JP2007161902A - テロメラーゼ阻害活性を有する新規化合物 - Google Patents
テロメラーゼ阻害活性を有する新規化合物 Download PDFInfo
- Publication number
- JP2007161902A JP2007161902A JP2005360820A JP2005360820A JP2007161902A JP 2007161902 A JP2007161902 A JP 2007161902A JP 2005360820 A JP2005360820 A JP 2005360820A JP 2005360820 A JP2005360820 A JP 2005360820A JP 2007161902 A JP2007161902 A JP 2007161902A
- Authority
- JP
- Japan
- Prior art keywords
- axinelloside
- shows
- spectrum
- cor
- telomerase
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Images



































Landscapes
- Polysaccharides And Polysaccharide Derivatives (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
【課題】本発明は新規テロメラーゼ阻害物質を提供することを目的とする。
【解決手段】本発明者らは、日本近海産の海洋無脊椎動物から、TRAP法を用いてテロメラーゼ阻害物質の探索を行ったところ、式根島産海綿Axinella infundibulaに顕著な活性を見いだし、その活性成分として新規硫酸化糖脂質のaxinelloside Aを単離・構造決定した。
【選択図】なし
【解決手段】本発明者らは、日本近海産の海洋無脊椎動物から、TRAP法を用いてテロメラーゼ阻害物質の探索を行ったところ、式根島産海綿Axinella infundibulaに顕著な活性を見いだし、その活性成分として新規硫酸化糖脂質のaxinelloside Aを単離・構造決定した。
【選択図】なし
Description
本発明はテロメラーゼ阻害活性を有する新規化合物に関する。
真核生物の染色体の3’末端はテロメアと呼ばれるTTAGGGの繰り返し配列を持ち、染色体が安定に存在するために重要な役割を果たしている。正常細胞は細胞分裂をするごとにテロメアが短くなり、一定以下の長さになると分裂を止めるのに対し、がん細胞ではテロメラーゼと呼ばれる逆転写酵素が働きテロメアを伸長させるため、増殖が止まることがない。テロメラーゼはほとんどの正常細胞で発現していないのに対し、約90%のヒトのがん細胞では発現しているという。そのため、テロメラーゼの阻害物質は、抗がん剤のターゲットとして有望視されている。
なお本発明者らはこれまでにテロメラーゼ阻害活性を有する天然化合物としてdictyodendrins A-E を見出している(非特許文献1)。
Warabi, K.; Matsunaga, S.; van Soest, R. W. M.; Fusetani, N. J. Org. Chem. 2003, 68, 2765-2770.
本発明は新規テロメラーゼ阻害物質を提供することを目的とする。
本発明者らは、日本近海産の海洋無脊椎動物から、TRAP (telomeric repeat amplification protocol) 法を用いてテロメラーゼ阻害物質の探索を行ったところ、式根島産海綿Axinella infundibulaに顕著な活性を見いだし、その活性成分として新規硫酸化糖脂質のaxinelloside Aを単離・構造決定した。
本発明は以下の発明を包含する。
1. 式(I):
[式中、
-COR1、-COR2、-COR3および-COR4は、それぞれ独立に、直鎖状または分岐鎖状の、飽和または不飽和の、1つ以上の水酸基で置換されていてもよいアシル基を表し、
-OXは-OSO3 -M+(M+は一価の陽イオンを表す) を表す。]
により表される化合物。
1. 式(I):
-COR1、-COR2、-COR3および-COR4は、それぞれ独立に、直鎖状または分岐鎖状の、飽和または不飽和の、1つ以上の水酸基で置換されていてもよいアシル基を表し、
-OXは-OSO3 -M+(M+は一価の陽イオンを表す) を表す。]
により表される化合物。
2. 式(I)における-OH基の水素の少なくとも1つがSO3 -M+(M+は一価の陽イオンを表す) に置換されている、1記載の化合物の誘導体化合物。
3. 式(I)における-OSO3 -M+基のSO3 -M+の少なくとも1つが水素に置換されている、1または2記載の化合物の誘導体化合物。
3. 式(I)における-OSO3 -M+基のSO3 -M+の少なくとも1つが水素に置換されている、1または2記載の化合物の誘導体化合物。
4. -COR1、-COR3および-COR4がそれぞれ
であり、
-COR2が
(式中、3位の不斉炭素原子はR配置を有する)
である(各式中*は酸素原子への結合位置を示す)、1〜3のいずれかに記載の化合物。
-COR2が
である(各式中*は酸素原子への結合位置を示す)、1〜3のいずれかに記載の化合物。
5. 1〜4のいずれかに記載の化合物(ただしM+は薬学的に許容される一価の陽イオンである)を有効成分として含有する医薬組成物。
6. 1〜4のいずれかに記載の化合物(ただしM+は薬学的に許容される一価の陽イオンである)を有効成分として含有するテロメラーゼ阻害剤。
7. 1〜4のいずれかに記載の化合物(ただしM+は薬学的に許容される一価の陽イオンである)を有効成分として含有する抗癌剤。
6. 1〜4のいずれかに記載の化合物(ただしM+は薬学的に許容される一価の陽イオンである)を有効成分として含有するテロメラーゼ阻害剤。
7. 1〜4のいずれかに記載の化合物(ただしM+は薬学的に許容される一価の陽イオンである)を有効成分として含有する抗癌剤。
本発明により、新規テロメラーゼ阻害物質が提供される。
式(I)において、-COR1、-COR3および-COR4は、それぞれ独立に、直鎖状または分岐鎖状の、飽和または不飽和の、1つ以上(例えば1〜3個)の水酸基で置換されていてもよいアシル基であり、なかでも直鎖状であることが好ましく、1〜3不飽和、特に1不飽和であることが好ましく、水酸基で置換されていないことが好ましい。不飽和結合として二重結合が含まれる場合、当該二重結合はシス配置を有していてもトランス配置を有していても良いが、トランス配置を有していることが好ましい。当該アシル基に不飽和結合が含まれる場合、不飽和結合の位置は特に限定されない。当該アシル基の炭素数は特に限定されないが、2〜22が好ましく、12〜20がより好ましく、14〜20がもっとも好ましい。本発明の最も好ましい形態では、-COR1、-COR3および-COR4は共に
(式中、二重結合はシス配置を有していてもトランス配置を有していてもよいが、トランス配置を有することが好ましく、
*は酸素原子への結合位置を示す)
で表される基である。
*は酸素原子への結合位置を示す)
で表される基である。
式(I)において、-COR2は、直鎖状または分岐鎖状の、飽和または不飽和の、1つ以上(例えば1〜3個)の水酸基で置換されていてもよいアシル基であり、なかでも直鎖状であることが好ましく、飽和であることが好ましく、1つの水酸基で置換されていることが好ましい。当該アシル基が1つの水酸基で置換されている場合その置換位置は特に限定されないが例えば3位である。当該アシル基の炭素数は特に限定されないが、2〜22が好ましく、12〜20がより好ましく、14〜20がもっとも好ましい。不飽和結合として二重結合が含まれる場合、当該二重結合はシス配置を有していてもトランス配置を有していても良いが、トランス配置を有していることが好ましい。本発明の最も好ましい形態では、-COR2は
(式中、3位の不斉炭素原子はS配置を有していてもR配置を有していてもよいが、R配置を有していることが好ましく、
*は酸素原子への結合位置を示す)
で表される基である。
*は酸素原子への結合位置を示す)
で表される基である。
本発明の式(I)で表される最も好ましい化合物は海綿Axinella infundibulaに含まれる新規硫酸化糖脂質のaxinelloside Aである。Axinelloside Aは、海綿Axinella infundibulaのメタノール、エタノールおよびアセトンによる抽出物を、水とジエチルエーテルで二相分配し、そのエーテル画分をn-ヘキサンと90% メタノールで二相分配し、さらに、n-ヘキサン画分をシリカゲルクロマトグラフィーとODS HPLC (Cosmosil 5C18-AR-II)で分離して得られる、テロメラーゼ阻害活性を有する画分に含まれる。当該テロメラーゼ阻害活性画分は更に精製することなくそれ自体をテロメラーゼ活性阻害剤として用いることもできるが、実施例に記載の通りリサイクルHPLCなどの常法により精製し、axinelloside Aを高純度化することが好ましい。
式(I)において、M+は一価の陽イオンを表する。一価の陽イオンとしては具体的には、ナトリウム、カリウム、アンモニウム等が挙げられる。
テロメラーゼ活性阻害剤として有用である化合物は、式(I)で表される化合物のみには限定されず、-OH基の水素の少なくとも1つ(例えば1〜5個)がSO3 -M+(M+は一価の陽イオンを表す) に置換された誘導体化合物であってもよいし、-OSO3 -M+基のSO3 -M+の少なくとも1つ(例えば1〜5個)が水素に置換された誘導体化合物であってもよい。
本発明の化合物は溶媒和物(例えば水和物)として提供されてもよい。
本発明はまた、式(I)の化合物を有効成分として含有する医薬組成物に関する。本発明の医薬組成物はテロメラーゼ阻害剤として、癌の予防または治療のために用いることができる。
本発明はまた、式(I)の化合物を有効成分として含有する医薬組成物に関する。本発明の医薬組成物はテロメラーゼ阻害剤として、癌の予防または治療のために用いることができる。
本発明の医薬組成物は、常套手段に従って製剤化することができる。例えば、必要に応じて糖衣や腸溶性被膜を施した錠剤、カプセル剤、エリキシル剤、マイクロカプセル剤などとして経口的に、あるいは水もしくはそれ以外の薬学的に許容し得る液との溶液剤、または懸濁液剤などの注射剤の形で非経口的に使用できる。例えば、本発明の化合物を薬学的に許容される担体、香味剤、賦形剤、ベヒクル、防腐剤、安定剤、結合剤などとともに製剤化することができる。本発明の医薬組成物中の有効成分量は、所定の治療または予防効果が達成できる量であれば特に限定されない。
錠剤、カプセル剤などに混和することができる添加剤としては、例えばゼラチン、コーンスターチ、トラガントガム、アラビアゴムのような結合剤、結晶性セルロースのような賦形剤、コーンスターチ、ゼラチン、アルギン酸などのような膨化剤、ステアリン酸マグネシウムのような潤滑剤、ショ糖、乳糖またはサッカリンのような甘味剤、ペパーミント、アカモノ油またはチェリーのような香味剤などが用いられる。調剤単位形態がカプセルである場合には、前記タイプの材料にさらに油脂のような液状担体を含有することができる。注射のための無菌組成物は、注射用水などの適当なベヒクル中に本発明の化合物を溶解または懸濁させるなどの通常の製剤方法にしたがって処方することができる。注射用の水性液としては、例えば、生理食塩水、ブドウ糖やその他の補助薬を含む等張液(例えば、D−ソルビトール、D−マンニトール、塩化ナトリウムなど)などがあげられ、適当な溶解補助剤、たとえばアルコール(たとえばエタノール)、ポリアルコール(たとえばプロピレングリコール、ポリエチレングリコール)、非イオン性界面活性剤(たとえばポリソルベート80(商標)、HCO−50)などと併用してもよい。油性液としてはゴマ油、大豆油などがあげられる。また、注射液には更に、緩衝剤(例えば、リン酸塩緩衝液、酢酸ナトリウム緩衝液)、無痛化剤(例えば、塩化ベンザルコニウム、塩酸プロカインなど)、安定剤(例えば、ヒト血清アルブミン、ポリエチレングリコールなど)、保存剤(例えば、ベンジルアルコール、フェノールなど)、酸化防止剤などを配合してもよい。調製された注射液は通常、適当なアンプルに充填される。このようにして得られる製剤は安全で低毒性であるので、例えばヒトや哺乳動物(例えば、マウス、ラット、モルモット、ウサギ、ニワトリ、ヒツジ、ブタ、ウシ、ネコ、イヌ、サル、マントヒヒ、チンパンジーなど)に対して投与することができる。本発明の化合物の投与量は、症状などにより差異はあるが、一般的に成人(体重60kgとして)においては、一日につき約0.1〜1000mg程度である。他の動物の場合も、60kg当たりに換算した量を投与することができる。
1) 実験
1-1) 一般的手順
NMRスペクトル測定のために、サンプルをCD3OD/D2O (9:1) 中に1 mM の濃度で溶解した。NMRスペクトルはBruker Avance 600 または Avance 800 spectrometerを用いて記録した。データ処理はSilicon Graphics O2 ワークステーション上でXWINNMR ソフトウェア (Bruker)を用いて行った。1Hおよび13C 化学シフトは、DSS (2,2-ジメチル-2-シラペンタン-5-スルホン酸)-トリメチルの一重線共鳴を基準(プロトンおよび炭素についての0 ppm)とした場合の、CD3OD/D2O (9:1) についてのδH 3.302 およびδC50.695の溶媒ピーク、ならびに、CD3OH/H2O (9:1) についてのδH3.310およびδC 50.806 の溶媒ピークを参照した。一次元1H スペクトルおよび13C スペクトルは、300Kの温度でそれぞれ32Kおよび64Kのデータポイントで記録した。DQF-COSYスペクトルは、t1側が1K、t2側が8Kのデータポイントを用いた位相敏感検波モードで測定した。NOESY実験における混合時間は300msとし、HOHAHAにおけるスピンロッキング時間は80 msとした。HMBCスペクトルおよびHMQCスペクトルは実質的に文献記載の方法で記録した。重水素同位体シフト実験のためには、CD3OH/H2O (9:1)中の1 mM axinelloside A の溶液をHMQC実験に用いた。またHOHAHA-HSQCスペクトルを60 msの混合時間により測定した。二次元測定は位相敏感検波モードを用いて行い、そのデータポイントはDQF-COSYを除き、t1側が1K、t2側が2Kを用いた。FABMSは、JEOL JMX-SX102/SX102 タンデム質量分析装置により、加速電圧10 kVにて得た。サンプルをn-PrOH/MeCN/H2O (2:3:3) および 5 mM 酢酸アンモニウムに溶解した後、ナノスプレーイオン源を備えたQ-Tof2質量分析装置 (Micromass UK Limited, Manchester, UK) を用い、負イオンモードによりESI-MS分析を行った。負イオンモードでリニアまたはリフレクターモードのMALDI-TOF-MSについては、Ultraflex II TOF/TOF 装置 (Bruker Daltonik GmbH, Bremen, Germany)を用い、マトリックス (α-シアノ-4-ヒドロキシ桂皮酸、シナピン酸、2,5-ジヒドロキシ安息香酸または6-アザ-2-チオチミン) と対イオン (0.1% TFA または 0.2 M NaCl) との9:1 (v / v) 混合物 1 μLを用いて行った。サンプルのテトラメチルアンモニウム塩の負イオンモードESI-MS分析は、ナノ-ESIプローブに適合したLTQ質量分析装置 (Thermo Electron, San Jose, CA, USA) を用いて行った。IRスペクトルはJASCO FT/IR-5300赤外分光光度計により記録した。旋光度はJASCO DIP-1000ディジタル旋光計により測定した。UVスペクトルはShimadzu Bio Spc-1600紫外可視分光光度計により記録した。
1-1) 一般的手順
NMRスペクトル測定のために、サンプルをCD3OD/D2O (9:1) 中に1 mM の濃度で溶解した。NMRスペクトルはBruker Avance 600 または Avance 800 spectrometerを用いて記録した。データ処理はSilicon Graphics O2 ワークステーション上でXWINNMR ソフトウェア (Bruker)を用いて行った。1Hおよび13C 化学シフトは、DSS (2,2-ジメチル-2-シラペンタン-5-スルホン酸)-トリメチルの一重線共鳴を基準(プロトンおよび炭素についての0 ppm)とした場合の、CD3OD/D2O (9:1) についてのδH 3.302 およびδC50.695の溶媒ピーク、ならびに、CD3OH/H2O (9:1) についてのδH3.310およびδC 50.806 の溶媒ピークを参照した。一次元1H スペクトルおよび13C スペクトルは、300Kの温度でそれぞれ32Kおよび64Kのデータポイントで記録した。DQF-COSYスペクトルは、t1側が1K、t2側が8Kのデータポイントを用いた位相敏感検波モードで測定した。NOESY実験における混合時間は300msとし、HOHAHAにおけるスピンロッキング時間は80 msとした。HMBCスペクトルおよびHMQCスペクトルは実質的に文献記載の方法で記録した。重水素同位体シフト実験のためには、CD3OH/H2O (9:1)中の1 mM axinelloside A の溶液をHMQC実験に用いた。またHOHAHA-HSQCスペクトルを60 msの混合時間により測定した。二次元測定は位相敏感検波モードを用いて行い、そのデータポイントはDQF-COSYを除き、t1側が1K、t2側が2Kを用いた。FABMSは、JEOL JMX-SX102/SX102 タンデム質量分析装置により、加速電圧10 kVにて得た。サンプルをn-PrOH/MeCN/H2O (2:3:3) および 5 mM 酢酸アンモニウムに溶解した後、ナノスプレーイオン源を備えたQ-Tof2質量分析装置 (Micromass UK Limited, Manchester, UK) を用い、負イオンモードによりESI-MS分析を行った。負イオンモードでリニアまたはリフレクターモードのMALDI-TOF-MSについては、Ultraflex II TOF/TOF 装置 (Bruker Daltonik GmbH, Bremen, Germany)を用い、マトリックス (α-シアノ-4-ヒドロキシ桂皮酸、シナピン酸、2,5-ジヒドロキシ安息香酸または6-アザ-2-チオチミン) と対イオン (0.1% TFA または 0.2 M NaCl) との9:1 (v / v) 混合物 1 μLを用いて行った。サンプルのテトラメチルアンモニウム塩の負イオンモードESI-MS分析は、ナノ-ESIプローブに適合したLTQ質量分析装置 (Thermo Electron, San Jose, CA, USA) を用いて行った。IRスペクトルはJASCO FT/IR-5300赤外分光光度計により記録した。旋光度はJASCO DIP-1000ディジタル旋光計により測定した。UVスペクトルはShimadzu Bio Spc-1600紫外可視分光光度計により記録した。
1-2) 生物材料
本発明の化合物を単離した海綿Axinella infundibulaは、伊豆諸島の式根島近海の水深15-20mの海底から採取された。また、その標本をアムステルダム大学動物学博物館(ZMA POR 15731)に登録した。
本発明の化合物を単離した海綿Axinella infundibulaは、伊豆諸島の式根島近海の水深15-20mの海底から採取された。また、その標本をアムステルダム大学動物学博物館(ZMA POR 15731)に登録した。
1-3) テロメラーゼ阻害アッセイ
テロメラーゼ陽性HeLa細胞の2 x 106 個細胞のペレットを、200μLのCHAPSバッファー (1 mM MgCl2, 1 mM EGTA, 10 mM Tris-HCl, pH 7.5, 0.1 mM ベンズアミド, 5 mM β-メルカプトエタノール, 0.5% CHAPS, および 10% グリセロール) により抽出した。テロメラーゼ活性はTRAP (telomeric repeat amplification protocol) 法を少し改変した方法により測定した。0.5μLの測定対象溶液を、8.3μLのTRAPバッファー溶液 (Kim, N. W.; Piatyszek, M. A.; Prowse, K. R.; Harley, C. B.; West, M. D.; Ho, P. L. C.; Coviello, G. M.; Wright, W. E.; Weinrich, S. L.; Shay, J. W. Science, 1994, 266, 2011-2015.)および0.5μLの細胞抽出液との混合物に添加し、混合物を氷上で20分間プレインキュベートした。25 ngのTSプライマーと、0.5 nmol のdNTP混合物とを含む0.7μLの溶液を添加した後、反応混合物を30℃で40分間インキュベートし、その後、80℃で10分間加熱し、テロメラーゼを失活させた。反応混合物を室温にまで冷却した後、0.15 U のTaKaRa Ex Taq (タカラバイオ株式会社) と20μLの溶液 (96 μL の TaKaRa Ex Taq バッファー、19.2 μL の dNTP 溶液 (2.5 mM)、2.59 μL の TS プライマー溶液 (1 μg/μL)、2.59 μL の ACX プライマー溶液 (1 μg/μL)、1.20 μL の NT プライマー溶液 (0.5 μg/μL), 12.0 μL の TSNT 溶液 (0.01 μmol) (Oda, M.; Ueno, T.; Kasai, N.; Takahashi, H.; Yoshida, H.; Sugawara, F.; Sakaguchi, K.; Hayashi, H.; Mizushina, Y. Biochem. J. 2002, 367, 329-334.) および 826.5 μL の水を混合して調製された溶液)とを添加した。その後PCRを30サイクル行った。30サイクルは、3 サイクル (94℃ 30秒間, 66℃ 30秒間, および 72℃ 30秒間), 3 サイクル (94℃ 30秒間, 62℃ 30秒間, および 72℃ 30秒間), ならびに 24 サイクル (94℃ 30秒間, 58℃ 30秒間, および 72℃ 30秒間)からなる。PCR産物は、7%/15% ポリアクリルアミドゲル上にて、Trisホウ酸バッファー (2.22 mM Tris-HCl, 2.22 mM H3BO3, および 0.055 mM EDTANa2) 中で、SDS PAGE (150V、90分) により分析した。ゲルはGel Starを用いて染色し、UVランプのもとで写真撮影した。
テロメラーゼ陽性HeLa細胞の2 x 106 個細胞のペレットを、200μLのCHAPSバッファー (1 mM MgCl2, 1 mM EGTA, 10 mM Tris-HCl, pH 7.5, 0.1 mM ベンズアミド, 5 mM β-メルカプトエタノール, 0.5% CHAPS, および 10% グリセロール) により抽出した。テロメラーゼ活性はTRAP (telomeric repeat amplification protocol) 法を少し改変した方法により測定した。0.5μLの測定対象溶液を、8.3μLのTRAPバッファー溶液 (Kim, N. W.; Piatyszek, M. A.; Prowse, K. R.; Harley, C. B.; West, M. D.; Ho, P. L. C.; Coviello, G. M.; Wright, W. E.; Weinrich, S. L.; Shay, J. W. Science, 1994, 266, 2011-2015.)および0.5μLの細胞抽出液との混合物に添加し、混合物を氷上で20分間プレインキュベートした。25 ngのTSプライマーと、0.5 nmol のdNTP混合物とを含む0.7μLの溶液を添加した後、反応混合物を30℃で40分間インキュベートし、その後、80℃で10分間加熱し、テロメラーゼを失活させた。反応混合物を室温にまで冷却した後、0.15 U のTaKaRa Ex Taq (タカラバイオ株式会社) と20μLの溶液 (96 μL の TaKaRa Ex Taq バッファー、19.2 μL の dNTP 溶液 (2.5 mM)、2.59 μL の TS プライマー溶液 (1 μg/μL)、2.59 μL の ACX プライマー溶液 (1 μg/μL)、1.20 μL の NT プライマー溶液 (0.5 μg/μL), 12.0 μL の TSNT 溶液 (0.01 μmol) (Oda, M.; Ueno, T.; Kasai, N.; Takahashi, H.; Yoshida, H.; Sugawara, F.; Sakaguchi, K.; Hayashi, H.; Mizushina, Y. Biochem. J. 2002, 367, 329-334.) および 826.5 μL の水を混合して調製された溶液)とを添加した。その後PCRを30サイクル行った。30サイクルは、3 サイクル (94℃ 30秒間, 66℃ 30秒間, および 72℃ 30秒間), 3 サイクル (94℃ 30秒間, 62℃ 30秒間, および 72℃ 30秒間), ならびに 24 サイクル (94℃ 30秒間, 58℃ 30秒間, および 72℃ 30秒間)からなる。PCR産物は、7%/15% ポリアクリルアミドゲル上にて、Trisホウ酸バッファー (2.22 mM Tris-HCl, 2.22 mM H3BO3, および 0.055 mM EDTANa2) 中で、SDS PAGE (150V、90分) により分析した。ゲルはGel Starを用いて染色し、UVランプのもとで写真撮影した。
1-4) Axinelloside Aの単離
凍結した海綿(湿重量3.9kg)の一部(1.2kg分)を順にMeOH (4.4 L), EtOH (2.9 L), およびアセトン (3.0 L) により抽出した。一方、前記海綿の残部(2.7kg分)を順にMeOH (6.0 L), EtOH (3.3 L), およびアセトン(3.0 L)により抽出した。得られた抽出物を合わせ、水(2.0 L)とEt2O (4.0 L)で二相分配した。Et2O 相を90%MeOH水溶液 (1.0 L) とn-ヘキサン(2.1 L)で二相分配した。テロメラーゼ阻害活性のあるn-ヘキサン画分 (12.2g) の一部 (7.2 g) を、シリカゲルカラムクロマトグラフィーにおいてCHCl3、CHCl3/MeOH (9:1)、CHCl3/MeOH/H2O (9:1:0.1)、CHCl3/MeOH/H2O (3:2:0.5)、MeOH、および n-BuOH/AcOH/H2O (4:1:2) により順次溶出させることにより分離した。一方、n-ヘキサン画分の残部は、シリカゲルカラムクロマトグラフィーにおいてCHCl3、CHCl3/MeOH (9:1)、CHCl3/MeOH (7:3)、CHCl3/MeOH (3:2)、CHCl3/MeOH/H2O (3:2:0.5)、CHCl3/MeOH/H2O (3:2:0.75)、MeOH、および n-BuOH/AcOH/H2O (4:1:2) により順次溶出させることにより分離した。テロメラーゼ阻害活性のある画分 (1035.6 mg) の一部 (696.1 mg) を、COSMOSIL 5C18-AR-II (ナカライテスク) によるC18逆相HPLCにより、50 mM AcONH4 含有 40% MeCNから50 mM AcONH4 含有60% MeCNへ勾配をかけながら溶出して分離した。テロメラーゼ阻害活性のある画分 (129.1 mg) を、Develosil C30-UG-5 (野村化学株式会社)による逆相HPLCにより、100 mM AcONH4 含有 n-PrOH/MeCN/H2O (3:1:2)で溶出して精製した。テロメラーゼ阻害活性のある画分 (95.0 mg) を更に、Develosil C30-UG-5によるリサイクル逆相HPLCにより、100 mM AcONH4含有n-PrOH/MeCN/H2O (3:1:2)で溶出して精製することにより、axinelloside A (27.6 mg, 湿重量基準で0.0011%) を得た。
Axinelloside A: 無色の無定形固体; [α]D 26 -10.0°(c 0.10, MeOH); UV (MeOH) λmax 210 nm; IR (フィルム) νmax 3464, 3144, 3045, 2926, 2847, 1711, 1643, 1406, 1224, 1049 cm-1。
凍結した海綿(湿重量3.9kg)の一部(1.2kg分)を順にMeOH (4.4 L), EtOH (2.9 L), およびアセトン (3.0 L) により抽出した。一方、前記海綿の残部(2.7kg分)を順にMeOH (6.0 L), EtOH (3.3 L), およびアセトン(3.0 L)により抽出した。得られた抽出物を合わせ、水(2.0 L)とEt2O (4.0 L)で二相分配した。Et2O 相を90%MeOH水溶液 (1.0 L) とn-ヘキサン(2.1 L)で二相分配した。テロメラーゼ阻害活性のあるn-ヘキサン画分 (12.2g) の一部 (7.2 g) を、シリカゲルカラムクロマトグラフィーにおいてCHCl3、CHCl3/MeOH (9:1)、CHCl3/MeOH/H2O (9:1:0.1)、CHCl3/MeOH/H2O (3:2:0.5)、MeOH、および n-BuOH/AcOH/H2O (4:1:2) により順次溶出させることにより分離した。一方、n-ヘキサン画分の残部は、シリカゲルカラムクロマトグラフィーにおいてCHCl3、CHCl3/MeOH (9:1)、CHCl3/MeOH (7:3)、CHCl3/MeOH (3:2)、CHCl3/MeOH/H2O (3:2:0.5)、CHCl3/MeOH/H2O (3:2:0.75)、MeOH、および n-BuOH/AcOH/H2O (4:1:2) により順次溶出させることにより分離した。テロメラーゼ阻害活性のある画分 (1035.6 mg) の一部 (696.1 mg) を、COSMOSIL 5C18-AR-II (ナカライテスク) によるC18逆相HPLCにより、50 mM AcONH4 含有 40% MeCNから50 mM AcONH4 含有60% MeCNへ勾配をかけながら溶出して分離した。テロメラーゼ阻害活性のある画分 (129.1 mg) を、Develosil C30-UG-5 (野村化学株式会社)による逆相HPLCにより、100 mM AcONH4 含有 n-PrOH/MeCN/H2O (3:1:2)で溶出して精製した。テロメラーゼ阻害活性のある画分 (95.0 mg) を更に、Develosil C30-UG-5によるリサイクル逆相HPLCにより、100 mM AcONH4含有n-PrOH/MeCN/H2O (3:1:2)で溶出して精製することにより、axinelloside A (27.6 mg, 湿重量基準で0.0011%) を得た。
Axinelloside A: 無色の無定形固体; [α]D 26 -10.0°(c 0.10, MeOH); UV (MeOH) λmax 210 nm; IR (フィルム) νmax 3464, 3144, 3045, 2926, 2847, 1711, 1643, 1406, 1224, 1049 cm-1。
1-5) axinelloside A の糖の組成分析
0.1 mg のaxinelloside Aを、90% HCOOH水溶液/10% TFA水溶液 (1:1, 0.5 mL)により、70℃にて18時間加水分解した。加水分解物を遠心エバポレーターにより乾燥させ、次いで減圧下で乾燥させた。加水分解物を水 (300μL) とn-ヘキサン (300μL x 3) で二相分配した。得られた水相中に30μLの1 M アンモニア水溶液と、300 μLのNaBH4(20 mg/mL) のDMSO溶液とを添加した。40℃で90分間反応させた後、氷浴中で60 μLのAcOHを添加することにより反応を停止した。反応混合物を遠心エバポレーターにより乾燥させ、次いで減圧下で乾燥させた。次にホウ酸をホウ酸メチルエステルとして気化させるために、残留物に AcOH/乾燥MeOH (1:99, 500μL x 5) を添加後、N2気流により溶媒を留去した。混合物中に200 μLの乾燥ピリジンと200μL のAc2O とを添加した。100℃にて50分間攪拌した後、反応混合物を室温にまで冷却し、次いでN2気流により乾燥させた。得られたaxinelloside Aのアルジトールアセテート誘導体をアセトン中に溶解し、溶液の一部をキャピラリーカラムDB-1701 (J & W SCIENTIFIC, φ 0.25 mm x 30 m) を用いたガスクロマトグラフィー (使用した温度プログラム: 150→250 ℃, 昇温速度 3℃/min) に供した。保持時間 (min): フコース (21.1), アラビノース (22.2), ガラクトース (30.4), およびscyllo-イノシトール (32.5)。フコース、アラビノース、ガラクトース、およびscyllo-イノシトールは5:1:5:1の比で検出された。
0.1 mg のaxinelloside Aを、90% HCOOH水溶液/10% TFA水溶液 (1:1, 0.5 mL)により、70℃にて18時間加水分解した。加水分解物を遠心エバポレーターにより乾燥させ、次いで減圧下で乾燥させた。加水分解物を水 (300μL) とn-ヘキサン (300μL x 3) で二相分配した。得られた水相中に30μLの1 M アンモニア水溶液と、300 μLのNaBH4(20 mg/mL) のDMSO溶液とを添加した。40℃で90分間反応させた後、氷浴中で60 μLのAcOHを添加することにより反応を停止した。反応混合物を遠心エバポレーターにより乾燥させ、次いで減圧下で乾燥させた。次にホウ酸をホウ酸メチルエステルとして気化させるために、残留物に AcOH/乾燥MeOH (1:99, 500μL x 5) を添加後、N2気流により溶媒を留去した。混合物中に200 μLの乾燥ピリジンと200μL のAc2O とを添加した。100℃にて50分間攪拌した後、反応混合物を室温にまで冷却し、次いでN2気流により乾燥させた。得られたaxinelloside Aのアルジトールアセテート誘導体をアセトン中に溶解し、溶液の一部をキャピラリーカラムDB-1701 (J & W SCIENTIFIC, φ 0.25 mm x 30 m) を用いたガスクロマトグラフィー (使用した温度プログラム: 150→250 ℃, 昇温速度 3℃/min) に供した。保持時間 (min): フコース (21.1), アラビノース (22.2), ガラクトース (30.4), およびscyllo-イノシトール (32.5)。フコース、アラビノース、ガラクトース、およびscyllo-イノシトールは5:1:5:1の比で検出された。
1-6) 糖の絶対配置の決定
0.5 mg のaxinelloside Aを、90% HCOOH水溶液/10% TFA水溶液 (1:1, 500 μL) により70℃にて一晩かけて加水分解した。加水分解物を遠心エバポレーターにより乾燥させ、次いで減圧下で乾燥させた。反応混合物を水 (400μL) とn-ヘキサン (400μL) で二相分配した。水相を更にCHCl3 (400 μL) で抽出した。水溶性画分に100μLの乾燥ピリジンに溶解した0.9 mg のL-システインメチルエステル塩酸塩を添加し、次いで混合物を60℃で1時間攪拌した。室温に冷却した後、反応混合物に100μL のトリメチルシリルイミダゾールを添加し、60℃にて30分間加温した。得られたメチル(4R)-チアゾリジン-4-カルボキシレート誘導体のトリメチルシリルエーテルをDB-1701を用いたガスクロマトグラフィー (使用した温度プログラム: 180 → 250 ℃, 昇温速度 3℃/min) に供した。保持時間 (min): D-アラビノース (23.4), L-フコース (26.2), D-ガラクトース (36.5)。
0.5 mg のaxinelloside Aを、90% HCOOH水溶液/10% TFA水溶液 (1:1, 500 μL) により70℃にて一晩かけて加水分解した。加水分解物を遠心エバポレーターにより乾燥させ、次いで減圧下で乾燥させた。反応混合物を水 (400μL) とn-ヘキサン (400μL) で二相分配した。水相を更にCHCl3 (400 μL) で抽出した。水溶性画分に100μLの乾燥ピリジンに溶解した0.9 mg のL-システインメチルエステル塩酸塩を添加し、次いで混合物を60℃で1時間攪拌した。室温に冷却した後、反応混合物に100μL のトリメチルシリルイミダゾールを添加し、60℃にて30分間加温した。得られたメチル(4R)-チアゾリジン-4-カルボキシレート誘導体のトリメチルシリルエーテルをDB-1701を用いたガスクロマトグラフィー (使用した温度プログラム: 180 → 250 ℃, 昇温速度 3℃/min) に供した。保持時間 (min): D-アラビノース (23.4), L-フコース (26.2), D-ガラクトース (36.5)。
1-7) axinelloside A からの (E)-2-ヘキサデセン酸の単離
2.2 mgのaxinelloside Aを、MeOH (300 μL) および 4 N LiOH水溶液 (99 μL)の混合物中で室温にて12時間加水分解した。反応混合物をMeOH (1 mL) で希釈し、次いで1 N HCl水溶液で中和した。混合物を遠心エバポレーターにより乾燥させた。加水分解物を水(1 mL) と AcOEt (1 mL x 4) で二相分配した。有機相を無水Na2SO4により乾燥し、0.6 mgの脂肪酸を得た。5.2 mg のaxinelloside A を、MeOH (675 μL) および 4 N LiOH水溶液 (225 μL) の混合物中で室温にて17時間加水分解した。反応混合物をMeOH (1 mL)で希釈し、次いで 1 N HCl水溶液で中和した。水(2 mL)で希釈した後、混合物を遠心エバポレーターにより乾燥し、凍結乾燥した。加水分解物を水(1.5 mL) とAcOEt (1.5 mL x 4)で二相分配した。有機相を無水Na2SO4により乾燥し、1.1 mgの脂肪酸を得た。得られたAcOEt-可溶性画分を合わせ、COSMOSIL 5C18-AR-II によるC18 逆相HPLCにより、200 mM NaClO4含有90% MeOH水溶液から、200 mM NaClO4含有97% MeOH水溶液へ勾配をかけながら溶出して精製して、1.0 mg の (E)-2-ヘキサデセン酸を得た。FABMS (マトリックス: トリエタノールアミン) m/z 253 [M-H]-; 1H NMR (600 MHz, CD3OD) δ 6.91 (1H, dt, J = 6.9, 15.6 Hz), 5.78 (1H, dt, J = 1.5, 15.6 Hz), 2.20 (2H, ddt, J = 1.5, 6.9, 7.1 Hz), 1.46 (2H, tt, J = 7.1, 7.3 Hz), 1.28 (20H, m), 0.98 (3H, t, J = 6.9 Hz)。
2.2 mgのaxinelloside Aを、MeOH (300 μL) および 4 N LiOH水溶液 (99 μL)の混合物中で室温にて12時間加水分解した。反応混合物をMeOH (1 mL) で希釈し、次いで1 N HCl水溶液で中和した。混合物を遠心エバポレーターにより乾燥させた。加水分解物を水(1 mL) と AcOEt (1 mL x 4) で二相分配した。有機相を無水Na2SO4により乾燥し、0.6 mgの脂肪酸を得た。5.2 mg のaxinelloside A を、MeOH (675 μL) および 4 N LiOH水溶液 (225 μL) の混合物中で室温にて17時間加水分解した。反応混合物をMeOH (1 mL)で希釈し、次いで 1 N HCl水溶液で中和した。水(2 mL)で希釈した後、混合物を遠心エバポレーターにより乾燥し、凍結乾燥した。加水分解物を水(1.5 mL) とAcOEt (1.5 mL x 4)で二相分配した。有機相を無水Na2SO4により乾燥し、1.1 mgの脂肪酸を得た。得られたAcOEt-可溶性画分を合わせ、COSMOSIL 5C18-AR-II によるC18 逆相HPLCにより、200 mM NaClO4含有90% MeOH水溶液から、200 mM NaClO4含有97% MeOH水溶液へ勾配をかけながら溶出して精製して、1.0 mg の (E)-2-ヘキサデセン酸を得た。FABMS (マトリックス: トリエタノールアミン) m/z 253 [M-H]-; 1H NMR (600 MHz, CD3OD) δ 6.91 (1H, dt, J = 6.9, 15.6 Hz), 5.78 (1H, dt, J = 1.5, 15.6 Hz), 2.20 (2H, ddt, J = 1.5, 6.9, 7.1 Hz), 1.46 (2H, tt, J = 7.1, 7.3 Hz), 1.28 (20H, m), 0.98 (3H, t, J = 6.9 Hz)。
1-8) Axinelloside A からの 3-ヒドロキシオクタデカン酸メチルの単離
0.5 mg の axinelloside A を 90% HCOOH水溶液/10% TFA水溶液 (1:1, 500 μL)により加水分解した。混合物を70℃にて一晩かけて加熱し、反応混合物を遠心エバポレーターにより乾燥した。加水分解物を水(400 μL)とn-ヘキサン(400 μL x 3)で二相分配した。水相を更にCHCl3(400 μL)により抽出した。n-ヘキサン相とCHCl3相とを合わせて0.1mgの脂肪酸混合物を得た。更に、1.5 mg の axinelloside A を上記と同様にして加水分解した。加水分解物を水(300 μL)とn-ヘキサン(300 μL x 3)で二相分配した。水相を更にAcOEt (300 μL x 2)により抽出した。n-ヘキサン相とAcOEt相とを合わせて0.3mgの脂肪酸混合物を得た。上記の2つの脂肪酸含有調製物を合わせ、次いで600μL の トルエン/MeOH (7:2)と6 μL の 2 N HCl水溶液との混合液中に溶解した。得られた溶液に、50μLの、2 M トリメチルシリルジアゾメタンのn-ヘキサン溶液を添加した。混合物を室温で1時間攪拌し、N2気流中で濃縮して、0.6 mg の未精製メチルエステルを得た。得られた0.6 mg の未精製メチルエステルを、シリカゲルカラムクロマトグラフィーにおいて n-ヘキサン、n-ヘキサン/AcOEt (95:5)、n-ヘキサン/AcOEt (9:1)、n-ヘキサン/AcOEt (8:2)、およびn-ヘキサン/AcOEt (7:3)により順次溶出させることにより精製して0.1 mgの 3-ヒドロキシオクタデカン酸のメチルエステルを得た。FABMS (マトリックス: NBA) m/z 315 [M+H]+ および 337 [M+Na]+; 1H NMR (CD3OD, 600 MHz) δ 3.96 (1H, m), 3.66 (3H, s), 2.47 (1H, dd, J = 4.2, 15.0 Hz), 2.38 (1H, dd, J = 8.5, 15.0 Hz), 1.45 (2H, m), 1.28 (26H, m), 0.89 (3H, t, J = 6.9 Hz)。
0.5 mg の axinelloside A を 90% HCOOH水溶液/10% TFA水溶液 (1:1, 500 μL)により加水分解した。混合物を70℃にて一晩かけて加熱し、反応混合物を遠心エバポレーターにより乾燥した。加水分解物を水(400 μL)とn-ヘキサン(400 μL x 3)で二相分配した。水相を更にCHCl3(400 μL)により抽出した。n-ヘキサン相とCHCl3相とを合わせて0.1mgの脂肪酸混合物を得た。更に、1.5 mg の axinelloside A を上記と同様にして加水分解した。加水分解物を水(300 μL)とn-ヘキサン(300 μL x 3)で二相分配した。水相を更にAcOEt (300 μL x 2)により抽出した。n-ヘキサン相とAcOEt相とを合わせて0.3mgの脂肪酸混合物を得た。上記の2つの脂肪酸含有調製物を合わせ、次いで600μL の トルエン/MeOH (7:2)と6 μL の 2 N HCl水溶液との混合液中に溶解した。得られた溶液に、50μLの、2 M トリメチルシリルジアゾメタンのn-ヘキサン溶液を添加した。混合物を室温で1時間攪拌し、N2気流中で濃縮して、0.6 mg の未精製メチルエステルを得た。得られた0.6 mg の未精製メチルエステルを、シリカゲルカラムクロマトグラフィーにおいて n-ヘキサン、n-ヘキサン/AcOEt (95:5)、n-ヘキサン/AcOEt (9:1)、n-ヘキサン/AcOEt (8:2)、およびn-ヘキサン/AcOEt (7:3)により順次溶出させることにより精製して0.1 mgの 3-ヒドロキシオクタデカン酸のメチルエステルを得た。FABMS (マトリックス: NBA) m/z 315 [M+H]+ および 337 [M+Na]+; 1H NMR (CD3OD, 600 MHz) δ 3.96 (1H, m), 3.66 (3H, s), 2.47 (1H, dd, J = 4.2, 15.0 Hz), 2.38 (1H, dd, J = 8.5, 15.0 Hz), 1.45 (2H, m), 1.28 (26H, m), 0.89 (3H, t, J = 6.9 Hz)。
1-9) 3-ヒドロキシオクタデカン酸メチルの絶対配置の決定
50 μgの3-ヒドロキシオクタデカン酸メチルに、100μLの混合物(ピリジン (250 μL), CH2Cl2 (250 μL), DMAP/ピリジン (5 μL, 2.3 mg/mL), および (-)-MTPACl (5 μL)を混合してられたもの)を添加した。混合物を室温で2時間反応させた。反応をNaHCO3飽和水溶液 (200 μL)を添加することにより停止し、反応混合物をAcOEt (200 μL x 3)により抽出した。AcOEt相を無水Na2SO4で乾燥し、ろ過後、N2気流中で溶媒を留去した。得られた反応生成物をシリカゲルカラムクロマトグラフィーにおいて n-ヘキサン, n-ヘキサン/AcOEt (95:5), n-ヘキサン/AcOEt (9:1), および n-ヘキサン/AcOEt (8:2) により順次溶出させることにより分離し、3-ヒドロキシオクタデカン酸メチルの(S)-MTPAエステルを得た。
50 μgの3-ヒドロキシオクタデカン酸メチルに、100μLの混合物(ピリジン (250 μL), CH2Cl2 (250 μL), DMAP/ピリジン (5 μL, 2.3 mg/mL), および (-)-MTPACl (5 μL)を混合してられたもの)を添加した。混合物を室温で2時間反応させた。反応をNaHCO3飽和水溶液 (200 μL)を添加することにより停止し、反応混合物をAcOEt (200 μL x 3)により抽出した。AcOEt相を無水Na2SO4で乾燥し、ろ過後、N2気流中で溶媒を留去した。得られた反応生成物をシリカゲルカラムクロマトグラフィーにおいて n-ヘキサン, n-ヘキサン/AcOEt (95:5), n-ヘキサン/AcOEt (9:1), および n-ヘキサン/AcOEt (8:2) により順次溶出させることにより分離し、3-ヒドロキシオクタデカン酸メチルの(S)-MTPAエステルを得た。
50 μgの3-ヒドロキシオクタデカン酸メチルを上記と同様の手順で(+)-MTPAClと反応させて3-ヒドロキシオクタデカン酸メチルの(R)-MTPAエステルを得た。(S)-MTPAエステルと(R)-MTPAエステルにおけるH2、H3、およびH4の化学シフト値 (それぞれδSとδR)、およびそのΔδ値 (δS-δR) を 1H NMRスペクトルおよびCOSYスペクトルの両方から得た。
1-10) イオンクロマトグラフィーによる硫酸含量の決定
55 μgのaxinelloside Aを90% HCOOH水溶液/10% TFA水溶液 (1:1, 500 μL) により70℃にて一晩かけて加水分解した。反応混合物を遠心エバポレーターにより濃縮した。残留物を400μLの水に溶解し、得られた溶液のうち10μLを、移動相にIC-MA3-1 (株式会社島津製作所)を用いたShim-pack IC-A3 (S) (株式会社島津製作所) によるイオンクロマトグラフィーにより分析した。Axinelloside Aの硫酸イオンの含量は、標準サンプルによる検量線を用いて推定した。
55 μgのaxinelloside Aを90% HCOOH水溶液/10% TFA水溶液 (1:1, 500 μL) により70℃にて一晩かけて加水分解した。反応混合物を遠心エバポレーターにより濃縮した。残留物を400μLの水に溶解し、得られた溶液のうち10μLを、移動相にIC-MA3-1 (株式会社島津製作所)を用いたShim-pack IC-A3 (S) (株式会社島津製作所) によるイオンクロマトグラフィーにより分析した。Axinelloside Aの硫酸イオンの含量は、標準サンプルによる検量線を用いて推定した。
2) 結果と考察
2-1) 単離と活性
海綿A. infundibula (3.9 kg)から axinelloside A (27.6 mg)を単離した (図1)。Axinelloside Aは、強いテロメラーゼ阻害活性(IC50 2 μg/mL)を示した。
2-1) 単離と活性
海綿A. infundibula (3.9 kg)から axinelloside A (27.6 mg)を単離した (図1)。Axinelloside Aは、強いテロメラーゼ阻害活性(IC50 2 μg/mL)を示した。
2-2) 構造解析
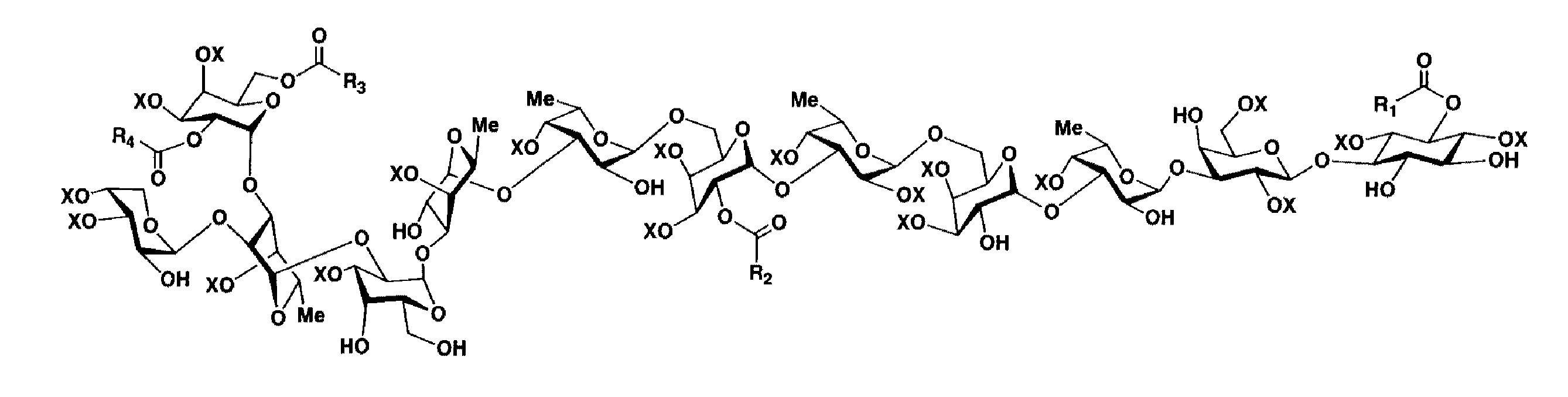
本化合物は、無色の非結晶性の粉末として得られ、水に難溶、メタノール、エタノールおよびDMSOに易溶であった。重メタノール中で良好な1H NMRスペクトルを与えたが、これに10%の重水を加えるとシグナルの分解能がさらに良くなったため、この溶媒中でNMR実験を進めることとした。1H NMRスペクトル(図2下)および13C NMRスペクトル(図2上)から、この化合物は、10個以上の糖および数個の脂肪酸を含んでいることがわかった。また、ESI-MSおよびMALDI-MSにおいて、それぞれ80 (SO3)および102 (NaSO3-H)マスユニット間隔でイオンピークを与えること、加水分解物のイオンクロマトグラフィーで著量の硫酸イオンが検出されたことなどから、分子中に多数の硫酸基が存在することが示唆された。以上のことから、axinelloside Aは、高分子量の硫酸化糖脂質と考えられた。
本化合物は、無色の非結晶性の粉末として得られ、水に難溶、メタノール、エタノールおよびDMSOに易溶であった。重メタノール中で良好な1H NMRスペクトルを与えたが、これに10%の重水を加えるとシグナルの分解能がさらに良くなったため、この溶媒中でNMR実験を進めることとした。1H NMRスペクトル(図2下)および13C NMRスペクトル(図2上)から、この化合物は、10個以上の糖および数個の脂肪酸を含んでいることがわかった。また、ESI-MSおよびMALDI-MSにおいて、それぞれ80 (SO3)および102 (NaSO3-H)マスユニット間隔でイオンピークを与えること、加水分解物のイオンクロマトグラフィーで著量の硫酸イオンが検出されたことなどから、分子中に多数の硫酸基が存在することが示唆された。以上のことから、axinelloside Aは、高分子量の硫酸化糖脂質と考えられた。
2-2-1) Axinelloside Aの糖組成
Axinelloside Aの酸加水分解物をアルジトールアセテート誘導体に導き、GCおよびGC / MS分析を行った。その結果、フコース、アラビノース、ガラクトース、およびscyllo-イノシトールが5:1:5:1の比で検出された。また、それらの糖の絶対配置は、酸加水分解物をmethyl (4R)-チアゾリジン-4-カルボキシレート誘導体化後、GC分析に付し、フコース、アラビノース、ガラクトースの絶対配置を、それぞれL, D, Dと決定した。
Axinelloside Aの酸加水分解物をアルジトールアセテート誘導体に導き、GCおよびGC / MS分析を行った。その結果、フコース、アラビノース、ガラクトース、およびscyllo-イノシトールが5:1:5:1の比で検出された。また、それらの糖の絶対配置は、酸加水分解物をmethyl (4R)-チアゾリジン-4-カルボキシレート誘導体化後、GC分析に付し、フコース、アラビノース、ガラクトースの絶対配置を、それぞれL, D, Dと決定した。
2-2-2) Axinelloside Aの構成脂肪酸の化学構造
Axinelloside Aの各種二次元NMRデータより、3つのα,β-不飽和脂肪酸エステルおよび1つのβ-ヒドロキシ脂肪酸エステルを同定した。α,β-不飽和脂肪酸については、3JH2,H3がおよそ15 Hzであることから、E-型の二重結合を持つことがわかった。脂肪酸ユニットの化学構造は、以下のように決定した。
Axinelloside Aの各種二次元NMRデータより、3つのα,β-不飽和脂肪酸エステルおよび1つのβ-ヒドロキシ脂肪酸エステルを同定した。α,β-不飽和脂肪酸については、3JH2,H3がおよそ15 Hzであることから、E-型の二重結合を持つことがわかった。脂肪酸ユニットの化学構造は、以下のように決定した。
まず、axinelloside Aをアルカリ加水分解し、ODS-HPLCにより、α,β-不飽和脂肪酸を単離した。負イオンモードのFAB-MSにおいて、m/z 253を与えることから、この脂肪酸を(E)-2-ヘキサデセン酸と同定した。次に、axinelloside Aを酸加水分解すると、負イオンモードのFAB-MS で2つのピーク(m/z 253および300)を与えた。m/z 253のピークは(E)-2-ヘキサデセン酸由来、m/z 300のイオンピークはβ-ヒドロキシ脂肪酸由来であるものと推定した。そこで、両者の混合物を(トリメチルシリル)ジアゾメタンでメチル化後、シリカゲルクロマトグラフィーで精製してβ-ヒドロキシ脂肪酸のメチルエステルを単離した。マススペクトルおよびNMRスペクトルデータを解析し、この物質を3-ヒドロキシオクタデカン酸メチルと同定した。C3位の絶対配置は、改良Mosher法を用いてR-配置と決定した。
2-2-3) 多糖由来のNMRシグナルの帰属
上述のようにして明らかにされた構成成分に関する情報を基に、axinelloside Aの各種二次元NMRデータの解析を行った。まず、DQF-COSY, HOHAHA, HMQCおよびHSQC-HOHAHAより、図3の太線で示した30個 (a-o, b'-k', m'-o') の部分構造を得た。それらの部分構造は、NOESYおよびHMBCデータを併せて解析することで、scyllo-イノシトール1個、D-アラビノース1個、D-ガラクトース5個、L-フコース5個および、3個の(E)-2-ヘキサデセン酸、1個の(R)-3-ヒドロキシオクタデカン酸に帰属された。糖のアノマー位の立体化学はカップリング定数の解析から、糖 B(3JH1,H2 = 7.7 Hz)がβ-配置で、他はすべてα-配置(3JH1,H2≒ 4.0 Hz)と決定した。
上述のようにして明らかにされた構成成分に関する情報を基に、axinelloside Aの各種二次元NMRデータの解析を行った。まず、DQF-COSY, HOHAHA, HMQCおよびHSQC-HOHAHAより、図3の太線で示した30個 (a-o, b'-k', m'-o') の部分構造を得た。それらの部分構造は、NOESYおよびHMBCデータを併せて解析することで、scyllo-イノシトール1個、D-アラビノース1個、D-ガラクトース5個、L-フコース5個および、3個の(E)-2-ヘキサデセン酸、1個の(R)-3-ヒドロキシオクタデカン酸に帰属された。糖のアノマー位の立体化学はカップリング定数の解析から、糖 B(3JH1,H2 = 7.7 Hz)がβ-配置で、他はすべてα-配置(3JH1,H2≒ 4.0 Hz)と決定した。
2-2-4) 糖鎖の配列ならびに脂肪酸結合部位の決定
糖鎖の配列は、A-H5/B-C1, B-H3/C-C1, C-H3/D-C1, E-H3/F-C1. G-H3/H-C1, H-H3/I-C1, I-H2/J-C1, J-H4/K-C1などに見られるHMBCクロスピーク (図 4)、およびD-H6s/E-H1, F-H6s/G-H1, J-H2/L-H1などに見られるNOESYクロスピーク(図 5)から、図 3に示すように決定した。
糖鎖の配列は、A-H5/B-C1, B-H3/C-C1, C-H3/D-C1, E-H3/F-C1. G-H3/H-C1, H-H3/I-C1, I-H2/J-C1, J-H4/K-C1などに見られるHMBCクロスピーク (図 4)、およびD-H6s/E-H1, F-H6s/G-H1, J-H2/L-H1などに見られるNOESYクロスピーク(図 5)から、図 3に示すように決定した。
4個の脂肪酸のうち、3個 [2個の(E)-2-ヘキサデセン酸と1個の3-ヒドロキシオクタデカン酸]については、A-H1, K-H2, F-H2からそれぞれの脂肪酸のカルボニル炭素へのHMBCクロスピーク(図 6)が観測されたことから、それらの結合位置を決定した。残る1個の(E)-2-ヘキサデセン酸の結合位置は、二次元NMRデータから明らかにできなかったが、糖 Kの6位プロトンの化学シフト値が4.40, 4.62 ppmと低磁場シフトしていることから、ここにエステル結合しているものと推定した。
2-2-5) 硫酸エステルの結合部位
Axinelloside Aは、イオンクロマトグラフィー(Shim-pack IC-A3 column)で、分子量の約24%に相当する硫酸を含むことが示された。これは1分子中に10-20個の硫酸基が含まれていることを示唆する。図 3に示した構造式中にオキシメチンおよびオキシメチレン炭素は合計30個ある。まず、水酸基の位置を決定するため、重水素同位体シフト実験(図7)を行った。90% CD3OD/D2Oと90% CD3OH/H2Oの二種類の溶媒中でそれぞれ13C-HMQCスペクトルを測定した(図7中、黒色のピークは90% CD3OH/H2O中でのHMQCスペクトルを示し、赤色のピークは90% CD3OD/D2O中でのHMQCスペクトルを示す)。13Cの化学シフト値の変化(ΔδC値= |δCOD - δCOH|)を調べた。ΔδC値が0.1 ppmより大きい炭素が、A-C3 (ΔδC= 0.17), A-C4 (0.17), B-C4 (0.15), C-C2 (0.16), D-C2 (0.14), G-C2 (0.14), H-C2 (0.14), I-C4 (0.14), I-C6 (0.17), L-C2 (0.14), R2-C3 (0.17)の11個見いだされ、これらの炭素には水酸基が結合するものと結論した。したがって、残りの19個は硫酸エステルで置換されているものと推定した(図 8)。
Axinelloside Aは、イオンクロマトグラフィー(Shim-pack IC-A3 column)で、分子量の約24%に相当する硫酸を含むことが示された。これは1分子中に10-20個の硫酸基が含まれていることを示唆する。図 3に示した構造式中にオキシメチンおよびオキシメチレン炭素は合計30個ある。まず、水酸基の位置を決定するため、重水素同位体シフト実験(図7)を行った。90% CD3OD/D2Oと90% CD3OH/H2Oの二種類の溶媒中でそれぞれ13C-HMQCスペクトルを測定した(図7中、黒色のピークは90% CD3OH/H2O中でのHMQCスペクトルを示し、赤色のピークは90% CD3OD/D2O中でのHMQCスペクトルを示す)。13Cの化学シフト値の変化(ΔδC値= |δCOD - δCOH|)を調べた。ΔδC値が0.1 ppmより大きい炭素が、A-C3 (ΔδC= 0.17), A-C4 (0.17), B-C4 (0.15), C-C2 (0.16), D-C2 (0.14), G-C2 (0.14), H-C2 (0.14), I-C4 (0.14), I-C6 (0.17), L-C2 (0.14), R2-C3 (0.17)の11個見いだされ、これらの炭素には水酸基が結合するものと結論した。したがって、残りの19個は硫酸エステルで置換されているものと推定した(図 8)。
2-2-6) マススペクトルによる構造の確認
硫酸基はイオン化の際に脱離しやすく、axinelloside Aは多数の硫酸基を含んでいるため、通常の質量分析法で分子イオンピークを検出することは困難であった。硫酸基が陽イオンと安定な塩を形成すると脱離が抑制されるとの報告があるため(Gunay, N. S.; Tadano-Aritomi, K.; Toida, T.; Ishizuka, I.; Linhardt, R. J. Anal. Chem. 2003, 75, 3226-3231)、対イオン等の測定条件を種々検討した。その結果、axinelloside Aの硫酸基を4級アンモニウム塩にした後にESI-QIT-MSを測定することで、上述の化学構造に相当する分子イオンピークを観測することができた。図9Aは、過剰のテトラメチルアンモニウムイオンを用いて得られたESI-QITマススペクトルを示す(M: m/z 5755; R: m/z 74 (Me4N))。一方、ナトリウム塩であっても、ATT (6-アザ-2-チオチミン) をマトリックスとし、MALDI-TOF-MSを測定すると、分子イオンピークを得ることができた。図9Bは、NaCl の存在下で6-アザ-2-チオチミンをマトリックスとして用いて得られたMALDI-TOF マススペクトルを示す。このようにして、NMRを用いて推定した化学構造が支持された。なお、axinelloside Aのナトリウム塩としての分子量(モノアイソトピック質量)は4784.2、分子式はC137H219O117S19Na19である。
硫酸基はイオン化の際に脱離しやすく、axinelloside Aは多数の硫酸基を含んでいるため、通常の質量分析法で分子イオンピークを検出することは困難であった。硫酸基が陽イオンと安定な塩を形成すると脱離が抑制されるとの報告があるため(Gunay, N. S.; Tadano-Aritomi, K.; Toida, T.; Ishizuka, I.; Linhardt, R. J. Anal. Chem. 2003, 75, 3226-3231)、対イオン等の測定条件を種々検討した。その結果、axinelloside Aの硫酸基を4級アンモニウム塩にした後にESI-QIT-MSを測定することで、上述の化学構造に相当する分子イオンピークを観測することができた。図9Aは、過剰のテトラメチルアンモニウムイオンを用いて得られたESI-QITマススペクトルを示す(M: m/z 5755; R: m/z 74 (Me4N))。一方、ナトリウム塩であっても、ATT (6-アザ-2-チオチミン) をマトリックスとし、MALDI-TOF-MSを測定すると、分子イオンピークを得ることができた。図9Bは、NaCl の存在下で6-アザ-2-チオチミンをマトリックスとして用いて得られたMALDI-TOF マススペクトルを示す。このようにして、NMRを用いて推定した化学構造が支持された。なお、axinelloside Aのナトリウム塩としての分子量(モノアイソトピック質量)は4784.2、分子式はC137H219O117S19Na19である。
図 10に示すように、陽イオン交換樹脂(Dowex 50WX8)を用いてH型としたaxinelloside AのMALDI-TOF-MSを測定すると、硫酸基18個が脱離したイオンピークMA (m/z 2922.4)に加え、axinelloside Aの糖鎖グリコシド結合がY-およびB-タイプで切断したフラグメントピークを観測できた。このことから、上記構造中の糖配列のみならず、脂肪酸の結合部位についても正しいことが確認された。図10中に、MA(M - 18NaSO3 + 18H - Na)- のY- および B-タイプのフラグメントの帰属結果を示す。スペクトル中の星印で示すピークはMB(M - 17NaSO3 + 17H - Na)- についてのフラグメントに対応する。ここで、フラグメントイオンは必ず1残基分の硫酸基を含むことから、硫酸基の脱離は、ランダムに起こっているものと考えられる。
3) その他の測定データ
図11はaxinelloside AのC30逆相カラムを用いたリサイクルHPLCのクロマトグラムを示す。HPLC 条件; カラム: Develosil C30 (野村化学株式会社, φ 20 mm x 250 mm), 溶媒: 100 mM NH4OAc 含有n-PrOH/MeCN/H2O (60/20/40), 流量: 9.45 mL/min, 検出: UV (220 nm)吸収。
図11はaxinelloside AのC30逆相カラムを用いたリサイクルHPLCのクロマトグラムを示す。HPLC 条件; カラム: Develosil C30 (野村化学株式会社, φ 20 mm x 250 mm), 溶媒: 100 mM NH4OAc 含有n-PrOH/MeCN/H2O (60/20/40), 流量: 9.45 mL/min, 検出: UV (220 nm)吸収。
図12はaxinelloside A酸加水分解物のアルジトールアセテート誘導体のGC/MS分析におけるガスクロマトグラムを示す。GC 条件; カラム: DB-1701 (φ 0.25 mm x 30 m), キャリヤーガス: He, カラム温度: 150 - 250 ℃, 昇温速度: 3 ℃/min, 検出: MS側での全イオン検出, 保持時間 (min): フコース (21.6), アラビノース (22.7), ガラクトース (30.8), scyllo-イノシトール (32.9). フコース, アラビノース, ガラクトース, scyllo-イノシトールは 5:1:5:1の比で検出された。
図13はaxinelloside A酸加水分解物のアルジトールアセテート誘導体のGC分析におけるガスクロマトグラムを示す。GC 条件; カラム: DB-1701 (φ 0.25 mm x 30 m), キャリヤーガス: He, カラム温度: 150 - 250 ℃, 昇温速度: 3 ℃/min, 検出: フレームイオン化, 保持時間 (min): フコース (21.1), アラビノース (22.2), ガラクトース (30.4), scyllo-イノシトール (32.5). フコース, アラビノース, ガラクトース, scyllo-イノシトールは 5:1:5:1の比で検出された。
図14は糖の標品のメチル(4R)-チアゾリジン-4-カルボキシレート誘導体のGC分析におけるガスクロマトグラムを示す。GC 条件; カラム: DB-1701 (φ 0.25 mm x 30 m), キャリヤーガス: He, カラム温度: 180 - 250 ℃, 昇温速度: 3 ℃/min, 検出: フレームイオン化, 保持時間(min): L-アラビノース(22.0), D-アラビノース (23.4), L-リキソース (22.6), D-リキソース(23.2), L-フコース (26.2), D-フコース (24.8), L-ガラクトース (31.7), D-ガラクトース(36.5)。
図15はaxinelloside A酸加水分解物のメチル(4R)-チアゾリジン-4-カルボキシレート誘導体のGC分析におけるガスクロマトグラムを示す。GC条件は図5と同様である。保持時間(min): D-アラビノース (23.4), L-フコース (26.2), D-ガラクトース (36.5)。
図16はaxinelloside Aアルカリ加水分解物より単離された(E)-2-ヘキサデセン酸の 1H NMR スペクトルを示す(溶媒:CD3OD)。
図17はaxinelloside Aアルカリ加水分解物より単離された (E)-2-ヘキサデセン酸の負イオンモードのFABMSを示す(マトリックス:トリエタノールアミン)。
図18はaxinelloside Aの酸加水分解物のn-ヘキサン画分の負イオンモードのFABMSを示す(マトリックス:トリエタノールアミン)。m/z 253 [M-H]-のイオンピークは (E)-2-ヘキサデセン酸と帰属された。m/z 299 [M-H]-のイオンピークは3-ヒドロキシオクタデカン酸と帰属された。
図19はaxinelloside Aの酸加水分解物より単離された3-ヒドロキシオクタデカン酸メチルの1H NMR スペクトルを示す(溶媒:CD3OD)。
図20は3-ヒドロキシオクタデカン酸メチルの正イオンモードのFABMSを示す(マトリックス:m-ニトロベンジルアルコール)。
図16はaxinelloside Aアルカリ加水分解物より単離された(E)-2-ヘキサデセン酸の 1H NMR スペクトルを示す(溶媒:CD3OD)。
図17はaxinelloside Aアルカリ加水分解物より単離された (E)-2-ヘキサデセン酸の負イオンモードのFABMSを示す(マトリックス:トリエタノールアミン)。
図18はaxinelloside Aの酸加水分解物のn-ヘキサン画分の負イオンモードのFABMSを示す(マトリックス:トリエタノールアミン)。m/z 253 [M-H]-のイオンピークは (E)-2-ヘキサデセン酸と帰属された。m/z 299 [M-H]-のイオンピークは3-ヒドロキシオクタデカン酸と帰属された。
図19はaxinelloside Aの酸加水分解物より単離された3-ヒドロキシオクタデカン酸メチルの1H NMR スペクトルを示す(溶媒:CD3OD)。
図20は3-ヒドロキシオクタデカン酸メチルの正イオンモードのFABMSを示す(マトリックス:m-ニトロベンジルアルコール)。
図21は3-ヒドロキシオクタデカン酸メチルおよびそのMTPA誘導体の1H NMR スペクトルを示す(溶媒:CD3OD)。A: 3-ヒドロキシオクタデカン酸メチル, B: (S)-MTPAエステル, C: (R)-MTPAエステル。スペクトル中の番号はプロトンの位置を示す。
図22は3-ヒドロキシオクタデカン酸メチルのMTPAエステルのΔδ値を示す。C3の絶対配置はRであると判明した。
図23は90% CD3OD/D2O中のaxinelloside AのDQF-COSYスペクトルを示す。
図24はaxinelloside AのDQF-COSYスペクトルの一部に帰属結果を表示したものである。
図25は90% CD3OD/D2O中のaxinelloside AのHOHAHAスペクトルを示す。
図22は3-ヒドロキシオクタデカン酸メチルのMTPAエステルのΔδ値を示す。C3の絶対配置はRであると判明した。
図23は90% CD3OD/D2O中のaxinelloside AのDQF-COSYスペクトルを示す。
図24はaxinelloside AのDQF-COSYスペクトルの一部に帰属結果を表示したものである。
図25は90% CD3OD/D2O中のaxinelloside AのHOHAHAスペクトルを示す。
図26はaxinelloside AのHOHAHAスペクトルの一部を示す。
図27は90% CD3OD/D2O中のaxinelloside AのNOESYスペクトルを示す。
図28は90% CD3OD/D2O中のaxinelloside AのHMQCスペクトルを示す。
図29はaxinelloside AのHMQCスペクトルの一部に帰属結果を表示したものである。
図30はaxinelloside AのHMQCスペクトルの一部に帰属結果を表示したものである。
図27は90% CD3OD/D2O中のaxinelloside AのNOESYスペクトルを示す。
図28は90% CD3OD/D2O中のaxinelloside AのHMQCスペクトルを示す。
図29はaxinelloside AのHMQCスペクトルの一部に帰属結果を表示したものである。
図30はaxinelloside AのHMQCスペクトルの一部に帰属結果を表示したものである。
図31は90% CD3OD/D2O中のaxinelloside AのHMBCスペクトルを示す。
図32はaxinelloside AのHMBCスペクトルの一部に残基間のクロスピークの帰属結果を表示したものである。
図33は90% CD3OD/D2O中のaxinelloside AのHSQC-HOHAHAスペクトルを示す。
図34はデコンボリューションにより得られたaxinelloside Aの負イオンモードESI-TOF-MSスペクトルを示す [溶媒: n-PrOH/MeOH/H2O (40:60:60) および 5 mM 酢酸アンモニウム]。m/z 4127 のピークは (M - 3SO3H + 2H)-と帰属された。
図35はaxinelloside Aの負イオンモードMALDI-TOF-MSスペクトルを示す(マトリックス:6-アザ-2-チオチミン)。(上図)NaCl添加時のスペクトル。(下図)無処理時のスペクトル。m/z 4761 (M-Na)- のピークはナトリウム塩としての分子質量を示す。
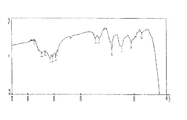
図36はaxinelloside AのIRスペクトル(film)を示す。
図32はaxinelloside AのHMBCスペクトルの一部に残基間のクロスピークの帰属結果を表示したものである。
図33は90% CD3OD/D2O中のaxinelloside AのHSQC-HOHAHAスペクトルを示す。
図34はデコンボリューションにより得られたaxinelloside Aの負イオンモードESI-TOF-MSスペクトルを示す [溶媒: n-PrOH/MeOH/H2O (40:60:60) および 5 mM 酢酸アンモニウム]。m/z 4127 のピークは (M - 3SO3H + 2H)-と帰属された。
図35はaxinelloside Aの負イオンモードMALDI-TOF-MSスペクトルを示す(マトリックス:6-アザ-2-チオチミン)。(上図)NaCl添加時のスペクトル。(下図)無処理時のスペクトル。m/z 4761 (M-Na)- のピークはナトリウム塩としての分子質量を示す。
図36はaxinelloside AのIRスペクトル(film)を示す。
表1に、axinelloside Aの1H および13C NMRケミカルシフト値、多重度、カップリング定数を示す。
Claims (7)
- 式(I):
-COR1、-COR2、-COR3および-COR4は、それぞれ独立に、直鎖状または分岐鎖状の、飽和または不飽和の、1つ以上の水酸基で置換されていてもよいアシル基を表し、
-OXは-OSO3 -M+(M+は一価の陽イオンを表す) を表す。]
により表される化合物。 - 式(I)における-OH基の水素の少なくとも1つがSO3 -M+(M+は一価の陽イオンを表す) に置換されている、請求項1記載の化合物の誘導体化合物。
- 式(I)における-OSO3 -M+基のSO3 -M+の少なくとも1つが水素に置換されている、請求項1または2記載の化合物の誘導体化合物。
- -COR1、-COR3および-COR4がそれぞれ
-COR2が
である(各式中*は酸素原子への結合位置を示す)、請求項1〜3のいずれか1項記載の化合物。 - 請求項1〜4のいずれか1項記載の化合物(ただしM+は薬学的に許容される一価の陽イオンである)を有効成分として含有する医薬組成物。
- 請求項1〜4のいずれか1項記載の化合物(ただしM+は薬学的に許容される一価の陽イオンである)を有効成分として含有するテロメラーゼ阻害剤。
- 請求項1〜4のいずれか1項記載の化合物(ただしM+は薬学的に許容される一価の陽イオンである)を有効成分として含有する抗癌剤。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2005360820A JP2007161902A (ja) | 2005-12-14 | 2005-12-14 | テロメラーゼ阻害活性を有する新規化合物 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2005360820A JP2007161902A (ja) | 2005-12-14 | 2005-12-14 | テロメラーゼ阻害活性を有する新規化合物 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| JP2007161902A true JP2007161902A (ja) | 2007-06-28 |
Family
ID=38245172
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2005360820A Pending JP2007161902A (ja) | 2005-12-14 | 2005-12-14 | テロメラーゼ阻害活性を有する新規化合物 |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP2007161902A (ja) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2011111804A1 (ja) | 2010-03-11 | 2011-09-15 | 学校法人早稲田大学 | 腔腸動物由来抗原虫化合物 |
-
2005
- 2005-12-14 JP JP2005360820A patent/JP2007161902A/ja active Pending
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2011111804A1 (ja) | 2010-03-11 | 2011-09-15 | 学校法人早稲田大学 | 腔腸動物由来抗原虫化合物 |
| US8722909B2 (en) | 2010-03-11 | 2014-05-13 | Waseda University | Antiprotozoal compound derived from coelenterata |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| Yoshikawa et al. | Medicinal foodstuffs. IX. The inhibitors of glucose absorption from the leaves of Gymnema sylvestre R. BR.(Asclepiadaceae): structures of gymnemosides a and b | |
| Ubukata et al. | Structure elucidation of liposidomycins, a class of complex lipid nucleoside antibiotics | |
| Pfeiffer et al. | Glucuronidation of zearalenone, zeranol and four metabolites in vitro: Formation of glucuronides by various microsomes and human UDP‐glucuronosyltransferase isoforms | |
| Kim et al. | Two new monogalactosyl diacylglycerols from brown alga Sargassum thunbergii | |
| Warabi et al. | Axinelloside A, an unprecedented highly sulfated lipopolysaccharide inhibiting telomerase, from the marine sponge, Axinella infundibula | |
| Fedina et al. | Synthesis of branched arabinofuranose pentasaccharide fragment of mycobacterial arabinans as 2-azidoethyl glycoside | |
| Kaeothip et al. | Preparation of p-nitrophenyl β-L-arabinofuranoside as a substrate of β-L-arabinofuranosidase | |
| Rombouts et al. | Mycobacterium marinum lipooligosaccharides are unique caryophyllose-containing cell wall glycolipids that inhibit tumor necrosis factor-α secretion in macrophages | |
| Pace-Asciak | Polyhydroxy cyclic ethers formed from tritiated arachidonic acid by acetone powders of sheep seminal vesicles | |
| Greimel et al. | Syntheses of phosphatidyl-β-D-glucoside analogues to probe antigen selectivity of monoclonal antibody ‘DIM21’ | |
| US20140024576A1 (en) | Heptose derivatives for use in the treatment of bacterial infections | |
| Nakao et al. | Penasulfate A, a new α-glucosidase inhibitor from a marine sponge Penares sp. | |
| Nudelman et al. | A novel tumor-associated, developmentally regulated glycolipid antigen defined by monoclonal antibody ACFH-18. | |
| JP2007161902A (ja) | テロメラーゼ阻害活性を有する新規化合物 | |
| Bassanini et al. | Laccase-catalyzed dimerization of glycosylated lignols | |
| WO2019030002A1 (en) | SYNTHESIS OF 1,3-OLEINE-2-PALMITIN (OPO) | |
| Charrier et al. | Preparation of silybin phase II metabolites: Streptomyces catalyzed glucuronidation | |
| Fontenelle et al. | The synthesis of tetrafluorinated aminosugars | |
| Morimoto et al. | Studies on glycolipids. VII. Isolation of two new sulfoquinovosyl diacylglycerols from the green alga Chlorella vulgaris | |
| JPH06510661A (ja) | ジーn−アセチルラクトサミニル構造で終わるモノフコシル化オリゴ糖の合成法 | |
| Inagaki et al. | Isolation and structure of a new ganglioside molecular species | |
| Miyamoto et al. | Two new gangliosides, acanthagangliosides I and J from the starfish Acanthaster planci | |
| Lee et al. | Metabolism and antioxidant effect of malaxinic acid and its corresponding aglycone in rat blood plasma | |
| Adhikari et al. | Structural elucidation of major degradation products of milbemycin oxime drug substance using LC-MS and NMR | |
| van de Poll et al. | The role of sulfation in the metabolic activation of N-hydroxy-4′-fluoro-4-acetylaminobiphenyl |