FR3055337A1 - Methode de preparation de milieux de culture riches pour le marquage d'acides amines incorpores dans des proteines - Google Patents
Methode de preparation de milieux de culture riches pour le marquage d'acides amines incorpores dans des proteines Download PDFInfo
- Publication number
- FR3055337A1 FR3055337A1 FR1657906A FR1657906A FR3055337A1 FR 3055337 A1 FR3055337 A1 FR 3055337A1 FR 1657906 A FR1657906 A FR 1657906A FR 1657906 A FR1657906 A FR 1657906A FR 3055337 A1 FR3055337 A1 FR 3055337A1
- Authority
- FR
- France
- Prior art keywords
- labeled
- amino acid
- hydrolyzate
- medium
- methyl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Withdrawn
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B59/00—Introduction of isotopes of elements into organic compounds ; Labelled organic compounds per se
- C07B59/008—Peptides; Proteins
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K1/00—General methods for the preparation of peptides, i.e. processes for the organic chemical preparation of peptides or proteins of any length
- C07K1/13—Labelling of peptides
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12P—FERMENTATION OR ENZYME-USING PROCESSES TO SYNTHESISE A DESIRED CHEMICAL COMPOUND OR COMPOSITION OR TO SEPARATE OPTICAL ISOMERS FROM A RACEMIC MIXTURE
- C12P21/00—Preparation of peptides or proteins
- C12P21/06—Preparation of peptides or proteins produced by the hydrolysis of a peptide bond, e.g. hydrolysate products
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Health & Medical Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Zoology (AREA)
- Genetics & Genomics (AREA)
- Molecular Biology (AREA)
- Biochemistry (AREA)
- Engineering & Computer Science (AREA)
- Wood Science & Technology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Microbiology (AREA)
- Biotechnology (AREA)
- General Engineering & Computer Science (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Analytical Chemistry (AREA)
- Biophysics (AREA)
- Medicinal Chemistry (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Preparation Of Compounds By Using Micro-Organisms (AREA)
Abstract
La présente invention concerne une méthode de préparation d'un hydrolysat de microorganisme dont au moins un acide aminé et/ou un précurseur d'acide aminé est marqué, comprenant une étape de d'obtention d'un hydrolysat de microorganisme en solution dans lequel tous les acides aminés se trouvent sous la forme libre, et une étape de remplacement dans cet hydolysat d'au moins un acide aminé et son cétoacide s'il est présent par le même acide aminé mais sous une forme marquée ou par son précurseur marqué. La présente invention concerne également des mélange d'enzymes pour mettre en œuvre cette méthode.
Description
Titulaire(s) : COMMISSARIAT A L'ENERGIE ATOMIQUE ET AUX ENERGIES ALTERNATIVES Etablissement public.
Demande(s) d’extension
Mandataire(s) : GEVERS & ORES Société anonyme.
METHODE DE PREPARATION DE MILIEUX DE CULTURE RICHES POUR LE MARQUAGE D'ACIDES AMINES INCORPORES DANS DES PROTEINES.
FR 3 055 337 - A1 (5/J La présente invention concerne une méthode de préparation d'un hydrolysat de microorganisme dont au moins un acide aminé et/ou un précurseur d'acide aminé est marqué, comprenant une étape de d'obtention d'un hydrolysat de microorganisme en solution dans lequel tous les acides aminés se trouvent sous la forme libre, et une étape de remplacement dans cet hydolysat d'au moins un acide aminé et son cétoacide s'il est présent par le même acide aminé mais sous une forme marquée ou par son précurseur marqué. La présente invention concerne également des mélange d'enzymes pour mettre en oeuvre cette méthode.

Méthode de préparation de milieux de culture riches pour le marquage d’acides aminés incorporés dans des protéines
La présente invention concerne la préparation de milieux de culture riche et leur utilisation pour le marquage des protéines produites in vitro par une cellule procaryote ou eucaryote, ou dans un système acellulaire, notamment le marquage spécifique des groupements méthyles des acides aminés à chaîne ramifiée de ces protéines.
L’étude des protéines de taille supérieure à plusieurs kilodaltons par résonance magnétique nucléaire (RMN) nécessite le remplacement des atomes de carbone-12, hydrogène, azote-14 par des isotopes qui ne sont pas naturellement abondants, tels que les isotopes carbone-13 (I3C), hydrogène-2 (2H ou D) et azote-15 (15N).
Le marquage isotopique spécifique des atomes de carbone et d’hydrogène des groupements méthyles des acides aminés à chaîne ramifiée dans le contexte de la production in vitro de protéines entièrement deutérées est réalisé principalement selon deux méthodes. Selon une première méthode, un milieu de culture minimum (milieu minimum), qui ne comprend que du glucose deutéré comme source carboné et du sulfate (ou chlorure) d'ammonium comme source d'azote dans du D2O, est utilisé. A ce milieu perdeutéré est additionné soit l'acide aminé marqué sur au moins un groupement méthyle soit un précurseur métabolique de cet acide aminé également marqué sur au moins un groupement méthyle. Selon une deuxième méthode, à ce milieu de culture est ajouté un mélange d'acides aminés deutérés issu de l'hydrolyse d’organismes unicellulaires tels que microalgues, bactéries ou levures et auquel sont additionnés les acides aminés souhaités marqués sur au moins un groupement méthyle.
L'inconvénient majeur des méthodes de marquage en milieu de culture minimum est que certaines protéines, en particulier celles d'origine eucaryote, ne sont que très faiblement exprimées voire ne sont pas exprimées du tout par les cellules bactériennes. Ces faibles niveaux de production empêchent alors l’étude de ces protéines. L'utilisation de milieux de culture riches - obtenus à partir d’un hydrolysat de microorganisme - avec addition d’acides aminés marqués présente l’inconvénient majeur que pour obtenir un enrichissement en acides aminés marqués à plus de 90 % de la protéine produite, compatible avec certaines expériences de RMN, telles que les expériences NOESY (Nuclear Overhauser Effect SpectroscopY), il faut que l’acide aminé non marqué présent dans le milieu soit dilué au moins d'un facteur 10 par l'ajout de précurseur ou d'acide aminé marqués. Dans ce cas, par exemple, futilisation d'un gramme d’hydrolysat d'algues ou de levures nécessitera au moins de l'ordre de 200 milligrammes d'isoleucine (Ile) marqué pour un marquage isotopique de l’isoleucine, de l'ordre de 500 mg de valine (Val) marqué pour un marquage isotopique de la valine et pour la leucine (Leu) de l'ordre de 600 mg pour un marquage isotopique de la leucine. Dans le cas de la valine, la leucine et de l'isoleucine, des acides aminés marqués stéréo- ou régio-sélectivement comme leurs cétoacides associés sont très coûteux. Pour l'alanine par exemple, Futilisation d’I gramme d’hydrolysat, en tant que supplémentation pour un milieu de culture riche, nécessitera de l'ordre du gramme d'alanine (Ala) marqué pour un marquage isotopique de l’alanine à plus de 90 % de marquage, ce qui est également coûteux. Le coût des acides aminés marqués nécessaires rend donc cette méthode extrêmement onéreuse. De plus, l'utilisation de précurseurs d’acide aminé marqués, du type acétolactate ou 2-cétobutyrate, utilisé dans des milieux minimum, ne permet pas, dans des milieux supplémentés avec des hydrolysats issus de microorganismes, d'obtenir des marquages à plus de 50 % quelle que soit la quantité de précurseurs utilisée. Enfin, constituer un mélange d'acides aminés deutérés en y rajoutant soit l'acide aminé marqué soit le précurseur marqué diminuerait le coût en l'acide aminé marqué mais cela serait compensé par celui des acides aminés deutérés. En outre, la plupart des autres acides aminés (que valine, leucine et isoleucine) ne sont pas ou entièrement deutérés ou disponibles.
Dans le cadre de leurs travaux, les Inventeurs se sont donnés pour objectif de diminuer le coût de préparation des milieux de culture deutérés supplémentés avec des hydrolysats de micro organisme (e.g., microalgues, bactéries ou levures) pour permettre le marquage spécifique de groupements méthyle dans le contexte de production in vitro de protéines entièrement deutérées, tout en augmentant le rendement de production (d’expression) des protéines perdeutérées et marquées stéréo-spécifiquement ou régio-sélectivement BC,H sur les méthyles des acides aminés à chaîne ramifiée et l'alanine.
Les Inventeurs ont utilisé comme source d'acides aminés pour les milieux de culture riche, un hydrolysat perdeutéré issu de la culture de microalgues en milieu deutéré. Us ont ensuite éliminé de cet hydrolysat les acides aminés leucine, valine, isoleucine d’une part ou alanine d’autre part, pour les remplacer par les acides aminés Leu, Val, Ile, Ala spécifiquement marqués ou par leurs précurseurs. Pour cela, plusieurs étapes ont été effectuées :
conversion totale de l'hydrolysat en acides aminés libres, dégradation des acides aminés méthylés Leu, Val, Ile d’une part ou Ala d’autre part présents dans cet hydrolysat ou de leur combinaison en leurs cétoacides associés par Putilisation d'enzymes spécifiques,
- élimination de ces cétoacides par conjugaison enzymatique, décarboxylation enzymatique ou spontanée ou par réduction chimique en hydroxyacides, complémentation du milieu constitué avec cet hydrolysat en l'acide aminé désiré (ou le précurseur) convenablement marqué, à un taux d'enrichissement supérieur à 50 % ou en une combinaison de ces acides aminés marqués (ou des précurseurs).
Le milieu marqué ainsi préparé permet d'une part, de produire en culture cellulaire (e.g., culture bactérienne) des protéines marquées de façon stéréo- ou régio-spécifique sur les groupements méthyle des chaînes ramifiées des acides aminés Leu, Val, Ile et Ala dont l'expression dans la fraction soluble est très faible sinon nulle en milieu minimum, et d'autre part, d'obtenir en général de meilleurs rendements d'expression et donc à production égale de baisser le coût de la préparation comme de supprimer les phases d'adaptation des bactéries en milieu minimum deutéré et donc de raccourcir le temps nécessaire à la production des protéines.
La présente invention a par conséquent pour objet une méthode de préparation d’un hydrolysat de microorganisme dont au moins un acide aminé et/ou un précurseur d’acide aminé est marqué, comprenant les étapes suivantes :
a) obtenir un hydrolysat de microorganisme en solution dans lequel tous les acides aminés se trouvent sous la forme libre,
b) remplacer dans l’hydolysat obtenu à l’étape a) au moins un acide aminé sous la forme L et son cétoacide s’il est présent par le même acide aminé mais sous une forme marquée ou par son précurseur marqué.
Selon un mode de mise en œuvre avantageux de la méthode de préparation d’un hydrolysat de microorganisme, l’étape b) peut être mise en œuvre selon les étapes suivantes :
bl) appauvrir l’hydrolysat obtenu à l’étape a) en au moins un acide aminé sous la forme L et son cétoacide s’il est présent, par élimination de l’hydrolysat dudit acide aminé et de son cétoacide, et b2) ajouter à l’hydrolysat appauvri obtenu à l’étape bl) ledit acide aminé éliminé mais sous une forme marquée et/ou son précurseur marqué.
La présente invention a aussi objet une méthode de préparation d’un milieu de culture riche, de préférence un milieu de culture riche pour la culture bactérienne, dont au moins un acide aminé et/ou un précurseur d’acide aminé est marqué, comprenant les étapes suivantes :
i) préparer un hydrolysat de microorganisme dont au moins un acide aminé et/ou un précurseur d’acide aminé est marqué selon l’une des deux méthodes de l’invention décrites cidessus, et
j) constituer un milieu de culture riche comprenant l’hydrolysat appauvri obtenu à l’étape i) et un milieu de culture minimum.
La présente invention a aussi objet une méthode de préparation d’un milieu de culture riche dont au moins un acide aminé et/ou un précurseur d’acide aminé est marqué, comprenant les étapes suivantes :
a) obtenir un hydrolysat de microorganisme en solution dans lequel tous les acides aminés se trouvent sous la forme libre, bl) appauvrir l’hydrolysat obtenu à l’étape a) en au moins un acide aminé sous la forme L et son cétoacide s’il est présent, par élimination de l’hydrolysat dudit acide aminé et de son cétoacide,
c) constituer un milieu de culture comprenant l’hydrolysat appauvri obtenu à l’étape b 1) et un milieu de culture minimum, mais ne contenant pas ledit acide aminé et son cétoacide éliminés,
d) ajouter au milieu de culture obtenu à l’étape c) ledit acide aminé éliminé mais sous une forme marquée et/ou son précurseur marqué de manière à obtenir ledit milieu de culture riche.
On entend par « milieu de culture riche » ou « milieu riche » au sens de la présente invention, un milieu liquide ou solide qui contient tous les éléments nécessaires à la croissance d’une cellule, notamment à la croissance de la plupart des bactéries. Le milieu de culture riche contient une source de carbone qui est généralement du glucose ou de l'acétate ou du glycérol, une source d’azote généralement sous forme minérale, de Peau, des sels minéraux nécessaires pour la croissance de la cellule (e.g, la croissance bactérienne) et une source d'acides aminés. Le milieu de culture riche permet la culture de cellules procaryote ou eucaryote, telles que les cellules de bactérie, levure, champignon, cellules d’insecte ou de mammifères. La source d'acides aminés du milieu de culture riche peut être un hydrolysat de levure, d’algues ou de bactéries, de préférence un hydrolysat d’algues. Par extension, on entend également par « milieu de culture riche » ou « milieu riche » au sens de la présente invention, un milieu liquide ou solide qui contient tous les éléments nécessaires à la production in vitro d’une protéine recombinante dans un système acellulaire.
On entend par « milieu de culture minimum » ou « milieu minimum » au sens de la présente invention, un milieu liquide ou solide qui contient uniquement les éléments essentiels à la croissance d’une cellule, c.à.d. une source de carbone qui est généralement du glucose (ou de l'acétate ou du glycérol), une source d'azote généralement sous forme minérale, de l'eau et des sels minéraux nécessaires pour la croissance de la cellule (e.g. la croissance bactérienne). Avantageusement, le milieu minimum est le milieu M9 (Sambrook et al., 1989) ou un milieu similaire, tel que ceux connus de l’homme de l’art (https://wikipédia.ors/wiki/milieu de culture #Notion de milieu minimum).
On entend par un « hydrolysat de microorganisme en solution » un hydrolysat obtenu par hydrolyse des protéines d’une culture de microorganisme. Le microorganisme peut être une levure, une algue, ou une bactérie, de préférence une algue.
Selon un mode de mise en œuvre avantageux de la méthode, l’hydrolysat de microorganisme en solution obtenu à l’étape a) est perdeutéré et/ou enrichi en carbone-13 (13C) et/ou en azote-15 (1SN), c’est-à-dire qu’au moins 90%, et par ordre de préférence croissant au moins 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98% et 99%, et de préférence encore 100% des atomes d’hydrogène (H) dudit hydrolysat sont remplacés par des atomes de deutérium (D) et/ou au moins 90%, et par ordre de préférence croissant au moins 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98% et 99%, et de préférence encore 100% des atomes de carbone dudit hydrolysat sont des atomes 13C et/ou au moins 90%, et par ordre de préférence croissant au moins 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98% et 99%, et de préférence encore 100% des atomes d’azote sont des atomes I5N respectivement.
L’obtention d’hydrolysats de microorganisme perdeutérés et/ou enrichis en carbone-13 et/ou en azote-15 est bien connue de l’homme du métier. Ces hydrolysats de microorganisme sont généralement obtenus par culture d’un microorganisme sur un milieu deutéré et/ou enrichi en carbone-13 et/ou en azote-15, et hydrolyse des microorganismes ainsi produits. Des hydrolysats pour les milieux riches perdeutérés sont disponibles commercialement, tels que les hydrolysats Celtone® (Cambridge Isotop Laboratories), Isogro® (Sigma-Aldrich) et OD® media (Silantes).
Lorsqu’un hydrolysat perdeutéré et/ou enrichi en carbone-13 et/ou en azote-15 est utilisé à l’étape a) alors l’acide aminé est remplacé à l’étape b) ou d) par un acide aminé ou son précurseur qui est marqué différemment que par un deutérium, carbone-13 et/ou en azote-15 respectivement.
En d’autres termes, lorsque l’hydrolysat utilisé à l’étape a) est perdeutéré (et donc les acides aminés ou leurs précurseurs contenus dans cet hydrolysat sont perdeutérés) l’acide aminé ajouté à l’étape b) est marqué différemment de l’acide aminé présent à l’étape a) et enlevé (remplacé).
Un hydrolysat dans lequel tous les acides aminés se trouvent sous la forme libre, est un hydrolysat dans lequel au moins 90%, et par ordre de préférence croissant au moins 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98% et 99%, et de préférence encore 100% des acides aminés se trouvent sous la forme libre.
Les hydrolysats de microorganisme disponibles commercialement sont généralement issus d'une hydrolyse incomplète des protéines et peptides de microorganismes et peuvent comprendre, outre des acides aminés sous la forme libre, des oligopeptides. Ces oligopeptides peuvent présenter l’inconvénient qu’ils pourraient être pris en charge par les machineries de dégradation cellulaire, conduisant à la libération d’acides aminés à l’intérieur de la cellule. Ce mécanisme pourrait alors conduire à la dilution du marquage des acides aminés lors de la synthèse in vitro des acides aminés.
Selon un mode de mise en œuvre avantageux de la méthode, l’hydrolysat de microorganismes en solution est traité par un mélange de protéases préalablement à l’étape b), de manière à hydrolyser complètement les protéines et peptides dudit milieu (c’est-à-dire au moins 90% des acides aminés se trouvant sous la forme libre après hydrolyse) tout en évitant la racémisation des acides aminés. Le mélange de protéases comprend avantageusement une protéase neutre, une chymotrypsine, une trypsine, une carboxypeptidase, une aminopeptidase, ainsi que des phosphatases neutres et alcalines. Un exemple d’un tel mélange de protéases est la Pronase® (Sigma-Aldrich), qui est issue des fluides extracellulaires de Streptomyces griseus, disponible commercialement. L’utilisation du mélange Pronase® pour hydrolyser complètement les protéines et peptides d’un milieu est décrite par D’Aniello et al., 1993.
Le mélange de protéases (e.g., la Pronase®) est ensuite inactivée et/ou éliminée de l’hydrolysat traité. L’inactivation du mélange de protéases présent dans l’hydrolysat traité et son élimination de l’hydrolysat traité peuvent être effectuées par toute méthode connues de l’homme du métier, L’hydrolysat traité peut subir un choc thermique de manière à inactiver le mélange de protéases, par exemple, pendant une demi-heure à une température de 85°C puis centrifugation. L’élimination du mélange de protéases peut être aussi effectuée par centrifugation sur une membrane d'ultrafiltration.
Un cétoacide est un composé organique portant à la fois une fonction carboxylique et une fonction cétone. On entend par un « cétoacide d’un acide aminé », le 2-cétoacide qui est associé à un acide aminé, où la fonction cétone est portée par le carbone voisin de celui portant la fonction carboxylique. A titre d’exemple, le cétoacide de la L-leucine est le 4-méthyl-2-oxopentanoate ou cétoisocaproate (KIC), le cétoacide de la L~valine est le 3-méthyl-2-oxobutanoate ou cétoisovalerate (KIV), le cétoacide de la L-isoleucine est le 3-méthyl-2-oxopentanoate (MOP), le cétoacide du L-glutamate est l’oxoglutarate, le cétoacide de la L-alanine est le pyruvate.
On entend par un « précurseur d’un acide aminé », un précurseur métabolique d’un acide aminé. Un précurseur d'un acide aminé est un composé organique apparaissant préalablement à l’acide aminé dans sa chaîne de biosynthèse et converti en cet acide aminé par une succession de réactions enzymatiques. Par exemple, le pyruvate est le précurseur pour l'alanine, qui est converti en l’alanine en une seule réaction enzymatique. L'acétolactate, le dihydroisovalérate ou le cétoisovalérate (3-méthyl-2-oxobutanoate) sont des précurseurs pour la valine et la leucine.
On entend par « appauvrir un hydrolysat en au moins un acide aminé et son cétoacide », un hydrolysat dans lequel au moins 90%, et par ordre de préférence croissant au moins 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98% et 99%, et de préférence encore 100% d’un acide aminé et de son cétoacide ont été éliminés.
L’appauvrissement d’un hydrolysat en au moins un acide aminé et éventuellement de son cétoacide selon l’étape bl) de la méthode selon la présente invention peut être effectué par différentes méthodes bien connues de l’homme du métier, telles que par chromatographie liquide puis reconstitution de l’hydrolysat appauvri en excluant un ou plusieurs acides aminés.
Selon un mode de mise en œuvre préféré de la méthode, l’hydrolysat obtenu à l’étape a) est appauvri en L-leucine, L-valine, L-isoleucine et leurs cétoacides respectifs s’ils sont présents (z.e., 4-inéthyl-2-oxopentanoate, 3-méthyl-2-oxobutanoate et 3-méthyl-2-oxopentanoate respectivement).
Selon un mode de mise en œuvre avantageux de ce mode de mise en œuvre préféré, l’hydrolysat obtenu à l’étape a) est traité par un mélange de trois enzymes constitué d’une isoleucine 2-épimérase (EC 5.1.1.21), une D-aminoacide oxydase (EC 1.4.3.3) et une catalase (EC 1,11.1.6). Avantageusement, l’isoleucine 2-épimérase, la D-aminoacide oxydase et la catalase sont ajoutées au milieu de culture simultanément. Ces trois enzymes sont ensuite éliminées de l’hydrolysat traité, éventuellement en ayant été inactivées préalablement à leur élimination.
L’isoleucine 2-épimerase catalyse réversiblement la conversion de la L-isoleucine - forme (2S,3S) - en D-allo-isoleucine - forme (2R,3S) -, l’isoleucine présentant 2 carbones asymétriques en positions 2 et 3. Cette enzyme convertit également réversiblement les formes L de la leucine, la valine, la méthionine, la phénylalanine en formes D, La D-amino oxydase transforme les acides aminés de la forme D en leurs cétoacides avec production d'eau oxygénée. La catalase dismute Peau oxygénée en oxygène moléculaire et en eau. Les réactions enzymatiques sont présentées ci-dessous :
L-leucine > D-leucine —» 4-méthyl-2-oxopentanoate (KIC)
L-valine D-valine —+ 3-méthyl-2-oxobutanoate (KIV)
L-isoleucine —> D-allo-isoleucine —* 3-méthyl-2-oxopentanoate (MOP)
Les D-aminoacide oxydases (EC 1.4.3.3) et catalases (EC 1.11.1.6) sont bien connues de l’homme du métier. Elles peuvent être d’origine procaryote - par exemple bactérienne - ou eucaryote, de préférence eucaryote. La D-AAO et la catalase sont disponibles commercialement. L’isoleucine 2-épimérase (EC 5.1.1.21) appartient à la famille des racémases dont le mode d'action est bien connu de l’homme du métier (Mutaguchi et al., 2013). Les réactions enzymatiques de ces enzymes sont également bien connues de l’homme du métier et dépendent de cofacteurs et d’effecteurs également bien connues de l’homme du métier : pyridoxal 5'phosphate (PLP), flavine adénine dinucléotide (FAD), pH généralement 7,5, tampon (e.g., tampon tris(hydroxymétyl)aminométhane ; Tris), température généralement 37°C.
L’isoleucine 2-épimérase peut être choisie parmi celles qui ont été identifiées chez les Lactobacillus, les Leuconostoc, les Oenococcus, les Weissella , les enterocoques et les streptocoques. Selon un mode de mise en œuvre préféré, l’isoleucine 2-épimérase est celle de
Lactobacillus buchneri JCM 1115, décrite par Mutaguchi et al. 2013, et dont la séquence d’acides aminés est disponible dans la base de données UniProt sous le numéro d’accession
M1GRN3.
La D-amino oxydase peut être choisie parmi les différentes protéines possédant une activité D-amino acide oxydase et issues d’organismes procaryotes ou eucaryotes (Pollegioni, L, et al. «. Physiological fonctions of D-amino oxidases : from yeast to humans » Cell. Mol. Life Sci (2007) 64: 1373). Selon un mode de mise en œuvre préféré, la D-amino oxydase est celle issue du rein de porc dont la séquence d’acides aminés est disponible dans la base de données UniProt sous le numéro d’accession P00371, et qui est également disponible commercialement.
La catalase peut être choisie parmi les trois familles de protéines possédant une activité catalase et issues d’organismes procaryotes (bactéries et archées) ou eucaryotes (Archives of Biochemistry and Biophysics. Volume 525, Issue 2, Pages 93-222 (2012). Catalasas and Hydrogen Peroxide Metobolism. (Spécial Issue). Edited by Christian Obinger). La première famille est mono-fonctionelle, dont le cofacteur est un hème et qui se trouve chez les eucaryotes (voir la base de données InterPro, numéro d’accession IPR018028), La seconde a comme cofacteur un hème et présente des activités péroxidase/catalases (voir la base de données InterPro, numéro d’accession IPR000763). Ces enzymes sont proches des péroxydases de plantes. La troisième contient du manganèse non-heminique et est trouvée chez les bactéries (voir la base de données InterPro, numéro d’accession IPR007760). Selon un mode de mise en œuvre préféré, la catalase est une catalase de la famille IPR018028. Selon un mode de mise en œuvre plus particulièrement préféré, la catalase est celle issue du foie de bœuf dont la séquence d’acides aminés est disponible dans la base de données UniProt sous le numéro d’accession P00432, et qui est également disponible commercialement.
L’inactivation d’enzymes présentes dans l’hydrolysat et leur élimination de cet hydrolysat peuvent être effectuées par toute méthode connue de l’homme du métier. L’hydrolysat peut subir un choc thermique de manière à inactiver les enzymes, par exemple, pendant une demi-heure à une température de 85°C. L’élimination des enzymes peut être effectuée par centrifogation sur membrane d’ultrafiltration.
Les cétoacides présents dans l’hydrolysat obtenu à l’étape a) et/ou produits lors du traitement enzymatique de l’hydrolysat peuvent être éliminés selon plusieurs méthodes bien ίο connues de l’homme du métier, telles que par dérivatisation chimique (Pailla et al., 2000), réduction chimique (Saegusa et al., 1977) ou décarboxylation enzymatique (Smit et al., 2005).
De préférence, les cétoacides sont éliminés de l’hydrolysat par réduction chimique des cétoacides en hydroxyacides par du borohydrure de sodium. Ce réactif est couramment utilisé pour ce type de réaction de réduction, et présente l’avantage qu’il est peu coûteux et qu’en outre la quantité nécessaire à la réduction des cétoacides présents n'est pas suffisamment importante pour diluer le marquage deutérium de façon significative. Les réductions chimiques sont présentées ci-dessous :
4-méthyl-2-oxopentanoate (KIC) —» 4-méthyl-2-hydroxypentanoate (HIC)
3-méthyl-2-oxobutanoate (KIV) —» 3-méthyl-2-hydroxybutanoate (HIV)
3-méthyl-2-oxopentanoate (MOP) —> 3-méthyl-2~hydroxypentanoate (MHP)
Selon un autre mode de mise en œuvre préféré de la méthode, l’hydrolysat obtenu à l’étape 10 a) est appauvri en L-alanine et son cétoacide s’il est présent.
Une méthode pour éliminer l’alanine d’un milieu liquide est décrite dans la Demande de Brevet CN 101824443.
Selon un mode de mise en œuvre avantageux de ce mode de mise en œuvre préféré, l’hydrolysat obtenu à l’étape a) est traité par un mélange de quatre enzymes constitué d’une alanine racémase (EC 5.1.1.1), une D-amino acide oxydase (D-AAO ; EC 1.4,3.3), une catalase (EC 1.11.1.6) et une acétohydroxyacide synthase (EC 2.2.1.6). L’alanine racémase, la D-amino acide oxydase et la catalase sont ajoutées simultanément. L’acétohydroxyacide synthase peut être ajoutée simultanément ou postérieurement aux 3 autres enzymes.
L’alanine racémase (EC 5.1.1.1) convertit la L-alanine en D-alanine. La D-alanine est ensuite désaminée et transformée en pyruvate par une D-amino acide oxydase (EC 1.4.3.3). Le pyruvate formé est ensuite condensé en acétolactate par Γacétohydroxyacide synthase (ou acétolactate synthase; AHAS II) (EC 2.2.1.6). Enfin, l'acétolactate formé est éliminé, par exemple par chauffage - entre 35°C et 55°C, de préférence à 40°C - en se transformant en acétoïne (3-hydroxybutanone) ou en diacétyle (butane-2,3-dione). Les réactions catalysées par la
D-AAO produisant du peroxyde d’hydrogène, une catalase (EC 1.11.1.6 ) est ajoutée au milieu pour l’éliminer.
Les enzymes ajoutées sont ensuite éliminées du milieu de culture traité, éventuellement en ayant été inactivées préalablement à leur élimination, tel que décrit ci-dessus.
Les alanine racémases (EC 5.1.1.1), D-amino acide oxydases (EC 1.4.3.3), catalases (EC 1.11.1.6) et acétohydroxyacide synthases (EC 2.2.1,6 ) sont bien connues de l’homme du métier. Elles peuvent être d’origine procaryote - par exemple bactérienne - ou eucaryote, de préférence procaryote pour l’alanine racémase et l’acétohydroxyacide synthase, de préférence eucaryote pour la D-amino acide oxydase et la catalase. La D-amino acid oxydase et la catalase sont disponibles commercialement. Les réactions enzymatiques de ces enzymes sont également bien connues de l’homme du métier et dépendent de cofacteurs et d’effecteurs également bien connues de l’homme du métier : pyridoxal phosphate, thiamine pyrophosphate, flavine adénine dinncléotide, pH généralement 7,5, tampon (e.g., tampon tris(hydroxymétyl)arninométhane ; Tris), température généralement 37°C.
L’alanine racémase peut être choisie parmi la famille des alanines racémases disponibles dans la base de données InterPro sous le numéro d’accession IPR00082. Selon un mode de mise en œuvre préféré, l’alanine racémase est celle de Geobacillus stearothermophilus (Shaw et al., 1997), dont la séquence d’acides aminés est disponible dans la base de données UniProt sous le numéro d’accès P10724.
La D-amino acide oxydase peut être choisie parmi la famille des D-amino acide oxydases disponibles dans la base de données InterPro sous le numéro d’accession IPR023209. Selon un mode de mise en œuvre préféré, la D-amino acide oxydase provient du rein de porc, dont la séquence d’acides aminés est disponible dans la base de données UniProt sous le numéro d’accès P0037, qui est disponible commercialement.
L’acétohydroxyacide synthase peut être choisie parmi la famille des acétohydroxyacide synthases disponibles dans la base de données InterPro sous le numéro d’accession IPR012846. Selon un mode de mise en œuvre préféré, l’acétohydroxyacide synthase est l'acétohydroxyacide synthase de type II d’Escherichia coli (Barack et al., 1987), dont la séquence d’acides aminés est disponible dans la base de données UniProt sous le numéro d’accès P00892.
La catalase est décrite ci-dessus.
Aux étapes b), b2) et d) de la méthode selon la présente invention, l’acide aminé (sous forme L) et éventuellement son cétoacide éliminés sont remplacés par le même acide aminé maïs sous une forme marquée ou par son précurseur marqué.
On entend par « acide aminé sous une forme marquée » (ou acide aminé marqué) ou par « précurseur marqué » au sens de la présente invention, un acide aminé ou un précurseur d’un acide aminé qui est enrichi en un isotope du carbone, hydrogène et/ou azote, tel que par exemple les isotopes carbone-13 (13C), hydrogène-2 (3H) et azote-15 (15N).
Les acides aminés marqués et leurs précurseurs marqués sont bien connus de l’homme du métier (Gardner et Kay, 1998 ; Ruschak et Kay, 2010 ; Kerfah et al., 2015). Ils sont disponibles commercialement. Des procédés pour marquer les groupements méthyle des valine, leucine et isoleucine sont décrits dans la Demande Internationale WO 2011/083356. Des procédés pour marquer des dérivés cétoacides alpha-hydroxy sont décrits dans la Demande Internationale WO 2014/024151.
Selon un mode de mise en œuvre particulier, les acides aminés et précurseurs comprenant un groupement méthyle sont marqués 13C,H sur un groupement méthyle soit (l3CH3, I3CH2D, 13CHD2 et bCD3).
Lorsque plusieurs acides aminés et éventuellement leurs cétoacides ont été éliminés de l’hydrolysat, un ou plusieurs acides aminés marqués correspondant aux acides aminés éliminés et/ou leurs précurseurs marqués peuvent être ajoutés à l’hydrolysat ou au milieu de culture constitué. Les acides aminés éliminés de l’hydrolysat qui ne sont pas remplacés par le même acide aminé mais sous une forme marquée ou par son précurseur marqué, sont remplacés par le même acide aminé non marqué et/ou son précurseur non marqué. Par exemple, lorsque la Lleucine, la L-valine et la L-isoleucine et leurs cétoacides respectifs sont éliminés de l’hydrolysat, alors ces trois acides aminés sous une forme marquée et/ou leurs précurseurs marqués peuvent être ajoutés à l’hydrolysat ou au milieu de culture constitué, ou bien seule l’isoleucine marquée et/ou son précurseur marqué sont ajoutés et la L-leucine et la L-valine non marqués sont également ajoutées, ou bien la L-isoieucine non marquée est ajoutée et la L-leucine et la L-valine marquées et/ou leurs précurseurs marqués sont également ajoutés.
Selon un mode de réalisation particulier de l’invention, lorsque l’hydrolysat obtenu à l’étape a) est appauvri en L-isoleucine et son cétoacide, le milieu appauvri obtenu à l’étape bl) ou le milieu de culture obtenu à l’étape c), de préférence le milieu de culture obtenu à l’étape c), est additionné d’un, précurseur de l’isoleucine choisi parmi le 2-oxobutanoate (pour le marquage des méthyles delta-1), le 2-hydroxy~2-éthyl-3-oxobutanoate (pour le marquage des méthyles gamma2 ou pour le marquage des méthyles delta-1), le 2,3-dihydroxy-3-méthylpentanoate (pour le marquage des méthyles gamma-2 ou pour le marquage des méthyles delta-1) et/ou le 3-méthyl-2oxopentanoate (pour le marquage des méthyles gamma-2 ou pour le marquage des méthyles delta-1). Le milieu de culture obtenu à l’étape c) peut aussi être additionné d'isoleucine marquée. Il peut également être additionné de L-isoleucine marquée pour le marquage des méthyles delta-1 ou gamma-2.
Selon un autre mode de réalisation particulier, lorsque l’hydrolysat obtenu à l’étape a) est appauvri en L-leucine et L-valine, le milieu appauvri à l’étape bl) ou le milieu de culture obtenu à l’étape c), de préférence le milieu de culture obtenu à l’étape c), est additionné d'un précurseur de la leucine et de la valine, choisi parmi le 2-hydroxy-2-méthyl~3~oxobutanoate (pour le marquage stéréospécifique des méthyles proR, proS), le 3-hydroxy-3-méthyl-2-oxobutanoate, le 2,3-dihydroxy-3-méthylbutanoate et/ou le 3-méthyl-2-oxobutanoate (cétoisovalérate) (pour un marquage astéréospécifique des méthyles), en présence ou non de L-leucine deutérée si le marquage des méthyles des valines est uniquement souhaité. Ce milieu peut être additionné de 4méthyl-2-oxopentanoate (cétoïsocaproate) pour le marquage uniquement des leucines. Ce milieu peut aussi être additionné de valine ou de leucine marquées. Il peut également être additioné de L-Valine et/ou L-Leucine marquée(s) pour le marquage des méthyles proS ou proR.
Selon un mode de réalisation particulier de l’invention, lorsque l'hydrolysat obtenu à l’étape a) est appauvri en L-alanine et son cétoacide, le milieu appauvri obtenu à l’étape bl) ou le milieu de culture obtenu à l’étape c), de préférence le milieu de culture obtenu à l’étape c), est additionné L-alanine marquée par le carbone-13 sur le groupement méthyle.
La quantité optimale d’acide aminé ou de précurseur marqués nécessaire pour obtenir une incorporation à plus de 90 % dans une protéine exprimée in vitro par une cellule peut être déterminée par une série de cultures dans lesquelles des quantités croissantes de cet acide aminé ou précurseur marqué sont ajoutées une heure avant induction pour des concentrations finales de 0, 100, 300, 400, 600, et 800 mg/L. Le niveau d’incorporation peut être estimée dans la protéine purifiée par des expériences 2D ,3C-HSQC.
A l’étape j) de la méthode selon l’invention, le milieu de culture riche peut être sous la forme liquide ou lyophilisée. L’hydrolysat appauvri obtenu à l’étape i) sous une forme lyophilisée peut être ajouté à un milieu minimum sous la forme liquide ou lyophilisée. L’hydrolysat appauvri obtenu à l’étape i) sous une forme liquide peut aussi être ajouté à un milieu minimum sous une forme liquide ou lyophilisée.
La présente invention concerne également un hydrolysat de microorganisme ou un milieu de culture riche comprenant un hydrolysat de microorganisme, dans lequel au moins un acide aminé et son céioacide s’il est présent sont remplacés à au moins 90%, et par- ordre de préférence croissant au moins 93%, 92%, 93%, 94%, 95%, 96%, 97%, 98% et 99%, et de préférence encore 100%, par le même acide aminé ou son précurseur mais sous une forme marquée, tels que défini ci-dessus. Ledit hydrolysai ou milieu est susceptible d’être obtenu par les méthodes de préparation d’un hydrolysat de microorganisme ou d’un milieu de culture riche dont au moins un acide aminé et/ou son précurseur est marqué telles que décrites ci-dessus.
L’hydrolysat ou milieu riche selon la présente invention peut être conservé congelé ou lyophilisé (sous la forme de poudre).
La présente invention concerne également une méthode de marquage d’une protéine par culture d’une cellule procaryote ou eucaryote, comprenant la culture de ladite cellule sur un milieu de culture riche susceptible d’être obtenu selon la présente invention dans lequel au moins un acide aminé et son cétoacide s’il est présent sont remplacés à au moins 90%, et par ordre de préférence croissant au moins 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98% et 99%, et de préférence encore 100%, par le même acide aminé mais sous une forme marquée ou par son précurseur marqué, tel que défini ci-dessus.
La cellule où se fait la production de la protéine peut être choisie parmi une cellule procaryote, de préférence une cellule bactérienne, telle qu’une cellule d’Escherichia coli. La cellule eucaryote peut être choisie parmi une cellule de levure, de champignon, de micro-algue, une cellule d’un animal, telle qu’une cellule de mammifère ou une cellule d’insecte.
La présente invention concerne également une méthode de marquage d’une protéine recombinante dans un système acelluîaire comprenant un milieu réactionnel, dans lequel le milieu réactionnel comprend un hydrolysat de microorganisme dont au moins un acide aminé et/ou un précurseur d’acide aminé est marqué selon la présente invention.
La présente invention concerne également une méthode pour appauvrir en L-leucine, Lvaline et L-isoleucine un milieu liquide, de préférence un hydrolysat de microorganisme en solution, comprenant de la L-leucine, L-valine, L-isoleucine sous forme libre, comprenant les étapes suivantes :
traitement dudit milieu par un mélange de trois enzymes constitué d’une isoleucine 2épimérase (EC 5.1.1.21), une D-aminoacîde oxydase (EC 1.4.3.3) et une catalase (EC 1.11.1.6) telles que définies ci-dessus, et éventuellement élimination de ces trois enzymes du milieu traité, éventuellement lesquelles enzymes ont été inactivées préalablement à leur élimination, telle que décrite ci-dessus, de préférence par un choc thermique de manière à inactiver les enzymes, suivi d’une centrifugation, et/ou
- élimination des cétoacides produits, telle que décrite ci-dessus, de préférence par réduction chimique des cétoacides en hydroxyacides par du borohydrure de sodium.
Avantageusement, ces trois enzymes sont ajoutées à l’hydrolysat simultanément.
La présente invention concerne également une méthode pour appauvrir en L-alanine un milieu liquide, de préférence un hydrolysat de microorganisme en solution, comprenant de la Lalanine sous forme libre, comprenant les étapes suivantes :
traitement dudit milieu liquide par un mélange de quatre enzymes constitué d’une alanine racémase (EC 5.1.1.1), une D-amino acide oxydase (EC 1.4.3.3), une catalase (EC 1.11.1.6) et une acétohydroxyacide synthase (EC 2.2.1.6), telles que définies ci-dessus, et éventuellement
- élimination de l’acétolactate formé, par exemple par chauffage - entre 35°C et 55°C, de préférence à 40°C - en se transformant en acétoïne (3-hydroxybutanone) ou en diacétyle (butane-2,3-dione), et/ou ~ élimination de ces quatre enzymes du milieu traité, éventuellement lesquelles enzymes ont été inactivées préalablement à leur élimination, telle que décrite ci-dessus, de préférence par un choc thermique de manière à inactiver les enzymes, suivi d’une centrifugation, et/ou
- élimination des cétoacides produits, telle que décrite ci-dessus, de préférence par réduction chimique des cétoacides en hydroxyacides par du borohydrure de sodium.
La présente invention concerne également un mélange d’enzymes choisi parmi un mélange d’enzymes constitué ou comprenant une isoleucine 2-épimérase (EC 5.1.1.21), une D-aminoacide oxydase (EC 1.4.3.3) et une catalase (EC 1.11.1.6) telles que définies ci-dessus, et un mélange d’enzymes constitué ou comprenant une alanine racémase (EC 5.1.1,1), une D-amino acide oxydase (EC 1.4.3.3), une catalase (EC 1.11.1.6) et optionnellement une acétohydroxyacide synthase (EC 2.2.1.6) telles que définies ci-dessus.
La présente invention concerne également un kit comprenant :
- un hydrolysat susceptible d’être obtenu à l’étape bl), dans lequel au moins un acide aminé et son cétoacide s’il est présent est éliminé, ou un milieu de culture susceptible d’être obtenu à l’étape c), dans lequel au moins un acide aminé et son cétoacide s’il est présent est éliminé, et
- ledit acide aminé éliminé mais sous une forme marquée et/ou son précurseur marqué, tels que définis ci-dessus.
Outre les dispositions qui précèdent, l’invention comprend encore d’autres dispositions, qui ressortiront de la description qui va suivre, qui se réfère à des exemples de mise en œuvre de la méthode objet de la présente invention ainsi qu’aux dessins annexés :
Figure 1 : Extraits des spectres I3C-HSQC correspondant à la zone des méthyles. Gauche : mélange des 6 acides aminés purs. Les acides aminés sont à l'abondance naturelle. Droit : hydrolysat de microalgues (milieu Isogro®, Sigma-Aldrich) enrichi 13C, les régions correspondant à chaque méthyle des acides aminés isoleucine, leucine, valine, thréonine, alanine et méthionine sont indiquées en pointillé. Les spectres C C-HSQC ont été obtenus grâce un spectromètre Avance III Bruker opérant à 600 MHz proton et équipé d'une cryosonde.
Figure 2 : Extraits des spectres 13C-HSQC correspondant à la zone des méthyles. Gauche : Isogro® enrichi l3C, les régions correspondant à chaque acide aminé sont indiquées en pointillé ; Droit : après traitement à la pronase. Seuls les signaux correspondant aux 6 acides aminés libres sont alors présents (voir Figure 1). Les spectres 13C-HSQC ont été obtenus grâce un spectromètre Avance III Bruker opérant à 600 MHz proton et équipé d'une cryosonde.
Figure 3 : Extrait des spectres 13C-HSQC correspondant à la zone des méthyles d'une solution d'isoleucine, leucine, valine, thréonine, alanine et méthionine en présence de leucine déshydro gênas e de BaciUlus cereus commerciale. La position des méthyles des cétoacides associés est indiquée en a : 3-méthyl-2-oxopentanoate (MOP), en b : 3~méthyl-2-oxobutanoate (cétoisovalerate : KIV), en c : 4-méthyl-2-oxopentanoate (cétoisocaproate : KIC). L'attribution des résonances des cétoacides d’après les données bmse000319, bmse000383 et bmseOOOlOlde la Biological Magnetic Résonance Data Bank (BMRB, http://www.bmrb.wisc.edu). Les spectres i3C-HSQC ont été obtenus grâce un spectromètre Avance III Bruker opérant à 600 MHz proton et équipé d'une cryosonde.
Figure 4 : Extrait des spectres î3C-HSQC correspondant à la zone des méthyles d'une solution de mélange d'acides aminés méthylés (A, T, Μ, I, L et V). Gauche: Témoin; Droite: en présence de 10 mg d'extrait bactérien de E. coli exprimant de la BCAT. Les conditions sont les suivantes: Tris 20mM pH 7.5, oxoglutarate 350mM, chaque acide aminé est à 35mM. La solution est incubée 4 H à 37°C. La position des méthyles des cétoacides associés est indiquée en a : 3méthyl-2-oxopentanoate (MOP), en b : 3-méthyl-2-oxobutanoate (KIV), en c : 4-méthyl-2oxopentanoate (KIC). L'attribution des résonances des cétoacides d’après les données bmse000319, bmseOOO383 et bmseOOOlOlde la Biological Magnetic Résonance Data Bank (BMRB, http://www.bmrb.wisc.edu). Les spectres 13C-HSQC ont été obtenus grâce un spectromètre Avance III Bruker opérant à 600 MHz proton et équipé d'une cryosonde.
Figure 5 : Activité de l'isoleucine 2-épimerase. Gauche : Extrait correspondant à la zone des méthyles du spectre l3C-HSQC d'une solution d'isoleucine en présence d'isoleucine épimerase. L'attribution des résonances de la D-allo-isoleucine est faite d’après les données HMDB00557 de 1 Human Metabolome Database (HMDB, http://www.hmdb.ca). Droit : Variation en fonction du temps de la concentration en L-isoleucine et en D-allo-isoleucine en présence d'isoleucine épimerase. L’équilibre s’établit à un rapport 6/4 comme décrit dans la littérature, les formes (2S,3S) et (2R,3S) n’étant pas équivalentes. Les spectres 13C-HSQC ont été obtenus grâce un spectromètre Avance III Bruker opérant à 600 MHz proton et équipé d'une cryosonde.
Figure 6 : Extraits des spectres 13C-HSQC correspondant à la zone des méthyles. Gauche : milieu Isogro® hydrolysé, les corrélations de chaque méthyle sont annotées. Droit : après traitement avec le mélange (système enzymatique) isoleucine-2-épimerase/D-amino acide oxydase/catalase. Les signaux correspondant aux méthyles des trois acides aminés Ile, Leu et Val ont disparu alors qu'apparaissent des signaux aux déplacements chimiques proton et carbone attendus pour les cétoacides associés. 3-méthyl~2-oxobutanoate (KIV) ; 3-méthyl-2oxopentanoate (MOP), 4-méthyl~2-oxopentanoate (KIC);. n ,i. : composé non identifié. Les spectres 13C-HSQC ont été obtenus grâce un spectromètre Avance III Bruker opérant à 600 MHz proton et équipé d'une cryosonde.
Figure 7 : Extraits des spectres !3C-HSQC correspondant à la zone des méthyles. Gauche : après traitement avec le mélange (système enzymatique) isoîeucine-2-épimerase/D-amino acide oxydase/catalase, les corrélations de chaque méthyle sont annotées. Droit : après traitement au NaBH4. Les signaux de l'alanine et de la thréonine ne sont pas affectés par ces traitements. Par contre, ceux correspondant aux méthyles des trois cétoacides ont disparu alors qu'apparaissent des signaux aux déplacements chimiques proton et carbone attendus pour les hydroxyacides associés. HIV : 3-méthyl-2-hydroxybutanoate ou hydroxyisovalerate; MHP : 3-méthyl-2hydroxypentanoate, HIC : 4-méthyl-2-hydroxypentanoate ou hydroxyisocaproate ; n.i. compose non identifié. L'attribution des résonances des hydroxyacides est faite d’après l'«Human Metabolome Database » N° HMDB00407 (hydroxyisovalerate) et HMDB00746 ( hydroxyisocaproate). Les signaux du 3-méthyl-2-hydroxypentanoate (présent sous ses formes (3S,2S et 3S,2R), ont été déterminé expérimentalement par déamination de l'isoleucine puis réduction au NaBH4. Les spectres BC-HSQC ont été obtenus grâce un spectromètre Avance III Bruker opérant à 600 MHz proton et équipé d'une cryosonde.
Figure 8 : Extraits des spectres lD-deuterium correspondant à la zone des méthyles. Gauche : milieu Celtone® hydrolyse perdeutéré issu de l'étape A, les corrélations de chaque méthyle sont annotées. Droit : après traitement avec le mélange (système enzymatique) isoîeucine-2-épimerase/D-amino acide oxydase/catalase. Les positions des fréquences des méthyles des différents acides aminés sont indiquées dans chaque spectre par des pointillés. Les signaux correspondant aux méthyles des valines, isoleucines et leucines ont disparu après traitement. Le spectre lD-deutérium a été obtenu grâce un spectromètre Avance III Bruker opérant à 600 MHz proton et équipé d'une cryosonde.
Figure 9 : Histogramme des quantités d'acides amines dans l'hydrolysat deutéré après traitement avec le mélange (système enzymatique) isoleucine 2-épimerase/D-AAO/catalase. Les résultats sont exprimés en pourcentage des quantités d'acides aminés dans l’hydrolysat avant traitement.
Figure 10 : A : Evolution du rapport des intensités des signaux des méthyles delta-1 des isoleucines et des méthyles epsilon des méthionines en fonction de la concentration en précurseur des isoleucines ajoutée dans le milieu. Carré : dans un milieu de culture riche non appauvri ; rond : dans un milieu de culture riche appauvri et additionné avec un précurseur. Le précurseur utilisé est du 2-oxobutanoate. La concentration en méthionine dans les cultures est de 200 mg/L. B : Evolution du rapport des intensités des signaux des méthyles des valines et des méthyles epsilon des méthionines en fonction de la concentration en précurseur des valines ajoutée dans le milieu. Carré : dans un milieu de culture riche non appauvri ; rond : dans un milieu culture riche appauvri et additionné avec un précurseur. Le précurseur utilisé est du 2-hydroxy-2-méthyl-3oxobutanoate. La concentration en méthionine dans les cultures est de 200 mg/L. Les spectres 13C-HSQC ont été obtenus à 30°C sur un spectromètre Avance III Bruker opérant à 600 MHz et équipé d'une cryosonde.
Figure 11 : Analyse par Gels SDS-page de l'expression en milieu minimum et en milieu riche appauvri et recomplété des proteines PBPkis et LDTmtî· Expression de ΡΒΡκη par E. coli dans des milieux deutérés (partie gauche de la figure). M: marqueurs de masse moléculaire, Al : fraction soluble de E. coli en milieu minimum après dialyse, A2 : fraction insoluble (précipité) de E. coli en milieu minimum après dialyse, A3 : fraction soluble de E. coli en milieu riche hydrolysé après dialyse, A4 : fraction insoluble(précipité) de E. coli en milieu riche hydrolyse après dialyse, A5 : PBP«i5 exprimée en milieu minimum (issu de la fraction insoluble), A6 : PBPr.15 exprimée en milieu riche hydrolysé (issu de la fraction soluble). Expression de LDTmtî en E. coli dans des milieux deutérés (partie droite de la Figure). M: marqueurs de masse moléculaire, B1 : fraction soluble de E. coli en milieu minimum, B2 : fraction insoluble de E. coli en milieu minimum, B3 : fraction soluble de E. coli en milieu riche hydrolysé, B4 : fraction insoluble de E. coli en milieu riche hydrolysé, B5 : fraction soluble de E. coli en milieu riche hydrolysé appauvri puis recomplété, B6 : fraction insoluble de E. coli en milieu riche hydrolysé appauvri puis recomplété.
Figure 12 : Extrait de la région des méthyles du spectre 13C-HSQC d'un échantillon marqué de LDTmtî- Le spectre C-HSQC a été obtenu grâce un spectromètre Avance III Bruker opérant à 600 MHz proton et équipé d'une cryosonde.
Figure 13 : Extraits des spectres C-HSQC correspondant à la zone des méthyles d'une part et à la région des protons alpha d'autre part. Gauche : mélange de méthionine et d'alanine, les corrélations correspondant à chaque acide aminé sont annotées ; Droit : après traitement avec le mélange (système enzymatique) alanine racémase/D-amino acide oxydase/catalase/acétolactate synthase et passage à 37°c pendant 24 heures. Seuls les signaux correspondant à la méthionine sont encore présents, La position des corrélations de l'alanine est indiquée par des rectangles en pointillé. Les signaux signalés par des astérisques correspondent aux produits issus de la dégradation de l'acétolactate.
Figure 14 : Extraits des spectres 13C-HSQC correspondant à la zone des méthyles. Gauche ; milieu commercial hydrolysé, les corrélations de chaque méthyle sont annotées. Droit : après traitement avec le mélange (système enzymatique) alanine racémase/D-amino acide oxydase/catalase/acétolactate synthase et passage à 37°C pendant 24 heures. Seul le signal correspondant au méthyle de l'alanine a disparu. La position de ce méthyle est indiquée par un rectangle en pointillé. Les signaux signalés par des astérisques correspondent aux produits issus de la dégradation de l’acétolactate.
Figure 15 : Extraits des spectres lD-deuterium correspondant à la zone des méthyles. Gauche : hydrolysat commercial perdeutéré après traitement avec de la Pronase®, les corrélations de chaque méthyle sont annotées. Droit : après traitement avec le mélange (système enzymatique) alanine racémase/D-amino acide oxydase/catalase/acétolactate synthase et passage à 37°C pendant 24 heures. Les fréquences des méthyles des différents acides aminés sont indiquées dans chaque spectre par le code de l'acide aminé à part celle de l'alanine qui l'est par une ligne pointillée. Seul le signal correspondant au méthyle de l'alanine a disparu après traitement.
Figure 16 : Histogramme des quantités d'acides aminés dans l’hydrolysat deutéré après traitement avec de l’alanine racémase/D-AAO/catalase. Les résultats sont exprimés en % des quantités d'acides aminés dans l’hydrolysat avant traitement.
Figure 17 : Evolution du rapport des intensités des signaux des méthyles beta de l'alanine et des méthyles epsilon des méthionines en fonction de la concentration en alanine ajoutée dans le milieu. Carré : dans un milieu de culture riche non appauvri ; cercle : dans un milieu de culture riche appauvri en alanine et additionné avec de l'alanine marquée. La concentration en méthionine dans les cultures est de 200 mg/L.
EXEMPLE : METHODE DE PREPARATION DE MILIEUX RICHES POUR LE
MARQUAGE SPECIFIQUE DES METHYLES DANS LES PROTEINES POUR LES
ETUDES DE RESONANCE MAGNETIQUE NUCLEAIRE BIOMOLE CUL AIRE
A) Obtention d’un milieu de culture riche dans lequel tous les acides aminés se trouvent sous la forme libre
Les sources d'acides aminés perdeutérés, Isogro® (Sigma-Aldrich) ou Celtone® (Cambridge isotop Laboratories), disponibles commercialement ont été utilisés. Ces milieux sont des hydrolysats d’algue. Les résultats sont semblables pour les deux hydrolysats commerciaux. Comme le montre la Figure 1, l’hydrolysat commercial présente pour chaque résonance des méthyles des Alanine, Valine, Leucine, Isoleucine et Thréonine des résonances associées indiquant l'existence de ces acides aminés engagés dans d'autres formes que ces acides aminés libres.
Traitement de l’hydrolysat commercial par la pronase.
lg de hydrolysat commercial Isogro® a été dissout dans 20ml de tampon Tris 20mM pH7, 5. Il a ensuite été incubé avec 31 mg de Pronase® commerciale (Sigma-Aldrich) sur la nuit à 37°C sous agitation. La solution a subit ensuite un choc thermique à 95°C pendant 30 min puis a été centrifugée à 40000g pendant 30min. 450 uL de la solution ont été additionnés de 50 uL D2O et ensuite le mélange a directement été analysé par résonance magnétique nucléaire grâce a une expérience de type 13C-HSQC.
Le traitement de ce milieu commercial avec la Pronase® permet de faire disparaître toutes les formes d’acides aminés autres que les acides aminés libres, comme le montre la Figure 2.
B) Remplacement dans l’hydrolysat obtenu à Pétape A) des acides aminés Leucine, Valine et Isoleucine et de leurs cétoacides par les précurseurs marqués de ces acides aminés.
1. Appauvrissement de l’hydrolysat obtenu à l’étape A) en acides aminés à chaîne ramifiée
Isoleucine, Leucine et Valine
L’élimination des acides aminés doit répondre à deux critères : le premier est une conversion complète (z'.e., au moins 90%) des acides aminés, le second est que le produit final de la conversion ne soit pas recyclable dans le métabolisme du microorganisme (e.g., bactérie) cultivé sur le milieu marqué.
a) Essai avec l'alanine déskydrogénase
La leucine déshydrogénase convertit in vivo la leucine, la valine et l'isoleucine en les cétoacides associés selon les réactions enzymatiques suivantes :
L-leucine + H2O + NAD+ —» 4-méthyl-2-oxopentanoate (KIC) + NH3 + NADH + H+
L-valine + H2O + NAD+ —> 3-méthyl-2-oxobutanoate (KIV) + NH3 + NADH + H+
L-isoleucine + H2O + NAD+ 3-méthyl-2-oxopentanoate (MOP) + NH3 + NADH + H*
Un mélange de 6 acides aminés a été préparé à des concentrations semblables à celles de l’Isogro® (isoleucine 2,6mg/mL, leucine 5,2mg/mL , valine 3,9mg/mL , thréonine 3,2mg/mL, méthionine l,95mg/mL et alanine 8,45mg/mL ). 100 μΐ ont été incubés avec 1 unité de leucine déshydrogénase en présence de lOmM β-NAD pour un volume final de 500 ul en tampon 20mM Tris pH 9 pendantl2 heures à 30°C. Le mélange a été ensuite directement analysé par résonance magnétique nucléaire grâce a une expérience de type 13C-HSQC.
Comme le montre la Figure 3, le traitement d'un mélange des 6 acides aminés méthylés par la leucine déshydrogénase conduit à l'apparition des cétoacides associés à la leucine, valine et isoleucine mais en quantités extrêmement faibles. Il est envisageable que cela soit dû à un recyclage du NADH en NAD+. L'importance d'un tel système de recyclage a été reconnu par Monti et al. (2011). Deux systèmes enzymatiques ont été décrits : les systèmes pyruvate/lactate déshydrogénase et 2-oxoglutarate/glutamate déshydrogénase mais chacun de ces systèmes conduit à introduire des composés non marqués dans le milieu, composés qui seraient réassimilables par les microorganismes cultivés et dilueraient les marquages. De plus, le pyruvate et le 2-oxoglutarate étant également des cétoacides, leur présence rendrait difficile l’élimination des cétoacides issus de l’Ile, Val et Leu.
Il a été testé des composés oxydants comme l'eau oxygénée ou le ferricyanure, des enzymes comme la flavine réductase de Streptomyces coelicolor mais quelles que soient les conditions expérimentales, les inventeurs n’ont pas réussi à obtenir une conversion significative des acides aminés en cétoacides. La leucine déshydrogénase n’a donc pas été retenue,
b) Essai de la voie «branched-chain aminoacid» transaminase d’Escherichia coli qui catalyse le transfert d'azote d'un cétoacide à un autre
Les « branched-chain amino acid » iransaminases (leucine transaminase et branched chain aminotransferass (BCAT)) convertissent in vivo la leucine, la valine et l’isoleucine en les cétoacides associés selon les réactions enzymatique suivantes
L-leucine + 2-oxoglutarate 4-méthyl-2-oxopentanoate (KIC) + glutamate
L-valine + 2-oxoglutarate —> 3-méthyl-2-oxobutanoate (KIV) + glutamate
L-isoleucine + 2-oxoglutarate —+ 3-méthyl-2-oxopentanoate (MOP) + glutamate mg d’Isogro® hydrolysé par la Pronase® ont été incubés avec 10 mg d'extrait brut de BCAT en présence d'une quantité d’oxoglutarate 10 fois égal au total des acides aminés hydrophobes Ile, Leu et Val présents dans le milieu. La BCAT a été obtenue par surexpression dans E.coli. Après une première centrifugation, les cellules ont été reprises par un tampon de lyse et cassées par sonication et centrifugées à 40000G pendant 30 minutes. Le surnageant contenant la BCAT a alors été recueilli puis lyophilisé et le lyophilisât utilisé sans autre purification. La réaction a été conduite pendant 4 heures à 37°C dans du 20mM Tris PH7,5. Le mélange a été ensuite directement analysé par résonance magnétique nucléaire grâce a une expérience de type 13C-HSQC.
Comme le montre la Figure 4, la BCAT en présence d'une concentration en oxoglutarate de dix fois celle des acides aminés convertit une partie des leucines, valines et isoleucines en cétoacides associés. L1 isoleucine est convertie à plus de 60 % comme l'est la leucine. Par contre la valine est faiblement convertie (de l'ordre de moins de 10 %). Il apparaît que la thréonine est également transformée ; la résonance du méthyle gamma de cet acide aminé n'étant plus observé. La conversion n’est donc ni totale, même avec une quantité d'oxoglutarate dix fois supérieure à celle des acides aminés, ni spécifique, ce qui rend cette voie inutilisable. De plus, il est nécessaire par la suite d'éliminer les cétoacides produits, La présence en quantité importante d'oxoglutarate qui est le cétoacide associé au glutamate rendrait cette élimination difficile.
En conclusion, les possibilités de conversion suggérées par le métabolisme cellulaire in vivo ne s’avèrent pas directement utilisables.
c) Essai avec le mélange (système enzymatique) isoleucine 2-épimerase, D-aminoacide oxydase et catalase
L’isoleucine 2-épimerase catalyse la conversion de la L-isoleucine - forme (2S,3S) - en Dallo-îsoleucine - forme (2R,3S) -, l’isoleucine présentant 2 carbones asymétriques en positions 2 et 3. Cette enzyme convertit d’après la littérature également les formes L de la leucine, la valine, la méthionine, la phénylalanine en formes D. La D~amino oxydase transforme les acides aminés de la forme D en leurs cétoacides avec production d'eau oxygénée. La catalase dismute l'eau oxygénée en oxygène moléculaire et en eau. Les réactions enzymatique sont les suivantes : L-leucine ~-+ D-leucine —> 4-méthyl-2-oxopentanoate (KIC)
L-valine —* D-valine —> 3-méthyl-2-oxobutanoate (KIV)
L-isoleucine —+ D-allo-isoleucine —» 3-méthyl-2-oxopentanoate (MOP)
i. Obtention du gène codant l'épimerase. Culture, expression et purification de l'épimerase Un gène optimisé codant l’épimérase a été fait synthétiser pour son expression dans E. coli.
La séquence du gène de la 2-isoleucine épimerase de L, buchneri JCM 1115 (séquence primaire n° M1GRN3, UniProt), après synthèse chimique (GeneCust, Luxembourg) est sous clonée dans le vecteur Pet-28a (Novagen) en fusion avec un tag polyhistidine. E. coli BL21 et DE3 ont été transformées avec le plasmide correspondant puis mises en culture dans du milieu Luria Bertani en présence de kanamycine. Une fois atteint une DO de 0,7, l'expression de l'épimerase est induite par addition de 1 mM IPTG sur la nuit à 20°C.
Purification : Les cellules sont re-suspendues avec du tampon 50mM phosphate de sodium,
0,5M NaCl, ImM B-mercapto-éthanol, 5mM imidazole, pH=8 (tampon A) en présence d’inhibiteurs de protéases. Les cellules sont alors lysées par trois cycles de sonication d’une minute puis centrifugées pendant 30 min à 40000g. La fraction soluble est recueillie et chargée sur une résine Ni-NTA (Qiagen) prééquilibrée avec le tampon A. La colonne est alors rincée avec le tampon A à 20mM imidazole puis avec le tampon A additionné de 50mM imidazole. Finalement, la protéine est éluée avec le tampon A additionné de 500 mM imidazole. La protéine est ensuite purifiée par chromatographie d'exclusion de taille sur Superdex S200. La protéine est éluée en un seul pic à un temps de rétention qui correspond à une protéine de 200 Kda.
La protéine obtenue est fonctionnelle comme le montre la Figure 5 et permet de convertir l'isoleucine en D-allo-isoleucine (la réaction étant réversible) comme montré ci-après (ii).
U. Conversion de l'isoleucine en D-allo-isoleucine
Un volume de 0,5ml d’isoleucine à 50mM est incubé avec 6,5 ug/ml d’isoleucine 2épimerase en présence de pyridoxal phosphate (Sigma) (0,lmM) dans le tampon 20mM Tris pH7,5 à 37°C, iii. Conversion de l'isoleucine, leucine et valine en leurs cétoacides
L’utilisation du mélange (système enzymatique) isoleucine 2-épimerase/D-AAO(D-acid amino oxydase)/catalase conduit à la conversion totale de l'isoleucine seule en le cétoacide associé : le 3-méthyl-2-oxopentanoate. (résultat non montré).
En mélange, les trois acides aminés isoleucine, leucine et valine sont également totalement convertis par le mélange (système enzymatique) isoleucine 2-épimerase/D-AAO/cataIase en leurs cétoacides associés (résultat non montré).
L’hydrolysat obtenu à l’étape A) ci-dessus, une fois traité avec le mélange des 3 enzymes isoleucine 2-épimerase/D-AAO/catalase, a également été totalement épuisé en ces trois acides aminés comme le montre la Figure 6 où les pics caractéristiques des méthyles des Ile, Leu, Val disparaissent et qu’apparaissent les signaux des cétoacides associés.
Après traitement par le mélange (système enzymatique) isoleucine 2-épimerase/DAAO/catalase, le milieu de culture riche a subi un choc thermique d’une demi-heure à 85°C puis une centrifugation pour éliminer les enzymes. Aucune activité résiduelle de conversion des acides aminés n'est alors observée (résultat non montré).
iv. Elimination des cétoacides produits
De nombreuses voies d'élimination des cétoacides sont envisageables, par extraction, par dérivatisation chimique, par réduction ou par décarboxylation enzymatique. La voie choisie ici est une réduction chimique des cétoacides en hydroxyacides par du borohydrure de sodium (NaBFU). 2g de milieu de culture riche Celtone ® hydrolysé préalablement par de la Pronase® contiennent l’équivalent de 3 mMoles de cétoacides issus de Leu, Val, Ile. Il faut 110 mg de NaBH4 pour réduire ces 3 mMoles, soit 11 mMoles d'H par rapport à 100 Moles de D dans un litre de D2O. Les réductions chimiques des cétoacides s’effectuent selon les réactions suivantes : 4-méthyL2-oxopentanoate (KIC) —* 4-méthyl-2-hydroxypentanoate (HIC)
3-méthyl~2-oxobutanoate (KIV) —> 3-méthyl-2-hydroxybutanoate (HIV)
3-méthyI-2-oxopentanoate (MOP) —> 3-méthyI-2-hydroxypentanoate (MHP)
Les résultats sont montrés à la Figure 7 Les signaux correspondant aux méthyles des trois 5 cétoacides ont disparu alors qu'apparaissent des signaux aux déplacements chimiques proton et carbone attendus pour les hydroxyacides associés indiquant la conversion totale des cétoacides présents dans la solution. Par contre, les signaux de l’alanine et de la thréonine ne sont pas affectés par ces traitements.
v. Appauvrissement de l’hydrolysat obtenu à l’étape A) en isoleucine, leucine et valine et 10 leurs cétoacides, par élimination dans cet hydrolysat de ces acides aminés et de leurs cétoacides g d’hydrolysat perdeutéré Isogro® préalablement traité par de la Pronase® a été incubé dans 20ml de tampon Tris 20mM pH7,5 avec 16ug/ml d’isoleucine 2-épimerase, 250unités de DAAO(D-acid amino oxydase), 250 unités de catalase, ImM FAD et ImM PLP pendant 24H à 37°C. Le milieu a subi ensuite un choc thermique pendant 30 minutes à 95°C puis a été centrifugé pendant 30 min à 40000g. Le milieu a ensuite été réduit avec du NaBH4 40 mM final afin de transformer les cétoacides en les hydroxyacides correpondants.
La substitution des protons par des deutériums peut théoriquement modifier l'activité des enzymes. Comme le montre la Figure 8, le mélange (système enzymatique) isoleucine 2épimerase/D-AAO/catalase permet également l’élimination complète de la leucine, valine et isoleucine dans un hydrolysat perdeutéré en solution dans du D2O. Ce mélange (système enzymatique) n’est donc pas affecté par le deutérium.
Pour vérifier que le traitement n'affecte essentiellement que la leucine, la valine et l'isoleucine, la concentration des différents acides aminés a été mesurée avant et après traitement. En effet, d’après Mutaguchi et al. (2013), la L-phénylalanine et la L-méthionine sont racémisées par l’isoleucine 2-épimerase mais avec une moindre activité. Comme attendu (voir Figure 9), l'isoleucine et la leucine sont pratiquement entièrement convertis (> 95 %), La valine est également convertie à plus de 90 %. De même, plus de 80 % de la phénylalanine et de la méthionine ont été transformés. La quantité de L-tryptophane était nulle dans l’hydrolysat d'algues perdeutéré Isogro® initial; celui-ci étant détruit lors de l'hydrolyse des microorganismes.
2, Ajout au l’hydrolysat appauvri obtenu à l’étape Bl) des précurseurs marqués de
Γisoleucine, leucine et valine et incorporation des acides aminés marqués isoleucine, leucine et valine dans les protéines exprimées par des bactéries cultivée sur le milieu de culture ainsi obtenu
Le milieu de culture constitué par du milieu minimum M9 additionné de l’hydrolysat appauvri en acides aminés contient des hydroxyacides dérivés des acides aminés. La viabilité d'une culture de E. coli et l'expression des protéines ont été testées dans ces conditions. Aucune différence significative de croissance de la culture bactérienne n'est observée entre le milieu constitué de milieu M9 (contenant deux grammes de glucose, un gramme de chlorure d’ammonium et d’oligoéléments) et de deux grammes de l’hydrolysat traité selon la présente Invention et donc appauvri en isoleucine, valine, leucine, phénylalanine et méthionine et ce même milieu M9 additionné de deux grammes de hydrolysat commercial (Celtone® ou Isogro ®) (résultat non montré). Cela confirme l’innocuité du traitement selon l’invention. Par rapport au milieu M9, l'augmentation de la masse cellulaire obtenue par addition de l’hydrolysat traité selon la présente invention est de l’ordre d'un tiers par rapport à celle obtenue dans un milieu M9 seul. Le niveau d'expression après induction d'une protéine témoin (ubiquitine) dans le milieu constitué de milieu M9 et de l’hydrolysat traité selon l’invention n'est pas modifié en comparaison d’un milieu riche classique constitué de milieu M9 additionné d'une source d'acides aminés commerciale - hydrolysat Celtone® ou Isogro® (résultat non montré).
L’hydrolysat appauvri obtenu à l’étape B1) ou le milieu de culture riche issu de l’ajout de cet hydrolysat à un milieu minimum de type M9 est ensuite additionné soit d'un précurseur de l'isoleucine choisi parmi le 2-oxobutanoate pour le marquage des méthyles delta-1 et le 2hydroxy-2-éthyl-3-oxobutanoate pour celui des méthyles gamma-2, soit d’un précurseur de la leucine et la valine choisi parmi le 2-hydroxy-2-méthyl-3-oxobutanoate, pour le marquage stéréospécifique des méthyles proR, proS et le 3-méthyl-2-oxobutanoate (ketoisovalerate) pour un marquage astéréospécifique des méthyles, en présence ou non de leucine deutérée si le marquage des méthyles des valines est uniquement souhaité.
Il a ensuite été comparé le niveau d'incorporation des précurseurs dans une protéine témoin avec un milieu riche classique et avec le milieu appauvri selon l’invention puis recomplété avec le précurseur approprié. Comme le montre la Figure 10A, le niveau d'incorporation des précurseurs reste faible inférieur à une soixantaine de pour cent - dans un milieu riche classique même à des concentrations élevées de précurseurs, fl est en particulier impossible d'obtenir des taux de marquage de plus de 90 % (nécessaires par exemple à des expériences de type NOESY). La culture et l'expression dans le milieu appauvri puis recomplété selon l’invention permet d'obtenir un tel niveau et cela pour des concentrations en précurseur proches de celles requises pour un milieu minimum M9, i.e., de l'ordre de 60 mg/L pour le 2-oxobutanoate et 300 mg/L pour les acétolactates (voir Figure 10B). Sans appauvrissement du milieu, le pourcentage d'incorporation est limité à 30 % pour la même quantité de précurseur et l'incorporation maximum observée en excès de précurseurs testées (oxobutyrate : 200mg ou 800 mg pour acétolactate) ne dépasse pas 50%.
Optimisation de l'incorporation des précurseurs dans les protéines
Le niveau d'incorporation du 2-oxobutanoate a été évalué en utilisant l'ubiquitine comme système modèle. Des cellules de la souche BL21(DE3) de E. colt ont été transformées avec le plasmide pET41c (Novagen) comprenant un gène codant pour rubiquitine humaine précédée d’un tag polyhistidine (pET41c~Hîs~Ubi). Ces cellules ont d'abord été adaptées en milieu deutéré. Pour cela, les cellules sont tout d’abord mises à pousser dans 5 ml de milieu de culture Luria-Bertani sur la journée. Elles sont ensuite transférées en milieu minimum M9/H2O. Le lendemain matin, les cellules sont de nouveau transférés dans du milieu minimum M9/50%D20:50%H20 et finalement le soir dans du milieu minimum M9/100%D20. Elles sont alors transférées dans un milieu M9/D2O contenant 1 g/L 15ND4C1, 2 g/L U-[2H], U-[13C] glucose et 2 g/L d'hydrolysat deutéré traité par de la Pronase® (étape A) puis appauvri en Ile, Val et Leu avec au départ une densité optique de 0,3 ; le volume utilisé correspond à 70 % du volume total de la culture. Quand la densité optique à 600 nm a atteint 1, une solution contenant le précurseur a été ajoutée. Après 1 heure, l'expression de la protéine a été induite par l'addition d'IPTG (isopropyl β-D-lthiogalactopyranoside) à une concentration de 1 mM. L'induction a été faite pendant 3 heures à 37°C. L'ubiquitine a été purifiée par chromatographie sur colonne Ni-NTA (Qiagen).
La quantité optimale de 2-oxobutanoate nécessaire pour obtenir une incorporation quasi complète dans la protéine surexprimée a été déterminée par une série de cultures (25 mL chacune) dans lesquelles des quantités croissantes de ce précurseur marqué 13C,H sur un méthyle sont ajoutées une heure avant induction pour des concentrations finales de 0,10, 20, 30, 50, 75, 100 et 200 mg/L Le niveau d'incorporation a été estimé dans la protéine purifiée par des expériences 2D I3C-HSQC. La quantification a été effectuée en mesurant le rapport de la moyenne de l’intégrale du signal des méthyles des isoleucines à la moyenne de l’intégrale du signal des méthyles des méthionines. L’addition de 100 mg de 2-oxobutanoate par litre de milieu épuisé permet une incorporation de plus de 90 % dans les chaînes latérales des isoleucines sans fuite détectable dans d'autres acides aminés.
Pour l'acétolactate, les cellules de la souche BL21(DE3) de E. coli ont été transformées avec le plasmide pET41c comprenant un gène codant pour rubiquitine humaine précédée d'un tag polyhistidine (pET41c-His-Ubi), Ces cellules ont d'abord été adaptées en milieu deutéré comme décrit ci-dessus puis transférées dans un milieu M9/D2O contenant 1 g/L 15ND4C1, 1.5 g/L U[2H], U-[I3C] glucose. Quand la densité optique à 600 nm a atteint 1,5, une solution contenant 1,5 g/L de d'hydrolysat deutéré traité par de la Pronase® (étape A) puis appauvri en Ile, Val et Leu est ajoutée avec l'acétolactate. Apres 1 heure, l'expression de la protéine a été induite par l'addition d’IPTG à une concentration de 1 mM, L'induction a été faite pendant 3 heures à 37°C. L'ubiquitine a été purifiée par chromatographie sur colonne Ni-NTA (Qiagen).
La quantité optimale d'acétolactate nécessaire pour obtenir une incorporation à plus de 90 % dans la protéine surexprimée a été déterminée par une série de cultures (20 mL chacune) dans lesquelles des quantités croissantes de ce précurseur marqué 13C,H sur un méthyle sont ajoutées une heure avant induction pour des concentrations finales de 0, 100, 300, 400, 600, et 800 mg/L. Le niveau d'incorporation a été estimée dans la protéine purifiée par des expériences 2D 13C-HSQC. La quantification a été effectuée en mesurant le rapport de la moyenne de l’intégrale du signal des méthyles des valines à la moyenne de l’intégrale du signal des méthyles des méthionines. L’addition de 300 mg d'acétolactate par litre de milieu appauvri permet une incorporation de plus de 90 % dans les chaînes latérales des valines sans fuite détectable dans d'autres acides aminés.
3. Expression de protéines non exprimables en milieu minimum par des bactéries
L'utilisation d'un milieu de culture riche obtenu à partir d’un hydrolysat appauvri puis recomplété en acides aminés marqués selon l’invention permet d'obtenir des augmentations de rendement en protéine soluble (en mg par litre) par rapport aux cultures en milieu minimum équivalentes à celles obtenues avec un milieu riche. Ces augmentations sont dues à soit une plus grande densité cellulaire lors de l'induction et donc une plus grande quantité de protéine produite, soit une augmentation de la fraction soluble de la protéine. Ces deux cas peuvent être rencontrés comme l'illustre la Figure 11 où sont présentés les résultats obtenus pour l’expression dans E. coli de deux protéines LDTmts - une transpeptîdase de Mycobacterium - et PBPkss - une penicillinbinding-protein issue de Streptomyces Kl 5.
Pour le milieu PBPkis, le milieu riche obtenu à partir d’un hydrolysat appauvri en isoleucine, leucine et valine par traitement avec le système enzymatique épimerase/DAAO/catalase puis réduction par le borohydrure selon l’invention, a été recomplété avec de la phénylalanine protonée, de l'oxoisovalérate et de la leucine deutérée et du 2-oxobutanoate marqué 13C,H en position 4. On observe (voir Figure 11) que, produite en milieu minimum, après dialyse de la fraction soluble contre le tampon sans sel, étape nécessaire pour la purification de la protéine, la quasi totalité de PBPrb précipite alors que, produite en milieu riche, la protéine se retrouve uniquement dans la fraction soluble (il n'y a aucune précipitation après dialyse).
Pour LDTmti, le milieu riche obtenu à partir d’un hydrolysat appauvri en isoleucine, leucine et valine par traitement avec le système enzymatique epimerase/D-AAO/catalase puis réduction par le borohydrure selon l’invention, a été recomplété avec de la phénylalanine protonée, de l'oxoisovalérate et de la leucine deutérée, de la méthionine deutérée et marquée bC,H en position ε et du 2-oxobutanoate marqué I3C,H en position 4. On observe (voir Figure 11) que l'expression de LDTmti dans le milieu riche appauvri puis recomplété est deux fois plus grande: 10,5 rng/L qu'en milieu minimum: 5,1 mg/L. Elle est équivalente à celle en milieu riche : 12 mg/L. Dans ces conditions, le marquage de LDTmti produite dans le milieu riche appauvri puis recomplété a été analysé par résonance magnétique nucléaire. Comme l'illustre la Figure 12, il a été observé uniquement le marquage des positions ôl des isoleucines et des méthionines sans fuite d'isotope vers d'autres acides aminés,
C) Remplacement dans le milieu de culture obtenu à l’étape A) de l’alanine et de son par un précurseur marqué,
a) Essai avec l'alanine déshydrogénase
Il a été testé l’élimination de l'alanine par l'utilisation de l’alanine déshydrogénase qui devrait la convertir en pyruvate selon :
L-alanine + H2O + NAD+ —> pyruvate + NH3 + NADH + IL
La conversion en pyruvate mesurée par la disparition des signaux de l'alanine est très faible quelles que soient les conditions utilisées (résultats non montrés). Cette voie a donc été abandonnée.
b) Essai avec la glutamate-pyruvate transaminase
Il a été testé l'utilisation de la glutamate-pyruvate transaminase de E. coli qui catalyse le transfert d'azote de l'alanine à l'oxoglutarate selon :
L-alanine + 2-oxoglutarate —» pyruvate + glutamate
Pour les mêmes raisons que pour la conversion des acides aminés à chaîne ramifiée (excès 5 de cétoacides, réaction à l’équilibre) décrites ci-dessus, cette voie a été également abandonnée.
c) Essai avec le mélange (système enzymatique) alanine racémase/D-amino acide oxydase/catalase/acétolactate synthase
Il a été développé un système enzymatique in vitro basé sur le schéma réactionnel suivant (Schéma 1 ci-dessous) : la L-alanine est convertie en D-alanine par l’alanine racémase de Bacillus stearothermophilus disponible commercialement (Sigma-Aldrich). La D-alanine est ensuite désaminée et transformée en pyruvate par la D-amino acide oxydase de rein de porc disponible commercialement (Sigma-Aldrich). Le pyruvate formé est alors condensé en acétolactate par la AHAS II (acétohydroxyacide synthase ou acétolactate synthase) de E. coli. L'acétolactate formé est éliminé par chauffage à 4Q°C en se transformant en acétoïne (3-hydroxybutanone) ou en diacétyle (butane-2,3-dione). Les réactions catalysées par la D-AAO produisant du péroxyde d’hydrogène, de la catalase est ajoutée au milieu pour l’éliminer.
Schéma 1 : Schéma réactionnel pour l’élimination de l'alanine, ala rac : alanine racemase; D-AAO : D-amino acid oxidase ; AHAS II‘. acétohydroxyacide synthase II ; cat : catalase
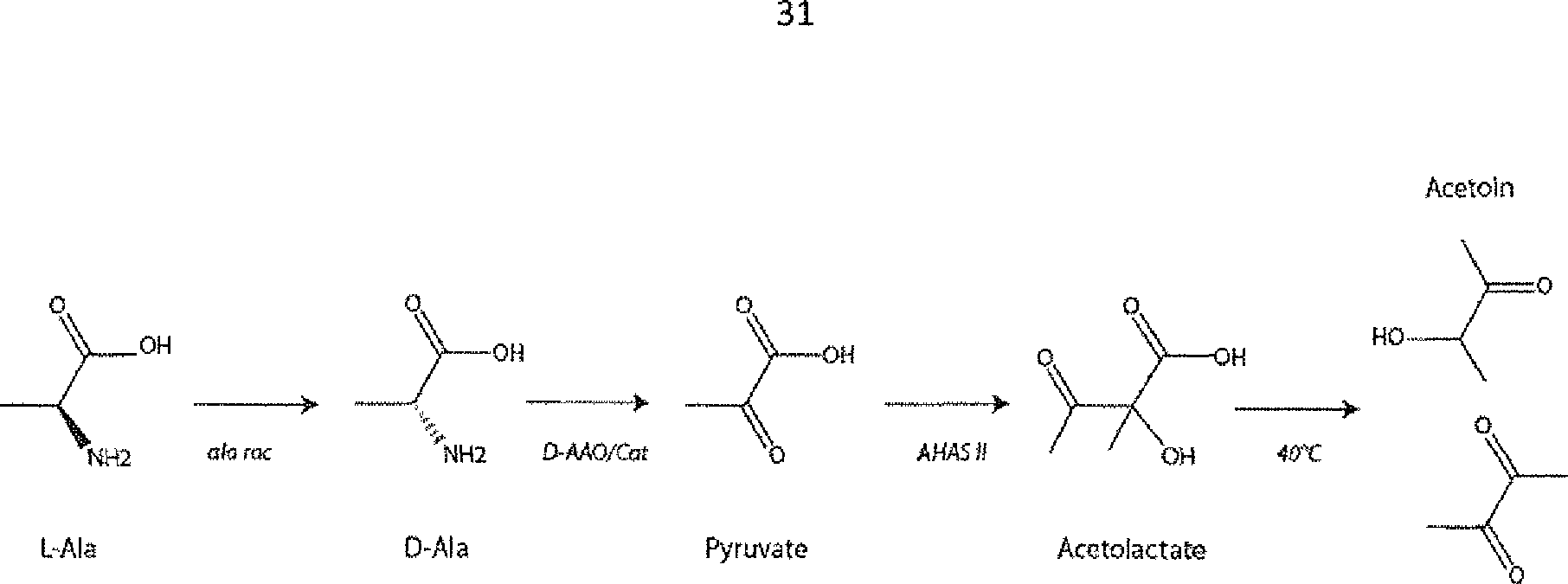
Diacetyl
500 μΐ d'une solution d’alanine et de méthionine (20mM respectivement) est incubée avec 3 unités de D-AAO ( Sigma-Aldrich), 3 unités de catalase (Sigma-Aldrich) ,lmg d’Acetolactate synthase, 6 unités d’alanine racémase (Sigma-Aldrich), ImM FAD (Flavine adénine dinucléotide), ImM PLP (pyridoxal phosphate), ImM ThPP (thiamine pyrophosphate)) dans du
Tris 20mM pH 7,5 pendant 24 Heures à 30°C.Le mélange est ensuite directement analysé par résonance magnétique nucléaire grâce à une expérience de type 13C-HSQC.
Comme la montre la Figure 13, la L-alanine a été convertie in vitro en produit de dégradation de l'acétolactate par le mélange (système enzymatique) ala rac/D-AAO/cat/AHASII de façon totale.
5,9 mg d’Isogro ou Celtone® 13C sont incubés avec 6 unités d’alanine racémase (SigmaAldrich), 3 unités de D-AAO ( Sigma-Aldrich), 1 mg d’acétolactate synthase, 5 unités de catalase (Sigma-Aldrich), ImM FAD (Flavine adénine dinucléotide), ImM PLP (pyridoxal phosphate), ImM ThPP (thiamine pyrophosphate) dans 500 ul de tampon 20mM Tris pH 7,5 pendant 24 Heures à 30°C. Le mélange est ensuite directement analysé par résonance magnétique nucléaire grâce à une expérience de type 13C-HSQC.
De la même façon, la L-alanine présente dans l’hydrolysat d'algue commercial (Celtone® ou Isogro®) a été éliminée par transformation en pyruvate puis en produits de dégradation de l'acétolactate par ce système, comme le montre la Figure 14.
Ce procédé a finalement été appliqué à un hydrolysat commercial d'algues entièrement deutéré (Celtone® ou Isogro®) et préalablement traité par de la Pronase. La deutériation complète des acides aminés pouvant influer sur l’affinité ou la cinétique de la réaction enzymatique, il était nécessaire de vérifier que ce procédé s'appliquait également.
0,5 g de hydrolysat commercial deutéré préalablement traité par de la pronase est incubé dans 20ml de tampon Tris 20mM pH 7,5 en présence de 3,2 mg d’alanine racémase, 100 unités de D-AAO (D-acid amino oxydase, Sigma-Aldricb), 100 unités de catalase (Sigma-Aldrich), 7,5 mg ALS (acétolactate synthase), ImM FAD (Flavine adénine dinucléotide), ImM PLP (pyridoxal phosphate), ImM ThPP (thiamine pyrophosphate) pendant 24H à 37°C. Le milieu a subi ensuite un choc thermique pendant 30 minutes à 95 degrés puis est centrifugé pendant 30 min à 40000g. Le mélange a été ensuite directement analysé par résonance magnétique nucléaire grâce à une expérience de type ld-deuterium.
Comme le montre la Figure 15 où sont présentés les spectres lD-deuterium du milieu deutéré contrôle et du milieu deutéré traité, le signal correspondant au méthyle de l'alanine a disparu alors que les autres signaux des méthyles ne sont pas affectés.
Pour vérifier que le traitement n'affecte essentiellement que l'alanine, la concentration des différents acides aminés a été mesurée avant et après traitement. Comme le montre la Figure 16, l'alanine est totalement éliminée du milieu. Seuls deux autres acides aminés sont faiblement convertis par le traitement : la L-serine et la L-cystéine dont les quantités initiales diminuent de 25 % à 35 %. En effet, une faible activité serine racémase a été rapportée pour l'alanine racémase de A stearothermophilus (Patrick et al., 2002). La quantité de tryptophane est nulle dans l’hydrolysat initial; celui-ci étant détruit lors de l'hydrolyse des microorganismes.
d) Ajout au milieu de culture appauvri obtenu à l’étape c) de l’alanine marquée et incorporation de l’alanine marquée dans les protéines exprimées par des bactéries cultivées sur le milieu de culture ainsi obtenu
De la même façon que pour le milieu riche obtenu à partir d’un hydrolysat appauvri en isoleucine, leucine et valîne, ci-dessus, aucune différence de croissance significative de la culture bactérienne n’a été observée entre un milieu M9 additionné de deux grammes d'hydrolys ai commercial (Celtone® ou Isogro®) et le même milieu additionné de deux grammes d'hydrolysat commercial (Celtone® ou Isogro®) appauvri en alanine (résultat non montré). Le niveau d'expression après induction d’une protéine témoin (ubiquitine) n’a pas été modifié en comparaison d'un milieu riche classique (résultat non montré).
Le niveau d'incorporation de l’alanine j-Af a été évalué en utilisant l'ubiquitine comme système modèle. Des cellules de la souche BL21(DE3) de E. coli ont été transformées avec un plasmide pET41c (Novagen) portant le gène codant pour l'ubiquitine humaine précédée d'un tag polyhistidine (pET4ic-His-Ubi). Ces cellules ont d'abord été adaptées en milieu deutéré comme décrit précédemment puis transférées dans un milieu M9/D2O contenant 1 g/L 15ND4C1, 2 g/L of U-[2H], U-[12C], glucose et 2 g/L de hydrolysat deutéré hydrolysé riche ou appauvri en alanine (le volume utilisé correspond à 70 % du volume total). Quand la densité optique à 600 nm atteint 1.5, le précurseur est dilué dans le volume restant du milieu puis ajouté à la culture. Après 1 heure, l'expression de la protéine est induite par l’addition d’IPTG à une concentration de 1 mM pendant 3 heures à 37°C. L'ubiquitine est ensuite purifiée par chromatographie sur colonne NiNTA (Qiagen).
La quantité optimale de d’alanine 3- CH3 nécessaire pour obtenir une incorporation quasi complète dans la protéine surexprimée a été déterminée par une série de cultures dans lesquelles des quantités croissantes de ce précurseur marqué 13C,H sur un méthyle sont ajoutés une heure avant induction pour des concentrations finales de (0, 100, 200, 400, 600, 800, 1000 et 1400 mg/L pour le milieu M9 additionné d'hydrolysat commercial non traité) et de (0, 10, 20, 30, 50, 75, 100, 150 et 250 mg/1 pour le milieu M9 additionné d'hydrolysat appauvri en alanine). Pour chaque culture, de la méthionine deutérée et marquée 13CH3 sur le méthyl ε a été ajoutée à 200 mg/L.
Le milieu ainsi appauvri a été ensuite additionné de 3- CH3-alanine. Il a été ensuite comparé le niveau d’incorporation de l'alanine dans l'ubiquitine avec un milieu M9 additionné de deux grammes d'hydrolysat commercial (Celtone® ou Isogro®) et avec le milieu M9 additionné de deux grammes d'hydrolysat commercial (Celtone® ou Isogro®) appauvri puis recomplété avec l’alanine marquée (Figure 17). Le niveau d'incorporation de l'alanine marquée dans un milieu riche classique nécessite pour atteindre 90 % une quantité de l’ordre du gramme d'alanine par litre. Dans un milieu minimum additionné d’hydrolysat appauvri puis recomplété, il ne faut que 300 mg/L d'alanine pour atteindre un tel niveau de marquage, une quantité proche de celle requise pour un milieu minimum M9.
REFERENCES
Barak Z. étal., 1987, J Bacteriol. 169:3750-6.
D’Aniello A., et al., 1993, Analytical Biochemistry, 213:290-295.
Gardner K.H. and Kay L.E., 1998, Annu Rev Biophys Biomol Struct. 27:357-406.
Kerfah R., et al., 2015, J. Curr Opin Struct Biol. 32:113-22.
Monti D., et al., 2011, Chemical Reviews, 111:4111-4140.
Mutaguchi Y., et al., 2013, Journal of Bacteriology, 195:5207-5215.
Pailla K., et al., 2000, Clinical Chemïstry, 46:848-853.
Patrick W.M., et al., 2002, ChemBioChem, 3:789-792.
Ruschak A.M. and Kay L.E., 2010, J Biomol NMR. 46:75-87.
Saegusa et al., 1977, The Journal of Organic Chemïstry, 42:2797-2798.
Sambrook, Fritsch and Maniatis. 1989, Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory, Cold Spring Harbor, N.Y.
Shaw J.P. étal., 1997, Bîochemistry, 36:1329-42,
Smit B.A., et al., 2005, Appl Environ Microbiol. 71:303-11,
Claims (11)
- REVENDICATIONS1. Méthode de préparation d’un hydrolysat de microorganisme dont au moins un acide aminé et/ou un précurseur d’acide aminé est marqué, comprenant les étapes suivantes :a) obtenir un hydrolysat de microorganisme en solution dans lequel tous les acides aminés se trouvent sous la forme libre,b) remplacer dans l’hydolysat obtenu à l’étape a) au moins un acide aminé sous la forme L et son cétoacide s’il est présent, par le même acide aminé mais sous une forme marquée ou par son précurseur marqué.
- 2. Méthode selon la revendication 1, caractérisée en ce que l’étape b) comprend les étapes suivantes :bl) appauvrir l’hydrolysat obtenu à l’étape a) en au moins un acide aminé sous la forme L et son cétoacide s’il est présent, par élimination de l’hydrolysat dudit acide aminé et de son cétoacide, et b2) ajouter à l’hydrolysat appauvri obtenu à l’étape bl) ledit acide aminé éliminé mais sous une forme marquée et/ou son précurseur marqué.
- 3. Méthode selon l’une quelconque des revendications 1 à 2, caractérisée en ce que ledit acide aminé remplacé est choisi parmi la L-leucine, la L-valine et la L-isoleucine.
- 4. Méthode selon la revendication 2 ou la revendication 3, caractérisée en ce que ledit acide aminé remplacé est choisi parmi la L-leucine, la L-valine et la L-isoleucine et en ce que l’étape bl) comprend les étapes suivantes :- traitement de l’hydrolysat obtenu à l’étape a) par un mélange de trois enzymes constitué d’une isoleucine 2-épimérase (EC 5.1.1.21), une D-aminoacide oxydase (EC 1.4.3.3) et une catalase (EC 1.11.1.6), et- élimination de l’hydrolysat traité desdites 3 enzymes et des cétoacides produits lors du traitement enzymatique de l’hydrolysat.
- 5. Méthode selon l’une quelconque des revendications 1 à 4, caractérisée en ce que ledit acide aminé et son cétoacide s’il est présent sont remplacés par le même acide aminé mais marqué par au moins un isotope choisi par le carbone-13, l’hydrogène-2 et l’azote-15.
- 6. Méthode selon l’une quelconque des revendications 1 à 5, caractérisée en ce que :- la L-isoleucine et son cétoacide s’il est présent sont remplacés par la L-isoleucine marquée, le 2-oxobutanoate marqué, le 2-hydroxy-2-éthyl-3-oxobutanoate marqué, le 2,3dihydroxy-3-méthylpentanoate marqué et/ou le 3-méthyl-2-oxopentanoate marqué, et/ou- la L-leucine et L-valine et leurs cétoacides s’ils sont présents sont remplacés par la Lleucine marquée, la L-valine marquée, le 2-hydroxy-2-méthyl-3-oxobutanoate marqué, le 3hydroxy-3-méthyl-2-oxobutanoate marqué, le 2,3-dihydroxy-3-méthylbutanoate marqué, le 3méthyl-2-oxobutanoate marqué et/ou le 4-méthyl-2-oxopentanoate marqué.
- 7. Méthode de préparation d’un milieu de culture riche, dont au moins un acide aminé et/ou un précurseur d’acide aminé est marqué, comprenant les étapes suivantes :i) préparer un hydrolysat de microorganisme dont au moins un acide aminé et/ou un précurseur d’acide aminé est marqué selon l’une quelconque des revendications 1 à 6, etj) constituer un milieu de culture riche comprenant l’hydrolysat obtenu à l’étape i) et un milieu de culture minimum.
- 8. Milieu de culture riche comprenant un hydrolysat de microorganisme ledit hydrolysat étant susceptible d’être obtenu selon la revendication 1 à 6.
- 9. Méthode de marquage d’une protéine par culture d’une cellule procaryote ou eucaryote, comprenant la culture de ladite cellule sur un milieu de culture riche selon la revendication 8.
- 10. Méthode de marquage d’une protéine recombinante dans un système acellulaire comprenant un milieu réactionnel, caractérisée en ce que le milieu réactionnel comprend un hydrolysat de microorganisme obtenu selon l’une quelconque des revendications 1 à 6.
- 11. Kit comprenant :- un hydrolysat appauvri susceptible d’être obtenu à l’étape bl) de la revendication 2, dans lequel au moins un acide aminé et son cétoacide s’il est présent est éliminé, ou un milieu de culture riche comprenant ledit hydrolysat appauvri et un milieu minimum, et- ledit acide aminé éliminé mais sous une forme marquée et/ou son précurseur marqué.oOICD
C\î oO 1 1 I Z3 <D CD CD LO όLOCD oC\i
Priority Applications (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| FR1657906A FR3055337A1 (fr) | 2016-08-24 | 2016-08-24 | Methode de preparation de milieux de culture riches pour le marquage d'acides amines incorpores dans des proteines |
| PCT/EP2017/071380 WO2018037097A1 (fr) | 2016-08-24 | 2017-08-24 | Méthode de préparation de milieux de culture riches pour le marquage d'acides aminés incorporés dans des protéines |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| FR1657906A FR3055337A1 (fr) | 2016-08-24 | 2016-08-24 | Methode de preparation de milieux de culture riches pour le marquage d'acides amines incorpores dans des proteines |
| FR1657906 | 2016-08-24 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| FR3055337A1 true FR3055337A1 (fr) | 2018-03-02 |
Family
ID=57590595
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| FR1657906A Withdrawn FR3055337A1 (fr) | 2016-08-24 | 2016-08-24 | Methode de preparation de milieux de culture riches pour le marquage d'acides amines incorpores dans des proteines |
Country Status (2)
| Country | Link |
|---|---|
| FR (1) | FR3055337A1 (fr) |
| WO (1) | WO2018037097A1 (fr) |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1999011589A1 (fr) * | 1997-09-02 | 1999-03-11 | Martek Biosciences Corporation | Proteines marquees 13c, 15n, 2h destinees a des determinations de structure par rmn et procede de preparation associe |
| EP1995324A1 (fr) * | 2007-05-22 | 2008-11-26 | ETH Zurich | Système acellulaire de synthèse de protéines deutériées pour études structurelles et fonctionnelles par spectroscopie RMN |
| CN101824443A (zh) * | 2009-03-06 | 2010-09-08 | 上海工业生物技术研发中心 | 一种去除制备中性氨基酸的反应体系中的丙氨酸的方法 |
| WO2011083356A1 (fr) * | 2010-01-06 | 2011-07-14 | Commissariat A L'energie Atomique Et Aux Energies Alternatives | Procédé de marquage isotopique spécifique de groupements méthyle de val, leu et ile |
| EP2695873A1 (fr) * | 2012-08-08 | 2014-02-12 | Commissariat A L'energie Atomique Et Aux Energies Alternatives | Dérivés de cétoacides alpha-hydroxylés chiraux étiquetés, procédé de préparation de ces dérivés et leur utilisation |
-
2016
- 2016-08-24 FR FR1657906A patent/FR3055337A1/fr not_active Withdrawn
-
2017
- 2017-08-24 WO PCT/EP2017/071380 patent/WO2018037097A1/fr not_active Ceased
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1999011589A1 (fr) * | 1997-09-02 | 1999-03-11 | Martek Biosciences Corporation | Proteines marquees 13c, 15n, 2h destinees a des determinations de structure par rmn et procede de preparation associe |
| EP1995324A1 (fr) * | 2007-05-22 | 2008-11-26 | ETH Zurich | Système acellulaire de synthèse de protéines deutériées pour études structurelles et fonctionnelles par spectroscopie RMN |
| CN101824443A (zh) * | 2009-03-06 | 2010-09-08 | 上海工业生物技术研发中心 | 一种去除制备中性氨基酸的反应体系中的丙氨酸的方法 |
| WO2011083356A1 (fr) * | 2010-01-06 | 2011-07-14 | Commissariat A L'energie Atomique Et Aux Energies Alternatives | Procédé de marquage isotopique spécifique de groupements méthyle de val, leu et ile |
| EP2695873A1 (fr) * | 2012-08-08 | 2014-02-12 | Commissariat A L'energie Atomique Et Aux Energies Alternatives | Dérivés de cétoacides alpha-hydroxylés chiraux étiquetés, procédé de préparation de ces dérivés et leur utilisation |
Non-Patent Citations (3)
| Title |
|---|
| ISABEL AYALA ET AL: "An efficient protocol for the complete incorporation of methyl-protonated alanine in perdeuterated protein", JOURNAL OF BIOMOLECULAR NMR., vol. 43, no. 2, 1 February 2009 (2009-02-01), NL, pages 111 - 119, XP055346837, ISSN: 0925-2738, DOI: 10.1007/s10858-008-9294-7 * |
| KERFAH RIME ET AL: "Scrambling free combinatorial labeling of alanine-[beta], isoleucine-[delta]1, leucine-proS and valine-proS methyl groups for the detection of long range NOEs.", JOURNAL OF BIOMOLECULAR NMR JAN 2015, vol. 61, no. 1, January 2015 (2015-01-01), pages 73 - 82, XP002767379, ISSN: 1573-5001 * |
| RIME KERFAH ET AL: "Methyl-specific isotopic labeling: a molecular tool box for solution NMR studies of large proteins", CURRENT OPINION IN STRUCTURAL BIOLOGY, vol. 32, 1 June 2015 (2015-06-01), GB, pages 113 - 122, XP055346812, ISSN: 0959-440X, DOI: 10.1016/j.sbi.2015.03.009 * |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2018037097A1 (fr) | 2018-03-01 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| Parmeggiani et al. | Synthetic and therapeutic applications of ammonia-lyases and aminomutases | |
| Ito et al. | Conserved pyridoxal protein that regulates Ile and Val metabolism | |
| US11371068B2 (en) | Extracellular heme production method using metabolically engineered microorganism | |
| Seto | The stickland reaction | |
| Pranawidjaja et al. | Analysis of heme biosynthetic pathways in a recombinant Escherichia coli | |
| Das et al. | Amino acid chirality: stereospecific conversion and physiological implications | |
| Takahashi | d-Aspartate oxidase: Distribution, functions, properties, and biotechnological applications | |
| Shimekake et al. | A novel thermostable d-amino acid oxidase of the thermophilic fungus Rasamsonia emersonii strain YA | |
| Seo et al. | Deracemization of unnatural amino acid: homoalanine using D-amino acid oxidase and ω-transaminase | |
| Shi et al. | Enhancement of substrate supply and ido expression to improve 4-hydroxyisoleucine production in recombinant Corynebacterium glutamicum ssp. lactofermentum | |
| EP2479272A1 (fr) | Procédé de production de monatine | |
| Adams | 13 Amino Acid Racemases and Epimerases | |
| Zuo et al. | Study of amino acids as nitrogen source in Chlamydomonas reinhardtii | |
| Zhu et al. | Combining fermentation to produce O-succinyl-l-homoserine and enzyme catalysis for the synthesis of l-methionine in one pot | |
| Yan et al. | In vitro reconstitution of a bacterial ergothioneine sulfonate catabolic pathway | |
| Qi et al. | Development of a multi-enzymatic desymmetrization and its application for the biosynthesis of L-norvaline from DL-norvaline | |
| Hu et al. | A novel type alanine dehydrogenase from Helicobacter aurati: molecular characterization and application | |
| KR102149044B1 (ko) | 2-히드록시 감마 부티로락톤 또는 2,4-디히드록시-부티레이트 의 제조 방법 | |
| Tavares et al. | The Methanosarcina mazei MM2060 gene encodes a bifunctional kinase/decarboxylase enzyme involved in cobamide biosynthesis | |
| Shin et al. | 5-Aminolevulinic acid biosynthesis in Escherichia coli coexpressing NADP-dependent malic enzyme and 5-aminolevulinate synthase | |
| FR3055337A1 (fr) | Methode de preparation de milieux de culture riches pour le marquage d'acides amines incorpores dans des proteines | |
| Yan et al. | Production of hydroxytyrosol through whole-cell bioconversion from L-DOPA using engineered Escherichia coli | |
| US20240010996A1 (en) | Bacterial unspecific peroxygenases (BUPO's) and methods and uses thereof | |
| JP2008109924A (ja) | 4−ヒドロキシ−l−イソロイシン又はその塩の製造法 | |
| KR102005664B1 (ko) | 아미노레불린산 생산용 형질전환메탄자화균 및 이의 용도 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PLFP | Fee payment |
Year of fee payment: 2 |
|
| PLSC | Publication of the preliminary search report |
Effective date: 20180302 |
|
| PLFP | Fee payment |
Year of fee payment: 3 |
|
| ST | Notification of lapse |
Effective date: 20200406 |