JP2004018480A - 5−置換−1,2,4−トリアゾール−3−カルボン酸エステル誘導体の製造方法 - Google Patents
5−置換−1,2,4−トリアゾール−3−カルボン酸エステル誘導体の製造方法 Download PDFInfo
- Publication number
- JP2004018480A JP2004018480A JP2002177822A JP2002177822A JP2004018480A JP 2004018480 A JP2004018480 A JP 2004018480A JP 2002177822 A JP2002177822 A JP 2002177822A JP 2002177822 A JP2002177822 A JP 2002177822A JP 2004018480 A JP2004018480 A JP 2004018480A
- Authority
- JP
- Japan
- Prior art keywords
- compound
- mol
- general formula
- solution
- optionally substituted
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
- 0 *c1nc(*)n[n]1 Chemical compound *c1nc(*)n[n]1 0.000 description 7
- SQZKDPNCFXYDCE-UHFFFAOYSA-N CCOC(C1OC1NNC(c1ccccc1)=N)=O Chemical compound CCOC(C1OC1NNC(c1ccccc1)=N)=O SQZKDPNCFXYDCE-UHFFFAOYSA-N 0.000 description 1
- SISQKQNJHCDULL-UHFFFAOYSA-N COC(c1ccccc1)=N Chemical compound COC(c1ccccc1)=N SISQKQNJHCDULL-UHFFFAOYSA-N 0.000 description 1
- CUPOOAWTRIURFT-UHFFFAOYSA-N N#Cc1ccc[s]1 Chemical compound N#Cc1ccc[s]1 CUPOOAWTRIURFT-UHFFFAOYSA-N 0.000 description 1
Landscapes
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Heterocyclic Compounds Containing Sulfur Atoms (AREA)
- Pyridine Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
Abstract
【課題】医薬品を製造するための中間体として有用な5−置換−1,2,4−トリアゾール−3−カルボン酸エステル誘導体ならびにアシルアミドラゾン誘導体、およびその製造方法を提供する。
【解決手段】一般式(I)で表わされる化合物およびそれらの製造方法を見出した。
【化1】
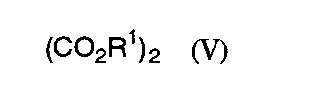
(式中、R1は置換されていてもよいアルキル、R2は置換されていてもよいアルキル、置換されていてもよいアラルキル、置換されていてもよいアリール、または置換されていてもよいヘテロアリール)
【選択図】 なし
【解決手段】一般式(I)で表わされる化合物およびそれらの製造方法を見出した。
【化1】
(式中、R1は置換されていてもよいアルキル、R2は置換されていてもよいアルキル、置換されていてもよいアラルキル、置換されていてもよいアリール、または置換されていてもよいヘテロアリール)
【選択図】 なし
Description
【0001】
【発明の属する技術分野】
本発明は、5−置換−1,2,4−トリアゾール−3−カルボン酸エステル誘導体、アシルアミドラゾン誘導体、およびそれらの製造方法に関する。
【0002】
【従来の技術】
J. Chem. Soc. (C), 1968, 824−830にはアシルヒドラジン誘導体とイミデート誘導体から種々の3,5−ジ置換−1,2,4−トリアゾール誘導体を製造する方法が記載されている。またWO00/39086には、HIVインテグレース阻害化合物を合成する際の重要中間体として5−(非置換または置換)−1,2,4−トリアゾール−3−カルボン酸エステル誘導体が記載されている。しかし、どちらの文献にも5−置換−1,2,4−トリアゾール−3−カルボン酸エステル誘導体の製造方法については記載されていない。
5−置換−1,2,4−トリアゾール−3−カルボン酸エステル誘導体の製造方法は、以下の文献に記載されている(J. Chem. Soc. Perkin Trans 1, 1977, 761−764、Collect. Czech. Chem. Commun., 49, 1984, 2492−2495、J. Heterocyclic Chem., 25, 1988, 651−654および WO00/75122)。J. Chem. Soc. Perkin Trans 1, 1977, 761−764には以下に示すアシルアミドラゾン誘導体(A)を経由して加熱閉環する製造方法が記載されている。また、Collect. Czech. Chem. Commun., 49, 1984, 2492−2495、J. Heterocyclic Chem., 25, 1988, 651−654および WO00/75122には以下に示すアシルアミドラゾン誘導体(B)を経由して加熱閉環する製造方法が記載されている。
【化16】
(式中、RAは置換されていてもよいアルキル;RBは水素原子、置換されていてもよいアルキル、置換されていてもよいアルケニル、置換されていてもよいアルキニル、置換されていてもよいアラルキル、置換されていてもよいアリール、または置換されていてもよいヘテロアリール)
しかし、上記の5−置換−1,2,4−トリアゾール−3−カルボン酸エステル誘導体の製造方法において、アシルアミドラゾン誘導体(A)を経由する場合は、反応条件がピリジン還流中と高温度であり、収率が低く、工業的製法として満足いく方法ではなかった。
アシルアミドラゾン誘導体(B)を経由する場合は、一般に反応温度が165℃以上と高くかつ生成する水を減圧下除去する必要があるなど、工業的に行うには問題があった。しかし、WO00/75122には120℃付近で反応させることができ、さらに生成する水を除去しなくてもよい製造方法が記載されていた。
【0003】
【発明が解決しようとする課題】
医薬品の中間体として有用である5−置換−1,2,4−トリアゾール−3−カルボン酸エステル誘導体、該誘導体を製造するにあたって有用な中間体であるアシルアミドラゾン誘導体、およびそれらを工業的かつ経済的に製造する方法が望まれている。
【0004】
【課題を解決するための手段】
上記の事情に鑑み、本発明者らは、鋭意検討した結果、以下に示す5−置換−1,2,4−トリアゾール−3−カルボン酸エステル誘導体、アシルアミドラゾン誘導体、およびそれらの製造方法を見出した。
【0005】
すなわち、本発明は、
1)一般式(IV):
【化17】
(式中、R1は置換されていてもよいアルキル;R2は置換されてもよいアルキル、置換されていてもよいアラルキル、置換されていてもよいアリール、または置換されていてもよいヘテロアリール)
で示される化合物またはその水和物を100℃以下で閉環させることを特徴とする、一般式(I):
【化18】
(式中、R1およびR2は前記と同意義である)
で示される化合物の製造方法。
2)一般式(II):
【化19】
(式中、R1は1)と同意義である)
で示される化合物と、一般式(III):
【化20】
(式中、R2は1)と同意義であり;R3は置換されていてもよいアルキル)
で示される化合物を縮合させて、一般式(IV):
【化21】
(式中、R1およびR2は1)と同意義である)
で示される化合物またはその水和物を得る工程;および
式(IV)で示される化合物またはその水和物を閉環させる工程
を包含する、一般式(I):
【化22】
(式中、R1およびR2は1)と同意義である)
で示される化合物の製造方法。
3)一般式(V):
【化23】
(式中、R1は1)と同意義である)
で示される化合物と、ヒドラジンを縮合させて一般式(II):
【化24】
(式中、R1は1)と同意義である)
で示される化合物を得る工程を包含する、2)記載の製造方法。
4)一般式(II):
【化25】
(式中、R1は1)と同意義である)
で示される化合物に、一般式(III):
【化26】
(式中、R2は1)と同意義であり;R3は2)と同意義である)
で示される化合物を縮合させて得られる、一般式(IV):
【化27】
(式中、R1およびR2は1)と同意義である)
で示される化合物の製造方法。
5)一般式(V):
【化28】
(式中、R1は1)と同意義である)
で示される化合物と、ヒドラジンを縮合させて、一般式(II):
【化29】
(式中、R1は1)と同意義である)
で示される化合物を得る工程を包含する、4)記載の製造方法。
6)R1が低級アルキルである1)から5)のいずれかに記載の製造方法。
7)R1がエチルである1)から5)のいずれかに記載の製造方法。
8)R2が低級アルキルである1)から7)のいずれかに記載の製造方法。
9)R3が低級アルキルである2)から8)のいずれかに記載の製造方法。
10)反応溶媒としてエタノールを用いる1)から9)のいずれかに記載の製造方法。
11)反応温度が90℃以下である1)から3)のいずれかに記載の製造方法。
12)一般式(IV−1):
【化30】
(式中、R1は1)と同意義であり;R4は置換されていてもよいヘテロアリール)
で示される化合物、もしくはその塩、またはその溶媒和物。
13)一般式(I−1):
【化31】
(式中、R1は1)と同意義であり;R4は12)と同意義である)
で示される化合物、もしくはその塩、またはその溶媒和物。
【0006】
本明細書中で用いられる「ハロゲン」とはフッ素、塩素、臭素およびヨウ素を意味する。
本明細書中、単独でもしくは他の用語と組み合わせて用いられる「アルキル」なる用語は、1〜10個の炭素原子を有する、直鎖または分枝鎖の1価の炭化水素基を包含する。例えば、メチル、エチル、n−プロピル、イソプロピル、n−ブチル、イソブチル、sec−ブチル、tert−ブチル、n−ペンチル、イソペンチル、neo−ペンチル、n−ヘキシル、イソヘキシル、n−ヘプチル、n−オクチル、n−ノニル、またはn−デシル等が挙げられる。なお本明細書中で用いている「低級アルキル」は、C1〜C6アルキルを意味する。
R1のアルキルとしては、C1〜C6アルキルが好ましい。さらにC1〜C4アルキルが好ましい。
R2またはR3のアルキルとしては、C1〜C6アルキルが好ましい。さらにC1〜C4アルキルが好ましい。
本明細書中で用いられる「置換されていてもよいアルキル」における置換基としては、ハロゲン(例えば、フッ素、塩素、臭素、またはヨウ素)、C2〜C4アルケニル(例えば、ビニル、アリル、2−ブテニル等)、C2〜C4アルキニル(例えば、エチニル、プロパルギル、2−ブチニル等)、C3〜C6シクロアルキル(例えば、シクロプロピル、シクロブチル、シクロペンチル、またはシクロヘキシル等)、ヒドロキシ、C1〜C4アルキルオキシ(例えば、メチルオキシまたは、エチルオキシ等)、置換されていてもよいアラルキルオキシ(例えば、ベンジルオキシまたは4−メチルオキシベンジルオキシ等)、メルカプト、C1〜C4アルキルチオ(例えば、メチルチオ等)、ニトロ、置換されていてもよいアミノ(例えば、アミノ、メチルアミノ、またはジメチルアミノ等)等が挙げられる。これら置換基は、全ての可能な位置で1個以上置換しうる。好ましい置換基としては、ハロゲン、C1〜C4アルキルオキシ、またはC1〜C4アルキルチオが挙げられる。
本明細書中、単独でもしくは他の用語と組み合わせて用いられる「アリール」なる用語は、単環状もしくは縮合環状芳香族炭化水素基を包含する。例えば、フェニル、1−ナフチル、2−ナフチル、またはアントリル等が挙げられる。
R2の好ましいアリールは、フェニルが挙げられる。
本明細書中、単独でもしくは他の用語と組み合わせて用いられる「ヘテロアリール」なる用語は、任意に選ばれる、酸素原子、硫黄原子又は窒素原子を環内に1個以上含む5〜6員の芳香環基を包含する。これらはさらに前記「アリール」、または他の「ヘテロアリール」と縮合していてもよい。ヘテロアリールが縮合環である場合は、結合可能なすべての位置で結合し得る。例えば、ピロリル(例えば、1−ピロリル等)、インドリル(例えば、3−インドリル等)、カルバゾリル(例えば、3−カルバゾリル等)、イミダゾリル(例えば、4−イミダゾリル等)、ピラゾリル(例えば、3−ピラゾリル等)、ベンゾイミダゾリル(例えば、2−ベンゾイミダゾリル等)、インダゾリル(例えば、3−インダゾリル等)、インドリジニル(例えば、6−インドリジニル等)、ピリジル(例えば、4−ピリジル等)、キノリル(例えば、5−キノリル等)、イソキノリル(例えば、3−イソキノリル等)、アクリジル(例えば、1−アクリジル等)、フェナントリジニル(例えば、2−フェナントリジニル等)、ピリダジニル(例えば、3−ピリダジニル等)、ピリミジニル(例えば、4−ピリミジニル等)、ピラジニル(例えば、2−ピラジニル等)、シンノリニル(例えば、3−シンノリニル等)、フタラジニル(例えば、2−フタラジニル等)、キナゾリニル(例えば、2−キナゾリニル等)、イソオキサゾリル(例えば、3−イソオキサゾリル等)、ベンゾイソオキサゾリル(例えば、3−ベンゾイソオキサゾリル等)、オキサゾリル(例えば、2−オキサゾリル等)、ベンゾオキサゾリル(例えば、2−ベンゾオキサゾリル等)、ベンゾオキサジアゾリル(例えば、4−ベンゾオキサジアゾリル等)、イソチアゾリル(例えば、3−イソチアゾリル等)、ベンゾイソチアゾリル(例えば、2−ベンゾイソチアゾリル等)、チアゾリル(例えば、4−チアゾリル等)、ベンゾチアゾリル(例えば、2−ベンゾチアゾリル等)、フリル(例えば、2−フリル等)、ベンゾフリル(例えば、3−ベンゾフリル等)、チエニル(例えば、2−チエニル等)、ベンゾチエニル(例えば、2−ベンゾチエニル等)、テトラゾリル、オキサジアゾリル(例えば、1,3,4−オキサジアゾリル等)、オキサゾリル、チアジアゾリル(例えば、1,3,4−チアジアゾリル等)、4H−1,2,4−トリアゾリル、キノキサリニル、または2−ピリドン−3−イル等が挙げられる。
R2またはR4の好ましいヘテロアリールとしては、2−フリル、2−チエニル、または4−ピリジルが挙げられる。
本明細書中、単独でもしくは他の用語と組み合わせて用いられる「アラルキル」なる用語は、前記「アルキル」に前記「アリ−ル」または前記「ヘテロアリール」が1または2ヶ所以上置換したものを包含する。例えば、ベンジル、フェニルエチル(例えば、1−フェニルエチル等)、フェニルプロピル(例えば、1−フェニルプロピルまたは3−フェニルプロピル等)、ナフチルメチル(例えば、1−ナフチルメチル等)、アントリルメチル(例えば、9−アントリルメチル等)、フリルメチル(例えば、2−フリルメチル等)、チエニルメチル(例えば、2−チエニルメチル等)、ピリジルメチル(例えば、4−ピリジルメチル等)、およびピリミジニルメチル(4−ピリミジニルメチル等)等が挙げられる。
R2の好ましいアラルキルとしては、ベンジル、1−フェニルエチル、2−フリルメチル、または2−チエニルメチルが挙げられる。
本明細書中で用いられる「置換されていてもよいアリール」、「置換されていてもよいヘテロアリール」および「置換されていてもよいアラルキル」における置換基としては、例えば、ハロゲン(例えば、フッ素、塩素、臭素、またはヨウ素)、低級アルキル(例えば、メチル、エチル、n−プロピル、イソプロピル、n−ブチル、イソブチル、sec−ブチル、tert−ブチル、n−ペンチル、イソペンチル、またはneo−ペンチル等)、C2〜C4アルケニル(例えば、ビニル、アリル、2−ブテニル等)、C2〜C4アルキニル(例えば、エチニル、プロパルギル、2−ブチニル等)、C3〜C6シクロアルキル(例えば、シクロプロピル、シクロブチル、シクロペンチル、またはシクロヘキシル等)、ハロ低級アルキル(例えば、トリフルオロメチル等)、ヒドロキシ、C1〜C4アルキルオキシ(例えば、メチルオキシ、またはエチルオキシ等)、C6〜C10アリ−ルオキシ(例えば、フェニルオキシ等)、メルカプト、C1〜C4アルキルチオ(例えば、メチルチオ等)、ニトロ、シアノ、置換されていてもよいアミノ(例えば、アミノ、メチルアミノ、ジメチルアミノ、ジエチルアミノ、またはホルミルアミノ等)、またはC1〜C4アルキルスルホニル(例えば、メチルスルホニル等)等が挙げられる。これら置換基は、全ての可能な位置で1個以上置換しうる。好ましい置換基としては、ハロゲン、低級アルキル、ハロ低級アルキル、またはC1〜C4アルキルオキシが挙げられる。
本明細書中で用いられる「置換されていてもよいアミノ」なる用語は、前記「アルキル」、前記「アラルキル」、前記「アリール」、または前記「ヘテロアリール」が同一であっても異なっていてもよく、1または2個置換されていてもよいアミノを意味する。例えば、アミノ、C1〜C6アルキルでモノ置換またはジ置換されているアミノ(例えば、モノメチルアミンまたはジメチルアミン等)、C1〜C4アシルでモノ置換されているアミノ(例えば、ホルミルアミノまたはアセチルアミノ等)を意味する。好ましくは、アミノまたはメチルアミノが挙げられる。
本明細書中で用いられる「保護基」なる用語は、反応後容易に脱離することができる基を意味する。例えば、C1〜C4アルキルオキシC1〜C4アルキル(例えば、メトキシメチル、エトキシメチル、ジアルキルオキキシメチル、または2−メトキシ−2−プロピル等)、C6〜C12アリールC1〜C4アルキル(例えば、トリチル等)、アリル、ヒドロキシメチル、C1〜C4アシル(例えば、ホルミル等)、C1〜C6アルキルオキシカルボニル(例えば、tert−ブトキシカルボニル等)、C6〜C12アリールC1〜C4アルキルオキシカルボニル(例えば、9−フルオレニルメトキシカルボニル等)、C1〜C4アルキルスルホニル(例えば、メタンスルホニル等)、C6〜C12アリールスルホニル(例えば、p−トルエンスルホニル等)、C1〜C4アルキルでモノ置換またはジ置換されていてもよいスルファモイル(例えば、N,N−ジメチルスルファモイル等)、またはTHP(テトラヒドロピラニル)等が挙げられる。
本明細書中で用いられる「ヒドラジン」なる用語は、ヒドラジン、ヒドラジンの水和物、およびヒドラジンまたはヒドラジン水和物を含有する溶液を意味する。
一般式(I)で示される化合物のR1およびR2において、好ましい置換基の群を(a)〜(e)で示す。
R1は、(a)置換されていてもよいアルキル。
R2は、(b)置換基されていてもよいアルキル、(c)置換基されていてもよいアラルキル、(d)置換されていてもよいアリール、または(e)置換されていてもよいヘテロアリール。
一般式(I)で示される化合物の好ましい一群としては、[R1, R2]=[a, b], [a, c], [a, d], [a, e]。
一般式(IV)で示される化合物のR1およびR2において、好ましい置換基の群を(a)〜(e)で示す。
R1は、(a)置換されていてもよいアルキル。
R2は、(b)置換基されていてもよいアルキル、(c)置換基されていてもよいアラルキル、(d)置換されていてもよいアリール、または(e)置換されていてもよいヘテロアリール。
一般式(IV)で示される化合物の好ましい一群としては、[R1, R2]=[a, b], [a, c], [a, d], [a, e]。
【0007】
【発明の実施の形態】
以下に本発明の製造方法について、第1工程〜第3工程に分けて説明する。
第1工程
【化32】
(式中、R1は1)と同意義である)
本工程は、一般式(V)で示される化合物とヒドラジンを縮合することによって、一般式(II)で示される化合物を製造する工程である。
一般式(V)で示される化合物としては、市販の化合物または公知の方法(例えば、J. Am. Chem. Soc., 60, 1938, 2790−2792等)に準じて合成した化合物を用いることができる。
ヒドラジンとしては、ヒドラジン水和物(例えば、ヒドラジン一水和物等)またはヒドラジン水溶液(例えば、35%ヒドラジン水溶液等)が挙げられ、特にヒドラジン一水和物または35%ヒドラジン水溶液が好ましい。
ヒドラジンは一般式(V)に対して0.5〜1.5当量、好ましくは0.9〜1.1当量用いることができる。
反応溶媒は、アルコール類(例えば、メタノール、エタノール、n−プロパノール、イソプロパノール、n−ブタノール、イソブタノール、sec−ブタノール、またはtert−ブタノール等)、エーテル類(例えば、ジエチルエーテル、THF(テトラヒドロフラン)、またはジオキサン等)、ニトリル類(例えば、アセトニトリルまたはプロピロニトリル等)、ハロゲン化炭化水素類(例えば、塩化メチレン、ジクロロエタン、またはクロロホルム等)、芳香族炭化水素類(例えば、ベンゼン、トルエンまたはキシレン等)、またはDMF(N,N−ジメチルホルムアミド)が挙げられる。これらの溶媒を単独あるいは混合して用いることができる。アルコール類が好ましく、さらにエタノールが好ましい。
反応温度としては−80〜80℃が挙げられる。−50〜30℃が好ましい。
反応時間としては0.5〜24時間が挙げられる。1〜6時間が好ましい。
得られた一般式(II)で示される化合物は、単離精製せずに第2工程に使用することができる。あるいは公知の手段(例えば、クロマトグラフィー、再結晶など)で精製することができる。副生成物として生成するシュウ酸ジヒドラジドを含有したまま第2工程さらに第3工程を行うことができる。
なお、35%ヒドラジン水溶液を用いた方が、ヒドラジン一水和物を用いた時に比べ、工業的に好ましい温度で反応させることができる。
第2工程
【化33】
(式中、R1およびR2は1)と同意義である;R3は2)と同意義である)
本工程は、一般式(II)で示される化合物と一般式(III)で示される化合物を縮合することによって、一般式(IV)で示される化合物を製造する工程である。
一般式(II)で示される化合物としては、上記の第1工程で合成された化合物または公知の方法(例えば、Chem. Ber. 123, 10, 1990, 2031−2037,等)に準じて合成した化合物を用いることができる。
一般式(III)で示される化合物としては、公知の方法(例えば、J. Hetrocyclic Chem., 25, 1988, 651−654等)に準じて合成した化合物を用いることができる。
【化34】
例えば、ニトリル類(例えば、アセトニトリル、プロピオニトリル、ベンゾニトリル、または4−シアノピリジン等)を酸条件下(例えば、塩酸または塩化水素)、アルコール類(例えば、メタノール、エタノール、またはイソプロパノール等)と縮合して合成する。反応溶媒は、酢酸エチル、トルエン、またはTHF(テトラヒドロフラン)等を単独であるいは混合して用いることができる。酸は、ニトリル類に対して、0.5〜5当量、好ましくは1〜2当量用いることができる。反応温度としては−20〜80℃が挙げられ、0〜50℃が好ましい。反応時間としては1時間〜14日間が挙げられ、12時間〜7日間が好ましい。得られた一般式(III)で示される化合物は、単離精製せずに次の反応に用いることができる。
なお、塩化水素を用いた方が塩酸を用いたときに比べて、より短期間で一般式(III)で示される化合物を得ることができる。
一般式(III)は、一般式(II)に対して0.5〜3当量、好ましくは1〜1.5当量用いられる。
反応溶媒は、アルコール類(例えば、メタノール、エタノール、n−プロパノール、イソプロパノール、n−ブタノール、イソブタノール、sec−ブタノール、またはtert−ブタノール等)、エーテル類(例えば、ジエチルエーテル、THF(テトラヒドロフラン)、またはジオキサン等)、ニトリル類(例えば、アセトニトリルまたはプロピロニトリル等)、ハロゲン化炭化水素類(例えば、塩化メチレン、ジクロロエタン、またはクロロホルム等)、芳香族炭化水素類(例えば、ベンゼン、トルエンまたはキシレン等)、またはDMF(N,N−ジメチルホルムアミド)等が挙げらる。これらの溶媒を単独あるいは混合して用いることができる。アルコール類または芳香族炭化水素類が好ましい。さらにエタノールまたはトルエンが好ましい。
反応温度としては−40〜100℃が挙げられる。−20〜50℃が好ましい。
反応時間としては0.5〜24時間が挙げられる。1〜6時間が好ましい。
得られた一般式(IV)で示される化合物は、単離精製せずに第3工程に使用することができる。または公知の手段(例えば、クロマトグラフィー、再結晶など)で精製することができる。
第3工程
【化35】
(式中、R1またはR2は1)と同意義である)
本工程は、一般式(IV)で示される化合物を閉環させることによって、一般式(I)で示される化合物を製造する工程である。
一般式(IV)で示される化合物としては、上記の第2工程で合成した化合物を用いることができる。
反応溶媒は、アルコール類(例えば、メタノール、エタノール、n−プロパノール、イソプロパノール、n−ブタノール、イソブタノール、sec−ブタノール、またはtert−ブタノール等)、エーテル類(例えば、ジエチルエーテル、THF(テトラヒドロフラン)、またはジオキサン等)、ニトリル類(例えば、アセトニトリルまたはプロピロニトリル等)、ハロゲン化炭化水素類(例えば、塩化メチレン、ジクロロエタン、またはクロロホルム等)、芳香族炭化水素類(例えば、ベンゼン、トルエンまたはキシレン等)、DMF(N,N−ジメチルホルムアミド)、アセトン、または水等が挙げられる。これらの溶媒を単独あるいは混合して用いることができる。
なお、アセトンを用いることにより第1工程で副生成したシュウ酸ジヒドラジドを除くことができるので、副生成したシュウ酸ジヒドラジドが含有している場合は、アセトンとアルコール類またはニトリル類の混合溶媒が好ましい。さらにアセトンとエタノールまたはアセトニトリルの混合溶媒が好ましい。シュウ酸ジヒドラジドが含有していない場合は、アルコール類またはニトリル類が好ましい。さらにエタノールまたはアセトニトリルが好ましい。
反応溶媒として水を単独で用いた場合は、一般式(I)で表わされる化合物の収率は若干低くなるが、副生するシュウ酸ジヒドラジドあるいはトリアジンジオンを水で再結晶することにより容易に母液へ取り除くことができる。
反応温度としては30〜100℃が挙げられる。50〜100℃が好ましい。さらに、80〜100℃が好ましい。
反応時間としては0.5〜24時間が挙げられる。1〜6時間が好ましい。
得られた一般式(I)で示される化合物は、公知の手段(例えば、クロマトグラフィー、再結晶など)で単離精製することができる。
なお、反応溶媒がアルコール類の場合、第1工程から第3工程までをワンポットで行うこともできる。アルコール類としては、エタノールが好ましい。
一般式(I)で示される化合物を出発原料として、WO00/39086およびWO00/75122に記載の方法に従って、一般式(XI)で表わされる化合物を合成することができる。
【化36】
(式中、R1およびR2は1)と同意義である;Xは同一でも異なっていてもよく水素原子、ハロゲン、低級アルキル、または低級アルキルオキシが1〜5個置換していることを示す;Qは保護基。)
実施例中、以下の略号を使用する。
Me:メチル
Et:エチル
n−Pr:n−プロピル
i−Pr:イソプロピル
Tr:トリチル
THP:テトラヒドロピラニル
THF:テトラヒドロフラン
DMF:N,N−ジメチルホルムアミド
DMSO:ジメチルスルホキシド
元素分析において、Calcd. forは理論値、Foundは実測値を意味する。
1H−NMRはδ値をppmで表わし、s=一重線、d=二重線、t=三重線、q=四重線、m=多重線、br=幅広線を意味する。J値はHzで表わした。
【実施例】
【0008】
参考例
参考例1:化合物(IIIa)の合成
【化37】
4mol/L塩酸の酢酸エチル溶液150 mL(0.6 mol)にエタノール32.1 mL(0.55 mol)及び、アセトニトリル(VIa)26.1 mL(0.5 mol)を滴下し、0℃で6日間攪拌した。溶媒を留去し、結晶性残査に炭酸カリウム水溶液(K2CO3 76.0 g, 0.55mol, H2O 400 mL)及び塩化メチレン(400 mL, 200 mL)を加えて中和、抽出した。有機層を飽和食塩水(200 mL)で洗浄後、Na2SO4で乾燥し、溶媒留去して化合物(IIIa)の粗製物を72.24 g(無色油状)得た。(NMRより化合物(IIIa)の含有量を測定した。33.11 g, 0.38 mol, 収率 76%)
参考例2:化合物(IIIa)の合成
【化38】
トルエン138 mL、エタノール44.5 mL(0.77 mol)及び、アセトニトリル(VIa)36.0 mL(0.69 mol)の混合液に塩化水素28 g(0.77 mol)を10〜20℃で導入し、20℃で1日間攪拌した。氷冷攪拌しながら、エタノール(69 mL) を加え、トリエチルアミン111 mL (0.80 mol)で中和した。析出したトリエチルアミン塩酸塩を濾別、トルエン 138 mLで洗浄し、化合物(IIIa)の粗製溶液を307.16 g(無色油状)得た。(NMRより化合物(IIIa)の含有量を測定した。50.68 g,
0.58 mol, 収率 84%)
参考例3:化合物(IIIb)の合成
【化39】
4mol/L塩酸の酢酸エチル溶液103 mL(0.41 mol)にエタノール22.3 mL(0.38mol)及び、プロピオニトリル(VIb)19.0 g(0.34 mol)を滴下し、0℃で1週間攪拌した。溶媒を留去し、結晶性残査に炭酸カリウム水溶液(K2CO3 52.44 g,0.38 mol, H2O 100 mL)及び塩化メチレン(200 mL, 100 mL)を加えて中和、抽出した。有機層を飽和食塩水(100 mL)で洗浄後、Na2SO4で乾燥し、溶媒を留去して化合物(IIIc)の粗製物を45.31 g(無色油状)得た。(NMRより化合物(IIIc)の含有量を測定した。21.66 g, 0.21 mol, 収率 62%)
参考例4:化合物(IIIc)の合成
【化40】
4mol/L塩酸の酢酸エチル溶液82.5 mL(0.33 mol)にエタノール17.7 mL(0.30 mol)及び、ブチロニトリル(VIc)19.0 g(0.27 mol)を滴下し、0℃で1週間攪拌した。溶媒を留去し、結晶性残査に炭酸カリウム水溶液(K2CO3 41.80 g,0.30 mol, H2O 100 mL)及び塩化メチレン(200 mL, 100 mL)を加えて中和、抽出した。有機層を飽和食塩水(100 mL)で洗浄後、Na2SO4で乾燥し、溶媒を留去して化合物(IIIc)の粗製物を39.11 g(無色油状)得た。(NMRより化合物(IIIc)の含有量を測定した。19.63 g, 0.17 mol, 収率 62%)
参考例5:化合物(IIId)の合成
【化41】
4mol/L塩酸の酢酸エチル溶液82.5 mL(0.33 mol)にエタノール17.7 mL(0.30 mol)及び、イソブチロニトリル(VId)19.0 g(0.27 mol)を滴下し、0℃で1週間攪拌した。溶媒を留去し、結晶性残査に炭酸カリウム水溶液(K2CO3 41.80 g, 0.30 mol, H2O 100 mL)及び塩化メチレン(200 mL, 100 mL)を加えて中和、抽出した。有機層を飽和食塩水(100 mL)で洗浄後、Na2SO4で乾燥し、溶媒を留去して化合物(IIId)の粗製物を27.89 g(無色油状)得た。(NMRより化合物(IIId)の含有量を測定した。17.63 g, 0.15 mol, 収率 56%)
参考例6:化合物(IIIe)の合成
【化42】
4mol/L塩酸の酢酸エチル溶液150 mL(0.6 mol)にメタノール22.3 mL(0.55 mol)及び、ベンゾニトリル(VIe)51.0 mL(0.5 mol)を滴下し、0℃で9日間攪拌した。溶媒を留去し、結晶性残査に炭酸カリウム水溶液(K2CO3 76.0 g, 0.55 mol, H2O 400 mL)及び塩化メチレン(400 mL, 200 mL)を加えて中和、抽出した。有機層を飽和食塩水(200 mL)で洗浄後、Na2SO4で乾燥し、溶媒を留去して化合物(IIIe)の粗製物を114.52 g(無色油状)得た。(NMRより化合物(IIIe)の含有量を測定した。18.31 g, 0.14 mol, 収率 27%)
参考例7:化合物(IIIf)の合成
【化43】
4mol/L塩酸の酢酸エチル溶液60 mL(0.24 mol)にエタノール12.9 mL(0.22 mol)及び、フェニルアセトニトリル(VIf)23.0 mL(0.2 mol)を滴下し、0℃で1週間攪拌した。溶媒を留去し、結晶性残査に炭酸カリウム水溶液(K2CO3 30.4 g, 0.22 mol, H2O 200 mL)及び塩化メチレン(200 mL, 100 mL)を加えて中和、抽出した。有機層を飽和食塩水(100 mL)で洗浄後、Na2SO4で乾燥し、溶媒を留去して化合物(IIIf)の粗製物を67.30 g(無色油状)得た。(NMRより化合物(IIIf)の含有量を測定した。27.38 g, 0.17 mol, 収率 84%)
参考例8:化合物(IIIg)の合成
【化44】
4mol/L塩酸の酢酸エチル溶液45 mL(0.18 mol)にエタノール9.6 mL(0.17 mol)及び、2−チオフェンカルボニトリル(VIg) 14.0 mL(0.15 mol)を滴下し、0℃で1週間攪拌した。溶媒を留去し、結晶性残査に炭酸カリウム水溶液(K2CO3 22.8 g, 0.17 mol, H2O 140 mL)及び塩化メチレン(140 mL, 70 mL)を加えて中和、抽出した。有機層を飽和食塩水(70 mL)で洗浄後、Na2SO4で乾燥し、溶媒留去して化合物(IIIg)の粗製物を45.93 g(無色油状)得た。(NMRより化合物(IIIg)の含有量を測定した。16.91 g, 0.11 mol, 収率 73%)
参考例9:化合物(IIIh)の合成
【化45】
4−シアノピリジン(VIh)(20.82 g, 0.20 mol)をメタノール(208 mL)に溶解させた後、28%ナトリウムメトキシドメタノール溶液3.86 g (0.020 mol)を滴下し、室温攪拌1日行った。氷酢酸1.15 mL (0.020 mol)で中和した後溶媒留去し、化合物(IIIh)の粗製物を31.16 g(薄茶色ガム状物質)得た。(NMRより化合物(IIIh)の含有量を測定した。24.51 g, 0.18 mol, 収率 90%)。
実施例1:化合物(Ia)の合成
【化46】
シュウ酸ジエチル(Va)13.6 mL(0.10 mol)のエタノール(108.8 mL)溶液にヒドラジン一水和物4.85 mL(0.10 mol)のエタノール(21.8 mL)溶液を−40〜−50℃で滴下した。室温攪拌1時間後、不溶物を濾別し、エタノール(68 mL)で不溶物を洗浄し化合物(IIa)の粗製物を得た。
化合物(IIa)の粗製物を氷冷し、参考例1で合成した化合物(IIIa)のエタノール(68 mL)溶液(内、化合物(IIIa)として10.89 g, 0.125 mol)を0〜5℃で滴下した後、氷冷攪拌1時間、還流2時間反応した。溶媒留去後エタノール−トルエンで再結晶し、白色結晶として化合物(Ia)を7.36 g(0.047 mol, 収率47%)得た。
融点: 189−191℃.
1H NMR (d6−DMSO, 300 MHz): 1.29 (3H, t, J= 7.2 Hz), 2.39 (3H, s), 4.30 (2H, q, J= 7.2 Hz).
元素分析:Calcd. for C6H9N3O2: C, 46.45; H, 5.85; N, 27.08. Found: C, 46.35; H, 5.75; N, 27.15; H2O, <0.1.
実施例2:化合物(IVa’)の合成
【化47】
シュウ酸ジエチル(Va)11.0 mL(0.081 mol)のエタノール(10 mL)溶液にヒドラジン一水和物3.57 mL(0.074 mol)のエタノール(40 mL)溶液を−10〜0℃で滴下した。氷冷攪拌1時間行なった後、不溶物を濾別し、エタノール(10 mL)で不溶物を洗浄し化合物(IIa)の粗製物を得た。
化合物(IIa)の粗製物を氷冷し、参考例1で合成した化合物(IIIa)のトルエン(60 mL)溶液(内、化合物(IIIa)として8.02 g, 0.092 mol)を0〜5℃で滴下した。氷冷攪拌1時間後析出晶を濾過し、白色結晶として化合物(IVa’)を9.98 g(0.052 mol, 収率71%)得た。
融点:98−100℃.
1H NMR (d6−DMSO, 300 MHz): 1.22 (3H, t, J= 7.2 Hz), 2.10 (3H, s), 4.10 (2H, brd, J= 7.2 Hz), 7.40 (1H, brs), 8.32 (1H, brs), 11.05 (1H, brs).
元素分析:Calcd. for C6H11N3O3・H2O: C, 37.69; H, 6.85; N, 21.98; H2O, 9.42. Found: C, 37.81; H, 7.04; N, 22.05; H2O, 9.42.
実施例3:化合物(Ia)の合成
【化48】
実施例2で合成した化合物(IVa’)9.98g(0.052 mol)をアセトニトリル(90 mL)中4時間還流した。エタノール(9.0 mL)を加えた後、氷冷し析出晶を濾過し、白色結晶として化合物(Ia)を5.50 g (0.035 mol, 収率 68%, ヒドラジン一水和物から通算収率48%) 得た。
実施例4:化合物(IVa’)の合成
【化49】
シュウ酸ジエチル(Va)75 mL(0.55 mol)のエタノール(50 mL)溶液に35wt%ヒドラジン水溶液45.79 g(0.50 mol)のエタノール(200 mL)溶液を−10〜−20℃で滴下した。飽和食塩水冷却下0.5時間攪拌し化合物(IIa)の粗製物を得た。化合物(IIa)の粗製物に参考例2で合成した粗製物(IIIa) 307.16 g(内、化合物(IIIa)として 50.68 g, 0.58 mol)を−10〜0℃で滴下し、トルエン (25mL)で洗浄した。飽和食塩水冷却下3時間攪拌後、析出晶を濾過し、白色結晶として化合物(IVa’)を77.64 g(0.41 mol, 収率81%,不純物としてシュウ酸ジヒドラジドを約5wt%含む)得た。
実施例5:化合物(Ia)の合成
【化50】
実施例4で合成した化合物(IVa’) 50.00g(0.26 mol, 不純物としてシュウ酸ジヒドラジドを約5wt%含む) をアセトニトリル(400 mL)、アセトン(40 mL)中50℃で1時間加温攪拌した。内温を70〜80℃に昇温した後、エタノール(40 mL)を加え、さらに3時間還流した。飽和食塩水冷却下2時間後、析出晶を濾過し、白色結晶として化合物(Ia)を28.37 g (0.18 mol, 収率 70%, 35wt%ヒドラジン水溶液から通算収率57%) 得た。
実施例6:化合物(Ib)の合成
【化51】
シュウ酸ジエチル(Va)24.2 mL(0.18 mol)のエタノール(48 mL)溶液にヒドラジン一水和物8.63 mL(0.18 mol)のエタノール(194 mL)溶液を−40〜−60℃で滴下した。室温攪拌1時間行った後、不溶物を濾別し、エタノール(121 mL)で不溶物を洗浄し化合物(IIa)の粗製物を得た。
化合物(IIa)の粗製物を氷冷し、参考例3で合成した化合物(IIIb)のエタノール(121 mL)溶液(内、化合物(IIIb)として21.66 g, 0.21 mol)を0〜5℃で滴下し、氷冷攪拌1日、還流2時間行った。溶媒留去後水で再結晶を行ない、白色結晶として化合物(Ib)を7.35 g得た。母液を酢酸エチルで抽出、濃縮後の結晶性残査をイソプロピルエーテルで洗浄し、白色結晶として化合物(Ib)の第二晶を 8.75 g(トータル 16.10 g, 0.095 mol, 収率 53%)得た。
融点:106−108℃.
1H NMR (d6−DMSO, 300 MHz): 1.24 (3H, t, J= 7.2 Hz), 1.29 (3H, t, J= 7.2 Hz), 2.75 (2H, q, J= 7.2 Hz) , 4.30 (2H, q, J= 7.2 Hz).
元素分析:Calcd. for C7H11N3O2: C, 49.70; H, 6.55; N, 24.84. Found: C, 49.72; H, 6.61; N, 24.74; H2O, <0.1.
実施例7:化合物(Ic)の合成
【化52】
シュウ酸ジエチル(Va)19.3 mL(0.14 mol)のエタノール(38.6 mL)溶液にヒドラジン一水和物6.89 mL(0.14 mol)のエタノール(154.4 mL)溶液を−40〜−60℃で滴下した。室温攪拌0.5時間行った後、不溶物を濾別し、エタノール(96.5mL)で不溶物を洗浄し化合物(IIa)の粗製物を得た。
化合物(IIa)の粗製物を氷冷し、参考例4で合成した化合物(IIIc)のエタノール(96.5 mL)溶液(内、化合物(IIIc)として19.63 g, 0.17 mol)を0〜5℃で滴下し、氷冷攪拌4時間、還流3時間行った。溶媒留去後、水で再結晶し、白色結晶として化合物(Ic)を15.98 g(0.087 mol, 収率61%)得た。
融点:117−118.5℃.
1H NMR (d6−DMSO, 300 MHz): 0.89 (3H, t, J= 7.2 Hz), 1.30 (3H, t, J= 7.2 Hz), 1.70 (2H, tq, J= 7.5, 7.2 Hz) , 2.71 (2H, t, J= 7.5 Hz) , 4.30 (2H,q, J= 7.2 Hz).
元素分析:Calcd. for C8H13N3O2: C, 52.45; H, 7.15; N, 22.94. Found: C, 52.47; H, 7.16; N, 22.98; H2O, 0.23.
実勢例8:化合物(Id)の合成
【化53】
シュウ酸ジエチル(Va)17.3 mL(0.13 mol)のエタノール(34.6 mL)溶液にヒドラジン一水和物6.19 mL(0.13 mol)のエタノール(138.4 mL)溶液を−40〜−60℃で滴下した。室温攪拌0.5時間行った後、不溶物を濾別し、エタノール(86.5 mL)で不溶物を洗浄し化合物(IIa)の粗製物を得た。
化合物(IIa)の粗製物を氷冷し、参考例5で合成した化合物(IIId)のエタノール(86.5 mL)溶液(内、化合物(IIId)として17.63 g, 0.15 mol)を0〜5℃で滴下し、氷冷攪拌5時間、還流2時間行った。溶媒留去後、酢酸エチルで抽出し、有機層の溶媒を濃縮後、結晶性残査をイソプロピルエーテルで洗浄し、白色結晶として化合物(Id)を14.93 g(0.081 mol, 収率 64%)得た。
融点:105−107℃.
1H NMR (d6−DMSO, 300 MHz): 1.27 (6H, d, J= 6.9 Hz), 1.30 (3H, t, J= 7.2 Hz), 3.02−3.18 (1H, m) , 4.31 (2H, q, J= 7.2 Hz).
元素分析:Calcd. for C8H13N3O2: C, 52.45; H, 7.15; N, 22.94. Found: C, 52.30; H, 7.15; N, 22.98; H2O, 0.30.
実施例9:化合物(Ie)の合成
【化54】
シュウ酸ジエチル(Va)15.3 mL(0.11mol)のエタノール(30.6 mL) 溶液にヒドラジン一水和物5.46 mL(0.11 mol)のエタノール(122.4 mL)溶液を−40〜−60℃で滴下した。室温攪拌0.5時間行った後、不溶物を濾別し、エタノール(76.5 mL)で不溶物を洗浄し化合物(IIa)の粗製物を得た。
化合物(IIa)の粗製物を氷冷し、参考例6で合成した化合物(IIIe)のエタノール(76.5 mL)溶液(内、化合物(IIIe)として18.31 g, 0.14 mol)を0〜10℃で滴下した後、室温攪拌1日、還流2時間行った。溶媒留去後アセトニトリル−トルエンに溶解させ不溶物を濾過した後、濾液をアセトニトリル−トルエンで再結晶し、白色結晶として化合物(Ie)を13.63 g(0.063 mol, 収率56%)得た。
融点:164−166℃.
1H NMR (d6−DMSO, 300 MHz): 1.35 (3H, d, J= 7.2 Hz), 4.38 (2H, q, J= 7.2 Hz), 7.48−7.60 (3H, m), 8.00−8.08 (2H, m).
元素分析: Calcd. for C11H11N3O2: C, 60.82; H, 5.10; N, 19.34. Found: C,60.90; H, 5.14; N, 19.42; H2O, <0.1.
実施例10:化合物(IVf)の合成
【化55】
シュウ酸ジエチル(Va)19.0 mL(0.14 mol)のエタノール(38 mL) 溶液にヒドラジン一水和物 6.79 mL(0.14 mol)のエタノール(152 mL)溶液を−40〜−60℃で滴下した。室温攪拌0.5時間行った後、不溶物を濾別し、エタノール(95 mL)で不溶物を洗浄し化合物(IIa)の粗製物を得た。
化合物(IIa)の粗製物を氷冷し、参考例7で合成した化合物(IIIf)のエタノール(95 mL)溶液(内、化合物(IIIf)として27.38 g, 0.17 mol)を0〜10℃で滴下した。氷冷攪拌1.5時間後析出晶を濾過し、白色結晶として化合物(IVf)を20.31 g得た。濾液を留去し、結晶性残査を酢酸エチルで洗浄し、白色結晶として化合物(IVf)の第2晶 8.50 g (total 28.81g, 0.12 mol, 収率 83%) 得た。
融点:115−117℃.
1H NMR (d6−DMSO, 300 MHz): 1.06 (3H, t, J= 6.9 Hz), 4.12 (2H, brs), 4.35
(2H, brs), 7.24−7.44 (5H, m).
実施例11:化合物(If)の合成
【化56】
実施例10で合成した化合物(IVf)28.81g(0.12 mol)をエタノール(576 mL)中5時間還流した。溶媒留去後エタノールで再結晶し、白色結晶として化合物(If)を16.29 g 得た。母液を溶媒留去後、トルエンに溶解させ熱時濾過した。濾液をトルエンで再結晶し、白色結晶として化合物(If)の第2晶を3.81 g(トータル20.10 g, 0.087 mol, 収率 75%, ヒドラジン一水和物から通算収率62%)得た。
融点:147.5−150℃.
1H NMR (d6−DMSO, 300 MHz): 1.28 (3H, t, J= 7.2 Hz), 4.12 (2H, s), 4.29 (2H, q, J= 7.2 Hz), 7.20−7.40 (5H, m).
元素分析:Calcd. for C12H13N3O2: C, 62.33; H, 5.67; N, 18.17. Found: C, 62.32; H, 5.58; N, 18.39; H2O, 0.18.
実施例12:化合物(Ig)の合成
【化57】
シュウ酸ジエチル(Va)12.4 mL(0.091mol)のエタノール溶液(24.8 mL) 溶液にヒドラジン一水和物4.40 mL(0.091 mol)のエタノール(99.2 mL)溶液を−40〜−60℃で滴下した。室温攪拌0.5時間行った後、不溶物を濾別し、エタノール(62.0 mL)で不溶物を洗浄し化合物(IIa)の粗製物を得た。
化合物(IIa)の粗製物を氷冷し、参考例8で合成した化合物(IIIg)のエタノール(62.0 mL)溶液(内、化合物(IIIg)として16.91 g, 0.11 mol)を0〜10℃で滴下した後、室温攪拌1日、還流5時間行った。溶媒留去後クロロホルムでカラムをかけた後、結晶性残査をイソプロピルエーテルで洗浄し、白色結晶として化合物(Ig)を7.38g(0.031 mol, 収率 34%)得た。
融点:176−178℃.
1H NMR (d6−DMSO, 300 MHz): 1.34 (3H, t, J= 7.2 Hz), 4.37 (2H, q, J= 7.2 Hz), 7.20−7.26 (1H, m), 7.72−7.80 (2H, m).
元素分析: Calcd. for C9H9N3O2S: C, 48.42; H, 4.06; N, 18.82; S, 14.36. Found: C, 48.31; H, 3.97; N, 18.66; S, 14.24; H2O, <0.1.
実施例13:化合物(IVh)の合成
【化58】
シュウ酸ジエチル(Va) 20.4 mL(0.15 mol)のエタノール(40.8 mL) 溶液にヒドラジン一水和物7.3 mL(0.15 mol)のエタノール(163.2 mL)溶液を−40〜−60℃で滴下した。室温攪拌0.5時間行った後、不溶物を濾別し、エタノール(102 mL)で不溶物を洗浄し化合物(IIa)の粗製物を得た。
化合物(IIa)の粗製物を氷冷し、参考例9で合成した化合物(IIIh)のエタノール(102 mL)溶液(内、化合物(IIIh)として24.51 g, 0.18 mol)を0〜10℃で滴下した。氷冷攪拌1日後濾過し、淡黄色結晶として化合物(IVh)を28.30 g(0.12 mol, 収率80%)得た。
融点: 212−216℃ (分解).
1H NMR (d6−DMSO, 300 MHz): 1.28 and 1.30 (total 3H, t, J= 7.2 Hz), 4.27 and 4.30 (total 2H, q, J= 7.2 Hz), 6.78 and 7.10 (total 2H, s), 7.60−7.78 (2H, m), 8.61−8.67 (2H, m).
実施例14:化合物(Ih)の合成
【化59】
実施例13で合成した化合物(IVh)28.30 g(0.12 mol)をエタノール(566 mL)中6時間還流した。不溶物を濾過した後、濾液を溶媒留去した。結晶性残査をイソプロピルエーテルで洗浄し、淡黄色結晶として化合物(Ih)を18.97 g (0.087 mol, 収率 73%, ヒドラジン一水和物から通算収率58%) 得た。
融点:175−177℃.
1H NMR (d6−DMSO, 300 MHz): 1.36 (3H, t, J= 7.2 Hz), 4.40 (2H, q, J= 7.2 Hz), 7.96−7.99 (2H, m), 8.74−8.77 (2H, m).
元素分析:Calcd. for C10H10N4O2: C, 55.04; H, 4.62; N, 25.67. Found: C, 54.76; H, 4.53; N, 25.61; H2O, 0.87.
参考例
参考例10:化合物(XIa)の合成
【化60】
化合物(Ia)0.68 g(4.4 mmol)のTHF(2.0 ml)懸濁液にp−トルエンスルホン酸一水和物(25 mg, 0.13 mmol)を加えた。反応液を攪拌しながら3,4−ジヒドロピラン−2H−ピラン(0.50 ml, 5.5 mmol)を室温で滴下し、その後2時間攪拌した。反応液を酢酸エチル(8 ml)で抽出し、飽和炭酸水素ナトリウム水溶液で洗浄後、Na2SO4で乾燥した。溶媒を留去後、残査をシリカゲルクロマトグラフィーで精製(酢酸エチルで溶出)し、化合物(VIIIa−1)を0.89 g(3.7
mmol, 収率 85%)得た。
化合物(IXa)0.81 g(3.7 mmol)と化合物(VIIIa−1)0.89 g(3.7 mmol)のTHF(8.0 ml)溶液に28%ナトリウムメトキシド/メタノール溶液(0.74 g, 3.7mmol)を氷冷下で滴下した。室温で14時間攪拌後、反応液に1.8%酢酸水溶液(23 ml)を加えた。反応液を酢酸エチル(30 ml)で抽出し、飽和炭酸水素ナトリウム水溶液で洗浄、水洗し、Na2SO4で乾燥した。溶媒を留去して化合物(XIa)を0.84 g(2.0 mmol, 収率 55%)得た。
1H NMR (d6−DMSO, 300 MHz): 2.43 (3H, s), 4.14 (2H, s), 6.46 (1H, d, J= 3.6 Hz), 6.89 (1H, s), 7.18 (2H, t, J= 9.0 Hz), 7.35 (2H, dd, J= 9.0, 5.7Hz), 7.50 (1H, d, J=3.6 Hz).
参考例11:化合物(XIa)の合成
【化61】
化合物(Ia)20.00 g(0.13 mol)にp−トルエンスルホン酸0.6 g(3 wt%)のTHF(70 mL)溶液を加え、氷冷下攪拌した。0〜5℃で2−メトキシプロペン(13.6 mL, 0.14 mol)を滴下後、氷冷下で3時間攪拌した。反応液に化合物(IXa)29.61 g(化合物(IXa)を95wt%含有、0.13 mol)、28%ナトリウムメトキシド/メタノール溶液(32.33 g, 0.17 mol)およびTHF(10 mL)を加えた。70℃で3時間、室温で16時間攪拌した。メタノール(80 mL)を加え、氷冷下で攪拌した。酢酸(11.07 mL, 0.19 mol)を0〜10℃で滴下し、さらに水(160 mL)を10〜20℃で滴下した。氷冷下2時間攪拌後、析出した結晶をろ取し、化合物(Xa−2)を黄土色結晶として45.41 g(0.11 mol, 収率 88%)得た。
化合物(Xa−2)20.00 g(0.050 mol)のアセトン(106 mL)、水(9.8 mL)混合溶液を65℃で5分間攪拌した。反応液に塩酸(濃塩酸 0.04 mL/水 1.6 mL)を加え、70℃で1時間攪拌した。内温40℃以上でアセトン(54 mL)を減圧留去した。45℃で1時間攪拌後、炭酸ナトリウム(26.5 mg)/水(54 mL)を40〜50℃で滴下した。室温で1時間、さらに氷冷下2時間攪拌した。析出した結晶をろ取し、化合物(XIa)を黄色結晶として15.63 g(0.048 mol, 収率 95%)得た。融点 : 195−197℃.
1H NMR (d6−DMSO, 300 MHz): 2.43 (3H, s), 4.14 (2H, s), 6.46 (1H, d, J= 3.6 Hz), 6.89 (1H, s), 7.18 (2H, t, J= 9.0 Hz), 7.35 (2H, dd, J= 9.0, 5.7Hz), 7.50 (1H, d, J=3.6 Hz).
元素分析:Calcd. for C17H14FN3O3: C, 62.38; H, 4.31; N, 12.84; F, 5.80. Found: C, 62.39; H, 4.28; N, 12.90; F, 5.66; H2O<0.1%.
参考例12:化合物(XIb)の合成
【化62】
化合物(Ib)0.74 g(4.4 mmol)のTHF(2.0 ml)懸濁液にp−トルエンスルホン酸一水和物(25 mg, 0.13 mmol)を加えた。反応液を攪拌しながら3,4−ジヒドロピラン−2H−ピラン(0.50 ml, 5.5 mmol)を室温で滴下し、その後2時間攪拌した。反応液を酢酸エチル(8 ml)で抽出し、飽和炭酸水素ナトリウム水溶液で洗浄後、Na2SO4で乾燥した。溶媒を留去後、残査をシリカゲルクロマトグラフィーで精製(酢酸エチルで溶出)し、化合物(VIIIb−1)を0.89 g(3.5
mmol, 収率 80%)得た。
化合物(IXa)0.77 g(3.5 mmol)と化合物(VIIIb−1)0.89 g(3.5 mmol)のTHF(8.0 ml)溶液に28%ナトリウムメトキシド/メタノール溶液(0.70 g, 3.5mmol)を氷冷下で滴下した。室温で14時間攪拌後、反応液に1.8%酢酸水溶液(22 ml)を加えた。反応液を酢酸エチル(30 ml)で抽出し、飽和炭酸水素ナトリウム水溶液で洗浄、水洗し、Na2SO4で乾燥した。溶媒を留去して化合物(XIb)を0.82 g(1.9 mmol, 収率 55%)得た。
融点 : 204−205℃.
1H NMR (d6−DMSO, 300 MHz): 1.27 (3H, t, J= 7.8Hz), 2.79 (2H, q, J= 7.8Hz), 4.15 (2H, s), 6.45 (1H, d, J= 3.6Hz), 6.88 (1H, s), 7.17 (2H, t, J= 9.3Hz), 7.25−7.40 (2H, m), 7.49 (1H, d, J= 3.6Hz).
元素分析:Calcd. for C18H16FN3O3・0.25H2O: C, 62.51; H, 4.81; N, 12.15; F,5.49. Found: C, 62.57; H, 4.68; N, 12.25; F, 5.30.
参考例13:化合物(XIb)の合成
【化63】
化合物(Ib)1.69 g(0.010 mol)にp−トルエンスルホン酸0.05 g(3 wt%)のTHF(5 mL)溶液を加え、氷冷下攪拌した。0〜5℃で2−メトキシプロペン(1.04 mL, 0.011 mol)を滴下後、氷冷下で3時間攪拌した。反応液に化合物(IXa)1.88 g(化合物(IXa)を95wt%含有、0.010 mol)、28%ナトリウムメトキシド/メタノール溶液(2.50 g, 0.013 mol)およびTHF(1 mL)を加えた。70℃で3時間、室温で16時間攪拌した。メタノール(6 mL)を加え、氷冷下で攪拌した。酢酸(0.86 mL, 0.015 mol)を0〜10℃で滴下し、さらに水(12 mL)を10〜20℃で滴下した。氷冷下2時間攪拌後、析出した結晶をろ取し、化合物(Xb−2)を2.48 g(0.006 mol, 収率 60%)得た。
化合物(Xb−2)2.48 g(0.006 mol)のアセトン(13 mL)、水(1.2 mL)混合溶液を65℃で5分間攪拌した。反応液に塩酸(濃塩酸 0.005 mL/水 0.19 mL)を加え、70℃で1時間攪拌した。内温40℃以上でアセトン(6 mL)を減圧留去した。45℃で1時間、つづけて室温で1時間、さらに氷冷下2時間攪拌した。析出した結晶をろ取し、化合物(XIb)を結晶として1.64 g(4.8 mmol, 収率 80%)得た。
融点 : 204−205℃.
1H NMR (d6−DMSO, 300 MHz): 1.27 (3H, t, J= 7.8Hz), 2.79 (2H, q, J= 7.8Hz), 4.15 (2H, s), 6.45 (1H, d, J= 3.6Hz), 6.88 (1H, s), 7.17 (2H, t, J= 9.3Hz), 7.25−7.40 (2H, m), 7.49 (1H, d, J= 3.6Hz).
参考例14:化合物(XIc)の合成
【化64】
60% 水素化ナトリウム (0.40g, 0.01mol)のDMF(20.0 ml)溶液に、室温下化合物(Ic)1.89g(0.01 mol)を加え、15分間攪拌した。トリチルクロリド(3.06g, 0.011 mol)を加え、さらに2時間攪拌した。反応液を飽和塩化アンモニウム水溶液に投入し、酢酸エチルにて抽出、水洗後、MgSO4で乾燥した。溶媒を留去後、結晶性残査をエーテル−ヘキサンにて洗浄し、化合物(VIIIc)の粗精製物を2.1g得た。
化合物(IXa)0.44 g(2.00 mmol)の THF(15.0 ml)溶液を−70℃まで冷却し、リチウムビストリメチルシリルアミド THF(1 M)溶液(2.60 ml, 2.60 mmol)を滴下した。反応液を一旦 0℃まで昇温し、再び−70℃まで冷却した。反応液に化合物(VIIIc)0.94 g(2.20 mmol)の THF(8.00 ml)溶液を−32〜−7℃で滴下し、室温まで昇温後1.5時間攪拌した。反応液を飽和塩化アンモニウム水溶液に投入し、酢酸エチルにて抽出、水洗後、MgSO4で乾燥した。溶媒を留去する事によって得られた化合物(Xc)の粗精製残渣(1.30 g)を次の反応に使用した。
化合物(Xc)1.30 gの粗精製物をジオキサン(15.0 ml)溶液に1.5 mol/L塩酸(5 ml)を加え、70 ℃で1時間攪拌した。次いで減圧下、ジオキサンを留去し、残査に水を加え、酢酸エチルで抽出した。酢酸エチル層を水洗、乾燥後、溶媒を留去した。残留物を酢酸エチルに溶解し、1 mol/L 水酸化ナトリウム水溶液で抽出した。アルカリ抽出液を酢酸エチルで2回洗浄後、6 mol/L塩酸で中和し、酢酸エチルで抽出した。抽出液を水洗、飽和食塩水で洗浄後、Na2SO4で乾燥した。溶媒を留去し、得られた粗結晶を少量の酢酸エチルで洗浄後、エーテルから再結晶することにより化合物(XIc)を化合物(IXa)から収率 14% (0.099 g, 0.28 mmol)で得た。
融点 : 167−168℃
1H NMR (d6−DMSO, 300 MHz): 0.92 (3H, t, J= 7.5Hz), 1.60−1.80 (2H, m), 2.73 (2H, q, J= 7.5Hz), 4.15 (2H, s), 6.45 (1H, d, J= 3.6Hz), 6.88 (1H, s), 7.17 (2H, t, J=9.3Hz), 7.25−7.40 (2H, m), 7.49 (1H, d, J= 3.6Hz).元素分析:Calcd. for C19H18FN3O3: C, 64.22; H, 5.11; N, 11.82; F, 5.35. Found: C, 64.05; H, 5.07; N, 11.80; F, 5.13.
参考例15:化合物(XId)の合成
【化65】
化合物(Id)1.83 g(0.010 mol)にp−トルエンスルホン酸0.05 g(3 wt%)のTHF(5 mL)溶液を加え、氷冷下攪拌した。0〜5℃で2−メトキシプロペン(1.04 mL, 0.011 mol)を滴下後、氷冷下で3時間攪拌した。反応液に化合物(IXa)1.88 g(化合物(IXa)を95wt%含有、0.010 mol)、28%ナトリウムメトキシド/メタノール溶液(2.50 g, 0.013 mol)およびTHF(1 mL)を加えた。70℃で3時間、室温で16時間攪拌した。メタノール(6 mL)を加え、氷冷下で攪拌した。酢酸(0.86 mL, 0.015 mol)を0〜10℃で滴下し、さらに水(12 mL)を10〜20℃で滴下した。氷冷下2時間攪拌後、析出した結晶をろ取し、化合物(Xd)を2.99 g(7.0 mmol, 収率 70%)得た。
化合物(Xb)2.99 g(7.0 mmol)のアセトン(15 mL)、水(1.4 mL)混合溶液を65℃で5分間攪拌した。反応液に塩酸(濃塩酸 0.006 mL/水 0.22 mL)を加え、70℃で1時間攪拌した。内温40℃以上でアセトン(7 mL)を減圧留去した。45℃で1時間、つづけて室温で1時間、さらに氷冷下2時間攪拌した。析出した結晶をろ取し、化合物(XIb)を結晶として1.74 g(4.9 mmol, 収率 70%)得た。
融点 : 146−147℃
1H NMR (d6−DMSO, 300 MHz): 1.30 (6H, d, J= 6.9Hz), 3.00−3.20 (1H, m), 4.15 (2H, s), 6.45 (1H, d, J= 3.6Hz), 6.87 (1H, s), 7.17 (2H, t, J= 9.3Hz), 7.25−7.40 (2H, m), 7.48 (1H, d, J= 3.6Hz).
元素分析:Calcd. for C19H18FN3O3・0.25H2O: C, 63.41; H, 5.18; N, 11.68; F,5.28. Found: C, 63.47; H, 5.09; N, 12.43; F, 4.85.
【発明の効果】
本発明により、医薬品製造のための中間体として有用な5−置換−1,2,4−トリアゾール−3−カルボン酸エステル誘導体およびアシルアミドラゾン誘導体を工業的に製造することができる。
【発明の属する技術分野】
本発明は、5−置換−1,2,4−トリアゾール−3−カルボン酸エステル誘導体、アシルアミドラゾン誘導体、およびそれらの製造方法に関する。
【0002】
【従来の技術】
J. Chem. Soc. (C), 1968, 824−830にはアシルヒドラジン誘導体とイミデート誘導体から種々の3,5−ジ置換−1,2,4−トリアゾール誘導体を製造する方法が記載されている。またWO00/39086には、HIVインテグレース阻害化合物を合成する際の重要中間体として5−(非置換または置換)−1,2,4−トリアゾール−3−カルボン酸エステル誘導体が記載されている。しかし、どちらの文献にも5−置換−1,2,4−トリアゾール−3−カルボン酸エステル誘導体の製造方法については記載されていない。
5−置換−1,2,4−トリアゾール−3−カルボン酸エステル誘導体の製造方法は、以下の文献に記載されている(J. Chem. Soc. Perkin Trans 1, 1977, 761−764、Collect. Czech. Chem. Commun., 49, 1984, 2492−2495、J. Heterocyclic Chem., 25, 1988, 651−654および WO00/75122)。J. Chem. Soc. Perkin Trans 1, 1977, 761−764には以下に示すアシルアミドラゾン誘導体(A)を経由して加熱閉環する製造方法が記載されている。また、Collect. Czech. Chem. Commun., 49, 1984, 2492−2495、J. Heterocyclic Chem., 25, 1988, 651−654および WO00/75122には以下に示すアシルアミドラゾン誘導体(B)を経由して加熱閉環する製造方法が記載されている。
【化16】
しかし、上記の5−置換−1,2,4−トリアゾール−3−カルボン酸エステル誘導体の製造方法において、アシルアミドラゾン誘導体(A)を経由する場合は、反応条件がピリジン還流中と高温度であり、収率が低く、工業的製法として満足いく方法ではなかった。
アシルアミドラゾン誘導体(B)を経由する場合は、一般に反応温度が165℃以上と高くかつ生成する水を減圧下除去する必要があるなど、工業的に行うには問題があった。しかし、WO00/75122には120℃付近で反応させることができ、さらに生成する水を除去しなくてもよい製造方法が記載されていた。
【0003】
【発明が解決しようとする課題】
医薬品の中間体として有用である5−置換−1,2,4−トリアゾール−3−カルボン酸エステル誘導体、該誘導体を製造するにあたって有用な中間体であるアシルアミドラゾン誘導体、およびそれらを工業的かつ経済的に製造する方法が望まれている。
【0004】
【課題を解決するための手段】
上記の事情に鑑み、本発明者らは、鋭意検討した結果、以下に示す5−置換−1,2,4−トリアゾール−3−カルボン酸エステル誘導体、アシルアミドラゾン誘導体、およびそれらの製造方法を見出した。
【0005】
すなわち、本発明は、
1)一般式(IV):
【化17】
で示される化合物またはその水和物を100℃以下で閉環させることを特徴とする、一般式(I):
【化18】
で示される化合物の製造方法。
2)一般式(II):
【化19】
で示される化合物と、一般式(III):
【化20】
で示される化合物を縮合させて、一般式(IV):
【化21】
で示される化合物またはその水和物を得る工程;および
式(IV)で示される化合物またはその水和物を閉環させる工程
を包含する、一般式(I):
【化22】
で示される化合物の製造方法。
3)一般式(V):
【化23】
で示される化合物と、ヒドラジンを縮合させて一般式(II):
【化24】
で示される化合物を得る工程を包含する、2)記載の製造方法。
4)一般式(II):
【化25】
で示される化合物に、一般式(III):
【化26】
で示される化合物を縮合させて得られる、一般式(IV):
【化27】
で示される化合物の製造方法。
5)一般式(V):
【化28】
で示される化合物と、ヒドラジンを縮合させて、一般式(II):
【化29】
で示される化合物を得る工程を包含する、4)記載の製造方法。
6)R1が低級アルキルである1)から5)のいずれかに記載の製造方法。
7)R1がエチルである1)から5)のいずれかに記載の製造方法。
8)R2が低級アルキルである1)から7)のいずれかに記載の製造方法。
9)R3が低級アルキルである2)から8)のいずれかに記載の製造方法。
10)反応溶媒としてエタノールを用いる1)から9)のいずれかに記載の製造方法。
11)反応温度が90℃以下である1)から3)のいずれかに記載の製造方法。
12)一般式(IV−1):
【化30】
で示される化合物、もしくはその塩、またはその溶媒和物。
13)一般式(I−1):
【化31】
で示される化合物、もしくはその塩、またはその溶媒和物。
【0006】
本明細書中で用いられる「ハロゲン」とはフッ素、塩素、臭素およびヨウ素を意味する。
本明細書中、単独でもしくは他の用語と組み合わせて用いられる「アルキル」なる用語は、1〜10個の炭素原子を有する、直鎖または分枝鎖の1価の炭化水素基を包含する。例えば、メチル、エチル、n−プロピル、イソプロピル、n−ブチル、イソブチル、sec−ブチル、tert−ブチル、n−ペンチル、イソペンチル、neo−ペンチル、n−ヘキシル、イソヘキシル、n−ヘプチル、n−オクチル、n−ノニル、またはn−デシル等が挙げられる。なお本明細書中で用いている「低級アルキル」は、C1〜C6アルキルを意味する。
R1のアルキルとしては、C1〜C6アルキルが好ましい。さらにC1〜C4アルキルが好ましい。
R2またはR3のアルキルとしては、C1〜C6アルキルが好ましい。さらにC1〜C4アルキルが好ましい。
本明細書中で用いられる「置換されていてもよいアルキル」における置換基としては、ハロゲン(例えば、フッ素、塩素、臭素、またはヨウ素)、C2〜C4アルケニル(例えば、ビニル、アリル、2−ブテニル等)、C2〜C4アルキニル(例えば、エチニル、プロパルギル、2−ブチニル等)、C3〜C6シクロアルキル(例えば、シクロプロピル、シクロブチル、シクロペンチル、またはシクロヘキシル等)、ヒドロキシ、C1〜C4アルキルオキシ(例えば、メチルオキシまたは、エチルオキシ等)、置換されていてもよいアラルキルオキシ(例えば、ベンジルオキシまたは4−メチルオキシベンジルオキシ等)、メルカプト、C1〜C4アルキルチオ(例えば、メチルチオ等)、ニトロ、置換されていてもよいアミノ(例えば、アミノ、メチルアミノ、またはジメチルアミノ等)等が挙げられる。これら置換基は、全ての可能な位置で1個以上置換しうる。好ましい置換基としては、ハロゲン、C1〜C4アルキルオキシ、またはC1〜C4アルキルチオが挙げられる。
本明細書中、単独でもしくは他の用語と組み合わせて用いられる「アリール」なる用語は、単環状もしくは縮合環状芳香族炭化水素基を包含する。例えば、フェニル、1−ナフチル、2−ナフチル、またはアントリル等が挙げられる。
R2の好ましいアリールは、フェニルが挙げられる。
本明細書中、単独でもしくは他の用語と組み合わせて用いられる「ヘテロアリール」なる用語は、任意に選ばれる、酸素原子、硫黄原子又は窒素原子を環内に1個以上含む5〜6員の芳香環基を包含する。これらはさらに前記「アリール」、または他の「ヘテロアリール」と縮合していてもよい。ヘテロアリールが縮合環である場合は、結合可能なすべての位置で結合し得る。例えば、ピロリル(例えば、1−ピロリル等)、インドリル(例えば、3−インドリル等)、カルバゾリル(例えば、3−カルバゾリル等)、イミダゾリル(例えば、4−イミダゾリル等)、ピラゾリル(例えば、3−ピラゾリル等)、ベンゾイミダゾリル(例えば、2−ベンゾイミダゾリル等)、インダゾリル(例えば、3−インダゾリル等)、インドリジニル(例えば、6−インドリジニル等)、ピリジル(例えば、4−ピリジル等)、キノリル(例えば、5−キノリル等)、イソキノリル(例えば、3−イソキノリル等)、アクリジル(例えば、1−アクリジル等)、フェナントリジニル(例えば、2−フェナントリジニル等)、ピリダジニル(例えば、3−ピリダジニル等)、ピリミジニル(例えば、4−ピリミジニル等)、ピラジニル(例えば、2−ピラジニル等)、シンノリニル(例えば、3−シンノリニル等)、フタラジニル(例えば、2−フタラジニル等)、キナゾリニル(例えば、2−キナゾリニル等)、イソオキサゾリル(例えば、3−イソオキサゾリル等)、ベンゾイソオキサゾリル(例えば、3−ベンゾイソオキサゾリル等)、オキサゾリル(例えば、2−オキサゾリル等)、ベンゾオキサゾリル(例えば、2−ベンゾオキサゾリル等)、ベンゾオキサジアゾリル(例えば、4−ベンゾオキサジアゾリル等)、イソチアゾリル(例えば、3−イソチアゾリル等)、ベンゾイソチアゾリル(例えば、2−ベンゾイソチアゾリル等)、チアゾリル(例えば、4−チアゾリル等)、ベンゾチアゾリル(例えば、2−ベンゾチアゾリル等)、フリル(例えば、2−フリル等)、ベンゾフリル(例えば、3−ベンゾフリル等)、チエニル(例えば、2−チエニル等)、ベンゾチエニル(例えば、2−ベンゾチエニル等)、テトラゾリル、オキサジアゾリル(例えば、1,3,4−オキサジアゾリル等)、オキサゾリル、チアジアゾリル(例えば、1,3,4−チアジアゾリル等)、4H−1,2,4−トリアゾリル、キノキサリニル、または2−ピリドン−3−イル等が挙げられる。
R2またはR4の好ましいヘテロアリールとしては、2−フリル、2−チエニル、または4−ピリジルが挙げられる。
本明細書中、単独でもしくは他の用語と組み合わせて用いられる「アラルキル」なる用語は、前記「アルキル」に前記「アリ−ル」または前記「ヘテロアリール」が1または2ヶ所以上置換したものを包含する。例えば、ベンジル、フェニルエチル(例えば、1−フェニルエチル等)、フェニルプロピル(例えば、1−フェニルプロピルまたは3−フェニルプロピル等)、ナフチルメチル(例えば、1−ナフチルメチル等)、アントリルメチル(例えば、9−アントリルメチル等)、フリルメチル(例えば、2−フリルメチル等)、チエニルメチル(例えば、2−チエニルメチル等)、ピリジルメチル(例えば、4−ピリジルメチル等)、およびピリミジニルメチル(4−ピリミジニルメチル等)等が挙げられる。
R2の好ましいアラルキルとしては、ベンジル、1−フェニルエチル、2−フリルメチル、または2−チエニルメチルが挙げられる。
本明細書中で用いられる「置換されていてもよいアリール」、「置換されていてもよいヘテロアリール」および「置換されていてもよいアラルキル」における置換基としては、例えば、ハロゲン(例えば、フッ素、塩素、臭素、またはヨウ素)、低級アルキル(例えば、メチル、エチル、n−プロピル、イソプロピル、n−ブチル、イソブチル、sec−ブチル、tert−ブチル、n−ペンチル、イソペンチル、またはneo−ペンチル等)、C2〜C4アルケニル(例えば、ビニル、アリル、2−ブテニル等)、C2〜C4アルキニル(例えば、エチニル、プロパルギル、2−ブチニル等)、C3〜C6シクロアルキル(例えば、シクロプロピル、シクロブチル、シクロペンチル、またはシクロヘキシル等)、ハロ低級アルキル(例えば、トリフルオロメチル等)、ヒドロキシ、C1〜C4アルキルオキシ(例えば、メチルオキシ、またはエチルオキシ等)、C6〜C10アリ−ルオキシ(例えば、フェニルオキシ等)、メルカプト、C1〜C4アルキルチオ(例えば、メチルチオ等)、ニトロ、シアノ、置換されていてもよいアミノ(例えば、アミノ、メチルアミノ、ジメチルアミノ、ジエチルアミノ、またはホルミルアミノ等)、またはC1〜C4アルキルスルホニル(例えば、メチルスルホニル等)等が挙げられる。これら置換基は、全ての可能な位置で1個以上置換しうる。好ましい置換基としては、ハロゲン、低級アルキル、ハロ低級アルキル、またはC1〜C4アルキルオキシが挙げられる。
本明細書中で用いられる「置換されていてもよいアミノ」なる用語は、前記「アルキル」、前記「アラルキル」、前記「アリール」、または前記「ヘテロアリール」が同一であっても異なっていてもよく、1または2個置換されていてもよいアミノを意味する。例えば、アミノ、C1〜C6アルキルでモノ置換またはジ置換されているアミノ(例えば、モノメチルアミンまたはジメチルアミン等)、C1〜C4アシルでモノ置換されているアミノ(例えば、ホルミルアミノまたはアセチルアミノ等)を意味する。好ましくは、アミノまたはメチルアミノが挙げられる。
本明細書中で用いられる「保護基」なる用語は、反応後容易に脱離することができる基を意味する。例えば、C1〜C4アルキルオキシC1〜C4アルキル(例えば、メトキシメチル、エトキシメチル、ジアルキルオキキシメチル、または2−メトキシ−2−プロピル等)、C6〜C12アリールC1〜C4アルキル(例えば、トリチル等)、アリル、ヒドロキシメチル、C1〜C4アシル(例えば、ホルミル等)、C1〜C6アルキルオキシカルボニル(例えば、tert−ブトキシカルボニル等)、C6〜C12アリールC1〜C4アルキルオキシカルボニル(例えば、9−フルオレニルメトキシカルボニル等)、C1〜C4アルキルスルホニル(例えば、メタンスルホニル等)、C6〜C12アリールスルホニル(例えば、p−トルエンスルホニル等)、C1〜C4アルキルでモノ置換またはジ置換されていてもよいスルファモイル(例えば、N,N−ジメチルスルファモイル等)、またはTHP(テトラヒドロピラニル)等が挙げられる。
本明細書中で用いられる「ヒドラジン」なる用語は、ヒドラジン、ヒドラジンの水和物、およびヒドラジンまたはヒドラジン水和物を含有する溶液を意味する。
一般式(I)で示される化合物のR1およびR2において、好ましい置換基の群を(a)〜(e)で示す。
R1は、(a)置換されていてもよいアルキル。
R2は、(b)置換基されていてもよいアルキル、(c)置換基されていてもよいアラルキル、(d)置換されていてもよいアリール、または(e)置換されていてもよいヘテロアリール。
一般式(I)で示される化合物の好ましい一群としては、[R1, R2]=[a, b], [a, c], [a, d], [a, e]。
一般式(IV)で示される化合物のR1およびR2において、好ましい置換基の群を(a)〜(e)で示す。
R1は、(a)置換されていてもよいアルキル。
R2は、(b)置換基されていてもよいアルキル、(c)置換基されていてもよいアラルキル、(d)置換されていてもよいアリール、または(e)置換されていてもよいヘテロアリール。
一般式(IV)で示される化合物の好ましい一群としては、[R1, R2]=[a, b], [a, c], [a, d], [a, e]。
【0007】
【発明の実施の形態】
以下に本発明の製造方法について、第1工程〜第3工程に分けて説明する。
第1工程
【化32】
本工程は、一般式(V)で示される化合物とヒドラジンを縮合することによって、一般式(II)で示される化合物を製造する工程である。
一般式(V)で示される化合物としては、市販の化合物または公知の方法(例えば、J. Am. Chem. Soc., 60, 1938, 2790−2792等)に準じて合成した化合物を用いることができる。
ヒドラジンとしては、ヒドラジン水和物(例えば、ヒドラジン一水和物等)またはヒドラジン水溶液(例えば、35%ヒドラジン水溶液等)が挙げられ、特にヒドラジン一水和物または35%ヒドラジン水溶液が好ましい。
ヒドラジンは一般式(V)に対して0.5〜1.5当量、好ましくは0.9〜1.1当量用いることができる。
反応溶媒は、アルコール類(例えば、メタノール、エタノール、n−プロパノール、イソプロパノール、n−ブタノール、イソブタノール、sec−ブタノール、またはtert−ブタノール等)、エーテル類(例えば、ジエチルエーテル、THF(テトラヒドロフラン)、またはジオキサン等)、ニトリル類(例えば、アセトニトリルまたはプロピロニトリル等)、ハロゲン化炭化水素類(例えば、塩化メチレン、ジクロロエタン、またはクロロホルム等)、芳香族炭化水素類(例えば、ベンゼン、トルエンまたはキシレン等)、またはDMF(N,N−ジメチルホルムアミド)が挙げられる。これらの溶媒を単独あるいは混合して用いることができる。アルコール類が好ましく、さらにエタノールが好ましい。
反応温度としては−80〜80℃が挙げられる。−50〜30℃が好ましい。
反応時間としては0.5〜24時間が挙げられる。1〜6時間が好ましい。
得られた一般式(II)で示される化合物は、単離精製せずに第2工程に使用することができる。あるいは公知の手段(例えば、クロマトグラフィー、再結晶など)で精製することができる。副生成物として生成するシュウ酸ジヒドラジドを含有したまま第2工程さらに第3工程を行うことができる。
なお、35%ヒドラジン水溶液を用いた方が、ヒドラジン一水和物を用いた時に比べ、工業的に好ましい温度で反応させることができる。
第2工程
【化33】
本工程は、一般式(II)で示される化合物と一般式(III)で示される化合物を縮合することによって、一般式(IV)で示される化合物を製造する工程である。
一般式(II)で示される化合物としては、上記の第1工程で合成された化合物または公知の方法(例えば、Chem. Ber. 123, 10, 1990, 2031−2037,等)に準じて合成した化合物を用いることができる。
一般式(III)で示される化合物としては、公知の方法(例えば、J. Hetrocyclic Chem., 25, 1988, 651−654等)に準じて合成した化合物を用いることができる。
【化34】
なお、塩化水素を用いた方が塩酸を用いたときに比べて、より短期間で一般式(III)で示される化合物を得ることができる。
一般式(III)は、一般式(II)に対して0.5〜3当量、好ましくは1〜1.5当量用いられる。
反応溶媒は、アルコール類(例えば、メタノール、エタノール、n−プロパノール、イソプロパノール、n−ブタノール、イソブタノール、sec−ブタノール、またはtert−ブタノール等)、エーテル類(例えば、ジエチルエーテル、THF(テトラヒドロフラン)、またはジオキサン等)、ニトリル類(例えば、アセトニトリルまたはプロピロニトリル等)、ハロゲン化炭化水素類(例えば、塩化メチレン、ジクロロエタン、またはクロロホルム等)、芳香族炭化水素類(例えば、ベンゼン、トルエンまたはキシレン等)、またはDMF(N,N−ジメチルホルムアミド)等が挙げらる。これらの溶媒を単独あるいは混合して用いることができる。アルコール類または芳香族炭化水素類が好ましい。さらにエタノールまたはトルエンが好ましい。
反応温度としては−40〜100℃が挙げられる。−20〜50℃が好ましい。
反応時間としては0.5〜24時間が挙げられる。1〜6時間が好ましい。
得られた一般式(IV)で示される化合物は、単離精製せずに第3工程に使用することができる。または公知の手段(例えば、クロマトグラフィー、再結晶など)で精製することができる。
第3工程
【化35】
本工程は、一般式(IV)で示される化合物を閉環させることによって、一般式(I)で示される化合物を製造する工程である。
一般式(IV)で示される化合物としては、上記の第2工程で合成した化合物を用いることができる。
反応溶媒は、アルコール類(例えば、メタノール、エタノール、n−プロパノール、イソプロパノール、n−ブタノール、イソブタノール、sec−ブタノール、またはtert−ブタノール等)、エーテル類(例えば、ジエチルエーテル、THF(テトラヒドロフラン)、またはジオキサン等)、ニトリル類(例えば、アセトニトリルまたはプロピロニトリル等)、ハロゲン化炭化水素類(例えば、塩化メチレン、ジクロロエタン、またはクロロホルム等)、芳香族炭化水素類(例えば、ベンゼン、トルエンまたはキシレン等)、DMF(N,N−ジメチルホルムアミド)、アセトン、または水等が挙げられる。これらの溶媒を単独あるいは混合して用いることができる。
なお、アセトンを用いることにより第1工程で副生成したシュウ酸ジヒドラジドを除くことができるので、副生成したシュウ酸ジヒドラジドが含有している場合は、アセトンとアルコール類またはニトリル類の混合溶媒が好ましい。さらにアセトンとエタノールまたはアセトニトリルの混合溶媒が好ましい。シュウ酸ジヒドラジドが含有していない場合は、アルコール類またはニトリル類が好ましい。さらにエタノールまたはアセトニトリルが好ましい。
反応溶媒として水を単独で用いた場合は、一般式(I)で表わされる化合物の収率は若干低くなるが、副生するシュウ酸ジヒドラジドあるいはトリアジンジオンを水で再結晶することにより容易に母液へ取り除くことができる。
反応温度としては30〜100℃が挙げられる。50〜100℃が好ましい。さらに、80〜100℃が好ましい。
反応時間としては0.5〜24時間が挙げられる。1〜6時間が好ましい。
得られた一般式(I)で示される化合物は、公知の手段(例えば、クロマトグラフィー、再結晶など)で単離精製することができる。
なお、反応溶媒がアルコール類の場合、第1工程から第3工程までをワンポットで行うこともできる。アルコール類としては、エタノールが好ましい。
一般式(I)で示される化合物を出発原料として、WO00/39086およびWO00/75122に記載の方法に従って、一般式(XI)で表わされる化合物を合成することができる。
【化36】
実施例中、以下の略号を使用する。
Me:メチル
Et:エチル
n−Pr:n−プロピル
i−Pr:イソプロピル
Tr:トリチル
THP:テトラヒドロピラニル
THF:テトラヒドロフラン
DMF:N,N−ジメチルホルムアミド
DMSO:ジメチルスルホキシド
元素分析において、Calcd. forは理論値、Foundは実測値を意味する。
1H−NMRはδ値をppmで表わし、s=一重線、d=二重線、t=三重線、q=四重線、m=多重線、br=幅広線を意味する。J値はHzで表わした。
【実施例】
【0008】
参考例
参考例1:化合物(IIIa)の合成
【化37】
参考例2:化合物(IIIa)の合成
【化38】
0.58 mol, 収率 84%)
参考例3:化合物(IIIb)の合成
【化39】
参考例4:化合物(IIIc)の合成
【化40】
参考例5:化合物(IIId)の合成
【化41】
参考例6:化合物(IIIe)の合成
【化42】
参考例7:化合物(IIIf)の合成
【化43】
参考例8:化合物(IIIg)の合成
【化44】
参考例9:化合物(IIIh)の合成
【化45】
実施例1:化合物(Ia)の合成
【化46】
化合物(IIa)の粗製物を氷冷し、参考例1で合成した化合物(IIIa)のエタノール(68 mL)溶液(内、化合物(IIIa)として10.89 g, 0.125 mol)を0〜5℃で滴下した後、氷冷攪拌1時間、還流2時間反応した。溶媒留去後エタノール−トルエンで再結晶し、白色結晶として化合物(Ia)を7.36 g(0.047 mol, 収率47%)得た。
融点: 189−191℃.
1H NMR (d6−DMSO, 300 MHz): 1.29 (3H, t, J= 7.2 Hz), 2.39 (3H, s), 4.30 (2H, q, J= 7.2 Hz).
元素分析:Calcd. for C6H9N3O2: C, 46.45; H, 5.85; N, 27.08. Found: C, 46.35; H, 5.75; N, 27.15; H2O, <0.1.
実施例2:化合物(IVa’)の合成
【化47】
化合物(IIa)の粗製物を氷冷し、参考例1で合成した化合物(IIIa)のトルエン(60 mL)溶液(内、化合物(IIIa)として8.02 g, 0.092 mol)を0〜5℃で滴下した。氷冷攪拌1時間後析出晶を濾過し、白色結晶として化合物(IVa’)を9.98 g(0.052 mol, 収率71%)得た。
融点:98−100℃.
1H NMR (d6−DMSO, 300 MHz): 1.22 (3H, t, J= 7.2 Hz), 2.10 (3H, s), 4.10 (2H, brd, J= 7.2 Hz), 7.40 (1H, brs), 8.32 (1H, brs), 11.05 (1H, brs).
元素分析:Calcd. for C6H11N3O3・H2O: C, 37.69; H, 6.85; N, 21.98; H2O, 9.42. Found: C, 37.81; H, 7.04; N, 22.05; H2O, 9.42.
実施例3:化合物(Ia)の合成
【化48】
実施例4:化合物(IVa’)の合成
【化49】
実施例5:化合物(Ia)の合成
【化50】
実施例6:化合物(Ib)の合成
【化51】
化合物(IIa)の粗製物を氷冷し、参考例3で合成した化合物(IIIb)のエタノール(121 mL)溶液(内、化合物(IIIb)として21.66 g, 0.21 mol)を0〜5℃で滴下し、氷冷攪拌1日、還流2時間行った。溶媒留去後水で再結晶を行ない、白色結晶として化合物(Ib)を7.35 g得た。母液を酢酸エチルで抽出、濃縮後の結晶性残査をイソプロピルエーテルで洗浄し、白色結晶として化合物(Ib)の第二晶を 8.75 g(トータル 16.10 g, 0.095 mol, 収率 53%)得た。
融点:106−108℃.
1H NMR (d6−DMSO, 300 MHz): 1.24 (3H, t, J= 7.2 Hz), 1.29 (3H, t, J= 7.2 Hz), 2.75 (2H, q, J= 7.2 Hz) , 4.30 (2H, q, J= 7.2 Hz).
元素分析:Calcd. for C7H11N3O2: C, 49.70; H, 6.55; N, 24.84. Found: C, 49.72; H, 6.61; N, 24.74; H2O, <0.1.
実施例7:化合物(Ic)の合成
【化52】
化合物(IIa)の粗製物を氷冷し、参考例4で合成した化合物(IIIc)のエタノール(96.5 mL)溶液(内、化合物(IIIc)として19.63 g, 0.17 mol)を0〜5℃で滴下し、氷冷攪拌4時間、還流3時間行った。溶媒留去後、水で再結晶し、白色結晶として化合物(Ic)を15.98 g(0.087 mol, 収率61%)得た。
融点:117−118.5℃.
1H NMR (d6−DMSO, 300 MHz): 0.89 (3H, t, J= 7.2 Hz), 1.30 (3H, t, J= 7.2 Hz), 1.70 (2H, tq, J= 7.5, 7.2 Hz) , 2.71 (2H, t, J= 7.5 Hz) , 4.30 (2H,q, J= 7.2 Hz).
元素分析:Calcd. for C8H13N3O2: C, 52.45; H, 7.15; N, 22.94. Found: C, 52.47; H, 7.16; N, 22.98; H2O, 0.23.
実勢例8:化合物(Id)の合成
【化53】
化合物(IIa)の粗製物を氷冷し、参考例5で合成した化合物(IIId)のエタノール(86.5 mL)溶液(内、化合物(IIId)として17.63 g, 0.15 mol)を0〜5℃で滴下し、氷冷攪拌5時間、還流2時間行った。溶媒留去後、酢酸エチルで抽出し、有機層の溶媒を濃縮後、結晶性残査をイソプロピルエーテルで洗浄し、白色結晶として化合物(Id)を14.93 g(0.081 mol, 収率 64%)得た。
融点:105−107℃.
1H NMR (d6−DMSO, 300 MHz): 1.27 (6H, d, J= 6.9 Hz), 1.30 (3H, t, J= 7.2 Hz), 3.02−3.18 (1H, m) , 4.31 (2H, q, J= 7.2 Hz).
元素分析:Calcd. for C8H13N3O2: C, 52.45; H, 7.15; N, 22.94. Found: C, 52.30; H, 7.15; N, 22.98; H2O, 0.30.
実施例9:化合物(Ie)の合成
【化54】
化合物(IIa)の粗製物を氷冷し、参考例6で合成した化合物(IIIe)のエタノール(76.5 mL)溶液(内、化合物(IIIe)として18.31 g, 0.14 mol)を0〜10℃で滴下した後、室温攪拌1日、還流2時間行った。溶媒留去後アセトニトリル−トルエンに溶解させ不溶物を濾過した後、濾液をアセトニトリル−トルエンで再結晶し、白色結晶として化合物(Ie)を13.63 g(0.063 mol, 収率56%)得た。
融点:164−166℃.
1H NMR (d6−DMSO, 300 MHz): 1.35 (3H, d, J= 7.2 Hz), 4.38 (2H, q, J= 7.2 Hz), 7.48−7.60 (3H, m), 8.00−8.08 (2H, m).
元素分析: Calcd. for C11H11N3O2: C, 60.82; H, 5.10; N, 19.34. Found: C,60.90; H, 5.14; N, 19.42; H2O, <0.1.
実施例10:化合物(IVf)の合成
【化55】
化合物(IIa)の粗製物を氷冷し、参考例7で合成した化合物(IIIf)のエタノール(95 mL)溶液(内、化合物(IIIf)として27.38 g, 0.17 mol)を0〜10℃で滴下した。氷冷攪拌1.5時間後析出晶を濾過し、白色結晶として化合物(IVf)を20.31 g得た。濾液を留去し、結晶性残査を酢酸エチルで洗浄し、白色結晶として化合物(IVf)の第2晶 8.50 g (total 28.81g, 0.12 mol, 収率 83%) 得た。
融点:115−117℃.
1H NMR (d6−DMSO, 300 MHz): 1.06 (3H, t, J= 6.9 Hz), 4.12 (2H, brs), 4.35
(2H, brs), 7.24−7.44 (5H, m).
実施例11:化合物(If)の合成
【化56】
融点:147.5−150℃.
1H NMR (d6−DMSO, 300 MHz): 1.28 (3H, t, J= 7.2 Hz), 4.12 (2H, s), 4.29 (2H, q, J= 7.2 Hz), 7.20−7.40 (5H, m).
元素分析:Calcd. for C12H13N3O2: C, 62.33; H, 5.67; N, 18.17. Found: C, 62.32; H, 5.58; N, 18.39; H2O, 0.18.
実施例12:化合物(Ig)の合成
【化57】
化合物(IIa)の粗製物を氷冷し、参考例8で合成した化合物(IIIg)のエタノール(62.0 mL)溶液(内、化合物(IIIg)として16.91 g, 0.11 mol)を0〜10℃で滴下した後、室温攪拌1日、還流5時間行った。溶媒留去後クロロホルムでカラムをかけた後、結晶性残査をイソプロピルエーテルで洗浄し、白色結晶として化合物(Ig)を7.38g(0.031 mol, 収率 34%)得た。
融点:176−178℃.
1H NMR (d6−DMSO, 300 MHz): 1.34 (3H, t, J= 7.2 Hz), 4.37 (2H, q, J= 7.2 Hz), 7.20−7.26 (1H, m), 7.72−7.80 (2H, m).
元素分析: Calcd. for C9H9N3O2S: C, 48.42; H, 4.06; N, 18.82; S, 14.36. Found: C, 48.31; H, 3.97; N, 18.66; S, 14.24; H2O, <0.1.
実施例13:化合物(IVh)の合成
【化58】
化合物(IIa)の粗製物を氷冷し、参考例9で合成した化合物(IIIh)のエタノール(102 mL)溶液(内、化合物(IIIh)として24.51 g, 0.18 mol)を0〜10℃で滴下した。氷冷攪拌1日後濾過し、淡黄色結晶として化合物(IVh)を28.30 g(0.12 mol, 収率80%)得た。
融点: 212−216℃ (分解).
1H NMR (d6−DMSO, 300 MHz): 1.28 and 1.30 (total 3H, t, J= 7.2 Hz), 4.27 and 4.30 (total 2H, q, J= 7.2 Hz), 6.78 and 7.10 (total 2H, s), 7.60−7.78 (2H, m), 8.61−8.67 (2H, m).
実施例14:化合物(Ih)の合成
【化59】
融点:175−177℃.
1H NMR (d6−DMSO, 300 MHz): 1.36 (3H, t, J= 7.2 Hz), 4.40 (2H, q, J= 7.2 Hz), 7.96−7.99 (2H, m), 8.74−8.77 (2H, m).
元素分析:Calcd. for C10H10N4O2: C, 55.04; H, 4.62; N, 25.67. Found: C, 54.76; H, 4.53; N, 25.61; H2O, 0.87.
参考例
参考例10:化合物(XIa)の合成
【化60】
mmol, 収率 85%)得た。
化合物(IXa)0.81 g(3.7 mmol)と化合物(VIIIa−1)0.89 g(3.7 mmol)のTHF(8.0 ml)溶液に28%ナトリウムメトキシド/メタノール溶液(0.74 g, 3.7mmol)を氷冷下で滴下した。室温で14時間攪拌後、反応液に1.8%酢酸水溶液(23 ml)を加えた。反応液を酢酸エチル(30 ml)で抽出し、飽和炭酸水素ナトリウム水溶液で洗浄、水洗し、Na2SO4で乾燥した。溶媒を留去して化合物(XIa)を0.84 g(2.0 mmol, 収率 55%)得た。
1H NMR (d6−DMSO, 300 MHz): 2.43 (3H, s), 4.14 (2H, s), 6.46 (1H, d, J= 3.6 Hz), 6.89 (1H, s), 7.18 (2H, t, J= 9.0 Hz), 7.35 (2H, dd, J= 9.0, 5.7Hz), 7.50 (1H, d, J=3.6 Hz).
参考例11:化合物(XIa)の合成
【化61】
化合物(Xa−2)20.00 g(0.050 mol)のアセトン(106 mL)、水(9.8 mL)混合溶液を65℃で5分間攪拌した。反応液に塩酸(濃塩酸 0.04 mL/水 1.6 mL)を加え、70℃で1時間攪拌した。内温40℃以上でアセトン(54 mL)を減圧留去した。45℃で1時間攪拌後、炭酸ナトリウム(26.5 mg)/水(54 mL)を40〜50℃で滴下した。室温で1時間、さらに氷冷下2時間攪拌した。析出した結晶をろ取し、化合物(XIa)を黄色結晶として15.63 g(0.048 mol, 収率 95%)得た。融点 : 195−197℃.
1H NMR (d6−DMSO, 300 MHz): 2.43 (3H, s), 4.14 (2H, s), 6.46 (1H, d, J= 3.6 Hz), 6.89 (1H, s), 7.18 (2H, t, J= 9.0 Hz), 7.35 (2H, dd, J= 9.0, 5.7Hz), 7.50 (1H, d, J=3.6 Hz).
元素分析:Calcd. for C17H14FN3O3: C, 62.38; H, 4.31; N, 12.84; F, 5.80. Found: C, 62.39; H, 4.28; N, 12.90; F, 5.66; H2O<0.1%.
参考例12:化合物(XIb)の合成
【化62】
mmol, 収率 80%)得た。
化合物(IXa)0.77 g(3.5 mmol)と化合物(VIIIb−1)0.89 g(3.5 mmol)のTHF(8.0 ml)溶液に28%ナトリウムメトキシド/メタノール溶液(0.70 g, 3.5mmol)を氷冷下で滴下した。室温で14時間攪拌後、反応液に1.8%酢酸水溶液(22 ml)を加えた。反応液を酢酸エチル(30 ml)で抽出し、飽和炭酸水素ナトリウム水溶液で洗浄、水洗し、Na2SO4で乾燥した。溶媒を留去して化合物(XIb)を0.82 g(1.9 mmol, 収率 55%)得た。
融点 : 204−205℃.
1H NMR (d6−DMSO, 300 MHz): 1.27 (3H, t, J= 7.8Hz), 2.79 (2H, q, J= 7.8Hz), 4.15 (2H, s), 6.45 (1H, d, J= 3.6Hz), 6.88 (1H, s), 7.17 (2H, t, J= 9.3Hz), 7.25−7.40 (2H, m), 7.49 (1H, d, J= 3.6Hz).
元素分析:Calcd. for C18H16FN3O3・0.25H2O: C, 62.51; H, 4.81; N, 12.15; F,5.49. Found: C, 62.57; H, 4.68; N, 12.25; F, 5.30.
参考例13:化合物(XIb)の合成
【化63】
化合物(Xb−2)2.48 g(0.006 mol)のアセトン(13 mL)、水(1.2 mL)混合溶液を65℃で5分間攪拌した。反応液に塩酸(濃塩酸 0.005 mL/水 0.19 mL)を加え、70℃で1時間攪拌した。内温40℃以上でアセトン(6 mL)を減圧留去した。45℃で1時間、つづけて室温で1時間、さらに氷冷下2時間攪拌した。析出した結晶をろ取し、化合物(XIb)を結晶として1.64 g(4.8 mmol, 収率 80%)得た。
融点 : 204−205℃.
1H NMR (d6−DMSO, 300 MHz): 1.27 (3H, t, J= 7.8Hz), 2.79 (2H, q, J= 7.8Hz), 4.15 (2H, s), 6.45 (1H, d, J= 3.6Hz), 6.88 (1H, s), 7.17 (2H, t, J= 9.3Hz), 7.25−7.40 (2H, m), 7.49 (1H, d, J= 3.6Hz).
参考例14:化合物(XIc)の合成
【化64】
化合物(IXa)0.44 g(2.00 mmol)の THF(15.0 ml)溶液を−70℃まで冷却し、リチウムビストリメチルシリルアミド THF(1 M)溶液(2.60 ml, 2.60 mmol)を滴下した。反応液を一旦 0℃まで昇温し、再び−70℃まで冷却した。反応液に化合物(VIIIc)0.94 g(2.20 mmol)の THF(8.00 ml)溶液を−32〜−7℃で滴下し、室温まで昇温後1.5時間攪拌した。反応液を飽和塩化アンモニウム水溶液に投入し、酢酸エチルにて抽出、水洗後、MgSO4で乾燥した。溶媒を留去する事によって得られた化合物(Xc)の粗精製残渣(1.30 g)を次の反応に使用した。
化合物(Xc)1.30 gの粗精製物をジオキサン(15.0 ml)溶液に1.5 mol/L塩酸(5 ml)を加え、70 ℃で1時間攪拌した。次いで減圧下、ジオキサンを留去し、残査に水を加え、酢酸エチルで抽出した。酢酸エチル層を水洗、乾燥後、溶媒を留去した。残留物を酢酸エチルに溶解し、1 mol/L 水酸化ナトリウム水溶液で抽出した。アルカリ抽出液を酢酸エチルで2回洗浄後、6 mol/L塩酸で中和し、酢酸エチルで抽出した。抽出液を水洗、飽和食塩水で洗浄後、Na2SO4で乾燥した。溶媒を留去し、得られた粗結晶を少量の酢酸エチルで洗浄後、エーテルから再結晶することにより化合物(XIc)を化合物(IXa)から収率 14% (0.099 g, 0.28 mmol)で得た。
融点 : 167−168℃
1H NMR (d6−DMSO, 300 MHz): 0.92 (3H, t, J= 7.5Hz), 1.60−1.80 (2H, m), 2.73 (2H, q, J= 7.5Hz), 4.15 (2H, s), 6.45 (1H, d, J= 3.6Hz), 6.88 (1H, s), 7.17 (2H, t, J=9.3Hz), 7.25−7.40 (2H, m), 7.49 (1H, d, J= 3.6Hz).元素分析:Calcd. for C19H18FN3O3: C, 64.22; H, 5.11; N, 11.82; F, 5.35. Found: C, 64.05; H, 5.07; N, 11.80; F, 5.13.
参考例15:化合物(XId)の合成
【化65】
化合物(Xb)2.99 g(7.0 mmol)のアセトン(15 mL)、水(1.4 mL)混合溶液を65℃で5分間攪拌した。反応液に塩酸(濃塩酸 0.006 mL/水 0.22 mL)を加え、70℃で1時間攪拌した。内温40℃以上でアセトン(7 mL)を減圧留去した。45℃で1時間、つづけて室温で1時間、さらに氷冷下2時間攪拌した。析出した結晶をろ取し、化合物(XIb)を結晶として1.74 g(4.9 mmol, 収率 70%)得た。
融点 : 146−147℃
1H NMR (d6−DMSO, 300 MHz): 1.30 (6H, d, J= 6.9Hz), 3.00−3.20 (1H, m), 4.15 (2H, s), 6.45 (1H, d, J= 3.6Hz), 6.87 (1H, s), 7.17 (2H, t, J= 9.3Hz), 7.25−7.40 (2H, m), 7.48 (1H, d, J= 3.6Hz).
元素分析:Calcd. for C19H18FN3O3・0.25H2O: C, 63.41; H, 5.18; N, 11.68; F,5.28. Found: C, 63.47; H, 5.09; N, 12.43; F, 4.85.
【発明の効果】
本発明により、医薬品製造のための中間体として有用な5−置換−1,2,4−トリアゾール−3−カルボン酸エステル誘導体およびアシルアミドラゾン誘導体を工業的に製造することができる。
Claims (13)
- 一般式(IV):
で示される化合物またはその水和物を100℃以下で閉環させることを特徴とする、一般式(I):
で示される化合物の製造方法。 - 一般式(II):
で示される化合物と、一般式(III):
で示される化合物またはその水和物を得る工程;および
式(IV)で示される化合物またはその水和物を閉環させる工程
を包含する、一般式(I):
で示される化合物の製造方法。 - 一般式(V):
で示される化合物と、ヒドラジンを縮合させて一般式(II):
で示される化合物を得る工程を包含する、請求項2記載の製造方法。 - 一般式(II):
で示される化合物に、一般式(III):
で示される化合物を縮合させて得られる、一般式(IV):
で示される化合物の製造方法。 - 一般式(V):
で示される化合物と、ヒドラジンを縮合させて、一般式(II):
で示される化合物を得る工程を包含する、請求項4記載の製造方法。 - R1が低級アルキルである請求項1から請求項5のいずれかに記載の製造方法。
- R1がエチルである請求項1から請求項5のいずれかに記載の製造方法。
- R2が低級アルキルである請求項1から請求項7のいずれかに記載の製造方法。
- R3が低級アルキルである請求項2から請求項8のいずれかに記載の製造方法。
- 反応溶媒としてエタノールを用いる請求項1から請求項9のいずれかに記載の製造方法。
- 反応温度が90℃以下である請求項1から請求項3のいずれかに記載の製造方法。
- 一般式(IV−1):
で示される化合物、もしくはその塩、またはその溶媒和物。 - 一般式(I−1):
で示される化合物、もしくはその塩、またはその溶媒和物。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2002177822A JP2004018480A (ja) | 2002-06-19 | 2002-06-19 | 5−置換−1,2,4−トリアゾール−3−カルボン酸エステル誘導体の製造方法 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2002177822A JP2004018480A (ja) | 2002-06-19 | 2002-06-19 | 5−置換−1,2,4−トリアゾール−3−カルボン酸エステル誘導体の製造方法 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| JP2004018480A true JP2004018480A (ja) | 2004-01-22 |
Family
ID=31175712
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2002177822A Pending JP2004018480A (ja) | 2002-06-19 | 2002-06-19 | 5−置換−1,2,4−トリアゾール−3−カルボン酸エステル誘導体の製造方法 |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP2004018480A (ja) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US12312508B2 (en) | 2019-11-27 | 2025-05-27 | Toyobo Mc Corporation | Polyolefin-based adhesive composition |
-
2002
- 2002-06-19 JP JP2002177822A patent/JP2004018480A/ja active Pending
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US12312508B2 (en) | 2019-11-27 | 2025-05-27 | Toyobo Mc Corporation | Polyolefin-based adhesive composition |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| AU2013272011B2 (en) | Processes to produce certain 2-(pyridine-3-yl)thiazoles | |
| RU2647853C2 (ru) | Способы получения определенных 2-(пиридин-3-ил)тиазолов | |
| US20050182055A1 (en) | Preparation process | |
| JP5476467B2 (ja) | 新規なビフェニルピリジンアミドおよびフェニルピリジンアミド | |
| CA2942871A1 (en) | Ror-gamma modulators and uses thereof | |
| CN106255681B (zh) | 用于制备取代的环丝氨酸的方法 | |
| JPH06505239A (ja) | 抗炎症剤として有用な4,6−ジ−第三ブチル−5−ヒドロキシ−1,3−ピリミジンの置換されたヘテロアリール類似体 | |
| JP5610599B2 (ja) | N−フェニル−n’−フェニルスルホニルピペラジン誘導体の製造方法 | |
| KR20100046026A (ko) | 4-페닐-6-(2,2,2-트리플루오로-1-페닐에톡시)피리미딘-기재화합물의 제조 방법 | |
| WO2012038904A1 (fr) | Derives de nicotinamide, leur preparation et leur application en therapeutique | |
| JPH0256471A (ja) | 新規な(ヘテロ)アリル置換ジアゾール誘導体、その製造方法および治療への適用 | |
| KR20030036929A (ko) | 신규한 아미노트리아졸론, 이의 제조 방법 및 이를함유하는 약제 조성물 | |
| JP2004018480A (ja) | 5−置換−1,2,4−トリアゾール−3−カルボン酸エステル誘導体の製造方法 | |
| JP2007246396A (ja) | 5−ジフルオロメトキシ−4−チオメチルピラゾール化合物の製造方法 | |
| US20220169640A1 (en) | Production method of quinolinecarboxamide derivative or production intermediate thereof | |
| WO2007068815A2 (fr) | Derives heterocycliques, leur preparation et leur application en therapeutique. | |
| WO2004113258A1 (ja) | 炭素−炭素結合生成反応 | |
| WO2007068814A1 (fr) | Derives diaryltriazolmethylamine, leur preparation et leur application en therapeutique. | |
| JP4075357B2 (ja) | 4,5−ジ置換−1,2,3−トリアゾール及びその製造法 | |
| JP2012136437A (ja) | ヘテロ5員環化合物 | |
| KR19990022022A (ko) | 치환된 5-하이드록시-옥사졸리딘-4-온의 제조방법 | |
| EA053007B1 (ru) | Способ получения замещенных халконов | |
| JP2003026640A (ja) | 芳香族ケトンの製造方法 | |
| MX2007012938A (es) | Metodos para sintesis de compuestos de dicarbamato e intermediarios en la formacion de los mismos. | |
| HU218210B (hu) | Eljárás szubsztituált indolonszármazékok előállítására |