JP2013104100A - 金属薄膜の成膜方法および金属薄膜成膜用原料 - Google Patents
金属薄膜の成膜方法および金属薄膜成膜用原料 Download PDFInfo
- Publication number
- JP2013104100A JP2013104100A JP2011248720A JP2011248720A JP2013104100A JP 2013104100 A JP2013104100 A JP 2013104100A JP 2011248720 A JP2011248720 A JP 2011248720A JP 2011248720 A JP2011248720 A JP 2011248720A JP 2013104100 A JP2013104100 A JP 2013104100A
- Authority
- JP
- Japan
- Prior art keywords
- thin film
- metal thin
- raw material
- forming
- chemical formula
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Images





Landscapes
- Chemical Vapour Deposition (AREA)
- Electrodes Of Semiconductors (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
【課題】基板温度の低温下を達成しつつ、成膜速度が向上された金属薄膜の成膜方法および金属薄膜成膜用原料を提供する。
【解決手段】アミジナート配位子を有する金属薄膜原料9と、窒素原子および水素原子を有する反応ガス23と、を用いて、前記金属薄膜原料と前記反応ガスを交互または同時に基板6表面に供給することにより、低不純物の金属薄膜を成膜する金属薄膜の成膜方法を提供する。また、窒素原子および水素原子を有する反応ガスとともに用いられることで金属薄膜を成膜する金属薄膜成膜用原料であって、アミジナート配位子を有する金属薄膜成膜用原料。
【選択図】図3
【解決手段】アミジナート配位子を有する金属薄膜原料9と、窒素原子および水素原子を有する反応ガス23と、を用いて、前記金属薄膜原料と前記反応ガスを交互または同時に基板6表面に供給することにより、低不純物の金属薄膜を成膜する金属薄膜の成膜方法を提供する。また、窒素原子および水素原子を有する反応ガスとともに用いられることで金属薄膜を成膜する金属薄膜成膜用原料であって、アミジナート配位子を有する金属薄膜成膜用原料。
【選択図】図3
Description
本発明は、金属薄膜の成膜方法および金属薄膜成膜用原料に関する。
近年、半導体素子やフラットパネルディスプレイ(FPD)、太陽電池といった技術分野での技術進歩は目覚しく、金属薄膜に関する研究開発も盛んに行われている。
特に、半導体デバイスの配線では、バリア層、ライナー層、シード層といった様々な金属薄膜の多層膜が導入されるようになり、これらの金属薄膜はスパッタリングを用いた物理的成膜手法によって形成されてきた。
特に、半導体デバイスの配線では、バリア層、ライナー層、シード層といった様々な金属薄膜の多層膜が導入されるようになり、これらの金属薄膜はスパッタリングを用いた物理的成膜手法によって形成されてきた。
しかしながら、半導体デバイスの配線スケールは年々縮小が進んでおり、トレンチ、ビア共にアスペクト比の高いものが設計されるようになった。
したがって、段差被覆性に劣るスパッタリングを用いて、高アスペクト比のトレンチやビア等に薄膜を形成すると、ボイド形成やバリア膜破れなどが生じ、信頼性劣化に繋がるという問題が生じていた。
したがって、段差被覆性に劣るスパッタリングを用いて、高アスペクト比のトレンチやビア等に薄膜を形成すると、ボイド形成やバリア膜破れなどが生じ、信頼性劣化に繋がるという問題が生じていた。
そこで、これらの問題を解決するため、例えばライナー膜などでは、スパッタリング等のPVD法(物理気相成長法)の代わりに、段差被覆性の優れたCVD法(化学気相成長法)によって金属薄膜を成膜することが検討されている(特許文献1)。
なお、図5に示すように、段差被覆性は、溝等の段差のある基板31に薄膜32を成膜した際に、溝等内での膜厚をdBとし、基板31の平坦部分の膜厚をdTとしたときの、dB/dTの値によって比較される。
例えば、図4(a)に示すように、CVD法によって成膜した場合は、dB/dTが1に近い値となり、基板31の平坦部分と同様に溝の奥まで均一に成膜されているといえ、段差被覆性は良好である。これに対し、図4(b)に示すように、PVD法によって成膜した場合は、dB/dTが小さい値となり、基板31の平坦部分よりも溝の奥の方が成膜度合いが小さく、溝の奥までは十分に成膜していないといえ、段差被覆性は悪い。
ところで、本願発明者らの研究によると、近年、半導体デバイスの配線形成工程で用いられるライナー層等および、その成膜工程については、(1)酸素を含有しないこと、(2)炭素、窒素等の含有量が少ないこと、(3)段差被覆性については、dB/dTが0.8以上であること、(4)成膜速度が0.2〜1nm/minであること、(5)成膜温度が400℃以下であること、の5つの要件を満たすことが求められてきている。
特に、酸素については、X線光電子分光法(XPS)によって測定した際に、検出下限以下であることが望まれており、また、炭素、窒素の含有量については、抵抗の上昇を抑えるためにも、原子数比が1%以下であることが望まれている。
しかしながら、従来のCVD法に用いられてきた薄膜成膜用原料は、基板表面での原料の自己分解反応や還元雰囲気として用いている水素の解離反応を利用しているため、400℃程度の高温が必要であり、成膜速度が遅く、孵化時間や原料と還元ガスの交互供給が必要となる、などの問題があった。
このような背景の下、基板温度の低温下を達成しつつ、成膜速度が向上された金属薄膜の成膜方法が要望されていたが、有効適切なものが提供されていないのが実情であった。
このような背景の下、基板温度の低温下を達成しつつ、成膜速度が向上された金属薄膜の成膜方法が要望されていたが、有効適切なものが提供されていないのが実情であった。
そこで、上記課題を解決するため、本発明は以下の構成を採用した。
すなわち、請求項1に係る発明は、下記化学式(1)で示されるアミジナート配位子を有する金属薄膜原料と、窒素原子および水素原子を有する反応ガスと、を用いて、前記金属薄膜原料と前記反応ガスを交互または同時に基板表面に供給することにより、低不純物の金属薄膜を成膜する金属薄膜の成膜方法である。
すなわち、請求項1に係る発明は、下記化学式(1)で示されるアミジナート配位子を有する金属薄膜原料と、窒素原子および水素原子を有する反応ガスと、を用いて、前記金属薄膜原料と前記反応ガスを交互または同時に基板表面に供給することにより、低不純物の金属薄膜を成膜する金属薄膜の成膜方法である。
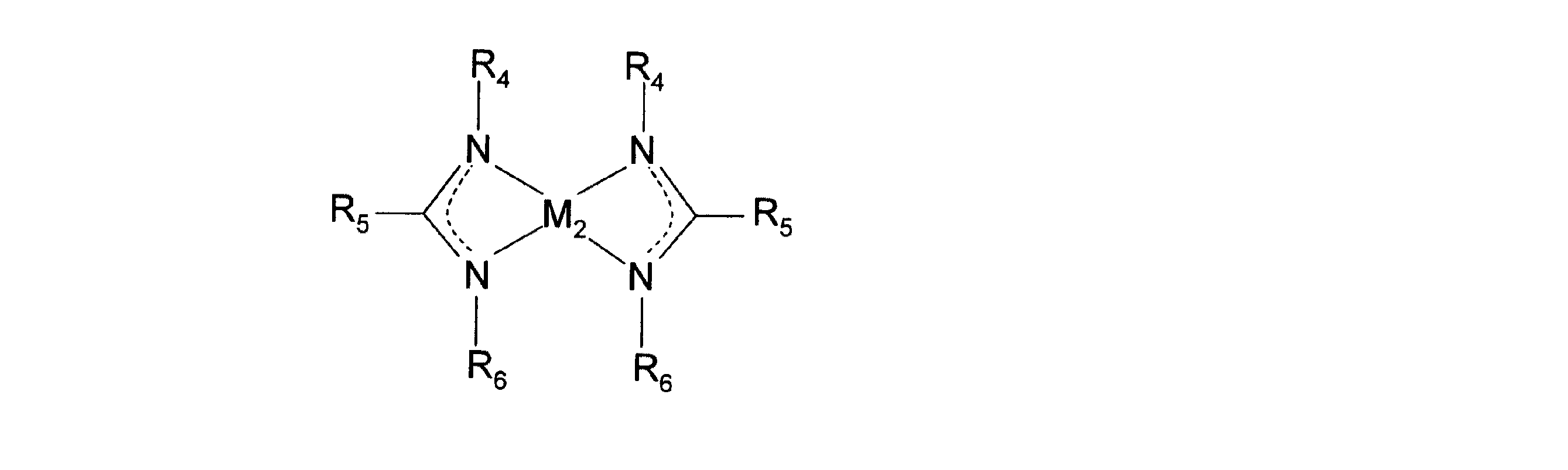
また、請求項2に係る発明は、前記化学式(1)において、R1〜R3が水素またはCnH2n+1(n=1〜4)の化学式で表される炭化水素であることを特徴とする請求項1に記載の金属薄膜の成膜方法である。
また、請求項3に係る発明は、前記化学式(1)において、M1がCo,Ni,Fe、またはMnであることを特徴とする請求項1または請求項2に記載の金属薄膜の成膜方法である。
また、請求項4に係る発明は、前記反応ガスが、アンモニア、メチルアミン、エチルアミン、プロピルアミン、イソプロピルアミン、ターシャリーブチルアミン、ノルマルブチルアミン、ジメチルアミン、ジエチルアミン、ヒドラジン、モノメチルヒドラジン、ジメチルヒドラジン、ジフェニルヒドラジン、およびエチレンイミンのうち、少なくとも1つ以上を含むことを特徴とする請求項1ないし請求項3のいずれか1項に記載の金属薄膜の成膜方法である。
また、請求項5に係る発明は、窒素原子および水素原子を有する反応ガスとともに用いられることで金属薄膜を成膜する金属薄膜成膜用原料であって、下記化学式(2)で示されるアミジナート配位子を有する金属薄膜成膜用原料である。
また、請求項6に係る発明は、前記化学式(2)において、R4〜R6が水素またはCnH2n+1(n=1〜4)の化学式で表される炭化水素であることを特徴とする請求項5に記載の金属薄膜成膜用原料である。
また、請求項7に係る発明は、前記化学式(2)において、M2がCo,Ni,Fe、またはMnであることを特徴とする請求項5または請求項6に記載の金属薄膜成膜用原料である。
また、請求項8に係る発明は、前記反応ガスが、アンモニア、メチルアミン、エチルアミン、プロピルアミン、イソプロピルアミン、ターシャリーブチルアミン、ノルマルブチルアミン、ジメチルアミン、ジエチルアミン、ヒドラジン、モノメチルヒドラジン、ジメチルヒドラジン、ジフェニルヒドラジン、およびエチレンイミンのうち、少なくとも1つ以上を含むことを特徴とする請求項5ないし請求項7のいずれか1項に記載の金属薄膜成膜用原料である。
本発明によれば、金属薄膜原料が、上記化学式(1)に示された構造を有しており、また、反応ガスに窒素原子が含まれている。この結果、金属薄膜の成膜時に、反応ガスに含まれる窒素原子が寄与することで、成膜反応が促進され、成膜時の基板温度を低温化することができるとともに、成膜速度を向上させることができる。また、この成膜反応の際、窒素原子は金属薄膜中に取り込まれることがないため、純金属に近い抵抗率をもった金属薄膜を得ることができる。
また、同様に、本発明によれば、金属薄膜成膜用原料は、上記化学式(2)で示された構造を有しているとともに、窒素原子が含まれている反応ガスとともに用いられる。この結果、本発明の金属薄膜成膜用原料を用いれば、金属薄膜の成膜時に、反応ガスに含まれる窒素原子が寄与するので、成膜反応が促進され、成膜時の基板温度を低温化することができるとともに、成膜速度を向上させることができる。また、この成膜反応の際、窒素原子は、金属薄膜中に取り込まれることがないため、純金属に近い抵抗率をもった金属薄膜を得ることができる。
以下、本発明を適用した一実施形態である金属薄膜成膜用原料および金属薄膜の成膜方法について、適宜図面を参照しつつ説明する。
<金属薄膜成膜用原料>
本実施形態の金属薄膜成膜用原料(以下では、金属薄膜原料ともいう。)は、有機金属原料であり、上記化学式(2)で示されるアミジナート配位子を有する構造となっている。
特に、上記化学式(2)においては、R4〜R6が水素またはCnH2n+1(n=1〜4)の化学式で表される炭化水素であることが好ましい。このような構造を採用することで、高蒸気圧が実現できることとなり、より好ましくなるからである。
また、上記化学式(2)においては、M2は、Co,Ni,Fe、またはMnであることが好ましい。
本実施形態の金属薄膜成膜用原料(以下では、金属薄膜原料ともいう。)は、有機金属原料であり、上記化学式(2)で示されるアミジナート配位子を有する構造となっている。
特に、上記化学式(2)においては、R4〜R6が水素またはCnH2n+1(n=1〜4)の化学式で表される炭化水素であることが好ましい。このような構造を採用することで、高蒸気圧が実現できることとなり、より好ましくなるからである。
また、上記化学式(2)においては、M2は、Co,Ni,Fe、またはMnであることが好ましい。
また、本実施形態の金属薄膜成膜用原料は、窒素原子および水素原子を有する反応ガスとともに用いられることで、金属薄膜を成膜する原料である。
反応ガスとしては、アンモニアーアミン系として、アンモニア、メチルアミン、エチルアミン、プロピルアミン、イソプロピルアミン、ターシャリーブチルアミン、ノルマルブチルアミン、ジメチルアミン、ジエチルアミンを用いることができ、ヒドラジン系として、ヒドラジン、モノメチルヒドラジン、ジメチルヒドラジン、ジフェニルヒドラジンを用いることができ、イミン系として、エチレンイミンを用いることができ、少なくともこれらを1つ以上含むものを用いることが好ましい。
反応ガスとしては、アンモニアーアミン系として、アンモニア、メチルアミン、エチルアミン、プロピルアミン、イソプロピルアミン、ターシャリーブチルアミン、ノルマルブチルアミン、ジメチルアミン、ジエチルアミンを用いることができ、ヒドラジン系として、ヒドラジン、モノメチルヒドラジン、ジメチルヒドラジン、ジフェニルヒドラジンを用いることができ、イミン系として、エチレンイミンを用いることができ、少なくともこれらを1つ以上含むものを用いることが好ましい。
特に、この中でもアンモニア、ヒドラジン、モノメチルヒドラジン、ジメチルヒドラジン、ジフェニルヒドラジンを用いることがより好ましい。このような反応ガスを用いることで、反応の活性化エネルギーの低減により、低温でも成膜が可能となり、より好ましくなるからである。
本実施形態の金属薄膜成膜用原料は、上記化学式(2)で示された構造を有しているとともに、窒素原子が含まれている反応ガスとともに用いられる。この結果、本実施形態の金属薄膜成膜用原料を用いれば、金属薄膜の成膜時に、反応ガスに含まれる窒素原子が寄与することで、成膜反応が促進され、成膜時の基板温度を低温化することができるとともに、成膜速度を向上させることができる。
より詳細に説明すると、一般に窒素原子を含む添加剤は、アミジナート配位子を有する原料と会合体を形成するという性質がある。
したがって、反応ガスに窒素原子が含有されており、かつ、金属薄膜成膜用原料が上記化学式(2)に示されるアミジナート配位子を有する場合、反応ガスとともに用いられた金属薄膜成膜用原料は、基板表面に吸着する前に、会合体を形成することになる。
そして、この会合体は、反応性の高い反応中間体であることから、成膜時の基板温度を低温化することができるとともに、成膜速度を向上させることができる。
したがって、反応ガスに窒素原子が含有されており、かつ、金属薄膜成膜用原料が上記化学式(2)に示されるアミジナート配位子を有する場合、反応ガスとともに用いられた金属薄膜成膜用原料は、基板表面に吸着する前に、会合体を形成することになる。
そして、この会合体は、反応性の高い反応中間体であることから、成膜時の基板温度を低温化することができるとともに、成膜速度を向上させることができる。
例えば、上記化学式(2)で示されるアミジナート配位子を有する金属薄膜成膜用原料と、水素(H2)を用いて金属薄膜を成膜する場合、図1に示すように、金属薄膜成膜用原料は、自己分解して基板に吸着することとなる。
したがって、この場合は、自己分解が反応の基点となるので、自己分解の反応熱に従った成膜速度の温度依存性を示すこととなる。なお、この自己分解の活性化エネルギーは30〜50kcal/molであり、反応熱は15〜30kcal/molである。
これに対し、上記化学式(2)に示されるアミジナート配位子を有する金属薄膜成膜用原料と、窒素原子を含有する反応ガスを用いて金属薄膜を成膜する場合は、図2に示すように、H2を用いた場合とは反応の機構が異なる。
すなわち、窒素原子を含有する反応ガスを用いた場合は、反応ガスと金属薄膜成膜用原料とが会合体を形成し、この会合体形成に引き続く水素転位が反応の基点となって、金属薄膜成膜用原料は基板に吸着することとなる。
したがって、この場合は、水素転位が反応の基点となるので、反応速度は転位反応の活性化エネルギーに従った成膜速度の温度依存性を示すこととなる。
なお、転位反応の活性化エネルギーは約3kcal/molであり、反応熱も約kcal/molである。
以上から、本実施形態の金属薄膜成膜用原料を用いれば、金属薄膜の成膜時に、基板温度を低温化することができるとともに、成膜速度を向上させることができる。
なお、転位反応の活性化エネルギーは約3kcal/molであり、反応熱も約kcal/molである。
以上から、本実施形態の金属薄膜成膜用原料を用いれば、金属薄膜の成膜時に、基板温度を低温化することができるとともに、成膜速度を向上させることができる。
また、この成膜反応の際、窒素原子は、金属薄膜中に取り込まれることがないため、低不純物で、純金属に近い抵抗率をもった金属薄膜を得ることができる。
なお、ここでいう低不純物とは、原子比で10%以下のことを指し、少なければ少ないほど好ましく、1%以下であることが好ましい。
なお、ここでいう低不純物とは、原子比で10%以下のことを指し、少なければ少ないほど好ましく、1%以下であることが好ましい。
<金属薄膜の成膜方法>
次に、本実施形態の金属薄膜の成膜方法について説明する。
本実施形態の金属薄膜の成膜方法は、上記化学式(1)で示されるアミジナート配位子を有する金属薄膜原料と、窒素原子および水素原子を有する反応ガスと、を用いて行う。
次に、本実施形態の金属薄膜の成膜方法について説明する。
本実施形態の金属薄膜の成膜方法は、上記化学式(1)で示されるアミジナート配位子を有する金属薄膜原料と、窒素原子および水素原子を有する反応ガスと、を用いて行う。
ここで、金属薄膜原料とは、上述した金属薄膜成膜用原料と同じものであり、上記化学式(1)においては、R1〜R3が水素またはCnH2n+1(n=1〜4)の化学式で表される炭化水素であることが好ましく、上記化学式(1)においては、M1は、Co,Ni,Fe、またはMnであることが好ましい。
次に、本実施形態の金属薄膜の成膜方法に用いる成膜装置について説明する。成膜装置1は、図3に示すように、減圧可能な真空チャンバー2と、有機金属化合物である金属薄膜原料を搬送可能な原料搬送手段3と、反応ガスを搬送可能な反応ガス搬送手段4と、を有している。
真空チャンバー2内には、載置台5が設けられており、この載置台5には基板6(成膜対象物)が自在に載置できるように構成されている。載置台5の内部にはヒーター(図示略)が設けられており、基板6を加熱できるように構成されている。
また、真空チャンバー2には、排気管(図示略)および開閉弁(図示略)を介して排気ポンプ7が接続されており、これにより排気可能なように構成されている。なお、真空チャンバー2内には、圧力計(図示略)が設けられており、これにより圧力が測定できるようになっている。
また、真空チャンバー2には、ガス噴出部8が設けられており、このガス噴出部8に、原料搬送手段3と反応ガス搬送手段4が接続されている。
また、真空チャンバー2には、ガス噴出部8が設けられており、このガス噴出部8に、原料搬送手段3と反応ガス搬送手段4が接続されている。
原料搬送手段3は、金属薄膜原料を供給可能であれば、どのような構成でもよく、本実施形態では金属薄膜原料が貯蔵されている原料貯液槽9と、原料貯液槽9の液相部分に接続された配管10と、原料貯液槽9の気相部分に接続された配管21と、配管21に設けられたバルブ22と、から概略構成されており、配管21はガス噴出部8に接続されている。
そして、配管10を介してヘリウムなどの不活性ガスや水素を原料貯液槽9に導入すると、金属薄膜原料が同伴されて配管21から導出される。
そして、配管10を介してヘリウムなどの不活性ガスや水素を原料貯液槽9に導入すると、金属薄膜原料が同伴されて配管21から導出される。
なお、本実施形態では、金属薄膜原料として常温で液体状のものを用いることを想定し、バブリングによる気化が可能な構成を採用したが、必ずしもこのような構成である必要はない。例えば、金属薄膜原料として常温で気体状のものを原料として用いてもよく、また液体状のものを用いた場合も気化器による気化や加熱による気化によってガス化してもよい。
以上のように構成された原料搬送手段3によって、原料貯液槽9の気相部分から取り出されたガス化した金属薄膜原料は、ガス噴出部8を介して真空チャンバー2内に導入され、基板6上に搬送される。
反応ガス搬送手段4は、反応ガス供給源23と、この反応ガス供給源23と接続された配管24と、配管24に取り付けられたバルブ25とから概略構成されており、配管24はガス噴出部8に接続されている。反応ガス供給源23は、反応ガスを供給可能であるのならば、どのような構成であっても構わない。
このように構成された反応ガス搬送手段4によって、反応ガス供給源23から導出した反応ガスは、ガス噴出部8を介して真空チャンバー2内に導入され、基板6上に搬送される。
次に、上記した成膜装置1を用いた金属薄膜の成膜方法について説明する。
本実施形態の金属薄膜の成膜方法は、原料搬送工程と、反応ガス搬送工程とを有しており、まずこれらについて説明する。
本実施形態の金属薄膜の成膜方法は、原料搬送工程と、反応ガス搬送工程とを有しており、まずこれらについて説明する。
原料搬送工程は、金属薄膜原料の蒸気を真空チャンバー2内に載置された基板6上に搬送する工程である。
金属薄膜原料は、常温で気体状であればそのまま用いることができ、液体状のものであれば、ヘリウムなどの不活性ガスを用いたバブリングによる気化、気化器による気化、または加熱による気化によってガス化して用いればよい。また、ヘリウムなどをキャリアガスとして、金属薄膜原料と併せて用いてもよい。
金属薄膜原料は、常温で気体状であればそのまま用いることができ、液体状のものであれば、ヘリウムなどの不活性ガスを用いたバブリングによる気化、気化器による気化、または加熱による気化によってガス化して用いればよい。また、ヘリウムなどをキャリアガスとして、金属薄膜原料と併せて用いてもよい。
例えば、常温で液体状の金属薄膜原料を、バブリングによって気化して用いる場合は、図3に示すように、配管10を介して金属薄膜原料が貯蔵されている原料貯液槽9の液相部分にヘリウムガスなどの不活性ガスや水素を導入する。その上で、原料貯液槽9の気相部分から蒸気化した金属薄膜原料を、配管21を介して抜き出せばよい。
気化した金属薄膜原料は、配管21を介してガス噴出部8に導入され、その後真空チャンバー2内に導入される。この際、バルブ22は開いた状態となっている。
そして、真空チャンバー2内に導入された金属薄膜原料は、図示略のヒーターによって加熱された基板6上に搬送される。
以上のようにして、金属薄膜原料は、真空チャンバー2内に載置された基板6上に搬送される。
そして、真空チャンバー2内に導入された金属薄膜原料は、図示略のヒーターによって加熱された基板6上に搬送される。
以上のようにして、金属薄膜原料は、真空チャンバー2内に載置された基板6上に搬送される。
なお、金属薄膜原料は、1種類である必要はなく、2種類以上であっても構わない。2種類以上用いる場合は、同時または順次に基板上に搬送すればよい。
反応ガス搬送工程は、反応ガスを、真空チャンバー2内に載置された基板6上に搬送する工程である。
反応ガスとしては、上述したように、アンモニアーアミン系として、アンモニア、メチルアミン、エチルアミン、プロピルアミン、イソプロピルアミン、ターシャリーブチルアミン、ノルマルブチルアミン、ジメチルアミン、ジエチルアミンを用いることができ、ヒドラジン系として、ヒドラジン、モノメチルヒドラジン、ジメチルヒドラジン、ジフェニルヒドラジンを用いることができ、イミン系として、エチレンイミンを用いることができ、少なくともこれらを1つ以上含むものを用いることが好ましく、アンモニア、ヒドラジン、モノメチルヒドラジン、ジメチルヒドラジン、ジフェニルヒドラジンを用いることがより好ましい。
反応ガスとしては、上述したように、アンモニアーアミン系として、アンモニア、メチルアミン、エチルアミン、プロピルアミン、イソプロピルアミン、ターシャリーブチルアミン、ノルマルブチルアミン、ジメチルアミン、ジエチルアミンを用いることができ、ヒドラジン系として、ヒドラジン、モノメチルヒドラジン、ジメチルヒドラジン、ジフェニルヒドラジンを用いることができ、イミン系として、エチレンイミンを用いることができ、少なくともこれらを1つ以上含むものを用いることが好ましく、アンモニア、ヒドラジン、モノメチルヒドラジン、ジメチルヒドラジン、ジフェニルヒドラジンを用いることがより好ましい。
反応ガス供給源23から供給される反応ガスは、配管24を介してガス噴出部8に導入され、その後真空チャンバー2内に導入される。この際、バルブ25は開いた状態となっている。
また、反応ガスを真空チャンバー2内に導入する際は、ガス噴出部8に設けられたシャワーヘッド26を介して導入してもよい。
そして、真空チャンバー2内に導入された反応ガスは、図示略のヒーターによって加熱された基板6上に搬送される。
そして、真空チャンバー2内に導入された反応ガスは、図示略のヒーターによって加熱された基板6上に搬送される。
次に、原料搬送工程と、反応性ガス搬送工程の関係について説明する。
本実施形態の金属薄膜の成膜方法は、CVD法による成膜方法でもALD法による成膜方法でも構わないが、成膜した金属薄膜の段差被覆性を考慮するとALD法による成膜方法の方が望ましい。
本実施形態の金属薄膜の成膜方法は、CVD法による成膜方法でもALD法による成膜方法でも構わないが、成膜した金属薄膜の段差被覆性を考慮するとALD法による成膜方法の方が望ましい。
CVD法による成膜方法の場合は、原料搬送工程と反応ガス搬送工程を同時に行えばよい。すなわち、金属薄膜原料と反応ガスを同時に基板6の表面に供給すればよい。
ALD法による成膜方法の場合は、原料搬送工程と反応ガス搬送工程を交互に行えばよい。すなわち、金属薄膜材料と反応ガスを交互に基板6の表面に供給すればよい。
ALD法による成膜方法の場合は、原料搬送工程と反応ガス搬送工程を交互に行えばよい。すなわち、金属薄膜材料と反応ガスを交互に基板6の表面に供給すればよい。
ALD法について詳しく説明すると、ヒーターによって加熱された基板6上に金属薄膜原料を搬送し、基板6上において金属薄膜原料を飽和吸着させた状態で、金属薄膜原料の供給を止め、その後基板6上に反応ガスを供給することで、金属薄膜原料を基板6上に成膜する。この工程を1サイクルとして、基板6上に所望の膜厚の金属薄膜が成膜されるまで、当該サイクルを複数回行う。
この際、金属薄膜原料を搬送しただけでは、基板6上に金属薄膜は成膜されず、反応ガスを搬送して初めて成膜される。そして、基板6上に飽和吸着している金属薄膜原料のみが成膜されるので、1原子ないし数原子単位で薄膜を成膜することになる。
したがって、所望の膜厚とするためには、原料搬送工程と反応ガス搬送工程を、それぞれ交互に繰り返すこととなるが、その際それぞれの工程の間において、真空チャンバー2内の残留ガスを除去するパージ工程を有することが好ましい。
このようなパージ工程を有することで、より精度よく金属薄膜を成膜することができる。
このようなパージ工程を有することで、より精度よく金属薄膜を成膜することができる。
原料搬送工程と反応ガス搬送工程の切替は、バルブ22、25の開閉で制御すればよい。
以上のようにして、原料搬送工程と反応ガス搬送工程を交互に行うことで、基板6上に金属薄膜が成膜される。
以上のようにして、原料搬送工程と反応ガス搬送工程を交互に行うことで、基板6上に金属薄膜が成膜される。
以上、本発明を実施形態に基づき説明したが、本発明は、上記実施形態に限定されるものではなく、その要旨を逸脱しない範囲で種々変更可能であることは言うまでもない。
以下、本発明を実施例および比較例により、更に詳細に説明する。ただし、本発明は以下の実施例等によって何ら限定されるものではない。
(実施例1〜4)
実施例1〜4では、図3に示した金属薄膜の成膜装置を用い、金属薄膜原料として下記化学式(3)に示すものを用い、CVD法によって基板(SiO2)上に金属薄膜を成膜した。
実施例1〜4では、図3に示した金属薄膜の成膜装置を用い、金属薄膜原料として下記化学式(3)に示すものを用い、CVD法によって基板(SiO2)上に金属薄膜を成膜した。
具体的には、金属薄膜原料が充填している原料貯液槽(有機金属ボトル)を50℃に加熱し、100sccmのヘリウムを用いてバブリングさせて、金属薄膜原料をチャンバー内の基板表面に搬送した。また、これと同時に、30sccmのアンモニアも基板表面に搬送した。
この際、チャンバー内の全圧を400Pa(=3Torr)に保ち、基板の温度を280℃、300℃、350℃、400℃に保った。
以上のようにして、金属薄膜であるコバルト膜を成膜した。このときの成膜速度を表1に示す。
以上のようにして、金属薄膜であるコバルト膜を成膜した。このときの成膜速度を表1に示す。
なお、成膜したコバルト膜について、不純物濃度をX線光電子分光法(XPS)によって測定したところ(ULVAC−Phi製、ESCA1600、Al線源を使用)、炭素、窒素、酸素のいずれもが1%未満であった。
(実施例5〜8)
実施例5〜8でも、実施例1〜4と同様に、図3に示した金属薄膜の成膜装置を用い、金属薄膜原料として上記化学式(3)に示すものを用いて、CVD法によって基板上に金属薄膜を成膜した。
実施例5〜8でも、実施例1〜4と同様に、図3に示した金属薄膜の成膜装置を用い、金属薄膜原料として上記化学式(3)に示すものを用いて、CVD法によって基板上に金属薄膜を成膜した。
具体的には、金属薄膜原料が充填している原料貯液槽(有機金属ボトル)を120℃に加熱し、100sccmのヘリウムを用いてバブリングさせて、金属薄膜原料をチャンバー内の基板表面に搬送した。また、これと同時に、30sccmのアンモニアも基板表面に搬送した。
この際、チャンバー内の全圧を400Pa(=3Torr)に保ち、基板の温度を150℃、200℃、250℃、300℃に保った。
以上のようにして、金属薄膜であるコバルト膜を成膜した。このときの成膜速度を表2に示す。
以上のようにして、金属薄膜であるコバルト膜を成膜した。このときの成膜速度を表2に示す。
なお、成膜したコバルト膜について、不純物濃度をX線光電子分光法(XPS)によって測定したところ(ULVAC−Phi製、ESCA1600、Al線源を使用)、炭素、窒素、酸素のいずれもが1%未満であった。
(実施例9)
実施例9でも、実施例1〜4と同様に、図3に示した金属薄膜の成膜装置を用い、金属薄膜原料として上記化学式(3)に示すものを用いて、CVD法によって基板上に金属薄膜を成膜した。
実施例9でも、実施例1〜4と同様に、図3に示した金属薄膜の成膜装置を用い、金属薄膜原料として上記化学式(3)に示すものを用いて、CVD法によって基板上に金属薄膜を成膜した。
具体的には、金属薄膜原料が充填している原料貯液槽(有機金属ボトル)を50℃に加熱し、100sccmのヘリウムを用いてバブリングさせて、金属薄膜原料をチャンバー内の基板表面に搬送した。また、これと同時に、30sccmのモノメチルヒドラジンも基板表面に搬送した。
この際、チャンバー内の全圧を400Pa(=3Torr)に保ち、基板の温度250℃に保った。
以上のようにして、金属薄膜であるコバルト膜を成膜した。
以上のようにして、金属薄膜であるコバルト膜を成膜した。
このときの成膜速度は、0.17nm/minであった。
なお、成膜したコバルト膜について、不純物濃度をX線光電子分光法(XPS)によって測定したところ(ULVAC−Phi製、ESCA1600、Al線源を使用)、炭素、窒素、酸素のいずれもが1%未満であった。
なお、成膜したコバルト膜について、不純物濃度をX線光電子分光法(XPS)によって測定したところ(ULVAC−Phi製、ESCA1600、Al線源を使用)、炭素、窒素、酸素のいずれもが1%未満であった。
(実施例10)
実施例10では、図3に示した金属薄膜の成膜装置を用い、金属薄膜原料として上記化学式(3)に示すものを用い、パージ材料としてヘリウムを用いて、ALD法によって基板上に金属薄膜を成膜した。
実施例10では、図3に示した金属薄膜の成膜装置を用い、金属薄膜原料として上記化学式(3)に示すものを用い、パージ材料としてヘリウムを用いて、ALD法によって基板上に金属薄膜を成膜した。
具体的には、金属薄膜原料が充填している原料貯液槽(有機金属ボトル)を50℃に加熱し、100sccmのヘリウムをバブリングさせることによって、金属薄膜原料を、全圧40Pa(=0.3Torr)に保ったチャンバー内の基板表面に、5秒間搬送した。
この際、基板の温度を350℃に保った。
この際、基板の温度を350℃に保った。
そして、金属薄膜原料を基板表面に吸着させた後、ヘリウムをパージ材料として1秒間流通させ、その後、100sccmのアンモニアを基板表面に5秒間搬送した。
アンモニアを基板表面に搬送した後は、ヘリウムをパージ材料として1秒間流通させ、その後に再び金属薄膜原料を基板表面に搬送した。
このようにして、金属薄膜原料の搬送、パージ材料の流通、アンモニアの搬送、パージ材料の流通の工程を1サイクルとして、2000サイクル、時間にして400分間繰り返してコバルト膜からなる金属薄膜を成膜した。
アンモニアを基板表面に搬送した後は、ヘリウムをパージ材料として1秒間流通させ、その後に再び金属薄膜原料を基板表面に搬送した。
このようにして、金属薄膜原料の搬送、パージ材料の流通、アンモニアの搬送、パージ材料の流通の工程を1サイクルとして、2000サイクル、時間にして400分間繰り返してコバルト膜からなる金属薄膜を成膜した。
成膜したコバルト膜の膜厚は60nmであった。また、1分あたりの成膜量は0.15nmであった。なお、成膜したコバルト膜について、不純物濃度をX線光電子分光法(XPS)によって測定したところ(ULVAC−Phi製、ESCA1600、Al線源を使用)、炭素、窒素、酸素のいずれもが1%未満であった。
(比較例1)
比較例1では、実施例1〜4と同様に、図3に示した金属薄膜の成膜装置を用い、金属薄膜原料として上記化学式(3)に示すものを用いて、CVD法によって基板上に金属薄膜を成膜した。
比較例1では、実施例1〜4と同様に、図3に示した金属薄膜の成膜装置を用い、金属薄膜原料として上記化学式(3)に示すものを用いて、CVD法によって基板上に金属薄膜を成膜した。
具体的には、金属薄膜原料が充填している原料貯液槽(有機金属ボトル)を50℃に加熱し、100sccmのヘリウムを用いてバブリングさせて、金属薄膜原料をチャンバー内の基板表面に搬送した。また、これと同時に、30sccmの水素も基板表面に搬送した。
この際、チャンバー内の全圧を400Pa(=3Torr)に保ち、基板の温度を280℃、300℃、350℃、400℃に保った。
120分間成膜を試みたが、いずれの温度においても、連続したコバルト膜を得ることはできなかった。
120分間成膜を試みたが、いずれの温度においても、連続したコバルト膜を得ることはできなかった。
(比較例2)
比較例2では、図3に示した金属薄膜の成膜装置を用い、金属薄膜原料として上記化学式(3)に示すものを用い、パージ材料としてヘリウムを用いて、ALD法によって基板上に金属薄膜を成膜した。
比較例2では、図3に示した金属薄膜の成膜装置を用い、金属薄膜原料として上記化学式(3)に示すものを用い、パージ材料としてヘリウムを用いて、ALD法によって基板上に金属薄膜を成膜した。
具体的には、金属薄膜原料が充填している原料貯液槽(有機金属ボトル)を100℃に加熱し、100sccmのヘリウムをバブリングさせることによって、金属薄膜原料を、全圧40Pa(=0.3Torr)に保ったチャンバー内の基板表面に、10秒間搬送した。この際、基板の温度を350℃に保った。
そして、金属薄膜原料を基板表面に吸着させた後、ヘリウムをパージ材料として5秒間流通させ、その後、100sccmの水素を基板表面に5秒間搬送した。
水素を基板表面に搬送した後は、ヘリウムをパージ材料として1秒間流通させ、その後に再び金属薄膜原料を基板表面に搬送した。
このようにして、金属薄膜原料の搬送、パージ材料の流通、水素の搬送、パージ材料の流通の工程を1サイクルとして、2000サイクル、時間にして700分間繰り返してコバルト膜からなる金属薄膜を成膜した。
水素を基板表面に搬送した後は、ヘリウムをパージ材料として1秒間流通させ、その後に再び金属薄膜原料を基板表面に搬送した。
このようにして、金属薄膜原料の搬送、パージ材料の流通、水素の搬送、パージ材料の流通の工程を1サイクルとして、2000サイクル、時間にして700分間繰り返してコバルト膜からなる金属薄膜を成膜した。
成膜したコバルト膜の膜厚は49nmであった。また、1分あたりの成膜量は0.07nmであった。なお、成膜したコバルト膜について、不純物濃度をX線光電子分光法(XPS)によって測定したところ(ULVAC−Phi製、ESCA1600、Al線源を使用)、炭素、窒素、酸素のいずれもが1%未満であった。
1・・・成膜装置、2・・・真空チャンバー、3・・・原料搬送手段、4・・・反応性ガス搬送手段、5・・・載置台、6・・・基板、7・・・排気ポンプ、8・・・ガス噴出部、9・・・原料貯液槽、10,21,24・・・配管、22,25・・・バルブ、23・・・反応性ガス供給源、26・・・シャワーヘッド
Claims (8)
- 下記化学式(1)で示されるアミジナート配位子を有する金属薄膜原料と、窒素原子および水素原子を有する反応ガスと、を用いて、
前記金属薄膜原料と前記反応ガスを交互または同時に基板表面に供給することにより、低不純物の金属薄膜を成膜する金属薄膜の成膜方法。
- 前記化学式(1)において、R1〜R3が水素またはCnH2n+1(n=1〜4)の化学式で表される炭化水素であることを特徴とする請求項1に記載の金属薄膜の成膜方法。
- 前記化学式(1)において、M1がCo,Ni,Fe、またはMnであることを特徴とする請求項1または請求項2に記載の金属薄膜の成膜方法。
- 前記反応ガスが、アンモニア、メチルアミン、エチルアミン、プロピルアミン、イソプロピルアミン、ターシャリーブチルアミン、ノルマルブチルアミン、ジメチルアミン、ジエチルアミン、ヒドラジン、モノメチルヒドラジン、ジメチルヒドラジン、ジフェニルヒドラジン、およびエチレンイミンのうち、少なくとも1つ以上を含むことを特徴とする請求項1ないし請求項3のいずれか1項に記載の金属薄膜の成膜方法。
- 窒素原子および水素原子を有する反応ガスとともに用いられることで金属薄膜を成膜する金属薄膜成膜用原料であって、下記化学式(2)で示されるアミジナート配位子を有する金属薄膜成膜用原料。
- 前記化学式(2)において、R4〜R6が水素またはCnH2n+1(n=1〜4)の化学式で表される炭化水素であることを特徴とする請求項5に記載の金属薄膜成膜用原料。
- 前記化学式(2)において、M2がCo,Ni,Fe、またはMnであることを特徴とする請求項5または請求項6に記載の金属薄膜成膜用原料。
- 前記反応ガスが、アンモニア、メチルアミン、エチルアミン、プロピルアミン、イソプロピルアミン、ターシャリーブチルアミン、ノルマルブチルアミン、ジメチルアミン、ジエチルアミン、ヒドラジン、モノメチルヒドラジン、ジメチルヒドラジン、ジフェニルヒドラジン、およびエチレンイミンのうち、少なくとも1つ以上を含むことを特徴とする請求項5ないし請求項7のいずれか1項に記載の金属薄膜成膜用原料。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2011248720A JP2013104100A (ja) | 2011-11-14 | 2011-11-14 | 金属薄膜の成膜方法および金属薄膜成膜用原料 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2011248720A JP2013104100A (ja) | 2011-11-14 | 2011-11-14 | 金属薄膜の成膜方法および金属薄膜成膜用原料 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| JP2013104100A true JP2013104100A (ja) | 2013-05-30 |
Family
ID=48623883
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2011248720A Pending JP2013104100A (ja) | 2011-11-14 | 2011-11-14 | 金属薄膜の成膜方法および金属薄膜成膜用原料 |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP2013104100A (ja) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2018088079A1 (ja) * | 2016-11-08 | 2018-05-17 | 株式会社Adeka | 化合物、薄膜形成用原料、薄膜の製造方法及びアミジン化合物 |
Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2006511716A (ja) * | 2002-11-15 | 2006-04-06 | プレジデント・アンド・フェロウズ・オブ・ハーバード・カレッジ | 金属アミジナートを用いる原子層の析出 |
| US20090208637A1 (en) * | 2006-06-15 | 2009-08-20 | Advanced Technology Materials, Inc. | Cobalt precursors useful for forming cobalt-containing films on substrates |
| WO2011027835A1 (ja) * | 2009-09-02 | 2011-03-10 | 株式会社アルバック | Co膜の形成方法 |
| WO2011027834A1 (ja) * | 2009-09-02 | 2011-03-10 | 株式会社アルバック | Co膜の形成方法及びCu配線膜の形成方法 |
| WO2011040385A1 (ja) * | 2009-09-29 | 2011-04-07 | 東京エレクトロン株式会社 | Ni膜の成膜方法 |
| US20110163062A1 (en) * | 2009-10-23 | 2011-07-07 | Gordon Roy G | Self-aligned barrier and capping layers for interconnects |
| JP2012229488A (ja) * | 2011-04-13 | 2012-11-22 | Ulvac Japan Ltd | Ni膜の形成方法 |
-
2011
- 2011-11-14 JP JP2011248720A patent/JP2013104100A/ja active Pending
Patent Citations (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2006511716A (ja) * | 2002-11-15 | 2006-04-06 | プレジデント・アンド・フェロウズ・オブ・ハーバード・カレッジ | 金属アミジナートを用いる原子層の析出 |
| US20090291208A1 (en) * | 2002-11-15 | 2009-11-26 | Gordon Roy G | Atomic layer deposition using metal amidinates |
| US20090208637A1 (en) * | 2006-06-15 | 2009-08-20 | Advanced Technology Materials, Inc. | Cobalt precursors useful for forming cobalt-containing films on substrates |
| WO2011027835A1 (ja) * | 2009-09-02 | 2011-03-10 | 株式会社アルバック | Co膜の形成方法 |
| WO2011027834A1 (ja) * | 2009-09-02 | 2011-03-10 | 株式会社アルバック | Co膜の形成方法及びCu配線膜の形成方法 |
| WO2011040385A1 (ja) * | 2009-09-29 | 2011-04-07 | 東京エレクトロン株式会社 | Ni膜の成膜方法 |
| US20110163062A1 (en) * | 2009-10-23 | 2011-07-07 | Gordon Roy G | Self-aligned barrier and capping layers for interconnects |
| JP2013508979A (ja) * | 2009-10-23 | 2013-03-07 | プレジデント アンド フェロウズ オブ ハーバード カレッジ | 相互接続用自己整合バリアおよびキャッピング層 |
| JP2012229488A (ja) * | 2011-04-13 | 2012-11-22 | Ulvac Japan Ltd | Ni膜の形成方法 |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2018088079A1 (ja) * | 2016-11-08 | 2018-05-17 | 株式会社Adeka | 化合物、薄膜形成用原料、薄膜の製造方法及びアミジン化合物 |
| US11161867B2 (en) | 2016-11-08 | 2021-11-02 | Adeka Corporation | Compound, raw material for forming thin film, method for manufacturing thin film, and amidine compound |
| US11618762B2 (en) | 2016-11-08 | 2023-04-04 | Adeka Corporation | Compound, raw material for forming thin film, method for manufacturing thin film, and amidine compound |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6942071B2 (ja) | 周期的堆積により基材上に金属性膜を形成する方法及び関連する半導体デバイス構造 | |
| US10995405B2 (en) | Deposition of molybdenum thin films using a molybdenum carbonyl precursor | |
| TWI655310B (zh) | 來自金屬脒鹽前驅物與鋁前驅物的金屬鋁合金膜 | |
| JP6041464B2 (ja) | 金属薄膜の製膜方法、および金属薄膜の製膜装置 | |
| US9121093B2 (en) | Bis-ketoiminate copper precursors for deposition of copper-containing films and methods thereof | |
| US11749523B2 (en) | Methods for preparing self-assembled monolayers | |
| WO2012012026A2 (en) | Metal film deposition | |
| JP6569831B1 (ja) | 酸化膜形成方法 | |
| TWI717669B (zh) | 氧化膜形成方法 | |
| CN106068335A (zh) | 锗或氧化锗的原子层沉积 | |
| TWI431146B (zh) | Method for forming tantalum nitride film and film forming device thereof | |
| US12065739B2 (en) | Vapor deposition processes | |
| CN108431295A (zh) | 形成含钴膜的组合物、其合成、以及在膜沉积中的用途 | |
| JP2013104100A (ja) | 金属薄膜の成膜方法および金属薄膜成膜用原料 | |
| JP5795520B2 (ja) | 金属薄膜材料および金属薄膜の成膜方法 | |
| WO2021144334A9 (en) | Ruthenium-containing films deposited on ruthenium-titanium nitride films and methods of forming the same | |
| TWI808951B (zh) | 用於過渡金屬的金屬、金屬氮化物,及金屬氧化物系膜的沉積之前驅物 | |
| US12297537B2 (en) | Compounds and methods for selectively forming metal-containing films | |
| US8088938B2 (en) | Low decomposition storage of a tantalum precursor | |
| TWI557256B (zh) | 來自金屬pcai前驅物與鋁前驅物的金屬鋁合金膜 | |
| WO2019030117A1 (en) | GE-CONTAINING CO-FILM FORMING MATERIAL, GE-CONTAINING CO-FILM AND CORRESPONDING FILM-FORMING METHOD | |
| TW201711963A (zh) | 鎂系構材形成材料、鎂系構材形成方法以及新穎化合物 | |
| JP2006100812A (ja) | 有機金属化学気相成長法用原料液及び該原料液を用いたHf−Si含有複合酸化物膜の製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20140718 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20150119 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20150224 |
|
| A02 | Decision of refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A02 Effective date: 20150728 |