JP2016158558A - Dnaとrnaの単離方法 - Google Patents
Dnaとrnaの単離方法 Download PDFInfo
- Publication number
- JP2016158558A JP2016158558A JP2015040090A JP2015040090A JP2016158558A JP 2016158558 A JP2016158558 A JP 2016158558A JP 2015040090 A JP2015040090 A JP 2015040090A JP 2015040090 A JP2015040090 A JP 2015040090A JP 2016158558 A JP2016158558 A JP 2016158558A
- Authority
- JP
- Japan
- Prior art keywords
- rna
- dna
- nucleic acid
- binding carrier
- supernatant
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Landscapes
- Measuring Or Testing Involving Enzymes Or Micro-Organisms (AREA)
Abstract
【課題】DNAおよびRNAを含む生体試料からDNAとRNAの両方を簡便且つ効率良く単離する方法を提供すること。
【解決手段】DNAおよびRNAを含む同一の生体試料からDNAとRNAを単離する方法であって、以下の工程(A)〜(F)を含む、DNAとRNAの単離方法。
(A)前記生体試料と第1の核酸結合性担体とを液相中で混合する工程
(B)工程(A)で得られた第1の核酸結合担体と上清とを分離する工程
(C)工程(B)で分離された第1の核酸結合担体から、DNAおよびRNAのうちいずれか一方を溶出させる工程
(D)工程(B)で分離された上清と第2の核酸結合性担体とを液相中で混合する工程
(E)工程(D)で得られた第2の核酸結合担体と上清とを分離する工程
(F)工程(E)で分離された第2の核酸結合担体から、RNAおよびDNAのうち工程(C)で溶出させなかった核酸を溶出させる工程
【選択図】なし
【解決手段】DNAおよびRNAを含む同一の生体試料からDNAとRNAを単離する方法であって、以下の工程(A)〜(F)を含む、DNAとRNAの単離方法。
(A)前記生体試料と第1の核酸結合性担体とを液相中で混合する工程
(B)工程(A)で得られた第1の核酸結合担体と上清とを分離する工程
(C)工程(B)で分離された第1の核酸結合担体から、DNAおよびRNAのうちいずれか一方を溶出させる工程
(D)工程(B)で分離された上清と第2の核酸結合性担体とを液相中で混合する工程
(E)工程(D)で得られた第2の核酸結合担体と上清とを分離する工程
(F)工程(E)で分離された第2の核酸結合担体から、RNAおよびDNAのうち工程(C)で溶出させなかった核酸を溶出させる工程
【選択図】なし
Description
本発明は、DNAとRNAの単離方法に関する。より詳細には、同一の生体試料からDNAとRNAの両方を単離する方法に関する。
DNAは生命情報の担い手であり、RNAはその情報を受け、生体内でタンパク質合成等に関わる重要な役割を担っている分子である。
生化学、遺伝子工学および臨床診断等の分野において、ノザンブロット解析、逆転写ポリメラーゼチェインリアション等の手法でRNA解析が行われており、斯かる解析を精度よく行うためには、高純度のRNAを使用することが必要である。
そのため、RNAを高純度で単離する方法として、リチウム塩やカオトロピック剤等を含有する酸性溶液とシリカ粒子を、RNAを含む生体試料と混合し、RNAをシリカ粒子に吸着させ、その後RNAを溶出させる方法が提案されている(特許文献1)。
生化学、遺伝子工学および臨床診断等の分野において、ノザンブロット解析、逆転写ポリメラーゼチェインリアション等の手法でRNA解析が行われており、斯かる解析を精度よく行うためには、高純度のRNAを使用することが必要である。
そのため、RNAを高純度で単離する方法として、リチウム塩やカオトロピック剤等を含有する酸性溶液とシリカ粒子を、RNAを含む生体試料と混合し、RNAをシリカ粒子に吸着させ、その後RNAを溶出させる方法が提案されている(特許文献1)。
また、RNAとともにDNAも分子生物学の分野において研究および臨床分析のために広く用いられる重要な分子である。DNAとRNAの両方を生体試料から得る方法として、生体試料を溶解液で処理した後、これを2つにわけて、一方の生体試料をpH7以上の緩衝液等で処理した後、これに含まれるRNAを担体に吸着させるなどしてRNAを得て、他方の生体試料をpH7以上の緩衝液等で処理した後、これに含まれるDNAを担体に吸着させるなどしてDNAを得る方法が知られている(特許文献2)。
しかしながら、特許文献2に記載のような方法では、抽出したDNAにRNAが混入しやすく、DNAを高純度化するためにはリボヌクレアーゼで処理することが必要であった。また、DNAとRNAを別々の生体試料から得ようとすると、DNA抽出過程におけるRNAのロスやRNA抽出過程におけるDNAのロスが生じ、DNAの収量とRNAの収量がいずれも少なくなるという問題もある。
したがって、本発明が解決しようとする課題は、DNAおよびRNAを含む生体試料からDNAとRNAの両方を簡便且つ効率良く単離する方法を提供することにある。
したがって、本発明が解決しようとする課題は、DNAおよびRNAを含む生体試料からDNAとRNAの両方を簡便且つ効率良く単離する方法を提供することにある。
上記課題は以下の手段により解決された。
<1>DNAおよびRNAを含む同一の生体試料からDNAとRNAを単離する方法であって、以下の工程(A)〜(F)を含む、DNAとRNAの単離方法。
(A)前記生体試料と第1の核酸結合性担体とを液相中で混合する工程
(B)工程(A)で得られた第1の核酸結合担体と上清とを分離する工程
(C)工程(B)で分離された第1の核酸結合担体から、DNAおよびRNAのうちいずれか一方を溶出させる工程
(D)工程(B)で分離された上清と第2の核酸結合性担体とを液相中で混合する工程
(E)工程(D)で得られた第2の核酸結合担体と上清とを分離する工程
(F)工程(E)で分離された第2の核酸結合担体から、RNAおよびDNAのうち工程(C)で溶出させなかった核酸を溶出させる工程
(A)前記生体試料と第1の核酸結合性担体とを液相中で混合する工程
(B)工程(A)で得られた第1の核酸結合担体と上清とを分離する工程
(C)工程(B)で分離された第1の核酸結合担体から、DNAおよびRNAのうちいずれか一方を溶出させる工程
(D)工程(B)で分離された上清と第2の核酸結合性担体とを液相中で混合する工程
(E)工程(D)で得られた第2の核酸結合担体と上清とを分離する工程
(F)工程(E)で分離された第2の核酸結合担体から、RNAおよびDNAのうち工程(C)で溶出させなかった核酸を溶出させる工程
<2>DNAおよびRNAを含む同一の生体試料からDNAとRNAを単離する方法であって、以下の工程(A)〜(F)を含む、DNAとRNAの単離方法。
(A)前記生体試料と第1の核酸結合性担体とを、リチウム塩が添加された液相中で混合する工程
(B)工程(A)で得られたRNA結合担体と上清とを分離する工程
(C)工程(B)で分離されたRNA結合担体から、RNAを溶出させる工程
(D)工程(B)で分離された上清と第2の核酸結合性担体とを、低級アルコールが添加された液相中で混合する工程
(E)工程(D)で得られたDNA結合担体と上清とを分離する工程
(F)工程(E)で分離されたDNA結合担体から、DNAを溶出させる工程
<3>工程(A)を、酸性条件下で行う、<2>の単離方法。
<4>前記リチウム塩が、リチウムの無機塩およびリチウムの有機塩から選ばれる1種以上である、<2>または<3>の単離方法。
<5>前記リチウム塩が、塩化リチウム、酢酸リチウム、クエン酸リチウム、炭酸リチウム、水酸化リチウムおよびホウ酸リチウムから選ばれる1種以上である、<2>〜<4>のいずれかの単離方法。
(A)前記生体試料と第1の核酸結合性担体とを、リチウム塩が添加された液相中で混合する工程
(B)工程(A)で得られたRNA結合担体と上清とを分離する工程
(C)工程(B)で分離されたRNA結合担体から、RNAを溶出させる工程
(D)工程(B)で分離された上清と第2の核酸結合性担体とを、低級アルコールが添加された液相中で混合する工程
(E)工程(D)で得られたDNA結合担体と上清とを分離する工程
(F)工程(E)で分離されたDNA結合担体から、DNAを溶出させる工程
<3>工程(A)を、酸性条件下で行う、<2>の単離方法。
<4>前記リチウム塩が、リチウムの無機塩およびリチウムの有機塩から選ばれる1種以上である、<2>または<3>の単離方法。
<5>前記リチウム塩が、塩化リチウム、酢酸リチウム、クエン酸リチウム、炭酸リチウム、水酸化リチウムおよびホウ酸リチウムから選ばれる1種以上である、<2>〜<4>のいずれかの単離方法。
<6>DNAおよびRNAを含む同一の生体試料からDNAとRNAを単離する方法であって、以下の工程(A)〜(F)を含む、DNAとRNAの単離方法。
(A)前記生体試料と第1の核酸結合性担体とを、ポリアルキレングリコールが添加された液相中で混合する工程
(B)工程(A)で得られたDNA結合担体と上清とを分離する工程
(C)工程(B)で分離されたDNA結合担体から、DNAを溶出させる工程
(D)工程(B)で分離された上清と第2の核酸結合性担体とを、低級アルコールまたはリチウム塩が添加された液相中で混合する工程
(E)工程(D)で得られたRNA結合担体と上清とを分離する工程
(F)工程(E)で分離されたRNA結合担体から、RNAを溶出させる工程
<7>前記低級アルコールまたはリチウム塩が、低級アルコールである、<6>の単離方法。
<8>前記ポリアルキレングリコールが、重量平均分子量6000〜10000のポリアルキレングリコールである、<6>または<7>の単離方法。
(A)前記生体試料と第1の核酸結合性担体とを、ポリアルキレングリコールが添加された液相中で混合する工程
(B)工程(A)で得られたDNA結合担体と上清とを分離する工程
(C)工程(B)で分離されたDNA結合担体から、DNAを溶出させる工程
(D)工程(B)で分離された上清と第2の核酸結合性担体とを、低級アルコールまたはリチウム塩が添加された液相中で混合する工程
(E)工程(D)で得られたRNA結合担体と上清とを分離する工程
(F)工程(E)で分離されたRNA結合担体から、RNAを溶出させる工程
<7>前記低級アルコールまたはリチウム塩が、低級アルコールである、<6>の単離方法。
<8>前記ポリアルキレングリコールが、重量平均分子量6000〜10000のポリアルキレングリコールである、<6>または<7>の単離方法。
<9>第1の核酸結合性担体および第2の核酸結合性担体が、磁性粒子である、<1>〜<8>のいずれかの単離方法。
<10>第1の核酸結合性担体および第2の核酸結合性担体が、核酸結合性官能基を有する非シリカ系磁性粒子である、<1>〜<9>のいずれかの単離方法。
<11>工程(A)の液相が、カオトロピック剤が添加された液相である、<1>〜<10>のいずれかの単離方法。
<12>前記カオトロピック剤が、グアニジウム塩、尿素、ヨウ化物、過塩素酸塩、チオシアン酸塩およびイソチオシアン酸塩から選ばれる1種以上である、<11>の単離方法。
<13>前記グアニジウム塩が、塩酸グアニジウム、酢酸グアニジウム、リン酸グアニジウム、チオシアン酸グアニジウム、イソチオシアン酸グアニジウム、硫酸グアニジウムおよび炭酸グアニジウムから選ばれる1種以上である、<12>の単離方法。
<14>核酸分解酵素を使用せずにDNAとRNAを単離する方法である、<1>〜<13>のいずれかの単離方法。
<10>第1の核酸結合性担体および第2の核酸結合性担体が、核酸結合性官能基を有する非シリカ系磁性粒子である、<1>〜<9>のいずれかの単離方法。
<11>工程(A)の液相が、カオトロピック剤が添加された液相である、<1>〜<10>のいずれかの単離方法。
<12>前記カオトロピック剤が、グアニジウム塩、尿素、ヨウ化物、過塩素酸塩、チオシアン酸塩およびイソチオシアン酸塩から選ばれる1種以上である、<11>の単離方法。
<13>前記グアニジウム塩が、塩酸グアニジウム、酢酸グアニジウム、リン酸グアニジウム、チオシアン酸グアニジウム、イソチオシアン酸グアニジウム、硫酸グアニジウムおよび炭酸グアニジウムから選ばれる1種以上である、<12>の単離方法。
<14>核酸分解酵素を使用せずにDNAとRNAを単離する方法である、<1>〜<13>のいずれかの単離方法。
本発明の単離方法によれば、DNAおよびRNAを含む生体試料からDNAとRNAの両方を簡便且つ効率良く単離できる。
本発明の単離方法は、DNAおよびRNAを含む同一の生体試料からDNAとRNAを単離する方法であって、以下の工程(A)〜(F)を含むものである。
(A)前記生体試料と第1の核酸結合性担体とを液相中で混合する工程
(B)工程(A)で得られた第1の核酸結合担体と上清とを分離する工程
(C)工程(B)で分離された第1の核酸結合担体から、DNAおよびRNAのうちいずれか一方を溶出させる工程
(D)工程(B)で分離された上清と第2の核酸結合性担体とを液相中で混合する工程
(E)工程(D)で得られた第2の核酸結合担体と上清とを分離する工程
(F)工程(E)で分離された第2の核酸結合担体から、RNAおよびDNAのうち工程(C)で溶出させなかった核酸を溶出させる工程
(A)前記生体試料と第1の核酸結合性担体とを液相中で混合する工程
(B)工程(A)で得られた第1の核酸結合担体と上清とを分離する工程
(C)工程(B)で分離された第1の核酸結合担体から、DNAおよびRNAのうちいずれか一方を溶出させる工程
(D)工程(B)で分離された上清と第2の核酸結合性担体とを液相中で混合する工程
(E)工程(D)で得られた第2の核酸結合担体と上清とを分離する工程
(F)工程(E)で分離された第2の核酸結合担体から、RNAおよびDNAのうち工程(C)で溶出させなかった核酸を溶出させる工程
〔工程(A)〕
工程(A)は、DNAおよびRNAを含む生体試料と第1の核酸結合性担体とを液相中で混合する工程である。斯かる工程(A)により、DNAおよびRNAのうちいずれか一方が第1の核酸結合性担体に結合し、第1の核酸結合担体が形成される。
工程(A)は、DNAおよびRNAを含む生体試料と第1の核酸結合性担体とを液相中で混合する工程である。斯かる工程(A)により、DNAおよびRNAのうちいずれか一方が第1の核酸結合性担体に結合し、第1の核酸結合担体が形成される。
生体試料は、DNAおよびRNAを含むものであれば特に限定されるものではなく、例えば、全血、血清、血漿、血液成分、各種血球、血餅、血小板等の血液組成成分の他、尿、糞便、精液、組織液、唾液や、これらから分離した細胞、培養細胞等が挙げられ、微生物やウイルス等でもよい。なお、生体試料は、安定化剤等で前処理したものでもよい。
また、DNAおよびRNAは、内在性でも外来性でもよく、また、生体外で酵素的に合成されたものでもよい。DNAとしては、例えば、ゲノムDNA、プラスミドDNA、合成オリゴDNA、PCR産物等が挙げられる。RNAとしては、例えば、mRNA、rRNA、tRNA、microRNA、合成RNA等が挙げられる。
生体試料の系中の濃度は、1〜100(w/v)%とするのが好ましく、10〜60(w/v)%とするのがより好ましい。
また、DNAおよびRNAは、内在性でも外来性でもよく、また、生体外で酵素的に合成されたものでもよい。DNAとしては、例えば、ゲノムDNA、プラスミドDNA、合成オリゴDNA、PCR産物等が挙げられる。RNAとしては、例えば、mRNA、rRNA、tRNA、microRNA、合成RNA等が挙げられる。
生体試料の系中の濃度は、1〜100(w/v)%とするのが好ましく、10〜60(w/v)%とするのがより好ましい。
第1の核酸結合性担体は、核酸を結合できる担体であれば特に限定されるものではなく、シリカやガラス、珪藻土、金属、金属酸化物等の無機物;樹脂等の有機物;これらの複合体のいずれでもよいが、非シリカ系担体が好ましい。樹脂は、多糖類(アガロース、デキストラン、セルロース等)等の天然高分子で構成されていても、スチレン系ポリマー、スチレン−ブタジエン系共重合体、(メタ)アクリル酸エステル系ポリマー、(メタ)アクリロニトリル系ポリマー、フッ素系ポリマー、ポリエチレン、ポリプロピレン、ポリエステル等の合成高分子で構成されていてもよいが、核酸結合性官能基を表面に有する樹脂が好ましい。
また、第1の核酸結合性担体としては、核酸結合性官能基を有するものが好ましい。核酸結合性官能基としては、核酸と結合可能な酸性基が好ましい。具体的には、カルボキシ基、スルホン酸基、リン酸基、フェノール性水酸基等が挙げられるが、解離定数の観点から、カルボキシ基、スルホン酸基が好ましく、カルボキシ基がより好ましい。
核酸結合性官能基の含有量は、単離効率の観点から、担体の固形分1mgあたり、好ましくは1〜2000nmol、より好ましくは5〜1000nmol、特に好ましくは10〜100nmolである。なお、核酸結合性官能基の含有量は、電気伝導度測定法等により測定できる。特に、第1の核酸結合性担体にDNAを結合させてDNA結合担体とする場合において、核酸結合性官能基の含有量を10〜100nmolとすることで、単離効率が大幅に改善される。
核酸結合性官能基の含有量は、単離効率の観点から、担体の固形分1mgあたり、好ましくは1〜2000nmol、より好ましくは5〜1000nmol、特に好ましくは10〜100nmolである。なお、核酸結合性官能基の含有量は、電気伝導度測定法等により測定できる。特に、第1の核酸結合性担体にDNAを結合させてDNA結合担体とする場合において、核酸結合性官能基の含有量を10〜100nmolとすることで、単離効率が大幅に改善される。
また、第1の核酸結合性担体の形態は特に限定されるものではなく、粒子、モノリス、膜、繊維、チップ、粉末、容器、プレート等のいずれでもよいが、取り扱いやすさの観点から、粒子が好ましい。本発明において、第1の核酸結合性担体としては、分離の容易性の観点から、磁性粒子が好ましい。磁性粒子としては、核酸結合性官能基を表面に有する樹脂中に磁性体を含む磁性粒子(以下、単に、樹脂中に磁性体を含む磁性粒子という)が好ましい。
磁性体は、強磁性、常磁性、超常磁性のいずれであってもよいが、集磁や再分散の容易性の観点から、超常磁性であることが好ましい。磁性体としては、フェライト、酸化鉄、鉄、酸化マンガン、マンガン、酸化ニッケル、ニッケル、酸化コバルト、コバルト等の金属、または合金が挙げられる。
磁性体は、強磁性、常磁性、超常磁性のいずれであってもよいが、集磁や再分散の容易性の観点から、超常磁性であることが好ましい。磁性体としては、フェライト、酸化鉄、鉄、酸化マンガン、マンガン、酸化ニッケル、ニッケル、酸化コバルト、コバルト等の金属、または合金が挙げられる。
また、樹脂中に磁性体を含む磁性粒子としては、具体的には、以下の(i)〜(iv)のような粒子が挙げられる。
(i)樹脂等の非磁性体を含む連続相中に磁性体微粒子が分散している粒子
(ii)磁性体微粒子の2次凝集体をコアとし、樹脂等の非磁性体をシェルとする粒子
(iii)樹脂等の非磁性体で構成される核粒子と、該核粒子の表面に設けられた磁性体微粒子を含む磁性体層(2次凝集体層)とを有する母粒子をコアとし、該母粒子の最外層に、樹脂等の非磁性体層がシェルとして設けられた粒子
(iv)粒子の最外層に樹脂等の非磁性体層がシェルとして設けられていてもよい、樹脂やシリカ等からなる多孔質粒子の孔内に磁性体微粒子が分散している粒子
なお、(i)〜(iv)の粒子はいずれも公知であり、常法に従い製造可能である。
(ii)磁性体微粒子の2次凝集体をコアとし、樹脂等の非磁性体をシェルとする粒子
(iii)樹脂等の非磁性体で構成される核粒子と、該核粒子の表面に設けられた磁性体微粒子を含む磁性体層(2次凝集体層)とを有する母粒子をコアとし、該母粒子の最外層に、樹脂等の非磁性体層がシェルとして設けられた粒子
(iv)粒子の最外層に樹脂等の非磁性体層がシェルとして設けられていてもよい、樹脂やシリカ等からなる多孔質粒子の孔内に磁性体微粒子が分散している粒子
なお、(i)〜(iv)の粒子はいずれも公知であり、常法に従い製造可能である。
また、第1の核酸結合性担体が粒子の場合の平均粒径(体積平均粒径)は、好ましくは0.1〜500μmであり、より好ましくは0.2〜100μmであり、特に好ましくは0.3〜50μmである。なお、平均粒径は、レーザー回析・散乱粒子径分布測定等により測定できる。
斯様な第1の核酸結合性担体の中でも、磁性粒子が好ましく、核酸結合性官能基を有する非シリカ系磁性粒子がより好ましい。
第1の核酸結合性担体の使用量は、生体試料100質量部に対し、通常、0.01〜100質量部程度であり、好ましくは0.1〜10質量部である。
工程(A)における液相としては、第1の核酸結合性担体にRNAを結合させてRNA結合担体とする場合は、リチウム塩が添加されたものが好ましい。また、第1の核酸結合性担体にDNAを結合させてDNA結合担体とする場合は、ポリアルキレングリコールが添加されたものが好ましい。
リチウム塩は、液相中にリチウムイオンを生じさせるものであれば特に限定されるものではなく、リチウムの無機塩でも、リチウムの有機塩でもよい。例えば、塩化リチウム、酢酸リチウム、クエン酸リチウム、炭酸リチウム、水酸化リチウム、ホウ酸リチウム等が挙げられ、塩化リチウム、酢酸リチウムが好ましい。リチウム塩は、1種を単独で使用しても2種以上を組み合わせて使用してもよい。
リチウム塩の系中の濃度は、通常1〜10Mであり、好ましくは3〜8Mである。
ポリアルキレングリコールとしては、例えば、ポリエチレングリコール、ポリプロピレングリコール、プルロニック系界面活性剤のようなポリエチレングリコールとポリプロピレングリコールとのABA型ブロック共重合体等が挙げられ、1種を単独で使用しても2種以上を組み合わせて使用してもよい。ポリアルキレングリコールの重量平均分子量としては、1000〜100000が好ましく、6000〜10000がより好ましい。斯かる重量平均分子量はNMR等で測定できる。
ポリアルキレングリコールの系中の濃度は、好ましくは5〜10(w/v)%である。
リチウム塩は、液相中にリチウムイオンを生じさせるものであれば特に限定されるものではなく、リチウムの無機塩でも、リチウムの有機塩でもよい。例えば、塩化リチウム、酢酸リチウム、クエン酸リチウム、炭酸リチウム、水酸化リチウム、ホウ酸リチウム等が挙げられ、塩化リチウム、酢酸リチウムが好ましい。リチウム塩は、1種を単独で使用しても2種以上を組み合わせて使用してもよい。
リチウム塩の系中の濃度は、通常1〜10Mであり、好ましくは3〜8Mである。
ポリアルキレングリコールとしては、例えば、ポリエチレングリコール、ポリプロピレングリコール、プルロニック系界面活性剤のようなポリエチレングリコールとポリプロピレングリコールとのABA型ブロック共重合体等が挙げられ、1種を単独で使用しても2種以上を組み合わせて使用してもよい。ポリアルキレングリコールの重量平均分子量としては、1000〜100000が好ましく、6000〜10000がより好ましい。斯かる重量平均分子量はNMR等で測定できる。
ポリアルキレングリコールの系中の濃度は、好ましくは5〜10(w/v)%である。
また、工程(A)における液相としては、カオトロピック剤が添加されたものが好ましい。カオトロピック剤としては、例えば、グアニジウム塩、尿素、ヨウ化物、過塩素酸塩、チオシアン酸塩、イソチオシアン酸塩が挙げられ、1種を単独で使用しても2種以上を組み合わせて使用してもよい。これらの中でも、カオトロピック剤としては、グアニジウム塩が好ましい。グアニジウム塩としては、塩酸グアニジウム、酢酸グアニジウム、リン酸グアニジウム、チオシアン酸グアニジウム、イソチオシアン酸グアニジウム、硫酸グアニジウム、炭酸グアニジウムが挙げられ、塩酸グアニジウム、酢酸グアニジウムが特に好ましい。
カオトロピック剤の系中の濃度は、好ましくは3〜8Mである。
カオトロピック剤の系中の濃度は、好ましくは3〜8Mである。
また、第1の核酸結合性担体にRNAを結合させてRNA結合担体とする場合は、工程(A)の混合を酸性条件下で行うのが好ましく、pH6.0以下で行うのがより好ましく、pH3.0〜6.0で行うのが更に好ましく、pH4.0〜5.5で行うのが特に好ましい。一方、第1の核酸結合性担体にDNAを結合させてDNA結合担体とする場合は、中性条件下またはアルカリ性条件下で行うのが好ましく、pH6.0〜9.0で行うのがより好ましく、pH6.5〜7.5で行うのが特に好ましい。pHの調整は、各種緩衝液を用いて行えばよい。
なお、本明細書においてpHは25℃におけるpHを意味する。
なお、本明細書においてpHは25℃におけるpHを意味する。
また、工程(A)における液相は、上記各成分の他に、水、界面活性剤等を含んでいてもよい。
界面活性剤としては、非イオン性界面活性剤、アニオン界面活性剤、カチオン界面活性剤等が挙げられるが、非イオン性界面活性剤が好ましい。非イオン性界面活性剤としては、例えば、ポリオキシエチレンオクチルフェニルエーテル(Triton X−100等)、ポリオキシエチレンモノラウリン酸ソルビタン(Tween 20等)が挙げられる。なお、界面活性剤は、1種を単独で使用しても2種以上を組み合わせて使用してもよい。
界面活性剤としては、非イオン性界面活性剤、アニオン界面活性剤、カチオン界面活性剤等が挙げられるが、非イオン性界面活性剤が好ましい。非イオン性界面活性剤としては、例えば、ポリオキシエチレンオクチルフェニルエーテル(Triton X−100等)、ポリオキシエチレンモノラウリン酸ソルビタン(Tween 20等)が挙げられる。なお、界面活性剤は、1種を単独で使用しても2種以上を組み合わせて使用してもよい。
工程(A)の混合をする際の温度は、通常10〜60℃であり、混合時間は、1〜10分間程度でよい。
〔工程(B)〕
工程(B)は、工程(A)で得られた第1の核酸結合担体と上清とを分離する工程である。具体的には、遠心分離や磁場を利用した分離などの常法の固液分離を行い、上清または第1の核酸結合担体を回収するなどすればよい。
また、工程(A)における液相が、リチウム塩が添加されたものである場合は、工程(B)により、RNA結合担体とDNAを含む上清とに分離される。一方、工程(A)における液相が、ポリアルキレングリコールが添加されたものである場合は、工程(B)により、DNA結合担体とRNAを含む上清とに分離される。
工程(B)は、工程(A)で得られた第1の核酸結合担体と上清とを分離する工程である。具体的には、遠心分離や磁場を利用した分離などの常法の固液分離を行い、上清または第1の核酸結合担体を回収するなどすればよい。
また、工程(A)における液相が、リチウム塩が添加されたものである場合は、工程(B)により、RNA結合担体とDNAを含む上清とに分離される。一方、工程(A)における液相が、ポリアルキレングリコールが添加されたものである場合は、工程(B)により、DNA結合担体とRNAを含む上清とに分離される。
工程(B)で分離した第1の核酸結合担体は、工程(C)に先立ち、洗浄しておくのが好ましい(以下、この工程を洗浄工程(α)という)。
洗浄工程(α)の手法は、通常、担体の形状により2種類に分けられる。磁性粒子のように担体が粒子状である場合には、洗浄液中に粒子を分散させて洗浄する方法が挙げられ、一方、担体がプレートのような形態である場合には、その表面に洗浄液を接触させて洗浄する方法が挙げられる。
担体が磁性粒子の場合の洗浄工程(α)としては、磁場を利用して磁性粒子を集めて磁性粒子と液相とを分離し、次いで磁性粒子を洗浄液中に再分散させる方法が好ましい。
なお、洗浄工程(α)の回数は、1回でも複数回でもよい。
洗浄工程(α)の手法は、通常、担体の形状により2種類に分けられる。磁性粒子のように担体が粒子状である場合には、洗浄液中に粒子を分散させて洗浄する方法が挙げられ、一方、担体がプレートのような形態である場合には、その表面に洗浄液を接触させて洗浄する方法が挙げられる。
担体が磁性粒子の場合の洗浄工程(α)としては、磁場を利用して磁性粒子を集めて磁性粒子と液相とを分離し、次いで磁性粒子を洗浄液中に再分散させる方法が好ましい。
なお、洗浄工程(α)の回数は、1回でも複数回でもよい。
洗浄液としては、リチウム塩、界面活性剤および低級アルコールから選ばれる1種以上を含む洗浄液が好ましい。特に、RNA結合担体を洗浄する場合は、リチウム塩および界面活性剤から選ばれる1種以上を含む洗浄液や、低級アルコールを含む洗浄液が好ましく、DNA結合担体を洗浄する場合は、低級アルコールを含む洗浄液が好ましい。
上記リチウム塩、界面活性剤としては、工程(A)で使用できるものと同様のものが挙げられる。また、低級アルコールとしては、メタノール、エタノール、プロパノール、イソプロパノール、1−ブタノール、2−ブタノール、1−ペンタノール、2−ペンタノール等が挙げられ、1種を単独で使用しても2種以上を組み合わせて使用してもよい。中でも、エタノールが好ましい。
また、RNA結合担体を洗浄する場合、洗浄液は、酸性であるのが好ましく、pH6.0以下であるのがより好ましく、pH3.0〜6.0であるのが更に好ましく、pH4.0〜5.5であるのが特に好ましい。
上記リチウム塩、界面活性剤としては、工程(A)で使用できるものと同様のものが挙げられる。また、低級アルコールとしては、メタノール、エタノール、プロパノール、イソプロパノール、1−ブタノール、2−ブタノール、1−ペンタノール、2−ペンタノール等が挙げられ、1種を単独で使用しても2種以上を組み合わせて使用してもよい。中でも、エタノールが好ましい。
また、RNA結合担体を洗浄する場合、洗浄液は、酸性であるのが好ましく、pH6.0以下であるのがより好ましく、pH3.0〜6.0であるのが更に好ましく、pH4.0〜5.5であるのが特に好ましい。
〔工程(C)〕
工程(C)は、工程(B)で分離された第1の核酸結合担体から、DNAおよびRNAのうちいずれか一方を溶出させる工程である。工程(C)は常法の核酸溶出に準じて行えばよい。溶出方法としては、例えば、工程(B)で分離された第1の核酸結合担体を、各種緩衝液や水等の水系媒体中で撹拌する方法が挙げられる。その後、遠心分離や磁場を利用した分離などの常法の固液分離を行い、必要に応じて水系媒体を除去することで、DNAおよびRNAのうちいずれか一方を高純度で単離することができる。
また、工程(A)における液相が、リチウム塩が添加されたものである場合は、工程(C)により、RNA結合担体からRNAが溶出する。一方、工程(A)における液相が、ポリアルキレングリコールが添加されたものである場合は、工程(C)により、DNA結合担体からDNA溶出する。
工程(C)は、工程(B)で分離された第1の核酸結合担体から、DNAおよびRNAのうちいずれか一方を溶出させる工程である。工程(C)は常法の核酸溶出に準じて行えばよい。溶出方法としては、例えば、工程(B)で分離された第1の核酸結合担体を、各種緩衝液や水等の水系媒体中で撹拌する方法が挙げられる。その後、遠心分離や磁場を利用した分離などの常法の固液分離を行い、必要に応じて水系媒体を除去することで、DNAおよびRNAのうちいずれか一方を高純度で単離することができる。
また、工程(A)における液相が、リチウム塩が添加されたものである場合は、工程(C)により、RNA結合担体からRNAが溶出する。一方、工程(A)における液相が、ポリアルキレングリコールが添加されたものである場合は、工程(C)により、DNA結合担体からDNA溶出する。
〔工程(D)〕
工程(D)は、工程(B)で分離された上清と第2の核酸結合性担体とを液相中で混合する工程である。斯かる工程(D)により、RNAおよびDNAのうち工程(C)で溶出させなかった核酸(工程(B)で分離された上清に含まれる核酸)が第2の核酸結合性担体に結合し、第2の核酸結合担体が形成される。
工程(D)は、工程(B)で分離された上清と第2の核酸結合性担体とを液相中で混合する工程である。斯かる工程(D)により、RNAおよびDNAのうち工程(C)で溶出させなかった核酸(工程(B)で分離された上清に含まれる核酸)が第2の核酸結合性担体に結合し、第2の核酸結合担体が形成される。
第2の核酸結合性担体としては、第1の核酸結合性担体と同様のものが挙げられる。中でも、磁性粒子が好ましく、核酸結合性官能基を有する非シリカ系磁性粒子がより好ましい。
第2の核酸結合性担体の使用量は、工程(B)で分離された上清に含まれる核酸が十分に結合できる量であればよく、例えば、上清100質量部に対し、通常、0.01〜100質量部程度であり、好ましくは0.1〜10質量部である。
第2の核酸結合性担体の使用量は、工程(B)で分離された上清に含まれる核酸が十分に結合できる量であればよく、例えば、上清100質量部に対し、通常、0.01〜100質量部程度であり、好ましくは0.1〜10質量部である。
工程(D)における液相としては、第2の核酸結合性担体にDNAを結合させてDNA結合担体とする場合は、低級アルコールが添加されたものが好ましい。また、第2の核酸結合性担体にRNAを結合させてRNA結合担体とする場合は、低級アルコールまたはリチウム塩が添加されたものが好ましく、低級アルコールが添加されたものがより好ましい。
リチウム塩としては、工程(A)で使用できるものと同様のものが挙げられる。
リチウム塩を使用する場合、リチウム塩の系中の濃度は、好ましくは3〜8Mである。
リチウム塩を使用する場合、リチウム塩の系中の濃度は、好ましくは3〜8Mである。
低級アルコールとしては、メタノール、エタノール、プロパノール、イソプロパノール、1−ブタノール、2−ブタノール、1−ペンタノール、2−ペンタノール等が挙げられ、1種を単独で使用しても2種以上を組み合わせて使用してもよい。これらの中でも、エタノール、プロパノール、イソプロパノールが好ましく、イソプロパノールが特に好ましい。
低級アルコールを使用する場合、低級アルコールの系中の濃度は、好ましくは30〜60(w/v)%である。
低級アルコールを使用する場合、低級アルコールの系中の濃度は、好ましくは30〜60(w/v)%である。
また、工程(D)における液相は、上記各成分の他に、水等を含んでいてもよい。
工程(D)の混合をする際の温度は、通常10〜60℃であり、混合時間は、1〜10分間程度でよい。
〔工程(E)〕
工程(E)は、工程(D)で得られた第2の核酸結合担体と上清とを分離する工程である。具体的には、遠心分離や磁場を利用した分離などの常法の固液分離を行い、上清または第2の核酸結合担体を回収するなどすればよい。
また、工程(A)における液相が、リチウム塩が添加されたものである場合は、工程(B)で分離された上清中のDNAが、工程(D)で担体に結合し、工程(E)により、DNA結合担体が分離される。一方、工程(A)における液相が、ポリアルキレングリコールが添加されたものである場合は、工程(B)で分離された上清中のRNAが、工程(D)で担体に結合し、工程(E)により、RNA結合担体が分離される。
工程(E)は、工程(D)で得られた第2の核酸結合担体と上清とを分離する工程である。具体的には、遠心分離や磁場を利用した分離などの常法の固液分離を行い、上清または第2の核酸結合担体を回収するなどすればよい。
また、工程(A)における液相が、リチウム塩が添加されたものである場合は、工程(B)で分離された上清中のDNAが、工程(D)で担体に結合し、工程(E)により、DNA結合担体が分離される。一方、工程(A)における液相が、ポリアルキレングリコールが添加されたものである場合は、工程(B)で分離された上清中のRNAが、工程(D)で担体に結合し、工程(E)により、RNA結合担体が分離される。
工程(E)で分離した第2の核酸結合担体は、工程(F)に先立ち、洗浄しておくのが好ましい(以下、この工程を洗浄工程(β)という)。
洗浄工程(β)の手法は、通常、担体の形状により2種類に分けられる。磁性粒子のように担体が粒子状である場合には、洗浄液中に粒子を分散させて洗浄する方法が挙げられ、一方、担体がプレートのような形態である場合には、その表面に洗浄液を接触させて洗浄する方法が挙げられる。
担体が磁性粒子の場合の洗浄工程(β)としては、磁場を利用して磁性粒子を集めて磁性粒子と液相とを分離し、次いで磁性粒子を洗浄液中に再分散させる方法が好ましい。
なお、洗浄工程(β)の回数は、1回でも複数回でもよい。
洗浄工程(β)の手法は、通常、担体の形状により2種類に分けられる。磁性粒子のように担体が粒子状である場合には、洗浄液中に粒子を分散させて洗浄する方法が挙げられ、一方、担体がプレートのような形態である場合には、その表面に洗浄液を接触させて洗浄する方法が挙げられる。
担体が磁性粒子の場合の洗浄工程(β)としては、磁場を利用して磁性粒子を集めて磁性粒子と液相とを分離し、次いで磁性粒子を洗浄液中に再分散させる方法が好ましい。
なお、洗浄工程(β)の回数は、1回でも複数回でもよい。
洗浄液としては、リチウム塩、界面活性剤および低級アルコールから選ばれる1種以上を含む洗浄液が好ましく、低級アルコールを含む洗浄液が好ましい。
上記リチウム塩、界面活性剤としては、工程(A)で使用できるものと同様のものが挙げられる。また、低級アルコールとしては、洗浄工程(α)で使用できる低級アルコールと同様のものが挙げられる。
上記リチウム塩、界面活性剤としては、工程(A)で使用できるものと同様のものが挙げられる。また、低級アルコールとしては、洗浄工程(α)で使用できる低級アルコールと同様のものが挙げられる。
〔工程(F)〕
工程(F)は、工程(E)で分離された第2の核酸結合担体から、RNAおよびDNAのうち工程(C)で溶出させなかった核酸を溶出させる工程である。工程(F)は常法の核酸溶出に準じて行えばよい。溶出方法としては、例えば、工程(E)で分離された第2の核酸結合担体を、各種緩衝液や水等の水系媒体中で撹拌する方法が挙げられる。その後、遠心分離や磁場を利用した分離などの常法の固液分離を行い、必要に応じて水系媒体を除去することで、RNAおよびDNAのうち工程(C)で溶出させなかった核酸を高純度で単離することができる。
工程(F)は、工程(E)で分離された第2の核酸結合担体から、RNAおよびDNAのうち工程(C)で溶出させなかった核酸を溶出させる工程である。工程(F)は常法の核酸溶出に準じて行えばよい。溶出方法としては、例えば、工程(E)で分離された第2の核酸結合担体を、各種緩衝液や水等の水系媒体中で撹拌する方法が挙げられる。その後、遠心分離や磁場を利用した分離などの常法の固液分離を行い、必要に応じて水系媒体を除去することで、RNAおよびDNAのうち工程(C)で溶出させなかった核酸を高純度で単離することができる。
そして、上記工程(A)〜(F)を含む本発明の単離方法によれば、DNAおよびRNAを含む生体試料からDNAとRNAの両方を簡便且つ効率良く単離できる。
本発明の単離方法で単離されたDNAにはRNAの混入が少なく、また、単離されたRNAへのDNAの混入も少ないため、本発明の単離方法によれば、核酸分解酵素(リボヌクレアーゼやデオキシリボヌクレアーゼ)を使用するなどして混入した核酸を分解させなくても、DNAとRNAの両方を高純度で得ることができる。
本発明の単離方法で単離されたDNAにはRNAの混入が少なく、また、単離されたRNAへのDNAの混入も少ないため、本発明の単離方法によれば、核酸分解酵素(リボヌクレアーゼやデオキシリボヌクレアーゼ)を使用するなどして混入した核酸を分解させなくても、DNAとRNAの両方を高純度で得ることができる。
以下、実施例を挙げて本発明を詳細に説明するが、本発明はこれら実施例に限定されるものではない。
〔実施例1〕
(1)HT29細胞の培養
牛胎児血清を1(w/v)%含有するMcCoy’s 5A培地(Thermo社製)にて、ヒト結腸腺ガン細胞であるHT29細胞を37℃で3日間培養した。その後、培地を取り除き、EDTA2mM含有PBSを1mL添加して37℃で10分間静置し、シャーレから細胞を剥がした。剥がした細胞をD−PBS(−)(日水製薬社製)に懸濁させた後、この懸濁液中の細胞数を血球計算盤を用いて計測し、マイクロチューブ(チューブAとする)内で、HT29細胞の濃度が1×106個/mLとなるようにD−PBS(−)を用いて希釈した。この細胞希釈液を、細胞希釈液Aとする。
(1)HT29細胞の培養
牛胎児血清を1(w/v)%含有するMcCoy’s 5A培地(Thermo社製)にて、ヒト結腸腺ガン細胞であるHT29細胞を37℃で3日間培養した。その後、培地を取り除き、EDTA2mM含有PBSを1mL添加して37℃で10分間静置し、シャーレから細胞を剥がした。剥がした細胞をD−PBS(−)(日水製薬社製)に懸濁させた後、この懸濁液中の細胞数を血球計算盤を用いて計測し、マイクロチューブ(チューブAとする)内で、HT29細胞の濃度が1×106個/mLとなるようにD−PBS(−)を用いて希釈した。この細胞希釈液を、細胞希釈液Aとする。
(2)RNA抽出
チューブA中で、手順(1)で得た細胞希釈液A(HT29細胞1×105個相当量)に、溶解液(グアニジン塩酸塩:6M、酢酸ナトリウム−塩酸緩衝液(pH4.0):0.2M、Triton X−100:0.1(w/v)%を含む)を200μL加え、細胞を完全に溶解させた。これに、磁性粒子(Dynabeads MyOne Calboxyl、Life Technologies社製)を1mg含む水懸濁液100μLと、8M塩化リチウム溶液(溶媒:水)200μLとを加え、混合した後、室温で10分間静置した。
次に、チューブAを磁気スタンドに設置し、磁性粒子を集磁させ、上清(上清Aとする)をマイクロピペットにて別のマイクロチューブ(チューブBとする)へと移した。
そして、チューブAに、洗浄液(塩化リチウム:3M、酢酸ナトリウム−塩酸緩衝液(pH4.0):0.2M、Triton X−100:0.1(w/v)%を含む)を500μL加え、磁気スタンドから外し、撹拌後、再度磁気スタンドに設置し、磁性粒子を集磁させ、上清をマイクロピペットにて除去した。この洗浄作業をもう一度繰り返したのち、70%エタノール1mLによる洗浄を3回繰り返したのち、液相を完全に除去した。チューブAに水50μLを加え、磁気スタンドから外し、撹拌することで磁性粒子を完全に分散させた。その後、チューブAを磁気スタンドに設置し、磁性粒子を集磁させ、上清(上清Bとする)をマイクロピペットで回収した。
チューブA中で、手順(1)で得た細胞希釈液A(HT29細胞1×105個相当量)に、溶解液(グアニジン塩酸塩:6M、酢酸ナトリウム−塩酸緩衝液(pH4.0):0.2M、Triton X−100:0.1(w/v)%を含む)を200μL加え、細胞を完全に溶解させた。これに、磁性粒子(Dynabeads MyOne Calboxyl、Life Technologies社製)を1mg含む水懸濁液100μLと、8M塩化リチウム溶液(溶媒:水)200μLとを加え、混合した後、室温で10分間静置した。
次に、チューブAを磁気スタンドに設置し、磁性粒子を集磁させ、上清(上清Aとする)をマイクロピペットにて別のマイクロチューブ(チューブBとする)へと移した。
そして、チューブAに、洗浄液(塩化リチウム:3M、酢酸ナトリウム−塩酸緩衝液(pH4.0):0.2M、Triton X−100:0.1(w/v)%を含む)を500μL加え、磁気スタンドから外し、撹拌後、再度磁気スタンドに設置し、磁性粒子を集磁させ、上清をマイクロピペットにて除去した。この洗浄作業をもう一度繰り返したのち、70%エタノール1mLによる洗浄を3回繰り返したのち、液相を完全に除去した。チューブAに水50μLを加え、磁気スタンドから外し、撹拌することで磁性粒子を完全に分散させた。その後、チューブAを磁気スタンドに設置し、磁性粒子を集磁させ、上清(上清Bとする)をマイクロピペットで回収した。
(3)DNA抽出
手順(2)にてチューブBへと移した上清A500μLに、2−プロパノール300μLと、磁性粒子(Dynabeads MyOne Calboxyl、Life Technologies社製)を1mg含む水懸濁液100μLとを加え、混合した後、室温で10分間静置した。次に、チューブBを磁気スタンドに設置し、磁性粒子を集磁させ、上清をマイクロピペットにて除去した。
そして、チューブBに、70%エタノールを1mL加え、磁気スタンドから外し、撹拌後、再度磁気スタンドに設置し、磁性粒子を集磁させ、上清をマイクロピペットにて除去した。この洗浄作業を計3回繰り返したのち、液相を完全に除去した。チューブBに水100μLを加え、磁気スタンドから外し、撹拌することで磁性粒子を完全に分散させた。その後、チューブBを磁気スタンドに設置し、磁性粒子を集磁させ、上清(上清Cとする)をマイクロピペットで回収した。
手順(2)にてチューブBへと移した上清A500μLに、2−プロパノール300μLと、磁性粒子(Dynabeads MyOne Calboxyl、Life Technologies社製)を1mg含む水懸濁液100μLとを加え、混合した後、室温で10分間静置した。次に、チューブBを磁気スタンドに設置し、磁性粒子を集磁させ、上清をマイクロピペットにて除去した。
そして、チューブBに、70%エタノールを1mL加え、磁気スタンドから外し、撹拌後、再度磁気スタンドに設置し、磁性粒子を集磁させ、上清をマイクロピペットにて除去した。この洗浄作業を計3回繰り返したのち、液相を完全に除去した。チューブBに水100μLを加え、磁気スタンドから外し、撹拌することで磁性粒子を完全に分散させた。その後、チューブBを磁気スタンドに設置し、磁性粒子を集磁させ、上清(上清Cとする)をマイクロピペットで回収した。
(4)逆転写PCRによるcDNAの合成
手順(2)で得られた上清Bに含まれるRNAから、トランスクリプターファーストストランドcDNA合成キット(Roche社製)を使用して逆転写PCRによりHT29細胞に定常的に発現しているcDNAを合成した。具体的手順を以下に示す。
まず、上清B10μLに、逆転写酵素、RNaseインヒビター、デオキシヌクレオチドミックス、オリゴ[dT]18プライマーおよびトランスクリプター反応用バッファーを、キットに添付されたマニュアル通りの濃度になるよう加え、液量が20μLとなるよう調整し、55℃で30分間処理した後、85℃で5分間更に処理し、cDNAを合成した後、氷上で冷却した。
手順(2)で得られた上清Bに含まれるRNAから、トランスクリプターファーストストランドcDNA合成キット(Roche社製)を使用して逆転写PCRによりHT29細胞に定常的に発現しているcDNAを合成した。具体的手順を以下に示す。
まず、上清B10μLに、逆転写酵素、RNaseインヒビター、デオキシヌクレオチドミックス、オリゴ[dT]18プライマーおよびトランスクリプター反応用バッファーを、キットに添付されたマニュアル通りの濃度になるよう加え、液量が20μLとなるよう調整し、55℃で30分間処理した後、85℃で5分間更に処理し、cDNAを合成した後、氷上で冷却した。
(5)抽出RNAの解析
手順(4)にて合成したcDNAを鋳型とし、リアルタイムPCRによりβアクチンmRNAを増幅し、検出を行った。また、手順(2)で得られた上清Bを検体とし、リアルタイムPCRによりβアクチンDNAを増幅し、βアクチンDNAの混入の有無を検証した。具体的手順を以下に示す。
まず、cDNA10μLまたは上清B10μLと、以下の組成のPCR Master Mix15μLを混合した。そして、7500 Real Time PCR System(Applied Biosystems社製)を用いて、以下の条件でリアルタイムPCRを行い、Ct値を算出した。結果を表1に示す。
PCR Master Mix組成:10×MgCl含有PCRバッファー 2.5μL、PCRグレードdNTPs 1μL、Fwプライマー 0.3μL、Rvプライマー 0.3μL、Taqmanプローブ 0.075μL、ROX dye 0.125μL、FastStart Taq DNA Polymerase 0.1μL、1%BSA 2.5μL、およびPCRグレード水 8.1μL
リアルタイムPCR条件:50℃−2min→95℃−10min→(95℃−15sec、60℃−1min)×40サイクル
手順(4)にて合成したcDNAを鋳型とし、リアルタイムPCRによりβアクチンmRNAを増幅し、検出を行った。また、手順(2)で得られた上清Bを検体とし、リアルタイムPCRによりβアクチンDNAを増幅し、βアクチンDNAの混入の有無を検証した。具体的手順を以下に示す。
まず、cDNA10μLまたは上清B10μLと、以下の組成のPCR Master Mix15μLを混合した。そして、7500 Real Time PCR System(Applied Biosystems社製)を用いて、以下の条件でリアルタイムPCRを行い、Ct値を算出した。結果を表1に示す。
PCR Master Mix組成:10×MgCl含有PCRバッファー 2.5μL、PCRグレードdNTPs 1μL、Fwプライマー 0.3μL、Rvプライマー 0.3μL、Taqmanプローブ 0.075μL、ROX dye 0.125μL、FastStart Taq DNA Polymerase 0.1μL、1%BSA 2.5μL、およびPCRグレード水 8.1μL
リアルタイムPCR条件:50℃−2min→95℃−10min→(95℃−15sec、60℃−1min)×40サイクル
(6)抽出DNAの解析
手順(3)で得られた上清Cに含まれるDNAを鋳型とし、リアルタイムPCRによりβグロビンDNAを増幅し、検出を行った。また、手順(3)で得られた上清CをDNase処理した後、逆転写PCRを行い、これを検体とし、リアルタイムPCRによりβグロビンRNAを増幅し、βグロビンRNAの混入の有無を検証した。なお、逆転写PCRおよびリアルタイムPCRは、使用するプライマーをβグロビン遺伝子を増幅できるプライマーに変更した以外は手順(4)および(5)と同様にして行った。
結果を表1に示す。
手順(3)で得られた上清Cに含まれるDNAを鋳型とし、リアルタイムPCRによりβグロビンDNAを増幅し、検出を行った。また、手順(3)で得られた上清CをDNase処理した後、逆転写PCRを行い、これを検体とし、リアルタイムPCRによりβグロビンRNAを増幅し、βグロビンRNAの混入の有無を検証した。なお、逆転写PCRおよびリアルタイムPCRは、使用するプライマーをβグロビン遺伝子を増幅できるプライマーに変更した以外は手順(4)および(5)と同様にして行った。
結果を表1に示す。
〔実施例2〕
実施例1の手順(2)および(3)で使用した磁性粒子をMagnosphere MS160 Carboxyl(JSRライフサイエンス社製)に変更した以外は、実施例1と同様の操作を行いCt値を算出した。結果を表1に示す。
実施例1の手順(2)および(3)で使用した磁性粒子をMagnosphere MS160 Carboxyl(JSRライフサイエンス社製)に変更した以外は、実施例1と同様の操作を行いCt値を算出した。結果を表1に示す。
〔実施例3〕
実施例1の手順(2)および(3)で使用した磁性粒子をAMPure XP(Beckman coulter社製)に変更した以外は、実施例1と同様の操作を行いCt値を算出した。結果を表1に示す。
実施例1の手順(2)および(3)で使用した磁性粒子をAMPure XP(Beckman coulter社製)に変更した以外は、実施例1と同様の操作を行いCt値を算出した。結果を表1に示す。
〔実施例4〕
実施例1の手順(2)および(3)で使用した磁性粒子をSeraMag speedbeads(GE healthcare社製)に変更した以外は、実施例1と同様の操作を行いCt値を算出した。結果を表1に示す。
実施例1の手順(2)および(3)で使用した磁性粒子をSeraMag speedbeads(GE healthcare社製)に変更した以外は、実施例1と同様の操作を行いCt値を算出した。結果を表1に示す。
〔実施例5〕
実施例1の手順(2)で使用した8M塩化リチウム溶液(溶媒:水)200μLを8M酢酸リチウム溶液(溶媒:水)200μLに変更した以外は、実施例1と同様の操作を行いCt値を算出した。結果を表1に示す。
実施例1の手順(2)で使用した8M塩化リチウム溶液(溶媒:水)200μLを8M酢酸リチウム溶液(溶媒:水)200μLに変更した以外は、実施例1と同様の操作を行いCt値を算出した。結果を表1に示す。
〔実施例6〕
(1)DNA抽出
マイクロチューブ(チューブCとする)中で、実施例1の手順(1)と同様にして得た細胞希釈液A(HT29細胞1×105個相当量)に、溶解液(グアニジン塩酸塩:6M、トリス−塩酸緩衝液(pH7.0):0.2M、Triton X−100:0.1(w/v)%を含む)を200μL加え、細胞を完全に溶解させた。これに、磁性粒子(Magnosphere MS160 Carboxyl、JSRライフサイエンス社製)を1mg含む水懸濁液100μLと、ポリエチレングリコール(Mw=1000)30(w/v)%溶液(溶媒:水)200μLとを加え、混合した後、室温で10分間静置した。
次に、チューブCを磁気スタンドに設置し、磁性粒子を集磁させ、上清(上清Dとする)をマイクロピペットにて別のマイクロチューブ(チューブDとする)へと移した。
そして、チューブCに、70%エタノール1mLを加え、磁気スタンドから外し、撹拌後、再度磁気スタンドに設置し、磁性粒子を集磁させ、上清をマイクロピペットにて除去した。この洗浄作業を計3回繰り返したのち、液相を完全に除去した。チューブCに水100μLを加え、磁気スタンドから外し、撹拌することで磁性粒子を完全に分散させた。その後、チューブCを磁気スタンドに設置し、磁性粒子を集磁させ、上清(上清Eとする)をマイクロピペットで回収した。
(1)DNA抽出
マイクロチューブ(チューブCとする)中で、実施例1の手順(1)と同様にして得た細胞希釈液A(HT29細胞1×105個相当量)に、溶解液(グアニジン塩酸塩:6M、トリス−塩酸緩衝液(pH7.0):0.2M、Triton X−100:0.1(w/v)%を含む)を200μL加え、細胞を完全に溶解させた。これに、磁性粒子(Magnosphere MS160 Carboxyl、JSRライフサイエンス社製)を1mg含む水懸濁液100μLと、ポリエチレングリコール(Mw=1000)30(w/v)%溶液(溶媒:水)200μLとを加え、混合した後、室温で10分間静置した。
次に、チューブCを磁気スタンドに設置し、磁性粒子を集磁させ、上清(上清Dとする)をマイクロピペットにて別のマイクロチューブ(チューブDとする)へと移した。
そして、チューブCに、70%エタノール1mLを加え、磁気スタンドから外し、撹拌後、再度磁気スタンドに設置し、磁性粒子を集磁させ、上清をマイクロピペットにて除去した。この洗浄作業を計3回繰り返したのち、液相を完全に除去した。チューブCに水100μLを加え、磁気スタンドから外し、撹拌することで磁性粒子を完全に分散させた。その後、チューブCを磁気スタンドに設置し、磁性粒子を集磁させ、上清(上清Eとする)をマイクロピペットで回収した。
(2)RNA抽出
手順(1)にてチューブDへと移した上清D500μLに、2−プロパノール300μLと、磁性粒子(Magnosphere MS160 Carboxyl、JSRライフサイエンス社製)を1mg含む水懸濁液100μLとを加え、混合した後、室温で10分間静置した。次に、チューブDを磁気スタンドに設置し、磁性粒子を集磁させ、上清をマイクロピペットにて除去した。
そして、チューブDに、70%エタノールを1mL加え、磁気スタンドから外し、撹拌後、再度磁気スタンドに設置し、磁性粒子を集磁させ、上清をマイクロピペットにて除去した。この洗浄作業を計3回繰り返したのち、液相を完全に除去した。チューブDに水100μLを加え、磁気スタンドから外し、撹拌することで磁性粒子を完全に分散させた。その後、チューブDを磁気スタンドに設置し、磁性粒子を集磁させ、上清(上清Fとする)をマイクロピペットで回収した。
手順(1)にてチューブDへと移した上清D500μLに、2−プロパノール300μLと、磁性粒子(Magnosphere MS160 Carboxyl、JSRライフサイエンス社製)を1mg含む水懸濁液100μLとを加え、混合した後、室温で10分間静置した。次に、チューブDを磁気スタンドに設置し、磁性粒子を集磁させ、上清をマイクロピペットにて除去した。
そして、チューブDに、70%エタノールを1mL加え、磁気スタンドから外し、撹拌後、再度磁気スタンドに設置し、磁性粒子を集磁させ、上清をマイクロピペットにて除去した。この洗浄作業を計3回繰り返したのち、液相を完全に除去した。チューブDに水100μLを加え、磁気スタンドから外し、撹拌することで磁性粒子を完全に分散させた。その後、チューブDを磁気スタンドに設置し、磁性粒子を集磁させ、上清(上清Fとする)をマイクロピペットで回収した。
(3)DNAおよびRNAの検出
手順(1)で得た上清Eを用いて、実施例1の手順(6)と同様にして、βグロビンDNAの検出とβグロビンRNAの混入の有無の検証とを行った。また、手順(2)で得た上清Fを用いて、実施例1の手順(4)および(5)と同様にして、βアクチンmRNAの検出とβアクチンDNAの混入の有無の検証とを行った。
これら結果を表1に示す。
手順(1)で得た上清Eを用いて、実施例1の手順(6)と同様にして、βグロビンDNAの検出とβグロビンRNAの混入の有無の検証とを行った。また、手順(2)で得た上清Fを用いて、実施例1の手順(4)および(5)と同様にして、βアクチンmRNAの検出とβアクチンDNAの混入の有無の検証とを行った。
これら結果を表1に示す。
〔実施例7〕
実施例6の手順(1)および(2)で使用した磁性粒子をDynabeads MyOne Carboxyl(Life Technologies社製)に変更した以外は、実施例6と同様の操作を行いCt値を算出した。結果を表1に示す。
実施例6の手順(1)および(2)で使用した磁性粒子をDynabeads MyOne Carboxyl(Life Technologies社製)に変更した以外は、実施例6と同様の操作を行いCt値を算出した。結果を表1に示す。
〔実施例8〕
実施例6の手順(1)および(2)で使用した磁性粒子をAMPure XP(Beckman coulter社製)に変更した以外は、実施例6と同様の操作を行いCt値を算出した。結果を表1に示す。
実施例6の手順(1)および(2)で使用した磁性粒子をAMPure XP(Beckman coulter社製)に変更した以外は、実施例6と同様の操作を行いCt値を算出した。結果を表1に示す。
〔実施例9〕
実施例6の手順(1)および(2)で使用した磁性粒子をSeraMag speedbeads(GE healthcare社製)に変更した以外は、実施例6と同様の操作を行いCt値を算出した。結果を表1に示す。
実施例6の手順(1)および(2)で使用した磁性粒子をSeraMag speedbeads(GE healthcare社製)に変更した以外は、実施例6と同様の操作を行いCt値を算出した。結果を表1に示す。
〔比較例1〕
実施例1の手順(1)で使用した8M塩化リチウム溶液(溶媒:水)を2−プロパノールに、実施例1の手順(2)で使用した2−プロパノールを8M塩化リチウム溶液(溶媒:水)にそれぞれ変更した以外は、実施例1と同様の操作を行いCt値を算出した。結果を表1に示す。
実施例1の手順(1)で使用した8M塩化リチウム溶液(溶媒:水)を2−プロパノールに、実施例1の手順(2)で使用した2−プロパノールを8M塩化リチウム溶液(溶媒:水)にそれぞれ変更した以外は、実施例1と同様の操作を行いCt値を算出した。結果を表1に示す。
以下の表1に示すとおり、実施例1〜9の方法により、DNAとRNAの両方を簡便且つ効率良く単離できた。
上記実施例および比較例で用いた各磁性粒子の特性を以下に示す。
Dynabeads MyOne Calboxyl(Life Technologies社製):カルボキシ基含有非シリカ系磁性粒子(平均粒径:1.0μm、表面カルボキシ基量:1000nmol/mg beads)
Magnosphere MS160 Carboxyl(JSRライフサイエンス社製):カルボキシ基含有非シリカ系磁性粒子(平均粒径:1.5μm、磁性体含有量:約28質量%、表面カルボキシ基量:30nmol/mg beads)
AMPure XP(Beckman coulter社製):カルボキシ基含有非シリカ系磁性粒子(平均粒径:1.0μm、表面カルボキシ基量:1000nmol/mg beads)
SeraMag speedbeads(GE healthcare社製):カルボキシ基含有非シリカ系磁性粒子(平均粒径:1.0μm、表面カルボキシ基量:1000nmol/mg beads)
Dynabeads MyOne Calboxyl(Life Technologies社製):カルボキシ基含有非シリカ系磁性粒子(平均粒径:1.0μm、表面カルボキシ基量:1000nmol/mg beads)
Magnosphere MS160 Carboxyl(JSRライフサイエンス社製):カルボキシ基含有非シリカ系磁性粒子(平均粒径:1.5μm、磁性体含有量:約28質量%、表面カルボキシ基量:30nmol/mg beads)
AMPure XP(Beckman coulter社製):カルボキシ基含有非シリカ系磁性粒子(平均粒径:1.0μm、表面カルボキシ基量:1000nmol/mg beads)
SeraMag speedbeads(GE healthcare社製):カルボキシ基含有非シリカ系磁性粒子(平均粒径:1.0μm、表面カルボキシ基量:1000nmol/mg beads)
〔比較例2〕
(1)DNAおよびRNAの抽出
特表2008−518618号公報の記載を参考にして、DNAおよびRNAの抽出を行った。具体的手順を以下に示す。
まず、実施例1の手順(1)と同様にして得た細胞希釈液A(HT29細胞1×105個相当量)に、溶解液(グアニジン塩酸塩:6M、トリス−塩酸緩衝液(pH7.0):0.2M、Triton X−100:0.1(w/v)%を含む)を200μL加え、細胞を完全に溶解させた。この細胞溶解液を100μLずつ別々のマイクロチューブ(チューブEおよびFとする)に分けた。
(1)DNAおよびRNAの抽出
特表2008−518618号公報の記載を参考にして、DNAおよびRNAの抽出を行った。具体的手順を以下に示す。
まず、実施例1の手順(1)と同様にして得た細胞希釈液A(HT29細胞1×105個相当量)に、溶解液(グアニジン塩酸塩:6M、トリス−塩酸緩衝液(pH7.0):0.2M、Triton X−100:0.1(w/v)%を含む)を200μL加え、細胞を完全に溶解させた。この細胞溶解液を100μLずつ別々のマイクロチューブ(チューブEおよびFとする)に分けた。
細胞溶解液100μLを含むチューブE中に、磁性粒子(Dynabeads MyOne Calboxyl、Life Technologies社製)を1mg含む水懸濁液100μLと、8M塩化リチウム溶液(溶媒:水)200μLとを加え、混合した後、室温で10分間静置した。
そして、チューブEに、洗浄液(塩化リチウム:3M、酢酸ナトリウム−塩酸緩衝液(pH4.0):0.2M、Triton X−100:0.1(w/v)%を含む)を500μL加え、磁気スタンドから外し、撹拌後、再度磁気スタンドに設置し、磁性粒子を集磁させ、上清をマイクロピペットにて除去した。この洗浄作業をもう一度繰り返したのち、70%エタノール1mLによる洗浄を3回繰り返したのち、液相を完全に除去した。チューブEに水50μLを加え、磁気スタンドから外し、撹拌することで磁性粒子を完全に分散させた。その後、チューブEを磁気スタンドに設置し、磁性粒子を集磁させ、上清(上清Gとする)をマイクロピペットで回収した。
そして、チューブEに、洗浄液(塩化リチウム:3M、酢酸ナトリウム−塩酸緩衝液(pH4.0):0.2M、Triton X−100:0.1(w/v)%を含む)を500μL加え、磁気スタンドから外し、撹拌後、再度磁気スタンドに設置し、磁性粒子を集磁させ、上清をマイクロピペットにて除去した。この洗浄作業をもう一度繰り返したのち、70%エタノール1mLによる洗浄を3回繰り返したのち、液相を完全に除去した。チューブEに水50μLを加え、磁気スタンドから外し、撹拌することで磁性粒子を完全に分散させた。その後、チューブEを磁気スタンドに設置し、磁性粒子を集磁させ、上清(上清Gとする)をマイクロピペットで回収した。
細胞溶解液100μLを含むチューブF中に、磁性粒子(Dynabeads MyOne Calboxyl、Life Technologies社製)を1mg含む水懸濁液100μLと、8M塩化リチウム溶液(溶媒:水)200μLとを加え、混合した後、室温で10分間静置した。
次に、チューブFを磁気スタンドに設置し、磁性粒子を集磁させ、上清(上清Hとする)をマイクロピペットにて別のマイクロチューブ(チューブGとする)へと移した。
チューブGへと移した上清H500μLに、2−プロパノール300μLと、磁性粒子(Dynabeads MyOne Calboxyl、Life Technologies社製)を1mg含む水懸濁液100μLとを加え、混合した後、室温で10分間静置した。次に、チューブGを磁気スタンドに設置し、磁性粒子を集磁させ、上清をマイクロピペットにて除去した。
そして、チューブGに、70%エタノールを1mL加え、磁気スタンドから外し、撹拌後、再度磁気スタンドに設置し、磁性粒子を集磁させ、上清をマイクロピペットにて除去した。この洗浄作業を計3回繰り返したのち、液相を完全に除去した。チューブGに水100μLを加え、磁気スタンドから外し、撹拌することで磁性粒子を完全に分散させた。その後、チューブGを磁気スタンドに設置し、磁性粒子を集磁させ、上清(上清Iとする)をマイクロピペットで回収した。
次に、チューブFを磁気スタンドに設置し、磁性粒子を集磁させ、上清(上清Hとする)をマイクロピペットにて別のマイクロチューブ(チューブGとする)へと移した。
チューブGへと移した上清H500μLに、2−プロパノール300μLと、磁性粒子(Dynabeads MyOne Calboxyl、Life Technologies社製)を1mg含む水懸濁液100μLとを加え、混合した後、室温で10分間静置した。次に、チューブGを磁気スタンドに設置し、磁性粒子を集磁させ、上清をマイクロピペットにて除去した。
そして、チューブGに、70%エタノールを1mL加え、磁気スタンドから外し、撹拌後、再度磁気スタンドに設置し、磁性粒子を集磁させ、上清をマイクロピペットにて除去した。この洗浄作業を計3回繰り返したのち、液相を完全に除去した。チューブGに水100μLを加え、磁気スタンドから外し、撹拌することで磁性粒子を完全に分散させた。その後、チューブGを磁気スタンドに設置し、磁性粒子を集磁させ、上清(上清Iとする)をマイクロピペットで回収した。
(2)DNAおよびRNAの検出
手順(1)で得た上清Gを用いて、実施例1の手順(4)および(5)と同様にして、βアクチンmRNAの検出とβアクチンDNAの混入の有無の検証とを行った。また、手順(1)で得た上清Iを用いて、実施例1の手順(6)と同様にして、βグロビンDNAの検出とβグロビンRNAの混入の有無の検証とを行った。
これら結果を表2に示す。なお、DNA、RNAどちらの抽出量も、実施例1〜9の方法で単離した場合と比較して半分以下であった。
手順(1)で得た上清Gを用いて、実施例1の手順(4)および(5)と同様にして、βアクチンmRNAの検出とβアクチンDNAの混入の有無の検証とを行った。また、手順(1)で得た上清Iを用いて、実施例1の手順(6)と同様にして、βグロビンDNAの検出とβグロビンRNAの混入の有無の検証とを行った。
これら結果を表2に示す。なお、DNA、RNAどちらの抽出量も、実施例1〜9の方法で単離した場合と比較して半分以下であった。
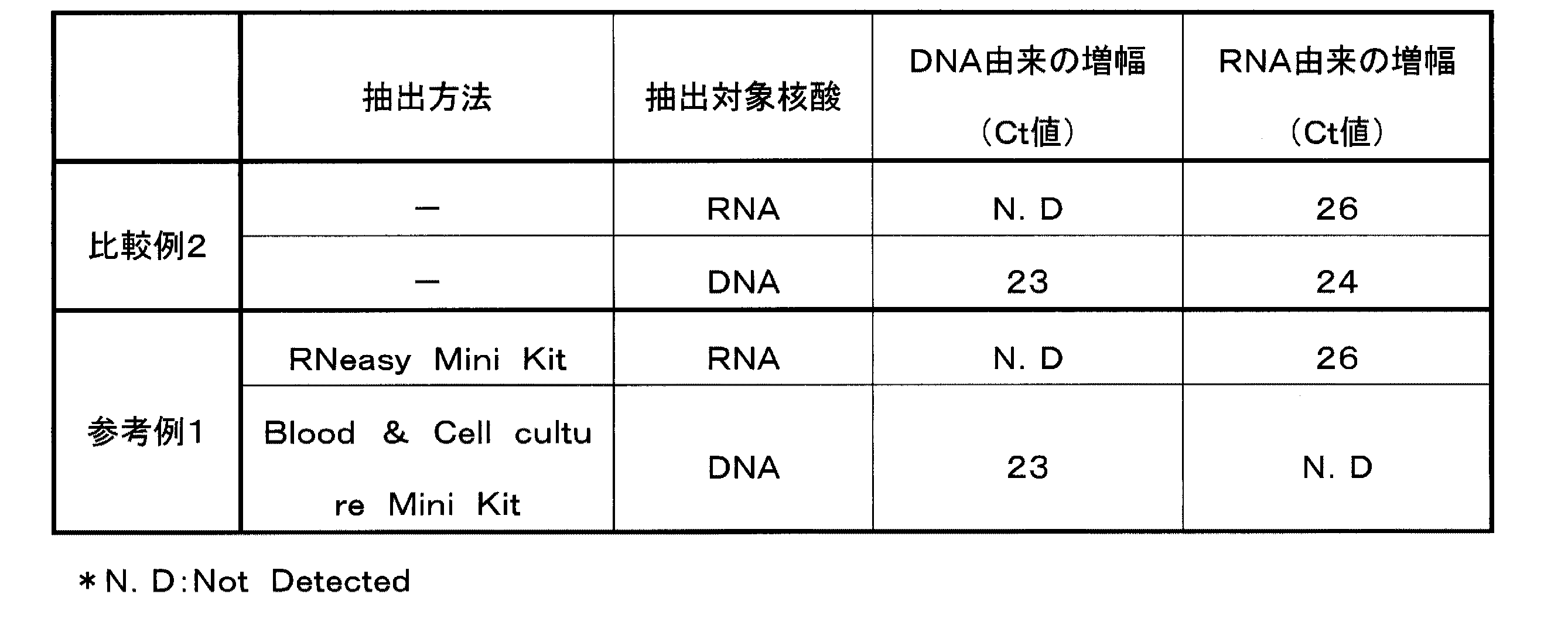
〔参考例1〕
(1)DNAおよびRNAの抽出
実施例1の手順(1)と同様にして得た細胞希釈液A(HT29細胞1×105個相当量)に、溶解液(グアニジン塩酸塩:6M、トリス−塩酸緩衝液(pH7.0):0.2M、Triton X−100:0.1(w/v)%を含む)を200μL加え、細胞を完全に溶解させた。この細胞溶解液を100μLずつ別々のマイクロチューブ(チューブHおよびIとする)に分けた。
チューブHに含まれる細胞溶解液100μLから、Blood & Cell culture DNA Mini Kit(QIAGEN社製)を用いてDNAを抽出(抽出物Aとする)した。また、チューブIに含まれる細胞溶解液100μLから、RNeasy Mini Kit(QIAGEN社製)を用いてRNAを抽出(抽出物Bとする)した。なお、Blood & Cell culture DNA Mini Kitは、RNaseを具備するものである。
(2)DNAおよびRNAの検出
手順(1)で得た抽出物Bを用いて、実施例1の手順(4)および(5)と同様にして、βアクチンmRNAの検出とβアクチンDNAの混入の有無の検証とを行った。また、手順(1)で得た抽出物Aを用いて、実施例1の手順(6)と同様にして、βグロビンDNAの検出とβグロビンRNAの混入の有無の検証とを行った。
これら結果を表2に示す。
(1)DNAおよびRNAの抽出
実施例1の手順(1)と同様にして得た細胞希釈液A(HT29細胞1×105個相当量)に、溶解液(グアニジン塩酸塩:6M、トリス−塩酸緩衝液(pH7.0):0.2M、Triton X−100:0.1(w/v)%を含む)を200μL加え、細胞を完全に溶解させた。この細胞溶解液を100μLずつ別々のマイクロチューブ(チューブHおよびIとする)に分けた。
チューブHに含まれる細胞溶解液100μLから、Blood & Cell culture DNA Mini Kit(QIAGEN社製)を用いてDNAを抽出(抽出物Aとする)した。また、チューブIに含まれる細胞溶解液100μLから、RNeasy Mini Kit(QIAGEN社製)を用いてRNAを抽出(抽出物Bとする)した。なお、Blood & Cell culture DNA Mini Kitは、RNaseを具備するものである。
(2)DNAおよびRNAの検出
手順(1)で得た抽出物Bを用いて、実施例1の手順(4)および(5)と同様にして、βアクチンmRNAの検出とβアクチンDNAの混入の有無の検証とを行った。また、手順(1)で得た抽出物Aを用いて、実施例1の手順(6)と同様にして、βグロビンDNAの検出とβグロビンRNAの混入の有無の検証とを行った。
これら結果を表2に示す。
表2に示すとおり、比較例2の方法では、抽出したDNAに多量のRNAの混入がみられ、このような方法でDNAを単離するためには参考例1で用いたようなRNaseの使用が必要で工程が増える。また、試料を半分に分ける必要があったため、抽出される核酸の量も微量だった。
Claims (14)
- DNAおよびRNAを含む同一の生体試料からDNAとRNAを単離する方法であって、以下の工程(A)〜(F)を含む、DNAとRNAの単離方法。
(A)前記生体試料と第1の核酸結合性担体とを液相中で混合する工程
(B)工程(A)で得られた第1の核酸結合担体と上清とを分離する工程
(C)工程(B)で分離された第1の核酸結合担体から、DNAおよびRNAのうちいずれか一方を溶出させる工程
(D)工程(B)で分離された上清と第2の核酸結合性担体とを液相中で混合する工程
(E)工程(D)で得られた第2の核酸結合担体と上清とを分離する工程
(F)工程(E)で分離された第2の核酸結合担体から、RNAおよびDNAのうち工程(C)で溶出させなかった核酸を溶出させる工程 - DNAおよびRNAを含む同一の生体試料からDNAとRNAを単離する方法であって、以下の工程(A)〜(F)を含む、DNAとRNAの単離方法。
(A)前記生体試料と第1の核酸結合性担体とを、リチウム塩が添加された液相中で混合する工程
(B)工程(A)で得られたRNA結合担体と上清とを分離する工程
(C)工程(B)で分離されたRNA結合担体から、RNAを溶出させる工程
(D)工程(B)で分離された上清と第2の核酸結合性担体とを、低級アルコールが添加された液相中で混合する工程
(E)工程(D)で得られたDNA結合担体と上清とを分離する工程
(F)工程(E)で分離されたDNA結合担体から、DNAを溶出させる工程 - 工程(A)を、酸性条件下で行う、請求項2に記載の単離方法。
- 前記リチウム塩が、リチウムの無機塩およびリチウムの有機塩から選ばれる1種以上である、請求項2または3に記載の単離方法。
- 前記リチウム塩が、塩化リチウム、酢酸リチウム、クエン酸リチウム、炭酸リチウム、水酸化リチウムおよびホウ酸リチウムから選ばれる1種以上である、請求項2〜4のいずれか1項に記載の単離方法。
- DNAおよびRNAを含む同一の生体試料からDNAとRNAを単離する方法であって、以下の工程(A)〜(F)を含む、DNAとRNAの単離方法。
(A)前記生体試料と第1の核酸結合性担体とを、ポリアルキレングリコールが添加された液相中で混合する工程
(B)工程(A)で得られたDNA結合担体と上清とを分離する工程
(C)工程(B)で分離されたDNA結合担体から、DNAを溶出させる工程
(D)工程(B)で分離された上清と第2の核酸結合性担体とを、低級アルコールまたはリチウム塩が添加された液相中で混合する工程
(E)工程(D)で得られたRNA結合担体と上清とを分離する工程
(F)工程(E)で分離されたRNA結合担体から、RNAを溶出させる工程 - 前記低級アルコールまたはリチウム塩が、低級アルコールである、請求項6に記載の単離方法。
- 前記ポリアルキレングリコールが、重量平均分子量6000〜10000のポリアルキレングリコールである、請求項6または7に記載の単離方法。
- 第1の核酸結合性担体および第2の核酸結合性担体が、磁性粒子である、請求項1〜8のいずれか1項に記載の単離方法。
- 第1の核酸結合性担体および第2の核酸結合性担体が、核酸結合性官能基を有する非シリカ系磁性粒子である、請求項1〜9のいずれか1項に記載の単離方法。
- 工程(A)の液相が、カオトロピック剤が添加された液相である、請求項1〜10のいずれか1項に記載の単離方法。
- 前記カオトロピック剤が、グアニジウム塩、尿素、ヨウ化物、過塩素酸塩、チオシアン酸塩およびイソチオシアン酸塩から選ばれる1種以上である、請求項11に記載の単離方法。
- 前記グアニジウム塩が、塩酸グアニジウム、酢酸グアニジウム、リン酸グアニジウム、チオシアン酸グアニジウム、イソチオシアン酸グアニジウム、硫酸グアニジウムおよび炭酸グアニジウムから選ばれる1種以上である、請求項12に記載の単離方法。
- 核酸分解酵素を使用せずにDNAとRNAを単離する方法である、請求項1〜13のいずれか1項に記載の単離方法。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2015040090A JP2016158558A (ja) | 2015-03-02 | 2015-03-02 | Dnaとrnaの単離方法 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2015040090A JP2016158558A (ja) | 2015-03-02 | 2015-03-02 | Dnaとrnaの単離方法 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| JP2016158558A true JP2016158558A (ja) | 2016-09-05 |
Family
ID=56843494
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2015040090A Pending JP2016158558A (ja) | 2015-03-02 | 2015-03-02 | Dnaとrnaの単離方法 |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP2016158558A (ja) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2021054209A1 (ja) * | 2019-09-19 | 2021-03-25 | 三洋化成工業株式会社 | 磁性粒子組成物、磁性粒子組成物の核酸分離用途での使用、磁性粒子組成物を得るためのキット、磁性粒子、カオトロピック塩及び分離精製方法 |
-
2015
- 2015-03-02 JP JP2015040090A patent/JP2016158558A/ja active Pending
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2021054209A1 (ja) * | 2019-09-19 | 2021-03-25 | 三洋化成工業株式会社 | 磁性粒子組成物、磁性粒子組成物の核酸分離用途での使用、磁性粒子組成物を得るためのキット、磁性粒子、カオトロピック塩及び分離精製方法 |
| JP7602467B2 (ja) | 2019-09-19 | 2024-12-18 | 三洋化成工業株式会社 | 磁性粒子組成物、磁性粒子組成物の核酸分離用途での使用、磁性粒子組成物を得るためのキット、磁性粒子、カオトロピック塩及び分離精製方法 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5576363B2 (ja) | 短鎖核酸を単離する方法 | |
| JP6689251B2 (ja) | Rnaを高収率で単離するための方法 | |
| EP3529374B1 (en) | Sequencing and analysis of exosome associated nucleic acids | |
| JP2022084700A (ja) | 生体液からの細胞外小胞の単離及びセルフリーdnaの同時単離のための自動及び手動方法 | |
| US9464316B2 (en) | Method for isolating nucleic acids comprising the use of ethylene glycol multimers | |
| US9416399B2 (en) | Method for purification of nucleic acids, particularly from fixed tissue | |
| US10844369B2 (en) | Isolation of cell-free nucleic acids from bodily fluid samples using solid chaotropic agents | |
| WO2016183292A1 (en) | Rapid methods for the extraction of nucleic acids from biological samples | |
| US20150299769A1 (en) | Method for lysing a fixed biological sample | |
| US20250263688A1 (en) | Method for isolating highly pure nucleic acid with magnetic particles | |
| JP2006517225A (ja) | 核酸抽出のための生物学的試料の化学処理および該処理用のキット | |
| US20230357747A1 (en) | Method for isolating rna with high yield | |
| JP7068183B2 (ja) | 単一洗浄溶出バッファー溶液を使用する核酸精製システム | |
| JP2016158558A (ja) | Dnaとrnaの単離方法 | |
| JP2001078761A (ja) | 核酸結合性磁性シリカ粒子担体 | |
| JP2016158559A (ja) | Rnaの単離方法 | |
| CN121620591A (zh) | 核酸制备 |