JP2016027005A - 2,3,3,3−テトラフルオロプロペンおよび1,1−ジフルオロエチレンの製造方法 - Google Patents
2,3,3,3−テトラフルオロプロペンおよび1,1−ジフルオロエチレンの製造方法 Download PDFInfo
- Publication number
- JP2016027005A JP2016027005A JP2012256211A JP2012256211A JP2016027005A JP 2016027005 A JP2016027005 A JP 2016027005A JP 2012256211 A JP2012256211 A JP 2012256211A JP 2012256211 A JP2012256211 A JP 2012256211A JP 2016027005 A JP2016027005 A JP 2016027005A
- Authority
- JP
- Japan
- Prior art keywords
- reactor
- methane
- tfe
- raw material
- tetrafluoropropene
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Images

Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C17/00—Preparation of halogenated hydrocarbons
- C07C17/26—Preparation of halogenated hydrocarbons by reactions involving an increase in the number of carbon atoms in the skeleton
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
【課題】熱分解を伴う1回の反応で、新冷媒として有用なHFO−1234yfを十分に高い収率で製造する経済的に有利な方法を提供する。
【解決手段】熱媒体存在下、R22および/またはTFEと、メタンとを含む原料組成物から、熱分解を伴う合成反応によりHFO−1234yfおよびVdFを製造する方法であって、(a)前記R22および/または前記TFEと前記メタンとを、予め混合してまたは別々に、反応器に供給し、該反応器内に所定の時間滞留させる工程と、(b)熱媒体を反応器に供給し、該反応器内で前記原料組成物と前記熱媒体とを接触させる工程を有する製造方法を提供する。
【選択図】図1
【解決手段】熱媒体存在下、R22および/またはTFEと、メタンとを含む原料組成物から、熱分解を伴う合成反応によりHFO−1234yfおよびVdFを製造する方法であって、(a)前記R22および/または前記TFEと前記メタンとを、予め混合してまたは別々に、反応器に供給し、該反応器内に所定の時間滞留させる工程と、(b)熱媒体を反応器に供給し、該反応器内で前記原料組成物と前記熱媒体とを接触させる工程を有する製造方法を提供する。
【選択図】図1
Description
本発明は、2,3,3,3−テトラフルオロプロペンおよび1,1−ジフルオロエチレンの製造方法に係り、特に、メタンを含む原料組成物から、1回の反応で2,3,3,3−テトラフルオロプロペンおよび1,1−ジフルオロエチレンを製造する方法に関する。
2,3,3,3−テトラフルオロプロペン(HFO−1234yf)は、オゾン層破壊係数が0(ゼロ)で地球温暖化係数が4と非常に低く、温室効果ガスである1,1,1,2−テトラフルオロエタン(HFC−134a)に代わる新しい冷媒として、近年大きな期待が寄せられている。なお、本明細書において、ハロゲン化炭化水素については、化合物名の後の括弧内にその化合物の略称を記すが、本明細書では必要に応じて化合物名に代えてその略称を用いる。
HFO−1234yfの製造方法としては、例えば、1,1−ジクロロ−2,2,3,3,3−ペンタフルオロプロパン(HCFC−225ca)を相間移動触媒の存在下にアルカリ水溶液で脱フッ化水素させて得られる1,1−ジクロロ−2,3,3,3−テトラフルオロプロペン(CFO−1214ya)を合成原料とし、水素により還元する方法が知られている。
しかし、このような方法では、多段階の反応を経るため設備コストが高くなる、中間生成物や最終生成物における蒸留・精製が難しい、などの問題がある。
特許文献1には、メタンと炭素数2のハイドロクロロカーボン(例えば、1−クロロ−1,2,2,2−テトラフルオロエタン)とを空気で希釈した混合物を、ニッケル等の特定の金属触媒の存在下600〜650℃に加熱して反応させ、HFO-1234yfを製造する方法が提示されている。
また、熱分解を伴う1回の反応でフルオロカーボンを製造する方法として、非特許文献1には、メタンとクロロジフルオロメタンを窒素で希釈した混合物を、反応器内で電気ヒータのような通常の加熱手段により400〜800℃の温度に加熱・分解して、フッ素樹脂原料等として工業的に有用な1,1−ジフルオロエチレン(VdF)を得る方法が提示されている。
しかしながら、特許文献1に示された方法では、特殊な金属触媒を調製する必要があり、工程が複雑化する、原料であるハイドロクロロカーボンに入手しにくいものが多い、などの問題があった。
また、非特許文献1には、入手の容易なメタンとクロロジフルオロメタンを用いて1,1−ジフルオロエチレン(VdF)を得る方法が提示されているが、この方法ではHFO-1234yfの製造には至っていなかった。
Ind.Eng.Chem.Res.2010,49,6010
本発明は、上記観点からなされたものであり、調達の容易な原料を使用し、熱分解を伴う1回の反応工程で、工業的に有用なHFO−1234yfおよびVdFを、十分に制御された状態でかつ効率よく製造する経済的に有利な方法を提供することを目的とする。
本発明は、熱媒体存在下、クロロジフルオロメタン(R22)および/またはテトラフルオロエチレン(TFE)と、メタンを含む原料組成物から、熱分解を伴う合成反応によりHFO−1234yfおよびVdFを製造する方法であって、(a)前記R22および/または前記TFEと、前記メタンとを、予め混合してまたは別々に反応器に供給し、該反応器内に所定の時間滞留させる工程と、(b)熱媒体を前記反応器に供給し、該反応器内で前記原料組成物と前記熱媒体とを接触させる工程とを有することを特徴とするHFO−1234yfおよびVdFの製造方法を提供する。
本発明の製造方法によれば、調達が容易なR22および/またはTFEと、メタンとを原料として、中間生成物を反応系から取り出すことなくそのまま反応させ、工業的に有用なHFO−1234yfおよびVdFを効率よく製造することができる。したがって、例えば、HCFC−225caを原料としてCFO−1214yaを経由してHFO−1234yfを製造する方法に比べて、原料および製造設備に要するコストを大幅に低減することができる。
また、本発明の製造方法によれば、製造(反応)条件の制御が容易であり、よって定量的なHFO−1234yfおよびVdFの製造が可能となり経済的なメリットが大きい。またさらに、副生物のリサイクルも可能であり、経済的な効果が大きい。
以下に、本発明の実施の形態について説明する。
本発明は、原料として、クロロジフルオロメタン(R22)とテトラフルオロエチレン(TFE)の少なくとも一方と、メタンを含む組成物を用い、熱媒体の存在下で、熱分解を伴う合成反応により、HFO−1234yfおよびVdFを製造する方法を提供する。そして、この製造方法は、
(a)前記R22とTFEの少なくとも一方とメタンとを、予め混合しまたは別々に、反応器に供給し、該反応器内に所定の時間滞留させる工程と、
(b)熱媒体を前記反応器に供給し、該反応器内で前記原料組成物と前記熱媒体とを接触させる工程とを有する。
本発明は、原料として、クロロジフルオロメタン(R22)とテトラフルオロエチレン(TFE)の少なくとも一方と、メタンを含む組成物を用い、熱媒体の存在下で、熱分解を伴う合成反応により、HFO−1234yfおよびVdFを製造する方法を提供する。そして、この製造方法は、
(a)前記R22とTFEの少なくとも一方とメタンとを、予め混合しまたは別々に、反応器に供給し、該反応器内に所定の時間滞留させる工程と、
(b)熱媒体を前記反応器に供給し、該反応器内で前記原料組成物と前記熱媒体とを接触させる工程とを有する。
本発明の製造方法は、連続式の製造方法であっても、バッチ式の製造方法であってもよい。連続式の製造方法において、R22および/またはTFEと、メタンとを含む原料組成物の反応器への供給と、熱媒体の反応器への供給は、いずれも連続的に行われ、(a)工程と(b)工程とは同時に行われる。
バッチ式の製造において、反応器への供給は、原料組成物と熱媒体のどちらが先であっても、あるいは同時であってもよい。すなわち、原料組成物と熱媒体のいずれか一方の供給の際に、他方が反応器内に供給されていない場合でも、先に供給された原料組成物または熱媒体の滞留中に、後から供給される成分が供給され、原料組成物と熱媒体とが反応器内で所定の時間接触すればよい。反応器への原料供給開始時の原料成分のカーボン化の可能性を低くするという観点からは、希釈ガスでもある熱媒体が先に反応器内に供給されている方が好ましい。
バッチ式の製造において、反応器への供給は、原料組成物と熱媒体のどちらが先であっても、あるいは同時であってもよい。すなわち、原料組成物と熱媒体のいずれか一方の供給の際に、他方が反応器内に供給されていない場合でも、先に供給された原料組成物または熱媒体の滞留中に、後から供給される成分が供給され、原料組成物と熱媒体とが反応器内で所定の時間接触すればよい。反応器への原料供給開始時の原料成分のカーボン化の可能性を低くするという観点からは、希釈ガスでもある熱媒体が先に反応器内に供給されている方が好ましい。
本発明の製造方法は、製造効率の点で連続式の方法であるのが好ましい。以下、特にバッチ式と断らない限り、本発明の方法を連続式の製造に適用する実施形態について説明するが、連続式に限定されるわけではない。
<HFO−1234yfの生成反応>
本発明において、原料組成物がR22とTFEの両方とメタンを含有する場合は、反応器内で以下の式(1)に示す熱分解および脱塩化水素を伴う合成反応が生起し、HFO−1234yfおよびVdFが生成する。
なお、原料組成物が、R22とTFEの両方を含有せず、一方の成分のみを含有する場合にも、同様の反応が生起し、HFO−1234yfとVdFが生成する。
本発明において、原料組成物がR22とTFEの両方とメタンを含有する場合は、反応器内で以下の式(1)に示す熱分解および脱塩化水素を伴う合成反応が生起し、HFO−1234yfおよびVdFが生成する。
なお、原料組成物が、R22とTFEの両方を含有せず、一方の成分のみを含有する場合にも、同様の反応が生起し、HFO−1234yfとVdFが生成する。
R22および/またはTFEと、メタンを含む原料組成物は、反応器内で熱分解および脱塩化水素反応により、ジフルオロカルベン(F2C:)およびTFEとメタンとを含む反応混合物を生成し、これらの反応混合物は、直接付加反応して、あるいは1種または2種以上の中間体を経て、テトラフルオロプロペン、特にHFO−1234yfへと転化されると考えられる。
<原料組成物>
本発明のHFO−1234yfおよびVdFの製造に用いられる原料組成物は、R22とTFEの少なくとも一方と、メタンをそれぞれ含有する。原料組成物は、前記2成分または3成分以外に、反応器内で熱媒体との接触により分解してジフルオロカルベン(F2C:)を発生し得る化合物、例えば、ヘキサフルオロプロペン(以下、HFPという。)、CTFE、トリフルオロエチレン、オクタフルオロシクロブタン(以下、RC318という。)、ヘキサフルオロプロペンオキサイド等を含有することができる。
本発明のHFO−1234yfおよびVdFの製造に用いられる原料組成物は、R22とTFEの少なくとも一方と、メタンをそれぞれ含有する。原料組成物は、前記2成分または3成分以外に、反応器内で熱媒体との接触により分解してジフルオロカルベン(F2C:)を発生し得る化合物、例えば、ヘキサフルオロプロペン(以下、HFPという。)、CTFE、トリフルオロエチレン、オクタフルオロシクロブタン(以下、RC318という。)、ヘキサフルオロプロペンオキサイド等を含有することができる。
原料成分の一つであるメタンの供給量と、R22とTFEの供給量の合計とのモル比(以下、メタン/(R22+TFE)と示す。)は、0.01〜10の範囲とするのが好ましい。0.2〜5の範囲がより好ましく、0.25〜4の範囲が特に好ましい。メタン/(R22+TFE)を0.01〜10とすることにより、メタンの転化率を上げ、HFO−1234yfを高い収率で製造することができる。
なお、原料組成物がR22とTFEの一方の成分のみを含有する場合には、他方の成分の供給量を0(ゼロ)とし、前記範囲になるようにメタンの供給量を調整する。また、原料組成物および熱媒体を、反応器内を連続的に流通させて反応を行わせる連続式製造の実施形態において、原料各成分および熱媒体の供給量は、単位時間当たりの供給量を示すものとする。
なお、原料組成物がR22とTFEの一方の成分のみを含有する場合には、他方の成分の供給量を0(ゼロ)とし、前記範囲になるようにメタンの供給量を調整する。また、原料組成物および熱媒体を、反応器内を連続的に流通させて反応を行わせる連続式製造の実施形態において、原料各成分および熱媒体の供給量は、単位時間当たりの供給量を示すものとする。
TFEの供給量と、R22とTFEの供給量の合計とのモル比(以下、TFE/(R22+TFE)と示す。)は、0〜1の範囲が可能であるが、経済性と安全性の観点から、0〜0.8の範囲がより好ましく、0〜0.5の範囲が特に好ましい。
反応器に供給するR22とTFEの温度は、反応性がある程度高いがカーボン化はしにくい温度にするという観点から、0〜600℃とするのが好ましい。また、F2C:を発生しうる前記含フッ素化合物を、R22および/またはTFEとともに供給する場合、その温度も0〜600℃とするのが好ましい。
反応性をより高めるという観点からは、R22とTFE、およびF2C:を発生しうる前記含フッ素化合物は、反応器に導入する前に常温(25℃)以上600℃以下に加熱することが好ましく、100〜500℃に加熱することがより好ましい。
また、反応器に供給するメタンの温度は、反応性の観点から0〜1200℃とするのが好ましい。反応性をより高めるという観点からは、メタンは、反応器に導入する前に常温(25℃)以上1200℃以下に加熱することが好ましく、100〜800℃に加熱することがより好ましい。
原料成分であるR22および/またはTFEと、メタン、さらに必要に応じて用いられるF2C:を発生し得る前記含フッ素化合物の反応器への供給は、別々であってもよいし、各成分を混合してから供給してもよい。各成分を混合してから供給する場合には、原料組成物をグループに分けて、例えば含フッ素化合物とそれ以外とに分けて、各グループで各成分を混合し、グループごとに反応器に別々に供給してもよいし、全成分を混合してから供給してもよい。上記供給温度の違いを考慮すれば、R22とTFE、およびF2C:を発生し得る前記含フッ素化合物を混合し、上記好ましい温度に調整して反応器に供給し、これとは別にメタンを上記好ましい温度に調整して反応器に供給することが好ましい。
なお、R22および/またはTFEと、メタン、さらには必要に応じて用いられるF2C:を発生し得る前記含フッ素化合物の各原料成分を、予め混合してから反応器に供給する場合、反応器の手前で分解・反応が進行してしまうことを防ぐという観点から、反応器に供給する際の温度は600℃未満にすることが好ましく、特に500℃未満にすることが好ましい。
また、メタンは、R22および/またはTFEの存在下においてのみ反応するので、バッチ式で製造する場合は、メタンと、R22および/またはTFEとを混合した後、混合ガスを反応器に供給するか、あるいは先にR22および/またはTFEを反応器に供給した後メタンを供給するように構成することが好ましい。なお、このように構成する場合、後述する熱媒体の反応器への供給のタイミングは特に限定されず、R22および/またはTFEの供給の後でメタンの供給の前でも、あるいはメタンの供給の後でもよい。
<熱媒体>
本発明における熱媒体は、前記原料組成物と反応器内で一定の時間接触するように、反応器に供給される。熱媒体は、反応器内の温度で熱分解が生じない媒体であり、具体的には100〜1200℃の温度で熱分解しない媒体であるのが好ましい。熱媒体としては、水蒸気、窒素、二酸化炭素から選ばれる1種または2種以上の気体が挙げられ、水蒸気を50体積%以上含み、残部が窒素および/または二酸化炭素である気体の使用が好ましい。前記式(1)の熱分解反応で生成するHClを塩酸にして除くために、熱媒体における水蒸気の含有割合は50体積%以上が好ましく、実質的に水蒸気のみ(100体積%)からなる気体の使用が特に好ましい。
本発明における熱媒体は、前記原料組成物と反応器内で一定の時間接触するように、反応器に供給される。熱媒体は、反応器内の温度で熱分解が生じない媒体であり、具体的には100〜1200℃の温度で熱分解しない媒体であるのが好ましい。熱媒体としては、水蒸気、窒素、二酸化炭素から選ばれる1種または2種以上の気体が挙げられ、水蒸気を50体積%以上含み、残部が窒素および/または二酸化炭素である気体の使用が好ましい。前記式(1)の熱分解反応で生成するHClを塩酸にして除くために、熱媒体における水蒸気の含有割合は50体積%以上が好ましく、実質的に水蒸気のみ(100体積%)からなる気体の使用が特に好ましい。
熱媒体の供給量は、熱媒体と原料組成物の供給量の合計に対して、20〜98体積%となる割合が好ましく、50〜95体積%がより好ましい。熱媒体および原料組成物の供給量の合計に対する熱媒体の供給量の割合を20体積%以上とすることで、高沸物の生成や原料のカーボン化を抑制しながら上記式(1)の熱分解反応を進行させて、HFO−1234yfおよびVdFを十分に高い収率で製造できるようになる。また、前記割合が98体積%を超えると、生産性が著しく低下するため、工業的に現実的でない。
このように供給される熱媒体と前記原料組成物との反応器内での接触時間は、0.01〜10秒間とするのが好ましく、0.2〜3.0秒間とするのがより好ましい。接触時間を0.01〜10秒間とすることで、HFO−1234yfおよびVdFの生成反応を十分に進行させ、かつ副生物の生成を抑えることができる。なお、熱媒体と原料組成物との接触時間は、連続式の製造方法では原料組成物の反応器内での滞留時間に相当し、原料組成物の反応器への供給量(流量)を調節することで制御できる。
<反応器>
反応器としては、後述する反応器内温度および圧力に耐えるものであれば、特に形状は限定されず、例えば円筒状の縦型反応器が挙げられる。反応器の材質としては、ガラス、鉄、ニッケル、または鉄、ニッケルを主成分とする合金等が挙げられる。
反応器としては、後述する反応器内温度および圧力に耐えるものであれば、特に形状は限定されず、例えば円筒状の縦型反応器が挙げられる。反応器の材質としては、ガラス、鉄、ニッケル、または鉄、ニッケルを主成分とする合金等が挙げられる。
(b)工程における反応器内の温度は、反応器に供給される原料組成物であるR22および/またはTFEと、メタンの供給温度以上の温度とし、かつ400〜1200℃とすることが好ましい。600〜900℃の範囲がさらに好ましく、710〜900℃の範囲が特に好ましい。反応器内の温度を400〜1200℃とすることで、前記式(1)で示される熱分解を伴う生成反応の反応率を高め、HFO−1234yfおよびVdFを十分に高い収率で得ることができる。
反応器内の温度は、反応器に供給される前記熱媒体の温度および圧力を調整することで制御することができる。また、前記反応器内の温度が特に好ましい温度範囲(710〜900℃)になるように、電気ヒータ等により反応器内を補助的に加熱することもできる。
反応器内の圧力は、ゲージ圧で0〜2MPaとすることが好ましく、0〜0.5MPaの範囲がさらに好ましい。
<反応装置>
本発明において、HFO−1234yfおよびVdFの製造に使用される反応装置の一例を、図1に示す。
この反応装置20は、電気ヒータ等の加熱手段を備えた反応器1を有する。反応器1には、第1の原料成分であるメタンの供給ライン2、第2の原料成分であるR22の供給ライン3、第3の原料成分であるTFEの供給ライン4、および水蒸気の供給ライン5が、以下に示すように接続されている。なお、R22とTFEの一方の成分のみを供給する場合には、他方の成分の供給ラインは使用されない構成となる。また、反応器1における加熱手段の設置は必須ではない。
本発明において、HFO−1234yfおよびVdFの製造に使用される反応装置の一例を、図1に示す。
この反応装置20は、電気ヒータ等の加熱手段を備えた反応器1を有する。反応器1には、第1の原料成分であるメタンの供給ライン2、第2の原料成分であるR22の供給ライン3、第3の原料成分であるTFEの供給ライン4、および水蒸気の供給ライン5が、以下に示すように接続されている。なお、R22とTFEの一方の成分のみを供給する場合には、他方の成分の供給ラインは使用されない構成となる。また、反応器1における加熱手段の設置は必須ではない。
メタンの供給ライン2、R22の供給ライン3およびTFEの供給ライン4には、それぞれ電気ヒータ等を備えた予熱器(プレヒータ)2a、3a、4aが設置されており、供給される各原料成分が所定の温度に予熱されてから反応器1に供給される。また、水蒸気の供給ライン5には、過熱水蒸気発生器5aが設置されており、過熱水蒸気と混合されることで、供給される水蒸気の温度および圧力が調整される。なお、予熱器(プレヒータ)2a、3a、4aの設置は必須ではない。
これらの供給ライン2、3、4はそれぞれ別々に反応器1に接続されていてもよいが、図1に示すように、それぞれの予熱器3a、4aを経た後にR22の供給ライン3とTFEの供給ライン4とを連結するとともに、この連結されたR22およびTFE原料供給ライン6に、予熱器2aを経た後のメタンの供給ライン2と過熱水蒸気発生器5aを経た後の水蒸気の供給ライン5をさらに連結してもよい。すなわち、まず予熱後のR22とTFEとを混合した後、このR22とTFEとの原料混合物に、予熱したメタンと所定の温度および圧力を有する水蒸気とをさらに混合し、このように全ての成分が混合されたものが、原料・水蒸気混合供給ライン7から反応器1に供給されるように構成することができる。
反応器1の出口には、熱交換器のような冷却手段8が設置された出口ライン9が接続されている。出口ライン9には、さらに、蒸気および酸性液回収槽10、アルカリ洗浄装置11および脱水塔12が順に設置されている。そして、脱水塔12により脱水された後、出口ガスの各成分がガスクロマトグラフィ(GC)のような分析装置により分析・定量されるようになっている。
<出口ガス成分>
本発明の製造方法においては、HFO−1234yfおよびVdFを前記出口ガスの成分として得ることができる。出口ガスに含有されるHFO−1234yfとVdF以外の化合物としては、R22、メタン、エチレン、TFE、HFP、CTFE、トリフルオロエチレン、RC318、3,3,3−トリフルオロプロペン(CF3CH=CH2:HFO−1243zf)等が挙げられる。これらの成分のうちで、メチレン基(=CH2)またはメチル基(−CH3)を有するメタンおよびエチレンは、原料成分のメタンに由来する化合物であり、フルオロ基(−F)を有するTFE、HFP、CTFE、トリフルオロエチレン、RC318、HFO−1243zfは、いずれも原料成分のうちのR22および/またはTFEに由来する化合物である。HFO−1234yfおよびVdFは、R22および/またはTFEに由来する化合物であるとともに、メタンに由来する化合物である。
本発明の製造方法においては、HFO−1234yfおよびVdFを前記出口ガスの成分として得ることができる。出口ガスに含有されるHFO−1234yfとVdF以外の化合物としては、R22、メタン、エチレン、TFE、HFP、CTFE、トリフルオロエチレン、RC318、3,3,3−トリフルオロプロペン(CF3CH=CH2:HFO−1243zf)等が挙げられる。これらの成分のうちで、メチレン基(=CH2)またはメチル基(−CH3)を有するメタンおよびエチレンは、原料成分のメタンに由来する化合物であり、フルオロ基(−F)を有するTFE、HFP、CTFE、トリフルオロエチレン、RC318、HFO−1243zfは、いずれも原料成分のうちのR22および/またはTFEに由来する化合物である。HFO−1234yfおよびVdFは、R22および/またはTFEに由来する化合物であるとともに、メタンに由来する化合物である。
出口ガスに含まれるHFO−1234yfとVdF以外の前記成分は、蒸留等の既知の手段により、望まれる程度に除去することができる。そして、分離されたTFEは、原料組成物の一部としてリサイクルが可能である。また、HFP、CTFE、トリフルオロエチレン、およびRC318も、ジフルオロカルベンラジカル(CF2:)を発生し得る化合物であり、原料組成物の一部としてリサイクルが可能である。
本発明の製造方法によれば、R22および/またはTFEと、メタンとを原料として、1回の反応で、地球温暖化係数(GWP)が4と小さく、新冷媒として有用なHFO−1234yfを十分に高い収率で製造することができる。例えば、本発明の製造方法は、従来公知のHFO−1234yfを製造する方法に比べて、原料および製造設備に要するコストを低減することができるばかりでなく、製造に必要なエネルギーを圧倒的に低減することができる。また、HFO−1234yfとともに、例えば水処理フィルターとして工業的に利用されているポリフッ化ビニリデンの原料であるVdFを製造することができ、地球環境を維持する上で重要な物質を、低エネルギーで安価に、かつ同時に製造することができる。
以下に、本発明を実施例によって具体的に説明するが、本発明はこれらの実施例によって限定されるものではない。
[実施例1]
図1に示す反応装置を用い、R22とメタンとからなる原料組成物(以下、原料ガスともいう。)から、以下に示すようにして粗HFO−1234yfと粗VdFを得た。
図1に示す反応装置を用い、R22とメタンとからなる原料組成物(以下、原料ガスともいう。)から、以下に示すようにして粗HFO−1234yfと粗VdFを得た。
炉内温度300℃に設定した電気炉内のステンレス製チューブに、メタンを連続的に導入し、メタンを300℃に加熱した。また、炉内温度300℃に設定した電気炉内の別のステンレス製チューブに、R22を連続的に導入し、R22を300℃に加熱した。
このように予め加熱されて上記温度に調整されたこれらの原料ガス成分(メタンおよびR22)と、炉内温度850℃に設定した電気炉によって加熱されたスチーム(水蒸気)とを、原料成分の供給量のモル比が、メタン/R22=0.5となり、かつガス供給量全体に対する水蒸気の供給割合が、体積%で、水蒸気/(メタン+R22+水蒸気)×100=90%となるようにして、内圧(ゲージ圧)0.04MPaで内温800℃に管理された反応器に供給した。以下、圧力はいずれもゲージ圧とする。
こうして、反応器内の原料ガスの滞留時間が1.0秒間となるように、原料ガスの流量(単位時間当たりの供給量)を制御し、ガスを反応器の出口より取り出した。反応器内温度の実測値は800℃であり、反応器内圧力の実測値は0.042MPaであった。なお、反応器の出口より取り出された出口ガスには、反応により生成または副生したガスの他に、未反応の原料ガスも含まれるが、以下の記載では出口ガスを生成ガスということもある。
次いで、反応器の出口より取り出したガスを、100℃以下に冷却し、蒸気および酸性液の回収とアルカリ洗浄を順に行ってから脱水処理した後、ガスクロマトグラフィで分析して、出口ガスに含まれるガス成分のモル組成を計算した。
そして、こうして求められた出口ガスのモル組成を基にして、メタンの転化率(反応率)、メタン由来の各成分の選択率、R22の転化率(反応率)、R22由来の各成分の選択率をそれぞれ求めた。これらの結果を、反応の条件とともに表1に示す。なお、メタンおよびR22のプレヒート温度は、プレヒート用の各電気炉における設定温度であり、水蒸気温度は、水蒸気加熱用の電気炉における設定温度である。また、水蒸気圧力は設定圧力である。
上記値は、それぞれ以下のことを意味するものである。
(メタン転化率(反応率))
メタン収率がX%であるとき、(100−X)%をメタンの転化率(反応率)という。反応したメタンの割合(モル%)を意味する。なお、メタン収率は、出口ガス中のメタン由来成分(メチレン基またはメチル基を持つ成分)のうちで、メタンが占める割合(モル%)をいう。
(メタン転化率(反応率))
メタン収率がX%であるとき、(100−X)%をメタンの転化率(反応率)という。反応したメタンの割合(モル%)を意味する。なお、メタン収率は、出口ガス中のメタン由来成分(メチレン基またはメチル基を持つ成分)のうちで、メタンが占める割合(モル%)をいう。
(メタン由来の各成分の選択率)
反応したメタンのうちで、メタン以外の各成分に転化したのは各々何%かをいう。各成分の選択率は、「メタン由来の各成分の収率」/「メタンの転化率(反応率)」で求められる。なお、メタン由来の各成分の収率は、出口ガス中のメタン由来成分のうちのメタン以外の各化合物の占める割合(モル%)をいう。
反応したメタンのうちで、メタン以外の各成分に転化したのは各々何%かをいう。各成分の選択率は、「メタン由来の各成分の収率」/「メタンの転化率(反応率)」で求められる。なお、メタン由来の各成分の収率は、出口ガス中のメタン由来成分のうちのメタン以外の各化合物の占める割合(モル%)をいう。
(R22転化率(反応率))
R22収率がX%であるとき、(100−X)%をR22の転化率(反応率)という。反応したR22の割合(モル%)を意味する。なお、R22収率は、出口ガス中のR22由来成分(フルオロ基を持つ成分)のうちで、R22の占める割合(モル%)をいう。
R22収率がX%であるとき、(100−X)%をR22の転化率(反応率)という。反応したR22の割合(モル%)を意味する。なお、R22収率は、出口ガス中のR22由来成分(フルオロ基を持つ成分)のうちで、R22の占める割合(モル%)をいう。
(R22由来の各成分の選択率)
反応したR22のうちで、R22以外の各成分に転化したのは各々何%かをいう。各成分の選択率は、「R22由来の各成分の収率」/「R22の転化率(反応率)」で求められる。なお、R22由来の各成分の収率は、出口ガス中のR22由来成分のうちのR22以外の各化合物の占める割合(モル%)をいう。
反応したR22のうちで、R22以外の各成分に転化したのは各々何%かをいう。各成分の選択率は、「R22由来の各成分の収率」/「R22の転化率(反応率)」で求められる。なお、R22由来の各成分の収率は、出口ガス中のR22由来成分のうちのR22以外の各化合物の占める割合(モル%)をいう。
[実施例2〜4]
反応器内の温度を表1に示す通りに変更した。それ以外は実施例1と同様な条件で、反応を行わせた。次いで、反応器の出口より取り出したガスを、実施例1と同様に処理した後、同様に分析を行った。結果を反応条件とともに表1に示す。
反応器内の温度を表1に示す通りに変更した。それ以外は実施例1と同様な条件で、反応を行わせた。次いで、反応器の出口より取り出したガスを、実施例1と同様に処理した後、同様に分析を行った。結果を反応条件とともに表1に示す。
[実施例5〜7]
ガス供給量全体に対する水蒸気の供給割合を、体積%(水蒸気/(メタン+R22+水蒸気)×100)で表2に示す通りに変更した。それ以外は実施例1と同様な条件で、反応を行わせた。次いで、反応器の出口より取り出したガスを、実施例1と同様に処理した後、同様に分析を行った。結果を反応条件とともに表2に示す。
ガス供給量全体に対する水蒸気の供給割合を、体積%(水蒸気/(メタン+R22+水蒸気)×100)で表2に示す通りに変更した。それ以外は実施例1と同様な条件で、反応を行わせた。次いで、反応器の出口より取り出したガスを、実施例1と同様に処理した後、同様に分析を行った。結果を反応条件とともに表2に示す。
[実施例8〜11]
R22の供給量に対するメタンの供給量のモル比(メタン/R22)を、表3に示す通りに変更した。それ以外は実施例1と同様な条件で反応を行わせた。次いで、反応器の出口より取り出したガスを、実施例1と同様に処理した後、同様に分析を行った。結果を反応条件とともに表3に示す。
R22の供給量に対するメタンの供給量のモル比(メタン/R22)を、表3に示す通りに変更した。それ以外は実施例1と同様な条件で反応を行わせた。次いで、反応器の出口より取り出したガスを、実施例1と同様に処理した後、同様に分析を行った。結果を反応条件とともに表3に示す。
[実施例12〜14]
反応器内の圧力を表4に示す通りに変更した。それ以外は実施例1と同様な条件で、反応を行わせた。次いで、反応器の出口より取り出したガスを、実施例1と同様に処理した後、同様に分析を行った。結果を反応条件とともに表4に示す。
反応器内の圧力を表4に示す通りに変更した。それ以外は実施例1と同様な条件で、反応を行わせた。次いで、反応器の出口より取り出したガスを、実施例1と同様に処理した後、同様に分析を行った。結果を反応条件とともに表4に示す。
[実施例15〜19]
反応器内の滞留時間を表5に示す通りに変更した。それ以外は実施例1と同様な条件で反応を行わせた。次いで、反応器の出口より取り出したガスを、実施例1と同様に処理した後、同様に分析を行った。結果を反応条件とともに表5に示す。
反応器内の滞留時間を表5に示す通りに変更した。それ以外は実施例1と同様な条件で反応を行わせた。次いで、反応器の出口より取り出したガスを、実施例1と同様に処理した後、同様に分析を行った。結果を反応条件とともに表5に示す。
表1〜5からわかるように、実施例1〜19のいずれにおいても、出口ガス中のHFO−1234yfおよびVdFの、メタン由来の選択率の合計が、80%以上と非常に高い値を示している。また、TFEのR22由来の選択率が高くなっていることから、メタンとの反応に関与しなかったR22の大半は、R22同士のホモカップリング反応により、TFEに転化していることがわかる。
また、表1〜5の各表から以下のことがわかる。
すなわち、表1から、反応温度が高いほどメタンの転化率は大きくなることがわかる。
表2から、水蒸気の割合が少なくなると、R22のTFEへの選択率が低下することがわかる。
表3から、(メタン/R22)が小さくなるにつれて、メタン由来のHFO−1234yfの選択率が増加し、メタン由来のVdFの選択率が低下することがわかる。このことから、(メタン/R22)が所定の範囲で、HFO−1234yfとVdFの併産バランスを取ることが可能であることがわかる。
表4から、反応器内の圧力に関係なく、メタン由来のHFO−1234yfおよびVdFの選択率の合計は、89%以上と非常に高いことがわかる。因みに、気相反応における圧力の増加は、実質的な生産能力の増加を意味する。
すなわち、表1から、反応温度が高いほどメタンの転化率は大きくなることがわかる。
表2から、水蒸気の割合が少なくなると、R22のTFEへの選択率が低下することがわかる。
表3から、(メタン/R22)が小さくなるにつれて、メタン由来のHFO−1234yfの選択率が増加し、メタン由来のVdFの選択率が低下することがわかる。このことから、(メタン/R22)が所定の範囲で、HFO−1234yfとVdFの併産バランスを取ることが可能であることがわかる。
表4から、反応器内の圧力に関係なく、メタン由来のHFO−1234yfおよびVdFの選択率の合計は、89%以上と非常に高いことがわかる。因みに、気相反応における圧力の増加は、実質的な生産能力の増加を意味する。
さらに表5から、滞留時間が長くなると、R22の転化率は92%で頭打ちだが、メタンの転化率が大きく向上することがわかる。これは、R22が転化して生成した成分が、メタンと反応しているためと考えることができる。この反応において、メタンと反応していると思われる化合物は、滞留時間の増加とともに、その出口ガス成分中からモル分率が減少しているTFEである、と考えられる。
以上の考察から、以下の実施例20〜25では、TFEを原料成分として用いた。
以上の考察から、以下の実施例20〜25では、TFEを原料成分として用いた。
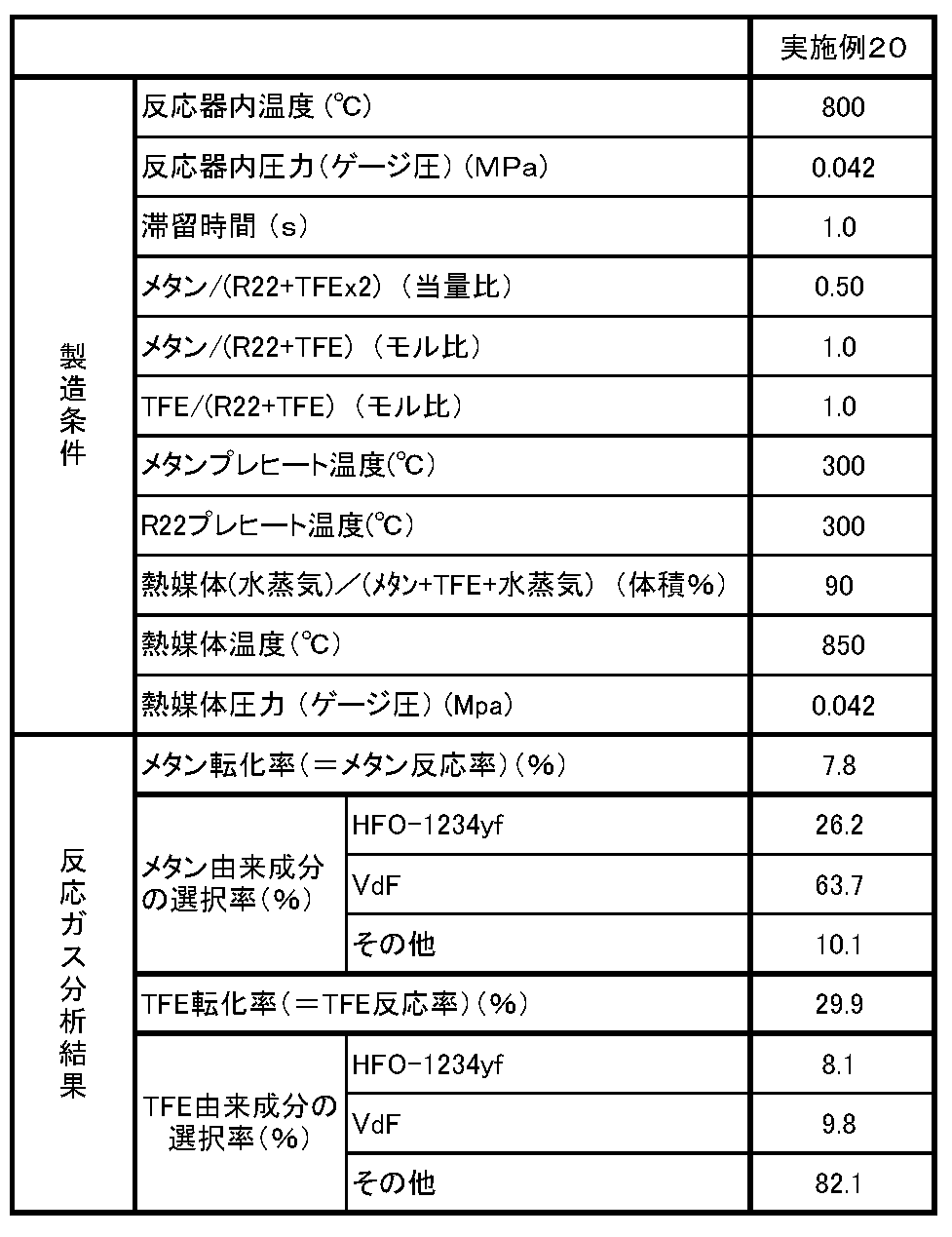
[実施例20]
実施例1と同様の反応装置を用い、TFEとメタンとからなる原料組成物から、以下に示すようにして粗HFO−1234yfと粗VdFを得た。
実施例1と同様の反応装置を用い、TFEとメタンとからなる原料組成物から、以下に示すようにして粗HFO−1234yfと粗VdFを得た。
炉内温度300℃に設定した電気炉内のステンレス製チューブに、メタンを連続的に導入し、メタンを300℃に加熱した。また、炉内温度300℃に設定した電気炉内のステンレス製チューブに、TFEを連続的に導入し、TFEを300℃に加熱した。
このように、予め加熱されて上記温度に調整されたこれらの原料ガス成分(メタンおよびTFE)と、炉内温度850℃に設定した電気炉によって加熱された水蒸気とを、原料成分の供給量のモル比が、メタン/TFE=1.0となり、かつガス供給量全体に対する水蒸気の供給量の割合が、体積%で、水蒸気/(メタン+TFE+水蒸気)×100=90%となるようにして、内圧(ゲージ圧)0.04MPaで内温800℃に管理された反応器に供給した。
こうして、反応器内の原料ガスの滞留時間が1秒間となるように、原料ガスの流量(単位時間当たりの供給量)を制御し、ガスを反応器の出口より取り出した。反応器内温度の実測値は800℃であり、反応器内圧力の実測値は0.042MPaであった。
次いで、反応器の出口より取り出したガスを、実施例1と同様に処理した後、同様に出口ガスに含まれるガス成分のモル組成を計算した。そして、こうして求められた出口ガスのモル組成を基にして、メタンの転化率(反応率)、メタン由来の各成分の選択率、TFEの転化率(反応率)、TFE由来の各成分の選択率をそれぞれ求めた。これらの結果を反応条件とともに表6に示す。
なお、メタンおよびTFEのプレヒート温度は、プレヒート用の各電気炉における設定温度であり、水蒸気温度は、水蒸気加熱用の電気炉における設定温度である。また、水蒸気圧力は設定圧力である。
なお、メタンおよびTFEのプレヒート温度は、プレヒート用の各電気炉における設定温度であり、水蒸気温度は、水蒸気加熱用の電気炉における設定温度である。また、水蒸気圧力は設定圧力である。
TFEの転化率(反応率)、およびTFE由来の各成分の選択率は、それぞれ以下のことを意味するものである。
(TFEの転化率(反応率))
TFE収率がX%であるとき、(100−X)%をTFEの転化率(反応率)という。反応したTFEの割合(モル%)を意味する。なお、TFE収率は、出口ガス中のTFE由来成分(フルオロ基を持つ成分)のうちで、TFEの占める割合(モル%)をいう。
TFE収率がX%であるとき、(100−X)%をTFEの転化率(反応率)という。反応したTFEの割合(モル%)を意味する。なお、TFE収率は、出口ガス中のTFE由来成分(フルオロ基を持つ成分)のうちで、TFEの占める割合(モル%)をいう。
(TFE由来の各成分の選択率)
反応したTFEのうちで、TFE以外の各成分に転化したのは各々何%かをいう。各成分の選択率は、「TFE由来の各成分の収率」/「TFEの転化率(反応率)」で求められる。なお、TFE由来の各成分の収率は、出口ガス中のTFE由来成分のうちのTFE以外の各化合物の占める割合(モル%)をいう。
反応したTFEのうちで、TFE以外の各成分に転化したのは各々何%かをいう。各成分の選択率は、「TFE由来の各成分の収率」/「TFEの転化率(反応率)」で求められる。なお、TFE由来の各成分の収率は、出口ガス中のTFE由来成分のうちのTFE以外の各化合物の占める割合(モル%)をいう。
[実施例21]
原料の供給ラインが3つある以外は実施例1と同様の反応装置を用いて、R22とメタンとTFEとからなる原料組成物から、以下に示すようにして粗HFO−1234yfと粗VdFを得た。
原料の供給ラインが3つある以外は実施例1と同様の反応装置を用いて、R22とメタンとTFEとからなる原料組成物から、以下に示すようにして粗HFO−1234yfと粗VdFを得た。
炉内温度300℃に設定した電気炉内のステンレス製チューブに、メタンを連続的に導入し、メタンを300℃に加熱(プレヒート)した。また、炉内温度300℃に設定した電気炉内のステンレス製チューブに、R22を連続的に導入し、R22を300℃にプレヒートした。さらに、炉内温度300℃に設定した電気炉内のステンレス製チューブに、TFEを連続的に導入し、TFEを300℃にプレヒートした。
こうしてプレヒートされたこれらの原料ガス成分(メタン、R22およびTFE)と、炉内温度850℃に設定した電気炉によって加熱された水蒸気とを、原料成分の供給量のモル比が、
TFE/R22=1(すなわち、TFE/(TFE+R22)=0.5)
メタン/(R22+TFE)=0.75
となり、かつガス供給量全体に対する水蒸気の供給割合が、体積%で、水蒸気/(メタン+R22+TFE+水蒸気)×100=90%となるようにして、内圧(ゲージ圧)0.04MPaで内温800℃に管理された反応器に供給した。
TFE/R22=1(すなわち、TFE/(TFE+R22)=0.5)
メタン/(R22+TFE)=0.75
となり、かつガス供給量全体に対する水蒸気の供給割合が、体積%で、水蒸気/(メタン+R22+TFE+水蒸気)×100=90%となるようにして、内圧(ゲージ圧)0.04MPaで内温800℃に管理された反応器に供給した。
なお、メタンと、R22とTFEの合計とのモル比(メタン/(R22+TFE)は、前記したように0.75であるが、原料組成物を構成する成分のうちで、フッ素を含む化合物としての働きの観点からは、TFEの1モルはR22の2モルに相当して2当量とカウントできるため、メタンと、R22およびTFEの合計との当量比(メタン/(R22+TFE×2)は、0.5となる。
こうして、反応器内の原料ガスの滞留時間が1.0秒間となるように、原料ガスの流量(単位時間当たりの供給量)を制御し、ガスを反応器の出口より取り出した。反応器内温度の実測値は800℃であり、反応器内圧力の実測値は0.042MPaであった。
次いで、反応器の出口より取り出したガスを、実施例1と同様に処理した後、同様に出口ガスに含まれるガス成分のモル組成を計算した。そして、こうして求められた出口ガスのモル組成を基にして、メタンの転化率(反応率)、メタン由来の各成分の選択率、R22および/またはTFEの転化率(反応率)、R22および/またはTFE由来の各成分の選択率をそれぞれ求めた。これらの結果を反応条件とともに表7に示す。
なお、メタン、R22およびTFEのプレヒート温度は、プレヒート用の各電気炉における設定温度であり、水蒸気温度は、水蒸気加熱用の電気炉における設定温度である。また、水蒸気圧力は設定圧力である。
なお、メタン、R22およびTFEのプレヒート温度は、プレヒート用の各電気炉における設定温度であり、水蒸気温度は、水蒸気加熱用の電気炉における設定温度である。また、水蒸気圧力は設定圧力である。
なお、R22およびTFEの転化率(反応率)と、R22および/またはTFE由来の各成分の選択率は、それぞれ以下のことを意味するものである。
(R22および/またはTFEの転化率(反応率))
出口ガス中のフッ素を含む化合物であるR22および/またはTFEに由来する成分(フルオロ基を持つ成分)のうちで、R22および/またはTFEの占める割合(R22および/またはTFE回収率)がX%であるとき、(100−X)%をR22および/またはTFEの転化率(反応率)という。反応したR22および/またはTFEの割合(モル%)を意味する。
出口ガス中のフッ素を含む化合物であるR22および/またはTFEに由来する成分(フルオロ基を持つ成分)のうちで、R22および/またはTFEの占める割合(R22および/またはTFE回収率)がX%であるとき、(100−X)%をR22および/またはTFEの転化率(反応率)という。反応したR22および/またはTFEの割合(モル%)を意味する。
(R22および/またはTFE由来の各成分の選択率)
反応したR22および/またはTFEのうちで、R22以外の各成分として得られたのは各々何%かをいう。各成分の選択率は、「R22および/またはTFE由来の各成分の収率」/「R22および/またはTFEの転化率(反応率)」で求められる。なお、R22および/またはTFE由来の各成分の収率は、出口ガス中のR22および/またはTFE由来成分のうちのR22以外の各成分の占める割合(モル%)をいう。
反応したR22および/またはTFEのうちで、R22以外の各成分として得られたのは各々何%かをいう。各成分の選択率は、「R22および/またはTFE由来の各成分の収率」/「R22および/またはTFEの転化率(反応率)」で求められる。なお、R22および/またはTFE由来の各成分の収率は、出口ガス中のR22および/またはTFE由来成分のうちのR22以外の各成分の占める割合(モル%)をいう。
ここで、原料ガスとしてTFEを含む実施例21〜25では、TFEは反応(=転化)しているがR22から生成もしているため、TFEだけの転化率(反応率)を求めることは不可能である。また、フルオロ基(−F)を有する生成物であるHFO−1234yfやVdFが、R22から生成したものであるか、TFEから生成したものであるかを求めることも不可能である。そのため、「原料のTFEは全てR22である」と仮定して、その原料R22が反応した割合をR22および/またはTFEの転化率(反応率)としている。また、前記原料R22から各成分に何%転化しているかを求め、R22および/またはTFE由来の各成分の選択率としている。
[実施例22〜25]
TFEの供給量と、TFEとR22の供給量の合計とのモル比(TFE/(TFE+R22))を、実施例22では0.1、実施例23では0.3、実施例24では0.7、実施例25では0.9とした。また、メタンと、R22およびTFEの合計との当量比(メタン/(R22+TFE×2)が0.5になるように、メタン/(TFE+R22)のモル比を、実施例22では0.55、実施例23では0.65、実施例24では0.85、実施例25では0.95とした。それ以外は実施例1と同様な条件で反応を行わせた。
TFEの供給量と、TFEとR22の供給量の合計とのモル比(TFE/(TFE+R22))を、実施例22では0.1、実施例23では0.3、実施例24では0.7、実施例25では0.9とした。また、メタンと、R22およびTFEの合計との当量比(メタン/(R22+TFE×2)が0.5になるように、メタン/(TFE+R22)のモル比を、実施例22では0.55、実施例23では0.65、実施例24では0.85、実施例25では0.95とした。それ以外は実施例1と同様な条件で反応を行わせた。
次いで、反応器の出口より取り出したガスを、実施例1と同様に処理した後、同様に分析を行った。結果を反応条件とともに表7に示す。
表6および表7からわかるように、実施例20〜25のいずれにおいても、出口ガス中のHFO−1234yfおよびVdFの、メタン由来の選択率の合計が、80%以上と非常に高い値を示しており、R22とTFEの少なくとも一方とメタンを含有する原料組成物を用いることで、HFO−1234yfおよびVdFを製造することができる。
これらの結果から、本発明の製造方法によれば、効率よくHFO−1234yfおよびVdFが得られるといえる。
なお、上記反応条件の各実施例において、同一条件での反応においては再現性よくほぼ同一の結果が得られることを確認した。これにより、本発明の製造方法によれば、反応条件の制御が容易であり、よって定量的なHFO−1234yfおよびVdFの製造が可能といえる。
なお、上記反応条件の各実施例において、同一条件での反応においては再現性よくほぼ同一の結果が得られることを確認した。これにより、本発明の製造方法によれば、反応条件の制御が容易であり、よって定量的なHFO−1234yfおよびVdFの製造が可能といえる。
本発明の製造方法によれば、調達が容易なR22および/またはTFEと、メタンとを原料として、中間生成物を反応系から取り出すことなく、そのまま反応させ、工業的に有用なHFO−1234yfおよびVdFを効率よく製造することができる。したがって、例えば、HCFC−225caを原料としてCFO−1214yaを経由してHFO−1234yfを製造する方法に比べて、原料および製造設備に要するコストを大幅に低減することができる。
また、本発明の製造方法によれば、製造(反応)条件の制御が容易であり、よって定量的なHFO−1234yfおよびVdFの製造が可能となり経済的なメリットが大きい。またさらに、副生物のリサイクルも可能であり、経済的な効果が大きい。
1…反応器、2…メタンの供給ライン、3…R22の供給ライン、4…TFEの供給ライン、5…水蒸気の供給ライン、2a,3a,4a…予熱器(プレヒータ)、5a…過熱水蒸気発生器、8…冷却手段、9…出口ライン、10…蒸気および酸性液回収槽、11…アルカリ洗浄装置、12…脱水塔、20…反応装置。
Claims (11)
- 熱媒体存在下、クロロジフルオロメタンおよび/またはテトラフルオロエチレンと、メタンを含む原料組成物から、熱分解を伴う合成反応により2,3,3,3−テトラフルオロプロペンおよび1,1−ジフルオロエチレンを製造する方法であって、
(a)前記クロロジフルオロメタンおよび/または前記テトラフルオロエチレンと、前記メタンとを、予め混合してまたは別々に反応器に供給し、該反応器内に所定の時間滞留させる工程と、
(b)熱媒体を前記反応器に供給し、該反応器内で前記原料組成物と前記熱媒体とを接触させる工程と
を有することを特徴とする2,3,3,3−テトラフルオロプロペンおよび1,1−ジフルオロエチレンの製造方法。 - 前記クロロジフルオロメタンと前記テトラフルオロエチレンとの合計1モルに対して、前記メタンを0.01〜10モルの割合で供給する、請求項1に記載の2,3,3,3−テトラフルオロプロペンおよび1,1−ジフルオロエチレンの製造方法。
- 前記テトラフルオロエチレンを、該テトラフルオロエチレンと前記クロロジフルオロメタンとの合計1モルに対して、0〜0.8モルの割合で供給する、請求項1または2に記載の2,3,3,3−テトラフルオロプロペンおよび1,1−ジフルオロエチレンの製造方法。
- 前記(b)工程における前記反応器内の温度は400〜1200℃である、請求項1〜3のいずれか1項に記載の2,3,3,3−テトラフルオロプロペンおよび1,1−ジフルオロエチレンの製造方法。
- 前記反応器に供給する前記メタンの温度は0〜1200℃である、請求項1〜4のいずれか1項に記載の2,3,3,3−テトラフルオロプロペンおよび1,1−ジフルオロエチレンの製造方法。
- 前記反応器に供給する前記クロロジフルオロメタンの温度は0〜600℃である、請求項1〜5のいずれか1項に記載の2,3,3,3−テトラフルオロプロペンおよび1,1−ジフルオロエチレンの製造方法。
- 前記反応器に供給する前記テトラフルオロエチレンの温度は0〜600℃である、請求項1〜6のいずれか1項に記載の2,3,3,3−テトラフルオロプロペンおよび1,1−ジフルオロエチレンの製造方法。
- 前記熱媒体は、100〜1200℃で熱分解しない媒体である、請求項1〜7のいずれか1項に記載の2,3,3,3−テトラフルオロプロペンおよび1,1−ジフルオロエチレンの製造方法。
- 前記熱媒体は、水蒸気、窒素、二酸化炭素から選ばれる1種または2種以上の気体である、請求項1〜8のいずれか1項に記載の2,3,3,3−テトラフルオロプロペンおよび1,1−ジフルオロエチレンの製造方法。
- 前記熱媒体の供給量は、該熱媒体と前記原料組成物の供給量の合計に対して20〜98体積%の割合である、請求項1〜9のいずれか1項に記載の2,3,3,3−テトラフルオロプロペンおよび1,1−ジフルオロエチレンの製造方法。
- 前記(b)工程において、前記反応器内での前記熱媒体と前記原料組成物との接触時間は0.01〜10秒間である、請求項1〜10のいずれか1項に記載の2,3,3,3−テトラフルオロプロペンおよび1,1−ジフルオロエチレンの製造方法。
Priority Applications (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2012256211A JP2016027005A (ja) | 2012-11-22 | 2012-11-22 | 2,3,3,3−テトラフルオロプロペンおよび1,1−ジフルオロエチレンの製造方法 |
| PCT/JP2013/080324 WO2014080779A1 (ja) | 2012-11-22 | 2013-11-08 | 2,3,3,3-テトラフルオロプロペンおよび1,1-ジフルオロエチレンの製造方法 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2012256211A JP2016027005A (ja) | 2012-11-22 | 2012-11-22 | 2,3,3,3−テトラフルオロプロペンおよび1,1−ジフルオロエチレンの製造方法 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| JP2016027005A true JP2016027005A (ja) | 2016-02-18 |
Family
ID=50775964
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2012256211A Pending JP2016027005A (ja) | 2012-11-22 | 2012-11-22 | 2,3,3,3−テトラフルオロプロペンおよび1,1−ジフルオロエチレンの製造方法 |
Country Status (2)
| Country | Link |
|---|---|
| JP (1) | JP2016027005A (ja) |
| WO (1) | WO2014080779A1 (ja) |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP3793968A4 (en) * | 2018-05-16 | 2022-03-09 | Srf Limited | PROCEDURE FOR PURIFICATION OF AN OLEFIN FEED WITH 1234YF |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US2931840A (en) * | 1958-11-25 | 1960-04-05 | Du Pont | Process for preparing 2, 3, 3, 3-tetrafluoropropene |
| US20120065436A1 (en) * | 2009-05-12 | 2012-03-15 | Daikin Industries, Ltd. | Process for preparing fluorine-containing propane |
| JP5201284B1 (ja) * | 2012-03-14 | 2013-06-05 | 旭硝子株式会社 | 2,3,3,3−テトラフルオロプロペンの製造方法 |
| JP5149456B1 (ja) * | 2012-03-14 | 2013-02-20 | 旭硝子株式会社 | 2,3,3,3−テトラフルオロプロペンおよび1,1−ジフルオロエチレンの製造方法 |
| JP2013227244A (ja) * | 2012-04-25 | 2013-11-07 | Asahi Glass Co Ltd | 2,3,3,3−テトラフルオロプロペンの製造方法 |
-
2012
- 2012-11-22 JP JP2012256211A patent/JP2016027005A/ja active Pending
-
2013
- 2013-11-08 WO PCT/JP2013/080324 patent/WO2014080779A1/ja not_active Ceased
Also Published As
| Publication number | Publication date |
|---|---|
| WO2014080779A1 (ja) | 2014-05-30 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5149456B1 (ja) | 2,3,3,3−テトラフルオロプロペンおよび1,1−ジフルオロエチレンの製造方法 | |
| JP5201284B1 (ja) | 2,3,3,3−テトラフルオロプロペンの製造方法 | |
| JP5975096B2 (ja) | 2,3,3,3−テトラフルオロプロペンおよび1,1−ジフルオロエチレンの製造方法 | |
| JP2013227244A (ja) | 2,3,3,3−テトラフルオロプロペンの製造方法 | |
| JP2016027004A (ja) | 2,3,3,3−テトラフルオロプロペンの製造方法 | |
| JP2013227245A (ja) | 2,3,3,3−テトラフルオロプロペンおよび1,1−ジフルオロエチレンの製造方法 | |
| JP2014101326A (ja) | 2,3,3,3−テトラフルオロプロペンの製造方法 | |
| JP2016027005A (ja) | 2,3,3,3−テトラフルオロプロペンおよび1,1−ジフルオロエチレンの製造方法 | |
| JP2014129260A (ja) | 2,3,3,3−テトラフルオロプロペンの製造方法 | |
| JP6197637B2 (ja) | (e)−1,3,3,3−テトラフルオロプロペンの製造方法 | |
| JP6549553B2 (ja) | フッ素化オレフィンの製造プロセス | |
| JP2015110533A (ja) | 2,3,3,3−テトラフルオロプロペンおよび1,1−ジフルオロエチレンの製造方法 | |
| JP2014129273A (ja) | 2,3,3,3−テトラフルオロプロペンの製造方法 | |
| JP2015117188A (ja) | トリフルオロエチレンの製造方法 | |
| JP6217750B2 (ja) | トリフルオロエチレンの製造方法 | |
| JP2014129259A (ja) | 2,3,3,3−テトラフルオロプロペンの製造方法 | |
| CN105339330B (zh) | 三氟乙烯的制造方法 | |
| JP2015010058A (ja) | トリフルオロエチレンの製造方法 | |
| JP2015117184A (ja) | トリフルオロエチレンの製造方法 | |
| JP6176182B2 (ja) | トリフルオロエチレンの製造方法 | |
| CN101495431A (zh) | 氟化有机化合物的制备方法 |