JP2012183823A - 水蒸気バリアーフィルムの製造方法、水蒸気バリアーフィルム及び電子機器 - Google Patents
水蒸気バリアーフィルムの製造方法、水蒸気バリアーフィルム及び電子機器 Download PDFInfo
- Publication number
- JP2012183823A JP2012183823A JP2012022853A JP2012022853A JP2012183823A JP 2012183823 A JP2012183823 A JP 2012183823A JP 2012022853 A JP2012022853 A JP 2012022853A JP 2012022853 A JP2012022853 A JP 2012022853A JP 2012183823 A JP2012183823 A JP 2012183823A
- Authority
- JP
- Japan
- Prior art keywords
- water vapor
- layer
- vapor barrier
- film
- barrier film
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
Images






Classifications
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02E—REDUCTION OF GREENHOUSE GAS [GHG] EMISSIONS, RELATED TO ENERGY GENERATION, TRANSMISSION OR DISTRIBUTION
- Y02E10/00—Energy generation through renewable energy sources
- Y02E10/50—Photovoltaic [PV] energy
Landscapes
- Electroluminescent Light Sources (AREA)
- Laminated Bodies (AREA)
- Photovoltaic Devices (AREA)
Abstract
【解決手段】基材1上に少なくとも2層の水蒸気バリアー層3(例えば、第1の水蒸気バリアー層と第2の水蒸気バリアー層)を備え、その水蒸気バリアー層3の間に、加水分解性化合物を含有する加水分解層4を少なくとも1層備える水蒸気バリアーフィルム10によって、水蒸気バリアー性とともに、折り曲げ耐性、平滑性の向上を図った。
【選択図】図1
Description
このようなガスバリアーフィルムを製造する方法としては、主に、プラズマCVD法(Chemical Vapor Deposition:化学気相成長法、化学蒸着法)によってフィルムなどの基材上にガスバリアー層を形成する方法や、ポリシラザンを主成分とする塗布液を基材上に塗布した後に表面処理を施してガスバリアー層を形成する方法、あるいはそれらを併用する方法が知られている。
しかしながら、これらの製造方法で形成されたガスバリアー層には、基材表面の突起やガスバリアー層中への異物の混入により生じた微細孔などの欠陥、ガスバリアー層の膨張・収縮により生じた微小なひび割れなどの欠陥、取り扱い時の折り曲げや接触などに起因した傷などの欠陥等が発生してしまうことがある。このような欠陥箇所が生じたガスバリアー層では、その欠陥箇所を通じて水蒸気等のガスが透過してしまい完全に遮断できていない。
例えば、特許文献1に記載の発明では、高い水蒸気バリアー性を奏するためにガスバリアー層の他に捕水層を設ける技術について開示されている。
また、特許文献2に記載の発明では、2層の無機ガスバリアー層を有する水蒸気バリアーフィルムにおいて、2層の無機ガスバリアー層の間に少なくとも1層のアルカリ土類金属一酸化物からなる吸湿性層を形成することで、水蒸気バリアー性能を高める技術について開示されている。
また、特許文献3に記載の発明では、透明基板上に少なくとも1層の金属窒化物膜が形成されてなる透明積層体において、その金属窒化物膜が、少なくとも酸素分子及び/又は水分子が存在する雰囲気中において酸化されうるものにした技術について開示されている。
また、特許文献4に記載の発明では、防湿性フィルムに吸湿性フィルムを挟み込むことで水蒸気バリアー性を向上させる技術について開示されている。
また、上記特許文献3の透明積層体(バリアーフィルム)は、水蒸気透過性の高い基材上に、水分によって酸化される金属窒化物膜が直接形成されているため、透明積層体の透明性が低いという欠点や、また基材上に直接形成された金属窒化物膜の固さに起因して屈曲時にクラックが発生しやすいという欠点があった。また、透明基板に含有する水分による酸化が進行しやすく、高湿保存下での水分との反応が迅速に進むため、高レベルの水蒸気バリアー性を得ることが困難であった。
また、上記特許文献4においては、吸湿性フィルムが防湿性フィルムに保護されているが、電子デバイスの封止工程において複数のフィルムを取り扱うことで、プロセスが複雑になるだけでなく、封止後のデバイスの厚みが厚くなり、フレキシブル性に劣るという問題があった。
基材上に少なくとも2層の水蒸気バリアー層を備え、
前記水蒸気バリアー層の間に、加水分解性化合物を含有する加水分解層を少なくとも1層備えることを特徴とする。
前記加水分解性化合物は加水分解によって、アルコール、アミン化合物、無機酸のうち少なくとも1つを放出する化合物であることを特徴とする。
前記加水分解性化合物は、金属アルコキシド、シラン化合物、シラザン化合物、金属ハロゲン化物のうち少なくとも1つを含むことを特徴とする。
60℃/90%RH雰囲気下、前記加水分解層における前記加水分解性化合物が加水分解して生成した化合物に由来する炭素または窒素原子組成比の半減期が、20時間以上1500時間以下であることを特徴とする。
前記基材に近い配置の前記水蒸気バリアー層は、金属酸化物、金属窒化物、金属酸窒化物のうち少なくとも1つを含む蒸着膜であることを特徴とする。
前記基材に近い配置の前記水蒸気バリアー層は、ポリシラザンを含有する塗膜に改質処理を施して形成された水蒸気バリアー層であることを特徴とする。
基材上に第1の水蒸気バリアー層を形成した後、加水分解性化合物を含有する塗布液を低湿度環境下で前記第1の水蒸気バリアー層上に塗布し乾燥して加水分解層を形成し、その加水分解層上に第2の水蒸気バリアー層を形成することを特徴とする。
前記第1の水蒸気バリアー層を蒸着法によって形成し、
前記第1の水蒸気バリアー層上に形成した前記加水分解層の上層側に改質処理を施して、その加水分解層上に重なった前記第2の水蒸気バリアー層を形成することを特徴とする。
前記第1の水蒸気バリアー層を塗布によって形成し、
前記第1の水蒸気バリアー層上に形成した前記加水分解層の上層側に改質処理を施して、その加水分解層上に重なった前記第2の水蒸気バリアー層を形成することを特徴とする。
基材上に第1の水蒸気バリアー層を形成した後、前記第1の水蒸気バリアー層上に加水分解性化合物を含有するバリアー層前駆体層を形成し、そのバリアー層前駆体層の上層側に改質処理を施すことで、前記第1の水蒸気バリアー層上に積層された加水分解層と、その加水分解層上に重なった第2の水蒸気バリアー層を形成することを特徴とする。
請求項1〜6の何れか一項に記載の水蒸気バリアーフィルムまたは請求項7〜10の何れか一項に記載の水蒸気バリアーフィルムの製造方法によって得られた水蒸気バリアーフィルムと、前記水蒸気バリアーフィルムによって封止された電子デバイスを備えることを特徴とする。
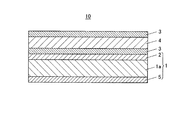
水蒸気バリアー層3は、例えば、スパッタリング法、プラズマCVD法などの蒸着法によって形成された金属酸化物、金属窒化物、金属酸窒化物等のうち少なくとも1つを含む蒸着膜からなるバリアー層であっても、あるいはポリシラザンを含む液体を塗布し乾燥した後に真空紫外光を照射して形成されたポリシラザン改質層からなるバリアー層であってもよい。
また、基材1の平滑性や基材1に対する水蒸気バリアー層3の密着性を向上させるための中間層として、平滑層2やアンカーコート層を基材表面に設けてもよい。
また、基材1に傷や汚れが付くことを防止するため耐傷層や、樹脂基材1aが加熱された際に内部から表面へモノマー、オリゴマー等の低分子量成分が析出する、いわゆるブリードアウトを抑制する目的でのブリードアウト防止層(保護層)5を基材表面に設けてもよい。
また、本発明に係る水蒸気バリアーフィルム11は、図2に示すように、基材1の一方の面に平滑層2を備え、その平滑層2上に、水蒸気バリアー層3、加水分解層4、水蒸気バリアー層3、加水分解層4、水蒸気バリアー層3を順に積層してなる5層構成のガスバリアー層を備えている。また基材1の他方の面にブリードアウト防止層5を備えている。
本実施形態の水蒸気バリアーフィルム(10、11)における基材1は、ガス透過性を有し、可撓性を有する折り曲げ可能なフィルム基材である。この基材1は、水蒸気バリアー性を有する蒸着層やポリシラザン改質層を保持することができるフィルム材料であれば特に限定されるものではない。
ここで、本発明におけるガス透過性を有する基材1とは、モコン法に従いMOCON社製PERMATRAN−W3/33を用いて、JIS規格のK7129法(温度40℃、湿度90%RH)に基づいて測定した水蒸気透過率が、モコン法の測定で0.5g/m2/日以上であるものと定義される。
なお、本発明に用いられる基材は、実質的に水蒸気バリアー性を有さない後述の樹脂基材1a単体でもよく、また、樹脂基材1aにアンカーコート層、平滑層2、ブリードアウト防止層5などの各種機能層を積層した基材1であってもよい。
但し、樹脂基材1a単体の水蒸気バリアー性(水蒸気透過率)が0.01g/m2/日よりも下回る基材を用いると、後述のように、基材1と接する水蒸気バリアー層3を介して放出される加水分解層4からのアミン化合物などの拡散除去が阻害されて、バリアーフィルムの長期保存性、耐熱性を低下させてしまう懸念がある。
これら樹脂フィルムのうち、コストや入手の容易性の点では、ポリエチレンテレフタレート(PET)、ポリブチレンテレフタレート、ポリエチレンナフタレート(PEN)、ポリカーボネート(PC)等のフィルムが好ましく用いられる。
また、デバイスを封止する加工工程で高温処理が必要な場合には、耐熱性と透明性を両立した透明ポリイミドのフィルム、例えば東洋紡株式会社製、透明ポリイミド系フィルム・タイプHMや、三菱瓦斯化学株式会社製、透明ポリイミド系フィルム・ネオプリムL L−3430などを好ましく用いることができる。
この樹脂基材1aの厚さは5〜500μm程度が好ましく、さらに好ましくは25〜250μmである。
また、樹脂基材1a(基材1)は透明であることが好ましい。樹脂基材1aが透明であって基材1上に形成する各種層も透明にすれば、光透過性を有する水蒸気バリアーフィルムとすることが可能となる。基材1が光透過性を有すれば、有機EL素子の発光光を透過させたり、太陽電池へ向かう太陽光を通過させたりすることが可能になるので、有機EL素子や太陽電池を封止する封止フィルム(透明基板)として好適に用いることができる。
また、上記の樹脂材料からなる樹脂基材1aは、従来公知の一般的な製法により製造することが可能である。例えば、材料となる樹脂を押し出し機により溶融し、環状ダイやTダイにより押し出して急冷することにより、実質的に無定形で配向していない未延伸の基材を製造することができる。また、未延伸の基材を一軸延伸、テンター式逐次二軸延伸、テンター式同時二軸延伸、チューブラー式同時二軸延伸等の公知の方法により、基材の流れ(縦軸)方向、または基材の流れ方向と直角(横軸)方向に延伸することにより延伸基材を製造することができる。この場合の延伸倍率は、基材の原料となる樹脂に合わせて適宜選択することできるが、縦軸方向及び横軸方向にそれぞれ2〜10倍であることが好ましい。
また、本実施形態における基材1(樹脂基材1a)の表面には、水蒸気バリアー層3(蒸着層やポリシラザン改質層)との密着性を向上させるためのアンカーコート層を形成してもよい。
このアンカーコート層に用いられるアンカーコート剤としては、ポリエステル樹脂、イソシアネート樹脂、ウレタン樹脂、アクリル樹脂、エチレンビニルアルコール樹脂、ビニル変性樹脂、エポキシ樹脂、変性スチレン樹脂、変性シリコン樹脂、及びアルキルチタネート、シロキサン系ポリマー等を、1種または2種以上併せて使用することができる。これらのアンカーコート剤には、従来公知の添加剤を加えることもできる。そして、上記のアンカーコート剤は、ロールコート、グラビアコート、ナイフコート、ディップコート、スプレーコート等の公知の方法により基材上にコーティングし、溶剤、希釈剤等を乾燥除去することによりアンカーコート層を形成することができる。
このアンカーコート剤の塗布量としては、乾燥状態で0.1〜5g/m2程度が好ましい。
また、本実施形態における基材1の表面には平滑層2を設けてもよい。平滑層2は樹脂基材1aの一方の面上に形成されている。
平滑層2は、微小な突起等が存在する樹脂基材1aの粗面を平坦化し、樹脂基材1a(基材1)表面の突起等によって基材1に成膜する蒸着層(水蒸気バリアー層3)などに凹凸やピンホールが生じないようにするために設けられる。このような平滑層2は、例えば、感光性樹脂を硬化させて形成される。
この平滑層2の形成に用いられる感光性樹脂としては、例えば、ラジカル反応性不飽和結合を有するアクリレート化合物を含有する樹脂組成物、アクリレート化合物とチオール基を有するメルカプト化合物を含有する樹脂組成物、エポキシアクリレート、ウレタンアクリレート、ポリエステルアクリレート、ポリエーテルアクリレート、ポリエチレングリコールアクリレート、グリセロールメタクリレート等の多官能アクリレートモノマーを溶解させた樹脂組成物等が挙げられる。また、上記のような樹脂組成物の任意の混合物を使用することも可能であり、光重合性不飽和結合を分子内に1個以上有する反応性モノマーを含有している感光性樹脂であれば特に制限はない。反応性モノマーは、1種または2種以上の混合物として、あるいは、その他の化合物との混合物として使用することができる。
また、感光性樹脂の組成物は、光重合開始剤を含有する。光重合開始剤は、1種または2種以上の組み合わせで使用することができる。
また、平滑層2を形成する際に必要に応じて、上記した感光性樹脂に酸化防止剤、紫外線吸収剤、可塑剤等の添加剤を加えることができる。また、形成した平滑層2への成膜性向上や、平滑層2に成膜された膜のピンホール発生防止等のために適切な樹脂や添加剤を使用してもよい。
なお、感光性樹脂を溶媒に溶解または分散させた塗布液を用いて平滑層2を形成する際に使用する溶媒としては、メタノール、エタノール、n−プロパノール、イソプロパノール、エチレングリコール、プロピレングリコール等のアルコール類、α−もしくはβ−テルピネオール等のテルペン類、アセトン、メチルエチルケトン、シクロヘキサノン、N−メチル−2−ピロリドン、ジエチルケトン、2−ヘプタノン、4−ヘプタノン等のケトン類、トルエン、キシレン、テトラメチルベンゼン等の芳香族炭化水素類、セロソルブ、メチルセロソルブ、エチルセロソルブ、カルビトール、メチルカルビトール、エチルカルビトール、ブチルカルビトール、プロピレングリコールモノメチルエーテル、プロピレングリコールモノエチルエーテル、ジプロピレングリコールモノメチルエーテル、ジプロピレングリコールモノエチルエーテル、トリエチレングリコールモノメチルエーテル、トリエチレングリコールモノエチルエーテル等のグリコールエーテル類、酢酸エチル、酢酸ブチル、セロソルブアセテート、エチルセロソルブアセテート、ブチルセロソルブアセテート、カルビトールアセテート、エチルカルビトールアセテート、ブチルカルビトールアセテート、プロピレングリコールモノメチルエーテルアセテート、プロピレングリコールモノエチルエーテルアセテート、2−メトキシエチルアセテート、シクロヘキシルアセテート、2−エトキシエチルアセテート、3−メトキシブチルアセテート等の酢酸エステル類、ジエチレングリコールジアルキルエーテル、ジプロピレングリコールジアルキルエーテル、3−エトキシプロピオン酸エチル、安息香酸メチル、N,N−ジメチルアセトアミド、N,N−ジメチルホルムアミド等を挙げることができる。
ここで、反応性シリカ粒子の平均粒子径としては、0.001〜0.1μmの平均粒子径であることが好ましい。平均粒子径をこのような範囲にすることにより、後述する平均粒子径1〜10μmの無機粒子からなるマット剤と組合せて用いることによって、防眩性と解像性とをバランスよく満たす光学特性と、ハードコート性とを兼ね備えた平滑層2を形成し易くなる。なお、このような効果をより得易くする観点からは、さらに平均粒子径が0.001〜0.01μmの反応性シリカ粒子を用いることがより好ましい。
本実施形態における平滑層2中には、上述の様な無機粒子を質量比として20%以上60%以下含有することが好ましい。20%以上添加することで、水蒸気バリアー層3との密着性が向上する。一方60%を超えると、フィルムを湾曲させたり、加熱処理を行った場合にクラックが生じたり、水蒸気バリアーフィルムの透明性や屈折率等の光学的物性に影響を及ぼすことがある。
なお、本実施形態では、重合性不飽和基修飾加水分解性シランが、加水分解性シリル基の加水分解反応によって、シリカ粒子との間に、シリルオキシ基を生成して化学的に結合しているようなものを、反応性シリカ粒子として用いることができる。加水分解性シリル基としては、例えば、アルコキシリル基、アセトキシリル基等のカルボキシリレートシリル基、クロシリル基等のハロゲン化シリル基、アミノシリル基、オキシムシリル基、ヒドリドシリル基等が挙げられる。重合性不飽和基としては、アクリロイルオキシ基、メタクリロイルオキシ基、ビニル基、プロペニル基、ブタジエニル基、スチリル基、エチニイル基、シンナモイル基、マレート基、アクリルアミド基等が挙げられる。
本実施形態において、平滑層2の厚さとしては、1〜10μm、好ましくは2〜7μmであることが望ましい。1μm以上にすることにより、平滑層2を有する水蒸気バリアーフィルムとしての平滑性を十分なものにし易くなり、10μm以下にすることにより、水蒸気バリアーフィルムの光学特性のバランスを調整し易くなると共に、平滑層2を水蒸気バリアーフィルムの一方の面にのみ設けた場合におけるその水蒸気バリアーフィルムのカールを抑え易くすることができるようになる。
本実施形態における水蒸気バリアー層3には、蒸着法によって成膜された水蒸気バリアー性を有する蒸着層や、ポリシラザンを含む液体を塗布し乾燥した塗膜に真空紫外光を照射することによって水蒸気バリアー性を有するポリシラザン改質層に転化した層を用いることができる。
基材1に水蒸気バリアー層3とする薄膜である蒸着層を形成する方法としては、物理気相成長法及び化学気相成長法が挙げられる。
物理気相成長法は、気相中で基材1の表面に物理的手法により目的とする物質(例えば、炭素膜等)の薄膜を堆積する方法であり、これらの方法としては、蒸着法(抵抗加熱法、電子ビーム蒸着法、分子線エピタキシー法)、イオンプレーティング法、スパッタリング法等がある。
一方、化学気相成長法(化学蒸着法、Chemical Vapor Deposition)は、気相中で基材1に、目的とする薄膜の成分を含む原料ガスを励起した放電ガスに混合して供給し、基材表面或いは気相での化学反応によって、基材1上に薄膜を堆積する方法である。特に、化学反応を活性化する目的で、プラズマなどを発生させる方法などがあり、熱CVD法、触媒化学気相成長法、光CVD法、プラズマCVD法、大気圧プラズマCVD法など公知のCVD方式等がある。
本発明においては、基材1に水蒸気バリアー層3としての蒸着層(蒸着膜)を形成する方法として、成膜速度や処理面積の観点からプラズマCVD法が好ましく、真空を必要としない大気圧プラズマCVD法がさらに好ましい。大気圧または大気圧近傍でのプラズマCVD処理を行う大気圧プラズマCVD法は、真空下のプラズマCVD法に比べ、減圧にする必要がなく生産性が高いだけでなく、プラズマ密度が高密度であるために成膜速度が速く、更には通常のCVD法の条件に比較して、大気圧下という高圧力条件ではガスの平均自由工程が非常に短いために極めて均質の膜が得られる。
本発明でいう大気圧もしくはその近傍の圧力とは、20kPa〜110kPa程度であり、本発明に記載の良好な効果を得るためには、93kPa〜104kPaであることが好ましい。また、本発明でいう励起したガスとは、エネルギーを得ることによって、ガス中の分子の少なくとも一部が、今ある状態からより高いエネルギー状態へ移ることをいい、励起ガス分子、ラジカル化したガス分子、イオン化したガス分子を含むガスがこれに該当する。
なお、本発明におけるプラズマCVD法により形成される金属酸化物、金属窒化物、金属酸窒化物を含有する蒸着層は、金属酸化物、金属窒化物、金属酸窒化物のうち少なくとも1つを含む蒸着膜であり、金属酸化物、金属窒化物、金属酸窒化物、金属炭化物等の複合化合物であってもよい。
このような無機膜(セラミック膜)の原料としては、典型または遷移金属元素を有していれば、常温常圧下で気体、液体、固体いずれの状態であっても構わない。気体の場合にはそのまま放電空間に導入できるが、液体、固体の場合は、加熱、バブリング、減圧、超音波照射等の手段により気化させて使用する。また、溶媒によって希釈して使用してもよく、溶媒はメタノール、エタノール、n−ヘキサンなどの有機溶媒及びこれらの混合溶媒を使用できる。なお、これらの希釈溶媒は、プラズマ放電処理中において分子状、原子状に分解されるため、成膜への影響は殆ど無視することができる。
また、金属元素を含む原料ガスを分解して無機化合物を得るための分解ガスとしては、水素ガス、メタンガス、アセチレンガス、一酸化炭素ガス、二酸化炭素ガス、窒素ガス、アンモニアガス、亜酸化窒素ガス、酸化窒素ガス、二酸化窒素ガス、酸素ガス、水蒸気、フッ素ガス、フッ化水素、トリフルオロアルコール、トリフルオロトルエン、硫化水素、二酸化硫黄、二硫化炭素、塩素ガスなどが挙げられる。
金属元素を含む原料ガスと、分解ガスを適宜選択することで、金属酸化物、また金属酸化物と金属炭化物、金属窒化物、金属酸窒化物、金属ハロゲン化物、金属硫化物等の混合物のセラミック膜を得ることができる。
このような放電ガスとしては、窒素ガス及び/または周期表の第18属原子、具体的には、ヘリウム、ネオン、アルゴン、クリプトン、キセノン、ラドン等が用いられる。これらの中でも、窒素、ヘリウム、アルゴンが好ましく用いられ、特に窒素がコストも安く好ましい。
放電ガスと反応性ガスを混合した混合ガスをプラズマ放電処理装置に供給することで蒸着層を形成する。放電ガスと反応性ガスの割合は、得ようとする膜の性質によって異なるが、混合ガス全体に対し、放電ガスの割合を50%以上として反応性ガスを供給することが好ましい。
本発明における水蒸気バリアー層3として用いるセラミック膜においては、セラミック膜が含有する無機化合物が、SiOx、SiOxCy(x=1.5〜2.0、y=0〜0.5)、またはSiOxNy(x=0.1〜2、y=0.1〜1.3)であることが好ましく、特にガスバリアー性、水分の透過性、光線透過性及び大気圧プラズマCVD適性の観点から、SiOxまたはSiOxNyであることが好ましい。
以上のように、上記したような原料ガス(反応性ガス)を放電ガスと共に使用することにより様々な無機膜(セラミック膜)を形成することができる。本発明の蒸着層は、これらの条件を変えた複数の層から構成されてもよく、また放電ガスと反応性ガスの比率や、放電の条件を連続的に変化させた、膜厚方向に不均質な膜から構成されてもよい。
CVD法(化学的気相成長法)は、揮発・昇華した有機金属化合物が高温の支持体(基材1)表面に付着し、熱により分解反応が起き、熱的に安定な無機物の薄膜が生成されるというものである。このような通常のCVD法(熱CVD法とも称する)では、通常500℃以上の基板温度が必要であるため、プラスチック製の基材1への製膜には使用することが難しい。
一方、プラズマCVD法は支持体(基材1)近傍の空間に電界を印加し、プラズマ状態となった気体が存在する空間(プラズマ空間)を発生させ、揮発・昇華した有機金属化合物がこのプラズマ空間に導入されて分解反応が起きた後に支持体(基材1)上に吹きつけられることにより、無機物の薄膜を形成するというものである。プラズマ空間内では、数%の高い割合の気体がイオンと電子に電離しており、ガスの温度は低く保たれるものの、電子温度は非常な高温のため、この高温の電子、あるいは低温ではあるがイオン・ラジカルなどの励起状態のガスと接するために、無機膜の原料である有機金属化合物は低温でも分解することができる。従って、無機物を製膜する支持体(基材1)についても低温化することができ、プラスチック製の基材1(樹脂フィルム)へも十分製膜することが可能な製膜方法である。このプラズマCVD法によれば、樹脂フィルム(基材1)上にセラミック膜を形成させたときの膜密度が緻密であり、安定した性能を有する薄膜が得られる。また、残留応力が圧縮応力で、0.01MPa以上、20MPa以下という範囲のセラミック膜が安定に得られることが特徴である。
なお、本発明に適用できるプラズマ放電処理装置としては、例えば、特開2004−68143号公報、同2003−49272号公報、国際公開第02/48428号パンフレット等に記載されている装置を挙げることができる。
また、本発明において、基材1に水蒸気バリアー層3としての蒸着層(蒸着膜)を形成する他の方法に、真空成膜法の一つであるスパッタリング法がある。
スパッタリング法とは、電場や磁場を利用してアルゴンガス等の不活性ガスの電離(プラズマ化)を行い、更に、電離したイオンを加速することにより得られる運動エネルギーによって、ターゲットの原子を叩き出す。そして、その叩き出された原子が対向する基材1上に堆積し、目的とする膜を形成する物理的プロセスである。スパッタリング法では、アルゴン等のスパッタガスを、電場や磁場を利用して電離(プラズマ化)し、加速することで、ターゲット表面に衝突させる。そして、プラズマ粒子が衝突したターゲットからはターゲット原子がはじき出され、このはじき出された原子が被処理体上に堆積してスパッタ膜が形成される。
本発明では、スパッタリング法のターゲットに二酸化珪素、窒化珪素、酸窒化珪素を使用し、ターゲットとスパッタ膜が形成される基材1の存在する雰囲気は、窒素含有ガス雰囲気、あるいは不活性ガス中に窒素含有ガスを添加した雰囲気等が挙げられ、特に窒素、一酸化二窒素、アンモニア、三フッ化窒素等の窒素含有ガスの含有する雰囲気が好ましく、水蒸気バリアー性を損ねることなく透明な水蒸気バリアー層3の形成することができる。水蒸気バリアー層3は、その他の成分として、アルミニウム、亜鉛、アンチモン、インジウム、セリウム、カルシウム、カドミウム、銀、金、クロム、珪素、コバルト、ジルコニウム、スズ、チタン、鉄、銅、ニッケル、白金、パラジウム・ビスマス、マグネシウム、マンガン、モリブデン、バナジウム、バリウム等の金属またはこれら酸化物または窒化物の無機材料を含有させることで、高い水蒸気バリアー性を維持させたり向上させたりすることができる。
なお、スパッタリング雰囲気下への窒素含有ガスの流量が少なすぎると、窒化酸化珪素膜の水蒸気バリアー層3の透明性が低下して好ましくない。また、一方でその窒素含有ガスの流量が多すぎると、不活性ガスの電離(プラズマ化)が生じにくくなり好ましくない。例えば、マグネトロンスパッタリング装置で、ターゲットに窒化珪素を使用し、30sccm流量のアルゴン(Ar)ガス、窒素(N2)ガスを導入し、周波数13.56MHzの高周波電力(投入電力1.2kW)を印加し、成膜圧力0.25Pa、膜厚120〜150nmで、窒化酸化珪素膜の水蒸気バリアー層3を形成する場合、窒素ガスの流量は、5〜15sccm程度が好ましい。これにより、透明性を損ねることなく、良好な水蒸気バリアー性を有する水蒸気バリアー層3が形成できる。ここで、上記の流量の単位[sccm]は真空成膜技術においてよく使われる単位であり、標準立方糎米(standard cubic centimeter)を意味し、標準条件に変換したガス流量を表す。この標準条件は、温度25℃及び圧力1013hPaと定義されている。
上述した各特性を有する窒化酸化珪素膜を5〜300nmの厚さという薄い厚さで形成した積層体は、窒化酸化珪素膜にクラックが入りづらいので、優れた水蒸気バリアー性を発揮する水蒸気バリアー層3として用いることができる。窒化酸化珪素膜が5nm未満の場合は、窒化酸化珪素膜が基材全面を覆うことができないことがあり、水蒸気バリアー性を向上させることができない。一方、窒化酸化珪素膜の厚さが300nmを超えると、クラックが入り易くなること、透明性や外観が低下すること、基材フィルムのカールが増大すること、さらに、量産し難く生産性が低下しコストが増大すること等の不具合が起こり易くなる。
また、本発明において、基材1に水蒸気バリアー層3を形成する他の方法として、ポリシラザン改質層を形成する方法が挙げられる。この方法は、ポリシラザンを含む液体を塗布し乾燥した後に、その乾燥した塗膜に真空紫外光を照射することによってポリシラザン改質層とした水蒸気バリアー層3を形成する方法である
本発明で用いられる「ポリシラザン」とは、珪素−窒素結合を持つポリマーで、Si−N、Si−H、N−H等の結合を有するSiO2、Si3N4及び両方の中間固溶体SiOxNy等のセラミック前駆体無機ポリマーである。
そのポリシラザンを含む液体を塗布する塗布方法としては、従来公知の適切な方法が採用され得る。具体例としては、スピンコート法、ロールコート法、フローコート法、インクジェット法、スプレーコート法、プリント法、ディップコート法、流延成膜法、バーコート法、グラビア印刷法等が挙げられる。塗布厚さは、目的に応じて適切に設定され得る。例えば、塗布厚さは、乾燥後の厚さが1nm〜100μm程度であることが好ましく、さらに好ましくは10nm〜10μm程度、最も好ましくは10nm〜1μm程度である。

本発明では、得られるガスバリアー膜(水蒸気バリアー層3)としての緻密性の観点から、R1、R2、及びR3の全てが水素原子であるパーヒドロポリシラザンが特に好ましい。
また、そのSiと結合する水素原子部分の一部がアルキル基等で置換されたオルガノポリシラザンは、メチル基等のアルキル基を有することにより下地である基材1との接着性が改善され、かつ硬くてもろいポリシラザンによるセラミック膜に靭性を持たせることができ、より膜厚(平均膜厚)を厚くした場合でもクラックの発生が抑えられる利点がある。そこで用途に応じて適宜、パーヒドロポリシラザンとオルガノポリシラザンを選択してよく、混合して使用することもできる。
低温でセラミック化するポリシラザンの他の例としては、上記一般式(1)で表される単位からなる主骨格を有するポリシラザンに、珪素アルコキシドを反応させて得られる珪素アルコキシド付加ポリシラザン(例えば、特開平5−238827号公報参照)、グリシドールを反応させて得られるグリシドール付加ポリシラザン(例えば、特開平6−122852号公報参照)、アルコールを反応させて得られるアルコール付加ポリシラザン(例えば、特開平6−240208号公報参照)、金属カルボン酸塩を反応させて得られる金属カルボン酸塩付加ポリシラザン(例えば、特開平6−299118号公報参照)、金属を含むアセチルアセトナート錯体を反応させて得られるアセチルアセトナート錯体付加ポリシラザン(例えば、特開平6−306329号公報参照)、金属微粒子を添加して得られる金属微粒子添加ポリシラザン(例えば、特開平7−196986号公報参照)等が挙げられる。
ポリシラザンを含有する塗布液を調製する有機溶媒としては、ポリシラザンと容易に反応するようなアルコール系や水分を含有するものを用いることは好ましくない。従って、具体的には、脂肪族炭化水素、脂環式炭化水素、芳香族炭化水素等の炭化水素溶媒、ハロゲン化炭化水素溶媒や、脂肪族エーテル、脂環式エーテル等のエーテル類が使用できる。詳しくは、ペンタン、ヘキサン、シクロヘキサン、トルエン、キシレン、ソルベッソ、ターベン等の炭化水素、塩化メチレン、トリクロロエタン等のハロゲン炭化水素、ジブチルエーテル、ジオキサン、テトラヒドロフラン等のエーテル類等がある。これらの有機溶媒は、ポリシラザンの溶解度や有機溶媒の蒸発速度等の特性にあわせて選択し、複数の有機溶媒を混合してもよい。
ポリシラザン含有の塗布液中におけるポリシラザン濃度は、目的とするポリシラザン改質層の膜厚や塗布液のポットライフによっても異なるが、0.2〜35質量%程度であることが好ましい。
ポリシラザン含有の塗布液中には、酸化珪素化合物への転化を促進するため、アミンや金属の触媒を添加することもできる。具体的には、AZエレクトロニックマテリアルズ(株)製のアクアミカ NAX120−20、NN110、NN310、NN320、NL110A、NL120A、NL150A、NP110、NP140、SP140等が挙げられる。
改質処理前または改質処理中にポリシラザン改質層中に入りうる水分の供給としては、例えば塗布基材表面からの移行、あるいは雰囲気中の水蒸気の吸収がある。基材側からポリシラザン改質層中に移行する水分の制御は、ポリシラザン含有の塗布液を塗布する前に基材を一定の温度湿度環境下で保存して、含水量を所望の値に制御することができる。所望の値は、後述の雰囲気中の湿度によって異なるが、通常、重量として1000ppm以下、好ましくは、300ppm以下である。
ポリシラザン含有の塗布液を基材上に塗布乾燥する工程においては、主に有機溶媒を取り除くため、乾燥条件を熱処理等の方法で適宜決めることができ、熱処理温度は迅速処理の観点から高い温度であることが好ましいが、樹脂フィルムである基材1に対する熱ダメージを考慮し、温度と処理時間を適宜決定することが好ましい。例えば、基材1として、ガラス転位温度(Tg)が70℃のポリエチレンテレフタレート基材を用いる場合には、熱処理温度は150℃以下を設定することができる。処理時間は溶媒が除去され、かつ基材1への熱ダメージが少なくなるように短時間に設定することが好ましく、熱処理温度が150℃以下であれば30分以内に設定することができる。
ポリシラザン含有の塗布液を基材上に塗布乾燥する工程における雰囲気は、比較的低湿に制御されていることが好ましいが、低湿度環境における湿度は温度により変化するので、温度と湿度の関係は露点温度の規定により好ましい形態が示される。好ましい露点温度は4℃以下(温度25℃/湿度25%)で、より好ましい露点温度は−8℃(温度25℃/湿度10%)以下、さらに好ましい露点温度は−31℃(温度25℃/湿度1%)以下である。また、水分を取り除きやすくするため、減圧乾燥してもよい。減圧乾燥における圧力は常圧〜0.1MPaを選ぶことができる。
本発明におけるポリシラザンの改質処理とは、ポリシラザン化合物の一部または全部が、酸化珪素または酸化窒化珪素への転化する反応をいう。
この改質処理は、ポリシラザンの転化反応に基づく公知の方法を選ぶことができる。ポリシラザン化合物の置換反応による酸化珪素膜または酸化窒化珪素膜の形成には450℃以上の高温が必要であり、樹脂フィルムを基材1に用いたフレキシブル基板においては、適応が難しい。従って、本発明の水蒸気バリアーフィルムを作製するに際しては、プラスチック基板への適応という観点から、より低温で、転化反応が可能な紫外光を使う転化反応が好ましい。
この紫外光照射により、セラミックス化に寄与するO2とH2Oや、紫外線吸収剤、ポリシラザン自身が励起、活性化される。そして、励起したポリシラザンのセラミックス化が促進され、得られるセラミックス膜が緻密になる。紫外光照射は、塗膜形成後であればいずれの時点で実施しても有効である。
本発明での真空紫外光照射処理には、常用されているいずれの紫外線発生装置を使用することが可能である。なお、本発明でいう紫外光とは、一般には、真空紫外光とよばれる10〜200nmの波長を有する電磁波を含む紫外光をいう。
基材1にプラスチックフィルムを用いた場合を例にとると、例えば、2kW(80W/cm×25cm)のランプを用い、基材表面の強度が20〜300mW/cm2、好ましくは50〜200mW/cm2になるように基材−紫外線照射ランプ間の距離を設定し、0.1秒〜10分間の照射を行うことができる。
一般に、紫外線照射処理時の基材温度が150℃以上になると、プラスチックフィルム等の場合には、基材1が変形したりその強度が劣化したりするなど、基材1の特性が損なわれることになる。しかしながら、ポリイミド等の耐熱性の高いフィルムなどの場合には、より高温での改質処理が可能である。従って、この紫外線照射時の基材温度としては、一般的な上限はなく、基材1の種類によって当業者が適宜設定することができる。また、紫外線照射雰囲気に特に制限はなく、空気中で実施すればよい。
このような紫外線の発生手段としては、例えば、メタルハライドランプ、高圧水銀ランプ、低圧水銀ランプ、キセノンアークランプ、カーボンアークランプ、エキシマランプ、UV光レーザー等が挙げられるが、特に限定されない。また、発生させた紫外線を改質前のポリシラザン層に照射する際には、効率向上と均一な照射を達成する観点から、発生源からの紫外線を反射板で反射させてから改質前のポリシラザン層に当てることが望ましい。
紫外線照射は、バッチ処理にも連続処理にも適合可能であり、使用する基材1の形状によって適宜選定することができる。ポリシラザン改質層を有する基材1が長尺フィルム状である場合には、これを搬送させながら上記のような紫外線発生源を具備した乾燥ゾーンで連続的に紫外線を照射することによりセラミックス化することができる。紫外線照射に要する時間は、使用する基材1やポリシラザン改質層の組成、濃度にもよるが、一般に0.1秒〜10分であり、好ましくは0.5秒〜3分である。
また、真空紫外光(VUV)を照射する際の、酸素濃度は300ppm〜10000ppm(1%)とすることが好ましく、更に好ましくは、500ppm〜5000ppmである。このような酸素濃度の範囲に調整することにより、酸素過多の水蒸気バリアー層3の生成を防止してバリアー性の劣化を防止することができる。
真空紫外光(VUV)照射時にこれら酸素以外のガスとしては乾燥不活性ガスを用いることが好ましく、特にコストの観点から乾燥窒素ガスにすることが好ましい。
酸素濃度の調整は照射庫内へ導入する酸素ガス、不活性ガスの流量を計測し、流量比を変えることで調整可能である。
なお、Xe、Kr、Ar、Ne等の希ガスの原子は化学的に結合して分子を作らないため、不活性ガスと呼ばれる。しかし、放電等によりエネルギーを得た希ガスの原子(励起原子)は他の原子と結合して分子を作ることができる。希ガスがキセノンの場合には、
e+Xe→e+Xe*
Xe*+Xe+Xe→Xe2 *+Xe
となり、励起されたエキシマ分子であるXe2 *が基底状態に遷移するときに172nmのエキシマ光(真空紫外光)を発光する。
エキシマランプの特徴としては、放射が一つの波長に集中し、必要な光以外がほとんど放射されないので効率が高いことが挙げられる。また、余分な光が放射されないので、対象物の温度を低く保つことができる。さらには始動・再始動に時間を要さないので、瞬時の点灯点滅が可能である。
また、効率よくエキシマ発光を得る方法としては、誘電体バリアー放電以外には無電極電界放電も知られている。無電極電界放電とは、容量性結合による放電であり、別名RF放電とも呼ばれる。ランプと電極及びその配置は、基本的には誘電体バリアー放電と同じでよいが、両極間に印加される高周波は数MHzで点灯される。無電極電界放電はこのように空間的にまた時間的に一様な放電が得られる。
そして、Xeエキシマランプは、波長の短い172nmの紫外線を単一波長で放射することから発光効率に優れている。この光は、酸素の吸収係数が大きいため、微量な酸素でラジカルな酸素原子種やオゾンを高濃度で発生することができる。また、有機物の結合を解離させる波長の短い172nmの光のエネルギーは能力が高いことが知られている。この活性酸素やオゾンと紫外線放射が持つ高いエネルギーによって、短時間でポリシラザン膜の改質を実現できる。従って、波長185nm、254nmの紫外線を発する低圧水銀ランプやプラズマ洗浄と比べて、高スループットに伴うプロセス時間の短縮や設備面積の縮小、熱によるダメージを受けやすい有機材料やプラスチック基板、樹脂フィルム等への照射を可能としている。
また、エキシマランプは光の発生効率が高いため、低い電力の投入で点灯させることが可能である。また、光による温度上昇の要因となる波長の長い光は発せず、紫外線領域で単一波長のエネルギーを照射するため、照射対象物の表面温度の上昇が抑えられる特徴を有する。このため、熱の影響を受けやすいとされるポリエチレンテレフタレート等の樹脂フィルムを基材1とする水蒸気バリアーフィルムへの照射に適している。
これとは逆に、本発明の水蒸気バリアーフィルムにおける水蒸気バリアー層3において、基材1から遠い配置の水蒸気バリアー層3は、加水分解層4が形成された後に積層されるため、ポリシラザン改質層からなる水蒸気バリアー層3であることが好ましい。これは、ポリシラザンを含む液体を塗布し乾燥した塗膜に真空紫外光を照射することにより形成する方法で設けられるポリシラザン改質層からなる水蒸気バリアー層3の方が、成膜時に加水分解層4に直接与える熱やその他のエネルギーが比較的少ないので、加水分解層4に与えるダメージが少なく、加水分解層4の機能を比較的良好に維持することができることによる。
本実施形態における加水分解層4は、加水分解性化合物を含有させて成膜した層(加水分解性化合物含有層)である。
本発明の加水分解層4に用いられる加水分解性化合物は、水・水蒸気と反応して加水分解を生じるものであり、好ましくは加水分解により、比較的低分子量のアルコール、アンモニアまたはアルキル基を有するアミン化合物、無機酸を放出する化合物である
このような加水分解性化合物として好ましく用いられるものには、金属アルコキシド、シラン化合物、シラザン化合物、金属ハロゲン化物等が挙げられる。
つまり、加水分解層4は、加水分解性化合物として金属アルコキシド、シラン化合物、シラザン化合物、金属ハロゲン化物のうち少なくとも1つを含み、加水分解によってアルコール、アミン化合物、無機酸のうち少なくとも1つを放出する機能層であることが好ましい。
そして、加水分解層4は、水蒸気バリアー層3に存在する欠陥(例えば、微細孔やひび割れ箇所)を通じて基材1側から侵入した水と反応して加水分解を起こし、水を分解して生成したアルコール、アミン化合物、無機酸などの生成物を、基材1側へ向けて拡散させて除去する機能を有している。
なお、本発明の加水分解層4の厚みは、通常1nm以上1000nm以下であり、好ましくは10nm以上500nm以下である。
例えば、テトラクロロシラン、メチルトリクロロシラン、ジメチルジクロロシラン、トリメチルクロロシラン、フェニルトリクロロシラン、メチルトリメトキシシラン、ジメチルジメトキシシラン、フェニルトリメトキシシラン、テトラエトキシシラン、メチルトリエトキシシラン、ジメチルジエトキシシラン、フェニルトリエトキシシラン、n−プロピルトリメトキシシラン、n−プロピルトリエトキシシラン、ヘキシルトリメトキシシラン、ヘキシルトリエトキシシラン、デシルトリメトキシシラン、デシルトリメトキシシラン、トリフルオロプロピルトリメトキシシラン、ヘキサメチルジシラザン、パーヒドロポリシラザン、メチルポリシラザン、ビニルトリメトキシシラン、ビニルトリエトキシシラン、2−(3,4−エポキシシクロヘキシル)エチルトリメトキシシラン、3−グリシドキシプロピルメチルジメトキシシラン、3−グリシドキシプロピルトリメトキシシラン、3−グリシドキシプロピルメチルジエトキシシラン、3−グリシドキシプロピルトリエトキシシラン、p−スチリルトリメトキシシラン、3−メタクリロキシプロピルメチルジメトキシシラン、3−メタクリロキシプロピルトリメトキシシラン、3−メタクリロキシプロピルメチルジエトキシシラン、3−メタクリロキシプロピルトリエトキシシラン、3−アクリロキシプロピルトリメトキシシラン、N−2−(アミノエチル)−3−アミノプロピルメチルジメトキシシラン、N−2−(アミノエチル)−3−アミノプロピルトリメトキシシラン、N−2−(アミノエチル)−3−アミノプロピルトリエトキシシラン、3−アミノプロピルトリメトキシシラン、3−アミノプロピルトリエトキシシラン、N−フェニル−3−アミノプロピルトリメトキシシラン、3−ウレイドプロピルトリエトキシシラン、3−クロロプロピルトリメトキシシラン、3−メルカプトプロピルメチルジメトキシシラン、3−メルカプトプロピルトリメトキシシラン、ビス(トリエトキシシリルプロピル)テトラスルフィド、3−イソシアネートプロピルトリエトキシシラン、テトライソシアネートシラン、メチルトリイソシアネートシラン等のシラン化合物などが挙げられる。
そして、加水分解性化合物としては特に、メタノールやエタノール等のアルコールや、アンモニアを加水分解時に生成する化合物が好ましく用いられる。加水分解生成物が低分子量であるほど、水蒸気バリアー層3の欠陥を通って加水分解層4から排出され除去されやすくなるためである。具体的には有機または無機のポリシラザンや1置換から4置換のメトキシ基、エトキシ基を有するアルコキシシラン類を好適に用いることができる。
加水分解性化合物が未改質ポリシラザンの場合、
−(SiH2NH)− + 2H2O → SiO2 + NH3 + 2H2
加水分解性化合物が金属アルコキシドの場合(M:金属、R:アルキル基)、
M(OR)n + xH2O → M(OH)x(OR)n−x + xROH
加水分解性化合物が金属ハロゲン化物の場合、
TiCl4 + 2H2O → TiO2 + 4HCl
SiCl4 + 2H2O → SiO2 + 4HCl
加水分解層4には、常温で固体の有機または無機の化合物を添加することができる。特に、加水分解性化合物が常温で液体の場合には、その添加物が加水分解層4に安定して保持されるものであれば公知の熱可塑性樹脂、熱または光硬化性樹脂、無機微粒子化合物を用いることができる。また加水分解の速度を調整する目的で、酸、または塩基等の触媒を添加することもできる。
但し、特開2009−90633号公報(特許文献1)、特開2006−82241号公報(特許文献2)に記載された様な吸湿性ポリマーやアルカリ土類金属一酸化物等、吸湿により体積の増大を伴う化合物は、多くとも30重量%未満、好ましくは10重量%以下に添加量を抑えることが必要である。
本発明の加水分解層4は、基材1上に設けられた少なくとも2層の水蒸気バリアー層3の間に介装されて積層されている。この構成によって、加水分解性化合物の不要な加水分解の進行を抑えることができる。
加水分解層4の形成方法としては、水分含有率の低い溶媒に加水分解性化合物を溶解、分散した液を低湿度環境下で塗布し乾燥することによって形成することが好ましい。ここで低湿度環境における湿度は温度により変化するので、温度と湿度の関係は露点温度の規定により好ましい形態が示される。好ましい露点温度は4℃以下(温度25℃/湿度25%)で、より好ましい露点温度は−8℃(温度25℃/湿度10%)以下、さらに好ましい露点温度は−31℃(温度25℃/湿度1%)以下である。
加水分解性化合物が液体であり、加水分解後に固体に変化するものであれば、加水分解性化合物液を塗布し、乾燥時に塗布液または雰囲気中に少量の水分を加えて、加水分解性化合物の一部と反応させて固化することにより、未反応の加水分解性化合物を保持した加水分解層4を形成することができる。その際の雰囲気中水分は、露点で10℃以下で−100℃以上、好ましくは0℃以下で−70℃以上が好ましい。高露点で雰囲気中の水分が多い場合には、塗布・乾燥工程で加水分解性化合物が必要以上に消費されるために本発明の効果が低下する。
本発明の加水分解層4およびその加水分解層4上に積層される水蒸気バリアー層3(第2の水蒸気バリアー層)に、ともにポリシラザンを用いる場合、加水分解層4の下に積層される第1の水蒸気バリアー層3を形成した後、ポリシラザンを含む液体を第1の水蒸気バリアー層3上に塗布し乾燥して、バリアー層前駆体層である膜を形成したのちに真空紫外光を照射することにより、膜(バリアー層前駆体層)の表面近傍のみを改質し、膜(バリアー層前駆体層)の内部のポリシラザンを一部または全部未反応のまま加水分解性化合物として残留させることで、その膜の内部領域を加水分解層4として存在させ、その加水分解層4上に改質層である水蒸気バリアー層3(第2の水蒸気バリアー層)を形成するようにして、加水分解層4と水蒸気バリアー層3(第2の水蒸気バリアー層)を1回の塗布工程で形成することができる。
このような構成を実現するためには、真空紫外光の照射時の雰囲気中の酸素濃度を0.5%以上5.0%以下の比較的高濃度にし、かつ照射時間を短時間で行うようにする方法をとる。こうすることでバリアー層前駆体層の表面近傍が優先的に酸化改質された水蒸気バリアー層3に形成される。一方、表面よりも内部の領域は酸素の供給が遮断されていることで、酸化改質反応が進行せずに加水分解性化合物が残留することで加水分解層4として形成される。
加水分解層4に含有される加水分解性化合物は、水蒸気バリアー層3の欠陥部分から浸透した水分と加水分解反応を起こし、その結果生じたアルコール、アンモニアまたはアルキル基を有するアミン化合物、無機酸等の加水分解生成物(副生成物)を加水分解層4から放出する。その副生成物は、時間とともに加水分解層4を拡散し、水蒸気バリアー層3の欠陥部分から拡散排出される。加水分解層4から副生成物が排出され、加水分解層4における副生成物含有量が減少することは、下記の測定方法で確認することができる。
例えば、加水分解層4中の加水分解性化合物の存在量は、スパッタ法を用いて、飛行時間型二次イオン質量分析法(TOF−SIMS)や、X線光電子分光法(XPS)などによって、加水分解性化合物が有する特有の原子組成の量の変化をもって測定することができる。この加水分解層4中の加水分解性化合物の減少量は、加水分解層4から副生成物が排出された量に対応するので、測定された加水分解層4中の加水分解性化合物の減少量に基づいて、加水分解層4から排出された副生成物の排出量を求めることができる。
この測定には、通常XPS法が好ましく用いられる。XPS法では、超高真空下におかれた固体表面に軟X線を照射し、光電効果により表面から放出される光電子の運動エネルギーを測定する。光電子の脱出深さが数nmであることから、固体最表面に近い層を構成する原子や分子に関する情報が得られる。イオン(Ar、Xe)照射によるスパッタリングを併用することにより、表面から深さ方向への組成、結合状態の変化などの情報を得ることができる。水蒸気バリアー層の深さ方向の測定は、汚れや異物の付着の無い状態の最表面を基点として、水蒸気バリアー層と基材1とが接する面までの厚み深さまで一定間隔にスパッタを行い、組成を分析する。測定間隔は、1nm以上50nm以下であることが好ましく、より好ましくは2nm以上30nm以下である。水蒸気バリアー層の厚みが測定間隔の整数倍と異なる場合は、水蒸気バリアー層の厚み以下で最も近い整数倍の測定箇所を組成の比較の終点とする。
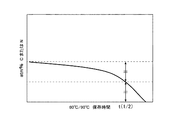
図3は、第2の水蒸気バリアー層/加水分解層/第1の水蒸気バリアー層における、膜厚方向の原子組成分布を模式的に表した図である。加水分解性化合物が含有する原子、例えばポリシラザンである場合は窒素原子、アルコキシシランの場合は炭素原子の膜厚方向に対する分布量を示している。本発明における半減期t(1/2)は、この分布を、60℃/90%RH雰囲気下で保存した時間に対する変化量を観察して、加水分解層4中の特定原子の存在量が、半分になった時間と定義する(図4参照)。具体的には、図3に示された加水分解層領域内の原子組成の分布曲線が形成する面積の比率変化で算出できる。
そして、本発明の水蒸気バリアーフィルムに用いる加水分解層4における、60℃/90%RH雰囲気下での炭素または窒素原子組成比の半減期t(1/2)であって、加水分解層4における加水分解性化合物が加水分解して生成した化合物に由来する炭素または窒素原子組成比の半減期t(1/2)が20時間以上1500時間以下であることが好ましい。
半減期がこれよりも短い場合には、加水分解性化合物で水分を捕捉することで維持できるガスバリアー効率が限定的である。また、半減期がこれよりも長い場合には、加水分解性化合物の水分捕捉能が必要十分でないか、バリアーフィルムのフレキシブル性が十分確保できないことがある。また、加水分解による副生成物が加水分解層4に貯留し過ぎてバリアーフィルムのガスバリアー性が悪化してしまうことがある。
なお、加水分解層4における炭素または窒素原子組成比の半減期t(1/2)は、例えば、水との反応性が高い加水分解性化合物と、水との反応性が低い加水分解性化合物との組成比を変化させることで調整することができる。
また、本実施形態における基材1の下面側の表面には、ブリードアウト防止層5を形成してもよい。
ブリードアウト防止層5は、平滑層2を有するフィルムを加熱した際に、樹脂基材1a中から表面に未反応のオリゴマー等が移行して、フィルム表面を汚染する現象を抑制する目的で、平滑層2を有する基材1の反対面に設けられる。ブリードアウト防止層5は、この機能を有していれば、基本的に平滑層2と同じ構成をとっても構わない。
ブリードアウト防止層5に含ませることが可能な重合性不飽和基を有する不飽和有機化合物としては、分子中に2個以上の重合性不飽和基を有する多価不飽和有機化合物、あるいは分子中に1個の重合性不飽和基を有する単価不飽和有機化合物等を挙げることができる。
その他の添加剤として、マット剤を含有してもよい。マット剤としては、平均粒子径が0.1〜5μm程度の無機粒子が好ましい。このような無機粒子としては、シリカ、アルミナ、タルク、クレイ、炭酸カルシウム、炭酸マグネシウム、硫酸バリウム、水酸化アルミニウム、二酸化チタン、酸化ジルコニウム等の1種または2種以上を併せて使用することができる。なお、無機粒子からなるマット剤は、ハードコート剤の固形分100質量部に対して2質量部以上、好ましくは4質量部以上、より好ましくは6質量部以上、20質量部以下、好ましくは18質量部以下、より好ましくは16質量部以下の割合で混合されていることが望ましい。
熱可塑性樹脂としては、アセチルセルロース、ニトロセルロース、アセチルブチルセルロース、エチルセルロース、メチルセルロース等のセルロース誘導体、酢酸ビニル及びその共重合体、塩化ビニル及びその共重合体、塩化ビニリデン及びその共重合体等のビニル系樹脂、ポリビニルホルマール、ポリビニルブチラール等のアセタール系樹脂、アクリル樹脂及びその共重合体、メタクリル樹脂及びその共重合体等のアクリル系樹脂、ポリスチレン樹脂、ポリアミド樹脂、線状ポリエステル樹脂、ポリカーボネート樹脂等が挙げられる。
熱硬化性樹脂としては、アクリルポリオールとイソシアネートプレポリマーとからなる熱硬化性ウレタン樹脂、フェノール樹脂、尿素メラミン樹脂、エポキシ樹脂、不飽和ポリエステル樹脂、シリコン樹脂等が挙げられる。
電離放射線硬化性樹脂としては、光重合性プレポリマーもしくは光重合性モノマー等の1種または2種以上を混合した電離放射線硬化塗料に、電離放射線(紫外線または電子線)を照射することで硬化するものを使用することができる。ここで光重合性プレポリマーとしては、1分子中に2個以上のアクリロイル基を有し、架橋硬化することにより3次元網目構造となるアクリル系プレポリマーが特に好ましく使用される。このアクリル系プレポリマーとしては、ウレタンアクリレート、ポリエステルアクリレート、エポキシアクリレート、メラミンアクリレート等が使用できる。また光重合性モノマーとしては、上記に記載した多価不飽和有機化合物等が使用できる。
光重合開始剤としては、アセトフェノン、ベンゾフェノン、ミヒラーケトン、ベンゾイン、ベンジルメチルケタール、ベンゾインベンゾエート、ヒドロキシシクロヘキシルフェニルケトン、2−メチル−1−(4−(メチルチオ)フェニル)−2−(4−モルフォリニル)−1−プロパン、α−アシロキシムエステル、チオキサンソン類等が挙げられる。
本実施形態におけるブリードアウト防止層5の厚さとしては、1〜10μm、好ましくは2〜7μmであることが望ましい。1μm以上にすることにより、水蒸気バリアーフィルムとしての耐熱性を十分なものにし易くなり、10μm以下にすることにより、水蒸気バリアーフィルムの光学特性のバランスを調整し易くなると共に、平滑層2を水蒸気バリアーフィルムの一方の面に設けた場合におけるその水蒸気バリアーフィルムのカールを抑え易くすることができるようになる。
本発明に係る水蒸気バリアーフィルム10(11)は、太陽電池、液晶表示素子、有機EL素子等の電子デバイスを封止する封止フィルムとして用いることができる。
この水蒸気バリアーフィルム10を封止フィルムとして用いた電子機器である有機ELパネル20の一例を図5に示す。
有機ELパネル20は、図5に示すように、水蒸気バリアーフィルム10と、水蒸気バリアーフィルム10上に形成されたITOなどの透明電極6と、透明電極6を介して水蒸気バリアーフィルム10上に形成された有機EL素子7と、その有機EL素子7を覆うように接着剤層8を介して配設された対向フィルム9等を備えている。なお、透明電極6は、有機EL素子7の一部を成すともいえる。
この水蒸気バリアーフィルム10における水蒸気バリアー層3(第2の水蒸気バリアー層)が形成された面に、透明電極6と有機EL素子7が形成されるようになっている。
また、対向フィルム9は、アルミ箔などの金属フィルムのほか、本発明に係る水蒸気バリアーフィルムを用いてもよい。対向フィルム9に水蒸気バリアーフィルムを用いる場合、水蒸気バリアー層3(第2の水蒸気バリアー層)が形成された面を有機EL素子7に向けて、接着剤層8によって貼付するようにすればよい。
有機ELパネル20において、水蒸気バリアーフィルム10(11)で封止される有機EL素子7について説明する。
以下に有機EL素子7の層構成の好ましい具体例を以下に示すが、本発明はこれらに限定されない。
(1)陽極/発光層/陰極
(2)陽極/正孔輸送層/発光層/陰極
(3)陽極/発光層/電子輸送層/陰極
(4)陽極/正孔輸送層/発光層/電子輸送層/陰極
(5)陽極/陽極バッファー層(正孔注入層)/正孔輸送層/発光層/電子輸送層/陰極バッファー層(電子注入層)/陰極
有機EL素子7における陽極(透明電極6)としては、仕事関数の大きい(4eV以上)金属、合金、電気伝導性化合物及びこれらの混合物を電極物質とするものが好ましく用いられる。このような電極物質の具体例としては、Au等の金属、CuI、インジウムチンオキシド(ITO)、SnO2、ZnO等の導電性透明材料が挙げられる。また、IDIXO(In2O3−ZnO)等非晶質で透明導電膜を作製可能な材料を用いてもよい。
陽極は、これらの電極物質を蒸着やスパッタリング等の方法により薄膜として形成し、その薄膜をフォトリソグラフィー法で所望の形状のパターンを形成してもよく、あるいはパターン精度をあまり必要としない場合は(100μm以上程度)、上記電極物質の蒸着やスパッタリング時に所望の形状のマスクを介してパターンを形成してもよい。
この陽極より発光を取り出す場合には、透過率を10%より大きくすることが望ましい。また、陽極としてのシート抵抗は数百Ω/□以下が好ましい。また、陽極の膜厚は材料にもよるが、通常10〜1000nm、好ましくは10〜200nmの範囲で選ばれる。
有機EL素子7における陰極としては、仕事関数の小さい(4eV以下)金属(電子注入性金属と称する)、合金、電気伝導性化合物及びこれらの混合物を電極物質とするものが用いられる。このような電極物質の具体例としては、ナトリウム、ナトリウム−カリウム合金、マグネシウム、リチウム、マグネシウム/銅混合物、マグネシウム/銀混合物、マグネシウム/アルミニウム混合物、マグネシウム/インジウム混合物、アルミニウム/酸化アルミニウム(Al2O3)混合物、インジウム、リチウム/アルミニウム混合物、希土類金属等が挙げられる。これらの中で、電子注入性及び酸化等に対する耐久性の点から、電子注入性金属とこれより仕事関数の値が大きく安定な金属である第二金属との混合物、例えば、マグネシウム/銀混合物、マグネシウム/アルミニウム混合物、マグネシウム/インジウム混合物、アルミニウム/酸化アルミニウム(Al2O3)混合物、リチウム/アルミニウム混合物、アルミニウム等が陰極として好適である。
陰極は、これらの電極物質を蒸着やスパッタリング等の方法により薄膜を形成させることにより作製することができる。また、陰極としてのシート抵抗は数百Ω/□以下が好ましい。また、陰極の膜厚は通常10nm〜5μm、好ましくは50〜200nmの範囲で選ばれる。なお、発光した光を透過させるため、有機EL素子7の陽極または陰極のいずれか一方が透明または半透明であれば、発光輝度が向上し好都合である。
また、陰極の説明で挙げた上記金属を1〜20nmの膜厚で作製した後に、陽極の説明で挙げた導電性透明材料をその上に作製することで、透明または半透明の陰極を作製することができ、これを応用することで陽極と陰極の両方が透過性を有する素子を作製することができる。
注入層には電子注入層と正孔注入層があり、電子注入層と正孔注入層を必要に応じて設け、陽極と発光層または正孔輸送層の間、及び陰極と発光層または電子輸送層との間に存在させる。
注入層とは、駆動電圧低下や発光輝度向上のために電極と有機層間に設けられる層のことで、「有機EL素子とその工業化最前線(1998年11月30日エヌ・ティー・エス社発行)」の第2編第2章「電極材料」(123〜166頁)に詳細に記載されており、正孔注入層(陽極バッファー層)と電子注入層(陰極バッファー層)とがある。
陽極バッファー層(正孔注入層)は、特開平9−45479号公報、特開平9−260062号公報、特開平8−288069号公報等にもその詳細が記載されており、具体例として、銅フタロシアニンに代表されるフタロシアニンバッファー層、酸化バナジウムに代表される酸化物バッファー層、アモルファスカーボンバッファー層、ポリアニリン(エメラルディン)やポリチオフェン等の導電性高分子を用いた高分子バッファー層等が挙げられる。
陰極バッファー層(電子注入層)は、特開平6−325871号公報、特開平9−17574号公報、特開平10−74586号公報等にもその詳細が記載されており、具体的には、ストロンチウムやアルミニウム等に代表される金属バッファー層、フッ化リチウムに代表されるアルカリ金属化合物バッファー層、フッ化マグネシウムに代表されるアルカリ土類金属化合物バッファー層、酸化アルミニウムに代表される酸化物バッファー層等が挙げられる。上記バッファー層(注入層)はごく薄い膜であることが望ましく、素材にもよるが、その膜厚は0.1nm〜5μmの範囲が好ましい。
有機EL素子7における発光層は、電極(陰極、陽極)または電子輸送層、正孔輸送層から注入されてくる電子及び正孔が再結合して発光する層であり、発光する部分は発光層の層内であっても発光層と隣接層との界面であってもよい。
有機EL素子7の発光層には、以下に示すドーパント化合物(発光ドーパント)とホスト化合物(発光ホスト)が含有されることが好ましい。これにより、より一層発光効率を高くすることができる。
(発光ドーパント)
発光ドーパントは、大きく分けて蛍光を発光する蛍光性ドーパントとリン光を発光するリン光性ドーパントの2種類がある。
蛍光性ドーパントの代表例としては、クマリン系色素、ピラン系色素、シアニン系色素、クロコニウム系色素、スクアリウム系色素、オキソベンツアントラセン系色素、フルオレセイン系色素、ローダミン系色素、ピリリウム系色素、ペリレン系色素、スチルベン系色素、ポリチオフェン系色素、または希土類錯体系蛍光体等が挙げられる。
リン光性ドーパントの代表例としては、好ましくは元素の周期表で8属、9属、10属の金属を含有する錯体系化合物であり、更に好ましくはイリジウム化合物、オスミウム化合物であり、中でも最も好ましいのはイリジウム化合物である。
発光ドーパントは複数種の化合物を混合して用いてもよい。
(発光ホスト)
発光ホスト(単にホストとも言う)とは、2種以上の化合物で構成される発光層中にて混合比(質量)の最も多い化合物のことを意味し、それ以外の化合物については「ドーパント化合物(単に、ドーパントとも言う)」という。例えば、発光層を化合物A、化合物Bという2種で構成し、その混合比がA:B=10:90であれば化合物Aがドーパント化合物であり、化合物Bがホスト化合物である。更に発光層を化合物A、化合物B、化合物Cの3種から構成し、その混合比がA:B:C=5:10:85であれば、化合物A、化合物Bがドーパント化合物であり、化合物Cがホスト化合物である。
発光ホストとしては構造的には特に制限はないが、代表的にはカルバゾール誘導体、トリアリールアミン誘導体、芳香族ボラン誘導体、含窒素複素環化合物、チオフェン誘導体、フラン誘導体、オリゴアリーレン化合物等の基本骨格を有するもの、またはカルボリン誘導体やジアザカルバゾール誘導体(ここで、ジアザカルバゾール誘導体とは、カルボリン誘導体のカルボリン環を構成する炭化水素環の少なくとも一つの炭素原子が窒素原子で置換されているものを表す。)等が挙げられる。中でも、カルボリン誘導体、ジアザカルバゾール誘導体等が好ましく用いられる。
そして、発光層は上記化合物を、例えば、真空蒸着法、スピンコート法、キャスト法、LB法、インクジェット法等の公知の薄膜化法により成膜して形成することができる。発光層としての膜厚は特に制限はないが、通常は5nm〜5μm、好ましくは5〜200nmの範囲で選ばれる。この発光層はドーパント化合物やホスト化合物が1種または2種以上からなる一層構造であってもよいし、あるいは同一組成または異種組成の複数層からなる積層構造であってもよい。
正孔輸送層とは正孔を輸送する機能を有する正孔輸送材料からなり、広い意味で正孔注入層、電子阻止層も正孔輸送層に含まれる。正孔輸送層は単層または複数層設けることができる。
正孔輸送材料としては、正孔の注入または輸送、電子の障壁性のいずれかを有するものであり、有機物、無機物のいずれであってもよい。例えば、トリアゾール誘導体、オキサジアゾール誘導体、イミダゾール誘導体、ポリアリールアルカン誘導体、ピラゾリン誘導体及びピラゾロン誘導体、フェニレンジアミン誘導体、アリールアミン誘導体、アミノ置換カルコン誘導体、オキサゾール誘導体、スチリルアントラセン誘導体、フルオレノン誘導体、ヒドラゾン誘導体、スチルベン誘導体、シラザン誘導体、アニリン系共重合体、また導電性高分子オリゴマー、特にチオフェンオリゴマー等が挙げられる。正孔輸送材料としては上記のものを使用することができるが、ポルフィリン化合物、芳香族第3級アミン化合物及びスチリルアミン化合物、特に芳香族第3級アミン化合物を用いることが好ましい。更にこれらの材料を高分子鎖に導入した、またはこれらの材料を高分子の主鎖とした高分子材料を用いることもできる。また、p型−Si、p型−SiC等の無機化合物も正孔注入材料、正孔輸送材料として使用することができる。
正孔輸送層は上記正孔輸送材料を、例えば、真空蒸着法、スピンコート法、キャスト法、インクジェット法を含む印刷法、LB法等の公知の方法により、薄膜化することにより形成することができる。正孔輸送層の膜厚については特に制限はないが、通常は5nm〜5μm程度、好ましくは5〜200nmである。この正孔輸送層は上記材料の1種または2種以上からなる一層構造であってもよい。
電子輸送層とは電子を輸送する機能を有する電子輸送材料からなり、広い意味で電子注入層、正孔阻止層も電子輸送層に含まれる。電子輸送層は単層または複数層設けることができる。
電子輸送材料としては、陰極より注入された電子を発光層に伝達する機能を有していればよく、その材料としては従来公知の化合物の中から任意のものを選択して用いることができ、例えば、ニトロ置換フルオレン誘導体、ジフェニルキノン誘導体、チオピランジオキシド誘導体、カルボジイミド、フレオレニリデンメタン誘導体、アントラキノジメタン及びアントロン誘導体、オキサジアゾール誘導体等が挙げられる。さらに、上記オキサジアゾール誘導体において、オキサジアゾール環の酸素原子を硫黄原子に置換したチアジアゾール誘導体、電子吸引基として知られているキノキサリン環を有するキノキサリン誘導体も、電子輸送材料として用いることができる。さらにこれらの材料を高分子鎖に導入した、またはこれらの材料を高分子の主鎖とした高分子材料を用いることもできる。また、8−キノリノール誘導体の金属錯体、例えば、トリス(8−キノリノール)アルミニウム(Alq)、トリス(5,7−ジクロロ−8−キノリノール)アルミニウム、トリス(5,7−ジブロモ−8−キノリノール)アルミニウム、トリス(2−メチル−8−キノリノール)アルミニウム、トリス(5−メチル−8−キノリノール)アルミニウム、ビス(8−キノリノール)亜鉛(Znq)等、及びこれらの金属錯体の中心金属がIn、Mg、Cu、Ca、Sn、GaまたはPbに置き替わった金属錯体も、電子輸送材料として用いることができる。その他、メタルフリーもしくはメタルフタロシアニン、またはそれらの末端がアルキル基やスルホン酸基等で置換されているものも、電子輸送材料として好ましく用いることができる。また、正孔注入層、正孔輸送層と同様に、n型−Si、n型−SiC等の無機半導体も電子輸送材料として用いることができる。
電子輸送層は上記電子輸送材料を、例えば、真空蒸着法、スピンコート法、キャスト法、インクジェット法を含む印刷法、LB法等の公知の方法により、薄膜化することにより形成することができる。電子輸送層の膜厚については特に制限はないが、通常は5nm〜5μm程度、好ましくは5〜200nmである。電子輸送層は上記材料の1種または2種以上からなる一層構造であってもよい。
有機EL素子7の作製方法について説明する。
ここでは有機EL素子7の一例として、陽極/正孔注入層/正孔輸送層/発光層/電子輸送層/電子注入層/陰極からなる有機EL素子の作製方法について説明する。
まず、水蒸気バリアーフィルム10上に所望の電極物質、例えば、陽極用物質からなる薄膜を1μm以下、好ましくは10〜200nmの膜厚になるように、例えば、蒸着やスパッタリング、プラズマCVD等の方法により形成させ、陽極を作製する。
次に、その上に有機EL素子材料である正孔注入層、正孔輸送層、発光層、電子輸送層、電子注入層の有機化合物薄膜を形成させる。この有機化合物薄膜の成膜方法としては、蒸着法、ウェットプロセス(スピンコート法、キャスト法、インクジェット法、印刷法)等があるが、均質な膜が得られやすく、且つピンホールが生成しにくい等の点から、真空蒸着法、スピンコート法、インクジェット法、印刷法が特に好ましい。更に層毎に異なる成膜法を適用してもよい。成膜に蒸着法を採用する場合、その蒸着条件は使用する化合物の種類等により異なるが、一般にボート加熱温度50〜450℃、真空度10−6〜10−2Pa、蒸着速度0.01〜50nm/秒、基板温度−50〜300℃、膜厚0.1nm〜5μm、好ましくは5〜200nmの範囲で適宜選ぶことが望ましい。
これらの層を形成後、その上に陰極用物質からなる薄膜を1μm以下好ましくは50〜200nmの範囲の膜厚になるように、例えば、蒸着やスパッタリング等の方法により形成させ、陰極を設けることにより所望の有機EL素子が得られる。
この有機EL素子7の作製は、一回の真空引きで一貫して陽極、正孔注入層から陰極まで作製するのが好ましいが、途中で取り出して異なる成膜法を施しても構わない。その際、作業を乾燥不活性ガス雰囲気下で行う等の配慮が必要となる。また、作製順序を逆にして、陰極、電子注入層、電子輸送層、発光層、正孔輸送層、正孔注入層、陽極の順に作製することも可能である。
このようにして得られた有機EL素子7を備える多色の表示装置(有機ELパネル20)に、直流電圧を印加する場合には、陽極をプラス、陰極をマイナスの極性として電圧2〜40V程度を印加すると発光が観測できる。また、交流電圧を印加してもよい。なお、印加する交流の波形は任意でよい。
以下、具体的な実施例を挙げて本発明の水蒸気バリアーフィルムを詳細に説明するが、本発明はこれらに限定されるものではない。なお、実施例において「部」あるいは「%」の表示を用いるが、特に断りがない限り「質量部」あるいは「質量%」を表す。
熱可塑性樹脂支持体であって、両面に易接着加工された75μm厚みのポリエステルフィルム(東洋紡績株式会社製、コスモシャインA4300)を樹脂基材1aとして用い、下記に示すように、片面にブリードアウト防止層5、反対面に平滑層2を作製したものを基材1として用いた。
(ブリードアウト防止層の形成)
上記樹脂基材1aの片面に、JSR株式会社製 UV硬化型有機/無機ハイブリッドハードコート材OPSTAR Z7535を、乾燥後の膜厚が4μmになるようにワイヤーバーで塗布した後、空気下で高圧水銀ランプ使用1.0J/cm2の硬化条件、80℃×3分の乾燥条件で硬化を行い、ブリードアウト防止層5を形成した。
(平滑層の形成)
続けて上記樹脂基材1aの反対面に、JSR株式会社製 UV硬化型有機/無機ハイブリッドハードコート材OPSTAR Z7501を、乾燥後の膜厚が4μmになるようにワイヤーバーで塗布した後、乾燥条件80℃×3分で乾燥し、その後、空気雰囲気下で高圧水銀ランプ使用1.0J/cm2の硬化条件で硬化を行い、平滑層2を形成した。
得られた平滑層2のJIS B 0601で規定される表面粗さで、最大断面高さRt(p)は16nmであった。
表面粗さは、AFM(原子間力顕微鏡)で、極小の先端半径の触針を持つ検出器で連続測定した凹凸の断面曲線から算出され、極小の先端半径の触針により測定方向が30μmの区間内を多数回測定し、微細な凹凸の振幅に関する平均の粗さである。
(基材の水蒸気透過率)
調湿後の上記基材1についての水蒸気透過率をモコン法に従い、MOCON社製PERMATRAN−W3/33を用いて、JIS規格のK7129法(温度40℃、湿度90%RH)に基づいて測定した。測定された水蒸気透過率は1g/m2/日を超えるレベルであった。
このように、樹脂基材1aにブリードアウト防止層5と平滑層2が形成された基材1を「B0」とした。
第1から第3の蒸着層はそれぞれ金属酸化物(酸化珪素)を含有しており、第1から第3の蒸着層の厚みはそれぞれ120nm、30nm、25nmの合計160nmであった。
・第1蒸着層
放電ガス :N2ガス
反応ガス1:水素ガスを全ガスに対し1%
反応ガス2:TEOS(テトラエトキシシラン)を全ガスに対し0.5%
成膜条件 ;
第1電極側 電源種類 応用電機製 80kHz
周波数 80kHz
出力密度 8W/cm2
電極温度 110℃
第2電極側 電源種類 パール工業製 13.56MHz CF−5000−13M
周波数 13.56MHz
出力密度 10W/cm2
電極温度 100℃
・第2蒸着層
放電ガス :N2ガス
反応ガス1:酸素ガスを全ガスに対し5%
反応ガス2:TEOSを全ガスに対し0.1%
成膜条件 ;
第1電極側 電源種類 ハイデン研究所 100kHz(連続モード) PHF−6k
周波数 100kHz
出力密度 10W/cm2
電極温度 120℃
第2電極側 電源種類 パール工業製 13.56MHz CF−5000−13M
周波数 13.56MHz
出力密度 10W/cm2
電極温度 100℃
・第3蒸着層
放電ガス :N2ガス
反応ガス1:水素ガスを全ガスに対し1%
反応ガス2:TEOSを全ガスに対し0.5%
成膜条件 ;
第1電極側 電源種類 応用電機製 80kHz
周波数 80kHz
出力密度 8W/cm2
電極温度 115℃
第2電極側 電源種類 パール工業製 13.56MHz CF−5000−13M
周波数 13.56MHz
出力密度 10W/cm2
電極温度 105℃
(試料「B1」の水蒸気透過率測定)
調湿後の上記試料「B1」についての水蒸気透過率をモコン法に従い、MOCON社製PERMATRAN−W3/33を用いて、JIS規格のK7129法(温度40℃、湿度90%RH)に基づいて測定した。測定された水蒸気透過率は0.01g/m2/日を下回るレベルであった。
上記、第1〜第3の蒸着層からなる第1の水蒸気バリアー層3が形成された試料「B1」の表面(水蒸気バリアー層3上)に、以下の条件でポリシラザンを主成分とする加水分解層4を形成した。
・加水分解層
パーヒドロポリシラザン(アクアミカ NN120−10、AZエレクトロニックマテリアルズ(株)製)の10質量%ジブチルエーテル溶液と、アミン触媒のN,N,N’,N’−テトラメチル−1,6−ジアミノヘキサンの10質量%ジブチルエーテル溶液を、99対1の割合で混合した液体を、ワイヤレスバーにて、乾燥後の(平均)膜厚が、0.10μmとなるように塗布し、温度25℃、露点−5℃の乾燥空気で1分間乾燥して塗布試料を得た(塗布工程)。得られた塗布試料を、温度85℃、露点−5℃の乾燥空気で2分間処理し、加水分解層4を形成した試料を得た(乾燥工程)。
さらに、その加水分解層4上に、以下の条件で第2の水蒸気バリアー層3を形成し、水蒸気バリアー層3間に加水分解層4が挟まれた水蒸気バリアーフィルムを作製した。これを試料1とした。
・第2の水蒸気バリアー層
パーヒドロポリシラザン(アクアミカ NN120−10、AZエレクトロニックマテリアルズ(株)製)の10質量%ジブチルエーテル溶液と、アミン触媒のN,N,N’,N’−テトラメチル−1,6−ジアミノヘキサンの10質量%ジブチルエーテル溶液を、99対1の割合で混合した液体を、ワイヤレスバーにて、乾燥後の(平均)膜厚が、0.08μmとなるように塗布し、温度25℃、露点−5℃の乾燥空気で1分間乾燥して塗布試料を得た(塗布工程)。得られた塗布試料を、温度85℃、露点−5℃の乾燥空気で2分間処理し、ポリシラザン塗膜を形成した試料を得た(乾燥工程)。
・エキシマ改質処理
ポリシラザン塗膜を乾燥した後の上記試料に対し、下記の装置、条件でエキシマ改質処理を施してポリシラザン塗膜を改質し、ポリシラザン改質層である第2の水蒸気バリアー層3を形成した。改質処理時の露点温度は−20℃で実施した(エキシマ処理工程)。
・改質処理装置
(株)エム・ディ・コム製エキシマ照射装置MODEL:MECL−M−1−200
波長:172nm
ランプ封入ガス:Xe
・改質処理条件
エキシマ光強度 :130mW/cm2(172nm)
試料と光源の距離 :2mm
ステージ加熱温度 :95℃
照射装置内の酸素濃度:0.3%
エキシマ光照射時のステージ搬送速度:10mm/秒
エキシマ光照射時のステージ搬送回数:6往復
上記、試料1の水蒸気バリアーフィルムの作製工程のうち、第1〜第3の蒸着層からなる第1の水蒸気バリアー層3を形成する工程を省略して、平滑層2上に直接、加水分解層4を形成した以外は、試料1と同様の方法で水蒸気バリアーフィルムを作製した。これを試料2とした。
上記、試料1の水蒸気バリアーフィルムの作製工程のうち、加水分解層4を形成する工程を省略した以外は、試料1と同様の方法で水蒸気バリアーフィルムを作製した。これを試料3とした。
上記、試料1の水蒸気バリアーフィルムの作製工程のうち、加水分解層4上の第2の水蒸気バリアー層3を形成する工程を省略した以外は、試料1と同様の方法で水蒸気バリアーフィルムを作製した。これを試料4とした。
上記、試料1の水蒸気バリアーフィルムにおいて、加水分解層4を下記の組成に変更して形成した以外は、試料1と同様の方法で水蒸気バリアーフィルムを作製した。これを試料5とした。
・加水分解層
ジルコウニウムテトラアセチルアセトネート(オルガチックスZC−150 マツモトファインケミカル(株)製)の10質量%トルエン溶液からなる液体を、ワイヤレスバーにて、乾燥後の(平均)膜厚が、0.10μmとなるように塗布し、温度25℃、露点−5℃の乾燥空気で1分間乾燥して塗布試料を得た(塗布工程)。得られた塗布試料を、温度85℃、露点−5℃の乾燥空気で2分間処理した試料を得た(乾燥工程)。
上記、試料1の水蒸気バリアーフィルムにおいて、加水分解層4を下記の組成に変更して形成した以外は、試料1と同様の方法で水蒸気バリアーフィルムを作製した。これを試料6とした。
・加水分解層
ポリヒドロキシチタンステアレート(オルガチックスTPHS マツモトファインケミカル(株)製)の10質量%トルエン溶液からなる液体を、ワイヤレスバーにて、乾燥後の(平均)膜厚が、0.10μmとなるように塗布し、温度25℃、露点−5℃の乾燥空気で1分間乾燥して塗布試料を得た(塗布工程)。得られた塗布試料を、温度85℃、露点−5℃の乾燥空気で2分間処理した試料を得た(乾燥工程)。
上記、試料1の水蒸気バリアーフィルムにおいて、加水分解層4を下記の組成に変更して形成した以外は、試料1と同様の方法で水蒸気バリアーフィルムを作製した。これを試料7とした。
・加水分解層
3−アミノプロピルトリメトキシシラン(KBM−903 信越化学工業(株)製)の10質量%トルエン溶液からなる液体を、ワイヤレスバーにて、乾燥後の(平均)膜厚が、0.10μmとなるように塗布し、温度25℃、露点−5℃の乾燥空気で1分間乾燥して塗布試料を得た(塗布工程)。得られた塗布試料を、温度85℃、露点−5℃の乾燥空気で2分間処理した試料を得た(乾燥工程)。
上記、試料1の水蒸気バリアーフィルムにおいて、加水分解層4を下記の組成に変更して形成した以外は、試料1と同様の方法で水蒸気バリアーフィルムを作製した。これを試料8とした。
・加水分解層
フェニルトリクロロシラン(KA−103 信越化学工業(株)製)の10質量%トルエン溶液と、パーヒドロポリシラザン(アクアミカ NN120−10、AZエレクトロニックマテリアルズ(株)製)の10質量%ジブチルエーテル溶液の6:4の重量比からなる液体を、ワイヤレスバーにて、乾燥後の(平均)膜厚が、0.10μmとなるように塗布し、温度25℃、露点−5℃の乾燥空気で1分間乾燥して塗布試料を得た(塗布工程)。得られた塗布試料を、温度85℃、露点−5℃の乾燥空気で2分間処理した試料を得た(乾燥工程)。
上記、試料1の水蒸気バリアーフィルムにおいて、加水分解層4上の第2の水蒸気バリアー層3を、加水分解層4下にプラズマCVD法で形成された第1〜第3の蒸着層からなる水蒸気バリアー層3と同じものに変更した以外は、試料1と同様の方法で水蒸気バリアーフィルムを作製した。これを試料9とした。
上記、試料1の水蒸気バリアーフィルムにおいて、加水分解層4下のプラズマCVD法による蒸着層からなる第1の水蒸気バリアー層3を、加水分解層4上に形成したポリシラザンのエキシマ改質処理層からなる水蒸気バリアー層3と同じものに変更した以外は、試料1と同様の方法で水蒸気バリアーフィルムを作製した。これを試料10とした。
上記、試料1の水蒸気バリアーフィルムにおいて、加水分解層4下のプラズマCVD法による蒸着層からなる第1の水蒸気バリアー層3を、下記のスパッタリング法による蒸着層からなる水蒸気バリアー層3に変更した以外は、試料1と同様の方法で水蒸気バリアーフィルムを作製した。これを試料11とした。
・スパッタリング法による水蒸気バリアー層の形成
スパッタ装置の真空槽内にブリードアウト防止層5と平滑層2が形成された基材1「B0」をセットして1×10−4Pa台まで真空引きし、放電ガスとしてアルゴンを分圧で0.04Pa導入し、反応ガスとして酸素を分圧で0.04Pa導入した。雰囲気圧力が安定したところで放電を開始しSiターゲット上にプラズマを発生させ、スパッタリングプロセスを開始した。プロセスが安定したところでシャッターを開き基材「B0」へのSiOx無機膜の形成を開始した。厚さ50nmの膜が堆積したところでシャッターを閉じて成膜を終了した。
上記、試料1の水蒸気バリアーフィルムの作製工程のうち、加水分解層4および加水分解層4上の第2の水蒸気バリアー層3を形成する工程を、下記の条件に変更して、加水分解層4および加水分解層4上の第2の水蒸気バリアー層3を1回の塗布工程で形成した以外は、試料1と同様の方法で水蒸気バリアーフィルムを作製した。これを試料12とした。
・加水分解層および加水分解層上の第2の水蒸気バリアー層の形成
パーヒドロポリシラザン(アクアミカ NN120−10、AZエレクトロニックマテリアルズ(株)製)の10質量%ジブチルエーテル溶液と、アミン触媒のN,N,N’,N’−テトラメチル−1,6−ジアミノヘキサンの10質量%ジブチルエーテル溶液を、99対1の割合で混合した液体を、ワイヤレスバーにて、乾燥後の(平均)膜厚が、0.16μmとなるように塗布し、温度25℃、露点−5℃の乾燥空気で1分間乾燥して塗布試料を得た(塗布工程)。得られた塗布試料を、温度85℃、露点−5℃の乾燥空気で2分間処理し、ポリシラザン塗膜(バリアー層前駆体層)を形成した試料を得た(乾燥工程)。
・エキシマ改質処理
ポリシラザン塗膜を乾燥した後の上記試料に対し、下記の装置、条件でエキシマ改質処理を施して、ポリシラザン塗膜の上層側にポリシラザン改質層である第2の水蒸気バリアー層3を形成するとともに、ポリシラザン塗膜の下層側にポリシラザンの少なくとも一部を未反応のまま加水分解性化合物として残留させた加水分解層4を形成した。改質処理時の露点温度は−20℃で実施した(エキシマ処理工程)。
・改質処理装置
(株)エム・ディ・コム製エキシマ照射装置MODEL:MECL−M−1−200
波長:172nm
ランプ封入ガス:Xe
・改質処理条件
エキシマ光強度 :130mW/cm2(172nm)
試料と光源の距離 :2mm
ステージ加熱温度 :95℃
照射装置内の酸素濃度:1.0%
エキシマ光照射時のステージ搬送速度:10mm/秒
エキシマ光照射時のステージ搬送回数:3往復
上記、試料1の水蒸気バリアーフィルムにおいて、加水分解層4を特開2006−82241号公報に記載の下記の組成からなる吸湿層に変更して形成した以外は、試料1と同様の方法で水蒸気バリアーフィルムを作製した。これを試料13とした。
・吸湿層の形成
過酸化ストロンチウムのターゲット(豊島製作所(株)製)を用い、Arガスを導入、放電電力100W、成膜圧力0.8Paで3分間のプレスパッタした後に、そのまま成膜を行った。吸湿層の膜厚は約20nmであった。
上記、試料1の水蒸気バリアーフィルムにおいて、加水分解層4を特開2009−90633号公報に記載の下記の組成からなる捕水層に変更して形成した以外は、試料1と同様の方法で水蒸気バリアーフィルムを作製した。これを試料14とした。
・捕水層の形成
光重合性アクリレートとしてトリプロピレングリコールジアクリレート(TPGDA、ダイセル・サイテック製)9g、および光重合開始剤(チバ・スペシャリティ・ケミカルズ製、イルガキュア907)0.1gを、メチルエチルケトン190gに溶解させた塗布液に、該捕水層形成後の膜中の含有量が30重量%となるように捕水剤としての無水硫酸バリウムを分散させた。この塗布液を、有機層と同様にワイヤーバーを用いて塗布したのち、酸素濃度0.1%以下の窒素パージ下で160W/cmの空冷メタルハライドランプ(アイグラフィックス(株)製)を用いて、照度350mW/cm2、照射量500mJ/cm2の紫外線を照射して紫外線硬化し、2000nmの厚さの捕水層を形成した。
上記、試料1の水蒸気バリアーフィルムにおいて、加水分解層4を特開2009−29070号公報に記載の下記の組成からなる金属窒化物層に変更して形成した以外は、試料1と同様の方法で水蒸気バリアーフィルムを作製した。これを試料15とした。
・金属窒化物膜
原料ガスとしてSiH4、NH3、H2を用い、RF電源を用いたプラズマCVD法によって、約90nmの窒化シリコン(SiN)からなる金属窒化物膜を形成した。
試料1において加水分解層4を形成する塗布液である、パーヒドロポリシラザンとアミン触媒の混合液に、水との加水分解性の低いメチルポリシラザン(アクアミカ LExp MHPS−20DB AZエレクトロニックマテリアルズ株式会社製、ジブチルエーテルの20%溶液)を混合して、塗布液におけるパーヒドロポリシラザンとアミン触媒の組成・混合比を変化させるか、あるいは試料「B1」の第2蒸着層の膜厚を増減することによって、ポリシラザンを用いた加水分解層4における窒素原子の半減期(図3、図4参照)を調整した以外は、試料1と同様の方法で水蒸気バリアーフィルムを作製し、それらの評価を行った。
試料1において加水分解層4を形成する塗布液である、パーヒドロポリシラザンとアミン触媒の混合液に、3−アミノプロピルトリメトキシシランと、N−フェニル−3−アミノプロピルトリメトキシシラン(KBM−573 信越化学工業(株)製)の10質量%トルエン溶液)を混合して、塗布液における組成・混合比を変化させるか、あるいは、試料「B1」の第2蒸着層の膜厚を増減することにより、アルコキシシランを用いた加水分解層4における炭素原子の半減期(図3、図4参照)を調整した以外は、試料1と同様の方法で水蒸気バリアーフィルムを作製し、それらの評価を行った
上記した基材1である「B0」に用いた樹脂基材1aの代わりに、熱可塑性樹脂支持体であって、両面に易接着加工が施された125μm厚みのポリエステルフィルム(東洋紡績株式会社製、コスモシャインA4300)を樹脂基材1aとして用い、「B0」と同様に、片面にブリードアウト防止層5を設け、反対面の平滑層2の代わりに下記のアンカーコート層を積層したものを基材1である「B2」とした。
(アンカーコート層の形成)
出光テクノファイン株式会社製、無機有機ハイブリッドコート剤#2087(変性ポリアルキルシロキサンとアクリル酸エステル共重合体のイソプロパノール、1−メトキシ−2−プロパノール、メタノール、エタノール、酢酸、水の固形分30%混合液)を乾燥後の膜厚が0.3μmとなるように樹脂基材1aに積層し、60℃で12時間加熱を行ってアンカーコート層を形成した。
得られたアンカーコート層表面のJIS B 0601で規定される表面粗さで、最大断面高さRt(p)は30nmであった。なお、表面粗さは、AFM(原子間顕微鏡)で、極小の先端半径の触針を持つ検出器で連続測定した凹凸の断面曲線から算出され、極小の先端半径の触針により測定方向が30μmの区間内を多数回測定し、微小な凹凸の振幅に関する平均粗さである。
試料10で用いた平滑層を有する基材「B0」を、アンカーコート層を有する基材「B2」に変更したした以外は同様の方法で水蒸気バリアーフィルム(試料32)を作製し、その評価を同様に行った。
試料1で用いた平滑層を有する基材「B0」を、アンカーコート層を有する基材「B2」に変更したした以外は同様の方法で水蒸気バリアーフィルム(試料33)を作製し、その評価を同様に行った。
上記のようにして得られた水蒸気バリアーフィルムの試料を、スパッタ法によって最外バリアー層(第2の水蒸気バリアー層3)表面から深さ方向へエッチングを行い、XPS表面分析装置を用いて、バリアー層最表面を0nmとして10nm毎のケイ素化合物を有する層の原子組成比を平滑層2表面まで測定した。
このうち、各試料の加水分解層4に該当する領域において加水分解で生成する副生成物が有する特定原子(具体的には下記)の組成比%を10nm毎の矩形面積で近似して積算した面積を算出した。
試料1(試料16〜23、33)、試料2、試料4、試料9、試料10(試料32)、試料11、試料12については、窒素原子%。
試料5、試料6、試料7、試料24〜31については、炭素原子%。
試料8については、塩素原子%。
XPS表面分析装置としては、特に限定なく、いかなる機種も使用することができるが、本実施例においてはVGサイエンティフィックス社製ESCA LAB−200Rを用いた。X線アノードにはMgを用い、出力600W(加速電圧15kV、エミッション電流40mA)で測定した。
具体的には、加水分解層の厚みが約80nmの試料1を例に取ると、60℃/90%RH雰囲気の環境で保存し、定期的に上記の測定を繰り返して、時間経過に伴う加水分解性化合物の残存量を測定して、図6に示したような加水分解層の厚み方向に対する窒素原子量の分布曲線を得た。図6の分布曲線を解析し、10nm毎の矩形(または三角形)で近似した面積を積算して、時間経過とともに囲まれた面積が半減するのに要する時間を図7に示すように算出した。
図8は、初期の加水分解層中の窒素原子量を100%として、各測定時間に対する残存量をプロットしたグラフであり、このグラフより半減期は500時間であると判断した。その他の試料についても同様な方法で半減期の算出をおこなった。その結果を表1〜4に示した。
水蒸気バリアーフィルムの性能評価は、水蒸気バリアー性(水蒸気透過率)、水蒸気バリアーフィルムに起因する有機EL素子の発光斑の2つの評価項目について行った。また、水蒸気バリアーフィルムを繰り返し折り返す屈曲処理前後での水蒸気バリアー性(WVTR)と有機EL素子の発光斑について評価することで、水蒸気バリアーフィルムの折り曲げ耐性についても評価した。
各試料の水蒸気バリアーフィルムを、半径が5mmの曲率になるように、180度の角度で100回の屈曲を繰り返す屈曲処理を行い、その屈曲処理前と屈曲処理後の各試料について、以下の方法で水蒸気バリアー性(WVTR)と有機EL素子の発光斑について評価した。
以下の測定方法に従って、各試料の水蒸気バリアーフィルムの水蒸気バリアー性(水蒸気透過率;WVTR)を評価した。
・装置
蒸着装置:日本電子(株)製真空蒸着装置JEE−400
恒温恒湿度オーブン:Yamato Humidic ChamberIG47M
水分と反応して腐食する金属:カルシウム(粒状)
水蒸気不透過性の金属:アルミニウム(φ3〜5mm、粒状)
・水蒸気バリアー性評価用セルの作製
真空蒸着装置(日本電子製真空蒸着装置 JEE−400)を用い、水蒸気バリアーフィルム試料1〜33の第2の水蒸気バリアー層3の表面に金属カルシウムを蒸着させた。その後、乾燥窒素ガス雰囲気下で、厚さ0.2mmの石英ガラスに封止用紫外線硬化樹脂(ナガセケムテックス製)を介して金属カルシウム蒸着面を対面させて接着し、紫外線を照射することで、評価用セルを作製した。
得られた試料(評価用セル)を40℃、90%RHの高温高湿下で保存し、特開2005−283561号公報に記載の方法に基づき、金属カルシウムの腐食量からセル内に透過した水分量を計算した。
なお、バリアーフィルム面以外からの水蒸気の透過がないことを確認するために、比較試料として水蒸気バリアーフィルム試料の代わりに、厚さ0.2mmの石英ガラス板を用いて金属カルシウムを蒸着した試料を、同様な40℃、90%RHの高温高湿下保存を行い、10000時間経過後でも金属カルシウム腐食が発生しないことを確認した。
こうして測定された各試料の水蒸気バリアーフィルムの水分量を下記の5段階に分類し、水蒸気バリアー性を評価した。
5:水分量が1×10−5g/m2/day未満
4:水分量が1×10−5g/m2/day以上、1×10−4g/m2/day未満
3:水分量が1×10−4g/m2/day以上、1×10−3g/m2/day未満
2:水分量が1×10−3g/m2/day以上、1×10−2g/m2/day未満
1:水分量が1×10−2g/m2/day以上
作製した試料1〜33の第2の水蒸気バリアー層3上に、以下の方法により透明導電膜を作製した。
・透明導電膜の形成
プラズマ放電装置としては電極が平行平板型のものを用い、この電極間に上記各試料の水蒸気バリアーフィルムを載置し、且つ混合ガスを導入して薄膜形成を行った。なお、アース(接地)電極としては、200mm×200mm×2mmのステンレス板に高密度、高密着性のアルミナ溶射膜を被覆し、その後、テトラメトキシシランを酢酸エチルで希釈した溶液を塗布乾燥後、紫外線照射により硬化させ封孔処理を行い、このようにして被覆した誘電体表面を研磨し、平滑にしてRmaxが5μmとなるように加工した電極を用いた。また、印加電極としては、中空の角型の純チタンパイプに対し、アース電極と同様の条件にて誘電体を被覆した電極を用いた。印加電極は複数作製し、アース電極に対向して設け放電空間を形成した。また、プラズマ発生に用いる電源としては、パール工業(株)製高周波電源CF−5000−13Mを用い、周波数13.56MHzで、5W/cm2の電力を供給した。
そして、電極間に以下の組成の混合ガスを流し、プラズマ状態とし、上記の水蒸気バリアーフィルムを大気圧プラズマ処理し、水蒸気バリアー層(セラミック膜)上に錫ドープ酸化インジウム(ITO)膜を100nmの厚さで成膜し、透明導電膜付の試料1〜33を得た。
放電ガス:ヘリウム 98.5体積%
反応性ガス1:酸素 0.25体積%
反応性ガス2:インジウムアセチルアセトナート 1.2体積%
反応性ガス3:ジブチル錫ジアセテート 0.05体積%
・有機EL素子の作製
得られた透明導電膜付の試料1〜33の100mm×100mmを基板とし、これにパターニングを行った後、このITO透明電極を設けた水蒸気バリアーフィルム基板をイソプロピルアルコールで超音波洗浄し、乾燥窒素ガスで乾燥した。この透明支持基板を市販の真空蒸着装置の基板ホルダーに固定し、一方、モリブデン製抵抗加熱ボートにα−NPD(下記の式(8))を200mg入れ、別のモリブデン製抵抗加熱ボートにホスト化合物としてCBP(下記の式(9))を200mg入れ、別のモリブデン製抵抗加熱ボートにバソキュプロイン(BCP(下記の式(10)))を200mg入れ、別のモリブデン製抵抗加熱ボートにIr−1(下記の式(11))を100mg入れ、更に別のモリブデン製抵抗加熱ボートにAlq3(下記の式(12))を200mg入れ、真空蒸着装置に取付けた。
引き続き、フッ化リチウム0.5nm及びアルミニウム110nmを蒸着して陰極を形成し、それぞれ透明導電膜付の試料1〜33を用いた有機EL素子試料1〜33を作製した。
・有機EL素子試料の封止
窒素ガス(不活性ガス)によりパージされた環境下で、有機EL素子試料1〜33のアルミニウム蒸着面と、厚さ100μmのアルミ箔を対面させる様にして、ナガセケムテックス社製エポキシ系接着剤を用いて接着させて封止を行った。
・有機EL素子試料の評価(ダークスポット、輝度ムラ)
封止された有機EL素子試料1〜33を40℃、90%RHの環境下で通電を行い、ダークスポットの発生等の発光ムラの状況を、0日から120日までの変化を観察した。
こうして観測された各試料の発光ムラを下記の5段階に分類し、評価した。
5:0日目でダークスポット、輝度ムラは観察されず、120日経過後に非発光領域が全発光面積の0.1%以下で、発生したダークスポットは全て目視では用意に観察できない大きさ(0.1mm径以下)であった。
4:0日目で発生したダークスポットは、全て目視では用意に観察できない大きさ(0.1mm以下)であり、輝度ムラは観察されず、120日経過後に非発光領域が全発光面積の0.2%以下で、発生したダークスポットは目視では用意に観察できない大きさ(0.1mm以下)を維持した。
3:0日目で発生したダークスポットは、全て目視では用意に観察できない大きさ(0.1mm以下)であり、120日経過後に非発光領域が全発光面積の2%を超えた。
2:0日目に目視で判別可能なダークスポット、輝度ムラが観察され、120日経過後に非発光領域が全発光面積の2%を超えた。
1:0日目に目視で判別可能なダークスポット、輝度ムラの非発光領域が全発光面積の1%を超えて観察され、120日以内に非発光領域が全発光面積の10%を超えた。
また、表2と表3に示すように、水蒸気バリアーフィルムの加水分解層4における、60℃/90%RH雰囲気下での炭素または窒素原子組成比の半減期t(1/2)であって、加水分解層4における加水分解性化合物が加水分解によって放出した化合物に由来する炭素または窒素原子組成比の半減期t(1/2)は、20時間以上1500時間以下であることが好ましいことがわかる。
また、表4に示すように、第1の水蒸気バリアー層が塗布で形成された場合には、基材表面の平滑性の変動に対して、水蒸気バリアー性、電子デバイスの耐久性の変動が少なく安定であることがわかる。
また、本発明の水蒸気バリアーフィルム10(11)の加水分解層4は、水分をそのまま吸着して保持するのではなく、加水分解により副生成物を生成し、その副生成物を加水分解層4中にほとんど蓄積することなく排出することで、加水分解層4の体積の増大を抑制することができるので、平滑性にも優れた水蒸気バリアーフィルムとすることができる。
1a 樹脂基材
2 平滑層
3 水蒸気バリアー層
4 加水分解層
5 ブリードアウト防止層
10、11 水蒸気バリアーフィルム
7 有機EL素子(電子デバイス)
20 有機ELパネル(電子機器)
Claims (11)
- 基材上に少なくとも2層の水蒸気バリアー層を備え、
前記水蒸気バリアー層の間に、加水分解性化合物を含有する加水分解層を少なくとも1層備えることを特徴とする水蒸気バリアーフィルム。 - 前記加水分解性化合物は加水分解によって、アルコール、アミン化合物、無機酸のうち少なくとも1つを放出する化合物であることを特徴とする請求項1に記載の水蒸気バリアーフィルム。
- 前記加水分解性化合物は、金属アルコキシド、シラン化合物、シラザン化合物、金属ハロゲン化物のうち少なくとも1つを含むことを特徴とする請求項1又は2に記載の水蒸気バリアーフィルム。
- 60℃/90%RH雰囲気下、前記加水分解層における前記加水分解性化合物が加水分解して生成した化合物に由来する炭素または窒素原子組成比の半減期が、20時間以上1500時間以下であることを特徴とする請求項1〜3の何れか一項に記載の水蒸気バリアーフィルム。
- 前記基材に近い配置の前記水蒸気バリアー層は、金属酸化物、金属窒化物、金属酸窒化物のうち少なくとも1つを含む蒸着膜であることを特徴とする請求項1〜4の何れか一項に記載の水蒸気バリアーフィルム。
- 前記基材に近い配置の前記水蒸気バリアー層は、ポリシラザンを含有する塗膜に改質処理を施して形成された層であることを特徴とする請求項1〜4の何れか一項に記載の水蒸気バリアーフィルム。
- 請求項1〜4の何れか一項に記載の水蒸気バリアーフィルムを製造する水蒸気バリアーフィルムの製造方法であって、
基材上に第1の水蒸気バリアー層を形成した後、加水分解性化合物を含有する塗布液を低湿度環境下で前記第1の水蒸気バリアー層上に塗布し乾燥して加水分解層を形成し、その加水分解層上に第2の水蒸気バリアー層を形成することを特徴とする水蒸気バリアーフィルムの製造方法。 - 前記第1の水蒸気バリアー層を蒸着法によって形成し、
前記第1の水蒸気バリアー層上に形成した前記加水分解層の上層側に改質処理を施して、その加水分解層上に重なった前記第2の水蒸気バリアー層を形成することを特徴とする請求項7に記載の水蒸気バリアーフィルムの製造方法。 - 前記第1の水蒸気バリアー層を塗布によって形成し、
前記第1の水蒸気バリアー層上に形成した前記加水分解層の上層側に改質処理を施して、その加水分解層上に重なった前記第2の水蒸気バリアー層を形成することを特徴とする請求項7に記載の水蒸気バリアーフィルムの製造方法。 - 請求項1〜6の何れか一項に記載の水蒸気バリアーフィルムを製造する水蒸気バリアーフィルムの製造方法であって、
基材上に第1の水蒸気バリアー層を形成した後、前記第1の水蒸気バリアー層上に加水分解性化合物を含有するバリアー層前駆体層を形成し、そのバリアー層前駆体層の上層側に改質処理を施すことで、前記第1の水蒸気バリアー層上に積層された加水分解層と、その加水分解層上に重なった第2の水蒸気バリアー層を形成することを特徴とする水蒸気バリアーフィルムの製造方法。 - 請求項1〜6の何れか一項に記載の水蒸気バリアーフィルムまたは請求項7〜10の何れか一項に記載の水蒸気バリアーフィルムの製造方法によって得られた水蒸気バリアーフィルムと、前記水蒸気バリアーフィルムによって封止された電子デバイスを備えることを特徴とする電子機器。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2012022853A JP5884531B2 (ja) | 2011-02-18 | 2012-02-06 | 水蒸気バリアーフィルムの製造方法、水蒸気バリアーフィルム及び電子機器 |
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2011032823 | 2011-02-18 | ||
| JP2011032823 | 2011-02-18 | ||
| JP2012022853A JP5884531B2 (ja) | 2011-02-18 | 2012-02-06 | 水蒸気バリアーフィルムの製造方法、水蒸気バリアーフィルム及び電子機器 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2012183823A true JP2012183823A (ja) | 2012-09-27 |
| JP5884531B2 JP5884531B2 (ja) | 2016-03-15 |
Family
ID=47014320
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2012022853A Expired - Fee Related JP5884531B2 (ja) | 2011-02-18 | 2012-02-06 | 水蒸気バリアーフィルムの製造方法、水蒸気バリアーフィルム及び電子機器 |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP5884531B2 (ja) |
Cited By (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2013168647A1 (ja) * | 2012-05-10 | 2013-11-14 | コニカミノルタ株式会社 | ガスバリア性フィルムの製造方法 |
| US20190030859A1 (en) * | 2016-03-31 | 2019-01-31 | Toppan Printing Co., Ltd. | Barrier film and method for manufacturing the same, wavelength conversion sheet and method for manufacturing the same, and optical laminate and method manufacturing the same |
| JP2019036598A (ja) * | 2017-08-10 | 2019-03-07 | 株式会社東芝 | 半導体素子およびその製造方法 |
| US20210313555A1 (en) * | 2013-10-22 | 2021-10-07 | Semiconductor Energy Laboratory Co., Ltd. | Power storage device electrode, method for manufacturing the same, power storage device, and electronic device |
| WO2022065498A1 (ja) * | 2020-09-28 | 2022-03-31 | 三菱ケミカル株式会社 | 画像表示用導光板 |
| JP2022055621A (ja) * | 2020-09-29 | 2022-04-08 | 三菱ケミカル株式会社 | 画像表示用導光板 |
| JP2022099540A (ja) * | 2020-12-23 | 2022-07-05 | 三菱ケミカル株式会社 | 画像表示用導光板 |
| JP2022129942A (ja) * | 2021-02-25 | 2022-09-06 | 三菱ケミカル株式会社 | 画像表示用導光板 |
| US11561328B2 (en) | 2016-08-12 | 2023-01-24 | Toppan Printing Co., Ltd. | Phosphor protection film, wavelength conversion sheet, and light-emitting unit |
| WO2024047806A1 (ja) * | 2022-08-31 | 2024-03-07 | 株式会社レニアス | 透明積層体の製造方法 |
| TWI922505B (zh) | 2020-09-28 | 2026-04-21 | 日商三菱化學股份有限公司 | 圖像顯示用導光板 |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2006297694A (ja) * | 2005-04-19 | 2006-11-02 | Konica Minolta Holdings Inc | ガスバリアフィルム、有機エレクトロルミネッセンス用樹脂基材、該樹脂基材を用いた有機エレクトロルミネッセンス素子及びガスバリアフィルムの製造方法 |
| JP2007023304A (ja) * | 2005-07-12 | 2007-02-01 | Konica Minolta Holdings Inc | 透明導電膜付ガスバリア性フィルムの製造方法及び有機エレクトロルミネッセンス素子の製造方法 |
| JP2007253590A (ja) * | 2006-03-27 | 2007-10-04 | Fujifilm Corp | ガスバリア性フィルム、基材フィルムおよび有機エレクトロルミネッセンス素子 |
| JP2008073993A (ja) * | 2006-09-22 | 2008-04-03 | Dainippon Printing Co Ltd | ガスバリア性積層フィルム |
| WO2011007543A1 (ja) * | 2009-07-17 | 2011-01-20 | 三井化学株式会社 | 積層体およびその製造方法 |
-
2012
- 2012-02-06 JP JP2012022853A patent/JP5884531B2/ja not_active Expired - Fee Related
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2006297694A (ja) * | 2005-04-19 | 2006-11-02 | Konica Minolta Holdings Inc | ガスバリアフィルム、有機エレクトロルミネッセンス用樹脂基材、該樹脂基材を用いた有機エレクトロルミネッセンス素子及びガスバリアフィルムの製造方法 |
| JP2007023304A (ja) * | 2005-07-12 | 2007-02-01 | Konica Minolta Holdings Inc | 透明導電膜付ガスバリア性フィルムの製造方法及び有機エレクトロルミネッセンス素子の製造方法 |
| JP2007253590A (ja) * | 2006-03-27 | 2007-10-04 | Fujifilm Corp | ガスバリア性フィルム、基材フィルムおよび有機エレクトロルミネッセンス素子 |
| JP2008073993A (ja) * | 2006-09-22 | 2008-04-03 | Dainippon Printing Co Ltd | ガスバリア性積層フィルム |
| WO2011007543A1 (ja) * | 2009-07-17 | 2011-01-20 | 三井化学株式会社 | 積層体およびその製造方法 |
Cited By (17)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9481010B2 (en) | 2012-05-10 | 2016-11-01 | Konica Minolta, Inc. | Method for producing gas barrier film |
| WO2013168647A1 (ja) * | 2012-05-10 | 2013-11-14 | コニカミノルタ株式会社 | ガスバリア性フィルムの製造方法 |
| US20210313555A1 (en) * | 2013-10-22 | 2021-10-07 | Semiconductor Energy Laboratory Co., Ltd. | Power storage device electrode, method for manufacturing the same, power storage device, and electronic device |
| US12218337B2 (en) * | 2013-10-22 | 2025-02-04 | Semiconductor Energy Laboratory Co., Ltd. | Power storage device electrode, method for manufacturing the same, power storage device, and electronic device |
| US12107198B2 (en) * | 2016-03-31 | 2024-10-01 | Toppan Printing Co., Ltd. | Barrier film and method for manufacturing the same, wavelength conversion sheet and method for manufacturing the same, and optical laminate and method for manufacturing the same |
| US20190030859A1 (en) * | 2016-03-31 | 2019-01-31 | Toppan Printing Co., Ltd. | Barrier film and method for manufacturing the same, wavelength conversion sheet and method for manufacturing the same, and optical laminate and method manufacturing the same |
| US11561328B2 (en) | 2016-08-12 | 2023-01-24 | Toppan Printing Co., Ltd. | Phosphor protection film, wavelength conversion sheet, and light-emitting unit |
| JP2019036598A (ja) * | 2017-08-10 | 2019-03-07 | 株式会社東芝 | 半導体素子およびその製造方法 |
| WO2022065498A1 (ja) * | 2020-09-28 | 2022-03-31 | 三菱ケミカル株式会社 | 画像表示用導光板 |
| CN116194285A (zh) * | 2020-09-28 | 2023-05-30 | 三菱化学株式会社 | 图像显示用导光板 |
| TWI922505B (zh) | 2020-09-28 | 2026-04-21 | 日商三菱化學股份有限公司 | 圖像顯示用導光板 |
| JP2022055621A (ja) * | 2020-09-29 | 2022-04-08 | 三菱ケミカル株式会社 | 画像表示用導光板 |
| JP7683182B2 (ja) | 2020-09-29 | 2025-05-27 | 三菱ケミカル株式会社 | 画像表示用導光板の製造方法 |
| JP2022099540A (ja) * | 2020-12-23 | 2022-07-05 | 三菱ケミカル株式会社 | 画像表示用導光板 |
| JP7631787B2 (ja) | 2020-12-23 | 2025-02-19 | 三菱ケミカル株式会社 | 画像表示用導光板 |
| JP2022129942A (ja) * | 2021-02-25 | 2022-09-06 | 三菱ケミカル株式会社 | 画像表示用導光板 |
| WO2024047806A1 (ja) * | 2022-08-31 | 2024-03-07 | 株式会社レニアス | 透明積層体の製造方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| JP5884531B2 (ja) | 2016-03-15 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5884531B2 (ja) | 水蒸気バリアーフィルムの製造方法、水蒸気バリアーフィルム及び電子機器 | |
| JP5857452B2 (ja) | バリアーフィルムの製造方法 | |
| JP5533585B2 (ja) | ガスバリアフィルムの製造方法、ガスバリアフィルム及び電子機器 | |
| JP5929775B2 (ja) | ガスバリア性フィルムおよびその製造方法、ならびに前記ガスバリア性フィルムを含む電子デバイス | |
| JP6287858B2 (ja) | ガスバリア性フィルム、その製造方法、およびこれを用いた電子デバイス | |
| JP5895687B2 (ja) | ガスバリア性フィルム | |
| JP2015003464A (ja) | ガスバリア性フィルム、その製造方法、およびこれを用いた電子デバイス | |
| JP5540949B2 (ja) | ガスバリア性フィルム、及び有機光電変換素子、有機エレクトロルミネッセンス素子 | |
| JP6229506B2 (ja) | ガスバリア性フィルム、およびこれを用いた電子デバイス | |
| JP2014201032A (ja) | ガスバリア性フィルムおよびその製造方法 | |
| CN105593013A (zh) | 气体阻隔性膜的制造方法 | |
| JPWO2010026869A1 (ja) | 複合フィルム、ガスバリアフィルム及びその製造方法並びに有機エレクトロルミネッセンス素子 | |
| JP2015103389A (ja) | 有機el素子 | |
| JP6070411B2 (ja) | ガスバリアー性フィルム、ガスバリアー性フィルムの製造方法及び有機エレクトロルミネッセンス素子 | |
| JP2012067193A (ja) | ガスバリア性フィルムの洗浄方法、ガスバリア性包装体及び有機電子デバイス | |
| JP5849790B2 (ja) | 水蒸気バリアーフィルムの製造方法、水蒸気バリアーフィルム及び電子機器 | |
| JPWO2010026852A1 (ja) | 樹脂フィルム及びその製造方法並びに有機エレクトロルミネッセンス素子 | |
| JP5761005B2 (ja) | 水蒸気バリアーフィルムの製造方法、水蒸気バリアーフィルム及び電子機器 | |
| JP2014201033A (ja) | ガスバリア性フィルムおよびその製造方法 | |
| JP5768652B2 (ja) | バリアーフィルムの製造方法 | |
| JP2013226732A (ja) | ガスバリアフィルムの製造方法 | |
| JPWO2015178069A6 (ja) | ガスバリア性フィルム | |
| JPWO2015178069A1 (ja) | ガスバリア性フィルム | |
| JP2014076590A (ja) | ガスバリアーフィルム及び電子デバイス | |
| JP6102986B2 (ja) | 水蒸気バリアーフィルムの製造方法、水蒸気バリアーフィルム、電子機器及び有機エレクトロルミネッセンスパネル |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20140513 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20150220 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20150303 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20151104 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20151216 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20160112 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20160125 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 5884531 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| LAPS | Cancellation because of no payment of annual fees |