JP2009504151A - 記憶Tリンパ球の視覚化、単離および遺伝子改変のための共通γ鎖サイトカインの使用 - Google Patents
記憶Tリンパ球の視覚化、単離および遺伝子改変のための共通γ鎖サイトカインの使用 Download PDFInfo
- Publication number
- JP2009504151A JP2009504151A JP2008525729A JP2008525729A JP2009504151A JP 2009504151 A JP2009504151 A JP 2009504151A JP 2008525729 A JP2008525729 A JP 2008525729A JP 2008525729 A JP2008525729 A JP 2008525729A JP 2009504151 A JP2009504151 A JP 2009504151A
- Authority
- JP
- Japan
- Prior art keywords
- cells
- specific
- antigen
- vitro method
- memory
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
- 210000003071 memory t lymphocyte Anatomy 0.000 title claims abstract description 103
- 102000004127 Cytokines Human genes 0.000 title claims description 72
- 108090000695 Cytokines Proteins 0.000 title claims description 72
- 238000002955 isolation Methods 0.000 title claims description 6
- 238000012239 gene modification Methods 0.000 title description 9
- 230000005017 genetic modification Effects 0.000 title description 9
- 235000013617 genetically modified food Nutrition 0.000 title description 9
- 108010066719 Interleukin Receptor Common gamma Subunit Proteins 0.000 title description 5
- 102000018682 Interleukin Receptor Common gamma Subunit Human genes 0.000 title description 5
- 238000012800 visualization Methods 0.000 title 1
- 210000004027 cell Anatomy 0.000 claims abstract description 381
- 239000000427 antigen Substances 0.000 claims abstract description 103
- 102000036639 antigens Human genes 0.000 claims abstract description 103
- 108091007433 antigens Proteins 0.000 claims abstract description 103
- 238000000338 in vitro Methods 0.000 claims abstract description 82
- 238000000034 method Methods 0.000 claims abstract description 73
- 210000001744 T-lymphocyte Anatomy 0.000 claims description 299
- 102000000588 Interleukin-2 Human genes 0.000 claims description 146
- 108010002350 Interleukin-2 Proteins 0.000 claims description 144
- 210000004698 lymphocyte Anatomy 0.000 claims description 104
- 230000015654 memory Effects 0.000 claims description 93
- 206010028980 Neoplasm Diseases 0.000 claims description 77
- 239000011324 bead Substances 0.000 claims description 70
- 101000914514 Homo sapiens T-cell-specific surface glycoprotein CD28 Proteins 0.000 claims description 61
- 102100027213 T-cell-specific surface glycoprotein CD28 Human genes 0.000 claims description 61
- 230000014509 gene expression Effects 0.000 claims description 56
- 230000004913 activation Effects 0.000 claims description 38
- 230000002062 proliferating effect Effects 0.000 claims description 35
- 238000000684 flow cytometry Methods 0.000 claims description 31
- 239000000523 sample Substances 0.000 claims description 28
- 108090000623 proteins and genes Proteins 0.000 claims description 25
- 239000000018 receptor agonist Substances 0.000 claims description 24
- 229940044601 receptor agonist Drugs 0.000 claims description 24
- 239000003446 ligand Substances 0.000 claims description 22
- 238000001514 detection method Methods 0.000 claims description 20
- 239000000409 cytokine receptor agonist Substances 0.000 claims description 16
- 108090000765 processed proteins & peptides Proteins 0.000 claims description 16
- 108010038498 Interleukin-7 Receptors Proteins 0.000 claims description 14
- 102000010782 Interleukin-7 Receptors Human genes 0.000 claims description 14
- 239000012472 biological sample Substances 0.000 claims description 13
- 238000003114 enzyme-linked immunosorbent spot assay Methods 0.000 claims description 13
- 108090000978 Interleukin-4 Proteins 0.000 claims description 12
- 102000004388 Interleukin-4 Human genes 0.000 claims description 12
- 238000002054 transplantation Methods 0.000 claims description 12
- 239000013598 vector Substances 0.000 claims description 12
- -1 IFN-g Proteins 0.000 claims description 11
- 238000003556 assay Methods 0.000 claims description 11
- 238000005259 measurement Methods 0.000 claims description 11
- 108010017535 Interleukin-15 Receptors Proteins 0.000 claims description 10
- 102000004556 Interleukin-15 Receptors Human genes 0.000 claims description 10
- 208000015181 infectious disease Diseases 0.000 claims description 10
- 102000005962 receptors Human genes 0.000 claims description 9
- 108020003175 receptors Proteins 0.000 claims description 9
- 238000011282 treatment Methods 0.000 claims description 9
- 210000004369 blood Anatomy 0.000 claims description 8
- 239000008280 blood Substances 0.000 claims description 8
- 201000011510 cancer Diseases 0.000 claims description 8
- 239000003550 marker Substances 0.000 claims description 8
- 238000011510 Elispot assay Methods 0.000 claims description 7
- 230000005784 autoimmunity Effects 0.000 claims description 7
- 230000003213 activating effect Effects 0.000 claims description 6
- 239000013566 allergen Substances 0.000 claims description 6
- 238000010790 dilution Methods 0.000 claims description 6
- 239000012895 dilution Substances 0.000 claims description 6
- 239000007787 solid Substances 0.000 claims description 6
- MZOFCQQQCNRIBI-VMXHOPILSA-N (3s)-4-[[(2s)-1-[[(2s)-1-[[(1s)-1-carboxy-2-hydroxyethyl]amino]-4-methyl-1-oxopentan-2-yl]amino]-5-(diaminomethylideneamino)-1-oxopentan-2-yl]amino]-3-[[2-[[(2s)-2,6-diaminohexanoyl]amino]acetyl]amino]-4-oxobutanoic acid Chemical compound OC[C@@H](C(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCCN=C(N)N)NC(=O)[C@H](CC(O)=O)NC(=O)CNC(=O)[C@@H](N)CCCCN MZOFCQQQCNRIBI-VMXHOPILSA-N 0.000 claims description 5
- 241000222120 Candida <Saccharomycetales> Species 0.000 claims description 5
- 102000003814 Interleukin-10 Human genes 0.000 claims description 5
- 108090000174 Interleukin-10 Proteins 0.000 claims description 5
- 108060008682 Tumor Necrosis Factor Proteins 0.000 claims description 5
- 102000000852 Tumor Necrosis Factor-alpha Human genes 0.000 claims description 5
- 230000036039 immunity Effects 0.000 claims description 5
- 230000001939 inductive effect Effects 0.000 claims description 5
- 230000007170 pathology Effects 0.000 claims description 5
- 239000013603 viral vector Substances 0.000 claims description 5
- 206010020751 Hypersensitivity Diseases 0.000 claims description 4
- 241000186359 Mycobacterium Species 0.000 claims description 4
- 241000223960 Plasmodium falciparum Species 0.000 claims description 4
- 241000223996 Toxoplasma Species 0.000 claims description 4
- 208000026935 allergic disease Diseases 0.000 claims description 4
- 230000007815 allergy Effects 0.000 claims description 4
- 210000003958 hematopoietic stem cell Anatomy 0.000 claims description 4
- 239000007788 liquid Substances 0.000 claims description 4
- 244000000010 microbial pathogen Species 0.000 claims description 4
- 230000002265 prevention Effects 0.000 claims description 4
- 102000004196 processed proteins & peptides Human genes 0.000 claims description 4
- 206010061598 Immunodeficiency Diseases 0.000 claims description 3
- 208000029462 Immunodeficiency disease Diseases 0.000 claims description 3
- 230000007813 immunodeficiency Effects 0.000 claims description 3
- 230000005298 paramagnetic effect Effects 0.000 claims description 3
- 229940002612 prodrug Drugs 0.000 claims description 3
- 239000000651 prodrug Substances 0.000 claims description 3
- 101710112752 Cytotoxin Proteins 0.000 claims description 2
- 238000012286 ELISA Assay Methods 0.000 claims description 2
- 102000001398 Granzyme Human genes 0.000 claims description 2
- 108060005986 Granzyme Proteins 0.000 claims description 2
- 108010002616 Interleukin-5 Proteins 0.000 claims description 2
- 102000000743 Interleukin-5 Human genes 0.000 claims description 2
- 102000004887 Transforming Growth Factor beta Human genes 0.000 claims description 2
- 108090001012 Transforming Growth Factor beta Proteins 0.000 claims description 2
- 239000000556 agonist Substances 0.000 claims description 2
- 238000001516 cell proliferation assay Methods 0.000 claims description 2
- 108091008034 costimulatory receptors Proteins 0.000 claims description 2
- 231100000599 cytotoxic agent Toxicity 0.000 claims description 2
- 239000002619 cytotoxin Substances 0.000 claims description 2
- 230000003278 mimic effect Effects 0.000 claims description 2
- 210000000056 organ Anatomy 0.000 claims description 2
- 229920001184 polypeptide Polymers 0.000 claims description 2
- 238000000159 protein binding assay Methods 0.000 claims description 2
- ZRKFYGHZFMAOKI-QMGMOQQFSA-N tgfbeta Chemical compound C([C@H](NC(=O)[C@H](C(C)C)NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H]([C@@H](C)O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H]([C@@H](C)O)NC(=O)[C@H](CC(C)C)NC(=O)CNC(=O)[C@H](C)NC(=O)[C@H](CO)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@@H](NC(=O)[C@H](C)NC(=O)[C@H](C)NC(=O)[C@@H](NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](N)CCSC)C(C)C)[C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](C)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](C)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](C)C(=O)N[C@@H](CC(C)C)C(=O)N1[C@@H](CCC1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(C)C)C(O)=O)C1=CC=C(O)C=C1 ZRKFYGHZFMAOKI-QMGMOQQFSA-N 0.000 claims description 2
- 241000233870 Pneumocystis Species 0.000 claims 2
- 201000000317 pneumocystosis Diseases 0.000 claims 2
- 102000000704 Interleukin-7 Human genes 0.000 description 235
- 108010002586 Interleukin-7 Proteins 0.000 description 235
- 102100036011 T-cell surface glycoprotein CD4 Human genes 0.000 description 180
- 241000699670 Mus sp. Species 0.000 description 97
- 102100034922 T-cell surface glycoprotein CD8 alpha chain Human genes 0.000 description 84
- 108090000172 Interleukin-15 Proteins 0.000 description 82
- 102000003812 Interleukin-15 Human genes 0.000 description 81
- 108010074328 Interferon-gamma Proteins 0.000 description 66
- 102100037850 Interferon gamma Human genes 0.000 description 65
- 210000001165 lymph node Anatomy 0.000 description 57
- 238000001727 in vivo Methods 0.000 description 44
- 210000003819 peripheral blood mononuclear cell Anatomy 0.000 description 43
- 102000017420 CD3 protein, epsilon/gamma/delta subunit Human genes 0.000 description 39
- 230000000638 stimulation Effects 0.000 description 39
- 101001018097 Homo sapiens L-selectin Proteins 0.000 description 30
- 102100033467 L-selectin Human genes 0.000 description 30
- 238000009825 accumulation Methods 0.000 description 30
- 239000012636 effector Substances 0.000 description 29
- 230000035755 proliferation Effects 0.000 description 27
- 108090001005 Interleukin-6 Proteins 0.000 description 24
- 102000004889 Interleukin-6 Human genes 0.000 description 24
- 101710122864 Major tegument protein Proteins 0.000 description 24
- 102100031545 Microsomal triglyceride transfer protein large subunit Human genes 0.000 description 24
- 101710199973 Tail tube protein Proteins 0.000 description 24
- 208000009329 Graft vs Host Disease Diseases 0.000 description 21
- 208000024908 graft versus host disease Diseases 0.000 description 21
- 238000010186 staining Methods 0.000 description 21
- 102100032912 CD44 antigen Human genes 0.000 description 19
- 101000868273 Homo sapiens CD44 antigen Proteins 0.000 description 19
- 230000000735 allogeneic effect Effects 0.000 description 19
- 229930105110 Cyclosporin A Natural products 0.000 description 18
- PMATZTZNYRCHOR-CGLBZJNRSA-N Cyclosporin A Chemical compound CC[C@@H]1NC(=O)[C@H]([C@H](O)[C@H](C)C\C=C\C)N(C)C(=O)[C@H](C(C)C)N(C)C(=O)[C@H](CC(C)C)N(C)C(=O)[C@H](CC(C)C)N(C)C(=O)[C@@H](C)NC(=O)[C@H](C)NC(=O)[C@H](CC(C)C)N(C)C(=O)[C@H](C(C)C)NC(=O)[C@H](CC(C)C)N(C)C(=O)CN(C)C1=O PMATZTZNYRCHOR-CGLBZJNRSA-N 0.000 description 18
- 108010036949 Cyclosporine Proteins 0.000 description 18
- 229960001265 ciclosporin Drugs 0.000 description 18
- 230000004083 survival effect Effects 0.000 description 18
- 210000000612 antigen-presenting cell Anatomy 0.000 description 17
- 238000002474 experimental method Methods 0.000 description 17
- 210000003491 skin Anatomy 0.000 description 17
- 101001057504 Homo sapiens Interferon-stimulated gene 20 kDa protein Proteins 0.000 description 16
- 101001055144 Homo sapiens Interleukin-2 receptor subunit alpha Proteins 0.000 description 16
- 102100027268 Interferon-stimulated gene 20 kDa protein Human genes 0.000 description 16
- 241000699666 Mus <mouse, genus> Species 0.000 description 16
- NHBKXEKEPDILRR-UHFFFAOYSA-N 2,3-bis(butanoylsulfanyl)propyl butanoate Chemical compound CCCC(=O)OCC(SC(=O)CCC)CSC(=O)CCC NHBKXEKEPDILRR-UHFFFAOYSA-N 0.000 description 15
- 102100021569 Apoptosis regulator Bcl-2 Human genes 0.000 description 15
- 101000971171 Homo sapiens Apoptosis regulator Bcl-2 Proteins 0.000 description 15
- 230000032823 cell division Effects 0.000 description 15
- QHNORJFCVHUPNH-UHFFFAOYSA-L To-Pro-3 Chemical compound [I-].[I-].S1C2=CC=CC=C2[N+](C)=C1C=CC=C1C2=CC=CC=C2N(CCC[N+](C)(C)C)C=C1 QHNORJFCVHUPNH-UHFFFAOYSA-L 0.000 description 14
- 230000003834 intracellular effect Effects 0.000 description 14
- 238000004519 manufacturing process Methods 0.000 description 14
- 230000006052 T cell proliferation Effects 0.000 description 13
- 239000000975 dye Substances 0.000 description 13
- 239000000543 intermediate Substances 0.000 description 13
- 230000001177 retroviral effect Effects 0.000 description 13
- 102000006601 Thymidine Kinase Human genes 0.000 description 12
- 108020004440 Thymidine kinase Proteins 0.000 description 12
- 238000004458 analytical method Methods 0.000 description 12
- 210000004443 dendritic cell Anatomy 0.000 description 12
- 230000028993 immune response Effects 0.000 description 12
- 238000002347 injection Methods 0.000 description 12
- 239000007924 injection Substances 0.000 description 12
- 239000002609 medium Substances 0.000 description 12
- 238000012360 testing method Methods 0.000 description 11
- 108700018351 Major Histocompatibility Complex Proteins 0.000 description 10
- 230000004044 response Effects 0.000 description 10
- 230000020382 suppression by virus of host antigen processing and presentation of peptide antigen via MHC class I Effects 0.000 description 10
- 230000009258 tissue cross reactivity Effects 0.000 description 10
- 102100027207 CD27 antigen Human genes 0.000 description 9
- 101000914511 Homo sapiens CD27 antigen Proteins 0.000 description 9
- 230000005867 T cell response Effects 0.000 description 9
- 230000012010 growth Effects 0.000 description 9
- 102100036301 C-C chemokine receptor type 7 Human genes 0.000 description 8
- 101000716065 Homo sapiens C-C chemokine receptor type 7 Proteins 0.000 description 8
- 108010038453 Interleukin-2 Receptors Proteins 0.000 description 8
- 238000012258 culturing Methods 0.000 description 8
- 230000000694 effects Effects 0.000 description 8
- 238000001943 fluorescence-activated cell sorting Methods 0.000 description 8
- 210000005259 peripheral blood Anatomy 0.000 description 8
- 239000011886 peripheral blood Substances 0.000 description 8
- 230000037452 priming Effects 0.000 description 8
- 238000012546 transfer Methods 0.000 description 8
- 210000004881 tumor cell Anatomy 0.000 description 8
- 230000004614 tumor growth Effects 0.000 description 8
- 238000004113 cell culture Methods 0.000 description 7
- 230000001419 dependent effect Effects 0.000 description 7
- 239000003112 inhibitor Substances 0.000 description 7
- 244000052769 pathogen Species 0.000 description 7
- 238000010361 transduction Methods 0.000 description 7
- 230000004580 weight loss Effects 0.000 description 7
- 101001043807 Homo sapiens Interleukin-7 Proteins 0.000 description 6
- 102000010789 Interleukin-2 Receptors Human genes 0.000 description 6
- 241000699660 Mus musculus Species 0.000 description 6
- 241000187479 Mycobacterium tuberculosis Species 0.000 description 6
- 210000001185 bone marrow Anatomy 0.000 description 6
- 230000030833 cell death Effects 0.000 description 6
- 230000004663 cell proliferation Effects 0.000 description 6
- 230000004186 co-expression Effects 0.000 description 6
- 230000016396 cytokine production Effects 0.000 description 6
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 6
- 238000009826 distribution Methods 0.000 description 6
- 102000052622 human IL7 Human genes 0.000 description 6
- 239000000463 material Substances 0.000 description 6
- 230000001105 regulatory effect Effects 0.000 description 6
- 230000035945 sensitivity Effects 0.000 description 6
- 210000001519 tissue Anatomy 0.000 description 6
- 230000026683 transduction Effects 0.000 description 6
- 238000011830 transgenic mouse model Methods 0.000 description 6
- 241000222122 Candida albicans Species 0.000 description 5
- 101001046686 Homo sapiens Integrin alpha-M Proteins 0.000 description 5
- 102100022338 Integrin alpha-M Human genes 0.000 description 5
- 230000006044 T cell activation Effects 0.000 description 5
- GLNADSQYFUSGOU-GPTZEZBUSA-J Trypan blue Chemical compound [Na+].[Na+].[Na+].[Na+].C1=C(S([O-])(=O)=O)C=C2C=C(S([O-])(=O)=O)C(/N=N/C3=CC=C(C=C3C)C=3C=C(C(=CC=3)\N=N\C=3C(=CC4=CC(=CC(N)=C4C=3O)S([O-])(=O)=O)S([O-])(=O)=O)C)=C(O)C2=C1N GLNADSQYFUSGOU-GPTZEZBUSA-J 0.000 description 5
- 230000000961 alloantigen Effects 0.000 description 5
- 210000001099 axilla Anatomy 0.000 description 5
- 201000010099 disease Diseases 0.000 description 5
- 239000012091 fetal bovine serum Substances 0.000 description 5
- 239000007850 fluorescent dye Substances 0.000 description 5
- 210000005105 peripheral blood lymphocyte Anatomy 0.000 description 5
- 230000009257 reactivity Effects 0.000 description 5
- 210000002966 serum Anatomy 0.000 description 5
- 210000004989 spleen cell Anatomy 0.000 description 5
- 201000008827 tuberculosis Diseases 0.000 description 5
- 238000002255 vaccination Methods 0.000 description 5
- WZUVPPKBWHMQCE-UHFFFAOYSA-N Haematoxylin Chemical compound C12=CC(O)=C(O)C=C2CC2(O)C1C1=CC=C(O)C(O)=C1OC2 WZUVPPKBWHMQCE-UHFFFAOYSA-N 0.000 description 4
- 101100005713 Homo sapiens CD4 gene Proteins 0.000 description 4
- 241001465754 Metazoa Species 0.000 description 4
- 230000006786 activation induced cell death Effects 0.000 description 4
- 230000008901 benefit Effects 0.000 description 4
- 230000000903 blocking effect Effects 0.000 description 4
- 230000022131 cell cycle Effects 0.000 description 4
- 210000004748 cultured cell Anatomy 0.000 description 4
- 230000034994 death Effects 0.000 description 4
- 238000001415 gene therapy Methods 0.000 description 4
- 238000009169 immunotherapy Methods 0.000 description 4
- PGHMRUGBZOYCAA-UHFFFAOYSA-N ionomycin Natural products O1C(CC(O)C(C)C(O)C(C)C=CCC(C)CC(C)C(O)=CC(=O)C(C)CC(C)CC(CCC(O)=O)C)CCC1(C)C1OC(C)(C(C)O)CC1 PGHMRUGBZOYCAA-UHFFFAOYSA-N 0.000 description 4
- PGHMRUGBZOYCAA-ADZNBVRBSA-N ionomycin Chemical compound O1[C@H](C[C@H](O)[C@H](C)[C@H](O)[C@H](C)/C=C/C[C@@H](C)C[C@@H](C)C(/O)=C/C(=O)[C@@H](C)C[C@@H](C)C[C@@H](CCC(O)=O)C)CC[C@@]1(C)[C@@H]1O[C@](C)([C@@H](C)O)CC1 PGHMRUGBZOYCAA-ADZNBVRBSA-N 0.000 description 4
- 230000007774 longterm Effects 0.000 description 4
- 238000007799 mixed lymphocyte reaction assay Methods 0.000 description 4
- 239000013642 negative control Substances 0.000 description 4
- 230000002093 peripheral effect Effects 0.000 description 4
- 230000003389 potentiating effect Effects 0.000 description 4
- 102000004169 proteins and genes Human genes 0.000 description 4
- 230000008093 supporting effect Effects 0.000 description 4
- 229960005486 vaccine Drugs 0.000 description 4
- 101800001467 Envelope glycoprotein E2 Proteins 0.000 description 3
- 101710091045 Envelope protein Proteins 0.000 description 3
- 101000738771 Homo sapiens Receptor-type tyrosine-protein phosphatase C Proteins 0.000 description 3
- 102100034349 Integrase Human genes 0.000 description 3
- 102100022339 Integrin alpha-L Human genes 0.000 description 3
- 108010064548 Lymphocyte Function-Associated Antigen-1 Proteins 0.000 description 3
- 102000043131 MHC class II family Human genes 0.000 description 3
- 108091054438 MHC class II family Proteins 0.000 description 3
- 101710188315 Protein X Proteins 0.000 description 3
- 102100037422 Receptor-type tyrosine-protein phosphatase C Human genes 0.000 description 3
- 101800001271 Surface protein Proteins 0.000 description 3
- 230000006907 apoptotic process Effects 0.000 description 3
- KQNZDYYTLMIZCT-KQPMLPITSA-N brefeldin A Chemical compound O[C@@H]1\C=C\C(=O)O[C@@H](C)CCC\C=C\[C@@H]2C[C@H](O)C[C@H]21 KQNZDYYTLMIZCT-KQPMLPITSA-N 0.000 description 3
- JUMGSHROWPPKFX-UHFFFAOYSA-N brefeldin-A Natural products CC1CCCC=CC2(C)CC(O)CC2(C)C(O)C=CC(=O)O1 JUMGSHROWPPKFX-UHFFFAOYSA-N 0.000 description 3
- 230000011712 cell development Effects 0.000 description 3
- 230000010261 cell growth Effects 0.000 description 3
- 238000002659 cell therapy Methods 0.000 description 3
- 238000006243 chemical reaction Methods 0.000 description 3
- 239000012141 concentrate Substances 0.000 description 3
- 230000001143 conditioned effect Effects 0.000 description 3
- BFMYDTVEBKDAKJ-UHFFFAOYSA-L disodium;(2',7'-dibromo-3',6'-dioxido-3-oxospiro[2-benzofuran-1,9'-xanthene]-4'-yl)mercury;hydrate Chemical compound O.[Na+].[Na+].O1C(=O)C2=CC=CC=C2C21C1=CC(Br)=C([O-])C([Hg])=C1OC1=C2C=C(Br)C([O-])=C1 BFMYDTVEBKDAKJ-UHFFFAOYSA-L 0.000 description 3
- 210000003162 effector t lymphocyte Anatomy 0.000 description 3
- 230000006870 function Effects 0.000 description 3
- IRSCQMHQWWYFCW-UHFFFAOYSA-N ganciclovir Chemical compound O=C1NC(N)=NC2=C1N=CN2COC(CO)CO IRSCQMHQWWYFCW-UHFFFAOYSA-N 0.000 description 3
- 229960002963 ganciclovir Drugs 0.000 description 3
- 210000004408 hybridoma Anatomy 0.000 description 3
- 238000011534 incubation Methods 0.000 description 3
- 230000008595 infiltration Effects 0.000 description 3
- 238000001764 infiltration Methods 0.000 description 3
- 238000001802 infusion Methods 0.000 description 3
- 230000005291 magnetic effect Effects 0.000 description 3
- 238000012423 maintenance Methods 0.000 description 3
- 239000000047 product Substances 0.000 description 3
- 230000002829 reductive effect Effects 0.000 description 3
- 230000000717 retained effect Effects 0.000 description 3
- 210000005212 secondary lymphoid organ Anatomy 0.000 description 3
- 230000003248 secreting effect Effects 0.000 description 3
- 230000011664 signaling Effects 0.000 description 3
- QFJCIRLUMZQUOT-HPLJOQBZSA-N sirolimus Chemical compound C1C[C@@H](O)[C@H](OC)C[C@@H]1C[C@@H](C)[C@H]1OC(=O)[C@@H]2CCCCN2C(=O)C(=O)[C@](O)(O2)[C@H](C)CC[C@H]2C[C@H](OC)/C(C)=C/C=C/C=C/[C@@H](C)C[C@@H](C)C(=O)[C@H](OC)[C@H](O)/C(C)=C/[C@@H](C)C(=O)C1 QFJCIRLUMZQUOT-HPLJOQBZSA-N 0.000 description 3
- 230000004936 stimulating effect Effects 0.000 description 3
- 239000006228 supernatant Substances 0.000 description 3
- 238000010257 thawing Methods 0.000 description 3
- 230000001225 therapeutic effect Effects 0.000 description 3
- 229960001005 tuberculin Drugs 0.000 description 3
- 230000035899 viability Effects 0.000 description 3
- YBJHBAHKTGYVGT-ZKWXMUAHSA-N (+)-Biotin Chemical compound N1C(=O)N[C@@H]2[C@H](CCCCC(=O)O)SC[C@@H]21 YBJHBAHKTGYVGT-ZKWXMUAHSA-N 0.000 description 2
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical group CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 2
- 208000023275 Autoimmune disease Diseases 0.000 description 2
- WSFSSNUMVMOOMR-UHFFFAOYSA-N Formaldehyde Chemical compound O=C WSFSSNUMVMOOMR-UHFFFAOYSA-N 0.000 description 2
- 241000222722 Leishmania <genus> Species 0.000 description 2
- 241000233872 Pneumocystis carinii Species 0.000 description 2
- 239000004698 Polyethylene Substances 0.000 description 2
- 210000000662 T-lymphocyte subset Anatomy 0.000 description 2
- 230000003187 abdominal effect Effects 0.000 description 2
- 238000000540 analysis of variance Methods 0.000 description 2
- 238000010171 animal model Methods 0.000 description 2
- 230000002424 anti-apoptotic effect Effects 0.000 description 2
- 230000000919 anti-host Effects 0.000 description 2
- 230000000890 antigenic effect Effects 0.000 description 2
- 230000007503 antigenic stimulation Effects 0.000 description 2
- 238000013459 approach Methods 0.000 description 2
- 230000000975 bioactive effect Effects 0.000 description 2
- 238000001574 biopsy Methods 0.000 description 2
- 229940095731 candida albicans Drugs 0.000 description 2
- 230000020411 cell activation Effects 0.000 description 2
- 230000003915 cell function Effects 0.000 description 2
- 239000006285 cell suspension Substances 0.000 description 2
- 230000008859 change Effects 0.000 description 2
- 238000012512 characterization method Methods 0.000 description 2
- 238000007796 conventional method Methods 0.000 description 2
- 102000003675 cytokine receptors Human genes 0.000 description 2
- 108010057085 cytokine receptors Proteins 0.000 description 2
- 238000003568 cytokine secretion assay Methods 0.000 description 2
- 230000007423 decrease Effects 0.000 description 2
- 230000003247 decreasing effect Effects 0.000 description 2
- 230000007812 deficiency Effects 0.000 description 2
- 230000003111 delayed effect Effects 0.000 description 2
- 231100000673 dose–response relationship Toxicity 0.000 description 2
- 230000003828 downregulation Effects 0.000 description 2
- 210000002615 epidermis Anatomy 0.000 description 2
- 238000011156 evaluation Methods 0.000 description 2
- 230000007717 exclusion Effects 0.000 description 2
- MHMNJMPURVTYEJ-UHFFFAOYSA-N fluorescein-5-isothiocyanate Chemical compound O1C(=O)C2=CC(N=C=S)=CC=C2C21C1=CC=C(O)C=C1OC1=CC(O)=CC=C21 MHMNJMPURVTYEJ-UHFFFAOYSA-N 0.000 description 2
- 230000006054 immunological memory Effects 0.000 description 2
- 230000001976 improved effect Effects 0.000 description 2
- 230000006698 induction Effects 0.000 description 2
- 230000000977 initiatory effect Effects 0.000 description 2
- 230000003993 interaction Effects 0.000 description 2
- 238000010212 intracellular staining Methods 0.000 description 2
- 210000005210 lymphoid organ Anatomy 0.000 description 2
- 230000007246 mechanism Effects 0.000 description 2
- 230000001404 mediated effect Effects 0.000 description 2
- 239000003068 molecular probe Substances 0.000 description 2
- 238000010172 mouse model Methods 0.000 description 2
- 210000000822 natural killer cell Anatomy 0.000 description 2
- 102000002574 p38 Mitogen-Activated Protein Kinases Human genes 0.000 description 2
- 230000001717 pathogenic effect Effects 0.000 description 2
- 239000008188 pellet Substances 0.000 description 2
- 230000002688 persistence Effects 0.000 description 2
- 230000002085 persistent effect Effects 0.000 description 2
- 230000004962 physiological condition Effects 0.000 description 2
- 239000013641 positive control Substances 0.000 description 2
- 230000001686 pro-survival effect Effects 0.000 description 2
- 230000005855 radiation Effects 0.000 description 2
- ZAHRKKWIAAJSAO-UHFFFAOYSA-N rapamycin Natural products COCC(O)C(=C/C(C)C(=O)CC(OC(=O)C1CCCCN1C(=O)C(=O)C2(O)OC(CC(OC)C(=CC=CC=CC(C)CC(C)C(=O)C)C)CCC2C)C(C)CC3CCC(O)C(C3)OC)C ZAHRKKWIAAJSAO-UHFFFAOYSA-N 0.000 description 2
- 102000037983 regulatory factors Human genes 0.000 description 2
- 108091008025 regulatory factors Proteins 0.000 description 2
- 230000028327 secretion Effects 0.000 description 2
- 229960002930 sirolimus Drugs 0.000 description 2
- 239000000243 solution Substances 0.000 description 2
- 210000000952 spleen Anatomy 0.000 description 2
- 239000008223 sterile water Substances 0.000 description 2
- 238000001890 transfection Methods 0.000 description 2
- 230000009261 transgenic effect Effects 0.000 description 2
- 241001430294 unidentified retrovirus Species 0.000 description 2
- 230000003827 upregulation Effects 0.000 description 2
- 210000003462 vein Anatomy 0.000 description 2
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Chemical compound O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 2
- 230000003442 weekly effect Effects 0.000 description 2
- QRXMUCSWCMTJGU-UHFFFAOYSA-L (5-bromo-4-chloro-1h-indol-3-yl) phosphate Chemical compound C1=C(Br)C(Cl)=C2C(OP([O-])(=O)[O-])=CNC2=C1 QRXMUCSWCMTJGU-UHFFFAOYSA-L 0.000 description 1
- STMRGLKPBJVVEG-UHFFFAOYSA-N 2-(2-oxopropyl)isoindole-1,3-dione Chemical compound C1=CC=C2C(=O)N(CC(=O)C)C(=O)C2=C1 STMRGLKPBJVVEG-UHFFFAOYSA-N 0.000 description 1
- 108090001008 Avidin Proteins 0.000 description 1
- 102100023995 Beta-nerve growth factor Human genes 0.000 description 1
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 1
- 108700031361 Brachyury Proteins 0.000 description 1
- 101150013553 CD40 gene Proteins 0.000 description 1
- 102000014914 Carrier Proteins Human genes 0.000 description 1
- 206010068051 Chimerism Diseases 0.000 description 1
- 241001227713 Chiron Species 0.000 description 1
- 208000015943 Coeliac disease Diseases 0.000 description 1
- 208000035473 Communicable disease Diseases 0.000 description 1
- 206010010144 Completed suicide Diseases 0.000 description 1
- 108020004414 DNA Proteins 0.000 description 1
- 108090000371 Esterases Proteins 0.000 description 1
- 208000020545 Exposure to communicable disease Diseases 0.000 description 1
- 102000006354 HLA-DR Antigens Human genes 0.000 description 1
- 108010058597 HLA-DR Antigens Proteins 0.000 description 1
- HTTJABKRGRZYRN-UHFFFAOYSA-N Heparin Chemical compound OC1C(NC(=O)C)C(O)OC(COS(O)(=O)=O)C1OC1C(OS(O)(=O)=O)C(O)C(OC2C(C(OS(O)(=O)=O)C(OC3C(C(O)C(O)C(O3)C(O)=O)OS(O)(=O)=O)C(CO)O2)NS(O)(=O)=O)C(C(O)=O)O1 HTTJABKRGRZYRN-UHFFFAOYSA-N 0.000 description 1
- 229920000209 Hexadimethrine bromide Polymers 0.000 description 1
- 102000018713 Histocompatibility Antigens Class II Human genes 0.000 description 1
- 108010027412 Histocompatibility Antigens Class II Proteins 0.000 description 1
- 241000282412 Homo Species 0.000 description 1
- 101001002657 Homo sapiens Interleukin-2 Proteins 0.000 description 1
- 101000581981 Homo sapiens Neural cell adhesion molecule 1 Proteins 0.000 description 1
- 101000634900 Homo sapiens Transcriptional-regulating factor 1 Proteins 0.000 description 1
- 208000022361 Human papillomavirus infectious disease Diseases 0.000 description 1
- 102100022297 Integrin alpha-X Human genes 0.000 description 1
- 108010002352 Interleukin-1 Proteins 0.000 description 1
- 102100030703 Interleukin-22 Human genes 0.000 description 1
- 108010002335 Interleukin-9 Proteins 0.000 description 1
- 102000000585 Interleukin-9 Human genes 0.000 description 1
- 238000012313 Kruskal-Wallis test Methods 0.000 description 1
- 101710128836 Large T antigen Proteins 0.000 description 1
- 208000016604 Lyme disease Diseases 0.000 description 1
- 238000000585 Mann–Whitney U test Methods 0.000 description 1
- 241001442495 Mantophasmatodea Species 0.000 description 1
- 102100024193 Mitogen-activated protein kinase 1 Human genes 0.000 description 1
- 101000746372 Mus musculus Granulocyte-macrophage colony-stimulating factor Proteins 0.000 description 1
- 101001002703 Mus musculus Interleukin-4 Proteins 0.000 description 1
- 101001043808 Mus musculus Interleukin-7 Proteins 0.000 description 1
- 108700037961 Mycobacterium tuberculosis CFP-10 Proteins 0.000 description 1
- 108700020164 Mycobacterium tuberculosis ESAT-6 Proteins 0.000 description 1
- 108010025020 Nerve Growth Factor Proteins 0.000 description 1
- 102100027347 Neural cell adhesion molecule 1 Human genes 0.000 description 1
- 239000012826 P38 inhibitor Substances 0.000 description 1
- 229930040373 Paraformaldehyde Natural products 0.000 description 1
- 241001494479 Pecora Species 0.000 description 1
- 102000003992 Peroxidases Human genes 0.000 description 1
- 108090000608 Phosphoric Monoester Hydrolases Proteins 0.000 description 1
- 102000004160 Phosphoric Monoester Hydrolases Human genes 0.000 description 1
- 108010047620 Phytohemagglutinins Proteins 0.000 description 1
- 239000012980 RPMI-1640 medium Substances 0.000 description 1
- 241001506137 Rapa Species 0.000 description 1
- 208000037323 Rare tumor Diseases 0.000 description 1
- 102000040739 Secretory proteins Human genes 0.000 description 1
- 108091058545 Secretory proteins Proteins 0.000 description 1
- 241000700584 Simplexvirus Species 0.000 description 1
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 1
- 101100289792 Squirrel monkey polyomavirus large T gene Proteins 0.000 description 1
- 230000033540 T cell apoptotic process Effects 0.000 description 1
- 108091008874 T cell receptors Proteins 0.000 description 1
- 102100027222 T-lymphocyte activation antigen CD80 Human genes 0.000 description 1
- 102100029446 Transcriptional-regulating factor 1 Human genes 0.000 description 1
- 102100040245 Tumor necrosis factor receptor superfamily member 5 Human genes 0.000 description 1
- 208000036142 Viral infection Diseases 0.000 description 1
- UYRDHEJRPVSJFM-VSWVFQEASA-N [(1s,3r)-3-hydroxy-4-[(3e,5e,7e,9e,11z)-11-[4-[(e)-2-[(1r,3s,6s)-3-hydroxy-1,5,5-trimethyl-7-oxabicyclo[4.1.0]heptan-6-yl]ethenyl]-5-oxofuran-2-ylidene]-3,10-dimethylundeca-1,3,5,7,9-pentaenylidene]-3,5,5-trimethylcyclohexyl] acetate Chemical compound C[C@@]1(O)C[C@@H](OC(=O)C)CC(C)(C)C1=C=C\C(C)=C\C=C\C=C\C=C(/C)\C=C/1C=C(\C=C\[C@]23[C@@](O2)(C)C[C@@H](O)CC3(C)C)C(=O)O\1 UYRDHEJRPVSJFM-VSWVFQEASA-N 0.000 description 1
- 208000036981 active tuberculosis Diseases 0.000 description 1
- 230000001154 acute effect Effects 0.000 description 1
- 208000009956 adenocarcinoma Diseases 0.000 description 1
- 230000000240 adjuvant effect Effects 0.000 description 1
- 238000011467 adoptive cell therapy Methods 0.000 description 1
- 230000002411 adverse Effects 0.000 description 1
- 108010050235 allogenic effect factor Proteins 0.000 description 1
- 108010004469 allophycocyanin Proteins 0.000 description 1
- 150000001413 amino acids Chemical class 0.000 description 1
- 230000003698 anagen phase Effects 0.000 description 1
- 239000003242 anti bacterial agent Substances 0.000 description 1
- 230000003092 anti-cytokine Effects 0.000 description 1
- 230000000259 anti-tumor effect Effects 0.000 description 1
- 229940088710 antibiotic agent Drugs 0.000 description 1
- 230000005975 antitumor immune response Effects 0.000 description 1
- 230000001640 apoptogenic effect Effects 0.000 description 1
- 206010003246 arthritis Diseases 0.000 description 1
- 210000003719 b-lymphocyte Anatomy 0.000 description 1
- 230000009286 beneficial effect Effects 0.000 description 1
- 108091008324 binding proteins Proteins 0.000 description 1
- 229960002685 biotin Drugs 0.000 description 1
- 235000020958 biotin Nutrition 0.000 description 1
- 239000011616 biotin Substances 0.000 description 1
- 230000000740 bleeding effect Effects 0.000 description 1
- 230000036760 body temperature Effects 0.000 description 1
- 201000008274 breast adenocarcinoma Diseases 0.000 description 1
- 230000024245 cell differentiation Effects 0.000 description 1
- 239000002771 cell marker Substances 0.000 description 1
- 230000010001 cellular homeostasis Effects 0.000 description 1
- 238000005119 centrifugation Methods 0.000 description 1
- 229930002875 chlorophyll Natural products 0.000 description 1
- 235000019804 chlorophyll Nutrition 0.000 description 1
- ATNHDLDRLWWWCB-AENOIHSZSA-M chlorophyll a Chemical compound C1([C@@H](C(=O)OC)C(=O)C2=C3C)=C2N2C3=CC(C(CC)=C3C)=[N+]4C3=CC3=C(C=C)C(C)=C5N3[Mg-2]42[N+]2=C1[C@@H](CCC(=O)OC\C=C(/C)CCC[C@H](C)CCC[C@H](C)CCCC(C)C)[C@H](C)C2=C5 ATNHDLDRLWWWCB-AENOIHSZSA-M 0.000 description 1
- 239000003593 chromogenic compound Substances 0.000 description 1
- 230000001684 chronic effect Effects 0.000 description 1
- 238000010367 cloning Methods 0.000 description 1
- 239000011248 coating agent Substances 0.000 description 1
- 238000000576 coating method Methods 0.000 description 1
- 210000002808 connective tissue Anatomy 0.000 description 1
- 230000002596 correlated effect Effects 0.000 description 1
- 230000004940 costimulation Effects 0.000 description 1
- 238000005138 cryopreservation Methods 0.000 description 1
- 239000012228 culture supernatant Substances 0.000 description 1
- 238000004163 cytometry Methods 0.000 description 1
- 210000000805 cytoplasm Anatomy 0.000 description 1
- 230000001934 delay Effects 0.000 description 1
- 230000000779 depleting effect Effects 0.000 description 1
- 206010012601 diabetes mellitus Diseases 0.000 description 1
- 238000003745 diagnosis Methods 0.000 description 1
- 230000004069 differentiation Effects 0.000 description 1
- 239000000539 dimer Substances 0.000 description 1
- 208000035475 disorder Diseases 0.000 description 1
- YQGOJNYOYNNSMM-UHFFFAOYSA-N eosin Chemical compound [Na+].OC(=O)C1=CC=CC=C1C1=C2C=C(Br)C(=O)C(Br)=C2OC2=C(Br)C(O)=C(Br)C=C21 YQGOJNYOYNNSMM-UHFFFAOYSA-N 0.000 description 1
- 210000001339 epidermal cell Anatomy 0.000 description 1
- 210000003743 erythrocyte Anatomy 0.000 description 1
- 150000002148 esters Chemical group 0.000 description 1
- 238000012921 fluorescence analysis Methods 0.000 description 1
- 239000012737 fresh medium Substances 0.000 description 1
- 230000005714 functional activity Effects 0.000 description 1
- 238000002825 functional assay Methods 0.000 description 1
- 238000010353 genetic engineering Methods 0.000 description 1
- ZDXPYRJPNDTMRX-UHFFFAOYSA-N glutamine Natural products OC(=O)C(N)CCC(N)=O ZDXPYRJPNDTMRX-UHFFFAOYSA-N 0.000 description 1
- 239000001963 growth medium Substances 0.000 description 1
- 229960002897 heparin Drugs 0.000 description 1
- 229920000669 heparin Polymers 0.000 description 1
- 238000010562 histological examination Methods 0.000 description 1
- 230000003284 homeostatic effect Effects 0.000 description 1
- 230000009097 homeostatic mechanism Effects 0.000 description 1
- 230000013632 homeostatic process Effects 0.000 description 1
- 230000009675 homeostatic proliferation Effects 0.000 description 1
- 210000005260 human cell Anatomy 0.000 description 1
- 238000010191 image analysis Methods 0.000 description 1
- 230000008073 immune recognition Effects 0.000 description 1
- 210000000987 immune system Anatomy 0.000 description 1
- 230000003053 immunization Effects 0.000 description 1
- 238000002649 immunization Methods 0.000 description 1
- 230000002055 immunohistochemical effect Effects 0.000 description 1
- 238000012744 immunostaining Methods 0.000 description 1
- 230000002458 infectious effect Effects 0.000 description 1
- 206010022000 influenza Diseases 0.000 description 1
- 230000005764 inhibitory process Effects 0.000 description 1
- 238000009830 intercalation Methods 0.000 description 1
- 230000004073 interleukin-2 production Effects 0.000 description 1
- 108010074108 interleukin-21 Proteins 0.000 description 1
- 230000017307 interleukin-4 production Effects 0.000 description 1
- 229940100994 interleukin-7 Drugs 0.000 description 1
- 230000031146 intracellular signal transduction Effects 0.000 description 1
- 238000010253 intravenous injection Methods 0.000 description 1
- 239000004816 latex Substances 0.000 description 1
- 229920000126 latex Polymers 0.000 description 1
- 208000032839 leukemia Diseases 0.000 description 1
- 230000000670 limiting effect Effects 0.000 description 1
- 230000005923 long-lasting effect Effects 0.000 description 1
- 210000000207 lymphocyte subset Anatomy 0.000 description 1
- 210000003563 lymphoid tissue Anatomy 0.000 description 1
- 229940124302 mTOR inhibitor Drugs 0.000 description 1
- 239000003628 mammalian target of rapamycin inhibitor Substances 0.000 description 1
- 230000006386 memory function Effects 0.000 description 1
- 230000005012 migration Effects 0.000 description 1
- 238000013508 migration Methods 0.000 description 1
- 210000001616 monocyte Anatomy 0.000 description 1
- 230000000877 morphologic effect Effects 0.000 description 1
- 201000006417 multiple sclerosis Diseases 0.000 description 1
- 229940053128 nerve growth factor Drugs 0.000 description 1
- 231100001160 nonlethal Toxicity 0.000 description 1
- 238000011369 optimal treatment Methods 0.000 description 1
- 239000012188 paraffin wax Substances 0.000 description 1
- 229920002866 paraformaldehyde Polymers 0.000 description 1
- UTIQDNPUHSAVDN-UHFFFAOYSA-N peridinin Natural products CC(=O)OC1CC(C)(C)C(=C=CC(=CC=CC=CC=C2/OC(=O)C(=C2)C=CC34OC3(C)CC(O)CC4(C)C)C)C(C)(O)C1 UTIQDNPUHSAVDN-UHFFFAOYSA-N 0.000 description 1
- 108040007629 peroxidase activity proteins Proteins 0.000 description 1
- 230000035479 physiological effects, processes and functions Effects 0.000 description 1
- 230000001885 phytohemagglutinin Effects 0.000 description 1
- 108010086652 phytohemagglutinin-P Proteins 0.000 description 1
- 238000002360 preparation method Methods 0.000 description 1
- 230000008569 process Effects 0.000 description 1
- 230000001737 promoting effect Effects 0.000 description 1
- 239000001397 quillaja saponaria molina bark Substances 0.000 description 1
- 230000036647 reaction Effects 0.000 description 1
- 230000001172 regenerating effect Effects 0.000 description 1
- 238000011069 regeneration method Methods 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 230000004043 responsiveness Effects 0.000 description 1
- 206010039073 rheumatoid arthritis Diseases 0.000 description 1
- 229930182490 saponin Natural products 0.000 description 1
- 150000007949 saponins Chemical class 0.000 description 1
- 229920006395 saturated elastomer Polymers 0.000 description 1
- 238000000926 separation method Methods 0.000 description 1
- 230000019491 signal transduction Effects 0.000 description 1
- 239000011780 sodium chloride Substances 0.000 description 1
- YEENEYXBHNNNGV-XEHWZWQGSA-M sodium;3-acetamido-5-[acetyl(methyl)amino]-2,4,6-triiodobenzoate;(2r,3r,4s,5s,6r)-2-[(2r,3s,4s,5r)-3,4-dihydroxy-2,5-bis(hydroxymethyl)oxolan-2-yl]oxy-6-(hydroxymethyl)oxane-3,4,5-triol Chemical compound [Na+].CC(=O)N(C)C1=C(I)C(NC(C)=O)=C(I)C(C([O-])=O)=C1I.O[C@H]1[C@H](O)[C@@H](CO)O[C@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 YEENEYXBHNNNGV-XEHWZWQGSA-M 0.000 description 1
- 241000894007 species Species 0.000 description 1
- 230000002269 spontaneous effect Effects 0.000 description 1
- 238000007447 staining method Methods 0.000 description 1
- 238000007619 statistical method Methods 0.000 description 1
- 210000000130 stem cell Anatomy 0.000 description 1
- 210000002536 stromal cell Anatomy 0.000 description 1
- 238000007920 subcutaneous administration Methods 0.000 description 1
- 230000002123 temporal effect Effects 0.000 description 1
- 238000002560 therapeutic procedure Methods 0.000 description 1
- 231100000331 toxic Toxicity 0.000 description 1
- 230000002588 toxic effect Effects 0.000 description 1
- 231100000419 toxicity Toxicity 0.000 description 1
- 230000001988 toxicity Effects 0.000 description 1
- 230000007704 transition Effects 0.000 description 1
- 238000010200 validation analysis Methods 0.000 description 1
- 230000009385 viral infection Effects 0.000 description 1
- 230000003612 virological effect Effects 0.000 description 1
- 238000005406 washing Methods 0.000 description 1
Images






































Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K48/00—Medicinal preparations containing genetic material which is inserted into cells of the living body to treat genetic diseases; Gene therapy
- A61K48/0091—Purification or manufacturing processes for gene therapy compositions
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K40/00—Cellular immunotherapy
- A61K40/10—Cellular immunotherapy characterised by the cell type used
- A61K40/11—T-cells, e.g. tumour infiltrating lymphocytes [TIL] or regulatory T [Treg] cells; Lymphokine-activated killer [LAK] cells
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K40/00—Cellular immunotherapy
- A61K40/40—Cellular immunotherapy characterised by antigens that are targeted or presented by cells of the immune system
- A61K40/41—Vertebrate antigens
- A61K40/42—Cancer antigens
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K48/00—Medicinal preparations containing genetic material which is inserted into cells of the living body to treat genetic diseases; Gene therapy
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
- A61P31/18—Antivirals for RNA viruses for HIV
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/04—Immunostimulants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/06—Animal cells or tissues; Human cells or tissues
- C12N5/0602—Vertebrate cells
- C12N5/0634—Cells from the blood or the immune system
- C12N5/0636—T lymphocytes
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/53—Immunoassay; Biospecific binding assay; Materials therefor
- G01N33/569—Immunoassay; Biospecific binding assay; Materials therefor for microorganisms, e.g. protozoa, bacteria, viruses
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/53—Immunoassay; Biospecific binding assay; Materials therefor
- G01N33/569—Immunoassay; Biospecific binding assay; Materials therefor for microorganisms, e.g. protozoa, bacteria, viruses
- G01N33/56966—Animal cells
- G01N33/56972—White blood cells
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/68—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving proteins, peptides or amino acids
- G01N33/6863—Cytokines, i.e. immune system proteins modifying a biological response such as cell growth proliferation or differentiation, e.g. TNF, CNF, GM-CSF, lymphotoxin, MIF or their receptors
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/20—Cytokines; Chemokines
- C12N2501/23—Interleukins [IL]
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/20—Cytokines; Chemokines
- C12N2501/23—Interleukins [IL]
- C12N2501/2302—Interleukin-2 (IL-2)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/20—Cytokines; Chemokines
- C12N2501/23—Interleukins [IL]
- C12N2501/2306—Interleukin-6 (IL-6)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/20—Cytokines; Chemokines
- C12N2501/23—Interleukins [IL]
- C12N2501/2307—Interleukin-7 (IL-7)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/20—Cytokines; Chemokines
- C12N2501/23—Interleukins [IL]
- C12N2501/2315—Interleukin-15 (IL-15)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/50—Cell markers; Cell surface determinants
- C12N2501/51—B7 molecules, e.g. CD80, CD86, CD28 (ligand), CD152 (ligand)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/50—Cell markers; Cell surface determinants
- C12N2501/515—CD3, T-cell receptor complex
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2506/00—Differentiation of animal cells from one lineage to another; Differentiation of pluripotent cells
- C12N2506/11—Differentiation of animal cells from one lineage to another; Differentiation of pluripotent cells from blood or immune system cells
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2510/00—Genetically modified cells
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02A—TECHNOLOGIES FOR ADAPTATION TO CLIMATE CHANGE
- Y02A50/00—TECHNOLOGIES FOR ADAPTATION TO CLIMATE CHANGE in human health protection, e.g. against extreme weather
- Y02A50/30—Against vector-borne diseases, e.g. mosquito-borne, fly-borne, tick-borne or waterborne diseases whose impact is exacerbated by climate change
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Engineering & Computer Science (AREA)
- Immunology (AREA)
- Chemical & Material Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Biomedical Technology (AREA)
- Molecular Biology (AREA)
- Hematology (AREA)
- Cell Biology (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Biotechnology (AREA)
- Medicinal Chemistry (AREA)
- Organic Chemistry (AREA)
- Genetics & Genomics (AREA)
- Urology & Nephrology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Epidemiology (AREA)
- Pharmacology & Pharmacy (AREA)
- Zoology (AREA)
- Biochemistry (AREA)
- Microbiology (AREA)
- Wood Science & Technology (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Analytical Chemistry (AREA)
- Virology (AREA)
- Food Science & Technology (AREA)
- Physics & Mathematics (AREA)
- General Physics & Mathematics (AREA)
- Pathology (AREA)
- Tropical Medicine & Parasitology (AREA)
- General Engineering & Computer Science (AREA)
- Communicable Diseases (AREA)
- Oncology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Manufacturing & Machinery (AREA)
Abstract
抗原特異的記憶T細胞の希少集団を増殖、検出または単離するインビトロ方法が記載されている。遺伝子改変された記憶T細胞集団を得るインビトロ方法も記載されている。こうして得られた細胞の使用も開示されている。
Description
抗原(Ag)特異的T細胞のレパートリーは、抗原が存在しなくてもその持続性および機能性を確保するホメオスタシス機構により厳重に制御されている。ナイーブT細胞は、Agに遭遇した後、急速にクローン増殖を起こし、エフェクターT細胞に分化する(1、2)。エフェクターT細胞の寿命は、さらにAgに遭遇すると生じうる細胞死(活性化誘導細胞死)により、または生存因子の欠如により制限される。免疫応答中に、記憶T細胞も産生される。記憶T細胞は、一生を通じて生存することができるため、再生病原体に対する長期防御をもたらす(3)。しかし、ほとんどの生物試料では抗原特異的記憶T細胞の発生頻度は、細胞内サイトカイン染色およびELISPOTなどの、Ag/MHC(主要組織適合複合体)四量体染色および機能アッセイの検出限界以下にとどまっている(4、5)。特にAg特異的CD4+ T細胞は、エクスビボではほとんどが検出不能であるため、インビトロでのAg誘導T細胞増殖を複数回行った後に分析される。しかし、インビトロでの再刺激は、細胞の末端分化を支持する可能性があり、その長期生存を妨害する。その結果、インビトロでAg再刺激されたT細胞は、インビボで見出される表現型を完全に代表しているとはいえない表現型を示す可能性もある。このため、Ag特異的T細胞のエクスビボでの検出を改善する代替戦略は、進行中の免疫応答をよりうまく特性化し、免疫関連障害患者の免疫能力を評価することが必要とされる。
幾つかの研究は、T細胞記憶の確立および維持が、細胞関連(Ag/MHC複合体)および可溶性(サイトカイン)誘導シグナルにより制御されることを示している(3、6、7)。自己および非自己Ag/MHC複合体によるTCRのトリガリングにより、ナイーブ細胞から記憶細胞への移行、記憶細胞の生存および増殖が制御される。記憶リンパ球のプールは、きわめて不均一である可能性がある。最近、2種類の記憶T細胞が同定された。エフェクター記憶T細胞(CD45RA-、CCR7-、CD62L-)、ならびに二次リンパ器官のT細胞領域へのホーミングに必要な2つの分子CCR7およびCD62Lの発現を特徴とする、CD45RA陰性細胞であるセントラル記憶T細胞である。抗原刺激があると、セントラル記憶T細胞は、IL-4およびIFN-γなどの低レベルのエフェクターサイトカインを産生するが、セントラル記憶T細胞の迅速で一貫した増殖を維持することができる、高レベルのIL-2を産生する。セントラル記憶T細胞は、抗原に遭遇すると、1)セントラル記憶T細胞のプールを増加させるための自己再生過程をもたらす増殖と、2)低増殖能を特徴とするが、刺激された非リンパ組織に移動し、免疫応答のエフェクター相を媒介することができる、エフェクター記憶T細胞の産生をもたらす分化とを起こす(8)。Agの除去は、過剰なTCR刺激および活性化誘導細胞死を回避するため、およびセントラル記憶T細胞の産生に重大な意味を持つ。適切なT細胞ホメオスタシスは、ヒトおよびマウスのTリンパ球の生存、増殖およびアポトーシスを厳重に制御するサイトカインにより、確保される。可溶性因子のうち、IL-2、IL-4、IL-7、IL-9、IL-15およびIL-21などの共通γ鎖結合サイトカインは、細胞の生存およびホメオスタティック増殖を促進する(2)。特に、IL-2は、抗原に遭遇すると、T細胞の増殖およびアポトーシスの両方を持続させる。TCRならびにIL-7により生成されたシグナルは、ナイーブ細胞および記憶細胞の増殖および生存を制御する(7、9〜14)。IL-7は、TCRと結合しないと、成熟したヒトナイーブCD4+ T細胞および記憶CD4+ T細胞のFas誘導細胞死に対する感受性を低下させる(15)。さらに、Bcl-2のアップレギュレーションを誘導することで、IL-7は、Agを経験したCD4+ T細胞の静止記憶細胞への移動を支持する(9、11)。最後に、IL-15は、抗アポトーシス活性を、ナイーブT細胞および記憶T細胞の増殖を促進する一貫した効果と組み合わせる(16)。このため、共通γ鎖結合サイトカインは、Ag特異的T細胞系のインビトロでの維持および増殖のため、Ag誘導細胞増殖と併用して以前から使用されてきた。幾つかの例では、共通γ鎖結合サイトカインは、Ag特異的T細胞の検出を改善するのにも使用された。
米国特許出願US2005/0074822は、共通γ鎖結合サイトカインの存在下、細胞が抗原に曝露される、抗原特異的T細胞集団の検出方法に言及している。この方法は、Ag特異的記憶細胞を増殖、または濃縮させることができない。
米国特許出願US2005/0074822
反対に、本発明では、本発明者らは、IL-7および/またはIL-15が、以下を可能にするかどうか調べた。
1)記憶T細胞のAgフリーの環境での蓄積を可能にし、病原体/腫瘍/アレルゲン/自己特異的T細胞を同定できるようにするかどうか(したがって、Ag誘導細胞増殖の非存在下で短期のインビトロ培養に使用して、インビボで初回抗原刺激された抗原特異的T細胞(それぞれがインビボで遭遇する抗原に特異的)の希少集団を濃縮することができるかどうか)。2)セントラル記憶細胞を、この機能表現型を維持しつつ増殖させることができるかどうか(したがって、ウイルスベクターによるセントラル記憶リンパ球の遺伝子改変を促進するのに使用することができるかどうか)。
1)記憶T細胞のAgフリーの環境での蓄積を可能にし、病原体/腫瘍/アレルゲン/自己特異的T細胞を同定できるようにするかどうか(したがって、Ag誘導細胞増殖の非存在下で短期のインビトロ培養に使用して、インビボで初回抗原刺激された抗原特異的T細胞(それぞれがインビボで遭遇する抗原に特異的)の希少集団を濃縮することができるかどうか)。2)セントラル記憶細胞を、この機能表現型を維持しつつ増殖させることができるかどうか(したがって、ウイルスベクターによるセントラル記憶リンパ球の遺伝子改変を促進するのに使用することができるかどうか)。
本発明を検証するため、本発明者らは、3つの無関係な前臨床動物モデルを利用し、さらにヒト試料での検証を行った。最初の2つのモデルは、腫瘍疾患と関連して、単一細胞レベルでのAg特異的T細胞(17)、および樹状細部ベースのワクチン接種(18)の計数を可能にする。これらのモデルは、Agフリーの環境におけるインビトロでのT細胞蓄積の概念検証を可能にする。第3のモデルは、免疫不全マウスにおけるヒトT細胞移植に基づくため、遺伝子改変されたセントラル記憶T細胞の免疫能を評価することができる。
モデル1。腫瘍特異的T細胞応答を試験するため、本発明者らは、最近開発された動物モデルを利用した(17)。このモデルでは、TS/A-LACK腫瘍(大形リーシュマニア由来抗原タンパク質LACKを発現するTS/A腺癌性腫瘍細胞)は、同系BALB/cマウスで増殖され、およびLACK特異的T細胞は、抹消リンパ器官(リンパ節、脾臓または血液)で、蛍光LACK-ペプチド/MHCクラスII多量体によるフローサイトメトリーにより試験された。このモデルでは、LACK特異的T細胞は、Ag誘導細胞内サイトカイン放出を独自に特徴とすることもできる。TS/A-LACK特異的T細胞は、LACK特異的CD4 T細胞応答のより容易な特性づけを可能にする、LACKに特異的なトランスジェニックTCR β鎖を発現する、BALB/cマウスおよび16.2β TCRトランスジェニックマウスにおいて追跡することができる。さらに、TS/A細胞は、本来、内因性MuLVのエンベロープタンパク質gp70を発現する。エンベロープタンパク質gp70の免疫優勢エピトープについては、以前に記述されている(AH-1、(19))。故に、LACK特異的CD4 T細胞応答に加えて、AH-1特異的T細胞応答も、TS/A-LACK腫瘍を有するマウスにおいて追跡することができる。
モデル2。ワクチン特異的T細胞応答を試験するため、本発明者らは、ウイルスのSV40由来抗原ペプチドTag IVでパルスした骨髄由来樹状細胞(DC)を使用し、同系C57BL/6マウスをワクチン接種した(18)。このモデルでは、Tag IV特異的CD8 T細胞応答を計数するため、Tag IV特異的T細胞もエクスビボの抗原特異的サイトカインアッセイにより特性化された。
Ag特異的CD4(LACK)およびCD8(AH-1、Tag IV)T細胞応答の試験は、Ag誘導細胞刺激の非存在下、IL-7の存在下で、提案された短期インビトロ培養を行った後にエクスビボで行われた。IL-7および、比較としてIL-2、またはIL-15、ならびに陰性対照として、IL-6、IL-10およびTNF-αの最適量が使用された。全実験条件において、本発明者らは、IL-7最適量での簡単な短期培養は、Ag誘導細胞増殖の必要性を回避しリンパ球本来の表現型を維持しつつ、インビボで初回抗原刺激されたAg特異的Tリンパ球を蓄積させることを見出した。最も重要なことは、幾つかの例ではIL-7における短期培養が、従来のアッセイでは検出できなかったAg特異的T細胞の希少集団を明らかにした点である。
ヒト試料における結果検証では、Tリンパ球は、健常ドナーおよびヒト型結核菌に感染した患者由来であり、抗原特異的サイトカインの放出によるIL-7誘導短期培養後にエクスビボで分析された。全例で、IL-7は、記憶T細胞のインビトロでの増殖および生存を持続させることで、抗原特異的なIL-2およびIFN-γを産生する中間記憶T細胞の蓄積を支持した。IL-7の効力は、インビボでの抗原遭遇、最適サイトカイン量、および高細胞濃度条件に依存し、抗LFA-1抗体およびシクロスポリンAにより抑制された。IL-7は、CD4記憶T細胞増殖に関して、IL-2およびIL-15より著しく効率的であった。
試験結果から以下のことが示される。
1)高細胞密度での短期培養、および最適IL-7、またはIL-15量は、インビボで初回抗原刺激された記憶CD4およびCD8T細胞集団の維持および選択的増殖に適している。これらの細胞は、IL-2およびIFN-g分泌能、ならびにIL-7(CD4)またはIL-15(CD8)に反応した急速な増殖能として最もよく定義される。
1)高細胞密度での短期培養、および最適IL-7、またはIL-15量は、インビボで初回抗原刺激された記憶CD4およびCD8T細胞集団の維持および選択的増殖に適している。これらの細胞は、IL-2およびIFN-g分泌能、ならびにIL-7(CD4)またはIL-15(CD8)に反応した急速な増殖能として最もよく定義される。
2)IL-7(およびある程度IL-15またはIL-2)での短期培養は、インビトロでのAg誘導増殖の必要性を回避しつつ、従来の方法では検出できない可能性のある、インビボで初回抗原刺激された希少なAg特異的CD4+またはCD8+T細胞の検出を可能にする。
3)IL-7(およびある程度IL-2またはIL-15)での培養は、Ag非依存的な方法で、インビボで初回抗原刺激されたAg特異的T細胞の頻度および合計数の両方を増強する。
4)IL-7(およびIL-2ではない)での短期培養は、リンパ球サブセットの全てを活性化状態から独自に保護し、細胞の末端分化を支持することなく、インビボで初回抗原刺激されたT細胞本来の表現型を維持する。
5)Ag非存在下でのIL-7での短期培養は、Ag特異的応答および長期生存できるCD4+エフェクターTリンパ球およびセントラル記憶Tリンパ球の蓄積を可能にする。
6)IL-7/IL-15増殖細胞は、ナイーブ動物に移された場合、腫瘍増殖を遅らせることができるため、臨床的関連がある。
既存のプロトコルに勝る本戦略案の利点は、以下の通りである。
A)TCRとの結合(すなわち、Agによる刺激)なしに、抗原特異的記憶T細胞用の生物試料を濃縮する可能性。既存の戦略とは異なり、このプロトコルは、細胞の表面表現型および機能表現型を変化させることはない。この新規なアプローチをペプチド/MHC IまたはMHC II多量体染色の普及している方法と併せることで、生物試料中のAg特異的T細胞を計数し、このインビボでの頻度を評価することが可能となろう。
A)TCRとの結合(すなわち、Agによる刺激)なしに、抗原特異的記憶T細胞用の生物試料を濃縮する可能性。既存の戦略とは異なり、このプロトコルは、細胞の表面表現型および機能表現型を変化させることはない。この新規なアプローチをペプチド/MHC IまたはMHC II多量体染色の普及している方法と併せることで、生物試料中のAg特異的T細胞を計数し、このインビボでの頻度を評価することが可能となろう。
B)従来の方法ではエクスビボで検出することができない、希少な抗原特異的T細胞を明らかにする可能性。これは、希少で抗原特異的な、インビボで初回抗原刺激されたCD4およびCD8 T細胞の計数が、診断および予後目的であるような、ならびに現在、頻回で時間のかかるインビトロでのAg誘導細胞増殖に依存しているような全ての臨床条件にとって、重大な意味を持つであろう。
C)エフェクター、セントラルおよび中間記憶Tリンパ球を増殖する可能性。現在、インビトロでのセントラル記憶リンパ球の維持に利用可能なプロトコルはない。本発明は、養子免疫療法戦略に対する影響を有する。実際、利用可能な戦略は、多数の短命エフェクター細胞の移植を必要とするのに対し、同等または改善した臨床結果が、限られた数の、再生可能な長命記憶IL-7/IL-15培養細胞の移植により実現される可能性がある。
全般に、本発明は、診断的および治療的意味の双方を有する。本発明は、一方では臨床的関連のある病原体/腫瘍特異的T細胞の希少集団の同定を目的とし、他方では現行の養子免疫療法戦略も改善するであろう。
確定したインビトロ培養は、HIV、CMV、RSV、インフルエンザ、HBV、HPV、癌、糖尿病、関節リウマチ、ライム病関節炎、多発性硬化症、セリアック病など、幾つかの感染症および免疫介在性疾患の研究に適用されることが期待される。
モデル3。本発明の別の態様は、セントラル記憶細胞が、共刺激およびガンマ-サイトカインによる培養物の存在下でのTCRのトリガリングにより、インビトロで増殖することができ、機能表現型を維持しつつウイルスベクターにより遺伝子改変することができるという概念を利用する。
Tリンパ球による細胞療法は、癌、感染症、免疫不全および自己免疫を治療する多大な可能性を有すると考えられる。さらに、Tリンパ球による細胞療法は、移植と関連して発生する免疫応答を調節するのに使用することができる。遺伝子改変は、Tリンパ球の治療間隔を、この効力増加および/または毒性制限により広げることを目的としている。これは、新規受容体、生物活性産物、耐性因子および制御因子をコードする遺伝子の移植により実現される。制御因子は、毒性/望ましくない効果が生じる場合は、遺伝子改変細胞をインビボで選択的に除外/不活化することが期待される。同種造血細胞移植(allo-HCT)と関連した自殺遺伝子療法は、制御因子によるT細胞の遺伝子改変が、いかに治療的利点をもたらすかの最も明白な例である。allo-HCTでは、ドナーT細胞による宿主抗原の免疫認識は、「諸刃の剣」であり、特定の有益な効果をもたらす:T細胞は、1)直接的な抗腫瘍効果を媒介し(移植片対白血病(GvL))、2)造血前駆細胞の移植を促進し、3)移植患者にインタクトな免疫システムを提供するため、移植後感染の発生および重症度を低減することができる。不幸なことに、ドナーT細胞は、健常な宿主組織に対して反応することもありえるため、致命的な移植片対宿主病(GvHD)につながる(20)。単純ヘルペスウイルスチミジンキナーゼ(TK)自殺遺伝子を発現するレトロウイルスによるT細胞の遺伝子改変は、選択的感受性をプロドラッグのガンシクロビル(GCV)に与える。患者では、TK+リンパ球の注入、およびそれに続くGCV投与は、T細胞効果を維持するための抗宿主反応性の時間的調節、およびGvHDの選択的制御をもたらす(21〜23)。
T細胞療法およびT細胞遺伝子療法の成功は、T細胞のインビボでの増殖能および長期生存能に依存する。この目的を達成するためには、T細胞が、二次リンパ器官へ正しくホーミングする必要がある。二次リンパ器官では、抗原との適切な遭遇が起こり、T細胞を誘導してエフェクター機能を獲得する。これらの特質は、成熟T細胞分化の初期段階で、および特にセントラル記憶区分で分離する傾向にあることがますます認識されるようになっている。ウイルスベクターによる遺伝子改変は、T細胞の生理機能を変更させうる。特に、レトロウイルスベクター(RV)を介した遺伝子改変は、細胞増殖を必要とする。これは、現在、多クローン刺激による活性化、および大量の組換えヒトIL-2の存在下での培養により実現される。本発明者らは、現行のプロトコル、すなわち、可溶性抗CD3抗体による活性化、およびIL-2存在下での培養により生成された遺伝子改変ヒトTリンパ球は、主にエフェクター記憶細胞であること、インビトロではエフェクター機能を容易に示すが、条件付けされた免疫不全宿主では生着不良であることを見出した。ヒトT細胞の増殖および持続は、効果的なT細胞ベースの遺伝子療法の重大な前提条件であるため、本発明は、遺伝子改変セントラル記憶T細胞を生成することができる、T細胞の培養および形質導入方法を提供する。この目的のため、本発明者らは、以下を組み合わせた。
-抗CD3抗体および抗CD28抗体結合ビーズによるT細胞の活性化
-少量でのIL-7およびIL-15による培養
-レトロウイルスベクターによる形質導入
-抗CD3抗体および抗CD28抗体結合ビーズによるT細胞の活性化
-少量でのIL-7およびIL-15による培養
-レトロウイルスベクターによる形質導入
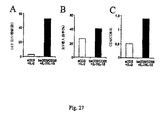
結果は、IL-7およびIL-15存在下でビーズにより遺伝子改変されたリンパ球の産生は、実現可能であること、これらの細胞は、i)CD45RA発現の非存在、およびCD62L発現の存在、ii)分子CD27およびCD28の共発現、ならびにiii)IFN-γおよび/またはIL-4の非存在下でのIL-2産生により定義される、生理学的なCD4/CD8比およびセントラル記憶機能表現型を有することを示している。
さらに、本発明者らは、条件付けされた免疫不全宿主に注入された遺伝子改変セントラル記憶T細胞は、i)エフェクター記憶遺伝子改変T細胞よりも有意に高レベルで生着および増殖し、ii)宿主抗原に対する免疫応答の誘導において、エフェクター記憶遺伝子改変リンパ球よりも強力であることを観察した。
これらの結果は、十分に機能的なセントラル記憶組換えリンパ球を、ヒト疾患治療のために入手および活用することができることを示している。
本発明では、十分に機能的なセントラル記憶組換えリンパ球は、末梢リンパ器官にホーミングし、インビボで抗原に再遭遇するとエフェクター細胞に分化することができる、長期生存能を有するセントラル記憶T細胞を意味する。
したがって、本発明の目的は、試料中の抗原特異的記憶T細胞の希少集団を増殖させるインビトロ方法であって、前記抗原特異的記憶T細胞の希少集団を選択的に増殖させることができる少なくとも1種のサイトカイン受容体アゴニストの有効量に、前記試料を曝露するステップを含む方法を提供することである。好ましくは、サイトカイン受容体アゴニストは、サイトカインまたはこの誘導体である。
好ましい実施形態では、少なくとも1種のサイトカイン受容体アゴニストは、IL-7受容体アゴニストまたはIL-15受容体アゴニストであり、好ましくは、IL-15受容体アゴニストまたはIL-7受容体アゴニストもそれぞれ存在している。
好ましい実施形態では、前記抗原特異的記憶T細胞の希少集団が、CD4+および/またはCD8+および/またはγδおよび/またはNKT T細胞集団を含む。
好ましい実施形態では、前記試料が、血液および生物起源の他の液体試料、固形組織試料、これら由来の細胞の組織培養物およびこの子孫、生物試料(すなわち、PBMCなど)からの単離細胞からなる群に属する生物試料である。
本発明のさらなる目的は、試料中の抗原特異的記憶T細胞の希少集団を検出するインビトロ方法であって、
a)上記に記載の通り、抗原特異的記憶T細胞の希少集団を選択的に増殖させることができる少なくとも1種のサイトカイン受容体アゴニストの有効量に、前記試料を曝露するステップと、
b)前記抗原特異的記憶T細胞の増殖した希少集団の1つに特異的なリガンドである、少なくとも1つのリガンドと共に、前記試料をインキュベートするステップと、
c)特異的リガンドに結合した抗原特異的記憶T細胞の増殖した希少集団を検出するステップとを含む方法を提供することである。
a)上記に記載の通り、抗原特異的記憶T細胞の希少集団を選択的に増殖させることができる少なくとも1種のサイトカイン受容体アゴニストの有効量に、前記試料を曝露するステップと、
b)前記抗原特異的記憶T細胞の増殖した希少集団の1つに特異的なリガンドである、少なくとも1つのリガンドと共に、前記試料をインキュベートするステップと、
c)特異的リガンドに結合した抗原特異的記憶T細胞の増殖した希少集団を検出するステップとを含む方法を提供することである。
好ましくは、前記特異的リガンドは、前記抗原特異的記憶T細胞の希少集団の1つに特異的な抗原またはこの誘導体であり、より好ましくは、特異的抗原は、マイコバクテリウム、ニューモシスティスカリニ、熱帯熱マラリア原虫、カンジダ、トキソプラズマ、CMV、EBV、BPV、HCV、HBV、HIVを含むが、これらに限定されない微生物病原体に関連している。あるいは、抗原は腫瘍関連抗原である。あるいは、抗原はアレルゲンである。あるいは、抗原は自己抗原である。
好ましい実施形態では、特異的抗原は、抗原MHC複合体またはこの誘導体として存在する。
好ましい実施形態では、前記抗原特異的記憶T細胞の増殖した希少集団の検出は、結合アッセイにより行われる。あるいは、前記抗原特異的記憶T細胞の増殖した希少集団の検出は、サイトカイン放出アッセイにより行われる。あるいは、前記抗原特異的記憶T細胞の増殖した希少集団の検出は、増殖アッセイにより行われる。
好ましい実施形態では、細胞は、試料を特異的リガンドと共にインキュベートする前に、蛍光生体染色色素で標識され、検出ステップは、色素希釈アッセイにより行われる。
上記に記載の通り、試料中の抗原特異的記憶T細胞の希少集団を検出する方法を実施するためのキットであって、少なくとも1種のサイトカイン受容体アゴニスト、抗原特異的記憶T細胞の希少集団に特異的な少なくとも1種のリガンド、検出手段を含むキットを提供することが、本発明のさらなる目的である。
試料中の抗原特異的記憶T細胞の希少集団を単離するインビトロ方法であって、
a)上記に記載の通り、抗原特異的記憶T細胞の希少集団を選択的に増殖させることができる少なくとも1種のサイトカイン受容体アゴニストの有効量に、前記試料を曝露するステップと、
b)前記抗原特異的記憶T細胞の増殖した希少集団の1つに特異的なリガンドである、少なくとも1つのリガンドと共に、前記試料をインキュベートするステップと、
c)特異的リガンドに結合した抗原特異的記憶T細胞の増殖した希少集団を単離するステップとを含む方法を提供することが、本発明のさらなる目的である。
a)上記に記載の通り、抗原特異的記憶T細胞の希少集団を選択的に増殖させることができる少なくとも1種のサイトカイン受容体アゴニストの有効量に、前記試料を曝露するステップと、
b)前記抗原特異的記憶T細胞の増殖した希少集団の1つに特異的なリガンドである、少なくとも1つのリガンドと共に、前記試料をインキュベートするステップと、
c)特異的リガンドに結合した抗原特異的記憶T細胞の増殖した希少集団を単離するステップとを含む方法を提供することが、本発明のさらなる目的である。
好ましくは、前記特異的リガンドは、前記抗原特異的記憶T細胞の希少集団の1つに特異的な抗原またはこの誘導体であり、より好ましくは、特異的抗原は、マイコバクテリウム、ニューモシスティスカリニ、熱帯熱マラリア原虫、カンジダ、トキソプラズマ、CMV、EBV、BPV、HCV、HBV、HIVを含むが、これらに限定されない微生物病原体に関連している。あるいは、抗原は腫瘍関連抗原である。あるいは、抗原は、アレルゲンである。あるいは、抗原は自己抗原である。
好ましい実施形態では、特異的抗原は、抗原MHC複合体またはこの誘導体として存在する。
好ましい実施形態では、前記抗原特異的記憶T細胞の増殖した希少集団の単離は、結合ステップにより行われる。あるいは、前記抗原特異的記憶T細胞の増殖した希少集団の単離は、IL-2、IFN-g、IL-4、IL-5、IL-10、TNF-アルファ、TGF-ベータ、グランザイムに関するELISPOTアッセイ、ELISAアッセイ、フローサイトメトリーサイトカイン検出アッセイを含むがこれらに限定されない、サイトカインおよびサイトトキシン産生物の測定により行われる。
免疫、感染、癌、アレルギー、自己免疫に関連した病態の診断的および/または予後的臨床試験のための、上記に記載のインビトロ方法を提供することが、本発明のさらなる目的である。
免疫、感染、癌、アレルギー、自己免疫に関連した病態の治療および/または予防のための、上記に記載の方法により単離された希少T細胞集団の使用を提供することが、本発明のさらなる目的である。特定の実施形態では、前記希少T細胞集団は遺伝子改変されている。
本発明のさらなる目的は、遺伝子改変された記憶T細胞集団を得るインビトロ方法であって、
a)リンパ球活性化を誘導することができる、アゴニスト抗体、組換えリガンドおよびこの誘導体を含むがこれらに限定されない、少なくとも2種の特異的活性化受容体アゴニストでリンパ球を活性化するステップと、
b)記憶T細胞集団を選択的に増殖させることができる、少なくとも1種のサイトカイン受容体アゴニストの有効量に、活性化リンパ球を曝露するステップと、
c)b)で得られた細胞に、適切なベクターを用いて外来遺伝子を挿入し発現させるステップとを含む方法を提供することである。
a)リンパ球活性化を誘導することができる、アゴニスト抗体、組換えリガンドおよびこの誘導体を含むがこれらに限定されない、少なくとも2種の特異的活性化受容体アゴニストでリンパ球を活性化するステップと、
b)記憶T細胞集団を選択的に増殖させることができる、少なくとも1種のサイトカイン受容体アゴニストの有効量に、活性化リンパ球を曝露するステップと、
c)b)で得られた細胞に、適切なベクターを用いて外来遺伝子を挿入し発現させるステップとを含む方法を提供することである。
好ましくは、記憶T細胞集団は、CD4+および/またはCD8+および/またはγδおよび/またはNKT T細胞集団を含む。
好ましくは、リンパ球は、血液および生物起源の他の液体試料、固形組織試料、これら由来の細胞の組織培養物およびこの子孫、生物試料からの単離細胞(すなわち、PBMCなど)からなる群に属する生物試料由来である。
好ましくは、特異的リンパ球活性化受容体アゴニストは、細胞模倣担体に結合しており、より好ましくは、細胞模倣担体は、常磁性ビーズである。
好ましい実施形態では、リンパ球活性化受容体アゴニストの1つは、CD3ポリペプチドに特異的であり、好ましくは、リンパ球活性化受容体アゴニストのもう1つは、共刺激受容体、すなわちCD28に特異的である。
好ましい実施形態では、少なくとも1種のサイトカイン受容体アゴニストは、IL-7受容体アゴニストまたはIL-15受容体アゴニストであり、好ましくは、IL-15受容体アゴニストまたはIL-7受容体アゴニストもそれぞれ存在している。
好ましい実施形態では、ベクターはウイルスベクターである。
好ましい実施形態では、外来遺伝子は、自殺遺伝子、および/またはマーカー遺伝子、および/または生物活性分子、および/または受容体、および/または細胞内に維持されるもしくは細胞外に放出される可溶性因子、および/またはプロドラッグに耐性を与える遺伝子をコードする。
本発明のさらなる目的は、癌、感染症、免疫不全もしくは自己免疫の治療および/または予防のため、または造血前駆細胞もしくは固形臓器の移植のため、上記に記載の方法により生成された、遺伝子改変記憶T細胞集団の使用を提供することである。
本発明を、以下の図面を参照し、非限定的な例によって説明する。
材料および方法
実験プロトコルは、サンラファエロ科学研究所倫理委員会(the Ethical Committee of the San Raffaele Scientific Institute)で承認され、このガイドラインに従って実施された。
実験プロトコルは、サンラファエロ科学研究所倫理委員会(the Ethical Committee of the San Raffaele Scientific Institute)で承認され、このガイドラインに従って実施された。
マウスおよび腫瘍細胞
7〜8週齢のBALB/cマウスおよびCD45.2+ C57BL/6マウスは、Charles River (Charles River Italia社、ミラノ、イタリア)から購入した。CD45.1+ C57BL/6、DO11.10および16.2βトランスジェニック(Tg)(BALB/cバックグラウンド)(25)マウスは、研究所の特定病原体のいない施設で飼育した。TS/AおよびTS/A-LACKマウス乳腺癌は、以前に記載されている(17、19、26)。対数増殖期の4×105腫瘍細胞を、同系マウス(BALB/c)の右側のPBS 100μlに皮下注射した。典型的には、実験ごとに1群あたり5匹のマウスが使用された。
7〜8週齢のBALB/cマウスおよびCD45.2+ C57BL/6マウスは、Charles River (Charles River Italia社、ミラノ、イタリア)から購入した。CD45.1+ C57BL/6、DO11.10および16.2βトランスジェニック(Tg)(BALB/cバックグラウンド)(25)マウスは、研究所の特定病原体のいない施設で飼育した。TS/AおよびTS/A-LACKマウス乳腺癌は、以前に記載されている(17、19、26)。対数増殖期の4×105腫瘍細胞を、同系マウス(BALB/c)の右側のPBS 100μlに皮下注射した。典型的には、実験ごとに1群あたり5匹のマウスが使用された。
T細胞初代培養
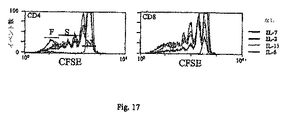
腫瘍細胞注入後20日目に、マウスを屠殺し、腫瘍増殖部位につながる、および遠位(非所属LN)の腋窩、上腕および鼠径抹消リンパ節(LN)を回収した。樹状細胞(DC)のワクチン接種実験では、マウスをDC投与後14日目に屠殺し、腋窩、上腕および鼠径LNを外科的に切除した。LN細胞は、組換えマウスIL-7(200ng/ml)、IL-2(20ng/ml)、IL-6(45ng/ml)、またはIL-15(100ng/ml)(全てPeprotech社製)の非存在下または存在下、完全培地において密度5×106にて24ウェルプレート中で培養した。並行培養では、細胞を、LACK由来MHC II制限ペプチド(5μm、(25))、および5×106の照射同系脾臓細部によりインビトロで刺激した。対照として、同様の培養を同系ナイーブマウスから用意した。幾つかの実験では、LN細胞を、メーカーの使用説明書に従い、最終濃度1μΜにて蛍光色素CFSE(5-(-6)-カルボキシフルオレセインジアセテート、スクシニミジルエステル)で標識した。
腫瘍細胞注入後20日目に、マウスを屠殺し、腫瘍増殖部位につながる、および遠位(非所属LN)の腋窩、上腕および鼠径抹消リンパ節(LN)を回収した。樹状細胞(DC)のワクチン接種実験では、マウスをDC投与後14日目に屠殺し、腋窩、上腕および鼠径LNを外科的に切除した。LN細胞は、組換えマウスIL-7(200ng/ml)、IL-2(20ng/ml)、IL-6(45ng/ml)、またはIL-15(100ng/ml)(全てPeprotech社製)の非存在下または存在下、完全培地において密度5×106にて24ウェルプレート中で培養した。並行培養では、細胞を、LACK由来MHC II制限ペプチド(5μm、(25))、および5×106の照射同系脾臓細部によりインビトロで刺激した。対照として、同様の培養を同系ナイーブマウスから用意した。幾つかの実験では、LN細胞を、メーカーの使用説明書に従い、最終濃度1μΜにて蛍光色素CFSE(5-(-6)-カルボキシフルオレセインジアセテート、スクシニミジルエステル)で標識した。
樹状細胞(DC)の調製
骨髄由来DCは、以前に記載した通りに得た(18)。簡単には、CD45.2+ C57BL/6骨髄前駆細胞を、25ng/ml組換えマウスGM-CSFおよび5ng/ml組換えマウスIL-4を含有するイスコフ完全培地(Pharmingen社、サンディエゴ、CA)において7日間増殖した。次いで、BMDCをLPS(1μg/ml、Sigma社、ミラノ、イタリア)の存在下、8時間、37℃で成熟させ、ラージT Ag由来Tag IVペプチド10μg/mlで1時間パルスした(18)。DCの成熟度および純度は、mAbを認識するCD11c、MHCクラスII、B7.1、B7.2およびCD40分子(全てPharmingen社製)で染色した後、フローサイトメトリーによりルーチンに評価した。2×105パルス成熟DCを、同系C57BL/6マウスの右側のPBS 200μlに皮下注射した。
骨髄由来DCは、以前に記載した通りに得た(18)。簡単には、CD45.2+ C57BL/6骨髄前駆細胞を、25ng/ml組換えマウスGM-CSFおよび5ng/ml組換えマウスIL-4を含有するイスコフ完全培地(Pharmingen社、サンディエゴ、CA)において7日間増殖した。次いで、BMDCをLPS(1μg/ml、Sigma社、ミラノ、イタリア)の存在下、8時間、37℃で成熟させ、ラージT Ag由来Tag IVペプチド10μg/mlで1時間パルスした(18)。DCの成熟度および純度は、mAbを認識するCD11c、MHCクラスII、B7.1、B7.2およびCD40分子(全てPharmingen社製)で染色した後、フローサイトメトリーによりルーチンに評価した。2×105パルス成熟DCを、同系C57BL/6マウスの右側のPBS 200μlに皮下注射した。
フローサイトメトリー分析
I-Ad/LACK多量体染色は、以前に記載されている。簡単には、I-Ad/LACK二量体(MHC II-ペプチド複合体、3μg/試料)を、室温で30分間、PBS中でアレクサ488結合タンパク質A(Molecular Probes Inc.社、ユージン、3μg/試料)を添加して多量体化する。遊離タンパク質A結合部位は、全(total)IgG(1μg/試料)を添加して飽和させた。6×105細胞を、先ず、ブロッキング緩衝液でインキュベートし(5%ラット血清+2.4G2抗-FcR mAb産生ハイブリドーマ細胞の95%培養上清、20分間)、次いで、多量体で染色した(0.5% BSAを補充したPBS中、4℃で1時間)。次いで、細胞を、抗CD4、抗CD44、抗CD11b、抗B220、抗CD8a mAb(PharMingen社、サンディエゴ、CA、USA)およびTO-PRO-3(1nM、Molecular Probes社)で染色した。3×105 CD4+または 103 CD4+ I-Ad/LACK+イベントを、全ての抗CD11b+、抗B220+、抗CD8a+およびTO-PRO-3+イベントを除外して回収した。指示された細胞を、抗CD4または抗CD8 mAb、ならびに抗CD44、抗CD127、抗CD25、抗CD132および抗CD62L mAb(抗CD127 Ab、A7R34クローン(Bioscience社製)以外全てPharMingen社製)で表面染色し、固定化し、透過処理し、メーカーの使用説明書に従って抗Bcl-2 mAbでさらに染色した。
I-Ad/LACK多量体染色は、以前に記載されている。簡単には、I-Ad/LACK二量体(MHC II-ペプチド複合体、3μg/試料)を、室温で30分間、PBS中でアレクサ488結合タンパク質A(Molecular Probes Inc.社、ユージン、3μg/試料)を添加して多量体化する。遊離タンパク質A結合部位は、全(total)IgG(1μg/試料)を添加して飽和させた。6×105細胞を、先ず、ブロッキング緩衝液でインキュベートし(5%ラット血清+2.4G2抗-FcR mAb産生ハイブリドーマ細胞の95%培養上清、20分間)、次いで、多量体で染色した(0.5% BSAを補充したPBS中、4℃で1時間)。次いで、細胞を、抗CD4、抗CD44、抗CD11b、抗B220、抗CD8a mAb(PharMingen社、サンディエゴ、CA、USA)およびTO-PRO-3(1nM、Molecular Probes社)で染色した。3×105 CD4+または 103 CD4+ I-Ad/LACK+イベントを、全ての抗CD11b+、抗B220+、抗CD8a+およびTO-PRO-3+イベントを除外して回収した。指示された細胞を、抗CD4または抗CD8 mAb、ならびに抗CD44、抗CD127、抗CD25、抗CD132および抗CD62L mAb(抗CD127 Ab、A7R34クローン(Bioscience社製)以外全てPharMingen社製)で表面染色し、固定化し、透過処理し、メーカーの使用説明書に従って抗Bcl-2 mAbでさらに染色した。
LACK特異的人工抗原提示細胞(aAPC)およびサイトカイン分泌アッセイ
5μmポリエチレンサルフェートラテックスビーズ(Interfacial Dynamics社)を、I-Ad/LACK多量体(20μg/ml)および抗CD28 mAb(37.51;2μg/ml)(LACK aAPC)で、または抗CD28 mAb単独(対照aAPC)でコーティングした。タンパク質のコーティングをフローサイトメトリー分析によりモニターした。典型的には、5×105LN細胞を、37℃にて5時間、5×106 aAPCと共に培養した。ブレフェルジンA(5μg/ml、Sigma社)を少なくとも2時間培養物に添加した。LACK aAPCにより誘導されたサイトカイン放出は、LACKをパルスした同系脾臓細胞により誘導されたものと同程度であった(不図示)。AH-1およびTag IV誘導サイトカイン産生の場合、脾臓細胞は、DO11.10およびCD45.1+ C57BL/6マウスから得られ、抗原提示細胞として使用された。脾臓細胞(3×107細胞/ml)を、1μM AH-1(19)および10μg/ml Tag IV(18)ペプチドで37℃にて1時間パルスし、次いで、腫瘍を有するマウスおよびDCワクチン接種マウスそれぞれのLN由来の同系LN細胞を刺激するのに使用した。その後、細胞を、上述の通りに抗CD8、およびIL-2およびIFN-γで染色し、APCオリジンのT細胞を除外するため抗クローン型KJ 1.26 mAb、および抗CD45.1+マーカーで染色した。次いで、CD4+、KJ 1.26-またはCD8+ CD45.1-イベントを、FACS Caliburで回収した。Ag特異的IL-2+/IFN-γ+ T細胞の合計数は、パーセンテージをトリパンブルー陰性LN細胞の合計数に乗じて決定した。
5μmポリエチレンサルフェートラテックスビーズ(Interfacial Dynamics社)を、I-Ad/LACK多量体(20μg/ml)および抗CD28 mAb(37.51;2μg/ml)(LACK aAPC)で、または抗CD28 mAb単独(対照aAPC)でコーティングした。タンパク質のコーティングをフローサイトメトリー分析によりモニターした。典型的には、5×105LN細胞を、37℃にて5時間、5×106 aAPCと共に培養した。ブレフェルジンA(5μg/ml、Sigma社)を少なくとも2時間培養物に添加した。LACK aAPCにより誘導されたサイトカイン放出は、LACKをパルスした同系脾臓細胞により誘導されたものと同程度であった(不図示)。AH-1およびTag IV誘導サイトカイン産生の場合、脾臓細胞は、DO11.10およびCD45.1+ C57BL/6マウスから得られ、抗原提示細胞として使用された。脾臓細胞(3×107細胞/ml)を、1μM AH-1(19)および10μg/ml Tag IV(18)ペプチドで37℃にて1時間パルスし、次いで、腫瘍を有するマウスおよびDCワクチン接種マウスそれぞれのLN由来の同系LN細胞を刺激するのに使用した。その後、細胞を、上述の通りに抗CD8、およびIL-2およびIFN-γで染色し、APCオリジンのT細胞を除外するため抗クローン型KJ 1.26 mAb、および抗CD45.1+マーカーで染色した。次いで、CD4+、KJ 1.26-またはCD8+ CD45.1-イベントを、FACS Caliburで回収した。Ag特異的IL-2+/IFN-γ+ T細胞の合計数は、パーセンテージをトリパンブルー陰性LN細胞の合計数に乗じて決定した。
ヒトT細胞培養およびELISPOTアッセイ
抹消血単核細胞(PBMC)は、結核患者(TB)および健常ドナーからFycoll-Hypaque密度勾配での血液遠心分離により得て、直ちに分析するかまたは今後の分析用に凍結した。必要であれば、細胞をCFSE(1μM)で染色した。細胞は、ヒトIL-7(200ng/ml)、IL-2(20ng/ml、IL-15(100ng/ml)もしくはIL-6(45ng/ml)の非存在下または存在下で7日間培養した。必要であれば、シクロスポリンA(CsA)(0.5μg/ml)または抗LFA-1(5μg/ml)ブロッキング抗体を添加した。次いで、細胞を回収し、CD4、CD8、CD3、CD56、CD45RA、CD62L mAbs(全てPharMingen社製)で染色し、フローサイトメトリーにより分析した。
抹消血単核細胞(PBMC)は、結核患者(TB)および健常ドナーからFycoll-Hypaque密度勾配での血液遠心分離により得て、直ちに分析するかまたは今後の分析用に凍結した。必要であれば、細胞をCFSE(1μM)で染色した。細胞は、ヒトIL-7(200ng/ml)、IL-2(20ng/ml、IL-15(100ng/ml)もしくはIL-6(45ng/ml)の非存在下または存在下で7日間培養した。必要であれば、シクロスポリンA(CsA)(0.5μg/ml)または抗LFA-1(5μg/ml)ブロッキング抗体を添加した。次いで、細胞を回収し、CD4、CD8、CD3、CD56、CD45RA、CD62L mAbs(全てPharMingen社製)で染色し、フローサイトメトリーにより分析した。
IFN-γ分泌に関するELISPOTアッセイは、以前に報告されているように行った(28)。簡単には、空気プラス5%CO2中で37℃にて18時間、自己照射PBMC(5×104細胞/ウェル)の存在下の抗IFN-γ捕捉mAb(B-B1; Diaclone社、ブザンソン、フランス)、およびMTPペプチドプールでプレコーティングした96ウェルプレート(MAIPS4510; Millipore社、ベッドフォード、マサチューセッツ州)に、細胞を5×104細胞/ウェルで二組播種した。ビオチン化抗IFN-γ検出mAb(B-G1; Diaclone社)を4時間添加した後、ストレプトアビジン-アルカリホスファターゼコンジュゲート(Amersham Pharmacia Biotech Europe GmbH、フライブルグ、ドイツ)を1時間添加した。洗浄工程後、ニトロブルーテトラゾリウム-BCIP(5-ブロモ-4-クロロ-3-インドリルホスフェート; Sigma社、セントルイス、ミズーリ州)発色基質を添加した。個々のスポット形成細胞(SFC)を、自動画像分析システムELISPOTリーダー(AID-GmbH、ストラスバーグ、ドイツ)を用いて計数した。特異的反応を検出するため、ヒト型結核菌のESAT-6およびCFP-10分泌タンパク質配列由来の、20アミノ酸長、70%超精製された6つの合成ヒト型結核菌ペプチド(MTP; Primm srl社、ミラノ、イタリア)プールを、ペプチドあたり最終濃度2μg/mlで使用した(28)。培地中のPBMC単独またはフィトヘマグルチニン(PHA-P; Sigma社)5μg/mlで刺激したPBMCを、陰性および陽性対照としてそれぞれ使用した。
幾つかの例では、MTP特異的IFN-γ放出を、細胞内サイトカイン分泌アッセイにより単一細胞レベルで分析した。簡単には、0.6×106 CFSE標識細胞を、HLA-DR制限MTP(4μg/ml)でパルスしたヒト抗CD28刺激mAb(2μg/ml)および3×106自己照射(5000rad)PBMCの存在下で、または陰性対照ではパルスせずに6時間再刺激した。最後の5時間で、ブレフェルジンA(10μg/ml)を細胞に添加した。その後、細胞を固定し、透過処理し、抗CD4、抗IL-2および抗IFN-γ mAbで染色し、FACS Caliburで分析した。
TB患者の分類
活動性TBを有するHIV血清反応陰性患者5例(臨床および培養により確認)を、サンラファエロ病院、感染症クリニック(the Clinic of Infectious Diseases、S. Raffaele Hospital)で回収した。患者は、Biocinetest-PPDツベルクリン(Chiron Italia srl社、ミラノ、イタリア)0.1ml(5ツベルクリン単位)を用いるマントー法により投与されたツベルクリン皮膚テスト(TST)を受けた。硬化径を48〜72時間後に評価した(硬化≧10mmを陽性に分類した)。治療を開始する前に、事前に書面によるインフォームドコンセントを得て、抹消血を回収した。健常ドナー(n=8)は、TB曝露、感染の既往がなく、TSTに陰性反応を示すHIV血清反応陰性者から選択した。
活動性TBを有するHIV血清反応陰性患者5例(臨床および培養により確認)を、サンラファエロ病院、感染症クリニック(the Clinic of Infectious Diseases、S. Raffaele Hospital)で回収した。患者は、Biocinetest-PPDツベルクリン(Chiron Italia srl社、ミラノ、イタリア)0.1ml(5ツベルクリン単位)を用いるマントー法により投与されたツベルクリン皮膚テスト(TST)を受けた。硬化径を48〜72時間後に評価した(硬化≧10mmを陽性に分類した)。治療を開始する前に、事前に書面によるインフォームドコンセントを得て、抹消血を回収した。健常ドナー(n=8)は、TB曝露、感染の既往がなく、TSTに陰性反応を示すHIV血清反応陰性者から選択した。
ヒトT細胞の活性化、培養およびレトロウイルス導入
抹消血単核細胞(PBMC)を、インフォームドコンセント後に得られた健常ドナーの軟膜からリンフォプレップ傾斜分離により単離した(Fresenius社、オスロ、ノルウェー)。PBMCは、抗生剤、グルタミンおよび10%加熱不活化FBS(BioWhittaker-Italia社、ミラノ、イタリア)を補充したRPMI1640培地(GIBCO-BRL, Gaitherburg, MD)で培養した。PBMCを、6ウェルプレート(1×106/ml)に播種し、抗aCD3(OKT3 30ng/ml、OrthoBiotech社、ラリタン、NJ)または常磁性baCD3/CD28(3:1 ビーズ/T細胞)(Dynabeads、Dynal Biotech社、またはXcyte Therapies Inc.社、ワシントン州シアトル、USA、Invitrogen社)で活性化した。T細胞を、培養前にbaCD3/CD28で濃縮した。抗aCD3により活性化した細胞は、ヒト組換えIL-2と共に600IU/mlで培養した(Chiron社、エメリービル、カリフォルニア州)。
抹消血単核細胞(PBMC)を、インフォームドコンセント後に得られた健常ドナーの軟膜からリンフォプレップ傾斜分離により単離した(Fresenius社、オスロ、ノルウェー)。PBMCは、抗生剤、グルタミンおよび10%加熱不活化FBS(BioWhittaker-Italia社、ミラノ、イタリア)を補充したRPMI1640培地(GIBCO-BRL, Gaitherburg, MD)で培養した。PBMCを、6ウェルプレート(1×106/ml)に播種し、抗aCD3(OKT3 30ng/ml、OrthoBiotech社、ラリタン、NJ)または常磁性baCD3/CD28(3:1 ビーズ/T細胞)(Dynabeads、Dynal Biotech社、またはXcyte Therapies Inc.社、ワシントン州シアトル、USA、Invitrogen社)で活性化した。T細胞を、培養前にbaCD3/CD28で濃縮した。抗aCD3により活性化した細胞は、ヒト組換えIL-2と共に600IU/mlで培養した(Chiron社、エメリービル、カリフォルニア州)。
baCD3/CD28により活性化した細胞を、1.サイトカイン非存在下、2.最小濃度5ng/mlのヒト組換えIL-7(Peprotech社、ロンドン、UK)を用いて、3.それぞれ最小濃度5ng/mlのヒト組換えIL-7およびIL-15(Peprotech社、ロンドン、UK)の両方を用いて、培養した。2日目および3日目に、細胞を、8μg/mlポリブレン(Sigma社、セントルイス、ミズーリ州)を用いて37℃で2時間、2400rpmにてスピノキュレーション(spinoculation)によりSFCMM3(39〜40)レトロウイルス上清を用いて導入した。SFCMM3レトロウイルスベクターは、LTRプロモーター下でTK自殺遺伝子を、およびSV40プロモーター下で神経成長因子の低親和性受容体(ΔLNGFR)の切断型をコードする(27)。レトロウイルス上清は、Molmed s.p.a.社より提供された。T細胞活性化後6日目に、刺激に使用した細胞大のビーズを、メーカー使用説明書に従ってT細胞から除去した。幾つかの実験では、T細胞活性化後7日目に、導入リンパ球を、以下のプロトコルにより陽性選択した(プロトコル名:導入リンパ球の陽性免疫選択)。
細胞は最大14日間培養した。14日目に、増殖(倍)を、トリパンブルー数を用いてフローサイトメトリーにより測定したLNGFR+細胞のパーセンテージを乗じて算出した。培地およびサイトカインは、当初のプロトコルに従い、3〜4日ごとに交換した。選択した時点で、増殖(倍)を、トリパンブルー数を用いてフローサイトメトリーにより測定したΔLNGFR+細胞のパーセンテージを乗じて算出した。
導入リンパ球の陽性免疫選択
細胞活性化後7日目に、導入リンパ球を陽性選択した。導入細胞は、この表面にΔLNGFrを発現したため、電磁ビーズで覆われた抗LNGFr抗体を用いて導入細胞を非導入リンパ球から分離した。
細胞活性化後7日目に、導入リンパ球を陽性選択した。導入細胞は、この表面にΔLNGFrを発現したため、電磁ビーズで覆われた抗LNGFr抗体を用いて導入細胞を非導入リンパ球から分離した。
細胞を、プレートからチューブに回収し、洗浄し(1500rpm、室温RTにて10分間)、WB中で最終濃度5×106/mlで再懸濁した。次いで、抗LNGFr抗体20.1を細胞懸濁液に添加し(1□g/20×106)、細胞をRTにて30分間、10rpmで回転させた。次いで、T細胞を1度洗浄し、WB中で25×106/mlで再懸濁した。次いで、ダイナビーズM-450ヒツジ抗マウスIgGを添加し(5×106ビーズ/106陽性細胞)、細胞を室温にて30分間、10rpmで回転させた。次いで、導入細胞を磁気選択した。このため、チューブを3分間磁石の近くに置き、陰性画分は廃棄した。この手順を合計3回繰り返した。最後に、ビーズに結合した細胞画分を磁石から除去し、洗浄し、濃度1×106細胞/mlで適切なサイトカインカクテルと共に新鮮培地において再懸濁した。
導入T細胞のフローサイトメトリー
表面表現型、導入効率、細胞周期、およびサイトカイン産生の分析には、フローサイトメトリーを用いた。以下の抗体(Pharmingen社)が使用された:ヒトCD4、CD8、CD45RA、CD27およびIFN-γに対してはFITC結合mAb、ヒトCD4、CD8、LNGFR、CD62L、CD28およびIL-4に対しては(PE)結合mAb、マウスCD45(Ly5.1)に対してはペリジニンクロロフィルタンパク質(PerCP)結合mAb、およびヒトCD3に対してはアロフィコシアニン(APC)結合mAb。アイソタイプをマッチングした蛍光色素に結合した無関係なmAb染色対照、およびデータをCellQuest Software(Becton Dickinson社)を用いて分析した後、試料は、Facscaliburフローサイトメーター(Becton Dickinson社、マウンテンビュー、カリフォルニア州)をくぐらせた。
表面表現型、導入効率、細胞周期、およびサイトカイン産生の分析には、フローサイトメトリーを用いた。以下の抗体(Pharmingen社)が使用された:ヒトCD4、CD8、CD45RA、CD27およびIFN-γに対してはFITC結合mAb、ヒトCD4、CD8、LNGFR、CD62L、CD28およびIL-4に対しては(PE)結合mAb、マウスCD45(Ly5.1)に対してはペリジニンクロロフィルタンパク質(PerCP)結合mAb、およびヒトCD3に対してはアロフィコシアニン(APC)結合mAb。アイソタイプをマッチングした蛍光色素に結合した無関係なmAb染色対照、およびデータをCellQuest Software(Becton Dickinson社)を用いて分析した後、試料は、Facscaliburフローサイトメーター(Becton Dickinson社、マウンテンビュー、カリフォルニア州)をくぐらせた。
CD127、CD122、CCR7、およびマウスCD45に対する蛍光色素結合抗体(Pharmingen社、サンディエゴ、CA、USA)も、リンパ球を染色するのに使用された。
サイトカイン産生
サイトカイン産生を測定するため、細胞を、24ウェルプレート(1×106/ml)に播種し、50ng/ml PMA(Sigma社)および1μg/mlイオノマイシン(Sigma社)で刺激した。4時間後、ブレフェルジンA(Sigma社)をさらに2時間添加した(10μg/ml)。次いで、細胞を適切な蛍光色素結合抗表面マーカー抗体で染色し、1%パラホルムアルデヒドで10分間4℃にて固定した。細胞内染色は、0.05%サポニン含有PBS 2% FBS(Sigma社)中でRTにて20分間インキュベートした後、適切な蛍光色素抗サイトカイン抗体により行った。
サイトカイン産生を測定するため、細胞を、24ウェルプレート(1×106/ml)に播種し、50ng/ml PMA(Sigma社)および1μg/mlイオノマイシン(Sigma社)で刺激した。4時間後、ブレフェルジンA(Sigma社)をさらに2時間添加した(10μg/ml)。次いで、細胞を適切な蛍光色素結合抗表面マーカー抗体で染色し、1%パラホルムアルデヒドで10分間4℃にて固定した。細胞内染色は、0.05%サポニン含有PBS 2% FBS(Sigma社)中でRTにて20分間インキュベートした後、適切な蛍光色素抗サイトカイン抗体により行った。
CFSE希釈アッセイおよび混合リンパ球反応(MLR)
T細胞同種反応性の分析は、リンパ球の初期培養後10日目に行った。導入T細胞を、CFSEで10日目に染色し、次いで、照射した同種PBMCと共に培養した。CFSEは、スクシニミジルエステル官能基および2つのアセテート部分を含有する蛍光分子からなる。CFSEは、細胞内に自由に拡散し、細胞内エステラーゼが、酢酸基を切断してこれを蛍光の、膜非透過性色素に変換する。色素は、隣接細胞に移動しない。CFSEは、細胞質において細胞により保持され、細胞機能に悪影響を及ぼさない。細胞分裂をするごとに、色素の相対強度は半減する。
T細胞同種反応性の分析は、リンパ球の初期培養後10日目に行った。導入T細胞を、CFSEで10日目に染色し、次いで、照射した同種PBMCと共に培養した。CFSEは、スクシニミジルエステル官能基および2つのアセテート部分を含有する蛍光分子からなる。CFSEは、細胞内に自由に拡散し、細胞内エステラーゼが、酢酸基を切断してこれを蛍光の、膜非透過性色素に変換する。色素は、隣接細胞に移動しない。CFSEは、細胞質において細胞により保持され、細胞機能に悪影響を及ぼさない。細胞分裂をするごとに、色素の相対強度は半減する。
CFSE染色法:
細胞をPBS中(血清非存在下)で2度洗浄し、2×107/mlに調節した。CFSEをPBS中で1μMに調節し、細胞懸濁液と1:1比で混合した。細胞を素早くボルテックスし、8分間RTにて混合した。次いで、FBSを1:1比で添加し、1分後、細胞を2000rpmで2分間遠心分離した。次いで、上清を捨て、細胞を10%FBS含有溶液(PBSまたは培地)で2度洗浄した。
細胞をPBS中(血清非存在下)で2度洗浄し、2×107/mlに調節した。CFSEをPBS中で1μMに調節し、細胞懸濁液と1:1比で混合した。細胞を素早くボルテックスし、8分間RTにて混合した。次いで、FBSを1:1比で添加し、1分後、細胞を2000rpmで2分間遠心分離した。次いで、上清を捨て、細胞を10%FBS含有溶液(PBSまたは培地)で2度洗浄した。
MLR
本方法の最後に、CFSE染色した導入T細胞(レスポンダー)を、2000 cGyを照射した同種PBMC(刺激因子)と共に1:1比にて24ウェルプレートで培養した。サイトカインは、細胞培養に添加しなかった。刺激因子の非存在下で培養したCFSE染色導入T細胞は、陰性対照として使用した。可溶性抗CD3抗体と共に培養した細胞は、陽性対照として使用した。
本方法の最後に、CFSE染色した導入T細胞(レスポンダー)を、2000 cGyを照射した同種PBMC(刺激因子)と共に1:1比にて24ウェルプレートで培養した。サイトカインは、細胞培養に添加しなかった。刺激因子の非存在下で培養したCFSE染色導入T細胞は、陰性対照として使用した。可溶性抗CD3抗体と共に培養した細胞は、陽性対照として使用した。
読み出し
刺激後、選択した時点で、細胞試料を回収し、蛍光色素結合抗表面マーカーモノクローナル抗体で染色した。FACS獲得直前に、1μl To-Pro-3溶液を、各FACS試料に添加した。To-Pro-3は、蛍光4において検出することができる、強い赤色の挿入DNA色素であり、FITC、PEおよびAPC結合抗体で細胞を共染色する可能性をもたらす。To-Pro-3の機能は、死細胞画分を明らかにすることである。
刺激後、選択した時点で、細胞試料を回収し、蛍光色素結合抗表面マーカーモノクローナル抗体で染色した。FACS獲得直前に、1μl To-Pro-3溶液を、各FACS試料に添加した。To-Pro-3は、蛍光4において検出することができる、強い赤色の挿入DNA色素であり、FITC、PEおよびAPC結合抗体で細胞を共染色する可能性をもたらす。To-Pro-3の機能は、死細胞画分を明らかにすることである。
インビボ分析
インビボでのヒトリンパ球の生物学を試験するには、通常、養子コンパートメント(scid、組換え活性化遺伝子-/-)ならびに先天性コンパートメント(NOD、共通γ鎖-/-)に免疫欠損を有するマウスが使用される。本発明者は、NOD/scidマウスを用いてインビボでのセントラル記憶遺伝子改変リンパ球の活性をテストした。6〜8週齢のメスのNOD/scidマウスは、Charles-River Italia社(カルコ、イタリア)から入手した。実験プロトコルは、本発明者の研究所の動物試験内部委員会(Institutional Animal Care and Use Committee [IACUC])により承認された。マウスは、以下のプロトコルに従い処理した。
インビボでのヒトリンパ球の生物学を試験するには、通常、養子コンパートメント(scid、組換え活性化遺伝子-/-)ならびに先天性コンパートメント(NOD、共通γ鎖-/-)に免疫欠損を有するマウスが使用される。本発明者は、NOD/scidマウスを用いてインビボでのセントラル記憶遺伝子改変リンパ球の活性をテストした。6〜8週齢のメスのNOD/scidマウスは、Charles-River Italia社(カルコ、イタリア)から入手した。実験プロトコルは、本発明者の研究所の動物試験内部委員会(Institutional Animal Care and Use Committee [IACUC])により承認された。マウスは、以下のプロトコルに従い処理した。
異種移植片対宿主疾患モデル
6〜8週齢のメスのNOD/scidマウスは、Charles-River Italia社(カルコ、イタリア)から入手した。注入1週間前に、マウスを層流アイソレーターから通常のケージへ移し、滅菌水および照射ペレットを適宜摂取する特定の病原体フリー条件下で維持した。実験前日、マウスに1mgブロッキング抗マウスIL-2Rβモノクローナル抗体を腹腔内投与し、残留NK活性を中和した。抗IL-2Rβ抗体は、Tanaka教授(大阪大学、日本)の好意により提供されたTMβ-1ハイブリドーマから、記述された通りに作製した。0日目、マウスに、単回線量350cGy(線形加速器からのガンマ線照射)を全身照射し、未修飾PBL、またはSFCMM3レトロウイルスベクターを導入されたヒトリンパ球を直ちに注入した(28)。未修飾PBLは、汚染単球、B細胞およびNK細胞をPan T細胞分離キット(Miltenyi社、ベルギッシュグラッドバッハ、ドイツ)で枯渇した後のPBMCから得た。細胞を、500μl X-VIVO15培地において再懸濁し、腹腔内に注入した。次いで、マウスを、体重減少の算出によりGvHDについてモニターした。瀕死のマウスは、道義上屠殺した。ヒトキメラ化を、尾静脈からの出血後、フローサイトメトリーにより週ごとに測定した。ヒトキメラ化は、以下のように算出した:ヒトキメラ化(%)=[huCD3+/(huCD3++mCD45+)]×100。
6〜8週齢のメスのNOD/scidマウスは、Charles-River Italia社(カルコ、イタリア)から入手した。注入1週間前に、マウスを層流アイソレーターから通常のケージへ移し、滅菌水および照射ペレットを適宜摂取する特定の病原体フリー条件下で維持した。実験前日、マウスに1mgブロッキング抗マウスIL-2Rβモノクローナル抗体を腹腔内投与し、残留NK活性を中和した。抗IL-2Rβ抗体は、Tanaka教授(大阪大学、日本)の好意により提供されたTMβ-1ハイブリドーマから、記述された通りに作製した。0日目、マウスに、単回線量350cGy(線形加速器からのガンマ線照射)を全身照射し、未修飾PBL、またはSFCMM3レトロウイルスベクターを導入されたヒトリンパ球を直ちに注入した(28)。未修飾PBLは、汚染単球、B細胞およびNK細胞をPan T細胞分離キット(Miltenyi社、ベルギッシュグラッドバッハ、ドイツ)で枯渇した後のPBMCから得た。細胞を、500μl X-VIVO15培地において再懸濁し、腹腔内に注入した。次いで、マウスを、体重減少の算出によりGvHDについてモニターした。瀕死のマウスは、道義上屠殺した。ヒトキメラ化を、尾静脈からの出血後、フローサイトメトリーにより週ごとに測定した。ヒトキメラ化は、以下のように算出した:ヒトキメラ化(%)=[huCD3+/(huCD3++mCD45+)]×100。
異種GvHDの分析
T細胞注入後1、2および3週間目に、マウスを秤量し、以下のスコアにより異種GvHDについて評価した:体重減少(体重減少<10%は0、10〜25%は1、>25%は2)、ハンチング(0〜2)、活動性(0〜2)、毛並み(0〜2)、および皮膚の完全性(0〜2)、最大インデックス10。体重減少は、最も客観的な基準であるとみなされたため、独立変数としても評価した(表1)。
T細胞注入後1、2および3週間目に、マウスを秤量し、以下のスコアにより異種GvHDについて評価した:体重減少(体重減少<10%は0、10〜25%は1、>25%は2)、ハンチング(0〜2)、活動性(0〜2)、毛並み(0〜2)、および皮膚の完全性(0〜2)、最大インデックス10。体重減少は、最も客観的な基準であるとみなされたため、独立変数としても評価した(表1)。

同種GvHDモデル
同種GvHDモデルでは、マウスにヒト皮膚を移植し、同種遺伝子改変リンパ球を注入して、ヒト皮膚へホーミングし同種GvH反応を媒介する能力を評価した。移植1週間前に、マウスを層流アイソレーターから通常のケージへ移し、滅菌水および照射ペレットを適宜摂取する特定の病原体フリー条件下で維持した。ヒトT細胞の注入約3週間前、マウスに12〜18mgアバチン/マウスで腹腔内麻酔を行った。次いで、マウスの背中の毛を剃り、背中の両側水平切開を行った。次いで、皮下ポケットを開き、ヒト腹部表皮の小片(皮膚脂肪および結合組織由来)を導入した。処置の最後に、創傷を縫合した。術中、マウスの体温は徐々に低下するため、加熱した箱の中に30分間入れ、最後にケージへ移した。
同種GvHDモデルでは、マウスにヒト皮膚を移植し、同種遺伝子改変リンパ球を注入して、ヒト皮膚へホーミングし同種GvH反応を媒介する能力を評価した。移植1週間前に、マウスを層流アイソレーターから通常のケージへ移し、滅菌水および照射ペレットを適宜摂取する特定の病原体フリー条件下で維持した。ヒトT細胞の注入約3週間前、マウスに12〜18mgアバチン/マウスで腹腔内麻酔を行った。次いで、マウスの背中の毛を剃り、背中の両側水平切開を行った。次いで、皮下ポケットを開き、ヒト腹部表皮の小片(皮膚脂肪および結合組織由来)を導入した。処置の最後に、創傷を縫合した。術中、マウスの体温は徐々に低下するため、加熱した箱の中に30分間入れ、最後にケージへ移した。
ヒトT細胞注入
ヒトリンパ球のNOD/scidマウスへの移植を促進するため、本発明者は、リンパ球移植の前に抗マウスIL-2受容体β(TMβ-1)抗体でNK細胞を機能的に不活性化した。抗体は、Tanaka教授(大阪大学、日本)の好意により提供されたTMβ-1ハイブリドーマから作製した。0日目、マウスに、単回線量300cGy(線形加速器からのガンマ線照射)を全身照射した。次いで、マウスを秤量し、初回刺激後9日目に収集した導入ヒトリンパ球を直ちに注入した。細胞は250μL食塩水で静注した。
ヒトリンパ球のNOD/scidマウスへの移植を促進するため、本発明者は、リンパ球移植の前に抗マウスIL-2受容体β(TMβ-1)抗体でNK細胞を機能的に不活性化した。抗体は、Tanaka教授(大阪大学、日本)の好意により提供されたTMβ-1ハイブリドーマから作製した。0日目、マウスに、単回線量300cGy(線形加速器からのガンマ線照射)を全身照射した。次いで、マウスを秤量し、初回刺激後9日目に収集した導入ヒトリンパ球を直ちに注入した。細胞は250μL食塩水で静注した。
T細胞移植の分析
T細胞注入後1、2および3週目に、約300μl血液/マウスを尾静脈の小切開から収集し、ヘパリン含有チューブに回収した。赤血球細胞を、ACKに3分間曝露して溶解し、次いで、パラグラフ「表面マーカーおよび細胞蛍光分析のための染色」に記載の通り、細胞蛍光分析のため染色した。
T細胞注入後1、2および3週目に、約300μl血液/マウスを尾静脈の小切開から収集し、ヘパリン含有チューブに回収した。赤血球細胞を、ACKに3分間曝露して溶解し、次いで、パラグラフ「表面マーカーおよび細胞蛍光分析のための染色」に記載の通り、細胞蛍光分析のため染色した。
同種GvHDの分析
T細胞注入後3週間目、または重度のGvHDの場合はこれ以前に、マウスを屠殺し、2片のヒト皮膚を両側から除去した。形態評価のため、ホルマリンで固定しパラフィンに包埋した皮膚を4μm厚の切片にカットし、ヘマトキシリンおよびエオシンで染色した。ヒトTリンパ球の存在については、Dako自動免疫染色装置を用いるアビジン/ビオチンペルオキシダーゼ複合体法により、1:100希釈でモノクローナル抗ヒトCD3抗体(Dako社、グロストラップ、デンマーク)による免疫組織化学的評価を実施した。染色反応を、四塩酸塩色素原法により明らかにし、切片をヘマトキシリンで対比染色した。画像は、Zeiss Axicam HRCで撮影した。
T細胞注入後3週間目、または重度のGvHDの場合はこれ以前に、マウスを屠殺し、2片のヒト皮膚を両側から除去した。形態評価のため、ホルマリンで固定しパラフィンに包埋した皮膚を4μm厚の切片にカットし、ヘマトキシリンおよびエオシンで染色した。ヒトTリンパ球の存在については、Dako自動免疫染色装置を用いるアビジン/ビオチンペルオキシダーゼ複合体法により、1:100希釈でモノクローナル抗ヒトCD3抗体(Dako社、グロストラップ、デンマーク)による免疫組織化学的評価を実施した。染色反応を、四塩酸塩色素原法により明らかにし、切片をヘマトキシリンで対比染色した。画像は、Zeiss Axicam HRCで撮影した。
統計分析
本研究で検討した変数ごとに、平均、中央値および標準偏差を算出した。統計分析は全て、Microsoft Excel 2003(Microsoft社、レドモンド、ワシントン州)およびこのアドイン型Statcel2(OMS出版、埼玉、日本)を用いて行った。パラメトリックデータについては、分散分析(ANOVA)後にシェッフェのF検定を実施し、ノンパラメトリックデータについては、クラスカルワリス検定後にマンホイットニーのU検定を実施した。
本研究で検討した変数ごとに、平均、中央値および標準偏差を算出した。統計分析は全て、Microsoft Excel 2003(Microsoft社、レドモンド、ワシントン州)およびこのアドイン型Statcel2(OMS出版、埼玉、日本)を用いて行った。パラメトリックデータについては、分散分析(ANOVA)後にシェッフェのF検定を実施し、ノンパラメトリックデータについては、クラスカルワリス検定後にマンホイットニーのU検定を実施した。
結果および考察
IL-7は、Ag誘導細胞増殖を必要とすることなく、希少な腫瘍特異的CD4+T細胞の検出を支持する
Ag特異的T細胞の計数は、Ag特異的T細胞の存在が、診断目的および予後目的であるような幾つかの臨床条件にとって重大な意味を持つ可能性がある。本発明者は、最近、大形リーシュマニア由来モデルAg LACKを発現するTS/A細胞(TS/A-LACK)を用いる、腫瘍疾患の前臨床マウスモデルを開発した。LACK特異的ナイーブCD4+T細胞は、未操作BALB/cマウスでは同定することができないのに対し(29)、本発明者は、最近、蛍光MHCクラスII/Ag多量体染色ならびにLACK特異的IL-2およびIFN-γ細胞内放出により、TS/A-LACK腫瘍を有するマウスにおいてLACK特異的Ag経験CD44high CD4+Tリンパ球を同定した(20)。
IL-7は、Ag誘導細胞増殖を必要とすることなく、希少な腫瘍特異的CD4+T細胞の検出を支持する
Ag特異的T細胞の計数は、Ag特異的T細胞の存在が、診断目的および予後目的であるような幾つかの臨床条件にとって重大な意味を持つ可能性がある。本発明者は、最近、大形リーシュマニア由来モデルAg LACKを発現するTS/A細胞(TS/A-LACK)を用いる、腫瘍疾患の前臨床マウスモデルを開発した。LACK特異的ナイーブCD4+T細胞は、未操作BALB/cマウスでは同定することができないのに対し(29)、本発明者は、最近、蛍光MHCクラスII/Ag多量体染色ならびにLACK特異的IL-2およびIFN-γ細胞内放出により、TS/A-LACK腫瘍を有するマウスにおいてLACK特異的Ag経験CD44high CD4+Tリンパ球を同定した(20)。
Ag特異的T細胞の検出およびクローニングを改善するため、本発明者は、記憶CD4+T細胞の生存を支持することが知られている(7、9〜14)IL-7は、腫瘍特異的CD4+T細胞の頻度を高めるかどうかを調べた。対照のTS/AおよびTS/A-LACK腫瘍細胞を、BALB/cマウスに皮下注射した。腫瘍細胞注入後20日目、全てのマウスが、測定可能な腫瘍を発現していた。マウスを屠殺し、腫瘍所属および腫瘍非所属LNを外科的に切除した。前者は、IL-2およびIFN-γを産生できる、LACK特異的Ag経験CD44high CD4+Tリンパ球集団を含有していたのに対し、後者は、腫瘍に気づかず、LACK特異的ナイーブCD4+Tリンパ球を示すままであった(31)。LACK特異的CD4+T細胞の頻度は、さらなるAg刺激を行わずに組換えIL-7の存在下で培養した7日後、エクスビボで分析した(図1)。エクスビボでは、TS/A-LACK腫瘍を有する動物の所属LNは、高レベルのCD44を発現するCD4+ I-Ad/LACK+細胞の低いながら有意な頻度を示し(図1A)、LACK刺激するとIL-2およびIFN-γを産生することができた(図1B)。IL-7単独で培養した7日後、CD4+ I-Ad/LACK+ CD44highおよびLACK特異的サイトカイン産生細胞の頻度(図1A、B)、ならびにこれらの合計数(不図示、および図2、4)は、著しく増加した。サイトカイン分泌細胞のうち、IL-2およびIFN-γ産生細胞(以前に多機能性として記載されている中間記憶Tリンパ球(30〜33)を代表している可能性のある)は、ほとんどが濃縮されていた。TS/A腫瘍を有する対照マウスの腫瘍所属LNにおけるLACK特異的T細胞、ならびにTS/AおよびTS/A-LACKマウスの非所属LN由来のLACK特異的T細胞の頻度は、エクスビボでナイーブBALB/cマウスにおいて見られたものと同程度であり(図1C、Dおよび不図示)、IL-7での培養後のバックグラウンド基準内にとどまっていた(図1C、D)。故に、IL-7は、インビボで初回刺激した腫瘍特異的Ag経験CD4+T細胞のLN培養物を濃縮し、それ故に、これらの計数を、インビトロでのAg誘導細胞増殖を必要とすることなく支持する。
IL-7およびIL-2は、腫瘍特異的CD4+T細胞の蓄積を支持するが、抗原刺激は腫瘍特異的CD4+T細胞の蓄積を支持しない
Agによる再刺激は、増殖のため、および幾つかの例ではAg特異的CD4+T細胞を同定するために最も多く使用されている。さらに、IL-7に加え、IL-2およびIL-15も、記憶T細胞増殖を制御する(13、35〜37)。故に、TS/A-LACK腫瘍を有するマウス由来のLN細胞を、非パルスの照射同系APC(APC)もしくはLACK由来ペプチドでパルスした照射同系APC(Ag/APC)の存在下、または対照として、最適量のIL-7、IL-2、IL-15およびIL-6の存在下で培養し、フローサイトメトリーにより分析した。CD4+ I-Ad/LACK+ CD44high T細胞の頻度は、エクスビボで見られるものと比較した場合、培養細胞でわずかに高かった(図1A)が、対照(APC)とAg刺激培養(Ag/APC)との間に差は検出されなかった(図2A)。反対に、IL-7における細胞の培養は、対照で見られたもの(不図示)およびIL-6誘導培養物を上回り、LACK特異的CD4+T細胞の頻度(図2A、B)および合計数(図2C、D)を増加させた。IL-7と同様、IL-2も、CD4+ I-Ad/LACK+ CD44high T細胞のLN培養物(図2A、C)、ならびにAg特異的刺激時にIL-2およびIFN-γを分泌することができるCD4+細胞のLN培養物を濃縮した(図2B、D)。IL-15誘導培養では、CD4+ I-Ad/LACK+ CD44high T細胞の頻度は、対照培養物の1つと同程度にとどまったが、幾つかの独立した実験では、サイトカインを分泌することができるLACK特異的CD4+T細胞を濃縮した。この場合もやはり、LACK特異的サイトカイン産生細胞のなかでは、IL-2/IFN-γ分泌CD4+T細胞が最も濃縮され、IL-7誘導培養をより支持した(図2C、D)。
Agによる再刺激は、増殖のため、および幾つかの例ではAg特異的CD4+T細胞を同定するために最も多く使用されている。さらに、IL-7に加え、IL-2およびIL-15も、記憶T細胞増殖を制御する(13、35〜37)。故に、TS/A-LACK腫瘍を有するマウス由来のLN細胞を、非パルスの照射同系APC(APC)もしくはLACK由来ペプチドでパルスした照射同系APC(Ag/APC)の存在下、または対照として、最適量のIL-7、IL-2、IL-15およびIL-6の存在下で培養し、フローサイトメトリーにより分析した。CD4+ I-Ad/LACK+ CD44high T細胞の頻度は、エクスビボで見られるものと比較した場合、培養細胞でわずかに高かった(図1A)が、対照(APC)とAg刺激培養(Ag/APC)との間に差は検出されなかった(図2A)。反対に、IL-7における細胞の培養は、対照で見られたもの(不図示)およびIL-6誘導培養物を上回り、LACK特異的CD4+T細胞の頻度(図2A、B)および合計数(図2C、D)を増加させた。IL-7と同様、IL-2も、CD4+ I-Ad/LACK+ CD44high T細胞のLN培養物(図2A、C)、ならびにAg特異的刺激時にIL-2およびIFN-γを分泌することができるCD4+細胞のLN培養物を濃縮した(図2B、D)。IL-15誘導培養では、CD4+ I-Ad/LACK+ CD44high T細胞の頻度は、対照培養物の1つと同程度にとどまったが、幾つかの独立した実験では、サイトカインを分泌することができるLACK特異的CD4+T細胞を濃縮した。この場合もやはり、LACK特異的サイトカイン産生細胞のなかでは、IL-2/IFN-γ分泌CD4+T細胞が最も濃縮され、IL-7誘導培養をより支持した(図2C、D)。
IL-7およびIL-2は、インビボで初回刺激した腫瘍特異的CD4+T細胞のAg非依存性自発的増殖および生存を維持する
本発明者は、次に、IL-7およびIL-2が、インビボで初回刺激したLACK特異的CD4+T細胞の蓄積を支持するメカニズムを調べた。先ず、本発明者は、インビトロでこれらの細胞の増殖を支持するこれらのサイトカインの能力を分析した。LN細胞をCFSE生体染色色素で標識し、組換えサイトカインの非存在下または存在下で1週間培養した。外因性サイトカインの非存在下では、ナイーブマウス由来LN細胞は、増殖せず、当初のCFSE含量を保持した(図3A、左のパネル)。反対に、TS/A-LACK腫瘍を有するマウス由来のCD4+ LN細胞の検出可能な集団は、増殖し、追加刺激の非存在下でインビトロでのCFSE含量を希釈した(図3A、右のパネル)。インビトロでのLACK再刺激時にIL-2およびIFN-γを分泌する能力により同定されるLACK特異的記憶T細胞は、主に急速増殖CFSEdim細胞において含有された(図3B、なし、MFI:66)。これは、最近のインビボでの腫瘍Ag遭遇による増殖に関与したことを示唆すものである。IL-7の存在下では、腫瘍所属LN由来の高頻度の細胞は、1〜3細胞分裂を終え、LACK特異的再刺激時にIL-2およびIFN-gを分泌する能力により同定されるLACK特異的T細胞は、著しく濃縮され、CFSE含量は低下していた(MFI:44)(図3BおよびC)。実際、LACK特異的CD4+T細胞は、3〜4回を超える細胞周期を繰り返していた。これは、LACK特異的CD4+T細胞と、緩徐な恒常的細胞分裂をしている細胞(4細胞周期未満)とを区別するものである。IL-7に誘導された恒常的細胞分裂は、ナイーブ対照マウスのLNにおいても見られた(図5C)。しかし、LACK特異的CD4+T細胞は、これらの培養物においては濃縮されなかった(不図示)。インビトロで自発的増殖すること、および再刺激時にIL-2およびIFN-gの両方を産生する能力があることから、腫瘍所属LNにおいて見られ、IL-7に反応してインビトロで蓄積するLACK特異的T細胞は、慢性感染患者のインビボでも見られる、最近初回刺激された非極性中間記憶T細胞を代表している可能性がある(32)。
本発明者は、次に、IL-7およびIL-2が、インビボで初回刺激したLACK特異的CD4+T細胞の蓄積を支持するメカニズムを調べた。先ず、本発明者は、インビトロでこれらの細胞の増殖を支持するこれらのサイトカインの能力を分析した。LN細胞をCFSE生体染色色素で標識し、組換えサイトカインの非存在下または存在下で1週間培養した。外因性サイトカインの非存在下では、ナイーブマウス由来LN細胞は、増殖せず、当初のCFSE含量を保持した(図3A、左のパネル)。反対に、TS/A-LACK腫瘍を有するマウス由来のCD4+ LN細胞の検出可能な集団は、増殖し、追加刺激の非存在下でインビトロでのCFSE含量を希釈した(図3A、右のパネル)。インビトロでのLACK再刺激時にIL-2およびIFN-γを分泌する能力により同定されるLACK特異的記憶T細胞は、主に急速増殖CFSEdim細胞において含有された(図3B、なし、MFI:66)。これは、最近のインビボでの腫瘍Ag遭遇による増殖に関与したことを示唆すものである。IL-7の存在下では、腫瘍所属LN由来の高頻度の細胞は、1〜3細胞分裂を終え、LACK特異的再刺激時にIL-2およびIFN-gを分泌する能力により同定されるLACK特異的T細胞は、著しく濃縮され、CFSE含量は低下していた(MFI:44)(図3BおよびC)。実際、LACK特異的CD4+T細胞は、3〜4回を超える細胞周期を繰り返していた。これは、LACK特異的CD4+T細胞と、緩徐な恒常的細胞分裂をしている細胞(4細胞周期未満)とを区別するものである。IL-7に誘導された恒常的細胞分裂は、ナイーブ対照マウスのLNにおいても見られた(図5C)。しかし、LACK特異的CD4+T細胞は、これらの培養物においては濃縮されなかった(不図示)。インビトロで自発的増殖すること、および再刺激時にIL-2およびIFN-gの両方を産生する能力があることから、腫瘍所属LNにおいて見られ、IL-7に反応してインビトロで蓄積するLACK特異的T細胞は、慢性感染患者のインビボでも見られる、最近初回刺激された非極性中間記憶T細胞を代表している可能性がある(32)。
IL-7に加え、IL-2も、CD4+T細胞画分のインビトロでの増殖を支持し、LACK特異的記憶リンパ球数を増加した。反対に、IL-15およびIL-6は、CD4+T細胞増殖も、またはLACK特異的CD4+T細胞の蓄積も、対照(なし)培養物において見られるもの以上に支持することはなかった(図3B、およびD)。故に、IL-7およびIL-2は、インビボでAgを経験した記憶T細胞のインビトロでの増殖を支持することができる。
並行実験では、本発明者は、かなりの頻度のI-Ad/LACK+ CD44+ナイーブT細胞を含有する、TS/AおよびTS/A-LACK腫瘍を有する16.2βトランスジェニックマウス由来のLN培養物を分析して、同様の結果が得られることを示した(25)。実際、腫瘍を有するBALB/cマウス由来培養物の場合のように、16.2βマウス由来の培養物は、高レベルのCD44を発現するLACK特異的CD4+を含有し(図4A)、IL-2およびIFN-γのLACK特異的放出が可能である(図4B、4C)。IL-7、IL-2および程度は下がるがIL-15は、これらの細胞培養物を濃縮したが、IL-6、TNF-αおよびIL-10は濃縮しなかった。IL-2およびIFN-γのLACK特異的放出が可能なLACK特異的CD4+T細胞は、この場合もやはりCFSE dim細胞内に含有されていた(図4E)。
この非関連モデルであっても、IL-7およびIL-2は、Ag経験CD4+T細胞のLN培養物を、Ag刺激を必要とすることなく、これらのインビトロでの増殖を維持することで濃縮する。
Ag経験CD4+T細胞の蓄積を促進するIL-7およびIL-2の相対力をさらに特性化するため、CFSE標識細胞を、増殖細胞の生細胞および死細胞を同定することができる、蛍光色素TO-PRO-3で染色した。IL-7は、培養物の生存能力を最もよく維持し、1週間後のTO-PRO-3+、死細胞は15%のみであった。IL-2において維持された細胞の最大47%、外因性サイトカインの非存在下、またはIL-15およびIL-6の存在下で培養された細胞の、それぞれ60%、57%、および73%で、TO-PRO-3+の結果が得られた(図5A)。さらに、IL-2存在下では、CFSE dim細胞の40%のみが色素を除外したのに対し、IL-7存在下では、CFSE dim細胞の最大72%がTO-PRO-3-のままであった。これは、IL-7単独存在下で活発に増殖している細胞は、IL-2単独において培養されたものと比較した場合、より生存能力があり、細胞死に対し低感受性である可能性があることを示している。
IL-7およびIL-2のT細胞生存を支持する能力は、抗アポトーシス因子Bcl-2発現を制御する能力に関連している(10、38)。故に、本発明者は、組換えサイトカインの非存在下または存在下で維持したCFSE標識LN培養物におけるBcl-2レベルを分析した。IL-15を除き、どの培養条件においても、CFSE dim細胞は、最適レベルのBcl-2を発現し(図5B)、インビボで初回刺激された細胞が、生存促進(pro-survival)シグナルを受け取ったことを示した。しかし、IL-7はCFSE dim細胞およびCFSE bright細胞の両方の生存を支持する点で独特であった。実際、CD4+T細胞の最大82.5%が、IL-7の存在下で最適なBcl-2レベルを発現したのに対し、対照培地、IL-2、IL-15およびIL-6において培養された細胞のそれぞれ41.3%、16.7%、38%、および10.5%のみが、高いBcl-2発現を維持した(図5B、C)。IL-2およびIL-15存在下では、大半の細胞においてBcl-2発現の減少すら見られた(図5C)。これらをまとめた所見は、IL-2またはIL-15と比較した場合、CD4+Tリンパ球の生存を支持するIL-7の優れた能力を支持し、IL-7が、CD4+Tリンパ球の全サブセットをこの活性化状態とは無関係に維持する点で独特でありうることを示唆している。
本発明者はさらに、IL-7が、インビボで産生された腫瘍特異的CD4T+細胞の全サブセットの維持においてIL-2より強力であるかどうか、その結果、IL-7は、希少なインビボAg経験CD4T+細胞を追跡し検出するための主要な興味深いツールとなるかどうかを評価するため、腫瘍所属LN由来培養物の表面表現型を分析した。TS/A-LACK腫瘍所属LN細胞を、CFSE生体染色色素で標識し、IL-7単独またはIL-2単独の存在下で1週間培養した。IL-7単独で維持されたCFSE dim細胞のうち、エクスビボで見られるものと同程度のCD4+T細胞画分が、CD44およびCD62Lの高発現または低発現を保持した(図6A、B)。これらの細胞は、ナイーブな、エフェクターリンパ球およびセントラル記憶リンパ球の代表である可能性がある(8、39〜41)。IL-7で処置したCFSE dim細胞は、以前に報告されたように、中間レベルのCD25およびCD132を発現し、CD127の表面発現をダウンレギュレートした(42)。IL-2単独で維持されたCFSE dim細胞のほとんどは、それぞれ十分に活性化された表面表現型を発現し(CD44high、CD25high、CD62Llow、Bcl-2low、図6A)、培養物は、CD44high T細胞が濃縮された(図6Bおよび7B)。16.2βトランスジェニックマウス由来の培養物では、IL-7およびIL-2の両方が、エクスビボで見られるものと同程度の表面表現型を有する、Ag経験CD4+T細胞を濃縮し(図7A)、IL-7のみが、I-Ad/LACK+およびI-Ad/LACK- CD4+T細胞のうちCD44 highとCD44 low細胞間の当初の比を維持した(図7B)。リンパ球の相対的発現量を維持する能力は、生物試料におけるAg特異的T細胞の頻度を測定するため、短期培養を利用しようとする場合に関係する可能性がある。さらに、Bcl-2+細胞のほとんどが、IL-2と共に培養された細胞では低レベルのLNホーミング分子CD62Lを発現したのに対し、IL-7存在下では、CFSE dim CD4+T細胞の最大53%が、Bcl-2およびCD62Lの最適発現を維持した(図6C)。
IL-7、IL-2およびIL-15は、Ag特異的記憶CD8+T細胞をAg非依存的なかたちで増殖する
インビボで産生されたAg特異的T細胞を明らかにするための、IL-7におけるAg非依存性短期培養の一般的有用性にさらに取り組むため、本発明者は、Ag特異的CD8+Tリンパ球を、腫瘍疾患とは異なる文脈で追跡できるかどうかを調べた。この主な目的は:1)インビボでAgを経験したCD8+T細胞(インビボでAgを経験したCD4+T細胞だけでなく)を追跡するための本発明の適用性を評価すること、2)抗腫瘍免疫応答の診断とは異なる、臨床状況(積極的ワクチン接種)において本発明の適用性を評価すること、である。
インビボで産生されたAg特異的T細胞を明らかにするための、IL-7におけるAg非依存性短期培養の一般的有用性にさらに取り組むため、本発明者は、Ag特異的CD8+Tリンパ球を、腫瘍疾患とは異なる文脈で追跡できるかどうかを調べた。この主な目的は:1)インビボでAgを経験したCD8+T細胞(インビボでAgを経験したCD4+T細胞だけでなく)を追跡するための本発明の適用性を評価すること、2)抗腫瘍免疫応答の診断とは異なる、臨床状況(積極的ワクチン接種)において本発明の適用性を評価すること、である。
IL-7が、前記1つの腫瘍疾患とは異なる臨床状況において、Ag特異的T細胞を明らかにするのに用いることができるかどうかを調べるため、本発明者は、樹状細胞(DC)ベースのワクチンにより誘導されたペプチド特異的CD8+T細胞を分析した。C57BL/6マウスに、SV40ラージT抗原由来のMHC-クラスI制限Tag IVペプチドでパルスした骨髄由来DC(DC-Tag)をワクチン接種した(18)。14日後、サイトカインで誘導した培養後のエクスビボでのAg特異的細胞内サイトカイン放出により、LN細胞を分析した。対照として、LN細胞を、ナイーブ未ワクチン接種C57BL/6マウスからも得た。Ag再刺激後にIFN-γのみまたはIL-2およびIFN-γを産生することができる、エクスビボ、Tag IV特異的CD8+T細胞は、ナイーブマウスでは検出できず、DCワクチン接種マウスでは低頻度で検出可能であった(それぞれ0%、0.37%)。Ag再刺激の非存在下で、IL-7、IL-2およびIL-15による培養7日後、Tag IV特異的CD8+T細胞の頻度(それぞれ、3.94%、1.83%および1.95%)および合計数は、DCワクチン接種マウス由来の培養物において著しく増加したが、ナイーブマウス由来の培養物では有意に増加することはなかった(不図示)。
同じ培養物において、本発明者は、合計CD8+T細胞と比較したTag IV特異的CD8+T細胞の相対的濃縮を分析した(図9)。全ての異なる条件のうち、IL-7、IL-2およびIL-15での培養は、CD8+T細胞の合計計数を増加し、エクスビボでの分析と比較した場合、絶対数は2倍になったが、IL-6単独または単純培地での培養は、CD8+T細胞の合計計数を増加しなかった(図9)。一方、CD8+T細胞の2倍の増加にもかかわらず、Tag IV特異的CD8+T細胞は、5〜7倍に増加した(図8)。これは、インビボでTag IVワクチンを経験したCD8T細胞が、他のCD8+T細胞集団のなかで大きな利点を有すること、IL-7、IL-2またはIL-15などの生存促進的サイトカイン存在下での培養により、選択的に濃縮されることを意味する。故に、IL-7は、Ag再刺激非存在下であっても、腫瘍誘導およびワクチン誘導CD8+T細胞反応を明らかにするのに使用することができる。
IL-7は、本来エクスビボでは検出できない抗原特異的CD8+T細胞を明らかにする
TS/A細胞は、自然には、免疫優勢エピトープが以前に記載された(AH-1、(19))、内因性MuLVのエンベロープタンパク質gp70を発現する。IL-7での培養が、希少なAg特異的CD8+T細胞の同定も助けるかどうかにさらに取り組むため、本発明者は、AH-1特異的CD8+T細胞反応を、Ag再刺激非存在下でIL-7、IL-2、IL-15およびIL-6を用いて、または用いずに培養した1週間後にエクスビボで比較した。リンパ球は、非パルスおよびAH-1パルス同系脾臓細胞による刺激時の、細胞内サイトカイン放出により分析した(図10)。腫瘍を有するマウスでは、サイトカイン産生細胞の頻度が、バックグラウンドレベル内にとどまり、ナイーブマウスにおいて見られるものと同程度であったため(不図示)、AH-1特異的CD8+T細胞はエクスビボでは検出できなかった(図10A)。7日後、TS/A-LACK腫瘍を有するLN由来の、組換えサイトカインを用いてまたは用いずに維持したLN培養物は、AH-1再刺激とは独立にIFN-γを産生する細胞の可変頻度を含有していた(図10B)。IL-7の存在下では、培養物は、Ag再刺激時にIFN-γのみ、またはIL-2およびIFN-γを産生することができるAH-1特異的CD8+T細胞が濃縮された(図10B)。IL-2もAH-1特異的CD8+T細胞の頻度および合計数を増加したのに対し(図10BおよびC)、頻度が、ナイーブマウス由来の培養物において見られるものと同程度、およびバックグラウンドレベル内にとどまったIL-15およびIL-6によって、これらが増加することはなかった(不図示)。故に、IL-7、およびIL-2は、インビトロでのAg誘導細胞増殖を必要とすることなく、エクスビボでは検出できない腫瘍特異的CD8+T細胞を明らかにすることができる。
TS/A細胞は、自然には、免疫優勢エピトープが以前に記載された(AH-1、(19))、内因性MuLVのエンベロープタンパク質gp70を発現する。IL-7での培養が、希少なAg特異的CD8+T細胞の同定も助けるかどうかにさらに取り組むため、本発明者は、AH-1特異的CD8+T細胞反応を、Ag再刺激非存在下でIL-7、IL-2、IL-15およびIL-6を用いて、または用いずに培養した1週間後にエクスビボで比較した。リンパ球は、非パルスおよびAH-1パルス同系脾臓細胞による刺激時の、細胞内サイトカイン放出により分析した(図10)。腫瘍を有するマウスでは、サイトカイン産生細胞の頻度が、バックグラウンドレベル内にとどまり、ナイーブマウスにおいて見られるものと同程度であったため(不図示)、AH-1特異的CD8+T細胞はエクスビボでは検出できなかった(図10A)。7日後、TS/A-LACK腫瘍を有するLN由来の、組換えサイトカインを用いてまたは用いずに維持したLN培養物は、AH-1再刺激とは独立にIFN-γを産生する細胞の可変頻度を含有していた(図10B)。IL-7の存在下では、培養物は、Ag再刺激時にIFN-γのみ、またはIL-2およびIFN-γを産生することができるAH-1特異的CD8+T細胞が濃縮された(図10B)。IL-2もAH-1特異的CD8+T細胞の頻度および合計数を増加したのに対し(図10BおよびC)、頻度が、ナイーブマウス由来の培養物において見られるものと同程度、およびバックグラウンドレベル内にとどまったIL-15およびIL-6によって、これらが増加することはなかった(不図示)。故に、IL-7、およびIL-2は、インビトロでのAg誘導細胞増殖を必要とすることなく、エクスビボでは検出できない腫瘍特異的CD8+T細胞を明らかにすることができる。
インターロイキン-7は、記憶CD4+T細胞の選択的増殖に対しシクロスポリンA感受性シグナルとの相乗作用をもたらす
インビボでの記憶CD4+T細胞の増殖および生存は、IL-7およびTCR誘導イベントの両方に依存する(9)。インビトロでは、ヒト記憶T細胞のTCR誘導増殖は、インタクトなERK活性を必要とするのに対し、サイトカインに誘導された恒常的細胞分裂は、p38に依存し、シクロスポリンA(CsA)に非感受性である(13)。故に、本発明者は、中間記憶T細胞のIL-7誘導蓄積に関する要件を、ブロッキング抗体または選択されたシグナル伝達経路阻害剤の存在下でリンパ球を培養して分析した。細胞は、ナイーブマウスまたはTS/A-LACK腫瘍を有する16.2βマウス(かなりの頻度のLACK特異的ナイーブCD4+T細胞を有するトランスジェニックマウス(25))のLNから得て、CFSEで標識し、IL-7最適量、および指示された阻害剤の存在下で培養した。対照として、ナイーブT細胞を、選択した阻害剤の非存在下または存在下、LACKでパルスした抗原提示細胞(APC)により刺激した(図11A、B)。Ag誘導T細胞増殖は、抗MHCクラスII mAb、およびCsAにより有意に阻害され、抗ICAM-1、抗LFA-1 mAbにより部分的に阻害された(図11A、B)。予想通り、ラパマイシン(mTOR阻害剤)のみが、Ag誘導T細胞分裂を遅延させた(図11A、B)のに対し、SB202190(p38阻害剤)は、以前に報告された通り(13)、Ag刺激T細胞分裂を阻害しなかった。IL-7存在下では、対照ナイーブマウス由来培養物におけるCD4+T細胞画分は、1〜2回の緩徐な恒常的細胞分裂を行っていた(図11C、なし、細線)のに対し、急速増殖するCD4+ CFSEdim細胞集団は、腫瘍所属LN由来培養物において蓄積した(図11C、なし、太線)。この場合もやはり、IFN-γ(図11E)およびIL-2(不図示)を放出可能なLACK特異的T細胞は、主に急速増殖CD4 T細胞で見られた。抗MHCクラスII mAbの、腫瘍所属LNのIL-7誘導培養物への添加は、急速増殖するCD4+ CFSEdim細胞の蓄積に若干の効果があり(図11D、太線)、LACK特異的IFN-γ産生細胞の頻度を50%低下させた(図11E)。抗ICAM-1、またはLFA-1 mAb、およびCsAは、急速分裂するCFSEdim CD4+T細胞、およびLACK特異的IFN-γ産生細胞のIL-7誘導蓄積を阻害し、IL-7誘導恒常的細胞分裂を変化させることなかった(図11D、太線および図11E)。SB202190、またはPP2のみが、急速増殖するLACK特異的CD4+TのIL-7誘導蓄積を部分的に阻害したのに対し(図11D、E)、ラパマイシンは、IL-7に誘導された緩徐および急速な細胞分裂(図11D、太線)、およびLACK特異的T細胞のIL-7誘導蓄積を完全に阻害した(図11E)。
インビボでの記憶CD4+T細胞の増殖および生存は、IL-7およびTCR誘導イベントの両方に依存する(9)。インビトロでは、ヒト記憶T細胞のTCR誘導増殖は、インタクトなERK活性を必要とするのに対し、サイトカインに誘導された恒常的細胞分裂は、p38に依存し、シクロスポリンA(CsA)に非感受性である(13)。故に、本発明者は、中間記憶T細胞のIL-7誘導蓄積に関する要件を、ブロッキング抗体または選択されたシグナル伝達経路阻害剤の存在下でリンパ球を培養して分析した。細胞は、ナイーブマウスまたはTS/A-LACK腫瘍を有する16.2βマウス(かなりの頻度のLACK特異的ナイーブCD4+T細胞を有するトランスジェニックマウス(25))のLNから得て、CFSEで標識し、IL-7最適量、および指示された阻害剤の存在下で培養した。対照として、ナイーブT細胞を、選択した阻害剤の非存在下または存在下、LACKでパルスした抗原提示細胞(APC)により刺激した(図11A、B)。Ag誘導T細胞増殖は、抗MHCクラスII mAb、およびCsAにより有意に阻害され、抗ICAM-1、抗LFA-1 mAbにより部分的に阻害された(図11A、B)。予想通り、ラパマイシン(mTOR阻害剤)のみが、Ag誘導T細胞分裂を遅延させた(図11A、B)のに対し、SB202190(p38阻害剤)は、以前に報告された通り(13)、Ag刺激T細胞分裂を阻害しなかった。IL-7存在下では、対照ナイーブマウス由来培養物におけるCD4+T細胞画分は、1〜2回の緩徐な恒常的細胞分裂を行っていた(図11C、なし、細線)のに対し、急速増殖するCD4+ CFSEdim細胞集団は、腫瘍所属LN由来培養物において蓄積した(図11C、なし、太線)。この場合もやはり、IFN-γ(図11E)およびIL-2(不図示)を放出可能なLACK特異的T細胞は、主に急速増殖CD4 T細胞で見られた。抗MHCクラスII mAbの、腫瘍所属LNのIL-7誘導培養物への添加は、急速増殖するCD4+ CFSEdim細胞の蓄積に若干の効果があり(図11D、太線)、LACK特異的IFN-γ産生細胞の頻度を50%低下させた(図11E)。抗ICAM-1、またはLFA-1 mAb、およびCsAは、急速分裂するCFSEdim CD4+T細胞、およびLACK特異的IFN-γ産生細胞のIL-7誘導蓄積を阻害し、IL-7誘導恒常的細胞分裂を変化させることなかった(図11D、太線および図11E)。SB202190、またはPP2のみが、急速増殖するLACK特異的CD4+TのIL-7誘導蓄積を部分的に阻害したのに対し(図11D、E)、ラパマイシンは、IL-7に誘導された緩徐および急速な細胞分裂(図11D、太線)、およびLACK特異的T細胞のIL-7誘導蓄積を完全に阻害した(図11E)。
これらをまとめた所見は、IL-7は、接着分子および/または自己ペプチド/MHC相互作用により媒介される可能性のある、細胞誘導CsA感受性シグナルとの相乗作用により、急速増殖するIL-2/IFN-γ+中間記憶CD4+T細胞の蓄積を誘導することができることを示している。
IL-7は、急速分裂する抹消血のヒトCD4記憶Tリンパ球の選択的蓄積を維持する
IL-7およびIL-15が、セントラル記憶およびエフェクターヒト記憶T細胞の緩徐な恒常的細胞分裂を維持することは、以前に報告されている(13)。本発明者は、高密度の培養条件およびIL-7最適量が、代わりに、抹消血由来Tリンパ球のうち急速に分裂する中間体記憶T細胞集団を明らかにするかどうかを調べた。このため、PBMCを、健常ドナーから得て、CFSEで標識し、IL-7最適量の非存在下または存在下、異なる細胞密度で7日間培養した(図12)。低い細胞密度(24ウェルあたり106細胞/ml)、およびIL-7非存在下では、全細胞が増殖せず、当初のCFSE含量を保持した。高い細胞密度(24ウェルあたり5×106細胞/ml)では、培養7日目に4回を超える細胞分裂を行うことができる急速増殖CFSEdim CD4+T細胞の、小さいが測定可能な集団が、ウシ胎児血清(図12、なし、上のパネル)または自己血清(図13)において維持された対照培養物中に現れた。IL-7存在下では、より高いCD4 T細胞画分が増殖し、1〜6細胞分裂を終えていた(図12、IL-7、下のパネル)。5回を超える細胞周期を繰り返すことができる急速増殖細胞は、高細胞密度の培養物(図12B)、およびIL-7最適量(図14)で最も明らかになった。これらの急速分裂細胞は、これらの大部分が、PMAおよびイオノマイシン刺激時にIFN-γ(図15)およびIL-2(不図示)を産生したため、中間体記憶細胞を大半が含有していた。IL-2/IFN-γ+T細胞は、50ng/mlより上の濃度で頻度(図16)、および合計数(不図示)のいずれにおいても最もよく蓄積した。
IL-7およびIL-15が、セントラル記憶およびエフェクターヒト記憶T細胞の緩徐な恒常的細胞分裂を維持することは、以前に報告されている(13)。本発明者は、高密度の培養条件およびIL-7最適量が、代わりに、抹消血由来Tリンパ球のうち急速に分裂する中間体記憶T細胞集団を明らかにするかどうかを調べた。このため、PBMCを、健常ドナーから得て、CFSEで標識し、IL-7最適量の非存在下または存在下、異なる細胞密度で7日間培養した(図12)。低い細胞密度(24ウェルあたり106細胞/ml)、およびIL-7非存在下では、全細胞が増殖せず、当初のCFSE含量を保持した。高い細胞密度(24ウェルあたり5×106細胞/ml)では、培養7日目に4回を超える細胞分裂を行うことができる急速増殖CFSEdim CD4+T細胞の、小さいが測定可能な集団が、ウシ胎児血清(図12、なし、上のパネル)または自己血清(図13)において維持された対照培養物中に現れた。IL-7存在下では、より高いCD4 T細胞画分が増殖し、1〜6細胞分裂を終えていた(図12、IL-7、下のパネル)。5回を超える細胞周期を繰り返すことができる急速増殖細胞は、高細胞密度の培養物(図12B)、およびIL-7最適量(図14)で最も明らかになった。これらの急速分裂細胞は、これらの大部分が、PMAおよびイオノマイシン刺激時にIFN-γ(図15)およびIL-2(不図示)を産生したため、中間体記憶細胞を大半が含有していた。IL-2/IFN-γ+T細胞は、50ng/mlより上の濃度で頻度(図16)、および合計数(不図示)のいずれにおいても最もよく蓄積した。
IL-7に加え、IL-2およびIL-15も、他でも報告されているように(13)、ヒトCD4+T細胞のインビトロでの増殖(図17A)、およびBcl-2発現のアップレギュレーション(図18)を維持した。しかし、IL-2およびIL-15に反応して増殖する細胞のほとんどが、緩徐な恒常的細胞分裂を1〜4回終えており、急速増殖記憶T細胞が蓄積することはなかった。CD8+T細胞を、同じ培養物において分析した場合、IL-7は大半が、CD8+Tリンパ球の一部分の緩徐な恒常的分裂を支持したのに対し、IL-2およびIL-15は、急速分裂CD8+Tリンパ球集団の蓄積を可能にし(図17B)、2つの異なるT細胞サブセットにおけるこれらのサイトカインの類似した役割を示した。
抹消血ヒトTリンパ球のIL-7に誘導されたT細胞増殖は、シクロスポリンAに感受性である
ヒト記憶T細胞の、IL-7およびIL-15に誘導された緩徐な恒常的細胞分裂は、CsAに感受性であり、代わりに、p38依存性シグナル伝達に依存することが報告された(13)。
ヒト記憶T細胞の、IL-7およびIL-15に誘導された緩徐な恒常的細胞分裂は、CsAに感受性であり、代わりに、p38依存性シグナル伝達に依存することが報告された(13)。
本発明者は、次に、中間記憶急速増殖ヒトT細胞のIL-7誘導増殖に必要なシグナル伝達を調べた。マウス細胞の場合のように、急速増殖CD4+T細胞のIL-7誘導蓄積は、CsA、およびRAPA、および程度は低いもののSB(図19A)およびPP2(不図示)に感受性であった。CsAは、以前の報告(13)と一貫して、サイトカインに誘導された緩徐な増殖を妨げた。急速増殖記憶細胞の増殖も、抗LFA-1 mAbの添加が、これらの蓄積を阻害したため、LFA-1依存性相互作用に依存していた(図19B)。高細胞密度条件、およびIL-7誘導培養物において増殖する急速増殖CD4 T細胞は、CD45RA-、CD62L-Tリンパ球(エフェクター記憶)、およびCD45RA-、CD62L+(セントラル記憶)CD4 T細胞リンパ球に代表された。注目すべきは、自発的に増殖するCD4 T細胞の最大画分であると思われるCD45RA-、CD62L+記憶T細胞は、ほとんどが、エクスビボでの増殖プロトコルにより濃縮され、CsA阻害に対し最も感受性であった(図19BおよびC)。
故に、本発明者は、健常ドナーの抹消血において、自発的にインビトロで急速増殖することができるCD45RA-、CD62L+記憶CD4+T細胞集団を同定した。これらの記憶CD4+T細胞の蓄積は、IL-7により支持され、細胞密度依存性であり、CsA感受性であった。
IL-7誘導短期培養は、ヒト被験者におけるヒト型結核菌特異的CD4+T細胞の計測を助ける
IL-7が、インビトロで増殖するようにインビボでプログラムされる可能性のある急速増殖記憶T細胞の、インビトロでの増殖を維持することを測定したことから、本発明者は、これが、免疫関連病態の臨床研究に利用されうるかどうかを調べた。このため、ヒト型結核菌感染(TB)患者の凍結保存されたPBMC試料を、解凍時またはIL-7最適量で培養した1週間後に、MTP特異的ELISPOT分析により分析した(43)。患者は、臨床歴および急性TB徴候(臨床および培養による確定)、TSTに対する陽性反応、ならびにELISPOT-IFN-γアッセイでのMTPに対する反応能力をもとに選択した。非感染健常ドナー由来の凍結保存PBMCも、対照として分析した。Pt#1は、(凍結保存:950 SFC×106PBMCの)解凍時に、MTP特異的再刺激した際、かなりの数のIFN-γ+スポットを示した(図20A)。対照培地(なし)で細胞を培養すると、IFN-γ+MTP特異的細胞数は倍増し(1890 SFC×106PBMC)、培養細胞におけるCD4+ T細胞の頻度の増加を反映していた(図20Aの括弧内の数字)。IL-7存在下では、IFN-γ+MTP特異的細胞数(3930 SFC×106PBMC)は、凍結保存/解凍試料において見られるものより4倍多く、対照培養物と比較した場合は2倍多かった。Pt#2およびPt#3は、試料採取時に検出可能なMTP特異的T細胞を有していたが(不図示)、凍結保存/解凍試料には有していなかった(図20B、20C)。代わりに、IL-7誘導培養すると、IFN-γ+MTP特異的スポットが明らかになった。IL-7は、分析した全てのTB患者でMTP特異的T細胞の絶対数の増加をもたらし、この結果、健常ドナーおよびTB患者の間の、MTP特異的IFN-γ産生細胞数の差は、有意性を得た(図21)。IL-2に反応して蓄積するMTP特異的T細胞は、ほとんどが、MTP誘導細胞内サイトカイン分泌により測定される、IL-2/IFN-γ+記憶CD4 T細胞に代表された(図22)。さらに、MTP特異的IL-2/IFN-γ+記憶CD4 T細胞の増殖は、CsA存在下で防がれた(図23)。
IL-7が、インビトロで増殖するようにインビボでプログラムされる可能性のある急速増殖記憶T細胞の、インビトロでの増殖を維持することを測定したことから、本発明者は、これが、免疫関連病態の臨床研究に利用されうるかどうかを調べた。このため、ヒト型結核菌感染(TB)患者の凍結保存されたPBMC試料を、解凍時またはIL-7最適量で培養した1週間後に、MTP特異的ELISPOT分析により分析した(43)。患者は、臨床歴および急性TB徴候(臨床および培養による確定)、TSTに対する陽性反応、ならびにELISPOT-IFN-γアッセイでのMTPに対する反応能力をもとに選択した。非感染健常ドナー由来の凍結保存PBMCも、対照として分析した。Pt#1は、(凍結保存:950 SFC×106PBMCの)解凍時に、MTP特異的再刺激した際、かなりの数のIFN-γ+スポットを示した(図20A)。対照培地(なし)で細胞を培養すると、IFN-γ+MTP特異的細胞数は倍増し(1890 SFC×106PBMC)、培養細胞におけるCD4+ T細胞の頻度の増加を反映していた(図20Aの括弧内の数字)。IL-7存在下では、IFN-γ+MTP特異的細胞数(3930 SFC×106PBMC)は、凍結保存/解凍試料において見られるものより4倍多く、対照培養物と比較した場合は2倍多かった。Pt#2およびPt#3は、試料採取時に検出可能なMTP特異的T細胞を有していたが(不図示)、凍結保存/解凍試料には有していなかった(図20B、20C)。代わりに、IL-7誘導培養すると、IFN-γ+MTP特異的スポットが明らかになった。IL-7は、分析した全てのTB患者でMTP特異的T細胞の絶対数の増加をもたらし、この結果、健常ドナーおよびTB患者の間の、MTP特異的IFN-γ産生細胞数の差は、有意性を得た(図21)。IL-2に反応して蓄積するMTP特異的T細胞は、ほとんどが、MTP誘導細胞内サイトカイン分泌により測定される、IL-2/IFN-γ+記憶CD4 T細胞に代表された(図22)。さらに、MTP特異的IL-2/IFN-γ+記憶CD4 T細胞の増殖は、CsA存在下で防がれた(図23)。
IL-7誘導短期培養は、カンジダ抗原特異的ヒトTリンパ球の計測を助ける
MTP特異的T細胞に加え、本発明者は、IL-7が、カンジダアルビカンス由来抗原に特異的なTリンパ球の同定を増大することができるかどうかも調べた。このため、Pt#1由来のPBMCを、非パルスまたはC.アルビカンス由来Agでパルスした照射自己PBMCで行ったELISPOTアッセイにより、解凍時または単純培地もしくはIL-7存在下での培養7日後に分析した。M.結核菌特異的T細胞反応の場合のように、IFN-γを放出することができるC.アルビカンス特異的T細胞も、IL-7での短期培養により濃縮された(図24A、B)。
MTP特異的T細胞に加え、本発明者は、IL-7が、カンジダアルビカンス由来抗原に特異的なTリンパ球の同定を増大することができるかどうかも調べた。このため、Pt#1由来のPBMCを、非パルスまたはC.アルビカンス由来Agでパルスした照射自己PBMCで行ったELISPOTアッセイにより、解凍時または単純培地もしくはIL-7存在下での培養7日後に分析した。M.結核菌特異的T細胞反応の場合のように、IFN-γを放出することができるC.アルビカンス特異的T細胞も、IL-7での短期培養により濃縮された(図24A、B)。
IL-7/IL-15と共に培養した記憶T細胞による養子細胞療法は、インビボでの腫瘍増殖を遅延させる
IL-7が、インビボで初回刺激した記憶CD4 T細胞の蓄積を決定すること、IL-15が、CD8記憶T細胞の増殖を最もよく誘導することを測定したことから、本発明者は、増殖した集団が臨床的関連を有するかどうか評価した。この点に取り組むため、リンパ節を、TS/A-LACK腫瘍を有するマウスから得て、最適細胞密度(5×106細胞/ml)および最適サイトカイン量(50ng/ml)で7日間培養した。対照として対照マウス由来のナイーブT細胞も、同じ条件で培養した。その後、CD4およびCD8 T細胞の同程度の頻度を有する107培養細胞を、ナイーブBALB/cマウスに養子移入した。48時間後、マウスを300,000TS/A-LACK細胞で曝露し、腫瘍増殖を経時的にモニターした。図25に示された通り、TS/A-LACK腫瘍は、対照マウスおよびサイトカインで処置したナイーブT細胞を養子移入したマウスにおいて急速に発症した。反対に、腫瘍増殖は、サイトカインで処置した腫瘍を有するマウス由来のT細胞を養子移入したマウスにおいて、有意に遅延した(図25)。これは、IL-7/IL-15由来培養物が、臨床的関連のある記憶T細胞集団の増殖を決定したことを示すものである。
IL-7が、インビボで初回刺激した記憶CD4 T細胞の蓄積を決定すること、IL-15が、CD8記憶T細胞の増殖を最もよく誘導することを測定したことから、本発明者は、増殖した集団が臨床的関連を有するかどうか評価した。この点に取り組むため、リンパ節を、TS/A-LACK腫瘍を有するマウスから得て、最適細胞密度(5×106細胞/ml)および最適サイトカイン量(50ng/ml)で7日間培養した。対照として対照マウス由来のナイーブT細胞も、同じ条件で培養した。その後、CD4およびCD8 T細胞の同程度の頻度を有する107培養細胞を、ナイーブBALB/cマウスに養子移入した。48時間後、マウスを300,000TS/A-LACK細胞で曝露し、腫瘍増殖を経時的にモニターした。図25に示された通り、TS/A-LACK腫瘍は、対照マウスおよびサイトカインで処置したナイーブT細胞を養子移入したマウスにおいて急速に発症した。反対に、腫瘍増殖は、サイトカインで処置した腫瘍を有するマウス由来のT細胞を養子移入したマウスにおいて、有意に遅延した(図25)。これは、IL-7/IL-15由来培養物が、臨床的関連のある記憶T細胞集団の増殖を決定したことを示すものである。
遺伝子改変セントラル記憶ヒトT細胞の産生
細胞増殖は、Tリンパ球のレトロウイルス導入に必要とされる。本発明者は、aCD3またはbaCD3/CD28でPBMCを活性化した。細胞を、baCD3/CD28で活性化しIL-7およびIL-15と共に培養、またはaCD3で活性化しIL-2と共に培養した(図27A)。細胞に、SFCMM3レトロウイルス性ベクターを、スピノキュレーションにより2日目に導入した。導入は、同時に同じプロトコルに従って実施した。IL-7およびIL-15存在下でのbaCD3/CD28による活性化は、IL-2存在下でのaCD3による活性化よりも高いT細胞増殖を促し(図27A)、高い導入効率をもたらした(図27B)。さらに、培養期間の最後に、生理学的CD4/CD8比を分析し、aCD3で活性化しIL-2と共に培養した細胞よりも、baCD3/CD28で活性化しIL-7およびIL-15と共に培養した導入細胞でよく維持されることを見出した(図27C)。
細胞増殖は、Tリンパ球のレトロウイルス導入に必要とされる。本発明者は、aCD3またはbaCD3/CD28でPBMCを活性化した。細胞を、baCD3/CD28で活性化しIL-7およびIL-15と共に培養、またはaCD3で活性化しIL-2と共に培養した(図27A)。細胞に、SFCMM3レトロウイルス性ベクターを、スピノキュレーションにより2日目に導入した。導入は、同時に同じプロトコルに従って実施した。IL-7およびIL-15存在下でのbaCD3/CD28による活性化は、IL-2存在下でのaCD3による活性化よりも高いT細胞増殖を促し(図27A)、高い導入効率をもたらした(図27B)。さらに、培養期間の最後に、生理学的CD4/CD8比を分析し、aCD3で活性化しIL-2と共に培養した細胞よりも、baCD3/CD28で活性化しIL-7およびIL-15と共に培養した導入細胞でよく維持されることを見出した(図27C)。
Tリンパ球のレトロウイルス導入に必要とされる多クローン活性化は、記憶細胞を濃縮する
レトロウイルス性ベクターを導入したヒトT細胞における記憶サブセットの相対分布を測定するため、本発明者は、CD45RA/CD62L共発現を分析した。14日目、aCD3で活性化しIL-2と共に培養した導入T細胞は、主にCD45RA-CD62L-、すなわちエフェクター記憶細胞であった。反対に、baCD3/CD28で活性化しIL-7およびIL-15と共に培養した導入CD4+T細胞は、CD45RA-CD62L+、すなわちセントラル記憶細胞が高度に濃縮されていた(図28A)。baCD3/CD28により活性化しIL-7およびIL-15と共に培養した細胞の場合、CD45RA-CD62L+セントラル記憶細胞の濃縮は、導入CD8+細胞についても観察された(図28B)。記憶表現型の定義を改善するため、本発明者は、CD28/CD27共発現も分析した。導入CD4+細胞では、2つの条件間でサブセットの相対分布に差はなかったが(図28C)、導入CD8+細胞では、baCD3/CD28による活性化ならびにIL-7およびIL-15による培養は、CD28+CD27+T細胞を促進した(図28D)。
レトロウイルス性ベクターを導入したヒトT細胞における記憶サブセットの相対分布を測定するため、本発明者は、CD45RA/CD62L共発現を分析した。14日目、aCD3で活性化しIL-2と共に培養した導入T細胞は、主にCD45RA-CD62L-、すなわちエフェクター記憶細胞であった。反対に、baCD3/CD28で活性化しIL-7およびIL-15と共に培養した導入CD4+T細胞は、CD45RA-CD62L+、すなわちセントラル記憶細胞が高度に濃縮されていた(図28A)。baCD3/CD28により活性化しIL-7およびIL-15と共に培養した細胞の場合、CD45RA-CD62L+セントラル記憶細胞の濃縮は、導入CD8+細胞についても観察された(図28B)。記憶表現型の定義を改善するため、本発明者は、CD28/CD27共発現も分析した。導入CD4+細胞では、2つの条件間でサブセットの相対分布に差はなかったが(図28C)、導入CD8+細胞では、baCD3/CD28による活性化ならびにIL-7およびIL-15による培養は、CD28+CD27+T細胞を促進した(図28D)。
遺伝子改変セントラル記憶ヒトT細胞の機能的相関
表面表現型により同定されるセントラルおよびエフェクター記憶ヒトTリンパ球は、エフェクターサイトカインを産生する能力に差がある。14日目、本発明者は、2つの遺伝子改変T細胞集団を再刺激し、サイトカイン産生についてこれらを分析した。aCD3で活性化しIL-2と共に培養したCD4+T細胞は、プロトタイプのエフェクターサイトカインIFN-γを効率的に産生した。著しく対照的に、baCD3/CD28、IL-7およびIL-15で刺激したCD4+T細胞の大多数は、非極性化細胞であり、IFN-γもIL-4も産生しなかった(図29A)。類似の結果は、CD8+T細胞で得られた(図29B)。
表面表現型により同定されるセントラルおよびエフェクター記憶ヒトTリンパ球は、エフェクターサイトカインを産生する能力に差がある。14日目、本発明者は、2つの遺伝子改変T細胞集団を再刺激し、サイトカイン産生についてこれらを分析した。aCD3で活性化しIL-2と共に培養したCD4+T細胞は、プロトタイプのエフェクターサイトカインIFN-γを効率的に産生した。著しく対照的に、baCD3/CD28、IL-7およびIL-15で刺激したCD4+T細胞の大多数は、非極性化細胞であり、IFN-γもIL-4も産生しなかった(図29A)。類似の結果は、CD8+T細胞で得られた(図29B)。
遺伝子改変セントラル記憶ヒトT細胞のGvHD能
さまざまな異種移植モデルが、ヒトリンパ球により誘導されるGvHDを研究するため提案されている。インビボでの2種類の自殺遺伝子改変セントラルおよびエフェクター記憶Tリンパ球の、相対的な抗宿主反応性を評価するため、これらの集団を、非致死的照射および抗NK抗体で馴化したNOD/scidマウスに注入した。対照マウスには、ヒト精製同系PBLを注入した。本発明者は、セントラル記憶遺伝子改変リンパ球が、このエフェクター記憶対応物よりも生着に有効であることを観察した(1週間目のヒトキメラ化:エフェクター記憶遺伝子改変細胞では平均0.45%、範囲0.2〜1.1vsセントラル記憶遺伝子改変細胞では平均4.5%、範囲4.1〜5.2、表2)。エフェクター記憶遺伝子改変T細胞を注入したマウス6例中5例が、1週間後にヒトキメラ化の低下を示し、GvHDを示さなかった。一方、持続的ヒトキメラ化は、セントラル記憶自殺遺伝子改変T細胞を注入したマウスの大半で観察され、6例中4マウスが重度のGvHDを発症した。
さまざまな異種移植モデルが、ヒトリンパ球により誘導されるGvHDを研究するため提案されている。インビボでの2種類の自殺遺伝子改変セントラルおよびエフェクター記憶Tリンパ球の、相対的な抗宿主反応性を評価するため、これらの集団を、非致死的照射および抗NK抗体で馴化したNOD/scidマウスに注入した。対照マウスには、ヒト精製同系PBLを注入した。本発明者は、セントラル記憶遺伝子改変リンパ球が、このエフェクター記憶対応物よりも生着に有効であることを観察した(1週間目のヒトキメラ化:エフェクター記憶遺伝子改変細胞では平均0.45%、範囲0.2〜1.1vsセントラル記憶遺伝子改変細胞では平均4.5%、範囲4.1〜5.2、表2)。エフェクター記憶遺伝子改変T細胞を注入したマウス6例中5例が、1週間後にヒトキメラ化の低下を示し、GvHDを示さなかった。一方、持続的ヒトキメラ化は、セントラル記憶自殺遺伝子改変T細胞を注入したマウスの大半で観察され、6例中4マウスが重度のGvHDを発症した。
遺伝子改変セントラル記憶ヒトT細胞の産生
臨床適用に適した幾つかの遺伝子改変セントラル記憶ヒトT細胞を得るための最低要件を決めるため、本発明者は、以下の5つのT細胞導入条件を比較した:
1.可溶性抗CD3抗体(OKT-3)+高用量のインターロイキン2(600UI/ml)、
2.サイトカインなしの抗CD3/CD28細胞大ビーズ、
3.抗CD3/CD28細胞大ビーズ+低用量のIL-2(200IU/ml)、
4.抗CD3/CD28細胞大ビーズ+低用量のIL-7(5ng/ml)、
5.抗CD3/CD28細胞大ビーズ+低用量のIL-7(5ng/ml)+低用量のIL-15(5ng/ml)。
臨床適用に適した幾つかの遺伝子改変セントラル記憶ヒトT細胞を得るための最低要件を決めるため、本発明者は、以下の5つのT細胞導入条件を比較した:
1.可溶性抗CD3抗体(OKT-3)+高用量のインターロイキン2(600UI/ml)、
2.サイトカインなしの抗CD3/CD28細胞大ビーズ、
3.抗CD3/CD28細胞大ビーズ+低用量のIL-2(200IU/ml)、
4.抗CD3/CD28細胞大ビーズ+低用量のIL-7(5ng/ml)、
5.抗CD3/CD28細胞大ビーズ+低用量のIL-7(5ng/ml)+低用量のIL-15(5ng/ml)。
細胞は、材料および方法に記載されたプロトコルに従って導入し、培養した。これらの実験は、以下の結果を生んだ。
1.抗CD3/CD28ビーズによる活性化は、可溶性抗CD3抗体による活性化よりも高い導入効率を可能にする
図30Aに示された通り、レトロウイルス導入効率は、可溶性抗CD3抗体による刺激後よりも細胞大電磁ビーズによる刺激後に有意に高かった。この結果は、培養カクテルでのサイトカインの使用とは無関係であった。
図30Aに示された通り、レトロウイルス導入効率は、可溶性抗CD3抗体による刺激後よりも細胞大電磁ビーズによる刺激後に有意に高かった。この結果は、培養カクテルでのサイトカインの使用とは無関係であった。
2.CD3/CD28ビーズによる活性化に続くサイトカイン(IL2、IL7+IL15またはIL7)存在下での培養は、導入Tリンパ球の生理学的CD4/CD8比を維持する
本発明者の導入プロトコルの、生理学的CD4/CD8比を維持する能力を評価するため、本発明者は、初回刺激後10日目に、異なるプロトコルで作製した導入細胞のCD4/CD8比を分析した。図30Bに示された通り、電磁ビーズによる刺激に続くサイトカイン(IL2、IL7またはIL7+IL15)での培養のみが、導入T細胞の生理学的CD4/CD8比を維持したのに対し、他の培養条件で観察された平均CD4/CD8比は、1/1を超えることはなかった。
本発明者の導入プロトコルの、生理学的CD4/CD8比を維持する能力を評価するため、本発明者は、初回刺激後10日目に、異なるプロトコルで作製した導入細胞のCD4/CD8比を分析した。図30Bに示された通り、電磁ビーズによる刺激に続くサイトカイン(IL2、IL7またはIL7+IL15)での培養のみが、導入T細胞の生理学的CD4/CD8比を維持したのに対し、他の培養条件で観察された平均CD4/CD8比は、1/1を超えることはなかった。
3.CD3/CD28ビーズによる活性化に続くサイトカイン(IL2、IL7+IL15またはIL7)存在下での培養は、他の培養条件よりも導入細胞の有意に高い増殖率を誘導する
臨床適用のためにデザインされたエクスビボでの遺伝子導入プロトコルは、実現可能性に関する関連基準を満たさなければならない。遺伝子療法アプローチの臨床解釈における主な実現可能性問題の1つは、インビトロでの細胞数および細胞発現に関する。図30Cに示された通り、統計的有意差は、他の条件(ビーズ単独およびOKT3+IL-2)と比べて、ビーズで刺激し、サイトカインで培養した遺伝子改変細胞の間で得られた細胞数において観察された。これらの結果は、抗CD3/CD28結合ビーズにより得られ、IL7、IL7+IL15またはIL2と共に培養された導入リンパ球が、臨床適用に適した数までインビトロで急速に増殖することを示している。
臨床適用のためにデザインされたエクスビボでの遺伝子導入プロトコルは、実現可能性に関する関連基準を満たさなければならない。遺伝子療法アプローチの臨床解釈における主な実現可能性問題の1つは、インビトロでの細胞数および細胞発現に関する。図30Cに示された通り、統計的有意差は、他の条件(ビーズ単独およびOKT3+IL-2)と比べて、ビーズで刺激し、サイトカインで培養した遺伝子改変細胞の間で得られた細胞数において観察された。これらの結果は、抗CD3/CD28結合ビーズにより得られ、IL7、IL7+IL15またはIL2と共に培養された導入リンパ球が、臨床適用に適した数までインビトロで急速に増殖することを示している。
4.抗CD3/CD28ビーズによる活性化は、主にセントラル記憶CD8+およびCD4+導入リンパ球を産生する
本発明者は、異なる培養条件で得られた導入細胞の免疫表現型を、初回刺激後10日目にCD3、CD4、CD8、CD45RAおよびCD62LについてFACS染色を介して調べた。本発明者は、抗CD3/CD28ビーズで刺激されたCD8+およびCD4+T両細胞のきわめて高い画分(約80%)が、CD45RA-CD62L+であることを観察した。このパターンは、セントラル記憶Tリンパ球と一致するものである。反対に、OKT3で刺激されたT細胞は、60% CD8+および45% CD4+エフェクター記憶T細胞(CD45RA-CD62L-)対30% CD8+および50% CD4+セントラル記憶TK+リンパ球を示した(図31)。
本発明者は、異なる培養条件で得られた導入細胞の免疫表現型を、初回刺激後10日目にCD3、CD4、CD8、CD45RAおよびCD62LについてFACS染色を介して調べた。本発明者は、抗CD3/CD28ビーズで刺激されたCD8+およびCD4+T両細胞のきわめて高い画分(約80%)が、CD45RA-CD62L+であることを観察した。このパターンは、セントラル記憶Tリンパ球と一致するものである。反対に、OKT3で刺激されたT細胞は、60% CD8+および45% CD4+エフェクター記憶T細胞(CD45RA-CD62L-)対30% CD8+および50% CD4+セントラル記憶TK+リンパ球を示した(図31)。
導入リンパ球培養間のγ鎖受容体発現
サイトカイン受容体発現は、T細胞刺激の間、厳重に制御されている。本発明者は、T細胞の機能および能力の指標として、異なるT細胞培養および導入プロトコル間のγ鎖サイトカイン受容体の発現動態を分析した。このため、本発明者は、初回刺激後異なる時点で、CD122(IL-2/15受容体共通β鎖)、CD25(IL-2受容体α鎖)およびCD127(IL-7受容体α鎖)に対する蛍光色素標識抗体で導入細胞を染色した後に、細胞蛍光分析を行った。
サイトカイン受容体発現は、T細胞刺激の間、厳重に制御されている。本発明者は、T細胞の機能および能力の指標として、異なるT細胞培養および導入プロトコル間のγ鎖サイトカイン受容体の発現動態を分析した。このため、本発明者は、初回刺激後異なる時点で、CD122(IL-2/15受容体共通β鎖)、CD25(IL-2受容体α鎖)およびCD127(IL-7受容体α鎖)に対する蛍光色素標識抗体で導入細胞を染色した後に、細胞蛍光分析を行った。
1.IL-2/IL-15受容体β鎖(CD122)発現は、異なる導入プロトコル間で変化することはない
免疫応答の過程で、IL-2/IL-15受容体β発現は、T細胞活性化後に増加し、次いで、記憶-細胞分裂相を通じて保持される中間レベルの発現まで減少する(13)。
免疫応答の過程で、IL-2/IL-15受容体β発現は、T細胞活性化後に増加し、次いで、記憶-細胞分裂相を通じて保持される中間レベルの発現まで減少する(13)。
図32は、培養13日間の導入リンパ球におけるCD122の発現動態を示す。本発明者は、5つのプロトコル間で培養されたT細胞におけるCD122発現に関し、いかなる違いも観察しなかった。全てのケースで、T細胞は、および特にCD4+細胞は、活性化後CD122をアップレギュレートし、約4日目にピークに達し、ほぼ100%の遺伝子改変細胞が分子を発現した。次いで、細胞はCD122発現を徐々にダウンレギュレートし、T細胞培養開始後13日目に刺激前に観察されたのと同じ発現レベルに達し、細胞は休止期に達した。
2.CD3/CD28ビーズによる刺激は、導入T細胞におけるIL-2受容体α発現を強化し延長させる
IL-2受容体α(CD25)は、Tリンパ球に関連のある活性化マーカーである。生理学的状態では、ナイーブT細胞はCD25を発現しないが、T細胞活性化によりこの発現は急速にアップレギュレートされ、通常は反応の増殖ピーク前に減少する。
IL-2受容体α(CD25)は、Tリンパ球に関連のある活性化マーカーである。生理学的状態では、ナイーブT細胞はCD25を発現しないが、T細胞活性化によりこの発現は急速にアップレギュレートされ、通常は反応の増殖ピーク前に減少する。
ビーズにより活性化した導入細胞では、その後の培養条件とは関係なく、フローサイトメトリー分析は、刺激後2日目にT細胞の大半でIL-2受容体α発現が有意に増加したことを示した。反対に、可溶性OKT3で活性化した導入細胞の40%のみが、受容体をアップレギュレートした(図33)。これは、T細胞の大半が、この刺激法では適切に活性化されなかったことを示すものである。CD25が発現される限り、T細胞は、IL-2に対して増殖することができるため、免疫応答に積極的に参加する。ビーズ活性化後に産生された導入細胞では、CD25発現は、最大13日まで細胞の大半で高いままであった。反対に、可溶性抗CD3抗体による刺激後にCD25をアップレギュレートした細胞は、ビーズで活性化した細胞より2日遅れて発現のピークに達し、次いでCD25分子を急速にダウンレギュレートした。
3.抗CD3/CD28ビーズ+IL-7で活性化した導入細胞は、長期生存記憶T細胞のマーカーである、IL-7受容体αの最大発現を示す
生理学的状態では、IL-7受容体α(CD127)は、ナイーブT細胞により構成的に発現される。この発現は、T細胞活性化によりダウンレギュレートされ(CD25と比べて鏡面的なかたちで)、このようなダウンレギュレーションが、細胞死を促進している可能性がある。反対に、CD127発現は、免疫応答が進むにつれて増加し、記憶T細胞において高レベルに達する。IL-7は、記憶Tリンパ球の強力な生存因子であり、IL-7による受容体の誘発が、T細胞の生存および増殖を促進し、さまざまな細胞内シグナル経路を介したアポトーシスから細胞を保護する。本発明者は、本発明者の導入細胞におけるCD127発現動態を分析した。IL-7受容体αは、刺激後強いダウンレギュレーションを起こした(約1日目〜6日目)。7日目から、この発現は徐々に増加し、興味深いことに、本発明者は、他の全ての条件と比較して、ビーズCD3/CD28およびIL-7により得られたCD127+導入細胞の割合に有意差を観察した(図34)。9日目から、細胞大ビーズおよびIL-7で刺激した、CD8+およびCD4+ TK+両細胞の80%超が、IL-7受容体α陽性であった。これは、IL-7が、活性化後、T細胞の大半で高レベルのD127発現の維持を担っており、故に、独占的な長期生存の利点を導入細胞にもたらすことを示唆するものである。
生理学的状態では、IL-7受容体α(CD127)は、ナイーブT細胞により構成的に発現される。この発現は、T細胞活性化によりダウンレギュレートされ(CD25と比べて鏡面的なかたちで)、このようなダウンレギュレーションが、細胞死を促進している可能性がある。反対に、CD127発現は、免疫応答が進むにつれて増加し、記憶T細胞において高レベルに達する。IL-7は、記憶Tリンパ球の強力な生存因子であり、IL-7による受容体の誘発が、T細胞の生存および増殖を促進し、さまざまな細胞内シグナル経路を介したアポトーシスから細胞を保護する。本発明者は、本発明者の導入細胞におけるCD127発現動態を分析した。IL-7受容体αは、刺激後強いダウンレギュレーションを起こした(約1日目〜6日目)。7日目から、この発現は徐々に増加し、興味深いことに、本発明者は、他の全ての条件と比較して、ビーズCD3/CD28およびIL-7により得られたCD127+導入細胞の割合に有意差を観察した(図34)。9日目から、細胞大ビーズおよびIL-7で刺激した、CD8+およびCD4+ TK+両細胞の80%超が、IL-7受容体α陽性であった。これは、IL-7が、活性化後、T細胞の大半で高レベルのD127発現の維持を担っており、故に、独占的な長期生存の利点を導入細胞にもたらすことを示唆するものである。
ビーズ+IL-7により産生された導入リンパ球は、最も高い自己反応性能を有する
以前に示された結果から、抗CD3/CD28電磁ビーズによる活性化およびサイトカイン(特にIL-7)のアジュバント効果は、高い生存能を有する、セントラル記憶導入Tリンパ球の産生にとって重要な因子であることが分かっている。本発明者の次なる目的は、これらのCM導入Tリンパ球が、実際に、強力で有効な免疫応答を引き出すことができるかどうかを調べることであった。本発明者は、導入細胞を同種抗原で刺激して、インビトロでこの問題に取り組み、以下の結果を得た。
以前に示された結果から、抗CD3/CD28電磁ビーズによる活性化およびサイトカイン(特にIL-7)のアジュバント効果は、高い生存能を有する、セントラル記憶導入Tリンパ球の産生にとって重要な因子であることが分かっている。本発明者の次なる目的は、これらのCM導入Tリンパ球が、実際に、強力で有効な免疫応答を引き出すことができるかどうかを調べることであった。本発明者は、導入細胞を同種抗原で刺激して、インビトロでこの問題に取り組み、以下の結果を得た。
1.ビーズ+IL-7により産生された導入リンパ球は、同種抗原で刺激した場合、最も高い増殖能を有する
各5つの条件により産生された導入T細胞を、CFSEで染色し、照射した同種PBMCで共培養した。1週間後、本発明者は、細胞数を計数し、FACSによるCFSE希釈を分析して分裂細胞の割合を評価した。図35Aに示された通り、本発明者は、CD3+およびCD8+T細胞において、IL-7含有条件と他のプロトコルとの間に統計的有意差を見出した。実際、ビーズCD3/CD28+IL-7により産生された導入細胞の高い割合(40%)が、同種刺激後1週間で分裂した。反対に、OKT3-により産生された導入細胞の20%のみが、同じ培養条件で分裂した。
各5つの条件により産生された導入T細胞を、CFSEで染色し、照射した同種PBMCで共培養した。1週間後、本発明者は、細胞数を計数し、FACSによるCFSE希釈を分析して分裂細胞の割合を評価した。図35Aに示された通り、本発明者は、CD3+およびCD8+T細胞において、IL-7含有条件と他のプロトコルとの間に統計的有意差を見出した。実際、ビーズCD3/CD28+IL-7により産生された導入細胞の高い割合(40%)が、同種刺激後1週間で分裂した。反対に、OKT3-により産生された導入細胞の20%のみが、同じ培養条件で分裂した。
2.ビーズ+IL-7により産生された導入リンパ球は、死に対する感受性が低い
Tリンパ球の恒常性を維持するため、同種抗原曝露に反応した大量のT細胞活性化は、通常、大規模なアポトーシスプログラムが続いて起こり、いわゆる「活性化誘導細胞死」(AICD)においてIL-2が中心的な役割を果たす。初回免疫応答後の有効で長期にわたる免疫記憶を開発するには、AICDに対抗するメカニズムが必要とされる。導入細胞のAICDに対する感受性を調べるため、本発明者は、死細胞に選択的に結合する蛍光色素、To-Pro 3により同種刺激CFSE+リンパ球を染色した。本発明者は、活性化誘導細胞死およびネグレクトによる死をそれぞれ評価するため、分裂および非分裂導入細胞集団における死細胞数を算出した(図35B)。IL-2において(活性化シグナルとは無関係に)培養した導入細胞は、AICDおよびネグレクトによる死に対し高い感受性をもたらした。反対に、ビーズCD3/CD28およびIL-7により産生された導入細胞は、未修飾リンパ球の死亡率と同程度の最も低い死亡率を示した。この観察は、ビーズCD3/CD28+IL-7により産生された遺伝子改変リンパ球の高い割合(CD8+の33%、CD4+の52%)におけるCD127の持続的発現と関係している可能性がある。
Tリンパ球の恒常性を維持するため、同種抗原曝露に反応した大量のT細胞活性化は、通常、大規模なアポトーシスプログラムが続いて起こり、いわゆる「活性化誘導細胞死」(AICD)においてIL-2が中心的な役割を果たす。初回免疫応答後の有効で長期にわたる免疫記憶を開発するには、AICDに対抗するメカニズムが必要とされる。導入細胞のAICDに対する感受性を調べるため、本発明者は、死細胞に選択的に結合する蛍光色素、To-Pro 3により同種刺激CFSE+リンパ球を染色した。本発明者は、活性化誘導細胞死およびネグレクトによる死をそれぞれ評価するため、分裂および非分裂導入細胞集団における死細胞数を算出した(図35B)。IL-2において(活性化シグナルとは無関係に)培養した導入細胞は、AICDおよびネグレクトによる死に対し高い感受性をもたらした。反対に、ビーズCD3/CD28およびIL-7により産生された導入細胞は、未修飾リンパ球の死亡率と同程度の最も低い死亡率を示した。この観察は、ビーズCD3/CD28+IL-7により産生された遺伝子改変リンパ球の高い割合(CD8+の33%、CD4+の52%)におけるCD127の持続的発現と関係している可能性がある。
この観察により、細胞死に対する高感受性をもたらす、ビーズCD3/CD28およびIL-2により産生された導入リンパ球は、CD127+細胞発現細胞の最も低い割合を示した(30%)。図36AのFACSプロットは、同種刺激後10日目のCFSE染色導入Tリンパ球におけるCD127検出の代表例を示す。
3.ビーズ+IL-7により産生された導入リンパ球は、同種刺激後にセントラル記憶表現型を維持する
免疫記憶は、抗原再遭遇時に、分裂し、病原体を直接除去することのできるエフェクター、および宿主を長期に防御することのできる記憶細胞の両方を産生する記憶細胞の、自己再生能により確保される。セントラル記憶遺伝子改変リンパ球が、この自己再生能を有するかどうかを検証するため、本発明者は、CFSE染色細胞におけるCCR7(セントラル記憶細胞マーカー)発現を、同種刺激後10日目に分析した。図36Aに示された通り、本発明者のデータは、ビーズCD3/CD28およびIL-7により産生されたCD3+遺伝子改変リンパ球(少なくとも1回分裂している)の59%が、CCR7発現陽性であったことを示している。反対に、OKT3により産生されたCD3+遺伝子改変リンパ球の17%のみが、CCR7を発現した。IL-7の存在下での培養は、ビーズCD3/CD28およびIL-2により産生されたCD3+遺伝子改変リンパ球の36%しか、同種刺激後のCCR7発現を維持しないため、セントラル記憶リンパ球の自己再生能の維持に不可欠であった。この結果により、初回刺激に使用したのと同じ培養条件に従って、細胞を、同じ同種刺激因子によりインビトロで再曝露した場合、CD3/CD28ビーズおよびIL-7により産生された導入細胞の70%超、およびOKT3により産生された導入細胞の50%のみが、翌週に増殖した(図36B)。
免疫記憶は、抗原再遭遇時に、分裂し、病原体を直接除去することのできるエフェクター、および宿主を長期に防御することのできる記憶細胞の両方を産生する記憶細胞の、自己再生能により確保される。セントラル記憶遺伝子改変リンパ球が、この自己再生能を有するかどうかを検証するため、本発明者は、CFSE染色細胞におけるCCR7(セントラル記憶細胞マーカー)発現を、同種刺激後10日目に分析した。図36Aに示された通り、本発明者のデータは、ビーズCD3/CD28およびIL-7により産生されたCD3+遺伝子改変リンパ球(少なくとも1回分裂している)の59%が、CCR7発現陽性であったことを示している。反対に、OKT3により産生されたCD3+遺伝子改変リンパ球の17%のみが、CCR7を発現した。IL-7の存在下での培養は、ビーズCD3/CD28およびIL-2により産生されたCD3+遺伝子改変リンパ球の36%しか、同種刺激後のCCR7発現を維持しないため、セントラル記憶リンパ球の自己再生能の維持に不可欠であった。この結果により、初回刺激に使用したのと同じ培養条件に従って、細胞を、同じ同種刺激因子によりインビトロで再曝露した場合、CD3/CD28ビーズおよびIL-7により産生された導入細胞の70%超、およびOKT3により産生された導入細胞の50%のみが、翌週に増殖した(図36B)。
ビーズ+IL-7により産生された導入細胞は、インビボで最も高い自己反応性能を有する
インビボでのセントラル記憶導入T細胞の有効性を評価するため、本発明者は、ヒト皮膚を移植されたNOD/Scidマウスをもとに、新規のキメラモデルを確立した。インビトロで得られた結果をもとに、本発明者は、ビーズCD3/CD28+IL-7、ビーズCD3/CD28+IL-2により産生された遺伝子改変セントラル記憶リンパ球の効力を調べること、ならびにOKT-3およびIL-2により産生された、および臨床試験で現在使用されている、遺伝子改変エフェクター記憶リンパ球によるこれらの細胞の機能的活性を比較することにした。導入細胞注入後、異種T細胞の反応性を、臨床的観察により判定したのに対し、同種GvHDは、組織学的に評価し、導入細胞によるヒト皮膚浸潤の分析と相関させた。これらの研究結果を以下にまとめる。
インビボでのセントラル記憶導入T細胞の有効性を評価するため、本発明者は、ヒト皮膚を移植されたNOD/Scidマウスをもとに、新規のキメラモデルを確立した。インビトロで得られた結果をもとに、本発明者は、ビーズCD3/CD28+IL-7、ビーズCD3/CD28+IL-2により産生された遺伝子改変セントラル記憶リンパ球の効力を調べること、ならびにOKT-3およびIL-2により産生された、および臨床試験で現在使用されている、遺伝子改変エフェクター記憶リンパ球によるこれらの細胞の機能的活性を比較することにした。導入細胞注入後、異種T細胞の反応性を、臨床的観察により判定したのに対し、同種GvHDは、組織学的に評価し、導入細胞によるヒト皮膚浸潤の分析と相関させた。これらの研究結果を以下にまとめる。
CD3/CD28およびIL-7により産生された遺伝子改変細胞は、皮膚移植されたNOD/Scidマウスにおいて急速に生着する
マウス抹消血におけるヒトTリンパ球数は、注入後1週間から2週間で増加した。ビーズにより産生された導入細胞は、OKT-3で産生された導入細胞より高度に生着した。この差は、T細胞注入後2週間目により明白であった(図37)。
マウス抹消血におけるヒトTリンパ球数は、注入後1週間から2週間で増加した。ビーズにより産生された導入細胞は、OKT-3で産生された導入細胞より高度に生着した。この差は、T細胞注入後2週間目により明白であった(図37)。
ビーズCD3/CD28およびIL-7刺激による活性化は異種抗原に対する最も高い反応性を導入細胞にもたらす。異種GvVDは、材料および方法に記載された臨床スコアに従い、体重減少を測定することでモニターした。注入されたNOD/Scidマウスは、徐々に体重が減少し、何例かは、実際に異種GvHDのため死亡し、または倫理的理由のため屠殺された。ほとんどの異種反応性T細胞は、抗CD3/CD28ビーズで刺激され、IL-7による培養後にCD3/CD28ビーズおよびIL-2により産生された導入細胞により培養されたものであった。可溶性抗CD3抗体による刺激は、これらのリンパ球を注入されたマウスが、著しい体重減少も、他の臨床的異種GvHD徴候も示さなかったため、異種反応性T細胞を強力に産生することはなかった(図38)。
ビーズCD3/CD28およびIL-7刺激による活性化は、同種抗原に対する最も高い反応性を導入細胞にもたらす
本発明者のNOD/Scidヒト皮膚キメラマウスモデルは、マウスの背中の2つの皮下ポケットに2片のヒト腹部表皮を挿入する皮膚移植を受け、続いて、遺伝子改変細胞を静注されたNOD/Scidマウスからなる。ヒト皮膚移植は、同種抗原に対するT細胞の反応性を、組織学的試験により調べることを可能にする。移植されたヒト皮膚は、実際、表皮細胞および間質細胞だけでなく、幾つかの抗原提示細胞も含有する。抗原提示細胞は、循環リンパ球を引きつけ、恐らくこの活性化を促進することができる。ヒトT細胞注入後3週間目に、本発明者は、NOD/Scidキメラマウスを屠殺し、ヒト皮膚を両側から除去した。続いて、生検は全て、ヘマトキシリン-エオシン(EE)染色および抗ヒトCD3染色を介して病理医により盲検分析された。本発明者は、「ビーズ+IL-7」で刺激したTリンパ球を注入されたマウスでのみ、ヒト皮膚の状況において大量のヒトT細胞浸潤を観察した。「OKT-3」条件では、T細胞の組織浸潤レベルは、あったとしても明らかにわずかであった。図39は、これらの結果の代表例を示す。
本発明者のNOD/Scidヒト皮膚キメラマウスモデルは、マウスの背中の2つの皮下ポケットに2片のヒト腹部表皮を挿入する皮膚移植を受け、続いて、遺伝子改変細胞を静注されたNOD/Scidマウスからなる。ヒト皮膚移植は、同種抗原に対するT細胞の反応性を、組織学的試験により調べることを可能にする。移植されたヒト皮膚は、実際、表皮細胞および間質細胞だけでなく、幾つかの抗原提示細胞も含有する。抗原提示細胞は、循環リンパ球を引きつけ、恐らくこの活性化を促進することができる。ヒトT細胞注入後3週間目に、本発明者は、NOD/Scidキメラマウスを屠殺し、ヒト皮膚を両側から除去した。続いて、生検は全て、ヘマトキシリン-エオシン(EE)染色および抗ヒトCD3染色を介して病理医により盲検分析された。本発明者は、「ビーズ+IL-7」で刺激したTリンパ球を注入されたマウスでのみ、ヒト皮膚の状況において大量のヒトT細胞浸潤を観察した。「OKT-3」条件では、T細胞の組織浸潤レベルは、あったとしても明らかにわずかであった。図39は、これらの結果の代表例を示す。
結論
T細胞は、一般に、病原体、腫瘍、および免疫不全に対する、および自己免疫障害における免疫産生に中心的な役割を果たすと認識されているが、ヒトにおける免疫能力の診断マーカーおよび予後マーカーとしてこれを使用することは困難であった。さらに、非極性多機能中間体およびセントラル記憶リンパ球の増殖に適した方法は、現在欠如している。
T細胞は、一般に、病原体、腫瘍、および免疫不全に対する、および自己免疫障害における免疫産生に中心的な役割を果たすと認識されているが、ヒトにおける免疫能力の診断マーカーおよび予後マーカーとしてこれを使用することは困難であった。さらに、非極性多機能中間体およびセントラル記憶リンパ球の増殖に適した方法は、現在欠如している。
本発明の報告に記載された結果は、
A)インビボで初回刺激された記憶T細胞のAg非依存的蓄積を可能にする、
B)リンパ球ポリクローナル刺激および遺伝子操作にもかかわらず、セントラル記憶リンパ球の増殖を導く
新たな培養条件を同定する。
A)インビボで初回刺激された記憶T細胞のAg非依存的蓄積を可能にする、
B)リンパ球ポリクローナル刺激および遺伝子操作にもかかわらず、セントラル記憶リンパ球の増殖を導く
新たな培養条件を同定する。
結果は、IL-7およびIL-15が、インビボで初回刺激されたAg(腫瘍/病原体/アレルゲン/自己抗原)特異的CD4+またはCD8+T細胞のために、抹消LN、血液および腫瘍などの生物試料を濃縮するのに使用できることを示している。IL-2と比較した場合、IL-7は、リンパ球本来の表現型および発現量をよく維持し、Tリンパ球サブセット全ての生存をよく支持し、希少なCD4+T記憶リンパ球の検出および増殖を可能にした。
慢性ウイルス感染、自己免疫疾患、ワクチン接種または免疫療法の状況において、Ag特異的CD4+またはCD8+T細胞を計数する能力は、患者の免疫能力または疾患の直接的指標を提供し、最適な治療を選択するうえで臨床家を助けるであろう。さらに、インビボで初回刺激された記憶T細胞を、この表現型を変えることなく濃縮する可能性は、インビボで初回刺激された記憶T細胞の特性化、および養子免疫療法戦略における免疫応答のためのこれらの利用を改善しうる。最後に、遺伝子改変セントラル記憶T細胞は、CD3/CD28活性化および恒常的なサイトカインによる培養に際し得ることができる。条件付けされた免疫不全宿主に注入された場合、遺伝子改変セントラル記憶T細胞は、i)エフェクター記憶遺伝子改変T細胞よりも有意に高いレベルで生着し増殖し、ii)宿主および同種抗原に対する免疫応答の誘導において、エフェクター記憶遺伝子改変リンパ球よりも強力である。
これらの結果は、十分に機能的なセントラル記憶遺伝子改変リンパ球は、ヒト疾患治療のために得ることができ、利用することができることを示すものである。
(参考文献)
(参考文献)
Claims (42)
- 試料中の抗原特異的記憶T細胞の希少集団を増殖させるインビトロ方法であって、前記抗原特異的記憶T細胞の希少集団を選択的に増殖させることができる少なくとも1種のサイトカイン受容体アゴニストの有効量に、前記試料を曝露するステップを含む方法。
- サイトカイン受容体アゴニストが、サイトカインまたはこの誘導体である、請求項1に記載のインビトロ方法。
- 少なくとも1種のサイトカイン受容体アゴニストが、IL-7受容体アゴニストまたはIL-15受容体アゴニストである、請求項1または2に記載のインビトロ方法。
- IL-15受容体アゴニストまたはIL-7受容体アゴニストもそれぞれ存在している、請求項3に記載のインビトロ方法。
- 前記抗原特異的記憶T細胞の希少集団が、CD4+および/またはCD8+および/またはγδおよび/またはNKT T細胞集団を含む、請求項1から4に記載のインビトロ方法。
- 前記試料が、血液および生物起源の他の液体試料、固形組織試料、これら由来の細胞の組織培養物およびこの子孫、生物試料からの単離細胞からなる群に属する生物試料である、請求項1から5に記載のインビトロ方法。
- 試料中の抗原特異的記憶T細胞の希少集団を検出するインビトロ方法であって、
a)前記請求項1から6のいずれか一項に記載の、抗原特異的記憶T細胞の希少集団を選択的に増殖させることができる少なくとも1種のサイトカイン受容体アゴニストの有効量に、前記試料を曝露するステップと、
b)前記抗原特異的記憶T細胞の増殖した希少集団の1つに特異的なリガンドである、少なくとも1つのリガンドと共に、前記試料をインキュベートするステップと、
c)特異的リガンドに結合した抗原特異的記憶T細胞の増殖した希少集団を検出するステップと
を含む方法。 - 前記特異的リガンドが、前記抗原特異的記憶T細胞の希少集団の1つに特異的な抗原またはこの誘導体である、請求項7に記載のインビトロ方法。
- 特異的抗原が、マイコバクテリウム、ニューモシスティスカリニ、熱帯熱マラリア原虫、カンジダ、トキソプラズマ、CMV、EBV、BPV、HCV、HBV、HIVを含むが、これらに限定されない微生物病原体に関連している、請求項8に記載のインビトロ方法。
- 抗原が、腫瘍関連抗原である、請求項8に記載のインビトロ方法。
- 抗原が、アレルゲンである、請求項8に記載のインビトロ方法。
- 抗原が、自己抗原である、請求項8に記載のインビトロ方法。
- 特異的抗原が、抗原MHC複合体またはこの誘導体として存在する、請求項8から12に記載のインビトロ方法。
- 前記抗原特異的記憶T細胞の増殖した希少集団の検出が、結合アッセイにより行われる、請求項7から13に記載のインビトロ方法。
- 前記抗原特異的記憶T細胞の増殖した希少集団の検出が、サイトカイン放出アッセイにより行われる、請求項7から13に記載のインビトロ方法。
- 前記抗原特異的記憶T細胞の増殖した希少集団の検出が、増殖アッセイにより行われる、請求項7から13に記載のインビトロ方法。
- 細胞が、試料を特異的リガンドと共にインキュベートする前に、蛍光生体染色色素で標識され、検出ステップが、色素希釈アッセイにより行われる、請求項7から13に記載のインビトロ方法。
- 請求項7から17に記載の、試料中の抗原特異的記憶T細胞の希少集団を検出する方法を実施するためのキットであって、少なくとも1種のサイトカイン受容体アゴニスト、抗原特異的記憶T細胞の希少集団に特異的な少なくとも1種のリガンド、検出手段を含むキット。
- 試料中の抗原特異的記憶T細胞の希少集団を単離するインビトロ方法であって、
a)前記請求項1から6のいずれか一項に記載の、抗原特異的記憶T細胞の希少集団を選択的に増殖させることができる少なくとも1種のサイトカイン受容体アゴニストの有効量に、前記試料を曝露するステップと、
b)前記抗原特異的記憶T細胞の増殖した希少集団の1つに特異的なリガンドである、少なくとも1つのリガンドと共に、前記試料をインキュベートするステップと、
c)特異的リガンドに結合した抗原特異的記憶T細胞の増殖した希少集団を単離するステップと
を含む方法。 - 前記特異的リガンドが、前記抗原特異的記憶T細胞の希少集団の1つに特異的な抗原またはこの誘導体である、請求項19に記載のインビトロ方法。
- 特異的抗原が、マイコバクテリウム、ニューモシスティスカリニ、熱帯熱マラリア原虫、カンジダ、トキソプラズマ、CMV、EBV、BPV、HCV、HBV、HIVを含むが、これらに限定されない微生物病原体に関連している、請求項19に記載のインビトロ方法。
- 抗原が、腫瘍関連抗原である、請求項19または20に記載のインビトロ方法。
- 抗原が、アレルゲンである、請求項19または20に記載のインビトロ方法。
- 抗原が、自己抗原である、請求項19または20に記載のインビトロ方法。
- 特異的抗原が、抗原MHC複合体またはこの誘導体として存在する、請求項19から24に記載のインビトロ方法。
- 前記抗原特異的記憶T細胞の増殖した希少集団の単離が、結合ステップにより行われる、請求項19から25に記載のインビトロ方法。
- 前記抗原特異的記憶T細胞の増殖した希少集団の単離が、IL-2、IFN-g、IL-4、IL-5、IL-10、TNF-アルファ、TGF-ベータ、グランザイムに関するELISPOTアッセイ、ELISAアッセイ、フローサイトメトリーサイトカイン検出アッセイを含むがこれらに限定されない、サイトカインおよびサイトトキシン産生物の測定により行われる、請求項19から25に記載のインビトロ方法。
- 免疫、感染、癌、アレルギー、自己免疫に関連した病態の診断的および/または予後的臨床試験のための、請求項1から27のいずれかに記載のインビトロ方法。
- 免疫、感染、癌、アレルギー、自己免疫に関連した病態の治療および/または予防のための、請求項18から26に記載の方法により単離された希少T細胞集団の使用。
- 前記希少T細胞集団が、遺伝子改変されている、請求項29に記載の希少T細胞集団の使用。
- 遺伝子改変された記憶T細胞集団を得るインビトロ方法であって、
a)リンパ球活性化を誘導することができる、アゴニスト抗体、組換えリガンドおよびこの誘導体を含むがこれらに限定されない、少なくとも2種の特異的活性化受容体アゴニストでリンパ球を活性化するステップと、
b)記憶T細胞集団を選択的に増殖させることができる、少なくとも1種のサイトカイン受容体アゴニストの有効量に、活性化リンパ球を曝露するステップと、
c)b)で得られた細胞に、適切なベクターを用いて外来遺伝子を挿入し発現させるステップと
を含む方法。 - 前記記憶T細胞の集団が、CD4+および/またはCD8+および/またはγδおよび/またはNKT T細胞集団を含む、請求項31に記載のインビトロ方法。
- 前記リンパ球が、血液および生物起源の他の液体試料、固形組織試料、これら由来の細胞の組織培養物およびこの子孫、生物試料からの単離細胞からなる群に属する生物試料由来である、請求項31または32に記載のインビトロ方法。
- 特異的リンパ球活性化受容体アゴニストが、細胞模倣担体に結合している、請求項31から33に記載のインビトロ方法。
- 細胞模倣担体が、常磁性ビーズである、請求項31から34に記載のインビトロ方法。
- リンパ球活性化受容体アゴニストの1つが、CD3ポリペプチドに特異的である、請求項31から35に記載のインビトロ方法。
- リンパ球活性化受容体アゴニストの1つが、共刺激受容体、すなわちCD28に特異的である、請求項31から36に記載のインビトロ方法。
- 少なくとも1種のサイトカイン受容体アゴニストが、IL-7受容体アゴニストまたはIL-15受容体アゴニストである、請求項31から37に記載のインビトロ方法。
- IL-15受容体アゴニストまたはIL-7受容体アゴニストもそれぞれ存在している、請求項31から38に記載のインビトロ方法。
- ベクターがウイルスベクターである、請求項31から39に記載のインビトロ方法。
- 外来遺伝子が、自殺遺伝子、および/またはマーカー遺伝子、および/または生物活性分子、および/または受容体、および/または細胞内に維持されるもしくは細胞外に放出される可溶性因子、および/またはプロドラッグに耐性を与える遺伝子をコードする、請求項31から40に記載のインビトロ方法。
- 癌、感染症、免疫不全もしくは自己免疫の治療および/または予防のための、または造血前駆細胞もしくは固形臓器の移植のための、請求項31から41に記載の方法により生成された遺伝子改変記憶T細胞集団の使用。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US70650305P | 2005-08-08 | 2005-08-08 | |
| PCT/IT2006/000600 WO2007017915A2 (en) | 2005-08-08 | 2006-08-03 | Use of common ϝ chain cytokines for the visualization, isolation and genetic modification of memory t lymphocytes. |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| JP2009504151A true JP2009504151A (ja) | 2009-02-05 |
Family
ID=37440920
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2008525729A Pending JP2009504151A (ja) | 2005-08-08 | 2006-08-03 | 記憶Tリンパ球の視覚化、単離および遺伝子改変のための共通γ鎖サイトカインの使用 |
Country Status (17)
| Country | Link |
|---|---|
| US (3) | US8999715B2 (ja) |
| EP (2) | EP1941028A2 (ja) |
| JP (1) | JP2009504151A (ja) |
| KR (1) | KR20080048455A (ja) |
| CN (1) | CN101273122A (ja) |
| AT (1) | ATE526395T1 (ja) |
| AU (1) | AU2006277577A1 (ja) |
| CA (1) | CA2618580A1 (ja) |
| CY (1) | CY1112293T1 (ja) |
| DK (1) | DK1956080T3 (ja) |
| ES (1) | ES2374504T3 (ja) |
| IL (1) | IL189143A0 (ja) |
| NO (1) | NO20081108L (ja) |
| PL (1) | PL1956080T3 (ja) |
| PT (1) | PT1956080E (ja) |
| SI (1) | SI1956080T1 (ja) |
| WO (1) | WO2007017915A2 (ja) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2019531722A (ja) * | 2016-09-16 | 2019-11-07 | ベイラー カレッジ オブ メディスンBaylor College Of Medicine | ウイルス特異的t細胞の活性化および拡大のためのプラットフォーム |
Families Citing this family (25)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN101824400B (zh) * | 2009-03-05 | 2012-08-08 | 中国科学院微生物研究所 | 一种放大增殖抗原特异性t细胞的方法 |
| US8956828B2 (en) | 2009-11-10 | 2015-02-17 | Sangamo Biosciences, Inc. | Targeted disruption of T cell receptor genes using engineered zinc finger protein nucleases |
| EP2831109B1 (en) | 2012-03-28 | 2017-12-06 | Gadeta B.V. | Combinatorial gamma 9 delta 2 t cell receptor chain exchange |
| JP6346266B2 (ja) | 2013-03-21 | 2018-06-20 | サンガモ セラピューティクス, インコーポレイテッド | 操作されたジンクフィンガータンパク質ヌクレアーゼを使用するt細胞受容体遺伝子の標的化された破壊 |
| CA3197849A1 (en) * | 2014-12-29 | 2016-07-07 | Novartis Ag | Methods of making chimeric antigen receptor-expressing cells |
| US20180171294A1 (en) * | 2015-03-26 | 2018-06-21 | The Trustees Of The University Of Pennsylvania | In vitro artificial lymph node method for sensitization and expansion of t cells for therapy and epitope mapping |
| SG11201708516YA (en) | 2015-04-17 | 2017-11-29 | David Maxwell Barrett | Methods for improving the efficacy and expansion of chimeric antigen receptor-expressing cells |
| AU2016297014B2 (en) | 2015-07-21 | 2021-06-17 | Novartis Ag | Methods for improving the efficacy and expansion of immune cells |
| WO2017034833A1 (en) * | 2015-08-21 | 2017-03-02 | Mayo Foundation For Medical Education And Research | Methods and materials for expanding antigen-specific t cells in culture |
| EA201891338A1 (ru) | 2015-12-04 | 2018-12-28 | Новартис Аг | Композиции и способы для иммуноонкологии |
| JP2019525898A (ja) | 2016-06-10 | 2019-09-12 | ガデタ・ベー・フェー | ヒト白血球抗原拘束ガンマデルタt細胞受容体及びその使用方法 |
| KR20190033066A (ko) * | 2016-06-24 | 2019-03-28 | 맥마스터 유니버시티 | 입양 세포 전달 및 종양붕괴 바이러스 병용 치료 |
| WO2018102761A1 (en) * | 2016-12-02 | 2018-06-07 | City Of Hope | Methods for manufacturing and expanding t cells expressing chimeric antigen receptors and other receptors |
| EP3572502B1 (en) * | 2017-01-20 | 2023-01-11 | Kyoto University | Method for producing cd8alpha +beta + cytotoxic t cells |
| WO2018175636A2 (en) | 2017-03-22 | 2018-09-27 | Novartis Ag | Compositions and methods for immunooncology |
| JP7486953B2 (ja) | 2017-05-18 | 2024-05-20 | ユーエムセー・ユトレヒト・ホールディング・ベー・フェー | 細胞標的化療法のための組成物および方法 |
| US11899017B2 (en) | 2017-07-28 | 2024-02-13 | Bristol-Myers Squibb Company | Predictive peripheral blood biomarker for checkpoint inhibitors |
| AU2019375997A1 (en) | 2018-11-08 | 2021-06-03 | Neximmune, Inc. | T cell compositions with improved phenotypic properties |
| CU24712B1 (es) * | 2019-03-15 | 2024-07-10 | Centre Hospitalier Univ Vaudois | Método para la expansión y diferenciación de linfocitos t y células nk en terapias de transferencia adoptiva |
| KR20220004028A (ko) | 2019-04-26 | 2022-01-11 | 알로젠 테라퓨틱스 인코포레이티드 | 동종 car t 세포를 제조하는 방법 |
| CN111077311B (zh) * | 2019-12-30 | 2023-05-09 | 北京立康生命科技有限公司 | 一种酶联免疫斑点检测试剂盒及其检测方法 |
| EP4039808A1 (en) | 2021-02-08 | 2022-08-10 | Ospedale San Raffaele S.r.l. | Guide rnas and uses thereof |
| AU2022227686A1 (en) | 2021-02-25 | 2023-07-27 | Lyell Immunopharma, Inc. | Ror1 targeting chimeric antigen receptor |
| CN113433308A (zh) * | 2021-06-02 | 2021-09-24 | 河北森朗泰禾生物科技有限公司 | 离体血液样品的分析方法和免疫力评估装置 |
| CN117192115A (zh) * | 2023-11-03 | 2023-12-08 | 赛德特(北京)生物工程有限公司 | 检测t淋巴细胞免疫活性的方法 |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7572631B2 (en) * | 2000-02-24 | 2009-08-11 | Invitrogen Corporation | Activation and expansion of T cells |
| US7364869B2 (en) | 2003-07-29 | 2008-04-29 | The J. David Gladstone Institutes | Method of detecting antigen-specific T lymphocytes |
| AU2005250408B2 (en) * | 2004-05-27 | 2010-09-23 | The Trustees Of The University Of Pennsylvania | Novel artificial antigen presenting cells and uses therefor |
| ES2686424T5 (es) * | 2010-05-04 | 2023-03-27 | Yeda Res & Dev | Inmunoterapia con células alogénicas redireccionadas |
-
2006
- 2006-08-03 EP EP06796252A patent/EP1941028A2/en not_active Withdrawn
- 2006-08-03 SI SI200631202T patent/SI1956080T1/sl unknown
- 2006-08-03 DK DK08102851.6T patent/DK1956080T3/da active
- 2006-08-03 EP EP08102851A patent/EP1956080B1/en active Active
- 2006-08-03 PL PL08102851T patent/PL1956080T3/pl unknown
- 2006-08-03 WO PCT/IT2006/000600 patent/WO2007017915A2/en not_active Ceased
- 2006-08-03 ES ES08102851T patent/ES2374504T3/es active Active
- 2006-08-03 CA CA002618580A patent/CA2618580A1/en not_active Abandoned
- 2006-08-03 JP JP2008525729A patent/JP2009504151A/ja active Pending
- 2006-08-03 PT PT08102851T patent/PT1956080E/pt unknown
- 2006-08-03 AT AT08102851T patent/ATE526395T1/de active
- 2006-08-03 KR KR1020087003174A patent/KR20080048455A/ko not_active Withdrawn
- 2006-08-03 AU AU2006277577A patent/AU2006277577A1/en not_active Abandoned
- 2006-08-03 CN CNA2006800351105A patent/CN101273122A/zh active Pending
-
2008
- 2008-01-31 IL IL189143A patent/IL189143A0/en unknown
- 2008-03-03 NO NO20081108A patent/NO20081108L/no not_active Application Discontinuation
-
2011
- 2011-09-22 US US13/240,009 patent/US8999715B2/en active Active
- 2011-12-22 CY CY20111101280T patent/CY1112293T1/el unknown
-
2015
- 2015-03-17 US US14/660,484 patent/US9974808B2/en active Active
-
2018
- 2018-05-04 US US15/970,945 patent/US11395835B2/en active Active
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2019531722A (ja) * | 2016-09-16 | 2019-11-07 | ベイラー カレッジ オブ メディスンBaylor College Of Medicine | ウイルス特異的t細胞の活性化および拡大のためのプラットフォーム |
| JP7002769B2 (ja) | 2016-09-16 | 2022-02-04 | ベイラー カレッジ オブ メディスン | ウイルス特異的t細胞の活性化および拡大のためのプラットフォーム |
Also Published As
| Publication number | Publication date |
|---|---|
| EP1956080A3 (en) | 2008-09-10 |
| CA2618580A1 (en) | 2007-02-15 |
| DK1956080T3 (da) | 2012-01-23 |
| US9974808B2 (en) | 2018-05-22 |
| IL189143A0 (en) | 2008-08-07 |
| US11395835B2 (en) | 2022-07-26 |
| CY1112293T1 (el) | 2015-12-09 |
| US8999715B2 (en) | 2015-04-07 |
| EP1941028A2 (en) | 2008-07-09 |
| KR20080048455A (ko) | 2008-06-02 |
| US20180333434A1 (en) | 2018-11-22 |
| PT1956080E (pt) | 2011-12-30 |
| PL1956080T3 (pl) | 2012-02-29 |
| ATE526395T1 (de) | 2011-10-15 |
| EP1956080B1 (en) | 2011-09-28 |
| US20120027802A1 (en) | 2012-02-02 |
| CN101273122A (zh) | 2008-09-24 |
| US20150306142A1 (en) | 2015-10-29 |
| SI1956080T1 (sl) | 2012-02-29 |
| WO2007017915A3 (en) | 2007-06-07 |
| ES2374504T3 (es) | 2012-02-17 |
| AU2006277577A1 (en) | 2007-02-15 |
| NO20081108L (no) | 2008-05-05 |
| WO2007017915A2 (en) | 2007-02-15 |
| EP1956080A2 (en) | 2008-08-13 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US11395835B2 (en) | Use of common gamma chain cytokines for the visualization, isolation and genetic modification of memory T lymphocytes | |
| US10577586B2 (en) | Compositions and methods for modulating an immune response | |
| AU637702B2 (en) | T-lymphocyte progenitor cell assay | |
| CA2133075A1 (en) | CD28 Pathway Immunoregulation | |
| AU2008201685A1 (en) | CD4+CD25+ regulatory T cells from human blood | |
| CA2940163C (en) | Tscm cells and methods for use | |
| CN108291203A (zh) | Cd8+cd45rc低treg的新亚群及其用途 | |
| Stockinger et al. | CD4 T-cell memory | |
| CN102112491A (zh) | 抗-cd8抗体阻断细胞毒素效应物的引发并导致调节性cd8+t细胞的产生 | |
| CN110713977B (zh) | 一种cd8 t细胞的培养扩增方法 | |
| US20210317410A1 (en) | Methods of making natural killer cells and uses thereof | |
| US20100035282A1 (en) | Use of common gamma chain cytokines for the visualization, isolation and genetic modification of memory t lymphocytes | |
| EP2001509A1 (en) | Pharmaceuticals for influencing the reaction of the human immune system | |
| JP2003033175A (ja) | 選択的免疫応答抑制を誘導する末梢血樹状細胞サブセット | |
| Prasad et al. | Propagation and characterisation of dendritic cells from G-CSF mobilised peripheral blood monocytes and stem cells in common marmoset monkeys | |
| HK1120548A (en) | USE OF COMMON γ CHAIN CYTOKINES FOR THE VISUALIZATION, ISOLATION AND GENETIC MODIFICATION OF MEMORY T LYMPHOCYTES | |
| US20150064206A1 (en) | Compositions for treating uveitis | |
| Chen | Roles of Transcription Factor T-Bet in Memory CD4+ T Cell Generation, Function, Homeostasis and Tissue | |
| Hoeppli | Tailoring the migration capacity of regulatory T cells to enhance their efficacy as a cellular therapy | |
| Smith | Mechanisms of Thymic Involution and Therapies to Prevent or Treat the loss of Thymic Epithelial Cells | |
| JP2019208501A (ja) | インビトロでの制御性t細胞の特異性評価方法 | |
| Marriott | Investigating the in vivo requirements for the generation and survival of CD4⁺ memory T cells | |
| Coleman | Characterization and manipulation of CD4+ CD25+ T regulatory cells from normal and SIV-infected African green monkeys and Rhesus macaques |