JP2012109365A - 有機光電変換素子および太陽電池 - Google Patents
有機光電変換素子および太陽電池 Download PDFInfo
- Publication number
- JP2012109365A JP2012109365A JP2010256423A JP2010256423A JP2012109365A JP 2012109365 A JP2012109365 A JP 2012109365A JP 2010256423 A JP2010256423 A JP 2010256423A JP 2010256423 A JP2010256423 A JP 2010256423A JP 2012109365 A JP2012109365 A JP 2012109365A
- Authority
- JP
- Japan
- Prior art keywords
- photoelectric conversion
- group
- layer
- electrode
- organic photoelectric
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Images


Classifications
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02E—REDUCTION OF GREENHOUSE GAS [GHG] EMISSIONS, RELATED TO ENERGY GENERATION, TRANSMISSION OR DISTRIBUTION
- Y02E10/00—Energy generation through renewable energy sources
- Y02E10/50—Photovoltaic [PV] energy
- Y02E10/52—PV systems with concentrators
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02E—REDUCTION OF GREENHOUSE GAS [GHG] EMISSIONS, RELATED TO ENERGY GENERATION, TRANSMISSION OR DISTRIBUTION
- Y02E10/00—Energy generation through renewable energy sources
- Y02E10/50—Photovoltaic [PV] energy
- Y02E10/549—Organic PV cells
Landscapes
- Electroluminescent Light Sources (AREA)
- Photovoltaic Devices (AREA)
Abstract
【解決手段】透明な基板上に、透明な第一の電極、p型有機半導体材料とn型有機半導体材料とを含有する光電変換層、および第二の電極をこの順に有する有機光電変換素子であって、該光電変換層が、該p型有機半導体材料として下記一般式1で表される部分構造を有する化合物を含有することを特徴とする有機光電変換素子。
【化1】

【選択図】なし
Description
なお外部量子効率×理論Jscの積分が短絡電流密度であるため、外部量子効率(EQE)および曲線因子(FF)が太陽電池の効率に非常に重要な要素であることが分かる。
6.前記一般式3のZ4で表される原子が、珪素原子である構造を有するp型有機半導体材料を含むことを特徴とする、前記1から5のいずれか1項に記載の有機光電変換素子。
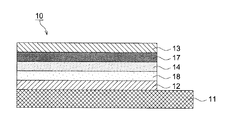
図1は、本発明の有機光電変換素子の構成の例を示す概略断面図である。
光電変換層は、p型有機半導体材料として上記一般式1で表される部分構造を有する化合物を含有する。
一般式1中、L1およびL2は、単結合または2価の連結基を表す。X1およびX2は、酸素原子または硫黄原子を表す。Y1およびY2は、−O−、−S−または−NR1−、を表す。R1は、アルキル基、フッ化アルキル基、シクロアルキル基、アリール基、ヘテロアリール基またはアルキルシリル基を表す。Z1およびZ2は、水素原子、ハロゲン原子、または酸素原子、窒素原子もしくは硫黄原子を介してL1に連結して環を形成する連結基を表す。
本発明に係る化合物のうち、一般式1においてZ1、Z2が水素原子のものは、Heterocycles1998p1415等を参考として合成することができる。また一般式2で表される化合物は、Chemistry of Heterocyclic Compounds(1975)p666、J.Org.Chem.(2004)p4867等を参考として合成することができる。
本発明に係る光電変換層に用いられるn型有機半導体材料としては、特に限定されないが、例えば、フラーレン、オクタアザポルフィリン等、p型有機半導体の水素原子をフッ素原子に置換したパーフルオロ体(パーフルオロペンタセンやパーフルオロフタロシアニン等)、ナフタレンテトラカルボン酸無水物、ナフタレンテトラカルボン酸ジイミド、ペリレンテトラカルボン酸無水物、ペリレンテトラカルボン酸ジイミド等の芳香族カルボン酸無水物やそのイミド化物を骨格として含む高分子化合物等を挙げることができる。
p型有機半導体材料とn型有機半導体材料とを含有する光電変換層の形成方法としては、蒸着法、塗布法(キャスト法、スピンコート法を含む)等を例示することができる。
本発明の有機光電変換素子は、光電変換層とカソードとの中間に電子輸送層を形成することで、光電変換層で発生した電荷をより効率的に取り出すことが可能となるため、これらの層を有していることが好ましい。
本発明の有機光電変換素子は、光電変換層とアノードとの中間には正孔輸送層を、光電変換層で発生した電荷をより効率的に取り出すことが可能となるため、これらの層を有していることが好ましい。
本発明の有機光電変換素子の構成としては、エネルギー変換効率の向上や、素子寿命の向上を目的に、各種中間層を素子内に有する構成としてもよい。
本発明の有機光電変換素子においては、第一の電極と第二の電極を有するが、タンデム構成をとる場合には、中間電極を用いることでタンデム構成を達成することができる。
本発明の第一の電極に用いられる、材料としては、例えば、インジウムチンオキシド(ITO)、AZO、FTO、SnO2、ZnO、酸化チタン等の透明金属酸化物、Ag、Al、Au、Pt等の非常に薄い金属層または金属ナノワイヤ、カーボンナノチューブ等のナノワイヤやナノ粒子を含有する層、PEDOT:PSS、ポリアニリン等の導電性高分子材料等を用いることができる。
第二の電極は導電材単独層であってもよいが、導電性を有する材料に加えて、これらを保持する樹脂を併用してもよい。
また、前記図3のようなタンデム構成の場合に必要となる中間電極の材料としては、透明性と導電性を併せ持つ化合物を用いた層であることが好ましく、前記アノードで用いたような材料(ITO、AZO、FTO、SnO2、ZnO、酸化チタン等の透明金属酸化物、Ag、Al、Au、Pt等の非常に薄い金属層または金属ナノワイヤ、カーボンナノチューブ等のナノワイヤやナノ粒子を含有する層、PEDOT:PSS、ポリアニリン等の導電性高分子材料等)を用いることができる。
本発明において、基板は透明な基板であるが、透明な、とは前述の電極の記載と同様の意味を有する。
本発明の有機光電変換素子は、太陽光のより効率的な受光を目的として、各種の光学機能層を有していてよい。光学機能層としては、例えば、反射防止膜、マイクロレンズアレイ等の集光層、カソードで反射した光を散乱させて再度発電層に入射させることができるような光拡散層などを設けてもよい。
本発明に係る各々の電極、光電変換層や、正孔輸送層、電子輸送層等をパターニングする方法やプロセスには特に制限はなく、公知の手法を適宜適用することができる。
本発明の太陽電池は、上記の有機光電変換素子を有する。
〔有機光電変換素子1の作製〕
特開2009−146981号を参考として、逆層型の有機光電変換素子を作製した。
先ず、100ml三口フラスコに2−メトキシエタノール12.5mlと、6.25mmolのチタニウムテトライソプロポキシドとを入れ、氷浴中で10分間冷却した。次に、12.5mmolのアセチルアセトンをゆっくり加えて、氷浴中で10分間撹拌した。次に、混合溶液を80℃で2時間加熱後、1時間還流した。最後に、室温まで冷却し、メトキシエタノールを用いて所定の濃度(150m)に調整した。TiOx前駆体を得た。なお、上記工程は全て窒素雰囲気で行った。
上記有機光電変換素子1の作製において、p型半導体材料を表1に記載の材料に変更し、さらにp:n比を表に記載の組成(全固形分濃度は1.8質量%に固定)に代えた以外は、比較の有機光電変換素子1と同様にして有機光電変換素子2〜6、11を得た。
上記有機光電変換素子1の作製において、p型半導体材料を表1に記載の材料に変更し、さらにp:n比を0.6質量%:1.2質量%(全固形分濃度は1.8質量%で同じ)に代えた以外は、比較の有機光電変換素子1と同様にして有機光電変換素子2〜7を得た。
p型半導体材料の構造を、B3LYP 6−31G*の手法に従って計算した(高分子材料の場合は3量体で計算)。得られた分子モデルから、最もπ共役平面でねじれた二面角を有する部位を検出し、その二面角を測定した。
上記作製した光電変換素子に、ソーラーシミュレーター(AM1.5Gフィルタ)の100mW/cm2の強度の光を照射し、有効面積を4.0mm2にしたマスクを受光部に重ね、短絡電流密度Jsc(mA/cm2)及び開放電圧Voc(V)、曲線因子(フィルファクター)FFを、同素子上に形成した4箇所の受光部をそれぞれ測定し、平均値を求めた。また、Jsc、Voc、FFから式1に従って光電変換効率η(%)を求めた。
実施例1で作製した有機光電変換素子17について、それぞれ同様の素材および組成を用いて以下のような順層型の素子を作製した。
実施例1と同じ透明基板を同様の工程で洗浄した後、ITO膜上に、導電性高分子であるBaytron P4083(スタルクヴィテック社製)を30nmの膜厚となるようにスピンコートした後、140℃で大気中10分間加熱乾燥した。
上記作製した光電変換素子に、ソーラーシミュレーター(AM1.5Gフィルタ)の100mW/cm2の強度の光を照射し、有効面積を4.0mm2にしたマスクを受光部に重ね、短絡電流密度Jsc(mA/cm2)及び開放電圧Voc(V)、曲線因子(フィルファクター)FFを、同素子上に形成した4箇所の受光部をそれぞれ測定し、平均値を求めた。また、Jsc、Voc、FFから式1に従ってエネルギー変換効率η(%)を求めた。
(耐久性評価)
温度80度、湿度80%に保持した容器内に保存し、定期的に取りだして電圧−電流特性を測定し、初期の変換効率を100として、初期の効率の80%の効率まで低下する時間をLT80として評価した。結果を表2に示す。
11 基板
12 第一の電極
13 第二の電極
14 光電変換層
14′ 第一の光電変換層
15 電荷再結合層
16 第二の光電変換層
17 正孔輸送層
18 電子輸送層
Claims (9)
- 透明な基板上に、透明な第一の電極、p型有機半導体材料とn型有機半導体材料とを含有する光電変換層、および第二の電極をこの順に有する有機光電変換素子であって、該光電変換層が、該p型有機半導体材料として下記一般式1で表される部分構造を有する化合物を含有することを特徴とする有機光電変換素子。
X1およびX2は、酸素原子または硫黄原子を表す。
Y1およびY2は、−O−、−S−または−NR1−、を表す。R1は、置換基を有してもよいアルキル基、フッ化アルキル基、シクロアルキル基、アシル基、アリール基、ヘテロアリール基またはアルキルシリル基を表す。
Z1およびZ2は、水素原子、ハロゲン原子、または酸素原子、窒素原子もしくは硫黄原子を介してL1に連結して環を形成する連結基を表す。 - 前記一般式1において、前記Y1およびY2が、前記−NR1−であることを特徴とする請求項1に記載の有機光電変換素子。
- 前記一般式1で表される構造が、下記一般式2で表される部分構造であることを特徴とする請求項2に記載の有機光電変換素子。
- 前記一般式2において、Z3が硫黄原子であることを特徴とする請求項3に記載の有機光電変換素子。
- 前記一般式1で表される構造を有する化合物が、下記一般式3で表される構造単位を含有する共重合体であることを特徴とする請求項1から4の何れか1項に記載の有機光電変換素子。
- 前記一般式3のZ4で表される原子が、珪素原子である構造を有するp型有機半導体材料を含むことを特徴とする、請求項1から5のいずれか1項に記載の有機光電変換素子。
- 前記一般式1で表される構造を有する化合物の数平均分子量が、15000〜50000であることを特徴とする請求項1から6のいずれか1項に記載の有機光電変換素子。
- 前記第一の電極が、カソードであり、第二の電極がアノードであることを特徴とする請求項1から7のいずれか1項に記載の有機光電変換素子。
- 請求項1から8のいずれか1項に記載の有機光電変換素子を具備することを特徴とする太陽電池。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2010256423A JP2012109365A (ja) | 2010-11-17 | 2010-11-17 | 有機光電変換素子および太陽電池 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2010256423A JP2012109365A (ja) | 2010-11-17 | 2010-11-17 | 有機光電変換素子および太陽電池 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| JP2012109365A true JP2012109365A (ja) | 2012-06-07 |
Family
ID=46494680
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2010256423A Pending JP2012109365A (ja) | 2010-11-17 | 2010-11-17 | 有機光電変換素子および太陽電池 |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP2012109365A (ja) |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20140183455A1 (en) * | 2012-12-27 | 2014-07-03 | Xerox Corporation | Pechmann dye based polymers and semiconductor compositions |
| WO2016005891A1 (en) * | 2014-07-10 | 2016-01-14 | Basf Se | Thienothiophene-isoindigo |
| JP2017512855A (ja) * | 2014-03-17 | 2017-05-25 | メルク パテント ゲーエムベーハー | 有機半導体化合物 |
| CN108727413A (zh) * | 2017-04-13 | 2018-11-02 | 中国科学院青岛生物能源与过程研究所 | 一种噻唑异靛蓝类化合物及其制备方法 |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2005136315A (ja) * | 2003-10-31 | 2005-05-26 | Akihiko Fujii | 有機太陽電池 |
| JP2008166339A (ja) * | 2006-12-27 | 2008-07-17 | Toray Ind Inc | 光起電力素子用材料および光起電力素子 |
| WO2009053291A1 (en) * | 2007-10-25 | 2009-04-30 | Basf Se | Ketopyrroles as organic semiconductors |
| WO2009117613A1 (en) * | 2008-03-19 | 2009-09-24 | The Regents Of The University Of Michigan | Organic thin films for infrared detection |
| WO2010051258A1 (en) * | 2008-10-27 | 2010-05-06 | The Regents Of The University Of Michigan | Inverted organic photosensitive devices |
-
2010
- 2010-11-17 JP JP2010256423A patent/JP2012109365A/ja active Pending
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2005136315A (ja) * | 2003-10-31 | 2005-05-26 | Akihiko Fujii | 有機太陽電池 |
| JP2008166339A (ja) * | 2006-12-27 | 2008-07-17 | Toray Ind Inc | 光起電力素子用材料および光起電力素子 |
| WO2009053291A1 (en) * | 2007-10-25 | 2009-04-30 | Basf Se | Ketopyrroles as organic semiconductors |
| WO2009117613A1 (en) * | 2008-03-19 | 2009-09-24 | The Regents Of The University Of Michigan | Organic thin films for infrared detection |
| WO2010051258A1 (en) * | 2008-10-27 | 2010-05-06 | The Regents Of The University Of Michigan | Inverted organic photosensitive devices |
Cited By (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20140183455A1 (en) * | 2012-12-27 | 2014-07-03 | Xerox Corporation | Pechmann dye based polymers and semiconductor compositions |
| US8865861B2 (en) * | 2012-12-27 | 2014-10-21 | Xerox Corporation | Pechmann dye based polymers and semiconductor compositions |
| JP2017512855A (ja) * | 2014-03-17 | 2017-05-25 | メルク パテント ゲーエムベーハー | 有機半導体化合物 |
| US10367143B2 (en) | 2014-03-17 | 2019-07-30 | Merck Patent Gmbh | Organic semiconducting compounds |
| WO2016005891A1 (en) * | 2014-07-10 | 2016-01-14 | Basf Se | Thienothiophene-isoindigo |
| CN106470998A (zh) * | 2014-07-10 | 2017-03-01 | 巴斯夫欧洲公司 | 噻吩并噻吩异靛 |
| JP2017523281A (ja) * | 2014-07-10 | 2017-08-17 | ビーエーエスエフ ソシエタス・ヨーロピアBasf Se | チエノチオフェン−イソインジゴ |
| KR101825896B1 (ko) | 2014-07-10 | 2018-02-05 | 바스프 에스이 | 티에노티오펜-이소인디고 |
| CN106470998B (zh) * | 2014-07-10 | 2019-05-31 | 巴斯夫欧洲公司 | 噻吩并噻吩异靛 |
| US10403822B2 (en) | 2014-07-10 | 2019-09-03 | Basf Se | Thienothiophene-isoindigo |
| CN108727413A (zh) * | 2017-04-13 | 2018-11-02 | 中国科学院青岛生物能源与过程研究所 | 一种噻唑异靛蓝类化合物及其制备方法 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5516153B2 (ja) | 有機光電変換素子、それを用いた太陽電池及び光センサアレイ | |
| JP5655568B2 (ja) | 有機光電変換素子、太陽電池及び光センサアレイ | |
| JP5447521B2 (ja) | 有機光電変換素子、それを用いた太陽電池および光センサアレイ | |
| JP5692228B2 (ja) | 有機光電変換素子およびそれを用いた太陽電池 | |
| JP5712769B2 (ja) | 有機光電変換素子及び太陽電池 | |
| JP5493496B2 (ja) | 有機光電変換素子、太陽電池及び光センサアレイ | |
| JP5920341B2 (ja) | 有機光電変換素子、その製造方法及び太陽電池 | |
| JP5493465B2 (ja) | 有機薄膜太陽電池 | |
| JP5699524B2 (ja) | 有機光電変換素子および太陽電池 | |
| JP5772836B2 (ja) | 有機光電変換層材料組成物、有機光電変換素子、有機光電変換素子の製造方法及び太陽電池 | |
| JP2014053383A (ja) | タンデム型の有機光電変換素子およびこれを用いた太陽電池 | |
| JP2011155185A (ja) | 有機光電変換素子、それを用いた太陽電池及び光センサアレイ | |
| JP2013089684A (ja) | 有機光電変換素子およびこれを用いた太陽電池 | |
| JP5686141B2 (ja) | 有機光電変換素子および太陽電池 | |
| JP5447513B2 (ja) | 有機光電変換素子、それを用いた太陽電池及び光センサアレイ | |
| JPWO2010090123A1 (ja) | 有機光電変換素子、それを用いた太陽電池、及び光センサアレイ | |
| JP2012109365A (ja) | 有機光電変換素子および太陽電池 | |
| JP2012049352A (ja) | 有機光電変換素子、それを用いた太陽電池、及び光センサアレイ | |
| JP5413055B2 (ja) | 有機光電変換素子、それを用いた太陽電池及び光センサアレイ | |
| JP5582042B2 (ja) | 有機光電変換素子および太陽電池 | |
| JP2011124469A (ja) | 有機光電変換素子、それを用いた太陽電池及び光センサアレイ | |
| JP5691810B2 (ja) | 共役系高分子およびこれを用いた有機光電変換素子 | |
| JP5691449B2 (ja) | 有機光電変換素子、それを用いた太陽電池、及び光センサアレイ | |
| JP5737377B2 (ja) | 有機薄膜太陽電池 | |
| JP2013089627A (ja) | 有機光電変換素子、およびそれを用いた有機太陽電池 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A711 | Notification of change in applicant |
Free format text: JAPANESE INTERMEDIATE CODE: A712 Effective date: 20121101 |
|
| A711 | Notification of change in applicant |
Free format text: JAPANESE INTERMEDIATE CODE: A712 Effective date: 20130415 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20130516 |
|
| RD02 | Notification of acceptance of power of attorney |
Free format text: JAPANESE INTERMEDIATE CODE: A7422 Effective date: 20130726 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20140527 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20140528 |
|
| A02 | Decision of refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A02 Effective date: 20141007 |