JP2012209031A - 金属化合物−導電剤複合体の製造方法、該方法により得られる金属化合物−導電剤複合体およびそれを用いてなるアルカリ金属二次電池用電極剤およびアルカリ金属二次電池 - Google Patents
金属化合物−導電剤複合体の製造方法、該方法により得られる金属化合物−導電剤複合体およびそれを用いてなるアルカリ金属二次電池用電極剤およびアルカリ金属二次電池 Download PDFInfo
- Publication number
- JP2012209031A JP2012209031A JP2011071953A JP2011071953A JP2012209031A JP 2012209031 A JP2012209031 A JP 2012209031A JP 2011071953 A JP2011071953 A JP 2011071953A JP 2011071953 A JP2011071953 A JP 2011071953A JP 2012209031 A JP2012209031 A JP 2012209031A
- Authority
- JP
- Japan
- Prior art keywords
- metal compound
- conductive agent
- solvent
- aqueous
- stirring
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Withdrawn
Links
Classifications
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02E—REDUCTION OF GREENHOUSE GAS [GHG] EMISSIONS, RELATED TO ENERGY GENERATION, TRANSMISSION OR DISTRIBUTION
- Y02E60/00—Enabling technologies; Technologies with a potential or indirect contribution to GHG emissions mitigation
- Y02E60/10—Energy storage using batteries
Landscapes
- Battery Electrode And Active Subsutance (AREA)
Abstract
【課題】
従来の固相合成法と比べて低温の溶媒中で達成可能な、電気化学的機能を有する金属化合物と導電剤とが確実に接合された金属化合物−導電剤複合体を形成する製造方法を提供する。
【解決手段】
不活性気体雰囲気かつ非密閉下で、金属化合物前駆体と導電剤から金属化合物−導電剤複合体を製造する方法であって、
(A)金属化合物前駆体を非水系第1溶媒に混合する工程、
(B)上記混合物を加熱して透明な溶液を得る工程、
(C)導電剤を混合する工程、
(D)非水系第2溶媒を混合して加熱する工程、
を有し、(A)(B)(D)の順になされ、かつ(C)は(D)の前の任意の時になされることを特徴とする金属化合物−導電剤複合体の製造方法。
【選択図】なし
従来の固相合成法と比べて低温の溶媒中で達成可能な、電気化学的機能を有する金属化合物と導電剤とが確実に接合された金属化合物−導電剤複合体を形成する製造方法を提供する。
【解決手段】
不活性気体雰囲気かつ非密閉下で、金属化合物前駆体と導電剤から金属化合物−導電剤複合体を製造する方法であって、
(A)金属化合物前駆体を非水系第1溶媒に混合する工程、
(B)上記混合物を加熱して透明な溶液を得る工程、
(C)導電剤を混合する工程、
(D)非水系第2溶媒を混合して加熱する工程、
を有し、(A)(B)(D)の順になされ、かつ(C)は(D)の前の任意の時になされることを特徴とする金属化合物−導電剤複合体の製造方法。
【選択図】なし
Description
本発明は、金属化合物−導電剤複合体の製造方法、該方法により得られる金属化合物−導電剤複合体およびそれを用いてなる二次電池に関するものである。より詳しくは、本発明は、電気化学的機能を有する金属化合物微粒子と導電剤とが確実に接合される合成反応を行う場合に、従来の固相合成法と比べて低温の溶媒中で遂行可能でかつ生成量が多い製造方法であって、また該金属化合物−導電剤複合体の形成において、導電剤上の金属化合物粒子が微細かつ均質で、かつ効率的に反応が遂行される金属化合物−導電剤複合体の製造方法と、該製法により得られる金属化合物−導電剤複合体、および該複合体を用いたアルカリ金属二次電池用電極剤に関するものである。
リチウム二次電池は、従来のニッケルカドミウム電池やニッケル水素電池に比べて、高電圧・高エネルギー密度が得られる電池として小型・軽量化が図れることから、携帯電話やラップトップパソコンなど情報関連のモバイル通信電子機器に広く用いられている。今後更に環境問題を解決する一つの手段として電気自動車・ハイブリッド電気自動車などに搭載する車載用途あるいは電動工具などの産業用途に利用拡大が進むと見られている一方、リチウム二次電池の更なる高容量化と高出力化が切望されている。
リチウム二次電池は少なくともリチウムイオンを可逆的に脱挿入可能な活物質を有する正極と負極、そして正極と負極を隔絶するセパレータを容器内に配置し、非水電解液を充填して構成されている。
正極はアルミニウム等の金属箔集電体に活物質、導電助剤および結着剤を含有する電極剤を塗布し加圧成形したものである。一般的に正極の活物質としては、コバルト酸リチウム(LiCoO2)、ニッケル酸リチウム(LiNiO2)、スピネル型マンガン酸リチウム(LiMn2O4)などに代表されるリチウムと遷移金属の複合酸化物(以後、リチウム金属酸化物と称することがある。)の粉体が用いられているほか、V2O5等の金属酸化物やTiS2、MoS2、NbSe2などの金属化合物等も利用されているが、特にコバルト金属酸化物は小型電池としての性能は優れている一方、クラーク数の低い、いわゆるレアアースを含有していてコスト面や安定供給面から避けられる傾向にあり、特に近年では鉄を含有したリン酸鉄リチウム(LiFePO4)等、資源的に豊富で安価な材料が開発・利用され始めている。
また負極は銅などの金属箔集電体に、正極同様に活物質や導電助剤および結着剤を含有する電極剤を塗布し加圧成形したものであり、一般に負極の活物質としては、金属リチウムやLi−Al合金等のリチウム合金、SiOやSiC、SiOC等を基本構成元素とするケイ素化合物、ポリアセチレンやポリピロール等のリチウムをドープした導電性高分子、リチウムイオンを結晶中に取り込んだ層間化合物や天然黒鉛、人造黒鉛、ハードカーボンなどの炭素材料等が用いられており、最近ではリチウムイオンとコンバージョン反応しうる酸化マンガン(MnO)や酸化コバルト(CoO)などの金属酸化物も検討され始めている。
これら正極あるいは負極の電極剤の調製は、従来、以下のように行われていた。すなわち、炭酸リチウム(Li2CO3)や水酸化リチウム(LiOH)等のリチウム化合物粉末と、炭酸マンガン(MnCO3)や酸化マンガン(MnO2,Mn2O3,Mn3O4)、酸化コバルト(CO3O4)等の金属化合物粉末とで構成される活物質前駆体数種の混合物を500℃〜1000℃で反応させる固相合成法により反応させて活物質を得て、さらに粉砕して得られた活物質粉末に活物質の重量比で数%〜数十%程度の導電助剤、例えば黒鉛粉末やカーボンブラック等の炭素粉を混ぜ、結着剤と呼ばれるPVdF(ポリフッ化ビリニデン)、PTFE(ポリテトラフルオロエチレン)あるいはSBR(スチレン−ブタジエンゴム)等の高分子材料とNMP(n−メチル−2−ピロリドン)等の溶媒を混練してペースト状にして集電体箔上に厚み10μm〜200μmに塗布、乾燥、プレス工程を経て形成する手法である。ただし、前記金属酸化物系正極あるいは負極活物質を採用する場合は、大きく3つの課題があった。
1点目は固相合成法が高温で遂行されることである。前述の通り固相合成法は反応前駆体を固体同士ないしはペースト状にして混ぜ合わせた後、一般的には水分を除去した空気あるいは窒素やアルゴンなどの不活性気体中で概ね3〜24時間の長時間、通常500〜1200℃の高温加熱でなされる。すなわち非常に高温の加熱のために特殊な加熱装置を必要としてかつエネルギー的負担が大きいことから、総じてコスト的に不利である。よって固相重合法に代わる効率的な合成手法が望まれている。
2点目は活物質粒子が大きいことである。電池としての充放電レート特性向上にはリチウム挿入・脱離促進のための反応面積増大あるいは活物質粒子内リチウム拡散パス(拡散経路)短小化が重要で、これは活物質粒子を微細にして比表面積を大きくすることによって達成しうる。しかし前述の固相合成法では得られる活物質が焼結して大粒径化するため、得られた化合物を活物質として用いるには粉砕して微細化する必要があり、しかも粉砕を行った場合であってもサブミクロン(数百nmオーダー)以下に粒子径を小さくするのは非常に困難であった。
3点目は活物質自体の導電性の低さである。従来の主たる活物質であるコバルト系活物質と比べて、最近使われつつある高容量型の金属酸化物系活物質の電気伝導率は10−1〜10−6S/cm2と非常に低いことから、該活物質と集電体間もしくは活物質相互間の電気伝導率を更に高めるべく導電助剤が添加されている。しかし該導電助剤は粒子の凝集力が強いことから導電助剤同士で凝集してしまい、活物質と導電助剤の均一分散あるいは良好な接触(電子が通る導電パス形成)を達成することが難しかったため、結果的に導電性向上は不十分で、本来活物質が持ちうる高い容量を最大限利用できない等の課題があった。
これらの課題、すなわち活物質を微細粒子化しつつ導電助剤を効果的に担持させて活物質の性能を十分に引き出すという課題を解決する試みが多数なされている。例えば、液体中で導電助剤の存在下、活物質の前駆体を合成・共沈させ、最終的に焼成によって活物質を合成して複合粒子を得る技術が開示されている(例えば、特許文献1あるいは2参照。)。これら技術においては、液相中で活物質の前駆体を合成させるものの、やはり最終的には400℃以上の焼成処理が必須で、本質的に焼成での固相反応のため活物質は大粒径化しやすいこと、また導電助剤が水系液相で凝集しやすいため活物質−導電助剤間の良好な接触が望めず、結果的に複合粒子中の活物質−導電助剤間の電気的接触はバラツキが生じやすく性能向上効果は乏しいものであった。
また活物質に導電性を付与すべく、活物質の表面上に炭素を担持させる技術が開示されている(例えば、特許文献3参照。)。該技術は、あらかじめ炭素前駆体となる有機物が炭素−リン酸鉄複合体とリン酸リチウムの共沈物に含有され、焼成工程において該有機物が炭素ネットワークを形成することによって活物質の電気伝導性を高めるものであるが、前述特許文献1あるいは2と同様、コスト的に不利な焼成処理を経た活物質は大粒子径化しやすい点、また活物質表面に存在する炭素ネットワークは確かに良好な接触が発現しやすい一方、該炭素化物は導電性獲得に不利な不純物を含み易く、結果として活物質の導電性を向上させるには限界があった。
一方で電極活物質と導電剤とが強固に接合される技術が開示されている(例えば特許文献4参照。)。この技術は、放電プラズマ焼結機と称する特殊装置を用いて300℃〜700℃の高温下、実質的に5MPa以上の高圧下でパルス通電させることにより電極活物質と導電剤の結合処理を行うもので、確かに効率的な導電剤と活物質との接合が可能である。しかし特殊な装置が必要で、かつ高温・高圧下の処理はで少量ずつ行われることからコスト的に不利で、また活物質同士も結合されて大粒径化しやすく、結果として電極材料の性能向上に必ずしも有利ではなかった。
あるいは、活物質を導電剤表面に形成させる技術が開示されている(例えば非特許文献1参照。)。該技術は水熱法と呼ばれる高温・高圧下・水溶液中での合成手法を採用し、酸化グラフェン上に酸化マンガンのナノ粒子を生成させて活物質と導電助剤の間の良好な接触を有する複合体を得るものである。しかし高圧下反応を行うために密閉系の特殊な製造設備が必要で汎用性に欠けかつ生産性に劣り、しかも水溶液を元にした反応であるためグラフェンの凝集が起こりやすく、ナノ粒子の酸化グラフェン上への生成率が低くなり、結果として導電性を向上させた均質な生成物を得るのは困難であった。
「Mn3O4−Graphene Hybrid as a High−Capacity Anode Material for Lithium Ion Batteries],Journal of American Chemical Society,2010年,第132号,p.13978−13980
本発明の目的は、上記従来技術の問題点を解消し、金属化合物−導電剤複合体の効率的な生成を達成するべく、非密閉系の非水系溶媒中における金属化合物−導電剤複合体の製造方法および該方法により得られる金属化合物−導電剤複合体を提供することにあり、本発明者らは、金属化合物が導電剤上に生成する時の環境をコントロールすることに着目し、鋭意検討を重ね、本発明に到達したものである。
本発明者らは、金属化合物が導電剤上に生成する際の反応環境をコントロールすることに着目し、鋭意検討を重ね、本発明に到達したものである。
すなわち本発明は、上記の課題を解決するため、以下の構成を採用するものである。
(1)不活性気体雰囲気かつ非密閉下で、金属化合物前駆体と導電剤から金属化合物−導電剤複合体を製造する方法であって、
[A]金属化合物前駆体を非水系第1溶媒に混合する工程、
[B]上記混合物を加熱して透明な溶液を得る工程、
[C]導電剤を混合する工程、
[D]非水系第2溶媒を混合して加熱する工程、
を有し、[A][B][D]の順になされ、かつ[C]は[D]の前の任意の時になされることを特徴とする金属化合物−導電剤複合体の製造方法。
[A]金属化合物前駆体を非水系第1溶媒に混合する工程、
[B]上記混合物を加熱して透明な溶液を得る工程、
[C]導電剤を混合する工程、
[D]非水系第2溶媒を混合して加熱する工程、
を有し、[A][B][D]の順になされ、かつ[C]は[D]の前の任意の時になされることを特徴とする金属化合物−導電剤複合体の製造方法。
(2)[A]〜[D]がその順でなされる前記(1)に記載の金属化合物−導電剤複合体の製造方法。
(3)上記[B]で金属化合物前駆体の分解温度以上に加熱する前記(1)または(2)に記載の金属化合物−導電剤複合体の製造方法。
(4)上記[D]で非水系第2溶媒を混合した後、撹拌することなく、あるいは撹拌媒の存在下、最大撹拌周速2m/秒以下で攪拌しながらまたは反応系内に撹拌体の存在無しで2m/秒以下で反応容器を運動させて撹拌しながら加熱する請求項1〜3のいずれか1項に記載の金属化合物−導電剤複合体の製造方法。
(5)上記[A]〜[D]の少なくとも1つで、工程の最初に減圧した後に不活性気体に置換する前記(1)〜(4)のいずれかに記載の金属化合物−導電剤複合体の製造方法。
(6)上記[A]でさらに金属化合物前駆体と実質的に反応しない非水系第3溶媒を加えて混合する前記(1)〜(5)のいずれかに記載の金属化合物−導電剤複合体の製造方法。
(7)上記[A]で非水系第1溶媒が飽和モノカルボン酸または不飽和モノカルボン酸、上記[D]で非水系第2溶媒が飽和脂肪族1級または2級アミン、または不飽和脂肪族1級または2級アミンから、それぞれ選ばれてなる前記(1)〜(6)のいずれかに記載の金属化合物−導電剤複合体の製造方法。
(8)前記(1)〜(7)のいずれかの製造方法により得られることを特徴とする金属化合物−導電剤複合体。
(9)前記(8)記載の金属化合物−導電剤複合体を少なくとも一部に用いてなることを特徴とするアルカリ金属二次電池用電極剤。
(10)前記(9)に記載のアルカリ金属二次電池用電極剤を少なくとも一部に用いてなることを特徴とするアルカリ金属二次電池。
本発明の金属化合物−導電剤複合体の製造方法は、不活性気体雰囲気かつ非密閉下で遂行される。不活性気体雰囲気中で行うことで製造中の合成反応を制御可能で、酸素や水分などが関与する副反応が抑制され、また非密閉下で行えることで手法の汎用性に富む。
そして本発明の金属化合物−導電剤複合体の製造方法は、金属化合物前駆体を非水系第1溶媒に混合する工程(A)を有する。非水系の溶媒を採用することにより、水の沸点よりも高温での反応を遂行可能で、また溶媒中に金属化合物前駆体を混合する手法は反応時の熱の均一伝達を達成して高効率の合成反応の発現を促し、かつ水による副反応も抑制される。
また本発明の金属化合物−導電剤複合体の製造方法は、前記金属化合物前駆体と非水系第1溶媒との混合物を加熱して透明な溶液を得る工程(B)を有する。通常、金属化合物前駆体は非水系第1溶媒と混合した際、該溶媒に溶解しないか殆ど溶解しない場合が多い。従って本発明で目的とする均質な金属化合物−導電剤複合体を得るには金属化合物前駆体を含む非水系第1溶媒を加熱することで該溶媒中に完全に溶解させて透明な溶液を得ることが必要である。
更に本発明の金属化合物−導電剤複合体の製造方法は、金属化合物−導電剤複合体を得るために導電剤を混合する工程(C)を有する。
加えて本発明の金属化合物−導電剤複合体の製造方法は、非水系第2溶媒を混合して加熱する工程(D)を有する。非水系第2溶媒を混合することで、非水系第1溶媒に含まれる金属化合物前駆体の溶解性に揺らぎを与え、金属化合物−導電剤複合体が得られかつ生成量が高くなるが、溶媒中で加熱することで加熱効率が高まり、結果として気体中加熱に比べて加熱温度を下げることができ、しかも金属化合物−導電剤複合体が効率的に得られる。
そして本発明の金属化合物−導電剤複合体の製造方法では、前記工程(A)〜(D)の工程が、(A)の次に(B)、(B)の次に(D)の順でなされ、(C)は(D)の前の任意の時になされることを必要な製造条件として採用しうる。すなわち、金属化合物前駆体を非水系第1溶媒に混合した後に、得られた混合物を加熱して透明な溶液を得て、そしてこの間の任意の時に導電剤を混合した後に、非水系第2溶媒を混合して加熱するという一連の工程によって、副反応の発現で妨げられること無しに、各工程で目的とする、固相反応法などの従来方法に対して低温でかつ高効率で遂行され、結果として本発明で目的とする金属化合物−導電剤複合体の製造においては、導電剤上に金属化合物が高効率、高生成量でよく得られる。
本発明の金属化合物−導電剤複合体の製造方法は不活性気体雰囲気を採用する。不活性気体雰囲気であることで酸素や水分などが関与する副反応が抑制され、目的とする金属化合物−導電剤複合体の製造が効率的に達成される。不活性気体としてはヘリウム、ネオン、アルゴン、クリプトン、キセノンなどの希ガスの他、窒素(N2)を用いうるが、より反応性が乏しいという点で好ましくはヘリウム、ネオン、アルゴン、窒素であり、容易に安価で入手可能という点で、アルゴン、窒素が特に好ましい。なお雰囲気中における不活性気体の割合(純度)は高いほど良く、98%以上が好ましく、99%以上がより好ましく、99.9%以上が特に好ましく、99.99%以上がもっとも好ましい。
また本発明の金属化合物−導電剤複合体の製造は非密閉下で行われる。非密閉下とは製造における工程で、工程が遂行される雰囲気の圧力が高まった場合に、雰囲気を構成する不活性気体が放出されて容器内雰囲気の圧力が減少して、または雰囲気の圧力が下がった場合に、雰囲気を構成する不活性気体が逐次補填されて容器内雰囲気の圧力が増加して、通常の大気圧(約0.1MPa)または大気圧に近い極微加圧の雰囲気圧力に一定に保たれる状況を指すものである。すなわち非密閉下が達成される製造装置は、工程が遂行される容器において雰囲気を構成する気体の入口から出口まで全く雰囲気の移動を遮るものがない大気圧のままの開放型装置でも良く、または後述する非水系の溶媒の蒸散を抑制するために0.4MPa以下の微加圧を達成しうる減圧弁などの雰囲気調整機構を備えた半開放型装置であっても良く、どちらでも手法の汎用性に富むものの、装置構造的に制御が容易という点で0.08MPa〜0.3MPaの雰囲気が好ましく、反応条件を安定して保持して効率的な合成が遂行されるという点で0.09MPa〜0.2MPaの雰囲気が特に好ましく、0.09MPa〜0.15MPaの雰囲気が最も好ましい。
本発明における金属化合物前駆体とは、後述する(A)〜(D)の工程を経て得られる金属化合物−導電剤複合体において導電剤と接合される金属化合物を形成する前駆化合物であり、化学反応を経て本発明で目的とする金属化合物に変化する。従って金属化合物前駆体には金属となる原子が単数もしくは複数含まれる。
該金属化合物前駆体中に選ばれてなる金属となる原子の種類は、本発明の目的の1つとしているアルカリ金属二次電池用電極剤に用いる場合やその他の目的など、それぞれに応じて適宜選択されればよいが、通常、後述する導電剤との複合体を形成するのが困難な金属化合物の構成元素が選ばれてなることが好ましい。また該金属化合物前駆体中に選ばれてなる金属となる原子の種類の数は1種類でも2種以上の複数種類でも良いが、均質な化合物が安定して得られるという点で6種類以下が好ましい。
これら金属化合物前駆体中に選ばれてなる金属となる原子としてはリチウム(Li)、ナトリウム(Na)、カリウム(K)、ルビジウム(Rb)、セシウム(Cs)、マグネシウム(Mg)、カルシウム(Ca)、ストロンチウム(Sr)、バリウム(Ba)、スカンジウム(Sc)、イットリウム(Y)、チタン(Ti)、ジルコニウム(Zr)、ハフニウム(Hf)、バナジウム(V)、ニオブ(Nb)、タンタル(Ta)、クロム(Cr)、モリブデン(Mo)、タングステン(W)、マンガン(Mn)、鉄(Fe)、コバルト(Co)、ロジウム(Rh)、イリジウム(Ir)、ニッケル(Ni)、パラジウム(Pd)、白金(Pt)、銅(Cu)、銀(Ag)、金(Au)、亜鉛(Zn)、カドミウム(Cd)、ガリウム(Ga)、インジウム(In)、タリウム(Tl)、ゲルマニウム(Ge)、スズ(Sn)、鉛(Pb)、ヒ素(As)、アンチモン(Sb)、ビスマス(Bi)、セレン(Se)、テルル(Te)等が挙げられる。
この中でも前記アルカリ金属二次電池用電極剤として用いるにあたって、好適な金属化合物に主として含有される場合に、電池の構成イオンとして存在するアルカリ金属としてリチウム、ナトリウム、カリウムが好ましく用いられ、その他に二次電池の電極剤中の活物質として良好な特性を発現しうるストロンチウム、バリウム、チタン、ジルコニウム、バナジウム、ニオブ、クロム、モリブデン、タングステン、マンガン、鉄、コバルト、ニッケル、白金、銅、銀、金、亜鉛、カドミウム、鉛が好ましく用いられる。そして優れた拡散性能を持つという点で、アルカリ金属であるリチウムとナトリウムが特に好ましく用いられ、また活物質として優れた電気化学特性を発現しうるチタン、バナジウム、モリブデン、タングステン、マンガン、鉄、コバルト、ニッケル、亜鉛が特に好ましく用いられる。
これら金属化合物前駆体中の金属原子は、化合物の一成分として用いられるが、その化合物としては、塩酸塩、弗酸塩(フッ化物)、硝酸塩、炭酸塩、硫酸塩、ギ酸塩、酢酸塩、シュウ酸塩などのカルボン酸塩、アセチルアセトン化合物、水酸化物が挙げられる。そして本発明において後述する非水系溶媒中でより反応性が高いという点で硝酸塩、炭酸塩、カルボン酸塩、アセチルアセトン化合物、水酸化物が好ましく用いられ、分解・脱離などの反応が起こりやすく、後述するように金属化合物を好適な大きさに制御が容易という点で、炭酸塩、カルボン酸塩、水酸化物が特に好ましく用いられ、より多くの化合物が入手しやすいという点でカルボン酸塩がもっとも好ましく用いられる。
ここで該カルボン酸塩を構成するカルボン酸としては多くの種類のカルボン酸が好適に用いられうる。具体的には、ギ酸、酢酸、酪酸などのモノカルボン酸、シュウ酸、マロン酸、コハク酸等のジカルボン酸、乳酸などのヒドロキシ酸、グルタミン酸などのアミノ酸といったものが挙げられ、より具体的には、ギ酸(またはメタン酸)、酢酸(またはエタン酸)、プロピオン酸(またはプロパン酸)、酪酸(またはブタン酸)、吉草酸(またはペンタン酸)、カプロン酸(またはヘキサン酸)、エナント酸(またはヘプタン酸)、カプリル酸(またはオクタン酸)、ペラルゴン酸(またはノナン酸)、カプリン酸(またはデカン酸)、ラウリン酸(またはドデカン酸)、ミリスチン酸(またはテトラデカン酸)、パルミチン酸(またはヘキサデカン酸)、マルガリン酸(またはヘプタデカン酸)、ステアリン酸(またはオクタデカン酸)等の飽和モノカルボン酸、オレイン酸、リノール酸、リノレン酸、アラキドン酸、ドコサヘキサエン酸、エイコサペンタエン酸、10−ウンデセン酸などの不飽和モノカルボン酸、シュウ酸(またはエタン二酸)、マロン酸(またはプロパン二酸)、コハク酸(またはブタン二酸)、グルタル酸(またはペンタン二酸)、アジピン酸(またはヘキサン二酸)、フマル酸やマレイン酸、エイコサン二酸などのジカルボン酸、安息香酸(またはベンゼンカルボン酸)、フタル酸、イソフタル酸、テレフタル酸、サリチル酸などの芳香族カルボン酸、乳酸やリンゴ酸、クエン酸などのヒドロキシ酸、その他にもアミノ酸など、多くのカルボン酸が好ましく用いられうる。
そしてこれらカルボン酸の中で、前述した分解・脱離などの反応が容易に起こるという点で、飽和モノカルボン酸、不飽和モノカルボン酸、ヒドロキシ酸がより好ましく、後述するように金属化合物の生成反応を阻害しにくく金属化合物が導電剤と複合化する際に適度な形状に制御しうる点で飽和モノカルボン酸、不飽和モノカルボン酸が特に好ましい。そして最も好ましいカルボン酸としては、ギ酸、酢酸、ステアリン酸、オレイン酸、リノール酸が挙げられる。
以上のように金属化合物前駆体は前述の金属原子と化合物の組み合わせで、目的とする組成に応じて適宜選択されるが、特に後述するアルカリ金属二次電池用電極剤に用いるのに好ましいものとして具体的には、まずアルカリ金属源として、塩化リチウム、臭化リチウム、フッ化リチウム、水酸化リチウム、炭酸リチウム、硝酸リチウム、硫酸リチウム、りん酸リチウム、ギ酸リチウム、酢酸リチウム、ステアリン酸リチウム、リチウムアセチルアセトナート、クエン酸リチウム、乳酸リチウム、シュウ酸リチウム、トリフルオロ酢酸リチウム、メタケイ酸リチウム等のリチウム化合物およびその水和物、あるいは同様のナトリウム化合物およびその水和物が挙げられる。
また二次電池中の電気化学反応で優れた特性を発現しうる金属源として、2価の塩化マンガン、2価の臭化マンガン、2価の炭酸マンガン、2価の硝酸マンガン、2価の硫酸マンガン、2価または3価のリン酸マンガン、二ギ酸マンガン、2価または3価の酢酸マンガン、2価のステアリン酸マンガン、2価または3価のマンガンアセチルアセトナート、2価の乳酸マンガン、シュウ酸マンガン、2価の安息香酸マンガン、3価のトリフルオロ酢酸マンガン等のマンガン化合物およびその水和物、あるいは同様のチタン化合物、バナジウム化合物、モリブデン化合物、タングステン化合物、鉄化合物、コバルト化合物、ニッケル化合物、亜鉛化合物およびそれらの水和物が挙げられる。
これら金属化合物前駆体は単独で用いても、また本発明の目的を損ねない範囲で複数種を同時に用いてもよい。また本発明で用いる金属化合物前駆体は水和水を有する化合物であっても良いが、水和物を含有しないことが好ましい。
本発明の製造方法では、工程(D)以前の任意の段階で、金属化合物合成に供される金属化合物を含まない前駆体が用いられてもよい。具体的にはリン酸、無水ケイ酸(SiO2)などである。
本発明における導電剤は金属微粒子をはじめとして多種多様のものを必要とされる性能や用いられる金属化合物に応じて適宜採用できるが、化学的に安定性が高いという点でカーボン系微粒子を用いることが好ましい。具体的なカーボン系微粒子として、導電性ファーネスブラック、導電性ケッチェンブラックあるいは導電性アセチレンブラック等のカーボンブラック、単層カーボンナノチューブ(以下カーボンナノチューブをCNTと略記することがある)や2層以上を有する複層CNT、気相成長炭素繊維(以下VGCFと略記することがある)、カップスタック型CNT、カーボンナノホーン等のカーボンチューブ、カーボン六員環が連続してシートを形成した単層グラフェンあるいは複数枚のグラフェンからなる複層グラフェンなどの他、ポリマー繊維を焼成して得られた後に破砕して得られるミルドカーボン繊維やポリマー繊維からなる不織布を焼成して得られるカーボン不織布シートおよび破砕して得られるミルドカーボン不織布、ポリマーシートを焼成して得られた後に破砕して得られるミルドカーボンシート、などが挙げられ、好適に用いられるが、導電性発現に関して近隣の導電剤同士が接近して安定した導電パス形成が可能となることから、繊維状あるいはシート状の構造を有するカーボン系微粒子がより好ましい。
すなわち単層CNTや複層CNT、VGCF、カップスタック型CNT、カーボンナノホーン等のカーボンチューブや、単層グラフェンや複層グラフェン、ミルドカーボン繊維、ミルドカーボン不織布シート、ミルドカーボンシートが好ましい導電剤であり、より高い導電性を有するという点で、単層CNTや複層CNT、VGCF、単層グラフェンや複層グラフェン、ミルドカーボン不織布シート、ミルドカーボンシートがより好ましく、比表面積が大きく金属化合物と密着性が高いという点で繊維状である複層CNTやVGCF、シート状である単層グラフェンや複層グラフェン、ミルドカーボン不織布シートが特に好ましい。ここでミルドカーボン不織布シートにおける不織布を形成するカーボン繊維の繊維径は2μm以下であることが好ましく、1μm以下であることがより好ましく、500nm以下であることが特に好ましく、300nm以下であることが最も好ましい。外カーボン繊維は細いほど好ましいものの、高い導電性を有しつつも構造を維持しうる強度を保持するために繊維径は1nm以上であることが好ましく、5nm以上であることがより好ましい。
なお比表面積が大きいという点では繊維状である複層CNTやVGCFの直径は2nm〜300nmであることが好ましく、5nm〜200nmであることがより好ましい。そしてこれら導電剤の導電性(体積抵抗率)は5000[Ω・cm]以下のものが好ましく用いられ、特に好ましい範囲としては、1.0×10−6[Ω・cm]〜500[Ω・cm]である。ここで該体積抵抗率は、下記実施例B.項の方法にて測定して求める。
本発明の金属化合物−導電剤複合体の製造方法は、金属化合物前駆体を非水系第1溶媒に混合する工程(A)を有する。非水系溶媒を採用することにより、前述の金属化合物を生成させる際に、水に依る副反応が起こらず、反応効率が高まるため好ましい。そして溶媒の種類によっては、水の沸点よりも高温での反応を遂行可能である。すなわち、従来行われてきた水熱法が高温反応を達成するために0.5MPa以上の密閉雰囲気とした容器中、高圧下で行っていたことと比較して、本発明では水の沸点よりも高い沸点を有する非水系溶媒を選択した場合、水熱法よりも遙かに高温ながら大気圧もしくは大気圧に近い低圧の穏やかな雰囲気下で複合化合成反応を達成しうるため好ましい。
また溶媒中に金属化合物前駆体を混合する点は、液相での反応自体が反応時の熱の均一伝達を達成して高効率の合成反応の発現を促すため、従来行われてきた固相反応法が高温気体中で、また水熱法が低温高圧の水蒸気中で、各々反応を行う際、共に不十分・不均一な熱の伝達だった点と比較して、本発明の手法は熱伝達良く好ましく遂行される。
なお本工程(A)において金属化合物前駆体を非水系第1溶媒に混合する手法は、撹拌翼や撹拌媒体を付属した撹拌棒による撹拌や、磁力により撹拌子を回転させる磁性撹拌、反応容器自体を直接旋回する旋回撹拌、一軸あるいは複数軸を有した混練機中に混合される物質を添加して、混練軸と混練機内壁面あるいは混練軸間での剪断により混合する混練撹拌、硬度の高い球状媒体(ビーズ)を一緒に撹拌するメディア分散、超音波を発振するプローブを混合する液中に浸して行う超音波混合など、混練物の性状に応じて適宜採用されるが、好ましくは撹拌棒による撹拌、混練撹拌、メディア分散であり、撹拌棒による撹拌と混練撹拌が特に好ましい。
前記非水系第1溶媒の種類としては、非水系であれば水の副反応を抑制しうるため特に制限されるものではないが、金属化合物前駆体と混合する際に平易な設備で混練可能であることから50℃以下の温度で液体である溶媒であることが好ましく、反応効率を高めるために後述するような工程(B)および工程(D)での加熱下での反応を行う際に加熱温度よりも沸点が高く液体のままで存在する溶媒であることが好ましく、および工程(B)にて金属化合物前駆体と反応または相互作用することにより金属化合物前駆体が消失して透明な溶液が得られる溶媒であることが好ましい。
これら条件を満たす好ましい非水系第1溶媒としては、具体的には、N−メチル−2-ピロリドン(NMP)、ジメチルスルホキシド(DMSO)、N,N−ジメチルフォルムアミド(DMF)、N,N−ジメチルアセトアミド(DMAc)等の非プロトン性極性溶媒、酪酸、吉草酸、カプロン酸、エナント酸、カプリル酸、ペラルゴン酸、カプリン酸、ウンデカン酸、ラウリン酸などの直鎖状、分岐状あるいは環状の飽和モノカルボン酸、ヘプテン酸、ミリストレイン酸、パルミトレイン酸、オレイン酸、リノール酸、α−リノレン酸、γ−リノレン酸、エライジン酸、バクセン酸、ガドレイン酸、エイコセン酸、エルカ酸、ネルボン酸、エイコサペンタエン酸、ドコサヘキサエン酸などの直鎖状、分岐状あるいは環状の不飽和モノカルボン酸、が挙げられ、疎水性で水の存在量が少ないという点で、飽和モノカルボン酸または不飽和モノカルボン酸がより好ましく、工程(B)で透明な溶液を得る際、後述するように金属化合物の微粒子が生成するのに適した構造を取りうる点で二重結合により屈曲した化学構造を有する不飽和モノカルボン酸であるミリストレイン酸、パルミトレイン酸、オレイン酸、バクセン酸、ガドレイン酸、エイコセン酸、エルカ酸、ネルボン酸が特に好ましく、オレイン酸が最も好ましい。
これら非水系第1溶媒は1種類を単独で用いても、複数種を選んで混合して用いてもよいが、副反応が起こることもあり得ることから1種単独で用いることが好ましい。なお、これら好ましいとされる非水系第1溶媒の沸点は150℃以上であることが好ましく、170℃以上であることが特に好ましい。また該沸点は高いほど好ましいが、概ね350℃以下である。また非水系第1溶媒は非水系であっても残存水が存在することがあるため、用いる溶媒中の水分は0.1wt%以下であることが好ましく、0.01wt%以下であることがより好ましい。
本発明の金属化合物前駆体を非水系第1溶媒に混合する工程(A)では、金属化合物前駆体を入手してそのまま非水系第1溶媒に混合して用いてもよいが、前述の通り水の副反応の影響を可能な限り除外するために、あらかじめ加熱乾燥によって乾燥した金属化合物前駆体を用いることが好ましい。この場合、加熱温度は80℃以上であることが好ましく、100℃以上であることがより好ましく、120℃以上であることが特に好ましい。
また乾燥雰囲気は大気(空気)中で加熱乾燥しても良いが、より効率的に水やその他不要物を除去するために、窒素(N2)やアルゴン、ヘリウムなどの不活性気体の雰囲気下あるいは真空ポンプなどによる真空下で乾燥することが好ましい。なおガスの流量としては多いほど効率的に乾燥されることから好ましく、0.1L/分以上であることが好ましく、1L/分以上であることがより好ましく、10L/分以上であることが特に好ましい。また真空乾燥における真空度は低いほど好ましく、10000Pa以下であることが好ましく、5000Pa以下であることがより好ましく、100Pa以下であることが特に好ましく、10Pa以下であることが最も好ましい。
また本発明の金属化合物−導電剤複合体の製造方法は、前記金属化合物前駆体と非水系第1溶媒との混合物を加熱して透明な溶液を得る工程(B)を有する。前述の金属化合物前駆体は非水系第1溶媒と混合した際、該溶媒に溶解しないか殆ど溶解しない場合が多い。従って本発明で目的とする均質な金属化合物−導電剤複合体を効率よく製造するには金属化合物前駆体を含む非水系第1溶媒を加熱することで該溶媒中に金属化合物前駆体を完全に溶解させて透明な溶液を得ることが必要である。
本発明の工程(B)における加熱方法としては、加熱源として電気を流すことにより発熱する発熱線を組み込んで媒体を加熱することで、金属化合物前駆体を含む非水系第1溶媒を加熱する方式が好ましく、例えば熱媒にシリコンオイルなどの耐熱性液体を用いたオイルバス、熱媒に気体を用いた熱風ヒーター、赤外線などの熱線による輻射熱で装置を暖める赤外線ヒーターの他、発熱線をバンド状に巻き付けて使うバンドヒーター、金属パイプの中に発熱線を組み込んだフレキヒーター、アルミニウム中に埋め込まれたアルミヒーター、アルミナ基板に組み込んだセラミックヒーター、シリコンゴム中に発熱線を組み込んだシリコンラバーヒーター、ガラス繊維に組み込まれたマントルヒーター、ポリイミドなどの樹脂中に発熱線を組み込んだポリマーヒーターなどが挙げられる。この中で、高い保温効果を有しながらも昇温や冷却により温度制御の応答が早く使いやすいことから、バンドヒーター、フレキヒーター、シリコンラバーヒーター、マントルヒーター、ポリマーヒーターがより好ましい。
本発明の当該工程(B)では加熱して透明な溶液を得る際、採用する金属化合物前駆体の種類によっては金属化合物前駆体の金属原子を除く化学構造部分が分解・脱離することで効率よく透明な溶液が得られることがある。従って工程(B)では金属化合物前駆体の分解温度以上に加熱することが好ましい。工程(B)での加熱温度は100℃以上であることが好ましく、120℃以上であることがより好ましく、140℃以上であることが特に好ましい。また加熱温度の上限としては非水系第1溶媒の種類によって決まり、320℃以下が好ましく、300℃以下がより好ましく、280℃以下であることが特に好ましい。
前記(A)の金属化合物前駆体を非水系第1溶媒に混合する工程と、続く(B)の上記混合物を加熱して透明な溶液を得る工程は、複数種の金属化合物前駆体を用いる場合、工程(A)で全ての金属化合物前駆体を一度に混合してもよいが、1種類ずつ添加する金属化合物前駆体の種類の数だけ(A)→(B)→(A)→(B)→・・・と工程操作を繰り返すことが好ましい。具体的には、例えば2種類の金属化合物前駆体a,bを用いる場合、金属化合物前駆体aを非水系第1溶媒に混合して工程(B)で加熱して透明な溶液を得たのち、金属化合物前駆体bを該透明な溶液に添加・混合して、再度加熱して透明な溶液を得ることが好ましい。なお複数種の非水系第1溶媒を採用する場合は、あらかじめ調製した非水系第1溶媒の混合溶媒に金属化合物前駆体を添加・混合して、加熱して透明な溶液を得ることが好ましい。
更に本発明の金属化合物−導電剤複合体の製造方法は、金属化合物−導電剤複合体を得るために導電剤を混合する工程(C)を有する。本工程(C)において導電剤を混合する手法は、撹拌翼や撹拌媒体を付属した撹拌棒による撹拌や、磁力により撹拌子を回転させる磁性撹拌、反応容器自体を直接旋回する旋回撹拌、一軸あるいは複数軸を有した混練機中に混合される物質を添加して、混練軸と混練機内壁面あるいは混練軸間での剪断により混合する混練撹拌、硬度の高い球状媒体(ビーズ)を一緒に撹拌するメディア分散、超音波を発振するプローブを混合する液中に浸して行う超音波混合などが適宜採用される。
しかし導電剤の粒子はそれ自身、相互作用が強いため凝集し易い傾向にあり、特に前述のように好ましいとする繊維状あるいはシート状の構造を有するカーボン系微粒子の導電剤の場合は微粒子同士が互いに絡み合っていて、より強い力で混合して分散させる必要がることから、混練撹拌、メディア分散、超音波混合がより好ましい手法であり、メディア分散と超音波分散が特に好ましい。この特に好ましいとされる超音波分散に関しては、印可周波数と印可出力が高いほど混合する力が強くなり好ましいものの、印可周波数としては20kHz以上が好ましく、30kHz以上がより好ましい。
また印可出力は50W以上が好ましく、100W以上がより好ましい。なお工程(C)における導電剤を混合する手法は、1種のみでも複数種を併用しても良い。また混合にかける時間は一般的に長い方が導電剤が均一に分散する傾向があり好ましいものの、一方で過度に長時間行うことで導電剤自体が劣化して導電性が劣ることもあるため、5分以上3時間以下が好ましく、10分以上1時間以下が特に好ましい。なお該導電剤については、前述の通り水の副反応の影響を可能な限り除外するために、あらかじめ加熱乾燥によって乾燥した導電剤を混合に用いることが好ましい。
加えて本発明の金属化合物−導電剤複合体の製造方法は、非水系第2溶媒を混合して加熱する工程(D)を有する。詳しいことはまだよく分かっていないが、工程(A),(B)で形成された非水系第1溶媒と金属化合物前駆体の透明な溶液に非水系第2溶媒を混合することで、非水系第1溶媒と金属化合物前駆体のイオン的、水素結合的あるいはファンデアワールス力的な相互作用に、非水系第2溶媒由来のイオン的、水素結合的あるいはファンデアワールス力的な刺激を与えて相互作用に揺らぎを与え、金属化合物の微粒子が均一に導電剤上に析出して複合体が得られると推定している。また加熱することで金属化合物−導電剤複合体が効率的に得られ、生成量が高くなる。
ここで該非水系第2溶媒の種類としては、非水系であれば前述の非水系第1溶媒と同様、水の副反応を抑制しうるため特に制限されるものではないが、金属化合物前駆体と非水系第1溶媒とからなる透明な溶液に更に添加・混合する際に平易な設備で混練可能であることが好ましいことから50℃以下の温度で液体である溶媒であることが好ましく、反応効率を高めるために後述の加熱下での反応を行う際に加熱温度よりも沸点が高く液体のままで存在する溶媒であることが好ましく、および工程(B)にて得られる金属化合物前駆体と非水系第1溶媒とからなる透明な溶液にイオン的、水素結合的あるいはファンデアワールス力的な刺激を与えて相互作用に揺らぎを与え、反応を促進することにより金属化合物が微粒子として析出せしめる溶媒であることが好ましい。
これら条件を満たす好ましい非水系第2溶媒としては、非水系第1溶媒と異なる種類のものであって、具体的には、N−メチル−2-ピロリドン(NMP)、ジメチルスルホキシド(DMSO)、N,N−ジメチルフォルムアミド(DMF)、N,N−ジメチルアセトアミド(DMAc)等の非プロトン性極性溶媒の他、アミン類が好ましい溶媒として挙げられ、具体的にはヘキシルアミン、ヘプチルアミン、オクチルアミン、ノニルアミン、デカンアミン、ジオクチルアミン、トリオクチルアミン、ピペラジン等の直鎖状、分岐状あるいは環状の飽和脂肪族1級、2級または3級アミンの他、オレイルアミン、リノールアミン、リノレンアミンなどの直鎖状、分岐状あるいは環状の不飽和脂肪族1級、2級または3級アミンが挙げられ、前述するように反応効率を高めて金属化合物の微粒子が生成するのに適している点で直鎖状の飽和脂肪族1級または2級アミン、あるいは不飽和脂肪族1級または2級アミンがより好ましい。これらの中でオレイルアミン、オクチルアミン、ジオクチルアミンは入手しやすいこともあり特に好ましい。
これら非水系第2溶媒は1種類を単独で用いても、複数種を選んで混合して用いてもよいが、副反応が起こることもあり得ることから1種単独で用いることが好ましい。なお、これら好ましいとされる非水系第2溶媒の沸点は150℃以上であることが好ましく、180℃以上であることがより好ましく、200℃以上であることが特に好ましい。また該沸点は高いほど好ましいが、概ね350℃以下である。また非水系第2溶媒は非水系であっても残存水が存在することがあるため、用いる溶媒中の水分は0.1wt%以下であることが好ましく、0.01wt%以下であることがより好ましい。
なお本工程(D)において金属化合物前駆体と非水系第1溶媒を混合後に加熱して得た透明な溶液と非水系第2溶媒とを混合する手法は、撹拌翼や撹拌媒体を付属した撹拌棒による撹拌や、磁力により撹拌子を回転させる磁性撹拌、反応容器自体を直接旋回する旋回撹拌、一軸あるいは複数軸を有した混練機中に混合される物質を添加して混練軸と混練機内壁面あるいは混練軸間での剪断により混合する混練撹拌、硬度の高い球状媒体(ビーズ)を一緒に撹拌するメディア分散、超音波を発振するプローブを混合する液中に浸して行う超音波混合など、混練物の性状に応じて適宜採用されるが、混合前後の液体の粘度変化に依らず効率よく撹拌が達成される点で好ましくは撹拌棒による撹拌、混練撹拌、メディア分散であり、撹拌棒による撹拌と混練撹拌が特に好ましい。
また本発明の当該工程(D)では、非水系第2溶媒を混合したのち加熱することで、前述のように金属化合物前駆体由来の金属化合物の微粒子が生成する。加熱温度は高いほど好ましく、150℃以上であることが好ましく、200℃以上であることがより好ましく、240℃以上であることが特に好ましい。また加熱温度の上限としては非水系第1溶媒および第2溶媒の種類によって決まるが、過度に高い場合、金属化合物の粒子が粗大化してしまい、本発明で目的とする微粒子状の金属化合物からなる金属化合物−導電剤複合体が得られないこともあるので、350℃以下が好ましく、320℃以下がより好ましい。また加熱する際に昇温速度が高いほど金属化合物が好ましい大きさでかつ揃った大きさになりやすいことから、5℃/分以上であることが好ましく、8℃/分以上であることがより好ましく、10℃/分以上であることが特に好ましい。
そして本工程(D)では、前述のように金属化合物の微粒子が導電剤上に析出することで金属化合物−導電剤複合体が生成するが、該工程(D)では混合したのちに加熱する際、撹拌することなく、または撹拌媒の存在下、最大撹拌周速2m/秒以下または反応系内に撹拌体の存在無しで3回/秒以下の頻度で混合物を撹拌しながら加熱することが好ましい。金属化合物の微粒子の析出は加熱によって達成しうるものの、過度に撹拌しながら加熱すると微粒子の析出が起こりにくいことがある。撹拌しない、または弱い撹拌とすることで適度に微粒子析出が促進され、好ましい大きさに揃った微粒子となる。
ここで撹拌媒としては撹拌軸棒に取り付けられた羽根状、翼状あるいは棒状などの構造を持つ物や攪拌子を指し、最大撹拌周速とは、前記撹拌媒の撹拌のための羽根や翼の撹拌軸棒の中心からの最遠位置が回転する際の速度を指し、1m/秒以下であることがより好ましい。
また反応系内に撹拌体の存在無しで混合物を撹拌するとは、混合物を内包する製造装置の容器中には撹拌するための媒体、例えば上記撹拌媒が無く、反応系の容器自体が円周運動ないしはそれに類する撹拌のための運動によって内部の非水系第1溶媒および第2溶媒と金属化合物前駆体とからなる混合物が撹拌されることを指す。そして該手法では2m/秒以下反応容器を運動することが好ましく、1m/秒以下であることがより好ましい。そしてこれらの加熱中の撹拌に関する手法の中で、撹拌媒の存在下、撹拌周速0.01m/秒〜0.5m/秒で撹拌しながら加熱することが特に好ましい。
本発明では、前記の工程(A)、工程(B)および工程(D)が、(A)(B)(D)の順になされ、かつ工程(C)は(D)の前の任意の時になされることを特徴としている。詳しいことはまだよく分かっていないものの、(A)(B)(D)の順になされることで、金属化合物の微細粒子の析出が安定かつ効率的に遂行される。
また工程(C)は(D)の前の任意の時になされるが、具体的には工程(A)の前に金属化合物前駆体と混合してなされても、あるいは非水系第1溶媒と混合してなされてもよく、あるいは工程(B)の前に金属化合物前駆体と非水系第1溶媒との混合物に、加熱する前に混合されてもよく、工程(D)の前に、金属化合物前駆体と非水系第1溶媒との混合物を加熱して得た透明な溶液に混合してもよい。
そして工程(B)で透明な溶液を得る工程でより均一な反応が起こりやすく、工程(D)で金属化合物の微粒子が効率的に生成するために好ましい方法として、前記工程(A)〜(D)の工程がその順でなされることをより好ましい製造条件として採用しうる。すなわち、金属化合物前駆体を非水系第1溶媒に混合した後に、得られた混合物を加熱して透明な溶液を得て、そして導電剤を混合した後に、非水系第2溶媒を混合して加熱するという一連の工程によって、副反応が最も抑制されつつ各工程で目的とする反応が遂行され、本発明で目的とする金属化合物−導電剤複合体の製造において、導電剤上に金属化合物が効率よく得られ、結果として金属化合物−導電剤複合体の生成量がより多くなりうる。
また本発明の導電剤を混合する工程(C)では、導電剤を入手してそのまま混合に用いてもよいが、前述の通り水やその他の物質に由来する副反応の影響を可能な限り除外するために、あらかじめ加熱乾燥によって乾燥した導電剤を用いることが好ましい。この場合、加熱温度は80℃以上であることが好ましく、100℃以上であることがより好ましく、120℃以上であることが特に好ましく、150℃以上であることが最も好ましい。
また乾燥雰囲気は大気(空気)中で加熱乾燥しても良いが、導電剤の劣化を抑制しつつより効率的に水やその他不要物を除去するために、窒素(N2)やアルゴン、ヘリウムなどの不活性気体の雰囲気下あるいは真空ポンプなどによる真空下で乾燥することが好ましい。なお不活性気体の雰囲気下において不活性ガスは流しながら乾燥することが好ましく、ガスの流量としては多いほど効率的に乾燥されることから好ましく、0.1L/分以上であることが好ましく、1L/分以上であることがより好ましく、10L/分以上であることが特に好ましい。また真空乾燥における真空度は低いほど好ましく、10000Pa以下であることが好ましく、5000Pa以下であることがより好ましく、100Pa以下であることが特に好ましく、10Pa以下であることが最も好ましい。
本発明の本発明の金属化合物−導電剤複合体の製造方法では、上記工程(A)〜(D)の少なくとも1つで、工程の最初に減圧した後に不活性気体に置換することを好ましい製造条件として採用しうる。減圧することで非水系第1溶媒、非水系第2溶媒に溶存していた、あるいは金属化合物前駆体や導電剤の表面に付着していた気体や揮発性物質が脱離し、混合物中の各々の成分と溶媒とのより密な接触が図られることで金属化合物の生成・析出、あるいは導電剤上への金属化合物の複合量がより増大しやすくなる。なお減圧した後に不活性気体に置換する一連の手順は1回のみ行ってもよいものの、十分に不活性気体に置換する目的で複数回繰り返すことが好ましい。
本発明の金属化合物−導電剤複合体の製造方法では、前記工程(A)で金属化合物前駆体と実質的に反応しない非水系第3溶媒を加えて混合することを好ましい製造条件として採用しうる。金属化合物前駆体と実質的に反応しない非水系第3溶媒を採用し、非水系第1溶媒との複合溶媒とすることで、金属化合物前駆体は実質的に非水系第1溶媒中のみに取り込まれ、非水系第1溶媒の使用量が削減可能となるほか、金属化合物前駆体と導電剤との反応効率が高まり、金属化合物−導電剤複合体の生成量が増大しうる。加えて非水系第3溶媒は安価なものが採用可能となりコスト面でも有利となりうる。なお第3溶媒を加える割合は非水系第1溶媒の同じ温度での体積に対して0.1〜10の比率であることが好ましく、0.25〜5の比率であることがより好ましく、0.5〜2の比率であることが特に好ましい。
該第3溶媒は非水系でかつ金属化合物前駆体と実質的に反応しない他、前述のように工程(B)あるいは工程(D)にて加熱した際にできる限り揮発しないことが好ましいことから、50℃以下に融点を持ち、かつ高沸点の溶媒が好ましく採用される。これら好ましい条件を満たす非水系第3溶媒の種類としては炭化水素系溶媒が好ましく、具体的にはオクタン、ノナン、デカン、ウンデカン、ドデカン、トリデカン、テトラデカン、ペンタデカン、ヘキサデカン、ヘプタデカン、オクタデカン、ノナデカン、イコサン、ヘンイコサン、シクロオクタン、イソデカン、などの直鎖状、分岐状、環状の飽和炭化水素や、1−ノネン、1−デセン、1−ウンデセン、1−ドデセン、1−トリデセン、1−テトラデセン、1−ペンタデセン、1−ヘキサデセン、1−オクタデセン、1−ノナデセンなどの直鎖状、分岐状、環状の不飽和炭化水素が好適に用いられる。
これら非水系第3溶媒は1種類を単独で用いても、複数種を選んで混合して用いてもよいが、副反応が起こることもあり得ることから1種単独で用いることが好ましい。これら好ましいとされる非水系第3溶媒を採用する場合、前述の工程(A)〜(D)にて金属化合物前駆体を混合させたりあるいは加熱したりする場合に用いられることから、非水系第3溶媒の沸点は120℃以上であることが好ましく、150℃以上であることがより好ましく、180℃以上であることが特に好ましく、200℃以上であることが最も好ましい。該沸点は高いほど高温反応に用いられうるため好ましいが、概ね350℃以下である。
また融点は50℃以下であることが好ましく、30℃以下であることがより好ましく、15℃以下であることが特に好ましく、0℃以下であることが最も好ましい。該融点は低いほど液体のままで取扱いが可能となるため好ましいが、概ね−30℃以上である。また非水系第3溶媒は非水系であっても残存水が存在することがあるため、用いる溶媒中の水分は0.1wt%以下であることが好ましく、0.01wt%以下であることがより好ましい。
工程(D)にて生成した金属化合物−導電剤複合体は、濾過、遠心分離、スプレードライ等種々の分離方法で非水系第1溶媒、非水系第2溶媒、あるいは非水系第3溶媒を除去して単離される。一例として、具体的には分離の際にn−ヘキサンなどの非極性溶媒で金属化合物−導電剤複合体の表面に存在する非水系第1溶媒、非水系第2溶媒、あるいは非水系第3溶媒を洗浄、分離除去して、エタノールなどの揮発性溶媒でn−ヘキサンを更に除去するなどの操作を繰り返すことにより金属化合物−導電剤複合体の粉末が得られる。
なお本発明の方法で得られる該金属化合物−導電剤複合体は粉末で得たのち、熱処理することが好ましい。得られた粉末を高温で熱処理することにより残存溶媒が除去されるほか、導電剤と複合した金属化合物結晶の結晶化度が高まり、また結晶サイズも大きくなることもあって後述するような用途で用いる際により性能が高まるため好ましい。該熱処理方法としてはヘリウム、ネオン、アルゴン、窒素などの不活性ガス雰囲気下での熱処理、あるいは真空下での熱処理が好ましく採用される。
本発明の金属化合物−導電剤複合体の製造方法により得られる金属化合物−導電剤複合体は、導電剤上に金属化合物の微細粒子が大量に接合しており、金属化合物由来の機能発現に好適な形態を有している。そして該金属化合物−導電剤複合体を少なくとも一部に用いたアルカリ金属二次電池用電極剤は、前述のように好ましい形態を有する金属化合物に由来して、アルカリ金属ないしそのイオンの挿入・脱離過程において金属化合物の結晶相に該アルカリ金属(イオン)がしっかりと固定されつつもスムーズな挿入・脱離が達成される。そして当該アルカリ金属二次電池用電極剤を少なくとも一部に用いてなるアルカリ金属二次電池となすことで、高容量化と高出力化を達成するし、材料特性の点では導電剤上の金属化合物の粒子径が小さいことにより充放電を繰り返した場合の劣化が少ない、すなわちサイクル特性にも優れることとなる。
以下、実施例により本発明を具体的かつより詳細に説明するが、本発明はこれらの実施例のみに制限されるものではない。なお実施例中の部は特に具体的な記載のない限り重量部を意味する。実施例中の物性値は、下記の方法によって測定した。
A.金属化合物の同定および結晶子サイズの解析方法
解析する試料は、窒素雰囲気下80℃で乾燥して測定に供した。試料をSi無反射板に載せ、Bruker AXS社製X線回折装置(D8ADVANCE)を用いて広角X線回折法(以下XRDと称することがある)により測定した。測定条件としては、X線源としてNiフィルターを使用したCuKα線を用い、出力40kV、40mA、スリット系としてDiv. Slit:0.3°を用い、検出器はLynxEye(高速検出器)を用い、測定範囲(2θ)5〜100°、ステップ幅(2θ)0.01712°、計数時間0.5秒/ステップで、2θ/θ連続スキャンのスキャン方式で測定した。
解析する試料は、窒素雰囲気下80℃で乾燥して測定に供した。試料をSi無反射板に載せ、Bruker AXS社製X線回折装置(D8ADVANCE)を用いて広角X線回折法(以下XRDと称することがある)により測定した。測定条件としては、X線源としてNiフィルターを使用したCuKα線を用い、出力40kV、40mA、スリット系としてDiv. Slit:0.3°を用い、検出器はLynxEye(高速検出器)を用い、測定範囲(2θ)5〜100°、ステップ幅(2θ)0.01712°、計数時間0.5秒/ステップで、2θ/θ連続スキャンのスキャン方式で測定した。
得られた広角X線回折パターンデータはJCPDS(Joint Committee on Powder Diffraction Standards)標準データと対比して金属化合物の種類を同定した。また結晶子サイズの算出は、以下に示すシェラーの式を用いて算出した。
結晶子サイズ(nm)=Kλ/βcosθ
β=(βe 2−β0 2)0.5
ここでK=0.9、λ=0.154056nm、βe:回折ピークの半値幅、β0:半値幅の補正値(0.07°)
B.導電剤の導電性(体積抵抗率)の測定方法
測定は、温度23℃、湿度55%の大気中で測定すべき試料を少なくとも該雰囲気中に1時間保持した後に行った。導電剤の試料1.0gを直径2cmの円筒管に入れた後、20kNの荷重で試料を圧縮したのち、電極間隔3.0mm、電極半径0.7mmの四探針プローブを用いて、三菱化学アナリテック社製ロレスタGP(MCP−T610)にて体積抵抗率を求めた。そして3つの異なる試料について各々1回ずつ測定して3回の平均値をその導電剤の導電性(体積抵抗率値)とした。
結晶子サイズ(nm)=Kλ/βcosθ
β=(βe 2−β0 2)0.5
ここでK=0.9、λ=0.154056nm、βe:回折ピークの半値幅、β0:半値幅の補正値(0.07°)
B.導電剤の導電性(体積抵抗率)の測定方法
測定は、温度23℃、湿度55%の大気中で測定すべき試料を少なくとも該雰囲気中に1時間保持した後に行った。導電剤の試料1.0gを直径2cmの円筒管に入れた後、20kNの荷重で試料を圧縮したのち、電極間隔3.0mm、電極半径0.7mmの四探針プローブを用いて、三菱化学アナリテック社製ロレスタGP(MCP−T610)にて体積抵抗率を求めた。そして3つの異なる試料について各々1回ずつ測定して3回の平均値をその導電剤の導電性(体積抵抗率値)とした。
C.溶媒の融点および沸点の測定。
パーキンエルマー社製示差走査熱量分析装置(DSC−2)を用い、試料10mgで測定した。融点(Tm)と沸点(Tb)の定義は、昇温速度5℃/分で室温(20℃)から350℃まで測定した際に観測される吸熱ピーク温度とし、室温(20℃)にて固体であるものについてのみTmを併せて測定した。
D.金属化合物前駆体の分解温度の測定
測定は、セイコーインスツルメント社製EXSTAR6000ユニットのTG/DTA6200を用いて、昇温速度10℃/分、窒素雰囲気下(窒素供給速度1cm3/分)で行い、減量曲線の微分曲線がピークを描く温度を求め、測定は3回行い、3回の平均値を分解温度とした。
測定は、セイコーインスツルメント社製EXSTAR6000ユニットのTG/DTA6200を用いて、昇温速度10℃/分、窒素雰囲気下(窒素供給速度1cm3/分)で行い、減量曲線の微分曲線がピークを描く温度を求め、測定は3回行い、3回の平均値を分解温度とした。
E.微細炭素繊維(CNF)の平均直径と平均長、グラフェンの最大幅の平均、平均厚みの確認、
CNFの平均直径、グラフェンの平均厚みについては、CNFまたはグラフェンを含有したエポキシ樹脂包埋ブロックをウルトラミクロトームにて切削して60〜100nmの厚さの超薄切片を作製し、透過型電子顕微鏡(TEM)観察装置(日立製作所社製、H−7100FA型)にて、加速電圧75kVで、倍率2万〜10万倍の任意の倍率で観察を行い、得られた写真を白黒にデジタル化した。
CNFの平均直径、グラフェンの平均厚みについては、CNFまたはグラフェンを含有したエポキシ樹脂包埋ブロックをウルトラミクロトームにて切削して60〜100nmの厚さの超薄切片を作製し、透過型電子顕微鏡(TEM)観察装置(日立製作所社製、H−7100FA型)にて、加速電圧75kVで、倍率2万〜10万倍の任意の倍率で観察を行い、得られた写真を白黒にデジタル化した。
CNFについては得られた写真を、コンピュータソフトウェアの三谷商事社製WinROOF(バージョン2.3)において、黒で見えるCNFを画像解析することによって写真上に存在する全てのCNF断面の面積をそれぞれ計算し、該面積値から略円形と判断して計算したCNFの直径の平均値を求めることで平均直径とした。グラフェンについては50個のグラフェン断面の最も厚い部分から平均厚みを算出した。
またCNF平均長およびグラフェンの最大幅の平均については、FEI Company社製 走査型電子顕微鏡(SEM) STRATA DB235を用いて、加速電圧2kVで、白金−パラジウム蒸着(蒸着膜圧:25〜50オングストローム)処理を行った後、CNFあるいはグラフェンが全て視野に入る倍率(2万倍)で確認した。任意のCNF50本について観察、測定して得られた平均値を平均長とし、任意のグラフェン50個について観察、最大長を測定して得られた平均値を最大幅の平均とした。
F.金属化合物−導電剤複合体の複合状態の確認
得られた粉末を用いて、前記E.と同じSEMを用いて、加速電圧2kVで、白金−パラジウム蒸着(蒸着膜圧:25〜50オングストローム)処理を行った後、倍率3万倍で観察し、金属化合物からなる微粒子が導電剤に複合化した状態を観察し、導電剤上にあることを確認した。
得られた粉末を用いて、前記E.と同じSEMを用いて、加速電圧2kVで、白金−パラジウム蒸着(蒸着膜圧:25〜50オングストローム)処理を行った後、倍率3万倍で観察し、金属化合物からなる微粒子が導電剤に複合化した状態を観察し、導電剤上にあることを確認した。
[実施例1](酸化マンガン(II)と微細炭素繊維との複合体の製造)
(工程A)金属化合物前駆体として125℃、0.1Paで3時間真空乾燥した純度95%以上の二ぎ酸マンガン(II)二水和物を5モル部を用い、非水系第1溶媒である純度99%以上のオレイン酸(融点16℃、沸点286℃)30モル部に室温で添加した。
(工程A)金属化合物前駆体として125℃、0.1Paで3時間真空乾燥した純度95%以上の二ぎ酸マンガン(II)二水和物を5モル部を用い、非水系第1溶媒である純度99%以上のオレイン酸(融点16℃、沸点286℃)30モル部に室温で添加した。
(工程B)室温で1Paへの減圧(真空脱気;1Paに到達後30秒保持)および0.1MPaとなるまで純度99.99%のアルゴンガス印可を3回繰り返して大気圧(0.1MPa)のアルゴンガスに置換した。続いて非密閉、大気圧の開放容器中アルゴンガスを100cm3/分の流量で流しながら、ポリテトラフルオロエチレン製の撹拌翼型撹拌棒で撹拌翼最速部が1.0m/秒となるように撹拌して二ぎ酸マンガンとオレイン酸とを混合したのち、そのまま撹拌を続けながら12℃/分の昇温速度で240℃まで加熱して、240℃に到達後60分間その温度を保持して、極薄く黄色に着色した透明な溶液を得た。
(工程C)透明な溶液を得て50℃まで冷却したのち、導電剤として、160℃、0.1Paで1時間真空乾燥した平均直径153nm、平均長7.2μm、体積抵抗率1.3×10−2Ω・cmのカーボンナノファイバー(CNF)を用い、計算上生成する酸化マンガン(II)の重量(5モル部;二ぎ酸マンガンと同モル部)の20分の1の質量となる量を前記透明な溶液に添加した。室温で1Paへの減圧(真空脱気;1Paに到達後30秒保持)および0.1MPaとなるまで純度99.99%のアルゴンガス印可を3回繰り返して大気圧(0.1MPa)のアルゴンガスに置換したのち、ヤマト科学株式会社製超音波洗浄器(型式:2510J−DTH、周波数42kHz、出力125W)で大気圧の開放容器中アルゴンガスを100cm3/分の流量で流しながら、15分間、超音波混合を行った。
(工程D)導電剤を混合したのち、透明な溶液と導電剤の混合物に、非水系第2溶媒として純度70%以上のオレイルアミン(融点22℃、沸点350℃)15モル部を室温で添加した。続いて室温で1Paへの減圧(真空脱気;1Paに到達後30秒保持)および0.1MPaとなるまで純度99.99%のアルゴンガス印可を3回繰り返して大気圧(0.1MPa)のアルゴンガスに置換した。置換後、非密閉、大気圧の開放容器中アルゴンガスを100cm3/分の流量で流しながら、ポリテトラフルオロエチレン製の撹拌翼型撹拌棒で撹拌翼最速部が0.5m/秒となるように撹拌して混合したのち、そのまま撹拌を続けながら15℃/分の昇温速度で260℃まで加熱した後260℃で5時間保持した。加熱終了後は放冷して濃褐色の不透明な液状の混合物を得た。
該液状の混合物に対し、該液状の混合物の体積の10倍量のn−ヘキサンを添加して、株式会社久保田製作所製高速冷却遠心機7780IIを用いて、重力の2000倍(2000×g)に相当する遠心力で遠心分離を行った。遠心分離後に上澄み液を除去して試料を得たのち、得られた試料に対して同様の遠心分離操作を合計3回行った。その後、n−ヘキサンとエタノールが同体積ずつ混合された混合液体を用いた以外は同様の遠心分離操作を更に2回行って、最終的に濃灰褐色の粉末試料を収率97%で得た。
得られた粉末試料はXRD解析を行うことで、酸化マンガン(II)(化学式:MnO)と炭素のみからなる複合体であることが分かり、またSEM観察の結果、酸化マンガンの微粒子の粒径が揃っていることが判明し、目的とする金属化合物−導電剤複合体が得られたことが判明した。
該金属化合物−導電剤複合体30部に結着剤としてポリ弗化ビニリデン3部、混合用溶媒としてN−メチル−2−ピロリドン60部を加えてメノウ製乳鉢で粗撹拌したのち、3本ロールミルを3回通すことで十分に混合された電極剤のペースト状物を得た。そして厚さ10μmの銅箔に、銅箔の一部は未塗布部分があってかつ乾燥後に平均25μmの厚さとなるように該電極剤ペースト状物をまず片面に塗布し、大気(空気)雰囲気下80℃で20分間乾燥し、続けて裏面にも同様に未塗布部分を設けかつペースト状物を同様の厚さになるよう塗布したのち、大気(空気)雰囲気下200℃で20分間乾燥して電極板を得た。
該電極板を未塗布部分が長さ2cm×幅1cm、塗布部分が長さ3cm×幅1cmとなるように切り出し、リチウム箔(厚さ30μm)を幅1cm×長さ5cmの大きさに切り出し、電解液としてLiPF6を1M含有するエチレンカーボネート:ジエチルカーボネート=7:3の溶媒を電解液として、水分露点−76℃、酸素濃度1ppmのアルゴンガス雰囲気下、ガラス容器中にこれらを封入して電気化学セルを作製した。
該電気化学セルを用いて、レート0.1C、上限電圧1.5V、下限電圧0.1V、理論容量756mAh/g、測定温度25℃、の条件下で充放電測定を3回行ったところ、放電時に1回目に720mAh/g、2回目に711mAh/g、3回目に701mAh/gの値をそれぞれ算出して得て、本発明の製造方法によって金属化合物−導電剤複合体が効率よく得られ、また該複合体がリチウムをアルカリ源としたアルカリ金属二次電池用電極剤として適用可能であることが示され、更にアルカリ金属二次電池が製造可能であることも示された。条件一覧を表1に示す。
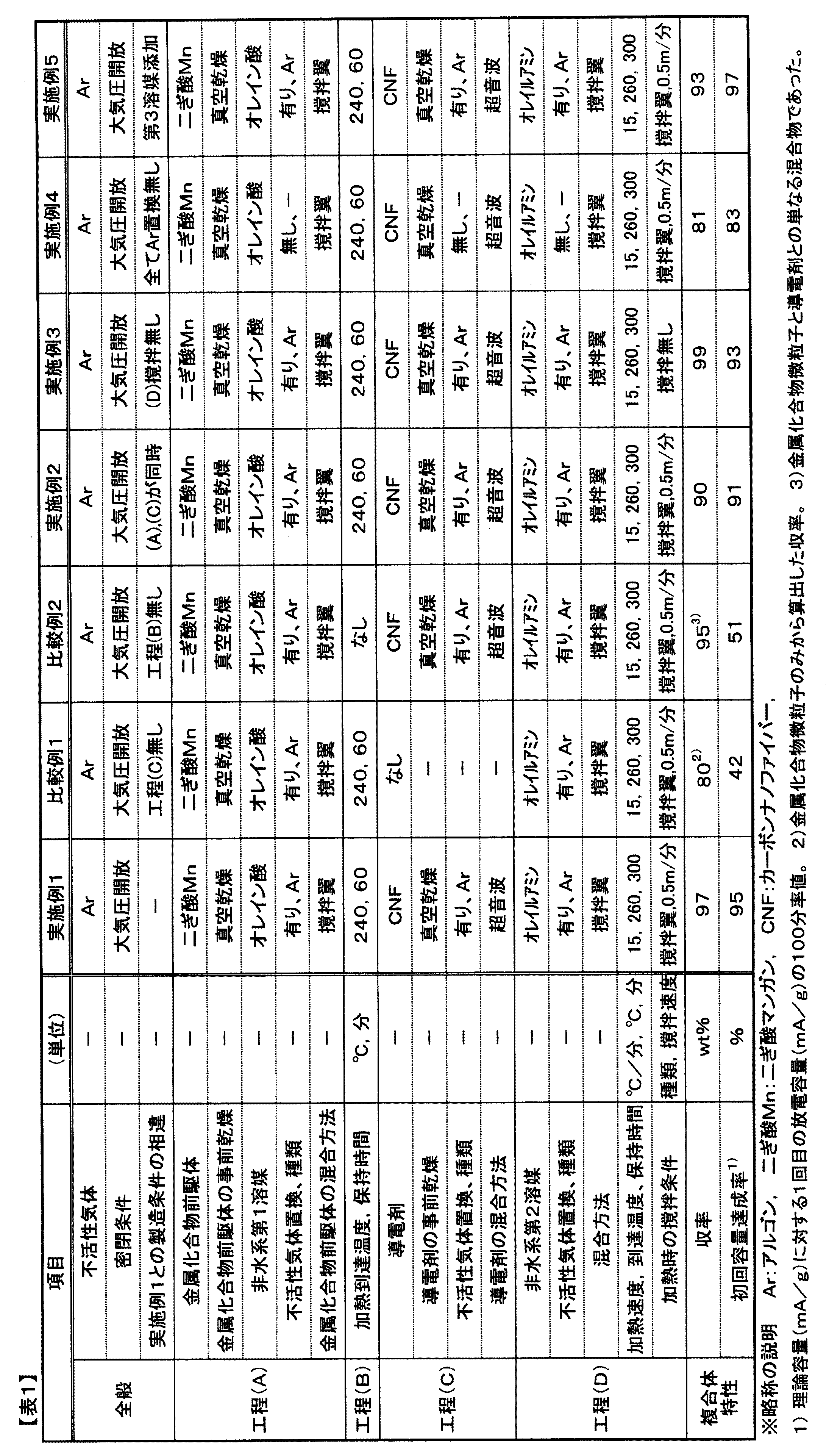
[比較例1]
実施例1において工程(C)の導電剤を混合する工程を適用しなかった以外は実施例1と同様の方法で工程(A),(B),(D)を行ったのち、同様の遠心分離操作を行って粉末を得て、XRDで酸化マンガン(II)であることを確認した。微粒子の収率は80%と実施例1に比べて低下したが、導電剤が存在しなかったことから金属化合物の微粒子の生成効率が低下したものと推測される。
実施例1において工程(C)の導電剤を混合する工程を適用しなかった以外は実施例1と同様の方法で工程(A),(B),(D)を行ったのち、同様の遠心分離操作を行って粉末を得て、XRDで酸化マンガン(II)であることを確認した。微粒子の収率は80%と実施例1に比べて低下したが、導電剤が存在しなかったことから金属化合物の微粒子の生成効率が低下したものと推測される。
該金属化合物微粒子28.5部に対し、実施例1と同じ導電剤(カーボンナノファイバー)を1.5部加えて、実施例1の金属化合物−導電剤複合体と同じ30部とした以外は実施例1と同様の方法、条件により結着剤と混合用溶媒も含めて十分に混合された電極剤ペースト状物を得た。そして実施例1と同様の方法により電極板を得た。そして該電極板を用いて実施例1と同様の方法によりガラス容器からなる電気化学セルを作製したのち実施例1と同様の方法・条件で充放電測定を行った。充放電測定3回の結果は、放電時に1回目に318mAh/g、2回目に286mAh/g、3回目に243mAh/gの値をそれぞれ算出して得たが、実施例1よりも低い容量を示し、金属化合物微粒子と導電剤とをただ単に混合・存在させるだけではアルカリ金属二次電池用電極剤としての性能が劣り、適用できないことが示された。条件一覧を表1に示す。
[比較例2]
実施例1において工程(B)の金属化合物前駆体と非水系第1溶媒の混合物を加熱する工程を適用しなかった以外は実施例1と同様の方法で、工程(A),(C),(D)を行ったのち、同様の遠心分離操作を行って金属化合物と導電剤からなる粉末を得て、XRDで酸化マンガン(II)であることを確認した。収率としては95%と実施例1に比べて僅かに低下したが、SEMで確認したところ実際に得られたものは金属化合物の粗大粒子が導電剤上に存在せず遊離した、単なる金属化合物粒子と導電剤との混合物であることが分かった。工程(B)で透明な溶液を得ないまま工程(C)および(D)を遂行したことから、金属化合物生成の際、粒子が遊離したまま過度に成長し、導電剤と複合化しなかったものと推測される。
実施例1において工程(B)の金属化合物前駆体と非水系第1溶媒の混合物を加熱する工程を適用しなかった以外は実施例1と同様の方法で、工程(A),(C),(D)を行ったのち、同様の遠心分離操作を行って金属化合物と導電剤からなる粉末を得て、XRDで酸化マンガン(II)であることを確認した。収率としては95%と実施例1に比べて僅かに低下したが、SEMで確認したところ実際に得られたものは金属化合物の粗大粒子が導電剤上に存在せず遊離した、単なる金属化合物粒子と導電剤との混合物であることが分かった。工程(B)で透明な溶液を得ないまま工程(C)および(D)を遂行したことから、金属化合物生成の際、粒子が遊離したまま過度に成長し、導電剤と複合化しなかったものと推測される。
該金属化合物粒子と導電剤の混合物30部に対し、実施例1と同様の方法、条件により結着剤と混合用溶媒も含めて十分に混合された電極剤ペースト状物を得た。そして実施例1と同様の方法により電極板を得た。そして該電極板を用いて実施例1と同様の方法によりガラス容器からなる電気化学セルを作製したのち実施例1と同様の方法・条件で充放電測定を行った。充放電測定3回の結果は、放電時に1回目に386mAh/g、2回目に355mAh/g、3回目に313mAh/gの値をそれぞれ算出して得たが、実施例1よりも低い容量を示し、工程(B)で透明な溶液を得ないまま続く工程(C),(D)を進めた場合、金属化合物粒子と導電剤との混合物となり、アルカリ金属二次電池用電極剤としての性能が劣り、適用できないことが示された。条件一覧を表1に示す。
[実施例2]
実施例1において工程(C)の導電剤を混合する工程を工程(A)と同時期とした、具体的には前記実施例の工程(A)でオレイン酸に室温で共に真空乾燥した二ぎ酸マンガン(II)とCNFとを添加し、真空脱気とアルゴンガス印可を3回繰り返してアルゴンガスに置換したのち、15分間の超音波混合を行い、続いて非密閉、大気圧の開放容器中アルゴンガス雰囲気下で撹拌翼型撹拌棒で撹拌して二ぎ酸マンガンとCNFとオレイン酸とを混合した以外は実施例1と同様の方法で工程(B),(D)を行ったのち、同様の遠心分離操作を行って粉体を得て、XRDで酸化マンガン(II)であることを確認した。SEMで金属化合物−導電剤複合体となっていることを確認して、かつ金属化合物−導電剤複合体の収率は90%と優れた結果であった。
実施例1において工程(C)の導電剤を混合する工程を工程(A)と同時期とした、具体的には前記実施例の工程(A)でオレイン酸に室温で共に真空乾燥した二ぎ酸マンガン(II)とCNFとを添加し、真空脱気とアルゴンガス印可を3回繰り返してアルゴンガスに置換したのち、15分間の超音波混合を行い、続いて非密閉、大気圧の開放容器中アルゴンガス雰囲気下で撹拌翼型撹拌棒で撹拌して二ぎ酸マンガンとCNFとオレイン酸とを混合した以外は実施例1と同様の方法で工程(B),(D)を行ったのち、同様の遠心分離操作を行って粉体を得て、XRDで酸化マンガン(II)であることを確認した。SEMで金属化合物−導電剤複合体となっていることを確認して、かつ金属化合物−導電剤複合体の収率は90%と優れた結果であった。
該金属化合物−導電剤複合体を用いて実施例1と同様の方法、条件により電極剤ペースト状物を得た。そして実施例1と同様の方法により電極板を得た。続いて該電極板を用いて実施例1と同様の方法によりガラス容器からなる電気化学セルを作製したのち実施例1と同様の方法・条件で充放電測定を行った。充放電測定3回の結果は、放電時に1回目に688mAh/g、2回目に633mAh/g、3回目に582mAh/gの値をそれぞれ算出し、実施例1と同様に優れた結果を得た。条件一覧を表1に示す。
[実施例3]
実施例1において工程(D)で加熱する際に撹拌しなかった以外は実施例1と同様の方法で工程(A),(B),(C),(D)を行ったのち、同様の遠心分離操作を行って粉体を得て、XRDで酸化マンガン(II)であることを確認した。SEMで金属化合物−導電剤複合体となっていることを確認したが、複合化した金属化合物粒子の中にわずかに粗大粒子が存在することを確認した。該金属化合物−導電剤複合体の収率は99%と極めて優れた結果であった。
実施例1において工程(D)で加熱する際に撹拌しなかった以外は実施例1と同様の方法で工程(A),(B),(C),(D)を行ったのち、同様の遠心分離操作を行って粉体を得て、XRDで酸化マンガン(II)であることを確認した。SEMで金属化合物−導電剤複合体となっていることを確認したが、複合化した金属化合物粒子の中にわずかに粗大粒子が存在することを確認した。該金属化合物−導電剤複合体の収率は99%と極めて優れた結果であった。
該金属化合物−導電剤複合体を用いて実施例1と同様の方法、条件により電極剤ペースト状物を得た。そして実施例1と同様の方法により電極板を得た。続いて該電極板を用いて実施例1と同様の方法によりガラス容器からなる電気化学セルを作製したのち実施例1と同様の方法・条件で充放電測定を行った。充放電測定3回の結果は、放電時に1回目に703mAh/g、2回目に640mAh/g、3回目に576mAh/gの値をそれぞれ算出し、実施例1と同様に優れた結果を得た。条件一覧を表1に示す。
[実施例4]
実施例1において、工程(A),(C),(D)で工程の最初に減圧(真空脱気)した後に不活性気体(アルゴンガス)を印可する、不活性気体置換を行わなかった以外は実施例1と同様の方法で工程(A),(B),(C),(D)を行ったのち、同様の遠心分離操作を行って粉体を得て、XRDで酸化マンガン(II)であることを確認した。SEMで金属化合物−導電剤複合体となっていることを確認した。該金属化合物−導電剤複合体の収率は81%と良好な結果であった。
実施例1において、工程(A),(C),(D)で工程の最初に減圧(真空脱気)した後に不活性気体(アルゴンガス)を印可する、不活性気体置換を行わなかった以外は実施例1と同様の方法で工程(A),(B),(C),(D)を行ったのち、同様の遠心分離操作を行って粉体を得て、XRDで酸化マンガン(II)であることを確認した。SEMで金属化合物−導電剤複合体となっていることを確認した。該金属化合物−導電剤複合体の収率は81%と良好な結果であった。
該金属化合物−導電剤複合体を用いて実施例1と同様の方法、条件により電極剤ペースト状物を得た。そして実施例1と同様の方法により電極板を得た。続いて該電極板を用いて実施例1と同様の方法によりガラス容器からなる電気化学セルを作製したのち実施例1と同様の方法・条件で充放電測定を行った。充放電測定3回の結果は、放電時に1回目に627mAh/g、2回目に552mAh/g、3回目に480mAh/gの値をそれぞれ算出し、良好な結果を得た。条件一覧を表1に示す。
[実施例5]
実施例1において、工程(A)で非水系第3溶媒として、純度99.5%以上の1−オクタデセン(融点15℃、沸点181℃)を15モル部添加し、オレイン酸を15モル部、オレイルアミン10モル部とした以外は実施例1と同様の方法で工程(A),(B),(C),(D)を行ったのち、同様の遠心分離操作を行って粉体を得て、XRDで酸化マンガン(II)であることを確認した。SEMで金属化合物−導電剤複合体となっていることを確認した。該金属化合物−導電剤複合体の収率は93%と実施例1と同様に優れた結果であった。
実施例1において、工程(A)で非水系第3溶媒として、純度99.5%以上の1−オクタデセン(融点15℃、沸点181℃)を15モル部添加し、オレイン酸を15モル部、オレイルアミン10モル部とした以外は実施例1と同様の方法で工程(A),(B),(C),(D)を行ったのち、同様の遠心分離操作を行って粉体を得て、XRDで酸化マンガン(II)であることを確認した。SEMで金属化合物−導電剤複合体となっていることを確認した。該金属化合物−導電剤複合体の収率は93%と実施例1と同様に優れた結果であった。
該金属化合物−導電剤複合体を用いて実施例1と同様の方法、条件により電極剤ペースト状物を得た。そして実施例1と同様の方法により電極板を得た。続いて該電極板を用いて実施例1と同様の方法によりガラス容器からなる電気化学セルを作製したのち実施例1と同様の方法・条件で充放電測定を行った。充放電測定3回の結果は、放電時に1回目に733mAh/g、2回目に674mAh/g、3回目に620mAh/gの値をそれぞれ算出し、優れた結果を得た。条件一覧を表1に示す。
[参考例1](グラフェンの製造方法)
氷冷した405部の98%硫酸を撹拌しながら、平均粒径8μmの天然黒鉛10部、純度99%以上の硝酸ナトリウム5部を加え、更に純度99.3%以上の過マンガン酸カリウム30部を少しずつ添加して加えたのち、20℃で4時間反応させた。反応物は460部の純水で氷冷しながら希釈した後15分間強撹拌し、更に680部の純水で希釈しながら30分間強撹拌したのち、濃度30%の過酸化水素水60部を添加して更に10分間強撹拌して反応を停止した。得られた混合物は実施例1で採用した遠心分離操作で5000×gの遠心力で20分間かけて分離して固体を得た後、pHが6以上となるまで純水での洗浄と20000×gでの遠心分離処理を繰り返して50℃で真空乾燥することで酸化グラフェンを得た。
氷冷した405部の98%硫酸を撹拌しながら、平均粒径8μmの天然黒鉛10部、純度99%以上の硝酸ナトリウム5部を加え、更に純度99.3%以上の過マンガン酸カリウム30部を少しずつ添加して加えたのち、20℃で4時間反応させた。反応物は460部の純水で氷冷しながら希釈した後15分間強撹拌し、更に680部の純水で希釈しながら30分間強撹拌したのち、濃度30%の過酸化水素水60部を添加して更に10分間強撹拌して反応を停止した。得られた混合物は実施例1で採用した遠心分離操作で5000×gの遠心力で20分間かけて分離して固体を得た後、pHが6以上となるまで純水での洗浄と20000×gでの遠心分離処理を繰り返して50℃で真空乾燥することで酸化グラフェンを得た。
該酸化グラフェンをアルゴンガス雰囲気下、1000℃で50時間、加熱還元することで還元されたグラフェンの粉体を得た。
[実施例6](リン酸鉄リチウムとグラフェンとの複合体の製造)
(工程A)金属化合物前駆体として125℃、0.1Paで3時間真空乾燥した純度95%以上の水酸化リチウム・1水和物を1モル部用い、非水系第1溶媒として実施例1で用いたオレイン酸30モル部に室温で添加した。室温で1Paへの減圧(真空脱気;1Paに到達後30秒保持)および0.1MPaとなるまで純度99.99%のアルゴンガス印可を3回繰り返して大気圧(0.1MPa)のアルゴンガスに置換した。続いて非密閉、大気圧の開放容器中アルゴンガスを100cm3/分の流量で流しながら、ポリテトラフルオロエチレン製の撹拌翼型撹拌棒で撹拌翼最速部が0.33m/秒となるように撹拌して水酸化リチウムとオレイン酸とを混合したのち、そのまま撹拌を続けながら10℃/分の昇温速度で140℃まで加熱して、140℃に到達後30分その温度を保持して、透明な溶液を得た。透明な溶液を得て50℃まで冷却したのち、2種類目の金属化合物前駆体として125℃、0.1Paで3時間真空乾燥した純度98%以上の硝酸マンガン・6水和物を1モル部用い、前記水酸化リチウムとオレイン酸とから得られた透明な溶液に添加した。
(工程A)金属化合物前駆体として125℃、0.1Paで3時間真空乾燥した純度95%以上の水酸化リチウム・1水和物を1モル部用い、非水系第1溶媒として実施例1で用いたオレイン酸30モル部に室温で添加した。室温で1Paへの減圧(真空脱気;1Paに到達後30秒保持)および0.1MPaとなるまで純度99.99%のアルゴンガス印可を3回繰り返して大気圧(0.1MPa)のアルゴンガスに置換した。続いて非密閉、大気圧の開放容器中アルゴンガスを100cm3/分の流量で流しながら、ポリテトラフルオロエチレン製の撹拌翼型撹拌棒で撹拌翼最速部が0.33m/秒となるように撹拌して水酸化リチウムとオレイン酸とを混合したのち、そのまま撹拌を続けながら10℃/分の昇温速度で140℃まで加熱して、140℃に到達後30分その温度を保持して、透明な溶液を得た。透明な溶液を得て50℃まで冷却したのち、2種類目の金属化合物前駆体として125℃、0.1Paで3時間真空乾燥した純度98%以上の硝酸マンガン・6水和物を1モル部用い、前記水酸化リチウムとオレイン酸とから得られた透明な溶液に添加した。
(工程B)続いて1Paへの減圧(真空脱気;1Paに到達後30秒保持)および0.1MPaとなるまで純度99.99%のアルゴンガス印可を3回繰り返して大気圧(0.1MPa)のアルゴンガスに置換した。その後非密閉、大気圧の開放容器中アルゴンガスを100cm3/分の流量で流しながら、ポリテトラフルオロエチレン製の撹拌翼型撹拌棒で撹拌翼最速部が0.33m/秒となるように撹拌して硝酸マンガンを混合したのち、そのまま撹拌を続けながら10℃/分の昇温速度で140℃まで加熱して、140℃に到達後60分間その温度を保持して透明な溶液を得た。
(工程C)透明な溶液を得て50℃まで冷却したのち、純度85%以上のリン酸を1モル部用い、前記水酸化リチウム、硝酸マンガンおよびオレイン酸とから得られた透明な溶液に添加し、非密閉、大気圧の開放容器中アルゴンガスを100cm3/分の流量で流しながら、ポリテトラフルオロエチレン製の撹拌翼型撹拌棒で撹拌翼最速部が0.33m/秒となるように1分間撹拌した。そして導電剤として、参考例1で得た、最大幅の平均が3.6μm、平均厚みが0.73nm、体積抵抗率2.3×10−1Ω・cmのグラフェンを用い、計算上生成するリン酸鉄リチウムの重量(1モル部)の20分の1の質量となる量を前記透明な溶液に添加した。室温で1Paへの減圧(真空脱気;1Paに到達後30秒保持)および0.1MPaとなるまで純度99.99%のアルゴンガス印可を3回繰り返して大気圧(0.1MPa)のアルゴンガスに置換したのち、実施例1で用いた超音波洗浄器で大気圧の開放容器中アルゴンガスを100cm3/分の流量で流しながら、15分間、超音波混合を行った。
(工程D)導電剤を混合したのち、透明な溶液と導電剤の混合物に、非水系第2溶媒として実施例1で用いたオレイルアミン15モル部を添加した。続いて実施例1と同様のアルゴンガス置換を行った後、非密閉、大気圧の開放容器中アルゴンガスを100cm3/分の流量で流しながら、ドイツ国ハイドルフ(Heidolph)社製の台座回転撹拌機UNIMAX1010を用いて、反応系内に撹拌体の存在無しで反応容器を2回/秒(0.4m/秒)で運動して混合物を撹拌して混合したのち、そのまま撹拌を続けながら15℃/分の昇温速度で270℃まで加熱した後270℃で5時間保持した。加熱終了後は放冷して濃褐色の不透明な液状の混合物を得た。なお運動速度は反応容器の中で最も運動速度の速い、反応溶液の外縁部がフラスコ内壁を回る位置(円周20cm)の速度とした。
該液状の混合物に対し、実施例1と同様の手法でn−ヘキサン、n−ヘキサンとエタノールの混合液体を用いて遠心分離操作を行って、最終的に濃灰褐色の粉末試料を収率95%で得た。
得られた粉末試料はXRD解析を行うことで、リン酸マンガンリチウム(化学式:LiMnPO4)と炭素のみからなる複合体であることが分かり、またSEM観察の結果、リン酸マンガンリチウムの微粒子の粒径が揃いかつグラフェン上に密に析出・複合化していることが判明し、目的とする金属化合物−導電剤複合体が得られたことが判明した。
該リン酸マンガンリチウム−グラフェン複合体30部を用いた以外は実施例1と同様の手法で電極剤ペースト状物を得て、さらに厚さ20μmのアルミニウム(Al)箔を用いた以外は実施例1と同様の手法で電極板を得たのち、ガラス容器の電気化学セルを作製した。
該リン酸マンガンリチウム−グラフェン複合体30部を用いた以外は実施例1と同様の手法で電極剤ペースト状物を得て、さらに厚さ20μmのアルミニウム(Al)箔を用いた以外は実施例1と同様の手法で電極板を得たのち、ガラス容器の電気化学セルを作製した。
該電気化学セルを用いて、レート0.1C、上限電圧4.5V、下限電圧3.0V、理論容量171mAh/g、測定温度25℃、の条件下で充放電測定を3回行ったところ、放電時に1回目に162mAh/g、2回目に154mAh/g、3回目に148mAh/gの値をそれぞれ算出して得て、本発明の製造方法によって金属化合物−導電剤複合体が効率よく得られ、また該複合体がリチウムをアルカリ源としたアルカリ金属二次電池用電極剤として適用可能であることが示され、更にアルカリ金属二次電池が製造可能であることも示された。
以上の如く、本発明の金属化合物−導電剤複合体の製造方法、該方法により得られる金属化合物−導電剤複合体に関して、電気的機能を有する金属化合物と導電剤とが確実に接合される合成反応を行う場合に、従来の固相合成法と比べて低温の溶媒中で製造が達成される方法であって、導電剤上での金属化合物粒子の生成が効率的に遂行されるため粒子生成量が多くかつ均質であるため、該複合体を用いたアルカリ金属二次電池用電極剤は、電池として機能する際の極初期の効率・再現性が高く、かつ電極剤としての性能を十分発揮する電極剤を構成しうるものである。
本発明の製造方法により得られた金属化合物−導電剤複合体はアルカリ金属二次電池用電極剤に適用した際に初期の効率および性能が高いことから、高性能の二次電池製造に好適に用いることができる。
Claims (10)
- 不活性気体雰囲気かつ非密閉下で、金属化合物前駆体と導電剤から金属化合物−導電剤複合体を製造する方法であって、
(A)金属化合物前駆体を非水系第1溶媒に混合する工程、
(B)上記混合物を加熱して透明な溶液を得る工程、
(C)導電剤を混合する工程、
(D)非水系第2溶媒を混合して加熱する工程、
を有し、(A)(B)(D)の順になされ、かつ(C)は(D)の前の任意の時になされることを特徴とする金属化合物−導電剤複合体の製造方法。 - (A)〜(D)がその順でなされる請求項1に記載の金属化合物−導電剤複合体の製造方法。
- 上記(B)で金属化合物前駆体の分解温度以上に加熱する請求項1または2のいずれか1項に記載の金属化合物−導電剤複合体の製造方法。
- 上記(D)で非水系第2溶媒を混合した後、撹拌することなく、あるいは撹拌媒の存在下、最大撹拌周速2m/秒以下で攪拌しながらまたは反応系内に撹拌体の存在無しで2m/秒以下で反応容器を運動させて撹拌しながら加熱する請求項1〜3のいずれか1項に記載の金属化合物−導電剤複合体の製造方法。
- 上記(A)〜(D)の少なくとも1つで、工程の最初に減圧した後に不活性気体に置換する請求項1〜4のいずれか1項に記載の金属化合物−導電剤複合体の製造方法。
- 上記(A)でさらに金属化合物前駆体と実質的に反応しない非水系第3溶媒を加えて混合する請求項1〜5のいずれか1項に記載の金属化合物−導電剤複合体の製造方法。
- 上記(A)で非水系第1溶媒が飽和モノカルボン酸または不飽和モノカルボン酸、上記(D)で非水系第2溶媒が飽和脂肪族1級または2級アミン、または不飽和脂肪族1級または2級アミンから、それぞれ選ばれてなる請求項1〜6のいずれか1項に記載の金属化合物−導電剤複合体の製造方法。
- 請求項1〜7のいずれか1項に記載の製造方法により得られることを特徴とする金属化合物−導電剤複合体。
- 請求項8に記載の金属化合物−導電剤複合体を少なくとも一部に用いてなることを特徴とするアルカリ金属二次電池用電極剤。
- 請求項9に記載のアルカリ金属二次電池用電極剤を少なくとも一部に用いてなるアルカリ金属二次電池。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2011071953A JP2012209031A (ja) | 2011-03-29 | 2011-03-29 | 金属化合物−導電剤複合体の製造方法、該方法により得られる金属化合物−導電剤複合体およびそれを用いてなるアルカリ金属二次電池用電極剤およびアルカリ金属二次電池 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2011071953A JP2012209031A (ja) | 2011-03-29 | 2011-03-29 | 金属化合物−導電剤複合体の製造方法、該方法により得られる金属化合物−導電剤複合体およびそれを用いてなるアルカリ金属二次電池用電極剤およびアルカリ金属二次電池 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| JP2012209031A true JP2012209031A (ja) | 2012-10-25 |
Family
ID=47188614
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2011071953A Withdrawn JP2012209031A (ja) | 2011-03-29 | 2011-03-29 | 金属化合物−導電剤複合体の製造方法、該方法により得られる金属化合物−導電剤複合体およびそれを用いてなるアルカリ金属二次電池用電極剤およびアルカリ金属二次電池 |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP2012209031A (ja) |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2012209032A (ja) * | 2011-03-29 | 2012-10-25 | Toray Ind Inc | 金属化合物−導電剤複合体およびそれを用いてなるリチウム二次電池、および金属化合物−導電剤複合体の製造方法 |
| WO2014077247A1 (ja) * | 2012-11-13 | 2014-05-22 | 日本ケミコン株式会社 | 電極材料の製造方法、電極材料及び該電極材料を備えた蓄電デバイス |
| WO2017073765A1 (ja) * | 2015-10-30 | 2017-05-04 | 宇部興産株式会社 | 蓄電デバイスの電極用リチウムナトリウムチタン複合酸化物粉末、及び活物質材料、並びにそれを用いた電極シートおよび蓄電デバイス |
| JPWO2017122805A1 (ja) * | 2016-01-15 | 2018-11-22 | 日本ゼオン株式会社 | 熱電変換素子用組成物、金属ナノ粒子が担持されたカーボンナノチューブの製造方法、熱電変換素子用成形体およびその製造方法、並びに熱電変換素子 |
-
2011
- 2011-03-29 JP JP2011071953A patent/JP2012209031A/ja not_active Withdrawn
Cited By (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2012209032A (ja) * | 2011-03-29 | 2012-10-25 | Toray Ind Inc | 金属化合物−導電剤複合体およびそれを用いてなるリチウム二次電池、および金属化合物−導電剤複合体の製造方法 |
| WO2014077247A1 (ja) * | 2012-11-13 | 2014-05-22 | 日本ケミコン株式会社 | 電極材料の製造方法、電極材料及び該電極材料を備えた蓄電デバイス |
| JP2014099301A (ja) * | 2012-11-13 | 2014-05-29 | Nippon Chemicon Corp | 電極材料の製造方法、電極材料及び該電極材料を備えた蓄電デバイス |
| WO2017073765A1 (ja) * | 2015-10-30 | 2017-05-04 | 宇部興産株式会社 | 蓄電デバイスの電極用リチウムナトリウムチタン複合酸化物粉末、及び活物質材料、並びにそれを用いた電極シートおよび蓄電デバイス |
| JPWO2017073765A1 (ja) * | 2015-10-30 | 2018-08-16 | 宇部興産株式会社 | 蓄電デバイスの電極用リチウムナトリウムチタン複合酸化物粉末、及び活物質材料、並びにそれを用いた電極シートおよび蓄電デバイス |
| JPWO2017122805A1 (ja) * | 2016-01-15 | 2018-11-22 | 日本ゼオン株式会社 | 熱電変換素子用組成物、金属ナノ粒子が担持されたカーボンナノチューブの製造方法、熱電変換素子用成形体およびその製造方法、並びに熱電変換素子 |
| EP3404728A4 (en) * | 2016-01-15 | 2019-10-09 | Zeon Corporation | COMPOSITION FOR A THERMOELECTRIC CONVERSION ELEMENT, METHOD FOR THE PRODUCTION OF CARBON NANOTONES CARRYING METALLIC NANOPARTICLES, THERMOELECTRIC CONVERSION MOLDING ELEMENTS, METHOD FOR THE MANUFACTURE THEREOF AND THERMOELECTRIC CONVERSION ELEMENT |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US10312519B2 (en) | Method for manufacturing electroconductive paste, and electroconductive paste | |
| Su et al. | Hierarchical Mn2O3 hollow microspheres as anode material of lithium ion battery and its conversion reaction mechanism investigated by XANES | |
| Chen et al. | SnO2‐based nanomaterials: synthesis and application in lithium‐ion batteries | |
| Roy et al. | Nanostructured anode materials for lithium ion batteries | |
| KR101521453B1 (ko) | 복합 탄소섬유 | |
| JP5664404B2 (ja) | 金属化合物−導電剤複合体およびそれを用いてなるリチウム二次電池、および金属化合物−導電剤複合体の製造方法 | |
| An et al. | Carbon nanofiber/cobalt oxide nanopyramid core–shell nanowires for high-performance lithium-ion batteries | |
| Li et al. | Growth of ultrafine SnO2 nanoparticles within multiwall carbon nanotube networks: non-solution synthesis and excellent electrochemical properties as anodes for lithium ion batteries | |
| WO2011162178A1 (ja) | リチウムイオン二次電池 | |
| JP6197454B2 (ja) | 金属酸化物ナノ粒子−導電剤複合体およびそれを用いてなるリチウムイオン二次電池及びリチウムイオンキャパシタ、ならびに金属酸化物ナノ粒子−導電剤複合体の製造方法 | |
| JP2010212309A (ja) | 電極材料及びこの電極材料を含有する電極 | |
| Zou et al. | Hydrothermally enhanced MnO/reduced graphite oxide composite anode materials for high performance lithium-ion batteries | |
| WO2012163426A1 (en) | Electrode material for lithium and lithium ion batteries | |
| JP2012252824A (ja) | 蓄電素子用電極の製造方法および蓄電素子 | |
| Guler et al. | Electrochemical performance of MWCNT reinforced ZnO anodes for Li-ion batteries | |
| An et al. | In situ preparation of 1D Co@ C composite nanorods as negative materials for alkaline secondary batteries | |
| Xiao et al. | Hydrothermal assembly of MnO-graphene core-shell nanowires with superior anode performance | |
| Shah et al. | Wet-chemical assisted synthesis of MnSe/ZnO nanostructures as low-resistance robust novel cathode material for advanced hybrid supercapacitors | |
| JPWO2019026690A1 (ja) | 非水系二次電池用電極活物質及びその製造方法 | |
| JP2012209031A (ja) | 金属化合物−導電剤複合体の製造方法、該方法により得られる金属化合物−導電剤複合体およびそれを用いてなるアルカリ金属二次電池用電極剤およびアルカリ金属二次電池 | |
| Feng et al. | Ce-doped Li4Ti5O12/C nanoparticles embedded in multiwalled carbon nanotube network as a high-rate and long cycle-life anode for lithium-ion batteries application | |
| Duan et al. | Hydrothermally prepared MnSe electrode as a promising pseudocapacitive material for high-performance supercapacitor | |
| KR20230170730A (ko) | 전극 활물질 전구체, 이의 제조 방법, 전극 활물질 및 전지 | |
| CN104603995B (zh) | 金属氧化物和导电性碳的复合材料的制造方法 | |
| JP2013030292A (ja) | オリビン系正極活物質及びその製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A300 | Withdrawal of application because of no request for examination |
Free format text: JAPANESE INTERMEDIATE CODE: A300 Effective date: 20140603 |