JP2004196678A - Pyrazole derivatives - Google Patents
Pyrazole derivatives Download PDFInfo
- Publication number
- JP2004196678A JP2004196678A JP2002364578A JP2002364578A JP2004196678A JP 2004196678 A JP2004196678 A JP 2004196678A JP 2002364578 A JP2002364578 A JP 2002364578A JP 2002364578 A JP2002364578 A JP 2002364578A JP 2004196678 A JP2004196678 A JP 2004196678A
- Authority
- JP
- Japan
- Prior art keywords
- group
- compound
- atom
- optionally substituted
- substituent
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
- 0 CC[C@](C)*(*)(C(B1)*)N(C)C1* Chemical compound CC[C@](C)*(*)(C(B1)*)N(C)C1* 0.000 description 4
- JTBZBRUNLMMZGB-GQCTYLIASA-N CCOC(C(CC(/C=C/c1c[n](C)c(cc2)c1cc2Cl)=O)=O)=O Chemical compound CCOC(C(CC(/C=C/c1c[n](C)c(cc2)c1cc2Cl)=O)=O)=O JTBZBRUNLMMZGB-GQCTYLIASA-N 0.000 description 1
- RSHFHAUQNQUMHN-HWKANZROSA-N CCOC(C1NNC(/C=C/c(cc2)cc(Cl)c2Cl)=C1)=O Chemical compound CCOC(C1NNC(/C=C/c(cc2)cc(Cl)c2Cl)=C1)=O RSHFHAUQNQUMHN-HWKANZROSA-N 0.000 description 1
- FQRDTPDNACEKLW-UHFFFAOYSA-N CCOC(CCc(cc1-c2ccc(cccc3)c3c2)n[n]1-c1cccc(Cl)c1)=O Chemical compound CCOC(CCc(cc1-c2ccc(cccc3)c3c2)n[n]1-c1cccc(Cl)c1)=O FQRDTPDNACEKLW-UHFFFAOYSA-N 0.000 description 1
- IHTFINBDKRJESJ-DUXPYHPUSA-N C[n]1c(ccc(Cl)c2)c2c(/C=C/C2=CC(CO)NN2)c1 Chemical compound C[n]1c(ccc(Cl)c2)c2c(/C=C/C2=CC(CO)NN2)c1 IHTFINBDKRJESJ-DUXPYHPUSA-N 0.000 description 1
- FBLHHVYNGQOVDT-BQYQJAHWSA-N Clc1cc(-[n]2nc(C3CCNCC3)cc2/C=C/c(c(Cl)ccc2)c2Cl)ccc1 Chemical compound Clc1cc(-[n]2nc(C3CCNCC3)cc2/C=C/c(c(Cl)ccc2)c2Cl)ccc1 FBLHHVYNGQOVDT-BQYQJAHWSA-N 0.000 description 1
- ZZRFPLYVZIEOGA-SREVYHEPSA-N Clc1cc(N2NC(C3CCNCC3)C=C2/C=C\C2=[Cl]C=CC=C2Cl)ccc1 Chemical compound Clc1cc(N2NC(C3CCNCC3)C=C2/C=C\C2=[Cl]C=CC=C2Cl)ccc1 ZZRFPLYVZIEOGA-SREVYHEPSA-N 0.000 description 1
Landscapes
- Plural Heterocyclic Compounds (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
【課題】多剤耐性菌に対して優れた抗菌剤の提供。
【解決手段】下記一般式(I)で表される化合物またはその製薬学的に許容される塩およびこれらを有効成分とする抗菌剤を提供することにある。
【化1】
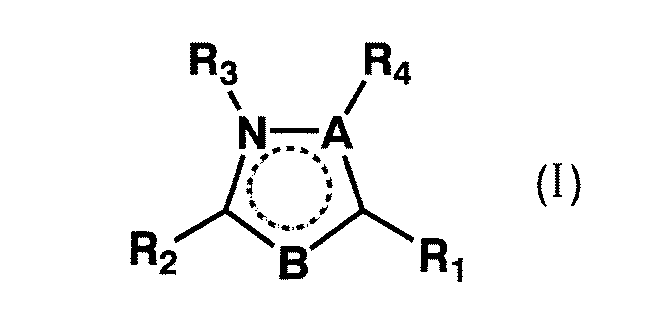
(式中、R1は一または二置換フェニル基等を意味し、R2はピペリジル基等を意味し、Aは窒素原子等を意味し、BはNH、酸素原子等を意味し、R3およびR4はどちらか一方が水素原子、アルキル基等を意味する。)
【効果】本発明の化合物は、極めて高い抗菌活性を有し、更には多剤耐性菌に対しても優れた抗菌活性を示すので、抗菌剤として有用である。
【選択図】 なし[PROBLEMS] To provide an excellent antibacterial agent against multidrug-resistant bacteria.
The object is to provide a compound represented by the following general formula (I) or a pharmaceutically acceptable salt thereof, and an antibacterial agent containing these as an active ingredient.
Embedded image
(Wherein, R 1 represents a mono- or di-substituted phenyl group and the like, R 2 represents a piperidyl group and the like, A represents a nitrogen atom and the like, B represents NH and an oxygen atom and the like, R 3 And one of R 4 represents a hydrogen atom, an alkyl group or the like.)
The compound of the present invention has an extremely high antibacterial activity and also exhibits excellent antibacterial activity against multidrug-resistant bacteria, so that it is useful as an antibacterial agent.
[Selection diagram] None
Description
【0001】
【発明の属する技術分野】
本発明は医薬として有用な新規な化合物に関する。更に詳しくは、本発明は、特に多剤耐性菌に対して優れた抗菌作用を有する新規な化合物またはその塩、ならびにその医薬組成物に関する。
【0002】
【従来の技術】
MRSAと略称されるメチシリン耐性黄色ブドウ球菌(Methicillin-Resistant Staphylococcus aureus)は、1961年に英国でその存在が初めて報告された難治性術後感染症、呼吸器や消化器における感染症の起因菌として知られている。MRSA感染症は、1960年代後半から1970年代の後半に欧米で広がり始め、日本では1980年代に院内感染の主要起因菌として世間の注目を集め、1990年代後半には全国的に蔓延した。現在、MRSAは、多剤に高度耐性化しており、MRSA感染症に対する抗菌剤であったバンコマイシン(Vancomycin)やアルベカシン(Arbekacin)の耐性株も出現している。
【0003】
また、医療界において高い関心を集めている多剤耐性菌としては、前述のMRSAのほかにペニシリン耐性肺炎球菌(Penicillin-Resistant Streptococcus
pneumoniae,PRSP)やバンコマイシン耐性腸球菌(Vancomycin-Resistant Enterococcus faecium,VRE)が知られている。肺炎球菌はその毒性が強いことから恐れられている肺炎起因菌であり、腸球菌はMRSAとともに臨床分離されることもある全身感染症および/または尿路感染症起因菌である。これらの多剤耐性菌による感染症に対しては、ニューキノロン系抗菌剤やオキサゾリジノン抗菌剤などが使用されているが、これらの抗菌活性はそれほど強くなく、またこれらの耐性株の出現も報告されている。
【0004】
そこで、本発明者らは、これらの多剤耐性菌に対して強い抗菌力を有する化合物の探索を行った結果、上記一般式(I)で表される化合物またはその製薬学的に許容される塩(以下、「本発明の化合物」ということもある)が優れた抗菌力を有することを見出して本発明を完成した。
【0005】
後記特許文献1(国際公開WO01/52845号公報)には、ピラゾール誘導体を含む下記一般式(A)で表される化合物が細菌感染治療に用いられることが記載されている。
【0006】
【化2】
R2は、水素原子であるか、またはR1が−CO2(C1-3脂肪族)等であるとき、更にハロゲン原子等から選ばれ、
A環は、チアゾール、オキサゾール、イミダゾールまたはピラゾール基から選ばれるヘテロ環であり、
ZはC−R3またはN−R3であり、
R3は、−(CH2)pN(R5)2等であり、
R4は、それぞれ水素原子、任意に置換しているC1-6脂肪族基等であり、
R5は、それぞれ水素原子、任意に置換しているC1-4脂肪族基等であり、
pは、0〜4の整数であり、
Arは、任意に置換しているアリール、ヘテロアリール等である。〕
【0007】
また、後記特許文献2(特表平8−502508号公報)には、下記一般式(B)で表される化合物が記載されており、該化合物はドーパミンレセプターサブタイプ用リガンドであってドーパミン系疾患の治療に有用であることが記載されている。
【0008】
【化3】
XおよびYは、一方が窒素を示し、他方は酸素、N−R2等を示し、
Qは、1個の窒素原子を唯一のヘテロ原子として含み、炭素原子によりXおよびY部分を含むヘテロ原子五員環に結合している置換単環式複素脂肪族五員又は六員環を示し、
R1は、水素原子、ハロゲン原子等を示し、
R2は、水素原子、C1-6アルキルを示し、
Aは式(i)、(ii)又は(iii)を示し、
【0009】
【化4】
R3、R4及びR5は独立して水素、炭化水素、−ORa、−NRaRb等を示し、
RaおよびRbは、独立して水素、複素環式基等を示す)
【0010】
本願発明の化合物の有する作用は、上記一般式(B)で表される化合物の用途であるドーパミン系疾患の治療とは全く異なるものである。
【0011】
また、後記特許文献3(特表平11−504010号公報)には、下記一般式(C)で表される化合物が記載されており、該化合物はアドレナリン性α1a受容体アンタゴニストであって良性の前立腺肥大の治療に有用であることが記載されている。
【0012】
【化5】
XおよびYは、一方が窒素を示し、他方は酸素、N−R2等を表し、
Qは、単一のヘテロ原子として窒素原子1個を含み且つ炭素原子を介してX及びY部分を含む5員複素環に結合している、置換された5員又は6員単環式へテロ脂肪族環を表し、
R1は、水素原子、ハロゲン原子等を表し、
R2は、水素原子またはC1-6アルキルを表し、
Aは式(i)、(ii)、(iii)、(iv):
【0013】
【化6】
R3、R4及びR5はそれぞれ独立に、水素、炭化水素、複素環式基、−ORa、−NRaRb等から選択され、
RaおよびRbは独立に、水素、複素環式基等を表し、
環Mは、場合によって、構造(iv)全体が、フェニル、ベンゾジオキサン等を含む、単環式、二環式若しくは多環式芳香族又はヘテロ芳香族環系であるような追加の環又は環系であり、該環系はそれぞれ、上記のようなR3、R4及びR5、並びにR18及びR19で置換され得、R18及びR19はそれぞれ独立に、C1-6アルキル等あってよい〕
【0014】
本願発明の化合物の有する作用は、上記一般式(C)で表される化合物の用途である良性の前立腺肥大の治療とは全く異なるものである。
【0015】
後記特許文献4(国際公開WO02/053559号公報)には、下記一般式(D)で表される化合物から誘導されるナトリウムチャンネルモデュレーターに関して記載されている。
【0016】
【化7】
R4は、アルキル基、ハロアルキル基、シクロアルキル基等を示し、
R2は、アルキル基、シクロアルキル基等を示し、
R3は、水素原子、アルキル基、シクロアルキル基または任意に置換されたアリール基等を示す)
【0017】
本願発明の化合物の有する作用は、上記一般式(D)で表される化合物の用途であるナトリウムチャンネルモデュレーターとは全く異なるものである。
【0018】
後記特許文献5(特表平10−509713号公報)には、下記一般式(E)で表される化合物がα−1Cアドレナリン作動性レセプターアンタゴニスト作用を有することが開示されている。
【0019】
【化8】
R2は、水素、C1-6アルキル、ヒドロキシC1-6アルキル等から選択され、
Wは、C1-6アルキレン鎖または窒素であり、
mは、独立して整数0または1であり、
Xは、CHまたは窒素であり、但し、Wが窒素であるときには、XはCHであり、
qは、独立して1,2,3または4からなる群から選択される整数であり、
但し、R1がフェニルであるかまたはハロゲン、メチルまたはメトキシで一または二置換されたフェニルであり、R2が水素またはメチルであり、R3が水素、ヒドロキシまたはC1-6アルコキシであり、qが1,2,3または4であり、Wが(CH2)であり、mが1であるときは、XはCHとなる。〕
【0020】
本発明の化合物の有する作用は、上記一般式(E)で表される化合物の用途であるα−1Cアドレナリン作動性レセプターアンタゴニスト作用とは全く異なるものである。
【0021】
以上の特許文献1〜5に記載の化合物〔上記一般式(A)〜(E)〕は本発明の化合物と類似する化合物が開示されているが、本発明の化合物は具体的には全く記載されていない。
【0022】
【特許文献1】
国際公開第01/52845号パンフレット
【特許文献2】
特表平8−502508号公報
【特許文献3】
特表平11−504010号公報
【特許文献4】
国際公開第02/053559号パンフレット
【特許文献5】
特表平10−509713号公報
【0023】
【発明が解決しようとする課題】
前述したように、現在関心の高い多剤耐性菌による感染症に対して使用できる強い抗菌活性を有する薬が求められている。したがって、本発明の目的は、多剤耐性菌に対して強い抗菌活性を有する化合物を提供することにある。
【0024】
【課題を解決するための手段】
本発明者らは、上記の課題を解決するために鋭意研究を重ねた結果、多剤耐性菌に対して強い抗菌活性を有する化合物を見出し、本発明を完成させた。
【0025】
本発明は細菌感染症の治療または予防に有用な、更に詳しくは、下記一般式(I)で表される化合物またはその製薬学的に許容される塩を有効成分とする抗菌剤に関する。
【0026】
すなわち、本発明は、下記一般式(I)
【0027】
【化9】
R2は、アミノアルキル基;メチルアミノアルキル基;ヒドロキシアルキルアミノアルキル基;アルコキシカルボニルアルキルアミノアルキル基;シクロヘキサン環上が置換されていてもよいシクロヘキシルアミノアルキル基(該置換基は、アルコキシカルボニル基またはヒドロキシアルキル基である);ピペリジル基;水酸基で置換されていてもよいピロリジノ基;ピペラジン環上がアルキルスルファモイル基で置換されていてもよいピペラジニルアルキル基またはアルコキシカルボニルピペリジノアルキル基を意味し、
Aは、窒素原子または炭素原子を意味し、
Bは、C−R5、NHまたは酸素原子を意味する。
但し、Aが窒素原子のときは、BはC−R5を意味し、Aが炭素原子のときは、BはNHまたは酸素原子を意味する。
また、R3およびR4は、どちらか一方が水素原子;アルキル基;1または2個のハロゲン原子で置換されていてもよいフェニル基;フェノキシフェニル基;ベンジルオキシフェニル基;ピリジル基;1もしくは2個のハロゲン原子またはトリフルオロメチル基で置換されていてもよいベンジル基あるいはベンジル基で置換されていてもよいピペリジル基で置換しており、他方は水素原子または置換基は存在しない。
R5は、水素原子、アルキル基またはハロゲン原子を意味し、
Aが窒素原子であって、かつBがC−R5であるとき、R1およびR5は一緒になってベンゼン環を形成していてもよく、形成されたベンゼン環は−O−R1’で置換していてもよく、R1’は、置換されていてもよいフェニルアルキル基(該置換基は、1もしくは2個のハロゲン原子,アルキル基またはアミノ基である)を意味する。〕
で表される化合物またはその製薬学的に許容される塩、およびその医薬組成物を提供する。
【0028】
本発明の化合物は上記一般式(I)で表されるが、好ましい化合物としては下記(1)〜(4)に記載の化合物が挙げられる。
【0029】
(1)Aが窒素原子であり、かつBがC−R5である請求項1記載の化合物またはその製薬学的に許容される塩。
(2)Aが炭素原子であり、かつBがNHである請求項1記載の化合物またはその製薬学的に許容される塩。
(3)Aが炭素原子であり、かつBが酸素原子である請求項1記載の化合物またはその製薬学的に許容される塩。
(4)Aが窒素原子であり、かつBがC−R5であって、R1およびR5が一緒になってベンゼン環を形成している請求項1記載の化合物またはその製薬学的に許容される塩。
【0030】
更に好ましい化合物として具体的な化合物またはその塩を下記に記載する。
【0031】
・5−〔(1E)−2−(5−クロロインドール−3−イル)ビニル〕−1−(3−クロロフェニル)−3−(4−ピペリジル)ピラゾール・1.25塩酸塩.・1−〔1−ベンジル−(4−ピペリジル)〕−5−〔4−(3,4−ジクロロベンジルオキシ)フェニル〕−3−(4−ピペリジル)ピラゾール・1塩酸塩.・1−(3−ブロモフェニル)−5−(2−ナフチル)−3−(4−ピペリジル)ピラゾール・1.65塩酸塩.
・1−(3−クロロフェニル)−3−(1−メチルアミノプロピル)−5−(4−フェノキシフェニル)ピラゾール・1.2塩酸塩.
・5−〔(1E)−2−(3,4−ジクロロフェニル)ビニル〕−3−(4−ピペリジル)ピラゾール・2塩酸塩.
・5−{(1E)―2−〔4−(3,4−ジクロロベンジルオキシ)フェニル〕ビニル}−3−(4−ピペリジル)ピラゾール・1.7塩酸塩.
・1−ブチル−5−{(1E)−2−〔4−(3,4−ジクロロベンジルオキシ)フェニル〕ビニル}−3−(4−ピペリジル)ピラゾール・1塩酸塩.
・5−〔(1E)−2−(5−クロロインドール−2−イル)ビニル〕−1−(3−クロロフェニル)−3−(4−ピペリジル)ピラゾール・1塩酸塩.
・5−メチルアミノメチル−3−(4−フェノキシフェニル)−1−(4−トリフルオロメチルベンジル)ピラゾール・1塩酸塩.
・1−(3−クロロフェニル)−5−〔(1E)−2−(3,4−ジクロロフェニル)ビニル〕−3−(4−ピペリジル)ピラゾール・1塩酸塩.
・1−(3−クロロフェニル)−3−メチルアミノメチル−5−(4−フェノキシフェニル)ピラゾール・1フマル酸塩.
・4−ブロモ−1−(3−クロロフェニル)−5−(2−ナフチル)−3−(4−ピペリジル)ピラゾール・1臭化水素酸塩.
・1−(3−クロロフェニル)−5−〔3−(2−ヒドロキシエチルアミノ)ブチル〕−3−(2−ナフチル)ピラゾール・1塩酸塩.
・5−〔(1E)−2−(5−クロロインドール−1−メチル−3−イル)ビニル〕−3−(3−メトキシカルボニルシクロヘキシルアミノメチル)ピラゾール・1塩酸塩.
・1−(3−クロロフェニル)−5−〔(1Z)−2−(3,4−ジクロロフェニル)ビニル〕−3−(4−ピペリジル)ピラゾール・1塩酸塩.
・5−〔(1Z)−2−(5−クロロインドール−1−メチル−3−イル)ビニル〕−3−(3−メトキシカルボニルシクロヘキシルアミノメチル)ピラゾール・1塩酸塩.
・5−(3−ベンジルオキシフェニル)−4−(3−クロロフェニル)−2−(4−ピペリジル)オキサゾール・1フマル酸塩.
・5−(3−ベンジルオキシフェニル)−4−(3−クロロフェニル)−2−(4−ピペリジル)イミダゾール・2塩酸塩.
・4−(3,4−ジクロロベンジルオキシ)−3−(3−メトキシカルボニルシクロヘキシルアミノメチル)−1H−インダゾール・1塩酸塩.
・4−(2,6−ジクロロフェネチルオキシ)−3−(3−メトキシカルボニルシクロヘキシルアミノメチル)−1H−インダゾール・1塩酸塩.
【0032】
一般式(I)で表される本発明の化合物は製薬学的に許容される塩を形成していてもよく、その塩は酸付加塩であり、例えば、塩酸,臭化水素酸,ヨウ化水素酸,硫酸,リン酸,硝酸等の無機酸との塩、酢酸,シュウ酸,フマル酸,マレイン酸,マロン酸,乳酸,リンゴ酸,コハク酸,クエン酸,酒石酸,安息香酸,メタンスルホン酸,p−トルエンスルホン酸,グルコン酸等の有機酸との塩が挙げられる。
【0033】
本発明において「アルキル基」および「アルキル」部分とは直鎖状または分枝状のC1-5のアルキル基を意味し、特にC1-3のアルキル基が好ましく、具体例として、メチル基、エチル基、プロピル基、イソプロピル基、n−ブチル基、イソブチル基、sec−ブチル基、tert−ブチル基、n−ペンチル基等が挙げられる。また、「アルコキシ基」および「アルコキシ」部分は、そのアルキル部分が上記の意味するアルキルオキシ基であり、例えばメトキシ、エトキシ、ブトキシ等が挙げられる。また、「ハロゲン原子」は、フッ素、塩素、臭素またはヨウ素を意味する。
【0034】
一般式(I)で表される本発明の化合物またはその塩は、水和物および/または溶媒和物として存在することもあるが、このような形態の化合物も本発明の化合物に包含される。また、光学異性体、立体異性体またはこれらの混合物として存在しており、これらの総てが本発明に包含される。
【0035】
上記一般式(I)中にある破線で描く円は、五員環内の任意の位置にある2個の非隣接二重結合を示す。
【0036】
具体的には、一般式(I)において、Aが窒素原子であり、かつBがC−R5である場合、またはAが窒素原子であり、かつBがC−R5であってR1とR5が一緒になってベンゼン環を形成している場合、R3またはR4のどちらか一方が水素原子のとき、互変異性体が存在するが、これらの互変異性体もまた本発明の化合物に含まれる。
【0037】
更に、一般式(I)において、Aが炭素原子であり、かつBがNHである場合、下記式で表されるようにBが−NH−の場合と=N−の場合の互変異性体が考えられるが、これらもまた本発明の化合物に含まれる。
【0038】
【化10】
次に、本発明の化合物の製造法について以下に説明する。
【0040】
一般式(I)で表される本発明の化合物において、Aが窒素原子であり、かつBがC−R5である本発明の化合物(Ia)は下記製造法1〜3により製造することができる。
【0041】
〔製造法1〕
本発明の化合物(Ia)は、下記反応式にしたがって製造することができる。
【0042】
【化11】
【0043】
すなわち、本発明の化合物(Ia)は、文献(Bioorg. Med. Chem., 1998, 6,743)記載の方法に準じて、1,3−ジオン誘導体(II)とR3’NHNH2を適当な溶媒中で反応させることにより製造することができる。また、化合物(II)において、R2に1級または2級のアミノ基を含む場合、R2のアミノ基を適当な保護基で保護した化合物を用いて、上記反応後脱保護することにより本発明の化合物(Ia)を製造することができる。
【0044】
反応温度は用いる原料化合物の種類などによって異なるが、通常−10〜150℃、好ましくは10〜100℃である。反応時間は通常約30分〜24時間、好ましくは約1〜10時間である。
【0045】
〔製造法2〕
また、本発明の化合物(Ia−1)および(Ia−2)は、下記反応式にしたがって製造することができる。
【0046】
【化12】
【0047】
〔工程1〕:1,3−ジオン誘導体(II)にヒドラジンを適当な溶媒中で反応させることにより化合物(Ia−2)を製造することができる。
【0048】
反応温度は用いる原料化合物の種類などによって異なるが、通常−10〜150℃、好ましくは10〜100℃である。反応時間は通常約30分〜24時間、好ましくは約1〜10時間である。
【0049】
〔工程2〕:工程1で得られた化合物(Ia−2)とR13Xを塩基の存在下、適当な溶媒中で反応させることにより化合物(Ia−1)を製造することができる。また、化合物(II)において、R2に1級または2級のアミノ基を含む場合、R2のアミノ基を適当な保護基で保護した化合物を用いて、工程1および2の反応後脱保護することにより本発明の化合物(Ia−1)を製造することができる。
【0050】
反応温度は用いる原料化合物の種類などによって異なるが、通常−10〜150℃、好ましくは10〜100℃である。反応時間は通常約30分〜24時間、好ましくは約1〜10時間である。
【0051】
工程2の反応で用いられる塩基としては、例えば水素化ナトリウム,水素化カリウムのような水素化アルカリ金属、水酸化ナトリウム,水酸化カリウムのような水酸化アルカリ金属、ナトリウムエトキシド,ナトリウムメトキシドのようなアルコキシアルカリ金属、炭酸ナトリウム,炭酸カリウムのような炭酸アルカリ金属またはピリジン,トリエチルアミン,ジイソプロピルエチルアミンのような有機塩基が挙げられる。溶媒としてはジエチルエーテル、テトラヒドロフラン、ジオキサン、ベンゼン、トルエン、ジメチルホルムアミドまたはメタノール,エタノール,イソプロパノール,tert−ブタノールなどのアルコール類等および水が挙げられる。これらの溶媒は単独または2種以上の混合溶媒として使用してもよい。
【0052】
〔製造法3〕
本発明の化合物(Ia−1)は、下記反応式にしたがって製造することもできる。
【0053】
【化13】
【0054】
すなわち、上記製造法2の工程1で得られる化合物(Ia−2)にR13B(OH)2および酢酸第二銅と適当な塩基存在下、適当な溶媒中で反応させることにより化合物(Ia−1)を製造することができる。また、化合物(Ia−2)において、R2に1級または2級のアミノ基を含む場合、製造法2における化合物(II)のR2のアミノ基を適当な保護基で保護した化合物を用いて、上記製造法2の工程1と同様に反応・処理し、次いで上記反応後脱保護をすることにより本発明の化合物(Ia−1)を製造することができる。
【0055】
反応温度は用いる原料化合物の種類などによって異なるが、通常−10〜150℃、好ましくは10〜100℃である。反応時間は通常約30分〜48時間、好ましくは約5〜24時間である。
【0056】
本反応で用いられる塩基としては、例えばピリジン、2,6−ルチジン、トリエチルアミン、ジイソプロピルエチルアミンのような有機塩基が挙げられる。溶媒としてはジクロロメタン、クロロホルム、ベンゼン、トルエンが挙げられる。これらの溶媒は単独または2種以上の混合溶媒として使用してもよい。
【0057】
上記製造法1および製造法2の工程1で用いられる反応は、通常反応に影響を及ぼさない適当な溶媒中で行われる。溶媒としては、例えば、メタノール,エタノール,イソプロパノール,tert−ブタノールなどのアルコール類、アセトン、アセトニトリル、N,N−ジメチルホルムアミド、ジメチルスルホキシド、ヘキサメチルホスホラミドおよび水等が挙げられる。これらの溶媒は単独にまたは2種以上の混合溶媒として使用してもよい。
【0058】
〔製造法4〕
一般式(I)で表される本発明の化合物において、Aが炭素原子であり、かつBが酸素原子である化合物(Ib)は、文献(J. Med. Chem., 1992, 35,3498)記載の方法に準じて下記反応式にしたがって製造することができる。
【0059】
【化14】
【0060】
〔工程1〕:本発明の化合物(V)は、2−ヒドロキシエタン−1−オン誘導体(IV)とR2COOHを1−(3−ジメチルアミノプロピル)−3−エチルカルボジイミド(WSC)・1塩酸塩の存在下、適当な溶媒中で反応させることにより製造することができる。
【0061】
反応温度は用いる原料化合物の種類などによって異なるが、通常−10〜150℃、好ましくは10〜100℃である。反応時間は通常約30分〜24時間、好ましくは約1〜10時間である。溶媒としてはジクロロメタン、クロロホルム、ジエチルエーテル、テトラヒドロフラン、ジオキサン、ベンゼン、トルエン、ジメチルホルムアミドが挙げられる。
【0062】
〔工程2〕:工程1で得られた化合物(V)を例えば酢酸アンモニウムと酢酸の存在下、適当な溶媒中で反応させることにより本発明の化合物(Ib)を製造することができる。また、R2COOHにおいて、R2に1級または2級のアミノ基を含む場合、R2のアミノ基を適当な保護基で保護した後、上記反応後脱保護をすることにより化合物(Ib)を製造することができる。
【0063】
反応温度は用いる原料化合物の種類などによって異なるが、通常−10〜150℃、好ましくは10〜100℃である。反応時間は通常約30分〜24時間、好ましくは約1〜10時間である。
【0064】
〔製造法5〕
一般式(I)で表される本発明の化合物において、Aが炭素原子であり、かつBがNHである化合物(Ic)は、文献(Tetrahedron Lett., 1994, 35, 1635)記載の方法に準じて下記反応式にしたがって製造することができる。
【0065】
【化15】
【0066】
すなわち、本発明の化合物(Ic)は、1,2−ジオン誘導体(VI)とR2CHOを例えば酢酸アンモニウムおよび酢酸存在下、適当な溶媒中で反応させることにより製造することができる。また、R2CHOにおいて、R2に1級または2級のアミノ基を含む場合、R2のアミノ基を適当な保護基で保護した後、上記反応後脱保護をすることにより化合物(Ic)を製造することができる。
【0067】
反応温度は用いる原料化合物の種類などによって異なるが、通常−10〜150℃、好ましくは10〜100℃である。反応時間は通常約30分〜12時間、好ましくは約1〜5時間である。
【0068】
一般式(I)で表される本発明の化合物において、Aが窒素原子であり、かつBがC−R5であってR1およびR5とが一緒になってベンゼン環を形成している化合物(Id−1)および化合物(Id−2)は、それぞれ下記製造法6および7により製造することができる。
【0069】
〔製造法6〕
一般式(I)で表される本発明の化合物において、R3またはR4が水素原子以外の基である本発明の化合物(Id−1)は、下記反応式に示す方法1または方法2によって製造することができる。
【0070】
【化16】
【0071】
〔方法1〕:本発明の化合物(Id−1)は、化合物(VII)とR1’Xを塩基の存在下、適当な溶媒中で反応させることにより製造することができる。
【0072】
反応温度は用いる原料化合物の種類などによって異なるが、通常−10〜150℃、好ましくは10〜100℃である。反応時間は通常約30分〜24時間、好ましくは約1〜10時間である。
【0073】
本反応で用いられる塩基としては、例えば水素化ナトリウム,水素化カリウムのような水素化アルカリ金属、水酸化ナトリウム,水酸化カリウムのような水酸化アルカリ金属、ナトリウムエトキシド,ナトリウムメトキシドのようなアルコキシアルカリ金属、炭酸ナトリウム,炭酸カリウムのような炭酸アルカリ金属またはピリジン,トリエチルアミン,ジイソプロピルエチルアミンのような有機塩基が挙げられる。溶媒としてはジエチルエーテル、テトラヒドロフラン、ジオキサン、ベンゼン、トルエン、ジメチルホルムアミドまたはメタノール,エタノール,イソプロパノール,tert−ブタノールなどのアルコール類等および水が挙げられる。これらの溶媒は単独または2種以上の混合溶媒として使用してもよい。
【0074】
〔方法2〕:本発明の化合物(Id−1)は、文献(J. Med. Chem., 1992, 35,1176)記載の方法に準じて製造することもできる。すなわち、本発明の化合物(Id−1)は、化合物(VII)と各種アルコールをジアルキルアゾジカルボキシレートおよびトリフェニルホスフィン存在下、適当な溶媒中で反応させることにより製造することができる。
【0075】
本反応で用いられる溶媒としては、ジエチルエーテル、テトラヒドロフラン、ジオキサン、ベンゼン、トルエン、ジメチルホルムアミド、ピリジン、ヘキサメチルホスホラストリアミド等が挙げられる。
【0076】
また、製造法6により製造される本発明の化合物(Id−1)のあるものは、後記の製造法7により製造される化合物(Id−2)を例えばアルキルハライド等の塩基の存在下、適当な溶媒中で反応させることにより製造することもできる。
【0077】
方法1および2において、化合物(VII)のR2に1級または2級のアミノ基を含む場合、R2のアミノ基を適当な保護基で保護した化合物を用いて、上記反応後脱保護することにより化合物(Id−1)を製造することができる。
【0078】
〔製造法7〕
一般式(I)で表される本発明の化合物において、R3またはR4が水素原子である本発明の化合物(Id−2)は、下記反応式にしたがって製造することができる。
【0079】
【化17】
【0080】
すなわち、本発明の化合物(Id−2)は、ピラゾール環部分を適当な保護基(R3’’)で保護した化合物(VII’)を上記製造法6にしたがって化合物(VIII)とし、次いで脱保護することにより製造することができる。また、化合物(VII’)において、R2に1級または2級のアミノ基を含む場合、R2のアミノ基を適当な保護基で保護した化合物を用いて、R3’’と同時にまたは別途脱保護することにより化合物(Id−2)を製造することができる。
【0081】
上記製造法1〜7により製造される式(Ia)、(Ib)、(Ic)、(Id−1)および(Id−2)で表される本発明の化合物は、クロマトグラフィー,再結晶など通常の化学操作により単離・精製することができる。また、式(Ia)、(Ib)、(Ic)、(Id−1)および(Id−2)で表される本発明の化合物は、遊離塩基または酸付加塩の形で得られるが、両者は通常の方法により互いに変換することができる。
【0082】
次に、上記製造法1〜7で用いられる原料化合物は下記の方法により製造することができる。
【0083】
前記製造法1および2で用いられる化合物(II)は、下記反応式に示される方法にしたがって製造することができる。
【0084】
【化18】
【0085】
すなわち、化合物(II)は化合物(IX)と化合物(X)を適当な縮合剤の存在下または非存在下および適当な塩基の存在下に適当な溶媒中で反応させることにより製造することができる。化合物(IX)において、R2に1級または2級のアミノ基を含む場合、R2のアミノ基を適当な保護基で保護し、化合物(II)を製造することができる。
【0086】
前記製造法4および5でそれぞれ用いられる化合物(IV)および化合物(VI)は、下記反応式に示される方法にしたがって製造することができる。
【0087】
【化19】
【0088】
すなわち、化合物(VI)は、文献(J. Chem. Soc. Perkin Trans. 1, 1997,16, 2429)記載の方法に準じて製造される化合物(XI)とR1CHOを塩基の存在下、適当な溶媒中で反応させ、更に酸で処理することによって化合物(IV)とし、次いで、適当な酸化剤で酸化することにより製造することできる。
【0089】
前記製造法6で用いられる化合物(VII)は、下記反応式で示される方法にしたがって製造することができる。
【0090】
【化20】
【0091】
すなわち、化合物(VII)は、文献(J. Med. Chem., 2000, 43, 2664)記載の方法に準じて製造される化合物(XII)とアミン類を塩基の存在下または非存在下、適当な溶媒中でそれぞれ反応させて化合物(XIII)とし、化合物(XIII)において、R2に1級または2級のアミノ基を含む場合は、R2を適当な保護基で保護し、次いでProを選択的に脱保護して製造することができる。
【0092】
また、前記製造法7で用いられる化合物(VII’)は、上記製造法6で用いられる化合物(VII)におけるR13の代わりに適当な保護基であるR3’’で保護したものを用いて、上記反応式と同様に反応・処理することによって製造することができる。
【0093】
本発明の化合物は、特にグラム陽性菌に対し優れた抗菌活性を有する。また、本発明の化合物は、現在臨床において問題視されている薬剤耐性菌であるMRSA、VRE、PRSP等に対しても感受性菌と同等の抗菌力を有しており、これらの菌に起因する感染症の予防・治療薬として使用することができる。
【0094】
本発明の化合物の投与量は、化合物の種類,投与方法,症状,年齢等により異なるが、通常、1日につき、体重60kgあたり約2〜5000mg、好ましくは約5〜500mg、特に好ましくは30〜300mgである。投与経路は、経口投与でもよいが、非経口投与、特に静脈内投与が推奨される。
【0095】
本発明の化合物は、上記の如き医薬用途に使用する場合、通常、製剤用担体と混合して調製された製剤の形で投与される。製剤用担体としては、製剤分野において常用され、かつ本発明の化合物と反応しない無毒性の物質が用いられる。具体的には、例えばクエン酸,グルタミン酸,グリシン,乳糖,イノシトール,ブドウ糖,マンニトール,デキストラン,ソルビトール,シクロデキストリン,デンプン,部分アルファー化デンプン,白糖,パラオキシ安息香酸メチル,パラオキシ安息香酸プロピル,メタケイ酸アルミン酸マグネシウム,合成ケイ酸アルミニウム,結晶セルロース,カルボキシメチルセルロースナトリウム,ヒドロキシプロピルデンプン,カルボキシメチルセルロースカルシウム,イオン交換樹脂,メチルセルロース,ゼラチン,アラビアゴム,プルラン,ヒドロキシプロピルセルロース,低置換度ヒドロキシプロピルセルロース,ヒドロキシプロピルメチルセルロース,ポリビニルピロリドン,ポリビニルアルコール,アルギン酸,アルギン酸ナトリウム,軽質無水ケイ酸,ステアリン酸マグネシウム,タルク,トラガント,ベントナイト,ビーガム,カルボキシビニルポリマー,酸化チタン,ソルビタン脂肪酸エステル,ラウリル硫酸ナトリウム,グリセリン,脂肪酸グリセリンエステル,精製ラノリン,グリセロゼラチン,ポリソルベート,マクロゴール,植物油,ロウ,プロピレングリコール,エタノール,ベンジルアルコール,塩化ナトリウム,水酸化ナトリウム,塩酸,水等が挙げられる。
【0096】
剤型としては、錠剤,カプセル剤,顆粒剤,散剤,シロップ剤,懸濁剤,注射剤,坐剤,点眼剤,軟膏剤,塗布剤,吸入剤等が挙げられる。これらの製剤は常法にしたがって調製することができる。なお、液体製剤にあっては、用時、水又は他の適当な媒体に溶解または懸濁する形であってもよい。また、錠剤及び顆粒剤は周知の方法でコーティングしてもよい。更に、これらの製剤は治療上価値ある他の成分を含有していてもよい。
【0097】
【実施例】
以下に参考例、実施例および試験例を挙げて本発明を更に具体的に説明するが、これらは本発明を限定するものではない。本発明の化合物は、元素分析値,マス・スペクトル,IRスペクトル,NMRスペクトル,高速液体クロマトグラフィー(HPLC)等により同定した。また、立体異性体の配置の決定は、NOESY(Nuclear Overhauser effect spectroscopy)により行った。
【0098】
明細書の記載を簡略化するために実施例および実施例中の表において以下に示すような略号を用いることもある。
【0099】
再結晶溶媒として用いられる記号としては、Eはエーテル、Iはジイソプロピルエーテル、ANはアセトニトリル、AEは酢酸エチル、ACはアセトン、CFはクロロホルム、MTはメタノール、ETはエタノールおよびIPはイソプロパノールを意味する。
【0100】
NMRに用いられる記号としては、sは一重線、dは二重線、tdは三重の二重線、tは三重線、qは四重線、quは五重線、mは多重線、brはなだらか、およびJは結合定数を意味する。
【0101】
参考例1:
5−クロロ−1−メチルインドール−3−カルボキシアルデヒド
【0102】
【化21】
5−クロロインドール−3−カルボキシアルデヒド5.50gのテトラヒドロフラン溶液50mlを水素化ナトリウム(60%オイルディスパージョン)1.43gのテトラヒドロフラン30ml懸濁液に、氷冷下滴下し、室温で30分間攪拌した。次に、ヨウ化メチル2.30mlを加え、室温で12時間攪拌した。10%クエン酸水溶液を加え、酢酸エチル80mlで抽出した。抽出液を飽和食塩水20mlで洗浄後、硫酸マグネシウムで乾燥後濃縮した。得られた粗結晶をジイソプロピルエーテルで洗浄し、目的物4.40gを得た。
【0104】
参考例2:
(1)3−〔(1E)−2−アセチルビニル〕−5−クロロインドール
【0105】
【化22】
5−クロロインドール−3−カルボキシアルデヒド2.00gのアセトン溶液7mlを水酸化ナトリウム1.35gのエタノール1mlと水15mlの混媒溶液に、氷冷下で滴下し、室温で1時間攪拌後、12時間加熱還流した。6mol/l塩酸を加え中和し、酢酸エチル30mlで抽出した。抽出液を硫酸マグネシウムで乾燥後濃縮し、残渣をシリカゲルカラムクロマトグラフィーに付し、クロロホルム−メタノール(50:1)で溶出・精製して目的物1.23gをアモルファスとして得た。
【0107】
参考例2における5−クロロインドール−3−カルボキシアルデヒドの代わりに対応するアルデヒドを用い、参考例2(1)に記載の方法にしたがって、同様に反応・処理して以下の化合物を得た。
【0108】
(2)3−〔(1E)−2−アセチルビニル〕−5−クロロ−1−メチルインドール
【0109】
【化23】
(3)2−〔(1E)−2−アセチルビニル〕−5−クロロインドール
【0111】
【化24】
(4)(3E)−4−〔4−(3,4−ジクロロベンジルオキシ)フェニル〕ブト−3−エン−2−オン
【0113】
【化25】
参考例3:
(1)1−〔4−(1−tert−ブトキシカルボニル)ピペリジル〕−3−〔(1E)−2−(5−クロロインドール−3−イル)ビニル〕プロパン−1,3−ジオン
【0115】
【化26】
1,1,1,3,3,3−ヘキサメチルジシラザン2.88mlのテトラヒドロフラン30ml溶液に、アルゴン雰囲気下、氷冷下で2.5mol/lのノルマルブチルリチウムのヘキサン溶液4.36mlを加え、同温で10分間攪拌した。次いで、参考例2(1)で得られた3−〔(1E)−2−アセチルビニル〕−5−クロロインドール1.00gのテトラヒドロフラン10ml溶液を−78℃で滴下し、同温で30分間攪拌した。1−tert−ブトキシカルボニルイソニペコチン酸1.10gと1,1’−カルボニルビス−1H−イミダゾール0.77gのテトラヒドロフラン溶液30mlを室温で45分間攪拌した溶液をアルゴン雰囲気下滴下し、室温に戻した後、同温で12時間攪拌した。10%クエン酸溶液50mlを加え、酢酸エチルで抽出し飽和食塩水で洗浄後、硫酸マグネシウムで乾燥後濃縮した。残渣をシリカゲルカラムクロマトグラフィーに付し、クロロホルム−メタノール(50:1)で溶出・精製し、目的物1.00gをアモルファスとして得た。
【0117】
対応する原料化合物を用いて、上記(1)と同様に反応・処理し、表1に示す化合物を得た。
【0118】
【表1】
参考例4:
(1)1−〔(1E)−2−(5−クロロ−1−メチルインドール−3−イル)ビニル〕−3−エトキシカルボニルプロパン−1,3−ジオン
【0120】
【化27】
ナトリウムエトキシド0.12gのジエチルエーテル20ml溶液に、アルゴン気流下、氷冷下、参考例2(2)で得られた3−〔(1E)−2−アセチルビニル〕−5−クロロ−1−メチルインドール0.5gを滴下し、次にシュウ酸ジエチル0.24mlを加え、室温にて12時間攪拌した。2mol/l塩酸を加えて酸性にし、酢酸エチル40mlで抽出し、飽和食塩水10mlで洗浄後、無水硫酸マグネシウムで乾燥し濃縮した。残渣をジイソプロピルエーテルで再結晶し、目的物0.20gを得た。
【0122】
対応する原料化合物を用いて、上記(1)と同様に反応・処理し、以下の化合物を得た。
【0123】
(2)1−〔(1E)−2−(3,4−ジクロロフェニル)ビニル〕−3−エトキシカルボニルプロパン−1,3−ジオン
【0124】
【化28】
参考例5:
(1)5−〔(1E)−2−(5−クロロ−1−メチルインドール−3−イル)ビニル〕−3−エトキシカルボニルピラゾール
【0126】
【化29】
参考例4(1)で得られた1−〔(1E)−2−(5−クロロ−1−メチルインドール−3−イル)ビニル〕−3−エトキシカルボニルプロパン−1,3−ジオン4.30gのエタノール100ml溶液にヒドラジン・1水和物1.02gと酢酸ナトリウム1.38gを加え、3時間加熱還流した。溶媒留去後、水20mlを加え、酢酸エチル100mlで抽出し飽和食塩水で洗浄後、硫酸マグネシウムで乾燥後濃縮した。残渣をシリカゲルカラムクロマトグラフィーに付し、酢酸エチル−ノルマルへキサン(2:1)で溶出・精製し、目的物2.90gをアモルファスとして得た。
【0128】
対応する原料化合物を用いて、上記(1)と同様に反応・処理し、以下の化合物を得た。
【0129】
(2)1−(3−クロロフェニル)−5−〔2−(エトキシカルボニル)エチル〕−3−(2−ナフチル)ピラゾール
【0130】
【化30】
(3)1−(3−クロロフェニル)−3−〔2−(エトキシカルボニル)エチル〕−5−(2−ナフチル)ピラゾール
【0132】
【化31】
(4)5−〔(1E)−2−(3,4−ジクロロフェニル)ビニル〕−3−エトキシカルボニルピラゾール
【0134】
【化32】
参考例6:
(1)5−〔(1E)−2−(5−クロロ−1−メチルインドール−3−イル)ビニル〕−3−ヒドロキシメチルピラゾール
【0136】
【化33】
参考例5(1)で得られた5−〔(1E)−2−(5−クロロ−1−メチルインドール−3−イル)ビニル〕−3−エトキシカルボニルピラゾール1.00gのテトラヒドロフラン30ml溶液を、水素化リチウムアルミニウム0.25gのテトラヒドロフラン30ml懸濁液に氷冷下滴下し、室温で3時間攪拌した。1mol/l硫酸水溶液を滴下し中和後、酢酸エチル50mlで抽出し、飽和食塩水で洗浄、硫酸マグネシウムで乾燥後濃縮した。残渣をノルマルへキサンより再結晶し、目的物0.63gを得た。
【0138】
対応する原料化合物を用いて、上記(1)と同様に反応・処理し、以下の化合物を得た。
(2)5−〔(1E)−2−(3,4−ジクロロフェニル)ビニル〕−3−ヒドロキシメチルピラゾール
【0139】
【化34】
参考例7:
(1)2−(3−クロロフェニル)−2−トリメチルシリルオキシエタンニトリル
【0141】
【化35】
3−クロロベンズアルデヒド16.5gのジクロロメタン100ml溶液に、氷冷下トリメチルシリルシアニド16.4mlを滴下し、次にヨウ化亜鉛50mgを加え室温で48時間攪拌した。溶媒留去後、残渣をシリカゲルカラムクロマトグラフィーに付し、クロロホルムで溶出・精製し、目的物を油状物として22.5gを得た。
【0143】
対応する原料化合物を用いて、上記(1)と同様に反応・処理し、次の化合物を得た。
【0144】
(2)2−(4−フェノキシフェニル)−2−トリメチルシリルオキシエタンニトリル
【0145】
【化36】
(3)2−(4−ブロモフェニル)−2−トリメチルシリルオキシエタンニトリル
【0147】
【化37】
(4)2−(3,5−ジベンジルオキシフェニル)−2−トリメチルシリルオキシエタンニトリル
【0149】
【化38】
参考例8:
(1)2−(3−ベンジルオキシフェニル)−1−(3−クロロフェニル)−2−ヒドロキシエタン−1−オン
【0151】
【化39】
1,1,1,3,3,3−ヘキサメチルジシラザン5.24mlのテトラヒドロフラン50ml溶液に、アルゴン雰囲気下、氷冷下で2.5mol/lのノルマルブチルリチウムのヘキサン溶液10.0mlを加え、同温で10分間攪拌した。次いで、参考例7(1)で得られた2−(3−クロロフェニル)−2−トリメチルシリルオキシエタンニトリル6.00gのテトラヒドロフラン15ml溶液を−78℃で滴下し、同温で30分間攪拌した。3−ベンジルオキシベンズアルデヒド5.31gのテトラヒドロフラン溶液20mlを同温で滴下し、室温に戻した後、同温で12時間攪拌した。10%クエン酸溶液50mlを加え、酢酸エチル100mlで抽出し飽和食塩水で洗浄後、硫酸マグネシウムで乾燥後濃縮した。残渣をメタノール10ml溶解し、5%硫酸水溶液12mlを加え、室温で30分間攪拌した。反応液をジエチルエーテル50mlで抽出し、0.5mol/l水酸化ナトリウム水溶液15mlで洗浄し、抽出液を硫酸マグネシウムで乾燥後濃縮した。残渣をシリカゲルカラムクロマトグラフィーに付し、クロロホルムで溶出・精製し、目的物8.60gをアモルファスとして得た。
【0153】
対応する原料化合物を用いて、上記(1)と同様に反応・処理し、表2に示す化合物を得た。
【0154】
【表2】
参考例9:
(1)2−(3−ベンジルオキシフェニル)−1−(3−クロロフェニル)エタン−1,2−ジオン
【0156】
【化40】
硫酸第二銅7.50gのピリジン40mlと水10mlの混合溶液を10分間加熱還流し、参考例8(1)で得られた2−(3−ベンジルオキシフェニル)−1−(3−クロロフェニル)−2−ヒドロキシエタン−1−オン8.3gを加え、1時間加熱還流した。溶媒留去後、水20mlを加えジエチルエーテル50mlで抽出し、硫酸マグネシウムで乾燥後濃縮した。残渣をシリカゲルカラムクロマトグラフィーに付し、クロロホルム−ノルマルへキサン(3:1)で溶出・精製し、目的物5.82gを油状物として得た。
【0158】
対応する原料化合物を用いて、上記(1)と同様に反応・処理し、表3に示す化合物を得た。
【0159】
【表3】
参考例10:
(1)3−ブロモメチル−1−(tert−ブトキシカルボニル)−4−(tert−ブチルジメチルシリルオキシ)−1H−インダゾール
【0161】
【化41】
文献(J. Med. Chem., 2000, 43, 2664)記載の方法にしたがって反応・処理して目的物を得た。
【0163】
対応する原料化合物を用い、上記(1)と同様に反応・処理して以下の化合物を得た。
【0164】
(2)3−ブロモメチル−1−(tert−ブトキシカルボニル)−4−(tert−ブチルジフェニルシリルオキシ)−1H−インダゾール
【0165】
【化42】
参考例11:
(1)3−アミノ−シクロヘキサンカルボン酸メチルエステル(シス・トランス混合物)・1塩酸塩
【0167】
【化43】
3−アミノシクロヘキサンカルボン酸(シス・トランス混合物)10.6gをメタノール75mlに加え、−10℃で塩化チオニル20mlを滴下し、室温で12時間攪拌した。溶媒留去し、アセトニトリルより結晶化し、目的物7.93gを得た。
【0169】
対応する原料化合物を用いて、上記(1)と同様に反応・処理し、以下の化合物を得た。
【0170】
(2)4−アミノブタン酸エチルエステル・1塩酸塩
【0171】
【化44】
参考例12:
(1)1−tert−ブトキシカルボニル−3−〔N−tert−ブトキシカルボニル−N−(3−メトキシカルボニルシクロヘキシル)〕アミノメチル−4−ヒドロキシ−1H−インダゾール
【0173】
【化45】
〔工程1〕:参考例10(2)で得られた3−ブロモメチル−1−(tert−ブトキシカルボニル)−4−(tert−ブチルジフェニルシリルオキシ)−1H−インダゾール2.00gをクロロホルム80mlとメタノール20mlの混媒に加え、室温下参考例11(1)で得られた3−アミノ−シクロヘキサンカルボン酸メチルエステル・1塩酸塩2.06gおよび10%水酸化ナトリウム水溶液4.24mlを加え、6時間加熱還流した。水50mlを加え、クロロホルム50mlで抽出し、抽出液を硫酸マグネシウムで乾燥後濃縮した。残渣をシリカゲルカラムクロマトグラフィーに付し、クロロホルム−メタノール(50:1)で溶出・精製し、1−(tert−ブトキシカルボニル)−4−(tert−ブチルジフェニルシリルオキシ)−3−(3−メトキシカルボニルシクロヘキシルアミノメチル)−1H−インダゾールをアモルファスとして1.44gを得た。
【0175】
〔工程2〕:上記工程1で得られた1−(tert−ブトキシカルボニル)−4−(tert−ブチルジフェニルシリルオキシ)−3−(3−メトキシカルボニルシクロヘキシルアミノメチル)−1H−インダゾール1.44gをアセトニトリル50mlに加え、室温下ジ−tert−ブチルジカーボネート0.54gを加え、室温で12時間攪拌した。溶媒留去し、残渣をシリカゲルカラムクロマトグラフィーに付し、クロロホルム−メタノール(50:1)で溶出・精製し、1−tert−ブトキシカルボニル−3−〔N−tert−ブトキシカルボニル−N−(3−メトキシカルボニルシクロヘキシル)〕アミノメチル−4−(tert−ブチルジフェニルシリルオキシ)−1H−インダゾール1.29gをアモルファスとして得た。
【0176】
〔工程3〕:上記工程2で得られた1−tert−ブトキシカルボニル−3−〔N−tert−ブトキシカルボニル−N−(3−メトキシカルボニルシクロヘキシル)〕アミノメチル−4−(tert−ブチルジフェニルシリルオキシ)−1H−インダゾール1.29gをテトラヒドロフラン50mlに加え、室温下1.0mol/lのテトラブチルアンモニウムフロリドのテトラヒドロフラン溶液17.4mlを加え、室温で4時間攪拌した。水50mlを加え、酢酸エチル50mlで抽出し、抽出液を硫酸マグネシウムで乾燥後濃縮した。残渣をシリカゲルカラムクロマトグラフィーに付し、クロロホルムで溶出・精製し、目的物0.76gをアモルファスとして得た。
【0177】
1H-NMR(CDCl3)δ:1.22-1.33(m,2H), 1.40(s,9H),1.66(s,9H),1.73-1.96(m,6H),2.30-2.39(m,1H), 3.64(s,3H), 3.66-3.72(m,1H), 4.70-4.81(m 2H),6.74(d,J=8.0Hz,1H), 7.32(t,J=8.0Hz,1H), 7.59(d,J=8.0Hz,1H).
【0178】
対応する原料化合物を用いて、上記(1)と同様に反応・処理し、表4に示す化合物を得た。
【0179】
【表4】
上記表4にあるR2’は、R2が1級または2級のアミノ基を含む場合、R2のアミノ基に適当な保護基を付加したものを意味する。
【0181】
実施例1:
5−〔(1E)−2−(5−クロロインドール−3−イル)ビニル〕−1−(3−クロロフェニル)−3−(4−ピペリジル)ピラゾール・1.25塩酸塩・1水和物
【0182】
【化46】
〔工程1〕:参考例3(1)で得られた1−〔4−(1−tert−ブトキシカルボニル)ピペリジル〕−3−〔(1E)−2−(5−クロロインドール−3−イル)ビニル〕プロパン−1,3−ジオン1.00gのエタノール30ml溶液に2mol/lの水酸化ナトリウム水溶液2.20mlと3−クロロフェニルヒドラジン・1塩酸塩0.83gを加え、12時間加熱還流した。反応液を濃縮後、10%クエン酸水溶液20mlを加え、酢酸エチル50mlで抽出し、飽和食塩水で洗浄、硫酸マグネシウムで乾燥後濃縮した。残渣をフラッシュシリカゲルカラムクロマトグラフィーに付し、クロロホルム−メタノール(50:1)で溶出・精製し、3−〔4−(1−tert−ブトキシカルボニル)ピペリジル〕−5−〔(1E)−2−(5−クロロインドール−3−イル)ビニル〕−1−(3−クロロフェニル)ピラゾール1.13gをアモルファスとして得た。
【0184】
〔工程2〕:上記工程1で得られた3−〔4−(1−tert−ブトキシカルボニル)ピペリジル〕−5−〔(1E)−2−(5−クロロインドール−3−イル)ビニル〕−1−(3−クロロフェニル)ピラゾール1.10gのジクロロメタン30ml溶液にトリフルオロ酢酸5mlを加え、室温で3時間攪拌した。溶媒留去後、残渣をCHP−20Pを用いた中圧カラムクロマトグラフィーに付して水−アセトニトリル(1:2)で溶出・精製し、減圧留去した。残渣をアセトニトリルから塩酸塩として再結晶し、目的物0.23gを得た。
融点158−161℃
【0185】
1H-NMR(DMSO-d6)δ:1.85-2.15(m,4H), 2.97-3.08(m,3H),3.26-3.34(m,2H),6.73(s,1H), 6.80(d,J=15.8Hz,1H), 7.13-7.77(m,9H),8.83(br,1H),9.02(br,1H),11.69(s,1H).
【0186】
上記の1H−NMRスペクトルデータおよびNOESYにより、標記化合物であることを決定した。
【0187】
対応する原料化合物を用いて実施例1と同様に反応・処理し、下記の表5〜9に示す実施例2〜34の化合物を得た。特に立体異性体が混在する場合は、上記で示したCHP−20Pを用いた中圧カラムクロマトグラフィーを用いた分離方法、もしくは分別再結晶を用いて、立体異性体をそれぞれ分離した。
【0188】
【表5】
【表6】
【表7】
【表8】
【表9】
実施例35:
5−メチルアミノメチル−3−(4−フェノキシフェニル)−1−(4−トリフルオロメチルベンジル)ピラゾール・1塩酸塩・1水和物
【0194】
【化47】
〔工程1〕:参考例3(20)で得られた1−(N−tert−ブトキシカルボニル)メチルアミノメチル−3−(4−フェノキシフェニル)プロパン−1,3−ジオン1.00gを用い、実施例1の工程1で用いた3−クロロフェニルヒドラジン・1塩酸塩の代わりにヒドラジン・1水和物を用いて同様に反応・処理し、3−(N−tert−ブトキシカルボニル)メチルアミノメチル−5−(4−フェノキシフェニル)ピラゾールの粗生成物1.20gを得た。
【0196】
〔工程2〕:上記工程1で得られた3−(N−tert−ブトキシカルボニル)メチルアミノメチル−5−(4−フェノキシフェニル)−ピラゾール1.20gのテトラヒドロフラン10ml溶液を、氷冷下水素化ナトリウム(60%オイルディスパージョン)のテトラヒドロフラン20ml懸濁液に加え、室温で30分間攪拌した。次に、4−トリフルオロベンジルブロミド0.94mlを加え、4時間加熱還流した。10%クエン酸水溶液20mlを加え、酢酸エチル50mlで抽出し飽和食塩水で洗浄後、硫酸マグネシウムで乾燥後濃縮した。残渣をシリカゲルカラムクロマトグラフィーに付し、ノルマルヘキサン−アセトン(10:1)で溶出・精製し、5−(N−tert−ブトキシカルボニル)メチルアミノメチル−3−(4−フェノキシフェニル)−1−(4−トリフルオロメチルベンジル)ピラゾール0.20gをアモルファスとして得た。
【0197】
〔工程3〕:上記工程2で得られた5−(N−tert−ブトキシカルボニル)メチルアミノメチル−3−(4−フェノキシフェニル)−1−(4−トリフルオロメチルベンジル)ピラゾール0.20gを用い、実施例1における工程2と同様に反応・処理後凍結乾燥し、目的物0.21gを得た。
【0198】
1H-NMR(DMSO-d6)δ:2.60(s,3H), 4.28(s,2H), 5.64(s,2H), 6.98(s,1H),7.03-7.06(m,4H), 7.16(t, J=7.2Hz,1H), 7.37-7.42(m,4H),7.73-7.77(m,4H),9.40(br,2H).
【0199】
上記の1H−NMRスペクトルデータおよびNOESYにより、標記化合物であることを決定した。
【0200】
対応する原料化合物を用いて実施例35と同様に反応・処理し、表10に示す実施例36〜44の化合物を得た。特に立体異性体が混在する場合は、CHP−20Pを用いた中圧カラムクロマトグラフィーを用いた分離方法、もしくは分別再結晶を用いて、立体異性体をそれぞれ分離した。
【0201】
【表10】
実施例45:
1−(3−クロロフェニル)−5−〔(1E)−2−(2,6−ジクロロフェニル)ビニル〕−3−(4−ピペリジル)ピラゾール・1塩酸塩
【0203】
【化48】
〔工程1〕:参考例3(11)で得られた 1−〔4−(1−tert−ブトキシカルボニル)ピペリジル〕−3−〔(1E)−2−(2,6−ジクロロフェニル)ビニル〕プロパン−1,3−ジオン2.00gを用い、実施例1の工程1で用いた3−クロロフェニルヒドラジン・1塩酸塩の代わりにヒドラジン・1水和物を用いて同様に反応・処理し、3−〔4−(1−tert−ブトキシカルボニル)ピペリジル〕−5−〔(1E)−2−(2,6−ジクロロフェニル)ビニル〕ピラゾール1.13gをアモルファスとして得た。
【0205】
〔工程2〕:上記工程1で得られた3−〔4−(1−tert−ブトキシカルボニル)ピペリジル〕−5−〔(1E)−2−(2,6−ジクロロフェニル)ビニル〕ピラゾール1.05gのジクロロメタン40ml溶液に、3−クロロフェニルボロン酸0.78g、酢酸第二銅(無水)0.68g、モレキュラーシーブ(4A)2.00gおよびピリジン0.40mlを加え、室温で24時間攪拌した。不溶物をろ別し、溶媒留去後、飽和炭酸水素ナトリウム水溶液30mlを加え、酢酸エチル50mlで抽出し、硫酸マグネシウムで乾燥後濃縮し、3−〔4−(1−tert−ブトキシカルボニル)ピペリジル〕−1−(3−クロロフェニル)−5−〔(1E)−2−(2,6−ジクロロフェニル)ビニル〕ピラゾールとその立体異性体の混合物0.98gをアモルファスとして得た。
【0206】
〔工程3〕:上記工程2で得られた、3−〔4−(1−tert−ブトキシカルボニル)ピペリジル〕−1−(3−クロロフェニル)−5−〔(1E)−2−(2,6−ジクロロフェニル)ビニル〕ピラゾールとその立体異性体の混合物0.96gを用い、実施例1の工程2と同様に反応・処理し、アセトニトリル−エタノールから塩酸塩として再結晶し、目的物0.51gを得た。
融点242−245℃
【0207】
1H-NMR(DMSO-d6)δ:1.82-2.22(m,4H), 2.98-3.38(m,5H), 6.90(s,1H),6.94(d,J=16.0Hz,1H), 7.22(d,J=16.0Hz,1H), 7.33-7.61(m,7H), 8.93(br,2H).
【0208】
上記の1H−NMRスペクトルデータおよびNOESYにより、標記化合物であることを決定した。
【0209】
対応する原料化合物を用いて実施例45と同様に反応・処理し、表11に示す実施例46〜49の化合物を得た。特に立体異性体が混在する場合は、CHP−20Pを用いた中圧カラムクロマトグラフィーを用いた分離方法、もしくは分別再結晶を用いて、立体異性体をそれぞれ分離した。
【0210】
【表11】
実施例50:
4−クロロ−1−(3−クロロフェニル)−5−(2−ナフチル)−3−(4−ピペリジル)ピラゾール・1塩酸塩
【0212】
【化49】
実施例45の工程1および工程2に記載の方法と同様にして、対応する原料化合物を用い処理して得られた3−〔4−(1−tert−ブトキシカルボニル)ピペリジル〕−1−(3−クロロフェニル)−5−(2−ナフチル)ピラゾール0.30gのクロロホルム20ml溶液に、氷冷下塩化スルフリル0.06mlを加え、1時間加熱還流した。飽和炭酸水素ナトリウム水溶液15mlを加え、クロロホルム20mlで抽出し、抽出液を硫酸マグネシウムで乾燥後濃縮し、エタノール−アセトニトリルから塩酸塩として再結晶し、目的物0.08gを得た。融点218−221℃
【0214】
1H-NMR(DMSO-d6)δ:2.02-2.06(m,4H), 3.08-3.41(m,5H), 7.09(d,J=4.0Hz,1H),7.27-7.44(m,5H), 7.56-7.63(m,2H), 7.94-8.01(m,3H), 8.76(br,2H).
【0215】
対応する試薬を用いて実施例50と同様に反応・処理し、実施例51および52の化合物を得た。
【0216】
実施例51:
4−ブロモ−1−(3−クロロフェニル)−5−(2−ナフチル)−3−(4−ピペリジル)ピラゾール・1臭化水素酸塩
融点243−246℃
【0217】
【化50】
実施例52:
1−(3−クロロフェニル)−4−フルオロ−5−(2−ナフチル)−3−(4−ピペリジル)ピラゾール・1塩酸塩
融点197−200℃
【0219】
【化51】
実施例53:
(a)1−(3−クロロフェニル)−5−〔3−(2−ヒドロキシエチルアミノ)プロピル〕−3−(2−ナフチル)ピラゾール・1塩酸塩
【0221】
【化52】
(b)1−(3−クロロフェニル)−3−〔3−(2−ヒドロキシエチルアミノ)プロピル〕−5−(2−ナフチル)ピラゾール・1塩酸塩
【0223】
【化53】
〔工程1〕:参考例5(2)および(3)で得られた 1−(3−クロロフェニル)−5−〔2−(エトキシカルボニル)エチル〕−3−(2−ナフチル)ピラゾールおよび1−(3−クロロフェニル)−3−〔2−(エトキシカルボニル)エチル〕−5−(2−ナフチル)ピラゾールの混合物1.50gにエタノールアミン4.48mlを加え、120℃で3時間攪拌した。10%クエン酸水溶液30mlを加え、クロロホルム50mlで抽出し、飽和食塩水で洗浄、硫酸マグネシウムで乾燥後濃縮した。残渣をシリカゲルカラムクロマトグラフィーに付し、クロロホルム−メタノール(25:1)で溶出・精製し、1−(3−クロロフェニル)−5−〔2−(2−ヒドロキシエチルアミノカルボニル)エチル〕−3−(2−ナフチル)ピラゾールおよび1−(3−クロロフェニル)−3−〔2−(2−ヒドロキシエチルアミノカルボニル)エチル〕−5−(2−ナフチル)ピラゾールの混合物として1.39gを得た(アモルファス)。
【0225】
〔工程2〕:上記工程1で得られた1−(3−クロロフェニル)−5−〔2−(2−ヒドロキシエチルアミノカルボニル)エチル〕−3−(2−ナフチル)ピラゾールおよび1−(3−クロロフェニル)−3−〔2−(2−ヒドロキシエチルアミノカルボニル)エチル〕−5−(2−ナフチル)ピラゾールの混合物0.95gのテトラヒドロフラン30ml溶液に1.0mol/lのジボランのテトラヒドロフラン溶液4.52mlを加え、室温で12時間攪拌した。次いで、30%塩酸−エタノール溶液3mlを加え、1時間加熱還流した。溶媒留去後、10mol/l水酸化ナトリウム水溶液10mlを加え、クロロホルム30mlで抽出し、飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥後濃縮した。残渣をCHP−20Pを用いた中圧カラムクロマトグラフィーに付して0.02mol/l塩酸水溶液−アセトニトリル(3:2)で溶出・分離精製後凍結乾燥し、目的物(a)0.04gおよび目的物(b)0.31gをそれぞれアモルファスとして得た。
【0226】
実施例53(a):
1H-NMR(DMSO-d6)δ:2.02-2.07(m,2H),2.87(t,J=7.2Hz,2H), 2.99(br,4H),3.65(br,1H), 5.26(br,1H), 7.04(s,1H), 7.49-7.62(m,5H), 7.74(br,1H),7.88-8.06(m,4H), 8.38(br,1H), 8.66(br,2H).
【0227】
実施例53(b):
1H-NMR(CDCl3)δ:2.38(br,2H), 2.91(t,J=7.1Hz,2H), 3.16(br,4H),3.96(br,2H), 6.49(s,1H), 7.07-7.26(m,4H), 7.45-7.53(m,3H),7.71-7.82(m,4H), 9.32(br,2H).
【0228】
上記の1H−NMRスペクトルデータおよびNOESYにより、標記化合物であることを決定した。
【0229】
実施例54:
5−〔(1E)−2−(5−クロロ−1−メチルインドール−3−イル)ビニル〕−3−(3−メトキシカルボニルシクロヘキシルアミノメチル)−ピラゾール・1塩酸塩
【0230】
【化54】
〔工程1〕:参考例6(1)で得られた5−〔(1E)−2−(5−クロロ−1−メチルインドール−3−イル)ビニル〕−3−ヒドロキシメチルピラゾール0.30gのジクロロメタン20ml溶液に、2.0mol/lオキサリルクロリドのジクロロメタン溶液0.63mlを滴下し、30分加熱還流した。溶媒留去し、3−クロロメチル−5−〔(1E)−2−(5−クロロ−1−メチルインドール−3−イル)ビニル〕ピラゾールの粗生成物0.40gを得た。
【0232】
〔工程2〕:上記工程1で得られた3−クロロメチル−5−〔(1E)−2−(5−クロロ−1−メチルインドール−3−イル)ビニル〕ピラゾールの粗生成物0.40gをクロロホルム20mlとメタノール5mlの混媒に加え、室温下参考例11(1)で得られた3−アミノシクロヘキサンカルボン酸メチルエステル・1塩酸塩1.00gおよび10%水酸化ナトリウム水溶液2.09mlを加え、4時間加熱還流した。水20mlを加え、クロロホルム20mlで抽出し、抽出液を硫酸マグネシウムで乾燥後濃縮した。残渣を表面を塩基処理したシリカゲルカラムクロマトグラフィーに付し、クロロホルム−メタノール(20:1)で溶出・分離精製し、減圧留去した。残渣をアセトニトリル−エーテルから塩酸塩として再結晶し、目的物のシス・トランスのうち低極性側の一方を0.10g得た。
融点175−178℃
【0233】
1H-NMR(DMSO-d6)δ:1.16-1.33(m,4H), 1.75-2.48(m,5H), 3.03(br,1H),3.60(s,3H), 3.81(s,3H), 4.13(br,2H), 6.61(s,1H), 6.87-6.97(m,1H),7.24(d,J=8.9Hz,1H), 7,32(d,J=16.6Hz,1H), 7.53(d,J=8.9Hz,1H),7.76(d,J=16.6Hz,1H), 7.92(br,1H), 8.11(br,2H), 9.31(br,1H).
【0234】
対応する原料化合物を用いて実施例54と同様に反応・処理し、表12に示す実施例55〜57の化合物を得た。
【0235】
【表12】
実施例58:
1−(3−クロロフェニル)−5−〔(1Z)−2−(2,6−ジクロロフェニル)ビニル〕−3−(4−ピペリジル)ピラゾール・1塩酸塩
【0237】
【化55】
実施例45で得られた1−(3−クロロフェニル)−5−〔(1E)−2−(2,6−ジクロロフェニル)ビニル〕−3−(4−ピペリジル)ピラゾール・1塩酸塩0.41gをメタノール400mlに溶解し、室温で5時間自然光を照射した。溶媒留去後、残渣をアセトニトリル−ジエチルエーテルから塩酸塩として再結晶して、目的物0.25gを得た。
融点82−85℃
【0239】
1H-NMR(CDCl3)δ:1.76-1.83(m,4H), 2.63-2.73(m,3H), 3.06-3.10(m,2H),5.54(s,1H), 6.51(d,J=12.1Hz,1H), 6.59(d,J=12.1Hz,1H), 7.20-7.26(m,1H),7.34-7.41(m,5H), 7.56(br,1H).
【0240】
上記の1H−NMRスペクトルデータにより、標記化合物であることを決定した。
【0241】
対応する原料化合物を用いて実施例58と同様に反応・処理し、表13に示す実施例59〜61の化合物を得た。
【0242】
【表13】
実施例62:
5−(3−ベンジルオキシフェニル)−4−(3−クロロフェニル)−2−(4−ピペリジル)オキサゾール・1フマル酸塩
【0244】
【化56】
〔工程1〕:参考例8(1)で得られた2−(3−ベンジルオキシフェニル)−1−(3−クロロフェニル)−2−ヒドロキシエタン−1−オン1.70g、N−(tert−ブトキシカルボニル)イソニペコチン酸1.22g、1−(3−ジメチルアミノプロピル)−3−エチルカルボジイミド・1塩酸塩0.94gおよび4−ジメチルアミノピリジン0.59gのジクロロメタン40ml溶液を室温で6時間攪拌した。反応液を10%クエン酸水溶液30mlを加え、クロロホルム50mlで抽出し硫酸マグネシウムで乾燥後濃縮し、2−(3−クロロフェニル)−2−オキソ−1−(3−ベンジルオキシフェニル)エチル−1−(1−tert−ブトキシカルボニル)ピペリジン−4−カルボキシレートの粗生成物1.20gを得た。
【0246】
〔工程2〕:上記工程1で得られた2−(3−クロロフェニル)−2−オキソ−1−(3−ベンジルオキシフェニル)エチル−1−(1−tert−ブトキシカルボニル)ピペリジン−4−カルボキシレートの粗生成物1.20gを酢酸40mlで溶解し、酢酸アンモニウム0.93gを加え、6時間加熱還流した。溶媒留去後、残渣をCHP−20Pを用いた中圧カラムクロマトグラフィーに付して水−アセトニトリル(15:85)で溶出・分離精製し、減圧留去した。エタノール−ジエチルエーテルから、フマル酸塩として再結晶し、目的物1.04gを得た。
融点123−126℃
【0247】
1H-NMR(CDCl3)δ:1.81-1.93(m,2H), 2.10-2.14(m,2H), 2.74-2.81(m,2H),3.00-3.05(m,2H), 3.19-3.23(m,2H), 5.01(s,2H), 6.95-6.97(m,1H),7.15-7.18(m,2H), 7.23-7.39(m,10H), 7.50-7.53(m,1H), 7.70(br,1H).
【0248】
対応する原料化合物を用いて実施例62と同様に反応・処理し、表14に示す実施例63および64の化合物を得た。
【0249】
【表14】
実施例65:
5−(3−ベンジルオキシフェニル)−4−(3−クロロフェニル)−2−(4−ピペリジル)イミダゾール・2塩酸塩・1.40水和物
【0251】
【化57】
参考例9(1)で得られた2−(3−ベンジルオキシフェニル)−1−(3−クロロフェニル)エタン−1,2−ジオン3.00g、N−(tert-ブトキシカルボニル)イソニペコチノアルデヒド1.82gおよび酢酸アンモニウム5.27gの酢酸50ml溶液を60℃で2時間攪拌した。溶媒留去後、水30mlを加えクロロホルム60mlで抽出し、飽和炭酸水素ナトリウム水溶液20mlで洗浄し、硫酸マグネシウムで乾燥後濃縮した。残渣をCHP−20Pを用いた中圧カラムクロマトグラフィーに付して水−アセトニトリル(1:4)で溶出・精製し、水−アセトニトリルより塩酸塩として再結晶し、目的物1.10gを得た。
融点140−141℃
【0253】
1H-NMR(DMSO-d6)δ:2.06-2.34(m,4H), 2.97-3.16(m,2H), 3.39-3.53(m,3H),5.11(s,2H), 7.03(d,J=8.0Hz,1H), 7.09-7.21(m,2H), 7.35-7.53(m,9H),7.64(br,1H), 9.24(br,2H).
【0254】
対応する原料化合物を用いて実施例65と同様に反応・処理し、表15に示す実施例66〜69の化合物を得た。
【0255】
【表15】
実施例70:
4−(3,4−ジクロロベンジルオキシ)−3−(3−メトキシカルボニルシクロヘキシルアミノメチル)−1H−インダゾール・1塩酸塩
【0257】
【化58】
〔工程1〕:参考例12(1)で得られた1−tert−ブトキシカルボニル−3−〔N−tert−ブトキシカルボニル−N−(3−メトキシカルボニルシクロヘキシル)〕アミノメチル−4−ヒドロキシ−1H−インダゾール1.63gを、氷冷下水素化ナトリウム(60%オイルディスパージョン)0.14gのジメチルホルムアミド50ml懸濁液に加え、室温で30分間攪拌した後、3,4−ジクロロベンジルクロリド0.52mlを加え、室温で12時間攪拌した。飽和塩化アンモニウム水溶液30mlを加え、ジエチルエーテル50mlで2回抽出し、抽出液を硫酸マグネシウムで乾燥後濃縮した。残渣をシリカゲルカラムクロマトグラフィーに付し、ノルマルヘキサン−酢酸エチル(4:1)で溶出・精製し、1−tert−ブトキシカルボニル−3−〔N−tert−ブトキシカルボニル−N−(3−メトキシカルボニルシクロヘキシル)〕アミノメチル−4−(3,4−ジクロロベンジルオキシ)−1H−インダゾール1.50gをアモルファスとして得た。
【0259】
〔工程2〕:上記工程1で得られた1−tert−ブトキシカルボニル−3−〔N−tert−ブトキシカルボニル−N−(3−メトキシカルボニルシクロヘキシル)〕アミノメチル−4−(3,4−ジクロロベンジルオキシ)−1H−インダゾール0.30gを用い、実施例1における工程2と同様に反応・処理し、アセトニトリル−ジエチルエーテルから塩酸塩として結晶化し、シス・トランスの混合物として目的物0.16gを得た。
【0260】
1H-NMR(DMSO-d6)δ:1.16-1.48(m,4H), 1.74-2.35(m,5H), 3.06(br,1H),3.60(s,3H), 4.53(br,2H), 5.26(s,2H), 6.66(d,J=8.0Hz,1H),7.14(d,J=8.0Hz,1H), 7.28-7.38(m,1H), 7.58(dd,J=1.7Hz,J=8.2Hz,1H),7.69(d,J=8.2Hz,1H), 7.88(d,J=1.7Hz,1H), 9.17(br,2H), 13.43(s,1H).
【0261】
対応する原料化合物を用いて実施例70と同様に反応・処理し、表16に示す実施例71〜80の化合物を得た。
【0262】
【表16】
【表17】
実施例81:
4−(2,6−ジクロロフェネチルオキシ)−3−(3−メトキシカルボニルシクロヘキシルアミノメチル)−1H−インダゾール・1塩酸塩
【0265】
【化59】
〔工程1〕:参考例12(1)で得られた1−tert−ブトキシカルボニル−3−〔N−tert−ブトキシカルボニル−N−(3−メトキシカルボニルシクロヘキシル)〕アミノメチル−4−ヒドロキシ−1H−インダゾール150mgおよび2−(2,6−ジクロロフェニル)エタノール63mgのテトラヒドロフラン20ml溶液に、トリフェニルホスフィン110mgおよびジイソプロピルアゾジカルボキシレート65μlを加え、2時間加熱還流した。溶媒留去後、残渣をフラッシュシリカゲルカラムクロマトグラフィーに付し、ノルマルヘキサン−酢酸エチル(4:1)で溶出・精製し、1−tert−ブトキシカルボニル−3−〔N−tert−ブトキシカルボニル−N−(3−メトキシカルボニルシクロヘキシル)〕アミノメチル−4−(2,6−ジクロロフェネチルオキシ)−1H−インダゾール110mgをアモルファスとして得た。
【0267】
〔工程2〕:上記工程1で得られた1−tert−ブトキシカルボニル−3−〔N−tert−ブトキシカルボニル−N−(3−メトキシカルボニルシクロヘキシル)〕アミノメチル−4−(2,6−ジクロロフェネチルオキシ)−1H−インダゾール110mgを用い、実施例1における工程2と同様に反応・処理し、アセトニトリル−ジエチルエーテルから塩酸塩として結晶化し、シス・トランスの混合物として目的物30mgを得た。
【0268】
1H-NMR(DMSO-d6)δ:1.19-1.52(m,4H), 1.84-2.50(m,5H), 3.22(br,1H),3.50(t,J=7.60Hz,2H), 3.60(s,3H), 4.35(t,J=7.6Hz,2H), 4.52(s,2H),6.69(d,J=8.0Hz,1H), 7.12(d,J=8.0Hz,1H), 7.29(t,J=8.0Hz,1H),7.35(t,J=8.0Hz,1H), 7.53(d,J=8.0Hz,2H), 9.13(s,2H), 13.37(s,1H).
【0269】
対応する原料化合物を用いて実施例81と同様に反応・処理し、表18に示す実施例82〜84の化合物を得た。
【0270】
【表18】
かくして製造された本発明の化合物について抗菌試験を行った。以下に、試験方法および結果について説明する。
【0272】
試験例:in vitroにおける抗菌活性(MIC:μg/ml)
【0273】
本発明の化合物について、The National Committee for Clinical LaboratoryStandards(NCCLS)の定める標準法に準じて、最小発育阻止濃度(MIC:μg/ml)を測定した。表19および20に代表的な本発明の化合物の抗菌結果を示した。
【0274】
【表19】
【表20】
菌株 A: Staphylococcus aureus FDA 209P
菌株 B: Staphylococcus aureus KMP9 (MRSA)*
菌株 C: Enterococcus faecium ATCC19434
菌株 D: Enterococcus faecium 1778 (VRE)*
菌株 E: Streptococcus pneumoniae ATCC49619
菌株 F: Streptococcus pneumoniae KT2524 (PRSP)*
*:臨床分離株である。
また、以上の菌株はすべてグラム陽性菌である。
【0277】
表19および20から明らかなように本発明の化合物は、多剤耐性菌(MRSA、VRE、PRSP等)に極めて優れた抗菌作用を有するので、抗菌剤として有用である。
【0278】
【発明の効果】
本発明の化合物は、特にグラム陽性菌に対し優れた抗菌活性を有する。また、本発明の化合物は、現在臨床において問題視されている薬剤耐性菌であるMRSA、VRE、PRSP等に対しても感受性菌と同等の抗菌力を有しており、これらの菌に起因する感染症の予防・治療薬として使用することができる。[0001]
TECHNICAL FIELD OF THE INVENTION
The present invention relates to novel compounds useful as medicaments. More specifically, the present invention relates to a novel compound having excellent antibacterial activity, particularly against multidrug-resistant bacteria, or a salt thereof, and a pharmaceutical composition thereof.
[0002]
[Prior art]
Methicillin-Resistant Staphylococcus aureus, abbreviated as MRSA, was first reported in the UK in 1961 as the cause of intractable postoperative infections and respiratory and digestive infections. Are known. MRSA infections began to spread in Europe and the United States from the late 1960s to the late 1970s, gained public attention in Japan in the 1980s as a major causative agent of hospital-acquired infections, and spread nationwide in the late 1990s. At present, MRSA is highly resistant to multiple drugs, and resistant strains of vancomycin and arbekacin, which were antibacterial agents against MRSA infection, have also appeared.
[0003]
In addition, the multidrug-resistant bacteria attracting high interest in the medical world include, in addition to the aforementioned MRSA, penicillin-resistant pneumococci (Penicillin-Resistant Streptococcus).
pneumoniae, PRSP) and Vancomycin-Resistant Enterococcus faecium (VRE) are known. Pneumococcus is a pneumonia-causing bacterium that is feared due to its high toxicity, and enterococci are systemic and / or urinary tract-causing bacteria that may be clinically isolated together with MRSA. New quinolone antibacterial agents and oxazolidinone antibacterial agents are used for infections caused by these multidrug-resistant bacteria, but their antibacterial activities are not so strong and the emergence of these resistant strains has been reported. I have.
[0004]
Then, the present inventors searched for a compound having a strong antibacterial activity against these multidrug-resistant bacteria, and as a result, the compound represented by the above general formula (I) or a pharmaceutically acceptable compound thereof was obtained. The inventors have found that a salt (hereinafter, also referred to as “the compound of the present invention”) has excellent antibacterial activity, and completed the present invention.
[0005]
Patent Literature 1 (International Publication WO 01/52845) describes that a compound represented by the following general formula (A) including a pyrazole derivative is used for treating bacterial infection.
[0006]
Embedded image
RTwoIs a hydrogen atom or R1Is -COTwo(C1-3(Aliphatic) and the like, further selected from halogen atoms and the like,
A ring is a hetero ring selected from thiazole, oxazole, imidazole or pyrazole groups,
Z is CRThreeOr NRThreeAnd
RThreeIs-(CHTwo)pN (RFive)TwoEtc.,
RFourRepresents a hydrogen atom and optionally substituted C1-6Aliphatic groups,
RFiveRepresents a hydrogen atom and optionally substituted C1-4Aliphatic groups,
p is an integer of 0 to 4,
Ar is an optionally substituted aryl, heteroaryl, or the like. ]
[0007]
Further, Patent Document 2 (Japanese Patent Application Laid-Open No. Hei 8-502508) describes a compound represented by the following general formula (B). This compound is a dopamine receptor subtype ligand, and It is described as being useful for treating diseases.
[0008]
Embedded image
X and Y each represents nitrogen, the other represents oxygen, NRTwoEtc.,
Q represents a substituted monocyclic heteroaliphatic 5- or 6-membered ring containing one nitrogen atom as the only heteroatom and bonded to the 5-membered heteroatom ring containing the X and Y moieties by a carbon atom. ,
R1Represents a hydrogen atom, a halogen atom, or the like,
RTwoIs a hydrogen atom, C1-6Represents alkyl,
A represents the formula (i), (ii) or (iii);
[0009]
Embedded image
RThree, RFourAnd RFiveIs independently hydrogen, hydrocarbon, -ORa, -NRaRbEtc.,
RaAnd RbRepresents independently hydrogen, a heterocyclic group, etc.)
[0010]
The action of the compound of the present invention is completely different from the use of the compound represented by the general formula (B) for the treatment of dopamine-related diseases.
[0011]
Further, Patent Document 3 (Japanese Patent Application Laid-Open No. 11-504010) describes a compound represented by the following general formula (C), and the compound has an adrenergic α1aIt is described as a receptor antagonist which is useful for treating benign prostatic hyperplasia.
[0012]
Embedded image
X and Y each represents nitrogen, the other represents oxygen, NRTwoEtc.,
Q is a substituted 5- or 6-membered monocyclic heterocycle containing one nitrogen atom as a single heteroatom and attached via a carbon atom to a 5-membered heterocycle containing X and Y moieties. Represents an aliphatic ring,
R1Represents a hydrogen atom, a halogen atom, or the like,
RTwoIs a hydrogen atom or C1-6Represents alkyl,
A represents the formula (i), (ii), (iii), (iv):
[0013]
Embedded image
RThree, RFourAnd RFiveIs independently hydrogen, hydrocarbon, heterocyclic group, -ORa, -NRaRbEtc.
RaAnd RbIndependently represents hydrogen, a heterocyclic group, or the like;
Ring M may optionally be an additional ring or ring such that the entire structure (iv) is a monocyclic, bicyclic or polycyclic aromatic or heteroaromatic ring system, including phenyl, benzodioxane, and the like. Wherein the ring systems are each RThree, RFourAnd RFive, And R18And R19With R18And R19Are each independently C1-6There may be alkyl etc.)
[0014]
The action of the compound of the present invention is completely different from the treatment of benign prostatic hyperplasia, which is a use of the compound represented by the general formula (C).
[0015]
Patent Document 4 (International Publication WO02 / 053559) described below describes a sodium channel modulator derived from a compound represented by the following general formula (D).
[0016]
Embedded image
RFourRepresents an alkyl group, a haloalkyl group, a cycloalkyl group, or the like;
RTwoRepresents an alkyl group, a cycloalkyl group, or the like;
RThreeRepresents a hydrogen atom, an alkyl group, a cycloalkyl group or an optionally substituted aryl group, etc.)
[0017]
The action of the compound of the present invention is completely different from that of the sodium channel modulator, which is a use of the compound represented by the general formula (D).
[0018]
Patent Document 5 (JP-T-10-509713) discloses that a compound represented by the following general formula (E) has an α-1C adrenergic receptor antagonistic action.
[0019]
Embedded image
RTwoIs hydrogen, C1-6Alkyl, hydroxy C1-6Selected from alkyl and the like,
W is C1-6An alkylene chain or nitrogen;
m is independently an integer 0 or 1,
X is CH or nitrogen, provided that when W is nitrogen, X is CH;
q is an integer independently selected from the group consisting of 1, 2, 3, or 4;
Where R1Is phenyl or phenyl mono- or di-substituted by halogen, methyl or methoxy;TwoIs hydrogen or methyl;ThreeIs hydrogen, hydroxy or C1-6Alkoxy, q is 1, 2, 3, or 4, and W is (CHTwo), And when m is 1, X is CH. ]
[0020]
The action of the compound of the present invention is completely different from the α-1C adrenergic receptor antagonist action, which is the use of the compound represented by the general formula (E).
[0021]
The compounds [General Formulas (A) to (E)] described in Patent Documents 1 to 5 above disclose compounds similar to the compounds of the present invention, but the compounds of the present invention are specifically described at all. It has not been.
[0022]
[Patent Document 1]
WO 01/52845 pamphlet
[Patent Document 2]
Japanese Patent Publication No. Hei 8-502508
[Patent Document 3]
Japanese Patent Publication No. 11-504010
[Patent Document 4]
WO 02/053559 pamphlet
[Patent Document 5]
Japanese Patent Publication No. 10-509713
[0023]
[Problems to be solved by the invention]
As described above, there is a need for a drug having strong antibacterial activity that can be used against infections caused by multidrug-resistant bacteria, which are currently of great interest. Therefore, an object of the present invention is to provide a compound having a strong antibacterial activity against multidrug-resistant bacteria.
[0024]
[Means for Solving the Problems]
The present inventors have conducted intensive studies to solve the above-mentioned problems, and as a result, found a compound having a strong antibacterial activity against multidrug-resistant bacteria, and completed the present invention.
[0025]
The present invention relates to an antibacterial agent useful for the treatment or prevention of bacterial infections, and more particularly to a compound represented by the following general formula (I) or a pharmaceutically acceptable salt thereof as an active ingredient.
[0026]
That is, the present invention relates to the following general formula (I)
[0027]
Embedded image
RTwoIs an aminoalkyl group; a methylaminoalkyl group; a hydroxyalkylaminoalkyl group; an alkoxycarbonylalkylaminoalkyl group; a cyclohexylaminoalkyl group which may be substituted on a cyclohexane ring (the substituent is an alkoxycarbonyl group or a hydroxyalkyl A piperidyl group; a pyrrolidino group which may be substituted with a hydroxyl group; a piperazinylalkyl group or an alkoxycarbonylpiperidinoalkyl group which may be substituted on the piperazine ring with an alkylsulfamoyl group. And
A represents a nitrogen atom or a carbon atom,
B is C—RFive, NH or an oxygen atom.
However, when A is a nitrogen atom, B is C—RFiveAnd when A is a carbon atom, B means NH or an oxygen atom.
Also, RThreeAnd RFourIs a hydrogen atom; an alkyl group; a phenyl group which may be substituted with one or two halogen atoms; a phenoxyphenyl group; a benzyloxyphenyl group; a pyridyl group; It is substituted with a benzyl group which may be substituted with a fluoromethyl group or a piperidyl group which may be substituted with a benzyl group, and the other has no hydrogen atom or substituent.
RFiveRepresents a hydrogen atom, an alkyl group or a halogen atom,
A is a nitrogen atom, and B is C—RFive, R1And RFiveMay together form a benzene ring, and the formed benzene ring is -OR1′, R1'Means an optionally substituted phenylalkyl group (the substituent is one or two halogen atoms, an alkyl group or an amino group). ]
Or a pharmaceutically acceptable salt thereof, and a pharmaceutical composition thereof.
[0028]
The compound of the present invention is represented by the above general formula (I), and preferred compounds include the following compounds (1) to (4).
[0029]
(1) A is a nitrogen atom and B is C—RFive2. The compound according to claim 1 or a pharmaceutically acceptable salt thereof.
(2) The compound according to claim 1, wherein A is a carbon atom and B is NH, or a pharmaceutically acceptable salt thereof.
(3) The compound according to claim 1, wherein A is a carbon atom and B is an oxygen atom, or a pharmaceutically acceptable salt thereof.
(4) A is a nitrogen atom and B is C—RFiveAnd R1And RFiveOr the pharmaceutically acceptable salt thereof according to claim 1, wherein together form a benzene ring.
[0030]
Specific compounds or salts thereof are described below as more preferred compounds.
[0031]
-5-[(1E) -2- (5-chloroindol-3-yl) vinyl] -1- (3-chlorophenyl) -3- (4-piperidyl) pyrazole.1.25 hydrochloride. 1- [1-benzyl- (4-piperidyl)]-5- [4- (3,4-dichlorobenzyloxy) phenyl] -3- (4-piperidyl) pyrazole monohydrochloride. • 1- (3-bromophenyl) -5- (2-naphthyl) -3- (4-piperidyl) pyrazole • 1.65 hydrochloride.
1- (3-chlorophenyl) -3- (1-methylaminopropyl) -5- (4-phenoxyphenyl) pyrazole 1.2 hydrochloride.
5-[(1E) -2- (3,4-dichlorophenyl) vinyl] -3- (4-piperidyl) pyrazole dihydrochloride.
-5-{(1E) -2- [4- (3,4-dichlorobenzyloxy) phenyl] vinyl} -3- (4-piperidyl) pyrazole 1.7 hydrochloride.
1-butyl-5-{(1E) -2- [4- (3,4-dichlorobenzyloxy) phenyl] vinyl} -3- (4-piperidyl) pyrazole monohydrochloride.
5-[(1E) -2- (5-chloroindol-2-yl) vinyl] -1- (3-chlorophenyl) -3- (4-piperidyl) pyrazole monohydrochloride.
5-methylaminomethyl-3- (4-phenoxyphenyl) -1- (4-trifluoromethylbenzyl) pyrazole monohydrochloride.
1- (3-chlorophenyl) -5-[(1E) -2- (3,4-dichlorophenyl) vinyl] -3- (4-piperidyl) pyrazole monohydrochloride.
1- (3-chlorophenyl) -3-methylaminomethyl-5- (4-phenoxyphenyl) pyrazole 1 fumarate.
4-bromo-1- (3-chlorophenyl) -5- (2-naphthyl) -3- (4-piperidyl) pyrazole monohydrobromide.
1- (3-chlorophenyl) -5- [3- (2-hydroxyethylamino) butyl] -3- (2-naphthyl) pyrazole monohydrochloride.
5-[(1E) -2- (5-chloroindol-1-methyl-3-yl) vinyl] -3- (3-methoxycarbonylcyclohexylaminomethyl) pyrazole monohydrochloride.
1- (3-chlorophenyl) -5-[(1Z) -2- (3,4-dichlorophenyl) vinyl] -3- (4-piperidyl) pyrazole monohydrochloride.
5-[(1Z) -2- (5-chloroindol-1-methyl-3-yl) vinyl] -3- (3-methoxycarbonylcyclohexylaminomethyl) pyrazole monohydrochloride.
5- (3-benzyloxyphenyl) -4- (3-chlorophenyl) -2- (4-piperidyl) oxazole / 1 fumarate.
5- (3-benzyloxyphenyl) -4- (3-chlorophenyl) -2- (4-piperidyl) imidazole dihydrochloride.
4- (3,4-dichlorobenzyloxy) -3- (3-methoxycarbonylcyclohexylaminomethyl) -1H-indazole monohydrochloride.
4- (2,6-dichlorophenethyloxy) -3- (3-methoxycarbonylcyclohexylaminomethyl) -1H-indazole monohydrochloride.
[0032]
The compound of the present invention represented by the general formula (I) may form a pharmaceutically acceptable salt, and the salt is an acid addition salt, for example, hydrochloric acid, hydrobromic acid, iodide Salts with inorganic acids such as hydrochloric acid, sulfuric acid, phosphoric acid, nitric acid, acetic acid, oxalic acid, fumaric acid, maleic acid, malonic acid, lactic acid, malic acid, succinic acid, citric acid, tartaric acid, benzoic acid, methanesulfonic acid , P-toluenesulfonic acid, salts with organic acids such as gluconic acid.
[0033]
In the present invention, "alkyl group" and "alkyl" moiety refer to a straight-chain or branched C1-5Means an alkyl group of1-3Are preferred, and specific examples include a methyl group, an ethyl group, a propyl group, an isopropyl group, an n-butyl group, an isobutyl group, a sec-butyl group, a tert-butyl group, and an n-pentyl group. In the “alkoxy group” and the “alkoxy” part, the alkyl part is the above-mentioned alkyloxy group, and examples thereof include methoxy, ethoxy, and butoxy. "Halogen atom" means fluorine, chlorine, bromine or iodine.
[0034]
The compound of the present invention represented by the general formula (I) or a salt thereof may exist as a hydrate and / or a solvate, and such a form of the compound is also included in the compound of the present invention. . Further, it exists as an optical isomer, a stereoisomer or a mixture thereof, and all of them are included in the present invention.
[0035]
The circle drawn by a broken line in the above general formula (I) indicates two non-adjacent double bonds at any positions in the five-membered ring.
[0036]
Specifically, in the general formula (I), A is a nitrogen atom, and B is C—RFiveOr A is a nitrogen atom and B is C—RFiveAnd R1And RFiveAre together forming a benzene ring,ThreeOr RFourWhen either one of is a hydrogen atom, tautomers exist, and these tautomers are also included in the compounds of the present invention.
[0037]
Further, in the general formula (I), when A is a carbon atom and B is NH, as shown by the following formula, tautomers when B is -NH- and when = N- Which are also included in the compounds of the present invention.
[0038]
Embedded image
Next, a method for producing the compound of the present invention will be described below.
[0040]
In the compound of the present invention represented by the general formula (I), A is a nitrogen atom, and B is C—RFiveThe compound (Ia) of the present invention can be produced by the following production methods 1 to 3.
[0041]
[Production method 1]
Compound (Ia) of the present invention can be produced according to the following reaction formula.
[0042]
Embedded image
[0043]
That is, the compound (Ia) of the present invention is described in the literature (Bioorg. Med. Chem., 1998,6, 743), a 1,3-dione derivative (II) and RThree'NHNHTwoCan be produced by reacting in a suitable solvent. In the compound (II), RTwoHaving a primary or secondary amino group inTwoThe compound (Ia) of the present invention can be produced by deprotection after the above reaction using a compound in which the amino group is protected with a suitable protecting group.
[0044]
The reaction temperature varies depending on the type of the starting compound used and the like, but is usually -10 to 150C, preferably 10 to 100C. The reaction time is generally about 30 minutes to 24 hours, preferably about 1 to 10 hours.
[0045]
[Production method 2]
Further, the compounds (Ia-1) and (Ia-2) of the present invention can be produced according to the following reaction formula.
[0046]
Embedded image
[0047]
[Step 1]: Compound (Ia-2) can be produced by reacting hydrazine with 1,3-dione derivative (II) in a suitable solvent.
[0048]
The reaction temperature varies depending on the type of the starting compound used and the like, but is usually -10 to 150C, preferably 10 to 100C. The reaction time is generally about 30 minutes to 24 hours, preferably about 1 to 10 hours.
[0049]
[Step 2]: Compound (Ia-2) obtained in Step 1 and R13Compound (Ia-1) can be produced by reacting X in a suitable solvent in the presence of a base. In the compound (II), RTwoHaving a primary or secondary amino group inTwoThe compound (Ia-1) of the present invention can be produced by performing deprotection after the reaction in Steps 1 and 2 using a compound in which the amino group of the above is protected with an appropriate protecting group.
[0050]
The reaction temperature varies depending on the type of the starting compound used and the like, but is usually -10 to 150C, preferably 10 to 100C. The reaction time is generally about 30 minutes to 24 hours, preferably about 1 to 10 hours.
[0051]
Examples of the base used in the reaction of step 2 include alkali metal hydrides such as sodium hydride and potassium hydride, alkali metal hydroxides such as sodium hydroxide and potassium hydroxide, sodium ethoxide and sodium methoxide. And the like. Alkoxy alkali metals such as sodium carbonate, potassium carbonate such as potassium carbonate, and organic bases such as pyridine, triethylamine and diisopropylethylamine. Examples of the solvent include diethyl ether, tetrahydrofuran, dioxane, benzene, toluene, dimethylformamide, alcohols such as methanol, ethanol, isopropanol and tert-butanol, and water. These solvents may be used alone or as a mixed solvent of two or more.
[0052]
[Production method 3]
Compound (Ia-1) of the present invention can also be produced according to the following reaction formula.
[0053]
Embedded image
[0054]
That is, the compound (Ia-2) obtained in step 1 of the above-mentioned production method 2 has R13B (OH)TwoCompound (Ia-1) can be produced by reacting with cupric acetate and a suitable base in the presence of a suitable base. In the compound (Ia-2), RTwoWhen the compound contains a primary or secondary amino group, the compound of formula (II)TwoThe compound (Ia-1) of the present invention is reacted and treated in the same manner as in Step 1 of the above-mentioned Production Method 2 using a compound in which the amino group of the above is protected with an appropriate protecting group, followed by deprotection after the reaction. Can be manufactured.
[0055]
The reaction temperature varies depending on the type of the starting compound used and the like, but is usually -10 to 150C, preferably 10 to 100C. The reaction time is generally about 30 minutes to 48 hours, preferably about 5 to 24 hours.
[0056]
Examples of the base used in this reaction include organic bases such as pyridine, 2,6-lutidine, triethylamine, and diisopropylethylamine. Examples of the solvent include dichloromethane, chloroform, benzene, and toluene. These solvents may be used alone or as a mixed solvent of two or more.
[0057]
The reaction used in Step 1 of Production Method 1 and Production Method 2 is generally performed in a suitable solvent that does not affect the reaction. Examples of the solvent include alcohols such as methanol, ethanol, isopropanol and tert-butanol, acetone, acetonitrile, N, N-dimethylformamide, dimethylsulfoxide, hexamethylphosphoramide and water. These solvents may be used alone or as a mixed solvent of two or more.
[0058]
[Production method 4]
In the compound of the present invention represented by the general formula (I), the compound (Ib) in which A is a carbon atom and B is an oxygen atom is described in the literature (J. Med. Chem., 1992,35, 3498) according to the following reaction formula.
[0059]
Embedded image
[0060]
[Step 1]: The compound (V) of the present invention is prepared by reacting a 2-hydroxyethane-1-one derivative (IV) with RTwoCOOH can be produced by reacting COOH in a suitable solvent in the presence of 1- (3-dimethylaminopropyl) -3-ethylcarbodiimide (WSC) monohydrochloride.
[0061]
The reaction temperature varies depending on the type of the starting compound used and the like, but is usually -10 to 150C, preferably 10 to 100C. The reaction time is generally about 30 minutes to 24 hours, preferably about 1 to 10 hours. Examples of the solvent include dichloromethane, chloroform, diethyl ether, tetrahydrofuran, dioxane, benzene, toluene, and dimethylformamide.
[0062]
[Step 2]: Compound (Ib) of the present invention can be produced by reacting compound (V) obtained in Step 1 with an appropriate solvent in the presence of, for example, ammonium acetate and acetic acid. Also, RTwoIn COOH, RTwoHaving a primary or secondary amino group inTwoCompound (Ib) can be produced by protecting the amino group of the above with an appropriate protecting group and then performing deprotection after the above reaction.
[0063]
The reaction temperature varies depending on the type of the starting compound used and the like, but is usually -10 to 150C, preferably 10 to 100C. The reaction time is generally about 30 minutes to 24 hours, preferably about 1 to 10 hours.
[0064]
[Production method 5]
In the compound of the present invention represented by the general formula (I), the compound (Ic) in which A is a carbon atom and B is NH is described in the literature (Tetrahedron Lett., 1994,35, 1635) according to the following reaction formula.
[0065]
Embedded image
[0066]
That is, the compound (Ic) of the present invention comprises a 1,2-dione derivative (VI) and RTwoIt can be produced by reacting CHO in a suitable solvent in the presence of, for example, ammonium acetate and acetic acid. Also, RTwoIn CHO, RTwoHaving a primary or secondary amino group inTwoCompound (Ic) can be produced by protecting the amino group of the above with an appropriate protecting group and then performing deprotection after the above reaction.
[0067]
The reaction temperature varies depending on the type of the starting compound used and the like, but is usually -10 to 150C, preferably 10 to 100C. The reaction time is generally about 30 minutes to 12 hours, preferably about 1 to 5 hours.
[0068]
In the compound of the present invention represented by the general formula (I), A is a nitrogen atom, and B is C—RFiveAnd R1And RFiveCompound (Id-1) and Compound (Id-2), which together form a benzene ring, can be produced by the following Production Methods 6 and 7, respectively.
[0069]
[Production method 6]
In the compound of the present invention represented by the general formula (I), RThreeOr RFourIs a group other than a hydrogen atom, the compound (Id-1) of the present invention can be produced by Method 1 or Method 2 shown in the following reaction formula.
[0070]
Embedded image
[0071]
[Method 1]: Compound (Id-1) of the present invention comprises compound (VII) and R1'X can be produced by reacting in a suitable solvent in the presence of a base.
[0072]
The reaction temperature varies depending on the type of the starting compound used and the like, but is usually -10 to 150C, preferably 10 to 100C. The reaction time is generally about 30 minutes to 24 hours, preferably about 1 to 10 hours.
[0073]
Examples of the base used in this reaction include alkali metal hydrides such as sodium hydride and potassium hydride, alkali metal hydroxides such as sodium hydroxide and potassium hydroxide, sodium ethoxide and sodium methoxide. Alkoxy alkali metals, alkali metal carbonates such as sodium carbonate and potassium carbonate or organic bases such as pyridine, triethylamine and diisopropylethylamine are exemplified. Examples of the solvent include diethyl ether, tetrahydrofuran, dioxane, benzene, toluene, dimethylformamide, alcohols such as methanol, ethanol, isopropanol and tert-butanol, and water. These solvents may be used alone or as a mixed solvent of two or more.
[0074]
[Method 2]: Compound (Id-1) of the present invention was prepared according to the literature (J. Med. Chem., 1992,35, 1176). That is, compound (Id-1) of the present invention can be produced by reacting compound (VII) with various alcohols in the presence of dialkylazodicarboxylate and triphenylphosphine in a suitable solvent.
[0075]
Examples of the solvent used in this reaction include diethyl ether, tetrahydrofuran, dioxane, benzene, toluene, dimethylformamide, pyridine, hexamethylphosphorous triamide and the like.
[0076]
Certain of the compounds (Id-1) of the present invention produced by the production method 6 can be prepared by converting the compound (Id-2) produced by the production method 7 described below in the presence of a base such as an alkyl halide. It can also be produced by reacting in a suitable solvent.
[0077]
In methods 1 and 2, R of compound (VII)TwoHaving a primary or secondary amino group inTwoCompound (Id-1) can be produced by deprotection after the above reaction using a compound in which the amino group of the above is protected with an appropriate protecting group.
[0078]
[Production method 7]
In the compound of the present invention represented by the general formula (I), RThreeOr RFourIs a hydrogen atom, the compound (Id-2) of the present invention can be produced according to the following reaction formula.
[0079]
Embedded image
[0080]
That is, the compound (Id-2) of the present invention has a pyrazole ring moiety having an appropriate protecting group (RThreeCompound (VII ') protected by' ') is converted to compound (VIII) according to the above-mentioned Production Method 6, followed by deprotection. In the compound (VII '), RTwoHaving a primary or secondary amino group inTwoUsing a compound in which the amino group of is protected with a suitable protecting group,ThreeCompound (Id-2) can be produced by simultaneous or separate deprotection.
[0081]
The compounds of the present invention represented by the formulas (Ia), (Ib), (Ic), (Id-1) and (Id-2) produced by the above production methods 1 to 7, can be used for chromatography, recrystallization and the like. It can be isolated and purified by ordinary chemical operations. The compounds of the present invention represented by formulas (Ia), (Ib), (Ic), (Id-1) and (Id-2) can be obtained in the form of a free base or an acid addition salt. Can be converted into each other by a usual method.
[0082]
Next, the starting compounds used in the above Production Methods 1 to 7 can be produced by the following method.
[0083]
Compound (II) used in Production Methods 1 and 2 can be produced according to the method shown in the following reaction formula.
[0084]
Embedded image
[0085]
That is, compound (II) can be produced by reacting compound (IX) with compound (X) in the presence or absence of a suitable condensing agent and in the presence of a suitable base in a suitable solvent. . In the compound (IX), RTwoHaving a primary or secondary amino group inTwoCan be protected with a suitable protecting group to produce compound (II).
[0086]
Compounds (IV) and (VI) used in Production Methods 4 and 5, respectively, can be produced according to the method shown in the following reaction formula.
[0087]
Embedded image
[0088]
That is, compound (VI) is described in the literature (J. Chem. Soc. Perkin Trans. 1, 1997,16, 2429) and the compound (XI) produced according to the method described in1Compound (IV) can be produced by reacting CHO in a suitable solvent in the presence of a base, further treating with an acid, and then oxidizing with a suitable oxidizing agent.
[0089]
Compound (VII) used in the above-mentioned Production method 6 can be produced according to the method shown by the following reaction formula.
[0090]
Embedded image
[0091]
That is, compound (VII) is described in the literature (J. Med. Chem., 2000,43, 2664), the compound (XII) and an amine are reacted in an appropriate solvent in the presence or absence of a base in a suitable solvent to give a compound (XIII). RTwoHaving a primary or secondary amino group inTwoCan be protected by an appropriate protecting group, and then Pro is selectively deprotected.
[0092]
The compound (VII ') used in the above-mentioned production method 7 is a compound represented by the formula (VII)13Is a suitable protecting group, RThreeThe compound protected by '' can be reacted and treated in the same manner as in the above reaction formula to produce the compound.
[0093]
The compounds of the present invention have excellent antibacterial activity, especially against Gram-positive bacteria. The compounds of the present invention also have the same antibacterial activity as susceptible bacteria against drug-resistant bacteria, such as MRSA, VRE, PRSP, etc., which are currently regarded as problematic in clinical practice. It can be used as a prophylactic / therapeutic agent for infectious diseases.
[0094]
The dose of the compound of the present invention varies depending on the type of the compound, the method of administration, the condition, the age, etc., but is usually about 2 to 5000 mg, preferably about 5 to 500 mg, particularly preferably 30 to 500 mg per 60 kg of body weight per day. 300 mg. The route of administration may be oral, but parenteral administration, especially intravenous administration, is recommended.
[0095]
When the compound of the present invention is used for pharmaceutical applications as described above, it is usually administered in the form of a preparation prepared by mixing with a carrier for preparation. As the carrier for the preparation, a non-toxic substance which is commonly used in the field of the preparation and which does not react with the compound of the present invention is used. Specifically, for example, citric acid, glutamic acid, glycine, lactose, inositol, glucose, mannitol, dextran, sorbitol, cyclodextrin, starch, partially pregelatinized starch, sucrose, methyl parahydroxybenzoate, propyl paraoxybenzoate, aluminum metasilicate Magnesium silicate, synthetic aluminum silicate, crystalline cellulose, sodium carboxymethylcellulose, hydroxypropyl starch, carboxymethylcellulose calcium, ion exchange resin, methylcellulose, gelatin, gum arabic, pullulan, hydroxypropylcellulose, low-substituted hydroxypropylcellulose, hydroxypropylmethylcellulose , Polyvinylpyrrolidone, polyvinyl alcohol, alginic acid, sodium alginate Light anhydrous silicic acid, magnesium stearate, talc, tragacanth, bentonite, veegum, carboxyvinyl polymer, titanium oxide, sorbitan fatty acid ester, sodium lauryl sulfate, glycerin, fatty acid glycerin ester, purified lanolin, glycerogelatin, polysorbate, macrogol, vegetable oil , Wax, propylene glycol, ethanol, benzyl alcohol, sodium chloride, sodium hydroxide, hydrochloric acid, water and the like.
[0096]
Dosage forms include tablets, capsules, granules, powders, syrups, suspensions, injections, suppositories, eye drops, ointments, coatings, inhalants, and the like. These preparations can be prepared according to a conventional method. In the case of a liquid preparation, it may be in the form of being dissolved or suspended in water or another appropriate medium at the time of use. Tablets and granules may be coated by a well-known method. In addition, these preparations may contain other therapeutically valuable ingredients.
[0097]
【Example】
Hereinafter, the present invention will be described more specifically with reference to Reference Examples, Examples, and Test Examples, but these do not limit the present invention. The compound of the present invention was identified by elemental analysis, mass spectrum, IR spectrum, NMR spectrum, high performance liquid chromatography (HPLC) and the like. The arrangement of the stereoisomers was determined by NOESY (Nuclear Overhauser effect spectroscopy).
[0098]
In order to simplify the description of the specification, the following abbreviations may be used in the examples and the tables in the examples.
[0099]
Symbols used as recrystallization solvents are E for ether, I for diisopropyl ether, AN for acetonitrile, AE for ethyl acetate, AC for acetone, CF for chloroform, MT for methanol, ET for ethanol and IP for isopropanol. .
[0100]
As symbols used in NMR, s is a singlet, d is a doublet, td is a triplet, t is a triplet, q is a quartet, qu is a quintet, m is a multiplet, br Smooth and J mean the coupling constant.
[0101]
Reference Example 1:
5-chloro-1-methylindole-3-carboxaldehyde
[0102]
Embedded image
A solution of 5.50 g of 5-chloroindole-3-carboxaldehyde in 50 ml of tetrahydrofuran was added dropwise to a suspension of 1.43 g of sodium hydride (60% oil dispersion) in 30 ml of tetrahydrofuran under ice-cooling, followed by stirring at room temperature for 30 minutes. . Next, 2.30 ml of methyl iodide was added, and the mixture was stirred at room temperature for 12 hours. A 10% aqueous citric acid solution was added, and the mixture was extracted with 80 ml of ethyl acetate. The extract was washed with 20 ml of saturated saline, dried over magnesium sulfate and concentrated. The obtained crude crystals were washed with diisopropyl ether to obtain 4.40 g of the desired product.
[0104]
Reference Example 2:
(1) 3-[(1E) -2-acetylvinyl] -5-chloroindole
[0105]
Embedded image
A solution of 2.00 g of 5-chloroindole-3-carboxaldehyde in 7 ml of acetone was added dropwise to a mixed solution of 1.35 g of sodium hydroxide in 1 ml of ethanol and 15 ml of water under ice-cooling. Heated to reflux for hours. The mixture was neutralized by adding 6 mol / l hydrochloric acid, and extracted with 30 ml of ethyl acetate. The extract was dried over magnesium sulfate and concentrated, and the residue was subjected to silica gel column chromatography, and eluted and purified with chloroform-methanol (50: 1) to obtain 1.23 g of the desired product as amorphous.
[0107]
Using the corresponding aldehyde in place of 5-chloroindole-3-carboxaldehyde in Reference Example 2 and reacting and treating in the same manner according to the method described in Reference Example 2 (1), the following compound was obtained.
[0108]
(2) 3-[(1E) -2-acetylvinyl] -5-chloro-1-methylindole
[0109]
Embedded image
(3) 2-[(1E) -2-acetylvinyl] -5-chloroindole
[0111]
Embedded image
(4) (3E) -4- [4- (3,4-dichlorobenzyloxy) phenyl] but-3-en-2-one
[0113]
Embedded image
Reference Example 3:
(1) 1- [4- (1-tert-butoxycarbonyl) piperidyl] -3-[(1E) -2- (5-chloroindol-3-yl) vinyl] propane-1,3-dione
[0115]
Embedded image
To a solution of 2,88 ml of 1,1,1,3,3,3-hexamethyldisilazane in 30 ml of tetrahydrofuran was added 4.36 ml of a 2.5 mol / l hexane solution of normal butyllithium in an argon atmosphere under ice-cooling. And stirred at the same temperature for 10 minutes. Next, a solution of 1.00 g of 3-[(1E) -2-acetylvinyl] -5-chloroindole obtained in Reference Example 2 (1) in 10 ml of tetrahydrofuran was added dropwise at -78 ° C, and the mixture was stirred at the same temperature for 30 minutes. did. A solution of 1.10 g of 1-tert-butoxycarbonylisonipecotic acid and 0.77 g of 1,1′-carbonylbis-1H-imidazole in 30 ml of tetrahydrofuran for 45 minutes at room temperature was added dropwise under an argon atmosphere. After returning, the mixture was stirred at the same temperature for 12 hours. 50 ml of a 10% citric acid solution was added, extracted with ethyl acetate, washed with saturated saline, dried over magnesium sulfate and concentrated. The residue was subjected to silica gel column chromatography, and eluted and purified with chloroform-methanol (50: 1) to obtain 1.00 g of the desired product as amorphous.
[0117]
Using the corresponding starting compounds, the compounds were reacted and treated in the same manner as in the above (1) to obtain the compounds shown in Table 1.
[0118]
[Table 1]
Reference example 4:
(1) 1-[(1E) -2- (5-chloro-1-methylindol-3-yl) vinyl] -3-ethoxycarbonylpropane-1,3-dione
[0120]
Embedded image
3-[(1E) -2-acetylvinyl] -5-chloro-1- obtained in Reference Example 2 (2) was added to a solution of 0.12 g of sodium ethoxide in 20 ml of diethyl ether under a stream of argon under ice cooling. 0.5 g of methylindole was added dropwise, and then 0.24 ml of diethyl oxalate was added, followed by stirring at room temperature for 12 hours. The mixture was acidified by adding 2 mol / l hydrochloric acid, extracted with 40 ml of ethyl acetate, washed with 10 ml of saturated saline, dried over anhydrous magnesium sulfate and concentrated. The residue was recrystallized from diisopropyl ether to obtain 0.20 g of the desired product.
[0122]
Using the corresponding starting compounds, the same reaction and treatment were carried out as in the above (1) to obtain the following compounds.
[0123]
(2) 1-[(1E) -2- (3,4-dichlorophenyl) vinyl] -3-ethoxycarbonylpropane-1,3-dione
[0124]
Embedded image
Reference example 5:
(1) 5-[(1E) -2- (5-chloro-1-methylindol-3-yl) vinyl] -3-ethoxycarbonylpyrazole
[0126]
Embedded image
4.30 g of 1-[(1E) -2- (5-chloro-1-methylindol-3-yl) vinyl] -3-ethoxycarbonylpropane-1,3-dione obtained in Reference Example 4 (1). To a solution of the above in 100 ml of ethanol was added 1.02 g of hydrazine monohydrate and 1.38 g of sodium acetate, and the mixture was refluxed for 3 hours. After evaporating the solvent, 20 ml of water was added, extracted with 100 ml of ethyl acetate, washed with saturated saline, dried over magnesium sulfate and concentrated. The residue was subjected to silica gel column chromatography, and eluted and purified with ethyl acetate-normal hexane (2: 1) to obtain 2.90 g of the desired product as amorphous.
[0128]
Using the corresponding starting compounds, the same reaction and treatment were carried out as in the above (1) to obtain the following compounds.
[0129]
(2) 1- (3-chlorophenyl) -5- [2- (ethoxycarbonyl) ethyl] -3- (2-naphthyl) pyrazole
[0130]
Embedded image
(3) 1- (3-chlorophenyl) -3- [2- (ethoxycarbonyl) ethyl] -5- (2-naphthyl) pyrazole
[0132]
Embedded image
(4) 5-[(1E) -2- (3,4-dichlorophenyl) vinyl] -3-ethoxycarbonylpyrazole
[0134]
Embedded image
Reference Example 6:
(1) 5-[(1E) -2- (5-chloro-1-methylindol-3-yl) vinyl] -3-hydroxymethylpyrazole
[0136]
Embedded image
A solution of 1.00 g of 5-[(1E) -2- (5-chloro-1-methylindol-3-yl) vinyl] -3-ethoxycarbonylpyrazole obtained in Reference Example 5 (1) in 30 ml of tetrahydrofuran was used. To a suspension of 0.25 g of lithium aluminum hydride in 30 ml of tetrahydrofuran was added dropwise under ice-cooling, followed by stirring at room temperature for 3 hours. A 1 mol / l aqueous solution of sulfuric acid was added dropwise to neutralize, and the mixture was extracted with 50 ml of ethyl acetate, washed with saturated saline, dried over magnesium sulfate, and concentrated. The residue was recrystallized from normal hexane to obtain 0.63 g of the desired product.
[0138]
Using the corresponding starting compounds, the same reaction and treatment were carried out as in the above (1) to obtain the following compounds.
(2) 5-[(1E) -2- (3,4-dichlorophenyl) vinyl] -3-hydroxymethylpyrazole
[0139]
Embedded image
Reference Example 7:
(1) 2- (3-chlorophenyl) -2-trimethylsilyloxyethanenitrile
[0141]
Embedded image
To a solution of 16.5 g of 3-chlorobenzaldehyde in 100 ml of dichloromethane was added dropwise 16.4 ml of trimethylsilyl cyanide under ice-cooling, then 50 mg of zinc iodide was added, and the mixture was stirred at room temperature for 48 hours. After evaporating the solvent, the residue was subjected to silica gel column chromatography, and eluted and purified with chloroform to obtain 22.5 g of the desired product as an oil.
[0143]
The corresponding compounds were reacted and treated in the same manner as in the above (1) to obtain the following compounds.
[0144]
(2) 2- (4-phenoxyphenyl) -2-trimethylsilyloxyethanenitrile
[0145]
Embedded image
(3) 2- (4-bromophenyl) -2-trimethylsilyloxyethanenitrile
[0147]
Embedded image
(4) 2- (3,5-dibenzyloxyphenyl) -2-trimethylsilyloxyethanenitrile
[0149]
Embedded image
Reference Example 8:
(1) 2- (3-benzyloxyphenyl) -1- (3-chlorophenyl) -2-hydroxyethane-1-one
[0151]
Embedded image
To a solution of 5.24 ml of 1,1,1,3,3,3-hexamethyldisilazane in 50 ml of tetrahydrofuran was added 10.0 ml of a 2.5 mol / l normal butyl lithium hexane solution under an argon atmosphere and ice cooling. And stirred at the same temperature for 10 minutes. Next, a solution of 6.00 g of 2- (3-chlorophenyl) -2-trimethylsilyloxyethanenitrile obtained in Reference Example 7 (1) in 15 ml of tetrahydrofuran was added dropwise at -78 ° C, and the mixture was stirred at the same temperature for 30 minutes. 20 ml of a tetrahydrofuran solution of 5.31 g of 3-benzyloxybenzaldehyde was added dropwise at the same temperature, and after returning to room temperature, the mixture was stirred at the same temperature for 12 hours. 50 ml of a 10% citric acid solution was added, extracted with 100 ml of ethyl acetate, washed with saturated saline, dried over magnesium sulfate and concentrated. The residue was dissolved in 10 ml of methanol, 12 ml of a 5% aqueous sulfuric acid solution was added, and the mixture was stirred at room temperature for 30 minutes. The reaction solution was extracted with 50 ml of diethyl ether, washed with 15 ml of a 0.5 mol / l aqueous sodium hydroxide solution, and the extract was dried over magnesium sulfate and concentrated. The residue was subjected to silica gel column chromatography, and eluted and purified with chloroform to obtain 8.60 g of the desired product as amorphous.
[0153]
Using the corresponding starting compounds, the compounds were reacted and treated in the same manner as in the above (1) to obtain the compounds shown in Table 2.
[0154]
[Table 2]
Reference Example 9:
(1) 2- (3-benzyloxyphenyl) -1- (3-chlorophenyl) ethane-1,2-dione
[0156]
Embedded image
A mixed solution of cupric sulfate (7.50 g) in pyridine (40 ml) and water (10 ml) was heated under reflux for 10 minutes, and the 2- (3-benzyloxyphenyl) -1- (3-chlorophenyl) obtained in Reference Example 8 (1) was obtained. 8.3 g of -2-hydroxyethan-1-one was added, and the mixture was heated under reflux for 1 hour. After evaporating the solvent, 20 ml of water was added, and the mixture was extracted with 50 ml of diethyl ether, dried over magnesium sulfate and concentrated. The residue was subjected to silica gel column chromatography, and eluted and purified with chloroform-normal hexane (3: 1) to obtain 5.82 g of the desired product as an oil.
[0158]
Using the corresponding starting compounds, the reaction and treatment were carried out in the same manner as in the above (1) to obtain the compounds shown in Table 3.
[0159]
[Table 3]
Reference example 10:
(1) 3-bromomethyl-1- (tert-butoxycarbonyl) -4- (tert-butyldimethylsilyloxy) -1H-indazole
[0161]
Embedded image
Literature (J. Med. Chem., 2000,43, 2664) to give the desired product.
[0163]
The corresponding compounds were reacted and treated in the same manner as in the above (1) to obtain the following compounds.
[0164]
(2) 3-bromomethyl-1- (tert-butoxycarbonyl) -4- (tert-butyldiphenylsilyloxy) -1H-indazole
[0165]
Embedded image
Reference Example 11:
(1) 3-amino-cyclohexanecarboxylic acid methyl ester (cis-trans mixture) monohydrochloride
[0167]
Embedded image
10.6 g of 3-aminocyclohexanecarboxylic acid (cis / trans mixture) was added to 75 ml of methanol, 20 ml of thionyl chloride was added dropwise at -10 ° C, and the mixture was stirred at room temperature for 12 hours. The solvent was distilled off and crystallized from acetonitrile to obtain 7.93 g of the desired product.
[0169]
Using the corresponding starting compounds, the same reaction and treatment were carried out as in the above (1) to obtain the following compounds.
[0170]
(2) 4-aminobutanoic acid ethyl ester / monohydrochloride
[0171]
Embedded image
Reference Example 12:
(1) 1-tert-butoxycarbonyl-3- [N-tert-butoxycarbonyl-N- (3-methoxycarbonylcyclohexyl)] aminomethyl-4-hydroxy-1H-indazole
[0173]
Embedded image
[Step 1]: 2.00 g of 3-bromomethyl-1- (tert-butoxycarbonyl) -4- (tert-butyldiphenylsilyloxy) -1H-indazole obtained in Reference Example 10 (2) is mixed with 80 ml of chloroform and methanol. 20 ml of the mixed medium, 2.06 g of 3-amino-cyclohexanecarboxylic acid methyl ester monohydrochloride obtained in Reference Example 11 (1) and 4.24 ml of a 10% aqueous sodium hydroxide solution were added at room temperature for 6 hours. Heated to reflux. 50 ml of water was added, extracted with 50 ml of chloroform, and the extract was dried over magnesium sulfate and concentrated. The residue was subjected to silica gel column chromatography, and eluted and purified with chloroform-methanol (50: 1) to give 1- (tert-butoxycarbonyl) -4- (tert-butyldiphenylsilyloxy) -3- (3-methoxy 1.44 g of carbonylcyclohexylaminomethyl) -1H-indazole was obtained as amorphous.
[0175]
[Step 2]: 1.44 g of 1- (tert-butoxycarbonyl) -4- (tert-butyldiphenylsilyloxy) -3- (3-methoxycarbonylcyclohexylaminomethyl) -1H-indazole obtained in the above step 1. Was added to 50 ml of acetonitrile, 0.54 g of di-tert-butyl dicarbonate was added at room temperature, and the mixture was stirred at room temperature for 12 hours. The solvent was distilled off, the residue was subjected to silica gel column chromatography, and eluted and purified with chloroform-methanol (50: 1) to give 1-tert-butoxycarbonyl-3- [N-tert-butoxycarbonyl-N- (3 -Methoxycarbonylcyclohexyl)] aminomethyl-4- (tert-butyldiphenylsilyloxy) -1H-indazole (1.29 g) was obtained as amorphous.
[0176]
[Step 3]: 1-tert-butoxycarbonyl-3- [N-tert-butoxycarbonyl-N- (3-methoxycarbonylcyclohexyl)] aminomethyl-4- (tert-butyldiphenylsilyl) obtained in the above step 2 1.29 g of oxy) -1H-indazole was added to 50 ml of tetrahydrofuran, and 17.4 ml of a 1.0 mol / l tetrabutylammonium fluoride solution in tetrahydrofuran was added at room temperature, followed by stirring at room temperature for 4 hours. 50 ml of water was added, extracted with 50 ml of ethyl acetate, and the extract was dried over magnesium sulfate and concentrated. The residue was subjected to silica gel column chromatography, and eluted and purified with chloroform to obtain 0.76 g of the desired product as amorphous.
[0177]
1H-NMR (CDClThree) δ: 1.22-1.33 (m, 2H), 1.40 (s, 9H), 1.66 (s, 9H), 1.73-1.96 (m, 6H), 2.30-2.39 (m, 1H), 3.64 (s, 3H) , 3.66-3.72 (m, 1H), 4.70-4.81 (m2H), 6.74 (d, J = 8.0Hz, 1H), 7.32 (t, J = 8.0Hz, 1H), 7.59 (d, J = 8.0Hz , 1H).
[0178]
Using the corresponding starting compounds, the reaction and treatment were carried out in the same manner as in the above (1) to obtain the compounds shown in Table 4.
[0179]
[Table 4]
R in Table 4 aboveTwo’Is RTwoContains a primary or secondary amino group,TwoIn which an appropriate protecting group is added to the amino group.
[0181]
Example 1:
5-[(1E) -2- (5-chloroindol-3-yl) vinyl] -1- (3-chlorophenyl) -3- (4-piperidyl) pyrazole • 1.25 hydrochloride monohydrate
[0182]
Embedded image
[Step 1]: 1- [4- (1-tert-butoxycarbonyl) piperidyl] -3-[(1E) -2- (5-chloroindol-3-yl) obtained in Reference Example 3 (1) Vinyl] propane-1,3-dione (1.00 g) in ethanol (30 ml) was mixed with 2 mol / l sodium hydroxide aqueous solution (2.20 ml) and 3-chlorophenylhydrazine monohydrochloride (0.83 g), and the mixture was heated under reflux for 12 hours. After concentrating the reaction solution, 20 ml of a 10% aqueous citric acid solution was added, extracted with 50 ml of ethyl acetate, washed with brine, dried over magnesium sulfate and concentrated. The residue was subjected to flash silica gel column chromatography, and eluted and purified with chloroform-methanol (50: 1) to give 3- [4- (1-tert-butoxycarbonyl) piperidyl] -5-[(1E) -2- 1.13 g of (5-chloroindol-3-yl) vinyl] -1- (3-chlorophenyl) pyrazole were obtained as amorphous.
[0184]
[Step 2]: 3- [4- (1-tert-butoxycarbonyl) piperidyl] -5-[(1E) -2- (5-chloroindol-3-yl) vinyl]-obtained in the above step 1. To a solution of 1.10 g of 1- (3-chlorophenyl) pyrazole in 30 ml of dichloromethane was added 5 ml of trifluoroacetic acid, and the mixture was stirred at room temperature for 3 hours. After evaporating the solvent, the residue was subjected to medium pressure column chromatography using CHP-20P, eluted and purified with water-acetonitrile (1: 2), and evaporated under reduced pressure. The residue was recrystallized from acetonitrile as a hydrochloride to obtain 0.23 g of the desired product.
158-161 ° C
[0185]
1H-NMR (DMSO-d6) δ: 1.85-2.15 (m, 4H), 2.97-3.08 (m, 3H), 3.26-3.34 (m, 2H), 6.73 (s, 1H), 6.80 (d, J = 15.8Hz, 1H), 7.13-7.77 (m, 9H), 8.83 (br, 1H), 9.02 (br, 1H), 11.69 (s, 1H).
[0186]
above1It was determined to be the title compound by 1 H-NMR spectrum data and NOESY.
[0187]
The reaction and treatment were carried out in the same manner as in Example 1 using the corresponding starting compounds to obtain the compounds of Examples 2 to 34 shown in Tables 5 to 9 below. In particular, when the stereoisomers are mixed, the stereoisomers were separated using the above-described separation method using medium pressure column chromatography using CHP-20P or fractional recrystallization.
[0188]
[Table 5]
[Table 6]
[Table 7]
[Table 8]
[Table 9]
Example 35:
5-methylaminomethyl-3- (4-phenoxyphenyl) -1- (4-trifluoromethylbenzyl) pyrazole monohydrochloride monohydrate
[0194]
Embedded image
[Step 1]: Using 1.00 g of 1- (N-tert-butoxycarbonyl) methylaminomethyl-3- (4-phenoxyphenyl) propane-1,3-dione obtained in Reference Example 3 (20), The same reaction and treatment were carried out using hydrazine monohydrate instead of 3-chlorophenylhydrazine monohydrochloride used in step 1 of Example 1, to give 3- (N-tert-butoxycarbonyl) methylaminomethyl- 1.20 g of a crude product of 5- (4-phenoxyphenyl) pyrazole was obtained.
[0196]
[Step 2]: A solution of 1.20 g of 3- (N-tert-butoxycarbonyl) methylaminomethyl-5- (4-phenoxyphenyl) -pyrazole obtained in Step 1 above in 10 ml of tetrahydrofuran is hydrogenated under ice cooling. The suspension was added to a suspension of sodium (60% oil dispersion) in 20 ml of tetrahydrofuran and stirred at room temperature for 30 minutes. Next, 0.94 ml of 4-trifluorobenzyl bromide was added, and the mixture was heated under reflux for 4 hours. 20 ml of 10% aqueous citric acid solution was added, extracted with 50 ml of ethyl acetate, washed with saturated saline, dried over magnesium sulfate and concentrated. The residue was subjected to silica gel column chromatography, and eluted and purified with normal hexane-acetone (10: 1) to give 5- (N-tert-butoxycarbonyl) methylaminomethyl-3- (4-phenoxyphenyl) -1-. 0.20 g of (4-trifluoromethylbenzyl) pyrazole was obtained as amorphous.
[0197]
[Step 3]: 0.20 g of 5- (N-tert-butoxycarbonyl) methylaminomethyl-3- (4-phenoxyphenyl) -1- (4-trifluoromethylbenzyl) pyrazole obtained in the above step 2 The reaction and treatment were carried out in the same manner as in Step 2 of Example 1 and lyophilized to obtain 0.21 g of the desired product.
[0198]
1H-NMR (DMSO-d6) δ: 2.60 (s, 3H), 4.28 (s, 2H), 5.64 (s, 2H), 6.98 (s, 1H), 7.03-7.06 (m, 4H), 7.16 (t , J = 7.2Hz, 1H), 7.37-7.42 (m, 4H), 7.73-7.77 (m, 4H), 9.40 (br, 2H).
[0199]
above1It was determined to be the title compound by 1 H-NMR spectrum data and NOESY.
[0200]
The reaction and treatment were carried out in the same manner as in Example 35 using the corresponding starting compounds to obtain the compounds of Examples 36 to 44 shown in Table 10. In particular, when the stereoisomers are mixed, the stereoisomers were separated by a separation method using medium pressure column chromatography using CHP-20P or a fractional recrystallization.
[0201]
[Table 10]
Example 45:
1- (3-chlorophenyl) -5-[(1E) -2- (2,6-dichlorophenyl) vinyl] -3- (4-piperidyl) pyrazole monohydrochloride
[0203]
Embedded image
[Step 1]: 1- [4- (1-tert-butoxycarbonyl) piperidyl] -3-[(1E) -2- (2,6-dichlorophenyl) vinyl] propane obtained in Reference Example 3 (11) Using 2.00 g of -1,3-dione, the same reaction and treatment was carried out using hydrazine monohydrate instead of 3-chlorophenylhydrazine monohydrochloride used in Step 1 of Example 1, and 1.13 g of [4- (1-tert-butoxycarbonyl) piperidyl] -5-[(1E) -2- (2,6-dichlorophenyl) vinyl] pyrazole was obtained as amorphous.
[0205]
[Step 2]: 1.05 g of 3- [4- (1-tert-butoxycarbonyl) piperidyl] -5-[(1E) -2- (2,6-dichlorophenyl) vinyl] pyrazole obtained in Step 1 above. To a solution of the above in 40 ml of dichloromethane, 0.78 g of 3-chlorophenylboronic acid, 0.68 g of cupric acetate (anhydrous), 2.00 g of molecular sieve (4A) and 0.40 ml of pyridine were added, and the mixture was stirred at room temperature for 24 hours. After filtering off the insoluble matter and distilling off the solvent, 30 ml of a saturated aqueous solution of sodium hydrogencarbonate was added, and the mixture was extracted with 50 ml of ethyl acetate, dried over magnesium sulfate and concentrated. 0.98 g of a mixture of 1- (3-chlorophenyl) -5-[(1E) -2- (2,6-dichlorophenyl) vinyl] pyrazole and its stereoisomer was obtained as amorphous.
[0206]
[Step 3]: 3- [4- (1-tert-butoxycarbonyl) piperidyl] -1- (3-chlorophenyl) -5-[(1E) -2- (2,6) obtained in the above step 2. -Dichlorophenyl) vinyl] pyrazole and 0.96 g of a stereoisomer mixture were reacted and treated in the same manner as in Step 2 of Example 1, and recrystallized from acetonitrile-ethanol as a hydrochloride to obtain 0.51 g of the desired product. Obtained.
Melting point 242-245 ° C
[0207]
1H-NMR (DMSO-d6) δ: 1.82-2.22 (m, 4H), 2.98-3.38 (m, 5H), 6.90 (s, 1H), 6.94 (d, J = 16.0 Hz, 1H), 7.22 (d , J = 16.0Hz, 1H), 7.33-7.61 (m, 7H), 8.93 (br, 2H).
[0208]
above1It was determined to be the title compound by 1 H-NMR spectrum data and NOESY.
[0209]
The reaction and treatment were carried out in the same manner as in Example 45 using the corresponding starting compounds, to obtain the compounds of Examples 46 to 49 shown in Table 11. In particular, when the stereoisomers are mixed, the stereoisomers were separated by a separation method using medium pressure column chromatography using CHP-20P or a fractional recrystallization.
[0210]
[Table 11]
Example 50:
4-chloro-1- (3-chlorophenyl) -5- (2-naphthyl) -3- (4-piperidyl) pyrazole monohydrochloride
[0212]
Embedded image
3- [4- (1-tert-butoxycarbonyl) piperidyl] -1- (3) obtained by treating with the corresponding starting compound in the same manner as described in Step 1 and Step 2 of Example 45. To a solution of 0.30 g of -chlorophenyl) -5- (2-naphthyl) pyrazole in 20 ml of chloroform was added 0.06 ml of sulfuryl chloride under ice-cooling, followed by heating under reflux for 1 hour. 15 ml of a saturated aqueous sodium hydrogen carbonate solution was added, extracted with 20 ml of chloroform, the extract was dried over magnesium sulfate, concentrated, and recrystallized from ethanol-acetonitrile as a hydrochloride to obtain 0.08 g of the desired product. 218-221 ° C
[0214]
1H-NMR (DMSO-d6) δ: 2.02-2.06 (m, 4H), 3.08-3.41 (m, 5H), 7.09 (d, J = 4.0 Hz, 1H), 7.27-7.44 (m, 5H), 7.56 -7.63 (m, 2H), 7.94-8.01 (m, 3H), 8.76 (br, 2H).
[0215]
The reaction and treatment were carried out in the same manner as in Example 50 using the corresponding reagents, whereby the compounds of Examples 51 and 52 were obtained.
[0216]
Example 51:
4-bromo-1- (3-chlorophenyl) -5- (2-naphthyl) -3- (4-piperidyl) pyrazole monohydrobromide
243-246 ° C
[0217]
Embedded image
Example 52:
1- (3-chlorophenyl) -4-fluoro-5- (2-naphthyl) -3- (4-piperidyl) pyrazole monohydrochloride
197-200 ° C
[0219]
Embedded image
Example 53:
(A) 1- (3-chlorophenyl) -5- [3- (2-hydroxyethylamino) propyl] -3- (2-naphthyl) pyrazole monohydrochloride
[0221]
Embedded image
(B) 1- (3-chlorophenyl) -3- [3- (2-hydroxyethylamino) propyl] -5- (2-naphthyl) pyrazole monohydrochloride
[0223]
Embedded image
[Step 1]: 1- (3-chlorophenyl) -5- [2- (ethoxycarbonyl) ethyl] -3- (2-naphthyl) pyrazole and 1- (3-chlorophenyl) -5- [2- (ethoxycarbonyl) ethyl] obtained in Reference Example 5 (2) and (3) To 1.50 g of a mixture of (3-chlorophenyl) -3- [2- (ethoxycarbonyl) ethyl] -5- (2-naphthyl) pyrazole was added 4.48 ml of ethanolamine, and the mixture was stirred at 120 ° C for 3 hours. 30 ml of a 10% aqueous citric acid solution was added, extracted with 50 ml of chloroform, washed with brine, dried over magnesium sulfate and concentrated. The residue was subjected to silica gel column chromatography, and eluted and purified with chloroform-methanol (25: 1) to give 1- (3-chlorophenyl) -5- [2- (2-hydroxyethylaminocarbonyl) ethyl] -3-. 1.39 g of a mixture of (2-naphthyl) pyrazole and 1- (3-chlorophenyl) -3- [2- (2-hydroxyethylaminocarbonyl) ethyl] -5- (2-naphthyl) pyrazole was obtained (amorphous ).
[0225]
[Step 2]: 1- (3-chlorophenyl) -5- [2- (2-hydroxyethylaminocarbonyl) ethyl] -3- (2-naphthyl) pyrazole obtained in the above step 1 and 1- (3- A mixture of 0.95 g of a mixture of chlorophenyl) -3- [2- (2-hydroxyethylaminocarbonyl) ethyl] -5- (2-naphthyl) pyrazole in 30 ml of tetrahydrofuran is 4.52 ml of a 1.0 mol / l solution of diborane in tetrahydrofuran. Was added and stirred at room temperature for 12 hours. Next, 3 ml of a 30% hydrochloric acid-ethanol solution was added, and the mixture was heated under reflux for 1 hour. After evaporating the solvent, 10 ml of a 10 mol / l aqueous sodium hydroxide solution was added, extracted with 30 ml of chloroform, washed with saturated saline, dried over anhydrous magnesium sulfate and concentrated. The residue was subjected to medium pressure column chromatography using CHP-20P, eluted with 0.02 mol / l aqueous hydrochloric acid-acetonitrile (3: 2), separated and purified, and then lyophilized to obtain 0.04 g of the desired product (a) and 0.31 g of the target product (b) was obtained as amorphous.
[0226]
Example 53 (a):
1H-NMR (DMSO-d6) δ: 2.02-2.07 (m, 2H), 2.87 (t, J = 7.2 Hz, 2H), 2.99 (br, 4H), 3.65 (br, 1H), 5.26 (br, 1H ), 7.04 (s, 1H), 7.49-7.62 (m, 5H), 7.74 (br, 1H), 7.88-8.06 (m, 4H), 8.38 (br, 1H), 8.66 (br, 2H).
[0227]
Example 53 (b):
1H-NMR (CDCl3) δ: 2.38 (br, 2H), 2.91 (t, J = 7.1 Hz, 2H), 3.16 (br, 4H), 3.96 (br, 2H), 6.49 (s, 1H), 7.07- 7.26 (m, 4H), 7.45-7.53 (m, 3H), 7.71-7.82 (m, 4H), 9.32 (br, 2H).
[0228]
above1It was determined to be the title compound by 1 H-NMR spectrum data and NOESY.
[0229]
Example 54:
5-[(1E) -2- (5-chloro-1-methylindol-3-yl) vinyl] -3- (3-methoxycarbonylcyclohexylaminomethyl) -pyrazole monohydrochloride
[0230]
Embedded image
[Step 1]: 0.30 g of 5-[(1E) -2- (5-chloro-1-methylindol-3-yl) vinyl] -3-hydroxymethylpyrazole obtained in Reference Example 6 (1) To a 20 ml solution of dichloromethane, 0.63 ml of a 2.0 mol / l oxalyl chloride solution in dichloromethane was added dropwise, and the mixture was heated under reflux for 30 minutes. The solvent was distilled off to obtain 0.40 g of a crude product of 3-chloromethyl-5-[(1E) -2- (5-chloro-1-methylindol-3-yl) vinyl] pyrazole.
[0232]
[Step 2]: 0.40 g of a crude product of 3-chloromethyl-5-[(1E) -2- (5-chloro-1-methylindol-3-yl) vinyl] pyrazole obtained in Step 1 above. Was added to a mixed solvent of 20 ml of chloroform and 5 ml of methanol, and 1.00 g of 3-aminocyclohexanecarboxylic acid methyl ester monohydrochloride obtained in Reference Example 11 (1) and 2.09 ml of a 10% aqueous sodium hydroxide solution were added at room temperature. The mixture was heated under reflux for 4 hours. 20 ml of water was added, extracted with 20 ml of chloroform, and the extract was dried over magnesium sulfate and concentrated. The residue was subjected to silica gel column chromatography whose surface was treated with a base, eluted with chloroform-methanol (20: 1), separated and purified, and evaporated under reduced pressure. The residue was recrystallized from acetonitrile-ether as a hydrochloride to obtain 0.10 g of one of the desired cis-trans on the low polarity side.
175-178 ° C
[0233]
1H-NMR (DMSO-d6) δ: 1.16-1.33 (m, 4H), 1.75-2.48 (m, 5H), 3.03 (br, 1H), 3.60 (s, 3H), 3.81 (s, 3H), 4.13 (br, 2H), 6.61 (s, 1H), 6.87-6.97 (m, 1H), 7.24 (d, J = 8.9Hz, 1H), 7,32 (d, J = 16.6Hz, 1H), 7.53 ( d, J = 8.9Hz, 1H), 7.76 (d, J = 16.6Hz, 1H), 7.92 (br, 1H), 8.11 (br, 2H), 9.31 (br, 1H).
[0234]
The reaction and treatment were carried out in the same manner as in Example 54 using the corresponding starting compounds, to give the compounds of Examples 55 to 57 shown in Table 12.
[0235]
[Table 12]
Example 58:
1- (3-chlorophenyl) -5-[(1Z) -2- (2,6-dichlorophenyl) vinyl] -3- (4-piperidyl) pyrazole monohydrochloride
[0237]
Embedded image
0.41 g of 1- (3-chlorophenyl) -5-[(1E) -2- (2,6-dichlorophenyl) vinyl] -3- (4-piperidyl) pyrazole monohydrochloride obtained in Example 45 was added. It was dissolved in 400 ml of methanol and irradiated with natural light at room temperature for 5 hours. After evaporating the solvent, the residue was recrystallized from acetonitrile-diethyl ether as a hydrochloride to obtain 0.25 g of the desired product.
82-85 ° C
[0239]
1H-NMR (CDCl3) δ: 1.76-1.83 (m, 4H), 2.63-2.73 (m, 3H), 3.06-3.10 (m, 2H), 5.54 (s, 1H), 6.51 (d, J = 12.1 Hz) , 1H), 6.59 (d, J = 12.1Hz, 1H), 7.20-7.26 (m, 1H), 7.34-7.41 (m, 5H), 7.56 (br, 1H).
[0240]
above1It was determined to be the title compound by 1 H-NMR spectrum data.
[0241]
The reaction and treatment were carried out in the same manner as in Example 58 using the corresponding starting compounds to obtain the compounds of Examples 59 to 61 shown in Table 13.
[0242]
[Table 13]
Example 62:
5- (3-benzyloxyphenyl) -4- (3-chlorophenyl) -2- (4-piperidyl) oxazole / 1 fumarate
[0244]
Embedded image
[Step 1]: 1.70 g of 2- (3-benzyloxyphenyl) -1- (3-chlorophenyl) -2-hydroxyethane-1-one obtained in Reference Example 8 (1), N- (tert- A solution of 1.22 g of butoxycarbonyl) isonipecotic acid, 0.94 g of 1- (3-dimethylaminopropyl) -3-ethylcarbodiimide monohydrochloride and 0.59 g of 4-dimethylaminopyridine in 40 ml of dichloromethane was stirred at room temperature for 6 hours. . The reaction solution was added with a 10% aqueous citric acid solution (30 ml), extracted with chloroform (50 ml), dried over magnesium sulfate, and concentrated. 1.20 g of a crude product of (1-tert-butoxycarbonyl) piperidine-4-carboxylate was obtained.
[0246]
[Step 2]: 2- (3-chlorophenyl) -2-oxo-1- (3-benzyloxyphenyl) ethyl-1- (1-tert-butoxycarbonyl) piperidine-4-carboxy obtained in the above step 1. 1.20 g of the crude product of the rate was dissolved in 40 ml of acetic acid, 0.93 g of ammonium acetate was added, and the mixture was refluxed for 6 hours. After evaporating the solvent, the residue was subjected to medium pressure column chromatography using CHP-20P, eluted with water-acetonitrile (15:85), separated and purified, and distilled under reduced pressure. It was recrystallized from ethanol-diethyl ether as a fumarate to obtain 1.04 g of the desired product.
123-126 ° C
[0247]
1H-NMR (CDCl3) δ: 1.81-1.93 (m, 2H), 2.10-2.14 (m, 2H), 2.74-2.81 (m, 2H), 3.00-3.05 (m, 2H), 3.19-3.23 (m, 2H), 5.01 (s, 2H), 6.95-6.97 (m, 1H), 7.15-7.18 (m, 2H), 7.23-7.39 (m, 10H), 7.50-7.53 (m, 1H), 7.70 (br, 1H).
[0248]
The reaction and treatment were carried out in the same manner as in Example 62 using the corresponding starting compounds to obtain the compounds of Examples 63 and 64 shown in Table 14.
[0249]
[Table 14]
Example 65:
5- (3-benzyloxyphenyl) -4- (3-chlorophenyl) -2- (4-piperidyl) imidazole.dihydrochloride.1.40 hydrate
[0251]
Embedded image
3.00 g of 2- (3-benzyloxyphenyl) -1- (3-chlorophenyl) ethane-1,2-dione obtained in Reference Example 9 (1), N- (tert-butoxycarbonyl) isonipecotino A solution of 1.82 g of aldehyde and 5.27 g of ammonium acetate in 50 ml of acetic acid was stirred at 60 ° C. for 2 hours. After evaporating the solvent, 30 ml of water was added and the mixture was extracted with 60 ml of chloroform, washed with 20 ml of a saturated aqueous solution of sodium hydrogen carbonate, dried over magnesium sulfate and concentrated. The residue was subjected to medium pressure column chromatography using CHP-20P, eluted and purified with water-acetonitrile (1: 4), and recrystallized from water-acetonitrile as a hydrochloride to obtain 1.10 g of the desired product. .
140-141 ° C
[0253]
1H-NMR (DMSO-d6) δ: 2.06-2.34 (m, 4H), 2.97-3.16 (m, 2H), 3.39-3.53 (m, 3H), 5.11 (s, 2H), 7.03 (d, J = 8.0Hz, 1H), 7.09-7.21 (m, 2H), 7.35-7.53 (m, 9H), 7.64 (br, 1H), 9.24 (br, 2H).
[0254]
The reaction and treatment were carried out in the same manner as in Example 65 using the corresponding starting compounds, to obtain the compounds of Examples 66 to 69 shown in Table 15.
[0255]
[Table 15]
Example 70:
4- (3,4-dichlorobenzyloxy) -3- (3-methoxycarbonylcyclohexylaminomethyl) -1H-indazole monohydrochloride
[0257]
Embedded image
[Step 1]: 1-tert-butoxycarbonyl-3- [N-tert-butoxycarbonyl-N- (3-methoxycarbonylcyclohexyl)] aminomethyl-4-hydroxy-1H obtained in Reference Example 12 (1). 1.63 g of indazole was added to a suspension of 0.14 g of sodium hydride (60% oil dispersion) in 50 ml of dimethylformamide under ice cooling, and the mixture was stirred at room temperature for 30 minutes, and then stirred with 0.34 g of 3,4-dichlorobenzyl chloride. 52 ml was added, and the mixture was stirred at room temperature for 12 hours. 30 ml of a saturated aqueous solution of ammonium chloride was added, and the mixture was extracted twice with 50 ml of diethyl ether. The extract was dried over magnesium sulfate and concentrated. The residue was subjected to silica gel column chromatography, eluted and purified with normal hexane-ethyl acetate (4: 1), and purified with 1-tert-butoxycarbonyl-3- [N-tert-butoxycarbonyl-N- (3-methoxycarbonyl). Cyclohexyl)] aminomethyl-4- (3,4-dichlorobenzyloxy) -1H-indazole (1.50 g) was obtained as amorphous.
[0259]
[Step 2]: 1-tert-butoxycarbonyl-3- [N-tert-butoxycarbonyl-N- (3-methoxycarbonylcyclohexyl)] aminomethyl-4- (3,4-dichloro obtained in the above step 1 Using 0.30 g of benzyloxy) -1H-indazole, the reaction and treatment were carried out in the same manner as in Step 2 of Example 1, and the resulting product was crystallized from acetonitrile-diethyl ether as a hydrochloride, thereby obtaining 0.16 g of the desired product as a cis-trans mixture. Obtained.
[0260]
1H-NMR (DMSO-d6) δ: 1.16-1.48 (m, 4H), 1.74-2.35 (m, 5H), 3.06 (br, 1H), 3.60 (s, 3H), 4.53 (br, 2H), 5.26 (s, 2H), 6.66 (d, J = 8.0Hz, 1H), 7.14 (d, J = 8.0Hz, 1H), 7.28-7.38 (m, 1H), 7.58 (dd, J = 1.7Hz, J = 8.2Hz, 1H), 7.69 (d, J = 8.2Hz, 1H), 7.88 (d, J = 1.7Hz, 1H), 9.17 (br, 2H), 13.43 (s, 1H).
[0261]
The reaction and treatment were carried out in the same manner as in Example 70 using the corresponding starting compounds to obtain the compounds of Examples 71 to 80 shown in Table 16.
[0262]
[Table 16]
[Table 17]
Example 81:
4- (2,6-dichlorophenethyloxy) -3- (3-methoxycarbonylcyclohexylaminomethyl) -1H-indazole monohydrochloride
[0265]
Embedded image
[Step 1]: 1-tert-butoxycarbonyl-3- [N-tert-butoxycarbonyl-N- (3-methoxycarbonylcyclohexyl)] aminomethyl-4-hydroxy-1H obtained in Reference Example 12 (1). To a solution of 150 mg of indazole and 63 mg of 2- (2,6-dichlorophenyl) ethanol in 20 ml of tetrahydrofuran were added 110 mg of triphenylphosphine and 65 μl of diisopropylazodicarboxylate, and the mixture was heated under reflux for 2 hours. After evaporating the solvent, the residue was subjected to flash silica gel column chromatography, eluted and purified with n-hexane-ethyl acetate (4: 1), and purified with 1-tert-butoxycarbonyl-3- [N-tert-butoxycarbonyl-N -(3-Methoxycarbonylcyclohexyl)] aminomethyl-4- (2,6-dichlorophenethyloxy) -1H-indazole 110 mg was obtained as an amorphous.
[0267]
[Step 2]: 1-tert-butoxycarbonyl-3- [N-tert-butoxycarbonyl-N- (3-methoxycarbonylcyclohexyl)] aminomethyl-4- (2,6-dichloro obtained in the above step 1. The reaction and treatment were carried out in the same manner as in Step 2 of Example 1 using 110 mg of phenethyloxy) -1H-indazole, and the product was crystallized as a hydrochloride from acetonitrile-diethyl ether to obtain 30 mg of the desired product as a cis-trans mixture.
[0268]
1H-NMR (DMSO-d6) δ: 1.19-1.52 (m, 4H), 1.84-2.50 (m, 5H), 3.22 (br, 1H), 3.50 (t, J = 7.60 Hz, 2H), 3.60 (s , 3H), 4.35 (t, J = 7.6Hz, 2H), 4.52 (s, 2H), 6.69 (d, J = 8.0Hz, 1H), 7.12 (d, J = 8.0Hz, 1H), 7.29 (t , J = 8.0Hz, 1H), 7.35 (t, J = 8.0Hz, 1H), 7.53 (d, J = 8.0Hz, 2H), 9.13 (s, 2H), 13.37 (s, 1H).
[0269]
The reaction and treatment were carried out in the same manner as in Example 81 using the corresponding starting compounds to obtain the compounds of Examples 82 to 84 shown in Table 18.
[0270]
[Table 18]
An antibacterial test was performed on the compound of the present invention thus produced. Hereinafter, the test method and the result will be described.
[0272]
Test example: Antibacterial activity in vitro (MIC: μg / ml)
[0273]
The minimum inhibitory concentration (MIC: μg / ml) of the compound of the present invention was measured according to the standard method defined by The National Committee for Clinical Laboratory Standards (NCCLS). Tables 19 and 20 show the antimicrobial results of representative compounds of the present invention.
[0274]
[Table 19]
[Table 20]
Strain A: Staphylococcus aureus FDA 209P
Strain B: Staphylococcus aureus KMP9 (MRSA)*
Strain C: Enterococcus faecium ATCC19434
Strain D: Enterococcus faecium 1778 (VRE)*
Strain E: Streptococcus pneumoniae ATCC49619
Strain F: Streptococcus pneumoniae KT2524 (PRSP)*
*: Clinical isolate.
All of the above strains are Gram-positive bacteria.
[0277]
As is clear from Tables 19 and 20, the compounds of the present invention have an extremely excellent antibacterial action against multidrug-resistant bacteria (MRSA, VRE, PRSP, etc.), and are therefore useful as antibacterial agents.
[0278]
【The invention's effect】
The compounds of the present invention have excellent antibacterial activity, especially against Gram-positive bacteria. The compounds of the present invention also have the same antibacterial activity as susceptible bacteria against drug-resistant bacteria, such as MRSA, VRE, PRSP, etc., which are currently regarded as problematic in clinical practice. It can be used as a prophylactic / therapeutic agent for infectious diseases.
Claims (7)
R2は、アミノアルキル基;メチルアミノアルキル基;ヒドロキシアルキルアミノアルキル基;アルコキシカルボニルアルキルアミノアルキル基;シクロヘキサン環上が置換されていてもよいシクロヘキシルアミノアルキル基(該置換基は、アルコキシカルボニル基またはヒドロキシアルキル基である);ピペリジル基;水酸基で置換されていてもよいピロリジノ基;ピペラジン環上がアルキルスルファモイル基で置換されていてもよいピペラジニルアルキル基またはアルコキシカルボニルピペリジノアルキル基を意味し、
Aは、窒素原子または炭素原子を意味し、
Bは、C−R5、NHまたは酸素原子を意味する。
但し、Aが窒素原子のときは、BはC−R5を意味し、Aが炭素原子のときは、BはNHまたは酸素原子を意味する。
また、R3およびR4は、どちらか一方が水素原子;アルキル基;1または2個のハロゲン原子で置換されていてもよいフェニル基;フェノキシフェニル基;ベンジルオキシフェニル基;ピリジル基;1もしくは2個のハロゲン原子またはトリフルオロメチル基で置換されていてもよいベンジル基あるいはベンジル基で置換されていてもよいピペリジル基で置換しており、他方は水素原子または置換基は存在しない。
R5は、水素原子、アルキル基またはハロゲン原子を意味し、
Aが窒素原子であって、かつBがC−R5であるとき、R1およびR5は一緒になってベンゼン環を形成していてもよく、形成されたベンゼン環は−O−R1’で置換していてもよく、R1’は、置換されていてもよいフェニルアルキル基(該置換基は、1もしくは2個のハロゲン原子,アルキル基またはアミノ基である)を意味する。〕
で表される化合物またはその製薬学的に許容される塩。The following general formula (I)
R 2 represents an aminoalkyl group; a methylaminoalkyl group; a hydroxyalkylaminoalkyl group; an alkoxycarbonylalkylaminoalkyl group; a cyclohexylaminoalkyl group optionally substituted on a cyclohexane ring (the substituent is an alkoxycarbonyl group or A piperidyl group; a pyrrolidino group optionally substituted with a hydroxyl group; a piperazinylalkyl group or an alkoxycarbonylpiperidinoalkyl group which may be substituted on the piperazine ring with an alkylsulfamoyl group. Means
A represents a nitrogen atom or a carbon atom,
B means C—R 5 , NH or an oxygen atom.
However, when A is a nitrogen atom, B means CR 5 , and when A is a carbon atom, B means NH or an oxygen atom.
R 3 or R 4 is a hydrogen atom; an alkyl group; a phenyl group optionally substituted with one or two halogen atoms; a phenoxyphenyl group; a benzyloxyphenyl group; a pyridyl group; It is substituted with two halogen atoms or a benzyl group which may be substituted with a trifluoromethyl group or a piperidyl group which may be substituted with a benzyl group, and the other has no hydrogen atom or substituent.
R 5 represents a hydrogen atom, an alkyl group or a halogen atom,
When A is a nitrogen atom and B is C—R 5 , R 1 and R 5 may together form a benzene ring, and the formed benzene ring is —O—R 1 R 1 ′ represents an optionally substituted phenylalkyl group (the substituent is one or two halogen atoms, an alkyl group or an amino group). ]
Or a pharmaceutically acceptable salt thereof.
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2002364578A JP2004196678A (en) | 2002-12-17 | 2002-12-17 | Pyrazole derivatives |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2002364578A JP2004196678A (en) | 2002-12-17 | 2002-12-17 | Pyrazole derivatives |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| JP2004196678A true JP2004196678A (en) | 2004-07-15 |
Family
ID=32762358
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2002364578A Pending JP2004196678A (en) | 2002-12-17 | 2002-12-17 | Pyrazole derivatives |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP2004196678A (en) |
Cited By (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2011147910A1 (en) * | 2010-05-27 | 2011-12-01 | Laboratorios Del Dr. Esteve, S.A. | Pyrazole compounds as sigma receptor inhibitors |
| JP2014514299A (en) * | 2011-04-08 | 2014-06-19 | ファイザー・インク | Imidazole, pyrazole and triazole derivatives useful as antibacterial agents |
| CN105272900A (en) * | 2015-01-21 | 2016-01-27 | 南通大学 | Bis(indolyl) derivatives, preparation method and applications |
| US9757358B2 (en) | 2010-02-04 | 2017-09-12 | Laboratorios Del Dr. Esteve, S.A. | Sigma ligands for potentiating the analgesic effect of opioids and opiates in post-operative pain and attenuating the dependency thereof |
| US9782483B2 (en) | 2010-05-21 | 2017-10-10 | Laboratories Del Dr. Esteve, S.A. | Sigma ligands for the prevention and/or treatment of emesis induced by chemotherapy or radiotherapy |
| US9789117B2 (en) | 2011-05-18 | 2017-10-17 | Laboratorios Del Dr. Esteve, S.A. | Use of sigma ligands in diabetes type-2 associated pain |
| US9789115B2 (en) | 2010-08-03 | 2017-10-17 | Laboratorios Del Dr. Esteve, S.A. | Use of sigma ligands in opioid-induced hyperalgesia |
| US9844516B2 (en) | 2010-02-04 | 2017-12-19 | Laboratorios De Dr. Esteve | Sigma ligands for use in the prevention and/or treatment of post-operative pain |
| US9914705B2 (en) | 2008-04-25 | 2018-03-13 | Laboratorios Del Dr. Esteve, S.A. | 1-aryl-3-aminoalkoxy pyrazoles as sigma ligands enhancing analgesic effect of opioids and attenuating the dependency thereof |
| US9931346B2 (en) | 2013-12-17 | 2018-04-03 | Laboratorios Del Dr. Esteve S.A. | Serotonin-norepinephrine reuptake inhibitors (SNRIs) and Sigma receptor ligands combinations |
| CN112778208A (en) * | 2021-01-08 | 2021-05-11 | 中南民族大学 | Dihydropyrazole MurA enzyme inhibitor molecule and preparation method and application thereof |
| US11345694B2 (en) | 2017-11-03 | 2022-05-31 | Discuva Ltd. | Antibacterial compounds |
| WO2022186362A1 (en) * | 2021-03-03 | 2022-09-09 | 白鳥製薬株式会社 | Method for producing pyrazole compound |
-
2002
- 2002-12-17 JP JP2002364578A patent/JP2004196678A/en active Pending
Cited By (20)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9914705B2 (en) | 2008-04-25 | 2018-03-13 | Laboratorios Del Dr. Esteve, S.A. | 1-aryl-3-aminoalkoxy pyrazoles as sigma ligands enhancing analgesic effect of opioids and attenuating the dependency thereof |
| US9757358B2 (en) | 2010-02-04 | 2017-09-12 | Laboratorios Del Dr. Esteve, S.A. | Sigma ligands for potentiating the analgesic effect of opioids and opiates in post-operative pain and attenuating the dependency thereof |
| US9844516B2 (en) | 2010-02-04 | 2017-12-19 | Laboratorios De Dr. Esteve | Sigma ligands for use in the prevention and/or treatment of post-operative pain |
| US9782483B2 (en) | 2010-05-21 | 2017-10-10 | Laboratories Del Dr. Esteve, S.A. | Sigma ligands for the prevention and/or treatment of emesis induced by chemotherapy or radiotherapy |
| US9181195B2 (en) | 2010-05-27 | 2015-11-10 | Laboratorios Del Dr. Esteve, S.A. | Sigma receptor inhibitors |
| EP2395003A1 (en) * | 2010-05-27 | 2011-12-14 | Laboratorios Del. Dr. Esteve, S.A. | Pyrazole compounds as sigma receptor inhibitors |
| CN103003267B (en) * | 2010-05-27 | 2018-05-25 | 埃斯特韦实验室有限公司 | Sigma receptor inhibitors |
| JP2016065092A (en) * | 2010-05-27 | 2016-04-28 | ラボラトリオス・デル・ドクター・エステベ・ソシエテ・アノニム | Pyrazole compound as sigma receptor inhibitor |
| AU2011257208B2 (en) * | 2010-05-27 | 2016-05-19 | Laboratorios Del Dr. Esteve, S.A. | Pyrazole compounds as sigma receptor inhibitors |
| WO2011147910A1 (en) * | 2010-05-27 | 2011-12-01 | Laboratorios Del Dr. Esteve, S.A. | Pyrazole compounds as sigma receptor inhibitors |
| JP2013528171A (en) * | 2010-05-27 | 2013-07-08 | ラボラトリオス・デル・ドクター・エステベ・ソシエテ・アノニム | Pyrazole compounds as sigma receptor inhibitors |
| CN103003267A (en) * | 2010-05-27 | 2013-03-27 | 埃斯特韦实验室有限公司 | Sigma receptor inhibitors |
| US9789115B2 (en) | 2010-08-03 | 2017-10-17 | Laboratorios Del Dr. Esteve, S.A. | Use of sigma ligands in opioid-induced hyperalgesia |
| JP2014514299A (en) * | 2011-04-08 | 2014-06-19 | ファイザー・インク | Imidazole, pyrazole and triazole derivatives useful as antibacterial agents |
| US9789117B2 (en) | 2011-05-18 | 2017-10-17 | Laboratorios Del Dr. Esteve, S.A. | Use of sigma ligands in diabetes type-2 associated pain |
| US9931346B2 (en) | 2013-12-17 | 2018-04-03 | Laboratorios Del Dr. Esteve S.A. | Serotonin-norepinephrine reuptake inhibitors (SNRIs) and Sigma receptor ligands combinations |
| CN105272900A (en) * | 2015-01-21 | 2016-01-27 | 南通大学 | Bis(indolyl) derivatives, preparation method and applications |
| US11345694B2 (en) | 2017-11-03 | 2022-05-31 | Discuva Ltd. | Antibacterial compounds |
| CN112778208A (en) * | 2021-01-08 | 2021-05-11 | 中南民族大学 | Dihydropyrazole MurA enzyme inhibitor molecule and preparation method and application thereof |
| WO2022186362A1 (en) * | 2021-03-03 | 2022-09-09 | 白鳥製薬株式会社 | Method for producing pyrazole compound |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5702392B2 (en) | Novel compound effective as xanthine oxidase inhibitor, process for producing the same, and pharmaceutical composition containing the same | |
| CN101228156B (en) | Fungicidal carboxamides | |
| AU2009331179B2 (en) | Novel bicyclic heterocyclic compound | |
| KR101941048B1 (en) | Aminoalkyl-substituted n-thienyl benzamide derivative | |
| CN101918376B (en) | 1,2,3-Triazole derivatives useful as stearoyl-CoA desaturase inhibitors | |
| TWI353834B (en) | Aryl- and heteroarylpiperidinecarboxylate derivati | |
| US9453014B2 (en) | Cyclic amide derivatives as inhibitors of 11-β-hydroxysteroid dehydrogenase and uses thereof | |
| JP5452594B2 (en) | Alkyl thiazole carbamate derivatives, their preparation, and their use as FAAH enzyme inhibitors | |
| CA2645712A1 (en) | Triazole derivative or salt thereof | |
| US20110319381A1 (en) | Derivatives of azaspiranyl-alkylcarbamates of 5-member heterocyclic compounds, preparation thereof and therapeutic use thereof | |
| CZ285799B6 (en) | Indole derivatives, process of their preparation, their use in the preparation of pharmaceutical preparations and pharmaceutical compositions containing thereof | |
| WO2007114338A1 (en) | Acid secretion inhibitor | |
| KR20180006450A (en) | A urea derivative or a pharmacologically acceptable salt thereof | |
| US6914058B2 (en) | Antibacterial compounds: process for their preparation and pharmaceutical compositions containing them | |
| CN1203589A (en) | new compound | |
| JP2000515133A (en) | Hypoglycemic and hypolipidemic compounds | |
| JP2004196678A (en) | Pyrazole derivatives | |
| JP5812985B2 (en) | 5-membered heterocyclic compounds cyclopenta [c] pyrrolylalkylcarbamate derivatives, their preparation and their therapeutic use | |
| EP2167083A2 (en) | Novel 1- heteroaryl-3-azabicycloý3.1.0¨hexanes, methods for their preparation and their use as medicaments | |
| JP2006528630A (en) | Diphenyl-substituted cycloalkanes, compositions containing such compounds, and methods of use | |
| WO2011148956A1 (en) | Fused imidazole derivative | |
| CA2743558A1 (en) | Carbamate derivatives of alkyl-heterocycles, preparation thereof and therapeutic use thereof | |
| CA2794176C (en) | Novel benzamide derivatives | |
| JP5511824B2 (en) | Novel oxazolidinone derivative having cyclic amidoxime or cyclic amidrazon group and pharmaceutical composition containing the same | |
| CN105399737B (en) | Oxazolidinone compounds and application thereof |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| RD01 | Notification of change of attorney |
Free format text: JAPANESE INTERMEDIATE CODE: A7421 Effective date: 20040506 |