FR2472571A1 - Procede pour la preparation de derives d'esters halogeno-vincaminiques et halogeno-apovincaminiques - Google Patents
Procede pour la preparation de derives d'esters halogeno-vincaminiques et halogeno-apovincaminiques Download PDFInfo
- Publication number
- FR2472571A1 FR2472571A1 FR8027509A FR8027509A FR2472571A1 FR 2472571 A1 FR2472571 A1 FR 2472571A1 FR 8027509 A FR8027509 A FR 8027509A FR 8027509 A FR8027509 A FR 8027509A FR 2472571 A1 FR2472571 A1 FR 2472571A1
- Authority
- FR
- France
- Prior art keywords
- general formula
- acid
- eburnan
- derivative
- homo
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
- 238000000034 method Methods 0.000 title claims abstract description 15
- 150000002148 esters Chemical class 0.000 title claims abstract description 12
- 238000002360 preparation method Methods 0.000 title claims abstract description 4
- 239000002253 acid Substances 0.000 claims abstract description 34
- 150000001875 compounds Chemical class 0.000 claims abstract description 27
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 claims abstract description 17
- 238000006243 chemical reaction Methods 0.000 claims abstract description 15
- 239000007800 oxidant agent Substances 0.000 claims abstract description 8
- 229910052736 halogen Inorganic materials 0.000 claims abstract description 4
- 230000007062 hydrolysis Effects 0.000 claims abstract 2
- 238000006460 hydrolysis reaction Methods 0.000 claims abstract 2
- 239000000243 solution Substances 0.000 claims description 32
- 150000003839 salts Chemical class 0.000 claims description 20
- NUJOXMJBOLGQSY-UHFFFAOYSA-N manganese dioxide Chemical compound O=[Mn]=O NUJOXMJBOLGQSY-UHFFFAOYSA-N 0.000 claims description 18
- 150000007513 acids Chemical class 0.000 claims description 14
- 230000003287 optical effect Effects 0.000 claims description 13
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 claims description 10
- 230000015572 biosynthetic process Effects 0.000 claims description 10
- AVXURJPOCDRRFD-UHFFFAOYSA-N Hydroxylamine Chemical compound ON AVXURJPOCDRRFD-UHFFFAOYSA-N 0.000 claims description 8
- 125000004432 carbon atom Chemical group C* 0.000 claims description 7
- 230000003647 oxidation Effects 0.000 claims description 5
- 238000007254 oxidation reaction Methods 0.000 claims description 5
- 125000000217 alkyl group Chemical group 0.000 claims description 3
- 239000003795 chemical substances by application Substances 0.000 claims description 3
- 229910052783 alkali metal Inorganic materials 0.000 claims description 2
- 150000008044 alkali metal hydroxides Chemical class 0.000 claims description 2
- 150000001340 alkali metals Chemical class 0.000 claims description 2
- 239000007864 aqueous solution Substances 0.000 claims description 2
- 150000007529 inorganic bases Chemical class 0.000 claims description 2
- 125000005843 halogen group Chemical group 0.000 claims 1
- 238000011282 treatment Methods 0.000 abstract description 5
- 150000002367 halogens Chemical class 0.000 abstract description 3
- 230000002159 abnormal effect Effects 0.000 abstract 1
- 229940125890 compound Ia Drugs 0.000 abstract 1
- 239000003814 drug Substances 0.000 abstract 1
- 239000013067 intermediate product Substances 0.000 abstract 1
- 230000003236 psychic effect Effects 0.000 abstract 1
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 72
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 63
- 239000000047 product Substances 0.000 description 30
- 239000000203 mixture Substances 0.000 description 26
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 18
- 238000002844 melting Methods 0.000 description 18
- 230000008018 melting Effects 0.000 description 18
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 17
- 238000002329 infrared spectrum Methods 0.000 description 15
- 239000011541 reaction mixture Substances 0.000 description 15
- 239000013078 crystal Substances 0.000 description 13
- 239000000706 filtrate Substances 0.000 description 13
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 12
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 10
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 9
- 239000007787 solid Substances 0.000 description 9
- 239000002904 solvent Substances 0.000 description 9
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 9
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 8
- 229960000583 acetic acid Drugs 0.000 description 8
- 239000012362 glacial acetic acid Substances 0.000 description 7
- 238000001819 mass spectrum Methods 0.000 description 7
- 229960002726 vincamine Drugs 0.000 description 7
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 6
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 6
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 6
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 6
- 150000001408 amides Chemical class 0.000 description 6
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 6
- 239000012074 organic phase Substances 0.000 description 6
- 239000003960 organic solvent Substances 0.000 description 6
- LPNYRYFBWFDTMA-UHFFFAOYSA-N potassium tert-butoxide Chemical compound [K+].CC(C)(C)[O-] LPNYRYFBWFDTMA-UHFFFAOYSA-N 0.000 description 5
- 239000002244 precipitate Substances 0.000 description 5
- 235000017557 sodium bicarbonate Nutrition 0.000 description 5
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 5
- 239000000725 suspension Substances 0.000 description 5
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 4
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 4
- 229910021529 ammonia Inorganic materials 0.000 description 4
- -1 npentyl Chemical group 0.000 description 4
- 230000000144 pharmacologic effect Effects 0.000 description 4
- 239000007858 starting material Substances 0.000 description 4
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 4
- 229960000744 vinpocetine Drugs 0.000 description 4
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 3
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 3
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 3
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical group [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 3
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 3
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 3
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 3
- 239000003637 basic solution Substances 0.000 description 3
- 238000002425 crystallisation Methods 0.000 description 3
- 230000008025 crystallization Effects 0.000 description 3
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 3
- 238000001914 filtration Methods 0.000 description 3
- 235000019253 formic acid Nutrition 0.000 description 3
- IXCSERBJSXMMFS-UHFFFAOYSA-N hydrogen chloride Substances Cl.Cl IXCSERBJSXMMFS-UHFFFAOYSA-N 0.000 description 3
- 229910000041 hydrogen chloride Inorganic materials 0.000 description 3
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 3
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 3
- 238000001953 recrystallisation Methods 0.000 description 3
- 229920006395 saturated elastomer Polymers 0.000 description 3
- 238000000926 separation method Methods 0.000 description 3
- 239000000126 substance Substances 0.000 description 3
- HVBSAKJJOYLTQU-UHFFFAOYSA-N 4-aminobenzenesulfonic acid Chemical compound NC1=CC=C(S(O)(=O)=O)C=C1 HVBSAKJJOYLTQU-UHFFFAOYSA-N 0.000 description 2
- AEMRFAOFKBGASW-UHFFFAOYSA-N Glycolic acid Chemical compound OCC(O)=O AEMRFAOFKBGASW-UHFFFAOYSA-N 0.000 description 2
- WTDHULULXKLSOZ-UHFFFAOYSA-N Hydroxylamine hydrochloride Chemical compound Cl.ON WTDHULULXKLSOZ-UHFFFAOYSA-N 0.000 description 2
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 2
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 2
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 2
- 239000003513 alkali Substances 0.000 description 2
- 239000012670 alkaline solution Substances 0.000 description 2
- 210000004556 brain Anatomy 0.000 description 2
- 230000031709 bromination Effects 0.000 description 2
- 238000005893 bromination reaction Methods 0.000 description 2
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Chemical compound BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 2
- 229950002641 brovincamine Drugs 0.000 description 2
- 239000012141 concentrate Substances 0.000 description 2
- 125000004122 cyclic group Chemical group 0.000 description 2
- XBDQKXXYIPTUBI-UHFFFAOYSA-N dimethylselenoniopropionate Natural products CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 2
- 238000004090 dissolution Methods 0.000 description 2
- 238000000921 elemental analysis Methods 0.000 description 2
- 238000010828 elution Methods 0.000 description 2
- 125000004185 ester group Chemical group 0.000 description 2
- 125000004494 ethyl ester group Chemical group 0.000 description 2
- 239000012467 final product Substances 0.000 description 2
- 229910052739 hydrogen Inorganic materials 0.000 description 2
- 229910052500 inorganic mineral Inorganic materials 0.000 description 2
- 239000000543 intermediate Substances 0.000 description 2
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 2
- 239000011707 mineral Substances 0.000 description 2
- 235000010755 mineral Nutrition 0.000 description 2
- VLTRZXGMWDSKGL-UHFFFAOYSA-N perchloric acid Chemical compound OCl(=O)(=O)=O VLTRZXGMWDSKGL-UHFFFAOYSA-N 0.000 description 2
- 229910052700 potassium Inorganic materials 0.000 description 2
- 239000011591 potassium Substances 0.000 description 2
- 238000003786 synthesis reaction Methods 0.000 description 2
- RXPRRQLKFXBCSJ-GIVPXCGWSA-N vincamine Chemical compound C1=CC=C2C(CCN3CCC4)=C5[C@@H]3[C@]4(CC)C[C@](O)(C(=O)OC)N5C2=C1 RXPRRQLKFXBCSJ-GIVPXCGWSA-N 0.000 description 2
- DNIAPMSPPWPWGF-GSVOUGTGSA-N (R)-(-)-Propylene glycol Chemical compound C[C@@H](O)CO DNIAPMSPPWPWGF-GSVOUGTGSA-N 0.000 description 1
- UWYVPFMHMJIBHE-OWOJBTEDSA-N (e)-2-hydroxybut-2-enedioic acid Chemical compound OC(=O)\C=C(\O)C(O)=O UWYVPFMHMJIBHE-OWOJBTEDSA-N 0.000 description 1
- SCYULBFZEHDVBN-UHFFFAOYSA-N 1,1-Dichloroethane Chemical compound CC(Cl)Cl SCYULBFZEHDVBN-UHFFFAOYSA-N 0.000 description 1
- ZXSQEZNORDWBGZ-UHFFFAOYSA-N 1,3-dihydropyrrolo[2,3-b]pyridin-2-one Chemical compound C1=CN=C2NC(=O)CC2=C1 ZXSQEZNORDWBGZ-UHFFFAOYSA-N 0.000 description 1
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 1
- SDTMFDGELKWGFT-UHFFFAOYSA-N 2-methylpropan-2-olate Chemical compound CC(C)(C)[O-] SDTMFDGELKWGFT-UHFFFAOYSA-N 0.000 description 1
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 description 1
- GDRVFDDBLLKWRI-UHFFFAOYSA-N 4H-quinolizine Chemical compound C1=CC=CN2CC=CC=C21 GDRVFDDBLLKWRI-UHFFFAOYSA-N 0.000 description 1
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical group [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 1
- 206010000117 Abnormal behaviour Diseases 0.000 description 1
- ZETHHMPKDUSZQQ-UHFFFAOYSA-N Betulafolienepentol Natural products C1C=C(C)CCC(C(C)CCC=C(C)C)C2C(OC)OC(OC)C2=C1 ZETHHMPKDUSZQQ-UHFFFAOYSA-N 0.000 description 1
- 0 C*[C@](C1)(CI(CCC*2CC3)I2C1=C3C(C=C1)=C(*)C=C*1S)O Chemical compound C*[C@](C1)(CI(CCC*2CC3)I2C1=C3C(C=C1)=C(*)C=C*1S)O 0.000 description 1
- KZBUYRJDOAKODT-UHFFFAOYSA-N Chlorine Chemical compound ClCl KZBUYRJDOAKODT-UHFFFAOYSA-N 0.000 description 1
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 1
- 206010019196 Head injury Diseases 0.000 description 1
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-Glutamic acid Natural products OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 1
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 1
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 1
- OTCCIMWXFLJLIA-UHFFFAOYSA-N N-acetyl-DL-aspartic acid Natural products CC(=O)NC(C(O)=O)CC(O)=O OTCCIMWXFLJLIA-UHFFFAOYSA-N 0.000 description 1
- OTCCIMWXFLJLIA-BYPYZUCNSA-N N-acetyl-L-aspartic acid Chemical compound CC(=O)N[C@H](C(O)=O)CC(O)=O OTCCIMWXFLJLIA-BYPYZUCNSA-N 0.000 description 1
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 1
- CTQNGGLPUBDAKN-UHFFFAOYSA-N O-Xylene Chemical compound CC1=CC=CC=C1C CTQNGGLPUBDAKN-UHFFFAOYSA-N 0.000 description 1
- 229930040373 Paraformaldehyde Natural products 0.000 description 1
- OFOBLEOULBTSOW-UHFFFAOYSA-N Propanedioic acid Natural products OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 1
- PCSMJKASWLYICJ-UHFFFAOYSA-N Succinic aldehyde Chemical compound O=CCCC=O PCSMJKASWLYICJ-UHFFFAOYSA-N 0.000 description 1
- 235000011054 acetic acid Nutrition 0.000 description 1
- 125000001931 aliphatic group Chemical group 0.000 description 1
- 150000001338 aliphatic hydrocarbons Chemical class 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- 235000001014 amino acid Nutrition 0.000 description 1
- 229940024606 amino acid Drugs 0.000 description 1
- 150000001413 amino acids Chemical class 0.000 description 1
- 229950006936 apovincamine Drugs 0.000 description 1
- 150000004945 aromatic hydrocarbons Chemical class 0.000 description 1
- 125000003118 aryl group Chemical group 0.000 description 1
- 239000002585 base Substances 0.000 description 1
- 229940076134 benzene Drugs 0.000 description 1
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid group Chemical group C(C1=CC=CC=C1)(=O)O WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- 229910052794 bromium Inorganic materials 0.000 description 1
- WYIJGAVIVKPUGJ-GIVPXCGWSA-N brovincamine Chemical compound BrC1=CC=C2C(CCN3CCC4)=C5[C@@H]3[C@]4(CC)C[C@](O)(C(=O)OC)N5C2=C1 WYIJGAVIVKPUGJ-GIVPXCGWSA-N 0.000 description 1
- 230000002308 calcification Effects 0.000 description 1
- 229910052799 carbon Inorganic materials 0.000 description 1
- 230000015556 catabolic process Effects 0.000 description 1
- 239000007795 chemical reaction product Substances 0.000 description 1
- 239000000460 chlorine Substances 0.000 description 1
- 229910052801 chlorine Inorganic materials 0.000 description 1
- 229940117975 chromium trioxide Drugs 0.000 description 1
- WGLPBDUCMAPZCE-UHFFFAOYSA-N chromium trioxide Inorganic materials O=[Cr](=O)=O WGLPBDUCMAPZCE-UHFFFAOYSA-N 0.000 description 1
- GAMDZJFZMJECOS-UHFFFAOYSA-N chromium(6+);oxygen(2-) Chemical compound [O-2].[O-2].[O-2].[Cr+6] GAMDZJFZMJECOS-UHFFFAOYSA-N 0.000 description 1
- 238000003776 cleavage reaction Methods 0.000 description 1
- 238000001816 cooling Methods 0.000 description 1
- 150000004292 cyclic ethers Chemical class 0.000 description 1
- ZHGASCUQXLPSDT-UHFFFAOYSA-N cyclohexanesulfonic acid Chemical compound OS(=O)(=O)C1CCCCC1 ZHGASCUQXLPSDT-UHFFFAOYSA-N 0.000 description 1
- 238000000354 decomposition reaction Methods 0.000 description 1
- 238000006731 degradation reaction Methods 0.000 description 1
- 230000018044 dehydration Effects 0.000 description 1
- 238000006297 dehydration reaction Methods 0.000 description 1
- HEOKFDGOFROELJ-UHFFFAOYSA-N diacetal Natural products COc1ccc(C=C/c2cc(O)cc(OC3OC(COC(=O)c4cc(O)c(O)c(O)c4)C(O)C(O)C3O)c2)cc1O HEOKFDGOFROELJ-UHFFFAOYSA-N 0.000 description 1
- RXPRRQLKFXBCSJ-UHFFFAOYSA-N dl-Vincamin Natural products C1=CC=C2C(CCN3CCC4)=C5C3C4(CC)CC(O)(C(=O)OC)N5C2=C1 RXPRRQLKFXBCSJ-UHFFFAOYSA-N 0.000 description 1
- 238000001035 drying Methods 0.000 description 1
- 230000000694 effects Effects 0.000 description 1
- 239000003480 eluent Substances 0.000 description 1
- CCIVGXIOQKPBKL-UHFFFAOYSA-N ethanesulfonic acid Chemical compound CCS(O)(=O)=O CCIVGXIOQKPBKL-UHFFFAOYSA-N 0.000 description 1
- 238000001704 evaporation Methods 0.000 description 1
- 230000008020 evaporation Effects 0.000 description 1
- 239000000284 extract Substances 0.000 description 1
- 229910052731 fluorine Inorganic materials 0.000 description 1
- 239000011737 fluorine Substances 0.000 description 1
- 239000012458 free base Substances 0.000 description 1
- 239000007789 gas Substances 0.000 description 1
- 229960002989 glutamic acid Drugs 0.000 description 1
- 229940093915 gynecological organic acid Drugs 0.000 description 1
- 230000026030 halogenation Effects 0.000 description 1
- 238000005658 halogenation reaction Methods 0.000 description 1
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 1
- 150000002443 hydroxylamines Chemical class 0.000 description 1
- 239000005457 ice water Substances 0.000 description 1
- 229910052740 iodine Inorganic materials 0.000 description 1
- 125000004491 isohexyl group Chemical group C(CCC(C)C)* 0.000 description 1
- 238000002955 isolation Methods 0.000 description 1
- 125000001972 isopentyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 239000007788 liquid Substances 0.000 description 1
- 229910052744 lithium Inorganic materials 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 239000011976 maleic acid Substances 0.000 description 1
- 238000004519 manufacturing process Methods 0.000 description 1
- 239000000155 melt Substances 0.000 description 1
- 230000003340 mental effect Effects 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- SYSQUGFVNFXIIT-UHFFFAOYSA-N n-[4-(1,3-benzoxazol-2-yl)phenyl]-4-nitrobenzenesulfonamide Chemical class C1=CC([N+](=O)[O-])=CC=C1S(=O)(=O)NC1=CC=C(C=2OC3=CC=CC=C3N=2)C=C1 SYSQUGFVNFXIIT-UHFFFAOYSA-N 0.000 description 1
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 229910017604 nitric acid Inorganic materials 0.000 description 1
- 229910052757 nitrogen Inorganic materials 0.000 description 1
- 239000012299 nitrogen atmosphere Substances 0.000 description 1
- 238000000655 nuclear magnetic resonance spectrum Methods 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- 150000002923 oximes Chemical class 0.000 description 1
- 125000004043 oxo group Chemical group O=* 0.000 description 1
- 229920002866 paraformaldehyde Polymers 0.000 description 1
- 239000012071 phase Substances 0.000 description 1
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 1
- WLJVXDMOQOGPHL-UHFFFAOYSA-N phenylacetic acid Chemical compound OC(=O)CC1=CC=CC=C1 WLJVXDMOQOGPHL-UHFFFAOYSA-N 0.000 description 1
- 229920000137 polyphosphoric acid Polymers 0.000 description 1
- 235000019260 propionic acid Nutrition 0.000 description 1
- 230000001681 protective effect Effects 0.000 description 1
- 238000000746 purification Methods 0.000 description 1
- 238000011403 purification operation Methods 0.000 description 1
- IUVKMZGDUIUOCP-BTNSXGMBSA-N quinbolone Chemical compound O([C@H]1CC[C@H]2[C@H]3[C@@H]([C@]4(C=CC(=O)C=C4CC3)C)CC[C@@]21C)C1=CCCC1 IUVKMZGDUIUOCP-BTNSXGMBSA-N 0.000 description 1
- 238000010992 reflux Methods 0.000 description 1
- 150000003870 salicylic acids Chemical class 0.000 description 1
- 230000007017 scission Effects 0.000 description 1
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 239000000741 silica gel Substances 0.000 description 1
- 229910002027 silica gel Inorganic materials 0.000 description 1
- LKZMBDSASOBTPN-UHFFFAOYSA-L silver carbonate Substances [Ag].[O-]C([O-])=O LKZMBDSASOBTPN-UHFFFAOYSA-L 0.000 description 1
- 229910001958 silver carbonate Inorganic materials 0.000 description 1
- SYXYWTXQFUUWLP-UHFFFAOYSA-N sodium;butan-1-olate Chemical compound [Na+].CCCC[O-] SYXYWTXQFUUWLP-UHFFFAOYSA-N 0.000 description 1
- 239000012265 solid product Substances 0.000 description 1
- 238000001228 spectrum Methods 0.000 description 1
- 230000000638 stimulation Effects 0.000 description 1
- 238000003756 stirring Methods 0.000 description 1
- 125000001424 substituent group Chemical group 0.000 description 1
- 229950000244 sulfanilic acid Drugs 0.000 description 1
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 1
- 238000004809 thin layer chromatography Methods 0.000 description 1
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 1
- 125000005270 trialkylamine group Chemical group 0.000 description 1
- 229940124549 vasodilator Drugs 0.000 description 1
- 239000003071 vasodilator agent Substances 0.000 description 1
- 238000005406 washing Methods 0.000 description 1
- 239000008096 xylene Substances 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D461/00—Heterocyclic compounds containing indolo [3,2,1-d,e] pyrido [3,2,1,j] [1,5]-naphthyridine ring systems, e.g. vincamine
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
PROCEDE DE PREPARATION DE DERIVES D'ESTERS HALOGENO-VINCAMINIQUES ET HALOGENO-APOVINCAMINIQUES REPONDANT AUX FORMULES GENERALES: (CF DESSIN DANS BOPI) ET OU: (CF DESSIN DANS BOPI) DANS LESQUELLES R ET RALKYLE EN C1-C6 ET XHALOGENE. ON TRAITE UN DERIVE HALOGENE D'HYDROXY-OXO-E-HOMOEBURNANE DE FORMULE GENERALE: (CF DESSIN DANS BOPI) PAR UN AGENT OXYDANT, ON FAIT REAGIR LE PRODUIT INTERMEDIAIRE OBTENU AVEC UN ALCOOL R-OH EN MILIEU ALCALIN, CE QUI DONNE UN COMPOSE IA QU'ON PEUT CONVERTIR, PAR HYDROLYSE PUIS REACTION AVEC UN ALCOOL R-OH EN PRESENCE D'UN ACIDE CONCENTRE DESHYDRATANT, EN UN COMPOSE IB. LES COMPOSES PREPARES SELON L'INVENTION SONT DES MEDICAMENTS UTILISES POUR LE TRAITEMENT DE CERTAINS ETATS PSYCHIQUES ANORMAUX.
Description
La présente invention a pour objet un procédé nouveau pour préparer les dérivés d'esters halogéno-vincaminiques et halogéno-apovincaminiques répondant aux formules générales
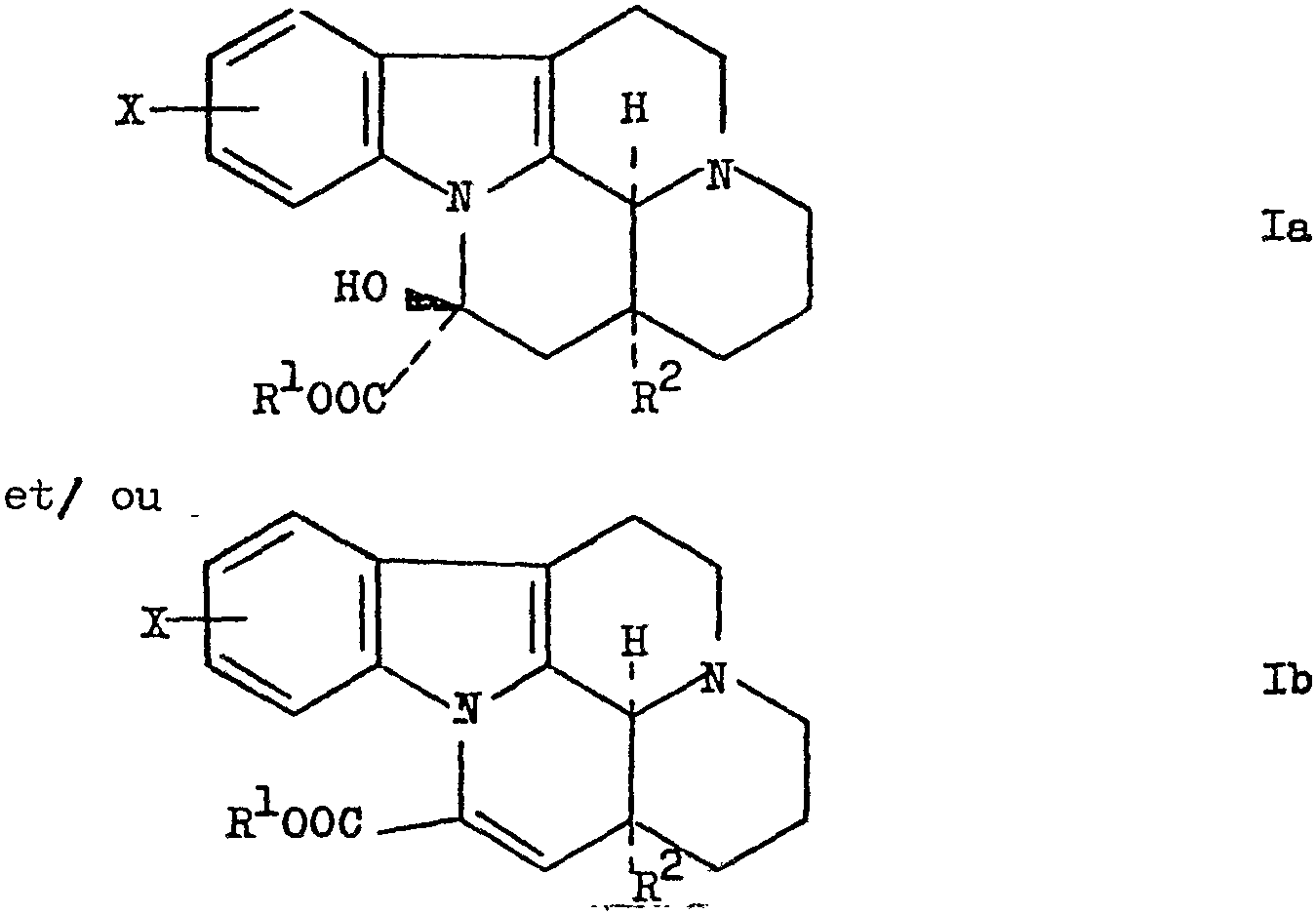
dans lesquelles R1 et -R2 représentent des groupes alkyles de 1 à 6 atomes de carbone et X représente un halogène, leurs sels formés par addition avec des acides acceptables pour l'usage pharmaceutique et leurs antipodes optiques, ce procédé étant caractérisé en ce que l'on traite un dérivé halogéné d'hydroxy-oxo-E-homo-éburnane répondant à la formule générale

dans laquelle R2 et X ont les sgnimications indiquées ci-dessus, ou ses épimères, antipodes optiques ou sels formés par addition avec des acides, par un agent oxydant et on fait réagir le dérivé hélogéné de dioxo-ehomo-ébur- nane ainsi obtenu, qui répond à la formule générale

dans laquelle R2 et X ont les significations indiquées cidessus, ou le cas échéant le dérivé d1hydroxyimino-oxo-é- burnane formé après réaction avec l'hydroxylamine ou un sel d'hydroxylamine, qui répond à la formule générale

dans laquelle R2 et X ont les significations indiquées ci-dessus, ou ses sels formés par addition avec des acides, le cas échéant après résolution, avec un alcanol répondant à la formule générale R1-OH dans laquelle R1 a les significations indiquées ci-dessus, en présence d'un agent alcalin, après quoi, si on le désire, on traite le dérivé d'ester halogéno-vincaminique formé, qui répond à la formule générale Ia dans laquelle R1, R2 et X ont les signification! indiquées ci-dessus, par un acide approprié à la formation de sels acceptables pour l'usage pharmaceutique, et/ou le résout ou on l'hydrolyse, et on fait réagir le dérivé d'acide halogéno-vincaminique obtenu, qui répond à la formule générale

dans laquelle R2 et X ont les significations indiquées cidessus, ou le dérivé dthydroxyimino-oxo-E-éburnane de formule générale IV dans laquelle R2 et X ont les significations indiquées ci-dessus, ou un sel de ce composé formé par addition avec un acide, si on le désire après résolution, avec un alcool de formule générale R1 -OH - dans laquelle R1, R2 et X ont les significations indiquées cidessus - en présence d'un acide concentré déshydratant, et, si on le désire, on traite le dérivé d'ester halogénoapovincaminique obtenu, qui répond à la formule générale Ib dans laquelle R1, R2 et X ont les significations indiquées ci-dessus, par un acide approprié à la formation de sels acceptables pour l'usage pharmaceutique, et/ou on le résout.
dans lesquelles R1 et -R2 représentent des groupes alkyles de 1 à 6 atomes de carbone et X représente un halogène, leurs sels formés par addition avec des acides acceptables pour l'usage pharmaceutique et leurs antipodes optiques, ce procédé étant caractérisé en ce que l'on traite un dérivé halogéné d'hydroxy-oxo-E-homo-éburnane répondant à la formule générale
dans laquelle R2 et X ont les sgnimications indiquées ci-dessus, ou ses épimères, antipodes optiques ou sels formés par addition avec des acides, par un agent oxydant et on fait réagir le dérivé hélogéné de dioxo-ehomo-ébur- nane ainsi obtenu, qui répond à la formule générale
dans laquelle R2 et X ont les significations indiquées cidessus, ou le cas échéant le dérivé d1hydroxyimino-oxo-é- burnane formé après réaction avec l'hydroxylamine ou un sel d'hydroxylamine, qui répond à la formule générale
dans laquelle R2 et X ont les significations indiquées ci-dessus, ou ses sels formés par addition avec des acides, le cas échéant après résolution, avec un alcanol répondant à la formule générale R1-OH dans laquelle R1 a les significations indiquées ci-dessus, en présence d'un agent alcalin, après quoi, si on le désire, on traite le dérivé d'ester halogéno-vincaminique formé, qui répond à la formule générale Ia dans laquelle R1, R2 et X ont les signification! indiquées ci-dessus, par un acide approprié à la formation de sels acceptables pour l'usage pharmaceutique, et/ou le résout ou on l'hydrolyse, et on fait réagir le dérivé d'acide halogéno-vincaminique obtenu, qui répond à la formule générale
dans laquelle R2 et X ont les significations indiquées cidessus, ou le dérivé dthydroxyimino-oxo-E-éburnane de formule générale IV dans laquelle R2 et X ont les significations indiquées ci-dessus, ou un sel de ce composé formé par addition avec un acide, si on le désire après résolution, avec un alcool de formule générale R1 -OH - dans laquelle R1, R2 et X ont les significations indiquées cidessus - en présence d'un acide concentré déshydratant, et, si on le désire, on traite le dérivé d'ester halogénoapovincaminique obtenu, qui répond à la formule générale Ib dans laquelle R1, R2 et X ont les significations indiquées ci-dessus, par un acide approprié à la formation de sels acceptables pour l'usage pharmaceutique, et/ou on le résout.
Dans les formules Ia et Ib, R1 et R2 représentent des groupes alkyles à chaîne droite ou ramifiée de 1 à 6 atomes de carbone, par exemple méthyle, éthyle, n-propyle, iso-propyle, n-butyle, .sec-butyle, tert-butyle, npentyle, iso-pentyle, n'hexyle ou iso-hexyle.
L'halogène dont il est question dans les formules Ia et Ib peut consister en un atome de fluor, de chlore, de brome ou d'iode.
Le substituant X des formules Ia et Ib peut être fixé sur un atome de carbone quelconque du noyau ben zénique, mais le procédé selon l'invention sert de préférence à préparer des dérivés d'esters 9, 10- et il-ha- logéno-vincaminiques.
Les composés répondant à la formule générale Ia, qui sont connus, présentent une activité pharmacologique intéressante; ils conviennent à l'utilisation pour le traitement des comportements anormaux résultant, avec l'âge, de la dégradation et de la calcification des vaisseaux du cerveau, pour le traitement des troubles de la conscience causés par les traumatismes crâniens, et pour la stimulation de la capacité mentale et de la fraîcheur d'esprit.
Certains des composés de formule générale Ia ont été décrits antérieurement dans le brevet de la RFA nO 2 458 164. Selon ce brevet, on prépare un mélange de 17-halogéno-vincamine et de 11-halogéno-apovincamine par scission acide de l'ester méthylique de l'acide 3-(1 éthyl-1 0-halogéno-octahydro-indolo-quinolizine-i -yl )-2- méthoxy-propénoSque. Après la réaction, il faut encore isoler la 11-halogéno-vincamine du mélange.Le produit de départ est préparé à partir de lthalogénotryptamine correspondante et du diacétal de l'ester éthylique de 1 'acide -aldéhyde ( p-t oluène-sulf onyloxy ) -propa-1 -y7- malonique par une synthèse en cinq stades opératoires, passant par des produits intermédiaires compliqués. D'après le meme brevet, on obtient par exemple la 11-bromo- vincamine par bromation de la vincamine qui présente également un intérêt pharmacologique mais on n'indique aucun rendement; d'après les essais de la demanderesse, ce rendement n'est pas suffisant : il est de 35%.
Les composés répondant à la formule générale Ib présentent également une activité pharmacologique intéressante : activité anti-hypoxémique et protectrice du cerveau.
Les composés répondant à la formule générale Ib dans laquelle R2 représente le groupe éthyle, X un atome de brome, ceux pour lesquels X est un atome de brome fixé en position 10 ou i1 du cycle, R1 représentant un groupe éthyle ou n-butyle et ceux pour lesquels X représente un atome de brome fixé en position 9 du cycle, R1 représentant le groupe n-butyle, sont connus; tous les autres composés répondant à la formule générale Ib sont nouveaux.
La préparation des composés connus est décrite dans la demande de brevet européen nO 1940. D'après la description de cette demande de brevet, on brome l'ester vincaminique correspondant, ce qui donne un mélange des esters 9-, 10- et 71-bromo-vincaminiques. On isole ensuite les isomères individuels du mélange et on élimine l'eau par l'acide formique. On ne donne aucune indication de rendement. Du point de vue industriel, le procédé présente des inconvénients importants. A la bromation, on n'obtient pas un produit unique mais un mélange d'isomères. Ce mélange doit encore être soumis à toutes les opérations de séparation. Par suite, le rendement en les isomères individuels est évidemment beaucoup plus bas que si l'on formait un seul isomère.Et lors de la déshydratation par l'acide formique, la séparation du produit à partir du mélange de réaction offre encore des difficultés.
Conformément à l'invention, les composés répondant aux formules générales Ia et Ib peuvent être obtenus à partir de produits de départ eux-mêmes faciles à préparer, en passant par des produits intermédiaires simples et dans deux stades de synthèse d'une mise en oeuvre simple, avec de bons rendements et dans de bons états de pureté.
Les composés répondant à la formule générale II ont également une activité pharmacologique: ils ont une activité vasodilatatrice et peuvent être préparés de manière simple avec de bons rendements.
Les composés de départ répondant à la formule générale II dans laquelle X est fixé en position 9 ou il du système cyclique peuvent être obtenus par halogénation directe du dérivé de 14-oxo-15-hydroxy-E-homo-éburnane non substitué correspondant (demande de brevet hongrois publiée sous nO RI-721).
Les composés de départ répondant à la formule générale II dans laquelle X est fixé en position 10 du système cyclique peuvent être obtenus par traitement alcalin de la 9-halogéno-1 -( 2-hydroxy-2-alcoxycarbonyl-é- thyl) -octahydro-indolo,3-a7 quinolizine correspondante (demande de brevet hongrois publiée sous nO RI - 723).
Les composés répondant à la formule générale II peuvent être oxydés au moyen d'un agent oxydant quelconque capable de convertir le groupe hydroxy en position 15 en groupe oxo sans modification indésirable à d'autres endroits de la molécule. Parmi les agents oxydants qui conviennent, on peut citer le bioxyde de manganèse, le carbonate d'argent déposé sur Celite, le trioxyde de chrome et d'autres encore.
De préférence, pour l'oxydation des composés de formule générale II, on utilise un agent oxydant actif déposé sur un support à grande surface, lui-même inactif dans la réaction, par exemple la Celite, et l'agent oxydant est de-préférence le bioxyde de manganèse actif (Tetrahedron 33, 1803 (1979)).
Le bioxyde de manganèse est de préférence utilisé en quantité de 8 à 10 fois et plus particulièrement de 10 fois la quantité du produit de départ.
L'oxydation des composés de formule générale II est réalisée dans un solvant organique aprotonique apolaire inerte qui ne participe pas à la réaction. Parmi ces solvants, on citera des hydrocarbures aliphatiques éventuellement halogénés comme le chloroforme, le dichlorométhane, etc., des hydrocarbures aromatiques comme le toluène, le xylène, etc., des éthers cycliques comme le dioxanne, le tétrahydrofuranne etc.
L'oxydation est effectuée à une température de 20 à 500C et de préférence à température ambiante.
Pour la réaction des composés de formule générale III formés par oxydation avec l'hydroxylamine, cette dernière est de préférence utilisée à l'état de sel, par exemple d'halogénhydrate, entre autres le chlorhydrate.
La réaction est avantageusement effectuée dans un excès d'hydroxylamine de 3 à 5 fois.
La formation d'oxime à l'aide de l'hydroxylamine est réalisée dans un solvant organique inerte. Du fait qu'au cours de la réaction il y a non seulement formation d'eau mais également libération d'un acide, il est recommandé d'ajouter au mélange de réaction un agent fixant les acides. On peut utiliser par exemple des trialkylamines comme la triéthylamine. On peut également utiliser un solvant qui, en raison de ses propriétés basiques, soit capable de fixer l'acide libéré. La pyridine par exemple constitue un tel solvant.
La réaction avec l'hydroxylamine est réalisée à chaud, à une température de 80 à 1100C.
Les composés répondant à la formule générale
III ou IV sont mis à réagir avec un alcool de formule générale R1 -OH dans laquelle R1 correspond au groupe ester à introduire dans la molécule, en présence d'un agent alcalin, de préférence un tert-alcoolate de métal alcalin tel que le tert-butylate de lithium, de potassium ou de sodium.
III ou IV sont mis à réagir avec un alcool de formule générale R1 -OH dans laquelle R1 correspond au groupe ester à introduire dans la molécule, en présence d'un agent alcalin, de préférence un tert-alcoolate de métal alcalin tel que le tert-butylate de lithium, de potassium ou de sodium.
Les composés répondant à la formule générale Ia sont hydrolysés à l'aide d'une base minérale, par exemple un hydroxyde alcalin comme l'hydroxyde de potassium, l'hydroxyde de sodium, dans une solution d'un alcanol en C1-C6.
Les composés de formule générale IV ou V sont mis à réagir avec un alcool de formule générale R1 -OH dans laquelle R1 correspond au groupe ester à introduire dans la molécule, en présence d'un acide minéral concentré déshydratant, par exemple l'acide sulfurique concencentré, l'acide polyphosphorique, etc., ou l'acide oxa lique anhydre, etc.
Les traitements du mélange de réaction dans une phase quelconque des opérations et l'isolement du produit final peuvent être réalisés selon des techniques connues et qui sont fonction de la nature des composants de départ, du produit final, du solvant, etc; ainsi par exemple, lorsque, à la fin de la réaction, le produit précipite. On peut séparer le précipité par filtration, si, par contre, le produit reste en solution, on peut l'isoler avantageusement en concentrant sous vide.
Le résidu sec est ensuite cristallisé dans un solvant organique inerte approprié. Le solvant est choisi en fonction des propriétés de dissolution et de cristallisation à l'égard de la substance à faire cristalliser.
Pour isoler le produit de réaction du mélange de réaction, on peut égale ent opére.r par extraction du produit à l'aide d'un solvant organique inerte comme le dichlorométhane, le dichloréthane, etc., en faisant suivre d'un séchage et d'une évaporation de la solution organique et le cas échéant d'une cristallisation du résidu. Le produit recherche peut également être précipité du mélange de réaction par addition d'un solvant organique inerte tel que l'éther, la substance étant ensuite isolée par filtration.
Les composés répondant aux formules Ia et Ib peuvent être mis à réagir avec des acides qui donnent des sels formés par addition et qui sont acceptables pour l'usage pharmaceutique.
Pour la formation des sels, on peut utiliser par exemple les acides suivants : les acides minéraux comme les hydracides halogénés, en particulier l'acide chlorhydrique ou l'acide bromhydrique, l'acide sulfurique, l'acide phosphorique, l'acide nitrique, les acides perhalogénés comme l'acide perchlorique, etc.; des acides organiques comme l'acide formique, l'acide acétique, l'acide propionique, l'acide glycolique, l'acide maléique, l'acide hydroxymaléique, les acides fumarique, succinique, tartrique, ascorbique, citrique, malique, salicylique, lactique, cinnamique, benzoïque, phénylacétique, p-aminobenzolque, p-hydroxybenzolque, p-aminosalicylique etc., des acides alcane-sulfoniques comme l'acide méthane-sulfonique, l'acide éthane-sulfonique, etc., des acides sulfoniques cycloaliphatiques et par exemple l'acide cyclohexyl-sulfonique, des acides aryl-sulfoniques et par exemple l'acide p-toluène-sulfonique, l'acide naphtylsulfonique, l'acide sulfanilique, etc., des aminoacides et par exemple l'acide aspartique, l'acide glutamique, l'acide N-acétyl-aspartique, 1 'acide N-acétyl-glutarique, etc.
La formation du sel est réalisée dans un solvant organique inerte, par exemple un alcool aliphatique contenant de 1 à 6 atomes de carbone, par dissolution des composés racémiques ou énantiomères de formule Ta ou Ib dans ce solvant et addition de l'acide, tel quel ou dans le même solvant, jusqu'à acidité modérée du mélange, (jusqu'à pH 5 à 6). Le sel formé par addition et qui précipite peut être séparé du mélange par une technique simple, par exemple par filtration.
Les composés racémiques répondant aux formules générales Ia et Ib sont résolus par des techniques connues mais on peut également utiliser comme composants de départ des isomères optiques des composés de formule générale II.
Les isomères optiques et racémates des composés répondant aux formules générales Ia et Ib et leurs sels formés par addition avec des acides peuvent, lorsqu'on le désire, être soumis à d'autres opérations de purification, par exemple à la recristallisation. La nature du solvant à utiliser pour la recristallisation est fonction des possibilités de dissolution et de cristallisation de la substance qu'on veut faire cristalliser. On peut utiliser des alcanols aliphatiques contenant de 1 à 6 atomes de carbone, l'acétonitrile, etc.
Le procédé selon l'invention permet de préparer les composés répondant aux formules générales Ia et Ib sous une forme identifiable. Les spectres infra-rouges et les valeurs du spectre de masse confirment sans équivoque la structure des composés.
Les exemples qui suivent illustrent l'invention sans toutefois en limiter la portée; dans ces exemples, les indications de parties et de pourcentages s'entendent en poids sauf mention contraire.
Exemple 1
9-bromo-14,75-dioxo-E-homo-éburnane(3 alpha, 17 alpha)
A une solution de 0,25 g (0,62 mmole) de 9 bromo-14-oxo-15-hydroxy-4-homo-éburnane (3 alpha, 17 alpha) dans 10 ml de dichlorométhane anhydre, on ajoute 2,0 g de bioxyde de manganèse actif (Merck) et on agite le mélange de réaction à température ambiante pendant 3 h. On filtre la partie solide de la suspension et on la lave à trois reprises avec 3 ml de dichlorométhane anhydre. On combine le filtrat avec le liquide de lave et on concentre sous vide. On fait cristalliser le résidu huileux dans 3 ml d'éther. On obtient 0,13 g du produit recherché à l'état cristallisé dans 3 ml d'éther.
9-bromo-14,75-dioxo-E-homo-éburnane(3 alpha, 17 alpha)
A une solution de 0,25 g (0,62 mmole) de 9 bromo-14-oxo-15-hydroxy-4-homo-éburnane (3 alpha, 17 alpha) dans 10 ml de dichlorométhane anhydre, on ajoute 2,0 g de bioxyde de manganèse actif (Merck) et on agite le mélange de réaction à température ambiante pendant 3 h. On filtre la partie solide de la suspension et on la lave à trois reprises avec 3 ml de dichlorométhane anhydre. On combine le filtrat avec le liquide de lave et on concentre sous vide. On fait cristalliser le résidu huileux dans 3 ml d'éther. On obtient 0,13 g du produit recherché à l'état cristallisé dans 3 ml d'éther.
On obtient 0,13 g du produit recherché à l'état cristallisé et fondant à 135 - 1370C, ce qui correspond à un rendement de 52,3%.
formule empirique : C20H21N2o2Br (poids molé
culaire : 401,31) spectre IR (KBR) : [# max 1730 cm-

culaire : 401,31) spectre IR (KBR) : [# max 1730 cm-
1695 cm- (CO d'amide)] spectre de masse : (m/e) : ( 402, 401, 400, 399,
387, 385, 375, 373, 371, 359,
357, 346, 344, 331, 329, 317,
315, 277, 275, 180, 167, 153,
140.)
Exemple 2
9-bromo-vincamine
On dissout 0,12 g (0,3 mmoles) de 9-bromo-1,4, 15-dioxo-E-homo-éburnane (3 alpha, 17 alpha) à 400C dans 5 ml de méthanol anhydre, on ajoute à la solution 10 mg de tert-butylate de potassium et on abandonne le mélange au repos pendant 2 h à température ambiante. On neutralise ensuite à pH 7 par l'acide acétique glacial et on concentre sous vide.On ajoute au résidu 15 ml d'une solution aqueuse de bicarbonate de sodium à 5% et on extrait le mélange à trois reprises par 10 ml de dichlorométhane. Les phases organiques combinées sont séchées sur sulfate de sodium anhydre,filtrées, et le filtrat concentré sous vide. Le résidu huileux est cristallisé dans 2 ml de méthanol.
387, 385, 375, 373, 371, 359,
357, 346, 344, 331, 329, 317,
315, 277, 275, 180, 167, 153,
140.)
Exemple 2
9-bromo-vincamine
On dissout 0,12 g (0,3 mmoles) de 9-bromo-1,4, 15-dioxo-E-homo-éburnane (3 alpha, 17 alpha) à 400C dans 5 ml de méthanol anhydre, on ajoute à la solution 10 mg de tert-butylate de potassium et on abandonne le mélange au repos pendant 2 h à température ambiante. On neutralise ensuite à pH 7 par l'acide acétique glacial et on concentre sous vide.On ajoute au résidu 15 ml d'une solution aqueuse de bicarbonate de sodium à 5% et on extrait le mélange à trois reprises par 10 ml de dichlorométhane. Les phases organiques combinées sont séchées sur sulfate de sodium anhydre,filtrées, et le filtrat concentré sous vide. Le résidu huileux est cristallisé dans 2 ml de méthanol.
On obtient 0,074 g du composé recherché à l'é- tat de cristaux blancs fondant à 210-2110C, ce qui correspond à un rendement de 57,1%.
Formule empirique : C21H25N203Br (poids molé
culaire : 433,35)
Spectre rR (KBR) : s max 1740 cm
culaire : 433,35)
Spectre rR (KBR) : s max 1740 cm
Spectre de masse : (m/e) : 434, 432, 419, 417,
375, 373, 345, 332, 330, 317,
315, 266, 195, 180, 167, 175.)
Exemple 3
(+)-9-bromo-1 4,1 5-dioxo-E-homo-éburnane (3 alpha, 17 alpha)
A une solution de 0,65 g (1,36 mmoles) de (+) 9-bromo-14-oxo-13-hydroxy-E-homo-éburnane (3 alpha, 17 alpha) dans 40 ml de dichlorométhane absolu, on ajoute 4,9 g de bioxyde de manganèse actif (Merck) et on agite le mélange de réaction à température ambiante pendant 1 h 30.On filtre la partie solide de la suspension, on la lave à trois reprises avec 5 ml de dichlorométhane anhydre et on concentre les filtrats combinés sous vide. Le résidu huileux est cristallisé dans 5 ml d'éther.
375, 373, 345, 332, 330, 317,
315, 266, 195, 180, 167, 175.)
Exemple 3
(+)-9-bromo-1 4,1 5-dioxo-E-homo-éburnane (3 alpha, 17 alpha)
A une solution de 0,65 g (1,36 mmoles) de (+) 9-bromo-14-oxo-13-hydroxy-E-homo-éburnane (3 alpha, 17 alpha) dans 40 ml de dichlorométhane absolu, on ajoute 4,9 g de bioxyde de manganèse actif (Merck) et on agite le mélange de réaction à température ambiante pendant 1 h 30.On filtre la partie solide de la suspension, on la lave à trois reprises avec 5 ml de dichlorométhane anhydre et on concentre les filtrats combinés sous vide. Le résidu huileux est cristallisé dans 5 ml d'éther.
On obtient 0,324 g de produit cristallisé fondant à 64-650C, soit un rendement de 59,1%.
Formule empirique : C20H21BrN20 (poids moléculaire
401,31)
Spectre IR (KBR) : [# max 1725 cm 1

401,31)
Spectre IR (KBR) : [# max 1725 cm 1
1965 cm- (C=P d'amide)] Rotation optique: [α]## = + 71,4 (c = 1 dans le
dichlorométhane)
Exemple 4
Chlorhydrate de la (+)-9-bromo-vincamine
On dissout 0,33 g (0,82 mmole) de (+)-9-bromo-14, 15-dioxo-E-homo-éburnane (3 alpha, 17 alpha) dans 15 ml de méthanol anhydre, on ajoute à la solution 30 mg de tert-butylate de potassium et on agite le mélange de réaction pendant 2 h à température ambiante. On neutralise ensuite à l'acide acétique glacial à pH 7 et on concentre sous vide.On ajoute au résidu 45 ml d'une solution aqueuse de bicarbonate de sodium à 5% et on extrait la solution basique par 15, 10 et 5 ml de dichlorométhane. On combine les phases organiques, on les sèche sur sulfate de sodium anhydre. On filtre et on concentre le filtrat sous vide. On-cristallise le résidu huileux dans 10 ml de méthanol saturé de gaz chlorhydrique.
dichlorométhane)
Exemple 4
Chlorhydrate de la (+)-9-bromo-vincamine
On dissout 0,33 g (0,82 mmole) de (+)-9-bromo-14, 15-dioxo-E-homo-éburnane (3 alpha, 17 alpha) dans 15 ml de méthanol anhydre, on ajoute à la solution 30 mg de tert-butylate de potassium et on agite le mélange de réaction pendant 2 h à température ambiante. On neutralise ensuite à l'acide acétique glacial à pH 7 et on concentre sous vide.On ajoute au résidu 45 ml d'une solution aqueuse de bicarbonate de sodium à 5% et on extrait la solution basique par 15, 10 et 5 ml de dichlorométhane. On combine les phases organiques, on les sèche sur sulfate de sodium anhydre. On filtre et on concentre le filtrat sous vide. On-cristallise le résidu huileux dans 10 ml de méthanol saturé de gaz chlorhydrique.
On obtient 0,21 g, soit un rendement de 54,4% de produit cristallisé blanc fondant à 223-225 C.
Formule empirique : C21H26BrClN203 (poids molécu
laire 469,82) Spectre IR (KBR) : [# max 1735 cm-1

Rotation optique : [α]## = + 14,8 (c = 1,52 de
D = + 14,80 (c = 1,52 de
la base libre dans le chloroforme
Exemple 5
10-bromo-14,15-dioxo-E-homo-éburnane (3 alpha, 17 alpha)
A une solution de 0,25 g (0,62 mmole) de 10-bromo 14-oxo-15-hydroxy-E-homo-éburnane (3 alpha, 17 alpha) dans 15 ml de dichlorométhane anhydre on ajoute 2,0 g de bioxyde de manganèse actif (Merck) tel quel ou déposé sur 4,1 g de
Celite et on agite le mélange à température ambiante pendant 7 h. On filtre la partie solide de la suspension, on lave avec 10 ml de dichlorométhane anhydre et on concentre les filtrats combinés sous vide. On cristallise le résidu solide dans 5 ml d'éther.
laire 469,82) Spectre IR (KBR) : [# max 1735 cm-1
Rotation optique : [α]## = + 14,8 (c = 1,52 de
D = + 14,80 (c = 1,52 de
la base libre dans le chloroforme
Exemple 5
10-bromo-14,15-dioxo-E-homo-éburnane (3 alpha, 17 alpha)
A une solution de 0,25 g (0,62 mmole) de 10-bromo 14-oxo-15-hydroxy-E-homo-éburnane (3 alpha, 17 alpha) dans 15 ml de dichlorométhane anhydre on ajoute 2,0 g de bioxyde de manganèse actif (Merck) tel quel ou déposé sur 4,1 g de
Celite et on agite le mélange à température ambiante pendant 7 h. On filtre la partie solide de la suspension, on lave avec 10 ml de dichlorométhane anhydre et on concentre les filtrats combinés sous vide. On cristallise le résidu solide dans 5 ml d'éther.
On obtient 0,15 g, soit un rendement de 60,3% du produit recherché à l'état cristallisé fondant à 172-1730C.
Formule empirique : C20H21BrN202 (poids moléculaire
401,31)
Spectre infra-rouge (KBR) : ro max 1705 cm
401,31)
Spectre infra-rouge (KBR) : ro max 1705 cm
1765 cm-i (CO d'amide) 7
Spectre de masse : (m/e) : 402, 401, 400, 399, 387,
385, 375, 373, 371, 359,
357, 346, 344, 331, 329,
317, 315, 277, 275, 180,
167, 153, 140.)
A partir du filtrat, on peut isoler 5% de 10-bromovincamine (3 alpha, 16 alpha).
Spectre de masse : (m/e) : 402, 401, 400, 399, 387,
385, 375, 373, 371, 359,
357, 346, 344, 331, 329,
317, 315, 277, 275, 180,
167, 153, 140.)
A partir du filtrat, on peut isoler 5% de 10-bromovincamine (3 alpha, 16 alpha).
Exemple 6 i 0-bromo-vincamine
On dissout 0,20 g (0,5 mmole) de 10-bromo-14,15-dioxo-E-homo-éburnane (3 alpha, 17 alpha) dans 10 ml de méthanol anhydre à 400 C et on ajoute à la solution 10 mg de tert-butylate de potassium; on abandonne au repos à température ambiante pendant 2 h.
On dissout 0,20 g (0,5 mmole) de 10-bromo-14,15-dioxo-E-homo-éburnane (3 alpha, 17 alpha) dans 10 ml de méthanol anhydre à 400 C et on ajoute à la solution 10 mg de tert-butylate de potassium; on abandonne au repos à température ambiante pendant 2 h.
On neutralise le mélange de réaction à pH 7 par l'acide acétique glacial et on concentre sous vide. Au résidu solide, on ajoute 15 ml de solution aqueuse de bicarbonate de sodium à 5% et on extrait la solution basique à trois reprises avec 10 ml de dichlorométhane. On sèche les phases organique s combinées sur sulfate de magnésium anhydre, on filtre et on concentre le filtrat sous vide. On cristallise le résidu dans 5 ml de méthanol.
On obtient 0,15 g soit un rendement de 69,5% du produit recherché à l'état de cristaux blancs fondant à 221-2220C.
Analyse élémentaire: C21H25BrN203 (poids molécu
laire 433,35)
calculé : C 58,20%; H : 5,81Só ; N : 6,46%
trouvé : C 58,15%; H : 5,76% ; N : 6,36%
Spectre IR (KBR) : max 1750 cm1
laire 433,35)
calculé : C 58,20%; H : 5,81Só ; N : 6,46%
trouvé : C 58,15%; H : 5,76% ; N : 6,36%
Spectre IR (KBR) : max 1750 cm1
Spectre de RMN(dans le dentérochloroforme)
g : 0,88 (t. 3H, CH3-CH2-) # : 3,77 (s, 3H,

s : 6,81 - 7,69 (m, 3H, H aromatique).
g : 0,88 (t. 3H, CH3-CH2-) # : 3,77 (s, 3H,
s : 6,81 - 7,69 (m, 3H, H aromatique).
Spectre de masse (m/e) : (434, 432, 419, 417, 375,
373, 345, 332, 330, 317,
315, 266, 195, 180, 167,
115.)
Exemple 7
11-bromo-14,15-dioxo-E-homo-éburnane (3 alpha, 17 alpha)
A une solution de 0,75 g (1,86 mmoles) de il-bromo- 14-oxo-15-hydroxy-E-homo-éburnane (3 alpha, 17 alpha) dans 20 ml de dichlorométhane anhydre on ajoute 6,0 g de bioxyde de manganèse actif (Merck) et on agite le mélange à température ambiante pendant 5 h. On filtre la partie solide de la suspension, on la lave avec 10 ml de dichlorométhane anhydre et on concentre les filtrats combinés sous vide. On cristallise le résidu huileux dans 10 ml d'éther.
373, 345, 332, 330, 317,
315, 266, 195, 180, 167,
115.)
Exemple 7
11-bromo-14,15-dioxo-E-homo-éburnane (3 alpha, 17 alpha)
A une solution de 0,75 g (1,86 mmoles) de il-bromo- 14-oxo-15-hydroxy-E-homo-éburnane (3 alpha, 17 alpha) dans 20 ml de dichlorométhane anhydre on ajoute 6,0 g de bioxyde de manganèse actif (Merck) et on agite le mélange à température ambiante pendant 5 h. On filtre la partie solide de la suspension, on la lave avec 10 ml de dichlorométhane anhydre et on concentre les filtrats combinés sous vide. On cristallise le résidu huileux dans 10 ml d'éther.
On obtient 0,484 g, soit un rendement de 64,9%, du produit recherché à l'état de cristaux fondant à 171-172 C.
Formule empirique -: C20H21N2 2Br (poids moléculaire
401 ,31).
401 ,31).
Spectre IR (KBR) : [# max 1725 cm 1

Spectre de masse : (m/e): (402, 401, 400, 399, 387,
385, 375, 373, 371, 359,
357, 346, 344, 331, 329,
317, 315, 277, 180, 167,
153, 140.)
Exemple 8 il -bromo-1 4-oxo-1 5-hydroxyimino-E-homo-éburnane
(3 alpha, 17 alpha)
A une solution de 100 mg (0,25 mmole) de ll-bromo- 14, 15-dioxo-E-homo-éburnane dans 0,5 ml de pyridine anhydre, on ajoute 100 mg (1,42 mmoles) de chlorhydrate d' hydroxylamine anhydre et on abandonne le mélange de réaction au repos pendant 2 h à température ambiante. On coule ensuite sur 2 ml d'eau glacée et on alcalinise à pH 9 par l'ammoniaque concentrée.
385, 375, 373, 371, 359,
357, 346, 344, 331, 329,
317, 315, 277, 180, 167,
153, 140.)
Exemple 8 il -bromo-1 4-oxo-1 5-hydroxyimino-E-homo-éburnane
(3 alpha, 17 alpha)
A une solution de 100 mg (0,25 mmole) de ll-bromo- 14, 15-dioxo-E-homo-éburnane dans 0,5 ml de pyridine anhydre, on ajoute 100 mg (1,42 mmoles) de chlorhydrate d' hydroxylamine anhydre et on abandonne le mélange de réaction au repos pendant 2 h à température ambiante. On coule ensuite sur 2 ml d'eau glacée et on alcalinise à pH 9 par l'ammoniaque concentrée.
On filtre le produit qui a précipité, on le lave avec 25 ml d'eau, on le sèche et on le cristallise-dans 2 ml de dichlorométhane.
On obtient 72 mg, soit un rendement de 69%, du produit recherché à l'état de-cristaux fondant à 140 145 C.
Spectre IR (KBR): : max 3320 cm 1 (CO d'amide7 1630 cm 1

On dissout 10 mg sur les 72 mg de base dans i ml de méthanol et on règle le pH de la solution à 5 à l'aide d'une solution méthanolique d'HCl. On filtre les cristaux qui précipitent, on les lave avec i ml de méthanol et on les sèche. On obtient 10 mg du chlorhydrate du produit ci-dessus, fondant à 2570C.
Exemple 9 Il -bromo-vincamine
On dissout 0, 11 g (0,27 mmole) de 11-bromo-14,15dioxo-E-homo-éburnane (3 alpha, 17 alpha) à 400C dans 5 ml de méthanol anhydre, on ajoute 10 mg de tert-butylate de potassium et on abandonne le mélange à température ambiante pendant 2 h.
On dissout 0, 11 g (0,27 mmole) de 11-bromo-14,15dioxo-E-homo-éburnane (3 alpha, 17 alpha) à 400C dans 5 ml de méthanol anhydre, on ajoute 10 mg de tert-butylate de potassium et on abandonne le mélange à température ambiante pendant 2 h.
On neutralise le mélange de réaction à pH 7 par l'acide acétique glacial et on concentre sous vide. On ajoute au résidu 15 ml d'une solution aqueuse de bicarbonate de sodium à 5% et on extrait la solution alcaline à trois reprises par 10 ml de dichlorométhane. On sèche les phases organique s combinées sur sulfate de magnésium anhydre, on filtre et on concentre le filtrat sous vide.
On cristallise le résidu solide dans 2 ml de méthanol.
On obtient 61,5 mg du produit recherché à l'état de cristaux blancs fondant à 215-2170C, soit un rendement 51,8%.
Formule empirique: C21H25N203Br (poids moléculaire
433,35).
433,35).
Spectre IR (KBR) : max 1735 cm1

Spectre de masse (m/e) : (434, 432, 419, 417, 375,
373, 345, 332, 330, 317,
315, 266, 195, 180, 167,
115)
Exemple 10
(+)-11-bromo-14,15-dioxo-E-homo-éburnane (3 alpha,
17 alpha)
A une solution de 1,0 g (2,48 mmoles) de (+)-11- bromo-14-oxo-15-hydroxy-E-homo-éburnane (3 alpha, 17 alpha) dans 70 ml de dichlorométhane anhydre, on ajoute 9,0 g de bioxyde de manganèse actif (Merck) et on agite le mélange de réaction pendant i h 30. On filtre la partie insoluble de la suspension, on lave à trois reprises avec 10 ml de dichlorométhane anhydre et on sèche les filtrats combinés sous vide. On cristallise le résidu dans 5 ml d'éther.
373, 345, 332, 330, 317,
315, 266, 195, 180, 167,
115)
Exemple 10
(+)-11-bromo-14,15-dioxo-E-homo-éburnane (3 alpha,
17 alpha)
A une solution de 1,0 g (2,48 mmoles) de (+)-11- bromo-14-oxo-15-hydroxy-E-homo-éburnane (3 alpha, 17 alpha) dans 70 ml de dichlorométhane anhydre, on ajoute 9,0 g de bioxyde de manganèse actif (Merck) et on agite le mélange de réaction pendant i h 30. On filtre la partie insoluble de la suspension, on lave à trois reprises avec 10 ml de dichlorométhane anhydre et on sèche les filtrats combinés sous vide. On cristallise le résidu dans 5 ml d'éther.
On obtient 0,65 g soit un rendement de 65,3% du produit recherché à l'état de cristaux fondant à 151 1530C.
Formule empirique : C20H21BrN202 (poids moléculaire
re 401,31) Spectre IR (KBR) : max 1715 cm-1
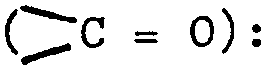
1685 cm-1
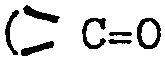
d'amide)7
Rotation optique : ri 7 D5 = +45,00 (c = 1 dans le dichlo
rométhane)
Exemple li (+)" bromo-14-oxo-15-hydroxyimino-E-homo- éburnane (3.alpha, 17 alpha)
A une solution de 100 mg (0,25 mmole) de (+)-11- bromo-14,15-dioxo-E-homo-éburnane (3 alpha, 17 alpha) dans 0,5 ml de pyridine anhydre, on ajoute 100 mg (1,42 mmoles) de chlorhydrate d'hydroxylamine anhydre et on abandonne le mélange de réaction pendant 2 h à température ambiante.
re 401,31) Spectre IR (KBR) : max 1715 cm-1
1685 cm-1
d'amide)7
Rotation optique : ri 7 D5 = +45,00 (c = 1 dans le dichlo
rométhane)
Exemple li (+)" bromo-14-oxo-15-hydroxyimino-E-homo- éburnane (3.alpha, 17 alpha)
A une solution de 100 mg (0,25 mmole) de (+)-11- bromo-14,15-dioxo-E-homo-éburnane (3 alpha, 17 alpha) dans 0,5 ml de pyridine anhydre, on ajoute 100 mg (1,42 mmoles) de chlorhydrate d'hydroxylamine anhydre et on abandonne le mélange de réaction pendant 2 h à température ambiante.
On coule la solution pyridique dans 2 ml d'eau glacée et on alcalinise à pH 9 par l'ammoniaque concentrée. On filtre le précipité amorphe, on le lave avec 5 ml d'eau et on le sèche. On purifie le produit solide sec par chromatographie sur couche mince. On utilise comme éluant un mélange de benzène et de méthanol 14 : 3 (plaque de gel de silice PF 254 + 366 en couche de 20 cm x 1,5 mm). On élue par un mélange dichlorométhane/méthanol, 20 : 3. Après l'élution on obtient 64 mg du produit recherché à l'état d'huile.
On dissout ce produit dans i ml de méthanol et on acidifie la solution à pH 5 à 0 C par le méthanol saturé de chlorure d'hydrogène. On filtre le sel qui a précipité, on le lave avec 1 ml de méthanol et on le sèche. On obtient 60 mg du chlorhydrate du produit ci-dessus fondant à 2350C.
Spectre IR (KBR): ro max 3460 cm1 (-OH); 1700 cm-

d'amide); 1622 cm-i

d'amide); 1622 cm-i
Rotation optique : ra 7 D) = + 45,60 (dans le diméthylformamide
Exemple 12 (+)-11-bromo-vincamine
On dissout 0,33 g (0,82 mmole) de (+)-11-bromo i4,15-dioxo-E-homo-éburnane (3 alpha, 17 alpha) dans 15 ml de méthanol anhydre et on ajoute 30 mg de tert-butylate de potassium; on agite le mélange pendant 1 h 30 à température ambiante. On neutralise à pH 7 par l'acide acétique glacial et on concentre sous vide.Au résidu solide, on ajoute 45 ml d'une solution aqueuse de bicarbonate de sodium à 5% et on extrait par 15, 10 et 5 ml de dichlorométhane. On sèche les phases organiques combinées sur sulfate de magnésium anhydre et on concentre le filtrat sous vide. On cristallise le résidu huileux dans 3 ml de méthanol.
Exemple 12 (+)-11-bromo-vincamine
On dissout 0,33 g (0,82 mmole) de (+)-11-bromo i4,15-dioxo-E-homo-éburnane (3 alpha, 17 alpha) dans 15 ml de méthanol anhydre et on ajoute 30 mg de tert-butylate de potassium; on agite le mélange pendant 1 h 30 à température ambiante. On neutralise à pH 7 par l'acide acétique glacial et on concentre sous vide.Au résidu solide, on ajoute 45 ml d'une solution aqueuse de bicarbonate de sodium à 5% et on extrait par 15, 10 et 5 ml de dichlorométhane. On sèche les phases organiques combinées sur sulfate de magnésium anhydre et on concentre le filtrat sous vide. On cristallise le résidu huileux dans 3 ml de méthanol.
On obtient 0,19 g, soit un rendement de 53,3% de produit à l'état de cristaux blancs fondant à 221 2230C.
Formule empirique: C21H25BrN203 (poids moléculaire
433,35)
Spectre IR (KBR): rO max 1730 cuiT1

433,35)
Spectre IR (KBR): rO max 1730 cuiT1
Rotation optique: rCn 7 D5 = + 8,40 (c = i dans le
chloroforme)
Exemple 13 (+)-10-bromo-vincamine
On dissout 1,2 g (26 mmoles) de (+)-10-bromo-14 oxo-15-hydroxyimino-E-homo-éburnane (3 alpha, 17 alpha) dans 240 ml d'acide acétique glacial et on ajoute à la solution 24 g d'acide p-toluène sulfonique anhydre et 36 g de paraformaldéhyde. On chauffe le mélange de réaction pendant 3 h 30 à 110 C à l'abir de l'humidité et on coule dans l'eau glacée.On alcalinise le mélange à pH 9 par l'ammoniaque concentrée, on filtre le (+)-10 bromo-14,15-dioxo-E-homo-éburnane (13 alpha, 17 alpha) qui a précipité, on lave à l'eau et on sèche sur P205 à ltexsiccateur. On obtient 8,4 g du produit recherché fondant à 1840C (décomposition), [ Spectre IR (KBR) 1720 (CO), 1690 cm- (CO d'amide)], qu'on peut utiliser tel quel, sans autre purification. Dans le stade de réaction subséquent, on dissout le produit dans 400 ml de dichlorométhane, on filtre et on élimine le solvant sous vide. Au résidu, on ajoute 40 ml de la solution de 10 g de tert-butylate de potassium dans 400 ml de méthanol anhydre. On chauffe le mélange à 400C et on abandonne au repos pendant 2 h.
chloroforme)
Exemple 13 (+)-10-bromo-vincamine
On dissout 1,2 g (26 mmoles) de (+)-10-bromo-14 oxo-15-hydroxyimino-E-homo-éburnane (3 alpha, 17 alpha) dans 240 ml d'acide acétique glacial et on ajoute à la solution 24 g d'acide p-toluène sulfonique anhydre et 36 g de paraformaldéhyde. On chauffe le mélange de réaction pendant 3 h 30 à 110 C à l'abir de l'humidité et on coule dans l'eau glacée.On alcalinise le mélange à pH 9 par l'ammoniaque concentrée, on filtre le (+)-10 bromo-14,15-dioxo-E-homo-éburnane (13 alpha, 17 alpha) qui a précipité, on lave à l'eau et on sèche sur P205 à ltexsiccateur. On obtient 8,4 g du produit recherché fondant à 1840C (décomposition), [ Spectre IR (KBR) 1720 (CO), 1690 cm- (CO d'amide)], qu'on peut utiliser tel quel, sans autre purification. Dans le stade de réaction subséquent, on dissout le produit dans 400 ml de dichlorométhane, on filtre et on élimine le solvant sous vide. Au résidu, on ajoute 40 ml de la solution de 10 g de tert-butylate de potassium dans 400 ml de méthanol anhydre. On chauffe le mélange à 400C et on abandonne au repos pendant 2 h.
On filtre les cristaux formés, on les lave avec un peu de méthanol froid et on les sèche.
On obtient 3,8 g, soit un rendement de 33% du produit recherché fondant à 220 - 2210C après recristallisation dans le méthanol.
Exemple 14
Acide (+)-10-bromo-vincaminique
On fait bouillir au reflux pendant 2 h à une solution de 2,5 g (5,77 mmoles) de la (+)-10-bromo-vincami- ne préparée dans l'exemple 13 et 3,4 g (soit 60 mmoles) d'hydroxyde de potassium dans 75 ml de méthanol. Après refroidissement, on acidifie la solution à pH 6 par l'acide acétique glacial et on fait cristalliser au réfrigérateur. On filtre les cristaux, on lave à deux reprises avec 10 ml d'eau et on sèche.
Acide (+)-10-bromo-vincaminique
On fait bouillir au reflux pendant 2 h à une solution de 2,5 g (5,77 mmoles) de la (+)-10-bromo-vincami- ne préparée dans l'exemple 13 et 3,4 g (soit 60 mmoles) d'hydroxyde de potassium dans 75 ml de méthanol. Après refroidissement, on acidifie la solution à pH 6 par l'acide acétique glacial et on fait cristalliser au réfrigérateur. On filtre les cristaux, on lave à deux reprises avec 10 ml d'eau et on sèche.
On obtient 1,9 g, soit un rendement de 78,5% d'acide (+)-iO-bromo-vincaminique à l'état de produit cristallisé blanc fondant à 234-2360C.
Formule empirique : C20H23BrN203 (poids moléculaire
419,32) Spectre IR (KBR): [# max 1635 cm

419,32) Spectre IR (KBR): [# max 1635 cm
Exemple 15
Chlorhydrate de l'ester éthylique de l'acide 1 O-bromo-apovincaminique
On chauffe au reflux pendant 10 h à une solution de 1,1 g (2,62 mmoles) de l'acide (+)-10-bromo-vincaminique préparé dans l'exemple 14 dans 40 ml d'éthanol anhydre et 3,0 ml d'acide sulfurique aqueux concentré. Lorsque la réaction est terminée, on concentre le mélange sous vide.
Chlorhydrate de l'ester éthylique de l'acide 1 O-bromo-apovincaminique
On chauffe au reflux pendant 10 h à une solution de 1,1 g (2,62 mmoles) de l'acide (+)-10-bromo-vincaminique préparé dans l'exemple 14 dans 40 ml d'éthanol anhydre et 3,0 ml d'acide sulfurique aqueux concentré. Lorsque la réaction est terminée, on concentre le mélange sous vide.
Au résidu, on ajoute 100 ml d'eau et on alcalinise la solu tion à pH 10 par une lessive de soude à 40%. On extrait la solution alcaline par 50, 45 et 30 ml de dichlorométhane, on sèche les extraits- organiques sur sulfate de magnésium anhydre et on concentre le filtrat sous vide.
On redissout le résidu solide dans 5 ml d'éthanol et on règle la solution à pH 5 par l'éthanol saturé d'HCl gazeux. On dilue la solution acide par 15 à 20 ml d'éther; des cristaux se séparent. On obtient 0,9 g, soit un rendement de 73,6%, de chlorhydrate de l'ester éthylique de l'acide (+)-10-bromo-apovincaminique à l'état de produit cristallisé blanc fondant à 221-2230C.
Analyse élémentaire: C22H26BrClN202 (poids molé
culaire 465,83)
calcule : C 56,73%, H 5,63 z6, N 6,01%
trouvé : C 56,58%, H 5,49%, N 5,86%.
culaire 465,83)
calcule : C 56,73%, H 5,63 z6, N 6,01%
trouvé : C 56,58%, H 5,49%, N 5,86%.
Spectre IR (KRr): rg max 1725 cm 1

1625 cm 1 1605 cm1

1625 cm 1 1605 cm1
Exemple 16
Ester éthylique de l'acide (+)-9-bromo-apovincaminique
On chauffe 3, 5 g de (+) -9-bromo-1 4-oxo-1 5-hydroxy-imino-
E-homo-éburnane dans un mélange de 70 ml d 'éthanol anhydre et 24,5 ml de solution aqueuse concentrée d'acide sulfurique pendant 6 h à 900C en atmosphère d'azote. On alcalinise ensuite le mélange de réaction à pH 9 par addition d'ammoniaque à 25%. On extrait la solution basique à trois reprises par 70 ml de dichlorométhane, on sèche la phase organique après séparation sur sulfate de sodium anhydre, on filtre et on concentre le filtrat.
Ester éthylique de l'acide (+)-9-bromo-apovincaminique
On chauffe 3, 5 g de (+) -9-bromo-1 4-oxo-1 5-hydroxy-imino-
E-homo-éburnane dans un mélange de 70 ml d 'éthanol anhydre et 24,5 ml de solution aqueuse concentrée d'acide sulfurique pendant 6 h à 900C en atmosphère d'azote. On alcalinise ensuite le mélange de réaction à pH 9 par addition d'ammoniaque à 25%. On extrait la solution basique à trois reprises par 70 ml de dichlorométhane, on sèche la phase organique après séparation sur sulfate de sodium anhydre, on filtre et on concentre le filtrat.
On obtient 2,5 g, soit un rendement de 75% du produit recherché à l'état d'huile mousseuse qui se solidifie et fond alors à 76-780C.
Rotation optique: r 7 25 = + 148,80 (c= 1 dans le
D
diméthylformamide).
D
diméthylformamide).
Exemple 17
Ester éthylique de l'acide (+)-11-bromo-apovinca- minique
On opère comme décrit dans l'exemple 16 mais en partant de 3,5 g du chlorhydrate de (+)-11-bromo-14-oxo15-hydroxy-imino-E-homo-éburnane. On obtient 3,0 g du produit recherché à l'état d'huile jaune clair. On triture 3,0 g de cette huile avec 5 ml d'éther, on filtre les cristaux et on sèche.
Ester éthylique de l'acide (+)-11-bromo-apovinca- minique
On opère comme décrit dans l'exemple 16 mais en partant de 3,5 g du chlorhydrate de (+)-11-bromo-14-oxo15-hydroxy-imino-E-homo-éburnane. On obtient 3,0 g du produit recherché à l'état d'huile jaune clair. On triture 3,0 g de cette huile avec 5 ml d'éther, on filtre les cristaux et on sèche.
Rendement : 2,1 g, soit 65% d'ester éthylique de l'acide ii-bromo-apovincaminique à l'état de cristaux blancs fondant à 158-1600C.
Rotation optique : rig 25 = + 80,20 (c = 1 dans le
diméthylformamide).
diméthylformamide).
Claims (5)
1) Procédé de préparation de dérivés d'esters halogéno-vincaminiques et halogéno-apovincaminiques répondant aux formules générales

dans lesquelles R1 et R2 représentent des groupes alkyles de 1 à 6 atomes de carbone et X représente un halogène, de leurs sels formés par addition avec des acides acceptables pour l'usage pharmaceutique et de leurs antipodes optiques, caractérisé en ce que l'on traite un dérivé halogéné d'hydroxy-oxo-E-homo-éburnane répondant à la formule générale

dans laquelle R2 et X ont les significations indiquées cidessus, ses épimères, antipodes optiques ou sels formés par addition avec des acides, par un agent oxydant, on fait réagir le dérivé halogéné de dioxo-E-homo-éburnane obtenu, qui répond à la formule générale

dans laquelle R2 et X ont les significations ci-dessus, ou le dérivé d'hydroxyimino-oxo-E-homo-éburnane de formule générale

dans laquelle R2 et X ont les significations indiquées cidessus, obtenu le cas échéant après réaction avec l'hydroxylamine ou un sel d'hydroxylamine, ou ses sels formés par addition avec des acides, le cas échéant après résolution, avec un alcool de formule générale R1OH dans laquelle R1 a les significations indiquées ci-dessus, en présence d'un agent alcalin, si on le désire, on traite le dérivé d'ester halogéno-vincaminique obtenu, qui répond à la formule générale Ia dans laquelle R1, R2 et X ont les significations indiquées ci-dessus, par un acide approprié à la formation d'un sel acceptable pour l'usage pharmaceutique, et/ou on le résout ou on l'hydrolyse et on fait réagir le dérivé d'acide halogéno-vincaminique obtenu, qui répond à la formule générale

dans laquelle R2 et X ont les significations indiquées cidessus, ou un dérivé d'hydroxyimino-oxo-E-homo-éburnane de formule générale IV dans laquelle R2 et X ont les significations indiquées ci-dessus, ou un sel de ce composé formé par addition avec un acide, si on le désire après résolution, avec un alcool de formule générale R1-OH dans laquelle R1 a les significations indiquées ci-dessus, en présence d'un acide concentré déshydratant et, si on le désire, on traite le dérivé d'ester halogéno-apovincaminique obtenu, qui répond à la formule générale Ib dans laquelle R1, R2 et X ont les significations indiquées cidessus, par un acide approprié à la formation d'un sel acceptable pour l'usage pharmaceutique, et/ou on le résout.
2) Procédé selon la revendication 1, caractérisé en ce que, pour l'oxydation du dérivé halogéné d'hydroxyoxo-E-homo-éburnane de formule générale II dans laquelle R2 et X ont les significations indiquées dans la revendication 1, on utilise comme agent oxydant du bioxyde de manganèse actif.
3) Procédé selon la revendication 1, caractérisé en ce que la réaction du dérivé halogéné de dioxo-E-homo-éburnane de formule générale III et du dérivé d'hydroxyimino-oxo-E-éburnane de formule générale IV, dans lesquelles R2 et X ont les significations indiquées ci-dessus, avec un alcool de formule générale R1-OH-dans laquelle R1 a les significations indiquées dans la revendication 1, est effectuée dans un milieu alcalin en présence d'un tert-alcoolate de métal alcalin.
4) Procédé selon la revendication 1, caractérisé en ce que l'hydrolyse des dérivés d'esters halogéno-vincaminiques de formule Ia dans laquelle R1, R2 et X ont les significations indiquées dans la revendication 1, est effectuée à l'aide d'une solution d'une base minérale, de préférence un hydroxyde de métal alcalin, dans un alcanol de i à 6 atomes de carbone.
5) Procédé selon la revendication 1, caractérisé en ce que la réaction des dérivés d'hydroxyimino-oxo-E-homoéburnane de formule générale IV et des dérivés d'acides halogéno-vincaminiques de formule générale V dans lesquelles R2 et X ont les significations indiquées ci-dessus, avec un alcool de formule générale R1-OH dans laquelle R1 a les significations indiquées ci-dessus,en présence d'un acide concentré déshydratant, est effectuée en présence d'une solution aqueuse concentrée d'acide sulfurique.
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| HURI000738 HU180930B (en) | 1979-12-28 | 1979-12-28 | Process for producing ester-derivatives of halogenovincaminic acid |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| FR2472571A1 true FR2472571A1 (fr) | 1981-07-03 |
| FR2472571B1 FR2472571B1 (fr) | 1985-04-26 |
Family
ID=11001119
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| FR8027509A Granted FR2472571A1 (fr) | 1979-12-28 | 1980-12-24 | Procede pour la preparation de derives d'esters halogeno-vincaminiques et halogeno-apovincaminiques |
Country Status (4)
| Country | Link |
|---|---|
| BE (1) | BE886867A (fr) |
| CH (1) | CH652126A5 (fr) |
| FR (1) | FR2472571A1 (fr) |
| HU (1) | HU180930B (fr) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| FR2467207A1 (fr) * | 1979-10-15 | 1981-04-17 | Richter Gedeon Vegyeszet | Nouveaux esters d'acide halogene-apovincaminique, procede pour leur preparation et preparations medicamenteuses les contenant |
| ES2052449A1 (es) * | 1992-12-22 | 1994-07-01 | Covex Sa | Nuevo procedimiento para la preparacion del apovincaminato de etilo. |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| HU185305B (en) * | 1981-08-23 | 1985-01-28 | Richter Gedeon Vegyeszet | Process for preparing vincine and apovincine |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| FR2081593A1 (en) * | 1970-03-31 | 1971-12-10 | Roussel Uclaf | Vincamine process |
-
1979
- 1979-12-28 HU HURI000738 patent/HU180930B/hu not_active IP Right Cessation
-
1980
- 1980-12-24 CH CH958280A patent/CH652126A5/de not_active IP Right Cessation
- 1980-12-24 FR FR8027509A patent/FR2472571A1/fr active Granted
- 1980-12-24 BE BE0/203324A patent/BE886867A/fr not_active IP Right Cessation
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| FR2081593A1 (en) * | 1970-03-31 | 1971-12-10 | Roussel Uclaf | Vincamine process |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| FR2467207A1 (fr) * | 1979-10-15 | 1981-04-17 | Richter Gedeon Vegyeszet | Nouveaux esters d'acide halogene-apovincaminique, procede pour leur preparation et preparations medicamenteuses les contenant |
| ES2052449A1 (es) * | 1992-12-22 | 1994-07-01 | Covex Sa | Nuevo procedimiento para la preparacion del apovincaminato de etilo. |
Also Published As
| Publication number | Publication date |
|---|---|
| CH652126A5 (en) | 1985-10-31 |
| BE886867A (fr) | 1981-04-16 |
| HU180930B (en) | 1983-05-30 |
| FR2472571B1 (fr) | 1985-04-26 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JPH0529216B2 (fr) | ||
| EP0001534B1 (fr) | Nouveaux dérivés du pyrrole, leur procédé de préparation et leurs applications en thérapeutique | |
| FR2620700A1 (fr) | Procede de synthese d'alpha amino acides n alkyles et leurs esters. application a la synthese de carboxyalkyl dipeptides | |
| EP0082049A2 (fr) | Nouveaux éthers dont les restes organiques comportent des atomes chiraux, leur préparation, leur application au dédoublement d'alcools ou de certains composés hémiacétaliques et les nouveaux produits dédoublés obtenus | |
| EP0004494A1 (fr) | Nouveaux dérivés de 1,3-dihydro 3-(1-(2-(2,3-dihydro 1,4-benzo-dioxin-2-yl)2-hydroxy éthyl)pipéridin-4-yl)2H-indol 2-one, leur procédé de préparation, leur application comme médicaments et les compositions pharmaceutiques les renfermant | |
| FR2468370A1 (fr) | Nouvelles pyridothienotriazines et leur procede de preparation | |
| EP0275742B1 (fr) | Dérivés 5-hydroxyéthylés de l'oxazolidinone-2, leurs procédés de préparation et leurs applications en thérapeutique | |
| FR2510575A1 (fr) | Nouveaux composes bicycliques, leur procede de preparation et composition pharmaceutique les renfermant | |
| FR2472571A1 (fr) | Procede pour la preparation de derives d'esters halogeno-vincaminiques et halogeno-apovincaminiques | |
| EP0998470B1 (fr) | Procede de preparation de derives amines d'alkyloxyfuranone, composes issus de ce procede et utilisation de ces composes | |
| CH615441A5 (fr) | ||
| FR2460951A1 (fr) | Procede de preparation de derives de 15-hydroxyimino-e-homoeburnane et composes obtenus par ce procede | |
| GB2036744A (en) | Eburnane derivatives | |
| FR2601008A1 (fr) | Procede de synthese stereospecifique de derives de l'indole | |
| FR2772029A1 (fr) | Procede de preparation de la mequitazine et nouvel intermediaire de synthese | |
| EP0053949A1 (fr) | Procédé nouveau de préparation des tétrahydro-5,6,7,7a-4H-thiéno(3,2-c)pyridinones-2 | |
| FR2510996A1 (fr) | Nouveaux derives du dibenz(cd,f)indole et leur preparation | |
| BE884145R (fr) | Composes indoliques nouveaux | |
| FR2511677A1 (fr) | Procede de preparation d'esters des acides alcoxyvincaminiques et alcoxyapovincaminiques | |
| FR2513638A1 (fr) | Procede de preparation d'esters d'acides vincaminiques | |
| EP0419297A1 (fr) | Nouveau procédé de séparation d'isomères optiques des dérivés de la dihydro-1,4 pyridine | |
| CH615171A5 (fr) | ||
| BE1004471A3 (fr) | Nouveaux derives diester d'octahydro-indolo-[2,3-a]-tetrahydropyranyl [2,3-c] quinolizine racemiques et optiquement actifs et procede pour les preparer. | |
| FR2597869A1 (fr) | Nouveaux derives d'ergolene, compositions pharmaceutiques les contenant et procede pour leur preparation | |
| FR2583755A1 (fr) | Derives de 10 a-methoxy-6-methyl-ergoline, leur preparation et leur application en tant que medicament |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| ST | Notification of lapse |