JP2005514031A - エポチロンをヒドロキシル化するための組成物および方法 - Google Patents
エポチロンをヒドロキシル化するための組成物および方法 Download PDFInfo
- Publication number
- JP2005514031A JP2005514031A JP2003558132A JP2003558132A JP2005514031A JP 2005514031 A JP2005514031 A JP 2005514031A JP 2003558132 A JP2003558132 A JP 2003558132A JP 2003558132 A JP2003558132 A JP 2003558132A JP 2005514031 A JP2005514031 A JP 2005514031A
- Authority
- JP
- Japan
- Prior art keywords
- epothilone
- seq
- acid sequence
- nucleic acid
- hydroxylase
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Withdrawn
Links
Images




Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N9/00—Enzymes; Proenzymes; Compositions thereof; Processes for preparing, activating, inhibiting, separating or purifying enzymes
- C12N9/0004—Oxidoreductases (1.)
- C12N9/0071—Oxidoreductases (1.) acting on paired donors with incorporation of molecular oxygen (1.14)
- C12N9/0077—Oxidoreductases (1.) acting on paired donors with incorporation of molecular oxygen (1.14) with a reduced iron-sulfur protein as one donor (1.14.15)
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/79—Transferrins, e.g. lactoferrins, ovotransferrins
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12P—FERMENTATION OR ENZYME-USING PROCESSES TO SYNTHESISE A DESIRED CHEMICAL COMPOUND OR COMPOSITION OR TO SEPARATE OPTICAL ISOMERS FROM A RACEMIC MIXTURE
- C12P17/00—Preparation of heterocyclic carbon compounds with only O, N, S, Se or Te as ring hetero atoms
- C12P17/18—Preparation of heterocyclic carbon compounds with only O, N, S, Se or Te as ring hetero atoms containing at least two hetero rings condensed among themselves or condensed with a common carbocyclic ring system, e.g. rifamycin
- C12P17/181—Heterocyclic compounds containing oxygen atoms as the only ring heteroatoms in the condensed system, e.g. Salinomycin, Septamycin
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2299/00—Coordinates from 3D structures of peptides, e.g. proteins or enzymes
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Zoology (AREA)
- Wood Science & Technology (AREA)
- General Health & Medical Sciences (AREA)
- Genetics & Genomics (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Biochemistry (AREA)
- Medicinal Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Microbiology (AREA)
- Molecular Biology (AREA)
- Biotechnology (AREA)
- General Engineering & Computer Science (AREA)
- General Chemical & Material Sciences (AREA)
- Communicable Diseases (AREA)
- Animal Behavior & Ethology (AREA)
- Toxicology (AREA)
- Biophysics (AREA)
- Biomedical Technology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Oncology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmacology & Pharmacy (AREA)
- Gastroenterology & Hepatology (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Preparation Of Compounds By Using Micro-Organisms (AREA)
- Micro-Organisms Or Cultivation Processes Thereof (AREA)
- Enzymes And Modification Thereof (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Oxygen Or Sulfur (AREA)
Abstract
エポチロンBヒドロキシラーゼおよびその突然変異体および変異体およびエポチロンBヒドロキシラーゼ遺伝子の下流に位置するフェレドキシンの単離核酸配列および該配列によってコードされるポリペプチドが提供される。ベクターおよび該ベクターを含む細胞もまた提供される。さらに、組換え微生物の製造方法、ヒドロキシアルキル含有エポチロンを生成するためにこれら組換え微生物を使用する方法、およびエポチロンBヒドロキシラーゼの突然変異体によって生成したエポチロンアナログも提供される。
Description
本発明は、エポチロンBヒドロキシラーゼおよびその突然変異体および変異体およびエポチロンBヒドロキシラーゼ遺伝子の下流に位置するフェレドキシンの単離核酸配列およびそれによってコードされるポリペプチドに関する。本発明はまた、エポチロンなどの末端アルキル基を有する小さな有機分子化合物をヒドロキシル化して末端ヒドロキシアルキル基を有する化合物を生成することのできる、エポチロンBヒドロキシラーゼまたはその突然変異体または変異体および/またはフェレドキシンを発現する組換え微生物に関する。そのような微生物を組換えにより製造する方法並びに末端ヒドロキシアルキル基を有する化合物の合成にこれら組換え微生物を用いる方法もまた提供される。本発明の組成物および方法は、医薬の分野で様々な有用性を有するエポチロンを製造するのに有用である。本発明のエポチロンBヒドロキシラーゼの突然変異体を用いて製造した新規なエポチロンアナログも記載する。
エポチロンは、医薬の分野で有用性の見出されたマクロライド系化合物である。たとえば、構造:

を有するエポチロンAおよびBは、パクリタキセル(タキソールR)と類似の微小管安定化作用を発揮し、それゆえ腫瘍細胞などの迅速に増殖する細胞や他の過剰増殖細胞性疾患に関連する細胞に対して細胞障害作用を発揮することがわかっている(Bollagら、Cancer Res., Vol. 55, No. 11, 2325-2333 (1995)を参照)。
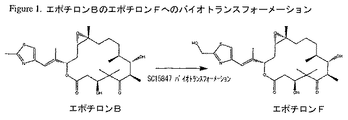
エポチロンAおよびBは、Hofleらによって初めて単離および特徴付けられたソランジウム・セルロスム(Sorangium cellulosum)によって産生される天然の抗癌剤である(DE4138042;WO93/10121; Angew. Chem. Int. Ed. Engl. Vol. 35, No13/14, 1567-1569 (1996);およびJ. Antibiot., Vol. 49, No. 6, 560-563 (1996))。その後、エポチロンAおよびBの全合成が、Balogら、Angew. Chem. Int. Ed. Engl. Vol. 35, No. 23/24, 2801-2803, 1996; Mengら、J. Am. Chem. Soc., Vol. 119, No. 42, 10073-10092 (1997); Nicolaouら、J. Am. Chem. Soc., Vol. 119, No. 34, 7974-7991 (1997); Schinzerら、Angew. Chem. Int. Ed. Engl. Vol. 36, No. 5, 523-524 (1997);およびYangら、Angew. Chem. Int. Ed. Engl., Vol. 36, No. 1/2, 166-168, 1997によって刊行されている。WO98/25929は、エポチロンA、エポチロンB、エポチロンのアナログおよびエポチロンアナログのライブラリーの化学合成法を開示している。エポチロンC、D、EおよびFの構造およびソランジウム・セルロスムDSM6773からの産生がWO98/22461に開示されている。図1は、WO00/39276に記載されているように、アクチノミセス種株SC15847(ATCC PT−1043)(その後、アミコラトプシス・オリエンタリス(Amycolatopsis orientalis)として同定された)におけるエポチロンBのエポチロンFへのバイオトランスフォーメーションの図解を示す。
シトクロムP450酵素は原核細胞および真核細胞において見出され、一酸化炭素と複合体を形成したときに450nmでの吸収極大により区別することのできるヘム結合ドメインを共通して有する。シトクロムP450酵素は、芳香族およびベンジル環およびアルカンを含む主として疎水性の基質に対して広スペクトルの酸化反応を行う。原核細胞においては、シトクロムP450酵素は、解毒系として、およびトルエン、ベンゼンおよび樟脳などの基質を代謝する最初の酵素過程として見出されている。シトクロムP450遺伝子はまた、ストレプトミセス・テンダエ(Streptomyces tendae)におけるnikkomycin(Bruntner, C.ら、1999, Mol. Gen. Genet. 262: 102-114)、ドキソルビシン(Dickens, M. L., Strohl, W. R., 1996, J. Bacteriol., 178: 3389-3395)、およびソランジウム・セルロスムのエポチロン生合成クラスター(Julien, B.ら、2000, Gene, 249: 153-160)などの二次代謝産物の生合成経路で見出されている。幾つかの例外を除いて、原核生物におけるシトクロムP450系は3つのタンパク質、フェレドキシンNADHまたはNADPH依存性レダクターゼ、鉄−硫黄フェレドキシンおよびシトクロムP450酵素からなっている(Lewis, D. F., Hlavica, P., 2000, Biochim. Biophys. Acta., 1460: 353-374)。電子は分子状酸素の開裂のため、フェレドキシンレダクターゼからフェレドキシンへ、最終的にシトクロムP450酵素に輸送される。
本発明の目的は、エポチロンBヒドロキシラーゼおよびその変異体または突然変異体をコードする単離核酸配列およびフェレドキシンまたはその変異体または突然変異体をコードする単離核酸配列を提供することである。
本発明の他の目的は、エポチロンBヒドロキシラーゼおよびその変異体または突然変異体のアミノ酸配列を含む単離ポリペプチドおよびフェレドキシンおよびその変異体または突然変異体のアミノ酸を含む単離ポリペプチドを提供することである。
本発明の他の目的は、エポチロンBヒドロキシラーゼのホモロジーモデルの構造座標を提供することである。構造座標は付表1に列挙してある。この本発明のモデルは、エポチロンBヒドロキシラーゼの生物学的機能のモデュレーター並びに特異性の変化したエポチロンBヒドロキシラーゼのさらなる突然変異体をデザインする手段を提供する。
本発明の他の目的は、エポチロンBヒドロキシラーゼまたはその変異体または突然変異体および/またはフェレドキシンまたはその変異体または突然変異体をコードする単離核酸配列を含むベクターを提供することである。好ましい態様において、これらベクターは、フェレドキシンをコードする核酸配列をさらに含む。
本発明の他の目的は、エポチロンBヒドロキシラーゼまたはその変異体または突然変異体および/またはフェレドキシンまたはその変異体または突然変異体をコードする単離核酸配列を含むベクターを含む宿主細胞を提供することである。
本発明の他の目的は、末端アルキル基を有する化合物、とりわけエポチロンをヒドロキシル化して末端ヒドロキシアルキル基を有する化合物を生成することのできる組換え微生物の製造方法を提供することである。
本発明の他の目的は、末端アルキル基を有する化合物、とりわけエポチロンをヒドロキシル化して末端ヒドロキシアルキル基を有する化合物を生成することのできる組換えにより生成した微生物を提供することである。
本発明の他の目的は、これら組換え微生物において化合物をヒドロキシル化する方法を提供することである。とりわけ本発明は、ヒドロキシアルキル含有エポチロンの製造方法を提供するものであるが、該ヒドロキシアルキル含有エポチロンは、抗癌剤として、および他のエポチロンアナログの調製における出発物質として有用である。
本発明の他の目的は、本明細書で24−OHエポチロンBまたは24−OH EpoBと称する、式A:
で示される化合物、並びに式Aの化合物を含む組成物および該組成物の製造方法を提供することである。
本発明は、単離核酸配列およびポリペプチドおよび末端炭素原子位置に所望の置換基を有する化合物を得る方法に関する。とりわけ本発明は、ヒドロキシアルキル含有エポチロンの組成物および製造方法を提供するものであるが、該ヒドロキシアルキル含有エポチロンは、抗癌剤として、および他のエポチロンアナログの調製における出発物質として有用である。
本明細書において「エポチロン」とは、本明細書において定義するエポチロンコアと側鎖基とを含む化合物をいう。本明細書において「エポチロンコア」とは、コア構造(本明細書に用いた環系位置の番号付けを示す):
を含む残基をいい、上記式において置換基は以下のとおりである:
Qは、
よりなる群から選ばれる;
Wは、OまたはNR6;
Xは、O、HおよびOR7よりなる群から選ばれる;
Mは、O、S、NR8、CR9R10;
B1およびB2は、OR11、OCOR12よりなる群から選ばれる;
R1−R5およびR12−R17は、H、アルキル、置換アルキル、アリール、およびへテロシクロよりなる群から選ばれ、R1およびR2がアルキルである場合はこれらは一緒になってシクロアルキルを形成することができる;
R6は、H、アルキル、および置換アルキルよりなる群から選ばれる;
R7およびR11は、H、アルキル、置換アルキル、トリアルキルシリル、アルキルジアリールシリルおよびジアルキルアリールシリルよりなる群から選ばれる;
R8は、H、アルキル、置換アルキル、R13C=O、R14OC=OおよびR15SO2よりなる群から選ばれる;および
R9およびR10は、H、ハロゲン、アルキル、置換アルキル、アリール、へテロシクロ、ヒドロキシ、R16C=O、およびR17OC=Oよりなる群から選ばれる。
Qは、
Wは、OまたはNR6;
Xは、O、HおよびOR7よりなる群から選ばれる;
Mは、O、S、NR8、CR9R10;
B1およびB2は、OR11、OCOR12よりなる群から選ばれる;
R1−R5およびR12−R17は、H、アルキル、置換アルキル、アリール、およびへテロシクロよりなる群から選ばれ、R1およびR2がアルキルである場合はこれらは一緒になってシクロアルキルを形成することができる;
R6は、H、アルキル、および置換アルキルよりなる群から選ばれる;
R7およびR11は、H、アルキル、置換アルキル、トリアルキルシリル、アルキルジアリールシリルおよびジアルキルアリールシリルよりなる群から選ばれる;
R8は、H、アルキル、置換アルキル、R13C=O、R14OC=OおよびR15SO2よりなる群から選ばれる;および
R9およびR10は、H、ハロゲン、アルキル、置換アルキル、アリール、へテロシクロ、ヒドロキシ、R16C=O、およびR17OC=Oよりなる群から選ばれる。
「側鎖基」とは、エポチロンAまたはBについて上記で定義した置換基Gまたは以下に示すG1およびG2をいう。
G1は、下記式V:
HO−CH2−(A1)n−(Q)m−(A2)o (V)
であり、
G2は、下記式VI:
CH3−(A1)n−(Q)m−(A2)o (VI)
(上記式中、A1およびA2は、任意に置換されたC1−C3アルキルおよびアルケニルよりなる群から独立に選ばれる;
Qは、1〜3の環および少なくとも一つの環中に少なくとも一つの炭素−炭素二重結合を含む任意に置換された環であり;
n、mおよびoは、0および1よりなる群から独立に選ばれた整数であり、n、mまたはoの少なくとも一つが1である)
である。
HO−CH2−(A1)n−(Q)m−(A2)o (V)
であり、
G2は、下記式VI:
CH3−(A1)n−(Q)m−(A2)o (VI)
(上記式中、A1およびA2は、任意に置換されたC1−C3アルキルおよびアルケニルよりなる群から独立に選ばれる;
Qは、1〜3の環および少なくとも一つの環中に少なくとも一つの炭素−炭素二重結合を含む任意に置換された環であり;
n、mおよびoは、0および1よりなる群から独立に選ばれた整数であり、n、mまたはoの少なくとも一つが1である)
である。
「末端炭素原子」または「末端アルキル基」とは、エポチロンコアに15位にて直接結合している残基の末端炭素原子または末端アルキル基あるいは15位に結合した側鎖基の末端炭素原子または末端アルキル基をいう。「アルキル基」は、本明細書に記載のアルキルおよび置換アルキルを包含することが理解されなければならない。
「アルキル」とは、炭素数が1〜20、好ましくは炭素数が1〜7の、任意に置換された直鎖または分枝鎖の飽和炭化水素基をいう。「低級アルキル」なる語は、炭素数1〜4の任意に置換されたアルキル基をいう。
「置換アルキル」とは、たとえば、ハロ、トリフルオロメチル、トリフルオロメトキシ、ヒドロキシ、アルコキシ、シクロアルキルオキシ、ヘテロシクロオキシ、オキソ、アルカノイル、アリールオキシ、アルカノイルオキシ、アミノ、アルキルアミノ、アリールアミノ、アラルキルアミノ、シクロアルキルアミノ、ヘテロシクロアミノ、二置換アミン(二つのアミノ置換基はアルキル、アリールまたはアラルキルから選ばれる)、アルカノイルアミノ、アロイルアミノ、アラルカノイルアミノ、置換アルカノイルアミノ、置換アリールアミノ、置換アラルカノイルアミノ、チオール、アルキルチオ、アリールチオ、アラルキルチオ、シクロアルキルチオ、ヘテロシクロチオ、アルキルチオノ、アリールチオノ、アラルキルチオノ、アルキルスルホニル、アリールスルホニル、アラルキルスルホニル、スルホンアミド(たとえば、SO2NH2)、置換スルホンアミド、ニトロ、シアノ、カルボキシ、カルバミル(たとえば、CONH2)、置換カルバミル(たとえば、CONHアルキル、CONHアリール、CONHアラルキル、または窒素原子上にアルキル、アリールまたはアラルキルから選ばれる2つの置換基が存在する場合)、アルコキシカルボニル、アリール、置換アリール、グアニジノおよびへテロシクロ(たとえば、インドリル、イミダゾリル、フリル、チエニル、チアゾリル、ピロリジル、ピリジル、ピリミジルなど)などの1ないし4の置換基で置換されたアルキル基をいう。上記において置換基がさらに置換されている場合、ハロゲン、アルキル、アルコキシ、アリールまたはアラルキルで置換されているであろう。
本発明の一つの側面によれば、末端アルキル基を有するエポチロンをヒドロキシル化して末端ヒドロキシアルキル基を有するエポチロンを生成できる酵素であるエポチロンBヒドロキシラーゼをコードする単離ポリヌクレオチドが提供される。
本発明の他の側面によれば、エポチロンBヒドロキシラーゼ遺伝子の下流に位置する遺伝子であるフェレドキシンをコードする単離ポリヌクレオチドが提供される。フェレドキシンは、シトクロムP450系のタンパク質である。
本明細書において使用する「ポリヌクレオチド」とは、これら酵素またはその活性断片をコードするあらゆる形態のDNAまたはRNA、たとえば、それぞれcDNAまたはゲノムDNAまたはmRNAを包含することを意味し、かかるポリヌクレオチドはクローニングにより得られ、またはよく知られた化学的方法により合成して製造される。DNAは二本鎖であっても一本鎖であってもよい。一本鎖DNAは、コードすなわちセンス鎖かまたは非コードすなわちアンチセンス鎖のいずれを含んでいてもよい。それゆえ、ポリヌクレオチドなる語はまた、本明細書に開示する配列に対して少なくとも60%またはそれ以上、好ましくは少なくとも80%のホモロジーを示し、上記ポリヌクレオチドにストリンジェントな条件下でハイブリダイズするポリヌクレオチドをも包含する。本明細書において「ストリンジェントな条件」なる語は、2×SSC緩衝液で60℃でのハイブリダイゼーション条件を意味する。さらに好ましいのは、配列番号1、30、32、34、36、37、38、39、40、41、42、60、62、64、66、68、70、72または74または配列番号3に示す核酸配列に、または配列番号1、30、32、34、36、37、38、39、40、41、42、60、62、64、66、68、70、72または74または配列番号3に示す核酸配列の相補的配列に65℃にて3×SSCのハイブリダイゼーション条件下で16時間ハイブリダイズすることができ、かつ配列番号1、30、32、34、36、37、38、39、40、41、42、60、62、64、66、68、70、72または74または配列番号3に示す核酸配列に、または配列番号1、30、32、34、36、37、38、39、40、41、42、60、62、64、66、68、70、72または74または配列番号3に示す核酸配列の相補的配列に55℃での0.5×SSCの洗浄条件下で30分間、ハイブリダイズしたままでいることができる単離核酸である。
一つの態様において、本発明のポリヌクレオチドは、配列番号1に示すゲノムDNAまたはその相同配列またはこのエポチロンBヒドロキシラーゼと同様の活性を有するポリペプチドをコードするその断片を含む。あるいは、本発明のポリヌクレオチドは、配列番号3に示すゲノムDNAまたはその相同配列またはこのフェレドキシンと同様の活性を有するポリペプチドをコードするその断片を含んでいてよい。遺伝暗号の縮重のため、本発明のポリヌクレオチドはまた、この酵素およびその誘導体、変異体または活性な断片をコードする他の核酸配列をも包含する。
本発明はまた、天然に存在する、すなわちアミコラトプシス・オリエンタリスおよびアミコラタ・オートロフィカ(Amicolata autorophica)などの微生物に、または核酸を単離することのできる土または他の採取源に存在するこれらポリヌクレオチドの変異体、またはよく知られた突然変異誘発法により調製した突然変異体にも関する。本発明の変異体の例示は配列番号36〜42に示してある。
本明細書において使用する「突然変異体」とは、配列番号1または配列番号3と比べて1またはそれ以上の点変異、または欠失または核酸の付加を有するが、配列番号1または配列番号3によってコードされるポリペプチドと同様の活性を有するポリペプチドまたは断片をコードしている核酸配列を含むことを意味する。好ましい態様において、基質特異性および/または酵素の収率の変化した突然変異体が製造される。エポチロンBヒドロキシラーゼ遺伝子に関して突然変異の好ましい領域は、該酵素の活性部位を含む約113アミノ酸をコードする核酸配列の領域である。配列番号1のアミノ酸位置GLU31、ARG67、ARG88、ILE92、ALA93、VAL106、ILE130、ALA140、MET176、PHE190、GLU231、SER294、PHE237、またはILE365に少なくとも一つのアミノ酸置換を有するポリペプチドをコードする突然変異体もまた好ましい。本発明のポリヌクレオチド突然変異体の例示を、配列番号30、32、34、60、62、64、66、68、70、72および74に示してある。
エポチロンBヒドロキシラーゼをコードする配列番号1の核酸配列のクローニングを、細菌からの6つのシトクロムP450遺伝子の核酸配列をアラインメントすることによりデザインしたPCRプライマーを用いて行った。以下のシトクロムP450遺伝子をアラインメントした:
配列1:遺伝子座:STMSUACB;受け入れ番号:M32238;文献:Omer, C. A., J. Bacteriol. 172: 3335-3345 (1990)
配列2:遺伝子座:STMSUBCB;受け入れ番号:M32239;文献:Omer, C. A., J. Bacteriol. 172: 3335-3345 (1990)
配列3:遺伝子座:AB018074(以前はSTMORFA);受け入れ番号:AB018074;文献:Ueda, K., J. Antibiot. 48: 638-646 (1995)
配列4:遺伝子座:SSU65940;受け入れ番号:U65940;文献:Motamedi, H., J. Bacteriol. 178: 5243-5248 (1996)
配列5:遺伝子座:STMOLEP;受け入れ番号:L37200;文献:Rodriguez, A. M., FEMS Microbiol. Lett. 127: 117-120 (1995)
配列6:遺伝子座:SERCP450A;受け入れ番号:M83110;文献:Andersen, J. F.およびHutchinson, C. R., J. Bacteriol. 174: 725-735 (1992)。
配列1:遺伝子座:STMSUACB;受け入れ番号:M32238;文献:Omer, C. A., J. Bacteriol. 172: 3335-3345 (1990)
配列2:遺伝子座:STMSUBCB;受け入れ番号:M32239;文献:Omer, C. A., J. Bacteriol. 172: 3335-3345 (1990)
配列3:遺伝子座:AB018074(以前はSTMORFA);受け入れ番号:AB018074;文献:Ueda, K., J. Antibiot. 48: 638-646 (1995)
配列4:遺伝子座:SSU65940;受け入れ番号:U65940;文献:Motamedi, H., J. Bacteriol. 178: 5243-5248 (1996)
配列5:遺伝子座:STMOLEP;受け入れ番号:L37200;文献:Rodriguez, A. M., FEMS Microbiol. Lett. 127: 117-120 (1995)
配列6:遺伝子座:SERCP450A;受け入れ番号:M83110;文献:Andersen, J. F.およびHutchinson, C. R., J. Bacteriol. 174: 725-735 (1992)。
Scientific and Educational Software(ダラム、ノースカロライナ、米国)からの整列(Align)プログラムであるMyers, E. W.およびW. Miller. 1988. CABIOS 4:1, 11-17のアルゴリズムの実行によりアラインメントを行った。I−ヘリックス(酸素結合ドメインを含む)中、K−ヘリックス中、および保存されたヘム結合ドメインを含むB−バルジとL−ヘリックスとのスパニング中に3つの高度に保存された領域が同定された。アラインメントで同定された3つの保存領域に対してプライマーをデザインした。プライマーのP450−1+(配列番号23)およびP450−1a+(配列番号24)はIヘリックスからデザインし、プライマーのP450−2+(配列番号25)はB−バルジおよびL−ヘリックス領域からデザインし、プライマーのP450−3−(配列番号27)はヘム結合タンパク質に対する逆相補鎖としてデザインした。
ついで、ポリメラーゼ連鎖反応(PCR)によりゲノム断片を増幅した。PCR増幅の後、反応生成物をゲル電気泳動により分離し、予期したサイズの断片を切り出した。Qiaquickゲル抽出法(Qiagen、サンタクラリタ、カリフォルニア、米国)を用いてアガロースゲルスライスからDNAを抽出した。ついで、PCRscript Ampクローニングキット(Stratagene、ラジョラ、カリフォルニア、米国)を用い、断片をPCRscriptベクター(Stratagene)にクローニングした。挿入物を含むコロニーを、100μg/mlのアンピシリンを含む1〜2mlのLBブロスに30〜37℃にて16〜24時間、230〜300rpmで選び取った。Mo Bioミニプラスミドプレプキット(Mo Bio、ソラノビーチ、カリフォルニア、米国)を用いてプラスミドの単離を行った。このプラスミドDNAをPCRおよびシークエンシングの鋳型として、および制限消化分析のために用いた。
クローニングしたPCR産物を、Applied Biosystems(フォスターシティー、カリフォルニア、米国)からのBig-Dyeシークエンシングキットを用いてシークエンシングし、ABI310シークエンサー(Applied Biosystems、フォスターシティー、カリフォルニア、米国)を用いて分析した。非重複(non-redundant)タンパク質データベースのAltschul, S. F.ら、Mol. Biol. 215: 403-410 (1990)のプロトコールを用い、挿入物の配列を用いてTblastXサーチを行った。既知のP450タンパク質に有意の類似性を有する独特の配列が保持された。このアプローチを用い、全部で9つの異なるP450配列がSC15847から、7つがゲノムDNA鋳型から、および2つがcDNAから同定された。2つのP450配列が該DNAおよびcDNA鋳型でともに見出された。分析した50のcDNAクローンのうち、2つの配列が優勢であり、それぞれ20クローンであった。ついで、これら2つの遺伝子をゲノムDNAからクローニングした。
ゲノムDNAの核酸配列をBig-Dyeシークエンシングシステム(Applied Biosystems)を用いて決定し、ABI310シークエンサーを用いて分析した。この配列を配列番号1に示す。404アミノ酸で予想分子量が44.7kDaのタンパク質をコードするオープンリーディングフレームがクローニングしたBglII断片内に認められた。このポリペプチドの推定アミノ酸配列を配列番号2に示す。このポリペプチドのアミノ酸配列は、ストレプトミセス・テンダエのNikFタンパク質(Bruntner, C.ら、1999, Mol. Gen. Genet. 262: 102-114)と51%の同一性を、ストレプトミセス・カーボフィルス(S. carbophilus)のSca−2タンパク質(Watanabe, I.ら、1995, Gene 163: 81-85)と48%の同一性を有することがわかった。これら両酵素ともにシトクロムP450ファミリー105に属する。すべてのシトクロムP450酵素のヘム結合ドメインに認められる不変のシステインは、残基356に見出される。このエポチロンBの遺伝子をebhと命名した。64アミノ酸の推定フェレドキシン遺伝子のATG開始コドンは、ebhの停止コドンの9塩基対下流に見出される。この酵素は、ストレプトミセス・グリセオウルス(S. griseoulus)のフェレドキシン遺伝子(O'Keefe, D. P.ら、1991, Biochemistry 30: 447-455)およびストレプトミセス・ノウルセイ(S. noursei)のフェレドキシン遺伝子(Brautaset, T.ら、2000, Chem. Biol. 7: 395-403)と50%の同一性を有することがわかった。このフェレドキシンをコードする核酸配列を配列番号3に示してあり、このフェレドキシンポリペプチドのアミノ酸配列を配列番号4に示してある。
ebh遺伝子配列はまた、他の微生物から変異体のシトクロムP450遺伝子を単離するのにも用いた。本発明の例示的変異体ポリヌクレオチドであるebh43491、ebh14930、ebh53630、ebh53550、ebh39444、ebh43333およびebh35165をそれらを単離した種とともに下記表1に示す。これら変異体の核酸配列は、配列番号36〜42にそれぞれ示してある。
表1:変異体ポリヌクレオチド
例示的変異体であるebh43491、ebh14930、ebh53630、ebh53550、ebh39444、ebh43333およびebh35165によってコードされるアミノ酸配列は、配列番号43〜49にそれぞれ示してある。表2は、これら例示的変異体のアミノ酸置換の要約を示す。
表2:アミノ酸置換
また、ebh遺伝子のコード領域に突然変異を導入して、改善された収率、および/またはバイオコンバージョンの速度、および/または変化した基質特異性を有する突然変異体を同定した。本発明の例示的な突然変異体の核酸配列を配列番号30、32、34、60、62、64、66、68、70、72および74に示す。
配列番号30の核酸配列は突然変異体ebh25−1をコードし、この突然変異体は変化した基質特異性を示す。この突然変異遺伝子を含むプラスミドpANT849ebh25−1を寄託してあり、ブダペスト条約の規定のもと、国際寄託当局により受託されている。寄託は、2002年11月 日にアメリカン・タイプ・カルチャー・コレクション(10801、ユニバーシティーブールバール、マナサス、バージニア20110−2209)に対して行った。ATCC受託番号は、 である。このプラスミドへの公的アクセスに対するすべての制限は、この特許出願が特許されると直ちに変更不能に解除されるであろう。寄託は、寄託日後の30年間の期間または最近の試料請求後の5年間または特許の有効期間のうちいずれか長い期間、公的寄託機関で維持されるであろう。上記プラスミドは、寄託のときには生存していた。寄託は、寄託機関によって生きた試料を分与することができないときには置き換えられるであろう。
突然変異25のスクリーニング(プライマーNPB29−mut25f(配列番号58)およびNPB29−mut25r(配列番号59))の際に同定されたこのストレプトミセス・リビダンス(S. lividans)形質転換体は、エポチロンBまたはエポチロンFとはHPLC溶出時間が異なる産物を生成することがわかった。この未知の試料をLC−MSにより分析したところ、分子量が523(M.W.)であることがわかったが、これはエポチロンBの単一のヒドロキシル化と一致していた。プラスミドDNAをストレプトミセス・リビダンスの培養液から単離し、プライマーNPB29−6f(配列番号28)およびNPB29−7r(配列番号29)を用いたPCR増幅の鋳型として用いた(実施例17参照)。予期された断片が得られ、Big-Dyeシークエンシングシステムを用いてシークエンシングした。ebh25−1突然変異体は、タンパク質のアミノ酸配列の変化となる2つの突然変異を有することがわかった(アスパラギン195がセリンに変わり、セリン294がプロリンに変わる)。コドン238で突然変異のためにターゲティングした位置は2つのヌクレオチド変化をもたらすことがわかったが、これはタンパク質のアミノ酸配列の変化という結果とはならなかった。配列番号30によってコードされる突然変異体ポリペプチドのアミノ酸配列を配列番号31に示す。
配列番号32の核酸配列は、突然変異体ebh10−53(改善されたバイオコンバージョン収率を示す)をコードする。突然変異10のスクリーニング(プライマーNPB29−mut10f(配列番号54)およびNPB29−mut10r(配列番号55))の際に同定されたこのストレプトミセス・リビダンス形質転換体は、エポチロンFの一層高い収率をもたらした。プラスミドDNAをストレプトミセス・リビダンスの培養液から単離し、プライマーNPB29−6f(配列番号28)およびNPB29−7r(配列番号29)を用いたPCR増幅の鋳型として用いた(実施例16参照)。予期された断片が得られ、Big-Dyeシークエンシングシステムを用いてシークエンシングした。ebh10−53突然変異体は、タンパク質のアミノ酸配列の変化となる2つの突然変異を有することがわかった(グルタミン酸231がアルギニンに変わり、フェニルアラニン190がチロシンに変わる)。位置231が突然変異誘発の標的であったが、残基190での変化は偶然の変化であり、突然変異誘発法の人工産物である。配列番号32によってコードされる突然変異体ポリペプチドのアミノ酸配列を配列番号33に示す。
配列番号34の核酸配列は、突然変異体ebh24−16(これも改善されたバイオコンバージョン収率を示す)をコードする。突然変異24のスクリーニング(プライマーNPB29−mut24f(配列番号56)およびNPB29−mut24r(配列番号57))の際に同定されたこのストレプトミセス・リビダンス形質転換体ebh24−16もまた、エポチロンFの一層高い収率をもたらした。プラスミドDNAをストレプトミセス・リビダンスの培養液から単離し、プライマーNPB29−6f(配列番号28)およびNPB29−7r(配列番号29)を用いたPCR増幅の鋳型として用いた。予期された断片が得られ、Big-Dyeシークエンシングシステムを用いてシークエンシングした。ebh24−16突然変異体は、タンパク質のアミノ酸配列の変化となる2つの突然変異を有することがわかった(フェニルアラニン237がアラニンに変わり、イソロイシン92がバリンに変わる)。位置237が突然変異誘発の標的であったが、残基92での変化は偶然の変化であり、突然変異誘発法の人工産物である。配列番号34によってコードされる突然変異体ポリペプチドのアミノ酸配列を配列番号35に示す。
配列番号60の核酸配列は、突然変異体ebh24−16d8(これも改善されたバイオコンバージョン収率を示す)をコードする。突然変異59のスクリーニング(プライマーNPB29mut59(配列番号77))の際に同定されたこのストレプトミセス・リモスス(S. rimosus)形質転換体ebh24−16d8もまた、エポチロンFの一層高い収率をもたらした。プラスミドDNAをストレプトミセス・リモススの培養液から単離し、プライマーNPB29−6f(配列番号28)およびNPB29−7r(配列番号29)を用いたPCR増幅の鋳型として用いた。予期された断片が得られ、Big-Dyeシークエンシングシステムを用いてシークエンシングした。ebh24−16d8突然変異体は、タンパク質のアミノ酸配列の変化となる1つの突然変異を有することがわかった(アルギニン67がグルタミンに変わる)。この変化は、突然変異誘発法の人工産物である。配列番号60によってコードされる突然変異体ポリペプチドのアミノ酸配列を配列番号61に示す。
配列番号62の核酸配列は、突然変異体ebh24−16c11(これも改善されたバイオコンバージョン収率を示す)をコードする。突然変異59のスクリーニング(プライマーNPB29mut59(配列番号77))の際に同定されたこのストレプトミセス・リモスス形質転換体ebh24−16c11もまた、エポチロンFの一層高い収率をもたらした。プラスミドDNAをストレプトミセス・リモススの培養液から単離し、プライマーNPB29−6f(配列番号28)およびNPB29−7r(配列番号29)を用いたPCR増幅の鋳型として用いた。予期された断片が得られ、Big-Dyeシークエンシングシステムを用いてシークエンシングした。ebh24−16c11突然変異体は、タンパク質のアミノ酸配列の変化となるさらに2つの突然変異を有することがわかった(アラニン93がグリシンに変わり、イソロイシン365がトレオニンに変わる)。位置93が突然変異誘発の標的であったが、365での変化は突然変異誘発法の人工産物である。配列番号62によってコードされる突然変異体ポリペプチドのアミノ酸配列を配列番号63に示す。
配列番号64の核酸配列は、突然変異体ebh24−16−16(これも改善されたバイオコンバージョン収率を示す)をコードする。ebh24−16のランダム突然変異体のスクリーニングの際に同定されたこのストレプトミセス・リモスス形質転換体ebh24−16−16もまた、エポチロンFの一層高い収率をもたらした。プラスミドDNAをストレプトミセス・リモススの培養液から単離し、プライマーNPB29−6f(配列番号28)およびNPB29−7r(配列番号29)を用いたPCR増幅の鋳型として用いた。予期された断片が得られ、Big-Dyeシークエンシングシステムを用いてシークエンシングした。ebh24−16−16突然変異体は、タンパク質のアミノ酸配列の変化となるさらに1つの突然変異を有することがわかった(バリン106がアラニンに変わる)。配列番号64によってコードされる突然変異体ポリペプチドのアミノ酸配列を配列番号65に示す。
配列番号66の核酸配列は、突然変異体ebh24−16−74(これも改善されたバイオコンバージョン収率を示す)をコードする。ebh24−16のランダム突然変異体のスクリーニングの際に同定されたこのストレプトミセス・リモスス形質転換体ebh24−16−74もまた、エポチロンFの一層高い収率をもたらした。プラスミドDNAをストレプトミセス・リモススの培養液から単離し、プライマーNPB29−6f(配列番号28)およびNPB29−7r(配列番号29)を用いたPCR増幅の鋳型として用いた。予期された断片が得られ、Big-Dyeシークエンシングシステムを用いてシークエンシングした。ebh24−16−74突然変異体は、タンパク質のアミノ酸配列の変化となるさらに1つの突然変異を有することがわかった(アルギニン88がヒスチジンに変わる)。配列番号66によってコードされる突然変異体ポリペプチドのアミノ酸配列を配列番号67に示す。
配列番号68の核酸配列は、突然変異体ebh24−M18(これも改善されたバイオコンバージョン収率を示す)をコードする。ebhのランダム突然変異体のスクリーニングの際に同定されたこのストレプトミセス・リモスス形質転換体ebhM−18もまた、エポチロンFの一層高い収率をもたらした。プラスミドDNAをストレプトミセス・リモススの培養液から単離し、プライマーNPB29−6f(配列番号28)およびNPB29−7r(配列番号29)を用いたPCR増幅の鋳型として用いた。予期された断片が得られ、Big-Dyeシークエンシングシステムを用いてシークエンシングした。ebhM−18突然変異体は、タンパク質のアミノ酸配列の変化となる2つの突然変異を有することがわかった(グルタミン酸31がリシンに変わり、メチオニン176がバリンに変わる)。配列番号68によってコードされる突然変異体ポリペプチドのアミノ酸配列を配列番号69に示す。
配列番号72の核酸配列は、突然変異体ebh24−16g8(これも改善されたバイオコンバージョン収率を示す)をコードする。突然変異50のスクリーニング(プライマーNPB29mut50(配列番号78))の際に同定されたこのストレプトミセス・リモスス形質転換体ebh24−16g8もまた、エポチロンFの一層高い収率をもたらした。プラスミドDNAをストレプトミセス・リモススの培養液から単離し、プライマーNPB29−6f(配列番号28)およびNPB29−7r(配列番号29)を用いたPCR増幅の鋳型として用いた。予期された断片が得られ、Big-Dyeシークエンシングシステムを用いてシークエンシングした。ebh24−16g8突然変異体は、タンパク質のアミノ酸配列の変化となるさらに2つの突然変異を有することがわかった(メチオニン176がアラニンに変わり、イソロイシン130がトレオニンに変わる)。位置176が突然変異誘発の標的であったが、130での変化は突然変異誘発法の人工産物である。配列番号72によってコードされる突然変異体ポリペプチドのアミノ酸配列を配列番号73に示す。
配列番号74の核酸配列は、突然変異体ebh24−16b9(これも改善されたバイオコンバージョン収率を示す)をコードする。突然変異50のスクリーニング(プライマーNPB29mut50(配列番号78))の際に同定されたこのストレプトミセス・リモスス形質転換体ebh24−16b9もまた、エポチロンFの一層高い収率をもたらした。プラスミドDNAをストレプトミセス・リモススの培養液から単離し、プライマーNPB29−6f(配列番号28)およびNPB29−7r(配列番号29)を用いたPCR増幅の鋳型として用いた。予期された断片が得られ、Big-Dyeシークエンシングシステムを用いてシークエンシングした。ebh24−16b9突然変異体は、タンパク質のアミノ酸配列の変化となるさらに2つの突然変異を有することがわかった(メチオニン176がセリンに変わり、アラニン140がトレオニンに変わる)。位置176が突然変異誘発の標的であったが、140での変化は突然変異誘発法の人工産物である。配列番号74によってコードされる突然変異体ポリペプチドのアミノ酸配列を配列番号75に示す。
これら9つの突然変異体遺伝子のプラスミドpANT849ebh−24−16、pANT849ebh−10−53、pANT849ebh−24−16d8、pANT849ebh−24−16c11、pANT849ebh−16−16、pANT849ebh−24−16−74、pANT849ebh−24−16b9、pANT849ebh−M18およびpANT849ebh−24−16g8からなる混合物を寄託してあり、ブダペスト条約の規定のもと、国際寄託当局により受託されている。寄託は、2002年11月 日にアメリカン・タイプ・カルチャー・コレクション(10801、ユニバーシティーブールバール、マナサス、バージニア20110−2209)に対して行った。ATCC受託番号は、 である。このプラスミドへの公的アクセスに対するすべての制限は、この特許出願が特許されると直ちに変更不能に解除されるであろう。寄託は、寄託日後の30年間の期間または最近の試料請求後の5年間または特許の有効期間のうちいずれか長い期間、公的寄託機関で維持されるであろう。上記プラスミドは、寄託のときには生存していた。寄託は、寄託機関によって生きた試料を分与することができないときには置き換えられるであろう。
それゆえ、本発明の他の側面によれば、エポチロンBヒドロキシラーゼおよびその変異体および突然変異体の単離ポリペプチドおよびフェレドキシンまたはその変異体の単離ポリペプチドが提供される。本発明の一つの態様において、「ポリペプチド」は配列番号2のアミノ酸配列、およびこのエポチロンBヒドロキシラーゼと本質的に同じ生物学的活性および/または機能を保持した断片または変異体を含むことを意味する。本発明の他の態様において、「ポリペプチド」は配列番号4のアミノ酸配列、およびこのフェレドキシンと本質的に同じ生物学的活性および/または機能を保持した断片または変異体を含むことを意味する。
本明細書において「変異体」とは、配列番号2または4と比較して保存的なアミノ酸置換のあるアミノ酸配列を有し、配列番号2または4と同じ生物学的活性および/または機能を示すことが証明されたポリペプチドを含むことを意味する。「保存的なアミノ酸置換」とは、脂肪族アミノ酸(Ala、Val、LeuおよびIleなど)、ヒドロキシル残基SerおよびThr、酸性残基AspおよびGlu、およびアミド残基AsnおよびGlnのうちのあるアミノ酸を別のアミノ酸で置き換えることを含むことを意味する。本発明の例示的な変異体のアミノ酸配列は配列番号43〜49に示してあり、これら例示的な変異体のアミノ酸置換は上記の表2に記載してある。
本明細書において「突然変異体」とは、配列番号1または3と比較して核酸の1またはそれ以上の点変異、または欠失または付加を有する核酸配列によってコードされているが、配列番号1または3によってコードされたポリペプチドと同様の活性を依然として有するポリペプチドを含むことを意味する。好ましい態様において、基質特異性および/またはそれによってコードされるポリペプチドからの収率を変えるような突然変異を核酸に生じさせる。エポチロンBヒドロキシラーゼ遺伝子に関して突然変異の好ましい領域は、該酵素の活性部位を含む約113アミノ酸残基をコードする核酸配列の領域である。同様に好ましいのは、配列番号1のアミノ酸位置GLU31、ARG67、ARG88、ILE92、ALA93、VAL106、ILE130、ALA140、MET176、PHE190、GLU231、SER294、PHE237、またはILE365に少なくとも一つのアミノ酸置換を有する突然変異である。本発明の例示的な突然変異体ebh25−1、ebh10−53、ebh24−16、ebh24−16d8、ebh24−16c11、ebh24−16−16、ebh24−16−74、ebh24−16g8、ebh24−16b9、およびそのような突然変異体をコードする核酸配列を、それぞれ配列番号31、33、35、61、63、65、67、69、71、73および75、および配列番号30、32、34、60、62、64、66、68、70、72および74に示す。
エポチロンBヒドロキシラーゼの三次元モデルもまた、相同タンパク質であるEryF(PDBコード1KIN鎖A)の既知構造に基づき、Greerら(Comparative modeling of homologous proteins. Methods In Enzymology 202239-52, 1991)、Leskら(Homology Modeling: Inferences from Tables of Aligned Sequences. Curr. Op. Struc. Biol. (2) 242-247, 1992)、およびCardozoら(Homology modeling by the ICM method. Proteins 23, 403-14, 1995)の一般的教示に従って構築した。これら配列のホモロジーは34%である。エポチロンBヒドロキシラーゼの配列(配列番号2)とEryFの配列(PDBコード1KIN鎖A;配列番号76)とのアラインメントを図3に示す。EryFとの配列アラインメントに基づくエポチロンBヒドロキシラーゼのホモロジーモデルを図4に示す。
EryF(PDBコード1JIN)と対比したエポチロンBヒドロキシラーゼモデルのエネルギープロットも作製し、図5に示す。51残基の平均ウインドウサイズをある残基位置で用いて、該残基を中央位置とした配列にある51残基のエネルギーの平均を計算した。図5に示すように、配列に沿ったすべてのエネルギーは0未満であり、図4および付表1に示すモデル構造が妥当であることを示していた。
図4のエポチロンBヒドロキシラーゼのホモロジーモデルに示す三次元構造は、付表1に示す構造座標によって定められる。「構造座標」とは、ホモロジーモデルの構築から生成したデカルト座標をいう。しかしながら、当業者には理解されるであろうように、あるタンパク質についての構造座標のセットは三次元での形状を定める点の相対的なセットである。それゆえ、全く異なる座標のセットが同様のまたは同一の形状を定め得ることがありうる。さらに、異なるアラインメント鋳型を用いおよび/またはホモロジーモデルを生成する際に異なる方法を用いた同様のホモロジーモデルの生成から生じるように、個々の座標の些細な変化は全体の形状にはわずかな影響しか及ぼさないであろう。座標における変化はまた、構造座標の数学的操作ゆえにも生じる。たとえば、付表1に示す構造座標は、構造座標の細分化(fractionalization)、構造座標のセットへの整数の加法または減法、構造座標の反転またはこれらの組み合わせによって操作することができるであろう。
それゆえ、ある分子またはその一部が上記エポチロンBヒドロキシラーゼの全体または一部と同じと考えられるほどに充分に類似しているか否かを決定するために種々のコンピューター分析が必要である。そのような分析は、SYBYLバージョン6.7またはINSIGHTII(Molecular Simulations Inc.、サンジエゴ、カリフォルニア)バージョン2000などの現行のソフトウエアアプリケーションで添付の使用者仕様の記載に従って行うことができる。
たとえば、プログラムSYBYLの重ね合わせツールは、異なる構造間および同じ構造の異なるコンホメーション間での比較を可能にする。SYBYLに用いられた構造を比較するための手順は4つのステップに分けられる:(1)比較すべき構造をローディングする;(2)これら構造における原子の等価性を定める;(3)適合操作を行う;ついで(4)結果を分析する。各構造を名称により確認する。一つの構造を標的(すなわち、固定した構造)とし、第二の構造(すなわち、移動させる構造)をソース構造とする。SYBYL内での原子の等価性は使用者の入力によって定められるので、本発明のこの側面の目的のためには、等価な原子は比較した2つの構造間のすべての保存された残基についてのタンパク質骨格原子(N、Cα、CおよびO)として定めることにする。さらに、リジッドな適合操作のみを考慮する。リジッドな適合法を使用する場合は、作業している構造を平行移動させ、回転させて標的構造との最適の適合が得られるようにする。適合操作は、等価な原子の特定したペアの適合の二乗平均差が絶対的な最小値となるよう、移動させる構造に適用すべき最適の平行移動および回転を計算するアルゴリズムを用いる。この数字(オングストロームで与えられる)はSYBYLにより報告されている。
本発明の目的のためには、付表1に列挙した構造座標によって記載された対応骨格原子上で重ね合わせたときに、保存された残基骨格原子(N、Cα、CおよびO)の二乗平均偏差が約4.0オングストローム未満であるエポチロンBヒドロキシラーゼのホモロジーモデルはすべて同一であると考えられる。さらに好ましくは、二乗平均偏差は約3.0オングストローム未満である。さらに好ましくは、二乗平均偏差は約2.0オングストローム未満である。
本発明の目的のためには、付表1に列挙した構造座標によって記載された対応骨格原子上で重ね合わせたときに、保存された残基骨格原子(N、Cα、CおよびO)の二乗平均偏差が約2.0オングストローム未満であるエポチロンBヒドロキシラーゼのホモロジーモデルはすべて同一であると考えられる。さらに好ましくは、二乗平均偏差は約1.0オングストローム未満である。
本発明の他の態様において、骨格原子が他の元素で置換され、対応の骨格原子上で重ね合わせたときに低い二乗平均偏差を有する構造モデルは同一であると考えられる。たとえば、もとの骨格炭素原子、および/または窒素原子、および/または酸素原子が他の元素で置換されており、付表1に列記する構造座標によって記載された対応の骨格原子上で重ね合わせたときに約4.0オングストローム、好ましくは約3.0オングストローム、さらに一層好ましくは約2オングストローム未満の二乗平均偏差を有するホモロジーモデルは同一であると考えられる。
「二乗平均偏差」とは、平均からの偏差の二乗の算術平均の平方根を意味する。それは、トレンドまたは対象からの偏差または変異を表現するための一つの方法である。本発明の目的のためには、「二乗平均偏差」は、本明細書に記載した構造座標によって定められる複合体のエポチロンBヒドロキシラーゼ部分の骨格の関連部分からのタンパク質の骨格の変異を定める。
本発明は、ホモロジーモデルによって具体化されるように、エポチロンBヒドロキシラーゼのさらなる突然変異体の構造ベースのデザインを可能にする。たとえば、本発明のホモロジーモデルを用い、エポチロンBヒドロキシラーゼの結合部位の10オングストローム内に存在する残基が今や定められた。これら残基は、付表1に示すように、LEU39、GLN43、ALA45、MET57、LEU58、HIS62、PHE63、SER64、SER65、ASP66、ARG67、GLN68、SER69、LEU74、MET75、VAL76、ALA77、ARG78、GLN79、ILE80、ASP84、LYS85、PRO86、PHE87、ARG88、PRO89、SER90、LEU91、ILE92、ALA93、MET94、ASP95、HIS99、ARG103、PHE110、ILE155、PHE169、GLN170、CYS172、SER173、SER174、ARG175、MET176、LEU177、SER178、ARG179、ARG186、PHE190、LEU193、VAL233、GLY234、LEU235、ALA236、PHE237、LEU238、LEU239、LEU240、ILE241、ALA242、GLY243、HIS244、GLU245、THR246、THR247、ALA248、ASN249、MET250、LEU283、THR287、ILE288、ALA289、GLU290、THR291、ALA292、THR293、SER294、ARG295、PHE296、ALA297、THR298、GLU312、GLY313、VAL314、VAL315、GLY316、VAL344、ALA345、PHE346、GLY347、PHE348、VAL350、HIS351、GLN352、CYS353、LEU354、GLY355、GLN356、LEU358、ALA359、GLU362、LYS389、ASP391、SER392、THR393、ILE394およびTYR395を含む。これら位置の1またはそれ以上に突然変異を有する突然変異体は変化した生物学的機能および/または特異性を示すことが予期され、それゆえ本発明の好ましい突然変異体の他の態様を構成する。好ましい突然変異体の他の態様は、該エポチロンBヒドロキシラーゼの骨格原子からの二乗平均偏差が約4.0オングストローム以下である分子である。
エポチロンBヒドロキシラーゼホモロジーモデルまたはその部分の構造座標は、機械読み取り可能な記憶媒体に入れておく。そのようなデータは、医薬の発見などの種々の目的に用いることができる。
従って、本発明の他の側面は、付表1に示す構造座標でコードしたデータ記憶材料を含む機械読み取り可能なデータ記憶媒体に関する。
エポチロンBヒドロキシラーゼの三次元モデル構造はまた、生物学的機能のモデュレーターおよび該酵素の潜在的な基質を同定するのに用いることもできる。そのようなモデュレーターを同定するのに種々の方法またはその組み合わせを用いることができる。
たとえば、付表1に従い、エポチロンBヒドロキシラーゼの結合部位に空間的に適合する被験化合物をモデル化することができる。アミノ酸のLEU39、GLN43、ALA45、MET57、LEU58、HIS62、PHE63、SER64、SER65、ASP66、ARG67、GLN68、SER69、LEU74、MET75、VAL76、ALA77、ARG78、GLN79、ILE80、ASP84、LYS85、PRO86、PHE87、ARG88、PRO89、SER90、LEU91、ILE92、ALA93、MET94、ASP95、HIS99、ARG103、PHE110、ILE155、PHE169、GLN170、CYS172、SER173、SER174、ARG175、MET176、LEU177、SER178、ARG179、ARG186、PHE190、LEU193、VAL233、GLY234、LEU235、ALA236、PHE237、LEU238、LEU239、LEU240、ILE241、ALA242、GLY243、HIS244、GLU245、THR246、THR247、ALA248、ASN249、MET250、LEU283、THR287、ILE288、ALA289、GLU290、THR291、ALA292、THR293、SER294、ARG295、PHE296、ALA297、THR298、GLU312、GLY313、VAL314、VAL315、GLY316、VAL344、ALA345、PHE346、GLY347、PHE348、VAL350、HIS351、GLN352、CYS353、LEU354、GLY355、GLN356、LEU358、ALA359、GLU362、LYS389、ASP391、SER392、THR393、ILE394およびTYR395によって定められるエポチロンBヒドロキシラーゼの結合領域の10オングストローム以内のアミノ酸の構造座標、および配位結合したヘム基HEM1もまた、そのようなモデュレーターの所望の構造的および化学的な特徴を同定するのに用いることができる。同定した構造的なまたは化学的な特徴は、ついで潜在的なエポチロンBヒドロキシラーゼのリガンドとしての化合物をデザインまたは選択するのに用いることができる。構造的および化学的な特徴とは、共有結合、ファンデアワールス相互作用、水素結合相互作用、電荷相互作用、疎水性相互作用、および双極子相互作用を含むことを意味するが、これらに限られるものではない。ついで、潜在的なエポチロンBヒドロキシラーゼリガンドとして同定された化合物は合成し、被験化合物のエポチロンBヒドロキシラーゼへの結合を特徴とするアッセイにおいて、または小さな分子の存在下でプロテアーゼ標的を変調するエポチロンBヒドロキシラーゼの能力を特徴付けることで、スクリーニングすることができる。潜在的なエポチロンBヒドロキシラーゼリガンドのスクリーニングに有用なアッセイの例としては、インシリコ(in silico)スクリーニング、インビトロアッセイおよび高処理量アッセイが挙げられるがこれらに限られるものではない。
この開示により当業者により理解されるであろうように、他の構造ベースのデザイン法を用いることができる。種々のコンピューターによる構造ベースのデザイン法が当該技術分野で開示されている。たとえば、エポチロンBヒドロキシラーゼの配列およびエポチロンBヒドロキシラーゼの構造(すなわち、付表1に示すエポチロンBヒドロキシラーゼの原子座標および/または上記で示した結合領域の10オングストローム内の原子座標)を入力することのできる多くのコンピューターモデリングシステムが利用できる。ついで、このコンピューターシステムは、1またはそれ以上のこれら領域の構造上の詳細を生成して潜在的なモデュレーターの相補的な構造の詳細を決定できるようにする。これらモデリングシステムにおけるデザインは、一般にエポチロンBヒドロキシラーゼに物理的および構造的に結合することのできる化合物に基づいている。さらに、該化合物は、エポチロンBヒドロキシラーゼとの結合を可能にするコンホメーションをとることができなければならない。幾つかのモデリングシステムでは、実際に合成および試験する前に潜在的なエポチロンBヒドロキシラーゼの基質またはモデュレーターの潜在的な抑制作用または結合作用を評価する。
所定のタンパク質標的に結合する能力について化学物質または断片をスクリーニングする方法もまたよく知られている。これら方法は、しばしばコンピュータースクリーン上で結合部位を目で調べることから開始する。ついで、選択した断片または化学物質をエポチロンBヒドロキシラーゼの結合領域に位置付ける。ドッキングをINSIGHTII、QUANTAおよびSYBYLなどのソフトウエアを用いて行い、ついでMMFF、CHARMMおよびAMBERなどの標準分子機械力場を用いてエネルギーの最小化および分子動力学を行う。本発明において有用な化学断片または化学物質の選択の助けとなるコンピュータープログラムの例としては、GRID(Goodford、1985)、AUTODOCK(Goodsell、1990)およびDOCK(Kuntzら、1982)が挙げられるが、これらに限られるものではない。
好ましい化学物質または断片を選択したら、お互い同士およびエポチロンBヒドロキシラーゼとの関係を視覚化することができ、ついで単一の潜在的なモデュレーターに組み立てることができる。個々の化学物質を組み立てるのに有用なプログラムとしては、CAVEAT(Bartlettら、1989)および3Dデータベースシステム(Martin、1992)が挙げられるが、これらに限られるものではない。
別法として、空白の結合部位を用いるかまたは既知のインヒビターの一部を含めてデノボで化合物をデザインすることができる。このタイプのデザイン法としては、LUDI(Bohm、1992)およびLeapFrog(Tripos Inc.、セントルイス、ミズーリ)が挙げられるが、これらに限られるものではない。
DOCK(Kuntzら、1982)などのプログラムをホモロジーモデルからの原子座標で用いて、活性部位中の結合領域に潜在的に結合し、それゆえ合成および試験に適した候補である潜在的なリガンドをデータベースまたは仮想データベースから同定することができる。
また、本発明のポリヌクレオチドを含むベクター、およびエポチロンBヒドロキシラーゼまたはその活性な断片および該酵素の変異体または突然変異体および/またはフェレドキシンまたはその活性な断片を産生すべく本発明のベクターで遺伝子操作した宿主細胞も本発明で提供される。一般に、ポリヌクレオチドを維持、増殖または発現してこれらポリペプチドを宿主細胞で産生するのに適したいかなるベクターもこの観点から発現に用いることができる。本発明のこの側面によれば、ベクターは、たとえば、プラスミドベクター、一本鎖または二本鎖ファージベクター、または一本鎖または二本鎖RNAまたはDNAウイルスベクターであってよい。ベクターは染色体外のものであってよく、または宿主染色体に組み込むべくデザインしたものであってよい。そのようなベクターとしては、染色体由来、エピソーム由来およびウイルス由来のベクター、たとえば、細菌プラスミド、バクテリオファージ、酵母エピソーム、酵母染色体配列、およびウイルス(バキュロウイルス、パポーバウイルス、SV40、ワクシニアウイルス、アデノウイルス、鶏痘ウイルス、偽狂犬病ウイルスおよびレトロウイルスなど)由来のベクター、およびそれらの組み合わせに由来するベクター、たとえばプラスミドとバクテリオファージの遺伝子配列、コスミドとファージミドに由来するベクターが挙げられるが、これらに限られるものではない。
原核生物宿主に有用な発現ベクターとしては、pBluescript、pGEX−2T、pUCベクター、pETベクター、ColE1、pCR1、pBR322、pMB9、pCW、pBMS200、pBMS2020、PIJ101、PIJ702、pANT849、pOJ260、pOJ446、pSET152、pKC1139、pKC1218、pFD666およびそれらの誘導体を含む、大腸菌、バシラスまたはストレプトミセスからのものなどの細菌プラスミド、広宿主範囲プラスミド、たとえば、RP4、ファージDNA、たとえば、ファージラムダの多数の誘導体、たとえばNM989、λGT10およびλGT11、および他のファージ、たとえばM13および繊維状一本鎖ファージDNAが挙げられるが、これらに限られるものではない。
酵母に使用するための本発明のベクターは、典型的に、酵母に使用するのに適した複製起点、および酵母で機能性の選択マーカーを含んでいるであろう。本発明において有用な酵母ベクターの例としては、酵母組み込み型プラスミド(たとえば、YIp5)および酵母自己複製型プラスミド(YRpおよびYEp系列プラスミド)、酵母動原体プラスミド(YCp系列プラスミド)、酵母人工染色体(YAC)(YLpと称する酵母の線状プラスミド、pGPD−2、2μプラスミドおよびその誘導体に基づく)、およびGietzら、Gene, 74: 527-34 (1988)に記載されているもの(YIplac、YEplacおよびYCplac)のような改良されたシャトルベクターが挙げられるが、これらに限られるものではない。
組換え発現に有用な哺乳動物ベクターは、ウイルス複製起点、たとえばSV40複製起点(COS1細胞やCOS7細胞などの大きなT抗原を発現する細胞株での複製用)、パピローマウイルス複製起点、または長期エピソーム複製のためのEBV複製起点(たとえば、EBV EBNA−1遺伝子産物およびアデノウイルスE1Aを構成的に発現する293−EBNA細胞での使用のため)を含んでいてよい。哺乳動物細胞での発現は、pSV2、pBC12BI、およびp91023、pCDNAベクター、並びに溶菌ウイルスベクター(たとえば、ワクシニアウイルス、アデノウイルス、およびバキュロウイルス)、エピソームウイルスベクター(たとえば、ウシパピローマウイルス)、およびレトロウイルスベクター(たとえば、マウスレトロウイルス)を含む(これらに限られるものではない)様々なプラスミドを用いて行うことができる。昆虫細胞に有用なベクターとしては、バキュロウイルスベクターおよびpVL941が挙げられる。
mRNAの転写を指令する適当なプロモーターの選択および発現ベクターの構築はよく知られている。しかしながら、一般に、発現構築物は転写の開始および終止のための部位、および転写された領域中に翻訳のためのリボソーム結合部位を含んでいるであろう。構築物によって発現された成熟転写物のコード部分は、始めに翻訳開始コドン、および翻訳すべきポリペプチドの末端にほぼ位置して終止コドンを含んでいるであろう。
原核生物に有用なプロモーターの例としては、ファージプロモーター、たとえば、ファージラムダpLプロモーター、trcプロモーター、trpプロモーターとlacプロモーターとに由来するハイブリッド、バクテリオファージT7プロモーター、TACまたはTRC系、ファージラムダの主要オペレーターおよびプロモーター領域、fdコートタンパク質の制御領域、snpAプロモーター、melCプロモーター、ermE*プロモーターまたはaraBADオペロンが挙げられるが、これらに限られるものではない。酵母に有用なプロモーターの例としては、CYC1プロモーター、GAL1プロモーター、GAL10プロモーター、ADH1プロモーター、酵母α−交配系(α-mating system)のプロモーター、およびGPDプロモーターが挙げられるが、これらに限られるものではない。哺乳動物発現ベクターで日常的に用いられるプロモーターの例としては、CMV前所期プロモーター、HSVチミジンキナーゼプロモーター、初期および後期SV40プロモーター、レトロウイルスLTRのプロモーター、たとえばラウス肉腫ウイルス(RSV)由来のもの、およびメタロチオネインプロモーター、たとえばマウスメタロチオネインIプロモーターが挙げられるが、これらに限られるものではない。
ポリヌクレオチドを含むベクターは、感染、形質導入、トランスフェクション、トランスベクション(transvection)および形質転換を含むよく知られた技術のいずれを用いても宿主細胞中に導入することができる。ポリヌクレオチドは、単独で、または、たとえば選択マーカーまたはフェレドキシンレダクターゼをコードするさらなるポリヌクレオチドとともに宿主に導入することができる。本発明の好ましい態様では、エポチロンBヒドロキシラーゼおよびフェレドキシンをコードするポリヌクレオチドを宿主細胞に導入する。種々の発現構築物のための宿主細胞がよく知られており、当業者は本発明のこの側面に従ってエポチロンBヒドロキシラーゼおよび/またはフェレドキシンを発現するための宿主細胞を日常的に選択することができる。本発明において有用な哺乳動物発現系の例としては、C127、3T3、CHO、HeLa、ヒト腎臓293およびBHK細胞株、およびサル腎臓繊維芽細胞のCOS−7細胞株が挙げられるが、これらに限られるものではない。
別法として、本明細書において例示するように、エポチロンBヒドロキシラーゼおよびフェレドキシンを微生物中で組換えにより発現することができる。
従って、本発明の他の側面は、エポチロンBヒドロキシラーゼ単独またはエポチロンBヒドロキシラーゼをフェレドキシンとともに発現し、末端アルキル基を有する化合物、とりわけエポチロンをヒドロキシル化して末端ヒドロキシアルキル基を有する化合物を生成することのできる、組換えにより生成した微生物に関する。組換えにより生成する微生物は、細菌細胞などの細胞をエポチロンBヒドロキシラーゼをコードする核酸配列を含むプラスミドで形質転換することにより生成する。好ましい態様において、エポチロンBヒドロキシラーゼまたはその突然変異体または変異体をコードする核酸とエポチロンBヒドロキシラーゼ遺伝子の下流に位置するフェレドキシンをコードする核酸配列とを含むプラスミドで細胞を形質転換する。これらプラスミドで形質転換して本発明の組換え微生物を生成することのできる微生物の例としては、大腸菌、バシラス・メガテリウム(Bacillus megaterium)、アミコラトプシス・オリエンタリス、ソランジウム・セルロスム、ロドコッカス・エリスロポリス(Rhodococcus erythropolis)、およびストレプトミセス・リビダンス、ストレプトミセス・ビルジニエ(Streptomyces virginiae)、ストレプトミセス・ベネズエレ(Streptomyces venezuelae)、ストレプトミセス・アルブス(Streptomyces albus)、ストレプトミセス・コエリコロール(Streptomyces coelicolor)、ストレプトミセス・リモススおよびストレプトミセス・グリセウス(Streptomyces griseus)などのストレプトミセス種が挙げられるが、これらに限られるものではない。
本発明の組換えにより生成した微生物は、末端ヒドロキシアルキル基を含む化合物、とりわけエポチロンの産生のための微生物プロセスまたは方法に有用である。一般に、ヒドロキシアルキル含有生成物は、資化しうる炭素および窒素源を含む水性栄養培地中の適当な基質の存在下、水中の好気性条件下で末端炭素原子またはアルキルを選択的にヒドロキシル化することのできる組換えにより生成した微生物または該微生物からの酵素を培養することにより生成することができる。
本発明の方法の基質として用いる適当なエポチロンは、本発明の酵素的ヒドロキシル化を受けることのできる末端炭素原子または末端アルキル基を有するそのような化合物であればいかなるものであってもよい。出発物質、すなわち基質は、ソランジウム・セルロスムなどの天然源から単離することもできるし、または合成により生成したエポチロンであってもよい。末端炭素原子または末端アルキル基を有し酵素的ヒドロキシル化を受けることのできる他の基質を本明細書に記載する方法によって用いることができる。たとえば、コンパクチンを基質として用いることができ、これはヒドロキシル化されると化合物プラバスタチンを生成する。アクチノマデュラ(Actinomadura)株によりコンパクチンをヒドロキシル化してプラバスタチンを生成する方法は、米国特許第5,942,423号および同第6,274,360号に記載されている。
たとえば、本発明の組換え微生物を用い、WO00/39276、米国特許出願第09/468,854号(1999年12月21日出願)(その本文を、あたかも詳細に示すごとく本明細書中に引用する)の記載に従って少なくとも1のエポチロンを調製することができる。
下記式I:
HO−CH2−(A1)n−(Q)m−(A2)o−E (I)
(式中、A1およびA2は、それぞれ独立に任意に置換されたC1−C3アルキルおよびアルケニルよりなる群から選ばれる;
Qは、1〜3の環および少なくとも1の環中に少なくとも一つの炭素−炭素二重結合を含む任意に置換された環系;
n、mおよびoは、0および1よりなる群から選ばれた整数、その際、mまたはnまたはoの少なくとも一つは1である;および
Eはエポチロンコアである)に示すエポチロンを調製することができる。
HO−CH2−(A1)n−(Q)m−(A2)o−E (I)
(式中、A1およびA2は、それぞれ独立に任意に置換されたC1−C3アルキルおよびアルケニルよりなる群から選ばれる;
Qは、1〜3の環および少なくとも1の環中に少なくとも一つの炭素−炭素二重結合を含む任意に置換された環系;
n、mおよびoは、0および1よりなる群から選ばれた整数、その際、mまたはnまたはoの少なくとも一つは1である;および
Eはエポチロンコアである)に示すエポチロンを調製することができる。
本発明の方法は、下記式II:
CH3−(A1)n−(Q)m−(A2)o−E (II)
(式中、A1、Q、A2、E、n、m、およびoは前記と同じ)で示される少なくとも1のエポチロンを、式IIの化合物のヒドロキシル化を選択的に触媒することができる組換えにより生成した微生物または該微生物からの酵素と接触させる工程、ついで該ヒドロキシル化を行う工程を含む。
CH3−(A1)n−(Q)m−(A2)o−E (II)
(式中、A1、Q、A2、E、n、m、およびoは前記と同じ)で示される少なくとも1のエポチロンを、式IIの化合物のヒドロキシル化を選択的に触媒することができる組換えにより生成した微生物または該微生物からの酵素と接触させる工程、ついで該ヒドロキシル化を行う工程を含む。
好ましい態様において、出発物質はエポチロンBである。エポチロンBは、DE4138042およびWO93/10121に記載されているように、ソランジウム・セルロスムSo ce90の発酵から得ることができる。この株は、ドイチェ・ザムルング・フォン・ミクロオルガニスメン(ジャーマン・コレクション・オブ・マイクロオーガニスムス)(DSM)に受託番号6773にて寄託してある。発酵プロセスはまた、Hofle, G.ら、Angew. Chem. Int. Ed. Engl., Vol. 35, No. 13/14, 1567-1569 (1996)にも記載されている。エポチロンBはまた、化学的手段、たとえば、Meng, D.ら、J. Am. Chem. Soc., Vol. 119, No. 42, 10073-10092 (1996); Nicolaou, K.ら、J. Am. Chem. Soc., Vol. 119, No. 34, 7974-7991 (1997)およびSchinzer, D.ら、Chem. Eur. J., Vol. 5, No. 9, 2483-2491 (1999)に記載の化学的手段により得ることができる。
本発明の方法に使用すべく選択した組換えにより生成した微生物の増殖は、適当な栄養培地の使用により当業者によって達成されうる。組換えにより生成した微生物の増殖のための適当な培地は、微生物細胞の増殖に必要な栄養素を提供するものを含む。たとえば、T. NagodawithanaおよびJ. M. Wasileski、第2章:“Media Design for Industrial Fermentations”、Nutritional Requirements of Commercially Important Microorganism、T. W. NagodawithanaおよびG. Reed編、Esteekay Associates, Inc.、ミルウォーキー、ウイスコンシン、18-45 (1998); T. L. MillerおよびB. W. Churchill、第10章:“Substrates for Large-Scale Fermentations”、Manual of Industrial Microbiology and Biotechnology、A. L. DemainおよびN. A. Solomon編、American Society for Microbiology、ワシントン、D.C., 122-136 (1986)を参照。増殖のための典型的な培地は、必要な炭素源、窒素源、および微量元素を含む。インデューサーを培地に加えることもできる。本明細書で使用するインデューサーなる語は、組換えにより生成した微生物細胞内で所望の酵素活性の生成を促進するあらゆる化合物を包含する。本明細書に使用する典型的なインデューサーとしては、基質を溶解するのに使用する溶媒、たとえば、ジメチルスルホキシド、ジメチルホルムアミド、ジオキサン、エタノールおよびアセトンが挙げられる。さらに、エポチロンBなどのある種の基質もまたインデューサーと考えられる。
炭素源としては、グルコース、ガラクトース、マルトース、ショ糖、マンニトール、ソルビトール、グリセリン、デンプンなどの糖;酢酸ナトリウム、クエン酸ナトリウムなどの有機酸;およびエタノール、プロパノールなどのアルコールが挙げられる。好ましい炭素源としては、グルコース、フルクトース、ショ糖、グリセリンおよびデンプンが挙げられるが、これらに限られるものではない。
窒素源としては、N−ZアミンA、トウモロコシ浸漬液、大豆粉、牛肉エキス、酵母エキス、トリプトン、ペプトン、綿実粕、ピーナッツ粕、グルタミン酸ナトリウムなどのアミノ酸、硝酸ナトリウム、硫酸アンモニウムなどが挙げられる。
微量元素としては、マグネシウム、マンガン、カルシウム、コバルト、ニッケル、鉄、ナトリウム塩およびカリウム塩が挙げられる。リン酸塩もまた微量にて、または好ましくは微量よりも多い量で加えることができる。
発酵に用いる培地は、1を超える炭素または窒素源または他の栄養素を含んでいてよい。
本発明の方法による組換えにより生成した微生物の増殖および/またはヒドロキシル化のため、培地のpHは約5〜約8であるのが好ましく、温度は約14℃〜約37℃であり、好ましくは温度は28℃である。反応時間は1〜100時間、好ましくは8〜72時間である。
培地を、高速液体クロマトグラフィー(HPLC)によりモニターしてバイオトランスフォーメーションが完了するのに必要な時間インキュベートする。典型的には、バイオトランスフォーメーションを完了するのに要する時間は、基質を添加してから12〜100時間、好ましくは約72時間である。培地を、150〜300rpm、好ましくは約250rpmで作動し、行程が2インチのロータリーシェーカー(New Brunswick Scientific Innova 5000)上に置く。
発酵ブロスからのヒドロキシアルキル含有生成物の回収は、他の知られた生物学的に活性な物質の回収に普通に用いられる通常の手段により行うことができる。そのような回収手段の例としては、酢酸エチルなどの通常の溶媒を用いた抽出による単離および精製によるもの;pH調整によるもの;通常の樹脂の処理によるもの、たとえば、陰イオンまたは陽イオン交換樹脂または非イオン性吸着樹脂の処理によるもの;通常の吸着剤の処理によるもの、たとえば、蒸留により、または結晶化によるもの;または再結晶によるものなどが挙げられるが、これらに限られるものではない。
上記でバイオトランスフォーメーション反応混合物から得た抽出物は、カラムクロマトグラフィーおよび分析的薄層クロマトグラフィーによりさらに単離および精製することができる。
本発明の組換えにより生成した微生物が末端アルキル基を有するエポチロンを末端ヒドロキシアルキル基を有するエポチロンにバイオトランスフォーメーションする能力が証明された。これら実験において、実施例11にさらに詳細に記載するようにebh遺伝子を有するプラスミドを含有するストレプトミセス・リビダンスクローンを含む培養液を、エポチロンB懸濁液とともに攪拌しながら30℃にて3日間インキュベートした。インキュベートの試料を等容量の25%メタノール:75%n−ブタノールで抽出し、回転攪拌し、ついで5分間沈殿させた。200μlの有機相をHPLCバイアルに移し、HPLC/MSにより分析した(実施例12)。エポチロンFの生成物ピークは15.9分の保持時間で溶出し、524のプロトン化分子量を有していた。エポチロンB基質は19.0分で溶出し、508のプロトン化分子量を有していた。ピークの保持時間および分子量は公知の標準を用いて確認した。
ebhを発現する細胞によるエポチロンBのバイオトランスフォーメーションの速度を、ebh突然変異体によるバイオトランスフォーメーションの速度と比較した。ebhを発現する細胞は、ストレプトミセス・リビダンス(pANT849−ebh)の凍結胞子調製物を含んでいる。突然変異体を発現する細胞は、ストレプトミセス・リビダンス(pANT849−ebh10−53)およびストレプトミセス・リビダンス(pANT849−ebh24−16)の凍結胞子調製物を含んでいる。ストレプトミセス・リビダンスTK24の凍結胞子調製物は対照として用いた。細胞を30℃にて数日間、プレインキュベートした。このプレインキュベーションの後、100%EtOH中のエポチロンBを各培養液に最終濃度0.05%w/vにて加えた。ついで、ストレプトミセス・リビダンス(pANT849−ebh24−16)の培養液(この場合、エポチロンBは48時間で完全にエポチロンFに変換された)を除いて、試料を、0、24、48および72時間の時点で採取した。試料をHPLCにより分析した。結果を0時間でのエポチロンBのパーセントとして計算した。
エポチロンB:
エポチロンF:
ebhを発現する細胞がコンパクチンをプラバスタチンにバイオトランスフォーメーションする能力についても調べた。これらの実験において、ストレプトミセス・リビダンス(pANT849)またはストレプトミセス・リビダンス(pANT849−ebh)の凍結胞子調製物を30℃で数日間増殖させた。プレインキュベーションの後、各細胞培養液のアリコートをポリプロピレン培養チューブに移し、コンパクチンを各培養チューブに加え、ついで培養チューブを30℃、250rpmにて24時間インキュベートした。ついで、培養ブロスのアリコートを抽出し、対照のストレプトミセス・リビダンス(pANT849)培養液と比較したコンパクチンおよびプラバスタチン値をHPLCにより測定した。
出発物質のコンパクチン濃度のパーセントとしてのコンパクチンおよびプラバスタチン:
上記で検討したように、突然変異体ebh25−1(配列番号30)は変化した基質特異性を示し、この突然変異体によるエポチロンBのバイオトランスフォーメーションはエポチロンBまたはエポチロンFとは異なるHPLC溶出時間を有する生成物という結果となる。この未知の試料をLC−MSにより分析したところ、分子量が523(M.W.)であり、エポチロンBの単一のヒドロキシル化と一致することがわかった。このバイオトランスフォーメーション生成物の構造を、MSおよびNMRデータに基づいて(エポチロンBのデータと比較して)24−ヒドロキシル−エポチロンBと決定した:
分子式:C27H41NO7S
分子量:523
質量スペクトル:ES+(m/z):524([M+H]+)、506
LC/MS/MS:+ESI(m/z):524、506、476、436、320
HRMS:[M+H]+として;計算値:524.2682;実測値:524.2701
HPLC(Rt):7.3分(分析的HPLC系で)
LC/NMR:ケミカルシフトを観察
Varian AS-600(プロトン:599.624MHz)
溶媒D2O/CD3CN(δ1.94):〜4/6
*1.8〜2.1ppmのピークは溶媒抑制(solvent suppression)のために観察されなかった。
分子量:523
質量スペクトル:ES+(m/z):524([M+H]+)、506
LC/MS/MS:+ESI(m/z):524、506、476、436、320
HRMS:[M+H]+として;計算値:524.2682;実測値:524.2701
HPLC(Rt):7.3分(分析的HPLC系で)
LC/NMR:ケミカルシフトを観察
Varian AS-600(プロトン:599.624MHz)
溶媒D2O/CD3CN(δ1.94):〜4/6
プロトンのケミカルシフトは以下のように割り当てられた:
*SSP:溶媒抑制のために観察されず。
従って、本発明の組成物および方法は、微小管安定化剤である公知の化合物並びに微小管安定化剤として有用であることが予期されるエポチロンアナログ(24−ヒドロキシル−エポチロンB(式A)など)および薬理学的に許容しうるその塩を含む新規な化合物を生成するのに有用である。これら組成物および方法を用いて生成した微小管安定化剤は、以下のものを含む(これらに限られるものではない)種々の癌および他の増殖性疾患の治療に有用である:
膀胱、乳房、直腸、腎臓、肝臓、肺、卵巣、膵臓、胃、子宮頸、甲状腺および皮膚のものを含む癌腫;扁平上皮細胞癌腫を含む;
白血病、急性リンパ球性白血病、急性リンパ芽球性白血病、B細胞リンパ腫、T細胞リンパ腫、ホジキンリンパ腫、非ホジキンリンパ腫、ヘアリーセルリンパ腫およびバーキットリンパ腫を含むリンパ系の造血性腫瘍;
急性および慢性の骨髄性白血病および前骨髄球性白血病を含む骨髄系の造血性腫瘍;
線維肉腫および横紋筋肉腫を含む間充織由来の腫瘍;
黒色腫、セミノーマ、奇形癌、神経芽細胞腫および神経膠芽細胞腫を含む他の腫瘍;
星状細胞種、神経芽細胞腫、神経膠芽細胞腫、および神経鞘腫を含む中枢および末梢神経系の腫瘍;
線維肉腫、横紋筋肉腫および骨肉腫を含む間充織由来の腫瘍;
黒色腫、色素性乾皮症、角化棘細胞腫、セミノーマ、甲状腺小胞癌および奇形癌を含む他の腫瘍。
膀胱、乳房、直腸、腎臓、肝臓、肺、卵巣、膵臓、胃、子宮頸、甲状腺および皮膚のものを含む癌腫;扁平上皮細胞癌腫を含む;
白血病、急性リンパ球性白血病、急性リンパ芽球性白血病、B細胞リンパ腫、T細胞リンパ腫、ホジキンリンパ腫、非ホジキンリンパ腫、ヘアリーセルリンパ腫およびバーキットリンパ腫を含むリンパ系の造血性腫瘍;
急性および慢性の骨髄性白血病および前骨髄球性白血病を含む骨髄系の造血性腫瘍;
線維肉腫および横紋筋肉腫を含む間充織由来の腫瘍;
黒色腫、セミノーマ、奇形癌、神経芽細胞腫および神経膠芽細胞腫を含む他の腫瘍;
星状細胞種、神経芽細胞腫、神経膠芽細胞腫、および神経鞘腫を含む中枢および末梢神経系の腫瘍;
線維肉腫、横紋筋肉腫および骨肉腫を含む間充織由来の腫瘍;
黒色腫、色素性乾皮症、角化棘細胞腫、セミノーマ、甲状腺小胞癌および奇形癌を含む他の腫瘍。
本発明の組成物および方法を用いて生成した微小管安定化剤はまた脈管形成をも抑制し、それによって腫瘍の増殖に影響を及ぼし、腫瘍および腫瘍関連疾患の治療をもたらすであろう。これら化合物のそのような抗脈管形成特性はまた、網膜血管新生に関連するある種の盲目、関節炎、とりわけ炎症性関節炎、多発性硬化症、再狭窄および乾癬を含む(これらに限られるものではない)、抗脈管形成剤が適用される他の状態の治療にも有用であろう。
本発明の組成物および方法を用いて生成した微小管安定化剤は、正常な発生およびホメオスタシスにとって重要な生理的細胞死のプロセスであるアポトーシスを誘発または抑制するであろう。アポトーシス経路の変化はヒトの様々な疾患の病因に貢献する。式I、式IIおよび式Aによって示される化合物などの本発明の化合物は、アポトーシスのモデュレーターとして、癌、前癌病変、免疫応答関連疾患、ウイルス感染症、筋骨格系の変性疾患および腎臓疾患を含む(これらに限られるものではない)、アポトーシスに異常を有するヒトの様々な疾患の治療に有用であろう。
いかなる機構または形態学にも限定されることを意図することなく、本発明の組成物および方法を用いて生成した微小管安定化剤はまた、癌や他の増殖性疾患以外の状態を治療するのにも用いることができる。そのような状態としては、ヘルペスウイルス、ポックスウイルス、エプスタインバーウイルス、シンドビスウイルスおよびアデノウイルスなどのウイルス感染症;全身性エリテマトーデス、免疫媒体糸球体腎炎、慢性関節リウマチ、乾癬、炎症性腸疾患および自己免疫真性糖尿病などの自己免疫疾患;アルツハイマー病、AIDS関連痴呆、パーキンソン病、筋萎縮性側索硬化症、色素性網膜炎、脊髄性筋萎縮症および小脳変性などの神経変性疾患;AIDS;骨髄異形成症候群;再生不良性貧血;虚血障害関連心筋梗塞;卒中および再潅流障害;再狭窄;不整脈;アテローム性動脈硬化症;毒素誘因またはアルコール誘因の肝疾患;慢性貧血および再生不良性貧血などの血液疾患;骨粗鬆症および関節炎などの筋骨格系の変性疾患;アスピリン感受性鼻副鼻腔炎;嚢胞性線維症;多発性硬化症;腎臓疾患;および癌の痛みが挙げられるが、これらに限られるものではない。
以下の制限されない実施例は本発明をさらに説明するために提供される。
実施例
実施例1:試薬
R2培地を以下のようにして調製した。
ショ糖(103g)、K2SO4(0.25g)、MgCl2・6H2O(10.12g)、グルコース(10g)、Difcoカザミノ酸(0.1g)および蒸留水(800ml)を含む溶液を調製した。ついで、この溶液の80mlを、2.2gのDifco Bactoアガーを入れた200ml容のねじぶたビンに注いだ。ビンにふたをし、オートクレーブにかけた。使用時には培地を再溶解し、以下のオートクレーブ溶液を列挙した順に加えた:
1mlのKH2PO4(0.5%)
8mlのCaCl2・2H2O(3.68%)
1.5mlのL−プロリン(20%)
10mlのTES緩衝液(5.73%、pH7.2に調整)
ZnCl2(40mg)、FeCl3・6H2O(200mg)、CuCl2・2H2O(10mg)、MnCl2・4H2O(10mg)、Na2B4O7・10H2O(10mg)および(NH4)6Mo7O24・H2Oを含む0.2mlの微量元素溶液
0.5mlのNaOH(1N)(滅菌は必要ない)
0.5mlの栄養要求体に必要な成長因子(ヒスチジン(50μg/ml);システイン(37μg/ml);アデニン、グアニン、チミジンおよびウラシル(7.5μg/ml);およびビタミン(0.5μg/ml))
実施例
実施例1:試薬
R2培地を以下のようにして調製した。
ショ糖(103g)、K2SO4(0.25g)、MgCl2・6H2O(10.12g)、グルコース(10g)、Difcoカザミノ酸(0.1g)および蒸留水(800ml)を含む溶液を調製した。ついで、この溶液の80mlを、2.2gのDifco Bactoアガーを入れた200ml容のねじぶたビンに注いだ。ビンにふたをし、オートクレーブにかけた。使用時には培地を再溶解し、以下のオートクレーブ溶液を列挙した順に加えた:
1mlのKH2PO4(0.5%)
8mlのCaCl2・2H2O(3.68%)
1.5mlのL−プロリン(20%)
10mlのTES緩衝液(5.73%、pH7.2に調整)
ZnCl2(40mg)、FeCl3・6H2O(200mg)、CuCl2・2H2O(10mg)、MnCl2・4H2O(10mg)、Na2B4O7・10H2O(10mg)および(NH4)6Mo7O24・H2Oを含む0.2mlの微量元素溶液
0.5mlのNaOH(1N)(滅菌は必要ない)
0.5mlの栄養要求体に必要な成長因子(ヒスチジン(50μg/ml);システイン(37μg/ml);アデニン、グアニン、チミジンおよびウラシル(7.5μg/ml);およびビタミン(0.5μg/ml))
R2培地と同様にしてR2YE培地を調製した。しかしながら、5mlのDifco酵母エキス(10%)を使用時に各100ml容フラスコに加えた。
P(プロトプラスト)緩衝液を以下のようにして調製した。
以下のものからなる基礎溶液を調製した:
ショ糖(103g)
K2SO4(0.25g)
MgCl2・6H2O(2.02g)
R2培地について記載した微量元素の溶液(2ml)
蒸留水(800mlまで)
以下のものからなる基礎溶液を調製した:
ショ糖(103g)
K2SO4(0.25g)
MgCl2・6H2O(2.02g)
R2培地について記載した微量元素の溶液(2ml)
蒸留水(800mlまで)
ついで、基礎溶液の80mlアリコートを分配し、オートクレーブにかけた。使用前に以下のものを列挙した順にて各フラスコに加えた:
1mlのKH2PO4(0.5%)
10mlのCaCl2・2H2O(3.68%)
TES緩衝液(5.75%、pH7.2に調整)
1mlのKH2PO4(0.5%)
10mlのCaCl2・2H2O(3.68%)
TES緩衝液(5.75%、pH7.2に調整)
以下の滅菌溶液を混合することによりT(形質転換)緩衝液を調製した:
25mlのショ糖(10.3%)
75mlの蒸留水
R2培地について記載した1mlの微量元素溶液
1mlのK2SO4(2.5%)
25mlのショ糖(10.3%)
75mlの蒸留水
R2培地について記載した1mlの微量元素溶液
1mlのK2SO4(2.5%)
ついで、この溶液の9.3mlに以下のものを加える:
0.2mlのCaCl2(5M)
トリスの1M溶液から調製し、マレイン酸を加えてpH8.0に調整した0.5mlのトリスマレイン酸緩衝液。
使用に際しては、上記溶液の3重量部を前もってオートクレーブ滅菌したPEG1000の1重量部に加える。
0.2mlのCaCl2(5M)
トリスの1M溶液から調製し、マレイン酸を加えてpH8.0に調整した0.5mlのトリスマレイン酸緩衝液。
使用に際しては、上記溶液の3重量部を前もってオートクレーブ滅菌したPEG1000の1重量部に加える。
以下の滅菌溶液を混合することによりL(溶解)緩衝液を調製した:
100mlのショ糖(10.3%)
10mlのTES緩衝液(5.73%、pH7.2に調整)
1mlのK2SO4(2.5%)
R2培地について記載した1mlの微量元素溶液
1mlのKH2PO4(0.5%)
0.1mlのMgCl2・6H2O(2.5M)
1mlのCaCl2(0.25M)
100mlのショ糖(10.3%)
10mlのTES緩衝液(5.73%、pH7.2に調整)
1mlのK2SO4(2.5%)
R2培地について記載した1mlの微量元素溶液
1mlのKH2PO4(0.5%)
0.1mlのMgCl2・6H2O(2.5M)
1mlのCaCl2(0.25M)
CRM培地
以下の成分を1リットルのdH2O中に含む溶液を調製した:グルコース(10g)、ショ糖(103g)、MgCl2・6H2O(10.12g)、BBLTM trypticase soy broth(15g)(Becton Dickinson Microbiology Systems、スパークス、メリーランド、米国)、およびBBLTM酵母エキス(5g)(Becton Dickinson Microbiology Systems)。この溶液を30分間オートクレーブにかけた。培養液をプラスミドとともに増殖させるためチオストレプトンを10μg/mlの濃度にて加えた。
以下の成分を1リットルのdH2O中に含む溶液を調製した:グルコース(10g)、ショ糖(103g)、MgCl2・6H2O(10.12g)、BBLTM trypticase soy broth(15g)(Becton Dickinson Microbiology Systems、スパークス、メリーランド、米国)、およびBBLTM酵母エキス(5g)(Becton Dickinson Microbiology Systems)。この溶液を30分間オートクレーブにかけた。培養液をプラスミドとともに増殖させるためチオストレプトンを10μg/mlの濃度にて加えた。
エレクトロポレーション緩衝液
30%(w/v)PEG1000、10%グリセリン、および6.5%ショ糖を含む溶液をdH2O中にて調製した。この溶液を0.22μmの酢酸セルロース膜での真空濾過により滅菌した。
30%(w/v)PEG1000、10%グリセリン、および6.5%ショ糖を含む溶液をdH2O中にて調製した。この溶液を0.22μmの酢酸セルロース膜での真空濾過により滅菌した。
実施例2:株SC15847からの染色体DNAの抽出
グアニジン−界面活性剤溶解法、DNAzol試薬(Invitrogen、カールスバード、カリフォルニア、米国)を用い、アミコラトプシス・オリエンタリスの土壌単離株の表示SC15847(ATCC PT−1043)からゲノムDNAを単離した。SC15847培養液をF7培地(グルコース2.2%、酵母エキス1.0%、麦芽エキス1.0%、ペプトン0.1%、pH7.0)中、28℃にて24時間増殖させた。20mlの培養液を遠心分離により回収し、20mlのDNAzolに再懸濁し、ピペッティングにより混合し、ついでBeckman TJ6遠心管中で10分間遠心分離にかけた。10mlの100%エタノールを加え、数回逆さまにし、室温にて3分間貯蔵した。DNAをガラスピペットに巻きつけ、100%エタノールで洗浄し、10分間空気乾燥した。ペレットを500μlの8mM NaOHに再懸濁し、溶解したら30μlの1M HEPES(pH7.2)で中和した。
グアニジン−界面活性剤溶解法、DNAzol試薬(Invitrogen、カールスバード、カリフォルニア、米国)を用い、アミコラトプシス・オリエンタリスの土壌単離株の表示SC15847(ATCC PT−1043)からゲノムDNAを単離した。SC15847培養液をF7培地(グルコース2.2%、酵母エキス1.0%、麦芽エキス1.0%、ペプトン0.1%、pH7.0)中、28℃にて24時間増殖させた。20mlの培養液を遠心分離により回収し、20mlのDNAzolに再懸濁し、ピペッティングにより混合し、ついでBeckman TJ6遠心管中で10分間遠心分離にかけた。10mlの100%エタノールを加え、数回逆さまにし、室温にて3分間貯蔵した。DNAをガラスピペットに巻きつけ、100%エタノールで洗浄し、10分間空気乾燥した。ペレットを500μlの8mM NaOHに再懸濁し、溶解したら30μlの1M HEPES(pH7.2)で中和した。
実施例3:PCR反応
200〜500ngのゲノムDNAかまたは0.1μlのcDNA、フォアウォードプライマーおよびリバースプライマー[フォアウォードプライマーはP450−1+(配列番号23)かまたはP450−1a+(配列番号24)かまたはP450−2+(配列番号25)のいずれかであり、リバースプライマーはP450−3−(配列番号27)かまたはP450−2−(配列番号26)のいずれかであった]を含むPCR反応液を50μlの容量で調製した。プライマーはすべて1.4〜2.0μMの最終濃度で加えた。PCR反応液は1μlのTaq酵素(2.5単位)(Stratagene)、5μlのTaq緩衝液および4μlの2.5mM dNTPを用い、dH2Oを50μlまで加えて調製した。サイクル反応は、GeneampR PCRシステムにて以下のプロトコールで行った:95℃にて5分間、5サイクル[95℃にて30秒、37℃にて15秒(30%ランプ(ramp))、72℃にて30秒]、35サイクル(94℃にて30秒、65℃にて15秒、72℃にて30秒)、72℃にて7分間。反応から予期されるサイズは、P450−1+(配列番号23)またはP450−1a+(配列番号24)とP450−3−(配列番号27)とのプライマーペアについては340bp、P450−1+(配列番号23)とP450−2−(配列番号26)とのプライマーペアについては240bp、およびP450−2+(配列番号25)とP450−3−(配列番号27)とのプライマーペアについては130bpである。
200〜500ngのゲノムDNAかまたは0.1μlのcDNA、フォアウォードプライマーおよびリバースプライマー[フォアウォードプライマーはP450−1+(配列番号23)かまたはP450−1a+(配列番号24)かまたはP450−2+(配列番号25)のいずれかであり、リバースプライマーはP450−3−(配列番号27)かまたはP450−2−(配列番号26)のいずれかであった]を含むPCR反応液を50μlの容量で調製した。プライマーはすべて1.4〜2.0μMの最終濃度で加えた。PCR反応液は1μlのTaq酵素(2.5単位)(Stratagene)、5μlのTaq緩衝液および4μlの2.5mM dNTPを用い、dH2Oを50μlまで加えて調製した。サイクル反応は、GeneampR PCRシステムにて以下のプロトコールで行った:95℃にて5分間、5サイクル[95℃にて30秒、37℃にて15秒(30%ランプ(ramp))、72℃にて30秒]、35サイクル(94℃にて30秒、65℃にて15秒、72℃にて30秒)、72℃にて7分間。反応から予期されるサイズは、P450−1+(配列番号23)またはP450−1a+(配列番号24)とP450−3−(配列番号27)とのプライマーペアについては340bp、P450−1+(配列番号23)とP450−2−(配列番号26)とのプライマーペアについては240bp、およびP450−2+(配列番号25)とP450−3−(配列番号27)とのプライマーペアについては130bpである。
実施例4:エポチロンBヒドロキシラーゼ遺伝子およびフェレドキシン遺伝子のクローニング
20μgのSC15847ゲノムDNAをBglII制限酵素で37℃にて6時間消化した。30kナノセプカラム(Gelman Sciences、アンアーバー、ミシガン、米国)を用いてDNAを濃縮し、酵素および緩衝液を除去した。反応液を40μlに濃縮し、200μlのTEで洗浄した。ついで、消化生成物を0.7%アガロースゲルで分離し、12〜15kbの範囲のゲノムDNAをゲルから切り出し、Qiagenゲル抽出法を用いて精製した。ついで、ゲノムDNAを、BamHIで消化およびSAPI酵素(Roche Molecular Biochemicals、インディアナポリス、インディアナ、カタログ#1758 250)を用いて脱リン酸化しておいたプラスミドpWB19N(米国特許第5,516,679号)にライゲートした。ライゲーション反応は、1UのT4DNAリガーゼ(Invitrogen)を用い、15μlの容量で室温にて1時間行った。1μlのライゲーション液を100μlの化学的にコンピテントなDH10B細胞(Invitrogen)に形質転換し、100μlを30μg/mlのネオマイシンを含む5つのLBアガープレートに37℃にて一夜プレーティングした。
20μgのSC15847ゲノムDNAをBglII制限酵素で37℃にて6時間消化した。30kナノセプカラム(Gelman Sciences、アンアーバー、ミシガン、米国)を用いてDNAを濃縮し、酵素および緩衝液を除去した。反応液を40μlに濃縮し、200μlのTEで洗浄した。ついで、消化生成物を0.7%アガロースゲルで分離し、12〜15kbの範囲のゲノムDNAをゲルから切り出し、Qiagenゲル抽出法を用いて精製した。ついで、ゲノムDNAを、BamHIで消化およびSAPI酵素(Roche Molecular Biochemicals、インディアナポリス、インディアナ、カタログ#1758 250)を用いて脱リン酸化しておいたプラスミドpWB19N(米国特許第5,516,679号)にライゲートした。ライゲーション反応は、1UのT4DNAリガーゼ(Invitrogen)を用い、15μlの容量で室温にて1時間行った。1μlのライゲーション液を100μlの化学的にコンピテントなDH10B細胞(Invitrogen)に形質転換し、100μlを30μg/mlのネオマイシンを含む5つのLBアガープレートに37℃にて一夜プレーティングした。
5つのナイロンメンブレンサークル(Roche Molecular Biochemicals、インディアナポリス、インディアナ)に番号を付け、方向に印を付けた。メンブレンをプレート上に2分間置き、ついで5分間乾燥させた。ついで、メンブレンをファットマンフィルターディスク上に置き、10%SDSで5分間、1.5M NaClとともに0.5N NaOHで5分間、1.0Mトリス(pH8.0)とともに1.5M NaClで5分間、および2×SSCで15分間飽和させた。フィルターを以前にサザーンハイブリダイゼーションについて記載されたようにしてハイブリダイズさせた。ハイブリダイズしたコロニーを30μg/mlのネオマイシンを含む2mlのTBに採り、37℃で一夜増殖させた。プラスミドDNAをミニプレップカラム法(Mo Bio)を用いて単離した。このプラスミドをNPB29−1と称した。
実施例5:DNAシークエンシングおよび分析
クローニングしたPCR産物を、蛍光−染料−標識(fluorescent-dye-labeled)ターミネーターサイクルシークエンシング、Big-Dyeシークエンシングキット(Applied Biosystems、フォスターシティー、カリフォルニア、米国)を用いてシークエンシングし、レーザー誘導蛍光キャピラリー電気泳動、ABI Prism 310シークエンサー(Applied Biosystems)を用いて分析した。
クローニングしたPCR産物を、蛍光−染料−標識(fluorescent-dye-labeled)ターミネーターサイクルシークエンシング、Big-Dyeシークエンシングキット(Applied Biosystems、フォスターシティー、カリフォルニア、米国)を用いてシークエンシングし、レーザー誘導蛍光キャピラリー電気泳動、ABI Prism 310シークエンサー(Applied Biosystems)を用いて分析した。
実施例6:全RNAの抽出
フェノールおよびグアニジンイソチオシアネートの単相溶液であるTrizol試薬(Invitrogen)を用いたChomczynskiおよびSacchi法の改変法を用い、全RNAをSC15847培養液から単離した。5mlのSC15847凍結ストック培養液を解凍し、500ml容エーレンマイヤーフラスコで100mlのF7培地に接種するのに用いた。培養液をシェーカーインキュベーター中、230rpmで30℃にて20時間、600nmでの光学濃度(OD600)が9.0となるまで増殖させた。培養液を16℃のシェーカーインキュベーターに230rpmで20分間置いた。25mgのエポチロンBを1mlの100%エタノールに溶解し、培養液に加えた。第二のmlのエタノールを用いて残留エポチロンBをチューブから濯ぎ、培養液に加えた。培養液を16℃、230rpmで30分間インキュベートした。30mlの培養液を50mlチューブに移し、150mgのリゾチームを培養液に加え、培養液を室温にて5分間インキュベートした。10mlの培養液を50mlファルコンチューブに入れ、TJ6遠心管中、4℃にて5分間遠心分離にかけた。2mlのクロロホルムを加え、チューブを15秒間激しく混合した。チューブを室温で2分間インキュベートし、TJ6遠心管の最高速度で10分間遠心分離にかけた。水性層を新たなチューブに移し、2.5mlのイソプロパノールを加えてRNAを沈殿させた。チューブを室温で10分間インキュベートし、4℃で10分間遠心分離にかけた。上澄み液を除去し、ペレットを70%エタノールで濯ぎ、真空下でしばらく乾燥させた。ペレットを150μlのRNアーゼ不含dH2Oに再懸濁した。このRNAに50μlの7.5M LiClを加え、−20℃にて30分間インキュベートした。RNAを遠心管で4℃にて10分間遠心分離にかけることによりペレット化した。ペレットを200μlの70%エタノールで濯ぎ、真空下でしばらく乾燥させ、150μlのRNアーゼ不含dH2Oに再懸濁した。
フェノールおよびグアニジンイソチオシアネートの単相溶液であるTrizol試薬(Invitrogen)を用いたChomczynskiおよびSacchi法の改変法を用い、全RNAをSC15847培養液から単離した。5mlのSC15847凍結ストック培養液を解凍し、500ml容エーレンマイヤーフラスコで100mlのF7培地に接種するのに用いた。培養液をシェーカーインキュベーター中、230rpmで30℃にて20時間、600nmでの光学濃度(OD600)が9.0となるまで増殖させた。培養液を16℃のシェーカーインキュベーターに230rpmで20分間置いた。25mgのエポチロンBを1mlの100%エタノールに溶解し、培養液に加えた。第二のmlのエタノールを用いて残留エポチロンBをチューブから濯ぎ、培養液に加えた。培養液を16℃、230rpmで30分間インキュベートした。30mlの培養液を50mlチューブに移し、150mgのリゾチームを培養液に加え、培養液を室温にて5分間インキュベートした。10mlの培養液を50mlファルコンチューブに入れ、TJ6遠心管中、4℃にて5分間遠心分離にかけた。2mlのクロロホルムを加え、チューブを15秒間激しく混合した。チューブを室温で2分間インキュベートし、TJ6遠心管の最高速度で10分間遠心分離にかけた。水性層を新たなチューブに移し、2.5mlのイソプロパノールを加えてRNAを沈殿させた。チューブを室温で10分間インキュベートし、4℃で10分間遠心分離にかけた。上澄み液を除去し、ペレットを70%エタノールで濯ぎ、真空下でしばらく乾燥させた。ペレットを150μlのRNアーゼ不含dH2Oに再懸濁した。このRNAに50μlの7.5M LiClを加え、−20℃にて30分間インキュベートした。RNAを遠心管で4℃にて10分間遠心分離にかけることによりペレット化した。ペレットを200μlの70%エタノールで濯ぎ、真空下でしばらく乾燥させ、150μlのRNアーゼ不含dH2Oに再懸濁した。
RNAをDNアーゼI(Ambion、オースチン、テキサス、米国)で処理した。25μlの全RNA(5.3μg/μl)、2.5μlのDNアーゼI緩衝液、1.0μlのDNアーゼIを加え、37℃で25分間インキュベートした。5μlのDNアーゼI不活化緩衝液を加え、2分間インキュベートし、1分間遠心分離にかけ、上澄み液を新たなチューブに移した。
実施例7:cDNA合成
Superscript II酵素(Invitrogen)を用いて全RNAからcDNAを合成した。1μlの全RNA(5.3μg/μl)、9μlのdH2O、1μlのdNTP混合物(10mM)、および1μlのランダムヘキサマーを用いて反応液を調製した。反応液を65℃で5分間インキュベートし、ついで氷上に置いた。ついで以下の成分を加えた:4μlの第一鎖緩衝液、1μlのRNアーゼインヒビター、2.0μlの0.1M DTT、および1μlのSuperscript II酵素。反応液を室温で10分間、42℃で50分間、ついで70℃で15分間インキュベートした。1μlのRNアーゼHを加え、37℃で20分間、70℃で15分間インキュベートし、4℃で貯蔵した。
Superscript II酵素(Invitrogen)を用いて全RNAからcDNAを合成した。1μlの全RNA(5.3μg/μl)、9μlのdH2O、1μlのdNTP混合物(10mM)、および1μlのランダムヘキサマーを用いて反応液を調製した。反応液を65℃で5分間インキュベートし、ついで氷上に置いた。ついで以下の成分を加えた:4μlの第一鎖緩衝液、1μlのRNアーゼインヒビター、2.0μlの0.1M DTT、および1μlのSuperscript II酵素。反応液を室温で10分間、42℃で50分間、ついで70℃で15分間インキュベートした。1μlのRNアーゼHを加え、37℃で20分間、70℃で15分間インキュベートし、4℃で貯蔵した。
実施例8:DNA標識
ゲノムDNAおよびcDNAからのP450特異的生成物の増幅に用いたPCR条件を用いてプラスミドpCRscript-29の挿入物を増幅した。プラスミドpCRscript-29は、プライマーのP450−1+(配列番号23)およびP450−3−(配列番号27)を用いてSC15847ゲノムDNAから増幅した340bpのPCR断片を含んでいる。2μlのプラスミドプレップを鋳型として用い、全部で25サイクル行った。増幅した生成物を、Qiaquickゲル抽出システム(Qiagen)を用いてゲル精製した。抽出したDNAをエタノール沈殿し、5μlのTEに再懸濁させ、収量は500ngと評価された。この断片を、chem link標識試薬(Roche Molecular Biochemicals、インディアナポリス、インディアナ、カタログ#1 836 463)を用いてジゴキシゲニンで標識した。5μlのPCR産物を0.5μlのDig-chem linkと混合し、dH2Oを20μlまで加えた。反応液を85℃で30分間インキュベートし、5μlの停止溶液を加えた。プローブ濃度は20ng/μlと評価された。
ゲノムDNAおよびcDNAからのP450特異的生成物の増幅に用いたPCR条件を用いてプラスミドpCRscript-29の挿入物を増幅した。プラスミドpCRscript-29は、プライマーのP450−1+(配列番号23)およびP450−3−(配列番号27)を用いてSC15847ゲノムDNAから増幅した340bpのPCR断片を含んでいる。2μlのプラスミドプレップを鋳型として用い、全部で25サイクル行った。増幅した生成物を、Qiaquickゲル抽出システム(Qiagen)を用いてゲル精製した。抽出したDNAをエタノール沈殿し、5μlのTEに再懸濁させ、収量は500ngと評価された。この断片を、chem link標識試薬(Roche Molecular Biochemicals、インディアナポリス、インディアナ、カタログ#1 836 463)を用いてジゴキシゲニンで標識した。5μlのPCR産物を0.5μlのDig-chem linkと混合し、dH2Oを20μlまで加えた。反応液を85℃で30分間インキュベートし、5μlの停止溶液を加えた。プローブ濃度は20ng/μlと評価された。
実施例9:サザーンDNAハイブリダイゼーション
10μlのゲノムDNA(0.5μg/ml)を、BamHI、BglII、EcoRI、HindIIIまたはNotIで消化し、12ボルトにて16時間分離した。ゲルを0.25N HClで10分間脱プリン化し(depurinated)、真空ブロッター(Bio-Rad Laboratories, Inc.、ヘルキュールズ、カリフォルニア、米国、カタログ#165−5000)を用いて0.4N NaOH5”Hgで90分間、ナイロンメンブレン(Roche Molecular Biochemicals)に真空下で移した。メンブレンを1M酢酸アンモニウムで濯ぎ、Stratalinker UV Crosslinker(Stratagene)を用いてUV架橋した。メンブレンを2×SSCで濯ぎ、室温で貯蔵した。
10μlのゲノムDNA(0.5μg/ml)を、BamHI、BglII、EcoRI、HindIIIまたはNotIで消化し、12ボルトにて16時間分離した。ゲルを0.25N HClで10分間脱プリン化し(depurinated)、真空ブロッター(Bio-Rad Laboratories, Inc.、ヘルキュールズ、カリフォルニア、米国、カタログ#165−5000)を用いて0.4N NaOH5”Hgで90分間、ナイロンメンブレン(Roche Molecular Biochemicals)に真空下で移した。メンブレンを1M酢酸アンモニウムで濯ぎ、Stratalinker UV Crosslinker(Stratagene)を用いてUV架橋した。メンブレンを2×SSCで濯ぎ、室温で貯蔵した。
メンブレンを20mlのDig Easy Hyb緩衝液(Roche Molecular Biochemicals)中、42℃で1時間プレハイブリダイズした。プローブを65℃で10分間変性させ、ついで氷上に置いた。Dig Easy Hyb中のおよその濃度が20ng/mlである5mlのプローブをメンブレンとともに42℃で一夜インキュベートした。メンブレンを室温で0.1%SDSを含む2×SCCで2回、ついで65℃で0.1%SDSを含む0.5×SSCで2回、洗浄した。メンブレンをGenius緩衝液1(10mMマレイン酸、15mM NaCl;pH7.5;0.3%v/v Tween 20)(Roche Molecular Biochemicals、インディアナポリス、インディアナ)で2分間平衡化し、ついで2%ブロッキング溶液(Genius緩衝液1中の2%ブロッキング試薬)(Roche Molecular Biochemicals、インディアナポリス、インディアナ)とともに室温で1時間インキュベートした。メンブレンを50mlのブロッキング溶液中の抗dig抗体の1:20,000希釈とともに30分間インキュベートした。メンブレンを50mlのGenius緩衝液1中で各15分間、2回洗浄した。メンブレンをGenius緩衝液3(10mMトリス−HCl、10mM NaCl;pH9.5)で2分間平衡化した。Genius緩衝液3中のCSPD(3−(4−メトキシスピロ{1,2−ジオキシエタン−3,2’−(5’−クロロ)トリシクロ[3.3.1.13,7]デカン}−4−イル)フェニルリン酸二ナトリウム)(Roche Molecular Biochemicals)の1:100希釈(1ml)をメンブレンに加えて室温で5分間インキュベートし、ついで37℃で15分間置いた。メンブレンをBiomax MLフィルム(Kodak、ロチェスター、ニューヨーク、米国)に1時間暴露した。
実施例10:大腸菌の形質転換
コンピテントな細胞をInvitrogenから購入した。大腸菌株DH10Bをゲノムクローニングの宿主として用いた。化学的にコンピテントな細胞を氷上で解凍し、100μlを氷上の17×100mmポリプロピレンチューブにアリコートした。1μlのライゲーション混合物を細胞に加え、氷上で30分間インキュベートした。細胞を42℃で45秒間インキュベートし、ついで氷上に1〜2分間置いた。0.9mlのSOC培地(Invitrogen)を加え、細胞を30〜37℃、200〜240rpmで1時間インキュベートした。細胞を選択培地(ネオマイシンまたはアンピシリンをそれぞれ30μg/mlまたは100μg/mlの濃度で含むLuriaアガー)上に置いた。
コンピテントな細胞をInvitrogenから購入した。大腸菌株DH10Bをゲノムクローニングの宿主として用いた。化学的にコンピテントな細胞を氷上で解凍し、100μlを氷上の17×100mmポリプロピレンチューブにアリコートした。1μlのライゲーション混合物を細胞に加え、氷上で30分間インキュベートした。細胞を42℃で45秒間インキュベートし、ついで氷上に1〜2分間置いた。0.9mlのSOC培地(Invitrogen)を加え、細胞を30〜37℃、200〜240rpmで1時間インキュベートした。細胞を選択培地(ネオマイシンまたはアンピシリンをそれぞれ30μg/mlまたは100μg/mlの濃度で含むLuriaアガー)上に置いた。
実施例11:ストレプトミセス・リビダンスTK24の形質転換
プラスミドpWB19NをHindIIIで消化およびSAPIで処理し、プラスミドpANT849(Keiserら、2000, Practical Streptomyces Genetics, John Innes)をHindIIIで消化することにより、プラスミドpWB19N849を構築した。これら2つの線状化した断片を1UのT4DNAリガーゼを用いて室温で1時間ライゲートした。1μlのライゲーション溶液を用いてXL−1 Blueエレクトロコンピテント細胞(Stratagene)を形質転換した。回収した細胞をLBネオマイシン(30μg/ml)に37℃にて一夜プレーティングした。コロニーを30μg/mlのネオマイシンを含む2mlのLBに採り、30℃で一夜インキュベートした。MoBioプラスミドミニプレップをすべての培養液について行った。pWB19NとpANT849とのライゲーションにより構築したプラスミドを0.7%アガロース上での電気泳動移動度により決定した。プラスミドpWB19N849をHindIIIおよびBglIIで消化して、BglIIおよびHindIIIで消化したプラスミドpANT849と等価な5.3kb断片を切り出した。この5.3kb断片をアガロースゲル上で精製し、Qiaquickゲル抽出システムを用いて抽出した。
プラスミドpWB19NをHindIIIで消化およびSAPIで処理し、プラスミドpANT849(Keiserら、2000, Practical Streptomyces Genetics, John Innes)をHindIIIで消化することにより、プラスミドpWB19N849を構築した。これら2つの線状化した断片を1UのT4DNAリガーゼを用いて室温で1時間ライゲートした。1μlのライゲーション溶液を用いてXL−1 Blueエレクトロコンピテント細胞(Stratagene)を形質転換した。回収した細胞をLBネオマイシン(30μg/ml)に37℃にて一夜プレーティングした。コロニーを30μg/mlのネオマイシンを含む2mlのLBに採り、30℃で一夜インキュベートした。MoBioプラスミドミニプレップをすべての培養液について行った。pWB19NとpANT849とのライゲーションにより構築したプラスミドを0.7%アガロース上での電気泳動移動度により決定した。プラスミドpWB19N849をHindIIIおよびBglIIで消化して、BglIIおよびHindIIIで消化したプラスミドpANT849と等価な5.3kb断片を切り出した。この5.3kb断片をアガロースゲル上で精製し、Qiaquickゲル抽出システムを用いて抽出した。
エポチロンBヒドロキシラーゼ遺伝子および下流のフェレドキシン遺伝子を含む1.469kb DNA断片をPCRを用いて増幅した。50μlのPCR反応液は、5μlのTaq緩衝液、2.5μlのグリセリン、1μlの20ng/μl NPB29−1プラスミド、0.4μlの25mM dNTP、各1.0μlのプライマーNPB29−6F(配列番号28)およびNPB29−7R(配列番号29)(5ピコモル/μl)、38.1μlのdH2Oおよび0.5μlのTaq酵素(Stratagene)からなっていた。反応をPerkin Elmer 9700で95℃で5分間、ついで30サイクル(96℃で30秒間、60℃で30秒間、72℃で2分間)、および72℃で7分間行った。PCR生成物をPCRクリーンアップ(cleanup)法でQiagenミニエルートカラムを用いて精製した。精製した生成物をBglIIおよびHindIIIで消化し、0.7%アガロースゲルで精製した。1.469kbのバンドをゲルから切り出し、Qiagenミニエルートカラムを用いて溶出した。このPCR生成物5μlを10μlのライゲーション反応液中、BglIIおよびHindIIIで消化したpANT849ベクターとライゲートした。反応液を室温で24時間インキュベートし、ついでストレプトミセス・リビダンスTK24プロトプラストに形質転換した。
20mlのYEME培地にストレプトミセス・リビダンスTK24の凍結胞子懸濁液を接種し、125ml容の両くぼみ(bi-indent)フラスコ中で48時間増殖させた。プロトプラストはPractical Streptomyces Geneticsの記載に従って調製した。ライゲーション反応液をプロトプラストと混合し、ついで500μlの形質転換緩衝液を加え、ついで5mlのP緩衝液を直ちに加えた。形質転換反応液を2,750rpmで7分間充分に(down)回転させ、100μlのP緩衝液に再懸濁し、一つのR2YEプレートにプレーティングした。このプレートを28℃で20時間インキュベートし、ついで250μg/mlのチオストレプトンを含む5mlの0.7%LBアガーを重層した。7日後、コロニーを50μg/mlのチオストレプトンを含むR2YEグリッドプレートに採った。コロニーを28℃でさらに5日間増殖させ、4℃で貯蔵した。
この組換え微生物はATCCに寄託してあり、PTA−4022と表示されている。
実施例12:ストレプトミセス・リモススの形質転換
PigacおよびSchrempf、Appl. Environ. Microb., Vol. 61, No. 1, 352-356 (1995)の手順を用いてストレプトミセス・リモススを形質転換した。ストレプトミセス・リモスス株R6 593を20mlのCRM培地中、30℃にてロータリーシェーカー(250rpm)上で培養した。細胞を24時間の時点で5,000rpm、4℃で5分間遠心分離にかけることにより回収し、20mlの10%ショ糖に4℃で再懸濁し、5,000rpm、4℃で5分間遠心分離にかけた。ペレットを10mlの15%グリセリンに4℃で再懸濁し、5,000rpm、4℃で5分間遠心分離にかけた。ペレットを2mlの15%グリセリンに100μg/mlのリゾチームとともに4℃で再懸濁し、37℃で30分間インキュベートし、5,000rpm、4℃で5分間遠心分離にかけ、2mlの15%グリセリンに4℃で再懸濁した。15%グリセリン洗浄を1回繰り返し、ペレットを1〜2mlのエレクトロポレーション緩衝液に再懸濁した。細胞を50〜200μlのアリコートとして−80℃にて貯蔵した。
PigacおよびSchrempf、Appl. Environ. Microb., Vol. 61, No. 1, 352-356 (1995)の手順を用いてストレプトミセス・リモススを形質転換した。ストレプトミセス・リモスス株R6 593を20mlのCRM培地中、30℃にてロータリーシェーカー(250rpm)上で培養した。細胞を24時間の時点で5,000rpm、4℃で5分間遠心分離にかけることにより回収し、20mlの10%ショ糖に4℃で再懸濁し、5,000rpm、4℃で5分間遠心分離にかけた。ペレットを10mlの15%グリセリンに4℃で再懸濁し、5,000rpm、4℃で5分間遠心分離にかけた。ペレットを2mlの15%グリセリンに100μg/mlのリゾチームとともに4℃で再懸濁し、37℃で30分間インキュベートし、5,000rpm、4℃で5分間遠心分離にかけ、2mlの15%グリセリンに4℃で再懸濁した。15%グリセリン洗浄を1回繰り返し、ペレットを1〜2mlのエレクトロポレーション緩衝液に再懸濁した。細胞を50〜200μlのアリコートとして−80℃にて貯蔵した。
ライゲーション液をストレプトミセス・リビダンスの形質転換について記載したのと同様にして調製した。ライゲーション反応液をインキュベートした後、容量をdH2Oで100μlとし、NaClを0.3Mまで加え、反応液を等容量の24:1:1フェノール:クロロホルム:イソアミルアルコールで抽出した。20μgのグリコーゲンを加え、ライゲートしたDNAを2容量の100%エタノールで−20℃にて30分間沈殿させた。DNAをミクロ遠心管で10分間ペレット化し、70%エタノールで1回洗浄し、speed-vac濃縮機で5分間乾燥させ、5μlのdH2Oに再懸濁した。
細胞の凍結アリコートの一つを室温で解凍し、エレクトロポレーションの各DNA試料について50μl/チューブに分けた。細胞を使用時まで氷上で貯蔵した。1〜2μlのdH2O中のDNAを加え、混合した。細胞とDNAとの混合物を、前もって氷上で冷却しておいたギャップが2mmのエレクトロキュベット(Bio-Rad Laboratories、リッチモンド、カリフォルニア、米国)に移した。Gene PulserTM(Bio-Rad Laboratories)を用い、2kV(10kV/cm)、25μF、400オームの設定で細胞をエレクトロポレーションした。細胞を0.75〜1.0mlのCRM(0〜4℃)で希釈し、15ml培養チューブに移し、攪拌しながら30℃で3時間インキュベートした。細胞を10〜30μg/mlのチオストレプトンを含むtrypticase soy brothアガープレートにプレーティングした。
実施例13:高速液体クロマトグラフィー
Waters 2690 Separation Moduleシステム(Waters Corp.、ミルフォード、マサチューセッツ、米国)およびSymmetryShield RP8、粒径3.5μmを充填したカラム4.6×150mm(Waters Corp.、ミルフォード、マサチューセッツ、米国)を用いて液体クロマトグラフィー分離を行った。勾配移動相プログラミングを1.0ml/分の流速で用いた。溶出液Aは、水/アセトニトリル(20:1)+10mM酢酸アンモニウムであった。溶出液Bは、アセトニトリル/水(20:1)であった。移動相を12%Bから28%Bへ6分間かけて線状勾配し、28%Bで4分間アイソクラチックとした。この次に20分間かけて28%Bから100%Bへの線状勾配および2分間かけて12%Bへの線状勾配を行い、12%Bで3分間保持した。
Waters 2690 Separation Moduleシステム(Waters Corp.、ミルフォード、マサチューセッツ、米国)およびSymmetryShield RP8、粒径3.5μmを充填したカラム4.6×150mm(Waters Corp.、ミルフォード、マサチューセッツ、米国)を用いて液体クロマトグラフィー分離を行った。勾配移動相プログラミングを1.0ml/分の流速で用いた。溶出液Aは、水/アセトニトリル(20:1)+10mM酢酸アンモニウムであった。溶出液Bは、アセトニトリル/水(20:1)であった。移動相を12%Bから28%Bへ6分間かけて線状勾配し、28%Bで4分間アイソクラチックとした。この次に20分間かけて28%Bから100%Bへの線状勾配および2分間かけて12%Bへの線状勾配を行い、12%Bで3分間保持した。
実施例14:質量分光測光
カラム溶出液をZMD質量分析計(Micromass、マンチェスター、英国)のエレクトロスプレーイオン源に直接導入した。この装置はTest Juice参照標準(Waters Corp.、ミルフォード、マサチューセッツ、米国)を用いて目盛りが付されており、手動ポンプ(syringe pump)(Harvard Apparatus、ホリストン、マサチューセッツ、米国)から10μl/分の流速で送達された。質量分析計は13.2の低い質量解析および11.2の高い質量解析で操作した。10スペクトル/秒の捕捉速度でm/z100〜600のスキャン範囲を用い、スペクトルを捕捉した。使用したイオン化法は正イオンエレクトロスプレー(ES)であった。スプレーヤーの電圧は2900Vに保持し、イオン源の円錐体(cone)電圧は17Vの電位で保持した。
カラム溶出液をZMD質量分析計(Micromass、マンチェスター、英国)のエレクトロスプレーイオン源に直接導入した。この装置はTest Juice参照標準(Waters Corp.、ミルフォード、マサチューセッツ、米国)を用いて目盛りが付されており、手動ポンプ(syringe pump)(Harvard Apparatus、ホリストン、マサチューセッツ、米国)から10μl/分の流速で送達された。質量分析計は13.2の低い質量解析および11.2の高い質量解析で操作した。10スペクトル/秒の捕捉速度でm/z100〜600のスキャン範囲を用い、スペクトルを捕捉した。使用したイオン化法は正イオンエレクトロスプレー(ES)であった。スプレーヤーの電圧は2900Vに保持し、イオン源の円錐体(cone)電圧は17Vの電位で保持した。
実施例15:他の微生物からシトクロムP450遺伝子を単離するためのebh遺伝子配列(配列番号1)の使用
ゲノムDNAをDNAzol試薬を用いて培養液のセット(ATCC43491、ATCC14930、ATCC53630、ATCC53550、ATCC39444、ATCC43333、ATCC35165)から単離した。このDNAを、ebh遺伝子の配列に対してデザインしたプライマーを使用するPCR反応に鋳型として用いた。3セットのプライマーを増幅に用いた:NPB29−6f(配列番号28)とNPB29−7r(配列番号29)、NPB29−16f(配列番号50)とNPB29−17r(配列番号51)、およびNPB29−19f(配列番号52)とNPB29−20r(配列番号53)。
ゲノムDNAをDNAzol試薬を用いて培養液のセット(ATCC43491、ATCC14930、ATCC53630、ATCC53550、ATCC39444、ATCC43333、ATCC35165)から単離した。このDNAを、ebh遺伝子の配列に対してデザインしたプライマーを使用するPCR反応に鋳型として用いた。3セットのプライマーを増幅に用いた:NPB29−6f(配列番号28)とNPB29−7r(配列番号29)、NPB29−16f(配列番号50)とNPB29−17r(配列番号51)、およびNPB29−19f(配列番号52)とNPB29−20r(配列番号53)。
200〜500ngのゲノムDNAおよびフォアウォードプライマーおよびリバースプライマーを含むPCR反応液を20μlの容量で調製した。プライマーはすべて1.4〜2.0μMの最終濃度で加えた。PCR反応液は0.2μlのAdvantageTM 2 Taq酵素(BD Biosciences Clontech、パロアルト、カリフォルニア、米国)、2μlのAdvantageTM 2 Taq緩衝液および0.2μlの2.5mM dNTPを用い、dH2Oを20μlまで加えて調製した。サイクル反応は、GeneampR 9700PCRシステムまたはMastercyclerR勾配(Eppendorf、ウエストベリー、ニューヨーク、米国)にて以下のプロトコールで行った:95℃にて5分間、35サイクル(96℃にて20秒、54〜69℃にて30秒、72℃にて2分間)、72℃にて7分間。PCR生成物の予期されるサイズは、NPB29−6f(配列番号28)とNPB29−7r(配列番号29)とのプライマーペアについては約1469bp、NPB29−16f(配列番号50)とNPB29−17r(配列番号51)とのプライマーペアについては1034bp、およびNPB29−19f(配列番号52)とNPB29−20r(配列番号53)とのプライマーペアについては1318bpである。PCR反応液を0.7%アガロースゲルで分析した。予期されたサイズのPCR産物をゲルから切り出し、Qiagenゲル抽出法を用いて精製した。精製した生成物をBig-Dyeシークエンシングキットを用いてシークエンシングし、ABI310シークエンサーを用いて分析した。
実施例16:プラスミドpPCRscript-ebhの構築
エポチロンBヒドロキシラーゼ遺伝子および下流のフェレドキシン遺伝子を含む1.469kb DNA断片をPCRを用いて増幅した。50μlのPCR反応液は、5μlのTaq緩衝液、2.5μlのグリセリン、1μlの20ng/μl NPB29−1プラスミド、0.4μlの25mM dNTP、各1.0μlのプライマーNPB29−6f(配列番号28)およびNPB29−7r(配列番号29)(5ピコモル/μl)、38.1μlのdH2Oおよび0.5μlのTaq酵素(Stratagene)からなっていた。反応をGeneampR 9700PCRシステムで以下の条件で行った:95℃で5分間、ついで30サイクル(96℃で30秒間、60℃で30秒間、72℃で2分間)、および72℃で7分間。PCR生成物をPCRクリーンアップ法でQiagen Qiaquickカラムを用いて精製した。精製した生成物をBglIIおよびHindIIIで消化し、0.7%アガロースゲルで精製した。1.469kbのバンドをゲルから切り出し、Qiagen Qiaquickゲル抽出法を用いて溶出した。ついで、これら断片を、PCRscript Ampクローニングキットを用いてpPCRscript Ampベクターにクローニングした。挿入物を含むコロニーを、100μg/mlのアンピシリンを含む1〜2mlのLB(Luria Broth)に37℃、230〜300rpmで16〜24時間採った。プラスミド単離をMo Bioミニプラスミドプレップキットを用いて行った。挿入物の配列を、Big-Dyeシークエンシングキットを用いたサイクルシークエンシングにより確認した。このプラスミドをpPCRscript-ebhと称した。
エポチロンBヒドロキシラーゼ遺伝子および下流のフェレドキシン遺伝子を含む1.469kb DNA断片をPCRを用いて増幅した。50μlのPCR反応液は、5μlのTaq緩衝液、2.5μlのグリセリン、1μlの20ng/μl NPB29−1プラスミド、0.4μlの25mM dNTP、各1.0μlのプライマーNPB29−6f(配列番号28)およびNPB29−7r(配列番号29)(5ピコモル/μl)、38.1μlのdH2Oおよび0.5μlのTaq酵素(Stratagene)からなっていた。反応をGeneampR 9700PCRシステムで以下の条件で行った:95℃で5分間、ついで30サイクル(96℃で30秒間、60℃で30秒間、72℃で2分間)、および72℃で7分間。PCR生成物をPCRクリーンアップ法でQiagen Qiaquickカラムを用いて精製した。精製した生成物をBglIIおよびHindIIIで消化し、0.7%アガロースゲルで精製した。1.469kbのバンドをゲルから切り出し、Qiagen Qiaquickゲル抽出法を用いて溶出した。ついで、これら断片を、PCRscript Ampクローニングキットを用いてpPCRscript Ampベクターにクローニングした。挿入物を含むコロニーを、100μg/mlのアンピシリンを含む1〜2mlのLB(Luria Broth)に37℃、230〜300rpmで16〜24時間採った。プラスミド単離をMo Bioミニプラスミドプレップキットを用いて行った。挿入物の配列を、Big-Dyeシークエンシングキットを用いたサイクルシークエンシングにより確認した。このプラスミドをpPCRscript-ebhと称した。
実施例17:改良した収量または変化した特異性のためのebh遺伝子の突然変異誘発
QuikchangeR XL Site-Directed Mutagenesis KitおよびQuikchangeR Multi Site-Directed Mutagenesisキット(ともにStratageneから)を用い、ebh遺伝子のコード領域に突然変異を導入した。これら両方法は、所望の突然変異を含む35〜45塩基の長さのDNAプライマー(配列番号54〜59および77)、メチル化した環状プラスミド鋳型およびPfuTurboRDNAポリメラーゼ(米国特許第5,545,552号および同第5,866,395号および同第5,948,663号)を用い、突然変異誘導プライマーによりもたらされる突然変異を導入したプラスミド鋳型のコピーを生成した。その後の制限エンドヌクレアーゼ酵素DpnIを用いた反応液の消化は、メチル化されたプラスミド鋳型を選択的に消化するが、メチル化されていない突然変異プラスミドは完全なままに残す。DpnI消化工程(インキュベート時間を1時間から3時間に長くした)を除き、すべての手順は製造業者の指示に従った。pPCRscript-ebhベクターを突然変異誘発の鋳型として用いた。
QuikchangeR XL Site-Directed Mutagenesis KitおよびQuikchangeR Multi Site-Directed Mutagenesisキット(ともにStratageneから)を用い、ebh遺伝子のコード領域に突然変異を導入した。これら両方法は、所望の突然変異を含む35〜45塩基の長さのDNAプライマー(配列番号54〜59および77)、メチル化した環状プラスミド鋳型およびPfuTurboRDNAポリメラーゼ(米国特許第5,545,552号および同第5,866,395号および同第5,948,663号)を用い、突然変異誘導プライマーによりもたらされる突然変異を導入したプラスミド鋳型のコピーを生成した。その後の制限エンドヌクレアーゼ酵素DpnIを用いた反応液の消化は、メチル化されたプラスミド鋳型を選択的に消化するが、メチル化されていない突然変異プラスミドは完全なままに残す。DpnI消化工程(インキュベート時間を1時間から3時間に長くした)を除き、すべての手順は製造業者の指示に従った。pPCRscript-ebhベクターを突然変異誘発の鋳型として用いた。
1〜2μlの反応液をXL1-BlueRエレクトロコンピテント細胞かまたはXL10-GoldRウルトラコンピテント細胞(Stratagene)のいずれかに形質転換した。細胞をLA(Luria Agar)100μg/mlアンピシリンプレート上に、プレート当たり100コロニーを超える密度でプレーティングし、30〜37℃で24〜48時間インキュベートした。プレート全体を100μg/mlのアンピシリンを含む5mlのLBに再懸濁した。細胞を遠心分離にかけ、ついでMo Bioミニプレップ法を用いてプラスミドを精製することにより、プラスミドを再懸濁細胞から直接単離した。ついで、このプラスミドをプライマーNPB29−6f(配列番号28)およびNPB29−7r(配列番号29)を用いたPCRの鋳型として用いて突然変異した発現カセットを増幅させた。制限酵素BglIIおよびHindIIIによる1.469kb PCR産物の消化を、これもBglIIおよびHindIIIで消化したベクターpANT849へのライゲーション用断片を調製するのに用いた。あるいは、再懸濁した細胞を用いて100μg/mlのアンピシリンを含む20〜50mlのLBに接種し、30〜37℃で18〜24時間増殖させた。培養液に対してQiagenミニプレップを行って所望の突然変異を含むプラスミドDNAを単離した。制限酵素BglIIおよびHindIIIによる消化を用い、BglIIおよびHindIII消化したプラスミドpANT849へのライゲーションのための突然変異した発現カセットを切り出した。記載に従ってストレプトミセス・リビダンスまたはストレプトミセス・リモススで突然変異体のスクリーニングを行った。
別法として、Leungらの方法(Technique-A Journal of Methods in Cell and Molecular Biology, Vol. 1, No. 1, 11-15 (1989))を用いてebh遺伝子のランダム突然変異ライブラリーを生成した。マンガンおよび/または低減したdATP濃度を用いてTaqポリメラーゼの突然変異誘発頻度を制御する。プラスミドpCRscript-ebhをNotIで消化してプラスミドを線状にした。ポリメラーゼ緩衝液を、0.166M (NH4)2SO4、0.67Mトリス−HCl(pH8.8)、61mM MgCl2、67μM EDTA(pH8.0)、1.7mg/mlウシ血清アルブミンを用いて調製した。PCR反応液を、10μlのNotI消化したpCRscript-ebh(0.1ng/μl)、10μlのポリメラーゼ緩衝液、1.0μlの1M β−メルカプトエタノール、10.0μlのDMSO、1.0μlのNPB29−6f(配列番号28)プライマー(100ピコモル/μl)、1.0μlのNPB29−7r(配列番号29)プライマー(100ピコモル/μl)、10μlの5mM MnCl2、10.0μlの10mM dGTP、10.0μlの2mM dATP、10mM dTTP、10.0μlの10mM dCTP、および2.0μlのTaqポリメラーゼを用いて調製した。dH2Oを加えて100μlとした。反応液も上記と同様にして調製したが、MnCl2は用いなかった。サイクリング反応をGeneAmpR PCRシステムで以下のプロトコールにて行った:95℃で1分間、25〜30サイクル(94℃で1分間、55℃で30秒間、72℃で4分間)、72℃で7分間。PCR反応液をQiagenスピンカラムを用いてアガロースゲル上で分離した。ついで、得られた断片をBglIIおよびHindIIIで消化し、Qiagenスピンカラムを用いて精製した。ついで、精製した断片をBglIIおよびHindIIIで消化したpANT849プラスミドにライゲートした。突然変異体のスクリーニングをストレプトミセス・リビダンスおよびストレプトミセス・リモススで行った。
特徴付けした突然変異体の一覧
実施例18:ebhおよびその突然変異体を発現する細胞におけるエポチロンB形質転換の比較
これら実験では、125ml容の両くぼみフラスコ中の20mlのYEME培地にストレプトミセス・リビダンスTK24、ストレプトミセス・リビダンス(pANT849−ebh)、ストレプトミセス・リビダンス(pANT849−ebh10−53)またはストレプトミセス・リビダンス(pANT849−ebh24−16)の200μlの凍結胞子調製液を接種し、230rpm、30℃で48時間インキュベートした。ストレプトミセス・リビダンス(pANT849−ebh)、ストレプトミセス・リビダンス(pANT849−ebh10−53)およびストレプトミセス・リビダンス(pANT849−ebh24−16)を接種した培地にチオストレプトン10μg/mlを加えた。4mlの培養液を125ml容エーレンマイヤーフラスコ中の20mlのR5培地に移し、230rpm、30℃で18時間インキュベートした。各培養液に100%EtOH中のエポチロンBを最終濃度0.05%w/vで加えた。ストレプトミセス・リビダンス(pANT849−ebh24−16)の培養液(この場合、エポチロンBは48時間で完全にエポチロンFに変換された)を除いて、試料を、0、24、48および72時間の時点で採取した。試料をHPLCにより分析した。結果を0時間でのエポチロンBのパーセントとして計算した。
これら実験では、125ml容の両くぼみフラスコ中の20mlのYEME培地にストレプトミセス・リビダンスTK24、ストレプトミセス・リビダンス(pANT849−ebh)、ストレプトミセス・リビダンス(pANT849−ebh10−53)またはストレプトミセス・リビダンス(pANT849−ebh24−16)の200μlの凍結胞子調製液を接種し、230rpm、30℃で48時間インキュベートした。ストレプトミセス・リビダンス(pANT849−ebh)、ストレプトミセス・リビダンス(pANT849−ebh10−53)およびストレプトミセス・リビダンス(pANT849−ebh24−16)を接種した培地にチオストレプトン10μg/mlを加えた。4mlの培養液を125ml容エーレンマイヤーフラスコ中の20mlのR5培地に移し、230rpm、30℃で18時間インキュベートした。各培養液に100%EtOH中のエポチロンBを最終濃度0.05%w/vで加えた。ストレプトミセス・リビダンス(pANT849−ebh24−16)の培養液(この場合、エポチロンBは48時間で完全にエポチロンFに変換された)を除いて、試料を、0、24、48および72時間の時点で採取した。試料をHPLCにより分析した。結果を0時間でのエポチロンBのパーセントとして計算した。
エポチロンB:
エポチロンF:
別法として、エポチロンBのエポチロンFへのバイオコンバージョンを、ebh遺伝子およびその変異体または突然変異体を含む発現プラスミドで形質転換したストレプトミセス・リモスス宿主細胞で行った。100μlの凍結したストレプトミセス・リモスス形質転換体培養液を10μg/mlのチオストレプトンを含む20mlのCRM培地に接種し、30℃、230〜300rpmで16〜24時間培養した。各培養液に100%エタノール中のエポチロンBを最終濃度0.05%w/vで加えた。反応液を典型的に30℃、230〜300rpmで20〜40時間インキュベートした。エポチロンBおよびエポチロンFの濃度をHPLC分析により決定した。
ストレプトミセス・リモススにおける突然変異体の評価
実施例19:コンパクチンのプラバスタチンへのバイオトランスフォーメーション
125ml容フラスコ中の10μg/mlのチオストレプトンを含む20mlのR2YE培地に、ストレプトミセス・リビダンス(pANT849)、ストレプトミセス・リビダンス(pANT849−ebh)の凍結胞子調製物(200μl)を接種し、230rpm、28℃で72時間インキュベートした。4mlの培養液を20mlのR2YE培地に接種し、230rpm、28℃で24時間増殖させた。1mlの培養液を15mlポリプロピレンチューブに移し、各培養液に10μlのコンパクチン(40mg/ml)を加え、28℃、250rpmで24時間インキュベートした。500μlの培養液ブロスを新たな15mlポリプロピレン培養チューブに移した。500μlの50mM水酸化ナトリウムを加え、回転攪拌した。3mlのメタノールを加えて回転攪拌し、チューブをTJ−6卓上遠心管中、3000rpmで10分間遠心分離にかけた。有機相をHPLCにより分析した。対照のストレプトミセス・リビダンス(pANT849)培養液と比較したコンパクチンおよびプラバスタチン値を評価した。
125ml容フラスコ中の10μg/mlのチオストレプトンを含む20mlのR2YE培地に、ストレプトミセス・リビダンス(pANT849)、ストレプトミセス・リビダンス(pANT849−ebh)の凍結胞子調製物(200μl)を接種し、230rpm、28℃で72時間インキュベートした。4mlの培養液を20mlのR2YE培地に接種し、230rpm、28℃で24時間増殖させた。1mlの培養液を15mlポリプロピレンチューブに移し、各培養液に10μlのコンパクチン(40mg/ml)を加え、28℃、250rpmで24時間インキュベートした。500μlの培養液ブロスを新たな15mlポリプロピレン培養チューブに移した。500μlの50mM水酸化ナトリウムを加え、回転攪拌した。3mlのメタノールを加えて回転攪拌し、チューブをTJ−6卓上遠心管中、3000rpmで10分間遠心分離にかけた。有機相をHPLCにより分析した。対照のストレプトミセス・リビダンス(pANT849)培養液と比較したコンパクチンおよびプラバスタチン値を評価した。
出発物質のコンパクチン濃度のパーセントとしてのコンパクチンおよびプラバスタチン:
実施例20:コンパクチンおよびプラバスタチンの検出のための高速液体クロマトグラフィー法
Hewlett Packard 1090 Series Separationシステム(Agilent Technologies、パロアルト、カリフォルニア、米国)およびSpherisorb ODS2、粒径5μmを充填したカラム50×46mm(Keystone Scientific, Inc.、ベルフォンテ、ペンシルベニア、米国)を用い、液体クロマトグラフィー分離を行った。勾配移動相プログラミングを2.0ml/分の流速で用いた。溶出液Aは、水、10mM酢酸アンモニウムおよび0.05%リン酸であった。溶出液Bは、アセトニトリルであった。移動相を20%Bから90%Bへ4分間かけて線状勾配した。
Hewlett Packard 1090 Series Separationシステム(Agilent Technologies、パロアルト、カリフォルニア、米国)およびSpherisorb ODS2、粒径5μmを充填したカラム50×46mm(Keystone Scientific, Inc.、ベルフォンテ、ペンシルベニア、米国)を用い、液体クロマトグラフィー分離を行った。勾配移動相プログラミングを2.0ml/分の流速で用いた。溶出液Aは、水、10mM酢酸アンモニウムおよび0.05%リン酸であった。溶出液Bは、アセトニトリルであった。移動相を20%Bから90%Bへ4分間かけて線状勾配した。
実施例21:突然変異体ebh25−1のバイオトランスフォーメーション産物の構造決定
分析的HPLCを、YMC充填ODS−AQカラム、4.6mm内径×15cm長とともにHewlett Packard 1100 Series Liquid chromatographを用いて行った。水(溶媒A)およびアセトニトリル(溶媒B)の勾配系を用いた:20%から90%Bへの線状勾配を10分間;90%から20%への線状勾配を2分間。流速は1ml/分であり、UV検出は254nmで行った。
分析的HPLCを、YMC充填ODS−AQカラム、4.6mm内径×15cm長とともにHewlett Packard 1100 Series Liquid chromatographを用いて行った。水(溶媒A)およびアセトニトリル(溶媒B)の勾配系を用いた:20%から90%Bへの線状勾配を10分間;90%から20%への線状勾配を2分間。流速は1ml/分であり、UV検出は254nmで行った。
分取HPLCを以下の装置および条件を用いて行った:
ポンプ:Varian ProStar Solvent Delivery Module(Varian Inc.、パロアルト、カリフォルニア、米国)。検出器:Gynkotek UVD340S
カラム:YMC ODS−Aカラム(30mm内径×100mm長、5μ粒径)
溶出の流速:30ml/分
溶出勾配:(溶媒A:水、溶媒B:アセトニトリル)、20%B、2分間;20%から60%Bへの線状勾配、18分間;60%B、2分間;60%から90%Bへの線状勾配、1分間;90%B、3分間;90%から20%Bへの線状勾配、2分間
検出:UV、210nm
ポンプ:Varian ProStar Solvent Delivery Module(Varian Inc.、パロアルト、カリフォルニア、米国)。検出器:Gynkotek UVD340S
カラム:YMC ODS−Aカラム(30mm内径×100mm長、5μ粒径)
溶出の流速:30ml/分
溶出勾配:(溶媒A:水、溶媒B:アセトニトリル)、20%B、2分間;20%から60%Bへの線状勾配、18分間;60%B、2分間;60%から90%Bへの線状勾配、1分間;90%B、3分間;90%から20%Bへの線状勾配、2分間
検出:UV、210nm
LC/NMRを以下のようにして行った:40μlの試料をYMC充填ODS−AQカラム(4.6mm内径×15cm長)に注入した。カラムを1ml/分の流速でD2O(溶媒A)およびアセトニトリル−d3(溶媒B)の勾配系を用いて溶出した:30%B、1分間;30%から80%Bへの線状勾配、11分間。溶出液をVarian AS-600 NMR分析計中のF19/H1 NMRプローブ(60μl活性容量)に流す前にUV検出セル(254nmでモニター)を通過させた。バイオトランスフォーメーション生成物は約7.5分で溶出されたので溶出流を手動で停止させてNMRデータ捕捉のために溶出液がNMRプローブ中に残るようにした。
単離および分析を以下のようにして行った。ブタノール/メタノール抽出液(約10ml)を窒素流下で蒸発乾固させた。残渣(38mg)に1mlのメタノールを加え、不溶性物質を遠心分離(13000rpm、2分)により除去した。上澄み液の0.1mlをLC/NMR研究に用い、残りの0.9mlを分取HPLC(0.2〜0.4ml/注入)に供した。2つの主要なピークが観察され、回収した:ピークAは14〜15分で溶出したのに対し、ピークBは16.5〜17.5分で溶出した。分析的HPLC分析は、ピークBが親化合物のエポチロンBであり(Rt8.5分)、ピークAがバイオトランスフォーメーション生成物である(Rt7.3分)ことを示していた。ピークAのフラクションをプールし、これらプールしたフラクションでMS分析データを得た。プールしたフラクションを蒸発させて小容量とし、ついで凍結乾燥して3mgの白色固体を得た。この白色固体(メタノールに溶解)のNMRおよびHPLC分析により、バイオトランスフォーメーション生成物が乾燥プロセスの際に部分的に分解したことが明らかになった。
付表1
Claims (41)
- エポチロンBヒドロキシラーゼまたはその突然変異体または変異体をコードする単離核酸配列。
- 配列番号1、30、32、34、36、37、38、39、40、41、42、60、62、64、66、68、72または74を含む、請求項1に記載の単離核酸配列。
- 配列番号1を含む、請求項1に記載の単離核酸配列。
- エポチロンBヒドロキシラーゼ酵素の活性部位に少なくとも一つのアミノ酸置換を有する突然変異体をコードする、請求項1に記載の単離核酸配列。
- 配列番号2のアミノ酸GLU31、ARG67、ARG88、ILE92、ALA93、VAL106、ILE130、ALA140、MET176、PHE190、GLU231、SER294、PHE237、またはILE365に少なくとも一つのアミノ酸置換を有する突然変異体をコードする、請求項1に記載の単離核酸配列。
- 配列番号2のアミノ酸LEU39、GLN43、ALA45、MET57、LEU58、HIS62、PHE63、SER64、SER65、ASP66、ARG67、GLN68、SER69、LEU74、MET75、VAL76、ALA77、ARG78、GLN79、ILE80、ASP84、LYS85、PRO86、PHE87、ARG88、PRO89、SER90、LEU91、ILE92、ALA93、MET94、ASP95、HIS99、ARG103、PHE110、ILE155、PHE169、GLN170、CYS172、SER173、SER174、ARG175、MET176、LEU177、SER178、ARG179、ARG186、PHE190、LEU193、VAL233、GLY234、LEU235、ALA236、PHE237、LEU238、LEU239、LEU240、ILE241、ALA242、GLY243、HIS244、GLU245、THR246、THR247、ALA248、ASN249、MET250、LEU283、THR287、ILE288、ALA289、GLU290、THR291、ALA292、THR293、SER294、ARG295、PHE296、ALA297、THR298、GLU312、GLY313、VAL314、VAL315、GLY316、VAL344、ALA345、PHE346、GLY347、PHE348、VAL350、HIS351、GLN352、CYS353、LEU354、GLY355、GLN356、LEU358、ALA359、GLU362、LYS389、ASP391、SER392、THR393、ILE394、またはTYR395に少なくとも一つのアミノ酸置換を有する突然変異体をコードする、請求項1に記載の単離核酸配列。
- 配列番号43、44、45、46、47、48または49を含む突然変異体をコードする、請求項1に記載の単離核酸配列。
- 請求項1に記載の単離核酸配列によってコードされるポリペプチド。
- 請求項2に記載の核酸配列または該核酸配列の相補的配列に3×SSC、65℃にて16時間のハイブリダイゼーション条件下でハイブリダイズすることのできる単離核酸分子であって、該核酸配列または該核酸配列の相補的配列に0.5×SSC、55℃にて30分間の洗浄条件下でハイブリダイズしたままであることができることを特徴とする単離核酸分子。
- 配列番号2を含む単離ポリペプチド。
- 配列番号2のエポチロンBヒドロキシラーゼ酵素の活性部位に少なくとも一つのアミノ酸置換を有するアミノ酸配列を含む、配列番号2のエポチロンBヒドロキシラーゼの単離した突然変異体ポリペプチド。
- 配列番号2のアミノ酸GLU31、ARG67、ARG88、ILE92、ALA93、VAL106、ILE130、ALA140、MET176、PHE190、GLU231、SER294、PHE237、またはILE365に少なくとも一つのアミノ酸置換を有するアミノ酸配列を含む、配列番号2のエポチロンBヒドロキシラーゼの単離した突然変異体ポリペプチド。
- 配列番号2のアミノ酸LEU39、GLN43、ALA45、MET57、LEU58、HIS62、PHE63、SER64、SER65、ASP66、ARG67、GLN68、SER69、LEU74、MET75、VAL76、ALA77、ARG78、GLN79、ILE80、ASP84、LYS85、PRO86、PHE87、ARG88、PRO89、SER90、LEU91、ILE92、ALA93、MET94、ASP95、HIS99、ARG103、PHE110、ILE155、PHE169、GLN170、CYS172、SER173、SER174、ARG175、MET176、LEU177、SER178、ARG179、ARG186、PHE190、LEU193、VAL233、GLY234、LEU235、ALA236、PHE237、LEU238、LEU239、LEU240、ILE241、ALA242、GLY243、HIS244、GLU245、THR246、THR247、ALA248、ASN249、MET250、LEU283、THR287、ILE288、ALA289、GLU290、THR291、ALA292、THR293、SER294、ARG295、PHE296、ALA297、THR298、GLU312、GLY313、VAL314、VAL315、GLY316、VAL344、ALA345、PHE346、GLY347、PHE348、VAL350、HIS351、GLN352、CYS353、LEU354、GLY355、GLN356、LEU358、ALA359、GLU362、LYS389、ASP391、SER392、THR393、ILE394、またはTYR395に少なくとも一つのアミノ酸置換を有するアミノ酸配列を含む、配列番号2のエポチロンBヒドロキシラーゼの単離した突然変異体ポリペプチド。
- 配列番号31、33、35、61、63、65、67、69、71、73または75を含む、エポチロンBヒドロキシラーゼの単離した突然変異体ポリペプチド。
- 配列番号43、44、45、46、47、48または49を含む、エポチロンBヒドロキシラーゼの単離した変異体ポリペプチド。
- フェレドキシンをコードする単離核酸配列。
- 配列番号3を含む、請求項16に記載の単離核酸配列。
- 請求項16に記載の単離核酸配列によってコードされるポリペプチド。
- 配列番号3に示す核酸配列または配列番号3に示す核酸配列の相補的配列に3×SSC、65℃にて16時間のハイブリダイゼーション条件下でハイブリダイズすることのできる単離核酸分子であって、配列番号3に示す核酸配列または配列番号3に示す核酸配列の相補的配列に0.5×SSC、55℃にて30分間の洗浄条件下でハイブリダイズしたままであることができることを特徴とする単離核酸分子。
- 請求項1に記載の単離核酸配列を含むベクター。
- フェレドキシンをコードする単離核酸配列をさらに含む、請求項20に記載のベクター。
- 請求項20に記載のベクターを含む宿主細胞。
- 請求項21に記載のベクターを含む宿主細胞。
- 末端アルキル基を有するエポチロンをヒドロキシル化して末端ヒドロキシアルキル基を有するエポチロンを生成する組換え微生物を製造する方法であって、微生物を請求項20または21に記載のベクターでトランスフェクションすることを含む方法。
- 末端アルキル基を有するエポチロンをヒドロキシル化して末端ヒドロキシアルキル基を有するエポチロンを生成する組換えにより生成した微生物。
- 配列番号1、30、32、34、36、37、38、39、40、41、42、60、62、64、66、68、72または74の核酸配列を発現する、請求項25に記載の組換えにより生成した微生物。
- 下記式I:
HO−CH2−(A1)n−(Q)m−(A2)o−E (I)
(式中、A1およびA2は、それぞれ独立に任意に置換されたC1−C3アルキルおよびアルケニルよりなる群から選ばれる;
Qは、1〜3の環および少なくとも1の環中に少なくとも一つの炭素−炭素二重結合を含む任意に置換された環系;
n、mおよびoは、0および1よりなる群から選ばれた整数、その際、mまたはnまたはoの少なくとも一つは1である;および
Eはエポチロンコアである)で示される少なくとも1のエポチロンを製造する方法であって、下記式II:
CH3−(A1)n−(Q)m−(A2)o−E (II)
(式中、A1、Q、A2、E、n、m、およびoは前記と同じ)で示される少なくとも1のエポチロンを、式IIの化合物のヒドロキシル化を選択的に触媒することができる組換えにより生成した微生物または該微生物からの酵素と接触させ、ついで該ヒドロキシル化を行うことを含む方法。 - 式A:
- 式A:
- 付表1に列挙した構造座標によって記載された対応骨格原子上で重ね合わせたときに、保存された残基骨格原子の二乗平均偏差が約4.0オングストローム未満である、エポチロンBヒドロキシラーゼのホモロジーモデル。
- エポチロンBヒドロキシラーゼと比較して変化した生物学的特性、機能、所望の生成物の収量、反応速度、基質特異性、または活性を有する突然変異体の製造方法であって、突然変異すべき配列番号2中のアミノ酸を同定し、ついで該同定したアミノ酸を突然変異させて突然変異体タンパク質を生成することを含む方法。
- 付表1に列挙した構造座標によって記載された対応骨格原子上で重ね合わせたときに、保存された残基骨格原子の二乗平均偏差が約4.0オングストローム未満であるエポチロンBヒドロキシラーゼのホモロジーモデルを、突然変異すべき配列番号2中のアミノ酸を同定するために用いる、請求項31に記載の方法。
- 同定したアミノ酸が、配列番号2のLEU39、GLN43、ALA45、MET57、LEU58、HIS62、PHE63、SER64、SER65、ASP66、ARG67、GLN68、SER69、LEU74、MET75、VAL76、ALA77、ARG78、GLN79、ILE80、ASP84、LYS85、PRO86、PHE87、ARG88、PRO89、SER90、LEU91、ILE92、ALA93、MET94、ASP95、HIS99、ARG103、PHE110、ILE155、PHE169、GLN170、CYS172、SER173、SER174、ARG175、MET176、LEU177、SER178、ARG179、ARG186、PHE190、LEU193、VAL233、GLY234、LEU235、ALA236、PHE237、LEU238、LEU239、LEU240、ILE241、ALA242、GLY243、HIS244、GLU245、THR246、THR247、ALA248、ASN249、MET250、LEU283、THR287、ILE288、ALA289、GLU290、THR291、ALA292、THR293、SER294、ARG295、PHE296、ALA297、THR298、GLU312、GLY313、VAL314、VAL315、GLY316、VAL344、ALA345、PHE346、GLY347、PHE348、VAL350、HIS351、GLN352、CYS353、LEU354、GLY355、GLN356、LEU358、ALA359、GLU362、LYS389、ASP391、SER392、THR393、ILE394、またはTYR395である、請求項31に記載の方法。
- 同定したアミノ酸が、配列番号2のGLU31、ARG67、ARG88、ILE92、ALA93、VAL106、ILE130、ALA140、MET176、PHE190、GLU231、SER294、PHE237、またはILE365である、請求項31に記載の方法。
- 該突然変異体タンパク質の所望の生成物の収量が、エポチロンBヒドロキシラーゼを用いて得られる所望の生成物の収量と比較して改善されている、請求項31に記載の方法。
- 該所望の生成物がエポチロンFである、請求項35に記載の方法。
- 該突然変異体の反応速度が、エポチロンBヒドロキシラーゼを用いて得られる反応速度と比較して改善されている、請求項31に記載の方法。
- 該突然変異体が、エポチロンBヒドロキシラーゼの基質特異性と比較して変化した基質特異性を示す、請求項31に記載の方法。
- アミノ酸SER294が突然変異している、請求項38に記載の方法。
- 該突然変異体が、エポチロンBヒドロキシラーゼと本質的に同様の生物学的活性または機能を示す、請求項31に記載の方法。
- 付表1に示す構造座標でコードしたデータ記憶材料を含む機械読み取り可能なデータ記憶媒体。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US34427101P | 2001-12-26 | 2001-12-26 | |
| PCT/US2002/040359 WO2003057830A2 (en) | 2001-12-26 | 2002-12-17 | Compositions and methods for hydroxylating epothilones |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2005514031A true JP2005514031A (ja) | 2005-05-19 |
| JP2005514031A5 JP2005514031A5 (ja) | 2005-12-22 |
Family
ID=23349792
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2003558132A Withdrawn JP2005514031A (ja) | 2001-12-26 | 2002-12-17 | エポチロンをヒドロキシル化するための組成物および方法 |
Country Status (6)
| Country | Link |
|---|---|
| EP (1) | EP1465908A4 (ja) |
| JP (1) | JP2005514031A (ja) |
| AU (1) | AU2002357872A1 (ja) |
| CA (1) | CA2471874A1 (ja) |
| TW (1) | TW200301697A (ja) |
| WO (1) | WO2003057830A2 (ja) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2019536479A (ja) * | 2016-11-15 | 2019-12-19 | ハイファ ディスカバリー リミテッド | アミコラトプシス・ルリダ由来シトクロムp450を用いた分岐状脂肪族または芳香族基質のヒドロキシ化法 |
Families Citing this family (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6204388B1 (en) | 1996-12-03 | 2001-03-20 | Sloan-Kettering Institute For Cancer Research | Synthesis of epothilones, intermediates thereto and analogues thereof |
| CA2273083C (en) | 1996-12-03 | 2012-09-18 | Sloan-Kettering Institute For Cancer Research | Synthesis of epothilones, intermediates thereto, analogues and uses thereof |
| US6884608B2 (en) * | 2001-12-26 | 2005-04-26 | Bristol-Myers Squibb Company | Compositions and methods for hydroxylating epothilones |
| US20060234337A1 (en) * | 2002-04-12 | 2006-10-19 | Akira Arisawa | Expression system for actinomycete-origin cytochrome p-450 in escherichia coli |
| GB201807815D0 (en) | 2018-05-14 | 2018-06-27 | Hypha Discovery Ltd | Hydroxylation techniques |
| GB201819209D0 (en) | 2018-11-26 | 2019-01-09 | Hypha Discovery Ltd | Biocatalytic techniques |
| GB201917077D0 (en) | 2019-11-22 | 2020-01-08 | Hypha Discovery Ltd | Biocatalytic techniques |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR20010092453A (ko) * | 1998-12-23 | 2001-10-25 | 스티븐 비. 데이비스 | 에포틸론 제조를 위한 미생물적 전환 방법 |
| US7070964B2 (en) * | 2001-11-15 | 2006-07-04 | Kosan Biosciences Incorporated | Epothilone compounds and methods for making the same |
-
2002
- 2002-12-17 CA CA002471874A patent/CA2471874A1/en not_active Abandoned
- 2002-12-17 EP EP02792416A patent/EP1465908A4/en not_active Withdrawn
- 2002-12-17 WO PCT/US2002/040359 patent/WO2003057830A2/en not_active Ceased
- 2002-12-17 JP JP2003558132A patent/JP2005514031A/ja not_active Withdrawn
- 2002-12-17 AU AU2002357872A patent/AU2002357872A1/en not_active Abandoned
- 2002-12-20 TW TW091136889A patent/TW200301697A/zh unknown
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2019536479A (ja) * | 2016-11-15 | 2019-12-19 | ハイファ ディスカバリー リミテッド | アミコラトプシス・ルリダ由来シトクロムp450を用いた分岐状脂肪族または芳香族基質のヒドロキシ化法 |
| JP7142813B2 (ja) | 2016-11-15 | 2022-09-28 | ハイファ ディスカバリー リミテッド | アミコラトプシス・ルリダ由来シトクロムp450を用いた分岐状脂肪族または芳香族基質のヒドロキシ化法 |
Also Published As
| Publication number | Publication date |
|---|---|
| TW200301697A (en) | 2003-07-16 |
| EP1465908A4 (en) | 2005-11-30 |
| WO2003057830A2 (en) | 2003-07-17 |
| AU2002357872A1 (en) | 2003-07-24 |
| EP1465908A2 (en) | 2004-10-13 |
| WO2003057830A3 (en) | 2004-08-05 |
| CA2471874A1 (en) | 2003-07-17 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US6884608B2 (en) | Compositions and methods for hydroxylating epothilones | |
| Peek et al. | Rifamycin congeners kanglemycins are active against rifampicin-resistant bacteria via a distinct mechanism | |
| JP5514185B2 (ja) | Nrps−pks遺伝子群ならびにその操作および有用性 | |
| Liu et al. | Cyclopiazonic acid biosynthesis in Aspergillus sp.: characterization of a reductase-like R* domain in cyclopiazonate synthetase that forms and releases cyclo-acetoacetyl-L-tryptophan | |
| EP0618972A1 (en) | CONSTITUTIVE EXPRESSION OF P450SOY AND FERREDOXIN-SOY IN $i(STREPTOMYCES), AND BIOTRANSFORMATION OF CHEMICALS BY RECOMBINANT ORGANISMS | |
| JP2002518004A (ja) | エポチロン生合成用遺伝子 | |
| Ren et al. | Non-modular fatty acid synthases yield distinct N-terminal acylation in ribosomal peptides | |
| CN110218244A (zh) | 化合物ilamycin F及其应用 | |
| Biermann et al. | Exploration, expansion and definition of the atropopeptide family of ribosomally synthesized and posttranslationally modified peptides | |
| Ye et al. | Combination of atmospheric and room temperature plasma (ARTP) mutagenesis, genome shuffling and dimethyl sulfoxide (DMSO) feeding to improve FK506 production in Streptomyces tsukubaensis | |
| JP2005514031A (ja) | エポチロンをヒドロキシル化するための組成物および方法 | |
| Renata | Exploration of iron-and α-ketoglutarate-dependent dioxygenases as practical biocatalysts in natural product synthesis | |
| JP6092104B2 (ja) | 修飾ペプチドディスプレイ | |
| Wang et al. | Characterization and structural analysis of a versatile aromatic prenyltransferase for imidazole-containing diketopiperazines | |
| Ding et al. | Characterization of a LanC-free pathway for the formation of an ll-MeLan residue and an allo AviMeCys residue in the newly identified class V lanthipeptide triantimycins | |
| Zolova et al. | Importance of the MbtH-like protein TioT for production and activation of the thiocoraline adenylation domain of TioK | |
| Weinig et al. | Markerless mutations in the myxothiazol biosynthetic gene cluster: a delicate megasynthetase with a superfluous nonribosomal peptide synthetase domain | |
| WO2011160573A1 (zh) | 一种生产高活性和高纯度利福霉素sv的方法 | |
| WO2004078978A1 (en) | Compositions and methods for hydroxylating epothilones | |
| US6699699B2 (en) | Mutated penicillin expandases | |
| Pal et al. | Examining Heterodimerization by Aryl C–N Coupling in Dynemicin Biosynthesis | |
| Hollands et al. | Analysis of the cryptic biosynthetic gene cluster encoding the RiPP curacozole reveals a phenylalanine-specific peptide hydroxylase | |
| US20230348539A1 (en) | A new antibiotic selectively kills gram-negative pathogens | |
| Yang et al. | The Myxobacterial Genus Archangium: A Prolific and Underexploited Source of Bioactive Secondary Metabolites | |
| Dreckmann et al. | Biosynthesis of the corallorazines, a widespread class of antibiotic cyclic lipodipeptides |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20051021 |
|
| A761 | Written withdrawal of application |
Free format text: JAPANESE INTERMEDIATE CODE: A761 Effective date: 20070521 |