ES2954474T3 - Sales de adición de ácido de succinato y fumarato de derivados de piperazina útiles como inhibidores de glucosidasa - Google Patents
Sales de adición de ácido de succinato y fumarato de derivados de piperazina útiles como inhibidores de glucosidasa Download PDFInfo
- Publication number
- ES2954474T3 ES2954474T3 ES19759533T ES19759533T ES2954474T3 ES 2954474 T3 ES2954474 T3 ES 2954474T3 ES 19759533 T ES19759533 T ES 19759533T ES 19759533 T ES19759533 T ES 19759533T ES 2954474 T3 ES2954474 T3 ES 2954474T3
- Authority
- ES
- Spain
- Prior art keywords
- mmol
- yield
- max
- mono
- vacuo
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 150000003839 salts Chemical class 0.000 title claims abstract description 51
- 239000002253 acid Substances 0.000 title claims description 43
- 102000004366 Glucosidases Human genes 0.000 title claims description 10
- 108010056771 Glucosidases Proteins 0.000 title claims description 10
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 title abstract description 30
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 title abstract description 15
- 150000004885 piperazines Chemical class 0.000 title abstract description 5
- 229940066771 systemic antihistamines piperazine derivative Drugs 0.000 title abstract description 5
- 239000003112 inhibitor Substances 0.000 title description 4
- KDYFGRWQOYBRFD-UHFFFAOYSA-L succinate(2-) Chemical compound [O-]C(=O)CCC([O-])=O KDYFGRWQOYBRFD-UHFFFAOYSA-L 0.000 title description 3
- 239000007787 solid Substances 0.000 claims abstract description 125
- 238000000034 method Methods 0.000 claims description 189
- 150000001875 compounds Chemical class 0.000 claims description 139
- 239000000203 mixture Substances 0.000 claims description 72
- 239000002904 solvent Substances 0.000 claims description 24
- 201000011240 Frontotemporal dementia Diseases 0.000 claims description 12
- 238000002360 preparation method Methods 0.000 claims description 12
- 239000003814 drug Substances 0.000 claims description 11
- 238000000634 powder X-ray diffraction Methods 0.000 claims description 10
- 208000003407 Creutzfeldt-Jakob Syndrome Diseases 0.000 claims description 7
- 208000034799 Tauopathies Diseases 0.000 claims description 7
- 206010012289 Dementia Diseases 0.000 claims description 6
- 206010002026 amyotrophic lateral sclerosis Diseases 0.000 claims description 6
- 201000002212 progressive supranuclear palsy Diseases 0.000 claims description 6
- 238000010438 heat treatment Methods 0.000 claims description 5
- 208000024827 Alzheimer disease Diseases 0.000 claims description 4
- 208000020406 Creutzfeldt Jacob disease Diseases 0.000 claims description 4
- 208000010859 Creutzfeldt-Jakob disease Diseases 0.000 claims description 4
- 208000000609 Pick Disease of the Brain Diseases 0.000 claims description 4
- 208000036757 Postencephalitic parkinsonism Diseases 0.000 claims description 4
- 208000018756 Variant Creutzfeldt-Jakob disease Diseases 0.000 claims description 4
- 208000018813 behavioral variant of frontotemporal dementia Diseases 0.000 claims description 4
- 208000017004 dementia pugilistica Diseases 0.000 claims description 4
- 201000010099 disease Diseases 0.000 claims description 4
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims description 4
- 230000004770 neurodegeneration Effects 0.000 claims description 4
- 239000006186 oral dosage form Substances 0.000 claims description 4
- 208000000170 postencephalitic Parkinson disease Diseases 0.000 claims description 4
- 208000024172 Cardiovascular disease Diseases 0.000 claims description 3
- 208000002339 Frontotemporal Lobar Degeneration Diseases 0.000 claims description 3
- 206010028980 Neoplasm Diseases 0.000 claims description 3
- 238000009835 boiling Methods 0.000 claims description 3
- 201000011510 cancer Diseases 0.000 claims description 3
- 206010012601 diabetes mellitus Diseases 0.000 claims description 3
- 208000019715 inherited Creutzfeldt-Jakob disease Diseases 0.000 claims description 3
- 208000010125 myocardial infarction Diseases 0.000 claims description 3
- 208000015122 neurodegenerative disease Diseases 0.000 claims description 3
- 208000002593 pantothenate kinase-associated neurodegeneration Diseases 0.000 claims description 3
- 208000023697 ABri amyloidosis Diseases 0.000 claims description 2
- 208000017227 ADan amyloidosis Diseases 0.000 claims description 2
- 208000014644 Brain disease Diseases 0.000 claims description 2
- 208000004051 Chronic Traumatic Encephalopathy Diseases 0.000 claims description 2
- 208000028698 Cognitive impairment Diseases 0.000 claims description 2
- 208000011990 Corticobasal Degeneration Diseases 0.000 claims description 2
- 208000009093 Diffuse Neurofibrillary Tangles with Calcification Diseases 0.000 claims description 2
- 201000010374 Down Syndrome Diseases 0.000 claims description 2
- 208000032274 Encephalopathy Diseases 0.000 claims description 2
- 201000004066 Ganglioglioma Diseases 0.000 claims description 2
- 208000003736 Gerstmann-Straussler-Scheinker Disease Diseases 0.000 claims description 2
- 206010072075 Gerstmann-Straussler-Scheinker syndrome Diseases 0.000 claims description 2
- 206010018341 Gliosis Diseases 0.000 claims description 2
- 208000023105 Huntington disease Diseases 0.000 claims description 2
- 201000000162 ITM2B-related cerebral amyloid angiopathy 1 Diseases 0.000 claims description 2
- 201000000194 ITM2B-related cerebral amyloid angiopathy 2 Diseases 0.000 claims description 2
- 208000001089 Multiple system atrophy Diseases 0.000 claims description 2
- 206010068871 Myotonic dystrophy Diseases 0.000 claims description 2
- 208000014060 Niemann-Pick disease Diseases 0.000 claims description 2
- 208000037658 Parkinson-dementia complex of Guam Diseases 0.000 claims description 2
- 208000027089 Parkinsonian disease Diseases 0.000 claims description 2
- 206010034010 Parkinsonism Diseases 0.000 claims description 2
- 208000010291 Primary Progressive Nonfluent Aphasia Diseases 0.000 claims description 2
- 102000029797 Prion Human genes 0.000 claims description 2
- 108091000054 Prion Proteins 0.000 claims description 2
- 208000024777 Prion disease Diseases 0.000 claims description 2
- 208000018642 Semantic dementia Diseases 0.000 claims description 2
- 208000037065 Subacute sclerosing leukoencephalitis Diseases 0.000 claims description 2
- 206010042297 Subacute sclerosing panencephalitis Diseases 0.000 claims description 2
- 208000026911 Tuberous sclerosis complex Diseases 0.000 claims description 2
- 210000000349 chromosome Anatomy 0.000 claims description 2
- 208000010877 cognitive disease Diseases 0.000 claims description 2
- 230000007850 degeneration Effects 0.000 claims description 2
- 201000006061 fatal familial insomnia Diseases 0.000 claims description 2
- 201000005649 gangliocytoma Diseases 0.000 claims description 2
- 201000008361 ganglioneuroma Diseases 0.000 claims description 2
- 230000007387 gliosis Effects 0.000 claims description 2
- 238000000338 in vitro Methods 0.000 claims description 2
- 230000002401 inhibitory effect Effects 0.000 claims description 2
- 206010023497 kuru Diseases 0.000 claims description 2
- 210000004498 neuroglial cell Anatomy 0.000 claims description 2
- 208000001282 primary progressive aphasia Diseases 0.000 claims description 2
- 230000000750 progressive effect Effects 0.000 claims description 2
- 208000009999 tuberous sclerosis Diseases 0.000 claims description 2
- 208000018737 Parkinson disease Diseases 0.000 claims 2
- 208000024571 Pick disease Diseases 0.000 claims 2
- KDYFGRWQOYBRFD-UHFFFAOYSA-N succinic acid Chemical compound OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 abstract description 20
- 239000001530 fumaric acid Substances 0.000 abstract description 10
- 239000001384 succinic acid Substances 0.000 abstract description 10
- 239000004615 ingredient Substances 0.000 abstract description 3
- 239000003316 glycosidase inhibitor Substances 0.000 abstract 1
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 213
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 209
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 153
- 239000011541 reaction mixture Substances 0.000 description 106
- 239000000243 solution Substances 0.000 description 97
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 92
- 239000000543 intermediate Substances 0.000 description 87
- 235000019439 ethyl acetate Nutrition 0.000 description 75
- 238000006243 chemical reaction Methods 0.000 description 73
- 239000013058 crude material Substances 0.000 description 70
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 67
- 238000005160 1H NMR spectroscopy Methods 0.000 description 66
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 66
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 66
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 64
- 238000003818 flash chromatography Methods 0.000 description 61
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 59
- 238000004809 thin layer chromatography Methods 0.000 description 58
- 238000004128 high performance liquid chromatography Methods 0.000 description 50
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 48
- 239000012044 organic layer Substances 0.000 description 46
- 239000011734 sodium Substances 0.000 description 46
- 239000010410 layer Substances 0.000 description 44
- 239000003480 eluent Substances 0.000 description 39
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 38
- -1 hexafluorophosphate Chemical compound 0.000 description 38
- 239000003208 petroleum Substances 0.000 description 37
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 35
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 33
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 33
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 33
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 29
- 125000004194 piperazin-1-yl group Chemical group [H]N1C([H])([H])C([H])([H])N(*)C([H])([H])C1([H])[H] 0.000 description 27
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 24
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 22
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 20
- 150000003462 sulfoxides Chemical class 0.000 description 20
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 19
- BXVSAYBZSGIURM-UHFFFAOYSA-N 2-phenoxy-4h-1,3,2$l^{5}-benzodioxaphosphinine 2-oxide Chemical compound O1CC2=CC=CC=C2OP1(=O)OC1=CC=CC=C1 BXVSAYBZSGIURM-UHFFFAOYSA-N 0.000 description 17
- 239000004480 active ingredient Substances 0.000 description 16
- 239000012267 brine Substances 0.000 description 16
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 16
- 125000004528 pyrimidin-5-yl group Chemical group N1=CN=CC(=C1)* 0.000 description 15
- 239000012071 phase Substances 0.000 description 14
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 14
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 13
- DLFVBJFMPXGRIB-UHFFFAOYSA-N Acetamide Chemical compound CC(N)=O DLFVBJFMPXGRIB-UHFFFAOYSA-N 0.000 description 12
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 12
- 150000003890 succinate salts Chemical class 0.000 description 12
- 238000004808 supercritical fluid chromatography Methods 0.000 description 12
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 11
- 238000005481 NMR spectroscopy Methods 0.000 description 11
- 229910021529 ammonia Inorganic materials 0.000 description 11
- 238000004458 analytical method Methods 0.000 description 11
- 125000001841 imino group Chemical group [H]N=* 0.000 description 11
- 239000000741 silica gel Substances 0.000 description 11
- 229910002027 silica gel Inorganic materials 0.000 description 11
- 239000000725 suspension Substances 0.000 description 11
- 150000001412 amines Chemical class 0.000 description 10
- 230000015572 biosynthetic process Effects 0.000 description 10
- 125000005555 sulfoximide group Chemical group 0.000 description 10
- SJRJJKPEHAURKC-UHFFFAOYSA-N N-Methylmorpholine Chemical compound CN1CCOCC1 SJRJJKPEHAURKC-UHFFFAOYSA-N 0.000 description 9
- 238000010828 elution Methods 0.000 description 9
- 238000007866 imination reaction Methods 0.000 description 9
- 150000002576 ketones Chemical class 0.000 description 9
- 239000000047 product Substances 0.000 description 9
- NRKYWOKHZRQRJR-UHFFFAOYSA-N 2,2,2-trifluoroacetamide Chemical compound NC(=O)C(F)(F)F NRKYWOKHZRQRJR-UHFFFAOYSA-N 0.000 description 8
- ZWEHNKRNPOVVGH-UHFFFAOYSA-N 2-Butanone Chemical compound CCC(C)=O ZWEHNKRNPOVVGH-UHFFFAOYSA-N 0.000 description 8
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 8
- 230000008859 change Effects 0.000 description 8
- 229940079593 drug Drugs 0.000 description 8
- 239000000706 filtrate Substances 0.000 description 8
- 238000002953 preparative HPLC Methods 0.000 description 8
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 7
- 230000000052 comparative effect Effects 0.000 description 7
- 238000003821 enantio-separation Methods 0.000 description 7
- 229910052760 oxygen Inorganic materials 0.000 description 7
- 125000006239 protecting group Chemical group 0.000 description 7
- 239000000126 substance Substances 0.000 description 7
- NHQDETIJWKXCTC-UHFFFAOYSA-N 3-chloroperbenzoic acid Chemical compound OOC(=O)C1=CC=CC(Cl)=C1 NHQDETIJWKXCTC-UHFFFAOYSA-N 0.000 description 6
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical class CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 6
- BZLVMXJERCGZMT-UHFFFAOYSA-N Methyl tert-butyl ether Chemical compound COC(C)(C)C BZLVMXJERCGZMT-UHFFFAOYSA-N 0.000 description 6
- UCKMPCXJQFINFW-UHFFFAOYSA-N Sulphide Chemical compound [S-2] UCKMPCXJQFINFW-UHFFFAOYSA-N 0.000 description 6
- 239000005457 ice water Substances 0.000 description 6
- 239000003446 ligand Substances 0.000 description 6
- 230000003647 oxidation Effects 0.000 description 6
- 238000007254 oxidation reaction Methods 0.000 description 6
- 238000000746 purification Methods 0.000 description 6
- SYBXSZMNKDOUCA-UHFFFAOYSA-J rhodium(2+);tetraacetate Chemical compound [Rh+2].[Rh+2].CC([O-])=O.CC([O-])=O.CC([O-])=O.CC([O-])=O SYBXSZMNKDOUCA-UHFFFAOYSA-J 0.000 description 6
- 239000012279 sodium borohydride Substances 0.000 description 6
- 229910000033 sodium borohydride Inorganic materials 0.000 description 6
- 239000007858 starting material Substances 0.000 description 6
- 238000003786 synthesis reaction Methods 0.000 description 6
- WSLDOOZREJYCGB-UHFFFAOYSA-N 1,2-Dichloroethane Chemical compound ClCCCl WSLDOOZREJYCGB-UHFFFAOYSA-N 0.000 description 5
- QCQCHGYLTSGIGX-GHXANHINSA-N 4-[[(3ar,5ar,5br,7ar,9s,11ar,11br,13as)-5a,5b,8,8,11a-pentamethyl-3a-[(5-methylpyridine-3-carbonyl)amino]-2-oxo-1-propan-2-yl-4,5,6,7,7a,9,10,11,11b,12,13,13a-dodecahydro-3h-cyclopenta[a]chrysen-9-yl]oxy]-2,2-dimethyl-4-oxobutanoic acid Chemical compound N([C@@]12CC[C@@]3(C)[C@]4(C)CC[C@H]5C(C)(C)[C@@H](OC(=O)CC(C)(C)C(O)=O)CC[C@]5(C)[C@H]4CC[C@@H]3C1=C(C(C2)=O)C(C)C)C(=O)C1=CN=CC(C)=C1 QCQCHGYLTSGIGX-GHXANHINSA-N 0.000 description 5
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 5
- XEEYBQQBJWHFJM-UHFFFAOYSA-N Iron Chemical compound [Fe] XEEYBQQBJWHFJM-UHFFFAOYSA-N 0.000 description 5
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical compound CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 5
- DKGAVHZHDRPRBM-UHFFFAOYSA-N Tert-Butanol Chemical compound CC(C)(C)O DKGAVHZHDRPRBM-UHFFFAOYSA-N 0.000 description 5
- 238000002441 X-ray diffraction Methods 0.000 description 5
- 239000013078 crystal Substances 0.000 description 5
- 238000002425 crystallisation Methods 0.000 description 5
- 230000008025 crystallization Effects 0.000 description 5
- JMMWKPVZQRWMSS-UHFFFAOYSA-N isopropyl acetate Chemical compound CC(C)OC(C)=O JMMWKPVZQRWMSS-UHFFFAOYSA-N 0.000 description 5
- 238000011170 pharmaceutical development Methods 0.000 description 5
- BDERNNFJNOPAEC-UHFFFAOYSA-N propan-1-ol Chemical compound CCCO BDERNNFJNOPAEC-UHFFFAOYSA-N 0.000 description 5
- 238000000425 proton nuclear magnetic resonance spectrum Methods 0.000 description 5
- 239000012453 solvate Substances 0.000 description 5
- PAQZWJGSJMLPMG-UHFFFAOYSA-N 2,4,6-tripropyl-1,3,5,2$l^{5},4$l^{5},6$l^{5}-trioxatriphosphinane 2,4,6-trioxide Chemical compound CCCP1(=O)OP(=O)(CCC)OP(=O)(CCC)O1 PAQZWJGSJMLPMG-UHFFFAOYSA-N 0.000 description 4
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 4
- OKKJLVBELUTLKV-MZCSYVLQSA-N Deuterated methanol Chemical compound [2H]OC([2H])([2H])[2H] OKKJLVBELUTLKV-MZCSYVLQSA-N 0.000 description 4
- PCLIMKBDDGJMGD-UHFFFAOYSA-N N-bromosuccinimide Chemical compound BrN1C(=O)CCC1=O PCLIMKBDDGJMGD-UHFFFAOYSA-N 0.000 description 4
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 4
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 4
- 239000002585 base Substances 0.000 description 4
- 150000001558 benzoic acid derivatives Chemical class 0.000 description 4
- 239000001913 cellulose Substances 0.000 description 4
- 229920002678 cellulose Polymers 0.000 description 4
- 238000004090 dissolution Methods 0.000 description 4
- 239000002552 dosage form Substances 0.000 description 4
- 150000002148 esters Chemical class 0.000 description 4
- 239000012458 free base Substances 0.000 description 4
- 239000011521 glass Substances 0.000 description 4
- 229910052739 hydrogen Inorganic materials 0.000 description 4
- 238000002844 melting Methods 0.000 description 4
- 230000008018 melting Effects 0.000 description 4
- 230000008569 process Effects 0.000 description 4
- 238000001953 recrystallisation Methods 0.000 description 4
- 230000002829 reductive effect Effects 0.000 description 4
- 238000010992 reflux Methods 0.000 description 4
- 230000002441 reversible effect Effects 0.000 description 4
- 239000011877 solvent mixture Substances 0.000 description 4
- 238000001179 sorption measurement Methods 0.000 description 4
- 238000003756 stirring Methods 0.000 description 4
- 125000000475 sulfinyl group Chemical group [*:2]S([*:1])=O 0.000 description 4
- 125000004496 thiazol-5-yl group Chemical group S1C=NC=C1* 0.000 description 4
- FYSNRJHAOHDILO-UHFFFAOYSA-N thionyl chloride Chemical compound ClS(Cl)=O FYSNRJHAOHDILO-UHFFFAOYSA-N 0.000 description 4
- 230000004584 weight gain Effects 0.000 description 4
- 235000019786 weight gain Nutrition 0.000 description 4
- MSXVEPNJUHWQHW-UHFFFAOYSA-N 2-methylbutan-2-ol Chemical compound CCC(C)(C)O MSXVEPNJUHWQHW-UHFFFAOYSA-N 0.000 description 3
- 125000003349 3-pyridyl group Chemical group N1=C([H])C([*])=C([H])C([H])=C1[H] 0.000 description 3
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 3
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 3
- QGJOPFRUJISHPQ-UHFFFAOYSA-N Carbon disulfide Chemical compound S=C=S QGJOPFRUJISHPQ-UHFFFAOYSA-N 0.000 description 3
- WMFOQBRAJBCJND-UHFFFAOYSA-M Lithium hydroxide Chemical compound [Li+].[OH-] WMFOQBRAJBCJND-UHFFFAOYSA-M 0.000 description 3
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 3
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 3
- 238000001069 Raman spectroscopy Methods 0.000 description 3
- 239000000443 aerosol Substances 0.000 description 3
- 238000013459 approach Methods 0.000 description 3
- BTANRVKWQNVYAZ-UHFFFAOYSA-N butan-2-ol Chemical compound CCC(C)O BTANRVKWQNVYAZ-UHFFFAOYSA-N 0.000 description 3
- HGXJOXHYPGNVNK-UHFFFAOYSA-N butane;ethenoxyethane;tin Chemical compound CCCC[Sn](CCCC)(CCCC)C(=C)OCC HGXJOXHYPGNVNK-UHFFFAOYSA-N 0.000 description 3
- 125000004432 carbon atom Chemical group C* 0.000 description 3
- 239000003638 chemical reducing agent Substances 0.000 description 3
- 238000010511 deprotection reaction Methods 0.000 description 3
- WQOXQRCZOLPYPM-UHFFFAOYSA-N dimethyl disulfide Chemical compound CSSC WQOXQRCZOLPYPM-UHFFFAOYSA-N 0.000 description 3
- 239000012154 double-distilled water Substances 0.000 description 3
- 238000011065 in-situ storage Methods 0.000 description 3
- 239000007924 injection Substances 0.000 description 3
- 238000002347 injection Methods 0.000 description 3
- 229910052742 iron Inorganic materials 0.000 description 3
- 229910052748 manganese Inorganic materials 0.000 description 3
- 239000011572 manganese Substances 0.000 description 3
- 238000004519 manufacturing process Methods 0.000 description 3
- 238000004949 mass spectrometry Methods 0.000 description 3
- 229910052751 metal Inorganic materials 0.000 description 3
- 239000002184 metal Substances 0.000 description 3
- 229910052757 nitrogen Inorganic materials 0.000 description 3
- 239000007800 oxidant agent Substances 0.000 description 3
- 230000001590 oxidative effect Effects 0.000 description 3
- 230000035699 permeability Effects 0.000 description 3
- 230000000704 physical effect Effects 0.000 description 3
- 238000012545 processing Methods 0.000 description 3
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 3
- 230000001105 regulatory effect Effects 0.000 description 3
- IOGXOCVLYRDXLW-UHFFFAOYSA-N tert-butyl nitrite Chemical compound CC(C)(C)ON=O IOGXOCVLYRDXLW-UHFFFAOYSA-N 0.000 description 3
- 238000001757 thermogravimetry curve Methods 0.000 description 3
- 150000003568 thioethers Chemical class 0.000 description 3
- 230000009466 transformation Effects 0.000 description 3
- 230000000007 visual effect Effects 0.000 description 3
- 230000004580 weight loss Effects 0.000 description 3
- KZPYGQFFRCFCPP-UHFFFAOYSA-N 1,1'-bis(diphenylphosphino)ferrocene Chemical compound [Fe+2].C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1 KZPYGQFFRCFCPP-UHFFFAOYSA-N 0.000 description 2
- BDNKZNFMNDZQMI-UHFFFAOYSA-N 1,3-diisopropylcarbodiimide Chemical compound CC(C)N=C=NC(C)C BDNKZNFMNDZQMI-UHFFFAOYSA-N 0.000 description 2
- RXYPXQSKLGGKOL-UHFFFAOYSA-N 1,4-dimethylpiperazine Chemical compound CN1CCN(C)CC1 RXYPXQSKLGGKOL-UHFFFAOYSA-N 0.000 description 2
- OKPBVDZHVNDNKR-UHFFFAOYSA-N 1-(1,3-benzothiazol-5-yl)ethanol Chemical compound CC(O)C1=CC=C2SC=NC2=C1 OKPBVDZHVNDNKR-UHFFFAOYSA-N 0.000 description 2
- FCKINXPLWQMSSB-UHFFFAOYSA-N 1-(1,3-benzothiazol-5-yl)ethanone Chemical compound CC(=O)C1=CC=C2SC=NC2=C1 FCKINXPLWQMSSB-UHFFFAOYSA-N 0.000 description 2
- VPTKWRFYAYTYDL-UHFFFAOYSA-N 1-(2-methyl-1,3-benzothiazol-5-yl)ethanol Chemical compound CC=1SC2=C(N=1)C=C(C=C2)C(C)O VPTKWRFYAYTYDL-UHFFFAOYSA-N 0.000 description 2
- KPMZOEHLPRSIEC-UHFFFAOYSA-N 1-(2-methyl-1,3-benzothiazol-5-yl)ethanone Chemical compound CC(=O)C1=CC=C2SC(C)=NC2=C1 KPMZOEHLPRSIEC-UHFFFAOYSA-N 0.000 description 2
- KBPLFHHGFOOTCA-UHFFFAOYSA-N 1-Octanol Chemical compound CCCCCCCCO KBPLFHHGFOOTCA-UHFFFAOYSA-N 0.000 description 2
- BBMCTIGTTCKYKF-UHFFFAOYSA-N 1-heptanol Chemical compound CCCCCCCO BBMCTIGTTCKYKF-UHFFFAOYSA-N 0.000 description 2
- WPVSIRHOGVUDLI-UHFFFAOYSA-N 2-methyl-5-[1-[4-(4-methylsulfanylphenyl)piperazin-1-yl]ethyl]-1,3-benzothiazole Chemical compound CC=1SC2=C(N=1)C=C(C=C2)C(C)N1CCN(CC1)C1=CC=C(C=C1)SC WPVSIRHOGVUDLI-UHFFFAOYSA-N 0.000 description 2
- BZVORZDCHXKUED-UHFFFAOYSA-N 2-methyl-5-[1-[4-(4-methylsulfinylphenyl)piperazin-1-yl]ethyl]-1,3-benzothiazole Chemical compound CC=1SC2=C(N=1)C=C(C=C2)C(C)N1CCN(CC1)C1=CC=C(C=C1)S(=O)C BZVORZDCHXKUED-UHFFFAOYSA-N 0.000 description 2
- CZMYKDAVWMRQLM-UHFFFAOYSA-N 5-[1-[4-(4-methylsulfanylphenyl)piperazin-1-yl]ethyl]-1,3-benzothiazole Chemical compound CSC1=CC=C(C=C1)N1CCN(CC1)C(C)C=1C=CC2=C(N=CS2)C=1 CZMYKDAVWMRQLM-UHFFFAOYSA-N 0.000 description 2
- WMGKLNGJADYXDA-UHFFFAOYSA-N 5-[1-[4-(4-methylsulfinylphenyl)piperazin-1-yl]ethyl]-1,3-benzothiazole Chemical compound CS(=O)C1=CC=C(C=C1)N1CCN(CC1)C(C)C=1C=CC2=C(N=CS2)C=1 WMGKLNGJADYXDA-UHFFFAOYSA-N 0.000 description 2
- DUDNTLGZXQFTGB-UHFFFAOYSA-N 5-[1-[4-(5-bromopyrimidin-2-yl)piperazin-1-yl]ethyl]-1,3-benzothiazole Chemical compound BrC=1C=NC(=NC=1)N1CCN(CC1)C(C)C=1C=CC2=C(N=CS2)C=1 DUDNTLGZXQFTGB-UHFFFAOYSA-N 0.000 description 2
- QPKDIPKASMXTHJ-UHFFFAOYSA-N CS(=O)c1cnc(Cl)nc1 Chemical compound CS(=O)c1cnc(Cl)nc1 QPKDIPKASMXTHJ-UHFFFAOYSA-N 0.000 description 2
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 2
- QOSSAOTZNIDXMA-UHFFFAOYSA-N Dicylcohexylcarbodiimide Chemical compound C1CCCCC1N=C=NC1CCCCC1 QOSSAOTZNIDXMA-UHFFFAOYSA-N 0.000 description 2
- CETBSQOFQKLHHZ-UHFFFAOYSA-N Diethyl disulfide Chemical compound CCSSCC CETBSQOFQKLHHZ-UHFFFAOYSA-N 0.000 description 2
- XTHFKEDIFFGKHM-UHFFFAOYSA-N Dimethoxyethane Chemical compound COCCOC XTHFKEDIFFGKHM-UHFFFAOYSA-N 0.000 description 2
- 239000007821 HATU Substances 0.000 description 2
- 238000004566 IR spectroscopy Methods 0.000 description 2
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 2
- PWHULOQIROXLJO-UHFFFAOYSA-N Manganese Chemical compound [Mn] PWHULOQIROXLJO-UHFFFAOYSA-N 0.000 description 2
- BAVYZALUXZFZLV-UHFFFAOYSA-N Methylamine Chemical compound NC BAVYZALUXZFZLV-UHFFFAOYSA-N 0.000 description 2
- 241001024304 Mino Species 0.000 description 2
- FXHOOIRPVKKKFG-UHFFFAOYSA-N N,N-Dimethylacetamide Chemical compound CN(C)C(C)=O FXHOOIRPVKKKFG-UHFFFAOYSA-N 0.000 description 2
- SECXISVLQFMRJM-UHFFFAOYSA-N N-Methylpyrrolidone Chemical compound CN1CCCC1=O SECXISVLQFMRJM-UHFFFAOYSA-N 0.000 description 2
- AMQJEAYHLZJPGS-UHFFFAOYSA-N N-Pentanol Chemical compound CCCCCO AMQJEAYHLZJPGS-UHFFFAOYSA-N 0.000 description 2
- YNPNZTXNASCQKK-UHFFFAOYSA-N Phenanthrene Natural products C1=CC=C2C3=CC=CC=C3C=CC2=C1 YNPNZTXNASCQKK-UHFFFAOYSA-N 0.000 description 2
- GLUUGHFHXGJENI-UHFFFAOYSA-N Piperazine Chemical compound C1CNCCN1 GLUUGHFHXGJENI-UHFFFAOYSA-N 0.000 description 2
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 2
- CZPWVGJYEJSRLH-UHFFFAOYSA-N Pyrimidine Chemical compound C1=CN=CN=C1 CZPWVGJYEJSRLH-UHFFFAOYSA-N 0.000 description 2
- 238000001237 Raman spectrum Methods 0.000 description 2
- 229910005965 SO 2 Inorganic materials 0.000 description 2
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 2
- DGEZNRSVGBDHLK-UHFFFAOYSA-N [1,10]phenanthroline Chemical compound C1=CN=C2C3=NC=CC=C3C=CC2=C1 DGEZNRSVGBDHLK-UHFFFAOYSA-N 0.000 description 2
- 230000002378 acidificating effect Effects 0.000 description 2
- 150000007513 acids Chemical class 0.000 description 2
- 150000001298 alcohols Chemical class 0.000 description 2
- XXROGKLTLUQVRX-UHFFFAOYSA-N allyl alcohol Chemical compound OCC=C XXROGKLTLUQVRX-UHFFFAOYSA-N 0.000 description 2
- CHKQALUEEULCPZ-UHFFFAOYSA-N amino 2,4,6-trimethylbenzenesulfonate Chemical compound CC1=CC(C)=C(S(=O)(=O)ON)C(C)=C1 CHKQALUEEULCPZ-UHFFFAOYSA-N 0.000 description 2
- BVCZEBOGSOYJJT-UHFFFAOYSA-N ammonium carbamate Chemical compound [NH4+].NC([O-])=O BVCZEBOGSOYJJT-UHFFFAOYSA-N 0.000 description 2
- 150000001502 aryl halides Chemical class 0.000 description 2
- 238000003556 assay Methods 0.000 description 2
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 2
- 125000001584 benzyloxycarbonyl group Chemical group C(=O)(OCC1=CC=CC=C1)* 0.000 description 2
- MUALRAIOVNYAIW-UHFFFAOYSA-N binap Chemical compound C1=CC=CC=C1P(C=1C(=C2C=CC=CC2=CC=1)C=1C2=CC=CC=C2C=CC=1P(C=1C=CC=CC=1)C=1C=CC=CC=1)C1=CC=CC=C1 MUALRAIOVNYAIW-UHFFFAOYSA-N 0.000 description 2
- 210000004556 brain Anatomy 0.000 description 2
- 239000002775 capsule Substances 0.000 description 2
- KXDHJXZQYSOELW-UHFFFAOYSA-N carbonic acid monoamide Natural products NC(O)=O KXDHJXZQYSOELW-UHFFFAOYSA-N 0.000 description 2
- 230000003197 catalytic effect Effects 0.000 description 2
- 239000003153 chemical reaction reagent Substances 0.000 description 2
- 238000004296 chiral HPLC Methods 0.000 description 2
- 150000004696 coordination complex Chemical class 0.000 description 2
- 239000010949 copper Substances 0.000 description 2
- 239000006184 cosolvent Substances 0.000 description 2
- MWKFXSUHUHTGQN-UHFFFAOYSA-N decan-1-ol Chemical compound CCCCCCCCCCO MWKFXSUHUHTGQN-UHFFFAOYSA-N 0.000 description 2
- BGRWYRAHAFMIBJ-UHFFFAOYSA-N diisopropylcarbodiimide Natural products CC(C)NC(=O)NC(C)C BGRWYRAHAFMIBJ-UHFFFAOYSA-N 0.000 description 2
- 238000009509 drug development Methods 0.000 description 2
- 229940126534 drug product Drugs 0.000 description 2
- 230000000694 effects Effects 0.000 description 2
- 238000000132 electrospray ionisation Methods 0.000 description 2
- 238000006911 enzymatic reaction Methods 0.000 description 2
- 150000002170 ethers Chemical class 0.000 description 2
- 125000005519 fluorenylmethyloxycarbonyl group Chemical group 0.000 description 2
- ZSIAUFGUXNUGDI-UHFFFAOYSA-N hexan-1-ol Chemical compound CCCCCCO ZSIAUFGUXNUGDI-UHFFFAOYSA-N 0.000 description 2
- 239000012442 inert solvent Substances 0.000 description 2
- PHTQWCKDNZKARW-UHFFFAOYSA-N isoamylol Chemical compound CC(C)CCO PHTQWCKDNZKARW-UHFFFAOYSA-N 0.000 description 2
- ZXEKIIBDNHEJCQ-UHFFFAOYSA-N isobutanol Chemical compound CC(C)CO ZXEKIIBDNHEJCQ-UHFFFAOYSA-N 0.000 description 2
- 238000002955 isolation Methods 0.000 description 2
- 210000001853 liver microsome Anatomy 0.000 description 2
- 230000014759 maintenance of location Effects 0.000 description 2
- 239000000463 material Substances 0.000 description 2
- 150000002739 metals Chemical class 0.000 description 2
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 2
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 2
- 239000002547 new drug Substances 0.000 description 2
- LQNUZADURLCDLV-UHFFFAOYSA-N nitrobenzene Chemical compound [O-][N+](=O)C1=CC=CC=C1 LQNUZADURLCDLV-UHFFFAOYSA-N 0.000 description 2
- ZWRUINPWMLAQRD-UHFFFAOYSA-N nonan-1-ol Chemical compound CCCCCCCCCO ZWRUINPWMLAQRD-UHFFFAOYSA-N 0.000 description 2
- 238000002414 normal-phase solid-phase extraction Methods 0.000 description 2
- 239000008194 pharmaceutical composition Substances 0.000 description 2
- 239000000825 pharmaceutical preparation Substances 0.000 description 2
- LCPDWSOZIOUXRV-UHFFFAOYSA-N phenoxyacetic acid Chemical compound OC(=O)COC1=CC=CC=C1 LCPDWSOZIOUXRV-UHFFFAOYSA-N 0.000 description 2
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 2
- 229920001592 potato starch Polymers 0.000 description 2
- 238000005932 reductive alkylation reaction Methods 0.000 description 2
- 238000006268 reductive amination reaction Methods 0.000 description 2
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 2
- 235000017557 sodium bicarbonate Nutrition 0.000 description 2
- 239000011780 sodium chloride Substances 0.000 description 2
- 239000007909 solid dosage form Substances 0.000 description 2
- 239000007790 solid phase Substances 0.000 description 2
- 238000002336 sorption--desorption measurement Methods 0.000 description 2
- 150000003457 sulfones Chemical class 0.000 description 2
- 239000000829 suppository Substances 0.000 description 2
- 239000000454 talc Substances 0.000 description 2
- 229910052623 talc Inorganic materials 0.000 description 2
- MZKYHMBHEQEXRC-UHFFFAOYSA-N tert-butyl 4-(4-methylsulfanylphenyl)piperazine-1-carboxylate Chemical compound C1=CC(SC)=CC=C1N1CCN(C(=O)OC(C)(C)C)CC1 MZKYHMBHEQEXRC-UHFFFAOYSA-N 0.000 description 2
- CWXPZXBSDSIRCS-UHFFFAOYSA-N tert-butyl piperazine-1-carboxylate Chemical compound CC(C)(C)OC(=O)N1CCNCC1 CWXPZXBSDSIRCS-UHFFFAOYSA-N 0.000 description 2
- VZGDMQKNWNREIO-UHFFFAOYSA-N tetrachloromethane Chemical compound ClC(Cl)(Cl)Cl VZGDMQKNWNREIO-UHFFFAOYSA-N 0.000 description 2
- 238000002076 thermal analysis method Methods 0.000 description 2
- 238000002411 thermogravimetry Methods 0.000 description 2
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 2
- GEXJFIOPGAASTP-UHFFFAOYSA-N $l^{1}-azanylethane Chemical compound CC[N] GEXJFIOPGAASTP-UHFFFAOYSA-N 0.000 description 1
- CYPYTURSJDMMMP-WVCUSYJESA-N (1e,4e)-1,5-diphenylpenta-1,4-dien-3-one;palladium Chemical compound [Pd].[Pd].C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1.C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1.C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1 CYPYTURSJDMMMP-WVCUSYJESA-N 0.000 description 1
- UFDULEKOJAEIRI-UHFFFAOYSA-N (2-acetyloxy-3-iodophenyl) acetate Chemical compound CC(=O)OC1=CC=CC(I)=C1OC(C)=O UFDULEKOJAEIRI-UHFFFAOYSA-N 0.000 description 1
- GKWZKYWXCHCDQB-UHFFFAOYSA-N 1-(4-methylsulfanylphenyl)piperazine hydrochloride Chemical compound Cl.CSc1ccc(cc1)N1CCNCC1 GKWZKYWXCHCDQB-UHFFFAOYSA-N 0.000 description 1
- 239000005968 1-Decanol Substances 0.000 description 1
- YEUYZNNBXLMFCW-UHFFFAOYSA-N 1-bromo-4-methylsulfanylbenzene Chemical compound CSC1=CC=C(Br)C=C1 YEUYZNNBXLMFCW-UHFFFAOYSA-N 0.000 description 1
- LMDZBCPBFSXMTL-UHFFFAOYSA-N 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide Chemical compound CCN=C=NCCCN(C)C LMDZBCPBFSXMTL-UHFFFAOYSA-N 0.000 description 1
- PZNPLUBHRSSFHT-RRHRGVEJSA-N 1-hexadecanoyl-2-octadecanoyl-sn-glycero-3-phosphocholine Chemical compound CCCCCCCCCCCCCCCCCC(=O)O[C@@H](COP([O-])(=O)OCC[N+](C)(C)C)COC(=O)CCCCCCCCCCCCCCC PZNPLUBHRSSFHT-RRHRGVEJSA-N 0.000 description 1
- DVWQNBIUTWDZMW-UHFFFAOYSA-N 1-naphthalen-1-ylnaphthalen-2-ol Chemical compound C1=CC=C2C(C3=C4C=CC=CC4=CC=C3O)=CC=CC2=C1 DVWQNBIUTWDZMW-UHFFFAOYSA-N 0.000 description 1
- 238000001644 13C nuclear magnetic resonance spectroscopy Methods 0.000 description 1
- BDKLKNJTMLIAFE-UHFFFAOYSA-N 2-(3-fluorophenyl)-1,3-oxazole-4-carbaldehyde Chemical compound FC1=CC=CC(C=2OC=C(C=O)N=2)=C1 BDKLKNJTMLIAFE-UHFFFAOYSA-N 0.000 description 1
- XNWFRZJHXBZDAG-UHFFFAOYSA-N 2-METHOXYETHANOL Chemical group COCCO XNWFRZJHXBZDAG-UHFFFAOYSA-N 0.000 description 1
- NAMYKGVDVNBCFQ-UHFFFAOYSA-N 2-bromopropane Chemical compound CC(C)Br NAMYKGVDVNBCFQ-UHFFFAOYSA-N 0.000 description 1
- VATKBTIEWMXJDM-UHFFFAOYSA-N 2-chloro-5-ethylsulfanylpyrimidine Chemical compound ClC1=NC=C(C=N1)SCC VATKBTIEWMXJDM-UHFFFAOYSA-N 0.000 description 1
- CDHDUSOAKGSHAD-UHFFFAOYSA-N 2-chloro-5-ethylsulfinylpyrimidine Chemical compound ClC1=NC=C(C=N1)S(=O)CC CDHDUSOAKGSHAD-UHFFFAOYSA-N 0.000 description 1
- GKQDGTBDVTVIDS-UHFFFAOYSA-N 2-chloro-5-methylsulfanylpyridine Chemical compound CSC1=CC=C(Cl)N=C1 GKQDGTBDVTVIDS-UHFFFAOYSA-N 0.000 description 1
- UGWJTFHCCZHCDI-UHFFFAOYSA-N 2-chloro-5-methylsulfanylpyrimidine Chemical compound CSC1=CN=C(Cl)N=C1 UGWJTFHCCZHCDI-UHFFFAOYSA-N 0.000 description 1
- KKYZKENWPRMGAH-UHFFFAOYSA-N 2-chloro-5-propylsulfanylpyrimidine Chemical compound CCCSC1=CN=C(Cl)N=C1 KKYZKENWPRMGAH-UHFFFAOYSA-N 0.000 description 1
- GZKOPIDFRJFMMW-UHFFFAOYSA-N 2-chloro-5-propylsulfinylpyrimidine Chemical compound ClC1=NC=C(C=N1)S(=O)CCC GZKOPIDFRJFMMW-UHFFFAOYSA-N 0.000 description 1
- DZBKIOJXVOECRA-UHFFFAOYSA-N 2-chloropyrimidin-5-amine Chemical compound NC1=CN=C(Cl)N=C1 DZBKIOJXVOECRA-UHFFFAOYSA-N 0.000 description 1
- 125000000954 2-hydroxyethyl group Chemical group [H]C([*])([H])C([H])([H])O[H] 0.000 description 1
- 125000004200 2-methoxyethyl group Chemical group [H]C([H])([H])OC([H])([H])C([H])([H])* 0.000 description 1
- NEUHZUKMEAYZGD-UHFFFAOYSA-N 2-methyl-5-(1-piperazin-1-ylethyl)-1,3-benzothiazole Chemical compound S1C2=C(N=C1C)C=C(C(C)N1CCNCC1)C=C2 NEUHZUKMEAYZGD-UHFFFAOYSA-N 0.000 description 1
- CSXZTVGZLVMVRY-UHFFFAOYSA-N 3,4-dibromo-1-naphthalen-1-ylnaphthalen-2-ol Chemical compound C1=CC=C2C(C3=C4C=CC=CC4=C(Br)C(Br)=C3O)=CC=CC2=C1 CSXZTVGZLVMVRY-UHFFFAOYSA-N 0.000 description 1
- FRIBMENBGGCKPD-UHFFFAOYSA-N 3-(2,3-dimethoxyphenyl)prop-2-enal Chemical compound COC1=CC=CC(C=CC=O)=C1OC FRIBMENBGGCKPD-UHFFFAOYSA-N 0.000 description 1
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 1
- ZQPANRJQVGSNOS-UHFFFAOYSA-N 5-(1-piperazin-1-ylethyl)-1,3-benzothiazole Chemical compound S1C=NC2=C1C=CC(=C2)C(C)N1CCNCC1 ZQPANRJQVGSNOS-UHFFFAOYSA-N 0.000 description 1
- KFDDRUWQFQJGNL-UHFFFAOYSA-N 5-bromo-1,3-benzothiazole Chemical compound BrC1=CC=C2SC=NC2=C1 KFDDRUWQFQJGNL-UHFFFAOYSA-N 0.000 description 1
- XPGIBDJXEVAVTO-UHFFFAOYSA-N 5-bromo-2-chloropyrimidine Chemical compound ClC1=NC=C(Br)C=N1 XPGIBDJXEVAVTO-UHFFFAOYSA-N 0.000 description 1
- OLQKNZNXLBILDD-UHFFFAOYSA-N 5-bromo-2-methyl-1,3-benzothiazole Chemical compound BrC1=CC=C2SC(C)=NC2=C1 OLQKNZNXLBILDD-UHFFFAOYSA-N 0.000 description 1
- QRXMUCSWCMTJGU-UHFFFAOYSA-N 5-bromo-4-chloro-3-indolyl phosphate Chemical compound C1=C(Br)C(Cl)=C2C(OP(O)(=O)O)=CNC2=C1 QRXMUCSWCMTJGU-UHFFFAOYSA-N 0.000 description 1
- CLRJRWBQIJXBIU-UHFFFAOYSA-N 6,7-dimethoxynaphthalen-1-amine;hydrochloride Chemical class Cl.C1=CC(N)=C2C=C(OC)C(OC)=CC2=C1 CLRJRWBQIJXBIU-UHFFFAOYSA-N 0.000 description 1
- AULWPXHFRBLPAE-UHFFFAOYSA-N 6-chloropyridine Chemical compound ClC1=C=CC=C[N]1 AULWPXHFRBLPAE-UHFFFAOYSA-N 0.000 description 1
- 241000416162 Astragalus gummifer Species 0.000 description 1
- 239000005711 Benzoic acid Substances 0.000 description 1
- 101150065749 Churc1 gene Proteins 0.000 description 1
- VBLITEVHYIURRX-UHFFFAOYSA-N ClC(C)C=1C=CC2=C(N=CS2)C=1 Chemical compound ClC(C)C=1C=CC2=C(N=CS2)C=1 VBLITEVHYIURRX-UHFFFAOYSA-N 0.000 description 1
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical compound [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 description 1
- XDTMQSROBMDMFD-UHFFFAOYSA-N Cyclohexane Chemical compound C1CCCCC1 XDTMQSROBMDMFD-UHFFFAOYSA-N 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- ZAFNJMIOTHYJRJ-UHFFFAOYSA-N Diisopropyl ether Chemical compound CC(C)OC(C)C ZAFNJMIOTHYJRJ-UHFFFAOYSA-N 0.000 description 1
- KRHYYFGTRYWZRS-UHFFFAOYSA-M Fluoride anion Chemical compound [F-] KRHYYFGTRYWZRS-UHFFFAOYSA-M 0.000 description 1
- 101000801643 Homo sapiens Retinal-specific phospholipid-transporting ATPase ABCA4 Proteins 0.000 description 1
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 1
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 1
- 241000124008 Mammalia Species 0.000 description 1
- 241001465754 Metazoa Species 0.000 description 1
- NQTADLQHYWFPDB-UHFFFAOYSA-N N-Hydroxysuccinimide Chemical compound ON1C(=O)CCC1=O NQTADLQHYWFPDB-UHFFFAOYSA-N 0.000 description 1
- 101100030361 Neurospora crassa (strain ATCC 24698 / 74-OR23-1A / CBS 708.71 / DSM 1257 / FGSC 987) pph-3 gene Proteins 0.000 description 1
- 230000006271 O-GlcNAcylation Effects 0.000 description 1
- CTQNGGLPUBDAKN-UHFFFAOYSA-N O-Xylene Chemical compound CC1=CC=CC=C1C CTQNGGLPUBDAKN-UHFFFAOYSA-N 0.000 description 1
- OFOBLEOULBTSOW-UHFFFAOYSA-N Propanedioic acid Natural products OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 1
- 102100033617 Retinal-specific phospholipid-transporting ATPase ABCA4 Human genes 0.000 description 1
- 238000005700 Stille cross coupling reaction Methods 0.000 description 1
- 229930006000 Sucrose Natural products 0.000 description 1
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 1
- 239000012317 TBTU Substances 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 1
- 229920001615 Tragacanth Polymers 0.000 description 1
- XSTXAVWGXDQKEL-UHFFFAOYSA-N Trichloroethylene Chemical group ClC=C(Cl)Cl XSTXAVWGXDQKEL-UHFFFAOYSA-N 0.000 description 1
- OKJPEAGHQZHRQV-UHFFFAOYSA-N Triiodomethane Natural products IC(I)I OKJPEAGHQZHRQV-UHFFFAOYSA-N 0.000 description 1
- CLZISMQKJZCZDN-UHFFFAOYSA-N [benzotriazol-1-yloxy(dimethylamino)methylidene]-dimethylazanium Chemical compound C1=CC=C2N(OC(N(C)C)=[N+](C)C)N=NC2=C1 CLZISMQKJZCZDN-UHFFFAOYSA-N 0.000 description 1
- WXIONIWNXBAHRU-UHFFFAOYSA-N [dimethylamino(triazolo[4,5-b]pyridin-3-yloxy)methylidene]-dimethylazanium Chemical compound C1=CN=C2N(OC(N(C)C)=[N+](C)C)N=NC2=C1 WXIONIWNXBAHRU-UHFFFAOYSA-N 0.000 description 1
- 238000009825 accumulation Methods 0.000 description 1
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 description 1
- 230000009471 action Effects 0.000 description 1
- 230000004913 activation Effects 0.000 description 1
- 239000008186 active pharmaceutical agent Substances 0.000 description 1
- 238000005917 acylation reaction Methods 0.000 description 1
- 229910001573 adamantine Inorganic materials 0.000 description 1
- 125000003158 alcohol group Chemical group 0.000 description 1
- 230000001476 alcoholic effect Effects 0.000 description 1
- 229910052783 alkali metal Inorganic materials 0.000 description 1
- 229910000288 alkali metal carbonate Inorganic materials 0.000 description 1
- 150000008041 alkali metal carbonates Chemical class 0.000 description 1
- 150000008044 alkali metal hydroxides Chemical class 0.000 description 1
- 150000001340 alkali metals Chemical class 0.000 description 1
- 229910001860 alkaline earth metal hydroxide Inorganic materials 0.000 description 1
- 125000000217 alkyl group Chemical group 0.000 description 1
- 125000005278 alkyl sulfonyloxy group Chemical group 0.000 description 1
- 150000001408 amides Chemical class 0.000 description 1
- 239000003708 ampul Substances 0.000 description 1
- 239000007900 aqueous suspension Substances 0.000 description 1
- 150000004945 aromatic hydrocarbons Chemical class 0.000 description 1
- 125000003118 aryl group Chemical group 0.000 description 1
- 125000005279 aryl sulfonyloxy group Chemical group 0.000 description 1
- 238000006254 arylation reaction Methods 0.000 description 1
- 238000011914 asymmetric synthesis Methods 0.000 description 1
- RQPZNWPYLFFXCP-UHFFFAOYSA-L barium dihydroxide Chemical compound [OH-].[OH-].[Ba+2] RQPZNWPYLFFXCP-UHFFFAOYSA-L 0.000 description 1
- 229910001863 barium hydroxide Inorganic materials 0.000 description 1
- 229960000686 benzalkonium chloride Drugs 0.000 description 1
- 235000010233 benzoic acid Nutrition 0.000 description 1
- CADWTSSKOVRVJC-UHFFFAOYSA-N benzyl(dimethyl)azanium;chloride Chemical compound [Cl-].C[NH+](C)CC1=CC=CC=C1 CADWTSSKOVRVJC-UHFFFAOYSA-N 0.000 description 1
- 230000004071 biological effect Effects 0.000 description 1
- 230000037396 body weight Effects 0.000 description 1
- JAMFGQBENKSWOF-UHFFFAOYSA-N bromo(methoxy)methane Chemical compound COCBr JAMFGQBENKSWOF-UHFFFAOYSA-N 0.000 description 1
- RDHPKYGYEGBMSE-UHFFFAOYSA-N bromoethane Chemical compound CCBr RDHPKYGYEGBMSE-UHFFFAOYSA-N 0.000 description 1
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 description 1
- 229910000024 caesium carbonate Inorganic materials 0.000 description 1
- AXCZMVOFGPJBDE-UHFFFAOYSA-L calcium dihydroxide Chemical compound [OH-].[OH-].[Ca+2] AXCZMVOFGPJBDE-UHFFFAOYSA-L 0.000 description 1
- 239000000920 calcium hydroxide Substances 0.000 description 1
- 229910001861 calcium hydroxide Inorganic materials 0.000 description 1
- 229910002091 carbon monoxide Inorganic materials 0.000 description 1
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 1
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 1
- 150000001735 carboxylic acids Chemical class 0.000 description 1
- 239000003054 catalyst Substances 0.000 description 1
- 239000000919 ceramic Substances 0.000 description 1
- 239000003610 charcoal Substances 0.000 description 1
- PBAYDYUZOSNJGU-UHFFFAOYSA-N chelidonic acid Natural products OC(=O)C1=CC(=O)C=C(C(O)=O)O1 PBAYDYUZOSNJGU-UHFFFAOYSA-N 0.000 description 1
- 125000003636 chemical group Chemical group 0.000 description 1
- 239000003795 chemical substances by application Substances 0.000 description 1
- 150000008280 chlorinated hydrocarbons Chemical class 0.000 description 1
- 238000004587 chromatography analysis Methods 0.000 description 1
- 239000011248 coating agent Substances 0.000 description 1
- 238000000576 coating method Methods 0.000 description 1
- 229940110456 cocoa butter Drugs 0.000 description 1
- 235000019868 cocoa butter Nutrition 0.000 description 1
- 229910052802 copper Inorganic materials 0.000 description 1
- 230000001351 cycling effect Effects 0.000 description 1
- HPXRVTGHNJAIIH-UHFFFAOYSA-N cyclohexanol Chemical compound OC1CCCCC1 HPXRVTGHNJAIIH-UHFFFAOYSA-N 0.000 description 1
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- XCIXKGXIYUWCLL-UHFFFAOYSA-N cyclopentanol Chemical compound OC1CCCC1 XCIXKGXIYUWCLL-UHFFFAOYSA-N 0.000 description 1
- 230000001419 dependent effect Effects 0.000 description 1
- 239000011903 deuterated solvents Substances 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- 238000010586 diagram Methods 0.000 description 1
- ZBCBWPMODOFKDW-UHFFFAOYSA-N diethanolamine Chemical compound OCCNCCO ZBCBWPMODOFKDW-UHFFFAOYSA-N 0.000 description 1
- YSAVZVORKRDODB-WDSKDSINSA-N diethyl tartrate Chemical compound CCOC(=O)[C@@H](O)[C@H](O)C(=O)OCC YSAVZVORKRDODB-WDSKDSINSA-N 0.000 description 1
- 238000000113 differential scanning calorimetry Methods 0.000 description 1
- 230000004069 differentiation Effects 0.000 description 1
- 238000002050 diffraction method Methods 0.000 description 1
- SBZXBUIDTXKZTM-UHFFFAOYSA-N diglyme Chemical compound COCCOCCOC SBZXBUIDTXKZTM-UHFFFAOYSA-N 0.000 description 1
- 238000007865 diluting Methods 0.000 description 1
- ALVPFGSHPUPROW-UHFFFAOYSA-N dipropyl disulfide Chemical compound CCCSSCCC ALVPFGSHPUPROW-UHFFFAOYSA-N 0.000 description 1
- BNIILDVGGAEEIG-UHFFFAOYSA-L disodium hydrogen phosphate Chemical compound [Na+].[Na+].OP([O-])([O-])=O BNIILDVGGAEEIG-UHFFFAOYSA-L 0.000 description 1
- SDIXRDNYIMOKSG-UHFFFAOYSA-L disodium methyl arsenate Chemical compound [Na+].[Na+].C[As]([O-])([O-])=O SDIXRDNYIMOKSG-UHFFFAOYSA-L 0.000 description 1
- 229940088679 drug related substance Drugs 0.000 description 1
- 238000001035 drying Methods 0.000 description 1
- 239000000975 dye Substances 0.000 description 1
- 238000005516 engineering process Methods 0.000 description 1
- 230000002255 enzymatic effect Effects 0.000 description 1
- 238000011067 equilibration Methods 0.000 description 1
- LYCAIKOWRPUZTN-UHFFFAOYSA-N ethylene glycol Natural products OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 1
- 239000000284 extract Substances 0.000 description 1
- 239000003889 eye drop Substances 0.000 description 1
- 229940012356 eye drops Drugs 0.000 description 1
- 239000012065 filter cake Substances 0.000 description 1
- 235000019253 formic acid Nutrition 0.000 description 1
- 238000009472 formulation Methods 0.000 description 1
- 239000007789 gas Substances 0.000 description 1
- 239000007903 gelatin capsule Substances 0.000 description 1
- 238000007429 general method Methods 0.000 description 1
- 125000001072 heteroaryl group Chemical group 0.000 description 1
- 125000000623 heterocyclic group Chemical group 0.000 description 1
- 239000008240 homogeneous mixture Substances 0.000 description 1
- 150000004677 hydrates Chemical class 0.000 description 1
- 229930195733 hydrocarbon Natural products 0.000 description 1
- 150000002430 hydrocarbons Chemical class 0.000 description 1
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 1
- JUINSXZKUKVTMD-UHFFFAOYSA-N hydrogen azide Chemical compound N=[N+]=[N-] JUINSXZKUKVTMD-UHFFFAOYSA-N 0.000 description 1
- DOUHZFSGSXMPIE-UHFFFAOYSA-N hydroxidooxidosulfur(.) Chemical compound [O]SO DOUHZFSGSXMPIE-UHFFFAOYSA-N 0.000 description 1
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 1
- WGCNASOHLSPBMP-UHFFFAOYSA-N hydroxyacetaldehyde Natural products OCC=O WGCNASOHLSPBMP-UHFFFAOYSA-N 0.000 description 1
- NPZTUJOABDZTLV-UHFFFAOYSA-N hydroxybenzotriazole Substances O=C1C=CC=C2NNN=C12 NPZTUJOABDZTLV-UHFFFAOYSA-N 0.000 description 1
- JBFYUZGYRGXSFL-UHFFFAOYSA-N imidazolide Chemical compound C1=C[N-]C=N1 JBFYUZGYRGXSFL-UHFFFAOYSA-N 0.000 description 1
- 150000002466 imines Chemical class 0.000 description 1
- 239000012535 impurity Substances 0.000 description 1
- 230000005764 inhibitory process Effects 0.000 description 1
- INQOMBQAUSQDDS-UHFFFAOYSA-N iodomethane Chemical compound IC INQOMBQAUSQDDS-UHFFFAOYSA-N 0.000 description 1
- 125000003253 isopropoxy group Chemical group [H]C([H])([H])C([H])(O*)C([H])([H])[H] 0.000 description 1
- 229940011051 isopropyl acetate Drugs 0.000 description 1
- FMKOJHQHASLBPH-UHFFFAOYSA-N isopropyl iodide Chemical compound CC(C)I FMKOJHQHASLBPH-UHFFFAOYSA-N 0.000 description 1
- GWYFCOCPABKNJV-UHFFFAOYSA-N isovaleric acid Chemical compound CC(C)CC(O)=O GWYFCOCPABKNJV-UHFFFAOYSA-N 0.000 description 1
- 239000008101 lactose Substances 0.000 description 1
- 239000007788 liquid Substances 0.000 description 1
- 238000004811 liquid chromatography Methods 0.000 description 1
- DLEDOFVPSDKWEF-UHFFFAOYSA-N lithium butane Chemical compound [Li+].CCC[CH2-] DLEDOFVPSDKWEF-UHFFFAOYSA-N 0.000 description 1
- 229910052749 magnesium Inorganic materials 0.000 description 1
- 239000011777 magnesium Substances 0.000 description 1
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 1
- 235000019341 magnesium sulphate Nutrition 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 239000011976 maleic acid Substances 0.000 description 1
- 239000011159 matrix material Substances 0.000 description 1
- 230000007246 mechanism Effects 0.000 description 1
- 229910044991 metal oxide Inorganic materials 0.000 description 1
- 150000004706 metal oxides Chemical class 0.000 description 1
- 229940098779 methanesulfonic acid Drugs 0.000 description 1
- 239000011259 mixed solution Substances 0.000 description 1
- MZRVEZGGRBJDDB-UHFFFAOYSA-N n-Butyllithium Substances [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 description 1
- KPSSIOMAKSHJJG-UHFFFAOYSA-N neopentyl alcohol Chemical compound CC(C)(C)CO KPSSIOMAKSHJJG-UHFFFAOYSA-N 0.000 description 1
- 150000002825 nitriles Chemical class 0.000 description 1
- 150000002828 nitro derivatives Chemical class 0.000 description 1
- 239000012299 nitrogen atmosphere Substances 0.000 description 1
- LYGJENNIWJXYER-UHFFFAOYSA-N nitromethane Chemical compound C[N+]([O-])=O LYGJENNIWJXYER-UHFFFAOYSA-N 0.000 description 1
- 238000010534 nucleophilic substitution reaction Methods 0.000 description 1
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 1
- 239000002674 ointment Substances 0.000 description 1
- 230000003287 optical effect Effects 0.000 description 1
- 238000005457 optimization Methods 0.000 description 1
- 150000007530 organic bases Chemical class 0.000 description 1
- 150000002894 organic compounds Chemical class 0.000 description 1
- 230000008520 organization Effects 0.000 description 1
- TWNQGVIAIRXVLR-UHFFFAOYSA-N oxo(oxoalumanyloxy)alumane Chemical compound O=[Al]O[Al]=O TWNQGVIAIRXVLR-UHFFFAOYSA-N 0.000 description 1
- UQPUONNXJVWHRM-UHFFFAOYSA-N palladium;triphenylphosphane Chemical compound [Pd].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 UQPUONNXJVWHRM-UHFFFAOYSA-N 0.000 description 1
- 230000036961 partial effect Effects 0.000 description 1
- WEYVCQFUGFRXOM-UHFFFAOYSA-N perazine Chemical compound C1CN(C)CCN1CCCN1C2=CC=CC=C2SC2=CC=CC=C21 WEYVCQFUGFRXOM-UHFFFAOYSA-N 0.000 description 1
- 229960002195 perazine Drugs 0.000 description 1
- 235000019271 petrolatum Nutrition 0.000 description 1
- 239000002798 polar solvent Substances 0.000 description 1
- 229910000027 potassium carbonate Inorganic materials 0.000 description 1
- RPDAUEIUDPHABB-UHFFFAOYSA-N potassium ethoxide Chemical compound [K+].CC[O-] RPDAUEIUDPHABB-UHFFFAOYSA-N 0.000 description 1
- LPNYRYFBWFDTMA-UHFFFAOYSA-N potassium tert-butoxide Chemical compound [K+].CC(C)(C)[O-] LPNYRYFBWFDTMA-UHFFFAOYSA-N 0.000 description 1
- 230000003389 potentiating effect Effects 0.000 description 1
- 239000000843 powder Substances 0.000 description 1
- 230000000069 prophylactic effect Effects 0.000 description 1
- 102000004169 proteins and genes Human genes 0.000 description 1
- 108090000623 proteins and genes Proteins 0.000 description 1
- 238000010926 purge Methods 0.000 description 1
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 1
- 150000003254 radicals Chemical class 0.000 description 1
- 239000000376 reactant Substances 0.000 description 1
- 230000035484 reaction time Effects 0.000 description 1
- 230000009257 reactivity Effects 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 238000006722 reduction reaction Methods 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 239000013557 residual solvent Substances 0.000 description 1
- 238000012552 review Methods 0.000 description 1
- 150000003303 ruthenium Chemical class 0.000 description 1
- 229920006395 saturated elastomer Polymers 0.000 description 1
- 238000010956 selective crystallization Methods 0.000 description 1
- 239000000377 silicon dioxide Substances 0.000 description 1
- 235000017281 sodium acetate Nutrition 0.000 description 1
- 229940087562 sodium acetate trihydrate Drugs 0.000 description 1
- RCOSUMRTSQULBK-UHFFFAOYSA-N sodium;propan-1-olate Chemical compound [Na+].CCC[O-] RCOSUMRTSQULBK-UHFFFAOYSA-N 0.000 description 1
- 239000008247 solid mixture Substances 0.000 description 1
- 239000012265 solid product Substances 0.000 description 1
- 239000008347 soybean phospholipid Substances 0.000 description 1
- 238000010561 standard procedure Methods 0.000 description 1
- 230000000707 stereoselective effect Effects 0.000 description 1
- 239000012258 stirred mixture Substances 0.000 description 1
- 238000003860 storage Methods 0.000 description 1
- 125000001424 substituent group Chemical group 0.000 description 1
- 239000005720 sucrose Substances 0.000 description 1
- 229940124530 sulfonamide Drugs 0.000 description 1
- 229910052717 sulfur Inorganic materials 0.000 description 1
- 125000004434 sulfur atom Chemical group 0.000 description 1
- 235000012222 talc Nutrition 0.000 description 1
- 239000011975 tartaric acid Substances 0.000 description 1
- 235000002906 tartaric acid Nutrition 0.000 description 1
- XDOOTDWGMYKOFH-UHFFFAOYSA-N tert-butyl 4-[1-(1,3-benzothiazol-5-yl)ethyl]piperazine-1-carboxylate Chemical compound S1C=NC2=C1C=CC(=C2)C(C)N1CCN(CC1)C(=O)OC(C)(C)C XDOOTDWGMYKOFH-UHFFFAOYSA-N 0.000 description 1
- 125000005931 tert-butyloxycarbonyl group Chemical group [H]C([H])([H])C(OC(*)=O)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 150000003512 tertiary amines Chemical class 0.000 description 1
- 238000010998 test method Methods 0.000 description 1
- 238000012360 testing method Methods 0.000 description 1
- FPGGTKZVZWFYPV-UHFFFAOYSA-M tetrabutylammonium fluoride Chemical compound [F-].CCCC[N+](CCCC)(CCCC)CCCC FPGGTKZVZWFYPV-UHFFFAOYSA-M 0.000 description 1
- 238000002560 therapeutic procedure Methods 0.000 description 1
- 239000000196 tragacanth Substances 0.000 description 1
- 235000010487 tragacanth Nutrition 0.000 description 1
- 229940116362 tragacanth Drugs 0.000 description 1
- 229910052723 transition metal Inorganic materials 0.000 description 1
- 150000003624 transition metals Chemical class 0.000 description 1
- UBOXGVDOUJQMTN-UHFFFAOYSA-N trichloroethylene Natural products ClCC(Cl)Cl UBOXGVDOUJQMTN-UHFFFAOYSA-N 0.000 description 1
- WTVXIBRMWGUIMI-UHFFFAOYSA-N trifluoro($l^{1}-oxidanylsulfonyl)methane Chemical group [O]S(=O)(=O)C(F)(F)F WTVXIBRMWGUIMI-UHFFFAOYSA-N 0.000 description 1
- 229910052721 tungsten Inorganic materials 0.000 description 1
- 238000000825 ultraviolet detection Methods 0.000 description 1
- 229910052720 vanadium Inorganic materials 0.000 description 1
- 239000008096 xylene Substances 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/506—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D277/00—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings
- C07D277/60—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings condensed with carbocyclic rings or ring systems
- C07D277/62—Benzothiazoles
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D277/00—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings
- C07D277/60—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings condensed with carbocyclic rings or ring systems
- C07D277/62—Benzothiazoles
- C07D277/64—Benzothiazoles with only hydrocarbon or substituted hydrocarbon radicals attached in position 2
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Diabetes (AREA)
- Biomedical Technology (AREA)
- Neurosurgery (AREA)
- Neurology (AREA)
- Hospice & Palliative Care (AREA)
- Psychiatry (AREA)
- Endocrinology (AREA)
- Hematology (AREA)
- Obesity (AREA)
- Urology & Nephrology (AREA)
- Heart & Thoracic Surgery (AREA)
- Emergency Medicine (AREA)
- Vascular Medicine (AREA)
- Cardiology (AREA)
- Epidemiology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Plural Heterocyclic Compounds (AREA)
- Preparation Of Compounds By Using Micro-Organisms (AREA)
Abstract
La invención se refiere a sales de adición de ácido succínico o sales de adición de ácido fumárico de derivados de piperazina de fórmula (I), así como a formas sólidas, tales como formas polimórficas, de los mismos, que son útiles como ingredientes farmacéuticos y en particular como inhibidores de glicosidasas. (Traducción automática con Google Translate, sin valor legal)
Description
DESCRIPCIÓN
Sales de adición de ácido de succinato y fumarato de derivados de piperazina útiles como inhibidores de glucosidasa Campo de la invención
La invención se refiere a sales de adición de ácido de ácido succínico o ácido fumárico con derivados de piperazina, así como formas sólidas, tales como formas polimórficas, de los mismos, que son útiles como ingredientes farmacéuticos y en particular como inhibidores de glucosidasa.
Antecedentes de la invención
Los derivados de piperazina de fórmula I

en donde X, Y y T se definen adicionalmente más adelante, son útiles como ingredientes farmacéuticos y muestran una alta actividad como inhibidores de glucosidasa. Los compuestos similares se desvelan, por ejemplo, en el documento WO2016/030443.
Aunque los compuestos de fórmula I tienen actividades farmacéuticas muy útiles como bases libres, no son ideales para la fabricación farmacéutica y, como tales, pueden no ser adecuados para ciertas formas de dosificación, especialmente formas de dosificación oral, debido a su comportamiento de disolución y estabilidad o reactividad desfavorables y otras propiedades en estado sólido.
Por lo tanto, existe la necesidad de proporcionar formas sólidas mejoradas que comprendan los compuestos de fórmula I, que exhiban propiedades mejoradas, se puedan fabricar fácilmente en formas de dosificación sólidas u otras formas de dosificación farmacéuticas, y muestren un comportamiento de disolución y estabilidad mejorados y/o sean menos reactivos en estado sólido.
Sumario de la invención
La materia objeto de la invención es como se expone en las reivindicaciones adjuntas.
Un aspecto de la invención se refiere a una sal de ácido mono-succínico de uno de los compuestos Ia, Ib, Ic y Id en una forma sólida que tiene el patrón de difracción de polvo de rayos X como se muestra en la Figura 1:


Un aspecto adicional de la invención se refiere a una sal de ácido mono-fumárico de uno de los compuestos la, Ib, Ic y Id en una forma sólida que tiene el patrón de difracción de polvo de rayos X como se muestra en la Figura 4:

Un aspecto adicional de la invención se refiere a un método para la preparación de una sal de ácido mono-succínico de la invención o una sal de ácido mono-fumárico de la invención que comprende las siguientes etapas:
a) suspender o disolver el compuesto seleccionado de fórmula I y el ácido respectivo en un disolvente o una mezcla disolvente adecuados;
b) calentar la mezcla obtenida en la etapa a) a una temperatura de entre aproximadamente 30 °C a aproximadamente el punto de ebullición del disolvente o la mezcla disolvente seleccionados y permitir que la mezcla se enfríe a temperatura ambiente;
c) opcionalmente repetir la etapa b) varias veces;
d) separar y secar el sólido obtenido de esta manera.
Un aspecto adicional de la invención se refiere a una forma de dosificación oral sólida que comprende una sal de ácido mono-succínico de la invención o una sal de ácido mono-fumárico de la invención.
Un aspecto adicional de la invención se refiere a una sal de ácido mono-succínico de acuerdo con la invención o una sal de ácido mono-fumárico de acuerdo con la invención para su uso como un medicamento.
Un aspecto adicional de la invención se refiere a una sal de ácido mono-succínico de acuerdo con la invención o una sal de ácido mono-fumárico de acuerdo con la invención para su uso en un método para el tratamiento de una afección seleccionada del grupo que consiste en enfermedades neurodegenerativas, diabetes, cáncer, enfermedades cardiovasculares y accidente cerebrovascular.
Las realizaciones preferidas se exponen en las reivindicaciones dependientes.
Se ha descubierto ahora que los compuestos de fórmula I muestran propiedades mejoradas en estado sólido, después de haber sido transformados en sales de adición de ácido de ácido succínico o ácido fumárico. En particular, las sales de adición de ácido pueden fabricarse fácilmente en formas de dosificación sólidas u otras formas de dosificación farmacéuticas, y muestran un comportamiento de disolución y una estabilidad mejorados y/o son menos reactivas en estado sólido. Las sales de adición de ácido de la presente invención también exhiben baja higroscopicidad.
También se ha descubierto que ciertas formas polimórficas de las sales de adición de ácido muestran propiedades aún más mejoradas, haciéndolas ideales para la fabricación farmacéutica, en particular para formas sólidas de dosificación oral. Por otra parte, las sales de adición de ácido de la presente invención que tienen una proporción molar de los compuestos de fórmula I al ácido respectivo de 1 a 1 son especialmente estables, solubles y/o muestran otras propiedades mejoradas.
Descripción detallada de la invención
La presente solicitud desvela la sal de adición de ácido de ácido succínico o ácido fumárico con compuestos de fórmula I (no de acuerdo con la invención a menos que esté abarcado por las reivindicaciones)

en donde
Y representa H o CH3,
T representa N o CH,
X representa uno de los siguientes grupos de sulfoximina:
S(O)(NR3')CH3, S(O)(NR3')CH2CH3, S(O)(NR3')CH2CH2OH, S(O)(NR3')CH2CH2OCH3, NS(O)(R3')CH3, NS(O)(R3')CH2CH3, NS(O)(R3')CH2CH2OH o NS(O)(R3')CH2CH2OCH3
y
R3' representa H o un grupo alquilo de cadena lineal o ramificada que tiene 1 a 12 átomos de carbono, en donde 1 a 3 grupos CH2 pueden reemplazarse por un grupo seleccionado de SO2, CO, O y en donde de 1 a 5 átomos de hidrógeno pueden reemplazarse por F, Cl, Br o I.
así como estereoisómeros, formas sólidas, tales como solvatos y formas polimórficas de los mismos.
"Formas sólidas" de acuerdo con la invención es preferentemente una expresión que abarca generalmente cualquier
estado sólido de un compuesto y/o sus sales y/o sus solvatos, preferentemente formas cristalinas, incluyendo formas polimórficas, pero también formas amorfas (véase: Aitipamula, S. et al. Cryst. Growth Des., 2012, 12 (5), pp 2147 2152).
El polimorfismo describe la aparición de diferentes formas sólidas o cristalinas de un solo compuesto y es una propiedad de ciertos compuestos y complejos. Por tanto, los polimorfos o formas polimórficas son sólidos distintos que comparten la misma fórmula molecular, pero cada polimorfo o forma polimórfica puede tener propiedades físicas distintas. Por lo tanto, un solo compuesto puede dar lugar a una diversidad de formas polimórficas donde cada forma tiene propiedades físicas diferentes y distintas, tales como diferentes perfiles de solubilidad, diferentes temperaturas de punto de fusión y/o diferentes picos de difracción de rayos X.
La aparición de una forma polimórfica puede estar determinada por las condiciones de cristalización, tales como la elección del disolvente o disolventes, la velocidad de adición del disolvente, la temperatura, la velocidad de agitación, el nivel de sobresaturación y el nivel de impurezas. Por tanto, diferentes procesos de cristalización pueden dar lugar a diferentes polimorfos. Los polimorfos también tienen diferentes estabilidades y pueden convertirse espontáneamente de una forma a otra.
La imprevisibilidad del polimorfismo, tanto en lo que respecta a la incertidumbre de que se puedan encontrar formas, como la falta de métodos estándar para preparar una nueva forma, se ha discutido, por ejemplo, en A. Goho, "Tricky Business," Science News, Vol. 166(8), 21 de agosto de 2004 y A. M. Rouhi, "The Right Stuff," Chemical and Engineering News, 24 de febrero de 2003, paginas 32-35.
Los polimorfos pueden distinguirse entre sí mediante una diversidad de técnicas analíticas. Los polimorfos exhiben distintas propiedades espectroscópicas y pueden identificarse usando espectroscopía infrarroja, espectroscopía Raman y espectroscopía RMN 13C. Debido al hecho de que cada forma de cristal difracta los rayos X de diferentes formas, la difractometría de rayos X en polvo (XRPD, por sus siglas en inglés) también puede usarse para la identificación y diferenciación de dos formas polimórficas. Adicionalmente, los métodos térmicos tales como la calorimetría diferencial de barrido (DSC, por sus siglas en inglés), el análisis térmico simultáneo (STA, por sus siglas en inglés) y el análisis termogravimétrico (TGA, por sus siglas en inglés) pueden proporcionar información única para un polimorfo particular. Las formas polimórficas de un compuesto también pueden distinguirse por otros métodos tales como espectrometría infrarroja. Para una revisión general de los polimorfos y las aplicaciones farmacéuticas de los polimorfos, véase G. M. Wall, Pharm Manuf. 3, 33 (1986); J. Haleblian y W. McCrone, J. Pharm. Sci., 58, 91 1 (1969); y J. Haleblian, J. Pharm. Sci., 64, 1269 (1975).
Las propiedades fisicoquímicas pueden variar mucho entre formas polimórficas individuales. Por ejemplo, la solubilidad y la velocidad de disolución pueden variar entre polimorfos, lo que conduce a posibles diferencias en la biodisponibilidad. Adicionalmente, las propiedades mecánicas tales como la fluidez y la compactibilidad, que afectan a las propiedades de procesamiento de un compuesto, pueden ser diferentes. La estabilidad, la impermeabilidad al vapor y la vida útil de un compuesto o formas de dosificación del mismo también pueden depender del polimorfo elegido.
En vista de las posibles diferencias entre las formas polimórficas de los mismos ingredientes farmacéuticos activos, existen amplios requisitos establecidos por las autoridades reguladoras de aprobación de fármacos para controlar el polimorfismo. En particular, generalmente se requiere que solo se preestablezca la misma forma polimórfica única reproducible en un medicamento dado o que se obtengan mezclas de formas polimórficas de manera consistente y reproducible, de tal manera que el medicamento permanezca siempre idéntico en todos los aspectos (ICH Topic Q 6 A, Specifications: Test Procedures and Acceptance Criteria for New Drug Substances and New Drug Products: Chemical Substances. Mayo de 2000 CPMP/ICH/367/96)
Las mezclas de formas polimórficas de un fármaco dado a menudo no son adecuadas para el desarrollo farmacéutico, ya que pueden estar compuestas o contener formas polimórficas que son inestables e influyen en la consistencia del producto de fármaco.
Por lo tanto, la industria farmacéutica invierte importantes recursos para descubrir una forma polimórfica estable única que sea adecuada para el desarrollo farmacéutico y el proceso reproducible para la fabricación específica de ese polimorfo estable único.
Las formas polimórficas, incluyendo los solvatos de la presente invención, proporcionan materiales que tienen propiedades de procesamiento deseables, tales como facilidad de manejo, facilidad de procesamiento, estabilidad de almacenamiento y facilidad de purificación, o como formas cristalinas intermedias deseables que faciliten la conversión a otras formas polimórficas con propiedades mejoradas. Además, la invención proporciona formas estables de principios activos, que preferentemente exhiben estabilidad termodinámica, solubilidad mejorada, inicio de acción rápido y biodisponibilidad mejorada. Las sales de adición de ácido preferidas de la presente invención se mejoran en al menos una de las propiedades mencionadas anteriormente.
La presente memoria descriptiva desvela además las sales de adición de ácido de los compuestos de fórmula I1, I2 y
I3 (no de acuerdo con la invención a menos que esté abarcado por las reivindicaciones):

en donde X e Y tienen el significado dado anteriormente y formas sólidas, tales como solvatos y formas polimórficas, de los mismos.
La presente invención desvela además las sales de adición de ácido de los compuestos de fórmula I1a y I1b, I2a y I2b y I3a y I3b (no de acuerdo con la invención a menos que esté abarcado por las reivindicaciones)


en donde X e Y tienen el significado dado anteriormente y formas sólidas, tales como solvatos y formas polimórficas, de los mismos.
La presente invención desvela además compuestos de fórmula I, en donde Y es H y T es N (no de acuerdo con la invención a menos que esté abarcado por las reivindicaciones). Incluso más preferidos son los compuestos de fórmula I1a1, I2a1 y I3a1 a continuación (no de acuerdo con la invención a menos que esté abarcado por las reivindicaciones):


en donde X tiene el significado dado anteriormente.
La presente memoria descriptiva desvela además sales de adición de ácido de compuestos de fórmula I, en donde X se selecciona del grupo

en donde R3' tiene el significado dado anteriormente (no de acuerdo con la invención a menos que esté abarcado por las reivindicaciones).
La presente memoria descriptiva desvela además sales de adición de ácido de compuestos de fórmula I, en donde R3' se selecciona entre H, CH3, CH2CH3, CH2CH2OH, CH2CH2OCH3 así como formas sólidas, tales como formas polimórficas, de los mismos (no de acuerdo con la invención a menos que esté abarcado por las reivindicaciones).
La presente memoria descriptiva desvela además sales de adición de ácido de ácido succínico o ácido fumárico, así como formas sólidas, tales como formas polimórficas de los mismos, de un compuesto de fórmula la, lb, lc, Id, le, If, Ig, Ih, li, Ij, Ik, IL (no de acuerdo con la invención a menos que esté abarcado por las reivindicaciones):


El compuesto la y sus formas sólidas son los más preferidos (no de acuerdo con la invención a menos que esté abarcado por las reivindicaciones).
Los compuestos de fórmula I se emplean preferentemente como un diastereómero y/o enantiómero único en un exceso enantiomérico, medido por métodos bien conocidos por un experto en la materia, del 10 % o más, preferentemente el 50 % o más, y más preferentemente más del 95 %, 96 %, 98 % o 99 %.
La nomenclatura como se usa en el presente documento para definir compuestos, especialmente los compuestos de acuerdo con la invención, se basa en general en las reglas de la organización lUPAC para compuestos químicos y especialmente compuestos orgánicos. Los compuestos de la invención han sido nombrados de acuerdo con los estándares usados en el programa AutoNom 2000 o ACD Lab Version 12.01 o Instant JChem Version: 15.12.7.0.
Las sales de adición de ácido succínico y ácido fumárico de la presente invención pueden obtenerse en diferentes relaciones molares. Se ha descubierto que las sales de adición de mono ácido de la presente invención, es decir, las sales de adición de ácido succínico o ácido fumárico que tienen una relación molar de los compuestos de fórmula I al ácido respectivo de 1 a 1, son especialmente preferidas y estables, solubles y/o muestran otras propiedades mejoradas.
Un método preferido de preparación de las sales de adición de ácido de los compuestos de fórmula I de acuerdo con la invención comprende las siguientes etapas (no de acuerdo con la invención a menos que esté abarcado por las reivindicaciones):
a) suspender o disolver el compuesto de fórmula I y el ácido succínico o ácido fumárico en un disolvente o mezcla de disolventes adecuados;
b) calentar la mezcla obtenida en la etapa a) a una temperatura entre aproximadamente 30 °C a aproximadamente el punto de ebullición del disolvente seleccionado, preferentemente entre aproximadamente 50 °C y aproximadamente 100 °C y lo más preferentemente a aproximadamente 60 °C, a aproximadamente 70 °C o a aproximadamente 80 °C y dejar que la mezcla se enfríe a temperatura ambiente;
c) opcionalmente repetir la etapa b) varias veces;
d) separar y secar el sólido obtenido de esta manera.
Los disolventes adecuados para el método de preparación de las sales de adición de ácido de la presente invención son preferentemente agua o alcoholes tales como metanol (MeOH), etanol (EtOH), 1-propanol, 2-propanol (IPA), 1-butanol, 2-butanol, 2-metil-1-propanol, 2-metil-2-propanol, 1-pentanol, 3-metil-1-butanol, 2,2-dimetil-1-propanol, ciclopentanol, 1-hexanol, ciclohexanol, 1 -heptanol, 1-octanol, 1-nonanol, 1-decanol, 2-propen-1-ol, cetonas, tales como acetona, ésteres, tales como acetato de etilo, acetonitrilo, éteres tales como tetrahidrofurano (THF),
hidrocarburos aromáticos, tales como tolueno y mezclas homogéneas de los disolventes anteriores, tales como MeOH/agua, por ejemplo, como mezcla 50/50 (v/v) o IPNagua, por ejemplo, como mezcla 90/10 (v/v) o MeCN/agua, por ejemplo, como mezcla 95/5 (v/v).
Las formulaciones farmacéuticas pueden administrarse en forma de unidades de dosificación, que comprenden una cantidad predeterminada de principio activo por unidad de dosificación. La concentración del ingrediente profiláctico o terapéuticamente activo en la formulación puede variar de aproximadamente el 0,1 al 100 % en peso. Preferentemente, las sales de adición de ácido de los compuestos de fórmula I o los farmacéuticamente se administran en dosis de aproximadamente 0,5 a 1000 mg, más preferentemente entre 1 y 700 mg, lo más preferentemente 5 y 100 mg por unidad de dosis, calculadas sobre la base respectiva. Generalmente, tal intervalo de dosis es apropiado para la incorporación diaria total. En otros términos, la dosis diaria está preferentemente entre aproximadamente 0,02 y 100 mg/kg de peso corporal.
Preferentemente, la dosis diaria, es decir, la suma de todas las dosis administradas a un paciente durante un día determinado, de un compuesto de fórmula I y preferentemente la está entre aproximadamente 20 y aproximadamente 300 mg, más preferentemente entre aproximadamente 100 mg y 300 mg calculados sobre la base respectiva, tal como aproximadamente 200, 225 o 270 administrados una vez al día, o 50, 70 o 100 mg administrados dos veces al día, preferentemente administrados por vía oral. Las sales de adición de ácido de la presente invención se administran preferentemente por vía oral.
La presente memoria descriptiva desvela las siguientes realizaciones relacionadas con el uso de sales de adición de ácido de la invención (no de acuerdo con la invención a menos que esté abarcado por las reivindicaciones):
1. Sales de adición de ácido de acuerdo con la invención para su uso en el tratamiento de una afección seleccionada de enfermedades neurodegenerativas, diabetes, cáncer, enfermedades cardiovasculares y accidente cerebrovascular.
2. Sales de adición de ácido de acuerdo con la realización 1 para su uso en el tratamiento de una afección, en donde la afección se selecciona del grupo de una o más tauopatías y enfermedad de Alzheimer, Demencia, Esclerosis lateral amiotrófica (ELA), Esclerosis lateral amiotrófica con deterioro cognitivo (ALSci, por sus siglas en inglés), Enfermedad del grano argirofílico, Variante conductual de la demencia frontotemporal (BvFTD, por sus siglas en inglés), Enfermedad de Bluit, Encefalopatía traumática crónica, Degeneración corticobasal (CBP, por sus siglas en inglés), Demencia pugilística, Ovillos neurofibrilares difusos con calcificación, síndrome de Down, Demencia británica familiar, Demencia danesa familiar, Demencia frontotemporal con parkinsonismo ligado al cromosoma 17 (FTDP-17), Degeneración lobar frontotemporal (FTLD, por sus siglas en inglés), Ganglioglioma, Gangliocitoma, Enfermedad de Gerstmann-Straussler-Scheinker, Tauopatía de la glía globular, Parkinsonismo de Guadalupe, Enfermedad de Hallervorden-Spatz (neurodegeneración con acumulación de hierro en el cerebro tipo 1), Encefalopatía por plomo, Lipofuscinosis, Meningioangiomatosis, Atrofia multisistémica, Distrofia miotónica, Enfermedad de Niemann-Pick (tipo C), Degeneración pálido-ponto-nigral, Complejo parkinsonismo-demencia de Guam, Enfermedad de Pick (PiD, por sus siglas en inglés), Demencia por enfermedad de Parkinson, Parkinsonismo postencefalítico (PEP, por sus siglas en inglés), afasia progresiva primaria, enfermedades priónicas (incluyendo la enfermedad de Creutzfeldt-Jakob (CJD, por sus siglas en inglés), Afasia progresiva no fluente, Variante de la enfermedad de Creutzfeldt-Jakob (vCJD), insomnio familiar fatal, Kuru, Gliosis supercortical progresiva, Parálisis supranuclear progresiva (PSP), Demencia semántica, Síndrome de Steele-Richardson-Olszewski, Panencefalitis esclerosante subaguda, Demencia por enredos, Esclerosis tuberosa, Enfermedad de Huntington y Enfermedad de Parkinson, preferentemente una o más tauopatías y enfermedad de Alzheimer. Las referencias a los métodos de tratamiento en los posteriores párrafos de esta descripción han de interpretarse como referencias a los compuestos, las composiciones farmacéuticas o los medicamentos de la presente invención para su uso en un método de tratamiento del cuerpo humano (o animal) mediante terapia.
3. Un método para tratar una tauopatía, en donde se administra una sal de adición de ácido de acuerdo con la invención a un mamífero que necesita dicho tratamiento.
4. Un método para inhibir una glucosidasa, en donde un sistema que expresa la glucosidasa se pone en contacto con una sal de adición de ácido de ácido clorhídrico, ácido maleico o ácido tartárico con sal de adición de ácido de acuerdo con la invención en condiciones in vitro de tal manera que se inhibe la glucosidasa.
Preparación de los compuestos de la fórmula I
Las formas preferidas de las sales de adición de ácido de la presente invención demuestran propiedades adecuadas para su uso como un fármaco. En particular, tales compuestos preferidos muestran una alta estabilidad en estado sólido, alta estabilidad en presencia de microsoma hepático, alta estabilidad a la oxidación y permeabilidad adecuada. Los compuestos preferidos adicionales de la presente invención demuestran su idoneidad como fármacos por actividad biológica potente, tal como el nivel de O-GlcNAcilación de proteínas totales medidas en extractos de cerebro. Los expertos en la materia conocen las pruebas relevantes para determinar tales parámetros, por ejemplo, estabilidad en estado sólido (Waterman K.C. (2007) Pharm Res 24(4); 780-790), estabilidad en presencia de microsoma hepático
(Obach R. S. (1999) Drug Metab Dispos 27(11); 1350-135) y la permeabilidad (por ejemplo, Caco-2 permeability assay, Calcagno A. M. (2006) Mol Pharm 3(1); 87-93). Los compuestos de la presente invención que muestran una alta potencia en los ensayos de inhibición de OGA y una o más de las propiedades anteriores son especialmente adecuados como fármaco para las indicaciones mencionadas en la presente memoria descriptiva.
Los compuestos de acuerdo con la fórmula (I) y los materiales de partida para su preparación, respectivamente, se producen mediante métodos conocidos en sí mismos, como se describe en la bibliografía, es decir, en condiciones de reacción que son conocidas y adecuadas para dichas reacciones.
También pueden usarse variantes que son conocidas en sí mismas, pero que no se mencionan con mayor detalle en el presente documento. Si se desea, los materiales de partida también pueden formarse in situ dejándolos en el estado no aislado en la mezcla de reacción bruta, pero convirtiéndolos inmediatamente en el compuesto de acuerdo con la invención. Por otro lado, es posible llevar a cabo la reacción por etapas.
Las siguientes abreviaturas se refieren respectivamente a las siguientes definiciones:
Ac (acetilo), ac (acuoso), h (hora), g (gramo), l (litro), mg (miligramo), MHz (Megahercio, μM (micromolar), min (minuto), mm (milímetro), mmol (milimol), mM (milimolar), p.f. (punto de fusión), equiv (equivalente), ml (mililitro), μl (microlitro), ACN (acetonitrilo), AcOh (ácido acético), BInA p (2,2'-bis(difenilfosfino)-1,1'- binaftaleno, Bo C (terc-butoxicarbonilo), CBZ (carbobenzoxi), CDCb (cloroformo deuterado), CD3OD (metanol deuterado), CH3CN (acetonitrilo), c-hex (ciclohexano), DCC (diciclohexil carbodiimida), DCM (diclorometano), dppf (1,1-bis(difenilfosfino)ferroceno), DIC (diisopropil carbodiimida), DIEA (diisopropiletilamina), DMF (dimetilformamida), DMSO (dimetilsulfóxido), DMSO-d6 (dimetilsulfóxido deuterado), EDC (1-(3-dimetil-amino-propil)-3-etilcarbodiimida), ESI (ionización por electropulverización), EtOAc (acetato de etilo), Et2O (éter dietílico), EtOH (etanol), FMOC (fluorenilmetiloxicarbonilo), HATU (hexafluorofosfato de dimetilamino-([1,2,3]triazolo[4,5-b]piridin-3-iloxi)-metilen]-dimetil-amonio), HPLC (cromatografía líquida de alta resolución), i-PrOH (2-propanol), K2CO3 (carbonato potásico), LC (cromatografía líquida), MD Autoprep (Autoprep dirigida a masas), MeOH (metanol), MgSO4 (sulfato de magnesio), MS (espectrometría de masas), MTBE (metil terc-butil éter), MTR (4-metoxi-2,3,6-trimetilbencenosulfonilo), MW (microondas), NBS (N-bromo succinimida), NaHCO3 (bicarbonato sódico), NaBH4 (borohidruro sódico), NMM (N-metil morfolina), RMN (resonancia magnética nuclear), POA (fenoxiacetato), Py (piridina), PyBOP® (hexafluorofosfato de benzotriazol-1-il-oxi-tris-pirrolidino-fosfonio), TA (temperatura ambiente), Tr. (tiempo de retención), SFC (cromatografía de fluidos supercríticos), SPE (extracción en fase sólida), T3P (anhídrido propilfosfónico), TBAF (fluoruro de tetra-nbutilamonio), TBTU (tetrafluoroborato de 2-(1-H-benzotriazol-1-il)-1,1,3,3-tetrametiluromio), TEA (trietilamina), TFA (ácido trifluoroacético), THF (tetrahidrofurano), TLC (cromatografía de capa fina), UV (ultravioleta).
En general, los compuestos de acuerdo con la Fórmula (I) y las fórmulas relacionadas de esta invención pueden prepararse a partir de materiales de partida fácilmente disponibles. Si tales materiales de partida no están disponibles en el mercado, pueden prepararse mediante técnicas sintéticas convencionales. En general, las rutas de síntesis para cualquier compuesto individual de Fórmula (I) y fórmulas relacionadas dependerán de los sustituyentes específicos de cada molécula, siendo estos factores apreciados por los expertos en la materia. Los siguientes métodos y procedimientos generales descritos en lo sucesivo en el presente documento en los ejemplos pueden emplearse para preparar compuestos de Fórmula (I) y fórmulas relacionadas. Las condiciones de reacción representadas en los siguientes esquemas, tales como temperaturas, disolventes o correactivos, se dan solo como ejemplos y no son restrictivas. Se apreciará que cuando se dan las condiciones experimentales típicas o preferidas (es decir, temperaturas de reacción, tiempo, moles de reactivos, disolventes, etc.), también pueden usarse otras condiciones experimentales a menos que se indique lo contrario. Las condiciones óptimas de reacción pueden variar con los reactivos o disolventes particulares usados, pero tales condiciones pueden determinarse por un experto en la materia, usando procedimientos de optimización rutinarios. Para conocer todos los métodos de protección y desprotección, véase Philip J. Kocienski, en "Protecting Groups", Georg Thieme Verlag Stuttgart, New York, 1994 y, Theodora W. Greene y Peter G. M. Wuts en "Protective Groups in Organic Synthesis", Wiley Interscience, 3a Edición 1999.
Un "grupo saliente" LG representa una porción química que puede eliminarse o reemplazarse por otro grupo químico. En toda la memoria descriptiva, el término grupo saliente representa preferentemente Cl, Br, I o un grupo OH modificado reactivamente, tal como, por ejemplo, un éster activado, una imidazolida o alquilsulfoniloxi que tiene de 1 a 6 átomos de carbono (preferentemente metilsulfoniloxi o trifluorometilsulfoniloxi) o arilsulfoniloxi que tiene de 6 a 10 átomos de carbono (preferentemente fenilo- o p-tolilsulfoniloxi). Cuando un grupo saliente LG se une a un anillo aromático o heteroaromático, el LG puede representar además SO2-alquilo o F. En la bibliografía se describen radicales de este tipo para la activación del grupo carboxilo en reacciones de acilación típicas (por ejemplo, en los trabajos convencionales, tales como Houben-Weyl, Methoden der organischen Chemie [Methods of Organic Chemistry], Georg-Thieme-Verlag, Stuttgart). Los ésteres activados se forman ventajosamente in situ, por ejemplo, mediante la adición de HOBt, N-hidroxisuccinimida o HATU.
El compuesto de fórmula (I) puede separarse su otro enantiómero correspondiente mediante cromatografía quiral o mediante resolución quiral, recristalización con el uso de un ácido ópticamente activo.
Preferentemente, se usan entre aproximadamente 0,5 y aproximadamente 2 equivalentes de ácido quiral para la cristalización selectiva. Los disolventes y mezclas de disolventes que se usan preferentemente para la resolución
quiral con ácidos quirales son H2O, MeCN (acetonitrilo), aproximadamente 2 a aproximadamente 50 % de H2O en EtOH (etanol), EtOH, 2 a 50 % de H2O en MeOH (metanol), MeOH, 2 a 50 % de H2O en IPA (alcohol isopropílico), IPA, 2 a 50 % de MeOH en MEK (metiletilcetona, 2-butanona), MEK, 2 a 50 % de MeOH en iPrOAc (acetato de isopropilo), iPrOAc, dioxano. Todos los porcentajes para las mezclas de disolventes se dan en porcentaje en volumen, si no se indica lo contrario.
Preferentemente, en la preparación se usan métodos conocidos por un experto en la materia. Otros métodos de preparación se describen a continuación en los ejemplos. Dependiendo de la naturaleza de Y, T y X, pueden seleccionarse diferentes estrategias sintéticas para la síntesis de compuestos de Fórmula (I). En el proceso ilustrado en los siguientes esquemas, Y, T y X son como se definieron anteriormente en la descripción a menos que se mencione lo contrario.
Los compuestos de Fórmula (I), en donde Y, T y X se definen como anteriormente, pueden prepararse a partir de compuestos alternativos de Fórmula (I), usando procedimientos de interconversión adecuados tales como los descritos a continuación en los ejemplos, o procedimientos de interconversión convencionales bien conocidos por un experto en la materia.
El compuesto de fórmula (I) puede separarse en compuestos de fórmula (IA) y (IB) por cromatografía quiral o por resolución quiral, recristalización con el uso de un ácido ópticamente activo, usando métodos conocidos por un experto en la materia y como se describe a continuación en los ejemplos (Esquema 1).
Esquema 1

Los compuestos de fórmula (I), en donde Y, T y X se definen como anteriormente, pueden prepararse mediante la adición de una amina de fórmula (II) a un heterociclo de fórmula (III), donde LG es un grupo saliente como se definió anteriormente. Esta adición puede realizarse bajo condiciones térmicas, calentando ambos compuestos a una temperatura entre 50 °C y 200 °C, usando calentamiento regular o irradiación por microondas, en presencia de una base, tal como pero no limitado a TEA, DIEA, K2CO3 o CS2CO3, en un disolvente polar, por ejemplo DMF, DMA o NMP. Alternativamente, esta adición puede catalizarse por un complejo metálico, tal como pero no limitado a PdCh, Pd(OAc)2, Pd2(dba)3 en presencia de un ligando, por ejemplo BINAP, o-To^P, X-Phos y una base, por ejemplo, NaOfBu, Cs2CO3 o K2CO3, en un disolvente o mezcla de disolventes adecuados, por ejemplo, dioxano, tolueno/MeOH, a una temperatura entre TA y 150 °C, preferentemente a TA, durante unas pocas horas, por ejemplo de una hora a 24 h (Esquema 2). La amina de fórmula (II) se obtiene después de la desprotección del compuesto (IVa). PG es un grupo protector adecuado, que es compatible con la química descrita a continuación, tal como, pero no limitado a, BOC. Puede eliminarse bajo condiciones ácidas, tales como, pero no limitado a, HCl en MeOH o dioxano o TFA en DCM, lo que produce el aislamiento de la amina (II).
Esquema 2

El compuesto de fórmula (IV), en donde PG es un grupo protector, puede prepararse a partir de la correspondiente cetona (IX) mediante aminación reductora con amina (VI), usando condiciones conocidas por los expertos en la materia, tales como, pero no limitado al uso de NaBH(OAc)3 como agente reductor, en presencia de un equivalente de AcOH en DCE. Alternativamente, la aminación reductora puede realizarse en dos etapas, con la primera formación de imina, que puede catalizarse por Ti(OiPr)4, seguida de reducción con un agente reductor adecuado, tal como pero no limitado a NaBH4 en MeOH (Abdel-Magid, AF et al. J. Org. Chem. 1996, 61,3849-3862). Alternativamente, la cetona (IX) puede reducirse en el alcohol correspondiente (VIII) usando agentes reductores habituales como NaBH4 en un disolvente alcohólico, tal como MeOH. Después, la funcionalidad alcohol puede transformarse en un grupo saliente
adecuado, tal como, pero no limitado a, Cl u OM, usando condiciones conocidas por un experto en la materia. La adición de amina (VI) al intermedio (VII) produciría la formación del compuesto (IV).
Esquema 3

Alternativamente, el compuesto de fórmula (X), en donde PG es un grupo protector adecuado, tal como, pero no limitado a BOC, puede prepararse a partir de la adición de amina (XI) a un heterociclo de fórmula (III), donde LG es un grupo saliente como se define anteriormente. Esta adición puede realizarse en condiciones térmicas o puede catalizarse por un complejo metálico, usando condiciones conocidas por un experto en la materia y como se describe a continuación en los ejemplos (Esquema 4).
PG es un grupo protector adecuado, que es compatible con la química descrita anteriormente, tal como, pero no limitado a, BOC. Puede eliminarse bajo condiciones ácidas, tales como, pero sin limitarse a HCl en MeOH o dioxano o TFA en DCM, lo que produce el aislamiento de la amina (XIV). Puede transformarse adicionalmente en el compuesto de fórmula (I) mediante alquilación reductora con cetona de fórmula (IX), siguiendo condiciones bien conocidas por un experto en la materia, como se describe en los ejemplos (Abdel-Magid, A. F. et al. J. Org. Chem. 1996, 61, 3849 3862). Alternativamente, la adición de amina (XIV) al compuesto (VII), preparado como se describe anteriormente y en los ejemplos, produciría la formación del compuesto de fórmula (I).
Esquema 4

La amina de fórmula (II) puede separarse en aminas de fórmula (IIa) y (IIb) mediante cromatografía quiral o resolución quiral mediante recristalización con un ácido ópticamente activo, usando métodos conocidos por un experto en la materia y como se describe a continuación en los ejemplos (Esquema 5).
Esquema 5

En otro proceso, la cetona de fórmula (IX) puede obtenerse mediante la reacción de acoplamiento cruzado de Stille entre haluro de arilo (XX) y tributil(1-etoxivinil)estaño en presencia de un catalizador, tal como, pero no limitado a Pd(PPha)2Cl2 en tolueno a temperaturas que varían entre Ta y 110 °C (Esquema 6).
Esquema 6

El grupo sulfoximina como se indica en la definición de X puede introducirse o generarse en cualquier fase de la síntesis de compuestos de fórmula (I), como se describe a continuación en los ejemplos.
Las rutas sintéticas generales para la preparación de sulfoximinas se describen en el Esquema 7, en donde G1 y G2 juntos representan el resto del compuesto de fórmula I:
Esquema 7

La síntesis típica de sulfoximinas comienza con la oxidación de un sulfuro (XXI) seguida de la iminación del sulfóxido resultante (XXII). La iminación catalizada por rodio generalmente da como resultado sulfoximinas W-protegidas (XXV) (R3’ = grupo protector) que después pueden desprotegerse, produciendo NH-sulfoximinas libres (XXIV) (R3’ = H). La iminación catalizada por metal de sulfóxidos (XXII) o sulfuro (XXI) también puede lograrse con metales alternativos, tales como, pero sin limitarse a, complejos de cobre, hierro, manganeso o rutenio. Las iminaciones de sulfóxido (XXII) usando ácido hidrazoico generado in situ, reactivos activados tales como O-mesitilenosulfonilhidroxilamina (MSH) u 0-(2,4-dinitrofenil)-hidroxilamina (DPH) o carbamato de amonio en presencia de diacetoxiyodobenceno [Phl(OAc)2], conducen directamente a la sulfoximina libre (XXIV) (R3’ = H). La sulfoximina W-funcionalizada XXV (R3’ í H) puede prepararse a partir de las NH-sulfoximinas libres (XXIV) (R3’ = H) mediante métodos tales como arilaciones catalizadas por Cu, sustituciones nucleofílicas o alquilación reductora. Alternativamente, las sulfoximinas (XXV) (R3’ t H) también pueden obtenerse por oxidación de sulfiliminas (XXIII), que son accesibles por iminación de sulfuros (XXI) o transformación de sulfóxidos (XXII) (Frings, M. et al. Eur. J. Med. Chem. 2017, 126, 225-245 y las referencias citadas).
Las sulfoximinas (XXIV) o (XXV) pueden separarse en compuestos de fórmula (XXIVa) y (XXIVb) o (XXVa) y (XXVb) mediante cromatografía quiral o mediante resolución quiral, recristalización con el uso de un ácido ópticamente activo, usando métodos conocidos por un experto en la materia y como se describe a continuación en los ejemplos (Esquema 8).
Alternativamente, el sulfóxido (XXII) puede separarse en compuestos de fórmula (XXIIa) y (XXIIb) por cromatografía quiral o por resolución quiral, usando métodos conocidos por un experto en la materia y como se describe a continuación en los ejemplos (Esquema 8).
Esquema 8
Cromatografía quiral
O, N -R3' 0 O N -R 3
V ' resolución quiral S '
G1" b ^ G 2 -------------------------  + G1" "G 2
+ G1" "G 2
(XXIV) (R j. J r _ H) (XXIVa) (R3’ = H) (XXIVb) (R3’ = H) o o o
(XXV) (R3’ no H) (XXVa) (R3' no H) (XXVb) (R3' no H)
iminacion iminacion
Cromatografía quiral
O o O O n n i¡
-S, resolución quiral
'G ¿ g ^ s "g 2 G1' ’-S>G2
(XXÍI) (XXIIa) (XXIlb)
El sulfóxido quiral de fórmula (XXIIa) y (XXIIb) puede transformarse en sulfoximina quiral de fórmula (XXIVa) y (XXIVb) respectivamente o (XXVa) y (XXVb) respectivamente. La transformación estereoespecífica con retención de la configuración puede lograrse mediante iminación catalizada por rodio y desprotección posterior (H. Okamura et al. Organic Letters 2004, 6, 1305-1307) o iminación con carbamato de amonio y Phl(OAc)2 en MeOH, dando directamente (XXIVa) y (XXIVb) (M. Zenzola et al. Angew. Chem. Int. Ed. 2016, 55, 7203-7207).
Las principales rutas a los sulfóxidos quirales se muestran en el Esquema 9. La resolución de una mezcla racémica (ruta i) es un posible método usado para producir sulfóxidos quirales, ya sea mediante un enfoque químico o una reacción enzimática. La transformación de un sulfinato diastereoquímicamente puro es una ruta alternativa que proporciona sulfóxidos con altos valores de exceso enantiomérico (ee) (ruta ii). La oxidación enantioselectiva de sulfuros proquirales (XXI) por métodos enzimáticos o no enzimáticos representa una forma relativamente directa (ruta iii) de preparar sulfóxidos enantioenriquecidos. Otro método de preparación (ruta iv) es modificar la estructura de algunos sulfóxidos quirales sin ninguna pérdida de estereoquímica en el átomo de azufre (Organosulfur chemistry in asymmetric synthesis, Takeshi Toru; 2008).
Esquema 9

Un enfoque para la oxidación enantioselectiva de sulfuros proquirales (XXI) en sulfóxido quiral de fórmula (XXIIa) o (XXIIb) es el uso de complejos de metales de transición quirales en combinación con un oxidante (ruta ii). Normalmente, implica un metal tal como, pero no limitado a Ti(i-PrO)4, en una cantidad estequiométrica o catalítica, un ligando quiral seleccionado de tartrato de dietilo, ácido madelico, binaftol, dibromo-binaftol, hidrobenzαna o cualquier otro ligando conocido por un experto en la materia, un oxidante, tal como pero no limitado a hidroperóxido de cumeno, hidroperóxido de tere-butilo, H2O2, con la adición opcional de agua o una amina terciaria, tal como i-Pr2NEt, N-metilmorfolina o 1,4-dimetil-piperazina (G. E. O'Mahony et al. Arkivoc 2011 (i) 1-110; J. Legros et ál. Adv. Synth. Catal. 2005, 347, 19-31). También pueden usarse metales alternativos, tales como, pero no limitados a, Mn, V, Fe o W en presencia de un ligando quiral (G. E. O'Mahony et al. Arkivoc 2011 (i) 1-110; J. Legros et ál. Adv. Synth. Catal. 2005, 347, 19-31). La oxidación catalítica quimioselectiva y enantioselectiva de sulfuros heteroaromáticos puede lograrse en diversos grados de selectividades con un complejo de manganeso que lleva un ligando quiral, tal como ligandos similares a las porfirinas, en presencia de un ácido carboxílico, tal como ácido adamantino carboxílico y un oxidante tal como pero no limitado a H2O2 (Dai, W. et al. ACS catal. 2017, 7, 4890-4895). Puede aplicarse una purificación adicional para lograr purezas enantioméricas adecuadas.
Alternativamente, la resolución cinética del sulfóxido de fórmula (XXII) en sulfona (XXVI) puede lograrse bajo condiciones similares y de acuerdo con métodos bien conocidos, dejando un sulfóxido enantioenriquecido sin cambios (G. E. O'Mahony et al. Arkivoc 2011 (i) 1-110).
Cuando una reacción se realiza preferentemente bajo condiciones básicas, puede seleccionarse una base adecuada de óxidos metálicos, por ejemplo óxido de aluminio, hidróxido de metal alcalino (hidróxido de potasio, hidróxido de sodio e hidróxido de litio, entre otros), hidróxido de metal alcalinotérreo (hidróxido de bario e hidróxido de calcio, entre otros), alcoholatos de metales alcalinos (etanolato de potasio y propanolato de sodio, entre otros), carbonatos de metales alcalinos (por ejemplo, bicarbonato de sodio) y varias bases orgánicas (por ejemplo, W,W-diisopropiletilamina, piperidina o dietanolamina, entre otras).
La reacción se lleva a cabo generalmente en un disolvente inerte. Los disolventes inertes adecuados son, por ejemplo, hidrocarburos, tales como hexano, éter de petróleo, benceno, tolueno o xileno; hidrocarburos clorados, tales como tricloroetileno, 1,2-dicloroetano, tetracloruro de carbono, cloroformo o diclorometano; alcoholes, tales como metanol, etanol, isopropanol, n-propanol, n-butanol o terc-butanol; éteres, tales como dietiléter, diisopropiléter, tetrahidrofurano (THF) o dioxano; éteres de glicol, tales como éter monometílico o monoetílico de etilenglicol, éter dimetílico de etilenglicol (diglima); cetonas, tales como acetona o butanona; amidas, tales como acetamida, dimetilacetamida o dimetilformamida (DMF); nitrilos, tales como acetonitrilo; sulfóxidos, tales como dimetilsulfóxido (DMSO); disulfuro de carbono; ácidos carboxílicos, tales como ácido fórmico, ácido acético o ácido trifluoroacético (TFA); compuestos nitro, tales como nitrometano o nitrobenceno; ésteres, como acetato de etilo, o mezclas de dichos disolventes. Se da preferencia particular a TFA, DMF, diclorometano, THF, H2O, metanol, terc-butanol, alcohol terc-amílico, trietilamina o dioxano.
Dependiendo de las condiciones usadas, el tiempo de reacción es de unos pocos minutos a 14 días, la temperatura de reacción está entre aproximadamente -80 °C y 140 °C, normalmente entre -50 °C y 120 °C, preferentemente entre -20 °C y 100 °C.
Breve descripción de las figuras
La presente invención se ilustra además con referencia a las figuras adjuntas:
Figura 1: Patrón de difracción de rayos X de polvo característico de la sal de succinato cristalina del Ejemplo 35, Forma 1
Figura 2: Espectro de RMN 1H característico de la sal de succinato cristalina del Ejemplo 35, Forma 1 Figura 3: Termograma STA característico de la sal de succinato cristalina del Ejemplo 35, Forma 1
Figura 4: Patrón de difracción de rayos X de polvo característico de la sal de fumarato cristalina del Ejemplo 35 Figura 5: Espectro de RMN 1H característico de la sal de fumarato cristalina del Ejemplo 35
Figura 6: Termograma STA característico de la sal de fumarato cristalina del Ejemplo 35
Figura 7: Patrón de difracción de rayos X de polvo característico de la sal de succinato cristalina del Ejemplo 35, Forma 2
Figura 8: Espectro de RMN 1H característico de la sal de succinato cristalina del Ejemplo 35, Forma 2 Figura 9: Patrón de difracción de rayos X de polvo característico de la sal de clorhidrato cristalina del Ejemplo 35 Figura 10: Termograma STA característico de la sal de clorhidrato cristalina del Ejemplo 35
Figura 11: Patrón de difracción de rayos X de polvo característico de la sal de benzoato cristalina del Ejemplo 35 Figura 12: Espectro característico de RMN 1H de la sal de benzoato cristalina del Ejemplo 35
PARTE EXPERIMENTAL
Preparación de los compuestos
Los compuestos de acuerdo con la Fórmula (I) pueden prepararse a partir de materiales de partida fácilmente disponibles mediante varios enfoques sintéticos, usando protocolos químicos tanto en fase de solución como en fase sólida o protocolos de solución mixta y fase sólida. Los ejemplos de rutas sintéticas se describen a continuación en los ejemplos. Todos los rendimientos informados son rendimientos no optimizados. A menos que se indique lo contrario, los compuestos de Fórmula (I) y fórmulas relacionadas obtenidos como una mezcla racémica pueden separarse para proporcionar una mezcla enriquecida enantioméricamente o un enantiómero puro.
Los materiales de partida disponibles en el mercado usados en la siguiente descripción experimental se adquirieron de Aldrich, Sigma, ACROS, ABCR, Combi-Blocks, Matrix, Apollo Scientific, Alfa Aesar, etc. a menos que se informe lo contrario.
Los datos de HPLC, MS y RMN proporcionados en los ejemplos descritos a continuación se obtienen como sigue: Los análisis de RMN 1H se llevaron a cabo usando BRUKe R RMN, modelo AV-II y AV-III 400 MHz FT-RMN. La señal residual de disolvente deuterado se usó como referencia interna. Los desplazamientos químicos (8) se informan en ppm en relación con la señal de disolvente residual (8 = 2,50 para RMN 1H en DMSO-d6, y 7,26 en CDCl3). s (singlete), d (doblete), t (triplete), q (cuadruplete), a (ancho), quint (quintuplete).
Condiciones del análisis CLEM:
Nombre del instrumento: Agilent Technologies 1290 infinity 11.
Método A: Método: A-TFA al 0,1 % en H2O, B-TFA al 0,1 % en ACN; caudal: 2,0 ml/min; columna: XBridge C8 (50 x 4,6 mm, 3,5 μm), modo ve
Método B: Método: A-NH4HCO310 mM en H2O, B-ACN; caudal: 1,0 ml/min; columna: XBridge C8 (50 x 4,6 mm, 3,5 μm), modo ve
Método C: Método: A-HCOOH al 0,1 % en H2O, B-ACN; caudal: 1,5 ml/min; columna: ZORBAX Eclipse XDB-C18 (50 x 4,6 mm, 3,5 μm), modo ve
Condiciones del análisis HPLC:
Nombre del instrumento: Instrumentos Agilent serie 1200 como sigue usando % con detección UV (maxplot).
Método A: Método: A-TFA al 0,1 % en H2O, B-TFA al 0,1 % en ACN; caudal: 2,0 ml/min; columna: XBridge C8 (50 x 4,6 mm, 3,5 μm).
Método B: Método: A- NH4HCO310 mM en H2O, B-ACN; caudal: 1,0 ml/min; columna: XBridge C8 (50 x 4,6 mm, 3,5 μm).
Condición del análisis HPLC quiral:
Nombre del instrumento: Agilent 1260 Infinity II
Método A: Fase móvil: 0,1 % DEA en n-Hexano: EtOH: 60:40, Caudal: 1,0 ml/min; columna: Chiralcell OD-H (250 x 4,6 mm, 5 μm).
Condición del análisis SFC quiral:
Nombre del instrumento: THAR-SFC 80 y THAR-SFC 200 (analítico)
La proporción entre CO2 y codisolvente varía entre 60:40 y 80:20
Método A: Fase móvil: Amoníaco 20 mM en IPA, caudal: 4 ml/min; columna: Chiralpak ADH (250 x 4,6 mm, 5 μm).
Método B: Fase móvil: Amoníaco 20 mM en metanol, caudal: 10 ml/min; columna: YMC Cellulose C (250 x 4,6 mm, 5 μm).
Método C: Fase móvil: Amoníaco 20 mM en IPA, caudal: 4 ml/min; columna: LuxA1 (250 x 4,6 mm, 5 μm).
Método D: Fase móvil: Amoníaco 20 mM en MeOH, caudal: 4 ml/min; columna: Chiralpak ADH (250 x 4,6 mm, 5 μm).
Método E: Fase móvil: IPA, caudal: 3 ml/min; columna: Lux A1 (250 x 4,6 mm, 5 μm).
Condición de análisis de HPLC prep:
Método A: A-TFA al 0,1 % en H2O, B-MeOH o CAN; columna: Sunfire C8 (19 * 250 mm, 5 |jm) o Sunfire C18 (30 * 250 mm, 10 jm).
Método B: A-10 mM NH4HCO3 en H2O, B-MeOH o ACN, Columna: Sunfire C8 (19 * 250 mm, 5 jm ) o Sunfire C18 (30 * 250 mm, 10jm).
Condición del análisis SFC quiral preparativa:
Nombre del instrumento: THAR-SFC 80, THAR-SFC 200 y PIC SFC 10-150
La proporción entre CO2 y codisolvente varía entre 60:40 y 80:20
Método A: Fase móvil: Amoníaco 20 mM en IPA; caudal: 3 ml/min; columna: Chiralpak ADH (250 * 30 mm, 5 jm).
Método B: Fase móvil: Amoníaco 20 mM en metanol; caudal: 5 ml/min; columna: YMC Cellulose C (250 * 30 mm, 5 jm ).
Método C: Fase móvil: Amoníaco 20 mM en IPA; caudal: 5 ml/min; columna: LuxA1 (250 * 30 mm, 5 jm).
Método D: Fase móvil: Amoníaco 20mM en MeOH; caudal: 4 ml/min; columna: Chiralpak ADH (250 * 30 mm, 5 jm).
Método E: Fase móvil: IPA, caudal: 100 ml/min; columna: Phenomenex Lux Amylose-1 (250 * 30 mm, 5 jm).
Condiciones generales de cromatografía ultrarrápida usadas para la purificación de intermedios o compuestos de Fórmula I: gel de sílice malla 230-400; gradientes usados como eluyente: EtOAc del 10 al 80 % en éter de petróleo o MeOH del 1 al 15 % en DCM.
La química de microondas se realizó en un reactor de microondas de un solo modo Initiator™ Sixty de Biotage. Rotación óptica específica
Nombre del instrumento: Autopol VI, de Rudolph Research Analytical, Hackettstown, NJ, EE.UU.
Síntesis:
Intermedio 1: 5-(1-cloroetil)benzordltiazol

Etapa 1: 1-(benzo[d]tiazol-5-il)etan-1-ona:

A una solución desgasificada de 5-bromo benzotiazol (Combi-Blocks, 750 g, 3,51 mol) en tolueno seco (6 l), se añadieron 1-etoxivinil tributilestaño (1,42 l, 4,21 mol) seguido de Pd(PPh3)2Ch (105,6 g, 150,7 mmol) a TAy la mezcla resultante se calentó a 90 °C durante 16 h. Una vez completada la reacción (monitorizada por TLC), la mezcla de reacción se enfrió a TA, se filtró a través de celite y se lavó con EtOAc (1 l). El filtrado se evaporó al vacío y se añadió una solución de HCl 5 N (2,5 l) a la mezcla bruta. La solución de color marrón claro resultante se agitó a TA durante 1,5 h, se neutralizó con la adición lenta de una solución saturada de NaHCO3 (12 l) durante 1 h a 0 °C y se extrajo con EtOAc ( 2 * 5 l). La capa orgánica combinada se lavó con solución de salmuera (2,5 l), se secó sobre Na2SO4 anhidro y se evaporó al vacío. El material bruto resultante se disolvió en DCM (750 ml), se le añadió hexano (3 l) y el sólido resultante se filtró y los sólidos se lavaron con MTBE (4 l). El filtrado combinado se concentró al vacío y el residuo se disolvió en EtOAc (2,5 l). Se añadió carbón vegetal (35 g) a la solución resultante. La capa orgánica se agitó durante 6 h a TA y se filtró y los sólidos se lavaron con EtOAc (1 l). La capa orgánica se concentró para producir el compuesto del título. Rendimiento: 79 % (475 g, sólido marrón claro) RMN 1H (400 MHz, DMSO-de): 89,53 (s, 1H), 8,69 (s, 1H), 8,32 (d, J = 8,4 Hz, 1H), 8,04 (dd, J = 8,4, 1,3 Hz, 1H), 2,71 (s, 3H). CLEM: (Método C) 178,0 (M+H), Tr. 1,4 min, 98,5
% (Máx). HPLC: (Método A) Tr. 2,6 min, 97,2 % (Máx).
Etapa 2: 1-(benzo[d]tiazol-5-il)etan-1-ol:

A una solución agitada de 1-(benzo[d]tiazol-5-il)etan-1-ona (475 g, 2,68 mol)) en MeOH (4,75 l), se añadió en porciones NaBH4 (152,28 g, 4,03 mol) a 0 °C y la mezcla de reacción se agitó a TA durante 1 h. La finalización de la reacción se monitorizó mediante TLC. Después, la mezcla de reacción se inactivó con agua helada (400 ml) a 0 °C y se concentró al vacío. A la mezcla bruta resultante se le añadió agua (2,5 l) y la capa acuosa se extrajo con EtOAc (2 x 2,5 l). La capa orgánica combinada se lavó con salmuera (2 l), se secó sobre Na2SO4 anhidro y se concentró al vacío. El sólido bruto resultante se trituró con hexano: éter dietílico (8:2 ) y se decantó para producir el compuesto del título.
Rendimiento: 93 % en bruto (440 g, sólido gomoso de color marrón pálido). RMN 1H (400 MHz, DMSO-d6): 89,37 (s, 1H), 8,09 (d, J= 8,4 Hz, 1H), 8,04 (s, 1H), 7,50 (d, J= 1,2 Hz, 1H), 5,32 (d, J= 4,0 Hz, 1H), 4,93-4,89 (m, 1H), 1,40 (d, J= 6,4 Hz, 3H). CLEM: (Método C) 180,0 (M+H), Tr. 1,2 min, 98,7 % (Máx). HPLC: (Método A) Tr. 2,2 min, 99,5 % (Máx).
Etapa 3: 5-(1-cloroetil)benzo[d]tiazol:

A una solución agitada de 1-(benzo[d]tiazol-5-il)etan-1-ol (440 g, 2,46 mol)) en DCM (4,4 l), se añadió gota a gota cloruro de tionilo (534 ml, 7,37 mol) durante 30 min a 0 °C y la mezcla de reacción se agitó durante 1 h a 0-10 °C. La finalización de la reacción se monitorizó mediante TLC. Después, la mezcla de reacción se evaporó al vacío. El material bruto resultante se codestiló con DCM seco (3 * 400 ml), se secó al vacío para producir el compuesto del título que se usó en la siguiente etapa sin purificación adicional. Rendimiento: 100 % bruto (488 g, sólido amarillo). RMN 1H (400 MHz, DMSO-d6): 810,79 (s, 1H), 8,52 (s, 1H), 8,16 (d, J= 8,4 Hz, 1H), 7,86 (d, J= 8,4 Hz, 1H), 5,30-5,24 (m, 1H), 1,91 (d, J = 6,8 Hz, 3H). CLEM: (Método C) 198,0 (M+H), Tr. 2,0 min, 50,1 % (Máx). HPLC: (Método A) Tr. 3,9 min, 66,8 % (Máx).
Intermedio 2: 5-(1-cloroetil)-2-metilbenzordltiazol

Etapa 1: 1-(2-metilbenzo[d]tiazol-5-il)etan- 1-ona

A una solución desgasificada de 5-bromo-2-metilbenzo[d]tiazol (10 g, 43,85 mmol, Combi block) en tolueno seco (40 ml), se añadieron Pd(PPh3)2Ch (3,07 g, 4,3 mmol) seguido de 1-etoxivinil tributilestaño (16,2 ml, 48,2 mmol) y la mezcla de reacción se calentó a 90 °C durante 16 h. La finalización de la reacción se monitorizó mediante TLC, la mezcla de reacción se enfrió después a 0 °C y se filtró a través de celite. El filtrado resultante se evaporó al vacío y después se añadió una solución de HCl 6 N (80 ml) al material bruto. La mezcla de reacción se agitó a TA durante 1 h, después se neutralizó usando NaHCO3 y la capa acuosa se extrajo con EtOAc (2 * 80 ml). La capa orgánica combinada se secó sobre Na2SO4 anhidro y se evaporó al vacío. El material bruto resultante se purificó mediante cromatografía en columna ultrarrápida (Biotage Isolera, eluyente: 60-80 % EtOAc en hexano). Rendimiento: 72 % (6 g, sólido amarillo)
RMN 1H (400 MHz, DMSO-de): 58,48 (s, 1H), 8,18 (d, J= 11,2 Hz, 1H), 7,95 (d, J= 11,2 Hz, 1H), 2,85 (s, 3H), 2,67 (s, 3H). CLEM: (Método A) 192,3 (M+H), Tr. 2,9 min, 96,8% (Máx).
Etapa 2: 1-(2-metilbenzo[d]tiazol-5-il)etan-1-ol

A una solución agitada de 1-(2-metilbenzo[d]tiazol-5-il)etan-1-ona (6 g, 31,31 mmol) en MeOH (30 ml), se añadió en porciones NaBH4 (2,37 g, 62,74 mmol) a 0 °C y la mezcla de reacción se agitó a TA durante 1 h. La finalización de la reacción se monitorizó mediante TLC, después la mezcla de reacción se inactivó con hielo y se evaporó al vacío. A la mezcla de reacción resultante, se le añadió agua (10 ml) y se extrajo con EtOAc (2 * 60 ml). La capa orgánica combinada se secó sobre Na2SO4 anhidro y se evaporó al vacío. El material bruto resultante se purificó mediante cromatografía ultrarrápida (Biotage Isolera, eluyente: 70-90 % EtOAc en hexano). Rendimiento: 87 % (5,3 g, sólido marrón). RMN 1H (400 MHz, DMSO-de): 57,94 (d, J= 8,4 Hz, 1H), 7,86 (s, 1H), 7,38 (dd, J= 8,2, 1,2 Hz, 1H), 5,28 (d, J= 4,4 Hz, 1H), 4,90-4,80 (m, 1H), 2,79 (s, 3H), 1,38 (d, J = 6,4 Hz, 3H). CLEM: (Método A) 194,2 (M+H), Tr. 2,5 min, 98,9 % (Máx).
Etapa 3: 5-(1-doroetil)-2-metilbenzo[d]tiazol

A una solución agitada de 1-(2-metilbenzo[d]tiazol-5-il)etan-1-ol (5,3 g, 27,4 mmol) en DCM seco (50 ml), se añadió cloruro de tionilo (4 ml, 54,8 mmol) gota a gota a 0 °C y se agitó a 25 °C durante 1 h. La finalización de la reacción se monitorizó mediante TLC, la mezcla de reacción se concentró después al vacío y se codestiló con tolueno (10 ml). El material bruto resultante se secó a alto vacío para producir el compuesto del título que se usó en la siguiente etapa sin purificación adicional. Rendimiento: 5,5 g (bruto), aceite marrón. RMN 1H (400 MHz, DMSO-de): 58,05-8,01 (m, 2H), 7,53 (dd, J = 8,4, 2,0 Hz, 1H), 5,51 (q, J = 6,8 Hz, 1H), 2,81 (s, 3H), 1,86 (d, J = 6,8 Hz, 3H). CLEM: (Método A) 212,2 (M+H), Tr. 4,26 min, 36,1 % (Máx).
Intermedio 6: 5-(1-(piperazin-1-il)etil)benzord1tiazol

Etapa 1: 4-(1-(benzo[d]tiazol-5-il)etil)piperazin-1-carboxilato de terc-butilo:

A una solución agitada de piperazin-1-carboxilato de terc-butilo (522 g, 2,97 mol) y TEA (2,5 l, 17,34 mol) en DMF (2 l), se añadió gota a gota el Intermedio 1 (488 g, 2,48 mol) en DMF (3 l) a TA en atmósfera de N2 y la mezcla de reacción se calentó a 60 °C durante 24 h. La finalización de la reacción se monitorizó mediante TLC. Después, la mezcla de reacción se enfrió a TA. A la mezcla resultante, se le añadió agua (10 l) y la capa acuosa se extrajo con EtOAc ( 6 * 2 l). La capa orgánica combinada se lavó con salmuera (2,5 l), se secó sobre Na2SO4 anhidro y se concentró al vacío.
El material bruto resultante se purificó mediante cromatografía ultrarrápida (gel de sílice: malla 60-120, eluyente: EtOAc al 40 % en éter de petróleo) para producir el compuesto del título. Rendimiento: 81 % (700 g, sólido gomoso de color marrón pálido). RMN 1H (400 MHz, DMSO-de): 69,39 (s, 1H), 8,11 (d, J= 8,4 Hz, 1H), 7,99 (s, 1H), 7,47 (d, J = 8,4 Hz, 1H), 3,45 (q, J = 6,8 Hz, 1H), 3,34-3,29 (m, 4H), 2,37- 2,27 (m, 4H), 1,41-1,18 (m, 12H). CLEM: (Método A) 348,1 (M+H), Tr. 1,6 min, 85,6 % (Máx). HPLC: (Método A) Tr. 2,89 min, 81,5 % (Máx)
Etapa 5: 5-(1-(piperazin-1-il)etil)benzo[d]tiazol

A una solución agitada de 4-(1-(benzo[d]tiazol-5-il)etil)piperazin-1-carboxilato de tere-butilo (700 g, 2,02 mol) en 1,4-dioxano (3 l), se añadió gota a gota una solución de Hcl en dioxano (3,50 l, 4 M) a 0 °C y la solución resultante se agitó a TA durante 6 h. Después de terminar la reacción (monitorizada por TLC), la mezcla de reacción se concentró al vacío y el material bruto resultante se trituró con EtOAc (2 * 1 l). La sal de clorhidrato se disolvió en agua (2,5 l) y la capa acuosa se lavó con EtOAc (3 * 2 l) y DCM (3 * 2 l). La capa acuosa resultante se basificó con NaOH 6 N (pH ~12) y se extrajo con EtOAc (3 * 2 l). La capa orgánica combinada se lavó con salmuera (500 ml), agua (500 ml), se secó sobre Na2SO4 anhidro y se concentró al vacío para producir el compuesto del título. Rendimiento: 70 % (350 g, sólido gomoso de color marrón pálido). RMN 1H (400 MHz, DMSO-de): 69,38 (s, 1H), 8,10 (d, J = 8,4 Hz, 1H), 7,98 (s, 1H), 7,46 (dd, J = 8,4, 1,2 Hz, 1H), 3,33 (m, 1H), 3,58 (q, J= 6,8 Hz, 1H), 2,71-2,68 (m, 4H), 2,37-2,27 (m, 4H), 1,19 (d, J= 6,8 Hz, 3H). CLEM: (Método A) 248,1 (M+H), Tr. 0,88 min, 97,3% (Máx) HPLC: (Método A) Tr. 1,6 min, 99,1 % (Máx).
Intermedio 7: (S)-5-(1-(piperazin-1-il)etil)benzord1tiazol o (R)-5-(1-(piperazin-1-il)etil)benzord1tiazol

A una mezcla agitada del intermedio 6 (100 g, 405,0 mmol) en EtOH (2 l, 20 V), se añadió ácido D-di-p-anisoiltartárico (42,31 g, 101,2 mmol) a TA y se calentó a 90 °C durante 20 min. (Nota: La formación de sal se observó lentamente después de 3 a 5 minutos después de la adición de ácido D-di-p-anisoiltartárico). Después, la mezcla de reacción se agitó a TA durante la noche. La mezcla resultante se filtró y la torta de filtración se lavó con EtOH (2 * 250 ml, 5 V), éter dietílico (250 ml) y se secó a alto vacío. Para aumentar el ee, la sal (66 g, 79 % ee) se calentó más a reflujo en EtOH (1 l, 10 V) durante 24 h y se agitó a TA durante la noche. La sal obtenida se filtró, se lavó con EtOH (200 ml, 2 V), éter dietílico (200 ml) y se secó a alto vacío. Se repitió el mismo procedimiento para lograr el ee del 96,1 % (21,2 g). Esta etapa se repitió en una escala de 300 g para obtener la sal (113,2 g).
Las sales obtenidas anteriormente (134,4 g) se disolvieron en agua (300 ml), se basificaron a pH ~14 con una solución de NaOH 6 N (350 ml) y la capa acuosa se extrajo con EtOAc (2 * 1 l). La capa de EtOAc combinada se lavó con solución de salmuera (2 * 1 l), agua (300 ml), se secó sobre Na2SO4 anhidro y se concentró al vacío para obtener el compuesto del título (proporción de enantiómeros 97,41:2,58 %). Rendimiento: 85 % (63,0 g, sólido gomoso de color marrón pálido). RMN 1H (400 MHz, DMSO-d6): 69,38 (s, 1H), 8,09 (d, J = 8,4 Hz, 1H), 7,97 (s, 1H), 7,45 (d, J = 8,0 Hz, 1H), 3,55 (q, J = 6,8 Hz, 1H), 2,67-2,66 (m, 4H), 2,34-2,25 (m, 4H), 1,34 (d, J = 6,8 Hz, 3H). CLEM: (Método A) 248,2 (M+H), Tr. 1,5 min, 98,5 % (Máx). HPLC: (Método A) Tr. 1,6 min, 98,7 % (Máx). HPLC quiral: (Método A) Tr.
11,1 min, 97,4% (Máx).
El agente de resolución quiral ácido D-di-p-anisoiltartárico se puede intercambiar por ácido D-di-p-toluiltartárico o (R)-(+)-clocifos para obtener productos idénticos.
El Intermedio 6, sal de clorhidrato (15,57 g, 48,60 mmol) se mezcló con acetato de sodio trihidratado (26,45 g, 194,4 mmol, 400 % en moles), R-(+)-clocifos (8,11 g, 29,32 mmol, 99 % ee, 60,3 % en moles), agua (120 ml) y etanol (16 ml). La mezcla, que se convirtió en una suspensión espesa con la agitación, se calentó, dando como resultado una solución transparente cuando alcanzó la temperatura de reflujo. Se dejó enfriar la solución con agitación y se añadieron algunos cristales de siembra (espátula pequeña, aproximadamente 10-20 mg) aproximadamente cada 5-10 minutos, hasta que comenzó la cristalización (entre 5 y 10 veces). La cristalización de la sal comenzó a aproximadamente 45 °C. La suspensión se agitó a 20 °C durante la noche, después se filtró, el sólido se lavó con agua/etanol 10/1 (55 ml) y agua (20 ml). Se secó durante 2 d a 20 °C (11,12 g, 21,22 mmol, 44 %). El ee fue del 97,5 %.
Esta sal se calentó a reflujo suave con agua (90 ml) y etanol (10 ml). Se añadió más etanol (2 ml) y la solución se dejó
enfriar a 20 °C y se agitó durante 6 h. El sólido resultante se filtró y se lavó con agua (50 ml). Después de secar a 20 °C durante 3 d, se aisló la sal de clorcifos (9,50 g, 18,13 mmol, 37 %). El ee fue del 100 %.
La sal de clorcifos obtenida anteriormente con 100 % de ee (8,50 g, 16,22 mmol) se agitó durante 1,5 h en una mezcla de tolueno (100 ml), agua (50 ml) e hidróxido de sodio (4,04 g, 101 mmol). Se añadió cloruro de sodio (20 g) y la mezcla se agitó durante 15 min, después se filtró. Las capas filtradas se separaron. El sólido aislado en el filtro y en la capa acuosa se agitó con tolueno (125 ml). Se filtró y se volvieron a separar las capas de filtrado. Las capas de tolueno combinadas se secaron y evaporaron a 60 °C para producir la amina libre deseada como un aceite solidificante (3,60 g, 14,55 mmol, 90 %, puro por RMN) y con 99,6 % de ee.
Intermedio 8: 2-metil-5-(1-(piperazin-1-il) etil)benzordlt¡azol

Etapa 4: 2-metil-5-(1-(piperazin-1-il)etil)benzo[d]tiazol

A una solución agitada de piperazina (13,6 g, 15,9 mmol) en DCM seco (80 ml), se añadió gota a gota el Intermedio 2 (4,2 g, 19,8 mmol) durante un período de 20 min y la mezcla de reacción se agitó a TA durante la noche. Una vez completada la reacción (monitorizada por TLC), se añadió agua (50 ml) a la mezcla resultante y se agitó durante 10 min. La capa orgánica se separó, se lavó con salmuera (50 ml), se secó sobre Na2SO4 anhidro y se concentró al vacío. El material bruto resultante se purificó mediante cromatografía ultrarrápida (Biotage Isolera, eluyente: metanol al 18 20 % en DCM) para producir el compuesto del título. Rendimiento: 16 % (870 mg, sólido gomoso de color marrón pálido). RMN 1H (400 MHz, DMSO-de): 88,32 (s, 1H), 7,95 (d, J= 8,6 Hz, 1H), 7,80 (s, 1H), 7,34 (d, J= 8,8 Hz, 1H), 3,52-3,48 (m, 1H), 2,78 (s, 3H), 2,70 (t, J = 6,0 Hz, 4H), 2,44-2,24 (m, 4H), 1,33 (d, J =8,8 Hz, 3H). CLEM: (Método A) 262,2 (M+H), Tr. 1,8 min, 97,3 % (Máx)
Intermedio 9: 2-Cloro-5-(metilsulfinil)pirimidina

Etapa 1: 2-cloro-5-(metiltio)pirimidina
A una solución agitada de 5-bromo-2-cloropirimidina (5 g, 25,8 mmol) y 1,2-dimetildisulfano (2,92 g, 31,02 mmol) en THF (15 ml), se añadió n-BuLi (16,0 ml, 25,8 mmol, 1,6 M en hexano) a -78 °C y se agitó durante 1 h a la misma temperatura. Una vez completada la reacción (monitorizada por TLC), la reacción se inactivó con la adición de NH4CI sat. (15 ml) y la capa acuosa se extrajo con EtOAc (50 ml). La capa orgánica se lavó con agua (10 ml), salmuera (10 ml) y se secó sobre Na2SO4 anhidro. El material bruto resultante se purificó mediante cromatografía ultrarrápida (gel de sílice: malla 60-120, eluyente: EtOAc al 15% en éter de petróleo) para producir el compuesto del título.
Rendimiento: 13 % (0,6 g sólido blanco) RMN 1H (400 MHz, CDCl3): 88,50 (s, 2H), 2,56 (s, 3H). CLEM: (Método A) 161,1 (M+H), Tr. 2,1 min, 95,2% (Máx). HPLC: (Método A) Tr. 2,4 min, 98,5% (Máx).
Etapa 2: 2-cloro-5-(metilsulfinil)pirimidina

A una solución agitada de 2-doro-5-(metiltio)pirimidina (0,6 g, 2,49 mmol) en DCM (2 ml, 10 V), se añadió en porciones m-CPBA (0,644 g, 3,23 mmol) a 0 °C durante 30 min. La finalización de la reacción se monitorizó mediante TLC, la mezcla de reacción se inactivó después con una solución de NaHCO3 al 10% y se extrajo con DCM (2 * 100 ml). La capa orgánica combinada se lavó con salmuera (30 ml), se secó sobre Na2SO4 anhidro y se concentró al vacío. El material bruto resultante se purificó mediante cromatografía ultrarrápida (Biotage Isolera, eluyente: EtOAc al 10-12 % en éter de petróleo) para producir el compuesto del título. Rendimiento: 33 % (330 mg, sólido blanquecino). RMN 1H (400 MHz, DMSO-da): 88,88 (s, 2H), 2,92 (s, 3H). CLEM: (Método A) 177,1 (M+H), Tr. 0,8 min, 99,1 % (Máx). HPLC:
(Método A) Tr. 1,9 min, 99,6 % (Máx).
Intermedio 10: N-((2-cloropirimidin-5-il)(metilHoxo)-Á6-sulfanilideno)-2.2.2-trifluoroacetamida

A una solución agitada del Intermedio 9 (0,9 g, 5,09 mmol) en DCM (18,0 ml, 20 V), se añadieron trifluoroacetamida (1,15 g, 10,19 mmol), MgO (0,8 g, 20,38 mmol), Rh2(OAc)4 (0,12 g, 0,25 mmol) y Phl(OAC)2 (2,46 g, 7,64 mmol) y la mezcla de reacción se agitó a TA durante la noche. La finalización de la reacción se monitorizó mediante TLC, la mezcla de reacción se filtró después a través de celite y el filtrado se concentró al vacío. El material bruto resultante se purificó mediante cromatografía ultrarrápida (Biotage Isolera, eluyente: EtOAc al 16-18% en éter de petróleo) para producir el compuesto del título. Rendimiento: 74 % (1,1 g sólido blanco) CLEM: (Método A) 288,1 (M+H), Tr. 3,8 min, 71,1 % (Max).
Intermedio 11 e intermedio 12: N-((2-cloropirimidin-5-il)-(R)-(metilHoxo)-A6-sulfanilideno)-2.2.2-trifluoroacetamida y N-((2-cloropirimidin-5-il)-(S)-(metil)(oxo)-A6-sulfanilideno)-2.2.2-trifluoroacetamida

Etapa 1:2-doro-5-(metilsulfínil)pirimidina:

El intermedio 9 (502 g, 2,84 mol) se separó mediante análisis SFC (Pic SFC 10-150; CO2: IPA (70:30); columna: Lux A1 (250 * 30); caudal: 100 ml/min; longitud de onda: 210 nm; tiempo del ciclo: 5 min; contrapresión: 10 MPa (100 bar), Método E). El primer pico de elución (250,0 l de IPA) se concentró a 40 °C. Rendimiento: 40 % (201,0 g, sólido blanco) RMN 1H (400 MHz, DMSO-d6): 88,88 (s, 2H), 2,92 (s, 3H). CLEM: (Método A) 177,0 (M+H), Tr. 0,7 min, 99,9 % (Máx).
HPLC: (Método B) Tr. 2,04 min, 99,8 % (Máx). SFC quiral: (Método E) Tr. 2,1 min, 100 % (Máx).
El segundo pico de elución (250,0 l de IPA) se concentró a 40 °C. Rendimiento: 36 % (180,0 g, sólido blanco) RMN 1H (400 MHz, DMSO-de): 89,04 (s, 2H), 2,98 (s, 3H). CLEM: (Método A) 177,0 (M+H), Tr. 0,8 min, 99,8 % (Máx).
HPLC: (Método A) Tr. 2,04 min, 99,8 % (Máx). SFC quiral: (Método E) Tr. 4,6 min, 99,7 %
Etapa 2: N-((2-doropirimidin-5-il)-(S)-(metil)(oxo)-\6-sulfaniliden)-2,2,2-trifiuoroacetamida y N-((2-dompirimidin-5-il)-(R)-(meW)(oxo)-h6-sulfaniliden)-2,2,2-trifiuoroacetamida
A la solución agitada del primer compuesto de elución aislado en la etapa 1 (0,5 g, 2,8 mmol) en DCM (5 ml), se añadió trifluroacetamida (0,64 g, 5,66 mmol), MgO (0,45 g, 11,3 mmol), Rh2(OAc)4 (0,062 g, 0,14 mmol) y phl(OAc)2 (1,36 g, 4,20 mmol) y la mezcla de reacción se agitó a TA durante la noche. La finalización de la reacción se monitorizó mediante TLC. Después, la mezcla de reacción se filtró a través de celite y se lavó con DCM. La capa orgánica se concentró al vacío y el material bruto resultante se purificó mediante cromatografía ultrarrápida (Biotage Isolera, eluyente: EtOAc al 25-28 % en éter de petróleo) para producir el Intermedio 11. Rendimiento: 86 % (0,69 g, sólido blanco) RMN 1H (400 MHz, DMSOd>): 89,39 (s, 2H), 3,98 (s, 3H). CLEM: (Método A) 191,9 (M-COCF3+H), Tr. 3,8 min, 73,8 %.
A una solución agitada del segundo compuesto de elución aislado en la etapa 1 (2,0 g, 0,01 mmol) en DCM (20 ml, 10 V), se añadió trifluoroacetamida (2,56 g, 0,226 mol), MgO (1,825 g, 0,045 mol), Rh2(OAC)4 (250 mg, 0,56 mol) y Phl(OAC)2 (5,49 g, 0,016 mol) y se agito durante la noche a TA. La finalización de la reacción se monitorizó mediante TLC, después la mezcla de reacción se filtró a través de celite. El filtrado se concentró al vacío, el material bruto resultante se purificó mediante cromatografía ultrarrápida (Biotage Isolera, eluyente: EtOAc al 15-25% en éter de petróleo) para producir el Intermedio 12. Rendimiento: 62 % (2,0 g, sólido blanco) 1H RMN (400 MHz, DMSO-d6): 8 9,39 (s, 2H), 4,04 (s, 3H). CLEM: (Método A) 288,1 (M+H), Tr. 1,9 min, 92,8 % (Máx). HPLC: (Método A) Tr. 3,8 min, 96,1 % (Máx).
Intermedio 13: N-((2-cloropirimidin-5-il)(etilHoxo)-Á6-sulfaniliden)-2.2.2-trifluoroacetamida

Etapa 1: 2-doro-5-(etiltio)pirimidina

A una solución agitada de nitrito de t-butilo (5,99 g, 58,13 mmol) y 1,2-dietildisulfano (9,4 g, 77,51 mmol) en DCM (200 ml), se añadió en porciones 2-cloropirimidin-5-amina (5 g, 38,75 mmol) a TA durante 30 min y la mezcla de reacción se agitó a TA durante la noche. Una vez completada la reacción (monitorizada por TLC), la mezcla de reacción se concentró para obtener el material bruto que se purificó mediante cromatografía ultrarrápida (gel de sílice: malla 60 120, eluyente: EtOAc al 5 % en éter de petróleo) para producir el compuesto del título. Rendimiento: 24 % (1,6 g sólido blanco) RMN 1H (400 MHz, DMSO-d6): 88,74 (s, 2H), 3,12- 3,08 (m, 2H), 1,26-1,22 (m, 3H).
Etapa 2: 2-cloro-5-(etilsulfinil)pirimidina

A una solución agitada de 2-cloro-5-(etiltio)pirimidina (1,6 g, 9,16 mmol) en DCM (32,0 ml, 20 V) enfriada a 0 °C, se añadió en porciones m-CPBA(2,05 g, 11,90 mmol) y la mezcla resultante se agitó a 0 °C durante 30 min. La finalización de la reacción se controló mediante TLC, la mezcla de reacción se inactivó después con una solución de NaHCO3 al 10% y se extrajo con DCM (2 x 100 ml). La capa orgánica combinada se lavó con salmuera (30 ml), se secó sobre Na2SO4 anhidro y se concentró al vacío. El material bruto resultante se purificó mediante cromatografía ultrarrápida (Biotage Isolera, eluyente: EtOAc al 10-12 % en éter de petróleo) para producir el compuesto del título. Rendimiento:
58% (1,0 g, sólido blanquecino). RMN 1H (400 MHz, DMSO-d6): 88,99 (s, 2H), 3,31 (q, J= 8,6 Hz, 2H), 1,11 (t, J = 8,6 Hz, 3H). CLEM: (Método A) 191,2 (M+H), Tr. 1,3 min, 98,7 % (Máx).
Etapa 3: N-((2-dompirimidin-5-il)(etil)(oxo)-Á6-sulfaniliden)-2,2,2-trifíuomacetamida

A una solución agitada de 2-doro-5-(etilsulfinil)pirimidina (0,95 g, 5,00 mmol) en DCM (18,0 ml, 20 V), se añadió trifluoroacetamida (1,13 g, 10,0 mmol), MgO (0,8 g, 20,0 mmol), Rh2(OAc)4 (0,11 g, 0,25 mmol) y Phl(OAc)2 (2,41 g, 7,5 mmol) y la mezcla de reacción se agitó a TA durante la noche. La finalización de la reacción se monitorizó mediante TLC, la mezcla de reacción se filtró después a través de celite y el filtrado se concentró al vacío. El material bruto resultante se purificó mediante cromatografía ultrarrápida (Biotage Isolera, eluyente: EtOAc al 16-18% en éter de petróleo) para producir el compuesto del título. Rendimiento: 63 % (1,1 g sólido blanco)
Intermedio 14: N-((2-clorop¡r¡m¡d¡n-5-¡l)(oxoHprop¡l)-Á6-sulfan¡l¡den)-2.2.2-tr¡fluoroacetam¡da

Etapa-1:2-doro-5-(propiltio)pirimidina

A una solución agitada de nitrito de t-butilo (6,9 ml, 57,91 mmol) y 1,2-dipropil disulfano (12 ml, 77,2 mmol) en DCE (200 ml), se añadió en porciones 2-cloropirimidin-5-amina (5,0 g, 38,61 mmol, Angene) a TA durante 30 min y la mezcla de reacción se agitó a TA durante la noche. La finalización de la reacción se monitorizó mediante TLC, después la mezcla de reacción se concentró a presión reducida. El material bruto resultante se purificó mediante cromatografía ultrarrápida (Biotage Isolera, eluyente: EtOAc al 20 % en éter de petróleo) para producir el compuesto del título.
Rend¡m¡ento: 25 % (2,0 g, sólido amarillo pálido). RMN 1H (400 MHz, DMSO-de): 88,72 (s, 2H), 2,68 (t, J = 9,2 Hz, 2H), 1,81-1,54 (m, 2H), 1,14-0,90 (m, 3H). CLEM: (Método A) 189 (M+H), Tr. 3,7 min, 94,5 % (Máx).
Etapa 2: 2-cloro-5-(propilsulfinil)pirimidina

A una solución agitada de 2-cloro-5-(propiltio)pirimidina (2,3 g, 12,7 mmol) en DCM (23 ml, 10 V), se añadió en porciones m-CPBA (Spectrochem, 1,89 g, 10,97 mmol) a 0 °C y se agitó durante 60 min a 0 °C. La finalización de la reacción se monitorizó mediante TLC, después la mezcla de reacción se inactivó con una solución de NaHCO3 al 10 % y se extrajo con DCM (2 * 50 ml). La capa de DCM combinada se lavó con salmuera (20 ml), se secó sobre Na2SO4 anhidro y se concentró al vacío. El material bruto resultante se purificó mediante cromatografía ultrarrápida (Biotage Isolera, eluyente: EtOAc al 60-70 % en éter de petróleo) para producir el compuesto del título. Rend¡m¡ento: 43 % (0,9 g, sólido amarillo pálido). RMN 1H (400 MHz, DMSO-de): 89,01 (s, 2H), 3,19-3,00 (m, 2H), 1,75-1,53 (m, 2H), 0,97 (t, J = 6,0 Hz, 3H).
Etapa 3: N-((2-doropirimidin-5-il)(oxo)(propil)-A6-sulfaniliden)-2,2,2-trifluoroacetamida

A una solución agitada de 2-doro-5-(propilsulfinil)pirimidina (0,9 g, 4,07 mmol) en DCM (20 ml, 10 V), se añadieron trifluoroacetamida (0,92 g, 8,10 mmol), MgO (1,56 g, 16,30 mmol), Rh2(OAc)4 (90,11 mg, 0,20 mmol) y Phl(OAc)2 (1,97 g, 6,11 mmol) a TA y la mezcla de reacción se agitó a TA durante la noche. La finalización de la reacción se monitorizó por TLC, después la mezcla de reacción se filtró a través de celite y se concentró al vacío. El material bruto resultante se purificó mediante cromatografía ultrarrápida (Biotage Isolera, eluyente: EtOAc al 16-18 % en éter de petróleo) para producir el compuesto del título. Rendimiento: 78 % (1,0 g, sólido blanquecino). RMN 1H (400 MHz, DMSO-cfe): 89,36 (s, 2H), 3,19-3,00 (m, 2H), 1,75-1,53 (m, 2H), 0,97 (t, J= 6,0 Hz, 3H).
Intermedio 15: N-((6-clorop¡r¡d¡n-3-¡l)(met¡lHoxo)-Á6-sulfanil¡den)-2.2.2-tr¡fluoroacetam¡da

Etapa 1: 2-doro-5-(metiltio)piridina

A una solución agitada de nitrito de t-butilo (6,01 g, 58,33 mmol) y dimetil disulfano (7,32 ml, 77,78 mmol) en DCE (50 ml), se añadió en porciones 6-cloropiridin-3-amina (5,0 g, 38,89 mmol) a TA durante 30 min y se agitó a TA durante la noche. La finalización de la reacción se monitorizó mediante TLC, después la mezcla de reacción se vertió en agua y la capa acuosa se extrajo con EtOAc (2 * 50 ml). La capa orgánica combinada se secó sobre Na2SO4 anhidro y se concentró al vacío. El material bruto resultante se purificó mediante cromatografía ultrarrápida (Biotage Isolera, eluyente: EtOAc al 50 % en éter de petróleo) para producir el compuesto del título. Rendimiento: 73 % (4,5 g, líquido incoloro). RMN 1H (400 MHz, DMSO-de): 89,36 (s, 2H), 3,19-3,00 (m, 2H), 1,75-1,53 (m, 2H), 0,97 (t, J= 6,0 Hz, 3H).
CLEM: (Método A) 160,2 (M+H), Tr. 2,3 min, 95,4 % (Máx).
Etapa 2: 2-doro-5-(metilsulfinil)piridina

A una solución agitada de 2-cloro-5-(metiltio)piridina (4,5 g, 28,19 mmol) en DCM (45 ml, 10 V) enfriada a 0 °C, se añadió en porciones m-CPBA (6,32 g, 36,64 mmol) y se agitó a 0 °C durante 60 min. La finalización de la reacción se monitorizó mediante TLC, después la mezcla de reacción se inactivó con una solución de NaHCO3 al 10 % y se extrajo con DCM (2 * 100 ml). La capa orgánica combinada se lavó con salmuera (30 ml), se secó sobre Na2SO4 anhidro y se concentró al vacío. El material bruto resultante se purificó mediante cromatografía ultrarrápida (Biotage Isolera, eluyente: EtOAc al 60-70 % en éter de petróleo) para producir el compuesto del título. Rendimiento: 72 % (3,5 g, sólido amarillo pálido). RMN 1H (400 MHz, DMSO-de): 88,69 (d, J = 3,2 Hz, 1H), 8,20-8,16 (m, 1H), 7,76 (s, 1H), 2,89 (s, 3H). CLEM: (Método A) 176,2 (M+H), Tr. 1,4 min, 96,3 % (Máx)
Etapa 3: N-((6-dompiridin-3-il)(meW)(oxo)-Á6-sulfaniliden)-2,2,2-trifíuomacetamida

A una solución agitada de 2-doro-5-(metilsulfinil)piridina (2,0 g, 11,42 mmol) en DCM (20 ml, 10 V), se añadieron trifluoroacetamida (2,58 g, 22,85 mmol), MgO (1,84 g, 45,68 mmol), Rh2(OAc)4 (252 mg, 0,57 mmol) y Phl(OAC)2 (5,52 g, 17,13 mmol) y la mezcla de reacción se agitó a TA durante la noche. Una vez completada la reacción (monitorizada por TLC), la mezcla de reacción se filtró a través de celite y el filtrado se concentró al vacío. El material bruto resultante se purificó mediante cromatografía ultrarrápida (Biotage Isolera, eluyente: EtOAc al 16-18% en éter de petróleo) para producir el compuesto del título. Rendimiento: 86% (2,8 g, sólido blanquecino). 1H RMN (400 MHz, DMSO- efe): 89,03 (s, 1H), 8,48-8,46 (m, 1H), 7,96-7,93 (m, 1H), 3,91 (s, 3H). CLEM: (Método B) 190,9 (M-CF3CO), Tr. 2,6 min, 96,4 % (Máx).
Intermedio 18: clorhidrato de 1-(4-(met¡lt¡o)fen¡l)p¡peraz¡na

Etapa 1: 4-(4-(metiltio)fenil)piperazina-1-carboxilato de terc-butilo

A una solución agitada desgasificada de (4-bromofenil)(metil)sulfano (5,0 g, 24,6 mmol), 1-Boc piperazina (4,6 g, 24,6 mmol), Davefos (2,63 g, 6,66 mmol, Combi-blocks) y KOtBu (4,7 g, 49,0 mmol) en 1,4 dioxano (10 ml), se añadió Pd(dba)3 (0,45 g, 0,4 mmol) a TA. La mezcla de reacción se calentó con irradiación de microondas a 120 °C durante 15 min. La finalización de la reacción se monitorizó mediante TLC, después la mezcla de reacción se evaporó a 50 °C a presión reducida. A la mezcla bruta resultante se le añadió agua (10 ml) y la capa acuosa se extrajo con EtOAc (2 * 50 ml). La capa orgánica combinada se secó sobre Na2SO4 anhidro y se concentró al vacío. El material bruto resultante se purificó mediante cromatografía ultrarrápida (Biotage Isolera, eluyente: EtOAc al 50 % en éter de petróleo) para producir el compuesto del título. Rendimiento: 88 % (6,0 g, sólido blanquecino). RMN 1H (400 MHz, DMSO-d6): 8 7,25-7,23 (m, 2H), 6,96-6,93 (m, 2H), 3,58-3,57 (m, 4H), 3,12-3,09 (m, 4H), 2,42 (s, 3H), 1,50 (s, 9H). CLEM: (Método A) 309,2 (M+H), Tr. 4,3 min, 98,7 % (Máx).
Etapa 2: clorhidrato de 1-(4-(metiltio)fenil)piperazina

A una solución agitada de 4-(4-(metiltio)fenil)piperazina-1-carboxilato de terc-butilo (6,0 g, 19,41 mmol) en 1,4 dioxano (20 ml), se añadió una solución de HCl en dioxano (4 M, 20 ml) a 0 °C y la mezcla de reacción se agitó durante 4 h a TA. La finalización de la reacción se monitorizó por TLC, después la mezcla de reacción se evaporó a presión reducida para producir el compuesto del título. Rendimiento: 89 % (4,8 g, sólido blanquecino). RMN 1H (400 MHz, DMSO-d6): 8 7,5 (m, 2H), 7,22-7,16 (m, 2H), 7,06-6,93 (m, 2H), 3,02-2,99 (m, 4H), 2,51-2,38 (m, 4H), 2,38 (s, 3H).
Ejemplo 22: (2-(4-(1-(benzord1tiazol-5-il)etil)piperazin-1-il)pmmidin-5-il)(imino)(metil)-Á6-sulfanona

A una solución agitada del intermedio 6 (0,12 g, 0,51 mmol) en DMF (1,2 ml, 10 V), se le añadieron TEA (0,23 ml, 1,68 mmol) y el intermedio 10 (0,16 g, 5,60 mmol) y la mezcla de reacción se agitó a TA durante la noche. La finalización de la reacción se monitorizó mediante TLC, después la mezcla de reacción se concentró al vacío. El material bruto resultante se purificó mediante cromatografía ultrarrápida (Biotage Isolera, eluyente: EtOAc al 40% en éter de petróleo) para obtener el intermedio puro N-((2-(4-(1-(benzo[d]tiazol-5-il)etilpiperazin-1-il)pirimidin-5-il)(metil)(oxo)-A6-sulfaniliden)-2,2,2-trifluoroacetamida. Rendimiento: 87 % (0,21 g, sólido blanquecino).
A este intermedio, se le añadieron metanol (2,2 ml, 20 V) y K2CO3(0,21 g, 1,68 mmol) y la mezcla resultante se agitó durante 20 min. Después de 20 min, la mezcla de reacción se filtró a través de celite y se concentró al vacío. A la mezcla resultante se le añadió agua (50 ml) y la capa acuosa se extrajo con DCM ( 2 * 100 ml). La capa orgánica combinada se secó sobre Na2SO4 anhidro y se concentró al vacío. El material bruto resultante se purificó mediante cromatografía ultrarrápida (Biotage Isolera, gradiente: metanol al 1-2 % en EtOAc) para producir el compuesto del título. Rendimiento: 15 % (30 mg, sólido blanco). RMN 1H (400 MHz, DMSO-de): 89,38 (s, 1H), 8,65 (s, 2H), 8,11 (d, J= 8,4 Hz, 1H), 8,02 (d, J= 1,2 Hz, 1H), 7,50 (dd, J= 8,2, 1,2 Hz, 1H), 4,22 (s, 1H), 3,85-3,83 (m, 4H), 3,68 (d, J = 6,4 Hz, 1H), 3,06 (d, J= 0,8 Hz, 3H), 2,52-2,32 (m, 4H), 1,41 (d, J = 6,8 Hz, 3H). CLEM: (Método A) 403,3 (M+H), Tr. 1,8 min, 97,5 % (Máx) HPLC: (Método A) Tr. 1,9 min, 95,9 % (Máx).
Ejemplo 23 (2-(4-(1-(benzord1tiazol-5-il)etil)piperazin-1-il)μmmidin-5-il)(etil)(imino)-Á6-sulfanona

A una solución agitada del intermedio 6 (0,25 g, 1,01 mmol) en DMF (2,50 ml, 10 V), se le añadieron TEA (0,4 ml, 3,03 mmol) y el intermedio 13 (0,30 g, 1,01 mmol) y la mezcla de reacción se agitó a TA durante la noche. La finalización de la reacción se monitorizó mediante TLC, después la mezcla de reacción se concentró al vacío. El material bruto resultante se purificó mediante cromatografía ultrarrápida (Biotage Isolera, eluyente: EtOAc al 40 % en éter de petróleo) para obtener el intermedio puro N-((2-(4-(1-(benzo[d]tiazol-5-il)etil)piperazin-1-il)pirimidin-5-il)(etil)(oxo)-A6-sulfaniliden)-2,2,2-trifluoroacetamida. Rendimiento: 88 % (0,45 g, sólido blanquecino).
A este intermedio, se le añadieron metanol (2,5 ml, 20 V) y K2CO3(0,40 g, 3,23 mmol) y la mezcla resultante se agitó durante 20 min. Después de 20 min, la mezcla de reacción se filtró a través de celite y se concentró al vacío. A la mezcla resultante se le añadió agua (50 ml) y la capa acuosa se extrajo con DCM (2 x 100 ml). La capa orgánica combinada se secó sobre Na2SO4 anhidro y se concentró al vacío. El material bruto resultante se purificó mediante cromatografía ultrarrápida (Biotage Isolera, gradiente: metanol al 1-2 % en EtOAc) para producir el compuesto del título. Rendimiento: 18 % (60 mg, sólido blanco). RMN 1H (400 MHz, DMSO- de): 89,38 (s, 1H), 8,58 (s, 2H), 8,12 (d, J= 8,4 Hz, 1H), 8,02 (s, 1H), 7,51-7,49 (m, 1H), 4,22 (s, 1H), 3,85-3,83 (m, 4H), 3,67 (d, J= 6,8 Hz, 1H), 3,12 (t, J = 7,6 Hz, 2H), 2,49-2,39 (m, 4H), 1,40 (d, J = 6,40 Hz, 3H), 1,07 (t, J = 7,20 Hz, 3H). CLEM: (Método A) 417,3 (M+H), Tr.
2,2 min, 99,6 % (Máx). HPLC: (Método A) Tr. 2,0 min, 97,1 % (Máx).
Ejemplo 24: (2-(4-(1-(benzord1tiazol-5-il)etil)piperazin-1-il)μmmidin-5-il)(imino)(proμM)-Á6-sulfanona
A una solución agitada del intermedio 6 (235 mg, 9,50 mmol) en DMF (2,5 ml, 10 V), se añadieron TEA (0,5 ml, 3,8 mmol) y el intermedio 14 (235 mg, 0,95 mmol) a TA y la mezcla de reacción se agitó durante la noche a TA. La finalización de la reacción se monitorizó mediante TLC, después la mezcla de reacción se evaporó a 50 °C al vacío. A la mezcla resultante, se le añadió agua (2 ml) y la capa acuosa se extrajo con EtOAc (2 x 50 ml). La capa orgánica combinada se secó sobre Na2SO4 anhidro y se concentró al vacío. El material bruto resultante se purificó mediante cromatografía ultrarrápida (gel de sílice: malla 230-400, eluyente: EtOAc al 50 % en éter de petróleo) para obtener el intermedio puro N-((2-(4-(1-(benzo[d]tiazol-5-il)etil)piperazin-1-il)pirimidin-5-il)(oxo)(propil)-A6-sulfaniliden)-2,2,2-trifluoroacetamida. Rendimiento: 28 % (l40 mg, sólido blanquecino).
A este intermedio se le añadió metanol (7 ml, 20 V) y K2CO3 (414 mg, 4,53 mmol) y se agitó a TA durante 20 min. Después de 20 min, la mezcla de reacción se filtró a través de celite y se concentró al vacío. A la mezcla resultante, se le añadió agua (20 ml) y la capa acuosa se extrajo con DCM (2 x 100 ml). La capa orgánica combinada se secó sobre Na2SO4 anhidro y se concentró al vacío. El material bruto resultante se purificó mediante cromatografía ultrarrápida (Biotage Isolera, gradiente: metanol al 1-2 % en DCM) para producir el compuesto del título. Rendimiento:
21 % (35 mg, sólido blanquecino). RMN 1H (400 MHz, DMSO-de): 89,39 (s, 1H), 8,59 (s, 2H), 8,13 (d, J= 8,0 Hz, 1H), 8,03 (s, 1H), 7,51 (d, J= 8,4 Hz, 1H), 4,22 (s, 1H), 3,85-3,83 (m, 4H), 3,68 (d, J= 6,0 Hz, 1H), 3,17-3,08 (m, 2H), 2,53 2,44 (m, 4H), 1,57-1,51 (m, 2H), 1,41 (d, J = 6,40 Hz, 3H), 0,88 (t, J = 7,20 Hz, 3H). CLEM: (Método A) 431,3 (M+H), Tr. 2,4 min, 97,2 % (Máx). HPLC: (Método A) Tr. 2,2 min, 97,6 % (Máx).
Ejemplo 25: (2-(4-(1-(benzord1tiazol-5-il)etil)piperazin-1-il)μmmidin-5-il)(metil)(metilimino)-Á6-sulfanona

A una solución agitada del ejemplo 22 (0,1 g, 0,25 mmol) en THF (1,0 ml. 10 V), se añadió NaH (60 %) (18 mg, 0,27 mmol) a 0 °C y se agitó durante 15 min. Después se añadió Mel (0,04 ml, 0,62 mmol) a la mezcla de reacción en un tubo sellado y se calentó durante la noche a 90 °C. La finalización de la reacción se monitorizó mediante TLC, después la mezcla de reacción se concentró al vacío. El material bruto resultante se purificó mediante cromatografía ultrarrápida (Biotage Isolera, gradiente: MeOH al 5-6 % en DCM) y se purificó adicionalmente por HPLC preparativa (Método B) para producir el compuesto del título. Rendimiento: 23 % (23 mg, sólido blanquecino). RMN 1H (400 m Hz , DMSO-de): 89,38 (s, 1H), 8,55 (s, 2H), 8,11 (d, J= 8,4 Hz, 1H), 8,02 (d, J = 1,2 Hz, 1H), 7,50 (dd, J = 8,2, 1,2 Hz, 1H), 3,86 3,70 (m, 4H), 3,69-3,65 (m, 1H), 3,10 (s, 3H), 2,56-2,51 (m, 2H), 2,50-2,32 (m, 5H), 1,41 (d, J= 6,8 Hz, 3H). CLEM:
(Método A) 416,8 (M+H), Tr. 1,94 min, 98,9 % (Máx). HPLC: (Método A) Tr. 1,9 min, 99,7 % (Máx).
Ejemplo 26: (6-(4-(1-(benzord1tiazol-5-il)etil)piperazin-1-il)piridin-3-il)(imino)(metil)-Á6-sulfanona

A una solución agitada del intermedio 6 (350 mg, 1,41 mmol) en DMF (3,5 ml), se le añadió TEA (0,6 ml, 4,25 mmol) y el intermedio 15 (446 mg, 1,56 mmol) a TA y la mezcla de reacción se agitó durante la noche. La finalización de la reacción se monitorizó mediante TLC, después la mezcla de reacción se evaporó a 50 °C al vacío. A la mezcla
resultante, se le añadió agua (10 ml) y la capa acuosa se extrajo con EtOAc (2 * 50 ml). La capa orgánica combinada se secó sobre Na2SO4 anhidro y se concentró al vacío. El material bruto resultante se purificó mediante cromatografía ultrarrápida (gel de sílice: malla 230-400, eluyente: EtOAc al 50 % en éter de petróleo) para obtener el intermedio puro N-((6-(4-(1-(benzo[d]tiazol-5-il)etil)piperazin-1-il)piridin-3-il)(metil)(oxo)-A6-sulfaniliden)-2,2,2-trifluoroacetamida. Rendimiento: 41 % (252 mg, sólido blanquecino).
A este intermedio se le añadió metanol (7 ml, 20 V) y K2CO3(414 mg, 4,53 mmol) y la mezcla resultante se agitó a TA durante 20 min. Después de 20 min, la mezcla de reacción se filtró a través de celite y se concentró al vacío. A la mezcla resultante se le añadió agua (50 ml) y la capa acuosa se extrajo con DCM (2 * 100 ml). La capa orgánica combinada se secó sobre Na2SO4 anhidro y se concentró al vacío. El material bruto resultante se purificó mediante cromatografía ultrarrápida (Biotage Isolera, gradiente: metanol al 1-2 % en DCM) para producir el compuesto del título.
Rendimiento: 30 % (168,89 mg, sólido blanquecino). RMN 1H (400 MHz, DMSO- de): 89,38 (s, 1H), 8,48 (d, J= 2,4 Hz, 1H), 8,12 (d, J = 8,4 Hz, 1H), 8,02 (d, J= 1,2 Hz, 1H), 7,85 (dd, J = 9,2, 2,4 Hz, 1H), 7,49 (dd, J = 6,8, 1,6 Hz, 1H), 6,87 (d, J = 9,2 Hz, 1H), 4,02 (s, 1H), 3,67-3,62 (m, 5H), 3,00 (s, 3H), 2,67-2,33 (m, 4H), 1,41 (d, J = 6,40 Hz, 3H).
CLEM: (Método A) 402,0 (M+H), Tr. 1,8 min, 97,7 % (Máx) HPLC: (Método A) Tr. 1,8 min, 97,6 % (Máx)
Ejemplo 27: (2-(4-(1-(benzord1tiazol-5-il)etil)piperazin-1-il)pirimidin-5-il)(etilimino)(metil)-A6-sulfanona

A una solución agitada del ejemplo 22 (0,12 g, 0,51 mmol) en DMF (1,2 ml, 10 V), se añadió NaH (60 %) (0,23 mg, 1,68 mmol) a 0 °C y se agitó durante 15 min. Después se añadió bromuro de etilo (0,16 g, 5,6 mmol) a la mezcla de reacción y se agitó durante la noche a TA. La finalización de la reacción se monitorizó mediante TLC, después la mezcla de reacción se concentró al vacío. El material bruto resultante se purificó mediante HPLC preparativa (Método B). Rendimiento: 15 % (30 mg, sólido blanco). RMN 1H (400 MHz, DMSO-de): 89,38 (s, 1H), 8,65 (s, 2H), 8,11 (d, J = 8,4 Hz, 1H), 8,02 (d, J= 1,2 Hz, 1H), 7,50 (dd, J= 8,2, 1,2 Hz, 1H), 3,85-3,83 (m, 4H), 3,68 (q, J= 6,4 Hz, 1H), 3,33 3,30 (m, 2H), 3,06 (s, 3H), 2,44-2,33 (m, 4H), 1,41 (d, J = 6,80 Hz, 3H) 1,08 (t, J = 6,4 Hz, 3H). CLEM: (Método A) 431,3 (M+H), Tr. 2,1 min, 99,7 % (Máx). HPLC: (Método A) Tr. 1,9 min, 95,9 % (Máx).
Ejemplo 28: (2-(4-(1-(benzord1tiazol-5-il)etil)piperazin-1-il)pirimidin-5-il)(isopropilimino)(metil)-A6-sulfanona

A una solución agitada del ejemplo 22 (0,15 g, 0,37 mmol) en DMF (3,0 ml, 10 V), se añadió NaH (60 %) (17 mg, 0,746 mmol) a 0 °C y se agitó durante 15 min. Después se añadió bromuro de isopropilo (91 mg, 0,74 mmol) a la mezcla de reacción y la mezcla de reacción se agitó durante la noche a TA. La finalización de la reacción se monitorizó mediante TLC, después la mezcla de reacción se concentró al vacío. El bruto se purificó mediante HPLC preparativa (Método de condición B). Rendimiento: 8 % (12,5 mg, sólido blanco). RMN 1H (400 MHz, DMSO-de): 89,39 (d, J = 2,0 Hz, 1H), 8,58 (d, J = 2,0 Hz, 2H), 8,12 (d, J = 8,8 Hz, 1H), 8,03 (s, 1H), 7,50 (d, J = 8,8 Hz, 1H), 4,11-4,10 (m, 4H), 3,85 (q, J = 6,8 Hz, 1H), 3,18- 3,09 (m, 4H), 2,52-2,33 (m, 4H), 1,42 (d, J = 6,4 Hz, 3H), 1,02 (d, J = 7,2 Hz, 6H). CLEM:
(Método A) 445,0 (M+H), Tr. 2,2 min, 96,5 % (Máx). HPLC: (Método A) Tr. 2,3 min, 97,4 % (Máx)
Ejemplo 29: (2-(4-(1-(Benzord1tiazol-5-il)etil)piperazin-1-il)pirimidin-5-il)((2-metoxietil) imino)(metil)-A6-sulfanona

A una solución agitada del ejemplo 22 (0,15 g, 0,51 mmol) en DMF (3,0 ml, 10 V), se añadió NaH (60 %) (0,13 mg, 0,55 mmol) a 0 °C y se agitó durante 15 min. Después se añadió bromuro de metoximetilo (103 mg, 0,74 mmol) y la mezcla de reacción se agitó durante la noche a TA. La finalización de la reacción se monitorizó mediante TLC, después la mezcla de reacción se concentró al vacío. El bruto resultante se purificó mediante HPLC preparativa (método B).
Rendimiento: 8 % (14,3 mg, sólido blanco). RMN 1H (400 MHz, DMSO- de): 89,38 (s, 1H), 8,59 (s, 2H), 8,11 (d, J = 8,4 Hz, 1H), 8,03 (s, 1H), 7,50 (dd, J = 8,4, 1,2 Hz, 1H), 3,85-3,82 (m, 4H), 3,68 (d, J = 6,4 Hz, 1H), 3,18 (s, 3H), 3,12 (s, 3H), 2,95-2,82 (m, 2H), 2,50-2,33 (m, 4H), 1,40 (d, J = 6,4 Hz, 3H). CLEM: (Método A) 460,9 (M+H), Tr. 2,1 min, 99,1 % (Máx). HPLC: (Método A) Tr. 2,1 min, 99,1 % (Máx).
Ejemplo 30: (6-(4-(1-(benzord1t¡azol-5-¡l)et¡l)p¡peraz¡n-1-¡l)p¡r¡d¡n-3-¡l)(met¡lHmet¡lim¡no)-Á6-sulfanona

A la solución agitada del ejemplo 26 (0,11 g, 0,27 mmol) en THF (2 ml), se añadió NaH (60 %) (0,03 g, 0,55 mmol) a 0 °C y se agitó durante 15 min. Después se añadió Mel (0,05 ml, 0,87 mmol) a la mezcla de reacción y se agitó durante la noche a TA. Una vez completada la reacción (monitorizada por TLC), la mezcla de reacción resultante se inactivó con agua helada (2 ml) y la capa acuosa se extrajo con EtOAc (2x15 ml). La capa orgánica combinada se secó sobre Na2SO4 anhidro y se evaporó al vacío. El material bruto resultante se purificó mediante HPLC preparativa (Método B) para producir el compuesto del título. Rend¡m¡ento: 23 % (27 mg, sólido blanquecino). RMN 1H (400 m Hz , DMSO-de): 89,39 (s, 1H), 8,37 (s, 1H), 8,13 (d, J = 8,4 Hz, 1H), 8,03 (s, 1H), 7,75 (d, J = 9,2 Hz, 1H), 7,51 (d, J = 8,4 Hz, 1H), 6,91 (d, J = 9,2 Hz, 1H), 3,64-3,57 (m, 5H), 3,05 (s, 3H), 2,44-2,41 (m, 7H), 1,42 (d, J = 6,4 Hz, 3H). CLEM: (Método A) 415,8 (M H), Tr. 1,9 min, 97,5 % (Máx) HPLC: (Método A) Tr. 2,1 min, 97,3 % (Máx)
Ejemplo 31: ((2-(4-(1-(benzord1t¡azol-5-¡l)et¡l)p¡peraz¡n-1-¡l)p¡r¡m¡d¡n-5-¡l)¡m¡no)d¡met¡l-Á6-sulfanona

Etapa 1: 5-(1-(4-(5-bromopirimidin-2-il)piperazin-1-il)etil)benzo[d]tiazol

A una solución agitada del intermedio 6 (0,5 g, 2,02 mmol) en DMF (10 ml), se añadieron TEA (0,84 ml, 6,06 mmol) y 5-bromo-2-doropirimidina (0,469 g, 2,42 mmol) a TA y se agitó durante la noche a 90 °C. Una vez completada la reacción (monitorizada por TLC), la mezcla de reacción se evaporó a 45 °C al vacío y la mezcla resultante se disolvió en DCM (10 ml). La capa orgánica se lavó con agua (5 ml), solución de salmuera (5 ml), se secó sobre Na2SO4 anhidro y se evaporó al vacío. El material bruto resultante se purificó mediante cromatografía en columna ultrarrápida (Biotage ¡solera, EtOAC al 60 % en éter de petróleo) para producir el compuesto del título. Rendimiento: 61 % (500 mg, sólido blanco). RMN 1H (400 MHz, DMSO-de): 89,38 (s, 1H), 8,42 (s, 2H), 8,11 (d, J = 8,4 Hz, 1H), 8,01 (s, 1H), 7,49 (d, J = 8,0 Hz, 1H), 3,69-3,64 (m, 5H), 2,40-2,33 (m, 4H), 1,40 (d, J = 6,4 Hz, 3H). CLEM: (Método A) 406,2 (M H), Tr. 3,0 min, 99,9 % (Máx).
Etapa 2: ((2-(4-(1-(benzo[d]tiazol-5-il)etil)pipemzin-1-il)pirimidin-5-il)imino)dimetil-A6-sulfanona

A una solución agitada de 5-(1-(4-(5-bromopirimidin-2-il)piperazin-1-il)etil)benzo[d]tiazol (300 mg, 0,74 mmol) en tolueno seco (6 ml), se añadió Pd(OAc)2 (6,6 mg, 0,03 mmol), Ru-phos (27,7 mg, 0,06 mmol), carbonato de cesio (727 mg, 2,23 mmol) y S,S-dimetil sulfimida (83,2 mg, 0,9 mmol) y se calentó durante la noche a 110 °C. La finalización de la reacción se monitorizó mediante TLC, después la mezcla de reacción se concentró al vacío. El material bruto resultante se purificó mediante cromatografía ultrarrápida (Biotage Isolera, eluyente: metanol al 8-10 % en CHCh) para producir el compuesto del título. Rendimiento: 4 % (10,7 mg, sólido marrón) RMN 1H (400 MHz, DMSO-d6): 89,39 (d, J = 8,0 Hz, 1H), 8,12 (d, J = 8,0 Hz, 1H), 8,03-8,01 (m, 3H), 7,51-7,49 (m, 1H), 3,64- 3,59 (m, 4H), 3,37-3,36 (m, 1H), 3,18 (s, 6H), 2,42-2,34 (m, 4H), 1,42 (d, J = 8,0 Hz, 3H).| CLEM: (Método A) 417,0 (M+H), Tr. 2,1 min, 98,5 % (Máx). HPLC: (Método A) Tr. 2,1 min, 98,8 % (Máx)
Ejemplo 33: (4-(4-(1-(benzord1t¡azol-5-¡l)et¡l)p¡peraz¡n-1-¡l)fen¡lH¡m¡no)(metil)-Á6-sulfanona

Etapa 1: 5-(1-(4-(4-(metiltio)fenil)piperazin-1-il)etil)benzo[d]tiazol

A una solución agitada del intermedio 18 (1,6 g, 6,56 mmol) y TEA (2,76 ml, 19,67 mmol) en DMF (10 ml), se añadió
el Intermedio 1 (1,29 g, 6,56 mmol) a TAy se agitó a 70 °C durante la noche. La finalización de la reacción se monitorizó mediante TLC, después la mezcla de reacción se evaporó a 50 °C al vacío. A la mezcla resultante, se le añadió agua (10 ml) y la capa acuosa se extrajo con EtOAc (2 x 50 ml). La capa orgánica combinada se secó sobre Na2SO4 anhidro y se concentró al vacío. El material bruto resultante se purificó mediante cromatografía ultrarrápida (Biotage Isolera, gradiente: MeOH al 1-2 % en DCM) para producir el compuesto del título. Rendimiento: 25 % (600 mg, sólido blanquecino). RMN 1H (400 MHz, DMSO-de): 89,94 (s, 1H), 8,13 (d, J = 8,4 Hz, 1H), 8,04 (s, 1H), 7,68 (d, J =8,8 Hz, 2H), 7,51 (d, J = 8,4 Hz, 1H), 7,01 (d, J = 9,2 Hz, 2H), 3,68-3,66 (m, 1H), 3,34-3,30 (m, 4H), 2,37 (s, 3H), 2,68-2,34 (m, 4H), 1,42 (d, J = 6,8 Hz, 3H). CLEM: (Método A) 369,9 (M H), Tr. 2,3 min, 83,3 % (Máx)
Etapa 2: 5-(1-(4-(4-(metilsulfinil)fenil)piperazin-1-il)etil)benzo[d]tiazol

A una solución agitada de 5-(1-(4-(4-(metiltio)fenil)piperazin-1-il)etil)benzo[d]tiazol (700 mg, 1,90 mmol) en DCM (7 ml, 10 V) a 0 °C, se añadió en porciones m-CPBA (722 mg, 2,09 mmol) y se agitó durante 1 h a 0 °C. Una vez completada la reacción (monitorizada por TLC), la mezcla de reacción se inactivó con una solución de NaHCO3 al 10 % y la capa acuosa se extrajo con DCM (2 x 100 ml). La capa orgánica combinada se lavó con salmuera (30 ml), se secó sobre Na2SO4 anhidro y se concentró al vacío. El material bruto resultante se purificó mediante cromatografía ultrarrápida (Biotage Isolera, eluyente: EtOAc al 60-70 % en éter de petróleo) para producir el compuesto del título. Rendimiento:
34 % (250 mg, sólido gomoso de color amarillo pálido). RMN 1H (400 MHz, DMSO-de): 89,94 (s, 1H), 8,13 (d, J = 8,4 Hz, 1H), 8,04 (s, 1H), 7,68 (d, J = 8,8 Hz, 2H), 7,51 (d, J = 8,4 Hz, 1H), 7,01 (d, J = 9,2 Hz, 2H), 3,68-3,66 (m, 1H), 3,34-3,30 (m, 4H), 2,65 (s, 3H), 2,68-2,34 (m, 4H), 1,42 (d, J = 6,80 Hz, 3H). CLEM: (Método A) 386,5 (M H), Tr. 1,7 min, 83,3 % (Máx)
Etapa 3: (4-(4-(1-(benzo[d]tiazol-5-il)etil)piperazin-1-il)fenil)(imino)(metil)-Á6-sulfanona

A una solución agitada de 5-(1-(4-(4-(metilsulfinil)fenil)piperazin-1-il)etil)benzo[d]tiazol (250 mg, 0,65 mmol) en DCM (5 ml, 20 V), se añadieron trifluoroacetamida (146 mg, 1,30 mmol), MgO (118 mg, 2,59 mmol), Rh2(OAc)4 (14,32 mg, 0,03 mmol) y Phl(OAc)2 (166 mg, 0,97 mmol) y la mezcla de reacción se agitó durante la noche a TA. La finalización de la reacción se monitorizó por TLC, después la mezcla de reacción se filtró a través de celite y se concentró al vacío. El material bruto resultante se purificó mediante cromatografía ultrarrápida (Biotage Isolera, eluyente: EtOAc al 55 60 % en éter de petróleo) para obtener el intermedio puro N-((4-(4-(1-(benzo[d]tiazol-5-il)etil)piperazin-1-il)fenil)(metil)(oxo)-A6-sulfonamida)-2,2,2-trifluoroacetamida. Rendimiento: 8 % (25 mg, sólido blanquecino).
A este intermedio, se añadieron metanol (10 ml, 20 V) y K2CO3 (89 mg, 0,648 mmol) y se agitó durante 20 min. Después de 20 min, la mezcla de reacción se filtró a través de celite y se concentró al vacío. A la mezcla resultante se le añadió agua (50 ml) y la capa acuosa se extrajo con DCM (2 x 100 ml). La capa orgánica combinada se secó sobre Na2SO4 anhidro y se concentró al vacío. El material bruto resultante se purificó mediante cromatografía ultrarrápida (Biotage Isolera, gradiente: metanol al 1-2 % en DCM) para producir el compuesto del título. Rendimiento: 6 % (4,86 mg, sólido blanquecino). RMN 1H (400 MHz, DMSO-d6): 89,94 (s, 1H), 8,13 (d, J = 8,4 Hz, 1H), 8,04 (s, 1H), 7,68 (d, J = 8,8 Hz, 2H), 7,51 (d, J = 8,4 Hz, 1H), 7,01 (d, J = 9,2 Hz, 2H), 3,89 (s, 1H), 3,68-3,66 (m, 1H), 3,34-3,30 (m, 4H), 2,97 (s, 3H), 2,68-2,34 (m, 4H), 1,42 (d, J = 6,80 Hz, 3H). CLEM: (Método A) 401,0 (M+H), Tr. 1,9 min, 98,2 % (Máx) HPLC: (Método A) Tr. 1,8 min, 96,9% (Máx)
Ejemplo 34: (2-(4-((S)-1-(benzord1tiazol-5-il)etil)piperazin-1-il)pmmidin-5-il)(iminoHmetil)-Á6-sulfanona o (2-(4-((R)-1-(benzord1tiazol-5-il)etil)piperazin-1-il)pirimidin-5-il)(imino)(metil)-Á6-sulfanona

A una solución agitada del intermedio 7 (400 mg, 1,41 mmol) en ACN (5 ml), se añadieron TEA (0,6 ml, 4,23 mmol) y el intermedio 10 (445 mg, 1,54 mmol) a TA y se agitó durante la noche. La finalización de la reacción se monitorizó mediante TLC, después la mezcla de reacción se evaporó a 50 °C al vacío. A la mezcla resultante, se le añadió agua (10 ml) y la capa acuosa se extrajo con EtOAc (2 x 50 ml). La capa orgánica combinada se secó sobre Na2SO4 anhidro y se concentró al vacío. El material bruto resultante se purificó mediante cromatografía ultrarrápida (gel de sílice: malla 230-400, eluyente: EtOAc al 50 % en éter de petróleo) para obtener el intermedio puro N-((6-(4-(1-(benzo[d]tiazol-5-il)etil)piperazin-1-il)piridin-3-il)(metil)(oxo)-A6-sulfaniliden)-2,2,2-trifluoroacetamida. Rendimiento: 39% (273 mg, sólido blanquecino).
A este intermedio, se añadieron metanol (7 ml, 20 V) y K2CO3(414 mg, 4,53 mmol) y se agitó a TA durante 20 min. Después de 20 min, la mezcla de reacción se filtró a través de celite y se concentró al vacío. A la mezcla resultante se añadió agua (50 ml) y la capa acuosa se extrajo con DCM (2 x 100 ml). La capa orgánica combinada se secó sobre Na2SO4 anhidro y se concentró al vacío. El material bruto resultante se purificó mediante cromatografía ultrarrápida (Biotage Isolera, gradiente: metanol al 1-2 % en DCM) para producir el compuesto del título. Rendimiento: 34 % (190 mg, sólido blanquecino). RMN 1H (400 MHz, DMSO-de): 89,39 (s, 1H), 8,65 (s, 2H), 8,12 (d, J =8,4 Hz, 1H), 8,03 (s, 1H), 7,51-7,49 (m, 1H), 4,24 (s, 1H), 3,86-3,83 (m, 4H), 3,69- 3,67 (m, 1H), 3,07 (s, 3H), 2,54-2,39 (m, 4H), 1,41 (d, J = 6,8 Hz, 3H). CLEM: (Método A) 402,8 (M+H), Tr. 1,8 min, 99,7%(Máx) HPLC: (Método A) Tr. 1,8 min, 99,8%(Máx).
Ejemplo 35: (S)-(2-(4-((S)-1-(benzord1tiazol-5-il)etil)piperazin-1-il)μmmidin-5-il)(imino)(metil)-Á6-sulfanonao (R)-(2-(4-((S)-1-(benzord1tiazol-5-il)etil)piperazin-1-il)μmmidin-5-il)(iminoHmetil)-Á6-sulfanona o (S)-(2-(4-((R)-1-(benzord1t¡azol-5-¡l)et¡l)p¡peraz¡n-1-¡l)p¡r¡m¡d¡n-5-¡l)(im¡no)(met¡l)-Á6-sulfanonao (R)-(2-(4-((R)-1-(benzord1tiazol-5-il)etil)piperazin-1-il)μmmidin-5-il)(imino)(metil)-Á6-sulfanona

A una solución agitada del intermedio 7 (400 mg, 1,41 mmol) en ACN (5 ml), se añadieron TEA (0,6 ml, 4,23 mmol) y el intermedio 11 (445 mg, 1,54 mmol) a TA y se agitó durante la noche. La finalización de la reacción se monitorizó mediante TLC, después la mezcla de reacción se evaporó a 50 °C al vacío. A la mezcla resultante, se añadió agua (10 ml) y la capa acuosa se extrajo con EtOAc (2 x 50 ml). La capa orgánica combinada se secó sobre Na2SO4 anhidro y se concentró al vacío. El material bruto resultante se purificó mediante cromatografía ultrarrápida (gel de sílice: malla 230-400, eluyente: EtOAc al 50 % en éter de petróleo) para obtener el intermedio puro. Rendimiento: 39 % (273 mg, sólido blanquecino).
A este intermedio se le añadió metanol (7 ml, 20 V) y K2CO3 (414 mg, 4,53 mmol) y se agitó a TA durante 20 min. Después de 20 min, la mezcla de reacción se filtró a través de celite y se concentró al vacío. A la mezcla resultante, se le añadió agua (50 ml) y la capa acuosa se extrajo con DCM (2 x 100 ml). La capa orgánica combinada se secó sobre Na2SO4 anhidro y se concentró al vacío. El material bruto resultante se purificó mediante cromatografía
ultrarrápida (Biotage Isolera, gradiente: metanol al 1-2 % en DCM) para producir el compuesto del título. Rendimiento:
10 % (15 mg, sólido blanquecino). RMN 1H (400 MHz, DMSO-de): 89,39 (s, 1H), 8,65 (s, 2H), 8,13 (d, J =8,0 Hz, 1H), 8,03 (s, 1H), 7,51-7,49 (m, 1H), 4,24 (s, 1H), 3,86-3,83 (m, 4H), 3,69-3,67 (m, 1H), 3,07 (s, 3H), 2,54-2,39 (m, 4H), 1,41 (d, J = 6,8 Hz, 3H). CLEM: (Método A) 403,3 (M H), Tr. 1,8 min, 99,6 % (Máx) HPLC: (Método A) Tr. 1,8 min, 99,2 % (Máx) SFC quiral: (Método B) Tr. 9,3 min, 99,9 % (Máx). [a]25D = -107,69, c 0,104 (MeOH).
Ejemplo 36: (S)-(2-(4-((S)-1-(benzord1tiazol-5-il)etil)piperazin-1-il)pirimidin-5-il)(imino)(metil)-Á6-sulfanonao (R)-(2-(4-((S)-1-(benzord1tiazol-5-il)etil)piperazin-1-il)pirimidin-5-il)(iminoHmetil)-Á6-sulfanona o (S)-(2-(4-((R)-1-(benzord1tiazol-5-il)etil)piperazin-1-il)pirimidin-5-il)(imino)(metil)-Á6-sulfanonao (R)-(2-(4-((R)-1-(benzord1tiazol-5-il)etil)piperazin-1-il)pirimidin-5-il)(imino)(metil)-Á6-sulfanona

A una solución agitada del intermedio 7 (400 mg, 1,41 mmol) en ACN (5 ml), se añadieron TEA (0,6 ml, 4 ,23 mmol) y el intermedio 12 (445 mg, 1,54 mmol) y la mezcla de reacción se agitó durante la noche a TA. La finalización de la reacción se monitorizó mediante TLC, después la mezcla de reacción se evaporó a 50 °C al vacío. A la mezcla resultante, se añadió agua (10 ml) y la capa acuosa se extrajo con EtOAc (2 x 50 ml). La capa orgánica combinada se secó sobre Na2SO4 anhidro y se concentró al vacío. El material bruto resultante se purificó mediante cromatografía ultrarrápida (gel de sílice: malla 230-400, eluyente: EtOAc al 50 % en éter de petróleo) para obtener el intermedio puro.
Rendimiento: 39 % (273 mg, sólido blanquecino).
A este intermedio, se añadieron metanol (7 ml, 20 V) y K2CO3(414 mg, 4,53 mmol) y se agitó durante 20 min. Después de 20 min, la mezcla de reacción se filtró a través de celite y se concentró al vacío. A la mezcla resultante se añadió agua (50 ml) y la capa acuosa se extrajo con DCM (2 x 100 ml). La capa orgánica combinada se secó sobre Na2SO4 anhidro y se concentró al vacío. El material bruto resultante se purificó mediante cromatografía ultrarrápida (Biotage Isolera, gradiente: metanol al 1-2 % en DCM) para producir el compuesto del título. Rendimiento: 14 % (22 mg, sólido blanquecino). RMN 1H (400 MHz, DMSO-de): 89,39 (t, J = 2,0 Hz, 1H), 8,65 (t, J = 2,0 Hz, 2H), 8,12 (d, J = 8,4 Hz, 1H), 8,03 (s, 1H), 7,50 (d, J = 8,4 Hz, 1H), 4,24 (s, 1H), 3,84-3,82 (m, 4H), 3,68 (d, J= 6,4 Hz, 1H), 3,07 (s, 3H), 2,51 2,34 (m, 4H), 1,41 (d, J = 6,4 Hz, 3H). CLEM: (Método A) 403,3 (M+H), Tr. 1,8 min, 93,9 % (Máx) HPLC: (Método A) Tr. 1,9 min, 94,5 % (Máx) SFC quiral: (Método B) Tr. 10,2 min, 98,8 % (Máx). [a]25d = -18,64, c 0,103 (MeOH).
Ejemplo 37: (S)-(2-(4-((S)-1-(benzord1tiazol-5-il)etil)piperazin-1-il)pirimidin-5-il)(metilHmetilimino)-Á6-sulfanona o (R)-(2-(4-((S)-1-(benzord1tiazol-5-il)etil)piperazin-1-il)pirimidin-5-il)(metilHmetilimino)-Á6-sulfanona o (S)-(2-(4-((R)-1-(benzord1tiazol-5-il)etil)piperazin-1-il)pirimidin-5-il)(metil)(metilimino)-Á6-sulfanona o (R)-(2-(4-((R)-1-(benzord1tiazol-5-il)etil)piperazin-1-il)pirimidin-5-il)(metil)(metilimino)-Á6-sulfanona

A una solución agitada del ejemplo 35 (150 mg, 0,372 mmol) en DMF (5 ml), se añadió NaH (60 %) (35,79 mg, 0,74 mmol) a 0 °C y se agitó durante 15 min. Después se añadió yodometano (0,05 ml, 0,74 mmol) a la mezcla de reacción y se agitó a t A durante 2 h. Una vez completada la reacción (monitorizada por TLC), la mezcla de reacción se inactivó con agua helada (10 ml) y la capa acuosa se extrajo con EtOAc (2 x 50 ml). La capa orgánica combinada se secó sobre Na2SO4 anhidro y se concentró al vacío. El material bruto resultante se purificó mediante cromatografía ultrarrápida (Biotage Isolera, gradiente: metanol al 1-2 % en DCM) para producir el compuesto del título. Rendimiento:
27 % (41,2 mg, sólido blanquecino). RMN 1H (400 MHz, DMSO-cfe): 89,39 (s, 1H), 8,55 (s, 2H), 8,12 (d, J = 8,0 Hz, 1H), 8,03 (s, 1H), 7,51 (d, J = 1,6, 8,0 Hz, 1H), 3,87-3,84 (m, 4H), 3,71-3,66 (m, 1H), 3,11 (s, 3H), 2,56-2,42 (m, 7H), 1,41 (d, J = 6,8 Hz, 3H). CLEM: (Método A) 417,0 (M+H), Tr. 1,9 min, 98,1 % (Máx) HPLC: (Método A) Tr. 1,9 min, 98,5 % (Máx)
Ejemplo 38: (R)-(2-(4-(1-(benzord1t¡azol-5-¡l)et¡l)p¡peraz¡n-1-¡l)p¡r¡m¡d¡n-5-¡l)(¡mino)(met¡l)-Á6-sulfanona o (S)-(2-(4-(1-(benzord1t¡azol-5-¡l)et¡l)p¡peraz¡n-1-¡l)p¡r¡m¡d¡n-5-¡l)(¡mino)(met¡l)-Á6-sulfanona

Etapa 1: N-((R)-(2-(4-(1-(benzo[d]tiazol-5-il)etil)piperazin-1-il)pirimidin-5-il)(metil)(oxo)-A6-sulfaniliden)-2,2,2-trifluoroacetamida o N-((S)-(2-(4-(1-(benzo[d]tiazol-5-il)etil)piperazin-1-il)pirimidin-5-il)(metil)(oxo)-A6-sulfaniliden)-2,2,2-trifluoroacetamida
A una solución agitada del intermedio 6 (0,47 g, 1,46 mmol) en ACN (2,0 ml), se añadieron TEA (0,88 ml, 5,8 mmol) y el intermedio 12 (464 mg 1,6 mmol) y la mezcla de reacción se agitó a TA durante 30 minutos. La finalización de la reacción se monitorizó mediante TLC, después la mezcla de reacción se concentró al vacío. El material bruto resultante se purificó mediante cromatografía ultrarrápida (Biotage Isolera, EtOAc al 60-80 % en éter de petróleo) para producir el compuesto del título. Rendimiento: 44 % (320 mg, sólido blanco). RMN 1H (400 MHz, DMSO-d6): 89,39 (s, 1H), 8,75 (s, 2H), 8,13 (d, J = 8,4 Hz, 1H), 8,04 (s, 1H), 7,51 (d, J= 8,4 Hz, 1H), 3,89 (t, J = 4,8 Hz, 4H), 3,76 (s, 3H), 3,70 (d, J = 6,8 Hz, 1H), 2,58-2,43 (m, 4H), 1,41 (d, J = 6,4 Hz, 3H). CLEM: (Método A) 268 (M+H), Tr. 1,9 min, 92,8 % (Máx). HPLC: (Método A) Tr. 3,8 min, 96,1 % (Máx).
Etapa 2: (R)-(2-(4-(1-(benzo[d]tiazol-5-il)etil)piperazin-1-il)pirimidin-5-il)(imino)(metil)-A6-sulfanona o (S)-(2-(4-(1-(benzo[d]tiazol-5-il)etil)piperazin-1-il)pirimidin-5-il)(imino)(metil)-A6-sulfanona
A una solución agitada del producto de la etapa 1 (310 mg, 0,62 mmol) en MeOH (2 ml) y DCM (1 ml), se le añadió K2CO3 (200 mg, 1,0 mmol) y se agitó durante 1 h. La finalización de la reacción se monitorizó mediante TLC, después la mezcla de reacción se concentró al vacío. El material bruto resultante se purificó mediante cromatografía ultrarrápida (Biotage Isolera, gradiente: metanol al 3-4 % en DCM) para producir el compuesto del título. Rendimiento: 84 % (210 g, sólido blanco). RMN 1H (400 MHz, DMSO-d6): 89,38 (s, 1H), 8,64 (s, 2H), 8,11 (d, J = 8,4 Hz, 1H), 8,02 (s, 1H), 7,49 (dd, J= 8,2, 1,2 Hz, 1H), 4,23 (s, 1H), 3,84 (t, J= 4,8 Hz, 4H), 3,06 (s, 3H), 2,54-2,41 (m, 4H), 1,40 (d, J = 6,4 Hz, 3H). CLEM: (Método A) 403,1 (M+H), Tr. 1,6 min, 99,9 % (Máx). HPLC: (Método A) Tr. 1,9 min, 99,7 % (Máx).
Ejemplo 39: (R)-(2-(4-(1-(benzord1t¡azol-5-¡l)et¡l)p¡peraz¡n-1-¡l)p¡r¡m¡d¡n-5-¡l)(¡m¡noHmetil)-Á6-sulfanona o (S)-(2-(4-(1-(benzord1t¡azol-5-¡l)et¡l)p¡peraz¡n-1-¡l)p¡r¡m¡d¡n-5-¡l)(¡m¡noHmetil)-Á6-sulfanona

A una solución agitada del intermedio 6 (0,47 g, 1,46 mmol) en ACN (2,0 ml), se añadieron TEA (0,88 ml, 5,80 mmol) y el intermedio 11 (464 mg 1,60 mmol) a TA y la mezcla de reacción se agitó durante 30 min a TA. La finalización de la reacción se monitorizó mediante TLC, después la mezcla de reacción se concentró al vacío. El material bruto resultante se purificó mediante cromatografía ultrarrápida (Biotage Isolera, EtOAc al 60-80 % en éter de petróleo) para producir el intermedio puro N-((R)-(2-(4-(1 -(benzo[d]tiazol-5-il)etil)piperazin-1-il)pirimidin-5-il)(metil)(oxo)-A6-sulfanilideno)-2,2,2-trifluoroacetamida o N-((S)-(2-(4-(1-(benzo[d]tiazol-5-il)etil)piperazin-1-il)pirimidin-5-il)(metil)(oxo)-A6-sulfanilideno)-2,2,2-trifluoroacetamida. Rend¡m¡ento: 44 % (320 mg, sólido blanco). RMN 1H (400 MHz, DMSO-de): 8 9,39 (s, 1H), 8,75 (s, 2H), 8,13 (d, J =8,4 Hz, 1H), 8,04 (s, 1H), 7,51 (d, J = 8,4 Hz, 1H), 3,89 (t, J = 4,8 Hz, 4H), 3,76 (s, 3H), 3,70 (d, J = 6,8 Hz, 1H), 2,58-2,43 (m, 4H), 1,41 (d, J = 6,4 Hz, 3H). CLEM: (Método A) 403. 1,0 (M+H), Tr.
1,9 min, 92,8 % (Máx). HPLC: (Método A) Tr. 3,8 min, 96,1 % (Máx).
A una solución agitada de este intermedio (310 mg, 0,62 mol) en MeOH (2 ml) y DCM (1 ml), se añadió K2CO3(200 mg, 1,0 mol) y se agitó durante 1 h a TA. La finalización de la reacción se monitorizó mediante TLC, después la mezcla de reacción se concentró al vacío. El material bruto resultante se purificó mediante cromatografía ultrarrápida (Biotage Isolera, gradiente: metanol al 3-4 % en DCM) para producir el compuesto del título. Rend¡m¡ento: 84 % (210 g, sólido blanco). RMN 1H (400 MHz, DMSO- de): 89,38 (s, 1H), 8,64 (s, 2H), 8,11 (d, J = 8,4 Hz, 1H), 8,02 (s, 1H), 7,49 (dd, J= 8,2, 1,2 Hz, 1H), 4,23 (s, 1H), 3,84 (t, J= 4,8 Hz, 4H), 3,06 (s, 3H), 2,54-2,41 (m, 4H), 1,40 (d, J = 6,4 Hz, 3H).
CLEM: (Método A) 403,1 (M+H), Tr. 1,6 min, 99,9 % (Máx). HPLC: (Método A) Tr. 1,9 min, 99,7 % (Máx).
Ejemplo 40: (S)-(2-(4-((R)-1-(benzord1t¡azol-5-¡l)et¡l)p¡peraz¡n-1-¡l)p¡r¡m¡d¡n-5-¡l)(¡m¡noHmet¡l)-Á6-sulfanona o (R)-(2-(4-((R)-1-(benzord1t¡azol-5-¡l)et¡l)p¡peraz¡n-1-¡l)p¡r¡m¡d¡n-5-¡l)(¡m¡noHmet¡l)-Á6-sulfanona o (S)-(2-(4-((S)-1-(benzord1t¡azol-5-¡l)et¡l)p¡peraz¡n-1-¡l)p¡r¡m¡d¡n-5-¡l)(¡m¡no)(met¡l)-Á6-sulfanona o (R)-(2-(4-((S)-1-(benzord1t¡azol-5-¡l)et¡l)p¡peraz¡n-1-¡l)p¡r¡m¡d¡n-5-¡l)(¡m¡no)(met¡l)-Á6-sulfanona

La mezcla de dos enantiómeros obtenida del ejemplo 39 se separó por SFC (Método H: amoníaco 20 mM en metanol, columna: YMC Cellulose C). El primer pico de elución se concentró para producir el compuesto del título.
Rend¡m¡ento: 21 % (35 mg, sólido blanquecino). RMN 1H: (400 MHz, DMSO-de): 89,38 (s, 1H), 8,65 (s, 2H), 8,12 (d, J= 8,0 Hz, 1H), 8,02 (s, 1H), 7,49 (d, J = 8,0 Hz, 1H), 4,23 (s, 1H), 3,84 (d, J = 4,4 Hz, 4H), 3,67 (d, J = 6,4 Hz, 1H), 3,06 (s, 3H), 2,44-2,40 (m, 2H), 1,41 (d, J = 6,40 Hz, 3H). CLEM: (Método A) 403,1 (M+H), Tr. 1,6 min, 99,3 % (Máx).
HPLC: (Método A) Tr. 1,8 min, 98,9 % (Máx). SFC qu¡ral: (Método B) Tr. 8,1 min, 100% (Máx).
Ejemplo 41: (S)-(2-(4-((R)-1-(benzord1t¡azol-5-¡l)et¡l)p¡peraz¡n-1-¡l)p¡r¡m¡d¡n-5-¡l)(¡m¡noHmet¡l)-Á6-sulfanona o (R)-(2-(4-((R)-1-(benzord1t¡azol-5-¡l)et¡l)p¡peraz¡n-1-¡l)p¡r¡m¡d¡n-5-¡l)(¡m¡noHmet¡l)-Á6-sulfanona o (S)-(2-(4-((S)-1-
(benzord1t¡azol-5-¡l)et¡l)p¡peraz¡n-1-¡l)p¡r¡m¡d¡n-5-il)(¡m¡no)(met¡l)-Á6-sulfanona o (R)-(2-(4-((S)-1-(benzord1tiazol-5-¡l)et¡l)p¡peraz¡n-1-¡l)p¡r¡m¡d¡n-5-¡l)(¡m¡no)(metil)-Á6-sulfanona
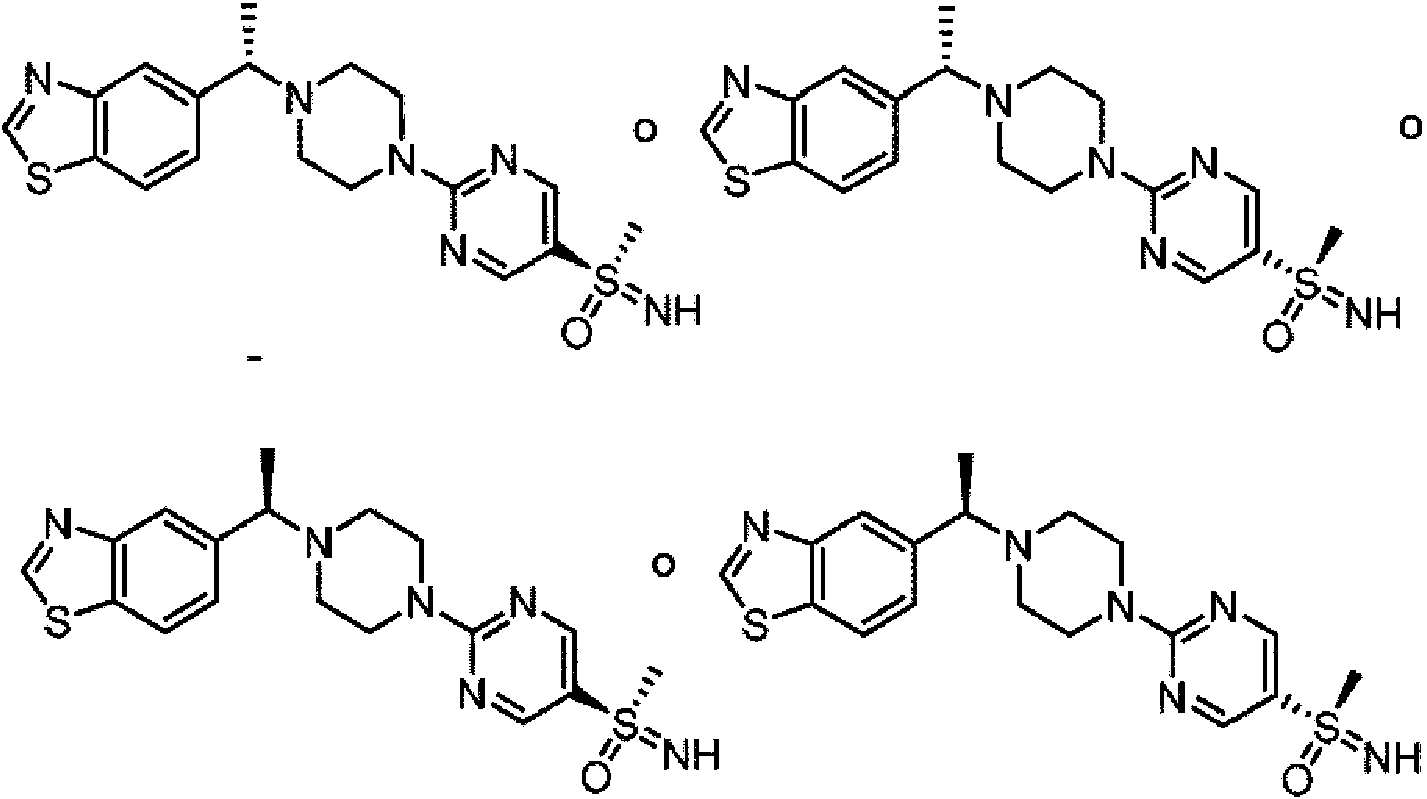
La mezcla de dos enantiómeros del ejemplo 38 se separó por SFC (Método H: amoníaco 20 mM en metanol, columna: YMC Cellulose C). El primer pico de elución se concentró para producir el compuesto del título. Rend¡m¡ento: 28 % (46 mg, sólido blanquecino). RMN 1H: (400 MHz, DMSO-de): 89,39 (d, J= 1,6 Hz, 1H), 8,66 (d, J= 1,6 Hz, 2H), 8,13 (q, J = 1,6 Hz, 1H), 8,03 (s, 1H), 7,51 (d, J= 8,4 Hz, 1H), 4,25 (s, 1H), 3,85 (m, 4H), 3,69 (d, J = 6,8 Hz, 1H), 3,08 (s, 3H), 2,45-2,34 (m, 2H), 1,42 (d, J = 6,40 Hz, 3H). CLEM: (Método A) 403,1 (M+H), Tr. 1,6 min, 99,7 % (Máx). HPLC:
(Método A) Tr. 1,9 min, 99,5 % (Máx). SFC qu¡ral: (Método B) Tr. 9,33 min, 100 % (Máx).
Ejemplo 42: lm¡no(met¡lH2-(4-(1-(2-met¡lbenzord1t¡azol-5-¡l)et¡l)p¡peraz¡n-1-¡l)p¡r¡m¡d¡n-5-¡l)-Á6-sulfanona
A una solución agitada del intermedio 8 (0,88 g, 3,80 mmol) en DMF (11,0 ml, 10 V), se le añadieron TEA (1,6 ml, 11,41 mmol) y el intermedio 10 (1,1 g, 3,80 mmol) y la mezcla de reacción se agitó durante la noche a TA. La finalización de la reacción se monitorizó mediante t Lc , después la mezcla de reacción se concentró al vacío. El material bruto resultante se purificó mediante cromatografía ultrarrápida (Biotage Isolera, eluyente: EtOAc al 60 % en éter de petróleo) para producir el intermedio puro 2,2,2-trifluoro-N-(metil(2-(4-(1-(2-metilbenzo[d]tiazol-5-il)etil)piperazin-1-il)pirimidin-5-il)(oxo)-A6-sulfanilideno)acetamida. Rend¡m¡ento: 22 % (246 mg, sólido blanco).
A este intermedio se le añadieron metanol (22,0 ml, 20 V) y K2CO3 (1,46 g, 11,41 mmol) y se agitó durante 20 min. Después de 20 min, la mezcla de reacción se filtró a través de celite y se concentró al vacío. A la mezcla resultante se añadió agua (50 ml) y la capa acuosa se extrajo con DCM (2 x 100 ml). La capa orgánica combinada se secó sobre Na2SO4 anhidro y se concentró al vacío. El material bruto resultante se purificó mediante cromatografía ultrarrápida (Biotage Isolera, gradiente: metanol al 1-2 % en EtOAc) para producir el compuesto del título. Rend¡m¡ento: 23 % (15 mg, sólido blanco). RMN 1H (400 MHz, DMSO-de): 88,65 (s, 2H), 7,97 (d, J = 8,0 Hz, 1H), 7,84 (s, 1H), 7,38 (d, J= 8,4 Hz, 1H), 4,23 (s, 1H), 3,84 (t, J= 4,8 Hz, 4H), 3,63 (d, J = 6,8 Hz, 1H), 3,06 (s, 3H), 2,60 (s, 3H), 2,43-2,39 (m, 4H), 1,39 (d, J = 6,8 Hz, 3H). CLEM: (Método A) 417,3 (M+H), Tr. 2,1 min, 97,3 % (Máx) HPLC: (Método A) Tr. 2,2 min, 97,1 % (Máx).
Ejemplo 43: et¡l(¡m¡noH2-(4-(1-(2-met¡lbenzord1t¡azol-5-¡l)et¡l)p¡peraz¡n-1-¡l)p¡r¡m¡d¡n-5-¡l)-Á6-sulfanona

A una solución agitada del intermedio 8 (0,25 g, 1,01 mmol) en DMF (2,50 ml, 10 V), se añadieron TEA (0,4 ml, 3,03 mmol) y el intermedio 13 (0,30 g, 1,01 mmol) a TA y se agitó durante la noche. La finalización de la reacción se monitorizó mediante TLC, después la mezcla de reacción se concentró al vacío. El material bruto resultante se purificó mediante cromatografía ultrarrápida (Biotage Isolera, eluyente: EtOAc al 40 % en éter de petróleo) para producir el intermedio N-(etil(2-(4-(1-(2-metilbenzo[d]tiazol-5-il)etil)piperazin-1-il)pirimidin-5-il)(oxo)-A6-sulfaniliden)-2,2,2-trifluoroacetamida. Rendimiento: 94 % (0,48 g, sólido gomoso de color amarillo pálido).
A este intermedio se le añadieron metanol (2,5 ml, 20 V) y K2CO3 (0,40 g, 3,23 mmol) y se agitó a TA durante 20 min. Después de 20 min, la mezcla de reacción se filtró a través de celite y se concentró al vacío. A la mezcla resultante se añadió agua (50 ml) y la capa acuosa se extrajo con DCM (2 x 100 ml). La capa orgánica combinada se secó sobre Na2SO4 anhidro y se concentró al vacío. El material bruto resultante se purificó mediante cromatografía ultrarrápida (Biotage Isolera, gradiente: metanol al 1-2% en EtOAc) para producir el compuesto del título. Rendimiento: 5 % (20 mg, sólido blanco). RMN 1H (400 MHz, DMSO-de): 88,58 (s, 2H), 7,97 (d, J = 8,4 Hz, 1H), 7,84 (s, 1H), 7,38 (d, J= 7,6 Hz, 1H), 4,22 (s, 1H), 3,83 (t, J = 4,4 Hz, 4H), 3,65- 3,60 (m, 1H), 3,17-3,08 (m, 2H), 2,78 (s, 3H), 2,52-2,32 (m, 4H), 1,38 (d, J = 6,4 Hz, 3H), 1,07 (t, J = 7,2 Hz, 3H). CLEM: (Método A) 431,3 (M+H), Tr. 2,5 min, 98,2 % (Máx). HPLC:
(Método A) Tr. 2,2 min, 98,3 % (Máx).
Ejemplo 44: lmino(2-(4-(1-(2-metilbenzord1tiazol-5-il)etil)piperazin-1-il)μmmidin-5-il)(proμM)-Á6-sulfanona
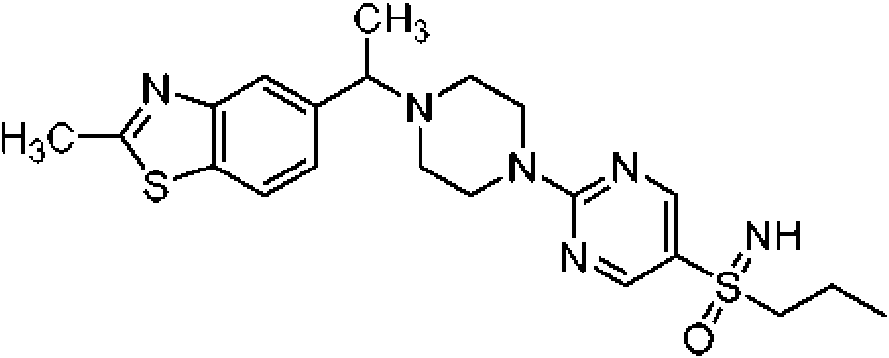
A una solución agitada del intermedio 8 (249 mg, 9,50 mmol) en DMF (2,5 ml), se le añadieron TEA (0,5 ml, 3,80 mmol) y el intermedio 14 (300 mg, 9,50 mmol) a TA y se agitó durante la noche. La finalización de la reacción se monitorizó mediante TLC, después la mezcla de reacción se evaporó a 50 °C al vacío. A la mezcla resultante, se añadió agua (2 ml) y la capa acuosa se extrajo con EtOAc (2 x 50 ml). La capa orgánica combinada se secó sobre Na2SO4 y se concentró al vacío. El material bruto resultante se purificó mediante cromatografía ultrarrápida (gel de sílice: malla 230-400, eluyente: EtOAc al 50 % en éter de petróleo) para producir el intermedio puro 2,2,2-trifluoro-N-((2-(4-(1-(2-metilbenzo[d]tiazol-5-il)etil)piperazin-1-il)pirimidin-5-il)(oxo)(propil)-A6-sulfaniliden)acetamida.
Rendimiento: 27 % (136 mg, sólido blanquecino).
A este intermedio se añadieron metanol (7 ml, 20 V) y K2CO3(414 mg, 4,53 mmol) y se agitó durante 20 min. Después de los 20 min, la mezcla de reacción se filtró a través de celite y se concentró al vacío. A la mezcla resultante, se añadió agua (20 ml) y la capa acuosa se extrajo con DCM (2 x 100 ml). La capa orgánica combinada se secó sobre Na2SO4 anhidro y se concentró al vacío. El material bruto resultante se purificó mediante cromatografía ultrarrápida (Biotage Isolera, gradiente: metanol al 1-2 % en DCM) para producir el compuesto del título. Rendimiento: 18 % (26,5 mg, sólido blanquecino). RMN 1H (400 MHz, DMSO-de): 88,59 (s, 2H), 7,97 (d, J= 8,0 Hz, 1H), 7,85 (s, 1H), 7,38 (d, J= 8,4 Hz, 1H), 4,22 (s, 1H), 3,85-3,83 (m, 4H), 3,66-3,61 (m, 1H), 3,12-3,08 (m, 2H), 2,79 (s, 3H), 2,49-2,39 (m, 2H), 1,57-1,51 (m, 2H), 1,39 (d, J = 6,40 Hz, 3H), 0,88 (t, J = 7,20 Hz, 3H). CLEM: (Método A) 445,2 (M+H), Tr. 2,2 min, 99,7 % (Máx). HPLC: (Método A) Tr. 2,4 min, 99,7 % (Máx).
Ejemplo 45: metil(2-(4-(1-(2-metilbenzord1tiazol-5-il)etil)piperazin-1-il)μmmidin-5-il)(metilimino)-Á6-sulfanona

A una solución agitada del ejemplo 42 (0,15 g, 0,36 mmol) en THF (1,5 ml, 10 V), se añadió NaH (60 %) (26 mg, 0,54 mmol) a 0 °C y se agitó durante 15 min. Después se añadió Mel (0,05 ml, 0,9 mmol) a la mezcla de reacción en un tubo sellado y se calentó durante la noche a 90 °C. Una vez completada la reacción (monitorizada por TLC), la mezcla de reacción se concentró al vacío y el material bruto resultante se purificó mediante cromatografía ultrarrápida (Biotage Isolera, eluyente: metanol al 5-6 % en DCM). El material obtenido se purificó adicionalmente mediante HPLC prep. (Método B) para producir el compuesto del título. Rendimiento: 9 % (13 mg, sólido blanquecino). RMN 1H (400 MHz, DMSO-de): 88,55 (s, 2H), 7,97 (d, J = 8,4 Hz, 1H), 7,84 (s, 1H), 7,39 (d, J = 8,4 Hz, 1H), 3,85-3,84 (m, 4H), 3,64-3,62 (m, 1H), 3,10 (s, 3H), 2,78 (s, 3H), 2,54-2,53 (m, 2H), 2,46 (s, 3H), 2,44-2,42 (m, 2H), 1,39 (d, J = 6,8 Hz, 3H). CLEM:
(Método A) 430,8 (M+H), Tr. 2,2 min, 98,7 % (Máx). HPLC: (Método A) Tr. 2,1 min, 99,3 % (Máx)
Ejemplo 46: ¡m¡no(met¡lH6-(4-(1-(2-met¡lbenzord1t¡azol-5-¡l)et¡l)p¡peraz¡n-1-¡l)p¡r¡d¡n-3-¡l)-Á6-sulfanona

A una solución agitada del intermedio 8 (350 mg, 1,34 mmol) en DMF (3,5 ml), se añadieron TEA (0,6 ml, 4,02 mmol) y el intermedio 15 (422 mg, 1,47 mmol) a TA y se agitó durante la noche. La finalización de la reacción se monitorizó mediante TLC, después la mezcla de reacción se evaporó a 50 °C al vacío. A la mezcla resultante, se añadió agua (10 ml) y la capa acuosa se extrajo con EtOAc (2 x 50 ml). La capa orgánica combinada se secó sobre Na2SO4 anhidro y se concentró al vacío. El material bruto resultante se purificó mediante cromatografía ultrarrápida (gel de sílice: malla 230-400, eluyente: EtOAc al 50 % en éter de petróleo) para producir el intermedio puro 2,2,2-trifluoro-N-(metil(6-(4-(1-(2-metilbenzo[d]tiazol-5-il)etil)piperazin-1-il)piridin-3-il)(oxo)-A6-sulfaniliden)acetamida. Rend¡m¡ento: 40% (241 mg, sólido blanquecino).
A este intermedio se añadieron metanol (7 ml, 20 V) y K2CO3 (414 mg, 4,53 mmol) y se agitó durante 20 min. Después de 20 min, la mezcla de reacción se filtró a través de celite y se concentró al vacío. A la mezcla resultante se añadió agua (50 ml) y la capa acuosa se extrajo con DCM (2 x 100 ml). La capa orgánica combinada se secó sobre Na2SO4 anhidro y se concentró al vacío. El material bruto resultante se purificó mediante cromatografía ultrarrápida (Biotage Isolera, gradiente: metanol al 1-2 % en DCM) para producir el compuesto del título. Rend¡m¡ento: 30 % (163,7 mg, sólido blanquecino). RMN 1H (400 MHz, DMSO-de): 88,48 (d, J = 2,4 Hz, 1H), 7,96 (d, J = 8,4 Hz, 1H), 7,86-7,84 (m, 2H), 7,38 (dd, J= 8,0, 1,2 Hz, 1H), 6,87 (d, J= 9,2 Hz, 1H), 4,02 (s, 1H), 3,63-3,59 (m, 5H), 3,00 (s, 3H), 2,78 (s, 3H), 2,54-2,49 (m, 2H), 2,43-2,37 (m, 2H), 1,38 (d, J= 6,8 Hz, 3H). CLEM: (Método A) 415,8 (M H), Tr. 2,1 min, 99,0 % (Máx) HPLC: (Método A) Tr. 2,1 min, 99,2 % (Máx)
Ejemplo 47: met¡l(6-(4-(1-(2-met¡lbenzord1t¡azol-5-¡l)et¡l)p¡peraz¡n-1-¡l)p¡r¡d¡n-3-¡l)(met¡l¡m¡no)-Á6-sulfanona

A la solución agitada del ejemplo 46 (0,11 g, 0,26 mmol) en THF (2 ml), se añadió NaH (60 %) (0,03 g, 0,52 mmol) a 0 °C y se agitó durante 15 min. Después se añadió Mel (0,05 ml, 0,79 mmol) a la mezcla de reacción y se agitó a TA durante la noche. Una vez completada la reacción (monitorizada por TLC), la mezcla de reacción se inactivó con agua helada (2 ml) y la capa acuosa se extrajo con EtOAc (2x15 ml). La capa orgánica combinada se secó sobre Na2SO4 anhidro y se evaporó al vacío. El material bruto resultante se purificó mediante HPLC prep. (Método B) para producir el compuesto del título. Rendimiento: 15 % (17 mg, sólido blanquecino). RMN 1H (400 MHz, DMSO-cfe): 88,37 (d, J = 2,4 Hz, 1H), 7,97 (d, J = 8,4 Hz, 1H), 7,85 (s, 1H), 7,76-7,73 (m, 1H), 7,39 (d, J= 8,4 Hz, 1H), 6,91 (d, J = 9,2 Hz, 1H), 3,63-3,58 (m, 5H), 3,04 (s, 3H), 2,79 (s, 3H), 2,51-2,48 (m, 2H), 2,44 (s, 3H), 2,44-2,40 (m, 2H), 1,39 (d, J= 6,80 Hz, 3H). CLEM: (Método A) 429,8 (M+H), Tr. 2,2 min, 96,2 % (Máx). HPLC: (Método A) Tr. 2,1 min, 99,6 % (Máx)
Ejemplo 48: (et¡l¡m¡noHmet¡lH2-(4-(1-(2-met¡lbenzord1t¡azol-5-¡l)et¡l)p¡peraz¡n-1-¡l)p¡r¡m¡d¡n-5-¡l)-Á6-sulfanona

A la solución agitada del ejemplo 42 (150 mg, 0,35 mmol) en THF (1,5 ml), se añadió NaH (60 %) (19 mg, 0,39 mmol) a 0 °C y se agitó durante 15 min. Después se añadió Etl (0,18 ml, 0,54 mmol, 3,0 M en THF) y se agitó a TA durante la noche. Una vez completada la reacción (monitorizada por TLC), la mezcla de reacción resultante se vertió en agua helada (2 x 50 ml) y la capa acuosa se extrajo con EtOAc (2 x 100 ml). La capa orgánica combinada se lavó con salmuera (50 ml), se secó sobre Na2SO4 anhidro y se concentró al vacío. El material bruto resultante se purificó mediante cromatografía ultrarrápida (Biotage Isolera, gradiente: MeOH al 3 % en DCM) para producir el compuesto del título. Rend¡m¡ento: 19 % (30 mg, sólido amarillo pálido). RMN 1H (400 MHz, DMSO-cfe): 88,56 (s, 2H), 7,97 (d, J= 8,0 Hz, 1H), 7,85 (s, 1H), 7,39 (dd, J= 8,2, 1,2 Hz, 1H), 3,84 (t, J = 4,8 Hz, 4H), 3,63 (d, J = 6,4 Hz, 1H), 3,10 (s, 3H), 2,84-2,73 (m, 5H), 2,55-2,39 (m, 4H), 1,39 (d, J = 6,4 Hz, 3H), 1,03 (t, J = 7,20 Hz, 3H). CLEM: (Método A) 445,0 (M+H), Tr. 2,3 min, 91,3 % (Máx) HPLC: (Método A) Tr. 2,2 min, 92,0 % (Máx)
Ejemplo 49: (¡soprop¡l¡m¡no)(met¡lH2-(4-(1-(2-met¡lbenzord1t¡azol-5-¡l)et¡l)p¡peraz¡n-1-¡l)p¡r¡m¡d¡n-5-¡l)-Á6-sulfanona

A la solución agitada del ejemplo 42 (150 mg, 0,35 mmol) en DMF (1,5 ml), se añadió NaH (60 %) (19 mg, 0,39 mmol) a 0 °C y se agitó durante 15 min. Después se añadió yoduro de isopropilo (0,1 ml, 1,05 mmol) y se agitó durante la noche a 60 °C. Una vez completada la reacción (monitorizada por t Lc ), la reacción se inactivó con agua helada (2 x 50 ml) y la capa acuosa se extrajo con EtOAc (2 x 100 ml). La capa orgánica combinada se lavó con salmuera (50 ml), se secó sobre Na2SO4 anhidro y se concentró al vacío. El material bruto resultante se purificó mediante HPLC prep. (Método A) para producir el compuesto del título. Rend¡m¡ento: 8 % (11 mg, sólido amarillo pálido). RMN 1H (400 MHz, DMSO-cfe): 88,58 (s, 2H), 7,97 (d, J= 8,4 Hz, 1H), 7,85 (s, 1H), 7,39 (dd, J = 8,2, 1,2 Hz, 1H), 3,84 (t, J = 5,2 Hz, 4H), 3,63 (d, J = 6,8 Hz, 1H), 3,15-3,08 (m, 4H), 2,79 (s, 3H), 2,50-2,33 (m, 4H), 1,39 (d, J= 6,4 Hz, 3H), 1,05 (d, J = 6.4 Hz, 3H), 0,98 (d, J = 6,4 Hz, 3H). CLEM: (Método A) 459 (M+H), Tr. 2,4 min, 99,3 % (Máx) HPLC: (Método A) Tr.
2.5 min, 99,3 % (Máx)
Ejemplo 50: ¡m¡no(met¡lH4-(4-(1-(2-met¡lbenzord1t¡azol-5-¡l)et¡l)p¡peraz¡n-1-¡l)fen¡l)-Á6-sulfanona
Etapa 1: 2-metil-5-(1-(4-(4-(metiltio) fenil)piperazin-1-il)etil)benzo[d]tiazol

A una solución agitada del intermedio 18 (1,72 g, 6,12 mmol) en DMF (10 ml), se añadieron TEA (3,45 ml, 24,4 mmol) y el intermedio 2 (1,30 g, 6,12 mmol) a TA y se agitó durante la noche a 70 °C. Una vez completada la reacción (monitorizada por TLC), la mezcla de reacción se evaporó a 50 °C al vacío. A la mezcla resultante, se añadió agua (10 ml) y la capa acuosa se extrajo con EtOAc (2 x 50 ml). La capa orgánica combinada se secó sobre Na2SO4 anhidro y se concentró al vacío. El material bruto resultante se purificó mediante cromatografía ultrarrápida (Biotage Isolera, gradiente: MeOH al 1-2% en DCM) para producir el compuesto del título. Rendimiento: 39% (900 mg, sólido blanquecino). RMN 1H (400 MHz, DMSO-de): 88,13 (d, J = 8,4 Hz, 1H), 8,04 (s, 1H), 7,68 (d, J = 8,8 Hz, 2H), 7,51 (d, J = 8,4 Hz, 1H), 7,01 (d, J = 9,2 Hz, 2H), 3,68-3,66 (m, 1H), 3,34-3,30 (m, 4H), 2,96 (s, 3H ) 2,37 (s, 3H), 2,68-2,34 (m, 4H), 1,42 (d, J = 6,8 Hz, 3H). CLEM: (Método A) 384,3 (M+H), Tr. 2,3 min, 83,3 % (Máx)
Etapa 2: 2-metil-5-(1-(4-(4-(metilsulfinil)fenil)piperazin-1-il)etil)benzo[d]tiazol

A una solución agitada de 2-metil-5-(1-(4-(4-(metiltio)fenil)piperazin-1-il)etil)benzo[d]tiazol (850 mg, 2,21 mmol) en DCM (7 ml, 10 V), se añadió en porciones m-CPBA (0,5 g, 2,88 mmol) a 0 °C durante 60 min. Una vez completada la reacción (monitorizada por TLC), la mezcla de reacción se inactivó con una solución de NaHCO3 al 10 % y la capa acuosa se extrajo con DCM (2 x 100 ml). La capa orgánica combinada se lavó con salmuera (30 ml), se secó sobre Na2SO4 anhidro y se concentró al vacío. El material bruto resultante se purificó mediante cromatografía ultrarrápida (Biotage Isolera, eluyente: EtOAc al 60-70 % en éter de petróleo) para producir el compuesto del título. Rendimiento: 51 % (450 mg, sólido amarillo pálido). RMN 1H (400 MHz, DMSO-de): 88,13 (d, J = 8,4 Hz, 1H), 8,04 (s, 1H), 7,68 (d, J= 8,8 Hz, 2H), 7,51 (d, J= 8,4 Hz, 1H), 7,01 (d, J = 9,2 Hz, 2H), 3,68-3,66 (m, 1H), 3,34-3,30 (m, 4H), 2,97 (s, 3H), 2,65 (s, 3H), 2,68-2,34 (m, 4H), 1,42 (d, J = 6,8 Hz, 3H). CLEM: (Método A) 400,3 (M H), Tr. 1,7 min, 83,3% (Máx)
Etapa 3: lmino(metil)(4-(4-(1-(2-metilbenzo[d]tiazol-5-il)etil)piperazin-1-il)fenil)-A6-sulfanona

A una solución agitada de 2-metil-5-(1-(4-(4-(metilsulfinil)fenil)piperazin-1-il)etil)benzo[d]tiazol (420 mg, 1,05 mmol) en DCM (8 ml, 20 V), se añadieron trifluoroacetamida (240 mg, 2,1 mmol), MgO (404 mg, 4,2 mmol), Rh2(OAC)4 (24 mg, 0,05 mmol) y Phl(OAc)2 (507 mg, 1,5 mmol) a TA y se agita durante la noche a la misma temperatura. Una vez completada la reacción (monitorizada por TLC), la mezcla de reacción se filtró a través de celite y se concentró al vacío. El material bruto resultante se purificó mediante cromatografía ultrarrápida (Biotage Isolera, eluyente: EtOAc al 55-60 % en éter de petróleo) para producir el intermedio puro 2,2,2-trifluoro-N-(metil(4-(4-(1-(2-metilbenzo[d]tiazol-5-il)etil)piperazin-1-il)fenil)(oxo)-A6-sulfaniliden)acetamida. Rendimiento: 40 % (210 mg, sólido blanquecino).
A este intermedio, se añadieron metanol (10 ml, 20 V) y K2CO3 (300 mg, 2,30 mmol) y se agitó durante 20 min.
Después de 20 min, la mezcla de reacción se filtró a través de celite y se concentró al vacío. A la mezcla resultante se añadió agua (50 ml) y la capa acuosa se extrajo con DCM (2 x 100 ml). La capa orgánica combinada se secó sobre Na2SO4 anhidro y se concentró al vacío. El material bruto resultante se purificó mediante cromatografía ultrarrápida (Biotage Isolera, gradiente: metanol al 1-2 % en DCM) para producir el compuesto del título. Rendimiento: 4 % (16 mg, sólido blanquecino). RMN 1H (400 MHz, DMSO-de): 87,98 (d, J = 8,0 Hz, 1H), 7,86 (s, 1H), 7,68 (d, J =8,8 Hz, 2H), 7,40 (d, J = 8,4 Hz, 1H), 7,01 (d, J = 8,8 Hz, 2H), 3,86 (s, 1H), 3,61 (d, J = 6,4 Hz, 1H), 3,34-3,28 (m, 4H), 2,97 (s, 3H), 2,80 (s, 3H), 2,59-2,46 (m, 4H), 1,40 (d, J = 6,4 Hz, 3H). CLEM: (Método A) 415,2 (M+H), Tr. 2,2 min, 96,5 % (Máx). HPLC: (Método A) Tr. 2,2 min, 96,0 % (Máx)
Ejemplos 51, 52, 53 y 54. (S)-¡m¡no(met¡lH2-(4-((S)-1-(2-met¡lbenzord1t¡azol-5-¡l)et¡l)p¡peraz¡n-1-¡l)p¡r¡mid¡n-5-¡l)-Á6-sulfanona y (R)-lm¡no(met¡lH2-(4-((S)-1-(2-met¡lbenzord1t¡azol-5-¡l)et¡l)p¡peraz¡n-1-¡l)p¡r¡m¡d¡n-5-¡l)-Á6-sulfanona y (S)-lm¡no(met¡lH2-(4-((R)-1-(2-met¡lbenzord1t¡azol-5-¡l)et¡l)p¡peraz¡n-1-¡l)p¡r¡m¡d¡n-5-¡l)-Á6-sulfanona y (R)-lm¡no(met¡lH2-(4-((R)-1-(2-met¡lbenzord1t¡azol-5-¡l)et¡l)p¡peraz¡n-1-¡l)p¡r¡m¡d¡n-5-¡l)-Á6-sulfanona

A una solución agitada del intermedio 8 (1,10 g, 4,20 mmol) en ACN (11 ml), se añadieron TEA (1,6 ml, 11,5 mmol) y el intermedio 10 (1,10 g, 4,00 mmol) a TA y la mezcla resultante se agitó durante la noche. Una vez completada la reacción (monitorizada por TLC), la mezcla resultante se concentró al vacío. El material bruto resultante se purificó mediante cromatografía ultrarrápida (Biotage Isolera, eluyente: EtOAc al 90-95 % en éter de petróleo) para producir el intermedio puro 2,2,2-trifluoro-N-(metil(2-(4-(1-(2-metilbenzo[d]tiazol-5-il)etil)piperazin-1-il)pirimidin-5-il)(oxo)-A6-sulfaniliden)acetamida. Rend¡m¡ento: 61 % (1,2 g, sólido blanquecino).
A este intermedio, se añadieron metanol (2,5 ml) y K2CO3(500 mg, 3,1 mmol) y se agitó durante 15 min. Después de 15 min, la mezcla de reacción se filtró a través de celite y se concentró al vacío. El material bruto resultante se purificó mediante cromatografía ultrarrápida (Biotage Isolera, eluyente: metanol al 3-4 % en DCM) para producir el compuesto del título en forma racémica. Los cuatro enantiómeros de este compuesto racémico se separaron por SFC (Método I: Fase móvil de purificación quiral: 40 % de amoníaco 20 mM en IPA, columna: LUX A1, caudal: 4,0 ml).
Análisis de la primera fracción de elución (ejemplo 51); Rend¡m¡ento: 12 % (55 mg, sólido blanquecino). RMN 1H (400 MHz, DMSO-de): 88,65 (s, 2H), 7,97 (d, J = 8,4 Hz, 1H), 7,84 (s, 1H), 7,39 (d, J = 8,0 Hz, 1H), 4,23 (s, 1H), 3,85-3,84 (m, 4H), 3,66-3,64 (m, 1H), 3,07 (s, 3H), 2,79 (s, 3H), 2,50-2,43 (m, 4H), 1,39 (d, J= 6,8 Hz, 3H). CLEM: (Método A) 416.8 (M+H), Tr. 2,1 min, 99,4 % (Máx) HPLC: (Método A) Tr. 2,0 min, 99,7 % (Máx). SFC qu¡ral: (Método C) Tr. 3,8 min, 100 % (Máx).
Análisis de la segunda fracción de elución (ejemplo 52); Rend¡m¡ento: 11 % (46 mg, sólido blanquecino). RMN 1H (400 MHz, DMSO-de): 88,65 (s, 2H), 7,97 (d, J= 8,4 Hz, 1H), 7,84 (s, 1H), 7,39 (d, J= 8,0 Hz, 1H), 4,23 (s, 1H), 3,85 3,84 (m, 4H), 3,66-3,64 (m, 1H), 3,07 (s, 3H), 2,79 (s, 3H), 2,50-2,43 (m, 4H), 1,39 (d, J = 6,80 Hz, 3H). CLEM: (Método A) 416,8 (M+H), Tr. 2,1 min, 99,2 % (Máx) HPLC: (Método A) Tr. 2,0 min, 99,7 % (Máx). SFC qu¡ral: (Método C) Tr.
4,5 min, 97,6 % (Máx)
Análisis de la tercera fracción de elución (ejemplo 53); Rend¡m¡ento: 15 % (65 mg, sólido blanquecino). RMN 1H (400 MHz, DMSO-de): 88,65 (s, 2H), 7,97 (d, J = 8,4 Hz, 1H), 7,84 (s, 1H), 7,39 (d, J = 8,0 Hz, 1H), 4,23 (s, 1H), 3,85-3,84 (m, 4H), 3,66-3,64 (m, 1H), 3,07 (s, 3H), 2,79 (s, 3H), 2,50-2,43 (m, 4H), 1,39 (d, J = 6,80 Hz, 3H). CLEM: (Método A) 416.8 (M+H), Tr. 2,1 min, 99,4 % (Máx) HPLC: (Método A) Tr. 2,0 min, 99,4 % (Máx). SFC qu¡ral: (Método C) Tr. 4,9 min, 97,4 % (Máx)
Análisis de la cuarta fracción de elución (ejemplo 54); Rend¡m¡ento: 17 % (75 mg, sólido blanquecino). RMN 1H (400
MHz, DMSO-de): 88,65 (s, 2H), 7,97 (d, J = 8,4 Hz, 1H), 7,84 (s, 1H), 7,39 (d, J = 8,0 Hz, 1H), 4,23 (s, 1H), 3,85-3,84 (m, 4H), 3,66-3,64 (m, 1H), 3,07 (s, 3H), 2,79 (s, 3H), 2,50-2,43 (m, 4H), 1,39 (d, J = 6,80 Hz, 3H). CLEM: (Método A) 416,8 (M+H), Tr. 2,1 min, 98,2 % (Máx) HPLC: (Método A) Tr. 2,0 min, 97,9 % (Máx). SFC quiral: (Método C) Tr. 8,5 min, 98,9 % (Máx).
Ejemplo B01: Preparaciones farmacéuticas
(A) Viales de inyección: Una solución de 100 g de un principio activo de acuerdo con la invención y 5 g de hidrogenofosfato disódico en 3 l de agua bidestilada se ajustó a pH 6,5 usando ácido clorhídrico 2 N, se filtró de forma estéril, se transfirió a viales de inyección, se liofilizó en condiciones estériles y se selló en condiciones estériles. Cada vial de inyección contenía 5 mg de principio activo.
(B) Supositorios: Se fundió una mezcla de 20 g de un principio activo de acuerdo con la invención con 100 g de lecitina de soja y 1400 g de manteca de cacao, se vertió en moldes y se dejó enfriar. Cada supositorio contenía 20 mg de principio activo.
(C) Solución: Se preparó una solución a partir de 1 g de un principio activo de acuerdo con la invención, 9,38 g de NaH2PO42 H2O, 28,48 g de Na2HPO412 H2O y 0,1 g de cloruro de benzalconio en 940 ml de agua bidestilada. El pH se ajustó a 6,8 y la solución se completó hasta 1 l y se esterilizó por irradiación. Esta solución puede usarse en forma de gotas para los ojos.
(D) Pomada: Se mezclaron 500 mg de un principio activo de acuerdo con la invención con 99,5 g de vaselina en condiciones asépticas.
(E) Comprimidos: Se comprimió una mezcla de 1 kg de un principio activo de acuerdo con la invención, 4 kg de lactosa, 1,2 kg de fécula de patata, 0,2 kg de talco y 0,1 kg de estearato de magnesio para dar comprimidos de manera convencional de tal manera que cada comprimido contenía 10 mg de principio activo.
(F) Comprimidos recubiertos: Los comprimidos se comprimieron de forma análoga a (E) y posteriormente se recubrieron de manera convencional con un recubrimiento de sacarosa, almidón de patata, talco, tragacanto y colorante.
(G) Cápsulas: Se introdujeron 2 kg de un principio activo de acuerdo con la invención en cápsulas de gelatina dura de manera convencional de tal manera que cada cápsula contenía 20 mg del principio activo.
(H) Ampollas: Una solución de 1 kg de un principio activo de acuerdo con la invención en 60 l de agua bidestilada se filtró de forma estéril, se transfirió a ampollas, se liofilizó en condiciones estériles y se selló en condiciones estériles. Cada ampolla contenía 10 mg de principio activo. (I)
(I) Aerosol para inhalación: Se disolvieron 14 g de un principio activo de acuerdo con la invención en 10 l de solución isotónica de NaCl y la solución se transfirió a envases de aerosol disponibles comercialmente con un mecanismo de bomba. La solución puede aplicarse en la boca o la nariz. Una dosis de aerosol (aproximadamente 0,1 ml) correspondió a una dosis de aproximadamente 0,14 mg.
Ejemplo C01: Métodos de caracterización de propiedades físicas
Difracción de rayos X de polvo (XRPD)
Aproximadamente 5-10 mg de muestra se comprimieron suavemente en el portamuestras de sílice de corte oblicuo simple de fondo cero XRPD. A continuación, la muestra se cargó en un difractómetro Philips X-Pert PRO y se analizó usando las siguientes condiciones experimentales.
Ánodo de tubo: Cu
Tensión del generador: 40 kV
Corriente del tubo: 40 mA
Longitud de onda alfa1: 1,5406 A
Longitud de onda alfa2: 1,5444 A
Ángulo de inicio [2 theta]: 5
Ángulo final [2 theta]: 50
Escaneo continuo
Para las sales novedosas sospechadas también se usó una velocidad de escaneo más lenta en un intervalo de 4 -40° 20.
Espectroscopia Raman
Las muestras se analizaron por un microscopio dispersivo Raman Nicolet Almega DXR para su espectro Raman usando las siguientes condiciones:
Tiempo de exposición: 1,0 s
N.° de Adquisición: 10
Apertura del espectrógrafo: Orificio 50 μm
Rejilla: 600 líneas / mm
Láser: He-Ne 633 nm 100 % de potencia
Después, los espectros Raman medidos se corrigieron mediante sustracción del valor inicial usando el software OMNIC™ v9.
Resonancia magnética nuclear (RMN)
Los compuestos se disolvieron en DMSO deuterado a una concentración de 7 mg/ml. Los espectros de RMN 1H se obtuvieron mediante Bruker Avance 400 (Bruker, Coventry, UK). Los archivos FID se procesaron mediante el software NMR MestReNova V8.0. Se analizaron los desplazamientos químicos y las integrales de los máximos.
Análisis térmico simultáneo (STA)
Aproximadamente 5 mg de muestra se pesaron con precisión en un crisol de cerámica y se colocaron en la cámara del analizador TGNDTA STA 6000 Perkin-Elmer a temperatura ambiente. A continuación, la muestra se calentó a una velocidad de 10 °C/min, normalmente de 30 °C a 300 °C, tiempo durante el cual se monitorizó el cambio de peso así como la señal DTA. El gas de purga usado fue nitrógeno a una caudal de 20 cm3/min.
Sorción de vapor gravimétrico (GVS)
Se colocaron aproximadamente 10 mg de muestra en una sartén de equilibrio de sorción de vapor de malla de alambre y se cargaron en una balanza de sorción de vapor "IgaSorp" (Hiden Analytical Instruments). Después, la muestra se secó manteniendo un ambiente de 0 % de humedad hasta que no se registró ningún cambio de peso adicional. Posteriormente, la muestra se sometió después a un perfil de rampa de 0 a 90 % de HR en aumentos del 10 % de HR, manteniendo la muestra en cada etapa hasta que se alcanzó el equilibrio (99 % finalización de la etapa). Al alcanzar el equilibrio, el % de HR dentro del aparato se elevó a la siguiente etapa y se repitió el procedimiento de equilibrio. Una vez completado el ciclo de sorción, la muestra se secó usando el mismo procedimiento. El ciclo de sorción / desorción se repitió generalmente una segunda vez en caso de que la muestra hubiera cambiado durante el primer ciclo. El cambio de peso durante los ciclos de adsorción/desorción se monitorizó después, permitiendo determinar la naturaleza higroscópica de la muestra.
Ejemplo C02: Métodos de preparación de la sal de succinato del Ejemplo 35.
Un método preferido de preparación de la sal de succinato fue el siguiente:
Se pesaron un compuesto del Ejemplo 35 (3,0 g) y ácido succínico (0,97 g) en un vial de vidrio de 20 ml y se añadió etanol (10 ml) a la mezcla sólida física. La suspensión resultante se calentó hasta que se obtuvo una solución transparente que después se sometió a un ciclo de temperatura entre 40 °C y temperatura ambiente durante 18 horas durante la noche. Durante este tiempo, los cristales se precipitaron y se acumularon. Se añadió más etanol (2 ml) para movilizar y el producto se filtró a temperatura ambiente, se lavó con etanol ( 2 x 5 ml) y se secó en un horno de vacío a 50 °C durante 24 horas hasta peso constante. (Rendimiento 3,1 g).
Ejemplo C03: Solubilidad acuosa visual
La sal de succinato se pesó en viales de vidrio y se añadió agua en porciones de 100 μl hasta 1 ml y después porciones de 0,5 ml. La sal de succinato tuvo buena solubilidad, disolviéndose completamente en 0,8 ml de agua (>12,5 mg/ml). Los datos característicos de la sal de succinato se resumen a continuación:
• El patrón XRPD se muestra en la Figura 1
• Los datos de RMN fueron consistentes con una monosal estequiométrica (Figura 2).
• STA no mostró pérdida de peso antes de la fusión con inicio a ~ 125,5 °C. La muestra no estaba hidratada ni solvatada (Figura 3).
• GVS indicó que la muestra era solo ligeramente higroscópica con una ganancia de peso reversible del 1,8 % al 80 % de HR.
• No hubo cambios en la XRPD después de la suspensión de agua a 10 mg / 200 μl.
• No hubo cambios en el patrón de rayos X después de la suspensión en IPA / agua.
• Esta sal tenía muy buena solubilidad acuosa estimada visualmente en ~ 12,5 mg / ml
• No tiene evidencia de tendencia a formar hidratos.
• Estas observaciones combinadas harían que la sal de succinato del compuesto del Ejemplo 35 sea un candidato adecuado para estudios y desarrollo adicionales.
Ejemplo C04: Sal de fumarato de un compuesto del Ejemplo 35
Se calentó un compuesto del Ejemplo 35 (300 mg) en etanol (2 ml) para disolver el sólido. Se añadió ácido fumárico (95 mg) a la solución caliente y la mezcla se agitó de manera eficiente. Se añadió más etanol (1 ml). Inicialmente se observó una solución transparente, pero se precipitó gradualmente un sólido a medida que la mezcla se enfriaba a temperatura ambiente. La suspensión resultante se sometió a ciclos de temperatura entre 40 °C y temperatura ambiente durante la noche (18-24 horas). El producto se filtró, se lavó con etanol (2 x 1 ml) y se secó en un horno de vacío a 45 °C hasta peso constante (Rendimiento 350 mg, 91 %).
Ejemplo C05: Solubilidad acuosa visual
La sal de fumarato se pesó en viales de vidrio y se añadió agua en porciones de 100 μl hasta 1 ml y en lo sucesivo porciones de 0,5 ml. El fumarato se disolvió completamente en 3 ml de agua (>3,3 mg/ml).
Los datos característicos de la sal de fumarato se resumen a continuación:
• El patrón XRPD se muestra en la Figura 4. No se ha encontrado forma polimórfica.
• Los datos de RMN fueron consistentes con una monosal estequiométrica (Figura 5).
• STA no mostró pérdida de peso conduciendo a la fusión con inicio a ~ 169 °C. La muestra no fue hidratada ni solvatada (Figura 6).
• GVS indicó que la muestra fue solo ligeramente higroscópica con una ganancia de peso marginal reversible de 2,1 % a 80 % de HR.
• No hubo cambio en XRPD después de la suspensión en agua a 10 mg / 200 μl.
• No hubo cambio en el patrón de rayos X después de la suspensión en IPA / agua
• La solubilidad acuosa del fumarato se midió a ~ 3,3 mg/ml.
Ejemplo C06: Ejemplos comparativos
Ejemplo comparativo C06-1: Preparación de sal de succinato Forma 2 de un compuesto del Ejemplo 35.
El succinato del Ejemplo 35 (200 mg) preparado en el ejemplo C02 (forma 1) se suspendió en 2-propanol (2 ml) y se calentó para disolverlo. Se añadieron unos pocos cristales obtenidos después de una cristalización lenta en 2-propanol o tetrahidrofurano a la solución caliente y se precipitaron los cristales a medida que la mezcla se enfrió a temperatura ambiente. Se dejó evaporar el exceso de disolvente bajo un flujo de nitrógeno y el producto se secó adicionalmente a 50 °C al vacío durante ~ 24 horas a peso constante.
• El patrón XRPD se muestra en la Figura 7
• Los datos de RMN fueron consistentes con una monosal estequiométrica (Figura 8).
• GVS indicó que la muestra era ligeramente higroscópica con una ganancia de peso reversible de 1,25 % a 80 % de HR. Además, hubo una conversión parcial a la Forma 1 evidente en el patrón de rayos X, obtenido después de GVS. Esto demuestra la inestabilidad termodinámica de la Forma 2.
• Las suspensiones con la Forma 1 y la Forma 2 de la sal respectiva en etanol, 2-propanol / agua 90/10, 2-propanol y tetrahidrofurano dieron como resultado una conversión completa a la Forma 1 a temperatura ambiente y a 40 °C. Esto demuestra nuevamente la inestabilidad termodinámica de la Forma 2.
• Por lo tanto, la Forma 2 no cumpliría con los requisitos reglamentarios actuales para el desarrollo de fármacos.
Ejemplo comparativo C06-2: Preparación de sal de clorhidrato de un compuesto del Ejemplo 35
Se calentó un compuesto del Ejemplo 35 (300 mg) en acetona (3 ml) de modo que el sólido se disolvió. Se añadió ácido clorhídrico (5,825 M, 135 μl), preparado diluyendo el ácido concentrado por dos, a la solución caliente y se mezcló bien. Se separó inicialmente un aceite que se raspó con una espátula dando como resultado que el producto se solidificara en 5-10 minutos. La suspensión resultante se sometió a ciclos de temperatura entre 40 °C y temperatura
ambiente durante la noche (18-24 horas). El producto se filtró, se lavó con acetona (2 x 1 ml) y se secó en un horno de vacío a 45 °C hasta peso constante. (Rendimiento 194 mg, 57 %).
Los datos característicos de la sal de clorhidrato se resumen a continuación:
• El patrón XRPD se muestra en la Figura 9.
• STA mostró una pérdida de peso de ~ 3,6 % entre 50 °C y 150 °C. Esto fue consistente con el valor teórico para una versión hidratada de la sal (Figura 10).
• GVS también indicó que la muestra era claramente higroscópica con una ganancia de peso reversible de 3,6 % a 70 % de HR y 4 % a 80 % de HR.
• La sal se convirtió en una materia aceitosa después de la suspensión en agua.
• Debido a las propiedades negativas anteriores, esta sal de clorhidrato no cumpliría con los requisitos reglamentarios actuales para el desarrollo de fármacos.
Ejemplo comparativo C06-3: Sal de benzoato de un compuesto del Ejemplo 35
Se calentó un compuesto del Ejemplo 35 (300 mg) a reflujo en acetona (2 ml). Aunque el sólido no se disolvió completamente, se añadió ácido benzoico (100 mg) a la mezcla caliente y se añadió más acetona (1 ml). La mezcla se calentó de nuevo a reflujo y se obtuvo una solución transparente. Esta solución se sometió a ciclos de temperatura entre 40 °C y temperatura ambiente durante la noche (18 horas), pero permaneció transparente. Se dejó evaporar aproximadamente la mitad del volumen de disolvente y se separó un sólido durante ~ 24 horas.
El producto se filtró, se lavó con acetona (2 x 1 ml) y se secó en un horno de vacío a 45 °C a peso constante. (Rendimiento 290 mg, 74 %).
Los datos característicos de la sal de benzoato se resumen a continuación:
• El patrón XRPD se muestra en la Figura 11.
• Los datos de RMN fueron consistentes con una monosal estequiométrica (Figura 12).
• Hubo un cambio en XRPD después de la suspensión en agua y en IPNagua, lo que indica que la forma polimórfica de esta sal no es termodinámicamente estable y, por lo tanto, no es adecuada para un desarrollo farmacéutico.
Ejemplo comparativo C06-4: Sal de mesilato de un compuesto del Ejemplo 35
Se calentó un compuesto del Ejemplo 35 (300 mg) en acetona (3 ml) de modo que el sólido se disolvió. Se añadió ácido metanosulfónico (27 μl) a una mezcla caliente dando como resultado la formación inicial de un aglomerado aceitoso. La mezcla se sometió a ultrasonidos durante aproximadamente 1 minuto y pareció separarse algo de sólido. La mezcla se sometió a ciclos de temperatura entre 40 °C y temperatura ambiente durante la noche (18 horas) y se dejó evaporar aproximadamente la mitad del volumen de disolvente. Resultó un producto sólido que se filtró y se secó en un horno de vacío a 45 °C a peso constante (Rendimiento 95 mg).
Los datos característicos de la sal mesilato se resumen a continuación:
• El patrón de rayos X del producto fue consistente con una mezcla de la base libre original y la sal amorfa, lo que indica que la sal de mesilato no puede aislarse de forma reproducible como una forma sólida. Por tanto, la sal de mesilato del Ejemplo 35 no es adecuada para el desarrollo farmacéutico.
Ejemplo comparativo C06-5: Solubilidad acuosa visual comparativa con la base libre del Ejemplo 35
La base libre del Ejemplo 35 (10 mg) se pesó en viales de vidrio y se añadió agua en porciones de 100 μl hasta 1 ml y en lo sucesivo porciones de 0,5 ml. La solubilidad se evaluó visualmente después de un breve período de equilibrio. La base libre no dio ningún indicio de que fuera soluble en absoluto en 8 ml de agua (<<1,25 mg/ml).
Las sales de compuestos de fórmula I con diversos ácidos distintos del ácido succínico y ácido fumárico no produjeron propiedades farmacéuticas adecuadas, es decir, no han sido solubles, estables o sólidas o tienen otras propiedades no adecuadas para el desarrollo farmacéutico, especialmente para formas sólidas de dosificación oral, tales como comprimidos, por ejemplo, comprimidos que están comprimidos.
Claims (8)
1. Una sal de ácido mono-succínico de uno de los compuestos la, Ib, Ic y Id en una forma sólida que tiene el patrón de difracción de polvo de rayos X característico como se muestra en la Figura 1:

2. Una sal de ácido mono-fumárico de uno de los compuestos Ia, Ib, Ic y Id en una forma sólida que tiene el patrón de difracción de polvo de rayos X característico como se muestra en la Figura 4:


3. Un método para la preparación de una sal de ácido mono-succínico de la reivindicación 1 o una sal de ácido monofumárico de la reivindicación 2 que comprende las siguientes etapas:
a) suspender o disolver el compuesto seleccionado de fórmula I y el ácido respectivo en un disolvente o mezcla de disolventes adecuados;
b) calentar la mezcla obtenida en la etapa a) a una temperatura entre aproximadamente 30 °C y aproximadamente el punto de ebullición del disolvente o mezcla de disolventes seleccionados y dejar que la mezcla se enfríe a temperatura ambiente;
c) opcionalmente repetir la etapa b) varias veces;
d) separar y secar el sólido obtenido de esta manera.
4. Una forma de dosificación oral sólida que comprende una sal de ácido mono-succínico de acuerdo con la reivindicación 1 o una sal de ácido mono-fumárico de acuerdo con la reivindicación 2.
5. Una sal de ácido mono-succínico de acuerdo con la reivindicación 1 o una sal de ácido mono-fumárico de acuerdo con la reivindicación 2 para su uso como un medicamento.
6. Una sal de ácido mono-succínico de acuerdo con la reivindicación 1 o una sal de ácido mono-fumárico de acuerdo con la reivindicación 2 para su uso en un método de tratamiento de una afección seleccionada del grupo que consiste en enfermedades neurodegenerativas, diabetes, cáncer, enfermedades cardiovasculares y accidente cerebrovascular.
7. Una sal de ácido mono-succínico de acuerdo con la reivindicación 1 o una sal de ácido mono-fumárico de acuerdo con la reivindicación 2 para su uso en un método de tratamiento de una afección de acuerdo con la reivindicación 6, en donde la afección se selecciona del grupo de una o más tauopatías y enfermedad de Alzheimer, Demencia, Esclerosis lateral amiotrófica (ELA), Esclerosis lateral amiotrófica con deterioro cognitivo (ALSci), Enfermedad del grano argirofílico, Variante conductual de la demencia frontotemporal (BvFTD), Enfermedad de Bluit, Encefalopatía traumática crónica, Degeneración corticobasal (CBP), Demencia pugilística, Ovillos neurofibrilares difusos con calcificación, síndrome de Down, Demencia británica familiar, Demencia danesa familiar, Demencia frontotemporal con parkinsonismo ligado al cromosoma 17 (FTDP-17), Degeneración lobar frontotemporal (FTLD), Ganglioglioma, Gangliocitoma, Enfermedad de Gerstmann-Straussler-Scheinker, Tauopatía de la glía globular, Parkinsonismo de Guadalupe, Enfermedad de Hallervorden-Spatz (neurodegeneración con acumulación de hierro en el cerebro tipo 1), Encefalopatía por plomo, Lipofuscinosis, Meningioangiomatosis, Atrofia multisistémica, Distrofia miotónica, Enfermedad de Niemann-Pick (tipo C), Degeneración palido-ponto-nigral, Complejo parkinsonismo-demencia de Guam, Enfermedad de Pick (PiD), Demencia por enfermedad de Parkinson, Parkinsonismo postncefalítico (PEP), Afasia progresiva primaria, Enfermedades priónicas (incluyendo la Enfermedad de Creutzfeldt-Jakob (CJD), Afasia progresiva no fluente, Variante de la enfermedad de Creutzfeldt-Jakob (vCJD), Insomnio familiar fatal, Kuru, Gliosis supercortical progresiva, Parálisis supranuclear progresiva (PSP), Demencia semántica, Síndrome de Steele-Richardson-Olszewski, Panencefalitis esclerosante subaguda, Demencia por ovillos, Esclerosis tuberosa, Enfermedad de Huntington y Enfermedad de Parkinson, preferentemente una o más tauopatías y enfermedad de Alzheimer.
8. Un método para inhibir una glucosidasa, en donde un sistema que expresa la glucosidasa se pone en contacto con una sal de ácido mono-succínico de acuerdo con la reivindicación 1 o una sal de ácido mono-fumárico de acuerdo con la reivindicación 2 en condiciones in vitro de tal manera que se inhiba la glucosidasa.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP18190155 | 2018-08-22 | ||
| PCT/EP2019/072474 WO2020039030A1 (en) | 2018-08-22 | 2019-08-22 | Succinate and fumarate acid addition salts of piperazine derivatives useful as glycosidase inhibitors |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2954474T3 true ES2954474T3 (es) | 2023-11-22 |
Family
ID=63363926
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES19759533T Active ES2954474T3 (es) | 2018-08-22 | 2019-08-22 | Sales de adición de ácido de succinato y fumarato de derivados de piperazina útiles como inhibidores de glucosidasa |
Country Status (18)
| Country | Link |
|---|---|
| US (1) | US12195455B2 (es) |
| EP (1) | EP3841093B1 (es) |
| JP (1) | JP7407171B2 (es) |
| KR (1) | KR102839500B1 (es) |
| CN (1) | CN113166083A (es) |
| AU (2) | AU2019324522B2 (es) |
| BR (1) | BR112021003022A2 (es) |
| CA (1) | CA3110032A1 (es) |
| EA (1) | EA202190275A1 (es) |
| ES (1) | ES2954474T3 (es) |
| HU (1) | HUE063226T2 (es) |
| IL (1) | IL280841B2 (es) |
| MA (1) | MA53429A (es) |
| MX (1) | MX2021002111A (es) |
| PL (1) | PL3841093T3 (es) |
| SG (1) | SG11202101651RA (es) |
| WO (1) | WO2020039030A1 (es) |
| ZA (1) | ZA202100981B (es) |
Families Citing this family (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP3868752A1 (en) | 2014-08-28 | 2021-08-25 | Asceneuron SA | Glycosidase inhibitors |
| US11261183B2 (en) | 2016-02-25 | 2022-03-01 | Asceneuron Sa | Sulfoximine glycosidase inhibitors |
| AU2017222958B2 (en) | 2016-02-25 | 2019-07-18 | Asceneuron S. A. | Glycosidase inhibitors |
| SG11201807012QA (en) | 2016-02-25 | 2018-09-27 | Asceneuron S A | Acid addition salts of piperazine derivatives |
| WO2019037860A1 (en) | 2017-08-24 | 2019-02-28 | Asceneuron S.A. | LINEAR INHIBITORS OF GLYCOSIDASE |
| WO2020039028A1 (en) | 2018-08-22 | 2020-02-27 | Asceneuron S. A. | Tetrahydro-benzoazepine glycosidase inhibitors |
| WO2020039029A1 (en) | 2018-08-22 | 2020-02-27 | Asceneuron S. A. | Spiro compounds as glycosidase inhibitors |
| WO2020039027A1 (en) | 2018-08-22 | 2020-02-27 | Asceneuron S. A. | Pyrrolidine glycosidase inhibitors |
| WO2025131275A1 (en) | 2023-12-20 | 2025-06-26 | Asceneuron Sa | (2-(4-(1-(benzo[d]thiazol-5-yl)ethyl)piperazin-1 -yl)pyrimidin-5-yl)(imino)(methyl)-lamda6-sulfanone for use in the treatment of colitis, parkinson disease, tauopathy, als and alzheimer's disease |
| CN117471001B (zh) * | 2023-12-26 | 2024-03-26 | 山东百诺医药股份有限公司 | 一种瑞卢戈利起始物料中n-溴代琥珀酰亚胺含量的测定方法 |
Family Cites Families (97)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| FR1311316A (fr) | 1961-04-12 | 1962-12-07 | Science Union Et Compagnie Soc | Nouveaux dérivés de la pipérazine et leurs préparations |
| NL127996C (es) | 1963-11-19 | |||
| US3485757A (en) | 1964-11-23 | 1969-12-23 | Atomic Energy Commission | Thermoelectric composition comprising doped bismuth telluride,silicon and boron |
| DE1595923A1 (de) | 1965-02-20 | 1969-11-27 | Merck Ag E | 1-Aralkyl-4-(thiazolyl-2)-piperazine und Verfahren zu ihrer Herstellung |
| GB1165283A (en) | 1967-01-17 | 1969-09-24 | Science Union & Cie | New Purine Derivatives and processes for prepararing them |
| US4600025A (en) | 1982-11-18 | 1986-07-15 | Grigg Ronald E | Smoking products comprising nicotine substitutes |
| EP0636130A1 (en) | 1992-04-15 | 1995-02-01 | Merck Sharp & Dohme Ltd. | Azacyclic compounds |
| IL118768A (en) | 1995-07-12 | 2000-10-31 | Akzo Nobel Nv | Diphenylmethane piperidine derivatives pharmaceutical compositions containing them and a method for their preparation |
| TW504510B (en) | 1996-05-10 | 2002-10-01 | Janssen Pharmaceutica Nv | 2,4-diaminopyrimidine derivatives |
| CA2282390A1 (en) | 1997-04-17 | 1998-10-22 | Yukio Fujisawa | Thermogenic composition and benzazepine thermogenics |
| CN1191248C (zh) | 1997-10-24 | 2005-03-02 | 纽罗根公司 | 作为多巴胺d 受体亚型配体的1-(2-萘基)和1-(2-氮杂萘基)-4-(1-苯基甲基)哌嗪 |
| CA2448080A1 (en) | 2001-05-22 | 2002-11-28 | Neurogen Corporation | Melanin concentrating hormone receptor ligands: substituted 1-benzyl-4-aryl piperazine analogues |
| HRP20040767A2 (en) | 2002-02-19 | 2004-12-31 | Teva Pharma | Desolvating solvates of atorvastatin hemi-calcium |
| US6982259B2 (en) | 2002-04-30 | 2006-01-03 | Schering Aktiengesellschaft | N-heterocyclic derivatives as NOS inhibitors |
| MXPA04012440A (es) | 2002-06-12 | 2005-04-28 | Abbott Lab | Antagonistas de receptor de hormona concentradora de melanina. |
| AU2003243921B2 (en) | 2002-06-27 | 2009-05-07 | Novo Nordisk A/S | Aryl carbonyl derivatives as therapeutic agents |
| DK1519939T5 (da) | 2002-07-05 | 2011-01-24 | Targacept Inc | N-Aryl-diazaspirocykliske forbindelser og fremgangsmåder til fremstilling og anvendelse deraf |
| JP4712384B2 (ja) | 2002-09-09 | 2011-06-29 | ジヤンセン・フアーマシユーチカ・ナームローゼ・フエンノートシヤツプ | Orl−1受容体介在障害の治療に有用なヒドロキシアルキル置換1,3,8−トリアザスピロ[4.5]デカン−4−オン誘導体 |
| MXPA05011223A (es) | 2003-04-18 | 2006-01-26 | Lilly Co Eli | Compuestos de (piperidiniloxi)fenilo, (piperidiniloxi)piridinilo, (piperidinilsulfanil)fenilo y (piperidinilsulfanil)piridinilo como agonistas 5-htif. |
| CA2526374A1 (en) | 2003-05-21 | 2004-12-02 | Banyu Pharmaceutical Co., Ltd. | 2-aminoquinoline derivatives |
| BRPI0506662B8 (pt) | 2004-01-06 | 2021-05-25 | Novo Nordisk As | compostos ativadores de glucoquinase |
| WO2005110982A2 (en) | 2004-04-07 | 2005-11-24 | Neurogen Corporation | Substituted 1-benzyl-4-substituted piperazine analogues |
| HU227119B1 (en) | 2004-07-29 | 2010-07-28 | Richter Gedeon Nyrt | Indole and benzimidazole carboxylic acid amide derivatives and pharmaceutical compositions containing them |
| HUP0401522A2 (en) | 2004-07-29 | 2006-04-28 | Richter Gedeon Vegyeszet | New 4-benzylidene-piperidine derivatives, pharmaceutical compositions containing the same and process for their preparation |
| AU2006220187B2 (en) | 2005-03-01 | 2012-11-08 | Simon Fraser University | Selective glycosidase inhibitors, methods of making inhibitors, and uses thereof |
| TW200724140A (en) | 2005-05-27 | 2007-07-01 | Eisai Co Ltd | Hydantoin compounds |
| WO2007008541A2 (en) | 2005-07-08 | 2007-01-18 | Kalypsys, Inc. | Cellular cholesterol absorption modifiers |
| JP2009500377A (ja) | 2005-07-08 | 2009-01-08 | ノボ・ノルデイスク・エー/エス | ジシクロアルキルウレア型グルコキナーゼ活性化剤 |
| CA2646755A1 (en) | 2006-03-31 | 2007-10-11 | Astrazeneca Ab | Bicyclic benzimidazole compounds and their use as metabotropic glutamate receptor potentiators |
| WO2007135398A1 (en) | 2006-05-22 | 2007-11-29 | Astrazeneca Ab | Indole derivatives |
| US20080051387A1 (en) | 2006-06-09 | 2008-02-28 | Yuelian Xu | Tetrahydropyrido[3,4-d]pyrimidines and related analogues |
| WO2008012623A1 (en) | 2006-07-25 | 2008-01-31 | Pfizer Products Inc. | Benzimidazolyl compounds as potentiators of mglur2 subtype of glutamate receptor |
| EP2057171A4 (en) | 2006-08-31 | 2010-04-21 | Univ Fraser Simon | SELECTIVE GLYCOSIDASE INHIBITORS AND APPLICATIONS THEREOF |
| US20100022517A1 (en) | 2006-12-18 | 2010-01-28 | Richards Lori A | Ophthalmic formulation of rho kinase inhibitor compound |
| US8148369B2 (en) | 2007-05-10 | 2012-04-03 | Janssen Pharmaceutica Nv | Fused pyrazine compounds useful for the treatment of degenerative and inflammatory diseases |
| WO2009011904A1 (en) | 2007-07-19 | 2009-01-22 | Renovis, Inc. | Compounds useful as faah modulators and uses thereof |
| US8404713B2 (en) | 2007-10-26 | 2013-03-26 | Janssen Pharmaceutica Nv | Quinolinone derivatives as PARP inhibitors |
| WO2009086303A2 (en) | 2007-12-21 | 2009-07-09 | University Of Rochester | Method for altering the lifespan of eukaryotic organisms |
| WO2009127943A1 (en) | 2008-04-17 | 2009-10-22 | Pfizer Inc. | Ether benzylidene piperidine 5-membered aryl carboxamide compounds useful as faah inhibitors |
| CA2717087A1 (en) | 2008-04-17 | 2009-10-22 | Pfizer Inc. | 4-[3-(aryloxy)benzylidene]-3-methyl piperidine 5-membered aryl carboxamides |
| US7863291B2 (en) | 2008-04-23 | 2011-01-04 | Bristol-Myers Squibb Company | Quinuclidine compounds as alpha-7 nicotinic acetylcholine receptor ligands |
| JPWO2009154132A1 (ja) | 2008-06-19 | 2011-12-01 | Msd株式会社 | スピロジアミン−ジアリールケトオキシム誘導体 |
| JP2010065024A (ja) | 2008-08-14 | 2010-03-25 | Ishihara Sangyo Kaisha Ltd | トリアゾロピリミジン誘導体又はその塩を含有する有害生物防除剤 |
| WO2010021381A1 (ja) | 2008-08-22 | 2010-02-25 | 武田薬品工業株式会社 | 縮合複素環誘導体およびその用途 |
| CA2735269C (en) | 2008-08-29 | 2018-10-16 | Saint Mary's University | Use of gluconacetobacter with reduced use of nitrogen fertilizer to improve beet crop production |
| CN102137841B (zh) | 2008-09-02 | 2014-05-14 | 日产化学工业株式会社 | 邻位取代卤代烷基磺酰苯胺衍生物及除草剂 |
| TW201030007A (en) | 2009-02-06 | 2010-08-16 | Gruenenthal Gmbh | Substituted spiro-amides as b1r modulators |
| AU2010221417A1 (en) | 2009-03-02 | 2011-09-22 | Sirtris Pharmaceuticals, Inc. | 8-substituted quinolines and related analogs as sirtuin modulators |
| WO2010108115A1 (en) | 2009-03-20 | 2010-09-23 | Sanford-Burnham Medical Research Institute | Allosteric jnk inhibitors |
| US20120010186A1 (en) | 2009-03-23 | 2012-01-12 | Merck Frosst Canada Ltd. | Heterocyclic compounds as inhibitors of stearoyl-coenzyme a delta-9 desaturase |
| JP2010270034A (ja) | 2009-05-20 | 2010-12-02 | Sumitomo Chemical Co Ltd | アミド化合物並びにその植物病害防除用途 |
| CA2765678A1 (en) | 2009-06-22 | 2010-12-29 | Christopher Blackburn | Substituted hydroxamic acids and uses thereof |
| DE102009049679A1 (de) | 2009-10-19 | 2011-04-21 | Merck Patent Gmbh | Pyrazolopyrimidinderivate |
| US9120781B2 (en) | 2010-05-11 | 2015-09-01 | Simon Fraser University | Selective glycosidase inhibitors and uses thereof |
| US20130196971A1 (en) | 2010-09-17 | 2013-08-01 | Christopher Joseph Aquino | Fatty acid synthase inhibitors |
| US8933040B2 (en) | 2010-11-08 | 2015-01-13 | Craig A. Coburn | Selective glycosidase inhibitors and uses thereof |
| AU2011328203B2 (en) | 2010-11-08 | 2015-03-19 | Janssen Pharmaceuticals, Inc. | 1,2,4-triazolo[4,3-a]pyridine derivatives and their use as positive allosteric modulators of mGluR2 receptors |
| WO2012061972A1 (en) | 2010-11-08 | 2012-05-18 | Alectos Therapeutics Inc. | Selective glycosidase inhibitors and uses thereof |
| DK2655388T3 (en) * | 2010-12-23 | 2016-09-19 | Alectos Therapeutics Inc | Selective glykosidaseinhibitorer and uses thereof |
| GB201103526D0 (en) | 2011-03-02 | 2011-04-13 | Summit Corp Plc | Selective glycosidase inhibitors and uses thereof |
| JPWO2012124744A1 (ja) | 2011-03-14 | 2014-07-24 | 大正製薬株式会社 | 含窒素縮合複素環化合物 |
| AU2012298949B2 (en) | 2011-08-25 | 2016-09-22 | Merck Patent Gmbh | Pyrano [3, 2 - D] [1, 3] thiazole as glycosidase inhibitors |
| US9221809B2 (en) | 2011-10-31 | 2015-12-29 | Merck Sharp & Dohme Corp. | Aminopyrimidinones as interleukin receptor-associated kinase inhibitors |
| AR092031A1 (es) | 2012-07-26 | 2015-03-18 | Merck Sharp & Dohme | Inhibidores del canal de potasio medular externo renal |
| WO2014023723A1 (en) | 2012-08-08 | 2014-02-13 | Novartis Ag | Subtituted azines as pesticides |
| US9522883B2 (en) | 2012-08-31 | 2016-12-20 | Alectos Therapeutics Inc. | Glycosidase inhibitors and uses thereof |
| PT2970272T (pt) | 2013-03-14 | 2019-06-05 | Merck Patent Gmbh | Inibidores de glicosidase |
| CN103435606A (zh) | 2013-08-22 | 2013-12-11 | 中国药科大学 | CDK2与GSK3β双重抑制剂及用途 |
| SI3077395T1 (en) | 2013-12-05 | 2018-03-30 | Pfizer Inc. | Pyrrolo(2,3-d)pyrimidinyl, pyrrolo(2,3-b)pyrazinyl and pyrrolo(2,3-d)pyridinyl acrylamides |
| EP2913330A1 (en) | 2014-02-27 | 2015-09-02 | Laboratoire Biodim | Condensed derivatives of imidazole useful as pharmaceuticals |
| HUE057317T2 (hu) | 2014-04-23 | 2022-04-28 | Dart Neuroscience Llc | Helyettesített [1,2,4]triazolo[1,5-A]pirimidin-7-IL vegyületeket tartalmazó készítmények mint PDE2 inhibítorok |
| EP3868752A1 (en) * | 2014-08-28 | 2021-08-25 | Asceneuron SA | Glycosidase inhibitors |
| BR112017028318B1 (pt) | 2015-07-02 | 2024-02-20 | Janssen Sciences Ireland Uc | Composto antibacteriano, seu uso, processo para sua preparação,produto que o contém, composição farmacêutica e combinação |
| ES2765738T3 (es) | 2015-11-02 | 2020-06-10 | Janssen Pharmaceutica Nv | Compuesto de [1,2,4]triazolo[1,5-a]pirimidin-7-ilo |
| WO2017087858A1 (en) | 2015-11-20 | 2017-05-26 | Abide Therapeutics, Inc. | Pyrazole compounds and methods of making and using same |
| WO2017087863A1 (en) | 2015-11-20 | 2017-05-26 | Abide Therapeutics, Inc. | Pyrazole compounds and methods of making and using same |
| EP3380471B8 (en) | 2015-11-25 | 2022-01-19 | Lieber Institute Inc. DBA Lieber Institute For Brain Development | Tetrahydro-8h-pyrido[1,2-a]pyrazine-8-ones as comt inhibitors for the treatment of neurodegenerative disorders |
| EP3389658B1 (en) | 2015-12-18 | 2020-11-25 | Merck Sharp & Dohme Corp. | Glycosidase inhibitors and uses thereof |
| US10344021B2 (en) | 2016-02-25 | 2019-07-09 | Asceneuron S A | Process for the separation of enantiomers of piperazine derivatives |
| CN108884078A (zh) * | 2016-02-25 | 2018-11-23 | 阿森纽荣股份公司 | 糖苷酶抑制剂 |
| US11261183B2 (en) | 2016-02-25 | 2022-03-01 | Asceneuron Sa | Sulfoximine glycosidase inhibitors |
| SG11201807012QA (en) | 2016-02-25 | 2018-09-27 | Asceneuron S A | Acid addition salts of piperazine derivatives |
| WO2018153508A2 (en) * | 2017-02-24 | 2018-08-30 | Asceneuron S.A. | Sulfoximine glycosidase inhibitors |
| AU2017222958B2 (en) * | 2016-02-25 | 2019-07-18 | Asceneuron S. A. | Glycosidase inhibitors |
| EP3472129A4 (en) | 2016-06-21 | 2019-12-04 | X4 Pharmaceuticals, Inc. | CXCR4 INHIBITORS AND USES THEREOF |
| US11186564B2 (en) | 2016-08-04 | 2021-11-30 | Sunovion Pharmaceuticals Inc. | Dual NAV1.2/5HT2a inhibitors for treating CNS disorders |
| US20190359609A1 (en) | 2016-12-16 | 2019-11-28 | Janssen Pharmaceutica Nv | Bicyclic oga inhibitor compounds |
| EP3555087A1 (en) | 2016-12-16 | 2019-10-23 | Janssen Pharmaceutica NV | Monocyclic oga inhibitor compounds |
| TWI654978B (zh) | 2017-01-27 | 2019-04-01 | 美商美國禮來大藥廠 | 5-甲基-1,2,4-二唑-3-基化合物 |
| AU2018216040A1 (en) | 2017-02-06 | 2019-06-20 | Janssen Pharmaceutica Nv | OGA inhibitor compounds |
| WO2018154133A1 (en) | 2017-02-27 | 2018-08-30 | Janssen Pharmaceutica Nv | [1,2,4]-triazolo [1,5-a]-pyrimidinyl derivatives substituted with piperidine, morpholine or piperazine as oga inhibitors |
| AR111693A1 (es) | 2017-05-25 | 2019-08-07 | Lilly Co Eli | Compuestos de 5-metil-1,3,4-oxadiazol-2-ilo con actividad inhibitoria de oga |
| WO2019037860A1 (en) | 2017-08-24 | 2019-02-28 | Asceneuron S.A. | LINEAR INHIBITORS OF GLYCOSIDASE |
| WO2020039029A1 (en) | 2018-08-22 | 2020-02-27 | Asceneuron S. A. | Spiro compounds as glycosidase inhibitors |
| WO2020039027A1 (en) | 2018-08-22 | 2020-02-27 | Asceneuron S. A. | Pyrrolidine glycosidase inhibitors |
| WO2020039028A1 (en) | 2018-08-22 | 2020-02-27 | Asceneuron S. A. | Tetrahydro-benzoazepine glycosidase inhibitors |
| US20220143042A1 (en) | 2019-02-22 | 2022-05-12 | Asceneuron Sa | Fused glycosidase inhibitors |
-
2019
- 2019-08-22 IL IL280841A patent/IL280841B2/en unknown
- 2019-08-22 EA EA202190275A patent/EA202190275A1/ru unknown
- 2019-08-22 BR BR112021003022-0A patent/BR112021003022A2/pt unknown
- 2019-08-22 JP JP2021509145A patent/JP7407171B2/ja active Active
- 2019-08-22 EP EP19759533.3A patent/EP3841093B1/en active Active
- 2019-08-22 AU AU2019324522A patent/AU2019324522B2/en active Active
- 2019-08-22 US US17/269,811 patent/US12195455B2/en active Active
- 2019-08-22 MA MA053429A patent/MA53429A/fr unknown
- 2019-08-22 HU HUE19759533A patent/HUE063226T2/hu unknown
- 2019-08-22 MX MX2021002111A patent/MX2021002111A/es unknown
- 2019-08-22 WO PCT/EP2019/072474 patent/WO2020039030A1/en not_active Ceased
- 2019-08-22 CN CN201980055121.7A patent/CN113166083A/zh active Pending
- 2019-08-22 ES ES19759533T patent/ES2954474T3/es active Active
- 2019-08-22 PL PL19759533.3T patent/PL3841093T3/pl unknown
- 2019-08-22 CA CA3110032A patent/CA3110032A1/en active Pending
- 2019-08-22 KR KR1020217008344A patent/KR102839500B1/ko active Active
- 2019-08-22 SG SG11202101651RA patent/SG11202101651RA/en unknown
-
2021
- 2021-02-12 ZA ZA2021/00981A patent/ZA202100981B/en unknown
-
2024
- 2024-10-28 AU AU2024227691A patent/AU2024227691A1/en active Pending
Also Published As
| Publication number | Publication date |
|---|---|
| SG11202101651RA (en) | 2021-03-30 |
| KR102839500B1 (ko) | 2025-07-28 |
| WO2020039030A1 (en) | 2020-02-27 |
| PL3841093T3 (pl) | 2023-12-11 |
| JP2021535103A (ja) | 2021-12-16 |
| HUE063226T2 (hu) | 2024-01-28 |
| US20210198250A1 (en) | 2021-07-01 |
| CN113166083A (zh) | 2021-07-23 |
| EA202190275A1 (ru) | 2021-08-02 |
| IL280841A (en) | 2021-04-29 |
| KR20210049849A (ko) | 2021-05-06 |
| MA53429A (fr) | 2022-03-30 |
| MX2021002111A (es) | 2021-07-16 |
| IL280841B2 (en) | 2024-12-01 |
| EP3841093B1 (en) | 2023-06-21 |
| BR112021003022A2 (pt) | 2021-05-11 |
| EP3841093A1 (en) | 2021-06-30 |
| IL280841B1 (en) | 2024-08-01 |
| US12195455B2 (en) | 2025-01-14 |
| JP7407171B2 (ja) | 2023-12-28 |
| AU2019324522A1 (en) | 2021-03-04 |
| EP3841093C0 (en) | 2023-06-21 |
| ZA202100981B (en) | 2025-10-29 |
| CA3110032A1 (en) | 2020-02-27 |
| AU2024227691A1 (en) | 2024-11-21 |
| AU2019324522B2 (en) | 2024-11-14 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2954474T3 (es) | Sales de adición de ácido de succinato y fumarato de derivados de piperazina útiles como inhibidores de glucosidasa | |
| EP3157904B1 (en) | Anti-infective compounds | |
| ES2879351T3 (es) | Sales de derivados de piperazina obtenidas por adición de ácidos | |
| AU2013366513B2 (en) | Novel benzimidazole derivatives as kinase inhibitors | |
| EP3613737B1 (en) | Novel inhibitor of cyclin-dependent kinase cdk9 | |
| KR20210126036A (ko) | 화합물, 조성물 및 방법 | |
| BR112020018562A2 (pt) | Processo preparativo | |
| CA2987570A1 (en) | 1,3,4-oxadiazole sulfamide derivative compounds as histone deacetylase 6 inhibitor, and the pharmaceutical composition comprising the same | |
| JP2019503362A (ja) | Dubの阻害剤としてのシアノピロリジン誘導体 | |
| CN101341138A (zh) | 作为gsk-3抑制剂的环状苯胺基-吡啶并三嗪类 | |
| CA3045855A1 (en) | Nitrogenous macrocyclic compound, preparation method therefor, pharmaceutical composition and application thereof | |
| AU2001236698A1 (en) | 2-benzothiazolyl urea derivatives and their use as protein kinase inhibitors | |
| AU2016382372B2 (en) | Sulfonamide derivative and preparation method and use thereof | |
| EP4618979A2 (en) | Compounds, compositions, and methods | |
| CN108463222B (zh) | 用于治疗疾病的杂环化合物 | |
| KR20190086445A (ko) | Dyrk1 억제제로서의 벤조사이아졸 유도체 | |
| WO2011127406A2 (en) | Acridines as inhibitors of haspin and dyrk kinases | |
| JP6858252B2 (ja) | ラパマイシンシグナル伝達経路阻害剤のメカニズム標的、及びその治療応用 | |
| ES3053759T3 (en) | Gpr52 modulator compounds | |
| AU2020410900B2 (en) | Compound used as RET kinase inhibitor and application thereof | |
| CN107428730B (zh) | 作为抗癌试剂的1,3,5-三嗪基pi3k抑制剂及其制备方法 | |
| CA3074981A1 (en) | Chemically activated water-soluble prodrug | |
| HK40057124B (en) | Succinate and fumarate acid addition salts of piperazine derivatives useful as glycosidase inhibitors | |
| HK40057124A (en) | Succinate and fumarate acid addition salts of piperazine derivatives useful as glycosidase inhibitors | |
| US20190135746A1 (en) | Indoline derivatives and method for using and producing the same |