ES2954596T3 - Profármacos unidos a lípidos - Google Patents
Profármacos unidos a lípidos Download PDFInfo
- Publication number
- ES2954596T3 ES2954596T3 ES16877035T ES16877035T ES2954596T3 ES 2954596 T3 ES2954596 T3 ES 2954596T3 ES 16877035 T ES16877035 T ES 16877035T ES 16877035 T ES16877035 T ES 16877035T ES 2954596 T3 ES2954596 T3 ES 2954596T3
- Authority
- ES
- Spain
- Prior art keywords
- alkyl
- mmol
- lipid
- dex
- optionally
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 239000000651 prodrug Substances 0.000 title abstract description 155
- 229940002612 prodrug Drugs 0.000 title abstract description 155
- 150000002632 lipids Chemical class 0.000 title abstract description 128
- 150000001875 compounds Chemical class 0.000 claims abstract description 101
- 238000011282 treatment Methods 0.000 claims abstract description 12
- 125000004432 carbon atom Chemical group C* 0.000 claims description 95
- 239000003814 drug Substances 0.000 claims description 89
- 229940079593 drug Drugs 0.000 claims description 87
- 229960003957 dexamethasone Drugs 0.000 claims description 38
- UREBDLICKHMUKA-CXSFZGCWSA-N dexamethasone Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1C[C@@H](C)[C@@](C(=O)CO)(O)[C@@]1(C)C[C@@H]2O UREBDLICKHMUKA-CXSFZGCWSA-N 0.000 claims description 37
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical group [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 claims description 27
- 125000004429 atom Chemical group 0.000 claims description 24
- 125000000250 methylamino group Chemical group [H]N(*)C([H])([H])[H] 0.000 claims description 24
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 claims description 21
- 239000008194 pharmaceutical composition Substances 0.000 claims description 11
- 206010028980 Neoplasm Diseases 0.000 claims description 8
- 201000011510 cancer Diseases 0.000 claims description 6
- 208000023275 Autoimmune disease Diseases 0.000 claims description 4
- 239000003937 drug carrier Substances 0.000 claims description 4
- 208000015181 infectious disease Diseases 0.000 claims description 4
- 238000000034 method Methods 0.000 abstract description 25
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 109
- 125000000217 alkyl group Chemical group 0.000 description 103
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical class CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 92
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 90
- -1 Calcitrol Chemical compound 0.000 description 83
- 239000000203 mixture Substances 0.000 description 76
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 54
- 235000019439 ethyl acetate Nutrition 0.000 description 54
- 239000000243 solution Substances 0.000 description 54
- 229910052786 argon Inorganic materials 0.000 description 45
- 229910052760 oxygen Inorganic materials 0.000 description 44
- 239000011541 reaction mixture Substances 0.000 description 43
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 42
- 239000010779 crude oil Substances 0.000 description 40
- 229910052799 carbon Inorganic materials 0.000 description 39
- 229960003668 docetaxel Drugs 0.000 description 37
- ZDZOTLJHXYCWBA-VCVYQWHSSA-N N-debenzoyl-N-(tert-butoxycarbonyl)-10-deacetyltaxol Chemical compound O([C@H]1[C@H]2[C@@](C([C@H](O)C3=C(C)[C@@H](OC(=O)[C@H](O)[C@@H](NC(=O)OC(C)(C)C)C=4C=CC=CC=4)C[C@]1(O)C3(C)C)=O)(C)[C@@H](O)C[C@H]1OC[C@]12OC(=O)C)C(=O)C1=CC=CC=C1 ZDZOTLJHXYCWBA-VCVYQWHSSA-N 0.000 description 36
- 239000002105 nanoparticle Substances 0.000 description 35
- 239000012230 colorless oil Substances 0.000 description 34
- 229910052717 sulfur Inorganic materials 0.000 description 34
- 230000015572 biosynthetic process Effects 0.000 description 33
- 238000003786 synthesis reaction Methods 0.000 description 32
- 238000009472 formulation Methods 0.000 description 31
- 239000011734 sodium Substances 0.000 description 29
- 125000004122 cyclic group Chemical group 0.000 description 28
- 239000007787 solid Substances 0.000 description 26
- 238000003818 flash chromatography Methods 0.000 description 24
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 23
- 150000003839 salts Chemical class 0.000 description 22
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 22
- 238000005160 1H NMR spectroscopy Methods 0.000 description 21
- 229960000549 4-dimethylaminophenol Drugs 0.000 description 21
- 241000699666 Mus <mouse, genus> Species 0.000 description 21
- 229910004298 SiO 2 Inorganic materials 0.000 description 21
- 210000004027 cell Anatomy 0.000 description 21
- 108090000371 Esterases Proteins 0.000 description 20
- 238000011534 incubation Methods 0.000 description 20
- 125000005647 linker group Chemical group 0.000 description 19
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 18
- 238000000746 purification Methods 0.000 description 17
- 238000003756 stirring Methods 0.000 description 17
- 102000004127 Cytokines Human genes 0.000 description 16
- 108090000695 Cytokines Proteins 0.000 description 16
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 16
- 239000003921 oil Substances 0.000 description 16
- 241000699670 Mus sp. Species 0.000 description 15
- 239000012267 brine Substances 0.000 description 15
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 15
- 238000005481 NMR spectroscopy Methods 0.000 description 14
- 239000000284 extract Substances 0.000 description 14
- GLVAUDGFNGKCSF-UHFFFAOYSA-N mercaptopurine Chemical compound S=C1NC=NC2=C1NC=N2 GLVAUDGFNGKCSF-UHFFFAOYSA-N 0.000 description 14
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 14
- 229910052757 nitrogen Inorganic materials 0.000 description 14
- 238000004704 ultra performance liquid chromatography Methods 0.000 description 14
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 12
- VPGRYOFKCNULNK-ACXQXYJUSA-N Deoxycorticosterone acetate Chemical compound C1CC2=CC(=O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H](C(=O)COC(=O)C)[C@@]1(C)CC2 VPGRYOFKCNULNK-ACXQXYJUSA-N 0.000 description 12
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 12
- APWFTHDYKJHNEV-NDEPHWFRSA-N NPC Chemical compound C1=C2C(CC)=C3CN(C(C4=C([C@@](C(=O)OC4)(O)CC)C=4)=O)C=4C3=NC2=CC=C1OC(=O)N1CCC(N)CC1 APWFTHDYKJHNEV-NDEPHWFRSA-N 0.000 description 12
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 12
- 239000002253 acid Substances 0.000 description 12
- 239000002953 phosphate buffered saline Substances 0.000 description 12
- PMATZTZNYRCHOR-CGLBZJNRSA-N Cyclosporin A Chemical compound CC[C@@H]1NC(=O)[C@H]([C@H](O)[C@H](C)C\C=C\C)N(C)C(=O)[C@H](C(C)C)N(C)C(=O)[C@H](CC(C)C)N(C)C(=O)[C@H](CC(C)C)N(C)C(=O)[C@@H](C)NC(=O)[C@H](C)NC(=O)[C@H](CC(C)C)N(C)C(=O)[C@H](C(C)C)NC(=O)[C@H](CC(C)C)N(C)C(=O)CN(C)C1=O PMATZTZNYRCHOR-CGLBZJNRSA-N 0.000 description 11
- 108010036949 Cyclosporine Proteins 0.000 description 11
- 229960001265 ciclosporin Drugs 0.000 description 11
- 230000003308 immunostimulating effect Effects 0.000 description 11
- 238000010348 incorporation Methods 0.000 description 11
- SHGAZHPCJJPHSC-NUEINMDLSA-N Isotretinoin Chemical compound OC(=O)C=C(C)/C=C/C=C(C)C=CC1=C(C)CCCC1(C)C SHGAZHPCJJPHSC-NUEINMDLSA-N 0.000 description 10
- 241001465754 Metazoa Species 0.000 description 10
- 101001043818 Mus musculus Interleukin-31 receptor subunit alpha Proteins 0.000 description 10
- 125000002252 acyl group Chemical group 0.000 description 10
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 10
- HVYWMOMLDIMFJA-DPAQBDIFSA-N cholesterol Chemical compound C1C=C2C[C@@H](O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H]([C@H](C)CCCC(C)C)[C@@]1(C)CC2 HVYWMOMLDIMFJA-DPAQBDIFSA-N 0.000 description 10
- 239000002158 endotoxin Substances 0.000 description 10
- 229960005280 isotretinoin Drugs 0.000 description 10
- 229920006008 lipopolysaccharide Polymers 0.000 description 10
- HPNSFSBZBAHARI-UHFFFAOYSA-N micophenolic acid Natural products OC1=C(CC=C(C)CCC(O)=O)C(OC)=C(C)C2=C1C(=O)OC2 HPNSFSBZBAHARI-UHFFFAOYSA-N 0.000 description 10
- HPNSFSBZBAHARI-RUDMXATFSA-N mycophenolic acid Chemical compound OC1=C(C\C=C(/C)CCC(O)=O)C(OC)=C(C)C2=C1C(=O)OC2 HPNSFSBZBAHARI-RUDMXATFSA-N 0.000 description 10
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 9
- 229930105110 Cyclosporin A Natural products 0.000 description 9
- QJJXYPPXXYFBGM-LFZNUXCKSA-N Tacrolimus Chemical compound C1C[C@@H](O)[C@H](OC)C[C@@H]1\C=C(/C)[C@@H]1[C@H](C)[C@@H](O)CC(=O)[C@H](CC=C)/C=C(C)/C[C@H](C)C[C@H](OC)[C@H]([C@H](C[C@H]2C)OC)O[C@@]2(O)C(=O)C(=O)N2CCCC[C@H]2C(=O)O1 QJJXYPPXXYFBGM-LFZNUXCKSA-N 0.000 description 9
- 230000015556 catabolic process Effects 0.000 description 9
- 238000003776 cleavage reaction Methods 0.000 description 9
- 238000006731 degradation reaction Methods 0.000 description 9
- 239000000706 filtrate Substances 0.000 description 9
- 230000002209 hydrophobic effect Effects 0.000 description 9
- 238000002347 injection Methods 0.000 description 9
- 239000007924 injection Substances 0.000 description 9
- 125000000896 monocarboxylic acid group Chemical group 0.000 description 9
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 9
- 230000007017 scission Effects 0.000 description 9
- 229960001967 tacrolimus Drugs 0.000 description 9
- QJJXYPPXXYFBGM-SHYZHZOCSA-N tacrolimus Natural products CO[C@H]1C[C@H](CC[C@@H]1O)C=C(C)[C@H]2OC(=O)[C@H]3CCCCN3C(=O)C(=O)[C@@]4(O)O[C@@H]([C@H](C[C@H]4C)OC)[C@@H](C[C@H](C)CC(=C[C@@H](CC=C)C(=O)C[C@H](O)[C@H]2C)C)OC QJJXYPPXXYFBGM-SHYZHZOCSA-N 0.000 description 9
- LTMHDMANZUZIPE-AMTYYWEZSA-N Digoxin Natural products O([C@H]1[C@H](C)O[C@H](O[C@@H]2C[C@@H]3[C@@](C)([C@@H]4[C@H]([C@]5(O)[C@](C)([C@H](O)C4)[C@H](C4=CC(=O)OC4)CC5)CC3)CC2)C[C@@H]1O)[C@H]1O[C@H](C)[C@@H](O[C@H]2O[C@@H](C)[C@H](O)[C@@H](O)C2)[C@@H](O)C1 LTMHDMANZUZIPE-AMTYYWEZSA-N 0.000 description 8
- FBOZXECLQNJBKD-ZDUSSCGKSA-N L-methotrexate Chemical compound C=1N=C2N=C(N)N=C(N)C2=NC=1CN(C)C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 FBOZXECLQNJBKD-ZDUSSCGKSA-N 0.000 description 8
- 239000002144 L01XE18 - Ruxolitinib Substances 0.000 description 8
- 239000004012 Tofacitinib Substances 0.000 description 8
- 125000000738 acetamido group Chemical group [H]C([H])([H])C(=O)N([H])[*] 0.000 description 8
- 125000000218 acetic acid group Chemical group C(C)(=O)* 0.000 description 8
- 125000005605 benzo group Chemical group 0.000 description 8
- 229960005156 digoxin Drugs 0.000 description 8
- LTMHDMANZUZIPE-PUGKRICDSA-N digoxin Chemical compound C1[C@H](O)[C@H](O)[C@@H](C)O[C@H]1O[C@@H]1[C@@H](C)O[C@@H](O[C@@H]2[C@H](O[C@@H](O[C@@H]3C[C@@H]4[C@]([C@@H]5[C@H]([C@]6(CC[C@@H]([C@@]6(C)[C@H](O)C5)C=5COC(=O)C=5)O)CC4)(C)CC3)C[C@@H]2O)C)C[C@@H]1O LTMHDMANZUZIPE-PUGKRICDSA-N 0.000 description 8
- LTMHDMANZUZIPE-UHFFFAOYSA-N digoxine Natural products C1C(O)C(O)C(C)OC1OC1C(C)OC(OC2C(OC(OC3CC4C(C5C(C6(CCC(C6(C)C(O)C5)C=5COC(=O)C=5)O)CC4)(C)CC3)CC2O)C)CC1O LTMHDMANZUZIPE-UHFFFAOYSA-N 0.000 description 8
- JYGXADMDTFJGBT-VWUMJDOOSA-N hydrocortisone Chemical compound O=C1CC[C@]2(C)[C@H]3[C@@H](O)C[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 JYGXADMDTFJGBT-VWUMJDOOSA-N 0.000 description 8
- 238000001727 in vivo Methods 0.000 description 8
- 229960000485 methotrexate Drugs 0.000 description 8
- 125000000951 phenoxy group Chemical group [H]C1=C([H])C([H])=C(O*)C([H])=C1[H] 0.000 description 8
- 229960005205 prednisolone Drugs 0.000 description 8
- OIGNJSKKLXVSLS-VWUMJDOOSA-N prednisolone Chemical compound O=C1C=C[C@]2(C)[C@H]3[C@@H](O)C[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 OIGNJSKKLXVSLS-VWUMJDOOSA-N 0.000 description 8
- XOFYZVNMUHMLCC-ZPOLXVRWSA-N prednisone Chemical compound O=C1C=C[C@]2(C)[C@H]3C(=O)C[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 XOFYZVNMUHMLCC-ZPOLXVRWSA-N 0.000 description 8
- 229960004618 prednisone Drugs 0.000 description 8
- 238000002360 preparation method Methods 0.000 description 8
- 229960000215 ruxolitinib Drugs 0.000 description 8
- HFNKQEVNSGCOJV-OAHLLOKOSA-N ruxolitinib Chemical compound C1([C@@H](CC#N)N2N=CC(=C2)C=2C=3C=CNC=3N=CN=2)CCCC1 HFNKQEVNSGCOJV-OAHLLOKOSA-N 0.000 description 8
- 239000002904 solvent Substances 0.000 description 8
- KDYFGRWQOYBRFD-UHFFFAOYSA-L succinate(2-) Chemical compound [O-]C(=O)CCC([O-])=O KDYFGRWQOYBRFD-UHFFFAOYSA-L 0.000 description 8
- 230000001225 therapeutic effect Effects 0.000 description 8
- UJLAWZDWDVHWOW-YPMHNXCESA-N tofacitinib Chemical compound C[C@@H]1CCN(C(=O)CC#N)C[C@@H]1N(C)C1=NC=NC2=C1C=CN2 UJLAWZDWDVHWOW-YPMHNXCESA-N 0.000 description 8
- 229960001350 tofacitinib Drugs 0.000 description 8
- XEEQGYMUWCZPDN-DOMZBBRYSA-N (-)-(11S,2'R)-erythro-mefloquine Chemical compound C([C@@H]1[C@@H](O)C=2C3=CC=CC(=C3N=C(C=2)C(F)(F)F)C(F)(F)F)CCCN1 XEEQGYMUWCZPDN-DOMZBBRYSA-N 0.000 description 7
- FALRKNHUBBKYCC-UHFFFAOYSA-N 2-(chloromethyl)pyridine-3-carbonitrile Chemical compound ClCC1=NC=CC=C1C#N FALRKNHUBBKYCC-UHFFFAOYSA-N 0.000 description 7
- FJHBVJOVLFPMQE-QFIPXVFZSA-N 7-Ethyl-10-Hydroxy-Camptothecin Chemical compound C1=C(O)C=C2C(CC)=C(CN3C(C4=C([C@@](C(=O)OC4)(O)CC)C=C33)=O)C3=NC2=C1 FJHBVJOVLFPMQE-QFIPXVFZSA-N 0.000 description 7
- APKFDSVGJQXUKY-KKGHZKTASA-N Amphotericin-B Natural products O[C@H]1[C@@H](N)[C@H](O)[C@@H](C)O[C@H]1O[C@H]1C=CC=CC=CC=CC=CC=CC=C[C@H](C)[C@@H](O)[C@@H](C)[C@H](C)OC(=O)C[C@H](O)C[C@H](O)CC[C@@H](O)[C@H](O)C[C@H](O)C[C@](O)(C[C@H](O)[C@H]2C(O)=O)O[C@H]2C1 APKFDSVGJQXUKY-KKGHZKTASA-N 0.000 description 7
- AXRYRYVKAWYZBR-UHFFFAOYSA-N Atazanavir Natural products C=1C=C(C=2N=CC=CC=2)C=CC=1CN(NC(=O)C(NC(=O)OC)C(C)(C)C)CC(O)C(NC(=O)C(NC(=O)OC)C(C)(C)C)CC1=CC=CC=C1 AXRYRYVKAWYZBR-UHFFFAOYSA-N 0.000 description 7
- 108010019625 Atazanavir Sulfate Proteins 0.000 description 7
- CYQFCXCEBYINGO-DLBZAZTESA-N Dronabinol Natural products C1=C(C)CC[C@H]2C(C)(C)OC3=CC(CCCCC)=CC(O)=C3[C@H]21 CYQFCXCEBYINGO-DLBZAZTESA-N 0.000 description 7
- VWUXBMIQPBEWFH-WCCTWKNTSA-N Fulvestrant Chemical compound OC1=CC=C2[C@H]3CC[C@](C)([C@H](CC4)O)[C@@H]4[C@@H]3[C@H](CCCCCCCCCS(=O)CCCC(F)(F)C(F)(F)F)CC2=C1 VWUXBMIQPBEWFH-WCCTWKNTSA-N 0.000 description 7
- ARFHIAQFJWUCFH-IZZDOVSWSA-N Nifurtimox Chemical compound CC1CS(=O)(=O)CCN1\N=C\C1=CC=C([N+]([O-])=O)O1 ARFHIAQFJWUCFH-IZZDOVSWSA-N 0.000 description 7
- 229930012538 Paclitaxel Natural products 0.000 description 7
- BUQLXKSONWUQAC-UHFFFAOYSA-N Parthenolide Natural products CC1C2OC(=O)C(=C)C2CCC(=C/CCC1(C)O)C BUQLXKSONWUQAC-UHFFFAOYSA-N 0.000 description 7
- PJSFRIWCGOHTNF-UHFFFAOYSA-N Sulphormetoxin Chemical compound COC1=NC=NC(NS(=O)(=O)C=2C=CC(N)=CC=2)=C1OC PJSFRIWCGOHTNF-UHFFFAOYSA-N 0.000 description 7
- CYQFCXCEBYINGO-UHFFFAOYSA-N THC Natural products C1=C(C)CCC2C(C)(C)OC3=CC(CCCCC)=CC(O)=C3C21 CYQFCXCEBYINGO-UHFFFAOYSA-N 0.000 description 7
- NAVMQTYZDKMPEU-UHFFFAOYSA-N Targretin Chemical compound CC1=CC(C(CCC2(C)C)(C)C)=C2C=C1C(=C)C1=CC=C(C(O)=O)C=C1 NAVMQTYZDKMPEU-UHFFFAOYSA-N 0.000 description 7
- BHTRKEVKTKCXOH-UHFFFAOYSA-N Taurochenodesoxycholsaeure Natural products OC1CC2CC(O)CCC2(C)C2C1C1CCC(C(CCC(=O)NCCS(O)(=O)=O)C)C1(C)CC2 BHTRKEVKTKCXOH-UHFFFAOYSA-N 0.000 description 7
- MCGSCOLBFJQGHM-SCZZXKLOSA-N abacavir Chemical compound C=12N=CN([C@H]3C=C[C@@H](CO)C3)C2=NC(N)=NC=1NC1CC1 MCGSCOLBFJQGHM-SCZZXKLOSA-N 0.000 description 7
- 229960004748 abacavir Drugs 0.000 description 7
- HXHWSAZORRCQMX-UHFFFAOYSA-N albendazole Chemical compound CCCSC1=CC=C2NC(NC(=O)OC)=NC2=C1 HXHWSAZORRCQMX-UHFFFAOYSA-N 0.000 description 7
- APKFDSVGJQXUKY-INPOYWNPSA-N amphotericin B Chemical compound O[C@H]1[C@@H](N)[C@H](O)[C@@H](C)O[C@H]1O[C@H]1/C=C/C=C/C=C/C=C/C=C/C=C/C=C/[C@H](C)[C@@H](O)[C@@H](C)[C@H](C)OC(=O)C[C@H](O)C[C@H](O)CC[C@@H](O)[C@H](O)C[C@H](O)C[C@](O)(C[C@H](O)[C@H]2C(O)=O)O[C@H]2C1 APKFDSVGJQXUKY-INPOYWNPSA-N 0.000 description 7
- 229960003942 amphotericin b Drugs 0.000 description 7
- YMARZQAQMVYCKC-OEMFJLHTSA-N amprenavir Chemical compound C([C@@H]([C@H](O)CN(CC(C)C)S(=O)(=O)C=1C=CC(N)=CC=1)NC(=O)O[C@@H]1COCC1)C1=CC=CC=C1 YMARZQAQMVYCKC-OEMFJLHTSA-N 0.000 description 7
- 229960001830 amprenavir Drugs 0.000 description 7
- 238000004458 analytical method Methods 0.000 description 7
- AXRYRYVKAWYZBR-GASGPIRDSA-N atazanavir Chemical compound C([C@H](NC(=O)[C@@H](NC(=O)OC)C(C)(C)C)[C@@H](O)CN(CC=1C=CC(=CC=1)C=1N=CC=CC=1)NC(=O)[C@@H](NC(=O)OC)C(C)(C)C)C1=CC=CC=C1 AXRYRYVKAWYZBR-GASGPIRDSA-N 0.000 description 7
- 229960003277 atazanavir Drugs 0.000 description 7
- 229960002938 bexarotene Drugs 0.000 description 7
- 210000004369 blood Anatomy 0.000 description 7
- 239000008280 blood Substances 0.000 description 7
- ISZNZKHCRKXXAU-UHFFFAOYSA-N chlorproguanil Chemical compound CC(C)\N=C(/N)N=C(N)NC1=CC=C(Cl)C(Cl)=C1 ISZNZKHCRKXXAU-UHFFFAOYSA-N 0.000 description 7
- 229950000764 chlorproguanil Drugs 0.000 description 7
- ZCIGNRJZKPOIKD-CQXVEOKZSA-N cobicistat Chemical compound S1C(C(C)C)=NC(CN(C)C(=O)N[C@@H](CCN2CCOCC2)C(=O)N[C@H](CC[C@H](CC=2C=CC=CC=2)NC(=O)OCC=2SC=NC=2)CC=2C=CC=CC=2)=C1 ZCIGNRJZKPOIKD-CQXVEOKZSA-N 0.000 description 7
- 229960002402 cobicistat Drugs 0.000 description 7
- CYQFCXCEBYINGO-IAGOWNOFSA-N delta1-THC Chemical compound C1=C(C)CC[C@H]2C(C)(C)OC3=CC(CCCCC)=CC(O)=C3[C@@H]21 CYQFCXCEBYINGO-IAGOWNOFSA-N 0.000 description 7
- NIJJYAXOARWZEE-UHFFFAOYSA-N di-n-propyl-acetic acid Natural products CCCC(C(O)=O)CCC NIJJYAXOARWZEE-UHFFFAOYSA-N 0.000 description 7
- HKXBNHCUPKIYDM-CGMHZMFXSA-N doxercalciferol Chemical compound C1(/[C@@H]2CC[C@@H]([C@]2(CCC1)C)[C@H](C)/C=C/[C@H](C)C(C)C)=C\C=C1\C[C@@H](O)C[C@H](O)C1=C HKXBNHCUPKIYDM-CGMHZMFXSA-N 0.000 description 7
- 229960000413 doxercalciferol Drugs 0.000 description 7
- 229960004242 dronabinol Drugs 0.000 description 7
- 229960005420 etoposide Drugs 0.000 description 7
- VJJPUSNTGOMMGY-MRVIYFEKSA-N etoposide Chemical compound COC1=C(O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@H](C)OC[C@H]4O3)O)[C@@H]3[C@@H]2C(OC3)=O)=C1 VJJPUSNTGOMMGY-MRVIYFEKSA-N 0.000 description 7
- 238000002474 experimental method Methods 0.000 description 7
- 229960002258 fulvestrant Drugs 0.000 description 7
- 230000014509 gene expression Effects 0.000 description 7
- 229960004580 glibenclamide Drugs 0.000 description 7
- ZNNLBTZKUZBEKO-UHFFFAOYSA-N glyburide Chemical compound COC1=CC=C(Cl)C=C1C(=O)NCCC1=CC=C(S(=O)(=O)NC(=O)NC2CCCCC2)C=C1 ZNNLBTZKUZBEKO-UHFFFAOYSA-N 0.000 description 7
- 229910052739 hydrogen Inorganic materials 0.000 description 7
- 230000001404 mediated effect Effects 0.000 description 7
- 229960001962 mefloquine Drugs 0.000 description 7
- 229960001428 mercaptopurine Drugs 0.000 description 7
- 108020004999 messenger RNA Proteins 0.000 description 7
- 229960002644 nifurtimox Drugs 0.000 description 7
- 229960001592 paclitaxel Drugs 0.000 description 7
- 229960000987 paricalcitol Drugs 0.000 description 7
- BPKAHTKRCLCHEA-UBFJEZKGSA-N paricalcitol Chemical compound C1(/[C@@H]2CC[C@@H]([C@]2(CCC1)C)[C@@H](\C=C\[C@H](C)C(C)(C)O)C)=C\C=C1C[C@@H](O)C[C@H](O)C1 BPKAHTKRCLCHEA-UBFJEZKGSA-N 0.000 description 7
- KTEXNACQROZXEV-PVLRGYAZSA-N parthenolide Chemical compound C1CC(/C)=C/CC[C@@]2(C)O[C@@H]2[C@H]2OC(=O)C(=C)[C@@H]21 KTEXNACQROZXEV-PVLRGYAZSA-N 0.000 description 7
- 229940069510 parthenolide Drugs 0.000 description 7
- OLBCVFGFOZPWHH-UHFFFAOYSA-N propofol Chemical compound CC(C)C1=CC=CC(C(C)C)=C1O OLBCVFGFOZPWHH-UHFFFAOYSA-N 0.000 description 7
- 229960004134 propofol Drugs 0.000 description 7
- WKSAUQYGYAYLPV-UHFFFAOYSA-N pyrimethamine Chemical compound CCC1=NC(N)=NC(N)=C1C1=CC=C(Cl)C=C1 WKSAUQYGYAYLPV-UHFFFAOYSA-N 0.000 description 7
- 229960000611 pyrimethamine Drugs 0.000 description 7
- 150000003431 steroids Chemical class 0.000 description 7
- 230000000638 stimulation Effects 0.000 description 7
- 229940014800 succinic anhydride Drugs 0.000 description 7
- 229960004673 sulfadoxine Drugs 0.000 description 7
- BHTRKEVKTKCXOH-LBSADWJPSA-N tauroursodeoxycholic acid Chemical compound C([C@H]1C[C@@H]2O)[C@H](O)CC[C@]1(C)[C@@H]1[C@@H]2[C@@H]2CC[C@H]([C@@H](CCC(=O)NCCS(O)(=O)=O)C)[C@@]2(C)CC1 BHTRKEVKTKCXOH-LBSADWJPSA-N 0.000 description 7
- RCINICONZNJXQF-MZXODVADSA-N taxol Chemical compound O([C@@H]1[C@@]2(C[C@@H](C(C)=C(C2(C)C)[C@H](C([C@]2(C)[C@@H](O)C[C@H]3OC[C@]3([C@H]21)OC(C)=O)=O)OC(=O)C)OC(=O)[C@H](O)[C@@H](NC(=O)C=1C=CC=CC=1)C=1C=CC=CC=1)O)C(=O)C1=CC=CC=C1 RCINICONZNJXQF-MZXODVADSA-N 0.000 description 7
- 229960001278 teniposide Drugs 0.000 description 7
- NRUKOCRGYNPUPR-QBPJDGROSA-N teniposide Chemical compound COC1=C(O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@@H](OC[C@H]4O3)C=3SC=CC=3)O)[C@@H]3[C@@H]2C(OC3)=O)=C1 NRUKOCRGYNPUPR-QBPJDGROSA-N 0.000 description 7
- MSRILKIQRXUYCT-UHFFFAOYSA-M valproate semisodium Chemical compound [Na+].CCCC(C(O)=O)CCC.CCCC(C([O-])=O)CCC MSRILKIQRXUYCT-UHFFFAOYSA-M 0.000 description 7
- 229960000604 valproic acid Drugs 0.000 description 7
- HBOMLICNUCNMMY-XLPZGREQSA-N zidovudine Chemical compound O=C1NC(=O)C(C)=CN1[C@@H]1O[C@H](CO)[C@@H](N=[N+]=[N-])C1 HBOMLICNUCNMMY-XLPZGREQSA-N 0.000 description 7
- 108091034117 Oligonucleotide Proteins 0.000 description 6
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 6
- SHGAZHPCJJPHSC-YCNIQYBTSA-N all-trans-retinoic acid Chemical compound OC(=O)\C=C(/C)\C=C\C=C(/C)\C=C\C1=C(C)CCCC1(C)C SHGAZHPCJJPHSC-YCNIQYBTSA-N 0.000 description 6
- 201000010099 disease Diseases 0.000 description 6
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 6
- 238000010494 dissociation reaction Methods 0.000 description 6
- 230000005593 dissociations Effects 0.000 description 6
- 230000000694 effects Effects 0.000 description 6
- 125000000524 functional group Chemical group 0.000 description 6
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Natural products C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 description 6
- 230000008629 immune suppression Effects 0.000 description 6
- 229960000951 mycophenolic acid Drugs 0.000 description 6
- 239000002245 particle Substances 0.000 description 6
- 150000003138 primary alcohols Chemical class 0.000 description 6
- ZAHRKKWIAAJSAO-UHFFFAOYSA-N rapamycin Natural products COCC(O)C(=C/C(C)C(=O)CC(OC(=O)C1CCCCN1C(=O)C(=O)C2(O)OC(CC(OC)C(=CC=CC=CC(C)CC(C)C(=O)C)C)CCC2C)C(C)CC3CCC(O)C(C3)OC)C ZAHRKKWIAAJSAO-UHFFFAOYSA-N 0.000 description 6
- 229920006395 saturated elastomer Polymers 0.000 description 6
- 229960002930 sirolimus Drugs 0.000 description 6
- QFJCIRLUMZQUOT-HPLJOQBZSA-N sirolimus Chemical compound C1C[C@@H](O)[C@H](OC)C[C@@H]1C[C@@H](C)[C@H]1OC(=O)[C@@H]2CCCCN2C(=O)C(=O)[C@](O)(O2)[C@H](C)CC[C@H]2C[C@H](OC)/C(C)=C/C=C/C=C/[C@@H](C)C[C@@H](C)C(=O)[C@H](OC)[C@H](O)/C(C)=C/[C@@H](C)C(=O)C1 QFJCIRLUMZQUOT-HPLJOQBZSA-N 0.000 description 6
- 229940034208 thyroxine Drugs 0.000 description 6
- 229960001727 tretinoin Drugs 0.000 description 6
- 238000001644 13C nuclear magnetic resonance spectroscopy Methods 0.000 description 5
- ABFPKTQEQNICFT-UHFFFAOYSA-M 2-chloro-1-methylpyridin-1-ium;iodide Chemical compound [I-].C[N+]1=CC=CC=C1Cl ABFPKTQEQNICFT-UHFFFAOYSA-M 0.000 description 5
- 102000004190 Enzymes Human genes 0.000 description 5
- 108090000790 Enzymes Proteins 0.000 description 5
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 5
- 229960002669 albendazole Drugs 0.000 description 5
- 150000001412 amines Chemical class 0.000 description 5
- 230000008901 benefit Effects 0.000 description 5
- 238000006065 biodegradation reaction Methods 0.000 description 5
- 235000012000 cholesterol Nutrition 0.000 description 5
- 235000014113 dietary fatty acids Nutrition 0.000 description 5
- KPUWHANPEXNPJT-UHFFFAOYSA-N disiloxane Chemical compound [SiH3]O[SiH3] KPUWHANPEXNPJT-UHFFFAOYSA-N 0.000 description 5
- 229930195729 fatty acid Natural products 0.000 description 5
- 239000000194 fatty acid Substances 0.000 description 5
- 150000004665 fatty acids Chemical class 0.000 description 5
- 239000000463 material Substances 0.000 description 5
- 125000004194 piperazin-1-yl group Chemical group [H]N1C([H])([H])C([H])([H])N(*)C([H])([H])C1([H])[H] 0.000 description 5
- 239000013612 plasmid Substances 0.000 description 5
- 229960005294 triamcinolone Drugs 0.000 description 5
- GFNANZIMVAIWHM-OBYCQNJPSA-N triamcinolone Chemical compound O=C1C=C[C@]2(C)[C@@]3(F)[C@@H](O)C[C@](C)([C@@]([C@H](O)C4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 GFNANZIMVAIWHM-OBYCQNJPSA-N 0.000 description 5
- 108091032973 (ribonucleotides)n+m Proteins 0.000 description 4
- FUFLCEKSBBHCMO-UHFFFAOYSA-N 11-dehydrocorticosterone Natural products O=C1CCC2(C)C3C(=O)CC(C)(C(CC4)C(=O)CO)C4C3CCC2=C1 FUFLCEKSBBHCMO-UHFFFAOYSA-N 0.000 description 4
- WHBHBVVOGNECLV-UHFFFAOYSA-N 11-deoxy-17-hydroxy-corticosterone Natural products O=C1CCC2(C)C3CCC(C)(C(CC4)(O)C(=O)CO)C4C3CCC2=C1 WHBHBVVOGNECLV-UHFFFAOYSA-N 0.000 description 4
- WHBHBVVOGNECLV-OBQKJFGGSA-N 11-deoxycortisol Chemical compound O=C1CC[C@]2(C)[C@H]3CC[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 WHBHBVVOGNECLV-OBQKJFGGSA-N 0.000 description 4
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 4
- PQSUYGKTWSAVDQ-ZVIOFETBSA-N Aldosterone Chemical compound C([C@@]1([C@@H](C(=O)CO)CC[C@H]1[C@@H]1CC2)C=O)[C@H](O)[C@@H]1[C@]1(C)C2=CC(=O)CC1 PQSUYGKTWSAVDQ-ZVIOFETBSA-N 0.000 description 4
- PQSUYGKTWSAVDQ-UHFFFAOYSA-N Aldosterone Natural products C1CC2C3CCC(C(=O)CO)C3(C=O)CC(O)C2C2(C)C1=CC(=O)CC2 PQSUYGKTWSAVDQ-UHFFFAOYSA-N 0.000 description 4
- KPYSYYIEGFHWSV-UHFFFAOYSA-N Baclofen Chemical compound OC(=O)CC(CN)C1=CC=C(Cl)C=C1 KPYSYYIEGFHWSV-UHFFFAOYSA-N 0.000 description 4
- CULUWZNBISUWAS-UHFFFAOYSA-N Benznidazole Chemical compound [O-][N+](=O)C1=NC=CN1CC(=O)NCC1=CC=CC=C1 CULUWZNBISUWAS-UHFFFAOYSA-N 0.000 description 4
- VOVIALXJUBGFJZ-KWVAZRHASA-N Budesonide Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@@H]2[C@@H]1[C@@H]1C[C@H]3OC(CCC)O[C@@]3(C(=O)CO)[C@@]1(C)C[C@@H]2O VOVIALXJUBGFJZ-KWVAZRHASA-N 0.000 description 4
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 4
- OMFXVFTZEKFJBZ-UHFFFAOYSA-N Corticosterone Natural products O=C1CCC2(C)C3C(O)CC(C)(C(CC4)C(=O)CO)C4C3CCC2=C1 OMFXVFTZEKFJBZ-UHFFFAOYSA-N 0.000 description 4
- MFYSYFVPBJMHGN-ZPOLXVRWSA-N Cortisone Chemical compound O=C1CC[C@]2(C)[C@H]3C(=O)C[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 MFYSYFVPBJMHGN-ZPOLXVRWSA-N 0.000 description 4
- MFYSYFVPBJMHGN-UHFFFAOYSA-N Cortisone Natural products O=C1CCC2(C)C3C(=O)CC(C)(C(CC4)(O)C(=O)CO)C4C3CCC2=C1 MFYSYFVPBJMHGN-UHFFFAOYSA-N 0.000 description 4
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 4
- HKVAMNSJSFKALM-GKUWKFKPSA-N Everolimus Chemical compound C1C[C@@H](OCCO)[C@H](OC)C[C@@H]1C[C@@H](C)[C@H]1OC(=O)[C@@H]2CCCCN2C(=O)C(=O)[C@](O)(O2)[C@H](C)CC[C@H]2C[C@H](OC)/C(C)=C/C=C/C=C/[C@@H](C)C[C@@H](C)C(=O)[C@H](OC)[C@H](O)/C(C)=C/[C@@H](C)C(=O)C1 HKVAMNSJSFKALM-GKUWKFKPSA-N 0.000 description 4
- FQISKWAFAHGMGT-SGJOWKDISA-M Methylprednisolone sodium succinate Chemical compound [Na+].C([C@@]12C)=CC(=O)C=C1[C@@H](C)C[C@@H]1[C@@H]2[C@@H](O)C[C@]2(C)[C@@](O)(C(=O)COC(=O)CCC([O-])=O)CC[C@H]21 FQISKWAFAHGMGT-SGJOWKDISA-M 0.000 description 4
- SMWDFEZZVXVKRB-UHFFFAOYSA-N Quinoline Chemical compound N1=CC=CC2=CC=CC=C21 SMWDFEZZVXVKRB-UHFFFAOYSA-N 0.000 description 4
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 4
- JLCPHMBAVCMARE-UHFFFAOYSA-N [3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-hydroxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methyl [5-(6-aminopurin-9-yl)-2-(hydroxymethyl)oxolan-3-yl] hydrogen phosphate Polymers Cc1cn(C2CC(OP(O)(=O)OCC3OC(CC3OP(O)(=O)OCC3OC(CC3O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c3nc(N)[nH]c4=O)C(COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3CO)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cc(C)c(=O)[nH]c3=O)n3cc(C)c(=O)[nH]c3=O)n3ccc(N)nc3=O)n3cc(C)c(=O)[nH]c3=O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)O2)c(=O)[nH]c1=O JLCPHMBAVCMARE-UHFFFAOYSA-N 0.000 description 4
- 229960002478 aldosterone Drugs 0.000 description 4
- HTIQEAQVCYTUBX-UHFFFAOYSA-N amlodipine Chemical compound CCOC(=O)C1=C(COCCN)NC(C)=C(C(=O)OC)C1C1=CC=CC=C1Cl HTIQEAQVCYTUBX-UHFFFAOYSA-N 0.000 description 4
- 229960000528 amlodipine Drugs 0.000 description 4
- 229960002170 azathioprine Drugs 0.000 description 4
- LMEKQMALGUDUQG-UHFFFAOYSA-N azathioprine Chemical compound CN1C=NC([N+]([O-])=O)=C1SC1=NC=NC2=C1NC=N2 LMEKQMALGUDUQG-UHFFFAOYSA-N 0.000 description 4
- 229960000794 baclofen Drugs 0.000 description 4
- 239000002585 base Substances 0.000 description 4
- 229960004001 benznidazole Drugs 0.000 description 4
- 229960002537 betamethasone Drugs 0.000 description 4
- UREBDLICKHMUKA-DVTGEIKXSA-N betamethasone Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1C[C@H](C)[C@@](C(=O)CO)(O)[C@@]1(C)C[C@@H]2O UREBDLICKHMUKA-DVTGEIKXSA-N 0.000 description 4
- 229960004436 budesonide Drugs 0.000 description 4
- 229940097217 cardiac glycoside Drugs 0.000 description 4
- 239000002368 cardiac glycoside Substances 0.000 description 4
- 238000006243 chemical reaction Methods 0.000 description 4
- OMFXVFTZEKFJBZ-HJTSIMOOSA-N corticosterone Chemical compound O=C1CC[C@]2(C)[C@H]3[C@@H](O)C[C@](C)([C@H](CC4)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 OMFXVFTZEKFJBZ-HJTSIMOOSA-N 0.000 description 4
- 229960004544 cortisone Drugs 0.000 description 4
- 238000000604 cryogenic transmission electron microscopy Methods 0.000 description 4
- ZESRJSPZRDMNHY-UHFFFAOYSA-N de-oxy corticosterone Natural products O=C1CCC2(C)C3CCC(C)(C(CC4)C(=O)CO)C4C3CCC2=C1 ZESRJSPZRDMNHY-UHFFFAOYSA-N 0.000 description 4
- 229960004486 desoxycorticosterone acetate Drugs 0.000 description 4
- 229960003654 desoxycortone Drugs 0.000 description 4
- 150000002009 diols Chemical class 0.000 description 4
- LOKCTEFSRHRXRJ-UHFFFAOYSA-I dipotassium trisodium dihydrogen phosphate hydrogen phosphate dichloride Chemical compound P(=O)(O)(O)[O-].[K+].P(=O)(O)([O-])[O-].[Na+].[Na+].[Cl-].[K+].[Cl-].[Na+] LOKCTEFSRHRXRJ-UHFFFAOYSA-I 0.000 description 4
- JUZYLCPPVHEVSV-LJQANCHMSA-N elvitegravir Chemical compound COC1=CC=2N([C@H](CO)C(C)C)C=C(C(O)=O)C(=O)C=2C=C1CC1=CC=CC(Cl)=C1F JUZYLCPPVHEVSV-LJQANCHMSA-N 0.000 description 4
- 229960003586 elvitegravir Drugs 0.000 description 4
- 229960005167 everolimus Drugs 0.000 description 4
- 229960002011 fludrocortisone Drugs 0.000 description 4
- AAXVEMMRQDVLJB-BULBTXNYSA-N fludrocortisone Chemical compound O=C1CC[C@]2(C)[C@@]3(F)[C@@H](O)C[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 AAXVEMMRQDVLJB-BULBTXNYSA-N 0.000 description 4
- 229960003469 flumetasone Drugs 0.000 description 4
- WXURHACBFYSXBI-GQKYHHCASA-N flumethasone Chemical compound C1([C@@H](F)C2)=CC(=O)C=C[C@]1(C)[C@]1(F)[C@@H]2[C@@H]2C[C@@H](C)[C@@](C(=O)CO)(O)[C@@]2(C)C[C@@H]1O WXURHACBFYSXBI-GQKYHHCASA-N 0.000 description 4
- 229960002714 fluticasone Drugs 0.000 description 4
- MGNNYOODZCAHBA-GQKYHHCASA-N fluticasone Chemical compound C1([C@@H](F)C2)=CC(=O)C=C[C@]1(C)[C@]1(F)[C@@H]2[C@@H]2C[C@@H](C)[C@@](C(=O)SCF)(O)[C@@]2(C)C[C@@H]1O MGNNYOODZCAHBA-GQKYHHCASA-N 0.000 description 4
- MNWFXJYAOYHMED-UHFFFAOYSA-M heptanoate Chemical compound CCCCCCC([O-])=O MNWFXJYAOYHMED-UHFFFAOYSA-M 0.000 description 4
- 229960000890 hydrocortisone Drugs 0.000 description 4
- 230000028993 immune response Effects 0.000 description 4
- 239000007943 implant Substances 0.000 description 4
- 238000000338 in vitro Methods 0.000 description 4
- VHOGYURTWQBHIL-UHFFFAOYSA-N leflunomide Chemical compound O1N=CC(C(=O)NC=2C=CC(=CC=2)C(F)(F)F)=C1C VHOGYURTWQBHIL-UHFFFAOYSA-N 0.000 description 4
- 229960000681 leflunomide Drugs 0.000 description 4
- OYHQOLUKZRVURQ-IXWMQOLASA-N linoleic acid Natural products CCCCC\C=C/C\C=C\CCCCCCCC(O)=O OYHQOLUKZRVURQ-IXWMQOLASA-N 0.000 description 4
- 229920002521 macromolecule Polymers 0.000 description 4
- 239000011777 magnesium Substances 0.000 description 4
- 229960004584 methylprednisolone Drugs 0.000 description 4
- 229960001664 mometasone Drugs 0.000 description 4
- QLIIKPVHVRXHRI-CXSFZGCWSA-N mometasone Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@]2(Cl)[C@@H]1[C@@H]1C[C@@H](C)[C@@](C(=O)CCl)(O)[C@@]1(C)C[C@@H]2O QLIIKPVHVRXHRI-CXSFZGCWSA-N 0.000 description 4
- 229940014456 mycophenolate Drugs 0.000 description 4
- 108020004707 nucleic acids Proteins 0.000 description 4
- 102000039446 nucleic acids Human genes 0.000 description 4
- 150000007523 nucleic acids Chemical class 0.000 description 4
- CTSLXHKWHWQRSH-UHFFFAOYSA-N oxalyl chloride Chemical compound ClC(=O)C(Cl)=O CTSLXHKWHWQRSH-UHFFFAOYSA-N 0.000 description 4
- 239000000546 pharmaceutical excipient Substances 0.000 description 4
- 238000011533 pre-incubation Methods 0.000 description 4
- GRJJQCWNZGRKAU-UHFFFAOYSA-N pyridin-1-ium;fluoride Chemical compound F.C1=CC=NC=C1 GRJJQCWNZGRKAU-UHFFFAOYSA-N 0.000 description 4
- 239000012453 solvate Substances 0.000 description 4
- 229930002534 steroid glycoside Natural products 0.000 description 4
- 150000008143 steroidal glycosides Chemical class 0.000 description 4
- 239000000126 substance Substances 0.000 description 4
- 239000000725 suspension Substances 0.000 description 4
- YAPQBXQYLJRXSA-UHFFFAOYSA-N theobromine Chemical compound CN1C(=O)NC(=O)C2=C1N=CN2C YAPQBXQYLJRXSA-UHFFFAOYSA-N 0.000 description 4
- 230000002588 toxic effect Effects 0.000 description 4
- 231100000419 toxicity Toxicity 0.000 description 4
- 230000001988 toxicity Effects 0.000 description 4
- 239000003039 volatile agent Substances 0.000 description 4
- BVHMITOWPMCRLY-HZJYTTRNSA-N (6z,9z)-18-chlorooctadeca-6,9-diene Chemical compound CCCCC\C=C/C\C=C/CCCCCCCCCl BVHMITOWPMCRLY-HZJYTTRNSA-N 0.000 description 3
- JXNPEDYJTDQORS-HZJYTTRNSA-N (9Z,12Z)-octadecadien-1-ol Chemical compound CCCCC\C=C/C\C=C/CCCCCCCCO JXNPEDYJTDQORS-HZJYTTRNSA-N 0.000 description 3
- MQGBAQLIFKSMEM-MAZCIEHSSA-N 1,2-dilinoleoylglycerol Chemical compound CCCCC\C=C/C\C=C/CCCCCCCC(=O)OCC(CO)OC(=O)CCCCCCC\C=C/C\C=C/CCCCC MQGBAQLIFKSMEM-MAZCIEHSSA-N 0.000 description 3
- ZOCUOMKMBMEYQV-GSLJADNHSA-N 9alpha-Fluoro-11beta,17alpha,21-trihydroxypregna-1,4-diene-3,20-dione 21-acetate Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1CC[C@@](C(=O)COC(=O)C)(O)[C@@]1(C)C[C@@H]2O ZOCUOMKMBMEYQV-GSLJADNHSA-N 0.000 description 3
- 108091006112 ATPases Proteins 0.000 description 3
- 102000057290 Adenosine Triphosphatases Human genes 0.000 description 3
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 3
- OYHQOLUKZRVURQ-HZJYTTRNSA-N Linoleic acid Chemical compound CCCCC\C=C/C\C=C/CCCCCCCC(O)=O OYHQOLUKZRVURQ-HZJYTTRNSA-N 0.000 description 3
- PCZOHLXUXFIOCF-UHFFFAOYSA-N Monacolin X Natural products C12C(OC(=O)C(C)CC)CC(C)C=C2C=CC(C)C1CCC1CC(O)CC(=O)O1 PCZOHLXUXFIOCF-UHFFFAOYSA-N 0.000 description 3
- SJRJJKPEHAURKC-UHFFFAOYSA-N N-Methylmorpholine Chemical compound CN1CCOCC1 SJRJJKPEHAURKC-UHFFFAOYSA-N 0.000 description 3
- ZQPPMHVWECSIRJ-UHFFFAOYSA-N Oleic acid Natural products CCCCCCCCC=CCCCCCCCC(O)=O ZQPPMHVWECSIRJ-UHFFFAOYSA-N 0.000 description 3
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 3
- GLUUGHFHXGJENI-UHFFFAOYSA-N Piperazine Chemical compound C1CNCCN1 GLUUGHFHXGJENI-UHFFFAOYSA-N 0.000 description 3
- 239000002202 Polyethylene glycol Substances 0.000 description 3
- OFOBLEOULBTSOW-UHFFFAOYSA-N Propanedioic acid Natural products OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 3
- 108060008682 Tumor Necrosis Factor Proteins 0.000 description 3
- 102000000852 Tumor Necrosis Factor-alpha Human genes 0.000 description 3
- GZOSMCIZMLWJML-VJLLXTKPSA-N abiraterone Chemical compound C([C@H]1[C@H]2[C@@H]([C@]3(CC[C@H](O)CC3=CC2)C)CC[C@@]11C)C=C1C1=CC=CN=C1 GZOSMCIZMLWJML-VJLLXTKPSA-N 0.000 description 3
- 229960000853 abiraterone Drugs 0.000 description 3
- 150000001721 carbon Chemical group 0.000 description 3
- 230000003833 cell viability Effects 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- 239000002299 complementary DNA Substances 0.000 description 3
- 229930182912 cyclosporin Natural products 0.000 description 3
- 150000005690 diesters Chemical class 0.000 description 3
- ZQPPMHVWECSIRJ-MDZDMXLPSA-N elaidic acid Chemical compound CCCCCCCC\C=C\CCCCCCCC(O)=O ZQPPMHVWECSIRJ-MDZDMXLPSA-N 0.000 description 3
- 150000002148 esters Chemical class 0.000 description 3
- 125000001924 fatty-acyl group Chemical group 0.000 description 3
- 229960000676 flunisolide Drugs 0.000 description 3
- 238000004128 high performance liquid chromatography Methods 0.000 description 3
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 3
- 230000003834 intracellular effect Effects 0.000 description 3
- 229960002857 isoflupredone Drugs 0.000 description 3
- QXJSBBXBKPUZAA-UHFFFAOYSA-N isooleic acid Natural products CCCCCCCC=CCCCCCCCCC(O)=O QXJSBBXBKPUZAA-UHFFFAOYSA-N 0.000 description 3
- 239000010410 layer Substances 0.000 description 3
- 235000020778 linoleic acid Nutrition 0.000 description 3
- JXNPEDYJTDQORS-UHFFFAOYSA-N linoleyl alcohol Natural products CCCCCC=CCC=CCCCCCCCCO JXNPEDYJTDQORS-UHFFFAOYSA-N 0.000 description 3
- 125000005645 linoleyl group Chemical group 0.000 description 3
- 238000011068 loading method Methods 0.000 description 3
- PCZOHLXUXFIOCF-BXMDZJJMSA-N lovastatin Chemical compound C([C@H]1[C@@H](C)C=CC2=C[C@H](C)C[C@@H]([C@H]12)OC(=O)[C@@H](C)CC)C[C@@H]1C[C@@H](O)CC(=O)O1 PCZOHLXUXFIOCF-BXMDZJJMSA-N 0.000 description 3
- 229960004844 lovastatin Drugs 0.000 description 3
- QLJODMDSTUBWDW-UHFFFAOYSA-N lovastatin hydroxy acid Natural products C1=CC(C)C(CCC(O)CC(O)CC(O)=O)C2C(OC(=O)C(C)CC)CC(C)C=C21 QLJODMDSTUBWDW-UHFFFAOYSA-N 0.000 description 3
- 230000014759 maintenance of location Effects 0.000 description 3
- 238000002156 mixing Methods 0.000 description 3
- 125000004433 nitrogen atom Chemical group N* 0.000 description 3
- 125000001117 oleyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])/C([H])=C([H])\C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 3
- 125000004430 oxygen atom Chemical group O* 0.000 description 3
- 239000012071 phase Substances 0.000 description 3
- 229920001223 polyethylene glycol Polymers 0.000 description 3
- 230000008569 process Effects 0.000 description 3
- 108090000623 proteins and genes Proteins 0.000 description 3
- MIXMJCQRHVAJIO-TZHJZOAOSA-N qk4dys664x Chemical compound O.C1([C@@H](F)C2)=CC(=O)C=C[C@]1(C)[C@@H]1[C@@H]2[C@@H]2C[C@H]3OC(C)(C)O[C@@]3(C(=O)CO)[C@@]2(C)C[C@@H]1O.C1([C@@H](F)C2)=CC(=O)C=C[C@]1(C)[C@@H]1[C@@H]2[C@@H]2C[C@H]3OC(C)(C)O[C@@]3(C(=O)CO)[C@@]2(C)C[C@@H]1O MIXMJCQRHVAJIO-TZHJZOAOSA-N 0.000 description 3
- 230000009467 reduction Effects 0.000 description 3
- 239000000523 sample Substances 0.000 description 3
- 238000007910 systemic administration Methods 0.000 description 3
- 231100000331 toxic Toxicity 0.000 description 3
- YNDXUCZADRHECN-JNQJZLCISA-N triamcinolone acetonide Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1C[C@H]3OC(C)(C)O[C@@]3(C(=O)CO)[C@@]1(C)C[C@@H]2O YNDXUCZADRHECN-JNQJZLCISA-N 0.000 description 3
- 229960002117 triamcinolone acetonide Drugs 0.000 description 3
- 239000003643 water by type Substances 0.000 description 3
- WBXMERNUPVKEID-MURFETPASA-N (11Z,14Z)-1-(dimethylamino)-2-hydroxy-2-(hydroxymethyl)icosa-11,14-dien-3-one Chemical compound C(CCCCCCC\C=C/C\C=C/CCCCC)(=O)C(CO)(CN(C)C)O WBXMERNUPVKEID-MURFETPASA-N 0.000 description 2
- YWWVWXASSLXJHU-AATRIKPKSA-N (9E)-tetradecenoic acid Chemical compound CCCC\C=C\CCCCCCCC(O)=O YWWVWXASSLXJHU-AATRIKPKSA-N 0.000 description 2
- WRIDQFICGBMAFQ-UHFFFAOYSA-N (E)-8-Octadecenoic acid Natural products CCCCCCCCCC=CCCCCCCC(O)=O WRIDQFICGBMAFQ-UHFFFAOYSA-N 0.000 description 2
- JFBCSFJKETUREV-UHFFFAOYSA-N 1,2 ditetradecanoylglycerol Chemical compound CCCCCCCCCCCCCC(=O)OCC(CO)OC(=O)CCCCCCCCCCCCC JFBCSFJKETUREV-UHFFFAOYSA-N 0.000 description 2
- AFSHUZFNMVJNKX-CLFAGFIQSA-N 1,2-dioleoylglycerol Chemical compound CCCCCCCC\C=C/CCCCCCCC(=O)OCC(CO)OC(=O)CCCCCCC\C=C/CCCCCCCC AFSHUZFNMVJNKX-CLFAGFIQSA-N 0.000 description 2
- NRJAVPSFFCBXDT-HUESYALOSA-N 1,2-distearoyl-sn-glycero-3-phosphocholine Chemical compound CCCCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCCCCCCCCCCCCCCCC NRJAVPSFFCBXDT-HUESYALOSA-N 0.000 description 2
- KBPLFHHGFOOTCA-UHFFFAOYSA-N 1-Octanol Chemical compound CCCCCCCCO KBPLFHHGFOOTCA-UHFFFAOYSA-N 0.000 description 2
- PAMIQIKDUOTOBW-UHFFFAOYSA-N 1-methylpiperidine Chemical compound CN1CCCCC1 PAMIQIKDUOTOBW-UHFFFAOYSA-N 0.000 description 2
- HZAXFHJVJLSVMW-UHFFFAOYSA-N 2-Aminoethan-1-ol Chemical compound NCCO HZAXFHJVJLSVMW-UHFFFAOYSA-N 0.000 description 2
- LQJBNNIYVWPHFW-UHFFFAOYSA-N 20:1omega9c fatty acid Natural products CCCCCCCCCCC=CCCCCCCCC(O)=O LQJBNNIYVWPHFW-UHFFFAOYSA-N 0.000 description 2
- HPMZBILYSWLILX-UMDUKNJSSA-N 3'''-O-acetyldigitoxin Chemical compound C1[C@H](OC(C)=O)[C@H](O)[C@@H](C)O[C@H]1O[C@@H]1[C@@H](C)O[C@@H](O[C@@H]2[C@H](O[C@@H](O[C@@H]3C[C@@H]4[C@]([C@@H]5[C@H]([C@]6(CC[C@@H]([C@@]6(C)CC5)C=5COC(=O)C=5)O)CC4)(C)CC3)C[C@@H]2O)C)C[C@@H]1O HPMZBILYSWLILX-UMDUKNJSSA-N 0.000 description 2
- NEVBISWWNWLRED-UHFFFAOYSA-N 3-dimethylsilyl-3-[(2-methylpropan-2-yl)oxy]propane-1,2-diol Chemical compound CC(C)(C)OC([SiH](C)C)C(O)CO NEVBISWWNWLRED-UHFFFAOYSA-N 0.000 description 2
- XMIIGOLPHOKFCH-UHFFFAOYSA-N 3-phenylpropionic acid Chemical compound OC(=O)CCC1=CC=CC=C1 XMIIGOLPHOKFCH-UHFFFAOYSA-N 0.000 description 2
- IEKLVCVEJCEIJD-LYRWTKHRSA-N 4-[2-[(8s,9r,10s,11s,13s,14s,16r,17r)-9-fluoro-11,17-dihydroxy-10,13,16-trimethyl-3-oxo-6,7,8,11,12,14,15,16-octahydrocyclopenta[a]phenanthren-17-yl]-2-oxoethoxy]-4-oxobutanoic acid Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1C[C@@H](C)[C@@](C(=O)COC(=O)CCC(O)=O)(O)[C@@]1(C)C[C@@H]2O IEKLVCVEJCEIJD-LYRWTKHRSA-N 0.000 description 2
- FQYRLEXKXQRZDH-UHFFFAOYSA-N 4-aminoquinoline Chemical compound C1=CC=C2C(N)=CC=NC2=C1 FQYRLEXKXQRZDH-UHFFFAOYSA-N 0.000 description 2
- QSBYPNXLFMSGKH-UHFFFAOYSA-N 9-Heptadecensaeure Natural products CCCCCCCC=CCCCCCCCC(O)=O QSBYPNXLFMSGKH-UHFFFAOYSA-N 0.000 description 2
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 2
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 2
- 239000005711 Benzoic acid Substances 0.000 description 2
- FERIUCNNQQJTOY-UHFFFAOYSA-N Butyric acid Chemical compound CCCC(O)=O FERIUCNNQQJTOY-UHFFFAOYSA-N 0.000 description 2
- RGHNJXZEOKUKBD-SQOUGZDYSA-N D-gluconic acid Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C(O)=O RGHNJXZEOKUKBD-SQOUGZDYSA-N 0.000 description 2
- XBPCUCUWBYBCDP-UHFFFAOYSA-N Dicyclohexylamine Chemical compound C1CCCCC1NC1CCCCC1 XBPCUCUWBYBCDP-UHFFFAOYSA-N 0.000 description 2
- ROSDSFDQCJNGOL-UHFFFAOYSA-N Dimethylamine Chemical compound CNC ROSDSFDQCJNGOL-UHFFFAOYSA-N 0.000 description 2
- QUSNBJAOOMFDIB-UHFFFAOYSA-N Ethylamine Chemical compound CCN QUSNBJAOOMFDIB-UHFFFAOYSA-N 0.000 description 2
- 108090000331 Firefly luciferases Proteins 0.000 description 2
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 2
- AEMRFAOFKBGASW-UHFFFAOYSA-N Glycolic acid Chemical compound OCC(O)=O AEMRFAOFKBGASW-UHFFFAOYSA-N 0.000 description 2
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 2
- 102000018251 Hypoxanthine Phosphoribosyltransferase Human genes 0.000 description 2
- 108010091358 Hypoxanthine Phosphoribosyltransferase Proteins 0.000 description 2
- XEEYBQQBJWHFJM-UHFFFAOYSA-N Iron Chemical compound [Fe] XEEYBQQBJWHFJM-UHFFFAOYSA-N 0.000 description 2
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 2
- 229910010082 LiAlH Inorganic materials 0.000 description 2
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 2
- BAVYZALUXZFZLV-UHFFFAOYSA-N Methylamine Chemical compound NC BAVYZALUXZFZLV-UHFFFAOYSA-N 0.000 description 2
- YNAVUWVOSKDBBP-UHFFFAOYSA-N Morpholine Chemical compound C1COCCN1 YNAVUWVOSKDBBP-UHFFFAOYSA-N 0.000 description 2
- JLTDJTHDQAWBAV-UHFFFAOYSA-N N,N-dimethylaniline Chemical compound CN(C)C1=CC=CC=C1 JLTDJTHDQAWBAV-UHFFFAOYSA-N 0.000 description 2
- MBBZMMPHUWSWHV-BDVNFPICSA-N N-methylglucamine Chemical compound CNC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO MBBZMMPHUWSWHV-BDVNFPICSA-N 0.000 description 2
- PVNIIMVLHYAWGP-UHFFFAOYSA-N Niacin Chemical compound OC(=O)C1=CC=CN=C1 PVNIIMVLHYAWGP-UHFFFAOYSA-N 0.000 description 2
- 239000005642 Oleic acid Substances 0.000 description 2
- 206010033128 Ovarian cancer Diseases 0.000 description 2
- 206010061535 Ovarian neoplasm Diseases 0.000 description 2
- 229910019142 PO4 Inorganic materials 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 2
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 2
- LCTONWCANYUPML-UHFFFAOYSA-N Pyruvic acid Chemical compound CC(=O)C(O)=O LCTONWCANYUPML-UHFFFAOYSA-N 0.000 description 2
- 238000011529 RT qPCR Methods 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 2
- 102000002689 Toll-like receptor Human genes 0.000 description 2
- 108020000411 Toll-like receptor Proteins 0.000 description 2
- 230000002378 acidificating effect Effects 0.000 description 2
- WNLRTRBMVRJNCN-UHFFFAOYSA-N adipic acid Chemical compound OC(=O)CCCCC(O)=O WNLRTRBMVRJNCN-UHFFFAOYSA-N 0.000 description 2
- 230000003110 anti-inflammatory effect Effects 0.000 description 2
- 230000001028 anti-proliverative effect Effects 0.000 description 2
- 238000013459 approach Methods 0.000 description 2
- JUHORIMYRDESRB-UHFFFAOYSA-N benzathine Chemical compound C=1C=CC=CC=1CNCCNCC1=CC=CC=C1 JUHORIMYRDESRB-UHFFFAOYSA-N 0.000 description 2
- 235000010233 benzoic acid Nutrition 0.000 description 2
- WERYXYBDKMZEQL-UHFFFAOYSA-N butane-1,4-diol Chemical compound OCCCCO WERYXYBDKMZEQL-UHFFFAOYSA-N 0.000 description 2
- RYYVLZVUVIJVGH-UHFFFAOYSA-N caffeine Chemical compound CN1C(=O)N(C)C(=O)C2=C1N=CN2C RYYVLZVUVIJVGH-UHFFFAOYSA-N 0.000 description 2
- 239000002775 capsule Substances 0.000 description 2
- 239000000969 carrier Substances 0.000 description 2
- 230000001413 cellular effect Effects 0.000 description 2
- 229960000541 cetyl alcohol Drugs 0.000 description 2
- 230000000973 chemotherapeutic effect Effects 0.000 description 2
- 238000004440 column chromatography Methods 0.000 description 2
- 229920001577 copolymer Polymers 0.000 description 2
- MWKFXSUHUHTGQN-UHFFFAOYSA-N decan-1-ol Chemical compound CCCCCCCCCCO MWKFXSUHUHTGQN-UHFFFAOYSA-N 0.000 description 2
- GHVNFZFCNZKVNT-UHFFFAOYSA-N decanoic acid Chemical compound CCCCCCCCCC(O)=O GHVNFZFCNZKVNT-UHFFFAOYSA-N 0.000 description 2
- 238000013461 design Methods 0.000 description 2
- 239000003085 diluting agent Substances 0.000 description 2
- XBDQKXXYIPTUBI-UHFFFAOYSA-N dimethylselenoniopropionate Natural products CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 2
- UKMSUNONTOPOIO-UHFFFAOYSA-N docosanoic acid Chemical compound CCCCCCCCCCCCCCCCCCCCCC(O)=O UKMSUNONTOPOIO-UHFFFAOYSA-N 0.000 description 2
- 239000003640 drug residue Substances 0.000 description 2
- 238000005538 encapsulation Methods 0.000 description 2
- 230000002255 enzymatic effect Effects 0.000 description 2
- 239000000499 gel Substances 0.000 description 2
- KWIUHFFTVRNATP-UHFFFAOYSA-N glycine betaine Chemical compound C[N+](C)(C)CC([O-])=O KWIUHFFTVRNATP-UHFFFAOYSA-N 0.000 description 2
- 230000012010 growth Effects 0.000 description 2
- BXWNKGSJHAJOGX-UHFFFAOYSA-N hexadecan-1-ol Chemical compound CCCCCCCCCCCCCCCCO BXWNKGSJHAJOGX-UHFFFAOYSA-N 0.000 description 2
- IPCSVZSSVZVIGE-UHFFFAOYSA-N hexadecanoic acid Chemical compound CCCCCCCCCCCCCCCC(O)=O IPCSVZSSVZVIGE-UHFFFAOYSA-N 0.000 description 2
- 239000001257 hydrogen Substances 0.000 description 2
- 230000001506 immunosuppresive effect Effects 0.000 description 2
- 230000015788 innate immune response Effects 0.000 description 2
- 150000007529 inorganic bases Chemical class 0.000 description 2
- SUMDYPCJJOFFON-UHFFFAOYSA-N isethionic acid Chemical compound OCCS(O)(=O)=O SUMDYPCJJOFFON-UHFFFAOYSA-N 0.000 description 2
- TWBYWOBDOCUKOW-UHFFFAOYSA-N isonicotinic acid Chemical compound OC(=O)C1=CC=NC=C1 TWBYWOBDOCUKOW-UHFFFAOYSA-N 0.000 description 2
- JVTAAEKCZFNVCJ-UHFFFAOYSA-N lactic acid Chemical compound CC(O)C(O)=O JVTAAEKCZFNVCJ-UHFFFAOYSA-N 0.000 description 2
- JJTUDXZGHPGLLC-UHFFFAOYSA-N lactide Chemical compound CC1OC(=O)C(C)OC1=O JJTUDXZGHPGLLC-UHFFFAOYSA-N 0.000 description 2
- JTEGQNOMFQHVDC-NKWVEPMBSA-N lamivudine Chemical compound O=C1N=C(N)C=CN1[C@H]1O[C@@H](CO)SC1 JTEGQNOMFQHVDC-NKWVEPMBSA-N 0.000 description 2
- 229960001627 lamivudine Drugs 0.000 description 2
- 231100000636 lethal dose Toxicity 0.000 description 2
- 239000007788 liquid Substances 0.000 description 2
- 210000002540 macrophage Anatomy 0.000 description 2
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 2
- 229960003746 metildigoxin Drugs 0.000 description 2
- IYJMSDVSVHDVGT-PEQKVOOWSA-N metildigoxin Chemical compound O1[C@H](C)[C@@H](OC)[C@@H](O)C[C@@H]1O[C@@H]1[C@@H](C)O[C@@H](O[C@@H]2[C@H](O[C@@H](O[C@@H]3C[C@@H]4[C@]([C@@H]5[C@H]([C@]6(CC[C@@H]([C@@]6(C)[C@H](O)C5)C=5COC(=O)C=5)O)CC4)(C)CC3)C[C@@H]2O)C)C[C@@H]1O IYJMSDVSVHDVGT-PEQKVOOWSA-N 0.000 description 2
- 150000007522 mineralic acids Chemical class 0.000 description 2
- 238000010172 mouse model Methods 0.000 description 2
- UTOPWMOLSKOLTQ-UHFFFAOYSA-N octacosanoic acid Chemical compound CCCCCCCCCCCCCCCCCCCCCCCCCCCC(O)=O UTOPWMOLSKOLTQ-UHFFFAOYSA-N 0.000 description 2
- 230000003287 optical effect Effects 0.000 description 2
- 150000007524 organic acids Chemical class 0.000 description 2
- SECPZKHBENQXJG-FPLPWBNLSA-N palmitoleic acid Chemical compound CCCCCC\C=C/CCCCCCCC(O)=O SECPZKHBENQXJG-FPLPWBNLSA-N 0.000 description 2
- 125000000913 palmityl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 2
- 238000007911 parenteral administration Methods 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 2
- 239000010452 phosphate Substances 0.000 description 2
- WTJKGGKOPKCXLL-RRHRGVEJSA-N phosphatidylcholine Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCCCCCCC=CCCCCCCCC WTJKGGKOPKCXLL-RRHRGVEJSA-N 0.000 description 2
- WLJVNTCWHIRURA-UHFFFAOYSA-N pimelic acid Chemical compound OC(=O)CCCCCC(O)=O WLJVNTCWHIRURA-UHFFFAOYSA-N 0.000 description 2
- 229920000642 polymer Polymers 0.000 description 2
- 239000002244 precipitate Substances 0.000 description 2
- PAHGJZDQXIOYTH-UHFFFAOYSA-N pristanic acid Chemical compound CC(C)CCCC(C)CCCC(C)CCCC(C)C(O)=O PAHGJZDQXIOYTH-UHFFFAOYSA-N 0.000 description 2
- 230000000750 progressive effect Effects 0.000 description 2
- 230000000770 proinflammatory effect Effects 0.000 description 2
- 230000000069 prophylactic effect Effects 0.000 description 2
- 238000011002 quantification Methods 0.000 description 2
- LISFMEBWQUVKPJ-UHFFFAOYSA-N quinolin-2-ol Chemical compound C1=CC=C2NC(=O)C=CC2=C1 LISFMEBWQUVKPJ-UHFFFAOYSA-N 0.000 description 2
- 230000004044 response Effects 0.000 description 2
- YGSDEFSMJLZEOE-UHFFFAOYSA-N salicylic acid Chemical compound OC(=O)C1=CC=CC=C1O YGSDEFSMJLZEOE-UHFFFAOYSA-N 0.000 description 2
- 238000013268 sustained release Methods 0.000 description 2
- 239000012730 sustained-release form Substances 0.000 description 2
- 230000009885 systemic effect Effects 0.000 description 2
- 239000003826 tablet Substances 0.000 description 2
- 230000008685 targeting Effects 0.000 description 2
- 238000012360 testing method Methods 0.000 description 2
- 229960004559 theobromine Drugs 0.000 description 2
- 230000004797 therapeutic response Effects 0.000 description 2
- DUYAAUVXQSMXQP-UHFFFAOYSA-M thioacetate Chemical compound CC([S-])=O DUYAAUVXQSMXQP-UHFFFAOYSA-M 0.000 description 2
- ZMZDMBWJUHKJPS-UHFFFAOYSA-N thiocyanic acid Chemical compound SC#N ZMZDMBWJUHKJPS-UHFFFAOYSA-N 0.000 description 2
- 150000003573 thiols Chemical class 0.000 description 2
- FYSNRJHAOHDILO-UHFFFAOYSA-N thionyl chloride Chemical compound ClS(Cl)=O FYSNRJHAOHDILO-UHFFFAOYSA-N 0.000 description 2
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 2
- 238000005809 transesterification reaction Methods 0.000 description 2
- GETQZCLCWQTVFV-UHFFFAOYSA-N trimethylamine Chemical compound CN(C)C GETQZCLCWQTVFV-UHFFFAOYSA-N 0.000 description 2
- ZDPHROOEEOARMN-UHFFFAOYSA-N undecanoic acid Chemical compound CCCCCCCCCCC(O)=O ZDPHROOEEOARMN-UHFFFAOYSA-N 0.000 description 2
- 229960000653 valrubicin Drugs 0.000 description 2
- ZOCKGBMQLCSHFP-KQRAQHLDSA-N valrubicin Chemical compound O([C@H]1C[C@](CC2=C(O)C=3C(=O)C4=CC=CC(OC)=C4C(=O)C=3C(O)=C21)(O)C(=O)COC(=O)CCCC)[C@H]1C[C@H](NC(=O)C(F)(F)F)[C@H](O)[C@H](C)O1 ZOCKGBMQLCSHFP-KQRAQHLDSA-N 0.000 description 2
- 229960002555 zidovudine Drugs 0.000 description 2
- PHOLEJIIASOWOL-XOGNUPMYSA-N (1S,4R,5S,8R,9R,11S,12S,13R,14R,16R,18S)-5,11-dihydroxy-9,16-dimethyl-10-oxo-8-(6-oxopyran-3-yl)-15,17,20-trioxahexacyclo[14.3.1.114,18.01,13.04,12.05,9]henicosane-13-carbaldehyde Chemical compound C[C@@]12[C@H](CC[C@]1(O)[C@@H]1CC[C@@]34C[C@@H]5C[C@@H](O[C@@](C)(O5)O3)[C@]4(C=O)[C@H]1[C@H](O)C2=O)c1ccc(=O)oc1 PHOLEJIIASOWOL-XOGNUPMYSA-N 0.000 description 1
- QBYIENPQHBMVBV-HFEGYEGKSA-N (2R)-2-hydroxy-2-phenylacetic acid Chemical compound O[C@@H](C(O)=O)c1ccccc1.O[C@@H](C(O)=O)c1ccccc1 QBYIENPQHBMVBV-HFEGYEGKSA-N 0.000 description 1
- GHYOCDFICYLMRF-UTIIJYGPSA-N (2S,3R)-N-[(2S)-3-(cyclopenten-1-yl)-1-[(2R)-2-methyloxiran-2-yl]-1-oxopropan-2-yl]-3-hydroxy-3-(4-methoxyphenyl)-2-[[(2S)-2-[(2-morpholin-4-ylacetyl)amino]propanoyl]amino]propanamide Chemical compound C1(=CCCC1)C[C@@H](C(=O)[C@@]1(OC1)C)NC([C@H]([C@@H](C1=CC=C(C=C1)OC)O)NC([C@H](C)NC(CN1CCOCC1)=O)=O)=O GHYOCDFICYLMRF-UTIIJYGPSA-N 0.000 description 1
- FBBMEIGVYZBQGH-HVFQMFNGSA-N (2S,3S)-1-(dimethylamino)-4-dimethylsilyl-5,5-dimethylhexane-2,3,4-triol Chemical compound C(C)(C)(C)C([C@H]([C@H](CN(C)C)O)O)(O)[SiH](C)C FBBMEIGVYZBQGH-HVFQMFNGSA-N 0.000 description 1
- QIFKBFBTVKHULG-PMACEKPBSA-N (2S,3S)-4-(dimethylamino)-2,3-bis(phenylmethoxy)butan-1-ol Chemical compound C(C1=CC=CC=C1)O[C@@H](CO)[C@H](CN(C)C)OCC1=CC=CC=C1 QIFKBFBTVKHULG-PMACEKPBSA-N 0.000 description 1
- KWMLJOLKUYYJFJ-GASJEMHNSA-N (2xi)-D-gluco-heptonic acid Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C(O)C(O)=O KWMLJOLKUYYJFJ-GASJEMHNSA-N 0.000 description 1
- RLCKHJSFHOZMDR-UHFFFAOYSA-N (3R, 7R, 11R)-1-Phytanoid acid Natural products CC(C)CCCC(C)CCCC(C)CCCC(C)CC(O)=O RLCKHJSFHOZMDR-UHFFFAOYSA-N 0.000 description 1
- IWZSHWBGHQBIML-ZGGLMWTQSA-N (3S,8S,10R,13S,14S,17S)-17-isoquinolin-7-yl-N,N,10,13-tetramethyl-2,3,4,7,8,9,11,12,14,15,16,17-dodecahydro-1H-cyclopenta[a]phenanthren-3-amine Chemical compound CN(C)[C@H]1CC[C@]2(C)C3CC[C@@]4(C)[C@@H](CC[C@@H]4c4ccc5ccncc5c4)[C@@H]3CC=C2C1 IWZSHWBGHQBIML-ZGGLMWTQSA-N 0.000 description 1
- ZTFGOPUOTATSAL-LZGKLYRASA-N (4r)-4-[(1r,3s,5s,8r,9s,10r,11r,13r,14s,17r)-1,5,11,14-tetrahydroxy-10-(hydroxymethyl)-13-methyl-3-[(2r,3r,4r,5r,6s)-3,4,5-trihydroxy-6-methyloxan-2-yl]oxy-2,3,4,6,7,8,9,11,12,15,16,17-dodecahydro-1h-cyclopenta[a]phenanthren-17-yl]oxolan-2-one Chemical compound O[C@@H]1[C@H](O)[C@@H](O)[C@H](C)O[C@H]1O[C@@H]1C[C@@]2(O)CC[C@H]3[C@@]4(O)CC[C@H]([C@H]5CC(=O)OC5)[C@@]4(C)C[C@@H](O)[C@@H]3[C@@]2(CO)[C@H](O)C1 ZTFGOPUOTATSAL-LZGKLYRASA-N 0.000 description 1
- FQKHBOPZYUBBBV-HEJPWNJWSA-N (6Z,9Z,19S,20S,29Z,32Z)-19-[(dimethylamino)methyl]-19,20-dihydroxy-20-(hydroxymethyl)octatriaconta-6,9,29,32-tetraene-18,21-dione Chemical compound C(CCCCCCC\C=C/C\C=C/CCCCC)(=O)[C@](CO)([C@](CN(C)C)(O)C(CCCCCCC\C=C/C\C=C/CCCCC)=O)O FQKHBOPZYUBBBV-HEJPWNJWSA-N 0.000 description 1
- FBWMYSQUTZRHAT-HZJYTTRNSA-N (9z,12z)-octadeca-9,12-dienoyl chloride Chemical compound CCCCC\C=C/C\C=C/CCCCCCCC(Cl)=O FBWMYSQUTZRHAT-HZJYTTRNSA-N 0.000 description 1
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 description 1
- BJEPYKJPYRNKOW-REOHCLBHSA-N (S)-malic acid Chemical compound OC(=O)[C@@H](O)CC(O)=O BJEPYKJPYRNKOW-REOHCLBHSA-N 0.000 description 1
- 102000040650 (ribonucleotides)n+m Human genes 0.000 description 1
- WBYWAXJHAXSJNI-VOTSOKGWSA-M .beta-Phenylacrylic acid Natural products [O-]C(=O)\C=C\C1=CC=CC=C1 WBYWAXJHAXSJNI-VOTSOKGWSA-M 0.000 description 1
- MQGBAQLIFKSMEM-UHFFFAOYSA-N 1,2-dilinoleoyl glycerol Natural products CCCCCC=CCC=CCCCCCCCC(=O)OCC(CO)OC(=O)CCCCCCCC=CCC=CCCCCC MQGBAQLIFKSMEM-UHFFFAOYSA-N 0.000 description 1
- UNILWMWFPHPYOR-KXEYIPSPSA-M 1-[6-[2-[3-[3-[3-[2-[2-[3-[[2-[2-[[(2r)-1-[[2-[[(2r)-1-[3-[2-[2-[3-[[2-(2-amino-2-oxoethoxy)acetyl]amino]propoxy]ethoxy]ethoxy]propylamino]-3-hydroxy-1-oxopropan-2-yl]amino]-2-oxoethyl]amino]-3-[(2r)-2,3-di(hexadecanoyloxy)propyl]sulfanyl-1-oxopropan-2-yl Chemical compound O=C1C(SCCC(=O)NCCCOCCOCCOCCCNC(=O)COCC(=O)N[C@@H](CSC[C@@H](COC(=O)CCCCCCCCCCCCCCC)OC(=O)CCCCCCCCCCCCCCC)C(=O)NCC(=O)N[C@H](CO)C(=O)NCCCOCCOCCOCCCNC(=O)COCC(N)=O)CC(=O)N1CCNC(=O)CCCCCN\1C2=CC=C(S([O-])(=O)=O)C=C2CC/1=C/C=C/C=C/C1=[N+](CC)C2=CC=C(S([O-])(=O)=O)C=C2C1 UNILWMWFPHPYOR-KXEYIPSPSA-M 0.000 description 1
- LDMOEFOXLIZJOW-UHFFFAOYSA-N 1-dodecanesulfonic acid Chemical compound CCCCCCCCCCCCS(O)(=O)=O LDMOEFOXLIZJOW-UHFFFAOYSA-N 0.000 description 1
- CMCBDXRRFKYBDG-UHFFFAOYSA-N 1-dodecoxydodecane Chemical compound CCCCCCCCCCCCOCCCCCCCCCCCC CMCBDXRRFKYBDG-UHFFFAOYSA-N 0.000 description 1
- WTJKGGKOPKCXLL-VYOBOKEXSA-N 1-hexadecanoyl-2-(9Z-octadecenoyl)-sn-glycero-3-phosphocholine Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCCCCCC\C=C/CCCCCCCC WTJKGGKOPKCXLL-VYOBOKEXSA-N 0.000 description 1
- ZONJATNKKGGVSU-UHFFFAOYSA-N 14-methylpentadecanoic acid Chemical compound CC(C)CCCCCCCCCCCCC(O)=O ZONJATNKKGGVSU-UHFFFAOYSA-N 0.000 description 1
- HACVUBOZWSMWSY-UHFFFAOYSA-N 18-butyl-2,3,4,5,6,7,8,9,10,11,12,13,14,14,15,15,16,16,17,17,18-henicosa(hex-2-enoyl)-19-oxotetracosa-2,4,6,8,10,12,20-heptaenoic acid Chemical compound C(C=CCCC)(=O)C(C(C(C(C(C(=C(C(=C(C(=C(C(=C(C(=C(C(=C(C(=O)O)C(C=CCCC)=O)C(C=CCCC)=O)C(C=CCCC)=O)C(C=CCCC)=O)C(C=CCCC)=O)C(C=CCCC)=O)C(C=CCCC)=O)C(C=CCCC)=O)C(C=CCCC)=O)C(C=CCCC)=O)C(C=CCCC)=O)C(C=CCCC)=O)(C(C=CCCC)=O)C(C=CCCC)=O)(C(C=CCCC)=O)C(C=CCCC)=O)(C(C=CCCC)=O)C(C=CCCC)=O)(C(C=CCCC)=O)C(C=CCCC)=O)(CCCC)C(C=CCCC)=O HACVUBOZWSMWSY-UHFFFAOYSA-N 0.000 description 1
- HXIYLSCCXCLSEF-UHFFFAOYSA-N 18-methyl-2-(14-methylpentadecyl)-3-oxononadecanoic acid Chemical compound C(CCCCCCCCCCCCCCC(C)C)(=O)C(C(=O)O)CCCCCCCCCCCCCC(C)C HXIYLSCCXCLSEF-UHFFFAOYSA-N 0.000 description 1
- OXQGTIUCKGYOAA-UHFFFAOYSA-N 2-Ethylbutanoic acid Chemical compound CCC(CC)C(O)=O OXQGTIUCKGYOAA-UHFFFAOYSA-N 0.000 description 1
- MSWZFWKMSRAUBD-IVMDWMLBSA-N 2-amino-2-deoxy-D-glucopyranose Chemical compound N[C@H]1C(O)O[C@H](CO)[C@@H](O)[C@@H]1O MSWZFWKMSRAUBD-IVMDWMLBSA-N 0.000 description 1
- TWJNQYPJQDRXPH-UHFFFAOYSA-N 2-cyanobenzohydrazide Chemical compound NNC(=O)C1=CC=CC=C1C#N TWJNQYPJQDRXPH-UHFFFAOYSA-N 0.000 description 1
- LZCSGXIVRQXJNJ-UHFFFAOYSA-N 2-decyl-3-oxotetradecanoic acid Chemical compound CCCCCCCCCCCC(=O)C(C(O)=O)CCCCCCCCCC LZCSGXIVRQXJNJ-UHFFFAOYSA-N 0.000 description 1
- BFSVOASYOCHEOV-UHFFFAOYSA-N 2-diethylaminoethanol Chemical compound CCN(CC)CCO BFSVOASYOCHEOV-UHFFFAOYSA-N 0.000 description 1
- 229940013085 2-diethylaminoethanol Drugs 0.000 description 1
- OXFWLMOOAQTRJI-UHFFFAOYSA-N 2-docosylhexacosanoic acid Chemical compound CCCCCCCCCCCCCCCCCCCCCCCCC(C(O)=O)CCCCCCCCCCCCCCCCCCCCCC OXFWLMOOAQTRJI-UHFFFAOYSA-N 0.000 description 1
- WTACYDSQHWXANE-UHFFFAOYSA-N 2-dodecylhexadecanoic acid Chemical compound CCCCCCCCCCCCCCC(C(O)=O)CCCCCCCCCCCC WTACYDSQHWXANE-UHFFFAOYSA-N 0.000 description 1
- BCXBRXMGDFMQHD-UHFFFAOYSA-N 2-hexadecyl-3-oxoicosanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(=O)C(C(O)=O)CCCCCCCCCCCCCCCC BCXBRXMGDFMQHD-UHFFFAOYSA-N 0.000 description 1
- ALHINGOCCWHAHD-UHFFFAOYSA-N 2-octadecyl-3-oxodocosanoic acid Chemical compound C(CCCCCCCCCCCCCCCCCCC)(=O)C(C(=O)O)CCCCCCCCCCCCCCCCCC ALHINGOCCWHAHD-UHFFFAOYSA-N 0.000 description 1
- RLCKHJSFHOZMDR-PWCSWUJKSA-N 3,7R,11R,15-tetramethyl-hexadecanoic acid Chemical compound CC(C)CCC[C@@H](C)CCC[C@@H](C)CCCC(C)CC(O)=O RLCKHJSFHOZMDR-PWCSWUJKSA-N 0.000 description 1
- AZKSAVLVSZKNRD-UHFFFAOYSA-M 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide Chemical compound [Br-].S1C(C)=C(C)N=C1[N+]1=NC(C=2C=CC=CC=2)=NN1C1=CC=CC=C1 AZKSAVLVSZKNRD-UHFFFAOYSA-M 0.000 description 1
- QCMHUGYTOGXZIW-UHFFFAOYSA-N 3-(dimethylamino)propane-1,2-diol Chemical compound CN(C)CC(O)CO QCMHUGYTOGXZIW-UHFFFAOYSA-N 0.000 description 1
- FPQQSJJWHUJYPU-UHFFFAOYSA-N 3-(dimethylamino)propyliminomethylidene-ethylazanium;chloride Chemical compound Cl.CCN=C=NCCCN(C)C FPQQSJJWHUJYPU-UHFFFAOYSA-N 0.000 description 1
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 description 1
- ZRPLANDPDWYOMZ-UHFFFAOYSA-N 3-cyclopentylpropionic acid Chemical compound OC(=O)CCC1CCCC1 ZRPLANDPDWYOMZ-UHFFFAOYSA-N 0.000 description 1
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 1
- AAOABJIKMCOCHX-HZJYTTRNSA-N 4-[(9Z,12Z)-octadeca-9,12-dienoxy]butane-1-thiol Chemical compound CCCCC\C=C/C\C=C/CCCCCCCCOCCCCS AAOABJIKMCOCHX-HZJYTTRNSA-N 0.000 description 1
- BRRUPRMXQXEZAR-KTKRTIGZSA-N 4-[(z)-octadec-9-enoxy]-4-oxobutanoic acid Chemical compound CCCCCCCC\C=C/CCCCCCCCOC(=O)CCC(O)=O BRRUPRMXQXEZAR-KTKRTIGZSA-N 0.000 description 1
- HVCNXQOWACZAFN-UHFFFAOYSA-N 4-ethylmorpholine Chemical compound CCN1CCOCC1 HVCNXQOWACZAFN-UHFFFAOYSA-N 0.000 description 1
- NLFOXHSVYRWVNA-HZJYTTRNSA-N 4-sulfanylbutyl (9Z,12Z)-octadeca-9,12-dienoate Chemical compound C(CCCCCCC\C=C/C\C=C/CCCCC)(=O)OCCCCS NLFOXHSVYRWVNA-HZJYTTRNSA-N 0.000 description 1
- AWQSAIIDOMEEOD-UHFFFAOYSA-N 5,5-Dimethyl-4-(3-oxobutyl)dihydro-2(3H)-furanone Chemical compound CC(=O)CCC1CC(=O)OC1(C)C AWQSAIIDOMEEOD-UHFFFAOYSA-N 0.000 description 1
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 1
- WREVVZMUNPAPOV-UHFFFAOYSA-N 8-aminoquinoline Chemical compound C1=CN=C2C(N)=CC=CC2=C1 WREVVZMUNPAPOV-UHFFFAOYSA-N 0.000 description 1
- YWWVWXASSLXJHU-UHFFFAOYSA-N 9E-tetradecenoic acid Natural products CCCCC=CCCCCCCCC(O)=O YWWVWXASSLXJHU-UHFFFAOYSA-N 0.000 description 1
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 1
- HPMZBILYSWLILX-UHFFFAOYSA-N Acetyl-digitoxine Natural products C1C(OC(C)=O)C(O)C(C)OC1OC1C(C)OC(OC2C(OC(OC3CC4C(C5C(C6(CCC(C6(C)CC5)C=5COC(=O)C=5)O)CC4)(C)CC3)CC2O)C)CC1O HPMZBILYSWLILX-UHFFFAOYSA-N 0.000 description 1
- LPMXVESGRSUGHW-UHFFFAOYSA-N Acolongiflorosid K Natural products OC1C(O)C(O)C(C)OC1OC1CC2(O)CCC3C4(O)CCC(C=5COC(=O)C=5)C4(C)CC(O)C3C2(CO)C(O)C1 LPMXVESGRSUGHW-UHFFFAOYSA-N 0.000 description 1
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical compound [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 description 1
- ATRRKUHOCOJYRX-UHFFFAOYSA-N Ammonium bicarbonate Chemical compound [NH4+].OC([O-])=O ATRRKUHOCOJYRX-UHFFFAOYSA-N 0.000 description 1
- 229910000013 Ammonium bicarbonate Inorganic materials 0.000 description 1
- 108020000948 Antisense Oligonucleotides Proteins 0.000 description 1
- 239000004475 Arginine Substances 0.000 description 1
- 235000021357 Behenic acid Nutrition 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-M Bicarbonate Chemical compound OC([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-M 0.000 description 1
- 241000283690 Bos taurus Species 0.000 description 1
- DPUOLQHDNGRHBS-UHFFFAOYSA-N Brassidinsaeure Natural products CCCCCCCCC=CCCCCCCCCCCCC(O)=O DPUOLQHDNGRHBS-UHFFFAOYSA-N 0.000 description 1
- VOZHMAYHYHEWBW-NVOOAVKYSA-N Bufotalin Chemical compound C=1([C@@H]2[C@@]3(C)CC[C@@H]4[C@@]5(C)CC[C@H](O)C[C@H]5CC[C@H]4[C@@]3(O)C[C@@H]2OC(=O)C)C=CC(=O)OC=1 VOZHMAYHYHEWBW-NVOOAVKYSA-N 0.000 description 1
- FURJIYVUHAAVPE-UHFFFAOYSA-N C(CCCC=C/CC=C/CC=C/CC=C/CCCCC)(=O)C(C(=O)O)CCC=C/CC=C/CC=C/CC=C/CCCCC Chemical compound C(CCCC=C/CC=C/CC=C/CC=C/CCCCC)(=O)C(C(=O)O)CCC=C/CC=C/CC=C/CC=C/CCCCC FURJIYVUHAAVPE-UHFFFAOYSA-N 0.000 description 1
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 1
- LSPHULWDVZXLIL-UHFFFAOYSA-N Camphoric acid Natural products CC1(C)C(C(O)=O)CCC1(C)C(O)=O LSPHULWDVZXLIL-UHFFFAOYSA-N 0.000 description 1
- 241000283707 Capra Species 0.000 description 1
- 239000005632 Capric acid (CAS 334-48-5) Substances 0.000 description 1
- WWZKQHOCKIZLMA-UHFFFAOYSA-N Caprylic acid Natural products CCCCCCCC(O)=O WWZKQHOCKIZLMA-UHFFFAOYSA-N 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-L Carbonate Chemical compound [O-]C([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-L 0.000 description 1
- 108010051152 Carboxylesterase Proteins 0.000 description 1
- 102000013392 Carboxylesterase Human genes 0.000 description 1
- 101150065749 Churc1 gene Proteins 0.000 description 1
- WBYWAXJHAXSJNI-SREVYHEPSA-N Cinnamic acid Chemical compound OC(=O)\C=C/C1=CC=CC=C1 WBYWAXJHAXSJNI-SREVYHEPSA-N 0.000 description 1
- SCULJPGYOQQXTK-OLRINKBESA-N Cinobufagin Chemical compound C=1([C@@H]2[C@@]3(C)CC[C@@H]4[C@@]5(C)CC[C@H](O)C[C@H]5CC[C@H]4[C@@]43O[C@@H]4[C@@H]2OC(=O)C)C=CC(=O)OC=1 SCULJPGYOQQXTK-OLRINKBESA-N 0.000 description 1
- SCULJPGYOQQXTK-UHFFFAOYSA-N Cinobufagin Natural products CC(=O)OC1C2OC22C3CCC4CC(O)CCC4(C)C3CCC2(C)C1C=1C=CC(=O)OC=1 SCULJPGYOQQXTK-UHFFFAOYSA-N 0.000 description 1
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical compound [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 description 1
- TYYDXNISHGVDGA-UHFFFAOYSA-N Corotoxigenin Natural products CC12CCC3C(CCC4CC(O)CCC34C=O)C1CCC2C5=CC(=O)OC5 TYYDXNISHGVDGA-UHFFFAOYSA-N 0.000 description 1
- RGHNJXZEOKUKBD-UHFFFAOYSA-N D-gluconic acid Natural products OCC(O)C(O)C(O)C(O)C(O)=O RGHNJXZEOKUKBD-UHFFFAOYSA-N 0.000 description 1
- PHOLEJIIASOWOL-UHFFFAOYSA-N Daigremontianin Natural products C1CC(C2C(C3(C=O)C4CC5CC3(OC(C)(O5)O4)CC2)C(O)C2=O)(O)C2(C)C1C=1C=CC(=O)OC=1 PHOLEJIIASOWOL-UHFFFAOYSA-N 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- BWLUMTFWVZZZND-UHFFFAOYSA-N Dibenzylamine Chemical compound C=1C=CC=CC=1CNCC1=CC=CC=C1 BWLUMTFWVZZZND-UHFFFAOYSA-N 0.000 description 1
- WDJUZGPOPHTGOT-OAXVISGBSA-N Digitoxin Natural products O([C@H]1[C@@H](C)O[C@@H](O[C@@H]2C[C@@H]3[C@@](C)([C@@H]4[C@H]([C@]5(O)[C@@](C)([C@H](C6=CC(=O)OC6)CC5)CC4)CC3)CC2)C[C@H]1O)[C@H]1O[C@@H](C)[C@H](O[C@H]2O[C@@H](C)[C@@H](O)[C@@H](O)C2)[C@@H](O)C1 WDJUZGPOPHTGOT-OAXVISGBSA-N 0.000 description 1
- SHIBSTMRCDJXLN-UHFFFAOYSA-N Digoxigenin Natural products C1CC(C2C(C3(C)CCC(O)CC3CC2)CC2O)(O)C2(C)C1C1=CC(=O)OC1 SHIBSTMRCDJXLN-UHFFFAOYSA-N 0.000 description 1
- URXZXNYJPAJJOQ-UHFFFAOYSA-N Erucic acid Natural products CCCCCCC=CCCCCCCCCCCCC(O)=O URXZXNYJPAJJOQ-UHFFFAOYSA-N 0.000 description 1
- OTMSDBZUPAUEDD-UHFFFAOYSA-N Ethane Chemical compound CC OTMSDBZUPAUEDD-UHFFFAOYSA-N 0.000 description 1
- PIICEJLVQHRZGT-UHFFFAOYSA-N Ethylenediamine Chemical compound NCCN PIICEJLVQHRZGT-UHFFFAOYSA-N 0.000 description 1
- 241000282326 Felis catus Species 0.000 description 1
- QITDIWRKOXBKAM-UHFFFAOYSA-N Gofrusid Natural products OC1C(O)C(O)C(C)OC1OC1CC(CCC2C3(CCC(C3(C)CCC32)C=2COC(=O)C=2)O)C3(C=O)CC1 QITDIWRKOXBKAM-UHFFFAOYSA-N 0.000 description 1
- 229910004373 HOAc Inorganic materials 0.000 description 1
- HKNQDCBTAOHPIM-UHFFFAOYSA-N Helveticosid Natural products CC1OCC(CC(O)C1O)OC2CCC3(C=O)C4CCC5(C)C(CCC5(O)C4CCC3(O)C2)C6=CC(=O)OC6 HKNQDCBTAOHPIM-UHFFFAOYSA-N 0.000 description 1
- QBILRDAMJUPXCX-AGAUEGNUSA-N Helveticoside Chemical compound C1[C@H](O)[C@H](O)[C@@H](C)O[C@H]1O[C@@H]1C[C@@]2(O)CC[C@H]3[C@@]4(O)CC[C@H](C=5COC(=O)C=5)[C@@]4(C)CC[C@@H]3[C@@]2(C=O)CC1 QBILRDAMJUPXCX-AGAUEGNUSA-N 0.000 description 1
- 101001124388 Homo sapiens NPC intracellular cholesterol transporter 1 Proteins 0.000 description 1
- 208000001953 Hypotension Diseases 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- 206010022004 Influenza like illness Diseases 0.000 description 1
- 102100034343 Integrase Human genes 0.000 description 1
- 102000003814 Interleukin-10 Human genes 0.000 description 1
- 108090000174 Interleukin-10 Proteins 0.000 description 1
- LPHGQDQBBGAPDZ-UHFFFAOYSA-N Isocaffeine Natural products CN1C(=O)N(C)C(=O)C2=C1N(C)C=N2 LPHGQDQBBGAPDZ-UHFFFAOYSA-N 0.000 description 1
- ODKSFYDXXFIFQN-BYPYZUCNSA-P L-argininium(2+) Chemical compound NC(=[NH2+])NCCC[C@H]([NH3+])C(O)=O ODKSFYDXXFIFQN-BYPYZUCNSA-P 0.000 description 1
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 1
- HNDVDQJCIGZPNO-YFKPBYRVSA-N L-histidine Chemical compound OC(=O)[C@@H](N)CC1=CN=CN1 HNDVDQJCIGZPNO-YFKPBYRVSA-N 0.000 description 1
- KDXKERNSBIXSRK-YFKPBYRVSA-N L-lysine Chemical compound NCCCC[C@H](N)C(O)=O KDXKERNSBIXSRK-YFKPBYRVSA-N 0.000 description 1
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 1
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 1
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 1
- 239000004472 Lysine Substances 0.000 description 1
- 231100000002 MTT assay Toxicity 0.000 description 1
- 238000000134 MTT assay Methods 0.000 description 1
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 1
- PWHULOQIROXLJO-UHFFFAOYSA-N Manganese Chemical compound [Mn] PWHULOQIROXLJO-UHFFFAOYSA-N 0.000 description 1
- 235000021360 Myristic acid Nutrition 0.000 description 1
- TUNFSRHWOTWDNC-UHFFFAOYSA-N Myristic acid Natural products CCCCCCCCCCCCCC(O)=O TUNFSRHWOTWDNC-UHFFFAOYSA-N 0.000 description 1
- OUSFTKFNBAZUKL-UHFFFAOYSA-N N-(5-{[(5-tert-butyl-1,3-oxazol-2-yl)methyl]sulfanyl}-1,3-thiazol-2-yl)piperidine-4-carboxamide Chemical compound O1C(C(C)(C)C)=CN=C1CSC(S1)=CN=C1NC(=O)C1CCNCC1 OUSFTKFNBAZUKL-UHFFFAOYSA-N 0.000 description 1
- UEEJHVSXFDXPFK-UHFFFAOYSA-N N-dimethylaminoethanol Chemical compound CN(C)CCO UEEJHVSXFDXPFK-UHFFFAOYSA-N 0.000 description 1
- HTLZVHNRZJPSMI-UHFFFAOYSA-N N-ethylpiperidine Chemical compound CCN1CCCCC1 HTLZVHNRZJPSMI-UHFFFAOYSA-N 0.000 description 1
- OPFJDXRVMFKJJO-ZHHKINOHSA-N N-{[3-(2-benzamido-4-methyl-1,3-thiazol-5-yl)-pyrazol-5-yl]carbonyl}-G-dR-G-dD-dD-dD-NH2 Chemical compound S1C(C=2NN=C(C=2)C(=O)NCC(=O)N[C@H](CCCN=C(N)N)C(=O)NCC(=O)N[C@H](CC(O)=O)C(=O)N[C@H](CC(O)=O)C(=O)N[C@H](CC(O)=O)C(N)=O)=C(C)N=C1NC(=O)C1=CC=CC=C1 OPFJDXRVMFKJJO-ZHHKINOHSA-N 0.000 description 1
- 102100029565 NPC intracellular cholesterol transporter 1 Human genes 0.000 description 1
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 1
- LPMXVESGRSUGHW-GHYGWZAOSA-N Ouabain Natural products O([C@@H]1[C@@H](O)[C@@H](O)[C@@H](O)[C@H](C)O1)[C@H]1C[C@@H](O)[C@@]2(CO)[C@@](O)(C1)CC[C@H]1[C@]3(O)[C@@](C)([C@H](C4=CC(=O)OC4)CC3)C[C@@H](O)[C@H]21 LPMXVESGRSUGHW-GHYGWZAOSA-N 0.000 description 1
- 235000021314 Palmitic acid Nutrition 0.000 description 1
- 235000021319 Palmitoleic acid Nutrition 0.000 description 1
- 241001494479 Pecora Species 0.000 description 1
- 229920003171 Poly (ethylene oxide) Polymers 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- IWYDHOAUDWTVEP-UHFFFAOYSA-N R-2-phenyl-2-hydroxyacetic acid Natural products OC(=O)C(O)C1=CC=CC=C1 IWYDHOAUDWTVEP-UHFFFAOYSA-N 0.000 description 1
- 108010092799 RNA-directed DNA polymerase Proteins 0.000 description 1
- XWOHXZNTRISJIU-NQLNTKRDSA-N S-[4-[(9Z,12Z)-octadeca-9,12-dienoxy]butyl] ethanethioate Chemical compound C(C)(=O)SCCCCOCCCCCCCC\C=C/C\C=C/CCCCC XWOHXZNTRISJIU-NQLNTKRDSA-N 0.000 description 1
- LSMIOFMZNVEEBR-KIZPAXIPSA-N Scilliroside Natural products O=C(O[C@@H]1C=2[C@@](C)([C@@H]3[C@](O)([C@]4(O)[C@@](C)([C@H](C5=COC(=O)C=C5)CC4)CC3)C1)CC[C@H](O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1)C=2)C LSMIOFMZNVEEBR-KIZPAXIPSA-N 0.000 description 1
- 229920002684 Sepharose Polymers 0.000 description 1
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 1
- ODJLBQGVINUMMR-UHFFFAOYSA-N Strophanthidin Natural products CC12CCC(C3(CCC(O)CC3(O)CC3)C=O)C3C1(O)CCC2C1=CC(=O)OC1 ODJLBQGVINUMMR-UHFFFAOYSA-N 0.000 description 1
- 244000166550 Strophanthus gratus Species 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-N Succinic acid Natural products OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 1
- UCKMPCXJQFINFW-UHFFFAOYSA-N Sulphide Chemical compound [S-2] UCKMPCXJQFINFW-UHFFFAOYSA-N 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 1
- XSTXAVWGXDQKEL-UHFFFAOYSA-N Trichloroethylene Chemical compound ClC=C(Cl)Cl XSTXAVWGXDQKEL-UHFFFAOYSA-N 0.000 description 1
- BAECOWNUKCLBPZ-HIUWNOOHSA-N Triolein Natural products O([C@H](OCC(=O)CCCCCCC/C=C\CCCCCCCC)COC(=O)CCCCCCC/C=C\CCCCCCCC)C(=O)CCCCCCC/C=C\CCCCCCCC BAECOWNUKCLBPZ-HIUWNOOHSA-N 0.000 description 1
- PHYFQTYBJUILEZ-UHFFFAOYSA-N Trioleoylglycerol Natural products CCCCCCCCC=CCCCCCCCC(=O)OCC(OC(=O)CCCCCCCC=CCCCCCCCC)COC(=O)CCCCCCCC=CCCCCCCCC PHYFQTYBJUILEZ-UHFFFAOYSA-N 0.000 description 1
- UWHZIFQPPBDJPM-FPLPWBNLSA-M Vaccenic acid Natural products CCCCCC\C=C/CCCCCCCCCC([O-])=O UWHZIFQPPBDJPM-FPLPWBNLSA-M 0.000 description 1
- 235000021322 Vaccenic acid Nutrition 0.000 description 1
- 241000251539 Vertebrata <Metazoa> Species 0.000 description 1
- 229930003448 Vitamin K Natural products 0.000 description 1
- 238000002441 X-ray diffraction Methods 0.000 description 1
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 1
- DOMHWKQEPDYUQX-LJAQBGIBSA-N [(2r,3r,4s,6r)-3-[(2s,4s,5r,6r)-5-[(2s,4s,5r,6r)-4,5-diformyloxy-6-methyloxan-2-yl]oxy-4-formyloxy-6-methyloxan-2-yl]oxy-6-[[(3s,5r,8r,9s,10s,13r,14s,16s,17r)-16-formyloxy-14-hydroxy-10,13-dimethyl-17-(5-oxo-2h-furan-3-yl)-1,2,3,4,5,6,7,8,9,11,12,15,16,17 Chemical compound C1[C@H](OC=O)[C@H](OC=O)[C@@H](C)O[C@H]1O[C@H]1[C@@H](OC=O)C[C@H](O[C@H]2[C@H](C[C@@H](O[C@@H]2C)O[C@@H]2C[C@@H]3[C@]([C@@H]4[C@H]([C@]5(C[C@@H]([C@@H]([C@@]5(C)CC4)C=4COC(=O)C=4)OC=O)O)CC3)(C)CC2)OC=O)O[C@@H]1C DOMHWKQEPDYUQX-LJAQBGIBSA-N 0.000 description 1
- JJFLSSAGKSQEHW-NQLNTKRDSA-N [(9z,12z)-octadeca-9,12-dienyl] methanesulfonate Chemical compound CCCCC\C=C/C\C=C/CCCCCCCCOS(C)(=O)=O JJFLSSAGKSQEHW-NQLNTKRDSA-N 0.000 description 1
- 238000010521 absorption reaction Methods 0.000 description 1
- 229960000583 acetic acid Drugs 0.000 description 1
- 235000011054 acetic acid Nutrition 0.000 description 1
- 229960003635 acetyldigitoxin Drugs 0.000 description 1
- 229960003304 acetyldigoxin Drugs 0.000 description 1
- HWKJSYYYURVNQU-DXJNJSHLSA-N acetyldigoxin Chemical compound C1[C@H](OC(C)=O)[C@H](O)[C@@H](C)O[C@H]1O[C@@H]1[C@@H](C)O[C@@H](O[C@@H]2[C@H](O[C@@H](O[C@@H]3C[C@@H]4[C@]([C@@H]5[C@H]([C@]6(CC[C@@H]([C@@]6(C)[C@H](O)C5)C=5COC(=O)C=5)O)CC4)(C)CC3)C[C@@H]2O)C)C[C@@H]1O HWKJSYYYURVNQU-DXJNJSHLSA-N 0.000 description 1
- NREAGDHHMSOWKZ-DXJNJSHLSA-N acetyldigoxin Chemical compound O([C@H]1[C@@H](O)C[C@@H](O[C@@H]1C)O[C@H]1[C@@H](O)C[C@@H](O[C@@H]1C)O[C@@H]1C[C@@H]2[C@]([C@@H]3[C@H]([C@]4(CC[C@@H]([C@@]4(C)[C@H](O)C3)C=3COC(=O)C=3)O)CC2)(C)CC1)[C@H]1C[C@H](O)[C@H](OC(C)=O)[C@@H](C)O1 NREAGDHHMSOWKZ-DXJNJSHLSA-N 0.000 description 1
- 229940022702 acetyldigoxins Drugs 0.000 description 1
- 230000009471 action Effects 0.000 description 1
- 230000004913 activation Effects 0.000 description 1
- 230000009056 active transport Effects 0.000 description 1
- 230000010933 acylation Effects 0.000 description 1
- 238000005917 acylation reaction Methods 0.000 description 1
- 239000001361 adipic acid Substances 0.000 description 1
- 235000011037 adipic acid Nutrition 0.000 description 1
- 229960000250 adipic acid Drugs 0.000 description 1
- 125000003158 alcohol group Chemical group 0.000 description 1
- 239000000783 alginic acid Substances 0.000 description 1
- 235000010443 alginic acid Nutrition 0.000 description 1
- 229920000615 alginic acid Polymers 0.000 description 1
- 229960001126 alginic acid Drugs 0.000 description 1
- 150000004781 alginic acids Chemical class 0.000 description 1
- 229910052783 alkali metal Inorganic materials 0.000 description 1
- 150000001340 alkali metals Chemical class 0.000 description 1
- 229910052784 alkaline earth metal Inorganic materials 0.000 description 1
- 150000001342 alkaline earth metals Chemical class 0.000 description 1
- 150000001336 alkenes Chemical class 0.000 description 1
- JAZBEHYOTPTENJ-JLNKQSITSA-N all-cis-5,8,11,14,17-icosapentaenoic acid Chemical compound CC\C=C/C\C=C/C\C=C/C\C=C/C\C=C/CCCC(O)=O JAZBEHYOTPTENJ-JLNKQSITSA-N 0.000 description 1
- MGVYFNHJWXJYBE-UHFFFAOYSA-N alpha-Acetyl-digoxin Natural products CC1OC(CC(O)C1O)OC2C(O)CC(OC3C(C)OC(CC3OC(=O)C)OC4CCC5(C)C(CCC6C5CCC7(C)C(C(O)CC67O)C8=CC(=O)OC8)C4)OC2C MGVYFNHJWXJYBE-UHFFFAOYSA-N 0.000 description 1
- AWUCVROLDVIAJX-UHFFFAOYSA-N alpha-glycerophosphate Natural products OCC(O)COP(O)(O)=O AWUCVROLDVIAJX-UHFFFAOYSA-N 0.000 description 1
- BJEPYKJPYRNKOW-UHFFFAOYSA-N alpha-hydroxysuccinic acid Natural products OC(=O)C(O)CC(O)=O BJEPYKJPYRNKOW-UHFFFAOYSA-N 0.000 description 1
- DTOSIQBPPRVQHS-PDBXOOCHSA-N alpha-linolenic acid Chemical compound CC\C=C/C\C=C/C\C=C/CCCCCCCC(O)=O DTOSIQBPPRVQHS-PDBXOOCHSA-N 0.000 description 1
- 235000020661 alpha-linolenic acid Nutrition 0.000 description 1
- 229910052782 aluminium Inorganic materials 0.000 description 1
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- 125000003368 amide group Chemical group 0.000 description 1
- 150000001414 amino alcohols Chemical class 0.000 description 1
- 125000003277 amino group Chemical group 0.000 description 1
- 229910021529 ammonia Inorganic materials 0.000 description 1
- 235000012538 ammonium bicarbonate Nutrition 0.000 description 1
- 239000001099 ammonium carbonate Substances 0.000 description 1
- 239000012491 analyte Substances 0.000 description 1
- 238000010171 animal model Methods 0.000 description 1
- 238000005349 anion exchange Methods 0.000 description 1
- 150000001450 anions Chemical class 0.000 description 1
- 230000000692 anti-sense effect Effects 0.000 description 1
- 239000000074 antisense oligonucleotide Substances 0.000 description 1
- 238000012230 antisense oligonucleotides Methods 0.000 description 1
- 239000007864 aqueous solution Substances 0.000 description 1
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 1
- 235000010323 ascorbic acid Nutrition 0.000 description 1
- 239000011668 ascorbic acid Substances 0.000 description 1
- 229960005070 ascorbic acid Drugs 0.000 description 1
- 235000003704 aspartic acid Nutrition 0.000 description 1
- 229960005261 aspartic acid Drugs 0.000 description 1
- 229940116226 behenic acid Drugs 0.000 description 1
- 125000003910 behenoyl group Chemical group O=C([*])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 230000009286 beneficial effect Effects 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-N benzenesulfonic acid Chemical compound OS(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-N 0.000 description 1
- 229940092714 benzenesulfonic acid Drugs 0.000 description 1
- 229960004365 benzoic acid Drugs 0.000 description 1
- RWCCWEUUXYIKHB-UHFFFAOYSA-N benzophenone Chemical compound C=1C=CC=CC=1C(=O)C1=CC=CC=C1 RWCCWEUUXYIKHB-UHFFFAOYSA-N 0.000 description 1
- 239000012965 benzophenone Substances 0.000 description 1
- 125000001231 benzoyloxy group Chemical group C(C1=CC=CC=C1)(=O)O* 0.000 description 1
- GONOPSZTUGRENK-UHFFFAOYSA-N benzyl(trichloro)silane Chemical compound Cl[Si](Cl)(Cl)CC1=CC=CC=C1 GONOPSZTUGRENK-UHFFFAOYSA-N 0.000 description 1
- MSWZFWKMSRAUBD-UHFFFAOYSA-N beta-D-galactosamine Natural products NC1C(O)OC(CO)C(O)C1O MSWZFWKMSRAUBD-UHFFFAOYSA-N 0.000 description 1
- OQFSQFPPLPISGP-UHFFFAOYSA-N beta-carboxyaspartic acid Natural products OC(=O)C(N)C(C(O)=O)C(O)=O OQFSQFPPLPISGP-UHFFFAOYSA-N 0.000 description 1
- 229960003237 betaine Drugs 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- 239000013060 biological fluid Substances 0.000 description 1
- 230000033228 biological regulation Effects 0.000 description 1
- 230000008499 blood brain barrier function Effects 0.000 description 1
- 210000001218 blood-brain barrier Anatomy 0.000 description 1
- PXOHOSHERMSUCD-RTBYDAEBSA-N bufanolide Chemical compound C([C@H]1[C@H]2[C@@H]([C@]3(CCCCC3CC2)C)CC[C@@]11C)CC1[C@H]1CCC(=O)OC1 PXOHOSHERMSUCD-RTBYDAEBSA-N 0.000 description 1
- 239000000872 buffer Substances 0.000 description 1
- 239000007975 buffered saline Substances 0.000 description 1
- KDYFGRWQOYBRFD-NUQCWPJISA-N butanedioic acid Chemical compound O[14C](=O)CC[14C](O)=O KDYFGRWQOYBRFD-NUQCWPJISA-N 0.000 description 1
- BMQGVNUXMIRLCK-OAGWZNDDSA-N cabazitaxel Chemical compound O([C@H]1[C@@H]2[C@]3(OC(C)=O)CO[C@@H]3C[C@@H]([C@]2(C(=O)[C@H](OC)C2=C(C)[C@@H](OC(=O)[C@H](O)[C@@H](NC(=O)OC(C)(C)C)C=3C=CC=CC=3)C[C@]1(O)C2(C)C)C)OC)C(=O)C1=CC=CC=C1 BMQGVNUXMIRLCK-OAGWZNDDSA-N 0.000 description 1
- 229960001573 cabazitaxel Drugs 0.000 description 1
- 229960001948 caffeine Drugs 0.000 description 1
- VJEONQKOZGKCAK-UHFFFAOYSA-N caffeine Natural products CN1C(=O)N(C)C(=O)C2=C1C=CN2C VJEONQKOZGKCAK-UHFFFAOYSA-N 0.000 description 1
- 229910052791 calcium Inorganic materials 0.000 description 1
- 239000011575 calcium Substances 0.000 description 1
- 238000011088 calibration curve Methods 0.000 description 1
- LSPHULWDVZXLIL-QUBYGPBYSA-N camphoric acid Chemical compound CC1(C)[C@H](C(O)=O)CC[C@]1(C)C(O)=O LSPHULWDVZXLIL-QUBYGPBYSA-N 0.000 description 1
- 230000009702 cancer cell proliferation Effects 0.000 description 1
- PMTSPAGBAFCORP-UHFFFAOYSA-N cannogenin-alpha-L-thevetoside Natural products OC1C(OC)C(O)C(C)OC1OC1CC(CCC2C3(CCC(C3(C)CCC32)C=2COC(=O)C=2)O)C3(C=O)CC1 PMTSPAGBAFCORP-UHFFFAOYSA-N 0.000 description 1
- 125000002843 carboxylic acid group Chemical group 0.000 description 1
- JIUWTCXNUNHEGP-GJHPUSIBSA-N cardenolide Chemical compound C1([C@H]2CC[C@@H]3[C@H]4[C@@H]([C@]5(CCCCC5CC4)C)CC[C@@]32C)=CC(=O)OC1 JIUWTCXNUNHEGP-GJHPUSIBSA-N 0.000 description 1
- 238000005341 cation exchange Methods 0.000 description 1
- 150000001768 cations Chemical class 0.000 description 1
- 238000004113 cell culture Methods 0.000 description 1
- 230000010261 cell growth Effects 0.000 description 1
- 108091092328 cellular RNA Proteins 0.000 description 1
- 230000004700 cellular uptake Effects 0.000 description 1
- 238000005119 centrifugation Methods 0.000 description 1
- 125000003312 cerotoyl group Chemical group O=C([*])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 230000008859 change Effects 0.000 description 1
- 238000012512 characterization method Methods 0.000 description 1
- 239000003153 chemical reaction reagent Substances 0.000 description 1
- 239000003795 chemical substances by application Substances 0.000 description 1
- OEYIOHPDSNJKLS-UHFFFAOYSA-N choline Chemical compound C[N+](C)(C)CCO OEYIOHPDSNJKLS-UHFFFAOYSA-N 0.000 description 1
- 229960001231 choline Drugs 0.000 description 1
- 238000004587 chromatography analysis Methods 0.000 description 1
- 229930016911 cinnamic acid Natural products 0.000 description 1
- 235000013985 cinnamic acid Nutrition 0.000 description 1
- SECPZKHBENQXJG-UHFFFAOYSA-N cis-palmitoleic acid Natural products CCCCCCC=CCCCCCCCC(O)=O SECPZKHBENQXJG-UHFFFAOYSA-N 0.000 description 1
- 238000007398 colorimetric assay Methods 0.000 description 1
- 230000000052 comparative effect Effects 0.000 description 1
- 229940125773 compound 10 Drugs 0.000 description 1
- 229940125797 compound 12 Drugs 0.000 description 1
- 229940125782 compound 2 Drugs 0.000 description 1
- 229940125810 compound 20 Drugs 0.000 description 1
- 229940126086 compound 21 Drugs 0.000 description 1
- 229940126214 compound 3 Drugs 0.000 description 1
- 229910052802 copper Inorganic materials 0.000 description 1
- 239000010949 copper Substances 0.000 description 1
- 230000008878 coupling Effects 0.000 description 1
- 238000010168 coupling process Methods 0.000 description 1
- 238000005859 coupling reaction Methods 0.000 description 1
- 238000002425 crystallisation Methods 0.000 description 1
- 230000008025 crystallization Effects 0.000 description 1
- 229940127089 cytotoxic agent Drugs 0.000 description 1
- 239000002254 cytotoxic agent Substances 0.000 description 1
- OBATZBGFDSVCJD-UHFFFAOYSA-N de-O-acetyl-lanatoside C Natural products CC1OC(OC2CC3C(C4C(C5(CCC(C5(C)C(O)C4)C=4COC(=O)C=4)O)CC3)(C)CC2)CC(O)C1OC(OC1C)CC(O)C1OC(OC1C)CC(O)C1OC1OC(CO)C(O)C(O)C1O OBATZBGFDSVCJD-UHFFFAOYSA-N 0.000 description 1
- 238000000354 decomposition reaction Methods 0.000 description 1
- 230000007423 decrease Effects 0.000 description 1
- 229940009976 deoxycholate Drugs 0.000 description 1
- KXGVEGMKQFWNSR-LLQZFEROSA-N deoxycholic acid Chemical compound C([C@H]1CC2)[C@H](O)CC[C@]1(C)[C@@H]1[C@@H]2[C@@H]2CC[C@H]([C@@H](CCC(O)=O)C)[C@@]2(C)[C@@H](O)C1 KXGVEGMKQFWNSR-LLQZFEROSA-N 0.000 description 1
- 230000001419 dependent effect Effects 0.000 description 1
- 229960001324 deslanoside Drugs 0.000 description 1
- OBATZBGFDSVCJD-LALPQLPRSA-N deslanoside Chemical compound O([C@H]1[C@@H](O)C[C@@H](O[C@@H]1C)O[C@H]1[C@@H](O)C[C@@H](O[C@@H]1C)O[C@H]1[C@@H](O)C[C@@H](O[C@@H]1C)O[C@@H]1C[C@@H]2[C@]([C@@H]3[C@H]([C@]4(CC[C@@H]([C@@]4(C)[C@H](O)C3)C=3COC(=O)C=3)O)CC2)(C)CC1)[C@@H]1O[C@H](CO)[C@@H](O)[C@H](O)[C@H]1O OBATZBGFDSVCJD-LALPQLPRSA-N 0.000 description 1
- 238000010586 diagram Methods 0.000 description 1
- ZBCBWPMODOFKDW-UHFFFAOYSA-N diethanolamine Chemical compound OCCNCCO ZBCBWPMODOFKDW-UHFFFAOYSA-N 0.000 description 1
- HPNMFZURTQLUMO-UHFFFAOYSA-N diethylamine Chemical compound CCNCC HPNMFZURTQLUMO-UHFFFAOYSA-N 0.000 description 1
- 230000029087 digestion Effects 0.000 description 1
- QONQRTHLHBTMGP-UHFFFAOYSA-N digitoxigenin Natural products CC12CCC(C3(CCC(O)CC3CC3)C)C3C11OC1CC2C1=CC(=O)OC1 QONQRTHLHBTMGP-UHFFFAOYSA-N 0.000 description 1
- 229960000648 digitoxin Drugs 0.000 description 1
- WDJUZGPOPHTGOT-XUDUSOBPSA-N digitoxin Chemical compound C1[C@H](O)[C@H](O)[C@@H](C)O[C@H]1O[C@@H]1[C@@H](C)O[C@@H](O[C@@H]2[C@H](O[C@@H](O[C@@H]3C[C@@H]4[C@]([C@@H]5[C@H]([C@]6(CC[C@@H]([C@@]6(C)CC5)C=5COC(=O)C=5)O)CC4)(C)CC3)C[C@@H]2O)C)C[C@@H]1O WDJUZGPOPHTGOT-XUDUSOBPSA-N 0.000 description 1
- SHIBSTMRCDJXLN-KCZCNTNESA-N digoxigenin Chemical compound C1([C@@H]2[C@@]3([C@@](CC2)(O)[C@H]2[C@@H]([C@@]4(C)CC[C@H](O)C[C@H]4CC2)C[C@H]3O)C)=CC(=O)OC1 SHIBSTMRCDJXLN-KCZCNTNESA-N 0.000 description 1
- 239000012153 distilled water Substances 0.000 description 1
- 150000004625 docetaxel anhydrous derivatives Chemical class 0.000 description 1
- 238000012377 drug delivery Methods 0.000 description 1
- 238000009510 drug design Methods 0.000 description 1
- 238000002651 drug therapy Methods 0.000 description 1
- 238000002296 dynamic light scattering Methods 0.000 description 1
- 235000020673 eicosapentaenoic acid Nutrition 0.000 description 1
- 229960005135 eicosapentaenoic acid Drugs 0.000 description 1
- JAZBEHYOTPTENJ-UHFFFAOYSA-N eicosapentaenoic acid Natural products CCC=CCC=CCC=CCC=CCC=CCCCC(O)=O JAZBEHYOTPTENJ-UHFFFAOYSA-N 0.000 description 1
- 125000004016 elaidoyl group Chemical group O=C([*])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])/C([H])=C([H])/C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 238000001493 electron microscopy Methods 0.000 description 1
- 239000002702 enteric coating Substances 0.000 description 1
- 238000009505 enteric coating Methods 0.000 description 1
- 230000007613 environmental effect Effects 0.000 description 1
- 238000007824 enzymatic assay Methods 0.000 description 1
- DPUOLQHDNGRHBS-KTKRTIGZSA-N erucic acid Chemical compound CCCCCCCC\C=C/CCCCCCCCCCCC(O)=O DPUOLQHDNGRHBS-KTKRTIGZSA-N 0.000 description 1
- 125000001901 erucoyl group Chemical group O=C([*])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])/C([H])=C([H])\C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- CCIVGXIOQKPBKL-UHFFFAOYSA-M ethanesulfonate Chemical compound CCS([O-])(=O)=O CCIVGXIOQKPBKL-UHFFFAOYSA-M 0.000 description 1
- 229940012017 ethylenediamine Drugs 0.000 description 1
- 238000000105 evaporative light scattering detection Methods 0.000 description 1
- 238000000605 extraction Methods 0.000 description 1
- 238000001125 extrusion Methods 0.000 description 1
- 238000001914 filtration Methods 0.000 description 1
- XSFJVAJPIHIPKU-XWCQMRHXSA-N flunisolide Chemical compound C1([C@@H](F)C2)=CC(=O)C=C[C@]1(C)[C@@H]1[C@@H]2[C@@H]2C[C@H]3OC(C)(C)O[C@@]3(C(=O)CO)[C@@]2(C)C[C@@H]1O XSFJVAJPIHIPKU-XWCQMRHXSA-N 0.000 description 1
- 239000006260 foam Substances 0.000 description 1
- 235000019253 formic acid Nutrition 0.000 description 1
- 239000001530 fumaric acid Substances 0.000 description 1
- 230000030279 gene silencing Effects 0.000 description 1
- 238000010363 gene targeting Methods 0.000 description 1
- 239000000174 gluconic acid Substances 0.000 description 1
- 235000012208 gluconic acid Nutrition 0.000 description 1
- 229960002442 glucosamine Drugs 0.000 description 1
- 230000009036 growth inhibition Effects 0.000 description 1
- 239000001963 growth medium Substances 0.000 description 1
- JAXFJECJQZDFJS-XHEPKHHKSA-N gtpl8555 Chemical compound OC(=O)C[C@H](N)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](C(C)C)C(=O)N1CCC[C@@H]1C(=O)N[C@H](B1O[C@@]2(C)[C@H]3C[C@H](C3(C)C)C[C@H]2O1)CCC1=CC=C(F)C=C1 JAXFJECJQZDFJS-XHEPKHHKSA-N 0.000 description 1
- 230000009931 harmful effect Effects 0.000 description 1
- 238000010842 high-capacity cDNA reverse transcription kit Methods 0.000 description 1
- HNDVDQJCIGZPNO-UHFFFAOYSA-N histidine Natural products OC(=O)C(N)CC1=CN=CN1 HNDVDQJCIGZPNO-UHFFFAOYSA-N 0.000 description 1
- XGIHQYAWBCFNPY-AZOCGYLKSA-N hydrabamine Chemical compound C([C@@H]12)CC3=CC(C(C)C)=CC=C3[C@@]2(C)CCC[C@@]1(C)CNCCNC[C@@]1(C)[C@@H]2CCC3=CC(C(C)C)=CC=C3[C@@]2(C)CCC1 XGIHQYAWBCFNPY-AZOCGYLKSA-N 0.000 description 1
- 150000004677 hydrates Chemical class 0.000 description 1
- 239000000017 hydrogel Substances 0.000 description 1
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 1
- XLYOFNOQVPJJNP-UHFFFAOYSA-M hydroxide Chemical compound [OH-] XLYOFNOQVPJJNP-UHFFFAOYSA-M 0.000 description 1
- NPZTUJOABDZTLV-UHFFFAOYSA-N hydroxybenzotriazole Substances O=C1C=CC=C2NNN=C12 NPZTUJOABDZTLV-UHFFFAOYSA-N 0.000 description 1
- 230000036543 hypotension Effects 0.000 description 1
- 238000011065 in-situ storage Methods 0.000 description 1
- 230000002779 inactivation Effects 0.000 description 1
- 239000000411 inducer Substances 0.000 description 1
- 238000002329 infrared spectrum Methods 0.000 description 1
- 238000001802 infusion Methods 0.000 description 1
- 239000003112 inhibitor Substances 0.000 description 1
- 239000003456 ion exchange resin Substances 0.000 description 1
- 229920003303 ion-exchange polymer Polymers 0.000 description 1
- 150000002500 ions Chemical class 0.000 description 1
- 229910052742 iron Inorganic materials 0.000 description 1
- 238000002955 isolation Methods 0.000 description 1
- 238000006317 isomerization reaction Methods 0.000 description 1
- JJWLVOIRVHMVIS-UHFFFAOYSA-N isopropylamine Chemical compound CC(C)N JJWLVOIRVHMVIS-UHFFFAOYSA-N 0.000 description 1
- ZLVXBBHTMQJRSX-VMGNSXQWSA-N jdtic Chemical compound C1([C@]2(C)CCN(C[C@@H]2C)C[C@H](C(C)C)NC(=O)[C@@H]2NCC3=CC(O)=CC=C3C2)=CC=CC(O)=C1 ZLVXBBHTMQJRSX-VMGNSXQWSA-N 0.000 description 1
- 239000004310 lactic acid Substances 0.000 description 1
- 235000014655 lactic acid Nutrition 0.000 description 1
- 239000008101 lactose Substances 0.000 description 1
- OYHQOLUKZRVURQ-AVQMFFATSA-N linoelaidic acid Chemical compound CCCCC\C=C\C\C=C\CCCCCCCC(O)=O OYHQOLUKZRVURQ-AVQMFFATSA-N 0.000 description 1
- 229960004488 linolenic acid Drugs 0.000 description 1
- KQQKGWQCNNTQJW-UHFFFAOYSA-N linolenic acid Natural products CC=CCCC=CCC=CCCCCCCCC(O)=O KQQKGWQCNNTQJW-UHFFFAOYSA-N 0.000 description 1
- 125000005644 linolenyl group Chemical group 0.000 description 1
- 239000002502 liposome Substances 0.000 description 1
- 229910052744 lithium Inorganic materials 0.000 description 1
- 230000004807 localization Effects 0.000 description 1
- 229910052749 magnesium Inorganic materials 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 239000011976 maleic acid Substances 0.000 description 1
- 239000001630 malic acid Substances 0.000 description 1
- 235000011090 malic acid Nutrition 0.000 description 1
- 229960002510 mandelic acid Drugs 0.000 description 1
- 229910052748 manganese Inorganic materials 0.000 description 1
- 239000011572 manganese Substances 0.000 description 1
- 238000005259 measurement Methods 0.000 description 1
- 230000007246 mechanism Effects 0.000 description 1
- 239000002609 medium Substances 0.000 description 1
- 229960003194 meglumine Drugs 0.000 description 1
- 125000000412 melissoyl group Chemical group O=C([*])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 238000002844 melting Methods 0.000 description 1
- 230000008018 melting Effects 0.000 description 1
- 229910052751 metal Inorganic materials 0.000 description 1
- 239000002184 metal Substances 0.000 description 1
- 229940098779 methanesulfonic acid Drugs 0.000 description 1
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 1
- WBYWAXJHAXSJNI-UHFFFAOYSA-N methyl p-hydroxycinnamate Natural products OC(=O)C=CC1=CC=CC=C1 WBYWAXJHAXSJNI-UHFFFAOYSA-N 0.000 description 1
- 239000011859 microparticle Substances 0.000 description 1
- 125000002320 montanoyl group Chemical group O=C([*])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- LNOPIUAQISRISI-UHFFFAOYSA-N n'-hydroxy-2-propan-2-ylsulfonylethanimidamide Chemical compound CC(C)S(=O)(=O)CC(N)=NO LNOPIUAQISRISI-UHFFFAOYSA-N 0.000 description 1
- UQEIFYRRSNJVDO-UHFFFAOYSA-N n,n-dibenzyl-2-phenylethanamine Chemical compound C=1C=CC=CC=1CN(CC=1C=CC=CC=1)CCC1=CC=CC=C1 UQEIFYRRSNJVDO-UHFFFAOYSA-N 0.000 description 1
- WQEPLUUGTLDZJY-UHFFFAOYSA-N n-Pentadecanoic acid Natural products CCCCCCCCCCCCCCC(O)=O WQEPLUUGTLDZJY-UHFFFAOYSA-N 0.000 description 1
- FUZZWVXGSFPDMH-UHFFFAOYSA-N n-hexanoic acid Natural products CCCCCC(O)=O FUZZWVXGSFPDMH-UHFFFAOYSA-N 0.000 description 1
- PSZYNBSKGUBXEH-UHFFFAOYSA-N naphthalene-1-sulfonic acid Chemical compound C1=CC=C2C(S(=O)(=O)O)=CC=CC2=C1 PSZYNBSKGUBXEH-UHFFFAOYSA-N 0.000 description 1
- KVBGVZZKJNLNJU-UHFFFAOYSA-N naphthalene-2-sulfonic acid Chemical compound C1=CC=CC2=CC(S(=O)(=O)O)=CC=C21 KVBGVZZKJNLNJU-UHFFFAOYSA-N 0.000 description 1
- 150000002790 naphthalenes Chemical class 0.000 description 1
- 239000007923 nasal drop Substances 0.000 description 1
- 229940100662 nasal drops Drugs 0.000 description 1
- 239000013642 negative control Substances 0.000 description 1
- 229960003512 nicotinic acid Drugs 0.000 description 1
- 239000011664 nicotinic acid Substances 0.000 description 1
- 235000001968 nicotinic acid Nutrition 0.000 description 1
- 229910017604 nitric acid Inorganic materials 0.000 description 1
- 229910000069 nitrogen hydride Inorganic materials 0.000 description 1
- 231100000028 nontoxic concentration Toxicity 0.000 description 1
- ABCVHPIKBGRCJA-UHFFFAOYSA-N nonyl 8-[(8-heptadecan-9-yloxy-8-oxooctyl)-(2-hydroxyethyl)amino]octanoate Chemical compound OCCN(CCCCCCCC(=O)OC(CCCCCCCC)CCCCCCCC)CCCCCCCC(=O)OCCCCCCCCC ABCVHPIKBGRCJA-UHFFFAOYSA-N 0.000 description 1
- 125000002347 octyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 239000002674 ointment Substances 0.000 description 1
- JRZJOMJEPLMPRA-UHFFFAOYSA-N olefin Natural products CCCCCCCC=C JRZJOMJEPLMPRA-UHFFFAOYSA-N 0.000 description 1
- 150000007530 organic bases Chemical class 0.000 description 1
- 239000012044 organic layer Substances 0.000 description 1
- 239000012074 organic phase Substances 0.000 description 1
- 239000003960 organic solvent Substances 0.000 description 1
- 230000003204 osmotic effect Effects 0.000 description 1
- LPMXVESGRSUGHW-HBYQJFLCSA-N ouabain Chemical compound O[C@@H]1[C@H](O)[C@@H](O)[C@H](C)O[C@H]1O[C@@H]1C[C@@]2(O)CC[C@H]3[C@@]4(O)CC[C@H](C=5COC(=O)C=5)[C@@]4(C)C[C@@H](O)[C@@H]3[C@@]2(CO)[C@H](O)C1 LPMXVESGRSUGHW-HBYQJFLCSA-N 0.000 description 1
- 229960003343 ouabain Drugs 0.000 description 1
- 235000006408 oxalic acid Nutrition 0.000 description 1
- KJIFKLIQANRMOU-UHFFFAOYSA-N oxidanium;4-methylbenzenesulfonate Chemical compound O.CC1=CC=C(S(O)(=O)=O)C=C1 KJIFKLIQANRMOU-UHFFFAOYSA-N 0.000 description 1
- 238000012856 packing Methods 0.000 description 1
- 125000001312 palmitoyl group Chemical group O=C([*])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- WLJNZVDCPSBLRP-UHFFFAOYSA-N pamoic acid Chemical compound C1=CC=C2C(CC=3C4=CC=CC=C4C=C(C=3O)C(=O)O)=C(O)C(C(O)=O)=CC2=C1 WLJNZVDCPSBLRP-UHFFFAOYSA-N 0.000 description 1
- FJKROLUGYXJWQN-UHFFFAOYSA-N papa-hydroxy-benzoic acid Natural products OC(=O)C1=CC=C(O)C=C1 FJKROLUGYXJWQN-UHFFFAOYSA-N 0.000 description 1
- 230000009057 passive transport Effects 0.000 description 1
- 239000006072 paste Substances 0.000 description 1
- 229920003175 pectinic acid Polymers 0.000 description 1
- 235000019371 penicillin G benzathine Nutrition 0.000 description 1
- 229960004180 peruvoside Drugs 0.000 description 1
- PMTSPAGBAFCORP-HBUONDEYSA-N peruvoside Chemical compound O[C@H]1[C@H](OC)[C@@H](O)[C@H](C)O[C@H]1O[C@@H]1C[C@@H](CC[C@H]2[C@]3(CC[C@@H]([C@@]3(C)CC[C@H]32)C=2COC(=O)C=2)O)[C@]3(C=O)CC1 PMTSPAGBAFCORP-HBUONDEYSA-N 0.000 description 1
- 239000008177 pharmaceutical agent Substances 0.000 description 1
- 239000000825 pharmaceutical preparation Substances 0.000 description 1
- 230000000144 pharmacologic effect Effects 0.000 description 1
- 229950010879 phenamine Drugs 0.000 description 1
- SHUZOJHMOBOZST-UHFFFAOYSA-N phylloquinone Natural products CC(C)CCCCC(C)CCC(C)CCCC(=CCC1=C(C)C(=O)c2ccccc2C1=O)C SHUZOJHMOBOZST-UHFFFAOYSA-N 0.000 description 1
- 230000004962 physiological condition Effects 0.000 description 1
- OXNIZHLAWKMVMX-UHFFFAOYSA-N picric acid Chemical compound OC1=C([N+]([O-])=O)C=C([N+]([O-])=O)C=C1[N+]([O-])=O OXNIZHLAWKMVMX-UHFFFAOYSA-N 0.000 description 1
- IUGYQRQAERSCNH-UHFFFAOYSA-N pivalic acid Chemical compound CC(C)(C)C(O)=O IUGYQRQAERSCNH-UHFFFAOYSA-N 0.000 description 1
- 230000036470 plasma concentration Effects 0.000 description 1
- 229920001515 polyalkylene glycol Polymers 0.000 description 1
- 229920000768 polyamine Polymers 0.000 description 1
- 229920002503 polyoxyethylene-polyoxypropylene Polymers 0.000 description 1
- 229910052700 potassium Inorganic materials 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- 230000003389 potentiating effect Effects 0.000 description 1
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 1
- MFDFERRIHVXMIY-UHFFFAOYSA-N procaine Chemical compound CCN(CC)CCOC(=O)C1=CC=C(N)C=C1 MFDFERRIHVXMIY-UHFFFAOYSA-N 0.000 description 1
- 229960004919 procaine Drugs 0.000 description 1
- VVWRJUBEIPHGQF-UHFFFAOYSA-N propan-2-yl n-propan-2-yloxycarbonyliminocarbamate Chemical compound CC(C)OC(=O)N=NC(=O)OC(C)C VVWRJUBEIPHGQF-UHFFFAOYSA-N 0.000 description 1
- 235000019260 propionic acid Nutrition 0.000 description 1
- 229940095574 propionic acid Drugs 0.000 description 1
- 125000006239 protecting group Chemical group 0.000 description 1
- 235000018102 proteins Nutrition 0.000 description 1
- 102000004169 proteins and genes Human genes 0.000 description 1
- 150000003212 purines Chemical class 0.000 description 1
- KOUKXHPPRFNWPP-UHFFFAOYSA-N pyrazine-2,5-dicarboxylic acid;hydrate Chemical compound O.OC(=O)C1=CN=C(C(O)=O)C=N1 KOUKXHPPRFNWPP-UHFFFAOYSA-N 0.000 description 1
- 229940107700 pyruvic acid Drugs 0.000 description 1
- IUVKMZGDUIUOCP-BTNSXGMBSA-N quinbolone Chemical compound O([C@H]1CC[C@H]2[C@H]3[C@@H]([C@]4(C=CC(=O)C=C4CC3)C)CC[C@@]21C)C1=CCCC1 IUVKMZGDUIUOCP-BTNSXGMBSA-N 0.000 description 1
- 238000001953 recrystallisation Methods 0.000 description 1
- 238000010992 reflux Methods 0.000 description 1
- BOLDJAUMGUJJKM-LSDHHAIUSA-N renifolin D Natural products CC(=C)[C@@H]1Cc2c(O)c(O)ccc2[C@H]1CC(=O)c3ccc(O)cc3O BOLDJAUMGUJJKM-LSDHHAIUSA-N 0.000 description 1
- 239000011347 resin Substances 0.000 description 1
- 229920005989 resin Polymers 0.000 description 1
- 230000000717 retained effect Effects 0.000 description 1
- WBHHMMIMDMUBKC-XLNAKTSKSA-N ricinelaidic acid Chemical compound CCCCCC[C@@H](O)C\C=C\CCCCCCCC(O)=O WBHHMMIMDMUBKC-XLNAKTSKSA-N 0.000 description 1
- 229960003656 ricinoleic acid Drugs 0.000 description 1
- FEUQNCSVHBHROZ-UHFFFAOYSA-N ricinoleic acid Natural products CCCCCCC(O[Si](C)(C)C)CC=CCCCCCCCC(=O)OC FEUQNCSVHBHROZ-UHFFFAOYSA-N 0.000 description 1
- 229960004889 salicylic acid Drugs 0.000 description 1
- NNNVXFKZMRGJPM-KHPPLWFESA-N sapienic acid Chemical compound CCCCCCCCC\C=C/CCCCC(O)=O NNNVXFKZMRGJPM-KHPPLWFESA-N 0.000 description 1
- LSMIOFMZNVEEBR-ICLSSMQGSA-N scilliroside Chemical compound C=1([C@@H]2[C@@]3(C)CC[C@H]4[C@@]([C@]3(CC2)O)(O)C[C@H](C2=C[C@@H](O[C@H]3[C@@H]([C@@H](O)[C@H](O)[C@@H](CO)O3)O)CC[C@@]24C)OC(=O)C)C=CC(=O)OC=1 LSMIOFMZNVEEBR-ICLSSMQGSA-N 0.000 description 1
- 238000000926 separation method Methods 0.000 description 1
- 210000002966 serum Anatomy 0.000 description 1
- 238000001542 size-exclusion chromatography Methods 0.000 description 1
- AWUCVROLDVIAJX-GSVOUGTGSA-N sn-glycerol 3-phosphate Chemical compound OC[C@@H](O)COP(O)(O)=O AWUCVROLDVIAJX-GSVOUGTGSA-N 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- 239000011780 sodium chloride Substances 0.000 description 1
- 238000010561 standard procedure Methods 0.000 description 1
- 239000008223 sterile water Substances 0.000 description 1
- 230000004936 stimulating effect Effects 0.000 description 1
- 238000003860 storage Methods 0.000 description 1
- ODJLBQGVINUMMR-HZXDTFASSA-N strophanthidin Chemical compound C1([C@H]2CC[C@]3(O)[C@H]4[C@@H]([C@]5(CC[C@H](O)C[C@@]5(O)CC4)C=O)CC[C@@]32C)=CC(=O)OC1 ODJLBQGVINUMMR-HZXDTFASSA-N 0.000 description 1
- 238000005556 structure-activity relationship Methods 0.000 description 1
- 125000001424 substituent group Chemical group 0.000 description 1
- 239000000758 substrate Substances 0.000 description 1
- 229940032330 sulfuric acid Drugs 0.000 description 1
- 239000011975 tartaric acid Substances 0.000 description 1
- 235000002906 tartaric acid Nutrition 0.000 description 1
- BCNZYOJHNLTNEZ-UHFFFAOYSA-N tert-butyldimethylsilyl chloride Chemical compound CC(C)(C)[Si](C)(C)Cl BCNZYOJHNLTNEZ-UHFFFAOYSA-N 0.000 description 1
- CBXCPBUEXACCNR-UHFFFAOYSA-N tetraethylammonium Chemical class CC[N+](CC)(CC)CC CBXCPBUEXACCNR-UHFFFAOYSA-N 0.000 description 1
- QEMXHQIAXOOASZ-UHFFFAOYSA-N tetramethylammonium Chemical class C[N+](C)(C)C QEMXHQIAXOOASZ-UHFFFAOYSA-N 0.000 description 1
- 125000003831 tetrazolyl group Chemical group 0.000 description 1
- 229940124597 therapeutic agent Drugs 0.000 description 1
- 231100001274 therapeutic index Toxicity 0.000 description 1
- 238000002560 therapeutic procedure Methods 0.000 description 1
- 238000004448 titration Methods 0.000 description 1
- 238000011200 topical administration Methods 0.000 description 1
- 231100000816 toxic dose Toxicity 0.000 description 1
- 231100000440 toxicity profile Toxicity 0.000 description 1
- UWHZIFQPPBDJPM-BQYQJAHWSA-N trans-vaccenic acid Chemical compound CCCCCC\C=C\CCCCCCCCCC(O)=O UWHZIFQPPBDJPM-BQYQJAHWSA-N 0.000 description 1
- 230000032258 transport Effects 0.000 description 1
- IMFACGCPASFAPR-UHFFFAOYSA-N tributylamine Chemical compound CCCCN(CCCC)CCCC IMFACGCPASFAPR-UHFFFAOYSA-N 0.000 description 1
- PHYFQTYBJUILEZ-IUPFWZBJSA-N triolein Chemical compound CCCCCCCC\C=C/CCCCCCCC(=O)OCC(OC(=O)CCCCCCC\C=C/CCCCCCCC)COC(=O)CCCCCCC\C=C/CCCCCCCC PHYFQTYBJUILEZ-IUPFWZBJSA-N 0.000 description 1
- 229940117972 triolein Drugs 0.000 description 1
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 1
- YFTHZRPMJXBUME-UHFFFAOYSA-N tripropylamine Chemical compound CCCN(CCC)CCC YFTHZRPMJXBUME-UHFFFAOYSA-N 0.000 description 1
- 210000002700 urine Anatomy 0.000 description 1
- 125000003847 vaccenoyl group Chemical group O=C([*])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])=C([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 235000013311 vegetables Nutrition 0.000 description 1
- 239000003981 vehicle Substances 0.000 description 1
- 230000035899 viability Effects 0.000 description 1
- 235000019168 vitamin K Nutrition 0.000 description 1
- 239000011712 vitamin K Substances 0.000 description 1
- 150000003721 vitamin K derivatives Chemical class 0.000 description 1
- 229940046010 vitamin k Drugs 0.000 description 1
- 238000005406 washing Methods 0.000 description 1
- 239000011701 zinc Substances 0.000 description 1
- 229910052725 zinc Inorganic materials 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07J—STEROIDS
- C07J5/00—Normal steroids containing carbon, hydrogen, halogen or oxygen, substituted in position 17 beta by a chain of two carbon atoms, e.g. pregnane and substituted in position 21 by only one singly bound oxygen atom, i.e. only one oxygen bound to position 21 by a single bond
- C07J5/0007—Normal steroids containing carbon, hydrogen, halogen or oxygen, substituted in position 17 beta by a chain of two carbon atoms, e.g. pregnane and substituted in position 21 by only one singly bound oxygen atom, i.e. only one oxygen bound to position 21 by a single bond not substituted in position 17 alfa
- C07J5/0023—Normal steroids containing carbon, hydrogen, halogen or oxygen, substituted in position 17 beta by a chain of two carbon atoms, e.g. pregnane and substituted in position 21 by only one singly bound oxygen atom, i.e. only one oxygen bound to position 21 by a single bond not substituted in position 17 alfa substituted in position 16
- C07J5/003—Normal steroids containing carbon, hydrogen, halogen or oxygen, substituted in position 17 beta by a chain of two carbon atoms, e.g. pregnane and substituted in position 21 by only one singly bound oxygen atom, i.e. only one oxygen bound to position 21 by a single bond not substituted in position 17 alfa substituted in position 16 by a saturated or unsaturated hydrocarbon group including 16-alkylidene substitutes
- C07J5/0038—Normal steroids containing carbon, hydrogen, halogen or oxygen, substituted in position 17 beta by a chain of two carbon atoms, e.g. pregnane and substituted in position 21 by only one singly bound oxygen atom, i.e. only one oxygen bound to position 21 by a single bond not substituted in position 17 alfa substituted in position 16 by a saturated or unsaturated hydrocarbon group including 16-alkylidene substitutes by an alkyl group
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/54—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic compound
- A61K47/543—Lipids, e.g. triglycerides; Polyamines, e.g. spermine or spermidine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/335—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin
- A61K31/337—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin having four-membered rings, e.g. taxol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/4353—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems
- A61K31/436—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems the heterocyclic ring system containing a six-membered ring having oxygen as a ring hetero atom, e.g. rapamycin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/519—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/56—Compounds containing cyclopenta[a]hydrophenanthrene ring systems; Derivatives thereof, e.g. steroids
- A61K31/57—Compounds containing cyclopenta[a]hydrophenanthrene ring systems; Derivatives thereof, e.g. steroids substituted in position 17 beta by a chain of two carbon atoms, e.g. pregnane or progesterone
- A61K31/573—Compounds containing cyclopenta[a]hydrophenanthrene ring systems; Derivatives thereof, e.g. steroids substituted in position 17 beta by a chain of two carbon atoms, e.g. pregnane or progesterone substituted in position 21, e.g. cortisone, dexamethasone, prednisone or aldosterone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/56—Compounds containing cyclopenta[a]hydrophenanthrene ring systems; Derivatives thereof, e.g. steroids
- A61K31/58—Compounds containing cyclopenta[a]hydrophenanthrene ring systems; Derivatives thereof, e.g. steroids containing heterocyclic rings, e.g. danazol, stanozolol, pancuronium or digitogenin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D295/00—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms
- C07D295/16—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms acylated on ring nitrogen atoms
- C07D295/18—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms acylated on ring nitrogen atoms by radicals derived from carboxylic acids, or sulfur or nitrogen analogues thereof
- C07D295/182—Radicals derived from carboxylic acids
- C07D295/185—Radicals derived from carboxylic acids from aliphatic carboxylic acids
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D305/00—Heterocyclic compounds containing four-membered rings having one oxygen atom as the only ring hetero atoms
- C07D305/14—Heterocyclic compounds containing four-membered rings having one oxygen atom as the only ring hetero atoms condensed with carbocyclic rings or ring systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D475/00—Heterocyclic compounds containing pteridine ring systems
- C07D475/06—Heterocyclic compounds containing pteridine ring systems with a nitrogen atom directly attached in position 4
- C07D475/08—Heterocyclic compounds containing pteridine ring systems with a nitrogen atom directly attached in position 4 with a nitrogen atom directly attached in position 2
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07J—STEROIDS
- C07J31/00—Normal steroids containing one or more sulfur atoms not belonging to a hetero ring
- C07J31/006—Normal steroids containing one or more sulfur atoms not belonging to a hetero ring not covered by C07J31/003
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07J—STEROIDS
- C07J41/00—Normal steroids containing one or more nitrogen atoms not belonging to a hetero ring
- C07J41/0033—Normal steroids containing one or more nitrogen atoms not belonging to a hetero ring not covered by C07J41/0005
- C07J41/005—Normal steroids containing one or more nitrogen atoms not belonging to a hetero ring not covered by C07J41/0005 the 17-beta position being substituted by an uninterrupted chain of only two carbon atoms, e.g. pregnane derivatives
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07J—STEROIDS
- C07J43/00—Normal steroids having a nitrogen-containing hetero ring spiro-condensed or not condensed with the cyclopenta(a)hydrophenanthrene skeleton
- C07J43/003—Normal steroids having a nitrogen-containing hetero ring spiro-condensed or not condensed with the cyclopenta(a)hydrophenanthrene skeleton not condensed
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07J—STEROIDS
- C07J5/00—Normal steroids containing carbon, hydrogen, halogen or oxygen, substituted in position 17 beta by a chain of two carbon atoms, e.g. pregnane and substituted in position 21 by only one singly bound oxygen atom, i.e. only one oxygen bound to position 21 by a single bond
- C07J5/0046—Normal steroids containing carbon, hydrogen, halogen or oxygen, substituted in position 17 beta by a chain of two carbon atoms, e.g. pregnane and substituted in position 21 by only one singly bound oxygen atom, i.e. only one oxygen bound to position 21 by a single bond substituted in position 17 alfa
- C07J5/0061—Normal steroids containing carbon, hydrogen, halogen or oxygen, substituted in position 17 beta by a chain of two carbon atoms, e.g. pregnane and substituted in position 21 by only one singly bound oxygen atom, i.e. only one oxygen bound to position 21 by a single bond substituted in position 17 alfa substituted in position 16
- C07J5/0069—Normal steroids containing carbon, hydrogen, halogen or oxygen, substituted in position 17 beta by a chain of two carbon atoms, e.g. pregnane and substituted in position 21 by only one singly bound oxygen atom, i.e. only one oxygen bound to position 21 by a single bond substituted in position 17 alfa substituted in position 16 by a saturated or unsaturated hydrocarbon group
- C07J5/0076—Normal steroids containing carbon, hydrogen, halogen or oxygen, substituted in position 17 beta by a chain of two carbon atoms, e.g. pregnane and substituted in position 21 by only one singly bound oxygen atom, i.e. only one oxygen bound to position 21 by a single bond substituted in position 17 alfa substituted in position 16 by a saturated or unsaturated hydrocarbon group by an alkyl group
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Epidemiology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Pain & Pain Management (AREA)
- Rheumatology (AREA)
- Molecular Biology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Biophysics (AREA)
- Steroid Compounds (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Medicinal Preparation (AREA)
Abstract
Esta invención proporciona profármacos unidos a lípidos que tienen estructuras como se establece en el presente documento. También se proporcionan usos de tales compuestos de profármacos unidos a lípidos para el tratamiento de diversas indicaciones, y métodos para preparar y usar profármacos unidos a lípidos. (Traducción automática con Google Translate, sin valor legal)
Description
DESCRIPCIÓN
Profármacos unidos a lípidos
Campo técnico
La presente invención se refiere a un conjugado de un compuesto terapéutico y un resto lipídico, a composiciones de este y a métodos para su uso en la administración de compuestos terapéuticos. En particular, la invención se refiere a profármacos unidos a lípidos, en donde el ligador unido covalentemente al profármaco es biodegradable. También se proporcionan métodos para fabricar y utilizar los conjugados.
Antecedentes
La selectividad del fármaco para un tejido diana es una consideración importante durante el diseño de fármacos. La administración selectiva de un fármaco a su diana puede permitir dosis más bajas y efectos secundarios reducidos. La selectividad para un tejido diana determinado puede ser particularmente importante cuando los agentes terapéuticos administrados son quimioterapéuticos para el tratamiento del cáncer. En particular, las terapias con fármacos citotóxicos se dirigen a células que se dividen rápidamente, pero a menudo pueden verse limitadas por los efectos secundarios tóxicos del quimioterapéutico en las células sanas. Además, otros restos terapéuticos pueden beneficiarse de una orientación selectiva y unas características farmacocinéticas mejoradas (es decir, estabilidad in vivo, captación celular (por ejemplo, lipofilia), etc.).
Los ácidos grasos se han utilizado para mejorar la selectividad de los fármacos para sus tejidos diana (por ejemplo, US 7.235.583; US 8.552.054; US 6.090.800; US 2012/0264810; US 2013/0330401). Con anterioridad se han conjugado ácidos grasos con fármacos para ayudar a los fármacos como conjugados a atravesar la barrera hematoencefálica (por ejemplo, US 4.933.324).
Las moléculas lipídicas, incluidos los ácidos grasos o las aminas grasas, también se han conjugado con fármacos para que los conjugados sean más lipofílicos que los fármacos no conjugados. Las aminas grasas son moléculas lipídicas que terminan en un grupo amino (a diferencia de los ácidos grasos, que terminan en un grupo ácido carboxílico). Los ácidos grasos se producen de forma natural, mientras que las aminas grasas no son un componente tisular común en los animales.
Sumario
La presente invención se basa en parte en el descubrimiento fortuito de que los conjugados de los compuestos terapéuticos y los restos lipídicos descritos en la presente, muestran una administración selectiva mejorada. Además, algunos de los conjugados de compuestos terapéuticos y restos lipídicos muestran una carga mejorada, una toxicidad reducida, una orientación mejorada, una potencia in vivo mejorada o una estabilidad in vivo mejorada. El alcance de la presente invención se define por el alcance de las reivindicaciones adjuntas.
En un aspecto, la presente invención se refiere a un compuesto, en donde el compuesto tiene la estructura de la Fórmula III:

en donde:
A-Z es dexametasona, en donde Z es un átomo electronegativo seleccionado de N u O, de tal manera que la dexametasona se derivó de un fármaco que tiene la Fórmula A-Z-H cuando Z ha perdido un H para unirse covalentemente a Y;
Y es CH2 , C(=O) o PO32-;
T5 son 0-6 átomos de carbono; y
L4 es una cadena de carbono lineal o ramificada C9-C29, que tiene uno o más enlaces dobles C=C cis o trans y opcionalmente sustituida con OH o tiene la Fórmula V

en donde:
T2 son 1-4 átomos de carbono;
T3 son 0-4 átomos de carbono
T4 son 0-4 átomos de carbono;
J1 es -O-C(=O)-L1;
J2 es -COOH, -OH, -NH3+, -NH2, -SH, -NMe2 o -NHMe; y
L1 es una cadena de carbono lineal o ramificada C9-C29, que opcionalmente tiene uno o más enlaces dobles C=C cis o trans y opcionalmente sustituida con OH.
En una forma de realización de la invención, el compuesto tiene la estructura de la Fórmula II:

en donde:
Y es CH2 , C(=O), o PO32-;
T1 son 0-4 átomos de carbono
T2 son 1-3 átomos de carbono; y
J2 es -COOH, -OH, -NH3+, -NH2,-NMe2 , o -NHMe.
En otra forma de realización de la invención, el compuesto tiene la estructura de la Fórmula III:

en donde:
Y es CH2 , C(=O) o PO32-; y
T5 son 0-4 átomos de carbono.
En formas de realización preferidas, Y es CH2 o C(=O); y T5 o T1 son 0-2 átomos de carbono.
Preferentemente, el compuesto de acuerdo con la presente invención se selecciona de uno o más de los siguientes:

y 
Aún más preferentemente, el compuesto de acuerdo con la presente invención es

En un aspecto adicional, la presente invención se refiere a una composición farmacéutica, la composición farmacéutica comprende un compuesto de acuerdo con la presente invención y un portador farmacéuticamente aceptable.
En un aspecto adicional, la presente invención se refiere a un compuesto de acuerdo con la presente invención para su uso en el tratamiento del cáncer, enfermedad autoinmune o infección.
Se proporciona para la comparación en la presente un compuesto que tiene la estructura de la Fórmula I:

en donde:
R1 puede ser H, un alquilo C1-C20 lineal, ramificado o cíclico, opcionalmente con uno o más enlaces dobles C=C cis o trans de geometría E o Z, en donde el alquilo C1-C10 puede estar opcionalmente sustituido con F, Cl, Br, I, OH, O-alquilo C1-C6 u O-acilo C1-C6, en donde el O-alquilo C1-C6 o el O-acilo C1-C6 pueden estar opcionalmente sustituidos con F, Cl, Br, I u OH;
R2 puede ser H, una amina primaria C1-C6, una amina secundaria C1-C6 o una amina terciaria C1-C6 , un alquilo C1-C10 lineal, ramificado o cíclico, opcionalmente con uno o más enlaces dobles C=C cis o trans de geometría E o Z, en donde el alquilo C1-C10 puede estar opcionalmente sustituido con F, Cl, Br, I, OH, O alquilo C1-C6 u O-acilo C1-C6, en donde el O-alquilo C1-C6 o el O-acilo C1-C6 pueden estar opcionalmente sustituidos con F, Cl, Br, I u OH;
X1 puede ser O, S o NR10, en donde R10 puede ser H, un alquilo C1-C10 lineal, ramificado o cíclico, opcionalmente con uno o más enlaces dobles C=C cis o trans de geometría E o Z, en donde el alquilo C1-C10 puede estar opcionalmente sustituido con F, Cl, Br, I, OH, O-alquilo C1-C6, u O-acilo C1-C6, en donde el O-alquilo C1-C6 o el O-acilo C1-C6 pueden estar opcionalmente sustituidos con F, Cl, Br, I u OH;
G1 puede ser

en donde:
A-Zg1 puede ser un resto de fármaco, en donde Zg1 puede ser un átomo electronegativo seleccionado de N y O, de tal manera que el resto de fármaco se derivó de un fármaco que tiene la Fórmula A-Zg1-H cuando Zg1 ha perdido un H para unirse covalentemente a Yg1;
Yg1 puede ser CH2 , O, S, C(=O), PO32', o NR11, en donde R11 puede ser H, un alquilo C1-C10 lineal, ramificado o cíclico, opcionalmente con uno o más enlaces dobles C=C cis o trans de geometría E o Z, en donde el alquilo C1-C10 puede estar opcionalmente sustituido con F, Cl, Br, I, OH, O-alquilo C1-C6, u O-acilo C1-C6 , y en donde el O-alquilo C1-C6 o el O-acilo C1-C6 pueden estar opcionalmente sustituidos con F, Cl, Br, I u OH;
Mg1 puede ser 0-12 átomos seleccionados de C, N, O o S, en donde dos o más átomos de C tienen opcionalmente uno o más enlaces dobles C=C cis o trans de geometría E o Z y en donde los átomos de C pueden estar opcionalmente sustituidos con F, Cl, Br, I, OH, =O, O-alquilo C1-C6, O-acilo C1-C6 o S-alquilo C1-C6 ;
alternativamente, G1 puede ser H, R13 o R13-C(=O)-, en donde R13 puede ser una cadena de carbono lineal o ramificada C9-C29, opcionalmente con uno o más enlaces dobles C=C cis o trans y opcionalmente sustituida con OH;
alternativamente, G1 puede ser un resto ionizable seleccionado de: -COOH; -NH+; -OH; -NH3+; -NH2+; -SH; -NMe2; -NHMe; y R18, en donde R18 puede ser alquilo C1-C10 opcionalmente sustituido con uno o más de COOH, -NH+, -OH, -NH3+, -NH2+, -SH, -NMe2 , -NHMe;
n1 puede ser 0, 1, 2, 3, 4 o 5;
R3 puede ser H, un alquilo C1-C10 lineal, ramificado o cíclico, opcionalmente con uno o más enlaces dobles C=C cis o trans de geometría E o Z, en donde el alquilo C1-C10 puede estar opcionalmente sustituido con F, Cl, Br, I, OH, O-alquilo C1-C6, u O-acilo C1-C6, en donde el O-alquilo C1-C6 o el O-acilo C1-C6 pueden estar opcionalmente sustituidos con F, Cl, Br, I u OH;
R4 puede ser H, un alquilo C1-C10 lineal, ramificado o cíclico, opcionalmente con uno o más enlaces dobles C=C cis o trans de geometría E o Z, en donde el alquilo C1-C10 puede estar opcionalmente sustituido con F, Cl, Br, I, OH, O-alquilo C1-C6, u O-acilo C1-C6, en donde el O-alquilo C1-C6 o el O-acilo C1-C6 pueden estar opcionalmente sustituidos con F, Cl, Br, I u OH;
n2 puede ser 0, 1, 2, 3, 4 o 5;
R5 puede ser H, amina primaria C1-C6, amina secundaria C1-C6 o amina terciaria C1-C6 , un alquilo C1-C10 lineal, ramificado o cíclico, opcionalmente con uno o más enlaces dobles C=C cis o trans de geometría E o Z, en donde el alquilo C1-C10 puede estar opcionalmente sustituido con F, Cl, Br, I, OH, O-alquilo C1-C6 u O-acilo C1-C6, en donde el O-alquilo C1-C6 o el O-acilo C1-C6 pueden estar opcionalmente sustituidos con F, Cl, Br, I u OH;
X2 puede ser O, S o NR14, en donde R14 puede ser H, un alquilo C1-C10 lineal, ramificado o cíclico, opcionalmente con uno o más enlaces dobles C=C cis o trans de geometría E o Z, en donde el alquilo C1-C10 puede estar opcionalmente sustituido con F, Cl, Br, I, OH, O-alquilo C1-C6, u O-acilo C1-C6, en donde el O-alquilo C1-C6 o el O-acilo C1-C6 pueden estar opcionalmente sustituidos con F, Cl, Br, I u OH;
G2 es

en donde:
A-Zg2 puede ser un resto de fármaco, en donde Zg2 puede ser un átomo electronegativo seleccionado de N y O, de tal manera que el resto de fármaco se derivó de un fármaco que tiene la Fórmula A-Zg2-H cuando Zg2 ha perdido un H para unirse covalentemente a Yg2;
YG2 puede ser CH2 , O, S, C(=O), PO32', o NR11 donde R11 puede ser H, un alquilo C1-C10 lineal, ramificado o cíclico, opcionalmente con uno o más enlaces dobles C=C cis o trans de geometría E o Z, en donde el alquilo C1-C10 puede estar opcionalmente sustituido con F, Cl, Br, I, OH, O-alquilo C1-C6, u O-acilo C1-C6, y donde el O-alquilo C1-C6 o el O-acilo C1-C6 pueden estar opcionalmente sustituidos con F, Cl, Br, I u OH; Mg2 puede ser 0-12 átomos seleccionados de C, N, O o S, en donde dos o más átomos de C tienen opcionalmente uno o más enlaces dobles C=C cis o trans de geometría E o Z y en donde los átomos de C pueden estar opcionalmente sustituidos con F, Cl, Br, I, OH, =O, O-alquilo C1-C6, O-acilo C1-C6 o S-alquilo C1-C6 ;
alternativamente, G2 puede ser H o R16-C(=O)-, en donde R16 puede ser una cadena de carbono lineal o ramificada C9-C29, opcionalmente con uno o más enlaces dobles C=C cis o trans y opcionalmente sustituida con OH;
alternativamente, G2 puede ser un resto ionizable seleccionado de: -COOH; -NH+; -OH; -NH3+; -NH2+; -SH; -NMe2; -NHMe; y R18, en donde R18 puede ser un alquilo C1-C10 opcionalmente sustituido con uno o más de COOH, -NH+, -OH, -NH3+, -NH2+, -SH, -NMe2 , -NHMe;
n3 puede ser 0, 1, 2, 3, 4 o 5;
R6 puede ser H, un alquilo C1-C10 lineal, ramificado o cíclico, opcionalmente con uno o más enlaces dobles C=C cis o trans de geometría E o Z, en donde el alquilo C1-C10 puede estar opcionalmente sustituido con F, Cl, Br, I, OH, O-alquilo C1-C6, u O-acilo C1-C6, en donde el O-alquilo C1-C6 o el O-acilo C1-C6 pueden estar opcionalmente sustituidos con F, Cl, Br, I u OH;
R7 puede ser H, un alquilo C1-C10 lineal, ramificado o cíclico, opcionalmente con uno o más enlaces dobles C=C cis o trans de geometría E o Z, en donde el alquilo C1-C10 puede estar opcionalmente sustituido con F, Cl, Br, I, OH, O-alquilo C1-C6, u O-acilo C1-C6, en donde el O-alquilo C1-C6 o el O-acilo C1-C6 pueden estar opcionalmente sustituidos con F, Cl, Br, I u OH;
n4 puede ser 0, 1, 2, 3, 4 o 5;
R8 puede ser H, amina primaria C1-C6, amina secundaria C1-C6 o amina terciaria C1-C6, alquilo C1-C10 lineal, ramificado o cíclico, opcionalmente con uno o más enlaces dobles C=C cis o trans de geometría E o Z, en donde el alquilo C1-C10 puede estar opcionalmente sustituido con F, Cl, Br, I, OH, O-alquilo C1-C6 u O-acilo C1-C6 , en donde el O-alquilo C1-C6 o el O-acilo C1-C6 pueden estar opcionalmente sustituidos con F, Cl, Br, I u OH;
X3 puede ser O, S o NR17, en donde R17 puede ser H, un alquilo C1-C10 lineal, ramificado o cíclico, opcionalmente con uno o más enlaces dobles C=C cis o trans de geometría E o Z, en donde el alquilo C1-C10 puede estar opcionalmente sustituido con F, Cl, Br, I, OH, O-alquilo C1-C6, u O-acilo C1-C6, en donde el O-alquilo C1-C6 o el O-acilo C1-C6 pueden estar opcionalmente sustituidos con F, Cl, Br, I u OH;
G3 puede ser
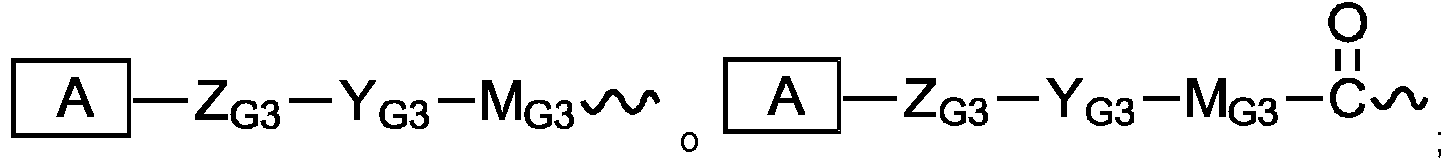
en donde:
A-Zg3 puede ser un resto de fármaco, en donde Zg3 puede ser un átomo electronegativo seleccionado de N y O, de tal manera que el resto de fármaco se derivó de un fármaco que tiene la Fórmula A-Zg3-H cuando Zg3 ha perdido un H para unirse covalentemente a Yg3 í
YG3 puede ser CH2 , O, S, C(=O), PO32", o NR11 donde R11 puede ser H, un alquilo C1-C10 lineal, ramificado o cíclico, que opcionalmente tiene uno o más enlaces dobles C=C cis o trans de geometría E o Z, en donde el alquilo C1-C10 puede estar opcionalmente sustituido con F, Cl, Br, I, OH, O-alquilo C1-C6, u 11O-acilo C1-C6, y donde el O-alquilo C1-C6 o el O-acilo C1-C6 pueden estar opcionalmente sustituidos con F,C l, '/ B vr, I u OH; Mg3 puede ser de 0-12 átomos seleccionados de C, N, O o S, en donde dos o más átomos de C tienen opcionalmente uno o más enlaces dobles C=C cis o trans de geometría E o Z y en donde los átomos de C pueden estar opcionalmente sustituidos con F, Cl, Br, I, un OH, =O, O-alquilo C1-C6, O-acilo C1-C6 o S-alquilo C1-C6 ;
alternativamente, G3 puede ser H, R19 o R19-C(=O)-, en donde R19 puede ser una cadena de carbono lineal o ramificada C9-C29, opcionalmente con al menos uno o más enlaces dobles C=C cis o trans y opcionalmente sustituida con OH;
alternativamente, G3 puede ser un resto ionizable seleccionado de: -COOH; -NH+; -OH; -NH3+; -NH2+, -SH; -NMe2; -NHMe; y R18, en donde R18 puede ser un alquilo C1-C10 opcionalmente sustituido con uno o más de COOH; -NH+; -OH; -NH3+; -NH2+, -SH; -NMe2 ; -NHMe;
R9 puede ser H, una amina primaria C1-C6, una amina secundaria C1-C6 o una amina terciaria C1-C6 , un alquilo lineal, ramificado o cíclico C1-C10, opcionalmente con uno o más enlaces dobles C=C cis o trans de geometría E o Z, en donde el alquilo C1-C10 puede estar opcionalmente sustituido con un F, Cl, Br, I, un OH, un O-alquilo C1-C6, o un O-acilo C1-C6, en donde el O-alquilo C1-C6 o el O-acilo C1-C6 pueden estar opcionalmente sustituidos con un F, Cl, Br, I u OH;
alternativamente, en donde n1, n2, n3, n4 puede ser 0 y R1 puede ser H, entonces R9 puede ser R20-C(=O)-, en donde R20 puede ser una cadena de carbono lineal o ramificada C7-C29, opcionalmente con uno o más enlaces dobles C=C cis o trans y opcionalmente sustituido con OH; y
siempre que al menos uno de G1, G2 o G3 pueda ser un resto de fármaco, en donde el resto de fármaco se selecciona de

o

respectivamente.
También se proporciona para comparación en la presente, un compuesto que tiene la estructura de la Fórmula I:

en donde:
R1 puede ser H o un alquilo lineal, ramificado C1-C20, opcionalmente con uno o más enlaces dobles C=C cis o trans de geometría E o Z, en donde el alquilo C1-C10 puede estar opcionalmente sustituido con un F, Cl, Br o I;
R2 puede ser H o un alquilo lineal, ramificado C1-C20, opcionalmente con uno o más enlaces dobles C=C cis o trans de geometría E o Z, en donde el alquilo C1-C10 puede estar opcionalmente sustituido con un F, Cl, Br, o I;
X1 puede ser O, S o NR10, en donde R10 puede ser H, un alquilo C1-C6 lineal, ramificado o cíclico, opcionalmente con uno o más enlaces dobles C=C cis o trans de geometría E o Z, en donde el alquilo C1-C6 puede estar opcionalmente sustituido con F, Cl, Br, I u OH;
G1 puede ser

en donde:
A-Zg1 puede ser un resto de fármaco, en donde Zg1 puede ser un átomo electronegativo seleccionado de N y O, de tal manera que el resto de fármaco se derivó de un fármaco que tiene la Fórmula A-Zg1-H cuando Zg1 ha perdido un H para unirse covalentemente a Yg1;
Yg1 puede ser CH2 , O, S, C(=O), PO32', o NR11 donde R11 puede ser H, un alquilo C1-C10 lineal, ramificado o cíclico, opcionalmente con uno o más enlaces dobles C=C cis o trans de geometría E o Z, en donde el alquilo C1-C10 puede estar opcionalmente sustituido con F, Cl, Br, I, OH, O-alquilo C1-C6, u O-acilo C1-C6, y donde el O-alquilo C1-C6 o el O-acilo C1-C6 pueden estar opcionalmente sustituidos con F, Cl, Br, I u OH; Mg1 puede ser 0-12 átomos seleccionados de C, N, O o S, en donde dos o más átomos de C tienen opcionalmente uno o más enlaces dobles C=C cis o trans de geometría E o Z y en donde los átomos de C pueden estar opcionalmente sustituidos con F, Cl, Br, I, OH, a =O, O-alquilo C1-C6 , O-acilo C1-C6 o S-alquilo C1-C6 ;
alternativamente, G1 puede ser H, R13 o R13-C(=O)-, en donde R13 puede ser una cadena de carbono lineal o ramificada C9-C29, con al menos un doble enlace C=C cis o trans y opcionalmente sustituido con OH; alternativamente, G1 puede ser un resto ionizable seleccionado de: -COOH; -NH+; -OH; -NH3+; -NH2+; -SH; -NMe2 ; -NHMe; y R18, en donde R18 puede ser un alquilo C1-C10 opcionalmente sustituido con uno o más de COOH, -NH+; -OH, -NH3+, -NH2+, -SH, -NMe2 , -NHMe;
n1 puede ser 0 , 1 o 2 ;
R3 puede ser H, OH, o un alquilo lineal y ramificado C1-C10, opcionalmente con uno o más enlaces dobles C=C cis o trans de geometría E o Z, en donde el alquilo C1-C10 puede estar opcionalmente sustituido con F, Cl, Br o I;
R4 puede ser H, OH, o un alquilo lineal o ramificado C1-C10, opcionalmente con uno o más enlaces dobles C=C cis o trans de geometría E o Z, en donde el alquilo C1-C10 puede estar opcionalmente sustituido con F, Cl, Br o I;
n2 puede ser 0 , 1 o 2 ;
R5 puede ser H, OH, o un alquilo lineal, ramificado C1-C10, opcionalmente con uno o más enlaces dobles C=C cis o trans de geometría E o Z, en donde el alquilo C1-C10 puede estar opcionalmente sustituido con F, Cl, Br, o I;
X2 puede ser O, S o NR10, en donde R10 puede ser H, un alquilo C1-C6 lineal, ramificado o cíclico, opcionalmente con uno o más enlaces dobles C=C cis o trans de geometría E o Z, en donde el alquilo C1-C6 puede estar opcionalmente sustituido con F, Cl, Br, I u OH;
G2 puede ser

en donde:
A-Zg2 puede ser un resto de fármaco, en donde Zg2 puede ser un átomo electronegativo seleccionado de N y O, de tal manera que el resto de fármaco se derivó de un fármaco que tiene la Fórmula A-Zg2-H cuando Zg2 ha perdido un H para unirse covalentemente a Yg2 í
Yg2 puede ser CH2 , O, S, C(=O), PO32', o NR11 donde R11 puede ser H, un alquilo C1-C10 lineal, ramificado o cíclico, opcionalmente con uno o más enlaces dobles C=C cis o trans de geometría E o Z, en donde el alquilo C1-C10 puede estar opcionalmente sustituido con F, Cl, Br, I, OH, O-alquilo C1-C6, u O-acilo C1-C6, y donde el O-alquilo C1-C6 o el O-acilo C1-C6 pueden estar opcionalmente sustituidos con F, Cl, Br, I u OH; Mg2 puede ser 0-12 átomos seleccionados de C, N, O o S, en donde dos o más átomos de C tienen opcionalmente uno o más enlaces dobles C=C cis o trans de geometría E o Z y en donde los átomos de C pueden estar opcionalmente sustituidos con F, Cl, Br, I, OH, =O, O-alquilo C1-C6, O-acilo C1-C6 o S-alquilo C1-C6 ;
alternativamente, G2 puede ser H o R16-C(=O)-, en donde R16 puede ser una cadena de carbono lineal o ramificada C9-C29, opcionalmente con al menos un doble enlace C=C cis o trans y opcionalmente sustituido con OH;
alternativamente, G2 puede ser un resto ionizable seleccionado de: -COOH; -NH+; -OH; -NH3+; -NH2+; -SH; -NMe2; -NHMe; y R18, en donde R18 puede ser un alquilo C1-C10 opcionalmente sustituido con uno o más de COOH, -NH+, -OH, -NH3+, -NH2+, -SH, -NMe2 , -NHMe;
n3 puede ser 0, 1o 2;
R6 puede ser H, OH, o un alquilo lineal y ramificado C1-C10, opcionalmente con uno o más enlaces dobles C=C cis o trans de geometría E o Z, en donde el alquilo C1-C10 puede estar opcionalmente sustituido con F, Cl, Br, o I;
R7 puede ser H, OH, o un alquilo lineal o ramificado C1-C10, opcionalmente con uno o más enlaces dobles C=C cis o trans de geometría E o Z, en donde el alquilo C1-C10 puede estar opcionalmente sustituido con F, Cl, Br o I;
n4 puede ser 0, 1o 2;
R8 puede ser H, OH, o un alquilo lineal, ramificado C1-C10, opcionalmente con uno o más enlaces dobles C=C cis o trans de geometría E o Z, en donde el alquilo C1-C10 puede estar opcionalmente sustituido con F, Cl, Br, o I;
X3 puede ser O, S o NR10, en donde R10 puede ser H, un alquilo C1-C6 lineal, ramificado o cíclico, opcionalmente con uno o más enlaces dobles C=C cis o trans de geometría E o Z, en donde el alquilo C1-C6 puede estar opcionalmente sustituido con F, Cl, Br, I u OH;
G3 puede ser

en donde:
A-Zg3 puede ser un resto de fármaco, en donde Zg3 puede ser un átomo electronegativo seleccionado de N y O, de tal manera que el resto de fármaco se derivó de un fármaco que tiene la Fórmula A-Zg3-H cuando Zg3 ha perdido un H para unirse covalentemente a Yg3;
YG3 puede ser CH2 , O, S, C(=O), PO32', o NR11 donde R11 puede ser H, un alquilo C1-C10 lineal, ramificado o cíclico, que opcionalmente tiene uno o más enlaces dobles C=C cis o trans de geometría E o Z, en donde el alquilo C1-C10 puede estar opcionalmente sustituido con F, Cl, Br, I, OH, O-alquilo C1-C6, u O-acilo C1-C6, y donde el O-alquilo C1-C6 o el O-acilo C1-C6 pueden estar opcionalmente sustituidos con F, Cl, Br, I u OH; Mg3 puede ser de 0-12 átomos seleccionados de C, N, O o S, en donde dos o más átomos C tienen opcionalmente uno o más enlaces dobles C=C cis o trans de geometría E o Z y donde los átomos C pueden estar opcionalmente sustituidos con F, Cl, Br, I, OH, =O, O-alquilo C1-C6, O-acilo C1-C6 o S-alquilo C1-C6 ; alternativamente, G3 puede ser H, R19 o R19-C(=O)-, en donde R19 puede ser una cadena de carbono lineal o ramificada C9-C29, opcionalmente con al menos un doble enlace C=C cis o trans y opcionalmente sustituida con OH;
alternativamente, G3 puede ser un resto ionizable seleccionado de: -COOH; -NH+; -OH; -NH3+; -NH2+; -SH; -NMe2; -NHMe; y R18, en donde R18 puede ser un alquilo C1-C10 opcionalmente sustituido con uno o más de COOH, -NH+, -OH, -NH3+, -NH2+, -SH, -NMe2 , -NHMe;
R9 puede ser H, amina primaria C1-C6 , amina secundaria C1-C6 o amina terciaria C1-C6 , alquilo lineal, ramificado o cíclico C1-C10, opcionalmente con uno o más enlaces dobles C=C cis o trans de geometría E o Z, en donde el alquilo C1-C10 puede estar opcionalmente sustituido con F, Cl, Br, I, OH, O-alquilo C1-C6 u O-acilo C1-C6, en donde el O-alquilo C1-C6 o el O-acilo C1-C6 pueden estar opcionalmente sustituidos con F, Cl, Br, I u OH;
alternativamente en donde n1, n2, n3, n4 puede ser 0 y R1 puede ser H, entonces R9 puede ser R20-C(=O)-, en donde R20 puede ser una cadena de carbono lineal o ramificada C7-C29, opcionalmente con uno o más enlaces dobles C=C cis o trans y opcionalmente sustituido con OH; y
siempre que al menos uno de G1, G2 o G3 pueda ser un resto de fármaco, en donde el resto de fármaco se
selecciona de:

respectivamente.
Para la comparación, también se proporciona un compuesto que tiene la estructura de la Fórmula III:

en donde:
A-Z puede ser un resto de fármaco, en donde Z puede ser un átomo electronegativo seleccionado de N u O, de tal manera que el resto de fármaco se derivó de un fármaco que tiene la Fórmula A-Z-H cuando Z ha perdido un H para unirse covalentemente a Y;
Y puede ser CH2 , C(=O), PO32 o NH;
T5 puede ser de 0-6 átomos de carbono; y 11
L4 puede ser una cadena de carbono lineal o ramificada C9-C29, opcionalmente con uno o más enlaces dobles C=C cis o trans y opcionalmente sustituida con OH o tiene la Fórmula V:

en donde
T2 pueden ser 1-4 átomos de carbono;
T3 pueden ser 0-4 átomos de carbono;
T4 pueden ser 0-4 átomos de carbono;
J1 puede ser -O-C(=O)-L1;
J2 puede ser -O-C(=O)-L2, -COOH, -NH+, -OH, -NH3+, -NH2+, -SH, -NMe2 , -NHMe, o

J3 puede ser NMe2 , -CH2NHMe o H;
J4 puede ser -O-C(=O)-L3;
L1 puede ser una cadena de carbono lineal o ramificada C9-C29, opcionalmente con uno o más enlaces dobles C=C cis o trans y puede estar opcionalmente sustituida con OH;
L2 puede ser una cadena de carbono lineal o ramificada C9-C29, opcionalmente con uno o más enlaces dobles C=C cis o trans y puede estar opcionalmente sustituida con OH; y
L3 puede ser una cadena de carbono lineal o ramificada C9-C29, opcionalmente con uno o más enlaces dobles C=C cis o trans y puede estar opcionalmente sustituida con OH;
alternativamente, Z, Y y T5 pueden estar ausentes y A puede ser un resto de fármaco, con un átomo electronegativo O precedido por un C(=O), que ha perdido un H para unirse covalentemente a L4.
Y puede ser CH2 , C(=O) o PO32'. Y puede ser CH2 o C(=O). Y puede ser PO32'. Y puede ser CH2. Y puede ser C(=O);
siempre que cuando A-Z sea un fármaco esteroide entonces R20 sea un C7-C29 tenga uno o más, cis o trans C=C enlaces dobles;
y siempre que el compuesto no sea

Para la comparación también se proporciona un compuesto que tiene la estructura de la Fórmula II:

en donde:
A-Z puede ser un resto de fármaco, en donde Z puede ser un átomo electronegativo seleccionado de N u O, de tal manera que el resto de fármaco se derivó de un fármaco que tiene la Fórmula A-Z-H cuando Z ha perdido un H para unirse covalentemente a Y;
Y puede ser CH2 , C(=O), PO32-, o NH;
T1 pueden ser de 0-6 átomos de carbono;
T2 pueden ser de 1-4 átomos de carbono;
T3 pueden ser de 0-4 átomos de carbono;
T4 pueden ser 0-4 átomos de carbono;
J1 puede ser -O-C(=O)-L1;
J2 puede ser -O-C(=O)-L2, -COOH, -NH+, -OH, -NH3+, -NH2+, -SH, -NMe2 , -NHMe, o

J3 puede ser NMe2 , -CH2NHMe o H;
J4 puede ser -O-C(=O)-L3
L1 puede ser una cadena de carbono lineal o ramificada C9-C29, opcionalmente con uno o más enlaces dobles C=C cis o trans y puede estar opcionalmente sustituida con OH;
L2 puede ser una cadena de carbono lineal o ramificada C9-C29, opcionalmente con uno o más enlaces dobles C=C cis o trans y puede estar opcionalmente sustituida con OH; y
L3 puede ser una cadena de carbono lineal o ramificada C9-C29, opcionalmente con uno o más enlaces dobles C=C cis o trans y puede estar opcionalmente sustituida con OH;
siempre que cuando A-Z sea un fármaco esteroide entonces R20 sea un C7-C29 tenga uno o más, cis o trans C=C enlaces dobles;
y siempre que el compuesto no sea

Para la comparación se proporciona un compuesto que tiene la estructura de la Fórmula III:

en donde:
A-Z puede ser un resto de fármaco, en donde Z puede ser un átomo electronegativo seleccionado de N u O, de tal manera que el resto de fármaco se derivó de un fármaco que tiene la Fórmula A-Z-H cuando Z ha perdido un H para unirse covalentemente a Y;
Y puede ser CH2 , C(=O), PO32-, o NH;
T5 puede ser de 0-6 átomos de carbono; y
L4 puede ser una cadena de carbono lineal o ramificada C9-C29, opcionalmente con uno o más enlaces dobles C=C cis o trans y opcionalmente sustituida con OH.
Para la comparación también se proporciona un compuesto que tiene la estructura de la Fórmula IV:

en donde:
A puede ser un resto de fármaco, con un átomo electronegativo O precedido por un C(=O), y que ha perdido un H para unirse covalentemente a L5; y
L5 puede ser una cadena de carbono lineal o ramificada C9-C29, opcionalmente con uno o más enlaces dobles C=C cis o trans y puede estar opcionalmente sustituida con OH.
T5 puede ser de 0 a 5 átomos de carbono. T5 puede ser de 0-4 átomos de carbono. T5 puede ser de 0-3 átomos de carbono. T5 puede ser de 0-2 átomos de carbono. T5 puede ser de 0-1 átomos de carbono. T5 puede ser de 0 átomos de carbono. T5 puede ser de 1-6 átomos de carbono. T5 puede ser de 1-5 átomos de carbono. T5 puede ser de 1-4 átomos de carbono. T5 puede ser de 1-3 átomos de carbono. T5 puede ser de 1-2 átomos de carbono. T5 puede ser de 2 átomos de carbono. T5 puede ser de 1 átomo de carbono. T5 puede ser de 3 átomos de carbono.
L4 puede ser una cadena de carbono lineal o ramificada C9-C29, opcionalmente con uno o más enlaces dobles C=C cis o trans y opcionalmente sustituida con OH. L4 puede ser una cadena de carbono lineal o ramificada C9-C29, opcionalmente con uno o más enlaces dobles C=C cis o trans. L4 puede ser una cadena de carbono lineal o ramificada C9-C25, opcionalmente con uno o más enlaces dobles C=C cis o trans y opcionalmente sustituida con OH. L4 puede ser una cadena de carbono lineal o ramificada C9-C22, opcionalmente con uno o más enlaces dobles C=C cis o trans y opcionalmente sustituida con OH. L4 puede ser una cadena de carbono lineal o ramificada C9-C20, opcionalmente con uno o más enlaces dobles C=C cis o trans y opcionalmente sustituida con OH. L4 puede ser una cadena de carbono lineal o ramificada C9-C18, opcionalmente con uno o más enlaces dobles C=C cis o trans y opcionalmente sustituida con OH. L4 puede ser una cadena de carbono lineal o ramificada C9-C16, opcionalmente con uno o más enlaces dobles C=C cis o trans y opcionalmente sustituida con OH.
L4 puede tener la Fórmula V:

T2 puede ser de 1-4 átomos de carbono. T2 puede ser de 1-3 átomos de carbono. T2 puede ser de 1-2 átomos de carbono. T2 puede ser de 2-4 átomos de carbono. T2 puede ser de 3-4 átomos de carbono. T2 puede ser de 1 átomo de carbono. T2 puede ser de 2 átomos de carbono. T2 puede ser de 3 átomos de carbono. T3 puede ser de 0-4 átomos de carbono. T3 puede ser de 0-3 átomos de carbono. T3 puede ser de 0-2 átomos de carbono. T3 puede ser de 0-1 átomos de carbono. T3 puede ser de 1-4 átomos de carbono. T3 puede tener 1-3 átomos de carbono. T3 puede ser de 1-2 átomos de carbono. T3 puede ser de 2 átomos de carbono. T3 puede ser de 3 átomos de carbono. T3 puede ser de 4 átomos de carbono. T4 puede ser de 0-4 átomos de carbono. T4 puede ser de 0-3 átomos de carbono. T4 puede ser de 0-2 átomos de carbono. T4 puede ser de 0-1 átomos de carbono. T4 puede ser de 1-4 átomos de carbono. T4 puede ser de 1-3 átomos de carbono. T4 puede ser de 1-2 átomos de carbono. T4 puede ser de 2 átomos de carbono. T4 puede ser de 3 átomos de carbono. T4 puede ser de 4 átomos de carbono. J1 puede ser -O-C(=O)-L1. J2 puede ser -O-C(=O)-L2, -COOH, -NH+, -OH, -NH3+, -NH2+, -SH, -NMe2 , -NHMe, o

J2 puede ser -O-C(=O)-L2, -COOH, -NH+, -OH, -NH3+, -NH2+, -SH, -NMe2 , o -NHMe. J2 puede ser -O-C(=O)-L2, -COOH, -NH+, -OH, -NH3+, -NH2+, -SH, o -NMe2. J2 puede ser

J2 puede ser -O-C(=O)-L2, -COOH, -NH+, -OH, -NH3+ o -NH2+. J2 puede ser -O-C(=O)-L2. J2 puede ser -O-C(=O)-L2, -COOH, -NH+, o -OH. J2 puede ser -COOH, -NH+ o -OH. J3 puede ser NMe2 , -CH2NHMe o H. J3 puede ser -CH2NHMe. J3 puede ser H. J3 puede ser NMe2. J4 puede ser -O-C(=O)-L3.
L1 puede ser una cadena de carbono lineal o ramificada C9-C29, opcionalmente con uno o más enlaces dobles C=C cis o trans.
L2 puede ser una cadena de carbono lineal o ramificada C9-C29, opcionalmente con uno o más enlaces dobles C=C cis o trans.
L3 puede ser una cadena de carbono lineal o ramificada C9-C29, opcionalmente con uno o más enlaces dobles C=C cis o trans.
T1 puede ser de 0-5 átomos de carbono. T1 puede ser 0-4 átomos de carbono. T1 puede ser de 0-3 átomos de carbono. T1 puede ser de 0-2 átomos de carbono. T1 puede ser de 0-1 átomos de carbono. T1 puede ser de 0 átomos de carbono. T1 puede ser de 1-6 átomos de carbono. T1 puede ser de 1-5 átomos de carbono. T1 puede ser de 1-4 átomos de carbono. T1 puede ser de 1-3 átomos de carbono. T1 puede ser de 1-2 átomos de carbono. T1 puede ser de 2 átomos de carbono. T1 puede ser de 1 átomo de carbono. T1 puede ser de 3 átomos de carbono. L5 puede ser una cadena de carbono lineal o ramificada C9-C29, opcionalmente con uno o más enlaces dobles C=C cis o trans. L5 puede ser una cadena de carbono lineal o ramificada C9-C29, opcionalmente con uno o más enlaces dobles C=C cis o trans y opcionalmente sustituida con OH. L5 puede ser una cadena de carbono lineal o ramificada C9-C29, opcionalmente con uno o más enlaces dobles C=C cis o trans. L5 puede ser una cadena de carbono lineal o ramificada C9-C25, opcionalmente con uno o más enlaces dobles C=C cis o trans y opcionalmente sustituida con OH. L5 puede ser una cadena de carbono lineal o ramificada C9-C22, opcionalmente con uno o más enlaces dobles C=C cis o trans y opcionalmente sustituida con OH. L5 puede ser una cadena de carbono lineal o ramificada C9-C20, opcionalmente con uno o más enlaces dobles C=C cis o trans y opcionalmente sustituida con OH. L5 puede ser una cadena de carbono lineal o ramificada C9-C18, opcionalmente con uno o más enlaces dobles C=C cis o trans y opcionalmente sustituida con OH. L5 puede ser una cadena de carbono lineal o ramificada C9-C16, opcionalmente con uno o más enlaces dobles C=C cis o trans y opcionalmente sustituida con OH.
Alternativamente, Z, Y y T5 pueden estar ausentes y A puede ser un resto de fármaco, con un átomo electronegativo O precedido por un C(=O), que ha perdido un H para unirse covalentemente a L4.
A puede seleccionarse de uno o más de Tacrolimus, Dexametasona, SN-38, Docetaxel, Metotrexato, NPC1I, Amprenavir, Anfotericina B, Bexaroteno, Calcitrol, Ciclosporina A, Digoxina, Doxercalciferol, Dronabinol, Etopósido, Fulvestrant, Paricalcitol, Tenipósido, Isotretinoína, Sirolimus, Tretinoína, Ácido valproico, Paclitaxel, Valrubicina, Propofol, Prednisona, Prednisolona, Ácido micofenólico, Lovastatina, Lamivudina, Zidovudina, Abacavir, Emitricitabina, Atazanavir, Cobicistat, Elvitegravir, Isonazid, Albendazol, Lisinapril, Amlodipino, Isotretinoína, Baclofeno, Benznidazol, Nifurtimox, Mefloquina, Sulfadoxina, Pirimetamina, Clorproguanil, 6-Mercaptopurina, 1-Tiroxina, Gliburida, MCC95 0 , Partenolida, Ácido tauroursodesoxicólico, Ruxolitinib y Tofacitinib. Alternativamente, A puede seleccionarse de uno o más de Docetaxel, Metotrexato, SN-38, NPC1I, Amprenavir, Anfotericina B,
Bexaroteno, Calcitrol, Ciclosporina A, Digoxina, Doxercalciferol, Dronabinol, Etopósido, Fulvestrant, Paricalcitol, Tenipósido, Isotretinoína, Tretinoína, Ácido valproico, Paclitaxel, Valrubicina, Propofol, Ácido micofenólico, Lovastatina, Lamivudina, Zidovudina, Abacavir, Emitricitabina, Atazanavir, Cobicistat, Elvitegravir, Isonazida, Albendazol, Lisinapril, Amlodipino, Isotretinoína, Baclofeno, Benznidazol, Nifurtimox, Mefloquina, Sulfadoxina, Pirimetamina, Clorproguanil, 6-mercaptopurina, 1-tiroxina, gliburida, MCCg50, Partenolida, Ácido tauroursodesoxicólico, Ruxolitinib, Tofacitinib, Ciclosporina, Tacrolimus, Everolimus, Sirolimus, Azatioprina, Leflunomida, Micofenolato, Dexametasona, Budesonida, Prednisona, Prednisolona, Metilprednisolona, Hidrocortisona, Cortisona, Fludrocortisona, Betametasona, Triamcinolona, Acetonuro de Triamcinolona, Flunisolida, Beclametasona, Fluticasona, Mometasona, Flumetasona, Isoflupredona, Corticosterona, Acetato de desoxicortona, Enantato de desoxicortona, 11-Desoxicorticosterona, 11-Desoxicortisol y Aldosterona.
Alternativamente, A puede seleccionarse de uno o más de Docetaxel, Metotrexato, SN-38, NPC1I, Amprenavir, Anfotericina B, Bexaroteno, Calcitrol, Ciclosporina A, Digoxina, Doxercalciferol, Dronabinol, Etopósido, Fulvestrant, Paricalcitol, Tenipósido, Isotretinoína, Tretinoína, Ácido valproico, Paclitaxel, Valrubicina, Propofol, Ácido micofenólico, Lovastatina, Lamivudina, Zidovudina, Abacavir, Emitricitabina, Atazanavir, Cobicistat, Elvitegravir, Isonazid, Albendazol, Lisinapril, Amlodipino, Isotretinoína, Baclofeno, Benznidazol, Nifurtimox, Mefloquina, Sulfadoxina, Pirimetamina, Clorproguanil, 6-Mercaptopurina, 1-Tiroxina, Gliburida, MCCg50, Partenolida, Ácido tauroursodesoxicólico, Ruxolitinib, Tofacitinib, Ciclosporina, Tacrolimus, Everolimus, Sirolimus, Azatioprina, Leflunomida y Micofenolato.
Alternativamente, A puede seleccionarse de uno o más de Docetaxel, Metotrexato, SN-38, NPC1I, Amprenavir, Anfotericina B, Bexaroteno, Calcitrol, Ciclosporina A, Digoxina, Doxercalciferol, Dronabinol, Etopósido, Fulvestrant, Paricalcitol, Tenipósido, Isotretinoína, Tretinoína, Ácido valproico, Paclitaxel, Valrubicina, Propofol, Ácido micofenólico, Lovastatina, Lamivudina, Zidovudina, Abacavir, Emitricitabina, Atazanavir, Cobicistat, Elvitegravir, Isonazid, Albendazol, Lisinapril, Amlodipino, Isotretinoína, Baclofeno, Benznidazol, Nifurtimox, Mefloquina, Sulfadoxina, Pirimetamina, Clorproguanil, 6-Mercaptopurina, 1-Tiroxina, Gliburida, MCCg50, Partenolida, Ácido tauroursodesoxicólico, Ruxolitinib y Tofacitinib.
Alternativamente, A puede seleccionarse de uno o más de Docetaxel, Metotrexato, SN-38, NPC1I, Amprenavir, Anfotericina B, Bexaroteno, Calcitrol, Ciclosporina A, Digoxina, Doxercalciferol, Dronabinol, Etopósido, Fulvestrant, Paricalcitol, Tenipósido, Isotretinoína, Tretinoína, Ácido valproico, Paclitaxel, Valrubicina, Propofol, Ácido micofenólico, Lovastatina, Lamivudina, Zidovudina, Abacavir, Emitricitabina, Atazanavir, Cobicistat, Elvitegravir, Isonazid, Albendazol, Lisinapril, Amlodipino, Isotretinoína, Baclofeno, Benznidazol, Nifurtimox, Mefloquina, Sulfadoxina, Pirimetamina, Clorproguanil, 6-Mercaptopurina, 1-Tiroxina, Gliburida, MCCg50, Partenolida, Ácido tauroursodesoxicólico, Ruxolitinib, Tofacitinib, Dexametasona, Budesonida, Prednisona, Prednisolona, Metilprednisolona, Hidrocortisona, Cortisona, Fludrocortisona, Betametasona, Triamcinolona, Acetonuro de triamcinolona, Flunisolida, Beclametasona, Fluticasona, Mometasona, Flumetasona, Isoflupredona, Corticosterona, Acetato de desoxicortona, Enantato de desoxicortona, 11-Desoxicorticosterona, 11-Desoxicortisol y Aldosterona. Alternativamente, A puede seleccionarse de uno o más de Ciclosporina, Tacrolimus, Everolimus, Sirolimus, Azatioprina, Leflunomida, Micofenolato, Dexametasona, Budesonida, Prednisona, Prednisolona, Metilprednisolona, Hidrocortisona, Cortisona, Fludrocortisona, Betametasona, Triamcinolona, Acetonuro de triamcinolona, Flunisolida, Beclametasona, Fluticasona, Mometasona, Flumetasona, Isoflupredona, Corticosterona, Acetato de desoxicortona, Enantato de desoxicortona, 11-Desoxicorticosterona, 11-Desoxicortisol y Aldosterona.
Pueden incorporarse fármacos alternativos basados en los métodos expuestos en la presente.
Yg1, Yg2 o YG3 pueden seleccionarse independientemente entre CH2 , O, S, C(=O), PO32', o NR11 donde R11 puede ser H, un alquilo C1-C8 lineal, ramificado o cíclico, puede tener opcionalmente uno o más enlaces dobles C=C cis o trans de geometría E o Z, en donde el alquilo C1-C8 puede estar opcionalmente sustituido con F, Cl, Br, I, OH, O alquilo C1-C6, u O-acilo C1-C6 , y donde el O-alquilo C1-C6 o el O-acilo C1-C6 pueden estar opcionalmente sustituidos con F, Cl, Br, I u OH. Y puede ser CH2 , O, S, C(=O), PO32', o NR11 en donde R11 puede ser H, un alquilo C1-C6 lineal, ramificado o cíclico, opcionalmente con uno o más enlaces dobles C=C cis o trans de geometría E o Z, en donde el alquilo C1-C6 puede estar opcionalmente sustituido con F, Cl, Br, I u OH. Yg1, Yg2 o Yg3 pueden seleccionarse, de modo independiente, de CH2 , O, S, C(=O), PO32' o NH. Yg1, Yg2 o Yg3 pueden seleccionarse, de modo independiente, de CH2 , O, S, C(=O) o NH. Yg1, Yg2 o Yg3 pueden seleccionarse, de modo independiente, de CH2 , C(=O) o PO32-.
M puede ser 0-10 átomos seleccionados de C, N, O o S, en donde dos o más átomos de C pueden tener opcionalmente uno o más enlaces dobles C=C cis o trans de geometría E o Z y donde los átomos de C o N pueden estar opcionalmente sustituidos con F, Cl, Br, I, OH, =O, O-alquilo C1-C6 , O-acilo C1-C6 o S-alquilo C1-C6. M puede ser 0-8 átomos seleccionados de C, N, O o S, en donde dos o más átomos de C pueden tener opcionalmente uno o más enlaces dobles C=C cis o trans de geometría E o Z y en donde los átomos de C o N pueden estar opcionalmente sustituidos con F, Cl, Br, I, OH, =O, O-alquilo C1-C6 , O-acilo C1-C6 o S-alquilo C1-C6. M puede ser 0-8 átomos seleccionados de C, N, O o S, en donde dos o más átomos C opcionalmente pueden tener uno o más enlaces dobles C=C cis o trans de geometría E o Z, en donde los átomos C o N opcionalmente pueden estar sustituidos con F, Cl, Br, o I. M pueden ser 0-6 átomos seleccionados de C, N, O o S, en donde dos o más átomos C opcionalmente tienen uno o más enlaces dobles C=C cis o trans de geometría E o Z, en donde los átomos C o N
opcionalmente pueden estar sustituidos con F, Cl, Br, o I. M pueden ser 0 a 5 átomos seleccionados de C, N, O o S, en donde dos o más átomos de C pueden tener opcionalmente uno o más enlaces dobles C=C cis o trans de geometría E o Z, en donde los átomos de C o N pueden estar opcionalmente sustituidos con F, Cl, Br o I. M puede ser 0-5 átomos seleccionados de C, N, O o S, en donde los átomos de C o N pueden estar opcionalmente sustituidos con F, Cl, Br o I.
G1 puede ser H, R13 o R13-C(=O)-, en donde R13 puede ser una cadena de carbono lineal o ramificada C9-C29, puede tener al menos un doble enlace C=C cis o trans. G1 puede ser H, R13 o R13-C(=O)-. G1 puede ser H o R13 G1 puede ser H o R13-C(=O)-. G1 puede ser H. G1 puede ser R13-C(=O)-. G1 puede ser R13. G1 puede ser un resto ionizable seleccionado de: -COo H; -NH+; -OH; -NH3+; -NH2+; -SH; -NMe2; y -NHMe. R13 puede ser una cadena de carbono lineal o ramificada C9-C29, puede tener al menos un doble enlace C=C cis o trans.
G2 puede ser H o R16-C(=O)-, en donde R16 es una cadena de carbono lineal o ramificada C9-C29, opcionalmente con al menos un doble enlace C=C cis o trans. G2 puede ser un resto ionizable seleccionado de: -COOH; -NH+; -OH; -NH3+; -NH2+; -SH; -NMe2; y -NHMe.
G3 puede ser H, R19 o R19-C(=O)-, en donde R19 puede ser una cadena de carbono lineal o ramificada C9-C29, opcionalmente con al menos un doble enlace C=C cis o trans. G3 puede ser H, R19 o R19-C(=O)-. G3 puede ser H o R19. G3 puede ser H o R19-C(=O)-. G3 puede ser H. G3 puede ser R19 o R19-C(=O)-. G3 puede ser R19. G3 puede ser R19-C(=O)-. R19 puede ser una cadena de carbono lineal o ramificada C9-C29, opcionalmente con al menos un doble enlace C=C cis o trans. G3 puede ser un resto ionizable seleccionado de: -COOH; -NH+; -OH; -NH3+; -NH2+; -SH; -NMe2 ; y-NHMe.
R1 puede ser H, OH, o un alquilo lineal y ramificado C1-C8, opcionalmente con uno o más enlaces dobles C=C cis o trans de geometría E o Z, en donde el alquilo C1-C8 puede estar opcionalmente sustituido con un F, Cl, Br o I. R1 puede ser H, OH, o un alquilo lineal, ramificado C1-C8, en donde el alquilo C1-C8 puede estar opcionalmente sustituido con un F, Cl, Br o I. R1 puede ser H, OH, o un alquilo lineal, ramificado C1-C8, puede tener opcionalmente uno o más enlaces dobles C=C cis o trans de geometría E o Z. Alternativamente, R1 puede ser H, OH, o un alquilo lineal, ramificado C1-C10, opcionalmente puede tener uno o más enlaces dobles C=C cis o trans de geometría E o Z, en donde el alquilo C1-C10 puede estar opcionalmente sustituido con un F, Cl, Br, o I. Alternativamente, R1 puede ser H, OH, o un alquilo lineal, ramificado C1-C10, opcionalmente puede tener uno o más enlaces dobles C=C cis o trans de geometría E o Z. Alternativamente, R1 puede ser H, OH, o un alquilo lineal, ramificado C1-C5, opcionalmente con uno o más enlaces dobles C=C cis o trans de geometría E o Z, en donde el alquilo C1-C10 puede estar opcionalmente sustituido con un F, Cl, Br, o I. R2 puede ser OH.
X1 puede ser O, S o NR10, en donde R10 puede ser H, un alquilo C1-C6 lineal, ramificado o cíclico, y opcionalmente puede tener uno o más enlaces dobles C=C cis o trans de geometría E o Z. X1 puede ser O, S o NR10, en donde R10 puede ser H, un alquilo C1-C6 lineal, ramificado o cíclico, en donde el alquilo C1-C6 puede estar opcionalmente sustituido con F, Cl, Br, I u OH. X1 puede ser O o S. n1 puede ser 1o 2. n1 puede ser 0.
R3 puede ser H, OH, o un alquilo lineal, ramificado C1-C10, opcionalmente puede tener uno o más enlaces dobles C=C cis o trans de geometría E o Z. R3 puede ser H, OH o un alquilo lineal y ramificado C1-C10, en donde el alquilo C1-C10 puede estar opcionalmente sustituido con F, Cl, Br o I. R3 puede ser H u OH.
R4 puede ser H, OH o un alquilo lineal y ramificado C1-C10, en donde el alquilo C1-C10 puede estar opcionalmente sustituido con F, Cl, Br o I. R4 puede ser H, OH o un alquilo lineal y ramificado C1-C10, y opcionalmente puede tener uno o más enlaces dobles C=C cis o trans de geometría E o Z. R4 puede ser H u OH.
n2 puede ser 1 o 2. n2 puede ser 0.
R5 puede ser H, OH o un alquilo lineal y ramificado C1-C10, y puede tener opcionalmente uno o más enlaces dobles C=C cis o trans de geometría E o Z. R5 puede ser H, OH o un alquilo lineal y ramificado C1-C10, en donde el alquilo C1-C10 puede estar opcionalmente sustituido con F, Cl, Br o I. R5 puede ser H u OH.
X2 puede ser O, S o NR10, en donde R10 puede ser H, lineal, en donde el alquilo C1-C6 puede estar opcionalmente sustituido con F, Cl, Br, I u OH. X2 puede ser O, S o NR10, en donde R10 puede ser H, un alquilo C1-C6 lineal, ramificado o cíclico, puede tener opcionalmente uno o más enlaces dobles C=C cis o trans de geometría E o Z. X2 puede ser O o S.
n3 puede ser 1 o 2. n3 puede ser 0.
R6 puede ser H, OH o un alquilo lineal y ramificado C1-C10, en donde el alquilo C1-C10 puede estar opcionalmente sustituido con F, Cl, Br o I. R6 puede ser H, OH o un alquilo lineal y ramificado C1-C10, y opcionalmente puede tener uno o más enlaces dobles C=C cis o trans de geometría E o Z. R6 puede ser H u OH.
R7 puede ser H, OH, o un alquilo lineal y ramificado C1-C10, en donde el alquilo C1-C10 puede estar opcionalmente
sustituido con F, Cl, Br o I. R7 puede ser H, OH, o un alquilo lineal y ramificado C1-C10, y puede tener opcionalmente uno o más enlaces dobles C=C cis o trans de geometría E o Z. R7 puede ser H u OH.
n4 puede ser 1 o 2. n4 puede ser 0.
R8 puede ser H, OH o un alquilo lineal y ramificado C1-C10, en donde el alquilo C1-C10 puede estar opcionalmente sustituido con F, Cl, Br o I. R8 puede ser H, OH o un alquilo lineal y ramificado C1-C10, y opcionalmente puede tener uno o más enlaces dobles C=C cis o trans de geometría E o Z. R8 puede ser H u OH.
X3 puede ser O, S o NR10, en donde R10 puede ser H, un alquilo C1-C6 lineal, ramificado o cíclico, en donde el alquilo C1-C6 puede estar opcionalmente sustituido con F, Cl, Br, I u OH. X3 puede ser O, S o NR10, en donde R10 es H, un alquilo C1-C6 lineal, ramificado o cíclico, y opcionalmente puede tener uno o más enlaces dobles C=C cis o trans de geometría E o Z. X3 puede ser O o S.
R9 puede ser R20-C(=O)-, en donde R20 puede ser una cadena de carbono lineal o ramificada C9-C29, opcionalmente con uno o más enlaces dobles C=C cis o trans y opcionalmente sustituida con OH. R9 puede ser R20-C(=O)-, en donde R20 puede ser una cadena de carbono lineal o ramificada C8-C29, opcionalmente con uno o más enlaces dobles C=C cis o trans y opcionalmente sustituida con OH. R9 puede ser R20-C(=O)-, en donde R20 puede ser una cadena de carbono lineal o ramificada C7-C29, opcionalmente con uno o más enlaces dobles C=C cis o trans y opcionalmente sustituida con OH. R9 puede ser R20-C(=O)-, en donde R20 puede ser una cadena de carbono lineal o ramificada C6-C29, opcionalmente con uno o más enlaces dobles C=C cis o trans y opcionalmente sustituida con OH. R9 puede ser R20-C(=O)-, en donde R20 puede ser una cadena de carbono lineal o ramificada C5-C29, opcionalmente con uno o más enlaces dobles C=C cis o trans y opcionalmente sustituida con OH. R9 puede ser H, amina primaria C1-C6 , amina secundaria C1-C6 o amina terciaria C1-C6 , alquilo C1-C10 lineal, ramificado o cíclico, en donde el alquilo C1-C10 puede estar opcionalmente sustituido con F, Cl, Br, I, OH, O-alquilo C1-C6 u O-acilo C1-C6 , en donde el O alquilo C1-C6 o el O-acilo C1-C6 pueden estar opcionalmente sustituidos con F, Cl, Br, I u OH. R9 puede ser H, amina primaria C1-C6, amina secundaria C1-C6 o amina terciaria C1-C6 , alquilo C1-C10 lineal, ramificado o cíclico, y puede tener opcionalmente uno o más enlaces dobles C=C cis o trans de geometría E o Z, en donde el alquilo C1-C10 puede estar opcionalmente sustituido con F, Cl, Br, I, OH, O-alquilo C1-C6 u O-acilo C1-C6 , en donde el O-alquilo C1-C6 o el O-acil C1-C6. R9 puede ser H.
El compuesto puede seleccionarse de uno o más de los siguientes, sin embargo, de los siguientes compuestos, solo los dos primeros son compuestos de la presente invención, los otros se proporcionan solo para comparación:


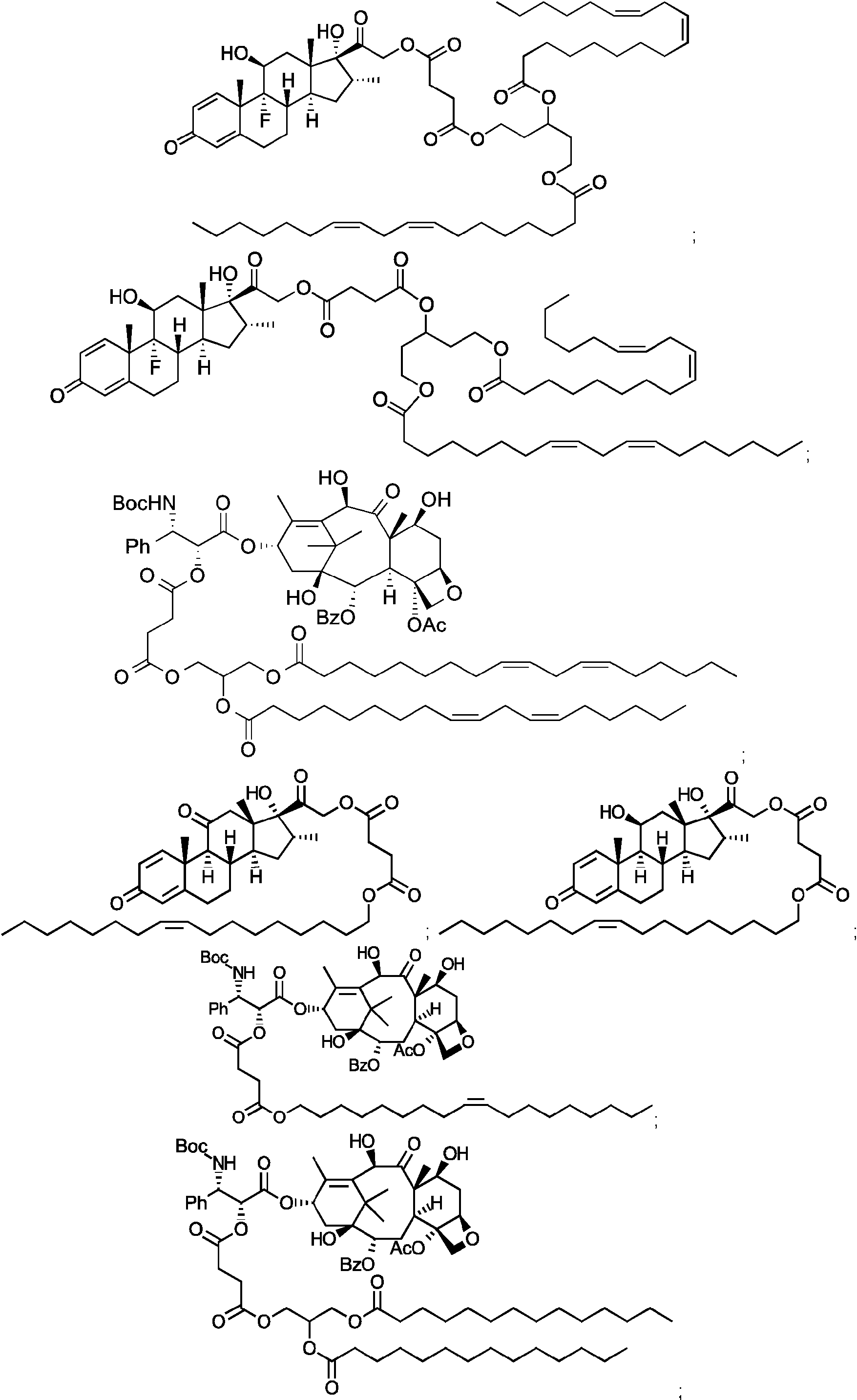
Alternativamente, el compuesto puede ser

De acuerdo con otra forma de realización, se proporciona una composición farmacéutica, la composición farmacéutica que comprende un compuesto descrito en la presente y un portador farmacéuticamente aceptable. De acuerdo con otra forma de realización, se proporciona un compuesto descrito en la presente para el tratamiento del cáncer, enfermedad autoinmune o infección.
De acuerdo con otra forma de realización, se proporciona una composición farmacéutica para el tratamiento del cáncer, enfermedad autoinmune o infección, que comprende el compuesto descrito en la presente y un portador farmacéuticamente aceptable.
De acuerdo con otra forma de realización, se proporciona un envase comercial que comprende (a) un compuesto descrito en la presente; y (b) instrucciones de uso.
De acuerdo con otra forma de realización, se proporciona un envase comercial que comprende (a) una composición farmacéutica descrita en la presente; y (b) instrucciones de uso.
Alternativamente, el compuesto o composición puede utilizarse para tratar cualquier indicación para la que el fármaco del resto de fármaco A-Z pueda utilizarse para tratar.
Breve descripción de los dibujos
La figura 1 muestra la ultraestructura de las formulaciones de profármacos unidos a lípidos, en las que las imágenes crioTEM mostraron estructuras vesiculares bicapa para las LNP que contenían LD02-DEX y LD03-DEX.
La figura 2 muestra la biodegradabilidad de la LNP LD03-DEX, en donde la escisión progresiva del profármaco LD03-DEX del profármaco ligado a lípidos está mediada por esterasas plasmáticas y aisladas a lo largo de 1 y 4 h y los datos se normalizan con respecto a la cantidad del profármaco respectivo en la mezcla de preincubación, con niveles por debajo de 1 que indican degradación (las barras de error representan la desviación estándar de al menos tres experimentos).
La figura 3 muestra la biodegradabilidad de los profármacos LD-DEX, en donde la escisión relativa del profármaco LD01, LD02, LD03 y LD07-DEX del profármaco ligado a lípidos tras 1 h de incubación en plasma o PBS con/sin enzima y los datos se normalizan con respecto a la cantidad del respectivo profármaco en la mezcla de preincubación, con niveles inferiores a 1 que indican degradación (las barras de error representan la desviación estándar de al menos tres experimentos).
La figura 4 muestra la biodegradabilidad de profármacos de LD-DEX en LNP, en donde los profármacos LD03, LD13, LD14, LD17, LD21 y LD24 - DEX se formularon en LNP y se incubaron durante 1h en PBS o plasma de ratón y la cantidad de cada profármaco se determinó mediante análisis de UPLC, y en donde los datos se normalizaron con respecto a la cantidad del profármaco respectivo en la mezcla de preincubación, con niveles inferiores a 1 que indican degradación (las barras de error representan la desviación estándar de al menos tres experimentos).
La figura 5 muestra la biodegradabilidad de profármacos de LD-DTX en LNP, en donde los profármacos LD10, LD18, LD22 y LD23 - DTX se formularon en (B) PC-Chol, (C) PC-TO o (A) LNP ionizable y se sometieron a 1 h de incubación en plasma de ratón y la cantidad de cada profármaco se determinó mediante análisis UPLC, y los datos se normalizaron con respecto a la cantidad del respectivo profármaco en la mezcla de preincubación (las barras de error representan la desviación estándar de al menos tres experimentos).
La figura 6 muestra la biodegradación de LD02 en LNP a lo largo del tiempo, en donde (A) muestra el profármaco LD02 formulado en LNP y luego sometido a incubación en plasma de ratón o esterasa porcina purificada a lo largo del tiempo y la cantidad de profármaco LD02 residual se analizó por UPLC a los tiempos 0, 2, 4 y 24 h; y en (B) se muestra un cromatograma UPLC de LD02 sin incubación con plasma o esterasa, LD02 incubada con esterasa durante 24 h, o LD02 incubada con plasma de ratón durante 24 h (Las flechas indican LD02 intacta o dexametasona (DEX)).
La figura 7 muestra la biodegradación de LD03 en LNP a lo largo del tiempo, en donde (A) muestra el profármaco LD03 formulado en LNP y luego sometido a incubación en plasma de ratón o esterasa porcina purificada a lo largo del tiempo y la cantidad de profármaco LD03 residual se analizó por UPLC a los tiempos 0, 2, 4 y 24 h; y en (B) se muestra un cromatograma UPLC de LD03 sin incubación con plasma o esterasa, LD03 incubada con esterasa durante 24 h, o LD03 incubada con plasma de ratón durante 24 h (las flechas indican LD02 intacta o dexametasona (DEX)).
La figura 8 muestra la biodegradación de LD18 en LNP a lo largo del tiempo, en donde (A) muestra el profármaco LD18 formulado en LNP y luego sometido a incubación en plasma de ratón o esterasa porcina purificada a lo largo del tiempo y la cantidad de profármaco LD18 residual se analizó por UPLC a los tiempos 0, 2, 4 y 24 h; y en (B) se muestra un cromatograma UPLC de LD18 sin incubación con plasma o esterasa, LD18 incubado con esterasa durante 24 h, o LD18 incubado con plasma de ratón durante 24 h (las flechas indican LD18 intacto o docetaxel (DTX)).
La figura 9 muestra la biodegradación de LD22 en LNP a lo largo del tiempo, en donde (A) se muestra el profármaco LD22 formulado en LNP y luego sometido a incubación en plasma de ratón o esterasa porcina purificada a lo largo del tiempo. La cantidad de profármaco LD22 residual se analizó por UPLC a los tiempos 0, 2, 4 y 24 h; y en (B) se muestra un cromatograma UPLC de LD22 sin incubación con plasma o esterasa, LD22 incubado con esterasa durante 24 h, o LD22 incubado con plasma de ratón durante 24 h (las flechas indican LD22 intacto o docetaxel (DTX)).
La figura 10 muestra que la disociación del profármaco LD-DEX de la LNP se correlaciona con la hidrofobicidad prevista LD02 y LD03 - los profármacos DEX se formularon en (A) PC-Chol LNP o (B) LNP ionizable, y luego se sometieron a incubación en plasma humano durante 24 h, y la LNP se aisló mediante cromatografía en columna antes de determinar la cantidad residual de cada profármaco en los tiempos 0, 3, 5 y 24 h mediante análisis UPLC - LD02 se disocia de la LNP a un ritmo más lento que el de LD03.
La figura 11 muestra que la disociación del profármaco LD-DTX de la LNP se correlaciona con la hidrofobicidad prevista LD18 y LD22 - los profármacos DTX se formularon en (A) PC-Chol LNP o (B) LNP ionizable, y después se sometieron a incubación en plasma humano durante 24 h, y la LNP se aisló mediante cromatografía en columna antes de determinar la cantidad residual de cada profármaco a los tiempos 0, 3, 5 y 24 h mediante análisis UPLC - LD18 se disocia de la LNP más rápidamente que LD22. La figura 12 muestra que los profármacos LD-DEX suprimen los efectos inmunoestimulantes de LNP-CpG en ratones, cuando se formuló LD01, LD02, LD03 o LD07 en LNP que contenían oligonucleótidos CpG inmunoestimulantes, se inyectó a los ratones con PBS o LNP-CpG con o sin profármacos LD-DEX a 10 mg/kg de CpG, se recogió sangre 2 h después de la inyección y se midieron los niveles de TNFα y KC/GRO; los datos se normalizaron con respecto a los niveles de citoquinas de los ratones tratados con LNP-CpG de control sin LD-DEX y los niveles inferiores a 1 indican supresión inmunitaria (las barras de error representan la desviación estándar derivada de al menos 3 animales).
La figura 13 muestra que los profármacos LD-DEX suprimen los efectos inmunoestimulantes de LNP-CpG en ratones, en donde LD03, LD13, LD14, LD17, lD21 o LD24 se formularon en LNP que contenían oligonucleótidos de ADN CpG inmunoestimulantes, se inyectó a los ratones con PBS o LNP-CpG con o sin profármacos LD-DEX a 10 mg/kg de CpG y se recogió sangre 2 h después de la inyección antes de medir los niveles de TNFα, IL6 e IL10; los datos se normalizaron con respecto a los niveles de citoquinas de los ratones tratados con LNP-CpG de control sin LD-DEX y los niveles inferiores a 1 indican supresión inmunitaria (las barras de error representan la desviación estándar derivada de al menos 3 animales). La figura 14 muestra que la LD03 suprime los efectos inmunoestimuladores del LNP-ARNm en ratones, en donde la formulación con o sin LD03 se inyectó en ratones a 3 mg/kg de ARNm, se recogió sangre 2 h después de la inyección y se determinaron los niveles de diversas citoquinas, en donde los datos se normalizaron con respecto a los niveles de citoquinas de ratones tratados con ARNm-LNP de control sin LD03 y los niveles inferiores a 1 indican supresión inmunitaria (las barras de error representan la desviación estándar derivada de al menos 3 animales).
La figura 15 muestra que LD03 suprime los efectos inmunoestimuladores de la LNP-ADNp en ratones, en donde se administró LNP-ADN plasmídico (LNP-ADNp) con o sin LD03 por vía intravenosa en ratones a 1 mg/kg de ADNp, se recogió sangre a las 2 h después de la inyección y se midieron los niveles de diversas citoquinas, los datos se normalizaron con respecto a los niveles de citoquinas de los ratones tratados con LNP-ADNp de control sin LD03 y los niveles inferiores a 1 indican supresión inmunitaria (las barras de error representan la desviación estándar derivada de al menos 4 animales).
La figura 16 muestra que los profármacos LD-DEX suprimen las respuestas inmunoestimuladoras en células Raw264.7 tratadas con lipopolisacárido (LPS), en donde las células se incubaron en medio que contenía 2 ng/mL de LPS y formulación de LNP que no contenía profármaco (control), LD03, LD07, LD13, LD14, LD17, LD21 y LD24 durante 4 h. A continuación, se cosecharon las células para aislar el ARN y se determinaron los niveles de IL1p mediante qRT-PCR, y los datos se normalizaron con respecto a las células tratadas con LNP de control sin profármaco (las barras de error representan la desviación estándar de al menos tres experimentos).
La figura 17 muestra que los profármacos LD-DTX inhiben el crecimiento de células de cáncer de ovario, en comparación con LD10, LD1 8 , LD22 o LD23, que se formuló en (D) PC-Chol, (C) PC-TO, (B) LNP ionizable o (A) profármaco libre de lípido, luego las células Ovcar3 se incubaron con PBS, docetaxel libre, libre LD-DTX profármacos, o LNP con o sin LD-DTX profármacos en 0 a 15 μM de docetaxel y la viabilidad celular se determinó después de 48 h y se informó como valor relativo normalizado a la viabilidad de las células tratadas con PBS (barras de error representan la desviación estándar de al menos tres experimentos).
Descripción detallada
Los expertos en la técnica apreciarán que el punto de unión covalente del resto a los compuestos como se describe en la presente, puede, por ejemplo y sin limitación, escindirse en condiciones específicas. Las condiciones especificadas pueden incluir, por ejemplo, y sin limitación, medios enzimáticos o no enzimáticos in vivo. La escisión del resto puede ocurrir, por ejemplo, y sin limitación, espontáneamente, o puede ser catalizada, inducida por otro agente, o un cambio en un parámetro físico o ambiental, por ejemplo, una enzima, luz, ácido, temperatura o pH. El resto puede ser, por ejemplo, y sin limitación, un grupo protector que actúa para enmascarar un grupo funcional, un grupo que actúa como sustrato para uno o más mecanismos de transporte activo o pasivo, o un grupo que actúa para impartir o mejorar una propiedad del compuesto, por ejemplo, solubilidad, biodisponibilidad o localización. En algunas formas de realización, los compuestos de la tabla 1 (excepto LD07-DEX) y los compuestos descritos en las reivindicaciones se divulgan para su uso en el tratamiento sistémico o el tratamiento localizado de al menos una indicación para la que el profármaco se considera adecuado. En algunas formas de realización, los compuestos de la tabla 1 (excepto LD07-DEX) y los compuestos descritos en las reivindicaciones pueden utilizarse en la preparación de un medicamento o una composición para el tratamiento sistémico o el tratamiento localizado de una indicación descrita en la presente.
Tabla 1: Compuestos de ejemplo


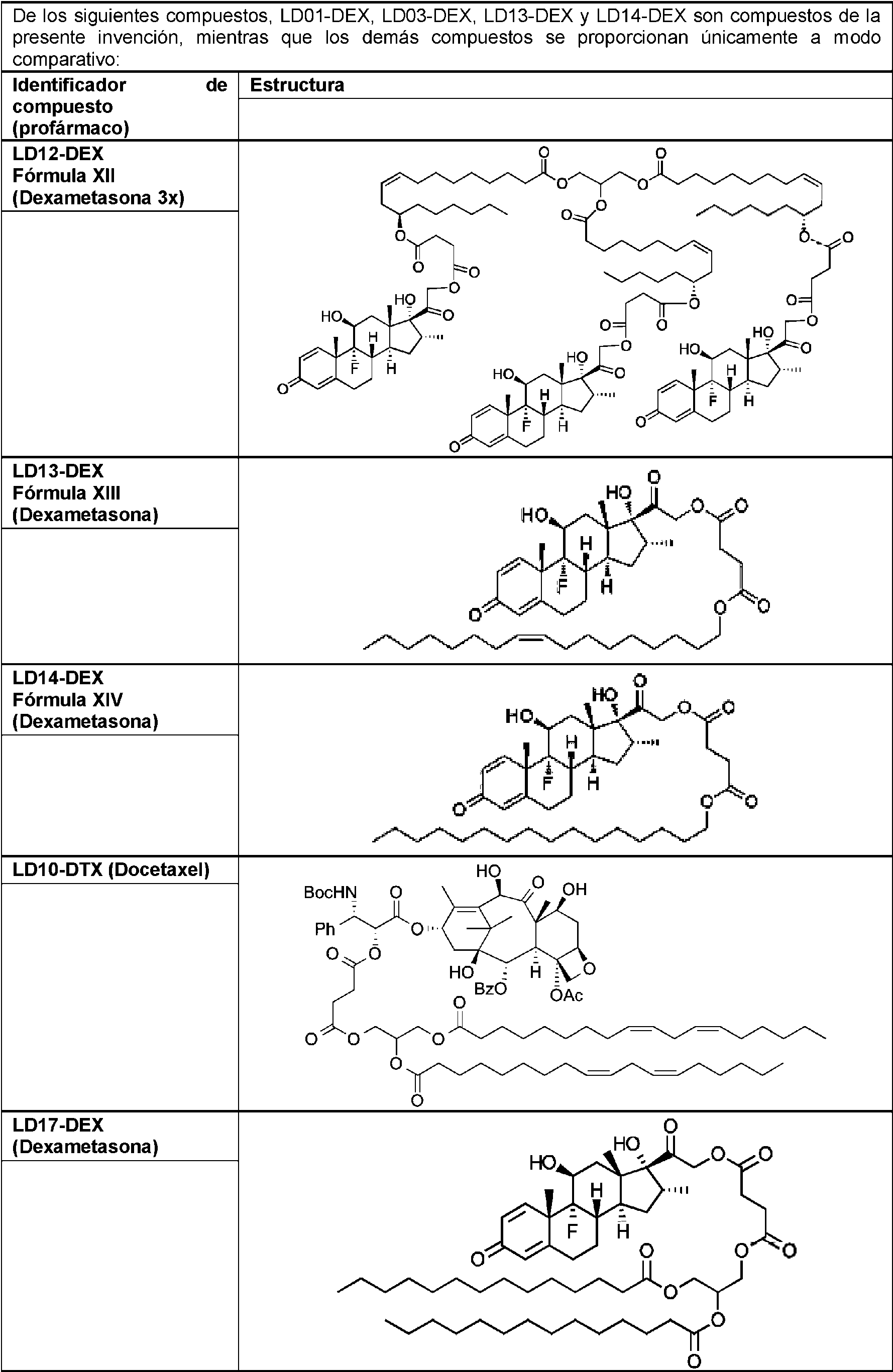


Los fármacos contemplados en la presente pueden describirse con la estructura general A-Z que se muestra a continuación, en donde Z puede representar un OH, o un NH, o un NH2, o un grupo COOH unido al marco molecular principal del fármaco representado por A. El fármaco también puede incorporar múltiples grupos Z, que pueden ser, independientemente, un OH, o un NH, o un NH2, o un COOH, como se ha indicado con anterioridad. Los profármacos unidos a lípidos preparados como se describe en la presente pueden administrarse a pacientes humanos con el fin de tratar una enfermedad, dolencia u otra condición fisiológica indeseable, que sea tratable o curable con el fármaco. Sin embargo, la presente invención se limita a compuestos como los definidos en las reivindicaciones, en donde la parte de fármaco de la estructura se limita a dexametasona.

Algunos ejemplos de fármacos incluyen, pero sin limitación a, Docetaxel, Metotrexato, SN-38, NPC1I, Amprenavir, Anfotericina B, Bexaroteno, Calcitrol, Ciclosporina A, Digoxina, Doxercalciferol, Dronabinol, Etopósido, Fulvestrant, Paricalcitol, Tenipósido, Isotretinoína, Tretinoína, Ácido valproico, Paclitaxel, Valrubicina, Propofol, Ácido
micofenólico, Lovastatina, Lamivudina, Zidovudina, Abacavir, Emitricitabina, Atazanavir, Cobicistat, Elvitegravir, Isonazida, Albendazol, Lisinapril, Amlodipino, Isotretinoína, Baclofeno, Benznidazol, Nifurtimox, Mefloquina, Sulfadoxina, Pirimetamina, Clorproguanil, 6-mercaptopurina, 1-tiroxina, gliburida, MCCg50, Partenolida, Ácido Tauroursodesoxicólico, Ruxolitinib, Tofacitinib, Ciclosporina, Tacrolimus, Everolimus, Sirolimus, Azatioprina, Leflunomida, Micofenolato, Dexametasona, Budesonida, Prednisona, Prednisolona, Metilprednisolona, Hidrocortisona, Cortisona, Fludrocortisona, Betametasona, Triamcinolona, Acetonuro de triamcinolona, Flunisolida, Beclametasona, Fluticasona, Mometasona, Flumetasona, Isoflupredona, Corticosterona, Acetato de desoxicortona, Enantato de desoxicortona, 11-Desoxicorticosterona, 11-Desoxicortisol y Aldosterona.
Alternativamente, ejemplos de fármacos incluyen, pero sin limitación a, Tacrolimus, SN-38, Metotrexato, Docetaxel, NPC1l, Amprenavir, Anfotericina B, Bexaroteno, Calcitrol, Ciclosporina A, Digoxina, Doxercalciferol, Dronabinol, Etopósido, Fulvestrant, Paricalcitol, Tenipósido, Isotretinoína, Sirolimus, Tretinoína, Ácido valproico, Paclitaxel, Valrubicina, Propofol, Ácido micofenólico, Lovastatina, Lamivudina, Zidovudina, Abacavir, Emitricitabina, Atazanavir, Cobicistat, Elvitegravir, Isonazid, Albendazol, Lisinapril, Amlodipino, Isotretinoína, Baclofeno, Benznidazol, Nifurtimox, Mefloquina, Sulfadoxina, Pirimetamina, Clorproguanil, 6-Mercaptopurina, 1-Tiroxina, Gliburida, MCCg50, Partenolida, Ácido tauroursodesoxicólico, Ruxolitinib y Tofacitinib.
Las formulaciones de profármacos unidos a lípidos descritas en la presente se basan en sustancias que se obtienen aprovechando el grupo o grupos Z para unir covalentemente el fármaco a un lípido apropiado. Generalmente, un conjugado lípido-fármaco apropiado como el descrito en la presente tiene un logP igual o superior a 5 (Viswanadhan, V. N.; Ghose, A. K.; Revankar, G. R.; Robins, R. K., J. Chem. Inf. Comput. Sci., 1989, 29, 163-172, o utilizando un programa como Marvin (ChemAxon)); y es, por lo tanto, poco soluble o insoluble en agua, pero soluble en solventes orgánicos comunes como etanol, cloroformo, acetato de etilo, diclorometano, isopropanol, etc.
La estructura molecular de dicho lípido puede incorporar grupos funcionales que permitan la unión covalente del fármaco al lípido. Ejemplos de tales grupos funcionales pueden ser OH, SH, NH, NH2, COOH.
En algunas formas de realización, el profármaco sería al menos débilmente hidrófobo. Además, en algunas formas de realización, el fármaco estaría unido a la fracción lipídica a través de un ligador biodegradable, como un enlace éster. Sin embargo, se contemplan otros ligadores biodegradables (por ejemplo, fosfato).
Un “resto de fármaco”, como se utiliza en la presente, es cualquier agente farmacéutico capaz de unirse covalentemente al lípido, tal como se describe en la presente. En algunas formas de realización, un resto de fármaco puede excluir los esteroides, los compuestos que contienen quinoleína o los restos de unión a Na+/K+ ATPasa. En algunas formas de realización, un resto de fármaco puede excluir los esteroides. En algunas formas de realización, un resto de fármaco puede excluir los compuestos que contienen quinoleína. En algunas formas de realización, un resto de fármaco puede excluir las moléculas de unión a Na+/K+ ATPasa. En algunas formas de realización, las moléculas de unión a Na+/K+ ATPasa pueden ser glucósidos cardíacos. En algunas formas de realización, un resto de fármaco puede excluir los glucósidos cardíacos. En algunas formas de realización, un resto de fármaco puede excluir los glucósidos cardíacos y los compuestos que contienen quinolona. El compuesto que contiene quinolona puede ser 4-aminoquinolina, 8-aminoquinolina o 4-metanolquinolina. Los glucósidos cardíacos pueden seleccionarse de uno o más de los siguientes: acetildigitoxinas; acetildigoxinas; cimarina; digitoxina; digitoxigenina; digoxigenina; digoxina; medigoxina; neoconvallósido; ouabaína; dihidroouabaína; estrofantinas; estrofantidina; bufenólidos; bufanólido; daigremontianina; cardenólido; helveticósido; k-estofantidina; latanósido C; procillardina A; arenobufagina; bufotalina; cinobufagina; marinobufagina; escillirósido; acetildigitoxina; acetildigoxina; deslanósido; medigoxina; gitoformato; digremontianina; peruvósido. En algunas formas de realización, si el resto de fármaco es un esteroide, el resto de fármaco esteroide (A-Z) puede unirse -Y-M-C(=O)- o -Y-M- a través del anillo de 5 miembros del esteroide.
Como se utiliza en la presente, un grupo acilo graso [R'-C(=O)], puede ser un R' con una cadena de carbono lineal o ramificada Cg-C29, que incorpore facultativamente enlaces dobles C=C de geometría E o Z y/o sustituyentes tales como grupos OH. Ejemplos de tales grupos acilo grasos (ácido parental) son - pero no se limitan a: Caprilo (ácido cáprico), Lauroilo (ácido láurico), Miristilo (ácido mirístico), Miristoleilo (ácido miristoleico), Palmitoilo (ácido palmítico), Isopalmitoilo (ácido isopalmítico), Palmitoleilo (ácido palmitoleico), Sapienoilo (ácido sapiénico), Estearoilo (ácido esteárico), Isoestearoilo (ácido isosteárico), Oleilo (ácido oleico), Elaidoilo (ácido elaídico), Vaccenoilo (ácido vaccénico), Linoleilo (ácido linoleico), Linoelaidoilo (ácido linoelaídico), Linolenilo (ácido linolénico), Ricinoleilo (ácido ricinoleico), Pristroilo (ácido pristánico), Araquidoilo (ácido araquídico), Araquidonoilo (ácido araquidónico), Eicosapentaenoilo (ácido eicosapentaenoico), Fitanoilo (ácido fitánico), Behenoilo (ácido behénico), Erucoilo (ácido erúcico), Docosahexaenoilo (ácido docosahexaenoico), Lignocerilo (ácido lignocérico), Cerotoilo (ácido ceroico), Montanoilo (ácido montanico), Melissoilo (ácido melísico). Alternativamente, un grupo acilo graso, según se describe en la presente, puede tener originalmente un grupo OH (por ejemplo: ricinoleilo), en donde el grupo OH se ha modificado covalentemente mediante la unión a un ligador o a un profármaco y ligador como se describe en la presente.
La hidrofobicidad relativa del fármaco y de las moléculas lipídicas, la presencia de grupos cargados/ionizables, el tipo de ligador biodegradable, etc., pueden adaptarse al profármaco concreto y/o a la diana concreta de los profármacos (o nanopartículas lipídicas (LNP)) unidos a lípidos descritos en la presente. Las propiedades hidrófobas
del profármaco (compuesto por el fármaco y las partes lipídicas) deberían permitir que estos profármacos se incorporen fácilmente mediante la simple adición del fármaco a la mezcla de formulación lipídica sin necesidad de modificar el proceso de formulación. Además, la hidrofobicidad también puede permitir la retención estable en formulaciones de profármacos unidos a lípidos, dotando al profármaco de un perfil de toxicidad mejorado en comparación. Algunas formulaciones de LNP presentan ocasionalmente liberación de fármaco tras la administración sistémica, lo que da lugar a propiedades farmacocinéticas indeseables [Charrois y Allen (2004); Cui et al. (2007); y Johnston et al. (2006)]. Por el contrario, la mayoría de los profármacos unidos a lípidos descritos en la presente deberían presentar una fuga mínima tras la administración sistémica hasta que sean absorbidos por las células diana. Por último, aunque la hidrofobicidad del profármaco debe ser suficiente para lograr una encapsulación/retención eficaz, es preferible que no dé lugar a que el profármaco quede blindado en el resto lipídico y, por lo tanto, no se libere cuando se encuentre en el entorno adecuado. En los casos en que el profármaco unido a lípidos tenga características hidrófobas, la distribución del profármaco en el lípido permite el acceso al ligador biodegradable. La liberación del fármaco activo puede producirse tras la absorción a través de la actividad de las enzimas intracelulares apropiadas para escindir el ligador biodegradable.
Estos profármacos unidos a lípidos no requieren carga activa (es decir, gradiente de pH) [Nichols y Deamer (1976); y Mayer et al. (1986)] para lograr una incorporación eficaz en el lípido. Dado que los profármacos no necesitan ser bases débiles, este enfoque permite la incorporación de fármacos en nanopartículas lipídicas (LNP), donde el fármaco no se encapsularía utilizando enfoques convencionales. Las ventajas de la administración de fármacos mediada por LNP se han descrito con detalle, como la mejora de la administración en los focos de la enfermedad y la facilitación de la absorción en las células diana, lo que se traduce en un aumento de la potencia y una reducción de la toxicidad (frente al fármaco libre) debido a los menores niveles de fármaco en los tejidos no diana [Allen y Cullis (2013)].
Como se usa en la presente, un resto ionizable puede ser cualquier átomo o molécula que adquiere una carga negativa o positiva al perder o ganar protones para formar iones. Ejemplos de restos ionizables son -NH, -NHMe, -NMe2 , -COOH, -OH, -NH+, -NH3+, -NH2+, -SH, etc. La presencia de un resto ionizable en un profármaco lipídico puede influir en el índice de hidrofobicidad del profármaco (por ejemplo, véase la tabla 2).
Los compuestos descritos en la presente pueden estar en forma libre o en forma de una sal de estos. En algunas formas de realización, los compuestos descritos en la presente pueden estar en forma de una sal farmacéuticamente aceptable, que es conocida en la técnica (Berge S. M. et al., J. Pharm. Sci. (1977) 66(1):1 -19). La sal farmacéuticamente aceptable, como se utiliza en la presente, incluye, por ejemplo, sales que tienen la actividad farmacológica deseada del compuesto original (sales que conservan la eficacia biológica y/o las propiedades del compuesto original y que no son indeseables desde el punto de vista biológico y/o de otro tipo). Los compuestos como se describen en la presente que tienen uno o más grupos funcionales capaces de formar una sal pueden, por ejemplo, formarse como una sal farmacéuticamente aceptable. Los compuestos que contienen uno o más grupos funcionales básicos pueden ser capaces de formar una sal farmacéuticamente aceptable con, por ejemplo, un ácido orgánico o inorgánico farmacéuticamente aceptable. Las sales farmacéuticamente aceptables pueden derivarse de, por ejemplo, y sin limitación, ácido acético, ácido adípico, ácido algínico, ácido aspártico, ácido ascórbico, ácido benzoico, ácido bencenosulfónico, ácido butírico, ácido cinámico, ácido cítrico, ácido canforico, ácido canforosulfónico, ácido ciclopentanopropiónico, ácido dietilacético, ácido diglucónico, ácido dodecilsulfónico, ácido etanosulfónico, ácido fórmico, ácido fumárico, ácido glucoheptanoico, ácido glucónico, ácido glicerofosfórico, ácido glicólico, ácido hemisulfónico, ácido heptanoico, ácido hexanoico, ácido clorhídrico, ácido bromhídrico, ácido yodhídrico, ácido 2-hidroxietanosulfónico, ácido isonicotínico, ácido láctico, ácido málico, ácido maleico, ácido malónico, ácido mandélico, ácido metanosulfónico, ácido 2-naftalenosulfónico, ácido naftalendisulfónico, ácido ptoluenosulfónico, ácido nicotínico, ácido nítrico, ácido oxálico, ácido pamóico, ácido pectínico, ácido 3-fenilpropiónico, ácido fosfórico, ácido pícrico, ácido pimélico, ácido piválico, ácido propiónico, ácido pirúvico, ácido salicílico, ácido succínico, ácido sulfúrico, ácido sulfámico, ácido tartárico, ácido tiociánico o ácido undecanoico. Los compuestos que contienen uno o más grupos funcionales ácidos pueden ser capaces de formar sales farmacéuticamente aceptables con una base farmacéuticamente aceptable, por ejemplo, y sin limitación, bases inorgánicas basadas en metales alcalinos o metales alcalinotérreos o bases orgánicas tales como compuestos de amina primaria, compuestos de amina secundaria, compuestos de amina terciaria, compuestos de amina cuaternaria, aminas sustituidas, aminas sustituidas naturales, aminas cíclicas o resinas básicas de intercambio iónico. Las sales farmacéuticamente aceptables pueden derivarse de, por ejemplo, y sin limitación, un hidróxido, carbonato o bicarbonato de un catión metálico farmacéuticamente aceptable como amonio, sodio, potasio, litio, calcio, magnesio, hierro, zinc, cobre, manganeso o aluminio, amoníaco, benzatina, meglumina, metilamina, dimetilamina, trimetilamina, etilamina, dietilamina, trietilamina, isopropilamina, tripropilamina, tributilamina, etanolamina, dietanolamina, 2-dimetilaminoetanol, 2-dietilaminoetanol, diciclohexilamina, lisina, arginina, histidina, cafeína, hidrabamina, colina, betaína, etilendiamina, glucosamina, glucamina, metilglucamina, teobromina, purinas, piperazina, piperidina, procaína, N-etilpiperidina, teobromina, compuestos de tetrametilamonio, compuestos de tetraetilamonio, piridina, N,N-dimetilanilina, N-metilpiperidina, morfolina, N-metilmorfolina, N-etilmorfolina, diciclohexilamina, dibencilamina, N,N-dibencilfenetilamina, 1-fenamina, N,N'-dibenciltilendiamina o resinas de poliamina. En algunas formas de realización, los compuestos descritos en la presente pueden contener tanto grupos ácidos como básicos y pueden estar en forma de sales internas o zwitteriones, por ejemplo, y sin limitación, betaínas. Las sales descritas en la presente pueden prepararse mediante procesos convencionales conocidos por
un experto en la técnica, por ejemplo, y sin limitación, haciendo reaccionar la forma libre con un ácido orgánico o un ácido o base inorgánicos, o mediante intercambio aniónico o catiónico a partir de otras sales. Los expertos en la técnica apreciarán que la preparación de sales puede tener lugar in situ durante el aislamiento y purificación de los compuestos o la preparación de sales puede tener lugar haciendo reaccionar por separado un compuesto aislado y purificado.
En algunas formas de realización, los compuestos y todas las diferentes formas de estos (por ejemplo, formas libres, sales, polimorfos, formas isoméricas) tal como se describen en la presente pueden estar en forma de adición de solvente, por ejemplo, solvatos. Los solvatos contienen cantidades estequiométricas o no estequiométricas de un solvente en asociación física con el compuesto o su sal. El solvente puede ser, por ejemplo, y sin limitación, un solvente farmacéuticamente aceptable. Por ejemplo, se forman hidratos cuando el solvente es agua o alcoholatos cuando el solvente es un alcohol.
En algunas formas de realización, los compuestos y todas sus diferentes formas (por ejemplo, formas libres, sales, solvatos, formas isoméricas) tal como se describen en la presente pueden incluir formas cristalinas y amorfas, por ejemplo, polimorfos, pseudopolimorfos, polimorfos conformacionales, formas amorfas, o una combinación de estos. Los polimorfos incluyen diferentes disposiciones de empaquetamiento cristalino de la misma composición elemental de un compuesto. Los polimorfos suelen tener diferentes patrones de difracción de rayos X, espectros infrarrojos, puntos de fusión, densidad, dureza, forma cristalina, propiedades ópticas y eléctricas, estabilidad y/o solubilidad. Los expertos en la técnica apreciarán que diversos factores, como el solvente de recristalización, la velocidad de cristalización y la temperatura de almacenamiento, pueden hacer que predomine una única forma cristalina.
En algunas formas de realización, los compuestos y todas sus diferentes formas (por ejemplo, formas libres, sales, solvatos, polimorfos) descritos en la presente incluyen isómeros tales como isómeros geométricos, isómeros ópticos basados en carbono asimétrico, estereoisómeros, tautómeros, enantiómeros individuales, diastereómeros individuales, racematos, mezclas diastereoméricas y combinaciones de estos, y no están limitados por la descripción de la Fórmula ilustrada por conveniencia.
En otras formas de realización, los compuestos pueden incluir compuestos dentro de una estructura genérica o Markush, según se determine a partir de las relaciones estructura-actividad identificadas a partir de los datos presentados en la tabla 2 y en los ejemplos siguientes.
En algunas formas de realización, las composiciones farmacéuticas descritas en la presente pueden comprender una sal de dicho compuesto, preferentemente una sal farmacéutica o fisiológicamente aceptable. Las preparaciones farmacéuticas comprenderán típicamente uno o más portadores, excipientes o diluyentes aceptables para el modo de administración de la preparación, ya sea por inyección, inhalación, administración tópica, lavado u otros modos adecuados para el tratamiento seleccionado. Los portadores, excipientes o diluyentes adecuados (utilizados indistintamente en la presente) son los conocidos en la técnica para su uso en dichos modos de administración. Las composiciones farmacéuticas adecuadas pueden formularse por medios conocidos en la técnica y su modo de administración y dosis pueden ser determinados por el experto en la técnica. Para la administración parenteral, un compuesto puede disolverse en agua estéril o solución salina o en un vehículo farmacéuticamente aceptable utilizado para la administración de compuestos no solubles en agua, como los utilizados para la vitamina K. Para la administración enteral, el compuesto puede administrarse en un comprimido, cápsula o disuelto en forma líquida. El comprimido o la cápsula pueden tener un recubrimiento entérico o una formulación de liberación sostenida. Se conocen muchas formulaciones adecuadas, como micropartículas poliméricas o proteicas que encapsulan un compuesto que se va a liberar, pomadas, pastas, geles, hidrogeles o soluciones que se pueden utilizar tópica o localmente para administrar un compuesto. Se puede emplear un parche o implante de liberación sostenida para proporcionar liberación durante un período de tiempo prolongado. En Remington: the Science & Practice of Pharmacy de Alfonso Gennaro, 20a ed., Lippencott Williams & Wilkins, (2000) se describen muchas técnicas conocidas por los expertos en la técnica. Las formulaciones para administración parenteral pueden contener, por ejemplo, excipientes, polialquilenglicoles como el polietilenglicol, aceites de origen vegetal o naftalenos hidrogenados. Para controlar la liberación de los compuestos pueden utilizarse polímeros de láctido biocompatibles y biodegradables, copolímeros de láctido/glicólido o copolímeros de polioxietileno polioxipropileno. Otros sistemas de administración parenteral potencialmente útiles para los compuestos moduladores son las partículas de copolímero de etilvinilacetato, las bombas osmóticas, los sistemas de infusión implantables y los liposomas. Las formulaciones para inhalación pueden contener excipientes, por ejemplo, lactosa, o pueden ser soluciones acuosas que contengan, por ejemplo, polioxietilen 9 lauril éter, glucocolato y desoxicolato, o pueden ser soluciones oleosas para administración en forma de gotas nasales, o como un gel.
Los compuestos o composiciones farmacéuticas descritos en la presente o para su uso descrito en la presente pueden administrarse mediante un dispositivo o aparato médico como un implante, injerto, prótesis, stent, etc. Asimismo, pueden idearse implantes destinados a contener y liberar dichos compuestos o composiciones. Un ejemplo sería un implante hecho de un material polimérico adaptado para liberar el compuesto durante un período de tiempo.
Una “cantidad eficaz” de una composición farmacéutica descrita en la presente incluye una cantidad
terapéuticamente eficaz o una cantidad profilácticamente eficaz. Una “cantidad terapéuticamente eficaz” se refiere a una cantidad eficaz, a las dosis y durante los períodos de tiempo necesarios, para lograr el resultado terapéutico deseado, por ejemplo, la reducción del tamaño del tumor, el aumento de la duración de la vida o el aumento de la esperanza de vida. Una cantidad terapéuticamente eficaz de un compuesto puede variar en función de factores como el estado de la enfermedad, la edad, el sexo y el peso del sujeto, y la capacidad del compuesto para provocar una respuesta deseada en el sujeto. Los regímenes de dosificación pueden ajustarse para proporcionar la respuesta terapéutica óptima. Una cantidad terapéuticamente eficaz es también aquella en donde cualquier efecto tóxico o perjudicial del compuesto se ve compensado por los efectos terapéuticamente beneficiosos. Una “cantidad profilácticamente eficaz” se refiere a una cantidad eficaz, a las dosis y durante los períodos de tiempo necesarios, para lograr el resultado profiláctico deseado, por ejemplo, tumores más pequeños, mayor duración de la vida, mayor esperanza de vida. Típicamente, una dosis profiláctica se utiliza en sujetos antes o en una fase más temprana de la enfermedad, de modo que una cantidad profilácticamente eficaz puede ser menor que una cantidad terapéuticamente eficaz.
Cabe señalar que los valores de dosificación pueden variar con la gravedad de la afección que se desea aliviar. Para cualquier sujeto en particular, los regímenes de dosificación específicos pueden ajustarse con el tiempo según la necesidad individual y el juicio profesional de la persona que administra o supervisa la administración de las composiciones. Los intervalos de dosificación establecidos en la presente son solo ejemplares y no limitan los intervalos de dosificación que pueden seleccionar los médicos. La cantidad de compuesto(s) activo(s) en la composición puede variar en función de factores como el estado de la enfermedad, la edad, el sexo y el peso del sujeto. Los regímenes de dosificación pueden ajustarse para proporcionar una respuesta terapéutica óptima. Por ejemplo, puede administrarse un único bolo, pueden administrarse varias dosis divididas a lo largo del tiempo o la dosis puede reducirse o aumentarse proporcionalmente según indiquen las exigencias de la situación terapéutica. Puede ser ventajoso formular las composiciones parenterales en forma de unidades de dosificación para facilitar la administración y uniformizar la dosificación.
En general, los compuestos descritos en la presente deben utilizarse sin causar toxicidad sustancial. La toxicidad de los compuestos descritos en la presente puede determinarse mediante técnicas estándar, por ejemplo, realizando pruebas en cultivos celulares o animales de experimentación y determinando el índice terapéutico, es decir, la relación entre la LD50 (la dosis letal para el 50 % de la población) y la LD100 (la dosis letal para el 100 % de la población). Sin embargo, en algunas circunstancias, como en el caso de enfermedades graves, puede ser apropiado administrar excesos sustanciales de las composiciones. Algunos compuestos descritos en la presente pueden ser tóxicos a determinadas concentraciones. Pueden utilizarse estudios de titulación para determinar las concentraciones tóxicas y no tóxicas.
Los compuestos descritos en la presente pueden administrarse a un sujeto. Tal como se utiliza en la presente, un “sujeto” puede ser un ser humano, un primate no humano, una rata, un ratón, una vaca, un caballo, un cerdo, una oveja, una cabra, un perro, un gato, etc. El sujeto puede ser sospechoso de tener o estar en riesgo de tener una indicación para la que un resto terapéutico puede proporcionar un beneficio.
Materiales y métodos
En los siguientes ejemplos, los compuestos LO01-DEX, LD03-DEX, LD13-DEX y LD14-DEX son compuestos de la presente invención, mientras que otros compuestos se proporcionan únicamente a efectos de comparación.
Química
Todos los reactivos y solventes se adquirieron a proveedores comerciales y se utilizaron sin más purificación, a menos que se indique lo contrario, excepto el THF (recién destilado de Na/benzofenona bajo Ar), y Et3N y CH2Cl2 (recién destilados de CaH2 bajo Ar). RMN Los desplazamientos químicos se indican en partes por millón (ppm) en la escala 8 y las constantes de acoplamiento, J, en hercios (Hz). Las multiplicidades se expresan como “s” (singlete), “d” (doblete), “dd” (doblete de dobletes), “dt” (doblete de tripletes), “ddd” (doblete de dobletes de dobletes), “t” (triplete), “td” (triplete de dobletes), “q” (cuartete), “quin” (quintete), “sex” (sextete), “m” (multiplete), y calificados además como “app” (aparente) y “br” (amplio).
Procedimientos de síntesis de lípidos
ESQUEMA 1: Esquema para la síntesis del lípido 3, 2-linoleoil-3-dimetilamino-1,2-propanodiol.

Se añadió TBSCI sólido (1,36 g, 9,00 mmol, 1,00 equiv.) a una solución de 3-dimetilaminopropano-1,2-diol (1,07 g, 9,00 mmol) e imidazol (613 mg, 9,00 mmol, 1,00 equiv.) en un matraz de base redonda bajo argón. Después de 2,5 h, la mezcla de reacción se diluyó con CH2Ch y se lavó con agua destilada (1*25 mL). La capa acuosa se extrajo con CH2Cl2 (1*15 mL) y las capas orgánicas combinadas se lavaron con salmuera (1*15 mL), se secaron sobre Na2SO4 y se concentraron para obtener un aceite claro e incoloro como éter silílico 1 (1,83 g, 87 % de rendimiento), que se utilizó sin purificación adicional.
Rf = 0,09 (SiO2, 50:50 EtOAc/hexanos);
1H RMN (300 MHz, CDCls): 83,80-3,68 (m, 1H), 3,60 (m, 1H), 2,39 (dd, J = 12,3, 9,0 Hz, 1H), 2,34-2,24 (m, 1H), 2,29 (s, 6H), 0,90 (s, 9H), 0,07 (s, 6H).
COMPUESTO 2: 1-(íer-butildimetilsilil)-2-linoleoil-3-dimetilaminopropano-1,2-diol:
El cloruro de linoleílo se preparó añadiendo cloruro de oxalilo (0,30 mL, 3,60 mmol, 1,80 equiv.) a una solución de benceno (5 mL) de ácido linoleico (673 mg, 2,40 mmol, 1,20 equiv. respecto al alcohol) y DMF (18 μL, 0,24 mmol, 0,12 equiv.) a temperatura ambiente en un matraz de base redonda bajo argón. Después de 3 h, los volátiles se eliminaron en un evaporador rotativo y el residuo se azeotrópico con benceno (2*5 mL), después se secó a alto vacío durante 3 h y se utilizó inmediatamente sin purificación adicional.
Se añadió una solución de CH2Cl2 (4 mL) de alcohol 1 (467 mg, 2,00 mmol, 1,00 equiv.) y Et3N (0,69 mL, 5,00 mmol, 2,50 equiv.) a una solución de CH2Cl2 (4 mL) helada y en agitación del cloruro de linoleoilo preparado con anterioridad en un matraz de base redonda bajo argón, seguido de DMAP sólido (293 mg, 2,40 mmol, 1,20 equiv.). La mezcla de reacción se dejó calentar durante 14 h, después se diluyó con CH2Cl2 , se lavó con NaHCO3 al 5 % acuoso (2*10 mL), se secó sobre Na2SO4 y se concentró en un evaporador rotativo para obtener el crudo como un semisólido marrón. El crudo se purificó por cromatografía en columna flash (130 mL SiO2, 79:20:1^-69:30:1 hexanos/EtOAc/Et3N) para obtener un aceite amarillo claro como el éster deseado 2 (828 mg, 84 % de rendimiento).
Rf = 0,39 (SO2, 50:50 EtOAc/hexanos);
1H RMN (300 MHz, CDCla): 85,37-5,25 (m, 4H), 5,09-4,95 (m, 1H), 3,76-3,63 (m, 2H), 2,77 (t, J = 5,8 Hz, 2H), 2,56-2,44 (m, 2H), 2,40-20 (m, 2H), 2,27 (s, 6H), 2,04 (q, J = 6,4 Hz, 4H), 1,62 (t, J = 7,0 Hz, 2H), 1,45 1,20 (m, 16H), 0,88 (app s, 12H), 0,04 (s, 6H).
COMPUESTO 3: 2-linoleoil-3-dimetilaminopropano-1,2-diol:
Se añadió una solución pura de HF-piridina (68 μL de HF al 70 % en piridina, 0,54 mmol, 3,00 equiv.) a una solución de piridina (44 μL, 0,54 mmol, 3,00 equiv.) y éter silílico 2 (90 mg, 0,18 mmol) en THF helado (1,2 mL) en un matraz de base redonda bajo argón. Después de 2 h, la mezcla de reacción se apagó con NaHCO3 al 5 % ac. La mezcla se extrajo con EtOAc (3*5 mL), después los extractos orgánicos combinados se lavaron con salmuera, se secaron sobre Na2SO4 y se concentraron en un evaporador rotativo para obtener el alcohol primario 3 (68 mg, rendimiento teórico de 69 mg) como un aceite claro e incoloro que se usó inmediatamente y sin purificación adicional. Cabe señalar que la transesterificación completa al alcohol primario se observó (TLC, 1H RMN) en ≤24 h cuando se almacenó puro a -20 °C.
ESQUEMA 2: Esquema para la síntesis del lipido 4, hemisuccinato de 2,3-dilinoleoil glicerol.

COMPUESTO 4: hemisuccinato de 2,3-dilinoleoil glicerol:
Se añadieron anhídrido succínico sólido (67 mg, 0,66 mmol, 2,0 equiv.) y DMAP (102 mg, 0,83 mmol, 2,5 equiv.) a una solución agitada de CH2Cl2 (3,5 mL) a temperatura ambiente de 1,2-dilinoleoil glicerol [Abe et al. (2011)] (205 mg, 0,33 mmol, 1,0 equiv.) en un matraz de base redonda bajo argón. Después de 12 horas, la reacción se apagó con HCl 1 M acuoso, se extrajo con CH2Cl2 (2*15 mL), los extractos orgánicos combinados se lavaron con HCl 1 M acuoso (1*15 mL), se secaron sobre Na2SO4 y se concentraron en un evaporador rotativo para obtener el hemisuccinato 4 (rendimiento cuantitativo) como un aceite amarillo pálido que se utilizó sin purificación adicional. ESQUEMA 3: Esquema para la síntesis del lípido 5, hemisuccinato de alcohol linoleílico.

Se añadieron anhídrido succínico sólido (500 mg, 5,00 mmol, 2,00 equiv.) y DMAP (764 mg, 6,25 mmol, 2,50 equiv.) a una solución de alcohol linoleílico (666 mg, 2,50 mmol) agitada en C ^C h (10 mL) a temperatura ambiente en un matraz de base redonda bajo argón. Después de 14 h, la mezcla de reacción se diluyó con CH2Ch, se lavó con HCl 1 M acuoso (2*10 mL), se secó sobre Na2SO4 y se concentró en un evaporador rotativo para obtener el hemisuccinato 5 (915 mg, rendimiento cuantitativo) como un semisólido blanco que se utilizó sin purificación adicional.
ESQUEMA 4: Esquema para la síntesis del lípido 10, 2,3-dilinoleoil-4-dimetilamino-1,2,3-propanotriol.

Se añadió una solución de Me2NH/THF (22,5 mL de 2,0 M en THF, 45,0 mmol, 3,00 equiv.) durante 5 min a una solución de (3R,4R)-bis(benzoiloxi)anhídrido succínico [Ohwada et al. (1990)] (4,68 g, 15,0 mmol) en un matraz de base redonda bajo argón. Después de 16 h, la mezcla de reacción se diluyó con Et2O y se apagó con NH4Cl acuoso saturado. La capa acuosa se extrajo con Et2O (1*25 mL), los extractos orgánicos combinados se lavaron con salmuera, se secaron sobre Na2SO4 y se concentraron en un evaporador rotativo para obtener el amidoácido intermedio como un aceite viscoso de color amarillo (5,36 g de rendimiento teórico).
Se añadió cloruro de tionilo (1,14 mL, 15,7 mmol, 1,05 equiv.) a una solución de MeOH (25 mL) helada y en agitación del amidoácido en un matraz de base redonda bajo argón y se dejó calentar la mezcla. Después de 14 h, se eliminaron los volátiles en un evaporador rotativo y se añadió NaHCO3 acuoso saturado. La mezcla se extrajo con Et2O (2*75 mL), los extractos orgánicos combinados se lavaron con NaHCO3 acuoso saturado (1*100 mL), se secaron sobre Na2SO4 y se concentraron en un evaporador rotativo para obtener el crudo como un aceite amarillo. El crudo se purificó mediante cromatografía flash en columna (220 mL SiO2, 60:40 EtOAc/hexanos) para obtener un aceite amarillo pálido como amidoéster 6 (4,44 g, 80 % de rendimiento).1
1H RMN (300 MHz, CDCh): 8 7,43-7,19 (m, 10H), 4,84 (d, J = 11,6, 1H), 3,68 (s, 3H), 3,02 (s, 3H, rotámeros), 2,91 (s, 3H, rotámeros);
13C RMN (300 MHz, CDCI3): 5170,1, 168,5, 137,1, 136,7, 128,3, 128,2, 128,1, 127,9, 80,1, 78,9, 73,5, 72,4, 52,1, 36,9, 36,4.
COMPUESTO 7: (2S,3S)-2,3-bis(benciloxi)-4-(dimetilamino)butan-1-ol:
Una solución de THF (15 mL) del amidoéster 6 (2,26 g, 6,08 mmol) se añadió durante 1 h desde un embudo de adición a una suspensión de THF (20 mL) a reflujo de LiAlH4 (1,15 g, 30,4 mmol, 5,00 equiv.) en un matraz de base redonda de dos cuellos con condensador y bajo argón. Después de 5 min adicionales, la mezcla de reacción se enfrió en un baño de hielo, se diluyó con Et2O, se apagó por el método de Fieser [Fieser y Fieser (1967)] (1,2 mL de agua, 1,2 mL de NaOH 1 M acuoso, 3,6 ml de agua), se sacó del baño de hielo y se agitó a temperatura ambiente hasta que los precipitados se volvieron blancos (~1 h). La mezcla se filtró a través de Celite y el filtrado se concentró en un evaporador rotativo para obtener un aceite claro e incoloro como el aminoalcohol 7 deseado (1,99 g, 99 % de rendimiento), que se utilizó sin más purificación.
1H RMN (300 MHz, CDCls): 57,41-7,24 (m, 10H), 6,04 (br s, 1H), 4,73 (d, J = 11,7, 1H), 4,64 (d, J = 11,7, 1H), 4,64 (s, 2H), 3,74 (br d, J = 3,6, 2H), 3,70 (dt, J = 6,9, 3,3, 1H), 3,62-3,55 (m, 1H), 2,61 (dd, J = 13,2, 6,9, 1H), 2,51 (dd, J = 13,3, 3,3, 1H), 2,30 (s, 6 H); 13C RMN (300 MHz, CDCla): 5 138,5, 138,2, 128,33, 128,29, 127,81, 127,76, 127,6, 127,5, 81,7, 79,2, 72,6, 72,2, 60,1, 59,0, 46,0.
COMPUESTO 8: (2S,3S)-1-(ter-butil)dimetilsilil-4-dimetilaminobutano-1,2,3-triol:
Se añadió TBSCl sólido (497 mg, 3,30 mmol, 1,10 equiv.) a una solución de alcohol primario 7 (1,00 g, 3,00 mmol) e imidazol (449 mg, 6,60 mmol, 2,20 equiv.) en DMF ( 6 mL) a temperatura ambiente y en un matraz de base redonda bajo argón. Después de 14 h, la mezcla de reacción se apagó con NaHCO3 al 5 % acuoso (25 mL) y se extrajo con Et2O (3x20 mL).
Los extractos orgánicos combinados se lavaron con agua (3x20 mL), salmuera, se secaron sobre Na2SO4 y se concentraron en un evaporador rotativo para obtener el crudo como un aceite claro e incoloro. El crudo se purificó por cromatografía en columna flash (100 mL SO2, 70:30:0^-55:40:5 hexanos/EtOAc/MeOH) para obtener un aceite claro e incoloro como el intermedio silil éter (1,17 g, 87 % de rendimiento).
Se añadió Pd(OH)2/C sólido (177 mg de 10 % en peso, 0,12 mmol, 0,10 equiv.) a una solución agitada a temperatura ambiente 7:3 THF/HOAc (8,5 mL) del triol bisbencilado O-TBS anterior (558 mg, 1,26 mmol) en un matraz de base redonda bajo argón. El globo de argón se sustituyó por un globo de hidrógeno y se burbujeó gas hidrógeno a través de la mezcla durante 5 min, antes de dejar que se agitara con hidrógeno a presión de globo durante 18 h. La mezcla se diluyó con EtOAc y se filtró a través de Celite. El pH del filtrado se elevó a pH 8-10 mediante la adición de Na2CO3 acuoso saturado, se extrajo con EtOAc (2 x10 mL), después los extractos orgánicos combinados se lavaron con salmuera, se secaron sobre Na2SO4 y se concentraron para obtener un aceite claro e incoloro como el diol 8 deseado (230 mg, 331 mg de rendimiento teórico), que se utilizó sin purificación adicional.
1H RMN (300 MHz, CDCh): 53,85-3,76 (m, 1H), 3,72 (dd, J = 5,9, 2,7, 2H), 3,55 (dt, J = 5,7, 2,5, 1H), 3,31 (br s, 2H), 2,60 (dd, J = 12,4, 8 ,8 , 1H), 2,37-2,28 (m,1H), 2,29 (s, 6 H), 0,89 (s, 9H), 0,08 (s,6 H); 13C RMN (300 MHz, CDCl3): 572,3, 67,4, 64,9, 62,0, 45,8, 25,8, 18,2, -5,46.
COMPUESTO 9: (2S,3S)-1-(ter-butil)dimetilsilil-2,3-dilinoleoil-4-dimetilaminobutano-1,2,3-triol:
Una porción del diol 8 se utilizó en la acilación posterior. El cloruro de linoleilo se preparó añadiendo cloruro de oxalilo (0,19 mL, 2,25 mmol) a una solución de ácido linoleico (422 mg, 1,50 mmol, 2,20 equiv. en relación con el diol) y DMF (12 μL, 0,15 mmol) a temperatura ambiente en un matraz de base redonda bajo argón. Después de 4 h, los volátiles se eliminaron en un evaporador rotativo y el residuo se azeotrópico con benceno (2x5 mL), después se secó a alto vacío durante 3 h y se utilizó inmediatamente sin purificación adicional. Se añadió una solución de CH2Cl2 (1,5 mL) del diol anterior (180 mg, 0,68 mmol, 1,00 equiv.) y Et3N (0,47 mL, 3,42 mmol, 5,00 equiv.) a una solución agitada y helada de CH2Cl2 (2 mL) del cloruro de linoleilo en un matraz de base redonda bajo argón, seguido de DMAP sólido (184 mg, 1,50 mmol, 2,20 equiv.). La mezcla de reacción se dejó calentar durante 14 h, se diluyó con CH2Cl2, se lavó con NaHCO3 al 5 % acuoso (2x10 mL), se secó sobre Na2SO4 y se concentró en un evaporador rotativo para obtener el crudo como un semisólido amarillo. El crudo se purificó mediante cromatografía en columna flash (50 mL SO2 , 90:10 hexanos/EtOAc) para obtener un aceite claro e incoloro como el diéster 9 deseado (319 mg, 59 % de rendimiento).
1H RMN (300 MHz, CDCh): 55,46-5,22 (m, 9H), 5,18-5,09 (m, 1H), 3,66 (dd, J = 5,6, 1,3, 2H), 2,77 (t, J = 5,8, 4H), 2,49-2,26 (m, 6 H), 2,22 (s, 6 H), 2,05 (app q, J = 13,0, 6 ,6 , 8 H), 1,70-1,47 (m, 6 H), 1,44-1,19 (m, 27H), 0,94-0,84 (m, 6 H), 0,87 (s, 9H), 0,03 (s, 6 H);
13C RMN (300 MHz, CDCh): 5173,0, 172,9, 130,2, 130,0, 128,0, 127,8, 72,9, 69,0, 61,3, 59,3, 45,9, 34,33,
34,28, 31,5, 29,6, 29,3, 29,21,29,19, 29,12, 29,07, 27,2, 25,7, 25,6, 25,0, 22,5, 18,1, 14,0, -5,6.
COMPUESTO 10: (2S,3S)-2,3-dilinoleoil-4-dimetilaminobutano-1,2,3-triol:
Se añadió una solución pura de HF-piridina (87 |jl de HF al 70 % en piridina, 0,70 mmol, 3,00 equiv.) a una solución agitada de THF a -10 °C (1,2 mL) que contenía piridina (57 jl, 0,70 mmol, 3,00 equiv.) y éter silílico 9 (185 mg, 0,23 mmol) en un matraz de base redonda bajo argón. Después de 20 min, la mezcla de reacción se transfirió a un baño de hielo, se agitó durante 2 h más y se apagó con NaHCO3 al 5 % ac. La mezcla se extrajo con EtOAc (3*5 mL), después los extractos orgánicos combinados se lavaron con salmuera, se secaron sobre Na2SO4 y se concentraron en un evaporador rotativo para obtener un aceite amarillo pálido como alcohol primario deseado 10 (rendimiento cuantitativo) que se utilizó inmediatamente sin purificación adicional. Cabe señalar que se observó la transesterificación completa al alcohol primario (1H RMN, TLC) en ≤24 h cuando se almacenó puro a -20 °C.
ESQUEMA 5: Esquema para la síntesis del lípido 13, 4-linoleoiloxibutano-1-tiol.

Se añadió una solución de 1,4-butanodiol en DMF (1 mL) (135 mg, 1,50 mmol, 1,20 equiv.) a una suspensión de NaH (80 mg, 2,50 mmol, 1,50 equiv.; previamente lavada con hexanos, 2 x 3 mL) en un matraz de base redonda bajo argón. A continuación se retiró el baño frío y la mezcla se agitó durante 30 min. Después se enfrió de nuevo en un baño de hielo y se añadió una solución de DMF (1 mL) de metanosulfonato de linoleilo (345 mg, 1,00 mmol), dejando que la mezcla se calentara durante 15 h. La mezcla de reacción se apagó añadiendo agua (10 mL), se extrajo con Et2O (3*5 mL), después los extractos combinados se lavaron con agua (3*5 mL), se secaron sobre Na2SO4 y se concentraron en un evaporador rotativo para obtener el crudo como un aceite amarillo pálido. La RMN de 1H puso de manifiesto que se había producido cierta isomerización de olefinas (~25 %) durante la reacción y los intentos de mitigarla o eliminarla no tuvieron éxito. El crudo se purificó mediante cromatografía en columna flash (50 mL SiO2, 80:20 hexanos/EtOAc) para obtener un aceite claro e incoloro como éter deseado 11 (276 mg, 82 % de rendimiento, mezcla ~3:1 de deseado/isomerizado).
1H RMN (300 MHz, CDCls): 8 5,49-5,22 (m, 4H), 3,65 (t, J = 5,7, 2H), 3,5-3,38 (m, 4H), 2,79 (t, J = 5,9, 2H), 2,06 (q, J = 6,4, 4H), 1,76-1,62 (m, 4H), 1,65-1,51 (m, 2H), 1,46-1,18 (m, 17H), 0,90 (t, J = 6,5, 3H);
13C RMN (300 MHz, CDCla): 8 130,15, 130,10, 127,93, 127,90, 71,2, 70,8, 62,7, 31,5, 30,4, 29,62, 29,59, 29,43, 29,41,29,3, 29,23, 29,18, 27,19, 27,16, 27,0, 26,1,25,6, 22,5, 14,0.
COMPUESTO 12: S-(4-linoleiloxibutil)tioacetato
Se añadió azodicarboxilato de diisopropilo puro (0,59 mL, 3,00 mmol, 2,00 equiv.) a una solución agitada y helada de THF ( 6 mL) de Ph3P (787 mg, 3,00 mmol, 2,00 equiv.) en un matraz de base redonda bajo argón, lo que dio lugar a la formación de un precipitado blanquecino, y el resultante se agitó durante 1 h. A continuación, se añadió una solución de THF (2 mL) de ácido tioacético (0,21 mL, 3,00 mmol, 2,00 equiv.) y alcohol S10 (508 mg, 1,50 mmol) a la mezcla helada anterior, que se dejó calentar hasta temperatura ambiente. Tras 16 h, se eliminaron los volátiles en un evaporador rotativo. El residuo se resuspendió en 80:20 hexanos/EtOAc, se agitó durante 15 min, momento en donde los sólidos blancos se eliminaron por filtración a través de Celite y el filtrado se concentró en un evaporador rotativo para obtener el crudo como un aceite amarillo. El crudo se purificó mediante cromatografía en columna flash (80 mL SiO2, 99:1^-95:5 hexanos/EtOAc) para obtener un aceite claro e incoloro como el tioacetato 12 deseado (549 mg, 92 % de rendimiento).
1H RMN (300 MHz, CDCls): 8 5,47-5,24 (m, 4H), 3,49-3,12 (m, 4H), 2,98-2,86 (m, 2H), 2,78 (t, J = 6,1, 2H), 2,33 (s, 3H), 2,06 (q, J = 6,3, 4H), 1,75-1,61 (m, 4H), 1,63-1,48 (m, 2H), 1,46-1,18 (m, 17H), 0,90 (t, J = 6,5, 3H).
COMPUESTO 13: 4-(linoleiloxi)butano-1-tiol
Se añadió gota a gota una solución de THF (1 mL) de tioacetato 12 (119 mg, 0,30 mmol) a una suspensión agitada y helada de THF (2 mL) de LiAlH4 (46 mg, 1,20 mmol, 4,00 equiv.) en un matraz de base redonda bajo argón, que se dejó calentar con el tiempo. Después de 4,5 h, la reacción se enfrió en un baño de hielo, se diluyó con Et2O y se apagó con HCl 1 M acuoso (3 mL). La capa acuosa se extrajo con Et2O (2*5 mL), después los extractos combinados se lavaron con salmuera (1 x 5 mL), se secaron sobre Na2SO4 y se concentraron en un evaporador rotativo para obtener un aceite claro y coloreado como el tiol 13 deseado (101 mg, 95 % de rendimiento) que se utilizó inmediatamente y sin purificación adicional.
ESQUEMA 6: Esquema para la síntesis del lípido 16, hemisuccinato de oleilo.

Se añadieron anhídrido succínico sólido (200 mg, 2,00 mmol, 2,00 equiv.) y DMAP (305 mg, 2,50 mmol, 2,50 equiv.) a una solución de alcohol palmítico (268 mg, 1,00 mmol) agitada en CH2Cl2 (5 mL) a temperatura ambiente en un matraz de base redonda bajo argón. Después de 14 h, la mezcla de reacción se diluyó con CH2Ch, se lavó con HCl 1 M acuoso (2*10 mL), se secó sobre Na2SO4 y se concentró en un evaporador rotativo para obtener el hemisuccinato 16 (385 mg, rendimiento cuantitativo) como un semisólido blanco que se utilizó sin purificación adicional. Rf 0,22 (SiO2, 70:30 hexanos/EtOAc).
ESQUEMA 7: Esquema para la síntesis del lípido 17, hemisuccinato de palmitilo.

Se añadieron anhídrido succínico sólido (200 mg, 2,00 mmol, 2,00 equiv.) y DMAP (305 mg, 2,50 mmol, 2,50 equiv.) a una solución de alcohol palmítico (242 mg, 1,00 mmol) agitada en CH2Cl2 (5 mL) a temperatura ambiente en un matraz de base redonda bajo argón. Después de 14 h, la mezcla de reacción se diluyó con CH2Ch, se lavó con HCl 1 M acuoso (2*10 mL), se secó sobre Na2SO4 y se concentró en un evaporador rotativo para obtener el hemisuccinato 17 (355 mg, rendimiento cuantitativo) como un semisólido blanco que se utilizó sin purificación adicional. Rf 0,15 (SO2, 70:30 hexanos/EtOAc).
ESQUEMA 8: Esquema para la síntesis del lípido 19, hemisuccinato de 2,3-dimiristoilglicerol.

Se añadió DCC (1,36 g, 6,60 mmol, 2,20 equiv.) a una suspensión helada de CH2Cl2 (15 mL) de 1 -(terbutil)dimetilsililglicerol (619 mg, 3,00 mmol) y ácido mirístico (1,51 g, 6,60 mmol, 2,20 equiv.) en un matraz de base redonda bajo argón, seguido de DMAP (806 mg, 6,60 mmol, 2,20 equiv.) y se dejó que la mezcla de reacción se calentara a temperatura ambiente durante 14 h. La mezcla de reacción se diluyó con EtOAc, se filtró a través de Celite y se concentró en un evaporador rotativo. 60 mmol, 2,20 equiv.) y la mezcla de reacción se dejó calentar a temperatura ambiente durante 14 h. La mezcla de reacción se diluyó con EtOAc, se filtró a través de Celite y se concentró en un evaporador rotativo. El crudo se suspendió en 80:20 hexanos/EtOAc y se filtró a través de SO2 (40 mL) para obtener un aceite claro e incoloro como diéster 18 (1,91 g, rendimiento cuantitativo).
Rf 0,79 (SiO2 , 80:20 hexanos/EtOAc); 1H RMN (300 MHz, CDCla): 8 5,13-5,04 (m, 1H), 4,36 (dd, J = 11,8, 3,8 Hz, 1H), 4,17 (dd, J = 11,9, 6,2 Hz, 1H), 3,73 (d, J = 5,5 Hz, 2H), 2,32 (t, J = 7,3 Hz, 4H), 1,69-1,56 (m, 4H), 1,27 (br s, 40H), 0,90 (br s, 15H), 0,07 (s, 6 H).
COMPUESTO 19: hemisuccinato de 2,3-dimiristoilglicerol
Se añadió una solución pura de HF-piridina (1,13 mL de HF al 70 % en piridina, 9,14 mmol, 3,00 equiv.) a una solución de THF (12 mL) enfriada con hielo y en agitación que contenía piridina (0,74 mL, 9,14 mmol, 3,00 equiv.) y
silil éter 18 (1,91 g, 3,04 mmol) en un matraz de base redonda bajo argón, y se dejó calentar hasta temperatura ambiente. Después de 5 h, la mezcla de reacción se neutralizó con NaHCO3 acuoso saturado y se extrajo con Et2O (3 x 25 mL). Los extractos orgánicos combinados se lavaron con H2O (1 x 25 mL), salmuera (1 x 25 mL), se secaron sobre Na2SO4 y se concentraron en un evaporador rotativo. El crudo se purificó por cromatografía flash en columna (100 mL SiO2 , 80:20 hexanos/EtOAc) para obtener un sólido blanco como alcohol intermedio (1,31 g, 84 % de rendimiento).
Rf = 0,26 (SiO2, 80:20 hexanos/EtOAc);
1H RMN (300 MHz, CDCla): 8 5,10 (quin, J = 5,0 Hz, 1H), 4,34 (dd, J = 11,9, 4,6 Hz, 1H), 4,25 (dd, J = 11,9, 5,6 Hz, 1H), 3,74 (t, J = 5,4 Hz, 2H), 2,35 (q, J = 7,3 Hz, 4H), 2,10 (br s, 1H), 1,63 (br s, 4H), 1,27 (br s, 40H), 0,90 (br t, J = 6,3 Hz, 6 H).
Una porción del alcohol anterior (513 mg, 1,00 mmol) se disolvió en CH2Cl2 (3 mL) en un matraz de base redonda bajo argón y se enfrió en un baño de hielo. Se añadieron secuencialmente anhídrido succínico (200 mg, 2,00 mmol, 2,00 equiv.) y DMAP (305 mg, 2,50 mmol, 2,50 equiv.) y la mezcla se dejó calentar a temperatura ambiente durante 14 h. La mezcla de reacción se diluyó con CH2Cl2, se lavó con HCl 1 M acuoso 1 M (3 □ 10 mL), se secó sobre Na2SO4 y se concentró en un evaporador rotativo para obtener un sólido blanco como hemisuccinato 19 (608 mg, rendimiento cuantitativo) que se utilizó sin purificación adicional.
Rf = 0,18 (SiO2, 50:50 hexanos/EtOAc).
ESQUEMA 9: Esquema para la síntesis del lípido 21, hemisuccinato de 2,3-dioleoil glicerol.

COMPUESTO 20: 1-(ter-butM)dimetilsiMl-2,3-dioleoM glicerol
Se añadió DCC (1,36 g, 6,60 mmol, 2,20 equiv.) a una solución helada de CH2Cl2 (10 mL) de 1-(terbutil)dimetilsililglicerol (619 mg, 3,00 mmol) y ácido oleico (1,86 g, 6,60 mmol, 2,20 equiv.) en un matraz de base redonda bajo argón, seguido de DMAP (806 mg, 6,60 mmol, 2,20 equiv.) y la mezcla de reacción se dejó calentar a temperatura ambiente durante 14 h. La mezcla de reacción se diluyó con Et2O, se filtró a través de Celite y se concentró en un evaporador rotativo. 60 mmol, 2 , 2 0 equiv.) y la mezcla de reacción se dejó calentar a temperatura ambiente durante 14 h. La mezcla de reacción se diluyó con Et2O, se filtró a través de Celite y se concentró en un evaporador rotativo. El crudo se suspendió en 95:5 hexanos/EtOAc y se filtró a través de SiO2 (40 mL) para obtener un aceite claro e incoloro como diéster 20 (2,04 g, 93 % de rendimiento).
Rf 0,84 (SiO2, 80:20 hexanos/EtOAc); 1H RMN (300 MHz, CDCla): 8 5,44-5,28 (m, 4H), 5,14-5,03 (m, 1H), 4,36 (dd, J = 11,9, 3,8 Hz, 1H), 4,17 (dd, J = 11,9, 6,3 Hz, 1H), 3,73 (d, J = 5,4 Hz, 2H), 2,32 (t, J = 7,7 Hz, 4H), 2,11-1,93 (m, 8 H), 1,71-1,56 (m, 4H), 1,42-1,22 (m, 40H), 0,90 (br s, 15H), 0,07 (s, 6 H).
COMPUESTO 21: hemisuccinato de 2,3-dioleoil glicerol
Se añadió una solución pura de HF-piridina (0,72 mL de HF al 70 % en piridina, 5,05 mmol, 3,00 equiv.) a una solución de THF ( 8 mL) helada y en agitación que contenía piridina (0,41 mL, 5,05 mmol, 3,00 equiv.) y éter silílico 20 (1,24 g, 1,68 mmol) en un matraz de base redonda bajo argón, y se dejó calentar a temperatura ambiente. Después de 4 h, la mezcla de reacción se neutralizó con NaHCO3 acuoso saturado y se extrajo con Et2O (3*10 mL). Los extractos orgánicos combinados se lavaron con H2O (1*25 mL), salmuera (1*25 mL), se secaron sobre Na2SO4 y se concentraron en un evaporador rotativo para obtener un aceite claro e incoloro como alcohol intermedio (1,08 g, rendimiento cuantitativo).
Rf = 0,39 (SiO2, 80:20 hexanos/EtOAc);
1H RMN (300 MHz, CDCh): 8 5,47-5,27 (m, 4H), 5,10 (quin, J = 5,0 Hz, 1H), 4,34 (dd, J = 12,0, 4,7 Hz, 1H), 4,25 (dd, J = 11,9, 5,6 Hz, 1H), 3,75 (t, J = 5,0 Hz, 2H), 2,35 (q, J = 7,4 Hz, 4H), 2,12-1,93 (m, 8 H), 1,73 1,52 (m, 4H), 1,43-1,19 (m, 40H), 0,90 (br t, J = 7,1 Hz, 6 H).
Una porción del alcohol anterior (714 mg, 1,15 mmol) se disolvió en CH2Cl2 ( 6 mL) en un matraz de base redonda bajo argón y se enfrió en un baño de hielo. Se añadieron secuencialmente anhídrido succínico (230 mg, 2,30 mmol, 2,00 equiv.) y DMAP (351 mg, 2,87 mmol, 2,50 equiv.) y la mezcla se dejó calentar a temperatura ambiente durante 14 h. La mezcla de reacción se diluyó con CH2O2, se lavó con HCl 1 M acuoso 1 M (3 x 10 mL), se secó sobre
Na2SO4 y se concentró en un evaporador rotativo para obtener un aceite claro e incoloro como hemisuccinato 21 (696 mg, 84 % de rendimiento) que se utilizó sin purificación adicional.
Rf = 0,22 (SiO2, 50:50 hexanos/EtOAc).
Procedimientos para la síntesis de conjugados de fármacos
ESQUEMA 10: Esquema para la síntesis de LD01-DEX, dexametasona-21-[4-(3-(dimetilamino)-2-((linoleoiloxi)propoxi)-4-oxobutanoato.

LD01-DEX: Dexametasona 21-[4-(3-(dimetilamino)-2-((linoleoiloxi)propoxi)-4-oxobutanoato]:
Se añadió dexametasona 21-hemisuccinatoi sólida (45 mg, 0,18 mmol, 0,50 equiv.) a una solución de DMF (1 mL) a temperatura ambiente de alcohol primario 3 (68 mg, 0,18 mmol, 2. 00 equiv.) y Et 3 N (38 j L, 0,27 mmol, 1,50 equiv.) en un matraz de base redonda bajo argón, seguido de yoduro de 2-cloro-1-metilpiridinio (28 mg, 0,11 mmol, 0,60 equiv.). Después de 14 h, la mezcla se apagó con NaHCO 3 al 5 %. La mezcla se extrajo con EtOAc (2*3 mL), después los extractos orgánicos combinados se lavaron con NaHCO 3 al 5 % acuoso (1*3 mL), salmuera, se secaron sobre Na 2 SO 4 y se concentraron en un evaporador rotativo para obtener el crudo como un aceite amarillo. El crudo se purificó mediante cromatografía en columna flash (20 mL SiO 2 , 97:3 CHCl 3 /MeOH) para obtener un aceite claro e incoloro como el conjugado deseado LD01-DEX (72 mg, 94 % de rendimiento).
Rf = 0,48 (SiO 2 , 90:10 CHCls/MeOH);
1
H RMN (300 MHz, CDCl
3
): 87,20 (d, J = 10,2 Hz, 1H), 6,31 (d, J = 10,2 Hz, 1H), 6,09 (s, 1H), 5,48-5,25 (m, 4H), 5,24-5,12 (m, 1H), 4,96 (dd, J = 12,0, 6,6 Hz, 1H), 4,83 (dd, J = 12,0, 6,6 Hz, 1H), 4,48-4,24 (m, 2H), 4,22-3,97 (m, 1H), 3,08 (m, 2H), 2,85-2,70 (m, 4H), 2,75-2,40 (m, 6H), 2,44-2,18 (m, 12H), 2,20-1,93 (m, 6H), 1,90-1,45 (m, 7H), 1,53 (s, 3H), 1,43-1,24 (m, 18H), 1,02 (s, 3H), 0,93-0,80 (m, 6H).
ESQUEMA 11: esquema para la síntesis de LD02-DEX, 2,3-dilinoleoiloxipropil (2-((8S,9R,10S,11 S,13S,14S,16R,17R)-9-fluoro-11,17-dihidroxi-10,13,16-trimetil-3-oxo-6,7,8,9,10,11,12,13,14,15,16,17-dodecahidro-3H-ciclopenta[a]fenantren-17-il)-2-oxoetil)succinato.

LD02-DEX: 2,3-dilinoleoiloxipropil (2-((8S,9R,10S,11S,13S,14S,16R,17R)-9-fluoro-11,17-dihidroxi-10,13,16-trimetil-3-oxo-6,7,8,9,10,11,12,13,14,15,16,17-dodecahidro-3H-ciclopenta[a]fenantren-17-il)-2-oxoetil)succinato:
Se añadió DCC sólido (28 mg, 0,13 mmol, 1,10 equiv.) a una solución agitada a temperatura ambiente de CH2Cl2 (1,5 mL) del hemisuccinato 4 (105 mg, 0,15 mmol, 1,10 equiv.) en un matraz de base redonda bajo argón. Después de agitar durante 5 min, se añadieron dexametasona sólida (52 mg, 0,13 mmol) y DMAP (24 mg, 0,20 mmol, 1,50 equiv.). La mezcla de reacción se dejó agitar durante 14 h, se diluyó con CH2O2, se filtró a través de Celite y el filtrado se concentró para obtener el crudo como un aceite amarillo pálido. El
crudo se purificó mediante cromatografía en columna flash (25 mL SÍO2, 80:20^70:30^-50:50 hexanos/EtOAc) para obtener un aceite claro e incoloro como el conjugado deseado LD02-DEX (67 mg, 46 % de rendimiento).
1H RMN (300 MHz, CDCh): 87,22 (dd, J = 10,2, 3,9, 1H), 6,32 (dd, J = 10,2, 1,7, 1H), 6,1 (s, 1H), 5,44-5,17 (m, 9H), 5,00-4,81 (m, 2H), 4,43-4,22 (m, 4H), 4,21-4,06 (m, 2H), 3,16-3,01 (m, 1H), 2,84-2,51 (m, 11H), 2,50-2,23 (m, 9H), 2,21-1,48 (m, 25H), 1,45-1,15 (m, 34H), 1,14-1,00 (m, 1H), 1,03 (s, 3H), 0,95-0,81 (m, 10H).
ESQUEMA 12: esquema para la síntesis de LD03-DEX, linoleil 2-((8S,9R,10S,11S,13S,14S,16R,17R)-9-fluoro-11,17-dihidroxi-10,13,16-trimetil-3-oxo-6,7,8,9,10,11,12,13,14,15,16,17-dodecahidro-3H-ciclopenta[a]fenantren-17-il)-2-oxoetil succinato.

LD03-DEX: linoleil 2-((8S,9R,10S,11S,13S,14S,16R,17R)-9-fluoro-11,17-dihidroxi-10,13,16-trimetil-3-oxo-6.7.8.9.10.11.12.13.14.15.16.17- dodecahidro-3H-ciclopenta[a]fenantren-17-il)-2-oxoetil succinato:
Se añadió DCC sólido (37 mg, 0,18 mmol, 1,10 equiv.) a una solución agitada a temperatura ambiente de CH2Cl2 (1,6 mL) de hemisuccinato 5 (65 mg, 0,18 mmol, 1,10 equiv.) en un matraz de base redonda bajo argón. Después de agitar durante 5 min, se añadieron dexametasona sólida (63 mg, 0,16 mmol) y DMAP (30 mg, 0,24 mmol, 1,50 equiv.). La mezcla de reacción se dejó agitar durante 14 h, se diluyó con CH2O2, se filtró a través de Celite y el filtrado se concentró para obtener el crudo como un aceite claro e incoloro. El crudo se purificó mediante cromatografía en columna flash (25 mL SO2, 70:30^60:40 hexanos/EtOAc) para obtener un aceite claro e incoloro como el conjugado deseado LD03-DEX (76 mg, 64 % de rendimiento).
1H RMN (300 MHz, CDCla): 87,25 (d, J = 10,2, 1H), 6,33 (d, J = 10,1, 1H), 6,1 (s, 1H), 5,45-5,25 (m, 4H), 4,93 (s, 2H), 4,45-4,27 (m, 1H), 4,08 (t, J = 6,7, 2H), 3,17-3,01 (m, 1H), 2,92 (s, 1H), 2,77 (t, J = 6,5, 4H), 2,72-2,52 (m, 4H), 2,50-2,24 (m, 4H), 2,23-2,07 (m, 1H), 2,05 (q, J = 6,6, 4H), 1,97-1,45 (m, 10H), 1,44-1,01 (m, 20H), 1,02 (s, 3H), 0,97-0,80 (m, 6H).
ESQUEMA 13: esquema para la síntesis de LD06-DEX, (2S,3S)-4-(dimetilamino)-2,3-bis(linoleoiloxi)butil (2-((8S,9R,10S,11S,13S,14S,16R,17R)-9-fluoro-11,17-dihidroxi-10,13,16-trimetil-3-oxo-6.7.8.9.10.11.12.13.14.15.16.17- dodecahidro-3H-ciclopenta[a]fenantren-17-il)-2-oxoetil)succinato.

LD06-DEX: (2S,3S)-4-(dimetilamino)-2,3-bis(linoleoiloxi)butil (2-((8S,9R,10S,11S,13S,14S,16R,17R)-9-fluoro-11,17-dihidroxi-10,13,16-trimetil-3-oxo-6,7,8,9,10,11,12,13,14,15,16,17-dodecahidro-3H-ciclopenta[a]fenantren-17-il)-2-oxoetil)succinato:
Se añadió dexametasona-21-hemisuccinato sólido [Mao et al. (2012)] (88 mg, 0,18 mmol, 1,10 equiv.) a una solución de DCC (37 mg, 0,18 mmol, 1,10 equiv.) en THF (0,8 mL) a temperatura ambiente en un matraz de base redonda bajo argón. Después de 10 min, la mezcla se enfrió en un baño de hielo, se añadió una solución de THF (0,8 mL) de alcohol 10 (109 mg, 0,16 mmol), DMAP (22 mg, 0,18 mmol, 1,10 equiv.) y EtaN (25 uL, 0,18 mmol, 1,10 equiv.) y la mezcla se dejó calentar durante la noche. Después de 20 h, la mezcla de reacción se diluyó con THF, se filtró a través de Celite y se concentró en un evaporador rotativo para obtener el crudo como un semisólido amarillo pálido. El crudo se purificó mediante cromatografía en
columna flash (50 mL SÍO
2
, 36:60:4 Et
2
O/hexanos/MeOH) para obtener un aceite claro e incoloro como el conjugado deseado LD06-DEX (103 mg, 55 % de rendimiento).
1
H RMN (300 MHz, CDCl
3
): 87,24 (d, J = 10,1, 1H), 6,34 (d, J = 10,1, 1H), 6,12 (s, 1H), 5,47-5,26 (m, 10H), 5,26 5,15 (m, 1H), 5,01 (d, J = 17,5, 1H), 4,86 (d, J = 17,5, 1H), 4,43-4,27 (m, 2H), 4,20-4,08 (m, 1H), 3,20-3,02 (m, 1H), 2,78 (app t, J = 5,6, 7H), 2,70-2,60 (m, 2H), 2,60-2,5 (m, 18H), 2,24 (s, 6H), 2,18-2,03 (m, 3H), 2,06 (app q, J = 13,1, 6,7, 8H), 1,90-1,49 (m, 13H), 1,47-1,16 (m, 39H), 1,05 (s, 3H), 0,90 (app t, J = 6,7, 11H).
ESQUEMA 14: esquema para la síntesis de LD07-DEX, (8S,9R,10S,11S,13S,14S,16R,17R)-9-fluoro-11,17-dihidroxi-10,13,16-trimetil-17-(2-((4-(linoleiloxi)butil)tio)acetil)-6,7,8,9,10,11,12,13,14,15,16,17-dodecahidro-3H-ciclopenta[a]fenantren-3-ona.

LD07-DEX: (8S,9R,10S,11S,13S,14S,16R,17R)-9-fluoro-11,17-dihidroxi-10,13,16-trimetil-17-(2-((4-(linoleiloxi)butil)tio)acetil)-6,7,8,9,10,11,12,13,14,15,16,17-dodecahidro-3H-ciclopenta[a]fenantren-3-ona:
Se añadió una solución de THF (0,6 mL) de tiol 13 (66 mg, 0,19 mmol, 1,90 equiv.) a una solución de dexametasona-21-bromuro [López y Simons (1991)] (48 mg, 0,10 mmol) en THF (0,4 mL) agitada y enfriada con hielo en un matraz de base redonda bajo argón. Después de 8 h, la mezcla de reacción se diluyó con THF, se filtró a través de Celite y se concentró en un evaporador rotativo para obtener el crudo como un aceite amarillo. El crudo se purificó mediante cromatografía en columna flash (25 mL SO2, 80:20^-65:35 hexanos/EtOAc) para obtener un aceite claro e incoloro como el sulfuro deseado LD07-DEX (48 mg, 62 % de rendimiento).
1H RMN (300 MHz, CDCl3): 87,18 (d, J = 10,2, 1H), 6,32 (dd, J = 10,1, 1,7, 1H), 6,11 (s, 1H), 5,47-5,23 (m, 4H), 4,34 (br d, J = 10,0, 1H), 3,52 (d, J = 13,4, 1H), 3,48-3,29 (m, 5H), 3,18 (d, J = 13,2, 1H), 3,14-2,99 (m, 1H), 2,77 (t, J = 6,0, 2H), 2,65 (s, 1H), 2,69-2,19 (m, 8H), 2,04 (q, J = 6,5, 4H), 1,90-1,46 (m, 17H), 1,46-1,15 (m, 23H), 1,05 (s, 3H), 0,95-0,82 (m, 7H).
ESQUEMA 15: esquema para la síntesis de LD13-DEX, 2-((8S,9R,10S,11S,13S,14S,16R,17R)-9-fluoro-11,17-dihidroxi-10,13,16-trimetil-3-oxo-6,7,8,9,10,11,12,13,14,15,16,17-dodecahidro-3H-ciclopenta[a]fenantren-17-il)-2-oxoetil succinato de oleilo.
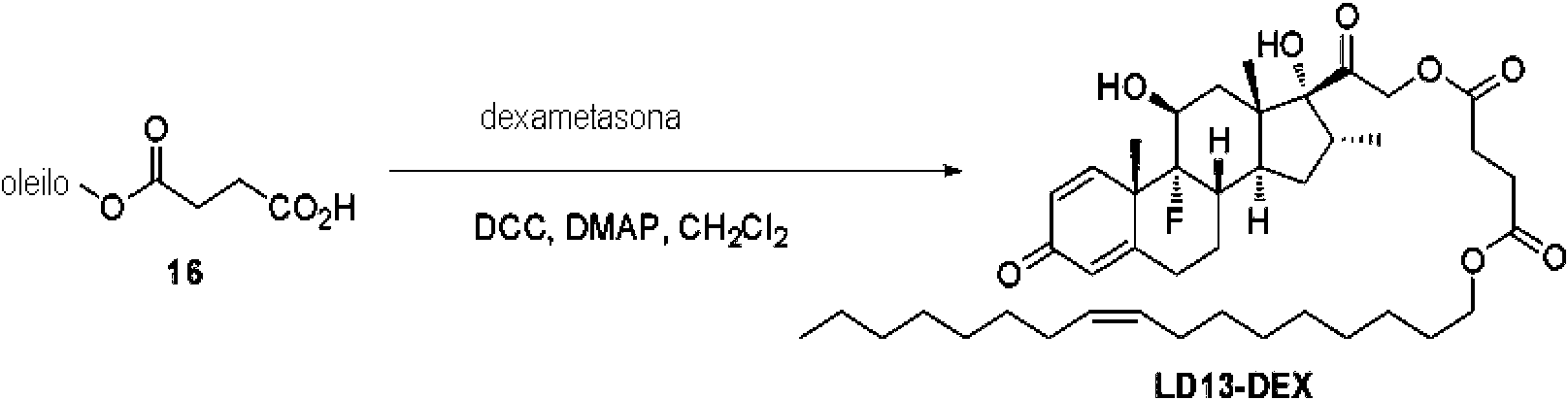
LD13-DEX: 2-((8S,9R,10S,11S,13S,14S,16R,17R)-9-fluoro-11,17-dihidroxi-10,13,16-trimetil-3-oxo-6,7,8,9,10,11,12,13,14,15,16,17-dodecahidro-3H-ciclopenta[a]fenantren-17-il)-2-oxoetil succinato de oleilo:
Se añadió DCC sólido (25 mg, 0,12 mmol, 1,20 equiv.) a una solución agitada de CH
2
Cl
2
(1 mL) a temperatura ambiente de hemisuccinato 16 (44 mg, 0,12 mmol, 1,20 equiv.) en un matraz de base redonda bajo argón. Después de agitar durante 5 min, se añadieron dexametasona sólida (39 mg, 0,10 mmol) y DMAP (18 mg, 0,15 mmol, 1,50 equiv.). La mezcla de reacción se dejó agitar durante 14 h, se diluyó con CH
2
Cl
2
, se filtró a través de Celite y el filtrado se concentró para obtener el crudo como un aceite claro e incoloro. El crudo se purificó mediante cromatografía en columna flash (20 mL SO
2
, 60:40^50:50 hexanos/EtOAc) para obtener un aceite claro e incoloro como el conjugado deseado LD13-DEX (50 mg, 67 % de rendimiento).
1H RMN (300 MHz, CDCI3): 57,22 (d, J = 9,7 Hz, 1H), 6,31 (dd, J = 10,2, 1,8 Hz, 1H), 6,08 (s, 1H), 5,40-5,25 (m, 2H), 4,99-4,83 (m, 2H), 4,41-4,27 (m, 1H), 4,06 (t, J = 6,8 Hz, 2H), 3,16-2,99 (m, 1H), 2,82 (br s, 1H), 2,79-2,69 (m, 2H), 2,68-2,58 (m, 3H), 2,49-2,24 (m, 4H), 2,22-2,06 (m, 1H), 2,06-1,92 (m, 4H), 1,94-1,46 (m, 10H), 1,40-1,08 (m, 22H), 1,01 (s, 3H), 0,95-0,78 (m, 6H).
ESQUEMA 16: esquema para la síntesis de LD14-DEX, 2-((8S,9R,10S,11S,13S,14S,16R,17R)-9-fluoro-11,17-dihidroxi-10,13,16-trimetil-3-oxo-6,7,8,9,10,11,12,13,14,15,16,17-dodecahidro-3H-ciclopenta[a]fenantren-17-il)-2-oxoetil succinato de palmitilo.
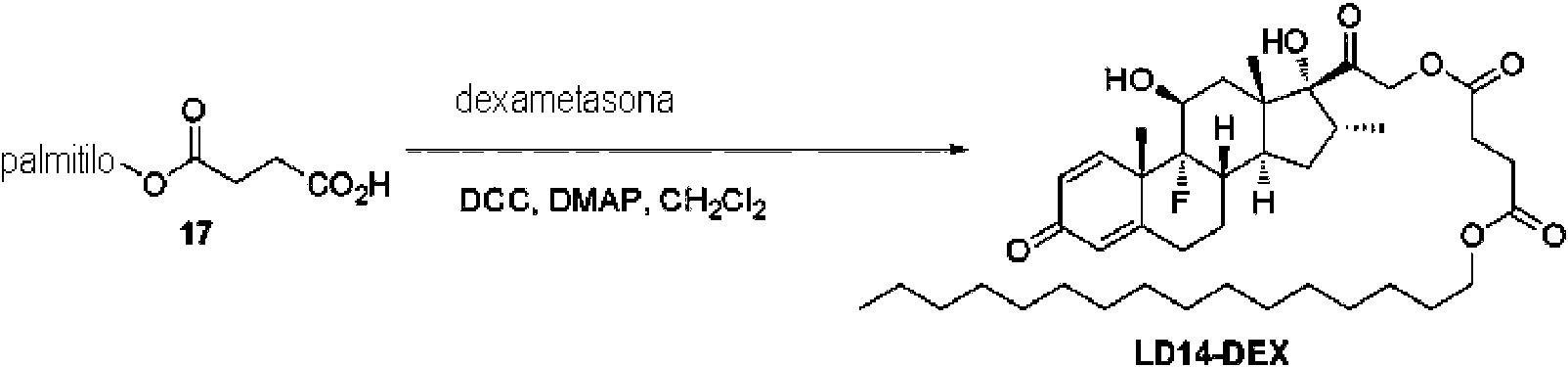
LD14-DEX: 2-((8S,9R,10S,11S,13S,14S,16R,17R)-9-fluoro-11,17-dihidroxi-10,13,16-trimetil-3-oxo-6,7,8,9,10,11,12,13,14,15,16,17-dodecahidro-3H-ciclopenta[a]fenantren-17-il)-2-oxoetil succinato de palmitilo:
Se añadió DCC sólido (25 mg, 0,12 mmol, 1,20 equiv.) a una solución agitada de CH
2
Cl
2
(1 mL) a temperatura ambiente de hemisuccinato 16 (41 mg, 0,12 mmol, 1,20 equiv.) en un matraz de base redonda bajo argón. Después de agitar durante 5 min, se añadieron dexametasona sólida (39 mg, 0,10 mmol) y DMAP (18 mg, 0,15 mmol, 1,50 equiv.). La mezcla de reacción se dejó agitar durante 14 h, se diluyó con CH
2
Cl
2
, se filtró a través de Celite y el filtrado se concentró para obtener el crudo como un aceite claro e incoloro. El crudo se purificó mediante cromatografía en columna flash (20 mL SO
2
, 60:40^50:50 hexanos/EtOAc) para obtener un aceite claro e incoloro como el conjugado deseado LD14-DEX (43 mg, 60 % de rendimiento).
1 H RMN (300 MHz, CDCla): 57,22 (d, J = 10,0 Hz, 1H), 6,31 (dd, J = 10,2, 1,8 Hz, 1H), 6,08 (s, 1H), 5,00-4,83 (m, 2H), 4,38-4,26 (m, 1H), 4,06 (t, J = 6,8 Hz, 2H), 3,16-3,00 (m, 1H), 2,84 (br s, 1H), 2,80-2,70 (m, 2H), 2,69-2,53 (m, 3H), 2,48 (br s, 1H), 2,42-2,24 (m, 3H), 2,22-2,06 (m, 1H), 1,95-1,42 (m, 10H), 1,39-1,17 (m, 26H), 1,16-1,02 (m, 2H), 1,01 (s, 3H), 0,95-0,80 (m, 6H).
ESQUEMA 17: esquema para la síntesis de LD17-DEX, (2-((8S,9R,10S,11S,13S,14S,16R,17R)-9-fluoro-11,17-dihidroxi-10,13,16-trimetil-3-oxo-6,7,8,9,10,11,12,13,14,15,16,17-dodecahidro-3H-ciclopenta[a]fenantren-17-il)-2-oxoetil)succinato de 2,3-dimiri stoilpropilo.

LD17-DEX: (2-((8S,9R,10S,11S,13S,14S,16R,17R)-9-fluoro-11,17-dihidroxi-10,13,16-trimetil-3-oxo-6,7,8,9,10,11,12,13,14,15,16,17-dodecahidro-3H-ciclopenta[a]fenantren-17-il)-2-oxoetil)succinato de 2,3-dimiristroilpropilo:
Se añadió Et
3
N (53 pL, 0,20 mmol, 2,50 equiv.), seguido del reactivo de Mukaiyama (51 mg, 0,20 mmol, 1,30 equiv.), a una solución helada de CH
2
Cl
2
(0,7 mL) de dexametasona (76 mg, 0,15 mmol) y hemisuccinato 19 (92 mg, 0,18 mmol, 1,20 equiv.) en un matraz de base redonda seco bajo argón. La mezcla de reacción se dejó calentar durante 14 h, después se diluyó con EtOAc, se lavó con NaHCO
3
al 5 % acuoso (1*5 mL), H
2
O (2*5 mL), salmuera (1*5 mL), se secó sobre Na
2
SO
4
y se concentró en un evaporador rotativo. El crudo se purificó mediante cromatografía en columna flash (20 mL SO
2
, 80:20^50:50 hexanos/EtOAc) para obtener un aceite claro e incoloro como conjugado deseado LD17-DEX (72 mg, 47 % de rendimiento).
Rf = 0,45 (SO 2 , 50:50 hexanos/EtOAc);
1H RMN (300 MHz, CDCI3): 57,23 (d, J = 10,0 Hz, 1H), 6,33 (dd, J = 10,2, 1,8 Hz, 1H), 6,11 (s, 1H), 5,30 5,17 (m, 1H), 5,07-4,83 (m, 2H), 4,45-4,08 (m, 5H), 3,19-3,03 (m, 1H), 2,85-2,49 (m, 7H), 2,47-2,25 (m, 7H), 2,23-2,07 (m, 1H), 1,96-1,54 (m, 8H), 1,38-1,15 (m, 43H), 1,04 (s, 3H), 0,98-0,79 (m, 9H).
ESQUEMA 18: Esquema para la síntesis de LD19-PDN, (2-((8S,9S,10R,13S,14S,17R)-17-hidroxi-10,13-dimetil-3,11-dioxo-6,7,8,9,10,11,12,13,14,15,16,17-dodecahidro-3H-cidopenta[a]fenantren-17-N)-2-oxoetil)succinato de oleilo.

LD19-PDN: (2-((8S,9S,10R,13S,14S,17R)-17-h¡droxi-10,13-d¡met¡l-3,11-d¡oxo-6,7,8,9,10,11,12,13,14,15,16,17-dodecahidro-3H-cidopenta[a]fenantren-17-il)-2-oxoetil)succinato de oleilo:
Se añadió DCC sólido (52 mg, 0,25 mmol, 1,20 equiv.) a una solución agitada de CH
2
Cl
2
(1 mL) a temperatura ambiente de hemisuccinato 16 (92 mg, 0,25 mmol, 1,20 equiv.) en un matraz de base redonda bajo argón. Después de agitar durante 5 min, se añadieron prednisona sólida (75 mg, 0,21 mmol) y DMAP (38 mg, 0,31 mmol, 1,50 equiv.). La mezcla de reacción se dejó agitar durante 14 h, se diluyó con CH
2
O
2
, se filtró a través de Celite y el filtrado se concentró para obtener el crudo como un aceite amarillo pálido. El crudo se purificó mediante cromatografía en columna flash (25 mL SO
2
, 70:30^50:50 hexanos/EtOAc) para obtener un aceite claro e incoloro como el conjugado deseado LD19-PDN (103 mg, 71 % de rendimiento).
Rf = 0,52 (SO 2 , 75:25 EtOAc/hexanos);
1 H RMN (300 MHz, CDCls): 57,69 (d, J = 10,1 Hz, 1H), 6,19 (dd, J = 10,3, 1,8 Hz, 1H), 6,07 (s, 1H), 5,42 5,25 (m, 2H), 5,07 (d, J = 17,1 Hz, 1H), 4,73 (d, J = 17,5 Hz, 1H), 4,07 (t, J = 6,7 Hz, 2H), 2,88 (d, J = 12,2 Hz, 1H), 2,83-2,69 (m, 3H), 2,69-2,59 (m, 2H), 2,51 (dt, J = 13,1, 4,3 Hz, 1H), 2,44-2,26 (m, 3H), 2,14-1,83 (m, 7H), 1,76-1,55 (m, 4H), 1,43 (s, 3H),1,38-1,16 (m, 25H), 1,16-0,98 (m, 1H), 0,87 (br t, J = 6,6 Hz, 3H), 0,67 (s, 3H).
ESQUEMA 19: Esquema para la síntesis de LD20-PDL, 2-((8S,9S,10R,11S,13S,14S,17R)-11,17-dihidroxi-10,13-d¡met¡l-3-oxo-6,7,8,9,10,11,12,13,14,15,16,17-dodecah¡dro-3H-c¡dopenta[a]fenantren-17-¡l)-2-oxoet¡l succinato de oleilo.

LD20-PDL: 2-((8S,9S,10R,11S,13S,14S,17R)-11,17-d¡h¡drox¡-10,13-d¡met¡l-3-oxo-6,7,8,9,10,11,12,13,14,15,16,17-dodecahidro^H-ciclopenta^fenantren-^-il^-oxoetil succinato de oleilo:
Se añadió DCC sólido (74 mg, 0,36 mmol, 1,20 equiv.) a una solución agitada de CH
2
Cl
2
(2 mL) a temperatura ambiente de hemisuccinato 16 (133 mg, 0,36 mmol, 1,20 equiv.) en un matraz de base redonda bajo argón. Después de agitar durante 5 min, se añadieron prednisolona sólida (75 mg, 0,30 mmol) y DMAP (55 mg, 0,45 mmol, 1,50 equiv.). La mezcla de reacción se dejó agitar durante 14 h, se diluyó con CH
2
Cl
2
, se filtró a través de Celite y el filtrado se concentró para obtener el crudo como un aceite amarillo pálido. El crudo se purificó mediante cromatografía en columna flash (30 mL SO
2
, 70:30^-50:50 hexanos/EtOAc) para obtener un aceite claro e incoloro como el conjugado deseado LD20-PDL (142 mg, 68 % de rendimiento).
Rf = 0,45 (SO 2 , 75:25 EtOAc/hexanos);
1 H RMN (CDCla, 300 MHz): 57,26 (d, J = 10,5 Hz, 1H), 6,27 (dd, J = 10,1, 1,8 Hz, 1H), 6,01 (s, 1H), 5,42 5,28 (m, 2H), 5,02 (d, J = 17,4 Hz, 1H), 4,89 (d, J = 17,3 Hz, 1H), 4,54-4,45 (m, 1H), 4,08 (t, J = 6,9 Hz, 2H), 2,76 (d, J = 6,3 Hz, 2H), 2,73-2,60 (m, 3H), 2,61-2,49 (m, 1H), 2,36-2,27 (m, 1H), 2,20-1,91 (m, 7H), 1,71 1,45 (m, 4H), 1,45 (s, 3H), 1,40-1,19 (m, 25H), 1,19-1,04 (m, 1H), 0,97 (s, 3H), 0,88 (br t, J = 6,6 Hz, 3H).
ESQUEMA 20: Esquema para la síntesis de LD10-DTX, (2R,3S)-1- 2aR,4S,4aS,6R,9S,11S,12S,12aR,12bS)-12bacetoxi-12-(benzoiloxi)-4,6,11-trihidroxi-4a,8,13,13-tetrametil-5-oxo-2a,3,4,4a,5,6,9,10,11,12,12a,12b-dodecahidro-1H-7,11-metanocidodeca[3,4]benzo[1,2-b]oxet-9-il)oxi)-3-((ter-butoxicarbonil)amino)-1-oxo-3-fenilpropan-2-il (2,3-bis((9Z,12Z)-octadeca-9,12-dienoiloxi)propil)succinato.

LD10-DTX: (2R,3S)-1-(((2aR,4S,4aS,6R,9S,11S,12S,12aR,12bS)-12b-acetoxi-12-(benzoiloxi)-4,6,11-trihidroxi-4a,8,13,13-tetrametil-5-oxo-2a,3,4,4a,5,6,9,10,11,12,12a,12b-dodecahidro-1H-7,11-metanociclodeca[3,4]benzo[1,2-b]oxet-9-il)oxi)-3-((ter-butoxicarbonil)amino)-1-oxo-3-fenilpropan-2-il (2,3-bis((9Z,12Z)-octadeca-9,12-dienoiloxi)propil)succinato:
Se añadió EDC-HCl (15 mg, 8,10*10
' 2
mmol, 1,10 equiv.) a una solución de CH
2
Cl
2
(1 mL) a temperatura ambiente de hemisuccinato 4 (58 mg, 8,10*10
' 2
mmol, 1,10 equiv.) e i-P^NEt (19 pL, 0,11 mmol, 1,50 equiv.) en un matraz de base redonda bajo argón. La mezcla se agitó durante 5 min, se enfrió a -10 °C, momento en donde se añadieron docetaxel (59 mg, 7,36 x 10
' 2
mmol) y HOBt (11 mg, 8,10 x 10
' 2
mmol, 1,10 equiv.) y se dejó calentar la mezcla durante 14 h. La mezcla de reacción se diluyó con EtOAc, se lavó con HCl 0,5 M acuoso (2*5 mL), H
2
O (1*5 mL), salmuera (1*5 mL), se secó sobre Na
2
SO
4
y se concentró en un evaporador rotativo. El crudo se purificó por cromatografía en columna flash (25 mL SO
2
, 80:20^-50:50 hexanos/EtOAc) para obtener un aceite claro e incoloro como conjugado deseado LD10-DTX (89 mg, 81 % de rendimiento).
Rf = 0,34 (SO 2 , 50:50 hexanos/EtOAc);
1 H RMN (300 MHz, CDCla): 88,13 (d, J = 7,5 Hz, 2H), 7,67-7,58 (m, 1H), 7,57-7,47 (m, 2H), 7,46-7,36 (m, 2H), 7,36-7,26 (m, 3H), 6,30-6,14 (m, 1H), 5,69 (d, J = 7,2 Hz, 1H), 5,50-5,16 (m, 12H), 4,97 (d, J = 8,4 Hz, 1H), 4,38-4,0 (m, 8H), 3,94 (d, J = 6,7 Hz, 1H), 2,83-2,52 (m, 9H), 2,43 (s, 3H), 2,32 (t, J = 7,4 Hz, 6H), 2,15 1,98 (m, 6H), 1,94 (s, 3H), 1,47-1,18 (m, 50H), 1,14 (s, 3H), 0,89 (br t, J = 6,6 Hz, 6H).
ESQUEMA 21: Esquema para la síntesis de LD18-DTX, (2R,3S)-1-(((2aR,4S,4aS,6R,9S,11S,12S,12aR,12bS)-12bacetoxi-12-(benzoiloxi)-4,6,11-trihidroxi-4a,8,13,13-tetrametil-5-oxo-2a,3,4,4a,5,6,9,10,11,12,12a,12b-dodecahidro-1H-7,11-metanociclodeca[3,4]benzo[1,2-b]oxet-9-il)oxi)-3-((ter-butoxicarbonil)amino)-1-oxo-3-fenilpropan-2-il (Z)-octadec-9-en-1-il succinato.

LD18-DTX: (2R,3S)-1-(((2aR,4S,4aS,6R,9S,11S,12S,12aR,12bS)-12b-acetoxi-12-(benzoiloxi)-4,6,11-trihidroxi-4a,8,13,13-tetrametil-5-oxo-2a,3,4,4a,5,6,9,10,11,12,12a,12b-dodecahidro-1H-7,11-metanociclodeca[3,4]benzo[1,2-b]oxet-9-il)oxi)-3-((ter-butoxicarbonil)amino)-1-oxo-3-fenilpropan-2-il (Z)-octadec-9-en-1-il succinato:
Se añadió Et3N (35 pL, 0,25 mmol, 2,50 equiv.), seguido del reactivo de Mukaiyama (33 mg, 0,13 mmol, 1,30 equiv.), a una solución helada de CH2Cl2 (1 mL) de docetaxel (81 mg, 0,10 mmol) y hemisuccinato 16 (44 mg, 0,12 mmol, 1,20 equiv.) en un matraz de base redonda seco bajo argón. La mezcla de reacción se dejó calentar durante 14 h y después se concentró en un evaporador rotativo. El crudo se purificó mediante cromatografía en columna flash (25 mL SO2, 80:20^50:50 hexanos/EtOAc) para obtener un aceite claro e incoloro como conjugado deseado LD18-DTX (95 mg, 82 % de rendimiento).
1H RMN (300 MHz, CDCI3): 58,13 (d, J = 7,5 Hz, 2H), 7,67-7,58 (m, 1H), 7,57-7,47 (m, 2H), 7,46-7,36 (m, 2H), 7,36 7,26 (m, 3H), 6,34-6,17 (m, 1H), 5,70 (d, J = 7,1 Hz, 1H), 5,49 (br s, 2H), 5,43-5,29 (m, 4H), 5,23 (s, 1H), 4,97 (d, J = 8,2 Hz, 1H), 4,38-4,07 (m, 6H), 4,05 (t, J = 6,8 Hz, 2H), 3,94 (d, J = 6,8 Hz, 1H), 2,77-2,52 (m, 6H), 2,44 (s, 3H), 2,06 (s, 3H), 2,08-1,98 (m, 4H), 1,68-1,52 (m, 3H), 1,43-1,19 (m, 43H), 1,13 (s, 3H), 0,89 (br t, J = 6,5 Hz, 3H).
ESQUEMA 22: Esquema para la síntesis de LD22-DTX, (2R,3S)-1-(((2aR,4S,4aS,6R,9S,11S,12S,12aR,12bS)-12bacetoxi-12-(benzoiloxi)-4,6,11-trihidroxi-4a,8,13,13-tetrametil-5-oxo-2a,3,4,4a,5,6,9,10,11,12,12a,12b-dodecahidro-1H-7,11-metanociclodeca[3,4]benzo[1,2-b]oxet-9-il)oxi)-3-((ter-butoxicarbonil)amino)-1-oxo-3-fenilpropan-2-il (2,3-bis(tetradecanoiloxi)propil)succinato.

LD22-DTX: (2R,3S)-1-(((2aR,4S,4aS,6R,9S,11S,12S,12aR,12bS)-12b-acetoxi-12-(benzoiloxi)-4,6,11-trihidroxi-4a,8,13,13-tetrametil-5-oxo-2a,3,4,4a,5,6,9,10,11,12,12a,12b-dodecahidro-1H-7,11-metanociclodeca[3,4]benzo[1,2-b]oxet-9-il)oxi)-3-((ter-butoxicarbonil)amino)-1-oxo-3-fenilpropan-2-il (2,3-bis(tetradecanoiloxi)propil)succinato:
Se añadió Et3N (35 pL, 0,25 mmol, 2,50 equiv.), seguido del reactivo de Mukaiyama (33 mg, 0,13 mmol, 1,30 equiv.), a una solución helada de CH2CI2 (1 mL) de docetaxel (81 mg, 0,10 mmol) y hemisuccinato 19 (74 mg, 0,12 mmol, 1,20 equiv.) en un matraz de base redonda seco bajo argón. La mezcla de reacción se dejó calentar durante 14 h y después se concentró en un evaporador rotativo. El crudo se purificó mediante cromatografía en columna flash (25 mL SO2, 80:20^50:50 hexanos/EtOAc) para obtener un aceite claro e incoloro como conjugado deseado LD22-DTX (63 mg, 45 % de rendimiento).
1H RMN (300 MHz, CDCh): 58,13 (d, J = 7,2 Hz, 2H), 7,67-7,58 (m, 1H), 7,57-7,47 (m, 2H), 7,46-7,36 (m, 2H), 7,36 7,26 (m, 3H), 6,30-6,14 (m, 1H), 5,70 (d, J = 7,2 Hz, 1H), 5,60-5,41 (m, 2H), 5,36 (br s, 1H), 4,97 (d, J = 8,4 Hz, 1H), 4,40-4,07 (m, 9H), 3,94 (d, J = 6,7 Hz, 1H), 2,80-2,51 (m, 6H), 2,43 (s, 3H), 2,32 (t, J = 7,4 Hz, 6H), 1,95-1,81 (m, 3H), 1,94 (s, 3H), 1,69-1,53 (m, 5H), 1,14 (s, 3H), 1,41-1,18 (m, 54H), 1,14 (s, 3H), 0,89 (brt, J = 6,6 Hz, 6H).
ESQUEMA 23: Esquema para la síntesis de LD23-DTX, (2R,3S)-1-(((2aR,4S,4aS,6R,9S,11S,12S,12aR,12bS)-12bacetoxi-12-(benzoiloxi)-4,6,11-trihidroxi-4a,8,13,13-tetrametil-5-oxo-2a,3,4,4a,5,6,9,10,11,12,12a,12b-dodecahidro-1H-7,11-metanociclodeca[3,4]benzo[1,2-b]oxet-9-il)oxi)-3-((ter-butoxicarbonil)amino)-1-oxo-3-fenilpropan-2-il (2,3-bis(oleoiloxi)propil)succinato.
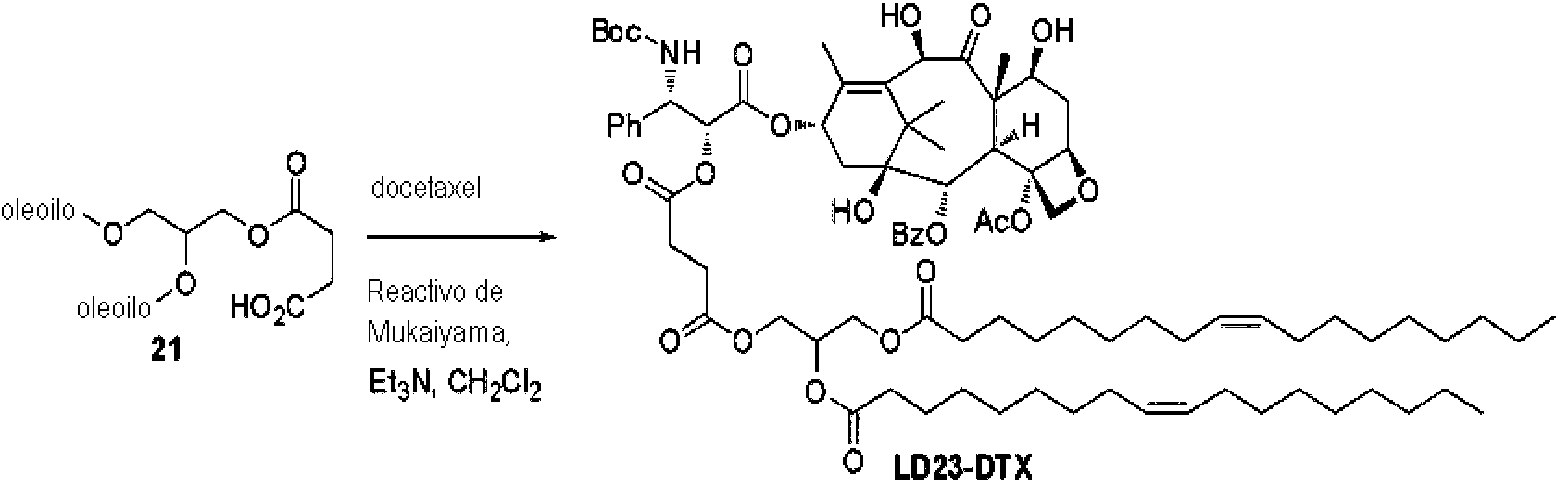
LD23-DTX: (2R,3S)-1-(((2aR,4S,4aS,6R,9S,11S,12S,12aR,12bS)-12b-acetoxi-12-(benzoiloxi)-4,6,11-trihidroxi-4a,8,13,13-tetrametil-5-oxo-2a,3,4,4a,5,6,9,10,11,12,12a,12b-dodecahidro-1H-7,11-metanociclodeca[3,4]benzo[1,2-b]oxet-9-il)oxi)-3-((ter-butoxicarbonil)amino)-1-oxo-3-fenilpropan-2-il (2,3-bis(oleoiloxi)propil)succinato:
Se añadió Et3N (26 pL, 0,18 mmol, 3,00 equiv.), seguido del reactivo de Mukaiyama (25 mg, 9,90*10'2 mmol, 1,60 equiv.), a una solución helada de CH2Cl2 (0,6 mL) de docetaxel (50 mg, 6,19*10'2 mmol) y hemisuccinato 21 (67 mg, 9,28*10'2 mmol, 1,50 equiv.) en un matraz de base redonda seco bajo argón. La mezcla de reacción se dejó calentar durante 14 h y después se concentró en un evaporador rotativo. El crudo se purificó mediante cromatografía en columna flash (25 mL SO2, 60:40 hexanos/EtOAc) para obtener un aceite claro e incoloro como conjugado deseado LD23-DTX (59 mg, 63 % de rendimiento).
1H RMN (300 MHz, CDCI3): 58,13 (d, J = 7,5 Hz, 2H), 7,67-7,58 (m, 1H), 7,57-7,47 (m, 2H), 7,46-7,36 (m, 2H), 7,36 7,26 (m, 3H), 6,30-6,14 (m, 1H), 5,69 (d, J = 7,2 Hz, 1H), 5,57-5,42 (m, 2H), 5,42-5,19 (m, 8H), 4,97 (d, J = 8,4 Hz, 1H), 4,38-4,07 (m, 8H), 3,94 (d, J = 6,7 Hz, 1H), 2,83-2,50 (m, 6H), 2,43 (s, 3H), 2,32 (t, J = 7,4 Hz, 6H), 2,11-1,81 (m, 8H), 1,94 (s, 3H), 1,69-1,53 (m, 5H), 1,45-1,17 (m, 56H), 1,13 (s, 3H), 0,89 (br t, J = 6,6 Hz, 6H).
ESQUEMA 24: Esquema para la síntesis de LD12-ABN, (3S,8R,9S,10R,13S,14S)-10,13-dimetil-17-(piridin-3-il)-2,3,4,7,8,9,10,11,12,13,14,15-dodecahidro-1H-ciclopenta[a]fenantren-3-il linoleil succinato.

LD12-ABN: (3S,8R,9S,10R,13S,14S)-10,13-dimetil-17-(piridin-3-N)-2,3,4,7,8,9,10,11,12,13,14,15-dodecahidro-1H-ciclopenta[a]fenantren-3-il linoleil succinato
Se añadió DCC (17 mg, 8,25*10'2 mmol, 1,10 equiv.) a una solución helada de CH2Cl2 (0,7 mL) de hemisuccinato 5 (30 mg, 8,25 x 10'2 mmol, 1,10 equiv.) en un matraz de base redonda bajo argón, seguido de abiraterona (26 mg, 7,5*10'2 mmol) y DMAP (10 mg, 8,25*10'2 mmol, 1,10 equiv.). La mezcla de reacción se dejó calentar durante 14 h, se filtró a través de Celite y se concentró en un evaporador rotativo. El crudo se purificó mediante cromatografía en columna flash (20 mL SO2, 70:30 hexanos/EtOAc) para obtener un aceite claro e incoloro como el conjugado deseado LD12-ABN (31 mg, 60 % de rendimiento). 1H RMN (300 MHz, CDCla): 58,61 (s, 1H), 8,45 (d, J = 3,9 Hz, 1H), 7,63 (dt, J = 8,0, 1,9 Hz, 1H), 7,21 (dd, J = 7,9, 4,8 Hz, 1H), 6,01-5,94 (m, 1H), 5,45-5,25 (m, 5H), 4,71-4,55 (m, 1H), 4,07 (t, J = 6,7 Hz, 2H), 2,76 (t, J = 6,0 Hz, 2H), 2,60 (s, 4H), 2,39-2,31 (m, 2H), 2,32-2,19 (m, 1H), 2,12-1,97 (m, 7H), 1,92-1,42 (m, 12H), 1,41-1,23 (m, 17H), 1,21 1,07 (m, 2H), 1,07 (s, 3H), 1,03 (s, 3H), 0,87 (br t, J = 6,7 Hz, 3H).
ESQUEMA 25: Esquema para la síntesis de LD11-NPC-I, 4-((2-((4-(2-(2-((3r,5r,7r)-adamantan-1-il)acetamido)acetil)piperazin-1-il)metil)fenoxi)metil)benzoato de linoleilo.

LD11-NPCi: 4-((2-((4-(2-(2-((3r,5r,7r)-adamantan-1-il)acetamido)acetil)piperazin-1-il)metil)fenoxi)metil)benzoato de linoleilo:
Se añadió DCC (18 mg, 8,65*10
' 2
mmol, 1,10 equiv.) a una solución helada de CH
2
Cl
2
(0,8 mL) de NPC
1
I [Lee et al. (2013)] (44 mg, 7,86*10
' 2
mmol) y Et
3
N (22 pL, 0,16 mmol, 2,00 equiv.) en un matraz de base redonda bajo argón, después se sacó del baño frío y se agitó durante 15 min. La mezcla de reacción se colocó de nuevo en el baño de hielo, después se añadieron alcohol linoleílico (23 mg, 8,65*10
' 2
mmol, 1,10 equiv.) y DMAP (11 mg, 8,65*10
' 2
mmol, 1,10 equiv.) y la mezcla se dejó calentar durante 14 h. La mezcla de reacción se diluyó con Et
2
O, se lavó con NaHCO
3
al 5 % acuoso (1*5 mL), salmuera (1*5 mL), se secó sobre Na
2
SO
4
y se concentró en un evaporador rotativo. El crudo se purificó mediante cromatografía en columna flash (20 mL SO
2
, 75:25:0^100:0:0^-98:0:2 EtOAc/hexanos/MeOH) para obtener un aceite claro e incoloro como conjugado deseado LD11-NPC
1
(26 mg, 41 % de rendimiento).
Rf = 0,40 (SiO 2 , 65:30:5 EtOAc/hexanos/MeOH); 1 H RMN (300 MHz, CDCla): 58,06 (d, J = 8,1 Hz, 2H), 7,49 (d, J = 8,1 Hz, 2H), 7,34 (d, J = 7,4 Hz, 1H), 7,22 (d, J = 7,4 Hz, 1H), 6,97 (t, J = 7,4 Hz, 1H), 6,90 (d, J = 8,1 Hz, 1H), 6,52 (br s, 1H), 5,45-5,26 (m, 4H), 5,14 (s, 2H), 4,31 (t, J = 6,7 Hz, 2H), 4,04 (d, J = 3,4 Hz, 2H), 3,67 (br s, 4H), 3,41 (br t, J = 4,4 Hz, 2H), 2,77 (t, J = 5,9 Hz, 2H), 2,52 (br t, J = 4,7 Hz, 4H), 2,11-1,90 (m, 9H), 1,85-1,53 (m, 6H), 1,50-1,18 (m, 18H), 0,88 (br t, J = 6,8 Hz, 3H).
ESQUEMA 26: Esquema para la síntesis de LD25-NPC-I, 4-((2-((4-(2-(2-((3r,5r,7r)-adamantan-1-il)acetamido)acetil)piperazin-1-il)metil)fenoxi)metil)benzoato de octilo.

LD25-NPCi : 4-((2-((4-(2-(2-((3r,5r,7r)-adamantan-1-il)acetamido)acetil)piperazin-1-il)metil)fenoxi)metil)benzoato de octilo:
Se añadió DCC (50 mg, 0,24 mmol, 1,20 equiv.) a una solución helada de CH2CI2 (1 mL) de NPC1 I (112 mg, 0,20 mmol) y Et3N (69 μL, 0,50 mmol, 2,50 equiv.) en un matraz de base redonda bajo argón, después se sacó del baño frío y se agitó durante 15 min. La mezcla de reacción se volvió a colocar en el baño de hielo, después se añadieron octanol (41 μL, 0,26 mmol, 1,30 equiv.) y DMAP (29 mg, 0,24 mmol, 1,20 equiv.) y la mezcla se dejó calentar durante 14 h. La mezcla de reacción se diluyó con Et2O, se lavó con NaHCO3 al 5 % acuoso (1*5 mL), salmuera (1*5 mL), se secó sobre Na2SO4 y se concentró en un evaporador rotativo. El crudo se purificó mediante cromatografía en columna flash (20 mL SO2 , 50:45:5 EtOAc/hexanos/MeOH) para obtener un aceite claro e incoloro como conjugado deseado LD11-NPC1 (73 mg, 54 % de rendimiento). Rf = 0,47 (SiO2, 50:40:10 EtOAc/hexanos/MeOH);
1H RMN (300 MHz, CDCla): 88,06 (d, J = 8,1 Hz, 2H), 7,49 (d, J = 8,1 Hz, 2H), 7,34 (d, J = 7,4 Hz, 1H), 7,22 (d, J = 7,4 Hz, 1H), 6,97 (t, J = 7,4 Hz, 1H), 6,90 (d, J = 8,1 Hz, 1H), 6,52 (br s, 1H), 5,15 (s, 2H), 4,33 (t, J = 6,6 Hz, 2H), 4,05 (d, J = 3,5 Hz, 2H), 3,66 (br s, 4H), 3,40 (br t, J = 4,2 Hz, 2H), 2,51 (br t, J = 4,3 Hz, 4H), 2,08-1,87 (m, 6H), 1,85-1,53 (m, 15H), 1,52-1,20 (m, 11H), 0,88 (br t, J = 6,8 Hz, 3H).
ESQUEMA 27: Esquema para la síntesis de LD2 6 -NPC1, 4-((2-((4-(2-(2-((3r,5r,7r)-adamantan-1-il)acetamido)acetil)piperazin-1-il)metil)fenoxi)metil)benzoato de decilo.

LD25-NPCi : 4-((2-((4-(2-(2-((3r,5r,7r)-adamantan-1-il)acetamido)acetil)piperazin-1-il)metil)fenoxi)metil)benzoato de decilo:
Se añadió DCC (50 mg, 0,24 mmol, 1,20 equiv.) a una solución helada de CH2Cl2 (1 mL) de NPC1 I (112 mg, 0,20 mmol) y Et3N (69 μL, 0,50 mmol, 2,50 equiv.) en un matraz de base redonda bajo argón, después se sacó del baño frío y se agitó durante 15 min. La mezcla de reacción se volvió a colocar en el baño de hielo, después se añadieron decanol (50 μL, 0,26 mmol, 1,30 equiv.) y DMAP (29 mg, 0,24 mmol, 1,20 equiv.) y se dejó calentar la mezcla durante 14 h. La mezcla de reacción se diluyó con Et2O, se lavó con NaHCO3 al 5 % acuoso (1*5 mL), salmuera (1*5 mL), se secó sobre Na2SO4 y se concentró en un evaporador rotativo. El crudo se purificó mediante cromatografía en columna flash (20 mL SiO2 , 50:45:5 EtOAc/hexanos/MeOH) para obtener un aceite claro e incoloro como conjugado deseado LD11-NPC1 (68 mg, 49 % de rendimiento). Rf = 0,47 (SiO2, 50:40:10 EtOAc/hexanos/MeOH);
1H RMN (300 MHz, CDCl3): 88,06 (d, J = 8,1 Hz, 2H), 7,49 (d, J = 8,1 Hz, 2H), 7,34 (d, J = 7,4 Hz, 1H), 7,22 (d, J = 7,4 Hz, 1H), 6,97 (t, J = 7,4 Hz, 1H), 6,90 (d, J = 8,1 Hz, 1H), 6,52 (br s, 1H), 5,15 (s, 2H), 4,33 (t, J = 6,6 Hz, 2H), 4,05 (d, J = 3,5 Hz, 2H), 3,66 (br s, 4H), 3,40 (br t, J = 4,2 Hz, 2H), 2,51 (br t, J = 4,3 Hz, 4H), 2,08-1,87 (m, 6H), 1,85-1,53 (m, 15H), 1,52-1,20 (m, 15H), 0,88 (br t, J = 6,8 Hz, 3H).
ESQUEMA 28. Esquema para la síntesis de NPC1 I, ácido 4-((2-((4-(2-(2-((3r,5r,7r)-adamantan-1-il)acetamido)acetil)piperazin-1-il)metil)fenoxi)metil)benzoico.

NPCi I: ácido 4-((2-((4-(2-(2-((3r,5r,7r)-adamantan-1-il)acetamido)acetil)piperazin-1-il)metil)fenoxi)metil)benzoico:
Se añadió NaOH 1 M acuoso (6,1 mL, 6,13 mmol, 2,00 equiv.) a una solución de MeOH (9 mL) a temperatura ambiente de NPC1 ((Lee et al. 2013) 1,76 g, 3,06 mmol) en un matraz de base redonda bajo argón y se agitó. Después de 14 h, el pH de la mezcla se ajustó a ≤ 2 mediante la adición de HCl 1 M acuoso y, a continuación, se extrajo con CHCh (3*15 mL). Los extractos orgánicos combinados se secaron sobre Na2SO4 y se concentraron en un evaporador rotativo para obtener una espuma blanca (1,60 g, 93 % de rendimiento), que se utilizó sin purificación adicional.
Rf = 0,28 (SiO2, 70:20:10 EtOAc/hexanos/MeOH); 1H RMN (300 MHz, CD3OD): 8 8,07 (d, J = 8,4 Hz, 2H), 7,61 (d, J = 8,4 Hz, 2H), 7,53-7,44 (m, 2H), 7,21 (d, J = 8,4 Hz, 1H), 7,10 (t, J = 7,5 Hz, 1H), 5,32 (s, 2H), 4,40 (s, 2H), 4,07 4,01 (m, 2H), 3,78 (br s, 4H), 2,02 (s, 2H), 2,00-1,90 (m, 5H), 1,79-1,61 (m, 16H).
ESQUEMA 29. Esquema para la síntesis de LD01-RXN y LD01-TFN
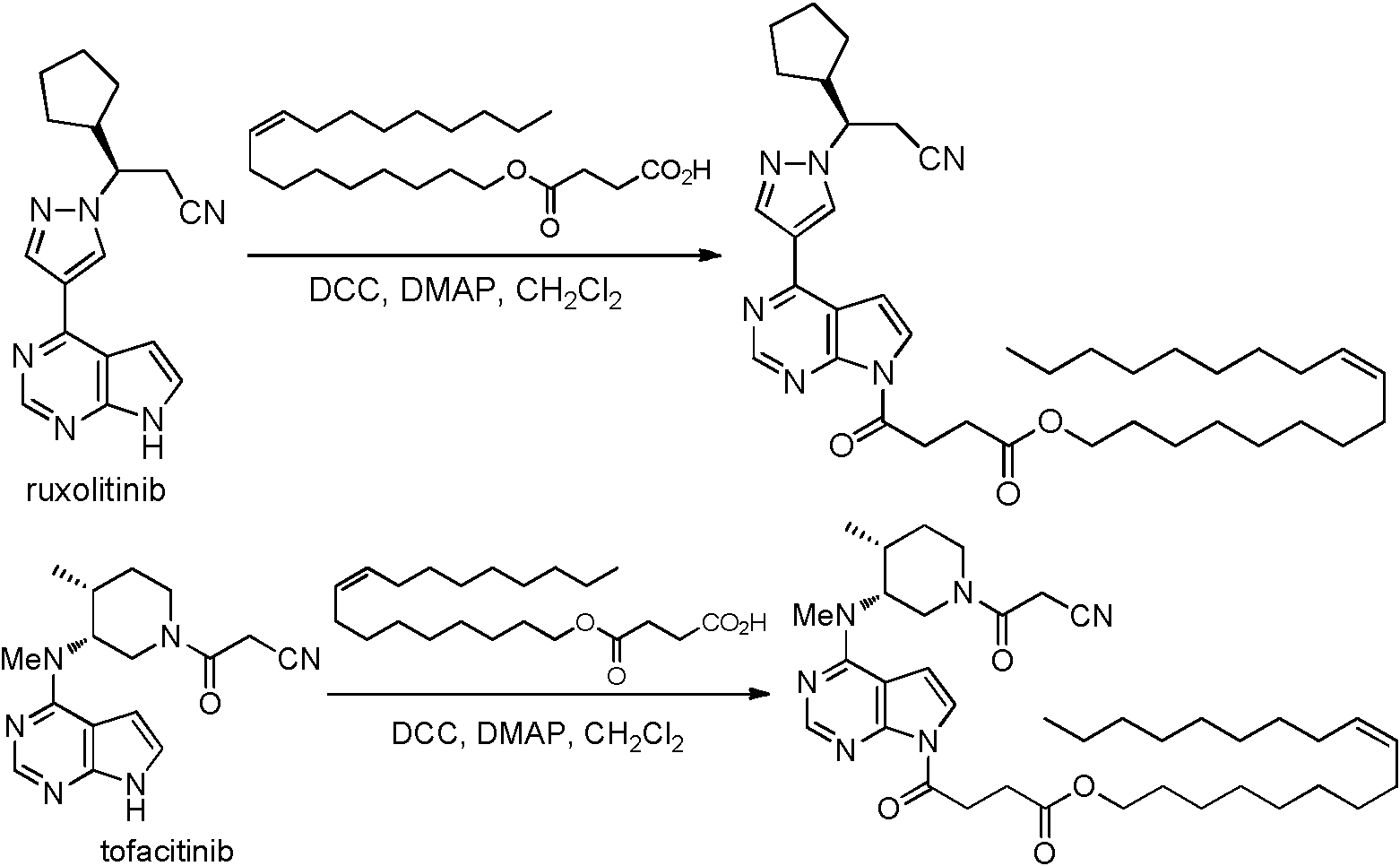
ESQUEMAS DE EJEMPLO


Formulación de DEX o DTX en nanopartículas de lípido
Las propiedades hidrófobas de los profármacos unidos a lípidos (compuestos por el fármaco y las moléculas lipídicas) permiten que los fármacos se incorporen fácilmente a las nanopartículas lipídicas simplemente añadiéndolos a la mezcla de formulación lipídica, sin necesidad de modificar más el proceso de formulación. Como resultado, las nanopartículas lipídicas que incorporan estos fármacos pueden fabricarse utilizando una amplia variedad de metodologías de formulación bien descritas, que incluyen, entre otras, la extrusión, la inyección de etanol y la mezcla en línea [Maclachlan y Cullis (2005); y Jeffs et al. (2005)].
Materiales: 1-palmitoil-2-oleoil-sn-glicero-3-fosfocolina (POPC) y 1,2-diestearoil-sn-glicero-3-fosfocolina (DSPC), 1,2-dimiristoil-sn-glicero-3-fosfoetanolamina-N-[metoxi(polietilenglicol)-2000] (PEG-DMPE), y 1,2-diestearoil-sn-glicero-3-fosfoetanolamina-N-[amino(polietilenglicol)-2000] (PEG-DSPE) se adquirieron a Avanti Polar Lipids™ (Alabaster, AL). El colesterol se obtuvo de Sigma™ (St Louis, MO). Los lípidos ionizables están disponibles comercialmente. Los profármacos LD-DEX o LD-DTX se sintetizaron como se ha descrito con anterioridad.
Preparación de los sistemas de LNP: Los sistemas PC-Chol LNP se prepararon mezclando rápidamente POPC, colesterol, profármaco ligado a lípidos y PEG-DMPE (en proporción molar de 49/39/10/2) disueltos en etanol con solución salina tamponada con fosfato utilizando un mezclador en T. Las formulaciones se dializaron con solución salina tamponada con fosfato para eliminar el etanol residual y elevar el pH a 7,4. Las formulaciones se dializaron contra solución salina tamponada con fosfato para eliminar el etanol residual y elevar el pH a 7,4. Los sistemas PC-TO y LNP ionizables se prepararon de la misma manera, excepto que las composiciones lipídicas para estos sistemas son: POPC, trioleína, profármaco ligado a lípidos y PEG-DSPE (en proporción molar de 19/70/10/1), y lípido ionizable, DSPC, colesterol, profármaco ligado a lípidos y PEG-DMPE (en proporción molar de 40/10/38,5/10/1,5), respectivamente.
Caracterización de las LNP: El tamaño de las partículas se determinó mediante dispersión dinámica de la luz utilizando un Malvern Zetasizer Nano ZS™ (Malvern, Reino Unido) tras el intercambio de tampones en solución salina tamponada con fosfato. Se utilizaron datos de tamaño y distribución ponderados por número. Las concentraciones de lípidos se determinaron midiendo el colesterol total mediante el kit de ensayo enzimático Cholesterol E de Wako Chemicals USA™ (Richmond, VA). La morfología de las formulaciones de LNP que contenían LD-DEX se analizó mediante microscopía electrónica de transmisión criogénica (cryoTEM) como se ha descrito previamente [Leung et al. (2012)].
Cuantificación de profármacos por HPLC: LD-DEX/DTX se cuantificaron mediante cromatografía líquida de ultra alta presión (UHPLC) utilizando un sistema Waters Acquity™ UPLC equipado con un detector de matriz de fotodiodos (PDA) y un detector de dispersión de luz evaporativa (ELS); se utilizó el software de adquisición de datos Empower™ versión 3.0 (Waters™, EE.UU.). Las separaciones se realizaron utilizando una columna Waters Acquity™ BEH C-is (1,7 |jm, 2,1*100 mm) a un caudal de 0,5 ml/min, con fases móviles A y B consistentes en metanol/10 mM de bicarbonato amónico (80/20 % v/v) y metanol/acetonitrilo (80/20 % v/v), respectivamente. Las fases móviles se administraron en un gradiente lineal programado (45 %- 80 % B en 1 min y luego a 99 % B en 2 min) a una temperatura de columna de 55°C. El analito se midió mediante PDA (a una longitud de onda de 239 nm) y detector ELS frente a curvas estándar de calibración. Para el análisis de la descomposición de LD-DEX/DTX en compuesto activo, se utilizaron gradientes lineales de 20/80 % a 100/0 % (acetonitrilo/agua, v/v) durante 7 min.
Cryo-TEM de LNP: Se aplicó una pequeña cantidad de formulaciones a ~20 mg/mL (concentración total de lípidos) a una rejilla de microscopía electrónica con una película de carbono perforada. La muestra se congeló rápidamente en etano líquido utilizando un sistema Vitrobot™ (FEI™, Hillsboro, OR). Se tomaron imágenes utilizando un TEM FEI G20 Lab6200 kV (FEI™, Hillsboro, OR) en condiciones criogénicas (88 K) a un aumento de 50.000* con una cámara CCD AMT HR. Se muestran imágenes representativas de LD02 y LD03 en LNP de PC-Chol fabricadas con 5 % en moles de PEG-DSPE.
Degradación in vitro de profármacos unidos a lípidos
Para determinar la biodegradabilidad de los profármacos de dexametasona o docetaxel (LD-DEX/DTX), se incubó 1 mg/mL de LNP en plasma de ratón (Cedarlane™, Burlington, Ontario) o PBS suplementado con 10U de esterasa purificada (Sigma-Aldrich™, St. Louis, MO) durante un máximo de 4 horas a 37 °C. Tras la incubación, se añadieron cuatro volúmenes de cloroformo/metanol (2:1) y la mezcla se mezcló en vórtex. Las muestras se centrifugaron a 13.000 g durante 5 minutos y se desechó la fase superior. La fase orgánica restante se secó al vacío y el extracto lipídico resultante se disolvió en metanol/acetonitrilo (1:1). La cantidad de compuesto LD-DEX/DTX parental se determinó mediante cromatografía líquida de ultra alta presión en un Waters Acquity™ H-Class UPLC System™ equipado con una columna BEH C18 y un detector de matriz de fotodiodos.
Disociación in vitro de profármacos unidos a lípidos
Las LNP se incubaron durante 0, 3, 5 y 24 ha 37 °C en 400 μL de plasma humano estéril (Cedarlane™, Burlington, Ontario) a una concentración final de lípidos de 1,2 mM. En los distintos puntos temporales, las muestras se cargaron en una columna Sepharose CL-4B de 1,5 * 27 cm (Sigma-Aldrich™, St. Louis, MO). Se recogieron fracciones de 30 * 2 mL. Se añadieron tres volúmenes de metanol/acetonitrilo (1:1) a cada fracción y se analizaron por UHPLC para determinar el LD-DEX/DTX residual como se ha descrito con anterioridad.
Supresión inmunitaria in vivo mediante profármacos unidos a lípidos (LD-DEX)
A ratones hembra Cs7Bl/6 de 6-8 semanas de edad (Charles River Laboratories™, Wilmington, MA) se les inyectaron por vía intravenosa nanopartículas lipídicas que contenían oligonucleótidos de ADN CpG (10 mg/kg), ARNm (3 mg/kg) o ADN plasmídico (ADNp) (1 mg/kg). Los animales fueron eutanasiados 2 horas después de la inyección y se les extrajo sangre por vía intracardiaca o safena. El plasma se separó de la sangre total por centrifugación y se analizó en busca de citoquinas proinflamatorias utilizando el kit Mesoscale Proinflammatory Multiplex™ (Rockville, MD) según las instrucciones del fabricante. Todos los procedimientos fueron aprobados por el Comité de Cuidado Animal de la Universidad de British Columbia y se realizaron de acuerdo con las directrices establecidas por el Consejo Canadiense de Cuidado Animal (Canadian Council on Animal Care).
Supresión inmunitaria in vitro mediante profármacos unidos a lípidos (LD-DEX)
Se sembraron células de macrófagos de ratón RAW 264.7 a 250.000 células por mL en placas de 12 pocillos y se incubaron a 37 °C durante la noche. Se disolvió lipopolisacárido (LPS) en solución salina tamponada con fosfato a una concentración de 2 μg/ml. Las células se trataron con nanopartículas lipídicas que contenían profármacos LD-DEX y 2 ng/ml de LPS durante 4 horas. Esta concentración de LPS se eligió debido a su capacidad para aumentar la expresión de IL1p aproximadamente 1000 veces en comparación con las células no tratadas. Al final del período de incubación, se aspiró el medio de cultivo y se extrajo el ARN celular utilizando el kit de extracción Purelink RNA™ según el protocolo del fabricante (Life Technologies™, Burlington, ON). La concentración del ARN extraído se determinó utilizando un Nanodrop Lite™ (Thermo Fisher™). 1 μg de ARN se convirtió en ADNc utilizando el kit de transcripción inversa de ADNc de alta capacidad (High Capacity cDNA Reverse Transcription kit) (Life Technologies™, Burlington, ON). Para validar la preparación de la muestra, se incluyeron un control sin molde y un control negativo de transcriptasa inversa. A continuación, el ADNc se cuantificó mediante qPCR comparativa en tiempo real, en donde los valores Ct (ciclo umbral) se convierten en cantidad relativa mediante el método 2-AACT. Los conjuntos de cebadores-sonda se diseñaron para unirse al ADNc de la interleucina-1p (IL1p). Se eligió la hipoxantina-guanina fosforribosiltransferasa (HPRT) como gen normalizador endógeno, conocido por mantener niveles constantes con tratamientos variables.
Inhibición del crecimiento in vitro mediante profármacos unidos a lípidos (LD-DTX)
Las células OVCAR3 se obtuvieron de ATCC (Manassas, VA). Las células se sembraron a 12.000 células por pocillo en placas de 96 pocillos y se trataron con docetaxel libre, profármacos, formulaciones de nanopartículas lipídicas con o sin profármacos de 0 a 15 μM de docetaxel durante 48 h. La viabilidad celular se determinó mediante un ensayo colorimétrico, basado en la sal de tetrazolio MTT (3-(4,5-dimetiltiazol-2-il)-2,5-bromuro de difeniltetrazolio), como se ha descrito previamente [Mossman 1983].
EJEMPLOS
Ejemplo 1: Efecto de la Hidrofobicidad en la Incorporación del profármaco LNP a la preparación de profármacos DEX o DTX:
Los profármacos unidos a lípidos descritos en la presente pueden estar compuestos por un fármaco conjugado con una o más moléculas lipídicas a través de un ligador. Un profármaco representativo a base de dexametasona (-DEX) compuesto de DEX unido a una cadena de acilo de linoleilo mediante un enlace éster es LD01-DEX. Un profármaco representativo a base de Docetaxel (-DTX) que contiene DTX unido a dos cadenas de acil linoleilo mediante un enlace éster hidrolizable es el LD10-DTX.
Para definir las propiedades/características importantes de estos profármacos que influyen en su utilidad, se produjeron una serie de profármacos basados en DEX o DTX. Los profármacos lipídicos ejemplificados muestran variaciones en la fracción lipídica (por ejemplo, el número y la longitud de las cadenas acilo), así como en el ligador (por ejemplo, la presencia o ausencia de un grupo cargado/ionizable, degradabilidad) para variar la liberación del fármaco activo del profármaco lipídico tras la administración in vivo. Los ejemplos de las series DEX y DTX se muestran en la tabla 2 y la tabla 3, respectivamente, que especifica la designación, las características y propiedades del lípido y el ligador, la estructura química y el índice de hidrofobicidad (LogP). En la tabla 4 se enumeran ejemplos de otros profármacos unidos a lípidos, como la abiraterona (-ABN), el tacrolimus (-TAC), la prednisona (-PDN), la prednisolona (-PDL) y el inhibidor de NPC1 NP3.47 (-NPC1B).
Tabla 2. Ejemplos de profármacos LD-DEX con ligadores y lípidos alternativos

Tabla 3. Ejemplos de profármacos LD-DTX con ligadores y lípidos alternativos


Tabla 4. Otros ejemplos de profármacos con ligadores y lípidos alternativos

Ejemplo 2: Eficacia de carga del fármaco
Los análisis de los profármacos lipídicos formulados como se ha descrito con anterioridad, confirmaron que la inclusión de hasta un 10 % en moles de profármacos basados en DEX en la mezcla lipídica de la formulación inicial no tuvo ningún impacto significativo en las características fisicoquímicas o de formulación, como el tamaño y la polidispersidad. Las imágenes CryoTEM de profármacos unidos a lípidos (es decir, LD02- DEX o LD03-DEX), dos profármacos de diferentes anclajes lipídicos e hidrofobicidades, mostraron que estos profármacos unidos a lípidos son vesículas bicapa (ver figura 1). Según la hipótesis, los análisis lipídicos por UHPLC de las LNP finales indicaron que la incorporación de profármacos basados en DEX dependía de la hidrofobicidad del profármaco basado en DEX. Por lo tanto, LD01-DEX, que tiene una hidrofobicidad relativamente baja, solo incorporó el 65-72 % en moles de la cantidad de entrada en la preparación final de nanopartículas lipídicas (tabla 5). LD06-DEX, que tiene una hidrofobicidad más alta que LD01- DEX, se incorporó más en LNP que LD01-DEX. Por el contrario, l D02-, 03-, 07-,
13-, 14-, 17-, 21-, y 24-DEX que fueron diseñadas para ser más hidrofóbicas se incorporaron eficientemente (91-100 %) en profármacos unidos a lípidos como se esperaba. Sin embargo, aunque la hidrofobicidad relativa es un componente esencial, no representa el único factor que influye en la incorporación del profármaco a las nanopartículas lipídicas. La menor eficiencia de atrapamiento observada para LD01-DEX o LD06-DEX en comparación con LD03-DEX o LD02-DEX puede atribuirse, al menos en parte, a la presencia de un grupo ionizable dentro del ligador de LD01-DEX, que actúa alterando la incorporación y disposición de los profármacos en las nanopartículas lipídicas.
De forma similar, los análisis por UHPLC de los profármacos basados en DTX mostraron una incorporación casi completa a las nanopartículas lipídicas (tabla 6). Esto se debe probablemente a la hidrofobicidad relativamente alta de estos compuestos.
Tabla 5. Eficacia de la incorporación del profármaco LD-DEX en nanopartículas lipídicas en función de la hidrofobicidad. El tamaño de las partículas se indica en nm. El índice de polidispersidad (PDI) se indica entre paréntesis.

Tabla 6. Eficacia de la incorporación del profármaco LD-DTX en nanopartículas lipídicas. El tamaño de las partículas in i n nm. El ín i li i r i PDI in i nr r n i .

Ejemplo 3: Efecto de la hidrofobicidad en la liberación del fármaco activo a partir de profármacos unidos a lípidos
La hidrofobicidad también puede ser suficiente para promover una encapsulación/retención eficaz, y lo ideal sería que no diera lugar a que el profármaco quedara sustancialmente incrustado o protegido de otro modo en el profármaco lipídico. El fármaco también puede distribuirse en los profármacos lipídicos para permitir el acceso al ligador biodegradable, la escisión del profármaco por enzimas intracelulares apropiadas y, en última instancia, la liberación del fármaco activo de los profármacos lipídicos. Los análisis por UHPLC mostraron que una incubación de 1 o 4 horas de LD03-DEX en plasma o PBS con esterasas aisladas producía una escisión progresiva del profármaco (figura 2). Como era de esperar, la escisión del profármaco fue suprimida por el calor, probablemente debido a la inactivación de la enzima.
Para comparar las tasas relativas de escisión de varios profármacos de DEX, se incubaron nanopartículas lipídicas que contenían profármacos durante 1 h en plasma a 37 °C, se extrajo el lípido mediante un método Bligh-Dyer & Folsch modificado y se sometió a análisis por UHPLC. Como puede observarse en la figura 3, LD01-DEX y LD03-DEX mostraron una escisión significativa durante este período de tiempo, mientras que, por el contrario, LD02-DEX prácticamente no mostró degradación. Como era de esperar, LD07-DEX, que contiene un ligador no degradable, tampoco mostró degradación. En la figura 4 se muestran más ejemplos de escisión de profármacos LD-DEX. Como era de esperar, los profármacos de LD-DEX con hidrofobicidades relativamente altas (como LD17-, LD21- y LD24-DEX) no fueron susceptibles de degradación en el plasma de ratón. En cambio, los profármacos LD13- y LD14-DEX, que tienen menor hidrofobicidad que LD17-DEX y mayor que LD03-DEX, se degradaron parcialmente en plasma. En la figura 5, se muestra la biodegradación de profármacos LD-DTX en nanopartículas lipídicas. Se formularon LD-DTX en varias formulaciones de nanopartículas lipídicas y se sometieron a incubación en plasma de ratón durante 1 h a 37 °C. A continuación, se extrajeron los lípidos mediante un método Bligh-Dyer & Folsch modificado y se sometieron a análisis UHPLC para su cuantificación. Como en el caso de los profármacos LD-DEX relativamente hidrofóbicos, los profármacos LD-DTX (LD10-, LD18-, LD22- y LD23-DTX) no se degradaron en gran medida tras 1 h de incubación en plasma de ratón.
Para demostrar que el fármaco activo puede liberarse del profármaco ligado a lípidos tras la digestión en el ligador biodegradable, se formularon profármacos representativos de las series LD-DEX y LD-DTX en nanopartículas lipídicas y se sometieron a incubación con esterasa purificada o plasma de ratón durante 24 h (figura 6 a figura 9). Como se muestra en las figura S 6 y 7, tanto el LD02-DEX como el LD03-DEX se degradaron progresivamente a lo largo de 24 horas. Como se ha demostrado con anterioridad, el LD03-DEX, que es menos hidrófobo que el LD02-DEX, se degradó a un ritmo más rápido que el LD02-DEX. Además, mientras que el pico del profármaco LD02 o LD03 disminuye con el tiempo, un pico correspondiente a la dexametasona (DEX) activa apareció en los cromatogramas tras 24 h de incubación en esterasa purificada o en plasma de ratón, lo que indica que la DEX activa se libera (figura 6 y figura 7). Del mismo modo, se observó docetaxel (DTX) activo en los cromatogramas de LD18- y LD22-DTX tras 24 h de incubación en esterasa o plasma (figura 8 y figura 9).
La hidrofobicidad también puede afectar a la disociación eficiente de las formulaciones lipídicas. Para detectar y cuantificar la disociación de los profármacos de las nanopartículas lipídicas, se incubaron en plasma humano durante 24 horas formulaciones que contenían profármacos unidos a lípidos y, a continuación, se sometieron a cromatografía de exclusión por tamaño para recuperar fracciones de nanopartículas lipídicas. Los lípidos se extrajeron de estas fracciones utilizando el método Bligh-Dyer & Folsch modificado y luego se analizaron por UHPLC para determinar la cantidad residual de profármaco (figura 10 y figura 11). En estos experimentos de disociación lipídica se utilizó plasma humano para eliminar la contribución de la degradación por esterasas, ya que se sabe que el plasma humano contiene una actividad carboxilesterasa sustancialmente menor que el plasma de ratón. Como se muestra en la figura 10, LD02-DEX (con un índice de hidrofobicidad mayor que LD03-DEX) se disoció de las nanopartículas lipídicas a un ritmo más lento que LD03-DEX. En la serie LD-DTX se observó un fenómeno similar. LD18-DTX (menos hidrófobo que LD22-DTX) se disoció de las nanopartículas lipídicas a un ritmo más rápido que LD22-DTX (figura 11).
Ejemplo 4: Diseño y síntesis de profármacos alternativos unidos a lípidos
Pueden sintetizarse profármacos lipídicos adicionales utilizando los métodos descritos en la presente, incluyendo profármacos que contengan otros fármacos activos como metotrexato (LD01-METH), tacrolimus (LD15-TAC), tofacitinib (LD01-TFN), cabazitaxel (LD01-CTX) y ruxolitinib (LD01-RXN). De los profármacos que se han fabricado y diseñado para ajustarse a estas “reglas” de diseño, se ha comprobado que todos se incorporan y retienen eficientemente en la LNP según lo previsto.
Ejemplo 5: Efectos antiinflamatorios de profármacos unidos a lípidos en modelos de ratón y celulares de estimulación inmunitaria
Para demostrar la utilidad de los profármacos LD-DEX, se utilizaron modelos de estimulación inmunitaria tanto en ratones como en células. En particular, los modelos experimentales de estimulación inmunitaria mediados por ácidos nucleicos son fáciles y proporcionan mediciones fiables de las respuestas inmunitarias.
Las macromoléculas basadas en ácidos nucleicos, como los oligonucleótidos antisentido para el silenciamiento
génico, los ARN para la regulación/expresión génica o el ADN plasmídico para la expresión/edición génica son terapias prometedoras. Sin embargo, son potentes inductores de la respuesta inmunitaria innata en vertebrados [Sakurai et al. (2008) y Barbalat et al. (2011)]. Las nanopartículas basadas en lípidos o polímeros se utilizan a menudo como sistemas de administración para proteger estas macromoléculas de la degradación en fluidos biológicos y transportarlas al lugar de la enfermedad y al sitio intracelular de acción. A pesar de sus evidentes ventajas, estos sistemas provocan síntomas similares a los de la gripe e hipotensión debido a la activación de los receptores tipo Toll y al aumento de los niveles séricos de citoquinas en animales y pacientes, incluso en el caso de cargas útiles diseñadas para minimizar el potencial inmunoestimulador [Barros et al. (2012); Kumar et al. (2014); y Abe et al. (2011)]. Por lo tanto, la administración sistémica de nanopartículas lipídicas que contienen macromoléculas basadas en ácidos nucleicos como antisentido, ARNm y ADN plasmídico puede ser un modelo valioso para demostrar los efectos antiinflamatorios de los profármacos LD-DEX in vivo.
Los profármacos LD-DEX se formularon en nanopartículas lipídicas que contenían un oligonucleótido de ADN que contenía CpG inmunoestimulador (figura 12, figura 13 y figura 15) o ARNm (figura 14) utilizando una técnica de mezcla rápida como se indica en Materiales y métodos. Mediante esta técnica, las macromoléculas de ácidos nucleicos se incrustan en el espacio acuoso interno de las nanopartículas lipídicas. Todas las formulaciones tenían diámetros de partícula de aproximadamente 50 nm con índices de polidispersidad (PdI) ≤ 0,1 indicando que la incorporación del LD-DEX en la LNP no afectaba significativamente al tamaño de partícula ni a la homogeneidad. Los ratones inyectados con nanopartículas lipídicas que contenían oligonucleótido de ADN CpG inmunoestimulador (LNP-CpG) mostraron niveles elevados de TNFoí y k C/GRO en plasma (figura 12) 2 h después de la administración. Por el contrario, la incorporación de LD01-, LD02- y LD03-DEX en nanopartículas lipídicas dio lugar a una marcada reducción de los niveles de citoquinas en comparación con el LNP-CpG de control. Como era de esperar, el LD07-DEX que contenía un ligador no hidrolizable no mostró ningún efecto inmunosupresor. La figura 13 muestra que otros profármacos LD-DEX que contienen un ligador hidrolizable (LD13-, LD14-, LD17-, LD21- y LD24-DEX) fueron capaces de suprimir los niveles plasmáticos de citoquinas (TNFα, IL- 6 e IL-10) en comparación con el LNP-CpG de control. Estos resultados demostraron que la dexametasona activa se liberaba de profármacos unidos a lípidos para reducir las respuestas inmunitarias causadas por oligonucleótidos de ADN CpG inmunoestimuladores que contenían nanopartículas lipídicas.
En la figura 14 se muestra un modelo de ratón de estimulación inmunitaria mediada por ARNm. LD03-DEX se formuló en nanopartículas lipídicas que contenían un ARNm no modificado de 1,7 kb que codificaba la luciferasa de luciérnaga. Se extrajo sangre 2 h después de la inyección y se determinaron los niveles plasmáticos de citoquinas. Los animales tratados con LD03-DEX mostraron niveles significativamente reducidos de una serie de citoquinas plasmáticas en comparación con el grupo de control inyectado con LNP-ARNm que no contenía el profármaco LD-DEX. Estos resultados indicaron que se liberó dexametasona activa a partir de profármacos unidos a lípidos para reducir las respuestas inmunitarias causadas por ARNm que contenían nanopartículas lipídicas.
Se sabe que el ADN plasmídico (ADNp) estimula la inmunidad innata mediante la estimulación de los receptores TLR. Para demostrar los efectos inmunosupresores del LD-DEX in vivo, se realizó un modelo de ratón de estimulación inmunitaria mediada por ADNp (figura 15). En este modelo, el ADNp que codifica la luciferasa de luciérnaga se formuló en nanopartículas lipídicas con o sin LD03-DEX, y las formulaciones resultantes se inyectaron en ratones. Los niveles plasmáticos de citoquinas se determinaron 2 h después de la inyección. Al igual que en los otros modelos de estimulación inmunitaria descritos con anterioridad, la LNP de control sin LD03-DEX fue capaz de estimular la expresión de varias citoquinas. En cambio, la LNP que contenía LD03-DEX suprimió eficazmente los niveles de citoquinas, lo que sugiere que la dexametasona activa se escindió del profármaco ligado a lípidos para reducir las respuestas inmunitarias mediadas por el ADNp.
En la figura 16 se muestra un modelo celular de estimulación inmunitaria mediada por lipopolisacárido (LPS). Se formuló un subconjunto de profármacos LD-DEX en nanopartículas lipídicas. Los macrófagos RAW264.7 incubados con LPS y la formulación de control (elaborada sin ningún profármaco) mostraron una expresión elevada de la citoquina IL1p. En cambio, los profármacos LD03-, LD13-, LD14-, lD17-, LD21- y LD24-DEX que contenían nanopartículas lipídicas fueron capaces de reducir la expresión de IL1p en comparación con la formulación de control sin ningún profármaco. Como era de esperar, LD07-DEX mostró una expresión de IL1p a un nivel similar al control.
EJEMPLO 6: Efectos antiproliferativos de profármacos unidos a lípidos en células cancerosas
Los efectos antiproliferativos de los profármacos de LD-DTX se demostraron en células de cáncer de ovario OVCAR3. Los profármacos de LD-dTx (LD10-, LD18-, LD22- o LD23-DTX) se formularon en varios tipos de nanopartículas lipídicas, tal como se describe en Materiales y métodos. Las células se trataron con diversas concentraciones de docetaxel libre, profármacos de LD-DTX libres o formulaciones con o sin LD-DTX durante 48 h y se analizó la viabilidad celular mediante el ensayo MTT. Como se muestra en la figura 17, todos los profármacos de LD-DTX fueron capaces de suprimir la proliferación de células cancerosas. Como era de esperar, la cantidad de docetaxel libre necesaria para suprimir el crecimiento celular fue muy inferior a la de todos los demás artículos de ensayo. Curiosamente, el LD18-DTX mostró la IC50 (concentración necesaria para inhibir el crecimiento en un 50 %) más bajo en comparación con otros profármacos. Esto se debe probablemente al hecho de que el LD18-DTX es el
menos hidrófobo, lo que permite que se disocie y/o que las esterasas accedan a él más fácilmente que a los demás profármacos.
Tabla 7: Valores de IC50 para LD10, LD18, LD22 y LD23

Los compuestos descritos en la presente pueden sintetizarse como se describe en la presente, utilizando métodos modificados descritos en la presente o mediante métodos conocidos por un experto en la técnica.
REFERENCIAS
1. Charrois, G.J. and T.M. Allen, Drug release rate influences the pharmacokinetics, biodistribution, therapeutic activity, and toxicity of pegylated liposomal doxorubicin formulations in murine breast cancer. Biochim Biophys Acta, 2004. 1663(1-2): p. 167-77.
2. Cui, J., et al., Direct comparison of two pegylated liposomal doxorubicin formulations: is AUC predictive for toxicity and efficacy? J Control Release, 2007. 118(2): p. 204-15.
3. Johnston, M.J., et al., Therapeutically optimized rates of drug release can be achieved by varying the drug-to-lipid ratio in liposomal vincristine formulations. Biochim Biophys Acta, 2006. 1758(1): p. 55-64.
4. Nichols, J.W. and D.W. Deamer, Catecholamine uptake and concentration by liposomes maintaining p/ gradients. Biochim Biophys Acta, 1976. 455(1): p.269-71.
5. Mayer, L.D., et al., Techniques for encapsulating bioactive agents into liposomes. Chem Phys Lipids, 1986. 40(2-4): p. 333-45.
6. Allen, T.M. and P.R. Cullis, Liposomal drug delivery systems: from concept to clinical applications. Advanced Drug Delivery Reviews, 2013. 65(1): p. 36-48.
7. Mao, L., et al., Conjugation of two complementary anti-cancer drugs confers molecular hidrogels as a co-delivery system. Chem Commun (Camb), 2012. 48(3): p. 395-7.
8. Maclachlan, I. and P. Cullis, “Diffusible-PEG-Lipid Stabilized Plasmid Lipid Particles”. Adv Genet, 2005. 53PA: p. 157-188.
9. Jeffs, L.B., et al., A scalable, extrusion-free method for efficient liposomal encapsulation of plasmid DNA. Pharm Res, 2005. 22(3): p. 362-72.
10. Bligh, E.G. and W.J. Dyer, A rapid method of total lipid extraction and purification. Can J Biochem Physiol, 1959. 37(8): p. 911-7.
11. Leung, A.K., et al., Lipid Nanoparticles Containing siRNA Synthesized by Microfluidic Mixing Exhibit an Electron-Dense Nanostructured Core. The journal of physical chemistry. C, Nanomaterials and interfaces, 2012. 116(34): p. 18440-18450.
12. Chatham, W.W. and R.P. Kimberly, Treatment of lupus with corticosteroids. Lupus, 2001. 10(3): p.
140-7.
13. Rhen, T. and J.A. Cidlowski, Antiinflammatory Action of Glucocorticoids - New Mechanisms for Old Drugs. New England Journal of Medicine, 2005. 353(16): p. 1711-1723.
14. Inaba, H. and C.-H. Pui, Glucocorticoid use in acute lymphoblastic leukemia: comparison of prednisone and dexamethasone. The Lancet Oncology, 2010. 11(11): p. 1096-1106.
15. Teuffel, O., et al., Dexamethasone versus prednisone for induction therapy in childhood acute lymphoblastic leukemia: a systematic review and meta-analysis. Leukemia, 2011.25(8): p. 1232-1238. 16. Wilson, K.D., et al., The combination of stabilized plasmid lipid particles and lipid nanoparticle encapsulated CpG containing oligodeoxinucleotides as a systemic genetic vaccine. The journal of gene medicine, 2009. 11(1): p. 14-25.
17. Miyabo, S., et al., A comparison of the bioavailability and potency of dexamethasone phosphate and sulphate in man. Eur J Clin Pharmacol, 1981.20(4): p.277-82.
18. Rohdewald, P., et al., Pharmacokinetics of dexamethasone and its phosphate ester. Biopharm Drug Dispos, 1987. 8(3): p. 205-12.
19. Mossman, T. Rapid Colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. Journal of Immunological Methods, 1983. 65: p. 55-63.
20. Sakurai, H., et al., Innate immune response induced by gene delivery vectors. International journal of pharmaceutics, 2008. 354: p. 9-15.
21. Barbalat, R., et al., Nucleic acid recognition by the innate immune system. Annu Rev Immunol, 2011. 29: p. 185-214.
22. Barros, S.A., et al., Safety profile of RNAi nanomedicines. Adv Drug Deliv Rev, 2012. 64: p. 1730 1737.
23. Abrams, M.T., et al., Evaluation of efficacy, biodistribution, and inflammation for a potent siRNA nanoparticle: effect of dexamethasone co-treatment. Mol Ther, 2010. 18: p. 171-180.
24. Kumar, V., et al., Shielding of lipid nanoparticles for siRNA delivery: impact on physicochemical properties, cytokine induction, and efficacy. Mol Ther Nucleic acids, 2014. 3, e210.
25. Abe, M.; Niibayashi, R.; Koubori, S.; Moriyama, I.; Miyoshi, H.; Biochemistry 2011, 50, 8383.
26. Ohwada, J.; Inouye, Y.; Kimura, M.; Kakisawa, H. Bull. Chem. Soc. Jpn. 1990, 63, 287.
27. Fieser, L; Fieser, M. Reagents for Organic Synthesis 1967, 581-595.
28. Lopez, S.; Simons, Jr.; S. S. J. Med. Chem. 1991, 34, 1726 and reference therein.
29. Lee, K.; Ren, T.; Cote, M.; Gholamreza, B.; Misasi, J.; Bruchez, A.; Cunningham, J. ACS Med. Chem. Lett. 2013, 4, 239.
Claims (8)
1. Un compuesto, en donde el compuesto tiene la estructura de la Fórmula III:

en donde:
A-Z es dexametasona, en donde Z es un átomo electronegativo seleccionado de N u O, de modo tal que la dexametasona se derivó de un fármaco que tiene la fórmula A-Z-H cuando Z ha perdido un H para unirse covalentemente a Y;
Y es CH2 , C(=O) o PO32-;
T5 son 0-6 átomos de carbono; y
L4 es una cadena de carbono lineal o ramificada C9-C29, que tiene uno o más enlaces dobles C=C cis o trans y opcionalmente sustituida con OH o tiene la Fórmula V

en donde,
T2 son 1-4 átomos de carbono;
T3 son 0-4 átomos de carbono;
T4 son 0-4 átomos de carbono;
J1 son -O-C(=O)-L1;
J2 es -COOH, -OH, -NH3+, -NH2 , -SH, -NMe2 o -NHMe; y
L1 es una cadena de carbono lineal o ramificada C9-C29, que opcionalmente tiene uno o más enlaces dobles C=C cis o trans y opcionalmente sustituida con OH.
2. El compuesto de acuerdo con la reivindicación 1, en donde el compuesto tiene la estructura de la Fórmula II:

en donde:
Y es CH2 , C(=O), o PO32-;
T1 son 0-4 átomos de carbono;
T2 son 1-3 átomos de carbono; y
J2 es -COOH, -OH, -NH3+, -NH2 , -NMe2, o -NHMe.
3. El compuesto de acuerdo con la reivindicación 1, en donde el compuesto tiene la estructura de la Fórmula III:

en donde:
Y es CH2 , C(=O) o PO32-; y
T5 son 0-4 átomos de carbono.
4. El compuesto de acuerdo con cualquiera de las reivindicaciones 1-3, en donde Y es CH2 o C(=O); y T5 o T1 son 0 - 2 átomos de carbono.
5. El compuesto de acuerdo con la reivindicación 1, en donde el compuesto se selecciona de uno o más de los siguientes:

y 
6. El compuesto de acuerdo con la reivindicación 1, que es el compuesto:

7. Una composición farmacéutica, en donde la composición farmacéutica comprende un compuesto de acuerdo con cualquiera de las reivindicaciones 1-6 y un portador farmacéuticamente aceptable.
8. Un compuesto de acuerdo con cualquiera de las reivindicaciones 1-6 para su uso en el tratamiento del cáncer, enfermedad autoinmune o infección.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201562387160P | 2015-12-23 | 2015-12-23 | |
| PCT/CA2016/000322 WO2017106957A1 (en) | 2015-12-23 | 2016-12-22 | Lipid-linked prodrugs |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2954596T3 true ES2954596T3 (es) | 2023-11-23 |
Family
ID=59088669
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES16877035T Active ES2954596T3 (es) | 2015-12-23 | 2016-12-22 | Profármacos unidos a lípidos |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US10780174B2 (es) |
| EP (1) | EP3394080B1 (es) |
| AU (1) | AU2016379454B2 (es) |
| CA (1) | CA3008909C (es) |
| ES (1) | ES2954596T3 (es) |
| WO (1) | WO2017106957A1 (es) |
Families Citing this family (24)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR102727059B1 (ko) | 2015-02-16 | 2024-11-05 | 더 유니버서티 어브 퀸슬랜드 | 설포닐우레아와 관련 화합물 및 그 용도 |
| RS60237B1 (sr) | 2015-11-24 | 2020-06-30 | Theravance Biopharma R&D Ip Llc | Prolekovi jak inhibitornih jedinjenja za lečenje gastorintestinalne inflamatorne bolesti |
| US11465992B2 (en) | 2017-07-07 | 2022-10-11 | Inflazome Limited | Sulfonamide carboxamide compounds |
| US11542255B2 (en) | 2017-08-15 | 2023-01-03 | Inflazome Limited | Sulfonylureas and sulfonylthioureas as NLRP3 inhibitors |
| WO2019034693A1 (en) | 2017-08-15 | 2019-02-21 | Inflazome Limited | SULFONYLURATES AND SULFONYLTHIOURES AS INHIBITORS OF NLRP3 |
| MX2020001776A (es) | 2017-08-15 | 2020-03-24 | Inflazome Ltd | Sulfonilureas y sulfoniltioureas como inhibidores de nlrp3. |
| KR20200087759A (ko) | 2017-11-09 | 2020-07-21 | 인플라좀 리미티드 | 신규한 설폰아마이드 카복스아마이드 화합물 |
| US12221434B2 (en) | 2017-11-09 | 2025-02-11 | Inflazome Limited | Sulfonamide carboxamide compounds |
| EP3759077A1 (en) | 2018-03-02 | 2021-01-06 | Inflazome Limited | Novel compounds |
| EP3679952A1 (en) * | 2019-01-10 | 2020-07-15 | Université de Strasbourg | Nanoemulsions encapsulating prodrugs |
| CA3130349A1 (en) * | 2019-02-28 | 2020-09-03 | Puretech Lyt, Inc. | Lipid prodrugs of jak inhibitors and uses thereof |
| US10792292B2 (en) * | 2019-03-06 | 2020-10-06 | Propella Therapeutics, Inc. | Abiraterone prodrugs |
| US20220226480A1 (en) * | 2019-03-22 | 2022-07-21 | Integrated Nanotherapeutics Inc. | Lipid conjugate prepared from scaffold moiety |
| US11628178B2 (en) | 2019-03-26 | 2023-04-18 | Epalex Corporation | Fospropofol methods and compositions |
| US11478490B1 (en) | 2021-03-30 | 2022-10-25 | Epalex Corporation | Fospropofol formulations |
| US11439653B1 (en) | 2021-03-30 | 2022-09-13 | Epalex Corporation | Fospropofol formulations |
| US11547714B2 (en) | 2020-02-05 | 2023-01-10 | Epalex Corporation | Fospropofol salts, methods and compositions |
| IL300557A (en) | 2020-08-18 | 2023-04-01 | Incyte Corp | Process and intermediates for preparing a JAK1 inhibitor |
| PE20231743A1 (es) | 2020-08-18 | 2023-10-31 | Incyte Corp | Proceso e intermediarios para preparar un inhibidor de jak |
| CN116490165A (zh) * | 2020-09-02 | 2023-07-25 | 普洛佩拉治疗公司 | 阿比特龙前药 |
| US11957696B2 (en) | 2021-02-15 | 2024-04-16 | Propella Therapeutics, Inc. | Abiraterone prodrugs |
| US12071439B2 (en) | 2021-07-12 | 2024-08-27 | Incyte Corporation | Process and intermediates for preparing a JAK inhibitor |
| CN117355527B (zh) * | 2021-07-20 | 2024-06-07 | 上海椿安生物医药科技有限公司 | 一种消炎偶联化合物药物及其制法和应用 |
| US20240374734A1 (en) * | 2021-09-08 | 2024-11-14 | Integrated Nanotherapeutics Inc. | Immunomodulatory Combinations of Antigen and Drug-Lipid Conjugate |
Family Cites Families (17)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4933324A (en) | 1988-02-26 | 1990-06-12 | Shashoua Victor E | Fatty acid-neuroactive drug conjugate as a prodrug |
| US5952499A (en) | 1995-01-16 | 1999-09-14 | Commonwealth Scientific And Industrial Research Organisation | Therapeutic compound-fatty acid conjugates |
| US6090800A (en) | 1997-05-06 | 2000-07-18 | Imarx Pharmaceutical Corp. | Lipid soluble steroid prodrugs |
| US7235583B1 (en) | 1999-03-09 | 2007-06-26 | Luitpold Pharmaceuticals, Inc., | Fatty acid-anticancer conjugates and uses thereof |
| EP1427407A4 (en) | 2001-03-23 | 2005-05-11 | Luitpold Pharm Inc | FAT AMIN DRUG CONJUGATES |
| EP2222278B1 (en) | 2007-11-28 | 2015-04-08 | Celator Pharmaceuticals, Inc. | Improved taxane delivery system |
| KR101975843B1 (ko) * | 2008-05-23 | 2019-05-09 | 더 유니버시티 오브 브리티쉬 콜롬비아 | 리포좀 나노입자에 사용하기 위한 변형된 약물 |
| US20100240883A1 (en) * | 2009-03-18 | 2010-09-23 | Nian Wu | Lipid-drug conjugates for drug delivery |
| WO2011036557A1 (en) | 2009-09-22 | 2011-03-31 | The University Of British Columbia | Compositions and methods for enhancing cellular uptake and intracellular delivery of lipid particles |
| US20130245253A1 (en) | 2010-03-26 | 2013-09-19 | Department Of Veterans Affairs | Conjugated Neuroactive Steroid Compositions And Methods Of Use |
| ES2691671T3 (es) * | 2010-06-24 | 2018-11-28 | Alkermes Pharma Ireland Limited | Profármacos de compuestos NH-acídicos: derivados de éster, carbonato, carbamato y fosfonato |
| HRP20181855T1 (hr) | 2012-06-08 | 2018-12-28 | Nitto Denko Corporation | Lipidi za formulacije namijenjene unosu terapijskog sredstva |
| WO2014111815A2 (en) * | 2013-01-18 | 2014-07-24 | Cortendo Ab (Publ) | Abiraterone and analogs thereof for the treatment of diseases associated with cortisol overproduction |
| RU2015138893A (ru) * | 2013-03-08 | 2017-04-13 | Аллерган, Инк. | Конъюгаты антибиотиков, связанные со стероидными препаратами |
| CN104211760B (zh) * | 2013-06-05 | 2017-05-24 | 首都医科大学 | 饱和脂肪链醇,地塞米松和Glu‑Asp‑Gly的缀合物,其制备,纳米结构及应用 |
| CN104211762B (zh) * | 2013-06-05 | 2017-05-24 | 首都医科大学 | 饱和脂肪链醇,地塞米松和His‑Gly‑Lys的缀合物,其制备,纳米结构及应用 |
| BR112016022499A2 (pt) | 2014-04-03 | 2017-08-15 | Invictus Oncology Pvt Ltd | Produtos terapêuticos combinatórios supramoleculares |
-
2016
- 2016-12-22 EP EP16877035.2A patent/EP3394080B1/en active Active
- 2016-12-22 ES ES16877035T patent/ES2954596T3/es active Active
- 2016-12-22 WO PCT/CA2016/000322 patent/WO2017106957A1/en not_active Ceased
- 2016-12-22 CA CA3008909A patent/CA3008909C/en active Active
- 2016-12-22 AU AU2016379454A patent/AU2016379454B2/en active Active
- 2016-12-22 US US16/063,666 patent/US10780174B2/en active Active
Also Published As
| Publication number | Publication date |
|---|---|
| WO2017106957A1 (en) | 2017-06-29 |
| EP3394080B1 (en) | 2023-06-07 |
| AU2016379454A8 (en) | 2018-08-09 |
| US10780174B2 (en) | 2020-09-22 |
| EP3394080A1 (en) | 2018-10-31 |
| WO2017106957A8 (en) | 2017-07-20 |
| EP3394080A4 (en) | 2019-10-09 |
| CA3008909C (en) | 2023-10-03 |
| US20190151458A1 (en) | 2019-05-23 |
| AU2016379454A1 (en) | 2018-08-02 |
| CA3008909A1 (en) | 2017-06-29 |
| AU2016379454B2 (en) | 2021-01-28 |
| EP3394080C0 (en) | 2023-06-07 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2954596T3 (es) | Profármacos unidos a lípidos | |
| CA2725535C (en) | Modified drugs for use in liposomal nanoparticles | |
| JP7610208B2 (ja) | 足場部分から調製した脂質結合体 |