JP2006036780A - ルテニウム錯体の製造方法 - Google Patents
ルテニウム錯体の製造方法 Download PDFInfo
- Publication number
- JP2006036780A JP2006036780A JP2005226885A JP2005226885A JP2006036780A JP 2006036780 A JP2006036780 A JP 2006036780A JP 2005226885 A JP2005226885 A JP 2005226885A JP 2005226885 A JP2005226885 A JP 2005226885A JP 2006036780 A JP2006036780 A JP 2006036780A
- Authority
- JP
- Japan
- Prior art keywords
- ruthenium
- group
- general formula
- represented
- pentadiene
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Images


















Landscapes
- Chemical Vapour Deposition (AREA)
Abstract
【課題】ペンタジエン誘導体とハロゲン化ルテニウム化合物を反応させ、オープン型ルテニウム錯体を製造する。
【解決手段】1,3−ペンタジエン誘導体とハロゲン化ルテニウム水和物を反応させ、一般式[3]
[式中、R2、R3、R4はH,アルキル基等を示す]で表されるオープン型ルテニウム錯体を製造する。
【選択図】なし
【解決手段】1,3−ペンタジエン誘導体とハロゲン化ルテニウム水和物を反応させ、一般式[3]
[式中、R2、R3、R4はH,アルキル基等を示す]で表されるオープン型ルテニウム錯体を製造する。
【選択図】なし
Description
本発明は、化学気相蒸着法(Chemical Vapor Deposition法;以下、CVD法とする)、塗布熱分解法等によるルテニウム含有薄膜の形成に有用な有機ルテニウム錯体、その製造方法、及び半導体メモリー等の電子素子等に使用されるルテニウム含有薄膜の製造方法に関する。
半導体メモリー素子の高集積化に伴いメモリーセルの微細化が進み、キャパシター絶縁膜に(Ba、Sr)TiO3等の強誘電体薄膜を使用することが検討されている。強誘電体薄膜を使用したキャパシターでは電極としてPt、Ru、Irといった貴金属が使用される。このうちRuは酸化物が導電性を持つ、微細加工性に優れるといった点から電極材料として最も有力視されており、Ru薄膜あるいはRuO2薄膜による電極が検討されている。高集積化したメモリー素子におけるこれらRu含有薄膜の形成方法としては段差被覆性、組成制御性に優れるといった点からCVD法が最適である。
このCVD法を用いて薄膜を形成させるための原料物質としては金属化合物の中でも融点が低く取り扱い性が容易である有機金属化合物が適していると考えられる。従来、ルテニウムまたはルテニウム酸化物薄膜を析出させる為の有機金属化合物としてはルテノセンあるいはトリ(ジピバロイルメタナート)ルテニウム(以後Ru(DPM)3)[特開平6−283438号公報]またはトリ(オクタン−2,4−ジオネート)ルテニウム(以後Ru(OD)3)[特開2000−212744号公報]が用いられていた。ルテノセンは、それぞれのシクロペンタジエン環を構成するのが炭素と水素のみであり、2つのシクロペンタジエン環の間にルテニウムが挟まれているサンドイッチ構造を有する。このルテノセンは大気中の安定性が高く、毒性も無いことからCVD原料としての適性を有するものの、常温では固体であり、融点が約200℃と比較的高い為、原料の気化および基盤への輸送が多少困難になるという問題点がある。
そこで最近では融点の低いルテニウム化合物についての研究が活発に行われている。このルテニウム含有有機金属化合物の低融点化の手法としては、ルテノセンのシクロペンタジエン環の少なくとも一つの水素原子をメチル基、エチル基等のアルキル基で置換したルテノセン誘導体とするものがある。例えば、特開平11−35589号公報ではルテノセン誘導体として、ビス(エチルシクロペンタジエニル)ルテニウム(以後Ru(EtCp)2)およびビス(イソプロピルシクロペンタジエニル)ルテニウムに代表される、ビス(アルキルシクロペンタジエニル)ルテニウムが開示されている。また特開2000−281694号公報ではアルキル置換ルテノセンをCVD材料として用いる事が開示されている。これらの金属化合物はいずれも常温で液体であり、その融点もルテノセンに比して低いことから、CVD法に適用する原料物質として必要な特性を具備するものであるとされている。しかしこれらビス(アルキルシクロペンタジエニル)ルテニウムは基本的にルテノセン構造を有しており、この構造は安定性が極めて高いことから錯体の分解温度が高く、必然的に成膜時の基盤温度を高くする必要があり、結果としてステップカバレッジが悪くなるという問題点を抱えていた。
一方、一分子のシクロペンタジエニル基を配位子とするハーフサンドイッチ構造を有する錯体の合成例としては、R.Gleiter等のOrganometallics,8,298(1989)に報告されている、(シクロペンタジエニル)(2,4−ジメチルペンタジエニル)ルテニウムがある。しかしながらこの錯体は融点が136〜137℃で、常温で固体となり、CVD材料として適当な材料であるとは言い難い。これまでに室温で液体の優れた気化特性を示すハーフサンドイッチ構造のルテニウム錯体の合成報告例はない。
また、ハーフサンドイッチ構造のルテノセンの合成法に関しては、ペンタジエン誘導体、シクロペンタジエン誘導体、亜鉛およびルテニウムを適当な溶媒中に一度に加え、適当な反応条件で反応させるのが一般的であるが、この方法では収率が極端に悪くなり、実用的でない。また、反応後の後処理として反応液を濃縮し、泥状混合物を得た後にその泥状混合物から適当な溶媒で目的物を抽出し、セライト濾過もしくはアルミナカラムを用いたカラムクロマトグラフィーを行い精製することで目的物を得る方法が一般的であった。しかしこの方法は反応終了後に濃縮していられる泥状化合物からの抽出や、セライト濾過もしくはカラムクロマトグラフィーなど工業上好ましくないプロセスを含んでおり、ハーフサンドイッチ構造有機ルテニウム化合物が工業的に有利になる為に、安定して高収率で目的物を得ることが出来る製造方法が望まれていた。
一方、塗布熱分解法による成膜は比較的集積度の低い素子の製造に使用されている。塗布熱分解法で使用される原料は膜厚を制御するため有機溶媒に溶解して使用することから有機溶媒に可溶で、さらに低温で分解するものが好ましい。しかし、このような特徴を有するルテニウム化合物はこれまでほとんどなかった。
従来、カルボニルビス(ジエン)ルテニウム錯体としてはカルボニルビス(1,3−ブタジエン)ルテニウム(D.Minniti and P.L.Timms,J.Organomet.Chem.,258,C12(1983))、カルボニルビス(2,3−ジメチル−1,3−ブタジエン)ルテニウム、カルボニルビス(1,3−シクロヘキサジエン)ルテニウム(D.N.Cox and R.Roulet,Helv.Chim.Acta,67,1365(1984))のみが知られている。これらの錯体の製造方法は−196℃でRuとジエンを反応させた後、COを加えるという低温での反応を必要とする方法であった。
CVD法では薄膜の原料となる錯体をガスとして供給する必要があるが、従来使用されている錯体のうちRu(DPM)3は融点168℃で高温なため、昇華によるガス化となる。昇華によるガス化では固体の表面積変化により原料ガス濃度に変化が生じ安定な供給量が得られないという問題がある。これに対し、錯体を有機溶媒に溶解して使用する方法が提案されている(特開平5−132776号公報)。しかし、この方法では溶媒と錯体の揮発性の差により溶媒のみが揮発したり、固体が析出するといった問題があり必ずしも安定な原料供給方法とはいえない。一方、Ru(OD)3およびRu(EtCp)2は室温で液体であり比較的高い蒸気圧を持つため原料の安定供給については問題ないが、どちらの錯体もRuと有機配位子の結合が安定で分解しにくいため、高温での成膜が必要である。
本発明は、ルテニウム錯体の製造方法を提供することを目的とする。
本発明者らは、先の課題を解決すべく検討を重ねた結果、ルテノセン構造を有する既知化合物の片方のシクロペンタジエニル環(以後Cp環)を直鎖型のペンタジエニルに変えることで分解温度が下がることを見出し、さらに鋭意検討を重ねた結果、Cp環に低級アルキル基を導入することで、室温で液体の融点を示し、良好な気化特性、分解特性を有する新規なルテニウム錯体を開発するに至った。また低分子量のジエンとカルボニル基を配位子としたカルボニルビス(ジエン)ルテニウム錯体により上記目的を達成できることを見出し、本発明を完成するに至った。
更に、ある種のペンタジエン誘導体とハロゲン化ルテニウム化合物を反応させることにより、オープン型ルテニウム錯体が得られることを見出し、本発明を完成するに至った。
即ち本発明は、一般式[5]
[式中、R2、R3、R4は同一または相異なって水素、ハロゲン、低級アシル基、低級アルコキシ基、低級アルコキシカルボニル基、または低級アルキル基を示す。]で表されるペンタジエンと亜鉛との混合液中に、一般式[6]
RuX3・nH2O [6]
[式中、Xはハロゲンを表し、nは0乃至10の数字を表す。]で表されるハロゲン化ルテニウム水和物を溶媒で希釈した溶液を滴下・反応させることを特徴とする、一般式[3]
RuX3・nH2O [6]
[式中、Xはハロゲンを表し、nは0乃至10の数字を表す。]で表されるハロゲン化ルテニウム水和物を溶媒で希釈した溶液を滴下・反応させることを特徴とする、一般式[3]
[式中、R2、R3、R4は前記と同様を示す]で表されるオープン型ルテニウム錯体の製造方法である。
また本発明は、上述の方法において、一般式[5]で表されるペンタジエンとして一般式[9]
[式中、R2は前記と同様を示す]で表されるペンタジエンを用いることを特徴とする、一般式[10]
[式中、R2は前記と同様を示す]で表されるオープン型ルテニウム錯体の製造方法である。
さらに本発明は、上述の方法において、一般式[5]または[9]で表されるペンタジエンとして2,4−ジメチル−1,3−ペンタジエンを用いることを特徴とする、ビス(2,4−ジメチルペンタジエニル)ルテニウム(II)の製造方法である。
また本発明は、上述の方法において、メタノールを一部または全部の溶媒として用いることを特徴とする方法である。
さらに本発明は、上述の方法において、一般式[5]もしくは[9]で表されるペンタジエン、または2,4−ジメチル−1,3−ペンタジエンの添加量が、一般式[6]で表されるハロゲン化ルテニウム水和物1モルに対して2乃至20モルであることを特徴とする方法である。
また本発明は、上述の方法において、亜鉛の添加量が、一般式[6]で表されるハロゲン化ルテニウム水和物1モルに対して1.5乃至100モルであることを特徴とする方法である。
さらに本発明は、上述の方法において、反応温度が−20乃至100℃であることを特徴とする方法である。
また本発明は、上述の方法において、反応を窒素ガスまたは不活性ガス雰囲気中で行うことを特徴とする方法である。
また本発明は、一般式[1]
[式中R1、R2、R3、R4は同一または相異なって水素、ハロゲン、低級アシル基、低級アルコキシ基、低級アルコキシカルボニル基、または低級アルキル基を示す。ただしR1〜R4全てが水素である場合、及び、R1が水素でR2〜R4のいずれか1つが水素で残りがメチル基である場合を除く。]で表されることを特徴とする、ハーフサンドイッチ構造有機ルテニウム化合物である。
また本発明は、上述のハーフサンドイッチ構造有機ルテニウム化合物を原料とし、化学気相蒸着法を用いて、加熱した基板上にルテニウム含有薄膜を製造することを特徴とする、ルテニウム含有薄膜の製造方法である。
更に本発明は、一般式[3]
[式中R2、R3、R4は同一または相異なって水素、ハロゲン、低級アシル基、低級アルコキシ基、低級アルコキシカルボニル基、または低級アルキル基を示す。]で表されるオープンルテノセンに、溶媒中で亜鉛存在下、一般式[4]
[式中R1は水素、ハロゲン、低級アシル基、低級アルコキシ基、低級アルコキシカルボニル基、または低級アルキル基を示す。]で表されるシクロペンタジエンを反応させることを特徴とする、一般式[1]で表されるハーフサンドイッチ構造有機ルテニウム化合物の製造方法である。
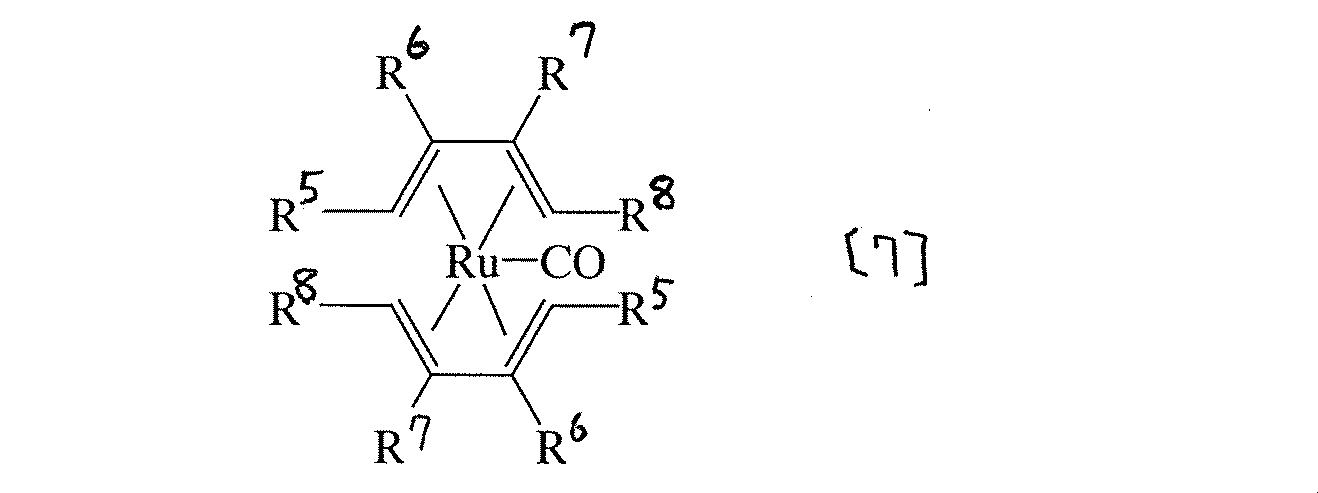
また本発明は、一般式[7]
(式中R5〜R8は水素または炭素数1〜6のアルキル、エーテル、エステル、アルデヒド、アルコール、ケトン、ハロゲン化アルキル、カルボン酸、アミン、アミドを示す。但し、R5〜R8全てが水素の場合、及びR5、R8が水素でR6、R7がメチルの場合を除く。)で表されることを特徴とする、カルボニルビス(ジエン)ルテニウム錯体である。
さらに本発明は、塩化ルテニウム・n水和物(nは1以上の数)とジエンを亜鉛粉末存在下アルコール中で反応させることを特徴とする、上述のカルボニルビス(ジエン)ルテニウム錯体の製造方法である。
また本発明は、上述のカルボニルビス(ジエン)ルテニウム錯体を原料として使用することを特徴とする、ルテニウム含有薄膜の製造方法である。以下、本発明について詳しく述べる。
最初に本明細書で用いられる用語の定義ならびにその具体例について説明する。本明細書中に記述の「低級」なる用語は特に断らない限り、この語が付与された基に於いて、炭素数1個以上6個以下の直鎖状、分岐状、または環状の炭化水素基を含有するものであることを示す。
よってR1、R2、R3またはR4において用いられる低級アルキル基としては、例えば、メチル基、エチル基、n−プロピル基、イソプロピル基、n−ブチル基、イソブチル基、sec−ブチル基、tert−ブチル基、ペンチル(アミル)基、イソペンチル基、ネオペンチル基、tert−ペンチル基、1−メチルブチル基、2−メチルブチル基、1,2−ジメチルプロピル基、ヘキシル基、イソヘキシル基、1−メチルペンチル基、2−メチルペンチル基、3−メチルペンチル基、1,1−ジメチルブチル基、2,2−ジメチルブチル基、1,3−ジメチルブチル基、2,3−ジメチルブチル基、3,3−ジメチルブチル基、1−エチルブチル基、2−エチルブチル基、1,1,2−トリメチルプロピル基、1,2,2−トリメチルプロピル基、1−エチル−1−メチルプロピル基、1−エチル−2−メチルプロピル基、シクロプロピル基、シクロブチル基、シクロペンチル基、シクロヘキシル基、シクロプロピルメチル基、シクロプルピルエチル基、およびシクロブチルメチル基等があげられる。より好ましくはメチル基、エチル基、プロピル基、イソプロピル基、シクロプロピル基である。
また、R1、R2、R3またはR4において用いられる低級アルコキシ基としては、具体的に例えばメトキシ基、エトキシ基、プロポキシ基、イソプロポキシ基、ブトキシ基、イソブトキシ基、sec−ブトキシ基、tert−ブトキシ基、ペンチルオキシ基、1−メチルブチルオキシ基、2−メチルブチルオキシ基、3−メチルブチルオキシ基、1,2−ジメチルプロピルオキシ基、ヘキシルオキシ基、1−メチルペンチルオキシ基、1−エチルプロピルオキシ基、2−メチルペンチルオキシ基、3−メチルペンチルオキシ基、4−メチルペンチルオキシ基、1,2−ジメチルブチルオキシ基、1,3−ジメチルブチルオキシ基、2,3−ジメチルブチルオキシ基、1,1−ジメチルブチルオキシ基、2,2−ジメチルブチルオキシ基、3,3−ジメチルブチルオキシ基等が挙げられる。より好ましくは、メトキシ基、エトキシ基、またはプロポキシ基である。
R1、R2、R3またはR4において用いられる低級アルコキシカルボニル基としては、例えばメトキシカルボニル基、エトキシカルボニル基、プロポキシカルボニル基、イソプロポキシカルボニル基、シクロプロポキシカルボニル基、ブトキシカルボニル基、イソブトキシカルボニル基、sec−ブトキシカルボニル基、tert−ブトキシカルボニル基等が挙げられる。より好ましくは、メトキシカルボニル基、エトキシカルボニル基、プロポキシカルボニル基、イソプロポキシカルボニル基またはシクロプロポキシカルボニル基である。
R1、R2、R3またはR4において用いられる低級アシル基としては、例えば、ホルミル基、アセチル基、プロピオニル基、ブチリル基、イソブチリル基、バレリル基、1−メチルプロピルカルボニル基、イソバレリル基、ペンチルカルボニル基、1−メチルブチルカルボニル基、2−メチルブチルカルボニル基、3−メチルブチルカルボニル基、1−エチルプロピルカルボニル基、2−エチルプロピルカルボニル基等を挙げることが出来る。より好ましくはホルミル基、アセチル基、プロピオニル基である。
また、R1、R2、R3またはR4においては上記した低級アルキル基、低級アルコキシ基、低級アルコキシカルボニル基および低級アシル基の他に、同一または異なって水素原子またはハロゲン原子が好ましく用いられる。ハロゲン原子の具体的な例として、フッ素、塩素、臭素またはヨウ素が挙げられ、より好ましくはフッ素および塩素である。
本発明は、上述のように一般式[1]で表されることを特徴とするハーフサンドイッチ構造ルテニウム化合物である。好ましくは一般式[2]
[式中R1、R2は同一または相異なって水素、ハロゲン、低級アシル基、低級アルコキシ基、低級アルコキシカルボニル基、または低級アルキル基を示す。但し、R1が水素でR2がメチル基の場合を除く。]で表されるハーフサンドイッチ構造有機ルテニウム化合物であり、更に好ましくはR1,R2共に低級アルキル基であり、特にR1はエチル基、R2はメチル基が好ましい。
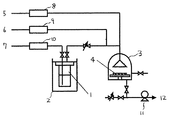
また本発明は上述のハーフサンドイッチ構造有機ルテニウム化合物を用いてCVD法でルテニウム含有薄膜を作る方法である。図1に装置の一例を示す。本発明のハーフサンドイッチ構造有機ルテニウム化合物を原料容器1に入れ、40〜120℃に保ち、この液に減圧下でキャリアーガス7をバブリングさせることによりハーフサンドイッチ構造有機ルテニウム化合物を蒸発させ、反応槽3に送る。加熱して200〜750℃に保持された基板4の上においてハーフサンドイッチ構造有機ルテニウム化合物を熱分解させるとルテニウム含有薄膜が生成する。
さらに本発明におけるCVD成膜法は、図1のようなバブリング法でもよいし、また、本発明の有機ルテニウム化合物をそのまま又は有機溶媒に溶かした溶液を気化器内に送って気化器内でガス化する溶液気化型でもよい。
また、本発明におけるCVD法で用いるハーフサンドイッチ構造有機ルテニウム化合物は、そのまま用いてもよいし、有機溶媒に溶解したハーフサンドイッチ構造有機ルテニウム化合物溶液として用いてもよい。この場合に用いられる有機溶媒としては、例えば、メタノール、エタノール、イソプロパノール等のアルコール類、酢酸エチル、酢酸ブチル、酢酸イソアミル等のエステル類、エチレングリコールモノエチルエーテル、エチレングリコールモノメチルエーテル、エチレングリコールモノブチルエーテル等のグリコールエーテル類、ジエチルエーテル、グライム、ジグライム、トリグライム、テトラヒドロフラン等のエーテル類、メチルブチルケトン、メチルイソブチルケトン、エチルブチルケトン、ジプロピルケトン、ジイソブチルケトン、メチルアミルケトン、シクロヘキサノン等のケトン類、ヘキサン、シクロヘキサン、ヘプタン、オクタン、ベンゼン、トルエン、キシレン等の炭化水素類が挙げられるが特に限定する物ではない。
本発明の一般式[1]で表される化合物は、一般式[3]で表されるオープン型ルテニウム錯体と一般式[4]で表されるシクロペンタジエンとを反応させることにより得ることができる。このとき、一般式[3]で表されるオープン型ルテニウム錯体は、一般式[5]で表されるペンタジエン誘導体と一般式[6]で表されるハロゲン化ルテニウム水和物を亜鉛存在下に反応させて得ることができる。これらの反応を反応式[I]に示す。従来これらハーフサンドイッチ構造有機ルテニウム化合物の製造方法の多くが、ペンタジエン誘導体とシクロペンタジエン誘導体を一度に加えて反応させる為に収率が芳しくなかったのに対し、このような製法によれば高収率で目的物を得ることが出来る。
[式中、Xはハロゲンを表し、nは0〜10の数を示す。R1、R2、R3、R4は前記と同じ内容を表す。]
この製法では、反応溶媒は特に限定されず、また生成物の回収・精製方法は特に限定されるものではない。しかしながらメタノールを一部又は全部の反応溶媒として用い、反応終了後にろ過して過剰の亜鉛を取り除いた後、メタノールと任意に交じり合わない溶媒を用いて一般式[1]で表されるハーフサンドイッチ構造有機ルテニウム化合物を抽出し、濃縮して得られる油状物を蒸留することにより、工業的に有利な工程を経て目的物を得ることが出来る。この際用いるメタノールと任意に交わらない溶媒としては、例えばペンタン、ヘキサン、ヘプタン、オクタン等、脂肪族炭化水素を挙げることが出来る。この中でも特にペンタン、ヘキサンは安価に入手可能であり、工業的に有利であるため好ましい。
この製法では、反応溶媒は特に限定されず、また生成物の回収・精製方法は特に限定されるものではない。しかしながらメタノールを一部又は全部の反応溶媒として用い、反応終了後にろ過して過剰の亜鉛を取り除いた後、メタノールと任意に交じり合わない溶媒を用いて一般式[1]で表されるハーフサンドイッチ構造有機ルテニウム化合物を抽出し、濃縮して得られる油状物を蒸留することにより、工業的に有利な工程を経て目的物を得ることが出来る。この際用いるメタノールと任意に交わらない溶媒としては、例えばペンタン、ヘキサン、ヘプタン、オクタン等、脂肪族炭化水素を挙げることが出来る。この中でも特にペンタン、ヘキサンは安価に入手可能であり、工業的に有利であるため好ましい。
本発明において反応に用いる亜鉛の量は特に限定されないが、一般式[6]で表される化合物又は一般式[3]で表される化合物1モルに対して1.0モル以上が好ましく、1.5モル以上用いるのが更に好ましい。大過剰量用いても経済的に不利なので1.5乃至100モル用いるのが有利である。一般式[5]で表される化合物と、一般式[6]で表される化合物を亜鉛の存在下反応させる際、一般式[5]で表される化合物を一般式[6]で表される化合物1モルに対して2モルあるいは過剰モル用いて反応させるのが好ましい。大過剰量用いても経済的に不利なので2乃至20モル用いるのが有利である。
一般式[5]で表される化合物と一般式[6]で表される化合物を亜鉛の存在下反応させる際、反応温度は−20乃至100℃で反応させるのが好ましい。さらに好ましくは−20乃至80℃である。一般式[3]で表される化合物と一般式[4]で表される化合物を亜鉛の存在下反応させる際、反応温度は−20乃至100℃で反応させるのが好ましい。さらに好ましくは−20乃至80℃である。
一般式[3]で表される化合物と一般式[4]で表される化合物を亜鉛の存在下反応させる際、一般式[4]で表される化合物を一般式[3]で表される化合物1モルに対して0.8乃至1.0モル用いて反応させるのが好ましい。0.8モル未満用いれば未反応で残る一般式[3]で表される化合物が多くなり、1.0モルを越えて用いれば副生物としてビス(エチルシクロペンタジエニル)ルテニウムが多く生成するために好ましい条件とは言えない。
本発明において、一般式[5]で表される化合物と一般式[6]で表される化合物とを反応させて、一般式[3]で表される化合物を製造した場合は、一般式[3]で表される化合物を単離することなくそのまま1ポットで一般式[4]で表される化合物と反応させて、一般式[1]で表される化合物を合成することが好ましい。
本発明における反応は、すべて窒素または不活性ガス雰囲気下で行うのが好ましい。不活性ガスとは例えばヘリウム、ネオン、アルゴン、クリプトン、キセノン、ラドンを挙げることが出来る。これらのうち安価に入手でき、空気より思いという点から窒素、アルゴンがさらに好ましい。
次に一般式[7]で表される化合物について説明する。このようなカルボニルビス(ジエン)ルテニウム錯体としてはカルボニルビス(1,3−ヘキサジエン)ルテニウム[R5〜R7=−H、R8=−C2H5]、カルボニルビス(2,4−ヘキサジエン)ルテニウム[R5,R8=−CH3、R6,R7=−H]、カルボニルビス(3−メチル−1,3−ペンタジエン)ルテニウム[R5,R6=−H、R7,R8=−CH3]、カルボニルビス(2,4−ヘキサジエナール)ルテニウム[R5=−CH3、R8=−C=O、R6,R7=−H]、カルボニルビス(2,4−ヘキサジエン−1−オル)ルテニウム[R5=−CH3、R8=−CH2OH、R6,R7=−H]、カルボニルビス(1−アセトキシ−1,3−ブタジエン)ルテニウム[R5=−O−COCH3、R6,R7,R8=−H]、カルボニルビス(2,4−ヘキサジエン酸)ルテニウム[R5=−CH3、R8=−COOH、R6,R7=−H]、カルボニルビス(2,4−ペンタジエン酸メチル)ルテニウム[R5=−COOCH3、R6,R7,R8=−H]等があげられる。
またカルボニルビス(2,4−ヘプタジエナール)ルテニウム[R5=−C2H5、R8=−C=O、R6,R7=−H]、カルボニルビス(2,6−ジメチル−2,4,6−オクタトリエン)ルテニウム[R5,R6=−CH3、R8=−C=C(CH3)2、R7=−H]、カルボニルビス(2,4−デカジエン酸エチル)ルテニウム[R5=−CH2CH2CH2CH2CH3、R8=−COOC2H5、R6,R7=−H]、カルボニルビス(ミルセン)ルテニウム[R6=−CH2CH2CH=C(CH3)2、R5,R7,R8=−H]、カルボニルビス(2,4−オクタジエナール)ルテニウム[R5=−CH2CH2CH3、R8=−C=O、R6,R7=−H]、カルボニルビス(ソルビン酸エチル)ルテニウム[R5=−CH3、R8=−COOC2H5、R6,R7=−H]、カルボニルビス(ソルビン酸メチル)ルテニウム[R5=−CH3、R8=−COOCH3、R6,R7=−H]、カルボニルビス(2,4−ヘプタジエン−6−オン)ルテニウム[R5=−CH3、R8=−COCH3、R6,R7=−H]等が挙げられる。
錯体の気化温度を下げる上では、R5〜R8のうち少なくとも1つが炭素数1〜2のアルキル基、エーテル、エステル、アルデヒド、アルコール、ケトン、ハロゲン化アルキル、カルボン酸、アミン、アミドで、他は水素であることが好ましく、更に好ましくはR5〜R8のうち少なくとも1つが炭素数1〜2のアルキル基で他は水素であり、このうちカルボニルビス(2−メチル−1,3−ペンタジエン)ルテニウム[R5,R7=−H、R6,R8=−CH3]が最も好ましい。
本発明の一般式[7]で表されるカルボニルビス(ジエン)ルテニウム錯体は塩化ルテニウム・n水和物とジエンを亜鉛存在下、アルコール中で反応させることにより製造することができる。この時、ジエンが不足の状態になると金属ルテニウムが生成して収率が低下するためジエン過剰で反応させることが好ましく、塩化ルテニウム・n水和物の10〜20倍モルのジエン量が好ましい。亜鉛粉末は塩化ルテニウム・n水和物の還元を十分に行なうため、10倍モル以上の過剰量が好ましい。また、これらを混合する際、亜鉛粉末を分散させたジエンまたはジエンのアルコール溶液中に塩化ルテニウム・n水和物のアルコール溶液を滴下すると高収率で目的とするカルボニルビス(ジエン)ルテニウム錯体を得ることができる。
使用されるジエンは分子内の連続した炭素鎖上に2つ以上の二重結合を持つ化合物であれば特に限定されない。反応中に二重結合の移動が起こるので1,4−ヘキサジエン、1,5−ヘキサジエン、2−メチル−1,4−ペンタジエン、3−メチル−1,4−ペンタジエン、2−メチル−1,5−ヘキサジエン等の非共役ジエンを使用してもよいが、下記一般式[8]で表わされる共役ジエンが好ましい。
(式中R5〜R8は水素または炭素数1〜6のアルキル、エーテル、エステル、アルデヒド、アルコール、ケトン、ハロゲン化アルキル、カルボン酸、アミン、アミドを示す。但し、R5〜R8全てが水素の場合、及びR5、R8が水素でR6、R7がメチルの場合を除く。)。
特に一般式[8]において、R5〜R8のうち少なくとも1つが炭素数1〜2のアルキル、エーテル、エステル、アルデヒド、アルコール、ケトン、ハロゲン化アルキル、カルボン酸、アミン、アミドであり、他は水素であることが好ましい。
このような共役ジエンとしては1,3−ヘキサジエン(R5〜R7=−H、R8=−C2H5)、2,4−ヘキサジエン(R5,R8=−CH3、R6,R7=−H)、3−メチル−1,3−ペンタジエン(R5,R6=−H、R7,R8=−CH3)、2,4−ヘキサジエナール(R5=−CH3、R8=−C=O、R6,R7=−H)、2,4−ヘキサジエン−1−オル(R5=−CH3、R8=−CH2OH、R6,R7=−H)、1−アセトキシ−1,3−ブタジエン(R5=−O−COCH3、R6,R7,R8=−H)、2,4−ヘキサジエン酸(R5=−CH3、R8=−COOH、R6,R7=−H)、2,4−ペンタジエン酸メチル(R5=−COOCH3、R6,R7,R8=−H)、2,4−ヘプタジエナール(R5=−C2H5、R8=−C=O、R6,R7=−H)があげられる。
また、2,6−ジメチル−2,4,6−オクタトリエン(R5,R6=−CH3、R8=−C=C(CH3)2、R7=−H)、2,4−デカジエン酸エチル(R5=−CH2CH2CH2CH2CH3、R8=−COOC2H5、R6,R7=−H)、ミルセン(R6=−CH2CH2CH=C(CH3)2、R5,R7,R8=−H)、2,4−オクタジエナール(R5=−CH2CH2CH3、R8=−C=O、R6,R7=−H)、ソルビン酸エチル(R5=−CH3、R8=−COOC2H5、R6,R7=−H)、ソルビン酸メチル(R5=−CH3、R8=−COOCH3、R6,R7=−H)、2,4−ヘプタジエン−6−オン(R5=−CH3、R8=−COCH3、R6,R7=−H)等が挙げられるが、2−メチル−1,3−ペンタジエン(R5,R7=−H、R6,R8=−CH3)が最も好ましい。
アルコールは室温(25℃)で液体であるものであれば特に限定されないが、アルコールから配位子となるCOが生成するため、反応速度の点からメタノール、エタノール、1−プロパノールが好ましい。また、使用するアルコールは1種類のみでなく、数種類の混合でもよい。亜鉛粉末を分散させたジエン又はジエンのアルコール溶液中に塩化ルテニウム・n水和物のアルコール溶液を滴下する場合には、ジエンを溶解に使用するアルコールと塩化ルテニウム・n水和物の溶解に使用するアルコールは同一である必要はなく異なっていても良い。反応温度は低温では反応速度が遅く、高温ではジエンの重合反応がおこるため、0〜80℃が好ましい。反応後、生成した錯体は亜鉛粉末をろ過により取り除いた後、反応混合物から直接あるいは反応混合物からアルコール、未反応のジエンを除去した残査から、ペンタン、ヘキサン等の溶媒により抽出することができる。抽出した液中には錯体以外に反応で生成したジエンの重合体等も含まれるため、クロマトグラフィー、蒸留等により精製し、錯体が得られる。
これらのカルボニルビス(ジエン)ルテニウム錯体を原料としてルテニウム含有薄膜を製造することができる。
CVD法によりカルボニルビス(ジエン)ルテニウム錯体を原料として基板上にルテニウムまたは酸化ルテニウムなどのルテニウム含有薄膜を製造する場合、ガス化して基板上に供給する。ガス化の方法としては加熱した液体の錯体中に不活性キャリアガスを導入し、キャリアガスに同伴させて基板の置かれた反応槽に導く方法、錯体を有機溶媒に溶かして溶液とし、溶液を気化器に送って気化器内でガス化して基板上の置かれた反応槽に導く方法等がある。
カルボニルビス(ジエン)ルテニウム錯体を有機溶媒に溶かして溶液として用いる場合、有機溶媒としては、例えば、メタノール、エタノール、イソプロパノ−ル等のアルコール類、酢酸エチル、酢酸ブチル、酢酸イソアミル等のエステル類、エチレングリコールモノメチルエーテル、エチレングリコールモノエチルエーテル、エチレングリコールモノブチルエーテル等のグリコールエーテル類、グライム、ジグライム、トリグライム、テトラヒドロフラン等のエーテル類、メチルブチルケトン、メチルイソブチルケトン、エチルブチルケトン、ジプロピルケトン、ジイソブチルケトン、メチルアミルケトン、シクロヘキサノン等のケトン類、ヘキサン、シクロヘキサン、ヘプタン、オクタン、トルエン、キシレン等の炭化水素類が挙げられるが特に限定されるものではない。
また、塗布熱分解法により基板上にルテニウム又は酸化ルテニウムなどのルテニウム含有薄膜を形成する場合も溶液で使用するが、このときも上記の有機溶媒を使用することができる。
本発明のルテニウム含有薄膜の製造に用いられるCVD法は熱CVD、プラズマCVD、光CVD等一般に使用されるCVD法であれば特に限定されない。塗布熱分解法の原料塗布方法としては例えばスピンコート法、ディップ法、噴霧法等が挙げられ、加熱方法としてはオーブン、ホットプレート等が使用できるが、塗布方法、加熱方法およびその組み合せは特に限定されない。
本発明の一般式[1]で表されるハーフサンドイッチ構造有機ルテニウム化合物は室温で液体であり、100℃付近で充分な蒸気圧を有しているので、CVD原料としてガスバブリングにより定量的に供給できる。また、従来の材料よりも低温で熱分解することができるので基板上にステップカバレッジに優れるRu含有薄膜を形成することが出来る。本発明により量産性に優れたCVD法でRu含有薄膜を形成できる。
本発明の有機ルテニウム化合物を用いてCVD法を行うことにより、CVDにおける酸素流量によってRu膜とRuO2膜とを作り分けることが可能である。また得られたRu含有薄膜は、緻密で、不純物が少なく、かつ結晶性にも優れたものであり、結果として抵抗率がバルクの値に近い良好な値を示すものを得ることができる。また本発明によるRu含有薄膜は、従来品を用いて作製した場合と比較して、緻密かつ薄膜表面が平坦なものを得ることができる。
本発明のハーフサンドイッチ構造有機ルテニウム化合物の製造方法によれば、従来の製造方法では低収率でしか得られなかったハーフサンドイッチ構造有機ルテニウム化合物を安定的に高収率で得られる。また、アルコールを濃縮することなく目的物を得ることもできるため、エネルギー的に有利に製造できる。このため、本発明のハーフサンドイッチ構造有機ルテニウム化合物の製造方法は、これらを製造する際の少量スケールでの製造のみならず、工業的な規模の製造に至るまで幅広く利用が可能である。
本発明のカルボニルビス(ジエン)ルテニウム錯体は、CVD法による成膜において従来使用さている錯体よりも低温での成膜が可能であり、且つ安定した原料供給のもとルテニウム含有薄膜の形成が可能である。また、熱分解温度が低く溶媒への溶解性が高いことから塗布熱分解法でもルテニウム含有薄膜の形成が可能である。さらに、溶媒中での反応触媒としての機能も期待できる。製造方法は従来知られているカルボニルビス(ジエン)ルテニウム錯体の製造方法に比べ、穏やかな条件であり製造に有利である。
また本発明によれば、一般式[3]で表されるオープン型ルテニウム錯体を、一般式[5]で表されるペンタジエンと一般式[6]で表されるハロゲン化ルテニウム水和物から製造することができる。
次に本発明を実施例によって詳細に説明するが、本発明はこの実施例にのみ限定されるものではない。
実施例1 (2,4−ジメチルペンタジエニル)(エチルシクロペンタジエニル)ルテニウムの合成および熱分解特性
四つ口フラスコに亜鉛400gを秤量し、容器をアルゴン置換して、2,4−ジメチル−1,3−ペンタジエン205mlを加えて懸濁液とした。3塩化ルテニウムn水和物(n=約3)30gをメタノール1000mlに溶かした溶液を40分かけて室温で滴下した。滴下終了後室温で30分間攪拌した後、60℃に昇温して2時間攪拌した。一旦放冷した後、エチルシクロペンタジエン12mlを投入しそのまま室温で30分攪拌、60℃に昇温して2時間攪拌した。反応終了後室温まで冷却し、グラスフィルターを用いて未反応の亜鉛を取り除いた後、ヘキサン750ml×1回、300ml×4回抽出した。抽出溶液を減圧下濃縮し、得られた油状物について減圧蒸留を行い、目的物である(2,4−ジメチルペンタジエニル)(エチルシクロペンタジエニル)ルテニウムを25.4g(収率76.3%)得た。
黄色油状物。
四つ口フラスコに亜鉛400gを秤量し、容器をアルゴン置換して、2,4−ジメチル−1,3−ペンタジエン205mlを加えて懸濁液とした。3塩化ルテニウムn水和物(n=約3)30gをメタノール1000mlに溶かした溶液を40分かけて室温で滴下した。滴下終了後室温で30分間攪拌した後、60℃に昇温して2時間攪拌した。一旦放冷した後、エチルシクロペンタジエン12mlを投入しそのまま室温で30分攪拌、60℃に昇温して2時間攪拌した。反応終了後室温まで冷却し、グラスフィルターを用いて未反応の亜鉛を取り除いた後、ヘキサン750ml×1回、300ml×4回抽出した。抽出溶液を減圧下濃縮し、得られた油状物について減圧蒸留を行い、目的物である(2,4−ジメチルペンタジエニル)(エチルシクロペンタジエニル)ルテニウムを25.4g(収率76.3%)得た。
黄色油状物。
1H−NMR(500MHz,CDCl3,dppm)
5.38(s,1H),4.63(t,J=2.0Hz,2H),4.52(t,J=2.0Hz,2H),2.70(d,J=2.5Hz,2H),2.15(q,J=7.5Hz,2H),1.93(s,6H),1.12(t,J=7.5Hz,3H),−0.09(d,J=2.5Hz,2H)。
IR(neat,cm−1)
3050,2960,2910,1475,1445,1430,1375,1030,860,800
MS(GC/MS,EI)
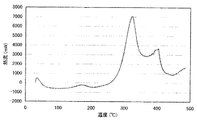
102Ruでの(2,4−ジメチルペンタジエニル)(エチルシクロペンタジエニル)ルテニウムの分子イオンピーク;m/z 290。なおこのMSのチャートを図2に示す。
5.38(s,1H),4.63(t,J=2.0Hz,2H),4.52(t,J=2.0Hz,2H),2.70(d,J=2.5Hz,2H),2.15(q,J=7.5Hz,2H),1.93(s,6H),1.12(t,J=7.5Hz,3H),−0.09(d,J=2.5Hz,2H)。
IR(neat,cm−1)
3050,2960,2910,1475,1445,1430,1375,1030,860,800
MS(GC/MS,EI)
102Ruでの(2,4−ジメチルペンタジエニル)(エチルシクロペンタジエニル)ルテニウムの分子イオンピーク;m/z 290。なおこのMSのチャートを図2に示す。
(分解特性)
また得られた(2,4−ジメチルペンタジエニル)(エチルシクロペンタジエニル)ルテニウムの分解特性を測定した結果を図3に示す。なお分解特性測定条件は以下の通りである。
測定方法:入力補償示差走査熱量測定(DSC)
測定条件: 参照 アルミナ
不活性ガス 窒素 50ml/min
昇温 10℃/min
比較例1 ビス(エチルシクロペンタジエニル)ルテニウムの分解特性
実施例1と同様の条件でビス(エチルシクロペンタジエニル)ルテニウムの分解特性を測定した。320℃付近より発熱反応が見られた。結果を図4に示す。
また得られた(2,4−ジメチルペンタジエニル)(エチルシクロペンタジエニル)ルテニウムの分解特性を測定した結果を図3に示す。なお分解特性測定条件は以下の通りである。
測定方法:入力補償示差走査熱量測定(DSC)
測定条件: 参照 アルミナ
不活性ガス 窒素 50ml/min
昇温 10℃/min
比較例1 ビス(エチルシクロペンタジエニル)ルテニウムの分解特性
実施例1と同様の条件でビス(エチルシクロペンタジエニル)ルテニウムの分解特性を測定した。320℃付近より発熱反応が見られた。結果を図4に示す。
実施例2 (2,4−ジメチルペンタジエニル)(メチルシクロペンタジエニル)ルテニウムの合成
50mlシュレンク管に亜鉛8.0gを秤量し、容器にアルゴンを置換して、2,4−ジメチル−1,3−ペンタジエン4mlを加えて懸濁液をした。3塩化ルテニウム水和物0.6gをエタノール20mlに溶かした溶液を50分かけて室温で滴下した。滴下終了後、室温で30分攪拌した後、70℃に昇温して2時間攪拌した。いったん放冷した後、メチルシクロペンタジエン240μlを投入し、そのまま室温で30分攪拌、70℃に昇温して2時間攪拌した。反応終了後室温まで冷却し、グラスフィルターを用いて未反応の亜鉛を取り除いた後、濃縮して泥状混合物を得た。得られた泥状混合物からペンタンで抽出し、抽出液について、アルミナを担体、ペンタンを溶離液としてカラムクロマトグラフィーを行い、目的物である、(2,4−ジメチルペンタジエニル)(メチルシクロペンタジエニル)ルテニウム0.28gを得た。
50mlシュレンク管に亜鉛8.0gを秤量し、容器にアルゴンを置換して、2,4−ジメチル−1,3−ペンタジエン4mlを加えて懸濁液をした。3塩化ルテニウム水和物0.6gをエタノール20mlに溶かした溶液を50分かけて室温で滴下した。滴下終了後、室温で30分攪拌した後、70℃に昇温して2時間攪拌した。いったん放冷した後、メチルシクロペンタジエン240μlを投入し、そのまま室温で30分攪拌、70℃に昇温して2時間攪拌した。反応終了後室温まで冷却し、グラスフィルターを用いて未反応の亜鉛を取り除いた後、濃縮して泥状混合物を得た。得られた泥状混合物からペンタンで抽出し、抽出液について、アルミナを担体、ペンタンを溶離液としてカラムクロマトグラフィーを行い、目的物である、(2,4−ジメチルペンタジエニル)(メチルシクロペンタジエニル)ルテニウム0.28gを得た。
1H−NMR(500MHz,CDCl3,δppm)
5.36(s、1H),4.61(t,J=2.0Hz,2H),4.57(t,J=2.0Hz,2H),2.67(d,J=2.5Hz,2H),1.93(s,6H),1.83(s,3H),−0.07(d,J=2.5Hz,2H)
MS(GC/MS,EI)
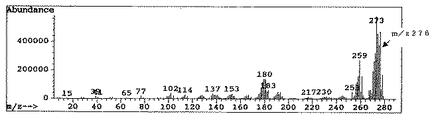
102Ruでの(2,4−ジメチルペンタジエニル)(メチルシクロペンタジエニル)ルテニウムの分子イオンピーク;m/z 276。なお、このMSチャートは図5に示す。
5.36(s、1H),4.61(t,J=2.0Hz,2H),4.57(t,J=2.0Hz,2H),2.67(d,J=2.5Hz,2H),1.93(s,6H),1.83(s,3H),−0.07(d,J=2.5Hz,2H)
MS(GC/MS,EI)
102Ruでの(2,4−ジメチルペンタジエニル)(メチルシクロペンタジエニル)ルテニウムの分子イオンピーク;m/z 276。なお、このMSチャートは図5に示す。
実施例3 (2,4―ジメチルペンタジエニル)(エチルシクロペンタジエニル)ルテニウムを原料としてCVD法によるルテニウム含有薄膜の製造
図1に示す装置を用い、基板としては表面にSiO2膜が100nm形成されたSi基板を用いた。原料容器1内に(2,4―ジメチルペンタジエニル)(エチルシクロペンタジエニル)ルテニウム約10gを入れてオイルバスで加熱、60℃恒温状態にした。真空ポンプ11、圧力調整弁を用いて反応槽3を10Torr、反応容器内を100Torrに調整した。キャリアガス7として窒素を用い、流量をマスフローコントローラーで100sccmに設定した。酸化ガス5として酸素を用い、カウンターガス6として窒素を用いた。酸化ガス流量を0、10、30、300sccmに設定し、カウンターガス流量を、酸化ガス流量との合計が500sccmとなるように設定した。基板4を400℃に設定し、加熱保持した状態で60分間成膜を行った。
図1に示す装置を用い、基板としては表面にSiO2膜が100nm形成されたSi基板を用いた。原料容器1内に(2,4―ジメチルペンタジエニル)(エチルシクロペンタジエニル)ルテニウム約10gを入れてオイルバスで加熱、60℃恒温状態にした。真空ポンプ11、圧力調整弁を用いて反応槽3を10Torr、反応容器内を100Torrに調整した。キャリアガス7として窒素を用い、流量をマスフローコントローラーで100sccmに設定した。酸化ガス5として酸素を用い、カウンターガス6として窒素を用いた。酸化ガス流量を0、10、30、300sccmに設定し、カウンターガス流量を、酸化ガス流量との合計が500sccmとなるように設定した。基板4を400℃に設定し、加熱保持した状態で60分間成膜を行った。
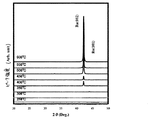
図6に酸素流量に対する基板上に成膜されたRu含有薄膜の膜厚を示す。また図6のA,B,Cで得られた膜について、図7にこれら膜のX線回折図形を示す。図7より、酸素流量が0sccmの場合(A)はRu膜が得られ、またそれ以上の酸素流量(B)(C)においてRuの酸化膜が得られることが明らかである。即ち、CVDにおける酸素流量により、Ru膜とRuO2膜とを作り分けることが可能であることが明らかとなった。
実施例4 (2,4―ジメチルペンタジエニル)(エチルシクロペンタジエニル)ルテニウムを原料としてCVD法によるルテニウム含有薄膜の製造
図1に示す装置を用い、基板としては表面にSiO2膜が100nm形成されたSi基板を用いた。原料容器1内に(2,4―ジメチルペンタジエニル)(エチルシクロペンタジエニル)ルテニウム約10gを入れてオイルバスで加熱、60℃恒温状態にした。真空ポンプ11、圧力調整弁を用いて反応槽3を10Torr、反応容器内を100Torrに調整した。キャリアガス7として窒素を用い、流量をマスフローコントローラーで100sccmに設定した。酸化ガス5として酸素を用い、カウンターガス6として窒素を用いた。酸化ガス流量を300sccmに設定し、カウンターガス流量を200sccmに設定した。基板4を170、200、300、400、500、600℃に設定し、加熱保持した状態で60分間成膜を行った。
図1に示す装置を用い、基板としては表面にSiO2膜が100nm形成されたSi基板を用いた。原料容器1内に(2,4―ジメチルペンタジエニル)(エチルシクロペンタジエニル)ルテニウム約10gを入れてオイルバスで加熱、60℃恒温状態にした。真空ポンプ11、圧力調整弁を用いて反応槽3を10Torr、反応容器内を100Torrに調整した。キャリアガス7として窒素を用い、流量をマスフローコントローラーで100sccmに設定した。酸化ガス5として酸素を用い、カウンターガス6として窒素を用いた。酸化ガス流量を300sccmに設定し、カウンターガス流量を200sccmに設定した。基板4を170、200、300、400、500、600℃に設定し、加熱保持した状態で60分間成膜を行った。
図8の(a)(黒丸)で膜の成長速度のアレニウスプロットを示す。200℃以上でRu含有薄膜を形成することができた。得られた膜の抵抗率を図9に示す。バルクの抵抗率(図9の矢印で表示)に近い良好な抵抗率が得られた。
実施例5 (2,4―ジメチルペンタジエニル)(エチルシクロペンタジエニル)ルテニウムを原料としてCVD法によるルテニウム含有薄膜の製造
図1に示す装置を用い、基板としては表面にSiO2膜が100nm形成されたSi基板を用いた。原料容器1内に(2,4―ジメチルペンタジエニル)(エチルシクロペンタジエニル)ルテニウム約10gを入れてオイルバスで加熱、60℃恒温状態にした。真空ポンプ11、圧力調整弁を用いて反応槽3を10Torr、反応容器内を100Torrに調整した。キャリアガス7として窒素を用い、流量をマスフローコントローラーで100sccmに設定した。酸化ガス5を使用せず、カウンターガス6として窒素を用い、カウンターガス流量を500sccmに設定した。基板4を250、275、300、325、350、375、400、450、500、550、600℃に設定し、加熱保持した状態で60分間成膜を行った。
図1に示す装置を用い、基板としては表面にSiO2膜が100nm形成されたSi基板を用いた。原料容器1内に(2,4―ジメチルペンタジエニル)(エチルシクロペンタジエニル)ルテニウム約10gを入れてオイルバスで加熱、60℃恒温状態にした。真空ポンプ11、圧力調整弁を用いて反応槽3を10Torr、反応容器内を100Torrに調整した。キャリアガス7として窒素を用い、流量をマスフローコントローラーで100sccmに設定した。酸化ガス5を使用せず、カウンターガス6として窒素を用い、カウンターガス流量を500sccmに設定した。基板4を250、275、300、325、350、375、400、450、500、550、600℃に設定し、加熱保持した状態で60分間成膜を行った。
図8の(b)(白丸)で膜の成長速度のアレニウスプロットを示す。275℃以上でRu膜を形成することができた。また図10に成膜されたRu含有薄膜のX線回折図を示す。400℃以上で結晶性のよい膜が得られた。図11に得られた膜の抵抗率を示す。バルクの抵抗率(図11の矢印で表示)に近い良好な抵抗率が得られた。図12,図14に基板温度300℃および600℃の成膜により得られた膜の断面のSEM写真を示す。いずれの温度においても緻密な膜が観察されたが、特に600℃の成膜ではより緻密な膜が観察された。また図16,図18に基板温度350℃および600℃の成膜により得られた膜の断面のAFM像を示す。AFMによる表面粗さの測定の結果、Ra(算術平均粗さ)及びRy(最大高さ)を表1(a)に示す。ここでRa及びRyはJIS B0601−1994・JIS B0031−1994に記載された方法である。本発明によるRu含有薄膜の表面は非常に平坦であることがわかる。
比較例2
比較例として、図1に示す装置を用い、原料容器1内に市販のビス(エチルシクロペンタジエニル)ルテニウム約10gを入れた以外はすべて実施例6と同様の条件において成膜を行った。図13,図15に基板温度300℃および600℃の成膜により得られた膜の断面のSEM写真を示す。いずれの場合もまくの緻密さに劣り、柱状の結晶成長が確認された。また図17,図19に基板温度350℃および600℃の成膜により得られた膜の断面のAFM像を示す。AFMによる表面粗さの測定の結果、Ra及びRyを表1(b)に示す。実施例と比較して表面粗さの大きいものであった。
比較例として、図1に示す装置を用い、原料容器1内に市販のビス(エチルシクロペンタジエニル)ルテニウム約10gを入れた以外はすべて実施例6と同様の条件において成膜を行った。図13,図15に基板温度300℃および600℃の成膜により得られた膜の断面のSEM写真を示す。いずれの場合もまくの緻密さに劣り、柱状の結晶成長が確認された。また図17,図19に基板温度350℃および600℃の成膜により得られた膜の断面のAFM像を示す。AFMによる表面粗さの測定の結果、Ra及びRyを表1(b)に示す。実施例と比較して表面粗さの大きいものであった。
実施例6 (2,4―ジメチルペンタジエニル)(エチルシクロペンタジエニル)ルテニウムを原料としてCVD法によるルテニウム含有薄膜の製造
図1に示す装置を用い、基板としては表面にSiO2膜が100nm形成されたSi基板を用いた。原料容器1内に(2,4―ジメチルペンタジエニル)(エチルシクロペンタジエニル)ルテニウム約10gを入れてオイルバスで加熱、60℃恒温状態にした。真空ポンプ11、圧力調整弁を用いて反応槽3を10Torr、反応容器内を100Torrに調整した。キャリアガス7として窒素を用い、流量をマスフローコントローラーで30、100sccmに設定した。酸化ガス5として酸素を用い、カウンターガス6として窒素を用いた。酸化ガス流量を300sccmに設定し、カウンターガス流量を200sccmに設定した。基板4を400℃に設定し、加熱保持した状態で60分間成膜を行った。キャリアガス流量と基板上に成膜されたRu含有薄膜の成膜速度の関係を図20に示す。キャリアガスの少ない領域においても十分成膜可能であった。
図1に示す装置を用い、基板としては表面にSiO2膜が100nm形成されたSi基板を用いた。原料容器1内に(2,4―ジメチルペンタジエニル)(エチルシクロペンタジエニル)ルテニウム約10gを入れてオイルバスで加熱、60℃恒温状態にした。真空ポンプ11、圧力調整弁を用いて反応槽3を10Torr、反応容器内を100Torrに調整した。キャリアガス7として窒素を用い、流量をマスフローコントローラーで30、100sccmに設定した。酸化ガス5として酸素を用い、カウンターガス6として窒素を用いた。酸化ガス流量を300sccmに設定し、カウンターガス流量を200sccmに設定した。基板4を400℃に設定し、加熱保持した状態で60分間成膜を行った。キャリアガス流量と基板上に成膜されたRu含有薄膜の成膜速度の関係を図20に示す。キャリアガスの少ない領域においても十分成膜可能であった。
実施例7 (2,4―ジメチルペンタジエニル)(エチルシクロペンタジエニル)ルテニウムを原料としてCVD法によるルテニウム含有薄膜の製造
図1に示す装置を用い、基板としては表面にSiO2膜が100nm形成されたSi基板を用いた。原料容器1内に(2,4―ジメチルペンタジエニル)(エチルシクロペンタジエニル)ルテニウム約10gを入れてオイルバスで加熱、60℃恒温状態にした。真空ポンプ11、圧力調整弁を用いて反応槽3を10Torr、反応容器内を100Torrに調整した。キャリアガス7として窒素を用い、流量をマスフローコントローラーで100sccmに設定した。酸化ガス5を使用せず、カウンターガス6として窒素を用い、カウンターガス流量を500sccmに設定した。基板4を350℃に設定し、加熱保持した状態で2.5、5、10、20、60、120分間成膜を行った。
図1に示す装置を用い、基板としては表面にSiO2膜が100nm形成されたSi基板を用いた。原料容器1内に(2,4―ジメチルペンタジエニル)(エチルシクロペンタジエニル)ルテニウム約10gを入れてオイルバスで加熱、60℃恒温状態にした。真空ポンプ11、圧力調整弁を用いて反応槽3を10Torr、反応容器内を100Torrに調整した。キャリアガス7として窒素を用い、流量をマスフローコントローラーで100sccmに設定した。酸化ガス5を使用せず、カウンターガス6として窒素を用い、カウンターガス流量を500sccmに設定した。基板4を350℃に設定し、加熱保持した状態で2.5、5、10、20、60、120分間成膜を行った。
図21の(a)(黒丸)で成膜時間と基板上に成膜されたRu含有薄膜の膜厚を示す。成膜時間2.5分以上でRu含有薄膜が得られ、時間に対し直線的に膜厚が増加した。本発明によるRu含有薄膜はインキュベーション時間(基板上に膜が形成されない時間)が認められず、Ru含有薄膜を容易に成膜することができる。
比較例3
比較例として、図1に示す装置を用い、原料容器1内に市販のビス(エチルシクロペンタジエニル)ルテニウム約10gを入れた以外はすべて実施例7と同様の条件において成膜を行った。図21の(b)(四角)で成膜時間と基板上に成膜されたRu含有薄膜の膜厚を示す。成膜時間20分まではインキュベ−ション時間が存在した。
比較例として、図1に示す装置を用い、原料容器1内に市販のビス(エチルシクロペンタジエニル)ルテニウム約10gを入れた以外はすべて実施例7と同様の条件において成膜を行った。図21の(b)(四角)で成膜時間と基板上に成膜されたRu含有薄膜の膜厚を示す。成膜時間20分まではインキュベ−ション時間が存在した。
実施例8 カルボニルビス(2−メチル−1,3−ペンタジエン)ルテニウムの合成
1000mlの4つ口フラスコに亜鉛粉末160gを入れ、攪拌装置、滴下ロート、冷却管、温度計を取り付け、フラスコ内をアルゴンで置換した。2−メチル−1,3−ペンタジエン60mlを加えて攪拌して亜鉛粉末を分散させた後、水浴で25℃以下に保ちながら滴下ロートより塩化ルテニウム・n水和物10gをエタノール400mlに溶解したものを1時間で滴下した。滴下終了後、70℃に加熱して3時間攪拌した。反応後の溶液から亜鉛粉末を濾過により除去し、減圧下でエタノール等を除去して得られた泥状物をペンタンで抽出した。抽出液をアルミナを充填剤、ペンタンを溶媒としたカラムクロマトグラフィーにより精製し、溶媒除去後、カルボニルビス(2−メチル−1,3−ペンタジエン)ルテニウムを黄色液体として9.99g(収率89%)得た。
1000mlの4つ口フラスコに亜鉛粉末160gを入れ、攪拌装置、滴下ロート、冷却管、温度計を取り付け、フラスコ内をアルゴンで置換した。2−メチル−1,3−ペンタジエン60mlを加えて攪拌して亜鉛粉末を分散させた後、水浴で25℃以下に保ちながら滴下ロートより塩化ルテニウム・n水和物10gをエタノール400mlに溶解したものを1時間で滴下した。滴下終了後、70℃に加熱して3時間攪拌した。反応後の溶液から亜鉛粉末を濾過により除去し、減圧下でエタノール等を除去して得られた泥状物をペンタンで抽出した。抽出液をアルミナを充填剤、ペンタンを溶媒としたカラムクロマトグラフィーにより精製し、溶媒除去後、カルボニルビス(2−メチル−1,3−ペンタジエン)ルテニウムを黄色液体として9.99g(収率89%)得た。
1H−NMR(CDCl3)δ3.91(d,2H,J=7.5Hz),2.04(s,6H),1.42(s,2H),1.27(d,6H,J=6Hz),1.17(dq,2H,J=14Hz),0.30(s,2H)。
13C−NMR(CDCl3)δ218.88(CO),91.51(C),89.47(CH),50.51(CH),37.58(CH2),22.84(CH3),18.78(CH3)。
IR 1967cm−1(CO)。
MS 294(M+)。
13C−NMR(CDCl3)δ218.88(CO),91.51(C),89.47(CH),50.51(CH),37.58(CH2),22.84(CH3),18.78(CH3)。
IR 1967cm−1(CO)。
MS 294(M+)。
実施例9 カルボニルビス(2−メチル−1,3−ペンタジエン)ルテニウムの熱分解特性(DSC測定)
実施例8で得られたカルボニルビス(2−メチル−1,3−ペンタジエン)ルテニウム6.7mgをステンレス製パンに取り、ステンレス製のふたで密封した。このサンプルを酸化アルミニウム18.8mgをリファレンスとして1分間に10℃の昇温速度で示差走査熱量測定装置(DSC)により加熱時の熱量変化を測定した。120℃付近より発熱反応が見られ錯体の分解が確認された。結果を図22に示す。
実施例8で得られたカルボニルビス(2−メチル−1,3−ペンタジエン)ルテニウム6.7mgをステンレス製パンに取り、ステンレス製のふたで密封した。このサンプルを酸化アルミニウム18.8mgをリファレンスとして1分間に10℃の昇温速度で示差走査熱量測定装置(DSC)により加熱時の熱量変化を測定した。120℃付近より発熱反応が見られ錯体の分解が確認された。結果を図22に示す。
実施例10
カルボニルビス(2−メチル−1,3−ペンタジエン)ルテニウムを原料としてCVD法によるルテニウム含有膜膜の製造
カルボニルビス(2−メチル−1、3−ペンタジエン)ルテニウムを原料として図1に示すCVD装置を用いて、原料温度50℃、キャリアガス(N2)流量100sccm、原料圧力400Torr、カウンターガス(N2)流量200sccm、配管温度80℃、基板温度300℃、チャンバー圧力10Torr、酸化ガス(O2)流量300sccmでSiO2膜付シリコンウエハ上に1時間成膜を行なった。膜組成をX線回折により確認したところRuO2であり、膜厚をSEMにより測定したところ500nmだった。
カルボニルビス(2−メチル−1,3−ペンタジエン)ルテニウムを原料としてCVD法によるルテニウム含有膜膜の製造
カルボニルビス(2−メチル−1、3−ペンタジエン)ルテニウムを原料として図1に示すCVD装置を用いて、原料温度50℃、キャリアガス(N2)流量100sccm、原料圧力400Torr、カウンターガス(N2)流量200sccm、配管温度80℃、基板温度300℃、チャンバー圧力10Torr、酸化ガス(O2)流量300sccmでSiO2膜付シリコンウエハ上に1時間成膜を行なった。膜組成をX線回折により確認したところRuO2であり、膜厚をSEMにより測定したところ500nmだった。
実施例11 カルボニルビス(2−メチル−1,3−ペンタジエン)ルテニウムを原料としてスピンコート法によるルテニウム含有膜膜の製造
カルボニルビス(2−メチル−1、3−ペンタジエン)ルテニウム1mlをヘプタン9mlに溶かし溶液を作製した。この溶液をシリコンウエハ上に500rpmで5秒、1000rpmで10秒スピンコートし、150℃のオーブン中で20分間加熱した。膜組成をX線回折により確認したところRuO2であり、膜厚をSEMにより測定したところ300nmだった。
カルボニルビス(2−メチル−1、3−ペンタジエン)ルテニウム1mlをヘプタン9mlに溶かし溶液を作製した。この溶液をシリコンウエハ上に500rpmで5秒、1000rpmで10秒スピンコートし、150℃のオーブン中で20分間加熱した。膜組成をX線回折により確認したところRuO2であり、膜厚をSEMにより測定したところ300nmだった。
比較例4
実施例10と同様の方法によりビス(エチルシクロペンタジエン)ルテニウム
を原料として成膜を行なったところRuを含む膜は得られなかった。
実施例10と同様の方法によりビス(エチルシクロペンタジエン)ルテニウム
を原料として成膜を行なったところRuを含む膜は得られなかった。
実施例12
亜鉛8gをシュレンク管に仕込み、アルゴン置換を行った後、エチルシクロペンタジエン240ml、2,4−ジメチル−1,3−ペンタジエン4mlを加えた。氷冷下に塩化ルテニウム水和物608mg(n=約3)を脱水エタノール25mlに溶解した溶液を1時間15分かけて滴下した後、氷冷下1時間30分攪拌、室温1時間攪拌した。反応終了後グラスフィルターを用いて未反応の亜鉛を取り除いた後、減圧下濃縮して泥状物を得た。この泥状物からペンタンで抽出を行い、抽出液についてカラムクロマトグラフィー(ペンタン/アルミナ)を行い、目的物の(2,4−ジメチルペンタジエニル)(エチルシクロペンタジエニル)ルテニウム106mg(収率16%)を得た。また、副生物としてビス(エチルシクロペンタジエニル)ルテニウムを70mg(収率11%)得た。
亜鉛8gをシュレンク管に仕込み、アルゴン置換を行った後、エチルシクロペンタジエン240ml、2,4−ジメチル−1,3−ペンタジエン4mlを加えた。氷冷下に塩化ルテニウム水和物608mg(n=約3)を脱水エタノール25mlに溶解した溶液を1時間15分かけて滴下した後、氷冷下1時間30分攪拌、室温1時間攪拌した。反応終了後グラスフィルターを用いて未反応の亜鉛を取り除いた後、減圧下濃縮して泥状物を得た。この泥状物からペンタンで抽出を行い、抽出液についてカラムクロマトグラフィー(ペンタン/アルミナ)を行い、目的物の(2,4−ジメチルペンタジエニル)(エチルシクロペンタジエニル)ルテニウム106mg(収率16%)を得た。また、副生物としてビス(エチルシクロペンタジエニル)ルテニウムを70mg(収率11%)得た。
1 原料容器
2 恒温槽
3 反応槽
4 基板
5 酸化ガス
6 カウンターガス
7 キャリアガス
8 マスフローコントローラー
9 マスフローコントローラー
10 マスフローコントローラー
11 真空ポンプ
12 排気
2 恒温槽
3 反応槽
4 基板
5 酸化ガス
6 カウンターガス
7 キャリアガス
8 マスフローコントローラー
9 マスフローコントローラー
10 マスフローコントローラー
11 真空ポンプ
12 排気
Claims (8)
- 一般式[5]
RuX3・nH2O [6]
[式中、Xはハロゲンを表し、nは0乃至10の数字を表す。]で表されるハロゲン化ルテニウム水和物を溶媒で希釈した溶液を滴下・反応させることを特徴とする、一般式[3]
- 請求項1に記載の方法において、一般式[5]で表されるペンタジエンとして一般式[9]
- 請求項1または2に記載の方法において、一般式[5]または[9]で表されるペンタジエンとして2,4−ジメチル−1,3−ペンタジエンを用いることを特徴とする、ビス(2,4−ジメチルペンタジエニル)ルテニウム(II)の製造方法。
- 請求項1〜3いずれかに記載の方法において、メタノールを一部または全部の溶媒として用いることを特徴とする方法。
- 請求項1〜4いずれかに記載の方法において、一般式[5]もしくは[9]で表されるペンタジエン、または2,4−ジメチル−1,3−ペンタジエンの添加量が、一般式[6]で表されるハロゲン化ルテニウム水和物1モルに対して2乃至20モルであることを特徴とする方法。
- 請求項1〜5いずれかに記載の方法において、亜鉛の添加量が、一般式[6]で表されるハロゲン化ルテニウム水和物1モルに対して1.5乃至100モルであることを特徴とする方法。
- 請求項1〜6いずれかに記載の方法において、反応温度が−20乃至100℃であることを特徴とする方法。
- 請求項1〜7いずれかに記載の方法において、反応を窒素ガスまたは不活性ガス雰囲気中で行うことを特徴とする方法。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2005226885A JP2006036780A (ja) | 2001-09-12 | 2005-08-04 | ルテニウム錯体の製造方法 |
Applications Claiming Priority (7)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2001276479 | 2001-09-12 | ||
| JP2001276478 | 2001-09-12 | ||
| JP2001276481 | 2001-09-12 | ||
| JP2001276480 | 2001-09-12 | ||
| JP2001359769 | 2001-11-26 | ||
| JP2002078970 | 2002-03-20 | ||
| JP2005226885A JP2006036780A (ja) | 2001-09-12 | 2005-08-04 | ルテニウム錯体の製造方法 |
Related Parent Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2002217225A Division JP4517565B2 (ja) | 2001-09-12 | 2002-07-25 | ルテニウム錯体、その製造方法、及び薄膜の製造方法 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| JP2006036780A true JP2006036780A (ja) | 2006-02-09 |
Family
ID=35902142
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2005226885A Pending JP2006036780A (ja) | 2001-09-12 | 2005-08-04 | ルテニウム錯体の製造方法 |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP2006036780A (ja) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2009007270A (ja) * | 2007-06-27 | 2009-01-15 | Tosoh Corp | ルテニウム化合物の製造方法および薄膜の製造方法 |
| WO2011080978A1 (ja) | 2009-12-28 | 2011-07-07 | 東ソー株式会社 | ルテニウム錯体混合物、その製造方法、成膜用組成物、ルテニウム含有膜及びその製造方法 |
-
2005
- 2005-08-04 JP JP2005226885A patent/JP2006036780A/ja active Pending
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2009007270A (ja) * | 2007-06-27 | 2009-01-15 | Tosoh Corp | ルテニウム化合物の製造方法および薄膜の製造方法 |
| WO2011080978A1 (ja) | 2009-12-28 | 2011-07-07 | 東ソー株式会社 | ルテニウム錯体混合物、その製造方法、成膜用組成物、ルテニウム含有膜及びその製造方法 |
| US8742153B2 (en) | 2009-12-28 | 2014-06-03 | Tosoh Corporation | Ruthenium complex mixture, method of producing the same, composition for film formation, ruthenium-containing film, and method of producing the same |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5104499B2 (ja) | カルボニルビス(ジエン)ルテニウム錯体、その製造方法、および薄膜の製造方法 | |
| JP4517565B2 (ja) | ルテニウム錯体、その製造方法、及び薄膜の製造方法 | |
| US8221837B2 (en) | Organometallic compounds and processes for preparation thereof | |
| JP4696454B2 (ja) | 新規有機イリジウム化合物、その製造方法、及び膜の製造方法 | |
| JP2006036780A (ja) | ルテニウム錯体の製造方法 | |
| WO2005103318A1 (en) | High nucleation density organometallic compounds | |
| US7667065B2 (en) | High nucleation density organometallic compounds | |
| KR101126141B1 (ko) | 유기 이리듐 화합물, 그의 제법 및 막의 제조방법 | |
| JP4553642B2 (ja) | 有機イリジウム化合物、その製法、及び膜の製造方法 | |
| EP1471567B1 (en) | Organometallic iridium compounds, processes for producing the same, and processes for producing thin films | |
| TWI227239B (en) | Ruthenium complex, process for producing the same and process for producing thin film | |
| CN100390186C (zh) | 有机金属铱化合物,制备它的方法,以及制备薄膜的方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A977 | Report on retrieval |
Effective date: 20070904 Free format text: JAPANESE INTERMEDIATE CODE: A971007 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20080212 |
|
| A02 | Decision of refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A02 Effective date: 20080819 |