JP2015125302A - トナー - Google Patents
トナー Download PDFInfo
- Publication number
- JP2015125302A JP2015125302A JP2013270112A JP2013270112A JP2015125302A JP 2015125302 A JP2015125302 A JP 2015125302A JP 2013270112 A JP2013270112 A JP 2013270112A JP 2013270112 A JP2013270112 A JP 2013270112A JP 2015125302 A JP2015125302 A JP 2015125302A
- Authority
- JP
- Japan
- Prior art keywords
- toner
- fine particles
- particles
- silica fine
- titanium oxide
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
Images



Landscapes
- Developing Agents For Electrophotography (AREA)
Abstract
【解決手段】外添剤として酸化チタン微粒子及びシリカ微粒子を有するトナーであって、長軸径が30〜500nm、短軸径が10〜80nm、長軸径/短軸径の比が2.0以上8.0以下であるルチル型の酸化チタン微粒子を、トナー粒子100質量部当たり0.1〜1.0質量部含有し、酸化チタンとシリカの含有量の比が、0.4〜30.0、X線光電子分光装置(ESCA)により求めた、トナー粒子の表面のシリカ微粒子による被覆率X1が、40.0〜75.0面積%、シリカ微粒子による理論被覆率をX2としたとき、X1/X2≧−0.0042×X1+0.62を満足し、トナー粒子への外添剤の埋め込み率が25〜60%である。
【選択図】なし
Description
一方、小型化の点においては、現像スリーブの小径化が重要な技術となる。トナーへの電荷付与は、主としてトナー規制部材(以下、現像ブレードという)によってトナーが規制された領域において、トナーと現像スリーブ等の摩擦帯電付与部材との摺擦に起因する摩擦帯電によって行なわれる。
例えば、トナーのチャージアップにより、トナーが現像スリーブに留まることで画像濃度が低下したり、トナーの帯電が不均一になることでかぶりといった画像欠陥が起こったりする場合がある。「かぶり」とは本来白抜けとなるべき非画像部にトナーが付着して濃度が高くなる現象を意味する。
また、チャージアップに起因する画像欠陥として、高温高湿環境においてベタ黒画像を出力すると、感光体にトナーが貼りついてしまい、トナーが転写材に効率的に転写されず、ベタ黒画像が白くぼやけてしまう画像弊害が発生する(以下、転写抜けという)。
転写材の電気的な体積抵抗値(以下、体積抵抗値という)は種々であり、特に高温高湿環境下では、転写材が水分を吸湿すると体積抵抗値は低くなり、吸湿していないと体積抵抗値は高くなる傾向にある。
しかし、吸湿紙などの体積抵抗値の低い転写材に対しては、転写材を介して紙搬送の上流に搬送をガイドする転写前ガイド部材や定着器などの本体へ転写電流が逃げてしまい、転写電流不足に起因する転写抜けが発生する場合がある。逆に体積抵抗値の低い転写材の電流の逃げを考慮して転写電流を高めに設定した場合、吸湿してない紙のように体積抵抗値の高い転写材では転写電流過多に起因する画像弊害が発生してしまう。特にトナーの耐久劣化が促進されてしまう耐久使用後半では、トナーの帯電が落ちる傾向にあり、このような転写抜けが顕著に現れてしまう。
そのため、転写電流が低い状態であっても、吸湿紙のような体積抵抗値の低い転写材で効率的に転写できるトナーが好ましい。
具体的には、使用環境によらず安定した画像濃度と、かぶりのない良好な画像が得られるトナーであり、画像形成装置の小型化及び厳しい耐久試験(繰り返し使用試験)においても、転写抜けの発生を抑制できるトナーを提供することにある。
結着樹脂及び着色剤を含有するトナー粒子と、
外添剤としての、酸化チタン微粒子及びシリカ微粒子と、
を有するトナーであって、
前記酸化チタン微粒子は、
長軸径が30nm以上500nm以下であり、
短軸径が10nm以上80nm以下であり、
長軸径/短軸径の比が2.0以上8.0以下である
ルチル型の酸化チタン微粒子であり、
前記酸化チタン微粒子の含有量が、前記トナー粒子100質量部当たり0.1質量部以上1.0質量部以下であり、
前記トナー中の、前記酸化チタン微粒子の含有量に対する前記シリカ微粒子の含有量の比が、0.4以上30.0以下であり
X線光電子分光装置(ESCA)により求めた、前記トナー粒子の表面の前記シリカ微粒子による被覆率X1が、40.0面積%以上75.0面積%以下であり、
前記シリカ微粒子による理論被覆率をX2としたとき、下記式1で示される拡散指数が、下記式2を満たし、
前記トナー粒子への前記外添剤の埋め込み率が、25%以上60%以下である
ことを特徴とするトナー。
(式1)拡散指数=X1/X2
(式2)拡散指数≧−0.0042×X1+0.62
結着樹脂及び着色剤を含有するトナー粒子と、
外添剤としての、酸化チタン微粒子及びシリカ微粒子と、
を有するトナーであって、
前記酸化チタン微粒子は、
長軸径が30nm以上500nm以下であり、
短軸径が10nm以上80nm以下であり、
長軸径/短軸径の比が2.0以上8.0以下である
ルチル型の酸化チタン微粒子であり、
前記酸化チタン微粒子の含有量が、前記トナー粒子100質量部当たり0.1質量部以上1.0質量部以下であり、
前記トナー中の、前記酸化チタン微粒子の含有量に対する前記シリカ微粒子の含有量の比が0.4以上30.0以下であり、
X線光電子分光装置(ESCA)により求めた、前記トナー粒子の表面の前記シリカ微粒子による被覆率X1が、40.0面積%以上75.0面積%以下であり、
前記シリカ微粒子による理論被覆率をX2としたとき、下記式1で示される拡散指数が下記式2を満たし、
前記トナー粒子への前記外添剤の埋め込み率が25%以上60%以下である
ことを特徴とするトナーである。
(式1)拡散指数=X1/X2
(式2)拡散指数≧−0.0042×X1+0.62
ここで、転写抜けの問題は以下の原因により発生すると考えている。
これにより、例えば、非画像領域へのかぶりといった画像欠陥を引き起こしやすくなるだけでなく、トナーと他の部材との付着力が高まることによる、様々な問題が発生しやすくなる。例えば、チャージアップしたトナーが現像スリーブ上に留まることにより、画像濃度が低下しやすくなる。
被覆率X1は、好ましくは、45.0面積%以上70.0面積%以下であり、より好ましくは、45.0面積%以上68.0面積%以下である。
ここで、被覆率X1が40.0面積%未満のとき、本発明の意図する効果が得られない。一方、被覆率X1が75.0面積%を超える場合には、低温定着性を阻害する場合がある。
上記のように被覆率X1と拡散指数を制御することにより、トナーの長期耐久使用に起因するトナー劣化を大幅に抑制することが可能である。
そのため、耐久使用初期に被覆率X1を調整するようにシリカ微粒子を添加しても、耐久使用後期にはシリカ微粒子による被覆率が低下しやすい。さらに凝集体であることにより、シリカ微粒子同士の力によりトナーにシリカ微粒子がより多く埋めこまれやすく、耐久使用初期と耐久使用後期のトナー物性が大きく異なりやすく、トナー劣化を引き起こしやすい。
この場合、シリカ微粒子がより一次粒子に近い状態でトナー粒子表面に付着しているため、耐久使用試験を行っても、トナー粒子表面からシリカ微粒子が脱離しにくい。また、シリカ微粒子がより一次粒子に近い状態であるため、シリカ微粒子同士が接触する確率が低くなるとともに、シリカ微粒子同士の力でトナー粒子に埋めこまれやすくなることも抑制することが可能である。
感光体とトナーの付着力を低減するには、感光体とトナー粒子の間に、無機微粒子である外添剤が介在することが非常に重要となる。上述のように、高度に均一拡散した外添剤が、ある特定の状態で埋めこまれていることにより、トナー粒子表面の状態がより均一化されると考えられる。その結果、トナーと感光体が接触した際に、外添剤が介在する確率を最大限に高くできるため、トナーと感光体の付着力が低減できると考えられる。
すると、埋没していない部分がトナー粒子表面で動くなどして、外添剤の付着していない部分が露出し、感光体と直接接触することになる確率が高くなる。その結果、感光体とトナーの付着力を低減させることができない。
逆に、外添剤の埋め込み率が60%を超える場合は、トナーの循環性が低下しやすく、一度、トナー粒子と感光体が直接接触する部分があった場合にはトナーが回転せずに、外添剤が介在することができずに、感光体からトナーが離れにくい場合がある。
被覆率と拡散指数については後に詳述する。
本発明者らは、シリカ微粒子が高度に均一拡散した状態で、特定の構造および形状の酸化チタン微粒子を添加することで、トナー粒子表面において、酸化チタン微粒子も高度に均一拡散させることが可能であることを見出した。その結果、酸化チタン微粒子によるトナーのチャージアップ抑制効果を十分に発揮させることでき、トナーと感光体との付着力を低減させることができ、高温高湿環境下における吸湿紙での転写抜け抑制に顕著な効果を得られることを同時に見出した。
特に、シリカ微粒子が凝集体の状態だと、酸化チタン微粒子の周りにシリカ微粒子が付着するなどして、トナーのチャージアップ抑制効果を十分に発揮しにくい。
すると、例えば、現像スリーブを小径化し、高温高湿環境下の長期耐久使用後に吸湿紙などの体積抵抗値が低い転写材を用いた画像形成においても、トナー全体にチャージを適正に持たせることが可能になり、転写抜けを抑制することができる。この場合、チャージアップ抑制効果を十分に発揮することができ、トナーのチャージアップに起因する問題を抑制することができる。
本発明において、酸化チタン微粒子は、長軸径が30nm以上500nm以下であり、短軸径が10nm以上80nm以下であり、長軸径/短軸径の比が2.0以上8.0以下であることが重要である。長軸径は好ましくは30nm以上300nm以下であり、より好ましくは30nm以上150nm以下である。短軸径は、好ましくは10nm以上50nm以下である。長軸径/短軸径の比は好ましくは2.5以上8.0以下である。
この範囲であることにより、酸化チタン微粒子が耐久使用試験においてもトナーに埋め込まれにくいため、耐久使用後半においてもチャージアップ抑制効果が得られやすい。
また、長軸径が500nm超又は短軸径が80nm超の場合、トナーから酸化チタン微粒子の遊離が顕著になり、十分なチャージアップ抑制の効果が得られにくい。
長軸径/短軸径の比が2.0よりも小さい場合は、球状に近くなるためにトナー粒子との接触面積が小さく、トナーの付着性の効果が少なりやすく、トナーから遊離しやすくなる。
一方、長軸径/短軸径の比が8.0よりも大きい場合、酸化チタン微粒子の形状が極端に棒型や針状になり、トナーや他の外添剤との接触面積が大きくなってスペーサー効果が小さくなりやすく、耐久性が悪化しやすい。
酸化チタンとしては、結晶系がルチル型とアナターゼ型のものがある。このうち、アナターゼ型の酸化チタンは体積抵抗値が107Ω・cm程度と一般的に低いため、特に高温高湿環境下では、摩擦帯電のリークが早くなりやすく、摩擦帯電安定化の面で十分ではない。これに対してルチル型酸化チタンは、体積抵抗値が1010Ω・cm〜1018Ω・cm程度と高いため、摩擦帯電量の安定化の面で極めて有効に働く。
本発明において使用するルチル型の酸化チタン微粒子の原材料及び製造方法は、特に制約されるものではないが、以下に製造例を示す。
R:アルコキシ基
M:1〜3の整数
Y:アルキル基、ビニル基、グリシドキシ基、メタクリル基を含む炭化水素基
N:1〜3の整数
で表されるものであり、例えば以下のものが挙げられる。ビニルトリメトキシシラン、ビニルトリエトキシシラン、γ−メタクリルオキシプロピルトリメトキシシラン、ビニルトリアセトキシシラン、メチルトリメトキシシラン、メチルトリエトキシシラン、イソブチルトリメトキシシラン、ジメチルジメトキシシラン、ジメチルジエトキシシラン、トリメチルメトキシシラン、ヒドロキシプロピルトリメトキシシラン、フェニルトリメトキシシラン、n−ヘキサデシルトリメトキシシラン、n−オクタデシルトリメトキシシラン、オクチルトリメトキシシラン、オクチルトリエトキシシランの如きシランカップリング剤。
疎水化度が20%未満の場合、高温高湿環境下での長期静置に起因する帯電量低下が大きくなり現像性の低下を招くことがある。また疎水化度が98%を超えると、酸化チタン自身の帯電コントロールが難しくなり、結果として低湿下でトナーがチャージアップしてしまう場合がある。
0.1質量部より少ない場合は、チャージアップ抑制の効果が十分に得にくく、特に耐久による転写悪化を抑制しづらい。1.0質量部よりも多い場合は、トナー粒子から遊離したものが多くなることによる部材汚染が発生しやすく、また低温定着性が低下する場合がある。
シリカ微粒子の含有量の比が0.4より小さい場合は、酸化チタン微粒子がシリカ微粒子に対して過剰になってしまうため、外添剤による埋め込み率の制御がしづらくなってしまう。
また、シリカ微粒子の含有量の比が30.0より大きい場合は、酸化チタン微粒子がシリカ微粒子に対して少なくなりすぎてしまうため、チャージアップ抑制の効果が得られにくくなってしまう。
酸化チタン微粒子の遊離率を上記範囲に制御する手段としては、外添混合処理時の動力や処理時間の調整が挙げられる。外添混合処理時の動力を下げるか、処理時間を短くすることで、遊離率を高くすることができる。また、外添混合処理時の動力を上げるか、処理時間を長くすることで遊離率を低くすることができる。
これまで述べてきたような均一帯電やチャージアップ抑制が、トナーが劣化した場合にも、適正に行われるためには、ブレードニップにおける摺擦が、耐久使用後期においても、トナー一粒一粒に行われるよう、トナーがほぐれやすいことが重要である。
この「トナーが劣化した際にも、トナーが一粒一粒にほぐれやすくなる現象」には、上述の被覆率及び拡散指数が密接に関係している。
本発明のトナーは、X線光電子分光装置(ESCA)により求めた、トナー粒子の表面のシリカ微粒子による被覆率X1が40.0面積%以上75.0面積%以下である。さらに、シリカ微粒子による理論被覆率をX2としたとき、下記式1で示される拡散指数が下記式2を満足することを特徴とするトナーである。
(式1)拡散指数=X1/X2
(式2)拡散指数≧−0.0042×X1+0.62
被覆率X1が40.0面積%以上75.0面積%以下の場合、耐久使用試験を通じて、トナーの流動性及び帯電性を良好な状態に制御できる。被覆率X1が40.0面積%未満の場合、後述するトナーのほぐれ易さを十分に得ることができない。このため、評価条件や環境によっては、トナーが劣化しやすく、流動性が悪化する。
(式4)理論被覆率x2(面積%)=31/2/(2Π)×(dT/dA)×(ρT/ρT)×C×100
dA:シリカ微粒子の個数平均粒径(d1)
dA:トナーの重量平均粒径(d4)
ρa:シリカ微粒子の真比重
ρt:トナーの真比重
C:シリカ微粒子の質量/トナーの質量(=シリカ微粒子の質量部数/(シリカ微粒子の質量部数+100)
(式5) 外添剤の埋め込み率(%)=100−(Bt−Bm)/Br×100
Bt:トナーのBET
Bm:トナー粒子のBET
Br:トナーに外添剤を単に添加した場合に上昇するBETの理論値
(BETとは、BET法で測定した窒素吸着による比表面積(m2/g)である)
(式6) Br=[(外添剤1のBET(B1)×外添剤1の質量部数/100)+(外添剤2のBET(B2)×外添剤2の質量部数/100)+・・・(外添剤nのBET(Bn)×外添剤Nの質量部数/100)]
(本発明においては、外添剤としてシリカ微粒子および酸化チタン微粒子を用いるため、外添剤1および2として、それぞれのBETおよび質量部数を用いる。)
外添剤のBET法で測定した窒素吸着による比表面積の測定は、JIS Z8830(2001年)に準じて行なう。測定装置については後述する。
拡散指数は、実測の被覆率X1と理論的な被覆率X2の乖離を示す。この乖離の程度は、トナー粒子表面から垂直方向に二層、三層と積層したシリカ微粒子の多さを示すと考えている。理想的には拡散指数は1になるが、これは、被覆率X1が理論被覆率X2と一致した場合であり、二層以上積層したシリカ微粒子が全く存在しない状態である。一方、シリカ微粒子が、凝集体としてトナー粒子の表面に存在すると、実測の被覆率と理論的な被覆率の乖離が生じ、拡散指数が低くなる。つまり、拡散指数は、凝集体として存在するシリカ微粒子の量を示すと言い換えることもできる。
これまで、トナーのほぐれ易さは、数nm程度の小粒径の外添剤を多量に外添して被覆率X1を上げることで、向上すると考えられてきた。一方、本発明者らの検討によると、被覆率X1を同じにして、拡散指数の異なるトナーのほぐれ易さを測定した場合、トナーのほぐれ易さに差が生じることが明らかとなった。さらに、加圧しながらほぐれ易さを測定した場合、さらに顕著な差が見られることも明らかとなった。
被覆率X1、及び、拡散指数が式2で示される範囲を同時に満たした場合、トナーのほぐれ易さが良好になる理由について、詳細は分かっていないが、本発明者らは次のように推測している。
特に、トナーが劣化した際には、少なからず、シリカ微粒子がトナー粒子表面に埋没してしまい、トナーの流動性が低下する。その時に、埋没していない凝集体として存在するシリカ微粒子同士による噛みあわせの影響が大きくなり、トナーのほぐれやすさを阻害すると推察される。
そのため、従来のトナーでは、ストレスを受けて劣化したトナーは、ブレードニップ内での循環性が悪く、トナー全体が適正に摩擦帯電されにくくなり、転写残トナーが多くなりやすかったが、本発明のトナーにおいてはその問題が解消された。
その結果、トナー全体が適正に帯電されることになり、不均一な帯電やチャージアップに伴う諸問題が大幅に改善することができる。
本発明における拡散指数の境界線は、被覆率X1が40.0面積%以上75.0面積%以下の範囲において、被覆率X1を変数とした関数である。この関数の算出は、シリカ微粒子、外添条件等を変化させて、被覆率X1と拡散指数を得た際、トナーが加圧時に十分にほぐれやすくなる現象から、経験的に得たものである。
ここで、拡散指数が被覆率X1に依存する理由に関して、詳細は分かっていないが、本発明者らは次のように推測している。加圧時のトナーのほぐれ易さを改善するためには、二次粒子として存在しているシリカ微粒子の量が少ない方が良いが、被覆率X1の影響も少なからず受ける。被覆率X1が増加するにつれて、トナーのほぐれ易さが徐々に良好になるため、二次粒子として存在するシリカ微粒子の量の許容量が増えることになる。
拡散指数が下記に示される式3の範囲にある場合、凝集体として存在するシリカ微粒子の量が多くなり、トナーの劣化を抑制しにくく、および、トナーのほぐれやすさを十分に向上させにくいため、本発明の意図する効果を十分に発揮できない。
(式3)拡散指数<−0.0042×X1+0.62
本発明のトナーに用いられる結着樹脂としては、ビニル系樹脂、ポリエステル系樹脂、エポキシ樹脂、ポリウレタン樹脂等が挙げられる。特に限定されずこれら従来公知の樹脂を用いることができる。なかでも帯電性と定着性の両立の観点から、ポリエステル樹脂もしくはビニル系樹脂を含有することが好ましい。
2価のアルコール成分としては、エチレングリコール、プロピレングリコール、1,3−ブタンジオール、1,4−ブタンジオール、2,3−ブタンジオール、ジエチレングリコール、トリエチレングリコール、1,5−ペンタンジオール、1,6−ヘキサンジオール、ネオペンチルグリコール、2−エチル−1,3−ヘキサンジオール、水素化ビスフェノールA、また(A)式で表されるビスフェノール及びその誘導体;

また(B)式で示されるジオール類;
3価以上の多価アルコール成分としては、例えばソルビトール、1,2,3,6−ヘキサンテトロール、1,4−ソルビタン、ペンタエリスリトール、ジペンタエリスリトール、トリペンタエリスリトール、1,2,4−ブタントリオール、1,2,5−ペンタントリオール、グリセロール、2−メチルプロパントリオール、2−メチル−1,2,4−ブタントリオール、トリメチロールエタン、トリメチロールプロパン、1,3,5−トリヒドロキシベンゼン等が挙げられる。
また、本発明における3価以上の多価カルボン酸成分としては、例えばトリメリット酸、ピロメリット酸、1,2,4−ベンゼントリカルボン酸、1,2,5−ベンゼントリカルボン酸、2,5,7−ナフタレントリカルボン酸、1,2,4−ナフタレントリカルボン酸、1,2,4−ブタントリカルボン酸、1,2,5−ヘキサントリカルボン酸、1,3−ジカルボキシル−2−メチル−2−メチレンカルボキシプロパン、テトラ(メチレンカルボキシル)メタン、1,2,7,8−オクタンテトラカルボン酸、エンポール三量体酸、及びこれらの無水物、低級アルキルエステル;(C)式で表されるテトラカルボン酸等、
及びこれらの無水物、低級アルキルエステル等の多価カルボン酸類及びその誘導体が挙げられる。
アルコール成分の含有量は、通常40〜60モル%、好ましくは45〜55モル%である。また、酸成分の含有量は、通常60〜40モル%、好ましくは55〜45モル%である。
該ポリエステル樹脂は通常一般に知られている縮重合によって得られる。
ビニル系樹脂を生成する為の重合性単量体(ビニル系モノマー)としては、次の様なものが挙げられる。
スチレン;o−メチルスチレン、m−メチルスチレン、p−メチルスチレン、p−メトキシスチレン、p−フェニルスチレン、p−クロルスチレン、3,4−ジクロルスチレン、p−エチルスチレン、2,4−ジメチルスチレン、p−n−ブチルスチレン、p−tertブチルスチレン、p−nヘキシルスチレン、p−nオクチルスチレン、p−nノニルスチレン、p−nデシルスチレン、p−nドデシルスチレンの如きスチレン及びその誘導体;エチレン、プロピレン、ブチレン、イソブチレンの如きスチレン不飽和モノオレフィン類;ブタジエン、イソプレンの如き不飽和ポリエン類;塩化ビニル、塩化ビニリデン、臭化ビニル、沸化ビニルの如きハロゲン化ビニル類;酢酸ビニル、プロピオン酸ビニル、ベンゾエ酸ビニルの如きビニルエステル類;メタクリル酸メチル、メタクリル酸エチル、メタクリル酸プロピル、メタクリル酸nブチル、メタクリル酸イソブチル、メタクリル酸nオクチル、メタクリル酸ドデシル、メタクリル酸2エチルヘキシル、メタクリル酸ステアリル、メタクリル酸フェニル、メタクリル酸ジメチルアミノエチル、メタクリル酸ジエチルアミノエチルの如きα−メチレン脂肪族モノカルボン酸エステル類;アクリル酸メチル、アクリル酸エチル、アクリル酸nブチル、アクリル酸イソブチル、アクリル酸プロピル、アクリル酸nオクチル、アクリル酸ドデシル、アクリル酸2−エチルヘキシル、アクリル酸ステアリル、アクリル酸2−クロルエチル、アクリル酸フェニルの如きアクリル酸エステル類;ビニルメチルエーテル、ビニルエチルエーテル、ビニルイソブチルエーテルの如きビニルエーテル類;ビニルメチルケトン、ビニルヘキシルケトン、メチルイソプロペニルケトンの如きビニルケトン類;n−ビニルピロール、n−ビニルカルバゾール、n−ビニルインドール、n−ビニルピロリドンの如きn−ビニル化合物;ビニルナフタリン類;アクリロニトリル、メタクリロニトリル、アクリルアミドの如きアクリル酸もしくはメタクリル酸誘導体が挙げられる。
さらに、2−ヒドロキシエチルアクリレート、2−ヒドロキシエチルメタクリレート、2−ヒドロキシプロピルメタクリレートなどのアクリル酸またはメタクリル酸エステル類;4−(1−ヒドロキシ−1−メチルブチル)スチレン、4−(1−ヒドロキシ−1−メチルヘキシル)スチレンの如きヒドロキシ基を有するモノマーが挙げられる。
これらの架橋剤は、架橋剤以外のモノマー成分100質量部に対して、通常、0.01〜10質量部(好ましくは0.03〜5質量部)用いることができる。
これらの架橋性モノマーのうち、結着樹脂に定着性、耐オフセット性の点から好適に用いられるものとして、芳香族ジビニル化合物(特にジビニルベンゼン)、芳香族基及びエーテル結合を含む鎖で結ばれたジアクリレート化合物類が挙げられる。
Tgが45℃未満の場合には、保存性が悪化しやすい傾向にある。また、Tgが70℃より高い場合には、低温定着性が悪化しやすい傾向にある。
シアン系着色剤としての有機顔料又は有機染料としては、銅フタロシアニン化合物及びその誘導体、アントラキノン化合物、塩基染料レーキ化合物が挙げられる。
マゼンタ系着色剤としての有機顔料又は有機染料としては、以下のものが挙げられる。縮合アゾ化合物、ジケトピロロピロール化合物、アントラキノン、キナクリドン化合物、塩基染料レーキ化合物、ナフトール化合物、ベンズイミダゾロン化合物、チオインジゴ化合物、ペリレン化合物。
黒色着色剤としては、カーボンブラック、上記イエロー系着色剤、マゼンタ系着色剤、及びシアン系着色剤を用い黒色に調色されたものが挙げられる。
着色剤を用いる場合、好ましくは重合性単量体又は結着樹脂100質量部に対し1質量部以上20質量部以下添加して用いられる。
本発明において用いられる磁性体としては、マグネタイト、マグヘマイト、フェライト等の酸化鉄;鉄、コバルト、ニッケルのような金属或はこれらの金属アルミニウム、コバルト、銅、鉛、マグネシウム、スズ、亜鉛、アンチモン、ベリリウム、ビスマス、カドミウム、カルシウム、マンガン、セレン、チタン、タングステン、バナジウムのような金属の合金及びその混合物等が挙げられる。
35質量%未満の場合には、現像スリーブ内のマグネットロールとの磁気引力が低下し、かぶりが悪化しやすい傾向にある。
一方、50質量%を超える場合には、現像性が低下することにより、濃度が低下する傾向にある。
次に、種晶を含むスラリー状の液に前に加えたアルカリの添加量を基準として約1当量の硫酸第一鉄を含む水溶液を加える。液のpHを5から10に維持しながら空気を吹き込みながら水酸化第一鉄の反応を進め、種晶を芯にして磁性酸化鉄を成長させる。この時、任意のpH及び反応温度、撹拌条件を選択することにより、磁性体の形状及び磁気特性をコントロールすることが可能である。
酸化反応が進むにつれて液のpHは酸性側に移行していくが、液のpHは5未満にしない方が好ましい。このようにして得られた磁性体を定法により濾過、洗浄、乾燥することにより磁性粉を得ることができる。
具体的には、再分散液を十分撹拌しながらシランカップリング剤を添加し、加水分解後温度を上げる、或いは、加水分解後に分散液のpHをアルカリ域に調整することでカップリング処理を行う。この中でも、均一な表面処理を行うという観点から、酸化反応終了後、濾過、洗浄後に乾燥させずそのままリスラリー化し、表面処理を行うことが好ましい。
RmSIYn (i)
[式中、Rはアルコキシ基を示し、mは1〜3の整数を示し、Yはアルキル基、ビニル基、エポキシ基、(メタ)アクリル基などの官能基を示し、nは1〜3の整数を示す。ただし、m+n=4である。]
cph2p+1−si−(ocqh2q+1)3 (ii)
[式中、pは2〜20の整数を示し、qは1〜3の整数を示す。]
上記シランカップリング剤を用いる場合、単独で処理する、或いは複数の種類を併用して処理することが可能である。複数の種類を併用する場合、それぞれのカップリング剤で個別に処理してもよいし、同時に処理してもよい。
用いるカップリング剤の総処理量は磁性体100質量部に対して0.9〜3.0質量部であることが好ましく、磁性体の表面積、カップリング剤の反応性等に応じて処理剤の量を調整することが重要である。
負帯電性のものとしては、例えば、有機金属錯体、キレート化合物が有効で、その例としては、モノアゾ金属錯体;アセチルアセトン金属錯体;芳香族ハイドロキシカルボン酸または芳香族ダイカルボン酸の金属錯体及びその金属塩、無水物、エステル類やビスフェノールの如きフェノール誘導体類が挙げられる。
負帯電用の荷電制御剤として好ましいものは、例えばSPILON BLACK TRH、T−77、T−95(保土谷化学工業(株))、BONTRON(登録商標)S−34、S−44、S−54、E−84、E−88、E−89(オリエント化学工業(株))が挙げられる。
これらの金属錯化合物は、単独でも或いは二種以上組み合わせて用いることが可能である。これらの荷電制御剤の使用量は、トナーの帯電量の点から、結着樹脂100質量部あたり0.1〜5.0質量部が好ましい。
酸化ポリエチレンワックスなどの脂肪族炭化水素系ワックスの酸化物、または、それらのブロック共重合物;カルナバワックス、サゾールワックス、モンタン酸エステルワックスなどの脂肪酸エステルを主成分とするワックス類;及び脱酸カルナバワックスなどの脂肪酸エステル類を一部または全部を脱酸化したものなどが挙げられる。さらに、パルミチン酸、ステアリン酸、モンタン酸などの飽和直鎖脂肪酸類;プラシジン酸、エレオステアリン酸、バリナリン酸などの不飽和脂肪酸類;ステアリルアルコール、アラルキルアルコール、ベヘニルアルコール、カルナウビルアルコール、セリルアルコール、メリシルアルコールなどの飽和アルコール類;長鎖アルキルアルコール類;ソルビトールなどの多価アルコール類;リノール酸アミド、オレイン酸アミド、ラウリン酸アミドなどの脂肪酸アミド類;メチレンビスステアリン酸アミド、エチレンビスカプリン酸アミド、エチレンビスラウリン酸アミド、ヘキサメチレンビスステアリン酸アミドなどの飽和脂肪酸ビスアミド類;エチレンビスオレイン酸アミド、ヘキサメチレンビスオレイン酸アミド、n,n’−ジオレイルアジピン酸アミド、n,n−ジオレイルセバシン酸アミドなどの不飽和脂肪酸アミド類;m−キシレンビスステアリン酸アミド、n,n−ジステアリルイソフタル酸アミドなどの芳香族系ビスアミド類;ステアリン酸カルシウム、ラウリン酸カルシウム、ステアリン酸亜鉛、ステアリン酸マグネシウムなどの脂肪酸金属塩(一般に金属石けんといわれているもの)、また、脂肪族炭化水素系ワックスにスチレンやアクリル酸などのビニル系モノマーを用いてグラフト化させたワックス類;また、ベヘニン酸モノグリセリドなどの脂肪酸と多価アルコールの部分エステル化物、また、植物性油脂の水素添加などによって得られるヒドロキシル基を有するメチルエステル化合物などが挙げられる。
これをアルミパン中に入れ、リファレンスとして空のアルミパンを用い、測定温度範囲30〜200℃の間で、昇温速度10℃/分、常温常湿下で測定を行う。2回目の昇温過程で、温度40℃〜100℃の範囲において最大吸熱ピークが得られるので、その時の温度をワックスの融点として用いる。
ワックスの量は、トナー製法にもよるが、結着樹脂100質量部あたり、通常1〜40質量部、好ましくは2〜30質量部である。
SICl4+2H2+02→SiO2+4HCl
この製造工程において、例えば塩化アルミニウム又は塩化チタンの如き他の金属ハロゲン化合物をケイ素ハロゲン化合物と共に用いることによってシリカと他の金属酸化物の複合微粒子を得ることも可能である。本発明にはそのような複合微粒子を用いることもできる。
本発明における、シリカ微粒子の一次粒子の個数平均粒径(D1)の測定法は後述する。
上記疎水化処理の方法としては、シリカ微粒子と反応あるいは物理吸着する、有機ケイ素化合物及び/又はシリコーンオイルで化学的に処理する方法が挙げられる。ケイ素ハロゲン化合物の蒸気相酸化により生成されたシリカ微粒子を有機ケイ素化合物で化学的に処理する方法が、好ましい方法として挙げられる。
シリコーンオイルの処理量は、シリカ微粒子100質量部に対し、通常1〜40質量部、好ましくは3〜35質量部である。上記範囲であれば良好な疎水性が得られやすい。
ここで、シリカ微粒子の添加量は、トナー粒子100質量部に対して、通常0.3質量部以上3.5質量部以下であり、好ましくは0.3質量部以上2.0質量部以下である。シリカ微粒子の添加量が上記範囲であることにより、被覆率、拡散指数、および外添剤の埋め込み率を適正に制御しやすい。
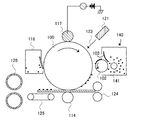
図3は、本発明に用いられる無機微粒子(シリカ微粒子及び酸化チタン微粒子)を外添混合する際に、用いることができる混合処理装置の一例を示す模式図である。
当該混合処理装置は、トナー粒子と無機微粒子に対して、狭いクリアランス部において、シェアがかかる構成になっているために、無機微粒子を二次粒子から一次粒子へとほぐしながら、トナー粒子表面に付着させることができる。無機微粒子を一次粒子へとほぐすことで、被覆率X1、拡散指数、および外添剤の埋め込み率を好ましい範囲に制御しやすくなる。
さらに、後述するように、回転体の軸方向において、トナー粒子と無機微粒子が循環しやすく、固着が進む前に十分に均一混合されやすい点で、被覆率X1、拡散指数及び外添剤の埋め込み率を本発明において好ましい範囲に制御しやすい。
本発明においては、上記のように公知の混合処理装置(ヘンシェルミキサー等)を用いることもできるが、上記のように、ヘンシェルミキサー等に対して、図3のようにシェアがかかる構成である装置による方が、本発明の外添状態に制御しやすい。
つまり、図3のような装置を用いずに、比較的シェアのかかりにくいヘンシェルミキサーなどを用いて、被覆率X1、拡散指数及び外添剤の埋め込み率を本発明において好ましい範囲に制御するためには、多段外添をしながら、処理時間としても著しく長くするような対応をしても達成しにくく、好ましくない。
上記無機微粒子を外添混合する混合処理装置は、少なくとも複数の攪拌部材3が表面に設置された回転体2と、回転体を回転駆動する駆動部8と、攪拌部材3と間隙を有して設けられた本体ケーシング1とを有する。
本体ケーシング1の内周部と、撹拌部材3との間隙(クリアランス)は、トナー粒子に均一にシェアを与え、無機微粒子を二次粒子から一次粒子へとほぐしながら、トナー粒子表面に付着しやすくするために、一定かつ微小に保つことが好ましい。
図4に示すように、複数の撹拌部材3の少なくとも一部が、回転体2の回転に伴って、トナー粒子及び無機微粒子を回転体の軸方向の一方向に送る送り用撹拌部材3aとして形成される。また、複数の撹拌部材3の少なくとも一部が、トナー粒子及び無機微粒子を、回転体2の回転に伴って、回転体の軸方向の他方向に戻す戻し用撹拌部材3bとして形成されている。
すなわち、図4に示すように、送り用撹拌部材3aの板面は送り方向13にトナー粒子を送るように傾斜している。一方、撹拌部材3bの板面は戻り方向12にトナー粒子及び無機微粒子を送るように傾斜している。
これにより、「送り方向13」への送りと、「戻り方向12」への送りとを繰り返し行いながら、トナー粒子の表面に無機微粒子の外添混合処理を行う。
図4に示す例では、撹拌部材3aと3bは等間隔で、計12枚形成されている。
さらに、図4において、Dは撹拌部材の幅、dは撹拌部材の重なり部分を示す間隔を示す。トナー粒子及び無機微粒子を、送り方向と戻り方向に効率よく送る観点から、図4における回転体2の長さに対して、Dは20%以上30%以下程度の幅であることが好ましい。図4においては、23%である例を示す。さらに撹拌部材3aと3bは撹拌部材3aの端部位置から垂直方向に延長線を引いた場合、撹拌部材3bと撹拌部材の重なり部分dをある程度有することが好ましい。
これにより、二次粒子となっている無機微粒子に効率的にシェアをかけることが可能である。Dに対するdは、10%以上30%以下であることがシェアをかける点で好ましい。
なお、羽根の形状に関しては、図4に示すような形状以外にも、送り方向及び戻り方向にトナー粒子を送ることができ、クリアランスを維持することができれば、曲面を有する形状や先端羽根部分が棒状アームで回転体2に結合されたパドル構造であってもよい。
図3に示す装置は、少なくとも複数の攪拌部材3が表面に設置された回転体2と、回転体2を回転駆動する駆動部8と、攪拌部材3と間隙を有して設けられた本体ケーシング1を有する。さらに、本体ケーシング1の内側及び回転体端部側面10にあって、冷熱媒体を流すことのできるジャケット4を有している。
さらに、図3に示す装置は、原料投入口5内に、原料投入口用インナーピース16が挿入されており、製品排出口6内に、製品排出口用インナーピース17が挿入されている。
本発明においては、まず、原料投入口5から原料投入口用インナーピース16を取り出し、トナー粒子を原料投入口5より処理空間9に投入する。次に無機微粒子を原料投入口5より処理空間9に投入し、原料投入口用インナーピース16を挿入する。次に、駆動部8により回転体2を回転させ(11は回転方向を示す)、上記で投入した処理物を、回転体2表面に複数設けられた撹拌部材3により撹拌、混合しながら外添混合処理する。
なお、投入する順序は、先に無機微粒子を原料投入口5より投入し、次に、トナー粒子を原料投入口5より投入しても構わない。また、ヘンシェルミキサーのような混合機であらかじめ、トナー粒子と無機微粒子を混合した後、混合物を、図3に示す装置の原料投入口5より投入しても構わない。
より具体的には、外添混合処理条件として、駆動部8の動力を、0.2W/g以上2.0W/g以下に制御することが、本発明で規定する被覆率X1、拡散指数及び外添剤の埋め込み率を得るうえで好ましい。また、駆動部8の動力を、0.6W/g以上1.6W/g以下に制御することが、より好ましい。
0.2W/gより動力が低い場合には、被覆率X1が高くなりにくく、拡散指数が低くなりすぎる傾向にある。一方、2.0W/gより高い場合には、拡散指数が高くなるが、外添剤が埋め込まれすぎてしまう傾向にある。
処理時間としては、特に限定されないが、好ましくは、3分以上10分以下である。処理時間が3分より短い場合には、被覆率X1及び拡散指数が低くなる傾向にある。
さらに、本発明において、特に好ましい処理方法は、外添混合処理操作の前に、プレ混合工程を持たせることである。プレ混合工程を入れることにより、シリカ微粒子および酸化チタン微粒子がトナー粒子表面上で高度に均一分散される。そのため、被覆率X1が高くなりやすく、拡散指数を高くしやすい。
より具体的には、プレ混合処理条件として、駆動部8の動力を、0.06W/g以上0.20W/g以下とし、処理時間を0.5分以上1.5分以下とすることが好ましい。プレ混合処理条件として、0.06W/gより負荷動力が低い、或いは処理時間が0.5分より短い場合には、プレ混合として十分な均一混合がなされにくい。一方、プレ混合処理条件として、0.20W/gより負荷動力が高い、或いは処理時間1.5分より長い場合には、十分な均一混合がなされる前に、トナー粒子表面にシリカ微粒子が固着されてしまう場合がある。
外添混合処理終了後、製品排出口6内の、製品排出口用インナーピース17を取り出し、駆動部8により回転体2を回転させ、製品排出口6からトナーを排出する。得られたトナーを、必要に応じて円形振動篩機等の篩機で粗粒等を分離し、トナーを得る。
本発明に係るトナー粒子は、好適には、重合性単量体及び着色剤を含有する重合性単量体組成物の粒子を水系媒体中で形成し、粒子中に含有される重合性単量体を重合させて得ることができる。重合性単量体には、結着樹脂の材料として前述したものを用いることができる。
本発明のトナーは、現像性や定着性のバランスの観点から、重量平均粒径(D4)が、通常5.0μm以上10.0μm以下であり、好ましくは、6.0μm以上9.0μm以下である。
さらに、加圧時のトナーのほぐれ易さという観点においても、トナー粒子の表面形状における噛み合わせ効果が発生しにくくなり、ほぐれ易さをさらに向上できるため、好ましい。前記水系媒体中でトナー粒子を製造した場合には、平均円形度を上記範囲に制御することが容易になる。粉砕法の場合は、熱球形化処理や、表面改質及び微粉除去を行うことで、上記範囲に制御することが可能である。
機械的衝撃力を加える手段としては、例えば川崎重工(株)製のクリプトロンシステムやターボ工業社製のターボミル等の機械衝撃式粉砕機を用いる方法が挙げられる。また、ホソカワミクロン(株)製のメカノフージョンシステムや奈良機械製作所製のハイブリダイゼーションシステム等の装置のように、圧縮力、摩擦力等の力によりトナー粒子に機械的衝撃力を加える方法が挙げられる。
この懸濁重合法で得られるトナー粒子(以後「重合トナー粒子」ともいう)は、個々のトナー粒子形状がほぼ球形に揃っているため、平均円形度が0.960以上という本発明に好適な物性要件を満たすトナー粒子が得られやすい。さらにこういったトナー粒子は帯電量の分布も比較的均一となるために画質の向上が期待できる。
本発明において、上記懸濁重合法に使用される重合開始剤としては、重合反応時における半減期が0.5時間以上30.0時間以下であるものが好ましい。また、重合開始剤の添加量は重合性単量体100質量部に対して0.5質量部以上20.0質量部以下であることが好ましい。
上記懸濁重合法において、重合反応時に上記架橋剤を添加しても良く、好ましい添加量としては、重合性単量体100質量部に対して0.1質量部以上10.0質量部以下である。
ここで架橋剤としては、主として2個以上の重合可能な二重結合を有する化合物が好ましい。例えば、前述のように、芳香族ジビニル化合物、二重結合を2個有するカルボン酸エステル、ジビニル化合物、及び3個以上のビニル基を有する化合物が好ましい。これらを単独で、又は2種以上の混合物として用いることができる。
重合開始剤添加の時期としては、重合性単量体中に他の添加剤を添加する時に同時に加えても良いし、水系媒体中に懸濁する直前に混合しても良い。また、造粒直後、重合反応を開始する前に重合性単量体又は溶媒に溶解した重合開始剤を加えることもできる。
上記分散安定剤として公知の界面活性剤、有機分散剤又は無機分散剤が使用できる。中でも無機分散剤は、有害な超微粉を生じにくく、その立体障害性により分散安定性を得ているので反応温度を変化させても安定性が崩れにくく、洗浄も容易でトナー粒子に悪影響を与えにくいため、好ましく使用できる。こうした無機分散剤の例としては、リン酸三カルシウム、リン酸マグネシウム、リン酸アルミニウム、リン酸亜鉛、ヒドロキシアパタイト等のリン酸多価金属塩、炭酸カルシウム、炭酸マグネシウム等の炭酸塩、メタケイ酸カルシウム、硫酸カルシウム、硫酸バリウム等の無機塩、水酸化カルシウム、水酸化マグネシウム、水酸化アルミニウム等の無機化合物が挙げられる。
上記重合性単量体の重合反応における、重合温度は40℃以上一般には50℃以上90℃以下の温度に設定される。
上記重合性単量体の重合終了後、得られた重合体粒子を公知の方法によって濾過、洗浄、乾燥することによりトナー粒子が得られる。このトナー粒子に、無機微粒子であるシリカ微粒子およびチタン酸ストロンチウム微粒子を外添混合してトナー粒子の表面に付着させることで、本発明のトナーを得る。
また、製造工程(無機微粒子の混合前)に分級工程を入れ、トナー粒子中に含まれる粗粉や微粉を除去することも可能である。
そして、レーザー発生装置121によりレーザー光123を静電潜像担持体100に照射することによって露光が行われ、目的の画像に対応した静電潜像が形成される。静電潜像担持体100上の静電潜像は現像器140によって一成分トナーで現像されてトナー画像を得、トナー画像は転写材を介して静電潜像担持体に当接された転写ローラー114により転写材上へ転写される。トナー画像を載せた転写材は定着器126へ運ばれ転写材上に定着される。また、静電潜像担持体上に残された一部のトナーはクリーニングブレードによりかき落とされ、廃トナー容器116に収納される。
<シリカ微粒子の定量方法>
(1)トナー中のシリカ微粒子の含有量の定量(標準添加法)
トナー3gを直径30mmのアルミリングに入れ、10トンの圧力でペレットを作製する。そして、波長分散型蛍光X線分析(XRF)により、ケイ素(Si)の強度を求める(Si強度−1)。なお、測定条件は使用するXRF装置で最適化されたものであれば良いが、一連の強度測定はすべて同一条件で行うこととする。トナーに、一次粒子の個数平均粒径が12nmのシリカ微粒子を、トナーに対して1.0質量%添加して、コーヒーミルにより混合する。
混合後、上記と同様にペレット化した後に、上記同様にSiの強度を求める(Si強度−2)。同様の操作を、シリカ微粒子を、トナーに対して2.0質量%、3.0質量%添加混合したサンプルにおいても、Siの強度を求める(Si強度−3,Si強度−4)。Si強度−1〜4を用いて、標準添加法によりトナー中のシリカ含有量(質量%)を計算する。
トナーが磁性体を含有する場合、次の工程を経て、シリカ微粒子の定量を行う。
トナー5gを、精密天秤を用いて200mLの蓋付きポリカップに秤量し、メタノールを100mL加え、超音波分散機で5分間分散させる。ネオジム磁石によりトナーを引き付け、上澄み液を捨てる。メタノールによる分散と上澄みを捨てる操作を3回繰り返した後、下記の材料を加え、軽く混合した後、24時間静置する。
・10%NaOH 100mL
・「コンタミノンN」(非イオン界面活性剤、陰イオン界面活性剤、有機ビルダーからなるpH7の精密測定器洗浄用中性洗剤の10質量%水溶液、和光純薬工業(株)製)
数滴
その後、再びネオジム磁石を用いて分離する。なお、この際にNaOHが残留しないように繰り返し蒸留水ですすぐ。回収された粒子を真空乾燥機により十分に乾燥させ、粒子Aを得る。上記操作により、外添されたシリカ微粒子は溶解、除去される。
3gの粒子Aを直径30mmのアルミリングに入れ、10トンの圧力でペレットを作製し、波長分散型蛍光X線分析(XRF)により、Siの強度を求める(Si強度−5)。Si強度−5とトナー中のシリカ含有量の定量で使用したSi強度−1〜4を利用して、粒子A中のシリカ含有量(質量%)を計算する。
5gの粒子Aに対して、100mLのテトラヒドロフランを加え、良く混合した後に超音波分散を10分間行う。磁石により磁性粒子を引き付け、上澄み液を捨てる。この作業を5回繰り返し、粒子Bを得る。この操作で、磁性体以外の樹脂等の有機成分はほぼ取り除くことができる。ただし、樹脂中のテトラヒドロフラン不溶解分が残存する可能性がある。このため、上記操作で得られた粒子Bを温度800℃まで加熱して残存する有機成分を燃焼させることが好ましく、加熱後に得られた粒子Cを、トナーに含有されていた磁性体と近似することができる。
粒子Cの質量を測定することにより、トナー中の磁性体含有量W(質量%)とすることができる。この際、磁性体の酸化増量分を補正するために、粒子Cの質量に0.9666(Fe2O3→Fe3O4)を乗じる。
各定量値を以下の式に代入することにより、外添されたシリカ微粒子量を算出する。
外添されたシリカ微粒子量(質量%)=トナー中のシリカ含有量(質量%)−粒子A中のシリカ含有量(質量%)
酸化チタン微粒子の定量は、上記シリカ微粒子の定量方法と同様に、標準添加法により定量可能である。この際、長軸径が145nm、短軸径が50nmの酸化チタン微粒子を用いた。波長分散型蛍光X線分析(XRF)により、Tiの強度を使用することにより、定量可能である。
トナー粒子の表面のシリカ微粒子による被覆率X1は、以下のようにして算出する。
下記装置を下記条件にて使用し、トナー粒子の表面の元素分析を行う。
・測定装置:Quantum2000(商品名、アルバックファイ(株)製)
・X線源:モノクロAl KΑ
・Xray Setting:100μmφ(25W(15KV))
・光電子取りだし角:45度
・中和条件:中和銃とイオン銃の併用
・分析領域:300×200ΜM
・Pass Energy:58.70eV
・ステップサイズ:1.25eV
・解析ソフト:Maltipak(PHI社)
次いで、上述のトナー粒子の表面の元素分析と同様にして、シリカ微粒子単体の元素分析を行い、ここで得られたSi元素の定量値をY2とする。
被覆率X1(面積%)=Y1/Y2×100
なお、本測定の精度を向上させるために、Y1及びY2の測定を、2回以上行うことが好ましい。
定量値Y2を求めるに際して、外添に使用されたシリカ微粒子を入手できれば、それを用いて測定を行えばよい。
また、トナー粒子の表面から分離したシリカ微粒子を測定試料とする場合、シリカ微粒子のトナー粒子からの分離は以下の手順で行う。
まず、イオン交換水100mLに、コンタミノンN(非イオン界面活性剤、陰イオン界面活性剤、有機ビルダーからなるpH7の精密測定器洗浄用中性洗剤の10質量%水溶液、和光純薬工業(株)製)を6mL入れ分散媒を作製する。この分散媒に、トナー5gを添加し、超音波分散機で5分間分散させる。その後、いわき産業(株)製「KM Shaker」(model: V.SX)にセットし、1分当たり350往復の条件で20分間振盪する。
その後、ネオジム磁石を用いてトナー粒子を拘束し、上澄みを採取する。この上澄みを乾燥させることにより、シリカ微粒子を採集する。十分な量のシリカ微粒子を採集することができない場合には、この作業を繰り返して行う。
この方法では、シリカ微粒子以外の外添剤が添加されている場合には、シリカ微粒子以外の外添剤も採集される。このような場合には、採集された外添剤から、遠心分離法などを利用して、シリカ微粒子を選別すればよい。
イオン交換水100mLにスクロース(キシダ化学(株)製)160gを加え、湯せんをしながら溶解させショ糖濃厚液を調製する。遠心分離用チューブに該ショ糖濃厚液31gと、6mLのコンタミノンNを入れ、分散液を作製する。この分散液にトナー1gを添加し、スパチュラなどでトナーのかたまりをほぐす。
遠心分離用チューブを上記シェイカー(KM Shaker)にて1分当たり350往復の条件で20分間振盪する。振盪後、溶液をスイングローター用ガラスチューブ(50mL)に入れ替えて、遠心分離機にて、3500rpm、30分の条件で遠心分離を行う。遠心分離後のガラスチューブ内においては、最上層にはトナーが存在し、下層の水溶液側にはシリカ微粒子が存在する。下層の水溶液を採取して、遠心分離を行い、ショ糖とシリカ微粒子とを分離し、シリカ微粒子を採集する。必要に応じて、遠心分離を繰り返し行い、分離を十分に行った後、分散液を乾燥し、シリカ微粒子を採集する。
磁性トナーの場合と同様に、シリカ微粒子以外の外添剤が添加されている場合には、シリカ微粒子以外の外添剤も採集される。そのため、採集された外添剤から、遠心分離法などを利用して、シリカ微粒子を選別する。
トナーの重量平均粒径(D4)は、以下のようにして算出する(トナー粒子の場合も同様に算出する)。測定装置としては、100μmのアパーチャチューブを備えた細孔電気抵抗法による精密粒度分布測定装置「コールター・カウンター Multisizer 3」(登録商標、ベックマン・コールター(株)製)を用いる。測定条件の設定及び測定データの解析は、付属の専用ソフト「ベックマン・コールター Multisizer 3 Version3.51」(ベックマン・コールター(株)製)を用いる。なお、測定は実効測定チャンネル数2万5千チャンネルで行う。
測定に使用する電解水溶液は、特級塩化ナトリウムをイオン交換水に溶解して濃度が約1質量%となるようにしたもの、例えば、「ISOTON II」(ベックマン・コールター(株)製)が使用できる。
専用ソフトの「標準測定方法(SOM)を変更」画面において、コントロールモードの総カウント数を50000粒子に設定し、測定回数を1回、Kd値は「標準粒子10.0ΜM」(ベックマン・コールター(株)製)を用いて得られた値を設定する。「閾値/ノイズレベルの測定ボタン」を押すことで、閾値とノイズレベルを自動設定する。また、カレントを1600μAに、ゲインを2に、電解液をISOTON IIに設定し、「測定後のアパーチャチューブのフラッシュ」にチェックを入れる。
専用ソフトの「パルスから粒径への変換設定」画面において、ビン間隔を対数粒径に、粒径ビンを256粒径ビンに、粒径範囲を2μmから60μmまでに設定する。
(1)Multisizer 3専用のガラス製250mL丸底ビーカーに前記電解水溶液約200mLを入れ、サンプルスタンドにセットし、スターラーロッドの撹拌を反時計回りで24回転/秒にて行う。そして、専用ソフトの「アパーチャのフラッシュ」機能により、アパーチャチューブ内の汚れと気泡を除去しておく。
(2)ガラス製の100mL平底ビーカーに前記電解水溶液約30mLを入れる。この中に分散剤として「コンタミノンN」(非イオン界面活性剤、陰イオン界面活性剤、有機ビルダーからなるpH7の精密測定器洗浄用中性洗剤の10質量%水溶液、和光純薬工業(株)製)をイオン交換水で約3質量倍に希釈した希釈液を約0.3mL加える。
(4)前記(2)のビーカーを前記超音波分散器のビーカー固定穴にセットし、超音波分散器を作動させる。そして、ビーカー内の電解水溶液の液面の共振状態が最大となるようにビーカーの高さ位置を調整する。
(6)サンプルスタンド内に設置した前記(1)の丸底ビーカーに、ピペットを用いてトナーを分散した前記(5)の電解水溶液を滴下し、測定濃度が約5%となるように調整する。そして、測定粒子数が50000個になるまで測定を行う。
(7)測定データを装置付属の前記専用ソフトにて解析を行い、重量平均粒径(D4)を算出する。なお、専用ソフトでグラフ/体積%と設定したときの、「分析/体積統計値(算術平均)」画面の「平均径」が重量平均粒径(D4)である。
シリカ微粒子の一次粒子の個数平均粒径及び酸化チタン微粒子の長軸径及び短軸径は、日立超高分解能電界放出形走査電子顕微鏡S−4800((株)日立ハイテクノロジーズ)にて撮影されるトナー粒子の表面のシリカ微粒子及び酸化チタン微粒子画像から算出される。S−4800の画像撮影条件は以下の通りである。
試料台(アルミニウム試料台15mm×6mm)に導電性ペーストを薄く塗り、その上にトナーを吹きつける。さらにエアブローして、余分なトナーを試料台から除去し十分乾燥させる。試料台を試料ホルダにセットし、試料高さゲージにより試料台高さを36mmに調節する。
シリカ微粒子の一次粒子の個数平均粒径及び酸化チタン微粒子の長軸径及び短軸径の算出は、S−4800の反射電子像観察により得られた画像を用いて行う。反射電子像は二次電子像と比べてのチャージアップが少ないため、粒径を精度良く測定することができる。
S−4800の筺体に取り付けられているアンチコンタミネーショントラップに液体窒素を溢れるまで注入し、30分間置く。S−4800の「PCSTEM」を起動し、フラッシング(電子源であるFEチップの清浄化)を行う。画面上のコントロールパネルの加速電圧表示部分をクリックし、[フラッシング]ボタンを押し、フラッシング実行ダイアログを開く。
フラッシング強度が2であることを確認し、実行する。フラッシングによるエミッション電流が20〜40μAであることを確認する。試料ホルダをS−4800筺体の試料室に挿入する。コントロールパネル上の[原点]を押し試料ホルダを観察位置に移動させる。
加速電圧表示部をクリックしてHV設定ダイアログを開き、加速電圧を[0.8kV]、エミッション電流を[20μA]に設定する。オペレーションパネルの[基本]のタブ内にて、信号選択を[SE]に設置し、SE検出器を[上(U)]および[+BSE]を選択し、[+BSE]の右の選択ボックスで[L.A.100]を選択し、反射電子像で観察するモードにする。
同じくオペレーションパネルの[基本]のタブ内にて、電子光学系条件ブロックのプローブ電流を[NORMAL]に、焦点モードを[UHR]に、WDを[3.0mm]に設定する。コントロールパネルの加速電圧表示部の[ON]ボタンを押し、加速電圧を印加する。
コントロールパネルの倍率表示部内をドラッグして、倍率を100000(100k)倍に設定する。操作パネルのフォーカスつまみ[COARSE]を回転させ、ある程度焦点が合ったところでアパーチャアライメントの調整を行う。コントロールパネルの[ALIGN]をクリックし、アライメントダイアログを表示し、[ビーム]を選択する。操作パネルのSTIGMA/ALIGNMENTつまみ(X,Y)を回転し、表示されるビームを同心円の中心に移動させる。
次に[アパーチャ]を選択し、STIGMA/ALIGNMENTつまみ(X,Y)を一つずつ回し、像の動きを止める又は最小の動きになるように合わせる。アパーチャダイアログを閉じ、オートフォーカスで、ピントを合わせる。この操作をさらに2度繰り返し、ピントを合わせる。
その後、トナー粒子の表面上の少なくとも300個のシリカ微粒子の粒径を測定して、平均粒径を求める。ここで、シリカ微粒子は凝集塊として存在するものもあるため、一次粒子と確認できるものの最大径を求め、得られた最大径を算術平均することによって、シリカ微粒子の一次粒子の個数平均粒径(D1)を得る。
また、同様にトナー粒子の表面上の少なくとも300個の酸化チタン微粒子の長軸と短軸を測定し、それらの個数平均を求めることで、長軸径と短軸径を得る。また、ここで求めた長軸径を短軸径で除すことで、長軸径/短軸径を得る。
トナー粒子の平均円形度は、フロー式粒子像分析装置「FPIA−3000」(シスメックス(株)製)によって、校正作業時の測定及び解析条件で測定する。
具体的な測定方法は、以下の通りである。まず、ガラス製の容器中にあらかじめ不純固形物などを除去したイオン交換水約20mLを入れる。この中に分散剤として「コンタミノンN」(非イオン界面活性剤、陰イオン界面活性剤、有機ビルダーからなるpH7の精密測定器洗浄用中性洗剤の10質量%水溶液、和光純薬工業(株)製)をイオン交換水で約3質量倍に希釈した希釈液を約0.2mL加える。
さらに測定試料を約0.02g加え、超音波分散器を用いて2分間分散処理を行い、測定用の分散液とする。その際、分散液の温度が10℃以上40℃以下となる様に適宜冷却する。超音波分散器としては、発振周波数50kHz、電気的出力150Wの卓上型の超音波洗浄器分散器(例えば「VS−150」((株)ヴェルヴォクリーア製))を用い、水槽内には所定量のイオン交換水を入れ、この水槽中に前記コンタミノンNを約2mL添加する。
測定にあたっては、測定開始前に標準ラテックス粒子(例えば、Duke Scientific社製の「RESEARCH AND TEST PARTICLES Latex Microsphere Suspensions 5200A」をイオン交換水で希釈)を用いて自動焦点調整を行う。その後、測定開始から2時間毎に焦点調整を実施することが好ましい。
なお、本発明においては、シスメックス(株)による校正作業が行われた、シスメックス(株)が発行する校正証明書の発行を受けたフロー式粒子像測定装置を使用する。解析粒子径を円相当径1.985μm以上39.69μm未満に限定した以外は、校正証明を受けた時の測定及び解析条件で測定を行う。
フラットシースフローセル内を通過する試料に対しては、1/60秒間隔でストロボ光が照射されており、流れている粒子を静止画像として撮影することが可能である。また、扁平な流れであるため、焦点の合った状態で撮像される。粒子像はCCDカメラで撮像され、撮像された画像は512×512画素の画像処理解像度(一画素あたり0.37×0.37μm)で画像処理され、各粒子像の輪郭抽出を行い、粒子像の投影面積Sや周囲長L等が計測される。
次に、上記面積Sと周囲長Lを用いて円相当径と円形度を求める。円相当径とは、粒子像の投影面積と同じ面積を持つ円の直径のことであり、円形度は、円相当径から求めた円の周囲長を粒子投影像の周囲長で割った値として定義され、次式で算出される。
円形度=2×(π×S)1/2/L
粒子像が円形の時に円形度は1.000になり、粒子像の外周の凹凸の程度が大きくなればなるほど円形度は小さい値になる。各粒子の円形度を算出後、円形度0.200〜1.000の範囲を800分割し、得られた円形度の相加平均値を算出し、その値を平均円形度とする。
シリカ微粒子の見掛け密度の測定は、100mLのメスシリンダーに、紙の上にのせた測定試料をゆっくり加えて100mLになるようにし、試料を加える前と後のメスシリンダーの質量差を求め次式によって算出する。なお、試料をメスシリンダーに加える場合、紙を叩いたりしないよう注意する。
見掛け密度(g/L)=(100mL投入した時点の質量(g))/0.1
トナー及びシリカ微粒子の真比重は、乾式自動密度計オートピクノメーター(ユアサアイオニクス(株)製)により測定した。条件は下記の通りである。
セル :SMセル(10mL)
サンプル量 :約2.0g(トナー)、0.05g(シリカ微粒子)
この測定方法は、気相置換法に基づいて、固体・液体の真比重を測定するものである。液相置換法と同様、アルキメデスの原理に基づいているが、置換媒体としてガス(アルゴンガス)を用いるため、微細孔への精度が高い。
サンプルの準備
遊離前トナー:後述する実施例で作製した各種トナーをそのまま用いた。
遊離後トナー:50mL容量のバイアルに「コンタミノンN」(非イオン界面活性剤、陰イオン界面活性剤、有機ビルダーからなるpH7の精密測定器洗浄用中性洗剤の2質量%水溶液20gを秤量し、トナー1gと混合する。いわき産業(株)製「KM Shaker」(model: V.SX)にセットし、speedを50に設定して30秒間振盪する。その後、遠心分離機(1000rpmにて5分間)にて、トナーと水溶液を分離する。上澄液を分離し、沈殿しているトナーを真空乾燥することで乾固させて、サンプルとする。
外添剤除去トナー:外添剤除去トナーとは、この試験において遊離しうる外添剤を除いた状態を意味する。サンプル調製方法はイソプロパノールの如きトナーを溶かさない溶媒中にトナーを入れ、超音波洗浄機にて10分振動を与える。その後、遠心分離機(1000rpmにて5分間)にて、トナーと溶液を分離する。上澄液を分離し、沈殿しているトナーを真空乾燥することで乾固させてサンプルとする。
これらの遊離外添剤除去前後のサンプルについて、波長分散型蛍光X線分析(XRF)により、Tiの強度を使用することにより、酸化チタン微粒子の定量を行い、どの程度遊離したかを求めた。
蛍光X線分析装置3080(理学電気(株))
試料プレス成型機MAEKAWA Testing Machine(MFG Co,LTD製)
(II)測定条件
測定電位、電圧 50kV、50〜70mA
2θ角度 a
結晶板 LiF
測定時間 60秒
(III)トナーからの遊離率の算出方法について
まず、上記方法にて遊離前トナー、遊離後トナーおよび外添剤除去トナーの元素の強度を求める。その後、下記式に基づき遊離率を算出する。
[式]酸化チタン微粒子の遊離率=100−(遊離後トナーのTi元素の強度−外添剤除去トナーのTi元素の強度)/(遊離前トナーのTi元素の強度−外添剤除去トナーのTi元素の強度)×100
上記BET法で測定した窒素吸着による比表面積の測定は、JIS Z8830(2001年)に準じて行った。測定装置としては、定容法によるガス吸着法を測定方式として採用している「自動比表面積・細孔分布測定装置 TRISTAR3000((株)島津製作所製)」を用いることができる。
(磁性体1)
硫酸第一鉄水溶液中に、下記の材料を混合し、水酸化第一鉄を含む水溶液を調製した。
・鉄元素に対して1.00から1.10当量の苛性ソーダ溶液
・鉄元素に対しリン元素換算で0.12質量%となる量のP2O5
・鉄元素に対してケイ素元素換算で0.60質量%となる量のSiO2
水溶液のpHを8.0とし、空気を吹き込みながら温度85℃で酸化反応を行い、種晶を有するスラリー液を調製した。
次いで、このスラリー液に当初のアルカリ量(苛性ソーダのナトリウム成分)に対し0.90から1.20当量となるよう硫酸第一鉄水溶液を加えた。その後、スラリー液をpH7.6に維持して、空気を吹込みながら酸化反応をすすめ、磁性酸化鉄を含むスラリー液を得た。濾過、洗浄した後、この含水スラリー液を一旦取り出した。この時、含水サンプルを少量採取し、含水量を計っておいた。
次に、この含水サンプルを乾燥せずに別の水系媒体中に投入し、撹拌すると共にスラリーを循環させながらピンミルにて再分散させ、再分散液のpHを約4.8に調整した。そして、撹拌しながらn−ヘキシルトリメトキシシランカップリング剤を磁性酸化鉄100質量部に対し1.7質量部(磁性酸化鉄の量は含水サンプルから含水量を引いた値として計算した)添加し、加水分解を行った。
その後、撹拌を十分行い、分散液のpHを8.6にして表面処理を行った。生成した疎水性磁性体をフィルタープレスにて濾過し、多量の水で洗浄した後に温度100℃で15分、90℃で30分乾燥し、得られた粒子を解砕処理して体積平均粒径が0.23μmの磁性体1を得た。
磁性体1の製造例において、リン元素を添加せずに、ケイ素元素換算で0.40質量%となる量のSiO2を混合した以外は同様にして、スラリー液を調製し、磁性体1の製造例と同様に、酸化反応をすすめ、磁性酸化鉄を含むスラリー液を得た。
濾過、洗浄、乾燥した後、得られた粒子を解砕処理して体積平均粒径が0.21μmの磁性体2を得た。
冷却管、撹拌機及び窒素導入管の付いた反応槽中に、下記成分を入れ、温度230℃で窒素気流下に生成する水を留去しながら10時間反応させた。
・ビスフェノールA プロピレンオキサイド2モル付加物 75質量部
・ビスフェノールA プロピレンオキサイド3モル付加物 25質量部
・テレフタル酸 110質量部
・チタン系触媒 0.25質量部
(チタニウムジヒドロキシビス(トリエタノールアミネート))
次いで5〜20mmHgの減圧下に反応させ、酸価が2mgKOH/g以下になった時点で180℃に冷却し、無水トリメリット酸8質量部を加え、常圧密閉下2時間反応後取り出し、室温まで冷却後、粉砕してポリエステル樹脂1を得た。得られたポリエステル樹脂1は、ゲルパーミェーションクロマトグラフィ(GPC)で測定されたメインピーク分子量(Mp)が9500であった。
イオン交換水720質量部に0.1M−Na3PO4水溶液450質量部を投入して60℃に加温した後、1.0M−CaCl2水溶液67.7質量部を添加して、分散安定剤を含む水系媒体を得た。
・スチレン 78.0質量部
・n−ブチルアクリレート 22.0質量部
・ジビニルベンゼン 0.6質量部
・モノアゾ染料の鉄錯体(T−77:保土谷化学工業(株)) 2.0質量部
・磁性体1 90.0質量部
・ポリエステル樹脂1 3.0質量部
上記処方をアトライター(三井三池化工機(株))を用いて均一に分散混合して重合性単量体組成物を得た。得られた重合性単量体組成物を60℃に加温し、フィッシャートロプシュワックス(融点:74℃、数平均分子量Mn:500)15.0質量部を添加混合し、溶解した。溶解後に重合開始剤としてジラウロイルパーオキサイド7.0質量部を溶解し、トナー組成物を得た。
上記水系媒体中に上記トナー組成物を投入し、温度60℃、N2雰囲気下においてTK式ホモミキサー(特殊機化工業(株))にて12500rpmで12分間撹拌し、造粒した。その後パドル撹拌翼で撹拌しつつ温度74℃で6時間反応させた。
反応終了後、懸濁液を冷却し、塩酸を加えて洗浄した後に濾過・乾燥してトナー粒子1を得た。得られたトナー粒子1の物性を表1に示す。
トナー粒子の製造例1において、ホモミキサーの回転数を12500rpmから10500rpm及び9500rpmへ低下させること以外は同様にして、それぞれトナー粒子2及び3を製造した。得られたトナー粒子2及び3の物性を表1に示す。
・スチレンアクリル共重合体 100質量部
(スチレンとn−ブチルアクリレートの質量比が78.0:22.0、メインピーク分子量Mpが10000)
・磁性体2 90質量部
・モノアゾ染料の鉄錯体(T−77:保土谷化学工業(株)) 2.0質量部
・フィッシャートロプシュワックス 4質量部
(融点:74℃、数平均分子量Mn:500)
このトナー粒子Aに対し熱球形化処理を行った。熱球形化処理はサーフュージングシステム(日本ニューマチック(株)製)を使用して行った。熱球形化装置の運転条件は、フィード量=5kg/時間、熱風温度C=260℃、熱風流量=6m3/分、冷風温度E=5℃、冷風流量=4m3/分、冷風絶対水分量=3g/m3、ブロワー風量=20m3/分、インジェクションエア流量=1m3/分、拡散エア=0.3m3/分とした。
上記条件の表面処理によって、重量平均粒径(d4)8.2μmであるトナー粒子4を得た。得られたトナー粒子4の物性を表1に示す。
トナー粒子の製造例4において、得られたトナー粒子Aを、表面改質装置ファカルティー(ホソカワミクロン(株)製)で表面改質及び微粉除去を行い、トナー粒子5を得た。表面改質装置ファカルティーを用いた表面改質及び微粉除去の条件は、以下のとおりとした。
・分散ローターの回転周速 200m/秒
・微粉砕品の投入量 1サイクル当たり6kg
・表面改質時間(サイクルタイム:原料供給が終了してから排出弁が開くまでの時間)
90秒間
またトナー粒子排出時の温度は45℃であった。得られたトナー粒子5の物性を表1に示す。なお、トナー粒子1〜5について、真密度を測定した結果、いずれも1.6g/cm3であった。
撹拌機付きオートクレーブに、未処理の乾式シリカ(平均1次粒径=9nm)を投入し、撹拌による流動化状態において、200℃に加熱した。
反応器内部を窒素ガスで置換して反応器を密閉し、乾式シリカ100質量部に対し、25質量部のヘキサメチルジシラザンを内部に噴霧し、シリカの流動化状態でシラン化合物処理を行なった。この反応を60分間継続した後、反応を終了した。反応終了後、オートクレーブを脱圧し、窒素ガス気流による洗浄を行い、疎水性シリカから過剰のヘキサメチルジシラザン及び副生物を除去した。
さらに、反応槽内を撹拌しながら乾式シリカ100質量部に対し、10質量部のジメチルシリコーンオイル(粘度=100mm2/秒)を噴霧し、30分間攪拌を続けた。その後、攪拌しながら300℃まで昇温させてさらに2時間攪拌して後に取り出し、解砕処理を実施し、シリカ微粒子1を得た。シリカ微粒子1の物性を表2に示す。
シリカ微粒子の製造例1において、使用する未処理シリカの粒径を変更し、解砕処理強度を適宜調整した以外は同様にして、シリカ微粒子2〜7を得た。シリカ微粒子2〜7の物性を表2に示す。なお、シリカ微粒子1〜7について、真密度を測定した結果、いずれも2.2g/cm3であった。
出発原料としてTiO2相当分を50質量%含有しているイルメナイト鉱石を使用した。この原料を温度150℃で2時間乾燥させた後、硫酸を添加して溶解させることによって、TiOSO2の水溶液を得た。この水溶液に炭酸ナトリウムを加えてpH9.0に調整してアルカリ中和を行い、濾過することにより白色沈殿物を得た。この白色沈殿物に純水を加えて温度約90℃に保ちながら2.5時間加熱処理して加水分解処理を行い、濾過・水洗浄を繰り返し行うことでアナターゼ型の酸化チタンを得た。
得られたアナターゼ型の酸化チタンを1100℃の高温加熱によって焼結させることによってルチル型の酸化チタンを得た。このルチル型の酸化チタンをジェットミルにて解砕処理を行い、酸化チタン微粒子を得た。この酸化チタン微粒子をエタノール中に分散させ、酸化チタン微粒子100質量部に対して、疎水化剤としてi−ブチルトリメトキシシランを固形分で10質量部を粒子の合一が生じないように十分に撹拌しながら滴下混合し、反応させて疎水化処理を行った。
さらに十分に撹拌しながら、スラリーのpHを6.5に調整した。これを濾過、乾燥した後、温度170℃で2時間加熱処理し、その後、酸化チタンの凝集体がなくなるまで繰り返しジェットミルにより解砕処理を行い、酸化チタン微粒子1を得た。
酸化チタン微粒子の製造例1において、加水分解する温度と時間の変更、ジェットミルでの解砕処理条件、疎水化処理で使用したi−ブチルトリメトキシシランの添加量を変更した以外は同様にして作製を行い、ルチル型の酸化チタン微粒子2〜16を得た。酸化チタン微粒子2〜16の物性を表3に示す。
酸化チタン微粒子1の製造例において、加水分解する温度と時間を変更し、1100℃の高温加熱による焼結温度を700℃に変更して、アナターゼ型の酸化チタンを得た。そしてジェットミルでの解砕処理条件、疎水化処理で使用したi−ブチルトリメトキシシランの添加量を変更した以外は同様にして、アナターゼ型の酸化チタン微粒子17を得た。酸化チタン微粒子17の物性を表3に示す。
トナー粒子の製造例1で得たトナー粒子1に対して、図3に示す装置を用いて、外添混合処理を行った。
本実施例においては、図3に示す装置で、本体ケーシング1の内周部の径が130mmであり、処理空間9の容積が2.0×10−3m3の装置を用い、駆動部8の定格動力を5.5kWとし、攪拌部材3の形状を図4のものとした。そして、図4における攪拌部材3aと攪拌部材3bの重なり幅dを攪拌部材3の最大幅Dに対して0.25Dとし、攪拌部材3と本体ケーシング1の内周とのクリアランスを3.0mmとした。
上記した装置構成で、トナー粒子1の100質量部と、シリコーンオイルとシランカップリング剤で疎水化処理したシリカ微粒子1の0.40質量部と、酸化チタン微粒子1の0.30質量部とを、図3に示す装置に投入した。
トナー粒子とシリカ微粒子および酸化チタン微粒子を投入後、トナー粒子とシリカ微粒子および酸化チタン微粒子を均一に混合するために、プレ混合を実施した。プレ混合の条件は、駆動部8の動力を0.10W/g(駆動部8の回転数150rpm)とし、処理時間を1分間とした。
(一段目の外添混合)
プレ混合終了後、外添混合処理を行った。外添混合処理条件は、駆動部8の動力を0.60W/g(駆動部8の回転数1400rpm)で一定となるように、攪拌部材3の最外端部周速を調整し、処理時間を3分間とした。
(二段目の外添混合)
その後シリカ微粒子1を0.10質量部添加(トナー粒子に対して、トータル0.50質量部)し、駆動部8の動力を0.60W/g(駆動部8の回転数1400rpm)で一定となるように、攪拌部材3の最外端部周速を調整し、さらに処理を2分間実施とした。
つまり、酸化チタン微粒子1の含有量(0.30質量部)に対する、シリカ微粒子1の含有量(0.50質量部)の比は、5/3=1.7とした。
実施例用トナー1の製造例において、表2、表3又は表4に示す、外添剤の種類及び添加部数、トナー粒子、外添装置、外添条件等へ変更した以外は同様にして、トナー2〜39、及び比較トナー1〜16を製造した。得られたトナー2〜39、及び比較トナー1〜16の外添条件を表4及び表5に、物性を表6にそれぞれ示す。
ここで、外添装置としてヘンシェルミキサーを使用する場合、ヘンシェルミキサーFM10C(三井三池化工機(株))を用いた。また、一部の製造例においては、プレ混合工程を行わなかった。
一段目の外添条件:動力は全て0.60(W/g)、回転数は全て1400(rpm)
二段目の外添条件:動力は全て0.60(W/g)、回転数は全て1400(rpm)
(画像形成装置)
画像形成装置として、直径10mmであるスリーブを搭載した、LBP−3100(キヤノン(株)製)を用い、外部電源を接続して転写バイアスを変えられるように改造した。転写バイアスを低くすると、上述したように高温高湿環境下で作られた吸湿紙のような体積抵抗の低い媒体では、転写抜けが発生しやすい。また、トナー充填部の容積を1.5倍に改造した。
容積を1.5倍にすることで、トナー劣化を促進し、画像濃度やかぶりを厳しく評価することができる。この改造機を用いて通常の転写バイアス(0.5kV)でトナー1を使用して、高温高湿環境下(温度32.5℃/相対湿度80%RH)にて、ベタ黒画像を1枚出力した。その後、印字率が2%の横線を1枚間欠モードで2000枚画出し試験を行い、2000枚の画出後、ベタ黒画像を1枚画出しした。その後、転写バイアスを0.3kVに設定し、後述する吸湿紙を用いてベタ黒画像を1枚出力した。
また、かぶりの評価は上述した改造機を用いて、常温常湿環境下(温度23℃/相対湿度50%RH)にて、ベタ白画像を一枚出力して評価を行った。
本発明の実施例及び比較例で行った各評価の評価方法とその判断基準について以下に述べる。
画像濃度はベタ黒画像部を形成し、このベタ黒画像の濃度をマクベス反射濃度計(マクベス社製)にて測定した。紙はOCE RED LABEL (A4,80g/cm2)を使用した。なお、紙は画出し直前に紙袋から開封した状態で用いた。
耐久使用初期(1枚目)における、ベタ黒画像の反射濃度の判断基準は以下の通りである。
A:非常に良好(1.45以上)
B:良好(1.40以上1.45未満)
C:普通(1.35以上1.40未満)
D:悪い(1.35未満)
耐久使用初期のベタ黒画像の反射濃度と、2000枚耐久使用後のベタ黒画像の反射濃度の差が、小さいほど良好とした。
A:非常に良好(差が0.10未満)
B:良好(差が0.10以上0.15未満)
C:普通(差が0.15以上0.20未満)
D:悪い(差が0.20以上)
ベタ白画像を出力して、その反射率を東京電色社製のREFLECTMETER MODEL TC−6DSを使用して測定した。一方、ベタ白画像形成前の転写紙(標準紙)についても同様に反射率を測定した。フィルターは、グリーンフィルターを用いた。ベタ白画像出力前後の反射率から、下記式を用いてかぶりを算出した。
かぶり(反射率)(%)=標準紙の反射率(%)−ベタ白画像サンプルの反射率(%)
なお、かぶりの判断基準は以下の通りである。
A:非常に良好(1.0%未満)
B:良好(1.0%以上1.5%未満)
C:普通(1.5%以上2.5%未満)
D:悪い(2.5%以上)
紙 OCE RED LABEL (A4,80g/cm2)を高温高湿環境下(温度32.5℃/相対湿度80%RH)に紙袋から開封した状態で48時間静置した。転写バイアスを0.3kVに変更して、上記の2000枚耐久使用後にベタ黒画像を出力し、目視で評価を行った。このように高温高湿環境下で静置した紙を評価することで、厳しく転写性を評価することができる。
A:非常に良好(転写抜け未発生)
B:濃度のムラが一部あるが、実用的には問題のない画像
C:濃度のムラが見られるが、実用的には問題の無い画像
D:明確な濃度ムラが見られる。実用上好ましくない画像
E:ベタ黒画像上に白く抜けた部分が見られる。実用好ましくない画像
トナー2〜39を用いたこと以外は実施例1と同様に画出し試験を行った。その結果、いずれのトナーも耐久試験前後で実用上問題ないレベル以上の画像が得られた。評価結果を表7に示す。
<比較例1〜16>
比較トナー1〜16を用いたこと以外は、実施例1と同様に画出し試験を行った。評価結果を表7に示す。
Claims (5)
- 結着樹脂及び着色剤を含有するトナー粒子と、
外添剤としての、酸化チタン微粒子及びシリカ微粒子と、
を有するトナーであって、
前記酸化チタン微粒子は、
長軸径が30nm以上500nm以下であり、
短軸径が10nm以上80nm以下であり、
長軸径/短軸径の比が2.0以上8.0以下である
ルチル型の酸化チタン微粒子であり、
前記酸化チタン微粒子の含有量が、前記トナー粒子100質量部当たり0.1質量部以上1.0質量部以下であり、
前記トナー中の、前記酸化チタン微粒子の含有量に対する前記シリカ微粒子の含有量の比が、0.4以上30.0以下であり、
X線光電子分光装置(ESCA)により求めた、前記トナー粒子の表面の前記シリカ微粒子による被覆率X1が、40.0面積%以上75.0面積%以下であり、
前記シリカ微粒子による理論被覆率をX2としたとき、下記式1で示される拡散指数が、下記式2を満たし、
前記トナー粒子への前記外添剤の埋め込み率が、25%以上60%以下である
ことを特徴とするトナー。
(式1)拡散指数=X1/X2
(式2)拡散指数≧−0.0042×X1+0.62 - 前記トナー粒子の平均円形度が、0.960以上である請求項1に記載のトナー。
- 前記酸化チタン微粒子の遊離率が、50%以下である請求項1又は2に記載のトナー。
- 前記シリカ微粒子の見掛け密度が、15g/L以上50g/L以下である請求項1〜3のいずれか1項に記載のトナー。
- 前記トナー粒子が、重合性単量体及び着色剤を含有する重合性単量体組成物の粒子を水系媒体中で形成し、前記粒子中に含有される前記重合性単量体を重合させて得られたトナー粒子である請求項1〜4のいずれか1項に記載のトナー。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2013270112A JP6272024B2 (ja) | 2013-12-26 | 2013-12-26 | トナー |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2013270112A JP6272024B2 (ja) | 2013-12-26 | 2013-12-26 | トナー |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| JP2015125302A true JP2015125302A (ja) | 2015-07-06 |
| JP2015125302A5 JP2015125302A5 (ja) | 2017-02-09 |
| JP6272024B2 JP6272024B2 (ja) | 2018-01-31 |
Family
ID=53536029
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2013270112A Active JP6272024B2 (ja) | 2013-12-26 | 2013-12-26 | トナー |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP6272024B2 (ja) |
Cited By (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2016038592A (ja) * | 2014-08-07 | 2016-03-22 | キヤノン株式会社 | トナー及びトナーの製造方法 |
| JP2017068006A (ja) * | 2015-09-30 | 2017-04-06 | コニカミノルタ株式会社 | 静電荷像現像用トナー、静電荷像現像用二成分現像剤 |
| JP2018072694A (ja) * | 2016-11-02 | 2018-05-10 | コニカミノルタ株式会社 | 静電荷像現像用トナー |
| JP2018084645A (ja) * | 2016-11-22 | 2018-05-31 | コニカミノルタ株式会社 | 静電潜像現像剤および静電潜像現像剤の製造方法 |
| CN108508719A (zh) * | 2017-02-28 | 2018-09-07 | 佳能株式会社 | 调色剂 |
| JP2018180146A (ja) * | 2017-04-07 | 2018-11-15 | コニカミノルタ株式会社 | 静電荷像現像用トナー及び静電荷像現像用トナーの製造方法 |
| JP2018194614A (ja) * | 2017-05-15 | 2018-12-06 | キヤノン株式会社 | トナー |
| EP3470927A4 (en) * | 2016-06-09 | 2020-01-08 | C/o Canon Kabushiki Kaisha | TONER |
| CN110998458A (zh) * | 2017-08-04 | 2020-04-10 | 佳能株式会社 | 调色剂 |
| JP7627171B2 (ja) | 2021-05-17 | 2025-02-05 | 東芝テック株式会社 | トナー、トナーカートリッジ、画像形成装置 |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH0566608A (ja) * | 1991-03-08 | 1993-03-19 | Canon Inc | 磁性トナー、画像形成方法、表面改質シリカ微粉末及びその製造方法 |
| JP2005055733A (ja) * | 2003-08-06 | 2005-03-03 | Canon Inc | トナー |
| JP2013134447A (ja) * | 2011-12-27 | 2013-07-08 | Canon Inc | 磁性トナー |
-
2013
- 2013-12-26 JP JP2013270112A patent/JP6272024B2/ja active Active
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH0566608A (ja) * | 1991-03-08 | 1993-03-19 | Canon Inc | 磁性トナー、画像形成方法、表面改質シリカ微粉末及びその製造方法 |
| JP2005055733A (ja) * | 2003-08-06 | 2005-03-03 | Canon Inc | トナー |
| JP2013134447A (ja) * | 2011-12-27 | 2013-07-08 | Canon Inc | 磁性トナー |
Cited By (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2016038592A (ja) * | 2014-08-07 | 2016-03-22 | キヤノン株式会社 | トナー及びトナーの製造方法 |
| JP2017068006A (ja) * | 2015-09-30 | 2017-04-06 | コニカミノルタ株式会社 | 静電荷像現像用トナー、静電荷像現像用二成分現像剤 |
| US9823596B2 (en) | 2015-09-30 | 2017-11-21 | Konica Minolta, Inc. | Electrostatic image developing toner and two-component electrostatic image developer |
| US10620556B2 (en) | 2016-06-09 | 2020-04-14 | Canon Kabushiki Kaisha | Toner |
| EP3470927A4 (en) * | 2016-06-09 | 2020-01-08 | C/o Canon Kabushiki Kaisha | TONER |
| JP2018072694A (ja) * | 2016-11-02 | 2018-05-10 | コニカミノルタ株式会社 | 静電荷像現像用トナー |
| JP2018084645A (ja) * | 2016-11-22 | 2018-05-31 | コニカミノルタ株式会社 | 静電潜像現像剤および静電潜像現像剤の製造方法 |
| CN108508719A (zh) * | 2017-02-28 | 2018-09-07 | 佳能株式会社 | 调色剂 |
| CN108508719B (zh) * | 2017-02-28 | 2022-11-15 | 佳能株式会社 | 调色剂 |
| JP2018180146A (ja) * | 2017-04-07 | 2018-11-15 | コニカミノルタ株式会社 | 静電荷像現像用トナー及び静電荷像現像用トナーの製造方法 |
| JP2018194614A (ja) * | 2017-05-15 | 2018-12-06 | キヤノン株式会社 | トナー |
| CN110998458A (zh) * | 2017-08-04 | 2020-04-10 | 佳能株式会社 | 调色剂 |
| JP7627171B2 (ja) | 2021-05-17 | 2025-02-05 | 東芝テック株式会社 | トナー、トナーカートリッジ、画像形成装置 |
| US12416873B2 (en) | 2021-05-17 | 2025-09-16 | Toshiba Tec Kabushiki Kaisha | Toner, toner cartridge, image forming apparatus |
Also Published As
| Publication number | Publication date |
|---|---|
| JP6272024B2 (ja) | 2018-01-31 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6399804B2 (ja) | トナー | |
| JP6272024B2 (ja) | トナー | |
| US9857707B2 (en) | Toner | |
| JP6429616B2 (ja) | 磁性トナー | |
| JP6910805B2 (ja) | トナー、画像形成装置及び画像形成方法 | |
| JP6497907B2 (ja) | トナー | |
| JP6486181B2 (ja) | 画像形成装置、プロセスカートリッジおよび画像形成方法 | |
| US9658546B2 (en) | Toner and method of producing toner | |
| JP6289432B2 (ja) | トナー及びトナーの製造方法 | |
| JP2019032364A (ja) | トナー | |
| JP5230297B2 (ja) | トナー | |
| JP6300508B2 (ja) | トナー、トナーの製造方法 | |
| JP6448395B2 (ja) | トナーおよびトナーの製造方法 | |
| JP6415171B2 (ja) | トナー | |
| JP2018036596A (ja) | トナー、トナーの製造方法、トナーを用いたプロセスカートリッジ及び画像形成方法 | |
| JP6184198B2 (ja) | トナー | |
| JP6272020B2 (ja) | トナーの製造方法 | |
| JP6762700B2 (ja) | トナー | |
| JP6381424B2 (ja) | 画像形成装置および画像形成方法 | |
| JP2008015230A (ja) | トナー | |
| JP5159497B2 (ja) | 磁性トナー | |
| JP5645583B2 (ja) | トナー |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20161222 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20161222 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20170726 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20170801 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20171205 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20171228 |
|
| R151 | Written notification of patent or utility model registration |
Ref document number: 6272024 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R151 |
|
| RD03 | Notification of appointment of power of attorney |
Free format text: JAPANESE INTERMEDIATE CODE: R3D03 |