JP2013018240A - ポリカーボネート樹脂成形品の製造方法 - Google Patents
ポリカーボネート樹脂成形品の製造方法 Download PDFInfo
- Publication number
- JP2013018240A JP2013018240A JP2011155044A JP2011155044A JP2013018240A JP 2013018240 A JP2013018240 A JP 2013018240A JP 2011155044 A JP2011155044 A JP 2011155044A JP 2011155044 A JP2011155044 A JP 2011155044A JP 2013018240 A JP2013018240 A JP 2013018240A
- Authority
- JP
- Japan
- Prior art keywords
- polycarbonate resin
- group
- substituted
- resin molded
- structural unit
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
- XGIZHBLCRCEGPN-UHFFFAOYSA-N Cc(cc(C1(C(C2)C(CCC3)C3C2C1)c(cc1)cc(C)c1O)cc1)c1O Chemical compound Cc(cc(C1(C(C2)C(CCC3)C3C2C1)c(cc1)cc(C)c1O)cc1)c1O XGIZHBLCRCEGPN-UHFFFAOYSA-N 0.000 description 1
Landscapes
- Injection Moulding Of Plastics Or The Like (AREA)
- Compositions Of Macromolecular Compounds (AREA)
Abstract
【解決手段】ポリカーボネート樹脂(a)と、該ポリカーボネート樹脂(a)とは異なる構造単位を有し、ISO 15184で規定される鉛筆硬度が該ポリカーボネート樹脂(a)の鉛筆硬度より低いポリカーボネート樹脂(b)とを、重量比で1:99〜45:55の範囲で含有するポリカーボネート樹脂組成物を、成形時における射出成形機の金型温度70℃以上120℃以下で射出成形するポリカーボネート樹脂成形品の製造方法。
【選択図】なし
Description
しかしながら、ポリカーボネート樹脂は、表面硬度が充分に優れているとはいえず、特に、ビスフェノールAを原料とする従来のポリカーボネート樹脂は、屋外で使用される用途においては硬度が不十分であるため、表面にハードコート処理等の付加的な処理を施すことがしばしば行われる。
かかる状況下、本発明の目的は、高硬度であり、色調、耐衝撃性に優れたポリカーボネート樹脂成形品の製造方法を提供することにある。
<1> ポリカーボネート樹脂(a)と、該ポリカーボネート樹脂(a)とは異なる構造単位を有し、ISO 15184で規定される鉛筆硬度が該ポリカーボネート樹脂(a)の鉛筆硬度より低いポリカーボネート樹脂(b)とを、重量比で1:99〜45:55の範囲で含有するポリカーボネート樹脂組成物を、成形時における射出成形機の金型温度70℃以上120℃以下で射出成形するポリカーボネート樹脂成形品の製造方法。
<2> 前記射出成形時における射出成形機のシリンダー温度が270℃以上340℃以下である前記<1>に記載のポリカーボネート樹脂成形品の製造方法。
<3> 前記射出成形時の射出速度が1mm/秒以上60mm/秒以下である前記<1>または<2>に記載のポリカーボネート樹脂成形品の製造方法。
<4> 前記ポリカーボネート樹脂(a)の鉛筆硬度がF以上である前記<1>乃至<3>のいずれかに記載のポリカーボネート樹脂成形品の製造方法。
<5> 前記ポリカーボネート樹脂(a)が、下記式(1)で表される化合物に由来する構造単位を含むポリカーボネート樹脂である前記<1>乃至<4>のいずれかに記載のポリカーボネート樹脂成形品の製造方法。
(一般式(1)中、R1及びR2は、それぞれ独立に、置換若しくは無置換の炭素数1〜炭素数20のアルキル基、又は置換若しくは無置換のアリール基を示し、R3及びR4は、それぞれ独立に、水素原子、置換若しくは無置換の炭素数1〜炭素数20のアルキル基、又は置換若しくは無置換のアリール基を示し、Xは、単結合、カルボニル基、置換若しくは無置換のアルキリデン基、置換若しくは無置換の硫黄原子、又は酸素原子を示す。)
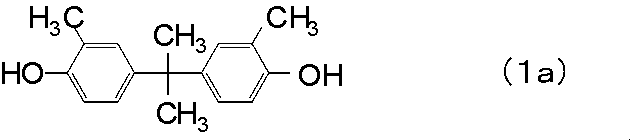
<6> 前記ポリカーボネート樹脂(a)が、下記一般式(1a)〜(1c)からなる群より選ばれた少なくとも1種の化合物に由来する構造単位を少なくとも含むポリカーボネート樹脂である前記<5>に記載のポリカーボネート樹脂成形品の製造方法。
<7> 前記ポリカーボネート樹脂(b)が、下記一般式(2)で表される化合物に由来する構造単位を主として含むポリカーボネート樹脂である前記<1>乃至<6>のいずれかに記載のポリカーボネート樹脂成形品の製造方法。
<9> 前記<1>乃至<8>のいずれかに記載のポリカーボネート樹脂成形品の製造方法により得られてなるポリカーボネート樹脂成形品。
なお、上記「異なる構造単位を有する」とは、
[I]ポリカーボネート樹脂(a)とポリカーボネート樹脂(b)のそれぞれが単独重合体(ホモポリマー)の場合には、“構造単位の種類が異なること”を意味し、
[II]ポリカーボネート樹脂(a)とポリカーボネート樹脂(b)の少なくともいずれか一方が共重合体(コポリマー)の場合には、
(A)構造単位の種類が異なること、
又は
(B)構造単位の種類が同一で、その構造単位の組成比が異なること、
を含む概念である。
すなわち、上記[I]の具体例は、ポリカーボネート樹脂(a)が構造単位Aからなるホモポリマーであり、ポリカーボネート樹脂(b)が構造単位Bからなるホモポリマーである場合である。
上記[II](A)の具体例は、ポリカーボネート樹脂(a)が構造単位Aと構造単位Cとからなるコポリマーであり、ポリカーボネート樹脂(b)が構造単位Bと構造単位Cとからなるコポリマーである場合である。
上記[II](B)の具体例は、ポリカーボネート樹脂(a)とポリカーボネート樹脂(b)のそれぞれが構造単位Aと構造単位Bとからなるが、ポリカーボネート樹脂(a)とポリカーボネート樹脂(b)における構造単位Aと構造単位Bの比率が相違する場合である。
ここで規定するポリカーボネート樹脂の鉛筆硬度は、その詳細は実施例における評価方法「(1)成形品の鉛筆硬度」にて説明する、射出成形品として測定された鉛筆硬度である。以下、本明細書において、特に説明のない限り、単に「鉛筆硬度」と記載した場合は、当該射出成形品の鉛筆硬度を意味する。
ポリカーボネート樹脂(a)の鉛筆硬度がポリカーボネート樹脂(b)の鉛筆硬度と同じかあるいは低い場合には、得られるポリカーボネート樹脂成形品の鉛筆硬度が低くなる可能性があり、成形品表面が傷つきやすい。
金型温度が低すぎると鉛筆硬度が低くなる可能性があり、金型温度が高すぎると成形時の冷却時間が必要以上に長くなり生産性が低くなる可能性がある。
より高品質な樹脂成形品を得るためには、成形時の金型温度は、好ましくは75℃以上115℃以下であり、より好ましくは、80℃以上110℃以下である。
(1)ポリカーボネート樹脂及びその製造方法
まず、本発明のポリカーボネート樹脂成型品を構成するポリカーボネート樹脂組成物の原料である、ポリカーボネート樹脂(a)及びポリカーボネート樹脂(b)として、好適なポリカーボネート樹脂についてそれぞれ説明する。
ポリカーボネート樹脂(a)としては、先ず、下記一般式(1)で表される化合物に由来する構造単位を少なくとも含むポリカーボネート樹脂が好適な例として挙げられる。
(一般式(1)中、R1及びR2は、それぞれ独立に、置換若しくは無置換の炭素数1〜炭素数20のアルキル基、又は置換若しくは無置換のアリール基を示し、R3及びR4は、それぞれ独立に、水素原子、置換若しくは無置換の炭素数1〜炭素数20のアルキル基、又は置換若しくは無置換のアリール基を示し、Xは、単結合、カルボニル基、置換若しくは無置換のアルキリデン基、置換若しくは無置換の硫黄原子、又は酸素原子を示す。)
R3及びR4の、置換若しくは無置換の炭素数1〜炭素数20のアルキル基としては、例えば、メチル基、エチル基、n−プロピル基、イソプロピル基、n−ブチル基、イソブチル基、sec−ブチル基、tert−ブチル基、n−ペンチル基、sec−ペンチル基、n−ヘキシル基、n−ヘプチル基、n−オクチル基等が挙げられ、置換若しくは無置換のアリール基としては、例えば、フェニル基、ベンジル基、トリル基、4−メチルフェニル基、ナフチル基等が挙げられる。
これらの中でも、R1及びR2は、メチル基、エチル基、n−プロピル基、4−メチルフェニル基が好ましく、特にメチル基が好ましい。R3及びR4は、水素原子、メチル基、エチル基、n−プロピル基、4−メチルフェニル基が好ましく、特に水素原子が好ましい。ここで、一般式(1)におけるR1、R2、R3、R4の結合位置は、それぞれのフェニル環上のXに対して2位、3位、5位及び6位から選ばれる任意の位置であり、好ましくは3位、5位である。
これらの中でも、R5及びR6は、メチル基、エチル基、n−プロピル基、4−メチルフェニル基が好ましく、特にメチル基が好ましい。
Zは一般式(1)において、2個のフェニル基と結合する炭素と結合して、置換若しくは無置換の二価の炭素環を形成する。二価の炭素環としては、例えば、シクロペンチリデン基、シキロヘキシリデン基、シクロヘプチリデン基、シクロドデシリデン基又はアダマンチリデン基等のシクロアルキリデン基(好ましくは炭素数4〜炭素数12)が挙げられ、置換されたものとしては、これらのメチル置換基、エチル置換基を有するもの等が挙げられる。これらの中でも、シクロヘキシリデン基、シクロヘキシリデン基のメチル置換体、シクロドデシリデン基が好ましい。
そのような構造単位としては、特に制限はないが、具体的には、2,2−ビス(4−ヒドロキシフェニル)プロパン(以下、「ビスフェノールA」と称する場合がある。)、無水糖アルコール等の脂環式ジヒドロキシ化合物、スピログリコール等の環状エーテル化合物に由来する構造単位が挙げられる。この中でもビスフェノールAに由来する構造単位が特に好ましい。
ポリカーボネート樹脂(a)の粘度平均分子量であるMv(a)は好ましくは10,000以上50,000以下であり、上限はより好ましくは40,000、更に好ましくは35,000である。下限はより好ましくは5,000、更に好ましくは7,000である。Mv(a)が高すぎると、ポリカーボネート樹脂組成物の溶融粘度が高くなり、ポリカーボネート樹脂成形品の表面硬度向上効果が小さくなる可能性がある。一方、Mv(a)が低すぎると、ポリカーボネート樹脂成形品の成形が困難に成る場合がある。
次に、ポリカーボネート樹脂(b)としては、下記一般式(2)〜(13)からなる群より選ばれた少なくとも1種の化合物に由来する構造単位を含むポリカーボネート樹脂が好適に用いられ、下記一般式(2)で示されるビスフェノールAに由来する構造単位を主として含むビスフェノールA型ポリカーボネート樹脂がより好適に用いられる。ここで、「ビスフェノールAに由来する構造単位を主として含む」とは、ポリカーボネート樹脂(b)を構成する構造単位のうち、50重量%以上、好ましくは80重量%以上、より好ましくは90重量%以上が、ビスフェノールAに由来する構造単位であることを意味する。
なお、ポリカーボネート樹脂(b)中の構造単位の含有量は、NMR法により求めることができる。具体的には、核磁気共鳴装置(NMR装置)を使用し、ポリカーボネート樹脂(b)の重クロロホルム溶液を1H−NMR測定した際に観測される、ポリカーボネート樹脂(b)を合成する際に使用したジヒドロキシ化合物に依存した特徴的なシグナルの面積強度比により、各構造単位のモル組成を求めることができる。この得られたモル組成と、各構造単位の式量より、各構造単位の重量比が求まる。
次に、本発明に係る上記ポリカーボネート樹脂(a)及びポリカーボネート樹脂(b)の製造方法について説明する。(以下、「ポリカーボネート樹脂(a)及びポリカーボネート樹脂(b)」を「ポリカーボネート樹脂」と総称する場合がある。)
本発明に係るポリカーボネート樹脂は、ジヒドロキシ化合物とカルボニル化合物とを用いて重合することにより得られる。具体的には、ジヒドロキシ化合物と塩化カルボニル(以下「CDC」もしくは「ホスゲン」と称することがある。)とを、任意に混合しない有機相と水相との界面にて反応させることによりポリカーボネート樹脂を製造する界面重縮合法(以下、「界面法」と称することがある。)と、ジヒドロキシ化合物とカルボニル化合物とをエステル交換反応触媒存在下、溶融状態にてエステル交換反応させることによりポリカーボネート樹脂を製造する溶融重縮合法(以下、「溶融法」と称することがある。)がある。
以下、界面法および溶融法のそれぞれについて、具体的に説明する。
界面法によるポリカーボネート樹脂の製造方法では、通常、ジヒドロキシ化合物のアルカリ水溶液を調製し、縮合触媒として、例えばアミン化合物の存在下で、ジヒドロキシ化合物とホスゲンとの界面重縮合反応を行い、次いで、中和、水洗、乾燥工程を経てポリカーボネート樹脂が得られる。具体的には、界面法によるポリカーボネート樹脂製造プロセスは、モノマー成分等の原料調製を行う原調工程、オリゴマー化反応が行われるオリゴマー化工程、オリゴマーを用いた重縮合反応が行われる重縮合工程、重縮合反応後の反応液をアルカリ洗浄、酸洗浄、水洗浄により洗浄する洗浄工程、洗浄された反応液を予濃縮しポリカーボネート樹脂を造粒後に単離するポリカーボネート樹脂単離工程、単離されたポリカーボネート樹脂の粒子を乾燥する乾燥工程を、少なくとも有している。以下、各工程について説明する。
原調工程では、原調タンクに、ジヒドロキシ化合物と、水酸化ナトリウム(NaOH)等のアルカリ金属化合物の水溶液又は水酸化マグネシウム等のアルカリ土類金属化合物の水溶液と、脱塩水(DMW)と、さらに必要に応じてハイドロサルファイト(HS)等の還元剤を含むジヒドロキシ化合物のアルカリ水溶液等の原料が調製される。
本発明に係るポリカーボネート樹脂の原料であるジヒドロキシ化合物としては、具体的には前記一般式(1)から(13)で表されるジヒドロキシ化合物等が挙げられる。
アルカリ金属化合物又はアルカリ土類金属化合物としては、通常、水酸化物が好ましく、例えば、水酸化ナトリウム、水酸化リチウム、水酸化カリウム、水酸化マグネシウム、水酸化カルシウム等が挙げられる。これらの中でも、水酸化ナトリウムが特に好ましい。
ジヒドロキシ化合物に対するアルカリ金属化合物又はアルカリ土類金属化合物の割合は、通常、1.0〜1.5(当量比)、好ましくは、1.02〜1.04(当量比)である。アルカリ金属化合物又はアルカリ土類金属化合物の割合が過度に多い又は過度に少ない場合は、後述するオリゴマー化工程において得られるカーボネートオリゴマーの末端基に影響し、その結果、重縮合反応が異常となる傾向がある。
次に、オリゴマー化工程では、所定の反応器において、原調工程で調製されたジヒドロキシ化合物のアルカリ水溶液とホスゲン(CDC)とを、塩化メチレン(CH2Cl2)等の有機溶媒の存在下で、ジヒドロキシ化合物のホスゲン化反応が行われる。
続いて、ジヒドロキシ化合物のホスゲン化反応が行われた混合液に、トリエチルアミン(TEA)等の縮合触媒と、p−t−ブチルフェノール(pTBP)等の連鎖停止剤が添加され、ジヒドロキシ化合物のオリゴマー化反応が行われる。
次に、ジヒドロキシ化合物のオリゴマー化反応液は、さらにオリゴマー化反応が進められた後、所定の静置分離槽に導入され、カーボネートオリゴマーを含有する有機相と水相とが分離され、分離された有機相は、重縮合工程に供給される。
ここで、ジヒドロキシ化合物のホスゲン化反応が行われる反応器にジヒドロキシ化合物のアルカリ水溶液が供給されてから静置分離槽に入るまでのオリゴマー化工程における滞留時間は、通常、120分以下、好ましくは、30分〜60分である。
オリゴマー化工程で使用するホスゲンは、通常、液状又はガス状で使用される。オリゴマー化工程におけるCDCの好ましい使用量は、反応条件、特に、反応温度及び水相中のジヒドロキシ化合物の濃度によって適宜選択され、特に限定されない。通常、ジヒドロキシ化合物の1モルに対し、CDC1モル〜2モル、好ましくは1.05モル〜1.5モルである。CDCの使用量が過度に多いと、未反応CDCが多くなり原単位が極端に悪化する傾向がある。また、CDCの使用量が過度に少ないと、クロロフォルメート基量が不足し、適切な分子量伸長が行われなくなる傾向がある。
オリゴマー化工程では、通常、有機溶媒を使用する。有機溶媒としては、オリゴマー化工程における反応温度及び反応圧力において、ホスゲン及びカーボネートオリゴマー、ポリカーボネート樹脂等の反応生成物を溶解し、水と相溶しない(または、水と溶液を形成しない)任意の不活性有機溶媒が挙げられる。
このような不活性有機溶媒として、例えば、ヘキサン、n−ヘプタン等の脂肪族炭化水素;ジクロロメタン、クロロホルム、四塩化炭素、ジクロロエタン、トリクロロエタン、テトラクロロエタン、ジクロロプロパン及び1,2−ジクロロエチレン等の塩素化脂肪族炭化水素;ベンゼン、トルエン及びキシレン等の芳香族炭化水素;クロロベンゼン、o−ジクロロベンゼン及びクロロトルエン等の塩素化芳香族炭化水素;その他、ニトロベンゼン及びアセトフェノン等の置換芳香族炭化水素等が挙げられる。
これらの中でも、ジクロロメタン又はクロロベンゼン等の塩素化された炭化水素が好適に使用される。これらの不活性有機溶媒は、単独であるいは他の溶媒との混合物として使用することができる。
オリゴマー化反応は、縮合触媒の存在下で行うことができる。縮合触媒の添加時期は、CDCを消費した後が好ましい。縮合触媒としては、二相界面縮合法に使用されている多くの縮合触媒の中から、任意に選択することができる。例えば、トリアルキルアミン、N−エチルピロリドン、N−エチルピペリジン、N−エチルモルホリン、N−イソプロピルピペリジン、N−イソプロピルモルホリン等が挙げられる。中でも、トリエチルアミン、N−エチルピペリジンが好ましい。
本実施の形態において、オリゴマー化工程では、通常、連鎖停止剤としてモノフェノールを使用する。モノフェノールとしては、例えば、フェノール;p−t−ブチルフェノール、p−クレゾール等の炭素数1〜炭素数20のアルキルフェノール;p−クロロフェノール、2,4,6−トリブロモフェノール等のハロゲン化フェノールが挙げられる。モノフェノールの使用量は、得られるカーボネートオリゴマーの分子量に応じ適宜選択され、通常、ジヒドロキシ化合物に対して、0.5モル%〜10モル%である。
カーボネート形成性化合物の共存下でモノフェノールを添加すると、モノフェノール同士の縮合物(炭酸ジフェニル類)が多く生成し、目標とする分子量のポリカーボネート樹脂が得られにくい傾向がある。モノフェノールの添加時期が極端に遅れると、分子量制御が困難となり、さらに、分子量分布の低分子側に特異な肩を有する樹脂となり、成形時には垂れを生じる等の弊害が生じる傾向がある。
また、オリゴマー化工程では、任意の分岐剤を使用することができる。このような分岐剤としては、たとえば、2,4−ビス(4−ヒドロキシフェニルイソプロピル)フェノール、2,6−ビス(2−ヒドロキシ−5−メチルベンジル)−4−メチルフェノール、2−(4−ヒドロキシフェニル)−2−(2,4−ジヒドロキシフェニル)プロパン、1,4−ビス(4,4’−ジヒドロキシトリフェニルメチル)ベンゼン等が挙げられる。また、2,4−ジヒドロキシ安息香酸、トリメシン酸、塩化シアヌル等も使用しうる。これらの中でも、少なくとも3個のフェノール性ヒドロキシル基を有する分岐剤が好適である。分岐剤の使用量は、得られるカーボネートオリゴマーの分岐度に応じ適宜選択され、通常、ジヒドロキシ化合物に対し、0.05モル%〜2モル%である。
このような乳濁液を形成する手段としては、例えば、所定の撹拌翼を有する撹拌機、ホモジナイザー、ホモミキサー、コロイドミル、フロージェットミキサー、超音波乳化機等の動的ミキサー、静的ミキサー等の混合機を使用するのが好ましい。乳濁液は、通常、0.01μm〜10μmの液滴径を有し、乳化安定性を有する。
乳濁液の乳化状態は、通常、ウェーバー数又はP/q(単位容積当たりの負荷動力値)で表される。ウェーバー数としては、好ましくは10,000以上、さらに好ましくは20,000以上、最も好ましくは35,000以上である。また、上限としては1,000,000以下程度で十分である。また、P/qとしては、好ましくは200kg・m/L以上、さらに好ましくは500kg・m/L以上、最も好ましくは1,000kg・m/L以上である。
次に、重縮合工程では、静置分離槽で水相と分離されたカーボネートオリゴマーを含有する有機相は、撹拌機を有するオリゴマー貯槽に移送される。オリゴマー貯槽には、トリエチルアミン(TEA)等の縮合触媒がさらに添加される。
続いて、オリゴマー貯槽内で撹拌された有機相は所定の重縮合反応槽に導入され、続いて、重縮合反応槽に、脱塩水(DMW)、塩化メチレン(CH2Cl2)等の有機溶媒及び水酸化ナトリウム水溶液が供給され、撹拌混合されてカーボネートオリゴマーの重縮合反応が行われる。
ここで、重縮合工程において、連続的にカーボネートオリゴマーの重縮合反応が行われる重縮合反応槽における滞留時間は、通常、12時間以下、好ましくは、0.5時間〜5時間である。
重縮合工程の好ましい態様としては、先ず、カーボネートオリゴマーを含む有機相と水相とを分離し、分離した有機相に必要に応じて不活性有機溶媒を追加し、カーボネートオリゴマーの濃度を調整する。この場合、重縮合反応によって得られる有機相中のポリカーボネート樹脂の濃度が5重量%〜30重量%となるように、不活性有機溶媒の量を調整する。次に、新たに水及びアルカリ金属化合物又はアルカリ土類金属化合物を含む水溶液を加え、さらに、重縮合条件を整えるために、好ましくは縮合触媒を添加し、界面重縮合法に従い重縮合反応を行う。重縮合反応における有機相と水相との割合は、容積比で有機相:水相=1:0.2〜1:1程度が好ましい。
アルカリ金属化合物又はアルカリ土類金属化合物の使用量が過度に多いと、副反応である加水分解反応が進む傾向がある。そのため、重縮合反応終了後における水相に含まれるアルカリ金属化合物又はアルカリ土類金属化合物の濃度が0.05N以上、好ましくは0.05N〜0.3N程度となるようにするのがよい。
重縮合工程における重縮合反応の温度は、通常、常温付近である。反応時間は0.5時間〜5時間、好ましくは1時間〜3時間程度である。
次に、重縮合反応槽における重縮合反応が完結した後、重縮合反応液は、公知の方法により、アルカリ洗浄液によるアルカリ洗浄、酸洗浄液による酸洗浄及び洗浄水による水洗浄が行われる。なお、洗浄工程の全滞留時間は、通常、12時間以下、好ましくは、0.5時間〜6時間である。
ポリカーボネート樹脂単離工程では、先ず、洗浄工程において洗浄されたポリカーボネート樹脂を含む重縮合反応液は、所定の固形分濃度に濃縮された濃縮液として調製される。濃縮液におけるポリカーボネート樹脂の固形分濃度は、通常、5重量%〜35重量%、好ましくは、10重量%〜30重量%である。
次に、濃縮液は、所定の造粒槽に連続的に供給され、所定の温度の脱塩水(DMW)と撹拌混合される。そして、水中で懸濁状態を保ちながら有機溶媒を蒸発させる造粒処理が行われ、ポリカーボネート樹脂粒状体を含む水スラリーが形成される。
ここで、脱塩水(DMW)の温度は、通常、37℃〜67℃、好ましくは、40℃〜50℃である。また、造粒槽内で行われる造粒処理によりポリカーボネート樹脂の固形化温度は、通常、37℃〜67℃、好ましくは、40℃〜50℃である。
造粒槽から連続的に排出されるポリカーボネート樹脂粉状体を含む水スラリーは、その後、所定の分離器に連続的に導入され、水スラリーから水が分離される。
乾燥工程では、分離器において、水スラリーから水が分離されたポリカーボネート樹脂粉状体が、所定の乾燥機に連続的に供給され、所定の滞留時間で滞留させた後、連続的に抜き出される。乾燥機としては、例えば流動床型乾燥機が挙げられる。なお、複数の流動床型乾燥機を直列につなぎ、連続的に乾燥処理を行ってもよい。
ここで、乾燥機は、通常、熱媒ジャケット等の加熱手段を有し、例えば、水蒸気にて、通常、0.1MPa−G〜1.0MPa−G、好ましくは、0.2MPa−G〜0.6MPa−Gに保持されている。これにより、乾燥機の中を流通する窒素(N2)の温度は、通常、100℃〜200℃、好ましくは、120℃〜180℃に保持されている。
次に、溶融法について説明する。
本発明に係るポリカーボネート樹脂の原料であるジヒドロキシ化合物としては、具体的には前記一般式(1)から(13)で表されるジヒドロキシ化合物等が挙げられる。
本発明に係るポリカーボネート樹脂の原料である炭酸ジエステルとしては、下記一般式(14)で示される化合物が挙げられる。
なお、A’上の置換基としては、ハロゲン原子、炭素数1〜炭素数10のアルキル基、炭素数1〜炭素数10のアルコキシ基、フェニル基、フェノキシ基、ビニル基、シアノ基、エステル基、アミド基、ニトロ基等が例示される。
これらの中でも、ジフェニルカーボネート(以下、「DPC」と称する場合がある。)、置換ジフェニルカーボネートが好ましい。これらの炭酸ジエステルは、単独又は2種以上を混合して用いることができる。
代表的なジカルボン酸又はジカルボン酸エステルとしては、例えば、テレフタル酸、イソフタル酸、テレフタル酸ジフェニル、イソフタル酸ジフェニル等が挙げられる。このようなジカルボン酸又はジカルボン酸エステルで置換した場合には、ポリエステルカーボネートが得られる。
ポリカーボネート樹脂の溶融法による製造方法において使用されるエステル交換触媒としては、通常、エステル交換法によりポリカーボネート樹脂を製造する際に用いられる触媒が挙げられ、特に限定されない。
一般的には、例えば、アルカリ金属化合物、アルカリ土類金属化合物、ベリリウム化合物、マグネシウム化合物、塩基性ホウ素化合物、塩基性リン化合物、塩基性アンモニウム化合物、アミン系化合物等の塩基性化合物が挙げられる。これらの中でも、実用的にはアルカリ金属化合物、アルカリ土類金属化合物が好ましい。これらのエステル交換触媒は、単独で使用してもよく、2種類以上を組み合わせて使用してもよい。
これらのアルカリ金属化合物の中でも、セシウム化合物が好ましく、特に、炭酸セシウム、炭酸水素セシウム、水酸化セシウムが好ましい。
本発明に於いては、エステル交換反応終了後に、エステル交換触媒を中和失活させるための触媒失活剤を添加しても良い。このような処理により得られたポリカーボネート樹脂の耐熱性、耐加水分解性が向上する。
このような触媒失活剤としては、スルホン酸やスルホン酸エステルのようなpKaが3以下の酸性化合物が好ましく、具体的にはベンゼンスルホン酸、p−トルエンスルホン酸、ベンゼンスルホン酸メチル、ベンゼンスルホン酸エチル、ベンゼンスルホン酸プロピル、ベンゼンスルホン酸ブチル、p−トルエンスルホン酸メチル、p−トルエンスルホン酸エチル、p−トルエンスルホン酸プロピル、並びにp−トルエンスルホン酸ブチルなどが挙げられる。
これらの中でも、p−トルエンスルホン酸並びにp−トルエンスルホン酸ブチルが好適に用いられる。
重縮合工程後、反応を停止させ、重縮合反応液中の未反応原料や反応副生物を脱揮除去する工程や、熱安定剤、離型剤、色剤等を添加する工程、ポリカーボネート樹脂を所定の粒径に形成する工程等を適宜追加してもよい。
次に、製造方法の各工程について説明する。
ポリカーボネート樹脂の原料として使用するジヒドロキシ化合物と炭酸ジエステル化合物とは、通常、窒素、アルゴン等の不活性ガスの雰囲気下、バッチ式、半回分式又は連続式の撹拌槽型の装置を用いて、原料混合溶融液として調製される。溶融混合の温度は、例えば、ジヒドロキシ化合物としてビスフェノールAを用い、炭酸ジエステル化合物としてジフェニルカーボネートを用いる場合は、通常120℃〜180℃、好ましくは125℃〜160℃の範囲から選択される。
以下、ジヒドロキシ化合物としてビスフェノールA、炭酸ジエステル化合物としてジフェニルカーボネートを原料として用いる場合を例として説明する。
この際、ジヒドロキシ化合物と炭酸ジエステル化合物との割合は、炭酸ジエステル化合物が過剰になるように調整され、ジヒドロキシ化合物1モルに対して、炭酸ジエステル化合物は、通常1.01モル〜1.30モル、好ましくは1.02モル〜1.20モルの割合になるように調整される。
ジヒドロキシ化合物と炭酸ジエステル化合物とのエステル交換反応による重縮合は、通常、2段階以上、好ましくは3段階〜7段階の多段方式で連続的に行われる。各段階の具体的な反応条件としては、温度:150℃〜320℃、圧力:常圧〜0.01Torr(1.3Pa)、平均滞留時間:5分〜150分の範囲である。
多段方式の各反応槽においては、エステル交換反応の進行とともに副生するフェノール等のモノヒドロキシ化合物をより効果的に系外に除去するために、上記の反応条件内で、段階的により高温、より高真空に設定する。
ここで、反応槽としては、例えば、撹拌槽型反応槽、薄膜反応槽、遠心式薄膜蒸発反応槽、表面更新型二軸混練反応槽、二軸横型撹拌反応槽、濡れ壁式反応槽、自由落下させながら重縮合する多孔板型反応槽、ワイヤーに沿わせて落下させながら重縮合するワイヤー付き多孔板型反応槽等が用いられる。
次にポリカーボネート樹脂組成物の製造方法について説明する。
本発明のポリカーボネート樹脂成形品の原料である、ポリカーボネート樹脂組成物を製造する方法については特に制限がないが、
1)ポリカーボネート樹脂(a)とポリカーボネート樹脂(b)とを溶融混練する方法;2)溶融状態のポリカーボネート樹脂(a)と溶融状態のポリカーボネート樹脂(b)とを溶融混練する方法;
3)ポリカーボネート樹脂(a)とポリカーボネート樹脂(b)とを溶液状態で混合する方法;
4)ポリカーボネート樹脂(a)とポリカーボネート樹脂(b)とをドライブレンドする方法;
等が挙げられる。
以下、各方法について説明する。
ポリカーボネート樹脂(a)のペレットもしくは粉粒体とポリカーボネート樹脂(b)のペレットもしくは粉粒体とを、例えばニーダーや二軸押出機、単軸押出機等の混合装置を用いて溶融混練する。ポリカーボネート樹脂(a)のペレットもしくは粉粒体とポリカーボネート樹脂(b)のペレットもしくは粉粒体は予め固体状態で混合し、その後混練されても良いし、またはどちらか一方を先に前記混合装置で溶融させ、そこへもう一方のポリカーボネート樹脂を添加し、混練しても良い。混練させる温度に特に規定はないが、240℃以上が好ましく、260℃以上より好ましく、280℃以上がさらに好ましい。また、350℃以下が好ましく、320℃以下が特に好ましい。混練させる温度が低いとポリカーボネート樹脂(a)とポリカーボネート樹脂(b)の混合が完全ではなく、ポリカーボネート樹脂成形品を製造した際に、鉛筆硬度等にばらつきが出る虞があり、好ましくない。また、混練する温度が高すぎると、ポリカーボネート樹脂成形品の色調が悪化する可能性があり、好ましくない。
溶融状態のポリカーボネート樹脂(a)と溶融状態のポリカーボネート樹脂(b)とを、例えば攪拌槽やスタティックミキサー、ニーダー、二軸押出機、単軸押出機等の混合装置を用いて混合する。このとき、例えば溶融重合法で得られたポリカーボネート樹脂であれば、冷却・固化することなく溶融状態で上記混合装置に導入しても良い。混合する温度としては特に規定はないが、150℃以上が好ましく、180℃以上がより好ましく、200℃以上がさらに好ましい。また、300℃以下が好ましく、250℃以下が特に好ましい。混合させる温度が低いとポリカーボネート樹脂(a)とポリカーボネート樹脂(b)の混合が完全ではなく、ポリカーボネート樹脂成形品を製造した際に、鉛筆硬度等にばらつきが出る虞があり、好ましくない。また、混合する温度が高すぎると、ポリカーボネート樹脂成形品の色調が悪化する可能性があり、好ましくない。
ポリカーボネート樹脂(a)とポリカーボネート樹脂(b)とを適当な溶媒に溶解して溶液とし、溶液状態で混合し、その後、ポリカーボネート樹脂組成物として単離する方法である。適当な溶媒としては、例えば、ヘキサン、n−ヘプタン等の脂肪族炭化水素;ジクロロメタン、クロロホルム、四塩化炭素、ジクロロエタン、トリクロロエタン、テトラクロロエタン、ジクロロプロパン及び1,2−ジクロロエチレン等の塩素化脂肪族炭化水素;ベンゼン、トルエン及びキシレン等の芳香族炭化水素;その他、ニトロベンゼン及びアセトフェノン等の置換芳香族炭化水素が挙げられる。これらの中でも、例えば、ジクロロメタン又はクロロベンゼン等の塩素化された炭化水素が好適に使用される。これらの溶媒は、単独であるいは他の溶媒との混合物として使用することができる。
混合装置としては、攪拌槽やスタティックミキサー等が挙げられる。また、混合温度としてはポリカーボネート樹脂(a)とポリカーボネート樹脂(b)とが溶解する条件であれば特に規定はなく、通常、使用する溶媒の沸点以下で実施される。
ポリカーボネート樹脂(a)のペレットもしくは粉粒体とポリカーボネート樹脂(b)のペレットもしくは粉粒体とをタンブラー、スーパーミキサー、ヘンシェルミキサーやナウターミキサー等を用いてドライブレンドする方法である。
なお、組成物を製造するにあたり、上記いずれの方法においても、顔料、染料、離型剤、熱安定剤等を本発明の目的を損なわない範囲において適宜添加することができる。
上述のポリカーボネート樹脂組成物から本発明のポリカーボネート樹脂成形品を製造するには、上述のように射出成形機が使用される。
そのため、前記射出成形時における射出成形機のシリンダー温度は、好ましくは270℃以上340℃以下であり、より好ましくは、280℃以上320℃以下である。このような温度範囲にシリンダー温度を設定することにより、十分な表面硬度を有し、色調も良好な樹脂成形品を得ることができる。
このような範囲に射出速度を設定することにより、表面外観に優れた樹脂成形品を得ることができる。
厚み3mm、縦60mm、横60mmのポリカーボネート樹脂又はポリカーボネート樹脂組成物のプレート(成形品)について、ISO 15184に準拠し、鉛筆硬度試験機(東洋精機株式会社製)を用いて、750g荷重にて測定した鉛筆硬度を求めた。
前記(1)で成形した成形品を用いて分光測色計(ミノルタ株式会社製CM−3700d)にてイエローインデックス(YI)を測定した。数値が小さいほど色調が良好であることを示す。
ポリカーボネート樹脂を塩化メチレンに溶解し(濃度6.0g/L)、ウベローデ粘度管を用いて20℃における比粘度(ηsp)を測定し、下記の式により粘度平均分子量(Mv)を算出した。
ηsp/C=[η](1+0.28ηsp)
[η]=1.23×10-4Mv0.83
(参考例1)PC(a1)の合成:BPCホモポリマーの合成(溶融法)
2,2−ビス(4−ヒドロキシ-3−メチルフェニル)プロパン(以下、「BPC」と略記する場合がある。)(本州化学社製)37.60kg(約147mol)とジフェニルカーボネート(DPC)32.20kg(約150mol)に、炭酸セシウムの水溶液を、炭酸セシウムがBPC1mol当たり2μmolとなるように添加して混合物を調整した。次に該混合物を、攪拌機、熱媒ジャケット、真空ポンプ、還流冷却器を具備した内容量200Lの第1反応器に投入した。
次に、第1反応器内を1.33kPa(10Torr)に減圧し、続いて、窒素で大気圧に復圧する操作を5回繰り返し、第1反応器の内部を窒素置換した。窒素置換後、熱媒ジャケットに温度230℃の熱媒を通じて第1反応器の内温を徐々に昇温させ、混合物を溶解させた。その後、55rpmで撹拌機を回転させ、熱媒ジャケット内の温度をコントロールして、第1反応器の内温を220℃に保った。そして、第1反応器の内部で行われるBPCとDPCのオリゴマー化反応により副生するフェノールを留去しながら、40分間かけて第1反応器内の圧力を絶対圧で101.3kPa(760Torr)から13.3kPa(100Torr)まで減圧した。
続いて、第1反応器内の圧力を13.3kPaに保持し、フェノールをさらに留去させながら、80分間、エステル交換反応を行った。
その後、系内を窒素で絶対圧で101.3kPaに復圧の上、ゲージ圧で0.2MPaまで昇圧し、予め200℃以上に加熱した移送配管を経由して、第1反応器内のオリゴマーを、第2反応器に圧送した。尚、第2反応器は内容量200Lであり、攪拌機、熱媒ジャケット、真空ポンプ並びに還流冷却管を具備しており、内圧は大気圧、内温は240℃に制御していた。
次に、第2反応器内に圧送したオリゴマーを16rpmで攪拌し、熱媒ジャケットにて内温を昇温し、第2反応器内を40分かけて絶対圧で101.3kPaから13.3kPaまで減圧した。その後、昇温を継続し、さらに40分かけて、内圧を絶対圧で13.3kPaから399Pa(3Torr)まで減圧し、留出するフェノールを系外に除去した。さらに、昇温を続け、第2反応器内の絶対圧が70Pa(約0.5Torr)に到達後、70Paを保持し、重縮合反応を行った。第2反応器内の最終的な内部温度は285℃であった。第2反応器の攪拌機が予め定めた所定の攪拌動力となったときに、重縮合反応を終了した。
次いで、第2反応器内を、窒素により絶対圧で101.3kPaに復圧の上、ゲージ圧で0.2MPaまで昇圧し、第2反応器の槽底からポリカーボネート樹脂をストランド状で抜き出し、水槽で冷却しながら、回転式カッターを使用してペレット化した。得られたポリカーボネート樹脂を用いて前記評価項目に記載の方法に準じて評価した鉛筆硬度は2H、粘度平均分子量は18,500であった。結果を表1にまとめて示す。
BPC(本州化学製)を360重量部、25重量%水酸化ナトリウム(NaOH)水溶液585.1重量部および水1721.5重量部をハイドロサルファイト0.41重量部の存在下に、40℃で溶解したのち20℃に冷却し、BPC水溶液を得た。このBPC水溶液8.87kg/時間と塩化メチレン4.50kg/時間とを、還流冷却器、攪拌機、冷媒ジャケットを有する1.8Lのガラス製第一反応器に導入し、ここに別途供給される常温のホスゲン0.672kg/時間とを接触させた。このときの反応温度は35℃に達した。次にこの反応液・反応ガスを反応器に取り付けてあるオーバーフロー管にて次の第一反応器と同じ形状を有する第二反応器(1.8L)に導入し、反応させた。第二反応器には、別途、分子量調整剤としてp−t−ブチルフェノール(8重量%塩化メチレン溶液)0.097kg/時間を導入した。次いで、第二反応器に取り付けてあるオーバーフロー管より反応液・反応ガスを第一反応器と同じ形状を有するオリゴマー化槽(4.5L)に導入した。オリゴマー化槽には別途触媒として2重量%トリメチルアミン水溶液0.020kg/時間を導入した。次いで、このようにして得られたオリゴマー化された乳濁液をさらに内容積5.4Lの分液槽(セトラー)に導き、水相と油槽を分離し、オリゴマーの塩化メチレン溶液を得た。
上記オリゴマーの塩化メチレン溶液のうち、2.60kgを内容積6.8Lのパドル翼付き反応槽に仕込み、これに希釈用塩化メチレン2.44kgを追加し、さらに25重量%水酸化ナトリウム水溶液0.278kg、水0.927kg、2重量%トリエチルアミン水溶液8.37g、p−t−ブチルフェノール25.8gを加え、10℃で攪拌し、180分間重縮合反応を行った。
上記重縮合反応液のうち、3.12kgを内容積5.4Lのパドル翼付き反応槽に仕込み、これに塩化メチレン2.54kg及び水0.575kgを加え、15分間攪拌した後、攪拌を停止し、水相と有機相を分離した。分離した有機相に、0.1N塩酸1.16kgを加え15分間攪拌し、トリエチルアミン及び少量残存するアルカリ成分を抽出した後、攪拌を停止し、水相と有機相を分離した。更に、分離した有機相に、純水1.16kgを加え、15分間攪拌した後、攪拌を停止し、水相と有機相を分離した。この操作を3回繰り返した。得られたポリカーボネート溶液を60〜75℃温水中にフィードすることで粉化し、乾燥し、粉末状ポリカーボネート樹脂を得た。得られたポリカーボネート樹脂を用いて前記評価項目に記載の方法に準じて評価した鉛筆硬度は2H、粘度平均分子量は9,500であった。結果を表1にまとめて示す。
原料としてBPCの代わりに1,1−ビス(4−ヒドロキシ−3−メチルフェニル)シクロヘキサン以下、「Bis−OCZ」と略記する場合がある。)(本州化学製)43.48kgを使用し、炭酸セシウムの水溶液を、炭酸セシウムがBis−OCZ1mol当たり5μmolとなるように添加した以外は参考例1と同様にして実施した。得られたポリカーボネート樹脂を用いて前記評価項目に記載の方法に準じて評価した鉛筆硬度は3H、粘度平均分子量は10,200であった。結果を表1にまとめて示す。
原料のジヒドロキシ化合物としてBPA20.62kg(約90mol)、1,1ビス(4−ヒドロキシ−3−メチルフェニル)シクロドデカン(以下、「CDOBC」と略記する場合がある。)(田岡化学社製)20.62kg(約54mol)を使用し、炭酸セシウムの水溶液を、炭酸セシウムがジヒドロキシ化合物1mol当たり1μmolとなるように添加して混合物を調整した以外は参考例1と同様にして実施した。得られたポリカーボネート樹脂を用いて前記評価項目に記載の方法に準じて評価した鉛筆硬度は2H、粘度平均分子量は11,700であった。結果を表1にまとめて示す。
原料としてBPC(本州化学製)3.39kg、BPA(三菱化学(株)製)30.47kgを使用した以外は参考例1と同様にして実施し、ポリカーボネート樹脂を得た。得られたポリカーボネート樹脂を用いて前記評価項目に記載の方法に準じて評価した鉛筆硬度はB、粘度平均分子量は21,000であった。結果を表1にまとめて示す。
(参考例6)PC(b1):BPAホモポリマー(溶融法)
PC(b1)として、BPAに由来する構造単位のみで構成された溶融法による市販のポリカーボネート樹脂(三菱エンジニアリングプラスチックス社製 M7022J)を用いた。該PC(b1)を用いて前記評価項目に記載の方法に準じて評価した鉛筆硬度は2B、の粘度平均分子量は20,600であった。結果を表1にまとめて示す。
ポリカーボネート樹脂(a),ポリカーボネート樹脂(b)として、それぞれPC(a1)とPC(b1)を表2に示す割合で1つのベント口を有する日本製鋼所製2軸押出機(LABOTEX30HSS−32)にて溶融混練し、該2軸押出機の出口からストランド状に押し出し、水で冷却固化させた後、回転式カッターでペレット化し、ポリカーボネート樹脂組成物を得た。このとき、該2軸押出機のバレル温度は280℃、該2軸押出機の出口におけるポリカーボネート樹脂温度は300℃であった。尚、溶融混練時は、該2軸押出機のベント口は真空ポンプに連結し、該ベント口での圧力が500Paになるように制御した。
得られたポリカーボネート樹脂組成物を、射出成形機(株式会社日本製鋼所製J50E2)を用い、表2に示す射出成形条件(シリンダー温度、金型温度、射出速度)下にて、厚み3mm、縦60mm、横60mmのポリカーボネート樹脂組成物のプレート(成形品)を射出成形した。
得られたポリカーボネート樹脂成形品を用いて、前記評価項目に記載の方法に準じて鉛筆硬度、イエローインデックス(YI)を評価した。その結果を表2に示した。
表2に示す2種類のポリカーボネート樹脂とした以外は実施例1と同様に実施し、実施例2〜7、比較例3、4のポリカーボネート樹脂組成物を得た。
得られたポリカーボネート樹脂組成物を、射出成形機(株式会社日本製鋼所製J50E2)を用い、表2に示す射出成形条件(シリンダー温度、金型温度、射出速度)下にて、厚み3mm、縦60mm、横60mmのポリカーボネート樹脂組成物のプレート(成形品)を射出成形した。
得られたポリカーボネート樹脂成形品を用いて、前記評価項目に記載の方法に準じて鉛筆硬度、イエローインデックス(YI)を評価した。その結果を表2に示した。
表2に示すポリカーボネート樹脂(a)、あるいはポリカーボネート樹脂(b)を、射出成形機(株式会社日本製鋼所製J50E2)を用い、表2に示す射出成形条件(シリンダー温度、金型温度、射出速度)下にて、厚み3mm、縦60mm、横60mmのポリカーボネート樹脂のプレート(成形品)を射出成形した。
得られたポリカーボネート樹脂成形品を用いて、前記評価項目に記載の方法に準じて鉛筆硬度、イエローインデックス(YI)を評価した。その結果を表2に示した。
<1> ポリカーボネート樹脂(a)と、該ポリカーボネート樹脂(a)とは異なる構造単位を有し、ISO 15184で規定される鉛筆硬度が該ポリカーボネート樹脂(a)の鉛筆硬度より低いポリカーボネート樹脂(b)とを、重量比で1:99〜45:55の範囲で含有するポリカーボネート樹脂組成物を、成形時における射出成形機の金型温度70℃以上120℃以下で射出成形するポリカーボネート樹脂成形品の製造方法。
<2> 前記射出成形時における射出成形機のシリンダー温度が270℃以上340℃以下である前記<1>に記載のポリカーボネート樹脂成形品の製造方法。
<3> 前記射出成形時の射出速度が1mm/秒以上60mm/秒以下である前記<1>または<2>に記載のポリカーボネート樹脂成形品の製造方法。
<4> 前記ポリカーボネート樹脂(a)の鉛筆硬度がF以上である前記<1>乃至<3>のいずれかに記載のポリカーボネート樹脂成形品の製造方法。
<5> 前記ポリカーボネート樹脂(a)が、下記式(1)で表される化合物に由来する構造単位を含むポリカーボネート樹脂である前記<1>乃至<4>のいずれかに記載のポリカーボネート樹脂成形品の製造方法。
(一般式(1)中、R1及びR2は、それぞれ独立に、置換若しくは無置換の炭素数1〜炭素数20のアルキル基、又は置換若しくは無置換のアリール基を示し、R3及びR4は、それぞれ独立に、水素原子、置換若しくは無置換の炭素数1〜炭素数20のアルキル基、又は置換若しくは無置換のアリール基を示し、Xは、単結合、カルボニル基、置換若しくは無置換のアルキリデン基、置換若しくは無置換の硫黄原子、又は酸素原子を示す。)
<6> 前記ポリカーボネート樹脂(a)が、下記一般式(1a)〜(1c)からなる群より選ばれた少なくとも1種の化合物に由来する構造単位を少なくとも含むポリカーボネート樹脂である前記<5>に記載のポリカーボネート樹脂成形品の製造方法。
<7> 前記ポリカーボネート樹脂(b)が、下記一般式(2)で表される化合物に由来する構造単位を主として含むポリカーボネート樹脂である前記<1>乃至<6>のいずれかに記載のポリカーボネート樹脂成形品の製造方法。
<9> 前記<1>乃至<8>のいずれかに記載のポリカーボネート樹脂成形品の製造方法により得られてなるポリカーボネート樹脂成形品。
Claims (9)
- ポリカーボネート樹脂(a)と、該ポリカーボネート樹脂(a)とは異なる構造単位を有し、ISO 15184で規定される鉛筆硬度が該ポリカーボネート樹脂(a)の鉛筆硬度より低いポリカーボネート樹脂(b)とを、重量比で1:99〜45:55の範囲で含有するポリカーボネート樹脂組成物を、成形時における射出成形機の金型温度70℃以上120℃以下で射出成形することを特徴とするポリカーボネート樹脂成形品の製造方法。
- 前記射出成形時における射出成形機のシリンダー温度が270℃以上340℃以下であることを特徴とする請求項1に記載のポリカーボネート樹脂成形品の製造方法。
- 前記射出成形時の射出速度が1mm/秒以上60mm/秒以下であることを特徴とする請求項1または2に記載のポリカーボネート樹脂成形品の製造方法。
- 前記ポリカーボネート樹脂(a)の鉛筆硬度がF以上であることを特徴とする請求項1乃至3のいずれか1項に記載のポリカーボネート樹脂成形品の製造方法。
- 前記ポリカーボネート樹脂(a)が、下記式(1)で表される化合物に由来する構造単位を含むポリカーボネート樹脂であることを特徴とする請求項1乃至4のいずれか1項に記載のポリカーボネート樹脂成形品の製造方法。
(一般式(1)中、R1及びR2は、それぞれ独立に、置換若しくは無置換の炭素数1〜炭素数20のアルキル基、又は置換若しくは無置換のアリール基を示し、R3及びR4は、それぞれ独立に、水素原子、置換若しくは無置換の炭素数1〜炭素数20のアルキル基、又は置換若しくは無置換のアリール基を示し、Xは、単結合、カルボニル基、置換若しくは無置換のアルキリデン基、置換若しくは無置換の硫黄原子、又は酸素原子を示す。) - 前記ポリカーボネート樹脂(a)が、下記一般式(1a)〜(1c)からなる群より選ばれた少なくとも1種の化合物に由来する構造単位を少なくとも含むポリカーボネート樹脂であることを特徴とする請求項5に記載のポリカーボネート樹脂成形品の製造方法。
- 前記ポリカーボネート樹脂(b)が、下記一般式(2)で表される化合物に由来する構造単位を主として含むポリカーボネート樹脂であることを特徴とする請求項1乃至6のいずれか1項に記載のポリカーボネート樹脂成形品の製造方法。
- 前記ポリカーボネート樹脂組成物のYIが、4.0以下であることを特徴とする請求項1乃至7のいずれか1項に記載のポリカーボネート樹脂成形品の製造方法。
- 請求項1乃至8のいずれか1項に記載のポリカーボネート樹脂成形品の製造方法により得られてなるポリカーボネート樹脂成形品。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2011155044A JP2013018240A (ja) | 2011-07-13 | 2011-07-13 | ポリカーボネート樹脂成形品の製造方法 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2011155044A JP2013018240A (ja) | 2011-07-13 | 2011-07-13 | ポリカーボネート樹脂成形品の製造方法 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| JP2013018240A true JP2013018240A (ja) | 2013-01-31 |
Family
ID=47690159
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2011155044A Pending JP2013018240A (ja) | 2011-07-13 | 2011-07-13 | ポリカーボネート樹脂成形品の製造方法 |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP2013018240A (ja) |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2020075611A1 (ja) * | 2018-10-12 | 2020-04-16 | 本州化学工業株式会社 | エポキシ樹脂組成物 |
| JPWO2021090823A1 (ja) * | 2019-11-06 | 2021-05-14 | ||
| JPWO2021090824A1 (ja) * | 2019-11-06 | 2021-05-14 |
Citations (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS6210160A (ja) * | 1985-07-08 | 1987-01-19 | Idemitsu Kosan Co Ltd | 光学機器用素材 |
| JPH08183852A (ja) * | 1994-12-28 | 1996-07-16 | Nippon G Ii Plast Kk | コポリカーボネート、コポリカーボネート組成物およびこれらの製造方法 |
| JPH11309761A (ja) * | 1998-04-30 | 1999-11-09 | Nippon Zeon Co Ltd | 導光板の製造方法 |
| JP2003535948A (ja) * | 2000-06-05 | 2003-12-02 | ゼネラル・エレクトリック・カンパニイ | 透明ポリカーボネートブレンドを含む情報記録媒体 |
| JP2009062501A (ja) * | 2007-09-10 | 2009-03-26 | Teijin Ltd | 光学素子成形品およびそのための成形材料 |
| JP2009063976A (ja) * | 2007-09-10 | 2009-03-26 | Teijin Ltd | 光学素子成形品およびそのための成形材料 |
| JP2010126605A (ja) * | 2008-11-26 | 2010-06-10 | Mitsubishi Gas Chemical Co Inc | 芳香族ポリカーボネート樹脂組成物、及び成形品 |
| JP2010168420A (ja) * | 2009-01-20 | 2010-08-05 | Teijin Chem Ltd | 芳香族ポリカーボネート樹脂組成物およびそれから形成された光学用成形品 |
| JP2010243659A (ja) * | 2009-04-02 | 2010-10-28 | Sumitomo Electric Fine Polymer Inc | 光学レンズ−ホルダー複合体の製造方法、及び光学レンズ−ホルダー複合体 |
| JP2011105931A (ja) * | 2009-10-22 | 2011-06-02 | Mitsubishi Chemicals Corp | ポリカーボネート樹脂 |
-
2011
- 2011-07-13 JP JP2011155044A patent/JP2013018240A/ja active Pending
Patent Citations (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS6210160A (ja) * | 1985-07-08 | 1987-01-19 | Idemitsu Kosan Co Ltd | 光学機器用素材 |
| JPH08183852A (ja) * | 1994-12-28 | 1996-07-16 | Nippon G Ii Plast Kk | コポリカーボネート、コポリカーボネート組成物およびこれらの製造方法 |
| JPH11309761A (ja) * | 1998-04-30 | 1999-11-09 | Nippon Zeon Co Ltd | 導光板の製造方法 |
| JP2003535948A (ja) * | 2000-06-05 | 2003-12-02 | ゼネラル・エレクトリック・カンパニイ | 透明ポリカーボネートブレンドを含む情報記録媒体 |
| JP2009062501A (ja) * | 2007-09-10 | 2009-03-26 | Teijin Ltd | 光学素子成形品およびそのための成形材料 |
| JP2009063976A (ja) * | 2007-09-10 | 2009-03-26 | Teijin Ltd | 光学素子成形品およびそのための成形材料 |
| JP2010126605A (ja) * | 2008-11-26 | 2010-06-10 | Mitsubishi Gas Chemical Co Inc | 芳香族ポリカーボネート樹脂組成物、及び成形品 |
| JP2010168420A (ja) * | 2009-01-20 | 2010-08-05 | Teijin Chem Ltd | 芳香族ポリカーボネート樹脂組成物およびそれから形成された光学用成形品 |
| JP2010243659A (ja) * | 2009-04-02 | 2010-10-28 | Sumitomo Electric Fine Polymer Inc | 光学レンズ−ホルダー複合体の製造方法、及び光学レンズ−ホルダー複合体 |
| JP2011105931A (ja) * | 2009-10-22 | 2011-06-02 | Mitsubishi Chemicals Corp | ポリカーボネート樹脂 |
Cited By (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2020075611A1 (ja) * | 2018-10-12 | 2020-04-16 | 本州化学工業株式会社 | エポキシ樹脂組成物 |
| CN112752781A (zh) * | 2018-10-12 | 2021-05-04 | 本州化学工业株式会社 | 环氧树脂组合物 |
| JPWO2020075611A1 (ja) * | 2018-10-12 | 2021-09-16 | 本州化学工業株式会社 | エポキシ樹脂組成物 |
| CN112752781B (zh) * | 2018-10-12 | 2023-08-15 | 本州化学工业株式会社 | 环氧树脂组合物 |
| JP7355304B2 (ja) | 2018-10-12 | 2023-10-03 | 本州化学工業株式会社 | エポキシ樹脂組成物 |
| JPWO2021090823A1 (ja) * | 2019-11-06 | 2021-05-14 | ||
| JPWO2021090824A1 (ja) * | 2019-11-06 | 2021-05-14 | ||
| WO2021090824A1 (ja) * | 2019-11-06 | 2021-05-14 | 三菱エンジニアリングプラスチックス株式会社 | レーザーダイレクトストラクチャリング用樹脂組成物、成形品、および、メッキ付き成形品の製造方法 |
| JP7632297B2 (ja) | 2019-11-06 | 2025-02-19 | 三菱ケミカル株式会社 | レーザーダイレクトストラクチャリング用樹脂組成物、成形品、および、メッキ付き成形品の製造方法 |
| US12509549B2 (en) | 2019-11-06 | 2025-12-30 | Mitsubishi Chemical Corporation | Resin composition for laser direct structuring, molded article, and, method for manufacturing plated molded article |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5668408B2 (ja) | ポリカーボネート樹脂 | |
| TWI494368B (zh) | A polycarbonate resin composition, a method for producing the same, and a molded product of the resin composition | |
| US9771477B2 (en) | Polycarbonate resin composition, method for producing same and molded article of this resin composition | |
| JP6252650B2 (ja) | ポリカーボネート樹脂組成物及びその製造方法 | |
| JP2011219636A (ja) | ポリカーボネート樹脂の製造方法およびポリカーボネート樹脂 | |
| JP6100416B2 (ja) | ポリカーボネート樹脂及びポリカーボネート樹脂組成物の製造方法 | |
| JP6252649B2 (ja) | ポリカーボネート樹脂組成物及びその製造方法 | |
| JP5989358B2 (ja) | ポリカーボネート樹脂成形体及びその製造方法 | |
| JP2013018240A (ja) | ポリカーボネート樹脂成形品の製造方法 | |
| JP5903904B2 (ja) | ポリカーボネート樹脂成形体及びその製造方法 | |
| JP2013072077A (ja) | ポリカーボネート樹脂の製造方法 | |
| JP6270304B2 (ja) | ポリカーボネート樹脂組成物及びその製造方法 | |
| JP2012177090A (ja) | ポリカーボネート樹脂組成物及びその製造方法 | |
| JP2012214620A (ja) | ポリカーボネート樹脂シート | |
| JP6248509B2 (ja) | ポリカーボネート樹脂組成物ならびに、ポリカーボネート樹脂およびポリカーボネート樹脂組成物の製造方法 | |
| JP6213025B2 (ja) | ポリカーボネート樹脂及びポリカーボネート樹脂組成物 | |
| JP2012177091A (ja) | ポリカーボネート樹脂組成物及びその製造方法 | |
| JP2013040309A (ja) | ポリカーボネート樹脂シート | |
| JP6402803B2 (ja) | ポリカーボネート樹脂 | |
| JP5796424B2 (ja) | ポリカーボネート樹脂積層体及びその製造方法 | |
| JP2011246628A (ja) | 芳香族ポリカーボネート樹脂の製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20140707 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20150306 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20150310 |
|
| A521 | Written amendment |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20150508 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20150728 |
|
| A02 | Decision of refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A02 Effective date: 20160119 |