FR2805815A1 - Derives de polyfluoroalkylimidazole, leur preparation et leur application en therapeutique - Google Patents
Derives de polyfluoroalkylimidazole, leur preparation et leur application en therapeutique Download PDFInfo
- Publication number
- FR2805815A1 FR2805815A1 FR0002596A FR0002596A FR2805815A1 FR 2805815 A1 FR2805815 A1 FR 2805815A1 FR 0002596 A FR0002596 A FR 0002596A FR 0002596 A FR0002596 A FR 0002596A FR 2805815 A1 FR2805815 A1 FR 2805815A1
- Authority
- FR
- France
- Prior art keywords
- methyl
- formula
- imidazol
- trifluoromethyl
- compound
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
- 108091005482 5-HT4 receptors Proteins 0.000 title claims description 7
- 101150060249 CHRM3 gene Proteins 0.000 title claims description 5
- 208000002551 irritable bowel syndrome Diseases 0.000 title claims description 3
- 239000002464 receptor antagonist Substances 0.000 title claims description 3
- 229940044551 receptor antagonist Drugs 0.000 title claims description 3
- 208000026139 Memory disease Diseases 0.000 title claims 2
- DHACXYJORXTASB-UHFFFAOYSA-N 4-[(1-phenylimidazol-2-yl)oxymethyl]piperidine Chemical class C1CNCCC1COC1=NC=CN1C1=CC=CC=C1 DHACXYJORXTASB-UHFFFAOYSA-N 0.000 title 1
- 208000029162 bladder disease Diseases 0.000 title 1
- 238000002360 preparation method Methods 0.000 claims abstract description 20
- 150000003839 salts Chemical class 0.000 claims abstract description 13
- -1 2-(1-substituted piperidin-4-ylmethoxy)-1-phenyl-1H-imidazole Chemical class 0.000 claims abstract description 10
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims abstract description 9
- 150000001204 N-oxides Chemical class 0.000 claims abstract description 6
- 125000003545 alkoxy group Chemical group 0.000 claims abstract description 6
- 150000004677 hydrates Chemical class 0.000 claims abstract description 4
- 150000001875 compounds Chemical class 0.000 claims description 98
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 claims description 45
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 33
- 238000000034 method Methods 0.000 claims description 23
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 claims description 15
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 claims description 15
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims description 12
- 229910052739 hydrogen Inorganic materials 0.000 claims description 10
- 239000001257 hydrogen Substances 0.000 claims description 10
- 125000004198 2-fluorophenyl group Chemical group [H]C1=C([H])C(F)=C(*)C([H])=C1[H] 0.000 claims description 9
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 claims description 8
- 125000005843 halogen group Chemical group 0.000 claims description 8
- 125000004178 (C1-C4) alkyl group Chemical group 0.000 claims description 6
- 239000000546 pharmaceutical excipient Substances 0.000 claims description 6
- 125000004207 3-methoxyphenyl group Chemical group [H]C1=C([H])C(*)=C([H])C(OC([H])([H])[H])=C1[H] 0.000 claims description 5
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 claims description 5
- 230000007170 pathology Effects 0.000 claims description 5
- 125000004482 piperidin-4-yl group Chemical group N1CCC(CC1)* 0.000 claims description 5
- 238000011282 treatment Methods 0.000 claims description 5
- 125000000229 (C1-C4)alkoxy group Chemical group 0.000 claims description 4
- 102000017925 CHRM3 Human genes 0.000 claims description 4
- 125000001028 difluoromethyl group Chemical group [H]C(F)(F)* 0.000 claims description 4
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims description 4
- 239000008194 pharmaceutical composition Substances 0.000 claims description 4
- 125000004208 3-hydroxyphenyl group Chemical group [H]OC1=C([H])C([H])=C([H])C(*)=C1[H] 0.000 claims description 3
- 206010020853 Hypertonic bladder Diseases 0.000 claims description 3
- 239000003814 drug Substances 0.000 claims description 3
- 125000006340 pentafluoro ethyl group Chemical group FC(F)(F)C(F)(F)* 0.000 claims description 3
- 230000001225 therapeutic effect Effects 0.000 claims description 3
- 125000006526 (C1-C2) alkyl group Chemical group 0.000 claims description 2
- 125000006656 (C2-C4) alkenyl group Chemical group 0.000 claims description 2
- 206010046543 Urinary incontinence Diseases 0.000 claims description 2
- 230000008901 benefit Effects 0.000 claims description 2
- 125000004093 cyano group Chemical group *C#N 0.000 claims description 2
- 125000002962 imidazol-1-yl group Chemical group [*]N1C([H])=NC([H])=C1[H] 0.000 claims description 2
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 claims description 2
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 claims description 2
- 230000008569 process Effects 0.000 claims description 2
- 102100025027 E3 ubiquitin-protein ligase TRIM69 Human genes 0.000 claims 1
- 101000830203 Homo sapiens E3 ubiquitin-protein ligase TRIM69 Proteins 0.000 claims 1
- 206010061876 Obstruction Diseases 0.000 claims 1
- 125000000217 alkyl group Chemical group 0.000 abstract description 5
- 239000000543 intermediate Substances 0.000 abstract description 2
- 125000005010 perfluoroalkyl group Chemical group 0.000 abstract description 2
- 125000001475 halogen functional group Chemical group 0.000 abstract 2
- 150000002460 imidazoles Chemical class 0.000 abstract 2
- 229940079865 intestinal antiinfectives imidazole derivative Drugs 0.000 abstract 2
- 229910006074 SO2NH2 Inorganic materials 0.000 abstract 1
- 125000003342 alkenyl group Chemical group 0.000 abstract 1
- 125000002431 aminoalkoxy group Chemical group 0.000 abstract 1
- 125000002541 furyl group Chemical group 0.000 abstract 1
- CBOIHMRHGLHBPB-UHFFFAOYSA-N hydroxymethyl Chemical compound O[CH2] CBOIHMRHGLHBPB-UHFFFAOYSA-N 0.000 abstract 1
- 125000002883 imidazolyl group Chemical group 0.000 abstract 1
- 125000001624 naphthyl group Chemical group 0.000 abstract 1
- 125000004076 pyridyl group Chemical group 0.000 abstract 1
- 125000000168 pyrrolyl group Chemical group 0.000 abstract 1
- 125000000565 sulfonamide group Chemical group 0.000 abstract 1
- 125000000335 thiazolyl group Chemical group 0.000 abstract 1
- 125000001544 thienyl group Chemical group 0.000 abstract 1
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 42
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 36
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 36
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 21
- 239000000203 mixture Substances 0.000 description 21
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 18
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 16
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Natural products C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 description 16
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 15
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 12
- 239000000243 solution Substances 0.000 description 12
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 11
- 239000002904 solvent Substances 0.000 description 11
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 10
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 10
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 10
- 239000000741 silica gel Substances 0.000 description 10
- 229910002027 silica gel Inorganic materials 0.000 description 10
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 9
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 9
- 238000003818 flash chromatography Methods 0.000 description 9
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 9
- 235000019341 magnesium sulphate Nutrition 0.000 description 9
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 9
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 8
- 239000008346 aqueous phase Substances 0.000 description 8
- 239000012074 organic phase Substances 0.000 description 8
- 239000011541 reaction mixture Substances 0.000 description 8
- 235000017557 sodium bicarbonate Nutrition 0.000 description 8
- 239000012267 brine Substances 0.000 description 7
- 238000006243 chemical reaction Methods 0.000 description 7
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 7
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 6
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 6
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 6
- 238000001914 filtration Methods 0.000 description 6
- 238000011534 incubation Methods 0.000 description 6
- 210000001519 tissue Anatomy 0.000 description 6
- 230000009471 action Effects 0.000 description 5
- 238000000605 extraction Methods 0.000 description 5
- 239000012299 nitrogen atmosphere Substances 0.000 description 5
- 238000000746 purification Methods 0.000 description 5
- 239000012429 reaction media Substances 0.000 description 5
- 238000010992 reflux Methods 0.000 description 5
- 229920006395 saturated elastomer Polymers 0.000 description 5
- 150000003457 sulfones Chemical class 0.000 description 5
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 5
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 4
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 description 4
- JRNVZBWKYDBUCA-UHFFFAOYSA-N N-chlorosuccinimide Chemical compound ClN1C(=O)CCC1=O JRNVZBWKYDBUCA-UHFFFAOYSA-N 0.000 description 4
- 239000002253 acid Substances 0.000 description 4
- 239000002585 base Substances 0.000 description 4
- 239000000872 buffer Substances 0.000 description 4
- 239000000460 chlorine Substances 0.000 description 4
- 229940050411 fumarate Drugs 0.000 description 4
- 229910052736 halogen Inorganic materials 0.000 description 4
- 150000002367 halogens Chemical class 0.000 description 4
- 210000004379 membrane Anatomy 0.000 description 4
- 239000012528 membrane Substances 0.000 description 4
- 102000005962 receptors Human genes 0.000 description 4
- 108020003175 receptors Proteins 0.000 description 4
- 230000002829 reductive effect Effects 0.000 description 4
- 230000004044 response Effects 0.000 description 4
- QZAYGJVTTNCVMB-UHFFFAOYSA-N serotonin Chemical compound C1=C(O)C=C2C(CCN)=CNC2=C1 QZAYGJVTTNCVMB-UHFFFAOYSA-N 0.000 description 4
- 238000003756 stirring Methods 0.000 description 4
- 108010001237 Cytochrome P-450 CYP2D6 Proteins 0.000 description 3
- 102100021704 Cytochrome P450 2D6 Human genes 0.000 description 3
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 3
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 3
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 3
- UCKMPCXJQFINFW-UHFFFAOYSA-N Sulphide Chemical compound [S-2] UCKMPCXJQFINFW-UHFFFAOYSA-N 0.000 description 3
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 3
- 239000004480 active ingredient Substances 0.000 description 3
- 150000001299 aldehydes Chemical class 0.000 description 3
- 125000001931 aliphatic group Chemical group 0.000 description 3
- PNEYBMLMFCGWSK-UHFFFAOYSA-N aluminium oxide Inorganic materials [O-2].[O-2].[O-2].[Al+3].[Al+3] PNEYBMLMFCGWSK-UHFFFAOYSA-N 0.000 description 3
- 239000005557 antagonist Substances 0.000 description 3
- 239000007864 aqueous solution Substances 0.000 description 3
- AIXAANGOTKPUOY-UHFFFAOYSA-N carbachol Chemical compound [Cl-].C[N+](C)(C)CCOC(N)=O AIXAANGOTKPUOY-UHFFFAOYSA-N 0.000 description 3
- 229960004484 carbachol Drugs 0.000 description 3
- 238000010586 diagram Methods 0.000 description 3
- 239000000706 filtrate Substances 0.000 description 3
- 235000019000 fluorine Nutrition 0.000 description 3
- 150000002431 hydrogen Chemical group 0.000 description 3
- 125000004356 hydroxy functional group Chemical group O* 0.000 description 3
- 210000001589 microsome Anatomy 0.000 description 3
- 229940124531 pharmaceutical excipient Drugs 0.000 description 3
- 239000008363 phosphate buffer Substances 0.000 description 3
- 239000002244 precipitate Substances 0.000 description 3
- 125000006239 protecting group Chemical group 0.000 description 3
- LOUPRKONTZGTKE-LHHVKLHASA-N quinidine Chemical compound C([C@H]([C@H](C1)C=C)C2)C[N@@]1[C@H]2[C@@H](O)C1=CC=NC2=CC=C(OC)C=C21 LOUPRKONTZGTKE-LHHVKLHASA-N 0.000 description 3
- 230000000862 serotonergic effect Effects 0.000 description 3
- 229910000104 sodium hydride Inorganic materials 0.000 description 3
- 230000009870 specific binding Effects 0.000 description 3
- 239000000725 suspension Substances 0.000 description 3
- 238000012360 testing method Methods 0.000 description 3
- UMGDCJDMYOKAJW-UHFFFAOYSA-N thiourea Chemical compound NC(N)=S UMGDCJDMYOKAJW-UHFFFAOYSA-N 0.000 description 3
- 230000009466 transformation Effects 0.000 description 3
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 2
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 2
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 2
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 2
- NDYWSWDCOWRLOQ-UHFFFAOYSA-N CCN(CC)SN(CC)CC.F.F.F Chemical compound CCN(CC)SN(CC)CC.F.F.F NDYWSWDCOWRLOQ-UHFFFAOYSA-N 0.000 description 2
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical group [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 2
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 2
- XDTMQSROBMDMFD-UHFFFAOYSA-N Cyclohexane Chemical compound C1CCCCC1 XDTMQSROBMDMFD-UHFFFAOYSA-N 0.000 description 2
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 2
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 2
- 102000014415 Muscarinic acetylcholine receptor Human genes 0.000 description 2
- 108050003473 Muscarinic acetylcholine receptor Proteins 0.000 description 2
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 2
- SECXISVLQFMRJM-UHFFFAOYSA-N N-Methylpyrrolidone Chemical compound CN1CCCC1=O SECXISVLQFMRJM-UHFFFAOYSA-N 0.000 description 2
- LZCOQTDXKCNBEE-XJMZPCNVSA-N N-methylscopolamine Chemical compound C1([C@@H](CO)C(=O)OC2C[C@@H]3[N+]([C@H](C2)[C@@H]2[C@H]3O2)(C)C)=CC=CC=C1 LZCOQTDXKCNBEE-XJMZPCNVSA-N 0.000 description 2
- FULZLIGZKMKICU-UHFFFAOYSA-N N-phenylthiourea Chemical compound NC(=S)NC1=CC=CC=C1 FULZLIGZKMKICU-UHFFFAOYSA-N 0.000 description 2
- 241000283973 Oryctolagus cuniculus Species 0.000 description 2
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 2
- 102000001708 Protein Isoforms Human genes 0.000 description 2
- 108010029485 Protein Isoforms Proteins 0.000 description 2
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 2
- OKJPEAGHQZHRQV-UHFFFAOYSA-N Triiodomethane Natural products IC(I)I OKJPEAGHQZHRQV-UHFFFAOYSA-N 0.000 description 2
- 230000003042 antagnostic effect Effects 0.000 description 2
- 230000027455 binding Effects 0.000 description 2
- 230000036983 biotransformation Effects 0.000 description 2
- ILAHWRKJUDSMFH-UHFFFAOYSA-N boron tribromide Chemical compound BrB(Br)Br ILAHWRKJUDSMFH-UHFFFAOYSA-N 0.000 description 2
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 2
- 229910052794 bromium Inorganic materials 0.000 description 2
- 239000002775 capsule Substances 0.000 description 2
- 125000004432 carbon atom Chemical group C* 0.000 description 2
- 210000004027 cell Anatomy 0.000 description 2
- 229910052801 chlorine Inorganic materials 0.000 description 2
- 230000008602 contraction Effects 0.000 description 2
- 238000007796 conventional method Methods 0.000 description 2
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 2
- 239000006185 dispersion Substances 0.000 description 2
- 230000000694 effects Effects 0.000 description 2
- 238000011067 equilibration Methods 0.000 description 2
- 210000003238 esophagus Anatomy 0.000 description 2
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 2
- 125000001153 fluoro group Chemical group F* 0.000 description 2
- 125000000524 functional group Chemical group 0.000 description 2
- AMWRITDGCCNYAT-UHFFFAOYSA-L hydroxy(oxo)manganese;manganese Chemical compound [Mn].O[Mn]=O.O[Mn]=O AMWRITDGCCNYAT-UHFFFAOYSA-L 0.000 description 2
- 239000003112 inhibitor Substances 0.000 description 2
- 238000007918 intramuscular administration Methods 0.000 description 2
- 238000001990 intravenous administration Methods 0.000 description 2
- INQOMBQAUSQDDS-UHFFFAOYSA-N iodomethane Chemical compound IC INQOMBQAUSQDDS-UHFFFAOYSA-N 0.000 description 2
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 2
- 239000007788 liquid Substances 0.000 description 2
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 2
- PPNAOCWZXJOHFK-UHFFFAOYSA-N manganese(2+);oxygen(2-) Chemical class [O-2].[Mn+2] PPNAOCWZXJOHFK-UHFFFAOYSA-N 0.000 description 2
- 230000003228 microsomal effect Effects 0.000 description 2
- 230000003551 muscarinic effect Effects 0.000 description 2
- 230000003387 muscular Effects 0.000 description 2
- 238000007254 oxidation reaction Methods 0.000 description 2
- 239000008188 pellet Substances 0.000 description 2
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 2
- AQHHHDLHHXJYJD-UHFFFAOYSA-N propranolol Chemical compound C1=CC=C2C(OCC(O)CNC(C)C)=CC=CC2=C1 AQHHHDLHHXJYJD-UHFFFAOYSA-N 0.000 description 2
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 2
- 238000000926 separation method Methods 0.000 description 2
- 239000011734 sodium Substances 0.000 description 2
- 239000012312 sodium hydride Substances 0.000 description 2
- 238000007920 subcutaneous administration Methods 0.000 description 2
- KZNICNPSHKQLFF-UHFFFAOYSA-N succinimide Chemical compound O=C1CCC(=O)N1 KZNICNPSHKQLFF-UHFFFAOYSA-N 0.000 description 2
- 238000003786 synthesis reaction Methods 0.000 description 2
- 238000002560 therapeutic procedure Methods 0.000 description 2
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 2
- 230000000699 topical effect Effects 0.000 description 2
- 238000005303 weighing Methods 0.000 description 2
- PUPZLCDOIYMWBV-UHFFFAOYSA-N (+/-)-1,3-Butanediol Chemical compound CC(O)CCO PUPZLCDOIYMWBV-UHFFFAOYSA-N 0.000 description 1
- FLQPYEOKVZYXRL-UHFFFAOYSA-N (1-benzylpiperidin-4-yl)methanol Chemical compound C1CC(CO)CCN1CC1=CC=CC=C1 FLQPYEOKVZYXRL-UHFFFAOYSA-N 0.000 description 1
- QBYIENPQHBMVBV-HFEGYEGKSA-N (2R)-2-hydroxy-2-phenylacetic acid Chemical compound O[C@@H](C(O)=O)c1ccccc1.O[C@@H](C(O)=O)c1ccccc1 QBYIENPQHBMVBV-HFEGYEGKSA-N 0.000 description 1
- QDZOEBFLNHCSSF-PFFBOGFISA-N (2S)-2-[[(2R)-2-[[(2S)-1-[(2S)-6-amino-2-[[(2S)-1-[(2R)-2-amino-5-carbamimidamidopentanoyl]pyrrolidine-2-carbonyl]amino]hexanoyl]pyrrolidine-2-carbonyl]amino]-3-(1H-indol-3-yl)propanoyl]amino]-N-[(2R)-1-[[(2S)-1-[[(2R)-1-[[(2S)-1-[[(2S)-1-amino-4-methyl-1-oxopentan-2-yl]amino]-4-methyl-1-oxopentan-2-yl]amino]-3-(1H-indol-3-yl)-1-oxopropan-2-yl]amino]-1-oxo-3-phenylpropan-2-yl]amino]-3-(1H-indol-3-yl)-1-oxopropan-2-yl]pentanediamide Chemical compound C([C@@H](C(=O)N[C@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(C)C)C(N)=O)NC(=O)[C@@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](CCCCN)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](N)CCCNC(N)=N)C1=CC=CC=C1 QDZOEBFLNHCSSF-PFFBOGFISA-N 0.000 description 1
- FELGMEQIXOGIFQ-CYBMUJFWSA-N (3r)-9-methyl-3-[(2-methylimidazol-1-yl)methyl]-2,3-dihydro-1h-carbazol-4-one Chemical compound CC1=NC=CN1C[C@@H]1C(=O)C(C=2C(=CC=CC=2)N2C)=C2CC1 FELGMEQIXOGIFQ-CYBMUJFWSA-N 0.000 description 1
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 description 1
- NGFOOROBRWVGMB-UHFFFAOYSA-N 1-[(3-methoxyphenyl)methyl]-4-[[1-phenyl-4-(trifluoromethyl)imidazol-2-yl]oxymethyl]piperidine Chemical compound COc1cccc(CN2CCC(COc3nc(cn3-c3ccccc3)C(F)(F)F)CC2)c1 NGFOOROBRWVGMB-UHFFFAOYSA-N 0.000 description 1
- DEASGFUXKKZMEV-UHFFFAOYSA-N 1-[(3-methoxyphenyl)methyl]piperidine Chemical compound COC1=CC=CC(CN2CCCCC2)=C1 DEASGFUXKKZMEV-UHFFFAOYSA-N 0.000 description 1
- MSRCZRWMUXRWKZ-UHFFFAOYSA-N 1-benzyl-4-[[1-(3-methoxyphenyl)-4-(trifluoromethyl)imidazol-2-yl]oxymethyl]piperidine Chemical compound COc1cccc(c1)-n1cc(nc1OCC1CCN(Cc2ccccc2)CC1)C(F)(F)F MSRCZRWMUXRWKZ-UHFFFAOYSA-N 0.000 description 1
- ONCKTBFVNYWXLB-UHFFFAOYSA-N 1-benzyl-4-[[1-phenyl-4-(trifluoromethyl)imidazol-2-yl]oxymethyl]piperidine Chemical compound FC(F)(F)c1cn(c(OCC2CCN(Cc3ccccc3)CC2)n1)-c1ccccc1 ONCKTBFVNYWXLB-UHFFFAOYSA-N 0.000 description 1
- JKMHFZQWWAIEOD-UHFFFAOYSA-N 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid Chemical compound OCC[NH+]1CCN(CCS([O-])(=O)=O)CC1 JKMHFZQWWAIEOD-UHFFFAOYSA-N 0.000 description 1
- QKNYBSVHEMOAJP-UHFFFAOYSA-N 2-amino-2-(hydroxymethyl)propane-1,3-diol;hydron;chloride Chemical compound Cl.OCC(N)(CO)CO QKNYBSVHEMOAJP-UHFFFAOYSA-N 0.000 description 1
- KMGUEILFFWDGFV-UHFFFAOYSA-N 2-benzoyl-2-benzoyloxy-3-hydroxybutanedioic acid Chemical compound C=1C=CC=CC=1C(=O)C(C(C(O)=O)O)(C(O)=O)OC(=O)C1=CC=CC=C1 KMGUEILFFWDGFV-UHFFFAOYSA-N 0.000 description 1
- OPMLQJNNYVVNGK-UHFFFAOYSA-N 2-methylsulfonyl-1-phenylimidazole-4-carbaldehyde Chemical compound CS(=O)(=O)c1nc(C=O)cn1-c1ccccc1 OPMLQJNNYVVNGK-UHFFFAOYSA-N 0.000 description 1
- 229940080296 2-naphthalenesulfonate Drugs 0.000 description 1
- WTVULBYVOQDNQD-UHFFFAOYSA-N 3-[2-[(1-benzylpiperidin-4-yl)methoxy]-5-chloro-4-(trifluoromethyl)imidazol-1-yl]phenol Chemical compound OC1=CC=CC(N2C(=C(N=C2OCC2CCN(CC=3C=CC=CC=3)CC2)C(F)(F)F)Cl)=C1 WTVULBYVOQDNQD-UHFFFAOYSA-N 0.000 description 1
- PDYSPQOTNBKSPP-UHFFFAOYSA-N 3-[[4-[[1-phenyl-4-(trifluoromethyl)imidazol-2-yl]oxymethyl]piperidin-1-yl]methyl]phenol Chemical compound Oc1cccc(CN2CCC(COc3nc(cn3-c3ccccc3)C(F)(F)F)CC2)c1 PDYSPQOTNBKSPP-UHFFFAOYSA-N 0.000 description 1
- UNMQKOWDQSVOAS-UHFFFAOYSA-N 3-[[4-[[5-chloro-1-(2-fluorophenyl)-4-(trifluoromethyl)imidazol-2-yl]oxymethyl]piperidin-1-yl]methyl]phenol Chemical compound Oc1cccc(CN2CCC(COc3nc(c(Cl)n3-c3ccccc3F)C(F)(F)F)CC2)c1 UNMQKOWDQSVOAS-UHFFFAOYSA-N 0.000 description 1
- PFANDRFFRGECHZ-UHFFFAOYSA-N 3-[[4-[[5-chloro-1-phenyl-4-(trifluoromethyl)imidazol-2-yl]oxymethyl]piperidin-1-yl]methyl]phenol Chemical compound Oc1cccc(CN2CCC(COc3nc(c(Cl)n3-c3ccccc3)C(F)(F)F)CC2)c1 PFANDRFFRGECHZ-UHFFFAOYSA-N 0.000 description 1
- ONZQYZKCUHFORE-UHFFFAOYSA-N 3-bromo-1,1,1-trifluoropropan-2-one Chemical compound FC(F)(F)C(=O)CBr ONZQYZKCUHFORE-UHFFFAOYSA-N 0.000 description 1
- YPUWCUFNSMMGIL-UHFFFAOYSA-N 4-(difluoromethyl)-2-methylsulfonyl-1-phenylimidazole Chemical compound CS(=O)(=O)c1nc(cn1-c1ccccc1)C(F)F YPUWCUFNSMMGIL-UHFFFAOYSA-N 0.000 description 1
- CVICEEPAFUYBJG-UHFFFAOYSA-N 5-chloro-2,2-difluoro-1,3-benzodioxole Chemical group C1=C(Cl)C=C2OC(F)(F)OC2=C1 CVICEEPAFUYBJG-UHFFFAOYSA-N 0.000 description 1
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 1
- 208000000884 Airway Obstruction Diseases 0.000 description 1
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 1
- 229930003347 Atropine Natural products 0.000 description 1
- WHCOJXXVZZQQMX-UHFFFAOYSA-N CC(C)O.NC(N)=S Chemical compound CC(C)O.NC(N)=S WHCOJXXVZZQQMX-UHFFFAOYSA-N 0.000 description 1
- UXVMQQNJUSDDNG-UHFFFAOYSA-L Calcium chloride Chemical compound [Cl-].[Cl-].[Ca+2] UXVMQQNJUSDDNG-UHFFFAOYSA-L 0.000 description 1
- 241000700198 Cavia Species 0.000 description 1
- 241000700199 Cavia porcellus Species 0.000 description 1
- 0 Cc([n]1C(C=CC=C*)=*)c(C=O)nc1S(C)(=O)=O Chemical compound Cc([n]1C(C=CC=C*)=*)c(C=O)nc1S(C)(=O)=O 0.000 description 1
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 1
- 241000699802 Cricetulus griseus Species 0.000 description 1
- 241000252206 Cypriniformes Species 0.000 description 1
- 108010015742 Cytochrome P-450 Enzyme System Proteins 0.000 description 1
- 102000003849 Cytochrome P450 Human genes 0.000 description 1
- 108010052832 Cytochromes Proteins 0.000 description 1
- 102000018832 Cytochromes Human genes 0.000 description 1
- ZAFNJMIOTHYJRJ-UHFFFAOYSA-N Diisopropyl ether Chemical compound CC(C)OC(C)C ZAFNJMIOTHYJRJ-UHFFFAOYSA-N 0.000 description 1
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 1
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 1
- MOZPSIXKYJUTKI-UHFFFAOYSA-N GR 113808 Chemical compound C12=CC=CC=C2N(C)C=C1C(=O)OCC1CCN(CCNS(C)(=O)=O)CC1 MOZPSIXKYJUTKI-UHFFFAOYSA-N 0.000 description 1
- 108010010803 Gelatin Proteins 0.000 description 1
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 1
- 229920000084 Gum arabic Polymers 0.000 description 1
- 241000282412 Homo Species 0.000 description 1
- 101100329196 Homo sapiens CYP2D6 gene Proteins 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical compound Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 1
- RKUNBYITZUJHSG-UHFFFAOYSA-N Hyosciamin-hydrochlorid Natural products CN1C(C2)CCC1CC2OC(=O)C(CO)C1=CC=CC=C1 RKUNBYITZUJHSG-UHFFFAOYSA-N 0.000 description 1
- 239000007836 KH2PO4 Substances 0.000 description 1
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 1
- 241001465754 Metazoa Species 0.000 description 1
- KQJQICVXLJTWQD-UHFFFAOYSA-N N-Methylthiourea Chemical compound CNC(N)=S KQJQICVXLJTWQD-UHFFFAOYSA-N 0.000 description 1
- 239000012425 OXONE® Substances 0.000 description 1
- 229920002873 Polyethylenimine Polymers 0.000 description 1
- 102100024304 Protachykinin-1 Human genes 0.000 description 1
- IWYDHOAUDWTVEP-UHFFFAOYSA-N R-2-phenyl-2-hydroxyacetic acid Natural products OC(=O)C(O)C1=CC=CC=C1 IWYDHOAUDWTVEP-UHFFFAOYSA-N 0.000 description 1
- 241000700159 Rattus Species 0.000 description 1
- 241000978776 Senegalia senegal Species 0.000 description 1
- 229920002472 Starch Polymers 0.000 description 1
- 101800003906 Substance P Proteins 0.000 description 1
- 229930006000 Sucrose Natural products 0.000 description 1
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 1
- HDYANYHVCAPMJV-LXQIFKJMSA-N UDP-alpha-D-glucuronic acid Chemical compound C([C@@H]1[C@H]([C@H]([C@@H](O1)N1C(NC(=O)C=C1)=O)O)O)OP(O)(=O)OP(O)(=O)O[C@H]1O[C@H](C(O)=O)[C@@H](O)[C@H](O)[C@H]1O HDYANYHVCAPMJV-LXQIFKJMSA-N 0.000 description 1
- HDYANYHVCAPMJV-UHFFFAOYSA-N Uridine diphospho-D-glucuronic acid Natural products O1C(N2C(NC(=O)C=C2)=O)C(O)C(O)C1COP(O)(=O)OP(O)(=O)OC1OC(C(O)=O)C(O)C(O)C1O HDYANYHVCAPMJV-UHFFFAOYSA-N 0.000 description 1
- UHNYWOSVMWQJQK-UHFFFAOYSA-N [1-[(3-methoxyphenyl)methyl]piperidin-4-yl]methanol Chemical compound COC1=CC=CC(CN2CCC(CO)CC2)=C1 UHNYWOSVMWQJQK-UHFFFAOYSA-N 0.000 description 1
- MOZPSIXKYJUTKI-RLXJOQACSA-N [1-[2-(methanesulfonamido)ethyl]piperidin-4-yl]methyl 1-(tritritiomethyl)indole-3-carboxylate Chemical compound C12=CC=CC=C2N(C([3H])([3H])[3H])C=C1C(=O)OCC1CCN(CCNS(C)(=O)=O)CC1 MOZPSIXKYJUTKI-RLXJOQACSA-N 0.000 description 1
- XJLXINKUBYWONI-DQQFMEOOSA-N [[(2r,3r,4r,5r)-5-(6-aminopurin-9-yl)-3-hydroxy-4-phosphonooxyoxolan-2-yl]methoxy-hydroxyphosphoryl] [(2s,3r,4s,5s)-5-(3-carbamoylpyridin-1-ium-1-yl)-3,4-dihydroxyoxolan-2-yl]methyl phosphate Chemical compound NC(=O)C1=CC=C[N+]([C@@H]2[C@H]([C@@H](O)[C@H](COP([O-])(=O)OP(O)(=O)OC[C@@H]3[C@H]([C@@H](OP(O)(O)=O)[C@@H](O3)N3C4=NC=NC(N)=C4N=C3)O)O2)O)=C1 XJLXINKUBYWONI-DQQFMEOOSA-N 0.000 description 1
- IRFQKISACZBIOF-UHFFFAOYSA-N [amino(methylsulfanyl)methylidene]-phenylazanium;iodide Chemical compound I.CSC(N)=NC1=CC=CC=C1 IRFQKISACZBIOF-UHFFFAOYSA-N 0.000 description 1
- 210000001015 abdomen Anatomy 0.000 description 1
- 239000000205 acacia gum Substances 0.000 description 1
- 235000010489 acacia gum Nutrition 0.000 description 1
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 description 1
- 150000007513 acids Chemical class 0.000 description 1
- 239000013543 active substance Substances 0.000 description 1
- 230000001800 adrenalinergic effect Effects 0.000 description 1
- 238000005273 aeration Methods 0.000 description 1
- 125000003158 alcohol group Chemical group 0.000 description 1
- 230000001476 alcoholic effect Effects 0.000 description 1
- AZDRQVAHHNSJOQ-UHFFFAOYSA-N alumane Chemical compound [AlH3] AZDRQVAHHNSJOQ-UHFFFAOYSA-N 0.000 description 1
- 229910052782 aluminium Inorganic materials 0.000 description 1
- 150000001412 amines Chemical class 0.000 description 1
- 239000007900 aqueous suspension Substances 0.000 description 1
- 229960005070 ascorbic acid Drugs 0.000 description 1
- 235000010323 ascorbic acid Nutrition 0.000 description 1
- 239000011668 ascorbic acid Substances 0.000 description 1
- 229960000396 atropine Drugs 0.000 description 1
- RKUNBYITZUJHSG-SPUOUPEWSA-N atropine Chemical compound O([C@H]1C[C@H]2CC[C@@H](C1)N2C)C(=O)C(CO)C1=CC=CC=C1 RKUNBYITZUJHSG-SPUOUPEWSA-N 0.000 description 1
- 238000001574 biopsy Methods 0.000 description 1
- 230000015572 biosynthetic process Effects 0.000 description 1
- 230000037396 body weight Effects 0.000 description 1
- 210000004556 brain Anatomy 0.000 description 1
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 239000001110 calcium chloride Substances 0.000 description 1
- 235000011148 calcium chloride Nutrition 0.000 description 1
- 229910001628 calcium chloride Inorganic materials 0.000 description 1
- 229910052799 carbon Inorganic materials 0.000 description 1
- MOIPGXQKZSZOQX-UHFFFAOYSA-N carbonyl bromide Chemical compound BrC(Br)=O MOIPGXQKZSZOQX-UHFFFAOYSA-N 0.000 description 1
- 239000003054 catalyst Substances 0.000 description 1
- 239000001913 cellulose Substances 0.000 description 1
- 229920002678 cellulose Polymers 0.000 description 1
- 238000005119 centrifugation Methods 0.000 description 1
- 239000007795 chemical reaction product Substances 0.000 description 1
- 239000003638 chemical reducing agent Substances 0.000 description 1
- 210000004978 chinese hamster ovary cell Anatomy 0.000 description 1
- LOUPRKONTZGTKE-UHFFFAOYSA-N cinchonine Natural products C1C(C(C2)C=C)CCN2C1C(O)C1=CC=NC2=CC=C(OC)C=C21 LOUPRKONTZGTKE-UHFFFAOYSA-N 0.000 description 1
- 238000003776 cleavage reaction Methods 0.000 description 1
- 239000006071 cream Substances 0.000 description 1
- 238000002425 crystallisation Methods 0.000 description 1
- 230000008025 crystallization Effects 0.000 description 1
- 238000006264 debenzylation reaction Methods 0.000 description 1
- 238000010511 deprotection reaction Methods 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-M dihydrogenphosphate Chemical compound OP(O)([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-M 0.000 description 1
- 239000003085 diluting agent Substances 0.000 description 1
- 238000007907 direct compression Methods 0.000 description 1
- 201000010099 disease Diseases 0.000 description 1
- 208000035475 disorder Diseases 0.000 description 1
- 239000002270 dispersing agent Substances 0.000 description 1
- 238000007908 dry granulation Methods 0.000 description 1
- 150000002148 esters Chemical group 0.000 description 1
- VICYTAYPKBLQFB-UHFFFAOYSA-N ethyl 3-bromo-2-oxopropanoate Chemical compound CCOC(=O)C(=O)CBr VICYTAYPKBLQFB-UHFFFAOYSA-N 0.000 description 1
- 238000001704 evaporation Methods 0.000 description 1
- 230000008020 evaporation Effects 0.000 description 1
- 238000002474 experimental method Methods 0.000 description 1
- 229910052731 fluorine Inorganic materials 0.000 description 1
- 239000011737 fluorine Substances 0.000 description 1
- 239000006260 foam Substances 0.000 description 1
- 239000012634 fragment Substances 0.000 description 1
- 239000012458 free base Substances 0.000 description 1
- 230000006870 function Effects 0.000 description 1
- 239000008273 gelatin Substances 0.000 description 1
- 229920000159 gelatin Polymers 0.000 description 1
- 235000019322 gelatine Nutrition 0.000 description 1
- 235000011852 gelatine desserts Nutrition 0.000 description 1
- 239000008103 glucose Substances 0.000 description 1
- PCHJSUWPFVWCPO-UHFFFAOYSA-N gold Chemical compound [Au] PCHJSUWPFVWCPO-UHFFFAOYSA-N 0.000 description 1
- 229910052737 gold Inorganic materials 0.000 description 1
- 239000010931 gold Substances 0.000 description 1
- 239000008187 granular material Substances 0.000 description 1
- 229940093915 gynecological organic acid Drugs 0.000 description 1
- 239000007902 hard capsule Substances 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-M hydrogensulfate Chemical compound OS([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-M 0.000 description 1
- 125000003037 imidazol-2-yl group Chemical group [H]N1C([*])=NC([H])=C1[H] 0.000 description 1
- MTNDZQHUAFNZQY-UHFFFAOYSA-N imidazoline Chemical compound C1CN=CN1 MTNDZQHUAFNZQY-UHFFFAOYSA-N 0.000 description 1
- 238000002329 infrared spectrum Methods 0.000 description 1
- 230000002401 inhibitory effect Effects 0.000 description 1
- 230000005764 inhibitory process Effects 0.000 description 1
- 150000007529 inorganic bases Chemical class 0.000 description 1
- 229910052500 inorganic mineral Inorganic materials 0.000 description 1
- 238000011835 investigation Methods 0.000 description 1
- 229910052740 iodine Inorganic materials 0.000 description 1
- 239000011630 iodine Substances 0.000 description 1
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- 239000008101 lactose Substances 0.000 description 1
- 230000000670 limiting effect Effects 0.000 description 1
- 239000012280 lithium aluminium hydride Substances 0.000 description 1
- SIAPCJWMELPYOE-UHFFFAOYSA-N lithium hydride Chemical compound [LiH] SIAPCJWMELPYOE-UHFFFAOYSA-N 0.000 description 1
- 229910000103 lithium hydride Inorganic materials 0.000 description 1
- 210000001853 liver microsome Anatomy 0.000 description 1
- 239000006210 lotion Substances 0.000 description 1
- 235000019359 magnesium stearate Nutrition 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 229960002510 mandelic acid Drugs 0.000 description 1
- 238000001819 mass spectrum Methods 0.000 description 1
- 239000000463 material Substances 0.000 description 1
- 230000007246 mechanism Effects 0.000 description 1
- 230000001404 mediated effect Effects 0.000 description 1
- 238000002844 melting Methods 0.000 description 1
- 230000008018 melting Effects 0.000 description 1
- 206010027175 memory impairment Diseases 0.000 description 1
- 230000002503 metabolic effect Effects 0.000 description 1
- 230000004060 metabolic process Effects 0.000 description 1
- 238000006241 metabolic reaction Methods 0.000 description 1
- 125000004170 methylsulfonyl group Chemical group [H]C([H])([H])S(*)(=O)=O 0.000 description 1
- 235000010755 mineral Nutrition 0.000 description 1
- 239000011707 mineral Substances 0.000 description 1
- 150000007522 mineralic acids Chemical class 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- 229910000402 monopotassium phosphate Inorganic materials 0.000 description 1
- 235000019796 monopotassium phosphate Nutrition 0.000 description 1
- 210000004400 mucous membrane Anatomy 0.000 description 1
- 239000000472 muscarinic agonist Substances 0.000 description 1
- KVBGVZZKJNLNJU-UHFFFAOYSA-M naphthalene-2-sulfonate Chemical compound C1=CC=CC2=CC(S(=O)(=O)[O-])=CC=C21 KVBGVZZKJNLNJU-UHFFFAOYSA-M 0.000 description 1
- 210000001577 neostriatum Anatomy 0.000 description 1
- 238000006386 neutralization reaction Methods 0.000 description 1
- 229910052757 nitrogen Inorganic materials 0.000 description 1
- 230000009871 nonspecific binding Effects 0.000 description 1
- 238000000655 nuclear magnetic resonance spectrum Methods 0.000 description 1
- 239000002674 ointment Substances 0.000 description 1
- 229960005343 ondansetron Drugs 0.000 description 1
- 210000000056 organ Anatomy 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- 239000003960 organic solvent Substances 0.000 description 1
- 230000002611 ovarian Effects 0.000 description 1
- 235000006408 oxalic acid Nutrition 0.000 description 1
- 230000001590 oxidative effect Effects 0.000 description 1
- 230000004783 oxidative metabolism Effects 0.000 description 1
- 229910052763 palladium Inorganic materials 0.000 description 1
- 238000007911 parenteral administration Methods 0.000 description 1
- 230000000144 pharmacologic effect Effects 0.000 description 1
- OXNIZHLAWKMVMX-UHFFFAOYSA-N picric acid Chemical compound OC1=C([N+]([O-])=O)C=C([N+]([O-])=O)C=C1[N+]([O-])=O OXNIZHLAWKMVMX-UHFFFAOYSA-N 0.000 description 1
- RJUAEBLXGFKZMS-UHFFFAOYSA-N piperidin-1-ylmethanol Chemical compound OCN1CCCCC1 RJUAEBLXGFKZMS-UHFFFAOYSA-N 0.000 description 1
- XBXHCBLBYQEYTI-UHFFFAOYSA-N piperidin-4-ylmethanol Chemical compound OCC1CCNCC1 XBXHCBLBYQEYTI-UHFFFAOYSA-N 0.000 description 1
- 229910000027 potassium carbonate Inorganic materials 0.000 description 1
- GNSKLFRGEWLPPA-UHFFFAOYSA-M potassium dihydrogen phosphate Chemical compound [K+].OP(O)([O-])=O GNSKLFRGEWLPPA-UHFFFAOYSA-M 0.000 description 1
- NTTOTNSKUYCDAV-UHFFFAOYSA-N potassium hydride Chemical compound [KH] NTTOTNSKUYCDAV-UHFFFAOYSA-N 0.000 description 1
- 229910000105 potassium hydride Inorganic materials 0.000 description 1
- OKBMCNHOEMXPTM-UHFFFAOYSA-M potassium peroxymonosulfate Chemical compound [K+].OOS([O-])(=O)=O OKBMCNHOEMXPTM-UHFFFAOYSA-M 0.000 description 1
- 230000003389 potentiating effect Effects 0.000 description 1
- 239000000843 powder Substances 0.000 description 1
- 238000001556 precipitation Methods 0.000 description 1
- 239000000047 product Substances 0.000 description 1
- 230000000069 prophylactic effect Effects 0.000 description 1
- 238000011321 prophylaxis Methods 0.000 description 1
- 229960003712 propranolol Drugs 0.000 description 1
- 102000004169 proteins and genes Human genes 0.000 description 1
- 108090000623 proteins and genes Proteins 0.000 description 1
- 229960001404 quinidine Drugs 0.000 description 1
- 230000009257 reactivity Effects 0.000 description 1
- 238000011084 recovery Methods 0.000 description 1
- 238000006268 reductive amination reaction Methods 0.000 description 1
- 230000000717 retained effect Effects 0.000 description 1
- 230000007017 scission Effects 0.000 description 1
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 229940076279 serotonin Drugs 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- 239000012279 sodium borohydride Substances 0.000 description 1
- 229910000033 sodium borohydride Inorganic materials 0.000 description 1
- 239000011780 sodium chloride Substances 0.000 description 1
- 239000012321 sodium triacetoxyborohydride Substances 0.000 description 1
- 239000007901 soft capsule Substances 0.000 description 1
- 239000008247 solid mixture Substances 0.000 description 1
- 238000013222 sprague-dawley male rat Methods 0.000 description 1
- 239000008107 starch Substances 0.000 description 1
- 235000019698 starch Nutrition 0.000 description 1
- 239000007858 starting material Substances 0.000 description 1
- 238000006467 substitution reaction Methods 0.000 description 1
- 229960002317 succinimide Drugs 0.000 description 1
- 239000005720 sucrose Substances 0.000 description 1
- 239000006228 supernatant Substances 0.000 description 1
- 239000003826 tablet Substances 0.000 description 1
- 239000000454 talc Substances 0.000 description 1
- 229910052623 talc Inorganic materials 0.000 description 1
- 235000012222 talc Nutrition 0.000 description 1
- 235000002906 tartaric acid Nutrition 0.000 description 1
- 239000011975 tartaric acid Substances 0.000 description 1
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 1
- WHRNULOCNSKMGB-UHFFFAOYSA-N tetrahydrofuran thf Chemical compound C1CCOC1.C1CCOC1 WHRNULOCNSKMGB-UHFFFAOYSA-N 0.000 description 1
- JOXIMZWYDAKGHI-UHFFFAOYSA-M toluene-4-sulfonate Chemical compound CC1=CC=C(S([O-])(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-M 0.000 description 1
- ITMCEJHCFYSIIV-UHFFFAOYSA-M triflate Chemical compound [O-]S(=O)(=O)C(F)(F)F ITMCEJHCFYSIIV-UHFFFAOYSA-M 0.000 description 1
- 238000005550 wet granulation Methods 0.000 description 1
- 239000000080 wetting agent Substances 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/10—Drugs for disorders of the urinary system of the bladder
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D233/00—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings
- C07D233/54—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members
- C07D233/66—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D233/84—Sulfur atoms
Landscapes
- Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical & Material Sciences (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Public Health (AREA)
- Medicinal Chemistry (AREA)
- Biomedical Technology (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Urology & Nephrology (AREA)
- Hospice & Palliative Care (AREA)
- Psychiatry (AREA)
- Pulmonology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
Abstract
Composés de formule générale (I) : (CF DESSIN DANS BOPI) dans laquelle, R1 est un groupe de formule CnHxFy où n = 1 ou 2, x = 0 à 3, y = 1 à 5 et x + y = 2n + 1 et R2 représentent un hydrogène, un groupe C1-4 alkyle, un atome d'halogène, R représente un groupe C2-4 alkényle ou un phényle éventuellement substitué par un, deux ou trois groupes R4,R3 et R4 représentent, indépendamment l'un de l'autre, un atome d'hydrogène, un atome d'halogène, un groupe C1-4 alkyle, hydroxy, C1-4 alcoxy, cyano, nitro, perfluoro-C1-2 alkyle, amino,ainsi que leurs sels, N-oxydes et hydrates.Application en thérapeutique.
Description
<Desc/Clms Page number 1>
Dérivés de polyfluoroalkylimidazole, leur préparation et leur application en thérapeutique La présente invention a pour objet des dérivés de polyfluoroalkylimidazole leur préparation et leur utilisation en thérapeutique.
En conséquence la présente invention a pour premier objet un composé de formule générale (I):

dans laquelle, R1 est un groupe de formule CnHxFy où n = 1 ou 2, x = 0 à 3, y = 1 à 5 et x+y = 2n+1 et R2 représente un hydrogène, un groupe C1-4 alkyle, un atome d'halogène, R représente un groupe C2-4 alkényle ou un phényle éventuellement substitué par un, deux ou trois groupes R4 ; R3 et R4 représentent, indépendamment l'un de l'autre, un atome d'hydrogène, un atome d'halogène, un groupe C1-4 alkyle, hydroxy, C1-4 alcoxy, cyano, nitro, perfluoro-C1-2 alkyle, amino, et ses sels, N-oxydes et hydrates.
dans laquelle, R1 est un groupe de formule CnHxFy où n = 1 ou 2, x = 0 à 3, y = 1 à 5 et x+y = 2n+1 et R2 représente un hydrogène, un groupe C1-4 alkyle, un atome d'halogène, R représente un groupe C2-4 alkényle ou un phényle éventuellement substitué par un, deux ou trois groupes R4 ; R3 et R4 représentent, indépendamment l'un de l'autre, un atome d'hydrogène, un atome d'halogène, un groupe C1-4 alkyle, hydroxy, C1-4 alcoxy, cyano, nitro, perfluoro-C1-2 alkyle, amino, et ses sels, N-oxydes et hydrates.
La demande de brevet W099/2571 0 divulgue des composés de formule (1) dans laquelle R1 et R2 représentent, indépendamment l'un de l'autre, un hydrogène, un groupe C1-6 alkyle, ou ensemble forment un groupe polyméthylène -(CH2)n-, n pouvant prendre les valeurs de 3 à 6, comme antagonistes des récepteurs muscariniques M3 et sérotoninergiques 5-HT4.
Or il a été constaté que les composés de la présente invention, dans lesquels R1 représente un groupe alkyle polyfluoré, manifestent une affinité supérieure pour les récepteurs précités. D'autre part, ils présentent une inertie accrue face aux mécanismes métaboliques oxydatifs par les microsomes.
<Desc/Clms Page number 2>
Les composés préférés selon la présente invention sont les composés pour lesquels : * R2 représente un atome d'hydrogène ou d'halogène et/ou * R3 représente un atome d'hydrogène, d'halogène, un hydroxy ou un groupe C1-C4 alkoxy de préférence C1-2 alkoxy et/ou * R4 représente un atome d'hydrogène, un hydroxy ou un groupe C1-4 alkoxy de préférence C1-2 alkoxy.
Parmis ceux-ci, les composés particulièrement préférés sont ceux pour lesquels R1 représente un CF3, CF2H ou C2F5.
A titre d'exemple, on peut citer les composés suivants :

1 : 1-(phénylméthyl)-4-[[[1-phényl-4-(trifluorométhyl)-1H-imidazol-2yl]oxy]méthyl]pipéridine 2 : 1-[(3-méthoxyphényl)méthyl]-4-[[[1-phényl-4-(trifluorométhyl)-9H-imidazol-2- yl]oxy]méthyl] pipéridine 3 : 3-[[4-[[[1-phényl-4-(trifluorométhyl)-1H-imidazol-2-yl]oxy]méthyl]pipéridin-1yl]méthyl]phénol

4 : 4-[[[1-(2-fluorophényl)-4-(trifluorométhyl)-9H-imidazol-2-yl]oxy]méthyl]-1- (phénylméthyl)pipéridine 5 : 4-[[[1-(2-fluorophényl)-4-(trifluorométhyl)-9H-imidazol-2-yl]oxy]méthyl]-1-[(3méthoxyphényl)méthyl]pipéridine 6 : 3-[[4-[[[1-(2-fluorophényl)-4-(trifluorométhyl)-9H-imidazol-2yl]oxy]méthyl]pipéridin-1-yl]méthyl]phénol 7 : 1-[(3-méthoxyphényl)méthyl]-4-[[[1-(3-méthoxyphényl)-4-(trifluorométhyl)-9Himidazol-2-yl]oxy]méthyl]pipéridine 8 : 3-[2-[[ 1-[ (3-hyd roxyphé nyl )méthyl] pi périd in-4-yl] méthoxy]-4-(trifluorométhyl)- 1-/-imidazol-1 -yl]phénol 9 : 4-[[[1-(3-méthoxyphényl)-4-(trifluorométhyl)-lH-imidazol-2-yl]oxy]méthyl]-l- (phénylméthyl)pipéridine 10 : 3-[2-[[1-(phénylméthyl}pipéridin-4-yl]méthoxy]-4-(trifluorométhyl)-9Himidazol-1-yl]phénol 11 : 3-[[4-[[[5-chloro-1-phényl-4-(trifluorométhyl)-lH-imidazol-2yl]oxy]méthyl]pipéridin-1-yl]méthyl]phénol 12 : 3-[[4-[[[5-chloro-1-(2-fIuorophényl)-4-(trifluorométhyl)-1 H-imidazol-2yl]oxy]méthyl]pipéridin-1-yl]méthyl]phénol 13 : 4-[[[5-chloro-1-(2-fluorophényl)-4-trifluorométhyl-9N-imidazol-2- yl]oxy]méthyl]-1-(phénylméthyl)pipéridine
1 : 1-(phénylméthyl)-4-[[[1-phényl-4-(trifluorométhyl)-1H-imidazol-2yl]oxy]méthyl]pipéridine 2 : 1-[(3-méthoxyphényl)méthyl]-4-[[[1-phényl-4-(trifluorométhyl)-9H-imidazol-2- yl]oxy]méthyl] pipéridine 3 : 3-[[4-[[[1-phényl-4-(trifluorométhyl)-1H-imidazol-2-yl]oxy]méthyl]pipéridin-1yl]méthyl]phénol
4 : 4-[[[1-(2-fluorophényl)-4-(trifluorométhyl)-9H-imidazol-2-yl]oxy]méthyl]-1- (phénylméthyl)pipéridine 5 : 4-[[[1-(2-fluorophényl)-4-(trifluorométhyl)-9H-imidazol-2-yl]oxy]méthyl]-1-[(3méthoxyphényl)méthyl]pipéridine 6 : 3-[[4-[[[1-(2-fluorophényl)-4-(trifluorométhyl)-9H-imidazol-2yl]oxy]méthyl]pipéridin-1-yl]méthyl]phénol 7 : 1-[(3-méthoxyphényl)méthyl]-4-[[[1-(3-méthoxyphényl)-4-(trifluorométhyl)-9Himidazol-2-yl]oxy]méthyl]pipéridine 8 : 3-[2-[[ 1-[ (3-hyd roxyphé nyl )méthyl] pi périd in-4-yl] méthoxy]-4-(trifluorométhyl)- 1-/-imidazol-1 -yl]phénol 9 : 4-[[[1-(3-méthoxyphényl)-4-(trifluorométhyl)-lH-imidazol-2-yl]oxy]méthyl]-l- (phénylméthyl)pipéridine 10 : 3-[2-[[1-(phénylméthyl}pipéridin-4-yl]méthoxy]-4-(trifluorométhyl)-9Himidazol-1-yl]phénol 11 : 3-[[4-[[[5-chloro-1-phényl-4-(trifluorométhyl)-lH-imidazol-2yl]oxy]méthyl]pipéridin-1-yl]méthyl]phénol 12 : 3-[[4-[[[5-chloro-1-(2-fIuorophényl)-4-(trifluorométhyl)-1 H-imidazol-2yl]oxy]méthyl]pipéridin-1-yl]méthyl]phénol 13 : 4-[[[5-chloro-1-(2-fluorophényl)-4-trifluorométhyl-9N-imidazol-2- yl]oxy]méthyl]-1-(phénylméthyl)pipéridine
<Desc/Clms Page number 3>
14 : 3-[5-Chloro-2-[[1-(phénylméthyl)pipéridin-4-yl]méthoxy]-4-(trifluorométhyl)- 1H-imidazol-1-yl] phénol 15 : 3-[5-Chloro-2-[[1-[(3-hydroxyphényl)méthyl]pipéridin-4-yl]méthoxy]-4-

(trifluorométhyl)-9H-imidazol-1-yl]phénol 16 : 4-[[[4-(pentafluoroéthyl)-1-phényl-9N-imidazol-2-yl)oxy]méthyl]-1- (phénylméthyl)pipéridine 17 : 4-[[[4-(difluorométhyl)-1-phényl-9H-imidazof-2-yl)oxy]méthyl]-1- (phénylméthyl)pipéridine Dans le cadre de la présente invention, on entend par : - C1-z (ou C2-z), où z peut prendre les valeurs de 2 à 4, une chaîne carbonée pouvant avoir de 1 ou 2 à z atomes de carbone, - alkyle, un groupe aliphatique saturé, linéaire ou ramifié ; parexemple, un groupe C1-4 alkyle représente une chaîne carbonée de 1 à 4 atomes de carbone, linéaire ou ramifiée, plus particulièrement un méthyle, éthyle, propyle, isopropyle, butyle , isobutyle, secbutyle, tertbutyle, de préférence un méthyle, éthyle, propyle, isopropyle, - alkényle, un groupe aliphatique mono ou poly-insaturé, linéaire ou ramifié, comprenant de préférence 1 ou 2 insaturations éthyléniques, - perfluoroalkyle, un alkyle dont tous les hydrogènes ont été substitués par des fluors, par exemple CF3, - polyfluoroalkyle, un alkyle dont une partie des hydrogènes a été substituée par des fluors, par exemple CF2H, - CnHxFy avec un n, x et y tels que définis ci-dessus, un perfluoroalkyle ou polyfluoroalkyle, tel que, par exemple, -CF3, -CF2H, -CH2F, -CF2CF3, -CFHCF3, ...
(trifluorométhyl)-9H-imidazol-1-yl]phénol 16 : 4-[[[4-(pentafluoroéthyl)-1-phényl-9N-imidazol-2-yl)oxy]méthyl]-1- (phénylméthyl)pipéridine 17 : 4-[[[4-(difluorométhyl)-1-phényl-9H-imidazof-2-yl)oxy]méthyl]-1- (phénylméthyl)pipéridine Dans le cadre de la présente invention, on entend par : - C1-z (ou C2-z), où z peut prendre les valeurs de 2 à 4, une chaîne carbonée pouvant avoir de 1 ou 2 à z atomes de carbone, - alkyle, un groupe aliphatique saturé, linéaire ou ramifié ; parexemple, un groupe C1-4 alkyle représente une chaîne carbonée de 1 à 4 atomes de carbone, linéaire ou ramifiée, plus particulièrement un méthyle, éthyle, propyle, isopropyle, butyle , isobutyle, secbutyle, tertbutyle, de préférence un méthyle, éthyle, propyle, isopropyle, - alkényle, un groupe aliphatique mono ou poly-insaturé, linéaire ou ramifié, comprenant de préférence 1 ou 2 insaturations éthyléniques, - perfluoroalkyle, un alkyle dont tous les hydrogènes ont été substitués par des fluors, par exemple CF3, - polyfluoroalkyle, un alkyle dont une partie des hydrogènes a été substituée par des fluors, par exemple CF2H, - CnHxFy avec un n, x et y tels que définis ci-dessus, un perfluoroalkyle ou polyfluoroalkyle, tel que, par exemple, -CF3, -CF2H, -CH2F, -CF2CF3, -CFHCF3, ...
- alcoxy, un groupe alkyloxy à chaîne aliphatique saturée, linéaire ou ramifiée, et - atome d'halogène, un fluor, un chlore, un brome ou un iode.
On entend par groupe partant, un groupe pouvant être facilement clivé d'une molécule, avec départ d'une paire électronique, par rupture d'une liaison hétérolytique. Ce groupe peut ainsi être remplacé facilement par un autre groupe lors d'une réaction de substitution par exemple. De tels groupes partants sont, par exemple, les halogènes, ou un groupe hydroxy activé tel qu'un mésyle, tosyle, triflate, acétyle, ...etc. Des exemples de groupes partants ainsi que des références de préparation sont données dans Advanced Organic Chemistry , J. March, 3rd Edition, Wiley Interscience, p 310-316.
<Desc/Clms Page number 4>
On entend par groupe protecteur, un groupement permettant d'empêcher la réactivité d'une fonction ou position, lors d'une réaction chimique pouvant l'affecter, et qui restitue la molécule après clivage selon des méthodes connues de l'homme du métier. Des exemples de groupes protecteurs ainsi que les méthodes de protection et déprotection sont données, entre autres, dans Protective groups in Organic Synthesis, Greene et al., 2nd Ed. (John Wiley & Sons, Inc., New York)..
On entend par groupe fonctionnel, un groupement pouvant être oxydé, réduit, substitué, alkylé, désalkylé ou subir toute autre transformation classique de chimie organique. Ces réactions de transformation fonctionnelle peuvent être réalisées par des méthodes classiques connues de l'homme du métier pour donner d'autres composés. De tels groupes sont par exemple R3 et R4.
Les composés de formule générale (I) peuvent comporter un ou plusieurs carbones asymétriques. Ils peuvent donc exister sous forme d'énantiomères ou de diastéréoisomères.
Ces énantiomères, diastéréoisomères ainsi que leurs mélanges, y compris les mélanges racémiques, font partie de l'invention.
Les composés de formule générale (I) peuvent se présenter sous forme de base libre ou de sels d'addition à des acides, qui font également partie de l'invention.
Ces sels, selon la présente invention, comprennent ceux avec des acides minéraux ou organiques qui permettent une séparation ou une cristallisation convenable des composés de formule (1), tels que l'acide picrique, l'acide oxalique ou un acide optiquement actif, par exemple un acide tartrique, un acide dibenzoyltartrique, un acide mandélique ou un acide camphosulfonique, et ceux qui forment des sels physiologiquement acceptables, tels que le chlorhydrate, le bromhydrate, le citrate, le sulfate, l'hydrogénosulfate, le dihydrogénophosphate, le maléate, le fumarate, le pamoate, le 2-naphtalènesulfonate, le paratoluènesulfonate. Mêmes si les sels pharmaceutiquement acceptables sont préférés, les autres sels font partis de la présente invention. Ces sels peuvent être préparés, selon des méthodes connues de l'homme du métier, par exemple, par réaction du composé de formule (1) sous forme de base avec l'acide dans un solvant approprié, tel qu'une solution alcoolique ou un solvant organique, puis séparation du milieu qui le contient par évaporation du solvant ou par filtration.
<Desc/Clms Page number 5>
Les composés de formule générale (I) peuvent se présenter également sous forme de dérivés N-oxydes qui font partie de la présente invention. Ces dérivés sont obtenus par réaction d'oxydation du composé de formule (I) selon des méthodes connues de l'homme du métier.
La présente invention a pour second objet des procédés de préparation des composés de formule (1).
Ainsi, ces composés peuvent être préparés par des procédés, illustrés dans les schémas qui suivent, dont les conditions opératoires sont classiques pour l'homme du métier.
Selon le schéma 1, la phénylthiourée (II) est transformée en isothiouronium (III) par la méthode décrite par J.N. Baxter dans J. Chem. Soc., 1956, 663 par action d'iodométhane dans un solvant tel que l'acétone ou l'éthanol, de préférence au reflux de ce dernier. L'isothiouronium est mis ensuite en réaction avec une bromocétone de formule (IV) dans un alcool tel que l'isopropanol et en présence d'une base telle que l'hydrogénocarbonate de sodium, de préférence deux équivalents, pour conduire au mélange d'imidazoline (V) et d'imidazole (VI). Ce mélange est ensuite déshydraté selon des méthodes connues de l'homme de métier, par exemple par action d'acide paratoluènesulfonique (APTS) dans un solvant tel que le toluène à reflux pour donner le composé de formule (VI). Ce composé est oxydé selon des méthodes connues de l'homme de métier par exemple au moyen de peroxy monosulfate de potassium commercialisé sous le nom d'Oxone en présence d'alumine humide, dans un solvant tel que le chloroforme, à une température comprise entre 40 et 65 C, pour conduire à la sulfone (VII). Cette sulfone est ensuite couplée avec une hydroxyméthylpipéridine de formule (VIII) en milieu basique pour donner un composé de formule (1). La réaction peut être réalisée en présence d'une base telle que l'hydrure de sodium ou de potassium dans un solvant tel que le diméthylformamide (DMF) ou la N-méthylpyrrolidone (NMP) à une température allant de -10 C à 20 C.
Les significations de R1, R2, R3 et R dans les composés de formule (11), (III), (IV), (V), (VI), (VII) et (VIII) sont telles que définies pour les composés de formule (I).
<Desc/Clms Page number 6>

Schéma 1 Alternativement, les composés de formule (1), dans laquelle R1 représente un groupe HF2C-, peuvent être obtenus comme indiqué dans le schéma 2.
L'isothiouronium (III) est mis en réaction avec le bromocétoester (IX) pour lequel A est un groupe C1-4 alkyle, en présence d'une base telle que l'hydrogénocarbonate de sodium (2 eq. ) dans un solvant tel que l'isopropanol, pour conduire à l'imidazole (X). La fonction ester de celui-ci est réduite en alcool, selon des méthodes connues de l'homme du métier par exemple, par action de l'hydrure mixte d'aluminium et de lithium dans un solvant tel que le tétrahydrofurane THF, pour conduire au sulfure (XI) qui est oxydé par l'Oxone en sulfone (XII) selon les conditions décrites précédemment. La fonction alcool de ce dernier est à son tour oxydée en aldéhyde (XIII). La réaction d'oxydation peut être réalisée selon des méthodes connues de l'homme du métier, par exemple au moyen d'oxyde de manganèse activé dans un solvant tel que le chloroforme ou le dichlorométhane. L'aldéhyde est alors mis en réaction avec du trifluorure de diéthylaminosulfure (DAST) pour conduire au dérivé polyfluoroalkyle (XIV). Enfin, comme dans le schéma 1, le dérivé (XIV) est mis en réaction avec la 4-hydroxyméthylpipéridine (VIII), pour conduire aux composés de formule (1).
<Desc/Clms Page number 7>
Les significations de R1, R2, R3 et R dans les composés de formule (III), (IV), (X), (XI), (XII), (XIII) et (XIV) sont telles que définies pour les composés de formule (1).

Schéma 2 Alternativement, les composés de formule (1), dans laquelle R2 représente un atome d'hydrogène, peuvent être modifiés selon le schéma 3 pour donner d'autres composés de formule (I) dans laquelle R2 est un halogène. Selon ce schéma, un composé de formule (I, R2 = H) est mis en réaction avec une Nhalosuccinimide (NXS) pour laquelle X est un halogène, de préférence un chlore ou un brome, dans un solvant tel que le tétrahydrofurane ou l'acétonitrile pour conduire à un dérivé de formule (I, R2 = X).
<Desc/Clms Page number 8>
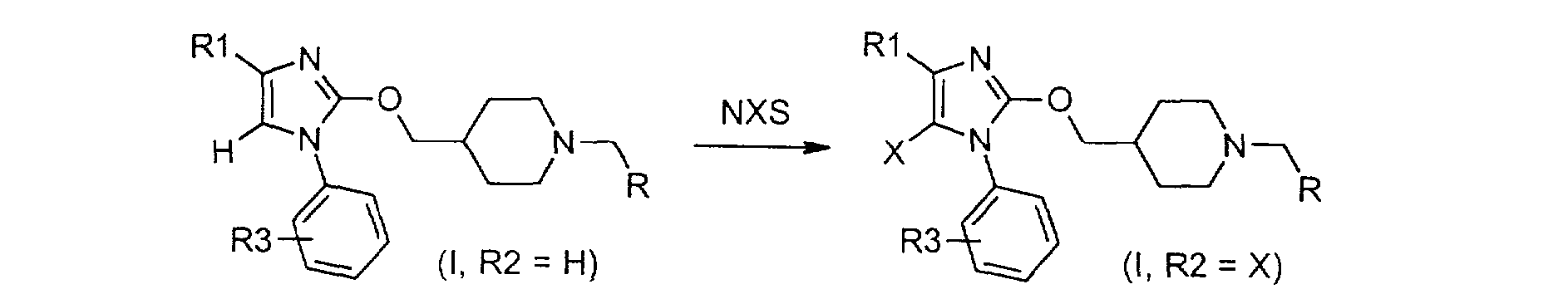
Schéma 3 D'autre part, les composés de formule (1), pour lesquels R est un phényle, éventuellement substitué par un ou plusieurs groupements électrodonneurs tel que des méthoxy, peuvent être débenzylés puis resubstitués pour donner d'autres composés de formule (I) dans laquelle R est un phényle substitué par un autre groupe R4 tel que défini pour les composés de formule (1). La réaction de débenzylation peut être réalisée selon des méthodes connues de l'homme du métier, par exemple, par action de l'hydrogène en présence d'un catalyseur palladié, pour conduire aux dérivés de formule (XV), selon le schéma 4. Les composés de formule (XV) peuvent être ensuite substitués, selon des méthodes connues de l'homme du métier, par exemple, par action d'un composé de formule RCH2Y, dans laquelle Y représente un groupe partant, de préférence un halogène, et R est tel que défini ci-dessus, en présence d'une amine acceptrice de proton, telle que la triéthylamine ou la diisopropyléthylamine, ou d'une base minérale telle que le carbonate de potassium et dans des solvants tels que l'éthanol, le diméthylformamide (DMF) ou le toluène, ou bien par amination réductrice au moyen d'un aldéhyde de formule RCHO dans laquelle R est tel que défini ci-dessus, en présence d'acide chlorhydrique et d'un réducteur tel le borohydrure de sodium, cyanoborohydrure de sodium, triacétoxyborohydrure de sodium, ou un complexe borane-amine, et dans le méthanol ou l'éthanol, pour conduire à de nouveaux composés de formule (1).
Les significations de R1, R2 et R3 des composés (1) et (XV) du schéma 4 sont telles que définies précédemment pour les composés de formule (1).
<Desc/Clms Page number 9>

Schéma 4 D'autre part, dans le cas de composés de formule (I) où R3 et/ou R4 sont des groupes fonctionnels, pouvant être substitués, oxydés, réduits, alkylés ou désalkylés, ces réactions de transformation fonctionnelle peuvent être réalisées par des méthodes classiques connues de l'homme du métier pour donner d'autres composés de formule (1).
Les composés de départ, notamment les composés de formule (II), (IV), (VIII), (IX), RCHO ou RCH2Y, sont disponibles dans le commerce ou décrits dans la littérature, ou peuvent être préparés par des méthodes qui y sont décrites ou qui sont connues de l'homme du métier.
Les composés de formule (V), (VI), (VII), (X), (XI), (XII), (XIII), (XIV) et (XV) dans laquelle R1, R2, R3 et R4 sont tels que définis pour les composés de formules (1), sont nouveaux et font également partie de l'invention. Ils sont utiles comme intermédiaires de synthèse pour préparer les composés de formule (1).
Les exemples suivants illustrent les procédés et techniques mises en oeuvre pour la préparation de cette invention, sans toutefois limiter l'étendue de la revendication. Les micro-analyses élémentaires et les spectres RMN, IR ou de masse confirment les structures des composés obtenus.
<Desc/Clms Page number 10>
Exemple 1 : Préparation de l'iodure de N-phényl-S-méthyl-isothiouronium (formule III; selon schéma 1) 30 g de phénylthiourée sont mis en suspension dans 500 mL d'acétone. On ajoute 12,3 mL d'iodométhane en controlant l'exothermicité, et agite durant 15 heures à température ambiante. Le mélange réactionnel est filtré et le filtrat est repris par 100 mL d'éther éthylique. le précipité formé est essoré et lavé deux fois avec 50 mL d'un mélange 1/1 d'éther éthylique et d'acétone, puis séché sous vide, pour conduire à 30 g d'iodure de N-phényl-S-méthyl-isothiouronium (PF=141 C).
Exemple 2 : Préparation de 2-méthylthio-1-phényl-4-(trifluorométhyl)-1 Himidazole (formule VI; selon schéma 1) On ajoute successivement dans 500 mL d'isopropanol, 10 g d'iodure de Nphényl-S-méthyl-isothiouronium, 6 g d'hydrogénocarbonate de sodium et 5 mL de 1-bromo-3,3,3-trifluoroacétone, et chauffe à 70-80 C durant 15 heures. Après concentration à sec, on reprend par 200 mL d'eau et épuise la phase aqueuse à l'acétate d'éthyle. Les phases organiques réunies sont lavées successivement à l'eau et à la saumure avant d'être séchées sur sulfate de magnésium, filtrées et concentrées à sec. On reprend alors le brut réactionnel dans 50 mL de toluène, et additionne 0,15 g d'acide paratoluènesulfonique (APTS), puis porte au reflux durant 5 heures. Après retour à température ambiante, on neutralise au moyen d'une solution aqueuse saturée en hydrogénocarbonate de sodium, et épuise la phase aqueuse à l'acétate d'éthyle. Les phases organiques réunies sont lavées successivement à l'eau et à la saumure avant d'être séchées sur sulfate de magnésium, filtrées et concentrées à sec. On purifie par chromatographie flash sur gel de silice en éluant avec un mélange 85/15 d'heptane et d'acétate d'éthyle, pour isoler 4g de composé attendu.

Exemple 3 : Préparation de 2-mésyl-1-phényl-4-(trifluorométhyl)-9H-imidazole (formule VI I; selon schéma 1) Dans 85 mL de chloroforme, on ajoute successivement, 3,9 g de 2-méthylthio-1-

phényl-4-(trifluorométhyl)-7/-/-imidazole, 28 g d'Oxone, et 15 g d'alumine (préalablement hydratée avec 3 mL d'eau). On porte le mélange réactionnel à 60 C durant 20 heures, puis le filtre à chaud, et lave deux fois le précipité avec
phényl-4-(trifluorométhyl)-7/-/-imidazole, 28 g d'Oxone, et 15 g d'alumine (préalablement hydratée avec 3 mL d'eau). On porte le mélange réactionnel à 60 C durant 20 heures, puis le filtre à chaud, et lave deux fois le précipité avec
<Desc/Clms Page number 11>
40 mL de chloroforme chaud. Le filtrat est concentré à sec puis trituré dans l'éther diisopropylique, essoré et séché sous vide, pour conduire à 3,35 g de composé attendu.

Exemple 4 : Préparation de 4-[[[I-phényi-4-(trifluorométhyl)-lH-imidazol-2- yl]oxy]méthyl]-1-[(3-méthoxyphényl)méthyl]pipéridine Formule (I); R1=CF3, R2=R3=H, R = phényle, R4= 3-OCH3, selon schéma 1 Sous atmosphère d'azote, on place 0,69 g d'hydrure de sodium (en dispersion à 60% dans l'huile), et additionne 20 mL de diméthylformamide (DMF) anhydre, puis 3,53 g de 4-hydroxyméthyl-1-[(3-méthoxyphényl)méthyl]pipéridine en solution dans 25 mL de DMF anhydre. On porte le mélange réactionnel à 50 C jusqu'à complétion du dégagement d'hydrogène, puis refroidit au bain de glace

avant d'ajouter 3,35 g de 2-mésyl-1-phényl-4-(trifluorométhyl)-9H-imidazole en solution dans 25 mL de DMF anhydre. Après une heure d'agitation à température ambiante, le milieu réactionnel est hydrolysé avec 150 mL d'eau. La phase aqueuse est épuisée à l'acétate d'éthyle. Les phases organiques réunies sont lavées successivement à l'eau et à la saumure avant d'être séchées sur sulfate de magnésium, filtrées et concentrées à sec. On purifie par chromatographie flash sur gel de silice en éluant avec un mélange 95/5 de dichlorométhane et de méthanol, pour isoler 4,4 g de composé attendu.

avant d'ajouter 3,35 g de 2-mésyl-1-phényl-4-(trifluorométhyl)-9H-imidazole en solution dans 25 mL de DMF anhydre. Après une heure d'agitation à température ambiante, le milieu réactionnel est hydrolysé avec 150 mL d'eau. La phase aqueuse est épuisée à l'acétate d'éthyle. Les phases organiques réunies sont lavées successivement à l'eau et à la saumure avant d'être séchées sur sulfate de magnésium, filtrées et concentrées à sec. On purifie par chromatographie flash sur gel de silice en éluant avec un mélange 95/5 de dichlorométhane et de méthanol, pour isoler 4,4 g de composé attendu.
Exemple 5 : Préparation de 4-éthylcarboxylate-2-méthylthio-1-phényl-H- imidazole (formule X ; selon schéma 2) Dans 1,25 L d'isopropanol, on mélange 25 g d' iodure de N-phényl-S-méthylisothiouronium, 15 g d'hydrogénocarbonate de sodium et 13,4 mL de bromopyruvate d'éthyle, et porte le mélange réactionnel à 80 C durant 20 heures. Après concentration à sec, on reprend par 250 mL d'eau et épuise la phase aqueuse par extractions successives à l'acétate d'éthyle. Les phases organiques réunies sont lavées successivement à l'eau et à la saumure avant d'être séchées sur sulfate de magnésium, filtrées et concentrées à sec. On purifie par chromatographie flash sur gel de silice en éluant avec un mélange 75/25 d'heptane et d'acétate d'éthyle, pour isoler 12,5 g de composé attendu (PF = 55 C).
<Desc/Clms Page number 12>
Exemple 6 : Préparation de 4-hydroxyméthyl-2-méthylthio-1-phényl-9H- imidazole (formule XI; selon schéma 2) Sous atmosphère d'azote, on recouvre 2 g d'hydrure mixte de lithium et d'aluminium par 100 mL de THF anhydre, et refroidit le mélange à -15 C. On coule lentement 12,5 g de 4-éthylcarboxylate-2-méthylthio-1-phényl-1Himidazole, préalablement solubilisés dans 140 mL de THF anhydre, et agite à température ambiante durant 1,5 heure. On refroidit le mélange réactionnel à 0 C et additionne successivement 2 mL d'eau, 2 mL de soude aqueuse à 15%, puis trois fois 2 mL d'eau. On agite à température ambiante durant 15 heures et empâte avec de la cellite. Après concentration à sec, on dépose cette pâte sur un gel de silice et élue avec un gradient de méthanol (de 0 à 2,5%) dans le dichlorométhane, pour conduire à 5,6 g de composé attendu (PF = 122 C).
Exemple 7 : Préparation de 4-hydroxyméthyl-2-mésyl-1-phényl-9H-imidazole (formule XII; selon schéma 2) Dans 73 mL de chloroforme, on ajoute successivement, 3,22 g de 4-

hydroxyméthyl-2-méthylthio-1-phényl-9N-imidazole, 27 g d'Oxone@, et 14,6 g d'alumine (préalablement hydratée avec 2,9 mL d'eau). On porte le mélange réactionnel à 50 C durant 16 heures, puis le filtre à chaud, et lave deux fois le précipité avec 20 mL d'un mélange 9/1 de THF et de méthanol. Le filtrat est concentré à sec puis purifié par chromatographie flash sur gel de silice en éluant avec de l'acétate d'éthyle pour conduire à 2,13 g de composé attendu.
hydroxyméthyl-2-méthylthio-1-phényl-9N-imidazole, 27 g d'Oxone@, et 14,6 g d'alumine (préalablement hydratée avec 2,9 mL d'eau). On porte le mélange réactionnel à 50 C durant 16 heures, puis le filtre à chaud, et lave deux fois le précipité avec 20 mL d'un mélange 9/1 de THF et de méthanol. Le filtrat est concentré à sec puis purifié par chromatographie flash sur gel de silice en éluant avec de l'acétate d'éthyle pour conduire à 2,13 g de composé attendu.
Exemple 8 : Préparation de 4-formyl-2-mésyl-1-phényl-?N-imidazole (formule XIII; selon schéma 2)

A 2,13 g de 4-hydroxyméthyl-2-mésyl-1-phényl-9H-imidazole, en solution dans 42 mL de dichlorométhane, on ajoute 0,74 g d'oxyde de manganèse activé, et porte au reflux durant 1 heure. On ajoute à nouveau 0,74 g d'oxyde de manganèse activé, et porte au reflux durant 1 heure. Cette addition est répétée cinq fois. Après filtration et concentration, on purifie par chromatographie flash sur gel de silice en éluant avec un mélange 1/1 de cyclohexane et de l'acétate d'éthyle pour conduire à 1,8 g de composé attendu.
A 2,13 g de 4-hydroxyméthyl-2-mésyl-1-phényl-9H-imidazole, en solution dans 42 mL de dichlorométhane, on ajoute 0,74 g d'oxyde de manganèse activé, et porte au reflux durant 1 heure. On ajoute à nouveau 0,74 g d'oxyde de manganèse activé, et porte au reflux durant 1 heure. Cette addition est répétée cinq fois. Après filtration et concentration, on purifie par chromatographie flash sur gel de silice en éluant avec un mélange 1/1 de cyclohexane et de l'acétate d'éthyle pour conduire à 1,8 g de composé attendu.
<Desc/Clms Page number 13>
Exemple 9 : Préparation de 4-(difluorométhyl)-2-mésyl-1-phényl-9N-imidazole (formule XIV; selon schéma 2) A 2,32 g de 4-formyl-2-mésyl-1-phényl-1H-imidazole, solubilisés dans 110 mL de dichlorométhane, on ajoute, sous atmosphère d'azote, 3,7 mL de trifluorure de diéthylaminosulfure (DAST) en solution dans 20 mL de dichlorométhane. Après trois heures d'agitation à température ambiante, on additionne à nouveau 3,7 mL de DAST en solution dans 20 mL de dichlorométhane, et poursuit l'agitation trois autres heures. On jette le milieu réactionnel sur un mélange de glace et d'hydrogénocarbonate de sodium et épuise la phase aqueuse par extractions successives au dichlorométhane. Les phases organiques réunies sont lavées successivement à l'eau et à la saumure avant d'être séchées sur sulfate de magnésium, filtrées et concentrées à sec. On purifie par chromatographie flash sur gel de silice en éluant avec un gradient d'acétate d'éthyle (de 0 à 20%) dans le cyclohexane, pour isoler 2. g de composé attendu.
Exemple 10 : Préparation de 4-[[[4-(diftuorométhyl)-1-phényl-1 H-imidazol-2- yl]oxy]méthyl]-1-(phénylméthyl)pipéridine Formule (1), R1=CF2H, R = phényle, R2=R3=R4=H ; selon schéma 2 Sous atmosphère d'azote, on place 16 mg d'hydrure de sodium (en dispersion à 95% dans l'huile), et additionne 1 mL de diméthylformamide (DMF) anhydre, puis 0,13 g de 4-hydroxyméthyl-1-(phénylméthyl)pipéridine en solution dans 2 mL de DMF anhydre. On porte le mélange réactionnel à 60 C jusqu'à complétion du dégagement d'hydrogène, puis refroidit au bain de glace avant
d'ajouter 0,12 g de 4-(difluorométhyl)-2-mésyl-1 -phényl- 1H-imidazole en solution dans 3 mL de DMF anhydre. Après 2 heures d'agitation à 45 C, le milieu réactionnel est hydrolysé avec 30 mL d'eau. La phase aqueuse est épuisée par extractions successives à l'acétate d'éthyle. Les phases organiques réunies sont lavées successivement à l'eau et à la saumure avant d'être séchées sur sulfate de magnésium, filtrées et concentrées à sec. On purifie par chromatographie flash sur gel de silice en éluant avec un gradient (0 à 5%) de méthanol dans le dichlorométhane, pour isoler 0,11g de composé attendu.
d'ajouter 0,12 g de 4-(difluorométhyl)-2-mésyl-1 -phényl- 1H-imidazole en solution dans 3 mL de DMF anhydre. Après 2 heures d'agitation à 45 C, le milieu réactionnel est hydrolysé avec 30 mL d'eau. La phase aqueuse est épuisée par extractions successives à l'acétate d'éthyle. Les phases organiques réunies sont lavées successivement à l'eau et à la saumure avant d'être séchées sur sulfate de magnésium, filtrées et concentrées à sec. On purifie par chromatographie flash sur gel de silice en éluant avec un gradient (0 à 5%) de méthanol dans le dichlorométhane, pour isoler 0,11g de composé attendu.
<Desc/Clms Page number 14>
Exemple 11 : Préparation de 4-[[[5-chloro-1-phényl-4-(trifluorométhyl)-1H- imidazol-2-yl]oxy]méthyi]-1-[(3-méthoxyphényl)méthyl]pipéridine Formule (1), R1=CF3, R2=CI, R3=H ; R = phényle, R4=3-OCH3, selon schéma 3

A 1,6 g de 4-[[[l-phényl-4-(trifluorométhyl)-lH-imidazol-2-yl]oxy]méthyl]-I-[(3- méthoxyphényl)méthyl]pipéridine, en solution dans 20 mL de THF anhydre, on ajoute sous azote 2,16 g de N-chlorosuccinimide (NCS), et chauffe le milieu réactionnel à 60 C durant 6 heures. On jette alors le milieu réactionnel sur un mélange de glace et d'une solution aqueuse saturée en hydrogénocarbonate de sodium. On épuise la phase aqueuse par extractions successives avec de l'acétate d'éthyle. Les phases organiques réunies sont concentrées à sec et reprises par de l'éther éthylique, ce qui entraîne la précipitation de la succinimide, qui est éliminée par plusieurs filtrations successives. Après concentration, le brut est purifié par chromatographie flash sur gel de silice, en éluant avec un gradient de méthanol (de 0 à 2,5%) dans le dichlorométhane, pour fournir 0,73 g de composé attendu.

A 1,6 g de 4-[[[l-phényl-4-(trifluorométhyl)-lH-imidazol-2-yl]oxy]méthyl]-I-[(3- méthoxyphényl)méthyl]pipéridine, en solution dans 20 mL de THF anhydre, on ajoute sous azote 2,16 g de N-chlorosuccinimide (NCS), et chauffe le milieu réactionnel à 60 C durant 6 heures. On jette alors le milieu réactionnel sur un mélange de glace et d'une solution aqueuse saturée en hydrogénocarbonate de sodium. On épuise la phase aqueuse par extractions successives avec de l'acétate d'éthyle. Les phases organiques réunies sont concentrées à sec et reprises par de l'éther éthylique, ce qui entraîne la précipitation de la succinimide, qui est éliminée par plusieurs filtrations successives. Après concentration, le brut est purifié par chromatographie flash sur gel de silice, en éluant avec un gradient de méthanol (de 0 à 2,5%) dans le dichlorométhane, pour fournir 0,73 g de composé attendu.
Exemple 12 : Préparation de 3-[[4-[[[5-chloro-1-phényl-4-(trifluorométhyl)-7/-/imidazol-2-yl]oxy] méthyl] pipéridin-1-yl]méthyl]phénol Formule (1), R1=CF3, R2=CI, R3=H; R = phényle, R4=3-OH On solubilise 0,9 g de 4-[[[5-chloro-1-phényl-4-(trifluorométhyl)-9H-imidazol-2- yl]oxy]méthyl]-1-[(3-méthoxyphényl)méthyl]pipéridine dans 20 mL de dichlorométhane (DCM), et ajoute 3,75 mL d'acide chlorhydrique 3,5N dans l'isopropanol. Après concentration à sec, on reprend par 20 mL de DCM et refroidit à -78 C, sous atmosphère d'azote. On coule ensuite 0,53 mL de tribromure de bore, et agite 0,25 heure à -78 C, avant de revenir à -15 C durant 1,75 heures. On jette alors le mélange réactionnel sur un mélange de glace et d'une solution aqueuse saturée en hydrogénocarbonate de sodium. On épuise la phase aqueuse par extractions successives avec de l'acétate d'éthyle.
Les phases organiques réunies sont lavées deux fois à l'eau puis à la saumure avant d'être séchées sur sulfate de magnésium. Après concentration à sec, le brut réactionnel est purifié par chromatographie flash sur gel de silice, en éluant avec un gradient (de 0 à 5%) de méthanol dans le dichlorométhane, pour fournir 0,63 g de composé attendu, sous forme de mousse amorphe beige.
<Desc/Clms Page number 15>
Les composés décrits dans le tableau suivant ont été préparés selon les méthodes décrites précédemment.

R=Ph entrée R1 R2 R3 R4 sel PF ( C)
1 CF3 H H H fuma rate 170
2 CF3 H H 3-OCH3 chlorhydrate 171
3 CF3 H H 3-OH chlorhydrate 196
4 CF3 H 2-F H chlorhydrate 173
5 CF3 H 2-F 3-OCH, chlorhydrate 155
6 CF3 H 2-F 3-OH chlorhydrate 190 7 CF3 H 3-OCH3 3-OCH3 chlorhydrate 150
8 CF3 H 3-OH 3-OH
9 CF3 H 3-OCH3 H chlorhydrate 151 10 CF3 H 3-OH H 11 CF3 CI H 3-OH fumarate 130
12 CF3 Cl 2-F 3-OH
13 CF3 CI 2-F H 14 CF3 Cl 3-OH H
15 CF3 CI 3-OH 3-OH
16 C2F5 H H H 17 CF2H H H H fumarate 150
18 CF3 CI H 3-OCH3
<Desc/Clms Page number 16>
Les composés de l'invention, de formule (1), ont fait l'objet d'essais pharmacologiques qui ont montré leur intérêt comme substances actives en thérapeutique.
Ils ont en particulier été testés quant à leurs effets inhibiteurs de la liaison de la [3H]-N-méthyl-scopolamine avec les récepteurs muscariniques de type M3 humains transfectés dans des cellules CHO (chinese hamster ovarian cells) (Buckley et al., Mol. Pharmacol. 35 : 469-476,1989).
Des membranes de cellules CHO, en solution dans un tampon TRIS-HCI 10mM, EDTA 2 mM pH 7,2, exprimant le sous-type de récepteur muscarinique humain M3 ont été fournis par la société Receptor Biology (Baltimore, USA).
10 à 30 g de membranes ont été incubées dans un tampon phosphate, pH 7,4 (Sigma, St Louis, MO) en présence de 0,5 nM de [3H]N-méthyl-scopolamine (NEN-Dupont, les Ulis, France), et d'un composé de l'invention, dans un volume total de 1 mL. La non-spécificité de la liaison a été déterminée par 0,5 M d'atropine (Sigma, St Louis, Mo). L'incubation (60 min à 25 C) a été stoppée par une filtration rapide sur filtres Whatmann GF/B par un dispositif de filtration Brandel. Les filtres ont été lavés trois fois par 4 mL de tampon phosphate froid, séchés et la radioactivité a été mesurée par scintillation liquide (scintillant Ultima Gold). La concentration de composé déplaçant de 50% la liaison spécifique (ICso) a été utilisée pour calculer les valeurs de Ki selon l'équation de ChengPrusoff. L'efficacité de chaque produit étudié est exprimée par le logarithme négatif de leur Ki (pKi).
Les pKi des composés de l'invention vis-à-vis des récepteurs M3 se situent entre 7 et 10.
Les composés de l'invention ont également été étudiés quant à leurs effets antagonistes vis-à-vis des contractions du détrusor de lapin femelle, médiées par les récepteurs M3.
Des lapins femelles (Néo-Zélandais, 3-4Kg ; fournisseur ESD) âgés de 20 semaines environ ont été sacrifiés par dislocation cervicale puis exsanguinés.
Après ouverture de l'abdomen, les vessies ont été prélevées puis mises rapidement dans une solution de Krebs bicarbonatée de composition (mM) : NaCI :114 ; KCI : 4,7 ; CaCI2 : 2,5 ; MgS04 : 1,2 ; KH2PO4: 1,2 ; NaHC03 : 25, ; acide ascorbique : 1,1 ; glucose : 11,7. Du propranolol (1 pM), du méthylsergide (1 M), de l'ondansetron (1 M), du GR113808 (1 M) ont été ajoutés au Krebs afin d'inhiber respectivement les récepteurs !]-adrénergiques et les différents
<Desc/Clms Page number 17>
sous-types de récepteurs sérotoninergiques 5-HT1/T-HT2, 5-HT3 et 5-HT4. Les vessies ont été nettoyées, dégraissées puis chaque face a été découpée en deux lambeaux longitudinaux d'environ 4 mm de large et 15 mm de long.
Les tissus ont été ensuite placés dans des cuves de 20 mL thermostatées à 37 C sous aération carbogène (95 % O2, 5% CO2) et ont été soumis à une tension basale de 1 g.
La tension a été mesurée par l'intermédiaire de jauges isométriques (Hugo Sacks, type 351) reliées à des coupleurs (Gould) qui transforment et amplifient les réponses qui seront tracées sur des enregistreurs potentiométriques 4 pistes (Gould) et reliées à un système d'acquisition de données (Jad, Notocord). Un temps d'équilibration d'environ 45 minutes a été observé pendant lequel le Krebs est renouvelé et la tension basale rectifiée.
Après une période d'équilibration de 30 minutes, une contraction initiale au carbachol (1 pM), puissant agoniste muscarinique, a été réalisée. Les tissus ont été ensuite rincés abondamment puis après une nouvelle période d'équilibration de 30 minutes, les tissus ont été incubés 30 minutes en présence ou non d'un composé de l'invention à étudier (concentration 0,1 ou 1 M) avant la réalisation d'une gamme concentration-réponse au carbachol par intervalle d'une demie unité de logarithme.
Les concentrations produisant la moitié de l'effet maximal (EC50 (uM)) ont été calculées pour chaque gamme (absence ou présence du composé à étudier), puis la puissance du composé à déplacer la courbe de réponse au carbachol à été déterminée par un calcul de l'affinité de l'antagoniste (pKb ou pA2 apparent) selon la méthode de Furchgott (Handbook of Expérimental Pharmacology, 1972, 283-335).
Les pKbdes composés de l'invention se situent entre 7 et 10.
Les composés de l'invention ont également été étudiés quant à leur affinité vis- à-vis des récepteurs 5-HT4 dans le striatum de cobaye, selon la méthode décrite par Grossman et coll., dans Br. J. Pharmacol., 109, 618-624 (1993).
On euthanasie des cobayes (Hartley, Charles River) de 300 à 400 g et on prélève leur cerveau. On excise les striata et on les congèle à -80 C. Le jour de l'expérience, on décongèle le tissu à +4 C dans 33 volumes de tampon HépèsNaOH 50 mM (pH=7,4 à 20 C) et on l'homogénéise à l'aide d'un broyeur Polytron. On centrifuge l'homogénat pendant 10 minutes à 48000xg, on
<Desc/Clms Page number 18>
récupère le culot, on le remet en suspension et on le centrifuge de nouveau dans les mêmes conditions. On suspend le culot final dans du tampon HépèsNaOH (30 mg de tissu frais/mL). Cette suspension membranaire est utilisée telle quelle.
On incube 100 L de la suspension membranaire à 0 C pendant 120 minutes, en présence de 0,1 nM de [3H]GR113808 (activité spécifique : 80-85 Ci/mmol), dans un volume final de 1 mL de tampon Hépès-NaOH (50 mM, pH = 7,4), en l'absence ou en présence du composé à tester. On arrête l'incubation par filtration sur filtres Whatman GF/B#, préalablement traités avec de la polyéthylèneimine 0,1 %, on rince chaque tube par 4mL de tampon à 0 C et on filtre de nouveau. On mesure la radioactivité retenue sur les filtres, par scintigraphie liquide. On détermine la liaison non spécifique en présence de sérotonine 30 M.
La liaison spécifique représente 90 % de la radioactivité totale récupérée sur le filtre.
Pour chaque concentration de composé étudié, on détermine le pourcentage d'inhibition de la liaison spécifique du [3H]GR118808 puis la concentration du composé testé qui inhibe 50 % de la liaison spécifique (Cl50).
Les Cl50 des composés de l'invention se situent entre 0,1 et 350 nM.
Enfin, les composés de l'invention ont été étudiés quant à leurs effets antagonistes vis-à-vis des récepteurs 5-HT4 dans l'oesophage de rat.
On utilise des rats mâles Sprague-Dawley pesant de 300 à 450 g. On prélève rapidement un fragment d'environ 1,5 cm de la partie terminale de l'oesophage, on élimine la couche musculaire, on ouvre longitudinalement la tunique muqueuse musculaire interne, on la monte dans une cuve à organe isolé contenant une solution de Krebs-Henseleit à 32 C oxygénée par un courant carbogène (95 % O2 et 5% CO2), et on la connecte à un transducteur isométrique sous une tension basale de 0,5 g.
Les composés sont étudiés à une concentration de 1 M. On mesure leur capacité à déplacer la relaxation introduite par la 5-HT (à des concentrations de 0,1 nM) du tissu oesophagien précontracté à la substance P 1 M.
Les composés de l'invention sont actifs dans ce test.
<Desc/Clms Page number 19>
Les résultats des tests biologiques montrent que les composés de l'invention sont des antagonistes des récepteurs muscariniques M3 et sérotoninergiques 5HT4.
L'implication de l'isoforme 2D6 des cytochromes humains (CYP2D6) dans le métabolisme microsomal des composés de l'invention a été évaluée après détermination de la biotransformation des composés en l'absence ou présence d'un inhibiteur spécifique du CYP2D6 après incubation avec des microsomes humains.
Les composés de l'invention sont incubés à la concentration de 1 M (dans du tampon phosphate, pH7,4) avec des microsomes humains (pool de biopsies humaines, 6 donneurs, 0,5,1 ou 2 mg de protéines / mL) en présence d'un système régénérateur d'équivalents réduits (NADP, 3mM ; MgS04, 10mM ; G-6P, 60mM, UDPGA, 8,6mM) à 37 C et en présence ou non de quinidine (3pM) (d'après : M.BOURIE, V.MEUNIER, Y. BERGER, G. FABRE, Cytochrome P450 isoform inhibitors as a tool for the investigation of metabolic reactions catalysed by human liver microsomes, Journal of Pharmacology and Expérimental Therapeutics, 1995, Vol. 277, n 1, p321-336.) dans un volume total de 1 mL.
Une aliquote du milieu d'incubation (100 l) est prélevée à différents temps compris entre 0 et 20 minutes après le début de l'incubation ; la réaction est stoppée par l'addition de 100uL d'acétonitrile. Après centrifugation et reprise, ie surnageant est ensuite analysé en LC/MS. Le composé inchangé est dosé en utilisant une gamme d'étalonnage externe traitée dans les mêmes conditions que les incubations.
Les résultats sont exprimés en pourcentage d'implication du CYP2D6 dans la biotransformation des composés.
L'implication du CYP2D6 humain dans le métabolisme oxydatif microsomal des composés de l'invention est faible.
Ils peuvent donc être utilisés dans le traitement des pathologies où un antagoniste des récepteurs 5-HT4 et M3 apporte un bénéfice thérapeutique. Par exemple, ils peuvent être utilisés dans le traitement du syndrome du colon irritable, des troubles de la mémoire, de l'obstruction des voies aériennes et des instabilités vésicales, en particulier l'incontinence urinaire d'urgence.
L'utilisation des composés selon l'invention pour la préparation d'un médicament destiné à traiter les pathologies ci-dessus mentionnées fait partie intégrante de l'invention.
<Desc/Clms Page number 20>
Selon un autre de ses aspects, la présente invention concerne des compositions pharmaceutiques renfermant en tant que principe actif, un composé selon l'invention.
Ainsi, ces compositions pharmaceutiques contiennent une dose efficace d'un composé selon l'invention ou d'un sel, N-oxyde ou hydrate pharmaceutiquement acceptable de celui-ci, et un ou plusieurs excipients pharmaceutiques convenables.
Lesdits excipients sont choisis selon la forme pharmaceutique et le mode d'administration souhaité.
Dans les compositions pharmaceutiques de la présente invention pour l'administration orale, sublinguale, sous-cutanée, intramusculaire, intra-veineuse, topique, intratrachéale, intranasale, transdermique ou rectale, le principe actif de formule (I) ci-dessus son sel ou hydrate éventuel, peut être administré sous forme unitaire d'administration, en mélange avec des excipients pharmaceutiques classiques, aux animaux et aux êtres humains pour la prophylaxie ou le traitement des troubles ou des maladies ci-dessus. Les formes unitaires d'administration appropriées comprennent les formes par voie orale telles que les comprimés, les gélules, les poudres, les granules et les solutions ou suspensions orales, les formes d'administration sublinguale, buccale, intratrachéale, intranasale, les formes d'administration sous-cutanée, intramusculaire ou intraveineuse et les formes d'administration rectale ou vaginale. Pour l'application topique, on peut utiliser les composés selon l'invention dans des crèmes, pommades ou lotions.
Afin d'obtenir l'effet prophylactique ou thérapeutique désiré, la dose de principe actif peut varier entre 0,1mg et 50 mg par kg de poids du corps et par jour. Bien que ces dosages soient des exemples de situation moyenne, il peut y avoir des cas particuliers où des dosages plus élevés ou plus faibles sont appropriés, de tels dosages appartiennent également à l'invention. Selon la pratique habituelle, le dosage approprié à chaque patient est déterminé par le médecin selon le mode d'administration, le poids et la réponse dudit patient.
Chaque dose unitaire peut contenir de 0,1à 1000 mg, de préférence de 0,1à 500 mg, de principe actif en combinaison avec un ou plusieurs excipients
<Desc/Clms Page number 21>
pharmaceutiques. Cette dose unitaire peut être administrée 1 à 5 fois par jour de façon à administrer un dosage journalier de 0,5 à 5000 mg, de préférence de 0,5 à 2500 mg.
Par exemple, lorsqu'on prépare une composition solide sous forme de comprimés, on mélange l'ingrédient actif principal avec un excipient pharmaceutique, tel que la gélatine, l'amidon, le lactose, le stéarate de magnésium, le talc, la gomme arabique ou analogues. On peut enrober les comprimés de saccharose, d'un dérivé cellulosique, ou d'autres matières. Les comprimés peuvent être réalisés par différentes techniques, compression directe, granulation sèche, granulation humide ou fusion à chaud.
Selon un deuxième exemple, on obtient une préparation en gélules en mélangeant l'ingrédient actif avec un diluant et en versant le mélange obtenu dans des gélules molles ou dures.
Pour une administration parentérale, on utilise des suspensions aqueuses, des solutions salines isotoniques ou des solutions stériles et injectables qui contiennent des agents de dispersion et/ou des mouillants pharmacologiquement compatibles, par exemple le propylène glycol ou le butylène glycol.
La présente invention selon un autre de ses aspects, concerne également une méthode de traitement des pathologies ci-dessus indiquées qui comprend l'administration d'un composé selon l'invention ou un de ses sels, N-oxydes ou hydrates.
Claims (13)
- R3 et R4 représentent, indépendamment l'un de l'autre, un atome d'hydrogène, un atome d'halogène, un groupe C1-4 alkyle, hydroxy, C1-4 alcoxy, cyano, nitro, perfluoro-C1-2 alkyle, amino, et ses sels, N-oxydes et hydrates.R représente un groupe C2-4 alkényle ou un phényle éventuellement substitué par un, deux ou trois groupes R4,R2 représentent un hydrogène, un groupe C1-4 alkyle, un atome d'halogène,dans laquelle, R1 est un groupe de formule CnHxFy où n = 1 ou 2, x = 0 à 3, y = 1 à 5 et x+y = 2n+1 et
 Revendications 1. Composé de formule (I):
Revendications 1. Composé de formule (I): - 2. Composé selon la revendication 1, caractérisé en ce que : * R2 représente un atome d'hydrogène ou d'halogène et/ou * R3 représente un atome d'hydrogène, d'halogène, un hydroxy ou un groupe C1-2 alkoxy et/ou * R4 représente un atome d'hydrogène, un hydroxy ou un groupe C1-2 alkoxy.
- 3. Composé selon revendication 1 ou 2, caractérisé en ce que : * R1 représente un groupe CF3, C2F5 ou CF2H.
- 4. Composé selon la revendication 1 caractérisé en ce qu'il consiste en le :
 1-(phénylméthyl)-4-[[[1-phényl-4-(trifluorométhyl)-H-imidazol-2yl]oxy]méthyl]pipéridine 1-[(3-méthoxyphényl)méthyl]-4-[[[1-phényl-4-(trifluorométhyl)-9H-imidazol-2yl]oxy]méthyl] pipéridine 3-[[4-[[[1-phényl-4-(trifluorométhyl)-1 H-imidazol-2-yl]oxy]méthyl]pipéridin-1yl]méthyl]phénol 4-[[[1-(2-fluorophényl)-4-(trifluorométhyl)-7H-imidazol-2-yl]oxy]méthyl]-1- (phénylméthyl)pipéridine<Desc/Clms Page number 23>dans laquelle R1, R2 et R3 sont tels que définis dans la revendication 1.
1-(phénylméthyl)-4-[[[1-phényl-4-(trifluorométhyl)-H-imidazol-2yl]oxy]méthyl]pipéridine 1-[(3-méthoxyphényl)méthyl]-4-[[[1-phényl-4-(trifluorométhyl)-9H-imidazol-2yl]oxy]méthyl] pipéridine 3-[[4-[[[1-phényl-4-(trifluorométhyl)-1 H-imidazol-2-yl]oxy]méthyl]pipéridin-1yl]méthyl]phénol 4-[[[1-(2-fluorophényl)-4-(trifluorométhyl)-7H-imidazol-2-yl]oxy]méthyl]-1- (phénylméthyl)pipéridine<Desc/Clms Page number 23>dans laquelle R1, R2 et R3 sont tels que définis dans la revendication 1.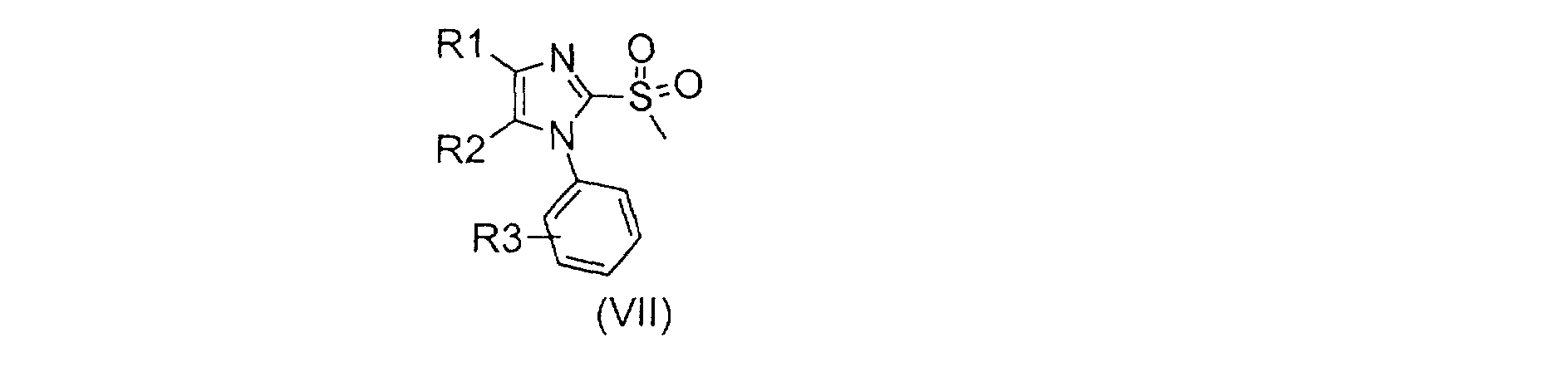 (trifluorométhyl)-'/H-imidazol-1 -yljphénol 4-[[[4-(pentafluoroéthyl)-1-phényl-9H-imidazol-2-yl)oxy]méthyl]-1- (phénylméthyl)pipéridine 4-[[[4-(difluorométhyl)-1-phényl-9H-imidazol-2-yl )oxy]méthyl]-1- (phénylméthyl)pipéridine
(trifluorométhyl)-'/H-imidazol-1 -yljphénol 4-[[[4-(pentafluoroéthyl)-1-phényl-9H-imidazol-2-yl)oxy]méthyl]-1- (phénylméthyl)pipéridine 4-[[[4-(difluorométhyl)-1-phényl-9H-imidazol-2-yl )oxy]méthyl]-1- (phénylméthyl)pipéridine - 5. Composé de formule (VII)
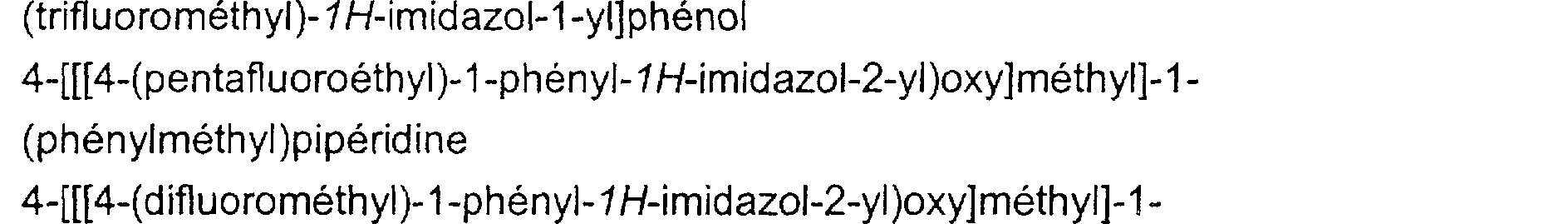 4-[[[1-(2-fluorophényl)-4-(trifluorométhyl)- /-/-imidazoi-2-yf]oxy]méthyt]-1 -[(3méthoxyphény()méthyl]pipéridine 3-[[4-[[[1 -(2-fluorophényl)-4-(trif!uorométhyl)- #/H-imidazol-2yl]oxy]méthyl]pipéridin-1-yi]méthyl]phénol 1-[(3-méthoxyphényl)méthyl]-4-[[[1-(3-méthoxyphényl)-4-(trifluorométhyl)-1Himidazol-2-yl]oxy]méthyl]pipéridine 3-[2-[[1-[{3-hydroxyphényl)méthyl]pipéridin-4-yl]méthoxy]-4-(trifluorométhyl)-9Himidazol-1-yl]phénol 4-[[[1-(3-méthoxyphényl)-4-(trifluorométhyl)-'//-/-imidazol-2-yl]oxy]méthyl]-1- (phénylméthyl)pipéridine 3-[2-[[1-(phénylméthyl)pipéridin-4-yl]méthoxy]-4-(trifluorométhyl)-9H-imidazol-1yl]phénol 3-[[4-[[[5-chloro-1-phényl-4-(trifiuorométhyl)-1 H-imidazol-2yl]oxy]méthyl]pipéridin-1-yl]méthyl]phénol 3-[[4-[[[5-chloro-1-(2-fluorophényl)-4-(trifluorométhyl)-9H-imidazol-2yl]oxy]méthyl]pipéridin-1-yl]méthyl]phénol 4-[[[5-chloro-1-(2-fluorophényl)-4-trifluorométhyl-TH-imidazol-2-yl]oxy]méthyl]-1- (phénylméthyl)pipéridine 3-[5-Chloro-2-[[1-(phénylméthyl)pipéridin-4-yl]méthoxy]-4-(trifluorométhyl )-1H~ imidazol-1-yl]phénol 3-[5-Chloro-2-[[1-[(3-hydroxyphényl)méthyl]pipéridin-4-yl]méthoxy]-4-
4-[[[1-(2-fluorophényl)-4-(trifluorométhyl)- /-/-imidazoi-2-yf]oxy]méthyt]-1 -[(3méthoxyphény()méthyl]pipéridine 3-[[4-[[[1 -(2-fluorophényl)-4-(trif!uorométhyl)- #/H-imidazol-2yl]oxy]méthyl]pipéridin-1-yi]méthyl]phénol 1-[(3-méthoxyphényl)méthyl]-4-[[[1-(3-méthoxyphényl)-4-(trifluorométhyl)-1Himidazol-2-yl]oxy]méthyl]pipéridine 3-[2-[[1-[{3-hydroxyphényl)méthyl]pipéridin-4-yl]méthoxy]-4-(trifluorométhyl)-9Himidazol-1-yl]phénol 4-[[[1-(3-méthoxyphényl)-4-(trifluorométhyl)-'//-/-imidazol-2-yl]oxy]méthyl]-1- (phénylméthyl)pipéridine 3-[2-[[1-(phénylméthyl)pipéridin-4-yl]méthoxy]-4-(trifluorométhyl)-9H-imidazol-1yl]phénol 3-[[4-[[[5-chloro-1-phényl-4-(trifiuorométhyl)-1 H-imidazol-2yl]oxy]méthyl]pipéridin-1-yl]méthyl]phénol 3-[[4-[[[5-chloro-1-(2-fluorophényl)-4-(trifluorométhyl)-9H-imidazol-2yl]oxy]méthyl]pipéridin-1-yl]méthyl]phénol 4-[[[5-chloro-1-(2-fluorophényl)-4-trifluorométhyl-TH-imidazol-2-yl]oxy]méthyl]-1- (phénylméthyl)pipéridine 3-[5-Chloro-2-[[1-(phénylméthyl)pipéridin-4-yl]méthoxy]-4-(trifluorométhyl )-1H~ imidazol-1-yl]phénol 3-[5-Chloro-2-[[1-[(3-hydroxyphényl)méthyl]pipéridin-4-yl]méthoxy]-4- <Desc/Clms Page number 24>
<Desc/Clms Page number 24> - 6. composé de formule (VI)
 dans laquelle R1, R2 et R3 sont tels que définis dans la revendication 1.
dans laquelle R1, R2 et R3 sont tels que définis dans la revendication 1. - 7. Composé de formule (XIV)
 dans laquelle R2 et R3 sont tels que définis dans la revendication 1.
dans laquelle R2 et R3 sont tels que définis dans la revendication 1. - 8. Composé de formule (XIII)
 dans laquelle R2 et R3 sont tels que définis dans la revendication 1.
dans laquelle R2 et R3 sont tels que définis dans la revendication 1. - 9. Procédé de préparation d'un composé de formule (I) selon l'une quelconque des revendications 1 à 4 dans lequel on fait réagir un composé de formule (VII)
 <Desc/Clms Page number 25>dans laquelle R est tel que définis dans la revendication 1.
<Desc/Clms Page number 25>dans laquelle R est tel que définis dans la revendication 1.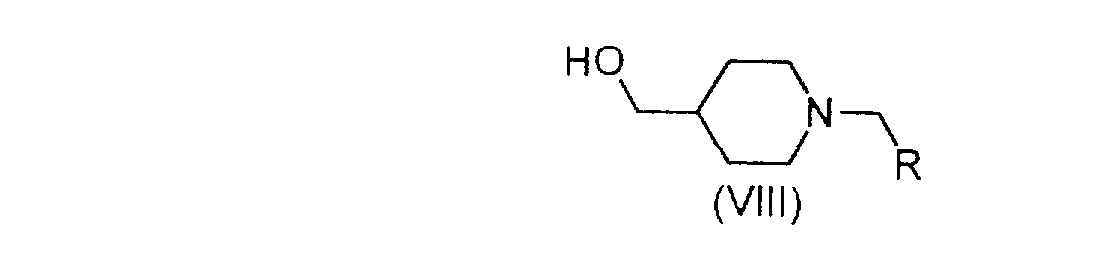 dans laquelle R1, R2 et R3 sont tels que définis dans la revendication 1, avec un composé de formule (VIII)
dans laquelle R1, R2 et R3 sont tels que définis dans la revendication 1, avec un composé de formule (VIII) - 10. Composition pharmaceutique caractérisée en ce qu'elle contient un composé de formule (I) selon l'une des revendications 1,2, 3 ou 4 en association avec un ou plusieurs excipients pharmaceutiquements acceptables.
- 11. Utilisation d'un composé de formule (1) selon l'une quelconque des revendications 1,2, 3 ou 4 pour la préparation d'un médicament destiné à traiter une pathologie où un antagoniste des récepteurs 5HT4 et M3 apporte un bénéfice thérapeutique.
- 12. Utilisation d'un composé de formule (I) selon la revendication 11 caractérisée en ce que la pathologie consiste en le traitement du syndrome du colon irritable, des troubles de la mémoire, de l'obstruction des voies aériennes et des instabilités vésicales.
- 13. Utilisation selon la revendication 12, caractérisé en ce que l'instabilité vésicale est l'incontinence urinaire d'urgence.
Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| FR0002596A FR2805815B1 (fr) | 2000-03-01 | 2000-03-01 | Derives de polyfluoroalkylimidazole, leur preparation et leur application en therapeutique |
| ARP010100874A AR027556A1 (es) | 2000-03-01 | 2001-02-27 | Derivados de polifluoroalquilimidazol, su preparacion y su utilizacion en terapeutica |
| PCT/FR2001/000591 WO2001064631A2 (fr) | 2000-03-01 | 2001-02-28 | Derives de polyfluoroalkylimidazole, leur preparation et leur application en therapeutique |
| AU2001237518A AU2001237518A1 (en) | 2000-03-01 | 2001-02-28 | Polyfluoroalkylimidazole derivatives, their preparation and therapeutic application |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| FR0002596A FR2805815B1 (fr) | 2000-03-01 | 2000-03-01 | Derives de polyfluoroalkylimidazole, leur preparation et leur application en therapeutique |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| FR2805815A1 true FR2805815A1 (fr) | 2001-09-07 |
| FR2805815B1 FR2805815B1 (fr) | 2002-05-17 |
Family
ID=8847574
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| FR0002596A Expired - Fee Related FR2805815B1 (fr) | 2000-03-01 | 2000-03-01 | Derives de polyfluoroalkylimidazole, leur preparation et leur application en therapeutique |
Country Status (4)
| Country | Link |
|---|---|
| AR (1) | AR027556A1 (fr) |
| AU (1) | AU2001237518A1 (fr) |
| FR (1) | FR2805815B1 (fr) |
| WO (1) | WO2001064631A2 (fr) |
Families Citing this family (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7737163B2 (en) | 2004-06-15 | 2010-06-15 | Pfizer Inc. | Benzimidazolone carboxylic acid derivatives |
| CA2569654C (fr) | 2004-06-15 | 2010-12-21 | Pfizer Inc. | Derives de benzimidazolone d'acide carboxylique |
| MY156662A (en) | 2007-11-01 | 2016-03-15 | Acucela Inc | Amine derivative compounds for treating ophthalmic diseases and disorders |
| GB0901487D0 (en) | 2009-01-30 | 2009-03-11 | Movetis N V | Asthma Therapy |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1998004546A1 (fr) * | 1996-07-25 | 1998-02-05 | Synthelabo | Derives de benzimidazole, leurs preparations et leurs applications en therapeutique |
| WO1999025710A1 (fr) * | 1997-11-19 | 1999-05-27 | Sanofi-Synthelabo | Derives d'imidazole, leur preparation et leur application en therapeutique |
| FR2772378A1 (fr) * | 1997-12-12 | 1999-06-18 | Synthelabo | Derives d'imidazole, leur preparation et leur application en therapeutique |
-
2000
- 2000-03-01 FR FR0002596A patent/FR2805815B1/fr not_active Expired - Fee Related
-
2001
- 2001-02-27 AR ARP010100874A patent/AR027556A1/es unknown
- 2001-02-28 AU AU2001237518A patent/AU2001237518A1/en not_active Abandoned
- 2001-02-28 WO PCT/FR2001/000591 patent/WO2001064631A2/fr not_active Ceased
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1998004546A1 (fr) * | 1996-07-25 | 1998-02-05 | Synthelabo | Derives de benzimidazole, leurs preparations et leurs applications en therapeutique |
| WO1999025710A1 (fr) * | 1997-11-19 | 1999-05-27 | Sanofi-Synthelabo | Derives d'imidazole, leur preparation et leur application en therapeutique |
| FR2772378A1 (fr) * | 1997-12-12 | 1999-06-18 | Synthelabo | Derives d'imidazole, leur preparation et leur application en therapeutique |
Also Published As
| Publication number | Publication date |
|---|---|
| AR027556A1 (es) | 2003-04-02 |
| FR2805815B1 (fr) | 2002-05-17 |
| WO2001064631A3 (fr) | 2002-03-28 |
| AU2001237518A1 (en) | 2001-09-12 |
| WO2001064631A2 (fr) | 2001-09-07 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP1499589B1 (fr) | Derives de n-¬phenyl(piperidin-2-yl)methyl|benzamide, leur preparation et leur application en therapeutique | |
| EP0760811B1 (fr) | Derives d'imidazole modulateurs du recepteur h3 de l'histamine | |
| JP4555071B2 (ja) | Nosインヒビターとしての1−置換イミダゾール誘導体 | |
| LU84011A1 (fr) | 2-phenoxyalkyl-1,2,4-triazol-3-ones antidepressives | |
| WO2005037785A2 (fr) | Derives de n-[phenyl(pyrrolidin-2-yl)methyl]benzamide, et n-[azepan-2-yl) phenylmethyl]benzamide, leur preparation et leur application en therapeutique | |
| EP0429341A2 (fr) | Dérivés hétérocycliques, leur préparation et les médicaments les contenant | |
| FR2587029A1 (fr) | Derives de benzimidazole, leur preparation et leur application en therapeutique | |
| EP0350403A1 (fr) | Dérivés de (aza)naphtalènesultame, leurs procédés de préparation et les médicaments les contenant | |
| RU2536688C2 (ru) | Новые бензамидные производные | |
| WO1999025710A1 (fr) | Derives d'imidazole, leur preparation et leur application en therapeutique | |
| FR2805815A1 (fr) | Derives de polyfluoroalkylimidazole, leur preparation et leur application en therapeutique | |
| WO1998004546A1 (fr) | Derives de benzimidazole, leurs preparations et leurs applications en therapeutique | |
| FR2811989A1 (fr) | Derives de polyfluoroalkytriazole, leur preparation et leur application en therapeutique | |
| EP1383762B1 (fr) | Tetrahydropyridyl-alkyl-heterocycles, procede pour leur preparation et compositions pharmaceutiques les contenants | |
| WO1999062900A1 (fr) | Derives d'oxindole utile comme antagonistes des recepteurs de neurokinines | |
| EP1960387B1 (fr) | Derives de isoquinoline et benzo[h]isoquinoline, leur preparation et leur utilisation en therapeutique en tant qu antagonistes du recepteur de l histamine h3 | |
| FR2805816A1 (fr) | Derives d'haloimidazole, leur preparation et leur application en therapeutique | |
| US9464058B2 (en) | Imidazolylketone derivatives | |
| EP1150972A1 (fr) | Derives de triazoles et tetrazoles, et leur utilisation en therapeutique | |
| FR2754261A1 (fr) | Derives de quinuclidine, leur preparation et leur application en therapeutique | |
| FR2835254A1 (fr) | Derives de thiazoles dans le traitement de maladies neurologiques | |
| FR2571722A1 (fr) | Derives de benzimidazolone, leur preparation et leur application en therapeutique | |
| FR2902790A1 (fr) | Derives d'urees de piperidine ou pyrrolidine,leur preparation et leur application en therapeutique | |
| FR2823750A1 (fr) | Tetrahydropyridyl-alkyl-heterocycles, procede pour leur preparation et compositions pharmaceutiques les contenant | |
| FR2584718A2 (fr) | Derives de benzimidazolone, leur preparation et leur application en therapeutique |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| ST | Notification of lapse |