FR2814166A1 - Derives 5-phenoxyindole et leurs applications therapeutiques - Google Patents
Derives 5-phenoxyindole et leurs applications therapeutiques Download PDFInfo
- Publication number
- FR2814166A1 FR2814166A1 FR0012052A FR0012052A FR2814166A1 FR 2814166 A1 FR2814166 A1 FR 2814166A1 FR 0012052 A FR0012052 A FR 0012052A FR 0012052 A FR0012052 A FR 0012052A FR 2814166 A1 FR2814166 A1 FR 2814166A1
- Authority
- FR
- France
- Prior art keywords
- phenoxy
- alkyl
- sep
- indole
- groups
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
- 208000019695 Migraine disease Diseases 0.000 title claims 2
- 208000017520 skin disease Diseases 0.000 title claims 2
- 201000002282 venous insufficiency Diseases 0.000 title claims 2
- YJBIMZVVUOJZSS-UHFFFAOYSA-N 5-phenoxy-1h-indole Chemical class C=1C=C2NC=CC2=CC=1OC1=CC=CC=C1 YJBIMZVVUOJZSS-UHFFFAOYSA-N 0.000 title abstract description 6
- 206010061218 Inflammation Diseases 0.000 title description 2
- 230000004054 inflammatory process Effects 0.000 title description 2
- 208000027520 Somatoform disease Diseases 0.000 title 1
- 208000014617 hemorrhoid Diseases 0.000 title 1
- 208000027753 pain disease Diseases 0.000 title 1
- 208000014001 urinary system disease Diseases 0.000 title 1
- 125000004193 piperazinyl group Chemical group 0.000 claims abstract description 3
- 125000002768 hydroxyalkyl group Chemical group 0.000 claims abstract 5
- 125000004423 acyloxy group Chemical group 0.000 claims abstract 4
- 125000000876 trifluoromethoxy group Chemical group FC(F)(F)O* 0.000 claims abstract 4
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 claims abstract 3
- 150000003839 salts Chemical class 0.000 claims abstract 3
- 125000003785 benzimidazolyl group Chemical group N1=C(NC2=C1C=CC=C2)* 0.000 claims abstract 2
- 125000004618 benzofuryl group Chemical group O1C(=CC2=C1C=CC=C2)* 0.000 claims abstract 2
- 125000001164 benzothiazolyl group Chemical group S1C(=NC2=C1C=CC=C2)* 0.000 claims abstract 2
- 125000004196 benzothienyl group Chemical group S1C(=CC2=C1C=CC=C2)* 0.000 claims abstract 2
- 125000004541 benzoxazolyl group Chemical group O1C(=NC2=C1C=CC=C2)* 0.000 claims abstract 2
- 125000000753 cycloalkyl group Chemical group 0.000 claims abstract 2
- 125000002541 furyl group Chemical group 0.000 claims abstract 2
- 125000001188 haloalkyl group Chemical group 0.000 claims abstract 2
- 125000002883 imidazolyl group Chemical group 0.000 claims abstract 2
- 125000001041 indolyl group Chemical group 0.000 claims abstract 2
- 125000005956 isoquinolyl group Chemical group 0.000 claims abstract 2
- 125000002757 morpholinyl group Chemical group 0.000 claims abstract 2
- 125000001715 oxadiazolyl group Chemical group 0.000 claims abstract 2
- 125000002971 oxazolyl group Chemical group 0.000 claims abstract 2
- 125000005936 piperidyl group Chemical group 0.000 claims abstract 2
- 125000003373 pyrazinyl group Chemical group 0.000 claims abstract 2
- 125000003072 pyrazolidinyl group Chemical group 0.000 claims abstract 2
- 125000003226 pyrazolyl group Chemical group 0.000 claims abstract 2
- 125000002098 pyridazinyl group Chemical group 0.000 claims abstract 2
- 125000004076 pyridyl group Chemical group 0.000 claims abstract 2
- 125000000714 pyrimidinyl group Chemical group 0.000 claims abstract 2
- 125000000719 pyrrolidinyl group Chemical group 0.000 claims abstract 2
- 125000000168 pyrrolyl group Chemical group 0.000 claims abstract 2
- 125000005493 quinolyl group Chemical group 0.000 claims abstract 2
- 125000001412 tetrahydropyranyl group Chemical group 0.000 claims abstract 2
- 125000003831 tetrazolyl group Chemical group 0.000 claims abstract 2
- 125000001113 thiadiazolyl group Chemical group 0.000 claims abstract 2
- 125000000335 thiazolyl group Chemical group 0.000 claims abstract 2
- 125000001544 thienyl group Chemical group 0.000 claims abstract 2
- -1 hydrogen halogen Chemical class 0.000 claims description 34
- 150000001875 compounds Chemical class 0.000 claims description 21
- SIKJAQJRHWYJAI-UHFFFAOYSA-N Indole Chemical compound C1=CC=C2NC=CC2=C1 SIKJAQJRHWYJAI-UHFFFAOYSA-N 0.000 claims description 13
- 229910052739 hydrogen Inorganic materials 0.000 claims description 12
- 239000001257 hydrogen Substances 0.000 claims description 9
- 229910052736 halogen Inorganic materials 0.000 claims description 7
- 150000002367 halogens Chemical class 0.000 claims description 6
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 5
- 125000003118 aryl group Chemical group 0.000 claims description 4
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 claims description 4
- 125000000623 heterocyclic group Chemical group 0.000 claims description 4
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 claims description 2
- 238000004519 manufacturing process Methods 0.000 claims 7
- 239000003814 drug Substances 0.000 claims 6
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 claims 4
- 125000004454 (C1-C6) alkoxycarbonyl group Chemical group 0.000 claims 3
- 125000006577 C1-C6 hydroxyalkyl group Chemical group 0.000 claims 3
- 125000004093 cyano group Chemical group *C#N 0.000 claims 3
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 claims 3
- 125000004191 (C1-C6) alkoxy group Chemical group 0.000 claims 2
- 125000001072 heteroaryl group Chemical group 0.000 claims 2
- 230000007170 pathology Effects 0.000 claims 2
- 125000004916 (C1-C6) alkylcarbonyl group Chemical group 0.000 claims 1
- 125000005913 (C3-C6) cycloalkyl group Chemical group 0.000 claims 1
- IQXXEPZFOOTTBA-UHFFFAOYSA-N 1-benzylpiperazine Chemical compound C=1C=CC=CC=1CN1CCNCC1 IQXXEPZFOOTTBA-UHFFFAOYSA-N 0.000 claims 1
- 208000002193 Pain Diseases 0.000 claims 1
- 239000002260 anti-inflammatory agent Substances 0.000 claims 1
- 229940124599 anti-inflammatory drug Drugs 0.000 claims 1
- 125000000532 dioxanyl group Chemical group 0.000 claims 1
- 125000005879 dioxolanyl group Chemical group 0.000 claims 1
- 206010027599 migraine Diseases 0.000 claims 1
- 125000000896 monocarboxylic acid group Chemical group 0.000 claims 1
- 239000008194 pharmaceutical composition Substances 0.000 claims 1
- 125000000217 alkyl group Chemical group 0.000 abstract description 13
- 125000003545 alkoxy group Chemical group 0.000 abstract 6
- 125000004453 alkoxycarbonyl group Chemical group 0.000 abstract 4
- 125000001475 halogen functional group Chemical group 0.000 abstract 3
- 125000004276 dioxalanyl group Chemical group 0.000 abstract 2
- 125000004448 alkyl carbonyl group Chemical group 0.000 abstract 1
- 125000006168 tricyclic group Chemical group 0.000 abstract 1
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 111
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 82
- 229910052799 carbon Inorganic materials 0.000 description 51
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 39
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 35
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 32
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 27
- 239000000243 solution Substances 0.000 description 22
- 239000000725 suspension Substances 0.000 description 21
- 239000003921 oil Substances 0.000 description 20
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 19
- 239000002244 precipitate Substances 0.000 description 19
- 239000000377 silicon dioxide Substances 0.000 description 19
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 18
- 239000000203 mixture Substances 0.000 description 18
- 239000003480 eluent Substances 0.000 description 17
- 239000000460 chlorine Substances 0.000 description 15
- 239000012074 organic phase Substances 0.000 description 15
- 238000003756 stirring Methods 0.000 description 15
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 14
- 239000000047 product Substances 0.000 description 14
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical group N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 12
- OKKJLVBELUTLKV-MZCSYVLQSA-N Deuterated methanol Chemical compound [2H]OC([2H])([2H])[2H] OKKJLVBELUTLKV-MZCSYVLQSA-N 0.000 description 12
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 description 12
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 12
- 229910021529 ammonia Inorganic materials 0.000 description 12
- VSJKWCGYPAHWDS-FQEVSTJZSA-N camptothecin Chemical compound C1=CC=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)[C@]5(O)CC)C4=NC2=C1 VSJKWCGYPAHWDS-FQEVSTJZSA-N 0.000 description 12
- 229910052731 fluorine Inorganic materials 0.000 description 12
- 239000007787 solid Substances 0.000 description 12
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 12
- 238000005160 1H NMR spectroscopy Methods 0.000 description 11
- 238000005481 NMR spectroscopy Methods 0.000 description 11
- 235000019439 ethyl acetate Nutrition 0.000 description 11
- RWRDLPDLKQPQOW-UHFFFAOYSA-N Pyrrolidine Chemical compound C1CCNC1 RWRDLPDLKQPQOW-UHFFFAOYSA-N 0.000 description 9
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 9
- 238000001644 13C nuclear magnetic resonance spectroscopy Methods 0.000 description 8
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 8
- 229920000742 Cotton Polymers 0.000 description 8
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 8
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 8
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 8
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 8
- YMWUJEATGCHHMB-DICFDUPASA-N dichloromethane-d2 Chemical compound [2H]C([2H])(Cl)Cl YMWUJEATGCHHMB-DICFDUPASA-N 0.000 description 8
- 229910052938 sodium sulfate Inorganic materials 0.000 description 8
- 235000011152 sodium sulphate Nutrition 0.000 description 8
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 7
- 239000006260 foam Substances 0.000 description 7
- 229910052757 nitrogen Inorganic materials 0.000 description 7
- 239000012047 saturated solution Substances 0.000 description 7
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 7
- 235000017557 sodium bicarbonate Nutrition 0.000 description 7
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 6
- KLWPJMFMVPTNCC-UHFFFAOYSA-N Camptothecin Natural products CCC1(O)C(=O)OCC2=C1C=C3C4Nc5ccccc5C=C4CN3C2=O KLWPJMFMVPTNCC-UHFFFAOYSA-N 0.000 description 6
- 101150050738 HTR1B gene Proteins 0.000 description 6
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 6
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 6
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 6
- 101100321769 Takifugu rubripes htr1d gene Proteins 0.000 description 6
- 239000000556 agonist Substances 0.000 description 6
- 210000004556 brain Anatomy 0.000 description 6
- 229940127093 camptothecin Drugs 0.000 description 6
- 239000013078 crystal Substances 0.000 description 6
- VSJKWCGYPAHWDS-UHFFFAOYSA-N dl-camptothecin Natural products C1=CC=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)C5(O)CC)C4=NC2=C1 VSJKWCGYPAHWDS-UHFFFAOYSA-N 0.000 description 6
- 239000000706 filtrate Substances 0.000 description 6
- PZOUSPYUWWUPPK-UHFFFAOYSA-N indole Natural products CC1=CC=CC2=C1C=CN2 PZOUSPYUWWUPPK-UHFFFAOYSA-N 0.000 description 6
- RKJUIXBNRJVNHR-UHFFFAOYSA-N indolenine Natural products C1=CC=C2CC=NC2=C1 RKJUIXBNRJVNHR-UHFFFAOYSA-N 0.000 description 6
- UWKQSNNFCGGAFS-XIFFEERXSA-N irinotecan Chemical compound C1=C2C(CC)=C3CN(C(C4=C([C@@](C(=O)OC4)(O)CC)C=4)=O)C=4C3=NC2=CC=C1OC(=O)N(CC1)CCC1N1CCCCC1 UWKQSNNFCGGAFS-XIFFEERXSA-N 0.000 description 6
- 238000000034 method Methods 0.000 description 6
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 6
- 241000283973 Oryctolagus cuniculus Species 0.000 description 5
- 239000002253 acid Substances 0.000 description 5
- 239000008346 aqueous phase Substances 0.000 description 5
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 5
- 239000012429 reaction media Substances 0.000 description 5
- 210000003752 saphenous vein Anatomy 0.000 description 5
- 229920006395 saturated elastomer Polymers 0.000 description 5
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 4
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 4
- 241000700159 Rattus Species 0.000 description 4
- 229910052786 argon Inorganic materials 0.000 description 4
- HUMNYLRZRPPJDN-UHFFFAOYSA-N benzaldehyde Chemical compound O=CC1=CC=CC=C1 HUMNYLRZRPPJDN-UHFFFAOYSA-N 0.000 description 4
- 125000001584 benzyloxycarbonyl group Chemical group C(=O)(OCC1=CC=CC=C1)* 0.000 description 4
- 239000007795 chemical reaction product Substances 0.000 description 4
- 230000003750 conditioning effect Effects 0.000 description 4
- 238000006073 displacement reaction Methods 0.000 description 4
- 238000001035 drying Methods 0.000 description 4
- 230000000694 effects Effects 0.000 description 4
- 238000001704 evaporation Methods 0.000 description 4
- 230000008020 evaporation Effects 0.000 description 4
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 4
- 229960004768 irinotecan Drugs 0.000 description 4
- 239000007788 liquid Substances 0.000 description 4
- 229910052760 oxygen Inorganic materials 0.000 description 4
- UYWQUFXKFGHYNT-UHFFFAOYSA-N phenylmethyl ester of formic acid Natural products O=COCC1=CC=CC=C1 UYWQUFXKFGHYNT-UHFFFAOYSA-N 0.000 description 4
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 4
- 229910052717 sulfur Inorganic materials 0.000 description 4
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 4
- CXUMKPRQWDUWQE-UHFFFAOYSA-N (3-phenoxyphenyl)hydrazine Chemical compound NNC1=CC=CC(OC=2C=CC=CC=2)=C1 CXUMKPRQWDUWQE-UHFFFAOYSA-N 0.000 description 3
- PZJHKWISMUCTGW-UHFFFAOYSA-N 5-ethyl-5-hydroxy-6-oxo-2h-pyran-2-carboxylic acid Chemical compound CCC1(O)C=CC(C(O)=O)OC1=O PZJHKWISMUCTGW-UHFFFAOYSA-N 0.000 description 3
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 3
- 102000017911 HTR1A Human genes 0.000 description 3
- 101150015707 HTR1A gene Proteins 0.000 description 3
- 101000610640 Homo sapiens U4/U6 small nuclear ribonucleoprotein Prp3 Proteins 0.000 description 3
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 3
- SJRJJKPEHAURKC-UHFFFAOYSA-N N-Methylmorpholine Chemical compound CN1CCOCC1 SJRJJKPEHAURKC-UHFFFAOYSA-N 0.000 description 3
- 101001110823 Saccharomyces cerevisiae (strain ATCC 204508 / S288c) 60S ribosomal protein L6-A Proteins 0.000 description 3
- 101000712176 Saccharomyces cerevisiae (strain ATCC 204508 / S288c) 60S ribosomal protein L6-B Proteins 0.000 description 3
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 3
- 102100040374 U4/U6 small nuclear ribonucleoprotein Prp3 Human genes 0.000 description 3
- 239000007864 aqueous solution Substances 0.000 description 3
- 239000002585 base Substances 0.000 description 3
- 235000015278 beef Nutrition 0.000 description 3
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 3
- 230000015572 biosynthetic process Effects 0.000 description 3
- 244000309464 bull Species 0.000 description 3
- 125000004432 carbon atom Chemical group C* 0.000 description 3
- 238000004587 chromatography analysis Methods 0.000 description 3
- 238000001914 filtration Methods 0.000 description 3
- 125000005842 heteroatom Chemical group 0.000 description 3
- 150000002431 hydrogen Chemical class 0.000 description 3
- 210000002414 leg Anatomy 0.000 description 3
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 3
- 229910052763 palladium Inorganic materials 0.000 description 3
- 238000002360 preparation method Methods 0.000 description 3
- VKKZSJQQRIGUIE-UHFFFAOYSA-N quinolin-3-ylmethanamine Chemical class C1=CC=CC2=CC(CN)=CN=C21 VKKZSJQQRIGUIE-UHFFFAOYSA-N 0.000 description 3
- 150000003248 quinolines Chemical class 0.000 description 3
- 238000010992 reflux Methods 0.000 description 3
- 230000004044 response Effects 0.000 description 3
- 229910000029 sodium carbonate Inorganic materials 0.000 description 3
- 239000011780 sodium chloride Substances 0.000 description 3
- 239000002904 solvent Substances 0.000 description 3
- 238000003786 synthesis reaction Methods 0.000 description 3
- UCFGDBYHRUNTLO-QHCPKHFHSA-N topotecan Chemical compound C1=C(O)C(CN(C)C)=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)[C@]5(O)CC)C4=NC2=C1 UCFGDBYHRUNTLO-QHCPKHFHSA-N 0.000 description 3
- JXGVXCZADZNAMJ-NSHDSACASA-N (2s)-1-phenylmethoxycarbonylpyrrolidine-2-carboxylic acid Chemical compound OC(=O)[C@@H]1CCCN1C(=O)OCC1=CC=CC=C1 JXGVXCZADZNAMJ-NSHDSACASA-N 0.000 description 2
- 125000004178 (C1-C4) alkyl group Chemical group 0.000 description 2
- SCYULBFZEHDVBN-UHFFFAOYSA-N 1,1-Dichloroethane Chemical compound CC(Cl)Cl SCYULBFZEHDVBN-UHFFFAOYSA-N 0.000 description 2
- GFTDGDAKGDUYGO-UHFFFAOYSA-N 2-(4-chlorobutyl)-1,3-dioxolane Chemical compound ClCCCCC1OCCO1 GFTDGDAKGDUYGO-UHFFFAOYSA-N 0.000 description 2
- NGNBDVOYPDDBFK-UHFFFAOYSA-N 2-[2,4-di(pentan-2-yl)phenoxy]acetyl chloride Chemical compound CCCC(C)C1=CC=C(OCC(Cl)=O)C(C(C)CCC)=C1 NGNBDVOYPDDBFK-UHFFFAOYSA-N 0.000 description 2
- 102000056834 5-HT2 Serotonin Receptors Human genes 0.000 description 2
- 108091005479 5-HT2 receptors Proteins 0.000 description 2
- ASXGJMSKWNBENU-UHFFFAOYSA-N 8-OH-DPAT Chemical compound C1=CC(O)=C2CC(N(CCC)CCC)CCC2=C1 ASXGJMSKWNBENU-UHFFFAOYSA-N 0.000 description 2
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 2
- 101100491335 Caenorhabditis elegans mat-2 gene Proteins 0.000 description 2
- BVKZGUZCCUSVTD-UHFFFAOYSA-L Carbonate Chemical compound [O-]C([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-L 0.000 description 2
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 2
- XDTMQSROBMDMFD-UHFFFAOYSA-N Cyclohexane Chemical compound C1CCCCC1 XDTMQSROBMDMFD-UHFFFAOYSA-N 0.000 description 2
- 206010015866 Extravasation Diseases 0.000 description 2
- 239000012448 Lithium borohydride Substances 0.000 description 2
- JLVHTNZNKOSCNB-YSVLISHTSA-N Mesulergine Chemical compound C1=CC([C@H]2C[C@@H](CN(C)[C@@H]2C2)NS(=O)(=O)N(C)C)=C3C2=CN(C)C3=C1 JLVHTNZNKOSCNB-YSVLISHTSA-N 0.000 description 2
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 2
- BKAYIFDRRZZKNF-VIFPVBQESA-N N-acetylcarnosine Chemical compound CC(=O)NCCC(=O)N[C@H](C(O)=O)CC1=CN=CN1 BKAYIFDRRZZKNF-VIFPVBQESA-N 0.000 description 2
- 208000007920 Neurogenic Inflammation Diseases 0.000 description 2
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 2
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 2
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical group [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 2
- 150000001412 amines Chemical class 0.000 description 2
- 150000008064 anhydrides Chemical class 0.000 description 2
- 239000002246 antineoplastic agent Substances 0.000 description 2
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical group [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 2
- AGEZXYOZHKGVCM-UHFFFAOYSA-N benzyl bromide Chemical compound BrCC1=CC=CC=C1 AGEZXYOZHKGVCM-UHFFFAOYSA-N 0.000 description 2
- 239000008280 blood Substances 0.000 description 2
- 210000004369 blood Anatomy 0.000 description 2
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 2
- 229910052794 bromium Inorganic materials 0.000 description 2
- PFKFTWBEEFSNDU-UHFFFAOYSA-N carbonyldiimidazole Chemical compound C1=CN=CN1C(=O)N1C=CN=C1 PFKFTWBEEFSNDU-UHFFFAOYSA-N 0.000 description 2
- 238000006243 chemical reaction Methods 0.000 description 2
- 229910052801 chlorine Inorganic materials 0.000 description 2
- IJOOHPMOJXWVHK-UHFFFAOYSA-N chlorotrimethylsilane Chemical compound C[Si](C)(C)Cl IJOOHPMOJXWVHK-UHFFFAOYSA-N 0.000 description 2
- 239000012230 colorless oil Substances 0.000 description 2
- 230000009989 contractile response Effects 0.000 description 2
- 238000001816 cooling Methods 0.000 description 2
- 238000000354 decomposition reaction Methods 0.000 description 2
- 230000008021 deposition Effects 0.000 description 2
- 239000000839 emulsion Substances 0.000 description 2
- 230000036251 extravasation Effects 0.000 description 2
- 150000004678 hydrides Chemical class 0.000 description 2
- 230000007062 hydrolysis Effects 0.000 description 2
- 238000006460 hydrolysis reaction Methods 0.000 description 2
- 230000002401 inhibitory effect Effects 0.000 description 2
- 239000000543 intermediate Substances 0.000 description 2
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 2
- 235000019341 magnesium sulphate Nutrition 0.000 description 2
- 239000012528 membrane Substances 0.000 description 2
- 229950008693 mesulergine Drugs 0.000 description 2
- 210000005036 nerve Anatomy 0.000 description 2
- 230000007383 nerve stimulation Effects 0.000 description 2
- CTSLXHKWHWQRSH-UHFFFAOYSA-N oxalyl chloride Chemical compound ClC(=O)C(Cl)=O CTSLXHKWHWQRSH-UHFFFAOYSA-N 0.000 description 2
- 239000001301 oxygen Substances 0.000 description 2
- QNGNSVIICDLXHT-UHFFFAOYSA-N para-ethylbenzaldehyde Natural products CCC1=CC=C(C=O)C=C1 QNGNSVIICDLXHT-UHFFFAOYSA-N 0.000 description 2
- 125000004194 piperazin-1-yl group Chemical group [H]N1C([H])([H])C([H])([H])N(*)C([H])([H])C1([H])[H] 0.000 description 2
- BASFCYQUMIYNBI-UHFFFAOYSA-N platinum Chemical compound [Pt] BASFCYQUMIYNBI-UHFFFAOYSA-N 0.000 description 2
- 229910000027 potassium carbonate Inorganic materials 0.000 description 2
- 230000008569 process Effects 0.000 description 2
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 2
- 230000009257 reactivity Effects 0.000 description 2
- 238000000926 separation method Methods 0.000 description 2
- 239000012321 sodium triacetoxyborohydride Substances 0.000 description 2
- 239000011593 sulfur Chemical group 0.000 description 2
- 229960000303 topotecan Drugs 0.000 description 2
- 125000005270 trialkylamine group Chemical group 0.000 description 2
- AALUTIYNYXEFNT-UHFFFAOYSA-N trimethylsilane hydroiodide Chemical compound C[SiH](C)C.I AALUTIYNYXEFNT-UHFFFAOYSA-N 0.000 description 2
- 238000005406 washing Methods 0.000 description 2
- CYPYTURSJDMMMP-WVCUSYJESA-N (1e,4e)-1,5-diphenylpenta-1,4-dien-3-one;palladium Chemical compound [Pd].[Pd].C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1.C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1.C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1 CYPYTURSJDMMMP-WVCUSYJESA-N 0.000 description 1
- BFNBYPAMZTYWBT-UHFFFAOYSA-N (4-phenoxyphenyl)hydrazine;hydrochloride Chemical compound Cl.C1=CC(NN)=CC=C1OC1=CC=CC=C1 BFNBYPAMZTYWBT-UHFFFAOYSA-N 0.000 description 1
- MLEGMEBCXGDFQT-UHFFFAOYSA-N 1-benzylpiperidin-2-one Chemical compound O=C1CCCCN1CC1=CC=CC=C1 MLEGMEBCXGDFQT-UHFFFAOYSA-N 0.000 description 1
- ZBPUNVFDQXYNDY-UHFFFAOYSA-N 2-(3-chloropropyl)-1,3-dioxolane Chemical compound ClCCCC1OCCO1 ZBPUNVFDQXYNDY-UHFFFAOYSA-N 0.000 description 1
- MIDXCONKKJTLDX-UHFFFAOYSA-N 3,5-dimethylcyclopentane-1,2-dione Chemical compound CC1CC(C)C(=O)C1=O MIDXCONKKJTLDX-UHFFFAOYSA-N 0.000 description 1
- ISDOYJZTKRDXLO-UHFFFAOYSA-N 3-(1-methyl-3,6-dihydro-2h-pyridin-4-yl)-5-phenoxy-1h-indole Chemical compound C1N(C)CCC(C=2C3=CC(OC=4C=CC=CC=4)=CC=C3NC=2)=C1 ISDOYJZTKRDXLO-UHFFFAOYSA-N 0.000 description 1
- CDWFDLYRKQOGNR-UHFFFAOYSA-N 3-(2-aminoethyl)-1h-indole-5-carboxylic acid Chemical compound C1=C(C(O)=O)C=C2C(CCN)=CNC2=C1 CDWFDLYRKQOGNR-UHFFFAOYSA-N 0.000 description 1
- SJJLZJQAQWGWRM-QFIPXVFZSA-N 3-[[(2s)-1-benzylpyrrolidin-2-yl]methyl]-5-phenoxy-1h-indole Chemical compound C([C@H]1CC=2C3=CC(OC=4C=CC=CC=4)=CC=C3NC=2)CCN1CC1=CC=CC=C1 SJJLZJQAQWGWRM-QFIPXVFZSA-N 0.000 description 1
- 125000000339 4-pyridyl group Chemical group N1=C([H])C([H])=C([*])C([H])=C1[H] 0.000 description 1
- 241000283690 Bos taurus Species 0.000 description 1
- RGHPCLZJAFCTIK-UHFFFAOYSA-N CC1NCCC1 Chemical compound CC1NCCC1 RGHPCLZJAFCTIK-UHFFFAOYSA-N 0.000 description 1
- IDZLYFSSURAWRT-UHFFFAOYSA-N CN(CC1)CCC1c1c[nH]c(cc2)c1cc2Oc1ccccc1 Chemical compound CN(CC1)CCC1c1c[nH]c(cc2)c1cc2Oc1ccccc1 IDZLYFSSURAWRT-UHFFFAOYSA-N 0.000 description 1
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 1
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 1
- 206010009944 Colon cancer Diseases 0.000 description 1
- 208000001333 Colorectal Neoplasms Diseases 0.000 description 1
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N DMSO Substances CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 1
- 229940123780 DNA topoisomerase I inhibitor Drugs 0.000 description 1
- QOSSAOTZNIDXMA-UHFFFAOYSA-N Dicylcohexylcarbodiimide Chemical compound C1CCCCC1N=C=NC1CCCCC1 QOSSAOTZNIDXMA-UHFFFAOYSA-N 0.000 description 1
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- 241001465754 Metazoa Species 0.000 description 1
- 206010028980 Neoplasm Diseases 0.000 description 1
- 206010030113 Oedema Diseases 0.000 description 1
- 239000005662 Paraffin oil Substances 0.000 description 1
- QGMRQYFBGABWDR-UHFFFAOYSA-M Pentobarbital sodium Chemical compound [Na+].CCCC(C)C1(CC)C(=O)NC(=O)[N-]C1=O QGMRQYFBGABWDR-UHFFFAOYSA-M 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- 241000700157 Rattus norvegicus Species 0.000 description 1
- WQDUMFSSJAZKTM-UHFFFAOYSA-N Sodium methoxide Chemical compound [Na+].[O-]C WQDUMFSSJAZKTM-UHFFFAOYSA-N 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 1
- 101000783611 Takifugu rubripes 5-hydroxytryptamine receptor 1D Proteins 0.000 description 1
- 241000906446 Theraps Species 0.000 description 1
- 239000000365 Topoisomerase I Inhibitor Substances 0.000 description 1
- 150000007513 acids Chemical class 0.000 description 1
- 230000009471 action Effects 0.000 description 1
- 230000004913 activation Effects 0.000 description 1
- 239000004480 active ingredient Substances 0.000 description 1
- 239000003513 alkali Substances 0.000 description 1
- 125000003282 alkyl amino group Chemical group 0.000 description 1
- 125000004414 alkyl thio group Chemical group 0.000 description 1
- 230000029936 alkylation Effects 0.000 description 1
- 238000005804 alkylation reaction Methods 0.000 description 1
- 125000003277 amino group Chemical group 0.000 description 1
- 230000003444 anaesthetic effect Effects 0.000 description 1
- 230000003110 anti-inflammatory effect Effects 0.000 description 1
- 125000003710 aryl alkyl group Chemical group 0.000 description 1
- 125000005199 aryl carbonyloxy group Chemical group 0.000 description 1
- 125000004429 atom Chemical group 0.000 description 1
- 229960000686 benzalkonium chloride Drugs 0.000 description 1
- WXNOJTUTEXAZLD-UHFFFAOYSA-L benzonitrile;dichloropalladium Chemical compound Cl[Pd]Cl.N#CC1=CC=CC=C1.N#CC1=CC=CC=C1 WXNOJTUTEXAZLD-UHFFFAOYSA-L 0.000 description 1
- CADWTSSKOVRVJC-UHFFFAOYSA-N benzyl(dimethyl)azanium;chloride Chemical compound [Cl-].C[NH+](C)CC1=CC=CC=C1 CADWTSSKOVRVJC-UHFFFAOYSA-N 0.000 description 1
- 125000006278 bromobenzyl group Chemical group 0.000 description 1
- 239000000872 buffer Substances 0.000 description 1
- 235000013736 caramel Nutrition 0.000 description 1
- 150000001718 carbodiimides Chemical class 0.000 description 1
- 125000005708 carbonyloxy group Chemical group [*:2]OC([*:1])=O 0.000 description 1
- 238000012754 cardiac puncture Methods 0.000 description 1
- 230000003197 catalytic effect Effects 0.000 description 1
- 239000003795 chemical substances by application Substances 0.000 description 1
- 125000001309 chloro group Chemical group Cl* 0.000 description 1
- 238000009833 condensation Methods 0.000 description 1
- 230000005494 condensation Effects 0.000 description 1
- GBRBMTNGQBKBQE-UHFFFAOYSA-L copper;diiodide Chemical compound I[Cu]I GBRBMTNGQBKBQE-UHFFFAOYSA-L 0.000 description 1
- 125000004122 cyclic group Chemical group 0.000 description 1
- 238000006356 dehydrogenation reaction Methods 0.000 description 1
- 238000010511 deprotection reaction Methods 0.000 description 1
- 125000006222 dimethylaminomethyl group Chemical group [H]C([H])([H])N(C([H])([H])[H])C([H])([H])* 0.000 description 1
- 239000012153 distilled water Substances 0.000 description 1
- 150000002148 esters Chemical class 0.000 description 1
- 238000000605 extraction Methods 0.000 description 1
- 239000011737 fluorine Substances 0.000 description 1
- 238000004108 freeze drying Methods 0.000 description 1
- ACGDKVXYNVEAGU-UHFFFAOYSA-N guanethidine Chemical compound NC(N)=NCCN1CCCCCCC1 ACGDKVXYNVEAGU-UHFFFAOYSA-N 0.000 description 1
- 229960003602 guanethidine Drugs 0.000 description 1
- 238000004128 high performance liquid chromatography Methods 0.000 description 1
- 229940088013 hycamtin Drugs 0.000 description 1
- 239000012442 inert solvent Substances 0.000 description 1
- 230000005764 inhibitory process Effects 0.000 description 1
- PNDPGZBMCMUPRI-UHFFFAOYSA-N iodine Chemical compound II PNDPGZBMCMUPRI-UHFFFAOYSA-N 0.000 description 1
- JEIPFZHSYJVQDO-UHFFFAOYSA-N iron(III) oxide Inorganic materials O=[Fe]O[Fe]=O JEIPFZHSYJVQDO-UHFFFAOYSA-N 0.000 description 1
- 210000004731 jugular vein Anatomy 0.000 description 1
- FRIJBUGBVQZNTB-UHFFFAOYSA-M magnesium;ethane;bromide Chemical compound [Mg+2].[Br-].[CH2-]C FRIJBUGBVQZNTB-UHFFFAOYSA-M 0.000 description 1
- 239000003550 marker Substances 0.000 description 1
- 125000004184 methoxymethyl group Chemical group [H]C([H])([H])OC([H])([H])* 0.000 description 1
- 125000002911 monocyclic heterocycle group Chemical group 0.000 description 1
- PSHKMPUSSFXUIA-UHFFFAOYSA-N n,n-dimethylpyridin-2-amine Chemical compound CN(C)C1=CC=CC=N1 PSHKMPUSSFXUIA-UHFFFAOYSA-N 0.000 description 1
- 125000001624 naphthyl group Chemical group 0.000 description 1
- 230000001272 neurogenic effect Effects 0.000 description 1
- 125000004433 nitrogen atom Chemical group N* 0.000 description 1
- 229910000069 nitrogen hydride Inorganic materials 0.000 description 1
- 239000003960 organic solvent Substances 0.000 description 1
- NXJCBFBQEVOTOW-UHFFFAOYSA-L palladium(2+);dihydroxide Chemical compound O[Pd]O NXJCBFBQEVOTOW-UHFFFAOYSA-L 0.000 description 1
- 210000005164 penile vein Anatomy 0.000 description 1
- 229960002275 pentobarbital sodium Drugs 0.000 description 1
- 230000000144 pharmacologic effect Effects 0.000 description 1
- 239000012071 phase Substances 0.000 description 1
- 229910052697 platinum Inorganic materials 0.000 description 1
- 125000004585 polycyclic heterocycle group Chemical group 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- 229910052700 potassium Inorganic materials 0.000 description 1
- 239000000843 powder Substances 0.000 description 1
- UORVCLMRJXCDCP-UHFFFAOYSA-M propynoate Chemical compound [O-]C(=O)C#C UORVCLMRJXCDCP-UHFFFAOYSA-M 0.000 description 1
- 125000006239 protecting group Chemical group 0.000 description 1
- 125000004309 pyranyl group Chemical group O1C(C=CC=C1)* 0.000 description 1
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 1
- JUJWROOIHBZHMG-UHFFFAOYSA-O pyridinium Chemical compound C1=CC=[NH+]C=C1 JUJWROOIHBZHMG-UHFFFAOYSA-O 0.000 description 1
- 238000010791 quenching Methods 0.000 description 1
- 230000000171 quenching effect Effects 0.000 description 1
- 239000000700 radioactive tracer Substances 0.000 description 1
- 239000011541 reaction mixture Substances 0.000 description 1
- 238000001953 recrystallisation Methods 0.000 description 1
- 238000007363 ring formation reaction Methods 0.000 description 1
- 238000007390 skin biopsy Methods 0.000 description 1
- 239000011734 sodium Substances 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- 238000002798 spectrophotometry method Methods 0.000 description 1
- 230000000638 stimulation Effects 0.000 description 1
- 229910021653 sulphate ion Inorganic materials 0.000 description 1
- KQKPFRSPSRPDEB-UHFFFAOYSA-N sumatriptan Chemical compound CNS(=O)(=O)CC1=CC=C2NC=C(CCN(C)C)C2=C1 KQKPFRSPSRPDEB-UHFFFAOYSA-N 0.000 description 1
- 229960003708 sumatriptan Drugs 0.000 description 1
- 150000003512 tertiary amines Chemical class 0.000 description 1
- 238000012360 testing method Methods 0.000 description 1
- 125000003944 tolyl group Chemical group 0.000 description 1
- 238000006257 total synthesis reaction Methods 0.000 description 1
- 125000004665 trialkylsilyl group Chemical group 0.000 description 1
- ITMCEJHCFYSIIV-UHFFFAOYSA-N triflic acid Chemical compound OS(=O)(=O)C(F)(F)F ITMCEJHCFYSIIV-UHFFFAOYSA-N 0.000 description 1
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 description 1
- 210000000689 upper leg Anatomy 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D209/00—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D209/02—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom condensed with one carbocyclic ring
- C07D209/04—Indoles; Hydrogenated indoles
- C07D209/10—Indoles; Hydrogenated indoles with substituted hydrocarbon radicals attached to carbon atoms of the hetero ring
- C07D209/14—Radicals substituted by nitrogen atoms, not forming part of a nitro radical
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/06—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pain & Pain Management (AREA)
- Pharmacology & Pharmacy (AREA)
- Rheumatology (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
Abstract
L'invention concerne de nouveaux composés de formule (CF DESSIN DANS BOPI) dans laquelle RI est choisi parmi les groupes : (CF DESSIN DANS BOPI) et leur utilisation dans le traitement des affections impliquant les nerfs sensitifs afférents.
Description
<Desc/Clms Page number 1>
PREPARATION DE LA CAMPTOTHECINE ET DE SES DERIVES La présente invention concerne la préparation de la camptothécine et de ses dérivés. Elle concerne plus particulièrement la préparation de la camptothécine, du topotécan et de l'irinotécan.
Il est connu selon le brevet européen EP 137145, cité ici à titre de référence, des dérivés de camptothécine de formule générale :

dans laquelle notamment R) est hydrogène, halogène ou alcoyle, X est un atome de chlore ou NR2R3 pour lequel R2 et R3 identiques ou différents peuvent représenter un atome d'hydrogène, un radical alcoyle éventuellement substitué, un carbocycle ou un hétérocycle éventuellement substitués, ou des radicaux alcoyle (éventuellement substitués) formant avec l'atome d'azote auquel ils sont attachés, un hétérocycle contenant éventuellement un autre hétéroatome choisi parmi 0, S et/ou NR4, R4 étant un atome d'hydrogène ou un radical alcoyle et dans laquelle le groupement X-CO-O- est situé en position 9,10 ou 11 du cycle A. Ces dérivés de camptothécine sont des agents anticancéreux, inhibiteurs de topoisomérase l, parmi lesquels l'irinotécan, pour lequel X-CO-0-est [4- (l-pipéridino)-l-pipéridino] carbonyloxy, est un principe actif particulièrement efficace sur les tumeurs solides et notamment le cancer colorectal.
dans laquelle notamment R) est hydrogène, halogène ou alcoyle, X est un atome de chlore ou NR2R3 pour lequel R2 et R3 identiques ou différents peuvent représenter un atome d'hydrogène, un radical alcoyle éventuellement substitué, un carbocycle ou un hétérocycle éventuellement substitués, ou des radicaux alcoyle (éventuellement substitués) formant avec l'atome d'azote auquel ils sont attachés, un hétérocycle contenant éventuellement un autre hétéroatome choisi parmi 0, S et/ou NR4, R4 étant un atome d'hydrogène ou un radical alcoyle et dans laquelle le groupement X-CO-O- est situé en position 9,10 ou 11 du cycle A. Ces dérivés de camptothécine sont des agents anticancéreux, inhibiteurs de topoisomérase l, parmi lesquels l'irinotécan, pour lequel X-CO-0-est [4- (l-pipéridino)-l-pipéridino] carbonyloxy, est un principe actif particulièrement efficace sur les tumeurs solides et notamment le cancer colorectal.
Il est encore connu selon la demande de brevet EP 74256, citée ici à titre de référence, d'autres dérivés de camptothécine qui sont mentionnés comme agents anticancéreux, notamment des dérivés de structure analogue à la structure donnée ci-dessus et dans laquelle X-CO-0-est remplacé par un radical-X'R'pour lequel X'est 0 ou S et R' est un atome d'hydrogène, un radical alcoyle ou acyle.
<Desc/Clms Page number 2>
D'autres dérivés de camptothécine ont aussi été décrits par exemple dans les brevets ou demandes de brevets, cités ici à titre de référence, EP 56692, EP 88642, EP 296612, EP 321122, EP 325247, EP 540099, EP 737686, WO 9003169, WO 9637496, WO 9638146, WO 9638449, WO 9700876, US 7104894, JP 57 116015, JP 57 116074, JP59005188, JP60019790, JP01 249777, JP 01246287, JP 91 012070 ou dans Canc. Res., 38 (1997) Abst. 1526 ou 95 (San Diego-12-16 avril), Canc. Res., 55 (3), 603-609 (1995) ou AFMC Int. Med. Chem.

Symp. (1997) Abst. PB-55 (Séoul-27 juillet-l août).
L'irinotécan (CPT-11) et ses dérivés sont habituellement préparés à partir de la camptothécine naturelle (US 4604463 ; S. SAWADA et coll., Chem. Pharm. Bull., 39, 2574-80 (1991), Chem. Pharm. Bull., 39,1446-54 (1991), Chem. Pharm. Bull., 39, 3183-88 (1991) et Ann. N. Y. Acad. Sci., 803,13-28 (1996). Les étapes comportent l'introduction d'une fonction hydroxyle en 9, une alcoylation en 11 et l'introduction du radical en position 9.
Dans la demande de brevet international WO 96/31513 a été décrite la préparation de dérivés de camptothécine et de mappicine par synthèse totale par préparation en premier lieu de l'enchaînement cyclique C-D ou C-D-E.
Tetrahedron, 53 (32), 11049-60 (1997) décrit également les synthèses totales de dérivés de camptothécine dans lesquelles les cycles A-B et D-E sont préparés préalablement, ou selon un autre aspect, les enchaînements C-D-E ou A-B-C.
Il a maintenant été trouvé, et c'est ce qui fait l'objet de la présente invention, que la camptothecine ou les dérivés de camptothécine de formule (I) suivante :

dans laquelle RI, R2, R3 représentent chacun un groupe identique ou différent choisi parmi :
dans laquelle RI, R2, R3 représentent chacun un groupe identique ou différent choisi parmi :
<Desc/Clms Page number 3>

. l'hydrogène, . un groupe hydroxy, * un atome d'halogène choisi parmi le fluor, le chlore, le brome ou l'iode, * les groupes alkoxy contenant 1 à 4 atomes de carbone linéaire ou ramifié, * les groupes alkylthio contenant 1 à 4 atomes de carbone linéraire ou ramifié, * les groupes alkyl (Cl-C4) amino éventuellement substitué par un ou plusieurs groupes alkyl en CI-C4, * les groupes aralkyles éventuellement substitués par un groupe alkyle en C1-C4, lesdits groupes aryles étant aussi éventuellement des hétérocycles contenant 1 à 3 hétéroatomes choisis parmi l'oxygène le soufre et l'azote * les groupes arylcarbonyloxy, lesdits groupes aryles étant aussi éventuellement des hétérocycles mono ou polycycliques contenant
1 à 3 hétéroatomes choisis parmi l'oxygène, le soufre et l'azote, pouvaient être obtenus par une synthèse convergente à partir d'un dérivé de la 3-aminométhyl quinoléine et de l'acide 5-hydroxy 5-éthyl 6-oxo 5,6-dihydro pyrane carboxylique avec des résultats particulièrement intéressants.
Les composés préférés et commerciaux synthétisés par le procédé de l'invention sont : 'la camptothécine pour laquelle RI, R2 et R3 représentent l'hydrogène * le topotécan ou Hycamtin@ pour lequel RI est l'hydrogène, R2 représente un groupe diméthylaminométhyl et R3 un groupe hydroxyl * l'irinotécan ou Campto@ pour lequel RI représente un groupe éthyle, R2 représente un groupe pipéridino piperidino carbonyloxy et R3 un hydrogène.
Le procédé selon l'invention consiste à condenser un dérivé de la 3-aminométhyl quinoléine et de l'acide 5-hydroxy 5-éthyl 6-oxo 5,6-dihydro pyrane carboxylique, suivi d'une étape d'éthynylation, éventuellement d'une étape d'hydrolyse, d'une
<Desc/Clms Page number 4>

double étape de cyclisation, d'une déhydrogénation et d'une étape de déprotection/décarbalkoxylation. Selon l'invention, l'acide 5-hydroxy 5-éthyl 6-oxo 5,6-dihydro pyrane carboxylique de structure :

dans laquelle G) représente l'hydrogène ou un groupe protecteur de la fonction hydroxyle notamment choisi parmi les groupes benzyle, para méthoxybenzyle, méthoxyméthyle, terbutyle et trialkylsilyle dont au moins un groupe alkyle a plus de deux atomes de carbone est condensé sur un dérivé de la 3-aminométhyl quinoléine de formule générale :

dans laquelle RI, R2 et R3 ont la même signification que dans la formule (I) ou représentent des radicaux protégés ou facilement convertibles en radicaux RI, Rz et R3 cités ci-avant, Y représente un groupe partant notamment choisi parmi les atomes
d'halogène, un radical OSOzR où R représente un groupe alkyle, tolyl, naphtyl, trifluorométhyl pour obtenir le dérivé de quinoléine de formule générale :

dans laquelle Gi, RI, R2, R3 et Y sont définis comme précédemment.
<Desc/Clms Page number 5>

On préfere parmi les groupements Gi, le groupe benzyle. On préfère parmi les groupes Y les halogènes choisis parmi le brome ou l'iode, parmi les groupes OSO2R, le trifluorométhylsulf onate.
La réaction s'effectue généralement selon les méthodes habituelles de condensation des acides sur les amines, notamment par action de l'acide ou d'un dérivé réactif ou activé de l'acide.
Lorsque l'on effectue la condensation d'un dérivé réactif de l'acide de formule générale (II), on opère avantageusement au moyen du chlorure d'acide, de l'anhydride, d'un anhydride mixte ou d'un ester réactif, ou d'un intermédaire acyle ammonium ou pyridinium.
Parmi les conditions réactionnelles on préfere utiliser une température comprise entre - 40 et +40 C. Parmi les solvants inertes utilisables, on préfère utiliser un solvant organique tel que notamment un solvant chloré (dichlorométhane, dichloroéthane, chloroforme par exemple). On peut éventuellement opérer en présence d'un accepteur d'acide tel qu'une base organique azotée comme par exemple la pyridine, la diméthylaminopyridine, la N-méthylmorpholine ou une trialcoylamine (notamment triéthylamine, diisopropyléthylamine). Il est également préférable d'opérer en présence d'un agent de condensation tel qu'un carbodiimide, [par exemple dicyclohexylcarbodiimide ou (diméthylamino-3 propyl)-l éthyl-3 carbodiimide], le N, N'-carbonyl- diimidazole ou l'éthoxy-2 éthoxycarbonyl-l dihydro-1, 2 quinoléine. De préférence on opère sous argon ou azote.
Il est entendu que, les radicaux amino, alcoylamino ou carboxy contenus dans RI, R2 et R3 comme la fonction hydroxy portée par le cycle pyrane sont de préférence préalablement protégés. Notamment, on protège selon les méthodes décrites par T. W. Greene et P. G. M. Wuts, Protective Groups in Organic Synthesis (3'led.), A. WileyInterscience Publication (1999).
Ensuite le dérivé de quinoléine de formule générale (IV) et l'ortho propiolate de trialkyl (C 1-C4 éventuellement substitué) ou le propiolate d'alkyle (C 1-C4 éventuellement substitué) sont mis en réaction en présence d'un complexe de palladium [comme par exemple le tris (dibenzylidèneacétone) dipalladium, le chlorure de bis (benzonitrile) palladium ou le dichloro bistriphénylphosphine palladium et d'iodure de cuivre et d'une base telle qu'une amine tertiaire (trialkylamine) ou d'un carbonate alcalin pour donner le dérivé de quinoléine de formule générale :
<Desc/Clms Page number 6>

Une solution de 5-phénoxyindole (4 g, 4. 78 mmol), MeONa (1. 24 g, 1. 2 éq) et 1-benzylpipéridone (5. 32 ml, 1. 5 éq) dans 100 ml de méthanol est portée à reflux pendant 96 h. Après retour à température ambiante, le milieu réactionnel est filtré. Le solide filtré est rincé avec 3x20 ml de MeOH. On obtient 4. 69 g de produit attendu sous forme d'un solide beige.
Rendement : 85% F : 168-170 C Rf : 0.1 (CH2Cl2/EtOAc, 7/3) SM (APc ! +) : m/z 381 (M+Hf RMN du'H (CD30DHz) : # (ppm) 8.32 (bd s, 1 H, NH) 7.57 (d, 1 H, H-4, JH4-H6 = 2. 2 Hz) 7.39-7. 21 (m, 8H, H-7, H-18, H-20, H-16, H-17, H-19, H-3', H-5') 7.15 (d, 1H, H-2, JH2-NH = 2. 5 Hz) 7.02-6. 90 (m, 4H, H-4', H-6, H-2', H-6') 6.07 (m, 1 H, H-9) 3.63 (s, 2H, H-14) 3.17 (m, 2H, H-10) 2. 70 (m, 2H, H-12) 2.55 (m, 2H, H-13) RMN du 13C (CD3OD) : # (ppm) 159.5 (1C, C-1') 149.8 (1C, C-5) 138.2 (1C, C-15) 133.8 (1 C, C-7a) 129.5 (1C, C-3a) 129.4 (2C, C-19, C-17) 129.0 (2C, C-3', C-5') 128.2 (2C, C-20, C-16) 127.1 (1C, C-18) 125.8 (1C, C-3) 122.5 (1 C, C-2) 121.8 (1 C, C-4')
<Desc/Clms Page number 7>
119.3 (1 C, C-9) 118. 1 (1C. C-8) 116.9 (2C, C-2', C-6') 116.0 (1C, C-6) 112. 1 (1C, C-7) 112. 0 (1C, C-4) 62.8 (1C, C-14) 53.2 (1C, C-10) 49.9 (1C, C-12) 28.9 (1C, C-13) IR (KBr) : v (cm-1) 3108 (NH), 3035,2797, 1572 (C=C), 1479,1217 (C-N-C), 819, 729 (Ph) Exemple 2 3- (N-benzytptpéridin-4-yl)-5-phénoxyindole

A une solution de 1.83 g (6.27 mmoles) de 3-pipéridin-4-yl-5-phénoxyindole et de 1.33 g (12. 5 mmoles, 2 éq) de carbonate de sodium dans 100 ml d'acétonitrile, on ajoute 0.895 ml (7.52 mmoles, 1. 2 éq) de bromure de benzyle puis on porte le mélange à reflux (77 C) durant 65 minutes. A température ambiante, on filtre le milieu réactionnel sur Whatman et on évapore le filtrat. On fait précipiter le produit dans un minimum d'éthanol. Le précipité obtenu est lavé avec du cyclohexane puis de l'éthanol et dissout dans un minimum d'éther pour être filtré sur millipore. On évapore la solution pour obtenir une mousse cristalline blanche que l'on met à sécher à l'étuve sous vide. On obtient ainsi 550 mg (1.44 mmoles) de 3- (N-benzylpipéridin-4-yl)-5-phénoxyindole sous forme de cristaux blancs.
A une solution de 1.83 g (6.27 mmoles) de 3-pipéridin-4-yl-5-phénoxyindole et de 1.33 g (12. 5 mmoles, 2 éq) de carbonate de sodium dans 100 ml d'acétonitrile, on ajoute 0.895 ml (7.52 mmoles, 1. 2 éq) de bromure de benzyle puis on porte le mélange à reflux (77 C) durant 65 minutes. A température ambiante, on filtre le milieu réactionnel sur Whatman et on évapore le filtrat. On fait précipiter le produit dans un minimum d'éthanol. Le précipité obtenu est lavé avec du cyclohexane puis de l'éthanol et dissout dans un minimum d'éther pour être filtré sur millipore. On évapore la solution pour obtenir une mousse cristalline blanche que l'on met à sécher à l'étuve sous vide. On obtient ainsi 550 mg (1.44 mmoles) de 3- (N-benzylpipéridin-4-yl)-5-phénoxyindole sous forme de cristaux blancs.
<Desc/Clms Page number 8>
Rendement : 23 % F : 53 oC (température de décomposition) Rf : 0.37 [phase amino (6/4 ; cyclohexane, acétate d'éthyle)] SM (APcl) + : m/z 383 (M+H)+ (APcl)-:m/z381(M-H)RMN du 1H (DMSO-d6) : 8 (ppm) 10.9 (si, 1 H, NHque) 7.4 à 7.22 (m, 8H, H-7, H-3'-, H-5', H-2", H-6", H-3", H-5", H-4") 7.20 (d, 1H, H-4, JH4-H6= 2 Hz) 7.15 (d, 1H, H-2) 7.00 (t, 1H, H-4', JH4'-H3'= 7.3 Hz) 6.87 (d, 2H, H-2', H-6', JH2'-H3'= 7.9 Hz)

6. 88 (dd, 1 H, H-6, JH6-H4= 2Hz, JH6-H7= 8. 6 Hz) 3.5 (s, 2H, CH2) 2.85 (d, 2H, H-c éq, JHc eq - Hc ax = 11. 2 Hz) 2.68 (m, 1 H, H-a, Ja-hub eq = 11. 7 Hz) 2.05 (t, 2H, H-c ax, JHc ax - Hc éq = 10. 8 Hz) 1.86 (d, 2H, H-b éq, Jhb éq - Hb ax = 12. 0 Hz)

1. 63 (m, 2H, H-b JHbax-Hbeq = 12. 0 Hz) RMN du 13C (DMSO-d6) : 8 (ppm) 159. 3 (1C, C-1') 147. 8 (1C, C-1") 138. 7 (1 C, C-5) 133.5 (1 C, C-7a) 129.7 ; 128.8 ; 128.1 (3*2C ; C-3' ; C-5' ; C-3" ; C-5" ; C-2" ; C-6") 126.9 (1C, C-3a) 126.1 (1C, C-4") 122.1 (1 C, C-2) 121.7 (1C, C-4') 119.9 (1C, C-3) 116.5 (2C, C-2', C-6') 114.5 (1 C, C-6) 112.5 (1C, C-7)
6. 88 (dd, 1 H, H-6, JH6-H4= 2Hz, JH6-H7= 8. 6 Hz) 3.5 (s, 2H, CH2) 2.85 (d, 2H, H-c éq, JHc eq - Hc ax = 11. 2 Hz) 2.68 (m, 1 H, H-a, Ja-hub eq = 11. 7 Hz) 2.05 (t, 2H, H-c ax, JHc ax - Hc éq = 10. 8 Hz) 1.86 (d, 2H, H-b éq, Jhb éq - Hb ax = 12. 0 Hz)
1. 63 (m, 2H, H-b JHbax-Hbeq = 12. 0 Hz) RMN du 13C (DMSO-d6) : 8 (ppm) 159. 3 (1C, C-1') 147. 8 (1C, C-1") 138. 7 (1 C, C-5) 133.5 (1 C, C-7a) 129.7 ; 128.8 ; 128.1 (3*2C ; C-3' ; C-5' ; C-3" ; C-5" ; C-2" ; C-6") 126.9 (1C, C-3a) 126.1 (1C, C-4") 122.1 (1 C, C-2) 121.7 (1C, C-4') 119.9 (1C, C-3) 116.5 (2C, C-2', C-6') 114.5 (1 C, C-6) 112.5 (1C, C-7)
<Desc/Clms Page number 9>
109.3 (1C, C-4) 62.6 (1 C, C-d) 53.7 (2C, C-c) 32.9 (1C, C-a) 32.7 (2C, C-b) IR (KBr) : v (cm-1) 3424 (NHIndolique) ; 3030 ; 2931-2919 ; 2849-2804-2763 ; 1595-1578 ; 1488-1471- 1450 ; 1223 (Ar-o-Ar)
<Desc/Clms Page number 10>
Exemple 3 5-phénoxy-3-(pipéridin-4-yl)-indole

Une suspension de 1g (2.63 mmol) de 3-(N-benzyl-1, 2,5, 6-tétrahydropyridiri-4-

yl)-5-phénoxyindole et de Pd (OH) 2/C est agitée pendant 18 h sous hydrogène (1 atm). Le milieu réactionnel est filtré sur Célite, le cake est rincé avec 100 ml d'EtOH. Après évaporation le brut réactionnel (620 mg) est purifié par

chromatographie sur silice (h = 15 cm, 0 = 3 cm) avec OCb-MeOH-NHa ; 8 : 2 : 1 % comme éluant. Le produit est salifié, lavé au CH2CI2 puis repassé sur une colonne identique. On obtient 77 mg de 5-phenoxy-3- (piperidin-4-yl)-indole.

Rendement : 81% F : décomposition vers 210 C, fusion > 260 C Rf : 0.2 (CH2Cl2/MeOH/NH3, 9/1/0. 5%) SM (APcl -) : m/z 291 (M-H)- RMN du'H (CD30D) : 8 (ppm) 7.29 (d, 1 H, H-7, JH7-H6 = 8.7 Hz) 7. 22-7. 13 (m, 3H, H-4, H-3', H-5') 7.07 (s, 1 H, H-2) 6.88 (m, 1 H, H-4', JH4'-H5'= 7. 3 Hz) 6. 81- 6. 73 (m, 3H, H-2', H-6', H-6) 3. 45 (bd d, 2H, H-10 ou H-12, JH10-H9 = 12. 6 Hz) 3. 46- 3. 06 (m, 3H, H-8, H-12 ou H-10) 2.22 (bd d, 2H, H-9 ou H-13, Jns-He = 13. 7 Hz) 2. 04-1. 86 (m, 2H, H-13 ou H-9) RMN du 13C (CD30D) : # (ppm) 163.1 (1C, C-1')
Une suspension de 1g (2.63 mmol) de 3-(N-benzyl-1, 2,5, 6-tétrahydropyridiri-4-
yl)-5-phénoxyindole et de Pd (OH) 2/C est agitée pendant 18 h sous hydrogène (1 atm). Le milieu réactionnel est filtré sur Célite, le cake est rincé avec 100 ml d'EtOH. Après évaporation le brut réactionnel (620 mg) est purifié par
chromatographie sur silice (h = 15 cm, 0 = 3 cm) avec OCb-MeOH-NHa ; 8 : 2 : 1 % comme éluant. Le produit est salifié, lavé au CH2CI2 puis repassé sur une colonne identique. On obtient 77 mg de 5-phenoxy-3- (piperidin-4-yl)-indole.
Rendement : 81% F : décomposition vers 210 C, fusion > 260 C Rf : 0.2 (CH2Cl2/MeOH/NH3, 9/1/0. 5%) SM (APcl -) : m/z 291 (M-H)- RMN du'H (CD30D) : 8 (ppm) 7.29 (d, 1 H, H-7, JH7-H6 = 8.7 Hz) 7. 22-7. 13 (m, 3H, H-4, H-3', H-5') 7.07 (s, 1 H, H-2) 6.88 (m, 1 H, H-4', JH4'-H5'= 7. 3 Hz) 6. 81- 6. 73 (m, 3H, H-2', H-6', H-6) 3. 45 (bd d, 2H, H-10 ou H-12, JH10-H9 = 12. 6 Hz) 3. 46- 3. 06 (m, 3H, H-8, H-12 ou H-10) 2.22 (bd d, 2H, H-9 ou H-13, Jns-He = 13. 7 Hz) 2. 04-1. 86 (m, 2H, H-13 ou H-9) RMN du 13C (CD30D) : # (ppm) 163.1 (1C, C-1')
<Desc/Clms Page number 11>

152. 2 (1C, C-5) 137. 2 (1 C, C-7a) 132.4 (2C, C-3', C-5') 129.9 (1 C, C-3a) 124. 9 (1C, C-2) 124.6 (1 C, C-4') 121.3 (1C, C-3) 119.7 (2C, C-2', C-6') 118. 2 (1C, C-6) 115.3 (1C, C-7) 112.3 (1C, C-4) 47.5 (2C, C-10, C-12) 34.5 (1C, C-8) 32.7 (2C, C-9, C-13) IR (KBr) : v (cm-') 3140 (NH), 3039 (NH), 2801, 1596, 1476, 1455,1402, 1218, 1174,941 Exemple 4 3-(N-méethylpiperidin-4-yl)-5-phénoxyindole

A une solution de 2.57 g (8. 44 mmoles) de 3- [1-méthyl- (1, 2,5, 6tétrahydro) pyridin-4-yl]-5-phenoxyindole dans 80 ml d'éthanol, on ajoute une quantité catalytique de palladium hydroxyde à 20 %. On chasse l'air avec un flux d'argon puis on fait buller jusqu'à saturation de l'hydrogène. On laisse sous agitation à température ambiante durant 20 heures. On filtre sur papier le palladium et on évapore le filtrat. On obtient un précipité blanc que l'on met à
<Desc/Clms Page number 12>
sécher à l'étuve sous vide à 50oC durant 2 heures. Le précipité est lavé avec 250 ml d'eau filtrée sous forte agitation durant 3 heures. Le milieu est filtré puis le précipité est remis dans 20 ml d'eau filtrée puis congelé avant d'être lyophilisé. On récupère 0.603 g (1.97 mmoles) de 3-(N-méthylpipéridin-4-yl)-5- phénoxyindole sous forme de cristaux blancs.
Rendement : 51% F : 149-150 C Rf : 0.53 ( (7/3+0. 3 (Toluène/éthanol/NH3,aq)) SM (APcl)' : m/z 307 (M+H) (APcl)-: m/z 305 (M-H)RMN du 1H CD3OD) : ö (ppm) 7.32 (d, 1H, H-7, JH7-H6 = 8.7 Hz) 7.3 à 7.21 (m, 3H, H-3', H-5', H-4) 7. 06 (s, 1H, H-2) 6.96 (tt, 1H, H-4', JH4'-H5'= 7. 3 Hz) 6.97 (m, 2H, H-2', H-6') 6.8 (dd, 1 H, H-6, JH6-H7 = 8. 7 Hz, JH6-H4=2. 2 Hz)

2. 96 (m, 2H, H-c1 (eqou ax) et/ou H-c2 (eq ou ax)) 2. 97 (m, 1 H, H-a) 2. 98 (s, 3H, CH3) 2. 99 (m, 2H, H-c1 (éqou ax) et/ou H-c2 (éqou ax)) 2. 0 (m, 2H, H-b1 (éq ou ax) et/ou H-b2 (éqouax)) 1.8 (m, 2H, H-b1 (6q ou ax) et/ou H-b2 (eq ou ax)) RMN du 13C (CD30D) : # (ppm) 161.3 (1C, C-1') 150.1 (1C, C-5) 135.4 (1C, C-7a) 130. 5 (2C, C-3', C-5') 128.5 (1C, C-3a) 122.7 (1C, C-4', C-2) 121.0 (1C, C-3) 117. 9 (2C, C-2', C-6') 116.0 (1C, C-6)
2. 96 (m, 2H, H-c1 (eqou ax) et/ou H-c2 (eq ou ax)) 2. 97 (m, 1 H, H-a) 2. 98 (s, 3H, CH3) 2. 99 (m, 2H, H-c1 (éqou ax) et/ou H-c2 (éqou ax)) 2. 0 (m, 2H, H-b1 (éq ou ax) et/ou H-b2 (éqouax)) 1.8 (m, 2H, H-b1 (6q ou ax) et/ou H-b2 (eq ou ax)) RMN du 13C (CD30D) : # (ppm) 161.3 (1C, C-1') 150.1 (1C, C-5) 135.4 (1C, C-7a) 130. 5 (2C, C-3', C-5') 128.5 (1C, C-3a) 122.7 (1C, C-4', C-2) 121.0 (1C, C-3) 117. 9 (2C, C-2', C-6') 116.0 (1C, C-6)
<Desc/Clms Page number 13>

113. 2 (1C, C-7) 110. 6 (1C, C-4) 57. 1 (2C, C-c) 46. 5 (1C, CH3) 34 (1 C, C-a) 33. 7 (2C, C-b) IR (KBr) : v (cm-1) 3127 (N-Ho ! e) ; 3099 ; 3042 ; 2936 ; 2918 ; 2851 ; 2787 ; 1579 ; 1486 ; 1460 (CH3) ; 1230 ou 1209 (Ar-0-Ar) Exemple 5 5-phénoxv-3- (3-N-pipéridinylpropyl) indole

A une suspension de 4.6 g (33.6 mmoles) de carbonate de potassium dans 7 ml (70.8 mmoles ; 2.1 éq) de pipéridine, on ajoute 5 ml (33.6 mmoles ; 1 éq) de 2- (4- chlorobutyl)-1, 3-dioxolane. On porte le mélange à reflux durant 125 minutes. A température ambiante, on verse le milieu réactionnel dans un bécher contenant 150 ml d'eau. On extrait la phase aqueuse avec 3 fois 150 ml d'acétate d'éthyle. Les phases organiques sont séchées sur sulfate de sodium, filtrées sur coton et évaporées. On obtient une huile jaune pâle que l'on place au réfrigérateur pour la nuit.
A une suspension de 6. 86 g (29 mmoles) de 5-phénoxyphénylhydrazine dans 250 ml d'une solution d'acide sulfurique à 4%, on ajoute l'huile obtenue précédemment. On laisse sous agitation à température ambiante durant 80 minutes avant de porter le mélange à 75 C durant 45 minutes. Puis on ajoute lentement et par petite quantité 250 ml d'une solution aqueuse saturée en hydrogénocarbonate de sodium. On extrait avec 1. 2 1 d'acétate d'éthyle. La
<Desc/Clms Page number 14>
phase organique est séchée sur sulfate de sodium, filtrée sur coton puis évaporée. On purifie le brut réactionnel sur colonne flash (support : silice ; conditionnement : 9/1 (CH2Cl2/MeOH) ; éluant : gradient de 9/1 (CH2Cl2/MeOH) à 7/3+0.2 (CH2CI2/MeOH/NH3. aq.)) pour obtenir une huile jaune que l'on fait précipiter dans l'éther. On recristallise le produit successivement dans l'éther puis l'heptane. Le précipité est ensuite purifié sur colonne à pression atmosphérique (support : silice ; conditionnement : 9/1/0. 2 (CH2CI2/MeOH/NH3. aq. ) ; éluant : identique) pour obtenir. 57 mg (0.17 mmoles) de 5-phénoxy-3- (3-Npipéridinylpropyl) indole sous forme de cristaux blancs.
Rendement : 20 % F : 109 - 110 C Rf : 0.4 (CH2CI2/MeOH/NH3. 32%, 90/10/0.2) SM (APc !) : m/z 335 (M+H) + (APcl)-:m/z333(M-H)RMN du'H (CD30D) : # (ppm) 7.31 (d, 1 H, H-7, JH7-H6 = 8., 7 HZ) 7.28 à 7.21 (m, 2H, H-3', H-5') 7.15 (d, 1H, H-4, JH4-H6 = 2. 0 HZ) 7.06 (s, 1 H, H-2)

6. 97 (tt, 1 H, H-4', JH4'-H5'= 7. 3 HZ, JH4'-H6'= 1. 0 HZ) 6.90 à 6.85 (m, 2H, H-2', H-6') 6.81 (dd, 1 H, H-6, JH6-H7 = 8.7 HZ, JH6-H4 = 2.2 HZ) 2.69 (t, 2H, H-a, JHA-HB = 7. 3 Hz) 2.42 à 2.34 (m, 6H, H-2", H-6", H-c) 1.94 à 1. 8 (m, 2H, H-b) 1.62 à 1.53 (m, 4H, H-3", H-5") 1. 46 à 1. 43 (m, 2H, H-4") RMN du C (CDsOD) : 8 (ppm) 161.2 (1 C, C-1') 150.4 (1C, C-5) 135.3 (1 C, C-7a) 130.5 (2C, C-3', C-5') 129.4 (1C, C-3a)
6. 97 (tt, 1 H, H-4', JH4'-H5'= 7. 3 HZ, JH4'-H6'= 1. 0 HZ) 6.90 à 6.85 (m, 2H, H-2', H-6') 6.81 (dd, 1 H, H-6, JH6-H7 = 8.7 HZ, JH6-H4 = 2.2 HZ) 2.69 (t, 2H, H-a, JHA-HB = 7. 3 Hz) 2.42 à 2.34 (m, 6H, H-2", H-6", H-c) 1.94 à 1. 8 (m, 2H, H-b) 1.62 à 1.53 (m, 4H, H-3", H-5") 1. 46 à 1. 43 (m, 2H, H-4") RMN du C (CDsOD) : 8 (ppm) 161.2 (1 C, C-1') 150.4 (1C, C-5) 135.3 (1 C, C-7a) 130.5 (2C, C-3', C-5') 129.4 (1C, C-3a)
<Desc/Clms Page number 15>
124.4 (1C, C-2) 122.8 (1C, C-4') 118.0 (2C, C-2', C-6') 116.1 (1 C, C-3) 115.9 (1C, C-6) 113.1 (1C, C-7) 110.1 (1C, C-4) 60.3 (1 C, C-c) 55.5 (2C, C-2", C-6") 28.1 (1C, C-b) 26.4 (2C, C-3", C-5") 25.2 (1C, C-4") 24.11 (1 C, C-a) IR (KBr) : v (cm-1) 3231 (N-Hindole) ; 1219 (Ar-o-Ar) ; 2948 ; 2800 ; 2763 ; 1487 ; 1454 Exemple 6 5-Phénoxy-3-[2- (piperazin-1-yl) éthyl]indole

A un mélange de 4g (13,3 mmol, 1éq.) de 2- {3- [ (4-tert-butyloxycarbonyl) pipérazinyl]propyl}-1,3-dioxolane en présence de 3,15 g (13,3 mmol, 1 éq. ) de chlorhydrat de 4-phénoxyphénylhydrazine sont ajoutés 110 mL d'une solution d'acide sulfurique à 4 %. La suspension obtenue est chauffée à 90 C pendant 2 heures puis refroidie à température ambiante. On obtient un précipité blanc. Le milieu est neutralisé par une solution saturée d'hydrogénocarbonate de sodium, puis extrait par du dichlorométhane. La phase organique est séchée sur sulfate de magnésium puis concentrée pour donner 2,41 g d'huile marron. Cette huile
A un mélange de 4g (13,3 mmol, 1éq.) de 2- {3- [ (4-tert-butyloxycarbonyl) pipérazinyl]propyl}-1,3-dioxolane en présence de 3,15 g (13,3 mmol, 1 éq. ) de chlorhydrat de 4-phénoxyphénylhydrazine sont ajoutés 110 mL d'une solution d'acide sulfurique à 4 %. La suspension obtenue est chauffée à 90 C pendant 2 heures puis refroidie à température ambiante. On obtient un précipité blanc. Le milieu est neutralisé par une solution saturée d'hydrogénocarbonate de sodium, puis extrait par du dichlorométhane. La phase organique est séchée sur sulfate de magnésium puis concentrée pour donner 2,41 g d'huile marron. Cette huile
<Desc/Clms Page number 16>

est purifiée sur colonne Büchi (support : silice 5-20 um, P=20 bars, éluant : dichlorométhane/méthanol/ammoniaque : 90/10/2) pour donner après lyophilisation 417 mg de 5-Phénoxy-3-[3-(pipérazin-1-yl)éthyl]indole sous forme de poudre blanche.
Rendement : 45 % F : 152 C Rf : 0, 30 (dichlorométhane/méthanol/ammoniaque : 92/8/1) SM (APcn : m/z : 322 (M+H) + RMN du 1H (CD30D) : # (ppm) 7,32 (d, 1 H, H-7, JH7-H6= 8, 7Hz) 7,25 (m, 2H, H-3', H-5') 7, 17 (d, 1 H, H-4, JH4-H6=2, 1Hz)

7, 10 (s, 1H, H-2) 6, 97 (t, 1 H, H-4', JH4'-H5'= 7, 3Hz) 6, 88 (d, 2H, H-2', H-6', JH2'-H3'= 7, 9Hz) 6,82 (dd, 1 H, H-6, JH6-H7 = 8, 7Hz, JH6-H4 = 2, 1 Hz) 2,88 (m, 6H, 2Ha, 4Hd) 2,60 (m, 6H, 2Hb, 4Hc) RMN du 13C (CD30D) : # (ppm)

161, 1 (1 C, C-1') 150,5 (1C, C-5) 135,2 (1C, C-7a) 130, 5 (2C, C-3', C-5') 129, 3 (1 C, C-3a) 124,7 (1C, C-2) 122, 8 (1 C, C-4') 118,1 (2C, C-2', C-6') 116, 0 (1C, C-6) 113,9 (1C, C-3) 113,2 (1C, C-7) 109,9 (1C, C-4) 61, 0 (1 C, C-b) 54,6 (2C, C-c)
7, 10 (s, 1H, H-2) 6, 97 (t, 1 H, H-4', JH4'-H5'= 7, 3Hz) 6, 88 (d, 2H, H-2', H-6', JH2'-H3'= 7, 9Hz) 6,82 (dd, 1 H, H-6, JH6-H7 = 8, 7Hz, JH6-H4 = 2, 1 Hz) 2,88 (m, 6H, 2Ha, 4Hd) 2,60 (m, 6H, 2Hb, 4Hc) RMN du 13C (CD30D) : # (ppm)
161, 1 (1 C, C-1') 150,5 (1C, C-5) 135,2 (1C, C-7a) 130, 5 (2C, C-3', C-5') 129, 3 (1 C, C-3a) 124,7 (1C, C-2) 122, 8 (1 C, C-4') 118,1 (2C, C-2', C-6') 116, 0 (1C, C-6) 113,9 (1C, C-3) 113,2 (1C, C-7) 109,9 (1C, C-4) 61, 0 (1 C, C-b) 54,6 (2C, C-c)
<Desc/Clms Page number 17>

46, 0 (2C, C-d) 23, 2 (1C, C-a) IR (KBr) : v (cm-1) 3415 à 2906 (NH), 1587 (NH), 1488 et 1466 (C-N), 1215 (Ar-O-Ar)

Exemple 7 S-Phénoxv-3-r2- (4-benzylpipérazi n -1-vl) éthvl] indole

A une solution de 930 mg (2,9 mmol, 1 éq. ) de 5-phénoxy-3-[2-(piperazin-1yl) ethyl] indole dans 35 mL d'acétonitrile est ajouté 768 mg (7,2 mmol, 2,5 éq. ) de carbonate de sodium. Après 30 minutes d'agitation à 50 C, 414 L (3,5 mmol, 1,2 éq. ) de bromobenzyle sont ajoutés rapidement. Le mélange est chauffé à reflux 2 heures, puis refroidi à température ambiante. Après filtration et lavage à l'acétonitrile, le filtrat est concentré pour donner 1,51 g de mousse orange. La mousse obtenue est purifiée sur Büchi (support : silice 5-20 m, éluant : dichlorométhane/méthanol/ammoniaque (90/10/2)) pour donner 950 mg de mousse blanche, qui est purifiée sur colonne Büchi (support : silice 5-20 um, P= 20 bars, éluant : dichlorométhane/méthanol/ammoniaque (96/4/1)) pour donner 394 mg de 5-phénoxy-3-[2- (4-benzylpipérazin-1-yl) éthyl]indole sous forme de

cristaux blancs. Rendement : 80 %

F : 48 C Rf : 0, 22 (dichlorométhane/méthanol/ammoniaque (96/4/1)) SM (APcn : m/z : 412 (M+Hf RMN du'H (CD30D) : 8 (ppm) 7,33-7, 21 (m, 8H, H-7, H-3', H-5', H-2", H-3", H-4", H-5", H-6")
<Desc/Clms Page number 18>

7, 16 (d, 1H, H-4, JH4-H6 = 2, 1 Hz) 7, 09 (s, 1 H, H-2) 6,96 (t, 1 H, H-4', JH4'-H5' = 7, 3 Hz) 6,87 (d, 2H, H-2', H-6', JH2'-H3'= JH5'-H6'= 8 Hz) 6,81 (dd, 1H, H-6, JH6-H7 = 8, 7 Hz, JH6-H4 2,2 Hz) 3,52 (s, 2H, (CH2Ph) 2, 95-2,40 (3 m, 12H, CH2) RMN du 13C (CD30D) : # (ppm) 161,5 (1C, C-1') 150, 9 (1C, C-5) 138,6 (1C, C-1") 135,5 (1C, C-7a) 131, 2 (2C, C-3", C-5") 130,9 (2C, C-3', C-5')
129,7 (2C, C-2", C-6")
129, 7 (1C, C-3a)
128,9 (1 C, C-4")
125, 1 (1 C, C-2)
123, 2 (1C, C-4')
118,5 (2C, C-2', C-6')
116,4 (1C, C-3)
114, 3 (1C, C-6)
113,6 (1C, C-7)
110, 3 (1C, C-4)
64,3 et 60,8 (2C, CH2Ph. C-b)
54,2 et 53,9 (4C, C-ci, C-C2, C-d1, C-d2)
23,8 (1C, C-a) IR (KBr) : v (cm-1)
3423 à 2800 (NH), 1578 (NH), 1218 (Ar-O-Ar)
<Desc/Clms Page number 19>
Exemple 8 5-Phénoxy-3-[3-(4-benzylpipérazin-1-yl)propyl]indole

A la 5-phénoxy-3-[3-(pipérazin-1-yl)propylindole] (2,12g ; 6,33 mmoles) en solution dans 130 mL d'acétonitrile est ajouté du carbonate de sodium (1,34g ; 12,66 mmoles, 2 éq. ) puis du bromure de benzyle (0,9 mL ; 7,59 mmoles : -1, 2 éq. ). Le mélange est porté à reflux pendant deux heures et demie, puis refroidi à température ambiante pour être filtré sur fritté. Le filtrat est évaporé pour obtenir 3g d'une mousse orange. Cette mousse est purifiée par chromatographie sur Büchi (support : silice 5-20 ! lm, P=20 bars, éluant : dichlorométhane/éthanol/ammoniaque : 98/2/2) pour obtenir 513 mg de produit qui est repurifié par une seconde chromatographie sur Büchi (support : silice 5- 20 ! lm, P=20 bars, éiuant : solvants HPLC

dichlorométhane/méthanol/ammoniaque : 98/2/1) pour donner 358 mg de 5phénoxy-3- [3- (4-benzy ! pipérazin-1-yi) propyi] indote sous forme d'une mousse blanche.
A la 5-phénoxy-3-[3-(pipérazin-1-yl)propylindole] (2,12g ; 6,33 mmoles) en solution dans 130 mL d'acétonitrile est ajouté du carbonate de sodium (1,34g ; 12,66 mmoles, 2 éq. ) puis du bromure de benzyle (0,9 mL ; 7,59 mmoles : -1, 2 éq. ). Le mélange est porté à reflux pendant deux heures et demie, puis refroidi à température ambiante pour être filtré sur fritté. Le filtrat est évaporé pour obtenir 3g d'une mousse orange. Cette mousse est purifiée par chromatographie sur Büchi (support : silice 5-20 ! lm, P=20 bars, éluant : dichlorométhane/éthanol/ammoniaque : 98/2/2) pour obtenir 513 mg de produit qui est repurifié par une seconde chromatographie sur Büchi (support : silice 5- 20 ! lm, P=20 bars, éiuant : solvants HPLC
dichlorométhane/méthanol/ammoniaque : 98/2/1) pour donner 358 mg de 5phénoxy-3- [3- (4-benzy ! pipérazin-1-yi) propyi] indote sous forme d'une mousse blanche.
Rendement : 49 % F : < 38 C Rf : 0,37 (dichlorométhane/méthanol/ammoniaque : 98/2/1) SM (APcl-) : m/z : 424 (M-H)- SM (APcl+) : m/z : 426 (M+H)' RMN du 1H (CD3OD) : (ppm)
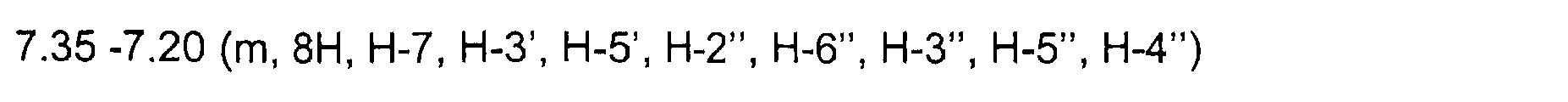
7. 35-7. 20 (m, 8H, H-7, H-3', H-5', H-2", H-6", H-3", H-5", H-4") 7.15 (d, 1H, H-4, JH-4, H-6 = 2. 1 Hz) 7. 06 (s, 1 H, H-2) 6.95 (t, 1H, H-4', JH4',H5' = JH-4',H-3' = 7.3 Hz) 6.87 (d, 2H, H-2', H-6', JH-2', H-3' = 8.0 Hz) 6.81 (dd, 1H, H-6, JH6, H7 = 8.7Hz, JH6, H4 = 2.1 Hz)
7. 35-7. 20 (m, 8H, H-7, H-3', H-5', H-2", H-6", H-3", H-5", H-4") 7.15 (d, 1H, H-4, JH-4, H-6 = 2. 1 Hz) 7. 06 (s, 1 H, H-2) 6.95 (t, 1H, H-4', JH4',H5' = JH-4',H-3' = 7.3 Hz) 6.87 (d, 2H, H-2', H-6', JH-2', H-3' = 8.0 Hz) 6.81 (dd, 1H, H-6, JH6, H7 = 8.7Hz, JH6, H4 = 2.1 Hz)
<Desc/Clms Page number 20>

3. 50 (s, 2H, GHZ-Ph) 2. 70 (t, 2H, H-a, H-a') 2.42 (m, 10H, H-c, H-c', H-d1, H-d1', H-d2, H-d2', H-e1, H-e1', H-e2, H-e2') 1.85 (m, 2H, H-b, H-b') RMN du 13C (CD30D) : # (ppm) 161.2 (1C, C-1') 150.3 (1C, C-5) 138.2 (1 C, C-1") 135.2 (1C, C-7a) 130.8 (2C, C-3" et C-5') 130.5 (2C, C-3'et C-5') 129.4 (1C, C-3a) 129.3 (2C, C-2" et C-6") 128.5 (1C, C-4") 124.5 (1 C, C-2)
122.7 (1C, C-4')
118.0 (2C, C-2'et C-6')
116.0 (1C, C-3)
115.9 (1C, C-6)
113.1 (1 C, C-7) 110. 1 (1C, C-4)
63.9 (1C, CH2-Ph)
59.3 (1 C, C-c)
53. 8 et 53.5 (4C, C-d1, C-d2, C-e1, C-e2)
28.2 (1C, C-b)
23.8 (1C, C-a)
IR (KBr) : v (cm-1)
3423 (NH), 1573 (NH), 1215 (Ar-O-Ar).
<Desc/Clms Page number 21>
Exemple 9

chlorhydrat du 5-phénoxy-3-F2- (pyrrolîdin-1-yliéthyllindole

A une solution de 5 ml (38 mmoles) de 2- (3-chloropropyl)-1, 3-dioxolane dans 5 ml (60 mmoles ; 1. 6 éq) de pyrrolidine, on ajoute 5. 23 g (38 mmoles ; 1 éq) de carbonate de potassium. On laisse agiter à température ambiante durant 19 heures. On porte le mélange à reflux durant 2 heures. A température ambiante, on ajoute 250 ml d'eau. On extrait la phase aqueuse avec 2 fois 250 ml de dichlorométhane. Les phases organiques sont séchées sur sulfate de sodium, filtrées sur coton et évaporées. On obtient une huile jaune pâle que l'on place au réfrigérateur pour la nuit.
A une suspension de 5. 91 g (25 mmoles, 0. 66 éq) de 5-phénoxyphénylhydrazine dans 200 ml d'une solution d'acide sulfurique à 4%, on ajoute l'huile obtenue précédemment. On laisse sous agitation à température ambiante durant 20 minutes avant de porter le mélange à 94 C durant 30 minutes. A température ambiante, on ajoute lentement et par petite quantité 200 ml d'une solution aqueuse saturée en hydrogénocarbonate de sodium. On extrait avec 3 fois 500 ml de dichlorométhane puis 500ml d'acétate d'éthyle. La phase organique est séchée sur sulfate de sodium, filtrée sur coton puis évaporée. On purifie le brut réactionnel sur colonne flash (support : silice ; conditionnement : 9/1/0. 2 (CH2CI2/MeOH/NH3) ; éluant : 9/1/0. 2 (CH2CI2/MeOH/NH3)) pour obtenir un précipité blanc qui correspond au sulfate du produit attendu. On lave le produit avec une solution de soude à 0. 1 N et on extrait avec du dichlorométhane. La phase organique est séchée sur sulfate de sodium, filtrée sur coton puis évaporée. On obtient un précipité jaune que l'on recristallise successivement dans l'heptane et le diéthyl éther. On dissout le précipité dans un mélange
<Desc/Clms Page number 22>
éther/dichlorométhane (50/50) puis on ajoute quelques gouttes d'éther saturé en HCI à froid afin d'obtenir le chlorhydrat. On récupère un précipité jaune que l'on recristallise dans l'acétone pour obtenir 98.6 mg (0.29 mmoles) du chlorhydrate du 5-phénoxy-3-[2-(pyrrolidin-1-yl)éthyl]indole sous forme de cristaux blancs.
Rendement : 14.8 % Rf : 0. 28 (CH2C) 2/MeOH/NH3. aq, 90/10/0. 2) SM (APclt : m/z 307 (M-HCI+H) + (APcl)-:m/z305(M-HCl-H)RMN du 1H (CD3OD) : ô (ppm) 7.38 (d, 1H, H-7, JH7-H6 = 8. 7 Hz) 7.29 à 7.21 (m, 4H, H-3', H-5', H-2, H-4) 6.97 (tt, 1 H, H-4', JH4'-H5' = 7.3 Hz) 6.9 à 6.84 (m, 3H, H-2', H-6', H-6) 3.5 à 3.35 (m, 4H, H-b, H-c) 3.15 (t, 2H, H-a, J=7. 5 Hz) 2.05 (m, 4H, H-d) RMN du 13C (CD30D) : (ppm) 161.0 (1C, C-1') 150.8 (1C, C-5) 135.3 (1C, C-7a) 130.6 (2C, C-3', C-5') 128.8 (1C, C-3a) 125.8 (1 C, C-2) 122.9 (1C, C-4') 118.0 (2C, C-2', C-6') 116.6 (1C, C-6) 113.6 (1C, C-7) 110.2 (1C, C-3) 119.8 (1C, C-4) 56.6 (1C, C-b) 55.2 (2C, C-c) 23.9 (2C, C-d) 23.1 (1C, C-a)
<Desc/Clms Page number 23>
IR (KBr) : v (cm-1) 3165 (N-Hindoie), 1223 (Ar-o-Ar) ; 2947 ; 2674 ; 2575 ; 2475,1595, 1574, 1477 ; 1454 Exemple 10 5-Phénoxy-[3-(pyrrolidin-1-yl)-propyl)]indole

A une solution de 5 ml (33 mmoles) de 2-(4-chlorobutyl)-1, 3-dioxolane dans 7 ml (84 mmoles ; 2.5 éq) de pyrrolidine, on ajoute 4.6 g (33 mmoles ; 1 éq) de carbonate de potassium. On porte le mélange à reflux (96 C durant 1.5 heures.
A température ambiante, on ajoute 70 ml d'eau. On extrait la phase aqueuse avec 3 fois 100 ml de dichlorométhane. Les phases organiques sont séchées sur sulfate de sodium, filtrées sur coton et évaporées. On obtient une huile jaune pâle que l'on place au réfrigérateur pour la nuit.
A une suspension de 4.4 g (19 mmoles, 0.66 éq) de 5-phénoxyphénylhydrazine dans 125 ml d'une solution d'acide sulfurique à 4%, on ajoute l'huile obtenue précédemment. On laisse sous agitation à température ambiante durant 20 minutes avant de porter le mélange à 80 C durant 1.5 heures. A température ambiante, on ajoute lentement et par petite quantité 125 ml d'une solution aqueuse saturée en hydrogénocarbonate de sodium. On extrait avec 2 L d' acétate d'éthyle. La phase organique est séchée sur sulfate de sodium, filtrée sur coton puis évaporée. On purifie le brut réactionnel sur colonne flash (support :

silice ; conditionnement : 9/1/0. 2 (CtC/MeOH/NHa) ; éluant : gradient de 9/1/0. 2 à 8/2/0. 2 (CH2Cl2/MeOH/NH3)) pour obtenir une huile jaune. On dissous l'huile dans le minimum d'éther et on évapore ; on obtient un précipité jaune.
silice ; conditionnement : 9/1/0. 2 (CtC/MeOH/NHa) ; éluant : gradient de 9/1/0. 2 à 8/2/0. 2 (CH2Cl2/MeOH/NH3)) pour obtenir une huile jaune. On dissous l'huile dans le minimum d'éther et on évapore ; on obtient un précipité jaune.
Après une recristallisation dans l'heptane, on passe de la base au chlorhydrat par ajout d'une solution d'éther saturée en acide chlorhydrique. On repasse à la
<Desc/Clms Page number 24>

base avec un lavage avec une solution d'hydroxyde de sodium 0. 1 N. On extrait avec 2 fois 50 ml d'acétate d'éthyle, la phase organique est séchée sur N32804 filtrée sur coton et évaporée pour obtenir une huile jaune que l'on fait précipiter dans un mélange 9/1 heptane/éther. Le précipité est filtré sur Whatman et séché d'abord à l'étuve sous vide à 50 C puis au lyophilisateur pour donner 430 mg (1. 34mmoles) de 5-phénoxy-[3-(pyrrolidin-1-yl)-propyl)]indole sous forme de cristaux blancs.
Rendement : 24 % F : 93.5-94. 2 C Rf : 0.42 (CH2Cl2/MeOH/NH3. aq, 90/10/0. 2) SM (APcl)+ : m/z 321 (M +H)+ (APcl)' : m/z 319 (M-H)- m/z 320 (M)- RMN du 1H (CD3OD) : # (ppm) 7.32 (d, 1 H, H-7, JH7-H6 = 8. 7 Hz) 7.25 (t, 2H, H-3', H-5', JH'3-H'2= 7.8 Hz) 7. 15 (d, 1H, H-4, JH4-H6 = 2. 1 Hz) 7.06 (s, 1 H, H-2) 6.97 (t, 1 H, H'-4, JH'4-H'3= 7.28 Hz) 6.88 (d, 2H, H'-2, H'-6, JH'2-H'3=7. 8 Hz) 6.79 (dd, 1H, H-6, JH7-H6= 8.7 Hz, JH4-H6= 2.1 Hz) 2.72 (t, 2H, H-a, JHa-Hb= 7.3 Hz) 2.53 (m, 6H, H-d, H-c) 1.90 (m, 2H, H-b) 1.78 (m, 4H, H-e) RMN du C (CD3OD) : # (ppm) 161.10 (1C, C-1') 151.9 (1C, C-5) 135.2 (1C, C-7a) 130.5 (2C, C-3', C-5') 129.3 (1C, C-3a) 124.4 (1C, C-2) 122.7 (1C, C-4')
<Desc/Clms Page number 25>
118. 0 (2C, C-2', C-6') 116.1 (1 C, C-3) 115.9 (1C, C-6) 113.1 (1C, C-7) 110.2 (1C, C-4) 57. 4 (1C, C-c) 54.9 (2C, C-d) 30. 4 (1C, C-b) 24.1 (3C, C-a, C-e) IR (KBr) : v (cm-1) 3429 (N-H, ndole) ; 1217 (Ar-o-Ar) ; 3086 ; 2916 ; 2804 ; 1453 Exemple 11 Intermédiaires :

(S)-3-FFN- (benzyloxycarbonyi) pyrrolidin-2ryllcarbonyil-5-phénoxyindole

A une solution incolore de 2,95 g (11,72 mmol) de carbobenzyloxy-L-proline 98% dans 9 mL de dichlorométhane anhydre sous argon, à température ambiante (22 C), on additionne 30 L de diméthylformamide anhydre puis lentement 1,57 mL (17, 58 mmol ; 1,5 éq) de chlorure d'oxalyle 98%. Après 4 heures sous agitation à température ambiante la solution jaune est évaporée sous pression réduite. Le résidu jaune est repris dans 12 mL d'hexane anhydre puis après évaporation sous pression réduite, on ajoute au résidu huileux jaune 12 mL de toluène anhydre.
(S)-3-FFN- (benzyloxycarbonyi) pyrrolidin-2ryllcarbonyil-5-phénoxyindole
A une solution incolore de 2,95 g (11,72 mmol) de carbobenzyloxy-L-proline 98% dans 9 mL de dichlorométhane anhydre sous argon, à température ambiante (22 C), on additionne 30 L de diméthylformamide anhydre puis lentement 1,57 mL (17, 58 mmol ; 1,5 éq) de chlorure d'oxalyle 98%. Après 4 heures sous agitation à température ambiante la solution jaune est évaporée sous pression réduite. Le résidu jaune est repris dans 12 mL d'hexane anhydre puis après évaporation sous pression réduite, on ajoute au résidu huileux jaune 12 mL de toluène anhydre.
<Desc/Clms Page number 26>

A une suspension gris clair de 3, 9 g (18, 66 mmol ; 2 éq) de 5-phénoxyindole dans 60 mL de toluène anhydre, sous argon à oac, on additionne lentement, 8, 20 mL (24, 61 mmoi ; 2, 1 éq) de bromure d'éthylmagnésium. Après 15 minutes à 0 C, on ajoute goutte à goutte, à la solution caramel obtenue, le chlorure d'acide de la carbobenzyloxy-L-proline. Le milieu est laissé sous vive agitation toute une nuit, puis le mélange réactionnel orangé est traité. Après ajout de 100 mL d'une solution saturée d'hydrogénocarbonate de sodium et 90 mL d'acétate d'éthyle, la suspension est laissée sous agitation pendant 1 heure puis filtrée sur fritté afin de casser l'émulsion. Après séparation, la phase aqueuse est extraite par 3x60 mL d'acétate d'éthyle. Les phases organiques jaunes sont réunies, séchées sur sulfate de magnésium, filtrées et évaporées sous pression réduite. Les 9, 9 g de résidu huileux marron clair sont triturés avec 300 mL de diéthyléther pour donner après filtration et séchage, 4, 20 g de 3- [ [N- (benzy ! oxycarbony)) pyrroiidin- (2S)yl] carbonyl]-5-phénoxyindole sous la forme d'un solide blanc.
Rendement : 81% F : 181, 1 C Rf : 0, 65 (CH2CI2/MeOH, 90/10) SM (APcl-) : m/z 439 (M-H)RMN du 1H (CD2CI2) : (ppm) 9, 46 (s, 1 H, H-1) 7, 99 (d, 1 H, H-4, JH4-H6= 2, 2 Hz) 7, 78-7, 83 (m, 2H, H-2, H-7) 7, 27-7, 43 (m, 6H, H-2"', H-6"', H3', H-5', H-4', H-4"') 6, 92-7, 13 (m, 5H, H-2', H-6', H-3"', H-5"', H6) 4, 91-5, 22 (m, 3H, CHz, H-2") 3, 40-3, 73 (m, 2H, 2H-5") 2, 20-2, 41 (m, 1 H, H-3") 1, 86-2, 09 (m, 3H, 2H-4"et H-3") RMN du C (CD2C < 2) : (ppm) 194, 0 (1 C, C=O) 158, 9 (1C, COO) 155, 4 (1C, C-1') 153, 0 (1C, C-5)
<Desc/Clms Page number 27>
137, 3 (lC, C-1"') 132,9 (lC, C-2) 132, 3 (1C, C-7a) 129,9 (2C, C-3' ; C-5' ou C-3"' ; C-5"') 128, 8 (2C, C-3' ; C-5' ou C-3'" ; C-5"') 128, 3 (1C, C-3a) 128,0 (2C, C-2"'et C-6"') 127,7 (1C, C-4'') 122,8 (1 C, C-4') 118, 3 (2C, C-2'et C-6') 117, 2 (1C, C-6) 115,2 (1C, C-3) 112, 9 (1C, C-7) 112,3 (1 C, C-4) 67, 1 (1C, C-2"ouCH)

62, 8 (1 CH20uC-2") 47, 5 (1C, C-5") 31, 5 (1C, C-3") 24, 3 (1C, C-4") Mo =-137 (c= 0. 265g/100m !, MeOH)

5-phénoxy-3-r (2S)- Dvrrolidin-2-ylméthvll-1 H-indole

A une suspension de 1,22 g (53,18 mmol ; 6 éq) de borohydrure de lithium 95% dans 30 ml de tétrahydrofurane anhydre, sous courant d'azote, on ajoute goutte à goutte, à température ambiante (22OC), 15,2 mL (0,115 mol ; 13 éq) de chlorure de triméthylsilane 98%. A la suspension blanche, laissée 5 minutes sous
62, 8 (1 CH20uC-2") 47, 5 (1C, C-5") 31, 5 (1C, C-3") 24, 3 (1C, C-4") Mo =-137 (c= 0. 265g/100m !, MeOH)
5-phénoxy-3-r (2S)- Dvrrolidin-2-ylméthvll-1 H-indole
A une suspension de 1,22 g (53,18 mmol ; 6 éq) de borohydrure de lithium 95% dans 30 ml de tétrahydrofurane anhydre, sous courant d'azote, on ajoute goutte à goutte, à température ambiante (22OC), 15,2 mL (0,115 mol ; 13 éq) de chlorure de triméthylsilane 98%. A la suspension blanche, laissée 5 minutes sous
<Desc/Clms Page number 28>

agitation à température ambiante, on ajoute 3, 90 g (8, 86 mmol ; 1 éq) de 3-[N- (benzyloxycarbonyl) pyrrolidin- (2S)-yl] carbonyl-5-phénoxy-1 H-indole en solution dans 20 mL de tétrahydrofurane anhydre. La suspension jaune pâle est laissée 15 heures sous agitation à température ambiante puis chauffée au reflux 1 heure.
Après refroidissement, l'excès d'hydrure est neutralisé avec 50 mL de méthanol et la solution jaune est laissée sous agitation pendant 30 minutes puis est évaporée sous pression réduite. Le résidu huileux marron est basifié à l'aide d'une solution de Nah03 saturée (pH = 7-8). On ajoute sous forte agitation 50 mL d'eau et 50 mL d'acétate d'éthyle. On observe la formation d'une émulsion et d'un précipité que l'on filtre sur fritté et qui conduit à 390 mg d'un solide blanc. La phase organique après séparation, puis concentration sous pression réduite-est filtrée sur fritté. Les 780 mg de précipité blanc cassé obtenus sont lavés avec 20 ml de méthanol pour conduire après filtration sur micropore à 522 mg du produit attendu sous la forme d'un solide blanc.
Rendement : 37% F : 55 C Rf : 0,52 (CH2CI2/MeOH, 95/5, silice greffée amino) SM (APcl+) : m/z 293 (M+H) + (APd-) : m/z 291 (M-H)' RMN du'H (CD2CI2) : 8 (ppm) 8, 65 (si, 1 H, H-1) 7,24-7, 35 (m, 4H, H-7, H-4, H-3', H-5) 7,07 (s, 1 H, H-2) 7,00 (t, 1 H, H-4'. JH4'-H3'= JH4'-H5'= 7,4 Hz) 6, 87-6,93 (m, 3H, H-6', H-2', H-6) 3,26-3, 45 (m, 1 H, H-2") 2,96-3, 02 (m, 1 H, H-5") 2, 73-2,93 (m, 3H, 2H-a, H-5") 2,05 (si, 1 H, H-1") 1,60-1, 92 (m, 3H, H-3", 2H-4") 1, 32-1,46 (m, 1 H, H-3") RMN du 13C (CD2CI2) : 8 (ppm) 159,9 (1C, C-1')
<Desc/Clms Page number 29>
149,6 (1 C, C-5) 133,7 (1C, C-7a) 129,8 (2C, C-3'et C-5') 128,8 (1C, C-3a) 124, 1 (1C, C-2) 122,1 (1C, C-4') 117, 3 (2C, C-2'et C-6') 115,7 (1C, C-6) 114,7 (1C, C-3) 112,3 (1C, C-7) 110,0 (1C, C-4) 59, 7 (1C, C-2") 46, 5 (1 C, C-5") 31, 9 (1 C-aouC-3") 31,9 (1C, C-3" ou C-a) 25, 5 (1 C, C-4") IR (KBr) : v (cm-1) 3410 (NHind), 1217 (Car-O-C), [α]D20 = +3 (c= 0. 265g/100ml, MeOH) Exemple 12
3 (2S) -1-benzylpvrrolidin-2-yl]méthyl} -5-phénoxy-1 H-indole

A une suspension blanche de 400 mg (1,37 mmol ; 1 éq) de 5-phénoxy-3-[(2S)pyrrolidin-2-ylméthyl]-1H-indole dans 75 ml de dichlorométhane, sous courant d'azote, on ajoute, à température ambiante (19 C), 80 @L (1,37 mmol ; 1 éq)
3 (2S) -1-benzylpvrrolidin-2-yl]méthyl} -5-phénoxy-1 H-indole
A une suspension blanche de 400 mg (1,37 mmol ; 1 éq) de 5-phénoxy-3-[(2S)pyrrolidin-2-ylméthyl]-1H-indole dans 75 ml de dichlorométhane, sous courant d'azote, on ajoute, à température ambiante (19 C), 80 @L (1,37 mmol ; 1 éq)
<Desc/Clms Page number 30>
d'acide acétique, puis 142 L (1, 37 mmol ; 1 éq) de benzaldéhyde. Enfin, 958mg (4, 38 mmol ; 3, 2 éq) de triacétoxyborohydrure de sodium est introduit au mélange et la solution blanchâtre est agitée pendant 18 heures.
Le milieu réactionnel est traité par 40 mL d'une solution saturée de chlorure de

sodium. La solution est basifiée par 60 mL d'une solution saturée de NaHCO3. Le mélange est extrait par 4x40 mL de dichlorométhane. La phase organique est séchée sur MgSO4, filtrée, puis concentrée sous pression réduite, pour conduire à 360 mg du produit attendu sous la forme d'une huile jaune. Les 360 mg sont purifiés sur chrompack (support : silice RP18 ; éluant : 80% acetonitril + 0,1 % d'ammoniaque à 28% ; 20 % H2O ; dépôt : solide ; pression : 1 bar ; débit 40 mL/min) pour conduire à une huile incolore. L'huile est triturée dans 20 ml' de diéthylether et après évaporation sous pression réduite, séchage sous vide pendant 2 jours on obtient 230 mg de 3-{[(2S)-1-benzylpyrrolidin-2-yl]méthyl}-5phénoxy-1 H-indole sous la forme d'un solide blanc.
sodium. La solution est basifiée par 60 mL d'une solution saturée de NaHCO3. Le mélange est extrait par 4x40 mL de dichlorométhane. La phase organique est séchée sur MgSO4, filtrée, puis concentrée sous pression réduite, pour conduire à 360 mg du produit attendu sous la forme d'une huile jaune. Les 360 mg sont purifiés sur chrompack (support : silice RP18 ; éluant : 80% acetonitril + 0,1 % d'ammoniaque à 28% ; 20 % H2O ; dépôt : solide ; pression : 1 bar ; débit 40 mL/min) pour conduire à une huile incolore. L'huile est triturée dans 20 ml' de diéthylether et après évaporation sous pression réduite, séchage sous vide pendant 2 jours on obtient 230 mg de 3-{[(2S)-1-benzylpyrrolidin-2-yl]méthyl}-5phénoxy-1 H-indole sous la forme d'un solide blanc.
Rendement : 69% Rf : 0,64 (95/5 Toluène/EtOH, silice greffée amino) SM (APcl+) : m/z 383 (M+H) + (APcl-) : m/z 381 (M-H)- RMN du 1H (CD2Cl2) : 8 (ppm) 8,18 (sl, 1H, H-1) 7, 16-7,36 (m, 9H, H-4"', H-4, H-5', H-3', H-3"', H-5"', H-6"', H-2"', H-7) 7, 13 (d, 1H, H-2,'JH2-Hi=1, 9 Hz) 7,02 (t, 1H, H-4', 3JH4'-H3'= 3JH4'-H5'=7, 4 Hz) 6, 88-6,96 (m, 3H, H-6', H-2', H-6)

4, 12 (d, 1H, H-b, 2JHb-Hb= 13 Hz, CH2-Ph) 3, 27 (d, 1H, H-b, 2JHb-Hb= 13 Hz, CH2-Ph) 3, 08 (dd, 1 H, H-a, 2JHa-Ha=13, 8 Hz, 3JHa-H2"=3, 2 Hz, (-Chb-ind)) 2, 86-2, 93 (m, 1 H, H-5") 2, 73-2, 83 (m, 1 H, H-2") 2,86-2, 93 (m, 1 H, H-a) 2, 11-2,21 (m, 1H. H-5") 1, 50-1,90 (m, 4H, H-3", H-3", H-4", H-4") RMN du 13C (CD2CI2) : 8 (ppm)
4, 12 (d, 1H, H-b, 2JHb-Hb= 13 Hz, CH2-Ph) 3, 27 (d, 1H, H-b, 2JHb-Hb= 13 Hz, CH2-Ph) 3, 08 (dd, 1 H, H-a, 2JHa-Ha=13, 8 Hz, 3JHa-H2"=3, 2 Hz, (-Chb-ind)) 2, 86-2, 93 (m, 1 H, H-5") 2, 73-2, 83 (m, 1 H, H-2") 2,86-2, 93 (m, 1 H, H-a) 2, 11-2,21 (m, 1H. H-5") 1, 50-1,90 (m, 4H, H-3", H-3", H-4", H-4") RMN du 13C (CD2CI2) : 8 (ppm)
<Desc/Clms Page number 31>

159, 9 (1C. C-1') 149, 6 (1C, C-5) 140, 6 (1 C, C-1"') 133,5 (1C, C-7a) 129, 8 (2C, C-3"' et C-5"' ou C-3'et C-5') 129, 1 (2C, C-3' et C-5' ou C-3"' et C-5"') 129,0 (1C, C-3a) 128,4 (2C, C-2"' et C-6"') - 127,0 (1C, C-4"') 124 (1C, C-2) 122, 1 (1C, C-4') 117,3 (2C, C-2'etC-6') 115,8 (1 C, C-6) 114,6 (1C, C-3) 112, 2 (1C, C-7) 110,2 (1C, C-4) 64,7 (1C, C-2") 59,3 (1C, C-b (CH2-Ph)) 54, 8 (1C, C-5") 31, 1 (1C, C-3") 30,5 (1C, C-a (CH2-ind)) 22,5 (1 C, C-4") IR (KBr) : v (cm-1) 3417 (NHind), 1209 (Car-O-C), [α]D20 = -45 (c= 0. 240g/100ml, MeOH0
<Desc/Clms Page number 32>
Exemple 13 5-phénoxy-3- [ (2R)- pyrrolidinylméthyll-1 H-indole
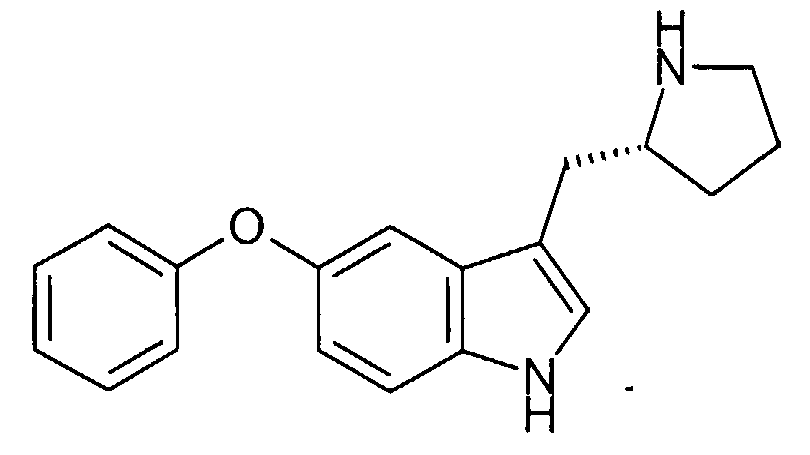
A une suspension de 830 mg (36 mmol ; 6 éq) de borohydrure de lithium dans 20 ml de tétrahydrofurane anhydre, on ajoute goutte à goutte, à température ambiante, 9,3 ml (72 mmol ; 12 éq) de chlorure de triméthylsilane. A la suspension blanche, laissée 5 minutes sous agitation à température ambiante, on ajoute 2,64 g (6 mmol ; 1 éq) de 3- [N- (Benzyioxycarbony !) pyrroiidin- (2R)- yl] carbonyl-5-phénoxy-1 H-indole en solution dans 20 ml de tétrahydrofurane anhydre. La suspension beige est laissée 3 heures sous agitation à température ambiante puis chauffée au reflux 3 heures. Après refroidissement, l'excès d'hydrure est neutralisé avec 20 ml de méthanol et la solution jaune est évaporée sous pression réduite. Le résidu huileux marron est dissous dans 50 ml d'eau puis extrait avec 4x50 ml d'acétate d'éthyle. Dans la phase organique verdâtre on observe la formation d'un précipité que l'on filtre sur fritté et qui conduit à 215 mg d'un solide gris. Le filtrat marron est concentré sous pression réduite puis filtré pour conduire à 380 mg d'un solide beige-gris. Les deux précipités ainsi obtenus contiennent le produit non pur. Le filtrat marron est évaporé sous pression réduite pour conduire à 2,49 g d'une huile marron contenant majoritairement du 3-[N- (Benzyloxycarbonyl) pyrrolidin- (2R)-yl] méthyl-5-phénoxy-1H-indole. L'amine de la pyrrolidine est déprotégée en présence d'iodure de triméthylsilane et de méthanol selon la méthode suivante. A l'huile marron dissoute dans 20 ml d'acétonitrile, on additionne, à température ambiante, 1,75 ml (11,69 mmol ; 2 éq) d'iodure de triméthylsilane. La suspension bordeaux est laissée sous agitation pendant 3 heures puis on ajoute 50 ml de méthanol. Après une heure d'agitation, la suspension bordeaux est évaporée sous pression réduite. L'huile bordeaux est dissoute dans 50 ml d'eau puis extraite avec 5x50 ml de diéthyléther. La phase
A une suspension de 830 mg (36 mmol ; 6 éq) de borohydrure de lithium dans 20 ml de tétrahydrofurane anhydre, on ajoute goutte à goutte, à température ambiante, 9,3 ml (72 mmol ; 12 éq) de chlorure de triméthylsilane. A la suspension blanche, laissée 5 minutes sous agitation à température ambiante, on ajoute 2,64 g (6 mmol ; 1 éq) de 3- [N- (Benzyioxycarbony !) pyrroiidin- (2R)- yl] carbonyl-5-phénoxy-1 H-indole en solution dans 20 ml de tétrahydrofurane anhydre. La suspension beige est laissée 3 heures sous agitation à température ambiante puis chauffée au reflux 3 heures. Après refroidissement, l'excès d'hydrure est neutralisé avec 20 ml de méthanol et la solution jaune est évaporée sous pression réduite. Le résidu huileux marron est dissous dans 50 ml d'eau puis extrait avec 4x50 ml d'acétate d'éthyle. Dans la phase organique verdâtre on observe la formation d'un précipité que l'on filtre sur fritté et qui conduit à 215 mg d'un solide gris. Le filtrat marron est concentré sous pression réduite puis filtré pour conduire à 380 mg d'un solide beige-gris. Les deux précipités ainsi obtenus contiennent le produit non pur. Le filtrat marron est évaporé sous pression réduite pour conduire à 2,49 g d'une huile marron contenant majoritairement du 3-[N- (Benzyloxycarbonyl) pyrrolidin- (2R)-yl] méthyl-5-phénoxy-1H-indole. L'amine de la pyrrolidine est déprotégée en présence d'iodure de triméthylsilane et de méthanol selon la méthode suivante. A l'huile marron dissoute dans 20 ml d'acétonitrile, on additionne, à température ambiante, 1,75 ml (11,69 mmol ; 2 éq) d'iodure de triméthylsilane. La suspension bordeaux est laissée sous agitation pendant 3 heures puis on ajoute 50 ml de méthanol. Après une heure d'agitation, la suspension bordeaux est évaporée sous pression réduite. L'huile bordeaux est dissoute dans 50 ml d'eau puis extraite avec 5x50 ml de diéthyléther. La phase
<Desc/Clms Page number 33>
organique après concentration sous pression réduite est filtrée sur fritté. Les 620 mg de précipité rouille obtenus sont lavés successivement avec 20 ml de diéthyléther et 20 ml de dichlorométhane pour conduire à 430 mg du produit attendu sous la forme d'un précipité jaune pale. L'ensemble des précipités (1,025 g) est réuni, lavé avec 50 ml de diéthyléther puis 50 ml de dichlorométhane pour conduire après séchage à l'étuve sous vide à 880 mg de produit sous la forme d'un solide beige d'une pureté de 98%. Les 880 mg sont purifiés sur chrompack (support : silice RP18 ; éluant : méthanol + 0,1 % d'ammoniaque à 28% ; dépôt : liquide dans l'éluant ; pression : 1 bar ; débit 40 ml/min) pour conduire à une huile incolore. L'huile est triturée dans 20 ml de diéthylether et après évaporation sous pression réduite, séchage sous vide 2 jours on obtient 500 mg de 5-phénoxy-3- [ (2R)- pyrrolidinylméthyl]-1 H-indole sous la forme d'un solide blanc.
Rendement : 50% F : 61 C Rf : 0,5 (CH2CI2/MeOH, 95/5, silice greffée amino) SM (APcl+) : m/z 293 (M+Hf RMN du H (DMSO-d6) : 8 (ppm) 10,88 (s, 1H, H-1) 7, 26-7, 36 (m, 3H, H-7, H-3', H-5') 7,18 (m, 2H, H-2, H-4) 7, 00 (t, 1 H, H-4', JH4'-H5'= JH4'-H3'= 7,3 Hz) 6, 88 (d, 2H, H-2', H-6', JH2'-H3'= JH6'-H5'= 7,9 Hz) 6, 79 (dd, 1 H, H-6, JH6-H7= 8, 6Hz, JH6-H4 =2,1 Hz) 3,04-3, 32 (m, 1 H, H-2) 2, 58-2, 89 (m, 4H, H-5, H-a, H-a, H-5) 1,51-1, 74 (m, 3H, H-3, 2H-4) 1,19-1, 33 (m, 1H, H-3) RMN du 13C (DMSO-d6) : 8 (ppm) 159,2 (1C, C-1') 148,1 (1C, C-5) 133,2 (1C, C-7a) 129,7 (2C, C-3'et C-5') 128,2 (1C, C-3a)
<Desc/Clms Page number 34>
124,4 (1C, C-2) 121, 8 (1C, C-4') 116,6 (2C, C-2'et C-6') 114,3 (1 C, C-6) 113,0 (1C, C-3) 112, 3 (1C, C-7) 109,1 (1 C, C-4) 59,1 (1C, C-2) 45,9 (1 C, C-5) 31,5 (1 C, C-a) 31, 3 (1C, C-3) 24,9 (1C, C-4) IR (KBr) : v (cm-1) 3420 (NHind), 3410 (NH), 1220 (Car-N), 1210 (Car-O-C), [α] D25 =-6 (c= 0. 28g/100mt, MeOH) Exemple 14

3-FF (2R)-l-benzyl pyrrol id in-2-yll méthyll-5-phénoxy-1 H-indole

A une suspension jaune de 500 mg (1,71 mmol ; 1 éq) de 5-phénoxy-3- [ (2R)- pyrrolidinylméthyl]-1 H-indole dans 75 ml de dichloroéthane anhydre, on ajoute, à température ambiante, sous azote, 178 u) (1,71 mmol ; 1 éq) de benzaldéhyde puis 100 l (1,71 mmol ; 1 éq) d'acide acétique. A la suspension jaune, laissée 15 minutes sous agitation à température ambiante, on additionne 1,20 g (5,48 mmol ; 3,2 éq) de triacétoxyborohydrure de sodium. La suspension est laissée toute la
3-FF (2R)-l-benzyl pyrrol id in-2-yll méthyll-5-phénoxy-1 H-indole
A une suspension jaune de 500 mg (1,71 mmol ; 1 éq) de 5-phénoxy-3- [ (2R)- pyrrolidinylméthyl]-1 H-indole dans 75 ml de dichloroéthane anhydre, on ajoute, à température ambiante, sous azote, 178 u) (1,71 mmol ; 1 éq) de benzaldéhyde puis 100 l (1,71 mmol ; 1 éq) d'acide acétique. A la suspension jaune, laissée 15 minutes sous agitation à température ambiante, on additionne 1,20 g (5,48 mmol ; 3,2 éq) de triacétoxyborohydrure de sodium. La suspension est laissée toute la
<Desc/Clms Page number 35>
nuit sous agitation à température ambiante. La suspension jaune pale est versée dans 50 ml d'une solution saturée en chlorure de sodium, basifiée avec 70 ml d'un solution saturée en hydrogénocarbonate de sodium puis extraite avec 4x50 ml de dichlorométhane. La phase organique jaune est séchée sur sulfate de sodium, filtrée et évaporée sous pression réduite pour conduire à 620 mg d'une huile marron. Les 620 mg de produit sont purifiés successsivement sur deux colonnes (support : silice greffée amino, 25-40 m ; éluant : 70/30 dichlorométhane/heptane ; dépôt : liquide dans l'éluant ; h=11 cm ; d=35 mm), (support : silice greffée amino, 25-40 um ; étuant : 60/40 dichlorométhane/heptane ; dépôt : liquide dans i'éiuant ; h=11-cm ; d=35 mm) puis sur Chrompack (support : silice RP18 ; éluant : 83/17 méthanol + 0, 1 % d'ammoniaque à 28%/eau puis 92/8 méthanol + 0,1 % d'ammoniaque à 28%/ eau ; dépôt : liquide dans le méthanol ; pression : 1, 5 bar ; débit 40 ml/min) pour conduire à 320 mg de 3-[[(2R)-1-benzylpyrrolidin-2-yl]méthyl]-5-phénoxy-1Hindole sous la forme d'un solide blanc sous vide et d'une huile beige au contact de l'air.
Rendement : 49% Rf : 0,32 (CH2CI2, silice greffée amino) SM (APcl+) : m/z 383 (M+H)' RMN du 1H (CD2Cl2) : 8 (ppm)

8, 18 (sl, 1H, H-1) 7, 20-7, 36 (m, 9H, H-3"', H-5"', H-4, H-3", H-5", H-2"', H-6"', H-4'', H-7) 7, 13 (d, 1H, H-2, JH2-Hi=1, 8 Hz) 7, 02 (t, 1 H, H-4, JH4'-H3'= JH4'-H5'=7, 3 Hz) 6, 88-6, 96 (m, 3H, H-2', H-6', H-6) 4,12 (d, 1H, H-b, 2JHb-Hb=13 Hz, CH2-Ph) 3,28 (d, 1H, H-b, Hb-Hb=13 Hz, CH2-Ph)

3, 08 (dd, 1 H, H-a, 2JHa-Ha=13, 8 Hz, 3j Ha-H2'=3, 2 Hz, (-CHz-ind)) 2, 76-2,93 (m, 2H, H-5", H-2") 2,61-2, 61 (m, 1H, H-a (-CH2-ind)) 2,17 (m, 1H, H-5") 1, 56-1, 82 (m, 4H, H-3", H-3", H-4", H-4") RMN du 13C (CD2CI2) : 8 (ppm)
8, 18 (sl, 1H, H-1) 7, 20-7, 36 (m, 9H, H-3"', H-5"', H-4, H-3", H-5", H-2"', H-6"', H-4'', H-7) 7, 13 (d, 1H, H-2, JH2-Hi=1, 8 Hz) 7, 02 (t, 1 H, H-4, JH4'-H3'= JH4'-H5'=7, 3 Hz) 6, 88-6, 96 (m, 3H, H-2', H-6', H-6) 4,12 (d, 1H, H-b, 2JHb-Hb=13 Hz, CH2-Ph) 3,28 (d, 1H, H-b, Hb-Hb=13 Hz, CH2-Ph)
3, 08 (dd, 1 H, H-a, 2JHa-Ha=13, 8 Hz, 3j Ha-H2'=3, 2 Hz, (-CHz-ind)) 2, 76-2,93 (m, 2H, H-5", H-2") 2,61-2, 61 (m, 1H, H-a (-CH2-ind)) 2,17 (m, 1H, H-5") 1, 56-1, 82 (m, 4H, H-3", H-3", H-4", H-4") RMN du 13C (CD2CI2) : 8 (ppm)
<Desc/Clms Page number 36>
159, 9 (1C, C-1') 149, 6 (1C, C-5) 140,6 (1C, C-1"') 133,5 (1C, C-7a) 129,8 (2C, C-3"' et C-5"' ou C-3'et C-5') 129,2 (2C, C-3'et C-5'ou C-3"'et C-5"') 129, 0 (1C, C-3a) 128,4 (2C, C-2"' et C-6"') 127,0 (1C, C-4"') 124, 0 (1 C, C-2)

122, 1 (1C, C-4') 117, 3 (2C, C-2'et C-6') 115, 8 (1C, C-6) 114, 6 (1C, C-3) 112, 2 (1C, C-7) 110, 2 (1C, C-4) 64, 7 (1C, C-2") 59, 3 (1C, CH2-Ph) 54, 8 (1 C, C-5") 31, 2 (1 C, C-3") 30, 5 (1C, CH2-ind) 22,5 (1C, C-4") IR (KBr) : v (cm-1) 3416 (NHind), 1222 (Car-O-C), [α]D25 = + 43 (c= 0. 29g/1 @, MeOH). On donnera ci-après les résultats des études pharmacologiques.
122, 1 (1C, C-4') 117, 3 (2C, C-2'et C-6') 115, 8 (1C, C-6) 114, 6 (1C, C-3) 112, 2 (1C, C-7) 110, 2 (1C, C-4) 64, 7 (1C, C-2") 59, 3 (1C, CH2-Ph) 54, 8 (1 C, C-5") 31, 2 (1 C, C-3") 30, 5 (1C, CH2-ind) 22,5 (1C, C-4") IR (KBr) : v (cm-1) 3416 (NHind), 1222 (Car-O-C), [α]D25 = + 43 (c= 0. 29g/1 @, MeOH). On donnera ci-après les résultats des études pharmacologiques.
1-Le pouvoir inhibiteur des molécules sur l'inflammation induite par stimulation du nerf saphène a été mesuré comme suit : Vingt-quatre heures après un traitement à la guanéthidine (20 mg/kg, sc) les rats Wistar mâles (220-250g) sont anesthésiés au pentobarbital sodique (60 mg/kg, ip). Les deux pattes postérieures sont rasées. Après incision dans la partie
<Desc/Clms Page number 37>
supérieure de la cuisse, le nerf saphène est dégagé, coupé, placé sur une électrode de platine et immergé dans une goutte d'huile de paraffine. Seule l'électrode positionnée au niveau de la patte droite est reliée à un stimulateur. Cette dernière représente la patte"stimulée"par opposition à la patte gauche

"sham". Le produit (à la dose de 5 ug/kg) ou le solvant correspondant (NaCI 9 %0- DMSO) sont administrés via la veine jugulaire, 15 minutes avant la stimulation électrique (SE). Un marquer plasmatique, le Bleu Evans (20 mg/kg iv) est administré par la veine du-pénis 10 minutes avant la SE. Le nerf saphène est stimulé selon les conditions suivantes : 3 V ; 5 Hz ; 1 ms ; 5 minutes (Stimulateur Harvard). A la fin de la SE, un prélèvement sanguin est réalisé par ponction cardiaque et la peau oedémateuse de chaque patte (visualisée par l'extravasation du Bleu Evans) prélevée et pesée ultérieurement. Les animaux sont sacrifiés par overdose d'anesthésique. Les prélèvements sanguins sont centrifugés (3000 tours/minute, pendant 15 minutes). Le plasma est ensuite dilué au 1/100, dans de l'eau distillée. Le marqueur plasmatique est extrait des biopsies de peau selon la méthode de Beach et Steinetz (J. Pharmacol. Exp.
"sham". Le produit (à la dose de 5 ug/kg) ou le solvant correspondant (NaCI 9 %0- DMSO) sont administrés via la veine jugulaire, 15 minutes avant la stimulation électrique (SE). Un marquer plasmatique, le Bleu Evans (20 mg/kg iv) est administré par la veine du-pénis 10 minutes avant la SE. Le nerf saphène est stimulé selon les conditions suivantes : 3 V ; 5 Hz ; 1 ms ; 5 minutes (Stimulateur Harvard). A la fin de la SE, un prélèvement sanguin est réalisé par ponction cardiaque et la peau oedémateuse de chaque patte (visualisée par l'extravasation du Bleu Evans) prélevée et pesée ultérieurement. Les animaux sont sacrifiés par overdose d'anesthésique. Les prélèvements sanguins sont centrifugés (3000 tours/minute, pendant 15 minutes). Le plasma est ensuite dilué au 1/100, dans de l'eau distillée. Le marqueur plasmatique est extrait des biopsies de peau selon la méthode de Beach et Steinetz (J. Pharmacol. Exp.
Therap., 1961,131, 400-406). Les peaux prélevées au niveau des pattes postérieures sont placées dans des tubes à col rodé contenant 3 ml d'acide chlorhydrique (36 %). Elles sont alors digérées par une hydrolyse de 2 heures, à 37 C. 3 ml de chlorure de benzalkonium (12,8 %) sont ensuite ajoutés. Après agitation et 30 minutes de repos, le traceur coloré est extrait par 7 ml de dichlorométhane. Les tubes sont agités lentement et périodiquement, pendant une heure. La phase aqueuse (supérieure) est éliminée par aspiration, à l'aide d'une trompe à vide, et la phase organique filtrée sur papier. Le Bleu Evans est dosé dans le plasma et après extraction des biospises de peau par méthode spectrophotométrique, à 620 nm. L'extravasation plasmatique développée au niveau de chaque patte est exprimée en al de plasma/g peau. L'oedème neurogène induit par la stimulation du nerf saphène est donnée comme étant la différence de volume de plasma entre la patte"stimulée"et de la patte

"contrôle". Les résultats obtenus le même jour avec un groupe de rat traités et un groupe de rat contrôles. Le pouvoir inhibiteur du composé testé est mesuré par le rapport volume- dème-moyen-rat traitée/volume- dème-moyen-rat contrôle.
"contrôle". Les résultats obtenus le même jour avec un groupe de rat traités et un groupe de rat contrôles. Le pouvoir inhibiteur du composé testé est mesuré par le rapport volume- dème-moyen-rat traitée/volume- dème-moyen-rat contrôle.
<Desc/Clms Page number 38>
2-La réponse contractile vasculaire de type 5HT1B a été mesurée sur des anneaux de veine saphène de lapin sub-maximalement contractés par un tampon physiologique contenant 40 mM de KCI à l'aide d'une installation EMKA technologie. Dans ces conditions, les réponse induites par les agonistes 5HT1 B tels que le sumatriptan et la 5-CT sont maximales. La potentiel agonist 5HT1B des molécules est obtenu par ajouts de doses croissantes de molécule et la concentration donnant 50% de la réponse maximale est utilisé ici comme indicateur (EC50 en M).
3-La fixation sur les récepteurs 5HT1 B/5HT1 D de membranes isolées de noyau caudé de boeuf a été mesurée en mesurant le déplacement de 5nM de 3H5CT par des concentrations croissantes de molécule en présence de 100 nu de 8-OH-DPAT pour masquer les récepteurs 5HT1A et de 100 nM de mesulergine pour masquer les récepteurs 5HT2. La concentration deplaçant 50 % de la fixation totale (IC50 exprimé en M) est utilisée ici comme valeur de réactivité.
4-La fixation sur les récepteurs 5HT1A de membranes isolées de cerveau de rat a été mesurée en mesurant le déplacement de 1 nM de 3H 8-OH-DPAT par des concentrations croissantes de molécule en présence de 100 nM de mesulergine pour masquer les récepteurs 5HT2.. La concentration déplaçant 50 % de la fixation totale (IC50 exprimé en M) est utilisée ici comme valeur de réactivité.
Les résultats sont rassemblés dans le tableau suivant.
Selon également donnés à titre comparatif les résultats pour deux

composés de référence de type 5-phénoxyindole.
composés de référence de type 5-phénoxyindole.
<Desc/Clms Page number 39>

<tb>
<tb>
<tb>
N <SEP> exemple <SEP> Inhib. <SEP> Inf. <SEP> Neuro <SEP> EC50 <SEP> Cont. <SEP> IC50 <SEP> 5HT1A <SEP> IC50 <SEP> 5HT1B/1D
<tb> > 1.10-4 <SEP> 3, <SEP> 0. <SEP> 10-5 <SEP> 4,0. <SEP> 10-6
<tb> 1 <SEP> 55% <SEP> > <SEP> 1.10-4 <SEP> 3,0. <SEP> 10-5 <SEP> 4,0. <SEP> 10-6
<tb> 2 <SEP> 39% <SEP> > <SEP> 1.10-4 <SEP> 5,5. <SEP> 10-66,0. <SEP> 10-6
<tb> 4 <SEP> 62% <SEP> 5. <SEP> 10-8 <SEP> 4,3. <SEP> 10-7 <SEP> 1,0. <SEP> 10-7
<tb> 5 <SEP> 1.10-7 <SEP> 3,8. <SEP> 106 <SEP> 4,1. <SEP> 10-7
<tb> 6 <SEP> 29% <SEP> 1. <SEP> 10-4 <SEP> 2,0. <SEP> 10-4 <SEP> 1,8. <SEP> 10-5
<tb> 8 <SEP> 20% <SEP> 1. <SEP> 10-7 <SEP> 1,8. <SEP> 10-6 <SEP> 1,1. <SEP> 10-7
<tb> 9 <SEP> 33% <SEP> 2,6. <SEP> 10-7 <SEP> 1,2. <SEP> 10-7 <SEP> 2,6. <SEP> 10-7
<tb> 11 <SEP> 56% <SEP> 1. <SEP> 10-5 <SEP> 2,8. <SEP> 10-6 <SEP> 2,5. <SEP> 10-6
<tb> 12 <SEP> 41% <SEP> > <SEP> 1.10-4 <SEP> 5,0. <SEP> 10-5
<tb> 13 <SEP> 24% <SEP> 7,5. <SEP> 10-8 <SEP> 1, <SEP> 8. <SEP> 10-6 <SEP> 2, <SEP> 3. <SEP> 10-7
<tb> 14 <SEP> 58% <SEP> 7,5. <SEP> 10-8 <SEP> 8, <SEP> 5. <SEP> 10-7 <SEP> 1,1. <SEP> 10-7
<tb> 1
<tb> a-1% <SEP> 1,4. <SEP> 10-7 <SEP> 3,4. <SEP> 10-7 <SEP> 7,3. <SEP> 10-8
<tb> b <SEP> 57% <SEP> 2,5. <SEP> 10'11, <SEP> 6. <SEP> 10-'1, <SEP> 3. <SEP> 10-8
<tb>
exemple a = 3- (N-méthyl-1, 2,5, 6-tétrahydropyrid-4-yl) -5-phénoxyindole exemple b = (R) 3-[(3-méthylpyrrolidin-2-yl)méthyl] -5-phénoxyindole Légende du tableau : Inhib. Inf. Neuro. = Inhibition de l'inflammation neurogène exprimée en % ; EC50 Cont. = EC50 de la réponse contractile de la veine saphène de lapin (réponse agoniste 5HT1 B) ; IC50 5HT1 A = IC50 du déplacement de la fixation de 3H 8-OH-DPAT sur les récepteurs 5HT1A de cerveau de rat ; IC50 5HT 1B/5HT1D = IC50 du déplacement de la fixation de 3H 5-carboxytryptamine sur les récepteurs 5HT1B/ 5HT1D de cerveau de b#uf.
<tb> > 1.10-4 <SEP> 3, <SEP> 0. <SEP> 10-5 <SEP> 4,0. <SEP> 10-6
<tb> 1 <SEP> 55% <SEP> > <SEP> 1.10-4 <SEP> 3,0. <SEP> 10-5 <SEP> 4,0. <SEP> 10-6
<tb> 2 <SEP> 39% <SEP> > <SEP> 1.10-4 <SEP> 5,5. <SEP> 10-66,0. <SEP> 10-6
<tb> 4 <SEP> 62% <SEP> 5. <SEP> 10-8 <SEP> 4,3. <SEP> 10-7 <SEP> 1,0. <SEP> 10-7
<tb> 5 <SEP> 1.10-7 <SEP> 3,8. <SEP> 106 <SEP> 4,1. <SEP> 10-7
<tb> 6 <SEP> 29% <SEP> 1. <SEP> 10-4 <SEP> 2,0. <SEP> 10-4 <SEP> 1,8. <SEP> 10-5
<tb> 8 <SEP> 20% <SEP> 1. <SEP> 10-7 <SEP> 1,8. <SEP> 10-6 <SEP> 1,1. <SEP> 10-7
<tb> 9 <SEP> 33% <SEP> 2,6. <SEP> 10-7 <SEP> 1,2. <SEP> 10-7 <SEP> 2,6. <SEP> 10-7
<tb> 11 <SEP> 56% <SEP> 1. <SEP> 10-5 <SEP> 2,8. <SEP> 10-6 <SEP> 2,5. <SEP> 10-6
<tb> 12 <SEP> 41% <SEP> > <SEP> 1.10-4 <SEP> 5,0. <SEP> 10-5
<tb> 13 <SEP> 24% <SEP> 7,5. <SEP> 10-8 <SEP> 1, <SEP> 8. <SEP> 10-6 <SEP> 2, <SEP> 3. <SEP> 10-7
<tb> 14 <SEP> 58% <SEP> 7,5. <SEP> 10-8 <SEP> 8, <SEP> 5. <SEP> 10-7 <SEP> 1,1. <SEP> 10-7
<tb> 1
<tb> a-1% <SEP> 1,4. <SEP> 10-7 <SEP> 3,4. <SEP> 10-7 <SEP> 7,3. <SEP> 10-8
<tb> b <SEP> 57% <SEP> 2,5. <SEP> 10'11, <SEP> 6. <SEP> 10-'1, <SEP> 3. <SEP> 10-8
<tb>
exemple a = 3- (N-méthyl-1, 2,5, 6-tétrahydropyrid-4-yl) -5-phénoxyindole exemple b = (R) 3-[(3-méthylpyrrolidin-2-yl)méthyl] -5-phénoxyindole Légende du tableau : Inhib. Inf. Neuro. = Inhibition de l'inflammation neurogène exprimée en % ; EC50 Cont. = EC50 de la réponse contractile de la veine saphène de lapin (réponse agoniste 5HT1 B) ; IC50 5HT1 A = IC50 du déplacement de la fixation de 3H 8-OH-DPAT sur les récepteurs 5HT1A de cerveau de rat ; IC50 5HT 1B/5HT1D = IC50 du déplacement de la fixation de 3H 5-carboxytryptamine sur les récepteurs 5HT1B/ 5HT1D de cerveau de b#uf.
II existe une corrélation entre les effets agonistes de types 5HT1 B sur la veine saphène de lapin et les IC50 vis à vis des récepteurs 5HT1B/5HT1D de cerveau de b#uf.
<Desc/Clms Page number 40>

Il n'existe pas de relation entre les effets anti-inflammatoires des composés selon l'invention et des composés a et b avec leurs effets agonistes de types 5HT1B sur la veine saphène de lapin et leurs IC50 vis à vis des récepteurs 5HT1B/5HT1D de cerveau de boeuf.
Les composés de la présente invention inhibent l'inflammation neurogène sans avoir d'effet agoniste substantiel sur la veine saphène de lapin et sans interagir fortement (IC50 > 1. 10-6 M) avec les récepteurs 5HT1B/5HT1D de cerveau de boeuf. Les effets des composés selon l'invention sont indépendants de la fixation/activation des récepteurs 5HT1 B ET 5HT1 D.
Claims (13)
1. Composés de formule :

R1 étant choisi parmi les groupes aryle mono-, bi-ou tricyclique en Cg-Ci4, les groupes hétéroaryle choisis parmi les groupes pyridyle, pyrimidinyle, pyridazinyl, pyrazinyl, thienyl, furyle, imidazolyl, pyrrolyle, oxazolyle, thiazolyle, pyrazolyle, tetrazolyle, oxadiazolyle, thiadiazolyle, quinolyle, isoquinolyle, benzothienyle, benzofuryle, indolyle, benzothiazolyle, benzimidazolyle, benzoxazolyle, les groupes cycloalkyle en C3-C6, les groupes hétérocycliques choisis parmi pyrrolidinyle, dioxolanyle, pyrazolidinyle, pipéridyle, morpholinyle, pipérazinyle, tétrahydropyranyle et dioxanyle, et les groupes haloalkyl en Ci-Ce,

<Desc/Clms Page number 42>

alkyle (Ci-C6), alcoxy (Ci-C6), a) coxy (Ci-Ce) carbonyte, hydroxyaiky ! e en Ci-Ce et alcoxy (Ci-C6) alkyle (Ci-C6), ni = 0, 2ou 3etn2=0, 1 ou 2, R'1, R'2 et R'3 étant choisis indépendamment parmi l'hydrogène, les halogènes, les groupes nitro, cyano, COOH, OCF3, S02 (alkyleCi-C4), alkyle en Ci-C6), alcoxy en Ci-Ce, alcoxy (Ci-C6) carbonyle, acyl (Ci-C6) oxy, hydroxyalkyl en Ci-Ce. a) coxy (Ci-Co) carbony ! e, hydroxyaikyie en Ci-C6 et alcoxy (Ci-Ce) alkyle (Ci-C6). et leurs sels pharmaceutiquement acceptables.
2. Composés selon la revendication 1, dans lesquels R est choisi parmi les groupes :

les groupes aryle et hétéroaryle pouvant être substitués par un ou plusieurs groupes choisis indépendamment parmi les halogènes, les groupes nitro, cyano, -COOH, -OCF3, -SO2(alkyle en Ci-C4), alkyle en Ci-Ce, alcoxy en Ci- C6, alcoxy (C1-C6)carbonyle, acyl(C1-C6)oxy, hydroxyalkyle en C1-C6, alcoxy (Ci-C6) alkyle (Ci-Ce), et alkyl (Ci-Ce) carbonyle, les groupes cycloalkyle pouvant être substitués par un ou plusieurs groupes choisis indépendamment parmi les halogènes, les groupes nitro, cyano,
3. Composés selon la revendication 1, dans lesquels R est choisi parmi les groupes :

<Desc/Clms Page number 43>
4. Composés selon la revendication 3, de configuration S.

5. Composés selon la revendication 1, choisis parmi les composés suivants : 3- (N-benzyl-1, 2, 5, 6-tétrahydropyridin-4-yi)-5-phénoxyindole 3 [N-Benzyl piperidin-4-yl]-5-phenoxyindoie 5-phenoxy-3- (piperidin-4-yl)-indole 3 [N-methylpiperidin-4-yl]-5-phenoxyindole 5-phénoxy-3 (3-N-pipéridinylpropyl) indole 5-Phénoxy-3-[2- (piperazin-1-yl) éthyl]indole 5-phénoxy-3- [3- (piperazin-1-yl) propyl] indole 5-phenoxy-3-[2- (4-benzylpiperazin-1-yl) ethyl]indole 5-Phénoxy-3-[3- (4-benzylpiperazin-1-yl) propyl]indole 5-phénoxy-3 [2- (pyrrolidin-1-yl) éthyl] indole 5-phenoxy- [3- (pyrro) idin-1-y !)-propy !)] indo) e 5-phénoxy-3- [ (2S)- pyrrolidin-2-ylméthyl]-1 H-indole 3- { [ (2S)-1-benzylpyrrolidin-2-yl] méthyl}-5-phénoxy-1 H-indole, et leurs sels pharmaceutiquement acceptables.
6. Composition pharmaceutique comprenant une quantité efficace d'un composé selon l'une quelconque des revendications 1 à 5.
7. Utilisation d'un composé selon l'une quelconque des revendications 1 à 5, pour la fabrication d'un médicament antiinflammatoire.
8. Utilisation d'un composé selon l'une quelconque des revendications 1 à 5, pour la fabrication d'un médicament pour le traitement de l'insuffisance veineuse.
9. Utilisation d'un composé selon l'une quelconque des revendications 1 à 5, pour la fabrication d'un médicament destiné au traitement des pathologies hémorroïdaires.
<Desc/Clms Page number 44>

10. Utilisation d'un composé selon l'une quelconque des revendications 1 à 5, pour la fabrication d'un médicament destiné au traitement des pathologies urologiques.
11. Utilisation d'un composé selon l'une quelconque des revendications 1 à 5, pour la fabrication d'un médicament destiné au traitement de la douleur.
12. Utilisation d'un composé selon l'une quelconque des revendications 1 à 5, pour la fabrication d'un médicament destiné au traitement de la migraine.
13. Utilisation d'un composé selon l'une quelconque des revendications 1 à 5, pour la fabrication d'un médicament destiné au traitement des troubles cutanés.
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| FR0012052A FR2814166B1 (fr) | 2000-09-21 | 2000-09-21 | Derives 5-phenoxyindole et leurs applications therapeutiques |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| FR0012052A FR2814166B1 (fr) | 2000-09-21 | 2000-09-21 | Derives 5-phenoxyindole et leurs applications therapeutiques |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| FR2814166A1 true FR2814166A1 (fr) | 2002-03-22 |
| FR2814166B1 FR2814166B1 (fr) | 2005-07-01 |
Family
ID=8854545
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| FR0012052A Expired - Fee Related FR2814166B1 (fr) | 2000-09-21 | 2000-09-21 | Derives 5-phenoxyindole et leurs applications therapeutiques |
Country Status (1)
| Country | Link |
|---|---|
| FR (1) | FR2814166B1 (fr) |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2007009485A1 (fr) * | 2005-07-22 | 2007-01-25 | Pharma C S.A. | Derives de 8-phenoxy-y-carboline substitues a activite 5ht1 |
| JP2012526837A (ja) * | 2009-05-14 | 2012-11-01 | ジヤンセン・フアーマシユーチカ・ナームローゼ・フエンノートシヤツプ | ロイコトリエンa4加水分解酵素のモジュレーターとしての縮合二環式ヘテロアリール部分を2つ有する化合物 |
| WO2025104491A1 (fr) * | 2023-11-14 | 2025-05-22 | Mindset Pharma Inc. | Dérivés indoles utilisés en tant qu'agents sérotoninergiques utiles pour le traitement de troubles associés |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1993011106A1 (fr) * | 1991-11-25 | 1993-06-10 | Pfizer, Inc. | Derives d'indole |
| WO1994014770A1 (fr) * | 1992-12-21 | 1994-07-07 | Smithkline Beecham Plc | Analogues de tryptamine a titre d'agonistes semblables a 5-ht¿1? |
| WO1998006715A1 (fr) * | 1996-08-09 | 1998-02-19 | Smithkline Beecham Corporation | Nouveaux composes piperazine |
-
2000
- 2000-09-21 FR FR0012052A patent/FR2814166B1/fr not_active Expired - Fee Related
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1993011106A1 (fr) * | 1991-11-25 | 1993-06-10 | Pfizer, Inc. | Derives d'indole |
| WO1994014770A1 (fr) * | 1992-12-21 | 1994-07-07 | Smithkline Beecham Plc | Analogues de tryptamine a titre d'agonistes semblables a 5-ht¿1? |
| WO1998006715A1 (fr) * | 1996-08-09 | 1998-02-19 | Smithkline Beecham Corporation | Nouveaux composes piperazine |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2007009485A1 (fr) * | 2005-07-22 | 2007-01-25 | Pharma C S.A. | Derives de 8-phenoxy-y-carboline substitues a activite 5ht1 |
| JP2012526837A (ja) * | 2009-05-14 | 2012-11-01 | ジヤンセン・フアーマシユーチカ・ナームローゼ・フエンノートシヤツプ | ロイコトリエンa4加水分解酵素のモジュレーターとしての縮合二環式ヘテロアリール部分を2つ有する化合物 |
| WO2025104491A1 (fr) * | 2023-11-14 | 2025-05-22 | Mindset Pharma Inc. | Dérivés indoles utilisés en tant qu'agents sérotoninergiques utiles pour le traitement de troubles associés |
| US12606543B2 (en) | 2023-11-14 | 2026-04-21 | Mindset Pharma Inc. | Indole derivatives as serotonergic agents useful for the treatment of disorders related thereto |
Also Published As
| Publication number | Publication date |
|---|---|
| FR2814166B1 (fr) | 2005-07-01 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| KR100191193B1 (ko) | 6환 화합물 | |
| ZA200602051B (en) | 4-pyrimidone derivatives and their use as peptidyl peptidase inhibitors | |
| CN109678815B (zh) | N-苄基苯甲酰胺类衍生物及其制备方法与制药用途 | |
| EP3466962A1 (fr) | Dérivé de phénylpropanamide et procédé de fabrication et application pharmaceutique associée | |
| WO2016001441A1 (fr) | Dérivés de flavaglines | |
| EP3917926A1 (fr) | Dérivés d'imidazoquinoline amine, composition pharmaceutique, utilisation de ceux-ci | |
| JPH11509541A (ja) | 新規なガランタミン誘導体、その製造方法、その医薬としての用途及びそれを含有する医薬組成物 | |
| EP0226508A1 (fr) | Dérivés de l'indolo(3,2-c)quinoléine, leur procédé de préparation et leur activité antitumorale | |
| US6268375B1 (en) | 10, 11-difluoromethylenedioxycamptothecin compounds with topoisomerase I inhibition | |
| WO2014049133A1 (fr) | Nouveaux effecteurs allostériques positifs du récepteur nicotinique de l'acétylcholine | |
| CN102190658A (zh) | 一类抗肿瘤海洋天然产物ecteinascidins的结构类似物 | |
| RU2412166C1 (ru) | Цитотоксические линейные гетероциклические производные антрацендиона, содержащие в боковой цепи циклические диамины, активные в отношении опухолевых клеток с множественной лекарственной устойчивостью | |
| EP2880024A1 (fr) | Derives de griseofulvine | |
| CN119278034A (zh) | 选择性组蛋白去乙酰化酶8(hdac8)降解剂和其使用方法 | |
| FR2933701A1 (fr) | Derives anticancereux, leur preparation et leur application en therapeutique | |
| WO2012007619A1 (fr) | Procédé d'obtention de dérivés solubles dans l'eau de 20(s) camptothécine en tant qu'agents antitumoraux | |
| FR2496104A1 (fr) | Nouveaux produits de la serie de la 3-(piperidin-4-yl) 2h-indol-2-one, et procede pour leur preparation | |
| CN113439084B (zh) | 倍癌霉素类似物 | |
| EP0412899A2 (fr) | Dérivés d'oxazolo pyridines, leur procédé de préparation et les compositions pharmaceutiques qui les contiennent | |
| FR2814166A1 (fr) | Derives 5-phenoxyindole et leurs applications therapeutiques | |
| WO2004069843A1 (fr) | Derives d'azepines tricycliques, leur procede de preparation et les compositions pharmaceutiques qui les contiennent | |
| CN117500787A (zh) | 二甲基色胺的新型前药和缀合物 | |
| FR2827864A1 (fr) | Nouveaux derives de benzo[b]pyrano[3,2-h]acridin-7-one, leur procede de preparation et les compositions pharmaceutiques qui les contiennent | |
| CN108948003B (zh) | 作为mTOR抑制剂的吡嗪并[2,3-c]喹啉-2(1H)-酮类化合物的制备及用途 | |
| JP2003506376A (ja) | 1,2−ジヒドロ−1−オキソ−ピラジノ[1,2−a]インドール誘導体 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| ST | Notification of lapse |
Effective date: 20110531 |