JP2012184408A - 変性ジエン系ゴム及びその製造方法並びにそれを用いたゴム組成物 - Google Patents
変性ジエン系ゴム及びその製造方法並びにそれを用いたゴム組成物 Download PDFInfo
- Publication number
- JP2012184408A JP2012184408A JP2012027339A JP2012027339A JP2012184408A JP 2012184408 A JP2012184408 A JP 2012184408A JP 2012027339 A JP2012027339 A JP 2012027339A JP 2012027339 A JP2012027339 A JP 2012027339A JP 2012184408 A JP2012184408 A JP 2012184408A
- Authority
- JP
- Japan
- Prior art keywords
- rubber
- acid
- diene rubber
- epoxidized
- group
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
Landscapes
- Compositions Of Macromolecular Compounds (AREA)
- Epoxy Resins (AREA)
- Addition Polymer Or Copolymer, Post-Treatments, Or Chemical Modifications (AREA)
Abstract
【解決手段】エポキシ化率が0.1%以上15%未満のエポキシ化ジエン系ゴムと、エポキシ基と反応する官能基を有するアルコキシシラン及びカルボン酸とを反応させた変性ジエン系ゴムである。また、エポキシ化率が0.1%以上15%未満のエポキシ化ジエン系ゴムと、エポキシ基と反応する官能基を有するアルコキシシラン及びカルボン酸とを機械的混練により反応させることを特徴とする変性ジエン系ゴムの製造方法である。
【選択図】なし
Description
先ず、参考例1として、実施例1乃至4、比較例2及び3に用いるエポキシ化ポリブタジエンゴムを作製した。具体的には、ポリブタジエン(宇部興産(株)製:UBEPOL BR150L)を100g取り、セパラブルフラスコのシクロへキサン1000ml中に投入して攪拌し、1晩(約8時間)をかけて溶解した。その後、ウォーターバスでセパラブルフラスコの温度を50℃とし、非イオン性界面活性剤テリック320(HUNTSMAN社製)1gおよび蟻酸4.25g投入して攪拌、続いて過酸化水素水(30%)10.5gを滴下ロートで投入して2時間攪拌を続けた。その後に加熱を止め、酸化防止剤(イルガノックス1520L)を0.1g投入し、ウォーターバス中に氷を投入して温度を室温まで下げた。次に、メタノール500mlを投入し、エポキシ化ポリブタジエンゴムを溶液から析出させた。溶媒を取り除いた後、2.5%炭酸ナトリウムを500ml投入し、10分間攪拌後、洗浄液を除去した。さらに、水500mlを投入し、10分間攪拌後、洗浄液を除去した。この水洗作業を3度行った。その後、反応液をテフロン(登録商標)コーティングしたバットに取り出し、90℃の真空乾燥機中に1時間置いて、乾燥させることで、エポキシ化ポリブタジエンゴムを得た。
次に、表1に示すとおり、参考例1において作製したエポキシ化ポリブタジエンゴムを100g取り、ロール(温度:60℃、ロール間隔:0.8mm)に巻きつけて混練した。ロールは、安田精機製6インチロール混練機を用いた。次に、ステアリン酸(花王社製)2.5g、GOPTMS(3−グリシジルオキシプロピルトリメトキシシラン)(東京化成製:試薬)1gをピペットでロール上のエポキシ化ポリブタジエンゴムに垂らし、さらに5分間練り込んだ。その後、真空乾燥機で真空下、90℃で1時間熱処理することで、実施例1に係る変性ポリブタジエンゴムを得た。この変性ポリブタジエンゴムを24時間静置した後、約5gを取りシクロヘキサン100mlに溶解し、エタノールで析出させる操作を2回行った。得られた試料を再び90℃、1時間乾燥させ、ICP分析によりSiの含量を測定した。結果を表3に示す。
実施例1において、ステアリン酸(花王社製)2.5gの代わりにマロン酸(和光純薬社製:試薬)1gを用いたこと以外は実施例1と同様にして、実施例2に係る変性ポリブタジエンゴムを得た。得られた試料について、実施例1と同様にICP分析によりSiの含量を測定した。結果を表3に示す。
実施例1において、ステアリン酸(花王社製)2.5gの代わりにマロン酸(和光純薬社製:試薬)2.5gを用いたこと以外は実施例1と同様にして、実施例3に係る変性ポリブタジエンゴムを得た。得られた試料について、実施例1と同様にICP分析によりSiの含量を測定した。結果を表3に示す。
実施例3において、変性ポリブタジエンゴムをシクロヘキサン100mlに溶解し、エタノールで析出させる操作を1回にしたこと以外は実施例3と同様にして、実施例4に係る変性ポリブタジエンゴムを得た。得られた試料について、実施例3と同様にICP分析によりSiの含量を測定した。結果を表3に示す。
ポリブタジエン(宇部興産(株)製:UBEPOL BR150L)を24時間静置した後、約5gを取りシクロヘキサン100mlに溶解し、エタノールで析出させる操作を2回行った。得られた試料を再び90℃、1時間乾燥させ、ICP分析によりSiの含量を測定した。結果を表3に示す。
参考例1において作製したエポキシ化ポリブタジエンゴムを24時間静置した後、約5gを取りシクロヘキサン100mlに溶解し、エタノールで析出させる操作を2回行った。得られた試料を再び90℃、1時間乾燥させ、ICP分析によりSiの含量を測定した。結果を表3に示す。
参考例1において作製したエポキシ化ポリブタジエンゴムを100g取り、ロール(温度:60℃、ロール間隔:0.8mm)に巻きつけて混練した。ロールは、安田精機製6インチロール混練機を用いた。次に、GOPTMS(3−グリシジルオキシプロピルトリメトキシシラン)(東京化成製:試薬)1gをピペットでロール上のエポキシ化ポリブタジエンゴムに垂らし、さらに5分間練り込んだ。その後、真空乾燥機で真空下、90℃で1時間熱処理することで、比較例3に係る変性ポリブタジエンゴムを得た。この変性ポリブタジエンゴムを24時間静置した後、約5gを取りシクロヘキサン100mlに溶解し、エタノールで析出させる操作を2回行った。得られた試料を再び90℃、1時間乾燥させ、ICP分析によりSiの含量を測定した。結果を表3に示す。
JIS K6251に準拠して測定し、引張強度(TB)、破壊伸度(EB)、(TB×EB)/2を下記計算式数2で指数表示した。(TB×EB)/2は破断までに費やされたエネルギーのおおよその大きさを示す。TB、EB、(TB×EB)/2は一般的に指数が大きい程、ゴム組成物として有利である。
反発弾性は、JIS K6251に規定されている測定法に従って、トリプソ式で測定し、下記計算式数2で指数表示した。一般的に指数が大きい程、ゴム組成物として有利である。
EPLEXOR 100N(GABO社製)を用いて、初期歪み10%、動歪み0.3%、周波数16Hz、温度50℃の測定条件で各配合物のtanδを測定し、比較例1のtanδを100とし、下記計算式数3で指数表示した。指数が大きいほど、転がり抵抗特性が優れることを示す。
次に、参考例2として、実施例5乃至10、比較例4に用いるエポキシ化ポリブタジエンゴムを作製した。具体的には、ポリブタジエン(宇部興産(株)製:UBEPOL BR150L)を100g取り、セパラブルフラスコのシクロへキサン1000ml中に投入して攪拌し、室温(25℃)で、1晩(約8時間)をかけて溶解した。その後、ウォーターバスでセパラブルフラスコの温度を40℃とし、非イオン性界面活性剤テリック320(HUNTSMAN社製)1gおよび蟻酸2.13g投入して攪拌、続いてセパラブルフラスコの温度を50℃まで上昇させ、過酸化水素水(30%)5.24gを滴下ロートで投入して2時間攪拌を続けた。その後に加熱を止め、酸化防止剤(イルガノックス1520L)を0.2g投入し、ウォーターバス中に氷を投入して温度を室温まで下げた。次に、エタノール500mlを投入し、エポキシ化ポリブタジエンゴムを溶液から析出させた。溶媒を取り除いた後、1.0%炭酸ナトリウムを500ml投入し、10分間攪拌後、洗浄液を除去した。さらに、水(pH=7.0)500mlを投入し、10分間攪拌後、洗浄液を除去した。この水洗作業を3度行った。その後、反応液をテフロン(登録商標)コーティングしたバットに取り出し、90℃の真空乾燥機中に3時間置いて、乾燥させることで、エポキシ化ポリブタジエンゴムを得た。
次に、表4に示すとおり、参考例2において作製したエポキシ化ポリブタジエンゴムを100g取り、ロール(温度:60℃、ロール間隔:0.8mm)に巻きつけて混練した。ロールは、安田精機製6インチロール混練機を用いた。次に、ステアリン酸(花王社製)1g、GOPTMS(3−グリシジルオキシプロピルトリメトキシシラン)(東京化成製:試薬)1gをピペットでロール上のエポキシ化ポリブタジエンゴムに垂らし、さらに5分間練り込んだ。その後、真空乾燥機で真空下、90℃で1時間熱処理することで、実施例5に係る変性ポリブタジエンゴムを得た。この変性ポリブタジエンゴムを24時間静置した後、約5gを取りシクロヘキサン120mlに溶解し、メタノール400mlで30分洗浄した。沈殿した試料を再びシクロヘキサン120mlに溶解し、老化防止剤(Irganox 1520L)0.05mlを加え、上記と同様にメタノールで洗浄することで、再度試料を析出させた。得られた試料を再び90℃、1時間乾燥させ、ICP分析によりSiの含量を測定した。結果を表6に示す。
実施例5において、ステアリン酸(花王社製)1gの代わりにマロン酸(和光純薬社製:試薬)1gを用いたこと以外は実施例5と同様にして、実施例6に係る変性ポリブタジエンゴムを得た。得られた試料について、実施例5と同様にICP分析によりSiの含量を測定した。結果を表6に示す。
実施例5において、ステアリン酸(花王社製)1gの代わりにマロン酸(和光純薬社製:試薬)2.5gを用いたこと以外は実施例5と同様にして、実施例7に係る変性ポリブタジエンゴムを得た。得られた試料について、実施例5と同様にICP分析によりSiの含量を測定した。結果を表6に示す。
参考例2において作製したエポキシ化ポリブタジエンゴムを100g取り、ロール(温度:60℃、ロール間隔:0.8mm)に巻きつけて混練した。ロールは、安田精機製6インチロール混練機を用いた。次に、GOPTMS(3−グリシジルオキシプロピルトリメトキシシラン)(東京化成製:試薬)1gをピペットでロール上のエポキシ化ポリブタジエンゴムに垂らし、さらに5分間練り込んだ。その後、真空乾燥機で真空下、90℃で1時間熱処理することで、比較例4に係る変性ポリブタジエンゴムを得た。この変性ポリブタジエンゴムを24時間静置した後、約5gを取りシクロヘキサン120mlに溶解し、メタノール400mlで30分洗浄した。沈殿した試料を再びシクロヘキサン120mlに溶解し、老化防止剤(Irganox 1520L)0.05mlを加え、上記と同様にメタノールで洗浄することで、再度試料を析出させた。得られた試料を再び90℃、1時間乾燥させ、ICP分析によりSiの含量を測定した。結果を表6に示す。
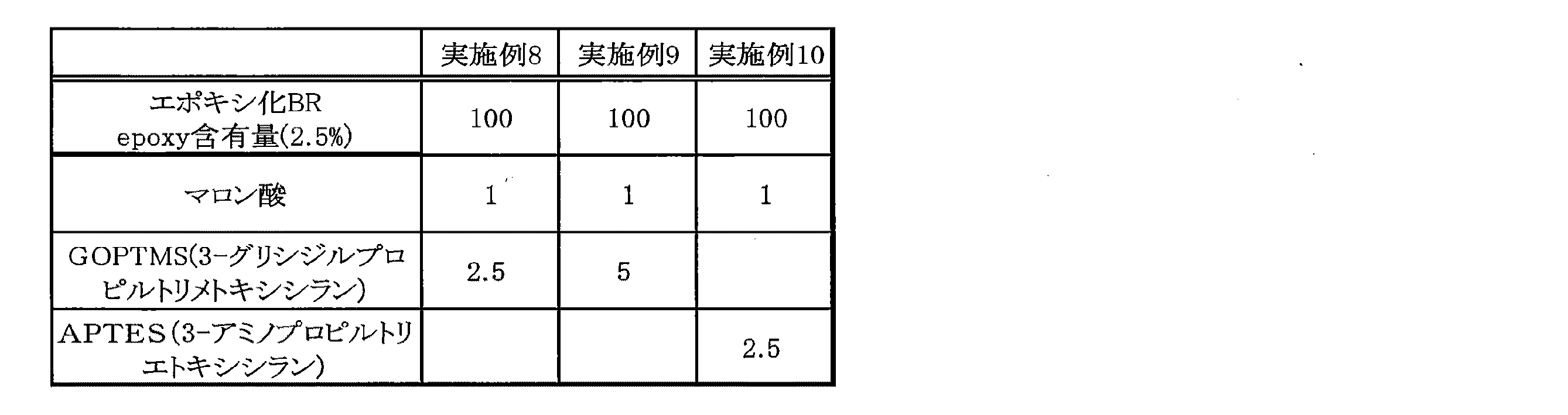
次に、表5に示すとおり、参考例2において作製したエポキシ化ポリブタジエンゴムを100g取り、ロール(温度:60℃、ロール間隔:0.8mm)に巻きつけて混練した。ロールは、安田精機製6インチロール混練機を用いた。次に、マロン酸(花王社製)1g、GOPTMS(3−グリシジルオキシプロピルトリメトキシシラン)(東京化成製:試薬)2.5gをピペットでロール上のエポキシ化ポリブタジエンゴムに垂らし、さらに5分間練り込んだ。その後、真空乾燥機で真空下、90℃で1時間熱処理することで、実施例8に係る変性ポリブタジエンゴムを得た。この変性ポリブタジエンゴムを24時間静置した後、約5gを取りシクロヘキサン120mlに溶解し、メタノール400mlで30分洗浄した。沈殿した試料を再びシクロヘキサン120mlに溶解し、老化防止剤(Irganox 1520L)0.05mlを加え、上記と同様にメタノールで洗浄することで、再度試料を析出させた。得られた試料を再び90℃、1時間乾燥させ、ICP分析によりSiの含量を測定した。結果を表6に示す。
実施例8において、GOPTMS(3−グリシジルオキシプロピルトリメトキシシラン)(東京化成製:試薬)を5g用いたこと以外は実施例8と同様にして、実施例9に係る変性ポリブタジエンゴムを得た。得られた試料について、実施例8と同様にICP分析によりSiの含量を測定した。結果を表6に示す。
実施例8において、GOPTMS(3−グリシジルオキシプロピルトリメトキシシラン)(東京化成製:試薬)2.5gの代わりにAPTES(3−アミノプロピルトリエトキシシラン)(東京化成製:試薬)2.5gを用いたこと以外は実施例8と同様にして、実施例10に係る変性ポリブタジエンゴムを得た。得られた試料について、実施例8と同様にICP分析によりSiの含量を測定した。結果を表6に示す。
Claims (4)
- エポキシ化率が0.1%以上15%未満のエポキシ化ジエン系ゴムと、エポキシ基と反応する官能基を有するアルコキシシラン及びカルボン酸とを反応させた変性ジエン系ゴム。
- 上記反応は、20〜140℃で反応させることを特徴とする請求項1記載の変性ジエン系ゴム。
- エポキシ化率が0.1%以上15%未満のエポキシ化ジエン系ゴムと、エポキシ基と反応する官能基を有するアルコキシシラン及びカルボン酸とを機械的混練により反応させることを特徴とする変性ジエン系ゴムの製造方法。
- 請求項1又は2記載の変性ジエン系ゴムとシリカとを含有することを特徴とする変性ジエン系ゴム組成物。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2012027339A JP5998505B2 (ja) | 2011-02-14 | 2012-02-10 | 変性ジエン系ゴムの製造方法及びゴム組成物の製造方法 |
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2011028590 | 2011-02-14 | ||
| JP2011028590 | 2011-02-14 | ||
| JP2012027339A JP5998505B2 (ja) | 2011-02-14 | 2012-02-10 | 変性ジエン系ゴムの製造方法及びゴム組成物の製造方法 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2012184408A true JP2012184408A (ja) | 2012-09-27 |
| JP5998505B2 JP5998505B2 (ja) | 2016-09-28 |
Family
ID=47014744
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2012027339A Expired - Fee Related JP5998505B2 (ja) | 2011-02-14 | 2012-02-10 | 変性ジエン系ゴムの製造方法及びゴム組成物の製造方法 |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP5998505B2 (ja) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2017154405A1 (ja) * | 2016-03-10 | 2017-09-14 | 信越化学工業株式会社 | 有機ケイ素化合物およびその製造方法 |
| JP2019077751A (ja) * | 2017-10-20 | 2019-05-23 | 株式会社ブリヂストン | ゴム組成物 |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2004359773A (ja) * | 2003-06-03 | 2004-12-24 | Sumitomo Rubber Ind Ltd | トレッド用ゴム組成物およびそれを用いた空気入りタイヤ |
| JP2005041960A (ja) * | 2003-07-25 | 2005-02-17 | Sumitomo Rubber Ind Ltd | タイヤトレッド用ゴム組成物およびそれを用いた空気入りタイヤ |
| JP2011006548A (ja) * | 2009-06-24 | 2011-01-13 | Bridgestone Corp | ポリマー組成物、ゴム組成物及びそれを用いたタイヤ |
| JP2011012161A (ja) * | 2009-07-01 | 2011-01-20 | Sumitomo Rubber Ind Ltd | タイヤ用ゴム組成物及び空気入りタイヤ |
-
2012
- 2012-02-10 JP JP2012027339A patent/JP5998505B2/ja not_active Expired - Fee Related
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2004359773A (ja) * | 2003-06-03 | 2004-12-24 | Sumitomo Rubber Ind Ltd | トレッド用ゴム組成物およびそれを用いた空気入りタイヤ |
| JP2005041960A (ja) * | 2003-07-25 | 2005-02-17 | Sumitomo Rubber Ind Ltd | タイヤトレッド用ゴム組成物およびそれを用いた空気入りタイヤ |
| JP2011006548A (ja) * | 2009-06-24 | 2011-01-13 | Bridgestone Corp | ポリマー組成物、ゴム組成物及びそれを用いたタイヤ |
| JP2011012161A (ja) * | 2009-07-01 | 2011-01-20 | Sumitomo Rubber Ind Ltd | タイヤ用ゴム組成物及び空気入りタイヤ |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2017154405A1 (ja) * | 2016-03-10 | 2017-09-14 | 信越化学工業株式会社 | 有機ケイ素化合物およびその製造方法 |
| JPWO2017154405A1 (ja) * | 2016-03-10 | 2018-09-27 | 信越化学工業株式会社 | 有機ケイ素化合物およびその製造方法 |
| US10323048B2 (en) | 2016-03-10 | 2019-06-18 | Shin-Etsu Chemical Co., Ltd. | Organosilicon compound and production process therefor |
| JP2019077751A (ja) * | 2017-10-20 | 2019-05-23 | 株式会社ブリヂストン | ゴム組成物 |
Also Published As
| Publication number | Publication date |
|---|---|
| JP5998505B2 (ja) | 2016-09-28 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN102245644B (zh) | 天然橡胶、其制造方法、橡胶组合物及使用它的充气轮胎、改性天然橡胶及其制造方法、以及胎面或包覆胎体帘线用橡胶组合物及使用它们的充气轮胎 | |
| JP4308292B2 (ja) | インナーライナー用ゴム組成物およびそれからなるインナーライナーを有するタイヤ | |
| JP5924562B1 (ja) | 変性ゴム、それを用いたタイヤ用ゴム組成物及びタイヤ | |
| CN1469898A (zh) | 采用有机硅烷四硫化物二氧化硅偶合剂在高混合温度下混炼的二氧化硅增强橡胶 | |
| CN1842542A (zh) | 共轭二烯烃(共)聚橡胶及其制造方法 | |
| JP5904430B1 (ja) | 変性ゴム及びその製造方法、ゴム組成物、並びにタイヤ | |
| JP2012012457A (ja) | S−(3−アミノプロピル)チオ硫酸および/またはその塩の使用方法、並びに、加硫ゴム組成物の発熱抑制方法 | |
| JP2011079978A (ja) | 変性ジエン系ゴムおよびゴム組成物 | |
| CN104995248B (zh) | 包含金属羧酸盐的橡胶组合物和其制备方法 | |
| JP5598365B2 (ja) | 変性ジエン系ゴムの製造方法 | |
| JP5998505B2 (ja) | 変性ジエン系ゴムの製造方法及びゴム組成物の製造方法 | |
| JP5488797B2 (ja) | エポキシ化ジエン系ゴムの製造方法およびゴム組成物 | |
| EP3842486A1 (en) | Rubber composition and tire | |
| JP6958013B2 (ja) | ゴム組成物の製造方法 | |
| JP5920107B2 (ja) | 変性ジエン系ゴム組成物およびその製造方法 | |
| JP2013177520A (ja) | タイヤ用ゴム組成物およびそれを用いた空気入りタイヤ | |
| JP2014201651A (ja) | 変性ジエン系ゴムおよびその製造方法、並びにそれを用いたゴム組成物 | |
| JP2016138198A (ja) | タイヤ用ゴム組成物およびタイヤ | |
| JP5962121B2 (ja) | 変性ジエン系ゴム及びその製造方法並びにそれを用いたゴム組成物 | |
| JP6497115B2 (ja) | タイヤ用ゴム組成物 | |
| JP5682273B2 (ja) | 変性単独重合体ジエン系ゴム及びその製造方法並びにそれを用いたゴム組成物 | |
| JP5541125B2 (ja) | エポキシ化ジエン系ゴムの製造方法及びエポキシ化ジエン系ゴムを含むゴム組成物 | |
| JP5912934B2 (ja) | タイヤ用ゴム組成物及び空気入りタイヤ | |
| JP5895517B2 (ja) | タイヤトレッド用ゴム組成物および空気入りタイヤ | |
| JP2018035326A (ja) | ゴム組成物 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20141226 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20151216 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20160105 |
|
| A521 | Written amendment |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20160303 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20160802 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20160815 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 5998505 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| LAPS | Cancellation because of no payment of annual fees |