/z = 471,2 [M+H]+.
Procedimiento A3
Ejemplo 22
(+)- o (-)-ds-6-(4-(5-dom-1-(oxetan-3-il)-1H-indol-3-il)piperidina-1-carbonil)hexahidm-2H-pirído [4,3-b][1,4]oxacin-3(4H)-ona
A una solución helada de carbonato de bis(triclorometilo) (26,6 mg, 89,6 jmol) en DCM (1 ml) se le añadieron NaHCO3 (32,3 mg, 384 jmol) y rac-cis-hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona (20 mg, 128 jmol, ChemBridge Corporation) y se agitó la mezcla a t. a. durante la noche. Se enfrió la suspensión a 0 °C antes de añadir 5-cloro-1 -(oxetan-3-il)-3-(piperidin-4-il)-1 H-indol (37,2 mg, 128 jmol, BB10) y DIPEA (49,7 mg, 67,1 jl, 384 jmol). Se agitó la mezcla a t. a. durante 1 hora. Se vertió la mezcla de reacción sobre agua y DCM y se separaron las capas. Se extrajo la capa acuosa dos veces con DCM. Se secaron las capas orgánicas sobre MgSO4, se filtró, se trató con gel de sílice y se evaporó. Se purificó el compuesto por cromatografía en gel de sílice en una columna de 4 g usando un sistema MPLC que se eluyó con un gradiente de DCM: MeOH (de 100: 0 a 90: 10) para obtener el compuesto deseado como un sólido incoloro (0,025 g; 41,3 %). EM (ESI): m/z = 473,2 [M+H]+.
Procedimiento A4
Ejemplo 67
(4aR,8aS)-6-(4-((3-ddopmpil-1,2,-4-oxadiazol-5-iQ(4-fíuorofenil)metil)pipendina-1-carbonil)hexahidm-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona
A una suspensión de (4aR,8aS)-3-oxohexahidro-2H-pirido[4,3-b][1,4]oxacina-6(5H)-carboxilato de 4-nitrofenilo (50 mg, 156 jmol, BB15a) y DIPEA (50,3 mg, 68 jl, 389 jmol) en ACN (0,7 ml) se le añadió una solución de clorhidrato de 3-ciclopropil-5-((4-fluorofenil)(piperidin-4-il)metil)-1,2,4-oxadiazol (55,2 mg, 163 jmol, BB39) en AC (0,7 ml) y se agitó la solución a t. a. durante la noche. Se agitó la solución amarillo claro a reflujo durante 23 h y a continuación se evaporó. Se disolvió el residuo en solución de Na2CO3 acuosa 2 M y EtOAc y se separaron las capas. Se extrajo dos veces la capa acuosa con EtOAc. Se lavaron una vez las capas orgánicas con agua, se secó sobre MgSO4, se filtró, se trató con gel de sílice y se evaporó. Se purificó el compuesto por cromatografía en gel de sílice en una columna de 4 g usando un sistema MPLC que se eluyó con un gradiente de n-heptano: EtOAc/EtOH 3/1 (de 70: 30 a 90: 10) para obtener el compuesto deseado como un sólido marrón claro (0,022 g; 29,2 %). EM (ESI): m/z = 484,2 [M+H]+.
Procedimiento A5
Ejemplo 111
(4aR,8aS)-6-(3-(4'-dom-[1,1'-bifenil]-4-il)acetidina-1-carbonil)hexahidm-2H-pindo[4,3-b][1,4]oxadn-3(4H)-ona
Se agitó una suspensión de ácido (4-clorofenil)borónico (15,9 mg, 101 jmol, CAS RN 1679-18-1), (4aR,8aS)-6-(3-(4-bromofenil)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona (40 mg, 101 jmol, ejemplo 110), carbonato de potasio (70,1 mg, 507 jmol), agua (100 jl) y tetraquis(trifenilfosfina)paladio(0) (5,86 mg, 5,07 jmol, CAS RN 14221-01-3) en THF (1 ml) a 80 °C durante 3 h. Se vertió la mezcla de reacción sobre agua y EtOAc y se separaron las capas. Se extrajo dos veces la capa acuosa con EtOAc. Se secaron las capas orgánicas sobre MgSO4, se filtró, se trató con gel de sílice y se evaporó. Se purificó el compuesto por cromatografía en gel de sílice en una columna de 4 g usando un sistema MPLC que se eluyó con un gradiente de n-heptano: EtOAc/EtOH (3/1 v/v) (de 70: 30 a 10: 90) para proporcionar el compuesto deseado como un sólido incoloro (0,020 g; 46,3%). EM (ESI): m/z = 426,16 [M+H]+.
Procedimiento A6
Ejemplo 128
(4aR,8aS)-6-(3-(4-(3-metoxiacetidin-1-il)fenil)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxadn-3(4H)-ona
A una solución de (4aR,8aS)-6-(3-(4-bromofenil)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona (50 mg, 127 |jmol, ejemplo 110) en terc-butanol (1 ml) bajo argón se le añadieron XPhos (5,44 mg, 11,4 |jmol, CAS RN 564483-18-7), aducto de tris(dibencilidenacetona)dipaladio(0)-cloroformo (3,94 mg, 3,8 jmol, CAS RN 52522-40-4), clorhidrato de 3-metoxiacetidina (23,5 mg, 190 jmol, CAS RN 148644-09-1) y carbonato de cesio (165 mg, 507 jmol) y se calentó la mezcla hasta 85 °C durante 21 h. Se filtró la mezcla y se evaporó el filtrado. Se purificó el producto en una HPLC preparativa (columna Gemini NX) usando un gradiente de ACN:agua (que contenía ácido fórmico al 0,1 %) (de 20:80 a 98: 2) para obtener el compuesto deseado como un sólido incoloro (0,006 g; 11,8 %). EM (ESI): m/z = 401,2 [M+H]+. De forma alternativa, también se puede llevar a cabo la reacción en un horno de microondas a 100 °C.
Procedimiento A7
Ejemplo 225
(4aR,8aS)-6-[3-[4-(oxetan-3-il)fenil]acetidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido]4,3-b][1,4]oxacin-3-ona
A un vial de 5 ml equipado con una barra agitadora se le añadió (Ir[dF(CF3)ppy]2(dtbpy))PF6 (1,42 mg, 1,27 jmol, CAS RN 870987-63-6), 3-bromooxetano (17,4 mg, 127 jmol, CAS RN 39267-79-3), (4aR,8aS)-6-(3-(4-bromofenil)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona (50 mg, 127 jmol, ejemplo 110), tris(trimetilsilil)silano (31,5 mg, 39,1 jl, 127 jmol, Ca s Rn 1873-77-4) y carbonato de sodio anhidro (26,9 mg, 254 jmol). Se selló el vial y se colocó bajo argón antes de añadir DME (1 ml). A un vial separado se le añadió complejo de cloruro de níquel(II)-éter dimetílico de etilenglicol (2,79 mg, 12,7 jmol, CAS RN 29046-78-4) y 4,4'-diterc-butil-2,2'-bipiridina (3,4 mg, 12,7 jmol, CAS AN 72914-19-3). Se selló el vial con precatalizador, se purgó con argón y a continuación se añadió DME (0,4 ml). Se sometió a ultrasonidos el vial con precatalizador durante 5 min, después de esto, se inyectaron 0,4 ml (0,5 % moles de catalizador, 0,01 eq) de este en el recipiente de reacción. Se desgasificó la mezcla de reacción con argón. Se agitó la reacción y se irradió con una lámpara de 420 nm durante 4 h. Se detuvo la reacción por exposición al aire, se filtró y se lavó con un pequeño volumen de EtOAc. Se trató el filtrado con gel de sílice y se evaporó. Se purificó el compuesto por cromatografía en gel de sílice en una columna de 4 g usando un sistema MPLC (ISCO) que se eluyó con un gradiente de n-heptano: EtOAc/EtOH 3/1 (v/v) (de 70: 30 a 10: 90) para proporcionar el compuesto deseado como un sólido amarillo claro (0,010 g; 21,2 %). EM (ESI): m/z = 372,3 [M+H]+.
Procedimiento A8
Ejemplo 159
2-[4-[1-[(4aR,8aS)-3-oxo-4,4a,5,7,8,8a-hexahidropindo[4,3-b][1,4]oxadna-6-carbonilo]acetidin-3-il]fenil]-5-dorobenzonitrilo
A una solución de ácido [4-[1-[(4aR,8aS)-3-oxo-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacina-6-carbonil]acetidin-3-il]fenil]borónico (100,0 mg, 0,280 mmol, BB38) y 2-bromo-5-clorobenzonitrilo (120,5 mg, 0,560 mmol, CAS RN 57381-37-0) y CsF (127 mg, 0,840 mmol) en Dm F (5 ml) se le añadió Pd(dppf)Cl2 (40,8 mg, 0,060 mmol), se agitó la mezcla a 90 °C en atmósfera de N2 durante 12 h. Se filtró la mezcla y se diluyó el filtrado con agua (20 ml) y se extrajo con EtOAc (10 ml tres veces), se lavó la fase orgánica combinada con salmuera, se secó sobre Na2SO4 y se concentró, se purificó el residuo por HPLC prep. (FA) y se liofilizó para obtener el compuesto deseado como sólido blanco (38,8 mg, 30,7 %). EM (ESI): m/z = 451,1 [M+H]+.
Procedimiento A9
Ejemplos 75 y 76
(4aR, 8aS)-6-(3-(R o S)-( 1-(2-cloro-4-(trifluorometil)fenoxi)etil)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona y
(4aR, 8aS)-6-(3-(S o R)-( 1-(2-cloro-4-(trifluorometil)fenoxi)etil)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona
A una solución de 2,2,2-trifluoroacetato de 3-(1-(2-cloro-4-(trifluorometil)fenoxi)etil)acetidina (BB94) (133,5 mg, 207 jmol) en una mezcla de CH3CN (517 jl) y 2-propanol (517 jl) se le añadió DIPeA (66,8 mg, 90,3 jl, 517 jmol) y (4aR,8aS)-3-oxohexahidro-2H-pirido[4,3-b][1,4]oxacina-6(5H)-carboxilato de 4-nitrofenilo (66,5 mg, 207 jmol, BB15a). Se agitó el vial de reacción a 90 °C durante 21 h. Se añadieron una porción adicional de (4aR,8aS)-3-oxohexahidro-2H-pirido[4,3-b][1,4]oxacina-6(5H)-carboxilato de 4-nitrofenilo (19,9 mg, 62,1 jmol, BB15a) y DIPEA
(8,02 mg, 10,8 jl, 62,1 |jmol) y se calentó la mezcla hasta 95 °C durante 23 h. Se sometió el material bruto a purificación por HPLc aquiral de fase inversa para proporcionar (4aR,8aS)-6-(3-(1-(2-cloro-4-(trifluorometil)fenoxi)etil)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona (88 mg, 92,1 %) como un sólido blanco. Se separaron los epímeros por HPLC quiral para proporcionar el ejemplo 75 (33,6 mg) y el ejemplo 76 (43,2 mg) como sólidos amarillos. EM (ESI): m/z = 462,2 [M+H]+ para ambos compuestos.
Procedimiento A10
Ejemplos 91, 96 y 97
(4aR,8aS)-6-(3-(1-(2-fíuom-4-(tnfíuorometil)fenoxi)etiQacetidina-1-carboniQhexahidro-2H-pindo[4,3-b][1,4]oxadn-3(4H)-ona y
(4aR, 8aS)-6-(3-((R o S)-1-(2-fluoro-4-(trifluorometil)fenoxi)etil)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona y
(4aR,8aS)-6-(3-((S o R)-1-(2-fluoro-4-(trifluorometil)fenoxi)etil)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona
A una solución de 2,2,2-trifluoroacetato de 3-(1-(2-fluoro-4-(trifluorometil)fenoxi)etil)acetidina, BB97 (51,4 mg, 136 |jmol) en CH3CN (681 |jl) se le añadió DIPEA (52,8 mg, 71,4 jil, 409 |jmol) y (4aR,8aS)-3-oxohexahidro-2H-pirido[4,3-b][1,4]oxacina-6(5H)-carboxilato de 4-nitrofenilo (43,8 mg, 136 jimol, BB15a). Se agitó el vial de reacción a 95 °C durante 3 h. Se sometió el material bruto a purificación por HPLC de fase inversa para proporcionar (4aR,8aS)-6-(3-(1-(2-fluoro-4-(trifluorometil)fenoxi)etil)acetidina-1-carbonil)hexahidro-2H-pi rido[4,3-b][1,4]oxacin-3(4H)-ona (ejemplo 91; 31 mg, 51 %) como un sólido blanquecino. EM (ESl): m/z = 446,3 [M+H]+. Se sometió este producto racémico a separación por SFC quiral para proporcionar (4aR,8aS)-6-(3-((R o S)-1-(2-fluoro-4-(trifluorometil)fenoxi)etil)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona (15,9 mg, ejemplo 96) y (4aR,8aS)-6-(3-((S o R)-1-(2-fluoro-4-(trifluorometil)fenoxi)etil)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona (15,2 mg, ejemplo 97) como sólidos blancos. EM (ESl): m/z = 446,2 [M+H]+ para ambos compuestos.
Procedimiento A11
Ejemplo 55
(4aR,8aS)-6-(3-(fíuombis(4-fíuorofenil)metil)acetidina-1-carbonil)hexahidm-2H-pirido[4,3-b][1,4]oxadn-3(4H)-ona
A una solución helada de trihidrofluoruro de trietilamina (31,8 mg, 32,1 jil, 197 jimol) en DCM (493 jil) bajo argón se le añadió tetrafluoroborato de (dietilamino)difluorosulfonio (33,9 mg, 148 jimol, CAS RN 63517-29-3) seguido de (4aR,8aS)-6-(3-(bis(4-fluorofenil)(hidroxi)metil)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona (ejemplo 54) (45,1 mg, 98,6 jimol) en DCM (493 jil), y se agitó la mezcla durante 7o min a 0 °C y a continuación a t. a. durante 25 min. Se detuvo la reacción a 0 °C con solución de NaHCO3 acuosa saturada y se almacenó en el frigorífico durante la noche. Se separaron las fases y se extrajo dos veces la fase acuosa con DCM. Se lavaron las capas orgánicas combinadas con salmuera, se secó sobre Na2SO4 y se concentró para dar un aceite pálido. Se sometió el material bruto a purificación por HPLC de fase inversa para proporcionar el compuesto deseado como un sólido blanco (20 mg, 41,5 %). EM (ESl): m/z = 504,18 [M+HCOO]-.
Procedimiento A12
Ejemplo 224
(4aR,8aS)-6-(3-(3-dom-4-(3-metilacetidin-1-il)fenil)acetidina-1-carbonil)hexahidm-2H-pirido[4,3-b][1,4]oxadn-3(4H)-ona
A una solución de (4aR,8aS)-6-(3-(4-bromo-3-clorofenil)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona (ejemplo 132, 0,020 g, 0,047 mmol) en dioxano (0,5 ml) se le añadió Pd2(dba)3 (0,004 g, 0,0047 mmol), Xantphos (0,003 g, 0,0047 mmol) y carbonato de cesio (0,030 g, 0,093 mmol). Se desgasificó la mezcla con argón, a continuación se añadió clorhidrato de 3-metilacetidina (CAS RN 1375472-05-1,0,010 g, 0,093 mmol) y se calentó la mezcla de reacción hasta 100 °C durante 16 h. Después de este tiempo, se añadieron clorhidrato de 3-metilacetidina (0,005 g, 0,047 mmol), Pd2(dba)3 (0,004 g, 0,0047 mmol), Xantphos (0,003 g, 0,0047 mmol) y carbonato de cesio (0,015 g, 0,047 mmol) y se calentó la mezcla de reacción a 100 °C durante otras 2 h. Se diluyó la mezcla con EtOAc y se lavó con H2O y salmuera. Se secó la fase orgánica sobre Na2SO4, se filtró y se concentró a vacío. Se purificó el residuo por HPLC prep. para dar el compuesto del título (0,007 g, 36 %) como un sólido amarillo. EM (ESl): m/z = 419,3 [M+H]+.
Procedimiento B1
Ejemplo 5
(+)-ds-6-]4-(6-fluoro-1H-indol-3-il)piperidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido]4,3-b][1,4]oxadn-3-ona Se separó el racemato (producto del ejemplo 7) en HPLC quiral preparativa (columna Reprosil Chiral NR) usando una mezcla isocrática de EtOH (que contenía un 0,05 % de NH4OAc):n-heptano (60:40). Se evaporaron las fracciones para obtener el compuesto deseado como un sólido amarillo claro (0,009 mg). EM (ESI): m/z = 401,2 [M+H]+.
Procedimiento B2
Ejemplo 11
(+)-ds-6-(4-(bis(4-fluorofenil)metil)piperadna-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxadn-3(4H)-ona Se separaron los enantiómeros del ejemplo 12 en HPLC quiral preparativa (columna Chiralcel OD) usando una mezcla isocrática de EtOH (que contenía un 0,05 % de NH4OAC):n-heptano (80: 20). Se evaporaron las fracciones para obtener el compuesto deseado como un sólido incoloro (0,014 g). EM (ESI): m/z = 471,3 [M+H]+.
Procedimiento B3
Ejemplos 31 y 32
(+)-cis-6-(4-((R o S)-1-(4-fíuorofenil)-3-hidroxipropiQpipendina-1-carboniQhexahidm-2H-pindo[4,3-b][1,4]oxadn-3(4H)-ona y
(-)-cis-6-(4-((S o R)-1-(4-fíuorofenil)-3-hidroxipropil)pipendina-1-carboniQhexahidm-2H-pindo[4,3-b][1,4]oxadn-3(4H)-ona
Se separaron los enantiómeros del ejemplo racémico 26 por HPLC quiral preparativa (columna Chiralpak AD) usando una mezcla isocrática de EtOH (que contenía un 0,05 % de NH4OAC):n-heptano (40: 60). Se evaporaron las fracciones. Se recogió el residuo en EtOH, DCM y n-heptano y se evaporó de nuevo para retirar el exceso de AcOH. Se purificaron los compuestos por cromatografía en gel de sílice en una columna de 4 g usando un sistema MPLC (ISCO) que se eluyó con un gradiente de DCM: MeOH (de 100: 0 a 80: 20) para proporcionar los productos deseados como gomas incoloras (0,004 g; 7,7 % y 0,010 g, 19 %). MS (ESI): m/z = 420,2 [M+H]+ para cada estereoisómero.
Procedimiento B4
Ejemplo 94
(4aR,8aS)-6-(4-((R o S)-(3-ddopropil-1,2,4-oxadiazol-5-il)(4-fíuorofenil)metiQpiperidina-1-carbonil)hexahidm-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona
El racemato del ejemplo 68 se separó por SFC quiral preparativa (columna IA, 12 nm, 5 |jm, 250 x 4,6 mm) usando una mezcla isocrática de MeOH:dióxido de carbono (25: 75). Se evaporaron las fracciones que contenían el para obtener el producto deseado como un sólido marrón claro (0,016 g, 40,0 %). EM (ESI): m/z = 484,2 [M+H]+. Procedimiento B5
Ejemplos 114 y 115
(4aR,8aS)-6-[3-[(1R)-1-[4-(trifluorometil)fenil]etoxi]acetidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona y (4aR,8aS)-6-[3-[(1S)-[4-(trifluorometil)fenil]etoxi]acetidina-1-carbonil]-4,4a,5,7,8,8ahexahidropirido[4,3-b][1,4]oxacin-3-ona
Se separaron los enantiómeros del ejemplo 90 por SFC quiral preparativa (columna OZ-H, 220 nm, 250 x 10 mm) usando una mezcla isocrática de MeOH:dióxido de carbono (20: 80). Se evaporaron las fracciones para dar los productos del título como sólidos amorfos incoloros (0,005 g). EM (ESI): m/z = 428,4 [M+H]+ para ambos enantiómeros.
Ejemplo 25
(+)- o (-)-ds-6-(4-(5-doro-1-(2-hidroxietil)-1H-indol-3-il)piperidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxadn-3(4H)-ona
A una solución de (+)- o (-)-cis-6-(4-(1-(2-((terc-butildimetilsilil)oxi)etil)-5-cloro-1H-indol-3-il)piperidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona (39 mg, 67,8 |jmol, ejemplo 24) en DCM (0,5 ml) se le añadió TFA (77,3 mg, 52,2 jl, 678 jmol) y se agitó la solución a t. a. durante 1,5 horas. A la solución amarillo claro se le añadieron THF (0,1 ml) y H2O (0,1 ml) y se continuó agitando a t. a. durante 96 horas. Se evaporó la mezcla de reacción. Se vertió el residuo en solución de NaHCO3 acuosa saturada y DCM y se separaron las capas. Se extrajo la capa acuosa dos veces con DCM. Se secaron las capas orgánicas sobre MgSO4, se filtró, se trató con gel de sílice y se evaporó. Se purificó el compuesto por cromatografía en gel de sílice en una columna de 4 g usando un sistema MPLC que se eluyó con un gradiente de DCM: MeOH (de 100: 0 a 90: 10) para proporcionar el compuesto deseado como un sólido incoloro (0,014 g; 44,8 %). EM (ESI): m/z = 461,2 [M+H]+.
Ejemplo 173
(4aR,8aS)-6-(3-(4'-dom-2'-(metilsulfonil)-[1,1'-bifenil]-4-il)acetidina-1-carbonil)hexahidm-2H-pirído[4,3-b][1,4]oxacin-3(4H)-ona
Se trató una solución de (4aR,8aS)-6-(3-(4'-cloro-2-fluoro-[1,1-bifenil]-4-il)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona (27 mg, 60,8 jmol, ejemplo 133) en DMSO (0,5 ml) con metanotiolato de sodio (4,26 mg, 60,8 jmol) y se agitó a 100 °C durante la noche. Se añadió otro lote de metanotiolato de sodio (4,26 mg, 60,8 jmol) y se continuó agitando a 100 °C durante 2 h. Después de enfriar, se vertió la mezcla sobre agua y se separaron las capas. Se extrajeron dos veces las capas orgánicas con EtOAc. Se secaron las capas orgánicas sobre MgSO4, se filtró y evaporó. Se disolvió el intermedio bruto obtenido (112 mg, aceite amarillo claro) en DCM (0,7 ml) y se añadió ácido m-cloroperbenzoico (98,2 mg, 195 jmol) a t. a. Se agitó la mezcla a t. a. y se continuó agitando a t. a. durante 1 h. Se vertió la mezcla de reacción en solución de NaHCO3 acuosa saturada y DCM y se separaron las capas. Se extrajo la capa acuosa dos veces con DCM. Se secaron las capas orgánicas sobre MgSO4, se filtró, se trató con gel de sílice y se evaporó. Se purificó el compuesto por cromatografía en gel de sílice en una columna de 4 g usando un sistema MPLC que se eluyó con un gradiente de DCM: MeOH (de 100: 0 a 90: 10) para obtener el sulfóxido intermedio deseado como una goma incolora (0,014 g). Se disolvió en DCM (0,7 ml) y se añadió ácido m-cloroperbenzoico (43,6 mg, 195 jmol). Se continuó agitando a t. a. durante 3 h. Se vertió la mezcla de reacción en solución de NaHCO3 acuosa saturada y DCM y se separaron las capas. Se extrajo la capa acuosa dos veces con DCM. Se secaron las capas orgánicas sobre MgSO4, se filtró, se trató con gel de sílice y se evaporó. Se purificó el compuesto por cromatografía en gel de sílice en una columna de 4 g usando un sistema MPLC que se eluyó con un gradiente de DCM: MeOH (de 100: 0 a 90: 10) para obtener el compuesto deseado como un sólido incoloro (0,004 g; 13,0%). EM (ESI): m/z = 504,1 [M+H]+.
Ejemplo 193
(4aR,8aS)-6-(3-(5-(2,4-didomfenil)pindin-2-il)acetidina-1-carbonil)hexahidm-2H-pirído[4,3-b][1,4]oxadn-3(4H)-ona
A una solución de (4aR,8aS)-6-(3-(5-cloropiridin-2-il)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona (63 mg, 180 jmol, ejemplo 181) en dioxano (0,5 ml) bajo argón se le añadieron triciclohexilfosfina (4,03 mg, 14,4 jmol, cAs RN 2622-14-2), aducto de tris(dibencilidenacetona)dipaladio(0)-cloroformo (7,44 mg, 7,18 jmol), ácido (2,4-diclorofenil)borónico (36 mg, 189 jmol, CAS RN 68716-47-2) y CS2CO3 (70,2 mg, 216 jmol) y se agitó la mezcla a 100 °C durante la noche. Se vertió la mezcla de reacción sobre agua y EtOAc y se separaron las capas. Se extrajo dos veces la capa acuosa con EtOAc. Se secaron las capas orgánicas combinadas sobre MgSO4, se filtró y evaporó. Se purificó el producto por HPLC preparativa (columna Gemini NX) usando un gradiente de ACN:agua (que contenía TEA al 0,1 %) (de 20:80 a 98: 2) para proporcionar el compuesto deseado como un sólido marrón claro (0,0038 g; 4,6 %). EM (ESI): m/z = 461,1 [M+H]+.
Ejemplo 218
(4aR,8aS)-6-[4-[fenil(piridacin-3-il)metil]piperidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona
Se agitó una solución de 3-[fenil(4-piperidil)metil]piridacina (70 mg, 0,28 mmol, BB86) y (4aR,8aS)-3-oxo-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacina-6-carboxilato de (4-nitrofenilo) (intermedio BB15a) (106 mg, 0,330 mmol) en piridina (3 ml) a 80 °C durante 16 h. Se filtró la solución y se concentró a vacío para dar un residuo, que se purificó por HPLC prep. (condiciones básicas) para proporcionar (4a
R
,8a
S
)-6-[4-[fenil(piridacin-3-il)metil]piperidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona (5,9 mg, 5 %) como un sólido gris. EM (ESI): m/z = 436,2 [M+H]+.
Ejemplo 247
(4aR,8aS)-6-[3-[4-(3-fluoropropil)fenil]acetidina-1-carbonil]-4,4a,5, 7, 8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona
A una solución agitada de 3-[4-(3-fluoropropil)fenil]acetidina (40 mg, 0,21 mmol, BB87) y DIPEA (0,07 ml, 0,41 mmol) en MeCN (0,9 ml) a 20 °C se le añadió (4aR,8aS)-3-oxo-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacina-6-carboxilato de (4-nitrofenilo) (66 mg, 0,210 mmol; BB15a). Se agitó la mezcla resultante a 80 °C durante 16 h. Se concentró la solución a vacío para dar un residuo, que se purificó por HPLC prep. (condición de TFA) para dar (4aR,8aS)-6-[3-[4-(3-fluoropropil)fenil]acetidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona (16 mg, 17 %, 84 % de pureza) como sólido blanco. Se combinó este material con el producto obtenido a partir de una segunda reacción a escala similar (0,23 mmol de 3-[4-(3-fluoropropil)fenil]acetidina), y se purificó por segunda vez por HPLC prep. (condiciones de TFA) para dar el compuesto del título como un sólido blanco. EM (ESI): m/z = 376,3 [M+H]+.
Ejemplo 252
(4aR,8aS)-6-[4-[[3-(2-aminoetoxi)fenil]-fenil-metil]piperidina-1-carbonil]-4,4a,5, 7, 8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona
A una solución de (2-(3-((1-((4aR,8aS)-3-oxooctahidro-2H-pirido[4,3-b][1,4]oxacina-6-carbonil)piperidin-4-il)(fenil)metil)fenoxi)etil)carbamato de terc-butilo (ejemplo 237; 178 mg, 300 |jmol) en DCM (2,5 ml) se le añadió TFA (274 mg, 185 jl, 2,4 mmol) y se agitó la mezcla de reacción a temperatura ambiente durante 3 horas. Se añadieron EtOAc y solución de KHCO3 acuosa. Se separaron las capas y se extrajo la fase acuosa con EtOAc. Se secaron las capas orgánicas combinadas sobre Na2SO4. Se retiró el disolvente a presión reducida para dar el compuesto del título (148 mg, cuant.) como una espuma amarillo claro. EM (ESI): m/z = 493,3 [M+H]+.
Ejemplo 255
N-[2-[3-[2-[3-[[1-[(4aR,8aS)-3-oxo-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacina-6-carbonil]-4-piperidil]-fenilmetil]fenoxi]etilamino]-3-oxo-propoxi]etil]carbamato de terc-butilo
Se añadió DIPEA (52,5 mg, 70,9 jl, 406 jmol) a una mezcla de (4aR,8aS)-6-(4-((3-(2-aminoetoxi)fenil)(fenil)metil)piperidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona (ejemplo 252; 50 mg, 101 jmol), ácido 3-(2-t-boc-aminoetoxi)propanoico (CAS RN 1260092-44-1; 23,7 mg, 101 jmol) y HATU (42,5 mg, 112 jmol) en DMF (203 jl). Se agitó la mezcla a temperatura ambiente durante 18 h. Se añadieron acetato de etilo y solución de KHCO3 acuosa. Se separaron las capas y se lavó dos veces la fase orgánica con agua. Se secó la capa orgánica sobre Na2SO4. Se retiró el disolvente a presión reducida para dar el compuesto del título (71 mg, cuant.) como aceite incoloro, EM (ESI): m/z = 708,4 [M+H]+.
Ejemplo 256
N-[2-[2-[3-[2-[3-[[1-[(4aR,8aS)-3-oxo-4,4a,5, 7, 8,8a-hexahidropirido[4,3-b][1,4]oxacina-6-carbonil]-4-piperidil]-fenilmetil]fenoxi]etilamino]-3-oxo-propoxi]etoxi]etil]carbamato de terc-butilo
Se añadió DIPEA (52,5 mg, 70,9 jl, 406 jmol) a una mezcla de (4aR,8aS)-6-(4-((3-(2-aminoetoxi)fenil)(fenil)metil)piperidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona (ejemplo 252; 50 mg, 101 jmol), ácido 13,13-dimetil-11-oxo-4-7,12-tioxa-10-azatetradecanoico (CAS RN 1365655-91-9; 28,1 mg, 101 jmol) y HATU (42,5 mg, 112 jmol) en DMF (135 jl). Se agitó la mezcla a temperatura ambiente durante 18 h. Se añadieron acetato de etilo y solución de KHCO3 acuosa. Se separaron las capas y se lavó dos veces la fase orgánica con agua. Se secó la capa orgánica sobre Na2SO4. Se retiró el disolvente a presión reducida para dar el compuesto del título (76 mg, cuant.) como aceite incoloro, EM (ESI): m/z = 774,4 [M+Na]+.
Ejemplo 258
(4aR, 8aS)-6-[3-(4-propoxifenil)acetidina-1-carbonil]-4,4a, 5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona
A una suspensión de (4aR,8aS)-6-(3-(4-hidroxifenil)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona (0,032 g, 96,6 jmol, BB145) y carbonato de potasio (16 mg, 116 jmol) en DMF (0,4 ml), se le añadió 1-yodopropano (19,7 mg, 11,3 jl, 116 jmol) y se agitó la mezcla a 50 °C en un tubo sellado durante la noche. Se filtró la mezcla y se lavó la torta del filtro con unas pocas gotas de DMF. Se purificó el producto en una HPLC preparativa (columna YMC Triart C18) usando un gradiente de ACN:agua (que contenía TEA al 0,1 %) (de 20:80 a 100: 0) para proporcionar el compuesto deseado como un sólido incoloro (0,013 g; 36,0 %). EM (ESI): m/z = 374,3 [M+H]+.
Ejemplo 260
2-[4-[1-[(4aR,8aS)-3-oxo-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacina-6-carbonil]acetidin-3-il]fenil]-N-etil-5-metil-benzamida
A una solución de (4aR,8aS)-6-[3-[4-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)fenil]acetidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona (100,0 mg, 0,230 mmol, BB91), Na2CO3 (48,03 mg, 0,450 mmol) y 2-bromo-N-etil-5-metil-benzamida (54,86 mg, 0,230 mmol, BB92) en 1,4-dioxano (4 ml) y agua (1 ml) se le añadió Pd(dppf)Cl2 (16,58 mg, 0,020 mmol) y se agitó la mezcla a 100 °C en atmósfera de N2. Después de agitar a esta temperatura durante 12 h, se vertió la mezcla en agua (20 ml) y se extrajo tres veces con EtOAc (10 ml). Se lavaron las capas orgánicas combinadas con salmuera y se secó sobre Na2SO4, se concentró, se purificó el residuo por cromatografía ultrarrápida inversa (FA al 0,1 % v/v) seguido de HPLC prep. (FA al 0,225 % v/v) para dar el producto deseado (13,8 mg, 12,5 %) como un sólido blanco. EM (ESI): m/z = 477,3 [M+H]+.
Si no se indica de otro modo, los siguientes ejemplos de la
tabla 2
se sintetizaron a partir del/de los bloque(s) constructivo(s) adecuado(s) de forma análoga a los procedimientos de reacción descritos en el presente documento.
Tabla 2

















Síntesis de bloques constructivos
BB1
4- Benzhidrilpiperidina-1-carboxilato de 4-nitrofenilo
A una solución de 4-benzhidrilpiperidina (50 mg, 199 |jmol, CAS RN 19841-73-7) en DCM (1,2 ml), se le añadió TEA (40,3 mg, 55,4 jl, 398 jmol). Al enfriar hasta 0 °C, se añadió carbonocloridato de 4-nitrofenilo (44,1 mg, 219 jmol, CAS RN 7693-46-1), se dejó que la mezcla de reacción se calentara hasta t. a. y se agitó durante 18 horas. Se diluyó la mezcla de reacción con DCM y posteriormente se lavó con H2O y solución de NaHCO3 acuosa sat., se lavaron las capas orgánicas combinadas con salmuera, se secó sobre Na2SO4, se filtró y se concentró a vacío. Se purificó el material bruto por cromatografía ultrarrápida (gel de sílice, 10 g, que se eluyó con EtOAc/nheptano 0-50 %), para dar el compuesto del título como un sólido amarillo claro (75 mg, 90,5 %). EM (ESI): m/z = 417,4 [M+H]+.
BB2
5- doro-1-(ddopmpilmetil)-3-(pipendin-4-il)-1H-indol (sal de FA)
Se agitó una mezcla de 4-[5-cloro-1 -(ciclopropilmetil)indol-3-il]piperidina-1-carboxilato de terc-butilo (400,0 mg, 1,03 mmol) en HCl/EtOAc (4 M, 15,0 ml) a 20 °C durante 15 horas. Se concentró la mezcla a vacío y se suspendió
el residuo en H2O (5 ml). Se ajustó el pH a pH=7 con amoníaco, a continuación se purificó por HPLC prep. (FA al 0,1
%
en H2O y ACN) para dar el compuesto del título como un sólido blanco (215,9 mg, 62,6 %). EM (ESI): m/z = 289.1 [M+H]+.
Intermedio: 4-(5-doro-1-(cidopropilmetil)-1H-indol-3-il)piperidina-1-carboxilato de terc-butilo
Se agitó una mezcla de (bromometil)ciclopropano (0,17 ml, 1,79 mmol; CAS RN 7051-34-5), 4-(5-cloro-1H-indol-3- il)piperidina-1-carboxilato de terc-butilo (500,0 mg, 1,49 mmol, BB9, etapa intermedia b), 15-corona-5 (0,06 ml, 0,300 mmol; CAS RN 33100-27-5) y terc-butóxido de sodio (287,01 mg, 2,99 mmol; CAS RN 865-48-5) en EtOH anhidro (5 ml) a 25 °C durante 12 horas. Se diluyó la mezcla con H2O (20 ml), se extrajo tres veces con EtOAc (40 ml cada vez) y se concentró a vacío. Se purificó el residuo por cromatografía en columna en gel de sílice ultrarrápida (malla 100-200) usando un gradiente de PE: EtOAc (de 50: 1 a 20: 1) para dar el compuesto del título como un sólido amarillo claro (400 mg, 68,8 %). EM (ESI): m/z = 411,1 [M+Na]+.
BB3
5-doro-1-metil-3-(piperidin-4-il)-1 H-indol
Se agitó una mezcla de 4-(5-cloro-1-metil-indol-3-il)piperidina-1-carboxilato de terc-butilo (480,0 mg, 1,38 mmol) en HCl/EtOAc (20,0 ml, 6 M) a 20 °C durante 15 horas. Se concentró la mezcla a vacío, se ajustó a pH~7 usando amoníaco acuoso y, a continuación, se concentró para dar un residuo que se purificó por HPLC prep. para dar el compuesto del título como un polvo incoloro (172,3 mg, 49,3 %). EM (e Si): m/z = 249,2 [M+H]+.
Intermedio: 4-(5-cloro-1-metil-indol-3-il)piperidina-1-carboxilato de terc-butilo
A una mezcla de NaH al 60 % en aceite de vaselina (119,46 mg, 2,99 mmol) en THF (20 ml) se le añadió una solución de 4-(5-cloro-1H-indol-3-il)piperidina-1-carboxilato de terc-butilo (500,0 mg, 1,49 mmol, BB9, etapa intermedia b) en THF (5 ml) a 0 °C. Se agitó la mezcla a 0 °C durante 0,5 horas, a continuación se añadió MeI (211,95 mg, 1,49 mmol). Se agitó la mezcla a 0 °C durante 1 hora, se vertió en H2O (5 ml) y se extrajo con EtOAc (20 ml x 3). Se lavaron las capas orgánicas combinadas con salmuera (10 ml), se secó sobre Na2SO4, se filtró y se concentró para dar el compuesto del título como sólido amarillo claro (480 mg, 92,1 %). EM (ESI): m/z = 349,1 [M-Boc+H]+.
BB4
4- (9H-fluoren-9-il)piperidina
A una solución de 4-(9H-fluoren-9-il)piridina (70 mg, 288 |jmol) en AcOH (1 ml) se le añadió óxido de platino(IV) (10 mg, 44 jmol). Se agitó la mezcla de reacción durante la noche en una atmósfera de hidrógeno de 20 bar a 40 °C. Se filtró la mezcla de reacción y se lavó el filtrado con AcOH. Se concentró la solución así obtenida a vacío. Se disolvió el residuo en DCM, y se lavó tres veces el DCM con solución de NaHCO3 saturada acuosa. Se secó la capa orgánica sobre MgSO4, se filtró y se concentró para dar el producto deseado. (0,060 g, 83 %). EM (ESI): m/z = 250,2 [M+H]+.
Intermedios:
a) 4-(9H-fluoren-9-il)piridina
A una solución de difenil(4-piridil)metanol (135 mg, 517 jmol) en FA (2 ml) se le añadió gota a gota ácido sulfúrico (0,8 ml). Se calentó la mezcla hasta 100 °C durante 15 minutos, se enfrió a t. a. y se vertió en 20 ml de solución de NaOH 5 N acuosa, lo que provocó la precipitación del producto. Se recogió el producto por filtración, para dar el producto como un sólido amarillo claro (75 mg, 60 %). EM (ESI): m/z = 244,2 [M+H]+.
b) Difenil(4-piridil)metanol
A una solución de fenil(piridin-4-il)metanona (0,52 g, 2,84 mmol, CAS RN 14548-46-0) en THF (10 ml) se le añadió gota a gota una solución 1 M de bromuro de fenilmagnesio en THF (9 ml, 9 mmol, CAS RN 100-58-3) a t. a. durante 2 horas. Se vertió la mezcla de reacción sobre solución de NH4Cl acuosa saturada y EtOAc y se separaron las capas. Se extrajo dos veces la capa acuosa con EtOAc. Se lavaron las capas orgánicas con salmuera, se secó sobre MgSO4, se filtró, se trató con gel de sílice y se evaporó. Se purificó el compuesto por cromatografía en gel de sílice en una columna de 4 g usando un sistema MPLC que se eluyó con un gradiente de DCM: MeOH (de 100: 0 a 90: 10) para proporcionar el compuesto deseado como un sólido incoloro (0,290 g; 39,0%). EM (ESI): m/z = 262.2 [M+H]+.
BB5
4-[1-(4-fluorofenil)-3-metoxi-propil]piperidina
A una solución de 4-(1-(4-fluorofenil)-3-metoxi-propil)piperidina-1-carboxilato de tere-butilo (95 mg, 270 |jmol) en dioxano (0,3 ml) se le añadió HCl 4 M en dioxano (338 jl, 1,35 mmol) y se agitó la mezcla a t. a. durante 2 h. Se evaporó por completo la solución amarillo claro. Se recogió el residuo en solución de NaHCO3 acuosa saturada y DCM y se separaron las capas. Se extrajo la capa acuosa dos veces con DCM. Se secaron las capas orgánicas sobre MgSO4, se filtró y evaporó para proporcionar el compuesto deseado como un aceite amarillo claro (0,057 g; 83,9 %). EM (ESI): m/z = 252,3 [M+H]+.
Intermedios:
a) 4-[1-(4-fluorofenil)-3-metoxi-propil]piperidina-1-earboxilato de tere-butilo
A una solución helada de 4-(1-(4-fluorofenil)-3-hidroxi-propil)piperidina-1-carboxilato de tere-butilo (95 mg, 282 jmol) en THF (1 ml) se le añadió NaH al 55 % en aceite de vaselina (13,5 mg, 310 jmol) y se agitó la mezcla a esta temperatura durante 30 minutos antes de añadir yodometano (47,9 mg, 21,1 jl, 337 jmol). Después de agitar a esta temperatura durante 30 minutos, se dejó que la solución turbia se calentara hasta t. a. Después de agitar a t. a. durante 4 horas, se vertió la mezcla de reacción sobre H2O y EtOAc y se separaron las capas. Se extrajo dos veces la capa acuosa con EtOAc. Se secaron las capas orgánicas sobre MgSO4, se filtró y se evaporó para obtener el compuesto deseado como un aceite amarillo claro (0,097 g; 98,0 %). EM (ESI): m/z = 252 [M-Boc+H]+.
b) 4-[1-(4-fluorofenil)-3-hidroxi-propil]piperidina-1-earboxilato de tere-butilo
A una solución de 4-[3-etoxi-1-(4-fluorofenil)-3-oxo-propil]piperidina-1-carboxilato de tere-butilo (148 mg, 390 jmol) en THF (1,5 ml) a 0 °C se le añadió en una porción borohidruro de litio (21,2 mg, 975 jmol) y se agitó la mezcla en un baño de hielo durante 1,25 horas. Se retiró el baño de hielo y se continuó agitando a t. a. durante 20 horas. Se vertió la mezcla de reacción sobre solución de NH4Cl acuosa saturada y EtOAc y se separaron las capas. Se extrajo dos veces la capa acuosa con EtOAc. Se secaron las capas orgánicas sobre MgSO4, se filtró, se trató con gel de sílice y se evaporó. Se purificó el compuesto por cromatografía en gel de sílice en una columna de 4 g usando un sistema MPLC que se eluyó con un gradiente de n-heptano: EtOAc (de 100: 0 a 0: 100) para proporcionar el compuesto deseado como un aceite incoloro (0,097 g; 73,7 %). EM (ESI): m/z = 282,3 [M-C4Hs+H]+.
e) 4-[3-etoxi-1-(4-fluorofenil)-3-oxo-propil]piperidina-1-earboxilato de tere-butilo
A una solución de (E) y (Z)-4-(3-etoxi-1-(4-fluorofenil)-3-oxoprop-1-en-1-il)piperidina-1-carboxilato de tere-butilo (1,74 g, 4,63 mmol) en EtOAc (17,1 ml) se le añadió Pd/C al 10 % (175 mg) y se agitó la suspensión a t. a. en atmósfera de hidrógeno a 1,5 bar durante 4 horas. Se filtró la mezcla de reacción sobre un microfiltro. Se purificó el compuesto por cromatografía en gel de sílice en una columna de 24 g usando un sistema MPLC que se eluyó con un gradiente de n-heptano: EtOAc (de 100: 0 a 50: 50) para obtener el compuesto deseado como un líquido incoloro (1,23 g; 69,8 %). EM (ESI): m/z = 280 [M-Boc+H]+.
d) 4-[(E) y (Z)-3-etoxi-1-(4-fluorofenil)-3-oxo-prop-1-enil]piperidina-1-earboxilato de tere-butilo
A una solución de 2-(dietoxifosforil)acetato de etilo (2,1 g, 1,86 ml, 9,36 mmol, cas RN 867-13-0) en 1,4-dioxano (9,73 ml) se le añadió gota a gota LiHMDS 1,0 M en hexanos (15 ml, 15 mmol) y se agitó la solución durante 15 minutos. Se añadió gota a gota a la mezcla una solución de 4-(4-fluorobenzoil)piperidina-1-carboxilato de
tere
-butilo (2,878 g, 9,36 mmol, CAS RN 160296-40-2) en 1,4-dioxano (9,73 ml) y se continuó agitando a reflujo durante dos días. Se vertió la mezcla de reacción en solución de NH4Cl acuosa saturada y EtOAc y se separaron las capas. Se extrajo dos veces la capa acuosa con EtOAc. Se lavaron una vez las capas orgánicas combinadas con H2O, se secó sobre MgSO4, se filtró, se trató con gel de sílice y se evaporó. Se purificó el compuesto por cromatografía en gel de sílice en una columna de 40 g usando un sistema MPLc que se eluyó con un gradiente de n-heptano: EtOAc (de 100: 0 a 50: 50) para proporcionar el compuesto deseado como un líquido incoloro (1,74 g; 49,4 %). EM (ESI): m/z = 278,2 [M-Boc+H]+.
BB6
4-[(4-fluorofenil)-metoxi-metil]piperidina
A una solución de 4-[(4-fluorofenil)-metoxi-metil]piperidina-1-carboxilato de tere-butilo (57 mg, 176 jmol) en dioxano (200 jl) se le añadió HCl 4 M en dioxano (220 jl, 880 jmol) y se agitó la mezcla a t. a. durante 2 horas. Se evaporó completamente la solución amarillo claro. Se recogió el residuo en solución de NaHCO3 acuosa saturada y DCM y se separaron las capas. Se extrajo la capa acuosa dos veces con DCM. Se secaron las capas orgánicas combinadas sobre MgSO4, se filtró y evaporó para proporcionar el compuesto deseado como un aceite amarillo claro (0,036 g; 91,5 %). EM (ESI): m/z = 224,2 [M+H]+.
Intermedios:
a) 4-[(4-fluorofenil)-metoxi-metil]piperidina-1-carboxilato de terc-butilo
A una solución helada de 4-((4-fluorofenil)(hidroxi)metil)piperidina-1-carboxilato de terc-butilo (60 mg, 194 |jmol, CAS RN 160296-41-3) en THF (1 ml) se le añadió NaH al 55 % en aceite de vaselina (9,31 mg, 213 jmol) y se agitó la mezcla a esta temperatura durante 30 minutos antes de añadir yodometano (33,1 mg, 14,6 jl, 233 jmol). Después de agitar a esta temperatura durante 30 minutos, se dejó que la solución turbia se calentara hasta t. a. Después de agitar durante 2 horas a t. a., se añadió otro lote de yodometano (33 mg, 14,6 jl, 233 jmol). Después de 4,5 horas, se vertió la mezcla de reacción sobre H2O y EtOAc y se separaron las capas. Se extrajo dos veces la capa acuosa con EtOAc. Se secaron las capas orgánicas combinadas sobre MgSO4, se filtró y evaporó para obtener el compuesto deseado como un aceite amarillo claro (0,057 g; 90,9 %). EM (ESI): m/z = 268,2 [M-C4H8+H]+.
BB7
Clorhidrato de 1-metil-5-(4-piperidil)indazol
A una solución de 4-(1-metil-1H-indazol-5-il)piperidina-1-carboxilato de terc-butilo (155 mg, 491 jmol) en dioxano (0,5 ml) se le añadió HCl 4 M en dioxano (614 jl, 2,46 mmol). Se agitó la suspensión incolora a t. a. durante 3,5 horas. Se añadió éter (4 ml) y se filtró la suspensión. Se lavó la torta del filtro con éter para obtener el producto deseado como un sólido amarillo claro (0,107 g; 86,5 %). EM (ESI): m/z = 216,2 [M+-HCl+H]+.
Intermedios:
a) 4-(1-metilindazol-5-il)piperidina-1-carboxilato de terc-butilo
A una solución de 4-(1-metil-1H-indazol-5-il)-3,6-dihidropiridin-1(2H)-carboxilato de terc-butilo (160 mg, 511 jmol) en MeOH (3 ml) y EtOAc (1 ml) bajo argón se le añadió Pd-C al 10 % (16,3 mg, 15,3 jmol) y se agitó la suspensión en una atmósfera de hidrógeno a 1,7 bar durante 2,5 horas. Se separaron por filtración los sólidos y se evaporó el filtrado para dar el compuesto deseado como un aceite incoloro (0,156 g; 96,9 %). EM (ESI): m/z = 316,3 [M+H]+. b) 4-(1-metil-1H-indazol-5-il)-3,6-dihidropiridin-1(2H)-carboxilato de terc-butilo
A una mezcla de 4-(((trifluorometil)sulfonil)oxi)-3,6-dihidropiridina-1(2H)-carboxilato de terc-butilo (200 mg, 604 jmol, CAS RN 138647-49-1) en DME (3 ml) y Na2CO3 acuoso 2 M (770 jl, 1,54 mmol) se le añadieron acetato de paladio(II) (2,71 mg, 12,1 jmol), trifenilfosfina (7,92 mg, 30,2 jmol) y 1-metil-5-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)-1H-indazol (171 mg, 664 jmol, CAS RN 1235469-00-7) y se calentó la mezcla de dos capas transparente hasta 90 °C durante 2 horas. Se vertió la mezcla de reacción sobre H2O y EtOAc y se separaron las capas. Se extrajo dos veces la capa acuosa con EtOAc. Se secaron las capas orgánicas combinadas sobre MgSO4, se filtró, se trató con gel de sílice y se evaporó. Se purificó el compuesto por cromatografía en gel de sílice en una columna de 12 g usando un sistema MPLC que se eluyó con un gradiente de n-heptano: EtOAc (de 100: 0 a 00: 100) para proporcionar el compuesto deseado como un sólido incoloro (0,163 g; 86,2%). EM (ESI): m/z = 314,3 [M+H]+.
BB8
Clorhidrato de (E o Z)-2-fenil-3-piperacin-1-il-prop-2-enonitrilo
Se trató (E o Z)-4-[2-ciano-2-fenil-vinil]piperacina-1-carboxilato de terc-butilo (300 mg, 951 jmol) con una solución de HCl 4 M en dioxano (3,6 g, 3 ml, 98,7 mmol). Se agitó la mezcla de reacción a t. a. durante 15 h, a continuación se concentró a vacío para dar un sólido amarillo claro (240 mg, 100 %); EM (ESI): m/z = 214,2[M+H]+.
Intermedio: (E o Z)-4-[2-ciano-2-fenil-vinil]piperacina-1-carboxilato de terc-butilo
Se disolvieron piperacina-1-carboxilato de terc-butilo (200 mg, 1,07 mmol, CAS RN 57260-71-6), 3-oxo-2-fenilpropanonitrilo (156 mg, 1,07 mmol, CAS RN 5841-70-3) y triacetoxiborohidruro de sodio (228 mg, 1,07 mmol, CAS r N 56553-60-7) en DCM (5 ml). Se agitó la mezcla de reacción a t. a. durante 48 horas. Se lavó la mezcla de reacción con H2O, NaHCO3 y salmuera. Se secó la fase orgánica sobre Na2SO4, se filtró y se concentró el filtrado a vacío para dar un sólido marrón (300 mg). EM (ESI): m/z = 314,2 [M+H]+.
BB9
Clorhidrato de 5-cloro-1-ciclopropil-3-(4-piperidil)indol
A una mezcla de 4-(5-cloro-1-ciclopropil-1 H-indol-3-il)piperidina-1 -carboxilato de terc-butilo (275,0 mg,
0,730 mmol) en EtOAc (5 ml) se le añadió HCl/EtOAc (4 M, 4 ml), y se agitó la mezcla a 20 °C durante 3 horas. Se concentró la mezcla y se diluyó el residuo con H2O (30 ml), se lavó con EtOAc (10 ml x 2). Se liofilizó la capa acuosa para dar el compuesto del título como sólido amarillo claro (189,4 mg, 81,3 %). RMN de 1H (400 MHz, DMSO-d6) 89,29 - 8,87 (m, 2H), 7,76 (d, J = 1,9 Hz, 1H), 7,53 (d, J = 8,7 Hz, 1H), 7,23 - 7,10 (m, 2H), 3,38 - 3,27 (m, 3H), 3,08 - 2,90 (m, 3H), 2,07 - 1,98 (m, 2H), 1,98 - 1,83 (m, 2H), 1,09 - 1,00 (m, 2H), 0,96 - 0,88 (m, 2H). EM (ESI): m/z = 275,1 [M+H]+.
Intermedios:
a) 4-(5-cloro-1-cidopropil-1H-indol-3-il)piperidina-1-carboxilato de terc-butilo
Se agitó una mezcla de 4-(5-cloro-1H-indol-3-il)piperidina-1-carboxilato de terc-butilo (500 mg, 1,49 mmol), ácido ciclopropilborónico (153,92 mg, 1,79 mmol; cAs RN 411235-57-9), solución de NaHMDS 1 M en THF (3 ml, 2,99 mmol; CAS RN 1070-89-9), DMAP (0,22 ml, 1,79 mmol) y acetato de cobre(II) (270,22 mg, 1,49 mmol; CAS RN 142-71-2) en tolueno (25 ml) a 95 °C durante 12 h (llevando un globo de oxígeno). Se vertió la mezcla en H2O (20 ml) y se extrajo con EtOAc (50 ml x 3). Se lavaron las capas orgánicas combinadas con salmuera (50 ml), se secó sobre Na2SO4, se filtró y concentró a vacío. Se purificó el residuo por cromatografía en columna en gel de sílice ultrarrápida (malla 100-200) usando un gradiente de PE: EtOAc (de 50: 1 a 20: 1) para dar el compuesto del título como aceite amarillo claro (275 mg, 0,730 mmol, 49,1 %). EM (ESI): m/z = 275,1 [M-Boc+H]+.
b) 4-(5-cloro-1H-indol-3-il)piperidina-1-carboxilato de terc-butilo
Se agitó una mezcla de 4-(5-cloro-1H-indol-3-il)-3,6-dihidro-2H-piridin-1-carboxilato de terc-butilo (1,003 g, 3,01 mmol) y óxido de platino(IV) (100,0 mg, 0,350 mmol, CAS RN 1314-15-4) en AcOH (10,0 ml) y EtOH (20 ml) a 40 °C durante 15 h en H2 (1520 mmHg). Se filtró la mezcla y se concentró el filtrado a vacío. Se trituró el residuo con PE (50 ml) y se filtró. Se recogió la torta del filtro y se secó a vacío para dar el compuesto del título como sólido gris claro (780 mg, 77,3 %). EM (ESI): m/z = 235,1 [M-Boc+H]+.
c) 4-(5-cloro-1H-indol-3-il)-3,6-dihidro-2H-piridin-1-carboxilato de terc-butilo
A una mezcla de 5-cloroindol (1 g, 6,6 mmol; CAS RN 17422-32-1) en MeOH (20 ml) se le añadió 4-oxopiperidina-1-carboxilato de terc-butilo (1,97 g, 9,9 mmol; CAS RN 79099-07-3). Se agitó la mezcla a 20 °C durante 1 hora y, a continuación, se añadió hidróxido de potasio (1,48 g, 26,39 mmol). Se agitó la mezcla a 70 °C durante 15 h. Se concentró la mezcla a vacío. Se trituró el residuo con una mezcla de PE y EtOAc (5:1, 50 ml) y se filtró. Se recogió la torta del filtro y se secó a vacío para dar el compuesto del título como sólido amarillo claro (2 g, 91,3 %). EM (ESI): m/z = 227,1 [M-C4Hs+H]+.
BB10
5-cloro-1-(oxetan-3-il)-3-(4-piperidil)indol
A una mezcla de 4-[5-cloro-1-(oxetan-3-il)-1H-indol-3-il]piperidina-1-carboxilato de terc-butilo (1 g, 2,56 mmol) en DCM (20 ml) se le añadió TFA (5,0 ml). Se agitó la mezcla a 20 °C durante 12 horas. Se concentró la mezcla a vacío, se purificó por HPLC prep. (FA al 0,1 % en H2O y ACN) para dar el compuesto del título como sólido amarillo claro (311,3 mg, 37,3 %). RMN de 1H (400 MHz, DMSO-d6) 87,76 (d, J = 1,8 Hz, 1H), 7,65 (s, 1H), 7,57 (d, J = 8,8 Hz, 1H), 7,17 (dd, J = 1,9, 8,7 Hz, 1H), 5,73 (quin, J = 7,0 Hz, 1H), 5,01 (t, J = 7,2 Hz, 2H), 4,90 (t, J = 6,5 Hz, 2H), 3,38 (d. a., J = 12,2 Hz, 2H), 3,14 - 3,00 (m, 3H), 2,07 (d. a., J = 13,2 Hz, 2H), 1,96 - 1,78 (m, 2H). EM (ESI): m/z = 291,1 [M+H]+.
Intermedio: 4-(5-cloro-1-(oxetan-3-il)-1H-indol-3-il)piperidina-1-carboxilato de terc-butilo
A una mezcla de 4-(5-cloro-1H-indol-3-il)piperidina-1-carboxilato de terc-butilo (1 g, 2,99 mmol, BB9, etapa intermedia b) en DMF (20 ml) se le añadió NaH al 60 % en aceite de vaselina (143,35 mg, 3,58 mmol) a 0 °C. Se agitó la mezcla a 20 °C durante 1 hora y, a continuación, se añadió 4-metilbencenosulfonato de oxetan-3-ilo (1022,55 mg, 4,48 mmol; CAS RN 26156-48-9) y se agitó la mezcla a 85 °C durante 12 horas. Se vertió la mezcla en H2O (60 ml) y se extrajo con EtOAc (50 ml x 3). Se lavaron las capas orgánicas combinadas con salmuera (50 ml), se secó sobre Na2SO4 y se concentró a vacío para dar el compuesto del título como sólido amarillo claro (1,1 g, 2,81 mmol, 94,2 %). EM (ESI): m/z = 291,1 [M-Boc+H]+.
BB11
Sal de ácido fórmico de 4-(bis(4-fluorofenil)metil)piperidina
A una solución de 4-[bis(4-fluorofenil)metileno]piperidina (700,0 mg, 1,75 mmol) en AcOH (50,0 ml) se le añadió Pd/C (1,0 g), se purgó la reacción con hidrógeno tres veces y se agitó a 90 °C (bajo globo de hidrógeno) durante 48 horas. Se filtró la mezcla de reacción y se concentró a vacío. Se purificó el producto bruto por HPLC prep. (FA
al 0,1
%
en agua y MeCN) para dar el compuesto del título como un sólido gris claro (263,5 mg, 32,1 %). RMN de 1H (DMSO-d6, 400 MHz) 58,39 (s, 1H), 7,45 - 7,37 (m, 4H), 7,15 - 7,07 (m, 4H), 3,68 (d, J = 11,2 Hz, 1H), 3,17 (d. a., J = 12,3 Hz, 2H), 2,80 - 2,68 (m, 1H), 2,46 (d. a., J = 11,2 Hz, 1H), 1,54 - 1,32 (m, 2H), 1,27 - 1,09 (m, 2H)). EM (ESI): m/z = 288,1 [M+H]+.
Intermedios:
a) 4-[bis(4-fluorofenil)metileno]piperidina
A una solución de bis(4-fluorofenil)(piperidin-4-il)metanol (900,0 mg, 2,59 mmol) en DCM (30 ml) se le añadió TFA (10,0 ml) y se agitó la mezcla de reacción a 20 °C durante 12 horas. Se concentró la mezcla de reacción a vacío para dar el compuesto del título bruto que se usó en la etapa siguiente sin purificación adicional. EM (ESI): m/z = 286.1 [M+H]+.
b) bis(4-fluorofenil) (piperidin-4-il)metanol
Se suspendió una mezcla de Mg (1,4 g, 58,29 mmol) y I2 (20,0 mg, 0,080 mmol) en THF (40 ml) a 10 °C. A la solución amarilla anterior se le añadió 1-bromo-4-fluorobenceno (1,0 g, 5,75 mmol, CAS RN 460-00-4) y se calentó la mezcla de reacción hasta 45 °C hasta que la solución se volvió transparente. Se añadió otro lote de 1-bromo-4-fluorobenceno (9,14 g, 52,54 mmol, CAS Rn 460-00-4) (disuelto en 10 ml de THF). Se agitó la mezcla de reacción a 45 °C durante 30 min. Se añadió N-BOC-piperidina-4-carboxilato de etilo (1,5 g, 5,83 mmol, CAS RN 142851-03 4) y se agitó la mezcla de reacción a 80 °C durante 12 horas. Se vertió la mezcla en solución ac. de NH4CI (50 ml) sat., se extrajo dos veces la mezcla con EtOAc (50 ml cada vez), se lavó la capa orgánica combinada con agua (30 ml x 2) y salmuera (30 ml), se secó sobre Na2SO4 y se concentró a vacío para dar el compuesto del título (900 mg, 38,2 %). RMN de 1H (400 MHz, DMSO-d6) 88,70 (s. a., 1H), 7,57 - 7,47 (m, 4H), 7,12 (t, J = 8,8 Hz, 4H), 5.73 (s, 1H), 3,23 (d. a., J = 12,3 Hz, 2H), 2,92 - 2,79 (m, 3H), 1,68 - 1,54 (m, 2H), 1,39 (d. a., J = 13,6 Hz, 2H). EM (ESI): m/z = 304,1 [M+H]+.
BB12
5-cloro-1-(oxetan-3-ilmetil)-3-(4-piperidil)indol
A una solución de 4-[5-cloro-1-(oxetan-3-ilmetil)indol-3-il]piperidina-1-carboxilato de terc-butilo (700,0 mg, 1.73 mmol) en DCM (20 ml) se le añadió TFA (3,38 ml). Se agitó la mezcla a 20 °C durante 12 h. Se concentró la mezcla a vacío, se purificó por HPLC prep. (FA al 0,1 % en H2O y ACN) para dar el compuesto del título como sólido amarillo claro (154,57 mg, 29,0 %). RMN de 1H (400 MHz, DMSO-d6) 88,60 (s. a., 1H), 7,71 (d, J = 1,7 Hz, 1H), 7,54 (d, J = 8,8 Hz, 1H), 7,33 (s, 1H), 7,13 (dd, J = 1,8, 8,7 Hz, 1H), 4,59 (dd, J = 6,2, 7,6 Hz, 2H), 4,47 - 4,33 (m, 4H), 3,43 - 3,37 (m, 3H), 3,08 - 2,99 (m, 3H), 2,04 (d, J = 13,0 Hz, 2H), 1,89 - 1,76 (m, 2H). EM (ESI): m/z = 305.2 [M+H]+.
Intermedios:
a) 4-[5-cloro-1-(oxetan-3-ilmetil)indol-3-il]piperidina-1-carboxilato de terc-butilo
A una solución de 4-(5-cloro-1H-indol-3-il)piperidina-1-carboxilato de terc-butilo (400,0 mg, 1,19 mmol, BB9, etapa intermedia b) en DMF (8 ml) se le añadió NaH al 60 % en aceite de vaselina (57,34 mg, 2,39 mmol) a 0 C. Se agitó la mezcla a 20 °C durante 1 hora. A continuación se añadió 4-metilbencenosulfonato de oxetan-3-ilmetilo (434,16 mg, 1,79 mmol) y se agitó la mezcla a 85 °C durante 12 horas. Se vertió la mezcla en H2O (60 ml) y se extrajo con EtOAc (50 ml x 3). Se lavaron las capas orgánicas combinadas con salmuera (50 ml), se secó sobre Na2SO4 y se concentró a vacío para dar el compuesto del título como aceite amarillo (629 mg, bruto). EM (ESI): m/z = 305,0 [M-Boc+H]+.
b) 4-metilbencenosulfonato de oxetan-3-ilmetilo
A una solución de 3-oxetanometanol (500,0 mg, 5,67 mmol; CAS RN 6246-06-6) en DCM (10 ml) se le añadió TEA (1,19 ml, 8,51 mmol), DMAP (69,33 mg, 0,570 mmol) y cloruro de 4-metilbenceno-1-sulfonilo (1293,77 mg, 6,81 mmol; CAS RN 98-59-9). Se agitó la mezcla a 20 °C durante 6 horas. Se diluyó la mezcla con una mezcla de EtOAc y solución de NH4Cl acuosa saturada (1:1, 20 ml). Se extrajo la mezcla con EtOAc (40 ml x 3). Se lavaron las capas orgánicas combinadas con salmuera, se secó sobre Na2SO4, se filtró y concentró para dar el compuesto del título como sólido marrón (1,25 g, 5,16 mmol, 78,7 %). EM (ESI): m/z = 243,1 [M+H]+.
BB13
1-(2-((terc-butildimetilsilil)oxi)etil)-5-cloro-3-(piperidin-4-il)-1H-indol
A una solución de 2-[5-cloro-3-(4-piperidil)indol-1-il]etanol (600,0 mg, 1,39 mmol) en piridina (5 ml) se le añadió tbutildimetilclorosilano (251,35 mg, 1,67 mmol; CAS RN 18162-48-6) y se agitó la mezcla a 20 °C durante 12 horas. Se vertió la mezcla en H2O (10 ml) y se extrajo con EtOAc (20 ml x 3). Se lavaron las capas orgánicas combinadas con salmuera (10 ml), se secó sobre Na2SO4, se filtró y concentró. Se trituró el residuo con PE (10 ml) y se filtró. Se lavó la torta del filtro con PE (5 ml x 2, se recogió el sólido y se secó a vacío para dar el compuesto del título como sólido amarillo claro (164,7 mg, 29,8 %). RMN de 1H (400 MHz, DMSO-d6) 88,97 (s. a., 1H), 7,72 (d, J = 2,0 Hz, 1H), 7,47 (d, J = 8,7 Hz, 1H), 7,20 (s, 1H), 7,10 (dd, J = 1,8, 8,7 Hz, 1H), 4,22 (t. a., J = 4,8 Hz, 2H), 3,84 (t, J = 4,9 Hz, 2H), 3,44 - 3,32 (m, 2H), 3,12 - 2,94 (m, 3H), 2,08 - 1,96 (m, 2H), 1,95 - 1,81 (m, 2H), 0,73 (s, 9H), -0,24 (s, 6H). EM (ESI): m/z = 393,1 [M+H]+.
Intermedios:
a) 2-[5-cloro-3-(4-piperidil)indol-1 -il]etanol
A una mezcla de 4-[5-cloro-1-(2-hidroxietil)indol-3-il]piperidina-1-carboxilato de terc-butilo (1,1 g, 2,9 mmol) en EtOAc (10 ml) se le añadió HCl/ETOAC (4 M, 0,73 ml). Se agitó la mezcla a 20 °C durante 3 horas y a continuación se concentró a vacío. Se diluyó el residuo con DCM (20 ml) y se lavó con solución de NaHCO3 acuosa saturada (10 ml). Se extrajo la capa acuosa con DCM: MeOH (de 5: 1 v/v; 20 ml x 3). Se secaron las capas orgánicas combinadas sobre Na2SO4 y se filtró. Se concentró el filtrado a vacío para dar el compuesto del título como aceite amarillo claro (750 mg, 59,8 %). EM (ESI): m/z = 279,1 [M+H]+.
b) 4-[5-cloro-1-(2-hidroxietil)indol-3-il]piperidina-1-carboxilato de terc-butilo
Se agitó una mezcla de 4-(5-cloro-1H-indol-3-il)piperidina-1-carboxilato de terc-butilo (1 g, 2,99 mmol, intermedio BB9, etapa b), 2-bromoetanol (0,32 ml, 4,48 mmol; CAS RN 540-51-2) e hidróxido de potasio (335,12 mg, 5,97 mmol) en DMF (10 ml) a 100 °C durante 12 horas. Se purificó la mezcla por HPLC prep. (amoníaco al 0,1 % en H2O y Ac N) para dar el compuesto del título como aceite amarillo claro (1,1 g, 97,2 %). Em (ESI): m/z = 323,1 [M-C4H8+H]+.
BB14a y BB14b
(+)-4a,5,6,7,8,8a-hexahidro-4H-pirido[4,3-b][1,4]oxacin-3-ona
y
(-)-cis-4a, 5,6,7,8,8a-hexahidro-4H-pirido[4,3-b][1,4]oxacin-3-ona
Se separaron los enantiómeros de diclorhidrato de rac-(4aR,8aS)-hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona (500 mg, 2,18 mmol, ChemBridge Corporation) por HPLC quiral preparativa (columna ReprosilChiral NR) usando una mezcla isocrática de EtOH (que contenía un 0,05 % de NH4OAc):n-heptano (30: 70).
Primer enantiómero en eluir: (+)-cis-4a,5,6,7,8,8a-hexahidro-4H-pirido[4,3-b][1,4]oxacin-3-ona. Sólido amarillo (0,150 g; 44,0 %). EM (ESI): m/z = 157,1 [M+H]+.
Segundo enantiómero en eluir: (-)-cis-4a,5,6,7,8,8a-hexahidro-4H-pirido[4,3-b][1,4]oxacin-3-ona. Sólido amarillo (0,152 g; 44,6 %). EM (ESI): m/z = 157,1 [M+H]+.
BB15a y BB15b
(4aR,8aS)-3-oxohexahidro-2H-pirido[4,3-b][1,4]oxacina-6(5H)-carboxilato de 4-nitrofenilo (BB15a)
y
(4aS,8aR)-3-oxohexahidro-2H-pirido[4,3-b][1,4]oxacina-6(5H)-carboxilato de 4-nitrofenilo (BB15b)
A una suspensión de rac-(4aR,8aS)-hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona; sal diclorhidrato (4,5 g, 19,6 mmol, BB1) en DCM seco (125 ml) a 0 °C se le añadió DiPEA (6,35 g, 8,58 ml, 49,1 mmol) seguido de carbonoclorato de 4-nitrofenilo (4,35 g, 21,6 mmol). Se agitó la mezcla de reacción a 0 °C durante 10 min y a t. a. durante 2 h. Se diluyó la reacción en bruto con DCM y se transfirió a un embudo de separación para su extracción con solución de Na2CO3 ac. sat. Se recogió la fase orgánica y se extrajo de nuevo la fase acuosa con DCM. Se secaron las fases orgánicas combinadas sobre Na2SO4 y se evaporó hasta sequedad para proporcionar 6,62 g de un producto racémico bruto (BB7) como un sólido amarillo. Se sometió el material bruto directamente a separación por SFC quiral para proporcionar el enantiómero BB15b (2,72 g, segundo enantiómero en eluir) como un sólido amarillo y el enantiómero BB15a (3,25 g, primer enantiómero en eluir) como un sólido beige claro pero contaminado con BB15b. Se llevó a cabo otra separación quiral por SFC para proporcionar 2,71 g de BB15a. EM (ESI): m/z = 322,2 [M+H]+ para ambos enantiómeros.
BB16
Clorhidrato de 3-(4-fluorofenil)-3-(piperidin-4-il)propan-1-ol
Se agitó una solución de 4-(1-(4-fluorofenil)-3-hidroxipropil)piperidina-1-carboxilato de tere-butilo (119 mg, 353 |jmol, BB5, intermedio b) en HCl 2 M en éter dietílico (1,76 ml, 3,52 mmol) a t. a. durante 4,5 h antes de añadir otro lote de HCl 2 M en éter dietílico (1,76 ml, 3,53 mmol). Se continuó agitando a t. a. durante la noche. Se homogeneizó la suspensión en un baño ultrasónico helado y se filtró. Se lavó la torta del filtro con un pequeño volumen de éter dietílico para obtener el compuesto deseado como un sólido incoloro (0,067 g; 69,4 %). EM (ESI): m/z = 238,2 [M+H]+.
BB17
Formiato de 4-[1-[4-(trifluorometil)fenil]etil]piperidina
Se agitó una solución de 4-[1-[4-(trifluorometil)fenil]etil]piperidina-1-carboxilato de tere-butilo (1,5 g, 4,2 mmol) en una mezcla de HCl/dioxano (50,0 ml, 1/1 v/v) a 20 °C durante 2 h. Se concentró la mezcla y se purificó el residuo por HPLC prep. (FA) y se liofilizó para obtener el compuesto deseado como aceite amarillo claro (838,4 mg, 77,1 %). EM (ESI): m/z = 258,1 [M+H]+.
Intermedios:
a) 4-[1-[4-(trifluorometil)fenil]etil]piperidina-1-earboxilato de tere-butilo
A una solución de 4-[1-[4-(trifluorometil)fenil]vinil]piperidina-1 -carboxilato de tere-butilo (2,0 g, 5,63 mmol) en EtOAc (100 ml) se le añadió Pd/C (1,0 g, 5,63 mmol), se agitó la mezcla a 20 °C en atmósfera de H2 (globo) durante 12 h. Se filtró la mezcla y se concentró el filtrado para obtener el compuesto deseado como un aceite incoloro (1,8 g, 89,5 %). EM (ESI): m/z = 302,2 [M-C4Hs+H]+.
b) 4-[1-[4-(trifluorometil)fenil]vinil]piperidina-1-earboxilato de tere-butilo
A una solución de bromuro de metiltrifenilfosfonio (10,0 g, 27,98 mmol, CAS RN 1779-49-3) en THF (250 ml) se le añadió gota a gota bis(trimetilsilil)amida de litio en THF (41,97 ml, 41,97 mmol) a 0 °C y se agitó la mezcla a 0 °C durante 1 h. A continuación se añadió gota a gota 4-[4-(trifluorometil)benzoil]piperidina-1-carboxilato de
tere
-butilo (10,0 g, 27,98 mmol, CAS RN 725229-27-6) en THF (50 ml) y se dejó que la mezcla se calentara hasta 20 °C durante 2 h. Se vertió la mezcla en solución de NH4CI ac. (500 ml) y se extrajo tres veces con EtOAc (200 ml cada vez). Se lavó la fase orgánica con salmuera y se secó sobre Na2SO4, se filtró y se purificó el filtrado en una columna en gel de sílice (PE: EtOAc = 20:1) para proporcionar el compuesto deseado como aceite amarillo claro. (5,1 g, 51,3 %). RMN de 1H (400 MHz, QUEROFORMA-d) 5 = 7,61 (d, J=8,2 Hz, 2H), 7,44 (d, J=8,1 Hz, 2H), 5,25 (s, 1H), 5,13 (s, 1H), 4,27-4,14 (m, 2H), 2,77 (t. a., J=12,3 Hz, 2H), 2,58 (t. a., J=11,6 Hz, 1H), 1,78 (d. a., J=13,0 Hz, 2H), 1,48 (s, 9H), 1,43-1,32 (m, 2H).
BB18
6-eloro-1-metil-3-(4-piperidil)indazol
Se calentó a reflujo una mezcla de 1-[4-(6-cloro-1-metil-indazol-3-il)-1-piperidil]etanona (3,95 g, 14 mmol) en solución de HCl al 25 % acuosa (25,75 ml) durante 4 h. Después de enfriar hasta t. a., se alcalinizó la mezcla usando una solución de NaOH acuosa concentrada. Se extrajo la capa acuosa dos veces con DCM. Se secaron las capas orgánicas sobre Na2SO4, se filtró y evaporó para obtener el compuesto deseado como un aceite rojo claro (3,6 g, 98,2 %). EM (ESI): m/z = 250,2 [M+H]+. Se usó el compuesto en la etapa siguiente sin purificación adicional.
Intermedio:
1-[4-(6-eloro-1-metil-indazol-3-il)-1-piperidil]etanona
En un tubo sellado se mezclaron 1-[4-(4-cloro-2-fluoro-benzoil)-1-piperidil]etanona (3,9 g, 14 mmol; Novel Chemical Solutions) y metilhidracina (0,94 ml, 18 mmol) y se calentó la mezcla a 120 °C durante 16 h. Después de enfriar, se alcalinizó la mezcla usando solución de NaHCO3 acuosa concentrada. Se extrajo la capa acuosa dos veces con DCM. Se secaron las capas orgánicas sobre Na2SO4, se filtró y evaporó para proporcionar el compuesto deseado como un aceite amarillo (3,97 g, 100 %). EM (ESI): m/z = 292,3 [M+H]+. Se usó el compuesto en la etapa siguiente sin purificación adicional.
BB19
Clorhidrato de 2-(4-fluorofenil)-2-(piperidin-4-il)acetonitrilo
Se obtuvo el producto de forma análoga a BB16 a partir de 4-(ciano(4-fluorofenil)metil)piperidina-1-carboxilato de terc-butilo (MFCD28112711, A Chemtek) como un sólido amarillo claro. (0,101 g, 87,1 %). EM (ESI): m/z = 219,1 [M+H]+.
BB20
Clorhidrato de 4-((4-fluorofenil)(2,2,2-trifluoroetoxi)metil)piperidina
Se obtuvo el producto de forma análoga a BB16 a partir de 4-((4-fluorofenil)(2,2,2-trifluoroetoxi)metil)piperidina-1-carboxilato de terc-butilo como una espuma incolora (0,151 g; 100,0 %). EM (ESI): m/z = 292,2 [M+H]+.
Intermedio:
4-((4-fluorofenil)(2,2,2-trifluoroetoxi)metil)piperidina-1-carboxilato de terc-butilo
A una solución amarilla de tri-n-butilfosfina (262 mg, 319 pl, 1,29 mmol) y dipiperidida azodicarboxílica (326 mg, 1,29 mmol) en tolueno (10 ml) se le añadió 4-((4-fluorofenil)(hidroxi)metil)piperidina-1-carboxilato de terc-butilo (200 mg, 646 pmol, CAS RN 160296-41-3) y se agitó la mezcla a t. a. durante 10 min. La adición de 2,2,2-trifluoroetanol (711 mg, 514 pl, 7,11 mmol) dio una suspensión. Después de calentar a reflujo durante 20 h, se enfrió la mezcla de reacción hasta t. a. Se añadió gel de sílice y se evaporó la mezcla. Se purificó el compuesto por cromatografía en gel de sílice en una columna de 12 g usando un sistema MPLC (ISCO) que se eluyó con un gradiente de n-heptano: EtOAc (de 100: 0 a 50: 50) para obtener el compuesto deseado como un aceite incoloro (0,180 g; 71,1 %). EM (ESI): m/z = 391,1 [M+H]+.
BB21
Clorhidrato de 5-(2-(4-fluorofenil)-2-(piperidin-4-il)etil)-3-metil-1,2,4-oxadiazol
Se obtuvo el producto de forma análoga a BB16 a partir de 4-(1-(4-fluorofenil)-2-(3-metil-1,2,4-oxadiazol-5-il)etil)piperidina-1-carboxilato de terc-butilo como un sólido incoloro (0,051 g; 98,3 %). EM (ESI): m/z = 290,2 [M+H]+.
Intermedios:
a) 4-(1-(4-fluorofenil)-2-(3-metil-1,2,4-oxadiazol-5-il)etil)piperidina-1-carboxilato de terc-butilo
A una mezcla de 4-(3-etoxi-1-(4-fluorofenil)-3-oxopropil)piperidina-1-carboxilato de terc-butilo (100 mg, 264 pmol) y (Z)-N'-hidroxiacetimidamida (39 mg, 527 pmol) en tolueno (2,5 ml) se le añadió K2CO3 (72,8 mg, 527 pmol) y se agitó la mezcla de reacción a 130 °C durante 14 días. La reacción no terminó, por lo que se añadieron (Z)-N'-hidroxiacetimidamida (39 mg, 527 pmol) y K2CO3 (72,8 mg, 527 pmol) y se agitó la MR durante el fin de semana a 130 °C. Se vertió la mezcla de reacción en solución de NH4Cl acuosa saturada y EtOAc y se separaron las capas. Se extrajo dos veces la capa acuosa con EtOAc. Se secaron las capas orgánicas sobre MgSO4, se filtró, se trató con gel de sílice y se evaporó. Se purificó el compuesto por cromatografía en gel de sílice en una columna de 4 g usando un sistema MPLC que se eluyó con un gradiente de n-heptano: EtOAc (de 100: 0 a 0: 100) para obtener el compuesto deseado como un sólido incoloro. EM (ESI): m/z = 290,3 [M-Boc+H]+.
b) 4-(3-etoxi-1-(4-fluorofenil)-3-oxopropil)piperidina-1-carboxilato de terc-butilo
En un matraz de fondo redondo de 25 ml, se combinó (E)-4-(3-etoxi-1-(4-fluorofenil)-3-oxoprop-1-en-1-il)piperidina-1-carboxilato de terc-butilo (1,747 g, 4,63 mmol) con EtOAc (17,1 ml) para dar una solución incolora. Se añadió Pd/C al 10 % (175 mg, 4,63 mmol) bajo argón. Se agitó la suspensión en atmósfera de hidrógeno a 1,5 bar durante 4 h. Se filtró la mezcla de reacción y se concentró el filtrado a vacío. Se purificó el compuesto por cromatografía en gel de sílice en una columna de 24 g usando un sistema MPLC (ISCO) que se eluyó con un gradiente de nheptano: EtOAc (de 100: 0 a 50: 50) para obtener el compuesto deseado como un aceite incoloro (1,22 g; 69,8 %). EM (ESI): m/z = 280 [M-Boc+H]+.
c) (E)-4-(3-etoxi-1-(4-fluorofenil)-3-oxoprop-1-en-1-il)piperidina-1-carboxilato de terc-butilo
A una solución de 2-(dietoxifosforil)acetato de etilo (2,1 g, 1,86 ml, 9,36 mmol) en 1,4-dioxano (9,73 ml) se le añadió gota a gota LiHMDS 1,0 M en hexanos (15 ml, 15 mmol) y se agitó la solución durante 15 min. Se añadió gota a gota a la mezcla 4-(4-fluorobenzoil)piperidina-1-carboxilato de terc-butilo (2,878 g, 9,36 mmol, CAS RN 160296 40-2) disuelto en 1,4-dioxano (9,73 ml) y se continuó agitando a reflujo durante dos días. Se vertió la mezcla de reacción sobre solución de NH4Cl acuosa saturada y EtOAc y se separaron las capas. Se extrajo dos veces la capa acuosa con EtOAc. Se lavaron una vez las capas orgánicas con agua, se secó sobre MgSO4, se filtró, se trató con gel de sílice y se evaporó. Se purificó el compuesto por cromatografía en gel de sílice en una columna de 40 g usando un sistema MPLC que se eluyó con un gradiente de n-heptano: EtOAc (de 100: 0 a 50: 50) para obtener el compuesto deseado como un líquido incoloro (1,74 g, 49,4 %). EM (ESI): m/z = 278,2 [M-Boc+H]+.
BB22
Formiato de 4-(1-(4-(trífíuorometil)fenil)ddopropil)piperídina
A una solución de 4-[1-[4-(trifluorometil)fenil]ciclopropil]piperidina-1 -carboxilato de terc-butilo (250,0 mg, 0,680 mmol) en DCM (5 ml) se le añadió TfA (1,0 ml, 0,680 mmol) y se agitó la mezcla a 20 °C durante 16 h. Se concentró la mezcla y se purificó el residuo por HPLC prep. (FA) para proporcionar el compuesto deseado como un aceite amarillo (181 mg, 81,5 %). EM (ESI): m/z = 270,1 [M+H]+.
Intermedios:
a) 4-[1-[4-(trifluorometil)fenil]ciclopropil]piperidina-1-carboxilato de terc-butilo
A una solución de 4-[2,2-dibromo-1-[4-(trifluorometil)fenil]ciclopropil]piperidina-1-carboxilato de terc-butilo (800,0 mg, 1,52 mmol) e isopropóxido de titanio(IV) (862,56 mg, 3,03 mmol) en THF (30 ml) se le añadió una solución 3 M de EtMgBr en t Hf (5,06 ml, 15,17 mmol) gota a gota a 0 °C. Se calentó la mezcla hasta 20 °C durante 16 h. Se vertió la mezcla en NH4CI (100 ml ac.) y se extrajo tres veces con EtOAc (30 ml cada vez). Se lavó la fase orgánica con salmuera y se secó sobre Na2SO4, se filtró y se concentró el filtrado. Se purificó el residuo por cromatografía ultrarrápida inversa (FA) para proporcionar el compuesto deseado como un aceite amarillo claro (250 mg, 44,6 %). EM (ESI): m/z = 314,1 [M-C4Hs+H]+.
b) 4-[2,2-dibromo-1-[4-(trífluommetil)fenil]ddopropil]piperidina-1-carboxilato de terc-butilo
A una mezcla agitada vigorosamente de 4-[1-[4-(trifluorometil)fenil]vinil]piperidina-1-carboxilato de terc-butilo (1,2 g, 3,38 mmol, BB17, intermedio b), bromoformo (2,56 g, 10,13 mmol, CAS RN 75-25-2) y cetrimida (0,37 g, 1,01 mmol, CAS RN 57-09-0) en DCM (10 ml) se le añadió 50 % de NaOH ac. (1,35 ml, 16,88 mmol) gota a gota. Se agitó vigorosamente la mezcla de reacción a 50 °C durante 16 h y, a continuación, se añadió H2O (20 ml). Se separó la fase orgánica y se extrajo tres veces la fase acuosa con DCM (10 ml cada vez). Se lavaron las fases orgánicas combinadas con H2O, HCl al 2 % y salmuera, y se secó (Na2SO4), se filtró y se concentró. Se purificó el residuo por TLC prep. (PE: EtOAc = 20: 1) para dar el compuesto deseado como aceite incoloro (900 mg, 50,5 %). EM (ESI): m/z = 471,9 [M-C4H8+H]+
BB23
Clorhidrato de 4-(1-(4-fluorofenil)-2-(2,2,2-trifluoroetoxi)etil)piperidina
Se obtuvo el producto de forma análoga a BB16 a partir de 4-(1 -(4-fluorofenil)-2-(2,2,2-trifluoroetoxi)etil)pi peridina-1-carboxilato de terc-butilo como una espuma incolora (0,173 g; 97,7 %). EM (e S i): m/z = 306,2 [M+H]+.
Intermedio:
4-(1-(4-fíuorofenil)-2-(2,2,2-trifíuoroetoxi)etil)piperidina-1-carboxilato de terc-butilo
A una solución amarilla de tri-n-butilfosfina (273 mg, 333 pl, 1,35 mmol) y dipiperidida azodicarboxílica (340 mg, 1,35 mm) en tolueno (11,1 ml) se le añadió 4-(1-(4-fluorofenil)-2-hidroxietil)piperidina-1 -carboxilato de terc-butilo (218 mg, 674 pmol, BB5, intermedio b) y se agitó la mezcla a t. a. durante 10 minutos. La adición de 2,2,2-trifluoroetanol (742 mg, 536 pl, 7,42 mmol) dio una suspensión. Tras el calentamiento de nuevo hasta 65 °C se formó una solución amarilla. Después de calentar a reflujo durante 17 horas, se enfrió la mezcla de reacción hasta t. a. Se añadió gel de sílice y se evaporó la mezcla.
Se purificó el compuesto por cromatografía en gel de sílice en una columna de 12 g usando un sistema MPLC (ISCO) que se eluyó con un gradiente de n-heptano: EtOAc (de 100: 0 a 50: 50) para obtener el compuesto deseado como un aceite incoloro (0,211 g; 77,2 %). e M (ESI): m/z = 350,1 [M-C4Hs+h ]+.
BB24
Formiato de 3-(1-(4-(trifluorometil)fenil)etil)acetidina
Se obtuvo el producto de forma análoga a BB22 a partir de 3-[1-[4-(trifluorometil)fenil]etil]acetidina-1-carboxilato de terc-butilo como aceite amarillo claro (151,5 mg, 27,4 %). EM (ESI): m/z = 230,1 [M+H]+.
Intermedios:
a) 3-[1-[4-(trifluorometil)fenil]etil]acetidin-1-carboxilato de terc-butilo
Se obtuvo el producto de forma análoga a BB17, intermedio a, a partir de 3-[1-[4-(trifluorometil)fenil]vinil]acetidina-1-carboxilato de terc-butilo como aceite incoloro (1 g, 99,4 %). e M (ESI): m/z = 274,0 [M-C4Hs+H]+.
b) 3-[1-[4-(trifluorometil)fenil]vinil]acetidina-1-carboxilato de terc-butilo
A una solución de bromuro de metiltrifenilfosfonio (1952,56 mg, 5,47 mmol, CAS RN 1779-49-3) en THF (25 ml) se le añadió bis(trimetilsilil)amida de litio/THF (9,11 ml, 9,11 mmol) gota a gota a 0 °C, se agitó la mezcla a 0 °C durante 1 h, a continuación se añadió gota a gota 3-[4-(trifluorometil)benzoil]acetidina-1-carboxilato de terc-butilo (1500,0 mg, 4,55 mmol, MFCD24368873, FCH Group) en THF (5 ml), se calentó la mezcla hasta 20 °C durante 2 h. Se vertió la mezcla en solución de NH4CI ac. (100 ml) y se extrajo tres veces con EtOAc (50 ml cada vez), se lavó la fase orgánica con salmuera y se secó sobre Na2SO4, se filtró y se purificó el filtrado por columna en gel de sílice (PE: EtOAc = 20:1) para dar el compuesto deseado como aceite amarillo claro (1000 mg, 67,0 %). RMN de 1H (400 MHz, CLOROFORMO-d) 8 = 7,61 (d, J=8,1 Hz, 2H), 7,42 (d, J=8,1 Hz, 2H), 5,61 (s, 1H), 5,32 (s, 1H), 4,20 (t, J=8,4 Hz, 2H), 3,89 (s. a., 2H), 3,83-3,73 (m, 1H), 1,46 (s, 9H).
BB25
Clorhidrato de 5-((4-fluorofenil) (piperidin-4-il)metil)-3-(trifluorometil)-1,2,4-oxadiazol
Se obtuvo el producto de forma análoga a BB16 a partir de 4-((4-fluorofenil)(3-(trifluorometil)-1,2,4-oxadiazol-5-il)metil)piperidina-1 -carboxilato de terc-butilo como un sólido incoloro (0,119 g; 90,1 %). EM (ESI): m/z = 330,2 [M+H]+.
Intermedio:
4-((4-fluorofenil)(3-(trifluorometil)-1,2,4-oxadiazol-5-il)metil)piperidina-1-carboxilato de terc-butilo
A una solución de ácido 2-(1-(terc-butoxicarbonil)piperidin-4-il)-2-(4-fluorofenil)acético (250 mg, 741 |jmol, MFCD17214722, A Chemtek) en Dm F (2 ml) se le añadió CDI (120 mg, 741 jmol) y se agitó la mezcla a t. a. durante 30 minutos. A la solución transparente se le añadió una solución de (Z)-2,2,2-trifluoro-N'-hidroxiacetimidamida (94,9 mg, 741 jmol) en DMF (0,5 ml) y se continuó agitando a t. a. durante 1 hora. Se calentó la mezcla de reacción en un microondas a 120 °C durante 30 minutos seguido de calentamiento a 150 °C durante 30 minutos. Se vertió la mezcla de reacción sobre agua y EtOAc y se separaron las capas. Se extrajo dos veces la capa acuosa con EtOAc. Se lavaron dos veces las capas orgánicas con agua, se secó sobre MgSO4, se filtró, se trató con gel de sílice y se evaporó. Se purificó el compuesto por cromatografía en gel de sílice en una columna de 12 g usando un sistema MPLC que se eluyó con un gradiente de n-heptano: EtOAc (de 100: 0 a 0: 100) para proporcionar el compuesto deseado como una espuma incolora (0,155 g; 48,7 %). EM (ESI): m/z = 428,3 [M-H]'.
BB26
(4aR,8aS)-6-[3-[(4-fluorofenil)-(2,2,2-trifluoroetoxi)metil]acetidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona
Se obtuvo el producto de forma análoga al procedimiento A4 usando 2,2,2-trifluoroacetato de 3-[(4-fluorofenil)-(2,2,2-trifluoroetoxi)metil]acetidina para obtener el producto deseado como sólido blanco. EM (ESI): m/z = 446,2 [M-C4H8+H]+. Se usó el producto en la etapa siguiente sin purificación adicional.
Intermedios:
a) 2,2,2-trifluoroacetato de 3-[(4-fluorofenil)-(2,2,2-trifluoroetoxi)metil]acetidina
A una solución de 3-[(4-fluorofenil)-(2,2,2-trifluoroetoxi)metil]acetidina-1-carboxilato de terc-butilo (1000,0 mg, 2,75 mmol) en DCM (20 ml) se le añadió TFA (2,0 ml, 2,75 mmol), se agitó la mezcla a 20 °C durante 12 h. Se concentró la mezcla, se purificó el residuo por HPLC prep. (TFA) para obtener el producto deseado como sólido blanquecino (608,8 mg, 57,6 %). EM (ESI): m/z = 264,1 [M+H]+.
b) 3-[(4-fluorofenil)-(2,2,2-trifluoroetoxi)metil]acetidina-1-carboxilato de terc-butilo
A una solución de 3-[(4-fluorofenil)-hidroxi-metil]acetidina-1-carboxilato de terc-butilo (3000,0 mg, 10,66 mmol), 2,2,2-trifluoroetanol (1,55 ml, 21,33 mmol) y trifenilfosfina (4195,52 mg, 16 mmol) en Th F (50 ml) se le añadió azodicarboxilato de diisopropilo (3,15 ml, 16 mmol) y se agitó la mezcla a 80 °C durante 12 h. Se concentró la mezcla y se purificó el residuo por ultrarrápida invertida (FA) para dar el producto deseado como aceite amarillo (1 g, 25,8 %). EM (ESI): m/z = 308,1 [M-C4Hs+H]+.
c) 3-[(4-fluorofenil)-hidroxi-metil]acetidina-1-carboxilato de terc-butilo
A una solución de 3-(4-fluorobenzoil)acetidina-1-carboxilato de terc-butilo (2 g, 7,16 mmol, MFCD24368873, FCH Group) en MeOH (30 ml) se le añadió NaBH4 (541,33 mg, 14,32 mmol) y se agitó la mezcla a 20 °C durante 1 h. Se concentró la mezcla y se disolvió en EtOAc (100 ml), se lavó con NH4Cl ac. y salmuera, se secó sobre Na2SO4 y se concentró para dar el producto deseado como aceite amarillo claro (2 mg, 99,3 %). Se usó el compuesto en la etapa siguiente sin ninguna purificación adicional.
BB27
(4aR,8aS)-6-[3-[1-(2-cloro-4-fluoro-fenil)etoxi]acetidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona
Se obtuvo el producto de forma análoga al procedimiento A4 usando 2,2,2-trifluoroacetato de 3-[1-(2-cloro-4-fluorofenil)etoxi]acetidina para dar el producto deseado como sólido blanquecino (230 mg, 59,8 %). EM (ESI): m/z = 412,2 [M+H]+.
Intermedios:
a) 2,2,2-trifluoroacetato de 3-[1-(2-cloro-4-fluoro-fenil)etoxi]acetidina
Se obtuvo el producto de forma análoga a BB26, intermedio a usando 3-[1-(2-cloro-4-fluoro-fenil)etoxi]acetidin-1-carboxilato de terc-butilo para dar el producto deseado como goma amarillo claro (1,4 g, 67,2 %). EM (ESI): m/z = 230,1 [M+H]+.
b) 3-[1-(2-cloro-4-fluoro-fenil)etoxi]acetidin-1-carboxilato de terc-butilo
Se añadió hidruro de sodio (1,54 g, 38,53 mmol) a 0 °C a una mezcla de 3-hidroxiacetidina-1-carboxilato de tercbutilo (5,34 g, 30,82 mmol, CAS RN 141699-55-0) en DMF (60 ml). Se agitó la mezcla a 20 °C durante 1 h. A continuación se añadió 1-(1-bromoetil)-2-cloro-4-fluoro-benceno (6,1 g, 25,68 mmol, CAS RN 1341821-29-1) y se agitó la mezcla a 20 °C durante 12 h. Se vertió la mezcla en agua con hielo (200 ml) y se extrajo tres veces con EtOAc (100 ml cada vez). Se lavó la capa orgánica combinada con salmuera (100 ml), se secó sobre Na2SO4 y se filtró. Se concentró el filtrado. Se purificó el residuo por HPLC prep. (FA) y se concentró a vacío para dar el producto deseado como aceite amarillo claro (2 g, 23,6 %). EM (ESI): m/z = 230 [M+H-Boc]+.
BB28
4-metilbencenosulfonato de 3-(2-cloro-[1,1 '-bifenil]-4-il)acetidina
A una solución de 3-(2-cloro-[1,1'-bifenil]-4-il)acetidina-1-carboxilato de terc-butilo (60 mg, 174 |jmol) en EtOAc (2 ml) se le añadió ácido 4-metilbencenosulfónico hidrato (39,8 mg, 209 jmol) y se calentó la mezcla a reflujo durante 1 hora. Se evaporó la solución transparente e incolora para obtener el producto deseado como un sólido incoloro (0,10 g; 100 %). EM (ESI): m/z = 244,2 [M+H]+.
Intermedio:
3- (2-cloro-[1,1 '-bifenil]-4-il)acetidin-1-carboxilato de terc-butilo
Se obtuvo el producto de forma análoga al procedimiento A5 a partir de ácido fenilborónico (CAS RN 98-80-6) y 3-(4-bromo-3-clorofenil)acetidina-1-carboxilato de terc-butilo (CAS RN 2222937-56-4 como un sólido incoloro. EM (ESI): m/z = 288,2 [M-C4Hs+H]+.
BB29
4- metilbencenosulfonato de 3-(4-bromo-3-clorofenil)acetidina
Se obtuvo el producto de forma análoga a BB28 a partir de 3-(4-bromo-3-clorofenil)acetidina-1-carboxilato de
terc
-butilo (CAS RN 2222937-56-4) como un sólido incoloro. EM (ESI): 248,1 [M+H]+.
BB30
4-metilbencenosulfonato de 3-(3-bromofenil)acetidina
Se obtuvo el producto de forma análoga a BB28 a partir de 3-(3-bromofenil)acetidina-1-carboxilato de terc-butilo (CAS RN 1203681-54-2) como un sólido incoloro. EM (ESI): 212,1 [M+H]+.
BB31
3- (4-terc-butilfenil)acetidina; ácido 4-metilbencenosulfónico
Se obtuvo el producto de forma análoga a BB28 a partir de 3-(4-(terc-butil)fenil)acetidina-1-carboxilato de
terc-
butilo (CAS RN 1629889-13-9) como un sólido incoloro. EM (ESI): 190,2 [M+H]+.
BB32
4- metilbencenosulfonato de 5-(acetidin-3-il)-2-cloropiridina
Se obtuvo el producto de forma análoga a BB28 a partir de 3-(6-cloropiridin-3-il)acetidina-1-carboxilato de
terc
-butilo (CAS RN 870689-19-3) como un sólido incoloro. EM (ESI): 169,1 [M+H]+.
BB33
4-metilbencenosulfonato de 3-(4-(trifluorometil)fenil)acetidina
Se obtuvo el producto de forma análoga a BB28 a partir de 3-(4-(trifluorometil)fenil)acetidina-1-carboxilato de
terc-
butilo (CAS RN 1638255-66-9) como un sólido incoloro. EM (ESI): 202,2 [M+H]+.
BB34
Clorhidrato de 3-((1,1,1-trifluoro-2-metilpropan-2-il)oxi)acetidina
A una solución de 1 -benzhidril-3-(( 1,1,1 -trifluoro-2-metilpropan-2-il)oxi)acetidina (114 mg, 326 |jmol) en etanol (0,5 ml) y EtOAc (0,5 ml) se le añadieron HCl 1 M en H2O (326 jl, 326 jmol) y Pd/C al 10 % (20 mg, 326 jmol) y se agitó la mezcla en atmósfera de hidrógeno a 1,7 bar durante 5 h. Se filtró la suspensión sobre un microfiltro y se evaporó el filtrado para obtener el compuesto deseado como un amorfo incoloro (0,105 g). EM (ESI): m/z = 184,2 [M+H]+. Usado sin purificación adicional en la etapa siguiente.
Intermedio:
Benzhidrilo-3-((1,1,1-trifluoro-2-metilpropan-2-il)oxi)acetidina
A una solución de 1,1,1-trifluoro-2-metil-propan-2-ol (807 mg, 690 jl, 6,3 mmol, CAS RN 507-52-8) en DMF (5 ml) bajo argón se le añadió hidruro de sodio al 60 % en aceite de vaselina (252 mg, 6,3 mmol) y se agitó la mezcla a t. a. durante 30 minutos. Después de la adición de metanosulfonato de (1-benzhidrilacetidin-3-il) (1 g, 3,15 mmol, CAS RN 33301-41-6), se calentó la mezcla de reacción en un horno de microondas a 130 °C durante 60 minutos. Se vertió la mezcla de reacción sobre agua y EtOAc y se separaron las capas. Se extrajo dos veces la capa acuosa con EtOAc. Se lavaron una vez las capas orgánicas con agua, se secó sobre MgSO4, se filtró, se trató con gel de sílice y se evaporó. Se purificó el compuesto por cromatografía en gel de sílice en una columna de 12 g usando un sistema MPLC que se eluyó con un gradiente de n-heptano: EtOAc (de 100: 0 a 50: 50) para producir el compuesto deseado como un sólido amarillo (0,324 g; 29,4 %). Em (ESI): m/z = 350,3 [M+H]+.
BB35
4-metilbencenosulfonato de 3-(4-( 1,1-difluoroetil)fenil)acetidina
Se obtuvo el producto de forma análoga a BB28 a partir de 3-(4-(1, 1 -difluoroetil)fenil)acetidin-1 -carboxilato de
terc-
butilo como un sólido incoloro (0,050 g; 35,9 %). EM (ESI): m/z = 198,2 [M+H]+.
Intermedio:
3-(4-( 1,1-difluoroetil)fenil)acetidina-1-carboxilato de terc-butilo
A una suspensión agitada de 2-(4-(1,1-difluoroetil)fenil)-4,4,5,5-tetrametil-1,3,2-dioxaborolano (568 mg, 2,12 mmol, CAS RN 1000994-94-4) en 2-propanol (1,5 ml) se le añadió una solución de 3-yodoacetidina-1-carboxilato de tercbutilo (300 mg, 1,06 mmol, c As Rn 254454-54-1) en 2-propanol (1,5 ml) a t. a., para dar una solución. A la mezcla se le añadió rac-(1R,2R)-2-aminociclohexan-1-ol (7,32 mg, 63,6 jmol, Ca s RN 13374-31-7), yoduro de níquel(II) (19,9 mg, 63,6 jmol) y bis(trimetilsilil)amida sódica (1,06 ml, 2,12 mmol) bajo argón. Se calentó la mezcla en un horno de microondas durante 30 minutos a 100 °C.
Se vertió la mezcla de reacción sobre agua y EtOAc y se separaron las capas. Se extrajo dos veces la capa acuosa con EtOAc. Se secaron las capas orgánicas sobre MgSO4, se filtró, se trató con gel de sílice y se evaporó. Se
purificó el compuesto por cromatografía en gel de sílice en una columna de 4 g usando un sistema MPLC que se eluyó con un gradiente de n-heptano: EtOAc (de 100: 0 a 70: 30) para proporcionar el compuesto deseado como un aceite incoloro (0,0112 g; 35,5 %). EM (ESI): m/z = 242,2 [M-C4Hs+H]+.
BB36
4-metilbencenosulfonato de 5-(4-(acetidin-3-il)fenil)-1-metil-1H-pirazol
Se obtuvo el producto de forma análoga a BB28 a partir de 3-(4-(1-metil-1H-pirazol-5-il)fenil)acetidin-1-carboxilato de terc-butilo como una goma marrón claro que se usó en el. EM (ESI): m/z = 214,1 [M+H]+. Se usó el producto en la etapa siguiente sin purificación adicional.
Intermedio:
3-(4-( 1-metil-1H-pirazol-5-il)fenil)acetidin-1-carboxilato de terc-butilo
Se obtuvo el producto de forma análoga a BB35, intermedio, a partir de ácido (4-(1-metil-1H-pirazol-5-il)fenil)borónico (CAS RN 1487353-57-0) como una goma marrón claro. EM (ESI): m/z = 314,3 [M+H]+. Se usó el producto en la etapa siguiente sin purificación adicional.
BB37
Trifluoroacetato de 3-[4-(2,2,2-trifluoroetoxi)fenil]acetidina
Se obtuvo el producto de forma análoga a BB26, intermedio a, a partir de 3-[4-(2,2,2-trifluoroetoxi)fenil]acetidin-1-carboxilato de terc-butilo como un aceite amarillo claro. EM (ESI): m/z = 232,1 [M+H]+. Se usó el producto en la etapa siguiente sin purificación adicional.
Intermedio:
3-[4-(2,2,2-trifluoroetoxi)fenil]acetidina-1-carboxilato de terc-butilo
A una solución de 1-bromo-4-(2,2,2-trifluoroetoxi)benceno (0,45 g, 1,77 mmol, CAS RN 106854-77-7), 1-BOC-3-yodoacetidina (0,5 g, 1,77 mmol, CAS RN 254454-54-1), 1,10-fenantrolina (63,65 mg, 0,350 mmol, CAS RN 5144 89-8), NaBF4 (96,95 mg, 0,880 mmol, CAS RN 13755-29-8), NiCh glyme (38,8 mg, 0,180 mmol, CAS RN 29046 78-4) y Mn en polvo (194,06 mg, 3,53 mmol) en MeOH (10 ml) se le añadió 4-etilpiridina (94,62 mg, 0,880 mmol, CAS Rn 536-75-4). Se agitó la mezcla a 60 °C en atmósfera de N2 durante 16 h. Se filtró la mezcla y se concentró el filtrado, se purificó el residuo por cromatografía ultrarrápida inversa (FA) para obtener el producto deseado como espuma amarillo claro (60 mg, 10,2 %). EM (ESI): m/z = 276,0 [M-C4H8+H]+.
BB38
Ácido [4-[1-[(4aR,8aS)-3-oxo-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacina-6-carbonil]acetidina-3-il]fenil]borónico
A una solución de (4aR,8aS)-6-[3-(4-bromofenil)acetidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona (300,0 mg, 0,760 mmol, ejemplo 110) y PdCl2(dppf).CH2Cl2 (53,34 mg, 0,080 mmol) se le añadió acetato de potasio (224,04 mg, 2,28 mmol) y bis(pinacolato)diboro (289,84 mg, 1,14 mmol, CAS RN 73183 34-3), se purgó la reacción con nitrógeno y se agitó a 90 °C durante 12 h. Se diluyó la reacción con agua y se extrajo con EtOAc tres veces, se lavó la capa orgánica combinada con agua y salmuera, se secó sobre sulfato de sodio y se concentró a vacío para obtener un residuo amarillo, que se purificó con cromatografía de fase inversa (FA) para obtener el producto deseado como sólido amarillo claro (230 mg, 84,1 %). EM (ESI): m/z = 360,5 [M+H]+. BB39
Clorhidrato de 3-ciclopropil-5-((4-fluorofenil)(piperidin-4-il)metil)-1,2,4-oxadiazol
Se obtuvo el producto de forma análoga a BB16 a partir de 4-((3-ciclopropil-1,2,4-oxadiazol-5-il)(4-fluorofenil)metil)piperidina-1-carboxilato de terc-butilo como una espuma incolora (0,170 g; 84,7 %). EM (ESI): m/z = 302,3 [M+H]+.
Intermedios:
a) 4-((3-ciclopropil-1,2,4-oxadiazol-5-il)(4-fluorofenil)metil)piperidina-1-carboxilato de terc-butilo
Se obtuvo el producto de forma análoga a BB25, intermedio de ácido 2-(1-(terc-butoxicarbonil)piperidin-4-il)-2-(4fluorofenil)acético y N'-hidroxicidopropanocarboxamidina como una espuma incolora (0,240 g; 80,7 %). EM (ESI): m/z = 346,2 [M-C4Hs+H]+.
b) Ácido 2-(1-(terc-butoxicarbonil)piperidin-4-il)-2-(4-fluorofenil)acético
A una solución turbia de bromhidrato de ácido 2-(4-fluorofenil)-2-(piperidin-4-il)acético (535 mg, 1,55 mmol) en NaOH 1 M en H2O (3,09 ml, 3,09 mmol) se le añadió gota a gota una solución de dicarbonato de di-terc-butilo (368 mg, 391 pl, 1,68 mmol) en DME (5 ml) y se agitó la mezcla a t. a. durante 3 h. Se evaporó el DME. Se recogió el residuo en aprox. 1,2 ml de ácido cítrico al 10 % en agua (pH aprox. de 4) y EtOAc y se separaron las capas. Se extrajo una vez la capa acuosa con EtOAc. Se secaron las capas orgánicas sobre MgSO4, se filtró y evaporó para obtener el compuesto deseado como un sólido marrón claro (0,520 g; 99,6 %). EM (ESI): m/z = 336,3 [M-H]\ c) Bromhidrato del ácido 2-(4-fluorofenil)-2-(piperidin-4-il)acético
Se agitó una solución de 4-(ciano(4-fluorofenil)metil)piperidina-1-carboxilato de terc-butilo (555 mg, 1,74 mmol, CAS RN 1824014-64-3) en HBr al 48 % en agua (8,27 g, 5,55 ml, 49,1 mmol) a reflujo durante 4,5 horas. Se evaporó la mezcla. Se suspendió el sólido marrón claro en 2-propanol (2 ml), se homogeneizó y filtró. Se lavó tres veces la torta del filtro con 2-propanol (1 ml cada vez). Se evaporaron completamente las aguas madres y se secó durante 2 horas a alto vacío en presencia de P2O5 para proporcionar el producto deseado como un sólido marrón claro (0,535 g; 96,5 %). EM (ESI): m/z = 238,2 [M-HBr+H]+.
BB40
4-metilbencenosulfonato de 3-(3-(trifluorometoxi)fenil)acetidina
Se obtuvo el producto de forma análoga a BB28 a partir de 3-(3-(trifluorometoxi)fenil)acetidina-1-carboxilato de terc-butilo (CAS RN 2222044-21-3) como un sólido incoloro. EM (ESI): 218,1 [M+H]+.
BB41
4-metilbencenosulfonato de [4-(acetidin-3-il)fenil]-pentafluoro-lambda6-sulfano
Se obtuvo el producto de forma análoga a BB28 a partir de 3-(4-(pentafluoro-16-sulfaneil)fenil)acetidina-1-carboxilato de terc-butilo como un sólido incoloro. EM (ESI): 260,1 [M+H]+.
Intermedio:
3-(4-(pentafluoro-16-sulfaneil)fenil)acetidina-1-carboxilato de terc-butilo
Se obtuvo el producto de forma análoga a BB35, intermedio, a partir de ácido (4-(pentafluoro-16-sulfaneil)fenil)borónico (CAS RN 871507-70-9) como un aceite incoloro. EM (ESI): m/z = 304,1 [M-C4Hs+H]+. BB42
bis(4-metilbencenosulfonato) de 2-(acetidin-3-il)-5-cloropiridina
Se obtuvo el producto de forma análoga a BB28 a partir de 3-(5-cloropiridin-2-il)acetidin-1-carboxilato de terc-butilo y dos veces la cantidad de p-TsOH como un sólido incoloro. EM (ESI): m/z = 169,1 [M+H]+.
Intermedio:
3- (5-cloropiridin-2-il)acetidin-1-carboxilato de terc-butilo
Se obtuvo el producto de forma análoga a BB37, intermedio, a partir de ácido (5-cloropiridin-2-il)borónico y 3-yodoacetidina-1-carboxilato de terc-butilo como un aceite incoloro (0,045 g; 11,2 %). EM (ESI): m/z = 213,1 [M-C4H8+H]+.
BB43
4- metilbencenosulfonato de 3-(2-fluoro-4-(trifluorometoxi)fenil)acetidina
Se obtuvo el producto de forma análoga a BB28 a partir de 3-(2-fluoro-4-(trifluorometoxi)fenil)acetidina-1-carboxilato de terc-butilo como un sólido incoloro. EM (ESI): m/z = 236,1 [M+H]+.
Intermedio:
3-(2-fluoro-4-(trifluorometoxi)fenil)acetidina-1-carboxilato de terc-butilo
Se obtuvo el producto de forma análoga a BB35, intermedio, a partir de ácido (2-fluoro-4-(trifluorometoxi)fenil)borónico (CAS RN 503309-10-2) como un aceite incoloro. EM (ESI): m/z = 280,1 [M-C4Hs+H]+. BB44
Trifluoroacetato de 3-[4-(2,2,2-trifluoroetil)fenil]acetidina
Se obtuvo el producto de forma análoga a BB26, intermedio a, a partir de 3-[4-(2,2,2-trifluoroetil)fenil]acetidina-1-carboxilato de terc-butilo como un aceite amarillo. EM (ESI): m/z = 216,2 [M+H]+.
Intermedio:
3-[4-(2,2,2-trifluoroetil)fenil]acetidina-1-carboxilato de terc-butilo
Se obtuvo el producto de forma análoga a BB37, intermedio a, a partir de 1-bromo-4-(2,2,2-trifluoroetil)benceno (CAS RN 155820-88-5) como un aceite amarillo. EM (ESI): m/z = 260,0 [M-C4Hs+H]+.
BB45
Trifluoroacetato de 3-[4-[1-(trifluorometil)ciclopropil]fenil]acetidina
Se obtuvo el producto de forma análoga a BB26, intermedio a, a partir de 3-[4-[1-(trifluorometil)ciclopropil]fenil]acetidina-1 -carboxilato de terc-butilo como un aceite amarillo. EM (ESI): m/z = 242,1 [M+H]+.
Intermedio:
3- [4-[1-(trifluorometil)ciclopropil]fenil]acetidina-1-carboxilato de terc-butilo
Se obtuvo el producto de forma análoga a BB37, intermedio, a partir de 1-bromo-4-[1-(trifluorometil)ciclopropil]benceno (CAS RN 1227160-18-0) como un aceite amarillo. Em (ESI): m/z = 286,0 [M-C4H8+H]+.
BB46
4- metilbencenosulfonato de 3-(3-fluoro-4-(trifluorometoxi)fenil)acetidina
Se obtuvo el producto de forma análoga a BB28 a, a partir de 3-(3-fluoro-4-(trifluorometoxi)fenil)acetidina-1-carboxilato de terc-butilo como un sólido incoloro. EM (ESI): m/z = 236,1 [M+H]+.
Intermedio:
3- (3-fluoro-4-(trifluorometoxi)fenil)acetidina-1-carboxilato de terc-butilo
Se obtuvo el producto de forma análoga a BB37, intermedio, a partir de 4-bromo-2-fluoro-1-(trifluorometoxi)benceno (CAS RN 105529-58-6) como un aceite incoloro. EM (ESI): m/z = 280,1 [M-C4Hs+H]+. BB47
4- metilbencenosulfonato de 3-(3-metil-4-(trifluorometoxi)fenil)acetidina
Se obtuvo el producto de forma análoga a BB28 a, a partir de 3-(3-metil-4-(trifluorometoxi)fenil)acetidina-1-carboxilato de terc-butilo como un sólido incoloro. EM (ESI): m/z = 232,1 [M+H]+.
Intermedio:
3- (3-metil-4-(trifluorometoxi)fenil)acetidina-1-carboxilato de terc-butilo
Se obtuvo el producto de forma análoga a BB37, intermedio, a partir de 4-bromo-2-metil-1-(trifluorometoxi)benceno (CAS RN 887268-26-0) como un aceite incoloro. EM (ESI): m/z = 276,2 [M-C4Hs+H]+.
BB48
4- metilbencenosulfonato de 3-(3,5-difluoro-4-(trifluorometoxi)fenil)acetidina
Se obtuvo el producto de forma análoga a BB28 a, a partir de 3-(3,5-difluoro-4-(trifluorometoxi)fenil)acetidina-1-carboxilato de terc-butilo como un sólido incoloro. EM (ESI): m/z = 254,1 [M+H]+.
Intermedio:
3- (3,5-difluoro-4-(trifluorometoxi)fenil)acetidina-1-carboxilato de terc-butilo
Se obtuvo el producto de forma análoga a BB37, intermedio a, a partir de 5-bromo-1,3-difluoro-2-(trifluorometoxi)benceno (CAS RN 115467-07-7) como un sólido incoloro. EM (ESI): m/z = 298,1 [M-C4Hs+H]+. BB49
4- metilbencenosulfonato de 3-(2-cloro-4-(trifluorometoxi)fenil)acetidina
Se obtuvo el producto de forma análoga a BB28 a, a partir de 3-(2-cloro-4-(trifluorometoxi)fenil)acetidina-1-carboxilato de terc-butilo como un sólido incoloro. EM (ESI): m/z = 252,1 [M+H]+.
Intermedio:
3- (2-cloro-4-(trifluorometoxi)fenil)acetidina-1-carboxilato de terc-butilo
Se obtuvo el producto de forma análoga a BB37, intermedio a, a partir de 1-bromo-2-cloro-4-(trifluorometoxi)benceno (CAS RN 892845-59-9) como un aceite incoloro. EM (ESI): m/z = 296,1 [M-C4Hs+H]+. BB50
4- metilbencenosulfonato de 3-(4-(bicido[1.1.1]pentan-1-il)fenil)acetidina
Se obtuvo el producto de forma análoga a BB28 a, a partir de 3-(4-(biciclo[1.1.1]pentan-1-il)fenil)acetidina-1-carboxilato de terc-butilo como un sólido incoloro. EM (ESI): m/z = 200,2 [M+H]+.
Intermedio:
3- (4-(biciclo[1.1.1]pentan-1-il)fenil)acetidina-1-carboxilato de terc-butilo
Se obtuvo el producto de forma análoga a BB37, intermedio a, a partir de 1-(4-bromofenil)biciclo[1.1.1]pentano (CAS RN 1823935-76-7) como un aceite incoloro. EM (ESI): m/z = 244,2 [M-C4Hs+H]+.
BB51
4- metilbencenosulfonato de 5-(acetidin-3-il)-2-(trifluorometoxi)benzonitrilo
Se obtuvo el producto de forma análoga a BB28 a partir de 3-(3-ciano-4-(trifluorometoxi)fenil)acetidina-1-carboxilato de terc-butilo como una goma marrón claro. e M (ESI): m/z = 243,1 [M+H]+. Se usó el compuesto en la etapa siguiente sin purificación adicional.
Intermedio:
3-(3-ciano-4-(trifluorometoxi)fenil)acetidina-1-carboxilato de terc-butilo
Se obtuvo el producto de forma análoga a BB37, intermedio a, a partir de 5-bromo-2-(trifluorometoxi)benzonitrilo (CAS RN 1210906-15-2) como un aceite amarillo claro. EM (ESI): m/z = 287,1 [M-C4Hs+H]+.
BB52
Clorhidrato de (1-(4-(acetidin-3-il)fenil)ciclopropil)metanol
A una solución de 3-(4-(1-(hidroximetil)ciclopropil)fenil)acetidina-1-carboxilato de terc-butilo (40 mg, 132 |jmol) en DCM (0,7 ml) se le añadió HCl en dioxano 4 M (330 jl, 1,32 mmol) y se agitó la mezcla a t. a. durante 3 horas. A la suspensión amarillo claro se le añadió éter dietílico (2 ml) y se evaporó la mezcla oleosa para obtener el compuesto deseado como un aceite amarillo claro. EM (ESI): m/z = 204,2 [M+H]+. Se usó el compuesto en la etapa siguiente sin purificación adicional.
Intermedios:
a) 3-(4-(1-(hidroximetil)dclopropil)fenil)acetidina-1-carboxilato de terc-butilo
A una solución helada de 3-(4-(1-(metoxicarbonil)cidopropil)fenil)acetidina-1-carboxilato de terc-butilo (238 mg, 718 |jmol) en THF (2 ml) se le añadió gota a gota una solución de LAH 1 M en THF (718 jl, 718 |jmol). Se agitó la solución a 0 °C durante 1,25 horas y a continuación se vertió sobre una solución de NH4Cl acuosa semisaturada y EtOAc y se separaron las capas. Se extrajo dos veces la capa acuosa con EtOAc. Se secaron las capas orgánicas sobre MgSO4, se filtró, se trató con gel de sílice y se evaporó. Se purificó el compuesto por cromatografía en gel de sílice en una columna de 4 g usando un sistema MPLC que se eluyó con un gradiente de n-heptano: EtOAc (de 100: 0 a 50: 50) para proporcionar el compuesto deseado como un aceite incoloro (0,151 g; 69,3 %). EM (ESI): m/z = 248,2 [M-C4Hs+H]+.
b) 3-(4-(1-(metoxicarbonil)ciclopropil)fenil)acetidina-1-carboxilato de terc-butilo
Se obtuvo el producto de forma análoga al procedimiento A7 a partir de 1-(4-bromofenil)ciclopropano-1-carboxilato de metilo (c As RN 638220-35-6) y 3-bromoacetidina-1-carboxilato de terc-butilo (CAS RN 1064194-10-0) como un sólido incoloro. EM (ESI): m/z = 276,2 [M-C4Hs+H]+.
BB53
Clorhidrato de 4-(acetidin-3-il)-1-metil-1H-indazol
Se obtuvo el producto de forma análoga a BB52 a, a partir de 3-(1-metil-1H-indazol-4-il)acetidina-1-carboxilato de terc-butilo como un sólido amarillo claro. EM (ESI): m/z = 188,2 [M+H]+.
Intermedios:
a) 3-(1-metil-1H-indazol-4-il)acetidina-1-carboxilato de terc-butilo
Se obtuvo el producto de forma análoga al procedimiento A7 a partir de 4-bromo-1-metil-1H-indazol (CAS RN 365427-30-1) y 3-bromoacetidina-1-carboxilato de terc-butilo (c As RN 1064194-10-0) como un aceite marrón claro. EM (ESI): m/z = 232,1 [M-C4H8+H]+.
BB54
Trifluoroacetato de 3-[4-(trifluorometoxi)fenil]pirrolidina
Se obtuvo el producto de forma análoga a BB26, intermedio a, a partir de 3-[4-(trifluorometoxi)fenil]pi rrolidina-1-carboxilato de terc-butilo como sólido marrón claro. EM (ESI): m/z = 232,6 [M+H]+.
Intermedios:
a) 3-[4-(trifluorometoxi)fenil]pirrolidina-1-carboxilato de terc-butilo
Se obtuvo el producto de forma análoga a BB17, intermedio a, a partir de 3-[4-(trifluorometoxi)fenil]-2,5-dihidropirrol-1-carboxilato de terc-butilo como aceite amarillo claro (200 mg, 66,3 %). EM (ESI): m/z = 276,5 [M-C4H8+H]+.
b) 3-[4-(trifluorometoxi)fenil]-2,5-dihidropirrol-1-carboxilato de terc-butilo
A una solución de 3-(((trifluorometil)sulfonil)oxi)-2,5-dihidro-1 H-pirrol-1 -carboxilato de terc-butilo (1 g, 3,15 mmol, CAS RN 630121-86-7) y ácido 4-(trifluorometoxi)fenilborónico (973,57 mg, 4,73 mmol, CAS r N 139301-27-2), Na2CO3 (668,1 mg, 6,3 mmol) en 1,4-dioxano (20 ml) y agua (5 ml) se le añadió [1,1'-bis(difenilfosfino)ferroceno]dicloropaladio(II) (461,24 mg, 0,630 mmol) y se agitó la mezcla a 100 °C en atmósfera de N2 durante 12 h. Se concentró la reacción y se purificó por cromatografía en gel de sílice (PE:EtOAc=20:1) para obtener el producto deseado como aceite amarillo claro (320 mg, 30,8 %). EM (ESI): m/z = 274,5 [M-C4Hs+H]+. BB55
Estannano de tributil-(3-cloro-2-piridilo)
A una solución de 2-bromo-3-cloropiridina (576,0 mg, 2,99 mmol, CAS RN 96424-68-9) en tolueno (20 ml) bajo N2 se le añadió gota a gota n-BuLi 2,5 M en hexano (1,32 ml, 3,29 mmol) a -78 °C. Se agitó la mezcla de reacción durante 2 h, seguido de la adición de cloruro de tributilestaño (1071,73 mg, 3,29 mmol, CAS RN 1461-22-9). Se agitó la mezcla de reacción durante 2 h a -78 °C, se calentó a t. a. y se agitó otras 12 h y, a continuación, se detuvo con una solución de NH4Cl saturada (50 ml). Se extrajo la mezcla con EtOAc (30 ml tres veces) y se lavaron las capas orgánicas combinadas con salmuera (15 ml), se secó (Na2SO4) y se filtró. Se concentró el filtrado a vacío
hasta obtener el producto deseado como un aceite amarillo claro (1,1 g, 91,3 %). EM (ESI): m/z = 404,1 [M+H]+. BB56
Clorhidrato de 6-(acetidin-3-il)-1-metil-1H-indazol
Se obtuvo el producto de forma análoga a BB52 a, a partir de 3-(1-metil-1H-indazol-6-il)acetidina-1-carboxilato de terc-butilo como un sólido incoloro. EM (ESI): m/z = 188,1 [M+H]+.
Intermedios:
a) 3-(1-metil-1H-indazol-6-il)acetidina-1-carboxilato de terc-butilo
Se obtuvo el producto de forma análoga al procedimiento A7 a partir de 6-bromo-1-metil-1H-indazol (CAS RN 365427-30-1) y 3-bromoacetidina-1-carboxilato de terc-butilo (CAS RN 1064194-10-0) como un aceite amarillo claro. EM (ESI): m/z = 288,2 [M+H]+.
BB57
(4aR, 8aS)-6-[3-[3-(trifluorometoxi)fenil]pirrolidina-1-carbonil]-4,4a, 5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona Se obtuvo el producto de forma análoga al procedimiento A4 usando 2,2,2-trifluoroacetato de 3-[3-(trifluorometoxi)fenil]pirrolidina para obtener el producto deseado como sólido amarillo claro. EM (ESI): m/z = 414,3 [M+H]+.
Intermedios:
a) Trifluoroacetato de 3-[3-(trifluorometoxi)fenil]pirrolidina
Se obtuvo el producto de forma análoga a BB26, intermedio a, a partir de 3-[3-(trifluorometoxi)fenil]pi rrolidina-1-carboxilato de terc-butilo como aceite marrón claro. EM (ESI): m/z = 232,6 [M+H]+.
b) 3-[3-(trifluorometoxi)fenil]pirrolidina-1-carboxilato de terc-butilo
Se obtuvo el producto de forma análoga a BB54, intermedio a, a partir de 3-[3-(trifluorometoxi)fenil]-2,5-dihidropirrol-1-carboxilato de terc-butilo para obtener el producto deseado como aceite amarillo claro. EM (ESI): m/z = 276,5 [M-C4Hs+H]+.
c) 3-[3-(trifluorometoxi)fenil]-2,5-dihidropirrol-1-carboxilato de terc-butilo
Se obtuvo el producto de forma análoga a BB54, intermedio b, a partir de ácido 3-(trifluorometoxi)fenilborónico (CAS RN 179113-90-7) para obtener el producto deseado como un aceite marrón claro. EM (ESI): m/z = 274,5 [M-C4Ha+H]+.
BB58
2,2,2-trifluoroacetato de 2-metil-3-(4-(trifluorometoxi)fenil)acetidina
Se obtuvo el producto de forma análoga a BB26, intermedio a, a partir de 2-metil-3-(4-(trifluorometoxi)fenil)acetidina-1-carboxilato de terc-butilo como un producto bruto. EM (ESI): m/z = 232,2 [M+H]+.
Intermedios:
a) 2-metil-3-(4-(trifluorometoxi)fenil)acetidina-1-carboxilato de terc-butilo
Se cargó un tubo de microondas con una solución de (Z)-3-(2-((4-metoxifenil)sulfonil)hidracinalideno)-2-metilacetidina-1-carboxilato de terc-butilo (265,5 mg, 719 |jmol) y ácido (4-(trifluorometoxi)fenil)borónico (222 mg, 1.08 mmol, CAS RN 139301-27-2) en dioxano (2,87 ml). A la solución se le añadió carbonato de cesio (351 mg, 1.08 mmol). Se desgasificó la MR con argón, se selló el vial y se calentó mientras se agitaba a 110 °C durante 18 h. Se enfrió la MR hasta t. a. antes de detenerla con 2 ml de solución de NaHCO3 acuosa saturada y se extrajo tres veces con DCM. Se secaron las capas orgánicas combinadas sobre MgSO4 y se concentró a vacío proporcionando el producto deseado como un aceite amarillo (71,1 mg; 29,9 %). EM (ESI): m/z = 276,2 [M-C4H8+H]+.
b) (Z)-3-(2-((4-metoxifenil)sulfonil)hidracinalideno)-2-metilacetidina-1-carboxilato de terc-butilo
Se calentó una solución de 4-metoxibencenosulfonhidracida (180 mg, 890 |jmol, CAS RN 1950-68-1) y 2-metil-3-oxoacetidina-1-carboxilato de terc-butilo (165 mg, 890 jmol, CAS RN 1408076-36-7) en DMSO d6 (593 jl) hasta 60 °C mientras se agitaba durante 1 h. Se enfrió la mezcla de reacción hasta t. a. y se vertió sobre H2O agitada, liberando un precipitado blanco. Se filtró el precipitado y se volvió a disolver en MeOH. Se retiró el disolvente a vacío proporcionando el producto deseado como un aceite amarillo (265,5 mg; 80,7 %). EM (ESI): m/z = 368,3 [M-H]-.
BB59
4-metilbencenosulfonato de 3-(3,3-dimetil-2,3-dihidrobenzofuran-6-il)acetidina
Se obtuvo el producto de forma análoga a BB28 a partir de 3-(3,3-dimetil-2,3-dihidrobenzofuran-6-il)acetidina-1-carboxilato de terc-butilo como un sólido incoloro. e M (ESI): m/z = 204,2 [M+H]+.
Intermedio:
3- (3,3-dimetil-2,3-dihidrobenzofuran-6-il)acetidina-1-carboxilato de terc-butilo
Se obtuvo el producto de forma análoga al procedimiento A7 a partir de 6-bromo-3,3-dimetil-2,3-dihidrobenzofurano (CAS RN 140896-85-1) como un sólido incoloro. EM (ESI): m/z = 248,2 [M-C4Hs+H]+.
BB60
4- metilbencenosulfonato de 3-(3-cloro-5-(2,2,2-trifluoroetoxi)fenil)pirrolidina
Se obtuvo el producto de forma análoga a BB28 a partir de 3-(3-cloro-5-(2,2,2-trifluoroetoxi)fenil)pirrolidina-1-carboxilato de terc-butilo como un aceite incoloro. EM (ESI): m/z = 280,1 [M+H]+.
Intermedios:
a) 3-(3-cloro-5-(2,2,2-trifluoroetoxi)fenil)pirrolidina-1-carboxilato de terc-butilo
Se obtuvo el producto de forma análoga al procedimiento A7 a partir de 1-bromo-3-cloro-5-(2,2,2-trifluoroetoxi)benceno como un aceite incoloro. EM (ESI): m/z = 324,1 [M-C4Hs+H]+.
b) 1-bromo-3-cloro-5-(2,2,2-trifluoroetoxi)benceno
A una solución de 3-bromo-5-clorofenol (330 mg, 1,59 mmol, CAS RN 56962-04-0) y trifluorometanosulfonato de 2,2,2-trifluoroetilo (554 mg, 2,39 mmol) en DMF (3 ml) se le añadió carbonato de potasio (440 mg, 3,18 mmol) y se agitó la mezcla a 50 °C durante el fin de semana. Después de enfriar, se vertió la mezcla de reacción sobre agua y EtOAc y se separaron las capas. Se extrajo dos veces la capa acuosa con EtOAc. Se lavaron dos veces las capas orgánicas con agua, se secó sobre MgSO4, se filtró y evaporó para proporcionar el compuesto deseado como un aceite incoloro (0,484 g; 100 %). Se usó el compuesto en la etapa siguiente sin purificación adicional. BB61
4-metilbencenosulfonato 2-(4-(acetidin-3-il)fenil)-2-metilpropanoato de metilo
Se obtuvo el producto de forma análoga a BB28 a partir de 3-(4-(1-metoxi-2-metil-1-oxopropan-2-il)fenil)acetidina-1-carboxilato de terc-butilo como una goma incolora. EM (ESI): m/z = 234,2 [M+H]+.
Intermedio:
3- (3,3-dimetil-2,3-dihidrobenzofuran-6-il)acetidina-1-carboxilato de terc-butilo
Se obtuvo el producto de forma análoga al procedimiento A7 a partir de 2-(4-bromofenil)-2-metilpropanoato de metilo (CAS RN 154825-97-5) como un aceite amarillo claro. EM (ESI): m/z = 278,2 [M-C4Hs+H]+.
BB62
4- metilbencenosulfonato de 3-(3,5-diclorofenil)pirrolidina
Se obtuvo el producto de forma análoga a BB28 a partir de 3-(3,5-diclorofenil)pirrolidin-1-carboxilato de terc-butilo como un aceite marrón. EM (ESI): m/z = 216,0 [M+H]+.
Intermedio:
3- (3,5-diclorofenil)pirrolidin-1-carboxilato de terc-butilo
Se obtuvo el producto de forma análoga al procedimiento A7 a partir de 1-bromo-3,5-diclorobenceno (CAS RN 19752-55-7) como un aceite incoloro. EM (e S i): m/z = 260,1 [M-C4H8+H]+.
BB63
2,2,2-trifluoroacetato de 2-[4-(acetidin-3-il)fenil]-5-(2,2-dimetilpropil)-1,3,4-oxadiazol
Se obtuvo el producto de forma análoga a BB26, intermedio a, a partir de 3-[4-[5-(2,2-dimetilpropil)-1,3,4-oxadiazol-2- il]fenil]acetidina-1-carboxilato de terc-butilo como un aceite marrón claro. EM (ESI): m/z = 272,6 [M+H]+.
Intermedios:
a) 3-[4-[5-(2,2-dimetilpropil)-1,3,4-oxadiazol-2-il]fenil]acetidina-1-carboxilato de terc-butilo
Se obtuvo el producto de forma análoga al procedimiento A7 a partir de 2-(4-bromofenil)-5-(2,2-dimetilpropil)-1,3,4-oxadiazol como un aceite marrón claro. e M (ESI): m/z = 372,5 [M+H]+.
b) 2-(4-bromofenil)-5-(2,2-dimetilpropil)-1,3,4-oxadiazol
A una solución de 4-bromo-N'-(3,3-dimetilbutanoil)benzohidracida (5 g, 15,96 mmol) en tolueno (102 ml) se le añadió ácido p-toluenosulfónico (5,5 g, 31,93 mmol), a continuación se agitó a 110 °C durante 12 h. La CLEM mostró que la reacción se había completado, se concentró la mezcla y se purificó el residuo por cromatografía en gel de sílice eluida con PE: EtOAc = 10: 1 para dar el producto deseado como un sólido amarillo claro (1 g, 21,2 %). EM (ESI): m/z = 295,4 [M+H]+.
c) 4-bromo-N-(3,3-dimetilbutanoil)benzohidracida
A una solución de 4-bromobenzohidracida (5,0 g, 23,25 mmol, CAS RN 5933-32-4) y DIPEA (12,39 ml, 69,75 mmol) en DCM (50 ml) se le añadió cloruro de 3,3-dimetilbutirilo (3,76 g, 27,9 mmol, CAS RN 7065-46-5) bajo 0 °C, se agitó la mezcla a 20 °C durante 12 h. Se extrajo la capa acuosa con EtOAc (200 ml, 3 veces) y agua (100 ml, 3 veces). Se lavó la capa orgánica separada con agua, se secó sobre Na2SO4 y se evaporó para dar el producto deseado como sólido amarillo claro (7 g, 96,1 %). EM (ESI): m/z = 315,4 [M+H]+.
BB64
4- metilbencenosulfonato de 3-(4-(terc-butil)-3-metoxifenil)acetidina
Se obtuvo el producto de forma análoga a BB28 a partir de 3-(4-(terc-butil)-3-metoxifenil)acetidina-1-carboxilato de terc-butilo como producto bruto. EM (ESI): m/z = 220,2 [M+H]+.
Intermedio:
3- (4-(terc-butil)-3-metoxifenil)acetidina-1-carboxilato de terc-butilo
Se obtuvo el producto de forma análoga al procedimiento A7 a partir de 4-bromo-1-(terc-butil)-2-metoxibenceno (CAS RN 30788-02-4) como un aceite amarillo claro. EM (ESI): m/z = 264,2 [M-C4H8+H]+.
BB65
4- metilbencenosulfonato de 5-(acetidin-3-il)-1-metil-1H-indazol
Se obtuvo el producto de forma análoga a BB28 a partir de 3-(1-metil-1H-indazol-5-il)acetidina-1-carboxilato de terc-butilo como producto bruto. EM (ESI): m/z = 188,1 [M+H]+.
Intermedio:
3-(1-metil-1H-indazol-5-il)acetidina-1-carboxilato de terc-butilo
Se obtuvo el producto de forma análoga al procedimiento A7 a partir de 5-bromo-1-metil-1H-indazol (CAS RN 465529-57-1) como un aceite amarillo. EM (ESI): m/z = 288,2 [M+H]+.
BB66
4-metilbencenosulfonato de 3-(4-propilfenil)acetidina
Se obtuvo el producto de forma análoga a BB28 a partir de 3-(4-propilfenil)acetidina-1-carboxilato de terc-butilo como producto bruto. EM (ESI): m/z = 176,1 [M+H]+.
Intermedio:
3- (4-propilfenil)acetidina-1-carboxilato de terc-butilo
Se obtuvo el producto de forma análoga al procedimiento A7 a partir de 1-bromo-4-propilbenceno (CAS RN 588 93-2) como un aceite amarillo claro. EM (ESI): m/z = 220,2 [M-C4Hs+H]+.
BB67
4- metilbencenosulfonato de 3-(4-(trifluorometoxi)-3-(trifluorometil)fenil)acetidina
Se obtuvo el producto de forma análoga a BB28 a partir de 3-(4-(trifluorometoxi)-3-(trifluorometil)fenil)acetidina-1-carboxilato de terc-butilo como producto bruto. EM (ESI): m/z = 286,1 [M+H]+.
Intermedio:
3- (4-(trifluorometoxi)-3-(trifluorometil)fenil)acetidina-1-carboxilato de terc-butilo
Se obtuvo el producto de forma análoga al procedimiento A7 a partir de 4-bromo-1-(trifluorometoxi)-2-(trifluorometil)benceno (CAS RN 933674-89-6) como un aceite amarillo claro. EM (ESI): m/z = 330,1 [M-C4H s +H]+. BB68
4- 2,2,2-trifluoroacetato de 3-[4-[[1-(trifluorometil)ciclopropil]metoxi]fenil]acetidina
Se obtuvo el producto de forma análoga a BB26, intermedio a, a partir de 3-[4-[[1-(trifluorometil)ciclopropil]metoxi]fenil]acetidina-1-carboxilato de terc-butilo como un aceite amarillo. EM (ESI): m/z = 272,1 [M+H]+.
Intermedio:
3-[4-[[1-(trifluorometil)ciclopropil]metoxi]fenil]acetidina-1-carboxilato de terc-butilo
Se obtuvo el producto de forma análoga al procedimiento A7 a partir de 1-bromo-4-[[1-(trifluorometil)ciclopropil]metoxi]benceno (CAS RN 1594130-28-5) como un aceite amarillo claro. EM (ESI): m/z = 316,1 [M-C4Hb+H]+.
BB69
2.2.2- trifluoroacetato de 3-[4-(2,2,2-trifluoro-1,1-dimetil-etil)fenil]acetidina
Se obtuvo el producto de forma análoga a BB26, intermedio a, a partir de 3-[4-(2,2,2-trifluoro-1, 1 -dimetiletil)fenil]acetidina-1-carboxilato de terc-butilo como un aceite amarillo. e M (ESI): m/z = 244,1 [M+H]+.
Intermedio:
3-[4-(2,2,2-trifluoro-1,1-dimetil-etil)fenil]acetidina-1-carboxilato de terc-butilo
Se obtuvo el producto de forma análoga al procedimiento A7 a partir de 1-bromo-4-(2,2,2-trifluoro-1,1-dimetiletil)benceno (Ca s RN 1225380-05-1) como un aceite amarillo claro. EM (ESI): m/z = 288,1 [M-C4Hs+H]+.
BB70
2.2.2- trifluoroacetato de 3-[4-(acetidin-3-il)fenil]-5-(2,2-dimetilpropil)-1,2,4-oxadiazol
Se obtuvo el producto de forma análoga a BB26, intermedio a, a partir de 3-[4-[5-(2,2-dimetilpropil)-1,2,4-oxadiazol-3-il]fenil]acetidina-1 -carboxilato de terc-butilo como un aceite amarillo claro. EM (ESI): m/z = 272,6 [M+H]+.
Intermedios:
a) 3-[4-[5-(2,2-dimetilpropil)-1,2,4-oxadiazol-3-il]fenil]acetidina-1-carboxilato de terc-butilo
A una solución de 3-[4-(N-hidroxicarbamimidoil)fenil]acetidina-1-carboxilato de terc-butilo (770,0 mg, 2,64 mmol) y DIPEA (1,41 ml, 7,93 mmol) en tolueno (6 ml) se le añadió cloruro de 3,3-dimetilbutirilo (426,88 mg, 3,17 mmol, CAS RN 7065-46-5) bajo 0D, se agitó la mezcla a 25 °C durante 10 min, a continuación se subió la temperatura hasta 80 °C y se agitó durante 12 h. Se evaporó la mezcla y se purificó el residuo por cromatografía en gel de sílice (PE: EtOAc = 10: 1) para dar el producto deseado como aceite marrón claro (620 mg, 63,2 %). EM (ESI): m/z = 316,5 [M-C4Hs+H]+.
b) 3-[4-(N-hidroxicarbamimidoil)fenil]acetidina-1-carboxilato de terc-butilo
A una solución de clorhidrato de hidroxilamina (349,71 mg, 5,03 mmol) en etanol (8 ml) se le añadió carbonato de sodio (266,69 mg, 2,52 mmol) en agua (2 ml) y se agitó a 20 °C durante 25 min. A continuación se añadió 3-(4-cianofenil)acetidina-1-carboxilato de terc-butilo (1000,0 mg, 3,87 mmol, CAS RN 206446-41-5), se agitó la mezcla a 95 °C durante 12 h.
Se diluyó la mezcla con agua, se concentró a vacío para retirar el EtOH, se repartió el residuo entre EtOAc (100 ml) con agua (100 ml x 3), a continuación se saturó con cloruro de sodio (50 ml), se secó sobre sulfato de sodio y se evaporó para dar el producto deseado como aceite amarillo claro (773 mg, 68,5 %). EM (ESI): m/z = 292,5 [M+H]+. BB71
(4aR,8aS)-6-[3-[4-(2,2,2-trifluoro-1-metil-etoxi)fenil]acetidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona
Se obtuvo el producto de forma análoga al procedimiento A4 a partir de 2,2,2-trifluoroacetato de 3-[4-(2,2,2-trifluoro-1-metil-etoxi)fenil]acetidina como un sólido amarillo claro. EM (ESI): m/z = 428,3 [M+H]+.
Intermedios:
a) 2,2,2-trifluoroacetato de 3-[4-(2,2,2-trifluoro-1-metil-etoxi)fenil]acetidina
Se obtuvo el producto de forma análoga a BB26, intermedio a, a partir de 3-[4-(2,2,2-trifluoro-1-metiletoxi)fenil]acetidina-1-carboxilato de terc-butilo como un aceite amarillo claro. EM (ESI): m/z = 246,1 [M+H]+. b) 3-[4-(2,2,2-trifluoro-1-metil-etoxi)fenil]acetidina-1-carboxilato de terc-butilo
Se obtuvo el producto de forma análoga al procedimiento A7 a partir de 1-bromo-4-(2,2,2-trifluoro-1-metiletoxi)benceno (CAS RN 1239611-43-8) como un sólido incoloro. EM (ESI): m/z = 290,1 [M-C4Hs+H]+.
BB72
4-metilbencenosulfonato de 3-(3-metoxi-4-metilfenil)acetidina
Se obtuvo el producto de forma análoga a BB28 a partir de 3-(3-metoxi-4-metilfenil)acetidina-1-carboxilato de tercbutilo como sólido incoloro. EM (ESI): m/z = 178,1 [M+H]+.
Intermedio:
3- (3-metoxi-4-metilfenil)acetidina-1-carboxilato de terc-butilo
Se obtuvo el producto de forma análoga al procedimiento A7 a partir de 4-bromo-2-metoxi-1-metilbenceno (CAS RN 67868-73-9) como un aceite marrón claro. EM (ESI): m/z = 222,1 [M-C4Hs+H]+.
BB73
Clorhidrato de 4-((4-fluorofenil)((1-metil-5-(trifluorometil)-1H-pirazol-3-il)oxi)metil)piperidina
Se obtuvo el producto de forma análoga a BB16 a partir de 4-((4-fluorofenil)((1-metil-5-(trifluorometil)-1 H-pirazol-3-il)oxi)metil)piperidina-1-carboxilato de terc-butilo como espuma incolora (0,194 g; 98,0 %). EM (ESI): m/z = 358,2 [M+H]+.
Intermedio:
4- ((4-fluorofenil)((1-metil-5-(trifluorometil)-1H-pirazol-3-il)oxi)metil)piperidina-1-carboxilato de terc-butilo A una solución amarilla de tri-n-butilfosfina (327 mg, 399 pl, 1,62 mmol) y dipiperidida azodicarboxílica (408 mg,
1,62 mmol) en tolueno (12,5 ml) se le añadió 4-((4-fluorofenil)(hidroxi)metil)piperidina-1-carboxilato de terc-butilo (250 mg, 808 |jmol, CAS RN 160296-41-3) y se agitó la mezcla a t. a. durante 20 min. Esto dio lugar a una solución amarillo pálido. La adición de 1-metil-5-(trifluorometil)-1 H-pirazol-3-ol (268 mg, 1,62 mmol, CAS RN 119022-51-4) dio una suspensión. Después de agitar a t. a. durante 75 min se instaló calentamiento. Después de agitar a 65 °C durante 19 h, se enfrió la mezcla de reacción a t. a. Se añadió gel de sílice y se evaporó la mezcla. Se purificó el compuesto por cromatografía en gel de sílice en una columna de 12 g usando un sistema MPLC (ISCO) que se eluyó con un gradiente de n-heptano: EtOAc (de 100: 0 a 50: 50) para obtener el compuesto deseado en forma de goma incolora (0,230 g; 62,2 %). EM (ESI): m/z = 358,2 [M-Boc+H]+.
BB74
3-[1-[4-trifluorometil)fenil]etoxi]acetidina
A una solución de 3-(1-(4-(trifluorometil)fenil)etoxi)acetidina-1-carboxilato de terc-butilo (0,04 g, 0,116 mmol) en DCM (0,6 ml) se le añadió TFA (0,178 ml, 2,32 mmol) y se agitó la mezcla de reacción a t. a. durante 30 min. Se retiró el disolvente a presión reducida y se recogió el residuo en EtOAc, se vertió en una solución de Na2CO3 ac. sat. (5 ml) y se extrajo dos veces la capa acuosa con EtOAc (10 ml cada vez). Se lavaron las capas orgánicas combinadas con salmuera, se secó sobre Na2SO4, se filtró y concentró a vacío para dar el compuesto del título bruto (0,025 g, 88 %) como una goma incolora; EM (ESI): m/z = 246,2 [M+H]+.
Intermedio:
3- [1-[4-(trifluorometil)fenil]etoxi]acetidina-1-carboxilato de terc-butilo
A una solución de 3-hidroxiacetidina-1-carboxilato de terc-butilo (0,1 g, 0,577 mmol) en DMF (5 ml) enfriada hasta 0 °C con un baño de hielo, se le añadió NaH (60 % en aceite de vaselina; 0,023 g, 0,577 mmol) y se agitó la mezcla de reacción a esta temperatura durante 15 min. A continuación, se añadió 1-(1-bromoetil)-4-(trifluorometil)benceno (0,161 g, 0,635 mmol, CAS RN 68120-42-3) y se dejó que la mezcla se calentara hasta t. a. y se continuó agitando durante la noche. Se vertió la mezcla en una solución ac. sat. de cloruro de amonio (15 ml) y se extrajo dos veces con EtOAc (20 ml cada vez). Se secaron las capas orgánicas sobre Na2SO4 y se concentró a vacío. Se purificó el residuo por cromatografía ultrarrápida en gel de sílice que se eluyó con un gradiente de EtOAc en n-heptano (de 0 % a 70 %) para dar el compuesto del título (0,045 g, 22,6 %) como un sólido amorfo incoloro. EM (ESl): m/z = 290,2 [M-C4Hs+H]+.
BB75
Clorhidrato de 4-[1-[4-(trifluorometil)fenil]etoxi]piperidina
Se obtuvo el producto de forma análoga a BH1, usando 4-hidroxipiperidina-1-carboxilato de terc-butilo (CAS RN 109384-19-2) en la etapa intermedia y, a continuación, una solución de HCl 4 M/dioxano en MeOH en lugar de TFA/DCM para la etapa de desprotección. Sólido incoloro; EM (ESl): 274,1 [M+H]+.
BB76
4- metilbencenosulfonato de 5-(acetidin-3-il)-2-metoxi-piridina
Se obtuvo el producto de forma análoga a BB28, usando 3-(6-metoxipiridin-3-il)acetidina-1-carboxilato de tercbutilo. Sólido incoloro. EM (ESl) = 165,1 [M+H]+.
Intermedio:
3- (6-metoxipiridin-3-il)acetidina-1-carboxilato de terc-butilo
Se obtuvo el producto de forma análoga a BB35, intermedio, usando ácido (6-metoxipiridin-3-il)borónico (CAS RN 163105-89-3), como un aceite incoloro. EM (ESl) = 265,2 [M+H]+.
BB77
4- metilbencenosulfonato de 3-[3-cloro-4-(trifluorometoxi)fenil]acetidina
Se obtuvo el producto de forma análoga a BB28, usando 3-[3-cloro-4-(trifluorometoxi)fenil]acetidina-1-carboxilato de terc-butilo, como un sólido incoloro; EM (ESI)= 252,1 [M+H]+.
Intermedio:
3-[3-cloro-4-(trifluorometoxi)fenil]acetidina-1-carboxilato de terc-butilo
Se obtuvo el producto de forma análoga a BB35, intermedio, usando ácido (3-doro-4-(trifluorometoxi)fenil)borónico (CAS RN 870822-79-0), como un líquido incoloro. EM (ESI) = 296,1 [M-C4Hs+H]+.
BB78
(4aR, 8aS)-6-[3-[3-bromo-4-(trifluorometoxi)fenil]acetidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona
Se obtuvo el producto de forma análoga al ejemplo 132, usando 4-metilbencenosulfonato de 3-[3-bromo-4-(trifluorometoxi)fenil]acetidina, como un sólido amarillo claro. EM (ESI) = 480,1 [M+H]+.
Intermedios:
a) 4-metilbencenosulfonato de 3-[3-bromo-4-(trifluorometoxi)fenil]acetidina
Se obtuvo el producto de forma análoga a BB28, usando 3-[3-bromo-4-(trifluorometoxi)fenil]acetidina-1-carboxilato de terc-butilo, como un sólido incoloro. EM (ESI) = 296,0 [M+H]+.
b) 3-[3-bromo-4-(trifluorometoxi)fenil]acetidina-1-carboxilato de terc-butilo
Se obtuvo el producto de forma análoga a BB35, intermedio, usando ácido (3-bromo-4-(trifluorometoxi)fenil)borónico (MFCD22580724; Apollo Scientific), como un aceite viscoso incoloro. EM (ESI) = 342,0 [M-C4H s +H]+.
BB79
4-metilbencenosulfonato de 3-[3-fluoro-4-(trifluorometil)fenil]acetidina
Se obtuvo el producto de forma análoga a BB28, usando 3-[3-fluoro-4-(trifluorometil)fenil]acetidina-1-carboxilato de terc-butilo. Sólido incoloro. EM (ESI): 220,1 [M+H]+.
Intermedio:
3-[3-fluoro-4-(trifluorometil)fenil]acetidina-1-carboxilato de terc-butilo
Se obtuvo el producto de forma análoga a BB37, intermedio, usando 4-bromo-2-fluoro-1-(trifluorometil)benceno, como un aceite viscoso incoloro. EM (ESI): 264,1 [M-C4Hs+H]+.
BB80
(4aR,8aS)-6-[4-[(3,4-dimetoxifenil)-(2-piridil)metil]piperidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona
Se agitó una solución de 2-[(3,4-dimetoxifenil)-(4-piperidil)metil]piridina (101 mg, 0,320 mmol), carbonato de cesio (97 mg, 0,30 mmol) y (4aR,8aS)-3-oxo-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacina-6-carboxilato de (4-nitrofenilo) (intermedio BB15a) (80 mg, 0,25 mmol) en DMF (5 ml) a 25 °C durante 16 h. Se vertió la solución en salmuera (10 ml) y se extrajo dos veces con EtOAc (10 ml cada vez). Se concentraron las capas orgánicas combinadas a vacío para dar un residuo, que se purificó por HPLC prep. (condición de TFA) para dar (4aR,8aS)-6-[4-[(3,4-dimetoxifenil)-(2-piridil)metil]piperidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona (60 mg, 49 %) como sólido blanco. EM (ESI): m/z = 495,3 [M+H]+.
Intermedios:
a) 4-(3,4-dimetoxibenzoil)piperidina-1-carboxilato de terc-butilo
A una solución a -78 °C de 4-bromoveratrol (12,4 g, 57,3 mmol) en THF (200 ml) se le añadió una solución de butillitio (28,6 ml, 71,6 mmol) y se agitó la solución a -78 °C durante 1 h. A continuación se añadió 4-[metoxi(metil)carbamoil]piperidina-1-carboxilato de terc-butilo (n.° CAS 139290-70-3) (13,0 g, 47,7 mmol) a -78 °C y se continuó agitando a -78 °C durante 5 h. Se vertió la solución en salmuera (20 ml) y se extrajo con EtOAc (2 x 10 ml). Se concentraron las capas orgánicas combinadas a vacío para dar un residuo que se purificó por cromatografía en columna ultrarrápida (PE: EtOAc = 2: 1) para dar el compuesto del título como sólido blanco. EM (ESI): m/z = 372,1 [M+Na]+.
b) 4-[C-(3,4-dimetoxifenil)-N-(p-tolilsulfonilamino)carbonimidoil]piperidina-1-carboxilato de terc-butilo Se agitó una solución de 4-metilbencenosulfonhidracida (6,40 g, 34,3 mmol) y 4-(3,4-dimetoxibenzoil)piperidina-1-carboxilato de terc-butilo (10,0 g, 28,6 mmol) en 1,4-dioxano (200 ml) a 80 °C durante 24 h. Se concentró la solución de reacción a vacío para dar un residuo que se purificó por cromatografía en columna ultrarrápida (éter de petróleo: EtOAc = 2: 1) para dar el compuesto del título (8,0 g, 54 %) como sólido amarillo claro. EM (ESI): m/z = 540,2 [M+Na]+.
c) 4-[(3,4-dimetoxifenil)-(2-piridil)metileno]piperidina-1-carboxilato de terc-butilo
Se agitó una mezcla de 2-bromopiridina (1,11 ml, 11,6 mmol), 4-[C-(3,4-dimetoxifenil)-N-(ptolilsulfonilamino)carbonimidoil]piperidina-1-carboxilato de
terc
-butilo (3,00 g, 5,8 mmol),
terc
-butóxido de litio (557 mg, 6,95 mmol) y cloruro de bis(trifenilfosfina)paladio(N) (407 mg, 0,580 mmol) en 1,4-dioxano (37 ml) a 90 °C durante 16 h bajo N2. Se filtraron los sólidos y se vertió la solución en salmuera (30 ml) y a continuación se extrajo con EtOAc (2 x 20 ml). Se concentraron las capas orgánicas combinadas a vacío. Se purificó el residuo por cromatografía en columna ultrarrápida (éter de petróleo: EtOAc = 1: 1) para dar el compuesto deseado como un aceite incoloro (280 mg, 12 %). EM (ESI): m/z = 411,3 [M+H]+.
d) 2-[(3,4-dimetoxifenil)-(4-piperidilideno)metil]piridina
Se agitó una solución de 4-[(3,4-dimetoxifenil)-(2-piridil)metileno]piperidina-1-carboxilato de terc-butilo (280 mg, 0,680 mmol) y ácido trifluoroacético (0,53 ml, 6,82 mmol) en Dc M (10 ml) a 25 °C durante 4 h. Se concentró la solución a vacío para dar el compuesto del título como aceite incoloro (200 mg, 94 %). EM (ESI): m/z = 311,1 [M+H]+.
e) 2-[(3,4-dimetoxifenil)-(4-piperidil)metil]piridina
Se agitó una solución de 2-[(3,4-dimetoxifenil)-(4-piperidilideno)metil]piridina (200 mg, 0,640 mmol) y Pd/C (69 mg, 0,060 mmol) en DMF (5 ml) a 25 °C durante 6 h bajo H2 (760 mmHg). Se concentró la solución a vacío para dar un residuo, que se purificó por HPLC prep. (condición de TFA) para dar el compuesto como aceite incoloro (150 mg, 71 %). EM (ESI): m/z = 313,3 [M+H]+.
BB81
d) (4aR, 8aS)-6-[4-[(3,4-dimetoxifenil)-(3-piridil)metil]piperidina-1-carbonil]-4,4a, 5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona
Se agitó una solución de 3-[(3,4-dimetoxifenil)-(4-piperidil)metil]piridina (126 mg, 0,400 mmol), carbonato de cesio (122 mg, 0,370 mmol) y (4aR,8aS)-3-oxo-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacina-6-carboxilato de (4-nitrofenilo) (intermedio BB15a) (100 mg, 0,310 mmol) en DMF (5 ml) a 25 °C durante 16 h. Se vertió la solución en salmuera (10 ml) y se extrajo con EtOAc (2 x 10 ml). Se concentraron las capas orgánicas combinadas a vacío para dar un residuo, que se purificó por HPLC prep. (condición de TFA) para dar el compuesto deseado como sólido blanco (80 mg, 52 %). EM (ESI): m/z = 495,3 [M+H]+.
Intermedios:
a) 4-[(3,4-dimetoxifenil)-(3-piridil)metileno]piperidina-1-carboxilato de terc-butilo
Se agitó una mezcla de 4-[C-(3,4-dimetoxifenil)-N-(p-tolilsulfonilamino)carbonimidoil]piperidina-1-carboxilato de terc-butilo (intermedio b, ejemplos 118/119) (2,00 g, 3,86 mmol), 3-bromopiridina (0,56 ml, 5,8 mmol), terc-butóxido de litio (464 mg, 5,8 mmol) y cloruro de bis(trifenilfosfina)paladio(N) (271 mg, 0,390 mmol) en DMF (30 ml) a 90 °C durante 16 h bajo N2. Se filtró la mezcla y se vertió el filtrado en salmuera (50 ml) y a continuación se extrajo con EtOAc (2 x 40 ml). Se concentraron las capas orgánicas combinadas a vacío para dar un residuo que se purificó por cromatografía en columna ultrarrápida (éter de petróleo: EtOAc = 2: 1). Se obtuvo 4-[(3,4-dimetoxifenil)-(3-piridil)metileno]piperidina-1 -carboxilato de terc-butilo como aceite incoloro (600 mg, 38 %). Em (ESI): m/z = 411,2 [M+H]+.
b) Trifluoroacetato de 3-[(3,4-dimetoxifenil)-(4-piperidilideno)metil]piridina
Se agitó una solución de 4-[(3,4-dimetoxifenil)-(3-piridil)metileno]piperidina-1-carboxilato de terc-butilo (600 mg, 1,46 mmol) y ácido trifluoroacético (1,13 ml, 15 mmol) en DCM (10 ml) a 25 °C durante 4 h. Se concentró la solución a vacío para dar 3-[(3,4-dimetoxifenil)-(4-piperidilideno)metil]piridina bruta (450 mg, 49 %, como sal de TFA) como un aceite marrón, que se usó directamente en la etapa siguiente. EM (ESI): m/z = 311,1 [M+H]+.
c) 3-[(3,4-dimetoxifenil)-(4-piperidil)metil]piridina
Se agitó una mezcla de 3-[(3,4-dimetoxifenil)-(4-piperidilideno)metil]piridina (450 mg, 0,710 mmol) y Pd/C (38 mg, 0,040 mmol) en DMF (10 ml) a 25 °C durante 16 h bajo H2 (760 mmHg). Se filtró la mezcla y se concentró a vacío
para dar un residuo, que se purificó por HPLC prep. (condición de TFA) para dar el compuesto deseado como aceite incoloro (220 mg, 99 %). EM (ESI): m/z = 313,2 [M+H]+.
BB82
(4aR,8aS)-6-[4-[(3,4-dimetoxifenil)-(4-piridil)metil]piperidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona
Se agitó una solución de 4-[(3,4-dimetoxifenil)-(4-piperidil)metil]piridina (100 mg, 0,320 mmol), carbonato de cesio (97 mg, 0,30 mmol) y (4aR,8aS)-3-oxo-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacina-6-carboxilato de (4-nitrofenilo) (intermedio BB15a) (80 mg, 0,25 mmol) en DMF (4 ml) a 25 °C durante 16 h. Se vertió la solución en salmuera (10 ml) y se extrajo dos veces con EtOAc (10 ml cada vez). Se concentraron las capas orgánicas combinadas a vacío para dar un residuo, que se purificó por HPLC prep. (condición de TFA) para dar el compuesto del título como sólido blanco (60 mg, 49 %). EM (ESI): m/z = 495,3 [M+H]+.
Intermedios:
a) 4-[(3,4-dimetoxifenil)-(4-piridil)metileno]piperidina-1-carboxilato de terc-butilo
Se agitó una mezcla de 4-bromopiridina (0,56 ml, 5,8 mmol), 4-[C-(3,4-dimetoxifenil)-N-(ptolilsulfonilamino)carbonimidoil]piperidina-1-carboxilato de terc-butilo (intermedio b, ejemplos 118/119) (2,00 g, 3,86 mmol), terc-butóxido de litio (464 mg, 5,8 mmol) y cloruro de bis(trifenilfosfina)paladio(N) (271 mg, 0,390 mmol) en DMF (30 ml) bajo N2 a 90 °C durante 16 h. Se filtró la mezcla y se vertió el filtrado en salmuera (30 ml) y a continuación se extrajo con EtOAc (2 x 20 ml). Se concentraron las capas orgánicas combinadas a vacío para dar un residuo que se purificó por cromatografía en columna ultrarrápida (éter de petróleo: EtOAc = 2:1). Se obtuvo 4-[(3,4-dimetoxifenil)-(4-piridil)metileno]piperidina-1-carboxilato de terc-butilo (400 mg, 25 %) como aceite incoloro. EM (ESI): m/z = 411,3 [M+H]+.
b) 4-[(3,4-dimetoxifenil)-(4-piperidilideno)metil]piridina
Se agitó una solución de 4-[(3,4-dimetoxifenil)-(4-piridil)metileno]piperidina-1-carboxilato de terc-butilo (400 mg, 0,970 mmol) y ácido trifluoroacético (0,75 ml, 9,7 mmol) en DCM (15 ml) a 25 °C durante 4 h. Se concentró la solución a vacío para dar 4-[(3,4-dimetoxifenil)-(4-piperidilideno)metil]piridina bruta (como sal de TFA; 250 mg, 83 %) como un aceite marrón, que se usó directamente en la etapa siguiente. EM (ESI): m/z = 311,2 [M+H]+.
c) 4-[(3,4-dimetoxifenil)-(4-piperidil)metil]piridina
Se agitó una mezcla de 4-[(3,4-dimetoxifenil)-(4-piperidilideno)metil]piridina (250 mg, 0,810 mmol) y Pd/C (43 mg, 0,040 mmol) en DMF (6 ml) a 25 °C durante 16 h bajo H2 (760 mmHg). Se filtró la mezcla y se concentró a vacío para dar un residuo, que se purificó por HPLC prep. (condición de TFA) para dar 4-[(3,4-dimetoxifenil)-(4-piperidil)metil]piridina (200 mg, 79 %) como aceite incoloro. EM (ESI): m/z = 313,2 [M+H]+.
BB83
(4aR,8aS)-6-[4-[(3-metilsulfonilfenil)-(3-piridil)metil]piperidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona
Se agitó una solución de 3-[(3-metilsulfonilfenil)-(4-piperidil)metil]piridina (110 mg, 0,330 mmol) y (4aR,8aS)-3-oxo-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacina-6-carboxylato de (4-nitrofenilo) (intermedio BB15a) (160 mg, 0,500 mmol) en piridina (5,5 ml) a 90 °C durante 16 h. Se concentró la solución a vacío para dar un residuo, que se purificó por HPLC prep. (condición de TFA) para dar (4a
R
,8a
S
)-6-[4-[(3-metilsulfonilfenil)-(3-piridil)metil]piperidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona (95 mg, 61 %) como sólido blanco. EM (ESI): m/z = 513,3 [M+H]+.
Intermedios:
a) 4-[N-(p-tolilsulfonilamino)-C-(3-piridil)carbonimidoil]piperidina-1-carboxilato de terc-butilo
Se agitó una solución de 4-metilbencenosulfonhidracida (1,54 g, 8,27 mmol) y 4-(piridin-3-carbonil)piperidina-1-carboxilato de terc-butilo (n.° CAS 148148-35-0) (2,00 g, 6,89 mmol) en 1,4-dioxano (200 ml) a 80 °C durante 16 h. Se concentró la mezcla de reacción a vacío para dar un residuo que se purificó por cromatografía en columna ultrarrápida (éter de petróleo: EtOAc = 2:1) para dar 4-[N-(p-tolilsulfonilamino)-C-(3-piridil)carbonimidoil]piperidina-1-carboxilato de terc-butilo (2,0 g, 63 %) como sólido amarillo claro EM (ESI): m/z = 403,1 [M-C4Hs+H]+.
b) 4-[(3-metilsulfonilfenil)-(3-piridil)metileno]piperidina-1-carboxilato de terc-butilo
Se agitó una solución de 3-bromofenilmetilsulfona (1,54 g, 6,54 mmol), 4-[N-(p-tolilsulfon¡lammo)-C-(3-piridil)carbonimidoil]piperidina-1-carboxilato de ferc-butilo (2,00 g, 4,36 mmol), ferc-butóxido de litio (524 mg, 6,54 mmol) y cloruro de bis^rifenilfosfinâ aladio^I) (918 mg, 1,31 mmol) en 1,4-dioxano (30 ml) a 90 °C durante 16 h. Se diluyó la reacción con EtOAc (20 ml) y a continuación se filtró a través de Celite. Se secó el filtrado sobre Na2SO4, se filtró y concentró a vacío. Se purificó el residuo por cromatografía en gel de sílice (éter de petróleo: EtOAc = de 20:1 a 1:1) para dar 4-[(3-met¡lsulfon¡lfen¡l)-(3-p¡r¡d¡l)met¡leno]p¡per¡d¡na-1-carbox¡lato de ferc-butilo (400 mg, 21 %) como aceite amarillo claro. EM (ESI): m/z = 429,2 [M+H]+.
c) 4-[(3-mefilsulfonilfenil)-(3-piridil)mefil]piperidina-1-carboxilafo de ferc-bufilo
Se agitó una mezcla de 4-[(3-met¡lsulfon¡lfen¡l)-(3-p¡r¡d¡l)met¡leno]p¡per¡d¡na-1-carbox¡lato de ferc-butilo (350 mg, 0,820 mmol) y Pd/C (43 mg, 0,410 mmol) en d Mf (10 ml) a 25 °C durante 16 h bajo H2 (760 mmHg). Se filtró la mezcla y se concentró a vacío para dar un residuo, que se purificó por HPLC prep. (condición básica) para dar 4-[(3-met¡lsulfon¡lfen¡l)-(3-p¡r¡d¡l)met¡l]p¡per¡d¡na-1-carbox¡lato de ferc-butilo (80 mg, 23 %) como aceite incoloro y la recuperación del material de partida (300 mg). El material de partida recuperado se sometió de nuevo a hidrogenación usando las mismas condiciones que anteriormente, proporcionando un segundo lote de producto (110 mg, 31 %) como aceite incoloro. Rendimiento total de 190 mg (54 %). EM (ESI): m/z = 375,0 [M-C4Hs+H]+.
d) 3-[(3-mefilsulfonilfenil)-(4-piperidil)mefil]piridina
Se agitó una solución de 4-[(3-met¡lsulfon¡lfen¡l)-(3-p¡r¡d¡l)met¡l]p¡per¡d¡na-1-carbox¡lato de ferc-butilo (220,0 mg, 0,510 mmol) y ácido trifluoroacético (0,2 ml, 2,55 mmol) en d Cm (4,4 ml) a 25 °C durante 4 h. Se concentró la solución a vacío para dar 3-[(3-met¡lsulfon¡lfen¡l)-(4-p¡per¡d¡l)met¡l]p¡r¡d¡na (como sal de TFA; 160 mg, cuant.) como aceite incoloro que se usó en la etapa siguiente sin purificación adicional.
BB84
(4aR,8aS)-6-[4-[(3-mefilsulfonilfenil)-(4-piridil)mefil]piperidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona
Se agitó una solución de 4-[(3-met¡lsulfon¡lfen¡l)-(4-p¡per¡d¡l)met¡l]p¡r¡d¡na (150 mg, 0,450 mmol) y (4aR,8aS)-3-oxo-4,4a,5,7,8,8a-hexah¡drop¡r¡do[4,3-b][1,4]oxac¡na-6-carbox¡lato de (4-nitrofenilo) (intermedio BB15a) (219 mg, 0,680 mmol) en piridina (6 ml) a 90 °C durante 16 h. Se concentró la solución a vacío para dar un residuo, que se purificó por HPLC prep. (condición de TFA) para dar (4a
R
,8a
S
)-6-[4-[(3-metilsulfonilfenil)-(4-piridil)metil]piperidina-1-carbon¡l]-4,4a,5,7,8,8a-hexah¡drop¡r¡do[4,3-b][1,4]oxac¡n-3-ona (60 mg, 26 %) como aceite incoloro. EM (ESI): m/z = 513,3 [M+H]+.
Infermedios:
a) 4-[N-(p-folilsulfonilamino)-C-(4-piridil)carbonimidoil]piperidina-1-carboxilafo de ferc-bufilo
Se agitó una solución de 4-metilbencenosulfonhidracida (1,31 g, 7,03 mmol) y 4-(p¡r¡d¡n-4-carbon¡l)p¡per¡d¡na-1-carboxilato de ferc-butilo (n.° CAS 1334415-27-8) (1,70 g, 5,85 mmol) en 1,4-dioxano (170 ml) a 80 °C durante 16 h. Se concentró la mezcla de reacción a vacío para dar un residuo que se purificó por cromatografía en columna ultrarrápida (éter de petróleo: EtOAc = 2:1) para dar 4-[N-(p-tol¡lsulfon¡lam¡no)-C-(4-p¡r¡d¡l)carbon¡m¡do¡l]p¡per¡d¡na-1-carboxilato de ferc-butilo (1,60 g, 60 %) como sólido amarillo claro. EM (ESI): m/z = 459,2 [M+H]+.
b) 4-[(3-mefilsulfonilfenil)-(4-piridil)mefileno]piperidina-1-carboxilafo de ferc-bufilo
Se agitó una solución de 3-bromofenilmetilsulfona (1,23 g, 5,23 mmol), 4-[N-(p-tolilsulfonilamino)-C-(4-p¡r¡d¡l)carbon¡m¡do¡l]p¡per¡d¡na-1 -carboxilato de ferc-butilo (1,60 g, 3,49 mmol), ferc-butóxido de litio (419 mg, 5,23 mmol) y cloruro de bis^rifenilfosfinâ aladio^I) (735 mg, 1,05 mmol) en 1,4-dioxano (32 ml) a 90 °C durante 16 h. Se diluyó la reacción con EtOAc (5 ml), a continuación se filtró a través de Celite. Se separó el filtrado, se secó sobre Na2SO4, se filtró y concentró a vacío. Se purificó el residuo por cromatografía en gel de sílice (éter de petróleo: EtOAc = de 20:1 a 1:1) para dar 4-[(3-met¡lsulfon¡lfen¡l)-(4-p¡r¡d¡l)met¡leno]p¡per¡d¡na-1-carbox¡lato de fercbutilo (420 mg, 28 %) como aceite incoloro. EM (ESI): m/z = 429,2 [m h ]+.
c) 4-[(3-mefilsulfonilfenil)-(4-piridil)mefil]piperidina-1-carboxilafo de ferc-bufilo
Se agitó una mezcla de 4-[(3-met¡lsulfon¡lfen¡l)-(4-p¡r¡d¡l)met¡leno]p¡per¡d¡na-1-carbox¡lato de ferc-butilo (320 mg, 0,750 mmol) y Pd/C (795 mg, 0,750 mmol) en DMf (6,4 ml) a 25 °C durante 16 h bajo H2 (760 mmHg). Se filtró la mezcla y se concentró a vacío para dar 4-[(3-met¡lsulfon¡lfen¡l)-(4-p¡r¡d¡l)met¡l]p¡per¡d¡na-1 -carboxilato de ferc-butilo (280 mg, 87 %) como aceite incoloro. EM (ESI): m/z = 431,2 [M+H]+.
d) 4-[(3-mefilsulfonilfenil)-(4-piperidil)mefil]piridina
Se agitó una solución de 4-[(3-metilsulfonilfenil)-(4-piridil)metil]piperidina-1-carboxilato de terc-butilo (280 mg, 0,650 mmol) y ácido trifluoroacético (0,25 ml, 3,2 mmol) en DCM (6 ml) a 25 °C durante 4 h. Se concentró la solución a vacío para dar un residuo, que se purificó por HPLC prep. (condición básica) para dar 4-[(3-metilsulfonilfenil)-(4-piperidil)metil]piridina (150 mg, 61 %) como aceite incoloro. EM (ESI): m/z = 331,2 [M+H]+.
BB85
(4aR, 8aS)-6-[4-[(3-metilsulfonilfenil)-(2-piridil)metil]piperidina-1-carbonil]-4,4a, 5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona
Se agitó una solución de oxona (256 mg, 0,420 mmol) y (4aR,8aS)-6-[4-[(3-metilsulfanilfenil)-(2-piridil)metil]piperidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona (100 mg, 0,210 mmol) en agua (5 ml) y MeCN (5 ml) a 25 °C durante 16 h. Se vertió la solución en Na2CO3 ac. sat. (10 ml) y se extrajo con EtOAc (2 x 20 ml). Se concentraron las capas orgánicas combinadas a vacío para dar un residuo, que se purificó por HPLC prep. (condición de TFA) para producir (4aR,8aS)-6-[4-[(3-metilsulfonilfenil)-(2-piridil)metil]piperidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona (60 mg, 56 %) como aceite incoloro. EM (ESI): m/z = 513,3 [M+H]+.
Intermedios:
a) 4-[hidroxi-(3-metilsulfanilfenil)-(2-piridil)metil]piperidina-1-carboxilato de terc-butilo
A una solución a -78 °C de 3-bromotioanisol (1,00 mg, 4,92 mmol) en THF (30 ml) se le añadió durante un tiempo de 0,5 h una solución de butillitio (2,36 ml, 5,91 mmol). A continuación se añadió 4-(piridina-2-carbonil)piperidina-1- carboxilato de terc-butilo (n.° CAS 416852-19-2) (1,43 g, 4,92 mmol) y se continuó agitando a -78 °C durante 4,5 h. Se detuvo la solución por NH4Cl ac. sat. (50 ml) y se extrajo con EtOAc (2 x 30 ml). Se concentraron las capas orgánicas combinadas a vacío para dar un residuo, que se purificó por HPLC prep. (condición de TFA) para proporcionar 4-[hidroxi-(3-metilsulfanilfenil)-(2-piridil)metil]piperidina-1 -carboxilato de terc-butilo (1,40 g, 69 %) como aceite incoloro. e M (ESI): m/z = 415,2 [M+H]+.
b) 2-[cloro-(3-metilsulfanilfenil)-(4-piperidil)metil]piridina
Se agitó una mezcla de 4-[hidroxi-(3-metilsulfanilfenil)-(2-piridil)metil]piperidina-1 -carboxilato de terc-butilo (300 mg, 0,720 mmol), dicloruro de azufre (86 mg, 0,72 mmol) y en Tol (5 ml) a 25 °C durante 6 h bajo N2. Se concentró la mezcla a vacío para dar 2-[cloro-(3-metilsulfanilfenil)-(4-piperidil)metil]piridina (220 mg, 91 %) como sólido amarillo, que se usó como materia prima en la etapa siguiente. EM (ESI): m/z = 333,1 [M+H]+.
c) 2-[(3-metilsulfanilfenil)-(4-piperidil)metil]piridina
Se agitó una mezcla de 2-[cloro-(3-metilsulfanilfenil)-(4-piperidil)metil]piridina (220 mg, 0,660 mmol), cloruro de amonio (35 mg, 0,66 mmol) y cinc (238 mg, 3,64 mmol) en MeoH (6 ml) a 25 °C durante 0,5 h. Se filtró la mezcla y se concentró a vacío para dar un residuo, que se purificó por HPLC prep. (condición básica) para proporcionar 2- [(3-metilsulfanilfenil)-(4-piperidil)metil]piridina (160 mg, 81 %) como aceite incoloro. EM (ESI): m/z = 299,1 [M+H]+.
d) (4aR, 8aS)-6-[4-[(3-metilsulfanilfenil)-(2-piridil)metil]piperidina-1-carbonil]-4,4a, 5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona
Se agitó una solución de 2-[(3-metilsulfanilfenil)-(4-piperidil)metil]piridina (160 mg, 0,540 mmol) y (4aR,8aS)-3-oxo-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacina-6-carboxilato de (4-nitrofenilo) (intermedio BB15a) (207 mg, 0,640 mmol) en piridina (10 ml) a 90 °C durante 16 h. Se filtró la solución y se concentró a vacío para dar un residuo, que se purificó por HPLC prep. (condición de TFA) para dar (4aR,8aS)-6-[4-[(3-metilsulfanilfenil)-(2-piridil)metil]piperidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona (200 mg, 78 %) como sólido blanco. EM (ESI): m/z = 481,2 [M+H]+.
BB86
Clorhidrato de 3-[fenil(4-piperidil)metil]piridacina
Se agitó una solución de 4-[fenil(piridacin-3-il)metil]piperidina-1-carboxilato de terc-butilo (150 mg, 0,420 mmol) y ácido trifluoroacético (0,1 ml, 1,3 mmol) en DCM (3 ml) a 25 °C durante 4 h. Se concentró la solución a vacío para dar un residuo, que se purificó por HPLC prep. (condición de HCl) para proporcionar 3-[fenil(4-piperidil)metil]piridacina (como sal de HCl, 70 mg, cuant.) como sólido gris. EM (ESI): m/z = 254,2 [M+H]+.
Intermedios:
4-(cloro-fenil-piridacin-3-il-metil)piperidina-1-carboxilato de terc-butilo
Se agitó una solución de 4-(piridacina-3-carbonil)piperidina-1-carboxilato de terc-butilo (n.° CAS 2044281-15-2) (250 mg, 0,860 mmol) y bromuro de fenilmagnesio (3 M, 0,34 ml, 1,03 mmol) en THF (6 ml) a 0 °C durante 3 h bajo N2. Se vertió la solución en salmuera (20 ml) y se extrajo con EtOAc (20 ml). Se concentró la capa orgánica a vacío para dar un residuo, que se purificó por HPLC prep. (condición de TFA) para dar 4-(hidroxi-fenil-piridacin-3-ilmetil)piperidina-1-carboxilato de terc-butilo (200 mg, 63 %) como sólido gris. EM (ESI): m/z = 314,1 [M-C4Hs+H]+.
4-(doro-fenil-piridadn-3-il-metil)piperidina-1-carboxilato de terc-butilo
Se agitó una mezcla de 4-(hidroxi-fenil-piridacin-3-il-metil)piperidina-1-carboxilato de terc-butilo (200 mg, 0,520 mmol) y dicloruro de azufre (306 mg, 2,59 mmol) en Tol (10 ml) a 25 °C durante 6 h. Se filtró la mezcla y se concentró a vacío para dar 4-(cloro-fenil-piridacin-3-il-metil)piperidina-1-carboxilato de terc-butilo (200 mg, 99 %) como sólido amarillo, que se usó como material bruto en la etapa siguiente. EM (ESI): m/z = 410,2 [M+Na]+.
4-[fenil(piridacin-3-il)metil]piperidina-1-carboxilato de terc-butilo
Se agitó una solución de 4-(cloro-fenil-piridacin-3-il-metil)piperidina-1-carboxilato de terc-butilo (200 mg, 0,52 mmol), cloruro de amonio (28 mg, 0,52 mmol) y cinc (185 mg, 2,84 mmol) en MeOH (4,7 ml) a 25 °C durante 0,5 h. Se concentró la mezcla a vacío para dar un residuo, que se purificó por HPLC prep. (condición de TFA) para proporcionar 4-[fenil(piridacin-3-il)metil]piperidina-1-carboxilato de terc-butilo (150 mg, 82 %) como sólido amarillo. EM (ESI): m/z = 298,1 [M-C4Hs+H]+.
BB87
3-[4-(3-fluoropropil)fenil]acetidina
Se agitó una solución de 3-[4-(3-fluoropropil)fenil]acetidina-1-carboxilato de terc-butilo (80 mg, 0,27 mmol) y ácido trifluoroacético (0,07 ml, 0,85 mmol) en DCM (2 ml) a 25 °C durante 4 h. Se concentró la mezcla a vacío para dar 3-[4-(3-fluoropropil)fenil]acetidina (40 mg, 76 %) como aceite incoloro, que se usó como tal en la etapa siguiente. EM (ESI): m/z = 194,1 [M+H]+.
Intermedios:
a) 2-[4-(3-fluoropropil) fenil]-4,4,5,5-tetrametil-1,3,2-dioxaborolano
Se agitó una solución de 1-bromo-4-(3-fluoropropil)benceno (n.° CAS 168104-62-9) (1,00 g, 4,61 mmol), bis(pinacolato)diboro (2,34 g, 9,21 mmol), [Pd(dppf)Ch]CH2Cl2 (376 mg, 0,460 mmol) y KOAc (1,35 g, 13,8 mmol) en 1,4-dioxano (40 ml) a 100 °C durante 16 h bajo N2. Se filtró la mezcla y se concentró para dar un residuo, que se purificó por cromatografía en columna ultrarrápida (éter de petróleo: EtOAc = 10:1) para dar 2-[4-(3-fluoropropil)fenil]-4,4,5,5-tetrametil-1,3,2-dioxaborolano (1,2 g, 99 %) como aceite incoloro. e M (ESI): m/z = 265,1 [M+H]+.
b) Ácido [4-(3-fluoropropil)fenil]borónico
A una solución de 2-[4-(3-fluoropropil)fenil]-4,4,5,5-tetrametil-1,3,2-dioxaborolano (600 mg, 2,27 mmol) en H2O (12 ml) y acetona (60 ml) se le añadió HCl (1 M, 1 ml). Se agitó la solución a 25 °C durante 16 h. Se vertió la solución en salmuera (40 ml) y se extrajo con EtOAc (2 x 20 ml). Se concentraron las capas orgánicas combinadas a vacío para dar ácido [4-(3-fluoropropil)fenil]borónico (300 mg, 73 %) como sólido amarillo claro. RMN de 1H (400 MHz, CLOROFORMO-d)
S
= 8,19 (d,
J
=7,9 Hz, 2H), 7,37 (d,
J
=7,8 Hz, 2H), 4,52 (dt,
J
=47,2, 5,9 Hz, 2H), 2,87 (t,
J
=7,1 Hz, 2H), 2,16-2,02 (m, 2H).
c) 3-[4-(3-fluoropropil)fenil]acetidina-1-carboxilato de terc-butilo
Se agitó una solución de ácido [4-(3-fluoropropil)fenil]borónico (145 mg, 0,790 mmol), 3-yodoacetidina-1-carboxilato de terc-butilo (n.° CAS 254454-54-1) (150 mg, 0,530 mmol), yoduro de níquel(II) (99 mg, 0,32 mmol) (1S,2S)-2-aminociclohexanol (37 mg, 0,32 mmol) y bis(trimetilsilil)amida de sodio (1,06 ml, 1,06 mmol) en 2-propanol (4 ml) a 80 °C durante 1 h en un tubo sellado. Se filtró la mezcla y se concentró a vacío para dar un residuo, que se purificó por HPLC prep. (Condición de TFA) para proporcionar 3-[4-(3-fluoropropil)fenil]acetidina-1- carboxilato de terc-butilo (82 mg, 46 %) como aceite incoloro. e M (ESI): m/z = 238,1 [M-C4Hs+H]+.
BB88
2- (acetidin-3-il)-5-(2,4-diclorofenil)-1,3,4-oxadiazol
Se suspendió el compuesto 3-[[(2,4-diclorobenzoil)amino]carbamoil]acetidina-1-carboxilato de terc-butilo
(800,0 mg, 2,06 mmol) en ácido polifosfórico (5 ml), se agitó la mezcla a 180 °C durante 2 h. Se vertió la mezcla en amoníaco helado (100 ml), se agitó durante 10 min, a continuación se extrajo tres veces con EtOAc (100 ml cada vez), se lavó la fase orgánica combinada con salmuera, se secó sobre Na2SO4 y se concentró. Se purificó el residuo por cromatografía en columna ultrarrápida inversa (FA al 0,1 % v/v) para dar el producto deseado (130 mg, 17 %) como un aceite marrón claro. EM (ESI): m/z = 270,4 [M+H]+.
Intermedio:
3-[[(2,4-diclorobenzoil)amino]carbamoil]acetidina-1-carboxilato de terc-butilo
A una solución de ácido 1-Boc-acetidina-3-carboxílico (490,68 mg, 2,44 mmol, CAS RN 142253-55-2) y 2,4-diclorobenzhidracida (500,0 mg, 2,44 mmol, CAS RN 5814-06-2), DIPEA (1,27 ml, 7,32 mmol) en DMF (20 ml) se le añadió T3P (1122,42 mg, 4,88 mmol, 50 % en EtOAc), se agitó la mezcla a 80 °C durante 12 h. Se diluyó la mezcla con EtOAc (50 ml), se lavó con NaHCO3 (100 ml) y salmuera, a continuación se secó sobre Na2SO4 y se evaporó para dar el producto bruto (800 mg, 84,5 %) como un aceite marrón claro. EM (ESI): m/z = 332,4 [M-C4H8+H]+.
BB89
3-(acetidin-3-il)-1-(2,4-diclorofenil)pirazol, sal de ácido trifluoroacético
A una solución de 3-[1-(2,4-diclorofenil)pirazol-3-il]acetidina-1-carboxilato de terc-butilo (350,0 mg, 0,950 mmol) en DCM (5 ml) se le añadió TFA (1,0 ml, 0,950 mmol) y se agitó la mezcla a 20 °C durante 12 h. Se concentró la mezcla para dar el producto deseado como un aceite amarillo (350 mg, 96,4 %). EM (ESI): m/z = 268,1 [M+H]+.
Intermedios:
a) 3-[metoxi(metil)carbamoil]acetidina-1-carboxilato de terc-butilo
A una solución de ácido 1-BOC-acetidina-3-carboxílico (10,0 g, 49,7 mmol, CAS RN 142253-55-2) en DCM (200 ml) se le añadió CDI (8,06 g, 49,7 mmol), se agitó la mezcla a 20 °C durante 1 h, a continuación se añadió Te a (13,85 ml, 99,39 mmol) y sal de HCl de O,N-dimetilhidroxilamina (5,82 g, 59,64 mmol, CAS RN 6638-79-5), se agitó la mezcla a 20 °C durante 15 h, a continuación se lavó la mezcla con Na2CO3 acuoso y salmuera, se secó sobre Na2SO4, se filtró y se concentró el filtrado para dar el producto bruto como aceite amarillo claro (12 g) que se usó en la etapa siguiente sin purificación adicional.
b) 3-acetilacetidina-1-carboxilato de terc-butilo
A una solución de 3-[metoxi(metil)carbamoil]acetidina-1-carboxilato de terc-butilo (1000,0 mg, 4,09 mmol) en THF (30 ml) se le añadió MeMgBr/THF (1,77 ml, 3 M) a 0 °C, se agitó la mezcla a 20 °C durante 2 h, a continuación se vertió la mezcla en solución de NH4Cl sat. (100 ml) y se extrajo con EtOAc (30 ml tres veces), se lavó la fase orgánica combinada con salmuera, se secó sobre Na2SO4 y se concentró para dar el producto deseado como aceite amarillo claro (810 mg, 99,3 %). RMN de 1H (400 MHz, CLOROFORMO-d) 8 = 4,05 (d. a., J = 7,4 Hz, 4H), 3,48-3,34 (m, 1H), 2,18 (d, J = 2,0 Hz, 2H), 1,44 (d, J = 2,0 Hz, 9H).
c) 3-[(E)-3-(dimetilamino)prop-2-enoil]acetidina-1-carboxilato de terc-butilo
Se agitó una solución de 3-acetilacetidina-1-carboxilato de terc-butilo (500,0 mg, 2,51 mmol) en N,N-dimetilformamida-dimetilacetal (10,0 ml) a 110 °C durante 12 h, se concentró la mezcla para dar el compuesto del título como aceite amarillo (640 mg, 2,52 mmol). EM (ESI): m/z = 199,2 [M-C4Hs+H]+.
d) 3-[1-(2,4-diclorofenil)pirazol-3-il]acetidina-1-carboxilato de terc-butilo
Se agitó una solución de 3-[(E)-3-(dimetilamino)prop-2-enoil]acetidina-1-carboxilato de terc-butilo (500,0 mg, 1,97 mmol) y clorhidrato de 2,4-diclorobenzohidracida (474,78 mg, 1,97 mmol) en EtOH (30 ml) a 80 °C durante 12 h, se concentró la mezcla, se purificó el residuo por columna en gel de sílice (PE: EtOAc = 20: 1 a 3: 1) para dar el producto deseado (450 mg, 62,2 %) como aceite amarillo. EM (ESI): m/z = 368,1 [M+H]+.
BB90
3-[4-(2,2-dimetilpropil)fenil]acetidina, sal del ácido trifluoroacético
A una solución de 3-[4-(2,2-dimetilpropil)fenil]acetidina-1-carboxilato de terc-butilo (500,0 mg, 1,65 mmol) en DCM (10 ml) se le añadió TFA (2,0 ml) y se agitó la mezcla a 20 °C durante 12 h. Se concentró la mezcla para dar el producto bruto como un aceite amarillo claro (520 mg). EM (ESI): m/z = 204,2 [M+H]+.
Intermedios:
a) 1-bromo-4-(2,2-dimetilpropil)benceno
A una solución de 2,2-dimetilpropilbenceno (500,0 mg, 3,37 mmol, CAS RN 1007-26-7) en ácido acético (10 ml) se le añadió bromo (0,17 ml, 3,37 mmol). Se agitó la mezcla a 20 °C en la oscuridad durante 12 h y, a continuación, se vertió la mezcla en solución de Na2SO3 ac. sat. (30 ml) y se extrajo tres veces con EtOAc (10 ml cada vez). Se lavaron las fases orgánicas combinadas con salmuera, se secaron sobre Na2SO4 y se concentraron para dar el producto bruto como un aceite amarillo claro (600 mg, 78,3 %) que se usó en la etapa siguiente sin purificación adicional.
b) 3-[4-(2,2-dimetilpropil)fenil]acetidina-1-carboxilato de terc-butilo
A un vial de 40 ml equipado con una barra agitadora se le añadió 3-bromoacetidina-1-carboxilato de terc-butilo (519,75 mg, 2,2 mmol, CAS RN 1064194-10-0), 1-bromo-4-(2,2-dimetilpropil))benceno (500,0 mg, 2,2 mmol), Ir[dF(CFa)ppy]2(dtbbpy)PF6 (24,68 mg, 0,020 mmol, CAS RN 870987-63-6), NiChglyme (2,42 mg, 0,010 mmol, CAS RN 29046-78-4), 4-terc-butil-2-(4-terc-butil-2-piridil)piridina (3,54 mg, 0,010 mmol, CAS RN 69641-93-6), bis(trimetilsililo)silil-trimetil-silano (547,37 mg, 2,2 mmol, Ca s RN 1873-77-4) y Na2CO3 (466,63 mg, 4,4 mmol), a continuación DME (20 ml). Se selló el vial y se colocó bajo nitrógeno. Se agitó la mezcla de reacción y se irradió con una lámpara LED azul de 34 W (7 cm de distancia), con ventilador de refrigeración para mantener la temperatura de reacción a 25 °C durante 14 h. Se filtró la mezcla y se concentró el filtrado. Se purificó el residuo por ultrarrápida inversa (FA al 0,1 % v/v) para dar el producto deseado (500 mg, 74,8 %) como un aceite de color amarillo claro. EM (ESI): m/z = 248,2 [M-C4Hs+H]+.
BB91
(4aR, 8aS)-6-[3-[4-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)fenil]acetidina-1-carbonil]-4,4a, 5,7,8,8ahexahidropirido[4,3-b][1,4]oxacin-3-ona
A una solución de (4aR,8aS)-6-[3-(4-bromofenil)acetidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona (200,0 mg, 0,510 mmol, ejemplo 110) y PdCh(dppf)DCM (35,56 mg, 0,050 mmol) se le añadió acetato de potasio (149,36 mg, 1,52 mmol) y bis(pinacolato)diboro (193,23 mg, 0,760 mmol), se purgó la mezcla de reacción con nitrógeno y se agitó a 90 °C durante 12 h. Se diluyó la mezcla de reacción con agua y se extrajo tres veces con EtOAc. Se lavaron las capas orgánicas combinadas con agua y salmuera, se secó sobre sulfato de sodio y se concentró a vacío para obtener un residuo amarillo, que se purificó por cromatografía ultrarrápida inversa (FA al 0,1 % v/v) para proporcionar el producto deseado (140 mg, 0,320 mmol, 62,5 %) como sólido amarillo claro. EM (ESI): m/z = 442,3 [M+H]+.
BB92
2-bromo-N-etil-5-metil-benzamida
A una solución de ácido 2-bromo-5-metilbenzoico (500,0 mg, 2,33 mmol, CAS RN 6967-82-4) y HOBT (534,11 mg, 3,49 mmol), EDCI (541,43 mg, 3,49 mmol) y DIPEA (1,21 ml, 6,98 mmol) en DMF (10 ml) se le añadió clorhidrato de etilamina (227,51 mg, 2,79 mmol) y se agitó la mezcla a 20 °C durante 12 h. A continuación se vertió la mezcla en solución de Na2CO3 ac. sat. (50 ml) y se extrajo tres veces con EtOAc (20 ml cada vez). Se lavaron las capas orgánicas combinadas con salmuera y se secó sobre Na2SO4 y se concentró para dar el producto deseado como sólido amarillo claro (450 mg, 79,9 %). EM (ESI): m/z = 242,0 [M+H]+.
BB93
Acetidin-3-ilbis(4-fluorofenil)metanol
Se cargó un matraz de fondo redondo de dos bocas de 100 ml seco equipado con condensador de reflujo bajo argón con THF (18,6 ml) y 3-metilacetidina-1,3-dicarboxilato de 1 -(terc-butilo) (800 mg, 746 pl, 3,72 mmol, CAS RN 610791-05-4). Se purgó la mezcla con argón durante 5 min y se enfrió a 0 °C. A continuación, se añadió bromuro de (4-fluorofenil)magnesio, solución 0,8 M en THF (18,6 ml, 14,9 mmol) durante 10 min. Después de completar la adición, se calentó la mezcla de reacción hasta 85 °C (baño de aceite) durante 17 h. Se detuvo la reacción con agua (5 ml), se diluyó con EtOAc (10 ml) y se agitó la suspensión resultante durante 15 min. Se añadió agua y se acidificó la mezcla con HCl 2 M (20 ml) hasta que la fase ac. se convirtió en una solución incolora. Se separaron las fases y se extrajo la fase acuosa con EtOAc (100 ml). Se ajustó el pH a 7 y se extrajo la fase acuosa cuatro veces con EtOAc. Se concentraron las capas orgánicas combinadas hasta aproximadamente 50 ml. Se formó un precipitado y se enfrió el matraz hasta 4 °C. Se filtró el precipitado y se lavó el sólido blanco con pequeñas porciones de EtOAc para proporcionar el compuesto del título (317 mg, 31 %). EM (ESI): m/z = 276,2 [M+H]+.
BB94
2.2.2- trifluoroacetato de 3-(1-(2-cloro-4-(trifluorometil)fenoxi)etil)acetidina
Se añadió ácido trifluoroacético (400 mg, 270 |jL, 3,51 mmol) a una solución de 3-(1-(2-cloro-4-(trifluorometil)fenoxi)etil)acetidina-1-carboxilato de terc-butilo (78,4 mg, 206 jmol)en DCM (1,03 ml) y se agitó la solución a t. a. durante 2 h. Se retiró el disolvente a presión reducida para dar el compuesto como un aceite amarillo claro (134 mg, 100 %). Se usó el producto bruto en la etapa siguiente sin purificación adicional. EM (ESI): m/z = 280.1 [M+H]+.
Intermedio
3-( 1-(2-cloro-4-(trifluorometil)fenoxi)etil)acetidina-1-carboxilato de terc-butilo
A una solución de 2-cloro-4-(trifluorometil)fenol (500 mg, 340 jl, 2,54 mmol), 3-(1-hidroxietil)acetidina-1-carboxilato de terc-butilo (512 mg, 2,54 mmol, CAS RN 1138331-90-4) y trifenilfosfina (734 mg, 2,8 mmol) en DCM (12,7 ml) se le añadió DIAD (566 mg, 544 jl, 2,8 mmol) gota a gota a 0 °C y se agitó la reacción a 0 °C durante 10 min y a t. a. durante 6 h. Se detuvo la mezcla de reacción por la adición de solución de NaHCO3 ac. sat. (20 ml). Se separaron las fases y se extrajo la fase ac. con DCM dos veces. Se secaron las capas orgánicas combinadas sobre Na2SO4, se filtró y almacenó a 4 °C durante cuatro días. Se concentró el producto bruto a sequedad, se inmovilizó en Isolute® y se purificó por cromatografía en columna ultrarrápida (que se eluyó con un gradiente de 0 a 30 % de EtOAc en n-heptano) para dar el compuesto del título como un aceite pálido (497 mg, 48,9 %). EM (ESI): m/z = 324.1 [M-C4Hs+H]+.
BB95
Clorhidrato de 4-(1-(2-doro-4-fíuorofenoxi)-2,2,2-trifíuoroetil)piperidina
Se añadió ácido clorhídrico (4 M en dioxano) (174 jl, 695 jmol) a una solución de 4-(1-(2-cloro-4-fluorofenoxi)-2.2.2- trifluoroetil)piperidina-1-carboxilato de terc-butilo (17,9 mg, 43,5 jmol) en dioxano (435 jl) y se agitó la solución durante 2 h a t. a. Se retiró el disolvente a presión reducida para dar el compuesto deseado como un aceite amarillo claro (15 mg, 95 %). Se usó el producto bruto en la etapa siguiente sin purificación adicional. EM (ESI): m/z = 312,1 [M+H]+.
Intermedios
a) 4-( 1-(2-doro-4-fíuorofenoxi)-2,2,2-trifíuoroetil)piperidina-1-carboxilato de terc-butilo
Se suspendieron 4-(2,2,2-trifluoro-1-(((trifluorometil)sulfonil)oxi)etil)piperidina-1-carboxilato de terc-butilo (126 mg, 303 jmol), 2-cloro-4-fluorofenol (48,9 mg, 36,4 jl, 334 jmol) y carbonato de cesio (109 mg, 334 jmol) en DMF (1,52 ml) y se agitó a t. a. durante 20 h. A continuación, se detuvo la mezcla de reacción con agua y se extrajo dos veces con EtOAc. Se lavaron las fases orgánicas combinadas con agua y salmuera, se secó sobre MgSO4 y se concentró a vacío. Se inmovilizó el residuo en Isolute® y se purificó por cromatografía en columna ultrarrápida (de 0 a 30 % de gradiente de EtOAc en n-heptano) para dar el compuesto del título como un aceite incoloro (20 mg, 15 %). EM (ESI): m/z = 356,1 [M-C4Hs+H]+.
b) 4-(2,2,2-trifluoro-1-(((trifluorometil)sulfonil)oxi)etil)piperidina-1-carboxilato de terc-butilo
Se cargó un vial de microondas con 4-(2,2,2-trifluoro-1-hidroxietil)piperidina-1-carboxilato de terc-butilo (100 mg, 353 jmol, CAS RN 184042-83-9). Se colocó el vial bajo argón, se añadió DCM (1,76 ml) y se enfrió hasta 0 °C. Se añadió piridina (33,5 mg, 34,3 jl, 424 jmol), seguido de anhídrido tríflico (110 mg, 65,6 jl, 388 jmol). Se agitó la mezcla de reacción a 0 °C durante 1 h y a continuación se detuvo con agua. Se separó la mezcla y se lavó la fase orgánica con agua y se extrajo tres veces con DCM. Se lavaron las capas orgánicas combinadas con salmuera, se secó sobre MgSO4 y se concentró a vacío dando el compuesto deseado como un aceite amarillo (126 mg, 85,9 %). Se usó el compuesto en la etapa siguiente sin purificación adicional.
BB96
3-(bis(4-fluorofenil)metil)acetidina
Se evacuó una solución de 3-(bis(4-fluorofenil)metileno)acetidina (20 mg, 77,7 jmol) en MeOH (777 jl) y se rellenó con argón cinco veces. Bajo una atmósfera de argón, se añadió Pd-C (4,14 mg, 3,89 jmol) y se reemplazó la atmósfera con hidrógeno tres veces. Se agitó la reacción en atmósfera de hidrógeno (globo) a 1 bar durante 19 h. Se reemplazó la atmósfera con argón y se filtró la mezcla de reacción sobre una capa de Dicalite. Se lavó la torta del filtro con MeOH. Se concentró el filtrado para dar el compuesto deseado como un sólido amarillo (20,1 mg, 94,7 %) que se usó en la etapa siguiente sin purificación adicional. EM (ESI): m/z = 260,2 [M+H]+.
Intermedio
3-(bis(4-fluorofenil)metileno)acetidina
A una suspensión de acetidin-3-ilbis(4-fluorofenil)metanol (244 mg, 886 |jmol, BB93) en DCM (2,22 ml) se le añadió TFA (2,22 ml) y se agitó la mezcla durante 3,5 h. Esto dio una solución homogénea. Se evaporó la reacción hasta sequedad, se diluyó el residuo resultante con EtOAc, se lavó dos veces con solución de bicarbonato de sodio acuosa saturada, a continuación con salmuera, se secó sobre Na2SO4 y se filtró. Se concentró el filtrado a vacío hasta sequedad y se trituró el residuo con EtOAc/n-heptano y se filtró. Esto dio el compuesto del título como un sólido blanco (20 mg, 8,3 %). EM (ESI): m/z = 258,2 [M+H]+.
BB97
2.2.2- trifluoroacetato de 3-(1-(2-fluoro-4-(trifluorometil)fenoxi)etil)acetidina
A una solución de 3-(1-(2-fluoro-4-(trifluorometil)fenoxi)etil)acetidina-1-carboxilato de terc-butilo (93 mg, 256 jmol) en DCM (1 ml) se le añadió ácido 2,2,2-trifluoroacético (467 mg, 316 jl, 4,1 mmol) y se agitó la reacción a t. a. durante 20 h. Se concentró la mezcla de reacción para dar el compuesto deseado como un aceite incoloro (112,8 mg, 99 %) que se usó en la etapa siguiente sin purificación adicional. EM (ESI): m/z = 264,1 [M+H]+.
Intermedio
a) 3-(1-(2-fíuoro-4-(trífíuorometil)fenoxi)etil)acetidina-1-carboxilato de terc-butilo
A una solución de 2-fluoro-4-(trifluorometil)fenol (100 mg, 69,9 jl, CAS RN 77227-78-2), 3-(1-hidroxietil)acetidina-1-carboxilato de terc-butilo (112 mg, 555 jmol, c As RN 1138331-90-4) y trifenilfosfina (160 mg, 611 jmol) en DCM (2,8 ml) se le añadió DIAD (124 mg, 119 jl, 611 jmol) gota a gota y se agitó la reacción a t. a. durante 1,5 h.
Se detuvo la mezcla de reacción por la adición de solución de NaHCO3 ac. sat. Se separaron las fases y se extrajo la fase acuosa con DCM. Se secaron las capas orgánicas combinadas sobre Na2SO4, se filtró y concentró a vacío. Se inmovilizó el residuo en Isolute y se purificó por cromatografía en columna ultrarrápida (de 0 a 20 % de gradiente de EtOAc en n-heptano) para dar el compuesto deseado como un aceite incoloro (93 mg, 43,8 %). EM (ESI): m/z = 308,1 [M-C4Hs+H]+.
BB98
2.2.2- trifluoroacetato de 3-(1-(4-trifluorometil)fenoxi)etil)acetidina
De forma análoga a BB97, se generó BB98 a partir de 4-(trifluorometil)fenol y 3-(1-hidroxietil)acetidina-1-carboxilato de terc-butilo. Se usó el compuesto en la etapa siguiente sin purificación adicional.
BB99
(S o R)-4-((4-fluorofenil)(3-metoxifenil)metil)piperidina
Se purificó una solución de 4-[(4-fluorofenil)-(3-metoxifenil)metil]piperidina (760 mg, 2,54 mmol) en MeOH (5 ml) por separación por SFC para dar 4-[(S o R)-(4-fluorofenil)-(3-metoxifenil)metil]piperidina (128 mg, 0,43 mmol, 17 %) como semisólido amarillo claro; e M (ESI): m/z = 300,1 [M+H]+; y 4-[(R o S)-(4-fluorofenil)-(3-metoxifenil)metil]piperidina (170 mg, 0,57 mmol, 22 %) como semisólido amarillo claro; EM (ESI): m/z = 300,1 [M+H]+.
Intermedios
a) 4-[(4-fluorofenil)(hidroxi)(3-metoxifenil)metil]piperidina-1-carboxilato de terc-butilo
A una solución de 3-bromoanisol (487 mg, 2,6 mmol) en THF (40 ml) se le añadió butillitio (1,5 ml, 3,75 mmol, 2,5 M) a -78 °C. Después de agitar durante 1 h, se añadió a la mezcla 4-(4-fluorobenzoil)piperidina-1-carboxilato de terc-butilo (CAS RN 160296-40-2; 800 mg, 2,6 mmol) y se continuó agitando a -78 °C durante 1 h. A continuación, se calentó la reacción hasta 25 °C y se agitó durante otras 13 h. Se detuvo la reacción con NH4Cl (ac. sat., 50 ml) y se extrajo con EtOAc (100 ml). Se separó la capa orgánica, se secó sobre Na2SO4, se filtró y concentró a vacío para dar el compuesto deseado (1100 mg, 63 %) como aceite amarillo. EM (ESI): m/z = 438,1 [M+Na]+.
b) Trifluoroacetato de 4-[(4-fluorofenil)(3-metoxifenil)metileno]piperidina
Se agitó una solución de 4-[(4-fluorofenil)(hidroxi)(3-metoxifenil)metil]piperidina-1-carboxilato de terc-butilo (1,1 g, 1,64 mmol) y ácido trifluoroacético (5,0 ml, 65 mmol) en DCM (10 ml) a 25 °C durante 5 h. Se concentró la reacción a vacío. Se disolvió el residuo en EtOAc (50 ml) y se lavó con Na2CO3 (ac., 10 %, 50 ml). Se separó la capa orgánica, se secó con Na2SO4, se filtró y concentró a vacío. Se purificó el aceite resultante por HPLC preparativa (TFA como aditivo) para dar 4-[(4-fluorofenil)(3-metoxifenil)metileno]piperidina (sal de TFA, 430 mg, 39 %) como un sólido blanco. EM (ESI): m/z = 298,1 [M+H]+.
c) Trifluoroacetato de 4-[(4-fluorofenil)(3-metoxifenil)metil]piperidina
Se agitó una solución de 4-[(4-fluorofenil)(3-metoxifenil)metileno]piperidina (410 mg, 1,38 mmol) y Pd/C (100 mg, 1,38 mmol) en THF (10 ml) a 25 °C durante 16 h bajo H2 (760 mmHg). Se filtró la mezcla de reacción a través de Celite, a continuación se concentró a vacío para dar 4-[(4-fluorofenil)(3-metoxifenil)metil]piperidina (sal de TFA, 260 mg, 59 %) como aceite amarillo. EM (ESI): m/z = 300,1 [M+H]+.
BB100
(R o S)-4-((4-fluorofenil)(3-metoxifenil)metil)piperidina
Se purificó una solución de 4-[(4-fluorofenil)-(3-metoxifenil)metil]piperidina (BB99, intermedio c, 760 mg, 2,54 mmol) en MeOH (5 ml) por separación por SFC para dar 4-[(S o R)-(4-fluorofenil)-(3-metoxifenil)metil]piperidina (128 mg, 17 %) como semisólido amarillo claro; CLEM: 300,1 [M+H]+; y 4-[(R o s )-(4-fluorofenil)-(3-metoxifenil)metil]piperidina o 4-[(S)-(4-fluorofenil)-(3-metoxifenil)metil]piperidina (170 mg, 22 %) como semisólido amarillo claro; Em (ESI): m/z = 300,1 [M+H]+.
BB101
(S o R)-4-((3-metoxifenil)(fenil)metil)piperidina
Se purificó una solución de 4-[(3-metoxifenil)-fenil-metil]piperidina (960 mg, 3,41 mmol) en MeOH (5 ml) por HPLC preparativa usando TFA como aditivo para dar 4-[-(3-metoxifenil)-fenil-metil]piperidina como su sal de TFA (1260 mg). Se purificó la sal de TFA de 4-[-(3-metoxifenil)-fenil-metil]piperidina (1260 mg) por separación por SFC para dar (S o R)-4-((3-metoxifenil)(fenil)metil)piperidina (443 mg, 45 %) como sólido amarillo claro; EM (ESI): m/z = 282,2 [M+H]+; y (R o S)-4-((3-metoxifenil)(fenil)metil)piperidina (383 mg, 39 %) como sólido amarillo; EM (ESI): m/z = 282,2 [M+H]+.
Intermedio
4-((3-metoxifenil)(fenil)metil)piperidina
Se preparó el compuesto del título de forma análoga al intermedio 1 -[(4-fluorofenil)-fenil-metil]piperacina (CAS RN 27064-89-7), comenzando a partir de 3-bromoanisol y 4-benzoilpiperidina-1-carboxilato de terc-butilo (CAS RN 922504-27-6) para dar el compuesto del título como semisólido amarillo claro. EM (ESI): m/z = 282,1 [M+H]+. BB102
(R o S)-4-((3-metoxifenil)(fenil)metil)piperidina
Se purificó una solución de 4-[(3-metoxifenil)-fenil-metil]piperidina (BB101, intermedio, 960 mg, 3,41 mmol) en MeOH (5 ml) por HPLC preparativa usando TFA como aditivo para dar 4-[-(3-metoxifenil)-fenil-metil]piperidina como su sal de TFA (1260 mg). Se purificó la sal de TFA de 4-[-(3-metoxifenil)-fenil-metil]piperidina (1260 mg) por separación por SFC para dar (S o R)-4-((3-metoxifenil)(fenil)metil)piperidina (443 mg, 45 %) como sólido amarillo claro; EM (ESI): m/z = 282,2 [M+H]+; y (R o S)-4-((3-metoxifenil)(fenil)metil)piperidina (383 mg, 39 %) como sólido amarillo; EM (ESI): m/z = 282,2 [M+H]+.
BB103
(S o R)-4-((3-(2-fluoroetoxi)fenil)(fenil)metil)piperidina
Se purificó una solución de 4-[[3-(2-fluoroetoxi)fenil]-fenil-metil]piperidina (800 mg, 2,55 mmol) en MeOH (5 ml) por separación por SFC para dar (S o R)-4-((3-(2-fluoroetoxi)fenil)(fenil)metil)piperidina (214 mg, 27 %) como sólido blanco; EM (ESI): m/z = 314,1 [M+H]+; y (R o S)-4-((3-(2-fluoroetoxi)fenil)(fenil)metil)piperidina (305 mg, 38 %) como sólido blanco; EM (ESI): m/z = 314,1 [M+H]+.
Intermedio
4-((3-(2-fluoroetoxi)fenil)(fenil)metil)piperidina
Se preparó el compuesto del título de forma análoga al intermedio 1-[(4-fluorofenil)-fenil-metil]piperacina (CAS RN 27064-89-7), comenzando a partir de 1-bromo-3-(2-fluoroetoxi)benceno (Ca s RN 132837-02-6) y 4-benzoilpiperidina-1-carboxilato de terc-butilo (CAS RN 922504-27-6) para dar el compuesto del título como aceite incoloro. EM (ESI): m/z = 314,1 [M+H]+.
BB104
(S o R)-4-((4-fluorofenil)(fenil)metil)piperidina
Se purificó una solución de sal de TFA de 4-[(4-fluorofenil)-fenil-metil]piperidina (690 mg, 1,8 mmol) en MeOH (10 ml) por separación por SFC para dar (S o R)-4-((4-fluorofenil)(fenil)metil)piperidina (172 mg, 35 %) como sólido amarillo claro; EM (ESI): m/z = 270,1 [M+H]+; y (R o S)-4-((4-fluorofenil)(fenil)metil)piperidina. Se disolvió (R o S)-4-((4-fluorofenil)(fenil)metil)piperidina en EtOAc (15 ml) y se lavó con Na2CO3 (5 ml, acuoso, 30 %) y agua (5 ml). Se separó la capa orgánica, se secó sobre Na2SO4, se filtró y concentró a vacío. Se liofilizó el residuo para dar (R o S)-4-((4-fluorofenil)(fenil)metil)piperidina (227 mg, 46 %) como sólido amarillo claro. EM (ESI): m/z = 270,1 [M+H]+.
Intermedios
a) 4-í(4-fluorofenil}-hidroxi-fenil-metillpiperidina-1-carboxilato de terc-butilo
A una solución de 4-bromofluorobenceno (2 g, 11,4 mmol) en THF (20 ml) enfriada hasta -78 °C, se le añadió nbutillitio (5,81 ml, 14,51 mmol) gota a gota y se agitó la mezcla de reacción durante 30 min. A continuación, se añadió a la mezcla una solución de 4-benzoilpiperidina-1-carboxilato de
terc
-butilo (CAS RN 922504-27-6; 3 g, 10,37 mmol) en THF (15 ml), que se agitó a -78 °C durante 2 horas. Se dejó que la mezcla se calentara hasta t. a., se vertió en una solución de NH4Cl acuosa sat. (50 ml) y se extrajo con EtOAc. Se secaron las capas orgánicas combinadas sobre Na2SO4, se filtró y concentró a vacío. Se purificó el residuo por HPLC preparativa (columna Gemini NX) para dar el compuesto del título (1,18 g, 29 %) como sólido incoloro. EM: 312,1 [M-C4Hs-H2O+H]+.
b) 4-[(4-fluorofenil)-fenil-metilenolpiperidina
A una solución de 4-[(4-fluorofenil)-hidroxi-fenil-metil]piperidina-1-carboxilato de terc-butilo (0,7 g, 1,82 mmol) en DCM (10 ml) se le añadió ácido trifluoroacético (1,4 ml, 18,16 mmol) y se agitó la reacción a t. a. durante 8 h. Se concentró la mezcla a vacío para dar el compuesto del título bruto (0,3 g, 62 %) como un sólido amarillo claro. EM: 268.0 [M+H]+.
c) Trifluoroacetato de 4-f(4-fluorofenil)-fenil-metillDiperidina
Se agitó una solución de 4-[(4-fluorofenil)-fenil-metileno]piperidina (1600 mg, 5,98 mmol) y Pd/C (300 mg, 5,98 mmol) en THF (100 ml) a t. a. durante 16 h en atmósfera de H2 (760 mmHg). Se filtró la suspensión y se concentró el filtrado a vacío. Se purificó el residuo por HPLC preparativa usando TFA como aditivo para obtener el compuesto del título como sal de ácido 2,2,2-trifluoroacético (700 mg, 29 %) como sólido blanco. EM (ESI): m/z = 270.1 [M+H]+.
BB105
(S o R)-4-((4-fluorofenil)(p-tolil)metil)piperidina
Se separó una solución de 4-[(4-fluorofenil)-(p-tolil)metil]piperidina (870 mg, 3,07 mmol) por SFC (procedimiento: columna DAICEL CHIRALPa K AD 250 mm* 30 mm, 5 pm, condición NH3^H2O al 0,1 % IPA, inicio B 35, fin B 35, tiempo de gradiente (min) 4,9 min; 110 min, tiempo de retención con B al 100 % (min) 0; caudal (ml/min) 60) para dar (R o S)-4-[(4-fluorofenil)(p-tolil)metil]piperidina (253 mg, 28 %; EM (ESI): m/z = 284,1 [M+H]+) y (S o R)-4-[(4-fluorofenil)(p-tolil)metil]piperidina (356 mg, 40 % de rendimiento; EM (ESI): m/z = 284,1 [M+H]+).
Intermedios
a) 4-((4-fluorofenil)(hidroxi)(p-tolil)metil)piperidina-1-carboxilato de terc-butilo
A una solución agitada a -78 °C de 4-bromotolueno (1,70 g, 9,94 mmol) en THF (40 ml) se le añadió una solución de butillitio (5,57 ml, 13,9 mmol). Después de agitar durante 1 h, se añadió a la mezcla 4-(4-fluorobenzoil)piperidina-1-carboxilato de terc-butilo (CAS RN 160296-40-2; 3,05 g, 9,94 mmol) y se continuó agitando a -78 °C durante 1 h. A continuación, se calentó la reacción hasta 25 °C y se agitó durante otras 13 h. Se detuvo la reacción con NH4Cl (ac. sat., 50 ml) y se extrajo con EtOAc (100 ml). Se separó la capa orgánica, se secó sobre Na2SO4, se filtró y concentró a vacío para dar el compuesto deseado como un aceite amarillo claro (2,85 g, 71 %). EM (ESI): m/z = 422,1 [M+Na]+.
b) 4-[(4-fluorofenil)-(p-tolil)metileno]piperidina
Se agitó una solución de 4-[(4-fluorofenil)-hidroxi-(p-tolil)metil]piperidina-1-carboxilato de tere-butilo (2,83 g, 7,08 mmol) y ácido trifluoroacético (11 ml, 142 mmol) en DCM (20 ml) a 25 °C durante 3 h. Se concentró la reacción a vacío. Se purificó el residuo por HPLC prep. (TFA como aditivo) para dar el compuesto del título como semisólido amarillo claro (1,36 mg, 67 %). EM (ESI): m/z = 282,1 [M+H]+.
e) Trifluoroaeetato de 4-[(4-fluorofenil)-(p-tolil)metil]piperidina
Se agitó una solución de 4-[(4-fluorofenil)-(p-tolil)metileno]piperidina (1,36 g, 4,83 mmol) y Pd/C (300 mg) en DMF (50 ml) a 25 °C durante 16 h bajo H2 (2280 mmHg). Se filtró la solución de reacción a través de Celite y se concentró a vacío. Se purificó el residuo por HPLC prep. (TFA como aditivo) para dar el compuesto deseado como semisólido blanco (sal de TFA, 870 mg, 45 %). EM (ESI): m/z = 284,1 [M+H]+.
BB106
(R o S)-4-((4-(2-fluoroetoxi)fenil)(fenil)metil)piperidina
Se purificó una solución de 4-[[4-(2-fluoroetoxi)fenil]-fenil-metil]piperidina (800 mg, 2,55 mmol) en MeOH (5 ml) y se separaron los enantiómeros por SFC para dar (S o R)-4-[[4-(2-fluoroetoxi)fenil]-fenil-metil]piperidina (279 mg, 0,89 mmol, 35 %) como sólido blanco; EM (ESI): m/z = 314,2 [M+H]+; y (R o S)-4-[[4-(2-fluoroetoxi)fenil]-fenilmetil]piperidina (373 mg, 47 %) como sólido blanco. EM (ESI): m/z = 314,1 [M+H]+.
Intermedio
4-((4-(2-fluoroetoxi)fenil)(fenil)metil)piperidina
Se preparó el compuesto del título de forma análoga al intermedio 1 -[(4-fluorofenil)-fenil-metil]piperacina (CAS RN 27064-89-7), comenzando a partir de 1-bromo-4-(2-fluoroetoxi)benceno (CAS RN 332-47-8) y 4-benzoilpiperidina-1-carboxilato de tere-butilo (CAS RN 922504-27-6) para dar el compuesto del título como aceite incoloro. EM (ESI): m/z = 314,1 [M+H]+.
BB107
(S o R)-4-[(4-fluorofenil)-(4-metoxifenil)metil]piperidina
Se purificó una solución de 4-[(4-fluorofenil)-(4-metoxifenil)metil]piperidina (930 mg, 3,11 mmol) en MeOH (5 ml) por separación por SFC para dar (S o R)-4-[(4-fluorofenil)-(4-metoxifenil)metil]piperidina (277 mg, 0,93 mmol, 27 %) como semisólido amarillo claro; EM (ESI): m/z = 300,1 [M+H]+; y (R o S)-4-[(4-fluorofenil)-(4-metoxifenil)metil]piperidina (291 mg, 30 %) como semisólido amarillo claro; e M (ESI): m/z = 300,1 [M+H]+.
Intermedio
4-[(4-fluorofenil)-(4-metoxifenil)metil]piperidina
Se preparó el compuesto del título de forma análoga al intermedio 1 -[(4-fluorofenil)-fenil-metil]piperacina (CAS RN 27064-89-7), comenzando a partir de 1-bromo-4-metoxibenceno (CAS RN 104-92-7) y 4-benzoilpiperidina-1-carboxilato de tere-butilo (CAS RN 922504-27-6) para dar el compuesto del título como semisólido incoloro. EM (ESI): m/z = 300,1 [M+H]+.
BB108
(R o S)-4-((4-fluorofenil)(p-tolil)metil)piperidina
Se separó una solución de 4-[(4-fluorofenil)-(p-tolil)metil]piperidina (870 mg, 3,07 mmol, BB105, intermedio c) por SFC (procedimiento: columna DAICEL CHIrAlPAK AD 250 mm* 30 mm, 5 pm, condición NH3^H2O al 0,1 % IpA, inicio B 35, fin B 35, tiempo de gradiente (min) 4,9 min; 110 min, tiempo de retención con B al 100 % (min) 0; caudal (ml/min) 60) para dar (R o S)-4-[(4-fluorofenil)(p-tolil)metil]piperidina (253 mg, 28 %); EM (ESI): m/z = 284,1 [M+H]+; y (S o R)-4-[(4-fluorofenil)(p-tolil)metil]piperidina (356 mg, 40 %). EM (ESI): m/z = 284,1 [M+H]+.
BB109
(S o R)-4-((3,4-dimetoxifenil)(4-fluorofenil)metil)piperidina
Se separó 4-((3,4-dimetoxifenil)(4-fluorofenil)metil)piperidina (900 mg) por SFC (procedimiento: columna DAICEL
CHIRALPAK AD (250 mm* 50 mm, 10 |jm), condición NH3^H2O al 0,1 % MeOH, inicio B 25, final B 25 tiempo de gradiente (min) 5,5 min:900 min, tiempo de retención con B al 100 % (min) 0, caudal (ml/min) 50 g/min) para dar (S o R)-4-((3,4-dimetoxifenil)(4-fluorofenil)metil)piperidina (278 mg, 29 %d). EM (ESI): m/z = 330,3 [M+H]+ y (R o S)-4-((3,4-dimetoxifenil)(4-fluorofenil)metil)piperidina (426 mg, 44 %; EM (ESI): m/z = 330,1 [M+H]+) como sólidos blanquecinos.
Intermedios
a) 4-((3,4-dimetoxifenil)(4-fluorofenil)(hidroxi)metil)piperidina-1-carboxilato de terc-butilo
A una solución de 4-bromoveratrol (CAS RN 2859-78-1; 1,27 g, 5,86 mmol) en THF (30 ml) se le añadió gota a gota butillitio (3,28 ml, 8,2 mmol) con agitación a -78 °C. Se continuó agitando a -78 °C durante 1 h. A continuación se añadió 4-(4-fluorobenzoil)piperidina-1-carboxilato de terc-butilo (CAS RN 160296-40-2; 1,80 g, 5,86 mmol) y se agitó la mezcla a -78 °C durante 5 h. Se vertió la mezcla en salmuera (50 ml) y se extrajo dos veces con EtOAc (30 ml cada vez). Se concentraron las capas orgánicas combinadas a vacío para dar el compuesto deseado como un aceite incoloro (2,4 g, 91 %). EM (ESI): m/z = 468,4 [M+Na]+.
b) Trifluoroacetato de 4-((3,4-dimetoxifenil)(4-fluorofenil)metileno)piperidina
Se agitó una mezcla de 4-((3,4-dimetoxifenil)(4-fluorofenil)(hidroxi)metil)piperidina-1-carboxilato de terc-butilo (2,4 g, 5,4 mmol) y ácido trifluoroacético (4,15 ml, 53,87 mmol) en DCM (20 ml) a 25 °C durante 4 h. Se concentró la mezcla de reacción a vacío para dar 4-((3,4-dimetoxifenil)(4-fluorofenil)metileno)piperidina (sal de TFA, 1,7 g, 96 %) como aceite marrón. EM (ESI): m/z = 328,3 [M+H]+.
c) 4-((3,4-dimetoxifenil)(4-fluorofenil)metil)piperidina
Se agitó una mezcla de 4-((3,4-dimetoxifenil)(4-fluorofenil)metileno)piperidina (1,7 g, 5,2 mmol, BB109, intermedio b) y Pd/C (276 mg, 0,260 mmol) en THF (10 ml) a 25 °C durante 16 h en H2 (760 mmHg). Se filtró la mezcla y se concentró a vacío para dar un residuo, que se purificó por HPLC preparativa (condiciones de TFA) para dar 4-((3,4-dimetoxifenil)(4-fluorofenil)metil)piperidina (900 mg, 52 %). como sólido blanco. EM (ESI): m/z = 330,3 [M+H]+. BB110
(R o S)-4-((3,4-dimetoxifenil)(fenil)metil)piperidina
Se purificó una solución de 4-[(3,4-dimetoxifenil)-fenil-metil]piperidina (900 mg, 2,89 mmol) en MeOH (5 ml) por separación por SFC para dar (S o R)-(3,4-dimetoxifenil)-fenil-metil]piperidina (326 mg, 36 %) como sólido blanco; e M (ESI): m/z = 312,3[M+H]+; y (R o S)-(3,4-dimetoxifenil)-fenil-metil]piperidina (222 mg, 24 %) como sólido blanco; EM (ESI): m/z = 312,3[M+H]+.
Intermedio
4-[(3,4-dimetoxifenil)-fenil-metil]piperidina
Se preparó el compuesto del título de forma análoga al intermedio 1 -[(4-fluorofenil)-fenil-metil]piperacina (CAS RN 27064-89-7), comenzando a partir de 4-bromo-1,2-dimetoxi-benceno (CAS RN 2859-78-1) y 4-benzoilpiperidina-1-carboxilato de terc-butilo (CAS RN 922504-27-6) para dar el compuesto del título como un sólido blanco. EM (ESI): m/z = 312,3 [M+H]+.
BB111
(R o S)-4-((4-fluorofenil)(fenil)metil)piperidina
Se purificó una solución de sal de TFA de 4-[(4-fluorofenil)-fenil-metil]piperidina (690 mg, 1,8 mmol) en MeOH (10 ml) por separación por SFC para dar (S o R)-4-((4-fluorofenil)(fenil)metil)piperidina (172 mg, 35 %) como un sólido amarillo claro. EM (ESI): m/z = 270,1 [M+H]+; y (R o S)-4-((4-fluorofenil)(fenil)metil)piperidina. Se disolvió (R o S)-4-((4-fluorofenil)(fenil)metil)piperidina en EtOAc (15 ml)y se lavó con solución de Na2CO3 acuosa (5 ml, 30 %) y agua (5 ml). Se separó la capa orgánica, se secó sobre Na2SO4, se filtró y concentró a vacío. Se liofilizó el residuo para dar (R o S)-4-((4-fluorofenil)(fenil)metil)piperidina (227 mg, 0,84 mmol, 46 %) como sólido amarillo claro; EM (ESI): m/z = 270,1 [M+H]+.
BB112
(S o R)-4-(fenil(m-tolil)metil)piperidina
Se purificó una solución de 4-(fenil(m-tolil)metil)piperidina (730 mg, 2,75 mmol) en MeOH (5 ml) por separación por
SFC para dar (S o R)-4-(fenil(m-tolil)metil)piperidina (221 mg, 0,83 mmol, 30 %) como semisólido amarillo. EM (ESI): m/z = 266,2 [M+H]+; y (R o S)-4-(fenil(m-tolil)metil)piperidina (228 mg, 31 %) como semisólido amarillo. EM (ESI): m/z = 266,2 [M+H]+.
Intermedio
4-(fenil(m-tolil)metil)piperidina
Se preparó el compuesto del título de forma análoga al intermedio 1-[(4-fluorofenil)-fenil-metil]piperacina (CAS RN 27064-89-7), comenzando a partir de 1-bromo-3-metil-benceno (CAS RN 591-17-3) y 4-benzoilpiperidina-1-carboxilato de tere-butilo (CAS RN 922504-27-6) para dar el compuesto del título como un sólido amarillo claro. EM (ESI): m/z = 266,1 [M+H]+.
BB113
(S o R)-4-((4-(2-fluoroetoxi)fenil)(fenil)metil)piperidina
Se purificó una solución de 4-[[4-(2-fluoroetoxi)fenil]-fenil-metil]piperidina (BB106, intermedio; 800 mg, 2,55 mmol) en MeOH (5 ml) y se separaron los enantiómeros por separación por SFC para dar (S o R)-4-[[4-(2-fluoroetoxi)fenil]-fenil-metil]piperidina (279 mg, 35 %) como sólido blanco, MS (ESI): m/z = 314,2 [M+H]+; y (R o S)-4-[(S)-[4-(2-fluoroetoxi)fenil]-fenil-metil]piperidina (373 mg, 47 %) como sólido blanco; EM (ESI): m/z = 314,1 [M+H]+.
BB114
(R o S)-4-(fenil(m-tolil)metil)piperidina
Se purificó una solución de 4-(fenil(m-tolil)metil)piperidina (BB112, intermedio, 730 mg, 2,75 mmol) en MeOH (5 ml) y se separaron los enantiómeros usando SFC para dar (S o R)-4-(fenil(m-tolil)metil)piperidina (221 mg, 30 %) como semisólido amarillo; EM (ESI): m/z = 266,2 [M+H]+; y (R o S)-4-(fenil(m-tolil)metil)piperidina (228 mg, 31 %) como semisólido amarillo. EM (ESI): m/z = 266,2 [M+H]+.
BB115
(S o R)-4-(fenil(p-tolil)metil)piperidina
Se purificó una solución de 4-(fenil(p-tolil)metil)piperidina (720 mg, 2,71 mmol) en MeOH (5 ml) y se separaron los enantiómeros usando SFC para dar ((S o R)-4-(fenil(p-tolil)metil)piperidina (277 mg, 35 %) como sólido blanquecino; EM (ESI): m/z = 266,2 [M+H]+; y (R o S)-4-(fenil(p-tolil)metil)piperidina (313 mg, 43 %) como sólido blanquecino MS (ESI): m/z = 266,2 [M+H]+.
Intermedio
4-(fenil(p-tolil)metil)piperidina
Se preparó el compuesto del título de forma análoga al intermedio 1 -[(4-fluorofenil)-fenil-metil]piperacina (CAS RN 27064-89-7), comenzando a partir de 1-bromo-4-metil-benceno (CAS RN 106-38-7) y 4-benzoilpiperidina-1-carboxilato de
tere
-butilo (CAS RN 922504-27-6) para dar el compuesto del título como sólido blanco, MS (ESI): m/z = 266,2 [M+H]+.
BB116
(R o S)-4-(fenil(p-tolil)metil)piperidina
Se purificó una solución de 4-(feni l (p-toli l)metil)pi pe ridina (BB117, intermedio, 720 mg, 2,71 mmol) en MeOH (5 ml) y se separaron los enantiómeros usando SFC para dar ((S o R)-4-(fenil(p-tolil)metil)piperidina (277 mg, 1,04 mmol, 35 %) como sólido blanquecino; MS (ESI): m/z = 266,2 [M+H]+; y (R o S)-4-(fenil(p-tolil)metil)piperidina (313 mg, 1,18 mmol, 43 %) como sólido blanquecino, MS (ESI): m/z = 266,2 [M+H]+.
BB117
3-benzhidrilaeetidina
Se agitó una solución de 3-(difenilmetileno)acetidina (800 mg, 3,62 mmol) y Pd/C (1923 mg) en THF (20 ml) a 25 °C en atmósfera de H2 (760 mmHg) durante 5 h. Se filtró la solución y se concentró a vacío para dar un residuo, que se purificó por HPLC preparativa (condición de TFA) para dar 3-benzhidrilacetidina (315 mg, 37 %) como sólido blanco. EM (ESI): m/z = 224,1 [M+H]+.
Intermedios
a) 3-[metoxi(metil)carbamoil]acetidina-1-carboxilato de terc-butilo
Se agitó una mezcla de ácido 1-boc-acetidina-3-carboxílico (CAS RN 142253-55-2; 50 g, 248 mmol), trietilamina (69,3 ml, 497 mmol), 1-hidroxibenzotriazol (33,5 g, 248 mmol) y EDCl (47,6 g, 248 mmol) y clorhidrato de
O,N-
dimetilhidroxilamina (24,24 g, 248,5 mmol) en DMF (1000 ml) a 25 °C durante 16 h. Se concentró la mezcla a vacío para dar un residuo, que se neutralizó con HCl (1 M) hasta pH = 7 y se extrajo tres veces con EtOAc (200 ml cada vez). Se lavaron dos veces las capas orgánicas combinadas con 200 ml de solución de NaHCO3 acuosa, se secó sobre Na2SO4 y se concentró a vacío para dar 3-[metoxi(metil)carbamoil]acetidina-1-carboxilato de terc-butilo (55 g, 72 %) como aceite incoloro. EM (ESI): m/z = 189,1 [M-C4Hs+H]+.
b) 3-benzoilacetidina-1-carboxilato de terc-butilo
A una solución de 3-[metoxi(metil)carbamoil]acetidina-1-carboxilato de terc-butilo (55 g, 225 mmol) en THF (600 ml) se le añadió bromuro de fenilmagnesio (82 ml, 248 mmol) con agitación a 0 °C y a continuación se agitó la solución a 0 °C durante 3 h. Se vertió la solución en solución de NH4Cl ac. sat. (300 ml) y se extrajo dos veces con EtOAc (150 ml cada vez). Se secaron as capas orgánicas combinadas sobre Na2SO4, se filtró y concentró a vacío para dar un residuo, que se purificó por cromatografía en columna (éter de petróleo: EtOAc 10: 1) para dar el compuesto deseado como sólido amarillo claro (28 g, 46 %). EM (ESI): m/z = 206,1 [M-C4Hs+H]+.
c) 3-(hidroxidifenilmetil)acetidina-1-carboxilato de terc-butilo
Se agitó una solución de bromuro de fenilmagnesio (3,06 ml, 9,18 mmol) y 3-benzoilacetidina-1-carboxilato de tercbutilo (2,0 g, 7,65 mmol) en THF (20 ml) a 0 °C durante 2 h. Se detuvo la solución por la adición de solución de NH4Cl ac. sat. (50 ml) y se extrajo dos veces con EtOAc (20 ml cada vez). Se concentraron las capas orgánicas combinadas a vacío para dar un residuo, que se purificó por cromatografía en columna ultrarrápida (PE: EtOAc 5: 1) para dar el compuesto deseado como sólido blanco (1,03 g, 39 %). EM (ESI): m/z = 362,2 [M+Na]+.
d) Trifluoroacetato de 3-(difenilmetileno)acetidina
Se agitó una solución de 3-(hidroxidifenilmetil)acetidina-1-carboxilato de terc-butilo (1,00 g, 2,95 mmol) y TFA (2,27 ml, 29,5 mmol) en DCM (15 ml) a 25 °C durante 4 h. Se concentró la solución a vacío para proporcionar el compuesto del título como aceite marrón (sal de TFA, 650 mg, 99 %). EM (ESI): m/z = 222,1 [M+h ]+.
BB118
(S o R)-4-((3,4-dimetoxifenil)(fenil)metil)piperidina
Se purificó una solución de 4-[(3,4-dimetoxifenil)-fenil-metil]piperidina (BB110, intermedio; 900 mg, 2,89 mmol) en MeOH (5 ml) y se separaron los enantiómeros usando SFC para producir (S o R)-(3,4-dimetoxifenil)-fenilmetil]piperidina (326 mg, 1,05 mmol, 36 %) como sólido blanco; EM (ESI): m/z = 312,3 [m +H]+; y (R o S)-(3,4-dimetoxifenil)-fenil-metil]piperidina (222 mg, 0,71 mmol, 24 %) como sólido blanco. EM (e S i): m/z = 312,3 [M+H]+.
BB119
(R o S)-4-((3,4-dimetoxifenil)(4-fluorofenil)metil)piperidina
Se separó 4-((3,4-dimetoxifenil)(4-fluorofenil)metil)piperidina (BB109, intermedio c; 900 mg) por SFC (procedimiento: columna DAICEL Ch IRALPAK Ad (250 mm* 50 mm, 10 |jm), condición NH3^H2O al 0,1 % MeOH, inicio B 25, fin B 25 tiempo de gradiente (min) 5,5 min:900 min, tiempo de retención con B al 100 % (min) 0, caudal (ml/min) 50 g/min) para dar (S o R)-4-((3,4-dimetoxifenil)(4-fluorofenil)metil)piperidina (278 mg, 29 %; EM (ESI): m/z = 330,3 [M+H]+) y (R o S)-4-((3,4-dimetoxifenil)(4-fluorofenil)metil)piperidina (426 mg, 44 %; EM (ESI): m/z = 330.1 [M+H]+) como sólidos blanquecinos.
BB120
(R o S)-4-[(4-fluorofenil)-(4-metoxifenil)metil]piperidina
Se purificó una solución de 4-[(4-fluorofenil)-(4-metoxifenil)metil]piperidina (BB107, intermedio; 930 mg, 3,11 mmol) en MeOH (5 ml) y se separaron los enantiómeros usando SFC para dar (S o R)-4-[(4-fluorofenil)-(4-metoxifenil)metil]piperidina (277 mg, 27 %) como semisólido amarillo claro; EM (ESI): m/z = 300,1 [M+H]+; y (R o S)-4-[(4-fluorofenil)-(4-metoxifenil)metil]piperidina (291 mg, 30 %) como semisólido amarillo claro; EM (ESI): m/z = 300.1 [M+H]+.
BB121
(R o S)-4-((4-(2-fluoroetoxi)fenil)(4-fluorofenil)metil)piperidina
Se purificó una solución de 4-[[4-(2-fluoroetoxi)fenil]-(4-fluorofenil)metil]piperidina (700 mg, 2,11 mmol) en MeOH (5 ml) y se separaron los enantiómeros usando SFC para dar (R o S)-4-[[4-(2-fluoroetoxi)fenil]-(4-fluorofenil)metil]piperidina (203 mg, 29 %) como aceite incoloro; EM (ESI): m/z = 332,2 [M+H]+; y (S o R)-4-[[4-(2-fluoroetoxi)fenil]-(4-fluorofenil)metil]piperidina (160 mg, 23 %) como aceite incoloro; EM (ESI): m/z = 332,2 [M+H]+.
Intermedio
4-[[4-(2-fluoroetoxi)fenil]-(4-fluorofenil)metil]piperidina
Se preparó el compuesto del título de forma análoga al intermedio 1 -[(4-fluorofenil)-fenil-metil]piperacina (CAS RN 27064-89-7), comenzando a partir de 1-bromo-4-(2-fluoroetoxi)benceno (CAS RN 332-47-8)y 4-(4-fluorobenzoil)piperidina-1-carboxilato de tere-butilo (CAS RN 160296-40-2) para dar el compuesto del título como aceite incoloro. EM (ESI): m/z = 332,1 [M+H]+.
BB122
(R o S)-4-((3-(2-fluoroetoxi)fenil)(fenil)metil)piperidina
Se purificó una solución de 4-[[3-(2-fluoroetoxi)fenil]-fenil-metil]piperidina (BB103, intermedio; 800 mg, 2,55 mmol) en MeOH (5 ml) y se separaron los enantiómeros usando SFC para dar (S o R)-4-((3-(2-fluoroetoxi)fenil)(fenil)metil)piperidina (214 mg, 27 %) como sólido blanco; EM (ESI): m/z = 314,1 [M+H]+; y (R o S)-4-((3-(2-fluoroetoxi)fenil)(fenil)metil)piperidina (305 mg, 38 %) como sólido blanco. EM (ESI): m/z = 314,1 [M+H]+. BB123
(S o R)-4-((4-(2-fluoroetoxi)fenil)(4-fluorofenil)metil)piperidina
Se purificó una solución de 4-[[4-(2-fluoroetoxi)fenil]-(4-fluorofenil)metil]piperidina (BB121, intermedio; 700 mg, 2,11 mmol) en MeOH (5 ml) y se separaron los enantiómeros por SFC para dar (R o S)-4-[[4-(2-fluoroetoxi)fenil]-(4-fluorofenil)metil]piperidina (203 mg, 29 %) como aceite incoloro; EM (ESI): m/z = 332,2 [M+H]+; y (S o R)-4-[[4-(2-fluoroetoxi)fenil]-(4-fluorofenil)metil]piperidina (160 mg, 23 %) como aceite incoloro. EM (ESI): m/z = 332,2 [M+H]+.
BB124
(2-(3-(fenil(piperidin-4-il)metil)fenoxi)etil)earbamato de tere-butilo
Se cargó una mezcla de 4-((3-(2-((tere-butoxicarbonil)amino)etoxi)fenil)(fenil)metileno)piperidina-1-carboxilato de bencilo (60 mg, 111 |jmol), metanol (3 ml) y AcOH (60 |jl) con Pt-Pd/C (60 mg, 2,5 %+2,5 %, 63 % de H2O) y se puso en una atmósfera de gas hidrógeno (50 bar). Se agitó la mezcla a 50 °C durante 2 h, se diluyó con EtoAc y se filtró. Se lavó el filtrado con KHCO3 y se secó la capa orgánica sobre Na2SO4. Se retiró el disolvente a presión reducida para dar el compuesto del título bruto (33 mg, 68 %) como espuma blanca que se usó en la etapa de reacción siguiente sin purificación adicional. EM (ESI): m/z = 411,3 [M+H]+.
Intermedios
a) (Z)-4-(fenil(2-tosilhidraeinailideno)metil)piperidina-1-earboxilato de beneilo
Se agitó una solución de 4-metilbencenosulfonhidracida (1,54 g, 8,29 mmol, CAS RN 1576-35-8;) y 4-benzoilpiperidina-1-carboxilato de tere-butilo (2 g, 6,91 mmol, CAS RN 922504-27-6) en 1,4-dioxano (150 ml) a 100 °C durante 16 h. Se añadieron EtOAc y agua. Se separaron las capas y se secó la capa orgánica sobre Na2SO4. Se retiró el disolvente a presión reducida y se purificó el residuo por cromatografía ultrarrápida (25 g de gel de sílice; gradiente de EtOAc (0-30 %) en n-heptano) para dar el compuesto del título (3,55 g, 82 %) como aceite marrón claro. EM (ESI): m/z = 492,2 [M+H]+.
b) 4-((3-(2-((tere-butoxiearbonil)amino)etoxi)fenil)(fenil)metileno)piperidina-1-earboxilato
Se agitó una mezcla de (Z)-4-(fenil(2-tosilhidracinailideno)metil)piperidina-1-carboxilato de bencilo (1 g mg, 2,03 mmol), cloruro de bis(trifenilfosfina)paladio(II)(143 mg, 203 jmol), tere-butóxido de litio (246 mg, 3,05 mmol) y N-boc-2-(3-bromofenoxi)etilamina (CAS RN 1098107-26-6; 772 mg, 2,44 jmol) en 1,4-dioxano (65 ml) a 80 °C durante 12 h. Se añadieron EtOAc y agua. Se separaron las capas y se secó la capa orgánica sobre Na2SO4. Se retiró el disolvente a presión reducida y se purificó el residuo por cromatografía ultrarrápida (80 g de gel de sílice; gradiente de EtOAc en n-heptano 0-30 %) para dar el compuesto del título (280 mg, 22 %) como espuma amarillo claro. EM (ESI): m/z = 443,2 [M-Boc+H]+.
BB125
Clorhidrato de 5-cloro-2-(piperidin-4-il)isoindolina
Se obtuvo el compuesto de forma análoga a BB52 a partir de 4-(5-cloroisoindolin-2-il)piperidina-1-carboxilato de terc-butilo para obtener el compuesto deseado como un sólido verde claro. EM (ESI): m/z = 237,2 [M+H]+.
Intermedio
4-(5-cloroisoindolin-2-il)piperidina-1-carboxilato de terc-butilo
Se obtuvo el compuesto de forma análoga a BB148, intermedio, a partir de clorhidrato de 5-cloroisoindolina (CAS RN 912999-79-2) para obtener el compuesto deseado como un sólido marrón claro. EM (ESI): m/z = 337,3 [M+H]+. BB126
Clorhidrato de (4aS,8aS)-hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona
Se agitó una suspensión blanca de (4aS,8aS)-3-oxohexahidro-2H-pirido[4,3-b][1,4]oxacina-6(5H)-carboxilato de terc-butilo (330 mg, 1,29 mmol) en cloruro de hidrógeno 2 M en éter (6,44 ml, 12,9 mmol) a t. a. durante 22 horas. Se filtró la suspensión incolora y se lavó con éter dietílico para obtener el producto deseado como un sólido incoloro (0,172 g; 69,3 %). EM (ESI): m/z = 157,0990 [M+H]+.
Intermedios
a) (4aS, 8aS)-3-oxohexahidro-2H-pirido[4,3-b][1,4]oxacina-6(5H)-carboxilato de terc-butilo
A una solución helada de (3S,4S)-3-(2-cloroacetamido)-4-hidroxipiperidina-1-carboxilato de terc-butilo (436 mg, 1,49 mmol) en DCM (7 ml) se le añadió gota a gota una solución de terc-butóxido de potasio (668 mg, 5,96 mmol) en 2-propanol (18 ml). Se retiró el baño de hielo y se agitó la mezcla a t. a. durante 24 horas mientras se obtenía una suspensión blanca. Se evaporó la mezcla. Se recogió el residuo en acetato de etilo y agua y se separaron las capas. Se extrajo dos veces la capa acuosa con acetato de etilo. Se secaron las capas orgánicas sobre MgSO4, se filtró, se trató con gel de sílice y se evaporó. Se purificó el compuesto por cromatografía en gel de sílice en una columna de 40 g usando un sistema MPLC que se eluyó con un gradiente de DCM: MeOH (de 100: 0 a 90: 10) para obtener el compuesto deseado como una espuma incolora (330 mg; 86,5 % de rendimiento). EM (ESI): m/z = 201,1 [M-C4H8+H]+.
b) (3S,4S)-3-(2-cloroacetamido)-4-hidroxipiperidina-1-carboxilato de terc-butilo
A una suspensión helada de (3S,4S)-3-amino-4-hidroxipiperidina-1-carboxilato de terc-butilo (500 mg, 2,31 mmol, CAS RN 1312812-78-4) y acetato de sodio trihidrato (629 mg, 4,62 mmol) en acetona (4 ml) y agua (300 |jl) se le añadió gota a gota una solución de cloruro de 2-cloroacetilo (261 mg, 184 jl, 2,31 mmol) en acetona (500 jl). Se agitó la mezcla a t. a. durante 3 horas antes de añadir gel de sílice. Se evaporó la suspensión. Se purificó el compuesto por cromatografía en gel de sílice en una columna de 12 g usando un sistema de MPLC (ISCO) que se eluyó con un gradiente de n-heptano:acetato de etilo (de 100: 0 a 0: 100) para obtener el compuesto deseado como una espuma incolora (436 mg; 64,4 %). EM (ESI): m/z = 291,3 [M+H]'.
BB127
Formiato de 4-[1-[4-(trifluorometil)fenil]etil]piperidina
Se agitó una solución de 4-[1 -[4-(trifluorometil)fenil]etil]piperidina-1 -carboxilato de terc-butilo (1500,0 mg, 4,2 mmol) en HCl en dioxano (50,0 ml, 200 mmol) a 20 °C durante 2 h. Se concentró la mezcla y se purificó el residuo por HPLC prep. (FA) y se liofilizó para dar el compuesto deseado como un aceite amarillo claro (838,4 mg, 77,1 %). EM (ESI): m/z = 258,1 [M+H]+.
Intermedios
a) 4-[1-[4-(trifluorometil)fenil]vinil]piperidina-1-carboxilato de terc-butilo
A una solución de bromuro de metiltrifenilfosfonio (499,8 mg, 1,4 mmol, CAS RN 1779-49-3) en THF (10 ml) se le añadió terc-butóxido de potasio (235,5 mg, 2,1 mmol) en porciones a 0 °C y se agitó la mezcla a 0 °C durante 30 min. Se añadió gota a gota 4-[4-(trifluorometil)benzoil]piperidina-1-carboxilato de terc-butilo (500,0 mg,
1,4 mmol, CAS RN 725229-27-6) en THF (5 ml) y se calentó la mezcla hasta 20 °C y se agitó a t. a. durante 12 h. Se vertió la mezcla en solución de NH4Cl ac. Saturada (50 ml) y se extrajo dos veces con EtOAc (20 ml cada vez). Se separaron las capas, se lavó la fase orgánica con salmuera, se secó sobre Na2SO4 y se filtró. Se purificó el filtrado en columna de gel de sílice (PE: EtOAc = 20:1) para dar el compuesto deseado como aceite amarillo claro (200 mg, 40,2 %). EM: EM (ESI): m/z = 300,0 [M-C4Hs+H]+.
b) 4-[1-[4-(trifluorometil)fenil]etil]piperidina-1-carboxilato de terc-butilo
A una solución de 4-[1-[4-(trifluorometil)fenil]vinil]piperidina-1 -carboxilato de terc-butilo (2,0 g, 5,63 mmol) en EtOAc (100 ml) se le añadió Pd/C (1,0 g, 5,63 mmol) y se agitó la mezcla a 20 °C en atmósfera de hidrógeno durante 12 h. Se filtró la mezcla y se concentró el filtrado para dar el producto bruto (1,8 g, 89,5 %) como un aceite incoloro que se usó en la etapa siguiente sin purificación adicional. EM (ESI): m/z = 302,2 [M-C4Hs+H]+.
BB128
Diclorhidrato de 3-(piperidin-4-il)-5-(trifluorometil)piridina
En un tubo de 25 ml, se disolvió 4-(5-(trifluorometil)piridin-3-il)piperidina-1-carboxilato de terc-butilo (110 mg, 333 |jmol) en DCM (1 ml) y se añadió HCl en éter 2 M (2 ml, 4 mmol). Se agitó la reacción durante 6 h a t. a. Se retiró el disolvente a vacío para obtener el producto bruto como una espuma blanca, 100 mg (99 %). Usado sin purificación adicional. EM (Es I): m/z = 231,1 [M+H]+.
Intermedios:
a) 4-(5-(trifluorometil)piridin-3-il)piperidina-1-carboxilato de terc-butilo
En un matraz de tres bocas de 25 ml seco, se combinó cinc en polvo (172 mg, 2,63 mmol) con 1 ml de DMA (sobre tamiz molecular) para dar una suspensión gris. Se agitó la mezcla a t. a. mientras se añadía una mezcla 7:5 v/v de clorotrimetilsilano (32 ul) y 1,2-dibromoetano (22 ul) como solución en DMA (1 ml) a una tasa para mantener la temperatura por debajo de 65 °C (ligeramente exotérmica). Se agitó la suspensión resultante durante 20 minutos. Se añadió lentamente a la mezcla una solución de 4-yodopiperidina-1-carboxilato de terc-butilo (688 mg, 2,21 mmol) en 2 ml de DMA. A continuación, se agitó la mezcla de reacción resultante durante 30 minutos a t. a. Se dejó que la reacción reposara durante 15 min sin agitar para su decantación.
En un matraz de tres bocas de 25 ml, se combinó 3-bromo-5-(trifluorometil)piridina (250 mg, 1,11 mmol) con 1,5 ml de DMA para dar una solución incolora, se añadieron yoduro de cobre(I) (21,1 mg, 111 jmol) y [1,1'-bis(difenilfosfino)ferroceno]dicloropaladio(II), complejo con diclorometano (45,2 mg, 55,3 jmol). Se desgasificó la mezcla de reacción con argón, se añadió la solución de reactivo de cinc recién preparada, se desgasificó de nuevo con argón y se agitó la mezcla de reacción durante la noche a 80 °C. Se detuvo la mezcla de reacción con 10 ml de solución de NH4Cl acuosa sat. y se extrajo dos veces con EtOAc (40 ml cada vez). Se lavaron las capas orgánicas con salmuera. Se combinaron las capas orgánicas, se secó sobre MgSO4 y se concentró a vacío. La purificación por cromatografía ultrarrápida en SiO2 usando n-heptano/EtOAc de 0 a 80 % en 35 min dio el producto deseado como aceite amarillo claro (112 mg, 31 %). EM (ESI): m/z = 331,2 [M+H]+.
BB129
Clorhidrato de 4-(difluoro(4-(trifluorometil)fenil)metil)piperidina
Se disolvió 4-(difluoro(4-(trifluorometil)fenil)metil)piperidina-1-carboxilato de terc-butilo (0,338 g, 891 jmol) en DCM (1 ml) y se añadió HCl en éter 2 M (4,45 ml, 8,91 mmol). Se agitó la reacción durante 16 h a t. a. Se retiró el disolvente a vacío para obtener el producto bruto como un sólido blanquecino, 246 mg (82 %). Usado sin purificación adicional. EM (ESI): m/z = 280,11 [M+H]+.
Intermedios:
a) 4-(4-(trifluorometil)benzoil)piperidina-1-carboxilato de terc-butilo
A una solución de clorhidrato de piperidin-4-il(4-(trifluorometil)fenil)metanona (0,500 g, 1,7 mmol), trietilamina (345 mg, 475 jl, 3,4 mmol) y DMAP (62,4 mg, 511 jmol) en acetonitrilo (10 ml), se le añadió a 0 °C bajo argón Boc2O (446 mg, 2,04 mmol). Se agitó la mezcla de reacción a t. a. durante 4 horas. Se vertió la mezcla de reacción en 15 ml de NaHCO3 saturado y se extrajo dos veces con EtOAc (25 ml cada vez). Se combinaron las capas orgánicas, se lavó con salmuera, se secó sobre Na2SO4 y se concentró a vacío. Se trituró el material bruto con nheptano para proporcionar el producto deseado como un sólido blanquecino (354 mg, 58 %), que se usó directamente para la etapa siguiente. EM (ESI): m/z = 302,1 [M-C4Hs+H]+.
b) 4-(difluoro(4-(trifluorometil)fenil)metil)piperidina-1-carboxilato de terc-butilo
A una solución de 4-(4-(trifluorometil)benzoil)piperidina-1-carboxilato de tere-butilo (0,350 g, 979 |jmol) en DCM (3 ml), se le añadió DAST (5,53 g, 34,3 mmol). Se agitó la mezcla de reacción a 45 °C durante tres días. Se vertió la mezcla de reacción en 15 ml de H2O y hielo y se extrajo dos veces con DCM (20 ml cada vez). Se combinaron las capas orgánicas, se lavó con salmuera, se secó sobre Na2SO4 y se concentró a vacío. Se purificó el material bruto por cromatografía ultrarrápida (gel de sílice, 20 g, de 0 % a 20 % de EtOAc en n-heptano) para proporcionar el compuesto deseado como un aceite amarillo (341 mg, 92 %). EM (ESI): m/z = 379,1 [M+H]+.
BB130
Diclorhidrato de 3-metoxi-5-(piperídin-4-il)pirídina
En un tubo de 25 ml, se disolvió 4-(5-metoxipiridin-3-il)piperidina-1 -carboxilato de tere-butilo (150 mg, 513 jmol) en DCM (1 ml) y se añadió HCl en éter 2 M (2,57 ml, 5,13 mmol). Se agitó la reacción durante 3 h a t. a. Se retiró el disolvente a vacío para obtener el producto bruto como una espuma amarillo claro, 136 mg (100 %). Usado sin purificación adicional. EM (ESI): m/z = 193,1 [M+H]+.
Intermedio:
4-(5-metoxipiridin-3-il)piperidina-1-earboxilato de tere-butilo
En un matraz de tres bocas de 25 ml seco, se combinó cinc en polvo (232 mg, 3,54 mmol) con 1 ml de DMA (sobre tamiz molecular) para dar una suspensión gris. Se agitó la mezcla a t. a. mientras se añadía una mezcla 7:5 v/v de clorotrimetilsilano (32 ul) y 1,2-dibromoetano (22 ul) como solución en DMA (1 ml) a una tasa para mantener la temperatura por debajo de 65 °C (ligeramente exotérmica). Se agitó la suspensión resultante durante 20 minutos. Se añadió lentamente a la mezcla una solución de 4-yodopiperidina-1-carboxilato de tere-butilo (927 mg, 2,98 mmol) en 2 ml de DMA. A continuación, se agitó la mezcla de reacción resultante durante 30 minutos a t. a. Se dejó que la reacción reposara durante 15 min sin agitar para su decantación.
En un matraz de tres bocas de 25 ml, se combinó 3-bromo-5-metoxipiridina (280 mg, 1,49 mmol) con 1,5 ml de DMA para dar una solución incolora, se añadieron yoduro de cobre(I) (28,4 mg, 149 jmol) y [1,1'-bis(difenilfosfino)ferroceno]dicloropaladio(II), complejo con diclorometano (60,8 mg, 74,5 jmol). Se desgasificó la mezcla de reacción con argón, se añadió la solución de reactivo de cinc recién preparada, se desgasificó de nuevo con argón y se agitó la mezcla de reacción durante 6 h a 80 °C. Se detuvo la mezcla de reacción con 10 ml de NH4Cl sat. y se extrajo dos veces con EtOAc (40 ml cada vez). Se lavaron las capas orgánicas con salmuera. Se combinaron las capas orgánicas, se secó sobre MgSO4 y se concentró a vacío. La purificación por cromatografía ultrarrápida en SiO2 usando n-heptano/EtOAc de 0 a 100 % en 35 min dio el producto deseado como aceite amarillo claro (150 mg, 34,5 %). EM (ESI): m/z = 293,2 [M+H]+.
BB131
Clorhidrato de 4-((4-elorofenil)difluorometil)piperidina
Se disolvió 4-(difluoro(4-(trifluorometil)fenil)metil)piperidina-1-carboxilato de tere-butilo (440 mg, 1,27 mmol) en DCM (2 ml) y se añadió HCl en éter 2 M (6,36 ml, 12,7 mmol). Se agitó la reacción durante 64 h a t. a. Se retiró el disolvente a vacío para obtener el producto bruto como un sólido blanquecino, 323 mg (90 %). Usado sin purificación adicional. EM (ESI): m/z = 246,1 [M+H]+.
Intermedio
4-(difluoro(4-(trifluorometil)fenil)metil)piperidina-1-earboxilato de tere-butilo
A una solución de 4-(4-clorobenzoil)piperidina-1-carboxilato de tere-butilo (0,500 g, 1,54 mmol) en DCM (3 ml), se le añadió DAST (8,71 g, 54 mmol). Se agitó la mezcla de reacción a 45 °C durante ocho días. Se vertió la mezcla de reacción en 15 ml de H2O y hielo y se extrajo dos veces con DCM (20 ml cada vez). Se combinaron las capas orgánicas, se lavó con salmuera, se secó sobre Na2SO4 y se concentró a vacío. Se purificó el material bruto por cromatografía ultrarrápida (gel de sílice, 20 g, de 0 % a 20 % de EtOAc en n-heptano) para proporcionar el producto deseado como aceite amarillo (440 mg, 82 %). EM (ESI): m/z = 345,1 [M+H]+.
BB132
Dielorhidrato de 2-(piperidin-4-il)-5-(trifluorometoxi)piridina
En un tubo de 25 ml, se disolvió 4-(5-(trifluorometoxi)piridin-2-il)piperidina-1 -carboxilato de tere-butilo (380 mg, 1,1 mmol) en DCM (2 ml) y se añadió HCl en éter 2 M (3,29 ml, 6,58 mmol). Se agitó la reacción a t. a. durante 4 h. Se retiró el disolvente a vacío para obtener el producto bruto como una espuma amarillo claro (349 mg, 99 %) que
se usó en la etapa siguiente sin purificación adicional. EM (ESI): m/z = 246,2 [M+H]+.
Intermedio:
4-(5-(trifluorometoxi)piridin-2-il)piperidina-1-carboxilato de terc-butilo
En un matraz de tres bocas de 25 ml seco, se combinó cinc en polvo (193 mg, 2,38 mmol) con 1,5 ml de DMA (sobre tamiz molecular) para dar una suspensión gris. Se agitó la mezcla a t. a. mientras se añadía una mezcla 7:5 v/v de clorotrimetilsilano (32 ul) y 1,2-dibromoetano (22 ul) como solución en DMA (1 ml) a una tasa para mantener la temperatura por debajo de 65 °C (ligeramente exotérmica). Se agitó la suspensión resultante durante 20 minutos. Se añadió lentamente a la mezcla una solución de 4-yodopiperidina-1-carboxilato de terc-butilo (771 mg, 2,48 mmol) en 2 ml de DMA. A continuación, se agitó la mezcla de reacción resultante durante 30 minutos a t. a. Se dejó que la reacción reposara durante 15 min sin agitar para su decantación.
En un matraz de tres bocas de 25 ml, se combinó 2-bromo-5-(trifluorometoxi)piridina (300 mg, 172 pl, 1,24 mmol) con 1,5 ml de DMA para dar una solución incolora, se añadieron yoduro de cobre(I) (23,6 mg, 124 pmol) y [1,1'-bis(difenilfosfino)ferroceno]dicloropaladio(II), complejo con diclorometano (50,6 mg, 62 pmol). Se desgasificó la mezcla de reacción con argón, se añadió la solución de reactivo de cinc recién preparada, se desgasificó de nuevo con argón y se agitó la mezcla de reacción durante 6 h a 80 °C. Se detuvo la mezcla de reacción con 10 ml de NH4Cl sat. y se extrajo dos veces con EtOAc (40 ml cada vez). Se lavaron las capas orgánicas con salmuera. Se combinaron las capas orgánicas, se secó sobre MgSO4 y se concentró a vacío. La purificación por cromatografía ultrarrápida en SiO2 usando n-heptano/EtOAc de 0 a 50 % en 35 min dio el producto deseado como aceite amarillo claro (387 mg, 90,1 %). EM (ESI): m/z = 347,1 [M+H]+.
BB133
Clorhidrato de 4-((2-cloro-4-fluorofenil)difluorometil)piperidina
En un tubo de 25 ml, se disolvió 4-((2-cloro-4-fluorofenil)difluorometil)piperidina-1-carboxilato de terc-butilo (0,363 g, 998 pmol) en DCM (2 ml) y se añadió HCl en éter 2 M (4,99 ml, 9,98 mmol). Se agitó la reacción a t. a. durante la noche. Se retiró el disolvente a vacío para obtener el producto bruto como un sólido marrón claro, 296 mg (99 %). Usado sin purificación adicional. EM (ESI): m/z = 264,08 [M+H]+.
Intermedio
4-((2-cloro-4-fluorofenil)difluorometil)piperidina-1-carboxilato de terc-butilo
A una solución de 4-(2-cloro-4-fluorobenzoil)piperidina-1-carboxilato de terc-butilo (0,48 g, 1,4 mmol) en DCM (4 ml), se le añadió DAST (7,92 g, 49,2 mmol). Se agitó la mezcla de reacción a 45 °C durante diez días. Se vertió la mezcla de reacción en 15 ml de H2O hielo y se extrajo con DCM (2 x 20 ml). Se combinaron las capas orgánicas, se lavó con salmuera, se secó sobre Na2SO4 y se concentró a vacío. Se purificó el material bruto por cromatografía ultrarrápida (gel de sílice, 20 g, de 0 % a 20 % de EtOAc en n-heptano): 363 mg (71 %), aceite marrón claro, producto deseado. EM (ESI): m/z = 363,1 [M+H]+.
BB134
Diclorhidrato de 3-etil-5-(piperidin-4-il)piridina
En un tubo de 25 ml, se disolvió 4-(5-etilpiridin-3-il)piperidina-1-carboxilato de terc-butilo (295 mg, 1,02 mmol) en DCM (3 ml) y se añadió HCl en éter 2 M (3,05 ml, 6,09 mmol). Se agitó la reacción durante 4 h a t. a. Se retiró el disolvente a vacío para obtener el producto bruto como un sólido amarillo, 266 mg (99 %). Usado sin purificación adicional. EM (ESI): m/z = 191,1 [M+H]+.
Intermedio
4-(5-etilpiridin-3-il)piperidina-1-carboxilato de terc-butilo
En un matraz de tres bocas de 25 ml seco, se combinó cinc en polvo (234 mg, 3,58 mmol) con 1,5 ml de DMA (sobre tamiz molecular) para dar una suspensión gris. Se agitó la mezcla a t. a. mientras se añadía una mezcla 7:5 v/v de clorotrimetilsilano (32 ul) y 1,2-dibromoetano (22 ul) como solución en DMA (1 ml) a una tasa para mantener la temperatura por debajo de 65 °C (ligeramente exotérmica). Se agitó la suspensión resultante durante 20 minutos. Se añadió lentamente a la mezcla una solución de 4-yodopiperidina-1-carboxilato de terc-butilo (937 mg, 3,01 mmol) en 2 ml de DMA. A continuación, se agitó la mezcla de reacción resultante durante 30 minutos a t. a. Se dejó que la reacción reposara durante 15 min sin agitar para su decantación.
En un matraz de tres bocas de 25 ml, se combinó 3-bromo-5-etilpiridina (280 mg, 1,5 mmol) con 1,5 ml de DMA
para dar una solución incolora, se añadieron yoduro de cobre(I) (28,7 mg, 150 |jmol) y [1,1'-bis(difenilfosfino)ferroceno]didoropaladio(N), complejo con diclorometano (61,5 mg, 75,2 jmol). Se desgasificó la mezcla de reacción con argón, se añadió la solución de reactivo de cinc recién preparada, se desgasificó de nuevo con argón y se agitó la mezcla de reacción durante 2,5 h a 80 °C. Se detuvo la mezcla de reacción con 10 ml de solución de NH4Cl acuosa sat. y se extrajo con EtOAc (40 ml cada vez). Se lavaron las capas orgánicas con salmuera. Se combinaron las capas orgánicas, se secó sobre MgSO4 y se concentró a vacío. La purificación por cromatografía ultrarrápida en SiO2 usando n-heptano/EtOAc de 0 a 60 % en 40 min dio el producto deseado como aceite amarillo claro (299 mg, 68,4 %). EM (ESI): m/z = 291,2 [M+H]+.
BB135
Diclorhidrato de 5-(1,1-difluoroetil)-2-(piperidin-4-il)piridina
En un tubo de 25 ml, se disolvió 4-(5-(1, 1 -difluoroetil)piridin-2-il)piperidina-1 -carboxilato de tere-butilo (416 mg, 1,27 mmol) en DCM (3 ml) y se añadió HCl en éter 2 M (3,82 ml, 7,65 mmol). Se agitó la reacción durante 4 h a t. a. Se retiró el disolvente a vacío para obtener el producto bruto como un sólido amarillo, 381 mg (99 %). Usado sin purificación adicional. EM (ESI): m/z = 227,2 [M+H]+.
Intermedio:
4-(5-( 1,1-difluoroetil)piridin-2-il)piperidina-1-earboxilato de tere-butilo
En un matraz de tres bocas de 25 ml seco, se combinó cinc en polvo (210 mg, 3,22 mmol) con 1,5 ml de DMA (sobre tamiz molecular) para dar una suspensión gris. Se agitó la mezcla a t. a. mientras se añadía una mezcla 7:5 v/v de clorotrimetilsilano (32 ul) y 1,2-dibromoetano (22 ul) como solución en DMA (1 ml) a una tasa para mantener la temperatura por debajo de 65 °C (ligeramente exotérmica). Se agitó la suspensión resultante durante 20 minutos. Se añadió lentamente a la mezcla una solución de 4-yodopiperidina-1-carboxilato de tere-butilo (841 mg, 2,7 mmol) en 2 ml de DMA. A continuación, se agitó la mezcla de reacción resultante durante 30 minutos a t. a. Se dejó que la reacción reposara durante 15 min sin agitar para su decantación.
En un matraz de tres bocas de 25 ml, se combinó 2-bromo-5-( 1, 1 -difluoroetil)piridina (300 mg, 1,35 mmol) con 1,5 ml de DMA para dar una solución incolora, se añadieron yoduro de cobre(I) (25,7 mg, 135 jmol) y [1,1'-bis(difenilfosfino)ferroceno]dicloropaladio(II), complejo con diclorometano (55,2 mg, 67,6 jmol). Se desgasificó la mezcla de reacción con argón, se añadió la solución de reactivo de cinc recién preparada, se desgasificó de nuevo con argón y se agitó la mezcla de reacción durante 2,5 h a 80 °C. Se detuvo la mezcla de reacción con 10 ml de NH4Cl sat. y se extrajo dos veces con EtOAc (40 ml cada vez). Se lavaron las capas orgánicas con salmuera. Se combinaron las capas orgánicas, se secó sobre MgSO4 y se concentró a vacío. La purificación por cromatografía ultrarrápida en SiO2 usando n-heptano/EtOAc de 0 a 50 % en 40 min dio el producto deseado como aceite amarillo (419 mg, 85 %). EM (ESI): m/z = 327,2 [M+H]+.
BB136
(4aR,8aS)-6-(4-(5-(1,1-difluoroetil)piridin-2-il)-3-metilpiperidina-1-earbonil)hexahidro-2H-pirido[4,3-b][1,4]oxaein-3(4H)-ona
Se preparó el compuesto del título de forma análoga al procedimiento A2 a partir de BB14a y diclorhidrato de 5-(1,1 -difluoroetil)-2-(3-metil-4-piperidil)piridina. EM (ESI): m/z = 423,4 [M+H]+.
Intermedios:
a) Dielorhidrato de 5-(1,1-difluoroetil)-2-(3-metil-4-piperidil)piridina
En un tubo de 25 ml, se disolvió 4-(5-(1,1-difluoroetil)piridin-2-il)-3-metilpiperidina-1-carboxilato de tere-butilo (265 mg, 778 jmol) en DCM (3 ml) y se añadió HCl en éter 2 M (3,11 ml, 6,23 mmol). Se agitó la reacción durante 4 h a t. a. Se retiró el disolvente a vacío para obtener el producto bruto como un sólido amarillo claro (242 mg, 99 %). Usado sin purificación adicional. e M (ESI): m/z = 241,2 [M+H]+.
b) 4-[5-(1,1-difluoroetil)-2-piridil]-3-metil-piperidina-1-earboxilato de tere-butilo
En un matraz de tres bocas de 25 ml seco, se combinó cinc en polvo (140 mg, 2,14 mmol) con 1,5 ml de DMA (sobre tamiz molecular) para dar una suspensión gris. Se agitó la mezcla a t. a. mientras se añadía una mezcla 7:5 v/v de clorotrimetilsilano (32 ul) y 1,2-dibromoetano (22 ul) como solución en DMA (1 ml) a una tasa para mantener la temperatura por debajo de 65 °C (ligeramente exotérmica). Se agitó la suspensión resultante durante 20 minutos. Se añadió lentamente a la mezcla una solución de 4-yodo-3-metilpiperidina-1-carboxilato de tere-butilo (586 mg, 1,8 mmol) en 2 ml de DMA. A continuación, se agitó la mezcla de reacción resultante durante 30 minutos a t. a. Se dejó que la reacción reposara durante 15 min sin agitar para su decantación.
En un matraz de tres bocas de 25 ml, se combinó 2-bromo-5-(1,1-difluoroetil)piridina (200 mg, 901 |jmol, CAS RN 1211521-60-6) con 1,5 ml de DMA para dar una solución incolora, se añadieron yoduro de cobre(I) (17,2 mg, 90,1 jmol) y [1,1'-bis(difenilfosfino)ferroceno]dicloropaladio(II), complejo con diclorometano (36,8 mg, 45 jmol). Se desgasificó la mezcla de reacción con argón, se añadió la solución de reactivo de cinc recién preparada, se desgasificó de nuevo con argón y se agitó la mezcla de reacción durante 3 h a 80 °C. Se detuvo la mezcla de reacción con 10 ml de NH4CI sat. y se extrajo dos veces con EtOAc (40 ml cada vez). Se lavaron las capas orgánicas con salmuera. Se combinaron las capas orgánicas, se secó sobre MgSO4 y se concentró a vacío. La purificación por cromatografía ultrarrápida en SiO2 usando n-heptano/EtOAc (de 0 a 50 % durante 35 min), dio el producto deseado como aceite amarillo (266 mg, 87 %). EM (ESI): m/z = 341,2 [M+H]+.
BB137
Clorhidrato de 1-(2,2,2-trifluoro-1-fenil-etil)piperacina
En un tubo de 25 ml, se disolvió 4-(2,2,2-trifluoro-1 -feniletil) piperacina-1-carboxilato de terc-butilo (155 mg, 450 jmol) en DCM (2 ml) y se añadió HCl en éter 2 M (1,8 ml, 3,6 mmol). Se agitó la reacción durante 4 h a t. a. Se retiró el disolvente a vacío para obtener el producto bruto como un sólido marrón claro, 127 mg (100 %). Usado sin purificación adicional. EM (ESI): m/z = 245,3 [M+H]+.
Intermedio
4-(2,2,2-trifluoro-1-fenil-etil)piperacina-1-carboxilato de terc-butilo
A un matraz de 100 ml seco con tapón de goma y burbujeador de N2 se le añadió piperacina-1-carboxilato de tercbutilo (1000 mg, 5,37 mmol), trietilamina (1,63 g, 2,25 ml, 16,1 mmol), 2,2,2-trifluoro-1-feniletan-1-ona (935 mg, 754 jl, 5,37 mmol) y DCM (33 ml). Se añadió tetracloruro de titanio 1 M en DCM (2,68 ml, 2,68 mmol) por medio de una jeringa. Se agitó la reacción durante 18 h, se detuvo cuidadosamente con una solución metanólica de NaCNBHa (cianoborohidruro de sodio (1,01 g, 16,1 mmol) en metanol (12,8 ml, 316 mmol) y se agitó durante 15 min. Se alcalinizó la reacción hasta pH 13 con NaOH 5 M (0,5 ml), se extrajo con DCM (2x 60 ml), se secó (Na2SO4) y se evaporó para proporcionar un aceite amarillo (2 g). La purificación por cromatografía ultrarrápida en SiO2 usando un gradiente de EtOAc en n-heptano (de 0 a 30 % durante 40 min), dio el producto deseado como un aceite amarillo claro (156 mg, 8,4 %). EM (ESI): m/z = 345,2 [M+H]+.
BB138
1-[1-(2,4-difluorofenil)-2,2,2-trifluoro-etil]piperacina; clorhidrato
En un tubo de 25 ml, se disolvió 4-(2,2,2-trifluoro-1-feniletil)piperacina-1-carboxilato de terc-butilo (330 mg, 868 jmol) en DCM (2 ml) y se añadió HCl en éter 2 M (2,6 ml, 5,1 mmol). Se agitó la reacción durante 4 h a t. a. Se retiró el disolvente a vacío para obtener el producto bruto como un aceite viscoso marrón, 266 mg (97 %). Usado sin purificación adicional. EM (ESI): m/z = 282,3 [M+H]+.
Intermedio
4-[1-(2,4-difíuorofenil)-2,2,2-trifíuoro-etil]piperacina-1-carboxilato de terc-butilo
A un matraz de 100 ml seco con tapón de goma y burbujeador de N2 se le añadió piperacina-1-carboxilato de tercbutilo (1000 mg, 5,37 mmol), trietilamina (1,63 g, 2,25 ml, 16,1 mmol), 1-(2,4-difluorofenil))-2,2,2-trifluoroetan-1-ona (1,13 g, 5,37 mmol) y DCM (33 ml). Se añadió tetracloruro de titanio 1 M en DCM (2,68 ml, 2,68 mmol) por medio de una jeringa. Se agitó la reacción durante 18 h, se detuvo cuidadosamente con una solución metanólica de NaCNBH3 (cianoborohidruro de sodio (1,01 g, 16,1 mmol) en metanol (4,3 ml, 107 mmol) y se agitó durante 6 h. Se alcalinizó la reacción con NaHCO3 sat. (10 ml). Se separó por filtración el material insoluble obtenido usando celite, se extrajo el filtrado con DCM (2x 60 ml), se secó (Na2SO4) y se evaporó para proporcionar un aceite amarillo (2,1 g).
La purificación por cromatografía ultrarrápida en SiO2 con n-heptano/EtOAc de 0 a 20 % en 40 min dio el producto deseado como un aceite amarillo (340 mg, 13,3 %). EM (ESI): m/z = 381,1 [M+H]+.
BB139
Diclorhidrato de 2-ciclopropil-4-(4-piperidil)piridina
En un tubo de 25 ml, se disolvió 4-(2-ciclopropilpiridin-4-il)piperidina-1 -carboxilato de terc-butilo (278 mg, 919 jmol) en DCM (2 ml) y se añadió HCl en éter 2 M (2,76 ml, 5,52 mmol). Se agitó la reacción durante 4 h a t. a. Se retiró el disolvente a vacío para obtener el producto bruto como un sólido amarillo claro, 250 mg (99 %). Usado sin
purificación adicional. EM (ESI): m/z = 202,4 [M+H]+.
Intermedio
4-(2-ciclopropil-4-piridil)piperidina-1-carboxilato de terc-butilo
En un matraz de tres bocas de 25 ml seco, se combinó cinc en polvo (196 mg, 3 mmol) con 1,5 ml de DMA (sobre tamiz molecular) para dar una suspensión gris. Se agitó la mezcla a t. a. mientras se añadía una mezcla 7:5 v/v de clorotrimetilsilano (32 ul) y 1,2-dibromoetano (22 ul) como solución en DMA (1 ml) a una tasa para mantener la temperatura por debajo de 65 °C (ligeramente exotérmica). Se agitó la suspensión resultante durante 20 minutos. Se añadió lentamente a la mezcla una solución de 4-yodopiperidina-1-carboxilato de terc-butilo (786 mg, 2,52 mmol) en 2 ml de DMA. A continuación, se agitó la mezcla de reacción resultante durante 30 minutos a t. a. Se dejó que la reacción reposara durante 15 min sin agitar para su decantación.
En un matraz de tres bocas de 25 ml se combinó 4-bromo-2-ciclopropilpiridina (250 mg, 1,26 mmol) con 1,5 ml de DMA para dar una solución incolora, se añadieron yoduro de cobre(I) (24 mg, 126 |jmol) y [1,1'-bis(difenilfosfino)ferroceno]dicloropaladio(II), complejo con diclorometano (51,1 mg, 63,1 jmol). Se desgasificó la mezcla de reacción con argón, se añadió la solución de reactivo de cinc recién preparada, se desgasificó de nuevo con argón y se agitó la mezcla de reacción durante 3 h a 80 °C. Se detuvo la mezcla de reacción con 10 ml de solución de NH4Cl acuosa sat. y se extrajo con EtOAc (40 ml cada vez). Se lavaron las capas orgánicas con salmuera. Se combinaron las capas orgánicas, se secó sobre MgSO4 y se concentró a vacío. La purificación por cromatografía ultrarrápida en SiO2 usando n-heptano/ de 0 a 60 % en 45 min dio el producto deseado como aceite amarillo claro (278 mg, 73 %). EM (ESI): m/z = 303,2 [M+H]+.
BB140
Diclorhidrato de 3-metil-5-(4-piperidil)-2-(trifluorometil)piridina
En un tubo de 25 ml, se disolvió 4-(5-metil-6-(trifluorometil)piridin-3-il)piperidina-1-carboxilato de terc-butilo (360 mg, 1,05 mmol) en DCM (3 ml) y se añadió HCl en éter 2 M (2,61 ml, 5,23 mmol). Se agitó la reacción durante 4 h a t. a. Se retiró el disolvente a vacío para obtener el producto bruto como un sólido blanco, 314 mg (95 %). Usado sin purificación adicional. EM (ESI): m/z = 245,2 [M+H]+.
Intermedio
4-[5-metil-6-(trifluorometil)-3-piridil]piperidina-1-carboxilato de terc-butilo
En un matraz de tres bocas de 25 ml seco, se combinó cinc en polvo (182 mg, 2,78 mmol) con 1,5 ml de DMA (sobre tamiz molecular) para dar una suspensión gris. Se agitó la mezcla a t. a. mientras se añadía una mezcla 7:5 v/v de clorotrimetilsilano (32 ul) y 1,2-dibromoetano (22 ul) como solución en DMA (1 ml) a una tasa para mantener la temperatura por debajo de 65 °C (ligeramente exotérmica). Se agitó la suspensión resultante durante 20 minutos. Se añadió lentamente a la mezcla una solución de 4-yodopiperidina-1-carboxilato de terc-butilo (726 mg, 2,33 mmol) en 2 ml de DMA. A continuación, se agitó la mezcla de reacción resultante durante 30 minutos a t. a. Se dejó que la reacción reposara durante 15 min sin agitar para su decantación.
En un matraz de tres bocas de 25 ml, se combinó 5-bromo-3-metil-2-(trifluorometil)piridina (280 mg, 1,17 mmol) con 1,5 ml de DMA para dar una solución incolora, se añadieron yoduro de cobre(I) (22,2 mg, 117 jmol) y [1,1'-bis(difenilfosfino)ferroceno]dicloropaladio(II), complejo con diclorometano (48 mg, 58 jmol). Se desgasificó la mezcla de reacción con argón, se añadió la solución de reactivo de cinc recién preparada, se desgasificó de nuevo con argón y se agitó la mezcla de reacción durante 2,5 h a 70 °C. Se detuvo la mezcla de reacción con 10 ml de NH4CI sat. y se extrajo dos veces con EtOAc (40 ml cada vez). Se lavaron las capas orgánicas con salmuera. Se combinaron las capas orgánicas, se secó sobre MgSO4 y se concentró a vacío. La purificación por cromatografía ultrarrápida en SiO2 usando n-heptano/EtOAc de 0 a 50 % en 40 min dio el producto deseado como un sólido cristalino blanco (360 mg, 90 %). EM (ESI): m/z = 345,2 [M+H]+.
BB141
Diclorhidrato de 4-(4-piperidil)-5,6,7,8-tetrahidroquinolina
En un tubo de 25 ml, se disolvió 4-(5,6,7,8-tetrahidroquinolin-4-il)piperidina-1-carboxilato de terc-butilo (255 mg, 806 jmol) en DCM (3 ml) y se añadió HCl en éter 2 M (2,01 ml, 4,03 mmol). Se agitó la reacción durante 8 h a t. a. Se retiró el disolvente a vacío para obtener el producto bruto como un sólido amarillo claro, 230 mg (99 %). Usado sin purificación adicional. EM (ESI): m/z = 217,4 [M+H]+.
Intermedio
4-(5,6,7,8-tetrahidroquinolin-4-il)piperidina-1-carboxilato de terc-butilo
En un matraz de tres bocas de 25 ml seco, se combinó cinc en polvo (177 mg, 2,71 mmol) con 1,5 ml de DMA (sobre tamiz molecular) para dar una suspensión gris. Se agitó la mezcla a t. a. mientras se añadía una mezcla 7:5 v/v de clorotrimetilsilano (32 ul) y 1,2-dibromoetano (22 ul) como solución en DMA (1 ml) a una tasa para mantener la temperatura por debajo de 65 °C (ligeramente exotérmica). Se agitó la suspensión resultante durante 20 minutos. Se añadió lentamente a la mezcla una solución de 4-yodopiperidina-1-carboxilato de terc-butilo (734 mg, 2,36 mmol) en 2 ml de DMA. A continuación, se agitó la mezcla de reacción resultante durante 30 minutos a t. a. Se dejó que la reacción reposara durante 15 min sin agitar para su decantación.
En un matraz de tres bocas de 25 ml, se combinó 4-bromo-5,6,7,8-tetrahidroquinolina (250 mg, 1,18 mmol) con 1,5 ml de DMA para dar una solución incolora, se añadieron yoduro de cobre(I) (22,4 mg, 118 |jmol) y [1,1'-bis(difenilfosfino)ferroceno]dicloropaladio(II), complejo con diclorometano (48 mg, 58 jmol). Se desgasificó la mezcla de reacción con argón, se añadió la solución de reactivo de cinc recién preparada, se desgasificó de nuevo con argón y se agitó la mezcla de reacción durante 2,5 h a 75 °C. Se detuvo la mezcla de reacción con 10 ml de NH4CI sat. y se extrajo dos veces con EtOAc (40 ml cada vez). Se lavaron las capas orgánicas con salmuera. Se combinaron las capas orgánicas, se secó sobre MgSO4 y se concentró a vacío. La purificación por cromatografía ultrarrápida en SiO2 usando un gradiente de EtOAc en n-heptano/ (de 0 a 70 % durante 40 min), dio el producto deseado como un aceite viscoso incoloro (255 mg, 68,4 %). EM (ESI): m/z = 317,3 [M+H]+.
BB142
Trifluoroacetato de 3-[2-fluoro-4-(trifluorometil)fenil]acetidina
A una solución de 3-(2-fluoro-4-(trifluorometil)fenil)acetidina-1-carboxilato de terc-butilo (0,078 g, 244 jmol) en DCM (2 ml) se le añadió TFA (94,1 jl, 1,22 mmol). Se agitó la mezcla de reacción resultante a t. a. durante 1 hora. Se concentró la mezcla de reacción a vacío (evaporada conjuntamente con toluol) para proporcionar el producto deseado como un aceite incoloro (85 mg, 100 %). EM (ESI): m/z = 220,1 [M+H]+.
Intermedio
3-[2-fluoro-4-(trifluorometil)fenil]acetidina-1-carboxilato de terc-butilo
A un vial de 20 ml equipado con una barra de agitación se le añadió fotocatalizador (Ir[dF(CF3)ppy]2(dtbpy))PF6 (6,93 mg, 6,17 jmol), 1-bromo-2-fluoro-4-(trifluorometil)benceno (150 mg, 88,5 jl, 617 jmol), 3-bromoacetidina-1-carboxilato de terc-butilo (219 mg, 152 jl, 926 jmol), tris(trimetilsilil)silano (153 mg, 617 jmol) y carbonato de sodio anhidro (131 mg, 1,23 mmol). Se selló el vial y se colocó bajo argón antes de añadir DME (5 ml). A un vial separado se le añadió NiCh glyme (1,36 mg, 6,17 jmol) y 4,4'-di-terc-butil-2,2'-dipiridilo (1,66 mg, 6,17 jmol). Se selló el vial con precatalizador, se purgó con argón, a continuación se le añadió DME (2 ml). Se sometió a ultrasonidos el vial con precatalizador durante 5 min, después de lo que, se inyectó con jeringa 1 ml (0,5 % mol de catalizador, 0,005 eq) en el recipiente de reacción. Se desgasificó la solución rociando con argón. Se agitó la reacción y se irradió con una lámpara de 420 nm durante 5 horas. Se detuvo la reacción por exposición al aire y se concentró a vacío. Se purificó el material bruto por cromatografía ultrarrápida (gel de sílice, 50 g, gradiente de 0 % a 50 % de EtOAc en n-heptano) y una segunda cromatografía ultrarrápida (gel de sílice, 50 g, de 0 a 20 % de EtOAc en nheptano). Se obtuvo el producto como un líquido incoloro (197 mg, 44 %). EM (ESI): m/z = 264,2 [M-C4Hs+H]+.
BB143
Clorhidrato de 4-(1-(4-fluorofenil)-1H-pirazol-3-il)piperidina
En un tubo de 25 ml, se disolvió 4-(1-(4-fluorofenil)-1 H-pirazol-3-il)piperidina-1 -carboxilato de terc-butilo (100 mg, 290 jmol) en DCM (3 ml) y se añadió HCl en éter 2 M (1,16 ml, 2,23 mmol). Se agitó la reacción a t. a. durante 6 h. Se retiró el disolvente a vacío para obtener el producto bruto como un sólido amarillo claro (81 mg, 99 %). Se usó el compuesto sin purificación adicional. EM (ESI): m/z = 246,2 [M+H]+.
Intermedios:
a) 4-(1H-pirazol-3-il)piperidina-1-carboxilato de terc-butilo
Se combinó 4-acetilpiperidina-1-carboxilato de terc-butilo (620 mg, 2,73 mmol) con N,N-dimetilformamidadimetilacetal (6,7 g, 54,6 mmol) y se calentó a 100 °C durante 15 h. CLEM mostró una reacción completa. Se concentró la mezcla de reacción directamente a vacío y se combinó el residuo con EtOH (8 ml) y diclorhidrato de hidracina (344 mg, 3,27 mmol). Se agitó la mezcla a reflujo durante 1,5 h. CLEM mostró de nuevo una reacción completa. Se concentró la mezcla de reacción para proporcionar el producto bruto como un sólido amarillo claro (650 mg, 95 %). Usado para la etapa siguiente sin purificación adicional.
b) 4-[1-(4-fluorofenil)pirazol-3-il]piperidina-1-carboxilato de terc-butilo
En un matraz de 100 ml purgado con argón, se suspendió 4-(1H-pirazol-3-il)piperidina-1-carboxilato de terc-butilo (300 mg, 1,19 mmol) en DMF (8 ml), se añadieron piridina (386 pl, 4,77 mmol), ácido (4-fluorofenil)borónico (217 mg, 1,55 mmol) y acetato de cobre(II) (325 mg, 1,79 mmol). Se agitó la solución verde resultante durante 60 h a t. a. Se retiró el disolvente a vacío, se extrajo el residuo con acetato de etilo/agua/NaCl sat., se secó sobre MgSO4, se filtró y se el disolvente a vacío. Se purificó el material bruto por cromatografía ultrarrápida (gel de sílice, gradiente de 0 % a 40 % de EtOAc en n-heptano durante 40 minutos). Se obtuvo el producto como un aceite viscoso incoloro (290 mg, 70 %). EM (ESI): m/z = 290,2 [M-C4Hs+H]+.
BB144
Clorhidrato de 4-(4-piperidil)-3-(trifluorometil)piridacina
En 25 ml se disolvió 4-(3-(trifluorometil)piridacin-4-il)piperidina-1 -carboxilato de terc-butilo (180 mg, 543 pmol) en DCM (1 ml) y se añadió HCl en éter 2 M (2,72 ml, 5,43 mmol). Se agitó la reacción durante 6 h a t. a. Se retiró el disolvente a vacío para obtener el producto bruto como un sólido amarillo, 145 mg (100 %). Usado sin purificación adicional. EM (ESI): m/z = 232,2 [M+H]+.
Intermedio
4-(3-(trifluorometil)piridacin-4-il)piperidina-1-carboxilato de terc-butilo
Se añadieron (1-(terc-butoxicarbonil)piperidin-4-il)trifluoroborato de potasio (649 mg, 2,23 mmol), nitrato de plata (68,8 mg, 405 pmol) y persulfato de potasio (2,74 g, 10,1 mmol) a un tubo de reacción equipado con una barra de agitación. Se añadieron sucesivamente 1,2-dicloroetano (2 ml), agua (2 ml), 3-(trifluorometil)piridacina (300 mg, 2,03 mmol) y TFA (462 mg, 312 pl, 4,05 mmol) y se selló el tubo. Se agitó la reacción vigorosamente a t. a. durante 24 h.
A continuación, se vertió la mezcla de reacción en 20 ml de una mezcla 1/1 v/v de NaHCO3 ac. sat. y NaS2O3 ac. al 5 % y se extrajo tres veces la solución resultante con DCM. Se secaron las capas orgánicas combinadas (MgSO4) y se evaporó para dar el producto bruto. La purificación por cromatografía ultrarrápida en SiO2 con nheptano/EtOAc de 0 a 80 % en 35 min dio el producto deseado como un aceite viscoso amarillo (180 mg, 80 % de pureza, 21,5 %). Regioisómero confirmado por RMN. EM (ESI): m/z = 332,2 [M+H]+.
BB145
(4aR,8aS)-6-(3-(4-hidroxifenil)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona
Se obtuvo el compuesto de forma análoga al procedimiento A4 a partir de 4-metilbencenosulfonato de 3-(4-hidroxifenil)acetidina. Sólido marrón claro. EM (ESI): m/z = 332,2 [M+H]+.
Intermedios
a) 4-metilbencenosulfonato de 3-(4-hidroxifenil)acetidina
Se agitó a reflujo una mezcla de 3-(4-(terc-butoxi)fenil)acetidina-1-carboxilato de terc-butilo (70 mg, 229 pmol) y ácido 4-metilbencenosulfónico hidrato (52,3 mg, 275 pmol) en EtOAc (1 ml) durante 30 min. Se evaporó la mezcla para proporcionar el producto deseado que se usó en la etapa siguiente sin purificación adicional.
b) 3-(4-(terc-butoxi)fenil)acetidina-1-carboxilato de terc-butilo
Se obtuvo el compuesto de forma análoga al procedimiento A7 a partir de 1-bromo-4-(terc-butoxi)benceno (CAS RN 60876-70-2) para proporcionar el compuesto deseado como un sólido incoloro. EM (ESI): m/z = 250,2 [M-C4H8+H]+.
BB146
4-metilbencenosulfonato de 3-(3,4-dimetilfenil)acetidina
Se obtuvo el compuesto de forma análoga a BB28 a partir de 3-(3,4-dimetilfenil)acetidina-1-carboxilato de tercbutilo para proporcionar el compuesto deseado como un sólido incoloro. EM (ESI): m/z = 162,1 [M+H]+.
Intermedio
3-(3,4-dimetilfenil)acetidina-1-carboxilato de terc-butilo
Se obtuvo el compuesto de forma análoga a BB35, intermedio, a partir de ácido (3,4-dimetilfenil)borónico para proporcionar el compuesto deseado como un aceite incoloro. EM (ESI): m/z = 206,1 [M-C4Hs+H]+.
BB147
4- metilbencenosulfonato de 3-(4-terc-butoxifenil)acetidina
Se obtuvo el compuesto de forma análoga a BB28 a partir de 3-(4-terc-butoxifenil)acetidina-1-carboxilato de
terc-
butilo (BB145, intermedio b) a t. a. como un sólido incoloro. EM (ESI): m/z = 206,2 [M+H]+.
BB148
5- cloro-1-(4-piperidil)indolina
Se obtuvo el compuesto de forma análoga a BB26, intermedio a, a partir de 4-(5-cloroindolin-1-il)piperidina-1-carboxilato de terc-butilo para proporcionar el compuesto deseado como un sólido gris. EM (ESI): m/z = 237,1 [M+H]+.
Intermedio
4-(5-cloroindolin-1 -il)piperidina-1 -carboxilato de terc-butilo
A una solución de 1-Boc-4-piperidona (972,84 mg, 4,88 mmol, CAS RN 79099-07-3) en MeOH (13 ml) se le añadió AcOH (605,43 mg, 9,76 mmol), 5-cloroindolina (500,0 mg, 3,25 mmol, CAS RN 25658-80-4) y NaBHa(CN) (613,63 mg, 9,76 mmol) a 25 °C y se agitó la mezcla de reacción durante 2 h. Se vertió la reacción en solución de NH4Cl acuosa sat. (20 ml) y se extrajo tres veces con EtOAc (20 ml cada vez). Se lavaron tres veces las capas orgánicas combinadas con agua (20 ml cada vez) y salmuera (20 ml), se secó sobre Na2SO4 y se concentró a vacío para proporcionar el producto como un sólido blanco (1 g, 118,3 %). EM (ESI): m/z = 337,1 [M+H]+. Se usó el producto bruto directamente en la etapa siguiente sin purificación adicional.
BB149
Clorhidrato de 4-cloro-2-(piperidin-4-il)isoindolina
Se obtuvo el compuesto de forma análoga a BB52 a partir de 4-(4-cloroisoindolin-2-il)piperidina-1-carboxilato de terc-butilo para proporcionar el compuesto deseado. Em (ESI): m/z = 237,2 [M+H]+. Usado en la etapa siguiente sin purificación adicional.
Intermedio
4-(4-cloroisoindolin-2-il)piperidina-1-carboxilato de terc-butilo
Se obtuvo el compuesto de forma análoga a BB148, intermedio, a partir de 4-cloroisoindolina (CAS RN 123594 04-7) para proporcionar el compuesto deseado como un aceite gris. EM (ESI): m/z = 337,3 [M+H]+.
BB150
5'-cloro-1'-(4-piperidil)espiro[ciclopropano-1,3'-indolina]
Se obtuvo el compuesto de forma análoga a BB26, intermedio, a partir de 4-(5'-cloroespiro[ciclopropano-1,3'-indolina]-1'-il)piperidina-1-carboxilato de terc-butilo para proporcionar el compuesto deseado como un sólido amarillo. EM (ESI): m/z = 263,1 [M+H]+.
Intermedio
4-(5'-cloroespiro[ciclopropano-1,3'-indolina]-1'-il)piperidina-1-carboxilato de terc-butilo
Se obtuvo el compuesto de forma análoga a BB148, intermedio, a partir de 5'-cloroespiro[ciclopropano-1,3'-indolina] (CAS RN 1538359-43-1) para proporcionar el compuesto deseado como un sólido rosa. EM (ESI): m/z = 307,1 [M-C4Hb+H]+.
BB151
Clorhidrato de 2-(acetidin-3-il)-4-cloroisoindolina
Se obtuvo el compuesto de forma análoga a BB52 a partir de 3-(4-doroisoindolin-2-il)acetidina-1-carboxilato de terc-butilo para proporcionar el compuesto deseado como un sólido verde claro. EM (ESI): m/z = 209,2 [M+H]+. Intermedio
3-(4-cloroisoindolin-2-il)acetidina-1-carboxilato de terc-butilo
Se obtuvo el compuesto de forma análoga a BB148, intermedio, a partir de clorhidrato de 4-cloroisoindolina (CAS RN 924304-73-4) para proporcionar el compuesto deseado como un sólido amorfo incoloro. EM (ESI): m/z = 309,2 [M+H]+.
BB152
2,2,2-trifluoroacetato de 3-[2-metoxi-4-(2,2,2-trifluoroetil)fenil]acetidina
Se obtuvo el compuesto de forma análoga a BB26, intermedio a, a partir de 3-[2-metoxi-4-(2,2,2-trifluoroetil)fenil]acetidina-1-carboxilato de terc-butilo para proporcionar el compuesto deseado como un aceite amarillo. EM (ESI): m/z = 246,1 [M+H]+.
Intermedios
a) 3-[2-metoxi-4-(2,2,2-trifluoroetil)fenil]acetidina-1-carboxilato de terc-butilo
A una solución de 3-[2-metoxi-4-[2,2,2-trifluoro-1-(p-tolilsulfoniloxi)etil]fenil]acetidina-1-carboxilato de terc-butilo (480,0 mg, 0,930 mmol) en EtOH (24 ml) se le añadió Pd/C (120,0 mg, 0,930 mmol) y se agitó la mezcla a 20 °C en atmósfera de H2 (globo) durante 24 h. Se filtró la mezcla y se concentró el filtrado. Se disolvió el residuo en EtOAc (30 ml) y se lavó con solución de Na2CO3 acuosa (20 ml) seguido de salmuera, se secó sobre Na2SO4 y se concentró para dar el producto deseado como espuma amarillo claro. (300 mg, 93,3 %). EM (ESI): m/z = 290,1 [M-C4H8+H]+.
b) 3-[2-metoxi-4-[2,2,2-trifluoro-1-(p-tolilsulfoniloxi)etil]fenil]acetidina-1-carboxilato de terc-butilo
Se obtuvo el compuesto de forma análoga al procedimiento A7 a partir de 4-metilbencenosulfonato de [1-(4-bromo-3- metoxi-fenil)-2,2,2-trifluoro-etilo] para obtener el compuesto deseado como un aceite amarillo claro. EM (ESI): m/z = 460,1 [M-C4H8+H]+.
c) 4-metilbencenosulfonato de [1-(4-bromo-3-metoxi-fenil)-2,2,2-trifluoro-etilo]
A una solución de 1-(4-bromo-3-metoxi-fenil)-2,2,2-trifluoro-etanol (1000,0 mg, 3,51 mmol), cloruro de ptoluenosulfonilo (668,81 mg, 3,51 mmol) y DMAP (20,0 mg, 0,180 mmol) en DCM (20 ml) se le añadió TEA (708,62 mg, 7,02 mmol) a 0 °C. Se agitó la mezcla a 20 °C durante 12 h. Se lavó la mezcla con agua y salmuera, se secó sobre Na2SO4 y se concentró para dar el producto deseado como aceite amarillo claro (1,5 g, 97,4 %). Se usó el compuesto en la etapa siguiente sin purificación adicional.
d) 1-(4-bromo-3-metoxi-fenil)-2,2,2-trifluoro-etanol
A una solución de 4-bromo-3-metoxi-benzaldehído (2,0 g, 9,3 mmol, CAS RN 43192-34-3) y trifluoroetiltrimetilsilano (1586,94 mg, 11,16 mmol) en THF (30 ml) se le añadió TBAF/THF (0,09 ml, 0,090 mmol) a 0 °C, se agitó la mezcla a 20 °C durante 12 h, a continuación se añadió lentamente HCl ac. 1 M (18,6 ml, 18,6 mmol). Se agitó la mezcla a 20 °C durante otras 2 h. Se vertió la mezcla en agua (50 ml) y se extrajo tres veces con EtOAc (30 ml cada vez). Se lavó la fase orgánica combinada con salmuera, se secó sobre Na2SO4 y se concentró para dar el producto deseado como aceite amarillo claro (2,5 g, 94,3 %). RMN de 1H (400 MHz, CLOROFORMO-d) 5 = 7,57 (d, J = 8,1 Hz, 1H), 7,05 (s, 1H), 6,94 (d, J = 8,1 Hz, 1H), 5,01 (q, J = 6,5 Hz, 1H), 3,93 (s, 3H).
BB153
4- metilbencenosulfonato de 3-[4-(2,2-dimetilpropoxi)fenil]acetidina
Se obtuvo el compuesto de forma análoga a BB28 a t. a. durante 5 horas a partir de 3-[4-(2,2-dimetilpropoxi)fenil]acetidina-1-carboxilato de terc-butilo para obtener el compuesto deseado como sólido blanco. Se usa tal cual para la etapa siguiente.
Intermedio
3-[4-(2,2-dimetilpropoxi)fenil]acetidina-1-carboxilato de terc-butilo
Se obtuvo el compuesto de forma análoga al procedimiento A7 a, a partir de 1-bromo-4-(neopentiloxi)benceno (CAS RN 528528-58-7) para proporcionar el compuesto deseado como un sólido incoloro. EM (ESI): m/z = 264,2 [M-C4H8+H]+.
BB154
rac-(4aS, 8aS)-hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona
Se disolvió rac-(4aS,8aS)-3-oxohexahidro-2H-pirido[4,3-b][1,4]oxacina-6(5H)-carboxilato de bencilo (125 mg, 431 |jmol) en MeOH (5 ml). Se desgasificó la solución de reacción a vacío y se rellenó con argón. Se añadió Pd-C (20 mg, 188 jmol) en atmósfera de argón. Se evacuó el argón de la mezcla de reacción y se rellenó el matraz de reacción con hidrógeno. Se agitó la mezcla de reacción a t. a. durante 15 h en atmósfera de hidrógeno. Se filtró la mezcla de reacción a través de un filtro de jeringa y se concentró a vacío para dar el producto deseado como un sólido blanco (62 mg, 92,2 %). EM (ESI): m/z = 157,098 [M+H]+.
Intermedios
a) rac-(4aS,8aS)-3-oxohexahidro-2H-pirido[4,3-b][1,4]oxacina-6(5H)-carboxilato de bencilo
A una solución agitada de rac-(3S,4S)-3-(2-cloroacetamido)-4-hidroxipiperidina-1-carboxilato de bencilo (385 mg, 1,18 mmol) en THF seco (4 ml) se le añadió NaH (67,9 mg, 1,7 mmol) a 0 °C. Se dejó que la mezcla alcanzara la t. a. y a continuación se agitó durante 90 min en atmósfera de argón. Se añadió H2O (5 ml) y se continuó agitando a t. a. durante 10 min. Se retiró el THF a vacío de la mezcla de reacción. Se trató el residuo con DCM y se lavó la fase orgánica con H2O y salmuera, se secó sobre Na2SO4, se filtró y a continuación se concentró a vacío. Se purificó el residuo por cromatografía ultrarrápida (columna de fase inversa de 12 g, gradiente 0-100 % de ACN (FA al 0,1 %) en agua (FA al 0,1 %) para dar el producto deseado como un sólido blanco (133 mg, 38,9 %). EM (ESI): m/z = 291,3 [M+H]+.
b) rac-(3S,4S)-3-(2-cloroacetamido)-4-hidroxipiperidina-1-carboxilato de bencilo
A una suspensión agitada de rac-(3S,4S)-3-amino-4-hidroxipiperidina-1-carboxilato de bencilo (317 mg, 1,27 mmol, sintetizado de acuerdo con la patente de EE. UU. 2011/59118 A1) y acetato de sodio (208 mg, 2,53 mmol, CAS RN 127-09-3) en una mezcla de acetona (4 ml)/H2O (0,5 ml) se le añadió gota a gota una solución de cloruro de cloroacetilo (150 mg, 107 jl, 1,33 mmol, CAS RN 79-04-9) en acetona (3 ml) entre 0-5 °C. Después de la adición, se agitó la mezcla de reacción a t. a. durante 1 h y posteriormente se evaporó hasta sequedad dando una goma amarilla. Se purificó el producto bruto por cromatografía en gel de sílice para dar el producto deseado como un sólido amarillo (385 mg, 93 %). EM (ESI): m/z = 325,2 [M-H]-.
Tabla 3



Ejemplo A
Un compuesto de fórmula (Ic)
se puede usar de una manera conocida
per se
como ingrediente activo para la producción de comprimidos de la siguiente composición:
Por comprimido
Ingrediente activo 200 mg
Celulosa microcristalina 155 mg
Almidón de maíz 25 mg
Talco 25 mg
Hidroxipropilmetilcelulosa 20 mg
425 mg
Ejemplo B
Un compuesto de fórmula (Ic)
se puede usar de manera conocida
per se
como ingrediente activo para la producción de cápsulas de la siguiente composición:
Por cápsula
Ingrediente activo 100.0 mg
Almidón de maíz 20.0 mg
Lactosa 95.0 mg
Talco 4,5 mg
Estearato de magnesio 0,5 mg
220.0 mg
Claims (15)
1. Un compuesto de fórmula (Ic)

o una sal farmacéuticamente aceptable del mismo, en la que:
L se selecciona de -C R 1R2-(CH2)p-, -(CH2)p-CR1R2-, -OCR3R4-, -CR 3R4O - y un enlace covalente;
m es 0, n es 0 o 1 y X es CR24; o
m es 1, n es 1 o 2 y X es CR24 o N;
p es 0, 1 o 2;
R1 se selecciona de halógeno, alcoxi Ci-e, alquilo Ci-e, halo-alquilo Ci-e, hidroxi-alquilo Ci-e, alcoxi Ci-e-alquil Ci-e-

halo-alcoxi Ci-e, halo-alcoxi Ci-e-alquil Ci-e-, ciano y un grupo ; y R2 se selecciona de hidrógeno, halógeno e hidroxi; o R1 y R2, tomados conjuntamente con el átomo de carbono al que están unidos, forman un cicloalquilo C3-12 o un heterociclilo C2-9;
R3 se selecciona de alquilo C1-6, halo-alquilo C1-6, cicloalquilo C3-12 y arilo C6-14; y R4 es hidrógeno; o
R3 y R4, tomados conjuntamente con el átomo de carbono al que están unidos, forman un cicloalquilo C3-12 o un heterociclilo C2-9;
R20 es hidrógeno o alquilo C1-6;
R21, R22 y R23 se seleccionan independientemente de hidrógeno, halógeno, ciano, hidroxi, alcoxi C1-6, halo-alcoxi C1-6, amino-alcoxi C1-6, alquilo C1-6, halo-alquilo C1-6, hidroxi-alquilo C1-6, (halo-alquil C1-6)-hidroxi-alquilo C1-6, alcoxicarbonil C1-6-alquil C1-6-, alcoxicarbonil C1-6-NH-alcoxi C1-6-, alcoxicarbonil C1-6-NH-(alcoxi C1-6)2-alquil C1-6-C(0)-NH-alcox¡ C1-6-, alcoxicarbonil Ci-6-NH-alcox¡ Ci-e-alquil Ci-6-C(0)-NH-alcox¡ C1-6-, SFs, (alquil Ci-e)3S¡-0-
alquil C1-6-, un grupo
 grupo
grupo

R24 se selecciona de hidrógeno, halógeno, hidroxi, halo-alquilo C1-6 y alquilo C1-6;
R25 y R26 se seleccionan independientemente de hidrógeno, halógeno, alquilo C1-6, alcoxi C1-6, halo-alquilo C1-6, halo-alcoxi C1-6, alquil C1-6-SO2- y cicloalquilo C3-12;
R27 y R28 se seleccionan independientemente de hidrógeno, halógeno, alcoxi C1-6, halo-alcoxi C1-6, alquilo C1-6, halo-alquilo C1-6, hidroxi-alquilo C1-6, halógeno, hidroxi, alquilsulfonilo C1-6, carbamoílo, ciano, cicloalquil-alcoxi C1-6-, alquil C1-6-NH-C(O)- y cicloalquilo;
A y B se seleccionan independientemente de arilo C6-14, heteroarilo C1-13, cicloalquilo C3-12 y heterociclilo C2-9; C1 es cicloalquilo C3-12 o heterociclilo C2-9;
C2 es arilo C6-14;
L2 se selecciona de un enlace covalente, -O -, -CH2O-, -CH2OCH2- y -CH2-;
L3 se selecciona de un enlace covalente, -O -, -CH2O-, -OCH2-, -CH2OCH2- y -CH2-; y
L3a se selecciona de un enlace covalente, -CH2O-, -OCH2-, -CH2OCH2- y -CH2-.
2. El compuesto de fórmula (Ic) de acuerdo con la reivindicación 1, o una sal farmacéuticamente aceptable del mismo, en la que:
m es 0, n es 0 o 1 y X es CR24; o
m es 1, n es 1 y X es CR24 o N; y
R20 y R24 son ambos hidrógeno.
3. El compuesto de fórmula (Ic) de acuerdo con una cualquiera de las reivindicaciones 1 y 2, o una sal farmacéuticamente aceptable del mismo, en la que:
L se selecciona de -CR 1R2-(CH2)p-, -OCR3R4-, -CR 3R4O - y un enlace covalente;
p es 0 o 1;
R1 se selecciona de halógeno, alcoxi C1-6, alquilo C1-6, halo-alquilo C1-6, hidroxi-alquilo C1-6, alcoxi Ci-e-alquil C1-6-,
halo-alcoxi C1-6, halo-alcoxi C1-6-alquil C1-6--, ciano y un grupo
 selecciona de hidrógeno, halógeno e hidroxi; o
selecciona de hidrógeno, halógeno e hidroxi; o
R1 y R2, tomados conjuntamente con el átomo de carbono al que están unidos, forman un cicloalquilo C3-12; R3 es alquilo C1-6 o halo-alquilo C1-6;
R4 es hidrógeno;
R21 se selecciona de hidrógeno, halógeno, hidroxi, alcoxi C1-6, halo-alcoxi C1-6, alquilo C1-6, halo-alquilo C1-6, hidroxialquilo C1-6, (halo-alquil Ci-6)-hidroxi-alquilo C1-6, alcoxicarbonil Ci-e-alquil C1-6-, alcoxicarbonil Ci-6-NH-alcox¡
C1-6-, SFs, (alquil Ci-e)3Si-0-alquil C1-6-, un grupo
 grupo
grupo

R22 se selecciona de hidrógeno, halógeno, alcoxi C1-6, halo-alcoxi C1-6, alquilo C1-6 y ciano;
R23 es hidrógeno o halógeno;
R25 se selecciona de hidrógeno, halógeno, alquilo C1-6, alcoxi C1-6, halo-alquilo C1-6, halo-alcoxi C1-6, alquil C1-6-SO2- y cicloalquilo C3-12;
R26 se selecciona de hidrógeno, alquilo C1-6 y alcoxi C1-6;
R27 se selecciona de hidrógeno, halógeno, alcoxi C1-6, halo-alcoxi C1-6, alquilo C1-6, halo-alquilo C1-6, hidroxi-alquilo C1-6, halógeno, hidroxi, alquilsulfonilo C1-6, carbamoílo, ciano, cicloalquil-alcoxi C1-6- y cicloalquilo;
R28 se selecciona de hidrógeno, alquilo C1-6 y halógeno;
A es arilo C6-14 o heteroarilo C1-13;
B es arilo C6-14 o heteroarilo C1-13;
L2 se selecciona de un enlace covalente, -O - y -CH2-;
L3 se selecciona de un enlace covalente, -CH2O- y -CH2-; y
L3a es un enlace covalente o -CH2-.
4. El compuesto de fórmula (Ic) de acuerdo con una cualquiera de las reivindicaciones 1 a 4, o una sal farmacéuticamente aceptable del mismo, en la que:
L se selecciona de -C R 1R2-(CH2)p-, -OCR3R4-, -CR 3R4O - y un enlace covalente;
p es 0;
R1 se selecciona de halógeno, alquilo Ci-e, hidroxi-alquilo Ci-e, alcoxi Ci-6-alquil Ci-e-, halo-alcoxi Ci-e y un grupo
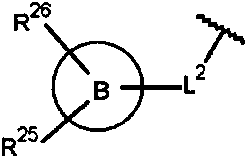
R2 es hidrógeno o halógeno;
R3 es alquilo C1-6;
R4 es hidrógeno;
R21 se selecciona de halógeno, alcoxi C1-6, halo-alcoxi C1-6, alquilo C1-6, halo-alquilo C1-6, SFs, arilo Ce-14 y un grupo

R22 se selecciona de hidrógeno, halógeno, alcoxi C1-6 y halo-alcoxi C1-6;
R23 es hidrógeno o halógeno;
R25 se selecciona de hidrógeno, halógeno, alcoxi C1-6 y cicloalquilo C3-12;
R26 es hidrógeno o alcoxi C1-6;
R27 se selecciona de hidrógeno, halo-alcoxi C1-6, alquilo C1-6, halo-alquilo C1-6, alquilo C1-6-NH-C(O)- y halógeno; R28 se selecciona de hidrógeno, alquilo C1-6 y halógeno;
A es arilo C6-14 o heteroarilo C1-13;
B es arilo C6-14 o heteroarilo C1-13;
C1 es cicloalquilo C3-12 o heterociclilo C2-9;
L2 es un enlace covalente; y
L3 es un enlace covalente o -CH2-.
5. El compuesto de fórmula (Ic) de acuerdo con una cualquiera de las reivindicaciones 1 a 4, o una sal farmacéuticamente aceptable del mismo, en la que:
L se selecciona de -CR 1R2-(CH2)p-, -OCR3R4-, -CR 3R4O - y un enlace covalente;
p es 0;

R2 es hidrógeno o fluoro;
R3 es metilo;
R4 es hidrógeno;
R21 se selecciona de fluoro, cloro, bromo, metilo, metoxi, ferc-butilo, propilo, trifluorometoxi, 2-fluoroetox¡, 2,2,2-

trifluoroetoxi, trifluorometilo, 2,2,2-trifluoroetilo, 1,1 -difluoroetilo, SFs, feniloy un grupo , en el que R27, R28, C1 y L3 son como se define en el presente documento;
R22 se selecciona de hidrógeno, fluoro, cloro, metoxi, metilo y trifluorometilo;
R23 es hidrógeno o fluoro;
R25 se selecciona de hidrógeno, metoxi, fluoro y ciclopropilo;
R26 es hidrógeno o metoxi;
R27 se selecciona de metilo, trifluorometoxi, trifluorometilo, 2,2,2-trifluoro-1-metil-etoxi, 2,2,2-trifluoro-1, 1-dimetiletoxi, 2,2,2-trifluoroetoxi, fluoro y cloro;
R28 se selecciona de hidrógeno, metilo, fluoro y cloro;
A se selecciona de fenilo, indol-3-ilo, 2-piridilo y 3-piridilo;
B es fenilo o 1,2,4-oxadiazol-5-ilo;
C1 se selecciona de acetidin-1-ilo, pirrolidin-1-ilo, ciclopropilo y oxetan-3-ilo;
L2 es un enlace covalente; y
L3 es un enlace covalente o -CH2-.
6. El compuesto de fórmula (Ic) de acuerdo con una cualquiera de las reivindicaciones 1 a 3, o una sal farmacéuticamente aceptable del mismo, en la que C2 es fenilo.
7. El compuesto de fórmula (Ic) de acuerdo con una cualquiera de las reivindicaciones 1 a 3, o una sal farmacéuticamente aceptable del mismo, en la que L3a es un enlace covalente.
8. El compuesto de fórmula (Ic) de acuerdo con la reivindicación 1, o una sal farmacéuticamente aceptable del mismo, en la que:
m es 0, n es 0 o 1 y X es CR24; o
m es 1, n es 1 y X es CR24 o N;
L se selecciona de -CR1R2-(CH2)p- , -OCHR3-, -CHR3O - y un enlace covalente;
p es 0 o 1;
R1 se selecciona de halógeno, alcoxi C1-6, alquilo C1-6, halo-alquilo C1-6, hidroxi-alquilo C1-6, alcoxi Ci-e-alquil C1-6-,

halo-alcoxi C1-6, halo-alcoxi Ci-e-alquil C1-6-, ciano y un grupo ; y R2 se selecciona de hidrógeno, halógeno e hidroxi; o R1 y R2, tomados conjuntamente con el átomo de carbono al que están unidos, forman un cicloalquilo C3-12;
R3 es alquilo C1-6 o halo-alquilo C1-6;
R20 es hidrógeno o alquilo C1-6;
R21 se selecciona de hidrógeno, halógeno, hidroxi, alcoxi Ci-6, halo-alcoxi Ci-6, alquilo Ci-6, halo-alquilo Ci-6, hidroxialquilo Ci-6, (halo-alquil Ci-6)-hidroxi-alquilo Ci-e, alcoxicarbonil Ci-e-alquil Ci-e-, alcoxicarbonil Ci-6-NH-alcox¡


C1-6-, SF5, (alquil C1-6)3Si-O-alquil C1-6-, un grupo y un grupo
R22 se selecciona de hidrógeno, halógeno, alcoxi C1-6, halo-alcoxi C1-6, alquilo C1-6 y ciano;
R23 es hidrógeno o halógeno;
R24 se selecciona de hidrógeno, halógeno, hidroxi y alquilo C1-6;
R25 se selecciona de hidrógeno, halógeno, alquilo C1-6, alcoxi C1-6, halo-alquilo C1-6, halo-alcoxi C1-6, alquil C1-6-SO2- y cicloalquilo C3-12;
R26 se selecciona de hidrógeno, alquilo C1-6 y alcoxi C1-6;
R27 se selecciona de hidrógeno, halógeno, alcoxi C1-6, halo-alcoxi C1-6, alquilo C1-6, halo-alquilo C1-6, hidroxi-alquilo C1-6, halógeno, hidroxi, alquilsulfonilo C1-6, carbamoílo, ciano, cicloalquil-alcoxi C1-6- y cicloalquilo;
R28 se selecciona de hidrógeno, alquilo C1-6 y halógeno;
A es arilo C6-14 o heteroarilo C1-13;
B es arilo C6-14 o heteroarilo C1-13;
C1 es cicloalquilo C3-12 o heterociclilo C2-9;
C2 es arilo C6-14;
L2 se selecciona de un enlace covalente, -O - y -CH2-;
L3 se selecciona de un enlace covalente, -CH2O- y -CH2-; y
L3a es un enlace covalente o -CH2-.
9. El compuesto de fórmula (Ic) de acuerdo con una cualquiera de las reivindicaciones 1 a 8, o una sal farmacéuticamente aceptable del mismo, seleccionado de:
rac-(4aR,8aS)-6-(4-benzhidrilpiperidina-1-carbonil)-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona; rac-(4aR,8aS)-6-(4-(5-cloro-1-(ciclopropilmetil)-1H-indol-3-il)piperidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
rac-(4aR,8aS)-6-(4-(5-cloro-1-metil-1H-indol-3-il)piperidina-1-carbonil)hexahidro-2H-pi rido[4,3-b][1,4]oxacin-3(4H)-ona;
rac-(4aR,8aS)-6-(4-(9H-fluoren-9-il)piperidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(+)-cis-6-[4-(6-fluoro-1H-indol-3-il)piperidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona; (4aR,8aS)-6-(4-(1-(4-fluorofenil)-3-metoxipropil)piperidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
rac-(4aR,8aS)-6-[4-(6-fluoro-1H-indol-3-il)piperidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona;
(4aR,8aS)-6-(4-((4-fluorofenil)(metoxi)metil)piperidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona; rac-(4aR,8aS)-6-(4-(5-fluorobenzo[d]isoxazol-3-il)piperidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacina-3(4H)-ona;
cis-6-(4-((4-clorofenil)(fenil)metil)piperacina-1-carbonil)hexahidro-2H-pi rido[4,3-b][1,4]oxacin-3(4H)-ona;
(+)-cis-6-(4-(bis(4-fluorofenil)metil)piperacina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
rac-(4aR,8aS)-6-[4-[bis(4-fluorofenil)metil]piperacina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona;
(-)-cis-6-(4-(bis(4-fluorofenil)metil)piperacina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona; rac-(4aR,8aS)-6-(4-(5-doro-1H-indol-3-il)piperacina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacina-3(4H)-ona; rac-(4aR,8aS)-6-(4-(1-feniletil)piperacina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
rac-(4aR,8aS)-6-(4-(1-metil-1H-indazol-5-il)piperidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacina-3(4H)-ona;
rac-(4aR,8aS)-6-[4-[(4-fluorofenil)-fenilmetil]piperacina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona;
(+)- o (-)-cis-6-(4-(5-doro-1-cidopropil-1H-indol-3-il)piperidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
rac-(4aR,8aS)-6-(4-(5-doro-1-(1-doro-3-hidroxipropan-2-il)-1H-indol-3-il)piperidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(+)- o (-)-cis-6-(4-(bis(4-fluorofenil)metil)piperidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona; (+)- o (-)-cis-6-(4-(5-doro-1-(oxetan-3-il)-1H-indol-3-il)piperidina-1-carbonil)hexahidro-2H-pirido [4,3-b][1,4]oxacin-3(4H)-ona;
(+)- o (-)-ds-6-(4-(5-doro-1-(oxetan-3-NmetN)-1 H-mdol-3-N)piperidma-1-carbonN)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(+)- o (-)-ds-6-(4-(1-(2-((terc-butNdimetNsNN)oxi)etN)-5-don>1H-mdol-3-N)piperidma-1-carbonNo)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(+)- o (-)-cis-6-(4-(5-doro-1-(2-hidroxietil)-1H-indol-3-il)piperidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
rac-cis-6-(4-(1-(4-fluorofenil)-3-hidroxipropil)piperidina-1-carbonil)hexahidro-2H-pi rido[4,3-b][1,4]oxacin-3(4H)-ona; rac-(4aR,8aS)-6-(4-(1-(4-(trifluorometil)fenil)etil)piperidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacina-3(4H)-ona;
rac-(4aR,8aS)-6-(4-(1-(2-doro-4-fluorofenoxi)etil)piperidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(4-(5-(trifluorometil)piridin-3-il)piperidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona; (4aR,8aS)-6-(4-(difluoro(4-(trifluorometil)fenil)metil)piperidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(+)-(4aR,8aS)-6-(4-((R o S)-1-(4-fluorofenN)-3-hidroxipropN)piperidma-1-carbonN)hexahidn>2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(-)-(4aR,8aS)--6-(4-((S o R)-1-(4-fluorofenil)-3-hidroxipropil)piperidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(+) o (-)-cis-6-(4-((S o R)-1-(2-doro-4-fluorofenoxi)etil)piperidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(+) o (-)-cis-6-(4-((R o S)-1-(2-doro-4-fluorofenoxi)etil)piperidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(+) o (-)-ds-6-(4-(5-metoxipmdm-3-N)piperidma-1-carbonN)hexahidn>2H-pirido[4,3-b][1,4]oxadn-3(4H)-ona;
(+) o (-)-cis-6-(4-(bis(4-fluorofenil)metil)piperidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(+) o (-)-cis-6-(4-((4-dorofenil)difluorometil)piperidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(+) o (-)-cis-6-(4-(5-(trifluorometoxi)piridin-2-il)piperidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(+) o (-)-cis-6-(4-((2-doro-4-fluorofenil)difluorometil)piperidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(+) o (-)-cis-6-(4-(5-etilpiridin-3-il)piperidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(+) o (-)-cis-6-(4-(5-(1,1-difluoroetil)piridin-2-il)piperidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(+) o (-)-ds-6-(4-(6-doro-1-metiM H-indazol-3-il)piperidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-((3R o 3S)-4-(5-(1,1-difluoroetN)pmdm-2-N)-3-metNpiperidma-1-carbonN)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-((3S o 3R)-4-(5-(1,1-difluoroetN)pmdm-2-N)-3-metNpiperidma-1-carbonN)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-((4R o 4S)-(5-(1,1-difluoroetil)piridin-2-il)-3-metilpiperidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(4-((R o S)-1-(4-(trifluorometil)fenil)etil)piperidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(4-((S o R)-1-(4-(trifluorometil)fenil)etil)piperidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
2-(4-fluorofenil)-2-(1-((4aR,8aS)-3-oxooctahidro-2H-pi rido[4,3-b][1,4]oxacina-6-carbonil)piperidin-4-il)acetonitrilo; (4aR,8aS)-6-(4-(2,2,2-trifluoro-1-feniletil)piperacina-1-carbonil)hexahidro-2H-pi rido[4,3-b][1,4]oxacin-3(4H)-ona; (4aR,8aS)-6-(4-((4-fluorofenil)(2,2,2-trifluoroetoxi)metil)piperidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(4-(1-(4-fluorofenil)-2-(3-metil-1,2,4-oxadiazol-5-il)etil)piperidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(4-(1-(4-(trifluorometil)fenil)cidopropil)piperidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(4-(1-(2,4-difluorofenil)-2,2,2-trifluoroetil)piperacina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-(bis(4-fluorofenil)(hidroxi)metil)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-(fluorobis(4-fluorofenil)metil)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona; (4aR,8aS)-6-(4-(1-(4-fluorofenil)-2-(2,2,2-trifluoroetoxi)etil)piperidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(4-((R o S)-(4-fluorofenil)(2,2,2-trifluoroetoxi)metil)piperidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(4-((S o R)-(4-fluorofenil)(2,2,2-trifluoroetoxi)metil)piperidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(4-((R o S)-1-(4-fluorofenil)-2-(3-metil-1,2,4-oxadiazol-5-il)etil)piperidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(4-((S o R)-1-(4-fluorofenil)-2-(3-metil-1,2,4-oxadiazol-5-il)etil)piperidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(4-(2-cidopropilpiridin-4-il)piperidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona; (4aR,8aS)-6-[4-[(S o R)-(4-fluorofenil)-(3-metoxifenil)metil]piperidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona;
(4aR,8aS)-6-(4-((R o S)-(4-fluorofenil)(3-metoxifenil)metil)piperidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(4-((S o R)-(3-metoxifenil)(fenil)metil)piperidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(4-((R o S)-(3-metoxifenil)(fenil)metil)piperidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3 (4H)-ona;
(4aR,8aS)-6-(4-((S o R)-(3-(2-fluoroetoxi)fenil)(fenil)metil)piperidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(4-((S)-(4-fluorofenil)(fenil)metil)piperidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacina-3(4H)-ona;
(4aR,8aS)-6-(4-((S o R)-(4-fluorofenil)(fenil)metil)piperidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-[4-[(S o R)-(4-fluorofenil)-(4-metilfenil)metil]piperidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona;
(4aR,8aS)-6-[4-[(R o S)-[4-(2-fluoroetoxi)fenil]-fenilmetil]piperidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona;
(4aR,8aS)-6-(4-((S o R)-(4-fluorofenil)(4-metoxifenil)metil)piperidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(4-((R o S)-(4-fluorofenil)(p-tolil)metil)piperidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(4-((S o R)-(3,4-dimetoxifenil)(4-fluorofenil)metil)piperidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(4-((R o S)-(3,4-dimetoxifenil)(fenil)metil)piperidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-(1-(2-doro-4-(trifluorometil)fenoxi)etil)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-(1-(2-doro-4-(tnfluorometN)fenoxi)etN)acetidma-1-carbonN)hexahidro-2H-pmdo[4,3-b][1,4]oxadn-3(4H)-ona;
(4aR,8aS)-6-(4-((R o S)-(4-fluorofenil)(fenil)metil)piperidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(4-((S o R)-fenN(m-toNl)metN)pipendma-1-carbonN)hexahidro-2H-pmdo[4,3-b][1,4]oxadn-3(4H)-ona; (4aR,8aS)-6-[4-[(S o R)-[4-(2-fluoroetoxi)fenil]-fenilmetil]piperidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona;
(4aR,8aS)-6-(4-((S o R)-((S)-3-metilcidohexa-2,4-dien-1-il)(fenil)metil)piperidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(4-(1-(2-doro-4-fluorofenoxi)-2,2,2-trifluoroetil)piperidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-(bis(4-fluorofenil)metil)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona; (4aR,8aS)-6-[3-[1-[4-(trifluorometil)fenil]etil]acetidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona;
(4aR,8aS)-6-[4-[(S o R)-fenN(p-tolN)metN]piperidma-1-carbonN]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxadn-3-ona;
(4aR,8aS)-6-(4-((R o S)-fenN(p-toNl)metN)piperidma-1-carbonN)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona; (4aR,8aS)-6-(3-benzhidrilacetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(4-((S o R)-(3,4-dimetoxifenil)(fenil)metil)piperidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(4-((R o S)-(3,4-dimetoxifenil)(4-fluorofenil)metil)piperidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(4-((R o S)-(4-fluorofenil)(4-metoxifenil)metil)piperidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-[3-[1-[4-(trifluorometil)fenil]etoxi]acetidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona;
(4aR,8aS)-6-(3-(1-(2-fluoro-4-(trifluorometil)fenoxi)etil)acetidina-1-carbonil)hexahidro-2H-pi rido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-(1-(4-(trifluorometil)fenoxi)etil)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-[4-[1-[4-(trifluorometil)fenil]etoxi]piperidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona;
(4aR,8aS)-6-(4-((R o SH3-ddopropiM,2,4-oxadiazol-5-N)(4-fluorofenN)metN)piperidma-1-carbonN)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(4-((S o RH3-ddopropiM,2,4-oxadiazol-5-N)(4-fluorofenN)metN)piperidma-1-carbonN)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-((R o S)-1-(2-fluoro-4-(trifluorometN)fenoxi)etN)acetidma-1-carbonN)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-((S o R)-1-(2-fluoro-4-(trifluorometN)fenoxi)etN)acetidma-1-carbonN)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-((R o S)-1-(4-(trifluorometil)fenoxi)etil)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-((S o R)-1-(4-(trifluorometil)fenoxi)etil)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(4-((R o S)-(4-(2-fluoroetoxi)fenil)(4-fluorofenil)metil)piperidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(4-(5-metil-6-(trifluorometil)piridin-3-il)piperidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4] oxacin-3(4H)-ona;
(4aR,8aS)-6-(4-((4-fluorofenil)(3-(trifluorometil)-1,2,4-oxadiazol-5-il)metil)piperidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-((S o R)-1-(4-(trifluorometil)fenil)etil)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-((R o S)-1-(4-(trifluorometil)fenil)etil)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-[3-(S o R)-[(4-fluorofenil)-(2,2,2-trifluoroetoxi)metil]acetidina-1-carbonil]-4,4a,5,7,8,8ahexahidropirido[4,3-b][1,4]oxacin-3-ona;
(4aR,8aS)-6-[3-(R o S)-[(4-fluorofenil)-(2,2,2-trifluoroetoxi)metil]acetidina-1-carbonil]-4,4a,5,7,8,8a-
hexahidropirido[4,3-b][1,4]oxacin-3-ona;
(4aR,8aS)-6-(4-(5,6,7,8-tetrahidroquinolin-4-il)piperidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(4-((R o S)-(3-(2-fluoroetoxi)fenil)(fenil)metil)piperidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(4-((S o R)-(4-(2-fluoroetoxi)fenil)(4-fluorofenil)metil)piperidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-(4-bromofenil)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-(4'-doro-[1,1'-bifenil]-4-il)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona; (4aR,8aS)-6-(3-(2'-doro-[1,1'-bifenil]-4-il)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona; (4aR,8aS)-6-(3-(2',4'-didoro-[1,1'-bifenil]-4-il)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-(1-(R o S)-(4-(trifluorometil)fenil)etoxi)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-(1-(S o R)-(4-(trifluorometil)fenil)etoxi)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-[3-(S o R)-[1-(2-doro-4-fluoro-fenil)etoxi]acetidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona;
(4aR,8aS)-6-[3-(R o S)-[1-(2-doro-4-fluoro-fenil)etoxi]acetidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona;
(4aR,8aS)-6-[4-[(R o S)-(3,4-dimetoxifenil)-(2-piridil)metil]piperidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona;
(4aR,8aS)-6-[4-[(S o R)-(3,4-dimetoxifenil)-(2-piridil)metil]piperidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona;
(4aR,8aS)-6-[3-[4-(trifluorometoxi)fenil]acetidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona; (4aR,8aS)-6-(3-(2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-(4-bromofenil)-3-fluoroacetidina-1-carbonil)hexahidro-2H-pi rido[4,3-b][1,4]oxacin-3(4H)-ona; (4aR,8aS)-6-(3-(4-bromofenil)-3-hidroxiacetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona; (4aR,8aS)-6-(3-(4-bromofenil)-3-metilacetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-(2'-(trifluorometil)-[1,1'-bifenil]-4-il)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-(2',4'-difluoro-[1,1'-bifenil]-4-il)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
4'-(1-((4aR,8aS)-3-oxooctahidro-2H-pirido[4,3-b][1,4]oxacina-6-carbonil)acetidin-3-il)-[1,1'-bifenil]-2-carbonitrilo; (4aR,8aS)-6-(3-(4-(3-metoxiacetidin-1-il)fenil)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-(4-(3-(trifluorometil)acetidin-1-il)fenil)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
2-[4-[1-[(4aR,8aS)-3-oxo-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacina-6-carbonil]acetidin-3-il]fenil]benzamida;
(4aR,8aS)-6-(3-(2-doro-[1,1'-bifenil]-4-il)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-(4-bromo-3-dorofenil)acetidina-1-carbonil)hexahidro-2H-pi rido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-[3-[4-(4-doro-2-fluoro-fenil)fenil]acetidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona;
(4aR,8aS)-6-[3-[4-(2-doro-4-fluoro-fenil)fenil]acetidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona;
(4aR,8aS)-6-[4-[(R o S)-(3,4-dimetoxifenil)-(3-piridil)metil]piperidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona;
(4aR,8aS)-6-[4-[(S o R)-(3,4-dimetoxifenil)-(3-piridil)metil]piperidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona;
(4aR,8aS)-6-(3-(2'-metil-[1,1'-bifenil]-4-il)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona; (4aR,8aS)-6-(3-(4-(3-((1,1,1-trifluoropropan-2-il)oxi)acetidin-1-il)fenil)acetidina-1-carbonil)hexahidro-2H-pi rido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-(3-bromofenil)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-(4-(ferc-butN)fenN)acetidma-1-carbonN)hexahidn>2H-pirido[4,3-b][1,4]oxadn-3(4H)-ona;
(4aR,8aS)-6-[3-(4-fenilfenil)acetidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona;
(4aR,8aS)-6-[3-[4-[2-(difluorometil)fenil]fenil]acetidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona;
(4aR,8aS)-6-(3-(3'-metil-[1,1'-bifenil]-4-il)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona; (4aR,8aS)-6-(3-(4'-metil-[1,1'-bifenil]-4-il)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona; (4aR,8aS)-6-(3-(6-doropiridin-3-il)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-(4-(trifluorometil)fenil)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-(4-(3-((1,1,1-trifluoro-2-metilpropan-2-il)oxi)acetidin-1-il)fenil)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-(4-(1,1-difluoroetil)fenil)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona; (4aR,8aS)-6-[4-[(R o S)-(3,4-dimetoxifenil)-(4-piridil)metil]piperidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona;
(4aR,8aS)-6-[4-[(S o R)-(3,4-dimetoxifenil)-(4-piridil)metil]piperidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona;
(4aR,8aS)-6-(3-(4-(3-metilacetidin-1-il)fenil)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona; (4aR,8aS)-6-(3-(4-(3,3-dimetilpirrolidin-1-il)fenil)acetidina-1-carbonil)hexahidro-2H-pi rido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-(4-(3-(trifluorometoxi)acetidin-1-il)fenil)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-(6-(2,4-didorofenil)piridin-3-il)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-(2'-doro-4'-(metilsulfonil)-[1,1'-bifenil]-4-il)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-(4-(3,3-difluoroacetidin-1-il)fenil)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(4-(4-bromofenil)piperidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-[3-[4-(2,2,2-trifluoroetoxi)fenil]acetidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona;
2-[4-[1-[(4aR,8aS)-3-oxo-4,4a,5,7,8,8a-hexahidmpindo[4,3-b][1,4]oxacma-6-carbonN]acetidm-3-N]fenN]-5-don> benzonitrilo;
(4aR,8aS)-6-[3-[4-[3-[(1S o 1R)-2,2,2-trifluoro-1-metN-etoxi]acetidin-1-N]fenN]acetidma-1-carbonN]-4,4a,5,7,8,8ahexahidropirido[4,3-b][l,4]oxacin-3-ona;
(4aR,8aS)-6-[3-[4-[3-[(1R o 1S)-2,2,2-trifluoro-1-metil-etoxi]acetidin-1-il]fenil]acetidina-1-carbonil]-4,4a,5,7,8,8ahexahidropirido[4,3-b][l,4]oxacin-3-ona;
(4aR,8aS)-6-(3-(4-(2-(trifluorometil)pirrolidin-1-il)fenil)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-(4-(3-(trifluorometil)pirrolidin-1-il)fenil)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-(3-bromofenil)pirrolidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-[3-[4-(2-etilpirrolidin-1-il)fenil]acetidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona;
2-[4-[1-[(4aR,8aS)-3-oxo-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacina-6-carbonil]acetidin-3-il]fenil]-3-clorobenzonitrilo;
(4aR,8aS)-6-(3-(4-(2-ciclopropilpirrolidin-1-il)fenil)acetidina-1-carbonil)hexahidro-2H-pi rido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(4-(4-bromofenil)piperacina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(4-(2',4'-dicloro-[1,1'-bifenil]-4-il)piperidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-(4-(3-azabiciclo[3.1.0]hexan-3-il)fenil)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-(3-(trifluorometoxi)fenil)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona; (4aR,8aS)-6-(3-(4-(3-(2,2,2-trifluoroetoxi)acetidin-1-il)fenil)acetidin-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-(4'-cloro-2'-(metilsulfonil)-[1,1'-bifenil]-4-il)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-(6-(2-(trifluorometil)pirrolidin-1-il)piridin-3-il)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-[4-[(R o S)-(3-metilsulfonilfenil)-(3-piridil)metil]piperidina-1-carbonil]-4,4a,5,7,8,8ahexahidropirido[4,3-b][1,4]oxacin-3-ona;
(4aR,8aS)-6-[4-[(S o R)-(3-metilsulfonilfenil)-(3-piridil)metil]piperidina-1-carbonil]-4,4a,5,7,8,8ahexahidropirido[4,3-b][1,4]oxacin-3-ona;
(4aR,8aS)-6-[4-[(R o S)-(3-metilsulfonilfenil)-(4-piridil)metil]piperidina-1-carbonil]-4,4a,5,7,8,8ahexahidropirido[4,3-b][1,4]oxacin-3-ona;
(4aR,8aS)-6-[4-[(S o R)-(3-metilsulfonilfenil)-(4-piridil)metil]piperidina-1-carbonil]-4,4a,5,7,8,8ahexahidropirido[4,3-b][1,4]oxacin-3-ona;
(4aR,8aS)-6-(3-(4-(5-azaespiro[2.4]heptan-5-il)fenil)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-(4-(pentafluoro-16-sulfanoil)fenil)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-(5-doropiridin-2-il)acetidina-1-carbonil)hexahidro-2H-pi rido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-(2-fluoro-4-(trifluorometoxi)fenil)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-[3-(6-metoxipiridin-3-il)acetidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona; (4aR,8aS)-6-(3-(4-bromofenil)pirrolidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-fenilacetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(4-fenilpiperidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-[3-[4-(2,2,2-trifluoroetil)fenil]acetidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona;
(4aR,8aS)-6-[3-[4-[1-(trifluorometil)cidopropil]fenil]acetidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona;
(4aR,8aS)-6-[4-[(R o S)-(3-metilsulfonilfenil)-(2-piridil)metil]piperidina-1-carbonil]-4,4a,5,7,8,8ahexahidropirido[4,3-b][1,4]oxacin-3-ona;
(4aR,8aS)-6-[4-[(S o R)-(3-metilsulfonilfenil)-(2-piridil)metil]piperidina-1-carbonil]-4,4a,5,7,8,8ahexahidropirido[4,3-b][1,4]oxacin-3-ona;
(4aR,8aS)-6-[3-[4-(6,6-difluoro-2-azaespi ro[3.3]heptan-2-il)fenil]acetidina-1-carbonil]-4,4a,5,7,8,8ahexahidropirido[4,3-b][1,4]oxacin-3-ona;
(4aR,8aS)-6-(3-(4-(8-oxa-3-azabicido[3.2.1]octan-3-il)fenil)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-(5-(2,4-didorofenil)piridin-2-il)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-(4-((R o S)-2-(trifluorometil)pirrolidin-1-il)fenil)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-(4-((S o R)-2-(trifluorometil)pirrolidin-1-il)fenil)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-[3-[4-(1-piperidil)fenil]acetidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona; (4aR,8aS)-6-(3-(4-((R o S)-3-(trifluorometil)pirrolidin-1-il)fenil)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-(4-((S o R)-3-(trifluorometil)pirrolidin-1-il)fenil)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-[3-[4-(3-hidroxi-3-metilacetidin-1-il)fenil]acetidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropi rido[4,3-b][1,4]oxacin-3-ona;
(4aR,8aS)-6-[3-[4-(3-fluoro-3-metil-acetidin-1-il)fenil]acetidin-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona;
(4aR,8aS)-6-(3-(4-(3-fluoroacetidin-1-il)fenil)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona; (4aR,8aS)-6-(3-(3-fluoro-4-(trifluorometoxi)fenil)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-(3-metil-4-(trifluorometoxi)fenil)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-(3,5-difluoro-4-(trifluorometoxi)fenil)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-(2-doro-4-(trifluorometoxi)fenil)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-(4-(bicido[1.1.1]pentan-1-il)fenil)acetidina-1-carbonil)hexahidro-2H-pi rido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-(5-(2-(trifluorometil)pirrolidin-1-il)piridin-2-il)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-(5-fluoro-1H-indol-3-il)pirrolidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3 (4H)-ona; (4aR,8aS)-6-(3-(1-bencil-5-doro-1H-indol-3-il)pirrolidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-(2-fluoro-4-(trifluorometil)fenil)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-(3-doro-4-(trifluorometoxi)fenil)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-[3-[4-[3-(cidopropilmetoxi)acetidin-1-il]fenil]acetidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona;
5-(1-((4aR,8aS)-3-oxooctahidro-2H-pirido[4,3-b][1,4]oxacina-6-carbonil)acetidin-3-il)-2-(trifluorometoxi)benzonitrilo;
(4aR,8aS)-6-(3-(4-(1-(hidroximetil)cidopropil)fenil)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-(1-metil-1H-indazol-4-il)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona; (4aR,8aS)-6-(3-(4-(3-fluoropirrolidin-1-il)fenil)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-(4-(2-metilacetidin-1-il)fenil)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona; (4aR,8aS)-6-[4-[fenil(piridacin-3-il)metil]piperidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona;
(4aR,8aS)-6-[3-[4-(trifluorometoxi)fenil]pirrolidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona;
(4aR,8aS)-6-(3-(1-metil-1H-indazol-6-il)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona; (4aR,8aS)-6-[3-(S o R)-[3-(trifluorometoxi)fenil]pirrolidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona;
(4aR,8aS)-6-[3-(R o S)-[3-(trifluorometoxi)fenil]pirrolidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona;
(4aR,8aS)-6-(3-(3-doro-4-hidroxifenil)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona; (4aR,8aS)-6-(3-(3-doro-4-(3-metilacetidin-1-il)fenil)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-[3-[4-(oxetan-3-il)fenil]acetidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona; (4aR,8aS)-6-(3-(3-doro-4-(3,3-difluoroacetidin-1-il)fenil)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-(3-(3-metilacetidin-1-il)-4-(trifluorometoxi)fenil)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-[(3S o R)-3-(3-bromofenil)pirrolidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona;
(4aR,8aS)-6-[(3R o S)-3-(3-bromofenil)pirrolidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona;
(4aR,8aS)-6-[3-[4-(3-azabicido[3.1.1]heptan-3-il)fenil]acetidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona;
(4aR,8aS)-6-(2-metil-3-(4-(trifluorometoxi)fenil)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-(3,3-dimetil-2,3-dihidrobenzofuran-6-il)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-(3-doro-5-(2,2,2-trifluoroetoxi)fenil)pirrolidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
2-metil-2-(4-(1-((4aR,8aS)-3-oxooctahidro-2H-pirido[4,3-b][1,4]oxacina-6-carbonil)acetidin-3-il)fenil)propanoato de metilo;
(4aR,8aS)-6-(3-(3,5-didorofenil)pirrolidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-(4-(3-metoxi-3-metilacetidin-1-il)fenil)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(2-(3-((1-((4aR,8aS)-3-oxoodahidro-2H-pirido[4,3-b][1,4]oxadna-6-carbonN)piperidm-4-il)(fenil)metil)fenoxi)etil)carbamato de ferc-butilo;
(4aR,8aS)-6-((R o S)-3-(3-don>5-(2,2,2-trifluoroetoxi)fenN)pirroNdma-1-carbonN)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-((S o R)-3-(3-cloro-5-(2,2,2-trifluoroetoxi)fenil)pirrolidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-(4-(ferc-butil)-3-metoxifenil)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona; (4aR,8aS)-6-(3-(1-metil-1H-indazol-5-il)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona; (4aR,8aS)-6-(3-(4-propilfenil)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-(4-(trifluorometoxi)-3-(trifluorometil)fenil)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-[3-[4-[[1-(trifluorometil)ciclopropil]metoxi]fenil]acetidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona;
(4aR,8aS)-6-[3-[4-(2,2,2-trifluoro-1,1-dimetil-etil)fenil]acetidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona;
(4aR,8aS)-6-[3-(R o S)-[4-(2,2,2-trifluoro-1-metil-etoxi)fenil]acetidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona;
(4aR,8aS)-6-[3-[4-(3-fluoropropil)fenil]acetidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona; (4aR,8aS)-6-[3-(S o R)-[4-(2,2,2-trifluoro-1-metil-etoxi)fenil]acetidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona;
(4aR,8aS)-6-[3-(4-ciclobutilfenil)acetidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona; (4aR,8aS)-6-[3-(3-metoxi-4-metil-fenil)acetidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona; (4aR,8aS)-6-(4-((4-fluorofenil)((1-metil-5-(trifluorometil)-1H-pirazol-3-il)oxi)metil)piperidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-[4-[[3-(2-aminoetoxi)fenil]-fenil-metil]piperidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona;
(4aR,8aS)-6-[3-[5-(2,4-diclorofenil)-1,3,4-oxadiazol-2-il]acetidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3
b][1,4]oxacin-3-ona;
(4aR,8aS)-6-[3-[3-fluoro-4-(trifluorometil)fenil]acetidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona;
N-[2-[3-[2-[3-[[1-[(4aR,8aS)-3-oxo-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacina-6-carbonil]-4-piperidil]-fenil-1-metil]fenoxi]etilamino]-3-oxo-propoxi]etil]carbamato de ferc-butilo;
N-[2-[2-[3-[2-[3-[[1-[(4aR,8aS)-3-oxo-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacina-6-carbonil]-4-piperidil]-fenilmetil]fenoxi]etilamino]-3-oxo-propoxi]etoxi]etil]carbamato de ferc-butilo;
(4aR,8aS)-6-[3-[1-(2,4-didorofenil)pirazol-3-il]acetidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona;
(4aR,8aS)-6-[3-(4-propoxifenil)acetidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona;
(4aR,8aS)-6-[3-(3,4-pimetilfenil)acetidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona;
2-[4-[1-[(4aR,8aS)-3-oxo-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacina-6-carbonil]acetidin-3-il]fenil]-N-etil-5-metil-benzamida;
(4aR,8aS)-6-[3-[4-(2,2-dimetilpropil)fenil]acetidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona;
(4aR,8aS)-6-[3-(4-ferc-butoxifenil)acetidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona; (4aR,8aS)-6-[4-(5-cloroindolin-1-il)piperidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona; (4aR,8aS)-6-[4-(4-cloroisoindolin-2-il)piperidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona; (4aR,8aS)-6-[4-(5'-cloroespi ro[ciclopropano-1,3'-indolina]-1'-il)piperidina-1-carbonil]-4,4a,5,7,8,8ahexahidropirido[4,3-b][1,4]oxacin-3-ona;
(4aR,8aS)-6-[3-(4-cloroisoindolin-2-il)acetidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona; (4aR,8aS)-6-[4-(5-cloroisoindolin-2-il)piperidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona; (4aS,8aS)-6-[4-[1-[2-[ferc-butil(dimetil)silil]oxietil]-5-cloro-indol-3-il]piperidina-1-carbonil]-4,4a,5,7,8,8ahexahidropirido[4,3-b][1,4]oxacin-3-ona;
(4aS,8aS)-6-[4-[5-cloro-1-(2-hidroxietil)indol-3-il]piperidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona;
(4aR,8aS)-6-(3-(5-(3-(trifluorometil)pirrolidin-1-il)piridin-2-il)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-(5-((R o S)-3-(trifluorometil)pirrolidin-1-il)piridin-2-il)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-(5-((S o R)-3-(trifluorometil)pirrolidin-1-il)piridin-2-il)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-[3-[6-[3-(trifluorometil)pirrolidin-1-il]-3-piridil]acetidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona;
(4aR,8aS)-6-[3-[6-[3-(R o S)-(trifluorometil)pirrolidin-1-il]-3-piridil]acetidina-1-carbonil]-4,4a,5,7,8,8ahexahidropirido[4,3-b][1,4]oxacin-3-ona;
(4aR,8aS)-6-[3-[6-[3S- o 3R-(trifluorometil)pirrolidin-1-il]-3-piridil]acetidina-1-carbonil]-4,4a,5,7,8,8ahexahidropirido[4,3-b][1,4]oxacin-3-ona;
(4aR,8aS)-6-[3-(4-tetrahidropiran-3-ilfenil)acetidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropi rido[4,3-b][1,4]oxacin-3-ona;
(4aR,8aS)-6-[3-[2-metoxi-4-(2,2,2-trifluoroetil)fenil]acetidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropi rido[4,3-b][1,4]oxacin-3-ona;
(4aR,8aS)-6-(3-(4-(neopentiloxi)fenil)acetidina-1-carbonil)hexahidro-2H-pi rido[4,3-b][1,4]oxacin-3(4H)-ona; rac-(4aR,8aS)-6-[4-(3-metoxi-1-fenil-propil)piperidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona;
rac-(4aR,8aS)-6-[4-[fenil(3-piridil)metil]piperidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona;
rac-(4aR,8aS)-6-[4-[(5-metil-1,2,4-oxadiazol-3-il)-fenil-metil]piperidina-1-carbonil]-4,4a,5,7,8,8ahexahidropirido[4,3-b][1,4]oxacin-3-ona;
rac-(4aR,8aS)-6-[4-(5-fluoro-1-metil-indol-3-il)piperidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona;
3-[5-doro-3-[1-[rac-(4aR,8aS)-3-oxo-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacina-6-carbonil]-4-piperidil]indol-1-il]acetidina-1-carboxilato de ferc-butilo;
3-[5-doro-3-[1-[rac-(4aR,8aS)-3-oxo-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacina-6-carbonil]-4-piperidil]indol-1-il]acetidina-1-carboxilato de ferc-butilo;
rac-(4aR,8aS)-6-[4-[1-(2-doro-4-fluoro-fenoxi)propil]piperidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropi rido[4,3-b][1,4]oxacin-3-ona;
rac-(4aR,8aS)-6-[4-[(2-cloro-4-fluoro-fenoxi)-ciclopropil-metil]piperidina-1-carbonil]-4,4a,5,7,8,8ahexahidropirido[4,3-b][1,4]oxacin-3-ona;
rac-(4aR,8aS)-6-[4-[(4-fluorofenoxi)-fenil-metil]piperidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona;
rac-(4aR,8aS)-6-[4-[5-(trifluorometil)-2-piridil]piperidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona;
rac-(4aR,8aS)-6-[4-[ciclopropilmetoxi-[4-(trifluorometil)fenil]metil]piperidina-1-carbonil]-4,4a,5,7,8,8ahexahidropirido[4,3-b][1,4]oxacin-3-ona;
rac-(4aR,8aS)-6-[4-[1-(4-fluorofenil)-2-hidroxi-etil]piperacina-1-carbonil]-4,4a,5,7,8,8a-hexahidropi rido[4,3-b][1,4]oxacin-3-ona;
rac-(4aR,8aS)-6-[4-[1-(4-fluorofenil)-2-metoxi-etil]piperacina-1-carbonil]-4,4a,5,7,8,8a-hexahidropi rido[4,3-b][1,4]oxacin-3-ona;
rac-(4aR,8aS)-6-[4-[1-(3,4-difluorofenil)-2-hidroxi-etil]piperacina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona;
rac-(4aR,8aS)-6-[4-[1-(3,4-difluorofenil)-2-metoxi-etil]piperacina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona;
rac-(4aR,8aS)-6-[4-[1-(4,4-difluoro-1-piperidil)ciclopropil]piperidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona;
rac-(4aR,8aS)-6-[4-[1-[4-(trifluorometil)imidazol-1-il]etil]piperidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona;
rac-(4aR,8aS)-6-[4-[1-[4-(trifluorometil)pirazol-1-il]ciclopropil]piperidina-1-carbonil]-4,4a,5,7,8,8ahexahidropirido[4,3-b][1,4]oxacin-3-ona;
rac-(4aR,8aS)-6-[4-[(4-ferc-butilpirazol-1-il)-(4-fluorofenil)metil]piperidina-1-carbonil]-4,4a,5,7,8,8ahexahidropirido[4,3-b][1,4]oxacin-3-ona;
rac-(4aR,8aS)-6-[4-[3-[4-(trifluorometil)fenil]oxetan-3-il]piperidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona;
rac-(4aR,8aS)-6-[4-[4-(trifluorometil)benzoil]piperidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona;
rac-(4aR,8aS)-6-[4-[1-[4-(trifluorometil)pirazol-1-il]etil]piperidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona;
rac-(4aR,8aS)-6-[4-[(4-fluorofenil)-morfolino-metil]piperidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropi rido[4,3-b][1,4]oxacin-3-ona;
rac-(4aR,8aS)-6-[4-[(4-terc-butNoxazol-2-N)-difluon>metN]piperidma-1-carbonN]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona;
rac-(4aR,8aS)-6-[4-[1-(4-dorofenil)-2-hidroxi-etil]piperacina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona;
rac-(4aR,8aS)-6-[4-[1-(4-dorofenil)-2-metoxi-etil]piperacina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona;
rac-(4aR,8aS)-6-[4-[2-hidroxi-1-[4-(trifluorometil)fenil]etil]piperacina-1-carbonil]-4,4a,5,7,8,8a-hexahidropi rido[4,3-b][1,4]oxacin-3-ona;
rac-(4aR,8aS)-6-[4-[2-metoxi-1-[4-(trifluorometil)fenil]etil]piperacina-1-carbonil]-4,4a,5,7,8,8a-hexahidropi rido[4,3-b][1,4]oxacin-3-ona;
rac-(4aR,8aS)-6-[4-(1-cidohexil-2-metoxi-etil)piperacina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona;
rac-(4aR,8aS)-6-[4-(1-cidohexil-2-hidroxi-etil)piperacina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona;
rac-(4aR,8aS)-6-[4-[1-(4-fluorofenil)cidopropil]piperacina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona;
rac-(4aR,8aS)-6-[4-[2-(4-fluorofenil)cidopropil]piperacina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona;
rac-(4aR,8aS)-6-[4-[1-(4-fluomfenN)ddobutN]piperadna-1-carbonN]-4,4a,5,7,8,8a-hexahidmpindo[4,3-b][1,4]oxacin-3-ona;
rac-(4aR,8aS)-6-[4-[cidopropil-(4-fluorofenil)metil]piperacina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona;
rac-(4aR,8aS)-6-[4-[cidohexil-(4-fluorofenil)metil]piperacina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona;
rac-(4aR,8aS)-6-[4-[(4-fluorofenil)-(4-piridil)metil]piperacina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona;
rac-(4aR,8aS)-6-[4-[2,2,2-trifluoro-1-(4-fluorofenil)etil]piperacina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona;
rac-(4aR,8aS)-6-[4-[(4-fluorofenil)-oxazol-5-il-metil]piperacina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona;
rac-(4aR,8aS)-6-[4-[bis(4-dorofenil)metil]piperacina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona;
rac-(4aS,8aS)-6-[4-[5-doro-1-(cidopropilmetil)indol-3-il]piperidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona;
rac-(4aS,8aS)-6-[4-[5-doro-1-(cidopropilmetil)indol-3-il]piperidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona;
rac-(4aR,8aR)-6-[4-[5-doro-1-(cidopropilmetil)indol-3-il]piperidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona;
rac-(4aR,8aS)-6-[4-[(4-dorofenil)-(4-fluorofenil)metil]piperacina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona;
rac-(4aR,8aS)-6-(4-benzhidrilpiperacina-1-carbonil)-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona;
rac-(4aR,8aS)-6-[4-[4-(2,2,2-trifluoroetil)fenil]piperacina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona;
rac-(4aR,8aS)-6-[4-[3-(2,2,2-trifluoroetil)fenil]piperacina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona;
rac-(4aR,8aS)-6-[4-[4-doro-3-(trifluorometil)fenil]piperacina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona;
rac-(4aR,8aS)-6-[4-[1-[4-(trifluorometil)fenil]etil]piperacina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona;
rac-(4aR,8aS)-6-[3-[1-(2-doro-4-fluoro-fenoxi)etil]acetidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona; y
rac-(4aR,8aS)-6-[3-[5-doro-1-(cidopropilmetil)indol-3-il]acetidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona.
10. El compuesto de fórmula (I) de acuerdo con una cualquiera de las reivindicaciones 1 a 8, o una sal farmacéuticamente aceptable del mismo, seleccionado de:
rac-cis-6-(4-(5-doro-1-(cidopropilmetil)-1H-indol-3-il)piperidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
rac-cis-6-(4-(5-cloro-1-metil-1H-indol-3-il)piperidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona; rac-cis-6-(4-(1-(4-fluorofenil)-3-metoxipropil)piperidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona; rac-cis-6-(4-((4-clorofenil)(fenil)metil)piperacina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona; (+)-cis-6-(4-(bis(4-fluorofenil)metil)piperacina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(+)- o (-)-cis-6-(4-(5-cloro-1-ciclopropil-1H-indol-3-il)piperidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(+)- o (-)-cis-6-(4-(5-cloro-1-(oxetan-3-il)-1H-indol-3-il)piperidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
rac-cis-6-(4-(1-(4-fluorofenil)-3-hidroxipropil)piperidina-1-carbonil)hexahidro-2H-pi rido[4,3-b][1,4]oxacin-3(4H)-ona; (4aR,8aS)-6-(4-(difluoro(4-(trifluorometil)fenil)metil)piperidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(+) o (-)-cis-6-(4-((R o S)-1-(2-cloro-4-fluorofenoxi)etil)piperidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(+) o (-)-cis-6-(4-((4-clorofenil)difluorometil)piperidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona; (+) o (-)-cis-6-(4-((2-cloro-4-fluorofenil)difluorometil)piperidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(4-((R o S)-1-(4-(trifluorometil)fenil)etil)piperidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(4-((S o R)-(4-fluorofenil)(2,2,2-trifluoroetoxi)metil)piperidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-[4-[(S o R)-(4-fluorofenil)-(3-metoxifenil)metil]piperidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona;
(4aR,8aS)-6-(4-((R o S)-(4-fluorofenil)(3-metoxifenil)metil)piperidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(4-((S o R)-(3-(2-fluoroetoxi)fenil)(fenil)metil)piperidina-1-carbonil)hexahidro-2H-pirido[4,3-
b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(4-((3-cidopropil-1,2,4-oxadiazol-5-il)(4-fluorofenil)metil)piperidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(4-((S o R)-(4-fluorofenil)(4-metoxifenil)metil)piperidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(4-((S o R)-(3,4-dimetoxifenil)(4-fluorofenil)metil)piperidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(4-((R o S)-(4-fluorofenil)(4-metoxifenil)metil)piperidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(4-((S o R)-(4-(2-fluoroetoxi)fenil)(4-fluorofenil)metil)piperidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-[3-[4-(trifluorometoxi)fenil]acetidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona; (4aR,8aS)-6-(3-(2'-trifluorometoxi)-[1,1'-bifenil]-4-il)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-(2'-(trifluorometil)-[1,1'-bifenil]-4-il)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-(2',4'-difluoro-[1,1'-bifenil]-4-il)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-(4-(3-(trifluorometil)acetidin-1-il)fenil)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-(4-bromo-3-dorofenil)acetidina-1-carbonil)hexahidro-2H-pi rido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-[3-[4-(4-doro-2-fluoro-fenil)fenil]acetidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona;
(4aR,8aS)-6-[3-[4-(2-doro-4-fluoro-fenil)fenil]acetidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona;
(4aR,8aS)-6-(3-(4-(3-((1,1,1-trifluoropropan-2-il)oxi)acetidin-1-il)fenil)acetidina-1-carbonil)hexahidro-2H-pi rido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-(4-(terc-butN)fenN)acetidma-1-carbonN)hexahidn>2H-pirido[4,3-b][1,4]oxadn-3(4H)-ona;
(4aR,8aS)-6-(3-(4-(trifluorometil)fenil)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-(4-(3-((1,1,1-trifluoro-2-metilpropan-2-il)oxi)acetidin-1-il)fenil)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-(4-(1,1-difluoroetil)fenil)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona; (4aR,8aS)-6-(3-(4-(3,3-dimetilpirrolidin-1-il)fenil)acetidina-1-carbonil)hexahidro-2H-pi rido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-(4-(3-(trifluorometoxi)acetidin-1-il)fenil)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-(6-(2,4-didorofenN)pmdm-3-N)acetidma-1-carbonN)hexahidn>2H-pirido[4,3-b][1,4]oxadn-3(4H)-ona;
(4aR,8aS)-6-[3-[4-(2,2,2-trifluoroetoxi)fenil]acetidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona;
(4aR,8aS)-6-[3-[4-[3-[(1S o 1R)-2,2,2-trifluoro-1-metil-etoxi]acetidin-1-il]fenil]acetidina-1-carbonil]-4,4a,5,7,8,8ahexahidropirido[4,3-b][l,4]oxacin-3-ona;
(4aR,8aS)-6-[3-[4-[3-[(1R o 1S)-2,2,2-trifluoro-1-metil-etoxi]acetidin-1-il]fenil]acetidina-1-carbonil]-4,4a,5,7,8,8ahexahidropirido[4,3-b][l,4]oxacin-3-ona;
(4aR,8aS)-6-(3-(4-(2-(trifluorometil)pirrolidin-1-il)fenil)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-(4-(3-(trifluorometil)pirrolidin-1-il)fenil)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-(4-(3-(2,2,2-trifluoroetoxi)acetidin-1-il)fenil)acetidin-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-(6-(2-(trifluorometil)pirrolidin-1-il)piridin-3-il)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-(4-(pentafluoro-16-sulfanoil)fenil)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-(2-fluoro-4-(trifluorometoxi)fenil)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-[3-[4-(2,2,2-trifluoroetil)fenil]acetidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona;
(4aR,8aS)-6-[3-[4-[1-(trifluorometil)cidopropil]fenil]acetidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropirido[4,3-b][1,4]oxacin-3-ona;
(4aR,8aS)-6-[3-[4-(6,6-difluoro-2-azaespi ro[3.3]heptan-2-il)fenil]acetidina-1-carbonil]-4,4a,5,7,8,8ahexahidropirido[4,3-b][1,4]oxacin-3-ona;
(4aR,8aS)-6-(3-(5-(2,4-didorofenil)piridin-2-il)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-(4-((R o S)-2-(trifluorometil)pirrolidin-1-il)fenil)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-(4-((S o R)-2-(trifluorometil)pirrolidin-1-il)fenil)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-(4-((R o S)-3-(trifluorometil)pirrolidin-1-il)fenil)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-(4-((S o R)-3-(trifluorometil)pirrolidin-1-il)fenil)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-(3-fluoro-4-(trifluorometoxi)fenil)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-(3-metil-4-(trifluorometoxi)fenil)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-(3,5-difluoro-4-(trifluorometoxi)fenil)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-(3-doro-4-(trifluorometoxi)fenil)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-((R o S)-3-(3-don>5-(2,2,2-trifluoroetoxi)fenN)pirroNdma-1-carbonN)hexahidro-2H-pmdo[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-((S o R)-3-(3-doro-5-(2,2,2-trifluoroetoxi)fenil)pirrolidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-(4-(terc-butN)-3-metoxifenN)acetidma-1-carbonN)hexahidro-2H-pmdo[4,3-b][1,4]oxadn-3(4H)-ona;
(4aR,8aS)-6-(3-(4-propilfenil)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-(4-(trifluorometoxi)-3-(trifluorometil)fenil)acetidina-1-carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-(5-((R o S)-3-(trifluorometil)pirrolidin-1-il)piridin-2-il)acetidina-1 -carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-(3-(5-((S o R)-3-(trifluorometil)pirrolidin-1-il)piridin-2-il)acetidina-1 -carbonil)hexahidro-2H-pirido[4,3-b][1,4]oxacin-3(4H)-ona;
(4aR,8aS)-6-[3-[6-[3-(R o S)-(trifluorometil)pirrolidin-1-il]-3-piridil]acetidina-1-carbonil]-4,4a,5,7,8,8ahexahidropirido[4,3-b][1,4]oxacin-3-ona;
(4aR,8aS)-6-[3-[6-[3-(S o R)-(trifluorometil)pirrolidin-1 -il]-3-piridil]acetidina-1-carbonil]-4,4a,5,7,8,8ahexahidropirido[4,3-b][1,4]oxacin-3-ona;
(4aR,8aS)-6-[3-(4-tetrahidropiran-3-ilfenil)acetidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropi rido[4,3-b][1,4]oxacin-3-ona;
(4aR,8aS)-6-[3-[2-metoxi-4-(2,2,2-trifluoroetil)fenil]acetidina-1-carbonil]-4,4a,5,7,8,8a-hexahidropi rido[4,3-b][1,4]oxacin-3-ona; y
(4aR,8aS)-6-(3-(4-(neopentiloxi)fenil)acetidina-1-carbonil)hexahidro-2H-pi rido[4,3-b][1,4]oxacin-3(4H)-ona.
11. Un procedimiento de fabricación de los compuestos de fórmula (Ic) de acuerdo con una cualquiera de las reivindicaciones 1 a 10, que comprende:
hacer reaccionar 4a,5,6,7,8,8a-hexahidro-4H-pirido[4,3-b][1,4]oxacin-3-ona (1),

con una amina heterocíclica 2a, en la que A, L, X, m, n y de R15 *20 a R23 son como se define en una cualquiera de las reivindicaciones 1 a 10

en presencia de una base y un reactivo formador de urea, para formar dicho compuesto de fórmula (Ic).
12. Un compuesto de fórmula (Ic) de acuerdo con una cualquiera de las reivindicaciones 1 a 10, o una sal farmacéuticamente aceptable del mismo, para su uso como sustancia terapéuticamente activa.
13. Una composición farmacéutica que comprende un compuesto de fórmula (Ic) de acuerdo con una cualquiera de las reivindicaciones 1 a 10, o una sal farmacéuticamente aceptable del mismo, y un vehículo terapéuticamente inerte.
14. Un compuesto de fórmula (Ic) de acuerdo con una cualquiera de las reivindicaciones 1 a 10, o una sal farmacéuticamente aceptable del mismo, para su uso en el tratamiento o profilaxis de neuroinflamación, enfermedades neurodegenerativas, dolor, cáncer y/o trastornos mentales en un mamífero.
15. El compuesto para su uso de acuerdo con la reivindicación 14, en el que dicha neuroinflamación, enfermedades neurodegenerativas, dolor, cáncer y/o trastornos mentales se seleccionan de esclerosis múltiple, enfermedad de Alzheimer, enfermedad de Parkinson, esclerosis lateral amiotrófica, traumatismo craneoencefálico, neurotoxicidad, apoplejía, epilepsia, ansiedad, migraña, depresión, carcinoma hepatocelular, carcinogénesis de colon, cáncer de ovario, dolor neuropático, neuropatía inducida por quimioterapia, dolor agudo, dolor crónico y/o espasticidad asociada con dolor en un mamífero.
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP18163273 | 2018-03-22 | ||
| CN2019075372 | 2019-02-18 | ||
| PCT/EP2019/057174 WO2019180185A1 (en) | 2018-03-22 | 2019-03-22 | Oxazine monoacylglycerol lipase (magl) inhibitors |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2942319T3 true ES2942319T3 (es) | 2023-05-31 |
Family
ID=65818027
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES19711920T Active ES2942319T3 (es) | 2018-03-22 | 2019-03-22 | Inhibidores de oxacina monoacilglicerol lipasa (MAGL) |
Country Status (27)
| Country | Link |
|---|---|
| US (2) | US20210107920A1 (es) |
| EP (1) | EP3768684B1 (es) |
| JP (1) | JP7308220B2 (es) |
| KR (1) | KR20200136425A (es) |
| CN (1) | CN111936503B (es) |
| AU (1) | AU2019238381B2 (es) |
| BR (1) | BR112020014992A2 (es) |
| CA (1) | CA3089443A1 (es) |
| CL (1) | CL2020002423A1 (es) |
| CO (1) | CO2020009567A2 (es) |
| CR (1) | CR20200416A (es) |
| DK (1) | DK3768684T3 (es) |
| ES (1) | ES2942319T3 (es) |
| FI (1) | FI3768684T3 (es) |
| HR (1) | HRP20230388T1 (es) |
| HU (1) | HUE061584T2 (es) |
| IL (1) | IL276031B2 (es) |
| LT (1) | LT3768684T (es) |
| MX (1) | MX2020008894A (es) |
| PE (1) | PE20201185A1 (es) |
| PH (1) | PH12020500661A1 (es) |
| PL (1) | PL3768684T3 (es) |
| RS (1) | RS64156B1 (es) |
| SG (1) | SG11202007608UA (es) |
| SI (1) | SI3768684T1 (es) |
| TW (1) | TWI818967B (es) |
| WO (1) | WO2019180185A1 (es) |
Families Citing this family (18)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| AR114136A1 (es) | 2017-10-10 | 2020-07-29 | Hoffmann La Roche | Compuestos heterocíclicos |
| WO2019105915A1 (en) | 2017-11-28 | 2019-06-06 | F. Hoffmann-La Roche Ag | New heterocyclic compounds |
| WO2019134985A1 (en) | 2018-01-08 | 2019-07-11 | F. Hoffmann-La Roche Ag | Octahydropyrido[1,2-alpha]pyrazines as magl inhibitors |
| PE20211089A1 (es) * | 2018-08-13 | 2021-06-14 | Hoffmann La Roche | Nuevos compuestos heterociclicos como inhibidores de la monoacilglicerol lipasa |
| WO2020104494A1 (en) | 2018-11-22 | 2020-05-28 | F. Hoffmann-La Roche Ag | New heterocyclic compounds |
| US20210094971A1 (en) * | 2019-09-09 | 2021-04-01 | Hoffmann-La Roche Inc. | Heterocyclic compounds |
| CN114269755A (zh) * | 2019-09-12 | 2022-04-01 | 豪夫迈·罗氏有限公司 | 作为magl抑制剂的4,4a,5,7,8,8a-六吡啶并[4,3-b][1,4]噁嗪-3-酮化合物 |
| PH12022550433A1 (en) * | 2019-09-23 | 2024-01-29 | Hoffmann La Roche | Heterocyclic compounds |
| WO2021058445A1 (en) * | 2019-09-24 | 2021-04-01 | F. Hoffmann-La Roche Ag | New heterocyclic monoacylglycerol lipase (magl) inhibitors |
| CR20220117A (es) * | 2019-09-24 | 2022-04-20 | F Horrmann La Roche Ag | Compuestos heterocíclicos |
| WO2021058443A1 (en) * | 2019-09-24 | 2021-04-01 | F. Hoffmann-La Roche Ag | Fluorescent probes for monoacylglycerol lipase (magl) |
| CA3190277A1 (en) | 2020-09-03 | 2022-03-10 | Joerg Benz | Heterocyclic compounds |
| EP4416131A4 (en) * | 2021-10-15 | 2025-07-30 | Genetolead Inc | RAS INHIBITORS, COMPOSITIONS AND METHODS OF USE THEREOF |
| CA3242372A1 (en) | 2021-12-29 | 2023-07-06 | Psy Therapeutics, Inc. | Inhibiting monoacylglycerol lipase (magl) |
| EP4543870A1 (en) * | 2022-06-24 | 2025-04-30 | F. Hoffmann-La Roche AG | New heterocyclic-carbonyl-cyclic compounds as magl inhibitors |
| TW202415651A (zh) | 2022-07-29 | 2024-04-16 | 日商住友製藥股份有限公司 | 含氮飽和雜環衍生物 |
| CN119894887A (zh) * | 2022-09-20 | 2025-04-25 | 豪夫迈·罗氏有限公司 | 用于magl的荧光探针 |
| EP4665718A1 (en) * | 2023-02-13 | 2025-12-24 | Apogee Pharmaceuticals, Inc. | Small molecules as monoacylglycerol lipase (magl) inhibitors, compositions and use thereof |
Family Cites Families (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7579495B2 (en) | 2003-12-19 | 2009-08-25 | Momentive Performance Materials Inc. | Active-releasing cyclic siloxanes |
| ATE450525T1 (de) | 2005-06-20 | 2009-12-15 | Schering Corp | Kohlenstoffgebundene substituierte piperidine und derivate daraus als histamin-h3-antagonisten |
| EP1984351A1 (en) | 2006-02-17 | 2008-10-29 | Memory Pharmaceuticals Corporation | Compounds having 5-ht6 receptor affinity |
| US7872028B2 (en) | 2006-04-05 | 2011-01-18 | Vitae Pharmaceuticals, Inc. | Diaminopropanol renin inhibitors |
| WO2011059118A1 (ko) | 2009-11-10 | 2011-05-19 | Kim Hyun Jeen | 후각인지능력 검사 시스템 |
| CN103781763B (zh) | 2011-05-16 | 2017-03-22 | 生态学有限公司 | 作为钾通道阻断剂的胺衍生物 |
| US9487495B2 (en) * | 2012-01-06 | 2016-11-08 | The Scripts Research Institute | Carbamate compounds and of making and using same |
| GB201209587D0 (en) | 2012-05-30 | 2012-07-11 | Takeda Pharmaceutical | Therapeutic compounds |
| WO2016109501A1 (en) | 2014-12-30 | 2016-07-07 | Karos Pharmaceuticals, Inc. | Amide compounds as tryptophan hydroxylase inhibitors |
| CA2979302A1 (en) | 2015-05-13 | 2016-11-17 | Selvita S.A. | Substituted quinoxaline derivatives |
| KR20180005250A (ko) | 2015-05-21 | 2018-01-15 | 글락소스미스클라인 인털렉츄얼 프로퍼티 디벨로프먼트 리미티드 | Pad4 억제제로서의 벤조이미다졸 유도체 |
| WO2016205590A1 (en) | 2015-06-18 | 2016-12-22 | Cephalon, Inc. | Substituted 4-benzyl and 4-benzoyl piperidine derivatives |
| CN105198903B (zh) * | 2015-11-09 | 2016-08-24 | 代先慧 | 一种治疗急性上呼吸道感染的药物组合物 |
| WO2017170830A1 (ja) | 2016-03-31 | 2017-10-05 | 武田薬品工業株式会社 | 複素環化合物 |
-
2019
- 2019-03-22 ES ES19711920T patent/ES2942319T3/es active Active
- 2019-03-22 PE PE2020001119A patent/PE20201185A1/es unknown
- 2019-03-22 CA CA3089443A patent/CA3089443A1/en active Pending
- 2019-03-22 MX MX2020008894A patent/MX2020008894A/es unknown
- 2019-03-22 HU HUE19711920A patent/HUE061584T2/hu unknown
- 2019-03-22 JP JP2020548747A patent/JP7308220B2/ja active Active
- 2019-03-22 AU AU2019238381A patent/AU2019238381B2/en not_active Ceased
- 2019-03-22 CR CR20200416A patent/CR20200416A/es unknown
- 2019-03-22 SI SI201930519T patent/SI3768684T1/sl unknown
- 2019-03-22 TW TW108110005A patent/TWI818967B/zh not_active IP Right Cessation
- 2019-03-22 KR KR1020207029587A patent/KR20200136425A/ko active Pending
- 2019-03-22 FI FIEP19711920.9T patent/FI3768684T3/fi active
- 2019-03-22 WO PCT/EP2019/057174 patent/WO2019180185A1/en not_active Ceased
- 2019-03-22 EP EP19711920.9A patent/EP3768684B1/en active Active
- 2019-03-22 SG SG11202007608UA patent/SG11202007608UA/en unknown
- 2019-03-22 CN CN201980018255.1A patent/CN111936503B/zh active Active
- 2019-03-22 HR HRP20230388TT patent/HRP20230388T1/hr unknown
- 2019-03-22 DK DK19711920.9T patent/DK3768684T3/da active
- 2019-03-22 RS RS20230303A patent/RS64156B1/sr unknown
- 2019-03-22 PL PL19711920.9T patent/PL3768684T3/pl unknown
- 2019-03-22 BR BR112020014992-6A patent/BR112020014992A2/pt not_active Application Discontinuation
- 2019-03-22 LT LTEPPCT/EP2019/057174T patent/LT3768684T/lt unknown
-
2020
- 2020-07-13 IL IL276031A patent/IL276031B2/en unknown
- 2020-07-31 CO CONC2020/0009567A patent/CO2020009567A2/es unknown
- 2020-09-08 PH PH12020500661A patent/PH12020500661A1/en unknown
- 2020-09-18 US US17/025,155 patent/US20210107920A1/en not_active Abandoned
- 2020-09-21 CL CL2020002423A patent/CL2020002423A1/es unknown
-
2022
- 2022-08-09 US US17/818,459 patent/US20230117324A1/en not_active Abandoned
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2942319T3 (es) | Inhibidores de oxacina monoacilglicerol lipasa (MAGL) | |
| JP7609854B2 (ja) | Magl阻害剤としての4,4a,5,7,8,8a-ヘキサピリド[4,3-b][1,4]オキサジン-3-オン化合物 | |
| RU2769507C1 (ru) | Новые гетероциклические соединения как ингибиторы моноацилглицеринлипазы | |
| KR20210044217A (ko) | 모노아실글리세롤 리파제 저해제로서의 신규 헤테로시클릭 화합물 | |
| US11981661B2 (en) | Heterocyclic compounds | |
| JP2024521618A (ja) | 複素環式化合物 | |
| CN114401968A (zh) | 新颖杂环单酰基甘油脂肪酶(magl)抑制剂 | |
| RU2794334C2 (ru) | Ингибиторы оксазинмоноацилглицеринлипазы (magl) | |
| HK40035599A (en) | Oxazine monoacylglycerol lipase (magl) inhibitors | |
| RU2801190C2 (ru) | Новые гетероциклические соединения в качестве ингибиторов моноацилглицеринлипазы | |
| HK40035599B (zh) | 恶嗪单醯甘油脂肪酶(magl)抑制剂 | |
| HK40065118A (en) | New heterocyclic monoacylglycerol lipase (magl) inhibitors | |
| HK40062310A (en) | 4,4a,5,7,8,8a-hexapyrido[4,3-b][1,4]oxazin-3-one compounds as magl inhibitors | |
| HK40082879A (zh) | 杂环化合物 |